
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 125
Platinum
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
First draft prepared by Dr. G. Rosner, Dr. H.P. König,
and Dr. D. Coenen-Stass, Fraunhofer Institute
of Toxicology and Aerosol Research, Germany
World Health Orgnization
Geneva, 1991
The International Programme on Chemical Safety (IPCS) is a joint
venture of the United Nations Environment Programme, the International
Labour Organisation, and the World Health Organization. The main
objective of the IPCS is to carry out and disseminate evaluations of
the effects of chemicals on human health and the quality of the
environment. Supporting activities include the development of
epidemiological, experimental laboratory, and risk-assessment methods
that could produce internationally comparable results, and the
development of manpower in the field of toxicology. Other activities
carried out by the IPCS include the development of know-how for coping
with chemical accidents, coordination of laboratory testing and
epidemiological studies, and promotion of research on the mechanisms
of the biological action of chemicals.
WHO Library Cataloguing in Publication Data
Platinum.
(Environmental health criteria: 125)
1. Platinum - adverse effects 2. Platinum - toxicity
3. Environmental exposure I.Series
ISBN 92 4 157125 X (LC Classification QD 181.P8)
ISSN 0250-863X
(c) World Health Organization 1991
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. For rights of reproduction or
translation of WHO publications, in part or in toto, application
should be made to the Office of Publications, World Health
Organization, Geneva, Switzerland. The World Health Organization
welcomes such applications.
The designations employed and the presentation of the material in
this publication do not imply the impression of any opinion whatsoever
on the part of the Secretariat of the World Health Organization
concerning the legal status of every country, territory, city, or area
or of its authorities, or concerning the delimitation of its frontiers
or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar nature
that are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR PLATINUM
1. SUMMARY AND CONCLUSIONS
1.1. Identity, physical and chemical properties,
analytical methods
1.2. Sources of human and environmental exposure
1.3. Environmental transport, distribution, and
transformation
1.4. Environmental levels and human exposure
1.5. Kinetics and metabolism
1.6. Effects on laboratory mammals and in vitro
test systems
1.7. Effects on humans
1.8. Effects on other organisms in the laboratory and field
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1. Identity
2.2. Physical and chemical properties
2.2.1. Platinum metal
2.2.2. Platinum compounds
2.3. Conversion factors
2.4. Analytical methods
2.4.1. Sampling
2.4.2. Sample pretreatment
2.4.3. Detection and measurement
2.4.3.1 Spectrophotometry
2.4.3.2 Radiochemical methods
2.4.3.3 X-ray fluorescence spectroscopy
2.4.3.4 Electron spectroscopy for
chemical analysis
2.4.3.5 Electrochemical analysis
2.4.3.6 Proton-induced X-ray emission
2.4.3.7 Liquid chromatography
2.4.3.8 Atomic absorption spectrometry
2.4.3.9 Inductively coupled plasma
2.4.3.10 Inductively coupled plasma -
mass spectrometry
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Anthropogenic sources
3.2.1. Production levels and processes
3.2.1.1 World production figures
3.2.1.2 Manufacturing processes
3.2.1.3 Emissions from stationary sources
3.2.1.4 Emissions from automobile catalysts
3.2.2. Uses
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1. Transport and distribution between media
4.2. Biotransformation
4.3. Ultimate fate following use
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Ambient air
5.1.2. Water and sediments
5.1.3. Soil
5.1.4. Food
5.1.5. Terrestrial and aquatic organisms
5.2. General population exposure
5.3. Occupational exposure during manufacture,
formulation or use
6. KINETICS AND METABOLISM
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1. Single exposure
7.2. Short-term exposure
7.3. Skin and eye irritation; skin and respiratory
sensitization
7.3.1. Skin irritation
7.3.2. Eye irritation
7.3.3. Skin sensitization
7.3.4. Skin and respiratory sensitization
7.3.5. Respiratory sensitization
7.3.6. Sensitization by other routes
7.4. Reproductive toxicity, embryotoxicity, and
teratogenicity
7.5. Mutagenicity and related end-points
7.6. Carcinogenicity and anticarcinogenicity
7.7. Other special studies
7.7.1. Effects on alveolar macrophages
7.7.2. Non-allergic mediator release
7.7.3. Effects on mitochondrial function
7.7.4. Effects on the nervous system
7.7.5. Side effects on cisplatin and its analogues
7.8. Factors modifying toxicity
8. EFFECTS ON HUMANS
8.1. General population exposure
8.1.1. Acute toxicity - poisoning
8.1.2. Effects of exposure to platinum
emitted from automobile catalysts
8.2. Occupational exposure
8.2.1. Case reports and cross-sectional studies
8.2.2. Allergenicity of platinum and
platinum compounds
8.2.3. Clinical manifestations
8.2.4. Immunological mechanism and diagnosis
8.2.5. Predisposing factors
8.3. Side effects of cisplatin
8.4. Carcinogenicity
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1. Microorganisms
9.2. Aquatic organisms
9.2.1. Plants
9.2.2. Animals
9.3. Terrestrial organisms
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1. Evaluation of human health risks
10.1.1. General population exposure
10.1.1.1 Exposure
10.1.1.2 Health effects
10.1.2. Occupational groups
10.1.2.1 Exposure
10.1.2.2 Health effects
10.2. Evaluation of effects on the environment
11. RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH AND
THE ENVIRONMENT
11.1. Pre-employment screening and medical evaluations
11.2. Substitution with non-allergenic substances
11.3. Employment screening and medical evaluations
11.4. Workplace hygiene
12. FURTHER RESEARCH
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
REFERENCES
RESUME
RESUMEN
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR PLATINUM
Members
Dr V. Bencko, Institute of Hygiene, Charles University, Prague,
Czechoslovakia
Dr R.E. Biagini, Division of Biomedical and Behavioral Sciences,
National Institute for Occupational Safety & Health,
Cincinnati, Ohio, USA (Joint Rapporteur)
Dr I. Farkas, National Institute of Hygiene, Budapest, Hungary
Dr U. Heinrich, Department of Environmental Hygiene, Fraunhofer
Institute of Toxicology and Aerosol Research, Hanover, Germany
Dr R. Hertel, Fraunhofer Institute of Toxicology and Aerosol
Research, Hanover, Germany
Professor G. Kazantzis, Centre for Environmental Technology, Royal
School of Mines, London, United Kingdom
Professor A. Massoud, Department of Community, Environmental and
Occupational Medicine, Faculty of Medicine, Ain Shams
University, Cairo, Egypt (Chairman)
Dr R. Merget, Department of Internal Medicine, Hospital of the
Johann Wolfgang Goethe University, Frankfurt am Main, Germany
Dr G. Rosner, Fraunhofer Institute of Toxicology and Aerosol
Research, Hanover, Germany (Joint Rapporteur)
Dr A.E. Soyombo, Environmental & Occupational Health Division,
Federal Ministry of Health, Lagos, Nigeria (Vice-Chairman)
Observers
Dr C.W. Bradford, Environmental, Health and Safety Services,
Johnson Matthey Technology Centre, Reading, United Kingdom
Dr W.E. Mayr, Industrial Toxicology Department, Degussa AG, Hanau-
Wolfgang, Germany
Secretariat
Dr P.G. Jenkins, International Programme on Chemical Safety,
Division of Environmental Health, World Health Organization,
Geneva, Switzerland
Dr E.M. Smith, International Programme on Chemical Safety, Division
of Environmental Health, World Health Organization, Geneva,
Switzerland
NOTE TO READERS OF THE CRITERIA DOCUMENTS
Every effort has been made to present information in the
criteria documents as accurately as possible without unduly delaying
their publication. In the interest of all users of the environmental
health criteria documents, readers are kindly requested to
communicate any errors that may have occurred to the Manager of the
International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland, in order that they may be
included in corrigenda, which will appear in subsequent volumes.
* * *
A detailed data profile can be obtained from the International
Register of Potentially Toxic Chemicals, Palais des Nations, 1211
Geneva 10, Switzerland (Telephone No. 7988400 or 7985850).
ENVIRONMENTAL HEALTH CRITERIA FOR PLATINUM
The WHO Task Group on Environmental Health Criteria for
Platinum met in Rome, Italy, from 3 to 7 December 1990. Dr A. Mochi
opened the meeting on behalf of the host country and Dr E. Smith
welcomed the participants on behalf of the heads of the three IPCS
cooperating organizations (UNEP/ILO/WHO). The Task Group reviewed
and revised the draft monograph and made an evaluation of the risks
for human health and the environment from exposure to platinum and
certain platinum salts.
The first draft of this document was prepared by Dr G. Rosner,
Dr H.P. König, and Dr D. Coenen-Stass, Fraunhofer Institute for
Toxicology and Aerosol Research, Hanover, Germany. The second draft
was prepared by Dr G. Rosner following circulation of the first
draft to IPCS contact points. Particularly valuable comments on the
draft were made by the European Chemical Industry Ecology and
Toxicology Centre (ECETOC), the US Environmental Protection Agency,
Food and Drug Administration, National Institute of Occupational
Safety and Health, and Centers for Disease Control, the United
Kingdom Department of Health, and the National Institute of Public
Health, Norway. Dr C.W. Bradford gave valuable assistance in
verifying the nomenclature of platinum compounds. Dr E.M. Smith and
Dr P.G. Jenkins, both members of the IPCS Central Unit, were
responsible for the overall scientific content and technical
editing, respectively, of this monograph. The efforts of all who
helped in the preparation and finalization of the document are
gratefully acknowledged.
* * *
Financial support for the meeting was provided by the Ministry
of the Environment of Italy. The Centro Italiano Studi e Indagini
undertook the organization and provision of meeting facilities.
Partial financial support for the publication of this monograph
was kindly provided by the United States Department of Health and
Human Services, through a contract from the National Institute of
Environmental Health Sciences, Research Triangle Park, North
Carolina, USA - a WHO Collaborating Centre for Environmental Health
Effects.
ABBREVIATIONS
AAS atomic absorption spectrometry
BSA bovine serum albumin
DC direct current
DNA deoxyribonucleic acid
ESCA electron spectroscopy for chemical analysis
ETV electrothermal vaporization
HSA human serum albumin
ICP inductively coupled plasma
Ig immunoglobulin
LC liquid chromatography
LC50 median lethal concentration
MeB12 methylcobalamin
MS mass spectrometry
OVA ovalbumin
PCA passive cutaneous anaphylaxis
PGM platinum-group metals
PIXE proton-induced X-ray emission
PSH platinum salt hypersensitivity
RAST radioallergosorbent test
TLV threshold limit value
TWA time-weighted average
UV ultraviolet
MOLECULAR FORMULAE OF PLATINUM COMPOUNDS
PtO platinum(II) oxide
PtO2 platinum(IV) oxide
PtCl2 platinum(II) chloride
PtCl4 platinum(IV) chloride
Pt(NO3)2 platinum(II) nitrate
Pt(SO4)2 platinum(IV) sulfate
H2[PtCl4] hydrogen tetrachloroplatinate(II)
H2[PtCl6] hydrogen hexachloroplatinate(IV)
(commonly known as hexachloroplatinic
acid)
H2[Pt(NO2)2SO4] hydrogen
dinitrosulfatoplatinate(II)
cis-[PtCl2(NH3)2] cis-
diamminedichloroplatinum(II)
(commonly known as cisplatin)
trans-[PtCl2(NH3)2] trans-
diamminedichloroplatinum(II)
[Pt(NH3)4]Cl2 tetraammineplatinum(II) chloride
[Pt(NO2)2(NH3)2] diamminedinitroplatinum(II)
[Pt(C5H7O2)2] bis(pentane-2,4-
dionato)platinum(II)
(commonly known as
bis(acetylacetonato)platinum(II))
[Pt{NH2)2CS}4]Cl2 tetrakis(thiourea)platinum(II)
dichloride
K2[PtCl4] potassium tetrachloroplatinate(II)
K2[PtCl6] potassium hexachloroplatinate(IV)
K2[Pt(CN)4] potassium tetracyanoplatinate(II)
K[PtCl3(NH3)] potassium amminetrichloroplati
nate(II)
K2[Pt(NO2)4] potassium tetranitroplatinate(II)
Na2[PtCl4] sodium tetrachloroplatinate(II)
Na2[PtCl6] sodium hexachloroplatinate(IV)
Na2[Pt(Oh)6] sodium hexahydroxyplatinate(IV)
Na[Pt(NH3)Cl3] sodium
amminetrichloroplatinate(II)
(NH4)2[PtCl4] ammonium
tetrachloroplatinate(II)
(NH4)2[PtCl6] ammonium hexachloroplatinate(IV)
Cs2[Pt(NO2)Cl3] cesium
trichloronitroplatinate(II)
Cs2[Pt(NO2)2Cl2] cesium
dichlorodinitroplatinate(II)
Cs2[Pt(NO2)3Cl] cesium
chlorotrinitroplatinate(II)
1. SUMMARY
1.1 Identity, physical and chemical properties, analytical methods
Platinum (Pt) is a malleable, ductile, silvery-white noble
metal with the atomic number 78 and an atomic weight of 195.09. It
occurs naturally mainly as the isotopes 194Pt (32.9%), 195Pt
(33.8%), and 196Pt (25.3%). In platinum compounds the maximum
oxidation state is +6, while the states +2 and +4 are the most
stable.
The metal does not corrode in air at any temperature, but can
be affected by halogens, cyanides, sulfur, molten sulfur compounds,
heavy metals, and hydroxides. Digestion with aqua regia or Cl2/HCl
(concentrated hydrochloric acid through which chlorine is bubbled)
produces hexachloroplatinic acid, H2[PtCl6], an important
platinum complex. When heated the ammonium salt of
hexachloroplatinic acid produces a grey platinum sponge. A
dispersive, black powder ("platinum black") results from reduction
in aqueous solution.
The chemistry of platinum compounds in aqueous solution is
dominated by the complex compounds. Many of the salts, particularly
those with halogen- or nitrogen-donor ligands, are water-soluble.
Platinum, like the other platinum-group metals, has a pronounced
tendency to react with carbon compounds, especially alkenes and
alkynes, forming Pt(II) coordination complexes.
There are various analytical methods for the determination of
platinum. Atomic absorption spectrometry (AAS) and plasma emission
spectroscopy provide high selectivity and specificity and are the
method of choice for analysing platinum in biotic and environmental
samples. With these methods detection limits of a few µg/kg or
µg/litre have been obtained for various media.
Inductively coupled argon plasma atomic emission spectroscopy
is superior to electrothermal AAS because of lower matrix effects
and the possibility of simultaneous multi-element analysis.
1.2 Sources of human and environmental exposure
The average concentration of platinum in the lithosphere or
rocky crust of the earth is estimated to be in the region of
0.001-0.005 mg/kg. Platinum is found either in the metallic form or
in a number of mineral forms. Economically important sources exist
in the Republic of South Africa and in the USSR. The platinum
content of these deposits is 1-500 mg/kg. In Canada, platinum-group
metals (platinum, palladium, iridium, osmium, rhodium, ruthenium)
are found in copper-nickel sulfide ores at an average concentration
of 0.3 mg/kg, but are concentrated to above 50 mg/kg during the
refining of copper and nickel. Small amounts are mined in the USA,
Ethiopia, the Philippines, and in Colombia.
World mine production of platinum-group metals, of which 40-50%
is platinum, has steadily increased during the last two decades. In
1971, production was 127 tonnes (51-64 tonnes of platinum).
Following the introduction of the automobile exhaust gas catalyst,
world mine production of platinum-group metals increased to
approximately 270 tonnes (108-135 tonnes of platinum) in 1987. In
1989, total platinum demand in the western world was approximately
97 tonnes.
The principal use of platinum derives from its exceptional
catalytic properties. Further industrial applications relate to
other outstanding properties, particularly resistance to chemical
corrosion over a wide temperature range, high melting point, high
mechanical strength, and good ductility. Platinum is also used in
jewellery and dentistry.
Specific complexes of platinum, particularly cis-
diamminedichloroplatinum(II) (cisplatin), are used
therapeutically.a
Data on emissions of platinum to the environment from
industrial sources are not available. During the use of platinum-
containing catalysts, some platinum may escape into the environment,
depending on the type of catalyst. Of the stationary catalysts used
in industry, only those used for ammonia oxidation emit significant
amounts of platinum.
Automobile catalysts are mobile sources of platinum. According
to limited data, platinum attrition from the old pellet-type
catalyst is between 0.8 and 1.9 µg per km travelled. About 10% of
the platinum is water-soluble.
a This monograph is specifically concerned with platinum and
selected platinum compounds of occupational and/or
environmental importance. A detailed discussion of the toxic
effects of the anticancer drug cisplatin and its analogues in
humans and animals is beyond the selected scope of the
Environmental Health Criteria series as these substances are
used primarily as therapeutic agents. In addition, their toxic
properties are exceptional compared to those of other platinum
compounds.
With the new generation of monolith-type catalyst, results from
engine test stand experiments with a three-way catalyst indicate
that total platinum emission is lower by a factor of 100-1000 than
in the case of pellet-type catalysts. At simulated speeds of 60,
100, and 140 km/h, total platinum emission was found to be between 3
and 39 ng/m3 in the exhaust gas, corresponding to about 2-39 ng
per km travelled. The mean aerodynamic diameter of emitted particles
was between 4 and 9 µm in different test runs. There is limited
evidence that most of the platinum emitted is in the form of the
metal or surface-oxidized particles.
1.3 Environmental transport, distribution, and transformation
Platinum-group metals are rare in the environment, in
comparison with other elements. In highly industrialized areas,
elevated amounts of platinum can be found in river sediments. It is
assumed that organic matter, e.g., humic and fulvic acids, binds
platinum, aided perhaps by appropriate pH and redox potential
conditions in the aquatic environment.
In soil, the mobility of platinum depends on the pH, redox
potential, chloride concentrations of soil water, and the mode of
occurrence of platinum in the primary rock. It is considered that
platinum will be mobile only in extremely acid conditions or in soil
water with a high chloride content.
In in vitro test systems it has been demonstrated that some
platinum(IV) complexes, in the presence of platinum(II), can be
methylated by bacterial methylcobalamin under abiotic conditions.
1.4 Environmental levels and human exposure
The data base concerning environmental concentrations is
extremely limited due to the very low levels of platinum in the
environment and the associated analytical problems.
Concentrations in ambient air samples taken near freeways in
the USA before the introduction of the automobile catalyst were
below the detection limit of 0.05 pg/m3. Some recent data from
Germany indicate that close to roads the platinum air concentrations
(particulate samples) range from < 1 pg/m3 to 13 pg/m3. In
rural areas the concentrations were of a similar order of magnitude
(< 0.6 to 1.8 pg/m3).
Ambient air concentrations of platinum close to roads resulting
from the introduction of pellet-type automobile catalysts have been
estimated on the basis of dispersion models and experimental
emission data. Estimated platinum concentrations near and on roads
ranged from 0.005 to 9 ng per m3 for total platinum. As the total
platinum emission from a monolith-type catalyst is lower, probably
by a factor of 100 to 1000, than that of a pellet-type catalyst, the
platinum concentrations for this type of catalyst would be in the
picogram to femtogram per m3 range.
In roadside dust deposited on broad-leaved plants at various
sites in California, concentrations of 37-680 µg per kg dry weight
were detected. Although the number of samples was limited, the
results indicate that automotive catalysts release platinum to the
roadside environment.
In plant chamber experiments, grass cultures exposed for four
weeks to slightly diluted exhaust gas from an engine equipped with a
three-way catalyst (simulated speed: 100 km/h) contained no platinum
at a detection limit of 2 ng/g dry weight.
Investigations of the platinum concentrations in Lake Michigan
sediments led to the conclusion that platinum has been deposited
there over the past 50 years at a fairly uniform rate.
Concentrations in sediment cores of 1 to 20 cm varied only between
0.3 and 0.43 µg/kg dry weight.
While no platinum levels have been reported for fresh waters,
high concentrations (730 to 31 220 µg/kg dry weight) have been found
in the sediments of a highly polluted cut-off channel of the Rhine
river, Germany.
Samples of limber pines contained platinum levels ranging
between non-detectable and 56 µg/kg (ash weight). However, the
content of the adjacent soils was in the same range, and no
accumulation tendency was indicated by these limited data.
In isolated samples of plants from an ultrabasic soil, platinum
levels of 100-830 µg/kg (dry weight) were found.
Sea-water samples have been found to contain between 37 and 332
pg/litre. In sediment cores from the Eastern Pacific, platinum
concentrations varied between 1.1 and 3 µg/kg (dry weight). The
highest concentration (21.9 µg per kg) was found in offshore ocean
sediments. In marine macroalgae, platinum concentrations of between
0.08 and 0.32 µg/kg dry weight have been found.
Blood platinum levels of 0.1 to 2.8 µg/litre have been found in
the general population. In sera from occupationally exposed workers,
levels of 150 to 440 µg per litre have been reported.
The data base for platinum concentrations at the workplace is
limited. Due to analytical shortcomings, older data (0.9 to 1700
µg/m3) are probably not reliable. However, from these data it can
be assumed that exposure to platinum salts was higher than the
occupational exposure limit of 2 µg/m3 currently adopted by most
countries. In recent workplace studies, concentrations either below
the detection limit of 0.05 µg/m3 or between 0.08 and 0.1 µg/m3
have been measured.
1.5 Kinetics and metabolism
Following a single inhalation exposure (48 min) to different
chemical forms of platinum (5-8 mg/m3), most of the inhaled
191Pt was rapidly cleared from the body. This was followed by a
slower clearance phase during the remaining post-exposure period.
Ten days after exposure to 191PtCl4, 191Pt(SO4)2,
191PtO2, and 191Pt metal, whole body retention of 191Pt was
approximately 1, 5, 8, and 6%, respectively, of the initial body
burden. Most of the 191Pt that was cleared from the lungs by
mucociliary action and swallowed was excreted via the faeces (half-
time, 24 h). A small fraction of the 191Pt was detected in the
urine, indicating that very little was absorbed in the lungs and the
gastrointestinal tract.
In a comparative study on the fate of 191PtCl4 in rats (25
µCi/animal) following different routes of exposure, retention was
highest after intravenous administration, followed by intratracheal
exposure. It was lowest after oral administration. Since only a
minute amount of the 191PtCl4 given orally was absorbed, most of
it passed through the gastrointestinal tract and was excreted via
the faeces. After 3 days, less than 1% of the initial dose was
detected in the whole body. Following intravenous administration,
191Pt was excreted in almost equal quantities in both faeces and
urine. Elimination was slower than after oral dosing. After 3 days
whole body retention was about 65%, and after 28 days it was still
14% of the initial dose. For comparison, after these periods about
22% and 8%, respectively, were retained by the body following
intratracheal administration.
Principal deposition sites are the kidneys, liver, spleen, and
adrenals. The high amount of 191Pt found in the kidney shows that
once platinum is absorbed most of it accumulates in the kidney and
is excreted in the urine. The lower level in the brain suggests that
platinum ions cross the blood-brain barrier only to a limited
extent.
In contrast to the water-soluble salts, the insoluble PtO2
was only taken up in minute amounts even though the salt was
administered in the diet at an extremely high level, which resulted
in a total platinum consumption of 4308 mg per rat over the 4-week
period.
For both the simple platinum salts and cisplatin, it has been
established that there is an initial rapid clearance followed by a
prolonged clearance phase during the remaining post-exposure period,
and that there is no evidence for markedly different retention
profiles. However, cisplatin is, due to high chloride concentrations
suppressing hydration, very stable in extracellular fluids. This
explains why it is excreted mainly in the unchanged form. Its
excretion, in contrast to that of the simple platinum salts, is
primarily via the urine.
1.6 Effects on laboratory mammals and in vitro test systems
The acute toxicity of platinum depends mainly on the platinum
species. Soluble platinum compounds are much more toxic than
insoluble ones. For example, oral toxicity to rats (LD50 values)
decreased in the following order: Na2[PtCl6] (25-50 mg/kg) >
(NH4)2[PtCl6] (195-200 mg/kg) > PtCl4 (240 mg/kg) >
Pt(SO4)2.4H2O (1010 mg/kg) > PtCl2 (> 2000 mg/kg) >
PtO2 (> 8000 mg/kg). For the two latter compounds no LD50 could
be calculated.
In skin testing of albino rabbits, PtO2, PtCl2,
K2[PtCl4], [Pt(NO2)2(NH3)2], Pt(C5H7O2)2 and
trans-[PtCl2(NH3)2] were graded as non-irritant.
(NH4)2[PtCl6], (NH4)2[PtCl4], Na2[PtCl6],
Na2[Pt(OH)6], K2[Pt(CN)4], [Pt(NH3)4]Cl2, and
cis-[PtCl2(NH3)2] appeared to be irritant, but to various
degrees.
In eye irritation tests all tested platinum compounds showed
irritating effects. Trans-[PtCl2(NH3)2] and
(NH4)2[PtCl4] were found to be corrosive.
Intense breathing difficulties were observed after the
intravenous injection of chloro-platinum complexes into guinea-pigs
and rats, presumably due to non-allergic histamine release. This
nonspecific histamine release has complicated the interpretation of
both animal and human studies with respect to the diagnosis of
allergic sensitization.
After subcutaneous and intravenous injection of Pt(SO4)2
three times a week for 4 weeks, there was no induction of an
allergic state, as measured by skin tests (guinea-pigs and rabbits),
passive transfer, and footpad tests (mice). Administration of
platinum-egg-albumin complex also failed to sensitize the
experimental animals.
Attempted sensitization of female hooded Lister rats with the
free salt of ammonium tetrachloroplatinate, (NH4)2[PtCl4],
applied via the intraperitoneal, intramuscular, intradermal,
subcutaneous, intratracheal, and footpad routes, together with
Bordetella pertussis adjuvant, was unsuccessful, as shown by the
direct skin test, passive cutaneous anaphylaxis (PCA) test or a
radio-allergosorbent test (RAST). However, with platinum-protein
conjugates positive PCA results have been reported.
In Cynomolgus monkeys (Macaca fasicularis) exposed to sodium
hexachloroplatinate, Na2[PtCl6], by nose-only inhalation at a
level of 200 µg/m3, 4 h/day, biweekly for 12 weeks, significantly
greater pulmonary deficits were observed by comparison with control
animals. With exposure to ammonium hexachloroplatinate,
(NH4)2[PtCl6], only concomitant exposure to ozone (2000
µg/m3) produced significant skin hypersensitivity and pulmonary
hyper-reactivity.
In oral studies with male Sprague-Dawley rats, the salts
PtCl4 (182 mg/litre drinking-water) and Pt(SO4)2.4H2O (248
mg/litre) did not affect normal weight gain within the observation
period of 4 weeks. With a 3-fold increase in platinum concentration,
weight gain was reduced by about 20% only during the first week,
paralleling a 20% decrease in feed and water consumption.
Only limited experimental data are available for platinum
effects on reproduction, embryotoxicity, and teratogenicity.
Pt(SO4)2 (200 mg Pt/kg) caused reduced offspring weight in Swiss
ICR mice from day 8 to 45 post-partum. The main effect of
Na2[PtCl6] (20 mg Pt/kg) was a reduced activity level of the
offspring of mothers exposed on the 12th day of gestation. Solid
platinum wire or foil is considered to be biologically inert and
adverse effects following implantation into the uterus of rats and
rabbits were probably due to the physical presence of a foreign
object.
After intravenous administration of 191PtCl4 to pregnant
rats (25 µCi/animal) on day 18 of gestation, the placental barrier
was crossed to a limited extent.
Several platinum compounds have been found to be mutagenic in a
number of bacterial systems. In comparative studies cisplatin was
several times more mutagenic than other tested platinum salts. In
in vitro studies with mammalian cells (CHO-HGPT-system), the
relative mutagenic activity of cis-PtCl2(NH3)2],
K[PtCl3(NH3)], and [Pt(NH3)3Cl]Cl was 100:9:0.3. The
mutagenicity of K2[PtCl4] and trans-[PtCl2(NH3)2] was
marginal, whereas [Pt(NH3)4]Cl2 was not mutagenic. No
mutagenic activity was observed for the compounds K2[PtCl4] and
[Pt(NH3)4]Cl2 in the Drosophila melanogaster sex-linked
recessive lethal test, a mouse micronucleus test, and the Chinese
hamster bone marrow test.
Except for cisplatin, no experimental data are available for
the carcinogenicity of platinum and platinum compounds. For
cisplatin there is sufficient evidence for carcinogenic effects on
animals. However, cisplatin and its analogues are rather exceptional
by comparison with other platinum compounds. This is reflected in
the unique mechanism for their anti-tumour activity. Intrastrand DNA
cross-links, formed only by the cis isomer at a certain position of
guanine, are regarded as reasons for this anti-tumour activity. It
appears that replication of DNA in cancer cells is impaired, while
in normal cells the cisplatin lesions on guanine are repaired before
replication.
1.7 Effects on humans
Exposure to platinum salts is mainly confined to occupational
environments, primarily to platinum metal refineries and catalyst
manufacture plants.
The compounds mainly responsible for platinum salt
hypersensitivitya are hexachloroplatinic acid, H2[PtCl6], and
some chlorinated salts such as ammonium hexachloroplatinate,
(NH4)2[PtCl6], potassium tetrachloroplatinate, K2[PtCl4],
potassium hexachloroplatinate, K2[PtCl6], and sodium
tetrachloroplatinate, Na2[PtCl4]. Complexes where there are no
halogen ligands coordinated to platinum ("non-halogenated
complexes"), such as K2[Pt(NO2)4], [Pt(NH3)4]Cl2 and
[Pt{(NH2)2CS}4]Cl2, and neutral complexes such as cis-
[PtCl2(NH3)2], are not allergenic, since they probably do not
react with proteins to form a complete antigen.
The signs and symptoms of hypersensitivity include urticaria,
contact dermatitis of the skin, and respiratory disorders ranging
from sneezing, shortness of breath, and cyanosis to severe asthma.
The latency period from the first contact with platinum to the
occurrence of the first symptoms varies from a few weeks to several
years. Once sensitization is established, symptoms tend to become
worse as long as the workers are exposed in the workplace but
usually disappear on removal from exposure. However, if long-
duration exposure occurs after sensitization, individuals may never
become completely free of symptoms.
Although no unequivocal exposure concentration-effect
relationship can be deduced from the available literature, the risk
of developing platinum salt sensitivity seems to be correlated with
exposure intensity. Metallic platinum seems to be non-allergenic.
With the exception of one single reported case of an alleged contact
dermatitis from a "platinum" ring, no allergic reactions have been
reported.
a The term "platinosis" is no longer used for platinum-salt-
related disease, as it implies a chronic fibrosing lung disease
such as silicosis. Instead, "platinum salt allergy", "allergy
to platinum compounds containing reactive halogen ligands", and
"platinum salt hypersensitivity" (PSH) have been used, the last
being preferred.
The clinical manifestations of platinum salt hypersensitivity
reflect a true allergic response. The mechanism appears to be a type
I (IgE mediated) response. The possibility of IgE antibodies to
platinum chloride complexes developing in sensitive people has been
assumed on the grounds of in vivo and in vitro tests. It is
believed that the platinum salts of low relative molecular mass act
as haptens that combine with serum proteins to form the complete
antigen.
Skin prick tests with dilute concentrations of soluble platinum
complexes appear to provide reproducible, reliable, reasonably
sensitive, and highly specific biological monitors of allergenicity.
The compounds used for routine screening of exposed workers are
(NH4)2[PtCl6], Na2[PtCl6], and Na2[PtCl4]. The
sensitivity and reliability of the skin prick test has not been
achieved by any in vitro test available. In enzyme immunoassays
and in radioallergosorbent tests (RAST), IgE antibodies specific to
platinum chloride complexes have been found. Although a correlation
with the results of prick tests was reported, the applicability of
RAST for screening purposes was questioned because of its
nonspecificity.
Only limited cross-reactivity between platinum and palladium
salts has been found in skin testing and RAST. Reactions to the
platinum-group metals other than platinum have only been seen in
individuals sensitive to platinum salts.
Smoking, atopy, and nonspecific pulmonary hyper-reactivity have
been associated with platinum salt hypersensitivity and could be
predisposing factors.
For the general population, there is a lack of data on the
actual exposure situation in countries where the automobile catalyst
has been introduced. The possible ambient air concentrations,
estimated on the basis of a few emission data and dispersion models,
are at least a factor of 10 000 lower than the occupational exposure
limit value of 1 mg/m3 adopted by some countries for platinum
metal as total inhalable dust. Since the emitted platinum is most
probably in the metallic form, the sensitizing potential of platinum
emissions from automotive catalysts is probably very low. Even if
part of the platinum emitted was soluble and potentially allergenic,
the safety margin to the occupational exposure limit for soluble
platinum salts (2 µg/m3) would be at least 2000.
In a preliminary immunological study, extracts of particulate
automobile exhaust samples were tested on three human volunteer
subjects using a skin prick test. No positive response was elicited.
No data are available to assess the carcinogenic risk of
platinum or its salts to humans. With regard to cisplatin, evidence
for human carcinogenicity is considered inadequate.
1.8 Effects on other organisms in the laboratory and field
Simple complexes of platinum have bactericidal effects. The
discovery that neutral complexes such as cisplatin selectively
inhibit cell division without reducing cell growth of a variety of
gram-positive, and especially, of gram-negative bacteria has led to
their application in medicine as anti-tumour agents.
Growth and yield of the green alga Euglena gracilis were
inhibited by the soluble hexachloroplatinic acid (250, 500, and 750
µg/litre) in a laboratory "microcosm". Cisplatin caused chlorosis
and stunted growth in the water hyacinth Eichhornia crassipes at a
concentration of 2.5 mg/litre.
A 3-week exposure to hexachloroplatinic acid, H2[PtCl6],
resulted in an LC50 value of 520 µg Pt per litre in the
invertebrate Daphnia magna. At concentrations of 14 and 82
µg/litre, reproduction, measured as total number of young, was
impaired by 16 and 50%, respectively.
After short-term exposure to tetrachloroplatinic acid,
H2[PtCl4], in a static bioassay, 24-, 48-, and 96-h LC50
values of 15.5, 5.2, and 2.5 mg Pt/litre, respectively, were found
for the coho salmon (Oncorhynchus kisutch). General swimming
activity and opercular movement were affected at 0.3 mg/litre.
Lesions in the gills and the olfactory organ were noted at 0.3
mg/litre or more. Concentrations of 0.03 and 0.1 mg/litre had no
effect.
There have been studies on the effects of platinum on
terrestrial plants, all conducted with soluble platinum chlorides.
The growth of beans and tomato plants in sand culture was inhibited
by hexachloroplatinic acid at concentrations of 3 x 10-5 to 15 x
10-5 mol/kg (5.9-29.3 mg/kg). Of nine horticultural crops grown in
hydroponic solution with platinum tetrachloride, PtCl4 (0.057,
0.57, and 5.7 mg Pt/litre), dry weights were significantly reduced
in tomato, bell pepper, and turnip tops, and in radish roots at the
highest concentration. At this level, the buds and immature leaves
of most species became chlorotic. In some of the species the low
levels of PtCl4 had a stimulatory effect on growth. In addition,
transpiration was suppressed at the highest platinum concentration,
probably due to increased stomatal resistance. Growth stimulation
was also observed at low levels of platinum (0.5 mg Pt/litre),
administered as potassium tetrachloroplatinate, K2[PtCl4], in
seedlings of the South African grass species Setaria verticillata
grown in nutrient solution. After two weeks, the length of the
longest roots had increased by 65%. At the highest concentration
applied, i.e. 2.5 mg Pt/litre, phytotoxic effects were seen in the
form of stunted root growth and chlorosis of the leaves.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1 Identity
Platinum is a malleable, ductile, silvery-white noble metal
with the atomic number 78 and an atomic weight of 195.09. It occurs
naturally mainly as the isotopes 194Pt (32.9%), 195Pt (33.8%),
and 196Pt (25.3%). In platinum compounds, the maximum oxidation
state is +6, while the states +2 and +4 are the most stable.
The most important platinum compounds are listed in Table 1.
2.2 Physical and chemical properties
2.2.1 Platinum metal
The metal does not corrode in air at any temperature, but can
be affected by halogens, cyanides, sulfur, molten sulfur compounds,
heavy metals, and hydroxides. Digestion with aqua regia or Cl2/HCl
(concentrated hydrochloric acid through which chlorine gas is
bubbled) leads to hexachloroplatinic acid, H2[PtCl6], an
important platinum complex.
Platinum has a coefficient of expansion almost equal to that of
sodium-calcium-silicate glass and the two materials can be used in
combination, e.g., in electrodes.
Some chemical and physical data on platinum and selected
compounds are listed in Table 2.
2.2.2 Platinum compounds
The chemistry of platinum compounds in aqueous solution is
dominated by the complex compounds. Many of the salts, particularly
those with halogen- or nitrogen-donor ligands, are water-soluble. In
biochemical processes, cis-trans effects in the quadratic
coordination of platinum play an important role. Platinum, like the
other platinum-group metals (PGM), has a marked tendency to react
with carbon compounds, especially alkenes and alkynes, forming
Pt(II) coordination complexes.
Table 1. Chemical names, synonyms, and formulae of elemental platinum and platinum compoundsa
Chemical name CAS registry numberb Synonyms Formula
Element
Platinum 7440-06-4 Pt
Binary compounds
Platinum(II) chloride 10025-65-7 platinous chloride PtCl2
Platinum(IV) chloride 13454-96-1 platinum tetrachloride PtCl4
Platinum(II) oxide n.a. platinous oxide PtO
Platinum(IV) oxide 1314-15-4 platinic oxide; platinum dioxide PtO2
Platinum sulfate n.a. - Pt(SO4)2.4H2O
Platinum nitratec n.a. - Pt(NO3)2
Coordination complexes
Hexachloroplatinic acid(IV) 16941-12-1 chloroplatinic acid; dihydrogen H2[PtCl6]
hexachloroplatinate
Sodium hexachloroplatinate(IV) 16923-58-3 disodium hexachloroplatinate; Na2[PtCl6]
sodium chloroplatinate
Potassium hexachloro- 16921-30-5 potassium chloroplatinate; platinic K2[PtCl6]
platinate(IV) potassium chloride
Potassium tetrachloro- 10025-99-7 platinum potassium chloride; K2[PtCl4]
platinate(II) potassium platinochloride
Ammonium tetrachloroplatinate(II) 13820-41-2 ammonium platinous chloride; (NH4)2[PtCl4]
ammonium chloroplatinite
Ammonium hexachloroplatinate(IV) 16919-58-7 ammonium platinic chloride; (NH4)2[PtCl6]
ammonium chloroplatinate; "yellow salt"
cis-Diamminedichloroplatinum(II) 15663-27-1 cisplatin; cis-platinum; DDP; CDDP; cis-[PtCl2(NH3)2]
CPDD; CACP; CPCC; Peyron's chloride
trans-Diamminedichloroplatinum(II) 14913-33-8 trans-dichlorodiammineplatinum(II) trans-[Pt(NH3)2Cl2]
a From: Windholz (1976); Weast & Astle (1981)
b n.a. = not available
c Kral & Peter (1977)
Table 2. Physical and chemical properties of platinum and selected platinum compoundsa
Relative
atomic/ Melting Boiling Relative Crystalline Solubilityd
Chemical name molecular pointb point density formc Cold Hot Other
mass (°C) (°C) (g/cm3) water water solvents
Platinum (Pt) 195.09 1772 3827 21.4520 silver-metallic ins ins sol aq.
(± 100) cubic cr. regia
Platinum(II) chloride 266.00 581b 6.05 olive-green, ins al, eth;
(PtCl2) (in Cl2) hexagonal cr. sl sol sol H5Cl,
NH4OH
Platinum(IV) 336.90 370b 4.303 brown-red cr. v sol v sol sl sol,
chloride (PtCl4) (in Cl2) al, NH3
Platinum(IV) oxide (PtO2) 227.03 450 10.2 black powder ins ins ins acid,
aq. regia
Platinum(II) oxide (PtO) 211.09 550b 14.9 violet-black ins ins sol HCl;
cr. ins aq.
regia
Platinum sulfate 459.27 yellow plates sol dec sol al,
(Pt(SO4)2.4H2O) eth, acid
Hexachloroplatinic 517.92 60 2.431 red-brown v sol v sol sol al, eth
acid(IV) deliquescent
(H2[PtCl6].6H2O) cr.
Sodium hexachloroplatinate(IV) 453.77 yellow, sol sol al
(Na2[PtCl6]) hygroscopic cr.
Table 2 (contd).
Relative
atomic/ Melting Boiling Relative Crystalline Solubilityd
Chemical name molecular pointb point density formc Cold Hot Other
mass (°C) (°C) (g/cm3) water water solvents
Potassium hexachloroplatinate(IV) 486.03 3.50 orange-yellow sl sol sol ins al
(K2[PtCl6]) cr. or yellow
powder
Potassium tetrachloroplatinate(II) 415.26 ruby-red cr. sol
(K2[PtCl4])
Ammonium tetrachloroplatinate(II) 373.00 dark ruby-red sol
((NH4)2[PtCl4]) cr.
Ammonium hexachloroplatinate(IV) 443.91 3.06 orange-red cr. v sol ins al
((NH4)2[PtCl6]) or yellow powder
cis-Diamminedichloroplatinum(II) 300.07 270b orange cr. sl sole
(cis-[PtCl2](NH3)2)
trans-Diamminedichloroplatinum(II) 300.07
(trans-[PtCl2](NH3)2)
a Compiled from: Windholz (1976); Weast & Astle (1981); Neumüller (1987).
b dec = decomposes
c cr. = crystals
d al = alcohol (ethanol); dec = decomposes; eth = ether; ins = insoluble; sl = slightly; sol = soluble; v = very
e Tobe & Khokhar (1977)
Platinum hexafluoride, PtF6, has the highest oxidation state
of the element and is a strong oxidizing agent; the noble gas xenon
can be oxidized to XeF2 and oxygen to O2+ (Hoppe, 1965).
Hexachloroplatinic acid, H2[PtCl6], is formed by the
reaction of platinum metal with aqua regia or Cl2/HCl. When
heated, the ammonium salt of this acid produces a grey platinum
sponge. A black powder ("platinum black") is produced by reduction
in aqueous solution. Depending on the pH value, hydroxides exchange
the halogen ligands with OH- in a stepwise manner, leading to
PtO2.nH2O after dehydration (n = 1, 2, 3, 4). Further heating
gives rise to PtO at 400 °C, which decomposes to platinum and O2
at 560 °C.
By heating hexachloroplatinic acid at 240 °C, PtCl2 can be
obtained. It has a hexameric structure (Pt6Cl12) in the solid
state and is soluble in benzene. This compound forms H2[PtCl4]
in HCl.
Platinum forms a large number of Pt(II) and Pt(IV) complexes
with the formulae:
Pt(IV): [PtX6-n(NH3)n]n-2 where n = 0-6; X = halogen ligand
Pt(II): [PtX4-n(NH3)n]n-2 where n = 0-4; X = halogen ligand
The chemical structures of two of the more important platinum
complexes are shown below.
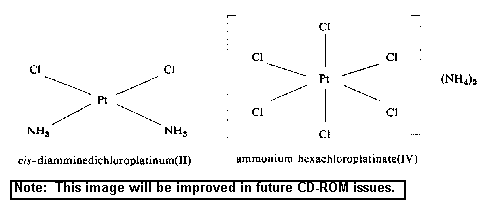 2.3 Conversion factors
Platinum 1 ppm = 7.98 mg/m3
1 mg/m3 = 0.13 ppm
2.4 Analytical methods
2.4.1 Sampling
Samples of ores, minerals, and preconcentrated technical
products can be obtained in a ground or powdered form. Metals and
alloys can be collected as chips and shavings. Platinum on alumina
pellets or monolithic supports must be comminuted before fusing or
digesting (Potter & Lange, 1981). Electronic scrap may contain
alloyed copper, nickel or lead. Melting with aluminium leads to a
brittle alloy, which can be easily crushed to a powder.
Blood samples may be frozen and lyophilized (Pera & Harder,
1977), homogenized with substances like TRITON-X 100(R) (Priesner
et al., 1981), and separated into plasma ultrafiltrate and proteins
(Bannister et al., 1978) or, if appropriate, analysed directly
without pretreatment.
With biological materials, homogeneous sampling is difficult
and often requires destructive methods resulting in the loss of all
information about the platinum species. Only the total content of
platinum and its isotopes can be determined.
For the analysis of platinum in urine, the untreated original
sample is usually unsuitable. Freeze-drying or a wet ashing
procedure with subsequent reduction of volume is necessary for most
analytical methods.
Other biological and environmental materials being investigated
for very low levels of platinum need to be sampled in large amounts,
with possible difficulty in homogenisation, digestion, storage, and
matrix effects.
2.4.2 Sample pretreatment
Determination of total platinum content in some materials
requires a digestion step, which is the pre-requisite for enrichment
and separation from other elements and organic substances. A modern
wet digestion procedure (Knapp, 1985) avoids contact with materials
other than quartz in order to reduce adsorption losses. In this way,
organic matter is destroyed most effectively and contamination with
platinum from other sources is minimized (Würfels et al., 1987).
In general, separation involves volatilization, distillation,
lyophilization, extraction, coprecipitation, flotation, sorption,
and other instrumental methods, such as electro-deposition,
chromatographic separations, and thermal pre-treatment in atomic
absorption spectroscopy (AAS) procedures (Knapp, 1984).
A selection of extraction and sorption techniques is shown in
Tables 3 and 4, respectively. For coprecipitation procedures,
details can be found in the reports of Fryer & Kerrich (1978),
Stockman (1983), Sighinolfi et al. (1984), Skogerboe et al. (1985),
Amosse et al. (1986), and Bankovsky et al. (1987).
2.4.3 Detection and measurement
2.4.3.1 Spectrophotometry
Unless the native soluble platinum compounds have an inherent
absorption spectrum, they can be treated with inorganic and organic
reagents to form coloured, soluble complexes that can be measured by
absorption spectrophotometry. Careful separation from other elements
is important (see section 2.4.2). The detection limits achieved are
in the low mg/kg (ppm) range (Jones et al., 1977; Brajter & Kozicka,
1979; Mojski & Kalinowski, 1980; Marone et al., 1981; Aneva et al.,
1986; Puri et al., 1986).
2.4.3.2 Radiochemical methods
Neutron-activation analysis is a very sensitive method for
determining submicrogram traces of platinum. It is at least one to
several orders of magnitude more sensitive than the best of the
spectrophotometric methods. For the determination of platinum a
sensitivity of 1 ng absolute was estimated on irradiation of a
sample for 1 month at a neutron flux of 10-2cm-2-second,
followed by a 2-h decay (NAS, 1977).
Radiochemical methods have been applied to the analysis of
platinum in various matrices. The detection limits are 1-2 µg/kg in
rock samples (Stockman, 1983), 30 µg per kg dry weight in plant
material (Valente et al., 1982), 1-3 µg/kg dry weight (0.3 ng
absolute) in plant material and animal tissue (Tjioe et al., 1984),
and 100 µg/kg in airborne particulate matter (Schutyser et al.,
1977).
2.4.3.3 X-ray fluorescence spectroscopy
This method permits the highly selective, sensitive, rapid, and
non-destructive analysis of platinum. Zolotov et al. (1983) obtained
a detection limit of 32 µg Pt per litre in aqueous solutions.
A new variant, total-reflection X-ray fluorescence
spectrometry, has the advantage of small sample size (5 to 40 µg)
with low absolute detection limits (Von Bohlen et al. 1987).
Table 3. Extraction procedures for separating platinum
Species Matrix Chemical modifier Extraction Elements Reference
medium separated
Pt(IV) aqueous 6 M HCl isopentanol Al, Ca, Mg, Aneva et al. (1986)
solutions Mn, Ni, Cr
4-methyl-2- Cu, Pb
pentanone (partially)
Pt(IV) aqueous dithio-oxamide tri-butyl Ir(III), Rh(III) Brajter & Kozicka (1979)
solutions phosphate
Pt(IV) plant- S-(1-decyl)- variety of co-extraction Jones et al. (1977)
processing N,N -diphenyl- organic liquids of noble metals
solutions isothiouronium bromide
Pt(IV) palladium(II) 1,5-diphenylthiocarbazone carbon Pd(II) Marczenko & Kus (1987)
chloride tetrachloride
Pt(IV) palladium triphenylphosphine dichloroethane Pd, Au Mojski & Kalinowski (1980)
metal oxide
Pt(IV) synthetic phenanthraquinonemonoxime molten Fe, Cu, Ni, V, Puri et al. (1986)
aqueous naphthalene Cr, Al, Au, Ag
solutions Ir, Rh, Pd
Pt(IV) aqueous potassium butylxanthate carbon - Singh & Garg (1987)
solutions tetrachloride
Pt(IV) automotive bis-(2-furyl)- trichloromethane V, Mo, W Wiele & Kuchenbecker (1974)
catalysts glyoxaldioxime
Pt(II), synthetic 1,4,7,10,13,16-hexa- 4-methyl-2- Fe(III) Arpadjan et al. (1987)
Pt(IV) aqueous azaoctadecane pentanone
solutions
Table 3 (contd).
Species Matrix Chemical modifier Extraction Elements Reference
medium separated
Pt(II) urine Diethylammonium- trichloromethane Ca, Zn, Fe(II) Borch et al. (1979)
diethyldithiocarbamate, and Mn(II)
NaSH
Pt(II) aqueous sodium co-extraction Mueller & Lovett (1987)
solutions diethyldithiocarbamate of Pd(II),
acetonitrile, NaCl Rh(II)
Pt(II) plasma sodium - Andrews et al. (1984)
ultrafiltrate diethyldithiocarbamate
methanol, H2O
Pt geological sodium tetraborate, molten lead - Millard (1987)
samples KCN
Pt geological KCN, KOH Ag, Au co-extraction Le Houillier & De Blois
samples of noble metals (1986)
Pt blood, hair, HCl, SnCl2 tri-n-octylamine, - Tillery & Johnson (1975)
faeces, urine xylene
Pt geological sodium nickel sulfide - Robert et al., (1971)
samples carbonate
and sodium
tetraborate
Table 4. Sorption techniques for preconcentrating platinum
Species Matrix Sorption medium Eluent Elements Reference
separated
Pt sea water Bio-Rad Ag-1-X2 0.1 M HCl, Ir Goldberg et al. (1986);
0.02 M thiourea Hodge et al. (1986)
Pt geological Srafion NMRR 0.01 M HCl, high selectivity Kritsotakis & Tobschall (1985)
samples 5% thiourea for transition
metals
Pt aqueous polyethenimine- Co(II), Zn, Cd, Geckeler et al. (1986)
solutions methylthiourea In(III), Na
suspended in water
at pH 1
Pt(II), aqueous Dowex 2X-8 75% NH3 in H2O Au Kahn & Van Loon (1978)
Pt(IV) solutions
Pt (IV) geological Bio-Rad Ag-50W-X8 0.1 M HCl - Coombes & Chow (1979)
samples
Pt (IV) geological P-TD 2 M HClO4 Al, Mg, Cu, Grote & Kettrup (1987)
samples Fe, Ni, Cr
Pt (IV) aqueous Hyphan 1 M HClO4 Na, K, Cs, Mg, Kenawy et al. (1987)
solutions Ca, Al
Pt (IV) geological Polyorgs digestion HClO4, coextraction Myasoedova et al. (1985)
samples, H2SO4, HNO3 noble metals
scaps
Pt (IV) aqueous (-CH2-S-)n(n approx. 1000) 6 M HCl Co, Ni, Pb, Fe, Zolotov et al. (1983)
solutions Zn, Cd
2.4.3.4 Electron spectroscopy for chemical analysis (ESCA)
ESCA is a technique typically applied in surface analysis
involving a few surface atomic layers (1-2 nm). This technique is
used for special purposes; for instance, Schlögl et al. (1987)
analysed microparticles from automotive exhaust gas catalysts (see
section 3.2.1.4).
2.4.3.5 Electrochemical analysis
Of the voltametric techniques available for element analysis,
polarography, in particular, has been applied for the determination
of platinum. Alexander et al. (1977a,b) described a pulse
polarography method for the analysis of platinum in ores after fire-
assay separation and preconcentration. By measuring the sensitive
catalytic polarographic wave generated by the Pt(II)-ethylenediamine
complex in alkali solutions a detection limit of 0.025 µg per kg was
obtained. A similar technique was applied to the analysis of urine
by Vrana et al. (1983), and the detection limit was 10 µg/litre.
However, these methods do not allow the direct determination of
platinum in complex solutions due to interferences from some heavy
metals and precipitation of platinum with other metals in the form
of their hydroxides. In this respect, inverse voltametry is
superior. Kritsotakis & Tobschall (1985) used the glassy carbon
electrode for the determination of platinum traces in synthetic
solutions. After preconcentration, 0.04 mg Pt/litre could be
determined. This detection limit is sufficient for determining
platinum in ores.
Using adsorptive cathodic stripping voltametry, Van den Berg &
Jacinto (1988) analysed sea-water samples (see section 5.1.2). The
detection limit was 7.8 pg Pt/litre.
Hoppstock et al. (1989) developed a sensitive volta-metric
method for determining platinum in the ng/kg range in biotic and
environmental materials. The overall recovery of platinum was
reported to be 97% or more.
Nygren et al. (1990) described an adsorptive volta-metric
method for the measurement of platinum in blood. The detection limit
for a 100-µl sample was 0.017 µg per litre.
2.4.3.6 Proton-induced X-ray emission (PIXE)
PIXE requires only small sample sizes (1-10 mg), but is a time-
consuming and labour-intensive method. Owing to the substantially
lower background, the detection limits are lower by a factor of 1000
than for X-ray fluorescence methods. Methods for analysing water
samples, air, and biological tissues have been described by Rickey
et al. (1979), Wolfe (1979), and Thompson et al. (1981).
2.4.3.7 Liquid chromatography (LC)
Marsh et al. (1984) published an adsorption chromatography
method in which the analyte was first separated with an ODS
Hypersil(R) column, reacted with NaHSO3, and then detected by UV
absorption. The detection limit for cisplatin was 40-60 µg/litre.
For the malonate derivates, Van der Vijgh et al. (1984) reported a
detection limit of 300-1200 µg/litre for human body fluids.
Ebina et al. (1983) analysed Pt(II) in aqueous solutions that
were modified with EDTA, ethanoic acid, and maleonitriledithiol. The
spectrophotometric detection limit for this partition ion-pair
method was 0.2 ng per litre.
Using an ion exchange chromatography method, Rocklin (1984)
separated Pt(IV) as the hexachlorocomplex on a polar anion exchange
column and determined the complex by UV. For samples digested in
aqua regia, a detection limit of 30 µg/litre can be obtained without
preconcentration and < 1 µg/litre after preconcentration.
2.4.3.8 Atomic absorption spectrometry (AAS)
AAS is a method of high selectivity and specificity and is
often the method of choice in analysing platinum in biological and
environmental samples. However, there are problems with background
radiation deriving from molecules and radicals, especially from
unseparated matrix. These interferences can be partly overcome by
background compensation through a radiation continuum or by the
application of the "Zeeman" effect. To determine platinum in the
range of the detection limit, an accurate separation from matrix is
essential.
For platinum determinations in biological materials, Farago &
Parsons (1982) recommended wet digestion in nitric acid and the
removal of residual nitrates by hydrochloric acid. Brown & Lee
(1986) proposed totally pyrolytic cuvettes for graphite furnace AAS,
thus achieving a greater sensitivity for refractory metals. These
results were confirmed by Schlemmer & Welz (1986). Although platinum
does not form a stable carbide, there was an effect on the wall
material of the carbon rod. Electro-graphite tubes coated with
pyrolytic graphite were found to be superior to glassy carbon tubes
(Welz & Schlemmer, 1987).
LeRoy et al. (1977) described a method for the detection of
platinum in biological samples that used controlled dehydration and
ashing with rapid sample evaporation to detect low levels of
platinum. This method did not suffer as much from matrix
interference as other AAS graphite furnace methods. The method can
be used to detect platinum down to approximately 30 µg/kg (30 ppb).
Hodge et al. (1986) determined platinum down to pg per litre
levels in marine waters, sediments, and organisms. Sea water was
extracted with an anion exchanger (Table 4), eluted, and purified by
acid digestion. In a second step, platinum was obtained from the
solution with an anion exchanger, stripped again from the bead, and
injected. Using a similar technique, Hodge & Stallard (1986)
determined platinum in roadside dust.
Jones (1976) digested urine and blood samples with nitric and
perchloric acids. The samples were diluted after cooling and
injected onto carbon rods. The minimum detectable platinum
concentration in 5-g samples was 30 µg per litre.
McGahan & Tyczkowska (1987) dried and ashed tissues and fluids
and diluted the residue with different acids before direct
injection. The detection limits were 6 µg per kg or 6 µg/litre.
Bannister et al. (1978) separated protein-bound platinum and
free circulating compounds by centrifugal ultra-filtration. In the
ultrafiltrate, platinum compounds were chelated with
ethylenediamine, extracted on a cation exchange paper disc, eluted,
and injected. The minimum working concentration was 35 µg/litre of
plasma.
Alt et al. (1988) described a simple and reliable method which
included high-pressure ashing (cf. Knapp, 1984), separation by
extraction, and detection by graphite furnace AAS. This method was
recommended for analysing biological and other materials down to the
µg/kg range.
König et al. (1989) determined platinum in the particulate
emissions in engine test-stand experiments (see section 3.2.1.4)
using a high-pressure digestion without a separation. The authors
studied the matrix influences with respect to the concomitant
elements and found interferences from A1, Pb, Ca, Zn, P and, most
severely, from Si, but under the controlled test conditions no
interference effects were observed. In particle-free condensates of
automotive exhaust gas, a detection limit of 0.1 ng/ml was achieved
by the method of signal addition described by Berndt et al. (1987).
2.4.3.9 Inductively coupled plasma (ICP)
The generation of plasmas is a further development of chemical
flame methods. They have a wide temperature range, a transparency
for the UV spectral lines, and are predominantly insensitive against
interfering chemical reactions in the excitation zone that occur
with chemical flames. Plasma excitation allows the determination of
several elements simultaneously and is, because of minor matrix
effects, easy to calibrate over many orders of magnitude. Two
methods of generating a plasma are currently used: firstly with
direct current (DC) and secondly with a high frequency current
(20-80 MHz, inductively coupled plasma, ICP). The ICP method works
with an argon plasma and temperatures of 4000-8000 K. Due to the
increasing ionization effects, the aerosol feeding is controlled by
cooling devices.
Boumans & Vrakking (1987) discussed standard values for a 50-
MHz ICP, considering effects of source characteristics, noise, and
spectral band-width, and obtained a detection limit for the platinum
spectral line at 214.42 nm of 7.2 µg/litre.
Maessen et al. (1986) studied the influence of chloroform on
the platinum signal at 203.65 nm. The detection limits by this
method were affected by chloroform and ranged from 30-400 µg/litre.
Wemyss & Scott (1978) determined platinum-group metals and gold
in ores after three different digestions. The method allowed
determination down to 0.13 mg/litre for the 299.8-nm line.
Fox (1984) reported interferences from aluminium and magnesium
in direct current methods. A buffer of lithium and lanthanum
compounds suppressed this effect.
Lo et al. (1987) described a simple method for determining
platinum in urine with a working range down to 50 µg/litre (50 ppb)
under direct application of acidified samples. Electrothermal
vaporization (ETV) was used for generating plasma-suitable aerosols
by Matusiewicz & Barnes (1983). They determined platinum at the
mg/litre level in human body fluids directly. A similar procedure
was used by Belliveau et al. (1986).
2.4.3.10 Inductively coupled plasma - mass spectrometry (ICP-MS)
Combining ICP with a mass spectrometer has new advantages in
analytical spectroscopy. Elemental ions generated from an aerosol or
an electrothermal vaporization unit are separated by a quadrupole
and detected as isotopes at low level. The ETV device allows
determination down to the pg/ml range.
Thompson & Houk (1986) used an ion-pair reversed-phase liquid
chromatography assay via a continuous flow ultrasonic nebulizer and
an ICP torch with a mass spectrometer. In synthetic solutions
detection limits of 7 µg/litre (7 ppb) were obtained.
Gregoire (1988) compared the results from the ICP-MS-ETV with
neutron activation analysis and the ICP-MS solution nebulization
method in the ng/ml concentration range and found good agreement.
For the analysis of air samples, the NIOSH Manual of Analytical
Methods (Eller, 1984a) describes a method based on inductively
coupled argon plasma atomic emission spectroscopy. The working range
is 0.005-2.0 mg/m3 with a 500-litre air sample. However, long
sampling periods are required for measuring soluble platinum
compounds in the workplace and the method does not distinguish
between soluble and insoluble platinum. Similar methods are
recommended for the analysis of platinum in blood and tissues
(Eller, 1984b) and in urine (Eller, 1984c).
The method recommended by the United Kingdom Health and Safety
Executive (1985) has a precision better than 8%, measured as a
coefficient of variation, for samples of a minimum of 120 litres in
the range 1-15 µg Pt/m3. The sensitivity of this method can be
improved by 100-1000 fold by using ICP-MS instead of carbon furnace
atomic absorption spectrometry.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
The six platinum-group metals, platinum, palladium, rhodium,
ruthenium, iridium, and osmium, were probably concentrated mainly in
the iron-nickel core during the earth's formation. This explains
their relatively low presence in the lithosphere (rocky crust) of
the earth (Goldschmidt, 1954) where the average concentration of
platinum ranges between 0.001 and 0.005 mg/kg (Mason, 1966, Bowen,
1979).
Platinum is found both in its metallic form and in a number of
minerals. The principal minerals are: sperrylite, PtAs2;
cooperite, (Pt,Pd)S; and braggite, (Pt,Pd,Ni)S. Primary deposits are
associated with ultrabasic, rather than silicic, rock formations.
Economically important sources exist in the Bushveld Igneous Rock
Complex in Transvaal, Republic of South Africa, and in the Noril'sk
region of Siberia, the Kola Peninsula, and in the Nishnij Tagil
region of the Urals, USSR. The platinum content in these deposits is
between 1 and 500 mg/kg. In the Sudbury district of Canada, platinum
metal is contained in copper-nickel sulfide ores at an average
concentration of 0.3 mg/kg but is concentrated to more than 50 mg/kg
during the refining of copper and nickel. In the USA, there is a
platinum-palladium mine in the Stillwater Complex area, Montana
(NAS, 1977; Renner, 1979).
Small amounts of platinum are also mined from secondary or
placer deposits in the USSR (Ural Mountains), Colombia, USA
(Alaska), Ethiopia, and the Philippines. In these deposits platinum
is present in the form of metallic alloys of varied composition
(NAS, 1977).
3.2 Anthropogenic sources
3.2.1 Production levels and processes
3.2.1.1 World production figures
World mine production of platinum-group metals, 40-50% of which
is platinum, has steadily increased during the last two decades. In
1971 production was 127 tonnes (51-64 tonnes platinum) and in 1972
it was 132 tonnes (53-66 tonnes platinum) (Butterman, 1975). In
1975, automobile exhaust gas catalysts were introduced in the USA in
order to meet the stringent emission limits for carbon monoxide,
hydrocarbons, and nitrogen oxides set by the Federal Clean Air Act.
In Japan, the automobile catalyst was introduced at the same time.
As a consequence, world production of PGM increased to 179 tonnes
(72-90 tonnes platinum) in 1975, reaching a plateau of between 200
and 203 tonnes per year (80-102 tonnes platinum) during the period
1977-1983 (Loebenstein, 1982, 1988).
From 1984 onwards world production increased, apparently in
response to the anticipated demand in Western Europe where
automobiles are being increasingly fitted with catalytic converters.
In 1987, world mine production of PGM amounted to about 270 tonnes
(108-135 tonnes platinum) (Loebenstein, 1988).
The future demand for platinum depends on improvements in
engine technology and emission control, but can be expected to
increase further during the coming years. Data on platinum demand
are presented in section 3.2.2.
3.2.1.2 Manufacturing processes
Most native placer platinum is recovered by dredging and, in
less developed areas, by small hand operations. The copper and
nickel sulfide ores are mined by large-scale underground methods and
concentrated by flotation (Stokinger, 1981).
The isolation of pure platinum metal from raw materials
involves two principal stages: (i) extraction of a concentrate of
precious metals from the ore; (ii) refining the concentrate to
separate the platinum-group metals from each other and purify them.
These processes require sophisticated chemical technology and
include precipitating crystallization and liquid-liquid extraction,
often combined with redox reactions to change the oxidation state of
the metals. Further processes involve halogenation and reduction
reactions at annealing temperatures and special distillations
(Renner, 1984).
Potential health hazards of exposure to soluble platinum salts
are encountered during the later stages of the refining process.
After dissolving platinum, palladium, and gold with aqua regia or
Cl2/HCl and the subsequent precipitation of gold by addition of
ferrous salts, ammonium chloride is added to precipitate ammonium
hexachloroplatinate, (NH4)2[PtCl6]. After several purification
processes there is a second precipitation of this complex salt,
which is then filtered off, dried and finally calcined to yield a
spongy mass of platinum metal having purity of 99.95-99.99%. This
can be further purified by a cationic exchange technique (NAS, 1977;
Stokinger, 1981).
Secondary sources in substantial quantities come from the
reclamation of scrap and used equipment, particularly industrial
catalysts. The recycling of platinum-group metals from automobile
catalysts is also increasing (see section 4.3). In principle, the
recycling of platinum involves the same wet-chemical and melting
processes that are applied to its production from ores (Renner,
1984).
3.2.1.3 Emissions from stationary sources
a) Production
Data on emissions of platinum during production are not
available.
b) Stationary catalysts
During the use of platinum-containing catalysts, platinum can
escape into the environment in variable amounts, depending on the
type of catalyst. Of the stationary catalysts used in industry, only
those employed for ammonia oxidation emit major amounts.
The loss of platinum from ammonia oxidation gauzes during
nitric acid production depends on the operating pressure. An average
figure is 0.15 g/tonne of nitric acid (Sperner & Hohmann, 1976). Of
this apparent loss, 70-85% is recovered on gold-palladium catchment
gauzes, reducing the loss to 0.03 g/tonne (Anon., 1990a). The
production of nitric acid in the USA in 1989 was 7 247 837 tonnes
(Anon., 1990b). Thus the amount of platinum "lost" in 1989 in the
USA is calcu-lated to be 217 kg. This is the maximum amount that
could be dissolved or suspended as a colloid in the nitric acid and,
thus, could be introduced into the environment if the nitric acid is
used in fertilizer production.
3.2.1.4 Emissions from automobile catalysts
Automobile catalysts are mobile sources of platinum. Although
these catalysts are designed to function for 80 000 km or more
(Koberstein, 1984), some loss of platinum can occur due to
mechanical and thermal impact. The data on platinum emissions from
automobile catalysts are very limited.
In the mid 1970s unrealistically high assumptions were made for
platinum loss. Brubaker et al. (1975) estimated the loss to be about
12 µg Pt/km, which would mean a total loss of approximately 1 g
after 80 000 km.
Experimental data show much lower emission rates. Malanchuk et
al. (1974) found a platinum concentration of 0.029 µg/m3 in an
inhalation chamber that was fed by catalysed engine exhaust. On the
basis of the chamber volume, flow rate, and the speed simulated on
the engine test stand, an emission rate of 0.39 µg/km was
calculated. In another US EPA study, Sigsby (1976) did not detect
platinum in particulate exhaust emissions (< 5 µm) at a detection
limit of 0.06 µg/g. In exhaust dilution tunnels, platinum was
detected in larger particles in the range of 0.034 to 635 µg/g
sample; whole or fragmented pellets contained the highest
concentrations.
Reliable emission data for the pellet-type catalyst come from a
study conducted by the General Motors Corporation (Hill & Mayer,
1977), in which emission rates as well as the soluble fraction were
determined by a radio-metric method. Platinum emission was found to
be 0.8 to 1.2 µg per km travelled in low-speed runs (starts and
stops, maximum speed 48 km/h) and 1.9 µg per kilometre travelled in
high-speed runs (96 km/h). It should be noted that these results
relate to the first 250 km of catalyst life. Lower loss rates would
be expected with increasing age of the catalyst. Of the particles
collected, 80% had particle diameters greater than 125 µm.
Experiments with an engine test stand using laboratory prepared
catalysts indicated that about 10% of the platinum emitted is water
soluble. However, the statistical significance of these results was
not reported. Even so, these emission data provide the best basis
for the estimation of expected ambient air concentrations resulting
from the introduction of pellet catalysts (see section 5.1.1).
However, this type of automobile catalyst is no longer used on new
cars in the USA, and has never been used in Europe where only
monolithic catalysts are on the market.
Emission data are available concerning the new generation
monolith-type catalyst. In Germany the Fraunhofer Institute of
Toxicology and Aerosol Research (König et al., 1989, König & Hertel,
1990) has conducted engine test stand experiments as part of a
programme of the Ministry of Research and Technology for assessing
the relative risk of this new man-made environmental source (GSF,
1990). First results indicated that platinum emission is lower by a
factor of 100 than in the case of pelleted catalysts: at a simulated
speed of 100 km/h, total loss from a three-way catalyst was
measured, using the AAS method, to be on average about 17 ng/m3 in
the exhaust gas (König et al., 1989). In further experiments this
value was validated (König & Hertel, 1990): the mean platinum
emission from two catalysts was found to be 12 and 8 ng/m3,
respectively. As shown in Table 5, platinum emission seems to be
temperature dependent. At an exhaust gas temperature of 690° C and a
simulated speed of 140 km/h, about 35-39 ng/m3 was found in the
exhaust gas. The mean aerodynamic diameter of the particles
collected after the muffler (silencer) on a Berner impactor varied
between 4 and 9 µm. Preliminary results indicated that approximately
10% of the total platinum penetrated a depth-type filter to be
trapped in the condensate (König et al., 1989), but this single
measurement could not be confirmed by subsequent determinations
where the platinum content in the condensate was below the detection
limit (0.1 ng/ml) (König & Hertel, 1990).
Schlögl et al. (1987) analysed microparticles emitted from
automobile exhaust and collected on several conducting surfaces. In
experiments with diesel and gasoline engines equipped with
catalysts, they found detectable traces of platinum. In diesel
engine exhaust it was presumed that most platinum would be in the
oxidation state 0 (platinum black). A small part was found to be
Pt(IV), probably in the oxide form. The platinum emission from
gasoline engines showed a photoemission spectrum indicating that
platinum is probably emitted mostly in the form of surface oxidized
particles.
Table 5. Mean platinum emissions from two monolith catalysts (1 and 2)
at different engine test stand runsa
Platinum emission
Simulated Number Exhaust gas Exhaust ng per km Mean aerodynamic
speed of samples temperature gas travelledb diameter (µm)
(km/h) (° C) (ng/m3)
(1) (2) (1) (2) (1) (2)
60 18 480 3 4 2 3 6 9
100 39 600 12 8 10 8 4 6
140 18 690 39 35 39 35 6 8
a Adapted from König et al. (in press)
b Calculated assuming that on average 10 m3 exhaust gas is emitted per litre
gasoline and a gasoline kilometrage of 7, 8, and 10 litres per 100 km travelled,
respectively.
3.2.2 Uses
The principal use of platinum derives from its special
catalytic properties. Further applications in industry are related
to other outstanding properties, particularly resistance to chemical
corrosion over a wide temperature range, high melting point, high
mechanical strength, and good ductility. Platinum has long been
known to have excellent catalytic properties. Before the
introduction of catalytic converters in automobiles, most of the
platinum was used as a catalyst in hydrogenation, dehydrogenation,
isomerization, cyclization, dehydration, dehalogenation, and
oxidation reactions. One of its major industrial uses is for
naphtha-reforming to upgrade catalytically the octane rating of
gasoline. Other catalytic uses are in ammonia oxidation to produce
nitric acid, hydrogen cyanide manufacture, the reduction of nitro
groups and, in the automobile catalyst application, the conversion
of carbon monoxide to carbon dioxide and nitrogen oxide to nitrogen
and water (NAS, 1977; Stokinger, 1981).
As shown in Table 6, in the USA in 1973, before the
introduction of the automobile catalyst, most of the platinum was
used for catalytic purposes in the chemical and petroleum industry.
In 1987 the use pattern had completely changed and 71% of the
platinum sold was used by the automobile industry. In 1987, a
typical USA car catalyst contained about 1.77 g of platinum and 10.6
million vehicles with catalysts were produced (Loebenstein, 1988);
this accounts for the 18.8 tonnes shown in Table 6.
Table 6. Platinum sales to various types of industry in the USA
before and after the introduction of automotive catalytic
convertersa
Industry 1973 1987
kg/year % of total kg/year % of total
Automobile - - 18 817 71.3
Chemical 7434 36.3 1920 7.5
Petroleum 3844 18.8 739 2.8
Dental and
medical 868 4.2 479 1.9
Electrical 3642 17.9 1821 7.1
Glass 2255 11.0 285 1.1
Jewellery and
decorative 697 3.4 177 0.7
Miscellaneous 1732 8.5 1430 5.6
Total 20 472 100 25 668 100
a From: Butterman (1975); Loebenstein (1988)
Tables 7 and 8 show the platinum demand by application in the
Western world, also reflecting the increased demand during recent
years. In 1989, total demand was 90 tonnes.
Platinum oxidation catalyst technology, developed to reduce
automobile exhaust emissions, has been extended to other
environmental control applications such as the reduction of carbon
monoxide and hydrocarbon emissions from large gas turbines (Jung &
Becker, 1987) and the transformation of hydrogen molecules into
active hydrogen atoms to reduce chlorohydrocarbons such as
trichloroethylene to ethane in water (Wang & Tan, 1987).
Table 7. Western-world platinum demand (kg/year) by applicationa
1980 1981 1982 1983 1984 1985 1986 1987 1988 1989
Automobile catalyst
gross 19 278 18 144 18 569 18 285 23 814 27 783 32 318 35 579 37 563 41 107
recovery 0 0 283 850 1276 1984 2551 3260 4536 4961
Chemical 7371 7087 7371 6946 7371 6379 5528 5528 4536 4536
Electrical 5953 5245 4819 4961 5386 5670 5103 5103 5245 5528
Glass 3969 2835 2410 2977 3969 3969 2551 3402 3685 3969
Investment
small 0 0 1276 2551 4819 7371 12 757 6095 9355 3685
large 4536 5528 3260 1843 4252 4819 3544 7796 8505 850
Jewellery 15 876 21 404 21 687 20 270 21 971 22 963 24 097 28 066 33 452 36 996
Petroleum 3685 3969 1843 567 425 425 567 1559 1417 2126
Other 5386 4678 4819 4252 3827 2835 3685 3402 3402 3260
Total 66 054 68 889 65 771 61 802 74 559 80 230 80 511 93 270 102 624 97 096
a From Johnson Matthey (1990)
Table 8. Regional platinum demand (kg/year) by applicationa
1980 1981 1982 1983 1984 1985 1986 1987 1988 1989
Japan
Automobile catalyst
grossb 5953 5386 4819 4819 4819 5953 7229 8788 9355 10 064
recoveryc 0 0 0 0 0 0 142 425 709 709
Chemical 283 283 283 283 425 425 425 425 425 425
Electrical 425 425 567 567 50 1134 1276 1276 1276 1417
Glass 1134 1417 1276 1701 2126 1701 850 1276 1276 1134
Investment
small 0 0 0 142 425 992 992 1701 3260 992
large 4536 5528 3260 1843 4252 4819 3544 7796 8505 850
Jewellery 12 474 17 718 17 577 15 876 17 718 19 136 20 979 25 515 30 050 32 602
Petroleum 425 425 425 425 567 425 0 0 0 0
Other 1417 1417 1559 1276 1134 850 567 425 425 425
Total 26 647 32 599 29 766 26 932 32 316 35 435 28 632 46 777 53 863 47 200
North America
Automobile catalyst
gross 12 474 12 190 12 899 12 757 18 002 19 845 21 120 19 561 19 561 20 412
recovery 0 0 0 850 1276 1984 2410 2835 3827 4252
Chemical 3260 1417 2268 2835 2835 2126 1843 1559 1559 1559
Electrical 4111 1984 1984 2551 2693 2268 1843 1843 1843 2126
Glass 1417 567 283 425 850 1134 709 709 709 850
Investment 0 0 1134 1134 850 3685 8505 2410 2410 1559
Jewellery 425 425 425 425 425 425 425 425 425 567
Petroleum 3969 1559 567 425 425 283 283 425 425 1134
Other 2126 1701 567 709 992 850 1417 1417 1417 1417
Total 27 782 19 843 20 127 20 411 25 796 28 632 33 735 25 514 24 522 25 372
Table 8 (contd).
1980 1981 1982 1983 1984 1985 1986 1987 1988 1989
Rest of Western world, including Europe
Automobile catalyst
gross 850 567 567 709 992 1984 3969 7229 8647 10 631
recovery 0 0 0 0 0 0 0 0 0 0
Chemical 3827 5386 4819 3827 4111 3827 3260 3544 2551 2551
Electrical 1417 2835 2268 1843 1843 2268 1984 1984 1984 1984
Glass 1417 850 850 850 992 1134 992 1417 1701 1984
Investment 0 0 142 1276 3544 2693 3260 1984 3685 1134
Jewellery 2977 3260 3685 3969 3827 3402 2693 2126 2977 3827
Petroleum 709 1984 850 283 567 283 283 1134 992 992
Other 1843 1559 2693 2268 1701 1134 1701 1559 1559 1417
Total 11 622 16 441 15 874 14 459 16 443 16 159 18 142 20 977 24 096 24 520
a From: Johnson Matthey (1990)
b Gross automobile catalyst demand is purchase of platinum by the auto industry for the manufacture of automobile catalysts.
c Automobile catalyst recovery is platinum recovered from catalytic converters removed from scrapped automobiles.
Platinum and platinum-rhodium alloys have many high-temperature
uses. Thermo-electrical applications arise from the simple and
stable relationship between resistance and temperature that platinum
exhibits over a wide temperature range. This explains its use in
platinum resistance thermometers, thermocouples, and strain gauges.
The high melting point of platinum and its resistance to oxidation
and many chemicals has led to its use in vessels in the glass-making
industry and in the fabrication of spinning jets and bushings for
the production of viscose rayon and fibreglass, respectively. It is
also used for laboratory ware, such as crucibles, combustion boats,
and the tips of tongs. Ships' hulls, propellers, and rudders are
protected against corrosion by "cathodic protection" using platinum-
clad anodes (NAS, 1977).
Platinum and/or its alloys have been used in electric contacts
for relays and switchgears for a variety of reasons, including
hardness and good conductivity. Many printed circuits are made using
preparations that contain platinum. Electrochemical platinum
electrodes have been used in preparative chemistry, since they
support many oxidative reactions although they resist oxidation
themselves (NAS, 1977).
A major use of platinum is in jewellery for making rings and
settings. Platinum is also used to produce a silvery lustre on
ceramic glazes (NAS, 1977).
In dentistry, platinum is used in gold-platinum-palladium
alloys to raise the melting-point range and increase the strength.
However, this use is decreasing, since platinum is being replaced by
other materials including palladium (Anusavice, 1985; NAS, 1977).
Platinum has an important role in neurological prostheses, i.e.
surgically implanted microelectronic devices, such as implants for
treating incontinence, or for recovering some use of paralysed limbs
following spinal accidents (Donaldson, 1987).
Platinum-iridium electrodes are used for long-term electrode
implantation for recording electrical activity and for stimulation
in human tissues and organs, e.g., pacemakers (Theopold et al.,
1981).
All these applications use platinum as a pure metal or in the
form of alloys, but soluble platinum salts are also used in the
manufacture of these products; e.g., hexachloroplatinic acid may be
used in platinizing alumina or charcoal in catalyst production. A
number of salts can be used in the electrodeposition of platinum,
e.g., sodium hexahydroxyplatinate(IV), Na2[Pt(OH)6].2H2O,
diamminedinitroplatinum(II), [Pt(NO2)2(NH3)2], hydrogen
dinitrosulfatoplatinate(II), H2[Pt(NO2)2SO4], and
tetraammineplatinum(II) compounds such as the hydrogenphosphate,
sulfamate, citrate, and tartrate (Baumgärtner & Raub, 1988; Skinner,
1989).
Complexes of platinum, particularly cis-
diamminedichloroplatinum(II) (cisplatin) (see footnote in section
1.2), have been used to treat cancer. In patients with testicular
cancers, remissions rates of more than 90% have been achieved
(Lippert & Beck, 1983).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1 Transport and distribution between media
By comparison with other elements, platinum-group metals are
distributed sparsely in the environment. Since platinum is so
valuable, great care is taken to avoid significant loss during
mining and refining processes, and during use and disposal of used
platinum-containing objects. Up to 1984, about 1050 tonnes of
platinum had been refined. Most of this has been used in the form of
the metal and platinum oxides, which are practically insoluble in
water, resistant to most chemical reactions in the biosphere, and do
not volatilize into air (Renner, 1984).
Part of the platinum released into the air from automobile
emissions (section 5) is deposited close to the roads and could be
washed off by rain into rivers and coastal marine waters (Hodge &
Stallard, 1986). However, only small amounts of platinum have been
detected in environmental samples (see sections 5.1.2. and 5.1.3.).
Large amounts of metals including platinum can be transported
in rivers draining major industrialized regions, leading to elevated
platinum concentrations in sediments (section 5.1.3).
Platinum forms soluble complexes with ammonia, cyanide, amines,
olefins, organic sulfides, and tertiary arsines. However, the level
of these ligands in natural waters is insufficient to make platinum
mobile (Fuchs & Rose, 1974).
Organic matter has a role as a vehicle for the transport of
platinum and for bringing about its precipitation or concentration.
There is a good correlation between high contents of platinum and
organic carbon in polluted stream sediments of the Ginsheimer-
Altrhine river, near Mainz, Germany (see section 5.1.2), and it is
assumed that organic matter such as humic and fulvic acids binds
platinum, aided perhaps by appropriate pH and redox potential
conditions in the aquatic environment (Dissanayake, 1983).
Detailed information about the geochemical behaviour of
platinum-group metals is available from the platinum mining area of
Stillwater, Montana, USA (Fuchs & Rose, 1974). The mobility of
platinum depends on pH, the redox potential, chloride concentrations
in soil water, and the mode of occurrence of platinum in the primary
rock. The relation between redox potentials and pH conditions
indicates that platinum behaviour also depends on the kind of ore it
is associated with. If bound in chromite, it has essentially no
mobility in weathering because of the resistant character of
chromite. On the other hand, platinum in the form of trace mineral
inclusions in sulfides is readily released by oxidation during
weathering. Calculated relations between pH and redox potential
indicate that increased chloride concentrations in soil water will
promote mobility. Thus, platinum will be mobile only in extremely
acid waters or those with a high chloride level (Fuchs & Rose,
1974).
In twigs from four limber pines (Pinus flexilis) in the
platinum mining area of Stillwater, the platinum concentrations were
the same as in the adjacent soil. It was concluded that limber pine
does not concentrate platinum, probably due to the limited mobility
of platinum (Fuchs & Rose, 1974). However, high concentrations of
platinum were found in the roots of nine horticultural crops
(cauliflower, radish, snapbean, sweet corn, pea, tomato, bell
pepper, broccoli, and turnip) grown in Hoagland's hydroponic culture
solution containing platinum tetrachloride concentrations of 0.057,
0.57, or 5.7 mg/litre (Pallas & Jones, 1978; see section 7.3). For
example, at the highest concentration, cauliflower and tomato roots
contained 1425 and 1710 mg Pt/kg, respectively. Only pepper,
cauliflower, and radish accumulated platinum in their tops, but to a
very limited extent. From the data of Pallas & Jones (1978) it is
not clear whether they differentiated between contamination of the
root surface and true uptake of platinum. However, these results
indicate that platinum can enter food crops but the bioavailability
essentially depends on the solubility of the platinum species. It
should be noted that the salt (PtCl4) used by Pallas & Jones
(1978) is soluble in water.
In the context of a German government programme (see section
3.2.1.4), Rosner et al. (1991) conducted engine test stand
experiments with a three-way-catalyst-equipped engine (monolith-type
catalyst) to determine platinum uptake by plants. Grass cultures
(Lolium multiflorum) were placed in continuously stirred tank
reactors and exposed to slightly diluted (1:10/20) exhaust gas for 4
weeks (8 h/day, 5 days/week). Using atomic absorption spectrometry
for the measurement of platinum emissions (see section 2.4.3.8,
König & Hertel, 1990), no platinum could be detected in the shoots
at a detection limit of 2 ng/g dry weight.
4.2 Biotransformation
By analogy, platinum compounds may undergo biotransformation
comparable to processes described for other metals. The
biomethylation of platinum compounds, i.e. [Pt(IV)Cl6]2-,
[Pt(IV)(CN)4Cl2]2-, [Pt(IV)(CN)5Cl]2-, and
[Pt(IV)(SO4)2], has been established only in in vitro test
systems (Taylor, 1976; Wood et al., 1978; Fanchiang et al., 1979;
Taylor et al., 1979; Fanchiang, 1985).
Methylcobalamin (MeB12) reacts with Pt(II) and Pt(IV)
complexes to give a methylated platinum compound. Agnes et al.
(1971) reported that this reaction requires the presence of platinum
in both oxidation states. Spectrophotometric measurements showed the
consumption of one mole of [Pt(IV)Cl6]2- per mole of MeB12,
[Pt(II)Cl4]2- being required only in catalytic quantities.
Aquocobalamin (aquo-B12) and methylplatinum were shown to be the
products of the reaction (Taylor & Hanna, 1977).
From these laboratory data produced under abiotic conditions it
is not, however, possible to conclude that microorganisms in the
environment are able to biomethylate platinum complexes.
4.3 Ultimate fate following use
The value of platinum-group metals has greatly increased and
methods for their recovery from spent catalysts are of economic
importance.
Platinum metal has been successfully recycled from used
chemical and petroleum catalysts for many years, but many companies
are still trying to find a successful formula for retrieving it from
automobile catalysts. The latter accounts for more than 30% of the
total platinum-group metal consumption in the USA. The US Office of
Technology calculated that if 50-60% of catalytic converters were
recovered for their metal value, about 7717 kg platinum per year
could be reclaimed in 1990. However, currently only between 25 to
40% of the used converters are being reclaimed (Agoos, 1986).
According to another estimate, 5443 kg of platinum was recovered in
1989 from automobile catalysts, of which 4666 kg was recovered in
the USA (Johnson Matthey, 1990).
In contrast to automobile catalysts, almost 100% of spent
reforming and gauze catalysts are collected for their metal value.
This is based on their much higher platinum metal content (Agoos,
1986).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Ambient air
Few measurements of platinum ambient air concentrations have
been reported. Results obtained before the introduction of cars with
catalytic converters can serve as a baseline. Air samples taken near
freeways in California, USA, and analysed using atomic absorption
spectrometry were below the detection limit of 0.05 pg/m3 (Johnson
et al., 1975; 1976).
No platinum could be detected in two air samples collected by
Ito & Kidani (1982) in an industrial area of Nagoya, Japan, in 1981.
Close to city roads in Frankfurt, Langenbrügge, Germany, the
platinum air concentrations (particulate samples) were measured in
1989 to be between < 1 and 13 pg/m3. In rural areas the
concentrations were < 0.6-1.8 pg/m3 (Tölg & Alt, 1990). At the
time of these measurements, few German cars were equipped with
catalysts. Thus, these levels virtually reflect background levels.
Rosner & Hertel (1986) estimated ambient air concentrations for
different scenarios, based on dispersion models used by US EPA
(Ingalls & Garbe, 1982) and on the emission data of Hill & Mayer
(1977) (see section 3.2.1.4). As shown in Table 9, total platinum
concentrations near and on roads could range from 0.005 to 9
ng/m3. Estimates for parking and personal garages were also made,
based on an assumed emission rate of 1 µg/min for total platinum,
but this is definitely an overestimate. It can be assumed that the
emission of platinum depends on the exhaust gas temperature. At
idling or very low speed conditions, emissions are expected to be
negligible (see section 3.2.1.4).
As described in section 3.2.1.4, emission data indicate that
the total platinum emission of a monolith-type catalyst is probably
lower by a factor of 100 than that of a pellet-type catalyst.
Assuming an average emission rate of approximately 20 ng/km (see
section 3.2.1.4) and applying the same dispersion models, the
theoretical ambient air concentrations would be lowered to the
picogram to femtogram per m3 range (see Table 9).
Table 9. Estimated ambient air concentrations of total platinum
at various exposure conditions, based on an emission rate
of 2 µg/km from the pelleted catalyst and 0.02 µg/km
from the monolithic three-way catalyst
Exposure situationa Ambient Pt concentration(ng/m3)
Pelleted Monolithic
catalyst catalyst
Roadway tunnel
Typical 4 0.04
Severe 9 0.09
Street canyon (sidewalk receptor)
Typical a) 800 vehicles per h 0.1 0.001
Typical b) 1600 vehicles per h 0.3 0.003
Severe a) 1200 vehicles per h 0.5 0.005
Severe b) 2400 vehicles per h 0.9 0.009
On expressway
Typical 0.7 0.007
Severe 1.6 0.016
Beside expressway (short-term)
Severe 1 m 1.3 0.013
10 m 1.1 0.011
100 m 0.3 0.003
1000 m 0.04 0.0004
Beside expressway (annual)
Severe 1 m 0.2 0.002
10 m 0.15 0.0015
100 m 0.04 0.0004
1000 m 0.005 0.00005
a Calculations based on dispersion models used by US EPA;
"Typical/severe" depends on wind conditions and road width
(Ingalls & Garbe, 1982)
Hodge & Stallard (1986) analysed roadside dust deposited in San
Diego, California, USA. At the edge of a major freeway (154 000
vehicles/day), dust samples contained the highest concentration (680
µg Pt/kg dry weight; 680 ppb). At a distance of about 34 m, the
platinum content of 100 µg/kg was about 7 times lower. At the edge
of another heavily used freeway (96 000 vehicles/day) platinum
content was 250 µg/kg, while with less heavy traffic (14 000
vehicles/day) 260 and 300 µg/kg were found in two dust samples. The
lowest concentrations, 37 and 60 µg/kg, were found in samples
collected from plants growing in the yards of houses located on
highly used road. The platinum concentration was not correlated with
the lead concentration. However, the samples with the highest
platinum concentrations also had the highest lead values. Although
the number of samples was limited, the results indicate that
automobile catalysts release platinum. However, it should be noted
that platinum emissions from pelleted catalysts were probably
responsible for the concentrations reported and that the use of
monolith catalysts should result in much lower platinum
concentrations in the roadside environment.
5.1.2 Water and sediments
In a study to determine baseline levels of platinum, Johnson et
al. (1976) analysed tap water samples collected in Lancaster and Los
Angeles, California, USA. No platinum was found at a detection limit
of 0.08 µg/litre. In tap water (probably only one sample) from
Liverpool, United Kingdom, a platinum content of 0.06 µg/litre was
determined by adsorptive cathodic stripping voltametry (Van den Berg
& Jacinto, 1988).
Investigations of platinum concentrations in Lake Michigan
sediments led to the conclusion that platinum has been deposited
over the past 50 years at a constant rate. Concentrations at
sediment depths of 1-20 cm varied between 0.3 and 0.43 µg/kg dry
weight (Goldberg et al., 1981). In comparison, lead concentrations
have markedly increased in the sediment due to increased emissions
from industry and motor traffic.
Lee (1983) noted a rapid increase in the palladium contents of
the sediments from the Palace Moat, Tokyo, Japan, between 1948 and
1973 and attributed it to the introduction of car catalysts.
However, this is not conclusive as the palladium content in the
sediment had already begun to increase in 1964-1965, before the
introduction of the catalytic converter, and even in 1973 only a few
cars were equipped with converters.
Dissanayake et al. (1984) determined platinum concentrations in
the sediments of a cut-off channel of the Rhine river near Mainz,
Germany. Sediment samples from this highly polluted river were
sieved and the < 2 µm fraction was analysed by flameless AAS. The
platinum concentrations in 12 samples collected at different sites
varied over a wide range. In four samples no platinum was detected,
while eight samples contained between 730 and 31 220 µg/kg (dry
weight). This is higher by a factor of up to 15 000 compared to
unpolluted average North Sea sediments. The high variation was
attributed to differences in pH and redox conditions. The extremely
high concentrations appeared at the interface between an extremely
reducing and an oxidizing aquatic environment that provided,
together with a pH of 6.6-7.8, optimum conditions for the formation
of metal-organic complexes. The sample containing 31 220 µg Pt/kg
also contained the highest concentration of palladium (4000 µg/kg).
The gold content (100-400 µg/kg) had a relatively uniform
distribution, but was also indicative of a high state of pollution.
Using a more sensitive graphite furnace AAS method, Goldberg et
al. (1986) detected very low platinum concentrations in sea water.
Samples of filtered water (0.45-µm filter) from the open Eastern
Pacific Ocean showed an increase in platinum concentration with
depth from surface values of around 100 to a value of 250 pg/litre
at 4500 m. Similar concentration profiles were obtained in
unfiltered sea water taken from the California Borderline region
(Hodge et al., 1985). Sea-water samples analysed by Van den Berg &
Jacinto (1988) were also within this concentration range. A deep-sea
and a shallow-water sample from the Indian Ocean contained 154 and
37 pg/litre, respectively, whereas sea water of coastal origin
contained 332 pg/litre. It should be noted that these were only
single samples.
In sediment cores from the Eastern Pacific taken to a depth of
6-22 cm in carbonate and siliceous ooze, platinum concentrations
varied between 1.1 and 3 µg/kg (dry weight basis). Lower
concentrations (0.3 µg/kg) were reported in the Santa Barbara Basin
(Hodge et al., 1985). The highest concentration (21.9 µg/kg) was
found in pelagic ocean sediments (Hodge et al., 1986).
In several investigations, the platinum content of seamount
ferromanganese nodules or crusts was studied. In deep-sea nodules
from the Northwest Pacific nodule belt, platinum concentrations from
< 5 to 145 µg/kg were found (Agiorgitis & Gundlach, 1978).
Platinum values in ferromanganese seamount crusts from the
Central Pacific were much higher and varied between 140 µg/kg at
3780 m and a maximum of 880 µg/kg at a depth of 1120 m (Halbach et
al., 1984). Both platinum and nickel concentrations correlated
positively with manganese content and led to the conclusion that
platinum and nickel are incorporated in the manganese oxide
fraction. It was suggested that the high platinum concentration in
the crusts is derived directly from sea water by a process of
specific adsorption onto colloidal particles of hydrous manganese
oxide, which has a negative surface charge in sea water.
In a further investigation, platinum concentrations in
ferromanganese minerals from various localities were found to vary
between 6 and 940 µg/kg (Goldberg et al., 1986). In manganese
nodules obtained at depths of between 1700 and 4200 m in the Pacific
Ocean, platinum concentrations varied between 138 and 940 µg/kg
(Hodge et al., 1986).
5.1.3 Soil
Few measurements of platinum in soil have been reported. In the
baseline study of Johnson et al. (1976), all surface soil samples
collected near freeways in California, USA, and in a mining area in
Sudbury, Canada, were below the detection limit of 0.8 µg/kg.
In the USA, the National Academy of Sciences (NAS, 1977)
estimated the accumulation of platinum in roadside environments on
the basis of an emission rate of 1.9 µg per km from cars equipped
with catalytic converters and a frequency of 5000 cars per day.
Assuming that all emitted platinum was localized near the freeway in
the topsoil (uniformly distributed about 30 cm deep over a width of
about 90 m and a length of 1.6 km, with a soil density of 1.5
g/cm3), a platinum concentration after 10 years of 8 µg/kg could
be expected.
5.1.4 Food
Hamilton & Minski (1972/1973) estimated a total daily platinum
intake of less than 1 µg/day, based on an analysis of a United
Kingdom total-diet sample and 1963 United Kingdom consumption and
population figures. No data were given on the platinum content of
the foods analysed.
5.1.5 Terrestrial and aquatic organisms
Fuchs & Rose (1974) analysed samples of twigs from four limber
pines (Pinus flexilis) in the Stillwater mining area, Montana,
USA. Three samples contained between 12 and 56 µg Pt/kg (ash
weight), while one contained platinum at a level below the detection
limit. The content of the adjacent soils was also in this range, so
that no evidence for accumulation could be derived from these
limited data (see also section 4.1).
Using neutron activation analysis (section 2.4.3.2) Valente et
al. (1982) measured the following platinum concentrations in
isolated samples of plants from an ultrabasic soil: Fragaria
virginiana, 830 µg/kg (dry weight); Prunella vulgaris, 440
µg/kg; Aspidotis densa, 100 µg/kg.
In marine macroalgae the following platinum concentrations (on
a dry weight basis) were found near La Jolla, California, USA (Hodge
et al., 1986): red algae Prionites australis and Opuntiella
californica, 0.19 and 0.08 µg/kg, respectively; brown algae
Macrocystis pyrifera and Pterygophora californica, 0.22 and 0.32
µg/kg, respectively.
5.2 General population exposure
Two studies were conducted in the USA to establish baseline
levels of platinum in the tissues and body fluids of the general
population prior to the introduction of automobile catalysts.
Johnson et al. (1975, 1976) analysed autopsy tissue samples
from 10 people, 12 to 75 years old, who died from a variety of
causes in Southern California. All samples taken from liver, kidney,
spleen, lung, muscle, and fat were below the detection limits
(0.2-2.6 µg/kg wet weight). Samples collected from 282 people from
Southern California living near a heavily used urban freeway (Los
Angeles) or in a desert area near Lancaster also showed platinum
concentrations below the detection limits (blood, < 31 µg/litre;
urine, < 0.6 µg/litre; hair, < 50 µg per kg; faeces, < 2 µg/kg).
Only in pooled blood samples were detectable concentrations
measured, i.e. 0.49 µg/litre in the Los Angeles group and 1.8
µg/litre in the Lancaster group.
In a second study, tissue samples were taken from autopsied
individuals from Southern California (95 people) and New York (2
people), who had not been knowingly exposed to platinum either
occupationally or by medical treatment (Duffield et al., 1976). In
42 individuals no platinum was detected. Of the 1313 samples
collected, only 62, i.e. 5%, had detectable concentrations of
platinum ranging from 0.003 to 1.46 mg/kg wet weight (mean 0.16
mg/kg, median 0.067 mg/kg). Table 10 shows the frequency of platinum
detection in the various tissue samples. The frequency of occurrence
was taken as a measure of the distribution of platinum among various
body organs. Platinum was frequently found in subcutaneous fat. This
is surprising, as most platinum compounds are regarded as lipid-
insoluble. Other target sites were kidney, pancreas, and liver.
However, the analytical accuracy has been questioned and
contamination of the samples suspected (NAS, 1977), because the
baseline levels found by Johnson et al. (1976) were at least one
order of magnitude lower. The problem of questionable analytical
reliability reflects the difficulties in interpreting data on trace
levels of platinum in the environment and in human tissues and body
fluids.
New data have been provided by Nygren et al. (1990). Using
absorptive voltametry (see section 2.4.3.5), the background levels
of platinum in human blood were found to be in the range of 0.1-2.8
µg/litre (median 0.6 µg per litre). These results were verified by
inductively coupled plasma mass spectrometry using gold as an
internal standard.
5.3 Occupational exposure during manufacture, formulation, or use
Occupational exposure occurs during the mining and processing
of platinum. However, the most common current occupational exposure
to soluble platinum compounds is through inhalation in platinum
refining and catalyst manufacture.
Table 10. Distribution of tissue samples with detectable platinuma
Number of Samples with detectable
samples platinum
analysed No. %
Subcutaneous fat 74 10 14
Kidney 91 11 12
Pancreas 84 10 12
Liver 90 10 11
Brain 9 1 11
Gonad 53 5 9
Adrenal 60 3 5
Muscle (psoas) 97 4 4
Aorta (descending) 92 3 3
Heart (left ventricle) 82 2 2
Spleen 52 1 2
Prostate/uterus 63 1 2
Thyroid 73 1 1
Lung 95 0 0
Vertebra (lumbar) 94 0 0
Rib (fifth) 97 0 0
Femur 57 0 0
Clavicle 30 0 0
Hair, scalp 9 0 0
Hair, pubic 1 0 0
1303 62 5
a From: Duffield et al. (1976)
Many countries have set occupational exposure limits. For
example, in the USA, the time-weighted Threshold Limit Value (TWA-
TLV) for daily occupational exposure has been established for
soluble platinum salts at 2 µg Pt/m3 (ACGIH, 1980, 1990). Many
countries have adopted this ACGIH value. In addition ACGIH (1980,
1990) recommended a Threshold Limit Value of 1 mg/m3 for platinum
metal. In the United Kingdom an occupational exposure limit (8-h
TWA) of 5 mg/m3 has been proposed for platinum metal as total
inhalable dust (Health and Safety Executive, 1990).
The published data base for platinum concentrations at the
workplace is meagre. Due to analytical shortcomings older data are
not considered reliable. In an early investigation (Fothergill et
al., 1945), a platinum content of less than 5 µg/m3 in the
atmosphere in the immediate neighbourhood of a refinery was measured
using particle filters. In the dry salts handling area, platinum
concentrations as high as 70 µg/m3 were found. In another
investigation (Hunter et al., 1945), the platinum content in the
atmosphere at various points in four refineries was estimated. At
most points concentrations varied between 1.6 and 5 µg/m3. Higher
concentrations were found in the neutralization of platinum salts
(20 µg/m3), sieving spongy platinum (400-900 µg/m3), and
crushing ammonium chloroplatinate (1700 µg/m3).
Workplace measurements in a catalyst production plant in the
USSR were reported to exceed an air concentration of 2 µg/m3 in
33% of the measurements (Gladkova et al., 1974).
In a cross-sectional survey (section 9.2), Bolm-Audorff et al.
(1988) reported workplace measurements at a platinum refinery in the
Federal Republic of Germany. In 1986, concentrations of between 0.08
and 0.1 µg/m3 were measured in the filter press area, but in other
working areas platinum salt exposure was generally below the
detection limit of 0.05 µg/m3. No data were given on the number of
samples.
The results obtained during a four-month period of measurements
in a US platinum refinery showed that workplace concentrations
exceeded the occupational limit of 2 µg/m3 between 50 and 75% of
the time (Brooks et al., 1990.
In samples of blood, urine, faeces, and hair from employees at
a Canadian mine near Sudbury, platinum concentrations were below the
limits of detection (0.1 µg per litre or 0.1 µg/kg). Tissue samples
from three out of nine autopsies had detectable platinum
concentrations in fat (4.5 µg/kg), lung (3.7 µg/kg), or muscle (25.0
µg per kg) (Johnson et al., 1976). However, since the three
detectable concentrations were in individuals who, like the other
six, showed no platinum concentrations in liver, kidney and spleen,
sample contamination was suggested (NAS, 1977). It was concluded
that people who work in mining areas probably do not incorporate
significant amounts of platinum into their body.
Blood samples collected from 61 refinery workers in New Jersey
contained no measurable platinum (less than 1.4 µg/litre) (Johnson
et al., 1976). However, platinum levels in 10% of the urine samples
were above the detection limit of 0.1 µg/litre, the maximum reported
value being 2.6 µg/litre.
Using the method of LeRoy et al. (1977), platinum serum levels
in 11 platinum refinery workers with positive skin tests were
analysed. These studies found serum platinum levels ranging from 150
to 440 µg/litre (mean = 240 µg/litre), the quantification limit
being 100 µg per litre (Biagini et al., 1985).
A special case of possible occupational exposure is the
handling of cisplatin and its analogues by pharmacy and nursing
staff and other hospital personnel. In a study with two pharmacists
(one male and one female) and eight female nurses, platinum levels
in urine (0.6-23.1 µg per litre) were at the limit of sensitivity of
the AAS method used and did not significantly differ from the
controls (2.6-15.0 µg/litre). By comparison, the urine of cisplatin-
treated patients contained on average 7 mg/litre (Venitt et al.
1984).
6. KINETICS AND METABOLISM
Most toxicokinetic data on platinum, both for experimental
animals and humans, have been derived from studies with platinum
complexes.
Moore et al. (1975c) studied the whole body retention, lung
clearance, distribution, and excretion of 191Pt in outbred albino
rats (Charles River CD-1 strain) after single nose-only inhalation
exposure to different chemical forms of platinum for 48 min.
Particle concentration in the nose-only exposure chambers was
approximately 5.0 mg per m3 with 191PtCl4, 5-7 mg/m3 with
191Pt(SO4)2, and 7-8 mg/m3 with PtO2 and 191Pt metal.
The aerodynamic diameter was given as 1.0 µm for 191PtCl4 and
191Pt(SO4)2; both aerosols were generated by a nebulizer. The
191PtO2 and 191Pt metal aerosols (aerodynamic diameter not
given) were generated by passing Pt(SO4)2 or PtCl4,
respectively, through a furnace tube and decomposing them at 600 °C.
Whole body counts, showed that most of the inhaled 191Pt was
rapidly cleared from the body, followed by a slower clearance phase
during the remaining post-exposure period. The whole body retention
of 191Pt was approximately 41, 33, 31, and 20%, respectively, of
the initial body burden 24 h after exposure to 191PtCl4,
191Pt(SO4)2, 191PtO2, and 191Pt metal. After ten days,
the body burden was only about 1, 5, 8, and 6%, respectively. This
shows that there was only a slight difference between the clearance
rates for the various chemical forms, although the clearance of
191PtCl4 seemed to be the fastest. Clearance from the lungs also
reflected the two-phase pharmacokinetics in the whole body, with a
fast clearance phase in the first 24 h followed by a slow phase with
a half-time of about 8 days.
Excretion data from the study by Moore et al. (1975c) indicate
that most of the 191Pt cleared from the lungs by mucociliary
action was swallowed and excreted via the faeces (half-time 24 h). A
small fraction of the 191Pt was detected in the urine, indicating
that little was absorbed by the lungs and the gastrointestinal
tract. However, no quantitative data were given.
As shown in Table 11, the portals of entry, lung and trachea,
contained most of the platinum, i.e. 93.5% and 3.9%, respectively,
of the total radioactivity (48 618 counts/g) 1 day after exposure.
Of the other tissues analysed, highest levels were found in the
kidney and bone, suggesting some accumulation in these organs. The
low percentages of 1.5% and 0.6%, respectively, on day 1, reflect
only a low accumulation tendency; no information on the statistical
significance of these figures was provided.
Table 11. Radioactive191Pt distribution in the rat following
inhalation exposure to platinum metal
(7-8 mg/m3, 48 min)a
Mean counts/g wet weight after exposure for
1 day 2 days 4 days 8 days
Blood 61 43 30 12
Trachea 1909 2510 738 343
Lung 45 462 28 784 28 280 23 543
Liver 52 46 37 17
Kidney 750 1002 906 823
Bone 281 258 231 156
Brain 5 3 1 0
Muscle 22 10 28 0
Spleen 39 73 23 5
Heart 37 58 23 5
a From: Moore et al. (1975c)
In a comparative study on the fate of 191PtCl4 (25 µCi per
animal) in rats following different routes of exposure (Moore et
al., 1975a,b) retention followed the classical pattern. The highest
retention was found after intravenous administration, the next
highest after intratracheal, and the lowest after oral
administration. For comparison, retention after inhalation was lower
than after intratracheal administration. However, the total dose was
much higher with inhalation (7000 µCi) than with intratracheal (25
µCi) administration. Only a minute amount of 191PtCl4 given
orally was absorbed. Most of it passed through the gastrointestinal
tract and was excreted via the faeces. After 3 days less than 1% of
the initial dose was detected in the whole body. Following
intravenous administration, 191Pt was excreted in almost equal
quantities in both faeces and urine but elimination was slower than
after oral dosing. After 3 days, whole body retention was about 65%
and after 28 days it was still 14% of the initial dose. By
comparison, following intratrachael administration about 22% and 8%,
respectively, were retained by the body after these periods (Moore
et al., 1975a,b).
In the same studies the tissue distribution of 191Pt was
determined. After the single oral dose, the kidney and liver
contained the highest concentrations, while in the other organs
there were no elevated levels. In contrast, after intravenous
administration 191Pt was found in all tissues (Table 12). The high
concentration of 191Pt found in the kidney shows that once
platinum is absorbed most of it collects in the kidney and is
excreted in the urine. The liver, spleen, and adrenal gland also
contained higher platinum concentrations than the blood. The lower
level in the brain suggests that platinum ions probably cross the
blood-brain barrier only to a limited extent (Moore et al.,
1975a,b).
This was confirmed by Lown et al. (1980) in male Swiss mice
given single intragastric doses of Pt(SO4)2 (144 or 213 mg Pt/kg
body weight). Platinum levels in the blood were several times higher
than in the brain. Clearance from the whole body was slower than in
the rat studies. This could be due to species-specific differences.
In addition, the mice received much higher doses than the rats. Lown
et al. (1980) noted an enhancing effect of the higher dose on
absorption.
In a long-term study, Holbrook (1977) found evidence that a
platinum-binding protein is induced. Male Sprague-Dawley rats
received platinum salts ad libitum either in the drinking-water or
in the dry feed. The sequential platinum contents in the tissues
analysed are shown in Table 13. The data demonstrate that the oral
administration of water-soluble platinum compounds, i.e. PtCl4 and
Pt(SO4)2, results in accumulation of platinum in some organs,
primarily the kidney. After 4 weeks, the platinum content of the
kidney was about 8-fold higher than that of the liver and spleen,
and at least 16-fold higher than in the blood and testis (except for
the highest dose of PtCl4). The total platinum intake after 4
weeks increased by 4.3 times and the platinum content in kidney,
spleen, and blood increased by at least 7 times as compared with the
1-week levels. It is notable that a more than 2-fold increase in the
intake of platinum (after a 4-week consumption of PtCl4 in the dry
feed; 743 vs. 1616 mg Pt/rat) did not lead to an increase in the
platinum content of the kidney, in contrast to the situation in the
liver and spleen. This observation was not corroborated with
Pt(SO4)2.
Table 12. Radioactive191Pt distribution (counts/g wet weight) in the rat following a
single intravenous dose of PtCl4(25 µCi/animal)a
Tissue 1 day 2 days 7 days 14 days
% counts/g % counts/g % counts/g % counts/g
Blood 0.91 22 147 0.81 19 732 0.52 12 774 0.32 7921
Heart 0.48 11 819 0.50 12 201 0.36 8 805 0.19 4593
Lung 0.75 18 432 0.66 16 139 0.46 11 180 0.24 5770
Liver 1.51 36 848 1.28 31 274 1.05 25 732 0.19 4733
Kidney 6.65 162 227 6.59 160 656 5.66 138 010 1.24 30 195
Spleen 1.68 41 085 1.89 45 840 2.29 55 764 0.86 20 973
Pancreas 0.91 22 208 0.80 19 487 0.60 14 802 0.16 3973
Bone 0.53 13 146 0.52 12 800 0.37 8932 0.22 5440
Brain 0.05 1150 0.10 2485 0.02 595 0.01 265
Fat 0.18 4487 0.18 4501 0.13 3201 0.02 429
Testes 0.17 4186 0.27 6540 0.16 3873 0.06 1431
Adrenal 1.86 45 439 1.74 42 363 1.09 26 667 0.25 6190
Muscle 0.19 4798 0.19 4671 0.14 3441 0.09 2146
Duodenal
segment 0.52 12 725 0.25 6044 0.16 4031 0.06 1410
a Adapted from: Moore et al. (1975a)
Table 13. Dietary levels, total platinum consumption, and platinum content of tissues
after oral administration of platinum salts to ratsa
Pt consumption Pt content (mg/kg wet weight; mean ± SE)b
Platinum Duration Dietary Total
salt (weeks) level (mg Pt/ Liver Kidney Spleen Testis Brain Blood
(as Pt) rat)
PtCl4 1 319c 59 2.2 4.8 0.24 0.23
± 0.2
PtCl4 4 319c 255 2.5 33.7 4.8 1.5 0.11 2.1
± 0.9 ± 3.5 ± 1.5 ± 0.5 ± 0.07 ± 0.4
PtCl4 4 1147d 743 3.2 33.5 3.1 1.1 < 0.02 1.5
± 0.9 ± 6.3 ± 0.9 ± 0.4 ± 0.4
PtCl4 4 2581d 1616 8.9 32.4 6.4 1.7 0.12 1.6
± 1.2 ± 4.6 ± 3.0 ± 0.3 ± 0.08 ± 0.2
PtCl4 13 106c 389 1.3 14.9 1.6 0.94 < 0.06 0.9
± 0.3 ± 0.4 ± 0.3 ± 0.20 ± 0.08
Pt(SO4)2.4H2O 1 106c 26 0.07 0.26 < 0.02 < 0.04 0.05
± 0.02
Pt(SO4)2.4H2O 1 319c 78 0.85 4.6 0.13 < 0.02 0.22
Pt(SO4)2.4H2O 4 1147d 716 3.5 43.4 3.2 1.1 0.33 1.6
± 0.4 ± 8.3 ± 0.5 ± 0.1 ± 0.18 ± 0.3
PtO2 4 5808d 4308 < 2.2 < 2.2 < 0.02 < 0.07 < 0.02 < 0.04
a Adapted from: Holbrook (1977)
b Standard error (SE) is given for four values; only the mean is given when two values
are available
c mg Pt/litre
d mg Pt/kg
In contrast to the water-soluble salts, insoluble PtO2 was
only taken up in minute amounts even though the salt was
administered in the diet at an extremely high level resulting in a
total consumption of 4308 mg Pt/rat over the 4-week period
(Holbrook, 1977).
Moore et al. (1975a) also administered 191PtCl4 (25
µCi/animal) intravenously to 15 pregnant rats on day 18 of gestation
to determine placental transfer after 24 h. High levels of 191Pt
radioactivity were found in the kidney (127 064 counts/g) and liver
(43 375 counts/g), compared with 10 568 counts/g in the blood.
Accumulation was also found in the placenta (27 750 counts/g).
191Pt was detected in the 60 fetuses examined, but only at very
low concentrations (an average of 432 counts/g). Thus, the placental
barrier is crossed to a limited extent.
In contrast to the simple platinum salts, the diammine
complexes such as cisplatin (see footnote in section 1.2) are
excreted primarily in the urine. In mice, Hoeschele & Van Camp
(1972) found about 90% of the intraperitoneally injected dose in the
urine within five days. Little or no excretion occurred via the
faeces. A high urinary recovery was also observed in rats and dogs
(Hoeschele & Van Camp, 1972; Lange et al., 1972; Litterst et al.,
1976a,b, 1979; Cvitkovic et al., 1977).
The excretion of both cis- and trans-
diamminedichloroplatinum(II) follows a biphasic pattern with a fast
initial alpha-phase and a second slow ß-phase. The variation in the
plasma half-lives is due to species differences and variations in
dose, route of administration, time points analysed, and analytical
method used (Litterst et al., 1979). The extremely rapid alpha-phase
accounts for early, high levels of platinum in kidney, liver, skin,
bone, ovary, and uterus. The prolonged ß-phase results in detectable
urine platinum concentrations 30 days after a single dose.
For both the simple platinum salts and cisplatin complexes, an
initial rapid clearance is followed by a prolonged clearance phase
during the remaining post-exposure period, and there is no evidence
for markedly different retention profiles between these two groups
of platinum compounds (Rosenberg, 1980).
All animal species studied show a similar organ distribution
pattern for cisplatin. An initial distribution to nearly all tissues
is followed by accumulation in the first hour mainly in kidney,
liver, muscle, and skin. By the end of the first day, plasma levels
decrease rapidly and there are elevated platinum levels in numerous
other tissues (Litterst et al., 1979).
Cisplatin is extensively bound to plasma proteins; 90% of it
may be bound 2 h after an intravenous injection. The bound portion
is no longer cytotoxic (Safirstein et al., 1983; Sternson et al.,
1984). In addition to its reactivity with plasma protein, renal
excretion leads to a very low concentration of free cisplatin in the
plasma and to a rapid accumulation in the kidney. Due to the
presence of high chloride ion concentrations, cisplatin is
relatively stable in extracellular fluids (see also section 7.6),
which explains why it is excreted mainly in the unchanged form in
human and rat urine (Safirstein et al., 1983).
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1 Single exposure
Acute toxicity data on platinum mainly relate to its
coordination complexes, the chloroplatinates and ammines. Hofmeister
(1882) was one of the first to test ammonium salts containing
divalent and tetravalent platinum with various numbers of ammine
ligands. He injected solutions of platinum complexes into the dorsal
lymphatic sac of single frogs and subcutaneously into the dorsal
skin of single rabbits. The symptoms observed included vomiting and
diarrhoea with bloody stools and a "curare-like" action of the
salts.
The acute toxicity of platinum depends considerably on the
species of platinum involved (Table 14). Soluble platinum compounds
are much more toxic. Hence, in the study of Holbrook (1976a) oral
toxicity to rats decreased in the following order: PtCl4 >
Pt(SO4)2.4H2O > PtCl2 > PtO2. For the two latter
compounds no LD50 could be derived.
Signs of poisoning observed, for example, with
(NH4)2[PtCl4], include hypokinesia, piloerection, diarrhoea,
clonic convulsions, laboured respiration, and cyanosis (Degussa,
1989a).
Hexachloroplatinic acid is highly nephrotoxic in rats. After an
intraperitoneal LD50 injection of 40-50 mg/kg, rats died of renal
failure, hypocalcaemia, and hyperkalaemia. The necrotizing renal
tubular lesions involved the entire renal cortex (Ward et al.,
1976).
In its metallic state, platinum has an extremely low acute
toxicity. Thus some alloys containing platinum are used in
protheses. Fine dust particles of metallic platinum, 1-5 µm in
diameter, orally administered to rats caused only slight necrotic
changes in the gastrointestinal epithelium, granular dystrophy of
hepatocytes, and swelling in the epithelium of the convoluted renal
tubules (Roshchin et al., 1979, 1984). The highest dose given was
not lethal. The dose was reported as "129 µA/kg" (25 167 µg per kg;
personal communication from Prof. A.V. Roshchin to IPCS dated 3
April 1991).
Due to the different absorption rates for platinum compounds,
the route of administration also affects the toxicity, the
intraperitoneal and intravenous routes leading to much higher
toxicity than the oral route (Table 14).
Table 14. Acute toxicity of platinum and platinum compounds after oral (p.o.),
intraperitoneal (i.p.), and intravenous (i.v.) administration to rats
Compound Route Sexa LD50 Reference
(mg/kg)
PtO2 p.o. m > 8000 Holbrook et al. (1976a,b)
PtCl2 p.o. m > 2000 Holbrook et al. (1976a,b)
PtCl2 p.o. m 3423b Roshchin et al. (1984)
PtCl2 i.p. m 670 Holbrook et al. (1976a,b)
PtCl4 p.o. m 240 Holbrook et al. (1976a,b)
PtCl4 p.o. m/f 276b Roshchin et al. (1984)
PtCl4 i.p. m 38 Holbrook et al. (1976a,b)
PtCl4 i.v. m 26.2 Moore et al. (1975b)
PtCl4 i.v. m 41.4 Moore et al. (1975b)
Pt(SO4)2.4 H2O p.o. m 1010 Holbrook et al. (1976a,b)
Pt(SO4)2.4 H2O i.p. m 310c Holbrook et al. (1976a,b)
Pt(SO4)2.4 H2O i.p. m 138-184c Holbrook et al. (1976a,b)
(NH4)2[PtCl6] p.o. m/f 195b Roshchin et al. (1984)
(NH4)2[PtCl6] p.o. m/f approx. 200 Johnson Matthey (1978a)
(NH4)2[PtCl4] p.o. m 212 Degussa (1989a)
(NH4)2[PtCl4] p.o. f 125 Degussa (1989a)
H2[PtCl6] i.p. m 40-50 Ward et al. (1976)
Na2[PtCl6] p.o. m/f 25-50 Johnson Matthey (1978b)
Na2[Pt(OH)6] p.o. m/f 500-2000 Johnson Matthey (1978c)
K2[PtCl4] p.o. m/f 50-200 Johnson Matthey (1981a,b)
K2[Pt(CN)4] p.o. m/f > 2000 Johnson Matthey (1977a)
[Pt(NH3)4]Cl2 p.o. m/f > 15 000 Johnson Matthey (1977b)
[Pt(NO2)2(NH3)2] p.o. m approx. 5000 Degussa (1989b)
[Pt(NO2)2(NH3)2] p.o. f > 5110 Degussa (1989b)
[Pt(C5H7O2)2] p.o. m/f > 500 Johnson Matthey (1976a)
cis-[PtCl2(NH3)2]d p.o. m/f approx. 20 Johnson Matthey (1977c)
cis-[PtCl2(NH3)2]d i.p. m 12 Kociba & Sleight (1971)
cis-[PtCl2(NH3)2]d i.p. m 7.7 Ward & Fauvie (1976)
cis-[PtCl2(NH3)2]d i.v. m 7.4 Ward et al. (1976)
trans-[PtCl2(NH3)2] p.o. m/f > 5110 Degussa (1989c)
a m = male; f = female
b Calculated from the original values given as mg A/kg (= milligramme atom/kg)
c Results from two different laboratories
d See footnote in section 1.2
7.2 Short-term exposure
Holbrook et al. (1975) conducted repeated-dose oral toxicity
studies on male Sprague-Dawley rats. The soluble salts PtCl4 and
Pt(SO4)2.4H2O were added to the drinking-water, which was
consumed ad libitum. Within the observation period of 4 weeks, a
concentration of 0.54 mmol/litre (182 mg PtCl4/litre or 248 mg
Pt(SO4)2.4H2O per litre) did not affect the normal weight
gain. A 3-fold increase in the platinum concentration to 1.63
mmol/litre reduced the weight gain by about 20% during the first
week only; this paralleled a 20% decrease in feed and fluid
consumption. The dietary administration of PtCl4 at concentrations
of 0.5 mmol/litre for approximately 30 days or 1.6 mmol/litre for 8
days (169 and 539 mg/litre, respectively) did not affect the weights
of any of the five organs investigated, i.e. liver, kidney, spleen,
heart, and testes. Similarly, the administration of 1.6 mmol per
litre of Pt(SO4)2.4H2O (734 mg/litre) for 8-9 days did not
significantly affect organ weights. Total platinum intake for each
of these three experimental conditions was approximately 50 mg per
rat. When 1.6 mmol PtCl4/litre (539 mg/litre) was given for about
30 days (total intake of about 250 mg Pt per rat), the kidney weight
increased by about 6-10%. No effects on the level of microsomal
protein or the activities of aniline hydroxylase and aminopyrine
demethylase in liver microsomes were found (Holbrook, 1976b).
7.3 Skin and eye irritation; skin and respiratory sensitization
7.3.1 Skin irritation
The dermal irritancy of several platinum compounds was tested
on albino rabbits using comparable procedures and evaluation
criteria. Platinum test materials were spread on abraded and intact
skin sites, located dorsolaterally on the animals' trunks. The skin
reactions were evaluated after 24, 48, and 72 h, and are summarized
in Table 15.
7.3.2 Eye irritation
Summarized data on eye irritation are presented in Table 15.
All tested platinum salts were either corrosive or irritating to
varying degrees.
7.3.3 Skin sensitization
In a study by Kolpakova & Kolpakov (1983), platinum
hydrochlorides administered intravenously to rabbits in repeated
doses induced sensitization confirmed by the basophil degranulation
test, neutrophil damage index, leucocyte agglomeration, neutrophil
alteration, and the drop skin and skin fenestra tests. These data
are unusual and have not been confirmed in other studies.
Table 15. Skin and eye irritation by platinum compoundsa
Skin irritation testb Eye irritation testc
Compound Primary Classification Reference Classification Reference
irritation
score
PtO2 0 non-irritant Campbell et al. (1975)
PtCl2 0.4d non-irritant Campbell et al. (1975)
PtCl4 2.2e irritant Campbell et al. (1975)
(NH4)2[PtCl6] 1.3 mild irritant Johnson Matthey (1978d)
(NH4)2[PtCl4] 2.7 slight irritant Degussa (1988a) corrosive Degussa (1988b)
Na2[PtCl6] 0.5 mild irritant Johnson Matthey (1978e) irritant Johnson Matthey (1978f)
Na2[Pt(OH)6] 5.4 severe irritant Johnson Matthey (1978g)
K2[PtCl4] 0f non-irritant Johnson Matthey (1981c) irritant Johnson Matthey (1981d)
K2[Pt(CN)4] 0.3 mild irritant Johnson Matthey (1977d) irritant Johnson Matthey (1978h)
[Pt(NH3)4]Cl2 2.8 moderate irritant Johnson Matthey (1977e) strongly irritant Johnson Matthey (1977f)
[Pt(NO2)2(NH3)2] 0 non-irritant Degussa (1989d) severely irritant Degussa (1989e)
[Pt(C5H7O2)2] 0 non-irritant Johnson Matthey (1976b) mildly irritant Johnson Matthey (1976c)
cis-[PtCl2(NH3)2] 0.13 mild irritant Johnson Matthey (1977g) severely irritant Johnson Matthey (1977h)
(toxic)
trans-[PtCl2(NH3)2] 0 non-irritant Degussa (1988c) corrosive Degussa (1988d)
a Adapted from Bradford (1988)
b The skin tests (patch tests on albino rabbits) were carried out according to the US Federal Register 1973 Skin Test (24 h-contact)
(Johnson Matthey) or according to OECD Test Guideline No. 404 (4-h contact) (Degussa). The method used by Campbell et al. (1975)
is comparable to these tests.
c The eye irritation tests on albino rabbits were carried out according to the US Federal Register 1973 Eye Test (Johnson Matthey)
or according to OECD Guideline No. 405 (Degussa).
d Average score from 0.2 (intact skin) and 0.6 (abraded skin); a score of 0-0.9 was considered as "non-irritant" by the authors.
e Average score from 1.8 (intact skin) and 2.6 (abraded skin); a score of 2 was considered as "irritant" by the authors.
f Using OECD Test Guideline No. 404 (4-h contact).
Taubler (1977) injected rabbits, guinea-pigs, and mice
subcutaneously and intravenously with PtSO4 (0.05-0.3 mg/litre
with and without NH4Cl) three times a week for 4 weeks. No
induction of an allergic state was found, as measured by skin tests
(guinea-pigs and rabbits), passive transfer, and footpad tests
(mice). Administration of platinum-egg-albumin complex also failed
to sensitize the experimental animals.
In a study by Murdoch & Pepys (1985), rats were immunized with
ovalbumin-platinum. Sera of the animals which were positive in the
passive cutaneous anaphylaxis (PCA) test and a radioallergosorbent
test (RAST) were pooled and used for PCA tests with other platinum
salts having differing ligands. A significant cross-reactivity
between ammonium tetrachloroplatinate(II), ammonium
hexachloroplatinate(IV), and the conjugated tetrachloroplatinate was
observed. There was very limited or no cross-reactivity with the
compounds cesium trichloronitroplatinate(II), cis-
diamminedichloroplatinum(II), potassium tetracyanoplatinate(II), and
tetraammineplatinum(II) chloride.
7.3.4 Skin and respiratory sensitization
Biagini et al. (1983) exposed two groups of Cynomolgus monkeys
(Macaca fasicularis) to disodium hexachloroplatinate,
Na2[PtCl6], by nose-only inhalation of 200 and 2000 µg/m3,
4h/day, biweekly for 12 weeks. Another group was exposed
percutaneously to the salt (20 mg/ml) applied biweekly to an open
patch area in the intrascapular region. Two weeks after termination
of exposure, bronchoprovocation challenges with Na2[PtCl6] and
pulmonary function tests were performed. Percutaneous application
did not affect post-challenge pulmonary function. The 200 µg/m3
group showed significantly greater pulmonary deficits as compared to
control animals. Average pulmonary flow resistance (RL) was
significantly increased, while forced expiratory volume in 0.5
seconds, corrected for vital capacity (FEV0.5/FVC), was decreased.
No dermal hypersensitivity was observed. The question of whether the
observed pulmonary hyper-reactivity is due to a
superpharmacological, irritant, local immune, or combination
mechanism is unresolved. The absence of hyper-reactivity in the
2000-µg/m3 group suggests a possible pulmonary tolerance
mechanism, tachyphylaxis, or delay in the development of symptoms at
higher sensitization concentrations.
7.3.5 Respiratory sensitization
In a 12-week inhalation experiment with Cynomolgus monkeys
exposed to either ammonium hexachloroplatinate (200 µg/m3) or
ozone (2000 µg/m3; 1 ppm) alone or as a combination of both,
Biagini et al. (1986) found significant allergic platinum dermal
hypersensitivity, based on concentrations necessary to give a
positive test, and pulmonary hyper-reactivity only with concomitant
exposure to ozone. Inflammation, epithelial damage, cell
recruitment, and modifications of cellular tight junctions caused by
ozone may increase the penetration of platinum into the pulmonary
epithelium and subepithelial tissue. This could lead to increased
protein binding sites or absorption of the platinum salts and
finally to the development of pulmonary hyper-reactivity and
allergic sensitization (Biagini et al., 1986).
7.3.6 Sensitization by other routes
Murdoch & Pepys (1984) investigated the immunological responses
to complex platinum salts in the female hooded Lister rat, a strain
that produces high and consistent levels of circulating IgE when
immunized with low doses of antigen together with Bordetella
pertussis adjuvant, and that reacts with enhanced synthesis of IgE
upon secondary boosting. Sensitization with the free salt of
ammonium tetrachloroplatinate, (NH4)2[PtCl4], was attempted
via the intraperitoneal, intramuscular, intradermal, subcutaneous,
intratracheal, and footpad routes over a wide range of doses (1 to
1000 µg). Both B. pertussis and/or aluminium hydroxide gel were
added as adjuvants. As shown by direct skin testing using the PCA or
RAST methods, no sensitization was achieved. However, sensitization
was obtained by intraperitoneal injection of the platinum salt
conjugated to ovalbumin (OVA). Antibodies were produced to Pt-OVA
and to OVA alone. Specific sensitization was demonstrated both by
PCA challenge with Pt-BSA (no positive PCA reactions were seen with
BSA alone) and by positive RAST, demonstrated by RAST inhibition
techniques with a Pt-BSA conjugate.
7.4 Reproductive toxicity, embryotoxicity, and teratogenicity
Only limited experimental data concerning the effects of
platinum on reproduction, embryotoxicity, and teratogenicity are
available. D'Agostino et al. (1984) studied the embryotoxic effects
of platinum compounds in Swiss ICR mice. Single doses of either
Pt(SO4)2.4H2O or Na2[PtCl6].6H2O were administered
intragastrically or subcutaneously, respectively, on the 7th and
12th day of gestation. The pups were cross-fostered to treated or
untreated dams at birth and were culled to three animals of each sex
per litter. In the Pt(SO4)2 study, the LD1 wdose of 200 mg
Pt/kg caused a reduced offspring weight from day 8 to day 45
postpartum. The major effect of disodium hexachloroplatinate (20 mg
Pt/kg) was a reduced activity level exhibited by the offspring of
dams exposed on the 12th day of gestation. The general activity was
quantified on an activity field consisting of concentric circles.
The number of lines crossed during 1 min comprised the activity
score. On days 60-65 postpartum, open-field behaviour (ambulation
and rearing), rotarod performance, and passive avoidance learning
were investigated in the adult offspring. No effects were found
after administration on the 7th day, but administration on day 12 of
gestation had significant behavioural effects.
Solid platinum, wire or foil, is considered to be biologically
inert and adverse effects on implantation are probable due to the
physical presence of a foreign object in the uterus (Barlow &
Sullivan, 1982).
Kraft et al. (1978) reported normal fertility in male rabbits
with open tube gold/platinum devices inserted into the vas deferens.
There was an initial decrease in sperm count and motility, but these
parameters returned to normal after three weeks. At 117-426 days
after insertion, 7 out of 9 animals were fertile in numerous
matings.
Effects on human sperm motility were investigated by Kesseru &
Leon (1974). Fresh sperm were incubated for up to 5 h in the
presence of strips of platinum or other metals. Motility after 2 and
5 h was 60 and 30%, respectively, compared to 10 and 0% for copper,
40 and 7% for silver, and 90 and 65% for gold.
Platinum wire inserted into the uterus of rats was reported to
reduce the implantation of fertilized ova. An 83% reduction in the
number of implantation sites in the affected uterine horn, compared
to the unoperated horn, was found in rats unilaterally implanted on
day 3 (Chang et al., 1970). Chang & Tatum (1975) found no effect on
embryonic or fetal survival if platinum wire was inserted after
implantation on day 6. Tobert & Davies (1977) showed a 37% reduction
in the number of implanting ova in the uteri of rabbits containing
platinum foil.
7.5 Mutagenicity and related end-points
The genotoxic effects of platinum compounds have been
investigated in bacterial systems, mammalian cell cultures and in
vitro studies.
In bacteria many of the tested platinum compounds were
moderately mutagenic. Cisplatin and some of its analogues showed the
greatest mutagenic potential; other platinum compounds were less
mutagenic.
In Ames tests, nearly all using the test strains Salmonella
typhimurium TA98 and TA100, positive results were reported
(Lecointe et al., 1977; Andersen, 1979; Suraikina et al., 1979; Life
Science Research, 1980a; Kanematsu et al., 1980). With
[Pt(NH3)4]Cl2, mutagenic potential was observed in strain
TA1537 with and without S-9 metabolic activation (Life Science
Research, 1980a).
The induction of reverse mutations in the plasmid-carrying
strains TA98 and TA100 indicated base-pair substitution and frame-
shift mutations (Lecointe et al., 1977; Suraikina et al., 1979;
Kanematsu et al., 1980).
(NH4)2[PtCl6] but not PtCl4 caused base-change mutation
in Escherichia coli B/r WP2 (Kanematsu et al., 1980).
The growth of a Rec strain of Bacillus subtilis was
significantly inhibited by (NH4)2[PtCl6] (0.1 mol/litre),
H2[PtCl6] (0.01 mol/litre), and PtCl4 (0.01 mol/litre)
(Kanematsu et al., 1980).
In a mutagenic test with the mouse lymphoma cell line L 5178Y,
cisplatin, transplatin, and PtCl4 produced significantly higher
mutation frequencies than occurred in the controls, but
[Pt(NO2)2](NH3)2 and PtCl2 did not (Sandhu, 1979).
Cellular resistance to the toxic effects of two platinum
complexes was introduced into Chinese hamster ovary (CHO) cells by
continuous exposure to K2[PtCl6] and Pt(SO4)2 for 5 and 4
months, respectively. These cell lines had resistant phenotypes
stable for at least 55 population doublings in the absence of a
platinum compound. The induced resistances were interpreted by the
authors to be a result of mutation and selection (Smith et al.,
1984).
In a micronucleus test in mice involving oral administration of
[Pt(NH3)4]Cl2, no significant increase in the incidence of
micronucleated polychromatic erythrocytes was found. Additionally,
[Pt(NH3)4]Cl2 did not markedly inhibit bone marrow cell
division at any level (Life Science Research, 1980b). Also, no
evidence of induced chromosomal damage leading to micronucleus
formation in polychromatic erythrocytes was observed after oral
administration of K2[PtCl4] in mice (Life Science Research,
1981a).
No significant increase in the incidence of aberrant metaphases
was found in bone marrow cells after subacute oral administration of
[Pt(NH3)4]Cl2 or K2[PtCl4] to Chinese hamsters (Life
Science Research, 1981b, 1982).
K2[PtCl4] and [Pt(NH3)4]Cl2 induced no increase in
the frequency of sex-linked recessive lethal mutations in
Drosophila melanogaster (Life Science Research, 1980c, 1981c).
In a structure-mutagenicity study with the CHO:HGPRT-system,
cis-[Pt(NH3)2Cl2] was the most potent of six platinum
compounds tested. Based on the slope of the mutation induction
curve, the approximate relative mutagenic activity of cis-
[Pt(NH3)2Cl2], K[Pt(NH3)Cl3], and [Pt(NH3)3Cl]Cl was
100:9:0.3. The mutation frequency for K2[PtCl4] and trans-
[Pt(NH3)2Cl2] was related to the concentration used, but was
not much greater than the maximum spontaneous mutation frequency. No
mutagenic activity was observed for [Pt(NH3)4]Cl2. The
relative cytotoxicity of the tested compounds was similar.
Additionally, it was found that cis- and trans-
[Pt(NH3)2Cl2] bind to DNA after entering the cell, but the
relative mutagenicities are not a consequence of different initial
levels of DNA binding (Johnson et al., 1980).
Dose-dependent forward mutations were induced by PtCl4 to 8-
azaguanine resistance (8-AGR/HGPRT locus) in Chinese hamster ovary
(CHO-S) cells. In addition there was an increased dose-related
frequency of CHO-AUXB1 reversion (Taylor et al., 1979)
Cisplatin, which is not reviewed in detail in this document
(see footnote in section 1.2), induces structural chromosomal
aberrations and sister chromatid exchanges in cells of rodents
treated in vivo, chromosomal aberrations, micronuclei, and sister
chromatid exchanges in both human and rodent cells in vitro, and
mutation and DNA damage in rodent cells in vitro. Cisplatin is
also mutagenic in Drosophila, fungal, and bacterial test systems
(IARC, 1987a).
7.6 Carcinogenicity and anticarcinogenicity
No experimental data are available on the carcinogenicity of
platinum and platinum compounds except for cisplatin (see footnote
in section 1.2). IARC (1987b) considered sufficient the evidence for
the carcinogenicity of cisplatin for animals (see chapter 13).
Cisplatin and its analogues, however, are exceptional compared
to the other platinum compounds. This is reflected by the unique
mechanism for their anti-neoplastic activity demonstrated in in
vitro studies (Rosenberg, 1980, 1985). At low doses cisplatin
produces specific inhibition of DNA synthesis (but not of RNA and
protein synthesis) by causing DNA lesions such as monofunctional
adducts, bifunctional binding to a single base moiety, and DNA
cross-links of inter- and intrastrand types (Harder & Rosenberg,
1970; Howle & Gale, 1970). There is sufficient evidence that the DNA
cross-links are responsible for cellular toxicity, but not for anti-
tumour activity. For the latter, another observation probably plays
the decisive role; only the cis isomer forms a closed ring chelate
of the aquated cisplatin with guanine at a certain position of
guanine. Thus, intrastrand DNA cross-linking is considered to be the
most important reason for anti-tumour activity. It appears that, due
to the cisplatin-induced DNA cross-links, the replication of DNA is
impaired in cancer cells, while in normal cells the cisplatin
lesions on guanine are repaired before replication (Rosenberg, 1985;
Pinto & Lippard, 1985).
The high chloride concentration of the extracellular fluid (112
mmol/litre) is sufficient to limit the substitution of water ligands
for chloride. However, within the cell the platinum complex is
exposed to low chloride concentrations (4.4 mmol/litre) and
hydrolysis of the chloride leaving groups can occur (Rosenberg,
1975), a process that has been shown to accelerate the rate of
reaction of platinum with DNA (Johnson et al., 1980) and to increase
its toxicity (Litterst, 1981). This hydration provides the only
known activation process required for cisplatin to react with
molecules in the cell, and metabolic activation is not required
(Rosenberg, 1980). The binding of cisplatin with plasma proteins, on
the other hand, is not inhibited by chloride and presumably involves
a different mechanism (De Conti et al., 1973), such as the strong
electrophiles on proteins (Cleare, 1977a).
7.7 Other special studies
7.7.1 Effects on alveolar macrophages
Rabbit alveolar macrophages exposed to the water-soluble
platinum(IV) chloride at a concentration of 0.4 mmol/litre (78 mg
Pt/litre) for a period of 20 h exhibited a 50% reduction in
macrophage viability. A reduction in phagocytic activity and a
decrease in total cellular adenosine triphosphate to 50% of the
value in control macrophages was observed at 0.21 and 0.25
mmol/litre (41 and 48 mg Pt/litre). Platinum(IV) oxide (PtO2) did
not dissolve in the culture medium and, hence, was ineffective at
concentrations as high as 500 mg/litre (Waters et al., 1975).
7.7.2 Non-allergic mediator release
Investigations in guinea-pigs, rats, and dogs showed an
increase in bronchomotility and histamine release after intravenous
treatment with disodium chloroplatinate, Na2[PtCl6] (Saindelle &
Ruff, 1969; Parrot et al., 1969).
Saindelle & Ruff (1969) noticed dyspnoea one minute after an
intravenous injection of disodium chloroplatinate (20 mg/kg) into
guinea-pigs. Within 5 min an intense attack of asthma occurred
resulting in death. Histamine release occurred following the
injection and the blood histamine level was greatly increased. The
injection of a smaller dose (1-2 mg/kg) resulted in bronchospasm
comparable to that caused by 3 µg/kg of histamine. Repeated
injections of histamine caused reproducible changes in bronchial
motility, whereas the platinum compound caused tachyphylaxis.
The intense breathing difficulties observed in these studies
were presumably due to non-allergenic histamine release. This
nonspecific histamine release has complicated the interpretation of
both animal and human studies with respect to the conclusion of
allergic sensitization.
7.7.3 Effects on mitochondrial function
No pronounced effects of platinum on the mitochondrial function
of liver, heart, lung or kidney cells were observed in an in vitro
test on succinate-stimulated O2 uptake 24 and 48 h after
intragastric administration of 40 and 80 µmol K2[PtCl6]/litre
(19 and 38 mg/litre) to Sprague-Dawley rats (Michael et al., 1976).
7.7.4 Effects on the nervous system
The open field behaviour of adult Swiss mice has been found to
be influenced by platinum salts administered intragastrically in the
form of a single dose or of repeated doses. A single dose of
Pt(SO4)2 at the LD25 level (213 mg Pt/kg body weight)
depressed ambulation significantly and rearing marginally. For
ambulation, this pattern persisted from 4 h to 7 days after
administration, although the effect was most obvious at 4 h.
Repeated doses of the same salt at the LD1 level (up to 10 doses
of 109 mg Pt/kg every 72 h) caused a marginal depression of activity
and exploratory behaviour (Lown et al., 1980). Also, a single dose
of Na2[PtCl6] depressed ambulation significantly (Massaro et
al., 1981).
During the course of a reproduction study, behavioural effects
were observed in the offspring of mice treated with sodium
hexachloroplatinate (see section 7.4; D'Agostino et al., 1984).
7.7.5 Side effects of cisplatin and its analoguesa
As discussed in section 8.3, the therapeutic use of cisplatin
in humans can be accompanied by several toxic side-effects. In
animal studies, only some of which are presented in this monograph,
similar effects were observed.
Ward et al. (1976) investigated the nephrotoxicity of cisplatin
and its analogues in male F-344 (Fischer CDF) rats. An intravenous
LD50 dose of cisplatin (7.4 mg Pt/kg body weight) caused an
increase in the blood urea nitrogen and creatinine levels reaching a
peak on days 4 to 5. Diarrhoea developed by day 3. Necrotizing
enteritis of the small intestine, caecum, and colon, cellular
depletion of bone marrow and thymus, and acute degenerative and
necrotizing renal tubular lesions also occurred (Ward et al., 1976).
Oxoplatinum ( cis-dichlorodiammine-trans-
dihydroxyplatinum(IV)) also caused marked nephrotoxicity after
intravenous administration (20 mg/kg as a single dose) to rats.
However, another cisplatin analogue, CBDCA ( cis-diammine-l,1-
cyclobutane dicarboxylate platinum(II)), did not result in
significant changes in renal function parameters (Laznickova et al.,
1989).
a See footnote in section 1.2.
Cisplatin has been found to cause bone marrow suppression. The
surviving fraction of haemopoietic bone-marrow system cells in mice
decreased from 1 to 0.03 after treatment with an LD50 dose of
cisplatin (Lelieveld et al., 1984). A 36-53% decrease in lymphocyte
and granulocyte counts was observed in mouse bone marrow after
intra-peritoneal treatment with 5 mg cisplatin/kg (Bodenner et al.,
1986).
Cisplatin administered intraperitoneally (6 mg/kg) has been
shown to affect gastric emptying in rats. There was a large increase
in the weight of the stomach due to retained food (Whitehouse &
Garrett, 1984).
In dogs, cisplatin given intravenously as a dose of 2 mg/kg
resulted in a complete interruption of interdigestive myoelectric
activity of the gastric antrum, duodenum, and jejunum (Chey et al.,
1988).
Ototoxicity has been demonstrated in guinea-pigs. A cisplatin
dose of 1.5 mg/kg administered intraperitoneally once a day caused
hearing loss beginning at about the ninth day of administration
(Hoeve et al., 1987).
7.8 Factors modifying toxicity
Physiological levels of selenium administered simultaneously
with food to mice markedly depressed the acute toxicity of some
platinum salts by forming inert complexes of high relative molecular
mass in the presence of protein (Imura, 1986).
8. EFFECTS ON HUMANS
8.1 General population exposure
8.1.1 Acute toxicity - poisoning
Except for one case of poisoning in 1896 (Hardman & Wright,
1896), no acute poisoning cases have been reported.
8.1.2 Effects of exposure to platinum emitted from automobile
catalysts
An immunological study conducted by Cleare (1977b) addressed
the question of whether the emitted platinum is allergenic. He
investigated the response of individuals, who were highly sensitive
to platinum salt (skin test positive at low platinum salt
concentrations), to extracts of particulate exhaust samples. The
total platinum content at the highest concentration was more than 5
µg/ml, which would normally be sufficient to elicit a response. Five
extracts tested on three people, using the skin prick test, did not
elicit a positive response.
8.2 Occupational exposure
The occupational hazards of platinum are principally confined
to some halogenated complex platinum salts (Rosner & Merget, 1990).
8.2.1 Case reports and cross-sectional studies
A report of health problems arising from occupational exposure
to platinum was produced by Karasek & Karasek (1911). They studied
workers in photographic studios in Chicago handling photographic
paper treated with complex platinum salts. The symptoms observed in
eight workers were pronounced irritation of the nose and throat
causing violent sneezing and coughing, together with difficulties in
breathing.
Hunter et al. (1945) conducted environmental and clinical
studies on workers in four British platinum refineries. Out of 91
workers exposed to complex salts of platinum, 52 showed symptoms
starting with repeated sneezing and rhinorrhoea, followed by
tightness of the chest, shortness of breath, cyanosis, and wheezing.
Scaly erythematous dermatitis of hands and forearms, sometimes also
affecting the face and neck, and urticaria were observed in 13
workers. The respiratory tract symptoms persisted during working
hours and for about one hour after leaving the factory. The latency
period from the first contact with platinum to the occurrence of the
first symptoms varied from a few months to six years. Once skin and
respiratory tract sensitization was established, symptoms tended to
become worse as long as the workers were exposed to platinum salts.
In the USA, Roberts (1951) studied 21 employees of a platinum
refinery for five years. All workers showed some form of platinum-
related disease for which Roberts (1951) introduced the term
"platinosis". According to his classification of this occupational
disease, 40% of the employees did not have typical symptoms but
exhibited the same inflammatory changes in the conjunctivae and the
mucous membranes of the upper respiratory tract as were seen in the
60% of the workers with definite symptoms.
These observations have been confirmed by other investigators.
The term "platinosis" is no longer used, as it implies a chronic
fibrosing lung disease. This had been assumed by Roberts (1951) but
has not been reported elsewhere. The terms "platinum salt allergy"
(Schultze-Werninghaus et al., 1978), "platinum salt sensitivity"
(Linnett, 1987), "allergy to platinum compounds containing reactive
halogen ligands" (Hughes, 1980) and "platinum salt hypersensitivity"
(PSH) have been used, with the latter being preferred.
The symptoms typical of platinum salt sensitization (Roshchin
et al., 1979; Health and Safety Executive, 1983; Brooks et al.,
1990) include watering of the eyes, sneezing, tightness of the
chest, wheezing, breathlessness, coughing, eczematous and urticarial
skin lesions, signs of mucous membrane inflammation.
In earlier studies, the prevalence of allergic symptoms due to
platinum salt exposure was as high as 80% (Table 16) although this
was not consistently confirmed by skin testing. Estimated workplace
exposure concentrations ranged from 0.9 to 1700 µg Pt/m3 (Hunter
et al., 1945). However, due to analytical deficiencies, these data
do not allow the quantification of these exposure situations. It can
be assumed that occupational exposure is much lower today due to
improved engineering control and occupational exposure limits.
Airborne dust analyses in a platinum refinery revealed levels
between 0.08 and 0.1 µg/m3 in the separation department; in other
areas the measurements were all below 0.05 µg/m3 (Bolm-Audorff et
al., 1988) or below 0.08 µg/m3 (Merget et al., 1988). Work-related
symptoms were reported in 8 to 23% of workers exposed to these
concentrations (Table 16). The risk of developing platinum salt
hypersensitivity seems to be correlated with the intensity of
exposure. In the surveys of Bolm-Audorff et al. (1988) and Merget et
al. (1988), the highest rates of prevalence occurred in the groups
exposed to the highest concentration.
Table 16. Prevalence of symptoms and positive skin tests
in refinery workers exposed to platinum salts
Total Workers Prevalence of Reference
workersa with symptomsb symptoms (%)c
91 (16) 52 (4) 57 (25) Hunter et al. (1945)
20 (19) 12 (8) 60 (42) Roberts (1951)
15 (nd) 12 (nd) 80 (nd) Massmann & Opitz (1954)
51 (nd) 35 (nd) 69 (nd) Hebert (1966)
107 (107) 31d 47e (15) 29d 44e (14) Biagini et al. (1985);
Brooks et al. (1990)
65 (64) 15 (12) 23 (19) Bolm-Audorff et al.
(1988)
24 (20) 2 (4) 8 (20) Merget et al. (1988)
a Values in parentheses are numbers of skin-tested workers
b Values in parentheses are numbers of workers with a positive
skin test
c Values in parentheses give the prevalence of positive skin
tests as a percentage of skin-tested workers
d Workers with upper respiratory tract symptoms
e Workers with lower respiratory tract symptoms
nd = not determined
8.2.2 Allergenicity of platinum and platinum
compounds
Metallic platinum seems to be non-allergenic. With the
exception of a single reported case of alleged contact dermatitis
from a "platinum" ring (Sheard, 1955), no allergic reactions have
been reported.
Halogenated platinum salts are among the most potent
sensitizers. The compounds mainly responsible for platinum
sensitization are hexachloroplatinic acid, H2[PtCl6], and the
chlorinated salts such as ammonium hexachloroplatinate,
(NH4)2[PtCl6], potassium tetrachloroplatinate, K2[PtCl4],
potassium hexachloroplatinate, K2[PtCl6], and sodium
tetrachloroplatinate, Na2[PtCl4]. Cleare et al. (1976)
investigated the allergenic potency of platinum complexes by means
of skin prick tests on platinum refinery workers who were known to
be sensitive to hexachloroplatinate. Their results suggest that
platinum allergy is confined to a small group of charged compounds
that contain reactive ligand systems, the most effective of which
are chloride ligands. The allergic response generally increases with
increasing number of chlorine atoms, as demonstrated by the
following sequence of potency:
(NH4)2[PtCl6] approx. (NH4)2[PtCl4] >
Cs2[Pt(NO2)Cl3] > Cs2[Pt(NO2)2Cl2] >
Cs2[Pt(NO2)3Cl]
Ionic platinum compounds containing bromide or iodide are also
allergenic, but are less effective.
Non-halogenated complexes such as [Pt{(NH2)2CS}4]Cl2,
K2[Pt(NO2)4], and [Pt(NH3)4]Cl2, and neutral complexes
such as cisplatin, cis-[PtCl2(NH3)2], are not allergenic,
probably because they do not react with proteins to form a complete
antigen. Anaphylactic shock reactions observed after the intravenous
administration of relatively high doses of cisplatin (Khan et al.,
1975; Von Hoff et al., 1979) were probably caused by contamination
with the potent hexa- or tetrachloroplatinate (Pepys, 1983).
8.2.3 Clinical manifestations
The latency period from the first exposure to platinum salts to
the occurrence of the first symptoms usually varies between three
months and three years (Parrot et al., 1969; Schultze-Werninghaus et
al., 1978; Ruff et al., 1979; Biagini et al., 1985), but is
sometimes only a few weeks (Roberts, 1951; Hughes, 1980; Merget et
al., 1988).
The dermatitis observed in the past (Roberts, 1951) is believed
to have been mainly of a primary irritant nature following exposure
to strong acids and alkalis. True contact dermatitis (i.e. allergic)
is extremely rare. However, contact urticaria is seen in sensitized
people following splashes with platinum salts and in some instances
this is the first indication of sensitization (Hughes, 1980).
The symptoms usually worsen with increasing duration of
exposure but generally disappear when the subject is removed from
exposure. The latter was shown by the follow-up study carried out on
platinum refinery workers who had to cease work with platinum salts
because of sensitization (Newman-Taylor, 1981). This study found no
evidence of long-term effects when workers giving a positive skin
prick test and showing symptoms of platinum sensitization were
removed immediately from contact with platinum salts. However,
Schultze-Werninghaus et al. (1989) reported that after long duration
exposure following sensitization individuals may never become
completely free of symptoms. Similarly, Biagini et al. (1985)
demonstrated the existence of positive platinum skin tests at very
low concentrations in workers who had been free of occupational
platinum exposure for periods of up to four years.
8.2.4 Immunological mechanism and diagnosis
The clinical manifestations of soluble platinum salt allergy
reflect a true allergic response based on the following clinical
criteria (Hughes, 1980; Biagini et al., 1985, 1986; Merget et al.,
1988; Schultze-Werninghaus et al., 1989):
* the appearance of sensitivity is preceded by a symptomless
exposure;
* not all exposed individuals become sensitized;
* the affected individuals become increasingly sensitive to
platinum and react even at very low levels of exposure;
* negative prick test results are obtained in atopic and non-
atopic controls.
The mechanism of platinum salt allergy appears to be a Type I
(IgE mediated) response. The possibility of the formation of IgE
antibodies to platinum chloride complexes in sensitized individuals
has been assumed on the grounds of allergy and serological tests. It
is believed that platinum salts of low relative molecular mass act
as haptens combining with serum proteins to form the complete
antigen. However, the actual immunological mechanism has not yet
been defined (Zachgo et al., 1985).
It has been demonstrated that platinum(II) reacts with the
sulfur atoms in the six methionine groups in human serum albumin
(HSA) and that methionine 123 is the primary binding site
(Grootveld, 1985).
Skin prick tests with freshly prepared solutions of soluble
platinum complexes appear to provide reproducible, reliable,
reasonably sensitive, and highly specific biological monitors of
allergenicity (Cleare et al., 1976; Dally et al., 1980). The
compounds used for routine screening of exposed workers are
(NH4)2[PtCl6], Na2[PtCl6], and Na2[PtCl4]. After
sensitization due to previous exposure, prick testing with
concentrations of the platinum compound in the range of 10-3 to
10-9 g/ml will produce immediate weal and flare reactions (Pepys
et al., 1972; Pickering, 1972; Hughes, 1980; Gallagher et al., 1982;
Biagini et al., 1985; Boggs, 1985; Jacobs, 1987; Linnett, 1987;
Murdoch & Pepys, 1987; Schultze-Werninghaus et al., 1989). At these
concentrations, nonspecific skin reactions were not found in atopic
or non-atopic controls (Pepys et al., 1972; Murdoch & Pepys, 1987;
Merget et al., 1988).
Passive transfer of immediate reactivity to intracutaneous
tests in humans was demonstrated in the Prausnitz-Küstner test by
Freedman & Krupey (1968). Schultze-Werninghaus et al. (1978)
observed positive reactions in passive cutaneous anaphylaxis (PCA)
in monkeys with serum from a platinum refinery worker. Similar tests
were performed by Pepys et al. (1979) and Biagini et al., (1985).
The results, however, were inconsistent, because positive as well as
negative Prausnitz-Küstner prick test or PCA reactions were elicited
in human recipients or in monkeys, respectively. Parish (1970) also
demonstrated the presence of heat-stable IgG antibodies by passive
cutaneous anaphylaxis on monkey skin. These results were confirmed
by Biagini et al. (1985).
The sensitivity and reliability of the skin prick test has not
been achieved in any in vitro test available. In enzyme
immunoassays (Zachgo et al., 1985; Merget et al., 1988) and in the
radioallergosorbent test (RAST) (Cromwell et al., 1979; Pepys et
al., 1979) IgE antibodies specific to platinum chloride complexes
were found. Although a good correlation with the results of prick
tests was reported (Cromwell et al., 1979), their practical
application for screening purposes was questioned because of the
lack of specificity (Boggs, 1985; Jacobs, 1987; Merget et al.,
1988). This was shown in a cross-sectional survey of platinum
refinery workers (Merget et al., 1988). Higher total serum IgE and
hexachloroplatinate-specific IgE levels in subjects with work-
related symptoms were noted. However, not all the individuals
allergic to platinum salt and some of the controls showed binding in
RAST. Similar effects were seen with in vitro histamine release
from basophils, which was relatively high in skin-test positive
workers but even higher in the atopic control group. Histamine
release with anti-IgE showed a similar pattern, indicating identical
binding sites of hexachloroplatinate and anti-IgE on the surface of
cutaneous mast cells and basophils.
Biagini et al. (1985) also found significantly higher mean
total serum IgE levels in platinum refinery workers. However, good
correlation between the RAST data and the skin test results was
seen. Of the workers with positive skin tests, 95% (20/22) showed
higher RAST binding than a control group, whereas only 8.5% (8/94)
of those with negative skin tests demonstrated positive RAST
results.
Since refinery workers are exposed to more than one platinum-
group metal salt, the question of cross-reactivity was investigated
by passive cutaneous anaphylaxis (PCA) tests. First results
indicated that platinum (Na2[PtCl6] and (NH4)2[PtCl6]) and
palladium (Na2[PdCl6]) appear to be equally effective as
eliciting agents. Five-fold concentrated sera from platinum refinery
workers produced positive PCA results in monkeys (Biagini et al.,
1982). No in situ reactions due to palladium salts were reported.
There was only limited cross-reactivity between platinum and
palladium salts in both skin test and RAST. Reactions to the
platinum-group metals other than platinum were only seen in
individuals sensitive to platinum salts (Murdoch et al., 1986;
Murdoch & Pepys, 1987).
Instillation in the nose of concentrations of 10-3 to 10-9
g per ml has been used in the past as another method of detecting
platinum salt sensitivity. A nasal reaction was considered positive
if itching, sneezing, nasal obstruction or discharge occurred singly
or in combination within 15 min of the challenge (Pepys et al.,
1972).
Inhalation tests with a mixture of ammonium hexachloroplatinate
and lactose dust gave immediate asthmatic reactions in sensitized
individuals and in one case a late asthmatic reaction occurred
(Pepys et al., 1972).
Merget et al. (1990) reported three cases of platinum refinery
workers with negative skin tests who showed non-specific hyper-
reactivity and a clearly positive immediate reaction in the
bronchial provocation test.
8.2.5 Predisposing factors
Dally et al. (1980) conducted a retrospective cohort analysis
in a group of 86 platinum workers entering a United Kingdom refinery
in 1973-1974. It was found that significantly more atopics left
employment, but this was apparently irrespective of the development
of platinum salt allergy. The incidence of the disease did not
differ significantly between the atopics (14/32 = 44%) and non-
atopics (21/54 = 39%), although Burge et al. (1979) demonstrated
that the atopics were sensitized more quickly. Thus, the increased
leaving rate of atopics cannot be regarded as proof for the atopic
status being a true predisposing factor, as suggested by Linnett
(1987). It may, at most, be considered a trend.
Merget et al. (1988) examined 27 refinery workers and found no
evidence to support tobacco smoking as a predisposing factor.
However, Linnett (1987) found a significant association between
smoking and the incidence of positive skin test results in life
table studies of 134 refinery workers. Also, in a longitudinal
cohort study on 91 platinum refinery workers (86 males, 5 females)
in the United Kingdom who started work in 1973-1974 and were
followed up until 1980, the risk of a positive skin test result was
found to be 4-5 times higher in smokers than in non-smokers
(Venables et al., 1989). Age, varying from 15-54 years in the
cohort, was a definite confounding factor. After taking account of
age, the risk of leaving refinery work was only 1.75 times greater
in smokers than in non-smokers. The risk from atopy was not
significant after taking smoking into consideration.
Brooks et al. (1990) further studied 107 current and 29
medically terminated workers, first described by Biagini (1985),
using platinum skin testing and cold air challenge for evaluation of
pulmonary hypersensitivity. Of these workers (74 current and 12
terminated workers), 63% underwent repeat platinum skin testing one
year later. Among current workers, there was a conversion to
positive platinum skin tests in five employees (with three of these
conversions occurring in workers who had positive cold air challenge
tests a year earlier). Thus, positive cold air challenge (airway
hyperactivity) appears to have value for predicting conversion to
positive skin test status with continued occupational exposure.
8.3 Side effects of cisplatina
The therapeutic use of cisplatin is often complicated by the
occurrence of side effects. Prominent among these are
nephrotoxicity, severe nausea and vomiting, myelotoxicity (bone
marrow suppression), and ototoxicity.
The most important toxic effect of cisplatin occurs in the
kidney, eventually becoming irreversible during continued treatment.
For instance, Lippman et al. (1973) found an approximately 50%
reduction in renal function in each of 16 patients after treatment
with total doses of 3.0-7.0 mg/kg body weight. Degeneration and
necrosis of the proximal convoluted tubules, dilation of distal
tubules, and glomerular abnormalities (elevation of the blood urea
nitrogen and serum creatine levels, decreased creatine clearance)
have been reported (Swierenga et al., 1987). A significant
protection of renal function can be obtained by forced hydration,
which flushes the drug through the kidney rapidly (Merrin, 1976).
The simultaneous intravenous administration of mannitol can
contribute to the prevention of cisplatin nephrotoxicity (Fillastre
& Raguenez-Viotte, 1989).
Gastrointestinal toxicity consists mainly of nausea and
vomiting lasting from 4 to 6 h and, occasionally in some sensitive
patients, anorexia for up to one week (Hill et al., 1975).
Ototoxicity is another serious side-effect, consisting of
tinnitus with or without clinical loss of hearing. Early in its
course it is almost exclusively associated with high-range hearing
loss in the 4000-8000 Hz range (Von Hoff et al., 1979).
Cisplatin can also cause peripheral neuropathy described as
sensory, affecting primarily large fibres (Mollman et al., 1988).
a See footnote in section 1.2.
Single cases of allergic reactions, angioneurotic oedema, rash,
asthma (Von Hoff et al., 1979), cardiac arrest (Vogl et al., 1980),
gingival discolouration (Ettinger & Freeman, 1979), and tetany due
to hypocalcaemia and hypomagnesaemia (Hayes et al., 1979) have all
been reported.
There are less toxic analogues, for example, cis-
diammine-1,1-cyclobutanedicarboxylato platinum(II) (Carboplatin,
JM8) and cis-dichloro- trans-dihydroxybisisopropylamine
platinum(IV) (JM9). These cause less kidney damage, nausea, and
vomiting. However, these analogues affect bone marrow and, in
addition to the negative effects of cisplatin, may inhibit the
formation of white cells, red cells, and blood platelets (Schacter &
Carter, 1986; Bradford, 1988).
8.4 Carcinogenicity
No data are available to assess the carcinogenic risk of
platinum or its salts to humans. With respect to cisplatin, IARC
(1987b) considered the evidence for carcinogenicity for humans to be
inadequate (see chapter 13).
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1 Microorganisms
Simple complexes of platinum have bactericidal effects. In
general, charged complexes in solutions, e.g., (NH4)2[PtCl6]
above a concentration of 1 mg/litre, are lethal for bacteria.
Neutral complexes are bactericidal only at considerably higher
concentrations (> 38 µmol per litre) (Rosenberg et al., 1967;
Shimazu & Rosenberg, 1973).
Rosenberg et al. (1965) reported the discovery of a unique
property of some simple platinum-group metal complexes. When culture
medium was subjected to an alternating current using platinum
electrodes, bacterial cell division was inhibited. The spent medium
itself was bactericidal. Detailed analysis revealed that the active
agent was the cis isomer of [PtCl2(NH3)2], i.e. cisplatin. It
was shown that such neutral platinum complexes, diluted in growth
media, selectively inhibit cell division without reducing the cell
growth of a variety of gram-positive and especially of gram-negative
bacteria. As a result the bacterial rods are forced to form long
filaments. This effect has been studied most intensively on
Escherichia coli with cisplatin. In this case filamentation is
reversed as soon as the bacterial filaments are transferred to a
fresh medium free of the drug (Rosenberg et al., 1967).
Hoffmann (1988) studied the effects of cisplatin and PtCl4 on
the in vitro metabolism of the yeast Saccharomyces cerevisiae.
Both compounds strongly inhibited DNA, RNA, and ribosome synthesis
in the mmol/litre range. The IC50 (median inhibitory
concentration) for the inhibition of DNA synthesis, for instance,
was 0.42 mmol/litre (126 mg/litre) for cisplatin and 0.2 mmol/litre
(67 mg per litre) for PtCl4.
9.2 Aquatic organisms
9.2.1 Plants
Barnes & Talbert (1984) studied the influence of
hexachloroplatinic acid (250, 500, and 750 µg/litre) on the growth
of the green alga Euglena gracilis using a laboratory "microcosm".
The growth recorded over 32 days was relatively slow, indicating
that the experiments were conducted under low nutrient conditions,
although these were not reported by the authors. For example, the
doubling time of the control culture was about 9 days. Although no
precise data were given, H2[PtCl6]reduced growth rate and yield
after 32 days in a dose-dependent manner.
After cisplatin was applied to the water hyacinth Eichhornia
crassipes at 2.5 mg/litre, chlorosis was evident and the plants
were stunted. At the 10-mg/litre level, some plant leaves were
necrotic and chlorotic, and the roots were darkened and stunted. The
most prominent symptom was the appearance of reddish-brown streaks
on the leaves. These were particularly noticeable on young leaves
and on the leaves of daughter plants (Farago & Parsons, 1985).
9.2.2 Animals
Biesinger & Christensen (1972), using Lake Superior water as a
medium, studied the effects of various metals on the survival,
growth, reproduction, and metabolism of the invertebrate Daphnia
magna. Chronic (3-week) exposure to hexachloroplatinic acid,
H2[PtCl6], resulted in an LC50 value of 520 µg Pt/litre (range
437-619 µg per litre). Biochemical measurements and reproductivity
were much more sensitive parameters than growth. A dose of 62
µg/litre caused a 12% reduction in weight gain, 13% reduction in
total protein, and 20% decrease in glutamic-oxalacetic transaminase
activity. At concentrations of 14 and 82 µg/litre, reproduction,
measured as total number of young, was impaired by 16 and 50%,
respectively.
Ferreira & Wolke (1979) investigated the effects of short-term
exposure to tetrachloroplatinic acid, H2[PtCl4], on the coho
salmon Oncorhynchus kisutch at 8.5°C and a water hardness of about
56 mg CaCO3/litre. In the static bioassay, 24-, 48-, and 96-h
LC50 values of 15.5, 5.2, and 2.5 mg Pt/litre, respectively, were
found. General swimming activity and opercular movement started to
be affected at 0.3 mg/litre. Lesions in the gills and the olfactory
organ were also noted at 0.3 mg/litre or more. Concentrations of
0.03 and 0.1 mg/litre had no effect.
9.3 Terrestrial organisms
A few studies have examined the effects of platinum on plants.
All were conducted with soluble platinum chlorides.
Hamner (1942) investigated the effect of hexachloroplatinic
acid, H2[PtCl6].6H2O, on the growth of beans and tomato plants
grown in sand culture. At concentrations of 3 x 10-5 to 15 x
10-5 mol/kg (5.9-29.3 mg/kg), growth was inhibited and the plants
showed smaller leaf areas, higher osmotic pressure, and lower
transpiration rates. They also resisted wilting longer than the
controls and were less succulent.
Tso et al. (1973) reported that platinum increased the nicotine
content of tobacco plants.
In a study by Pallas & Jones (1978) on the uptake of platinum
by nine horticultural crops (see section 4.1), effects on growth
were observed. Radish (Raphanus sativus), cauliflower ( Brassica
oleracea cv. Snowball), snapbean (Phaseolus vulgaris), sweet
corn (Zea mays), pea (Pisum sativum), tomato (Lycopersicon
esculentum), bell pepper (Capsicum annuum), broccoli ( Brassica
oleraceaw cv. Crusader), and turnips (Brassica rapa) were grown
in hydroponic solution at 25/20 °C, 60/90% relative humidity, and
320/400 µl CO2/litre air for 14/10 h photo- and nyctoperiods,
respectively. When the seedlings reached an early maturity stage,
such as flowering in the case of peas, snapbeans, cauliflower,
tomato, and broccoli, root expansion in the case of turnip and
radish, and considerable leafiness in the case of corn, platinum
tetrachloride, PtCl4, was added to fresh nutrient solution to give
concentrations of 0.057, 0.57, and 5.7 mg Pt/litre. After a 7-day
treatment, roots and tops were harvested and dried at 80 °C. As
shown in Table 17, dry weights were significantly reduced in tomato,
bell pepper, and turnip tops, and in radish roots at the highest
platinum concentration. At this level, the buds and immature leaves
of most species became chlorotic. In some of the species the low
levels of PtCl4 had a stimulatory effect on growth. In addition,
transpiration was suppressed at the highest platinum concentration,
probably due to increased stomatal resistance. Photosynthesis was
also apparently reduced, consistent with the observed growth
depression. On the other hand, the stimulation of transpiration and
growth observed at the two lower concentrations, as compared to the
control plants, explains the stimulated growth.
A stimulation of growth was also observed in seedlings of
Setaria verticillata (L. P. Beauv) treated with a low level of
platinum (0.5 mg Pt/litre) administered as potassium
tetrachloroplatinate, K2[PtCl4] (Farago & Parsons, 1986). This
South African grass species was grown in nutrient solution, and
after two weeks, the length of the longest roots had increased by
65%. At the higher concentration applied, i.e. 2.5 mg Pt/litre,
phytotoxic effects were seen in the form of stunted root growth,
i.e. root length about 75% compared to control, and chlorosis of the
leaves. As platinum was shown to accumulate in the roots and, at the
higher level, also in the shoots (see section 4.1), the potential
use of this grass species, either for the colonisation of flotation
tailings (a waste product from the concentration of precious metal
ores) or for the removal of platinum from the tailings, was
investigated. However, due to a substantial lack of essential
macronutrients in the tailings, growth of S. verticillata was very
poor. No platinum was detected in the plants.
Table 17. Dry weight (g) of tops and roots after a 7-day treatment with platinum tetrachloridea
Pt levels Bean Broccoli Cauliflower Corn Pea Pepper Radish Tomato Turnip
(mg/litre)
Tops
0 5.39 24.15 41.53 33.15 6.13 23.44 1.30 32.22 19.33
0.057 5.79 19.50 46.30 35.05 7.21 18.50 1.33 37.97 20.78
0.57 4.98 15.53 43.44 35.21 6.87 11.90 1.23 40.62 17.55
5.7 4.01 18.24 40.96 25.80 5.22 14.18 0.91 28.18 9.55
Roots
0 3.11 5.45 6.09 9.96 2.86 4.82 2.17 5.96 4.80
0.057 2.80 4.77 6.56 10.96 2.60 4.82 2.24 7.64 5.51
0.57 2.70 4.02 5.68 10.73 2.81 3.32 2.17 6.68 4.91
5.7 2.31 5.36 6.57 8.36 2.20 4.90 1.19 6.00 3.82
a Adapted from: Pallas & Jones (1978)
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of human health risks
10.1.1 General population exposure
10.1.1.1 Exposure
There is lack of data on the actual exposure situation in
countries where automobile exhaust gas catalysts have been
introduced. Therefore, estimates of possible ambient air
concentrations of platinum are based on emission data and dispersion
models.
Loss of platinum from the pellet-type catalyst, which has never
been used in Europe and is no longer used on new cars in the USA,
was found to be up to approximately 2 µg per km travelled. Of the
particles emitted, 80% had particle diameters greater than 125 µm.
Since no determination of the particle size distribution was
performed, the percentage of the respirable portion is not known.
About 10% of the platinum emission was found to be water-soluble. In
general, these data are based on single or only a few measurements
and have not been validated.
Recent emission data from the new-generation monolith-type
catalyst indicate that the emission of platinum is lower by a factor
of 100-1000 than that of the pelleted catalyst. Emissions were on
average between 2 and 39 ng per km travelled at simulated speeds of
between 60 and 140 km/h. The mean aerodynamic diameter of the
particles emitted was 4-9 µm. These data have been validated by
repeated measurements using two monolith-type catalysts. However,
other types of catalysts should be investigated to substantiate
these emission data. In addition, because of the inadequate data
base, the speciation is not known exactly, although there is an
indication that the platinum emitted is in the metallic form or
consists of surface-oxidized particles.
The striking difference between the emission pattern of the two
catalyst types may be attributable to their basically different
design.
The possible ambient air concentrations of platinum, estimated
on the basis of these emission data and dispersion models, range
between 0.005 and 9 ng/m3 for the pellet-type catalyst and between
0.05 and 90 pg/m3 for the monolith-type catalyst. These
concentrations are lower by factors of 1 x 105 to 2 x 108 and 1
x 107 to 2 x 1010, respectively, than the occupational exposure
limit of 1 mg/m3 established by some countries for platinum metal
as total inhalable dust.
Assuming that 10% of the platinum emission from pelleted
catalysts contains potentially allergenic soluble platinum
compounds, the safety factor to the occupational exposure limit for
soluble platinum salts (2 µg/m3) would be 2 x 103 to 4 x 106.
However, there is no evidence that the soluble fraction of the
platinum emissions is allergenic.
10.1.1.2 Health effects
Since platinum is most probably not emitted in the form of
halogenated soluble salts, the sensitization risk from car catalyst
platinum is very low. There is no substantial evidence of any
biological effects from automobile platinum emissions. There are
also no data to substantiate the possibility that very finely
dispersed metallic platinum could be biologically active upon
inhalation.
10.1.2 Occupational groups
10.1.2.1 Exposure
Occupational exposure to platinum occurs in various workplaces
including mining. However, only exposure to certain halogenated
soluble salts through inhalation of dusts and skin contact is of
toxicological relevance. These compounds are mainly encountered
during platinum refining and catalyst manufacture.
There are only limited data to quantify workplace exposure. An
occupational exposure limit of 2 µg/m3 for soluble platinum salts
has been adopted by several countries. There are again limited data
suggesting that the exposure limit may sometimes be exceeded in
practice. Before the allergenic potential of soluble platinum salts
was established, workplace concentrations exceeding the present
occupational exposure limit by up to one order of magnitude were
found. However, it should be noted that analytical accuracy was not
very reliable.
Occupational exposure to the anti-tumour agents cisplatin and
its analogues during manufacturing and use is of importance.
However, a review and an evaluation of the health effects of these
compounds are beyond the scope of this document as these substances
are used primarily as therapeutic agents. In addition, their
toxicological properties are exceptional compared to other platinum
compounds.
10.1.2.2 Health effects
The acute toxicity of platinum salts for animals is low and
depends on their solubility. Insoluble compounds such at PtCl2 and
PtO2 have an extremely low acute toxicity and this would also be
expected for metallic platinum.
By far the most significant health effect from exposure to
soluble platinum salts is sensitization.
Some halogenated platinum salts are highly allergenic in
humans. The compounds mainly responsible for platinum salt
hypersensitivity (PSH) are hexachloroplatinic acid, H2[PtCl6],
ammonium hexachloroplatinate, (NH4)2[PtCl6], and potassium
tetra- and hexachloroplatinate, K2[PtCl4] and K2[PtCl6].
Except for one unsubstantiated case of alleged contact dermatitis in
connection with a "platinum" ring, there is no evidence for
sensitization from metallic platinum.
The mechanism of platinum salt allergy appears to be a type I
(IgE mediated) response as established through in vivo and in
vitro tests. There is evidence that platinum salts of low relative
molecular mass act as haptens combining with serum proteins to form
the complete antigen.
The signs and symptoms of allergic reactions due to platinum
salt exposure include urticaria, itching of skin, eyes, and nose,
watering of the eyes, sneezing, rhinorrhoea, coughing, wheezing, and
dyspnoea. The latent period from the first contact with platinum
salts to the occurrence of the first symptoms varies from a few
weeks to several years. Once sensitivity is established, even minute
amounts can elicit immediate and/or late onset of signs and
symptoms. The symptoms persist during exposure and usually disappear
on removal from exposure. However, if long-duration exposure occurs
after sensitization, individuals may never become completely free of
symptoms.
The diagnosis of platinum salt hypersensitivity is usually
based on a history of work-related symptoms and a positive platinum
skin prick test. The combination of these has been shown to be
reasonably sensitive and specific for the diagnosis of platinum salt
hypersensitivity. In vitro tests appear to be useful for
epidemiological evaluation, but lack specificity for individual
dignosis. Symptoms usually worsen as long as the workers remain in
the contaminated environment.
There is good evidence for the association of smoking or
pulmonary hyper-reactivity and sensitization. The evidence for atopy
as a predisposing factor is equivocal. This may be due to bias from
pre-employment screening.
Despite the occupational exposure limit of 2 µg/m3, wthe
prevalence of positive skin prick tests was found to be between 14
and 20% in workers exposed to levels of between < 0.05 and 0.1 µg
Pt/m3. Since these data were derived from area samples, short
sharp exposures above this limit could also have been responsible
for the sensitization observed. The present occupational exposure
limit might not be sufficient to prevent platinum salt
hypersensitization, although it is difficult to reach a firm
conclusion because of the lack of adequate data. To minimize the
risk, workplace exposure should be as low as practicable.
10.2 Evaluation of effects on the environment
Compared to that of other metals, the total production of
platinum is low, amounting annually to approximately 100 tonnes.
There are no data on platinum emissions during production. However,
because of the high value of platinum, losses are assumed to be low.
During the use of platinum-containing catalysts, platinum can escape
into the environment in small amounts, depending on the type of
catalyst. Of the stationary catalysts used in industry, only those
used for ammonia oxidation emit a quantifiable amount of platinum.
This is present in the nitric acid produced, which may be used for
conversion to nitrate fertilizers. In the USA the annual loss of
platinum is estimated to be around 200 kg. Since part of this amount
is distributed fairly uniformly all over the country, a rise in the
background level of platinum in soil would probably not be detected
because of the very low likely concentration.
Platinum emission from automobile catalysts also contributes to
a diffuse contamination of the environment. On the basis of the
emission data derived from the new-generation monolithic-type
catalysts, total platinum loss from mobile sources would be less
than that from nitric acid production. For example, at an assumed
average emission rate of 20 ng platinum per km travelled, 100
million cars equipped with catalytic converters would emit
approximately 20 kg per year for an average kilometreage of 10 000
km per year and per car. This implies that the contamination of the
environment with platinum is very low or negligible.
In comparison, the total loss of platinum from the older design
pellet-type catalytic converter would have been higher by a factor
of 100, i.e. 2000 kg per year, most of the platinum being emitted in
the form of larger particles that would be deposited close to the
roads. This would also explain platinum levels of up to 0.7 mg/kg
dry weight found in roadside dust samples near major free-ways in
the USA.
There is limited evidence that most of the platinum emitted is
in the metallic form, and thus will probably not be bioavailable in
the soil. Biomethylation of soluble platinum(IV) compounds has been
demonstrated in the presence of platinum(II). However, from these
laboratory data produced under abiotic conditions, it is not
possible to conclude that microorganisms in the environment are able
to biomethylate platinum complexes.
Analysis of Lake Michigan sediments led to the conclusion that
platinum has been deposited over the past 50 years at a constant
rate, while lead concentrations have markedly increased. However,
since the car catalyst was introduced in the USA only a few years
before these measurements were performed, these data are
insufficient for firm conclusions to be drawn.
No data on the effects of platinum compounds on environmental
microorganisms are available. However, from the bactericidal
activity of platinum complexes it can be assumed that these
compounds could influence, at appropriate concentrations, microbial
communities in the environment or, for example, in sewage treatment
plants.
Aquatic and terrestrial plants are affected by platinum
compounds at concentrations in the mg/litre or mg/kg range. Although
there is a lack of definite data on platinum levels in the
environment, it is probable that platinum and platinum compounds do
not present a risk to naturally occurring organisms at the low
concentrations expected to occur in the environment.
11. RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH AND THE
ENVIRONMENT
11.1 Pre-employment screening and medical evaluations
To screen workers at risk of developing platinum salt
hypersensitivity (PSH), the following should be carried out for all
employees potentially exposed to soluble platinum salts:
* a questionnaire with particular attention being paid to
previous respiratory disease, allergy, smoking habits, and
employment history;
* a complete medical examination, including lung function tests
(spirometry, flow volume), tests of bronchial reactivity (cold
air, methacholine, histamine, etc.), and an immunological
profile including total serum IgE;
* a skin prick test for atopic status using a battery of antigens
to include house dust mite, tree and grass pollen or other
equivalent common aeroallergens;
* skin prick tests with freshly prepared, properly buffered
saline solutions (e.g., 5% v/v glycerol/water containing 2.5 g
NaCl/litre, 1.37 g NaHCO3/litre, and 2 g phenol per litre) of
(NH4)2[PtCl6], Na2[PtCl4], and Na2[PtCl6].
Concentrations used for testing may vary from 10-9 to 10-3
g/ml depending on specific situations. All tests should be
performed in duplicate and should include a positive and
negative saline control.
11.2 Substitution with non-allergenic substances
An attempt should be made to substitute, whenever practicable,
non-allergenic for allergenic platinum compounds during refining,
manufacturing, and use.
11.3 Employment screening and medical evaluations
a) To detect sensitization during employment, skin prick tests
should be performed on all potentially exposed people at least
once a year. There is no convincing evidence that repeated
platinum skin testing could cause sensitization. Quarterly
testing intervals might be considered during the first two
years of employment, as sensitization more often occurs during
this period.
b) If medical symptoms or signs suggest the development of PSH,
the worker should be removed from any risk of exposure as soon
as possible. To detect functional changes in the respiratory
tract, lung function assessment, as described in section 11.1,
should also be performed at appropriate intervals.
11.4 Workplace hygiene
a) Since PSH can occur despite time-weighted average workplace
concentrations being consistently below the ACGIH threshold
limit value (TLV) of 2 µg/m3, the most effective prevention
is the improvement of control measures. This includes enclosed
processing and optimal ventilation in order to reduce exposure
to platinum salt aerosols and dusts to the lowest practicable
limit.
b) It has been suggested that high but short-lived platinum
concentrations resulting from spills or accidents are of
importance with respect to sensitization. Since the correlation
between the platinum exposure concentration and the development
of sensitization is unknown, a recommendation for a reduction
in the occupational exposure limit cannot be justified.
However, it is recommended that the commonly used occupational
exposure limit of 2 µg/m3 be changed from an 8-h time-
weighted average (TWA) to a ceiling value and that personal
sampling devices be used in conjunction with area sampling to
determine more correctly the true platinum exposure.
c) Engineering controls should always be in place to minimize
exposure. However, in some circumstances the use of protective
clothing, including specially designed airstream helmets, may
be necessary.
d) Workers should be provided with clean overalls solely for use
in the workplace, and showering facilities. Outdoor clothes
should not be worn in the workplace.
12. FURTHER RESEARCH
a) As there appears to be a lack of information concerning the
concentration-response relationship for the development of PSH
in experimental animals, studies should be performed to
investigate the effect of exposure concentration on
sensitization and to define the thresholds for sensitization
and elicitation.
b) The effect of predisposing factors such as pulmonary hyper-
reactivity should be investigated in greater detail to
determine their applicability for screening and identifying
individuals at risk of developing PSH.
c) The use of provocation challenge with soluble platinum salts as
an indicator of sensitization should be investigated to
determine if it is a more sensitive indicator than skin prick
tests.
d) The majority of human occupational studies regarding PSH were
performed as cross-sectional studies at platinum refineries.
Due to the inherent lack of sensitivity of this type of study
with respect to past exposures and workers leaving employment
because of disease, longitudinal studies should be performed to
determine the true incidence of PSH in worker populations. In
addition, human studies should be designed to study, for
instance, exposure concentration effects on sensitization and
determine thresholds for sensitization and elicitation.
e) The extent of occupational and environmental exposure to
cisplatin is not known at the present time. It is recommended
that studies be initiated to determine exposure during the
manufacture and use of this compound.
f) Further measurements of the quantities and speciation of
platinum emitted from automobile catalysts should be performed.
g) The toxic effects of finely divided metallic platinum on humans
and animals have not been studied adequately. Adequate
inhalation studies are initially required, and further tests
may be necessary.
h) Quality control programmes should be initiated to ensure the
accuracy and precision of sampling methods and analyses and to
facilitate comparability.
i) Platinum-containing exhaust emissions from automobile catalysts
most probably do not pose an adverse health effect for the
general population. However, to be on the safe side, the
possibility should be kept under review.
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
The carcinogenicity of platinum and platinum compounds has not
been evaluated by international bodies, except for cisplatin, which
has not been covered in detail in this Environmental Health Criteria
monograph (see also footnote in section 1.2).
The International Agency for Research on Cancer (IARC, 1987b)
considered the evidence for carcinogenicity of cisplatin for animals
to be sufficient, but that for humans inadequate. Cisplatin is
classified in Group 2A, i.e. probably carcinogenic to humans.
REFERENCES
ACGIH (1980) Documentation of the threshold limit values, 4th ed.,
Cincinnati, Ohio, American Conference of Governmental Industrial
Hygienists, pp. 343-344.
ACGIH (1990) Threshold limit values for chemical substances and
physical agents and biological exposure indices for 1990-1991,
Cincinnati, Ohio, American Conference of Governmental Industrial
Hygienists, p. 31.
AGIORGITIS, G. & GUNDLACH, H. (1978) [Platinum contents of deep sea
manganese nodules.] Naturwissenschaften, 65: 534 (in German).
AGNES, G., HILL, H.A.O., PRATT, J.M., RIDSDALE, S.C., KENNEDY, F.S.,
& WILLIAMS, R.J.P. (1971) Methyl transfer from methyl vitamin B12.
Biochim. Biophys. Acta, 252: 207-211.
AGOOS, A. (1986) Metal reclaimers find slim pickings. Chem. Week,
138: 21-23.
ALEXANDER, P.W., HOH, R., & SMYTHE, L.E. (1977a) Trace determination
of platinum, I. A catalytic method using pulse polarography.
Talanta, 24: 543- 548.
ALEXANDER, P.W., HOH, R., & SMYTHE, L.E. (1977b) Trace determination
of platinum, II. Analysis in ores by pulse polarography after fire-
assay collection. Talanta, 24: 549-554.
ALT, F., JERONO, U., MESSERSCHMIDT, J., & TÖLG, G. (1988) The
determination of platinum in biotic and environmental materials: I.
µg/kg to g/kg- range. Mikrochim. Acta, 3: 299-304.
AMOSSE, J., FISCHER, W., ALLIBERT, M., & PIBOULE, M. (1986) Méthode
de dosage d'ultra-traces de platine, palladium, rhodium et or dans
les roches silicatées par spectrophotométrie d'absorption atomique
électrothermique. Analusis, 14: 26-31.
ANDERSEN, K.S. (1979) Platinum(II) complexes generate frame-shift
mutations in test strains of Salmonella typhimurium. Mutat. Res.,
67: 209-214.
ANDREWS, P.A., WUNG, W.E., & HOWELL, S.B. (1984) A high-performance
liquid chromatographic assay with improved selectivity for cisplatin
and active platinum (II) complexes in plasma ultrafiltrate. Anal.
Biochem., 43: 46-56.
ANEVA, Z., ARPADJAN, S., ALEXANDROV, S., & KOVATCHEVA, K. (1986)
Synergetic extraction of platinum(IV) from dilute hydrochloric acid
by isoamyl alcohol-methylisobutylketone mixture. Mikrochim. Acta, 1:
341-350.
ANON. (1990a) Nitric acid catalysts: the economic significance of
composition, operation conditions and recovery techniques. Nitrogen,
183: 27-32.
ANON. (1990b) Growth slackens appreciably as 1980s end. Chem. Eng.
News, 18 June: 36-43.
ANUSAVICE, K.J. (1985) Noble metal alloys for metal-ceramic
restorations. Dent. Clin. North Am., 29: 789-803.
ARPADJAN, S., MITEWA, M., & BONTCHEV, P.R. (1987) Liquid-liquid
extraction of metal ions by the 6-membered N-containing macrocycle
hexacyclen. Talanta, 34: 953-956.
BANKOVSKY, Y.A., VIRCAVS, M.V., VEVERIS, O.E., PELNE, A.R., &
VIRCAVA, D.K. (1987) Preconcentration of microamounts of elements in
natural waters with 8-mercaptoquinoline and bis(8-quinolyl)
disulphide for their atomic-absorption determination. Talanta, 34:
179-182.
BANNISTER, S.J., CHANG, Y., STERNSON, L.A., & REPTA, A.J. (1978)
Atomic absorption spectrophotometry of free circulating platinum
species in plasma derived from cis-dichlorodiammineplatinum(II).
Clin. Chem., 24: 877-880.
BARLOW, S.M. & SULLIVAN, F.M. (1982) Reproductive hazards of
industrial chemicals, London, Academic Press, pp. 431-436.
BARNES, G.D. & TALBERT, L.D. (1984) The effect of platinum on
population and absorbance of Euglena gracilis, Klebs utilizing a
method with atomic absorption and coulter counter analysis. J.
Mississipi Acad. Sci., 29: 143-150.
BAUMGÄRTNER, M.E. & RAUB, Ch.J. (1988) The electrodeposition of
platinum and platinum alloys. Platinum Met. Rev., 32: 188-197.
BELLIVEAU, J.F., MATOOK, G.M., O'LEARY, G.P., Jr, CUMMINGS, F.J.,
HILLSTROM, M., & CALABRESI, P. (1986) Microanalysis for platinum and
magnesium in body fluids and brain tissue of mice treated with cis-
platinum using graphite filament plasma emission spectroscopy. Anal.
Lett., 19: 135- 149.
BERNDT, H., SCHALDACH, G., & KLOCKENKÄMPER, R. (1987) Improvement of
the detection power in electrothermal atomic absorption spectrometry
by summation of signals. Determination of traces of metals in
drinking water and urine. Anal. Chim. Acta, 200: 573-579.
BIAGINI, R.E., CLARK, J.C., GALLAGHER, J.S., BERNSTEIN, I.L., &
MOORMAN, W.M. (1982) Passive transfer in the monkey of human
immediate hyper- sensitivity to complex salts of platinum and
palladium. Fed. Proc., 41: 827.
BIAGINI, R.E., MOORMAN, W.J., SMITH, R.J., LEWIS, T.R., & BERNSTEIN,
I.L. (1983) Pulmonary hyperreactivity in cynomolgus monkeys (Macaca
fasicularis) from nose-only inhalation exposure to disodium
hexachloroplatinate, Na2PtCl6. Toxicol. appl. Pharmacol., 69:
377-384.
BIAGINI, R.E., BERNSTEIN, I.L., GALLAGHER, J.S., MOORMAN, W.J.,
BROOKS, S., & GANN, P.H. (1985) The diversity of reaginic immune
responses to platinum and palladium metallic salts. J. Allergy clin.
Immunol., 76: 794-802.
BIAGINI, R.E., MOORMAN, W.J., LEWIS, T.R., & BERNSTEIN, I.L. (1986)
Ozone enhancement of platinum asthma in a primate model. Am. Rev.
Respir. Dis., 134: 719-725.
BIESINGER, K.E. & CHRISTENSEN, G.M. (1972) Effects of various metals
on survival, growth, reproduction, and metabolism of Daphnia magna.
J. Fish Res. Board Can., 29: 1691-1700.
BODENNER, D.L., DEDON, P.C., KENG, P.C., KATZ, J.C., & BORCH, R.F.
(1986) Selective protection against cis-diamminedichloroplatinum
(II)-induced toxicity in kidney, gut, and bone marrow by
diethyldithiocarbamate. Cancer Res., 46: 2751-2755.
BOGGS, P.B. (1985) Platinum allergy. Cutis, 35: 318-320.
BOLM-AUDORFF, U., BIENFAIT, H.-G., BURKHARD, J., BURY, A.-H.,
MERGET, R., PRESSEL, G., & SCHULTZE-WERNINGHAUS, G. (1988) [On the
frequency of respiratory allergies in a platinum processing
factory.] In: Baumgartner, E., Brenner, W., Dierich, M.P., &
Rutenfranz, J., ed. [Industrial change - Occupational medicine
facing new questions. Report of the 28th Annual Meeting of the
German Society for Occupational Medicine, Innsbruck, Austria, 4-7
May 1988] Stuttgart, Gentner Verlag, pp. 411-416 (in German).
BORCH, R.F., MARKOVITZ, J.H., & PLEASANTS, M.E. (1979) A new method
for the HPLC analysis of Pt(II) in urine. Anal. Lett., 12: 917-926.
BOUMANS, P.W.J.M. & VRAKKING, J.J.A.M. (1987) Detection of about 350
prominent lines of 65 elements observed in 50 and 27 MHz inductively
coupled plasmas (ICP): effects of source characteristics, noise and
spectral bandwidth - "Standard" values for the 50 MHz ICP.
Spectrochim. Acta, 42B: 553-579.
BOWEN, H.J.M. (1979) Environmental chemistry of the elements,
London, Academic Press, p. 37.
BRADFORD, C.W. (1988) Platinum. In: Seiler, H.G. & Sigel, H., ed.
Handbook of toxicity of inorganic compounds, New York, Basel, Marcel
Dekker, pp. 533-539.
BRAJTER, K. & KOZICKA, U. (1979) Extractive spectrophotometric
determination of platinum, rhodium and iridium. Talanta, 26:
417-419.
BROOKS, S.M., BAKER, D.B., GANN, P.H., JARABEK, A.M., HERTZBERG, V.,
GALLAGHER, J., BIAGINI, R.E., & BERNSTEIN, I.L. (1990) Cold air
challenge and platinum skin reactivity in platinum refinery workers.
Bronchial reactivity precedes skin prick response. Chest, 79:
1401-1407.
BROWN, A.A. & LEE, M. (1986) Peak profile and appearance times using
totally pyrolytic cuvettes in graphite furnace atomic absorption
spectrometry. Anal. Chem., 323: 697-702.
BRUBAKER, P.E., MORAN, J.P., BRIDBORD, K., & HUETER, F.G. (1975)
Noble metals: a toxicological appraisal of potential new
environmental contaminants. Environ. Health Perspect., 10: 39-56.
BURGE, P.S., HARRIES, M.G., O'BRIEN, I.M., & PEPYS, J. (1979)
Evidence for specific hypersensitivity in occupational asthma due to
small molecular weight chemicals and an organic (locust) allergen.
In: Pepys, J. & Edwards, A.M., ed. The mast cell - its role in
health and disease, Tunbridge Wells, Kent, United Kingdom, Pitman
Medical Publishing Co., Ltd, pp. 301-308.
BUTTERMAN, W.C. (1975) Platinum-group metals. Minerals Yearbook 1973
Metals, Minerals, and Fuels, Washington, DC, US Department of the
Interior, Bureau of Mines, pp. 1037-1049.
CAMPBELL, K.I., GEORGE, E.L., HALL, L.L., & STARA, J.F. (1975)
Dermal irritancy of metal compounds. Arch. environ. Health, 30:
168-170.
CHANG, C.C. & TATUM, H.J. (1975) Effect of intrauterine copper wire
on resorption of foetuses in rats. Contraception, 11: 79-84.
CHANG, C.C., TATUM, H.J., & KINCL, F.A. (1970) The effect of
intrauterine copper and other metals on implantation in rats and
hamsters. Fertil. Steril. 21(3): 274-278.
CHEY, R.D., LEE, K.Y, ASBURY, R., & CHEY, W.Y. (1988) Effect of
cisplatin on myoelectric activity of the stomach and small intestine
in dogs. Dig. Dis. Sci., 33: 338-344.
CLEARE, M.J. (1977a) Some aspects of platinum complex chemistry and
their relation to anti-tumor activity. J. clin. Hematol. Oncol., 7:
1-21.
CLEARE, M.J. (1977b) Immunological studies on platinum complexes and
their possible relevance to autocatalysts. Proceedings of the
International Automotive Engineering Congress and Exposition, Cobo
Hall, Detroit, 28 February - 4 March 24, 1977, Detroit, Michigan
Society of Automotive Engineers, 12 pp (SAE Report No. 770061).
CLEARE, M.J., HUGHES, E.G., JACOBY, B., & PEPYS, J. (1976) Immediate
(type I) allergic responses to platinum compounds. Clin. Allergy, 6:
183-195.
COOMBES, R.J. & CHOW, A. (1979) A comparison of methods for the
determination of platinum in ores. Talanta, 26: 991-998.
CROMWELL, O., PEPYS, J., PARISH, W.E, & HUGHES, E.G. (1979) Specific
IgE antibodies to platinum salts in sensitized workers. Clin.
Allergy, 9: 109-117.
CVITKOVIC, E., SPAULDING, J., BETHUNE, V., MARTIN, J., & WHITMORE,
W.F. (1977) Improvement of cis-dichlorodiammineplatinum (NSC
119875): therapeutic index in an animal model. Cancer, 39:
1357-1361.
D'AGOSTINO, R.B., LOWN, B.A., MORGANTI, J.B., CHAPIN, E., & MASSARO,
E.J. (1984) Effects on the development of offspring of female mice
exposed to platinum sulfate or sodium hexachloroplatinate during
pregnancy or lactation. J. Toxicol. environ. Health, 13: 879-891.
DALLY, M.B., HUNTER, J.V., HUGHES, E.G., STEWART, M., & NEWMAN
TAYLOR, A.J. (1980) Hypersensitivity to platinum salts: a population
study. Am. Rev. respir. Dis., 121(4/Part 2): 230.
DE CONTI, R.C., TOFTNESS, B.R., LANGE, R.C., & CREASEY, W.A. (1973)
Clinical and pharmacological studies with cis-diammine-
dichloroplatinum (II). Cancer Res., 33: 1310-1315.
DEGUSSA (1988a) Ammonium-tetrachloroplatinate(II) - Acute toxicity.
Testing the primary irritancy after single application to the skin
of the rabbit (patch test), Hanau, Germany, Degussa AG (Unpublished
report No. 863897).
DEGUSSA (1988b) Ammonium-tetrachloroplatinate(II) - Acute toxicity.
Testing the primary irritancy after single application to the eye of
the rabbit, Hanau, Germany, Degussa AG (Unpublished report No.
863908).
DEGUSSA (1988c) Trans-diammine-dichloroplatinum(II) - Acute
toxicity. Testing the primary irritancy after single application to
the skin of the rabbit, Hanau, Germany, Degussa AG (unpublished
report No. 864011).
DEGUSSA (1988d) Trans-diammine-dichloroplatinum(II) - Acute
toxicity. Testing the primary irritancy after single application to
the eye of the rabbit, Hanau, Germany, Degussa AG (Unpublished
report No. 864022).
DEGUSSA (1989a) Ammoniumtetrachloroplatinate(II) - Acute toxicity
after single oral administration in rats, Hanau, Germany, Degussa AG
(Unpublished report No. 863886).
DEGUSSA (1989b) Dinitrodiammine-platinum(II) - Acute toxicity after
single oral administration in rats, Hanau, Germany, Degussa AG
(Unpublished report No. 863910).
DEGUSSA (1989c) Trans-diammine-dichloroplatinum(II) - Acute toxicity
after single oral administration in rats, Hanau, Germany, Degussa AG
(Unpublished report No. 864000).
DEGUSSA (1989d) Dinitrodiammine-platinum(II) - Acute toxicity.
Testing the primary irritancy after single application to the skin
of the rabbit (patch test), Hanau, Germany, Degussa AG (Unpublished
report No. 863921).
DEGUSSA (1989e) Dinitrodiammine-platinum(II) - Acute toxicity.
Testing the primary irritancy after single application to the eye of
the rabbit), Hanau, Germany, Degussa AG (Unpublished report No.
863932).
DISSANAYAKE, C.B. (1983) Metal-organic interactions in environmental
pollution. Int. J. environ. Stud., 22: 25-42.
DISSANAYAKE, C.B., KRITSOTAKIS, K., & TOBSCHALL, H.J. (1984) The
abundance of Au, Pt, Pd, and the mode of heavy metal fixation in
highly polluted sediments from the Rhine River near Mainz, West
Germany. Int. J. environ. Stud., 22: 109-119.
DONALDSON, P.E.K. (1987) The role of platinum metals in neurological
prostheses. Platinum Met. Rev., 31: 2-7.
DUFFIELD, F.V.P., YOAKUM, A., BUMGARNER, J., & MORAN, J. (1976)
Determination of human body burden baseline data of platinum through
autopsy tissue analysis. Environ. Health Perspect., 15: 131-134.
EBINA, T., SUZUKI, H., & YOTSUYANAGI, T. (1983) [Spectrophotometric
determination of palladium(II) and platinum(II) with
maleonitriledithiol by reversed phase ion-pair chromatography.]
Bunseki Kagaku, 32: 575-580 (in Japanese).
ELLER, P.M., ed. (1984a) Elements (ICP). In: NIOSH manual of
analytical methods, 3rd ed., Cincinnati, Ohio, National Institute
for Occupational Safety and Health, Vol. 1, pp. 7300-1-7300-5.
ELLER, P.M., ed. (1984b) Elements in blood or tissue. In: NIOSH
manual of analytical methods, 3rd ed., Cincinnati, Ohio, National
Institute for Occupational Safety and Health, Vol.2, pp.
8005-1-8005-5.
ELLER, P.M., ed. (1984c) Metals in urine. In: NIOSH manual of
analytical methods, 3rd ed., Cincinnati, Ohio, National Institute
for Occupational Safety and Health, Vol. 2, pp. 8310-1-8310-6.
ETTINGER, L.J. & FREEMAN, A.I. (1979) The gingival platinum line. A
new finding following cis-dichlorodiammine platinum(II) treatment.
Cancer, 44: 1882-1884.
FANCHIANG, Y.-T. (1985) Reactions of alkylcobalamins with platinum
complexes. Coord. Chem. Rev., 68: 131-144.
FANCHIANG, Y-T., RIDLEY, W.P., & WOOD, J.M. (1979) Methylation of
platinum complexes by methylcobalamin. J. Am. Chem. Soc., 101:
1442-1447.
FARAGO, M.E. & PARSONS, P.J. (1982) Determination of platinum,
palladium and rhodium by atomic-absorption spectroscopy with
electrothermal atomisation. Analyst, 107: 1218-1228.
FARAGO, M.E. & PARSONS, P.J. (1985) Effects of platinum metals on
plants. Trace Subst. environ. Health, 19: 397-407.
FARAGO, M.E. & PARSONS, P.J. (1986) The effect of platinum, applied
as potassium tetrachloroplatinate, on Setaria verticillata (L) P.
Beauv. and its growth on flotation tailings. Environ. Technol.
Lett., 7: 147-154.
FERREIRA, P.F. & WOLKE, R.E. (1979) Acute toxicity of platinum to
coho salmon (Oncorhynchus kisutch). Mar. Pollut. Bull., 10: 79-83.
FILLASTRE, J.P. & RAGUENEZ-VIOTTE, G. (1989) Cisplatin
nephrotoxicity. Toxicol. Lett., 46: 163-175.
FOTHERGILL, S.J.R., WITHERS, D.F., & CLEMENTS, F.S. (1945)
Determination of traces of platinum and palladium in the atmosphere
of a platinum refinery. Br. J. ind. Med., 2: 99-101.
FOX, R.L. (1984) Enhancement errors in the determination of platinum
group metals in alumina-based matrices by direct-current plasma
emission spectrometry. Appl. Spectrosc., 38: 644-647.
FREEDMAN, S.O. & KRUPEY, J. (1968) Respiratory allergy caused by
platinum salts. J. Allergy, 42: 233-237.
FRYER, B.J. & KERRICH, R. (1978) Determination of precious metals at
ppb levels in rocks by a combined wet chemical and flameless atomic
absorption method. At. Absorpt. Newsl., 17: 4-6.
FUCHS, W.A. & ROSE, A.W. (1974) The geochemical behavior of platinum
and palladium in the weathering cycle in the Stillwater Complex,
Montana. Econ. Geol., 69: 332-346.
GALLAGHER, J.S., BAKER, D., GANN, P.H., JARABEK, A.M., BROOKS, S.M.,
& BERNSTEIN, I.L. (1982) A cross sectional investigation of workers
exposed to platinum salts. J. Allergy clin. Immunol., 69: 134.
GECKELER, K.E., BAYER, E., SPIVAKOV, B.Y., SHKINEV, V.M., &
VOROB'EVA, G.A. (1986) Liquid-phase polymer-based retention; a new
method for separation and preconcentration of elements. Anal. Chim.
Acta, 189: 285-292.
GLADKOVA, E.V., ODINTSOVA, F.P., VOLKOVA, I.D., & VINOGRADOVA, V.K.
(1974) [Health status of workers engaged in the production of the
platinum catalyst.] Gig. Tr. prof.Zabol., 3: 10-13 (in Russian).
GOLDBERG, E.D., HODGE, V.F., GRIFFIN, J.J., KOIDE, M., & EDGINGTON,
D.N. (1981) The impact of fossil fuel combustion on the sediments of
Lake Michigan. Environ. Sci. Technol., 15: 466-471.
GOLDBERG, E.D., HODGE, V., KAY, P., STALLARD, M., & KOIDE, M. (1986)
Some comparative marine chemistries of platinum and iridium. Appl.
Geochem., 1: 227-232.
GOLDSCHMIDT, V.M. (1954) Geochemistry, London, Oxford University
Press, 730 pp.
GREGOIRE, D.C. (1988) Determination of platinum, palladium,
ruthenium and iridium geological materials by inductively coupled
plasma mass spectrometry with sample introduction by electrothermal
vaporization. J. anal. at. Spectrom., 3: 309-314.
GROOTVELD, M.C. (1985) Studies on the reaction mechanisms of metal
complexes of biological and immunochemical interest, London,
University of London, Department of Chemistry, 341 pp (Thesis).
GROTE, M. & KETTRUP, A. (1987) Ion-exchange resins containing S-
bonded dithizone and dehydrodithizone as functional groups. Part 3.
Determination of gold, platinum and palladium in geological samples
by means of a dehydrodithizone resin and plasma emission
spectrometry. Anal. Chim. Acta, 201: 95-107.
GSF (1990) [Noble metal emissions - interim report; Research
Programme of the Federal Ministry of Research and Technology],
Munich, Association for Radiation and Environmental Research (GSF),
45 pp. (in German).
HALBACH, P., PUTEANUS, D., & MANHEIM, F.T. (1984) Platinum
concentrations in ferromanganese seamount crusts from the central
Pacific. Naturwissenschaften, 71: 577-579.
HAMILTON, E.L. & MINSKI, M.J. (1972/1973) Abundance of the chemical
elements in man's diet and possible relations with environmental
factors. Sci. total Environ., 1: 375-394.
HAMNER, C.L. (1942) Effects of platinum chloride on bean and tomato.
Bot. Gaz., 104: 161-166.
HARDER, H.C. & ROSENBERG, B. (1970) Inhibitory effects of anti-tumor
platinum compounds on DNA, RNA and protein syntheses in mammalian
cells in vitro. Int. J. Cancer, 6: 207-216.
HARDMAN, R.S. & WRIGHT, C.H. (1896) A case of poisoning by
chloroplatinite of potassium. Br. med. J., 1: 529.
HAYES, F.A., GREEN, A.A., SENZER, N., & PRATT, C.B. (1979) Tetany: a
complication of cis-dichlorodiammine-platinum(II) therapy. Cancer
Treat. Rep., 63: 547-548.
HEALTH AND SAFETY EXECUTIVE (1983) The medical monitoring of workers
exposed to platinum `salts', London, Her Majesty's Stationery
Office, 2 pp (MS/22).
HEALTH and SAFETY EXECUTIVE (1985) Platinum metal and soluble
inorganic compounds of platinum in air. Laboratory method using
carbon furnace atomic absorption spectrometry, London, Her Majesty's
Stationery Office, 4 pp (MDHS 46).
HEALTH AND SAFETY EXECUTIVE (1990) Occupational exposure limits
1990. Guidance note EH 40190, London, Her Majesty's Stationery
Office.
HEBERT, R. (1966) Affections provoquées par les composés du platine.
Arch. Mal. prof. Méd. Trav. Sécur. soc., 27: 877-886.
HILL, J.M., LOEB, E., MACLELLAN, A., HILL, N.O., KHAN, A., & KING,
J.J. (1975) Clinical studies of platinum coordination compounds in
the treatment of various malignant diseases. Cancer Chemother. Rep.,
59(Part 1): 647-659.
HILL, R.F. & MAYER, W.J. (1977) Radiometric determination of
platinum and palladium attrition from automotive catalysts. IEEE
Trans. nucl. Sci., NS-24: 2549-2554.
HODGE, V.F. & STALLARD, M.O. (1986) Platinum and palladium in
roadside dust. Environ. Sci. Technol., 20: 1058-1060.
HODGE, V.F., STALLARD, M., KOIDE, M., & GOLDBERG, E.D. (1985)
Platinum and the platinum anomaly in the marine environment. Earth
planet. Sci. Lett., 72: 158-162.
HODGE, V.F., STALLARD, M., KOIDE, M., & GOLDBERG, E.D. (1986)
Determination of platinum and iridium in marine waters, sediments,
and organisms. Anal. Chem., 58: 616-620.
HOESCHELE, J.D. & VAN CAMP, L. (1972) Whole-body counting and the
distribution of cis195m[Pt(NH3)2Cl2] in the major organs of
Swiss white mice. In: Hejzlar, M., ed. Advances in antimicrobial and
antineoplastic chemotherapy, Baltimore, University Park Press, Vol.
2, pp. 241-242.
HOEVE, L.J., CONIJIN, E.A.J.G., MERTENS ZUR BORG, I.R.A.M., &
RODENBURG, M. (1987) Development of cis-platinum-induced hearing
loss in guinea pigs. Arch. Otorhinolaryngol., 244: 265-268.
HOFFMANN, R.L. (1988) The effect of cisplatin and platinum (IV)
chloride on cell growth, RNA, protein, ribosome and DNA synthesis in
yeast. Toxicol. environ. Chem., 17: 139-151.
HOFMEISTER, F. (1882) [On the physiologic effects of the platinum
bases.] Naunyn-Schmiedeberg's Arch. exp. Pathol. Pharmakol., 16:
393-439 (in German).
HOLBROOK, D.J., Jr (1976a) Assessment of toxicity of automotive
metallic emissions, Vol. I: Assessment of fuel additives emission
toxicity via selected assays of nucleic and protein synthesis,
Research Triangle Park, North Carolina, US Environmental Protection
Agency, Office of Research and Development, Health Effects Research
Laboratories, 67 pp (EPA/600/1-76/010a).
HOLBROOK, D.J., Jr (1976b) Assessment of toxicity of automotive
metallic emissions, Vol. II: Relative toxicities of automotive
metallic emissions against lead compounds using biochemical
parameters, Research Triangle Park, North Carolina, US Environmental
Protection Agency, Office of Research and Development, Health
Effects Research Laboratories, 69 pp. (EPA/600/1-76/010b).
HOLBROOK, D.J., Jr (1977) Content of platinum and palladium in rat
tissue: correlation of tissue concentration of platinum and
palladium with biochemical effects, Research Triangle Park, North
Carolina, US Environmental Protection Agency, Office of Research and
Development, Health Effects Research Laboratory, 21 pp.
(EPA/600/1-77/051).
HOLBROOK, D.J., Jr, WASHINGTON, M.E., LEAKE, H.B., & BRUBAKER, P.E.
(1975) Studies on the evaluation of the toxicity of various salts of
lead, manganese, platinum, and palladium. Environ. Health Perspect.,
10: 95- 101.
HOPPE, R. (1965) [Chemistry of the noble gases.] Fortschr. chem.
Forsch., 5: 213-346 (in German).
HOPPSTOCK, K., ALT, F., CAMMANN, K., & WEBER, G. (1989)
Determination of platinum in biotic and environmental materials.
Part II: A sensitive voltammetric method. Fresenius Z. anal. Chem.,
335: 813-816.
HOWLE, J.A. & GALE, G.R. (1970) Cis-dichlorodiammineplatinum (II)
persistent and selective inhibition of deoxyribonucleic acid
synthesis in vivo. Biochem. Pharmacol., 19: 2757-2762.
HUGHES, E.G. (1980) Medical surveillance of platinum refinery
workers. J. Soc. Occup. Med., 30: 27-30.
HUNTER, D., MILTON, R., & PERRY, K.M.A. (1945) Asthma caused by the
complex salts of platinum. Br. J. ind. Med., 2: 92-98.
IARC (1987a) Cisplatin. In: Genetic and related effects: an updating
of selected IARC Monographs from Volumes 1 to 42, Lyon,
International Agency for Research on Cancer, pp. 178-181 (IARC
Monographs on the Evaluation of Carcinogenic Risks to Humans, Suppl.
6).
IARC (1987b) Cisplatin (Group 2A). In: Overall evaluations of
carcinogenicity: an updating of IARC Monographs Volumes 1 to 42,
Lyon, International Agency for Research on Cancer, pp. 170-171 (IARC
Monographs on the Evaluation of Carcinogenic Risks to Humans, Suppl.
7).
IMURA, N. (1986) The role of micronutrient selenium in the
manifestation of toxicity of heavy metals. Toxicol. Lett., 31: 11.
INGALLS, M.N. & GARBE, R.J. (1982) Ambient pollutant concentrations
from mobile sources in microscale situations, Warrendale,
Pennsylvania, Society of Automotive Engineers, Inc., 16 pp (SAE
Technical Paper Series. No. 820787).
ITO, E. & KIDANI, Y. (1982) Determination of platinum in the
environmental samples by graphite furnace atomic absorption
spectrophotometry. Bunseki Kagaku, 31: 381-388.
JACOBS, L. (1987) Platinum salt sensitivity. Nurs. RSA Verpleging
(Republic of South Africa), 2: 34-37.
JOHNSON, D.E., TILLERY, J.B., & PREVOST, R.J. (1975) Levels of
platinum, palladium, and lead in populations of Southern California.
Environ. Health Perspect., 12: 27-33.
JOHNSON, D.E., PREVOST, R.J., TILLERY, J.B., CAMAN, D.E., &
HOSENFELD, J.M. (1976) Baseline levels of platinum and palladium in
human tissue, San Antonio, Texas, Southwest Research Institute, 252
pp. (EPA/600/1- 76/019).
JOHNSON, N.P., HOESCHELE, J.D., RAHN, R.O., O'NEIL, P.J., & HSIE,
A.W. (1980) Mutagenicity, cytotoxicity and DNA binding of
platinum(II)-chloroammines in chinese hamster ovary cells. Cancer
Res., 40: 1463-1468.
JOHNSON MATTHEY (1976a) Acute oral toxicity study CB10:
Bis(acetylacetonate) platinum(II), Reading, United Kingdom, Johnson
Matthey Research Centre (Unpublished report No. 8/7610)
JOHNSON MATTHEY (1976b) Skin irritation study, CB10:
Bis(acetylacetonate) platinum(II), Reading, United Kingdom, Johnson
Matthey Research Centre (Unpublished report No. 8/7610).
JOHNSON MATTHEY (1976c) Eye irritation study CB10:
Bis(acetylacetonate) platinum(II), Reading, United Kingdom, Johnson
Matthey Research Centre (Unpublished report No.8/7610).
JOHNSON MATTHEY (1977a) Acute oral toxicity study, CB20: Potassium
tetracyanoplatinate(II), Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 86/7709).
JOHNSON MATTHEY (1977b) Acute oral toxicity study, CB18: Tetrammine
platinous chloride, Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 248/7702).
JOHNSON MATTHEY (1977c) Acute oral toxicity study, CB18:
Cis[PtCl2(NH3)2], Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 216/7707).
JOHNSON MATTHEY (1977d) Primary skin irritation study, CB20:
Potassium tetracyanoplatinate(II), Reading, United Kingdom, Johnson
Matthey Research Centre (Unpublished report No. 88/7709).
JOHNSON MATTHEY (1977e) Skin irritation study, CB13: Tetrammine
platinous chloride, Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 113/7701).
JOHNSON MATTHEY (1977f) Eye irritation study, CB13: Tetrammine
platinous chloride, Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 252/7702).
JOHNSON MATTHEY (1977g) Primary skin irritation study, CB18:
Cis[PtCl2(NH3)2], Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 217/7707).
JOHNSON MATTHEY (1977h) Eye irritation/toxicity study, sample CB18:
Cis[PtCl2(NH3)2], Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 337/7802).
JOHNSON MATTHEY (1978a) Acute oral toxicity study in the rat, CB14:
Ammonium hexachloroplatinate, batch No. 57662, Reading, United
Kingdom, Johnson Matthey Research Centre (Unpublished report No.
106/7808).
JOHNSON MATTHEY (1978b) Acute oral toxicity study, CB24: Sodium
chloroplatinate, Reading, United Kingdom, Johnson Matthey Research
Centre (Unpublished report No. 359/7711).
JOHNSON MATTHEY (1978c) Acute oral toxicity study in the rat, CB70:
Sodium hexachloroplatinate, Batch No. 031074, Reading, United
Kingdom, Johnson Matthey Research Centre (Unpublished report No.
42/7809).
JOHNSON MATTHEY (1978d) Primary skin irritation study, sample CB14:
Ammonium hexachloroplatinate, batch No. 57662, Reading, United
Kingdom, Johnson Matthey Research Centre (Unpublished report No.
109/7808).
JOHNSON MATTHEY (1978e) Primary skin irritation study, sample CB24:
Sodium chloroplatinate, batch No. 031003, Reading, United Kingdom,
Johnson Matthey Research Centre (Unpublished report No. 245/7802).
JOHNSON MATTHEY (1978f) Eye irritation study, sample CB24: Sodium
chloroplatinate, batch No. 031003, Reading, United Kingdom, Johnson
Matthey Research Centre (Unpublished report No. 240/7802).
JOHNSON MATTHEY (1978g) Primary skin irritation study U.S. Federal
Register 1973, sample CB70: Sodium hexahydroxyplatinate, batch No.
031074, Reading, United Kingdom, Johnson Matthey Research Centre
(Unpublished report No. 43/7809).
JOHNSON MATTHEY (1978h) Eye irritation study, sample CB20: Potassium
tetracyanoplatinate(II), Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 318/7802).
JOHNSON MATTHEY (1981a) An assessment of the acute oral toxicity of
potassium tetracyanoplatinate(II)-MS 332 in the rat, experiment No.
24/8112, Reading, United Kingdom, Johnson Matthey Research Centre
(Unpublished report).
JOHNSON MATTHEY (1981b) An assessment of the acute oral toxicity of
potassium tetracyanoplatinate(II)-MS 332 in the rat, experiment No.
216/8111, Reading, United Kingdom, Johnson Matthey Research Centre
(Unpublished report).
JOHNSON MATTHEY (1981c) OECD skin irritation test: determination of
the degree of primary cutaneous irritation caused by potassium
tetracyanoplatinate(II)-MS 332 in the rabbit, experiment No.
631/8111, Reading, United Kingdom, Johnson Matthey Research Centre
(Unpublished report).
JOHNSON MATTHEY (1981d) OECD eye irritation test: determination of
the degree of ocular irritation caused by potassium
tetrachloroplatinate(II)-MS 332 in the rabbit, experiment No.
278/8112, Reading, United Kingdom, Johnson Matthey Research Centre
(Unpublished report).
JOHNSON MATTHEY (1990) Platinum 1990, London, Johnson Matthey Public
Limited Company, 64 pp.
JONES, A.H. (1976) Determination of platinum and palladium in blood
and urine by flameless atomic absorption spectrophotometry. Anal.
Chem., 48: 1472-1474.
JONES, E.A., WARSHAWSKY, A., DIXON, K., NICOLAS, D.J., & STEELE,
T.W. (1977) The group extraction of noble metals with s-(1-Decyl)-
N,N'-diphenylisothiouronium bromide and their determination in the
organic extract by atomic-absorption spectrometry. Anal. Chim. Acta,
94: 257-268.
JUNG, H.J. & BECKER, E.R. (1987) Emission control for gas turbines.
Platinum-rhodium catalysts for carbon monoxide and hydrocarbon
removal. Platinum Met. Rev., 31: 162-170.
KAHN, N. & VAN LOON, J.V. (1978) Direct atomic absorption
spectrophotometric analysis of anion complexes of platinum and gold
after their concentration and separation from aqueous solutions by
anion exchange chromatography. Anal. Lett., 12: 991-1000.
KANEMATSU, N., HARA, M., & KADA, T. (1980) Rec assay and
mutagenicity studies on metal compounds. Mutat. Res., 77: 109-116.
KARASEK, S.R. & KARASEK, M. (1911) The use of platinum paper. Report
of (Illinois) Commission on Occupational Diseases to His Excellency
Governor Charles S. Deneen, January 1911, Chicago, Warner Printing
Company, p. 97
KENAWY, I.M., KHALIFA, M.E., & EL-DEFRAWY, M.M. (1987)
Preconcentration and determination (AAS) of trace Ag(I), Au(III),
Pd(II) and Pt(IV) using a cellulose ion-exchanger (Hyphan).
Analusis, 15: 314-317.
KESSERÜ, E. & LEON, F., (1974) Effect of different solid metals and
metallic pairs on human sperm motility. Int. J. Fertil., 19: 81-84.
KHAN, A., HILL, J.M., GRATER, W., LOEB, E., MACLELLAN, A., & HILL,
N. (1975) Atopic hypersensitivity to cis-dichlorodiammineplatinum
(II) and other platinum complexes. Cancer Res., 35: 2766-2770.
KNAPP, G. (1984) [Routes to powerful methods of elemental trace
analysis in environmental samples.] Fresenius Z. anal. Chem., 317:
213-219 (in German).
KNAPP, G. (1985) Sample preparation techniques - An important part
in trace element analysis for environmental research and monitoring.
Int. J. environ. anal. Chem., 22: 71-83.
KOBERSTEIN, E. (1984) [Catalysts for purifying automotive exhaust
gas.] Chem. Zeit, 18: 37-45 (in German).
KOIBA, R.J. & SLEIGHT, S.D. (1971) Acute toxicologic and pathologic
effects of cis-diamminedichloroplatinum (NSC-119875) in the male
rat. Cancer Chemother. Rep., 55(Part 1): 1-8.
KOLPAKOVA, A.F. & KOLPAKOV, F.I. (1983) [Comparative study of
sensitizing characteristics of the platinum group metals.] Gig. Tr.
prof. Zabol., 7: 22-24 (in Russian).
KÖNIG, H.P. & HERTEL, R.F. (1990) [Search and identification of
gaseous noble metal emissions from catalysts. Investigations into
the hazardous potential of catalyst-born nobel metal emissions],
Hanover, Fraunhofer Institute of Toxicology and Aerosol Research,
108 pp (Unpublished final research report to the Federal Ministry of
Research and Technology) (in German).
KÖNIG, H.P., KOCK, H., & HERTEL, R.F. (1989) [Analytical
determination of platinum with regard to the car catalyst issue.]
In: 5th Colloquium on Trace Analysis by Atomic Absorption
Spectrometry, Konstanz, 3-7 April, 1989], Überlingen, Perkin-Elmer
GmbH, pp. 647-656 (in German).
KÖNIG, H.P., HERTEL, F.R., KOCH, W., & ROSNER, G. (in press)
Determination of platinum emissions from a three-way catalyst
equipped gasoline engine. Atmospheric environment.
KRAFT, L.A., POLIDORO, J.P., CULVER, R.M., & HAHN, D.W. (1978)
Intravas device studies in rabbits: II. Effect on sperm output,
fertility and histology of the reproductive tract. Contraception,
18: 239-251.
KRAL, H. & PETER, K. (1977) [Method for the production of
platinum(II)-nitrate and its use for platinum catalysts for the
catalytic after-burning of automobile exhaust gas], Münich, German
Patent Office (Patent No. 2233677) (in German).
KRITSOTAKIS, K. & TOBSCHALL, H.J. (1985) [Trace determination of
platinum by differential-pulse anodic stripping voltammetry (DPASV)
using the glassy carbon electrode.] Fresenius Z. anal. Chem., 320:
156-158 (in German).
LANGE, R.C., SPENCER, R.P., & HARDER, H.C. (1972) Synthesis and
distribution of a radiolabeled antitumor agent: cis-
diamminedichloroplatinum (II). J. nucl. Med., 13: 328-330.
LAZNICKOVA, A., SEMECKY, V., LAZNICEK, M., ZUBER, V., KOKSAL, J., &
KVETINA, J. (1989) Effect of oxoplatinum and CBDCA on renal
functions in rats. Neoplasma, 36: 161-169.
LECOINTE, P., MACQUET, J.P., BUTOUR, J.L., & PAOLETTI, C. (1977)
Relative efficiencies of a series of square-planar platinum(II)
compounds on Salmonella mutagenesis. Mutat. Res., 48: 139-144.
LEE, D.S. (1983) Palladium and nickel in north-east Pacific waters.
Nature(Lond.), 305: 47-48.
LE HOUILLIER, R. & DE BLOIS, C. (1986) Alkyl cyanide medium for the
determination of precious metals by atomic absorption spectrometry.
Analyst, 111: 291-294.
LELIEVELD, P., VAN DER VIJGH, J.F., VELDHUIZEN, R.W., VAN VELZEN,
D., VAN PUTTEN, L.M., ATASSI, G., & DANGUY, A. (1984) Preclinical
studies on toxicity, antitumour activity and pharmacokinetics of
cisplatin and three recently developed derivatives. Eur. J. Cancer
clin. Oncol., 20: 1087-1104.
LEROY, A.F., WEHLING, M.L., SPONSELLER, H.L., FRIAUF, W.S., SOLOMON,
R.E., & DEDRICK, R.L., (1977) Analysis of platinum biological
materials by flameless atomic absorption spectrophotometry. Biochem.
Med., 18: 184-191.
LIFE SCIENCE RESEARCH (1980a) Pt(NH3)4Cl2; assessment of its
mutagenic potential in histidine auxotrophs of Salmonella
typhimurium, Eye, Suffolk, Life Science Research, (Unpublished
report No. 80/JOM001/005).
LIFE SCIENCE RESEARCH (1980b) Pt(NH3)4Cl2; assessment of
clastogenic action on bone marrow erythrocytes in the micronucleus
test, Eye, Suffolk, Life Science Research (Unpublished report No.
80/JOM004/182).
LIFE SCIENCE RESEARCH (1980c) Pt(NH3)4Cl2; assessment of its
mutagenic potential in Drosophila melanogaster, using the sex-
linked recessive lethal test, Eye, Suffolk, Life Science Research
(Unpublished report No. 80/JOM002/260).
LIFE SCIENCE RESEARCH (1981a) Potassium chloroplatinate: assessment
of clastogenic action on bone marrow erythrocytes in the
micronucleous test, Eye, Suffolk, Life Science Research (Unpublished
report No. 81/JOM013/241).
LIFE SCIENCE RESEARCH (1981b) TPC: assessment of its action on bone
marrow cell chromosomes following sub-acute oral administration in
the Chinese hamster, Eye, Suffolk, Life Science Research
(Unpublished report No. 80/JOM008/497).
LIFE SCIENCE RESEARCH (1981c) Potassium chloroplatinate(II):
assessment of its mutagenic potential in Drosophila melanogaster,
using the sex-linked recessive lethal test, Eye, Suffolk, Life
Science Research (Unpublished report No. 81/JOM012/474).
LIFE SCIENCE RESEARCH (1982) Potassium chloroplatinate:
investigation of effects on bone marrow chromosomes of the Chinese
hamster after sub-acute oral administration, Eye, Suffolk, Life
Science Research (Unpublished report).
LINNETT, P.J. (1987) Platinum salt sensitivity. A review of the
health aspects of platinum refining in South Africa. J. Mine Med.
Off. Assoc. S. Afr., 63: 24-28.
LIPPERT & BECK (1983) [Platinum complexes in cancer therapy.] Chem.
Zeit, 6: 190-199 (in German).
LIPPMAN, A.J., HELSON, C., HELSON, L., & KRAKOFF, I.H. (1973)
Clinical trials of cis-diamminedichloroplatinum (NSC 119875). Cancer
Chemother. Rep., 57: 191-200.
LITTERST, C.L. (1981) Alterations in the toxicity of cis-
dichlorodiammineplatinum-II and in tissue localization of platinum
as a function of NaCl concentration in the vehicle of
administration. Toxicol. appl. Pharmacol., 61: 99-108.
LITTERST, C.L., TORRES, I.J., & GUARINO, A.M. (1976a) Plasma levels
& organ distribution of platinum in the rat, dog & dogfish shark
following single intravenous administration of cis-
dichlorodiammineplatinum(II). J. clin. Hematol. Oncol., 7: 169-179.
LITTERST, C.L., GRAM, T.E., DEDRICK, R.L., LEROY, A.F., & GUARINO,
A.M. (1976b) Distribution and disposition of platinum following
intravenous administration of cis-diamminedichloroplatinum(II) (NSC
119875) to dogs. Cancer Res., 36: 2340-2344.
LITTERST, C.L., LEROY, A.F., & GUARINO, A.M. (1979) Disposition and
distribution of platinum following parenteral administration of cis-
dichlorodiammineplatinum-II to animals. Cancer Treat. Rep., 63:
1485-1492.
LO, F.B., ARAI, D.K., & NAZAR, M.A. (1987) Determination of platinum
in urine by inductively coupled plasma atomic emission spectrometry.
J. anal. Toxicol., 11: 242-246.
LOEBENSTEIN, J.R. (1982) Platinum-group metals. In: Minerals
yearbook, centennial edition 1981. Vol. I. Metals and minerals,
Washington, DC, US Department of the Interior, Bureau of Mines, pp.
667-678.
LOEBENSTEIN, J.R. (1988) Platinum-group metals. In: Minerals
yearbook 1987. Vol. I. Metals and minerals, Washington, DC, US
Department of the Interior, Bureau of Mines, pp. 689-700.
LOWN, B.A., MORGANTI, J.B., STINEMAN, C.H., D'AGOSTINO, R.B., &
MASSARO, E.J. (1980) Tissue organ distribution and behavior effects
of platinum following acute and repeated exposure of the mouse to
platinum sulfate. Environ. Health Perspect., 34: 203-212.
MCGAHAN, M.C. & TYCZKOWSKA, K. (1987) The determination of platinum
in biological materials by electrothermal atomic absorption
spectroscopy. Spectrochim. Acta, 42B: 665-668.
MAESSEN, F.J.M., KREUNING, G., & BALKE, J. (1986) Experimental
control of the solvent load of inductively coupled argon plasmas and
effects of the chloroform plasma load on their analytical
performance. Spectrochim. Acta, 41B: 3-25.
MALANCHUK, M., BARKELEY, N., CONTNER, G., RICHARDS, M., SLATER, R.,
BURKART, J., & YANG, Y. (1974) Exhaust emissions from catalyst-
equipped engines. In: Environmental Toxicology Research Laboratory,
NERC, ed. Interim report: 1. Studies on toxicology of catalytic
trace metal components. 2. Toxicology of automotive emissions with
and without catalytic converters, Cincinnati, Ohio, Environmental
Toxicology Research Laboratory, NERC, pp. 91-98.
MARCZENKO, Z. & KUS, S. (1987) Spectrophotometric determination of
traces of platinum in palladium with dithizone after matrix
precipitation as a compound with ammonia and iodide. Anal. Chim.
Acta, 196: 317-322.
MARONE, C., OLSINA, R., & SALAS, O. (1981) Spectrometric
determination of platinum in a spent reforming catalyst. Cron.
chim., 66: 22-28.
MARSH, K.C., STERNSON, L.A., & REPTA, A.J. (1984) Post-column
reaction detector for platinum(II) antineoplastic agents. Anal.
Chem., 56: 491-497.
MASON, B. (1966) Principles of geochemistry, 3rd ed., New York, John
Wiley & Sons, 310 pp.
MASSARO, E.J., LOWN, B.A., MORGANTI, J.B., STINEMAN, C.H., &
D'AGOSTINO, R.B. (1981) Sensitive biochemical and behavioral
indicators of trace substance exposure: Part II. Platinum, Research
Triangle Park, US Environmental Protection Agency, 3 pp
(EPA-600/S1-81-015).
MASSMANN, W. & OPITZ, H. (1954) [On platinum allergy.] Zentralbl.
Arbeitsmed. Arbeitsschutz, 4: 1-4 (in German).
MATUSIEWICZ, H. & BARNES, R.M. (1983) Determination of metal
chemotherapeutic agents (Al, Au, Li, and Pt) in human body fluids
using electrothermal vaporization with inductively coupled plasma
atomic emission spectroscopy. In: Chemical toxicology and clinical
chemistry of metals, Oxford, United Kingdom, International Union of
Pure and Applied Chemistry, pp. 49-52.
MERGET, R., SCHULTZE-WERNINGHAUS, G., MUTHORST, T., FRIEDRICH, W., &
MEIER-SYDOW, J. (1988) Asthma due to the complex salts of platinum -
a cross-sectional survey of workers in a platinum refinery. Clin.
Allergy, 18: 569-580.
MERGET, R., ZACHGO, W., SCHULTZE-WERNINGHAUS, G., BERGMANN, E.-M.,
BOLM-AUDORFF, U., FRIEDRICH, W., BURY, A.H., & MEIER-SYDOW, J.
(1990) [Quantitative skin test and inhalative provocation test in
asthma associated with platinum compounds.] Pneumologie, 44: 226-228
(in German).
MERRIN, C.A. (1976) A new method to prevent toxicity with high doses
of cis- diammineplatinum (therapeutic efficacy in previously treated
widespread and recurrent testicular tumors). In: Proceedings of the
12th Annual Meeting of the American Association for Cancer
Research/American Society of Clinical Oncology, Toronto, 4-5 May,
1976, Chicago, Illinois, American Society of Clinical Oncology, p.
243 (ASCO Abstracts, No. C-26).
MICHAEL, L.W., FINELLI, V.N., ELIA, V.J., PETERING, H.G., LEE, S.D.,
& CAMPBELL, K.I. (1976) Interaction of metal binding agents from
combustion products with trace metals. In: Proceedings of the
Platinum Research Review Conference, Quail Roost Conference Center,
Rougemont, North Carolina, 3-5 December 1975, Research Triangle
Park, North Carolina, US Environmental Protection Agency, pp.
37A-53A.
MILLARD, H.T. (1987) Neutron activation determination of iridium,
gold, platinum, and silver in geologic samples. J. radioanal. nucl.
Chem., 113: 125-132.
MOJSKI, M. & KALINOWSKI, K. (1980) Extractive separation of platinum
from macroamounts of palladium using triphenylphosphine oxide and
its successive spectrophotometric determination by the stannous
chloride method. Microchem. J., 25: 507-513.
MOLLMAN, J.E., HOGAN, W.M., GLOVER, D.J., & MCCLUSKEY, L.F. (1988)
Unusual presentation of cis-platinum neuropathy. Neurology, 38:
488-490.
MOORE, W., Jr, HYSELL, D., CROCKER, W., & STARA, J. (1975a)
Biological fate of a single adminstration of 191Pt in rats
following different routes of exposure. Environ. Res., 9: 152-158.
MOORE, W., Jr, HYSELL, D., HALL, L., CAMPBELL, K., & STARA, J.
(1975b) Preliminary studies on the toxicity and metabolism of
palladium and platinum. Environ. Health Perspect., 10: 63-71.
MOORE, W., Jr, MALANCHUK, M., CROCKER, W., HYSELL, D., COHEN, A., &
STARA, J.F. (1975c) Whole body retention in rats of different
191Pt compounds following inhalation exposure. Environ. Health
Perspect., 12: 35-39.
MUELLER, B.J. & LOVETT, R.J. (1987) Salt-induced phase separation
for the determination of metals as their diethyldithiocarbamate
complexes by high-performance liquid chromatography. Anal. Chem.,
59: 1405-1409.
MURDOCH, R.D. & PEPYS, J. (1984) Immunological responses to complex
salts of platinum. I. Specific IgE antibody production in the rat.
Clin. exp. Immunol., 57: 107-114.
MURDOCH, R.D. & PEPYS, J. (1985) Cross reactivity studies with
platinum group metal salts in platinum-sensitised rats. Int. Arch.
Allergy appl. Immunol., 77: 456-458.
MURDOCH, R.D. & PEPYS, J. (1987) Platinum group metal sensitivity:
reactivity to platinum group metal salts in platinum halide salt-
sensitive workers. Ann. Allergy, 59: 464-469.
MURDOCH, R.D., PEPYS, J., & HUGHES, E.G. (1986) IgE antibody
responses to platinum group metals: a large-scale refinery survey.
Br. J. ind. Med., 43: 37-43.
MYASOEDOVA, G.V., ANTOKOL'SKAYA, I.I., & SAVVIN, S.B. (1985) New
chelating sorbents for noble metals. Talanta, 32: 1105-1112.
NAS (1977) Platinum-group metals, Washington, DC, National Academy
of Science, 232 pp.
NEUMÜLLER, O.-A. (1987) [Römpps lexicon on chemistry], Stuttgart, W.
Keller & Co., p. 3256 (in German).
NEWMAN-TAYLOR, A.J. (1981) Follow-up study of a group of platinum
refinery workers, London, Brompton Hospital (Unpublished report).
NYGREN, O., VAUGHAN, G.T., FLORENCE, T.M., MORRISON, G.M., WARNER,
I., & DALE, L.S. (1990) Determination of platinum in blood by
adsorptive voltammetry. Anal. Chem., 62(15): 1637-1640.
PALLAS, J.E., Jr & JONES, J.B., Jr (1978) Platinum uptake by
horticultural crops. Plant Soil, 50: 207-212.
PARISH, W.E. (1970) Short-term anaphylactic IgG antibodies in human
sera. Lancet, 2: 591-592.
PARROT, J.L., HEBERT, R., SAINDELLE, A., & RUFF, F. (1969) Platinum
and platinosis. Allergy and histamine release due to some platinum
salts. Arch. environ. Health, 19: 685-691.
PEPYS, J. (1983) Allergy of the respiratory tract to low molecular
weight chemical agents. In: Born, G.V.R., Farah, A., Herken, H., &
Welch, A.D., ed. Handbook of experimental pharmacology, Berlin,
Springer-Verlag, pp. 163- 185.
PEPYS, J., PICKERING, C.A.C., & HUGHES, E.G. (1972) Asthma due to
inhaled chemical agents - complex salts of platinum. Clin. Allergy,
2: 391-396.
PEPYS, J., PARISH, W.E., CROMWELL, O., & HUGHES, E.G. (1979) Passive
transfer in man and the monkey of type I allergy due to heat labile
and heat stable antibody to complex salts of platinum. Clin.
Allergy, 9: 99-108.
PERA, M.F., Jr & HARDER, H.C. (1977) Analysis of platinum in
biological material by flameless atomic absorption spectrometry.
Clin. Chem., 23: 1245- 1249.
PICKERING, C.A.C. (1972) Inhalation tests with chemical allergens:
complex salts of platinum. Proc. R. Soc. Med., 65: 272-274.
PINTO, A.L. & LIPPARD, S.J. (1985) Binding of the antitumour drug
cis-diamminedichloroplatinum(II) (cisplatin) to DNA. Biochim.
Biophys. Acta, 780: 167-180.
POTTER, N.M. & LANGE, W.H. (1981) Determination of noble metals and
their distribution in automotive catalyst materials. Am. Lab., 1:
81-91.
PRIESNER, D., STERNSON, L.A., & REPTA, A.J. (1981) Analysis of total
platinum in tissue samples by flameless atomic absorption
spectrophotometry. Elimination of the need for sample digestion.
Anal. Lett., 14: 1255-1268.
PURI, B.K., WASEY, A., KATYAL, M., & SATAKE, M. (1986)
Spectrophotometric determination of platinum after extraction of its
phenanthraquinone monoximate into molten naphtalene. Analyst, 111:
743-745.
RENNER, H. (1979)a [Platinum metals and alloys.] In: Bartholomé,
E., Biekert, E., Hellmann, H., & Ley, H., ed. [Ullmann's
encyclopedia of technical chemistry], 4th ed., Weinheim, Verlag
Chemie, Vol. 18, pp. 697-705 (in German).
RENNER, H. (1984)b [Platinum metals.] In: Merian, E., Geldmacher-
von-Mallinckrodt, M., Machata, G., Nürnberg, H.W., Schlipköter,
H.W., & Stumm, W., ed. [Metals in the environment. Distribution,
analysis, and biological relevance], Weinheim, Verlag Chemie, pp.
499-510 (in German).
RICKEY, F.A., SIMMS, P.C., & MUELLER, K.A. (1979) PIXE analysis of
water with detection limits in the ppb range. IEEE Trans. nucl.
Sci., NS-26(1): 1347-1351.
ROBERT, R.V.D., VAN WYK, E., & PALMER, R. (1971) Concentration of
the noble metals by a fire-assay technique using sulphide as the
collector, Johannesburg, South Africa, National Institute for
Metallurgy (Report No. 1371).
ROBERTS, A.E. (1951) Platinosis. A five-year study of the effects of
soluble platinum salts on employees in a platinum laboratory and
refinery. Arch. ind. Hyg. occup. Med., 4: 549-559.
ROCKLIN, R.D. (1984) Determination of gold, palladium, and platinum
at the parts-per-billion level by ion chromatography. Anal. Chem.,
56: 1959-1962.
ROSENBERG, B. (1975) Possible mechanisms for the antitumor activity
of platinum coordination complexes. Cancer Chemother. Rep., 59(Part
1): 589-598.
ROSENBERG, B. (1980) Clinical aspects of platinum anticancer drugs.
In: Metal ions in biological systems, New York, Basel, Marcel
Dekker, Inc., Vol. 12, pp. 127-196.
ROSENBERG, B. (1985) Fundamental studies with cisplatin. Cancer, 55:
2303-2316.
a English edition in preparation:
RENNER, H. (in press) Platinum metals, compounds and alloys.
In: Ullmanns Encyclopaedia of Industrial Chemistry, 5th ed.,
Weinheim, VCH Verlagsgesellschaft.
b English edition in preparation:
RENNER, H. & SCHMUCKLER, G. (in press) Platinum-group metals.
In: Merian, E., ed. Metals and their compounds in the
environment, Weinheim, VCH Verlagsgesellschaft.
ROSENBERG, B., VAN CAMP, L., & KRIGAS, T. (1965) Inhibition of cell
division in Escherichia coli by electrolysis products from a
platinum electrode. Nature (Lond.), 205: 698-699.
ROSENBERG, B., RENSHAW, E., VAN CAMP, L., HARTWICK, J., & DROBNIK,
J. (1967) Platinum-induced filamentous growth in Escherichia coli.
J. Bacteriol., 93: 716-721.
ROSHCHIN, A.V., VESELOV, V.G., & PANOVA, A.I. (1979) [Toxicology of
platinum and platinum-group metals.] Gig. Tr. prof. Zabol., 9: 4-9
(in Russian).
ROSHCHIN, A.V., VESELOV, V.G., & PANOVA, A.I. (1984) Industrial
toxicology of metals of the platinum group. J. Hyg. Epidemiol.
Microbiol. Immunol., 28: 17-24.
ROSNER, G. & HERTEL, R.F. (1986) [Health risk assessment of platinum
emissions from automotive exhaust gas catalysts.] Staub-Reinhalt.
Luft, 46: 281-285 (in German).
ROSNER, G. & MERGET, R. (1990) Allergenic potential of platinum
compounds. In: Dayan, A.D., Hertel, R.F., Heseltine, E., Kazantzis,
G., Smith, E.M., & Van der Venne, M.T., ed. Immunotoxicity of metals
and immunotoxicology, New York, London, Plenum Press, pp. 93-102.
ROSNER, G., KÖNIG, H.P., KOCH, W., KOCK, H., HERTEL, R.F., & WINDT,
H. (1991) [Engine test stand experiments to assess platinum uptake
by plants.] Angew. Bot., 65: 127-132 (in German).
RUFF, F., DI MATTEO, G., DUPUIS, J.P., HEBERT, R., & PARROT, J.-L.
(1979) Réactions bronchopulmonaires et asthme dus au platine:
incidences chez les ouvriers du platine en région parisienne. Rev.
fr. Mal. Respir., 7: 206-208.
SAFIRSTEIN, R., DAYE, M., & GUTTENPLAN, J.B. (1983) Mutagenic
activity and identification of excreted platinum in human and rat
urine and rat plasma after administration of cisplatin. Cancer
Lett., 18: 329-338.
SAINDELLE, A. & RUFF, F. (1969) Histamine release by sodium
chloroplatinate. Br. J. Pharmacol., 35: 313-321.
SANDHU (1979) Evaluation of the mutagenic potential of platinum
compounds, Durham, North Carolina Central University, 36 pp
(EPA-600/1-79-033).
SCHACTER, L. & CARTER, S.K. (1986) The use of platinum to treat
cancer. In: Precious metals: Proceedings of the 9th International
Precious Metals Institute Conference, Lake Tahoe, Nevada, 9-12 June,
1986, Allentown, Pennsylvania, International Precious Metals
Institute, pp. 359-367.
SCHLEMMER, G. & WELZ, B. (1986) Influence of the tube surface on
atomization behavior in a graphite tube furnace. Fresenius Z. anal.
Chem., 323: 703-709.
SCHLÖGL, R., INDLEKOFER, G., & OELHAFEN, P. (1987) [Micro particle
emission from combustion engines with exhaust gas purification - X-
ray photoelectron spectroscopy in environmental analysis.] Angew.
Chem., 99: 312-322 (in German).
SCHULTZE-WERNINGHAUS, G., ROESCH, A., WILHELMS, O.-H., GONSIOR, E.,
& MEIER-SYDOW, J. (1978) [Bronchial asthma due to occupational
allergy of immediate type (I) to platinum salts.] Dtsch. med.
Wochenschr., 23: 972-975 (in German).
SCHULTZE-WERNINGHAUS, G., MERGET, R., ZACHGO, W., MUTHORST, T.,
MAHLESA, D., LISSON, R., & BOLM-AUDORFF, U. (1989) [Platinum salts
as occupational allergens - a review.] Allergologie, 12: 152-157 (in
German).
SCHUTYSER, P., GOVAERTS, A., DAMS, R., & HOSTE, J. (1977) Neutron
activation analysis of platinum metals in airborne particulate
matter. J. radioanal. Chem., 37: 651-660.
SHEARD, C. (1955) Contact dermatitis from platinum and related
metals. Arch. Dermatol. Syphilol., 71: 357-360.
SHIMAZU, M. & ROSENBERG, B. (1973) A similar action to UV-
irradiation and a preferential inhibition of DNA synthesis in E.
coli by antitumor platinum compounds. J. Antibiot., 26: 243-245.
SIGHINOLFI, G.P., GORGONI, C., & MOHAMED, A.H. (1984) Comprehensive
analysis of precious metals in some geological standards by
flameless a.a. spectroscopy. Geostand. Newsl., 8: 25-29.
SIGSBY, J. (1976) Measurement of platinum from catalyst-equipped
vehicles, combustion and attrition products. In: Proceedings of the
Platinum Research Review Conference, Quail Roost Conference Center,
Rougemont, North Carolina, 3-5 December 1975, Research Triangle
Park, North Carolina, US Environmental Protection Agency, pp.
25A-31A.
SINGH, N. & GARG, A.K. (1987) Spectrophotometric determination of
platinum(IV) with potassium butyl xanthate. Analyst, 112: 693-695.
SKINNER, P.E. (1989) Improvements in platinum plating. Platinum Met.
Rev., 33(3): 102-105.
SKOGERBOE, R.K., HANAGAN, W.A., & TAYLOR, H.E. (1985) Concentration
of trace elements in water samples by reductive precipitation. Anal.
Chem., 57: 2815-2818.
SMITH, B.L., HANNA, M.L., & TAYLOR, R.T. (1984) Induced resistance
to platinum in Chinese hamster ovary cells. J. environ. Sci. Health,
A19: 267-298.
SPERNER, F. & HOHMANN, W. (1976) Rhodium-platinum gauzes for ammonia
oxidation. Platinum Met. Rev., 20: 12-20.
STERNSON, L.A., REPTA, A.J., SHIH, H., HIMMELSTEIN, K.J., & PATTON,
T.F. (1984) Disposition of cisplatin vs total platinum in animals
and man. Dev. Oncol., 17: 126-137.
STOCKMAN, H.W. (1983) Neutron activation determination of noble
metals in rock: a rapid radiochemical separation based on tellurium
coprecipitation. J. radioanal. Chem., 78: 307-317.
STOKINGER, H.E. (1981) Platinum-group metals - platinum, Pt,
palladium, Pd, iridium, Ir, osmium, Os, rhodium, Rh, ruthenium, Ru.
In: Clayton, G.D. & Clayton, F.E., ed. Patty's industrial hygiene
and toxicology, New York, John Wiley & Sons, pp. 1853-1871.
SURAIKINA, T.I., ZAKHAROVA, I.A., MASHKOVSKII, Y., & FONSHTEIN, L.M.
(1979) Study of the mutagenic action of platinum and palladium
compounds on bacteria. Cytol. Genet. (USSR), 13: 50-54 (Translation
from: Tsitol. i. Genet. 13: 486-491).
SWIERENGA, S.H., GILMAN, J.P., & MCLEAN, J.R. (1987) Cancer risk
from inorganics. Cancer Metastasis Rev., 6: 113-154.
TAUBLER, J.H. (1977) Allergic response to platinum and palladium
complexes - determination of no-effect level, Research Triangle
Park, North Carolina, US Environmental Protection Agency, Office of
Research and Development, Health Effects Research Laboratories, pp.
1-81 (EPA/600/1-77-039).
TAYLOR, R.T. (1976) Comparative methylation chemistry of platinum,
palladium, lead and manganese, Research Triangle Park, North
Carolina, US Environmental Protection Agency, Office of Research and
Development, Health Effects Research Laboratories, 25 pp
(EPA/600/1-76-016).
TAYLOR, R.T. & HANNA, M.L. (1977) Methylation of platinum by methyl
cobalamin. In: Drucker, H. & Wildung, R.E., ed. Biological
implications of metals in the environment. Proceedings of the 15th
Annual Hanford Life Science Symposium, Richland, Washington, 29
September - 1 October, 1975, Springfield, Virginia, US Department of
Commerce, National Technical Information Service, pp. 36-51 (Energy
Research and Development Administration Symposium Series, Vol. 42).
TAYLOR, R.T., HAPPE, J.A., HANNA, M., & WU, R. (1979) Platinum
tetrachloride: mutagenicity and methylation with methylcobalamin. J.
environ. Sci. Health, A14: 87-109.
THEOPOLD, H.-M., ZOLLNER, M., SCHORN, K., SPAHMANN, J., & SCHERER,
H. (1981) [Tissue reactions with platinum-iridium electrodes.]
Laryng.-Rhinol., 60: 534-537 (in German).
THOMPSON, B.C., RAYBURN, L.A., & HOLADAY, S.R. (1981) Detection of
platinum in brain tissue by PIXE. IEEE Trans. nucl. Sci., NS-28:
1396-1397.
THOMPSON, J.J. & HOUK, R.S. (1986) Inductively coupled plasma mass
spectrometric determination for multielement flow injection analysis
and elemental speciation by reversed-phase liquid chromatography.
Anal. Chem., 58: 2541-2548.
TILLERY, J.B. & JOHNSON, D.E. (1975) Determination of platinum,
palladium, and lead in biological samples by atomic absorption
spectrophotometry. Environ. Health Perspect., 12: 19-26.
TJIOE, P.S., VOLKERS, K.J., KROON, J.J., DE GOEIJ, J.J.M., & THE,
S.K. (1984) Determination of gold and platinum traces in biological
materials as a part of a multielement radiochemical activation
analysis system. Int. J. environ. anal. Chem., 17: 13-24.
TOBE, M.L. & KHOKHAR, A.R. (1977) Structure, activity, reactivity
and solubility relationships of platinum diammine complexes. J.
clin. Hematol. Oncol., 7: 114-137.
TOBERT, A.J. & DAVIES, D.R. (1977) Effect of copper and platinum
intrauterine devices on endometrial morphology and implantation in
the rabbit. J. Reprod. Fertil., 50: 53-59.
TÖLG, G. & ALT, F. (1990) [Contribution to an improvement in extreme
trace analysis of metallic platinum in biotic and environmental
material.] In: [Abstract prepared for the Third Conference of the
BMFT Study Group on Precious Metal Emissions, 15 October, 1990],
Dortmund, Institute of Spectrochemistry, 7 pp (Unpublished report)
(in German).
TSO, T.C., SOROKIN, T.P., & ENGELHAUPT, M.E. (1973) Effects of some
rare elements on nicotine content of the tobacco plant. Plant
Physiol., 51: 805-806.
VALENTE, I.M., MINSKI, M.J., & PETERSON, P.J. (1982) Neutron
activation analysis of noble metals in vegetation. J. radioanal.
Chem., 71: 115-127.
VAN DEN BERG, C.M.G. & JACINTO, G.S. (1988) The determination of
platinum in sea water by adsorptive cathodic stripping voltammetry.
Anal. Chim. Acta, 211: 129-139.
VAN DER VIJGH, W.J.F., ELFERINK, F., & POSTMA, G.J. (1984)
Determination of ethylenediamineplatinum(II) malonate in infusion
fluids, human plasma and urine by high-performance liquid
chromatography. J. Chromatogr., 310: 335-342.
VENABLES, K.M., DALLY, M.B., NUNN, A.J., STEVENS, J.F., STEPHENS,
R., FARRER, N., HUNTER, J.V., STEWART, M., HUGHES, E.G., & NEWMAN-
TYLOR, A.J. (1989) Smoking and occupational allergy in workers in a
platinum refinery. Br. Med. J., 299: 939-942.
VENITT, S., CROFTON-SLEIGH, C., HUNT, J., SPEECHLEY, V., & BRIGGS,
K. (1984) Monitoring exposure of nursing and pharmacy personnel to
cytotoxic drugs: urinary mutation assays and urinary platinum as
markers of absorption. Lancet, January 14: 74-77.
VOGL, S.E., ZARAVINOS, T., & KAPLAN, B.H. (1980) Toxicity of cis-
diamminedichloroplatinum II given in a two-hour outpatient regimen
of diuresis and hydration. Cancer, 45: 11-15.
VON BOHLEN, A., ELLER, R., KLOCKENKÄMPER, R., & TÖLG, G. (1987)
Microanalysis of solid samples by total-reflection X-ray
fluorescence spectrometry. Anal. Chem., 59: 2551-2555.
VON HOFF, D.D., SCHILSKY, R., REICHERT, C.M., REDDICK, R.L.,
ROZENCWEIG, M., YOUNG, R.C., & MUGGIA, F.M. (1979) Toxic effects of
cis- dichlorodiammine-platinum(II) in man. Cancer Treat. Rep., 63:
1527-1531.
VRANA, O., KLEINWÄCHTER, V., & BRABEC, V. (1983) Determination of
platinum in urine by differential pulse polarography. Talanta, 30:
288-290.
WANG, T.C. & TAN, C.K. (1987) Photodegradation of trichloroethylene
in microheterogeneous aqueous systems. Environ. Int., 13: 359-362.
WARD, J.M. & FAUVIE, K.A. (1976) The nephrotoxic effects of cis-
diamminedichloroplatinum(II) (NSC-II 9875) in male F344 rats.
Toxicol. appl. Pharmacol., 38: 535-547.
WARD, J.M., YOUNG, D.M., FAUVIE, K.A., WOLPERT, M.K., DAVIS, R., &
GUARINO, A.M. (1976) Comparative nephrotoxity of platinum cancer
chemotherapeutic agents. Cancer Treat. Rep., 60: 1675-1678.
WATERS, M.D., VAUGHAN, T.O., ABERNETHY, D.J., GARLAND, H.R., COX,
C.C., & COFFIN, D.L. (1975) Toxicity of platinum (IV) salts for
cells of pulmonary origin. Environ. Health Perspect., 12: 45-56.
WEAST, R.C. & ASTLE, M.J. (1981) CRC handbook of chemistry and
physics, 62nd ed. Boca Raton, Florida, CRC Press, pp. B-128-B-129.
WELZ, B. & SCHLEMMER, G. (1987) Glassy carbon tubes for
electrothermal atomic absorption spectrometry. Fresenius Z. anal.
Chem., 327: 246-252.
WEMYSS, R.B., & SCOTT, R.H. (1978) Simultaneous determination of
platinum group metals and gold in ores and related plant materials
by inductively-coupled plasma-optical emission spectrometry. Anal.
Chem., 50: 1694-1697.
WHITEHOUSE, M.W. & GARRETT, I.R. (1984) Heavy metal (Au, Pt)
nephropathy: studies in normal and inflamed rats. In: Rainsford,
K.D. & Velo, G.P., ed. Advances in inflammation research, New York,
Raven Press, Vol. 6, pp 291-294.
WIELE, H. & KUCHENBECKER, H. (1974) [Spectrophotometric
determination of rhenium in the presence of platinum in bimetallic
catalysts.] Erdöl, Kohle, Erdgas, Petrochem., 27: 90-92 (in German).
WINDHOLZ, M. (1976) The Merck index, 9th ed., Rahway, New Jersey,
Merck & Co., pp. 75, 977-991, 1114.
WOLFE, G.W. (1979) PIXE analysis of atmospheric gases. IEEE Trans.
nucl. Sci., NS-26: 1389-1391.
WOOD, J.M., FANCHIANG, Y.-T., & RIDLEY, W.P. (1978) The biochemistry
of toxic elements. Q. Rev. Biophys., 11: 467-479.
WÜRFELS, M., JACKWERTH, E., & STOEPPLER, M. (1987) [On the problem
of disturbances of inverse voltammetric trace analysis after
pressure decomposition of biological samples.] Fresenius Z. anal.
Chem., 329: 459-461 (in German).
ZACHGO, W., MERGET, R., & SCHULTZE-WERNINGHAUS, G. (1985) [Proof of
specific IgE against low molecular weight substances (platinum
salts).] Atemweg. Lungenkr., 11: 267-268 (in German).
ZOLOTOV, YU.A., PETRUKHIN, O.M., MALOFEEVA, G.I., MARCHEVA, E.V.,
SHIRYAEVA, O.A., SHESTAKOV, V.A., MISKAR'YANTS, V.G., NEFEDOV, V.I.,
MURINOV, YU.I., & NIKITIN, YU.E. (1983) Determinations of platinum
metals by X-ray fluorescence, atomic emission and atomic absorption
spectrometry after preconcentration with a polymeric thioether.
Anal. Chim. Acta, 148: 135-157.
RESUME
1. Identité, propriétés physiques et chimiques et méthodes
d'analyse
Le platine (Pt) est un métal noble, malléable et ductile de
couleur blanc argent dont le numéro atomique est 78 et le poids
atomique 195,9. On le trouve principalement à l'état naturel sous la
forme des isotopes 194Pt (32,9 %), 195Pt (33,8 %) et 196Pt
(25,3 %). Son état d'oxydation maximum est de +6, les états +2 et +4
étant les plus stables.
Le métal ne se corrode pas à l'air quelle que soit la
température mais il peut être attaqué par les halogènes, les
cyanures, le soufre et ses composés en fusion, les métaux lourds et
les hydroxydes alcalins. L'attaque par l'eau régale ou par Cl2/HCl
(acide chlorhydrique concentré dans lequel on fait barboter du
chlore) produit l'acide hexachloroplatinique, H2(PtCl6), un
important complexe du platine. Lorsqu'il est chauffé, le sel
d'ammonium de l'acide hexachloroplatinique produit une substance
grisâtre appelée "mousse de platine". La réduction d'une solution
aqueuse d'acide hexachloro-platinique donne une poudre noire
dispersée appelée "noir de platine".
En solution aqueuse, les espèces chimiques dominantes sont des
complexes. Beaucoup de ces sels complexes sont solubles dans l'eau
en particulier ceux qui contiennent des ligands donneurs comme les
halogènes ou l'azote. Le platine, comme les autres métaux du même
groupe, ont une forte tendance à réagir sur les composés carbonés,
en particulier les alcènes et les alcynes pour former des complexes
de coordination de Pt(II).
On dispose de diverses méthodes d'analyse pour le dosage du
platine. La spectométrie d'absorption atomique et la spectroscopie
d'émission en plasma sont très sélectives et spécifiques et
constituent les méthodes de choix pour le dosage du platine dans les
échantillons d'origine biologique ou environnementale. Dans
différents milieux, on a obtenu avec ces méthodes des limites de
détection de l'ordre du µg par kg ou par litre.
La spectroscopie d'émission atomique en plasma induite par
haute fréquence (PIHF) est supérieure à l'absorption atomique
électrothermique du fait que les effets de matrice sont plus faibles
et qu'il y a possibilité d'exécuter simultanément l'analyse d'autres
éléments.
2. Sources d'exposition humaine et environnementale
La concentration moyenne du platine dans la lithosphère, c'est-
à-dire la croûte rocheuse de la terre, est estimée à 0,001-0,005
mg/kg. On trouve le platine soit à l'état natif (métallique) soit en
combinaison, dans un certain nombre de minéraux. Les sources
d'importance économique se trouvent en République d'Afrique du Sud
et en URSS. La teneur en platine de ces gisements est de 1-500
mg/kg. Au Canada, les métaux du groupe du platine (platine,
palladium, iridium, osmium, rhodium et ruthénium) sont présents dans
les minerais sulfurés de cuivre et de nickel à une concentration
moyenne de 0,3 mg/kg; l'affinage du cuivre et du nickel porte cette
concentration à plus de 50 mg/kg. De petites quantités sont
extraites de mines situées aux Etats-Unis d'Amérique, en Ethiopie,
aux Philippines et en Colombie.
La production minière mondiale des métaux du groupe du platine,
constituée à 40-50% de platine, augmente régulièrement depuis les
deux dernières décennies. En 1971, la production était de 127
tonnes, dont 51-64 tonnes de platine. Depuis l'apparition du pot
d'échappement catalytique, la production minière mondiale de ces
métaux est passée à environ 270 tonnes (dont 108-153 tonnes de
platine) en 1987. En 1989, la demande totale de platine dans le
monde occidental était d'environ 97 tonnes.
La principale utilisation du platine tient à ses propriétés
catalytiques exceptionnelles. Les autres applications industrielles
sont basées sur d'autres propriétés remarquables de ce métal, en
particulier sa résistance à la corrosion chimique dans un grand
intervalle de température, son point de fusion élevé, sa ductilité
et sa grande résistance mécanique. Le platine est également utilisé
en joaillerie et en art dentaire.
Certains complexes du platine, en particulier le cis-
diamminedichloroplatine(II) ou cisplatine sont utilisés en
thérapeutique.a
On ne dispose pas de données sur les émissions de platine
d'origine industrielle. Lors de l'utilisation de catalyseurs à base
de platine, une certaine quantité de métal peut s'échapper dans
l'environnement selon le type de catalyseur. Parmi les catalyseurs
fixes utilisés dans l'industrie, seuls ceux qui servent à
l'oxydation de l'ammoniac dégagent des quantités importantes de
platine.
a Cette monographie traite spécialement du platine et de
certains de ses dérivés importants du point de vue
professionnel ou écologique. Une étude détaillée des effets
toxiques du cisplatine en tant que médicament anti-cancéreux et
de ses analogues chez l'homme et l'animal sortirait du cadre de
cette série car il s'agit de produits utilisés essentiellement
comme agents thérapeutiques. En outre leurs propriétés toxiques
sont exceptionnelles comparées à celles des autres dérivés du
platine.
Les catalyseurs de véhicules automobiles constituent des
sources mobiles de platine. Selon des données limitées, l'attrition
des anciens catalyseurs en granulés se situe entre 0,8 et 1,9 µg/par
km parcouru. Environ 10% de ce platine est soluble dans l'eau.
Les résultats fournis par des mesures au banc d'essai montrent
que les pots catalytiques à trois voies utilisant des catalyseurs
monolithiques de la nouvelle génération réduisent les émissions
totales de platine d'un facteur de 100-1000 par rapport aux
catalyseurs en granulés. A des vitesses simulées de 60, 100 et 140
km/heure on a constaté que l'émission totale de platine se situait
entre 3 et 39 ng/m3 dans les gaz d'échappement, ce qui correspond
environ à 2-39 ng par kilomètre parcouru. Le diamètre aérodynamique
moyen des particules émises variait, lors des différents essais,
entre 4 et 9 µm. D'après quelques indices, on peut penser que la
majeure partie du platine est émise sous forme de métal ou de
particules oxydées en surface.
3. Transport, répartition et transformation dans l'environnement
Les métaux du groupe du platine sont rares dans le milieu
ambiant, comparativement aux autres éléments. Dans les zones très
industrialisées, on peut trouver d'importantes quantités de platine
dans les sédiments des cours d'eau. On pense que les matières
organiques, par exemple les acides humiques et fulviques, se lient
au platine, cette réaction étant sans doute facilitée par des
conditions convenables de pH et de potentiel redox dans le milieu
aquatique.
Dans le sol, la mobilité du platine dépend du pH, du potentiel
redox, de la teneur en chlore de l'eau qui imprègne le sol et de
l'état naturel du platine dans les roches primaires. On estime que
le platine n'est mobilisé que dans des conditions d'acidité extrême
ou lorsque l'eau du sol est très riche en chlore.
On a montré qu' in vitro certains complexes du platine(IV)
pouvaient être méthylés en présence de platine(II) par la
méthylcobalamine bactérienne dans des conditions abiotiques.
4. Concentrations dans l'environnement et exposition humaine
La base de données relatives aux concentrations dans
l'environnement est très limitée en raison de la très faible teneur
de celui-ci en platine et des problèmes d'analyse que cela pose.
Les concentrations de platine dans des échantillons d'air
ambiant prélevés à proximité d'autoroutes aux Etats-Unis d'Amérique
avant l'introduction du pot catalytique se situaient en dessous de
la limite de détection de 0,05 pg/m3. Un certain nombre de données
récentes en provenance d'Allemagne indiquent qu'à proximité des
routes, des concentrations de platine dans l'air ambiant
(échantillons de matière particulaire) vont de moins de 1 pg/m3 à
13 pg/m3. Dans les zones rurales, ces concentrations étaient du
même ordre de grandeur (moins de 0,6 à 1,8 pg/m3).
A proximité immédiate des routes, les concentrations de platine
dans l'air ambiant qui résultent de l'introduction de catalyseurs en
granulés ont été évaluées à partir de modèles de dispersion et sur
la base des données expérimentales relatives aux émissions. Etant
donné que l'émission totale de platine d'un catalyseur de type
monolithique est plus faible, sans doute d'un facteur allant de 100
à 1000, que celle d'un catalyseur en granulés, les concentrations en
platine provenant de ce type de catalyseur devraient être de l'ordre
du picogramme au femtogramme par mètre cube.
En divers endroits de Californie, on a trouvé, dans la
poussière déposée sur les plantes à larges feuilles, des
concentrations de 37-680 µg/kg de poids sec. Le nombre
d'échantillons était limité mais les résultats montrent tout de même
que le pot catalytique libère du platine dans l'environnement
immédiat des routes.
Des cultures de graminées ont été exposées dans des serres
expérimentales pendant quatre semaines à des gaz d'échappement
légèrement dilués provenant d'un moteur équipé d'un catalyseur à
trois voies (vitesse simulée 100 km/heure): à la limite de détection
de 2ng par gramme de poids sec, on n'a pas trouvé de platine.
Des analyses effectuées sur les sédiments du lac Michigan ont
montré que du platine s'y était déposé depuis une cinquantaine
d'années à un rythme assez uniforme. Des concentrations dans des
carottes de 1 à 20 cm étaient comprises entre 0,3 et 0,4 µg/kg de
poids sec seulement.
On ne signale pas la présence de platine dans les eaux douces,
en revanche de fortes concentrations (730 à 31 220 µg/kg de poids
sec) ont été mesurées dans les sédiments d'un canal très pollué du
Rhin en Allemagne.
Dans des échantillons de bois de Pinus flexilis on a trouvé
des concentrations de platine allant de 0 (non décelable) à 56 µg/kg
(poids des cendres). Toutefois la teneur du sol voisin était du même
ordre et ces données plutôt limitées n'indiquent aucune tendance à
l'accumulation.
Dans des échantillons isolés de végétaux provenant d'un sol
extrêmement basique, on a mesuré des teneurs en platine allant de
100 à 830 µg/kg (poids sec).
Dans des échantillons d'eau de mer, on a relevé des
concentrations allant de 37 à 332 pg/litre. Des carottes de sédiment
prélevées dans le Pacifique oriental présentaient des teneurs en
platine allant de 1,1 à 3 µg/kg (poids sec). La concentration la
plus élevée (21,9 µg/kg) a été mesurée dans des sédiments océaniques
à distance du littoral. Les algues macroscopiques marines présentent
des teneurs en platine allant de 0,08 à 0,32 µg/kg de poids sec.
Dans la population générale le taux de platine sanguin se situe
entre 0,1 et 2,8 µg/litre. Dans le sérum de travailleurs exposés au
platine de par leur activité professionnelle, on a relevé des
concentrations de 150 à 440 µg/litre.
La base de données relative aux concentrations de platine sur
les lieux de travail est limitée. En raison de problèmes d'analyse,
les données anciennes (0,9 à 1700 µg/m3) ne sont probablement pas
fiables. Toutefois on peut déduire de ces données que l'exposition
aux sels de platine était à l'époque plus forte que la limite
d'exposition professionnelle de 2 µg/m3 qui est actuellement en
vigueur dans la plupart des pays. Des études récentes effectuées sur
les lieux de travail font état de concentrations qui sont, soit
inférieures à la limite de détection de 0,05 µg/m3, soit comprises
entre 0,08 et 0,1 µg/m3.
5. Cinétique et métabolisme
Une seule exposition de 48 minutes par la voie respiratoire à
du platine sous différentes formes chimiques (5-8 mg/m3) a montré
que le 191Pt inhalé était rapidement éliminé de l'organisme. On
observe ensuite une phase d'élimination plus lente au cours de la
période suivant l'exposition. Dix jours après exposition à du
191PtCl4, du 191Pt(SO4), du 191PtO2 et du 191Pt sous
forme métallique, la rétention du 191Pt dans l'ensemble de
l'organisme était respectivement de 1, 5, 8 et 6% de la dose
initiale. La majeure partie du 191Pt a été éliminée des poumons
par l'action de l'ascenseur mucociliaire puis avalée et excrétée
dans les matières fécales (temps de demi-élimination, 24 h.). Une
petite fraction du 191Pt a été décelée dans les urines, ce qui
indique que la résorption est très faible au niveau des poumons et
des voies digestives.
Lors d'une étude sur la destinée comparée du 191PtCl4
administré à des rats selon différentes voies à raison de 25uCi par
animal, on a constaté que c'était la voie intraveineuse qui
entraînait la rétention la plus forte, suivie par la voie
intratrachéenne. Elle était minimale après administration par voie
orale. Etant donné que seule une très faible partie du produit
administré par voie orale a été résorbée, l'essentiel a traversé les
voies digestives et a été excrété dans les matières fécales. Au bout
de trois jours, on ne décelait plus dans l'ensemble de l'organisme
que moins de 1% de la dose initiale. Après administration
intraveineuse, le 191Pt se retrouvait en quantités pratiquement
égales dans les matières fécales et dans l'urine. L'élimination
était plus faible qu'après administration orale. Au bout de cette
même période le taux de rétention dans l'ensemble de l'organisme
était d'environ 65% et au bout de 28 jours il se situait encore à
14% de la dose initiale. A titre de comparaison, au bout de ces deux
intervalles de temps environ 22 et 8% respectivement de la dose
initiale demeuraient dans l'organisme après administration intra-
trachéenne.
Les principaux sites d'accumulation sont les reins, le foie, la
rate et les glandes surrénales. La forte quantité de 191Pt
retrouvée dans les reins montre qu'une fois absorbé, le platine
s'accumule en majeure partie dans ces organes d'où il est excrété
dans l'urine. La quantité plus faible trouvée dans le cerveau montre
que les ions platine ne traversent qu'en faible proportion la
barrière hémo-méningée.
Contrairement aux sels solubles dans l'eau, le PtO2, qui est
insoluble, n'a été résorbé qu'en quantités très faibles, même après
administration dans la nourriture à dose très élevée, c'est-à-dire
correspondant à une dose totale de platine de 4308 mg par rat sur
une période de quatre semaines.
Qu'il s'agisse des sels simples ou du cisplatine, il est établi
que l'élimination se fait en deux phases : une phase initiale rapide
suivie d'une phase prolongée au cours de la période suivant
l'exposition, et que rien ne permet de penser que les modalités de
rétention soient très différentes. Toutefois le cisplatine est très
stable dans les liquides extracellulaire en raison de la forte
concentration en ions chlorure qui suppriment l'hydratation. Ainsi
s'explique que cette substance soit excrétée presque entièrement
sous forme inchangée. Contrairement au cas des sels simples, elle
est excrétée principalement dans les urines.
6. Effets sur les mammifères de laboratoire et les systèmes
d'épreuve in vitro
La toxicité aiguë du platine est principalement fonction de la
forme sous laquelle il se trouve. Les sels solubles sont beaucoup
plus toxiques que les sels insolubles. Par exemple la toxicité par
voie orale pour les rats (DL50) décroît dans l'ordre suivant:
Na2[PtCl6] (25-50 mg/kg) > (NH4)2[PtCl6] (195-200 mg/kg)
> PtCl4 (240 mg/kg) > Pt(SO4)2.4H2O (1010 mg/kg) >
PtCl2(> 2000 mg/kg) > PtO2 (> 8000 mg/kg). En ce qui concerne
ces deux derniers composés on n'a pas pu calculer la valeur de la
DL50.
Des tests cutanés pratiqués sur des lapins albinos ont montré
que PtO2, PtCl2, K2[PtCl4], [Pt(NO2)2(NH3)2],
Pt(C5H7O2)2 et trans-[PtCl2(NH3)2] pouvaient être
considérés comme non irritants. Par contre (NH4)2[PtCl6],
(NH4)2[PtCl4], Na2[PtCl6], Na2[Pt(OH)6],
K2[Pt(CN)4], [Pt(NH3)4]Cl2, et cis-[PtCl2(NH3)2]
se sont révélés irritants mais à des degrés divers.
Des tests d'irritation oculaire ont montré que tous les
composés testés avaient une action irritante. Le trans-
[PtCl2(NH3)2] ainsi que (NH4)2[PtCl4] se sont révélés
corrosifs.
De très sérieuses difficultés respiratoires ont été observées
après injection intraveineuse de complexes chloroplatiniques à des
cobayes et à des rats, vraisemblablement par suite d'une libération
d'histamine d'origine non allergique. Cette libération aspécifique
d'histamine complique l'interprétation des études sur l'animal et
sur l'homme en ce qui concerne le diagnostic de la sensibilisation
allergique. Après injection sous-cutanée et intraveineuse de
Pt(SO4)2, trois fois par semaine pendant quatre semaines, on n'a
pas constaté l'apparition d'un état allergique, à en juger d'après
les résultats des épreuves cutanées effectuées sur des cobayes et
des lapins, le transfert passif et les tests sur le coussinet
plantaire de la souris. L'administration d'un complexe
platine/albumine d'oeuf n'a pas non plus entraîné de sensibilisation
chez les animaux de laboratoire.
On a essayé sans succès de sensibiliser des rats Lister
femelles avec du tétrachloroplatinate d'ammonium,
(NH4)2[PtCl4] par administration intrapéritonéale,
intramusculaire, intradermique, sous-cutanée, intratrachéenne, et
dans le coussinet plantaire en présence de Bordetella pertussis
comme adjuvant; la sensibilisation a été évaluée par un test cutané
direct, un test d'anaphylaxie cutanée passive (PCA) ou un test avec
radio-allergo-absorbant (RAST). Toutefois le test PCA a donné un
résultat positif après administration de conjugués
platine/protéines.
Chez des singes Cynomolgus (Macaca fascicularis) exposés à de
l'hexachloroplatinate de sodium, Na2[PtCl6] exclusivement en
inhalations nasales à la dose de 200 µg par m3, 4 h par jour, deux
fois par semaine pendant 12 semaines, on a observé un déficit
pulmonaire sensiblement plus élevé que chez les témoins. Dans le cas
de l'hexachloroplatinate d'ammonium, il a fallu exposer les animaux
simultanément à de l'ozone (200 µ/m3) pour obtenir une
hypersensibilisation cutanée et une hyperréactivité pulmonaire
significatives.
Des études sur des rats Sprague-Dawley mâles ont montré que des
sels comme PtCl4 (182 mg/litre d'eau de boisson) et comme
Pt(SO4).4H2O (248 mg/litre) n'affectaient pas le gain de poids
au cours de la période d'observation de 4 semaines. En triplant la
concentration de platine, il y a eu une réduction de 20% du gain de
poids, mais seulement pendant la première semaine, parallèlement à
une diminution de 20% de la prise de nourriture et de boisson.
On ne dispose que de données expérimentales limitées à propos
des effets du platine sur la reproduction, et plus particulièrement
à propos de ses éventuels effets embryotoxiques et tératogènes. Le
Pt(SO4)2 (200 mg Pt/kg) a provoqué la mise bas de souriceaux
Swiss ICR de poids réduit du jour 8 au jour 45 du post-partum. Le
principal effet de Na2[PtCl6] (20 mg Pt/kg) a consisté dans une
réduction de l'activité de la progéniture lorsque les mères avaient
été exposées le douzième jour de la gestation. Les fils et les
feuilles de platine sont considérés comme inertes biologiquement et
les effets nocifs constatés après implantation dans l'utérus de
rattes et de lapines étaient probablement dus à la présence physique
d'un corps étranger.
Après administration à des rattes gravides d'une dose de
191Pt égale à 25 microcuries par animal, le 18ème jour de la
gestation, on a constaté un passage limité à travers la barrière
foeto-placentaire.
Plusieurs dérivés du platine se sont révélés mutagènes dans un
certain nombre de systèmes bactériens. Lors d'études comparatives on
a constaté que la mutagénicité du cisplatine était plusieurs fois
supérieure à celle des autres composés. Des études in vitro ont
montré que, dans le système cellulaire mammalien CHO-HGPT,
l'activité mutagène relative s'établissait selon la proportion
100:9:0,3 respectivement pour les composés suivants: cis-
[PtCl2(NH3)2], K[PtCl3(NH3)], et [Pt(NH3)3Cl]Cl. La
mutagénicité de K2[PtCl4] et du trans-[PtCl2(NH3)] était
marginale, tandis que le [Pt(NH3)4]Cl2 n'était pas mutagène.
Les composés K2[PtCl4] et [Pt(NH3)4]Cl2 ne l'étaient pas
non plus dans les tests suivants: mutation récessive léthale liée au
sexe chez Drosophila melanogaster, recherche de micronoyaux dans
des cellules de souris et le test sur moelle osseuse de hamster. On
ne possède de données expérimentales sur la cancérogénicité des
dérivés du platine que dans le cas du cisplatine pour lequel les
preuves d'une activité cancérogène chez l'animal sont suffisantes.
Cependant le cisplatine et ses analogues font plutôt figure
d'exception si on les compare aux autres dérivés du platine. Cela
transparaît dans leur activité antitumorale, dont le mécanisme est
très particulier. On pense que celle-ci est, semble-t-il, due à la
formation de ponts intercaténaires qui ne se produit qu'en présence
de l'isomèreciset pour une certaine position de la guanine. Les
cellules tumorales ne sont plus en mesure de se répliquer, alors que
les cellules normales conservent leur capacité de réplication après
avoir réparé les lésions provoquées par le cisplatine.
7. Effets sur l'homme
L'exposition aux sels de platine se limite essentiellement aux
ambiances de travail, et plus précisément aux ateliers d'affinage du
platine et aux unités de production de catalyseurs.
Les composés principalement responsables de l'hyper-sensibilité
aux sels de platinea sont l'acide hexachloroplatinique
H2[PtCl6]et un certain nombre de sels comme
l'hexachloroplatinate d'ammonium (NH4)2[PtCl6], le
tétrachloroplatinate de potassium, K2[PtCl4], et le
tétrachloroplatinate de sodium, Na2[PtCl4]. Les complexes dans
lesquels il n'y a pas d'halogènes coordonnés au platine (complexes
non halogénés), comme K2[Pt(NO2)4], [Pt(NH3)4]Cl2 et
[Pt{(NH2)2CS}4]Cl2, de même que les complexes neutres comme
le cis-[PtCl2(NH3), ne sont pas allergéniques car ils
réagissent avec les protéines pour former un antigène complet.
Les symptômes de cette hypersensibilité sont les suivants:
urticaire, dermatite de contact, ainsi qu'un certain nombre de
troubles respiratoires, comme reniflement, essouflement, cyanose et
asthme grave. La période de latence entre le premier contact avec
des sels de platine et l'apparition des symptômes dure de quelques
semaines à plusieures années. Après sensibilisation les symptômes
ont tendance à s'aggraver aussi longtemps que les travailleurs sont
exposés sur leur lieu de travail, mais ils disparaissent, en
général, dès que cesse l'exposition. Toutefois, si une exposition de
longue durée fait suite à la sensibilisation, les symptômes risquent
de ne jamais disparaître complètement.
Bien qu'il soit impossible de tirer des données publiées une
relation dose-effet qui ne soit pas ambiguë, il semble que le risque
d'apparition d'une hyper-sensibilité aux sels de platine soit en
corrélation avec l'intensité de l'exposition. Le platine métallique
ne semble pas être allergénique. A l'exception d'un cas unique de
dermatite de contact, aucune réaction allergique n'a été signalée.
Les manifestations cliniques de l'hypersensibilité aux sels de
platine sont celles d'une véritable réaction allergique. Le
mécanisme de cette réaction est du type 1 (médiation par les IgE).
Sur la base d'épreuves in vivo et in vitro on pense que chez les
sujets sensibles il se forme des anticorps IgE dirigés contre les
complexes chloroplatiniques. Les sels de platine de faible masse
moléculaire relative se comportent comme des haptènes qui se
combinent aux protéines pour former des antigènes complets.
a Le terme platinose n'est plus usité pour désigner les
affections provoquées par les sels de platine, car il implique
une fibrose pulmonaire chronique du type silicose. Il est
préférable d'utiliser le terme allergie aux sels de platine, ou
allergie aux composés du platine contenant des ligands
halogénés réactifs ou mieux, hypersensibilité aux sels de
platine.
Les tests cutanés avec des sels de platine dilués permettent de
surveiller les réactions allergiques de manière reproductible,
fiable, assez sensible et très spécifique. Pour les contrôles de
routine en milieu professionnel on utilise les composés suivants:
(NH4)2[PtCl6], Na2[PtCl6] et Na2[PtCl4]. Il n'existe
pas d'épreuve in vitro dont la sensibilité et la fiabilité
approchent celles des tests cutanés. Des tests immunoenzymatiques ou
par immunoallergosorption ont permis de mettre en évidence des
anticorps IgE spécifiques dirigés contre les complexes chlorés du
platine. Il y avait corrélation avec les résultats des tests cutanés
mais l'utilisation du test RAST reste problématique en raison de son
manque de spécificité.
Les tests cutanés et l'épreuve RAST montrent qu'il n'existe
qu'une faible réactivité croisée entre les sels de platine et les
sels de palladium. Des réactions d'hypersensibilité à d'autres
métaux du groupe du platine ont également été observées, mais
seulement chez des personnes allergiques aux sels de platine.
Le tabagisme, l'atopie et l'hyperréactivité pulmonaire
aspécifique ont été associés à l'hypersensibilité aux sels de
platine et pourraient être des facteurs prédisposants.
En ce qui concerne la population générale, on manque de données
sur l'exposition effective dans les pays où le pot catalytique est
devenu obligatoire. Les concentrations dans l'air ambiant estimées
d'après de nouvelles données sur les émissions et sur la base de
modèles de dispersion sont probablement inférieures d'au moins un
facteur 10 000 à la limite d'exposition professionnelle de 1 mg/m3
adoptée par certains pays pour le platine total inhalable sous forme
de poussières. Etant donné que le platine est très probablement
présent dans les émissions sous forme métallique, le potentiel de
sensibilisation du platine émis par les pots catalytiques est
probablement très faible. Même si une partie du platine émis est
soluble et potentiellement allergénique, la marge de sécurité par
rapport à la limite d'exposition professionnelle pour les sels
solubles de platine (2 µg/m3) serait d'au moins 2000.
Lors d'une étude immunologique préliminaire, on a pratiqué des
tests cutanés sur trois volontaires au moyen d'extraits de matières
particulaires émises par des véhicules à moteur. On n'a pas observé
de réponse positive.
On ne dispose d'aucune donnée sur les risques de
cancérogénicité pour l'homme attribuables au platine et à ses sels.
Pour ce qui est du cisplatine les preuves de cancérogénicité sont
jugées insuffisantes.
8. Effets sur d'autres organismes au laboratoire et dans la nature
Les complexes simples du platine ont des effets bactéricides.
En observant que les complexes neutres comme le cisplatine
inhibaient sélectivement la division cellulaire sans réduction de la
croissance chez diverses bactéries gram-positives mais aussi, et
surtout, gram-négatives on a eu l'idée de les utiliser comme agents
anticancéreux.
On a observé qu'au sein d'un "microcosme" de laboratoire, la
croissance des algues vertes du genre euglène était inhibée en
présence d'acide hexachloroplatinique soluble aux concentrations de
250, 500, et 750 µg/litre. Le cisplatine a provoqué une chlorose et
un ralentissement de la croissance chez la jacinthe d'eau Eichornia
crassipesà la concentration de 2,5 mg/litre.
Après 3 semaines d'exposition à de l'acide
hexachloroplatinique, H2[PtCl6], on a observé chez la daphnie
une mortalité correspondant à une CL50 de 520 µg de Pt par litre.
Aux concentrations de 14 et 82 µg/litre, la reproduction (nombre de
jeunes daphnies) était réduite dans la proportion de 16 et 50%
respectivement.
Après exposition de saumons (Oncorhyncus kisutch) à de
l'acide tétrachloroplatinique pendant une brève période de temps
dans des conditions statiques, on a observé que les valeurs de la
CL50 étaient respectivement égales à 15,5, 5,2 et 2,5 mg Pt/litre
au bout de 24, 48, et 96 h. On constatait, à la dose de 0,3
mg/litre, une diminution globale de l'activité natatoire et du
mouvement des opercules. Aux concentrations supérieures à cette
valeur, des lésions apparaissaient au niveau des branchies et de
l'organe olfactif. Les concentrations de 0,03 et 0,1 mg/litre
étaient sans effet.
Toutes les études consacrées aux effets du platine sur les
plantes terrestres concernent uniquement les chlorures solubles. A
des concentrations allant de 3.10-5 à 15.10-5 mol/kg (5,9-29,3
mg/kg) il y a eu inhibition de la croissance de plants de haricots
et de tomates en sol sableux. Neuf variétés horticoles en culture
hydroponique ont présenté une réduction de leur poids à sec après
adjonction de tétrachlorure de platine aux concentrations
respectives de 0,057, 0,57, et 5,7 mg Pt/litre; il s'agissait de
tomates, de poivrons, de fanes de navets et pour la concentration la
plus élevée, de radis. A cette concentration les bourgeons et les
feuilles immatures devenaient chlorotiques chez la plupart des
espèces. En revanche, chez certaines espèces, le tétrachlorure de
platine stimulait la croissance. Par ailleurs, la teneur la plus
élevée en platine supprimait la transpiration, sans doute par
accroissement de la résistance des stomates. On constatait également
une stimulation de la croissance aux faibles teneurs en platine (0,5
mg/litre), lorsque l'on ajoutait au milieu nutritif d'une graminée
sud-africaine (Setaria vertillata) dutétrachloroplatinate de
potassium. Au bout de deux semaines, la racine la plus longue avait
poussé de 65%. A la concentration utilisée, soit 2,5 mg Pt/litre, on
observait des effets phytotoxiques tels que rabougrissement des
racines et chlorose foliaire.
RESUMEN
1. Identidad, propiedades físicas y químicas, métodos analíticos
El platino (Pt) es un metal noble maleable, dúctil, de color
plateado blanquecino; su número atómico es 78 y su peso atómico
195,09. Sus isótopos naturales más abundantes son 194Pt (32,9%),
195Pt (33,8%), y 196Pt (25,3%). En los compuestos de platino, el
estado de oxidación máxima es +6; los estados +2 y +4 son los más
estables.
Aunque el metal no se corroe en el aire a ninguna temperatura,
es sensible a los halógenos, los cianuros, el azufre, los compuestos
de azufre fundentes, los metales pesados y los hidróxidos de
álcalis. La digestión con agua regia o Cl2/HCl (ácido clorhídrico
concentrado por el que se burbujea cloro) produce ácido
hexacloroplatínico, H2[PtCl6], un importante complejo de
platino. Cuando se calienta, la sal amónica del ácido
hexacloroplatínico produce una esponja gris de platino. La reducción
en solución acuosa produce un polvo dispersivo de color negro
("negro de platino").
Las propiedades químicas de los compuestos del platino en
solución acuosa se ven dominadas por los compuestos complejos.
Muchas de las sales, particularmente las que llevan ligandos
donadores de halógeno o de nitrógeno, son solubles en agua. El
platino, al igual que los otros metales de su grupo, tiene una
pronunciada tendencia a reaccionar con los compuestos del carbono,
especialmente los alquenos y los alquinos, formando complejos de
coordinación Pt(II).
Existen diversos métodos analíticos para la determinación del
platino. La espectrometría de absorción atómica (EAA) y la
espectroscopia de emisión de plasma son sumamente selectivas y
específicas y constituyen el método de elección para analizar el
platino presente en muestras biológicas y medioambientales. Con esos
métodos se han alcanzado en diversos medios límites de detección del
orden de unos cuantos µg/kg o µg/litro.
La espectroscopia de emisión atómica con plasma de argón
acoplado por inducción es preferible a la EAA electrotérmica por sus
menores efectos matriciales y por la posibilidad de analizar
simultáneamente muchos elementos.
2. Fuentes de la exposición humana y ambiental
Se calcula que la concentración media de platino en la
litosfera o corteza terrestre es del orden de 0,001-0,005 mg/kg. El
platino se encuentra en forma metálica o en varias formas minerales.
Existen fuentes económicamente importantes en la República de
Sudáfrica y en la URSS. El contenido de platino de esos depósitos es
de 1-500 mg/kg. En el Canadá, los metales del grupo del platino
(platino, paladio, iridio, osmio, rodio, rutenio) se encuentran en
menas de sulfuro de cuproníquel con una concentración media de 0,3
mg/kg, pero esa concentración supera los 50 mg/kg durante el afinado
del cobre y el níquel. En los EE.UU., Etiopía, Filipinas y en
Columbia se extraen pequeñas cantidades.
La producción minera mundial de metales del grupo del platino,
de la cual el 40-50% corresponde al platino, ha aumentado
uniformemente durante los últimos 20 años. En 1971, la producción
fue de 127 toneladas (51-64 toneladas de platino). A raíz de la
introducción del catalizador de los gases de escape en los
automóviles, la producción minera mundial de metales del grupo del
platino aumentó hasta aproximadamente 270 toneladas (108-135
toneladas de platino) en 1987. En 1989, la demanda total de platino
en el mundo occidental fue de unas 97 toneladas.
El uso principal del platino deriva de sus excepcionales
propiedades catalíticas. Las demás aplicaciones industriales
aprovechan otras notables propiedades, en particular la resistencia
a la corrosión química en un amplio intervalo de temperaturas, su
elevado punto de fusión, su gran resistencia mecánica y su buena
ductilidad. El platino se usa asimismo en joyería y odontología.
Ciertos complejos de platino, en particular el cis-
diaminodicloroplatino(II) (cisplatino), tienen aplicaciones
terapéuticas.a
No se dispone de datos sobre las emisiones de platino al medio
ambiente a partir de fuentes industriales. El uso de catalizadores
con platino puede entrañar la liberación de ese elemento al medio
ambiente, según el tipo de catalizador. De los catalizadores
estacionarios utilizados en la industria, sólo los empleados para la
oxidación del amoniaco emiten cantidades significativas de platino.
Los catalizadores utilizados en automoción son fuentes móviles
de platino. Se dispone de datos limitados que indican que el
desgaste de platino a partir del antiguo catalizador en pastilla es
a La presente monografía se ocupa específicamente del platino y
de ciertos compuestos del platino de importancia ocupacional
y/o ambiental. No entra en el ámbito restringido de la serie de
Criterios de Salud Ambiental el estudio pormenorizado de los
efectos tóxicos del fármaco anticanceroso cisplatino y de sus
análogos en el hombre y los animales, puesto que esas
sustancias se usan principalmente como agentes terapéuticos.
Además, sus propiedades tóxicas son excepcionales en
comparación con las de otros compuestos de platino.
de 0,8 a 1,9 µg por km recorrido. Alrededor del 10% del platino es
soluble en agua.
Con la nueva generación de catalizadores de tipo monolítico,
los resultados de experimentos en plataforma de pruebas de motores
con un catalizador de tres vías indican que la emisión total de
platino es inferior por un factor de 100-1000 a la producida en
catalizadores en pastilla. Con velocidades simuladas de 60, 100 y
140 km/h, se encontró que la emisión total de platino era de 3 a 39
ng/m3 en los gases de escape, lo que corresponde a unos 2-39 ng
por km recorrido. El diámetro aerodinámico medio de las partículas
emitidas era de 4 a 9 µm en las distintas pruebas. Existen pruebas
limitadas de que la mayor parte del platino emitido se encuentra en
forma metálica o en partículas de superficie oxidada.
3. Transporte, distribución y transformación en el medio ambiente
Los metales del grupo del platino son escasos en el medio
ambiente en comparación con otros elementos. En zonas muy
industrializadas, pueden encontrarse cantidades elevadas de platino
en los sedimentos fluviales. Se supone que la materia orgánica, por
ejemplo los ácidos húmicos y fúlvicos, enlaza platino, proceso que
tal vez se vea favorecido por condiciones apropiadas de pH y de
potencial redox en el medio acuático.
En el suelo, la movilidad del platino depende del pH, el
potencial redox, las concentraciones de cloruros en las aguas
subterráneas y la forma en que se encuentra el platino en la roca
primitiva. Se considera que el platino sólo será móvil en
condiciones extremadamente ácidas o en aguas subterráneas con
elevado contenido de cloro.
En los sistemas de ensayo in vitro se ha demostrado que
algunos complejos de platino(IV), en presencia de platino(II),
pueden sufrir metilación por la metilcobalamina bacteriana en
condiciones abióticas.
4. Niveles medioambientales y exposición humana
Se dispone de muy pocos datos en cuanto a las concentraciones
medioambientales debido a los reducidos niveles de platino en el
medio ambiente y los problemas analíticos que ello acarrea.
Las concentraciones en muestras de aire obtenidas en las
proximidades de autopistas en los Estados Unidos antes de la
introducción del catalizador en los automóviles se encontraban por
debajo del límite de detección de 0,05 pg/m3. Algunos datos
obtenidos recientemente en Alemania indican que en las cercanías de
las carreteras las concentraciones de platino en el aire (muestras
particuladas) varían entre < 1 pg/m3 y 13 pg/m3. En zonas
rurales, las concentraciones se encontraban en un orden de magnitud
similar (< 0,6 a 1,8 pg/m3).
Las concentraciones de platino en el aire cercano a carreteras
tras la introducción de los catalizadores de pastilla en los
automóviles se han calculado basándose en modelos de dispersión y
datos experimentales de emisión. Las concentraciones estimadas de
platino en las carreteras y en las zonas próximas variaron entre
0,005 y 9 ng/m3 para el platino total. Puesto que la emisión total
de platino en un catalizador de tipo monolítico es inferior,
probablemente por un factor de 100 a 1000, que en un catalizador de
pastilla, las concentraciones de platino emitidas en ese tipo de
catalizador se encontrarían en el margen de picogramos a femtogramos
por m3.
En el polvo depositado en las plantas de hoja ancha que bordean
las carreteras en distintos lugares de California, se detectaron
concentraciones de 37-680 µg por kg de peso seco. Aunque el número
de muestras era limitado, los resultados indican que los
catalizadores de automóviles liberan platino al medio ambiente
próximo a las carreteras.
En experimentos en cámara vegetal, los cultivos herbáceos
expuestos durante cuatro semanas a gases de escape ligeramente
diluidos procedentes de un motor equipado con un catalizador de 3
vías (velocidad simulada: 100 km/h) no contenían platino con un
límite de detección de 2 ng/g de peso seco.
Los estudios de las concentraciones de platino en sedimentos
del Lago Michigan llevaron a la conclusión de que el platino se ha
ido depositando en ellos durante los últimos 50 años a una velocidad
uniforme. Las concentraciones en calas de sedimento de 1 a 20 cm
variaron sólo entre 0,3 y 0,43 µg/kg de peso seco.
Mientras que no se han comunicado niveles de platino en aguas
dulces, se han encontrado elevadas concentraciones (730 a 31 220
µg/kg de peso eco) en los sedimentos de un canal de corta sumamente
contaminado en el río Rin (Alemania).
En muestras de Pinus flexilisse encontraron niveles de
platino entre el límite de detección y 56 µg/kg (peso de ceniza). No
obstante, el contenido de los suelos adyacentes se encontraba entre
los mismos valores; estos datos limitados no indicaban tendencia
alguna de acumulación.
En muestras aisladas de vegetales procedentes de un suelo
ultrabásico, se encontraron niveles de platino de 100-830 µg/kg
(peso seco).
En muestras de agua marina se han encontrado entre 37 y 332
pg/litro. En calas de sedimento obtenidas en el Pacífico oriental,
las concentraciones de platino variaron entre 1,1 y 3 µg/kg (peso
seco). La concentración más elevada (21,9 µg por kg) se encontró en
sedimentos oceánicos alejados del litoral. En macroalgas marinas se
han encontrado concentraciones de platino entre 0,08 y 0,32 µg/kg de
peso seco.
En la población general se han medido niveles sanguíneos de
platino de 0,1 a 2,8 µg/litro. En suero de trabajadores expuestos
por su profesión, se han comunicado niveles de 150 a 440 µg por
litro.
Se dispone de datos limitados sobre las concentraciones de
platino en el lugar de trabajo. Es probable que los datos antiguos
(0,9 a 1700 µg/m3) no sean de fiar debido a las deficiencias del
análisis. No obstante, esos datos permiten suponer que el nivel de
exposición a sales de platino era superior al límite de exposición
profesional de 2 µg/m3 adoptado actualmente en la mayoría de los
países. En recientes estudios realizados en los lugares de trabajo,
se han medido concentraciones inferiores al límite de detección de
0,05 µg/m3 o entre 0,08 y 0,1 µg/m3.
5. Cinética y metabolismo
Tras una exposición única por inhalación (48 minutos) a
distintas formas químicas del platino (5-8 mg/m3), la mayor parte
del 191Pt inhalado fue rápidamente eliminado del organismo. A
continuación se observó una fase más lenta de eliminación durante el
resto del periodo posterior a la exposición. A los diez días de la
exposición a 191PtCl4, 191Pt(SO4)2, 191PtO2, y 191Pt
metálico, la retención total de 191Pt por el organismo fue de
aproximadamente 1, 5, 8 y 6%, respectivamente, de la carga inicial
del organismo. La mayor parte del 191Pt eliminado de los pulmones
por mecanismos mucociliares e ingerido se excretó con las heces
(semivida: 24 h). Una pequeña fracción del 191Pt se detectó en la
orina, lo que indica que la absorción en los pulmones y el tracto
gastrointestinal fue muy reducida.
En un estudio comparativo sobre el destino del 191PtCl4 en
ratas (25 µCi/animal) tras la exposición por distintas vías, la
retención fue máxima tras la administración intravenosa, seguida por
la exposición intratraqueal. La mínima se registró tras la
administración oral. Puesto que sólo fue absorbida una cantidad
minúscula del 191PtCl4 administrada por vía oral, la mayoría
atravesó el tracto gastrointestinal y se excretó con las heces. Al
cabo de tres días, menos del 1% de la dosis inicial se detectó en
todo el cuerpo. Tras la administración intravenosa, el 191Pt se
excretó en cantidades casi iguales tanto en las heces como en la
orina. La eliminación fue más lenta que en el caso de la
administración oral. A los tres días la retención en todo el
organismo era de alrededor del 65%, y al cabo de 28 días aún era del
14% de la dosis inicial. A título de comparación, al cabo de
periodos iguales alrededor del 22% y del 8%, respectivamente,
quedaron retenidos por el organismo tras la administración
intratraqueal.
Los principales lugares de depósito son el riñón, el hígado, el
bazo y las glándulas suprarrenales. La elevada cantidad de 191Pt
encontrada en el riñón demuestra que una vez que el platino es
absorbido, la mayor parte se acumula en él y se excreta en la orina.
El nivel más bajo en el cerebro sugiere que los iones de platino
atraviesan la barrera hematoencefálica sólo en grado limitado.
A diferencia de las sales hidrosolubles, el PtO2, que es
insoluble, sólo fue captado en cantidades insignificantes a pesar de
que la sal se administró con la dieta en concentraciones sumamente
elevadas, que representaron un consumo total de platino de 4308 mg
por rata durante el periodo de cuatro semanas.
Tanto en el caso de las sales simples de platino como en el
cisplatino, se ha determinado que existe un periodo de eliminación
rápida seguido de una fase prolongada de eliminación durante el
resto del periodo posterior a la exposición, y que no existen
pruebas de que los perfiles de retención sean notablemente
diferentes. No obstante, el cisplatino es sumamente estable en los
fluidos extracelulares debido a que las elevadas concentraciones de
cloruro suprimen la hidratación. Ello explica que se excrete
principalmente en la forma no alterada. Su excreción, a diferencia
de la de las sales simples de platino, tiene lugar principalmente
con la orina.
6. Efectos en mamíferos de laboratorio y en sistemas de ensayo in
vitro
La toxicidad aguda del platino depende principalmente de la
especie de platino. Los compuestos solubles son mucho más tóxicos
que los insolubles. Por ejemplo, la toxicidad por vía oral en la
rata (valores de la LD50) disminuyo en el orden siguiente:
Na2[PtCl6] (25-50 mg/kg) > (NH4)2[PtCl6] (195-200 mg/kg)
> PtCl4 (240 mg/kg) > Pt(SO4)2.4H2O (1010 mg/kg) >
PtCl2 (> 2000 mg/kg) > PtO2 (> 8000 mg/kg). No pudo alcularse
la DL50 correspondiente a los dos últimos compuestos.
En las pruebas cutáneas realizadas en conejos albinos, los
compuestos PtO2, PtCl2, K2[PtCl4], [Pt(NO2)2(NH3)2],
Pt(C5H7O2)2 y trans-[PtCl2(NH3)2] se clasificaron
como no irritantes. Los compuestos (NH4)2[PtCl6],
(NH4)2[PtCl4], Na2[PtCl6], Na2[Pt(OH)6],
K2[Pt(CN)4], [Pt(NH3)4]Cl2, y cis-[PtCl2(NH3)2]
resultaron irritantes en diversos grados.
En los ensayos de irritación ocular todos los compuestos de
platino ensayados dieron resultados positivos. El trans-
[PtCl2(NH3)2] y el (NH4)2[PtCl4] resultaron ser
corrosivos.
Tras la inyección intravenosa de complejos de cloroplatino en
cobayos y ratas, se observaron dificultades respiratorias intensas,
probablemente debidas a la liberación analérgica de histamina. Esta
liberación inespecífica de histamina ha complicado la interpretación
de los estudios en animales y en el hombre en relación con el
diagnóstico de la sensibilización alérgica.
Tras la inyección subcutánea e intravenosa de Pt(SO4)2 tres
veces a la semana durante cuatro semanas, no se observó inducción de
un estado alérgico, de acuerdo con las pruebas cutáneas (cobayos y
conejos), la transferencia pasiva y los ensayos en la almohadilla
plantar (ratones). La administración del complejo platino-huevo-
albúmina tampoco sensibilizó a los animales de experimentación.
No se consiguió sensibilizar a hembras de rata encapuchada de
Lister con la sal libre de tetracloroplatinato de amonio,
(NH4)2[PtCl4], aplicada por las vías intraperitoneal,
intramuscular, intradérmica, subcutánea, intratraqueal y por la
almohadilla plantar, conBordetella pertussisw como coadyuvante, de
acuerdo con los resultados de la prueba cutánea directa, la prueba
de anafilaxis cutánea pasiva o un ensayo de radioalergosorbencia
(RAST). No obstante, se han comunicado resultados positivos de
anafilaxis cutánea pasiva con conjugados platino-proteína.
En monos Cynomolgus (Macaca fasicularis) expuestos a
hexacloroplatinato de sodio, Na2[PtCl6], por inhalación
exclusivamente nasal de una concentración de 200 µg/m3 wdurante 4
horas al día, dos veces a la semana durante 12 semanas, se
observaron insuficiencias pulmonares significativamente mayores que
en los animales testigo. Con la exposición a hexacloroplatinato de
amonio, (NH4)2[PtCl6], sólo la exposición simultánea a ozono
(2000 µg/m3) produjo hipersensibilidad cutánea e hiperreactividad
pulmonar significativas.
En estudios de administración oral a machos de rata Sprague-
Dawley, las sales PtCl4 (182 mg/l de agua de bebida) y
Pt(SO4)2.4H2O (248 mg/litro) no ejercieron efecto alguno en la
adquisición normal de peso durante el periodo de observación de 4
semanas. Al triplicar la concentración de platino, la adquisición de
peso se redujo en un 20% sólo durante la primera semana,
paralelamente a una disminución del 20% del consumo de alimento y
agua.
Sólo se dispone de datos experimentales limitados sobre los
efectos del platino en la reproducción, la embriotoxicidad y la
teratogenicidad. El Pt(SO4)2 (200 mg Pt/kg) redujo el peso de
las crías en ratones suizos ICR desde el día 8 al 45 después del
parto. El principal efecto del Na2[PtCl6] (20 mg Pt/kg) fue un
nivel de actividad menor en las crías de madres expuestas el duo-
décimo día de gestación. Se considera que el alambre y las láminas
de platino sólido son biológicamente inertes; los efectos adversos
observados a raíz de la implantación en el útero de ratas y ratones
se debieron probablemente a la presencia física de un objeto
extraño.
Tras la administración intravenosa de 191PtCl4 a ratas
gestantes (25 µCi/animal) el día 18 de la gestación, una cantidad
limitada del compuesto atravesó la barrera placentaria.
Se ha observado que varios compuestos de platino son
mutagénicos en diversos sistemas bacterianos. En estudios
comparativos, el cisplatino era varias veces más mutagénico que
otras sales de platino ensayadas. En estudios realizados in vitro
con células de mamífero (sistema CHO-HGPT), la actividad mutagénica
relativa de los compuestos cis-[PtCl2(NH3)2],
K[PtCl3(NH3)] y [Pt(NH3)3Cl]Cl fue 100:9:0,3. La
mutagenicidad del K2[PtCl4] y el trans-[PtCl2(NH3)2] era
marginal, mientras que el [Pt(NH3)4]Cl2 no era mutagénico. No
se observó actividad mutagénica en los compuestos K2[PtCl4] y
[Pt(NH3)4]Cl2, en el ensayo de letalidad recesiva ligada al
sexo en Drosophila melanogaster, en un ensayo de micronúcleo de
ratón ni en el ensayo en médula ósea de hámster chino.
Salvo en el caso del cisplatino, no se dispone de datos
experimentales relativos a la carcinogenicidad del platino y sus
compuestos. Existen prueba suficientes de la carcinogenicidad del
cisplatino en los animales. No obstante, el cisplatino y sus
análogos son excepcionales en comparación con otros compuestos de
platino; ello se refleja en su mecanismo característico de actividad
anti-tumoral. La constitución de enlaces cruzados entre las hebras
de ADN, formados sólo por el isómero cis en determinada posición de
la guanina, se considera el motivo de esa actividad antitumoral.
Parece ser que la replicación del ADN es defectuosa en las células
cancerosas, mientras que en las normales las lesiones causadas por
el cisplatino en la guanina se reparan antes de la replicación.
7. Efectos en el ser humano
La exposición a sales de platino se limita principalmente al
medio ocupacional, en particular a las refinerías de platino
metálico y las plantas de fabricación de catalizadores.
Los princiaples compuestos responsables de la hiper-
sensibilidad a las sales de platino son el ácido hexacloroplatínico,
H2[PtCl6], y algunas sales cloradas como el hexacloroplatinato
de amonio, (NH4)2[PtCl6], el tetracloroplatinato de potasio,
K2[PtCl4], el hexacloroplatinato de potasio, K2[Pt6] y el
tetracloroplatinato de sodio, Na2[PtCl4].a Los complejos en
los que no hay ligandos halógenos coordinados al platino ("complejos
no halogenados"), como el K2[Pt(NO2)4], [Pt(NH3)4]Cl2 y
[Pt{(NH2)2CS}4]Cl2, así como los complejos neutros como el
cis-[PtCl2(NH3)2], no son alergénicos, puesto que
probablemente no reaccionan con proteínas para formar un antígeno
completo.
Entre los signos y síntomas de hipersensibilidad figuran
urticaria, dermatitis de contacto de la piel, y trastornos
respiratorios que pueden ir desde estornudos, disnea y cianosis a
crisis graves de asma. El periodo de latencia desde el primer
contacto con el platino hasta la aparición de los primeros síntomas
varía desde unas pocas semanas a varios años. Una vez que la
sensibilización está establecida, los síntomas tienden a empeorar
durante el tiempo que los individuos sigan expuestos en el lugar de
trabajo, pero suelen desaparecer al cesar la exposición. No
obstante, si se produce una exposición prolongada después de la
sensibilización, es posible que los individuos nunca queden
completamente exentos de síntomas.
Aunque a partir de los datos disponibles no puede deducirse una
relación inequívoca entre la concentración y el efecto, el riesgo de
desarrollar sensibilidad a las sales de platino parece guardar
relación con la intensidad de la exposición. El platino metálico
parece no ser alergénico. A excepción de un solo caso comunicado de
supuesta dermatitis de contacto provocada por un anillo "de
platino", no se han notificado reacciones alérgicas.
Las manifestaciones clínicas de la hipersensibilidad a las
sales de platino reflejan una auténtica respuesta alérgica. El
mecanismo parece ser una respuesta de tipo I (medida por la IgE). La
posibilidad de que se desarrollen anticuerpos IgE a complejos de
cloruro de platino en personas sensibles se ha supuesto basándose en
los ensayos in vivo e in vitro. Se cree que las sales de platino
de baja masa molecular relativa actúan como haptenos que se combinan
con las proteínas séricas para formar el antígeno completo.
a Se ha abandonado el término "platinosis" para describir las
enfermedades relacionadas con las sales de platino, puesto que
implica una enfermedad pulmonar fibrosante crónica del tipo de
la silicosis. En su lugar, se han utilizado "alergia a las
sales de platino", "alergia a los compuestos de platino que
contienen ligandos halógenos reactivos" e "hipersensibilidad a
sales de platino" (HSP), siendo preferible el último.
Las pruebas de punción cutánea con concentraciones diluidas de
complejos solubles de platino parecen proporcionar indicadores
biológicos de la alergenicidad reproducibles, fiables,
razonablemente sensibles y sumamente específicos. Los compuestos
utilizados para las pruebas periódicas de detección de alergias en
los trabajadores expuestos son (NH4)2[PtCl6], Na2[PtCl6] y
Na2[PtCl4]. La sensibilidad y fiabilidad de las pruebas de
punción cutánea no tienen igual en ninguno de los ensayos in vitro
disponibles. En los inmunoensayos enzimáticos y las pruebas de
radioalergosorbencia (RAST) se han encontrado anticuerpos IgE
específicos de los complejos del cloruro de platino. Aunque se
comunicó la existencia de correlación con los resultados de las
pruebas de punción cutánea, la aplicabilidad de la prueba RAST para
los exámenes de detección se puso en tela de juicio a causa de su
falta de especificidad.
Se ha encontrado una reactividad cruzada limitada entre las
sales de platino y de paladio en los ensayos cutáneos y el RAST. Las
reacciones a los metales del grupo del platino distintos de éste
sólo se han observado en individuos sensibles a las sales de
platino.
El tabaquismo, la atopia y la hiperreactividad pulmonar
inespecífica se han asociado a la hipersensibilidad a las sales de
platino y pueden ser factores predisponentes.
En cuanto a la población general, no se dispone de bastantes
datos sobre la situación real en materia de exposición en los países
en los que se ha introducido el catalizador en los automóviles. Las
concentraciones atmosféricas posibles, calculadas teniendo en cuenta
algunos datos de emisión y modelos de dispersión, son inferiores por
un factor al menos de 10 000 al límite de exposición ocupacional de
1 mg/m3 adoptado por algunos países para el platino metálico como
polvo inhalable total. Puesto que el platino emitido se encuentra
con toda probabilidad en forma metálica, el potencial de
sensibilización de las emisiones de platino a partir de los
catalizadores de los automóviles es probablemente muy bajo. Aunque
parte del platino emitido fuera soluble y potencialmente alergénico,
el margen de seguridad respecto del límite de exposición profesional
para las sales de platino solubles (2 µg/m3) sería de al menos
2000.
En un estudio inmunológico preliminar, se ensayaron extractos
de muestras particuladas procedentes de escapes de automóviles en
tres voluntarios humanos mediante una prueba de punción cutánea. No
se obtuvo respuesta positiva.
No se dispone de datos para evaluar el riesgo carcinogénico del
platino ni de sus sales para el ser humano. En cuanto al cisplatino,
las pruebas disponibles en materia de carcinogenicidad humana se
consideran insuficientes.
8. Efectos en otros organismos en el laboratorio y sobre el terreno
Los complejos simples de platino tienen efectos bactericidas.
El descubrimiento de que los complejos neutros como el cisplatino
inhiben selectivamente la división celular sin reducir el
crecimiento celular de diversidad de bacterias gram-positivas y
especialmente gram-negativas ha llevado a su aplicación en medicina
como agentes antitumorales.
El crecimiento y la cosecha del alga verde Euglena gracilis
fueron inhibidos por el ácido hexacloroplatínico soluble (250, 500 y
750 µg/litro) en un "microcosmos" experimental. El cisplatino
ocasionó clorosis y retraso del crecimiento en el jacinto acuático
Eichhornia crassipes con una concentración de 2,5 mg/litro.
En el invertebrado Daphnia magna, la exposición durante tres
semanas a ácido hexacloroplatínico (H2[PtCl6]), dio un valor de
CL50 de 520 µg Pt por litro. Con concentraciones de 14 y 82
µg/litro, se observaron efectos en la reproducción que se
manifestron en reducciones del 16 y el 50%, respectivamente, del
número total de crías.
Tras la exposición a corto plazo al ácido tetracloroplatínico,
H2[PtCl4], en un bioensayo estático, los valores de la CL50 a
las 24, 48 y 96 horas fueron de 15,5, 5,2 y 2,5 mg Pt/litro,
respectivamente, en el salmón Oncorhynchus kisutch. Con 0,3
mg/litro se observaron efectos en a actividad natatoria general y el
movimiento opercular. Con 0,3 mg/litro o más se observaron lesiones
en las branquias y el órgano olfatorio. No se observaron efectos con
concentraciones de 0,03 y 0,1 mg/litro.
Se han estudiado los efectos del platino en plantas terrestres;
todos ellos se realizaron con cloruros de platino solubles. El ácido
hexacloroplatínico inhibió el crecimiento de las plantas de judías y
tomates en cultivo arenoso con concentraciones de 3 x 10-5 a 15 x
10-5 mol/kg (5,9-29,3 mg/kg). De nueve especies hortícolas
cultivadas en solución hidropónica con tetracloruro de platino,
PtCl4 (0,057, 0,57 y 5,7 mg Pt/litro), se observaron reducciones
significativas del peso seco en el tomate, el pimiento y las hojas
de nabo, así como en las raíces de rábano con la concentración más
elevada. Con esa concentración, los brotes y las hojas inmaduras de
la mayoría de las especies sufrieron clorosis. En algunas de las
especies, con niveles bajos de PtCl4 se observó un efecto del
estímulo del crecimiento. Además, se suprimió la transpiración con
la concentración más elevada de platino, probablemente debido a una
mayor resistencia de los estomas. También se observó estímulo del
crecimiento con niveles reducidos de platino (0,5 mg Pt/litro),
administrado en forma de tetracloroplatinato de potasio
K2[PtCl4], en plantones de la herbácea sudafricana Setaria
verticillata cultivada en solución de nutrientes. Al cabo de dos
semanas, las raíces más largas habían sufrido un aumento de longitud
del 65%. Con la concentración más elevada que se aplicó, es decir
2,5 mg Pt/litro, se observaron efectos fitotóxicos en forma de
retraso del crecimiento radicular y clorosis foliar.
2.3 Conversion factors
Platinum 1 ppm = 7.98 mg/m3
1 mg/m3 = 0.13 ppm
2.4 Analytical methods
2.4.1 Sampling
Samples of ores, minerals, and preconcentrated technical
products can be obtained in a ground or powdered form. Metals and
alloys can be collected as chips and shavings. Platinum on alumina
pellets or monolithic supports must be comminuted before fusing or
digesting (Potter & Lange, 1981). Electronic scrap may contain
alloyed copper, nickel or lead. Melting with aluminium leads to a
brittle alloy, which can be easily crushed to a powder.
Blood samples may be frozen and lyophilized (Pera & Harder,
1977), homogenized with substances like TRITON-X 100(R) (Priesner
et al., 1981), and separated into plasma ultrafiltrate and proteins
(Bannister et al., 1978) or, if appropriate, analysed directly
without pretreatment.
With biological materials, homogeneous sampling is difficult
and often requires destructive methods resulting in the loss of all
information about the platinum species. Only the total content of
platinum and its isotopes can be determined.
For the analysis of platinum in urine, the untreated original
sample is usually unsuitable. Freeze-drying or a wet ashing
procedure with subsequent reduction of volume is necessary for most
analytical methods.
Other biological and environmental materials being investigated
for very low levels of platinum need to be sampled in large amounts,
with possible difficulty in homogenisation, digestion, storage, and
matrix effects.
2.4.2 Sample pretreatment
Determination of total platinum content in some materials
requires a digestion step, which is the pre-requisite for enrichment
and separation from other elements and organic substances. A modern
wet digestion procedure (Knapp, 1985) avoids contact with materials
other than quartz in order to reduce adsorption losses. In this way,
organic matter is destroyed most effectively and contamination with
platinum from other sources is minimized (Würfels et al., 1987).
In general, separation involves volatilization, distillation,
lyophilization, extraction, coprecipitation, flotation, sorption,
and other instrumental methods, such as electro-deposition,
chromatographic separations, and thermal pre-treatment in atomic
absorption spectroscopy (AAS) procedures (Knapp, 1984).
A selection of extraction and sorption techniques is shown in
Tables 3 and 4, respectively. For coprecipitation procedures,
details can be found in the reports of Fryer & Kerrich (1978),
Stockman (1983), Sighinolfi et al. (1984), Skogerboe et al. (1985),
Amosse et al. (1986), and Bankovsky et al. (1987).
2.4.3 Detection and measurement
2.4.3.1 Spectrophotometry
Unless the native soluble platinum compounds have an inherent
absorption spectrum, they can be treated with inorganic and organic
reagents to form coloured, soluble complexes that can be measured by
absorption spectrophotometry. Careful separation from other elements
is important (see section 2.4.2). The detection limits achieved are
in the low mg/kg (ppm) range (Jones et al., 1977; Brajter & Kozicka,
1979; Mojski & Kalinowski, 1980; Marone et al., 1981; Aneva et al.,
1986; Puri et al., 1986).
2.4.3.2 Radiochemical methods
Neutron-activation analysis is a very sensitive method for
determining submicrogram traces of platinum. It is at least one to
several orders of magnitude more sensitive than the best of the
spectrophotometric methods. For the determination of platinum a
sensitivity of 1 ng absolute was estimated on irradiation of a
sample for 1 month at a neutron flux of 10-2cm-2-second,
followed by a 2-h decay (NAS, 1977).
Radiochemical methods have been applied to the analysis of
platinum in various matrices. The detection limits are 1-2 µg/kg in
rock samples (Stockman, 1983), 30 µg per kg dry weight in plant
material (Valente et al., 1982), 1-3 µg/kg dry weight (0.3 ng
absolute) in plant material and animal tissue (Tjioe et al., 1984),
and 100 µg/kg in airborne particulate matter (Schutyser et al.,
1977).
2.4.3.3 X-ray fluorescence spectroscopy
This method permits the highly selective, sensitive, rapid, and
non-destructive analysis of platinum. Zolotov et al. (1983) obtained
a detection limit of 32 µg Pt per litre in aqueous solutions.
A new variant, total-reflection X-ray fluorescence
spectrometry, has the advantage of small sample size (5 to 40 µg)
with low absolute detection limits (Von Bohlen et al. 1987).
Table 3. Extraction procedures for separating platinum
Species Matrix Chemical modifier Extraction Elements Reference
medium separated
Pt(IV) aqueous 6 M HCl isopentanol Al, Ca, Mg, Aneva et al. (1986)
solutions Mn, Ni, Cr
4-methyl-2- Cu, Pb
pentanone (partially)
Pt(IV) aqueous dithio-oxamide tri-butyl Ir(III), Rh(III) Brajter & Kozicka (1979)
solutions phosphate
Pt(IV) plant- S-(1-decyl)- variety of co-extraction Jones et al. (1977)
processing N,N -diphenyl- organic liquids of noble metals
solutions isothiouronium bromide
Pt(IV) palladium(II) 1,5-diphenylthiocarbazone carbon Pd(II) Marczenko & Kus (1987)
chloride tetrachloride
Pt(IV) palladium triphenylphosphine dichloroethane Pd, Au Mojski & Kalinowski (1980)
metal oxide
Pt(IV) synthetic phenanthraquinonemonoxime molten Fe, Cu, Ni, V, Puri et al. (1986)
aqueous naphthalene Cr, Al, Au, Ag
solutions Ir, Rh, Pd
Pt(IV) aqueous potassium butylxanthate carbon - Singh & Garg (1987)
solutions tetrachloride
Pt(IV) automotive bis-(2-furyl)- trichloromethane V, Mo, W Wiele & Kuchenbecker (1974)
catalysts glyoxaldioxime
Pt(II), synthetic 1,4,7,10,13,16-hexa- 4-methyl-2- Fe(III) Arpadjan et al. (1987)
Pt(IV) aqueous azaoctadecane pentanone
solutions
Table 3 (contd).
Species Matrix Chemical modifier Extraction Elements Reference
medium separated
Pt(II) urine Diethylammonium- trichloromethane Ca, Zn, Fe(II) Borch et al. (1979)
diethyldithiocarbamate, and Mn(II)
NaSH
Pt(II) aqueous sodium co-extraction Mueller & Lovett (1987)
solutions diethyldithiocarbamate of Pd(II),
acetonitrile, NaCl Rh(II)
Pt(II) plasma sodium - Andrews et al. (1984)
ultrafiltrate diethyldithiocarbamate
methanol, H2O
Pt geological sodium tetraborate, molten lead - Millard (1987)
samples KCN
Pt geological KCN, KOH Ag, Au co-extraction Le Houillier & De Blois
samples of noble metals (1986)
Pt blood, hair, HCl, SnCl2 tri-n-octylamine, - Tillery & Johnson (1975)
faeces, urine xylene
Pt geological sodium nickel sulfide - Robert et al., (1971)
samples carbonate
and sodium
tetraborate
Table 4. Sorption techniques for preconcentrating platinum
Species Matrix Sorption medium Eluent Elements Reference
separated
Pt sea water Bio-Rad Ag-1-X2 0.1 M HCl, Ir Goldberg et al. (1986);
0.02 M thiourea Hodge et al. (1986)
Pt geological Srafion NMRR 0.01 M HCl, high selectivity Kritsotakis & Tobschall (1985)
samples 5% thiourea for transition
metals
Pt aqueous polyethenimine- Co(II), Zn, Cd, Geckeler et al. (1986)
solutions methylthiourea In(III), Na
suspended in water
at pH 1
Pt(II), aqueous Dowex 2X-8 75% NH3 in H2O Au Kahn & Van Loon (1978)
Pt(IV) solutions
Pt (IV) geological Bio-Rad Ag-50W-X8 0.1 M HCl - Coombes & Chow (1979)
samples
Pt (IV) geological P-TD 2 M HClO4 Al, Mg, Cu, Grote & Kettrup (1987)
samples Fe, Ni, Cr
Pt (IV) aqueous Hyphan 1 M HClO4 Na, K, Cs, Mg, Kenawy et al. (1987)
solutions Ca, Al
Pt (IV) geological Polyorgs digestion HClO4, coextraction Myasoedova et al. (1985)
samples, H2SO4, HNO3 noble metals
scaps
Pt (IV) aqueous (-CH2-S-)n(n approx. 1000) 6 M HCl Co, Ni, Pb, Fe, Zolotov et al. (1983)
solutions Zn, Cd
2.4.3.4 Electron spectroscopy for chemical analysis (ESCA)
ESCA is a technique typically applied in surface analysis
involving a few surface atomic layers (1-2 nm). This technique is
used for special purposes; for instance, Schlögl et al. (1987)
analysed microparticles from automotive exhaust gas catalysts (see
section 3.2.1.4).
2.4.3.5 Electrochemical analysis
Of the voltametric techniques available for element analysis,
polarography, in particular, has been applied for the determination
of platinum. Alexander et al. (1977a,b) described a pulse
polarography method for the analysis of platinum in ores after fire-
assay separation and preconcentration. By measuring the sensitive
catalytic polarographic wave generated by the Pt(II)-ethylenediamine
complex in alkali solutions a detection limit of 0.025 µg per kg was
obtained. A similar technique was applied to the analysis of urine
by Vrana et al. (1983), and the detection limit was 10 µg/litre.
However, these methods do not allow the direct determination of
platinum in complex solutions due to interferences from some heavy
metals and precipitation of platinum with other metals in the form
of their hydroxides. In this respect, inverse voltametry is
superior. Kritsotakis & Tobschall (1985) used the glassy carbon
electrode for the determination of platinum traces in synthetic
solutions. After preconcentration, 0.04 mg Pt/litre could be
determined. This detection limit is sufficient for determining
platinum in ores.
Using adsorptive cathodic stripping voltametry, Van den Berg &
Jacinto (1988) analysed sea-water samples (see section 5.1.2). The
detection limit was 7.8 pg Pt/litre.
Hoppstock et al. (1989) developed a sensitive volta-metric
method for determining platinum in the ng/kg range in biotic and
environmental materials. The overall recovery of platinum was
reported to be 97% or more.
Nygren et al. (1990) described an adsorptive volta-metric
method for the measurement of platinum in blood. The detection limit
for a 100-µl sample was 0.017 µg per litre.
2.4.3.6 Proton-induced X-ray emission (PIXE)
PIXE requires only small sample sizes (1-10 mg), but is a time-
consuming and labour-intensive method. Owing to the substantially
lower background, the detection limits are lower by a factor of 1000
than for X-ray fluorescence methods. Methods for analysing water
samples, air, and biological tissues have been described by Rickey
et al. (1979), Wolfe (1979), and Thompson et al. (1981).
2.4.3.7 Liquid chromatography (LC)
Marsh et al. (1984) published an adsorption chromatography
method in which the analyte was first separated with an ODS
Hypersil(R) column, reacted with NaHSO3, and then detected by UV
absorption. The detection limit for cisplatin was 40-60 µg/litre.
For the malonate derivates, Van der Vijgh et al. (1984) reported a
detection limit of 300-1200 µg/litre for human body fluids.
Ebina et al. (1983) analysed Pt(II) in aqueous solutions that
were modified with EDTA, ethanoic acid, and maleonitriledithiol. The
spectrophotometric detection limit for this partition ion-pair
method was 0.2 ng per litre.
Using an ion exchange chromatography method, Rocklin (1984)
separated Pt(IV) as the hexachlorocomplex on a polar anion exchange
column and determined the complex by UV. For samples digested in
aqua regia, a detection limit of 30 µg/litre can be obtained without
preconcentration and < 1 µg/litre after preconcentration.
2.4.3.8 Atomic absorption spectrometry (AAS)
AAS is a method of high selectivity and specificity and is
often the method of choice in analysing platinum in biological and
environmental samples. However, there are problems with background
radiation deriving from molecules and radicals, especially from
unseparated matrix. These interferences can be partly overcome by
background compensation through a radiation continuum or by the
application of the "Zeeman" effect. To determine platinum in the
range of the detection limit, an accurate separation from matrix is
essential.
For platinum determinations in biological materials, Farago &
Parsons (1982) recommended wet digestion in nitric acid and the
removal of residual nitrates by hydrochloric acid. Brown & Lee
(1986) proposed totally pyrolytic cuvettes for graphite furnace AAS,
thus achieving a greater sensitivity for refractory metals. These
results were confirmed by Schlemmer & Welz (1986). Although platinum
does not form a stable carbide, there was an effect on the wall
material of the carbon rod. Electro-graphite tubes coated with
pyrolytic graphite were found to be superior to glassy carbon tubes
(Welz & Schlemmer, 1987).
LeRoy et al. (1977) described a method for the detection of
platinum in biological samples that used controlled dehydration and
ashing with rapid sample evaporation to detect low levels of
platinum. This method did not suffer as much from matrix
interference as other AAS graphite furnace methods. The method can
be used to detect platinum down to approximately 30 µg/kg (30 ppb).
Hodge et al. (1986) determined platinum down to pg per litre
levels in marine waters, sediments, and organisms. Sea water was
extracted with an anion exchanger (Table 4), eluted, and purified by
acid digestion. In a second step, platinum was obtained from the
solution with an anion exchanger, stripped again from the bead, and
injected. Using a similar technique, Hodge & Stallard (1986)
determined platinum in roadside dust.
Jones (1976) digested urine and blood samples with nitric and
perchloric acids. The samples were diluted after cooling and
injected onto carbon rods. The minimum detectable platinum
concentration in 5-g samples was 30 µg per litre.
McGahan & Tyczkowska (1987) dried and ashed tissues and fluids
and diluted the residue with different acids before direct
injection. The detection limits were 6 µg per kg or 6 µg/litre.
Bannister et al. (1978) separated protein-bound platinum and
free circulating compounds by centrifugal ultra-filtration. In the
ultrafiltrate, platinum compounds were chelated with
ethylenediamine, extracted on a cation exchange paper disc, eluted,
and injected. The minimum working concentration was 35 µg/litre of
plasma.
Alt et al. (1988) described a simple and reliable method which
included high-pressure ashing (cf. Knapp, 1984), separation by
extraction, and detection by graphite furnace AAS. This method was
recommended for analysing biological and other materials down to the
µg/kg range.
König et al. (1989) determined platinum in the particulate
emissions in engine test-stand experiments (see section 3.2.1.4)
using a high-pressure digestion without a separation. The authors
studied the matrix influences with respect to the concomitant
elements and found interferences from A1, Pb, Ca, Zn, P and, most
severely, from Si, but under the controlled test conditions no
interference effects were observed. In particle-free condensates of
automotive exhaust gas, a detection limit of 0.1 ng/ml was achieved
by the method of signal addition described by Berndt et al. (1987).
2.4.3.9 Inductively coupled plasma (ICP)
The generation of plasmas is a further development of chemical
flame methods. They have a wide temperature range, a transparency
for the UV spectral lines, and are predominantly insensitive against
interfering chemical reactions in the excitation zone that occur
with chemical flames. Plasma excitation allows the determination of
several elements simultaneously and is, because of minor matrix
effects, easy to calibrate over many orders of magnitude. Two
methods of generating a plasma are currently used: firstly with
direct current (DC) and secondly with a high frequency current
(20-80 MHz, inductively coupled plasma, ICP). The ICP method works
with an argon plasma and temperatures of 4000-8000 K. Due to the
increasing ionization effects, the aerosol feeding is controlled by
cooling devices.
Boumans & Vrakking (1987) discussed standard values for a 50-
MHz ICP, considering effects of source characteristics, noise, and
spectral band-width, and obtained a detection limit for the platinum
spectral line at 214.42 nm of 7.2 µg/litre.
Maessen et al. (1986) studied the influence of chloroform on
the platinum signal at 203.65 nm. The detection limits by this
method were affected by chloroform and ranged from 30-400 µg/litre.
Wemyss & Scott (1978) determined platinum-group metals and gold
in ores after three different digestions. The method allowed
determination down to 0.13 mg/litre for the 299.8-nm line.
Fox (1984) reported interferences from aluminium and magnesium
in direct current methods. A buffer of lithium and lanthanum
compounds suppressed this effect.
Lo et al. (1987) described a simple method for determining
platinum in urine with a working range down to 50 µg/litre (50 ppb)
under direct application of acidified samples. Electrothermal
vaporization (ETV) was used for generating plasma-suitable aerosols
by Matusiewicz & Barnes (1983). They determined platinum at the
mg/litre level in human body fluids directly. A similar procedure
was used by Belliveau et al. (1986).
2.4.3.10 Inductively coupled plasma - mass spectrometry (ICP-MS)
Combining ICP with a mass spectrometer has new advantages in
analytical spectroscopy. Elemental ions generated from an aerosol or
an electrothermal vaporization unit are separated by a quadrupole
and detected as isotopes at low level. The ETV device allows
determination down to the pg/ml range.
Thompson & Houk (1986) used an ion-pair reversed-phase liquid
chromatography assay via a continuous flow ultrasonic nebulizer and
an ICP torch with a mass spectrometer. In synthetic solutions
detection limits of 7 µg/litre (7 ppb) were obtained.
Gregoire (1988) compared the results from the ICP-MS-ETV with
neutron activation analysis and the ICP-MS solution nebulization
method in the ng/ml concentration range and found good agreement.
For the analysis of air samples, the NIOSH Manual of Analytical
Methods (Eller, 1984a) describes a method based on inductively
coupled argon plasma atomic emission spectroscopy. The working range
is 0.005-2.0 mg/m3 with a 500-litre air sample. However, long
sampling periods are required for measuring soluble platinum
compounds in the workplace and the method does not distinguish
between soluble and insoluble platinum. Similar methods are
recommended for the analysis of platinum in blood and tissues
(Eller, 1984b) and in urine (Eller, 1984c).
The method recommended by the United Kingdom Health and Safety
Executive (1985) has a precision better than 8%, measured as a
coefficient of variation, for samples of a minimum of 120 litres in
the range 1-15 µg Pt/m3. The sensitivity of this method can be
improved by 100-1000 fold by using ICP-MS instead of carbon furnace
atomic absorption spectrometry.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
The six platinum-group metals, platinum, palladium, rhodium,
ruthenium, iridium, and osmium, were probably concentrated mainly in
the iron-nickel core during the earth's formation. This explains
their relatively low presence in the lithosphere (rocky crust) of
the earth (Goldschmidt, 1954) where the average concentration of
platinum ranges between 0.001 and 0.005 mg/kg (Mason, 1966, Bowen,
1979).
Platinum is found both in its metallic form and in a number of
minerals. The principal minerals are: sperrylite, PtAs2;
cooperite, (Pt,Pd)S; and braggite, (Pt,Pd,Ni)S. Primary deposits are
associated with ultrabasic, rather than silicic, rock formations.
Economically important sources exist in the Bushveld Igneous Rock
Complex in Transvaal, Republic of South Africa, and in the Noril'sk
region of Siberia, the Kola Peninsula, and in the Nishnij Tagil
region of the Urals, USSR. The platinum content in these deposits is
between 1 and 500 mg/kg. In the Sudbury district of Canada, platinum
metal is contained in copper-nickel sulfide ores at an average
concentration of 0.3 mg/kg but is concentrated to more than 50 mg/kg
during the refining of copper and nickel. In the USA, there is a
platinum-palladium mine in the Stillwater Complex area, Montana
(NAS, 1977; Renner, 1979).
Small amounts of platinum are also mined from secondary or
placer deposits in the USSR (Ural Mountains), Colombia, USA
(Alaska), Ethiopia, and the Philippines. In these deposits platinum
is present in the form of metallic alloys of varied composition
(NAS, 1977).
3.2 Anthropogenic sources
3.2.1 Production levels and processes
3.2.1.1 World production figures
World mine production of platinum-group metals, 40-50% of which
is platinum, has steadily increased during the last two decades. In
1971 production was 127 tonnes (51-64 tonnes platinum) and in 1972
it was 132 tonnes (53-66 tonnes platinum) (Butterman, 1975). In
1975, automobile exhaust gas catalysts were introduced in the USA in
order to meet the stringent emission limits for carbon monoxide,
hydrocarbons, and nitrogen oxides set by the Federal Clean Air Act.
In Japan, the automobile catalyst was introduced at the same time.
As a consequence, world production of PGM increased to 179 tonnes
(72-90 tonnes platinum) in 1975, reaching a plateau of between 200
and 203 tonnes per year (80-102 tonnes platinum) during the period
1977-1983 (Loebenstein, 1982, 1988).
From 1984 onwards world production increased, apparently in
response to the anticipated demand in Western Europe where
automobiles are being increasingly fitted with catalytic converters.
In 1987, world mine production of PGM amounted to about 270 tonnes
(108-135 tonnes platinum) (Loebenstein, 1988).
The future demand for platinum depends on improvements in
engine technology and emission control, but can be expected to
increase further during the coming years. Data on platinum demand
are presented in section 3.2.2.
3.2.1.2 Manufacturing processes
Most native placer platinum is recovered by dredging and, in
less developed areas, by small hand operations. The copper and
nickel sulfide ores are mined by large-scale underground methods and
concentrated by flotation (Stokinger, 1981).
The isolation of pure platinum metal from raw materials
involves two principal stages: (i) extraction of a concentrate of
precious metals from the ore; (ii) refining the concentrate to
separate the platinum-group metals from each other and purify them.
These processes require sophisticated chemical technology and
include precipitating crystallization and liquid-liquid extraction,
often combined with redox reactions to change the oxidation state of
the metals. Further processes involve halogenation and reduction
reactions at annealing temperatures and special distillations
(Renner, 1984).
Potential health hazards of exposure to soluble platinum salts
are encountered during the later stages of the refining process.
After dissolving platinum, palladium, and gold with aqua regia or
Cl2/HCl and the subsequent precipitation of gold by addition of
ferrous salts, ammonium chloride is added to precipitate ammonium
hexachloroplatinate, (NH4)2[PtCl6]. After several purification
processes there is a second precipitation of this complex salt,
which is then filtered off, dried and finally calcined to yield a
spongy mass of platinum metal having purity of 99.95-99.99%. This
can be further purified by a cationic exchange technique (NAS, 1977;
Stokinger, 1981).
Secondary sources in substantial quantities come from the
reclamation of scrap and used equipment, particularly industrial
catalysts. The recycling of platinum-group metals from automobile
catalysts is also increasing (see section 4.3). In principle, the
recycling of platinum involves the same wet-chemical and melting
processes that are applied to its production from ores (Renner,
1984).
3.2.1.3 Emissions from stationary sources
a) Production
Data on emissions of platinum during production are not
available.
b) Stationary catalysts
During the use of platinum-containing catalysts, platinum can
escape into the environment in variable amounts, depending on the
type of catalyst. Of the stationary catalysts used in industry, only
those employed for ammonia oxidation emit major amounts.
The loss of platinum from ammonia oxidation gauzes during
nitric acid production depends on the operating pressure. An average
figure is 0.15 g/tonne of nitric acid (Sperner & Hohmann, 1976). Of
this apparent loss, 70-85% is recovered on gold-palladium catchment
gauzes, reducing the loss to 0.03 g/tonne (Anon., 1990a). The
production of nitric acid in the USA in 1989 was 7 247 837 tonnes
(Anon., 1990b). Thus the amount of platinum "lost" in 1989 in the
USA is calcu-lated to be 217 kg. This is the maximum amount that
could be dissolved or suspended as a colloid in the nitric acid and,
thus, could be introduced into the environment if the nitric acid is
used in fertilizer production.
3.2.1.4 Emissions from automobile catalysts
Automobile catalysts are mobile sources of platinum. Although
these catalysts are designed to function for 80 000 km or more
(Koberstein, 1984), some loss of platinum can occur due to
mechanical and thermal impact. The data on platinum emissions from
automobile catalysts are very limited.
In the mid 1970s unrealistically high assumptions were made for
platinum loss. Brubaker et al. (1975) estimated the loss to be about
12 µg Pt/km, which would mean a total loss of approximately 1 g
after 80 000 km.
Experimental data show much lower emission rates. Malanchuk et
al. (1974) found a platinum concentration of 0.029 µg/m3 in an
inhalation chamber that was fed by catalysed engine exhaust. On the
basis of the chamber volume, flow rate, and the speed simulated on
the engine test stand, an emission rate of 0.39 µg/km was
calculated. In another US EPA study, Sigsby (1976) did not detect
platinum in particulate exhaust emissions (< 5 µm) at a detection
limit of 0.06 µg/g. In exhaust dilution tunnels, platinum was
detected in larger particles in the range of 0.034 to 635 µg/g
sample; whole or fragmented pellets contained the highest
concentrations.
Reliable emission data for the pellet-type catalyst come from a
study conducted by the General Motors Corporation (Hill & Mayer,
1977), in which emission rates as well as the soluble fraction were
determined by a radio-metric method. Platinum emission was found to
be 0.8 to 1.2 µg per km travelled in low-speed runs (starts and
stops, maximum speed 48 km/h) and 1.9 µg per kilometre travelled in
high-speed runs (96 km/h). It should be noted that these results
relate to the first 250 km of catalyst life. Lower loss rates would
be expected with increasing age of the catalyst. Of the particles
collected, 80% had particle diameters greater than 125 µm.
Experiments with an engine test stand using laboratory prepared
catalysts indicated that about 10% of the platinum emitted is water
soluble. However, the statistical significance of these results was
not reported. Even so, these emission data provide the best basis
for the estimation of expected ambient air concentrations resulting
from the introduction of pellet catalysts (see section 5.1.1).
However, this type of automobile catalyst is no longer used on new
cars in the USA, and has never been used in Europe where only
monolithic catalysts are on the market.
Emission data are available concerning the new generation
monolith-type catalyst. In Germany the Fraunhofer Institute of
Toxicology and Aerosol Research (König et al., 1989, König & Hertel,
1990) has conducted engine test stand experiments as part of a
programme of the Ministry of Research and Technology for assessing
the relative risk of this new man-made environmental source (GSF,
1990). First results indicated that platinum emission is lower by a
factor of 100 than in the case of pelleted catalysts: at a simulated
speed of 100 km/h, total loss from a three-way catalyst was
measured, using the AAS method, to be on average about 17 ng/m3 in
the exhaust gas (König et al., 1989). In further experiments this
value was validated (König & Hertel, 1990): the mean platinum
emission from two catalysts was found to be 12 and 8 ng/m3,
respectively. As shown in Table 5, platinum emission seems to be
temperature dependent. At an exhaust gas temperature of 690° C and a
simulated speed of 140 km/h, about 35-39 ng/m3 was found in the
exhaust gas. The mean aerodynamic diameter of the particles
collected after the muffler (silencer) on a Berner impactor varied
between 4 and 9 µm. Preliminary results indicated that approximately
10% of the total platinum penetrated a depth-type filter to be
trapped in the condensate (König et al., 1989), but this single
measurement could not be confirmed by subsequent determinations
where the platinum content in the condensate was below the detection
limit (0.1 ng/ml) (König & Hertel, 1990).
Schlögl et al. (1987) analysed microparticles emitted from
automobile exhaust and collected on several conducting surfaces. In
experiments with diesel and gasoline engines equipped with
catalysts, they found detectable traces of platinum. In diesel
engine exhaust it was presumed that most platinum would be in the
oxidation state 0 (platinum black). A small part was found to be
Pt(IV), probably in the oxide form. The platinum emission from
gasoline engines showed a photoemission spectrum indicating that
platinum is probably emitted mostly in the form of surface oxidized
particles.
Table 5. Mean platinum emissions from two monolith catalysts (1 and 2)
at different engine test stand runsa
Platinum emission
Simulated Number Exhaust gas Exhaust ng per km Mean aerodynamic
speed of samples temperature gas travelledb diameter (µm)
(km/h) (° C) (ng/m3)
(1) (2) (1) (2) (1) (2)
60 18 480 3 4 2 3 6 9
100 39 600 12 8 10 8 4 6
140 18 690 39 35 39 35 6 8
a Adapted from König et al. (in press)
b Calculated assuming that on average 10 m3 exhaust gas is emitted per litre
gasoline and a gasoline kilometrage of 7, 8, and 10 litres per 100 km travelled,
respectively.
3.2.2 Uses
The principal use of platinum derives from its special
catalytic properties. Further applications in industry are related
to other outstanding properties, particularly resistance to chemical
corrosion over a wide temperature range, high melting point, high
mechanical strength, and good ductility. Platinum has long been
known to have excellent catalytic properties. Before the
introduction of catalytic converters in automobiles, most of the
platinum was used as a catalyst in hydrogenation, dehydrogenation,
isomerization, cyclization, dehydration, dehalogenation, and
oxidation reactions. One of its major industrial uses is for
naphtha-reforming to upgrade catalytically the octane rating of
gasoline. Other catalytic uses are in ammonia oxidation to produce
nitric acid, hydrogen cyanide manufacture, the reduction of nitro
groups and, in the automobile catalyst application, the conversion
of carbon monoxide to carbon dioxide and nitrogen oxide to nitrogen
and water (NAS, 1977; Stokinger, 1981).
As shown in Table 6, in the USA in 1973, before the
introduction of the automobile catalyst, most of the platinum was
used for catalytic purposes in the chemical and petroleum industry.
In 1987 the use pattern had completely changed and 71% of the
platinum sold was used by the automobile industry. In 1987, a
typical USA car catalyst contained about 1.77 g of platinum and 10.6
million vehicles with catalysts were produced (Loebenstein, 1988);
this accounts for the 18.8 tonnes shown in Table 6.
Table 6. Platinum sales to various types of industry in the USA
before and after the introduction of automotive catalytic
convertersa
Industry 1973 1987
kg/year % of total kg/year % of total
Automobile - - 18 817 71.3
Chemical 7434 36.3 1920 7.5
Petroleum 3844 18.8 739 2.8
Dental and
medical 868 4.2 479 1.9
Electrical 3642 17.9 1821 7.1
Glass 2255 11.0 285 1.1
Jewellery and
decorative 697 3.4 177 0.7
Miscellaneous 1732 8.5 1430 5.6
Total 20 472 100 25 668 100
a From: Butterman (1975); Loebenstein (1988)
Tables 7 and 8 show the platinum demand by application in the
Western world, also reflecting the increased demand during recent
years. In 1989, total demand was 90 tonnes.
Platinum oxidation catalyst technology, developed to reduce
automobile exhaust emissions, has been extended to other
environmental control applications such as the reduction of carbon
monoxide and hydrocarbon emissions from large gas turbines (Jung &
Becker, 1987) and the transformation of hydrogen molecules into
active hydrogen atoms to reduce chlorohydrocarbons such as
trichloroethylene to ethane in water (Wang & Tan, 1987).
Table 7. Western-world platinum demand (kg/year) by applicationa
1980 1981 1982 1983 1984 1985 1986 1987 1988 1989
Automobile catalyst
gross 19 278 18 144 18 569 18 285 23 814 27 783 32 318 35 579 37 563 41 107
recovery 0 0 283 850 1276 1984 2551 3260 4536 4961
Chemical 7371 7087 7371 6946 7371 6379 5528 5528 4536 4536
Electrical 5953 5245 4819 4961 5386 5670 5103 5103 5245 5528
Glass 3969 2835 2410 2977 3969 3969 2551 3402 3685 3969
Investment
small 0 0 1276 2551 4819 7371 12 757 6095 9355 3685
large 4536 5528 3260 1843 4252 4819 3544 7796 8505 850
Jewellery 15 876 21 404 21 687 20 270 21 971 22 963 24 097 28 066 33 452 36 996
Petroleum 3685 3969 1843 567 425 425 567 1559 1417 2126
Other 5386 4678 4819 4252 3827 2835 3685 3402 3402 3260
Total 66 054 68 889 65 771 61 802 74 559 80 230 80 511 93 270 102 624 97 096
a From Johnson Matthey (1990)
Table 8. Regional platinum demand (kg/year) by applicationa
1980 1981 1982 1983 1984 1985 1986 1987 1988 1989
Japan
Automobile catalyst
grossb 5953 5386 4819 4819 4819 5953 7229 8788 9355 10 064
recoveryc 0 0 0 0 0 0 142 425 709 709
Chemical 283 283 283 283 425 425 425 425 425 425
Electrical 425 425 567 567 50 1134 1276 1276 1276 1417
Glass 1134 1417 1276 1701 2126 1701 850 1276 1276 1134
Investment
small 0 0 0 142 425 992 992 1701 3260 992
large 4536 5528 3260 1843 4252 4819 3544 7796 8505 850
Jewellery 12 474 17 718 17 577 15 876 17 718 19 136 20 979 25 515 30 050 32 602
Petroleum 425 425 425 425 567 425 0 0 0 0
Other 1417 1417 1559 1276 1134 850 567 425 425 425
Total 26 647 32 599 29 766 26 932 32 316 35 435 28 632 46 777 53 863 47 200
North America
Automobile catalyst
gross 12 474 12 190 12 899 12 757 18 002 19 845 21 120 19 561 19 561 20 412
recovery 0 0 0 850 1276 1984 2410 2835 3827 4252
Chemical 3260 1417 2268 2835 2835 2126 1843 1559 1559 1559
Electrical 4111 1984 1984 2551 2693 2268 1843 1843 1843 2126
Glass 1417 567 283 425 850 1134 709 709 709 850
Investment 0 0 1134 1134 850 3685 8505 2410 2410 1559
Jewellery 425 425 425 425 425 425 425 425 425 567
Petroleum 3969 1559 567 425 425 283 283 425 425 1134
Other 2126 1701 567 709 992 850 1417 1417 1417 1417
Total 27 782 19 843 20 127 20 411 25 796 28 632 33 735 25 514 24 522 25 372
Table 8 (contd).
1980 1981 1982 1983 1984 1985 1986 1987 1988 1989
Rest of Western world, including Europe
Automobile catalyst
gross 850 567 567 709 992 1984 3969 7229 8647 10 631
recovery 0 0 0 0 0 0 0 0 0 0
Chemical 3827 5386 4819 3827 4111 3827 3260 3544 2551 2551
Electrical 1417 2835 2268 1843 1843 2268 1984 1984 1984 1984
Glass 1417 850 850 850 992 1134 992 1417 1701 1984
Investment 0 0 142 1276 3544 2693 3260 1984 3685 1134
Jewellery 2977 3260 3685 3969 3827 3402 2693 2126 2977 3827
Petroleum 709 1984 850 283 567 283 283 1134 992 992
Other 1843 1559 2693 2268 1701 1134 1701 1559 1559 1417
Total 11 622 16 441 15 874 14 459 16 443 16 159 18 142 20 977 24 096 24 520
a From: Johnson Matthey (1990)
b Gross automobile catalyst demand is purchase of platinum by the auto industry for the manufacture of automobile catalysts.
c Automobile catalyst recovery is platinum recovered from catalytic converters removed from scrapped automobiles.
Platinum and platinum-rhodium alloys have many high-temperature
uses. Thermo-electrical applications arise from the simple and
stable relationship between resistance and temperature that platinum
exhibits over a wide temperature range. This explains its use in
platinum resistance thermometers, thermocouples, and strain gauges.
The high melting point of platinum and its resistance to oxidation
and many chemicals has led to its use in vessels in the glass-making
industry and in the fabrication of spinning jets and bushings for
the production of viscose rayon and fibreglass, respectively. It is
also used for laboratory ware, such as crucibles, combustion boats,
and the tips of tongs. Ships' hulls, propellers, and rudders are
protected against corrosion by "cathodic protection" using platinum-
clad anodes (NAS, 1977).
Platinum and/or its alloys have been used in electric contacts
for relays and switchgears for a variety of reasons, including
hardness and good conductivity. Many printed circuits are made using
preparations that contain platinum. Electrochemical platinum
electrodes have been used in preparative chemistry, since they
support many oxidative reactions although they resist oxidation
themselves (NAS, 1977).
A major use of platinum is in jewellery for making rings and
settings. Platinum is also used to produce a silvery lustre on
ceramic glazes (NAS, 1977).
In dentistry, platinum is used in gold-platinum-palladium
alloys to raise the melting-point range and increase the strength.
However, this use is decreasing, since platinum is being replaced by
other materials including palladium (Anusavice, 1985; NAS, 1977).
Platinum has an important role in neurological prostheses, i.e.
surgically implanted microelectronic devices, such as implants for
treating incontinence, or for recovering some use of paralysed limbs
following spinal accidents (Donaldson, 1987).
Platinum-iridium electrodes are used for long-term electrode
implantation for recording electrical activity and for stimulation
in human tissues and organs, e.g., pacemakers (Theopold et al.,
1981).
All these applications use platinum as a pure metal or in the
form of alloys, but soluble platinum salts are also used in the
manufacture of these products; e.g., hexachloroplatinic acid may be
used in platinizing alumina or charcoal in catalyst production. A
number of salts can be used in the electrodeposition of platinum,
e.g., sodium hexahydroxyplatinate(IV), Na2[Pt(OH)6].2H2O,
diamminedinitroplatinum(II), [Pt(NO2)2(NH3)2], hydrogen
dinitrosulfatoplatinate(II), H2[Pt(NO2)2SO4], and
tetraammineplatinum(II) compounds such as the hydrogenphosphate,
sulfamate, citrate, and tartrate (Baumgärtner & Raub, 1988; Skinner,
1989).
Complexes of platinum, particularly cis-
diamminedichloroplatinum(II) (cisplatin) (see footnote in section
1.2), have been used to treat cancer. In patients with testicular
cancers, remissions rates of more than 90% have been achieved
(Lippert & Beck, 1983).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1 Transport and distribution between media
By comparison with other elements, platinum-group metals are
distributed sparsely in the environment. Since platinum is so
valuable, great care is taken to avoid significant loss during
mining and refining processes, and during use and disposal of used
platinum-containing objects. Up to 1984, about 1050 tonnes of
platinum had been refined. Most of this has been used in the form of
the metal and platinum oxides, which are practically insoluble in
water, resistant to most chemical reactions in the biosphere, and do
not volatilize into air (Renner, 1984).
Part of the platinum released into the air from automobile
emissions (section 5) is deposited close to the roads and could be
washed off by rain into rivers and coastal marine waters (Hodge &
Stallard, 1986). However, only small amounts of platinum have been
detected in environmental samples (see sections 5.1.2. and 5.1.3.).
Large amounts of metals including platinum can be transported
in rivers draining major industrialized regions, leading to elevated
platinum concentrations in sediments (section 5.1.3).
Platinum forms soluble complexes with ammonia, cyanide, amines,
olefins, organic sulfides, and tertiary arsines. However, the level
of these ligands in natural waters is insufficient to make platinum
mobile (Fuchs & Rose, 1974).
Organic matter has a role as a vehicle for the transport of
platinum and for bringing about its precipitation or concentration.
There is a good correlation between high contents of platinum and
organic carbon in polluted stream sediments of the Ginsheimer-
Altrhine river, near Mainz, Germany (see section 5.1.2), and it is
assumed that organic matter such as humic and fulvic acids binds
platinum, aided perhaps by appropriate pH and redox potential
conditions in the aquatic environment (Dissanayake, 1983).
Detailed information about the geochemical behaviour of
platinum-group metals is available from the platinum mining area of
Stillwater, Montana, USA (Fuchs & Rose, 1974). The mobility of
platinum depends on pH, the redox potential, chloride concentrations
in soil water, and the mode of occurrence of platinum in the primary
rock. The relation between redox potentials and pH conditions
indicates that platinum behaviour also depends on the kind of ore it
is associated with. If bound in chromite, it has essentially no
mobility in weathering because of the resistant character of
chromite. On the other hand, platinum in the form of trace mineral
inclusions in sulfides is readily released by oxidation during
weathering. Calculated relations between pH and redox potential
indicate that increased chloride concentrations in soil water will
promote mobility. Thus, platinum will be mobile only in extremely
acid waters or those with a high chloride level (Fuchs & Rose,
1974).
In twigs from four limber pines (Pinus flexilis) in the
platinum mining area of Stillwater, the platinum concentrations were
the same as in the adjacent soil. It was concluded that limber pine
does not concentrate platinum, probably due to the limited mobility
of platinum (Fuchs & Rose, 1974). However, high concentrations of
platinum were found in the roots of nine horticultural crops
(cauliflower, radish, snapbean, sweet corn, pea, tomato, bell
pepper, broccoli, and turnip) grown in Hoagland's hydroponic culture
solution containing platinum tetrachloride concentrations of 0.057,
0.57, or 5.7 mg/litre (Pallas & Jones, 1978; see section 7.3). For
example, at the highest concentration, cauliflower and tomato roots
contained 1425 and 1710 mg Pt/kg, respectively. Only pepper,
cauliflower, and radish accumulated platinum in their tops, but to a
very limited extent. From the data of Pallas & Jones (1978) it is
not clear whether they differentiated between contamination of the
root surface and true uptake of platinum. However, these results
indicate that platinum can enter food crops but the bioavailability
essentially depends on the solubility of the platinum species. It
should be noted that the salt (PtCl4) used by Pallas & Jones
(1978) is soluble in water.
In the context of a German government programme (see section
3.2.1.4), Rosner et al. (1991) conducted engine test stand
experiments with a three-way-catalyst-equipped engine (monolith-type
catalyst) to determine platinum uptake by plants. Grass cultures
(Lolium multiflorum) were placed in continuously stirred tank
reactors and exposed to slightly diluted (1:10/20) exhaust gas for 4
weeks (8 h/day, 5 days/week). Using atomic absorption spectrometry
for the measurement of platinum emissions (see section 2.4.3.8,
König & Hertel, 1990), no platinum could be detected in the shoots
at a detection limit of 2 ng/g dry weight.
4.2 Biotransformation
By analogy, platinum compounds may undergo biotransformation
comparable to processes described for other metals. The
biomethylation of platinum compounds, i.e. [Pt(IV)Cl6]2-,
[Pt(IV)(CN)4Cl2]2-, [Pt(IV)(CN)5Cl]2-, and
[Pt(IV)(SO4)2], has been established only in in vitro test
systems (Taylor, 1976; Wood et al., 1978; Fanchiang et al., 1979;
Taylor et al., 1979; Fanchiang, 1985).
Methylcobalamin (MeB12) reacts with Pt(II) and Pt(IV)
complexes to give a methylated platinum compound. Agnes et al.
(1971) reported that this reaction requires the presence of platinum
in both oxidation states. Spectrophotometric measurements showed the
consumption of one mole of [Pt(IV)Cl6]2- per mole of MeB12,
[Pt(II)Cl4]2- being required only in catalytic quantities.
Aquocobalamin (aquo-B12) and methylplatinum were shown to be the
products of the reaction (Taylor & Hanna, 1977).
From these laboratory data produced under abiotic conditions it
is not, however, possible to conclude that microorganisms in the
environment are able to biomethylate platinum complexes.
4.3 Ultimate fate following use
The value of platinum-group metals has greatly increased and
methods for their recovery from spent catalysts are of economic
importance.
Platinum metal has been successfully recycled from used
chemical and petroleum catalysts for many years, but many companies
are still trying to find a successful formula for retrieving it from
automobile catalysts. The latter accounts for more than 30% of the
total platinum-group metal consumption in the USA. The US Office of
Technology calculated that if 50-60% of catalytic converters were
recovered for their metal value, about 7717 kg platinum per year
could be reclaimed in 1990. However, currently only between 25 to
40% of the used converters are being reclaimed (Agoos, 1986).
According to another estimate, 5443 kg of platinum was recovered in
1989 from automobile catalysts, of which 4666 kg was recovered in
the USA (Johnson Matthey, 1990).
In contrast to automobile catalysts, almost 100% of spent
reforming and gauze catalysts are collected for their metal value.
This is based on their much higher platinum metal content (Agoos,
1986).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Ambient air
Few measurements of platinum ambient air concentrations have
been reported. Results obtained before the introduction of cars with
catalytic converters can serve as a baseline. Air samples taken near
freeways in California, USA, and analysed using atomic absorption
spectrometry were below the detection limit of 0.05 pg/m3 (Johnson
et al., 1975; 1976).
No platinum could be detected in two air samples collected by
Ito & Kidani (1982) in an industrial area of Nagoya, Japan, in 1981.
Close to city roads in Frankfurt, Langenbrügge, Germany, the
platinum air concentrations (particulate samples) were measured in
1989 to be between < 1 and 13 pg/m3. In rural areas the
concentrations were < 0.6-1.8 pg/m3 (Tölg & Alt, 1990). At the
time of these measurements, few German cars were equipped with
catalysts. Thus, these levels virtually reflect background levels.
Rosner & Hertel (1986) estimated ambient air concentrations for
different scenarios, based on dispersion models used by US EPA
(Ingalls & Garbe, 1982) and on the emission data of Hill & Mayer
(1977) (see section 3.2.1.4). As shown in Table 9, total platinum
concentrations near and on roads could range from 0.005 to 9
ng/m3. Estimates for parking and personal garages were also made,
based on an assumed emission rate of 1 µg/min for total platinum,
but this is definitely an overestimate. It can be assumed that the
emission of platinum depends on the exhaust gas temperature. At
idling or very low speed conditions, emissions are expected to be
negligible (see section 3.2.1.4).
As described in section 3.2.1.4, emission data indicate that
the total platinum emission of a monolith-type catalyst is probably
lower by a factor of 100 than that of a pellet-type catalyst.
Assuming an average emission rate of approximately 20 ng/km (see
section 3.2.1.4) and applying the same dispersion models, the
theoretical ambient air concentrations would be lowered to the
picogram to femtogram per m3 range (see Table 9).
Table 9. Estimated ambient air concentrations of total platinum
at various exposure conditions, based on an emission rate
of 2 µg/km from the pelleted catalyst and 0.02 µg/km
from the monolithic three-way catalyst
Exposure situationa Ambient Pt concentration(ng/m3)
Pelleted Monolithic
catalyst catalyst
Roadway tunnel
Typical 4 0.04
Severe 9 0.09
Street canyon (sidewalk receptor)
Typical a) 800 vehicles per h 0.1 0.001
Typical b) 1600 vehicles per h 0.3 0.003
Severe a) 1200 vehicles per h 0.5 0.005
Severe b) 2400 vehicles per h 0.9 0.009
On expressway
Typical 0.7 0.007
Severe 1.6 0.016
Beside expressway (short-term)
Severe 1 m 1.3 0.013
10 m 1.1 0.011
100 m 0.3 0.003
1000 m 0.04 0.0004
Beside expressway (annual)
Severe 1 m 0.2 0.002
10 m 0.15 0.0015
100 m 0.04 0.0004
1000 m 0.005 0.00005
a Calculations based on dispersion models used by US EPA;
"Typical/severe" depends on wind conditions and road width
(Ingalls & Garbe, 1982)
Hodge & Stallard (1986) analysed roadside dust deposited in San
Diego, California, USA. At the edge of a major freeway (154 000
vehicles/day), dust samples contained the highest concentration (680
µg Pt/kg dry weight; 680 ppb). At a distance of about 34 m, the
platinum content of 100 µg/kg was about 7 times lower. At the edge
of another heavily used freeway (96 000 vehicles/day) platinum
content was 250 µg/kg, while with less heavy traffic (14 000
vehicles/day) 260 and 300 µg/kg were found in two dust samples. The
lowest concentrations, 37 and 60 µg/kg, were found in samples
collected from plants growing in the yards of houses located on
highly used road. The platinum concentration was not correlated with
the lead concentration. However, the samples with the highest
platinum concentrations also had the highest lead values. Although
the number of samples was limited, the results indicate that
automobile catalysts release platinum. However, it should be noted
that platinum emissions from pelleted catalysts were probably
responsible for the concentrations reported and that the use of
monolith catalysts should result in much lower platinum
concentrations in the roadside environment.
5.1.2 Water and sediments
In a study to determine baseline levels of platinum, Johnson et
al. (1976) analysed tap water samples collected in Lancaster and Los
Angeles, California, USA. No platinum was found at a detection limit
of 0.08 µg/litre. In tap water (probably only one sample) from
Liverpool, United Kingdom, a platinum content of 0.06 µg/litre was
determined by adsorptive cathodic stripping voltametry (Van den Berg
& Jacinto, 1988).
Investigations of platinum concentrations in Lake Michigan
sediments led to the conclusion that platinum has been deposited
over the past 50 years at a constant rate. Concentrations at
sediment depths of 1-20 cm varied between 0.3 and 0.43 µg/kg dry
weight (Goldberg et al., 1981). In comparison, lead concentrations
have markedly increased in the sediment due to increased emissions
from industry and motor traffic.
Lee (1983) noted a rapid increase in the palladium contents of
the sediments from the Palace Moat, Tokyo, Japan, between 1948 and
1973 and attributed it to the introduction of car catalysts.
However, this is not conclusive as the palladium content in the
sediment had already begun to increase in 1964-1965, before the
introduction of the catalytic converter, and even in 1973 only a few
cars were equipped with converters.
Dissanayake et al. (1984) determined platinum concentrations in
the sediments of a cut-off channel of the Rhine river near Mainz,
Germany. Sediment samples from this highly polluted river were
sieved and the < 2 µm fraction was analysed by flameless AAS. The
platinum concentrations in 12 samples collected at different sites
varied over a wide range. In four samples no platinum was detected,
while eight samples contained between 730 and 31 220 µg/kg (dry
weight). This is higher by a factor of up to 15 000 compared to
unpolluted average North Sea sediments. The high variation was
attributed to differences in pH and redox conditions. The extremely
high concentrations appeared at the interface between an extremely
reducing and an oxidizing aquatic environment that provided,
together with a pH of 6.6-7.8, optimum conditions for the formation
of metal-organic complexes. The sample containing 31 220 µg Pt/kg
also contained the highest concentration of palladium (4000 µg/kg).
The gold content (100-400 µg/kg) had a relatively uniform
distribution, but was also indicative of a high state of pollution.
Using a more sensitive graphite furnace AAS method, Goldberg et
al. (1986) detected very low platinum concentrations in sea water.
Samples of filtered water (0.45-µm filter) from the open Eastern
Pacific Ocean showed an increase in platinum concentration with
depth from surface values of around 100 to a value of 250 pg/litre
at 4500 m. Similar concentration profiles were obtained in
unfiltered sea water taken from the California Borderline region
(Hodge et al., 1985). Sea-water samples analysed by Van den Berg &
Jacinto (1988) were also within this concentration range. A deep-sea
and a shallow-water sample from the Indian Ocean contained 154 and
37 pg/litre, respectively, whereas sea water of coastal origin
contained 332 pg/litre. It should be noted that these were only
single samples.
In sediment cores from the Eastern Pacific taken to a depth of
6-22 cm in carbonate and siliceous ooze, platinum concentrations
varied between 1.1 and 3 µg/kg (dry weight basis). Lower
concentrations (0.3 µg/kg) were reported in the Santa Barbara Basin
(Hodge et al., 1985). The highest concentration (21.9 µg/kg) was
found in pelagic ocean sediments (Hodge et al., 1986).
In several investigations, the platinum content of seamount
ferromanganese nodules or crusts was studied. In deep-sea nodules
from the Northwest Pacific nodule belt, platinum concentrations from
< 5 to 145 µg/kg were found (Agiorgitis & Gundlach, 1978).
Platinum values in ferromanganese seamount crusts from the
Central Pacific were much higher and varied between 140 µg/kg at
3780 m and a maximum of 880 µg/kg at a depth of 1120 m (Halbach et
al., 1984). Both platinum and nickel concentrations correlated
positively with manganese content and led to the conclusion that
platinum and nickel are incorporated in the manganese oxide
fraction. It was suggested that the high platinum concentration in
the crusts is derived directly from sea water by a process of
specific adsorption onto colloidal particles of hydrous manganese
oxide, which has a negative surface charge in sea water.
In a further investigation, platinum concentrations in
ferromanganese minerals from various localities were found to vary
between 6 and 940 µg/kg (Goldberg et al., 1986). In manganese
nodules obtained at depths of between 1700 and 4200 m in the Pacific
Ocean, platinum concentrations varied between 138 and 940 µg/kg
(Hodge et al., 1986).
5.1.3 Soil
Few measurements of platinum in soil have been reported. In the
baseline study of Johnson et al. (1976), all surface soil samples
collected near freeways in California, USA, and in a mining area in
Sudbury, Canada, were below the detection limit of 0.8 µg/kg.
In the USA, the National Academy of Sciences (NAS, 1977)
estimated the accumulation of platinum in roadside environments on
the basis of an emission rate of 1.9 µg per km from cars equipped
with catalytic converters and a frequency of 5000 cars per day.
Assuming that all emitted platinum was localized near the freeway in
the topsoil (uniformly distributed about 30 cm deep over a width of
about 90 m and a length of 1.6 km, with a soil density of 1.5
g/cm3), a platinum concentration after 10 years of 8 µg/kg could
be expected.
5.1.4 Food
Hamilton & Minski (1972/1973) estimated a total daily platinum
intake of less than 1 µg/day, based on an analysis of a United
Kingdom total-diet sample and 1963 United Kingdom consumption and
population figures. No data were given on the platinum content of
the foods analysed.
5.1.5 Terrestrial and aquatic organisms
Fuchs & Rose (1974) analysed samples of twigs from four limber
pines (Pinus flexilis) in the Stillwater mining area, Montana,
USA. Three samples contained between 12 and 56 µg Pt/kg (ash
weight), while one contained platinum at a level below the detection
limit. The content of the adjacent soils was also in this range, so
that no evidence for accumulation could be derived from these
limited data (see also section 4.1).
Using neutron activation analysis (section 2.4.3.2) Valente et
al. (1982) measured the following platinum concentrations in
isolated samples of plants from an ultrabasic soil: Fragaria
virginiana, 830 µg/kg (dry weight); Prunella vulgaris, 440
µg/kg; Aspidotis densa, 100 µg/kg.
In marine macroalgae the following platinum concentrations (on
a dry weight basis) were found near La Jolla, California, USA (Hodge
et al., 1986): red algae Prionites australis and Opuntiella
californica, 0.19 and 0.08 µg/kg, respectively; brown algae
Macrocystis pyrifera and Pterygophora californica, 0.22 and 0.32
µg/kg, respectively.
5.2 General population exposure
Two studies were conducted in the USA to establish baseline
levels of platinum in the tissues and body fluids of the general
population prior to the introduction of automobile catalysts.
Johnson et al. (1975, 1976) analysed autopsy tissue samples
from 10 people, 12 to 75 years old, who died from a variety of
causes in Southern California. All samples taken from liver, kidney,
spleen, lung, muscle, and fat were below the detection limits
(0.2-2.6 µg/kg wet weight). Samples collected from 282 people from
Southern California living near a heavily used urban freeway (Los
Angeles) or in a desert area near Lancaster also showed platinum
concentrations below the detection limits (blood, < 31 µg/litre;
urine, < 0.6 µg/litre; hair, < 50 µg per kg; faeces, < 2 µg/kg).
Only in pooled blood samples were detectable concentrations
measured, i.e. 0.49 µg/litre in the Los Angeles group and 1.8
µg/litre in the Lancaster group.
In a second study, tissue samples were taken from autopsied
individuals from Southern California (95 people) and New York (2
people), who had not been knowingly exposed to platinum either
occupationally or by medical treatment (Duffield et al., 1976). In
42 individuals no platinum was detected. Of the 1313 samples
collected, only 62, i.e. 5%, had detectable concentrations of
platinum ranging from 0.003 to 1.46 mg/kg wet weight (mean 0.16
mg/kg, median 0.067 mg/kg). Table 10 shows the frequency of platinum
detection in the various tissue samples. The frequency of occurrence
was taken as a measure of the distribution of platinum among various
body organs. Platinum was frequently found in subcutaneous fat. This
is surprising, as most platinum compounds are regarded as lipid-
insoluble. Other target sites were kidney, pancreas, and liver.
However, the analytical accuracy has been questioned and
contamination of the samples suspected (NAS, 1977), because the
baseline levels found by Johnson et al. (1976) were at least one
order of magnitude lower. The problem of questionable analytical
reliability reflects the difficulties in interpreting data on trace
levels of platinum in the environment and in human tissues and body
fluids.
New data have been provided by Nygren et al. (1990). Using
absorptive voltametry (see section 2.4.3.5), the background levels
of platinum in human blood were found to be in the range of 0.1-2.8
µg/litre (median 0.6 µg per litre). These results were verified by
inductively coupled plasma mass spectrometry using gold as an
internal standard.
5.3 Occupational exposure during manufacture, formulation, or use
Occupational exposure occurs during the mining and processing
of platinum. However, the most common current occupational exposure
to soluble platinum compounds is through inhalation in platinum
refining and catalyst manufacture.
Table 10. Distribution of tissue samples with detectable platinuma
Number of Samples with detectable
samples platinum
analysed No. %
Subcutaneous fat 74 10 14
Kidney 91 11 12
Pancreas 84 10 12
Liver 90 10 11
Brain 9 1 11
Gonad 53 5 9
Adrenal 60 3 5
Muscle (psoas) 97 4 4
Aorta (descending) 92 3 3
Heart (left ventricle) 82 2 2
Spleen 52 1 2
Prostate/uterus 63 1 2
Thyroid 73 1 1
Lung 95 0 0
Vertebra (lumbar) 94 0 0
Rib (fifth) 97 0 0
Femur 57 0 0
Clavicle 30 0 0
Hair, scalp 9 0 0
Hair, pubic 1 0 0
1303 62 5
a From: Duffield et al. (1976)
Many countries have set occupational exposure limits. For
example, in the USA, the time-weighted Threshold Limit Value (TWA-
TLV) for daily occupational exposure has been established for
soluble platinum salts at 2 µg Pt/m3 (ACGIH, 1980, 1990). Many
countries have adopted this ACGIH value. In addition ACGIH (1980,
1990) recommended a Threshold Limit Value of 1 mg/m3 for platinum
metal. In the United Kingdom an occupational exposure limit (8-h
TWA) of 5 mg/m3 has been proposed for platinum metal as total
inhalable dust (Health and Safety Executive, 1990).
The published data base for platinum concentrations at the
workplace is meagre. Due to analytical shortcomings older data are
not considered reliable. In an early investigation (Fothergill et
al., 1945), a platinum content of less than 5 µg/m3 in the
atmosphere in the immediate neighbourhood of a refinery was measured
using particle filters. In the dry salts handling area, platinum
concentrations as high as 70 µg/m3 were found. In another
investigation (Hunter et al., 1945), the platinum content in the
atmosphere at various points in four refineries was estimated. At
most points concentrations varied between 1.6 and 5 µg/m3. Higher
concentrations were found in the neutralization of platinum salts
(20 µg/m3), sieving spongy platinum (400-900 µg/m3), and
crushing ammonium chloroplatinate (1700 µg/m3).
Workplace measurements in a catalyst production plant in the
USSR were reported to exceed an air concentration of 2 µg/m3 in
33% of the measurements (Gladkova et al., 1974).
In a cross-sectional survey (section 9.2), Bolm-Audorff et al.
(1988) reported workplace measurements at a platinum refinery in the
Federal Republic of Germany. In 1986, concentrations of between 0.08
and 0.1 µg/m3 were measured in the filter press area, but in other
working areas platinum salt exposure was generally below the
detection limit of 0.05 µg/m3. No data were given on the number of
samples.
The results obtained during a four-month period of measurements
in a US platinum refinery showed that workplace concentrations
exceeded the occupational limit of 2 µg/m3 between 50 and 75% of
the time (Brooks et al., 1990.
In samples of blood, urine, faeces, and hair from employees at
a Canadian mine near Sudbury, platinum concentrations were below the
limits of detection (0.1 µg per litre or 0.1 µg/kg). Tissue samples
from three out of nine autopsies had detectable platinum
concentrations in fat (4.5 µg/kg), lung (3.7 µg/kg), or muscle (25.0
µg per kg) (Johnson et al., 1976). However, since the three
detectable concentrations were in individuals who, like the other
six, showed no platinum concentrations in liver, kidney and spleen,
sample contamination was suggested (NAS, 1977). It was concluded
that people who work in mining areas probably do not incorporate
significant amounts of platinum into their body.
Blood samples collected from 61 refinery workers in New Jersey
contained no measurable platinum (less than 1.4 µg/litre) (Johnson
et al., 1976). However, platinum levels in 10% of the urine samples
were above the detection limit of 0.1 µg/litre, the maximum reported
value being 2.6 µg/litre.
Using the method of LeRoy et al. (1977), platinum serum levels
in 11 platinum refinery workers with positive skin tests were
analysed. These studies found serum platinum levels ranging from 150
to 440 µg/litre (mean = 240 µg/litre), the quantification limit
being 100 µg per litre (Biagini et al., 1985).
A special case of possible occupational exposure is the
handling of cisplatin and its analogues by pharmacy and nursing
staff and other hospital personnel. In a study with two pharmacists
(one male and one female) and eight female nurses, platinum levels
in urine (0.6-23.1 µg per litre) were at the limit of sensitivity of
the AAS method used and did not significantly differ from the
controls (2.6-15.0 µg/litre). By comparison, the urine of cisplatin-
treated patients contained on average 7 mg/litre (Venitt et al.
1984).
6. KINETICS AND METABOLISM
Most toxicokinetic data on platinum, both for experimental
animals and humans, have been derived from studies with platinum
complexes.
Moore et al. (1975c) studied the whole body retention, lung
clearance, distribution, and excretion of 191Pt in outbred albino
rats (Charles River CD-1 strain) after single nose-only inhalation
exposure to different chemical forms of platinum for 48 min.
Particle concentration in the nose-only exposure chambers was
approximately 5.0 mg per m3 with 191PtCl4, 5-7 mg/m3 with
191Pt(SO4)2, and 7-8 mg/m3 with PtO2 and 191Pt metal.
The aerodynamic diameter was given as 1.0 µm for 191PtCl4 and
191Pt(SO4)2; both aerosols were generated by a nebulizer. The
191PtO2 and 191Pt metal aerosols (aerodynamic diameter not
given) were generated by passing Pt(SO4)2 or PtCl4,
respectively, through a furnace tube and decomposing them at 600 °C.
Whole body counts, showed that most of the inhaled 191Pt was
rapidly cleared from the body, followed by a slower clearance phase
during the remaining post-exposure period. The whole body retention
of 191Pt was approximately 41, 33, 31, and 20%, respectively, of
the initial body burden 24 h after exposure to 191PtCl4,
191Pt(SO4)2, 191PtO2, and 191Pt metal. After ten days,
the body burden was only about 1, 5, 8, and 6%, respectively. This
shows that there was only a slight difference between the clearance
rates for the various chemical forms, although the clearance of
191PtCl4 seemed to be the fastest. Clearance from the lungs also
reflected the two-phase pharmacokinetics in the whole body, with a
fast clearance phase in the first 24 h followed by a slow phase with
a half-time of about 8 days.
Excretion data from the study by Moore et al. (1975c) indicate
that most of the 191Pt cleared from the lungs by mucociliary
action was swallowed and excreted via the faeces (half-time 24 h). A
small fraction of the 191Pt was detected in the urine, indicating
that little was absorbed by the lungs and the gastrointestinal
tract. However, no quantitative data were given.
As shown in Table 11, the portals of entry, lung and trachea,
contained most of the platinum, i.e. 93.5% and 3.9%, respectively,
of the total radioactivity (48 618 counts/g) 1 day after exposure.
Of the other tissues analysed, highest levels were found in the
kidney and bone, suggesting some accumulation in these organs. The
low percentages of 1.5% and 0.6%, respectively, on day 1, reflect
only a low accumulation tendency; no information on the statistical
significance of these figures was provided.
Table 11. Radioactive191Pt distribution in the rat following
inhalation exposure to platinum metal
(7-8 mg/m3, 48 min)a
Mean counts/g wet weight after exposure for
1 day 2 days 4 days 8 days
Blood 61 43 30 12
Trachea 1909 2510 738 343
Lung 45 462 28 784 28 280 23 543
Liver 52 46 37 17
Kidney 750 1002 906 823
Bone 281 258 231 156
Brain 5 3 1 0
Muscle 22 10 28 0
Spleen 39 73 23 5
Heart 37 58 23 5
a From: Moore et al. (1975c)
In a comparative study on the fate of 191PtCl4 (25 µCi per
animal) in rats following different routes of exposure (Moore et
al., 1975a,b) retention followed the classical pattern. The highest
retention was found after intravenous administration, the next
highest after intratracheal, and the lowest after oral
administration. For comparison, retention after inhalation was lower
than after intratracheal administration. However, the total dose was
much higher with inhalation (7000 µCi) than with intratracheal (25
µCi) administration. Only a minute amount of 191PtCl4 given
orally was absorbed. Most of it passed through the gastrointestinal
tract and was excreted via the faeces. After 3 days less than 1% of
the initial dose was detected in the whole body. Following
intravenous administration, 191Pt was excreted in almost equal
quantities in both faeces and urine but elimination was slower than
after oral dosing. After 3 days, whole body retention was about 65%
and after 28 days it was still 14% of the initial dose. By
comparison, following intratrachael administration about 22% and 8%,
respectively, were retained by the body after these periods (Moore
et al., 1975a,b).
In the same studies the tissue distribution of 191Pt was
determined. After the single oral dose, the kidney and liver
contained the highest concentrations, while in the other organs
there were no elevated levels. In contrast, after intravenous
administration 191Pt was found in all tissues (Table 12). The high
concentration of 191Pt found in the kidney shows that once
platinum is absorbed most of it collects in the kidney and is
excreted in the urine. The liver, spleen, and adrenal gland also
contained higher platinum concentrations than the blood. The lower
level in the brain suggests that platinum ions probably cross the
blood-brain barrier only to a limited extent (Moore et al.,
1975a,b).
This was confirmed by Lown et al. (1980) in male Swiss mice
given single intragastric doses of Pt(SO4)2 (144 or 213 mg Pt/kg
body weight). Platinum levels in the blood were several times higher
than in the brain. Clearance from the whole body was slower than in
the rat studies. This could be due to species-specific differences.
In addition, the mice received much higher doses than the rats. Lown
et al. (1980) noted an enhancing effect of the higher dose on
absorption.
In a long-term study, Holbrook (1977) found evidence that a
platinum-binding protein is induced. Male Sprague-Dawley rats
received platinum salts ad libitum either in the drinking-water or
in the dry feed. The sequential platinum contents in the tissues
analysed are shown in Table 13. The data demonstrate that the oral
administration of water-soluble platinum compounds, i.e. PtCl4 and
Pt(SO4)2, results in accumulation of platinum in some organs,
primarily the kidney. After 4 weeks, the platinum content of the
kidney was about 8-fold higher than that of the liver and spleen,
and at least 16-fold higher than in the blood and testis (except for
the highest dose of PtCl4). The total platinum intake after 4
weeks increased by 4.3 times and the platinum content in kidney,
spleen, and blood increased by at least 7 times as compared with the
1-week levels. It is notable that a more than 2-fold increase in the
intake of platinum (after a 4-week consumption of PtCl4 in the dry
feed; 743 vs. 1616 mg Pt/rat) did not lead to an increase in the
platinum content of the kidney, in contrast to the situation in the
liver and spleen. This observation was not corroborated with
Pt(SO4)2.
Table 12. Radioactive191Pt distribution (counts/g wet weight) in the rat following a
single intravenous dose of PtCl4(25 µCi/animal)a
Tissue 1 day 2 days 7 days 14 days
% counts/g % counts/g % counts/g % counts/g
Blood 0.91 22 147 0.81 19 732 0.52 12 774 0.32 7921
Heart 0.48 11 819 0.50 12 201 0.36 8 805 0.19 4593
Lung 0.75 18 432 0.66 16 139 0.46 11 180 0.24 5770
Liver 1.51 36 848 1.28 31 274 1.05 25 732 0.19 4733
Kidney 6.65 162 227 6.59 160 656 5.66 138 010 1.24 30 195
Spleen 1.68 41 085 1.89 45 840 2.29 55 764 0.86 20 973
Pancreas 0.91 22 208 0.80 19 487 0.60 14 802 0.16 3973
Bone 0.53 13 146 0.52 12 800 0.37 8932 0.22 5440
Brain 0.05 1150 0.10 2485 0.02 595 0.01 265
Fat 0.18 4487 0.18 4501 0.13 3201 0.02 429
Testes 0.17 4186 0.27 6540 0.16 3873 0.06 1431
Adrenal 1.86 45 439 1.74 42 363 1.09 26 667 0.25 6190
Muscle 0.19 4798 0.19 4671 0.14 3441 0.09 2146
Duodenal
segment 0.52 12 725 0.25 6044 0.16 4031 0.06 1410
a Adapted from: Moore et al. (1975a)
Table 13. Dietary levels, total platinum consumption, and platinum content of tissues
after oral administration of platinum salts to ratsa
Pt consumption Pt content (mg/kg wet weight; mean ± SE)b
Platinum Duration Dietary Total
salt (weeks) level (mg Pt/ Liver Kidney Spleen Testis Brain Blood
(as Pt) rat)
PtCl4 1 319c 59 2.2 4.8 0.24 0.23
± 0.2
PtCl4 4 319c 255 2.5 33.7 4.8 1.5 0.11 2.1
± 0.9 ± 3.5 ± 1.5 ± 0.5 ± 0.07 ± 0.4
PtCl4 4 1147d 743 3.2 33.5 3.1 1.1 < 0.02 1.5
± 0.9 ± 6.3 ± 0.9 ± 0.4 ± 0.4
PtCl4 4 2581d 1616 8.9 32.4 6.4 1.7 0.12 1.6
± 1.2 ± 4.6 ± 3.0 ± 0.3 ± 0.08 ± 0.2
PtCl4 13 106c 389 1.3 14.9 1.6 0.94 < 0.06 0.9
± 0.3 ± 0.4 ± 0.3 ± 0.20 ± 0.08
Pt(SO4)2.4H2O 1 106c 26 0.07 0.26 < 0.02 < 0.04 0.05
± 0.02
Pt(SO4)2.4H2O 1 319c 78 0.85 4.6 0.13 < 0.02 0.22
Pt(SO4)2.4H2O 4 1147d 716 3.5 43.4 3.2 1.1 0.33 1.6
± 0.4 ± 8.3 ± 0.5 ± 0.1 ± 0.18 ± 0.3
PtO2 4 5808d 4308 < 2.2 < 2.2 < 0.02 < 0.07 < 0.02 < 0.04
a Adapted from: Holbrook (1977)
b Standard error (SE) is given for four values; only the mean is given when two values
are available
c mg Pt/litre
d mg Pt/kg
In contrast to the water-soluble salts, insoluble PtO2 was
only taken up in minute amounts even though the salt was
administered in the diet at an extremely high level resulting in a
total consumption of 4308 mg Pt/rat over the 4-week period
(Holbrook, 1977).
Moore et al. (1975a) also administered 191PtCl4 (25
µCi/animal) intravenously to 15 pregnant rats on day 18 of gestation
to determine placental transfer after 24 h. High levels of 191Pt
radioactivity were found in the kidney (127 064 counts/g) and liver
(43 375 counts/g), compared with 10 568 counts/g in the blood.
Accumulation was also found in the placenta (27 750 counts/g).
191Pt was detected in the 60 fetuses examined, but only at very
low concentrations (an average of 432 counts/g). Thus, the placental
barrier is crossed to a limited extent.
In contrast to the simple platinum salts, the diammine
complexes such as cisplatin (see footnote in section 1.2) are
excreted primarily in the urine. In mice, Hoeschele & Van Camp
(1972) found about 90% of the intraperitoneally injected dose in the
urine within five days. Little or no excretion occurred via the
faeces. A high urinary recovery was also observed in rats and dogs
(Hoeschele & Van Camp, 1972; Lange et al., 1972; Litterst et al.,
1976a,b, 1979; Cvitkovic et al., 1977).
The excretion of both cis- and trans-
diamminedichloroplatinum(II) follows a biphasic pattern with a fast
initial alpha-phase and a second slow ß-phase. The variation in the
plasma half-lives is due to species differences and variations in
dose, route of administration, time points analysed, and analytical
method used (Litterst et al., 1979). The extremely rapid alpha-phase
accounts for early, high levels of platinum in kidney, liver, skin,
bone, ovary, and uterus. The prolonged ß-phase results in detectable
urine platinum concentrations 30 days after a single dose.
For both the simple platinum salts and cisplatin complexes, an
initial rapid clearance is followed by a prolonged clearance phase
during the remaining post-exposure period, and there is no evidence
for markedly different retention profiles between these two groups
of platinum compounds (Rosenberg, 1980).
All animal species studied show a similar organ distribution
pattern for cisplatin. An initial distribution to nearly all tissues
is followed by accumulation in the first hour mainly in kidney,
liver, muscle, and skin. By the end of the first day, plasma levels
decrease rapidly and there are elevated platinum levels in numerous
other tissues (Litterst et al., 1979).
Cisplatin is extensively bound to plasma proteins; 90% of it
may be bound 2 h after an intravenous injection. The bound portion
is no longer cytotoxic (Safirstein et al., 1983; Sternson et al.,
1984). In addition to its reactivity with plasma protein, renal
excretion leads to a very low concentration of free cisplatin in the
plasma and to a rapid accumulation in the kidney. Due to the
presence of high chloride ion concentrations, cisplatin is
relatively stable in extracellular fluids (see also section 7.6),
which explains why it is excreted mainly in the unchanged form in
human and rat urine (Safirstein et al., 1983).
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1 Single exposure
Acute toxicity data on platinum mainly relate to its
coordination complexes, the chloroplatinates and ammines. Hofmeister
(1882) was one of the first to test ammonium salts containing
divalent and tetravalent platinum with various numbers of ammine
ligands. He injected solutions of platinum complexes into the dorsal
lymphatic sac of single frogs and subcutaneously into the dorsal
skin of single rabbits. The symptoms observed included vomiting and
diarrhoea with bloody stools and a "curare-like" action of the
salts.
The acute toxicity of platinum depends considerably on the
species of platinum involved (Table 14). Soluble platinum compounds
are much more toxic. Hence, in the study of Holbrook (1976a) oral
toxicity to rats decreased in the following order: PtCl4 >
Pt(SO4)2.4H2O > PtCl2 > PtO2. For the two latter
compounds no LD50 could be derived.
Signs of poisoning observed, for example, with
(NH4)2[PtCl4], include hypokinesia, piloerection, diarrhoea,
clonic convulsions, laboured respiration, and cyanosis (Degussa,
1989a).
Hexachloroplatinic acid is highly nephrotoxic in rats. After an
intraperitoneal LD50 injection of 40-50 mg/kg, rats died of renal
failure, hypocalcaemia, and hyperkalaemia. The necrotizing renal
tubular lesions involved the entire renal cortex (Ward et al.,
1976).
In its metallic state, platinum has an extremely low acute
toxicity. Thus some alloys containing platinum are used in
protheses. Fine dust particles of metallic platinum, 1-5 µm in
diameter, orally administered to rats caused only slight necrotic
changes in the gastrointestinal epithelium, granular dystrophy of
hepatocytes, and swelling in the epithelium of the convoluted renal
tubules (Roshchin et al., 1979, 1984). The highest dose given was
not lethal. The dose was reported as "129 µA/kg" (25 167 µg per kg;
personal communication from Prof. A.V. Roshchin to IPCS dated 3
April 1991).
Due to the different absorption rates for platinum compounds,
the route of administration also affects the toxicity, the
intraperitoneal and intravenous routes leading to much higher
toxicity than the oral route (Table 14).
Table 14. Acute toxicity of platinum and platinum compounds after oral (p.o.),
intraperitoneal (i.p.), and intravenous (i.v.) administration to rats
Compound Route Sexa LD50 Reference
(mg/kg)
PtO2 p.o. m > 8000 Holbrook et al. (1976a,b)
PtCl2 p.o. m > 2000 Holbrook et al. (1976a,b)
PtCl2 p.o. m 3423b Roshchin et al. (1984)
PtCl2 i.p. m 670 Holbrook et al. (1976a,b)
PtCl4 p.o. m 240 Holbrook et al. (1976a,b)
PtCl4 p.o. m/f 276b Roshchin et al. (1984)
PtCl4 i.p. m 38 Holbrook et al. (1976a,b)
PtCl4 i.v. m 26.2 Moore et al. (1975b)
PtCl4 i.v. m 41.4 Moore et al. (1975b)
Pt(SO4)2.4 H2O p.o. m 1010 Holbrook et al. (1976a,b)
Pt(SO4)2.4 H2O i.p. m 310c Holbrook et al. (1976a,b)
Pt(SO4)2.4 H2O i.p. m 138-184c Holbrook et al. (1976a,b)
(NH4)2[PtCl6] p.o. m/f 195b Roshchin et al. (1984)
(NH4)2[PtCl6] p.o. m/f approx. 200 Johnson Matthey (1978a)
(NH4)2[PtCl4] p.o. m 212 Degussa (1989a)
(NH4)2[PtCl4] p.o. f 125 Degussa (1989a)
H2[PtCl6] i.p. m 40-50 Ward et al. (1976)
Na2[PtCl6] p.o. m/f 25-50 Johnson Matthey (1978b)
Na2[Pt(OH)6] p.o. m/f 500-2000 Johnson Matthey (1978c)
K2[PtCl4] p.o. m/f 50-200 Johnson Matthey (1981a,b)
K2[Pt(CN)4] p.o. m/f > 2000 Johnson Matthey (1977a)
[Pt(NH3)4]Cl2 p.o. m/f > 15 000 Johnson Matthey (1977b)
[Pt(NO2)2(NH3)2] p.o. m approx. 5000 Degussa (1989b)
[Pt(NO2)2(NH3)2] p.o. f > 5110 Degussa (1989b)
[Pt(C5H7O2)2] p.o. m/f > 500 Johnson Matthey (1976a)
cis-[PtCl2(NH3)2]d p.o. m/f approx. 20 Johnson Matthey (1977c)
cis-[PtCl2(NH3)2]d i.p. m 12 Kociba & Sleight (1971)
cis-[PtCl2(NH3)2]d i.p. m 7.7 Ward & Fauvie (1976)
cis-[PtCl2(NH3)2]d i.v. m 7.4 Ward et al. (1976)
trans-[PtCl2(NH3)2] p.o. m/f > 5110 Degussa (1989c)
a m = male; f = female
b Calculated from the original values given as mg A/kg (= milligramme atom/kg)
c Results from two different laboratories
d See footnote in section 1.2
7.2 Short-term exposure
Holbrook et al. (1975) conducted repeated-dose oral toxicity
studies on male Sprague-Dawley rats. The soluble salts PtCl4 and
Pt(SO4)2.4H2O were added to the drinking-water, which was
consumed ad libitum. Within the observation period of 4 weeks, a
concentration of 0.54 mmol/litre (182 mg PtCl4/litre or 248 mg
Pt(SO4)2.4H2O per litre) did not affect the normal weight
gain. A 3-fold increase in the platinum concentration to 1.63
mmol/litre reduced the weight gain by about 20% during the first
week only; this paralleled a 20% decrease in feed and fluid
consumption. The dietary administration of PtCl4 at concentrations
of 0.5 mmol/litre for approximately 30 days or 1.6 mmol/litre for 8
days (169 and 539 mg/litre, respectively) did not affect the weights
of any of the five organs investigated, i.e. liver, kidney, spleen,
heart, and testes. Similarly, the administration of 1.6 mmol per
litre of Pt(SO4)2.4H2O (734 mg/litre) for 8-9 days did not
significantly affect organ weights. Total platinum intake for each
of these three experimental conditions was approximately 50 mg per
rat. When 1.6 mmol PtCl4/litre (539 mg/litre) was given for about
30 days (total intake of about 250 mg Pt per rat), the kidney weight
increased by about 6-10%. No effects on the level of microsomal
protein or the activities of aniline hydroxylase and aminopyrine
demethylase in liver microsomes were found (Holbrook, 1976b).
7.3 Skin and eye irritation; skin and respiratory sensitization
7.3.1 Skin irritation
The dermal irritancy of several platinum compounds was tested
on albino rabbits using comparable procedures and evaluation
criteria. Platinum test materials were spread on abraded and intact
skin sites, located dorsolaterally on the animals' trunks. The skin
reactions were evaluated after 24, 48, and 72 h, and are summarized
in Table 15.
7.3.2 Eye irritation
Summarized data on eye irritation are presented in Table 15.
All tested platinum salts were either corrosive or irritating to
varying degrees.
7.3.3 Skin sensitization
In a study by Kolpakova & Kolpakov (1983), platinum
hydrochlorides administered intravenously to rabbits in repeated
doses induced sensitization confirmed by the basophil degranulation
test, neutrophil damage index, leucocyte agglomeration, neutrophil
alteration, and the drop skin and skin fenestra tests. These data
are unusual and have not been confirmed in other studies.
Table 15. Skin and eye irritation by platinum compoundsa
Skin irritation testb Eye irritation testc
Compound Primary Classification Reference Classification Reference
irritation
score
PtO2 0 non-irritant Campbell et al. (1975)
PtCl2 0.4d non-irritant Campbell et al. (1975)
PtCl4 2.2e irritant Campbell et al. (1975)
(NH4)2[PtCl6] 1.3 mild irritant Johnson Matthey (1978d)
(NH4)2[PtCl4] 2.7 slight irritant Degussa (1988a) corrosive Degussa (1988b)
Na2[PtCl6] 0.5 mild irritant Johnson Matthey (1978e) irritant Johnson Matthey (1978f)
Na2[Pt(OH)6] 5.4 severe irritant Johnson Matthey (1978g)
K2[PtCl4] 0f non-irritant Johnson Matthey (1981c) irritant Johnson Matthey (1981d)
K2[Pt(CN)4] 0.3 mild irritant Johnson Matthey (1977d) irritant Johnson Matthey (1978h)
[Pt(NH3)4]Cl2 2.8 moderate irritant Johnson Matthey (1977e) strongly irritant Johnson Matthey (1977f)
[Pt(NO2)2(NH3)2] 0 non-irritant Degussa (1989d) severely irritant Degussa (1989e)
[Pt(C5H7O2)2] 0 non-irritant Johnson Matthey (1976b) mildly irritant Johnson Matthey (1976c)
cis-[PtCl2(NH3)2] 0.13 mild irritant Johnson Matthey (1977g) severely irritant Johnson Matthey (1977h)
(toxic)
trans-[PtCl2(NH3)2] 0 non-irritant Degussa (1988c) corrosive Degussa (1988d)
a Adapted from Bradford (1988)
b The skin tests (patch tests on albino rabbits) were carried out according to the US Federal Register 1973 Skin Test (24 h-contact)
(Johnson Matthey) or according to OECD Test Guideline No. 404 (4-h contact) (Degussa). The method used by Campbell et al. (1975)
is comparable to these tests.
c The eye irritation tests on albino rabbits were carried out according to the US Federal Register 1973 Eye Test (Johnson Matthey)
or according to OECD Guideline No. 405 (Degussa).
d Average score from 0.2 (intact skin) and 0.6 (abraded skin); a score of 0-0.9 was considered as "non-irritant" by the authors.
e Average score from 1.8 (intact skin) and 2.6 (abraded skin); a score of 2 was considered as "irritant" by the authors.
f Using OECD Test Guideline No. 404 (4-h contact).
Taubler (1977) injected rabbits, guinea-pigs, and mice
subcutaneously and intravenously with PtSO4 (0.05-0.3 mg/litre
with and without NH4Cl) three times a week for 4 weeks. No
induction of an allergic state was found, as measured by skin tests
(guinea-pigs and rabbits), passive transfer, and footpad tests
(mice). Administration of platinum-egg-albumin complex also failed
to sensitize the experimental animals.
In a study by Murdoch & Pepys (1985), rats were immunized with
ovalbumin-platinum. Sera of the animals which were positive in the
passive cutaneous anaphylaxis (PCA) test and a radioallergosorbent
test (RAST) were pooled and used for PCA tests with other platinum
salts having differing ligands. A significant cross-reactivity
between ammonium tetrachloroplatinate(II), ammonium
hexachloroplatinate(IV), and the conjugated tetrachloroplatinate was
observed. There was very limited or no cross-reactivity with the
compounds cesium trichloronitroplatinate(II), cis-
diamminedichloroplatinum(II), potassium tetracyanoplatinate(II), and
tetraammineplatinum(II) chloride.
7.3.4 Skin and respiratory sensitization
Biagini et al. (1983) exposed two groups of Cynomolgus monkeys
(Macaca fasicularis) to disodium hexachloroplatinate,
Na2[PtCl6], by nose-only inhalation of 200 and 2000 µg/m3,
4h/day, biweekly for 12 weeks. Another group was exposed
percutaneously to the salt (20 mg/ml) applied biweekly to an open
patch area in the intrascapular region. Two weeks after termination
of exposure, bronchoprovocation challenges with Na2[PtCl6] and
pulmonary function tests were performed. Percutaneous application
did not affect post-challenge pulmonary function. The 200 µg/m3
group showed significantly greater pulmonary deficits as compared to
control animals. Average pulmonary flow resistance (RL) was
significantly increased, while forced expiratory volume in 0.5
seconds, corrected for vital capacity (FEV0.5/FVC), was decreased.
No dermal hypersensitivity was observed. The question of whether the
observed pulmonary hyper-reactivity is due to a
superpharmacological, irritant, local immune, or combination
mechanism is unresolved. The absence of hyper-reactivity in the
2000-µg/m3 group suggests a possible pulmonary tolerance
mechanism, tachyphylaxis, or delay in the development of symptoms at
higher sensitization concentrations.
7.3.5 Respiratory sensitization
In a 12-week inhalation experiment with Cynomolgus monkeys
exposed to either ammonium hexachloroplatinate (200 µg/m3) or
ozone (2000 µg/m3; 1 ppm) alone or as a combination of both,
Biagini et al. (1986) found significant allergic platinum dermal
hypersensitivity, based on concentrations necessary to give a
positive test, and pulmonary hyper-reactivity only with concomitant
exposure to ozone. Inflammation, epithelial damage, cell
recruitment, and modifications of cellular tight junctions caused by
ozone may increase the penetration of platinum into the pulmonary
epithelium and subepithelial tissue. This could lead to increased
protein binding sites or absorption of the platinum salts and
finally to the development of pulmonary hyper-reactivity and
allergic sensitization (Biagini et al., 1986).
7.3.6 Sensitization by other routes
Murdoch & Pepys (1984) investigated the immunological responses
to complex platinum salts in the female hooded Lister rat, a strain
that produces high and consistent levels of circulating IgE when
immunized with low doses of antigen together with Bordetella
pertussis adjuvant, and that reacts with enhanced synthesis of IgE
upon secondary boosting. Sensitization with the free salt of
ammonium tetrachloroplatinate, (NH4)2[PtCl4], was attempted
via the intraperitoneal, intramuscular, intradermal, subcutaneous,
intratracheal, and footpad routes over a wide range of doses (1 to
1000 µg). Both B. pertussis and/or aluminium hydroxide gel were
added as adjuvants. As shown by direct skin testing using the PCA or
RAST methods, no sensitization was achieved. However, sensitization
was obtained by intraperitoneal injection of the platinum salt
conjugated to ovalbumin (OVA). Antibodies were produced to Pt-OVA
and to OVA alone. Specific sensitization was demonstrated both by
PCA challenge with Pt-BSA (no positive PCA reactions were seen with
BSA alone) and by positive RAST, demonstrated by RAST inhibition
techniques with a Pt-BSA conjugate.
7.4 Reproductive toxicity, embryotoxicity, and teratogenicity
Only limited experimental data concerning the effects of
platinum on reproduction, embryotoxicity, and teratogenicity are
available. D'Agostino et al. (1984) studied the embryotoxic effects
of platinum compounds in Swiss ICR mice. Single doses of either
Pt(SO4)2.4H2O or Na2[PtCl6].6H2O were administered
intragastrically or subcutaneously, respectively, on the 7th and
12th day of gestation. The pups were cross-fostered to treated or
untreated dams at birth and were culled to three animals of each sex
per litter. In the Pt(SO4)2 study, the LD1 wdose of 200 mg
Pt/kg caused a reduced offspring weight from day 8 to day 45
postpartum. The major effect of disodium hexachloroplatinate (20 mg
Pt/kg) was a reduced activity level exhibited by the offspring of
dams exposed on the 12th day of gestation. The general activity was
quantified on an activity field consisting of concentric circles.
The number of lines crossed during 1 min comprised the activity
score. On days 60-65 postpartum, open-field behaviour (ambulation
and rearing), rotarod performance, and passive avoidance learning
were investigated in the adult offspring. No effects were found
after administration on the 7th day, but administration on day 12 of
gestation had significant behavioural effects.
Solid platinum, wire or foil, is considered to be biologically
inert and adverse effects on implantation are probable due to the
physical presence of a foreign object in the uterus (Barlow &
Sullivan, 1982).
Kraft et al. (1978) reported normal fertility in male rabbits
with open tube gold/platinum devices inserted into the vas deferens.
There was an initial decrease in sperm count and motility, but these
parameters returned to normal after three weeks. At 117-426 days
after insertion, 7 out of 9 animals were fertile in numerous
matings.
Effects on human sperm motility were investigated by Kesseru &
Leon (1974). Fresh sperm were incubated for up to 5 h in the
presence of strips of platinum or other metals. Motility after 2 and
5 h was 60 and 30%, respectively, compared to 10 and 0% for copper,
40 and 7% for silver, and 90 and 65% for gold.
Platinum wire inserted into the uterus of rats was reported to
reduce the implantation of fertilized ova. An 83% reduction in the
number of implantation sites in the affected uterine horn, compared
to the unoperated horn, was found in rats unilaterally implanted on
day 3 (Chang et al., 1970). Chang & Tatum (1975) found no effect on
embryonic or fetal survival if platinum wire was inserted after
implantation on day 6. Tobert & Davies (1977) showed a 37% reduction
in the number of implanting ova in the uteri of rabbits containing
platinum foil.
7.5 Mutagenicity and related end-points
The genotoxic effects of platinum compounds have been
investigated in bacterial systems, mammalian cell cultures and in
vitro studies.
In bacteria many of the tested platinum compounds were
moderately mutagenic. Cisplatin and some of its analogues showed the
greatest mutagenic potential; other platinum compounds were less
mutagenic.
In Ames tests, nearly all using the test strains Salmonella
typhimurium TA98 and TA100, positive results were reported
(Lecointe et al., 1977; Andersen, 1979; Suraikina et al., 1979; Life
Science Research, 1980a; Kanematsu et al., 1980). With
[Pt(NH3)4]Cl2, mutagenic potential was observed in strain
TA1537 with and without S-9 metabolic activation (Life Science
Research, 1980a).
The induction of reverse mutations in the plasmid-carrying
strains TA98 and TA100 indicated base-pair substitution and frame-
shift mutations (Lecointe et al., 1977; Suraikina et al., 1979;
Kanematsu et al., 1980).
(NH4)2[PtCl6] but not PtCl4 caused base-change mutation
in Escherichia coli B/r WP2 (Kanematsu et al., 1980).
The growth of a Rec strain of Bacillus subtilis was
significantly inhibited by (NH4)2[PtCl6] (0.1 mol/litre),
H2[PtCl6] (0.01 mol/litre), and PtCl4 (0.01 mol/litre)
(Kanematsu et al., 1980).
In a mutagenic test with the mouse lymphoma cell line L 5178Y,
cisplatin, transplatin, and PtCl4 produced significantly higher
mutation frequencies than occurred in the controls, but
[Pt(NO2)2](NH3)2 and PtCl2 did not (Sandhu, 1979).
Cellular resistance to the toxic effects of two platinum
complexes was introduced into Chinese hamster ovary (CHO) cells by
continuous exposure to K2[PtCl6] and Pt(SO4)2 for 5 and 4
months, respectively. These cell lines had resistant phenotypes
stable for at least 55 population doublings in the absence of a
platinum compound. The induced resistances were interpreted by the
authors to be a result of mutation and selection (Smith et al.,
1984).
In a micronucleus test in mice involving oral administration of
[Pt(NH3)4]Cl2, no significant increase in the incidence of
micronucleated polychromatic erythrocytes was found. Additionally,
[Pt(NH3)4]Cl2 did not markedly inhibit bone marrow cell
division at any level (Life Science Research, 1980b). Also, no
evidence of induced chromosomal damage leading to micronucleus
formation in polychromatic erythrocytes was observed after oral
administration of K2[PtCl4] in mice (Life Science Research,
1981a).
No significant increase in the incidence of aberrant metaphases
was found in bone marrow cells after subacute oral administration of
[Pt(NH3)4]Cl2 or K2[PtCl4] to Chinese hamsters (Life
Science Research, 1981b, 1982).
K2[PtCl4] and [Pt(NH3)4]Cl2 induced no increase in
the frequency of sex-linked recessive lethal mutations in
Drosophila melanogaster (Life Science Research, 1980c, 1981c).
In a structure-mutagenicity study with the CHO:HGPRT-system,
cis-[Pt(NH3)2Cl2] was the most potent of six platinum
compounds tested. Based on the slope of the mutation induction
curve, the approximate relative mutagenic activity of cis-
[Pt(NH3)2Cl2], K[Pt(NH3)Cl3], and [Pt(NH3)3Cl]Cl was
100:9:0.3. The mutation frequency for K2[PtCl4] and trans-
[Pt(NH3)2Cl2] was related to the concentration used, but was
not much greater than the maximum spontaneous mutation frequency. No
mutagenic activity was observed for [Pt(NH3)4]Cl2. The
relative cytotoxicity of the tested compounds was similar.
Additionally, it was found that cis- and trans-
[Pt(NH3)2Cl2] bind to DNA after entering the cell, but the
relative mutagenicities are not a consequence of different initial
levels of DNA binding (Johnson et al., 1980).
Dose-dependent forward mutations were induced by PtCl4 to 8-
azaguanine resistance (8-AGR/HGPRT locus) in Chinese hamster ovary
(CHO-S) cells. In addition there was an increased dose-related
frequency of CHO-AUXB1 reversion (Taylor et al., 1979)
Cisplatin, which is not reviewed in detail in this document
(see footnote in section 1.2), induces structural chromosomal
aberrations and sister chromatid exchanges in cells of rodents
treated in vivo, chromosomal aberrations, micronuclei, and sister
chromatid exchanges in both human and rodent cells in vitro, and
mutation and DNA damage in rodent cells in vitro. Cisplatin is
also mutagenic in Drosophila, fungal, and bacterial test systems
(IARC, 1987a).
7.6 Carcinogenicity and anticarcinogenicity
No experimental data are available on the carcinogenicity of
platinum and platinum compounds except for cisplatin (see footnote
in section 1.2). IARC (1987b) considered sufficient the evidence for
the carcinogenicity of cisplatin for animals (see chapter 13).
Cisplatin and its analogues, however, are exceptional compared
to the other platinum compounds. This is reflected by the unique
mechanism for their anti-neoplastic activity demonstrated in in
vitro studies (Rosenberg, 1980, 1985). At low doses cisplatin
produces specific inhibition of DNA synthesis (but not of RNA and
protein synthesis) by causing DNA lesions such as monofunctional
adducts, bifunctional binding to a single base moiety, and DNA
cross-links of inter- and intrastrand types (Harder & Rosenberg,
1970; Howle & Gale, 1970). There is sufficient evidence that the DNA
cross-links are responsible for cellular toxicity, but not for anti-
tumour activity. For the latter, another observation probably plays
the decisive role; only the cis isomer forms a closed ring chelate
of the aquated cisplatin with guanine at a certain position of
guanine. Thus, intrastrand DNA cross-linking is considered to be the
most important reason for anti-tumour activity. It appears that, due
to the cisplatin-induced DNA cross-links, the replication of DNA is
impaired in cancer cells, while in normal cells the cisplatin
lesions on guanine are repaired before replication (Rosenberg, 1985;
Pinto & Lippard, 1985).
The high chloride concentration of the extracellular fluid (112
mmol/litre) is sufficient to limit the substitution of water ligands
for chloride. However, within the cell the platinum complex is
exposed to low chloride concentrations (4.4 mmol/litre) and
hydrolysis of the chloride leaving groups can occur (Rosenberg,
1975), a process that has been shown to accelerate the rate of
reaction of platinum with DNA (Johnson et al., 1980) and to increase
its toxicity (Litterst, 1981). This hydration provides the only
known activation process required for cisplatin to react with
molecules in the cell, and metabolic activation is not required
(Rosenberg, 1980). The binding of cisplatin with plasma proteins, on
the other hand, is not inhibited by chloride and presumably involves
a different mechanism (De Conti et al., 1973), such as the strong
electrophiles on proteins (Cleare, 1977a).
7.7 Other special studies
7.7.1 Effects on alveolar macrophages
Rabbit alveolar macrophages exposed to the water-soluble
platinum(IV) chloride at a concentration of 0.4 mmol/litre (78 mg
Pt/litre) for a period of 20 h exhibited a 50% reduction in
macrophage viability. A reduction in phagocytic activity and a
decrease in total cellular adenosine triphosphate to 50% of the
value in control macrophages was observed at 0.21 and 0.25
mmol/litre (41 and 48 mg Pt/litre). Platinum(IV) oxide (PtO2) did
not dissolve in the culture medium and, hence, was ineffective at
concentrations as high as 500 mg/litre (Waters et al., 1975).
7.7.2 Non-allergic mediator release
Investigations in guinea-pigs, rats, and dogs showed an
increase in bronchomotility and histamine release after intravenous
treatment with disodium chloroplatinate, Na2[PtCl6] (Saindelle &
Ruff, 1969; Parrot et al., 1969).
Saindelle & Ruff (1969) noticed dyspnoea one minute after an
intravenous injection of disodium chloroplatinate (20 mg/kg) into
guinea-pigs. Within 5 min an intense attack of asthma occurred
resulting in death. Histamine release occurred following the
injection and the blood histamine level was greatly increased. The
injection of a smaller dose (1-2 mg/kg) resulted in bronchospasm
comparable to that caused by 3 µg/kg of histamine. Repeated
injections of histamine caused reproducible changes in bronchial
motility, whereas the platinum compound caused tachyphylaxis.
The intense breathing difficulties observed in these studies
were presumably due to non-allergenic histamine release. This
nonspecific histamine release has complicated the interpretation of
both animal and human studies with respect to the conclusion of
allergic sensitization.
7.7.3 Effects on mitochondrial function
No pronounced effects of platinum on the mitochondrial function
of liver, heart, lung or kidney cells were observed in an in vitro
test on succinate-stimulated O2 uptake 24 and 48 h after
intragastric administration of 40 and 80 µmol K2[PtCl6]/litre
(19 and 38 mg/litre) to Sprague-Dawley rats (Michael et al., 1976).
7.7.4 Effects on the nervous system
The open field behaviour of adult Swiss mice has been found to
be influenced by platinum salts administered intragastrically in the
form of a single dose or of repeated doses. A single dose of
Pt(SO4)2 at the LD25 level (213 mg Pt/kg body weight)
depressed ambulation significantly and rearing marginally. For
ambulation, this pattern persisted from 4 h to 7 days after
administration, although the effect was most obvious at 4 h.
Repeated doses of the same salt at the LD1 level (up to 10 doses
of 109 mg Pt/kg every 72 h) caused a marginal depression of activity
and exploratory behaviour (Lown et al., 1980). Also, a single dose
of Na2[PtCl6] depressed ambulation significantly (Massaro et
al., 1981).
During the course of a reproduction study, behavioural effects
were observed in the offspring of mice treated with sodium
hexachloroplatinate (see section 7.4; D'Agostino et al., 1984).
7.7.5 Side effects of cisplatin and its analoguesa
As discussed in section 8.3, the therapeutic use of cisplatin
in humans can be accompanied by several toxic side-effects. In
animal studies, only some of which are presented in this monograph,
similar effects were observed.
Ward et al. (1976) investigated the nephrotoxicity of cisplatin
and its analogues in male F-344 (Fischer CDF) rats. An intravenous
LD50 dose of cisplatin (7.4 mg Pt/kg body weight) caused an
increase in the blood urea nitrogen and creatinine levels reaching a
peak on days 4 to 5. Diarrhoea developed by day 3. Necrotizing
enteritis of the small intestine, caecum, and colon, cellular
depletion of bone marrow and thymus, and acute degenerative and
necrotizing renal tubular lesions also occurred (Ward et al., 1976).
Oxoplatinum ( cis-dichlorodiammine-trans-
dihydroxyplatinum(IV)) also caused marked nephrotoxicity after
intravenous administration (20 mg/kg as a single dose) to rats.
However, another cisplatin analogue, CBDCA ( cis-diammine-l,1-
cyclobutane dicarboxylate platinum(II)), did not result in
significant changes in renal function parameters (Laznickova et al.,
1989).
a See footnote in section 1.2.
Cisplatin has been found to cause bone marrow suppression. The
surviving fraction of haemopoietic bone-marrow system cells in mice
decreased from 1 to 0.03 after treatment with an LD50 dose of
cisplatin (Lelieveld et al., 1984). A 36-53% decrease in lymphocyte
and granulocyte counts was observed in mouse bone marrow after
intra-peritoneal treatment with 5 mg cisplatin/kg (Bodenner et al.,
1986).
Cisplatin administered intraperitoneally (6 mg/kg) has been
shown to affect gastric emptying in rats. There was a large increase
in the weight of the stomach due to retained food (Whitehouse &
Garrett, 1984).
In dogs, cisplatin given intravenously as a dose of 2 mg/kg
resulted in a complete interruption of interdigestive myoelectric
activity of the gastric antrum, duodenum, and jejunum (Chey et al.,
1988).
Ototoxicity has been demonstrated in guinea-pigs. A cisplatin
dose of 1.5 mg/kg administered intraperitoneally once a day caused
hearing loss beginning at about the ninth day of administration
(Hoeve et al., 1987).
7.8 Factors modifying toxicity
Physiological levels of selenium administered simultaneously
with food to mice markedly depressed the acute toxicity of some
platinum salts by forming inert complexes of high relative molecular
mass in the presence of protein (Imura, 1986).
8. EFFECTS ON HUMANS
8.1 General population exposure
8.1.1 Acute toxicity - poisoning
Except for one case of poisoning in 1896 (Hardman & Wright,
1896), no acute poisoning cases have been reported.
8.1.2 Effects of exposure to platinum emitted from automobile
catalysts
An immunological study conducted by Cleare (1977b) addressed
the question of whether the emitted platinum is allergenic. He
investigated the response of individuals, who were highly sensitive
to platinum salt (skin test positive at low platinum salt
concentrations), to extracts of particulate exhaust samples. The
total platinum content at the highest concentration was more than 5
µg/ml, which would normally be sufficient to elicit a response. Five
extracts tested on three people, using the skin prick test, did not
elicit a positive response.
8.2 Occupational exposure
The occupational hazards of platinum are principally confined
to some halogenated complex platinum salts (Rosner & Merget, 1990).
8.2.1 Case reports and cross-sectional studies
A report of health problems arising from occupational exposure
to platinum was produced by Karasek & Karasek (1911). They studied
workers in photographic studios in Chicago handling photographic
paper treated with complex platinum salts. The symptoms observed in
eight workers were pronounced irritation of the nose and throat
causing violent sneezing and coughing, together with difficulties in
breathing.
Hunter et al. (1945) conducted environmental and clinical
studies on workers in four British platinum refineries. Out of 91
workers exposed to complex salts of platinum, 52 showed symptoms
starting with repeated sneezing and rhinorrhoea, followed by
tightness of the chest, shortness of breath, cyanosis, and wheezing.
Scaly erythematous dermatitis of hands and forearms, sometimes also
affecting the face and neck, and urticaria were observed in 13
workers. The respiratory tract symptoms persisted during working
hours and for about one hour after leaving the factory. The latency
period from the first contact with platinum to the occurrence of the
first symptoms varied from a few months to six years. Once skin and
respiratory tract sensitization was established, symptoms tended to
become worse as long as the workers were exposed to platinum salts.
In the USA, Roberts (1951) studied 21 employees of a platinum
refinery for five years. All workers showed some form of platinum-
related disease for which Roberts (1951) introduced the term
"platinosis". According to his classification of this occupational
disease, 40% of the employees did not have typical symptoms but
exhibited the same inflammatory changes in the conjunctivae and the
mucous membranes of the upper respiratory tract as were seen in the
60% of the workers with definite symptoms.
These observations have been confirmed by other investigators.
The term "platinosis" is no longer used, as it implies a chronic
fibrosing lung disease. This had been assumed by Roberts (1951) but
has not been reported elsewhere. The terms "platinum salt allergy"
(Schultze-Werninghaus et al., 1978), "platinum salt sensitivity"
(Linnett, 1987), "allergy to platinum compounds containing reactive
halogen ligands" (Hughes, 1980) and "platinum salt hypersensitivity"
(PSH) have been used, with the latter being preferred.
The symptoms typical of platinum salt sensitization (Roshchin
et al., 1979; Health and Safety Executive, 1983; Brooks et al.,
1990) include watering of the eyes, sneezing, tightness of the
chest, wheezing, breathlessness, coughing, eczematous and urticarial
skin lesions, signs of mucous membrane inflammation.
In earlier studies, the prevalence of allergic symptoms due to
platinum salt exposure was as high as 80% (Table 16) although this
was not consistently confirmed by skin testing. Estimated workplace
exposure concentrations ranged from 0.9 to 1700 µg Pt/m3 (Hunter
et al., 1945). However, due to analytical deficiencies, these data
do not allow the quantification of these exposure situations. It can
be assumed that occupational exposure is much lower today due to
improved engineering control and occupational exposure limits.
Airborne dust analyses in a platinum refinery revealed levels
between 0.08 and 0.1 µg/m3 in the separation department; in other
areas the measurements were all below 0.05 µg/m3 (Bolm-Audorff et
al., 1988) or below 0.08 µg/m3 (Merget et al., 1988). Work-related
symptoms were reported in 8 to 23% of workers exposed to these
concentrations (Table 16). The risk of developing platinum salt
hypersensitivity seems to be correlated with the intensity of
exposure. In the surveys of Bolm-Audorff et al. (1988) and Merget et
al. (1988), the highest rates of prevalence occurred in the groups
exposed to the highest concentration.
Table 16. Prevalence of symptoms and positive skin tests
in refinery workers exposed to platinum salts
Total Workers Prevalence of Reference
workersa with symptomsb symptoms (%)c
91 (16) 52 (4) 57 (25) Hunter et al. (1945)
20 (19) 12 (8) 60 (42) Roberts (1951)
15 (nd) 12 (nd) 80 (nd) Massmann & Opitz (1954)
51 (nd) 35 (nd) 69 (nd) Hebert (1966)
107 (107) 31d 47e (15) 29d 44e (14) Biagini et al. (1985);
Brooks et al. (1990)
65 (64) 15 (12) 23 (19) Bolm-Audorff et al.
(1988)
24 (20) 2 (4) 8 (20) Merget et al. (1988)
a Values in parentheses are numbers of skin-tested workers
b Values in parentheses are numbers of workers with a positive
skin test
c Values in parentheses give the prevalence of positive skin
tests as a percentage of skin-tested workers
d Workers with upper respiratory tract symptoms
e Workers with lower respiratory tract symptoms
nd = not determined
8.2.2 Allergenicity of platinum and platinum
compounds
Metallic platinum seems to be non-allergenic. With the
exception of a single reported case of alleged contact dermatitis
from a "platinum" ring (Sheard, 1955), no allergic reactions have
been reported.
Halogenated platinum salts are among the most potent
sensitizers. The compounds mainly responsible for platinum
sensitization are hexachloroplatinic acid, H2[PtCl6], and the
chlorinated salts such as ammonium hexachloroplatinate,
(NH4)2[PtCl6], potassium tetrachloroplatinate, K2[PtCl4],
potassium hexachloroplatinate, K2[PtCl6], and sodium
tetrachloroplatinate, Na2[PtCl4]. Cleare et al. (1976)
investigated the allergenic potency of platinum complexes by means
of skin prick tests on platinum refinery workers who were known to
be sensitive to hexachloroplatinate. Their results suggest that
platinum allergy is confined to a small group of charged compounds
that contain reactive ligand systems, the most effective of which
are chloride ligands. The allergic response generally increases with
increasing number of chlorine atoms, as demonstrated by the
following sequence of potency:
(NH4)2[PtCl6] approx. (NH4)2[PtCl4] >
Cs2[Pt(NO2)Cl3] > Cs2[Pt(NO2)2Cl2] >
Cs2[Pt(NO2)3Cl]
Ionic platinum compounds containing bromide or iodide are also
allergenic, but are less effective.
Non-halogenated complexes such as [Pt{(NH2)2CS}4]Cl2,
K2[Pt(NO2)4], and [Pt(NH3)4]Cl2, and neutral complexes
such as cisplatin, cis-[PtCl2(NH3)2], are not allergenic,
probably because they do not react with proteins to form a complete
antigen. Anaphylactic shock reactions observed after the intravenous
administration of relatively high doses of cisplatin (Khan et al.,
1975; Von Hoff et al., 1979) were probably caused by contamination
with the potent hexa- or tetrachloroplatinate (Pepys, 1983).
8.2.3 Clinical manifestations
The latency period from the first exposure to platinum salts to
the occurrence of the first symptoms usually varies between three
months and three years (Parrot et al., 1969; Schultze-Werninghaus et
al., 1978; Ruff et al., 1979; Biagini et al., 1985), but is
sometimes only a few weeks (Roberts, 1951; Hughes, 1980; Merget et
al., 1988).
The dermatitis observed in the past (Roberts, 1951) is believed
to have been mainly of a primary irritant nature following exposure
to strong acids and alkalis. True contact dermatitis (i.e. allergic)
is extremely rare. However, contact urticaria is seen in sensitized
people following splashes with platinum salts and in some instances
this is the first indication of sensitization (Hughes, 1980).
The symptoms usually worsen with increasing duration of
exposure but generally disappear when the subject is removed from
exposure. The latter was shown by the follow-up study carried out on
platinum refinery workers who had to cease work with platinum salts
because of sensitization (Newman-Taylor, 1981). This study found no
evidence of long-term effects when workers giving a positive skin
prick test and showing symptoms of platinum sensitization were
removed immediately from contact with platinum salts. However,
Schultze-Werninghaus et al. (1989) reported that after long duration
exposure following sensitization individuals may never become
completely free of symptoms. Similarly, Biagini et al. (1985)
demonstrated the existence of positive platinum skin tests at very
low concentrations in workers who had been free of occupational
platinum exposure for periods of up to four years.
8.2.4 Immunological mechanism and diagnosis
The clinical manifestations of soluble platinum salt allergy
reflect a true allergic response based on the following clinical
criteria (Hughes, 1980; Biagini et al., 1985, 1986; Merget et al.,
1988; Schultze-Werninghaus et al., 1989):
* the appearance of sensitivity is preceded by a symptomless
exposure;
* not all exposed individuals become sensitized;
* the affected individuals become increasingly sensitive to
platinum and react even at very low levels of exposure;
* negative prick test results are obtained in atopic and non-
atopic controls.
The mechanism of platinum salt allergy appears to be a Type I
(IgE mediated) response. The possibility of the formation of IgE
antibodies to platinum chloride complexes in sensitized individuals
has been assumed on the grounds of allergy and serological tests. It
is believed that platinum salts of low relative molecular mass act
as haptens combining with serum proteins to form the complete
antigen. However, the actual immunological mechanism has not yet
been defined (Zachgo et al., 1985).
It has been demonstrated that platinum(II) reacts with the
sulfur atoms in the six methionine groups in human serum albumin
(HSA) and that methionine 123 is the primary binding site
(Grootveld, 1985).
Skin prick tests with freshly prepared solutions of soluble
platinum complexes appear to provide reproducible, reliable,
reasonably sensitive, and highly specific biological monitors of
allergenicity (Cleare et al., 1976; Dally et al., 1980). The
compounds used for routine screening of exposed workers are
(NH4)2[PtCl6], Na2[PtCl6], and Na2[PtCl4]. After
sensitization due to previous exposure, prick testing with
concentrations of the platinum compound in the range of 10-3 to
10-9 g/ml will produce immediate weal and flare reactions (Pepys
et al., 1972; Pickering, 1972; Hughes, 1980; Gallagher et al., 1982;
Biagini et al., 1985; Boggs, 1985; Jacobs, 1987; Linnett, 1987;
Murdoch & Pepys, 1987; Schultze-Werninghaus et al., 1989). At these
concentrations, nonspecific skin reactions were not found in atopic
or non-atopic controls (Pepys et al., 1972; Murdoch & Pepys, 1987;
Merget et al., 1988).
Passive transfer of immediate reactivity to intracutaneous
tests in humans was demonstrated in the Prausnitz-Küstner test by
Freedman & Krupey (1968). Schultze-Werninghaus et al. (1978)
observed positive reactions in passive cutaneous anaphylaxis (PCA)
in monkeys with serum from a platinum refinery worker. Similar tests
were performed by Pepys et al. (1979) and Biagini et al., (1985).
The results, however, were inconsistent, because positive as well as
negative Prausnitz-Küstner prick test or PCA reactions were elicited
in human recipients or in monkeys, respectively. Parish (1970) also
demonstrated the presence of heat-stable IgG antibodies by passive
cutaneous anaphylaxis on monkey skin. These results were confirmed
by Biagini et al. (1985).
The sensitivity and reliability of the skin prick test has not
been achieved in any in vitro test available. In enzyme
immunoassays (Zachgo et al., 1985; Merget et al., 1988) and in the
radioallergosorbent test (RAST) (Cromwell et al., 1979; Pepys et
al., 1979) IgE antibodies specific to platinum chloride complexes
were found. Although a good correlation with the results of prick
tests was reported (Cromwell et al., 1979), their practical
application for screening purposes was questioned because of the
lack of specificity (Boggs, 1985; Jacobs, 1987; Merget et al.,
1988). This was shown in a cross-sectional survey of platinum
refinery workers (Merget et al., 1988). Higher total serum IgE and
hexachloroplatinate-specific IgE levels in subjects with work-
related symptoms were noted. However, not all the individuals
allergic to platinum salt and some of the controls showed binding in
RAST. Similar effects were seen with in vitro histamine release
from basophils, which was relatively high in skin-test positive
workers but even higher in the atopic control group. Histamine
release with anti-IgE showed a similar pattern, indicating identical
binding sites of hexachloroplatinate and anti-IgE on the surface of
cutaneous mast cells and basophils.
Biagini et al. (1985) also found significantly higher mean
total serum IgE levels in platinum refinery workers. However, good
correlation between the RAST data and the skin test results was
seen. Of the workers with positive skin tests, 95% (20/22) showed
higher RAST binding than a control group, whereas only 8.5% (8/94)
of those with negative skin tests demonstrated positive RAST
results.
Since refinery workers are exposed to more than one platinum-
group metal salt, the question of cross-reactivity was investigated
by passive cutaneous anaphylaxis (PCA) tests. First results
indicated that platinum (Na2[PtCl6] and (NH4)2[PtCl6]) and
palladium (Na2[PdCl6]) appear to be equally effective as
eliciting agents. Five-fold concentrated sera from platinum refinery
workers produced positive PCA results in monkeys (Biagini et al.,
1982). No in situ reactions due to palladium salts were reported.
There was only limited cross-reactivity between platinum and
palladium salts in both skin test and RAST. Reactions to the
platinum-group metals other than platinum were only seen in
individuals sensitive to platinum salts (Murdoch et al., 1986;
Murdoch & Pepys, 1987).
Instillation in the nose of concentrations of 10-3 to 10-9
g per ml has been used in the past as another method of detecting
platinum salt sensitivity. A nasal reaction was considered positive
if itching, sneezing, nasal obstruction or discharge occurred singly
or in combination within 15 min of the challenge (Pepys et al.,
1972).
Inhalation tests with a mixture of ammonium hexachloroplatinate
and lactose dust gave immediate asthmatic reactions in sensitized
individuals and in one case a late asthmatic reaction occurred
(Pepys et al., 1972).
Merget et al. (1990) reported three cases of platinum refinery
workers with negative skin tests who showed non-specific hyper-
reactivity and a clearly positive immediate reaction in the
bronchial provocation test.
8.2.5 Predisposing factors
Dally et al. (1980) conducted a retrospective cohort analysis
in a group of 86 platinum workers entering a United Kingdom refinery
in 1973-1974. It was found that significantly more atopics left
employment, but this was apparently irrespective of the development
of platinum salt allergy. The incidence of the disease did not
differ significantly between the atopics (14/32 = 44%) and non-
atopics (21/54 = 39%), although Burge et al. (1979) demonstrated
that the atopics were sensitized more quickly. Thus, the increased
leaving rate of atopics cannot be regarded as proof for the atopic
status being a true predisposing factor, as suggested by Linnett
(1987). It may, at most, be considered a trend.
Merget et al. (1988) examined 27 refinery workers and found no
evidence to support tobacco smoking as a predisposing factor.
However, Linnett (1987) found a significant association between
smoking and the incidence of positive skin test results in life
table studies of 134 refinery workers. Also, in a longitudinal
cohort study on 91 platinum refinery workers (86 males, 5 females)
in the United Kingdom who started work in 1973-1974 and were
followed up until 1980, the risk of a positive skin test result was
found to be 4-5 times higher in smokers than in non-smokers
(Venables et al., 1989). Age, varying from 15-54 years in the
cohort, was a definite confounding factor. After taking account of
age, the risk of leaving refinery work was only 1.75 times greater
in smokers than in non-smokers. The risk from atopy was not
significant after taking smoking into consideration.
Brooks et al. (1990) further studied 107 current and 29
medically terminated workers, first described by Biagini (1985),
using platinum skin testing and cold air challenge for evaluation of
pulmonary hypersensitivity. Of these workers (74 current and 12
terminated workers), 63% underwent repeat platinum skin testing one
year later. Among current workers, there was a conversion to
positive platinum skin tests in five employees (with three of these
conversions occurring in workers who had positive cold air challenge
tests a year earlier). Thus, positive cold air challenge (airway
hyperactivity) appears to have value for predicting conversion to
positive skin test status with continued occupational exposure.
8.3 Side effects of cisplatina
The therapeutic use of cisplatin is often complicated by the
occurrence of side effects. Prominent among these are
nephrotoxicity, severe nausea and vomiting, myelotoxicity (bone
marrow suppression), and ototoxicity.
The most important toxic effect of cisplatin occurs in the
kidney, eventually becoming irreversible during continued treatment.
For instance, Lippman et al. (1973) found an approximately 50%
reduction in renal function in each of 16 patients after treatment
with total doses of 3.0-7.0 mg/kg body weight. Degeneration and
necrosis of the proximal convoluted tubules, dilation of distal
tubules, and glomerular abnormalities (elevation of the blood urea
nitrogen and serum creatine levels, decreased creatine clearance)
have been reported (Swierenga et al., 1987). A significant
protection of renal function can be obtained by forced hydration,
which flushes the drug through the kidney rapidly (Merrin, 1976).
The simultaneous intravenous administration of mannitol can
contribute to the prevention of cisplatin nephrotoxicity (Fillastre
& Raguenez-Viotte, 1989).
Gastrointestinal toxicity consists mainly of nausea and
vomiting lasting from 4 to 6 h and, occasionally in some sensitive
patients, anorexia for up to one week (Hill et al., 1975).
Ototoxicity is another serious side-effect, consisting of
tinnitus with or without clinical loss of hearing. Early in its
course it is almost exclusively associated with high-range hearing
loss in the 4000-8000 Hz range (Von Hoff et al., 1979).
Cisplatin can also cause peripheral neuropathy described as
sensory, affecting primarily large fibres (Mollman et al., 1988).
a See footnote in section 1.2.
Single cases of allergic reactions, angioneurotic oedema, rash,
asthma (Von Hoff et al., 1979), cardiac arrest (Vogl et al., 1980),
gingival discolouration (Ettinger & Freeman, 1979), and tetany due
to hypocalcaemia and hypomagnesaemia (Hayes et al., 1979) have all
been reported.
There are less toxic analogues, for example, cis-
diammine-1,1-cyclobutanedicarboxylato platinum(II) (Carboplatin,
JM8) and cis-dichloro- trans-dihydroxybisisopropylamine
platinum(IV) (JM9). These cause less kidney damage, nausea, and
vomiting. However, these analogues affect bone marrow and, in
addition to the negative effects of cisplatin, may inhibit the
formation of white cells, red cells, and blood platelets (Schacter &
Carter, 1986; Bradford, 1988).
8.4 Carcinogenicity
No data are available to assess the carcinogenic risk of
platinum or its salts to humans. With respect to cisplatin, IARC
(1987b) considered the evidence for carcinogenicity for humans to be
inadequate (see chapter 13).
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1 Microorganisms
Simple complexes of platinum have bactericidal effects. In
general, charged complexes in solutions, e.g., (NH4)2[PtCl6]
above a concentration of 1 mg/litre, are lethal for bacteria.
Neutral complexes are bactericidal only at considerably higher
concentrations (> 38 µmol per litre) (Rosenberg et al., 1967;
Shimazu & Rosenberg, 1973).
Rosenberg et al. (1965) reported the discovery of a unique
property of some simple platinum-group metal complexes. When culture
medium was subjected to an alternating current using platinum
electrodes, bacterial cell division was inhibited. The spent medium
itself was bactericidal. Detailed analysis revealed that the active
agent was the cis isomer of [PtCl2(NH3)2], i.e. cisplatin. It
was shown that such neutral platinum complexes, diluted in growth
media, selectively inhibit cell division without reducing the cell
growth of a variety of gram-positive and especially of gram-negative
bacteria. As a result the bacterial rods are forced to form long
filaments. This effect has been studied most intensively on
Escherichia coli with cisplatin. In this case filamentation is
reversed as soon as the bacterial filaments are transferred to a
fresh medium free of the drug (Rosenberg et al., 1967).
Hoffmann (1988) studied the effects of cisplatin and PtCl4 on
the in vitro metabolism of the yeast Saccharomyces cerevisiae.
Both compounds strongly inhibited DNA, RNA, and ribosome synthesis
in the mmol/litre range. The IC50 (median inhibitory
concentration) for the inhibition of DNA synthesis, for instance,
was 0.42 mmol/litre (126 mg/litre) for cisplatin and 0.2 mmol/litre
(67 mg per litre) for PtCl4.
9.2 Aquatic organisms
9.2.1 Plants
Barnes & Talbert (1984) studied the influence of
hexachloroplatinic acid (250, 500, and 750 µg/litre) on the growth
of the green alga Euglena gracilis using a laboratory "microcosm".
The growth recorded over 32 days was relatively slow, indicating
that the experiments were conducted under low nutrient conditions,
although these were not reported by the authors. For example, the
doubling time of the control culture was about 9 days. Although no
precise data were given, H2[PtCl6]reduced growth rate and yield
after 32 days in a dose-dependent manner.
After cisplatin was applied to the water hyacinth Eichhornia
crassipes at 2.5 mg/litre, chlorosis was evident and the plants
were stunted. At the 10-mg/litre level, some plant leaves were
necrotic and chlorotic, and the roots were darkened and stunted. The
most prominent symptom was the appearance of reddish-brown streaks
on the leaves. These were particularly noticeable on young leaves
and on the leaves of daughter plants (Farago & Parsons, 1985).
9.2.2 Animals
Biesinger & Christensen (1972), using Lake Superior water as a
medium, studied the effects of various metals on the survival,
growth, reproduction, and metabolism of the invertebrate Daphnia
magna. Chronic (3-week) exposure to hexachloroplatinic acid,
H2[PtCl6], resulted in an LC50 value of 520 µg Pt/litre (range
437-619 µg per litre). Biochemical measurements and reproductivity
were much more sensitive parameters than growth. A dose of 62
µg/litre caused a 12% reduction in weight gain, 13% reduction in
total protein, and 20% decrease in glutamic-oxalacetic transaminase
activity. At concentrations of 14 and 82 µg/litre, reproduction,
measured as total number of young, was impaired by 16 and 50%,
respectively.
Ferreira & Wolke (1979) investigated the effects of short-term
exposure to tetrachloroplatinic acid, H2[PtCl4], on the coho
salmon Oncorhynchus kisutch at 8.5°C and a water hardness of about
56 mg CaCO3/litre. In the static bioassay, 24-, 48-, and 96-h
LC50 values of 15.5, 5.2, and 2.5 mg Pt/litre, respectively, were
found. General swimming activity and opercular movement started to
be affected at 0.3 mg/litre. Lesions in the gills and the olfactory
organ were also noted at 0.3 mg/litre or more. Concentrations of
0.03 and 0.1 mg/litre had no effect.
9.3 Terrestrial organisms
A few studies have examined the effects of platinum on plants.
All were conducted with soluble platinum chlorides.
Hamner (1942) investigated the effect of hexachloroplatinic
acid, H2[PtCl6].6H2O, on the growth of beans and tomato plants
grown in sand culture. At concentrations of 3 x 10-5 to 15 x
10-5 mol/kg (5.9-29.3 mg/kg), growth was inhibited and the plants
showed smaller leaf areas, higher osmotic pressure, and lower
transpiration rates. They also resisted wilting longer than the
controls and were less succulent.
Tso et al. (1973) reported that platinum increased the nicotine
content of tobacco plants.
In a study by Pallas & Jones (1978) on the uptake of platinum
by nine horticultural crops (see section 4.1), effects on growth
were observed. Radish (Raphanus sativus), cauliflower ( Brassica
oleracea cv. Snowball), snapbean (Phaseolus vulgaris), sweet
corn (Zea mays), pea (Pisum sativum), tomato (Lycopersicon
esculentum), bell pepper (Capsicum annuum), broccoli ( Brassica
oleraceaw cv. Crusader), and turnips (Brassica rapa) were grown
in hydroponic solution at 25/20 °C, 60/90% relative humidity, and
320/400 µl CO2/litre air for 14/10 h photo- and nyctoperiods,
respectively. When the seedlings reached an early maturity stage,
such as flowering in the case of peas, snapbeans, cauliflower,
tomato, and broccoli, root expansion in the case of turnip and
radish, and considerable leafiness in the case of corn, platinum
tetrachloride, PtCl4, was added to fresh nutrient solution to give
concentrations of 0.057, 0.57, and 5.7 mg Pt/litre. After a 7-day
treatment, roots and tops were harvested and dried at 80 °C. As
shown in Table 17, dry weights were significantly reduced in tomato,
bell pepper, and turnip tops, and in radish roots at the highest
platinum concentration. At this level, the buds and immature leaves
of most species became chlorotic. In some of the species the low
levels of PtCl4 had a stimulatory effect on growth. In addition,
transpiration was suppressed at the highest platinum concentration,
probably due to increased stomatal resistance. Photosynthesis was
also apparently reduced, consistent with the observed growth
depression. On the other hand, the stimulation of transpiration and
growth observed at the two lower concentrations, as compared to the
control plants, explains the stimulated growth.
A stimulation of growth was also observed in seedlings of
Setaria verticillata (L. P. Beauv) treated with a low level of
platinum (0.5 mg Pt/litre) administered as potassium
tetrachloroplatinate, K2[PtCl4] (Farago & Parsons, 1986). This
South African grass species was grown in nutrient solution, and
after two weeks, the length of the longest roots had increased by
65%. At the higher concentration applied, i.e. 2.5 mg Pt/litre,
phytotoxic effects were seen in the form of stunted root growth,
i.e. root length about 75% compared to control, and chlorosis of the
leaves. As platinum was shown to accumulate in the roots and, at the
higher level, also in the shoots (see section 4.1), the potential
use of this grass species, either for the colonisation of flotation
tailings (a waste product from the concentration of precious metal
ores) or for the removal of platinum from the tailings, was
investigated. However, due to a substantial lack of essential
macronutrients in the tailings, growth of S. verticillata was very
poor. No platinum was detected in the plants.
Table 17. Dry weight (g) of tops and roots after a 7-day treatment with platinum tetrachloridea
Pt levels Bean Broccoli Cauliflower Corn Pea Pepper Radish Tomato Turnip
(mg/litre)
Tops
0 5.39 24.15 41.53 33.15 6.13 23.44 1.30 32.22 19.33
0.057 5.79 19.50 46.30 35.05 7.21 18.50 1.33 37.97 20.78
0.57 4.98 15.53 43.44 35.21 6.87 11.90 1.23 40.62 17.55
5.7 4.01 18.24 40.96 25.80 5.22 14.18 0.91 28.18 9.55
Roots
0 3.11 5.45 6.09 9.96 2.86 4.82 2.17 5.96 4.80
0.057 2.80 4.77 6.56 10.96 2.60 4.82 2.24 7.64 5.51
0.57 2.70 4.02 5.68 10.73 2.81 3.32 2.17 6.68 4.91
5.7 2.31 5.36 6.57 8.36 2.20 4.90 1.19 6.00 3.82
a Adapted from: Pallas & Jones (1978)
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of human health risks
10.1.1 General population exposure
10.1.1.1 Exposure
There is lack of data on the actual exposure situation in
countries where automobile exhaust gas catalysts have been
introduced. Therefore, estimates of possible ambient air
concentrations of platinum are based on emission data and dispersion
models.
Loss of platinum from the pellet-type catalyst, which has never
been used in Europe and is no longer used on new cars in the USA,
was found to be up to approximately 2 µg per km travelled. Of the
particles emitted, 80% had particle diameters greater than 125 µm.
Since no determination of the particle size distribution was
performed, the percentage of the respirable portion is not known.
About 10% of the platinum emission was found to be water-soluble. In
general, these data are based on single or only a few measurements
and have not been validated.
Recent emission data from the new-generation monolith-type
catalyst indicate that the emission of platinum is lower by a factor
of 100-1000 than that of the pelleted catalyst. Emissions were on
average between 2 and 39 ng per km travelled at simulated speeds of
between 60 and 140 km/h. The mean aerodynamic diameter of the
particles emitted was 4-9 µm. These data have been validated by
repeated measurements using two monolith-type catalysts. However,
other types of catalysts should be investigated to substantiate
these emission data. In addition, because of the inadequate data
base, the speciation is not known exactly, although there is an
indication that the platinum emitted is in the metallic form or
consists of surface-oxidized particles.
The striking difference between the emission pattern of the two
catalyst types may be attributable to their basically different
design.
The possible ambient air concentrations of platinum, estimated
on the basis of these emission data and dispersion models, range
between 0.005 and 9 ng/m3 for the pellet-type catalyst and between
0.05 and 90 pg/m3 for the monolith-type catalyst. These
concentrations are lower by factors of 1 x 105 to 2 x 108 and 1
x 107 to 2 x 1010, respectively, than the occupational exposure
limit of 1 mg/m3 established by some countries for platinum metal
as total inhalable dust.
Assuming that 10% of the platinum emission from pelleted
catalysts contains potentially allergenic soluble platinum
compounds, the safety factor to the occupational exposure limit for
soluble platinum salts (2 µg/m3) would be 2 x 103 to 4 x 106.
However, there is no evidence that the soluble fraction of the
platinum emissions is allergenic.
10.1.1.2 Health effects
Since platinum is most probably not emitted in the form of
halogenated soluble salts, the sensitization risk from car catalyst
platinum is very low. There is no substantial evidence of any
biological effects from automobile platinum emissions. There are
also no data to substantiate the possibility that very finely
dispersed metallic platinum could be biologically active upon
inhalation.
10.1.2 Occupational groups
10.1.2.1 Exposure
Occupational exposure to platinum occurs in various workplaces
including mining. However, only exposure to certain halogenated
soluble salts through inhalation of dusts and skin contact is of
toxicological relevance. These compounds are mainly encountered
during platinum refining and catalyst manufacture.
There are only limited data to quantify workplace exposure. An
occupational exposure limit of 2 µg/m3 for soluble platinum salts
has been adopted by several countries. There are again limited data
suggesting that the exposure limit may sometimes be exceeded in
practice. Before the allergenic potential of soluble platinum salts
was established, workplace concentrations exceeding the present
occupational exposure limit by up to one order of magnitude were
found. However, it should be noted that analytical accuracy was not
very reliable.
Occupational exposure to the anti-tumour agents cisplatin and
its analogues during manufacturing and use is of importance.
However, a review and an evaluation of the health effects of these
compounds are beyond the scope of this document as these substances
are used primarily as therapeutic agents. In addition, their
toxicological properties are exceptional compared to other platinum
compounds.
10.1.2.2 Health effects
The acute toxicity of platinum salts for animals is low and
depends on their solubility. Insoluble compounds such at PtCl2 and
PtO2 have an extremely low acute toxicity and this would also be
expected for metallic platinum.
By far the most significant health effect from exposure to
soluble platinum salts is sensitization.
Some halogenated platinum salts are highly allergenic in
humans. The compounds mainly responsible for platinum salt
hypersensitivity (PSH) are hexachloroplatinic acid, H2[PtCl6],
ammonium hexachloroplatinate, (NH4)2[PtCl6], and potassium
tetra- and hexachloroplatinate, K2[PtCl4] and K2[PtCl6].
Except for one unsubstantiated case of alleged contact dermatitis in
connection with a "platinum" ring, there is no evidence for
sensitization from metallic platinum.
The mechanism of platinum salt allergy appears to be a type I
(IgE mediated) response as established through in vivo and in
vitro tests. There is evidence that platinum salts of low relative
molecular mass act as haptens combining with serum proteins to form
the complete antigen.
The signs and symptoms of allergic reactions due to platinum
salt exposure include urticaria, itching of skin, eyes, and nose,
watering of the eyes, sneezing, rhinorrhoea, coughing, wheezing, and
dyspnoea. The latent period from the first contact with platinum
salts to the occurrence of the first symptoms varies from a few
weeks to several years. Once sensitivity is established, even minute
amounts can elicit immediate and/or late onset of signs and
symptoms. The symptoms persist during exposure and usually disappear
on removal from exposure. However, if long-duration exposure occurs
after sensitization, individuals may never become completely free of
symptoms.
The diagnosis of platinum salt hypersensitivity is usually
based on a history of work-related symptoms and a positive platinum
skin prick test. The combination of these has been shown to be
reasonably sensitive and specific for the diagnosis of platinum salt
hypersensitivity. In vitro tests appear to be useful for
epidemiological evaluation, but lack specificity for individual
dignosis. Symptoms usually worsen as long as the workers remain in
the contaminated environment.
There is good evidence for the association of smoking or
pulmonary hyper-reactivity and sensitization. The evidence for atopy
as a predisposing factor is equivocal. This may be due to bias from
pre-employment screening.
Despite the occupational exposure limit of 2 µg/m3, wthe
prevalence of positive skin prick tests was found to be between 14
and 20% in workers exposed to levels of between < 0.05 and 0.1 µg
Pt/m3. Since these data were derived from area samples, short
sharp exposures above this limit could also have been responsible
for the sensitization observed. The present occupational exposure
limit might not be sufficient to prevent platinum salt
hypersensitization, although it is difficult to reach a firm
conclusion because of the lack of adequate data. To minimize the
risk, workplace exposure should be as low as practicable.
10.2 Evaluation of effects on the environment
Compared to that of other metals, the total production of
platinum is low, amounting annually to approximately 100 tonnes.
There are no data on platinum emissions during production. However,
because of the high value of platinum, losses are assumed to be low.
During the use of platinum-containing catalysts, platinum can escape
into the environment in small amounts, depending on the type of
catalyst. Of the stationary catalysts used in industry, only those
used for ammonia oxidation emit a quantifiable amount of platinum.
This is present in the nitric acid produced, which may be used for
conversion to nitrate fertilizers. In the USA the annual loss of
platinum is estimated to be around 200 kg. Since part of this amount
is distributed fairly uniformly all over the country, a rise in the
background level of platinum in soil would probably not be detected
because of the very low likely concentration.
Platinum emission from automobile catalysts also contributes to
a diffuse contamination of the environment. On the basis of the
emission data derived from the new-generation monolithic-type
catalysts, total platinum loss from mobile sources would be less
than that from nitric acid production. For example, at an assumed
average emission rate of 20 ng platinum per km travelled, 100
million cars equipped with catalytic converters would emit
approximately 20 kg per year for an average kilometreage of 10 000
km per year and per car. This implies that the contamination of the
environment with platinum is very low or negligible.
In comparison, the total loss of platinum from the older design
pellet-type catalytic converter would have been higher by a factor
of 100, i.e. 2000 kg per year, most of the platinum being emitted in
the form of larger particles that would be deposited close to the
roads. This would also explain platinum levels of up to 0.7 mg/kg
dry weight found in roadside dust samples near major free-ways in
the USA.
There is limited evidence that most of the platinum emitted is
in the metallic form, and thus will probably not be bioavailable in
the soil. Biomethylation of soluble platinum(IV) compounds has been
demonstrated in the presence of platinum(II). However, from these
laboratory data produced under abiotic conditions, it is not
possible to conclude that microorganisms in the environment are able
to biomethylate platinum complexes.
Analysis of Lake Michigan sediments led to the conclusion that
platinum has been deposited over the past 50 years at a constant
rate, while lead concentrations have markedly increased. However,
since the car catalyst was introduced in the USA only a few years
before these measurements were performed, these data are
insufficient for firm conclusions to be drawn.
No data on the effects of platinum compounds on environmental
microorganisms are available. However, from the bactericidal
activity of platinum complexes it can be assumed that these
compounds could influence, at appropriate concentrations, microbial
communities in the environment or, for example, in sewage treatment
plants.
Aquatic and terrestrial plants are affected by platinum
compounds at concentrations in the mg/litre or mg/kg range. Although
there is a lack of definite data on platinum levels in the
environment, it is probable that platinum and platinum compounds do
not present a risk to naturally occurring organisms at the low
concentrations expected to occur in the environment.
11. RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH AND THE
ENVIRONMENT
11.1 Pre-employment screening and medical evaluations
To screen workers at risk of developing platinum salt
hypersensitivity (PSH), the following should be carried out for all
employees potentially exposed to soluble platinum salts:
* a questionnaire with particular attention being paid to
previous respiratory disease, allergy, smoking habits, and
employment history;
* a complete medical examination, including lung function tests
(spirometry, flow volume), tests of bronchial reactivity (cold
air, methacholine, histamine, etc.), and an immunological
profile including total serum IgE;
* a skin prick test for atopic status using a battery of antigens
to include house dust mite, tree and grass pollen or other
equivalent common aeroallergens;
* skin prick tests with freshly prepared, properly buffered
saline solutions (e.g., 5% v/v glycerol/water containing 2.5 g
NaCl/litre, 1.37 g NaHCO3/litre, and 2 g phenol per litre) of
(NH4)2[PtCl6], Na2[PtCl4], and Na2[PtCl6].
Concentrations used for testing may vary from 10-9 to 10-3
g/ml depending on specific situations. All tests should be
performed in duplicate and should include a positive and
negative saline control.
11.2 Substitution with non-allergenic substances
An attempt should be made to substitute, whenever practicable,
non-allergenic for allergenic platinum compounds during refining,
manufacturing, and use.
11.3 Employment screening and medical evaluations
a) To detect sensitization during employment, skin prick tests
should be performed on all potentially exposed people at least
once a year. There is no convincing evidence that repeated
platinum skin testing could cause sensitization. Quarterly
testing intervals might be considered during the first two
years of employment, as sensitization more often occurs during
this period.
b) If medical symptoms or signs suggest the development of PSH,
the worker should be removed from any risk of exposure as soon
as possible. To detect functional changes in the respiratory
tract, lung function assessment, as described in section 11.1,
should also be performed at appropriate intervals.
11.4 Workplace hygiene
a) Since PSH can occur despite time-weighted average workplace
concentrations being consistently below the ACGIH threshold
limit value (TLV) of 2 µg/m3, the most effective prevention
is the improvement of control measures. This includes enclosed
processing and optimal ventilation in order to reduce exposure
to platinum salt aerosols and dusts to the lowest practicable
limit.
b) It has been suggested that high but short-lived platinum
concentrations resulting from spills or accidents are of
importance with respect to sensitization. Since the correlation
between the platinum exposure concentration and the development
of sensitization is unknown, a recommendation for a reduction
in the occupational exposure limit cannot be justified.
However, it is recommended that the commonly used occupational
exposure limit of 2 µg/m3 be changed from an 8-h time-
weighted average (TWA) to a ceiling value and that personal
sampling devices be used in conjunction with area sampling to
determine more correctly the true platinum exposure.
c) Engineering controls should always be in place to minimize
exposure. However, in some circumstances the use of protective
clothing, including specially designed airstream helmets, may
be necessary.
d) Workers should be provided with clean overalls solely for use
in the workplace, and showering facilities. Outdoor clothes
should not be worn in the workplace.
12. FURTHER RESEARCH
a) As there appears to be a lack of information concerning the
concentration-response relationship for the development of PSH
in experimental animals, studies should be performed to
investigate the effect of exposure concentration on
sensitization and to define the thresholds for sensitization
and elicitation.
b) The effect of predisposing factors such as pulmonary hyper-
reactivity should be investigated in greater detail to
determine their applicability for screening and identifying
individuals at risk of developing PSH.
c) The use of provocation challenge with soluble platinum salts as
an indicator of sensitization should be investigated to
determine if it is a more sensitive indicator than skin prick
tests.
d) The majority of human occupational studies regarding PSH were
performed as cross-sectional studies at platinum refineries.
Due to the inherent lack of sensitivity of this type of study
with respect to past exposures and workers leaving employment
because of disease, longitudinal studies should be performed to
determine the true incidence of PSH in worker populations. In
addition, human studies should be designed to study, for
instance, exposure concentration effects on sensitization and
determine thresholds for sensitization and elicitation.
e) The extent of occupational and environmental exposure to
cisplatin is not known at the present time. It is recommended
that studies be initiated to determine exposure during the
manufacture and use of this compound.
f) Further measurements of the quantities and speciation of
platinum emitted from automobile catalysts should be performed.
g) The toxic effects of finely divided metallic platinum on humans
and animals have not been studied adequately. Adequate
inhalation studies are initially required, and further tests
may be necessary.
h) Quality control programmes should be initiated to ensure the
accuracy and precision of sampling methods and analyses and to
facilitate comparability.
i) Platinum-containing exhaust emissions from automobile catalysts
most probably do not pose an adverse health effect for the
general population. However, to be on the safe side, the
possibility should be kept under review.
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
The carcinogenicity of platinum and platinum compounds has not
been evaluated by international bodies, except for cisplatin, which
has not been covered in detail in this Environmental Health Criteria
monograph (see also footnote in section 1.2).
The International Agency for Research on Cancer (IARC, 1987b)
considered the evidence for carcinogenicity of cisplatin for animals
to be sufficient, but that for humans inadequate. Cisplatin is
classified in Group 2A, i.e. probably carcinogenic to humans.
REFERENCES
ACGIH (1980) Documentation of the threshold limit values, 4th ed.,
Cincinnati, Ohio, American Conference of Governmental Industrial
Hygienists, pp. 343-344.
ACGIH (1990) Threshold limit values for chemical substances and
physical agents and biological exposure indices for 1990-1991,
Cincinnati, Ohio, American Conference of Governmental Industrial
Hygienists, p. 31.
AGIORGITIS, G. & GUNDLACH, H. (1978) [Platinum contents of deep sea
manganese nodules.] Naturwissenschaften, 65: 534 (in German).
AGNES, G., HILL, H.A.O., PRATT, J.M., RIDSDALE, S.C., KENNEDY, F.S.,
& WILLIAMS, R.J.P. (1971) Methyl transfer from methyl vitamin B12.
Biochim. Biophys. Acta, 252: 207-211.
AGOOS, A. (1986) Metal reclaimers find slim pickings. Chem. Week,
138: 21-23.
ALEXANDER, P.W., HOH, R., & SMYTHE, L.E. (1977a) Trace determination
of platinum, I. A catalytic method using pulse polarography.
Talanta, 24: 543- 548.
ALEXANDER, P.W., HOH, R., & SMYTHE, L.E. (1977b) Trace determination
of platinum, II. Analysis in ores by pulse polarography after fire-
assay collection. Talanta, 24: 549-554.
ALT, F., JERONO, U., MESSERSCHMIDT, J., & TÖLG, G. (1988) The
determination of platinum in biotic and environmental materials: I.
µg/kg to g/kg- range. Mikrochim. Acta, 3: 299-304.
AMOSSE, J., FISCHER, W., ALLIBERT, M., & PIBOULE, M. (1986) Méthode
de dosage d'ultra-traces de platine, palladium, rhodium et or dans
les roches silicatées par spectrophotométrie d'absorption atomique
électrothermique. Analusis, 14: 26-31.
ANDERSEN, K.S. (1979) Platinum(II) complexes generate frame-shift
mutations in test strains of Salmonella typhimurium. Mutat. Res.,
67: 209-214.
ANDREWS, P.A., WUNG, W.E., & HOWELL, S.B. (1984) A high-performance
liquid chromatographic assay with improved selectivity for cisplatin
and active platinum (II) complexes in plasma ultrafiltrate. Anal.
Biochem., 43: 46-56.
ANEVA, Z., ARPADJAN, S., ALEXANDROV, S., & KOVATCHEVA, K. (1986)
Synergetic extraction of platinum(IV) from dilute hydrochloric acid
by isoamyl alcohol-methylisobutylketone mixture. Mikrochim. Acta, 1:
341-350.
ANON. (1990a) Nitric acid catalysts: the economic significance of
composition, operation conditions and recovery techniques. Nitrogen,
183: 27-32.
ANON. (1990b) Growth slackens appreciably as 1980s end. Chem. Eng.
News, 18 June: 36-43.
ANUSAVICE, K.J. (1985) Noble metal alloys for metal-ceramic
restorations. Dent. Clin. North Am., 29: 789-803.
ARPADJAN, S., MITEWA, M., & BONTCHEV, P.R. (1987) Liquid-liquid
extraction of metal ions by the 6-membered N-containing macrocycle
hexacyclen. Talanta, 34: 953-956.
BANKOVSKY, Y.A., VIRCAVS, M.V., VEVERIS, O.E., PELNE, A.R., &
VIRCAVA, D.K. (1987) Preconcentration of microamounts of elements in
natural waters with 8-mercaptoquinoline and bis(8-quinolyl)
disulphide for their atomic-absorption determination. Talanta, 34:
179-182.
BANNISTER, S.J., CHANG, Y., STERNSON, L.A., & REPTA, A.J. (1978)
Atomic absorption spectrophotometry of free circulating platinum
species in plasma derived from cis-dichlorodiammineplatinum(II).
Clin. Chem., 24: 877-880.
BARLOW, S.M. & SULLIVAN, F.M. (1982) Reproductive hazards of
industrial chemicals, London, Academic Press, pp. 431-436.
BARNES, G.D. & TALBERT, L.D. (1984) The effect of platinum on
population and absorbance of Euglena gracilis, Klebs utilizing a
method with atomic absorption and coulter counter analysis. J.
Mississipi Acad. Sci., 29: 143-150.
BAUMGÄRTNER, M.E. & RAUB, Ch.J. (1988) The electrodeposition of
platinum and platinum alloys. Platinum Met. Rev., 32: 188-197.
BELLIVEAU, J.F., MATOOK, G.M., O'LEARY, G.P., Jr, CUMMINGS, F.J.,
HILLSTROM, M., & CALABRESI, P. (1986) Microanalysis for platinum and
magnesium in body fluids and brain tissue of mice treated with cis-
platinum using graphite filament plasma emission spectroscopy. Anal.
Lett., 19: 135- 149.
BERNDT, H., SCHALDACH, G., & KLOCKENKÄMPER, R. (1987) Improvement of
the detection power in electrothermal atomic absorption spectrometry
by summation of signals. Determination of traces of metals in
drinking water and urine. Anal. Chim. Acta, 200: 573-579.
BIAGINI, R.E., CLARK, J.C., GALLAGHER, J.S., BERNSTEIN, I.L., &
MOORMAN, W.M. (1982) Passive transfer in the monkey of human
immediate hyper- sensitivity to complex salts of platinum and
palladium. Fed. Proc., 41: 827.
BIAGINI, R.E., MOORMAN, W.J., SMITH, R.J., LEWIS, T.R., & BERNSTEIN,
I.L. (1983) Pulmonary hyperreactivity in cynomolgus monkeys (Macaca
fasicularis) from nose-only inhalation exposure to disodium
hexachloroplatinate, Na2PtCl6. Toxicol. appl. Pharmacol., 69:
377-384.
BIAGINI, R.E., BERNSTEIN, I.L., GALLAGHER, J.S., MOORMAN, W.J.,
BROOKS, S., & GANN, P.H. (1985) The diversity of reaginic immune
responses to platinum and palladium metallic salts. J. Allergy clin.
Immunol., 76: 794-802.
BIAGINI, R.E., MOORMAN, W.J., LEWIS, T.R., & BERNSTEIN, I.L. (1986)
Ozone enhancement of platinum asthma in a primate model. Am. Rev.
Respir. Dis., 134: 719-725.
BIESINGER, K.E. & CHRISTENSEN, G.M. (1972) Effects of various metals
on survival, growth, reproduction, and metabolism of Daphnia magna.
J. Fish Res. Board Can., 29: 1691-1700.
BODENNER, D.L., DEDON, P.C., KENG, P.C., KATZ, J.C., & BORCH, R.F.
(1986) Selective protection against cis-diamminedichloroplatinum
(II)-induced toxicity in kidney, gut, and bone marrow by
diethyldithiocarbamate. Cancer Res., 46: 2751-2755.
BOGGS, P.B. (1985) Platinum allergy. Cutis, 35: 318-320.
BOLM-AUDORFF, U., BIENFAIT, H.-G., BURKHARD, J., BURY, A.-H.,
MERGET, R., PRESSEL, G., & SCHULTZE-WERNINGHAUS, G. (1988) [On the
frequency of respiratory allergies in a platinum processing
factory.] In: Baumgartner, E., Brenner, W., Dierich, M.P., &
Rutenfranz, J., ed. [Industrial change - Occupational medicine
facing new questions. Report of the 28th Annual Meeting of the
German Society for Occupational Medicine, Innsbruck, Austria, 4-7
May 1988] Stuttgart, Gentner Verlag, pp. 411-416 (in German).
BORCH, R.F., MARKOVITZ, J.H., & PLEASANTS, M.E. (1979) A new method
for the HPLC analysis of Pt(II) in urine. Anal. Lett., 12: 917-926.
BOUMANS, P.W.J.M. & VRAKKING, J.J.A.M. (1987) Detection of about 350
prominent lines of 65 elements observed in 50 and 27 MHz inductively
coupled plasmas (ICP): effects of source characteristics, noise and
spectral bandwidth - "Standard" values for the 50 MHz ICP.
Spectrochim. Acta, 42B: 553-579.
BOWEN, H.J.M. (1979) Environmental chemistry of the elements,
London, Academic Press, p. 37.
BRADFORD, C.W. (1988) Platinum. In: Seiler, H.G. & Sigel, H., ed.
Handbook of toxicity of inorganic compounds, New York, Basel, Marcel
Dekker, pp. 533-539.
BRAJTER, K. & KOZICKA, U. (1979) Extractive spectrophotometric
determination of platinum, rhodium and iridium. Talanta, 26:
417-419.
BROOKS, S.M., BAKER, D.B., GANN, P.H., JARABEK, A.M., HERTZBERG, V.,
GALLAGHER, J., BIAGINI, R.E., & BERNSTEIN, I.L. (1990) Cold air
challenge and platinum skin reactivity in platinum refinery workers.
Bronchial reactivity precedes skin prick response. Chest, 79:
1401-1407.
BROWN, A.A. & LEE, M. (1986) Peak profile and appearance times using
totally pyrolytic cuvettes in graphite furnace atomic absorption
spectrometry. Anal. Chem., 323: 697-702.
BRUBAKER, P.E., MORAN, J.P., BRIDBORD, K., & HUETER, F.G. (1975)
Noble metals: a toxicological appraisal of potential new
environmental contaminants. Environ. Health Perspect., 10: 39-56.
BURGE, P.S., HARRIES, M.G., O'BRIEN, I.M., & PEPYS, J. (1979)
Evidence for specific hypersensitivity in occupational asthma due to
small molecular weight chemicals and an organic (locust) allergen.
In: Pepys, J. & Edwards, A.M., ed. The mast cell - its role in
health and disease, Tunbridge Wells, Kent, United Kingdom, Pitman
Medical Publishing Co., Ltd, pp. 301-308.
BUTTERMAN, W.C. (1975) Platinum-group metals. Minerals Yearbook 1973
Metals, Minerals, and Fuels, Washington, DC, US Department of the
Interior, Bureau of Mines, pp. 1037-1049.
CAMPBELL, K.I., GEORGE, E.L., HALL, L.L., & STARA, J.F. (1975)
Dermal irritancy of metal compounds. Arch. environ. Health, 30:
168-170.
CHANG, C.C. & TATUM, H.J. (1975) Effect of intrauterine copper wire
on resorption of foetuses in rats. Contraception, 11: 79-84.
CHANG, C.C., TATUM, H.J., & KINCL, F.A. (1970) The effect of
intrauterine copper and other metals on implantation in rats and
hamsters. Fertil. Steril. 21(3): 274-278.
CHEY, R.D., LEE, K.Y, ASBURY, R., & CHEY, W.Y. (1988) Effect of
cisplatin on myoelectric activity of the stomach and small intestine
in dogs. Dig. Dis. Sci., 33: 338-344.
CLEARE, M.J. (1977a) Some aspects of platinum complex chemistry and
their relation to anti-tumor activity. J. clin. Hematol. Oncol., 7:
1-21.
CLEARE, M.J. (1977b) Immunological studies on platinum complexes and
their possible relevance to autocatalysts. Proceedings of the
International Automotive Engineering Congress and Exposition, Cobo
Hall, Detroit, 28 February - 4 March 24, 1977, Detroit, Michigan
Society of Automotive Engineers, 12 pp (SAE Report No. 770061).
CLEARE, M.J., HUGHES, E.G., JACOBY, B., & PEPYS, J. (1976) Immediate
(type I) allergic responses to platinum compounds. Clin. Allergy, 6:
183-195.
COOMBES, R.J. & CHOW, A. (1979) A comparison of methods for the
determination of platinum in ores. Talanta, 26: 991-998.
CROMWELL, O., PEPYS, J., PARISH, W.E, & HUGHES, E.G. (1979) Specific
IgE antibodies to platinum salts in sensitized workers. Clin.
Allergy, 9: 109-117.
CVITKOVIC, E., SPAULDING, J., BETHUNE, V., MARTIN, J., & WHITMORE,
W.F. (1977) Improvement of cis-dichlorodiammineplatinum (NSC
119875): therapeutic index in an animal model. Cancer, 39:
1357-1361.
D'AGOSTINO, R.B., LOWN, B.A., MORGANTI, J.B., CHAPIN, E., & MASSARO,
E.J. (1984) Effects on the development of offspring of female mice
exposed to platinum sulfate or sodium hexachloroplatinate during
pregnancy or lactation. J. Toxicol. environ. Health, 13: 879-891.
DALLY, M.B., HUNTER, J.V., HUGHES, E.G., STEWART, M., & NEWMAN
TAYLOR, A.J. (1980) Hypersensitivity to platinum salts: a population
study. Am. Rev. respir. Dis., 121(4/Part 2): 230.
DE CONTI, R.C., TOFTNESS, B.R., LANGE, R.C., & CREASEY, W.A. (1973)
Clinical and pharmacological studies with cis-diammine-
dichloroplatinum (II). Cancer Res., 33: 1310-1315.
DEGUSSA (1988a) Ammonium-tetrachloroplatinate(II) - Acute toxicity.
Testing the primary irritancy after single application to the skin
of the rabbit (patch test), Hanau, Germany, Degussa AG (Unpublished
report No. 863897).
DEGUSSA (1988b) Ammonium-tetrachloroplatinate(II) - Acute toxicity.
Testing the primary irritancy after single application to the eye of
the rabbit, Hanau, Germany, Degussa AG (Unpublished report No.
863908).
DEGUSSA (1988c) Trans-diammine-dichloroplatinum(II) - Acute
toxicity. Testing the primary irritancy after single application to
the skin of the rabbit, Hanau, Germany, Degussa AG (unpublished
report No. 864011).
DEGUSSA (1988d) Trans-diammine-dichloroplatinum(II) - Acute
toxicity. Testing the primary irritancy after single application to
the eye of the rabbit, Hanau, Germany, Degussa AG (Unpublished
report No. 864022).
DEGUSSA (1989a) Ammoniumtetrachloroplatinate(II) - Acute toxicity
after single oral administration in rats, Hanau, Germany, Degussa AG
(Unpublished report No. 863886).
DEGUSSA (1989b) Dinitrodiammine-platinum(II) - Acute toxicity after
single oral administration in rats, Hanau, Germany, Degussa AG
(Unpublished report No. 863910).
DEGUSSA (1989c) Trans-diammine-dichloroplatinum(II) - Acute toxicity
after single oral administration in rats, Hanau, Germany, Degussa AG
(Unpublished report No. 864000).
DEGUSSA (1989d) Dinitrodiammine-platinum(II) - Acute toxicity.
Testing the primary irritancy after single application to the skin
of the rabbit (patch test), Hanau, Germany, Degussa AG (Unpublished
report No. 863921).
DEGUSSA (1989e) Dinitrodiammine-platinum(II) - Acute toxicity.
Testing the primary irritancy after single application to the eye of
the rabbit), Hanau, Germany, Degussa AG (Unpublished report No.
863932).
DISSANAYAKE, C.B. (1983) Metal-organic interactions in environmental
pollution. Int. J. environ. Stud., 22: 25-42.
DISSANAYAKE, C.B., KRITSOTAKIS, K., & TOBSCHALL, H.J. (1984) The
abundance of Au, Pt, Pd, and the mode of heavy metal fixation in
highly polluted sediments from the Rhine River near Mainz, West
Germany. Int. J. environ. Stud., 22: 109-119.
DONALDSON, P.E.K. (1987) The role of platinum metals in neurological
prostheses. Platinum Met. Rev., 31: 2-7.
DUFFIELD, F.V.P., YOAKUM, A., BUMGARNER, J., & MORAN, J. (1976)
Determination of human body burden baseline data of platinum through
autopsy tissue analysis. Environ. Health Perspect., 15: 131-134.
EBINA, T., SUZUKI, H., & YOTSUYANAGI, T. (1983) [Spectrophotometric
determination of palladium(II) and platinum(II) with
maleonitriledithiol by reversed phase ion-pair chromatography.]
Bunseki Kagaku, 32: 575-580 (in Japanese).
ELLER, P.M., ed. (1984a) Elements (ICP). In: NIOSH manual of
analytical methods, 3rd ed., Cincinnati, Ohio, National Institute
for Occupational Safety and Health, Vol. 1, pp. 7300-1-7300-5.
ELLER, P.M., ed. (1984b) Elements in blood or tissue. In: NIOSH
manual of analytical methods, 3rd ed., Cincinnati, Ohio, National
Institute for Occupational Safety and Health, Vol.2, pp.
8005-1-8005-5.
ELLER, P.M., ed. (1984c) Metals in urine. In: NIOSH manual of
analytical methods, 3rd ed., Cincinnati, Ohio, National Institute
for Occupational Safety and Health, Vol. 2, pp. 8310-1-8310-6.
ETTINGER, L.J. & FREEMAN, A.I. (1979) The gingival platinum line. A
new finding following cis-dichlorodiammine platinum(II) treatment.
Cancer, 44: 1882-1884.
FANCHIANG, Y.-T. (1985) Reactions of alkylcobalamins with platinum
complexes. Coord. Chem. Rev., 68: 131-144.
FANCHIANG, Y-T., RIDLEY, W.P., & WOOD, J.M. (1979) Methylation of
platinum complexes by methylcobalamin. J. Am. Chem. Soc., 101:
1442-1447.
FARAGO, M.E. & PARSONS, P.J. (1982) Determination of platinum,
palladium and rhodium by atomic-absorption spectroscopy with
electrothermal atomisation. Analyst, 107: 1218-1228.
FARAGO, M.E. & PARSONS, P.J. (1985) Effects of platinum metals on
plants. Trace Subst. environ. Health, 19: 397-407.
FARAGO, M.E. & PARSONS, P.J. (1986) The effect of platinum, applied
as potassium tetrachloroplatinate, on Setaria verticillata (L) P.
Beauv. and its growth on flotation tailings. Environ. Technol.
Lett., 7: 147-154.
FERREIRA, P.F. & WOLKE, R.E. (1979) Acute toxicity of platinum to
coho salmon (Oncorhynchus kisutch). Mar. Pollut. Bull., 10: 79-83.
FILLASTRE, J.P. & RAGUENEZ-VIOTTE, G. (1989) Cisplatin
nephrotoxicity. Toxicol. Lett., 46: 163-175.
FOTHERGILL, S.J.R., WITHERS, D.F., & CLEMENTS, F.S. (1945)
Determination of traces of platinum and palladium in the atmosphere
of a platinum refinery. Br. J. ind. Med., 2: 99-101.
FOX, R.L. (1984) Enhancement errors in the determination of platinum
group metals in alumina-based matrices by direct-current plasma
emission spectrometry. Appl. Spectrosc., 38: 644-647.
FREEDMAN, S.O. & KRUPEY, J. (1968) Respiratory allergy caused by
platinum salts. J. Allergy, 42: 233-237.
FRYER, B.J. & KERRICH, R. (1978) Determination of precious metals at
ppb levels in rocks by a combined wet chemical and flameless atomic
absorption method. At. Absorpt. Newsl., 17: 4-6.
FUCHS, W.A. & ROSE, A.W. (1974) The geochemical behavior of platinum
and palladium in the weathering cycle in the Stillwater Complex,
Montana. Econ. Geol., 69: 332-346.
GALLAGHER, J.S., BAKER, D., GANN, P.H., JARABEK, A.M., BROOKS, S.M.,
& BERNSTEIN, I.L. (1982) A cross sectional investigation of workers
exposed to platinum salts. J. Allergy clin. Immunol., 69: 134.
GECKELER, K.E., BAYER, E., SPIVAKOV, B.Y., SHKINEV, V.M., &
VOROB'EVA, G.A. (1986) Liquid-phase polymer-based retention; a new
method for separation and preconcentration of elements. Anal. Chim.
Acta, 189: 285-292.
GLADKOVA, E.V., ODINTSOVA, F.P., VOLKOVA, I.D., & VINOGRADOVA, V.K.
(1974) [Health status of workers engaged in the production of the
platinum catalyst.] Gig. Tr. prof.Zabol., 3: 10-13 (in Russian).
GOLDBERG, E.D., HODGE, V.F., GRIFFIN, J.J., KOIDE, M., & EDGINGTON,
D.N. (1981) The impact of fossil fuel combustion on the sediments of
Lake Michigan. Environ. Sci. Technol., 15: 466-471.
GOLDBERG, E.D., HODGE, V., KAY, P., STALLARD, M., & KOIDE, M. (1986)
Some comparative marine chemistries of platinum and iridium. Appl.
Geochem., 1: 227-232.
GOLDSCHMIDT, V.M. (1954) Geochemistry, London, Oxford University
Press, 730 pp.
GREGOIRE, D.C. (1988) Determination of platinum, palladium,
ruthenium and iridium geological materials by inductively coupled
plasma mass spectrometry with sample introduction by electrothermal
vaporization. J. anal. at. Spectrom., 3: 309-314.
GROOTVELD, M.C. (1985) Studies on the reaction mechanisms of metal
complexes of biological and immunochemical interest, London,
University of London, Department of Chemistry, 341 pp (Thesis).
GROTE, M. & KETTRUP, A. (1987) Ion-exchange resins containing S-
bonded dithizone and dehydrodithizone as functional groups. Part 3.
Determination of gold, platinum and palladium in geological samples
by means of a dehydrodithizone resin and plasma emission
spectrometry. Anal. Chim. Acta, 201: 95-107.
GSF (1990) [Noble metal emissions - interim report; Research
Programme of the Federal Ministry of Research and Technology],
Munich, Association for Radiation and Environmental Research (GSF),
45 pp. (in German).
HALBACH, P., PUTEANUS, D., & MANHEIM, F.T. (1984) Platinum
concentrations in ferromanganese seamount crusts from the central
Pacific. Naturwissenschaften, 71: 577-579.
HAMILTON, E.L. & MINSKI, M.J. (1972/1973) Abundance of the chemical
elements in man's diet and possible relations with environmental
factors. Sci. total Environ., 1: 375-394.
HAMNER, C.L. (1942) Effects of platinum chloride on bean and tomato.
Bot. Gaz., 104: 161-166.
HARDER, H.C. & ROSENBERG, B. (1970) Inhibitory effects of anti-tumor
platinum compounds on DNA, RNA and protein syntheses in mammalian
cells in vitro. Int. J. Cancer, 6: 207-216.
HARDMAN, R.S. & WRIGHT, C.H. (1896) A case of poisoning by
chloroplatinite of potassium. Br. med. J., 1: 529.
HAYES, F.A., GREEN, A.A., SENZER, N., & PRATT, C.B. (1979) Tetany: a
complication of cis-dichlorodiammine-platinum(II) therapy. Cancer
Treat. Rep., 63: 547-548.
HEALTH AND SAFETY EXECUTIVE (1983) The medical monitoring of workers
exposed to platinum `salts', London, Her Majesty's Stationery
Office, 2 pp (MS/22).
HEALTH and SAFETY EXECUTIVE (1985) Platinum metal and soluble
inorganic compounds of platinum in air. Laboratory method using
carbon furnace atomic absorption spectrometry, London, Her Majesty's
Stationery Office, 4 pp (MDHS 46).
HEALTH AND SAFETY EXECUTIVE (1990) Occupational exposure limits
1990. Guidance note EH 40190, London, Her Majesty's Stationery
Office.
HEBERT, R. (1966) Affections provoquées par les composés du platine.
Arch. Mal. prof. Méd. Trav. Sécur. soc., 27: 877-886.
HILL, J.M., LOEB, E., MACLELLAN, A., HILL, N.O., KHAN, A., & KING,
J.J. (1975) Clinical studies of platinum coordination compounds in
the treatment of various malignant diseases. Cancer Chemother. Rep.,
59(Part 1): 647-659.
HILL, R.F. & MAYER, W.J. (1977) Radiometric determination of
platinum and palladium attrition from automotive catalysts. IEEE
Trans. nucl. Sci., NS-24: 2549-2554.
HODGE, V.F. & STALLARD, M.O. (1986) Platinum and palladium in
roadside dust. Environ. Sci. Technol., 20: 1058-1060.
HODGE, V.F., STALLARD, M., KOIDE, M., & GOLDBERG, E.D. (1985)
Platinum and the platinum anomaly in the marine environment. Earth
planet. Sci. Lett., 72: 158-162.
HODGE, V.F., STALLARD, M., KOIDE, M., & GOLDBERG, E.D. (1986)
Determination of platinum and iridium in marine waters, sediments,
and organisms. Anal. Chem., 58: 616-620.
HOESCHELE, J.D. & VAN CAMP, L. (1972) Whole-body counting and the
distribution of cis195m[Pt(NH3)2Cl2] in the major organs of
Swiss white mice. In: Hejzlar, M., ed. Advances in antimicrobial and
antineoplastic chemotherapy, Baltimore, University Park Press, Vol.
2, pp. 241-242.
HOEVE, L.J., CONIJIN, E.A.J.G., MERTENS ZUR BORG, I.R.A.M., &
RODENBURG, M. (1987) Development of cis-platinum-induced hearing
loss in guinea pigs. Arch. Otorhinolaryngol., 244: 265-268.
HOFFMANN, R.L. (1988) The effect of cisplatin and platinum (IV)
chloride on cell growth, RNA, protein, ribosome and DNA synthesis in
yeast. Toxicol. environ. Chem., 17: 139-151.
HOFMEISTER, F. (1882) [On the physiologic effects of the platinum
bases.] Naunyn-Schmiedeberg's Arch. exp. Pathol. Pharmakol., 16:
393-439 (in German).
HOLBROOK, D.J., Jr (1976a) Assessment of toxicity of automotive
metallic emissions, Vol. I: Assessment of fuel additives emission
toxicity via selected assays of nucleic and protein synthesis,
Research Triangle Park, North Carolina, US Environmental Protection
Agency, Office of Research and Development, Health Effects Research
Laboratories, 67 pp (EPA/600/1-76/010a).
HOLBROOK, D.J., Jr (1976b) Assessment of toxicity of automotive
metallic emissions, Vol. II: Relative toxicities of automotive
metallic emissions against lead compounds using biochemical
parameters, Research Triangle Park, North Carolina, US Environmental
Protection Agency, Office of Research and Development, Health
Effects Research Laboratories, 69 pp. (EPA/600/1-76/010b).
HOLBROOK, D.J., Jr (1977) Content of platinum and palladium in rat
tissue: correlation of tissue concentration of platinum and
palladium with biochemical effects, Research Triangle Park, North
Carolina, US Environmental Protection Agency, Office of Research and
Development, Health Effects Research Laboratory, 21 pp.
(EPA/600/1-77/051).
HOLBROOK, D.J., Jr, WASHINGTON, M.E., LEAKE, H.B., & BRUBAKER, P.E.
(1975) Studies on the evaluation of the toxicity of various salts of
lead, manganese, platinum, and palladium. Environ. Health Perspect.,
10: 95- 101.
HOPPE, R. (1965) [Chemistry of the noble gases.] Fortschr. chem.
Forsch., 5: 213-346 (in German).
HOPPSTOCK, K., ALT, F., CAMMANN, K., & WEBER, G. (1989)
Determination of platinum in biotic and environmental materials.
Part II: A sensitive voltammetric method. Fresenius Z. anal. Chem.,
335: 813-816.
HOWLE, J.A. & GALE, G.R. (1970) Cis-dichlorodiammineplatinum (II)
persistent and selective inhibition of deoxyribonucleic acid
synthesis in vivo. Biochem. Pharmacol., 19: 2757-2762.
HUGHES, E.G. (1980) Medical surveillance of platinum refinery
workers. J. Soc. Occup. Med., 30: 27-30.
HUNTER, D., MILTON, R., & PERRY, K.M.A. (1945) Asthma caused by the
complex salts of platinum. Br. J. ind. Med., 2: 92-98.
IARC (1987a) Cisplatin. In: Genetic and related effects: an updating
of selected IARC Monographs from Volumes 1 to 42, Lyon,
International Agency for Research on Cancer, pp. 178-181 (IARC
Monographs on the Evaluation of Carcinogenic Risks to Humans, Suppl.
6).
IARC (1987b) Cisplatin (Group 2A). In: Overall evaluations of
carcinogenicity: an updating of IARC Monographs Volumes 1 to 42,
Lyon, International Agency for Research on Cancer, pp. 170-171 (IARC
Monographs on the Evaluation of Carcinogenic Risks to Humans, Suppl.
7).
IMURA, N. (1986) The role of micronutrient selenium in the
manifestation of toxicity of heavy metals. Toxicol. Lett., 31: 11.
INGALLS, M.N. & GARBE, R.J. (1982) Ambient pollutant concentrations
from mobile sources in microscale situations, Warrendale,
Pennsylvania, Society of Automotive Engineers, Inc., 16 pp (SAE
Technical Paper Series. No. 820787).
ITO, E. & KIDANI, Y. (1982) Determination of platinum in the
environmental samples by graphite furnace atomic absorption
spectrophotometry. Bunseki Kagaku, 31: 381-388.
JACOBS, L. (1987) Platinum salt sensitivity. Nurs. RSA Verpleging
(Republic of South Africa), 2: 34-37.
JOHNSON, D.E., TILLERY, J.B., & PREVOST, R.J. (1975) Levels of
platinum, palladium, and lead in populations of Southern California.
Environ. Health Perspect., 12: 27-33.
JOHNSON, D.E., PREVOST, R.J., TILLERY, J.B., CAMAN, D.E., &
HOSENFELD, J.M. (1976) Baseline levels of platinum and palladium in
human tissue, San Antonio, Texas, Southwest Research Institute, 252
pp. (EPA/600/1- 76/019).
JOHNSON, N.P., HOESCHELE, J.D., RAHN, R.O., O'NEIL, P.J., & HSIE,
A.W. (1980) Mutagenicity, cytotoxicity and DNA binding of
platinum(II)-chloroammines in chinese hamster ovary cells. Cancer
Res., 40: 1463-1468.
JOHNSON MATTHEY (1976a) Acute oral toxicity study CB10:
Bis(acetylacetonate) platinum(II), Reading, United Kingdom, Johnson
Matthey Research Centre (Unpublished report No. 8/7610)
JOHNSON MATTHEY (1976b) Skin irritation study, CB10:
Bis(acetylacetonate) platinum(II), Reading, United Kingdom, Johnson
Matthey Research Centre (Unpublished report No. 8/7610).
JOHNSON MATTHEY (1976c) Eye irritation study CB10:
Bis(acetylacetonate) platinum(II), Reading, United Kingdom, Johnson
Matthey Research Centre (Unpublished report No.8/7610).
JOHNSON MATTHEY (1977a) Acute oral toxicity study, CB20: Potassium
tetracyanoplatinate(II), Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 86/7709).
JOHNSON MATTHEY (1977b) Acute oral toxicity study, CB18: Tetrammine
platinous chloride, Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 248/7702).
JOHNSON MATTHEY (1977c) Acute oral toxicity study, CB18:
Cis[PtCl2(NH3)2], Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 216/7707).
JOHNSON MATTHEY (1977d) Primary skin irritation study, CB20:
Potassium tetracyanoplatinate(II), Reading, United Kingdom, Johnson
Matthey Research Centre (Unpublished report No. 88/7709).
JOHNSON MATTHEY (1977e) Skin irritation study, CB13: Tetrammine
platinous chloride, Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 113/7701).
JOHNSON MATTHEY (1977f) Eye irritation study, CB13: Tetrammine
platinous chloride, Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 252/7702).
JOHNSON MATTHEY (1977g) Primary skin irritation study, CB18:
Cis[PtCl2(NH3)2], Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 217/7707).
JOHNSON MATTHEY (1977h) Eye irritation/toxicity study, sample CB18:
Cis[PtCl2(NH3)2], Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 337/7802).
JOHNSON MATTHEY (1978a) Acute oral toxicity study in the rat, CB14:
Ammonium hexachloroplatinate, batch No. 57662, Reading, United
Kingdom, Johnson Matthey Research Centre (Unpublished report No.
106/7808).
JOHNSON MATTHEY (1978b) Acute oral toxicity study, CB24: Sodium
chloroplatinate, Reading, United Kingdom, Johnson Matthey Research
Centre (Unpublished report No. 359/7711).
JOHNSON MATTHEY (1978c) Acute oral toxicity study in the rat, CB70:
Sodium hexachloroplatinate, Batch No. 031074, Reading, United
Kingdom, Johnson Matthey Research Centre (Unpublished report No.
42/7809).
JOHNSON MATTHEY (1978d) Primary skin irritation study, sample CB14:
Ammonium hexachloroplatinate, batch No. 57662, Reading, United
Kingdom, Johnson Matthey Research Centre (Unpublished report No.
109/7808).
JOHNSON MATTHEY (1978e) Primary skin irritation study, sample CB24:
Sodium chloroplatinate, batch No. 031003, Reading, United Kingdom,
Johnson Matthey Research Centre (Unpublished report No. 245/7802).
JOHNSON MATTHEY (1978f) Eye irritation study, sample CB24: Sodium
chloroplatinate, batch No. 031003, Reading, United Kingdom, Johnson
Matthey Research Centre (Unpublished report No. 240/7802).
JOHNSON MATTHEY (1978g) Primary skin irritation study U.S. Federal
Register 1973, sample CB70: Sodium hexahydroxyplatinate, batch No.
031074, Reading, United Kingdom, Johnson Matthey Research Centre
(Unpublished report No. 43/7809).
JOHNSON MATTHEY (1978h) Eye irritation study, sample CB20: Potassium
tetracyanoplatinate(II), Reading, United Kingdom, Johnson Matthey
Research Centre (Unpublished report No. 318/7802).
JOHNSON MATTHEY (1981a) An assessment of the acute oral toxicity of
potassium tetracyanoplatinate(II)-MS 332 in the rat, experiment No.
24/8112, Reading, United Kingdom, Johnson Matthey Research Centre
(Unpublished report).
JOHNSON MATTHEY (1981b) An assessment of the acute oral toxicity of
potassium tetracyanoplatinate(II)-MS 332 in the rat, experiment No.
216/8111, Reading, United Kingdom, Johnson Matthey Research Centre
(Unpublished report).
JOHNSON MATTHEY (1981c) OECD skin irritation test: determination of
the degree of primary cutaneous irritation caused by potassium
tetracyanoplatinate(II)-MS 332 in the rabbit, experiment No.
631/8111, Reading, United Kingdom, Johnson Matthey Research Centre
(Unpublished report).
JOHNSON MATTHEY (1981d) OECD eye irritation test: determination of
the degree of ocular irritation caused by potassium
tetrachloroplatinate(II)-MS 332 in the rabbit, experiment No.
278/8112, Reading, United Kingdom, Johnson Matthey Research Centre
(Unpublished report).
JOHNSON MATTHEY (1990) Platinum 1990, London, Johnson Matthey Public
Limited Company, 64 pp.
JONES, A.H. (1976) Determination of platinum and palladium in blood
and urine by flameless atomic absorption spectrophotometry. Anal.
Chem., 48: 1472-1474.
JONES, E.A., WARSHAWSKY, A., DIXON, K., NICOLAS, D.J., & STEELE,
T.W. (1977) The group extraction of noble metals with s-(1-Decyl)-
N,N'-diphenylisothiouronium bromide and their determination in the
organic extract by atomic-absorption spectrometry. Anal. Chim. Acta,
94: 257-268.
JUNG, H.J. & BECKER, E.R. (1987) Emission control for gas turbines.
Platinum-rhodium catalysts for carbon monoxide and hydrocarbon
removal. Platinum Met. Rev., 31: 162-170.
KAHN, N. & VAN LOON, J.V. (1978) Direct atomic absorption
spectrophotometric analysis of anion complexes of platinum and gold
after their concentration and separation from aqueous solutions by
anion exchange chromatography. Anal. Lett., 12: 991-1000.
KANEMATSU, N., HARA, M., & KADA, T. (1980) Rec assay and
mutagenicity studies on metal compounds. Mutat. Res., 77: 109-116.
KARASEK, S.R. & KARASEK, M. (1911) The use of platinum paper. Report
of (Illinois) Commission on Occupational Diseases to His Excellency
Governor Charles S. Deneen, January 1911, Chicago, Warner Printing
Company, p. 97
KENAWY, I.M., KHALIFA, M.E., & EL-DEFRAWY, M.M. (1987)
Preconcentration and determination (AAS) of trace Ag(I), Au(III),
Pd(II) and Pt(IV) using a cellulose ion-exchanger (Hyphan).
Analusis, 15: 314-317.
KESSERÜ, E. & LEON, F., (1974) Effect of different solid metals and
metallic pairs on human sperm motility. Int. J. Fertil., 19: 81-84.
KHAN, A., HILL, J.M., GRATER, W., LOEB, E., MACLELLAN, A., & HILL,
N. (1975) Atopic hypersensitivity to cis-dichlorodiammineplatinum
(II) and other platinum complexes. Cancer Res., 35: 2766-2770.
KNAPP, G. (1984) [Routes to powerful methods of elemental trace
analysis in environmental samples.] Fresenius Z. anal. Chem., 317:
213-219 (in German).
KNAPP, G. (1985) Sample preparation techniques - An important part
in trace element analysis for environmental research and monitoring.
Int. J. environ. anal. Chem., 22: 71-83.
KOBERSTEIN, E. (1984) [Catalysts for purifying automotive exhaust
gas.] Chem. Zeit, 18: 37-45 (in German).
KOIBA, R.J. & SLEIGHT, S.D. (1971) Acute toxicologic and pathologic
effects of cis-diamminedichloroplatinum (NSC-119875) in the male
rat. Cancer Chemother. Rep., 55(Part 1): 1-8.
KOLPAKOVA, A.F. & KOLPAKOV, F.I. (1983) [Comparative study of
sensitizing characteristics of the platinum group metals.] Gig. Tr.
prof. Zabol., 7: 22-24 (in Russian).
KÖNIG, H.P. & HERTEL, R.F. (1990) [Search and identification of
gaseous noble metal emissions from catalysts. Investigations into
the hazardous potential of catalyst-born nobel metal emissions],
Hanover, Fraunhofer Institute of Toxicology and Aerosol Research,
108 pp (Unpublished final research report to the Federal Ministry of
Research and Technology) (in German).
KÖNIG, H.P., KOCK, H., & HERTEL, R.F. (1989) [Analytical
determination of platinum with regard to the car catalyst issue.]
In: 5th Colloquium on Trace Analysis by Atomic Absorption
Spectrometry, Konstanz, 3-7 April, 1989], Überlingen, Perkin-Elmer
GmbH, pp. 647-656 (in German).
KÖNIG, H.P., HERTEL, F.R., KOCH, W., & ROSNER, G. (in press)
Determination of platinum emissions from a three-way catalyst
equipped gasoline engine. Atmospheric environment.
KRAFT, L.A., POLIDORO, J.P., CULVER, R.M., & HAHN, D.W. (1978)
Intravas device studies in rabbits: II. Effect on sperm output,
fertility and histology of the reproductive tract. Contraception,
18: 239-251.
KRAL, H. & PETER, K. (1977) [Method for the production of
platinum(II)-nitrate and its use for platinum catalysts for the
catalytic after-burning of automobile exhaust gas], Münich, German
Patent Office (Patent No. 2233677) (in German).
KRITSOTAKIS, K. & TOBSCHALL, H.J. (1985) [Trace determination of
platinum by differential-pulse anodic stripping voltammetry (DPASV)
using the glassy carbon electrode.] Fresenius Z. anal. Chem., 320:
156-158 (in German).
LANGE, R.C., SPENCER, R.P., & HARDER, H.C. (1972) Synthesis and
distribution of a radiolabeled antitumor agent: cis-
diamminedichloroplatinum (II). J. nucl. Med., 13: 328-330.
LAZNICKOVA, A., SEMECKY, V., LAZNICEK, M., ZUBER, V., KOKSAL, J., &
KVETINA, J. (1989) Effect of oxoplatinum and CBDCA on renal
functions in rats. Neoplasma, 36: 161-169.
LECOINTE, P., MACQUET, J.P., BUTOUR, J.L., & PAOLETTI, C. (1977)
Relative efficiencies of a series of square-planar platinum(II)
compounds on Salmonella mutagenesis. Mutat. Res., 48: 139-144.
LEE, D.S. (1983) Palladium and nickel in north-east Pacific waters.
Nature(Lond.), 305: 47-48.
LE HOUILLIER, R. & DE BLOIS, C. (1986) Alkyl cyanide medium for the
determination of precious metals by atomic absorption spectrometry.
Analyst, 111: 291-294.
LELIEVELD, P., VAN DER VIJGH, J.F., VELDHUIZEN, R.W., VAN VELZEN,
D., VAN PUTTEN, L.M., ATASSI, G., & DANGUY, A. (1984) Preclinical
studies on toxicity, antitumour activity and pharmacokinetics of
cisplatin and three recently developed derivatives. Eur. J. Cancer
clin. Oncol., 20: 1087-1104.
LEROY, A.F., WEHLING, M.L., SPONSELLER, H.L., FRIAUF, W.S., SOLOMON,
R.E., & DEDRICK, R.L., (1977) Analysis of platinum biological
materials by flameless atomic absorption spectrophotometry. Biochem.
Med., 18: 184-191.
LIFE SCIENCE RESEARCH (1980a) Pt(NH3)4Cl2; assessment of its
mutagenic potential in histidine auxotrophs of Salmonella
typhimurium, Eye, Suffolk, Life Science Research, (Unpublished
report No. 80/JOM001/005).
LIFE SCIENCE RESEARCH (1980b) Pt(NH3)4Cl2; assessment of
clastogenic action on bone marrow erythrocytes in the micronucleus
test, Eye, Suffolk, Life Science Research (Unpublished report No.
80/JOM004/182).
LIFE SCIENCE RESEARCH (1980c) Pt(NH3)4Cl2; assessment of its
mutagenic potential in Drosophila melanogaster, using the sex-
linked recessive lethal test, Eye, Suffolk, Life Science Research
(Unpublished report No. 80/JOM002/260).
LIFE SCIENCE RESEARCH (1981a) Potassium chloroplatinate: assessment
of clastogenic action on bone marrow erythrocytes in the
micronucleous test, Eye, Suffolk, Life Science Research (Unpublished
report No. 81/JOM013/241).
LIFE SCIENCE RESEARCH (1981b) TPC: assessment of its action on bone
marrow cell chromosomes following sub-acute oral administration in
the Chinese hamster, Eye, Suffolk, Life Science Research
(Unpublished report No. 80/JOM008/497).
LIFE SCIENCE RESEARCH (1981c) Potassium chloroplatinate(II):
assessment of its mutagenic potential in Drosophila melanogaster,
using the sex-linked recessive lethal test, Eye, Suffolk, Life
Science Research (Unpublished report No. 81/JOM012/474).
LIFE SCIENCE RESEARCH (1982) Potassium chloroplatinate:
investigation of effects on bone marrow chromosomes of the Chinese
hamster after sub-acute oral administration, Eye, Suffolk, Life
Science Research (Unpublished report).
LINNETT, P.J. (1987) Platinum salt sensitivity. A review of the
health aspects of platinum refining in South Africa. J. Mine Med.
Off. Assoc. S. Afr., 63: 24-28.
LIPPERT & BECK (1983) [Platinum complexes in cancer therapy.] Chem.
Zeit, 6: 190-199 (in German).
LIPPMAN, A.J., HELSON, C., HELSON, L., & KRAKOFF, I.H. (1973)
Clinical trials of cis-diamminedichloroplatinum (NSC 119875). Cancer
Chemother. Rep., 57: 191-200.
LITTERST, C.L. (1981) Alterations in the toxicity of cis-
dichlorodiammineplatinum-II and in tissue localization of platinum
as a function of NaCl concentration in the vehicle of
administration. Toxicol. appl. Pharmacol., 61: 99-108.
LITTERST, C.L., TORRES, I.J., & GUARINO, A.M. (1976a) Plasma levels
& organ distribution of platinum in the rat, dog & dogfish shark
following single intravenous administration of cis-
dichlorodiammineplatinum(II). J. clin. Hematol. Oncol., 7: 169-179.
LITTERST, C.L., GRAM, T.E., DEDRICK, R.L., LEROY, A.F., & GUARINO,
A.M. (1976b) Distribution and disposition of platinum following
intravenous administration of cis-diamminedichloroplatinum(II) (NSC
119875) to dogs. Cancer Res., 36: 2340-2344.
LITTERST, C.L., LEROY, A.F., & GUARINO, A.M. (1979) Disposition and
distribution of platinum following parenteral administration of cis-
dichlorodiammineplatinum-II to animals. Cancer Treat. Rep., 63:
1485-1492.
LO, F.B., ARAI, D.K., & NAZAR, M.A. (1987) Determination of platinum
in urine by inductively coupled plasma atomic emission spectrometry.
J. anal. Toxicol., 11: 242-246.
LOEBENSTEIN, J.R. (1982) Platinum-group metals. In: Minerals
yearbook, centennial edition 1981. Vol. I. Metals and minerals,
Washington, DC, US Department of the Interior, Bureau of Mines, pp.
667-678.
LOEBENSTEIN, J.R. (1988) Platinum-group metals. In: Minerals
yearbook 1987. Vol. I. Metals and minerals, Washington, DC, US
Department of the Interior, Bureau of Mines, pp. 689-700.
LOWN, B.A., MORGANTI, J.B., STINEMAN, C.H., D'AGOSTINO, R.B., &
MASSARO, E.J. (1980) Tissue organ distribution and behavior effects
of platinum following acute and repeated exposure of the mouse to
platinum sulfate. Environ. Health Perspect., 34: 203-212.
MCGAHAN, M.C. & TYCZKOWSKA, K. (1987) The determination of platinum
in biological materials by electrothermal atomic absorption
spectroscopy. Spectrochim. Acta, 42B: 665-668.
MAESSEN, F.J.M., KREUNING, G., & BALKE, J. (1986) Experimental
control of the solvent load of inductively coupled argon plasmas and
effects of the chloroform plasma load on their analytical
performance. Spectrochim. Acta, 41B: 3-25.
MALANCHUK, M., BARKELEY, N., CONTNER, G., RICHARDS, M., SLATER, R.,
BURKART, J., & YANG, Y. (1974) Exhaust emissions from catalyst-
equipped engines. In: Environmental Toxicology Research Laboratory,
NERC, ed. Interim report: 1. Studies on toxicology of catalytic
trace metal components. 2. Toxicology of automotive emissions with
and without catalytic converters, Cincinnati, Ohio, Environmental
Toxicology Research Laboratory, NERC, pp. 91-98.
MARCZENKO, Z. & KUS, S. (1987) Spectrophotometric determination of
traces of platinum in palladium with dithizone after matrix
precipitation as a compound with ammonia and iodide. Anal. Chim.
Acta, 196: 317-322.
MARONE, C., OLSINA, R., & SALAS, O. (1981) Spectrometric
determination of platinum in a spent reforming catalyst. Cron.
chim., 66: 22-28.
MARSH, K.C., STERNSON, L.A., & REPTA, A.J. (1984) Post-column
reaction detector for platinum(II) antineoplastic agents. Anal.
Chem., 56: 491-497.
MASON, B. (1966) Principles of geochemistry, 3rd ed., New York, John
Wiley & Sons, 310 pp.
MASSARO, E.J., LOWN, B.A., MORGANTI, J.B., STINEMAN, C.H., &
D'AGOSTINO, R.B. (1981) Sensitive biochemical and behavioral
indicators of trace substance exposure: Part II. Platinum, Research
Triangle Park, US Environmental Protection Agency, 3 pp
(EPA-600/S1-81-015).
MASSMANN, W. & OPITZ, H. (1954) [On platinum allergy.] Zentralbl.
Arbeitsmed. Arbeitsschutz, 4: 1-4 (in German).
MATUSIEWICZ, H. & BARNES, R.M. (1983) Determination of metal
chemotherapeutic agents (Al, Au, Li, and Pt) in human body fluids
using electrothermal vaporization with inductively coupled plasma
atomic emission spectroscopy. In: Chemical toxicology and clinical
chemistry of metals, Oxford, United Kingdom, International Union of
Pure and Applied Chemistry, pp. 49-52.
MERGET, R., SCHULTZE-WERNINGHAUS, G., MUTHORST, T., FRIEDRICH, W., &
MEIER-SYDOW, J. (1988) Asthma due to the complex salts of platinum -
a cross-sectional survey of workers in a platinum refinery. Clin.
Allergy, 18: 569-580.
MERGET, R., ZACHGO, W., SCHULTZE-WERNINGHAUS, G., BERGMANN, E.-M.,
BOLM-AUDORFF, U., FRIEDRICH, W., BURY, A.H., & MEIER-SYDOW, J.
(1990) [Quantitative skin test and inhalative provocation test in
asthma associated with platinum compounds.] Pneumologie, 44: 226-228
(in German).
MERRIN, C.A. (1976) A new method to prevent toxicity with high doses
of cis- diammineplatinum (therapeutic efficacy in previously treated
widespread and recurrent testicular tumors). In: Proceedings of the
12th Annual Meeting of the American Association for Cancer
Research/American Society of Clinical Oncology, Toronto, 4-5 May,
1976, Chicago, Illinois, American Society of Clinical Oncology, p.
243 (ASCO Abstracts, No. C-26).
MICHAEL, L.W., FINELLI, V.N., ELIA, V.J., PETERING, H.G., LEE, S.D.,
& CAMPBELL, K.I. (1976) Interaction of metal binding agents from
combustion products with trace metals. In: Proceedings of the
Platinum Research Review Conference, Quail Roost Conference Center,
Rougemont, North Carolina, 3-5 December 1975, Research Triangle
Park, North Carolina, US Environmental Protection Agency, pp.
37A-53A.
MILLARD, H.T. (1987) Neutron activation determination of iridium,
gold, platinum, and silver in geologic samples. J. radioanal. nucl.
Chem., 113: 125-132.
MOJSKI, M. & KALINOWSKI, K. (1980) Extractive separation of platinum
from macroamounts of palladium using triphenylphosphine oxide and
its successive spectrophotometric determination by the stannous
chloride method. Microchem. J., 25: 507-513.
MOLLMAN, J.E., HOGAN, W.M., GLOVER, D.J., & MCCLUSKEY, L.F. (1988)
Unusual presentation of cis-platinum neuropathy. Neurology, 38:
488-490.
MOORE, W., Jr, HYSELL, D., CROCKER, W., & STARA, J. (1975a)
Biological fate of a single adminstration of 191Pt in rats
following different routes of exposure. Environ. Res., 9: 152-158.
MOORE, W., Jr, HYSELL, D., HALL, L., CAMPBELL, K., & STARA, J.
(1975b) Preliminary studies on the toxicity and metabolism of
palladium and platinum. Environ. Health Perspect., 10: 63-71.
MOORE, W., Jr, MALANCHUK, M., CROCKER, W., HYSELL, D., COHEN, A., &
STARA, J.F. (1975c) Whole body retention in rats of different
191Pt compounds following inhalation exposure. Environ. Health
Perspect., 12: 35-39.
MUELLER, B.J. & LOVETT, R.J. (1987) Salt-induced phase separation
for the determination of metals as their diethyldithiocarbamate
complexes by high-performance liquid chromatography. Anal. Chem.,
59: 1405-1409.
MURDOCH, R.D. & PEPYS, J. (1984) Immunological responses to complex
salts of platinum. I. Specific IgE antibody production in the rat.
Clin. exp. Immunol., 57: 107-114.
MURDOCH, R.D. & PEPYS, J. (1985) Cross reactivity studies with
platinum group metal salts in platinum-sensitised rats. Int. Arch.
Allergy appl. Immunol., 77: 456-458.
MURDOCH, R.D. & PEPYS, J. (1987) Platinum group metal sensitivity:
reactivity to platinum group metal salts in platinum halide salt-
sensitive workers. Ann. Allergy, 59: 464-469.
MURDOCH, R.D., PEPYS, J., & HUGHES, E.G. (1986) IgE antibody
responses to platinum group metals: a large-scale refinery survey.
Br. J. ind. Med., 43: 37-43.
MYASOEDOVA, G.V., ANTOKOL'SKAYA, I.I., & SAVVIN, S.B. (1985) New
chelating sorbents for noble metals. Talanta, 32: 1105-1112.
NAS (1977) Platinum-group metals, Washington, DC, National Academy
of Science, 232 pp.
NEUMÜLLER, O.-A. (1987) [Römpps lexicon on chemistry], Stuttgart, W.
Keller & Co., p. 3256 (in German).
NEWMAN-TAYLOR, A.J. (1981) Follow-up study of a group of platinum
refinery workers, London, Brompton Hospital (Unpublished report).
NYGREN, O., VAUGHAN, G.T., FLORENCE, T.M., MORRISON, G.M., WARNER,
I., & DALE, L.S. (1990) Determination of platinum in blood by
adsorptive voltammetry. Anal. Chem., 62(15): 1637-1640.
PALLAS, J.E., Jr & JONES, J.B., Jr (1978) Platinum uptake by
horticultural crops. Plant Soil, 50: 207-212.
PARISH, W.E. (1970) Short-term anaphylactic IgG antibodies in human
sera. Lancet, 2: 591-592.
PARROT, J.L., HEBERT, R., SAINDELLE, A., & RUFF, F. (1969) Platinum
and platinosis. Allergy and histamine release due to some platinum
salts. Arch. environ. Health, 19: 685-691.
PEPYS, J. (1983) Allergy of the respiratory tract to low molecular
weight chemical agents. In: Born, G.V.R., Farah, A., Herken, H., &
Welch, A.D., ed. Handbook of experimental pharmacology, Berlin,
Springer-Verlag, pp. 163- 185.
PEPYS, J., PICKERING, C.A.C., & HUGHES, E.G. (1972) Asthma due to
inhaled chemical agents - complex salts of platinum. Clin. Allergy,
2: 391-396.
PEPYS, J., PARISH, W.E., CROMWELL, O., & HUGHES, E.G. (1979) Passive
transfer in man and the monkey of type I allergy due to heat labile
and heat stable antibody to complex salts of platinum. Clin.
Allergy, 9: 99-108.
PERA, M.F., Jr & HARDER, H.C. (1977) Analysis of platinum in
biological material by flameless atomic absorption spectrometry.
Clin. Chem., 23: 1245- 1249.
PICKERING, C.A.C. (1972) Inhalation tests with chemical allergens:
complex salts of platinum. Proc. R. Soc. Med., 65: 272-274.
PINTO, A.L. & LIPPARD, S.J. (1985) Binding of the antitumour drug
cis-diamminedichloroplatinum(II) (cisplatin) to DNA. Biochim.
Biophys. Acta, 780: 167-180.
POTTER, N.M. & LANGE, W.H. (1981) Determination of noble metals and
their distribution in automotive catalyst materials. Am. Lab., 1:
81-91.
PRIESNER, D., STERNSON, L.A., & REPTA, A.J. (1981) Analysis of total
platinum in tissue samples by flameless atomic absorption
spectrophotometry. Elimination of the need for sample digestion.
Anal. Lett., 14: 1255-1268.
PURI, B.K., WASEY, A., KATYAL, M., & SATAKE, M. (1986)
Spectrophotometric determination of platinum after extraction of its
phenanthraquinone monoximate into molten naphtalene. Analyst, 111:
743-745.
RENNER, H. (1979)a [Platinum metals and alloys.] In: Bartholomé,
E., Biekert, E., Hellmann, H., & Ley, H., ed. [Ullmann's
encyclopedia of technical chemistry], 4th ed., Weinheim, Verlag
Chemie, Vol. 18, pp. 697-705 (in German).
RENNER, H. (1984)b [Platinum metals.] In: Merian, E., Geldmacher-
von-Mallinckrodt, M., Machata, G., Nürnberg, H.W., Schlipköter,
H.W., & Stumm, W., ed. [Metals in the environment. Distribution,
analysis, and biological relevance], Weinheim, Verlag Chemie, pp.
499-510 (in German).
RICKEY, F.A., SIMMS, P.C., & MUELLER, K.A. (1979) PIXE analysis of
water with detection limits in the ppb range. IEEE Trans. nucl.
Sci., NS-26(1): 1347-1351.
ROBERT, R.V.D., VAN WYK, E., & PALMER, R. (1971) Concentration of
the noble metals by a fire-assay technique using sulphide as the
collector, Johannesburg, South Africa, National Institute for
Metallurgy (Report No. 1371).
ROBERTS, A.E. (1951) Platinosis. A five-year study of the effects of
soluble platinum salts on employees in a platinum laboratory and
refinery. Arch. ind. Hyg. occup. Med., 4: 549-559.
ROCKLIN, R.D. (1984) Determination of gold, palladium, and platinum
at the parts-per-billion level by ion chromatography. Anal. Chem.,
56: 1959-1962.
ROSENBERG, B. (1975) Possible mechanisms for the antitumor activity
of platinum coordination complexes. Cancer Chemother. Rep., 59(Part
1): 589-598.
ROSENBERG, B. (1980) Clinical aspects of platinum anticancer drugs.
In: Metal ions in biological systems, New York, Basel, Marcel
Dekker, Inc., Vol. 12, pp. 127-196.
ROSENBERG, B. (1985) Fundamental studies with cisplatin. Cancer, 55:
2303-2316.
a English edition in preparation:
RENNER, H. (in press) Platinum metals, compounds and alloys.
In: Ullmanns Encyclopaedia of Industrial Chemistry, 5th ed.,
Weinheim, VCH Verlagsgesellschaft.
b English edition in preparation:
RENNER, H. & SCHMUCKLER, G. (in press) Platinum-group metals.
In: Merian, E., ed. Metals and their compounds in the
environment, Weinheim, VCH Verlagsgesellschaft.
ROSENBERG, B., VAN CAMP, L., & KRIGAS, T. (1965) Inhibition of cell
division in Escherichia coli by electrolysis products from a
platinum electrode. Nature (Lond.), 205: 698-699.
ROSENBERG, B., RENSHAW, E., VAN CAMP, L., HARTWICK, J., & DROBNIK,
J. (1967) Platinum-induced filamentous growth in Escherichia coli.
J. Bacteriol., 93: 716-721.
ROSHCHIN, A.V., VESELOV, V.G., & PANOVA, A.I. (1979) [Toxicology of
platinum and platinum-group metals.] Gig. Tr. prof. Zabol., 9: 4-9
(in Russian).
ROSHCHIN, A.V., VESELOV, V.G., & PANOVA, A.I. (1984) Industrial
toxicology of metals of the platinum group. J. Hyg. Epidemiol.
Microbiol. Immunol., 28: 17-24.
ROSNER, G. & HERTEL, R.F. (1986) [Health risk assessment of platinum
emissions from automotive exhaust gas catalysts.] Staub-Reinhalt.
Luft, 46: 281-285 (in German).
ROSNER, G. & MERGET, R. (1990) Allergenic potential of platinum
compounds. In: Dayan, A.D., Hertel, R.F., Heseltine, E., Kazantzis,
G., Smith, E.M., & Van der Venne, M.T., ed. Immunotoxicity of metals
and immunotoxicology, New York, London, Plenum Press, pp. 93-102.
ROSNER, G., KÖNIG, H.P., KOCH, W., KOCK, H., HERTEL, R.F., & WINDT,
H. (1991) [Engine test stand experiments to assess platinum uptake
by plants.] Angew. Bot., 65: 127-132 (in German).
RUFF, F., DI MATTEO, G., DUPUIS, J.P., HEBERT, R., & PARROT, J.-L.
(1979) Réactions bronchopulmonaires et asthme dus au platine:
incidences chez les ouvriers du platine en région parisienne. Rev.
fr. Mal. Respir., 7: 206-208.
SAFIRSTEIN, R., DAYE, M., & GUTTENPLAN, J.B. (1983) Mutagenic
activity and identification of excreted platinum in human and rat
urine and rat plasma after administration of cisplatin. Cancer
Lett., 18: 329-338.
SAINDELLE, A. & RUFF, F. (1969) Histamine release by sodium
chloroplatinate. Br. J. Pharmacol., 35: 313-321.
SANDHU (1979) Evaluation of the mutagenic potential of platinum
compounds, Durham, North Carolina Central University, 36 pp
(EPA-600/1-79-033).
SCHACTER, L. & CARTER, S.K. (1986) The use of platinum to treat
cancer. In: Precious metals: Proceedings of the 9th International
Precious Metals Institute Conference, Lake Tahoe, Nevada, 9-12 June,
1986, Allentown, Pennsylvania, International Precious Metals
Institute, pp. 359-367.
SCHLEMMER, G. & WELZ, B. (1986) Influence of the tube surface on
atomization behavior in a graphite tube furnace. Fresenius Z. anal.
Chem., 323: 703-709.
SCHLÖGL, R., INDLEKOFER, G., & OELHAFEN, P. (1987) [Micro particle
emission from combustion engines with exhaust gas purification - X-
ray photoelectron spectroscopy in environmental analysis.] Angew.
Chem., 99: 312-322 (in German).
SCHULTZE-WERNINGHAUS, G., ROESCH, A., WILHELMS, O.-H., GONSIOR, E.,
& MEIER-SYDOW, J. (1978) [Bronchial asthma due to occupational
allergy of immediate type (I) to platinum salts.] Dtsch. med.
Wochenschr., 23: 972-975 (in German).
SCHULTZE-WERNINGHAUS, G., MERGET, R., ZACHGO, W., MUTHORST, T.,
MAHLESA, D., LISSON, R., & BOLM-AUDORFF, U. (1989) [Platinum salts
as occupational allergens - a review.] Allergologie, 12: 152-157 (in
German).
SCHUTYSER, P., GOVAERTS, A., DAMS, R., & HOSTE, J. (1977) Neutron
activation analysis of platinum metals in airborne particulate
matter. J. radioanal. Chem., 37: 651-660.
SHEARD, C. (1955) Contact dermatitis from platinum and related
metals. Arch. Dermatol. Syphilol., 71: 357-360.
SHIMAZU, M. & ROSENBERG, B. (1973) A similar action to UV-
irradiation and a preferential inhibition of DNA synthesis in E.
coli by antitumor platinum compounds. J. Antibiot., 26: 243-245.
SIGHINOLFI, G.P., GORGONI, C., & MOHAMED, A.H. (1984) Comprehensive
analysis of precious metals in some geological standards by
flameless a.a. spectroscopy. Geostand. Newsl., 8: 25-29.
SIGSBY, J. (1976) Measurement of platinum from catalyst-equipped
vehicles, combustion and attrition products. In: Proceedings of the
Platinum Research Review Conference, Quail Roost Conference Center,
Rougemont, North Carolina, 3-5 December 1975, Research Triangle
Park, North Carolina, US Environmental Protection Agency, pp.
25A-31A.
SINGH, N. & GARG, A.K. (1987) Spectrophotometric determination of
platinum(IV) with potassium butyl xanthate. Analyst, 112: 693-695.
SKINNER, P.E. (1989) Improvements in platinum plating. Platinum Met.
Rev., 33(3): 102-105.
SKOGERBOE, R.K., HANAGAN, W.A., & TAYLOR, H.E. (1985) Concentration
of trace elements in water samples by reductive precipitation. Anal.
Chem., 57: 2815-2818.
SMITH, B.L., HANNA, M.L., & TAYLOR, R.T. (1984) Induced resistance
to platinum in Chinese hamster ovary cells. J. environ. Sci. Health,
A19: 267-298.
SPERNER, F. & HOHMANN, W. (1976) Rhodium-platinum gauzes for ammonia
oxidation. Platinum Met. Rev., 20: 12-20.
STERNSON, L.A., REPTA, A.J., SHIH, H., HIMMELSTEIN, K.J., & PATTON,
T.F. (1984) Disposition of cisplatin vs total platinum in animals
and man. Dev. Oncol., 17: 126-137.
STOCKMAN, H.W. (1983) Neutron activation determination of noble
metals in rock: a rapid radiochemical separation based on tellurium
coprecipitation. J. radioanal. Chem., 78: 307-317.
STOKINGER, H.E. (1981) Platinum-group metals - platinum, Pt,
palladium, Pd, iridium, Ir, osmium, Os, rhodium, Rh, ruthenium, Ru.
In: Clayton, G.D. & Clayton, F.E., ed. Patty's industrial hygiene
and toxicology, New York, John Wiley & Sons, pp. 1853-1871.
SURAIKINA, T.I., ZAKHAROVA, I.A., MASHKOVSKII, Y., & FONSHTEIN, L.M.
(1979) Study of the mutagenic action of platinum and palladium
compounds on bacteria. Cytol. Genet. (USSR), 13: 50-54 (Translation
from: Tsitol. i. Genet. 13: 486-491).
SWIERENGA, S.H., GILMAN, J.P., & MCLEAN, J.R. (1987) Cancer risk
from inorganics. Cancer Metastasis Rev., 6: 113-154.
TAUBLER, J.H. (1977) Allergic response to platinum and palladium
complexes - determination of no-effect level, Research Triangle
Park, North Carolina, US Environmental Protection Agency, Office of
Research and Development, Health Effects Research Laboratories, pp.
1-81 (EPA/600/1-77-039).
TAYLOR, R.T. (1976) Comparative methylation chemistry of platinum,
palladium, lead and manganese, Research Triangle Park, North
Carolina, US Environmental Protection Agency, Office of Research and
Development, Health Effects Research Laboratories, 25 pp
(EPA/600/1-76-016).
TAYLOR, R.T. & HANNA, M.L. (1977) Methylation of platinum by methyl
cobalamin. In: Drucker, H. & Wildung, R.E., ed. Biological
implications of metals in the environment. Proceedings of the 15th
Annual Hanford Life Science Symposium, Richland, Washington, 29
September - 1 October, 1975, Springfield, Virginia, US Department of
Commerce, National Technical Information Service, pp. 36-51 (Energy
Research and Development Administration Symposium Series, Vol. 42).
TAYLOR, R.T., HAPPE, J.A., HANNA, M., & WU, R. (1979) Platinum
tetrachloride: mutagenicity and methylation with methylcobalamin. J.
environ. Sci. Health, A14: 87-109.
THEOPOLD, H.-M., ZOLLNER, M., SCHORN, K., SPAHMANN, J., & SCHERER,
H. (1981) [Tissue reactions with platinum-iridium electrodes.]
Laryng.-Rhinol., 60: 534-537 (in German).
THOMPSON, B.C., RAYBURN, L.A., & HOLADAY, S.R. (1981) Detection of
platinum in brain tissue by PIXE. IEEE Trans. nucl. Sci., NS-28:
1396-1397.
THOMPSON, J.J. & HOUK, R.S. (1986) Inductively coupled plasma mass
spectrometric determination for multielement flow injection analysis
and elemental speciation by reversed-phase liquid chromatography.
Anal. Chem., 58: 2541-2548.
TILLERY, J.B. & JOHNSON, D.E. (1975) Determination of platinum,
palladium, and lead in biological samples by atomic absorption
spectrophotometry. Environ. Health Perspect., 12: 19-26.
TJIOE, P.S., VOLKERS, K.J., KROON, J.J., DE GOEIJ, J.J.M., & THE,
S.K. (1984) Determination of gold and platinum traces in biological
materials as a part of a multielement radiochemical activation
analysis system. Int. J. environ. anal. Chem., 17: 13-24.
TOBE, M.L. & KHOKHAR, A.R. (1977) Structure, activity, reactivity
and solubility relationships of platinum diammine complexes. J.
clin. Hematol. Oncol., 7: 114-137.
TOBERT, A.J. & DAVIES, D.R. (1977) Effect of copper and platinum
intrauterine devices on endometrial morphology and implantation in
the rabbit. J. Reprod. Fertil., 50: 53-59.
TÖLG, G. & ALT, F. (1990) [Contribution to an improvement in extreme
trace analysis of metallic platinum in biotic and environmental
material.] In: [Abstract prepared for the Third Conference of the
BMFT Study Group on Precious Metal Emissions, 15 October, 1990],
Dortmund, Institute of Spectrochemistry, 7 pp (Unpublished report)
(in German).
TSO, T.C., SOROKIN, T.P., & ENGELHAUPT, M.E. (1973) Effects of some
rare elements on nicotine content of the tobacco plant. Plant
Physiol., 51: 805-806.
VALENTE, I.M., MINSKI, M.J., & PETERSON, P.J. (1982) Neutron
activation analysis of noble metals in vegetation. J. radioanal.
Chem., 71: 115-127.
VAN DEN BERG, C.M.G. & JACINTO, G.S. (1988) The determination of
platinum in sea water by adsorptive cathodic stripping voltammetry.
Anal. Chim. Acta, 211: 129-139.
VAN DER VIJGH, W.J.F., ELFERINK, F., & POSTMA, G.J. (1984)
Determination of ethylenediamineplatinum(II) malonate in infusion
fluids, human plasma and urine by high-performance liquid
chromatography. J. Chromatogr., 310: 335-342.
VENABLES, K.M., DALLY, M.B., NUNN, A.J., STEVENS, J.F., STEPHENS,
R., FARRER, N., HUNTER, J.V., STEWART, M., HUGHES, E.G., & NEWMAN-
TYLOR, A.J. (1989) Smoking and occupational allergy in workers in a
platinum refinery. Br. Med. J., 299: 939-942.
VENITT, S., CROFTON-SLEIGH, C., HUNT, J., SPEECHLEY, V., & BRIGGS,
K. (1984) Monitoring exposure of nursing and pharmacy personnel to
cytotoxic drugs: urinary mutation assays and urinary platinum as
markers of absorption. Lancet, January 14: 74-77.
VOGL, S.E., ZARAVINOS, T., & KAPLAN, B.H. (1980) Toxicity of cis-
diamminedichloroplatinum II given in a two-hour outpatient regimen
of diuresis and hydration. Cancer, 45: 11-15.
VON BOHLEN, A., ELLER, R., KLOCKENKÄMPER, R., & TÖLG, G. (1987)
Microanalysis of solid samples by total-reflection X-ray
fluorescence spectrometry. Anal. Chem., 59: 2551-2555.
VON HOFF, D.D., SCHILSKY, R., REICHERT, C.M., REDDICK, R.L.,
ROZENCWEIG, M., YOUNG, R.C., & MUGGIA, F.M. (1979) Toxic effects of
cis- dichlorodiammine-platinum(II) in man. Cancer Treat. Rep., 63:
1527-1531.
VRANA, O., KLEINWÄCHTER, V., & BRABEC, V. (1983) Determination of
platinum in urine by differential pulse polarography. Talanta, 30:
288-290.
WANG, T.C. & TAN, C.K. (1987) Photodegradation of trichloroethylene
in microheterogeneous aqueous systems. Environ. Int., 13: 359-362.
WARD, J.M. & FAUVIE, K.A. (1976) The nephrotoxic effects of cis-
diamminedichloroplatinum(II) (NSC-II 9875) in male F344 rats.
Toxicol. appl. Pharmacol., 38: 535-547.
WARD, J.M., YOUNG, D.M., FAUVIE, K.A., WOLPERT, M.K., DAVIS, R., &
GUARINO, A.M. (1976) Comparative nephrotoxity of platinum cancer
chemotherapeutic agents. Cancer Treat. Rep., 60: 1675-1678.
WATERS, M.D., VAUGHAN, T.O., ABERNETHY, D.J., GARLAND, H.R., COX,
C.C., & COFFIN, D.L. (1975) Toxicity of platinum (IV) salts for
cells of pulmonary origin. Environ. Health Perspect., 12: 45-56.
WEAST, R.C. & ASTLE, M.J. (1981) CRC handbook of chemistry and
physics, 62nd ed. Boca Raton, Florida, CRC Press, pp. B-128-B-129.
WELZ, B. & SCHLEMMER, G. (1987) Glassy carbon tubes for
electrothermal atomic absorption spectrometry. Fresenius Z. anal.
Chem., 327: 246-252.
WEMYSS, R.B., & SCOTT, R.H. (1978) Simultaneous determination of
platinum group metals and gold in ores and related plant materials
by inductively-coupled plasma-optical emission spectrometry. Anal.
Chem., 50: 1694-1697.
WHITEHOUSE, M.W. & GARRETT, I.R. (1984) Heavy metal (Au, Pt)
nephropathy: studies in normal and inflamed rats. In: Rainsford,
K.D. & Velo, G.P., ed. Advances in inflammation research, New York,
Raven Press, Vol. 6, pp 291-294.
WIELE, H. & KUCHENBECKER, H. (1974) [Spectrophotometric
determination of rhenium in the presence of platinum in bimetallic
catalysts.] Erdöl, Kohle, Erdgas, Petrochem., 27: 90-92 (in German).
WINDHOLZ, M. (1976) The Merck index, 9th ed., Rahway, New Jersey,
Merck & Co., pp. 75, 977-991, 1114.
WOLFE, G.W. (1979) PIXE analysis of atmospheric gases. IEEE Trans.
nucl. Sci., NS-26: 1389-1391.
WOOD, J.M., FANCHIANG, Y.-T., & RIDLEY, W.P. (1978) The biochemistry
of toxic elements. Q. Rev. Biophys., 11: 467-479.
WÜRFELS, M., JACKWERTH, E., & STOEPPLER, M. (1987) [On the problem
of disturbances of inverse voltammetric trace analysis after
pressure decomposition of biological samples.] Fresenius Z. anal.
Chem., 329: 459-461 (in German).
ZACHGO, W., MERGET, R., & SCHULTZE-WERNINGHAUS, G. (1985) [Proof of
specific IgE against low molecular weight substances (platinum
salts).] Atemweg. Lungenkr., 11: 267-268 (in German).
ZOLOTOV, YU.A., PETRUKHIN, O.M., MALOFEEVA, G.I., MARCHEVA, E.V.,
SHIRYAEVA, O.A., SHESTAKOV, V.A., MISKAR'YANTS, V.G., NEFEDOV, V.I.,
MURINOV, YU.I., & NIKITIN, YU.E. (1983) Determinations of platinum
metals by X-ray fluorescence, atomic emission and atomic absorption
spectrometry after preconcentration with a polymeric thioether.
Anal. Chim. Acta, 148: 135-157.
RESUME
1. Identité, propriétés physiques et chimiques et méthodes
d'analyse
Le platine (Pt) est un métal noble, malléable et ductile de
couleur blanc argent dont le numéro atomique est 78 et le poids
atomique 195,9. On le trouve principalement à l'état naturel sous la
forme des isotopes 194Pt (32,9 %), 195Pt (33,8 %) et 196Pt
(25,3 %). Son état d'oxydation maximum est de +6, les états +2 et +4
étant les plus stables.
Le métal ne se corrode pas à l'air quelle que soit la
température mais il peut être attaqué par les halogènes, les
cyanures, le soufre et ses composés en fusion, les métaux lourds et
les hydroxydes alcalins. L'attaque par l'eau régale ou par Cl2/HCl
(acide chlorhydrique concentré dans lequel on fait barboter du
chlore) produit l'acide hexachloroplatinique, H2(PtCl6), un
important complexe du platine. Lorsqu'il est chauffé, le sel
d'ammonium de l'acide hexachloroplatinique produit une substance
grisâtre appelée "mousse de platine". La réduction d'une solution
aqueuse d'acide hexachloro-platinique donne une poudre noire
dispersée appelée "noir de platine".
En solution aqueuse, les espèces chimiques dominantes sont des
complexes. Beaucoup de ces sels complexes sont solubles dans l'eau
en particulier ceux qui contiennent des ligands donneurs comme les
halogènes ou l'azote. Le platine, comme les autres métaux du même
groupe, ont une forte tendance à réagir sur les composés carbonés,
en particulier les alcènes et les alcynes pour former des complexes
de coordination de Pt(II).
On dispose de diverses méthodes d'analyse pour le dosage du
platine. La spectométrie d'absorption atomique et la spectroscopie
d'émission en plasma sont très sélectives et spécifiques et
constituent les méthodes de choix pour le dosage du platine dans les
échantillons d'origine biologique ou environnementale. Dans
différents milieux, on a obtenu avec ces méthodes des limites de
détection de l'ordre du µg par kg ou par litre.
La spectroscopie d'émission atomique en plasma induite par
haute fréquence (PIHF) est supérieure à l'absorption atomique
électrothermique du fait que les effets de matrice sont plus faibles
et qu'il y a possibilité d'exécuter simultanément l'analyse d'autres
éléments.
2. Sources d'exposition humaine et environnementale
La concentration moyenne du platine dans la lithosphère, c'est-
à-dire la croûte rocheuse de la terre, est estimée à 0,001-0,005
mg/kg. On trouve le platine soit à l'état natif (métallique) soit en
combinaison, dans un certain nombre de minéraux. Les sources
d'importance économique se trouvent en République d'Afrique du Sud
et en URSS. La teneur en platine de ces gisements est de 1-500
mg/kg. Au Canada, les métaux du groupe du platine (platine,
palladium, iridium, osmium, rhodium et ruthénium) sont présents dans
les minerais sulfurés de cuivre et de nickel à une concentration
moyenne de 0,3 mg/kg; l'affinage du cuivre et du nickel porte cette
concentration à plus de 50 mg/kg. De petites quantités sont
extraites de mines situées aux Etats-Unis d'Amérique, en Ethiopie,
aux Philippines et en Colombie.
La production minière mondiale des métaux du groupe du platine,
constituée à 40-50% de platine, augmente régulièrement depuis les
deux dernières décennies. En 1971, la production était de 127
tonnes, dont 51-64 tonnes de platine. Depuis l'apparition du pot
d'échappement catalytique, la production minière mondiale de ces
métaux est passée à environ 270 tonnes (dont 108-153 tonnes de
platine) en 1987. En 1989, la demande totale de platine dans le
monde occidental était d'environ 97 tonnes.
La principale utilisation du platine tient à ses propriétés
catalytiques exceptionnelles. Les autres applications industrielles
sont basées sur d'autres propriétés remarquables de ce métal, en
particulier sa résistance à la corrosion chimique dans un grand
intervalle de température, son point de fusion élevé, sa ductilité
et sa grande résistance mécanique. Le platine est également utilisé
en joaillerie et en art dentaire.
Certains complexes du platine, en particulier le cis-
diamminedichloroplatine(II) ou cisplatine sont utilisés en
thérapeutique.a
On ne dispose pas de données sur les émissions de platine
d'origine industrielle. Lors de l'utilisation de catalyseurs à base
de platine, une certaine quantité de métal peut s'échapper dans
l'environnement selon le type de catalyseur. Parmi les catalyseurs
fixes utilisés dans l'industrie, seuls ceux qui servent à
l'oxydation de l'ammoniac dégagent des quantités importantes de
platine.
a Cette monographie traite spécialement du platine et de
certains de ses dérivés importants du point de vue
professionnel ou écologique. Une étude détaillée des effets
toxiques du cisplatine en tant que médicament anti-cancéreux et
de ses analogues chez l'homme et l'animal sortirait du cadre de
cette série car il s'agit de produits utilisés essentiellement
comme agents thérapeutiques. En outre leurs propriétés toxiques
sont exceptionnelles comparées à celles des autres dérivés du
platine.
Les catalyseurs de véhicules automobiles constituent des
sources mobiles de platine. Selon des données limitées, l'attrition
des anciens catalyseurs en granulés se situe entre 0,8 et 1,9 µg/par
km parcouru. Environ 10% de ce platine est soluble dans l'eau.
Les résultats fournis par des mesures au banc d'essai montrent
que les pots catalytiques à trois voies utilisant des catalyseurs
monolithiques de la nouvelle génération réduisent les émissions
totales de platine d'un facteur de 100-1000 par rapport aux
catalyseurs en granulés. A des vitesses simulées de 60, 100 et 140
km/heure on a constaté que l'émission totale de platine se situait
entre 3 et 39 ng/m3 dans les gaz d'échappement, ce qui correspond
environ à 2-39 ng par kilomètre parcouru. Le diamètre aérodynamique
moyen des particules émises variait, lors des différents essais,
entre 4 et 9 µm. D'après quelques indices, on peut penser que la
majeure partie du platine est émise sous forme de métal ou de
particules oxydées en surface.
3. Transport, répartition et transformation dans l'environnement
Les métaux du groupe du platine sont rares dans le milieu
ambiant, comparativement aux autres éléments. Dans les zones très
industrialisées, on peut trouver d'importantes quantités de platine
dans les sédiments des cours d'eau. On pense que les matières
organiques, par exemple les acides humiques et fulviques, se lient
au platine, cette réaction étant sans doute facilitée par des
conditions convenables de pH et de potentiel redox dans le milieu
aquatique.
Dans le sol, la mobilité du platine dépend du pH, du potentiel
redox, de la teneur en chlore de l'eau qui imprègne le sol et de
l'état naturel du platine dans les roches primaires. On estime que
le platine n'est mobilisé que dans des conditions d'acidité extrême
ou lorsque l'eau du sol est très riche en chlore.
On a montré qu' in vitro certains complexes du platine(IV)
pouvaient être méthylés en présence de platine(II) par la
méthylcobalamine bactérienne dans des conditions abiotiques.
4. Concentrations dans l'environnement et exposition humaine
La base de données relatives aux concentrations dans
l'environnement est très limitée en raison de la très faible teneur
de celui-ci en platine et des problèmes d'analyse que cela pose.
Les concentrations de platine dans des échantillons d'air
ambiant prélevés à proximité d'autoroutes aux Etats-Unis d'Amérique
avant l'introduction du pot catalytique se situaient en dessous de
la limite de détection de 0,05 pg/m3. Un certain nombre de données
récentes en provenance d'Allemagne indiquent qu'à proximité des
routes, des concentrations de platine dans l'air ambiant
(échantillons de matière particulaire) vont de moins de 1 pg/m3 à
13 pg/m3. Dans les zones rurales, ces concentrations étaient du
même ordre de grandeur (moins de 0,6 à 1,8 pg/m3).
A proximité immédiate des routes, les concentrations de platine
dans l'air ambiant qui résultent de l'introduction de catalyseurs en
granulés ont été évaluées à partir de modèles de dispersion et sur
la base des données expérimentales relatives aux émissions. Etant
donné que l'émission totale de platine d'un catalyseur de type
monolithique est plus faible, sans doute d'un facteur allant de 100
à 1000, que celle d'un catalyseur en granulés, les concentrations en
platine provenant de ce type de catalyseur devraient être de l'ordre
du picogramme au femtogramme par mètre cube.
En divers endroits de Californie, on a trouvé, dans la
poussière déposée sur les plantes à larges feuilles, des
concentrations de 37-680 µg/kg de poids sec. Le nombre
d'échantillons était limité mais les résultats montrent tout de même
que le pot catalytique libère du platine dans l'environnement
immédiat des routes.
Des cultures de graminées ont été exposées dans des serres
expérimentales pendant quatre semaines à des gaz d'échappement
légèrement dilués provenant d'un moteur équipé d'un catalyseur à
trois voies (vitesse simulée 100 km/heure): à la limite de détection
de 2ng par gramme de poids sec, on n'a pas trouvé de platine.
Des analyses effectuées sur les sédiments du lac Michigan ont
montré que du platine s'y était déposé depuis une cinquantaine
d'années à un rythme assez uniforme. Des concentrations dans des
carottes de 1 à 20 cm étaient comprises entre 0,3 et 0,4 µg/kg de
poids sec seulement.
On ne signale pas la présence de platine dans les eaux douces,
en revanche de fortes concentrations (730 à 31 220 µg/kg de poids
sec) ont été mesurées dans les sédiments d'un canal très pollué du
Rhin en Allemagne.
Dans des échantillons de bois de Pinus flexilis on a trouvé
des concentrations de platine allant de 0 (non décelable) à 56 µg/kg
(poids des cendres). Toutefois la teneur du sol voisin était du même
ordre et ces données plutôt limitées n'indiquent aucune tendance à
l'accumulation.
Dans des échantillons isolés de végétaux provenant d'un sol
extrêmement basique, on a mesuré des teneurs en platine allant de
100 à 830 µg/kg (poids sec).
Dans des échantillons d'eau de mer, on a relevé des
concentrations allant de 37 à 332 pg/litre. Des carottes de sédiment
prélevées dans le Pacifique oriental présentaient des teneurs en
platine allant de 1,1 à 3 µg/kg (poids sec). La concentration la
plus élevée (21,9 µg/kg) a été mesurée dans des sédiments océaniques
à distance du littoral. Les algues macroscopiques marines présentent
des teneurs en platine allant de 0,08 à 0,32 µg/kg de poids sec.
Dans la population générale le taux de platine sanguin se situe
entre 0,1 et 2,8 µg/litre. Dans le sérum de travailleurs exposés au
platine de par leur activité professionnelle, on a relevé des
concentrations de 150 à 440 µg/litre.
La base de données relative aux concentrations de platine sur
les lieux de travail est limitée. En raison de problèmes d'analyse,
les données anciennes (0,9 à 1700 µg/m3) ne sont probablement pas
fiables. Toutefois on peut déduire de ces données que l'exposition
aux sels de platine était à l'époque plus forte que la limite
d'exposition professionnelle de 2 µg/m3 qui est actuellement en
vigueur dans la plupart des pays. Des études récentes effectuées sur
les lieux de travail font état de concentrations qui sont, soit
inférieures à la limite de détection de 0,05 µg/m3, soit comprises
entre 0,08 et 0,1 µg/m3.
5. Cinétique et métabolisme
Une seule exposition de 48 minutes par la voie respiratoire à
du platine sous différentes formes chimiques (5-8 mg/m3) a montré
que le 191Pt inhalé était rapidement éliminé de l'organisme. On
observe ensuite une phase d'élimination plus lente au cours de la
période suivant l'exposition. Dix jours après exposition à du
191PtCl4, du 191Pt(SO4), du 191PtO2 et du 191Pt sous
forme métallique, la rétention du 191Pt dans l'ensemble de
l'organisme était respectivement de 1, 5, 8 et 6% de la dose
initiale. La majeure partie du 191Pt a été éliminée des poumons
par l'action de l'ascenseur mucociliaire puis avalée et excrétée
dans les matières fécales (temps de demi-élimination, 24 h.). Une
petite fraction du 191Pt a été décelée dans les urines, ce qui
indique que la résorption est très faible au niveau des poumons et
des voies digestives.
Lors d'une étude sur la destinée comparée du 191PtCl4
administré à des rats selon différentes voies à raison de 25uCi par
animal, on a constaté que c'était la voie intraveineuse qui
entraînait la rétention la plus forte, suivie par la voie
intratrachéenne. Elle était minimale après administration par voie
orale. Etant donné que seule une très faible partie du produit
administré par voie orale a été résorbée, l'essentiel a traversé les
voies digestives et a été excrété dans les matières fécales. Au bout
de trois jours, on ne décelait plus dans l'ensemble de l'organisme
que moins de 1% de la dose initiale. Après administration
intraveineuse, le 191Pt se retrouvait en quantités pratiquement
égales dans les matières fécales et dans l'urine. L'élimination
était plus faible qu'après administration orale. Au bout de cette
même période le taux de rétention dans l'ensemble de l'organisme
était d'environ 65% et au bout de 28 jours il se situait encore à
14% de la dose initiale. A titre de comparaison, au bout de ces deux
intervalles de temps environ 22 et 8% respectivement de la dose
initiale demeuraient dans l'organisme après administration intra-
trachéenne.
Les principaux sites d'accumulation sont les reins, le foie, la
rate et les glandes surrénales. La forte quantité de 191Pt
retrouvée dans les reins montre qu'une fois absorbé, le platine
s'accumule en majeure partie dans ces organes d'où il est excrété
dans l'urine. La quantité plus faible trouvée dans le cerveau montre
que les ions platine ne traversent qu'en faible proportion la
barrière hémo-méningée.
Contrairement aux sels solubles dans l'eau, le PtO2, qui est
insoluble, n'a été résorbé qu'en quantités très faibles, même après
administration dans la nourriture à dose très élevée, c'est-à-dire
correspondant à une dose totale de platine de 4308 mg par rat sur
une période de quatre semaines.
Qu'il s'agisse des sels simples ou du cisplatine, il est établi
que l'élimination se fait en deux phases : une phase initiale rapide
suivie d'une phase prolongée au cours de la période suivant
l'exposition, et que rien ne permet de penser que les modalités de
rétention soient très différentes. Toutefois le cisplatine est très
stable dans les liquides extracellulaire en raison de la forte
concentration en ions chlorure qui suppriment l'hydratation. Ainsi
s'explique que cette substance soit excrétée presque entièrement
sous forme inchangée. Contrairement au cas des sels simples, elle
est excrétée principalement dans les urines.
6. Effets sur les mammifères de laboratoire et les systèmes
d'épreuve in vitro
La toxicité aiguë du platine est principalement fonction de la
forme sous laquelle il se trouve. Les sels solubles sont beaucoup
plus toxiques que les sels insolubles. Par exemple la toxicité par
voie orale pour les rats (DL50) décroît dans l'ordre suivant:
Na2[PtCl6] (25-50 mg/kg) > (NH4)2[PtCl6] (195-200 mg/kg)
> PtCl4 (240 mg/kg) > Pt(SO4)2.4H2O (1010 mg/kg) >
PtCl2(> 2000 mg/kg) > PtO2 (> 8000 mg/kg). En ce qui concerne
ces deux derniers composés on n'a pas pu calculer la valeur de la
DL50.
Des tests cutanés pratiqués sur des lapins albinos ont montré
que PtO2, PtCl2, K2[PtCl4], [Pt(NO2)2(NH3)2],
Pt(C5H7O2)2 et trans-[PtCl2(NH3)2] pouvaient être
considérés comme non irritants. Par contre (NH4)2[PtCl6],
(NH4)2[PtCl4], Na2[PtCl6], Na2[Pt(OH)6],
K2[Pt(CN)4], [Pt(NH3)4]Cl2, et cis-[PtCl2(NH3)2]
se sont révélés irritants mais à des degrés divers.
Des tests d'irritation oculaire ont montré que tous les
composés testés avaient une action irritante. Le trans-
[PtCl2(NH3)2] ainsi que (NH4)2[PtCl4] se sont révélés
corrosifs.
De très sérieuses difficultés respiratoires ont été observées
après injection intraveineuse de complexes chloroplatiniques à des
cobayes et à des rats, vraisemblablement par suite d'une libération
d'histamine d'origine non allergique. Cette libération aspécifique
d'histamine complique l'interprétation des études sur l'animal et
sur l'homme en ce qui concerne le diagnostic de la sensibilisation
allergique. Après injection sous-cutanée et intraveineuse de
Pt(SO4)2, trois fois par semaine pendant quatre semaines, on n'a
pas constaté l'apparition d'un état allergique, à en juger d'après
les résultats des épreuves cutanées effectuées sur des cobayes et
des lapins, le transfert passif et les tests sur le coussinet
plantaire de la souris. L'administration d'un complexe
platine/albumine d'oeuf n'a pas non plus entraîné de sensibilisation
chez les animaux de laboratoire.
On a essayé sans succès de sensibiliser des rats Lister
femelles avec du tétrachloroplatinate d'ammonium,
(NH4)2[PtCl4] par administration intrapéritonéale,
intramusculaire, intradermique, sous-cutanée, intratrachéenne, et
dans le coussinet plantaire en présence de Bordetella pertussis
comme adjuvant; la sensibilisation a été évaluée par un test cutané
direct, un test d'anaphylaxie cutanée passive (PCA) ou un test avec
radio-allergo-absorbant (RAST). Toutefois le test PCA a donné un
résultat positif après administration de conjugués
platine/protéines.
Chez des singes Cynomolgus (Macaca fascicularis) exposés à de
l'hexachloroplatinate de sodium, Na2[PtCl6] exclusivement en
inhalations nasales à la dose de 200 µg par m3, 4 h par jour, deux
fois par semaine pendant 12 semaines, on a observé un déficit
pulmonaire sensiblement plus élevé que chez les témoins. Dans le cas
de l'hexachloroplatinate d'ammonium, il a fallu exposer les animaux
simultanément à de l'ozone (200 µ/m3) pour obtenir une
hypersensibilisation cutanée et une hyperréactivité pulmonaire
significatives.
Des études sur des rats Sprague-Dawley mâles ont montré que des
sels comme PtCl4 (182 mg/litre d'eau de boisson) et comme
Pt(SO4).4H2O (248 mg/litre) n'affectaient pas le gain de poids
au cours de la période d'observation de 4 semaines. En triplant la
concentration de platine, il y a eu une réduction de 20% du gain de
poids, mais seulement pendant la première semaine, parallèlement à
une diminution de 20% de la prise de nourriture et de boisson.
On ne dispose que de données expérimentales limitées à propos
des effets du platine sur la reproduction, et plus particulièrement
à propos de ses éventuels effets embryotoxiques et tératogènes. Le
Pt(SO4)2 (200 mg Pt/kg) a provoqué la mise bas de souriceaux
Swiss ICR de poids réduit du jour 8 au jour 45 du post-partum. Le
principal effet de Na2[PtCl6] (20 mg Pt/kg) a consisté dans une
réduction de l'activité de la progéniture lorsque les mères avaient
été exposées le douzième jour de la gestation. Les fils et les
feuilles de platine sont considérés comme inertes biologiquement et
les effets nocifs constatés après implantation dans l'utérus de
rattes et de lapines étaient probablement dus à la présence physique
d'un corps étranger.
Après administration à des rattes gravides d'une dose de
191Pt égale à 25 microcuries par animal, le 18ème jour de la
gestation, on a constaté un passage limité à travers la barrière
foeto-placentaire.
Plusieurs dérivés du platine se sont révélés mutagènes dans un
certain nombre de systèmes bactériens. Lors d'études comparatives on
a constaté que la mutagénicité du cisplatine était plusieurs fois
supérieure à celle des autres composés. Des études in vitro ont
montré que, dans le système cellulaire mammalien CHO-HGPT,
l'activité mutagène relative s'établissait selon la proportion
100:9:0,3 respectivement pour les composés suivants: cis-
[PtCl2(NH3)2], K[PtCl3(NH3)], et [Pt(NH3)3Cl]Cl. La
mutagénicité de K2[PtCl4] et du trans-[PtCl2(NH3)] était
marginale, tandis que le [Pt(NH3)4]Cl2 n'était pas mutagène.
Les composés K2[PtCl4] et [Pt(NH3)4]Cl2 ne l'étaient pas
non plus dans les tests suivants: mutation récessive léthale liée au
sexe chez Drosophila melanogaster, recherche de micronoyaux dans
des cellules de souris et le test sur moelle osseuse de hamster. On
ne possède de données expérimentales sur la cancérogénicité des
dérivés du platine que dans le cas du cisplatine pour lequel les
preuves d'une activité cancérogène chez l'animal sont suffisantes.
Cependant le cisplatine et ses analogues font plutôt figure
d'exception si on les compare aux autres dérivés du platine. Cela
transparaît dans leur activité antitumorale, dont le mécanisme est
très particulier. On pense que celle-ci est, semble-t-il, due à la
formation de ponts intercaténaires qui ne se produit qu'en présence
de l'isomèreciset pour une certaine position de la guanine. Les
cellules tumorales ne sont plus en mesure de se répliquer, alors que
les cellules normales conservent leur capacité de réplication après
avoir réparé les lésions provoquées par le cisplatine.
7. Effets sur l'homme
L'exposition aux sels de platine se limite essentiellement aux
ambiances de travail, et plus précisément aux ateliers d'affinage du
platine et aux unités de production de catalyseurs.
Les composés principalement responsables de l'hyper-sensibilité
aux sels de platinea sont l'acide hexachloroplatinique
H2[PtCl6]et un certain nombre de sels comme
l'hexachloroplatinate d'ammonium (NH4)2[PtCl6], le
tétrachloroplatinate de potassium, K2[PtCl4], et le
tétrachloroplatinate de sodium, Na2[PtCl4]. Les complexes dans
lesquels il n'y a pas d'halogènes coordonnés au platine (complexes
non halogénés), comme K2[Pt(NO2)4], [Pt(NH3)4]Cl2 et
[Pt{(NH2)2CS}4]Cl2, de même que les complexes neutres comme
le cis-[PtCl2(NH3), ne sont pas allergéniques car ils
réagissent avec les protéines pour former un antigène complet.
Les symptômes de cette hypersensibilité sont les suivants:
urticaire, dermatite de contact, ainsi qu'un certain nombre de
troubles respiratoires, comme reniflement, essouflement, cyanose et
asthme grave. La période de latence entre le premier contact avec
des sels de platine et l'apparition des symptômes dure de quelques
semaines à plusieures années. Après sensibilisation les symptômes
ont tendance à s'aggraver aussi longtemps que les travailleurs sont
exposés sur leur lieu de travail, mais ils disparaissent, en
général, dès que cesse l'exposition. Toutefois, si une exposition de
longue durée fait suite à la sensibilisation, les symptômes risquent
de ne jamais disparaître complètement.
Bien qu'il soit impossible de tirer des données publiées une
relation dose-effet qui ne soit pas ambiguë, il semble que le risque
d'apparition d'une hyper-sensibilité aux sels de platine soit en
corrélation avec l'intensité de l'exposition. Le platine métallique
ne semble pas être allergénique. A l'exception d'un cas unique de
dermatite de contact, aucune réaction allergique n'a été signalée.
Les manifestations cliniques de l'hypersensibilité aux sels de
platine sont celles d'une véritable réaction allergique. Le
mécanisme de cette réaction est du type 1 (médiation par les IgE).
Sur la base d'épreuves in vivo et in vitro on pense que chez les
sujets sensibles il se forme des anticorps IgE dirigés contre les
complexes chloroplatiniques. Les sels de platine de faible masse
moléculaire relative se comportent comme des haptènes qui se
combinent aux protéines pour former des antigènes complets.
a Le terme platinose n'est plus usité pour désigner les
affections provoquées par les sels de platine, car il implique
une fibrose pulmonaire chronique du type silicose. Il est
préférable d'utiliser le terme allergie aux sels de platine, ou
allergie aux composés du platine contenant des ligands
halogénés réactifs ou mieux, hypersensibilité aux sels de
platine.
Les tests cutanés avec des sels de platine dilués permettent de
surveiller les réactions allergiques de manière reproductible,
fiable, assez sensible et très spécifique. Pour les contrôles de
routine en milieu professionnel on utilise les composés suivants:
(NH4)2[PtCl6], Na2[PtCl6] et Na2[PtCl4]. Il n'existe
pas d'épreuve in vitro dont la sensibilité et la fiabilité
approchent celles des tests cutanés. Des tests immunoenzymatiques ou
par immunoallergosorption ont permis de mettre en évidence des
anticorps IgE spécifiques dirigés contre les complexes chlorés du
platine. Il y avait corrélation avec les résultats des tests cutanés
mais l'utilisation du test RAST reste problématique en raison de son
manque de spécificité.
Les tests cutanés et l'épreuve RAST montrent qu'il n'existe
qu'une faible réactivité croisée entre les sels de platine et les
sels de palladium. Des réactions d'hypersensibilité à d'autres
métaux du groupe du platine ont également été observées, mais
seulement chez des personnes allergiques aux sels de platine.
Le tabagisme, l'atopie et l'hyperréactivité pulmonaire
aspécifique ont été associés à l'hypersensibilité aux sels de
platine et pourraient être des facteurs prédisposants.
En ce qui concerne la population générale, on manque de données
sur l'exposition effective dans les pays où le pot catalytique est
devenu obligatoire. Les concentrations dans l'air ambiant estimées
d'après de nouvelles données sur les émissions et sur la base de
modèles de dispersion sont probablement inférieures d'au moins un
facteur 10 000 à la limite d'exposition professionnelle de 1 mg/m3
adoptée par certains pays pour le platine total inhalable sous forme
de poussières. Etant donné que le platine est très probablement
présent dans les émissions sous forme métallique, le potentiel de
sensibilisation du platine émis par les pots catalytiques est
probablement très faible. Même si une partie du platine émis est
soluble et potentiellement allergénique, la marge de sécurité par
rapport à la limite d'exposition professionnelle pour les sels
solubles de platine (2 µg/m3) serait d'au moins 2000.
Lors d'une étude immunologique préliminaire, on a pratiqué des
tests cutanés sur trois volontaires au moyen d'extraits de matières
particulaires émises par des véhicules à moteur. On n'a pas observé
de réponse positive.
On ne dispose d'aucune donnée sur les risques de
cancérogénicité pour l'homme attribuables au platine et à ses sels.
Pour ce qui est du cisplatine les preuves de cancérogénicité sont
jugées insuffisantes.
8. Effets sur d'autres organismes au laboratoire et dans la nature
Les complexes simples du platine ont des effets bactéricides.
En observant que les complexes neutres comme le cisplatine
inhibaient sélectivement la division cellulaire sans réduction de la
croissance chez diverses bactéries gram-positives mais aussi, et
surtout, gram-négatives on a eu l'idée de les utiliser comme agents
anticancéreux.
On a observé qu'au sein d'un "microcosme" de laboratoire, la
croissance des algues vertes du genre euglène était inhibée en
présence d'acide hexachloroplatinique soluble aux concentrations de
250, 500, et 750 µg/litre. Le cisplatine a provoqué une chlorose et
un ralentissement de la croissance chez la jacinthe d'eau Eichornia
crassipesà la concentration de 2,5 mg/litre.
Après 3 semaines d'exposition à de l'acide
hexachloroplatinique, H2[PtCl6], on a observé chez la daphnie
une mortalité correspondant à une CL50 de 520 µg de Pt par litre.
Aux concentrations de 14 et 82 µg/litre, la reproduction (nombre de
jeunes daphnies) était réduite dans la proportion de 16 et 50%
respectivement.
Après exposition de saumons (Oncorhyncus kisutch) à de
l'acide tétrachloroplatinique pendant une brève période de temps
dans des conditions statiques, on a observé que les valeurs de la
CL50 étaient respectivement égales à 15,5, 5,2 et 2,5 mg Pt/litre
au bout de 24, 48, et 96 h. On constatait, à la dose de 0,3
mg/litre, une diminution globale de l'activité natatoire et du
mouvement des opercules. Aux concentrations supérieures à cette
valeur, des lésions apparaissaient au niveau des branchies et de
l'organe olfactif. Les concentrations de 0,03 et 0,1 mg/litre
étaient sans effet.
Toutes les études consacrées aux effets du platine sur les
plantes terrestres concernent uniquement les chlorures solubles. A
des concentrations allant de 3.10-5 à 15.10-5 mol/kg (5,9-29,3
mg/kg) il y a eu inhibition de la croissance de plants de haricots
et de tomates en sol sableux. Neuf variétés horticoles en culture
hydroponique ont présenté une réduction de leur poids à sec après
adjonction de tétrachlorure de platine aux concentrations
respectives de 0,057, 0,57, et 5,7 mg Pt/litre; il s'agissait de
tomates, de poivrons, de fanes de navets et pour la concentration la
plus élevée, de radis. A cette concentration les bourgeons et les
feuilles immatures devenaient chlorotiques chez la plupart des
espèces. En revanche, chez certaines espèces, le tétrachlorure de
platine stimulait la croissance. Par ailleurs, la teneur la plus
élevée en platine supprimait la transpiration, sans doute par
accroissement de la résistance des stomates. On constatait également
une stimulation de la croissance aux faibles teneurs en platine (0,5
mg/litre), lorsque l'on ajoutait au milieu nutritif d'une graminée
sud-africaine (Setaria vertillata) dutétrachloroplatinate de
potassium. Au bout de deux semaines, la racine la plus longue avait
poussé de 65%. A la concentration utilisée, soit 2,5 mg Pt/litre, on
observait des effets phytotoxiques tels que rabougrissement des
racines et chlorose foliaire.
RESUMEN
1. Identidad, propiedades físicas y químicas, métodos analíticos
El platino (Pt) es un metal noble maleable, dúctil, de color
plateado blanquecino; su número atómico es 78 y su peso atómico
195,09. Sus isótopos naturales más abundantes son 194Pt (32,9%),
195Pt (33,8%), y 196Pt (25,3%). En los compuestos de platino, el
estado de oxidación máxima es +6; los estados +2 y +4 son los más
estables.
Aunque el metal no se corroe en el aire a ninguna temperatura,
es sensible a los halógenos, los cianuros, el azufre, los compuestos
de azufre fundentes, los metales pesados y los hidróxidos de
álcalis. La digestión con agua regia o Cl2/HCl (ácido clorhídrico
concentrado por el que se burbujea cloro) produce ácido
hexacloroplatínico, H2[PtCl6], un importante complejo de
platino. Cuando se calienta, la sal amónica del ácido
hexacloroplatínico produce una esponja gris de platino. La reducción
en solución acuosa produce un polvo dispersivo de color negro
("negro de platino").
Las propiedades químicas de los compuestos del platino en
solución acuosa se ven dominadas por los compuestos complejos.
Muchas de las sales, particularmente las que llevan ligandos
donadores de halógeno o de nitrógeno, son solubles en agua. El
platino, al igual que los otros metales de su grupo, tiene una
pronunciada tendencia a reaccionar con los compuestos del carbono,
especialmente los alquenos y los alquinos, formando complejos de
coordinación Pt(II).
Existen diversos métodos analíticos para la determinación del
platino. La espectrometría de absorción atómica (EAA) y la
espectroscopia de emisión de plasma son sumamente selectivas y
específicas y constituyen el método de elección para analizar el
platino presente en muestras biológicas y medioambientales. Con esos
métodos se han alcanzado en diversos medios límites de detección del
orden de unos cuantos µg/kg o µg/litro.
La espectroscopia de emisión atómica con plasma de argón
acoplado por inducción es preferible a la EAA electrotérmica por sus
menores efectos matriciales y por la posibilidad de analizar
simultáneamente muchos elementos.
2. Fuentes de la exposición humana y ambiental
Se calcula que la concentración media de platino en la
litosfera o corteza terrestre es del orden de 0,001-0,005 mg/kg. El
platino se encuentra en forma metálica o en varias formas minerales.
Existen fuentes económicamente importantes en la República de
Sudáfrica y en la URSS. El contenido de platino de esos depósitos es
de 1-500 mg/kg. En el Canadá, los metales del grupo del platino
(platino, paladio, iridio, osmio, rodio, rutenio) se encuentran en
menas de sulfuro de cuproníquel con una concentración media de 0,3
mg/kg, pero esa concentración supera los 50 mg/kg durante el afinado
del cobre y el níquel. En los EE.UU., Etiopía, Filipinas y en
Columbia se extraen pequeñas cantidades.
La producción minera mundial de metales del grupo del platino,
de la cual el 40-50% corresponde al platino, ha aumentado
uniformemente durante los últimos 20 años. En 1971, la producción
fue de 127 toneladas (51-64 toneladas de platino). A raíz de la
introducción del catalizador de los gases de escape en los
automóviles, la producción minera mundial de metales del grupo del
platino aumentó hasta aproximadamente 270 toneladas (108-135
toneladas de platino) en 1987. En 1989, la demanda total de platino
en el mundo occidental fue de unas 97 toneladas.
El uso principal del platino deriva de sus excepcionales
propiedades catalíticas. Las demás aplicaciones industriales
aprovechan otras notables propiedades, en particular la resistencia
a la corrosión química en un amplio intervalo de temperaturas, su
elevado punto de fusión, su gran resistencia mecánica y su buena
ductilidad. El platino se usa asimismo en joyería y odontología.
Ciertos complejos de platino, en particular el cis-
diaminodicloroplatino(II) (cisplatino), tienen aplicaciones
terapéuticas.a
No se dispone de datos sobre las emisiones de platino al medio
ambiente a partir de fuentes industriales. El uso de catalizadores
con platino puede entrañar la liberación de ese elemento al medio
ambiente, según el tipo de catalizador. De los catalizadores
estacionarios utilizados en la industria, sólo los empleados para la
oxidación del amoniaco emiten cantidades significativas de platino.
Los catalizadores utilizados en automoción son fuentes móviles
de platino. Se dispone de datos limitados que indican que el
desgaste de platino a partir del antiguo catalizador en pastilla es
a La presente monografía se ocupa específicamente del platino y
de ciertos compuestos del platino de importancia ocupacional
y/o ambiental. No entra en el ámbito restringido de la serie de
Criterios de Salud Ambiental el estudio pormenorizado de los
efectos tóxicos del fármaco anticanceroso cisplatino y de sus
análogos en el hombre y los animales, puesto que esas
sustancias se usan principalmente como agentes terapéuticos.
Además, sus propiedades tóxicas son excepcionales en
comparación con las de otros compuestos de platino.
de 0,8 a 1,9 µg por km recorrido. Alrededor del 10% del platino es
soluble en agua.
Con la nueva generación de catalizadores de tipo monolítico,
los resultados de experimentos en plataforma de pruebas de motores
con un catalizador de tres vías indican que la emisión total de
platino es inferior por un factor de 100-1000 a la producida en
catalizadores en pastilla. Con velocidades simuladas de 60, 100 y
140 km/h, se encontró que la emisión total de platino era de 3 a 39
ng/m3 en los gases de escape, lo que corresponde a unos 2-39 ng
por km recorrido. El diámetro aerodinámico medio de las partículas
emitidas era de 4 a 9 µm en las distintas pruebas. Existen pruebas
limitadas de que la mayor parte del platino emitido se encuentra en
forma metálica o en partículas de superficie oxidada.
3. Transporte, distribución y transformación en el medio ambiente
Los metales del grupo del platino son escasos en el medio
ambiente en comparación con otros elementos. En zonas muy
industrializadas, pueden encontrarse cantidades elevadas de platino
en los sedimentos fluviales. Se supone que la materia orgánica, por
ejemplo los ácidos húmicos y fúlvicos, enlaza platino, proceso que
tal vez se vea favorecido por condiciones apropiadas de pH y de
potencial redox en el medio acuático.
En el suelo, la movilidad del platino depende del pH, el
potencial redox, las concentraciones de cloruros en las aguas
subterráneas y la forma en que se encuentra el platino en la roca
primitiva. Se considera que el platino sólo será móvil en
condiciones extremadamente ácidas o en aguas subterráneas con
elevado contenido de cloro.
En los sistemas de ensayo in vitro se ha demostrado que
algunos complejos de platino(IV), en presencia de platino(II),
pueden sufrir metilación por la metilcobalamina bacteriana en
condiciones abióticas.
4. Niveles medioambientales y exposición humana
Se dispone de muy pocos datos en cuanto a las concentraciones
medioambientales debido a los reducidos niveles de platino en el
medio ambiente y los problemas analíticos que ello acarrea.
Las concentraciones en muestras de aire obtenidas en las
proximidades de autopistas en los Estados Unidos antes de la
introducción del catalizador en los automóviles se encontraban por
debajo del límite de detección de 0,05 pg/m3. Algunos datos
obtenidos recientemente en Alemania indican que en las cercanías de
las carreteras las concentraciones de platino en el aire (muestras
particuladas) varían entre < 1 pg/m3 y 13 pg/m3. En zonas
rurales, las concentraciones se encontraban en un orden de magnitud
similar (< 0,6 a 1,8 pg/m3).
Las concentraciones de platino en el aire cercano a carreteras
tras la introducción de los catalizadores de pastilla en los
automóviles se han calculado basándose en modelos de dispersión y
datos experimentales de emisión. Las concentraciones estimadas de
platino en las carreteras y en las zonas próximas variaron entre
0,005 y 9 ng/m3 para el platino total. Puesto que la emisión total
de platino en un catalizador de tipo monolítico es inferior,
probablemente por un factor de 100 a 1000, que en un catalizador de
pastilla, las concentraciones de platino emitidas en ese tipo de
catalizador se encontrarían en el margen de picogramos a femtogramos
por m3.
En el polvo depositado en las plantas de hoja ancha que bordean
las carreteras en distintos lugares de California, se detectaron
concentraciones de 37-680 µg por kg de peso seco. Aunque el número
de muestras era limitado, los resultados indican que los
catalizadores de automóviles liberan platino al medio ambiente
próximo a las carreteras.
En experimentos en cámara vegetal, los cultivos herbáceos
expuestos durante cuatro semanas a gases de escape ligeramente
diluidos procedentes de un motor equipado con un catalizador de 3
vías (velocidad simulada: 100 km/h) no contenían platino con un
límite de detección de 2 ng/g de peso seco.
Los estudios de las concentraciones de platino en sedimentos
del Lago Michigan llevaron a la conclusión de que el platino se ha
ido depositando en ellos durante los últimos 50 años a una velocidad
uniforme. Las concentraciones en calas de sedimento de 1 a 20 cm
variaron sólo entre 0,3 y 0,43 µg/kg de peso seco.
Mientras que no se han comunicado niveles de platino en aguas
dulces, se han encontrado elevadas concentraciones (730 a 31 220
µg/kg de peso eco) en los sedimentos de un canal de corta sumamente
contaminado en el río Rin (Alemania).
En muestras de Pinus flexilisse encontraron niveles de
platino entre el límite de detección y 56 µg/kg (peso de ceniza). No
obstante, el contenido de los suelos adyacentes se encontraba entre
los mismos valores; estos datos limitados no indicaban tendencia
alguna de acumulación.
En muestras aisladas de vegetales procedentes de un suelo
ultrabásico, se encontraron niveles de platino de 100-830 µg/kg
(peso seco).
En muestras de agua marina se han encontrado entre 37 y 332
pg/litro. En calas de sedimento obtenidas en el Pacífico oriental,
las concentraciones de platino variaron entre 1,1 y 3 µg/kg (peso
seco). La concentración más elevada (21,9 µg por kg) se encontró en
sedimentos oceánicos alejados del litoral. En macroalgas marinas se
han encontrado concentraciones de platino entre 0,08 y 0,32 µg/kg de
peso seco.
En la población general se han medido niveles sanguíneos de
platino de 0,1 a 2,8 µg/litro. En suero de trabajadores expuestos
por su profesión, se han comunicado niveles de 150 a 440 µg por
litro.
Se dispone de datos limitados sobre las concentraciones de
platino en el lugar de trabajo. Es probable que los datos antiguos
(0,9 a 1700 µg/m3) no sean de fiar debido a las deficiencias del
análisis. No obstante, esos datos permiten suponer que el nivel de
exposición a sales de platino era superior al límite de exposición
profesional de 2 µg/m3 adoptado actualmente en la mayoría de los
países. En recientes estudios realizados en los lugares de trabajo,
se han medido concentraciones inferiores al límite de detección de
0,05 µg/m3 o entre 0,08 y 0,1 µg/m3.
5. Cinética y metabolismo
Tras una exposición única por inhalación (48 minutos) a
distintas formas químicas del platino (5-8 mg/m3), la mayor parte
del 191Pt inhalado fue rápidamente eliminado del organismo. A
continuación se observó una fase más lenta de eliminación durante el
resto del periodo posterior a la exposición. A los diez días de la
exposición a 191PtCl4, 191Pt(SO4)2, 191PtO2, y 191Pt
metálico, la retención total de 191Pt por el organismo fue de
aproximadamente 1, 5, 8 y 6%, respectivamente, de la carga inicial
del organismo. La mayor parte del 191Pt eliminado de los pulmones
por mecanismos mucociliares e ingerido se excretó con las heces
(semivida: 24 h). Una pequeña fracción del 191Pt se detectó en la
orina, lo que indica que la absorción en los pulmones y el tracto
gastrointestinal fue muy reducida.
En un estudio comparativo sobre el destino del 191PtCl4 en
ratas (25 µCi/animal) tras la exposición por distintas vías, la
retención fue máxima tras la administración intravenosa, seguida por
la exposición intratraqueal. La mínima se registró tras la
administración oral. Puesto que sólo fue absorbida una cantidad
minúscula del 191PtCl4 administrada por vía oral, la mayoría
atravesó el tracto gastrointestinal y se excretó con las heces. Al
cabo de tres días, menos del 1% de la dosis inicial se detectó en
todo el cuerpo. Tras la administración intravenosa, el 191Pt se
excretó en cantidades casi iguales tanto en las heces como en la
orina. La eliminación fue más lenta que en el caso de la
administración oral. A los tres días la retención en todo el
organismo era de alrededor del 65%, y al cabo de 28 días aún era del
14% de la dosis inicial. A título de comparación, al cabo de
periodos iguales alrededor del 22% y del 8%, respectivamente,
quedaron retenidos por el organismo tras la administración
intratraqueal.
Los principales lugares de depósito son el riñón, el hígado, el
bazo y las glándulas suprarrenales. La elevada cantidad de 191Pt
encontrada en el riñón demuestra que una vez que el platino es
absorbido, la mayor parte se acumula en él y se excreta en la orina.
El nivel más bajo en el cerebro sugiere que los iones de platino
atraviesan la barrera hematoencefálica sólo en grado limitado.
A diferencia de las sales hidrosolubles, el PtO2, que es
insoluble, sólo fue captado en cantidades insignificantes a pesar de
que la sal se administró con la dieta en concentraciones sumamente
elevadas, que representaron un consumo total de platino de 4308 mg
por rata durante el periodo de cuatro semanas.
Tanto en el caso de las sales simples de platino como en el
cisplatino, se ha determinado que existe un periodo de eliminación
rápida seguido de una fase prolongada de eliminación durante el
resto del periodo posterior a la exposición, y que no existen
pruebas de que los perfiles de retención sean notablemente
diferentes. No obstante, el cisplatino es sumamente estable en los
fluidos extracelulares debido a que las elevadas concentraciones de
cloruro suprimen la hidratación. Ello explica que se excrete
principalmente en la forma no alterada. Su excreción, a diferencia
de la de las sales simples de platino, tiene lugar principalmente
con la orina.
6. Efectos en mamíferos de laboratorio y en sistemas de ensayo in
vitro
La toxicidad aguda del platino depende principalmente de la
especie de platino. Los compuestos solubles son mucho más tóxicos
que los insolubles. Por ejemplo, la toxicidad por vía oral en la
rata (valores de la LD50) disminuyo en el orden siguiente:
Na2[PtCl6] (25-50 mg/kg) > (NH4)2[PtCl6] (195-200 mg/kg)
> PtCl4 (240 mg/kg) > Pt(SO4)2.4H2O (1010 mg/kg) >
PtCl2 (> 2000 mg/kg) > PtO2 (> 8000 mg/kg). No pudo alcularse
la DL50 correspondiente a los dos últimos compuestos.
En las pruebas cutáneas realizadas en conejos albinos, los
compuestos PtO2, PtCl2, K2[PtCl4], [Pt(NO2)2(NH3)2],
Pt(C5H7O2)2 y trans-[PtCl2(NH3)2] se clasificaron
como no irritantes. Los compuestos (NH4)2[PtCl6],
(NH4)2[PtCl4], Na2[PtCl6], Na2[Pt(OH)6],
K2[Pt(CN)4], [Pt(NH3)4]Cl2, y cis-[PtCl2(NH3)2]
resultaron irritantes en diversos grados.
En los ensayos de irritación ocular todos los compuestos de
platino ensayados dieron resultados positivos. El trans-
[PtCl2(NH3)2] y el (NH4)2[PtCl4] resultaron ser
corrosivos.
Tras la inyección intravenosa de complejos de cloroplatino en
cobayos y ratas, se observaron dificultades respiratorias intensas,
probablemente debidas a la liberación analérgica de histamina. Esta
liberación inespecífica de histamina ha complicado la interpretación
de los estudios en animales y en el hombre en relación con el
diagnóstico de la sensibilización alérgica.
Tras la inyección subcutánea e intravenosa de Pt(SO4)2 tres
veces a la semana durante cuatro semanas, no se observó inducción de
un estado alérgico, de acuerdo con las pruebas cutáneas (cobayos y
conejos), la transferencia pasiva y los ensayos en la almohadilla
plantar (ratones). La administración del complejo platino-huevo-
albúmina tampoco sensibilizó a los animales de experimentación.
No se consiguió sensibilizar a hembras de rata encapuchada de
Lister con la sal libre de tetracloroplatinato de amonio,
(NH4)2[PtCl4], aplicada por las vías intraperitoneal,
intramuscular, intradérmica, subcutánea, intratraqueal y por la
almohadilla plantar, conBordetella pertussisw como coadyuvante, de
acuerdo con los resultados de la prueba cutánea directa, la prueba
de anafilaxis cutánea pasiva o un ensayo de radioalergosorbencia
(RAST). No obstante, se han comunicado resultados positivos de
anafilaxis cutánea pasiva con conjugados platino-proteína.
En monos Cynomolgus (Macaca fasicularis) expuestos a
hexacloroplatinato de sodio, Na2[PtCl6], por inhalación
exclusivamente nasal de una concentración de 200 µg/m3 wdurante 4
horas al día, dos veces a la semana durante 12 semanas, se
observaron insuficiencias pulmonares significativamente mayores que
en los animales testigo. Con la exposición a hexacloroplatinato de
amonio, (NH4)2[PtCl6], sólo la exposición simultánea a ozono
(2000 µg/m3) produjo hipersensibilidad cutánea e hiperreactividad
pulmonar significativas.
En estudios de administración oral a machos de rata Sprague-
Dawley, las sales PtCl4 (182 mg/l de agua de bebida) y
Pt(SO4)2.4H2O (248 mg/litro) no ejercieron efecto alguno en la
adquisición normal de peso durante el periodo de observación de 4
semanas. Al triplicar la concentración de platino, la adquisición de
peso se redujo en un 20% sólo durante la primera semana,
paralelamente a una disminución del 20% del consumo de alimento y
agua.
Sólo se dispone de datos experimentales limitados sobre los
efectos del platino en la reproducción, la embriotoxicidad y la
teratogenicidad. El Pt(SO4)2 (200 mg Pt/kg) redujo el peso de
las crías en ratones suizos ICR desde el día 8 al 45 después del
parto. El principal efecto del Na2[PtCl6] (20 mg Pt/kg) fue un
nivel de actividad menor en las crías de madres expuestas el duo-
décimo día de gestación. Se considera que el alambre y las láminas
de platino sólido son biológicamente inertes; los efectos adversos
observados a raíz de la implantación en el útero de ratas y ratones
se debieron probablemente a la presencia física de un objeto
extraño.
Tras la administración intravenosa de 191PtCl4 a ratas
gestantes (25 µCi/animal) el día 18 de la gestación, una cantidad
limitada del compuesto atravesó la barrera placentaria.
Se ha observado que varios compuestos de platino son
mutagénicos en diversos sistemas bacterianos. En estudios
comparativos, el cisplatino era varias veces más mutagénico que
otras sales de platino ensayadas. En estudios realizados in vitro
con células de mamífero (sistema CHO-HGPT), la actividad mutagénica
relativa de los compuestos cis-[PtCl2(NH3)2],
K[PtCl3(NH3)] y [Pt(NH3)3Cl]Cl fue 100:9:0,3. La
mutagenicidad del K2[PtCl4] y el trans-[PtCl2(NH3)2] era
marginal, mientras que el [Pt(NH3)4]Cl2 no era mutagénico. No
se observó actividad mutagénica en los compuestos K2[PtCl4] y
[Pt(NH3)4]Cl2, en el ensayo de letalidad recesiva ligada al
sexo en Drosophila melanogaster, en un ensayo de micronúcleo de
ratón ni en el ensayo en médula ósea de hámster chino.
Salvo en el caso del cisplatino, no se dispone de datos
experimentales relativos a la carcinogenicidad del platino y sus
compuestos. Existen prueba suficientes de la carcinogenicidad del
cisplatino en los animales. No obstante, el cisplatino y sus
análogos son excepcionales en comparación con otros compuestos de
platino; ello se refleja en su mecanismo característico de actividad
anti-tumoral. La constitución de enlaces cruzados entre las hebras
de ADN, formados sólo por el isómero cis en determinada posición de
la guanina, se considera el motivo de esa actividad antitumoral.
Parece ser que la replicación del ADN es defectuosa en las células
cancerosas, mientras que en las normales las lesiones causadas por
el cisplatino en la guanina se reparan antes de la replicación.
7. Efectos en el ser humano
La exposición a sales de platino se limita principalmente al
medio ocupacional, en particular a las refinerías de platino
metálico y las plantas de fabricación de catalizadores.
Los princiaples compuestos responsables de la hiper-
sensibilidad a las sales de platino son el ácido hexacloroplatínico,
H2[PtCl6], y algunas sales cloradas como el hexacloroplatinato
de amonio, (NH4)2[PtCl6], el tetracloroplatinato de potasio,
K2[PtCl4], el hexacloroplatinato de potasio, K2[Pt6] y el
tetracloroplatinato de sodio, Na2[PtCl4].a Los complejos en
los que no hay ligandos halógenos coordinados al platino ("complejos
no halogenados"), como el K2[Pt(NO2)4], [Pt(NH3)4]Cl2 y
[Pt{(NH2)2CS}4]Cl2, así como los complejos neutros como el
cis-[PtCl2(NH3)2], no son alergénicos, puesto que
probablemente no reaccionan con proteínas para formar un antígeno
completo.
Entre los signos y síntomas de hipersensibilidad figuran
urticaria, dermatitis de contacto de la piel, y trastornos
respiratorios que pueden ir desde estornudos, disnea y cianosis a
crisis graves de asma. El periodo de latencia desde el primer
contacto con el platino hasta la aparición de los primeros síntomas
varía desde unas pocas semanas a varios años. Una vez que la
sensibilización está establecida, los síntomas tienden a empeorar
durante el tiempo que los individuos sigan expuestos en el lugar de
trabajo, pero suelen desaparecer al cesar la exposición. No
obstante, si se produce una exposición prolongada después de la
sensibilización, es posible que los individuos nunca queden
completamente exentos de síntomas.
Aunque a partir de los datos disponibles no puede deducirse una
relación inequívoca entre la concentración y el efecto, el riesgo de
desarrollar sensibilidad a las sales de platino parece guardar
relación con la intensidad de la exposición. El platino metálico
parece no ser alergénico. A excepción de un solo caso comunicado de
supuesta dermatitis de contacto provocada por un anillo "de
platino", no se han notificado reacciones alérgicas.
Las manifestaciones clínicas de la hipersensibilidad a las
sales de platino reflejan una auténtica respuesta alérgica. El
mecanismo parece ser una respuesta de tipo I (medida por la IgE). La
posibilidad de que se desarrollen anticuerpos IgE a complejos de
cloruro de platino en personas sensibles se ha supuesto basándose en
los ensayos in vivo e in vitro. Se cree que las sales de platino
de baja masa molecular relativa actúan como haptenos que se combinan
con las proteínas séricas para formar el antígeno completo.
a Se ha abandonado el término "platinosis" para describir las
enfermedades relacionadas con las sales de platino, puesto que
implica una enfermedad pulmonar fibrosante crónica del tipo de
la silicosis. En su lugar, se han utilizado "alergia a las
sales de platino", "alergia a los compuestos de platino que
contienen ligandos halógenos reactivos" e "hipersensibilidad a
sales de platino" (HSP), siendo preferible el último.
Las pruebas de punción cutánea con concentraciones diluidas de
complejos solubles de platino parecen proporcionar indicadores
biológicos de la alergenicidad reproducibles, fiables,
razonablemente sensibles y sumamente específicos. Los compuestos
utilizados para las pruebas periódicas de detección de alergias en
los trabajadores expuestos son (NH4)2[PtCl6], Na2[PtCl6] y
Na2[PtCl4]. La sensibilidad y fiabilidad de las pruebas de
punción cutánea no tienen igual en ninguno de los ensayos in vitro
disponibles. En los inmunoensayos enzimáticos y las pruebas de
radioalergosorbencia (RAST) se han encontrado anticuerpos IgE
específicos de los complejos del cloruro de platino. Aunque se
comunicó la existencia de correlación con los resultados de las
pruebas de punción cutánea, la aplicabilidad de la prueba RAST para
los exámenes de detección se puso en tela de juicio a causa de su
falta de especificidad.
Se ha encontrado una reactividad cruzada limitada entre las
sales de platino y de paladio en los ensayos cutáneos y el RAST. Las
reacciones a los metales del grupo del platino distintos de éste
sólo se han observado en individuos sensibles a las sales de
platino.
El tabaquismo, la atopia y la hiperreactividad pulmonar
inespecífica se han asociado a la hipersensibilidad a las sales de
platino y pueden ser factores predisponentes.
En cuanto a la población general, no se dispone de bastantes
datos sobre la situación real en materia de exposición en los países
en los que se ha introducido el catalizador en los automóviles. Las
concentraciones atmosféricas posibles, calculadas teniendo en cuenta
algunos datos de emisión y modelos de dispersión, son inferiores por
un factor al menos de 10 000 al límite de exposición ocupacional de
1 mg/m3 adoptado por algunos países para el platino metálico como
polvo inhalable total. Puesto que el platino emitido se encuentra
con toda probabilidad en forma metálica, el potencial de
sensibilización de las emisiones de platino a partir de los
catalizadores de los automóviles es probablemente muy bajo. Aunque
parte del platino emitido fuera soluble y potencialmente alergénico,
el margen de seguridad respecto del límite de exposición profesional
para las sales de platino solubles (2 µg/m3) sería de al menos
2000.
En un estudio inmunológico preliminar, se ensayaron extractos
de muestras particuladas procedentes de escapes de automóviles en
tres voluntarios humanos mediante una prueba de punción cutánea. No
se obtuvo respuesta positiva.
No se dispone de datos para evaluar el riesgo carcinogénico del
platino ni de sus sales para el ser humano. En cuanto al cisplatino,
las pruebas disponibles en materia de carcinogenicidad humana se
consideran insuficientes.
8. Efectos en otros organismos en el laboratorio y sobre el terreno
Los complejos simples de platino tienen efectos bactericidas.
El descubrimiento de que los complejos neutros como el cisplatino
inhiben selectivamente la división celular sin reducir el
crecimiento celular de diversidad de bacterias gram-positivas y
especialmente gram-negativas ha llevado a su aplicación en medicina
como agentes antitumorales.
El crecimiento y la cosecha del alga verde Euglena gracilis
fueron inhibidos por el ácido hexacloroplatínico soluble (250, 500 y
750 µg/litro) en un "microcosmos" experimental. El cisplatino
ocasionó clorosis y retraso del crecimiento en el jacinto acuático
Eichhornia crassipes con una concentración de 2,5 mg/litro.
En el invertebrado Daphnia magna, la exposición durante tres
semanas a ácido hexacloroplatínico (H2[PtCl6]), dio un valor de
CL50 de 520 µg Pt por litro. Con concentraciones de 14 y 82
µg/litro, se observaron efectos en la reproducción que se
manifestron en reducciones del 16 y el 50%, respectivamente, del
número total de crías.
Tras la exposición a corto plazo al ácido tetracloroplatínico,
H2[PtCl4], en un bioensayo estático, los valores de la CL50 a
las 24, 48 y 96 horas fueron de 15,5, 5,2 y 2,5 mg Pt/litro,
respectivamente, en el salmón Oncorhynchus kisutch. Con 0,3
mg/litro se observaron efectos en a actividad natatoria general y el
movimiento opercular. Con 0,3 mg/litro o más se observaron lesiones
en las branquias y el órgano olfatorio. No se observaron efectos con
concentraciones de 0,03 y 0,1 mg/litro.
Se han estudiado los efectos del platino en plantas terrestres;
todos ellos se realizaron con cloruros de platino solubles. El ácido
hexacloroplatínico inhibió el crecimiento de las plantas de judías y
tomates en cultivo arenoso con concentraciones de 3 x 10-5 a 15 x
10-5 mol/kg (5,9-29,3 mg/kg). De nueve especies hortícolas
cultivadas en solución hidropónica con tetracloruro de platino,
PtCl4 (0,057, 0,57 y 5,7 mg Pt/litro), se observaron reducciones
significativas del peso seco en el tomate, el pimiento y las hojas
de nabo, así como en las raíces de rábano con la concentración más
elevada. Con esa concentración, los brotes y las hojas inmaduras de
la mayoría de las especies sufrieron clorosis. En algunas de las
especies, con niveles bajos de PtCl4 se observó un efecto del
estímulo del crecimiento. Además, se suprimió la transpiración con
la concentración más elevada de platino, probablemente debido a una
mayor resistencia de los estomas. También se observó estímulo del
crecimiento con niveles reducidos de platino (0,5 mg Pt/litro),
administrado en forma de tetracloroplatinato de potasio
K2[PtCl4], en plantones de la herbácea sudafricana Setaria
verticillata cultivada en solución de nutrientes. Al cabo de dos
semanas, las raíces más largas habían sufrido un aumento de longitud
del 65%. Con la concentración más elevada que se aplicó, es decir
2,5 mg Pt/litro, se observaron efectos fitotóxicos en forma de
retraso del crecimiento radicular y clorosis foliar.