
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 124
LINDANE
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
First draft prepared by Dr M. Herbst, International Centre
for the Study of Lindane, Lyon, France and
Dr G.J. Van Esch, Bilthoven, The Netherlands
World Health Orgnization
Geneva, 1991
The International Programme on Chemical Safety (IPCS) is a
joint venture of the United Nations Environment Programme, the
International Labour Organisation, and the World Health
Organization. The main objective of the IPCS is to carry out and
disseminate evaluations of the effects of chemicals on human health
and the quality of the environment. Supporting activities include
the development of epidemiological, experimental laboratory, and
risk-assessment methods that could produce internationally
comparable results, and the development of manpower in the field of
toxicology. Other activities carried out by the IPCS include the
development of know-how for coping with chemical accidents,
coordination of laboratory testing and epidemiological studies, and
promotion of research on the mechanisms of the biological action of
chemicals.
WHO Library Cataloguing in Publication Data
Lindane.
(Environmental health criteria ; 124)
1.Benzene hexachloride - adverse effects 2.Benzene hexachloride
- toxicity 3.Environmental exposure 4.Environmental poluutants
I.Series
ISBN 92 4 157124 1 (NLM Classification: WA 240)
ISSN 0250-863X
The World Health Organization welcomes requests for permission
to reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made
to the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1991
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material
in this publication do not imply the expression of any opinion
whatsoever on the part of the Secretariat of the World Health
Organization concerning the legal status of any country, territory,
city or area or of its authorities, or concerning the delimitation
of its frontiers or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar
nature that are not mentioned. Errors and omissions excepted, the
names of proprietary products are distinguished by initial capital
letters.
CONTENTS
1. SUMMARY AND EVALUATION; CONCLUSIONS; RECOMMENDATIONS
1.1. Summary and evaluation
1.1.1. General properties
1.1.2. Environmental transport, distribution and
transformation
1.1.3. Environmental levels and human exposure
1.1.4. Kinetics and metabolism
1.1.5. Effects on organisms in the environment
1.1.6. Effects on experimental animals and in vitro
1.1.7. Effects on humans
1.2. Conclusions
1.2.1. General population
1.2.2. Subpopulations at special risk
1.2.3. Occupational exposure
1.2.4. Environmental effects
1.3. Recommendations
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL
METHODS
2.1. Identity
2.1.1. Primary constituent
2.1.2. Technical product
2.2. Physical and chemical properties
2.3. Conversion factors
2.4. Analytical methods
2.4.1. Sampling
2.4.2. Analytical methods
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Man-made sources
3.2.1. Production levels and processes
3.2.1.1 Manufacturing process
3.2.1.2 World-wide production figures
3.2.2. Emissions
3.2.3. Uses
3.2.4. Extent of use
3.2.5. Formulations
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1. Transport and distribution between media
4.1.1. Volatilization
4.1.2. Precipitation
4.1.3. Movement in soils
4.1.4. Uptake and translocation in plants
4.2. Biotransformation
4.2.1. Degradation
4.2.1.2 Degradation under humid conditions
4.2.1.2 Degradation under submerged conditions
4.2.2. Degradation under field conditions
4.2.3. Hydrolytic degradation
4.2.4. Photolytic degradation (laboratory studies)
4.2.5. Biodegradation in water
4.2.6. Microbial degradation (field studies)
4.2.7. Bioaccumulation/Biomagnification
4.2.7.1 n-Octanol/water partition coefficient
4.2.7.2 Aquatic environment
4.2.7.3 Terrestrial environment
4.2.7.4 Bioconcentration in humans
4.2.7.5 Field studies
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Air
5.1.2. Water
5.1.2.1 Rain and snow
5.1.2.2 Fresh water
5.1.2.3 Sea water
5.1.3. Soil
5.1.3.1 Sediment
5.1.3.2 Dumping grounds and sewage sludge
5.1.4. Drinking-water, food and feed
5.1.4.1 Drinking-water
5.1.4.2 Cereals, fruits, pulses, vegetables,
and vegetable oil
5.1.4.3 Meat, fat, milk, and eggs
5.1.4.4 Animal feed
5.1.4.5 Miscellaneous products
5.1.5. Terrestrial and aquatic organisms
5.1.5.1 Plants
5.1.5.2 Aquatic organisms
5.1.5.3 Terrestrial organisms
5.2. Exposure of the general population
5.2.1. Total-diet studies
5.2.2. Intake with drinking-water and air
5.2.3. Concentrations in human samples
5.2.3.1 Blood
5.2.3.2 Adipose tissue
5.2.3.3 Breast milk
6. KINETICS AND METABOLISM
6.1. Absorption
6.1.1. Oral administration - experimental animals
6.1.2. Dermal application - experimental animals
6.1.3. Other routes - experimental animals
6.2. Distribution
6.2.1. Oral administration - experimental animals
6.2.2. Inhalation - experimental animals
6.2.3. Other routes
6.3. Metabolic transformation
6.3.1. Enzymatic involvement
6.3.2. Identification of metabolites
6.3.3. Metabolites identified in humans
6.4. Elimination and excretion in expired air, faeces, and
urine
6.4.1. Oral administration
6.4.1.1 Rat
6.4.1.2 Rabbit
6.4.2. Other routes
6.4.2.1 Mouse
6.4.2.2 Rat
6.4.2.3 Human
6.5. Retention and turnover (experimental animals)
6.6. Biotransformation
6.6.1. Plants
6.6.2. Microorganisms
6.6.2.1 Anaerobic conditions
6.6.2.2 Aerobic conditions
6.7. Isomerization
7. EFFECTS ON LABORATORY MAMMALS AND IN IN-VITRO TEST SYSTEMS
7.1. Single exposure
7.1.1. Oral
7.1.2. Intraperitoneal and intramuscular
7.1.3. Inhalation
7.1.4. Dermal
7.2. Short-term exposure
7.2.1. Oral
7.2.1.1 Mouse
7.2.1.2 Rat
7.2.1.3 Dog
7.2.1.4 Pig
7.2.2. Inhalation
7.2.2.1 Mouse
7.2.2.2 Rat
7.2.3. Dermal
7.3. Skin and eye irritation; sensitization
7.3.1. Primary skin irritation
7.3.2. Primary eye irritation
7.3.3. Sensitization
7.4. Long-term exposure
7.4.1. Oral
7.4.2. Appraisal of acute and short- and long-term
studies
7.5. Reproduction, embryotoxicity, and teratogenicity
7.5.1. Reproduction
7.5.2. Embryotoxicity and teratogenicity
7.5.2.1 Oral administration
7.5.2.2 Subcutaneous injection
7.5.3. Reproductive behaviour
7.5.4. Appraisal of reproductive toxicology
7.6. Mutagenicity and related end-points
7.6.1. DNA damage
7.6.2. Mutation
7.6.3. Chromosomal effects
7.6.4. Miscellaneous tests
7.6.5. Appraisal of mutagenicity and related end-
points
7.7. Carcinogenicity
7.7.1. Mouse
7.7.2. Rat
7.7.3. Initiationpromotion
7.7.4. Mode of action
7.7.5. Appraisal of carcinogenicity
7.8. Special studies
7.8.1. Immunosuppression
7.8.2. Behavioural studies
7.8.3. Neurotoxicity
7.8.3.1 Dose-response studies using intact
animals
7.8.3.2 Studies on mechanism
7.8.3.3 Summary
7.9. Factors that modify toxicity; toxicity of metabolites
8. EFFECTS ON HUMANS
8.1. Exposure of the general population
8.1.1. Acute toxicity, poisoning incidents
8.1.2. Effects of short- and long-term exposures -
controlled human studies
8.1.2.1 Oral administration
8.1.2.2 Dermal application
8.1.3. Epidemiological studies (general population)
8.2. Occupational exposure
8.2.1. Toxic effects
8.2.2. Irritation and sensitization
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1. Microorganisms
9.1.1. Bacteria
9.1.2. Algae
9.1.2.1 Blue-green algae
9.1.2.2 Freshwater algae
9.1.2.3 Marine algae
9.1.3. Dinoflagellates, flagellates, and ciliates
9.2. Aquatic organisms
9.2.1. Invertebrates
9.2.1.1 Crustacea
9.2.1.2 Aquatic arthropods
9.2.1.3 Molluscs
9.2.2. Fish
9.2.2.1 Acute toxicity
9.2.2.2 Short- and long-term toxicity
9.2.2.3 Reproduction
9.2.3. Amphibia
9.2.3.1 Acute toxicity
9.2.3.2 Effects on hatching and larval
development
9.3. Terrestrial organisms
9.3.1. Honey-bees
9.3.2. Birds
9.3.2.1 Acute toxicity
9.3.2.2 Short-term toxicity
9.3.2.3 Reproduction
9.3.3. Mammals
9.4. Appraisal
10. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
APPENDIX I
REFERENCES
RESUMÉ
RESUMEN
WHO TASK GROUP MEETING ON ENVIRONMENTAL HEALTH CRITERIA FOR LINDANE
Members
Dr S. Dobson, Pollution and Ecotoxicology Section, Institute of
Terrestrial Ecology, Monkswood Experimental Station, Abbots
Ripton, Huntingdon, United Kingdom
Dr G.J. van Esch, Bilthoven, the Netherlands (Joint Rapporteur)
Dr M. Herbst, Biological Research, ASTA Pharma AG, Frankfurt,
Germany (Joint Rapporteur)
Professor J.S. Kagan, Department of General Toxicology and
Experimental Pathology, All-Union Scientific Research Instiute
of Hygiene and Toxicology of Pesticides, Polymers, and
Plastics, Kiev, USSR (Vice-Chairman)
Dr S.G.A. Magwood, Pesticides Division, Environmental Health Centre,
Health and Welfare Canada, Tunney's Pasture, Ottawa, Ontario,
Canada
Professor W.-O. Phoon, National Institute of Occupational Health and
Safety, University of Sydney, Sydney, Australia (Chairman)
Dr J.F. Risher, US Environmental Protection Agency, Environmental
Criteria and Assessment Office, Cincinnati, Ohio, USA
Dr Y. Saito, Division of Foods, National Institute of Hygienic
Sciences, Setagaya-ku, Tokyo, Japan
Dr V. Turusov, Laboratory of Carcinogenic Substances, All-Union
Cancer Research Centre, Moscow, USSR
Representatives of Non-Governmental Organizations
Dr P.G. Pontal, International Group of National Associations of
Manufacturers of Agrochemical Products (GIFAP), Brussels,
Belgium
Observers
Dr A.V. Bolotny, All-Union Scientific Research Institute of Hygiene
and Toxicology of Pesticides, Polymers, and Plastics, Kiev,
USSR
Dr D. Demozay, International Centre for the Study of Lindane (CIEL),
Rhône-Poulenc Agrochimie, Lyon, Franch.
Secretariat
Dr G.J. Burin, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland
Dr K.W. Jager, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland (Secretary)
Dr V.A. Rezepov, Centre for International Projects, USSR State
Committee for Environmental Protection, Moscow, USSR
NOTE TO READERS OF THE CRITERIA DOCUMENTS
Every effort has been made to present information in the
criteria documents as accurately as possible without unduly delaying
their publication. In the interest of all users of the environmental
health criteria documents, readers are kindly requested to
communicate any errors that may have occurred to the Manager of the
International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland, in order that they may be
included in corrigenda, which will appear in subsequent volumes.
* * *
A detailed data profile and a legal file can be obtained from
the International Register of Potentially Toxic Chemicals, Palais
des Nations, 1211 Geneva 10, Switzerland (Telephone No. 7988400 or
7985850)
* * *
The proprietary information contained in this document cannot
replace documentation for registration purposes, because the latter
has to be closely linked to the source, the manufacturing route, and
the purity/impurities of the substance to be registered. The data
should be used in accordance with paragraphs 82-84 and
recommendations paragraph 90 of the Second FAO Government
Consultation (1982).
ENVIRONMENTAL HEALTH CRITERIA FOR LINDANE
The WHO Task Group on environmental health criteria for lindane
met in Moscow, USSR, on 20-24 November 1989. The meeting was
convened with the financial assistance of the United Nations
Environment Programme (UNEP) and was hosted by the Centre for
International Projects (CIP), USSR State Committee for Environmental
Protection. On behalf of the CIP, Dr V.A. Rezepov opened the meeting
and welcomed the participants. Dr K.W. Jager welcomed the
participants on behalf of the three cooperating organizations of the
IPCS (UNEP, ILO, WHO). The Task Group reviewed and revised the draft
document and made an evaluation of the risks to human health and the
environment from exposure to lindane.
The first drafts of this monograph were prepared by Dr M.
Herbst (on behalf of the International Centre for the Study of
Lindane (CIEL)) and Dr G.J. van Esch (on behalf of the IPCS). The
second draft was prepared by Dr G.J. van Esch, incorporating
comments received following circulation of the first draft to the
IPCS contact points for Environmental Health Criteria publications.
The help of the CIEL in making available their proprietary
toxicological information on lindane to the IPCS and the Task Group
is gratefully acknowledged. This enabled the Task Group to make its
evaluation on the basis of more complete data than would otherwise
have been possible.
The efforts of all who helped in the preparation and
finalization of the document are also gratefully acknowledged. Dr
K.W. Jager of the IPCS Central Unit was responsible for the
technical development of this monograph and Mrs E. Heseltine of St
Léon-sur-Vézère, France, for the editing.
1. SUMMARY AND EVALUATION; CONCLUSIONS; RECOMMENDATIONS
1.1 Summary and evaluation
1.1.1 General properties
Technical-grade hexachlorocyclohexane (HCH) consists of 65-70%
alpha-HCH, 7-10% beta-HCH, 14-15% gamma-HCH, and approximately 10%
of other isomers and compounds. Lindane contains > 99% gamma-HCH.
It is a solid, with a low vapour pressure, and is poorly soluble in
water but very soluble in organic solvents, such as acetone, and in
aromatic and chlorinated solvents. The n-octanol/water partition
coefficient (log Pow) is 3.2-3.7.
Lindane can be determined separately from the other isomers of
HCH after extraction by liquid/liquid partition, column
chromatography and detection by gas chromatography with electron
capture. As these analytical methods are highly sensitive, residues
of lindane can be identified at a level of nanograms per kilogram or
per litre.
Lindane has been used as a broad-spectrum insecticide since the
early 1950s for agricultural and nonagricultural purposes, which
include treatment of seeds and soil, application on trees, timber
and stored materials, treatment of animals against ectoparasites and
in public health.
1.1.2 Environmental transport, distribution and transformation
Lindane is strongly adsorbed on soils that contain a large
amount of organic matter; furthermore, it can move downward through
the soil with water from rainfall or artificial irrigation.
Volatilization appears to be an important route of its dissipation
under the high-temperature conditions of tropical regions.
Lindane undergoes rapid degradation (dechlorination) in the
presence of ultra-violet irradiation, to form
pentachlorocyclohexenes (PCCHs) and tetrachlorocyclohexenes (TCCHs).
When lindane undergoes environmental degradation under humid or
submerged conditions and in field conditions, its half-time varies
from a few days to three years, depending on type of soil, climate,
depth of application and other factors. In agricultural soils common
in Europe, its half-time is 40-70 days. The biodegradation of
lindane is much faster in unsterilized than in sterilized soils.
Anaerobic conditions are the most favourable for its microbial
metabolization. Lindane present in water is degraded mostly by
microorganisms in sediments to form the same degradation products.
Limited amounts of lindane and gamma-PCCHs are taken up by and
translocated into plants, especially in soils with a high content of
organic matter. Residues are found mainly in the roots of plants,
and little, if any, is translocated into stems, leaves or fruits.
Rapid bioconcentration takes place in microorganisms, invertebrates,
fish, birds and humans, but biotransformation and elimination are
relatively rapid when exposure is discontinued. In aquatic
organisms, uptake from water is more important than uptake from
food. The bioconcentration factors in aquatic organisms under
laboratory conditions ranged from approximately 10 up to 6000; under
field conditions, the bioconcentration factors ranged from 10 to
2600.
1.1.3 Environmental levels and human exposure
Lindane has been found in the air above the oceans at
concentrations of 0.039-0.68 ng/m3 and has been measured at up to
11 ng/m3 in the air in some countries. The estimated
concentrations in surface water in a number of European countries
were mainly below 0.1 µg/litre. The concentration in the River Rhine
and its tributaries in 1969-74 varied between 0.01 and 0.4 µg/litre;
after 1974, it was below 0.1 µg/litre. Levels of 0.001-0.02 µg/litre
have been reported in seawater. The concentrations of lindane in
soil are generally low - in the range 0.001-0.01 mg/kg, except in
areas where waste is disposed of.
Fish and shellfish have been found to contain gamma-HCH at
concentrations ranging from none detected up to 2.5 mg/kg on a fat
basis, depending on whether they live in fresh or seawater and
whether they have a low or high fat content. Levels of about 330 and
440 µg/kg (wet weight) were found in adipose tissue of polar bears
in 1982 and 1984, respectively. The concentration of lindane in the
livers of birds of prey varied between 0.01 and 0.1 mg/kg. Eggs of
sparrow-hawks collected in 1972-73 in the Federal Republic of
Germany contained levels of 0.6 up to 11.1 mg/kg (on a fat basis).
The concentration of lindane in the livers of predatory birds
varied between 0.01 and 0.1 mg/kg. Eggs of sparrow- hawks collected
in 1972-73 in the Federal Republic of Germany contained levels of
0.6 up to 11.1 mg/kg (on a fat basis). The concentrations of lindane
in drinking-water are generally below 0.001 µg/litre, and in
industrialized countries more than 90% of human intake of lindane
originates from food. Over the last 25 years, selected food items
have been analysed for lindane in a large number of countries. The
concentrations found in cereals, fruits, vegetables, pulses, and
vegetable oils ranged from not detected up to 0.5 mg/kg product, and
those in milk, fat, meat, and eggs from not detected up to 1.0 mg/kg
product (on a fat basis). In only a few instances were higher
concentrations found. The concentrations in fish were generally far
lower than 0.05 mg/kg product (on a fat basis). In total-diet and
market-basket studies to estimate daily human intake of lindane, a
clear difference was observed with time: intake in the period around
1970 was up to 0.05 µg/kg body weight per day, whereas by 1980
intake had decreased to 0.003 µg/kg body weight per day or lower. In
the USA, the daily intake of gamma-HCH between 1976 and 1979
decreased from 0.005 to 0.001 µg/kg body weight per day for infants
and from 0.01 to 0.006 µg/kg body weight per day for toddlers.
Determinations of the lindane content in body tissues in the
general population have been made in a number of countries. The
content in blood in the Netherlands was in the order of < 0.1-0.2
µg/litre, but much higher concentrations were found in several
countries where technical-grade HCH was used. The mean
concentrations in human adipose tissue in various countries ranged
from < 0.01 up to 0.2 mg/kg on a fat basis. The concentrations of
lindane in human milk are generally rather low, at average
concentrations of < 0.001 up to 0.1 mg/kg on a fat basis; however,
there has been a clear decrease over time.
Lindane is thus distributed all over the world and can be
detected in air, water, soil, sediment, aquatic and terrestrial
organisms, and food, although the concentrations in these different
compartments are generally low and are gradually decreasing. Humans
are exposed daily via food, and lindane has been found in blood,
adipose tissue, and breast milk; the levels of intake, however, are
also decreasing.
1.1.4 Kinetics and metabolism
In rats, lindane is absorbed rapidly from the gastrointestinal
tract and distributed to all organs and tissues within a few hours.
The highest concentrations are found in adipose tissues and skin; in
various studies, the fat:blood ratio was about 150-200, the
liver:blood ratio, 5.3-9.6 and the brain:blood ratio, 4-6.5. The
same fat:blood ratio was found in rats exposed by inhalation. These
ratios vary with sex, being higher in females. Uptake of lindane
through the skin after dermal application is slow and occurs to a
very limited extent; this may explain the low toxicity of lindane
after dermal exposure.
Lindane is metabolized mainly in the liver by four enzymatic
reactions: dehydrogenation to gamma-HCH, dehydrochlorination to
gamma-PCCH, dechlorination to gamma-TCCH and hydroxylation to
hexachlorocyclohexanol. The end-products of biotransformation are
di-, tri-, tetra-, penta-, and hexachloro- compounds. These
metabolites are excreted mainly via the urine in the free form or
conjugated with glucuronic acid, sulfuric acid or N-acetylcystein.
The elimination is relatively fast, with half-times in rats of 3-4
days. Bacteria and fungi metabolize lindane to TCCH and PCCH. The
rate of metabolic transformation in plants is low, and the main
degradation pathway proceeds via PCCH to tri- and tetrachlorophenol
and conjugates with beta-glucose and other, unknown compounds. There
is no evidence that lindane is isomerized to alpha-HCH.
1.1.5 Effects on organisms in the environment
Lindane is not very toxic for bacteria, algae, or protozoa: 1
mg/litre was generally the no-observed effect level (NOEL). Its
action on fungi is variable, with NOELs varying from 1 to 30
mg/litre depending on the species. It is moderately toxic for
invertebrates and fish, the L(E)C50 values for these organisms being
20-90 µg/litre. In short-term and long-term studies with three
species of fish, the NOEL was 9 µg/litre; no effect on reproduction
was seen with levels of 2.1-23.4 µg/litre. The LC50 values for both
freshwater and marine crustacea varied between 1 and 1100 µg/litre.
Reproduction in Daphnia magna was depressed in a dose-dependent
fashion; the NOEL was in the range 11-19 µg/litre. Reproduction of
molluscs was not adversely effected by a dose of 1 mg/litre.
The LD50 for honey-bees was 0.56 µg/bee.
Acute oral LD50 values for a number of bird species were
between 100 and 1000 mg/kg body weight. In short-term studies with
birds, doses of 4-10 mg/kg diet had no effect, even on egg-shell
quality. Laying ducks treated with doses of lindane up to 20 mg/kg
body weight, however, had decreased egg production.
Bats exposed to wood shavings that initially contained 10-866
mg/m2 lindane, resulting from application at the recommended rate,
all died within 17 days. No effect on mortality or reproductive
success was seen in small field mammals given 20 mg/kg diet (the
highest dose tested). No data were available on effects on
populations and ecosystems.
1.1.6 Effects on experimental animals and in vitro
The acute oral toxicity of lindane is moderate: the LD50 for
mice and rats is in the range 60-250 mg/kg body weight, depending on
the vehicle used. The dermal LD50 for rats is approximately 900
mg/kg body weight. Toxicity was manifested by signs of central
nervous system stimulation.
Lindane does not irritate or sensitize the skin; it is slightly
irritating to the eye.
In a 90-day study in rats, the NOEL was 10 mg/kg diet
(equivalent to 0.5 mg/kg body weight). At 50 and 250 mg/kg diet, the
weights of the liver, kidneys, and thyroid were increased; at 250
mg/kg diet, an increase was seen in liver enzyme activity. This
increase in enzyme activity accelerates the breakdown of both
lindane and other compounds. In another 90-day study in rats, 4
mg/kg diet (equivalent to 0.2 mg/kg body weight) was considered to
be the no-adverse-effect level (NOAEL); renal and hepatic toxicity
were observed at concentrations of 20 mg/kg diet and higher. No
neurological effect was observed in a 30-day feeding study in rats
with 240 mg/kg diet (equivalent to 12 mg/kg body weight); however,
when this dose was given by gavage, neurological effects were seen.
A short-term toxicity study in mice was considered to be inadequate
to establish a NOEL.
Administration of lindane to dogs at 15 mg/kg in the diet
(equivalent to 0.6 mg/kg body weight) for 63 weeks had no toxic
effect. In a two-year study of the toxicity of this compound in
dogs, in which a large number of parameters were measured, no
treatment-related abnormality was apparent at doses of 50 mg/kg diet
(equivalent to 2 mg/kg body weight) and lower. In the group given
100 mg/kg diet, however, levels of alkaline phosphatase were
increased; and with 200 mg/kg diet, abnormalities in
electroencephalogram tracings indicative of non-specific neuronal
irritation were observed.
In rats exposed by inhalation to lindane at 0.02-4.54 mg/m3
for 6 h/day for 3 months, the highest dose induced increases in
hepatic cytochrome P450 values; the NOAEL was found to be 0.6
mg/m3. In two long-term studies in rats, carried out many years
ago, doses of 10-1600 mg/kg diet were tested. In one of these
studies, 50 mg/kg diet (equivalent to 2.5 mg/kg body weight) was
found to be the NOAEL. At 100 mg/kg diet, an increase in liver
weight, hepatocellular hypertrophy, fatty degeneration, and necrosis
were found. In the other study, 25 mg/kg diet (equivalent to 1.25
mg/kg body weight) had no effect, but hepatocellular hypertrophy and
fatty degeneration were seen with 50 mg/kg diet.
Lindane has been investigated for its effects on all aspects of
reproduction (in rats over three generations) and for its
embryotoxicity and teratogenicity after oral, subcutaneous and
intraperitoneal administration in mice, rats, dogs, and pigs. It had
no teratogenic effect after oral or parenteral administration (extra
ribs were regarded as variations). Fetotoxic and/or maternal toxic
effects were observed with doses of 10 mg/kg body weight and above
given by oral gavage; 5 mg/kg body weight is considered to be the
NOAEL. Lindane had no effect on reproduction or maturation in the
three-generation study in rats at doses of up to 100 mg/kg diet; but
with 50 mg/kg diet, morphological changes in the liver indicating
enzyme induction occurred in the offspring of the third generation.
The NOEL in this test was 25 mg/kg diet (equivalent to 1.25 mg/kg
body weight).
The NOEL for neurotoxicity in a 22-day study in rats was 2.5
mg/kg body weight.
The mutagenicity of lindane has been studied adequately. In
extensive investigations of its ability to induce gene mutations in
bacteria and in mammalian cells, and for its capacity to induce
sex-linked recessive lethal mutations in Drosophila melanogaster,
negative results were obtained consistently. Lindane also gave
negative results in tests for chromosomal damage and sister
chromatid exchange in mammalian cells in vitro and in vivo . The
results of assays for DNA damage in bacteria and for covalent
binding to DNA in the liver of rats and mice in vivo following
oral administration were also negative. In the very few studies in
which positive results were obtained, either the study design was
invalid or the purity of the compound tested was not reported.
Overall, however, lindane appears to have no mutagenic potential.
Studies to define the carcinogenic potential of lindane have
been carried out in mice and rats using dose levels of up to 600
mg/kg diet in mice and up to 1600 mg/kg diet in rats. Hyperplastic
nodules and/or hepatocellular adenomas were found in mice given
doses of 160 mg/kg diet or more; in some studies, the dose levels
exceeded the maximum tolerated dose. Two studies in mice with dose
levels of up to 160 mg/kg diet and one in rats with 640 mg/kg diet
showed no increase in the incidence of tumours.
The results of studies on initiation-promotion of
carcinogenicity, on the mode of action, and on mutagenicity indicate
that the tumorigenic response observed with gamma-HCH in mice is
mediated by a nongenetic mechanism.
1.1.7 Effects on humans
Several cases of fatal poisoning and of non-fatal illness
caused by lindane have been reported, which were either accidental,
intentional (suicide), or due to gross neglect of safety precautions
or improper uses of medical products containing lindane. Symptoms
included nausea, restlessness, headache, vomiting, tremor, ataxia,
and tonic-clonic convulsions and/or changes in the
electroencephalographic pattern. These effects were reversible after
discontinuation of exposure or symptomatic treatment.
Notwithstanding extensive use over 40 years, very few cases of
poisoning in the occupational setting have been reported. In workers
exposed for long periods during either manufacture or application of
lindane, the only sign found was increased activity of drug
metabolizing enzymes in the liver. There is no evidence for the
relationship suggested in some publications between exposure to
lindane and the occurrence of blood dyscrasias. A few acute and
short-term studies in humans indicate that a dose of approximately
1.0 mg/kg body weight does not induce poisoning; however, a dose of
15-17 mg/kg body weight resulted in severe toxic symptoms.
Approximately 10% of a dermally applied dose is absorbed,
although more passes through damaged skin.
1.2 Conclusions
1.2.1 General population
Lindane is circulating in the environment and is present in
food chains, so that humans will continue to be exposed. The daily
intake and total exposure of the general population are decreasing
gradually, however; they are clearly below the advised acceptable
daily intake and are of no concern to public health.
1.2.2 Subpopulations at special risk
The presence of lindane in breast milk results in exposure of
breast-fed babies to levels that are generally below the acceptable
daily intake and therefore of no concern to health. Although lower
levels of exposure would be preferred, the present levels are not a
limiting factor for the practice of natural breast-feeding.
Prescriptions should be followed strictly with regard to the
therapeutic use of lindane against scabies and to control body lice.
1.2.3 Occupational exposure
As long as the recommended precautions to minimize exposure are
observed, lindane can be handled safely.
1.2.4 Environmental effects
Lindane is toxic to bats that roost in close contact with wood
treated according to the recommended conditions of application.
Apart from the results of studies of spills into the aquatic
environment, there is no evidence to suggest that the presence of
lindane in the environment poses a significant hazard to populations
of other organisms.
1.3 Recommendations
1. In order to minimize environmental pollution by other isomers
of HCH, lindane (> 99% gamma-HCH) must be used instead of
technical-grade HCH.
2. In order to avoid environmental pollution, by-products of and
effluents from the manufacture of lindane should be disposed of
in an appropriate way.
3. In disposing of lindane, care should be taken to avoid
contamination of natural waters and soil.
4. As for other pesticides, proper instructions about application
procedures and safety precautions should be given to people who
handle lindane.
5. Long-term carcinogenicity tests conducted according to
present-day standards should be conducted.
6. Monitoring of the daily intake of lindane by the general
population should continue.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL
METHODS
2.1 Identity
2.1.1 Primary constituent
Common name: Lindane
Chemical structure:1
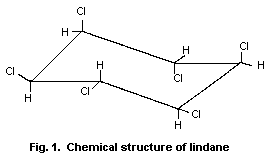 Fig. 1. Chemical structure of lindane
Chemical formula: C6H6Cl6
Relative molecular mass: 290.8 (290.9)
CAS chemical name: 1alpha,2alpha,3ß,5alpha,6ß-
hexachlorocyclohexane
CAS registry number: 58-89-9
RTECS registry number: GV4900000
Synonym: Hexachlorocyclohexane (gamma-isomer)
According to IUPAC rules, the designation 'benzene
hexachloride' is incorrect; nevertheless, it is still widely used,
especially in the form of its abbreviation, BHC. This is therefore
another common name approved by the ISO. The compound is called
gamma-HCH by the WHO, but gamma-BHC by the FAO (FAO, 1973). The
synonym hexachlorocyclohexane (gamma isomer) is used by the
Environmental Protection Agency and the American Conference of
Governmental Industrial Hygienists in the USA. The definitions of
these different appellations are given in Table 1.
1 See Appendix I
Table 1. Definitions of appellations of lindane
Name Definition Remarks
Lindane product containing not less ISO-AFNOR name for a product
than 99% gamma-HCH (not yet recognized by BSI)
Lindane = gamma-HCH Common name used for
gamma-HCH in the USSR only
gamma-HCH gamma isomer of 1,2,3,4,5,6- ISO-AFNOR common name
hexachlorocyclohexane
gamma-BHC gamma isomer of 1,2,3,4,5,6- ISO BSI common name in
benzene hexachloride English-speaking countries
(recognized by ISO as
synonym of gamma-HCH)
2.1.2 Technical product
Common trade names: A great number of products containing lindane
are on the market; no attempt has been made to
list the hundreds of trade names here (see
Hudson et al., 1984; Hill & Camardese, 1986;
International Register for Potentially Toxic
Chemicals, 1989).
Purity: The FAO (1973) requires that lindane "... shall
consist, essentially, of gamma-BHC as white or
nearly white granules, flakes or powder, free
from extraneous impurities or added modifying
agents and with not more than a faint odour."
The FAO further requires that it contain not
less than 99.0% gamma-HCH and that the
melting-point be at least 112 °C, which is not
depressed when the sample is mixed with an equal
amount of pure gamma-HCH.
In some processes for manufacturing lindane, low levels of
dioxin may be formed (US Environmental Protection Agency, 1985).
Under appropriate manufacturing conditions, however, no
2,3,7,8-tetrachlorodibenzodioxin or 2,3,7,8-tetrachlorodibenzofuran
is detected in HCH, lindane, trichlorobenzene, industrial liquid or
gaseous effluents at the analytical limit of detection of 1 µg/kg
letter from D. Demosay, Rhône-Poulenc, to IPCS dated 17 November
1989.
2.2 Physical and chemical properties
Lindane is a colourless, crystalline solid with either a faint
or no smell (the characteristic smell of technical-grade HCH is
attributed to impurities, particularly heptachlorocyclohexane).
Melting-point: 112.8 °C
Boiling-point: 288 °C
Vapour pressure: 0.434 x 10-5 kPa (3.26 x 10-5 mmHg) at 20 °C
60.6 x 10-5 kPa (45.6 x 10-5 mmHg) at 40 °C
Density: 1.85
Solubility: nearly insoluble in water at 20 °C (10
mg/litre); moderately soluble in ethanol (6.7%);
slightly soluble in mineral oils;
soluble in acetone and in aromatic and
chlorinated solvents
Stability: stable to light, air, heat, carbon dioxide, and
strong acids; dehydrochlorinates in the presence
of alkali or on prolonged exposure to heat with
the formation of trichlorobenzenes, phosgene,
and hydrochloric acid. It is incompatible with
strong bases and powdered metals, such as iron,
zinc, and aluminium, and with oxidizing agents;
can undergo oxidation when in contact with
ozone.
Corrosivity: corrosive to aluminium
Inflammability: not inflammable
n-Octanol/water 3.2-3.7 (see section 4.2.7.1) (Demozay &
partition Marechal, 1972; Dutch Chemical Industry
coefficient Association 1980; American Conference of
(log Pow): Governmental Industrial Hygienists, 1986;
Rhône-Poulenc Agrochimie, 1986)
2.3 Conversion factors
1 ppm = 12.1 mg/m3
1 mg/m3 = 0.083 ppm
2.4 Analytical methods
2.4.1 Sampling
Sampling procedures and methods for preparing samples of
formulations and for analysing residues have been described by
Mestres (1974), the Deutsche Forschungsgemeinschaft (1979), the
Association of Official Analytical Chemists (1980), and Hildebrandt
et al. (1986).
2.4.2 Analytical methods
Products are analysed by a cryoscopic method (Raw, 1970; FAO,
1973; WHO, 1985). Formulated products can be analysed by determining
hydrolysable chlorine (Raw, 1970; FAO, 1973). Since the latter
method is not specific, other methods, such as gas chromatography,
are used to obtain sufficient separation of the HCH isomers.
Residues in food and in soil can be determined after adequate
clean-up by gas chromatography and other chromatographic methods
(Nash et al., 1973; Eichler, 1977; Association of Official
Analytical Chemists, 1980; DeutscheForschungsgemeinschaft, 1983).
The principle of the method is extraction of a sample with organic
solvents (acetonitrile, hexane/acetone, acetone, and others). Fat is
extracted from fatty foods and partitioned between petroleum ether
and acetonitrile by extracting aliquots or an entire solution of
acetonitrile into petroleum ether. Residues are purified by
chromatography on a Florisil colum, and eluted with a mixture of
petroleum ether and ethylether. Concentrated residues are measured
by gas chromatography with electron capture detection.
The method described by the Deutsche Forschungsgemeinschaft
(1979) for fruits and vegetables is based on extraction of samples
with acetone and extraction of the aliquot with dichloromethane. The
residue obtained after evaporation of the solvent is cleaned by
co-distillation, and the distillate is analysed by gas
chromatography with electron capture detection. The limit of
determination depends on the method, the substrate and the sample
size; the lower limit of determination is 0.001-0.01 mg/kg.
Palmer & Kolmodin-Hedman (1972) analysed air samples by gas
chromatography with electron capture detection, and alpha-, beta-,
and gamma-HCH were determined in serum by gas chromatography after a
deproteinization extraction step (Palmer & Kolmodin-Hedman, 1972;
Angerer & Barchet, 1983).
Wittlinger & Ballschmiter (1987) provided an extensive
description of analytical methods for HCHs in air, involving
sampling by adsorption, extraction and preseparation, and
determination by high-resolution gas chromatography. Sampling was
performed by pumping air through a glass-fibre filter and then
through a silica-gel layer, using an internal standard. The sample
was extracted with dichloromethane and the extract evaporated. The
preseparation was done on silica gel, and the aliquot was eluted in
a mixture of hexane and dichloromethane. High-resolution capillary
gas chromatography, electron capture detection and a mass selective
detector were used for determination.
Eder et al. (1987) described three analytical methods for the
determination of HCHs in sediments: moist samples are extracted with
a solvent or a mixture of solvents, concentrated or fractionated and
determined by gas chromatography and electron capture detection.
Greve (1972) described a method for the determination of
organochlorine pesticides in water based on gas chromatography of a
petroleum ether extract after clean-up over Florisil or silica gel.
The limit of detection for lindane is 0.01 µg/litre.
Methods used for the determination of lindane in samples of
soil, animal, and vegetable products in the USSR are described by
Izmerov (1983). These methods are based on extraction with organic
solvents, purification and concentration of the extracts and
determination by gas-liquid chromatography with electron-capture
detection.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Lindane is not known to occur as a natural product.
3.2 Man-made sources
3.2.1 Production levels and processes
3.2.1.1 Manufacturing process
HCH was discovered in 1825, but its insecticidal properties
were first patented only in the 1940s. It has been produced
commercially since 1949.
Technical-grade HCH is synthesized from benzene and chlorine in
the presence of ultra-violet light and comprises 65-70% alpha-HCH,
7-10% beta-HCH, 14-15% lindane (gamma-HCH), approximately 7%
delta-HCH, 1-2% epsilon-HCH, and 1-2% other components. By-products
can be minimized by careful control of the reaction conditions.
Lindane (> 99% gamma-HCH) can be purified by multiple extractions
with methanol.
The extraction of lindane from HCH produces 85%
non-insecticidal HCH isomers, which can be used as intermediates in
the production of trichlorobenzene and hydrochloric acid after
cracking in an integrated installation. Trichlorobenzene is used in
the synthesis of other chemicals (van Velsen, 1986; Rhône-Poulenc
Agrochimie, 1986).
3.2.1.2 World-wide production figures
Lindane is produced in Austria, France, and Spain and in China,
India, Turkey, and the USSR. Before 1984, lindane was also
manufactured in the German Democratic Republic, Poland, Yugoslavia,
Romania, and Hungary; since then, all production has been stopped in
Germany, Japan, the Netherlands, the United Kingdom, and the USA.
Although in most developed countries use of technical-grade HCH
has been prohibited, it is still used elsewhere on a large scale:
total consumption of technical-grade HCH in India in 1986-87 was
approximately 27 000 tonnes (International Atomic Energy Agency,
1988).
3.2.2 Emissions
According to De Bruijn (1979), approximately 0.1% of the
lindane processed reaches the waste-water of a formulating plant.
Treatment of the waste-water, however, leads to solid waste, which
should be incinerated. In the past, it was often dumped in the
environment and could be dispersed from (open) chemical dumping
grounds to more remote soils by the wind.
Lindane enters the environment following application of
lindane-containing pesticides. Emissions can cross national
boundaries in water and air. For instance, the total trans-frontier
flux of lindane into the Netherlands via the surface water of the
River Rhine was approximately 1.8 tonnes per year (average for
1980-83) and that via the River Meuse, 0.2 tonnes per year (Slooff &
Matthijsen, 1988).
3.2.3 Uses
Lindane is a broad-spectrum insecticide, which has been used
since 1949 for agricultural as well as non-agricultural purposes.
Approximately 80% of the total production is used in agriculture
(Demozay & Marechal, 1972), mostly for seed and soil treatment. Wood
and timber protection is the major non-agricultural use. Lindane is
also used against ectoparasites in veterinary and pharmaceutical
products (Rhône-Poulenc Agrochimie, 1986).
3.2.4 Extent of use
Lindane is used worldwide, with the major exception of Japan,
where all uses of HCH were cancelled in 1971 mainly because of
environmental pollution with alpha- and beta-HCH resulting from
extensive use of technical-grade HCH. At that time, no clear
difference was made between the risks presented by the individual
HCH isomers, and lindane was banned as well. In almost all other
countries, lindane is registered for one or more applications,
although the use pattern differs from one country to another.
In 1979, the US Department of Agriculture and the Environmental
Protection Agency summarized the percentage uses of lindane in the
USA as follows: seed treatment 48%, hardwood lumber 23%, livestock
16%, pets 3%, pecans 3%, pineapples 2%, ornamentals 2%, household
1%, cucurbits 1%, forestry 0.5%, and structures 1%. In France and
Germany, 70-80% of all lindane used agriculturally is for soil
treatment, to protect maize and sugar beets, and 15-20% is used for
seed treatment. De Bruijn (1979) reported an estimate of the pattern
of use of lindane in the European Economic Community.
3.2.5 Formulations
Formulation facilities exist in many countries. Lindane is made
in numerous forms, the most important of which are: wettable powders
(up to 90% active ingredient); emulsifiable concentrates (not more
than 20% active ingredient); flowable suspensions (in water);
solutions in organic solvents (up to 50% active ingredient); dusts
and powders (0.5-2% active ingredient); granules and coarse dusts
(3-4% active ingredient); ready-for-use baits; aerosols; and special
formulations for use in human and veterinary medicine (Demozay &
Marechal, 1972).
Lindane dissolved in organic solvents may be used in 'thermal
foggers' in glasshouses or atomized in open areas; such solutions
are appropriate for aerial application (5-10 litres/ha of
formulations containing 5-10% active ingredient). Concentrated
solutions containing an anti-vaporization component have been
applied using an ultra-low volume method at 0.5-1 litre/ha. Various
fumigation preparations for indoor use have been sold, including
fumigation strips, tablets, and smoke generators. These contained
virtually pure lindane to which a small quantity of binding material
was added. Because of its versatility and relatively low acute
toxicity, lindane is often used in mixed formulations with other
insecticides and fungicides (Demozay & Marechal, 1972).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1 Transport and distribution between media
4.1.1 Volatilization
Some of the active ingredient of lindane volatilizes after it
has been applied to control insect pests, especially on leaves.
Starr & Johnson (1968) demonstrated that 20% of an applied dose had
evaporated 96 h after bean plants had been sprayed with lindane at
16 °C. The evaporation was dependent on temperature and on the
humidity of the air.
Some of the lindane that reaches the soil may also vaporize as
degradation products. Cliath & Spencer (1972) showed the presence of
vapours of the metabolite PCCH, which has a vapour pressure
approximately 14 times higher than that of lindane.
In a model test, four soil types, ranging from a loamy sand to
a clay, were treated with 14C-lindane to give a concentration of
10 mg/kg soil; water was then added to the samples, they were
air-dried at 33 °C and at 55 °C, and volatilization was measured by
trapping the vapours. Three cycles of about 14 days each were
followed. Lindane volatilized from the soils only with water, and no
further volatilization occurred after the soils had reached the dry
state. The four soil types were associated with different
volatilization rates: the highest occurred in loamy sand. In the
analysis of vaporized material, unchanged lindane and its
degradation products were not differentiated; however, considerable
degradation of lindane was found in the soils, and PCCH was
identified as a metabolite. At least some of the vaporized material
may therefore have consisted of degradation products (Guenzi &
Beard, 1970).
4.1.2 Precipitation
Evaporation and adsorption to solid particles are important
processes in the distribution of lindane. Reverse processes such as
deposition from the air and remobilization from silt and sediment
also play a part. Buscher et al. (1964) demonstrated that aeration
of aqueous solutions of lindane resulted in a loss of 10% over three
days, which was ascribed to a co-distillation process as it was
greater than could be explained by evaporation alone. MacKay &
Walkoff (1973) confirmed that evaporation is an important process in
the loss of HCH. Lichtenstein & Schulz (1970) found that 16.5% HCH
was lost from a non-aerated aqueous solution in 24 h at 30 °C.
The amount of lindane that is distributed by dry deposition
depends on the nature of the surface above which the organic
components are present. The half-time for dry deposition of HCHs
(height of mixing layer, 1000 m) in the Netherlands was calculated
to be 2-8 days. On this basis, a rough estimate of the annual flux
to soil and water in that country would be 0.5-1.5 tonnes from an
outdoor air concentration of 0.4 ng/m3 (Slooff & Matthijsen,
1988).
4.1.3 Movement in soils
Movement of a substance through the soil profile depends on its
adsorption-desorption characteristics in soil/water systems and, to
some extent, on its volatility in the soil pores and its diffusion.
The adsorption-desorption characteristics of lindane have been the
object of a number of studies (Kay & Elrick, 1967; Mills & Biggar,
1969; Baluja et al., 1975; Portmann, 1979; Wahid & Sethunathan,
1979, 1980; Wirth, 1985), all of which showed that lindane is
strongly adsorbed to organic soil material and weakly adsorbed to
inorganic matter. In the absence of organic matter, the clay content
and free iron oxide are implicated in the sorption of lindane (Wahid
& Sethunathan, 1979). It can be concluded that the mobility of
lindane is very low in soils with a high content of organic matter
but might be higher in soils containing little organic matter.
No consensus has been reached in the literature about the
possibility that lindane can be remobilized by desorption from
polluted soil. Generally, HCH isomers are strongly adsorbed. Under
certain conditions - high concentrations of lindane in highly
permeable soils with a low organic carbon content (< 0.1%) - a
small percentage of the compound may be washed out and reach the
groundwater. Nevertheless, the low rate of transport of lindane
makes the probability that it will reach groundwater low or very low
(Slooff & Matthijsen, 1988).
The diffusion of lindane through soil was investigated by
Ehlers et al. (1969a,b) and by Shearer et al. (1973). Diffusion was
strongly influenced by the water content of the soil, by the bulk
density and by temperature. The diffusion coefficient is nearly zero
in soil containing 1% water, but with a water content of 3%, lindane
is displaced from the adsorbing surface so that the diffusion
coefficient becomes maximal; a further increase in water content
reduces the diffusion coefficient. The diffusion of lindane in soils
can thus vary between a 'vapour' and a 'non-vapour' phase, depending
on the concentration of lindane, the length of time and the water
content of the soil.
Leaching of three formulations of lindane was investigated in a
series of model studies by Heupt (1974) in different soil types. The
test system consisted of 30-cm columns filled with soil to which
lindane formulations were applied at application rates corresponding
to 6 kg of active ingredient per hectare. Rainfall was simulated at
a rate of 200 mm within two days. No lindane was found in the eluate
at the limit of detection of 1 µg/litre. In field tests by Cliath &
Spencer (1971, 1972), lindane was worked into topsoil of two plots
of sandy loam and two of silty clay to a depth of 0-7.5 or 7.5-15
cm, corresponding to an application rate of 21 kg/ha. One of the
plots of each soil type received additional irrigation. Almost no
movement of lindane was found in the dry plots at the end of the
two-year observation period. In the irrigated plots, a broadening of
the lindane-containing zone and downward movement to a depth of 60
cm were observed, especially in sandy loam; in clay soil, lindane
had almost no mobility.
In a series of dissipation studies with 14C-labelled lindane
in soils, coordinated by FAO and the International Atomic Energy
Agency, it was demonstrated that persistent pesticides such as
lindane dissipate much faster in the tropics than in temperate
climates, probably owing to a large extent to volatilization
(International Atomic Energy Agency, 1988), as had been found by
Edwards (1973a,b, 1977). Table 2 summarizes the results of these
studies.
Table 2. Field half-times of gamma-HCH in soils (0-10-cm depth)a
Country Half-time (days)b Time required for
initial loss of 50%
Overall First phase Second phase of radioactivity (days)
India 138 (124-147) 41 (30-50) 188 (83-362) 30-45
Ecuador 150-171 54-60 120-160 40-50
Kenya 5-8 - - 3-4
a Adapted from International Atomic Energy Agency (1988)
b In temperate soils, the mean half-time was 438 (401-1022) days
(Edwards, 1973a, b)
4.1.4 Uptake and translocation in plants
One of the first investigations on the absorption of lindane by
various seeds was reported by Bradbury & Whitaker (1956). Lindane
was taken up from a nutrient solution by the roots of wheat
seedlings at a rate of up to 100 mg/kg (fresh weight) within seven
days. Subsequent investigations demonstrated that uptake by plants
is dependent on a variety of factors.
The influence of soil type was investigated by Bradbury
(1963). Seedlings grown from seed dressed with lindane and planted
in sand had residue levels about two-fold higher than those of
plants grown in compost. A further study was reported of the fate of
14C-lindane in loam and sandy soil and in oat plants grown in
these soils for 13 days. The loam soil was treated with about 7.3
mg/kg active ingredient and the sandy soil with about 3 mg/kg.
Residues were found to be more persistent in loam (53.5% of
radiolabel) than in sandy soil (33.8%), but oats grown in sandy soil
took up more residues than those grown in loam (loam soil: roots,
0.5%; tops, 0.3%; sandy soil: roots, 2.5%; tops, 1.2%).
14C-Lindane was the major constituent of the soil residues soluble
in organic solvents. A major metabolite, which was probably
gamma-PCCH, represented 11% of the organic-soluble radiolabelled
residues in loam soil; 2,4,5-trichlorophenol accounted for 2.7% of
these residues. The authors concluded that the three major factors
that determine the environmental fate of 14C-lindane and other
insecticides are the insecticide itself, its solubility in water and
the type of soil to which it is applied. Compounds with greater
aqueous solubility are more mobile, are taken up by plants to a
greater extent, and appear to be more susceptible to degradation
than compounds less soluble in water. In soils with little organic
matter, insecticide residues are more mobile and hence more
susceptible to volatilization, uptake by plants, and degradation
than in more adsorbent soils such as loam (Fuhremann & Lichtenstein,
1980).
The rate of application to soil was found to be a further
important factor in determining residue levels. Transfer of lindane
from soil into rice plants was almost proportional to the level of
contamination of the soil (Kawahara, 1972), but only at low levels
of contamination. Charnetski & Lichtenstein (1973) reported a good
correlation between the content of 14C-lindane in sand (at up to 6
mg/kg, which is about 12 times the concentration expected after
normal application) and that in pea plants grown for six days; at
concentrations greater than 10 mg/kg of soil, there was no further
increase in the residue levels.
Uptake of lindane after application to leaves is lower than
that resulting from application to soil. In lettuce and endives
treated with 14C-lindane and grown for 21 and 37 days,
respectively, only 4.5-13.9% of the applied radioactivity was found
at the time of harvest, and most of the lindane had evaporated into
the atmosphere (Kohli et al., 1976a).
Differences in residue levels are also dependent on the plant
species. Of a series of edible crops grown in soil containing
lindane at an initial concentration of 5 mg/kg (about 10 times the
normal application rate), carrots had higher levels than beans,
tomatoes, or potatoes (San Antonio, 1959). More lindane was absorbed
from soil with an initial concentration of 2.6 mg/kg by radishes,
turnips, and spinach than by Chinese cabbage (Kawahara et al.,
1971). The amounts of residues of HCH isomers in turnips were
proportional to the initial concentrations of the isomers in the
soil (0.05, 0.1, 0.5, 1, 5, 10, or 50 mg/kg soil). The soil:plant
residue ratios were in the range 10-20:1 (Kawahara & Nakamura,
1972).
The translocation of lindane and its metabolites in plants has
also been investigated in detail (San Antonio, 1959; Bradbury, 1963;
Itokawa et al., 1970; Kawahara, 1972; Kawahara & Nakamura, 1972;
Charnetski & Lichtenstein, 1973; Balba & Saha, 1974; Eichler, 1980;
Korte, 1980). Neither lindane taken up from soil nor its metabolites
were evenly distributed within the plants: Comparatively high
residue levels were always detected in the roots, whereas only small
amounts were translocated into stems, leaves, and fruits. Paasivirta
et al. (1988) showed that in water-plants, lindane concentrations
are similar in roots and leaves.
4.2 Biotransformation
4.2.1 Degradation
4.2.1.1 Degradation under humid conditions
The half-times of lindane found by different investigators vary
considerably, depending on the type of soil to which it is applied
and, possibly, temperature. Lindane incubated in a sandy-loam soil
with a water capacity of 28% and 60% saturation at room temperature
had a half-time of approximately 40 days (Heeschen et al., 1980).
The half-times of lindane in model tests were 4-6 weeks in humid
sand with a high content of organic matter and 30 weeks in sandy
loam (Heupt, 1979). The half-times in aerobic and anaerobic
conditions ranged from 12 to 174 days and 100 to 720 days,
respectively; in aerobic field conditions, the half-time was 88-1146
days (Edwards, 1966; Kohnen et al., 1975; Kampe, 1980; Rao &
Davidson, 1982; MacRae et al., 1984).
Assuming that lindane is not washed out below the level of
ploughed furrows (approximately 20 cm), a half-life of 350 days will
result in persistence of 50% of a dose one year after application
(Slooff & Matthijsen, 1988). One month after double treatment of
potato, beet, and maize crops with lindane, the gamma-HCH content in
sandy loam soil was 0.32 mg/kg in the field occupied by maize and
0.68-0.70 mg/kg in the fields with potatoes and beet. After nine
months, the lindane content in the beet fields had decreased 14
times and that in the maize fields by only 1.3 times (Kovaleva &
Talanov, 1973; see Izmerov, 1983).
4.2.1.2 Degradation under submerged conditions
Half-time values for lindane of a few to about 120 h were
determined after incubation in various submerged soil samples. More
rapid degradation occurred in soils with a high amino acid content,
and the rate also clearly depended on the number of degrading
microorganisms present (Ohisa & Yamaguchi, 1979). The rapidity with
which lindane was degraded under flooded conditions varied in soil
samples from different locations in Japan. Enrichment of the soils
with peptone and exclusion of oxygen increased the degradation rate
(Ohisa & Yamaguchi, 1978a).
Half-time values of 10-30 days were observed in a comparison of
four Philippine rice soils under flooded conditions at a temperature
of 30 °C. Lindane was degraded faster at higher temperatures
(Yoshida & Castro, 1970). In a similar study with five Indian rice
soils at 28 °C, 14C-labelled lindane was degraded at half-times of
between 10 days and more than 41 days. Addition of rice straw
enhanced the degradation (Siddaramappa & Sethunathan, 1975).
Tsukano (1973) found a half-time for lindane of 10-14 days in
soil samples mixed with water. The degradation was almost completely
inhibited after addition of sodium azide to the soils, indicating
that the degradation observed in non-sterilized soils was due to
microbial activity.
4.2.2 Degradation under field conditions
Nash (1983) used a microagroecosystem in which moist fallow
sandy loam was placed in a glass chamber at a depth of 15 cm, plants
were grown in the chamber and lindane was applied to the surface. A
half-time of 1-4 days was found for dissipation of lindane in the
soil.
In April 1954, formulations containing lindane were applied to
a sandy loam soil at rates of 2.25 and 4.5 kg/ha on field plots in
the Rhine valley, and loss of active ingredient was followed during
the subsequent 1.5 years using a biological test method. The
insecticidal activity disappeared rapidly during the following
vegetation period but remained almost constant in winter; further
degradation was observed during the second vegetation period. At the
end of the observation period, 3.5-5.5% of the lindane applied at
2.25 kg/ha remained, and 17-19.5% of that applied at 4.5 kg/ha: the
speed of degradation was therefore greater at the lower application
rate. Degradation was virtually identical when the lindane was
worked into the soil to a depth of 1-2 cm and when it was introduced
to a depth of 10 cm (Schmitt, 1956).
In a field test in Miami, Florida, USA, on silt loam and muck
soils, lindane was applied at the extremely high rates of 11.2 or
112.1 kg/ha. The initial half-time at the lower rate was 15.5 months
in muck soil and 4.75 months in loam soil. Degradation was slower at
the higher rate: the initial half-times were 28.8 months in muck
soil and 11.1 months in loam soil (Lichtenstein & Schulz, 1958a). In
an earlier study on the same field plots with the same application
rates, however, Lichtenstein & Schulz (1958b) found that most of the
material detected chemically was inactive in the bioassay and
therefore did not represent lindane. They concluded that the
breakdown of lindane is faster than it appeared to be using their
analytical method.
In an extensive study, sandy loam, silt loam, and muck soils on
plots in three midwestern states of the USA were treated with
lindane in 1954 at application rates of 1.1, 11.2, and 112.1 kg/ha
to a depth of 15.2 cm. After a 4.5-year follow-up, no lindane was
detected on plots treated with 1.1 kg/ha; but after application at
the higher rates (far in excess of normal rates), about 36% of the
applied dose remained. Two major factors that affect the persistence
of lindane in soils appear to be the amount of organic matter in the
soil and the climatic conditions of the area (Lichtenstein et al.,
1960).
The rates of loss of lindane were calculated by Wheatley (1965)
in 10 long-term field studies in the United Kingdom. When lindane
was applied to the soil surface, there was a 50% loss within 4-6
weeks and a 90% loss within 30-40 weeks. When the lindane was mixed
into the soil, a 50% loss was observed after 15-20 weeks and 90%
within 2-3 years. No lindane was recovered 13 years after the last
application of lindane to a loam soil in Nova Scotia at a rate of
0.84-1.7 kg/ha (Stewart & Fox, 1971). Cliath & Spencer (1971)
treated two test plots in California, USA, with 21 kg/ha, which is
an application rate about 20 times above normal. A half-time of 8
months was found in sandy loam and 10 months in silty clay.
After application of lindane on three test plots of light sandy
soil in the Netherlands for 15 years, to give total amounts of 6.5,
13.0, and 24.3 kg/ha, only 3, 4, and 8% of the applied amount,
respectively, was recovered in layers to a depth of 20 cm (Voerman &
Besemer, 1970). A further follow-up of these plots for four years
showed rapid disappearance on the two locations with the lower
application rates; slower degradation was seen on the plot that had
received the highest application, where lindane was found to a depth
of 40 cm (Voerman & Besemer, 1975). Admixture of a 5% lindane dust
to the top 15-cm layer of a test plot at a rate of 10 kg of active
ingredient per hectare in India led to an initial concentration of
3.2 mg/kg soil. After an observation period of 180 days, 97.7% of
the applied lindane had disappeared. The initial half-time of
lindane in this study was about 30 days (Agnihotri et al., 1977).
The degradation of gamma-HCH was also determined in a variety
of studies in which technical-grade HCH was applied to soils. In
most of these investigations, the application rates were extremely
high, and in some, applications were made once a year for several
years (Lichtenstein & Polivka, 1959; Stewart & Chisholm, 1971;
Shiota & Kanda, 1972; Nash et al., 1973; Jackson et al., 1974;
Suzuki et al., 1975). Under these conditions, gamma-HCH disappeared
slowly from the soils and sometimes persisted for long periods.
The distribution of HCHs was studied in soil treated with
BHC-20 (containing 70% alpha-HCH, 6.5% beta-HCH, 13.5% gamma-HCH,
and 5% delta-HCH) in an agricultural area. The concentrations
changed with time after application; the mean value for gamma-HCH
was 16 µg/kg. The organic carbon content of the soil appeared to be
of primary importance, and the significant decrease in isomer
concentration observed with greater soil moisture was attributed to
microbial degradation, which is favoured by these conditions
(Chessells et al., 1988).
Kathpal et al. (1988) studied the behaviour of a formulation
consisting of a mixture of five HCH isomers in paddy soils under
subtropical conditions in India. The recommended application rate of
2.5 kg active ingredient per hectare and a rate of 5.0 kg/ha were
used. Gamma-HCH had dissipated by 50-63% within three months under
paddy, and average residues in soil at harvest were 0.3-0.34 mg/kg.
Dissipation after nine months (two crop seasons) was 98%. The
persistence under paddy in this study was fairly high, probably
owing to the anerobic conditions, which slow microbial degradation.
The paddy plants absorbed gamma-HCH from the soil: the residues at
harvest were about 1.0 mg/kg in plants and 0.03-0.06 mg/kg in seeds.
4.2.3 Hydrolytic degradation
Determination of the hydrolytic stability of a substance
provides an indication of whether this process can contribute to the
disappearance of the substance from the aquatic environment and, to
a certain extent, from soil. In a model experiment, the half-time of
lindane at 22 °C was 47.9 h at pH 9 and 100.7 h at pH 7; no
measurable hydrolysis occurred at pH 5 (Heupt, 1983).
4.2.4 Photolytic degradation (laboratory studies)
As lindane has measurable volatility and can be found at low
levels in air, its degradation in sunlight has been studied.
Carbon dioxide was formed after 14C-lindane was adsorbed onto
silica-gel plates at a concentration of 33 µg/kg and irradiated with
artificial sunlight (> 290 nm) in the presence of air; 6.4% of the
carbon was oxidized within 17 h. This photo-induced oxidation was
enhanced when the lindane was exposed to pure oxygen during
irradiation (Kotzias et al., 1981). No measurable degradation (less
than 0.5%) was observed 2000 h after exposure of lindane to the
light of a Xenon lamp in a Xenotest 150 on the wall of a quartz
vessel (solid phase). When the irradiation was performed in aqueous
solution, about 4% of the applied lindane was degraded after 2000 h.
The main degradation product was PCCH (Gardais & Scherrer, 1979).
Irradiation of lindane with ultra-violet light (254 nm) is
obviously more effective for degradation of the compound than
irradiation with light of longer wavelengths. Hamada et al. (1981,
1982) found rapid degradation of lindane in both the crystalline
state and in solution with 2-propanol under these conditions, with
PCCHs and TCCHs as reaction products. Eichler (1977) also found
rapid degradation of lindane in the solid or gaseous form and in
aqueous solution in the presence of ultra-violet irradiation, with
half-times of 12-24 h for the first two phases and 1-2 days for the
latter two.
4.2.5 Biodegradation in water
In a study of the degradation of lindane in a biological
purification plant, 75% of the compound was degraded within 6 h
(Eichler et al., 1976).
Newland et al. (1969) investigated the degradation of gamma-HCH
in simulated lake impoundments. Sediments from Lake Tomahawk,
Wisconsin, USA, were added to solutions of 5 mg/litre 14C-labelled
lindane and equilibrated for 18 h, and aerobic and anaerobic tests
were run for approximately 88 days. Initially, about 45% of the
applied lindane was adsorbed to the sediment (200 g per 3-litre
solution). Under aerobic conditions, about 16% of the added lindane
was degraded by the end of the observation period, whereas more than
97% was degraded under anaerobic conditions. When lindane
degradation was tested in samples of surface water from two
different regions for periods of 3, 6, or 12 weeks, decreases of up
to 90% of the initial concentration were found. Most of the lindane
was metabolized by microorganisms in the sediments: In samples of
sediment and water autoclaved prior to treatment and incubation, up
to 95% of the applied lindane was still present (Oloffs et al.,
1973).
In a field test in rice fields in the Camargue, France, a
formulation containing lindane was applied at a rate that resulted
in an initial concentration in water of 54.8 mg/m3. Rapid
disappearance was observed, for a half-time of about 1.5 days, and
within 10 days the concentration had dropped to the background value
of 0.08 mg/m3 (Podlejski & Dervieux, 1978).
The degradation of lindane was also tested in the water of a
drainage canal in the Holland Marsh, Ontario, Canada, in distilled
water, and in both water types after sterilization. The half-time of
lindane in the natural water was about six weeks, but a very low
disappearance rate was seen in the distilled and sterilized water,
indicating the importance of microbial action for degradation of
lindane in water (Sharom et al., 1980).
An aquatic model ecosystem, with pond water, sludge, aquatic
plants, and fish, was used to study the decomposition and migration
of lindane. In water without hydrobionts, the half-time was 50 days.
When sludge and aquatic plants were present, the half-time was 34
days, and that in the presence of fish was 2 days (Vrochinsky, 1973;
see Izmerov, 1983).
4.2.6 Microbial degradation
A variety of experiments on the degradation of lindane was
performed with mixed populations of the microorganisms that occur in
different types of soil, in aquatic sediments (Newland et al., 1969;
Benezet & Matsumura, 1973), and in other types of soil under
aerated, submerged, and strictly anaerobic conditions (Macrae et
al., 1967; Yule et al., 1967; Kohnen et al., 1975; Mathur & Saha,
1975, 1977; Tu, 1975; Haider, 1979). The fact that lindane was
removed faster from non-sterile than from autoclaved soil
demonstrated that its degradation in soil is due to microbial
activity (Macrae et al., 1967; Kohnen et al., 1975).
The microorganisms shown by screening experiments to be capable
of metabolizing and degrading lindane are as follows (Tu, 1976;
Jagnow et al., 1977):
Bacteria Fungi Algae
Arthrobacter sp. Penicillium sp. Chlamydomonas sp.
Bacillus sp. Rhizopus sp. Chlorella sp.
Citrobacter sp.
Clostridium sp.
Enterobacter sp.
Micromonospora sp.
Pseudomonas sp.
Thermoactinomycetes sp
In addition, lindane was metabolized in cell-free preparations of
Clostridium sp. in vitro (Heritage & Macrae, 1977a; Ohisa et
al., 1980).
Lindane is degraded by soil microorganisms under aerobic as
well as under anaerobic conditions, but anaerobic conditions are the
most favourable for its metabolism (Newland et al., 1969; Haider &
Jagnow, 1975; Vonk & Quirijns, 1979). In an anaerobically grown
culture of Clostridium sphenoides supplemented with lindane at 5
mg/litre, none was found, even after 2 h (Heritage & Macrae, 1979).
Several species of soil bacteria that have been shown to degrade
lindane effectively are described in detail in section 6.6.2.
In field studies in which gamma-HCH was applied at excessive
doses, it was degraded more slowly than at doses closer to those
used for normal agricultural applications. Introduction of HCH at up
to 224 kg/ha, corresponding to 33.6 kg gamma-HCH per hectare,
exceeded the degradation capacity of soil microorganisms for a long
period (Nash et al., 1973). In addition, the analytical methods used
might have resulted in an overestimation of the actual gamma-HCH
concentration, as concluded by Lichtenstein & Schulz (1958b).
Therefore, studies in which technical-grade HCH is applied,
especially at excessive rates, cannot be used to evaluate the
degradability of lindane in soil.
4.2.7 Bioaccumulation/Biomagnification
4.2.7.1 n-Octanol/water partition coefficient
The n-octanol/water partition coefficient (Pow) of lindane
was determined in several studies, with good agreement, covering the
narrow range of log Pow = 3.29-3.72 (Kurihara et al., 1973;
Platford, 1981; Darskus, 1982; Geyer et al., 1982; Hermens &
Leeuwangh, 1982; Geyer et al., 1984). These values indicate that
lindane can become enriched in lipid-containing biological
compartments.
4.2.7.2 Aquatic environment
The bioconcentration factor for lindane was found to be
dependent on the concentration to which the organisms, such as
algae, crustaceae, and fish, were exposed (Bauer, 1972; Ernst, 1975;
Schimmel et al., 1977; Trautmann & Streit, 1979; Marcelle & Thome,
1983): The highest bioconcentration factors were seen with the
lowest exposure concentrations. For example, Marcelle & Thome (1983)
determined the residues of lindane in brain, liver, and muscle of
the gudgeon (Gobio gobio) after exposure to concentrations of
0.22-142 µg/litre lindane in water. At the lowest concentration, the
bioconcentration factors in brain, liver, and muscle were about 600,
200, and 100, respectively, but they decreased to values of less
than 50 at higher concentrations.
Mouvet (1985) transplanted the freshwater aquatic moss
Cinclidotus danubicus from an uncontaminated area to a river that
received the effluent from an insecticide factory and determined
gamma-HCH concentrations in water and moss 13, 24, and 51 days after
the transplant. A three-fold increase in the gamma-HCH level was
found, with a bioconcentration factor of 294.
In a variety of aquatic organisms exposed to contaminated
water, the bioconcentration factor for lindane ranged from 13 to
1000 on a wet weight basis (Table 3).
Table 3. Bioconcentration factors of lindane in laboratory
experiments; test organisms were exposed to contaminated
water for the specified time
System Exposure Exposure Bioconcentration Reference
time concentration factora
(µg/litre)
Algae
Cladophora sp. up to 80.0 180 (d) Bauer (1972)
48 h 3.9 341 (d)
Nitzschia 24 h 6.1 1500-4700 (v) Trautmann & Streit
actinastroides 4400-12 400 (d) (1979)
Molluscs
Aplysia punctata 3-6 days 9000 201-436 (w) Chabert & Vicente
(1978)
Mya arenaria 5 days 5 40 Butler (1971)
Mercenaria 5 days 13
mercenaria
Mytilus edulis ns 2.61 74 (w) Ernst (1975)
0.02 242 (w)
Mytilus edulis ns 2-5 139 (w) Ernst (1979)
Venerupis japonica 3 days 1 121 (ns) Yamato et al.
(1983)
Annelidae
Lanice conchilega ns 2-5 1240 (w) Ernst (1979)
Crustacea
Penaeus duorarum 96 h 0.23 143 (ns) Schimmel et al.
Palaemonetes pugio 96 h 1.0 80 (ns) (1977)
Table 3 (contd)
System Exposure Exposure Bioconcentration Reference
time concentration factora
(µg/litre)
Insects
Sigara striata and 1 day 10 70-100 Kopf & Schwoerbel
Sigara lateralis (1980)
Fish
Lagodon 96 h 23.0 287 (ns) Schimmel et al.
rhomboides (1977)
Cyprinodon 96 h 108.7 727 (ns)
variegatus
Leuciscus idus ns 10-500 765 (ns) Sugiura et al.
Cyprinus carpio 281 (ns) (1979)
Salmo truttafario 442 (ns)
Poecilia 938 (ns)
reticulata
Poecilia 4 days 1 697 (ns) Yamato et al.
reticulata (1983)
Salmo gairdneri 27 days 30-290 319 Ramamoorthy
(1985)
a Calculated on the basis of: wet weight (w), dry weight (d), volume (v); ns, not specified
Another approach to the study of the bioconcentration of
lindane is the use of systems that simulate natural conditions,
taking into account sedimentary absorption processes and the
influence of contaminated food. The bioconcentration factors for
brine shrimp, mosquito larvae, and the brook silverside
(Haludesthes sicculus sicculus) exposed to lindane applied to the
sand of a test aquarium were 95, 220-383, and 600-1613,
respectively, depending on the food chains used (Matsumura &
Benezet, 1973). Marcelle & Thome (1984) investigated the
bioconcentration of lindane in the gudgeon (Gobio gobio) in
relation to the route of exposure. Fish were exposed either to
contaminated water alone or additionally to contaminated food. After
18 days, the group fed contaminated food had a 2.5-fold higher level
of lindane residues in liver than the group exposed to contaminated
water alone. Within three days after cessation of exposure, 98.4% of
the lindane residues had been excreted.
The uptake, transport, and bioconcentration of lindane were
also studied in a freshwater food chain, which consisted of
Chlorella sp., Daphnia magna, and Gasterosteus aculeatus
(algae-crustacea-fish). Uptake from water was more rapid than uptake
from food and depended on the duration of the experiment and the
feeding rate. The increase in lindane residues in the last link of
the food chain (fish) was not directly proportional to the
concentration found in the primary links (Hansen, 1980).
4.2.7.3 Terrestrial environment
The bioconcentration of lindane was investigated in a
terrestrial food chain, which consisted of soil, barley,
caterpillar, and quail. Doses up to 400 times the standard
agricultural dose (50 and 200 mg/kg soil) were applied to the soil.
Although lindane was found in all of the links of the food chain,
the concentrations decreased progressively (Dugast, 1980).
Feeding hens diets containing lindane at 0.05, 0.15, or 0.45
mg/kg for 20 weeks resulted in constant values of 0.01, 0.03, and
0.09 mg/kg of eggs, demonstrating a dose-related accumulation of
lindane (Cummings et al., 1966).
Several studies are available on the bioconcentration of
lindane in rats. After seven rats had received daily doses of 2 or 4
mg/kg body weight for up to 12 weeks, gamma-HCH was found at a
concentration of about 8 mg/kg in adipose tissues of the group that
had recived the high dose (Jacobs et al., 1974). In another
experiment, four generations of rats were fed a diet containing 20%
fat and a mixture of insecticides including lindane at levels of
0.07-0.8 mg/kg. Even in the F3 generation, the levels of gamma-HCH
residues in adipose tissues were of the same order of magnitude
(< 0.05-0.56 mg/kg) as those of lindane in the diet (Adams et al.,
1974). No accumulation occurred, even in four consecutive
generations.
Accumulation factors have been determined from feeding studies
in rats (Fitzhugh et al., 1950; Oshiba, 1972; Baron et al., 1975;
Suter et al., 1983). In comparison to the concentration of lindane
in the diet, the highest reported bioconcentration factor was about
2 for adipose tissue. The average bioconcentration factor for
adipose tissues in rats derived from all these studies is 1; the
bioconcentration factors for other tissues are considerably lower.
4.2.7.4 Bioconcentration in humans
Geyer et al. (1986) examined data on environmental chemicals
detectable in adipose tissue and/or breast milk of
non-occupationally exposed humans and concluded that, in
industrialized countries, more than 90% of human exposure to HCHs
originates from food. Mean concentrations of gamma-HCH in human
adipose tissue in Czechoslovakia, the Federal Republic of Germany,
and the Netherlands were 0.086, 0.024-0.061, and 0.01-0.02 mg/kg,
respectively, on a fat basis. The mean bioconcentration factor,
calculated on the basis of the concentration in the diet (2.3, 5.0,
and 0.62-0.9 µg/kg, respectively) and levels in adipose tissue, was
18.6 ± 9 on a lipid basis (range, 10.4-32.5). Greve & Wegman (1985)
found an accumulation factor (adipose tissue/blood) of 70 for
gamma-HCH in humans.
4.2.7.5 Field studies
The bioconcentration of lindane was investigated by
environmental monitoring in aquatic ecosystems. The residue levels
found in different organisms were related to the environmental
background levels, and these data were used to calculate the
bioconcentration factors.
The bioconcentration factor for gamma-HCH in sea water and
bladder wrack (Fucus vesiculosus) in the Husum estuary and the
adjacent North Friesian Wadden Sea in the Netherlands was about 150
(Herrmann et al., 1984). On the basis of the data given in section
5.1.5.2 on the occurrence of gamma-HCH in muscle and fat of bream
collected in the River Elbe, a bioconcentration factor of 10 000 to
50 000 was calculated (Arbeitgemeinschaft für die Reinhaltung der
Elbe, 1982).
Frisque et al. (1983) studied the accumulation of lindane by
bryophytes (Cinclidotus danubicus and C. nigricans) in the Meuse
River and found a concentration factor of 300-350. The average level
in the river was 0.067 µg/litre. Hartley & Johnston (1983) found a
bioconcentration factor for the freshwater clam Corbicula
manilensis of 2610 on a lipid basis; and Cosson Mannevy & Marchand
(1980) found a mean factor of 26 198 (on a dry-weight basis) in
Mytilus edulis.
On the basis of the concentrations of gamma-HCH in sea water,
sediments, and fish from the Mediterranean Sea, El-Dib & Badawy
(1985) calculated a bioconcentration factor of about 1000 (on a
lipid basis). Tanabe et al. (1984) reported bioconcentration factors
for total HCHs in a trophic chain in the western North Pacific. As
the contribution of gamma-HCH to the residue levels was determined,
the bioconcentration factors for this isomer can be estimated to be
about 40, 40, 100, and 1850 for zooplankton, myctophid, squid, and
dolphin, respectively.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
An average of 0.23 ng/m3 (0.039-0.68 ng/m3) gamma-HCH was
found in 24 samples of air taken from over the western Pacific, the
eastern Indian Ocean, and the Antarctic Ocean (Tanabe et al., 1982).
Levels of gamma-HCH in the air of various regions of the USA
were within a similar range (US Environmental Protection Agency,
1976). The levels were below 1 ng/m3 in most samples, and values
up to 16.2 ng/m3 were found in only two regions.
Gamma-HCH was found at an average concentration of 0.14 ng/m3
in the air of unpolluted areas in the Federal Republic of Germany in
1972; in polluted areas (the Ruhr), a level of 0.8 ng/m3 was found
in 1976/77 (Deutsche Forschungsgemeinschaft, 1983; Hildebrandt et
al., 1986). It occurred at 0.52-11 ng/m3 in a location with heavy
traffic near Ulm in the Federal Republic of Germany and at 0.18-1.1
ng/m3 in a rural area. The authors concluded that the
concentrations in the lower troposphere under different
meteorological conditions reflect regional input and long-range
transport (Wittlinger & Ballschmiter, 1987).
The average concentration of gamma-HCH in 55 air samples taken
near Delft, the Netherlands, in 1979-80 was 0.36 ng/m3 (maximum,
3.4 ng/m3); in three other locations in the Netherlands, average
levels of 0.2-0.9 ng/m3 were found. In six houses built on former
dumping grounds, the average concentration of gamma-HCH was 6
ng/m3 (range, 1-14 ng/m3), whereas in the space beneath the
floor the level was below the detection limit (1 ng/m3). Outdoor
concentrations in this area were 0.3-0.4 ng/m3. In another study,
the concentrations of gamma-HCH in the space beneath the floor of
houses were 90 ng/m3. Much higher levels were found in houses
treated with lindane-containing products for the control of woodworm
or of long-horned beetle. Peak levels of 51-61 µg/m3 were found
four weeks after application; these decreased gradually to 8-24
µg/m3 after 10 weeks. After indoor application of lindane for wood
preservation, levels of 50 µg/m3 were common, with peak levels of
up to 100 mg/m3 (Sloof & Matthijsen, 1988).
5.1.2 Water
5.1.2.1 Rain and snow
Levels of 0.001-0.005 µg/litre were found in rain-water
analysed in the Federal Republic of Germany in 1970-72 (Mestres,
1974); in 1983, gamma-HCH was found at an average of 0.06 (range,
0.01-0.18) µg/litre in rain-water near de Bilt, the Netherlands
(Slooff & Matthijsen, 1988).
Strachan et al. (1980) found traces of gamma-HCH in 17 samples
of snow collected from the Canadian side of the Great Lakes in 1976
and 5-12 ng/litre in 81 samples of rain-water collected in 1976 and
1977.
5.1.2.2 Fresh water
Water samples from selected rivers in Yorkshire, United
Kingdom, analysed for gamma-BHC in 1966 contained levels of
0.001-0.18 µg/litre; in 1968, however, the highest value was 0.622
µg/litre. Water samples from six other rivers, also analysed in
1968, contained mean values of 0.011-0.030 µg/litre, and the highest
levels found were 0.020-0.098 µg/litre (Lowden et al., 1969).
River water samples analysed in 1969-72 in Belgium, France, the
Federal Republic of Germany, the Netherlands, and Italy contained
less than 0.1 µg/litre and usually less than 0.05 µg/litre. In 1826
water samples taken at 99 sites in the Netherlands in 1966-77, the
highest concentrations of gamma-HCH were found in those from the
River Rhine and its tributaries. The concentrations of gamma-HCH
over the period 1969-74 varied between 0.01 and 0.4 µg/litre, but in
1974-77, the concentrations were all below 0.1 µg/litre (Mestres,
1974). Gamma-HCH concentrations have been measured in the Rivers
Rhine, Meuse, and West-Scheldt and in other surface waters in the
Netherlands since 1969. Since 1974-75, the levels have been below
0.05 µg/litre in the Rhine and about 0.05 µg/litre in the
West-Scheldt; in the Meuse, the concentrations were more variable
and ranged from 0.01 to 1.0 µg/litre. In agricultural and
horticultural areas, the levels were 0.01-1.0 µg/litre, with
incidental peaks up to 0.5 µg/litre, probably due to use of lindane.
The average concentration of dissolved gamma-HCH in the Meuse-Rhine
estuary in 1974 was 20 ng/litre and that of suspended gamma-HCH
between 1 and 20 ng/litre. In coastal waters of the Netherlands, the
concentration of dissolved gamma-HCH was 0.9-4.6 ng/litre and that
of bound gamma-HCH, 3.1-8.7 ng/litre (Sloof & Matthijsen, 1988).
A sampling trip along the River Rhine, from Rheinfelden in
Switzerland to Rotterdam in the Netherlands, proved that the source
of alpha-, beta-, and gamma-HCH was located in the upper reaches of
the River. In the Meuse, lindane levels in 1969-77 were all below
0.1 µg/litre (Wegman & Greve, 1980). In an extensive programme in
1982 to determine pollution in Dutch surface waters at 45 locations,
gamma-HCH concentrations were generally between 0.01 and 0.1
µg/litre (Wammes et al., 1983).
The mean concentration of gamma-HCH in the River Elbe, from
Schnackenburg to the North Sea, in 1981-82 was 0.021
(< 0.001-0.051) µg/litre; during February-November 1988, the
concentrations were 0.005-0.044 µg/litre (Arbeitsgemeinschaft für
die Reinhaltung der Elbe, 1988). More figures for Germany are given
by Wirth (1985). Gamma-HCH was found at three locations in the River
Rhine at 0.02 µg/litre and in six side-rivers at 0.01-0.06 µg/litre.
These levels had decreased markedly since 1975 (Landesamt für Wasser
und Abfall, 1988).
5.1.2.3 Sea water
Atlas & Giam (1981), Bidleman & Leonard (1982), Oehme & Stray
(1982), and Oehme & Mano (1984) analysed water from such widely
differing areas as the Eniwetok Atoll in the North Pacific, the
Arabian Sea, the Persian Gulf, the Red Sea, Lillestrøm, Norway, Bear
Island, and Spitzbergen in the Arctic Ocean. The gamma-HCH
concentrations were in the range 0.01-0.05 ng/litre, except in the
Arabian Sea, the Persian Gulf, and at Lillestrøm, where levels up to
0.67 ng/litre were found (Slooff & Matthijsen, 1988). Levels of
0.0001-0.004 µg/litre gamma-HCH were measured in the Western
Pacific, the Eastern Indian, and Antarctic Oceans (Tanabe et al.,
1982). No gamma-HCH was found in 60 water samples from the Japan Sea
and Pacific Ocean (detection limit, 0.1 µg/litre) (A. Hamada, letter
to M. Mercier, dated 28 July 1989; T. Onishi, letter to M. Mercier,
dated 24 July 1989). The levels detected in water from the North Sea
and the Arctic Sea are of the order of 0.001-0.02 µg/litre (Deutsche
Forschungsgemeinschaft, 1983). The maximal level of gamma-HCH in
North Sea water in 1972 was 0.028 µg/litre; 5-10% of the samples
contained gamma-HCH (Mestres, 1974). The level of gamma-HCH in
surface-water of the North Sea in June-July 1986 ranged from 1.0 to
4.0 ng/litre. The highest concentrations were found close to the
coast (Umweltbundesamt, 1988-89).
5.1.3 Soil
Traces of gamma-HCH are transmitted to soil by precipitation;
the resulting contamination is generally below the limit of
detection (0.0001-0.001 mg/kg). Application of lindane in
agricultural areas can result in higher concentrations: levels in
some German districts were mainly in the range 0.001-0.01 mg/kg, but
in certain fields up to 0.6 mg/kg was found (Fricke, 1972).
Edelman (1984) analysed 96 samples of the upper 10 cm of soil
from 38 natural reserves in the Netherlands for gamma-HCH: 59
samples contained < 1 µg/kg, 21 contained 1-10, 9 had 10-20, and 7
had 20-80 µg/kg (Slooff & Matthijsen, 1988). In the National Soils
Monitoring Program of the US Environmental Protection Agency (Carey
et al., 1979), several thousand samples from cropland sites were
analysed for residues; no gamma-HCH residues were detected in more
than 99% of the samples. In the Ukraine, however, 36 of 136 soil
samples taken at various locations contained lindane at levels of
0.1-5 mg/kg (Talanov, 1977; see Izmerov, 1983).
In a study on the application of lindane dust by aircraft on
mosquito breeding sites at 1.3 kg/ha, the gamma-HCH content of the
soil was 1 mg/kg; after one year, the level was 0.01 mg/kg
(Vroschinsky, 1973; see Izmerov, 1983).
5.1.3.1 Sediment
Gamma-HCH was present in three of six samples of sediment taken
from Nyumba Ya Mungu Lake in the United Republic of Tanzania in
1986, at a concentration of 1-4 µg/kg dry weight (Paasivirta et al.,
1988).
Martin & Hartmann (1985) found gamma-HCH at levels above the
detection limit (5 µg/litre) in less than 4% of 117 samples of
sediment taken in 1980-82 from riverine and pothole wetlands in
north-central USA. In less than 4% of the samples, gamma-HCH was
present at above the detection level of 5 µg/kg.
In Japan, gamma-HCH was found in 9 out of 60 samples of
sediment at a concentration of 10 µg/kg in 1974 (A. Hamada,letter to
M. Mercier, dated 28 July 1989; T. Onoshi, letter to M. Mercier,
dated 24 July 1989).
The median levels of gamma-HCH in sediments from eight rivers,
harbours, and sites close to dumping places in the Netherlands were
15-342 µg/kg dry matter (Slooff & Matthijsen, 1988).
5.1.3.2 Dumping grounds and sewage sludge
The soil at various locations in the Netherlands is polluted
with HCHs as a result of spillage during production, storage, and
handling of this chemical during the 1950s. The concentrations found
range up to a few thousand milligrams of HCHs per kilogram of dry
soil. Further pollution has been caused by the dumping of chemical
waste, sometimes in order to level the ground; this waste can be
dispersed from dumping areas by leaching or wind erosion. In certain
polluted areas, high concentrations of HCHs (mainly alpha- and
beta-HCHs) were found at depths of more than 2 m below ground level.
In 18 locations in the Netherlands, the average concentrations of
gamma-HCH in sewage sludge in 1981 were 8-50 µg/kg dry matter.
Groundwater was also found to be polluted, but this was restricted
to the vicinity of the production areas; horizontal transportation
of HCHs in groundwater appeared to be limited (Slooff & Matthijsen,
1988).
Fieggen (1983) found gamma-HCH in sewage sludge at mean values
of 25 µg/kg dry matter in 1975, 43 µg/kg in 1978, and 12 µg/kg in
1981.
5.1.4 Drinking-water, food and feed
Although in most countries nowadays only lindane is used,
residues of alpha- and beta-HCH can still be found in crops and
animal products originating from regions where technical-grade HCH
(containing all of the HCH isomers) is still in use.
5.1.4.1 Drinking-water
Gamma-HCH was found at 0.0001-0.001 µg/litre in water from 19
lakes in Germany and at levels below 0.001 µg/litre (0.0001-0.0008
µg/litre) in the drinking-water derived from them (Bernhardt &
Ziemons, 1974). In the USA, only 3% of drinking-water samples
examined contained gamma-HCH, in a range of 0.001 to about 0.1
µg/litre (US Environmental Protection Agency, 1976). In Ottawa,
Canada, drinking-water samples collected in 1976 contained 0.4-11
ng/litre (Williams et al., 1978).
5.1.4.2 Cereals, fruits, pulses, vegetables, and vegetable oil
The large body of information on gamma-HCH residue levels in
crops grown and treated with this chemical according to Good
Agricultural Practice has been reviewed comprehensively by the
FAO/WHO Joint Meeting on Pesticide Residues and summarized in
published monographs (FAO/WHO, 1967, 1968, 1969, 1970, 1974, 1975,
1976, 1978, 1980).
In samples of ready-to-eat foods collected from 30 markets in
27 US cities in 1966-67, gamma-HCH levels were 0.003-0.009
(occasionally 0.06) mg/kg in grains and cereals, 0.002-0.027 mg/kg
in garden fruits, 0.001-0.005 mg/kg in potatoes, 0.002-0.007 mg/kg
in leafy vegetables, and 0.004-0.012 mg/kg product in oils, fat, and
shortening (Martin & Duggan, 1968). In 1967-68, residues of
gamma-HCH were found at 0.002-0.006 in leafy and root vegetables, at
0.002-0.003 in garden fruits, and at 0.029-0.085 mg/kg product in
oils, fat, and shortening (Corneliussen, 1969).
In monitoring studies carried out on grain in the Federal
Republic of Germany at one-year intervals since 1975, gamma-HCH
residues in wheat and barley were 0.001 mg/kg or less (Ocker, 1983).
More than 800 samples of cereal and cereal products analysed in
Germany in 1975-78 and 1979-83 contained mean concentrations of
0.0009-0.04, but cereal products had up to 0.11 mg/kg. The mean
concentration of gamma-HCH in 200 samples of wheat and rye collected
in 1986 and 1987 was 0.06 mg/kg, with a maximum of 0.3 mg/kg
(Umweltbundesamt, 1988-89).
Of 281 samples of wheat analysed for the presence of gamma-HCH
in the United Kingdom between October 1978 and April 1979, 71
contained levels in the range 0.002-0.04 mg/kg. Gamma-HCH was also
found in one sample of polished rice from Spain, at a concentration
of 0.008 mg/kg (Steering Group on Food Surveillance, 1982).
Gamma-HCH was found in 16% of samples of imported maize in the
United Kingdom in the range none detected to 0.007 mg/kg, and in 28
samples of different types of pulses at none detected to 0.05 mg/kg.
Of retail cereal products, only bran and wheat contained detectable
levels (0.01 mg/kg product) of gamma-HCH in 1982 (Steering Group on
Food Surveillance, 1986). In 1986-87, 31 of 142 samples of pulses
contained residues; in nine, levels of < 0.01-0.4 mg/kg were found.
Peanut butter and vegetable oils contained 0.01 mg/kg (Steering
Group on Food Surveillance, 1989).
About 80-90% of samples of fruit, potatoes, and other
vegetables analysed in the Federal Republic of Germany contained no
detectable residues of gamma-HCH (Weigert et al., 1983). The
remaining 10-20% had mean levels up to 0.01 mg/kg, with no
significant difference between 360 samples originating from
conventional agriculture and 360 samples from 'alternative'
agriculture (Vetter et al., 1983). In 1976-78 and 1980, the mean
concentrations of gamma-HCH were < 0.001-0.002 mg/kg product in
more than 400 samples of fruit, potatoes, and other vegetables. In
the Netherlands, residues in fruit and vegetables were generally in
the range 0-0.1 mg/kg, although some leafy crops, such as endive,
lettuce, celery, and leek, contained levels up to 5 mg/kg. Samples
of wheat contained only 0-0.05 mg/kg, with a few measurements up to
0.2 mg/kg (FAO/WHO, 1978). In France, gamma-HCH residues were found
in wheat at 0.01-0.02 mg/kg, and at low levels in other commodities,
such as carrots and endives (Laugel, 1981). Engst et al. (1967)
found that the gamma-HCH content of carrots grown from seed treated
with this compound decreased continuously during the first 120 days.
At normal harvesting time, the early varieties contained 3-6 mg/kg
product, the mid-season varieties about 2 mg, and the late
varieties, 0.4 mg/kg. When the carrots were harvested after 200
days, 0.3-0.7 mg/kg was present (independently of variety). Even
after 6 months' storage, low residues were still present.
5.1.4.3 Meat, fat, milk, and eggs
Martin & Duggan (1968) found gamma-HCH at levels of 0.09 mg/kg
in dairy products and at 0.01-0.03 mg/kg (with a peak of 0.374
mg/kg) in samples of meat, fish, and poultry collected from 30
markets in 27 cities in the USA in 1966-67. Residue levels in
samples of meat, fish, and poultry in 1967-68 were 0.003-0.026 mg/kg
(Corneliussen, 1969). No gamma-HCH or levels of 0.01-0.1 mg/kg were
found in 99% of samples of cow's milk and manufactured milk products
from Illinois (USA) (Wedberg et al., 1978). In milk samples
collected during Spring 1983 from 359 bulk transporters,
representing 16 municipalities of Ontario, Canada, gamma-HCH was
found in 68% of the samples at a mean concentration of 4.0 µg/kg
butter fat (Frank et al., 1985). Six samples of cow's milk from six
locations in Switzerland contained 3.0-5.1 mg/kg on a fat basis
(Rappe et al., 1987).
In about 25% of 976 samples of meat and poultry products
(including eggs) collected in the United Kingdom in 1984-86,
gamma-HCH was present at a mean concentration of 0.01-0.02 mg/kg.
The highest level, 3.7 mg/kg, was found in lamb. Processed meat and
poultry products (631 samples collected in 1985-87) contained mean
concentrations of 0.01-0.06 mg/kg product. About half of 849 samples
of retail milk and dairy products collected in 1984-87 contained
gamma-HCH at concentrations of 0.01-0.03 mg/kg; the highest level,
0.7 mg/kg, was found in milk (Steering Group on Food Surveillance,
1989). Imported meat products were also analysed in the United
Kingdom for the presence of alpha-, beta-, and gamma-HCH. No
detectable residue of gamma-HCH was found in beef or pork products:
processed pork contained none detectable to 0.03 mg/kg. Processed
poultry contained none detectable to 0.04 mg/kg (Steering Group on
Food Surveillance, 1986). In 1967-70, in the Ukraine, gamma-HCH was
found in cows' milk at an average concentration of 0.6 mg/litre
(Medvedev & Perepechkina, 1973; see Izmerov, 1983). In the USSR, the
following concentrations were found: milk and milk products, 0.055 ±
0.005 mg/kg; poultry and fish, 0.068 ± 0.021 mg/kg; butter, 0.003 ±
0.002 mg/kg; vegetables and fruits, 0.008 ± 0.003 mg/kg; groats and
flour, 0.005 ± 0.002 mg/kg (Sizova & Bogomolova, 1976; see Izmerov,
1983).
Concentrations of gamma-HCH were measured in 1250 samples of
milk and other dairy products in France in 1970-77 and in 1981. In
the first period, the gamma-HCH concentration was < 0.1 mg/kg of
fat; by 1981, the levels had declined to < 0.03 mg/kg of fat
(Laugel, 1981; Rhône-Poulenc Agrochimie, 1986). Higher levels (mean,
0.85 mg/kg) were found in animal fat, but meat and eggs generally
contained no detectable residue (Laugel, 1981). The mean levels of
gamma-HCH found in a large number of samples of various food items
in Germany (Hildebrandt et al., 1986) are shown in Table 4.
The levels of gamma-HCH in food items analysed in France were
0.006-0.01 mg/kg in 113 samples of vegetables, 0.005-0.04 mg/kg in
192 samples of fish and seafood, 0.005-0.041 mg/kg in 154 samples of
preserved meat, 0.007-0.017 mg/kg in 104 samples of cereal products,
0.007-0.034 mg/kg in 120 samples of butter and cheese, 0.005-0.059
mg/kg in 25 samples of oil and fat, and 0.006-0.021 mg/kg in 26
samples of fruit (Rhône-Poulenc Agrochimie, 1986).
Table 4. Levels of gamma-HCH (mg/kg) in food items in Germany
Food item 1973-78 1979-83 1973-83
Meata 0.004-0.04
Meat productsa 0.006-0.055
(maximum, 0.52)
Animal fata 0.007-0.09
(maximum, 0.5)
Gamea 0.042-4.072
Poultrya 0.01-0.05 0.004-0.046
(maximum, 0.471)
Chicken eggs < 0.001-0.01
Chicken eggsa,c 0.001-0.02
(maximum, 1.9)
Milk and milk productsa 0.05 0.01-0.02
Cow's milka,b 0.03 0.01
Vegetable oil and 0.01-0.02
margarinea
Oil seeds, nuts, pulses 0.001-0.127
Fish and fish products 0.01-0.02 0.002-0.009
Shell-fish and molluscs < 0.001-0.020
a From Hildebrandt et al. (1986); on fat basis
b From Anon. (1984)
c From Koelling (1978)
Skaftason & Johannesson (1979) found a mean value of 13 µg/kg
in 35 samples of butter from Iceland in 1968-70. Of 32 samples
analysed in 1974-78, only five contained gamma-HCH, at a mean value
of 7 ± 2 µg/kg. The mean concentration in meat, poultry and eggs in
the Netherlands in 1976-78 was 0.002 mg/kg (range, 0.001-0.004
mg/kg) (De Vos et al., 1984); the levels in dairy products were
similar.
Fifteen of 105 chicken eggs from seven areas in Kenya had a
median concentration of 0.01 mg/kg (range, 0.01-0.04 mg/kg) (Kahunyo
et al., 1988). Ten samples each from two lots of lamb and beef were
collected randomly from markets in Bagdad, Iraq, in 1983 and
analysed for the presence of gamma-HCH. An average concentration of
0.225 (0.004-1.611) mg/kg was found in lamb, and 0.116 (0.005-0.83)
mg/kg was found in beef (Al-Omar et al., 1985).
5.1.4.4 Animal feed
Of 114 samples of animal feed analysed in the United Kingdom in
1982-85, 49 contained gamma-HCH at concentrations up to 2.3 mg/kg
product (Steering Group on Food Surveillance, 1986).
5.1.4.5 Miscellaneous products
Lanolin produced from crude wool grease may contain gamma-HCH:
a level of 1.2 mg/kg was found in the USA (Anon. 1989); and Meemken
et al. (1982) found average levels of 2.4 and 2.1 mg/kg in 1976 and
1981, respectively, in Germany. Concentrations of 0.001-0.23 mg/kg
were found in cosmetic creams made from the lanolin.
5.1.5 Terrestrial and aquatic organisms
5.1.5.1 Plants
Gamma-HCH was present in most of 13 samples of three types of
moss and four types of lichen collected on the Antarctic Peninsula
(Graham Land) in 1985 at a mean concentration of 0.84 mg/kg (range,
0.4-1.7 µg/kg) (Bacci et al., 1986).
In 1984, near Florence and Siena, Italy, far from primary
sources of pollution, leaves from ten species of tree and two
species of lichen were found to contain average levels of 8.2
(range, 2-14) and 10 (9-11) µg/kg dry weight, respectively.
Gamma-HCH levels in plant species collected in 14 countries ranged
from 0.2 to 700 µg/kg dry weight (Gaggi et al., 1986).
5.1.5.2 Aquatic organisms
Freshwater mussels (Anodonta piscinalis) were used to monitor
bioaccumulation of pollutants at 17 sampling sites in a river basin
in Finland between 1984 and 1987. One to three mussels were used per
sampling site. Gamma-HCH was found in concentrations varying from
none detected to 553 µg/kg on a fat basis; however, a clear decrease
was seen over the period of study (Herve et al., 1988).
Cowan (1981) studied the extent of pollution by HCHs of
Scottish coastal waters using Mytilus edulis as the biological
indicator. The gamma-HCH levels at 118 sites were < 6-53 µg/kg dry
weight, which are similar to those found in Germany, the
Netherlands, Spain, and the United Kingdom. The fish and shellfish
sampling programme of the Ministry of Agriculture, Fisheries, and
Food in the United Kingdom in 1977-84 was implemented mainly in
areas around the coasts of England and Wales. The range found for
gamma-HCH was < 0.001 (none detected) to 0.075 mg/kg wet weight;
the level in fish muscle was < 0.001 mg/kg wet weight (Franklin,
1987).
The average concentration of gamma-HCH was measured in 10
marine organisms collected along the Mediterranean coast of Spain
during 1985. Mytilus galloprovincialis, Venus gallina, Sardina
pilchardus, and Mullus surmuletus contained 0.1-1.7 µg/kg fresh
weight (maximum, 16 µg/kg) (Pastor et al., 1988).
Bream collected in rivers and lakes at 15 locations in Germany
contained average gamma-HCH concentrations of 106-696 µg/kg on a fat
basis (Umweltbundesamt, 1988-89), while bream collected in the River
Elbe, between Schnackenburg and the North Sea, contained average
concentrations up to 0.031 mg/kg in muscle and up to 2.6 mg/kg in
adipose tissue (Arbeitgemeinschaft für die Reinhaltung der Elbe,
1982). In 1970-72, different types of fish, mussels, and shrimps
were analysed for gamma-HCH. Fish with a low fat content, collected
in the Atlantic Ocean and the North Sea, contained 0.004-0.008 mg/kg
fresh weight, and fat fish contained 0.01 (0.01-0.026) mg/kg fresh
weight. Fat fish caught in the Baltic contained higher levels - up to
0.2 mg/kg fresh weight. Mussels and shrimp caught in the North Sea
contained none to 0.009 mg/kg fresh weight; mussels from the Baltic
coast contained 0.009-0.011 mg/kg. In 1973-76, similar values were
found, except that the fat fish had lower levels. Marine organisms
from the Baltic Sea generally contained higher levels of gamma-HCH
than those from the North Sea. Freshwater fish from industrially
contaminated areas contained higher levels (Hildebrandt et al.,
1986). Gamma-HCH was detected at levels up to 7.0 µg/kg (mean, 2.5
µg/kg) in the muscle of flounders collected off the coast of the
North Sea in Germany in 1986 (Umweltbundesamt, 1988-89).
5.1.5.3 Terrestrial organisms
Earthworms: Gamma-HCH was found in the soil of ten arable and
two orchard sites in the United Kingdom at 0.01 and 0.08 mg/kg soil,
respectively, and in worms living in the two soils at 0.05 and 0.3
mg/kg (Advisory Committee on Pesticides and Other Toxic Chemicals,
1969).
Birds: Bednarek et al. (1975) determined total HCH isomers at
levels of 0.03-0.63 mg/kg total egg (or 0.6-11.1 mg/kg on a fat
basis) in eggs of birds of prey, such as the sparrowhawk (Accipiter
nisus), in two areas of Germany in 1972-73. Eggs of sandwich terns
collected in the Elbe estuary contained arithmetic means (10 eggs)
of 0.006 mg/kg fresh weight in 1981, 0.002 in 1985, 0.003 in 1986,
and 0.028 in 1987 (Umweltbundesamt, 1988-89). The concentrations
found in the livers of avian predators in the United Kingdom are
shown in Table 5.
The mean levels of gamma-HCH detected in 23 barn owls (Tyto
alba Scop.) obtained in Leon, Spain, were 0.03 (0.003-0.083) mg/kg
wet weight in muscle, 0.036 (0.002-0.208) in liver, 0.051
(0.009-0.144) in fat, 0.012 (0.002-0.031) in brain, and 0.081
(0.005-0.343) in kidneys (Sierra & Santiago, 1987).
Table 5. Residues of gamma-HCH in livers of avian predators in the
United Kingdoma
Bird Date No. of gamma-HCH
samples (mg/kg)
Sparrowhawk 1963 11 0.01
Kestrel 1963 20 0.04
1964 28 0.1
1965 60 0.03
Tawny owl 1964 14 0.01
Heron (adults) 1964 17 0.005
Great crested grebe 1963/66 15 0.03
a From Advisory Committee on Pesticides and Other Toxic Chemicals
(1969)
Faladysz & Szefer (1982) examined adipose tissue from seven
species of diving ducks at their winter quarters in the southern
Baltic. Residues of gamma-HCH were detected in only 4 of 129 samples
from three species of duck examined (range, 0.001-0.51 mg/kg on a
fat basis).
Mammals: No gamma-HCH (< 0.01 mg/kg) was found in muscle
tissue from 51 North American wolves captured in 1969-71 in sparsely
populated forest regions (Schneeweis et al., 1974). Norstrom et al.
(1988) determined the contamination of the marine ecosystem of the
Canadian Arctic and sub-Arctic by organochlorine compounds by
analysing adipose tissue and liver from 6-20 polar bears (Ursus
maritimus) per zone collected from 12 zones between 1982 and 1984.
The levels were 0.30-0.87 mg/kg on a fat basis; the highest levels
were found in zones receiving continental run-off.
Mean concentrations of gamma-HCH in 86 samples of kidney fat
from roe-deer (Capreolus capreolus) collected in five locations in
Germany in 1985-86 were 8-12 µg/kg, with a maximum of 1020 µg/kg
(Umweltbundesamt, 1988-89).
5.2 Exposure of the general population
The data presented above demonstrate that the main source of
exposure of the general population is food.
5.2.1 Total-diet studies
In total-diet studies carried out in the United Kingdom between
1966 and 1985, 22-25 samples of foods in 20-24 groups were purchased
in 21 towns throughout the country and prepared by cooking. The
calculated mean levels of gamma-HCH residues in the total diet were
0.004 mg/kg in 1966-67, 0.0035 in 1970-71, 0.003 in 1974-75, 0.0025
in 1975-77, 0.002 in 1979-80, 0.0015 in 1981, and 0.0005 in 1984-85,
resulting in dietary intakes of 6.6, 5.5, 4.4, 3.9, 3.0, 2.0, and
0.5 µg/person per day (Egan & Hubbard, 1975; Steering Group on Food
Surveillance, 1982, 1986, 1989).
The average daily intake of gamma-HCH in the USA was estimated
on the basis of residues found in 30 market-basket composites
collected in 30 cities over the period 1964-80, as shown in Table 6.
Infant foods collected in the United Kingdom in 1985-87
generally contained very low levels of gamma-HCH (range, < 0.002 to
< 0.01 mg/kg product) (Steering Group on Food Surveillance, 1989).
Residues of gamma-HCH were also measured in food composites from 10
cities of the USA in 1974-75 (Johnson et al., 1979). Levels of
0.008-0.012 mg/kg food were found in diets of six-month-old infants,
and 0.001-0.007 mg/kg in the diets of two-year-old toddlers. Similar
samples collected in 1976-79 in 10 cities consisted of about 50
items of infant food and 110 items of food for toddlers. The daily
intake of gamma-HCH was 0.005 µg/kg body weight for infants and 0.01
for toddlers in 1976, 0.006 and 0.008 in 1977, 0.003 and 0.005 in
1978, and 0.001 and 0.006 in 1979 (Gartrell et al., 1985b).
Table 6. Average daily intake of gamma-HCH in the USA, 1964-80a
Year gamma-HCH intake (µ/kg
body weight per day)
1964-69 0.05
1965-70 0.02-0.07b
1973 0.0032
1974 0.0084
1975 0.0031
1976 0.0025
1977 0.0039
1978 0.0024
1979 0.0038
1980 0.0028
a From Johnson & Manske, 1976; US Environmental Protection
Agency, 1980; Gartrell et al., 1985a
b From Duggan & Corneliussen (1972)
Total-diet studies conducted by the US Food and Drug
Administration before 1982 were based on a 'composite sample
approach', regardless of the diet involved. Later studies were based
on dietary information obtained through surveys, so that the 'total
diet' of the US population could be represented by a relatively
small number of food items for a large number of age-sex groups
(Gunderson, 1988). The average intake of gamma-HCH in the diet of
14-16-year-old boys (mean body weight, 60 kg), estimated using the
more recent methods, is shown in Table 7 (S. I. Shibko, letter to
IPCS, dated 29 June1989). The daily intakes in 1982-84 in different
age groups were 0.0019 µg/kg body weight per day for 6-11-month-old
children, 0.0079 for two-year-old children, 0.0031 for
14-16-year-old girls, 0.0034 for 14-16-year-old boys, 0.0020 for
25-30-year-old women, 0.0025 for 25-30-year-old men, 0.0016 for
60-65-year-old women, and 0.0018 for 60-65-year-old men (Gunderson,
1988). The concentrations for these eight groups in 1984-86, 1987,
and 1988 were < 0.01 µg/day for 6-11-month-old infants, < 0.04 for
two-year-old children, and < 0.1 for the other six groups (S. I.
Shibko, letter to IPCS, dated 29 June 1989).
Table 7. Average daily intake of gamma-HCH in 14-16 year-old boys
in the USAa
Year Intake
µg/day µg/kg body weight
per day
1982-84 0.204 0.0034
1984-86 0.078 0.0013
1987 0.108 0.0018
1988 0.084 0.0014
a From S.I. Shibko, letter to IPCS, dated 29 June 1989
In total-diet studies in Germany, fruit, potatoes, and other
vegetables ready for consumption contained 0.001 mg/kg product
(Kampe & Andre, 1980). In 17 food groups in Spain, the gamma-HCH
concentration ranged from none detected to 0.019 mg/kg product; the
level in fat was up to 0.268 mg/kg. The daily intake amounted to
0.0138 mg/person in 1971-72 (Carrasco et al., 1976). In a total-diet
study in the Netherlands in 1977, the average concentration of
gamma-HCH in 100 samples was 0.03 mg/kg on a fat basis; the highest
level was 0.14 mg/kg (Greve & van Hulst, 1977). In another study in
the Netherlands, a mean daily intake of 0.002 mg/person was
determined for 1976-78 (De Vos et al., 1984).
Data from Canada, Guatemala, Japan, the United Kingdom, and the
USA indicate a very low daily intake of gamma-HCH over the years
1971-84. The median values ranged from 0.01 to 0.05 µg/kg body
weight (Gorchev & Jelinek, 1985). The daily intake of lindane in the
USSR was calculated from a market-basket survey to be about 0.005
mg/day. Cooking reduced this level by a factor of 4.3 (Sizova &
Bogomolova, 1976; see Izmerov, 1983).
5.2.2 Intake with drinking-water and air
Edwards (1981) calculated the daily intake of gamma-HCH with
drinking-water to be 0.4 ng per person, assuming a daily consumption
of 2 litre of water; the median daily intake via air was also
calculated to be 17 ng per person, indicating that only small
quantities of gamma-HCH are ingested with water and air.
Guicherit & Schulting (1985) measured the concentration of
gamma-HCH in the atmosphere and calculated that the daily average
intake of a 70-kg Dutch person by inhalation would be 7.2 ng.
Another calculation of the average human intake with air, on the
basis of ambient concentrations, was 12 ng/day, which represents
about 1% of the total daily intake by all routes. The daily intake
of gamma-HCH in the USA was estimated to be 0.002 µg/kg body weight
by air and 0.07 µg/kg by the oral route (Hildebrandt et al., 1986).
5.2.3 Concentrations in human samples
The concentrations of gamma-HCH in human samples are a good
indication of the total exposure of the general population.
5.2.3.1 Blood
Gamma-HCH was detected at a geometric mean of 0.4 µg/litre
(range, 0.1-4.1 µg/litre) in the blood of 49 of 62 people in
Louisiana, USA (Selby et al., 1969). In a follow-up study, a
geometric mean of 0.4 µg/litre (range, 0.1-6.0 µg/litre was found in
47 out of 53 blood samples from pregnant women. Polishuk et al.
(1970) found a mean concentration of 0.4 ± 0.8 µg/litre in the blood
of 24 pregnant women and 0.3 ± 0.6 µg/litre in the blood of 23
infants living in Israel. Wassermann et al. (1982) found a mean of
4.3 ± 4.8 µg/litre in serum of 10 women in Israel with a normal
pregnancy. In a group of 17 women with an abnormal pregnancy
(premature birth), a mean concentration of 15.0 ± 7.2 µg/litre was
detected. Bercovici et al. (1983) found a concentration of 8.0 ± 4.5
µg/litre in the serum of seven Israeli women with a normal pregnancy
and a mean concentration of 8.5 ± 7.8 µg/litre in 17 women with a
'missed abortion'.
Reiner et al. (1977) found a mean concentration of 4.1 ± 0.6
µg/litre (range, 0.5-15.0) in 23 of 147 serum/plasma samples from
people living in a town in Yugoslavia. Similar levels were found in
other parts of the country (Krauthacker et al., 1980).
Siddique et al. (1981) found gamma-HCH at a mean concentration
of 25.0 ± 16.0 µg/litre (range, 8.0-47.0) in the blood of 15 people
in India, and Saxena et al. (1981) found a mean concentration of
19.0 ± 12.4 µg/litre (range, 2.4-135.0) in the blood of 100 pregnant
women, aged 18-34 years, in the Indian countryside. Nonvegetarian
women had higher blood levels than vegetarian women. Kaphalia & Seth
(1983) found blood levels of 12.0 (none detected to 71.0) µg/litre
in 48 men (aged 20-40 years), 12.00 (range, 5.0-24.0) µg/litre in 16
women (aged 10-30 years) and 16.0 (range, 3.0-64.0) µg/litre in 16
children (< 10 years) in India.
Eckenhausen et al. (1981) found a geometric mean of 0.9
µg/litre of gamma-HCH (range, < 0.4-3.8) in 28 out of 48 pregnant
women in the Netherlands. After delivery, a geometric mean of 0.5
(range, 0.2-19.0) µg/litre was measured in 24 out of 66 women, and a
mean of 0.5 (range, < 0.3-34) µg/litre in 33 out of 86 babies.
Blood samples from Dutch citizens were analysed in 1978 (70
samples), 1980 (48 samples), 1981 (127 samples), and 1982 (54
samples); the concentration of gamma-HCH was in the range < 0.1-0.2
µg/litre blood (Greve & van Harten, 1983; Greve & Wegman, 1985).
Blok et al. (1984) measured the levels of gamma-HCH in the blood of
65 healthy volunteers (34 women and 31 men) in the Netherlands and
found residues in approximately two-thirds of the people studied.
The median concentration in both men and women was 0.2 µg/litre
(range, none detected to 0.6 µg/litre). Bertram et al. (1980) found
a median concentration of 1.18 µg/litre (range, none detected to
2.94) in 118 whole-blood samples in Germany.
In 8 of 35 serum samples from mothers in Norway and in 6 of 35
corresponding samples of umbilical cord serum, the levels of
gamma-HCH ranged from 0.2 to 27 µg/kg wet weight. In serum samples
from immigrant mothers and in 5 of 7 corresponding umbilical cord
serum samples, the levels were 0.1-3.4 µg/kg wet weight (Skaare et
al., 1988).
5.2.3.2 Adipose tissu
Mes et al. (1982) analysed 99 samples of adipose tissue from
autopsied accident victims in different areas of Canada and found an
average concentration of gamma-HCH of 0.003 mg/kg (wet weight)
(range, 0.001-0.03 mg/kg). Nearly all of the samples (90%) contained
gamma-HCH.
In 567 samples of adipose tissues from Dutch citizens analysed
for the presence of gamma-HCH in 1968-83, the concentration varied
from < 0.01 to 0.04 mg/kg; the highest levels were found for
1968-76 (Greve & van Harten, 1983; Greve & Wegman, 1985). Bertram et
al. (1980) found a median concentration of 0.05 mg/kg (range, none
detected to 0.44) in 72 samples of adipose tissue from people in
Germany. In specimens of subcutaneous fat taken in 1982-83 from 48
children (34 < 1 year; 14 aged 2 years), the concentration of
gamma-HCH was 0.04 mg/kg fat (range, 0.01-0.21 mg/kg). The average
concentration was highest in infants aged 0-6 weeks, at 0.07 mg/kg
fat (range, 0.02-0.21 mg/kg) (Niessen et al., 1984). The results of
nine studies carried out in Germany in 1969-83 (598 samples) gave a
mean concentration for gamma-HCH of 0.01-0.34 mg/kg on a fat basis
(Hildebrandt et al., 1986).
Twenty-nine samples of adipose tissue were taken at necropsy
and 24 at surgery in the Poznan district in Poland and compared with
100 samples from residents of the Warsaw region. The mean
concentration of gamma-HCH in Poznan was 0.020 ± 0.018 and that in
Warsaw, 0.022 ± 0.003 mg/kg (Szymczynski et al., 1986).
The mean concentration of gamma-HCH in 360 samples of adipose
tissue collected in eight regions of Japan in 1974 was 0.035 mg/kg
(Takabatake, 1978).
5.2.3.3 Breast milk
Breast milk is a major route of elimination of organochlorine
pesticides and polychlorinated biphenyls in women.
In a Swedish study, the levels of gamma-HCH in mothers' milk
were found to be related to their dietary habits: levels in
lacto-vegetarians were lower than those in subjects who ate a mixed
diet, and these were lower than those found in mothers who ate a
mixed diet which regularly included fatty fish from the Baltic Sea
(Noren, 1983).
A significant correlation was found between the concentration
of gamma-HCH in breast milk and the amounts of meat products and
animal fat in the diet. In addition, concentrations of gamma-HCH in
breast milk appeared to be higher in rural areas than in urban areas
(Cetinkaya et al., 1984).
Variations in residue levels in human milk during a lactation
period of up to 9 months were investigated in five women aged 23-36
years in Germany: gamma-HCH concentrations were 0.004-0.022 mg/kg on
a fat basis, and no essential change in residue level occurred over
the lactation period (Fooken & Butte, 1987).
More than 7100 samples of breast milk were analysed in Germany
between 1969 and 1984 by 20 authors, and the results were summarized
by Hildebrandt et al. (1986). The mean concentration of gamma-HCH
was 0.01-0.11 mg/kg on a fat basis, but a mean concentration of 0.45
mg/kg was found for a group of 137 samples. A slow decrease in the
mean concentration was observed between 1978 and 1984. The average
concentration in human milk (2709 samples) in Germany in 1979-81 was
0.06 mg/kg on a fat basis (Fooken & Butte, 1987); in 1981-83, the
average level in 132 samples of breast milk was 0.032 mg/kg milk fat
(Cetinkaya et al., 1984). The results of other studies were
summarized by the Deutsche Forschunsgemeinschaft (1978, 1983). The
results for other countries are comparable to those for Germany,
although higher levels (mean, 0.33 mg/kg) were measured in
Czechoslovakia in 1971-73 (Deutsche Forschungsgemeinschaft, 1983).
Average concentrations in human milk in France between 1970 and 1975
were in the order of 0.06-0.07 mg/kg (fat basis) (Rhône-Poulenc
Agrochimie, 1986).
Tuinstra (1971) analysed 40 breast milk samples from young
mothers (18-32 years of age) in the Netherlands and found a median
concentration of 0.01 mg/kg on a fat basis (range, none detected to
0.04 mg/kg on a fat basis). The median concentration of gamma-HCH in
278 samples of human milk collected in 11 maternity centres in the
Netherlands was < 0.01 mg/kg on a fat basis; the highest value
found was 0.08 mg/kg (Greve & Wegman, 1985).
Mes et al. (1986) studied 210 breast milk samples from five
regions across Canada and found gamma-HCH at a mean concentration of
5 µg/kg (on a fat basis). Davies & Mes (1987) studied 18 breast milk
samples from Canadian, Indian, and Inuit mothers in Canada whose
fish consumption was comparable to the national rate. The level of
gamma-HCH was 7 µg/kg in milk fat of the indigenous population, in
comparison with 5 µg/kg in a national survey.
Vukavic et al. (1986) measured gamma-HCH in 59 samples of
colostrum collected in Autumn 1982 (26 samples) and Spring 1983 (33
samples) in Yugoslavia from healthy nursing mothers on the third day
after delivery. The concentrations of gamma-HCH were significantly
higher in Autumn (1.71 ± 0.44 µg/litre) than in Spring (0.67 ± 0.12
µg/litre).
Breast milk samples from four women in Iraq, examined once a
week for 20 weeks, contained average levels of 0.009, 0.005, 0.134,
and 0.005 mg/kg whole milk. Gamma-HCH levels in placental tissue
from these four donors were 0.004, 0.011, 0.013, and 0.006 mg/kg,
respectively. Fluctuations in the residue levels were seen to be due
to variations in the daily dietary intake and variations in the fat
content of the breast milk (Al-Omar et al., 1986).
6. KINETICS AND METABOLISM
6.1 Absorption
6.1.1 Oral administration - experimental animals
The uptake of lindane by rats or mice has been studied after
oral administration. Direct information on the velocity of uptake
from the gastrointestinal tract is available, which can be
supplemented by information from studies in which excretion of
orally administered radioactive lindane was followed.
Lindane taken up from the intestines is transferred almost
exclusively to the blood. No significant amount was found in the
lymph of rats after injection of 0.05 or 0.1 µmol into the loops of
the small intestines in vivo. Absorption was rapid: 29-53% of the
injected material was absorbed from the intestinal loops within the
first 30 min (Turner & Shanks, 1980). Uptake of lindane from the
intestines of rats given 12.5 mg in oil during five days was less
effective in animals depleted of their intestinal microorganisms by
maintenance under aseptic conditions than that in conventional rats.
The asepticized rats also excreted more unchanged lindane in the
faeces than conventional animals (Macholz et al., 1983).
6.1.2 Dermal application - experimental animals
Hawkins & Reifenrath (1984) developed an apparatus to determine
the evaporation and percutaneous penetration of
hexachloro-[U-14C]-cyclohexane (lindane) in vitro, applying a
dose of 4 µg/cm2 on pig skin. Evaporation accounted for 26 ± 5%;
skin oxidation for 43 ± 17%; and percutaneous penetration for only
0.7 ± 0.3% of the applied radiolabel. Reifenrath et al. (1984) also
evaluated models consisting of human or pig skin grafted onto the
congenitally athymic nude mouse, hairless dogs and weanling
Yorkshire pigs for predicting skin penetration in humans. A
radioactive dose of 0.05 µCi of [U-14C]-labelled lindane (98%) was
applied topically to 1.27 cm2 (4 µg/cm2) of each model, and
radiolabel (percentage of applied dose) was measured in urine and
faeces, skin scrub, application site, and carcass. Incomplete
excretion of the label following topical application was corrected
for by parental (subcutaneous) administration of 2 µCi in propylene
glycol. The results showed significant correlations between the
values for human skin and those for human skin grafted on athymic
mice and for weanling Yorkshire pigs, but no correlation was found
between the values for humans and those for the hairless dog or for
pig skin grafted on athymic nude mice.
Dermal absorption has also been investigated in rats and
rabbits. Groups of 24 male Charles-River Crl:CD(SD)BR rats and male
Hra:(NZW)SPF rabbits were given a single dermal application of
lindane (20% emulsifiable concentrate to which 14C-lindane had
been added) on clipped skin at doses of 0.1, 1, or 10 mg/rat and
0.5, 5, and 50 mg/rabbit, corresponding to 0.02, 0.2, and 2
mg/cm2, respectively. Urine and faeces were collected 0.5, 1, 2,
4, 10, or 24 h after application from four animals per dose level;
and four animals per group were bled and sacrificed 0.5, 2, 4, 10,
and 24 h after application of the test material. The ratio between
the quantity absorbed at a dose of 1 mg and that absorbed at 10 mg,
as well as that between 0.2 and 1 mg/rat at equal exposure duration,
decreased with increasing exposure time and concentration. On a
group basis, the total recoveries were 75-85% for rats and 75-82%
for rabbits. A significant fraction of the applied dose was found in
the urine: 16, 15, and 12%, respectively, at application levels of
0.1, 1, and 10 mg/rat. The corresponding values for rabbits were 46,
29, and 41% for doses of 0.5, 5, and 50 mg/rabbit. Much lower levels
were found in the faeces. Total adsorption (24 h) increased in rats
from 5% of the applied amount at the highest dose to 28% at the
lowest exposure. For rabbits, penetration was more rapid, and
adsorption at 24 h ranged from 17 to 56% of the applied dose. The
applied doses per unit surface area were approximately the same,
permitting a comparison of penetration rates. The average
penetration rates after 24 h for rats were 0.2, 2, and 4 µg/cm2
per hour for groups treated with 0.02, 0.2, and 2 mg/cm2. The
penetration rates after 24 h for rabbits were 0.5, 3, and 14
µg/cm2 per hour for the groups treated with 0.02, 0.2, and 2
mg/cm2. These studies indicate that appreciable absorption of
lindane takes place after dermal applicatioon (Bosch, 1987, 1987b).
6.1.3 Other routes - experimental animals
When doses of 40 or 80 mg/kg body weight of a mixture of 14C-
and 36Cl-gamma-HCH in rapeseed oil were injected intraperitoneally
into rats, 25% was absorbed within 1 h and at least 90% after 1 day.
Four days after the injection, only traces of lindane were left in
the abdominal cavity. One day after the injection, about 40% of the
applied dose was found in the organs and tissues (Koransky et al.,
1963).
6.2 Distribution
6.2.1 Oral administration - experimental animals
After uptake, lindane is distributed to all organs and tissues
in the body of laboratory animals, at measurable concentrations
within a few hours.
When lindane was administered orally to rats at doses of 1, 10,
or 100 mg/kg diet for up to 56 days, the highest concentrations were
found in adipose tissue. The fat:blood ratio in this study was very
close to 150 at all times, whereas the liver:blood ratio was
3.4-3.5. Lindane concentrations in organs reached a maximum after
2-3 weeks and slowly decreased thereafter. The authors did not
differentiate between males and females (Oshiba, 1972).
Twenty-four hours after oral administration of 14C-labelled
lindane at 8 mg/kg body weight in sunflower oil to rats for 10 days,
more than 35% of the administered activity was deposited in fat.
Muscle and kidneys contained 3.5 and 3.7%, respectively, and all
other organs that were analysed contained less than 1%; 17.4% was
found in urine and 13.8% in faeces. In total, only 78.7% was
recovered; however, skin was not analysed in this study. (Other
studies have demonstrated that a significant amount of lindane
administered to rats and mice is deposited in the skin, so that some
of the missing 21.3% of the total applied dose could have been
there.) A fter an additional 48 h, the concentrations in all organs
were reduced to about one-half of the values seen after 24 h,
showing that no single organ retained lindane significantly longer
than the others. Urine contained 24.5% and faeces, 20.9% (Seidler et
al., 1975).
After single oral doses to rats, the fat:blood ratio ranged
between 145 and 206 and the brain:blood ratio between 4 and 6.5
(Vohland et al., 1981).
After continuous dietary administration of lindane at doses of
0.2, 0.8, 4.0, 20, or 100 mg/kg diet for 13 weeks to Wistar KFM-Han
rats, the highest concentrations were reached in the fatty tissue.
At the highest dose, the fat:blood ratio was 44 in males and 69 in
females and the liver:blood ratio was 5.3 in males and 9.6 in
females. After six weeks with no further administration, lindane
concentrations in organs were close to the control values (Suter et
al., 1983).
The distribution of lindane in brain after oral administration
at 30 mg/kg or intravenous administration at 0.3 mg/kg was studied
using autoradiography-imaging analysis and dissection-liquid
scintillation counting techniques. The two routes of administration
gave similar results. A heterogeneous distribution of label in brain
regions was observed: the radiolabel concentration in the white
matter was higher than that in thalamus, mid-brain, pons and medulla
at different times relative to the mean value for whole brain. The
affinity of lindane for white matter and myelinated structures was
related to its lipophilic behaviour (Sanfeliu et al., 1988).
Mosha et al. (1986) studied the distribution and elimination of
gamma-HCH in adult female goats. Eight goats were administered a
daily dose of 6 mg/kg body weight by gavage for five consecutive
days. Blood and milk were collected before exposure and during 10-60
days after exposure, and organs and tissues were collected and
analysed. One goat was used as a control. The blood concentration of
gamma-HCH was approximately 0.1 mg/litre during the dosing period
and decreased gradually thereafter; none was detectable after day
20. The concentrations in milk were about eight times higher than
those in blood but decreased in parallel. The concentration in fat
samples on day 7 was 1.4 mg/kg, but those in other tissues were
about 0.1 mg/kg.
6.2.2 Inhalation - experimental animals
After rats were exposed to lindane at doses of 0.02, 0.1, 0.5,
or 5 mg/m3 for 90 days by inhalation, the highest concentrations
were found in fatty tissues. At 5 mg/m3, the fat:serum ratio was
150 in males and 464 in females; at 0.5 mg/kg, it was 161 in males
and 245 in males; at 0.1 mg/kg, 137 in males and 429 in males; and
at 0.02 mg/kg, 92 in males and 377 in females. At the 5 mg/m3
exposure level, the liver:blood ratios were 1.9 in males and 4.2 in
females and the brain:serum ratios were 9 in males and 23 in
females. These values suggest that higher concentrations are reached
in fat after inhalation than after oral administration, whereas
liver appear to be somewhat lower after inhalation. After a recovery
period of four weeks, concentrations in all organs had decreased to
the control values (Oldiges et al., 1983).
6.2.3 Other routes
One day after intraperitoneal injection to rats of a mixture of
14C- and 36Cl-lindane in rapeseed oil, the highest contents were
those of skin and fat - 15.7% and 10.7%, respectively. Less than 1%
was found in all other organs, including the central nervous system
(Koransky et al., 1963). When lindane or deuterated lindane was
administered intraperitoneally to rats at doses of 10 mg/kg body
weight, about 40 mg/kg fat were found after one day in both males
and females. At that time, the blood concentration in males was 0.2
mg/litre; 1-2 mg/kg were present in brain and 0.7 mg/kg in skeletal
muscle. Deuterated lindane was found at 110 mg/kg in depot fat of
males, and the levels in brain and muscle were about twice those of
undeuterated lindane (Stein et al., 1980).
Mottram et al. (1983) studied the metabolic fate of lindane in
three groups of two white female pigs. The pigs were sprayed once
with either 5.6 g or 1.4 g of an anti-louse spray, which represented
16 and 4 times, respectively, the normal dose of 350 mg/pig. Five
animals served as controls. Rapid accumulation of lindane occurred,
and several metabolites were found in adipose tissue. The main
metabolite was 1,2,4-trichlorobenzene. The residues were eliminated
rapidly from adipose tissue, so that 30 days after treatment, the
residual concentration in pigs sprayed with a dose 16 times the
recommended rate was no greater than that in the untreated controls.
Residue levels were also investigated in four lactating goats
following oral and topical application of labelled lindane (Wilkes
et al., 1987a,b). Two Alpine goats were housed in metabolic cages
and administered lindane (purity not stated) spiked with
14C-lindane in the diet at doses of 1 and 10 mg/kg twice daily for
four days; they were sacrificed 12-14 h after the last dose. One
Alpine goat received two topical applications at a seven-day
interval of a lindane solution (purity unspecified) containing 11.0
mg/ml to a shaved area that represented about 25% of the body
surface area, to simulate total body spray; it was sacrificed 48 h
after the last application. A Nubian goat was not only shaved over
the same extent but also had its remaining hair clipped to
approximately 3 cm, and the whole surface of the animal was treated
with lindane, to simulate dip treatment; this animal was sacrificed
24 h later. After sacrifice, radiolabel was measured in tissues and
in the intestinal contents; radiolabel in exhaled carbon dioxide was
measured on one occasion.
Total recovery of radiolabel was low: approximately 50% in the
study by oral administration and 16-30% in the study by dermal
application. To determine the reason for the losses after oral
administration, the fate of labelled lindane in rumen fluid at 37 °C
was investigated in vitro. About 55% of the radiolabel was
recovered as 14CO2, and 38% remained in the rumen fluid. The
authors stated that their results "clearly show that volatile
14C-labelled organics were evolved from 14C-lindane fortified
rumen fluid," and "that the losses in the in vivo studies were due
to volatile 14C-lindane metabolites." Attempts to trap the
labelled volatile compounds, however, proved unsuccessful.
From 35 to 46% of the radiolabel administered orally was
excreted in the urine over a period of 4 days, and 10-12% of the
dermily applied dose was found in urine over 8-9 days. Much lower
activities were found in faeces, and insignificant amounts in
expired air. The amount of radiolabel in whole milk after oral
administration reached a plateau after 2-3 days, corresponding to a
total concentration of 0.4 ppm (6-8 ppm in the fat) at the lower
dose (2 mg/kg per day) and 3 ppm (about 50 ppm in the fat) at the
higher exposure level (20 mg/kg per day). Significant activity was
also found in the milk after dermal administration, corresponding to
levels of 0.1-0.7 ppm in whole milk.
6.3 Metabolic transformation
The metabolism of lindane is initiated by one of four possible
reactions:
- Dehydrogenation leads to the formation of gamma-HCH;
- Dehydrochlorination leads to the formation of gamma-PCCH;
- Dechlorination leads to the formation of gamma-
tetrachlorohexene;
- Hydroxylation leads to the formation of
hexachlorocyclohexanol.
These compounds must be considered as intermediates, and the initial
reactions are followed by a series of further dehydrogenating,
dechlorinating, dehydrochlorinating, and hydroxylating steps.
A large number of metabolites and end-products occur during the
metabolism of lindane (see section 6.3.2). Detailed descriptions and
schemes of the metabolic pathway of lindane leading to the various
isomeric metabolites have been proposed (Engst et al., 1970, 1977,
1978a, 1979a; Kurihara & Nakajima, 1980). These pathways involve
metabolites that have not as yet been detected. These missing
metabolites may be very unstable compounds which are rapidly
transformed to other intermediates and thus escape detection.
Another possibility is that conjugates are formed which are tightly
bound to proteins via sulfur, and these could be detected only
after the complex is hydrolysed.
The essential steps in the metabolism of lindane are known, and
these are shown, with the main metabolites, in Figure 2 and are
discussed below.
6.3.1 Enzymatic involvement
Lindane is converted by enzymatic reactions, mainly in the
liver. One group of enzymes involved in the biotransformation of
lindane are microsomal, e.g., cytochrome P-450-dependent
monoxygenases. Five groups of male Wistar rats were injected
intraperitoneally with gamma-HCH at 25 mg/kg body weight on four
consecutive days to investigate the induction of cytochrome P-450 in
liver microsomes. gamma-HCH was found to be a 'mixed type' inducer
which mediates the induction of cytochrome P-450 b/e, c and d forms
(Kumar & Dwivedi, 1988). These enzymes are involved in
hydroxylation, dehydrogenation, and dechlorination. Other hepatic
cytosolic enzymes are involved in the dehydrochlorination reaction.
The intermediate metabolites or end-products of the
biotransformation may result as a consequence of the four enzymatic
reactions listed above.
A cytochrome P-450-dependent dehydrogenation reaction was
described in rat liver microsomes in vitro. Incubation of lindane
with rat liver homogenates resulted in the formation of
hexachlorocyclohex-1-ene, and the authors proposed that this
dehydrogenation is an important initial step in the metabolism
leading to the detoxication of lindane (Chadwick et al., 1975).
Stein et al. (1977) found that at least two independent
pathways were involved in the metabolism of lindane. The first is
the possible formation of unstable intermediates, such as
hexachlorocyclohexanol, after an initial hydroxylation leading to
the main metabolite, 2,4,6-trichlorophenol (2,4,6-TCP), and
involving cytochrome P-450. The second pathway includes
dehydrogenation of lindane to 1,2,3,4,5,6-HCCH, subsequent
hydroxylation and dehydrochlorination to 2,3,4,6-tetrachlorophenol.
TCP and tetrachlorophenol are formed in vitro in a ratio of about
2:1.
Fig. 1. Chemical structure of lindane
Chemical formula: C6H6Cl6
Relative molecular mass: 290.8 (290.9)
CAS chemical name: 1alpha,2alpha,3ß,5alpha,6ß-
hexachlorocyclohexane
CAS registry number: 58-89-9
RTECS registry number: GV4900000
Synonym: Hexachlorocyclohexane (gamma-isomer)
According to IUPAC rules, the designation 'benzene
hexachloride' is incorrect; nevertheless, it is still widely used,
especially in the form of its abbreviation, BHC. This is therefore
another common name approved by the ISO. The compound is called
gamma-HCH by the WHO, but gamma-BHC by the FAO (FAO, 1973). The
synonym hexachlorocyclohexane (gamma isomer) is used by the
Environmental Protection Agency and the American Conference of
Governmental Industrial Hygienists in the USA. The definitions of
these different appellations are given in Table 1.
1 See Appendix I
Table 1. Definitions of appellations of lindane
Name Definition Remarks
Lindane product containing not less ISO-AFNOR name for a product
than 99% gamma-HCH (not yet recognized by BSI)
Lindane = gamma-HCH Common name used for
gamma-HCH in the USSR only
gamma-HCH gamma isomer of 1,2,3,4,5,6- ISO-AFNOR common name
hexachlorocyclohexane
gamma-BHC gamma isomer of 1,2,3,4,5,6- ISO BSI common name in
benzene hexachloride English-speaking countries
(recognized by ISO as
synonym of gamma-HCH)
2.1.2 Technical product
Common trade names: A great number of products containing lindane
are on the market; no attempt has been made to
list the hundreds of trade names here (see
Hudson et al., 1984; Hill & Camardese, 1986;
International Register for Potentially Toxic
Chemicals, 1989).
Purity: The FAO (1973) requires that lindane "... shall
consist, essentially, of gamma-BHC as white or
nearly white granules, flakes or powder, free
from extraneous impurities or added modifying
agents and with not more than a faint odour."
The FAO further requires that it contain not
less than 99.0% gamma-HCH and that the
melting-point be at least 112 °C, which is not
depressed when the sample is mixed with an equal
amount of pure gamma-HCH.
In some processes for manufacturing lindane, low levels of
dioxin may be formed (US Environmental Protection Agency, 1985).
Under appropriate manufacturing conditions, however, no
2,3,7,8-tetrachlorodibenzodioxin or 2,3,7,8-tetrachlorodibenzofuran
is detected in HCH, lindane, trichlorobenzene, industrial liquid or
gaseous effluents at the analytical limit of detection of 1 µg/kg
letter from D. Demosay, Rhône-Poulenc, to IPCS dated 17 November
1989.
2.2 Physical and chemical properties
Lindane is a colourless, crystalline solid with either a faint
or no smell (the characteristic smell of technical-grade HCH is
attributed to impurities, particularly heptachlorocyclohexane).
Melting-point: 112.8 °C
Boiling-point: 288 °C
Vapour pressure: 0.434 x 10-5 kPa (3.26 x 10-5 mmHg) at 20 °C
60.6 x 10-5 kPa (45.6 x 10-5 mmHg) at 40 °C
Density: 1.85
Solubility: nearly insoluble in water at 20 °C (10
mg/litre); moderately soluble in ethanol (6.7%);
slightly soluble in mineral oils;
soluble in acetone and in aromatic and
chlorinated solvents
Stability: stable to light, air, heat, carbon dioxide, and
strong acids; dehydrochlorinates in the presence
of alkali or on prolonged exposure to heat with
the formation of trichlorobenzenes, phosgene,
and hydrochloric acid. It is incompatible with
strong bases and powdered metals, such as iron,
zinc, and aluminium, and with oxidizing agents;
can undergo oxidation when in contact with
ozone.
Corrosivity: corrosive to aluminium
Inflammability: not inflammable
n-Octanol/water 3.2-3.7 (see section 4.2.7.1) (Demozay &
partition Marechal, 1972; Dutch Chemical Industry
coefficient Association 1980; American Conference of
(log Pow): Governmental Industrial Hygienists, 1986;
Rhône-Poulenc Agrochimie, 1986)
2.3 Conversion factors
1 ppm = 12.1 mg/m3
1 mg/m3 = 0.083 ppm
2.4 Analytical methods
2.4.1 Sampling
Sampling procedures and methods for preparing samples of
formulations and for analysing residues have been described by
Mestres (1974), the Deutsche Forschungsgemeinschaft (1979), the
Association of Official Analytical Chemists (1980), and Hildebrandt
et al. (1986).
2.4.2 Analytical methods
Products are analysed by a cryoscopic method (Raw, 1970; FAO,
1973; WHO, 1985). Formulated products can be analysed by determining
hydrolysable chlorine (Raw, 1970; FAO, 1973). Since the latter
method is not specific, other methods, such as gas chromatography,
are used to obtain sufficient separation of the HCH isomers.
Residues in food and in soil can be determined after adequate
clean-up by gas chromatography and other chromatographic methods
(Nash et al., 1973; Eichler, 1977; Association of Official
Analytical Chemists, 1980; DeutscheForschungsgemeinschaft, 1983).
The principle of the method is extraction of a sample with organic
solvents (acetonitrile, hexane/acetone, acetone, and others). Fat is
extracted from fatty foods and partitioned between petroleum ether
and acetonitrile by extracting aliquots or an entire solution of
acetonitrile into petroleum ether. Residues are purified by
chromatography on a Florisil colum, and eluted with a mixture of
petroleum ether and ethylether. Concentrated residues are measured
by gas chromatography with electron capture detection.
The method described by the Deutsche Forschungsgemeinschaft
(1979) for fruits and vegetables is based on extraction of samples
with acetone and extraction of the aliquot with dichloromethane. The
residue obtained after evaporation of the solvent is cleaned by
co-distillation, and the distillate is analysed by gas
chromatography with electron capture detection. The limit of
determination depends on the method, the substrate and the sample
size; the lower limit of determination is 0.001-0.01 mg/kg.
Palmer & Kolmodin-Hedman (1972) analysed air samples by gas
chromatography with electron capture detection, and alpha-, beta-,
and gamma-HCH were determined in serum by gas chromatography after a
deproteinization extraction step (Palmer & Kolmodin-Hedman, 1972;
Angerer & Barchet, 1983).
Wittlinger & Ballschmiter (1987) provided an extensive
description of analytical methods for HCHs in air, involving
sampling by adsorption, extraction and preseparation, and
determination by high-resolution gas chromatography. Sampling was
performed by pumping air through a glass-fibre filter and then
through a silica-gel layer, using an internal standard. The sample
was extracted with dichloromethane and the extract evaporated. The
preseparation was done on silica gel, and the aliquot was eluted in
a mixture of hexane and dichloromethane. High-resolution capillary
gas chromatography, electron capture detection and a mass selective
detector were used for determination.
Eder et al. (1987) described three analytical methods for the
determination of HCHs in sediments: moist samples are extracted with
a solvent or a mixture of solvents, concentrated or fractionated and
determined by gas chromatography and electron capture detection.
Greve (1972) described a method for the determination of
organochlorine pesticides in water based on gas chromatography of a
petroleum ether extract after clean-up over Florisil or silica gel.
The limit of detection for lindane is 0.01 µg/litre.
Methods used for the determination of lindane in samples of
soil, animal, and vegetable products in the USSR are described by
Izmerov (1983). These methods are based on extraction with organic
solvents, purification and concentration of the extracts and
determination by gas-liquid chromatography with electron-capture
detection.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Lindane is not known to occur as a natural product.
3.2 Man-made sources
3.2.1 Production levels and processes
3.2.1.1 Manufacturing process
HCH was discovered in 1825, but its insecticidal properties
were first patented only in the 1940s. It has been produced
commercially since 1949.
Technical-grade HCH is synthesized from benzene and chlorine in
the presence of ultra-violet light and comprises 65-70% alpha-HCH,
7-10% beta-HCH, 14-15% lindane (gamma-HCH), approximately 7%
delta-HCH, 1-2% epsilon-HCH, and 1-2% other components. By-products
can be minimized by careful control of the reaction conditions.
Lindane (> 99% gamma-HCH) can be purified by multiple extractions
with methanol.
The extraction of lindane from HCH produces 85%
non-insecticidal HCH isomers, which can be used as intermediates in
the production of trichlorobenzene and hydrochloric acid after
cracking in an integrated installation. Trichlorobenzene is used in
the synthesis of other chemicals (van Velsen, 1986; Rhône-Poulenc
Agrochimie, 1986).
3.2.1.2 World-wide production figures
Lindane is produced in Austria, France, and Spain and in China,
India, Turkey, and the USSR. Before 1984, lindane was also
manufactured in the German Democratic Republic, Poland, Yugoslavia,
Romania, and Hungary; since then, all production has been stopped in
Germany, Japan, the Netherlands, the United Kingdom, and the USA.
Although in most developed countries use of technical-grade HCH
has been prohibited, it is still used elsewhere on a large scale:
total consumption of technical-grade HCH in India in 1986-87 was
approximately 27 000 tonnes (International Atomic Energy Agency,
1988).
3.2.2 Emissions
According to De Bruijn (1979), approximately 0.1% of the
lindane processed reaches the waste-water of a formulating plant.
Treatment of the waste-water, however, leads to solid waste, which
should be incinerated. In the past, it was often dumped in the
environment and could be dispersed from (open) chemical dumping
grounds to more remote soils by the wind.
Lindane enters the environment following application of
lindane-containing pesticides. Emissions can cross national
boundaries in water and air. For instance, the total trans-frontier
flux of lindane into the Netherlands via the surface water of the
River Rhine was approximately 1.8 tonnes per year (average for
1980-83) and that via the River Meuse, 0.2 tonnes per year (Slooff &
Matthijsen, 1988).
3.2.3 Uses
Lindane is a broad-spectrum insecticide, which has been used
since 1949 for agricultural as well as non-agricultural purposes.
Approximately 80% of the total production is used in agriculture
(Demozay & Marechal, 1972), mostly for seed and soil treatment. Wood
and timber protection is the major non-agricultural use. Lindane is
also used against ectoparasites in veterinary and pharmaceutical
products (Rhône-Poulenc Agrochimie, 1986).
3.2.4 Extent of use
Lindane is used worldwide, with the major exception of Japan,
where all uses of HCH were cancelled in 1971 mainly because of
environmental pollution with alpha- and beta-HCH resulting from
extensive use of technical-grade HCH. At that time, no clear
difference was made between the risks presented by the individual
HCH isomers, and lindane was banned as well. In almost all other
countries, lindane is registered for one or more applications,
although the use pattern differs from one country to another.
In 1979, the US Department of Agriculture and the Environmental
Protection Agency summarized the percentage uses of lindane in the
USA as follows: seed treatment 48%, hardwood lumber 23%, livestock
16%, pets 3%, pecans 3%, pineapples 2%, ornamentals 2%, household
1%, cucurbits 1%, forestry 0.5%, and structures 1%. In France and
Germany, 70-80% of all lindane used agriculturally is for soil
treatment, to protect maize and sugar beets, and 15-20% is used for
seed treatment. De Bruijn (1979) reported an estimate of the pattern
of use of lindane in the European Economic Community.
3.2.5 Formulations
Formulation facilities exist in many countries. Lindane is made
in numerous forms, the most important of which are: wettable powders
(up to 90% active ingredient); emulsifiable concentrates (not more
than 20% active ingredient); flowable suspensions (in water);
solutions in organic solvents (up to 50% active ingredient); dusts
and powders (0.5-2% active ingredient); granules and coarse dusts
(3-4% active ingredient); ready-for-use baits; aerosols; and special
formulations for use in human and veterinary medicine (Demozay &
Marechal, 1972).
Lindane dissolved in organic solvents may be used in 'thermal
foggers' in glasshouses or atomized in open areas; such solutions
are appropriate for aerial application (5-10 litres/ha of
formulations containing 5-10% active ingredient). Concentrated
solutions containing an anti-vaporization component have been
applied using an ultra-low volume method at 0.5-1 litre/ha. Various
fumigation preparations for indoor use have been sold, including
fumigation strips, tablets, and smoke generators. These contained
virtually pure lindane to which a small quantity of binding material
was added. Because of its versatility and relatively low acute
toxicity, lindane is often used in mixed formulations with other
insecticides and fungicides (Demozay & Marechal, 1972).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1 Transport and distribution between media
4.1.1 Volatilization
Some of the active ingredient of lindane volatilizes after it
has been applied to control insect pests, especially on leaves.
Starr & Johnson (1968) demonstrated that 20% of an applied dose had
evaporated 96 h after bean plants had been sprayed with lindane at
16 °C. The evaporation was dependent on temperature and on the
humidity of the air.
Some of the lindane that reaches the soil may also vaporize as
degradation products. Cliath & Spencer (1972) showed the presence of
vapours of the metabolite PCCH, which has a vapour pressure
approximately 14 times higher than that of lindane.
In a model test, four soil types, ranging from a loamy sand to
a clay, were treated with 14C-lindane to give a concentration of
10 mg/kg soil; water was then added to the samples, they were
air-dried at 33 °C and at 55 °C, and volatilization was measured by
trapping the vapours. Three cycles of about 14 days each were
followed. Lindane volatilized from the soils only with water, and no
further volatilization occurred after the soils had reached the dry
state. The four soil types were associated with different
volatilization rates: the highest occurred in loamy sand. In the
analysis of vaporized material, unchanged lindane and its
degradation products were not differentiated; however, considerable
degradation of lindane was found in the soils, and PCCH was
identified as a metabolite. At least some of the vaporized material
may therefore have consisted of degradation products (Guenzi &
Beard, 1970).
4.1.2 Precipitation
Evaporation and adsorption to solid particles are important
processes in the distribution of lindane. Reverse processes such as
deposition from the air and remobilization from silt and sediment
also play a part. Buscher et al. (1964) demonstrated that aeration
of aqueous solutions of lindane resulted in a loss of 10% over three
days, which was ascribed to a co-distillation process as it was
greater than could be explained by evaporation alone. MacKay &
Walkoff (1973) confirmed that evaporation is an important process in
the loss of HCH. Lichtenstein & Schulz (1970) found that 16.5% HCH
was lost from a non-aerated aqueous solution in 24 h at 30 °C.
The amount of lindane that is distributed by dry deposition
depends on the nature of the surface above which the organic
components are present. The half-time for dry deposition of HCHs
(height of mixing layer, 1000 m) in the Netherlands was calculated
to be 2-8 days. On this basis, a rough estimate of the annual flux
to soil and water in that country would be 0.5-1.5 tonnes from an
outdoor air concentration of 0.4 ng/m3 (Slooff & Matthijsen,
1988).
4.1.3 Movement in soils
Movement of a substance through the soil profile depends on its
adsorption-desorption characteristics in soil/water systems and, to
some extent, on its volatility in the soil pores and its diffusion.
The adsorption-desorption characteristics of lindane have been the
object of a number of studies (Kay & Elrick, 1967; Mills & Biggar,
1969; Baluja et al., 1975; Portmann, 1979; Wahid & Sethunathan,
1979, 1980; Wirth, 1985), all of which showed that lindane is
strongly adsorbed to organic soil material and weakly adsorbed to
inorganic matter. In the absence of organic matter, the clay content
and free iron oxide are implicated in the sorption of lindane (Wahid
& Sethunathan, 1979). It can be concluded that the mobility of
lindane is very low in soils with a high content of organic matter
but might be higher in soils containing little organic matter.
No consensus has been reached in the literature about the
possibility that lindane can be remobilized by desorption from
polluted soil. Generally, HCH isomers are strongly adsorbed. Under
certain conditions - high concentrations of lindane in highly
permeable soils with a low organic carbon content (< 0.1%) - a
small percentage of the compound may be washed out and reach the
groundwater. Nevertheless, the low rate of transport of lindane
makes the probability that it will reach groundwater low or very low
(Slooff & Matthijsen, 1988).
The diffusion of lindane through soil was investigated by
Ehlers et al. (1969a,b) and by Shearer et al. (1973). Diffusion was
strongly influenced by the water content of the soil, by the bulk
density and by temperature. The diffusion coefficient is nearly zero
in soil containing 1% water, but with a water content of 3%, lindane
is displaced from the adsorbing surface so that the diffusion
coefficient becomes maximal; a further increase in water content
reduces the diffusion coefficient. The diffusion of lindane in soils
can thus vary between a 'vapour' and a 'non-vapour' phase, depending
on the concentration of lindane, the length of time and the water
content of the soil.
Leaching of three formulations of lindane was investigated in a
series of model studies by Heupt (1974) in different soil types. The
test system consisted of 30-cm columns filled with soil to which
lindane formulations were applied at application rates corresponding
to 6 kg of active ingredient per hectare. Rainfall was simulated at
a rate of 200 mm within two days. No lindane was found in the eluate
at the limit of detection of 1 µg/litre. In field tests by Cliath &
Spencer (1971, 1972), lindane was worked into topsoil of two plots
of sandy loam and two of silty clay to a depth of 0-7.5 or 7.5-15
cm, corresponding to an application rate of 21 kg/ha. One of the
plots of each soil type received additional irrigation. Almost no
movement of lindane was found in the dry plots at the end of the
two-year observation period. In the irrigated plots, a broadening of
the lindane-containing zone and downward movement to a depth of 60
cm were observed, especially in sandy loam; in clay soil, lindane
had almost no mobility.
In a series of dissipation studies with 14C-labelled lindane
in soils, coordinated by FAO and the International Atomic Energy
Agency, it was demonstrated that persistent pesticides such as
lindane dissipate much faster in the tropics than in temperate
climates, probably owing to a large extent to volatilization
(International Atomic Energy Agency, 1988), as had been found by
Edwards (1973a,b, 1977). Table 2 summarizes the results of these
studies.
Table 2. Field half-times of gamma-HCH in soils (0-10-cm depth)a
Country Half-time (days)b Time required for
initial loss of 50%
Overall First phase Second phase of radioactivity (days)
India 138 (124-147) 41 (30-50) 188 (83-362) 30-45
Ecuador 150-171 54-60 120-160 40-50
Kenya 5-8 - - 3-4
a Adapted from International Atomic Energy Agency (1988)
b In temperate soils, the mean half-time was 438 (401-1022) days
(Edwards, 1973a, b)
4.1.4 Uptake and translocation in plants
One of the first investigations on the absorption of lindane by
various seeds was reported by Bradbury & Whitaker (1956). Lindane
was taken up from a nutrient solution by the roots of wheat
seedlings at a rate of up to 100 mg/kg (fresh weight) within seven
days. Subsequent investigations demonstrated that uptake by plants
is dependent on a variety of factors.
The influence of soil type was investigated by Bradbury
(1963). Seedlings grown from seed dressed with lindane and planted
in sand had residue levels about two-fold higher than those of
plants grown in compost. A further study was reported of the fate of
14C-lindane in loam and sandy soil and in oat plants grown in
these soils for 13 days. The loam soil was treated with about 7.3
mg/kg active ingredient and the sandy soil with about 3 mg/kg.
Residues were found to be more persistent in loam (53.5% of
radiolabel) than in sandy soil (33.8%), but oats grown in sandy soil
took up more residues than those grown in loam (loam soil: roots,
0.5%; tops, 0.3%; sandy soil: roots, 2.5%; tops, 1.2%).
14C-Lindane was the major constituent of the soil residues soluble
in organic solvents. A major metabolite, which was probably
gamma-PCCH, represented 11% of the organic-soluble radiolabelled
residues in loam soil; 2,4,5-trichlorophenol accounted for 2.7% of
these residues. The authors concluded that the three major factors
that determine the environmental fate of 14C-lindane and other
insecticides are the insecticide itself, its solubility in water and
the type of soil to which it is applied. Compounds with greater
aqueous solubility are more mobile, are taken up by plants to a
greater extent, and appear to be more susceptible to degradation
than compounds less soluble in water. In soils with little organic
matter, insecticide residues are more mobile and hence more
susceptible to volatilization, uptake by plants, and degradation
than in more adsorbent soils such as loam (Fuhremann & Lichtenstein,
1980).
The rate of application to soil was found to be a further
important factor in determining residue levels. Transfer of lindane
from soil into rice plants was almost proportional to the level of
contamination of the soil (Kawahara, 1972), but only at low levels
of contamination. Charnetski & Lichtenstein (1973) reported a good
correlation between the content of 14C-lindane in sand (at up to 6
mg/kg, which is about 12 times the concentration expected after
normal application) and that in pea plants grown for six days; at
concentrations greater than 10 mg/kg of soil, there was no further
increase in the residue levels.
Uptake of lindane after application to leaves is lower than
that resulting from application to soil. In lettuce and endives
treated with 14C-lindane and grown for 21 and 37 days,
respectively, only 4.5-13.9% of the applied radioactivity was found
at the time of harvest, and most of the lindane had evaporated into
the atmosphere (Kohli et al., 1976a).
Differences in residue levels are also dependent on the plant
species. Of a series of edible crops grown in soil containing
lindane at an initial concentration of 5 mg/kg (about 10 times the
normal application rate), carrots had higher levels than beans,
tomatoes, or potatoes (San Antonio, 1959). More lindane was absorbed
from soil with an initial concentration of 2.6 mg/kg by radishes,
turnips, and spinach than by Chinese cabbage (Kawahara et al.,
1971). The amounts of residues of HCH isomers in turnips were
proportional to the initial concentrations of the isomers in the
soil (0.05, 0.1, 0.5, 1, 5, 10, or 50 mg/kg soil). The soil:plant
residue ratios were in the range 10-20:1 (Kawahara & Nakamura,
1972).
The translocation of lindane and its metabolites in plants has
also been investigated in detail (San Antonio, 1959; Bradbury, 1963;
Itokawa et al., 1970; Kawahara, 1972; Kawahara & Nakamura, 1972;
Charnetski & Lichtenstein, 1973; Balba & Saha, 1974; Eichler, 1980;
Korte, 1980). Neither lindane taken up from soil nor its metabolites
were evenly distributed within the plants: Comparatively high
residue levels were always detected in the roots, whereas only small
amounts were translocated into stems, leaves, and fruits. Paasivirta
et al. (1988) showed that in water-plants, lindane concentrations
are similar in roots and leaves.
4.2 Biotransformation
4.2.1 Degradation
4.2.1.1 Degradation under humid conditions
The half-times of lindane found by different investigators vary
considerably, depending on the type of soil to which it is applied
and, possibly, temperature. Lindane incubated in a sandy-loam soil
with a water capacity of 28% and 60% saturation at room temperature
had a half-time of approximately 40 days (Heeschen et al., 1980).
The half-times of lindane in model tests were 4-6 weeks in humid
sand with a high content of organic matter and 30 weeks in sandy
loam (Heupt, 1979). The half-times in aerobic and anaerobic
conditions ranged from 12 to 174 days and 100 to 720 days,
respectively; in aerobic field conditions, the half-time was 88-1146
days (Edwards, 1966; Kohnen et al., 1975; Kampe, 1980; Rao &
Davidson, 1982; MacRae et al., 1984).
Assuming that lindane is not washed out below the level of
ploughed furrows (approximately 20 cm), a half-life of 350 days will
result in persistence of 50% of a dose one year after application
(Slooff & Matthijsen, 1988). One month after double treatment of
potato, beet, and maize crops with lindane, the gamma-HCH content in
sandy loam soil was 0.32 mg/kg in the field occupied by maize and
0.68-0.70 mg/kg in the fields with potatoes and beet. After nine
months, the lindane content in the beet fields had decreased 14
times and that in the maize fields by only 1.3 times (Kovaleva &
Talanov, 1973; see Izmerov, 1983).
4.2.1.2 Degradation under submerged conditions
Half-time values for lindane of a few to about 120 h were
determined after incubation in various submerged soil samples. More
rapid degradation occurred in soils with a high amino acid content,
and the rate also clearly depended on the number of degrading
microorganisms present (Ohisa & Yamaguchi, 1979). The rapidity with
which lindane was degraded under flooded conditions varied in soil
samples from different locations in Japan. Enrichment of the soils
with peptone and exclusion of oxygen increased the degradation rate
(Ohisa & Yamaguchi, 1978a).
Half-time values of 10-30 days were observed in a comparison of
four Philippine rice soils under flooded conditions at a temperature
of 30 °C. Lindane was degraded faster at higher temperatures
(Yoshida & Castro, 1970). In a similar study with five Indian rice
soils at 28 °C, 14C-labelled lindane was degraded at half-times of
between 10 days and more than 41 days. Addition of rice straw
enhanced the degradation (Siddaramappa & Sethunathan, 1975).
Tsukano (1973) found a half-time for lindane of 10-14 days in
soil samples mixed with water. The degradation was almost completely
inhibited after addition of sodium azide to the soils, indicating
that the degradation observed in non-sterilized soils was due to
microbial activity.
4.2.2 Degradation under field conditions
Nash (1983) used a microagroecosystem in which moist fallow
sandy loam was placed in a glass chamber at a depth of 15 cm, plants
were grown in the chamber and lindane was applied to the surface. A
half-time of 1-4 days was found for dissipation of lindane in the
soil.
In April 1954, formulations containing lindane were applied to
a sandy loam soil at rates of 2.25 and 4.5 kg/ha on field plots in
the Rhine valley, and loss of active ingredient was followed during
the subsequent 1.5 years using a biological test method. The
insecticidal activity disappeared rapidly during the following
vegetation period but remained almost constant in winter; further
degradation was observed during the second vegetation period. At the
end of the observation period, 3.5-5.5% of the lindane applied at
2.25 kg/ha remained, and 17-19.5% of that applied at 4.5 kg/ha: the
speed of degradation was therefore greater at the lower application
rate. Degradation was virtually identical when the lindane was
worked into the soil to a depth of 1-2 cm and when it was introduced
to a depth of 10 cm (Schmitt, 1956).
In a field test in Miami, Florida, USA, on silt loam and muck
soils, lindane was applied at the extremely high rates of 11.2 or
112.1 kg/ha. The initial half-time at the lower rate was 15.5 months
in muck soil and 4.75 months in loam soil. Degradation was slower at
the higher rate: the initial half-times were 28.8 months in muck
soil and 11.1 months in loam soil (Lichtenstein & Schulz, 1958a). In
an earlier study on the same field plots with the same application
rates, however, Lichtenstein & Schulz (1958b) found that most of the
material detected chemically was inactive in the bioassay and
therefore did not represent lindane. They concluded that the
breakdown of lindane is faster than it appeared to be using their
analytical method.
In an extensive study, sandy loam, silt loam, and muck soils on
plots in three midwestern states of the USA were treated with
lindane in 1954 at application rates of 1.1, 11.2, and 112.1 kg/ha
to a depth of 15.2 cm. After a 4.5-year follow-up, no lindane was
detected on plots treated with 1.1 kg/ha; but after application at
the higher rates (far in excess of normal rates), about 36% of the
applied dose remained. Two major factors that affect the persistence
of lindane in soils appear to be the amount of organic matter in the
soil and the climatic conditions of the area (Lichtenstein et al.,
1960).
The rates of loss of lindane were calculated by Wheatley (1965)
in 10 long-term field studies in the United Kingdom. When lindane
was applied to the soil surface, there was a 50% loss within 4-6
weeks and a 90% loss within 30-40 weeks. When the lindane was mixed
into the soil, a 50% loss was observed after 15-20 weeks and 90%
within 2-3 years. No lindane was recovered 13 years after the last
application of lindane to a loam soil in Nova Scotia at a rate of
0.84-1.7 kg/ha (Stewart & Fox, 1971). Cliath & Spencer (1971)
treated two test plots in California, USA, with 21 kg/ha, which is
an application rate about 20 times above normal. A half-time of 8
months was found in sandy loam and 10 months in silty clay.
After application of lindane on three test plots of light sandy
soil in the Netherlands for 15 years, to give total amounts of 6.5,
13.0, and 24.3 kg/ha, only 3, 4, and 8% of the applied amount,
respectively, was recovered in layers to a depth of 20 cm (Voerman &
Besemer, 1970). A further follow-up of these plots for four years
showed rapid disappearance on the two locations with the lower
application rates; slower degradation was seen on the plot that had
received the highest application, where lindane was found to a depth
of 40 cm (Voerman & Besemer, 1975). Admixture of a 5% lindane dust
to the top 15-cm layer of a test plot at a rate of 10 kg of active
ingredient per hectare in India led to an initial concentration of
3.2 mg/kg soil. After an observation period of 180 days, 97.7% of
the applied lindane had disappeared. The initial half-time of
lindane in this study was about 30 days (Agnihotri et al., 1977).
The degradation of gamma-HCH was also determined in a variety
of studies in which technical-grade HCH was applied to soils. In
most of these investigations, the application rates were extremely
high, and in some, applications were made once a year for several
years (Lichtenstein & Polivka, 1959; Stewart & Chisholm, 1971;
Shiota & Kanda, 1972; Nash et al., 1973; Jackson et al., 1974;
Suzuki et al., 1975). Under these conditions, gamma-HCH disappeared
slowly from the soils and sometimes persisted for long periods.
The distribution of HCHs was studied in soil treated with
BHC-20 (containing 70% alpha-HCH, 6.5% beta-HCH, 13.5% gamma-HCH,
and 5% delta-HCH) in an agricultural area. The concentrations
changed with time after application; the mean value for gamma-HCH
was 16 µg/kg. The organic carbon content of the soil appeared to be
of primary importance, and the significant decrease in isomer
concentration observed with greater soil moisture was attributed to
microbial degradation, which is favoured by these conditions
(Chessells et al., 1988).
Kathpal et al. (1988) studied the behaviour of a formulation
consisting of a mixture of five HCH isomers in paddy soils under
subtropical conditions in India. The recommended application rate of
2.5 kg active ingredient per hectare and a rate of 5.0 kg/ha were
used. Gamma-HCH had dissipated by 50-63% within three months under
paddy, and average residues in soil at harvest were 0.3-0.34 mg/kg.
Dissipation after nine months (two crop seasons) was 98%. The
persistence under paddy in this study was fairly high, probably
owing to the anerobic conditions, which slow microbial degradation.
The paddy plants absorbed gamma-HCH from the soil: the residues at
harvest were about 1.0 mg/kg in plants and 0.03-0.06 mg/kg in seeds.
4.2.3 Hydrolytic degradation
Determination of the hydrolytic stability of a substance
provides an indication of whether this process can contribute to the
disappearance of the substance from the aquatic environment and, to
a certain extent, from soil. In a model experiment, the half-time of
lindane at 22 °C was 47.9 h at pH 9 and 100.7 h at pH 7; no
measurable hydrolysis occurred at pH 5 (Heupt, 1983).
4.2.4 Photolytic degradation (laboratory studies)
As lindane has measurable volatility and can be found at low
levels in air, its degradation in sunlight has been studied.
Carbon dioxide was formed after 14C-lindane was adsorbed onto
silica-gel plates at a concentration of 33 µg/kg and irradiated with
artificial sunlight (> 290 nm) in the presence of air; 6.4% of the
carbon was oxidized within 17 h. This photo-induced oxidation was
enhanced when the lindane was exposed to pure oxygen during
irradiation (Kotzias et al., 1981). No measurable degradation (less
than 0.5%) was observed 2000 h after exposure of lindane to the
light of a Xenon lamp in a Xenotest 150 on the wall of a quartz
vessel (solid phase). When the irradiation was performed in aqueous
solution, about 4% of the applied lindane was degraded after 2000 h.
The main degradation product was PCCH (Gardais & Scherrer, 1979).
Irradiation of lindane with ultra-violet light (254 nm) is
obviously more effective for degradation of the compound than
irradiation with light of longer wavelengths. Hamada et al. (1981,
1982) found rapid degradation of lindane in both the crystalline
state and in solution with 2-propanol under these conditions, with
PCCHs and TCCHs as reaction products. Eichler (1977) also found
rapid degradation of lindane in the solid or gaseous form and in
aqueous solution in the presence of ultra-violet irradiation, with
half-times of 12-24 h for the first two phases and 1-2 days for the
latter two.
4.2.5 Biodegradation in water
In a study of the degradation of lindane in a biological
purification plant, 75% of the compound was degraded within 6 h
(Eichler et al., 1976).
Newland et al. (1969) investigated the degradation of gamma-HCH
in simulated lake impoundments. Sediments from Lake Tomahawk,
Wisconsin, USA, were added to solutions of 5 mg/litre 14C-labelled
lindane and equilibrated for 18 h, and aerobic and anaerobic tests
were run for approximately 88 days. Initially, about 45% of the
applied lindane was adsorbed to the sediment (200 g per 3-litre
solution). Under aerobic conditions, about 16% of the added lindane
was degraded by the end of the observation period, whereas more than
97% was degraded under anaerobic conditions. When lindane
degradation was tested in samples of surface water from two
different regions for periods of 3, 6, or 12 weeks, decreases of up
to 90% of the initial concentration were found. Most of the lindane
was metabolized by microorganisms in the sediments: In samples of
sediment and water autoclaved prior to treatment and incubation, up
to 95% of the applied lindane was still present (Oloffs et al.,
1973).
In a field test in rice fields in the Camargue, France, a
formulation containing lindane was applied at a rate that resulted
in an initial concentration in water of 54.8 mg/m3. Rapid
disappearance was observed, for a half-time of about 1.5 days, and
within 10 days the concentration had dropped to the background value
of 0.08 mg/m3 (Podlejski & Dervieux, 1978).
The degradation of lindane was also tested in the water of a
drainage canal in the Holland Marsh, Ontario, Canada, in distilled
water, and in both water types after sterilization. The half-time of
lindane in the natural water was about six weeks, but a very low
disappearance rate was seen in the distilled and sterilized water,
indicating the importance of microbial action for degradation of
lindane in water (Sharom et al., 1980).
An aquatic model ecosystem, with pond water, sludge, aquatic
plants, and fish, was used to study the decomposition and migration
of lindane. In water without hydrobionts, the half-time was 50 days.
When sludge and aquatic plants were present, the half-time was 34
days, and that in the presence of fish was 2 days (Vrochinsky, 1973;
see Izmerov, 1983).
4.2.6 Microbial degradation
A variety of experiments on the degradation of lindane was
performed with mixed populations of the microorganisms that occur in
different types of soil, in aquatic sediments (Newland et al., 1969;
Benezet & Matsumura, 1973), and in other types of soil under
aerated, submerged, and strictly anaerobic conditions (Macrae et
al., 1967; Yule et al., 1967; Kohnen et al., 1975; Mathur & Saha,
1975, 1977; Tu, 1975; Haider, 1979). The fact that lindane was
removed faster from non-sterile than from autoclaved soil
demonstrated that its degradation in soil is due to microbial
activity (Macrae et al., 1967; Kohnen et al., 1975).
The microorganisms shown by screening experiments to be capable
of metabolizing and degrading lindane are as follows (Tu, 1976;
Jagnow et al., 1977):
Bacteria Fungi Algae
Arthrobacter sp. Penicillium sp. Chlamydomonas sp.
Bacillus sp. Rhizopus sp. Chlorella sp.
Citrobacter sp.
Clostridium sp.
Enterobacter sp.
Micromonospora sp.
Pseudomonas sp.
Thermoactinomycetes sp
In addition, lindane was metabolized in cell-free preparations of
Clostridium sp. in vitro (Heritage & Macrae, 1977a; Ohisa et
al., 1980).
Lindane is degraded by soil microorganisms under aerobic as
well as under anaerobic conditions, but anaerobic conditions are the
most favourable for its metabolism (Newland et al., 1969; Haider &
Jagnow, 1975; Vonk & Quirijns, 1979). In an anaerobically grown
culture of Clostridium sphenoides supplemented with lindane at 5
mg/litre, none was found, even after 2 h (Heritage & Macrae, 1979).
Several species of soil bacteria that have been shown to degrade
lindane effectively are described in detail in section 6.6.2.
In field studies in which gamma-HCH was applied at excessive
doses, it was degraded more slowly than at doses closer to those
used for normal agricultural applications. Introduction of HCH at up
to 224 kg/ha, corresponding to 33.6 kg gamma-HCH per hectare,
exceeded the degradation capacity of soil microorganisms for a long
period (Nash et al., 1973). In addition, the analytical methods used
might have resulted in an overestimation of the actual gamma-HCH
concentration, as concluded by Lichtenstein & Schulz (1958b).
Therefore, studies in which technical-grade HCH is applied,
especially at excessive rates, cannot be used to evaluate the
degradability of lindane in soil.
4.2.7 Bioaccumulation/Biomagnification
4.2.7.1 n-Octanol/water partition coefficient
The n-octanol/water partition coefficient (Pow) of lindane
was determined in several studies, with good agreement, covering the
narrow range of log Pow = 3.29-3.72 (Kurihara et al., 1973;
Platford, 1981; Darskus, 1982; Geyer et al., 1982; Hermens &
Leeuwangh, 1982; Geyer et al., 1984). These values indicate that
lindane can become enriched in lipid-containing biological
compartments.
4.2.7.2 Aquatic environment
The bioconcentration factor for lindane was found to be
dependent on the concentration to which the organisms, such as
algae, crustaceae, and fish, were exposed (Bauer, 1972; Ernst, 1975;
Schimmel et al., 1977; Trautmann & Streit, 1979; Marcelle & Thome,
1983): The highest bioconcentration factors were seen with the
lowest exposure concentrations. For example, Marcelle & Thome (1983)
determined the residues of lindane in brain, liver, and muscle of
the gudgeon (Gobio gobio) after exposure to concentrations of
0.22-142 µg/litre lindane in water. At the lowest concentration, the
bioconcentration factors in brain, liver, and muscle were about 600,
200, and 100, respectively, but they decreased to values of less
than 50 at higher concentrations.
Mouvet (1985) transplanted the freshwater aquatic moss
Cinclidotus danubicus from an uncontaminated area to a river that
received the effluent from an insecticide factory and determined
gamma-HCH concentrations in water and moss 13, 24, and 51 days after
the transplant. A three-fold increase in the gamma-HCH level was
found, with a bioconcentration factor of 294.
In a variety of aquatic organisms exposed to contaminated
water, the bioconcentration factor for lindane ranged from 13 to
1000 on a wet weight basis (Table 3).
Table 3. Bioconcentration factors of lindane in laboratory
experiments; test organisms were exposed to contaminated
water for the specified time
System Exposure Exposure Bioconcentration Reference
time concentration factora
(µg/litre)
Algae
Cladophora sp. up to 80.0 180 (d) Bauer (1972)
48 h 3.9 341 (d)
Nitzschia 24 h 6.1 1500-4700 (v) Trautmann & Streit
actinastroides 4400-12 400 (d) (1979)
Molluscs
Aplysia punctata 3-6 days 9000 201-436 (w) Chabert & Vicente
(1978)
Mya arenaria 5 days 5 40 Butler (1971)
Mercenaria 5 days 13
mercenaria
Mytilus edulis ns 2.61 74 (w) Ernst (1975)
0.02 242 (w)
Mytilus edulis ns 2-5 139 (w) Ernst (1979)
Venerupis japonica 3 days 1 121 (ns) Yamato et al.
(1983)
Annelidae
Lanice conchilega ns 2-5 1240 (w) Ernst (1979)
Crustacea
Penaeus duorarum 96 h 0.23 143 (ns) Schimmel et al.
Palaemonetes pugio 96 h 1.0 80 (ns) (1977)
Table 3 (contd)
System Exposure Exposure Bioconcentration Reference
time concentration factora
(µg/litre)
Insects
Sigara striata and 1 day 10 70-100 Kopf & Schwoerbel
Sigara lateralis (1980)
Fish
Lagodon 96 h 23.0 287 (ns) Schimmel et al.
rhomboides (1977)
Cyprinodon 96 h 108.7 727 (ns)
variegatus
Leuciscus idus ns 10-500 765 (ns) Sugiura et al.
Cyprinus carpio 281 (ns) (1979)
Salmo truttafario 442 (ns)
Poecilia 938 (ns)
reticulata
Poecilia 4 days 1 697 (ns) Yamato et al.
reticulata (1983)
Salmo gairdneri 27 days 30-290 319 Ramamoorthy
(1985)
a Calculated on the basis of: wet weight (w), dry weight (d), volume (v); ns, not specified
Another approach to the study of the bioconcentration of
lindane is the use of systems that simulate natural conditions,
taking into account sedimentary absorption processes and the
influence of contaminated food. The bioconcentration factors for
brine shrimp, mosquito larvae, and the brook silverside
(Haludesthes sicculus sicculus) exposed to lindane applied to the
sand of a test aquarium were 95, 220-383, and 600-1613,
respectively, depending on the food chains used (Matsumura &
Benezet, 1973). Marcelle & Thome (1984) investigated the
bioconcentration of lindane in the gudgeon (Gobio gobio) in
relation to the route of exposure. Fish were exposed either to
contaminated water alone or additionally to contaminated food. After
18 days, the group fed contaminated food had a 2.5-fold higher level
of lindane residues in liver than the group exposed to contaminated
water alone. Within three days after cessation of exposure, 98.4% of
the lindane residues had been excreted.
The uptake, transport, and bioconcentration of lindane were
also studied in a freshwater food chain, which consisted of
Chlorella sp., Daphnia magna, and Gasterosteus aculeatus
(algae-crustacea-fish). Uptake from water was more rapid than uptake
from food and depended on the duration of the experiment and the
feeding rate. The increase in lindane residues in the last link of
the food chain (fish) was not directly proportional to the
concentration found in the primary links (Hansen, 1980).
4.2.7.3 Terrestrial environment
The bioconcentration of lindane was investigated in a
terrestrial food chain, which consisted of soil, barley,
caterpillar, and quail. Doses up to 400 times the standard
agricultural dose (50 and 200 mg/kg soil) were applied to the soil.
Although lindane was found in all of the links of the food chain,
the concentrations decreased progressively (Dugast, 1980).
Feeding hens diets containing lindane at 0.05, 0.15, or 0.45
mg/kg for 20 weeks resulted in constant values of 0.01, 0.03, and
0.09 mg/kg of eggs, demonstrating a dose-related accumulation of
lindane (Cummings et al., 1966).
Several studies are available on the bioconcentration of
lindane in rats. After seven rats had received daily doses of 2 or 4
mg/kg body weight for up to 12 weeks, gamma-HCH was found at a
concentration of about 8 mg/kg in adipose tissues of the group that
had recived the high dose (Jacobs et al., 1974). In another
experiment, four generations of rats were fed a diet containing 20%
fat and a mixture of insecticides including lindane at levels of
0.07-0.8 mg/kg. Even in the F3 generation, the levels of gamma-HCH
residues in adipose tissues were of the same order of magnitude
(< 0.05-0.56 mg/kg) as those of lindane in the diet (Adams et al.,
1974). No accumulation occurred, even in four consecutive
generations.
Accumulation factors have been determined from feeding studies
in rats (Fitzhugh et al., 1950; Oshiba, 1972; Baron et al., 1975;
Suter et al., 1983). In comparison to the concentration of lindane
in the diet, the highest reported bioconcentration factor was about
2 for adipose tissue. The average bioconcentration factor for
adipose tissues in rats derived from all these studies is 1; the
bioconcentration factors for other tissues are considerably lower.
4.2.7.4 Bioconcentration in humans
Geyer et al. (1986) examined data on environmental chemicals
detectable in adipose tissue and/or breast milk of
non-occupationally exposed humans and concluded that, in
industrialized countries, more than 90% of human exposure to HCHs
originates from food. Mean concentrations of gamma-HCH in human
adipose tissue in Czechoslovakia, the Federal Republic of Germany,
and the Netherlands were 0.086, 0.024-0.061, and 0.01-0.02 mg/kg,
respectively, on a fat basis. The mean bioconcentration factor,
calculated on the basis of the concentration in the diet (2.3, 5.0,
and 0.62-0.9 µg/kg, respectively) and levels in adipose tissue, was
18.6 ± 9 on a lipid basis (range, 10.4-32.5). Greve & Wegman (1985)
found an accumulation factor (adipose tissue/blood) of 70 for
gamma-HCH in humans.
4.2.7.5 Field studies
The bioconcentration of lindane was investigated by
environmental monitoring in aquatic ecosystems. The residue levels
found in different organisms were related to the environmental
background levels, and these data were used to calculate the
bioconcentration factors.
The bioconcentration factor for gamma-HCH in sea water and
bladder wrack (Fucus vesiculosus) in the Husum estuary and the
adjacent North Friesian Wadden Sea in the Netherlands was about 150
(Herrmann et al., 1984). On the basis of the data given in section
5.1.5.2 on the occurrence of gamma-HCH in muscle and fat of bream
collected in the River Elbe, a bioconcentration factor of 10 000 to
50 000 was calculated (Arbeitgemeinschaft für die Reinhaltung der
Elbe, 1982).
Frisque et al. (1983) studied the accumulation of lindane by
bryophytes (Cinclidotus danubicus and C. nigricans) in the Meuse
River and found a concentration factor of 300-350. The average level
in the river was 0.067 µg/litre. Hartley & Johnston (1983) found a
bioconcentration factor for the freshwater clam Corbicula
manilensis of 2610 on a lipid basis; and Cosson Mannevy & Marchand
(1980) found a mean factor of 26 198 (on a dry-weight basis) in
Mytilus edulis.
On the basis of the concentrations of gamma-HCH in sea water,
sediments, and fish from the Mediterranean Sea, El-Dib & Badawy
(1985) calculated a bioconcentration factor of about 1000 (on a
lipid basis). Tanabe et al. (1984) reported bioconcentration factors
for total HCHs in a trophic chain in the western North Pacific. As
the contribution of gamma-HCH to the residue levels was determined,
the bioconcentration factors for this isomer can be estimated to be
about 40, 40, 100, and 1850 for zooplankton, myctophid, squid, and
dolphin, respectively.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
An average of 0.23 ng/m3 (0.039-0.68 ng/m3) gamma-HCH was
found in 24 samples of air taken from over the western Pacific, the
eastern Indian Ocean, and the Antarctic Ocean (Tanabe et al., 1982).
Levels of gamma-HCH in the air of various regions of the USA
were within a similar range (US Environmental Protection Agency,
1976). The levels were below 1 ng/m3 in most samples, and values
up to 16.2 ng/m3 were found in only two regions.
Gamma-HCH was found at an average concentration of 0.14 ng/m3
in the air of unpolluted areas in the Federal Republic of Germany in
1972; in polluted areas (the Ruhr), a level of 0.8 ng/m3 was found
in 1976/77 (Deutsche Forschungsgemeinschaft, 1983; Hildebrandt et
al., 1986). It occurred at 0.52-11 ng/m3 in a location with heavy
traffic near Ulm in the Federal Republic of Germany and at 0.18-1.1
ng/m3 in a rural area. The authors concluded that the
concentrations in the lower troposphere under different
meteorological conditions reflect regional input and long-range
transport (Wittlinger & Ballschmiter, 1987).
The average concentration of gamma-HCH in 55 air samples taken
near Delft, the Netherlands, in 1979-80 was 0.36 ng/m3 (maximum,
3.4 ng/m3); in three other locations in the Netherlands, average
levels of 0.2-0.9 ng/m3 were found. In six houses built on former
dumping grounds, the average concentration of gamma-HCH was 6
ng/m3 (range, 1-14 ng/m3), whereas in the space beneath the
floor the level was below the detection limit (1 ng/m3). Outdoor
concentrations in this area were 0.3-0.4 ng/m3. In another study,
the concentrations of gamma-HCH in the space beneath the floor of
houses were 90 ng/m3. Much higher levels were found in houses
treated with lindane-containing products for the control of woodworm
or of long-horned beetle. Peak levels of 51-61 µg/m3 were found
four weeks after application; these decreased gradually to 8-24
µg/m3 after 10 weeks. After indoor application of lindane for wood
preservation, levels of 50 µg/m3 were common, with peak levels of
up to 100 mg/m3 (Sloof & Matthijsen, 1988).
5.1.2 Water
5.1.2.1 Rain and snow
Levels of 0.001-0.005 µg/litre were found in rain-water
analysed in the Federal Republic of Germany in 1970-72 (Mestres,
1974); in 1983, gamma-HCH was found at an average of 0.06 (range,
0.01-0.18) µg/litre in rain-water near de Bilt, the Netherlands
(Slooff & Matthijsen, 1988).
Strachan et al. (1980) found traces of gamma-HCH in 17 samples
of snow collected from the Canadian side of the Great Lakes in 1976
and 5-12 ng/litre in 81 samples of rain-water collected in 1976 and
1977.
5.1.2.2 Fresh water
Water samples from selected rivers in Yorkshire, United
Kingdom, analysed for gamma-BHC in 1966 contained levels of
0.001-0.18 µg/litre; in 1968, however, the highest value was 0.622
µg/litre. Water samples from six other rivers, also analysed in
1968, contained mean values of 0.011-0.030 µg/litre, and the highest
levels found were 0.020-0.098 µg/litre (Lowden et al., 1969).
River water samples analysed in 1969-72 in Belgium, France, the
Federal Republic of Germany, the Netherlands, and Italy contained
less than 0.1 µg/litre and usually less than 0.05 µg/litre. In 1826
water samples taken at 99 sites in the Netherlands in 1966-77, the
highest concentrations of gamma-HCH were found in those from the
River Rhine and its tributaries. The concentrations of gamma-HCH
over the period 1969-74 varied between 0.01 and 0.4 µg/litre, but in
1974-77, the concentrations were all below 0.1 µg/litre (Mestres,
1974). Gamma-HCH concentrations have been measured in the Rivers
Rhine, Meuse, and West-Scheldt and in other surface waters in the
Netherlands since 1969. Since 1974-75, the levels have been below
0.05 µg/litre in the Rhine and about 0.05 µg/litre in the
West-Scheldt; in the Meuse, the concentrations were more variable
and ranged from 0.01 to 1.0 µg/litre. In agricultural and
horticultural areas, the levels were 0.01-1.0 µg/litre, with
incidental peaks up to 0.5 µg/litre, probably due to use of lindane.
The average concentration of dissolved gamma-HCH in the Meuse-Rhine
estuary in 1974 was 20 ng/litre and that of suspended gamma-HCH
between 1 and 20 ng/litre. In coastal waters of the Netherlands, the
concentration of dissolved gamma-HCH was 0.9-4.6 ng/litre and that
of bound gamma-HCH, 3.1-8.7 ng/litre (Sloof & Matthijsen, 1988).
A sampling trip along the River Rhine, from Rheinfelden in
Switzerland to Rotterdam in the Netherlands, proved that the source
of alpha-, beta-, and gamma-HCH was located in the upper reaches of
the River. In the Meuse, lindane levels in 1969-77 were all below
0.1 µg/litre (Wegman & Greve, 1980). In an extensive programme in
1982 to determine pollution in Dutch surface waters at 45 locations,
gamma-HCH concentrations were generally between 0.01 and 0.1
µg/litre (Wammes et al., 1983).
The mean concentration of gamma-HCH in the River Elbe, from
Schnackenburg to the North Sea, in 1981-82 was 0.021
(< 0.001-0.051) µg/litre; during February-November 1988, the
concentrations were 0.005-0.044 µg/litre (Arbeitsgemeinschaft für
die Reinhaltung der Elbe, 1988). More figures for Germany are given
by Wirth (1985). Gamma-HCH was found at three locations in the River
Rhine at 0.02 µg/litre and in six side-rivers at 0.01-0.06 µg/litre.
These levels had decreased markedly since 1975 (Landesamt für Wasser
und Abfall, 1988).
5.1.2.3 Sea water
Atlas & Giam (1981), Bidleman & Leonard (1982), Oehme & Stray
(1982), and Oehme & Mano (1984) analysed water from such widely
differing areas as the Eniwetok Atoll in the North Pacific, the
Arabian Sea, the Persian Gulf, the Red Sea, Lillestrøm, Norway, Bear
Island, and Spitzbergen in the Arctic Ocean. The gamma-HCH
concentrations were in the range 0.01-0.05 ng/litre, except in the
Arabian Sea, the Persian Gulf, and at Lillestrøm, where levels up to
0.67 ng/litre were found (Slooff & Matthijsen, 1988). Levels of
0.0001-0.004 µg/litre gamma-HCH were measured in the Western
Pacific, the Eastern Indian, and Antarctic Oceans (Tanabe et al.,
1982). No gamma-HCH was found in 60 water samples from the Japan Sea
and Pacific Ocean (detection limit, 0.1 µg/litre) (A. Hamada, letter
to M. Mercier, dated 28 July 1989; T. Onishi, letter to M. Mercier,
dated 24 July 1989). The levels detected in water from the North Sea
and the Arctic Sea are of the order of 0.001-0.02 µg/litre (Deutsche
Forschungsgemeinschaft, 1983). The maximal level of gamma-HCH in
North Sea water in 1972 was 0.028 µg/litre; 5-10% of the samples
contained gamma-HCH (Mestres, 1974). The level of gamma-HCH in
surface-water of the North Sea in June-July 1986 ranged from 1.0 to
4.0 ng/litre. The highest concentrations were found close to the
coast (Umweltbundesamt, 1988-89).
5.1.3 Soil
Traces of gamma-HCH are transmitted to soil by precipitation;
the resulting contamination is generally below the limit of
detection (0.0001-0.001 mg/kg). Application of lindane in
agricultural areas can result in higher concentrations: levels in
some German districts were mainly in the range 0.001-0.01 mg/kg, but
in certain fields up to 0.6 mg/kg was found (Fricke, 1972).
Edelman (1984) analysed 96 samples of the upper 10 cm of soil
from 38 natural reserves in the Netherlands for gamma-HCH: 59
samples contained < 1 µg/kg, 21 contained 1-10, 9 had 10-20, and 7
had 20-80 µg/kg (Slooff & Matthijsen, 1988). In the National Soils
Monitoring Program of the US Environmental Protection Agency (Carey
et al., 1979), several thousand samples from cropland sites were
analysed for residues; no gamma-HCH residues were detected in more
than 99% of the samples. In the Ukraine, however, 36 of 136 soil
samples taken at various locations contained lindane at levels of
0.1-5 mg/kg (Talanov, 1977; see Izmerov, 1983).
In a study on the application of lindane dust by aircraft on
mosquito breeding sites at 1.3 kg/ha, the gamma-HCH content of the
soil was 1 mg/kg; after one year, the level was 0.01 mg/kg
(Vroschinsky, 1973; see Izmerov, 1983).
5.1.3.1 Sediment
Gamma-HCH was present in three of six samples of sediment taken
from Nyumba Ya Mungu Lake in the United Republic of Tanzania in
1986, at a concentration of 1-4 µg/kg dry weight (Paasivirta et al.,
1988).
Martin & Hartmann (1985) found gamma-HCH at levels above the
detection limit (5 µg/litre) in less than 4% of 117 samples of
sediment taken in 1980-82 from riverine and pothole wetlands in
north-central USA. In less than 4% of the samples, gamma-HCH was
present at above the detection level of 5 µg/kg.
In Japan, gamma-HCH was found in 9 out of 60 samples of
sediment at a concentration of 10 µg/kg in 1974 (A. Hamada,letter to
M. Mercier, dated 28 July 1989; T. Onoshi, letter to M. Mercier,
dated 24 July 1989).
The median levels of gamma-HCH in sediments from eight rivers,
harbours, and sites close to dumping places in the Netherlands were
15-342 µg/kg dry matter (Slooff & Matthijsen, 1988).
5.1.3.2 Dumping grounds and sewage sludge
The soil at various locations in the Netherlands is polluted
with HCHs as a result of spillage during production, storage, and
handling of this chemical during the 1950s. The concentrations found
range up to a few thousand milligrams of HCHs per kilogram of dry
soil. Further pollution has been caused by the dumping of chemical
waste, sometimes in order to level the ground; this waste can be
dispersed from dumping areas by leaching or wind erosion. In certain
polluted areas, high concentrations of HCHs (mainly alpha- and
beta-HCHs) were found at depths of more than 2 m below ground level.
In 18 locations in the Netherlands, the average concentrations of
gamma-HCH in sewage sludge in 1981 were 8-50 µg/kg dry matter.
Groundwater was also found to be polluted, but this was restricted
to the vicinity of the production areas; horizontal transportation
of HCHs in groundwater appeared to be limited (Slooff & Matthijsen,
1988).
Fieggen (1983) found gamma-HCH in sewage sludge at mean values
of 25 µg/kg dry matter in 1975, 43 µg/kg in 1978, and 12 µg/kg in
1981.
5.1.4 Drinking-water, food and feed
Although in most countries nowadays only lindane is used,
residues of alpha- and beta-HCH can still be found in crops and
animal products originating from regions where technical-grade HCH
(containing all of the HCH isomers) is still in use.
5.1.4.1 Drinking-water
Gamma-HCH was found at 0.0001-0.001 µg/litre in water from 19
lakes in Germany and at levels below 0.001 µg/litre (0.0001-0.0008
µg/litre) in the drinking-water derived from them (Bernhardt &
Ziemons, 1974). In the USA, only 3% of drinking-water samples
examined contained gamma-HCH, in a range of 0.001 to about 0.1
µg/litre (US Environmental Protection Agency, 1976). In Ottawa,
Canada, drinking-water samples collected in 1976 contained 0.4-11
ng/litre (Williams et al., 1978).
5.1.4.2 Cereals, fruits, pulses, vegetables, and vegetable oil
The large body of information on gamma-HCH residue levels in
crops grown and treated with this chemical according to Good
Agricultural Practice has been reviewed comprehensively by the
FAO/WHO Joint Meeting on Pesticide Residues and summarized in
published monographs (FAO/WHO, 1967, 1968, 1969, 1970, 1974, 1975,
1976, 1978, 1980).
In samples of ready-to-eat foods collected from 30 markets in
27 US cities in 1966-67, gamma-HCH levels were 0.003-0.009
(occasionally 0.06) mg/kg in grains and cereals, 0.002-0.027 mg/kg
in garden fruits, 0.001-0.005 mg/kg in potatoes, 0.002-0.007 mg/kg
in leafy vegetables, and 0.004-0.012 mg/kg product in oils, fat, and
shortening (Martin & Duggan, 1968). In 1967-68, residues of
gamma-HCH were found at 0.002-0.006 in leafy and root vegetables, at
0.002-0.003 in garden fruits, and at 0.029-0.085 mg/kg product in
oils, fat, and shortening (Corneliussen, 1969).
In monitoring studies carried out on grain in the Federal
Republic of Germany at one-year intervals since 1975, gamma-HCH
residues in wheat and barley were 0.001 mg/kg or less (Ocker, 1983).
More than 800 samples of cereal and cereal products analysed in
Germany in 1975-78 and 1979-83 contained mean concentrations of
0.0009-0.04, but cereal products had up to 0.11 mg/kg. The mean
concentration of gamma-HCH in 200 samples of wheat and rye collected
in 1986 and 1987 was 0.06 mg/kg, with a maximum of 0.3 mg/kg
(Umweltbundesamt, 1988-89).
Of 281 samples of wheat analysed for the presence of gamma-HCH
in the United Kingdom between October 1978 and April 1979, 71
contained levels in the range 0.002-0.04 mg/kg. Gamma-HCH was also
found in one sample of polished rice from Spain, at a concentration
of 0.008 mg/kg (Steering Group on Food Surveillance, 1982).
Gamma-HCH was found in 16% of samples of imported maize in the
United Kingdom in the range none detected to 0.007 mg/kg, and in 28
samples of different types of pulses at none detected to 0.05 mg/kg.
Of retail cereal products, only bran and wheat contained detectable
levels (0.01 mg/kg product) of gamma-HCH in 1982 (Steering Group on
Food Surveillance, 1986). In 1986-87, 31 of 142 samples of pulses
contained residues; in nine, levels of < 0.01-0.4 mg/kg were found.
Peanut butter and vegetable oils contained 0.01 mg/kg (Steering
Group on Food Surveillance, 1989).
About 80-90% of samples of fruit, potatoes, and other
vegetables analysed in the Federal Republic of Germany contained no
detectable residues of gamma-HCH (Weigert et al., 1983). The
remaining 10-20% had mean levels up to 0.01 mg/kg, with no
significant difference between 360 samples originating from
conventional agriculture and 360 samples from 'alternative'
agriculture (Vetter et al., 1983). In 1976-78 and 1980, the mean
concentrations of gamma-HCH were < 0.001-0.002 mg/kg product in
more than 400 samples of fruit, potatoes, and other vegetables. In
the Netherlands, residues in fruit and vegetables were generally in
the range 0-0.1 mg/kg, although some leafy crops, such as endive,
lettuce, celery, and leek, contained levels up to 5 mg/kg. Samples
of wheat contained only 0-0.05 mg/kg, with a few measurements up to
0.2 mg/kg (FAO/WHO, 1978). In France, gamma-HCH residues were found
in wheat at 0.01-0.02 mg/kg, and at low levels in other commodities,
such as carrots and endives (Laugel, 1981). Engst et al. (1967)
found that the gamma-HCH content of carrots grown from seed treated
with this compound decreased continuously during the first 120 days.
At normal harvesting time, the early varieties contained 3-6 mg/kg
product, the mid-season varieties about 2 mg, and the late
varieties, 0.4 mg/kg. When the carrots were harvested after 200
days, 0.3-0.7 mg/kg was present (independently of variety). Even
after 6 months' storage, low residues were still present.
5.1.4.3 Meat, fat, milk, and eggs
Martin & Duggan (1968) found gamma-HCH at levels of 0.09 mg/kg
in dairy products and at 0.01-0.03 mg/kg (with a peak of 0.374
mg/kg) in samples of meat, fish, and poultry collected from 30
markets in 27 cities in the USA in 1966-67. Residue levels in
samples of meat, fish, and poultry in 1967-68 were 0.003-0.026 mg/kg
(Corneliussen, 1969). No gamma-HCH or levels of 0.01-0.1 mg/kg were
found in 99% of samples of cow's milk and manufactured milk products
from Illinois (USA) (Wedberg et al., 1978). In milk samples
collected during Spring 1983 from 359 bulk transporters,
representing 16 municipalities of Ontario, Canada, gamma-HCH was
found in 68% of the samples at a mean concentration of 4.0 µg/kg
butter fat (Frank et al., 1985). Six samples of cow's milk from six
locations in Switzerland contained 3.0-5.1 mg/kg on a fat basis
(Rappe et al., 1987).
In about 25% of 976 samples of meat and poultry products
(including eggs) collected in the United Kingdom in 1984-86,
gamma-HCH was present at a mean concentration of 0.01-0.02 mg/kg.
The highest level, 3.7 mg/kg, was found in lamb. Processed meat and
poultry products (631 samples collected in 1985-87) contained mean
concentrations of 0.01-0.06 mg/kg product. About half of 849 samples
of retail milk and dairy products collected in 1984-87 contained
gamma-HCH at concentrations of 0.01-0.03 mg/kg; the highest level,
0.7 mg/kg, was found in milk (Steering Group on Food Surveillance,
1989). Imported meat products were also analysed in the United
Kingdom for the presence of alpha-, beta-, and gamma-HCH. No
detectable residue of gamma-HCH was found in beef or pork products:
processed pork contained none detectable to 0.03 mg/kg. Processed
poultry contained none detectable to 0.04 mg/kg (Steering Group on
Food Surveillance, 1986). In 1967-70, in the Ukraine, gamma-HCH was
found in cows' milk at an average concentration of 0.6 mg/litre
(Medvedev & Perepechkina, 1973; see Izmerov, 1983). In the USSR, the
following concentrations were found: milk and milk products, 0.055 ±
0.005 mg/kg; poultry and fish, 0.068 ± 0.021 mg/kg; butter, 0.003 ±
0.002 mg/kg; vegetables and fruits, 0.008 ± 0.003 mg/kg; groats and
flour, 0.005 ± 0.002 mg/kg (Sizova & Bogomolova, 1976; see Izmerov,
1983).
Concentrations of gamma-HCH were measured in 1250 samples of
milk and other dairy products in France in 1970-77 and in 1981. In
the first period, the gamma-HCH concentration was < 0.1 mg/kg of
fat; by 1981, the levels had declined to < 0.03 mg/kg of fat
(Laugel, 1981; Rhône-Poulenc Agrochimie, 1986). Higher levels (mean,
0.85 mg/kg) were found in animal fat, but meat and eggs generally
contained no detectable residue (Laugel, 1981). The mean levels of
gamma-HCH found in a large number of samples of various food items
in Germany (Hildebrandt et al., 1986) are shown in Table 4.
The levels of gamma-HCH in food items analysed in France were
0.006-0.01 mg/kg in 113 samples of vegetables, 0.005-0.04 mg/kg in
192 samples of fish and seafood, 0.005-0.041 mg/kg in 154 samples of
preserved meat, 0.007-0.017 mg/kg in 104 samples of cereal products,
0.007-0.034 mg/kg in 120 samples of butter and cheese, 0.005-0.059
mg/kg in 25 samples of oil and fat, and 0.006-0.021 mg/kg in 26
samples of fruit (Rhône-Poulenc Agrochimie, 1986).
Table 4. Levels of gamma-HCH (mg/kg) in food items in Germany
Food item 1973-78 1979-83 1973-83
Meata 0.004-0.04
Meat productsa 0.006-0.055
(maximum, 0.52)
Animal fata 0.007-0.09
(maximum, 0.5)
Gamea 0.042-4.072
Poultrya 0.01-0.05 0.004-0.046
(maximum, 0.471)
Chicken eggs < 0.001-0.01
Chicken eggsa,c 0.001-0.02
(maximum, 1.9)
Milk and milk productsa 0.05 0.01-0.02
Cow's milka,b 0.03 0.01
Vegetable oil and 0.01-0.02
margarinea
Oil seeds, nuts, pulses 0.001-0.127
Fish and fish products 0.01-0.02 0.002-0.009
Shell-fish and molluscs < 0.001-0.020
a From Hildebrandt et al. (1986); on fat basis
b From Anon. (1984)
c From Koelling (1978)
Skaftason & Johannesson (1979) found a mean value of 13 µg/kg
in 35 samples of butter from Iceland in 1968-70. Of 32 samples
analysed in 1974-78, only five contained gamma-HCH, at a mean value
of 7 ± 2 µg/kg. The mean concentration in meat, poultry and eggs in
the Netherlands in 1976-78 was 0.002 mg/kg (range, 0.001-0.004
mg/kg) (De Vos et al., 1984); the levels in dairy products were
similar.
Fifteen of 105 chicken eggs from seven areas in Kenya had a
median concentration of 0.01 mg/kg (range, 0.01-0.04 mg/kg) (Kahunyo
et al., 1988). Ten samples each from two lots of lamb and beef were
collected randomly from markets in Bagdad, Iraq, in 1983 and
analysed for the presence of gamma-HCH. An average concentration of
0.225 (0.004-1.611) mg/kg was found in lamb, and 0.116 (0.005-0.83)
mg/kg was found in beef (Al-Omar et al., 1985).
5.1.4.4 Animal feed
Of 114 samples of animal feed analysed in the United Kingdom in
1982-85, 49 contained gamma-HCH at concentrations up to 2.3 mg/kg
product (Steering Group on Food Surveillance, 1986).
5.1.4.5 Miscellaneous products
Lanolin produced from crude wool grease may contain gamma-HCH:
a level of 1.2 mg/kg was found in the USA (Anon. 1989); and Meemken
et al. (1982) found average levels of 2.4 and 2.1 mg/kg in 1976 and
1981, respectively, in Germany. Concentrations of 0.001-0.23 mg/kg
were found in cosmetic creams made from the lanolin.
5.1.5 Terrestrial and aquatic organisms
5.1.5.1 Plants
Gamma-HCH was present in most of 13 samples of three types of
moss and four types of lichen collected on the Antarctic Peninsula
(Graham Land) in 1985 at a mean concentration of 0.84 mg/kg (range,
0.4-1.7 µg/kg) (Bacci et al., 1986).
In 1984, near Florence and Siena, Italy, far from primary
sources of pollution, leaves from ten species of tree and two
species of lichen were found to contain average levels of 8.2
(range, 2-14) and 10 (9-11) µg/kg dry weight, respectively.
Gamma-HCH levels in plant species collected in 14 countries ranged
from 0.2 to 700 µg/kg dry weight (Gaggi et al., 1986).
5.1.5.2 Aquatic organisms
Freshwater mussels (Anodonta piscinalis) were used to monitor
bioaccumulation of pollutants at 17 sampling sites in a river basin
in Finland between 1984 and 1987. One to three mussels were used per
sampling site. Gamma-HCH was found in concentrations varying from
none detected to 553 µg/kg on a fat basis; however, a clear decrease
was seen over the period of study (Herve et al., 1988).
Cowan (1981) studied the extent of pollution by HCHs of
Scottish coastal waters using Mytilus edulis as the biological
indicator. The gamma-HCH levels at 118 sites were < 6-53 µg/kg dry
weight, which are similar to those found in Germany, the
Netherlands, Spain, and the United Kingdom. The fish and shellfish
sampling programme of the Ministry of Agriculture, Fisheries, and
Food in the United Kingdom in 1977-84 was implemented mainly in
areas around the coasts of England and Wales. The range found for
gamma-HCH was < 0.001 (none detected) to 0.075 mg/kg wet weight;
the level in fish muscle was < 0.001 mg/kg wet weight (Franklin,
1987).
The average concentration of gamma-HCH was measured in 10
marine organisms collected along the Mediterranean coast of Spain
during 1985. Mytilus galloprovincialis, Venus gallina, Sardina
pilchardus, and Mullus surmuletus contained 0.1-1.7 µg/kg fresh
weight (maximum, 16 µg/kg) (Pastor et al., 1988).
Bream collected in rivers and lakes at 15 locations in Germany
contained average gamma-HCH concentrations of 106-696 µg/kg on a fat
basis (Umweltbundesamt, 1988-89), while bream collected in the River
Elbe, between Schnackenburg and the North Sea, contained average
concentrations up to 0.031 mg/kg in muscle and up to 2.6 mg/kg in
adipose tissue (Arbeitgemeinschaft für die Reinhaltung der Elbe,
1982). In 1970-72, different types of fish, mussels, and shrimps
were analysed for gamma-HCH. Fish with a low fat content, collected
in the Atlantic Ocean and the North Sea, contained 0.004-0.008 mg/kg
fresh weight, and fat fish contained 0.01 (0.01-0.026) mg/kg fresh
weight. Fat fish caught in the Baltic contained higher levels - up to
0.2 mg/kg fresh weight. Mussels and shrimp caught in the North Sea
contained none to 0.009 mg/kg fresh weight; mussels from the Baltic
coast contained 0.009-0.011 mg/kg. In 1973-76, similar values were
found, except that the fat fish had lower levels. Marine organisms
from the Baltic Sea generally contained higher levels of gamma-HCH
than those from the North Sea. Freshwater fish from industrially
contaminated areas contained higher levels (Hildebrandt et al.,
1986). Gamma-HCH was detected at levels up to 7.0 µg/kg (mean, 2.5
µg/kg) in the muscle of flounders collected off the coast of the
North Sea in Germany in 1986 (Umweltbundesamt, 1988-89).
5.1.5.3 Terrestrial organisms
Earthworms: Gamma-HCH was found in the soil of ten arable and
two orchard sites in the United Kingdom at 0.01 and 0.08 mg/kg soil,
respectively, and in worms living in the two soils at 0.05 and 0.3
mg/kg (Advisory Committee on Pesticides and Other Toxic Chemicals,
1969).
Birds: Bednarek et al. (1975) determined total HCH isomers at
levels of 0.03-0.63 mg/kg total egg (or 0.6-11.1 mg/kg on a fat
basis) in eggs of birds of prey, such as the sparrowhawk (Accipiter
nisus), in two areas of Germany in 1972-73. Eggs of sandwich terns
collected in the Elbe estuary contained arithmetic means (10 eggs)
of 0.006 mg/kg fresh weight in 1981, 0.002 in 1985, 0.003 in 1986,
and 0.028 in 1987 (Umweltbundesamt, 1988-89). The concentrations
found in the livers of avian predators in the United Kingdom are
shown in Table 5.
The mean levels of gamma-HCH detected in 23 barn owls (Tyto
alba Scop.) obtained in Leon, Spain, were 0.03 (0.003-0.083) mg/kg
wet weight in muscle, 0.036 (0.002-0.208) in liver, 0.051
(0.009-0.144) in fat, 0.012 (0.002-0.031) in brain, and 0.081
(0.005-0.343) in kidneys (Sierra & Santiago, 1987).
Table 5. Residues of gamma-HCH in livers of avian predators in the
United Kingdoma
Bird Date No. of gamma-HCH
samples (mg/kg)
Sparrowhawk 1963 11 0.01
Kestrel 1963 20 0.04
1964 28 0.1
1965 60 0.03
Tawny owl 1964 14 0.01
Heron (adults) 1964 17 0.005
Great crested grebe 1963/66 15 0.03
a From Advisory Committee on Pesticides and Other Toxic Chemicals
(1969)
Faladysz & Szefer (1982) examined adipose tissue from seven
species of diving ducks at their winter quarters in the southern
Baltic. Residues of gamma-HCH were detected in only 4 of 129 samples
from three species of duck examined (range, 0.001-0.51 mg/kg on a
fat basis).
Mammals: No gamma-HCH (< 0.01 mg/kg) was found in muscle
tissue from 51 North American wolves captured in 1969-71 in sparsely
populated forest regions (Schneeweis et al., 1974). Norstrom et al.
(1988) determined the contamination of the marine ecosystem of the
Canadian Arctic and sub-Arctic by organochlorine compounds by
analysing adipose tissue and liver from 6-20 polar bears (Ursus
maritimus) per zone collected from 12 zones between 1982 and 1984.
The levels were 0.30-0.87 mg/kg on a fat basis; the highest levels
were found in zones receiving continental run-off.
Mean concentrations of gamma-HCH in 86 samples of kidney fat
from roe-deer (Capreolus capreolus) collected in five locations in
Germany in 1985-86 were 8-12 µg/kg, with a maximum of 1020 µg/kg
(Umweltbundesamt, 1988-89).
5.2 Exposure of the general population
The data presented above demonstrate that the main source of
exposure of the general population is food.
5.2.1 Total-diet studies
In total-diet studies carried out in the United Kingdom between
1966 and 1985, 22-25 samples of foods in 20-24 groups were purchased
in 21 towns throughout the country and prepared by cooking. The
calculated mean levels of gamma-HCH residues in the total diet were
0.004 mg/kg in 1966-67, 0.0035 in 1970-71, 0.003 in 1974-75, 0.0025
in 1975-77, 0.002 in 1979-80, 0.0015 in 1981, and 0.0005 in 1984-85,
resulting in dietary intakes of 6.6, 5.5, 4.4, 3.9, 3.0, 2.0, and
0.5 µg/person per day (Egan & Hubbard, 1975; Steering Group on Food
Surveillance, 1982, 1986, 1989).
The average daily intake of gamma-HCH in the USA was estimated
on the basis of residues found in 30 market-basket composites
collected in 30 cities over the period 1964-80, as shown in Table 6.
Infant foods collected in the United Kingdom in 1985-87
generally contained very low levels of gamma-HCH (range, < 0.002 to
< 0.01 mg/kg product) (Steering Group on Food Surveillance, 1989).
Residues of gamma-HCH were also measured in food composites from 10
cities of the USA in 1974-75 (Johnson et al., 1979). Levels of
0.008-0.012 mg/kg food were found in diets of six-month-old infants,
and 0.001-0.007 mg/kg in the diets of two-year-old toddlers. Similar
samples collected in 1976-79 in 10 cities consisted of about 50
items of infant food and 110 items of food for toddlers. The daily
intake of gamma-HCH was 0.005 µg/kg body weight for infants and 0.01
for toddlers in 1976, 0.006 and 0.008 in 1977, 0.003 and 0.005 in
1978, and 0.001 and 0.006 in 1979 (Gartrell et al., 1985b).
Table 6. Average daily intake of gamma-HCH in the USA, 1964-80a
Year gamma-HCH intake (µ/kg
body weight per day)
1964-69 0.05
1965-70 0.02-0.07b
1973 0.0032
1974 0.0084
1975 0.0031
1976 0.0025
1977 0.0039
1978 0.0024
1979 0.0038
1980 0.0028
a From Johnson & Manske, 1976; US Environmental Protection
Agency, 1980; Gartrell et al., 1985a
b From Duggan & Corneliussen (1972)
Total-diet studies conducted by the US Food and Drug
Administration before 1982 were based on a 'composite sample
approach', regardless of the diet involved. Later studies were based
on dietary information obtained through surveys, so that the 'total
diet' of the US population could be represented by a relatively
small number of food items for a large number of age-sex groups
(Gunderson, 1988). The average intake of gamma-HCH in the diet of
14-16-year-old boys (mean body weight, 60 kg), estimated using the
more recent methods, is shown in Table 7 (S. I. Shibko, letter to
IPCS, dated 29 June1989). The daily intakes in 1982-84 in different
age groups were 0.0019 µg/kg body weight per day for 6-11-month-old
children, 0.0079 for two-year-old children, 0.0031 for
14-16-year-old girls, 0.0034 for 14-16-year-old boys, 0.0020 for
25-30-year-old women, 0.0025 for 25-30-year-old men, 0.0016 for
60-65-year-old women, and 0.0018 for 60-65-year-old men (Gunderson,
1988). The concentrations for these eight groups in 1984-86, 1987,
and 1988 were < 0.01 µg/day for 6-11-month-old infants, < 0.04 for
two-year-old children, and < 0.1 for the other six groups (S. I.
Shibko, letter to IPCS, dated 29 June 1989).
Table 7. Average daily intake of gamma-HCH in 14-16 year-old boys
in the USAa
Year Intake
µg/day µg/kg body weight
per day
1982-84 0.204 0.0034
1984-86 0.078 0.0013
1987 0.108 0.0018
1988 0.084 0.0014
a From S.I. Shibko, letter to IPCS, dated 29 June 1989
In total-diet studies in Germany, fruit, potatoes, and other
vegetables ready for consumption contained 0.001 mg/kg product
(Kampe & Andre, 1980). In 17 food groups in Spain, the gamma-HCH
concentration ranged from none detected to 0.019 mg/kg product; the
level in fat was up to 0.268 mg/kg. The daily intake amounted to
0.0138 mg/person in 1971-72 (Carrasco et al., 1976). In a total-diet
study in the Netherlands in 1977, the average concentration of
gamma-HCH in 100 samples was 0.03 mg/kg on a fat basis; the highest
level was 0.14 mg/kg (Greve & van Hulst, 1977). In another study in
the Netherlands, a mean daily intake of 0.002 mg/person was
determined for 1976-78 (De Vos et al., 1984).
Data from Canada, Guatemala, Japan, the United Kingdom, and the
USA indicate a very low daily intake of gamma-HCH over the years
1971-84. The median values ranged from 0.01 to 0.05 µg/kg body
weight (Gorchev & Jelinek, 1985). The daily intake of lindane in the
USSR was calculated from a market-basket survey to be about 0.005
mg/day. Cooking reduced this level by a factor of 4.3 (Sizova &
Bogomolova, 1976; see Izmerov, 1983).
5.2.2 Intake with drinking-water and air
Edwards (1981) calculated the daily intake of gamma-HCH with
drinking-water to be 0.4 ng per person, assuming a daily consumption
of 2 litre of water; the median daily intake via air was also
calculated to be 17 ng per person, indicating that only small
quantities of gamma-HCH are ingested with water and air.
Guicherit & Schulting (1985) measured the concentration of
gamma-HCH in the atmosphere and calculated that the daily average
intake of a 70-kg Dutch person by inhalation would be 7.2 ng.
Another calculation of the average human intake with air, on the
basis of ambient concentrations, was 12 ng/day, which represents
about 1% of the total daily intake by all routes. The daily intake
of gamma-HCH in the USA was estimated to be 0.002 µg/kg body weight
by air and 0.07 µg/kg by the oral route (Hildebrandt et al., 1986).
5.2.3 Concentrations in human samples
The concentrations of gamma-HCH in human samples are a good
indication of the total exposure of the general population.
5.2.3.1 Blood
Gamma-HCH was detected at a geometric mean of 0.4 µg/litre
(range, 0.1-4.1 µg/litre) in the blood of 49 of 62 people in
Louisiana, USA (Selby et al., 1969). In a follow-up study, a
geometric mean of 0.4 µg/litre (range, 0.1-6.0 µg/litre was found in
47 out of 53 blood samples from pregnant women. Polishuk et al.
(1970) found a mean concentration of 0.4 ± 0.8 µg/litre in the blood
of 24 pregnant women and 0.3 ± 0.6 µg/litre in the blood of 23
infants living in Israel. Wassermann et al. (1982) found a mean of
4.3 ± 4.8 µg/litre in serum of 10 women in Israel with a normal
pregnancy. In a group of 17 women with an abnormal pregnancy
(premature birth), a mean concentration of 15.0 ± 7.2 µg/litre was
detected. Bercovici et al. (1983) found a concentration of 8.0 ± 4.5
µg/litre in the serum of seven Israeli women with a normal pregnancy
and a mean concentration of 8.5 ± 7.8 µg/litre in 17 women with a
'missed abortion'.
Reiner et al. (1977) found a mean concentration of 4.1 ± 0.6
µg/litre (range, 0.5-15.0) in 23 of 147 serum/plasma samples from
people living in a town in Yugoslavia. Similar levels were found in
other parts of the country (Krauthacker et al., 1980).
Siddique et al. (1981) found gamma-HCH at a mean concentration
of 25.0 ± 16.0 µg/litre (range, 8.0-47.0) in the blood of 15 people
in India, and Saxena et al. (1981) found a mean concentration of
19.0 ± 12.4 µg/litre (range, 2.4-135.0) in the blood of 100 pregnant
women, aged 18-34 years, in the Indian countryside. Nonvegetarian
women had higher blood levels than vegetarian women. Kaphalia & Seth
(1983) found blood levels of 12.0 (none detected to 71.0) µg/litre
in 48 men (aged 20-40 years), 12.00 (range, 5.0-24.0) µg/litre in 16
women (aged 10-30 years) and 16.0 (range, 3.0-64.0) µg/litre in 16
children (< 10 years) in India.
Eckenhausen et al. (1981) found a geometric mean of 0.9
µg/litre of gamma-HCH (range, < 0.4-3.8) in 28 out of 48 pregnant
women in the Netherlands. After delivery, a geometric mean of 0.5
(range, 0.2-19.0) µg/litre was measured in 24 out of 66 women, and a
mean of 0.5 (range, < 0.3-34) µg/litre in 33 out of 86 babies.
Blood samples from Dutch citizens were analysed in 1978 (70
samples), 1980 (48 samples), 1981 (127 samples), and 1982 (54
samples); the concentration of gamma-HCH was in the range < 0.1-0.2
µg/litre blood (Greve & van Harten, 1983; Greve & Wegman, 1985).
Blok et al. (1984) measured the levels of gamma-HCH in the blood of
65 healthy volunteers (34 women and 31 men) in the Netherlands and
found residues in approximately two-thirds of the people studied.
The median concentration in both men and women was 0.2 µg/litre
(range, none detected to 0.6 µg/litre). Bertram et al. (1980) found
a median concentration of 1.18 µg/litre (range, none detected to
2.94) in 118 whole-blood samples in Germany.
In 8 of 35 serum samples from mothers in Norway and in 6 of 35
corresponding samples of umbilical cord serum, the levels of
gamma-HCH ranged from 0.2 to 27 µg/kg wet weight. In serum samples
from immigrant mothers and in 5 of 7 corresponding umbilical cord
serum samples, the levels were 0.1-3.4 µg/kg wet weight (Skaare et
al., 1988).
5.2.3.2 Adipose tissu
Mes et al. (1982) analysed 99 samples of adipose tissue from
autopsied accident victims in different areas of Canada and found an
average concentration of gamma-HCH of 0.003 mg/kg (wet weight)
(range, 0.001-0.03 mg/kg). Nearly all of the samples (90%) contained
gamma-HCH.
In 567 samples of adipose tissues from Dutch citizens analysed
for the presence of gamma-HCH in 1968-83, the concentration varied
from < 0.01 to 0.04 mg/kg; the highest levels were found for
1968-76 (Greve & van Harten, 1983; Greve & Wegman, 1985). Bertram et
al. (1980) found a median concentration of 0.05 mg/kg (range, none
detected to 0.44) in 72 samples of adipose tissue from people in
Germany. In specimens of subcutaneous fat taken in 1982-83 from 48
children (34 < 1 year; 14 aged 2 years), the concentration of
gamma-HCH was 0.04 mg/kg fat (range, 0.01-0.21 mg/kg). The average
concentration was highest in infants aged 0-6 weeks, at 0.07 mg/kg
fat (range, 0.02-0.21 mg/kg) (Niessen et al., 1984). The results of
nine studies carried out in Germany in 1969-83 (598 samples) gave a
mean concentration for gamma-HCH of 0.01-0.34 mg/kg on a fat basis
(Hildebrandt et al., 1986).
Twenty-nine samples of adipose tissue were taken at necropsy
and 24 at surgery in the Poznan district in Poland and compared with
100 samples from residents of the Warsaw region. The mean
concentration of gamma-HCH in Poznan was 0.020 ± 0.018 and that in
Warsaw, 0.022 ± 0.003 mg/kg (Szymczynski et al., 1986).
The mean concentration of gamma-HCH in 360 samples of adipose
tissue collected in eight regions of Japan in 1974 was 0.035 mg/kg
(Takabatake, 1978).
5.2.3.3 Breast milk
Breast milk is a major route of elimination of organochlorine
pesticides and polychlorinated biphenyls in women.
In a Swedish study, the levels of gamma-HCH in mothers' milk
were found to be related to their dietary habits: levels in
lacto-vegetarians were lower than those in subjects who ate a mixed
diet, and these were lower than those found in mothers who ate a
mixed diet which regularly included fatty fish from the Baltic Sea
(Noren, 1983).
A significant correlation was found between the concentration
of gamma-HCH in breast milk and the amounts of meat products and
animal fat in the diet. In addition, concentrations of gamma-HCH in
breast milk appeared to be higher in rural areas than in urban areas
(Cetinkaya et al., 1984).
Variations in residue levels in human milk during a lactation
period of up to 9 months were investigated in five women aged 23-36
years in Germany: gamma-HCH concentrations were 0.004-0.022 mg/kg on
a fat basis, and no essential change in residue level occurred over
the lactation period (Fooken & Butte, 1987).
More than 7100 samples of breast milk were analysed in Germany
between 1969 and 1984 by 20 authors, and the results were summarized
by Hildebrandt et al. (1986). The mean concentration of gamma-HCH
was 0.01-0.11 mg/kg on a fat basis, but a mean concentration of 0.45
mg/kg was found for a group of 137 samples. A slow decrease in the
mean concentration was observed between 1978 and 1984. The average
concentration in human milk (2709 samples) in Germany in 1979-81 was
0.06 mg/kg on a fat basis (Fooken & Butte, 1987); in 1981-83, the
average level in 132 samples of breast milk was 0.032 mg/kg milk fat
(Cetinkaya et al., 1984). The results of other studies were
summarized by the Deutsche Forschunsgemeinschaft (1978, 1983). The
results for other countries are comparable to those for Germany,
although higher levels (mean, 0.33 mg/kg) were measured in
Czechoslovakia in 1971-73 (Deutsche Forschungsgemeinschaft, 1983).
Average concentrations in human milk in France between 1970 and 1975
were in the order of 0.06-0.07 mg/kg (fat basis) (Rhône-Poulenc
Agrochimie, 1986).
Tuinstra (1971) analysed 40 breast milk samples from young
mothers (18-32 years of age) in the Netherlands and found a median
concentration of 0.01 mg/kg on a fat basis (range, none detected to
0.04 mg/kg on a fat basis). The median concentration of gamma-HCH in
278 samples of human milk collected in 11 maternity centres in the
Netherlands was < 0.01 mg/kg on a fat basis; the highest value
found was 0.08 mg/kg (Greve & Wegman, 1985).
Mes et al. (1986) studied 210 breast milk samples from five
regions across Canada and found gamma-HCH at a mean concentration of
5 µg/kg (on a fat basis). Davies & Mes (1987) studied 18 breast milk
samples from Canadian, Indian, and Inuit mothers in Canada whose
fish consumption was comparable to the national rate. The level of
gamma-HCH was 7 µg/kg in milk fat of the indigenous population, in
comparison with 5 µg/kg in a national survey.
Vukavic et al. (1986) measured gamma-HCH in 59 samples of
colostrum collected in Autumn 1982 (26 samples) and Spring 1983 (33
samples) in Yugoslavia from healthy nursing mothers on the third day
after delivery. The concentrations of gamma-HCH were significantly
higher in Autumn (1.71 ± 0.44 µg/litre) than in Spring (0.67 ± 0.12
µg/litre).
Breast milk samples from four women in Iraq, examined once a
week for 20 weeks, contained average levels of 0.009, 0.005, 0.134,
and 0.005 mg/kg whole milk. Gamma-HCH levels in placental tissue
from these four donors were 0.004, 0.011, 0.013, and 0.006 mg/kg,
respectively. Fluctuations in the residue levels were seen to be due
to variations in the daily dietary intake and variations in the fat
content of the breast milk (Al-Omar et al., 1986).
6. KINETICS AND METABOLISM
6.1 Absorption
6.1.1 Oral administration - experimental animals
The uptake of lindane by rats or mice has been studied after
oral administration. Direct information on the velocity of uptake
from the gastrointestinal tract is available, which can be
supplemented by information from studies in which excretion of
orally administered radioactive lindane was followed.
Lindane taken up from the intestines is transferred almost
exclusively to the blood. No significant amount was found in the
lymph of rats after injection of 0.05 or 0.1 µmol into the loops of
the small intestines in vivo. Absorption was rapid: 29-53% of the
injected material was absorbed from the intestinal loops within the
first 30 min (Turner & Shanks, 1980). Uptake of lindane from the
intestines of rats given 12.5 mg in oil during five days was less
effective in animals depleted of their intestinal microorganisms by
maintenance under aseptic conditions than that in conventional rats.
The asepticized rats also excreted more unchanged lindane in the
faeces than conventional animals (Macholz et al., 1983).
6.1.2 Dermal application - experimental animals
Hawkins & Reifenrath (1984) developed an apparatus to determine
the evaporation and percutaneous penetration of
hexachloro-[U-14C]-cyclohexane (lindane) in vitro, applying a
dose of 4 µg/cm2 on pig skin. Evaporation accounted for 26 ± 5%;
skin oxidation for 43 ± 17%; and percutaneous penetration for only
0.7 ± 0.3% of the applied radiolabel. Reifenrath et al. (1984) also
evaluated models consisting of human or pig skin grafted onto the
congenitally athymic nude mouse, hairless dogs and weanling
Yorkshire pigs for predicting skin penetration in humans. A
radioactive dose of 0.05 µCi of [U-14C]-labelled lindane (98%) was
applied topically to 1.27 cm2 (4 µg/cm2) of each model, and
radiolabel (percentage of applied dose) was measured in urine and
faeces, skin scrub, application site, and carcass. Incomplete
excretion of the label following topical application was corrected
for by parental (subcutaneous) administration of 2 µCi in propylene
glycol. The results showed significant correlations between the
values for human skin and those for human skin grafted on athymic
mice and for weanling Yorkshire pigs, but no correlation was found
between the values for humans and those for the hairless dog or for
pig skin grafted on athymic nude mice.
Dermal absorption has also been investigated in rats and
rabbits. Groups of 24 male Charles-River Crl:CD(SD)BR rats and male
Hra:(NZW)SPF rabbits were given a single dermal application of
lindane (20% emulsifiable concentrate to which 14C-lindane had
been added) on clipped skin at doses of 0.1, 1, or 10 mg/rat and
0.5, 5, and 50 mg/rabbit, corresponding to 0.02, 0.2, and 2
mg/cm2, respectively. Urine and faeces were collected 0.5, 1, 2,
4, 10, or 24 h after application from four animals per dose level;
and four animals per group were bled and sacrificed 0.5, 2, 4, 10,
and 24 h after application of the test material. The ratio between
the quantity absorbed at a dose of 1 mg and that absorbed at 10 mg,
as well as that between 0.2 and 1 mg/rat at equal exposure duration,
decreased with increasing exposure time and concentration. On a
group basis, the total recoveries were 75-85% for rats and 75-82%
for rabbits. A significant fraction of the applied dose was found in
the urine: 16, 15, and 12%, respectively, at application levels of
0.1, 1, and 10 mg/rat. The corresponding values for rabbits were 46,
29, and 41% for doses of 0.5, 5, and 50 mg/rabbit. Much lower levels
were found in the faeces. Total adsorption (24 h) increased in rats
from 5% of the applied amount at the highest dose to 28% at the
lowest exposure. For rabbits, penetration was more rapid, and
adsorption at 24 h ranged from 17 to 56% of the applied dose. The
applied doses per unit surface area were approximately the same,
permitting a comparison of penetration rates. The average
penetration rates after 24 h for rats were 0.2, 2, and 4 µg/cm2
per hour for groups treated with 0.02, 0.2, and 2 mg/cm2. The
penetration rates after 24 h for rabbits were 0.5, 3, and 14
µg/cm2 per hour for the groups treated with 0.02, 0.2, and 2
mg/cm2. These studies indicate that appreciable absorption of
lindane takes place after dermal applicatioon (Bosch, 1987, 1987b).
6.1.3 Other routes - experimental animals
When doses of 40 or 80 mg/kg body weight of a mixture of 14C-
and 36Cl-gamma-HCH in rapeseed oil were injected intraperitoneally
into rats, 25% was absorbed within 1 h and at least 90% after 1 day.
Four days after the injection, only traces of lindane were left in
the abdominal cavity. One day after the injection, about 40% of the
applied dose was found in the organs and tissues (Koransky et al.,
1963).
6.2 Distribution
6.2.1 Oral administration - experimental animals
After uptake, lindane is distributed to all organs and tissues
in the body of laboratory animals, at measurable concentrations
within a few hours.
When lindane was administered orally to rats at doses of 1, 10,
or 100 mg/kg diet for up to 56 days, the highest concentrations were
found in adipose tissue. The fat:blood ratio in this study was very
close to 150 at all times, whereas the liver:blood ratio was
3.4-3.5. Lindane concentrations in organs reached a maximum after
2-3 weeks and slowly decreased thereafter. The authors did not
differentiate between males and females (Oshiba, 1972).
Twenty-four hours after oral administration of 14C-labelled
lindane at 8 mg/kg body weight in sunflower oil to rats for 10 days,
more than 35% of the administered activity was deposited in fat.
Muscle and kidneys contained 3.5 and 3.7%, respectively, and all
other organs that were analysed contained less than 1%; 17.4% was
found in urine and 13.8% in faeces. In total, only 78.7% was
recovered; however, skin was not analysed in this study. (Other
studies have demonstrated that a significant amount of lindane
administered to rats and mice is deposited in the skin, so that some
of the missing 21.3% of the total applied dose could have been
there.) A fter an additional 48 h, the concentrations in all organs
were reduced to about one-half of the values seen after 24 h,
showing that no single organ retained lindane significantly longer
than the others. Urine contained 24.5% and faeces, 20.9% (Seidler et
al., 1975).
After single oral doses to rats, the fat:blood ratio ranged
between 145 and 206 and the brain:blood ratio between 4 and 6.5
(Vohland et al., 1981).
After continuous dietary administration of lindane at doses of
0.2, 0.8, 4.0, 20, or 100 mg/kg diet for 13 weeks to Wistar KFM-Han
rats, the highest concentrations were reached in the fatty tissue.
At the highest dose, the fat:blood ratio was 44 in males and 69 in
females and the liver:blood ratio was 5.3 in males and 9.6 in
females. After six weeks with no further administration, lindane
concentrations in organs were close to the control values (Suter et
al., 1983).
The distribution of lindane in brain after oral administration
at 30 mg/kg or intravenous administration at 0.3 mg/kg was studied
using autoradiography-imaging analysis and dissection-liquid
scintillation counting techniques. The two routes of administration
gave similar results. A heterogeneous distribution of label in brain
regions was observed: the radiolabel concentration in the white
matter was higher than that in thalamus, mid-brain, pons and medulla
at different times relative to the mean value for whole brain. The
affinity of lindane for white matter and myelinated structures was
related to its lipophilic behaviour (Sanfeliu et al., 1988).
Mosha et al. (1986) studied the distribution and elimination of
gamma-HCH in adult female goats. Eight goats were administered a
daily dose of 6 mg/kg body weight by gavage for five consecutive
days. Blood and milk were collected before exposure and during 10-60
days after exposure, and organs and tissues were collected and
analysed. One goat was used as a control. The blood concentration of
gamma-HCH was approximately 0.1 mg/litre during the dosing period
and decreased gradually thereafter; none was detectable after day
20. The concentrations in milk were about eight times higher than
those in blood but decreased in parallel. The concentration in fat
samples on day 7 was 1.4 mg/kg, but those in other tissues were
about 0.1 mg/kg.
6.2.2 Inhalation - experimental animals
After rats were exposed to lindane at doses of 0.02, 0.1, 0.5,
or 5 mg/m3 for 90 days by inhalation, the highest concentrations
were found in fatty tissues. At 5 mg/m3, the fat:serum ratio was
150 in males and 464 in females; at 0.5 mg/kg, it was 161 in males
and 245 in males; at 0.1 mg/kg, 137 in males and 429 in males; and
at 0.02 mg/kg, 92 in males and 377 in females. At the 5 mg/m3
exposure level, the liver:blood ratios were 1.9 in males and 4.2 in
females and the brain:serum ratios were 9 in males and 23 in
females. These values suggest that higher concentrations are reached
in fat after inhalation than after oral administration, whereas
liver appear to be somewhat lower after inhalation. After a recovery
period of four weeks, concentrations in all organs had decreased to
the control values (Oldiges et al., 1983).
6.2.3 Other routes
One day after intraperitoneal injection to rats of a mixture of
14C- and 36Cl-lindane in rapeseed oil, the highest contents were
those of skin and fat - 15.7% and 10.7%, respectively. Less than 1%
was found in all other organs, including the central nervous system
(Koransky et al., 1963). When lindane or deuterated lindane was
administered intraperitoneally to rats at doses of 10 mg/kg body
weight, about 40 mg/kg fat were found after one day in both males
and females. At that time, the blood concentration in males was 0.2
mg/litre; 1-2 mg/kg were present in brain and 0.7 mg/kg in skeletal
muscle. Deuterated lindane was found at 110 mg/kg in depot fat of
males, and the levels in brain and muscle were about twice those of
undeuterated lindane (Stein et al., 1980).
Mottram et al. (1983) studied the metabolic fate of lindane in
three groups of two white female pigs. The pigs were sprayed once
with either 5.6 g or 1.4 g of an anti-louse spray, which represented
16 and 4 times, respectively, the normal dose of 350 mg/pig. Five
animals served as controls. Rapid accumulation of lindane occurred,
and several metabolites were found in adipose tissue. The main
metabolite was 1,2,4-trichlorobenzene. The residues were eliminated
rapidly from adipose tissue, so that 30 days after treatment, the
residual concentration in pigs sprayed with a dose 16 times the
recommended rate was no greater than that in the untreated controls.
Residue levels were also investigated in four lactating goats
following oral and topical application of labelled lindane (Wilkes
et al., 1987a,b). Two Alpine goats were housed in metabolic cages
and administered lindane (purity not stated) spiked with
14C-lindane in the diet at doses of 1 and 10 mg/kg twice daily for
four days; they were sacrificed 12-14 h after the last dose. One
Alpine goat received two topical applications at a seven-day
interval of a lindane solution (purity unspecified) containing 11.0
mg/ml to a shaved area that represented about 25% of the body
surface area, to simulate total body spray; it was sacrificed 48 h
after the last application. A Nubian goat was not only shaved over
the same extent but also had its remaining hair clipped to
approximately 3 cm, and the whole surface of the animal was treated
with lindane, to simulate dip treatment; this animal was sacrificed
24 h later. After sacrifice, radiolabel was measured in tissues and
in the intestinal contents; radiolabel in exhaled carbon dioxide was
measured on one occasion.
Total recovery of radiolabel was low: approximately 50% in the
study by oral administration and 16-30% in the study by dermal
application. To determine the reason for the losses after oral
administration, the fate of labelled lindane in rumen fluid at 37 °C
was investigated in vitro. About 55% of the radiolabel was
recovered as 14CO2, and 38% remained in the rumen fluid. The
authors stated that their results "clearly show that volatile
14C-labelled organics were evolved from 14C-lindane fortified
rumen fluid," and "that the losses in the in vivo studies were due
to volatile 14C-lindane metabolites." Attempts to trap the
labelled volatile compounds, however, proved unsuccessful.
From 35 to 46% of the radiolabel administered orally was
excreted in the urine over a period of 4 days, and 10-12% of the
dermily applied dose was found in urine over 8-9 days. Much lower
activities were found in faeces, and insignificant amounts in
expired air. The amount of radiolabel in whole milk after oral
administration reached a plateau after 2-3 days, corresponding to a
total concentration of 0.4 ppm (6-8 ppm in the fat) at the lower
dose (2 mg/kg per day) and 3 ppm (about 50 ppm in the fat) at the
higher exposure level (20 mg/kg per day). Significant activity was
also found in the milk after dermal administration, corresponding to
levels of 0.1-0.7 ppm in whole milk.
6.3 Metabolic transformation
The metabolism of lindane is initiated by one of four possible
reactions:
- Dehydrogenation leads to the formation of gamma-HCH;
- Dehydrochlorination leads to the formation of gamma-PCCH;
- Dechlorination leads to the formation of gamma-
tetrachlorohexene;
- Hydroxylation leads to the formation of
hexachlorocyclohexanol.
These compounds must be considered as intermediates, and the initial
reactions are followed by a series of further dehydrogenating,
dechlorinating, dehydrochlorinating, and hydroxylating steps.
A large number of metabolites and end-products occur during the
metabolism of lindane (see section 6.3.2). Detailed descriptions and
schemes of the metabolic pathway of lindane leading to the various
isomeric metabolites have been proposed (Engst et al., 1970, 1977,
1978a, 1979a; Kurihara & Nakajima, 1980). These pathways involve
metabolites that have not as yet been detected. These missing
metabolites may be very unstable compounds which are rapidly
transformed to other intermediates and thus escape detection.
Another possibility is that conjugates are formed which are tightly
bound to proteins via sulfur, and these could be detected only
after the complex is hydrolysed.
The essential steps in the metabolism of lindane are known, and
these are shown, with the main metabolites, in Figure 2 and are
discussed below.
6.3.1 Enzymatic involvement
Lindane is converted by enzymatic reactions, mainly in the
liver. One group of enzymes involved in the biotransformation of
lindane are microsomal, e.g., cytochrome P-450-dependent
monoxygenases. Five groups of male Wistar rats were injected
intraperitoneally with gamma-HCH at 25 mg/kg body weight on four
consecutive days to investigate the induction of cytochrome P-450 in
liver microsomes. gamma-HCH was found to be a 'mixed type' inducer
which mediates the induction of cytochrome P-450 b/e, c and d forms
(Kumar & Dwivedi, 1988). These enzymes are involved in
hydroxylation, dehydrogenation, and dechlorination. Other hepatic
cytosolic enzymes are involved in the dehydrochlorination reaction.
The intermediate metabolites or end-products of the
biotransformation may result as a consequence of the four enzymatic
reactions listed above.
A cytochrome P-450-dependent dehydrogenation reaction was
described in rat liver microsomes in vitro. Incubation of lindane
with rat liver homogenates resulted in the formation of
hexachlorocyclohex-1-ene, and the authors proposed that this
dehydrogenation is an important initial step in the metabolism
leading to the detoxication of lindane (Chadwick et al., 1975).
Stein et al. (1977) found that at least two independent
pathways were involved in the metabolism of lindane. The first is
the possible formation of unstable intermediates, such as
hexachlorocyclohexanol, after an initial hydroxylation leading to
the main metabolite, 2,4,6-trichlorophenol (2,4,6-TCP), and
involving cytochrome P-450. The second pathway includes
dehydrogenation of lindane to 1,2,3,4,5,6-HCCH, subsequent
hydroxylation and dehydrochlorination to 2,3,4,6-tetrachlorophenol.
TCP and tetrachlorophenol are formed in vitro in a ratio of about
2:1.
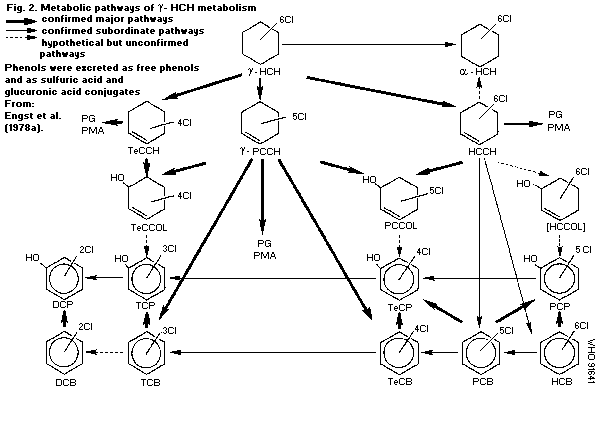 In the presence of oxygen and NADPH in vitro, rat liver
microsomes metabolize lindane mainly to 2,4,6-TCP (Tanaka et al.,
1977, 1979). 3,4,5,6-Tetrachlorocyclohexene was identified as an
intermediate after lindane was incubated under N2 with liver
preparations (Chadwick et al., 1978; Kurihara et al., 1979).
Experiments with rat liver preparations in vitro demonstrated the
importance of glutathione in the metabolism of lindane. Glutathione
enhanced the conversion of lindane to dichlorophenol (DCP) by a
factor of 3-4, but conjugates were formed only in the presence of
liver cytosol protein as a source of glutathione transferase. The
initial step appears to be dehydrochlorination to 1,3,4,5,6-PCCH,
followed by conjugation and further dehydrochlorination to DCP -
mainly 2,4-DCP. The DCPs found in the urine of rats are a mixture of
different isomers. The rate of formation of S-(dichlorophenyl)
glutathione from HCH in rat liver cytosol apparently depends on
gradual monodehydrochlorination, and the enzymatic transfer of
reduced L-glutathione (GSH) onto PCCH is not preceded by a second
dehydrochlorination (Stein et al., 1977; Portig et al., 1979; Tanaka
et al., 1979).
When a 1:1 mixture of lindane and the corresponding
hexadeuterated compound was fed to Wistar rats, the ratio of MCP,
DCP, and TCP, excreted as mercapturic acids, showed an isotopic
effect. The rate-limiting step for the formation of DCP and TCP is
either a dehydrochlorination or a dehydrogenation of lindane,
whereas formation of MCP must be initiated by dechlorination to
tetrachlorocyclohexane, followed by conjugation with glutathione
(Kurihara et al., 1980).
Lindane was converted mainly to gamma-HCCH by rat liver
microsomes, and significant amounts of 2,4,6-TCP and
2,3,4,6-tetrachlorophenol were detected. Human liver microsomes
converted lindane into four major metabolites:
gamma-1,2,3,4,5,6-hexachlorocyclohex-1-ene; gamma-1,3,4,5,6-PCCH;
beta-1,3,4,5,6-PCCH, and 2,4,6-TCP. Smaller amounts of
2,3,4,6-tetrachlorophenol and pentachlorobenzene were found
(Fitzloff et al., 1982).
Human and rat liver microsomes converted the lindane
metabolites gamma-PCCH and 3,4,6/5-PCCH to 1,2,4-tetrachlorobenzene,
1,2,3,4-tetrachlorobenzene, 2,4,5-TCP, 3,4,5/6-pentachloro-2-
cyclohexen-1-ol and beta-PCCH-oxide or 3,4,6/5-PCCH-oxide. The
identity of the beta-PCCH-oxide was confirmed by column
chromatography and gas-liquid chromatography-mass spectrometry. It
is stable to hydrolysis by microsomal epoxide hydrolase
(E.C.3.3.2.3.) and under various aqueous acid conditions. Its
toxicological role is still unknown. Although this compound is
structurally related to epichlorhydrin and epoxides, it was not
mutagenic to Salmonella typhimurium strain TM 677 (Fitzloff & Pan,
1984).
Two groups have reported the formation of trace amounts of
chlorinated benzenes from lindane in rats. Hexachlorobenzene was
found in faeces (Gopalaswamy & Aiyar, 1984) and pentachlorobenzene
in brain (Vohland et al., 1981). The two studies are consistent in
so far as the identified amounts of chlorobenzenes are extremely low
and near the detection limit; however, they are also contradictory,
because hexachlorobenzene was found exclusively in the first study
and pentachlorobenzene in the second. It is impossible to clarify
whether artefacts were measured in these studies, e.g., enrichment
of impurities in the starting material. If indeed chlorinated
benzenes are formed from lindane, the amounts obtained are
insignificant compared to those of other metabolites.
6.3.2 Identification of metabolites
Metabolites of lindane have been identified in a large number
of studies, in vivo in body fluids, urine, faeces, organs, and
tissues, and in vitro. Most of the in-vivo studies were carried
out with rats, but similar results were obtained in other animal
species.
The following metabolites have been identified:
Cycloalkenes: 1,2,3,4,5,6-hexachlorocyclohexene (HCCH);
1,3,4,5,6-pentachlorocyclohexene (PCCH); 3,4,5,6-
tetrachlorocyclohexene; 2,3,4,5,6-pentachloro-2-cyclohexen-1-ol; and
2,3,4,6- and 2,4,5,6-tetrachloro-2-cyclohexene-1-ol (Chadwick &
Freal, 1972a; Freal & Chadwick, 1973; Chadwick et al., 1975; Engst
et al., 1976; Kujawa et al., 1977; Tanaka et al., 1977; Chadwick et
al., 1978; Engst et al., 1978b; Kurihara et al., 1979; Vohland et
al., 1981; Fitzloff et al., 1982; Mottram et al., 1983; Sanfeliu et
al., 1988).
Chlorobenzenes: 1,2,3,4,5,6-hexachlorobenzene, 1,2,3,4,5-
pentachlorobenzene and 1,2,3,5-, 1,2,4,5-, and 1,2,3,4-
tetrachlorobenzene (Aiyar, 1980; Voh land et al., 1981; Gopalaswamy
& Aiyar, 1984; Artigas et al., 1988). Mono-, di-, tri-, and
tetrachlorobenzenes have been reported in pigs (Mottram et al.,
1983).
Chlorophenols: 2,3,4,5,6-pentachlorophenol; 2,3,4,5-,
2,3,4,6-, and 2,3,5,6-tetrachlorophenol; 2,3,5-, 2,4,5-, and
2,4,6-trichlorophenol (TCP); and 2,4- and 3,4-dichlorophenol (DCP)
(Grover & Sims, 1965; Chadwick & Freal, 1972a,b; Freal & Chadwick,
1973; Kurihara & Nakajima, 1974; Chadwick et al., 1975; Engst et
al., 1976; Kujawa et al., 1977; Stein et al., 1977; Tanaka et al.,
1977; Engst et al., 1978b; Tanaka et al., 1979; Aiyar, 1980;
Fitzloff et al., 1982).
Conjugates of these compounds:
- 2,3- and 2,6-dichlorophenol were conjugated with glutathione
(Portig et al., 1979);
- 2,4-DCP; 2,3,5-, 2,4,5-, and 2,4,6-TCP; 2,3,4,5-, 2,3,4,6-, and
2,3,5,6-tetrachlorophenol; 2,3,4,5,6-PCP and
tetrachloro-2-cyclohexene-1-ol were conjugated with glucuronic
acid (Grover & Sims, 1965; Kurihara & Nakajima, 1974; Engst
et al., 1976; Chadwick et al., 1981);
- 2,3,5-, 2,4,5-, and 2,4,6-TCP; 2,3,4,5- and 2,3,4,6-
tetrachlorophenol; 2,3,4,6- tetrachloro-2-cyclohexene-1-ol; and
2,3,4,5,6-pentachloro-2-cyclohexen-1-ol were conjugated with
sulfate (Grover & Sims, 1965; Kurihara & Nakajima, 1974;
Chadwick et al., 1981); and
- 4-monochlorophenol; 2,4- and 3,4-DCP and 2,3,5- and 2,4,5-TCP
were conjugated with mercapturic acids (Grover & Sims, 1965;
Kurihara et al., 1979, 1980).
6.3.3 Metabolites identified in humans
Engst et al. (1978b) analysed urine from workers apparently
exposed to technical-grade HCH (during manufacture?) and found
alpha-, beta-, gamma-, and delta-HCH, traces of hexa- and
pentachlorobenzene, gamma- and delta-PCCH, pentachlorophenol,
2,3,4,5-, 2,3,4,6-, and 2,3,5,6-tetrachlorophenol, several
trichlorophenols, as well as glucuronides of some of these
metabolites. The PCCHs, tetrachlorophenol, hexachlorobenzene, and
pentachlorophenol were also identified in blood.
The urine of 21 men working in the production of gamma-HCH with
a purity of 99.8% from technical-grade HCH (16% alpha-, 7% beta-,
and 45% gamma-HCH) was examined for the presence of chlorinated
phenols. External and internal exposure was estimated from
measurements of the concentrations of HCH isomers in the air of the
workroom and in serum samples. The men had been employed for periods
ranging from a few months up to 30 years (mean, 10.6 years,) and
they were aged 24-62 years (mean, 46 years). Fourteen mono-, di-,
tri-, and tetrachlorophenols and seven dihydroxychlorobenzenes of
unknown configuration were identified in urine. The main metabolites
were 2,4,6-, 2,3,5-, and 2,4,5-trichlorophenol, which were excreted
in nearly equal quantities. The mean concentrations of alpha-,
beta-, and gamma-HCH in the serum of exposed workers were 49 (range,
11-138), 82 (17-434) and 52 (9-188) µg/litre; the levels in controls
were < 1.0 µg/litre. The air concentrations of alpha-, beta-, and
gamma-HCH were 2-4, 1-3, and 23-63 µg/m3 (Angerer et al., 1983).
6.4 Elimination and excretion in expired air, faeces, and urine
6.4.1 Oral administration
In mammals, including human beings, lindane is excreted very
rapidly in urine and faeces after metabolic degradation; only small
quantities are eliminated unchanged (Seidler et al., 1975). As
lindane is subjected to four types of reaction -
dehydrochlorination, dechlorination, dehydrogenation, and oxidation
- many intermediate metabolites are found, the nature of which
depends on the initial reactions. Nevertheless, the excreted
metabolites are all various isomers of dichloro-, trichloro-, and
tetrachlorophenols, which are excreted either free or in a
conjugated form with glucuronic or sulfuric acid or
N-acetylcysteine (Rhône-Poulenc Agrochimie, 1986).
6.4.1.1 Rat
Sprague-Dawley rats were fed diets containing lindane at 400
mg/kg diet for 5 weeks. Within 24 h, mainly 2,3,4,6- and
2,3,4,5-tetrachlorophenols, 2,3,5-, 2,4,5-, and
2,4,6-trichlorophenols and 3,4-dichlorophenol were found in the free
form in urine and faeces, at 27.1%, 4.3%, 8.4%, 14.7%, and 51.1%,
respectively (Chadwick & Freal, 1972a; Chadwick et al., 1975).
After 14C-lindane was administered orally to rats at 8 mg/kg
body weight for 10 days, 23% of the metabolites in faeces and urine
were in the free form, and 77% in conjugated form, partly as
glucuronides (Seidler et al., 1975).
Metabolites were extracted from the urine of male Wistar rats
that had received 19 daily oral doses of 8 mg/kg body weight. After
hydrolysis of conjugates, the metabolites found were 2,4,6-TCP,
2,3,4,6- and 2,3,5,6-tetrachlorophenol, tetrachlorocyclohexenol,
pentachlorocyclohexenol, and pentachlorophenol (Engst et al., 1976).
Formation of 2,4,5,6- and 2,3,4,6-tetrachlorocyclohexenol was
confirmed by Chadwick et al. (1978) in a study with Sprague-Dawley
rats fed diets containing lindane at 400 mg/kg diet for one month.
6.4.1.2 Rabbit
Five rabbits fed gelatin capsules containing 14C-lindane at
3-12 mg/animal twice weekly for 26 weeks excreted 54% of the
radiolabel in urine and 13% in faeces. About 56% of the urinary
metabolites were soluble; those that were identified were 2,3,5-,
2,4,5-, and 2,4,6-trichlorophenol, 2,3,4,6- tetrachlorophenol, 2,3-
and 2,4-dichlorophenol and 2,3,4,5- tetrachlorophenol. The presence
of seven chlorophenols and six chlorobenzenes was indicated
(Karapally et al., 1973).
6.4.2 Other routes
6.4.2.1 Mouse
Lindane metabolites were analysed in the urine of mice after a
single intraperitoneal injection of 14C-lindane at 16 or 21
µg/mouse. Within three days, 57% of the total radiolabel had been
excreted in urine, mainly as conjugates with glucuronic or sulfuric
acid. About 25% of the excreted conversion products were 2,4,6-TCP,
and 4-6% was 2,4-DCP; ; 41-46% of the chlorophenols were conjugated
and 3% in the free form (Kurihara & Nakajima, 1974).
6.4.2.2 Rat
Intraperitoneal administration to rats of lindane in arachis
oil at daily doses of 40 mg/kg body weight (total, 4 g) was followed
by urinary excretion of 2,3,5- and 2,4,5-TCP, either in free form or
as sulfuric and glucuronic acid conjugates (Grover & Sims, 1965).
One day after intraperitoneal injection of a mixture of 14C-
and 36Cl-labelled lindane to rats, 18.97% of the radiolabel was
found in the excreta; 7.39% was still not absorbed, indicating again
that elimination of lindane begins during the absorption phase.
After 4 days, 52% of the total activity was found in the excreta.
The resulting half-time was about 4 days (Koransky et al., 1963). An
even shorter half-time of 1-2 days was seen in depot fat in another
study after intraperitoneal injection of 10 mg/kg to rats (Stein et
al., 1980).
After intraperitoneal administration of 40 mg/kg body weight to
rats, 20% of the total dose left the body via the faeces and 80%
via the urine (Koransky et al., 1963, 1964). In another study,
however, the amounts of radiolabel excreted by rats in urine and
faeces were about equal (Seidler et al., 1975). Only traces of
unchanged lindane were found in faeces and urine. of the chlorine
derived from lindane that is excreted in the urine, about 60% is
inorganic and 40% is organic (Koransky et al., 1964).
Mono-, di-, and trichlorophenyl mercapturic acids were found to
be the main metabolites after intraperitoneal administration of
lindane at 17.2 and 34.4 µmol to male Wistar rats, accounting for
more than 60% of the urinary metabolites (Kurihara et al., 1979).
6.4.2.3 Human
The urinary excretion of radiolabel after intravenous
administration of 14C-labelled lindane to six human subjects at 1
µCi in propylene glycol was 24.6% ± 6.1 of the administered dose
within five days. About 80% was excreted in first 24 h. The
half-time was 26 h (Feldmann & Maibach, 1974).
6.5 Retention and turnover (experimental animals)
The highest concentrations of gamma-HCH in the bodies of mice
were found 3 h after oral administration of a single dose of 1.2
mg/mouse. After 72 h, 270 µg of the original 1200 µg/animal
remained. A half-time of 2-3 days can be deduced from these results
(van Asperen, 1958).
When lindane was fed to rats for 56 days at doses of 1, 10, or
100 mg/kg diet, organ contents increased to a maximum within 2-3
weeks, depending on the dose. From that time on, the concentrations
in all organs decreased slowly, and equilibria were approached by
the end of the administration period. The concentration ratios
between different organs and blood remained constant throughout the
time of administration, and when treatment was stopped the
concentrations in all organs, including adipose tissue, decreased
rapidly. Similar results were seen after starvation for 6 days,
whereas diets rich in fat or protein accelerated the reduction of
the lindane content in organs and tissues. The kinetics of excretion
indicate a half-time of 3-4 days for oral administration of a single
dose of 14C-lindane (Oshiba, 1972).
After continuous feeding of lindane in corn oil at 50 mg/kg
diet for 60 days to Osborne-Mendel rats, a constant equilibrium
concentration of about 50 mg/kg was reached in adipose tissue within
9 days. After cessation of lindane administration, the concentration
dropped to values between 2.5 and 11.5 mg/kg tissue within 9 days
(Baron et al., 1975). One-half of an applied dose was excreted from
the bodies of rats within 3-4 days (Seidler et al., 1975).
After oral administration of gamma-HCH as a single dose of 60
mg/kg to rats, a maximal concentration of 8.8 ± 1.1 mg/kg tissue was
reached in the brain after 12-24 h. This concentration decreased
with a half-time of 1.5 days (Vohland et al., 1981). Oral
administration of lindane in the diet for 13 weeks at doses up to
100 mg/kg diet resulted in concentrations in fat that were lower
than those administered in the diet. The difference was more
pronounced at higher doses. After administration of 100 mg/kg diet,
11.4 mg/kg were found in fat (Suter et al., 1983).
All studies in which lindane was fed continuously to rats
showed that this compound does not accumulate in significant amounts
in the body. The highest accumulation factor found for fatty tissues
was about 2, and an average accumulation factor for fatty tissues of
about 1 can be deduced from the published data. The corresponding
factors for other tissues are considerably lower.
6.6 Biotransformation
6.6.1 Plants
In order to study the metabolism of lindane in wheat, plants
were grown from seed containing 480 mg/kg of 14C-lindane. In
seedlings, 35.5% of the radiolabel was associated with unmetabolized
lindane, 29.1% with the group of chlorobenzenes and 26.3% with
chlorophenols. In mature plants, the extractable residues consisted
of 5.4% lindane in roots and 21.4%in straw, up to 13.9%
chlorobenzenes and up to 53% chlorophenols. The chlorobenzenes
extracted from wheat roots were mostly tri- and tetrachlorobenzenes.
The concentrations of di- and pentachlorobenzene and of gamma-PCCH
were low (Balba & Saha, 1974).
Lindane and possible metabolites were determined in white
cabbage after leaf application and in carrots grown in treated soil.
In cabbage, a maximum of 0.04 mg/kg of lindane residues could be
detected at the time of harvest. In the carrots, residue levels of
0.4-1.0 mg/kg were found. In the second year after treatment,
residue levels were < 0.005 mg/kg. Up to 0.05 mg/kg of gamma-PCCH
and traces of 1,2,4-tri- and 1,2,3,4- tetrachlorobenzene were found.
Hexachlorobenzene was not detected in any of the samples (Eichler,
1975, 1980).
Itokawa et al. (1970) investigated the fate of 14C-lindane in
spinach and carrots grown in treated soil: 30-70% of the total
residues in the different plant parts were lindane. Five metabolites
were identified but not further characterized.
Lindane was converted to 36% soluble and 30% unextractable
residues under outdoor conditions after foliar application to
endives and lettuce. About 97% of the soluble fraction was found to
consist of chlorophenols in free or conjugated form. Minor
quantities of various chlorobenzenes were found. The conversion to
unextractable residues was dependent on weather conditions; the
composition of the unextractable residues was not analysed in detail
(Kohli et al., 1976a).
After lettuce plants were grown in nutrient solution containing
14C-lindane at 1.45 mg/kg for 4 weeks, the radiolabel extracted
from the plants consisted of about 77% lindane and 20% polar and 3%
non-polar residues. 2,3,4,6-Tetrachlorophenol, conjugated tetra- and
pentachlorophenol, and unidentified metabolites were found in the
polar fraction. The non-polar fraction contained tri- and
pentachlorobenzene as well as gamma-PCCH and HCCH (Kohli et al.,
1976b).
The metabolism of lindane was investigated in a variety of
plant-cell tissue cultures with high metabolic activity and in
lettuce plants grown in nutrient solution (Stöckigt, 1976; Stöckigt
& Ries, 1976). Tobacco tissue cultures were found to produce trace
amounts of 1,2,4-trichlorobenzene, while in carrot cultures,
1,2,3,4-tetrachlorobenzene and several isomers of trichlorophenol
conjugated with beta-glucose were found. This investigation also
demonstrated that intact lettuce plants cannot produce
pentachlorophenol or chlorobenzenes. Metabolism of lindane to carbon
dioxide was not detected.
Moza et al. (1974) applied gamma-PCCH, a plausible metabolic
intermediate of lindane, to young maize and pea plants in nutrient
solution. Various chlorobenzenes and chlorophenols were formed. The
most abundant metabolites in maize were 1,2,4,5-tetrachlorobenzene
and 2,3,5- and 2,4,5-trichlorophenol, and the most abundant in pea
plants was 1,2,4,5-tetrachlorobenzene.
Pea plants were grown under laboratory conditions in nutrient
solution containing 14C-lindane. After transfer into lindane-free
medium, either lindane or its metabolites were released from roots,
and to a lesser extent from other parts of the plant, within one day
(Charnetski & Lichtenstein, 1973).
The results described above are reflected in Figure 3, which
does not, however, include the conversion of lindane to traces of
hexachlorobenzene or pentachlorophenol or to the corresponding
alpha- and beta-HCH isomers (Kohli et al., 1976a,b; Steinwandter,
1976).
Appraisal
Lindane is not absorbed by the leaves of plants, and its poor
absorption by roots rapidly reaches a plateau. Most of the lindane
applied to plants is removed by evaporation or leaching. The rate of
metabolic transformation is low. The main degradation pathway
proceeds via formation of gamma-PCCH to tri- and tetrachlorophenol
in free or conjugated form. Other metabolites that have been
described occasionally, such as hexachlorocyclohexene and
chlorobenzenes, are present in only negligible quantities.
In the presence of oxygen and NADPH in vitro, rat liver
microsomes metabolize lindane mainly to 2,4,6-TCP (Tanaka et al.,
1977, 1979). 3,4,5,6-Tetrachlorocyclohexene was identified as an
intermediate after lindane was incubated under N2 with liver
preparations (Chadwick et al., 1978; Kurihara et al., 1979).
Experiments with rat liver preparations in vitro demonstrated the
importance of glutathione in the metabolism of lindane. Glutathione
enhanced the conversion of lindane to dichlorophenol (DCP) by a
factor of 3-4, but conjugates were formed only in the presence of
liver cytosol protein as a source of glutathione transferase. The
initial step appears to be dehydrochlorination to 1,3,4,5,6-PCCH,
followed by conjugation and further dehydrochlorination to DCP -
mainly 2,4-DCP. The DCPs found in the urine of rats are a mixture of
different isomers. The rate of formation of S-(dichlorophenyl)
glutathione from HCH in rat liver cytosol apparently depends on
gradual monodehydrochlorination, and the enzymatic transfer of
reduced L-glutathione (GSH) onto PCCH is not preceded by a second
dehydrochlorination (Stein et al., 1977; Portig et al., 1979; Tanaka
et al., 1979).
When a 1:1 mixture of lindane and the corresponding
hexadeuterated compound was fed to Wistar rats, the ratio of MCP,
DCP, and TCP, excreted as mercapturic acids, showed an isotopic
effect. The rate-limiting step for the formation of DCP and TCP is
either a dehydrochlorination or a dehydrogenation of lindane,
whereas formation of MCP must be initiated by dechlorination to
tetrachlorocyclohexane, followed by conjugation with glutathione
(Kurihara et al., 1980).
Lindane was converted mainly to gamma-HCCH by rat liver
microsomes, and significant amounts of 2,4,6-TCP and
2,3,4,6-tetrachlorophenol were detected. Human liver microsomes
converted lindane into four major metabolites:
gamma-1,2,3,4,5,6-hexachlorocyclohex-1-ene; gamma-1,3,4,5,6-PCCH;
beta-1,3,4,5,6-PCCH, and 2,4,6-TCP. Smaller amounts of
2,3,4,6-tetrachlorophenol and pentachlorobenzene were found
(Fitzloff et al., 1982).
Human and rat liver microsomes converted the lindane
metabolites gamma-PCCH and 3,4,6/5-PCCH to 1,2,4-tetrachlorobenzene,
1,2,3,4-tetrachlorobenzene, 2,4,5-TCP, 3,4,5/6-pentachloro-2-
cyclohexen-1-ol and beta-PCCH-oxide or 3,4,6/5-PCCH-oxide. The
identity of the beta-PCCH-oxide was confirmed by column
chromatography and gas-liquid chromatography-mass spectrometry. It
is stable to hydrolysis by microsomal epoxide hydrolase
(E.C.3.3.2.3.) and under various aqueous acid conditions. Its
toxicological role is still unknown. Although this compound is
structurally related to epichlorhydrin and epoxides, it was not
mutagenic to Salmonella typhimurium strain TM 677 (Fitzloff & Pan,
1984).
Two groups have reported the formation of trace amounts of
chlorinated benzenes from lindane in rats. Hexachlorobenzene was
found in faeces (Gopalaswamy & Aiyar, 1984) and pentachlorobenzene
in brain (Vohland et al., 1981). The two studies are consistent in
so far as the identified amounts of chlorobenzenes are extremely low
and near the detection limit; however, they are also contradictory,
because hexachlorobenzene was found exclusively in the first study
and pentachlorobenzene in the second. It is impossible to clarify
whether artefacts were measured in these studies, e.g., enrichment
of impurities in the starting material. If indeed chlorinated
benzenes are formed from lindane, the amounts obtained are
insignificant compared to those of other metabolites.
6.3.2 Identification of metabolites
Metabolites of lindane have been identified in a large number
of studies, in vivo in body fluids, urine, faeces, organs, and
tissues, and in vitro. Most of the in-vivo studies were carried
out with rats, but similar results were obtained in other animal
species.
The following metabolites have been identified:
Cycloalkenes: 1,2,3,4,5,6-hexachlorocyclohexene (HCCH);
1,3,4,5,6-pentachlorocyclohexene (PCCH); 3,4,5,6-
tetrachlorocyclohexene; 2,3,4,5,6-pentachloro-2-cyclohexen-1-ol; and
2,3,4,6- and 2,4,5,6-tetrachloro-2-cyclohexene-1-ol (Chadwick &
Freal, 1972a; Freal & Chadwick, 1973; Chadwick et al., 1975; Engst
et al., 1976; Kujawa et al., 1977; Tanaka et al., 1977; Chadwick et
al., 1978; Engst et al., 1978b; Kurihara et al., 1979; Vohland et
al., 1981; Fitzloff et al., 1982; Mottram et al., 1983; Sanfeliu et
al., 1988).
Chlorobenzenes: 1,2,3,4,5,6-hexachlorobenzene, 1,2,3,4,5-
pentachlorobenzene and 1,2,3,5-, 1,2,4,5-, and 1,2,3,4-
tetrachlorobenzene (Aiyar, 1980; Voh land et al., 1981; Gopalaswamy
& Aiyar, 1984; Artigas et al., 1988). Mono-, di-, tri-, and
tetrachlorobenzenes have been reported in pigs (Mottram et al.,
1983).
Chlorophenols: 2,3,4,5,6-pentachlorophenol; 2,3,4,5-,
2,3,4,6-, and 2,3,5,6-tetrachlorophenol; 2,3,5-, 2,4,5-, and
2,4,6-trichlorophenol (TCP); and 2,4- and 3,4-dichlorophenol (DCP)
(Grover & Sims, 1965; Chadwick & Freal, 1972a,b; Freal & Chadwick,
1973; Kurihara & Nakajima, 1974; Chadwick et al., 1975; Engst et
al., 1976; Kujawa et al., 1977; Stein et al., 1977; Tanaka et al.,
1977; Engst et al., 1978b; Tanaka et al., 1979; Aiyar, 1980;
Fitzloff et al., 1982).
Conjugates of these compounds:
- 2,3- and 2,6-dichlorophenol were conjugated with glutathione
(Portig et al., 1979);
- 2,4-DCP; 2,3,5-, 2,4,5-, and 2,4,6-TCP; 2,3,4,5-, 2,3,4,6-, and
2,3,5,6-tetrachlorophenol; 2,3,4,5,6-PCP and
tetrachloro-2-cyclohexene-1-ol were conjugated with glucuronic
acid (Grover & Sims, 1965; Kurihara & Nakajima, 1974; Engst
et al., 1976; Chadwick et al., 1981);
- 2,3,5-, 2,4,5-, and 2,4,6-TCP; 2,3,4,5- and 2,3,4,6-
tetrachlorophenol; 2,3,4,6- tetrachloro-2-cyclohexene-1-ol; and
2,3,4,5,6-pentachloro-2-cyclohexen-1-ol were conjugated with
sulfate (Grover & Sims, 1965; Kurihara & Nakajima, 1974;
Chadwick et al., 1981); and
- 4-monochlorophenol; 2,4- and 3,4-DCP and 2,3,5- and 2,4,5-TCP
were conjugated with mercapturic acids (Grover & Sims, 1965;
Kurihara et al., 1979, 1980).
6.3.3 Metabolites identified in humans
Engst et al. (1978b) analysed urine from workers apparently
exposed to technical-grade HCH (during manufacture?) and found
alpha-, beta-, gamma-, and delta-HCH, traces of hexa- and
pentachlorobenzene, gamma- and delta-PCCH, pentachlorophenol,
2,3,4,5-, 2,3,4,6-, and 2,3,5,6-tetrachlorophenol, several
trichlorophenols, as well as glucuronides of some of these
metabolites. The PCCHs, tetrachlorophenol, hexachlorobenzene, and
pentachlorophenol were also identified in blood.
The urine of 21 men working in the production of gamma-HCH with
a purity of 99.8% from technical-grade HCH (16% alpha-, 7% beta-,
and 45% gamma-HCH) was examined for the presence of chlorinated
phenols. External and internal exposure was estimated from
measurements of the concentrations of HCH isomers in the air of the
workroom and in serum samples. The men had been employed for periods
ranging from a few months up to 30 years (mean, 10.6 years,) and
they were aged 24-62 years (mean, 46 years). Fourteen mono-, di-,
tri-, and tetrachlorophenols and seven dihydroxychlorobenzenes of
unknown configuration were identified in urine. The main metabolites
were 2,4,6-, 2,3,5-, and 2,4,5-trichlorophenol, which were excreted
in nearly equal quantities. The mean concentrations of alpha-,
beta-, and gamma-HCH in the serum of exposed workers were 49 (range,
11-138), 82 (17-434) and 52 (9-188) µg/litre; the levels in controls
were < 1.0 µg/litre. The air concentrations of alpha-, beta-, and
gamma-HCH were 2-4, 1-3, and 23-63 µg/m3 (Angerer et al., 1983).
6.4 Elimination and excretion in expired air, faeces, and urine
6.4.1 Oral administration
In mammals, including human beings, lindane is excreted very
rapidly in urine and faeces after metabolic degradation; only small
quantities are eliminated unchanged (Seidler et al., 1975). As
lindane is subjected to four types of reaction -
dehydrochlorination, dechlorination, dehydrogenation, and oxidation
- many intermediate metabolites are found, the nature of which
depends on the initial reactions. Nevertheless, the excreted
metabolites are all various isomers of dichloro-, trichloro-, and
tetrachlorophenols, which are excreted either free or in a
conjugated form with glucuronic or sulfuric acid or
N-acetylcysteine (Rhône-Poulenc Agrochimie, 1986).
6.4.1.1 Rat
Sprague-Dawley rats were fed diets containing lindane at 400
mg/kg diet for 5 weeks. Within 24 h, mainly 2,3,4,6- and
2,3,4,5-tetrachlorophenols, 2,3,5-, 2,4,5-, and
2,4,6-trichlorophenols and 3,4-dichlorophenol were found in the free
form in urine and faeces, at 27.1%, 4.3%, 8.4%, 14.7%, and 51.1%,
respectively (Chadwick & Freal, 1972a; Chadwick et al., 1975).
After 14C-lindane was administered orally to rats at 8 mg/kg
body weight for 10 days, 23% of the metabolites in faeces and urine
were in the free form, and 77% in conjugated form, partly as
glucuronides (Seidler et al., 1975).
Metabolites were extracted from the urine of male Wistar rats
that had received 19 daily oral doses of 8 mg/kg body weight. After
hydrolysis of conjugates, the metabolites found were 2,4,6-TCP,
2,3,4,6- and 2,3,5,6-tetrachlorophenol, tetrachlorocyclohexenol,
pentachlorocyclohexenol, and pentachlorophenol (Engst et al., 1976).
Formation of 2,4,5,6- and 2,3,4,6-tetrachlorocyclohexenol was
confirmed by Chadwick et al. (1978) in a study with Sprague-Dawley
rats fed diets containing lindane at 400 mg/kg diet for one month.
6.4.1.2 Rabbit
Five rabbits fed gelatin capsules containing 14C-lindane at
3-12 mg/animal twice weekly for 26 weeks excreted 54% of the
radiolabel in urine and 13% in faeces. About 56% of the urinary
metabolites were soluble; those that were identified were 2,3,5-,
2,4,5-, and 2,4,6-trichlorophenol, 2,3,4,6- tetrachlorophenol, 2,3-
and 2,4-dichlorophenol and 2,3,4,5- tetrachlorophenol. The presence
of seven chlorophenols and six chlorobenzenes was indicated
(Karapally et al., 1973).
6.4.2 Other routes
6.4.2.1 Mouse
Lindane metabolites were analysed in the urine of mice after a
single intraperitoneal injection of 14C-lindane at 16 or 21
µg/mouse. Within three days, 57% of the total radiolabel had been
excreted in urine, mainly as conjugates with glucuronic or sulfuric
acid. About 25% of the excreted conversion products were 2,4,6-TCP,
and 4-6% was 2,4-DCP; ; 41-46% of the chlorophenols were conjugated
and 3% in the free form (Kurihara & Nakajima, 1974).
6.4.2.2 Rat
Intraperitoneal administration to rats of lindane in arachis
oil at daily doses of 40 mg/kg body weight (total, 4 g) was followed
by urinary excretion of 2,3,5- and 2,4,5-TCP, either in free form or
as sulfuric and glucuronic acid conjugates (Grover & Sims, 1965).
One day after intraperitoneal injection of a mixture of 14C-
and 36Cl-labelled lindane to rats, 18.97% of the radiolabel was
found in the excreta; 7.39% was still not absorbed, indicating again
that elimination of lindane begins during the absorption phase.
After 4 days, 52% of the total activity was found in the excreta.
The resulting half-time was about 4 days (Koransky et al., 1963). An
even shorter half-time of 1-2 days was seen in depot fat in another
study after intraperitoneal injection of 10 mg/kg to rats (Stein et
al., 1980).
After intraperitoneal administration of 40 mg/kg body weight to
rats, 20% of the total dose left the body via the faeces and 80%
via the urine (Koransky et al., 1963, 1964). In another study,
however, the amounts of radiolabel excreted by rats in urine and
faeces were about equal (Seidler et al., 1975). Only traces of
unchanged lindane were found in faeces and urine. of the chlorine
derived from lindane that is excreted in the urine, about 60% is
inorganic and 40% is organic (Koransky et al., 1964).
Mono-, di-, and trichlorophenyl mercapturic acids were found to
be the main metabolites after intraperitoneal administration of
lindane at 17.2 and 34.4 µmol to male Wistar rats, accounting for
more than 60% of the urinary metabolites (Kurihara et al., 1979).
6.4.2.3 Human
The urinary excretion of radiolabel after intravenous
administration of 14C-labelled lindane to six human subjects at 1
µCi in propylene glycol was 24.6% ± 6.1 of the administered dose
within five days. About 80% was excreted in first 24 h. The
half-time was 26 h (Feldmann & Maibach, 1974).
6.5 Retention and turnover (experimental animals)
The highest concentrations of gamma-HCH in the bodies of mice
were found 3 h after oral administration of a single dose of 1.2
mg/mouse. After 72 h, 270 µg of the original 1200 µg/animal
remained. A half-time of 2-3 days can be deduced from these results
(van Asperen, 1958).
When lindane was fed to rats for 56 days at doses of 1, 10, or
100 mg/kg diet, organ contents increased to a maximum within 2-3
weeks, depending on the dose. From that time on, the concentrations
in all organs decreased slowly, and equilibria were approached by
the end of the administration period. The concentration ratios
between different organs and blood remained constant throughout the
time of administration, and when treatment was stopped the
concentrations in all organs, including adipose tissue, decreased
rapidly. Similar results were seen after starvation for 6 days,
whereas diets rich in fat or protein accelerated the reduction of
the lindane content in organs and tissues. The kinetics of excretion
indicate a half-time of 3-4 days for oral administration of a single
dose of 14C-lindane (Oshiba, 1972).
After continuous feeding of lindane in corn oil at 50 mg/kg
diet for 60 days to Osborne-Mendel rats, a constant equilibrium
concentration of about 50 mg/kg was reached in adipose tissue within
9 days. After cessation of lindane administration, the concentration
dropped to values between 2.5 and 11.5 mg/kg tissue within 9 days
(Baron et al., 1975). One-half of an applied dose was excreted from
the bodies of rats within 3-4 days (Seidler et al., 1975).
After oral administration of gamma-HCH as a single dose of 60
mg/kg to rats, a maximal concentration of 8.8 ± 1.1 mg/kg tissue was
reached in the brain after 12-24 h. This concentration decreased
with a half-time of 1.5 days (Vohland et al., 1981). Oral
administration of lindane in the diet for 13 weeks at doses up to
100 mg/kg diet resulted in concentrations in fat that were lower
than those administered in the diet. The difference was more
pronounced at higher doses. After administration of 100 mg/kg diet,
11.4 mg/kg were found in fat (Suter et al., 1983).
All studies in which lindane was fed continuously to rats
showed that this compound does not accumulate in significant amounts
in the body. The highest accumulation factor found for fatty tissues
was about 2, and an average accumulation factor for fatty tissues of
about 1 can be deduced from the published data. The corresponding
factors for other tissues are considerably lower.
6.6 Biotransformation
6.6.1 Plants
In order to study the metabolism of lindane in wheat, plants
were grown from seed containing 480 mg/kg of 14C-lindane. In
seedlings, 35.5% of the radiolabel was associated with unmetabolized
lindane, 29.1% with the group of chlorobenzenes and 26.3% with
chlorophenols. In mature plants, the extractable residues consisted
of 5.4% lindane in roots and 21.4%in straw, up to 13.9%
chlorobenzenes and up to 53% chlorophenols. The chlorobenzenes
extracted from wheat roots were mostly tri- and tetrachlorobenzenes.
The concentrations of di- and pentachlorobenzene and of gamma-PCCH
were low (Balba & Saha, 1974).
Lindane and possible metabolites were determined in white
cabbage after leaf application and in carrots grown in treated soil.
In cabbage, a maximum of 0.04 mg/kg of lindane residues could be
detected at the time of harvest. In the carrots, residue levels of
0.4-1.0 mg/kg were found. In the second year after treatment,
residue levels were < 0.005 mg/kg. Up to 0.05 mg/kg of gamma-PCCH
and traces of 1,2,4-tri- and 1,2,3,4- tetrachlorobenzene were found.
Hexachlorobenzene was not detected in any of the samples (Eichler,
1975, 1980).
Itokawa et al. (1970) investigated the fate of 14C-lindane in
spinach and carrots grown in treated soil: 30-70% of the total
residues in the different plant parts were lindane. Five metabolites
were identified but not further characterized.
Lindane was converted to 36% soluble and 30% unextractable
residues under outdoor conditions after foliar application to
endives and lettuce. About 97% of the soluble fraction was found to
consist of chlorophenols in free or conjugated form. Minor
quantities of various chlorobenzenes were found. The conversion to
unextractable residues was dependent on weather conditions; the
composition of the unextractable residues was not analysed in detail
(Kohli et al., 1976a).
After lettuce plants were grown in nutrient solution containing
14C-lindane at 1.45 mg/kg for 4 weeks, the radiolabel extracted
from the plants consisted of about 77% lindane and 20% polar and 3%
non-polar residues. 2,3,4,6-Tetrachlorophenol, conjugated tetra- and
pentachlorophenol, and unidentified metabolites were found in the
polar fraction. The non-polar fraction contained tri- and
pentachlorobenzene as well as gamma-PCCH and HCCH (Kohli et al.,
1976b).
The metabolism of lindane was investigated in a variety of
plant-cell tissue cultures with high metabolic activity and in
lettuce plants grown in nutrient solution (Stöckigt, 1976; Stöckigt
& Ries, 1976). Tobacco tissue cultures were found to produce trace
amounts of 1,2,4-trichlorobenzene, while in carrot cultures,
1,2,3,4-tetrachlorobenzene and several isomers of trichlorophenol
conjugated with beta-glucose were found. This investigation also
demonstrated that intact lettuce plants cannot produce
pentachlorophenol or chlorobenzenes. Metabolism of lindane to carbon
dioxide was not detected.
Moza et al. (1974) applied gamma-PCCH, a plausible metabolic
intermediate of lindane, to young maize and pea plants in nutrient
solution. Various chlorobenzenes and chlorophenols were formed. The
most abundant metabolites in maize were 1,2,4,5-tetrachlorobenzene
and 2,3,5- and 2,4,5-trichlorophenol, and the most abundant in pea
plants was 1,2,4,5-tetrachlorobenzene.
Pea plants were grown under laboratory conditions in nutrient
solution containing 14C-lindane. After transfer into lindane-free
medium, either lindane or its metabolites were released from roots,
and to a lesser extent from other parts of the plant, within one day
(Charnetski & Lichtenstein, 1973).
The results described above are reflected in Figure 3, which
does not, however, include the conversion of lindane to traces of
hexachlorobenzene or pentachlorophenol or to the corresponding
alpha- and beta-HCH isomers (Kohli et al., 1976a,b; Steinwandter,
1976).
Appraisal
Lindane is not absorbed by the leaves of plants, and its poor
absorption by roots rapidly reaches a plateau. Most of the lindane
applied to plants is removed by evaporation or leaching. The rate of
metabolic transformation is low. The main degradation pathway
proceeds via formation of gamma-PCCH to tri- and tetrachlorophenol
in free or conjugated form. Other metabolites that have been
described occasionally, such as hexachlorocyclohexene and
chlorobenzenes, are present in only negligible quantities.
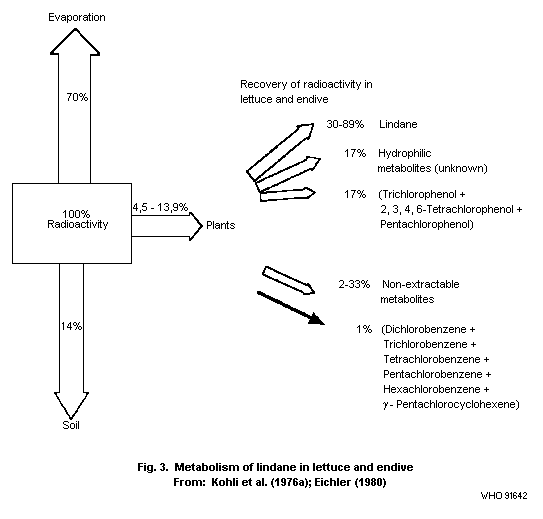 6.6.2 Microorganisms
The metabolism of lindane has also been investigated in
bacteria, fungi, and algae. Chlorocycloalkenes, chlorobenzenes, and
chlorophenols were found to be metabolic intermediates, and carbon
dioxide to be the end-product. Volatile, chlorine-free hydrocarbons
were also found (Haider & Jagnow, 1975; Haider et al., 1975).
Mixed populations of bacteria metabolize lindane to gamma-PCCH,
alpha-, beta-, or gamma-3,4,5,6-tetrachloro-1- cyclohexene (TCCH),
pentachlorobenzene, 1,2,3,4-, 1,2,3,5-, or
1,2,4,5-tetrachlorobenzene, 1,2,4- or 1,3,5-trichlorobenzene, 1,2-
and 1,4- dichlorobenzene, as well as carbon dioxide (Yule et al.,
1967; Haider et al., 1974; Kohnen et al., 1975; Mathur & Saha, 1975;
Tu, 1975; Haider et al., 1976; Jagnow et al., 1977; Mathur & Saha,
1977; Vonk & Quirijns, 1979).
Metabolites of lindane were identified as PCCH and TCCH in
populations of Escherichia coli (Francis et al., 1975; Vonk &
Quirijns, 1979); PCCH, tetrachloro-1-cyclohexene, 1,2,3,4-
tetrachlorobenzene, and carbon dioxide in Pseudomonas sp. (Benezet
& Matsumura, 1973; Matsumura et al., 1976; Engst et al., 1979a); and
TCCH, 1,2,4-trichlorobenzene, and 1,4-dichlorobenzene in
Clostridium sp. (Heritage & MacRae, 1977a,b; Ohisa & Yamaguchi,
1978b; Heritage & MacRae, 1979; Ohisa et al., 1980, 1982).
Quantitative data on the metabolism of lindane in bacteria are
given by MacRae et al. (1967), Benezet & Matsumura (1973), Haider et
al. (1974), Haider & Jagnow (1975), Kohnen et al. (1975), Mathur &
Saha (1975, 1977), and Haider (1979). The most abundant metabolites
are PCCH and TCCH (up to 45.8% and 21.7%, respectively, of the
initial dose of lindane). Chlorobenzenes may occur in only small or
trace amounts. Carbon dioxide is formed under aerobic or submerged
incubation conditions, and up to 20% of an initial dose of lindane
was converted to carbon dioxide within 140 days (Kohnen et al.,
1975). Strictly anaerobic conditions resulted in rapid release of
chloride from lindane and in its conversion to volatile chlorine-
free metabolites. Within 5 days, up to 90% of the applied dose had
been released as volatile, chlorine-free hydrocarbons (Haider &
Jagnow, 1975).
Lindane was shown to be effectively metabolized in the algae
Chlorella and Chlamydomonas (Sweeney, 1969; Elsner et al.,
1972); 1,3,4,5,6-PCCH was reported to occur as a metabolite, but no
quantitative data are available.
Depending on the availability of oxygen, lindane may follow
various metabolic pathways in bacteria (Fig. 4).
6.6.2 Microorganisms
The metabolism of lindane has also been investigated in
bacteria, fungi, and algae. Chlorocycloalkenes, chlorobenzenes, and
chlorophenols were found to be metabolic intermediates, and carbon
dioxide to be the end-product. Volatile, chlorine-free hydrocarbons
were also found (Haider & Jagnow, 1975; Haider et al., 1975).
Mixed populations of bacteria metabolize lindane to gamma-PCCH,
alpha-, beta-, or gamma-3,4,5,6-tetrachloro-1- cyclohexene (TCCH),
pentachlorobenzene, 1,2,3,4-, 1,2,3,5-, or
1,2,4,5-tetrachlorobenzene, 1,2,4- or 1,3,5-trichlorobenzene, 1,2-
and 1,4- dichlorobenzene, as well as carbon dioxide (Yule et al.,
1967; Haider et al., 1974; Kohnen et al., 1975; Mathur & Saha, 1975;
Tu, 1975; Haider et al., 1976; Jagnow et al., 1977; Mathur & Saha,
1977; Vonk & Quirijns, 1979).
Metabolites of lindane were identified as PCCH and TCCH in
populations of Escherichia coli (Francis et al., 1975; Vonk &
Quirijns, 1979); PCCH, tetrachloro-1-cyclohexene, 1,2,3,4-
tetrachlorobenzene, and carbon dioxide in Pseudomonas sp. (Benezet
& Matsumura, 1973; Matsumura et al., 1976; Engst et al., 1979a); and
TCCH, 1,2,4-trichlorobenzene, and 1,4-dichlorobenzene in
Clostridium sp. (Heritage & MacRae, 1977a,b; Ohisa & Yamaguchi,
1978b; Heritage & MacRae, 1979; Ohisa et al., 1980, 1982).
Quantitative data on the metabolism of lindane in bacteria are
given by MacRae et al. (1967), Benezet & Matsumura (1973), Haider et
al. (1974), Haider & Jagnow (1975), Kohnen et al. (1975), Mathur &
Saha (1975, 1977), and Haider (1979). The most abundant metabolites
are PCCH and TCCH (up to 45.8% and 21.7%, respectively, of the
initial dose of lindane). Chlorobenzenes may occur in only small or
trace amounts. Carbon dioxide is formed under aerobic or submerged
incubation conditions, and up to 20% of an initial dose of lindane
was converted to carbon dioxide within 140 days (Kohnen et al.,
1975). Strictly anaerobic conditions resulted in rapid release of
chloride from lindane and in its conversion to volatile chlorine-
free metabolites. Within 5 days, up to 90% of the applied dose had
been released as volatile, chlorine-free hydrocarbons (Haider &
Jagnow, 1975).
Lindane was shown to be effectively metabolized in the algae
Chlorella and Chlamydomonas (Sweeney, 1969; Elsner et al.,
1972); 1,3,4,5,6-PCCH was reported to occur as a metabolite, but no
quantitative data are available.
Depending on the availability of oxygen, lindane may follow
various metabolic pathways in bacteria (Fig. 4).
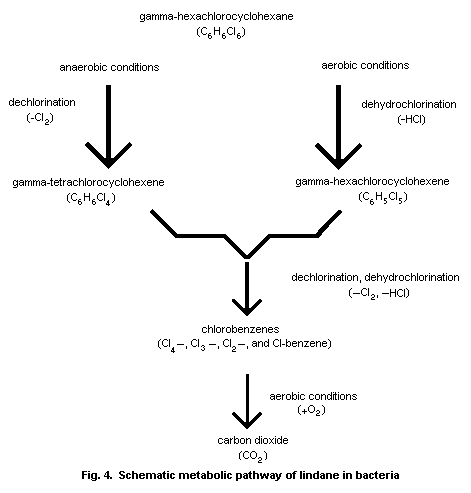 Unspecified fungi were also able to metabolize lindane,
although at a lower rate than bacteria. The following metabolites
were identified: gamma-PCCH; hexachlorobenzene; pentachlorobenzene;
TCCH; 1,2,3,4-, 1,2,3,5-, and 1,2,4,5- tetrachlorobenzene; 1,2,3-,
1,2,4-, and 1,3,5-trichlorobenzene; 1,2- and 1,4- dichlorobenzene;
pentachlorophenol; 2,3,4,5-, 2,3,4,6-, and
2,3,5,6-tetrachlorophenol; 2,3,4- and 2,4,6-trichlorophenol, and
carbon dioxide. Metabolic intermediates such as PCCH were found at
up to 1% of the initial dose of lindane. About 1% of the intial dose
was converted to carbon dioxide after an incubation period of 52
days (Engst et al., 1974; Kujawa et al., 1976; Engst et al., 1977).
6.6.2.1 Anaerobic conditions
The influence of growth conditions on the metabolic route of
lindane in bacteria was demonstrated in the facultative anaerobe E.
coli as well as with mixed populations of soil microorganisms
(Mathur & Saha, 1977; Vonk & Quirijns, 1979). These reports and
others demonstrate the predominant formation of gamma-TCCH under
anaerobic growth conditions. Anaerobic metabolism consists of a
series of dechlorinating steps, leading to rapid formation of
chlorine-free, volatile hydrocarbons and chloride (Haider & Jagnow,
1975; Jagnow et al., 1977). Carbon dioxide is not formed under
anaerobic conditions.
A possible metabolic pathway under anaerobic conditions was
proposed by Ohisa et al. (1980). Lindane is dechlorinated by a
cytochrome P-450-dependent reaction to TCCH, followed by
dechlorination to the unstable dichlorocyclohexadiene and
dehydrochlorination to monochlorobenzene. The degradation of lindane
serves as an energy source for the cells (Ohisa et al., 1982). A
relationship between the metabolization of lindane and the Stickland
reaction (a coupled oxidation-reduction reaction between pairs of
amino acids) has been discussed (Ohisa & Yamaguchi, 1979; Ohisa et
al., 1980, 1982). The ability to degrade lindane is linked to a
bacterial enzyme system that catalyses the evolution of hydrogen
during fermentation (Jagnow et al., 1977).
6.6.2.2 Aerobic conditions
Under aerobic conditions, the metabolism of lindane in bacteria
is initiated predominantly by dehydrochlorination to gamma-PCCH
(Vonk & Quirijns, 1979). Further intermediates are chlorobenzenes,
and the end-product is carbon dioxide. No phenolic intermediate was
observed under submerged conditions (Mathur & Saha, 1975).
6.7 Isomerization
The detection of beta-HCH in the tissues of rats fed gamma-HCH
led to the conclusion that isomerization of lindane had occurred
(Kamada, 1971); however, the purity of the gamma-HCH used was not
given in this report, and it is possible that impurities were
measured.
Studies in rats by Copeland & Chadwick (1979) and Eichler et
al. (1983) demonstrated that lindane did not undergo
bioisomerization. Gopalaswamy & Aiyar (1984) reported
biotransformation of gamma-HCH to hexachlorobenzene in male rats.
The results of a study by Chadwick & Copeland (1985), however, using
six young female Fischer 344 rats administered lindane in arachis
oil at 20 mg/kg body weight daily for six days (control animals
received the vehicle only), indicated that no significant
biotransformation of lindane to hexachlorobenzene occurred in these
animals. The gamma-HCH contents of adipose tissue on a fat basis
were 0.04 ± 0.003 mg/kg in controls and 129 ± 6 mg/kg in
lindane-treated rats.
The possibility of isomerization of lindane to alpha- and
beta-HCH was also investigated in mixed populations of soil
microorganisms and other defined bacterial strains.
Newland et al. (1969) studied the degradation of lindane in
simulated lake impoundments and found traces of alpha-HCH under
anaerobic incubation conditions. Benezet & Matsumura (1973)
described the formation of small amounts of alpha-HCH from lindane
incubated either with aquatic sediments or with suspension cultures
of Pseudomonas putida supplemented with NAD. Matsumura et al.
(1976) described an NAD-dependent pathway in P. putida leading to
the formation of alpha-HCH under anaerobic conditions. Three percent
of the initial radioactivity of 14C-lindane was found at a
location with the same Rf value as alpha-HCH after separation of the
metabolites by thin-layer chromatography.
Engst et al. (1979b) indicated isomerization of lindane to
alpha- and beta-HCH in anaerobically grown cultures of P.
aeruginosa, in a study in which metabolites were analysed by gas
chromatography. Lindane was not reported to be isomerized to alpha-
or beta-HCH in fungi (Engst et al., 1977).
Vonk & Quirijns (1979) found a conversion rate of lindane to
alpha-HCH of 0.2% after anaerobic incubation of lindane for 4 or 8
weeks with either sandy or silt loam soil samples and E. coli. No
alpha-HCH was formed in control experiments in which sterile
nutrient medium was incubated with gamma-HCH for 28 days. Growing
mycelium of Aspergillus niger produced no alpha-HCH. In this
study, metabolites were identified by gas-liquid chromatography and
verified by mass spectrometry.
Haider (1979, 1983) tested anaerobic, semi-anaerobic, and
aerobic incubation of radioactive labelled lindane with
Citrobacter, Serratia, Clostridium, Klebsiella, Pseudomonas, and
E. coli. Incubation did not increase the level of alpha-HCH. The
results of the experiment with Pseudomonas under anaerobic
conditions were not reported.
Deo et al. (1981) studied the interconversion of gamma-HCH in a
sterile aquatic solution over periods of 1 day up to 4 weeks.
Gas-liquid chromatography indicated a slow interconversion of
gamma-HCH with time. Solvent extracts were tested for their toxicity
by topical application onto 2-day-old Drosophila melanogaster. The
observed decrease in toxicity of the gamma-HCH solution with time
may have been due to both degradation and isomerization to less
toxic isomers, such as alpha-HCH.
Taken together, the results of tests conducted under anaerobic
conditions show that only a very small amount of lindane, if any, is
converted to alpha-HCH, and there is no conclusive indication of
isomerization to beta-HCH.
7. EFFECTS ON LABORATORY MAMMALS AND IN IN-VITRO TEST SYSTEMS
Lindane has been tested for acute and for short- and long-term
toxicity in a number of animal species. Some of the earlier studies
were undertaken using material of unspecified purity; and in some
others, technical-grade HCH was used that contained various
quantities of alpha- and beta-HCH, in addition to lindane. Those
studies that are relevant to this review have been included.
7.1 Single exposure
The acute toxicity of lindane has been investigated in numerous
studies in a variety of species and strains of laboratory animals
via several routes of application. The reported LD50 values for
lindane given by different routes of administration are of the same
order of magnitude in the various species, and no sex-dependent
difference was seen. A marked difference in acute oral toxicity
results from the type of vehicle used: oily solutions of lindane
were more toxic than suspensions in water, and when mineral oils
were used as carriers fewer toxic effects were seen than with
vegetable oils (Muralidhara et al., 1979). Young animals were
generally more sensitive than adults. Lindane was more toxic to
animals suffering from protein deficiency than to rats with a normal
protein supply (Chen, 1968).
7.1.1 Oral
The LD50 values for the mouse, rat, guinea-pig, and rabbit
are summarized in Table 8.
The choice of vehicle used for administering lindane in studies
of its acute oral toxicity is important: the LD50 after
administration in an oily solution or of an emulsifiable concentrate
was 88 mg/kg body weight, but that for wettable powders, granules,
flowable concentrations and aqueous suspensions was in the order of
170 mg/kg or even higher.
Single oral doses of 40 mg/kg body weight dissolved in oil were
lethal to dogs; dogs that received 30 mg/kg survived but had
convulsions (Barke, 1950). McNamara and Krop (1948) found that a
dose of 100 mg/kg body weight was lethal to all of three treated
dogs, and 50 mg/kg caused death in four out of seven animals. These
data indicate that the lethal dose for dogs of lindane administered
as an oily solution is about 40-50 mg/kg body weight.
Table 8. Reported oral LD50 values for g-HCH in experimental
animals
Species LD50(mg/kg) References
Rats (male, female, or 90-270 Slade (1945); Woodard &
males and females) Hagan (1947);
Riemshneider (1949);
Antonovic (1958); Gaines
(1960); Edson et al.
(1966); Muacevic (1966,
1970, 1971a,b); Chen
(1968); Schafer (1972);
Frohberg et al. (1972b)
Mice (different strains) 55-250 Woodard & Hagan (1947);
Graeve & Herrnring
(1951); Nurmatov (1965);
Frohberg et al. (1972a);
Paul et al. (1980);
Wolfe & Ralph (1980)
Guinea-pig 100 Cameron (1945)
Rabbit 90-200 Cameron (1945); Nurmatov
(1965)
In an incident in which eight cows ingested a powder containing
19.1% gamma-HCH, those that ate 112 g or more of the powder died,
while those given 70 g survived. These findings indicate that, for
cows, the fatal dose was between 70 and 112 g or 140-225 mg/kg body
weight of the powder (equivalent to 28-45 mg/kg body weight
(McParland et al., 1973).
7.1.2 Intraperitoneal and intramuscular
Mice of the NMRI-EMD strain (SPF) were administered lindane as
a 0.5% suspension in 0.5% carboxymethylcellulose solution and were
observed for 14 days. The intraperitoneal and intramuscular LD50
values were found to be 97 and 152 mg/kg body weight, respectively
(Frohberg et al., 1972a). In rats, only the intraperitoneal LD50
has been determined: it was found to be 69 mg/kg body weight in
Wistar-AF/HAN-EMD administered the compound in
carboxymethylcellulose (Frohberg et al., 1972b).
7.1.3 Inhalation
Wistar (HAN/Boe) rats were exposed by whole-body exposure to
lindane at (analytical) concentrations of 0, 273, or 603 mg/m3 for
4 h. The average particle size was 0.4 µm, and the animals were
observed for 14 days. Neither deaths nor abnormalities were found
(Oldiges et al., 1980). The
The 4-h acute LC50 for a lindane (99.6%) aerosol was determined
by exposing four groups of five males and five female KFM-HAN Wistar
rats by inhalation to aerosols containing lindane at 0.1, 0.38,
0.64, or 2.1 mg/litre; 50% or more of the particles had a diameter
of less than 7 µm. The observation time was 22 days. At toxic doses,
signs of neurotoxicity (curved body posture, paddling movements and
spasms) were observed. The acute 4-h LC50 was found to be about
1600 mg/m3 for animals of each sex (Ullman et al., 1986d).
7.1.4 Dermal
The acute dermal toxicity for rabbits was 200-300 mg/kg
(Medvedev, 1974; see International Register for Potentially Toxic
Chemicals, 1983).
Sherman strain rats were given one dermal application of
lindane (99%) dissolved in xylene, and no attempt was made to remove
the compound during the observation time of 14 days. The LD50 was
1000 mg/kg body weight for males and 900 mg/kg body weight for
females (Gaines, 1960).
Male New Zealand rabbits, both young adult (2-3 kg) and just
weaned (1 kg) were shaved only, shaved and depilated or shaved,
depilated and 'stripped', and a commercial preparation of 1% lindane
and 99% inert material was applied once to the entire body except
the head, limbs, and perineal surface at a dose of 6 ml/kg body
weight (equivalent to a dose of lindane of 60 mg/kg body weight, a
dose reportedly used in infants). The lindane was allowed to remain
on the skin during the experiment. Two of four adult rabbits that
were treated after having been shaved, depilated, and 'stripped'
exhibited excitement after about 24 h. Adult rabbits that had been
shaved only showed no effect. Weanling rabbits exhibited severe
anorexia and convulsions, and death occurred in some cases. The
effects were more pronounced in weanlings with inflamed or damaged
skin. The concentrations of lindane in whole blood of weanlings when
convulsions occurred (about 24 h after treatment) were 0.7-2.5 µg/ml
(Hanig et al., 1976).
7.2 Short-term exposure
7.2.1 Oral
7.2.1.1 Mouse
In young dd mice fed diets containing lindane at 0, 2, 4, or 10
mg/kg diet for three months, no effect on growth and no
histopathological change in the main organs were seen at any dose
level (Chen & Liang, 1956). Similarly, Kitamura et al. (1970) saw no
difference in behaviour, food consumption or body weight gain from
that in controls in ICR mice fed diets containing lindane at 0.1, 1,
10, and 100 mg/kg diet for 36 days. No histopathological examination
was carried out.
7.2.1.2 Rat
Short-term studies carried out by Slade (1945) and Laug (1948)
were more or less inadequate for an evaluation.
Doisy & Bocklage (1949, 1950) fed lindane-containing diets to
weanling rats for four weeks; doses of 400, 600, and 800 mg/kg diet
caused high mortality rates. Food intake and weight gain were
markedly reduced, especially in the group receiving 800 mg/kg diet.
The animals showed irritability, hyperactivity, and convulsions. A
dose of 200 mg/kg diet was without effect. Young rats were more
susceptible than adults.
In a three-month toxicity study, groups of 15 male and 15
female Wistar KFM-Han (outbred) SPF rats were fed diets containing
lindane (99.85%) at 0, 0.2, 0.8, 4, 20, or 100 mg/kg diet. After 12
weeks of treatment, most of the animals were sacrificed; the
remaining rats were placed on a control diet for six weeks and then
sacrificed. Lindane had no effect on mortality, food consumption,
haematological parameters, the results of urinalysis, or clinical
symptoms, although rats fed 100 mg/kg diet gained 8.4-14.9% less
weight than controls. Liver cytochrome P-450 levels were increased
in females given diets containing lindane at 0.8 mg/kg diet or more
and in high-dose males at the termination of dosing; these values
returned to the control levels during the recovery period. Such
increases in cytochrome P-450 activity are regarded as an adaptation
phenomenon due to induction of the microsomal detoxifying enzymes.
Slight, dose-related, reversible increases in absolute and/or
relative weights of livers and kidneys were observed in male and
female rats fed lindane at 20 or 100 mg/kg diet, and
histopathological examination revealed changes in these animals.
Those in the liver included dose-dependent, minimal-to-slight
centrilobular hepatocellular hypertrophy at the end of the
application period. After the recovery period, liver weights were
found to be normal, and no centrilobular hypertrophy was seen. In
the kidneys, minimal-to-slight, unicellular and multicellular
necrosis of epithelial cells was observed in proximal convoluted
tubules, and basophilic tubules, interstitial nephritis and hyaline
droplets were seen in epithelial cells of the convoluted tubules.
After the recovery period, the tubular degeneration was no longer
present, but the nephritis and basophilic tubules were still present
in the animals that had received 100 mg/kg. No effects were observed
with doses of 4 mg/kg diet (equivalent to 0.3 mg/kg body weight) and
below (Suter et al., 1983).
In a 12-week study with groups of 10 male and 10 female Wistar
RIV:TOX (C-S) rats, four weeks old at the beginning of the
experiment, lindane (99.8%) was administered in the diet at
concentrations of 0, 2, 10, 50, and 250 mg/kg. At the highest dose,
increases were seen in the induction of enzymes, such as
aminopyrine- N-demethylase and ethoxyresorufine- O-deethylase, but
cytochrome P-450 and aryl hydroxylase activity were not increased.
At the two highest dose levels, the weights of livers, kidneys, and
thyroid were increased. The no-effect level of lindane in this study
was 10 mg/kg diet (equivalent to 0.75 mg/kg body weight) (van Velsen
et al., 1984).
Young male Wistar rats were fed gamma-HCH at a dose of 0 (five
rats) or 800 mg/kg diet (eight rats) for two weeks, and urinary
excretion of body constituents that reflect renal function was
measured. Glucosuria and increased excretion of creatinine and urea
were found, and hypertrophy and degeneration of the renal tubular
epithelia were observed histologically (Srinivasan et al., 1984). In
young male Wistar rats administered gamma-HCH at 800 mg/kg diet for
two weeks, liver weights were increased, but no difference was found
in moisture, nitrogen, protein, or glycogen levels. The fat and DNA
content of the liver were found to be increased, but the DNA content
per unit issue was decreased. The predominant change in the liver
was hypertrophy. Testicular weight was no different from that in
control animals, but the protein content was higher, and the DNA
content was lower. The histological changes observed were tubular
atrophy and spermatogenic arrest; the interstitial space was found
to be oedematous (Srinivasan et al., 1988).
Liver function was studied in male Wistar rats fed a control
diet (6-8 rats) or gamma-HCH at 800 mg/kg diet (8-12 rats).
Gamma-HCH produced noticeable hepatocellular effects, as indicated
by increased activity of serum aminotransferases, hepatic
glucose-6-phosphate dehydrogenase and aldolase and decreased
activity of liver glucose-6-phosphatase. Liver mitochondrial
dinitrophenol/Mg++/Ca++-activated ATPase activity was decreased,
and levels of microsomal Na+, K+-ATPases were lower in treated
than control animals (Srinivasan & Radhakrishnamurty, 1988).
In a preliminary study, 24 Fischer-344 weanling, female rats,
received daily oral doses of either arachis oil or lindane at 0.069
mmol/kg body weight for 189 days. Lindane induced a significant
increase in body weight after 112 days of treatment. In a subsequent
dose-response study, female Fischer-344 rats, 21 days of age, were
gavaged daily with arachis oil (six rats) or lindane at 5 (six
rats), 10(eight rats), 20 (12 rats), or 40 mg/kg body weight (12
rats). At 20 mg/kg, lindane induced an increase in body weight after
10 weeks of treatment. At 40 mg/kg, 7 out of the 12 rats died; the
other animals had increased body weight gain. Greater food
consumption was observed, and obesity was induced, as indicated by
the Lee index. In addition, lindane caused delay in vaginal opening,
disrupted oestrous cycling, reduced pituitary and uterine weights
and elevated food consumption during pro-oestrous. This response
suggests that, by inducing alterations in the reproductive function
of female rats and by interfering with hormonal regulation of the
energy balance, lindane may be anti-oestrogenic rather than
oestrogenic as previously proposed (Chadwick et al., 1988).
7.2.1.3 Dog
Lehman (1952) found a high mortality rate in dogs given lindane
at daily doses of 10 or 15 mg/kg body weight on five days per week
over a period of 2 to 221 days. (No details available). Lehman
(1965) reported a study initially conducted by Fitzhugh et al., in
which dogs (two males and two females per group) were exposed to
lindane at 0 or 15 ppm (equivalent to approximately 0.6 mg/kg body
weight) in the diet for a total of 63 weeks. No effect was observed
on mortality, organ weights, body weight gain, haematological
parameters, or histological appearance.
During a two-year toxicity study, groups of four male and four
female beagle dogs were fed lindane (99%) at 0, 25, 50, or 100 mg/kg
diet. The amounts of lindane that were actually ingested were 0.83,
1.60, and 2.92 mg/kg body weight per day. Convulsions seen
occasionally in control and low-dose animals were not related to the
treatment. No treatment-related change was observed in body weight,
food or water consumption, ophthalmological parameters,
electroencephalographic traces, results of haematological
examinations, urinalysis, and liver function tests, or organ
weights. At autopsy, somewhat darker colouration and a brittle
consistency of the liver were seen at 100 mg/kg. In addition,
alkaline phosphatase activity was increased in the highest dose
group. No treatment-related abnormality was apparent with 25 or 50
mg/kg diet.
A supplementary group of four male and four female dogs was
administered lindane at 200 mg/kg diet for 32 weeks. High-voltage
slow-wave activity changes, possibly indicative of nonspecific
neuronal irritation, were recorded in electroencephalographic
tracings at this dose level. No such effect was observed in the
two-year study at 100 mg/kg diet (Rivett et al., 1978).
7.2.1.4 Pig
Schnell (1965) fed diets containing lindane (99.5%) at 0, 5,
10, 20, 40, or 80 mg/kg diet to groups of five pigs over a period of
nine months. No clinical symptom was seen in any animal during the
test period. Food and water intake remained normal, and
haematological investigations and histopathological examination of
the liver, spleen, kidneys, adrenals, heart, and brain revealed no
substance-related change, even at the highest dose level tested.
7.2.2 Inhalation
7.2.2.1 Mouse
Balaschow (1964) exposed mice for 6 h/day to a lindane aerosol
containing a nominal concentration of 1 mg/m3 for 2.5 months.
During the first two weeks, white blood cell counts showed the
presence of leukocytosis, with a shift to the left; from the end of
the first month, leukopenia with a shift to the right was observed,
and toxic granulations and vacuoles appeared in the nuclei and
cytoplasm of some leukocytes. Later, a reduced mitotic rate was
observed. The relationship between the different cell types in the
bone marrow was undisturbed.
Four groups of 45 male and 45 female CD-1 (Charles River) mice
were exposed by whole-body inhalation to aerosols (geometric mean
particle diameter, about 3 µm) containing lindane (purity at least
99.6%) at 0, 0.3, 1.0, or 5-10 g/m3 for 6 h/day, on five days per
week for 14 weeks. The test dose levels were selected on the basis
of a preliminary range-finding study, which showed that males did
not develop major signs of toxicity after five exposures for 6 h/day
to lindane at 1.0 and 10.0 mg/m3. During the main study, however,
an unexpectedly high mortality rate was seen in females exposed to
10 mg/m3; after the first five exposures, therefore, the
concentration for the high-dose group was lowered to 5 mg/m3.
Subgroups of 15 mice of each sex in each group were sacrificed after
7, 14, and 20 weeks. The group sacrificed at 20 weeks was a recovery
group, which was not exposed to lindane after exposure week 14. Ten
of the 15 mice in each subset were used for pathological evaluation,
and the remaining five were examined for serum lindane levels. The
lindane aerosol was highly toxic to female mice at 5 mg/m3, and
probably also at 1 mg/m3. The no-observed-effect level was
concluded to be 0.3 mg/m3 (Klonne & Kintigh, 1988).
7.2.2.2 Rat
Groups of 12 male and 12 female Wistar Han/Boe SPF rats were
exposed by whole-body inhalation to lindane (99.9%) at nominal
concentrations of 0, 0.02, 0.12, 0.6, or 4.5 mg/m3 (average
particle size, 0.92 µm) for 6 h/day for three months. The two groups
that received 0 or 4.54 mg/m3 were used to investigate recovery.
Slight diarrhoea and ruffled fur were observed temporarily in the
high-dose animals only. Measurements of body and organ weights and
of food and water intake, clinical chemistry, and histopathology
showed no treatment-related change. Hepatic cytochrome P-450 values
were increased at the end of the exposure period in animals at the
highest dose, but all values returned to that of the control during
the six-week recovery period. Increased kidney weights and cloudy
swelling of the tubular epithelium were observed especially in males
at the two highest dose levels, but, again, all values were
comparable with those in controls at the end of the recovery period.
The no-effect-level was probably 0.6 mg/m3 (Oldiges et al., 1983).
7.2.3 Dermal
Four groups of 49 male and 49 female Charles River rats (Crt:
(WI)BR strain) were exposed dermally to lindane (purity at least
99.5%) at dose levels of 0, 10, 60, or 400 mg/kg per day, selected
on the basis of a preliminary range-finding study, on five
consecutive days per week. The test substance was applied as a
suspension in aqueous carboxymethyl cellulose at a constant volume
of 4 ml/kg to a clipped area of the skin on the dorsal area and was
retained for 6 h with a dressing consisting of a gauze pad
heat-welded to plastic-backed aluminium foil. In the main phase of
the study, 23 animals of each sex in each group were treated for 13
weeks before sacrifice; one group of 13 animals/sex were sacrificed
after 6 weeks of treatment; a third (recovery phase) group,
consisting of 13 animals of each sex, was retained in the study for
an additional six weeks. At sacrifice, three animals in each group
were selected for determination of tissue levels of lindane (Brown,
1988).
The toxic effects induced by subchronic dermal exposure to 60
and 400 mg/kg consisted of pathological lesions of the kidneys in
males (increased organ weight, hyaline droplet formation, tubular
degeneration with necrosis, basophilic tubules, casts) and
hypertrophy of the liver in males and females. Whereas the effects
on the liver were reversible, some of the histopathological changes
in the kidneys persisted (tubular degeneration with necrosis,
granular casts) after the recovery period. Although there was
evidence of increased intensity of hyaline droplet formation at the
lowest dose tested (10 mg/kg per day), this effect was very slight;
that level could therefore be considered to be the
no-observed-effect level. The use in this study of semi-quantitative
methods for determination of blood levels, protein, and turbidity
makes it difficult to come to any definite conclusion; however, the
results of the urinalysis did not provide evidence that lindane
adversely affects kidney function.
7.3 Skin and eye irritation; sensitization
7.3.1 Primary skin irritation
Application of 0.5 g of lindane to the intact skin of New
Zealand white rabbits, in a study performed in compliance with the
guidelines of the Organization for Economic Co-operation and
Development and the US Environmental Protection Agency, did not
cause irritation (Ullmann et al., 1986a).
7.3.2 Primary eye irritation
Lindane placed in the conjunctival sac of the left eye of New
Zealand white rabbits at 0.1 g was slightly irritating (Ullmann et
al., 1986b).
7.3.3 Sensitization
The allergic potential of lindane was tested in a
Magnusson-Kligman maximization test (according to the guidelines of
the Organization for Economic Co-operation and Development) on
Durkin-Hartley albino guinea-pigs. Ten males and ten females
received lindane (99.6%) and five males and five females received
the vehicle, ethanol. No difference was seen between the test group
and the controls after the first and second challenge application 24
and 48 h later, and it was concluded that lindane has no skin
sensitizing (contact allergenic) potential in these guinea-pigs
(Ullmann et al., 1986b).
Ullmann et al. (1987a) conducted a further maximization test
(following the guidelines of the Organization for Economic
Co-operation and Development) with Dunkin-Hartley albino guinea-pigs
to test the contact hypersensitization potential of a lindane
formulation. Ten males and ten females received intradermal
injections of 5% 'Nexit fluessig' (containing 25.9% lindane) in
saline, and five males and five females received the saline vehicle.
No sensitization reaction was observed after the first and second
challenge application, 24 and 48 h later.
Comparable experiments were carried out with two other
formulations, 'Nexit stark', a powder containing 78.9% lindane
(Ullmann et al., 1987b), and 'Agronex Saatgutpuder', a powder
containing 20.1% (Ullmann et al., 1987c). Each was administered as
intradermal injections of 0.1% in saline. No sensitization reaction
was observed after two challenge reactions 24 and 48 h later.
7.4 Long-term exposure
7.4.1 Oral
In two long-term studies, lindane powder was mixed into the
diet of Wistar rats (10 males and 10 females) at 10, 100, or 800
mg/kg diet and as an oily solution at 5, 10, 50, 100, 400, 800, or
1600 mg/kg diet, either as the gamma isomer or as technical HCH
(containing only 13% of the gamma isomer). Two control groups were
used. At 100 mg/kg diet, liver weight was increased, and
histopathological examination revealed hepatocellular hypertrophy,
fatty degeneration and necrosis as well as nephritic reactions
(granular degeneration and calcification in male rats). These
findings were more pronounced at the 400, 800, and 1600 mg/kg
dietary levels. At these concentrations, the life span of animals in
groups treated with the oily solution was shortened by 20-40%. The
no-effect level in this experiment was 50 mg/kg diet (Fitzhugh et
al., 1950; Lehman, 1952).
Similar results were obtained in another lifetime study, in
which groups of 10 male and 10 female rats received lindane at 25,
50, or 100 mg/kg diet. The dose of 25 mg/kg had no effect on the
liver, but hepatocellular hypertrophy was observed with 50 mg/kg and
slight fatty liver-cell degeneration was described in the group
receiving 100 mg/kg diet (Truhaut, 1954).
7.4.2 Appraisal of acute and short- and long-term studies
The acute oral toxicity (LD50) of lindane in different
species, depending on the vehicle used, ranges from 56 to 480 mg/kg
body weight. Preparations in oil were more toxic than aqueous
solutions or suspensions. The ranges for rats and mice were similar
(88-270 and 56-246 mg/kg, respectively). The dermal LD50 for rats
is approximately 900 mg/kg body weight, but smaller amounts (60
mg/kg as a 1% cream) caused convulsions, anorexia, and deaths in
weanling rabbits. No skin irritation or sensitization was observed,
and eye irritation was slight.
Although older, long-term studies in the rat suggest a
no-observed-adverse-effect level of 25 mg/kg diet, contemporary
short-term studies in rats indicate that this level is10 mg/kg diet,
equivalent to 0.75 mg/kg body weight on the basis of increased
hepatic, renal, and thyroid weights, increased cytochrome P-450
activity and histopathological findings in liver and kidneys.
7.5 Reproduction, embryotoxicity, and teratogenicity
7.5.1 Reproduction
Trifonova et al. (1970) found no reduction in the fertilization
rate of female rats after oral treatment for 90 days with lindane at
approximately 5 mg/kg body weight. When the dose was doubled over a
test period of 138 days, the fertilization rate was reduced.
(Details not given.)
A three-generation test was carried out in which 10 male and 10
female CD-rats were administered lindane at concentrations of 25,
50, or 100 mg/kg diet continuously. The treatment had no influence
on fertility, litter size, breeding rate, weight of newborn animals,
lactation, malformation rate, or maturation. The liver weights of
young animals of the F3b generation were increased, especially
among females. Histopathological examination of the liver showed
enlarged hepatocytes and vacuolization in animals treated with 50
and 100 mg/kg diet (Palmer et al., 1978a).
7.5.2 Embryotoxicity and teratogenicity
7.5.2.1 Oral administration
Mouse: Lindane was administered orally to seven groups of 25
pregnant NMRI-EMD (SPF) female mice at 0, 12, 30, or 60 mg/kg body
weight in 0.5% carboxymethyl cellulose, on either days 6-15 or days
11-12 of pregnancy. In the group receiving the highest dose, fetal
mortality was increased and fetal weights were decreased. A slight,
non-dose-related increase in malformation rate was found in the
mid-dose group (4.2%, as compared to 1.9% in controls). At the
highest dose, increased maternal mortality (48%) and reduced body
weight gain were observed. The treatment had no effect on the number
of implantations per dam, the percentages of early and late
resorptions, the number of runts or the malformation rate (Frohberg
& Bauer, 1972b).
Rat: Groups of 20 female CFY-rats received lindane at 5, 10,
or 20 mg/kg body weight by gavage during days 6-15 of pregnancy. In
the groups given 10 and 20 mg/kg, maternal toxicity (reduced food
intake and reduced weight gain) was observed, and two female rats
given 20 mg/kg died. At the same dose, there was a dose-related
increase in the incidence of offspring with extra (14th) ribs, which
was statistically significant. Other anomalies and litter parameters
were comparable to those in the controls, and there was no evidence
of embryo- or fetoxicity (Palmer et al., 1978b).
Khera et al. (1979) gave female Wistar rats (20 animals per
group) a lindane formulation (50% in corn oil) at 3.12, 6.25, or
12.5 mg/kg body weight (expressed as 100% lindane) by intubation on
days 6-15 of gestation. No effect was seen on the number of living
fetuses per litter, the number of dead plus resorbed fetuses or mean
fetal weight at 22 days of gestation. No malformation other than the
usual range of developmental variants was observed in any group. A
slight increase in the frequency of anomalies of the ribs and
reduced cranial ossification were seen in the fetuses exposed to
6.25 mg/kg; these effects were confined to two litters and were
probably not dose-related.
Female rats that received lindane orally at a dose of 25 mg/kg
body weight daily during pregnancy had higher post-implantation
embryonal mortality than aontrols, and at 12.5 mg/kg, no mortality
was found. Neither dose level induced teratological abnormalities
(Mametkuliev, 1976; see International Register for Potentially Toxic
Chemicals, 1983).
Rabbit: Lindane was administered by intragastric intubation
to New Zealand white rabbits (13 animals per group) on days 6-18 of
gestation at doses of 5, 10, and 20 mg/kg body weight. All treated
animals showed slight tachypnoea and lethargy during the treatment
period, and body weight gain and food intake were reduced.
Pre-implantation loss was significantly higher in the group given 20
mg/kg, but, as treatment did not start until day 6 of gestation,
this effect is unlikely to have been due to lindane.
Post-implantation loss and the incidence of resorptions were
increased at 5 and 20 mg/kg. The number of offspring with extra
(13th) ribs was significantly lower in animals given 5 mg/kg and
significantly higher in rabbits at 20 mg/kg than in controls. Fetal
and litter weights were unaffected, and the incidence of other
anomalies was similar to that in controls (Palmer et al., 1978b).
Dog: An increased frequency of stillbirths, unrelated to
dosage or period of administration, was seen in beagle dogs fed
lindane at 0 (five dogs), 7.5 (13 dogs) or 15 mg/kg body weight (14
dogs) from day 1 or 5 throughout gestation. No significant
teratogenic effect was observed. The number of living pups was
similar in control and test groups (Earl et al., 1973).
Pig: Groups of six female pigs received lindane at 0, 50, or
500 mg/kg diet from 30 days prior to mating until day 30 of
gestation. No treatment-related effect was found on number of
embryos, embryo weight, or rate of ovulation (Duee et al., 1975).
Cow: In an accidental poisoning incident, four pregnant cows
received lindane at 13.4 g (28 mg/kg body weight) 6-17 weeks
pre-partum. All had convulsions and muscular tremors in the ensuing
48 h but recovered with veterinary treatment. All calved on time and
produced normal, healthy calves. Four non-pregnant cows died after
receiving 21 g of lindane, which suggests that the minimum lethal
dose is 28-45 mg/kg body weight (McParland et al., 1973).
7.5.2.2 Subcutaneous injection
Mouse: Lindane was administered subcutaneously (in a 0.5%
carboxymethyl cellulose solution) to groups of 25 pregnant NMRI
miceat 6 mg/kg body weight on either days 11-13 or days 6-15 of
pregnancy. Except for a slight increase in the frequency of runts in
the latter group, no effect was seen on the number of implantations
or of living embryos per dam or on the percentage of absorptions or
resorptions; no treatment-related malformation was reported
(Frohberg & Bauer, 1972a).
Rat: Groups of 20 Sprague-Dawley rats received lindane at
doses of 0, 5, 15, or 30 mg/kg body weight by subcutaneous injection
on days 6-15 of gestation. Maternal toxicity was observed in the
mid- and high-dose groups. No effect attributable to the
administration of lindane was noted on pregnancy rates, maternal
gross pathology or reproduction, or offspring viability and
development. No teratogenic effect was found at necropsy or in
visceral and skeletal examinations (Reno, 1976a; Hazelton
Laboratories, 1976a).
Rabbit: Lindane was injected subcutaneously at 0, 5, 15, 30
or 45 mg/kg body weight into pregnant rabbits on days 6-18 of
pregnancy, except that the highest dose was given on days 6-9 and
then reduced to 30 mg/kg body weight. No embryotoxic or teratogenic
effect was found in fetuses exposed to the two lower doses. At the
two higher dose levels, increased maternal toxicity was found. At
the highest dose, the number of resorptions was increased, and 14
out of 15 animals died (Reno, 1976b; Hazelton Laboratories, 1976b).
7.5.3 Reproductive behaviour
Adult female Fischer (CDF-344) rats were injected
intraperitoneally on the morning of pro-oestrus with lindane at 25,
33, 50, or 75 mg/kg body weight in sesame oil, and in the evening,
they were examined for lordosis behaviour with a sexually
experienced male. A dose-dependent reduction in sexual receptivity
was seen with increasing doses of lindane: treated animals required
a greater number of mounts before the first lordosis response was
observed, and they may have required more sensory stimulation to
elicit the lordosis reflex. Most of the females also failed to
exhibit proceptive behaviour (darting and hopping) during the mating
test. This inhibition resembles the rapid effects of another
chlorinated pesticide, chlordecone, and does not appear to depend
upon disruption by lindane of the inhibition of the central nervous
system by gamma-aminobutyric acid (GABA). The results substantiate
previous suggestions that the ability of chlorinated pesticides to
interfere with intracellular oestradiol receptors cannot explain
their rapid attenuation of reproductive behaviour (Uphouse, 1987).
7.5.4 Appraisal of reproductive toxicology
Lindane was investigated in tests covering all aspects of
reproduction (three-generation studies in rats) and in tests for
embryotoxicity and teratogenicity by oral, subcutaneous and
intraperitoneal administration in mice, rats, dogs, and pigs).
Lindane did not exhibit teratogenic properties after oral or
parenteral application (extra ribs were regarded as variations).
Fetal and/or maternal toxic effects were observed in rats with doses
of 10 mg/kg body weight and higher given by oral gavage; 5 mg/kg is
therefore considered to be the NOAEL.
No effect on reproduction or maturation was seen in the
three-generation study at doses of lindane up to 100 mg/kg diet, but
morphological signs suggesting liver enzyme induction occurred with
doses from 50 mg/kg diet in the third generation. The no-effect
level in this test was 25 mg/kg diet (equivalent to approximately
1.25 mg/kg body weight).
7.6 Mutagenicity and related end-points
Lindane was tested in mutagenicity tests with a variety of
end-points. The relevant experiments are summarized in Tables 9, 10,
and 11. Those studies that were not performed according to protocols
which comply to the present international standards are considered
to be of limited relevance; some studies used lindane preparations
of less than 99% purity or of unknown purity. The results are so
consistent, however, that the limitations of some studies did not
vitiate a final assessment.
7.6.1 DNA damage
The ability of lindane to damage DNA was tested in Bacillus
subtilis and in Escherichia coli WP2 in the rec assay, and
tests for unscheduled DNA synthesis tests were performed in primary
rat hepatocytes and human fibroblasts. No mutagenic potential was
detected.
Sina et al. (1983) developed a sensitive alkaline elution assay
in non-radiolabelled rat hepatocytes to measure DNA single-strand
breaks induced by chemicals. This assay is used to predict
carcinogenic/mutagenic activity. Lindane at doses of 0.03 and 0.3
mmol/litre induced DNA damage, increasing with dose.
After oral administration of lindane to rats and mice, a very
low covalent binding index (0.02-0.01) was calculated, suggesting
that no significant binding to DNA had occurred.
The incorporation of orally admini stered radiolabelled
thymidine into liver DNA was determined in SIV-50-SD-rats 24 h after
a single oral dose by gavage of 0.01, 0.1, or 1.0 mmol/kg gamma-HCH.
No effect on liver DNA synthesis was seen (Büsser & Lutz, 1987).
7.6.2 Mutation
The ability of lindane to induce gene mutation has been
investigated extensively in S. typhimurium and E. coli, using an
adequate range of strains to cover both base-pair and frame-shift
mutations. Most of the tests were performed both with and without
metabolic activation by 9000 x g preparations from the livers of
induced rats or mice (Table 9).
Negative reults were obtained in the host-mediated assay using
mice and S. typhimurium or Serratia marescens. Furthermore, a
test for point mutations in V79 Chinese hamster cells, the hprt
test for forward mutations, indicated no mutagenic effect of
lindane. A test for sex-linked recessive lethal mutation in
Drosophila melanogaster also gave a negative result (Table 9).
D. melanogaster were also used to test for dominant lethal
mutation. Groups of 25 males and 25 females aged 6-24 h were
transferred to food containing HCH (Gammexane) at 20 mg/kg food
medium, and their progeny were raised on this food. Five males and
five females of the F1 generation (the 'toxic generation') were
raised on normal food and were allowed to mate with each other and
lay eggs for 24 h in 10 oviposition jars. From this generation of
flies, three successive mutation-generations were raised on normal
food, and the numbers of larvae hatched from eggs laid on each of
the first 10 days after enclosion were again recorded. The
percentage of larvae hatched from the total number of eggs laid,
cumulated over the entire period was significantly decreased in the
second and third generations. These results suggest that the
preparation tested is mutagenic (Sinha & Sinha, 1983).
A test for induction of reverse mutations in Saccharomyces
cerevisiae gave inconclusive results.
7.6.3 Chromosomal effects
Most of the cytogenetic tests performed with lindane both in
vivo and in vitro did not indicate mutagenic properties of
lindane. In only une study were there positive findings, but the
purity of the material tested was not given and the description of
the test was poor. Lindane therefore apparently does not induce
chromosomal breakage (Table 10).
Table 9. Result of mutagenicity tests of gamma-HCH
Test system Dose Type of test Metabolic Result Reference
(Organisms/Cells) activation
Bacillus subtilis
H17 rec+ 0.02 ml of solution plate none - Shirasu et al. (1976)
M45 rec- containing 1 mg/ml plate none -
in DMSO
Escherichia coli
WP2 try- approx. 1 mg plate none - Ashwood-Smith et al. (1972)
WP2 4 gradient plates, plate S9-mix - Probst et al. (1981)
WP2 uvr A- covering 10 000-fold plate S9-mix -
concentration range
WP2 urv A 6-7 dose levels up to plate none - Oesch (1980)
5000 µg S9-mix -
Salmonella typhimurium
TA98, TA100, TA1535, 1-1000 µg plate S9-mix - van Dijck & van de Voorde
TA1537, TA1538, (1976)
TA1950, TA1978
TA98, TA100, 93, 139, 208 µg plate none - Röhrborn (1977a)
TA1535, TA1538
TA100, TA1535, 6-7 dose levels up to plate none - Oesch (1980)
TA1537, TA98 5000 µg plate S9-mix -
TA100, TA1535, 8 dose levels, 0 up to plate none - Haworth et al. (1983)
TA1537, TA98 333 µg S9-mix -
Table 9 (contd)
Test system Dose Type of test Metabolic Result Reference
(Organisms/Cells) activation
Salmonella typhimurium contd)
TA100, TA1535, 4, 20, 100, 500, or plate S9 - Anderson & Styles (1978)
TA1538, TA98 2500 µg in DMSO
G46, TA98, TA100, concentration gradient plate S9-mix - Probst et al. (1981)
TA1537, TA1538,
C3076, TA1535, D3052
Host-mediated assay
Salmonella typhimurium G46 25 mg/kg bw mouse nr - Buselmaier et al. (1972)
(subcutaneous) (NMRI)
Serratia marescens 25 mg/kg bw mouse nr - Buselmaier et al. (1972)
a 21. leu- (subcutaneous) (NMRI)
Mammalian cells
hprt locus
V79 Chinese hamster cells 0.5-500 µg/ml plate S9-mix - Glatt & Oesch (1984);
0.5-250 µg/ml plate S9-mix - Oesch & Glatt (1984)
Sex-linked recessive
lethal test
Drosophila melanogaster 0.001% injected nr - Benes & Sram (1969)
(aqueous sol.) into
abdomen
(0.2 µl)
DMSO, dimethyl sulfoxide; nr, not relevant; bw, body weight
Table 10. Results of tests for other genetic effects
End-point Dose Effects Result Reference
Chromosomal aberrations in vitro
Chinese hamster fibroblast cell line 2.1 mg/mla (in ethanol) Chromatid gaps, chromatid Equivocal Ishidate & Odashima
(CHL) and chromosomal breaks (1977)
Lymphocytes from human peripheral 0.1, 0.5, 1.0, 5.0, or Chromosomal breakage Equivocal b Tzoneva-Maneva et al.
blood (different donors) 10 µg/ml only at toxic dosages (1971)
(5 and 10 µg/ml)
Chromosomal aberrations in vivo
Chinese hamster bone-marrow cells 0.125, 1.25, or 12.5 mg/kg Increase in chromosomal (-) Röhrborn (1976, 1977a)
body weight orally for gaps at highest dose
5 days level
Syrian hamster bone-marrow cells 64, 128, 280, or 640 mg/kg No chromosomal - Dzwonkowska & Hubner
body weight aberration (1986)
Rat bone-marrow cells 1.5, 7.0, or 15 mg/kg - - Gencik (1977)
body weight orally for
12 weeks
Human lymphocytes occupational exposure - Desi (1972)
(no further details)
Sister chromatid exchange in vivo
Mouse bone-marrow cells (strain CF1) Male/female: 2/1.6, 10/8, or
50/40 mg/kg body weight - - Guenard et al. (1984a)
as a single oral
application
Mouse bone-marrow cells Single intraperitoneal - - Guenard et al. (1984b)
(strain CF1) injection of 1.3, 6.4, or
32.1 mg/kg body weight
Table 10 (contd)
End-point Dose Effects Result Reference
Micronucleus test in vivo
Mouse erythroblasts (CBA male mice) 75 mg/kg body weight - - Jenssen & Ramel (1980)
Dominant lethal test
Rats (males; strain Chbb = THOM) 1.5, 7.0, or 15 mg/kg body Röhrborn (1977b)
weight daily for 8 weeks
orally (in olive oil)c
Rat (males; strain Wistar) 1.5, 7.0, or 15 mg/kg body Questionable Cerey et al. (1975)
weight in olive oil positiveb
Mouse (males; strain ICR/Ha Swiss) 15, 75, 200, or 1000 mg/kg - - Epstein et al. (1972)
body weight once
intraperitoneally
15 mg/kg body weight - Equivocal Epstein et al. (1972)
five times, orally
Mouse (males; strain NMRI-EMD) Single intraperitoneal - - Frohberg & Bauer (1972c)
injection of 12.5, 25, or
50 mg/kg body weight
a Maximal effective dose
b Inadequate study; protocol does not comply with international standards
c Males dosed continuously during the whole mating period (8 weeks)
Table 11. Results of tests for DNA damage
End-point Dose Type of test Metabolic Result Reference
activation
Unscheduled DNA synthesis
(transformed SV-40) human 1, 1000 µM (in acetone) Tissue culture None - Ahmed et al. (1977)
fibroblast (cell-line VA-4) fluid S9-mix -
Unscheduled DNA synthesis and 500 µg/ml Tissue culture - 50-70% Rocchi et al. (1980)
repair capacity after damage inhibition
by UV-rays (human lymphocytes)
Primary rat hepatocytes 100 nmol/ml Plate - - Probst et al. (1981)
(in DMSO)
Covalent DNA binding
Male mouse (strain NMRI, CF1 12-13 mg/kg body Liver DNA Not relevant - Sagelsdorff et al. (1983)
and C6B3F1) weight orally and 8.7 (covalent
up to 23 mg/kg body binding
weight orally index,
0.02-0.1)
Lindane was tested for its ability to induce sister chromatid
exchange in vivo (in mice by oral and intraperitoneal
administration) and in vitro (in Chinese hamster ovary cells); no
effect was seen. No mutagenic effect was observed in a test for
micronucleus formation in the bone marrow of mice treated in vivo,
and lindane did not induce chromosomal damage in vivo.
Two of three tests for induction of dominant lethal mutation in
rats gave clearly negative results, and the other gave a
questionably positive response. The significance of the latter test
must be regarded as low, because the purity of the material tested
was not given and the test was not performed in compliance with an
acceptable standard.
7.6.4 Miscellaneous tests
Lindane tested in a MO4 cell culture at doses of 1, 10, and
100 µg/ml induced no multinucleation or major toxicity (de Brabander
et al., 1976).
Lindane at a concentration of 101.8 µg/ml did not induce
6-thioguanine-resistant mutations in Chinese hamster V79 cells.
Concentrations of 100 and 200 µg/ml were significantly cytotoxic;
the concentration that allowed 10% survival was 120 µg/ml. At 11.6
µg/ml, lindane weakly inhibited metabolic cooperation between 6-TGs
and 6-TGr V79 cells. It was concluded from these studies that
lindane is not mutagenic in this test system; however, it inhibits
metabolic cooperation, mimicking the powerful tumour promotor
12- O-tetradecanoylphorbol 13-acetate in this assay system
(Tsushimoto et al., 1983).
The morphology of primary monkey kidney cells was examined 24 h
after addition of lindane (99.8% in 1% dimethylformamide) to the
growth medium, and readings were made daily for three days. Lindane
applied at concentrations above 10 mg/litre induced marked cellular
damage, and 250 mg/litre had cytotoxic effects (Desi et al., 1977).
7.6.5 Appraisal of mutagenicity and related end-points
The mutagenicity of lindane has been adequately studied. This
compound has been extensively investigated for its ability to induce
gene mutation in both bacteria and mammalian cells, and for its
activity in the assay for sex-linked recessive lethal mutation in
D. melanogaster. Negative results were obtained consistently. Its
ability to induce chromosomal damage and sister chromatid exchange
has been investigated in mammalian cells both in vitro and in
vivo, again with negative results. Both assays for DNA damage in
bacteria and studies in vivo to investigate covalent binding to
DNA in the liver of rats and mice following oral administration also
gave negative results. The few studies in which positive results
were obtained involved invalid study designs or lindane of unknown
purity.
Overall, lindane appears not to have mutagenic potential.
7.7 Carcinogenicity
7.7.1 Mouse
Gamma-HCH was fed to 20 male ICR/JCL mice (five weeks old) at
300 or 600 mg/kg diet for 26 weeks. Increased liver weights were
reported in the group receiving the higher dose. Five of 10 mice in
this dose group had type 0 or type I liver lesions. Type 0 lesions
were characterized as areas of atypical, small liver cells, uniform
in size and with a small nucleus, which normally forms round spots
and is readily distinguishable from the surrounding liver tissue.
Type I lesions were described as 'benign liver tumours' (Goto et
al., 1972).
Groups of 20 male mice (eight weeks old) were fed gamma-HCH at
100, 250, or 500 mg/kg diet for 24 weeks. The highest dose level
resulted in increased liver weight. No nodular hyperplasia or
hepatocellular tumour was observed (Ito et al., 1973b).
Hanada et al. (1973) treated 10-11 dd mice of each sex with
lindane at 100, 300, or 600 mg/kg diet for 32 weeks and killed the
survivors 5-6 weeks after the end of exposure. Hepatomas were found
in 1/3 females and 3/4 males that ingested 600 mg/kg diet and
survived for 36-38 weeks; none were found in animals fed 100 or 300
mg/kg diet. At the two higher doses, most animals had atypical
proliferations in the liver. alpha-Fetoprotein could not be
identified in the serum of the animals with hepatomas.
In an experiment lasting 110 weeks, 30 male and 30 female CF1
mice were fed lindane (> 99.5%) at 400 mg/kg diet. A group of
controls comprising 45 male and 44 female mice were fed a standard
diet. Benign and malignant liver tumours were diagnosed in 24% of
male controls and 23% of female controls and 93% of treated males
and 69% of treated females. Significant mortality (15%) occurred
during the early phase of the study in the treated group (Thorpe &
Walker, 1973). In a complete reexamination of all slides, the
reviewer concluded that lindane had not affected the incidence of
hepatocellular carcinomas inanimals of either sex but had enhanced
the incidence of hepatocellular adenoma (and hyperplastic nodules)
in male mice. In this strain, therefore, lindane had a tumorigenic
effect only in male mice (Vesselinovitch & Carlborg, 1983).
Herbst et al. (1975) and Weisse & Herbst (1977) studied the
carcinogenic potential of lindane at 12.5, 25, or 50 mg/kg diet
administered for 80 weeks to 50 male and 50 female Chbi:NMRI mice (a
strain with a low (2%) spontaneous rate of hepatomas). The control
group consisted of 100 males and 100 females. No evidence of
substance-related tumour formation was seen in animals of either sex
at any dose level. Electron microscopic examination showed no fine
structural hepatocellular alterations.
Groups of 50 B6C3F1 hybrid mice of each sex were fed lindane
at 80 or 160 mg/kg diet for 80 weeks and were killed 10-11 weeks
after the end of treatment. Hepatocellular carcinomas were found in
5/49 pooled male controls, 2/10 matched male controls, 19/49 males
fed 80 mg/kg diet and 9/46 males fed 160 mg/kg diet, and in 2/47
pooled female controls, no matched female controls, 2/47 females fed
80 mg/kg diet, and 3/46 females fed 160 mg/kg diet. Only the
incidence of hepatocellular carcinomas in the males at the lower
dose was significantly different from that in controls. It was
concluded that lindane is not carcinogenic in this test system (US
National Cancer Institute, 1977). In a reexamination of the slides,
the reviewer was in full agreement with the conclusions of the
original authors (Vesselinovitch & Carlborg, 1983).
Wolff & Morrissey (1986) administered diets containing lindane
at 160 mg/kg diet for 24 months to three phenotypes of (YS x VY)
F1 hybrid mice: obese yellow Avy/a, lean pseudoagouti Avy/a, and
lean black a/a. Hepatocellular adenomas were found in 35% of yellow
A vy/a mice (9% in controls) and in 12% of pseudoagouti Avy/a mice
(5% in controls); no increase in the incidence of liver tumours was
seen in the black a/a mice.
7.7.2 Rat
Groups of 10 male and 10 female Wistar rats were fed for life
on diets containing 10, 100, or 800 mg/kg diet of powdered lindane
or 5, 10, 50, 100, 400, 800, and 1600 mg/kg diet of lindane in corn
oil. The life span of the animals was shortened by 20-40% in a
dose-dependent manner with administration of 400, 800, and 1600
mg/kg diet, except in those given 800 mg/kg diet of powdered
lindane. No increase in tumour incidence was reported in the 200
treated rats (Fitzhugh et al., 1950) (see also section 7.4.1).
In a lifetime study, groups of 10 rats of each sex received
lindane at 0, 25, 50, or 100 mg/kg diet. No tumour formation was
found (Truhaut, 1954). (Details not given.)
Groups of 18-24 male W rats received lindane (99%) at 500 mg/kg
diet for 24 or 48 weeks. High mortality was seen; none of the six or
eight surviving animals had developed a liver tumour by 24 or 48
weeks, respectively (Ito et al., 1975).
Groups of 50 Osborne-Mendel rats of each sex were administered
lindane for 80 weeks and were then transferred to the control diet
for an additional 28-30 weeks; survivors were killed at 108-110
weeks. The males received 320 or 640 mg/kg diet for 38 weeks,
lowered thereafter to 160 and 320 mg/kg diet; the females received
320 and 640 mg/kg for two weeks, then 160 and 320 mg/kg for 49 weeks
followed by 80 and 160 mg/kg diet for 29 weeks. Matched controls
consisted of 10 animals per sex; these were combined for statistical
evaluation with 45 untreated male and female rats from other
bioassays. No increase in tumour rate was seen in treated groups of
either sex (US National Cancer Institute, 1977).
7.7.3 Initiation-promotion
The tumour-initiating activity of gamma-HCH was studied by
observing the appearance of phenotypically altered foci in female
Wistar rats (Schröter et al., 1987). Groups of 3-8 rats were
operated to remove the median and right liver lobes; they were then
administered gamma-HCH at 30 mg/kg body weight daily for two weeks,
followed by phenobarbital at 50 mg/kg body weight daily for 15
weeks. Liver foci were identified by means of the
gamma-glutamyltransferase reaction and morphological alterations. No
evidence of initiating activity was found.
Promoting activity was studied by administering
N-nitrosomorpholine as a single dose of 250 mg/kg body weight by
gavage, followed by 4, 15, and 20 weeks' administration of gamma-HCH
at 0.1, 0.5, 2.5, 10.0, or 30.0 mg/kg body weight per day. Both the
number and the size of altered foci were enhanced by doses of 2-3
mg/kg. The authors concluded that gamma-HCH could be classified as a
tumour promotor.
In an experiment using male dd mice (26-30 per group, eight
weeks old), administration of Kanechlor-500 at 500 mg/kg diet
induced nodular hyperplasia and hepatocellular carcinoma in the
livers of mice after 32 weeks' exposure. Administration of lindane
(99% pure) at 50, 100, or 250 mg/kg dietwith or without the
polychlorinated biphenyl at 250 mg/kg diet induced none of those
lesions after 24 weeks. Lindane was therefore neither tumorigenic a
promoter in this experiment (Ito et al., 1973a).
7.7.4 Mode of action
Considerable work has been done using mice generated
genetically from (C3H x VY)F1 or (YS x VY)F1 mice. The resulting
Avy/Avy, Avy/a and A/a crosses contain a genomic locus known as the
Agouti locus, which has been linked to tumorigenicity in these mice.
Treatment of Agouti mice with lindane at 160 mg/kg diet has been
found to saturate the lindane elimination pathways and thereby
result in an increased burden of lindane and its metabolites. This
excessive build-up could explain the tumorigenicity of lindane, at
least when given at 'excessive' levels (Wolff, 1986; Wolff et al.,
1986).
The tumour response to lindane has been characterized in (YS x
VY) F1 hybrid mice (Table 12). Lindane increased the incidence of
benign tumours only in the Avy/a genotype, while the 'normal' A/a
mice had no tumours. This finding indicates the existence of a
genetic predisposing factor, which may be of some importance in
evaluating the hazard of exposure to lindane. A phenotypic factor is
apparently involved, as mice of the obese yellow phenotype had a
greater tumour response in the liver than their isogenic siblings,
pseudoagouti mice; such factors may themselves result in more
tumours. Tumour incidence was not increased in normal black mice,
but the incidence of benign tumours of the liver and lung was
increased in Y genotype mice. The time of tumour onset was as early
as 18 months in pseudoagouti mice, but the normal black mice had no
tumours in the 24-month test period. Avy/a yellow mice thus have a
proclivity to form hepatocellular adenomas and lung tumours, which
is augmented (and not caused exclusively) by exposure to lindane.
The pseudoagouti and normal black mice have a low rate of
spontaneous tumours in the liver and lung, but only the pseudoagouti
respond to lindane. Thus, some genetically derived mice form benign
tumours, but the 'normal' A/a controls do not. Holder & Stöhrer
(1989) concluded that these findings are of limited applicability to
the situation in humans.
Table 12. Carcinogenic responses in normal (A/a), pseudoagouti (Avy/a), and
agouti (Avy/a) mice after 24 months of dietary exposure to lindane a
Tumour Phenotypeb No lindane 160 ppm lindane p value
No. % No. %
Liver adenoma B 6/96 6 3/96 3 -
PS 5/95 5 11/95 12* 0.11
Y 8/93 9 33/94 35* 8.2E-06
Hepatocellular B 3/96 3 1/96 1 -
carcinoma PS 2/95 2 5/95 5 -
Y 12/93 13** 16/94 27 -
Combined liver B 9/96 9 4/96 4 -
tumours PS 7/95 7 16/95 17* 0.036
Y 20/93 22** 49/94 52* 1.1E-05
Hyperplasia of lung B 10/96 10 79/96 82* <E-08
Clara cells PS 10/95 10 71/94 76* <E-08
Y 14/95 15 68/95 52* <E-08
Lung tumourc B 2/29 2 1/96 3 -
PS 2/95 6 5/94 14 0.0692
Y 12/95 4 16/95 19* 0.0012
Table 12 (continued)
* Statistically significant dose-related response compared to non-treated
comparable controls of the same phenotype
** Liver response increased in obese yellow (Avy/a) mice compared to A/a black
controls even in absence of treatment
a From Holder & Stöhrer (1989)
b B, black normal controls; PS, pseudoagouti; Y, yellow
c Not malignant; origin of cells uncertain
Tumour promotion was tested as a mechanism of action for
lindane in both the yellow and pseudoagouti variants of the (C3H x
VY) F1 mouse using phenobarbital, which increases the incidence of
benign tumours in the liver of yellow mice, as the tumour-promoter.
Lindane at a dose of 160 mg/kg diet may exceed saturation of the
metabolic mechanisms, especially in yellow mice with the (C3H x
VY)F1 genotype. Chadwick et al. (1987) found reduced elimination
of lindane in yellow and pseudoagouti mice and explained their
findings as follows: The yellow mouse carrying the Avy locus has a
propensity for tumorigenicity, which is enhanced by the yellow obese
phenotype. The lungs and livers of these animals therefore are very
likely to contain cells that are already transformed, whereas in
normal black mice there may be none or very few transformed cells.
Hence, Avy strain mice would be expected to respond to a tumour
promoter, whereas black mice would not; this was also the pattern of
tumour response observed. The authors concluded that, because
phenobarbital promotes tumours in this strain of mice, lindane is
also a promoter.
Oesch et al. (1982) studied the specific activities in CF1
and B6C3F1 mice and Osborne-Mendel rats of some of the enzymes
thought to be involved in lindane metabolism. Lindane was
administered at 51-360 mg/kg diet for three days or three months. No
clear change was seen in animals treated for three days, but changes
in enzyme activity were noted after three months' treatment. In the
CF1 strain (sensitive to liver tumour induction), a large increase
in liver weight was observed; this was not the case in B6C3F1
mice. In the Osborne-Mendel rats, a smaller increase was found.
Glutathione- S-transferase activity was increased in CF1 mice and
to a lesser extent in B6C3F1 mice and the rats. Increased
glutathione- S-transferase activity may lead to rapid conjugation
of glutathione with reactive metabolites, as, for example, epoxides
derived from lindane. Rat liver microsomes had more
UDP-glucuronosyltransferase activity than those from mouse liver.
This increased activity in rats could also lead to rapid conjugation
of phenols derived from lindane. The most striking difference,
however, was that CF1 mice had more monooxygenase activity and
less epoxide hydroxylase activity than rats; whether either of these
changes would result in an accumulation of reactive epoxides from
lindane remains to be elucidated.
Iverson et al. (1984) studied the ability of 14C-gamma-HCH to
bind to liver macromolecules of untreated and
phenobarbital-pretreated male HPB black mice in vivo and in
vitro. There was preferential binding of gamma-HCH to protein but
not to DNA.
These studies in mice indicate that lindane does not behave as
an initiator, in that it does not induce the preneoplastic foci seen
with known carcinogens, such as N-nitrosoporpholine and
N-methyl- N-nitrosourea (Holder & Stöhrer, 1989). Lindane can,
however, act as a tumour promoter, in that it caused outgrowth of
foci and increased the areas of the foci, indicative of
preneoplastic conditions. Whether these foci actually go on to form
tumours was not determined.
The notion that lindane has some characteristics in common with
tumour promoters is corroborated by the finding that it inhibits
cell-to-cell communication of the low-molecular-weight compound,
tritiated uridine. Trosko (1982) found that such inhibition occurred
when cells were pretreated with a variety of free-radical scavengers
and suggested that the inhibition might involve a free-radical
generating process. Some tumour promoters have been suggested to act
by a mechanism involving free radicals (Kensler & Trush, 1984; Rao &
Reddy, 1987).
If lindane acts by the tumour promotion mechanism suggested by
formation of gamma-glutamyltransferase-positive foci in the liver
and inhibition of cell-to-cell communication, it is likely to be a
dose-rate-limited process because of its known reversibility. That
is, the compound must be administered at above a certain amount and
rate or its carcinogenic effects are reversible and cease to be
manifested. Such a mechanism would therefore probably result in a
sigmoid response in models.
Zeilmaker & Yamasaki (1986) studied the effect of lindane on
gap-junctional intercellular communication in cultured Chinese
hamster V79 cells, grown as monolayer using a microinjection/dye
transfer technique. Intercellular communication via gap junctions is
thought to play a crucial role in cell proliferation and
differentiation and in tissue homeostasis, and consequently in
carcinogenesis. Lindane inhibited junctional communication in a
dose-response relationship (0-20 µg/ml) after a 60-h exposure, but
inhibition was seen after 24 h incubation only with the highest dose
level. In an earlier study, lindane strongly inhibited metabolic
cooperation between V79 cells at a non-toxic dose of 10 µg/ml
(Tsushimoto et al., 1983).
Another explanation for the finding that Avy/a yellow mice have
a predisposition for tumorigenicity but not necessarily for
carcinogenicity is their reduced immunocompetence, as evidenced by
decreased antibody response to T-cell-dependent immunogen tetanus
toxoid, enhanced antibody response to T-cell-independent immunogen
type III pneumococcal polysaccharide, decreased rates of carbon
clearance and increased rates of immunoglobulin A formation. The
pseudoagouti mice did not have reduced immunocompetence and had
reactions similar to those of normal black A/a mice in these
immunological tests (Holder & Stöhrer, 1989).
The lindane metabolite, 2,4,6-trichlorophenol (TCP),
constitutes a significant proportion of the urinary metabolites of
lindane and is considered to be a carcinogen. However, direct
measurements of comparative potency indicate that TCP contributes
only a small fraction of the 'lindane cancer potency' and therefore
may not add significantly to the quantitative impact of lindane. The
notion that TCP adds quantitatively to the carcinogenicity of
lindane per se remains a major element in the evaluation of the
carcinogenic hazard of lindane to humans (Holder & Stöhrer, 1989).
7.7.5 Appraisal of carcinogenicity
Studies to define the carcinogenic potential of lindane have
been conducted with mice and rats, at doses of up to 600 mg/kg diet
in mice and up to 1600 mg/kg diet in rats. In some studies, the dose
levels exceeded the maximum tolerated dose. Hyperplastic nodules
and/or hepatocellular adenomas were found in studies with mice at
doses from 160 mg/kg diet. Two studies using mice and one study
using rats, with dose levels of up to 160 mg/kg diet in mice and 640
mg/kg diet in rats, showed no increase in the incidence of tumours.
The results of studies on initiation-promotion, on mode of action,
and on mutagenicity indicate that the tumorigenic effect of
gamma-HCH in mice results from non-genetic mechanisms.
7.8 Special studies
7.8.1 Immunosuppression
Desi (1976) and Desi et al. (1978) reported the results of a
subacute study in which groups of 30-36 male rabbits were treated
orally, five times per week for 5-6 weeks, with doses of lindane
representing 0, 1/5, 1/10, 1/20, and 1/40 of the oral LD50, which
was 60 mg/kg body weight. Once a week, different doses of S.
typhimurium 'Ty 2' vaccine were injected intravenously. The
humoral immune response was determined by the tube agglutination
test. A linear regression was found between the dose of lindane and
reduction in antibody titres, in a time-dependent manner. The lowest
dose, 1.5 mg/kg body weight (1/40 of the oral LD50) caused no
immunosuppression.
7.8.2 Behavioural studies
The learning rate in a maze and responses to conditioning in a
Skinner box were studied after feeding lindane at daily doses of
2.5, 5, 10, or 50 mg/kg body weight to Wistar rats for 40 days. In
the maze, no effect was seen with 2.5 mg/kg, but at 5 mg/kg there
was stimulation accompanied by an increased error rate in maze
running activity; at 10 and 50 mg/kg, the animals became sedated and
committed more errors than the controls. In the Skinner box,
stimulation was seen with 2.5 and 5 mg/kg. At 10 mg/kg, no
difference was seen from the controls, whereas animals treated at 50
mg/kg were less active than the controls (Desi, 1974).
7.8.3 Neurotoxicity
Lindane produces a variety of neurological effects, both
central and peripheral, in mammals. The induced increase in neuronal
excitability and the underlying mechanisms of action have been
investigated both in vivo and in vitro.
7.8.3.1 Dose-response studies using intact animals
The effects of lindane on body temperature, food intake, and
body weight were studied in Wistar rats given single or repeated
non-convulsant oral doses. Groups of eight male and eight female
rats were given lindane as a single oral dose of 30 mg/kg in olive
oil. Controls received olive oil alone. Further groups of eight
males and eight females received 10 mg/kg and two groups of male
rats received 30 mg/kg once daily for seven days at either
thermoneutral ambient temperature or cold ambient temperature
(4 °C). The single dose of 30 mg/kg significantly decreased core
temperature 5 h later; this lindane-induced hypothermia was strongly
potentiated by cold stress in rats kept at 4 °C. A decrease in body
weight gain was also observed. No hypothermic effect was seen with
10 mg/kg (Camon et al., 1988a).
The relationship between the brain concentration of lindane and
its convulsant effect was studied in male Wistar rats administered
lindane (99.5%) dissolved in olive oil daily by gavage at doses of
5, 12, or 20 mg/kg body weight for 12 days. The mean plateau
concentration in brain was achieved within 5-8 days. There was a
strong correlation between the doses administered and the
concentration in brain at the plateau. A convulsant response was not
seen with 5 mg/kg, but tonic convulsions occurred at the two higher
doses. The rate of response (percentage of rats with convulsions)
was also correlated with the log of the concentration of lindane in
brain. The concentration in brain decreased after 12 days of daily
administration of doses of 5 and 12 mg/kg, but not with 20 mg/kg
(Tusell et al., 1988).
Camon et al. (1988b) investigated the effect of convulsant and
non-convulsant doses of lindane on regional glucose uptake in the
brain. Male Wistar rats received intraperitoneal injections of
3H-2-deoxyglucose, and the amount of label in different brain
structures was assayed by liquid-scintillation counting in 18
dissected brain regions. Lindane at a single convulsant dose (150
mg/kg orally) increased 2-deoxyglucose uptake in olfactory
tubercules, hypothalamus, hippocampus, paraflocculi, and the
post-medulla. With a single, non-convulsant dose of 30 mg/kg, the
uptake of 2-deoxyglucose was less affected; after treatment with 10
mg/kg per day for one week, 2-deoxyglucose uptake was observed in
superior colliculi but was decreased in the parietal cortex. The
increased uptake in limbic regions seen at the convulsive dose
correlates with the experimentally observed association between
signs of poisoning induced by lindane and damage to the limbic
system.
Intraperitoneal injections of gamma-HCH (99.0%) in corn oil at
80-480 mg/kg body weight increased the accumulation of cerebellar
cyclic GM in male CD-1 mice. Furthermore, it inhibited the binding
of 3H-tert-butylbicyclo- ortho-benzoate (a ligand for the
GABA-A receptor-linked chloride channel) in mouse cerebellum
(Fishman & Gianutsos, 1987).
Fishman & Gianutsos (1988) gave male CD-1 mice gamma-HCH at
single intraperitoneal doses of 80-400 mg/kg body weight in corn
oil. At the lowest dose, gamma-HCH increased the lethality and the
frequency of tonic/clonic seizures induced by intraperitoneal
injection of 50 mg/kg pentylenetetrazole or 20 mg/kg picrotoxin but
had no effect on locomotor activity.
Sunol et al. (1988) studied the effect of administering lindane
by gavage at 150 mg/kg body weight in olive oil on the GABAergic and
dopaminergic systems, by measuring the concentrations of GABA,
dopamine and its metabolites in seven brain areas at the onset of
seizures. All animals suffered tonic convulsions 18.3 ± 1.4 min
after lindane administration. The concentration of GABA was
decreased only in the colloculi and not in the other areas. Dopamine
concentrations were increased in the mesencephalon, and those of its
metabolite, DOPAC, were also increased in the mesencephalon and the
striatum (abstract only).
In studies by Desi (1983), adult female CFY rats were given a
daily dose of lindane (99.5%) at 2.5 mg/kg body weight. This dose
level had no functional, neurological, electroencephalographic, or
psychophysiological effect, used as early signs of disturbances of
the nervous system. A dose of 5.0 mg/kg body weight altered the
electrical activity of the brain, as indicated by changes in the
complex electroencephalograph, the number of changing
electroencepahlographic bands and the index number. In behavioural
experiments, running speed and number of errors indicated an
inhibitory effect of lindane at 5.0 mg/kg body weight on learning
capacity; this result was not seen with 2.5 mg/kg.
Müller et al. (1981) studied the electroneurophysiological
effects of various HCH isomers on groups of 15 male Wistar rats by
feeding them diets containing each isomer for 30 days. Conduction
velocity delay was observed in the animals fed the gamma isomer at a
daily dose of 25.4 mg/kg, but not at 12.3 or 1.3 mg/kg. The greatest
delay was induced by the lindane metabolite
gamma-pentachlorocyclohexene (38-783 mg/kg body weight).
Lindane was reported to lower the threshold for kindled
seizures (resulting from repetitive stimulation of the limbic system
within the brain) in rats (Joy et al., 1982, 1983). In these
experiments, stimulating electrodes were implanted in the amygdala
and other part of the limbic system, and the animals were stimulated
with a one-second train of pulses at 60 Hz on each day of the study.
In this procedure, no response is elicited initially; however, with
repeated stimulation, the procedure induces increasing levels of
electrical seizure, with clonic seizures resulting after many
trials. A developing after-discharge becomes progressively longer,
and the severity of the accompanying motor signs becomes more
pronounced, until kindling is completed. The subject then exhibits
stable convulsive responses for weeks or months afterwards. The
duration of the electrical seizure and the severity of the
behavioural response were found to increase much more rapidly when
lindane was administered at daily oral doses of 1, 3, or 10 mg/kg
body weight 3 h before each kindling trial. These effects were found
to be dose-dependent, and a threshold exposure of 0.5 mg/kg per day
was calculated. Rats administered lindane at this concentration were
found to develop brain levels of lindane which fluctuated between
0.2 and 0.4 µg/g.
Joy & Albertson (1987a,b) demonstrated that lindane alters
dentate gyrus granule response to perforant path input in the intact
rat in a manner indistinguishable from picrotoxin or
pentylenetetrazol, which are known GABA-mediated chloride channel
antagonists. In this study, 19 male Sprague-Dawley rats were
anaesthetized with urethane, electrodes for stimulating and
recording responses from the dentate gyrus of the hippocampus were
implanted, and the animals were placed in a stereotaxic device.
Lindane was then administered intraperitoneally in dimethylsulfoxide
to each animal at sequential doses of 5, 10, 20, and 40 mg/kg body
weight. Single or paired electrical stimuli were presented at
different intensities and at different intervals to evaluate the
effects of lindane on inhibition and facilitation. These studies
demonstrated a dose-dependent change in perforant path granule cell
function, manifested as an increase in the excitability of the
granule cell to other stimuli. Lindane was also found to induce a
small but statistically significant, dose-dependent increase in
presynaptic inhibition, as well as a significant increase in
postsynaptic inhibition. A dose-dependent effect on GABA-mediated
inhibition was measurable at exposures that were not convulsant in
unanaesthetized animals. The results of this in-vivo study
indicate that inhibition of GABA-mediated chloride channels in the
brain is probably an important mechanism by which lindane produces
neuronal hyperexcitability and convulsions.
7.8.3.2 Studies on mechanism
Although the precise mechanism by which lindane exerts its
neurotoxic action is not fully resolved, studies using preparations
of synaptosomes (pinched-off nerve endings) and of cholinergic
neuromuscular junctions, as well as studies using intact animals,
have provided insight into this issue. The results of representative
studies with each type of preparation are summarized in Table 13.
TABLE 13. Known effects of lindane on central and peripheral nerves or muscle tissue
Effect Species Type of preparation Reference
Inhibition of Rat in vivo Joy & Albertson (1987 a,b)
GABA-mediated chloride
ion uptake in CNS
Rat Brain synaptosomes/ Eldefrawi et al. (1985)
neuronal membrane Matsumura & Tanaka (1984)
Abalis et al. (1985, 1986)
Mouse Brain synaptosomes Fishman & Gianutsos (1988)
Increased availability Rat Brain synaptosomes Hawkinson et al. (1989)
of intracellular Ca++
Rat Neuroblastoma cells Joy et al. (1987)
Rat Neurohybridoma cells Joy et al. (1988)
Frog Neuromuscular junction Joy et al. (1987)
Vogel et al. (1985)
Publicover & Duncan (1979)
Decreased conduction Rat Tail nerve Müller et al. (1981)
velocity
Mitochondrial damage Frog Skeletal muscle Publicover et al. (1979)
Na+-K+-ATPase inhibition Frog Skeletal muscle Pandy et al. (1985)
Rat Liver mitochondria Srinivasan &
Radhakrishnamurty (1988)
TABLE 13. (Continued)
Effect Species Type of preparation Reference
Ca++-Mg++-ATPase Beef Brain Uchida et al. (1974)
inhibition
Rat Liver mitochondria Srinivasen &
Radhakrishnamurty (1988)
Lindane was shown to inhibit the uptake of chloride ions at
inhibitory synapses in the brain (Matsumura et al., 1984; Abalis et
al., 1985, 1986; Fishman & Gianutsos, 1988), and it is this mode of
action that is now widely considered to account primarily for the
convulsant activity of this insecticide. Because of its structural
similarity to picrotoxinin, lindane has a good geometric fit to the
picrotoxinin-binding site at the outer end of the chloride channel.
Once bound, the lindane is believed to block the action of the
neurotransmitter GABA, which mediates the entry of the Cl- necessary
for inhibitory neuronal function (Matsumura, 1985). More recent
studies by Joy & Albertson (1987a,b) provide evidence of such
inhibition of GABA in rats in vivo, demonstrating that: (a) this
mechanism operates at clinically relevant exposure levels (5-40
mg/kg); (b) the magnitude of this effect is dose-dependent; and (c)
this effect can be clearly measured at subconvulsant exposure levels
in unanaesthetized subjects.
Lindane has also been demonstrated to increase the excitability
of presynaptic cholinergic neurons at central (Hawkinson et al.,
1989) and peripheral (Vogel et al., 1985; Publicover & Duncan, 1979)
synapses. Using a nerve-muscle preparation, a universally accepted
model for a cholinergic synapse, Publicover & Duncan demonstrated
that concentrations of lindane as low as 5 x 10-5 mol/litre
increase the spontaneous release of acetylcholine. Similar results
were reported by Vogel et al. (1985), who observed increases up to
100 fold in spontaneous release of neurotransmitter in the presence
of lindane at 10-4 mol/litre. This increase was found in both
studies to be somewhat dependent upon the concentration of
extracellular calcium ions. In the presence of normal (approx. 1-2
mmol/litre) Ca++, the increase in spontaneous transmitter release
was 2.5-20 times greater than that measured when the extracellular
Ca++ level was buffered to 10-7 mol/litre, the homeostatically
maintained intracellular Ca++ concentration. An expanded report of
the study of Vogel et al. (1985) was made by Joy et al. (1987), who
additionally described a similar lindane-induced excitability in
neuroblastoma cells. Hutchison verified the involvement of Ca++ in
the enhanced release of acetylcholine using a rat brain synaptosome
preparation, and he postulated that the increased excitability of
cholinergic synapses (widely dispersed throughout the brain) may
contribute to the convulsant effect of lindane.
Pandy et al. (1985) reported that lindane weakly inhibits
Na+-K+-ATPase activity in skeletal muscle, and they suggested
that lindane can inhibit Ca++-Mg++, Ca++-, and mg++-ATPase.
Uchida et al. (1974) reported decreased Ca++-Mg++-ATPase
activity in beef brain. Since all of these ATPases are involved in
the maintenance of intracellular Ca++ concentration and have been
identified in a neuronal membrane preparation (Yamaguchi et al.,
1979), it is possible that the disruption of homeostatic calcium
regulation by lindane contributes to its excitatory action on the
central nervous system.
7.8.3.3 Summary
Chronic exposure to low levels of lindane can result in
proconvulsant activity, as demonstrated experimentally using the
kindling model of experimental epilepsy. Lindane has also been shown
to cause convulsions in rats administered oral doses of 12 mg/kg
daily for 12 days; a dose of 5 mg/kg body weight did not induce
convulsions within that period. The convulsive effect has been
suggested to be associated with inhibition of GABA-mediated chloride
channels in the brain, as demonstrated experimentally in mammals
both in vivo and in vitro. Changes in the electroencephalogram
and decrements in a variety of behavioural parameters have been
observed with lindane at a dose of 5 mg/kg/body weight per day for
40 days, but not at 2.5 mg/kg for 22 days. A delay in peripheral
nerve conduction velocity was observed in rats administered a
dietary concentrations of 25 mg/kg body weight, but not at 12 mg/kg.
7.9 Factors that modify toxicity; toxicity of metabolites
Pretreatment of rats with lindane minimized or inhibited the
convulsive effects of pentazole, picrotoxin, loramine, strychnine,
mintracol, cyclohexane sulfonamide and electro-shock (Herken,
1950a,b,c, 1951; Coper et al., 1951; Kewitz et al., 1952; Lange,
1965). It was demonstrated that premedication for two weeks with
daily doses of 2 mg lindane not only accelerated the metabolism of
other chemicals, but also caused an acceleration of its own
breakdown: in Fischer rats given two oral administrations of lindane
in oil at 2 mg/kg, there was increased excretion of glucuronic acid
conjugates (Chadwick et al., 1971).
The smallest single oral dose of lindane that reduced
pentobarbital sleeping time in FW-49 rats was 5 mg/kg body weight
(Schwabe & Wendling, 1967). The smallest single intraperitoneal dose
of lindane that shortened hexabarbital anaesthesia was 15 mg/kg body
weight. A similar result was obtained in Sprague-Dawley rats fed a
diet containing lindane at 0.5 mg/kg; the effect was more distinct
with a dose of 4 mg/kg diet (Kolmodin-Hedman et al., 1971).
8. EFFECTS ON HUMANS
8.1 Exposure of the general population
8.1.1 Acute toxicity, poisoning incidents
Several cases of fatal poisoning and numerous cases of
non-fatal illness caused by or ascribed to lindane have been
reported. These incidents were either accidental, intentional
(suicide) or due to gross neglect of safety precautions. In many of
these cases, the effects ascribed to lindane were more likely to
have been due, in total or in part, to other substances. A critical
review of these cases is provided by Hayes (1982).
The toxic or lethal dose appears to vary considerably with the
carrier and/or the degree of homogenization of the product. Under
certain conditions, 10-20 mg/kg body weight can present a lethal
hazard to humans, but higher concentrations can be tolerated when
followed by timely and appropriate medication. Starr & Clifford
(1972) described a case of acute lindane intoxication in a
2.5-year-old child who had severe epileptiform convulsions after
ingesting presumably two 0.78-g pellets containing 95% lindane. The
child recovered under medical care.
A suicide attempt was reported by Ohly (1973). The person
ingested about 100 ml of a 25.5% lindane emulsion concentrate, which
corresponds to a dose of about 309 mg/kg body weight. No vomiting or
diarrhoea was seen, but severe convulsions occurred within the first
24 h after lindane ingestion. Elevated serum levels of
glutamic-oxaloacetic transaminase, lactic dehydrogenase,
glutamic-pyruvate transaminase, and creatine phosphokinase activity,
in conjunction with the results of a liver biopsy, suggested fatty
degeneration and severe toxic damage to the liver. After five weeks
of clinical treatment, the patient recovered completely.
A number of cases of poisoning cases have been described after
medical treatment or abuse (Davies et al., 1983; Kurt et al., 1986;
Petring et al., 1986; Berry et al., 1987).
Clinical signs of intoxication can appear from a few minutes to
some hours after intake of lindane, depending on the route of
administration, the formulation, the concentration of lindane, and
the quantity involved. In mild cases, indisposition, nausea,
dizziness, restlessness, frontal headaches, and sometimes vomiting
occur. Muscular fasciculation, disturbances of equilibrium, ataxia,
and tremor may appear. Pains in the upper abdomen are frequently
coupled with diarrhoea and uncontrolled micturition. Clonic-tonic
convulsions of some minutes' duration may occur, and these may recur
after several hours or even days in response to optical, tactile,
and acoustic stimuli. In fatal cases, death follows several hours to
several days after intake. The cause of death is usually central
respiratory failure or acute circulatory collapse, often after
convulsions (Hayes, 1982; Jaeger et al., 1984).
8.1.2 Effects of short- and long-term exposures - controlled human
studies
8.1.2.1 Oral administration
Lindane given orally as a vermicide at a dose of 45 mg to an
adult patient (26 years old) in poor condition induced convulsions,
nausea, and vomiting. Recovery took place within 3-4 h. Of 15
patients in the same trial, who repeatedly received up to 30
mg/person for up to three days, six complained of nausea; no other
symptom was reported (Graeve & Herrnring, 1949).
Severe toxic symptoms were described in healthy volunteers
after oral intake of 15-17 mg/kg body weight of lindane in a liquid
carrier (Hofer, 1953; Schmiedeberg & Wasserburger, 1953). Reports of
toxic effects after administration of lindane against scabies
indicate that children are more sensitive to lindane than adults,
and rare cases of aplastic anaemia have been reported (G. Volans,
National Poison Unit, London, letter to IPCS, 1989).
8.1.2.2 Dermal application
Clinical reports after pharmaceutical use of lindane
cutaneously suggest that exposures somewhat higher than 5 mg/kg body
weight per day do not usually result in acute neurotoxic symptoms.
No cause-effect relationship was found between lindane and blood
dyscrasias (Ginsburg et al., 1977; Kramer et al., 1980; Morgan et
al., 1980).
Three groups, each consisting of one male and one female
volunteer, received an application of 30 g of a 0.3% commercial
lindane emulsion over the entire body (except the head and the angle
of the elbow) on three consecutive days. The first group removed the
emulsion by washing with soap and water 3 h after application; the
second removed it 10 h after application by washing with water only
at body temperature, and the third group by washing with soap and
water 10 h after application. A group of scabies patients were given
an application of 50 g of the emulsion over the entire body, except
the angles of the elbows. Blood samples were taken regularly from
all participants in the trial over a period of 1-144 h. The highest
average serum concentration found approximately 5.5 h after
application in the healthy volunteers was slightly less than 5
µg/litre, while in scabies patients an average of about 200 µg/litre
was found after 4 h. It was concluded that scabies patients deposit
greater concentrations of lindane in their bodies than do healthy
persons; the levels were higher in women than in men (Lange et al.,
1981; Zesch et al., 1982).
Feldmann & Maibach (1974) studied the absorption and excretion
of 14C-labelled lindane dissolved in acetone, applied at 4
µg/cm2 to the forearms of six human subjects. The usual dose of
radioactivity was 1, 2, or 5 µCi. Data obtained after intravenous
dosing was used to correct the results obtained on skin penetration
for incomplete urinary recovery. Total excretion of 14C after
topical application was 9.3% ± 3.7 of the dose in five days. Skin
absorption is incomplete because the chemical is lost from the skin
surface by washing, evaporation or gradual exfoliation of outer
layers of the stratum corneum. The amount absorbed into the body
depends on the relationship between the speed with which it
penetrates the skin and the speed with which it is lost from the
skin surface.
Serum concentrations of gamma-HCH were determined in nine
children (3.5-18 years old) following application of a 1% gamma-HCH
shampoo to treat pediculosis capitis (head lice). The shampoo was
applied vigorously to dry hair in a sufficient amount to saturate
the hair and scalp thoroughly. After 10 min, small quantities of
water were added until a lather formed, and shampooing was continued
for 4 min; thereafter, the hair was rinsed and blown dry. Gamma-HCH
was present in the serum of all children 2-24 h following the
application. A maximal concentration of 1.4 µg/litre was found after
2-4 h; this level decreased within 24 h to 0.41 µg/litre.
Re-treatment increased the maximal level to 3.6 µg/litre (Ginsburg &
Lowry, 1983).
Nitsche et al. (1985) applied emulsions of 0.3 and 1.0%
14C-lindane to defined areas of 2 cm2 intact or stripped skin.
They found that the flux of lindane in the skin was time-dependent:
Generally, the concentration increased with the depth of the layer.
Increased availability of lindane, induced by absence of the stratum
corneum and a long application period, resulted in preferential
accumulation in the epidermis, with none in the subcutaneous fat.
When intact skin was washed with soap and water 3 h after the
application, the concentration of lindane in the layer below the
stratum corneum 7 h later was higher than that in skin that had not
been washed. Washing of intact skin after a short penetration period
(3 h) resulted in introduction of lindane; this phenomenon was not
seen with stripped skin. Lindane could be more effectively removed
from the stratum corneum with soap and water than with water alone.
8.1.3 Epidemiological studies (general population)
The rate of mortality from liver cancer in the USA was related
to the 'domestic disappearance' of organochlorine pesticides. In
1962, 18 and 15 years after the introduction of DDT and technical
HCH, respectively, by which time any increase in the mortality rate
from primary liver cancer would be manifest, the number of cases of
primary liver cancer as a percentage of the total number of deaths
from liver cancer began a gradual, steady decline, from 61.3% in
1962 to 56.9% in 1972. The death rate (per 100 000 per year) from
primary liver cancer during this period declined from 3.46 to 3.18
(Deichmann & MacDonald, 1977).
A considerable number of case reports have been published in
which different blood dyscrasias were described in people who had
been exposed to lindane or to lindane and other chemicals (Mendeloff
& Smith, 1955; Albahary et al., 1957; Jedlicka et al., 1958;
Stieglitz et al., 1967; West, 1967; Hoshizaki et al., 1969;
Vodopick, 1975). The issuance by the US Environmental Protection
Agency (1977) of a 'rebuttable presumption against registration and
continued reregistration' of lindane in 1977 was triggered in part
by the problem of blood dyscrasias. Studies conducted over periods
of several weeks to several years, however, have given no indication
that there might be a cause-effect relationship between exposure to
lindane and blood dyscrasias (Milby & Samuels, 1971; Samuels &
Milby, 1971; Morgan et al., 1980; Wang & Grufferman, 1981), and this
was the final conclusion of the US Environmental Protection Agency
(1984a).
8.2 Occupational exposure
8.2.1 Toxic effects
Evaluation of the effects of gamma-HCH in occupationally
exposed workers is seriously hampered by the fact that most of the
studies are of workers who were exposed during manufacturing and
handling of lindane, or in the handling or spraying of
technical-grade HCH among other pesticides. All of these groups are
also potentially exposed to other HCH isomers, to impurities and to
other (process) chemicals. It is, therefore, difficult to relate the
effects found in these studies to any individual substance. The only
such studies mentioned here are those that were considered to be
useful for the evaluation.
Kolmodin-Hedman (1974) investigated blood levels of gamma-HCH
in 54 spraymen exposed to 4% lindane and other insecticides in the
form of aerosols and mists and who also occasionally diluted stock
solutions. Their exposure varied from once daily to once weekly, and
the length of exposure was 1-20 years; they did not always wear
protective gloves and respirators. Spraymen exposed to lindane had
mean plasma levels of gamma-HCH of 6.4, 7.5, and 9.9 µg/litre; a
maximal concentration of 87.0 µg/litre was found. Antipyrine
half-lives in exposed subjects and in controls were compared to
investigate whether lindane induces drug metabolism in humans. In 26
workers exposed mainly to lindane, the mean antipyrine plasma
half-life was significantly shorter than that in 33 controls: 7.7 ±
2.6 h compared with 13.1 ± 7.5 h, respectively. Induction occurred
with plasma levels above 10 g/litre; workers who were exposed to
lindane had shorter antipyrine and phenylbutazone half-lives when
their serum levels were above this value. Hyperlipoproteinaemia
(defined as a serum cholesterol level above 800 mg/litre and a
phospholipid level above 1400 mg/litre in the HDL fraction of the
serum lipoproteins) was found in 40% of the spraymen
(Kolmodin-Hedman, 1974, 1984).
Three workers mixed rapeseed manually with 75% lindane powder,
which also contained 10% thiram, and usually closed the sacks by
hand. This mixing procedure was repeated up to 80 times during a
workshift. The working period comprised the spring months of each
year, and the total exposure period varied between one and five
years. Gloves and masks were not always used, so dermal and
respiratory exposures were intensive. The plasma levels during
exposure in the three people who prepared rapeseed were 102, 100,
and 4.2 µg/litre; the last person had less frequent exposure than
the other two. The plasma level in the person who had 100 µg/litre
during exposure had decreased to the level before exposure within
five months (Kolmodin-Hedman, 1974, 1984).
Workers engaged in the production of lindane and exposed for at
least six months (8 h/day; wearing face masks in an air-ventilated
location) were tested for the presence of chromatid-type and labile
chromosome-type aberrations in their lymphocytes. The frequency of
stable chromosomal aberrations did not differ significantly from
that in normal controls (Kiraly et al., 1979).
Herbst (1976) examined workers engaged in the production of
lindane in three factories. The people were exposed not only to the
gamma isomer but also to the alpha, beta, delta, and epsilon
isomers. The average length of service was slightly more than 10
years. Of the 118 persons examined, 115 were men and three were
women, and the average age was 39 years. No abnormality was detected
in the haematopoietic system, the liver, the kidneys, or the nervous
system.
A series of reports were made on groups of 54-60 male workers
(24-62 years of age) in a lindane-producing factory, with a
geometric mean duration of exposure of 7.2 years (range, 1-30 years)
(Baumann et al., 1981; Brassow et al., 1981; Tomczak et al., 1981).
The lindane concentrations in serum were in the range 5-188
µg/litre; that of alpha-HCH was 10-273 µg/litre, and that of
beta-HCH, 17-760 µg/litre. None of the controls had a HCH
concentration in serum above the limit of detection (O.7µg/litre).
The time-weighted average threshold limit value of 0.5 mg/m3 was
not exceeded at any of the workplaces (range, 0.004-0.15 mg/m3);
the level of alpha-HCH was 0.002-1.99 mg/m3 and that of beta-HCH,
0.001-0.38 mg/m3. Only small deviations were found in some
laboratory tests: higher polymorphonuclear leukocyte count, lower
lymphocyte count, higher reticulocyte count, lower prothrombin
level, lower blood concentrations of creatinine and uric acid; these
findings were regarded as of no pathological significance. No
significant difference was observed in total red cell, white cell,
and platelet counts, in haemoglobin content, or in levels of
gamma-glutamine transferase, glutamic-oxaloacetic transaminase,
glutamic-pyruvic transaminase, lactic dehydrogenase, cholinesterase,
triglyceride, cholesterol or urea. No sign of health impairment was
observed. Examination of reflexes, sensitivity, amplitude and
frequency of fore-finger tremor and manual skills showed no
significant difference between the HCH-exposed and the control
groups. In addition, no pathological result was obtained in tests
for electromyography, maximal motor nerve conduction velocity in
ulnar nerves, or neuromuscular conduction. Furthermore,
electroencephalographic recordings showed no specific pathological
sign. The authors concluded that, even after decades of occupational
exposure to HCH, no sign of neurological impairment or perturbation
of neuromuscular function had occurred. Serum levels of luteinizing
hormone were higher in HCH-exposed workers than in controls (8.8 vs
5.7 mIU/ml). Levels of follicle stimulating hormone were
insignificantly higher and testosterone levels were insignificantly
lower in exposed men than in controls.
In malaria-control workers, who sprayed technical-grade HCH for
16 weeks, the serum level of gamma-HCH increased from a mean of
0.009 to 0.037 mg/litre in previously unexposed workers, and from
0.009 to 0.034 mg/litre in workers who had been exposed during three
previous spraying seasons (Gupta et al., 1982). In comparison with
the alpha and beta isomers, the gamma isomer was the least
cumulative and the least persistent in serum. These findings, as
well as those of Milby et al. (1968), suggest that gamma-HCH levels
in blood are mainly a reflection of recent exposure to lindane.
Nigam et al. (1986) studied 64 employees at a manufacturing
plant who were directly or indirectly associated with the production
of HCH. The exposed group comprised 19 workers who handled and
packaged the insecticide, 26 plant operators and supervisors who
were exposed indirectly to HCH, and 19 members of the maintenance
staff who visited the plant frequently. The control group consisted
of 14 workers who had no occupational contact with HCH. The length
of exposure varied from 0 to 30 years. The mean serum concentrations
of lindane in the four groups were: control, 0.0007, maintenance
staff, 0.0227, indirect exposure, 0.016, and handlers, 0.0571
mg/litre. Alpha-, beta-, and delta-HCH were also present; the total
HCH concentrations in serum were 0.0514 mg/litre in the controls,
0.1436 mg/litre in the maintenance staff, 0.2656 mg/litre in the
indirectly exposed workers, and 0.604 mg/litre in the handlers. Most
of the directly and indirectly exposed workers had paraesthesia of
the face and extremities, headache, and giddiness, and some had
symptoms of malaise, vomiting, tremors, apprehension, confusion,
loss of sleep, impaired memory and loss of libido. The same symptoms
were found in the group of maintenance workers but were less severe
and occurred in fewer cases.
Chattopadhyay et al. (1988) studied 45 male workers exposed to
HCH during its manufacture and compared them with 22 matched
controls. Paraesthesia of the face and extremities, headache,
giddiness, vomiting, apprehension, and loss of sleep, as well as
some changes in liver function were reported. These changes were
found to be more closely related to intensity of exposure (as
measured by HCH levels in blood serum) than to duration of exposure.
The measured exposures to total HCH were 13 to 20 times higher than
those in the control groups (details not given). Of the total HCH,
60-80% was gamma-HCH.
Plasma levels of gamma-HCH and urinary levels of three TCPs
were measured in 45 forestry workers who were engaged in dipping
conifer seedlings in a gamma-HCH solution, transporting the dipped
seedlings to planting sites or planting the seedlings. Protective
clothing was supplied. The work started in April, and until June the
workers had plasma concentrations below the detection limit (5
nmol/litre). From June onwards, there was an upward trend in the
number of workers who had gamma-HCH levels in plasma of up to 40
nmol/litre. In July, levels of 59, 75, and 123 nmol/litre were found
in three workers, and the latter two had symptoms of poisoning that
included feeling unwell with a flu-like illness, fatigue, sore
throat, and nausea. No sign of hepatotoxicity was observed. People
with levels greater than 70 nmol/litre were removed from further
exposure. When exposure ceased at the end of July and the people who
had had elevated levels were monitored in August, 80% of the workers
had no detectable gamma-HCH in their plasma; the group mean
concentration was 16 nmol/litre. By September, all plasma levels had
returned to the pre-exposure level, below the detection limit. The
mean half-life of gamma-HCH in plasma was calculated to be about
eight days. 2,4,6-, 2,3,5-, and 2,4,5-TCP were the major metabolites
of gamma-HCH (Drummond et al., 1988).
Neurological studies on 37 workers exposed to lindane over a
period of two years revealed three with serious
electroencephalographic disturbances; minor symptoms and signs were
seen in 14 of the workers. No change was observed in the
electroencephalographic patterns of 21 of the exposed individuals.
Blood levels of gamma-HCH were 0.002-0.34 mg/litre. The frequency of
clinical symptoms and electroencephalographic changes was higher
among individuals whose blood contained 0.02 mg/litre or more of
gamma-HCH (Czeglédi-Janko & Avar, 1970; American Conference of
Governmental Industrial Hygienists, Inc., 1986).
Tolot et al. (1969) and Schüttmann (1972) reported peripheral
neuropathies after contact with technical-grade HCH or lindane.
8.2.2 Irritation and sensitization
Behrbohm & Brandt (1959) described 26 cases of allergic and
toxic dermatitis in workers exposed during the manufacture of
technical-grade HCH. Patch testing with pure alpha-, beta-, gamma-,
and delta-HCH gave negative results, but positive reactions were
obtained with residual fractions. Baumgartner (1953) also described
skin sensitization in four workers involved in lindane manufacture.
Cases of allergic disease (rhinitis, conjunctivitis, and eczema)
were also reported among workers in the USSR exposed to lindane
(Krzhyzhanovskaya, 1973, and Bezuglikh et al., 1976; see Izmerov,
1983).
Patch tests were performed with 1% lindane dissolved in a
petroleum solvent on the upper back of 200 subjects, 105 men and 95
women, aged 18-76 years; 50 of them (34 men and 16 women) were
agricultural workers, 24 (18 men and six women) had worked on the
land in the past, and the other 53 men and 73 women had never used
pesticides. Results were read after 48 and 72 h. A positive reaction
was found in none of the 200 subjects tested (Lisi et al., 1986).
In a study to establish the optimal test concentration of lindane
and the frequency of irritant and sensitization reactions, Lisi et al.
(1987) tested 335 men and women, of whom 70 were employed in
agriculture, 25 had been employed in agriculture in the past and 240
had never been exposed to pesticides. The results of the patch test,
using 1% lindane in a petroleum solvent, were all negative, and no
sensitization reaction was observed.
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1 Microorganisms
9.1.1 Bacteria
The effect of lindane at concentrations up to 100 times the
recommended dose for field application (1 mg/kg of soil) was studied
on organic mineralization and on nitrification rates in alluvial
soil around Delhi, India. Lindane inhibited the evolution of CO2
from soil at a concentration of 100 mg/kg but not or only slightly
with 1 and 10 mg/kg over an incubation period of 120 days.
Nitrification by the bacterial species Nitrobacter and
Nitrosomonas was decreased with the dose of 100 mg/kg, and 10
mg/kg induced inhibition during the first three weeks after
application. Nitrification was restored within 35 days (Gaur &
Misra, 1977).
The influence of technical-grade HCH on the nitrifying
characteristics of Nitrosomonas and Nitrobacter species isolated
from an alluvial soil was studied in artificial growth media and in
a flooded, autoclaved soil. The concentrations tested were 0, 5, 20,
and 50 mg/kg soil or culture medium; the observation period was
10-40 days. The rate of nitrification was slower in autoclaved soil
than in culture media. Technical-grade HCH inhibited nitrification
at concentrations of 5 mg/kg and more (Ray, 1983).
Oxidation of substrate nitrogen was evaluated on the basis of
the appearance of nitrite in the medium in the presence of
Nitrosomonas europaea and on the disappearance of nitrite in the
presence of Nitrobacter agilis. N. agilis was sensitive to lindane
at concentrations as low as 1 µg/ml, and the highest concentration
tested, 1000 µg/litre, delayed nitrate production. In N. europaea,
lindane induced complete inhibition at a concentration of 10 µg/ml
within 6-7 days (Garretson & San Clemente, 1968).
9.1.2 Algae
9.1.2.1 Blue-green algae
Bringmann & Kühn (1978) saw no inhibition of growth of
Microcystis aeruginosa at concentrations of lindane up to 0.3
mg/litre, and Palmer & Maloney (1955) found no inhibition of the
growth of Microcystis aeruginosa or Cylindrospermum licheniforme
at 2 mg/litre. Both concentrations are higher than those that would
result in adverse effects in fish or crustaceans.
Concentrations of 50, 60, and 80 µg/ml of lindane were lowered
in the presence of 25 species of blue-green algae, thus reducing its
toxicity (Das & Singh, 1977).
9.1.2.2 Freshwater algae
Lindane was lethal to Scenedesmus acutus (age of culture 1,
3, or 5 days) at a concentration of 5 mg/litre after 5 days'
exposure. At concentrations of 1-5 mg/litre, 50% growth reduction
occurred (Krishnakumari, 1977). Jeanne-Levain (1974) also observed
about 50% inhibition of growth of Dunaliella bioculata with
lindane at 5 mg/litre; 10 mg/litre completely inhibited growth.
Similar findings were described by Jeanne (1979). No observable
inhibition of growth was found by Bringmann & Kühn (1978) in
Scenedesmus quadricauda at concentrations up to 1.9 mg/litre; and
Palmer & Maloney (1955) saw no inhibition in Scenedesmus obliquus,
Chlorella variegata, Gomphonema parvulum, or Nitzschia palea at
the only dose tested, 2 mg/litre.
9.1.2.3 Marine algae
Growth inhibition was studied in three species of marine algae
by Ukeles (1962), using concentrations of 1-9 mg/litre. No
inhibitory effect was found in Protococcus sp. at up to the
highest concentration tested or in Pheodactylum tricornutum at up
to 1 mg/litre. Chlorella sp. were slight inhibited at the lowest
concentration tested, but the dose-response relationship indicated
that the threshold was not far below this dose. In tests with green
algae and diatoms, growth was not inhibited by lindane at
concentrations below about 1 mg/litre.
9.1.3 Dinoflagellates, flagellates, and ciliates
Representative members of these three groups were exposed to
lindane at concentrations of 0.5-60 mg/litre (Jeanne-Levain, 1974).
Lethality was reached in Amphidinium carteri (dinoflagellate) at 2
mg/litre and in Tetrahymena pyriformis (ciliate) at > 10
mg/litre; no lethal effect was observed in Euglena gracilis
(flagellate) at up to the highest dose of 60 mg/litre. All doses had
inhibitory effects.
Ukeles (1962) observed inhibition of the growth of the
flagellates Monochrysis lutheri and Dunaliella euchlora with
concentrations of lindane > 5 mg/litre.
9.2 Aquatic organisms
9.2.1 Invertebrates
Green et al. (1986) investigated a range of 10 common European
freshwater macroinvertebrates in a continuous flow system to
establish their response to lindane at concentrations ranging from
25 ± 4.8 to 430 ± 32 µg/litre under similar test conditions: water
temperature, 11 ± 1 °C; pH, 7.5-8.0; dissolved oxygen, > 96%; and
hardness of water (as CaCO3), 92.9 ± 6.3 mg. The 96-h LC50 varied
from 4.5 up to > 430 µg/litre (Table 14). The ephemeropteran
Baetis rhodani and the plecopterans Leuctra moselyi and
Protonemura meyeri were the most sensitive species; Physa
fontinalis and Polycelis tenuis were the most tolerant.
Table 14. LC50 values for invertebrates
Taxon Species 96-h LC50 (µ/litre)
Insecta
Plecoptera Pteronarcys california 4.5
Leuctra moselyi < 130
Protonemura meyeri < 130
Ephemeroptera Baetis rhodani 54
Diptera Chironomus riparius 235
Trichoptera Hudropsyche angustipennis 330
Crustacea
Amphipoda Gammerus rhodani 48
Gammerus facia 10
Gammerus pulex 225
Isopoda Asellus aquaticus 375
Mollusca
Gastropoda Physa fontinalis > 430
Platyhelminthes
Tricladia Polycelis tenui s > 430
Annelida
Oligocheata Limnodrilus hoffmeisteri > 430
9.2.1.1 Crustaceans
The sensitivity of both freshwater and seawater crustaceans to
the acute toxicity of lindane is summarized in Table 15. In general,
freshwater crustaceans were much less sensitive to lindane than
those living in seawater. The LC50 values for freshwater
crustaceans are comparable to or about one order of magnitude higher
than the median values for fish, whereas seawater crustaceans are
generally an order of magnitude more sensitive than fish.
Daphnia magna were exposed to lindane at 11-210 µg/litre
continuously for 64 days, thus covering three succeeding generations
(Macek et al., 1976). A clear dose-dependent depression of
reproduction was found at higher concentrations. The authors
assessed the NOEL to be between 11 and 19 µg/litre. In the same
study, Gammarus fasciatus was exposed to lindane at concentrations
of 1.2-17.7 µg/litre. The NOEL for survival and reproductive success
was 4.3 µg/litre. Gammarus fasciatus is thus more sensitive to
both the acute and chronic effects of lindane.
9.2.1.2 Aquatic arthropods
The results of studies on the acute toxicity of lindane in
freshwater insects (mainly nymphs) are summarized in Table 15.
Aquatic insects can be seen to be sensitive to lindane, with
considerable differences between the tested species. The NOEL for
continuous exposure of two successive generations of Chironomus
tentans was 2.2 µg/litre (Macek et al., 1976).
The 48-h LC50 for the water mite Hydrachna trilobata Viets,
an aquatic arachnida, was 0.05 mg/litre (Nair, 1981).
9.2.1.3 Molluscs
The acute effects of lindane have been investigated in bivalves
and gastropods; no data were available on cephalopods. The LC50
values determined (Table 16) are all > 1 mg/litre, and therefore at
least one order of magnitude higher than those reported for fish,
indicating that molluscs are much less sensitive to the acute action
of lindane.
Butler (1963b) determined shell growth in oysters exposed to
various concentrations of lindane. When sufficiently irritated,
oysters close their shells, do not feed and therefore do not grow.
Even at the lowest dose tested, 1 mg/litre, a decreased shell growth
of 43% was observed.
The freshwater snail Lymnea stagnalis was exposed to lindane
at concentrations of 1 or 2 mg/litre for periods up to seven weeks,
and therefore at least two successive generations. No increase in
mortality over that in controls was observed at either
concentration. A slight decrease in shell growth was found at 2
mg/litre, and egg production was reduced at 1 and 2 mg/litre.
Embryonic development was also disturbed at both concentrations in a
dose-dependent manner. A NOEL for reproduction was not obtained in
this study, but the results suggest that it is close to 1 mg/litre
(Bluzat & Seuge, 1979; Seuge & Bluzat, 1979a,b, 1982).
In a study on the effects of lindane on egg production in the
mud snail Nassa obsoleta (Eisler, 1970a), the number of egg cases
deposited by day 33 after treatment following initial exposure for
96 h was measured. A clear NOEL was found at 1 mg/litre; the next
dose tested (10 mg/litre) induced a clear decrease.
Table 15. Acute toxicity of gamma-HCH to freshwater and marine crustaceans
Species Size (g, mm, Temperature Test 96-h LC50 Reference
cm, age) (°C) conditions (µg/litre)
Freshwater
Daphnia magna 24-h old 20 Static 516 Randall et al. (1979)
Static 485 Juhnkje & Lüdemann (1978)
18-h old 20 Static 1100 Sanders & Cope (1966)
Daphnia pulex 18-h old 21 Static 460 Sanders & Cope (1966)
Gammarus fasciatus Static 39 Juhnke & Lüdemann (1978)
Gammarus pulex 1.0-1.5 cm 15 Statica 34 Abel (1980)
Gammarus lacustris 2 months? 21 Static 48 Sanders (1969)
Simocephalus serrulatus 18-h old 21 Static 880 Sanders & Cope (1966)
Marine
Pink shrimp (Penaeus duorarum) 30-52 mm 24-26 Flow-through 0.17 Schimmel et al. (1977)
Brown shrimp (Penaeus aztecus) Adult 30 Flow-through 0.4 Butler (1963b)
Brown shrimp (Crangon crangon) - 15 Static 1-3.3 Portmann (1970)
Grass shrimp (Palaemonetes pugio) - 23-27 Flow-through 4.4 Schimmel et al. (1977)
Grass shrimp (Palaemonetes vulgaris) 0.47 g 20 Static 10 Eisler (1969)
Sand shrimp (Crangon septemspinosa) 0.25 g 20 Static 5 Eisler (1969)
Hermit crab (Pagurus longicarpus) 0.28 g 20 Static 5 Eisler (1969)
Mysid shrimp (Mysidopsis bahia) 8-9.5 mm 23-25 Flow-through 6.3 Schimmel et al. (1977)
a Water renewed regularly
Table 16. Acute toxicity of gamma-HCH to aquatic invertebrates
Species Freshwater/ Size (g, mm, Temperature Test 96-h LC50 Reference
Marine water cm, age) (°C) conditions (mg/litre)
Bivalves
Mytulis galtoprovincialis Marine 50-70 mm 17 Static 5.5 Rao (1981)
Mercenaria mercenaria Marine Eggs 20 Static > 10 Eister (1970a)
Marine Eggs 24 Static > 10a Davis & Hidu (1969)
Crassostrea virginica Marine Eggs 24 Static 9.1a Davis & Hidu (1969)
Cardium edule Marine - 15 Static 10a Portmann (1970)
Gastropods
Physa acuta Freshwater - 20-28 Static 8.1a Hashimoto & Nishiuchi (1981)
Semisulcospira libertina Freshwater - 20-28 Static 6.2a Hashimoto & Nishiuchi (1981)
Indoplanorbis exastus Freshwater - 20-28 Static 7.1a Hashimoto & Nishiuchi (1981)
Lymnea stagnalis Freshwater Staticb 7.3a Bluzat & Seuge (1979)
Insects
Cloeon diprerum Freshwater - 20-28 Static 0.15a Hashimoto & Nishiuchi (1981)
Chironomus tentans Freshwater Static 0.207a Juhnke & Lüdemann (1978)
Pteronarcys californica Freshwater 30-35 mm 15.5 Static 0.0045 Sanders & Cope (1968)
a 48-h LC50
b Water renewed regularly
Davis & Hidu (1969) found 67% survival of the larvae of the
hard clam Mercenaria mercenaria over 12 days after exposure to
lindane at a concentration of 10 mg/litre. For oysters (Crassostrea
virginica), 50% survival was seen after exposure at 9.1 mg/litre
over 48 h.
Overall, these studies show that reproduction of molluscs is
not adversely affected at concentrations just below 1 mg/litre, a
level much higher than the NOELs for fish and crustaceans.
9.2.2 Fish
9.2.2.1 Acute toxicity
The results of studies on the acute toxicity of lindane in fish
have been reported in the literature since 1959, although in most of
these no data were given on the purity of the lindane used. Since
there is no significant difference in the values reported around
1960 and those reported in 1975-80, however, data on the acute
toxicity of lindane are summarized here regardless of whether the
purity of the compound tested was reported.
LC50 values for lindane in several fish species are
summarized in Table 17. Most values fall within a range of 0.02-0.09
mg/litre, the majority being around 0.05 mg/litre. Only Macek &
McAllister (1970) found an extraordinarily low LC50 value for
brown trout of 0.002 mg/litre.
The symptoms of acute poisoning are mainly gross irritability,
loss of equilibrium, changes in pigmentation and localized
peripheral haemorrhage. Irritability appears within the first
minutes of exposure and is accompanied by loss of equilibrium and
disturbed swimming motion. Poisoned fish show signs of respiratory
distress. Haemorrhages appear at sub-lethal doses 2-4 days after the
beginning of exposure.
A clear temperature dependence of the LC50 was found in
bluegills (Cope, 1965; Macek et al., 1969), lindane being more toxic
at higher temperatures, although the range of LC50 values was the
same. Macek et al. (1969) found LC50 values of 0.054 mg/litre at
12.7 °C and 0.037 mg/litre at 23.8 °C.
In two studies, wild populations of mosquito fish were compared
to laboratory strains and to each other with respect to the toxic
action of lindane (Culley & Ferguson, 1969). Wild populations from
areas that had previously been treated with lindane tolerated higher
concentrations of the substance: one wild population had a LC50
value of 3.104 mg/litre, whereas a susceptible population from a
different region had a value of 0.074 mg/litre. These results
suggest that wild populations can adapt to lindane when exposed
repeatedly. Boyd & Ferguson (1964) found a similar effect.
9.2.2.2 Long-term toxicity
Macek et al. (1969) exposed bluegills to lindane at 0.6-9.1
µg/litre for 18 months. No adverse effect was observed at any of the
tested concentrations; the NOEL was therefore considered to be
> 9.1 µg/litre. In the same study, fathead minnows (Pimephales
promelas) were exposed to lindane at 1.4-23.5 µg/litre for 43
weeks. A statistically significant increase in mortality was
observed at the highest dose. Growth of the surviving fish was not
adversely affected, and spawning appeared to be normal in all test
groups. The NOEL in this experiment was considered to be 9.1
µg/litre.
Macek et al. (1969) exposed brook trout (Salvelinus
fontinalis) to lindane at concentrations of 1.0-16.6 µg/litre for
261 days. Only slight effects on growth were observed at the end of
the exposure period. Although no statistical assessment of spawning
was performed, fish exposed to the highest test level were adversely
affected in this respect, and the NOEL was set at 8.8 µg/litre.
The results of these studies indicate that concentrations not
far below those inducing 50% mortality are well tolerated for long
periods (up to 18 months). A 5-10-fold difference can be seen
between the maximum dose tolerated in long-term tests and the LC50
in the three species tested (Macek et al., 1976).
9.2.2.3 Reproduction
The reproductive effects of lindane were tested in bluegills,
fathead minnows, and brook trout (Macek et al., 1969). Spawning,
hatchability of eggs, and survival of the fry appeared not to be
adversely affected by concentrations of up to 9.1 µg/litre in
guppies, up to 23.4 µg/litre in fathead minnows, and up to 2.1
µg/litre in brook trout.
9.2.3 Amphibia
9.2.3.1 Acute toxicity
All of the published studies on the acute toxicity of lindane
in amphibia (Table 18) were undertaken with tadpoles. Tests on
tadpoles are considered to be the most reliable for assessing
possible adverse effects to aquatic organisms since these larvae
live exclusively in an aquatic environment, whereas adults spend a
major part of their lifetime outside the water. The results show
that larvae of amphibia are less sensitive to lindane than fish.
Table 17. Acute toxicity of gamma-HCH to fish
Species Freshwater/ Size (g, Temperature Test conditions 96-h LC50 Reference
Marine water mm, cm) (°C) (µg/litre)
Rainbow trout (Salmo gairdneri) Freshwater 0.69 g 12 Statica 32 McLeay (1976)
Freshwater 3.2 g 20 Static 38 Katz (1961)
0.6-1.7 g 13 Static 27 Macek & McAllister (1970)
0.7 g 13 Static 22 Cope (1965)
3-cm fry 12 Flow-through 22 Tooby & Durbin (1975)
Yearling 12 Flow-through 30 Tooby & Durbin (1975)
2.6 g 15 No detail 34b Dion (1984)
Brown trout (Salmo trutta) Freshwater 0.6-1.7 g 13 Static 2 Macek & McAllister (1970)
1.1 g 15 No detail 38b Dion (1984)
Coho salmon (Oncorhynchus kisutch) Freshwater 2.7-4.1 g 20 Static 50 Katz (1961)
0.6-1.7 g 13 Static 41 Macek & McAllister (1970)
Chinook salmon (Oncorhynchus Freshwater 1.5-5 g 20 Static 40 Katz (1961)
tschawytscha)
Bluegill (Lepomis macrochirus) Freshwater 0.6-1.7 g 18 Static 68 Macek & McAllister (1970)
1.0 g 18 Static 53 Cope (1965)
0.26 g 19 Static 57 Randall et al. (1979)
- 25 Static 77 Henderson et al. (1959)
0.6-1.5 g 18 Static 51 Macek et al. (1969)
Redear sunfish (Lepomis microlophus) Freshwater 0.6-1.7 g 18 Static 83 Macek & McAllister (1970)
Threespine stickleback (Gasterosteus Freshwater 0.38-0.77 g Room Static 44 and Katz (1961)
aculeatus) temperature 50c
Carp (Cyprimus carpio) Freshwater 0.6-1.7 g 18 Static 90 Macek & McAllister (1970)
6.8 cm 17-19 Static 280b Lüdemann & Neumann (1960a)
- - Static 310b Hashimoto & Nishiuchi (1981)
Table 18. Acute toxicity (48-h LC50) of lindane in tadpodes of freshwater amphibia under static conditions
Species Size (g, mm, cm, age) Temperature (°C) LC50 (mg/litre) Reference
Pseudacris triserata 7-days old 15.5 3.8 Sanders (1970)
Bufo woodhousii 4-5-weeks old 15.5 5.4 Sanders (1970)
Bufo bufo japonicus - - 24 Hashimoto & Nishiuchi (1981)
Bufo bufo L. 25-30 mm 18-21 0.3 Lüdemann & Neumann (1960b)
9.2.3.2 Effects on hatching and larval development
Marchal-Segault & Ramade (1981) exposed eggs and larvae of
Xenopus laevis to lindane at concentrations of 0.5-2 mg/litre in
tap water. Hatchability was reduced by the highest dose only;
however, development of the larvae was disturbed at all
concentrations testing, as seen by lowered body weights, longer
periods (four weeks) from hatching to metamorphosis, and
morphological abnormalities. Altered function of the
hypothalamo-hypophyseal axis, which regulates growth and
metamorphosis, and dysfunction of the intermediate lobe of the
hypophysis, which controls pigmentation, were suggested. A NOEL
could not be established.
9.3 Terrestrial organisms
9.3.1 Honey-bees
Atkins et al. (1973) estimated the LD50 in the honey-bee to
be 0.56 µg.
9.3.2 Birds
9.3.2.1 Acute toxicity
Some of the studies of the acute toxicity of lindane in birds
were undertaken soon after its introduction as an insecticide, and,
in these, the quality of lindane used is not specified.
Nevertheless, these studies are included in this review, as well as
studies in which no precise LD50 could be obtained.
The results are summarized in Table 19. The values obtained
cover a wide range, but most are in the order of 100 mg/kg body
weight. Common symptoms of poisoning are vomiting and loss of
appetite, loss of weight, and hyperexcitability; central nervous
system symptoms occur as incoordination, convulsions, and tremors
(Rosenberg et al.,1953; Dahlen & Haugen, 1954; Adamic, 1958; Turtle
et al., 1963; Dittrich, 1966).
In hens, lethal oral doses of 330-1440 mg/kg body weight caused
inflammation of the gastrointestinal tract, degeneration of the
liver and kidneys, and changes in ganglionic cells (Adamic, 1958).
Similar findings were obtained in bobwhite quails and mourning doves
(Rosenberg et al., 1953; Dahlen & Haugen, 1954) at 120-210 mg/kg
body weight. In doves, doses of > 300 mg/kg body weight caused
mainly liver atrophy, congested lungs and kidneys, and haemorrhage.
Table 19. Toxicity of lindane to birds
Species Parameter Concentration (mg/kg)a Reference
Bobwhite quail (Colinus virginianus) 5-d LC50 882 (755-1041) Hill et al. (1975)
acute LD50 120-130 (male) Dahlen & Haugen (1954)
acute LD50 190-210 (female) Dahlen & Haugen (1954)
Japanese quail (Cotumix coturnix japonica) 5-d LC50 490 (408-589) Hill & Camardese (1986)
205 and 425 (347-520) Clausing et al. (1980)
Ring-necked pheasant (Phasianus colchicus) 5-d LC50 561 (445-590) Hill et al. (1975)
Mallard (Anas platyrhynchos) acute LD50 > 2000 (male) Hudson et al. (1984)
5-d LC50 > 5000 Hill et al. (1975)
Starling (Stumus vulgaris) acute LD50 100 Schafer (1972)
Red-winged blackbird (Agelaius phoeniceus) acute LD50 75 Schafer (1972)
Common grackle (Quiscalus quiscula) acute LD50 > 100 Schafer (1972)
House sparrow (Passer domesticus) acute LD50 56 (320-100) Schafer (1972)
Common crow (Corvus brachyrhynchos) acute LD50 > 100 Schafer (1972)
Mourning dove (Streptopelia risoria) acute LD50 350-400 Dahlen & Haugen (1954)
Feral pigeon (Columba livia) acute LD50 > 600 Turtle et al. (1963)
a Acute LD50 expressed as milligrams per kilogram body weight in a single oral dose; otherwise,
concentration expressed as milligrams per kilogram food (i.e., birds were fed with a dosed diet
for 5 days followed by a "clean" diet for 3 days)
The concentrations of lindane in the diet that caused 50%
mortality in young and adult bobwhite quail, ring-necked pheasants
and mallard ducks within < 10 days and < 100 days (Dewitt et al.,
1963) are summarized in Table 20.
Table 20. Oral LD50 of lindane in birdsa
Species Lindane intake (mg/kg body weight)
< 10 days < 100 days
Young bobwhite 1070 930
Adult bobwhite - 1050
Young ring-necked pheasant 175 > 1800
Adult ring-necked pheasant - > 630
Young mallard 415 -
Adult mallard 1000 -
a From Dewitt et al. (1963)
9.3.2.2 Short-term toxicity
Chen & Liang (1956) fed white leghorn chickens and hybrid
native ducks diets containing lindane at 2, 4, or 10 mg/kg of diet
for three months. No adverse effect was observed in either species
at any dose.
When laying hens were fed diets containing lindane at 0.01,
0.1, 1, or 10 mg/kg of diet for 60 days, no effect was observed on
body weight gain, mortality, clinical symptoms, or egg production.
The authors concluded that lindane does not adversely affect
reproduction in hens at doses up to 10 mg/kg of diet (Ware & Naber,
1961).
Harrison et al. (1963) fed diets containing lindane at 4, 16,
or 64 mg/kg to white Leghorn x Australorp chickens for 27 days.
Increased mortality was seen in the highest dose group, and the two
higher doses resulted in enlarged livers. No pathological change was
found in the animals given the lowest dose, but in the two higher
dose groups dose-dependent liver hypertrophy was observed. Tissues
were not examined microscopically. The NOEL in this experiment was
concluded to be 4 mg/kg of diet.
The 30-day oral LD50 for male mallard ducks (12 animals) was
30 mg/kg body weight; as the acute LD50 was > 2000 mg/kg body
weight, the toxic action of lindane appears to be cumulative (Hudson
et al., 1984).
The LC50 values for lindane given in the diet for five days
were 882 mg/kg of diet in bobwhite quail (aged 9 days), 425 mg/kg of
diet in Japanese quail (aged 7 days), 561 mg/kg of diet in
ring-necked pheasant (aged 10 days), and > 5000 mg/kg of diet in
mallard ducks (aged 15 days) (Hill et al., 1975).
(a) Effect on egg-shell quality: Whitehead et al. (1972a,b,
1974) found that the shells of hens' eggs were not adversely
affected by administration of lindane in amounts up to 200 mg/kg of
diet; however, egg production was reduced at 100 and 200 mg/kg of
diet. Doses of up to 100 mg/kg of diet had no effect on
hatchability, egg weight, yolk weight, shell thickness, calcium
content, shearing strength or structure. The NOEL was 10 mg/kg of
diet. Similar findings were obtained in Japanese quail.
(b) Field experience: A population of Canada geese (Branta
canadensis) living in the Pacific Northwest of the USA was
observed from 1978 through 1981. Lowered reproductive success,
increased mortality among adults and a population decline in this
region were associated with the use of heptachlor for treating wheat
seed. This hypothesis was supported by the results of analyses of
eggs and tissues. In 1979, heptachlor was replaced by lindane for
use in this area; the reproductive success of the geese increased,
mortality decreased, and the population increased. There was no
evidence of either biomagnification of lindane from treated seed to
goose tissues or eggs or of induction of adverse effects by lindane
(Blus et al., 1984).
9.3.2.3 Reproduction
Lindane (99.8% in olive oil) was administered by stomach tube
to four groups of laying ducks (Anas platyrhynchus domesticus),
comprising one drake and four ducks, at doses of 0 or 20 mg/kg body
weight daily, three times per week, or twice a week for eight weeks.
Egg laying stopped immediately in the groups treated daily and three
times weekly and was irregular when it resumed, with drastically
reduced clutch sizes. The effect in the group treated twice weekly
was marginal. At the end of treatment, the laying frequencies for
the four groups were 50%, 8.3%, 11.7%, and 40%, respectively.
Hepatic, plasma, and ovarian vitellogenin levels were reduced in the
groups treated daily and three times per week; the ovaries of the
birds in these groups lacked mature vitellogenic follicles, and the
thecal layer of moderately differentiated oocytes became highly
atrophic. Levels of liver RNA were markedly reduced. A single
injection of stilboestrol at 50 mg/kg body weight restored egg
laying and the other parameters to normal within 24 h, suggesting
that lindane imposed its effects by inducing oestradiol
insufficiency (Chakravarty et al., 1986).
9.3.3 Mammals
The toxicity of lindane to bats has been studied because of its
use in timber treatment. Racey & Swift (1986) exposed pipistrelle
bats (Pipistrellus pipistrellus) to 1% lindane, both in
combination with 5% pentachlorophenol in an organic solvent and
alone. When it was applied in combination with pentachlorophenol, at
the rates recommended by the manufacturer, to wooden roosting boxes
six weeks before bats used them, the animals were killed within
seven days; when the combination was applied 14 months before use,
the animals died within 23 days. When lindane was applied alone two
weeks before exposure of bats in the boxes, all animals died within
four days. These results were significant at the 0.1% level.
Boyd et al. (1988) exposed pipistrelle bats to wood blocks
coated with lindane at 9.9 mg/m2 for 44 days and then for a further
44 days to blocks coated with lindane at 866 mg/m2, 24 h after
coating the blocks. Significant mortality ( p < 0.007) was
recorded. In a second experiment, all bats exposed to lindane at
either 147 or 211 mg/m2 died within 17 days, whereas no death
occurred among controls exposed to the solvent only.
Turner (1979) studied the distribution and concentration of
gamma-HCH in maternal and fetal tissues of a 6.5-year-old desert
bighorn (Ovis canadensis cremnobates). The maternal organs and
tissues and the tissues of the term ram fetus contained residue
levels ranging from none detected to 0.01 mg/kg on a fat basis.
Residues of 0.01 mg/kg were present in adipose tissues, muscle,
liver, gonads, and placenta. Placental transfer of gamma-HCH thus
appears to be very low.
9.4 Appraisal
The toxicity of lindane to organisms in the environment must be
assessed on the basis of the results of laboratory toxicity tests
and of the probable bioavailability of lindane to similar organisms
exposed in the wild. The strong adsorption of lindane to particles
might be expected to reduce its toxic effects below that seen in
laboratory studies of microorganisms in culture and of aquatic
organisms in water without sediment; however, there is insufficient
information to substantiate this hypothesis. No information was
available on the toxicity of lindane to organisms that feed on or
live in sediment.
Low levels of residues in birds in the wild, coupled with the
reported low toxicity of lindane in laboratory tests, make it
unlikely that it affects birds in the wild.
Bats are killed by applications of lindane to wood at normal
rates and are affected by residues of past wood treatment. Since
many bat species are declining in numbers or are extremely rare,
lindane must be regarded as a major hazard and its use avoided in
areas where bats might be found. Other mammals are unlikely to be
adversely affected by this compound.
10. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
The International Agency for Research on Cancer (1987)
evaluated the hexachlorocyclohexanes and concluded that there is
sufficient evidence for the carcinogenicity to experimental animals
of the technical grade and the alpha isomer; such evidence was
considered to be limited for the beta and gamma isomers. There is
considered to be inadequate evidence for their carcinogenicity in
humans. Hexachlorocyclohexanes were thus classified into group 2B,
possibly carcinogenic to humans.
WHO (1990) classified technical-grade lindane as 'moderately
hazardous' in normal use, on the basis of an LD50 of 88 mg/kg.
The WHO/FAO (1975) issued a data sheet on lindane (No. 12), dealing
with labelling, safe handling, transport, storage, disposal,
decontamination, training, and medical supervision of workers,
first-aid and medical treatment.
Lindane was evaluated by the FAO/WHO Joint Meeting on Pesticide
Residues in 1966, 1967, 1968, 1969, 1973, 1974, 1975, 1977, 1979,
and 1989 (FAO/WHO, 1967, 1968, 1969, 1970, 1974, 1975, 1976, 1978,
1980, 1990). A maximal acceptable daily intake of lindane in humans
was established at 0-0.008 mg/kg body weight by the 1989 Joint
Meeting (WHO, 1990). This value is based on a NOAEL of 10 mg/kg in
the diet, equivalent to 0.75 mg/kg body weight per day in rats and
1.6 mg/kg body weight per day in dogs. Maximum residue limits have
been recommended for more than 35 commodities, ranging from 0.01
mg/kg in milk to 3 mg/kg on strawberries; a limit of 0.5 mg/kg was
recommended for most fruit and vegetables (Codex Alimentarius
Commission, 1986; Table 21).
Table 21. Maximum residue limits (MRL) for gamma-HCH of the
Codex Alimentarius Commission (1986)
Crop/Commodity MRL (mg/kg)
Apples 0.5
Beans (dried) 1
Brussels sprouts 0.5
Cabbage 0.5
Cabbage, Savoy 0.5
Carrots 0.2 Eb
Cattle, carcase meat (in the carcase fat) 2
Cauliflower 0.5
Cereal grains (including rice) 0.5
Cherries 0.5
Cocoa beans 1
Cocoa butter 1
Cocoa mass 1
Cranberries 3
Currants (red) 0.5
Eggs (on a shell-free basis) 0.1 E
Endive 2
Grapes 0.5
Kohlrabi 1
Lettuce 2
Milk 0.01
Pears 0.5
Peas 0.1
Pigs, carcase meat (in the carcase fat) 2
Plums 0.5
Potatoes 0.05a
Poultry (in the carcase fat) 0.7 E
Radishes 1
Rapeseed 0.05a
Sheep, carcase meat (in the carcase fat) 2
Strawberries 3
Sugar beets (roots) 0.1
Sugar beets (tops) 0.1
Spinach 2
Tomatoes 2
a Level at or about the limit of determination
b E, Extraneous residue limit
APPENDIX I. CHEMICAL STRUCTURE
The basic structure of HCH is a closed chain of six carbon
atoms. The structure can have two spatial forms, a cis and a
trans configuration. Each carbon atom is bound to a hydrogen and a
chlorine atom, and one of these substituents forms a plane with the
two connecting carbon atoms. Since this plane is parallel to the
'equator' of the molecule, this atom is said to be in the equatorial
position. The bond with the other atom is parallel to the 'axis' of
the molecule and is thus in the axial position. Owing to the size of
the chlorine atom, the carbon atoms are not free to rotate, so the
positions of the chlorine and hydrogen atoms are fixed: one is
always in the equatorial position and the other in the axial
position.
The various combinations of the spatial orientations of the
hydrogen and chlorine atoms on each of the carbon atoms of
cyclohexane result in different isomeric compounds. Theoretically,
17 isomers of HCH are possible; but, owing to spatial
incompatibilities and thermodynamic instability, only nine isomers
have been detected. They all have the trans configuration. In the
beta isomer, all of the chlorine atoms are in the equatorial
position. The positions in the major isomers of HCH are shown in
Table 22 (Demozay & Marechal, 1972; Van Velsen, 1986).
Table 22. Positions of chlorine atoms in the major isomers of
HCHa
Isomer Chlorine positionb Physical structure
alphac AAEEEE Monoclinic prisms
ß EEEEEE Octahedral cubic
lamda AAAEEE Monoclinic crystals
delta AEEEEE Crystals/fine patelets
epsilon AEEAEE Monoclinic needles
or hexagonal
monoclinic crystals
a From van Velsen (1986)
b A, axial position; E, equatorial position
c Racemate of two optical isomers
REFERENCES
Abalis IM, Eldefrawi ME, & Eldefrawi AT (1985) High-affinity
stereospecific binding of cyclodiene insecticides and gamma-
hexachlorocyclohexane to gamma-aminobutyric acid receptors of rat
brain. Pestic Biochem Physiol 24: 95-102.
Abalis IM, Eldefrawi ME, & Eidefrawi AT (1986) Effects of
insecticides on GABA-induced chloride influx into rat brain
microsacs. J Toxicol Environ Health 18: 13-23.
Abel PD (1980) Toxicity of gamma-hexachlorocyclohexane (lindane) to
Gammarus pulex: Mortality in relation to concentration and
duration of exposure. Freshwater Biol 10: 251-259.
Adamic S (1958) [The influence of gamma isomers of
hexachlorocyclohexane on hen and duck.] Veterinarija (Sarajevo) 7:
329-334 (in Yugoslav).
Adams M, Coon FB, & Poling CE (1974) Insecticides in the tissues of
four generations of rats fed different dietary fats containing a
mixture of chlorinated hydrocarbon insecticides. J Agric Food Chem
22(1): 69-75.
Advisory Committee on Pesticides and Other Toxic Chemicals (1969)
Further review of certain persistent organochlorine pesticides used
in Groat Britain. London, Her Majesty's Stationery Office.
Agnihotri NP, Pandey SY, Jain HK, & Srivastava KP (1977) Persistence
of aldrin, dieldrin, lindane, heptachlor, and pp'-DDT in soil. J
Entomol Res 1(1): 89-91.
Ahmed FE, Hart RW, & Lewis NJ (1977) Pesticide induced DNA damage
and its repair in cultured human cells. Mutat Res 42: 161-174.
Aiyar AS (1980) Biological implications of pesticides: studies with
lindane. Manage Environ pp. 182-192.
Alabaster JS (1969) Survival of fish in 164 herbicides,
insecticides, fungicides, wetting agents, and miscellaneous
substances. Int Pest Control 11(2): 29-35.
Albahary C, Dubrisay J, & Guerin (1957) Pancytopenic rebelle au
lindane [isomère gamma de l'hexachlorocyclohexane]. Arch Mal Prof
Méd Trav Sécur Soc 18: 687-691.
Al-Omar MA, Al-Bassomy M, Al-Ogaily NV, & Al-Din Shebl D (1985)
Residue levels of organochlorine insecticides in lamb and beef from
Bagdad. Bull Environ Contam Toxicol 34: 509-512.
Al-Omar MA, AbduI-Jalil FH, Al-Ogaily NH, Tawfiq SJ, & Al-Bassomy MA
(1986) A follow-up study of maternal milk contamination with
organochlorine insecticide residues. Environ Pollut A42: 790-791.
American Conference of Governmental Industrial Hygienists Inc.
(1986) Documentation of the threshold limit values and biological
exposure indices, 5th ed. Cincinnati, Ohio, p. 348.
Anderson D & Styles JA (1978) The bacterial mutation test. Br J
Cancer 37: 924-930.
Angerer J & Barchet R (1983) [alpha, ß- and gamma-HCH in serum.] In:
Henschler D, ed. [Analytical methods for the testing of working
media prejudicial to health. Analyses in biological material, 7th
instalment.] Weinheim, Verlag Chemie, Vol. 2 (in German).
Angerer J, Maasz R, & Heinrich R (1983) Occupational exposure to
hexachlorocyclohexane. VI. Metabolism of gamma-hexachlorocyclohexane
in man. Int Arch Occup Environ Health 52: 59-67.
Anon (1984) [Organochlorine compounds including PCB: 1984 nitrition
report.] Frankfurt am Main, German Nutrition Association (in
German).
Anon (1989) FDA industry trying to cut pesticide contamination of
lanolin. Pest Toxic Chem News, 3 May, pp. 26-27.
Antonovic EA (1958) Evaluation of the toxicity of gamma-isomer of
hexachlorocyclohexane and its standardization in foodstuffs. Vopr
Pitan 17(6): 54-59.
Arbeitsgemeinschaft für die Reinhaltung der Elbe (1982) [Chlorinated
hydrocarbons; data for the River Elbe. Report on the result of the
programme for the measurement of chlorinated hydrocarbons in the
section of the Elbe between Schnackenburg and the North Sea, 1980-
1982]. Hamburg, Study Group for Keeping the Elbe Clean, pp. 64-65,
84, 86-94 (in German).
Arbeitsgemeinschaft für die Reinhaltung der Elbe (1988) [Water
quality data for the Elbe from Schnackenburg to the sea. Numerical
table, 1988.] Hamburg, Study Group for Keeping the Elbe Clean, p.
158 (in German).
Artigas F, Martinez E, Camon L, Gelpi E, & Rodriguez-Farre E (1988)
Brain metabolites of lindane and related isomers: Identification by
negative ion mass spectrometry. Toxicology 49: 57-63.
Ashwood-Smith MJ, Trevino J, & Ring R (1972) Mutagenicity of
dichlorvos. Nature 240: 418-419.
van Asperen K (1958) [Distribution and metabolism of
hexachlorocyclohexane in mammals.] In: [Proceedings of the IVth
International Congress on Crop Protection, Hamburg, 6-15 September
1957.] Braunschweig, ACO Druck GmbH, Vol. 2, pp. 1619-1623 (in
German).
Association of Official Government Chemists (1980) AOAC Methods: 29.
Pesticide residues. Multiresidue methods: general method for
organochlorine and organophosphorus pesticides (1), Washington, DC,
pp. 466-475.
Atkins EL, Greywood EA, & MacDonald RL (1973) Toxicity of pesticides
and other agricultural chemicals to honey bees. Riverside,
California, University of California, Agricultural Extension
(Unpublished report M-16).
Atlas E & Giam CS (1981) Global transport of organic pollutants:
Ambient concentrations in the remote marine atmosphere. Science 211:
163-165.
Bacci E, Calamari D, Gaggi C, Fanelli R, Focardi S, & Morosini M
(1986) Chlorinated hydrocarbons in lichen and moss samples from the
Antarctic Peninsula. Chemosphere 15(6): 747-754.
Balaschow WE (1964) [Haematopoietic disorders following the action
of the gamma-isomer of hexachlorocyclohexane.] Gig Tr Prof Zabol 8:
55-56 (in Russian).
Balba MH & Saha JG (1974) Metabolism of lindane-14C by wheat
plants grown from treated seed. Environ Lett 7(3): 181-194.
Baluja G, Murado MA, & Tejedor MC (1975) Adsorption-desorption of
lindane and aldrin by soils as affected by soil main components.
Environ Qual Saf Suppl. 3: 243-249.
Barke A (1950) [Toxicity of the gamma-isomers of
hexachlorocyclohexane.] Tierärtztl Umsch 5: 61-63 (in German).
Baron RL, Copeland F, & Walton MS (1975) Pharmacokinetics of
organochlorine pesticides in mammalian adipose tissue. Environ Qual
Saf Suppl. 3: 855-860.
Bauer U (1972) [Cncentration of insecticides, chlorinated
hydrocarbons and PCB in algae. Schr Reihe Verh Wasser- Boden-Lufthyg
37: 211-219 (in German).
Baumann K, Behling, K, Brassow HL, & Stapel K (1981) Occupational
exposure to hexachlorocyclohexane. III. Neurophysiological findings
and neuromuscular function in chronically exposed workers. Int Arch
Occup Environ Health 48: 165-172.
Baumgartner O (1953) [Sensitization to hexachlorocyclohexane.]
Schweiz Med Wochenschr 83(45): 1093-1094 (in German).
Bednarek W, Hansdorf W, Jörissen V, Schulte E, & Wegener H (1975)
[The effects of chemical pollution of the environment on birds of
prey in two test areas of Westphalia.] J Ornitol 116(2): 181-194 (in
German).
Behrbohm P & Brandt B (1959) [Allergic and toxic dermatitis in the
manufacturing and processing of hexachlorocyclohexane.] Arch
Gewerbepathol Gewerbehyg 17: 365-383 (in German).
Benes V & Sram R (1969) Mutagenic activity of some pesticides in
Drosophila melanogaster. Ind Med Surg 38(12): 442-444.
Benezet HJ & Matsumura F (1973) Isomerization of gamma-BHC to alpha-
BHC in the environment. Nature 243: 480-481.
Bercovici B, Wassermann M, Cucos S, Ron M, Wassermann D, & Pines A
(1983) Serum levels of polychlorinated biphenyls and some
organochlorine insecticides in women with recent and former missed
abortion. Environ Res 30: 169-174.
Bernhardt H & Ziemons E (1974) [Pesticide content of 19 German
reservoirs. German Association of Gas and Water Experts.] Wasser 5:
1-104 (in German).
Berry DH, Brewster MA, Watson R, & Neuberg RW (1987) Untoward
effects associated with lindane abuse. Am J Dis Child 141(2): 125-
126.
Bertram H., Kemper FH, & Zenzen C (1980) [Occurrence of HCH isomers
in man.] In: Deutsche Forschungsgemeinschaft, ed.
[Hexachlorcyclohexane as a harmful substance in foods. Papers from
two symposia of the Senate Committee on the Testing of Residues in
Foods, held on 28-29 November 1979 and 6 March 1980.] Weinheim,
Verlag Chemie, pp. 155-163 (in German).
Bidleman TF & Leonard R (1982) Aerial transport of pesticides over
the northern Indian Ocean and adjacent seas. Atmos Environ 16: 1099-
1107.
Blok SMG, Greve PA, Sangster B, Savelkoul TJF, & Wegman RCC (1984)
[Investigation of normally occurring values of a number of
organochlorine pesticides and related compounds and their
metabolites, of polychlorobiphenyls and of chlorophenols in the
blood or plasma of healthy volunteers.] Bilthoven, National
Institute of Public Health and Environmental Hygiene (Report No.
638101001) (in Dutch).
Blus LJ, Henny C J, Lenhart D J, & Kaiser TE (1984) Effects of
heptachlor- and lindane-treated seed on Canada geese. J Wildl Manage
48(4): 1097-1110.
Bluzat R & Seuge J (1979) Etude de la toxicité chronique de deux
insecticides (carbaryl et lindane) de la generation F1 de Lymnea
stagnalis L. (mollusque gasteropode pulmone). I. Croissance des
coquilles. Hydrobiologia 65(3): 245-255.
Bosch AL (1987a) Dermal absorption of 14C-lindane in male rabbits.
Madison, Wisconsin, Hazleton Laboratories America, Inc. (Unpublished
report No. 6188-103, submitted to WHO by CIEL).
Bosch AL (1987b) Dermal absorption of 14C-lindane in male rats.
Madison, Wisconsin, Hazleton Laboratories America, Inc. (Unpublished
report No. 6188-104, submitted to WHO by CIEL).
Boulekbache H (1980) Study of the toxicity of the alpha-, beta-, and
gamma isomers of hexachlorocyclohexane to fish, using the guppy as
the test species. Paris, Université de Paris VII, Laboratoire
d'Anatomie comparée (Unpublished report).
Boyd CE & Ferguson DE (1964) Susceptibility and resistance of
mosquito fish to several insecticides. J Econ Entomol 57: 430-431.
Boyd IL, Myhill DG, & Mitchell-Jones AJ (1988) Uptake of gamma-HCH
(lindane) by Pipistrelle bats and its effect on survival. Environ
Pollut 51: 95-111.
Bradbury FR (1963) The systemic action of benzene hexachloride seed
dressings. Ann Appl Biol 52: 361-370.
Bradbury FR & Whitaker WO (1956) The systemic action of
benzenehexachloride in plants: Quantitative measurements. J Sci Food
Agric 7: 248-253.
Brassow HL, Baumann K, & Lehnert G (1981) Occupational exposure to
hexachlorocyclohexane. II. Health conditions of chronically exposed
workers. Int Arch Occup Environ Health 48: 81-87.
Bringmann G & Kühn R (1978) [Threshold values for the harmful effect
of water-endangering substances on blue algae (Microcystis
aeruginosa) and green algae (Scenedesmus quadricauda) in the
cell reproduction inhibition test. Jahresber Wasser 50: 45-60 (in
German).
Brown D (1988) Lindane: 13-week dermal toxicity study (with interim
kill and recovery period) in the rat (HUK Project No. 580/2).
Harrogate, North Yorkshire, Hazleton, UH (Unpublished report
submitted to WHO by CIEL).
Buscher CA, Dougherty JH, & Skrinde RJ (1964) Chemical oxidation of
selected organic pesticides. J Water Pollut Control Fed 36: 1005.
Buselmaier W, Röhrborn G, & Propping P (1972) Mutagenicity
investigations with pesticides in the host-mediated assay and the
dominant lethal test in mice. Biol Zbl 91: 311-325 (in German).
Büsser MT & Lutz WK (1987) Stimulation of DNA synthesis in rat and
mouse liver by various tumor promoters. Carcinogenesis 8(10): 1433-
1437.
Butler PA (1963a) Commercial fisheries investigations. Washington,
DC, US Fish and Wildlife Service (Circular 199), pp. 5-28.
Butler PA (1963b) Commercial fisheries investigations. Washington,
DC, US Fish and Wildlife Service (Circular 167), pp. 11-25.
Butler PA (1971) Influence of pesticides on marine ecosystems. Proc
R Soc Lond B177: 321-329.
Cameron GR (1945) Risks to man and animals from the use of 2,2-
bis(p-chlorophenyl),1,1,1-trichlorethane (DDT): with a note on the
toxicology of gamma-benzenehexachloride (666, gammexane). Br Med
Bull 3: 783-785.
Camon L, Martinez E, Artigas F, Sola C, & Rodriguez-Farre E (1988a)
The effect of non-convulsant doses of lindane on temperature and
body weight. Toxicology 49: 389-394.
Camon L, Sola C, Martinez E, Sanfeliu C, & Rodriguez-Farre E (1988b)
Cerebral glucose uptake in lindane-treated rats. Toxicology 49: 381-
387.
Carey AE, Gowen JA, Tai H, Mitchell G, & Wiersma GB (1979) Pesticide
residue levels in soils and crops from 37 states, 1972-National
Soils Monitoring Program (IV). Pestic Monit J 12(4): 209-229.
Carrasco JM, Cunat P, Martinez M, & Primo E (1976) Pesticide
residues in total diet samples, Spain 1971-1972. Pestic Monit J
10(1): 18-23.
Cerey K, Szokolayova J, Rosival L, & Gencik A (1975) Detection of
mutagenic actions of lindane by means of the dominant lethal test.
In: VIII International Plant Protection Congress. Reports and
information. Section IV: Plant protection in relation to human
health and environmental pollution. Moscow, pp. 34-43.
Cetinkaya M, Gabel B, Podbielski A, & Thiemann W (1984) [On the
correlation of nutrition and living habits of feeding mothers and
contamination of human milk with nonvolatile chlorinated organic
chemicals.] Akt Ernähr 9(4): 157-162 (in German).
Chabert D & Vicente N (1978) Contamination de mollusques
méditerranéens par un biocide organo-chloré: le lindane. Rev Int
Océanog Méd 49: 45-48.
Chadwick RW & Copeland MF (1985) Investigation of HCB as a
metabolite from female rats treated daily for six days with lindane.
J Anal Toxicol 9: 262-266.
Chadwick RW & Freal JJ (1972a) The identification of five unreported
lindane metabolites recovered from rat urine. Bull Environ Contam
Toxicol 7(2/3): 137-146.
Chadwick RW & Freal JJ (1972b) Comparative acceleration of lindane
metabolism to chlorophenols by pretreatrnent of rats with lindane or
with DDT and lindane. Food Cosmet Toxicol 10: 789-795.
Chadwick RW, Cranmer MF, & Peoples AJ (1971) Comparative stimulation
of gamma-HCH metabolism by pretreatment of rats with gamma-HCH, DDT,
and DDT + gamma-HCH. Toxicol Appl Pharmacol 18: 685-695.
Chadwick RW, Chuang LT, & Williams K (1975) Dehydrogenation. A
previously unreported pathway of lindane metabolism in mammals.
Pestic Biochem Physiol 5: 575-586.
Chadwick RW, Freal JJ, Sovocool GW, Bryden CC, & Copeland MF (1978)
The identification of three previously unreported lindane
metabolites recovered from mammals. Chemosphere 8: 633-640.
Chadwick RW, Copeland MF, Mole ML, Nesnow S, & Cooke N (1981)
Comparative effect of pretreatment with phenobarbital, Arochlor
1254, and ß-naphthaflavone on the metabolism of lindane. Pestic
Biochem Physiol 15: 120-136.
Chadwick RW, Copeland MF, Wolff GL, Stead AG, Mole ML, & Whitehouse
DA (1987) Saturation of lindane metabolism in chronically treated
(YS x VY)F1 hybrid mice. J Toxicol Environ Health 20: 411-434,
Chadwick RW, Cooper RL, Chang J, Rehnberg GL, & McElroy WK (1988)
Possible antiestrogenic activity of lindane in female rats. J
Biochem Toxicol 3: 147-158.
Chakravarty S, Mandal A, & Lahiri P (1986) Effect of lindane on
clutch size and level of egg protein in domestic duck. Toxicology
39: 93-103.
Chametski WA & Lichtenstein EP (1973) Penetration and translocation
of 14C-lindane in pea plants. J Econ Entomol 66(2): 344-349.
Chattopadhyay P, Karnik AB, Thakore KN, Lakkad BC, Nigam SK &
Kashyap SK (1988) Health effects among workers involved in the
manufacture of hexachlorocyclohexane. J Soc Occup Med 38: 77--81.
Chen C-P (1968) The effect of a protein-deficient diet on the acute
oral toxicity of lindane. Kingston, Ontario, Queen's University
(Thesis).
Chen T & Liang C (1956) Oral toxicity of lindane and its tolerance
in poultry and mice. J Agric Assoc China 15: 78-90.
Chessells MJ, Hawker DW, Connell DW, & Papajcsik IA (1988) Factors
influencing the distribution of lindane and isomers in soil of an
agricultural environment. Chemosphere 17(9): 1741-1749.
Clausing P, Grün G, & Beitz H (1980) [Possibilities for
investigating and avoiding damage to bird life by pesticides.]
Nachtrichtenbl Pflanzenschutzdienst DDR 34(7): (in German).
Cliath MM & Spencer WF (1971) Division S-6-soil and water management
and conservation. Soil Sci Soc Am Proc 35: 791-795.
Cliath MM & Spencer WF (1972) Dissipation of pesticides from soil by
volatilization of degradation products. I. Lindane and DDT. Environ
Sci Technol 6(10): 910-914.
Codex Alimentarius Commission (1986) Guide to Codex recommendations
concerning pesticide residues, Part 2. Maximum limits for pesticide
residues, 3rd prelim. issue, Rome, Food and Agricultural
Organization of the United Nations (CAC/Vol. 8-Ed 2-1986).
Cope OB (1965) Effects of pesticides on fish and wildlife (1964
research finding). Washington, DC, US Fish and Wildlife Service
(Circular 226), pp. 51-63.
Copeland MF & Chadwick RW (1979) Bioisomerization of lindane in
rats. J Environ Pathol Toxicol 2: 737-749.
Coper H, Herken H, & Klempau I (1951) [Antagonism and synergism of
ß- and gamma-hexachlorocyclohexane.] Kiln Wochenschr 29(13/14): 264-
265 (in German).
Corneliussen PE (1969) Residues in food and feed. Pesticide residues
in total diet samples (IV). Pestic Monit J 2(4): 140-152.
Cosson Mannevy MA & Marchand MH (1980) Bioaccumulation des éléments
chimiques chez les organismes marins: limites des études in vitro.
Exposé fait au symposium de Valbonne, 1-2 October 1980. (Unpublished
report Celamerck C80 P007C submitted to WHO by CIEL).
Cowan AA (1981) Organochlorine compounds in mussels from Scottish
coastal waters. Environ Pollut B2: 129-143.
Culley DD Jr & Ferguson DE (1969) Patterns of insecticide resistance
in the mosquito fish, (Gambusia affinis). J Fish Res Board Can 26:
2395-2401.
Cummings JG, Zee KT, Turner V, & Quinn F (1966) Residues in eggs
from low level feeding of five chlorinated hydrocarbon insecticides
to hens. J Assoc Off Anal Chem 49(2): 354-364.
Czeglédi-Janko G & Avar P (1970) Occupational exposure to lindane;
clinical and laboratory findings. Br J Ind Med 27: 283-286.
Dahlen JH & Haugen AO (1954) Acute toxicity of certain insecticides
to the bobwhite quail and mourning dove. J Wildl Manage 18: 477-481.
Darskus R (1982) Water solubility and partition coefficient
n-octano/water for CM active ingredients. (Unpublished report
Celamerck 111AC-114-006 submitted to WHO by CIEL).
Das B & Singh PK (1977) Detoxication of the pesticide
benzenehexachloride by blue-green algae. Microbios Lett 4: 99-102.
Davies D & Mes J (1987) Comparison of the residue levels of some
organochlorine compounds in breast milk of the general and
indigenous Canadian populations. Bull Environ Contam Toxicol 39:
743-749.
Davies JE, Dedhia HV, Morgade C, Barquet A, & Maibach HI (1983)
Lindane poisoning. Arch Dermatol 119(2): 142-144.
Davis HC & Hidu H (1969) Effects of pesticides on embryonic
development of clams and oysters and on survival and growth of
larvae. Fish Bull 67; 393-404.
De Brabander M, van de Veire R, Aerts F, Geuens S, & Hoebeke J
(1976) New culture model facilitating rapid quantitative testing of
mitotic spindle inhibition in mammalian cells. J Natl Cancer Inst
56(2): 357-363.
De Bruijn J (1979) Reduction of chlordane, DDT, heptachlor,
hexachlorobenzene and hexachlorocyclohexane isomers contained in
effluents. Brussels, Commission of the European Communities (Report
ENV/223/74-E Rev.2).
Deichmann WB & MacDonald WE (1977) Organochlorine pesticides and
liver cancer deaths in the United States, 1930-1972. Ecotoxicol
Environ Saf 1: 89-110.
Demozay D & Marechal G (1972) Physical and chemical properties. In:
Ulmann, E., ed., Lindane: Monograph of an insecticide. Freiburg im
Breisgau, K. Schillinger Verlag, pp. 14-21.
Deo PG, Hasan SB, & Majumder SK (1981) Interconversions and toxicity
changes in hexachlorocyclohexane isomers on dispersion in water. J
Environ Sci Health B16(6): 691-701.
Desi I (1972) In: Proceedings of the International Symposium on
Lindane, Vienna, 9 June. Budapest, National Institute of Public
Health. (Unpublished report submitted to WHO by CIEL).
Desi I (1974) Neurotoxicological effect of small quantities of
lindane. Int Arch Arbeitsmed 33: 152-162.
Desi I (1976) Lindane: Toxicological studies. In: Proceedings of the
Symposium on Lindane, Lyon-Chazay, 29 April 1976. Brussels, Centre
International d'Etudes du Lindane, pp. 67-69.
Desi I (1983) Neurotoxicological investigation of pesticides in
animal experiments, Neurobehav Toxicol Teratol 5: 503-515.
Desi I, Dura G, & Szolobodnyik J (1977) Testing of pesticide
toxicity in tissue culture. J Toxicol Environ Health 2: 1053-1066.
Desi I, Varga L, & Farkas I (1978) Studies on the immunosuppressive
effect of organochlorine and organophosphoric pesticides in subacute
experiments. J Hyg Epidemiol Microbiol Immunol 22(1): 115-122.
Deutsche Forschungsgemeinschaft (1978) [Residues in breast milk-
situation and assessment]. Boppard, Harald Boldt Verlag (in German).
Deutsche Forschungsgemeinschaft (1979) [Residue analysis of
pesticides (5th instalment): Organochlorine pesticides]. Weinheim,
Verlag Chemie, pp. 12-18 (in German).
Deutsche Forschungsgemeinschaft (1983) [Hexachlorcyclohexane as a
harmful substance in foods. Papers from two symposia of the Senate
Committee on the Testing of Residues in Foods, held on 28-29
November 1979 and 6 March 1980]. Weinheim, Verlag Chemie (in
German).
Dewitt JB, Stickel WH, & Springer PF (1963) Wildlife studies,
Patuxent Wildlife Research Center, 1961-1962. US Fish and Wildlife
Service (Circular 167), pp. 74-96.
van Dijck P & van de Voorde H (1976) Mutagenicity versus
carcinogenicity of organochlorine insecticides. Meded Fac
Landbouwwet Rijksuniv Gent 41(2): 1491-1498.
Dion BM (1984) Toxicité aigue de l'isomère gamma de
l'hexachlorocyclohexane à l'égard des truites arc-en-ciel et fario.
(Unpublished report Celamerck 111AC-442-006 submitted to WHO by
CIEL).
Dittrich V (1966) Investigation on the acute oral toxicity of
different pesticides on nestlings of Parus ater and adults of
Passer domesticus. Z Angew Entomol 57: 430-437.
Doisy EA & Bocklage BC (1949) Chronic toxicity of gamma isomer of
hexachlorocyclohexane in the albino rat. Proc Soc Exp Biol Med 71:
490-493.
Doisy EA & Bocklage BC (1950) Inositol and the toxicity of four
isomers of benzenehexachloride for the rat. Proc Soc Exp Biol Med
74: 613-616.
Drummond L, Gillanders EM, & Wilson HK (1988) Plasma gamma-
hexachlorocyclohexane concentrations in forestry workers exposed to
lindane. Br J Ind Med 45: 493-497.
Duee PH, Bories G, Froc J, Hascoet M, Henry Y, Peleran JC, & Consell
G (1975) Influence d'une teneur élevée en pesticide (lindane) dans
l'aliment sur le taux d'ovulation et la mortalité embryonnaire chez
la truie. J Rech Porcine Fr: 331-335.
Dugast P (1980) Cheminement du lindane dans une châine trophique
terrestre de laboratoire. Meded Fac Landbouwwet Rijksuniv Gent
45(4): 905-914.
Duggan RE & Corneliussen PE (1972) Dietary intake of pesticide
chemicals in the United States (Ill), June 1968-April 1970. Pestic
Monit J 5(4): 331-341.
Dutch Chemical Industry Association (1980) 1alpha, 2alpha, 3ß,
4alpha, 5alpha, 6ß-hexachlorocyclohexane. In: Handling chemicals
safely, 2nd ed. Amsterdam, Dutch Association of Safety Experts and
Dutch Safety Institute, p. 529.
Dzwonkowska A & Hubner H (1986) Induction of chromosomal aberrations
in the Syrian hamster by insecticides tested in vivo.
Arch Toxicol 58: 152-156.
Earl FL, Miller E & van Loon EJ (1973) Reproductive, teratogenic and
neonatal effects of some pesticides and related compounds in beagle
dogs and miniature swine. In: Deichmann WB, ed., Pesticides and
environment: a continuing controversy. New York, Intercontinental
Medical Book Corporation, pp. 253-266.
Eckenhausen FW, Bennett D, Beynon KI, & Elgar KE (1981)
Organochlorine pesticide concentrations in perinatal samples from
mothers and babies. Arch Environ Health 36(2): 81-92.
Edelman T (1984) [Background values of a number of inorganic and
organic substances in the soil of the Netherlands; an initial
reconnaissance]. The Hague, State Publishing House (Soil protection,
report VROM 34) (in Dutch).
Eder G, Sturm R, & Ernst W (1987) Chlorinated hydrocarbons in
sediments of the Elbe River and the estuary. Chemosphere 16(10-12):
2487-2496.
Edson EF, Sanderson DM, & Noakes DN (1966) Acute toxicty data for
pesticides. World Rev Pestic Control 5(3): 143-151.
Edwards CA (1966) Insecticide residues in soils. Residue Rev 13: 83-
132.
Edwards CA (1973a) Persistent pesticides in the environment.
Cleveland, Ohio, CRC Press.
Edwards CA, ed. (1973b) Environmental pollution by pesticides. New
York, Plenum Press.
Edwards CA (1977) Environmental aspects of the usage of pesticides
in developing countries. Med. Fac. Landbouwwet Rijksuniv Gent 42/2:
853-868.
Edwards CA (1981) Lindane exposure analysis (August 1980). in:
Response of the Centre International d'Etudes du Lindane to EPA's
preliminary Notice of Determination and Position Document 2/3 on
lindane. Brussels, Centre International d'Etudes du Lindane, Vol. 1.
Egan H & Hubbard AW (1975) Analytical surveys of food. Br Med Bull
31(3): 201-208.
Ehlers W, Farmer WJ, Spencer WF, & Letey J (1969a) Lindane diffusion
in soils. II. Water content, bulk density and temperature effects.
Soil Sci Soc Am Proc 33: 505-508.
Ehlers W, Letey, J, Spencer WF, & Farmer WJ (1969b) Lindane
diffusion in soils. I. Theoretical consideration and mechanism of
movement. Soil Sci Soc Am Proc 33: 501-504.
Eichler D (1975) Investigations of residue samples for possible
metabolites of lindane. (Unpublished report Celamerck 111AA-641-003
submitted to WHO by CIEL).
Eichler D (1977) Experiments on the decomposition of lindane when
exposed to UV-light. (Unpublished report Celamerck 111AC-143-002
submitted to WHO by CIEL).
Eichler D (1980) [Biotic and abiotic breakdown and conversion
behaviour, including isomerization (plant).]. In: Deutsche
Forschungsgemeinschaft, ed. [Hexachlorcyclohexane as a harmful
substance in foods. Papers from two symposia of the Senate Committee
on the Testing of Residues in Foods, held on 28-29 November 1979 and
6 March 1980]. Weinheim, Verlag Chemie, pp. 65-72 (in German).
Eichler D, Heupt W, & Fischer O (1976) [Attempts at lindane
breakdown in biomes]. (Unpublished report Celamerck no. 111AX-912002
submitted to WHO by CIEL) (in German).
Eichler D, Heupt W, & Paul W (1983) Comparative study on the
distribution of alpha- and gamma-hexachlorocyclohexane in the rat
with particular reference to the problem of isomerization.
Xenobiotica 13(11): 639-647.
Eisler R (1969) Acute toxicities of insecticides to marine decapod
crustaceans. Crustaceana Int J Crustacean Res 16(3): 302-310.
Eisler R (1970a) Latent effects of insecticide intoxication to
marine molluscs. Hydrobiologia 36(3-4): 345-352.
Eisler R (1970b) Acute toxicities of organochlorine and
organophosphorus insecticides to estuarine fishes. Washington, DC,
US Department of Interior, Bureau of Sport Fisheries and Wildlife
(Tech. paper 45), pp. 3-12.
Eisler R (1970c) Factors affecting pesticide-induced toxicity in an
estuarine fish. Washington DC, US Department of Interior, Bureau of
Sport Fisheries and Wildlife (Technical paper 45), pp. :2-20.
Eldefrawi ME, Sherby SM, Abalis IM, & Eldefrawi AT (1985)
Interaction of pyrethroid and cyclodiene insecticides with nicotinic
acetyicholine and GABA receptors. Neurotoxicology 6: 47-62.
El-Dib MA & Badawy MI (1985) Organochlorine insecticides and PCBs in
water, sediment and fish from the Mediterranean Sea. Bull Environ
Contam Toxicol 34: 216-227.
Eisner E, Bieniek D, Klein W, & Korte F (1972) [Contributions to
ecological chemistry. LII. Distribution and conversion of aldrin-
14C, heptachlor-14C and lindane-14C in the green alga
Chlorella pyrenoidosa.] Chemosphere 6: 247-250 (in German).
Engst R, Blazovich M, & Knoll R (1967) [Occurrence of lindane in
carrots and its influence on the carotene content]. Nahrung 11(5):
389-399 (in German).
Engst R, Macholz RM, & Kujawa M (1970) Recent state of lindane
metabolism. Residue Rev 68: 59-84.
Engst R, Macholz RM, & Kujawa M (1974) [Lindane metabolism.
Degradation of lindane by mould cultures. Unconjugated metabolites].
Nahrung 18(8): 737-745 (in German).
Engst R, Macholz RM, Kujawa M, Lewerenz HJ, & Plass R (1976) The
metabolism of lindane and its metabolites gamma-2,3,4,5,6-
pentachlorocyclohexene, pentachlorobenzene and pentachlorophenol in
rats and the pathways of lindane-metabolism. J Environ Sci Health
Bl1(2): 95-117.
Engst R, Macholz RM, & Kujawa M (1977) The metabolism of lindane in
a culture of mould and the degradation scheme of lindane. Chemospere
7: 401-418.
Engst R, Macholz RM, & Kujawa M (1978a) [Confirmations of the
degradation scheme of gamma-hexachlorocyclohexane]. Nahrung 22(6):
K29-K32 (in German).
Engst R, Macholz RM, & Kujawa M (1978b) [Metabolites of
hexachlorocyclohexane (HCH) isomers in human blood]. Pharmazie
33(2/3): 109-111 (in German).
Engst R, Macholz RM, & Kujawa M (1979a) Recent state of lindane
metabolism, Part II. Residue Rev 75: 71-95.
Engst R, Fritsche W, Knoll R, Kujawa M, Macholz RM, & Straube G
(1979b) Interim results of studies of microbial isomerization of
gamma-hexachlorocyclohexane. Bull Environ Contam Toxicol 22: 699-
707.
Epstein SS, Arnold E, Andrea J, Bass W, & Bishop Y (1972) Detection
of chemical mutagens by the dominant lethal assay in the mouse.
Toxicol Appl Pharmacol 23: 288-325.
Ernst W (1975) [Uptake, excretion and conversion of lindane-14C by
Mytilus edulis (common mussel)]. Chemosphare 6: 375-380 (in
German).
Ernst W (1979) Factors affecting the evaluation of chemicals in
laboratory experiments using marine organisms. Ecotoxicol Environ
Saf 3: 90-98.
Faladysz J & Szeler P (1982) Chlorinated hydrocarbons in diving
ducks wintering in Gdansk Bay, Baltic Sea. Sci Total Environ 24:
119-127.
FAO (1973) Organochlorine insecticides 1973 (BHC, BHC + DDT, gamma-
BHC, campheclor, chlordane, DDT, endosuifan, endrin, heod,
heptachlor, HHDN). Rome, Food and Agricultural Organization of the
United Nations, pp. 29-37 (FAO Specifications for Pesticides).
FAO/WHO (1967) Evaluation of some pesticide residues in food.
Geneva, World Health Organization (FAO: PL/CP/15; WHO Food
Add./67.32).
FAO/WHO (1968) 1967 Evaluation of some pesticide residues in food.
Geneva, World Health Organization (FAO: PL: 1967/M/11/1; WHO Food
Add./68.30).
FAO/WHO (1969) 1968 Evaluation of some pesticide residues in food.
Geneva, World Health Organization (FAO: PL:1968/M/9/1; WHO Food
Add./69.35).
FAO/WHO (1970) 1969 Evaluations of some pesticide residues in food.
Geneva, World Health Organization (FAO: PL:1969/M/17/1; WHO Food
Add./70.38).
FAO/WHO (1974) 1973 Evaluations of some pesticide residues in food.
Geneva, World Health Organization (AGP: 1973/M/9/1; WHO Pesticide
Residues Series No. 3).
FAO/WHO (1975) 1974 Evaluations of some pesticide residues in food.
Geneva, World Health Organization (AGP: 1974/M/11; WHO Pesticide
Residues Series No. 4).
FAO/WHO (1976) 1975 Evaluations of some pesticide residues in food.
Geneva, World Health Organization (AGP: 1975/M/13; WHO Pesticide
Residues series No. 5).
FAO/WHO (1978) 1977 Evaluations of some pesticide residues in food.
Rome, Food and Agriculture Organization of the United Nations (FAO
Plant Production and Protection Paper 10 Sup).
FAO/WHO (1980) 1979 Evaluations of some pesticide residues in food.
Rome, Food and Agriculture Organization of the United Nations (FAO
Plant Production and Protection Paper 20 Sup).
FAO/WHO (1990) Pesticide residues in food. Report of the 1989 Joint
Meeting of the FAO Panel of Experts on Pesticide Residues in Food
and the Environment and the WHO Expert Group on Pesticide Residues.
Rome, Food and Agriculture Organization of the United Nations (FAO
Plant Production and Protection Paper 99).
Feldmann RJ & Maibach HI (1974) Percutaneous penetration of some
pesticides and herbicides in man. Toxicol Appl Pharmacol 28: 126-
132.
Fieggen W (1983) [Report on investigation of purification mud on
pesticides and PCBs in 1981]. The Hague, Union of Polder Boards (in
Dutch).
Fishmen BE & Gianutsos G (1987) Opposite effects of different
hexachlorocyclohexane (lindane) isomers on cerebellar cyclic GMP:
relation of cyclic GMP accumulation to seizure activity. Life Sci
41: 1703-1709.
Fishman BE & Gianutsos G (1988) CNS biochemical and pharmacological
effects of the isomers of hexachlorocyclohexane (lindane) in the
mouse. Toxicol Appl Pharmacol 93: 146-153.
Fitzhugh OG, Nelson AA, & Frawley JP (1950) The chronic toxicities
of technical benzenehexachloride and its alpha, beta- and gamma
isomers. J Pharm Exp Ther 100(1): 59-66.
Fitzloff JF & Pan JC (1984) Epoxidation of the lindane metabolite,
gamma-PCCH, by human and rat liver microsomes. Xenobiotica 14(7):
599-604.
Fitzloff J., Portig J, & Stein K (1982) Lindane metabolism by human
and rat liver microsomes. Xenobiotica 12(3): 197-202.
Fooken C & Butte W (1987) Organochlorine pesticides and
polychlorinated biphenyls in human milk during lactation.
Chemosphere 16(6): 1301-1309.
Foulquier L, Reynier B, Granby A, & Bovard P (1971) Toxicité du
lindane sur l'anguille, France. CR Acad Agric 57(12): 1060-1068.
Francis AJ, Spanggord RJ, & Ouchi GI (1975) Degradation of lindane
by Escherichia coli. Appl Microbiol 29(4): 567-568.
Frank R, Braun HE, Sirons GH, Rasper J, & Ward GG (1985)
Organochlorine and organophosphorus insecticides and industrial
pollutants in the milk supplies of Ontario, 1983. J Food Prod 48(6):
499-504.
Franklin A (1987) The concentration of metals, organochlorine
pesticide and PCB residues in marine fish and shell fish: results
from MAFF fish and shell fish monitoring programmes, 1977-1984.
Lowestoft, Ministry of Agriculture, Fisheries and Food, Directorate
of Fisheries Research (Aquatic Environment Monitoring Report No.
16).
Freal JJ & Chadwick RW (1973) Metabolism of hexachlorocyclohexane to
chlorophenols and effect of isomer pretreatment on lindane
metabolism in rat. J Agric Food Chem 21(3): 424-427.
Fricke G (1972) [Contamination of soils with chlorinated hydrocarbon
insecticides, comparison of 1969 with 1972 (Communication 1, field
vegetable growing). Gesunde Pfianz 24(11): 177-179 (in German).
Frisque GE, Galoux M, & Bernes A (1983) Accumulation de deux
micropollutants (les polychlorobiphenyles et le gamma-HCH) par des
bryophytes aquatiques de la Meuse. Meded Fac Landbouwwet Rijksuniv
Gent 48(4): 971-983.
Frohberg H & Bauer A (1972a) Lindane. Testing for teratogenic
effects in mice following subcutaneous injection. Darmstadt, E.
Merck (Unpublished report 4/24/72 submitted to WHO by CIEL).
Frohberg H & Bauer A (1972b) Lindane. Testing for teratogenic
effects in mice following oral administration. Darmstadt, E. Merck
(Unpublished report 4/107/72 submitted to WHO by CIEL).
Frohberg H & Bauer A (1972c) [Lindane: Testing for mutagenic effect.
Dominant lethality test on male mice]. Darmstadt, E. Merck
(Unpublished report 4/8/72 submitted to WHO by CIEL).
Frohberg H, Wolf HP, & von Eberstein M (1972a) [Testing for acute
toxicity in rats after oral administration, intraperitoneal
injection and intramuscular injection]. Darmstadt, E. Merck
(Unpublished report submitted to WHO by CIEL) (in German).
Frohberg H, von Eberstein M, Engemann J, & Weisse G (1972b) [Testing
for acute toxicity in rats after oral administration and
intraperitoneal injection]. Darmstadt, E. Merck (Unpublished report
submitted to WHO by CIEL) (in German).
Fuhremann TW & Lichtenstein EP (1980) A comparative study of the
persistence, movement, and metabolism of six carbon-14 insecticides
in soils and plants. J Agric Food Chem 28: 446-452.
Gaggi C, Bacci E, Calamari D, & Fanelli R (1986) Chlorinated
hydrocarbons in plant foliage: An indication of the tropospheric
contamination level. Chemosphere 14(11-12): 1673-1686.
Gaines TB (1960) The acute toxicity of pesticides to rats. Toxicol
Appl Pharmacol 2: 88-99.
Gardais M & Scherrer M (1979) Photodecomposition of lindane (studies
realized in Rhône-Poulenc (CRD)). Lyon, Rhône-Poulenc (Unpublished
report submitted to WHO by CIEL).
Gartrell MJ, Craun JC, Podrebarac DS, & Gunderson EL (1985a)
Pesticides, selected elements and other chemicals in adult total
diet samples, October 1978-September 1979. J Assoc Off Anal Chem
68(5): 862-875.
Gartrell MJ, Craun JC, Podrebarac DS, & Gunderson EL (1985b)
Pesticides, selected elements and other chemicals in infant and
toddler total diet samples, October 1978-September 1979. J.Assoc Off
Anal Chem 68(5): 842-861.
Garretson AL & San Clemente CL (1968) Inhibition of nitrifying
chemolithotrophic bacteria by several insecticides. J Econ Entomol
61(1): 285-288.
Gaur AC & Misra KC (1977) Effect of simazine, lindane and ceresan on
soil respiration and nitrification rates. Plant Soil 46: 5-15.
Gencik A (1977) [Cytogenic examination of the bone marrow of rats
after administration of lindane]. Bratislava Lek Listy 67(5): 579-
582 (in Slovak).
Geyer H, Sheehan P, Kotzias D, Freitag D, & Korte F (1982)
Prediction of ecotoxicological behaviour of chemicals. Relationship
between physico-chemical properties and bioaccumulation of organic
chemicals in the mussel Mytilus edulis. Chemosphere 11(11): 1121-
1134.
Geyer H, Politzki G, & Freitag D (1984) Prediction of
ecotoxicological behaviour of chemicals. Relationship between n-
octanol/water partition coefficient and bioaccumulation of organic
chemicals by alga Chlorella. Chemosphere 13(2): 269-284.
Geyer H, Scheunert I, & Korte F (1986) Bioconcentration potential of
organic environmental chemicals in humans. Regul Toxicol Pharmacol
6: 313-347.
Ginsburg CM & Lowry W (1983) Absorption of gamma benzene
hexachloride following application of Kwell shampoo. Pediatr
Dermatol 1(1): 74-76.
Ginsburg CM, Lowry W, & Reisch J (1977) Absorption of lindane (gamma
benzene hexachloride) in infants and children. J Pediatr 91(6): 998-
1000.
Glatt HR & Oesch F (1984) Mammalian cell (V79) mutagenicity test on
lindane. Brussels, CIEL (Unpublished report Celamerck No. 111AC-457-
019, submitted to WHO by CIEL).
Gopalswamy UV & Aiyar AS (1984) Biotransformation of lindane in the
rat. Bull Environ Contam Toxicol 32: 148-156.
Gorchev HG & Jelinek CF (1985) A review of the dietary intakes of
chemical contaminants. World Health Organ Bull 63(5): 945-962.
Goto M, Hattod M, & Miyagawa T (1972) [Contribution to ecological
chemistry. II. Hepatoma formation in mice after administration of
high doses of HCH isomers]. Chemosphere 6: 279-282 (in German).
Graeve K & Herrnring G (1949) [The application of the gamma isomers
of hexachlorocyclohexane as an anthelmintic]. Klin Wochenschr 27:
318 (in German).
Graeve K & Herrnring G (1951) [The toxicity of gamma-
hexachlorocyclohexane]. Arch int pharmacodyn 85(1/2): 64-72 (in
German).
Green DWJ, Kendall A, Williams A, & Pascoe D (1986) Studies on the
acute toxicity of pollutants to freshwater macroinvertebrates. 4.
Lindane (gamma-hexachlorocyclohexane). Arch Hydrobiol 106(2): 263-
273.
Greve PA (1972) Potentially hazardous substances in surface waters.
Part I. Pesticides in the River Rhine. Sci Total Environ 1: 173-180.
Greve PA & van Harten DC (1983) [Relationship between levels of
organochlorine pesticides and PCBs in fat and blood in human
subjects]. Bilthoven, National Institute of Public Health and
Environmental Hygiene (Unpublished report No. 638219001) (in Dutch).
Greve PA & van Hulst S (1977) [Organochlorine pesticides and PCBs in
total diets]. Bilthoven, National Institute of Public Health and
Environmental Hygiene (Unpublished report No. 192/77 Tox-ROB) (in
Dutch).
Greve PA & Wegman RCC (1985) Organochlorine compounds in human milk;
data from a recent investigation in the Netherlands. Working paper
for the WHO consultation on organochlorine compounds in human milk
and related hazards, Bilthoven, 9-11 January 1985. Bilthoven,
Institute of Public Health and Environmental Hygiene.
Grover PL & Sims P (1965) The metabolism of gamma-2, 3,4,5,6-
pentachlorocyclo-1-ene and gamma-hexachlorocyclohexane in rats.
Biochem J 96: 521-525.
Guenard J, Eichelberger HP, & Terrier C (1984a) In vivo sister
chromatid exchange assay in CF1-mouse bone marrow cells with
lindane (oral application). Itingen, Switzerland, Research and
Consulting Company AG. (Unpublished report No. 025705, Celamerck No.
111AC-457-020, submitted to WHO by CIEL).
Guenard J, Eichelberger HP, & Terrier C (1984b) In vivo sister
chromatid exchange assay in CF1 mouse bone marrow cells with
lindane (intraperitoneal injection). Itingen, Switzerland, Research
and Consulting Company AG. (Unpublished report No. 025716, Celamerck
No. 111AC-457-021, submitted to WHO by CIEL).
Guenzi WD & Beard WE (1970) Volatilization of lindane and DDT from
soils. Soil Sci Soc Am Proc 34: 443-447.
Guicherit R & Schulting FL (1985) The occurrence of organic
chemicals in the atmosphere of the Netherlands. Sci Total Environ
43: 193-219.
Gunderson EL (1988) FDA Total Diet Study, April 1982-April 1984,
dietary intakes of pesticides, selected elements and other
chemicals. J Assoc Off Anal Chem 71(6): 1200-1209.
Gupta SK, Parikh JR, Shah M, Chatterjee SK & Kashyap SK (1982)
Changes in serum HCH residues in malaria spraymen after short-term
occupational exposure. Arch Environ Health 37(1): 41-44.
Haider K (1979) Degradation and metabolization of lindane and other
hexachlorocyclohexane isomers by anaerobic and aerobic soil
microorganisms. Z Naturtorsch 34C: 1066-1069.
Haider K (1983) [Breakdown and conversion of gamma-HCH and other HCH
isomers by soil microorganisms]. In: [Hexachlorcyclohexane as a
harmful substance in foods]. Bonn, German Research Council, pp. 73-
78 (DFG research report) (in German).
Haider K & Jagnow G (1975) [Degradation of 14C-, 3H- and 36Cl-
labelled gamma-hexachlorocyclohexan by anaerobic soil
microorganisms]. Arch Microbiol 104(2): 113-121 (in German).
Haider K, Jagnow G, Kohnen R, & Lim SU (1974) [Degradation of
chlorinated benzenes, phenols and cyclohexane derivatives by benzene
and phenol using soil bacteria under aerobic conditions]. Arch
Microbiol 96: 183-200 (in German).
Haider K, Jagnow, G, & Rohr K (1975) [Anaerobic degradation of
gamma-hexachlorocyclohexane by a mixed bacterial flora from arable
soils and from the cow rumen]. Landwirtsch Forsch 32(2): 147-152 (in
German).
Haider K, Jagnow G, & Rohr R (1976) [Anaerobic breakdown of gamma-
hexachlorocyclohexane by mixed bacterial flora from soil and cow's
rumen]. Landwirtsch Forsch 32(11): 147-152 (in German).
Hamada M, Kawano E, Kawamura S, & Shiro M (1981) Radiation- and
photo-induced degradation of five isomers of 1,2,3,4,5,6-
hexachlorocyclohexane. Agric Biol Chem 45(3): 659-665.
Hamada M, Kawano E, Kawamura S, & Shiro M (1982) A new isomer of
1,2,3,4,5-pentachlorocyclohexane from UV-irradiation products of
(alpha-, ß-, and delta-isomers of 1,2,3,4,5,6-hexachlorocyclohexane.
Agric Biol Chem 46(1): 153-157.
Hanada M, Yutani C, & Miyaji T (1973) Induction of hepatoma in mice
by benzenehexachloride. Gann 64(5): 511-513.
Hanig JP, Yoder PD, & Krop S (1976) Convulsions in weanling rabbits
after a single topical application of 1% lindane. Toxicol Appl
Pharmacol 36: 463-469.
Hansen PD (1980) Uptake and transfer of the chlorinated hydrocarbon
lindane (gamma-BHC) in a laboratory freshwater food chain. Environ
Pollut (Ser A), 21: 97-108.
Harrison DL, Poole WSH, & Mol JCM (1963) Observations on feeding
lindane-fortified mash to chickens. N Z Vet J 11(6): 137-140.
Hartley DM & Johnston JB (1983) Use of the freshwater clam
Corbicula manilensis as a monitor for organochlorine pesticides.
Bull Environ Contam Toxicol 31: 33-40.
Hashimoto Y & Nishiuchi Y (1981) [Establishment of bioassay methods
for the evaluation of acute toxicity of pesticides to aquatic
organisms]. J Pestic Sci (Jpn), 6: 257-264 (in Japanese).
Hawkins GS & Reifenrath WG (1984) Development of an in vitro model
for determining the fate of chemicals applied to skin. Fundam Appl
Toxicol 4: 133-144.
Hawkinson JE, Shull LR, & Joy RM (1989) Effects of lindane on
calcium fluxes in synaptosomes. Neurotoxicology 10: 29-40.
Haworth S, Lawlor T, Mortelmans K, Speck W, & Zeiger E (1983)
Salmonella mutagenicity test results for 250 chemicals. Environ
Mutagen Suppl 1: 3-142.
Hayes WJ Jr (1982) Benzene hexachioride. In: Hayes WJ Jr (ed.)
Pesticides studied in man. Baltimore, Williams and Wilkins, pp. 211-
228.
Hazleton Laboratories America Inc (1976a) Teratology studies in
rats-lindane (gamma benzene hexachloride, USP). Madison, Wisconsin.
(Unpublished report submitted to WHO by CIEL).
Hazleton Laboratories America Inc (1976b) Teratology studies in
rabbits-lindane (gamma benzene hexachloride, USP). Madison,
Wisconsin. (Unpublished report submitted to WHO by CIEL).
Heeschen W, Nyhuis H & Blüthgen, A (1980) [Investigations on the
significance of the transformation and degradation of benzene
hexachloride (BHC) in the lactating cow and in the surroundings for
the BHC-contamination of milk]. Milchwissenschaft 35(4): 221-224 (in
German).
Henderson C, Pickering QH, & Tarzwell CM (1959) Relative toxicity of
ten chlorinated hydrocarbon insecticides to four carbon insecticides
to species of fish. Trans Am Fish Soc 88: 23-32.
Herbst M (1976) Toxicology of lindane. In: Proceedings of the
symposium on lindane, Lyon-Chazay, 29 April 1976. Brussels, CIEL,
pp. 43-69.
Herbst M, Weisse I, & Keel LMER (1975) A contribution to the
question of the possible hepato-carcinogenic effects of lindane.
Toxicology 4: 91-96.
Heritage AD & MacRae LC (1977a) Identification of intermediates
formed during the degradation of hexachlorocyclohexanes by
Clostridium sphenoïdes. Appl Environ Microbiol 33(6): 1295-1297.
Heritage AD & MacRae LC (1977b) Degradation of lindane by cell-free
preparations of Clostridium sphenoïdes. Appl Environ Microbiol
34(2): 222-224.
Heritage AD & MacRae LC (1979) Degradation of hexachlorocyclohexanes
and structurally related substances by Clostridium sphenoïdes.
Aust J Biol Sci 32: 493-500.
Herken H (1950a) [Functional disorders of the central nervous system
caused by hexachlorocyclohexane]. Naunyn-Schmiedeberger Arch Exp
Pathel Pharmacol 212: 158-159 (in German).
Herken H (1950b) [Functional disorclers of the central nervous
system following the action of hexachlorocyclohexane]. Arztliche
Wochenschr 5(13/14): 193-195 (in German).
Herken H (1950c) [Alterations in cell function in the nervous system
caused by hexachlorocyclohexane]. Naunyn-Schmiedeberger Arch Exp
Pathel Pharmacol 211: 143-152 (in German).
Herken H (1951) [Disruption of the nervous system with
hexachlorcyclohexane]. Arzneimittelforschung 1: 356-359 (in German).
Hermens J & Leeuwangh P (1982) Joint toxicity of mixtures of 8 and
24 chemicals to the guppy (Poecilia reticulata). Ecotoxicol
Environ Saf 6: 302-310.
Herrmann R, Thomas W, & Huebrier D (1984) [Behaviour of organic
micropollutants (PAH, PCB and BHC) and of a fecal sterol in the
Husum estuary and in the adjacent North Frisian Wadden Sea]. Dtsch
Gewässerkd Mitt 4: 101-107 (in German).
Herve S, Heinonen P, Paukku R, Knuutila M, Koistinen J, & Paasivirta
J (1988) Mussel incubation method for monitoring organochlorine
pollutants in water courses. Four-year application in Finland.
Chemosphere 17(10): 1945-1961.
Heupt W (1974) [Seepage behaviour of pesticides]. (Unpublished
report Celamerck-Ingelheim No. 11110-922-001-11110-922-009,
submitted to WHO by CIEL) (in German).
Heupt W (1979) [Behaviour of the active ingredients of pesticides in
the soil]. (Unpublished report Celamerck No. 111 AA-921-008/009,
submitted to WHO by CIEL) (in German).
Heupt W (1983) Determination of the hydrolytic stability of lindane.
(Unpublished report Celamerck Ingelheim, submitted to WHO by CIEL).
Hildebrandt G, Jeep H, Hurka H, Stuke T, Heitmann M, & Boiselle C
(1986) [Breast milk as a biological indicator: study on the organic
halogen burden in breast milk and foodstuffs]. Berlin, Schmidt E.
Verlag. (Report 5/86 of the Federal Office for the Environment) (in
German).
Hill EF & Camardese MB (1986) Lethal dietary toxicities of
environmental contaminants and pesticides to Coturnix. Washington,
DC, US Department of Interior, Fish and Wildlife Service, p. 89
(Fish and Wildlife Technical Report No. 2).
Hill EF, Heath RG, Spano JW, & Williams JD (1975) Lethal dietary
toxicities of environmental pollutants to birds. Washington, DC, US
Department of Interior Fish and Wildlife Service (Special Scientific
Report. Wildlife No. 191).
Hofer K (1953) [Investigations on the efficacy of lindane
preparations in cow-sheds and on the influence of gamma-HCH on the
ripening of Emmental cheese]. University of Munich, Institute o!
Food Science, Veterinary Faculty (Thesis) (in German).
Holder JW & Stöhrer G (1989) Review of studies relating to lindane
carcinogenicity. Washington, DC, US Environmental Protection Agency,
Office of Health and Environmental Assessment, Cancer Assessment
Group (Unpublished report).
Hoshizaki H, Niki Y, Tajima H, Terada Y, & Kasahara A (1969) A case
of leukemia following exposure to insecticide. Acta haematol jpn
32(14): 672-677.
Hudson RH, Tucker RK, & Haegele MA (1984) Handbook of toxicology of
pesticides to wildlife, 2nd ed. Washington DC, US Department of
Interior, Fish and Wildlife Service, p. 49 (Resource Publication No.
153).
International Agency for Research on Cancer (1987) Overall
evaluations of carcinogenicity: an updating of IARC Monographs
Volumes 1 to 42. Lyon, pp. 220-222 (IARC monographs on the
evaluation of carcinogenic risks to humans. Suppl 7).
International Atomic Energy Agency (1988) isotope techniques for
studying the fate of persistent pesticides in the tropics. Vienna
(IAEA-TELDOC-476).
International Register for Potentially Toxic Chemicals (1989) Data
profile on lindane. Geneva, United Nations Environment Programme.
Ishidate M, Jr & Odashima S (1977) Chromosome tests with 134
compounds on Chinese hamster cells in vitro-A screening for
chemical caminogens. Mutat Res 48: 337-354.
Ito N, Nagasaki H, Arai M, Makiura S, Sugihara S, & Hirao K (1973a)
Histopathologic studies on liver tumorigenesis induced in mice by
technical polychlorinated biphenyls and its promoting effect on
liver tumours induced by benzenehexachloride. J Natl Cancer Inst
51(5): 1637-1646.
Ito N, Nagasaki H, Arai M, Sugihara S, & Makiura S (1973b)
Histologic and ultrastructural studies on the hepatocarcinogenicity
of benzenehexachloride in mice. J Natl Cancer Inst 51(3): 817-826.
Ito N, Nagasaki H, Aoe H, Sugihara S, Miyata Y, Arai M, & Shirai T
(1975) Brief communication: Development of hepatocellular carcinomas
in rats treated with benzenehexachloride. J Natl Cancer Inst 54(3):
801-805.
Itokawa H, Schallah A, Weisgerber I, Klein W, & Korte F (1970)
[Contributions to ecological chemistry. XXII. Metabolism and residue
behaviour of lindane-14C in higher plants]. Tetrahedron 26: 763-
773 (in German).
Iverson F, Ryan JJ, Lizotte R, & Hierlihy SL (1984) In vivo and
in vitro binding of alpha and gamma-hexachlorocyclohexane to mouse
liver macromolecules. Toxicol Lett 20(3): 331-335.
Izmerov NF ed. (1983) Lindane. Moscow, Centre of International
Projects, GKNT (IRPTC Scientific reviews of Soviet literature on
toxicity and hazards of chemicals No. 40).
Jackson MD, Sheets TJ, & Moffett CL (1974) Persistence and movement
of BHC in a watershed. Mount Mitchell State Park, North Carolina,
1967-1972. Pestic Monit J 8(3): 202-208.
Jacobs A, Blangetti M, & Hellmund E (1974) [Storage of chlorinated
harmful substances found in Rhine water in the fatty tissue of
rats]. Wasser 43: 259-274 (in German).
Jaeger U, Podczek A, Haubenstock A, Pirich K, Donner A, & Hruby K
(1984) Acute oral poisoning with lindane-solvent mixtures. Vet Hum
Toxicol 26(1): 11-14.
Jagnow G, Haider K, & Ellwardt PC (1977) Anaerobic dechlorination
and degradation of hexachlorocyclohexane isomers by anaerobic and
facultative anaerobic bacteria. Arch Microbiol 115: 285-292.
Jeanne N (1979) Effets du lindane sur la division, le cycle
cellulaire et les biosynthèses de deux algues unicellulaires. Can J.
Bot 57: 1464-1472.
Jeanne-Levain N (1974) Etude des effets du lindane sur la croissance
et le developpement de quelques organismes unicellulaires. Bull Soc
Zool Fr 99(1): 105-109.
Jedlicka VL, Hermanska Z, Smida I, & Kouba A (1958) Paramyeloblastic
leukaemia appearing simultaneously in two blood cousins after
simultaneous contact with gammexane (hexachlorocyclohexane). Acta
med scand 161(6): 147-151.
Jenssen D & Ramel C (1980) The micronucleus test as part of a short-
term mutagenicity test program for the prediction of carcinogenicity
evaluated by 143 agents tested. Mutat Res 75: 191-202.
Johnson RD & Manske DD (1976) Residues in food and feed. Pesticide
residues in total diet samples (IX). Pestic Monit J 9(4): 157-169.
Johnson RD, Manske DD, New DH, & Podrebarac DS (1979) Pesticides and
other chemical residues in infant and toddler total diet samples,
August 1974-July 1975. Pestic Monit J 13(3): 87-98.
Joy RM & Albertson TE (1987a) Factors responsible for increased
excitability of dentate gyrus granule cells during exposure to
lindane. Neurotoxicology 8(4): 517-528.
Joy RM & Albertson TE (1987b) Interactions of lindane with
synaptically mediated inhibition and facilitation in the dentate
gyrus. Neurotoxicology 8(4): 529-542.
Joy RM, Stark LG, & Albertson TE (1982) Proconsulvant effects of
lindane: enhancement of amygdaloid kindling in the rat. Neurobehav
Toxicol Teratol 4: 347-354.
Joy RM, Stark LG, & Albertson TE (1983) Proconvulsant action of
lindane compared at two different kindling sites in the rat-amygdala
and hippocampus. Neurobehav Toxicol Teratol 5: 461-465.
Joy, RM, Burns VW, & Stark LG (1988) Increases in free intracellular
Ca++ accompany exposure of neurohybridoma cells to the insecticide
lindane. Toxicology 8(1): 44.
Juhnke J & Lüdemann D (1978) [Results of the testing of 200 chemical
compounds for acute toxicity to fish in the golden orfe test].
Wasser-Abwasser-Forsch 11(5): 161-164 (in German).
Kahunyo JM, Froslie A, & Maitai CK (1988) Organochlorine pesticide
residues in chicken eggs: a survey. J Toxicol Environ Health 24:
543-550.
Kamada T (1971) [Part I. Accumulation of BHC (alpha, ß-, gamma-, and
delta-) isomers in rat bodies and excretion into urine following
oral administration]. Nippon Eiseigaku Zasshi 26(4): 358-364 (in
Japanese).
Kampe W (1980) [Occurrence of hexachlorocyclohexane in the soil].
In: [Hexachlorocyclohexane as a harmful substance in foodstuffs].
Weinheim, Verlag Chemie, pp. 18-23 (Research Report) (in German).
Kampe W & Andre W (1980) [Heavy metals, nitrate and chlorinated
hydrocarbons in the vegetable components of total weekly food
consumption]. Ernähr Umsch 27(11): 356-364 (in German).
Kaphalia BS & Seth TD (1983) Chlorinated pesticide residues in blood
plasma and adipose tissue of normal and exposed human population.
Indian J Med Res 77: 245-247.
Karapally JC, Saha JG, & Lee YW (1973) Metabolism of lindane-14C
in the rabbit: Ether-soluble urinary metabolites. J Agric Food Chem
21(5): 811-818.
Kathpal TS, Yadav GS, Kushawa KS, & Singh G (1988) Research
communication: persistence behavior of HCH in rise soil and its
uptake by rice plants. Ecotoxicol Environ Saf 15: 336-338.
Katz M (1961) Acute toxicity of some organic insecticides to three
species of salmonids and to the three-spine stickleback. Trans Am
Fish Soc 90(3): 264-268.
Kawahara T (1972) [Studies on organochlorine pesticide residues in
crops and soils. Report XV. Absorption of BHC by stems and leaves of
rice plants]. Noyaku Kensasho Hokohu 12: 31-34 (in Japanese, with
English summary).
Kawahara T & Nakamura T (1972) [Studies on the residual
organochlorine pesticides in crops and soils. Report XXl. Absorption
and translocation of chlorinated hydrocarbons in potted turnips].
Bull Agric Chem Insp Stn 12: 52-56 (Abstract) (in Japanese).
Kawahara T, Konuma S, Wada T, Kureha Y, & Nakamura H (1971)
[Absorption and translocation of chlorinated hydrocarbons into crops
in the high-cooling region]. Noyaku Kensasho Hokohu 11: 67-72 (in
Japanese, with English summary).
Kay BD & Elrick DE (1967) Adsorption and movement of lindane in
soils. Soil Sci 104(5): 314-322.
Kensler TW & Trush MA (1984) The role of oxygen radicals in tumour
promoters. Environ Mutagen 6: 593-616.
Kewitz H & Reinert H (1952) [Demonstration of functional changes to
the central nervous system through electrically and chemically
induced spasma]. Naunyn-Schmiedebergs Arch Exp Pathol Pharmakol 215:
93-99 (in Germen).
Khera KS, Whalen C, Trivett G, & Angers G (1979) Teratogenicity
studies on pesticidal formulations of dimethoate, diuron, and
lindane in rats. Bull Environ Contam Toxicol 22: 522-529.
Kiraly J, Szentesi I, Ruzicska M, & Czeize A (1979) Chromosome
studies in workers producing organophosphate insecticides. Arch
Environ Contam Toxicol 8: 309-319.
Kitamura S, Sumino K, & Hayakawa K (1970) [Decomposition of gamma-
BHC in vivo]. Jpn J Public Health 17(11): 108-109 (in Japanese).
Klone PR & Kintigh WJ (1988) Lindane technical-fourteen week dust
aerosol inhalation study on mice. Bushy Run Research Center
(Unpublished report Met Path No. 14014; Laboratory Project No. ID
BRRC/51-524).
Koelling K (1978) [Residues in poultry and eggs. Report of the
symposium, 28 Mai 1975]. Boppard, Harald Boldt Verlag, pp. 9-159 (in
German).
Kohli J, Weisgerber I, Klein W, & Korte F (1976a) Contributions to
ecological chemistry: CVII. Fate of lindane-14C in lettuce,
endives, and soil under outdoor conditions. J Environ Sci Health
B11(1): 23-32.
Kohli J, Weisgerber I, & Klein W (1976b) Balance of conversion of
[14C]-lindane in lettuce in hydroponic culture. Pestic Biochem
Physiol 6: 91-97.
Kohnen R, Haider K, & Jagnow G (1975) Investigations on the
microbial degradation of lindane in submerged and aerated moist
soil. Environ Qual Saf 3: 222-225.
Kolmodin-Hedman B (1974) Exposure to lindane and DDT and its effects
on drug metabolism and serum lipoproteins. Stockholm, Departments of
Clinical Pharmacology (at Huddinge Hospital) and Pharmacology,
Karotinska Institute, Toxicology (Swedish Medical Research Council)
and Division of Occupational Medicine, National Board of
Occupational Safety and Health (Thesis).
Kolmodin-Hedman B (1984) Pesticides. In: Aitio A, Riihimaki V, &
Vainio H, ed. Biological monitoring and surveillance of workers
exposed to chemicals. Washington, DC, Hemisphere Publishing Corp.,
pp. 209-218.
Kolmodin-Hedman B, Alexanderson B, & Sjöqvist F (1971) Effect of
exposure to lindane on drug metabolism: Decreased hexobarbital
sleeping-times and increased antipyrine disappearance rate in rats.
Toxicol Appl Pharmacol 20(3): 299-307.
Kopf W & Schwoerbel J (1980) [Accumulation studies of the
insecticide lindane (gamma-HCH, BHC) by aquatic insects of the genus
Sigara (Hemiptera, Corixidae)]. Arch Hydrobiol Suppl 59(1): 32-
42 (in German).
Koransky W, Portig J, & Münch G (1963) [Absorption, distribution and
excretion of alpha- and gamma-hexachlorocyclohexane]. Naunyn-
Schmiedebergs Arch Exp Pathol Pharmakol 244: 564-575 (in German).
Koransky W, Portig J, Vohland HW, & Klempau I (1964) [Elimination of
alpha- and gamma-hexachlorocyclohexane and the way it is influenced
by liver microsomal enzymes]. Naunyn-Schmiedebergs Arch Exp Pathol
Pharmakol 247: 49-60 (in German).
Korn S & Earnest R (1974) Acute toxicity of twenty insecticides to
striped bass, Morone saxatilis. California Fish Game 60(3): 128-131.
Korte F (1980) [Occurrence of HCH-hexachlorcyclohexane as a harmful
substance in foods. Papers from two symposia of the Senate Committee
on the Testing of Residues in Foods, held on 28-29 November 1979 and
6 March 1980]. Weinhelm, Verlag Chemie, pp. 46-64 (in German).
Kotzias D, Nitz S, & Korte F (1981) [Light-induced total breakdown
of organic molecules adsorbed on silica gel]. Chemosphere 10(4):
415-422 (in German).
Kramer MS, Hutchinson TA, Rudnick SA, Leventhal JM, & Feinstein AR
(1980) Operational criteria for adverse drug reactions in evaluating
suspected toxicity of a popular scabicide. Clin Pharmacol Ther 27:
149-154.
Krauthacker B, Alebic-Kolbah T, Kralj M, Tjalcevic B, & Reiner E
(1980) Organochlorine pesticides in blood serum of the general
Yugoslav population and in occupationally exposed workers. Int Arch
Occup Environ Health 45: 217-220.
Krishnakumari MK (1977) Sensitivity of the alga Scenedesrnus acutus
to some pesticides. Life Sci 20: 1525-1532.
Kujawa M, Härtig M, Macholz RM, & Engst R (1976) [The degradation of
14C-lindane by a mould culture]. Nahrung 20(2): 181-183 (in
German).
Kujawa M, Engst R, & Macholz R (1977) On the metabolism of lindane.
In: Proceedings of International Symposium on Industrial Toxicology,
Environmental Pollution and Human Health, pp. 661-672.
Kumar, A. & Dwivedi PP (1988) Relative induction of molecular forms
of cytochrome P-450 in gamma-hexachlorocyclohexane exposed rat liver
microsomes. Arch Toxicol 62: 479-481.
Kurihara N & Nakajima M (1974) Studies on BHC isomers and related
compounds. VIII. Urinary metabolites produced from gamma- and ß-BHC
in the mouse: Chlorophenol conjugates. Pestic Biochem Physiol 4:
220-231.
Kurihara N, Uchido M, Fujita T, & Nakajima M (1973) Studies on BHC
isomers and related compounds. V. Some physicochemical properties of
BHC-isomers(1). Pestic Biochem Physiol 2: 383-390.
Kurihara N, Tanaka K, & Nakajima M (1979) Mercapturic acid formation
from lindane in rats. Pestic Biochem Physiol 10: 137-150.
Kurihara N, Suzuki T, & Nakajima M (1980) Deuterium isotope effects
on the formation of mercapturic acids from lindane in rats. Pestic
Biochem Physiol 14: 41-49.
Kurt TL, Dost R, Gilliland M, Reed G, & Petty C (1986) Accidental
Kwell (lindane) ingestions. Vet Hum Toxicol 28(6): 569-571.
(Published erratum appears in Vet Hum Toxicol 29(1): 24, 1987.)
Landesamt für Wasser und Abfall (1988) [Water quality report 1987].
Dusseldorf, North Rhine-Westphalia Office for Water and Refuse (in
German).
Lange P (1965) [Studies on the convulsive process and the effect of
convulsion-inhibiting substances thereon during progressive infusion
of convulsive poisons in mice]. Acta Biol Med Ger 15: 109-119 (in
German).
Lange M, Nitzsche K, & Zesch A (1981) Percutaneous absorption of
lindane in healthy volunteers and scabies patients. Dependency of
penetration kinetics in serum upon frequency of application, time
and mode of washing. Arch Dermatol Res 271: 387-399.
Laug EP (1948) Tissue distribution of a toxicant following oral
ingestion of the gamma isomer of benzene hexachloride by rats. J
Pharmacol Exp Ther 93: 277-281.
Laugel P (1981) [The residue situation in France and ways of
overcoming it]. Lebensmitteichem Gerichtl Chem 35: 29-32 (in
German).
Lehman AJ (1952) Chemicals in foods: A report to the association of
Food and Drug officials on current developments. Part II.
Pesticides. Section III. Subacute and chronic toxicity. Q Bull Assoc
Food Drug Off US 16(2): 47-53.
Lehman AJ (1965) Summaries of pesticide toxicity. Part I.
Chlorinated organic compounds. Topeka, Kansas, Associations with
Food and Drug Officials of the United States, pp. 27-29.
Lichtenstein EP & Polivka JB (1959) Persistence of some chlorinated
hydrocarbon insecticides in turf soils. J Econ Entomol 52(2): 289-
293.
Lichtenstein EP & Schulz KR (1958a) Persistence of some chlorinated
hydrocarbon insecticides as influenced by soil types, rate of
application and temperature. J Econ Entomol 52(1): 124-131.
Lichtenstein EP & Schulz KR (1958b) Breakdown of lindane and aldrin
in soils. J Econ Entomol 52(1): 118-124.
Lichtenstein EP & Schulz KR (1970) Volatilization of insecticides
from various substrates. J Agric Food Chem 18: 814.
Lichtenstein EP, de Peu LJ, Eshbaugh EL, & Sleesman JP (1960)
Persistence of DDT, aldrin, and lindane in some Midwestern soils. J
Econ Entomol 53(1): 136-142.
Lisi P, Caraffini S, & Assalve D (1986) A test series for pesticide
dermatitis. Contact Derm 15: 266-269.
Lisi P, Caraffini S, & Assalve D (1987) Irritation and sensitization
potential of pesticides. Contact Derm 17: 212-218.
Lowden GF, Saunders CL, & Edwards RW (1969) Organochlorine
insecticides in water (Part II). J Water Treat Exam 18: 275-287.
Lüdemann D & Neumann H (1960a) [Tests on the acute and toxic effects
of modern contact insecticides on one-year-old carp (Cyprinus
carpio L.)]. Z Angew Zool 47: 11-33 (in German).
Lüdemann D & Neumann H (1960b) [Tests on the acute and toxic effects
of modern contact insecticides on freshwater animals]. Z. Angew Zool
47: 303-321 (in German).
Macek KJ & McAllister WA (1970) Insecticide susceptibility of some
common fish family representatives. Trans Am Fish Soc 99: 20-27.
Macek KJ, Hutchinson C, & Cope OB (1969) The effects of temperature
on the susceptibility of bluegills and rainbow trout to selected
pesticides. Bull Environ Contam Toxicol 4(3): 174-183.
Macek KJ, Buxton KS, Derr SK, Dean JW, & Sauter S (1976) Chronic
toxicity of lindane to selected aquatic invertebrates and fish.
Washington, DC, US Environmental Protection Agency (Ecological
Research Series, EPA-600/3-76-046).
Macholz RM, Knoll R, Lewerenz H J, Plass R, & Schulze J (1983)
[Metabolism of gamma-hexachlorocyclohexane (HCH) in germ-free and
conventional rats]. Zentralbl Pharmakol 122(2): 221 (in German).
MacKay D & Walkoff AW (1973) Rate of evaporation of low-solubility
contaminants from water bodies to atmosphere. Environ Sci Technol 7:
611.
MacRae IC, Raghu K, & Castro TF (1967) Persistence and
biodegradation of four common isomers of benzenehexachloride in
submerged soils. J Agric Food Chem 15(5): 911-914.
MacRae JC, Yamaya Y, & Yoshida T (1984) Persistence of HCH-isomers
in soil suspensions. Soil Biol Biochem 16: 285-286.
Marcelle C & Thome JP (1983) Acute toxicity and bioaccumulation of
lindane in gudgeon, Gobio gobio (L.). Bull Environ Contam Toxicol
31: 453-458.
Marcelle C & Thome JP (1984) Relative importance of dietary and
environmental sources of lindane in fish. Bull Environ Contam
Toxicol 33: 423-429.
Marchal-Segault D & Ramade F (1981) The effects of lindane, an
insecticide, on hatching and post-embryonic development of Xenopus
laevis (Daudin). Anuran amphibian. Environ Res 24: 250-258.
Martin RJ & Duggan RE (1968) Pesticide residues in total diet
samples (III). Pestic Monit J 1(4): 1-20.
Martin DB & Hartmann WA (1985) Organochlorine pesticides and
polychlorinated biphenyls in sediment and fish from wetlands in the
North-Central United States. J Assoc Off Anal Chem 69(4): 712-717.
Mathur SP & Saha JG (1975) Microbial degradation of lindane-14C in
a flooded sandy loam soil. Soil Sci 120(4): 301-307.
Mathur SP & Saha JG (1977) Degradation of lindane-14C in a mineral
soil and in an organic soil. Bull Environ Contam Toxicol 17(4): 424-
430.
Matsumura F (1985) Toxicology of insecticides, 2nd ed. New York,
Plenum Press, pp. 128-143.
Matsumura F & Benezet HJ (1973) Studies on the bioaccumulation and
microbial degradation of 2,3,7,8,-tetrachlorodibenzo-p-dioxin.
Environ Health Perspect 5: 253-258.
Matsumura F & Tanaka K (1984) Molecular basis of neuroexcitability
actions of cyclodiene-type insecticides. In: Narahashi T, ed.,
Cellular and molecular neurotoxicology. New York, Raven Press, pp.
225-240.
Matsumura F, Benezet HJ, & Patil KC (1976) Factors affecting
microbial metabolism of gamma-BHC. J Pestic Sci 1: 3-8.
McLeay DJ (1976) A rapid method for measuring the acute toxicity of
pulpmill effluents and other toxicants to salmonid fish at ambient
room temperature. J Fish Res Board Canada 33; 1303-1311,
McNamara B & Krop S (1948) The treatment of acute poisoning produced
by gamma hexachlorocyclohexane. J Pharmacol 92: 147-152.
McParland PJ, McCracken RM, O'Hare MB, & Raven AM (1973)
Benzenehexachloride poisoning in cattle. Vet Rec 93: 369-371.
Meemken HA, Habersaat K, & Groebel W (1982) [Burden of breast milk
with chlorinated hydrocarbons from creams containing anhydrous
lanolin]. Lebensmittelchem Gerichtl Chem 36: 51-53 (in German).
Mendeloff AI & Smith DE (1955) Exposure to insecticides, bone marrow
failure, gastrointestinal bleeding, and uncontrollable infections.
Am J Med 19(8): 274-284.
Mes J, Davies DJ, & Turton D (1982) Polychlorinated biphenyl and
other chlorinated hydrocarbon residues in adipose tissue of
Canadians. Bull Environ Contam Toxicol 28: 97-104.
Mes J, Davies DJ, Turton D, & Sun WF (1986) Levels and trends of
chlorinated hydrocarbon contaminants in the breast milk of Canadian
women. Food Addit Contam 3(4): 313-322.
Mestres R (1974) Report of a group of experts on the content of
organohalogen compounds detected between 1968 and 1972 in water, air
and foodstuffs and the methods of analysis used in the nine Member
States of the European Community. Presented at the European
Colloquium on problems raised by the contamination of man and his
environment by persistent pesticides and organo-halogenated
compounds, Luxembourg, 14-16 May 1974. Luxembourg, Commission of the
European Communities (Paper No. 1).
Milby TH & Samuels AJ (1971) Human exposure to lindane-comparison of
an exposed and unexposed population. J Occup Med 13(5): 256-258.
Milby TH, Samuels AJ, & Ottoboni F (1968) Human exposure to lindane;
blood lindane levels as a function of exposure. J Occup Med 10(10):
584-587.
Mills AC & Biggar JW (1969) Solubility-temperature effect on the
adsorption of gamma- and beta-BHC from aqueous and hexane solutions
by soil materials. Soil Sci Soc Am Proc 33: 210-216.
Morgan DP, Stockdale EM, Roberts R J, & Walter AW (1980) Anemia
associated with exposure to lindane. Arch Environ Health 35(5): 307-
310.
Mosha RD, Gyrd-Hansen N, & Kraul I (1986) Distribution and
elimination of lindane in goats. Bull Environ Contam Toxicol 36:
516-522.
Mottram DS, Psomas IE, & Patterson RLS (1983) Chlorinated residues
in the adipose tissue of pigs treated with gamma-
hexachlorocyclohexane. J Sci Food Agric 34: 378-387.
Mouvet C (1985) Monitoring of polychlorinated biphenyls (PCBs) and
hexachlorocyclohexanes (HCH) in fresh water using the aquatic moss
Cinclidotus danubicus. Sci Total Environ 44: 253-267.
Moza P, Mostafa I, & Klein W (1974) [ntributions to ecological
chemistry. LXXXIX. Exploratory investigations of the metabolism of
gamma-pentachlorcyclohex-1-ene in higher plants grown
hydroponically]. Chemosphere 6: 255-258 (in German).
Muacevic G (1966) [Oral toxicity in male rats]. Ingelheim am Rhein,
Boehringer & Son (Unpublished report submitted to WHO by CIEL) (in
German).
Muacevic G (1970) [LD50 determination in female rats]. Ingelheim
am Rhein, Boehringer & Son (Unpublished report submitted to WHO by
CIEL) (in German).
Muacevic G (1971a) [LD50 determination in male rats]. Ingelheim am
Rhein, Boehringer & Son (Unpublished report submitted to WHO by
CIEL) (in German).
Muacevic G (1971b) [LD50 determination in the rat]. Ingelheim am
Rhein, Boehringer & Son (Unpublished report submitted to WHO by
CIEL) (in German).
Müller D, Klepel H, Macholz RM, Lewerenz HJ, & Engst R (1981)
Electroneurophysiological studies on neurotoxic effects of
hexachlorocyclohexane isomers and gamma-pentachlorocyclohexene. Bull
Environ Contam Toxicol 27: 704-706.
Muralidhara MK, Krishnakumari MK, & Majumder SK (1979) Effects of
carriers on the oral toxicity of lindane (gamma-BHC) to albino rats.
J Food Sci Technol 16: 105-107.
Nair GA (1981) Toxic effects of certain biocides on a freshwater
mite, Hydrachna trilobata Viets (Arachnida; Hydrachnoidea;
Hydrachnidae). J Environ Biol 2(2): 91-96.
Nash RG (1983) Determining environmental fate of pesticides with
microagroecosystems. Residue Rev 85: 199-215.
Nash RG, Harris WG, Ensor PD, & Woolson EA (1973) Comparative
extraction of chlorinated hydrocarbon insecticides from soils 20
years after treatment. J Assoc Off Anal Chem 56(3): 728-732.
Newland LW, Chesters G, & Lee GB (1969) Degradation of gamma-BHC in
simulated lake impoundments as affected by aeration. J Water Pollut
Control Fed 41: R174-R188.
Niessen KH, Ramolla J, Binder M, Brümann G, & Holmann U (1984)
Chlorinated hydrocarbons in adipose tissue of infants and toddlers:
inventory and studies on their association with intake of mothers'
milk. Eur J Pediatr 142: 238-243.
Nigam SK, Karnik AB, Majumder SK, Visweswarriah K, Suryanarayana
Raju G, Muktha Bai K, Lakkad BC, Thakore KN, & Chatterjee BB (1986)
Serum hexachlorocyclohexane residues in workers engaged at a HCH
manufacturing plant. Int Arch Occup Environ Health 57: 315-320.
Nitsche K, Lange M, Bauer E, & Zesch A (1985) Quantitative
distribution of locally applied lindane in human skin and
subcutaneous fat in vitro. Dermatosen 32(5): 161-165.
Noren K (1983) Levels of organochlorine contaminants in human milk
in relation to the dietary habits of the mothers. Acta Paediatr
Scand 72: 811-816.
Norstrom RJ, Simon M, Muir DCG, & Schweinsburg RE (1988)
Organochlorine contaminants in Arctic marine food chains:
identification, geographical distribution and temporal trends in
polar bears. Environ Sci Technol 22: 1063-1071.
Nurmatov RS (1965) [Effect of trichlormetaphos-3 and lindane on
animals]. Veterinarija 44(8): 85-87 (in Russian).
Ocker HD (1983) [Contaminants in cereal grains-state of knowledge
and list of deficiencies]. Getreide-Mehl-Brot 37: 3-7 (in German).
Oehme M & Mano S (1984) The long-range transport of organic
pollutants in the Arctic. Fresenius Z Anal Chem 319: 141-146.
Oehme M & Stray H (1982) Quantitative determination of ultra-traces
of chlorinated compounds in high-volume air samples from the Arctic
using polyurethane foam as collecting medium. Fresenius Z Anal Chem
311: 655-673.
Oesch F (1980) Bacterial mutagenicity tests of lindane with mouse
liver preparations as metabolizing systems. University of Mainz
(Unpublished report Celamerck No. 111AA-457-006, submitted to WHO by
CIEL).
Oesch F & Glatt HR (1984) Mammalian cell (V79) mutagenicity test on
lindane. University of Mainz (Unpublished report Celamerck No.
111AC-457-019/SP 540-VT21, submitted to WHO by CIEL).
Oesch F, Friedberg T, Herbst M, Paul W, Wilhelm N, & Bentley P
(1982) Effects of lindane treatment on drug metabolizing enzymes and
liver weight of CF1 mice in which it evoked hepatomas and in non-
susceptible rodents. Chem-biol Interactions 40: 1-14.
Ohisa N & Yamaguchi M (1978a) Degradation of gamma-BHC in flooded
soils enriched with peptone. Agric Biol Chem 42(11): 1983-1987.
Ohisa N & Yamaguchi M (1978b) Gamma BHC degradation accompanied by
the growth of Clostridium rectum isolated from paddy field soil.
Agric Biol Chem 42(10): 1819-1823.
Ohisa N & Yamaguchi M (1979) Clostridium species and gamma-BHC
degradation in paddy soil. Soil Biol Biochem 11: 645-649.
Ohisa N, Yamaguchi M, & Kurihara N (1980) Lindane degradation by
cell free extracts of Clostridium rectum. Arch Microbiol 125: 221-
225.
Ohisa N, Kurihara N, & Nakajima M (1982) ATP synthesis associated
with the conversion of hexachlorocyclohexane related compounds. Arch
Microbiol 131: 330-333.
Ohly A (1973) Toxicology of lindane. In: Ulmann, E. ed., Lindane,
Vol. I, Supplement 1974. Brussels, CIEL, p. 27.
Oldiges H, Takenaka S, & Hochrainer D (1980) [Inhalation test with
lindane (gamma-hexachlorocyclohexane) to determine the LC50.
(Unpublished report Celamerck No. 111AA-423-001 submitted to WHO by
CIEL) (in German).
Oldiges H, Hertel R, Kördel W, Hochrainer D, & Mohr U (1983) [90-Day
inhalation with lindane]. Schmallenberg, Fraunhofer Institut für
Toxikologie und Aerosolforschung (Unpublished report Celamerck No.
111AC-435-004, submitted to WHO by CIEL) (in German).
Oloffs PC, Albright LJ, Szeto SY, & Lau J (1973) Factors affecting
the behaviour of five chlorinated hydrocarbons in two natural waters
and their sediments. J Fish Res Board Canada 30(11): 161 9-1623.
Oshiba K (1972) Experimental studies on the fate of ß- and gamma-BHC
in vivo following daily administration. Osaka Shiritsu Daigaku
Igaku Zasshi 21(1-3): 1-19.
Paasivirta J, Palm H, Paukka R, Akhabuhaya J, & Lodenius M (1988)
Chlorinated insecticide residues in Tanzanian environment,
Tanzadrin. Chemosphere 17(10): 2055-2062.
Palmer CM & Maloney TE (1955) Preliminary screening for potential
algicides. Ohio J Sci 55(1): 1-8.
Palmer L & Kolmodin-Hedman B (1972) Improved quantitative gas
chromatographic method for the analysis of small amounts of
chlorinated hydrocarbon pesticides in human plasma. J Chromatogr 74:
21-30.
Palmer AK, Cozen DD, Spicer EJF, & Worden AN (1978a) Effects of
lindane upon reproductive function in a 3-generation study in rats.
Toxicology 10: 45-54.
Palmer AK, Bottomley AM, Worden AN, Frohberg H, & Bauer A (1978b)
Effect of lindane on pregnancy in the rabbit and rat. Toxicology 9:
239-247.
Pandy RN, Zaidi SIM, & Kidawl AM (1985) Effect of pesticides on frog
skeletal muscle sarcolemmal enzymes. J Environ Biol 6(1): 7-9.
Panwar RS, Gupta RA, Joshi HC, & Kapoor D (1982) Toxicity of some
chlorinated hydrocarbons and organophosphorus insecticides to
gastropod, Viviparus bengalensis Swainson. J Environ Biol 3(1):
31-36.
Pastor A, Hernandez F, Medina J, Melero R, Lopez FJ, & Conesa M
(1988) Organochlorine pesticides in marine organisms from the
Castelion and Valencia coasts of Spain. Mar Pollut Bull 19(5): 235-
238.
Paul W, Knappen F, & Stötzer H (1980) [Testing for acute toxicity
after oral administration in an oily solution to Chbi: NMRI (SPF)
mice]. Ingelheim am Rhein, Boehringer & Son (Unpublished report
submitted to WHO by CIEL) (in German).
Petring OU, Adelhoj B, & Jorgensen FR (1986) Acute poisoning after
oral intake of lindane. Ugeskr Laeg 148(50): 3377.
Platford RF (1981) The environmental significance of surface films.
II. Enhanced partitioning of lindane in thin films of octanol on the
surface of water. Chemosphere 10(7): 719-722.
Podlejski J & Dervieux AM (1978) Persistance de quatre produits
phytosanitaires dans les eaux de rivières en Camargue:
caractérisation de leurs effets sur l'écosystème. Trav Soc Pharm
Montpellier 38(2): 153-164.
Polishuk ZW, Wassermann M, Wassermann D, Groner Y, Lazarovici S, &
Tomatis L (1970) Effects of pregnancy on storage of organochlorine
insecticides. Arch Environ Health 20: 215-217.
Portig J, Kraus P, Stein K, Koransky W, Noack G, Gross B, & Sodomann
S (1979) Glutathione conjugate formation from hexachlorocyclohexane
and pentachlorocyclohexene by rat liver in vitro. Xenobiotica 9:
353-378.
Portmann JE (1970) Results of acute toxicity test with marine
organisms, using a standard method. In: Ruivo, M. ed., Marine
pollution and sea life. London, Fishing News (Books) Ltd, pp. 212-
217.
Portmann JE (1979) Evaluation of the impact on the aquatic
environment of hexachlorocyclohexane (HCH-isomers). Brussels,
Commission of the European Communities (Contract No. U/78/180-SEC
(78)792).
Probst GS, McMahon RE, Hill LE, Thompson CZ, Epp JK, & Neal SB
(1981) Chemically-induced unscheduled DNA synthesis in primary rat
hepatocyte cultures: A comparison with bacterial mutagenicity using
218 compounds. Environ Mutagen 3: 11-32.
Publicover SJ & Duncan CJ (1979) The action of lindane in
accelerating the spontaneous release of neurotransmitter at the frog
neuromuscular junction. Naunyn-Schmledebergs Arch Pharmacol 381:
179-182.
Publicover SJ, Duncan CJ, & Smith JL (1979) The action of lindane in
causing ultrastructural damage in frog skeletal muscle. Comp Biochem
Physiol C64: 237-241.
Racey PA & Swift SM (1986) The residual effects of remedial timber
treatments on bats. Biol Conserv 35: 205-214.
Ramamoorthy S (1985) Competition of fate processes in the
bioconcentration of lindane. Bull Environ Contam Toxicol 34: 349-
358.
Randall WF, Dennis WH, & Warner MC (1979) Acute toxicity of
dechlorinated DDT, chlordane, and lindane to bluegill (Lepomis
macrochirus) and Daphnia magna. Bull Environ Contam Toxicol 21:
849-854.
Rao MB (1981) Effect of gamma-hexachloran and Sevin on the survival
of the black sea mussel, Mytilus galloprovincialis Lam.
Hydrobiologia 78: 33-37.
Rao PSC & Davidson JM (1982) Retention and transformation of
selected pesticides and phosphorus in soil-water system: a critical
review. Springfield, Virginia, US Department of Commerce, National
Technical Information Services (EPA Report No. 600/3-82-060, PB 92-
256884).
Rao SM & Reddy JK (1987) Peroxisome proliferation and
hepatocarcinogenesis. Carcinogenesis 8(5): 631-636.
Rappe C, Nygren M, Lindström G, Buser HR, Blaser O, & Wüthrich C
(1987) Polychlorinated dibenzofurans and dibenzo-p-dioxins and other
chlorinated contaminants in cow milk from various locations in
Switzerland. Environ Sci Technol 21: 964-970.
Raw GR, ed. (1970) ClPAC Handbook. Vol. 1. Analysis of technical and
formulated pesticides. Collaborative International Pesticides
Analytical Council Ltd, New York, Academic Press, pp. 71-78, 129-
131.
Ray RC (1983) Toxicity of the pesticides hexachlorocyclohexane and
Benomyl to nitrifying bacteria in flooded autoclaved soil and in
culture media. Environ Pollut A32: 147-155.
Reltenrath WG, Chellquist EM, Shipwash EA, Jederberg WW, & Krueger
GG (1984) Percutaneous penetration in the hairless dog, weanling pig
and grafted athymic nude mouse: evaluation of models for predicting
skin penetration in man. Br J Dermatol 27 (Suppl. III): 123-135.
Reiner E, Krauthacker B, Stipcevic M, & Stefanac Z (1977) Blood
levels of chlorinated hydrocarbon residues in the population of a
continental town in Croatia (Yugoslavia). Pestic Monit J 11: 54-55.
Reno FE (1976a) Teratology study in rats: Lindane (gamma benzene
hexachloride, USP). Madison, Wisconsin, Hazelton Laboratories
America Inc. (Unpublished report submitted to WHO by CIEL).
Reno FE (1976b) Teratology study in rabbits: Lindane (gamma benzene
hexachloride, USP). Madison, Wisconsin, Hazelton Laboratories
America Inc. (Unpublished report submitted to WHO by CIEL).
Rhöne-Poulenc Agrochimie (1986) Lindane. Information kit. Lyon.
Riemschneider R (1949) [A contribution to the toxicity of contact
insecticides]. Anz Schädlingskd 22(1): 1-3 (in German).
Rivett KF, Chesterman H, Kellett DN, Newman AJ, & Worden AN (1978)
Effects of feeding lindane to dogs for periods of up to 2 years.
Toxicology 9: 273-289.
Rocohi P, Perocco P, Alberghini W, Fini A, & Prodi G (1980) Effect
of pesticides on scheduled and unscheduled DNA synthesis of rat
thymocytes and human lymphocytes. Arch Toxicol 45: 101-108.
Röhrborn G (1976) Cytogenetic analysis of bone marrow of Chinese
harnster (Cricetulus grisens) after sub-acute treatment with
lindane. (Unpublished report Celamerck No. 111AA-457-008, submitted
to WHO by CIEL).
Röhrborn G (1977a) Statement on the potential mutagenicity of
lindane (gamma-hexachlorocyclohexane). (Unpublished report Celamerck
No. 111AA-457-018, submitted to WHO by CIEL).
Röhrborn G (1977b) Dominant lethal test after treatment of male rats
with lindane. (Unpublished report Celamerck No. 111AA-457-004,
submitted to WHO by CIEL).
Rosenberg LE, Rudd RL, Devaul J, Lundberg DE, Recca JM, & Smith MB
(1953) The effects of economic poisons on wildlife. Quarterly
progress report of the Federal Aid in Wildlife Restoration Act pp.
2, 7, 11-12, 19, 43-45 (Project No. W-45R-1) (Unpublished report).
Sagelsdorff P, Lutz WK, & Schlatter C (1983) The relevance of
covalent binding to mouse liver DNA to the carcinogenic action of
hexachlorocyclohexane isomers. Carcinogenesis 4(10): 1267-1273.
Samuels AJ & Milby TH (1971) Human exposure to lindane. Clinical,
haematological, and biochemical effects. J Occup Med 13(3): 147-151.
San Antonio JP (1959) Demonstration of lindane and a lindane
metabolite in plants by paper chromatography. J Agric Food Chem
7(5): 322-325.
Sanders HO (1969) Toxicity of pesticides to the crustacean Gammarus
lacustrus. Washington, DC, US Department of Interior, Bureau of
Sport Fisheries and Wildlife (Technical Paper No. 25), pp. 2-18.
Sanders HO (1970) Pesticide toxicities to tadpoles of the western
chorus frog Pseudacris triseriata and Fowler's toad Bufo
woodhousii fowleri. Copeia 2: 246-251.
Sanders HO & Cope OB (1966) Toxicities of several pesticides to two
species of Cladocerans. Trans Am Fish Soc 95: 165-169.
Sanders HO & Cope OB (1968) The relative toxicities of several
pesticides to naiads of three species of stoneflies. Limnolog
Oceanogr 13: 112-117.
Sanfeliu C, Camon L, Martinez E, Sola C, Artigas F, & Rodriguez-
Farre E (1988) Regional distribution of lindane in rat brain.
Toxicology 49: 189-196.
Saxena MC, Siddique MKJ, Bhargava AK, Krishna Murti CR, & Kutty D
(1981) Placental transfer of pesticides in humans. Arch Toxicol 48:
127-134.
Schafer EW (1972) The acute oral toxicity of 369 pesticidal,
pharmaceutical and other chemicals to wild birds. Toxicol Appl
Pharmacol 21: 315-330
Schimmel SC, Patrick JM, Jr, & Forester J (1977) Toxicity and
bioconcentration of BHC and lindane in selected animals. Arch
Environ Contam Toxicol 6: 355-363.
Schmiedeberg J & Wasserberger HJ (1953) [Acute toxicity of
hexachlorocyclohexane for man]. Anz Schädlingskd 26(9): 129-133 (in
German).
Schmitt F (1956) [Experimental investigations on the behaviour of
hexachlorocyclohexane and aldrin in the soil, with special reference
to duration of activity, effects on small animal fauna, and
impairment of the taste of geocarpic fruits]. Hohenheim,
Agricultural College (Thesis) (in German).
Schneeweis JC, Greichus TA, & Linder RL (1974) Organochlorine
pesticide residue levels in North American timber wolves-1969-71.
Pestic Monit J 8(2): 142-143.
Schnell U (1965) [Investigations on the chronic toxicity of lindane
for pigs, with special reference to investigations for residues in
fat and liver]. Berlin, Humboldt University (Thesis) (in German).
Schröter C, Parzefall W, Schröter H, & Schulte-Hermann R (1987)
Dose-response studies on the effects of alph-, ß- and gamma-
hexachlorocyclohexane on putative preneoplastic foci,
monooxygenases, and growth in rat liver. Cancer Res 47: 80-88.
Schüttmann W (1972) Clinical observations on the long-term toxicity
of organochlorine pesticides. Z Gesamte Hyg 17: 12-18 (in German).
Schwabe U & Wendling I (1967) [Stimulation of drug metabolism by low
doses of DDT and other chlorlnated hydrocarbon insecticides].
Arzneimittelforschung 17(5): 614-618 (in German).
Seidler H, Machoiz RM, Härtig M, Kujawa M, & Engst R (1975) [Studies
on the metabolism of certain insecticides and fingicides in the rat.
Part IV. Distribution, degradation and excretion of 14C-labelled
lindane]. Nahrung 19(5/6): 473-482 (in German).
Selby LA, Newell KW, Hauser GA, & Junker G (1969) Comparison of
chlorinated hydrocarbon pesticides in maternal blood and placental
tissues. Environ Res 2: 247-255.
Seuge J & Bluzat R (1979a) Toxicité chronique du carbaryl et du
lindane chez le mollusque d'eau douce Lymnea stagnails L. Water
Res 13(3): 285-293.
Seuge J & Bluzat R (1979b) Etude de la toxicité chronique de deux
insecticides (carbaryl et lindane) à la génération F1 de Lymnea
stagnails L. (mollusque gastéropode pulmone). II. Conséquences sur
le potentiel reproducteur. Hydrobiologia 66(1): 25-31.
Seuge J & Bluzat R (1982) Influence d'une intoxication par le
lindane en fonction de la dureté de l'eau chez Lymnea stagnails
(Mollusca pulmonata). Malacologia 22(1-2): 1 5-18.
Sharom MS, Miles JRW, Harris CR, & McEwen FL (1980) Persistence of
12 insecticides in water. Water Res 14: 1089-1093.
Shearer RC, Letey J, Farmer WJ, & Klute A (1973) Lindane diffusion
in soil. Soil Sci Soc Am Proc 37: 189-193.
Shiota Y & Kanda S (1972) [Fate of benzene hexachlonde in paddy
soils]. Aichi-Ken Nogyo Sogo Shikenjho, Kenkyu Hokoku 1972A/4: 128-
133 (in Japanese).
Shirasu Y, Moriya M, Karo K, Furuhashi A, & Kada T (1976)
Mutagenicity screening of pesticides in the microbial system. Mutat
Res 40: 1 9-30.
Siddaramappa R & Sethunathan N (1975) Persistence of gamma-BHC and
beta-BHC in Indian rice soils under flooded conditions. Pestic Sci
6: 395-403.
Siddique MKJ, Saxena MC, Mishra UK, Krishnamurti CR, & Nag D (1981)
Long-term occupational exposure to DDT. Int Arch Occup Environ
Health 48: 301-308.
Sierra M & Santiago D (1987) Organochlorine pesticide levels in barn
owls collected in Leon, Spain. Bull Environ Contam Toxicol 38: 261-
265.
Sina JF, Bean CL, Dysart GR, Taylor VI, & Bradley MO (1983)
Evaluation of the alkaline elution/rat hepatocyte assay as a
predictor of carcinogenic/mutagenic potential. Mutat Res 113: 357-
391.
Sinha RRP & Sinha SP (1983) Induction of dominant lethal mutations
in Drosophila melanogaster by three pesticides: Gammexane, Sevin,
and Folidol. Comp Physiol Ecol 8(2): 87-89.
Skaare JU, Tuveng JM, & Sande HA (1988) Organochlorine pesticides
and polychlorinated biphenyls in maternal adipose tissue, blood,
milk, and cord blood from mothers and their infants living in
Norway. Arch Environ Toxicol 17: 55-63.
Skaftason JF & Johannesson T (1979) Organochlorine compounds (DDT,
hexachlorocylohexane, hexachlorobenzene) in Icelandic animal body
fat and butter fat: Local and global sources of contamination.
Letter to the Editor. Acta Pharmacol Toxicol 44: 156-157.
Slade RE (1945) The gamma-isomer of hexachlorocyclohexane
(gammexane). Chem Ind 65: 314-319.
Slooff W & Matthijsen AJCM (1988) Integrated criteria document:
hexachlorocyclohexanes. Bilthoven, National Institute of Public
Health and Environmental Protection (Report No. 758473011).
Srinivasan K & Radhakrishnamurty R (1988) Biochemical changes
produced by ß- and gamma-hexachlorocyclohexane isomers in albino
rats. J Environ Sci Health B23(4); 367-386.
Srinivasan K, Ramesh HP, & Radhakrishnamurty R (1984) Renal tubular
dysfunction caused by dietary hexachlorocyclohexane (HCH) isomers. J
Environ Sci Health B19(4/5): 453-466.
Srinivasan K, Ramesh HP, & Radhakrishnamurty R (1988) Changes
induced by hexachlorocyclohexane isomers in rat livers and testes.
Bull Environ Contam Toxicol 41: 531-539.
Starr HG & Clifford NJ (1972) Acute lindane intoxication. A case
study. Arch Environ Health 25: 374-375.
Starr RI & Johnson RE (1968) Laboratory method for determining the
rate of volatilization of insecticides from plants. J Agric Food
Chem 16(3): 411-414.
Steering Group on Food Surveillance (1982) Report of the Working
Party on Pesticide Residues (1977-1981). London, Her Majesty's
Stationery Office (Food Surveillance Paper No. 9).
Steering Group on Food Surveillance (1986) Report of the Working
Party on Pesticide Residues (1982-1985). London, Her Majesty's
Stationery Office (Food Surveillance Paper No. 16).
Steering Group on Food Surveillance (1989) Report of the Working
Party on Pesticide Residues (1985-1988). London, Her Majesty's
Stationery Office (Food Surveillance Paper No. 25).
Stein K, Portig J, & Koransky W (1977) Oxidative transformation of
hexachlorocyclohexane in rats and with rat liver microsomes. Naunyn-
Schmiedebergs Arch Pharmacol 298: 115-128.
Stein K, Portig J, Fuhrmann H, Koransky W, & Noack G (1980) Steric
factors in the pharmacokinetics of lindane and alpha-
hexachlorocyclohexane in rats. Xenobiotica 10(1): 65-77.
Steinwandter H (1976) [Lindane metabolism in plants. II. Formation
of alpha-HCH]. Chemosphere 4: 221-225 (in German).
Stewart DKR & Chisholm D (1971) Long-term persistence of BHC, DDT,
and chlordane in a sandy loam soil. Can J Soil Sci 51: 379-383.
Stewart DKR & Fox CJS (1971) Persistence of organochlorine
insecticides and their metabolites in Nova Sootian soils. J Econ
Entomol 64(2): 367-371.
Stieglitz R, Stobbe H, & Schüttmann W (1967) [Bone marrow damage
following exposure to the insecticide gamma-hexachlorocyclohexane
(lindane)]. Acta Haematol 38: 337-350 (in German).
Stöckigt J (1976) Investigations of the metabolism of gamma-HCH in
plant tissue. (Unpublished report Celamerck No. 111AA-641-004,
submitted to WHO by CIEL).
Stöckigt J & Ries B (1976) Catabolism of gamma-hexachlorocyclohexane
(lindane) by plants and cell cultures, a comparison. Bochum, Chair
of Plant Physiology, Ruhr University (Unpublished report Celamerck
No. 111AA-641-005, submitted to WHO by CIEL).
Strachan WMJ, Huneault H, Schertzer WM, & Elder FC (1980)
Organochlorines in precipitation in the Great Lakes region. In:
Afghan BK & MacKay D ed., Hydrocarbons and halogenated hydrocarbons
in the aquatic environment. New York, Plenum Press, pp. 387-396.
Sugiura K, Washino T, Hattori M, Sato E, & Goto M (1979)
Accumulation of organochlorine compounds in fishes. Difference of
accumulation factors by fishes. Chemosphere 6: 359-364.
Sunol C, Tusell JM, Gelp E, & Rodriguez-Farre E (1988) Convulsant
effect of lindane and regional brain concentration of GABA and
dopamine. Toxicology 49(2/3): 247-252.
Suter P, Horst K, Luetkemeier H, Herbst M, Terrier C, Lind H, &
Ellgehausen H (1983) Three months toxicity study in rats with
lindane: Part 1 & 2. Itingen, Research and Consulting Company AG
(Unpublished report Celamerck No. 111AA-433-007/009, submitted to
WHO by CIEL).
Suzuki M, Yamato Y, & Watanabe T (1975) Persistence of BHC
(1,2,3,4,5,6-hexachlorocyclohexane) and dieldrin residues in field
soils. Bull Environ Contam Towicol 14(5): 520-529.
Sweeney RA (1969) Metabolism of lindane by unicellular algae. In:
Proceedings of the 12th Conference on Great Lakes Research,
International Associationforf Great Lakes Research, pp. 98-102.
Szymczynski GA, Waliszewski SM, Tuszewski M, & Pyda P (1986)
Chlorinated pesticides levels in human adipose tissue in the
district of Poznan. J Environ Sci Health A21(1): 5-14.
Takabatake E (1978) Levels of organochlorine compounds and heavy
metals in tissues of the general population in Japan. Geogr Med 8:
1-23.
Tanabe S, Tatsukawa R, Kawano M, & Hidaka H (1982) Global
distribution and atmospheric transport of chlorinated hydrocarbons:
HCH(BHC) isomers and DDT compounds in the Western Pacific, Eastern
Indian, and Antarctic Oceans. J Oceanogr Soc Jpn 38(3): 137-148.
Tanabe S, Tanaka H, & Tatsukawa R (1984) Polychlorobiphenyls, Sigma-
DDT, and hexachlorocyclohexane isomars in the Western North Pacific
ecosystem. Arch Environ Contam Toxicol 13: 731-738.
Tanaka K, Kudhara N, & Nakajima M (1977) Pathways of chlorophenol
formation in oxidative biodegradation of BHC. Agric Biol Chem 41(4):
723-725.
Tanaka K, Kurihara N, & Nakajima M (1979) Oxidative metabolism of
lindane and its isomers with microsomes from rat liver and housefly
abdomen. Pestic Biochem Physiol 10: 96-103.
Thorpe E & Walker AIT (1973) The toxicology of dieldrin (HEOD). II.
Comparative long-term oral toxicity studies in mice with dieldrin,
DDT, phenobarbitone, ß-BHC and gamma-BHC. Food Cosmet Toxicol 11:
433-442.
Tolot F, Longlet JP, & Berthelon J (1969) Polyneurite due au
lindane. J Méd Lyon 50: 747-754.
Tomczak S, Baumann K, & Lehnert G (1981) Occupational exposure to
hexachlorocyclohexane. IV. Sex hormone alterations in HCH-exposed
workers. Int Arch Occup Environ Health 48: 283-287.
Tooby TE & Durbin FJ (1975) Lindane residue accumulation and
elimination in rainbow trout (Salmo gairdnerii Richardson) and
roach (Rutilus rutilus L.). Environ Pollut 8(2): 79-89.
Trautmann A & Streit B (1979) [Sorption of lindane (gamma-
hexachlorocyclohexane) in Nitzschia actinastroïdes (Lemm.) v. Goor
(dimethoate) at different growth conditions]. Arch Hydrobiol Suppl
55(3/4): 324-348 (in German).
Trifonova TK, Gladenko IN, & Schukla WD (1970) [Effect of gamma-BHC
and Sevin on reproduction]. Veterinarja 47: 91-93 (in Russian).
Trosko JE (1982) Inhibition of cell-to-cell communication by tumour
promoters. Carcinogenesis 7: 565-585.
Truhaut R (1954) Communication to the International Symposium on
Prevention of Cancer, São Paulo, 1954. In: Evaluation of the
toxicity of pesticide residues in food. Report of a Joint Meeting of
the FAO Committee on Pesticides in Agriculture and te WHO Expert
Committee on Pesticide Residues, Geneva, 30 September-7 October
1963. Rome, Food and Agriculture Organization of the United Nations,
p. 48 (FAO Meeting Report No. PL/1963/13. WHO/Food Add., 23 (1964)).
Tsukano Y (1973) Factors affecting disappearance o! BHC isomers from
rice field soil. Jpn Agric Res Q 7(2): 93-97.
Tsushimoto G, Chang CC, Trosko JE, & Matsumura F (1983) Cytotoxic,
mutagenic, and cell-cell communication inhibitory properties of DDT,
lindane, and chlordane on Chinese hamster cells in vitro. Arch
Environ Contam Toxicol 12: 721-730.
Tu CM (1975) Interaction between lindane and microbes in soils. Arch
Microbiol 105(2): 131-134.
Tu CM (1976) Utilization and degradation of lindane by soil
microorganisms. Arch Microbiol 108: 259-263.
Tuinstra LGMT (1971) Organochlorine insecticide residues in human
milk in the Leiden region. Neth Milk Dairy J 25: 24-32.
Turner JC (1979) Transplacental movement of organochlorine pesticide
residues in desert bighorn sheep. Bull Environ Contam Toxicol 21:
116-124.
Turner JC & Shanks V (1980) Absorption of some organochlorine
compounds by the rat small intestine- in vivo. Bull Environ Contam
Toxicol 24: 652-655.
Turtle EE, Taylor A, Wright EN, Thearle RJP, Egan H, Evans WH, &
Soutar NM (1963) The effects on birds of certain chlorinated
insecticides used as seed dressings. J Sci Food Agric 14: 567-577.
Tussel JM, Sunol C, Gelpi E, & Rodriguez-Farre E (1988) Effect of
lindane at repeated low doses. Toxicology 49: 375-379.
Tzoneva-Maneva MT, Kaloyaneva F, & Georgieva V (1971) Influence of
diazinon and lindane on the mitotic activity and the caryotype of
human lymphocytes, cultivated in vitro. Bibl Haematol 38(1): 344-
347.
Uchida M, Kurihare N, Fujita T, & Nakajima M (1974) BHC isomers and
related compounds. VII. Inhibitory effects of BHC isomers on sodium-
potassium ion-dependent ATPase, yeast growth, and nerve conduction.
Pestic Biochem Physiol 4(3): 260-265.
Ukeles R (1962) Growth of pure cultures of marine phytoplankton in
the presence of toxicants. Appl Microbiol 10: 532-537.
Ullmann L, Sacher R, & Porrizello T (1986a) Primary skin irritation
study with lindane in rabbits (4-hour occlusive application).
(Unpublished report Celamerck No 111AD-465-003, submitted to WHO by
CIEL).
Ullmann L, Claire R, & Bognar G (1986b) Test for delayed contact
hypersensitivity in the albino guinea pig with lindane (maximization
test). Itingen, Research and Consulting Company AG (Unpublished
report No. 061650, Celamerck No. 111AC-467-001, submitted to WHO by
CIEL).
Ullmann L, Mohler H, & Gembardt G (1986c) Four-hour acute aerosol
inhalation toxicity study with lindane in rats. Itingen, Research
and Consulting Company AG (Unpublished report No. 061637, submitted
to WHO by CIEL).
Ullmann L, Claire R, & Bognar G (1987a) Contact hypersensitivity to
Nexit Fluessig (CME 11129) in albino guinea pigs (maximization
test). Itingen, Research and Consulting Company AG (Unpublished
report Celamerck No. 11129-467-004, submitted to WHO by CIEL).
Ullmann J, Claire R, & Bognar G (1987b) Contact hypersensitivity to
Nexit Stark (CME 11125) in albino guinea pigs (maximization test).
Itingen, Research and Consulting Company AG (Unpublished report
Celamerck No. 11125-467-002, submitted to WHO by CIEL).
Ullmann J, Claire R, & Bognar G (1987c) Contact hypersensitivity to
Agronex Saatgutpuder (CME 11102) in albino guinea pigs (maximization
test). Itingen, Research and Consulting Company AG (Unpublished
report Celamerck No. 11102-467-003, submitted to WHO by CIEL).
Umweltbundesamt (1988-89) [Data on the environment 1988/89. Berlin,
Schmidt E. Verlag, pp. 403, 410-412, 416-417, 521-522, 524, 526 (in
German).
Uphouse L (1987) Decreased rodent sexual receptivity after lindane.
Toxicol Lett 39: 7-14.
US Environmental Protection Agency (1976) National Interim Primary
Drinking Water Regulations, Subpart B-Maximum contaminant levels.
Washington, DC, Office of Water Supply (Report No. EPA-570/9-76-
003).
US Environmental Protection Agency (1977) Notice of rebuttable
presumption against registration and continued registration of
pesticide products containing lindane. Fed Reg 42(33): 9816-9947.
US Environmental Protection Agency (1980) Lindane position document
2/3. Washington, DC, Office of Pesticide Programs.
US Environmental Protection Agency (1984) Health effects assessment
for lindane. Cincinnatti, Ohio (Report No. EPA/540/1-86/056).
US Environmental Protection Agency (1985) Guidance for the
reregistration of pesticide products containing lindane as the
active ingredient. Washington, DC (Report No. RS-85).
US National Cancer Institute (1977) Bioassay of lindane for possible
carcinogenicity. Washington, DC, US Department of Health, Education,
and Weltare (Technical Report Series No. 14).
van Velsen FL (1986) The oestrogenicity of the environmental
contaminant ß-hexachlorocyclohexane. University of Utrecht (Thesis).
van Velsen FL, Franken MAM, van Leeuwen FXR, & Loeber JG (1984)
[Study of semi-chronic oral toxicity of gamma-HCH in the rat].
Bilthoven, National Institute of Public Health and Environmental
Hygiene (Unpublished report No. 618209001) (in Dutch).
Vesselinovitch SD & Carlborg FW (1983) Lindane bioassay studies and
human cancer risk assessment. Toxicol Pathol 11(1): 12-22.
Vetter H, Kampe W, & Ranfft, K (1983) [Results of three years of
comparative tests on vegetables, fruit and bread from modern and
alternative producers]. Darmstadt, Association of German Institutes
for Agricultural Testing and Research, pp. 1-11,143 (VDLUFA Report
No. 7) (in German).
Vodopick H (1975) Erythropoietic hypoplasia after exposure to gamma-
benzene hexachloride. J Am Med Assoc 234(8): 850-851.
Voerman S & Besemer AFH (1970) Residues of dieldrin, lindane, DDT,
and parathion in a light sandy soil after repeated application
throughout a period of 15 years. J Agric Food Chem 18(4): 717-719.
Voerman S & Besemer AFH (1975) Persistence of dieldrin, lindane, and
DDT in a light sandy soil and their uptake by grass. Bull Environ
Contam. Toxicol 13(4): 501-505.
Vogel SM, Joy RM, & Narahashi T (1985) Lindane exerts both
presynaptic and postsynaptic actions at frog neuromuscular junction.
In: 15th Annual Meeting of the Society for Neuroscience, Part 2.
Dallas, Texas, 20-25 October 1985. Soc Neurosci Abstr 11: 1004.
Vohland HW, Portig J & Stein K (1981) Neuropharmacological effects
of isomers of hexachlorocyclohexane. Toxicol Appl Pharmacol 57(3):
425-438.
Vonk JW & Quirijns JK (1979) Anaerobic formation of (alpha-
hexachlorocyclohexane from gamma-hexachlorocyclohexane in soil and
by Escherichia coli. Pestic Biochem Physiol 12: 68-74.
de Vos RH, van Dokkum W, Olthof PDA, Quirijns JK, Muys T, & van der
Poll JM (1984) Pesticides and other chemical residues in Dutch total
diet samples (June 1976-July 1978). Food Chem Toxicol 22(1): 11-21.
Vukavic T, Pavkov S, Cusic S, Roncevic N, Vojinovic M, & Tokovic B
(1986) Pesticide residues in human colostrum: seasonal variations,
Yugoslavia. Arch Environ Contam Toxicol 15: 525-528.
Wahid PA & Sethunathan N (1979) Sorption and desorption of (alpha,
ß, and gamma isomers of hexachlorocyclohexane in soils. J Agric Food
Chem 27(5): 1050-1053.
Wahid PA & Sethunathan N (1980) Sorption and desorption of lindane
by anaerobic and aerobic soils. J Agric Food Chem 28: 623-625.
Wammes JY, Wegman RCC, Hofstee AWM, Janssens H, Rieffe DW, Marsman
JA, Groenemeijer GS, & van den Broek HH (1983) [Investigation into
the presence of pesticides and related compounds in surface waters].
Bilthoven, National Institute of Public Health and Environmental
Hygiene (Unpublished report No. 218102003) (in Dutch).
Wang HH & Grufferman S (1981) Aplastic anemia and occupational
pesticide exposure: a case-control study. J Occup Med 23(5): 364-
366.
Ware GW & Naber EC (1961) Lindane in eggs and chicken tissues. J
Econ Entomol 54(4): 675-677.
Wassermann M, Ron M, Bercovici B, Wassermann D, Cucos S, & Pines A
(1982) Premature delivery and organochlorine compounds.
Polychlorinated biphenyls and some organochlorine insecticides.
Environ Res 28: 106-112.
Wedberg JL, Moore S, III, Amore FJ, & McAvoy H (1978) Residues in
food and feed. Organochlorine insecticide residues in bovine milk
and manufactured milk products in Illinois, 1971-76. Pest Monit J
11(4): 161-164.
Wegman RCC & Greve PA (1980) Halogenated hydrocarbons in Dutch water
samples over the years 1969-1977. In: Afghan BK & MacKay D (eds.),
Hydrocarbons and halogenated hydrocarbons in the aquatic
environment. New York, Plenum Press, pp. 405-415.
Weigert P, MUller J, Hans R, & Zufelde KP (1983) [Pesticide residues
in foodstuffs. Communication I: Organochlorine compounds]. Berlin,
Dietrich Reimer Verlag (ZEBS-Berichte 3) (in German).
Weisse I & Herbst M (1977) Carcinogenicity study of lindane in the
mouse. Toxicology 7: 233-238.
Wellborn TL, Jr (1971) Toxicity of some compounds to striped bass
fingerlings. Progr Fish-Cult 33: 32-36.
West I (1967) Lindane and haematologic reactions. Arch Environ
Health 15: 97-101.
Wheatley GA (1965) The assessment and persistence of residues of
organochlorine insecticides in soils and their uptake by crops. Proc
Assoc Appl Biol 55: 325-329.
Whitehead CC, Downing AG, & Pettigrew RJ (1972a) The effects of
lindane on laying hens. Br Poult Sci 13: 293-299.
Whitehead CC, Downie JN, & Phillips JA (1972b) BHC not found to
reduce the shell quality of hen's eggs. Nature 239: 411-412.
Whitehead CC, Downie JN, & Phillips JA (1974) Some characteristics
of the egg shells of quail fed gamma-BHC. Pestic Sci 5: 275-279.
WHO/FAO (1975) Data sheets on pesticides: No. 1- Lindane. Geneva
(Unpublished document VBC/DS/75.12).
WHO (1985) Specifications for pesticides used in public health:
insecticides-molluscicides-repellents - methods, 6th ed., Geneva.
WHO (1990) The WHO recommended classification of pesticides by
hazard and guidelines to classification 1990-1991. Geneva
(Unpublished document WHO/PCS/90.1 Rev. 1).
Wilkes LC, Tapprich BL, & Mulkey NS (1987a) Determination of 14C-
residues following oral administration of 14C-lindane to lactating
goats (in vivo and in vitro studies). Monument, California,
Development Corporation (Unpublished report No. ADC 957a, submitted
to WHO by CIEL).
Wilkes LC, Mulkey NS, Hallenbeck SA, Piznik M, & Wargo JP (1987b)
Metabolism study of 14C-lindane fed or topically applied to
lactating goats. Monument, California, Development Corporation
(Unpublished report No. ADC 957b, submitted to WHO by CIEL).
Williams DT, Benoit, FM, McNeil EE, & Otson R (1978) Organochlorine
pesticide levels in Ottawa drinking water, 1976. Pestic Monit J
12(3): 163.
Wirth H (1985) The adsorbtion of 14C-labelled gamma-HCH (lindane)
at levels in the range of ng/litre to soil components. Geesthacht,
GKSS Research Institute (Report No. GKSS 85/E/30).
Wittlinger R & Ballschmiter K (1987) Global baseline pollution
studies. XI: Congener specific determination of polychlorinated
biphenyls (PCBs) and occurrence of alpha- and gamma-
hexachlorocyclohexane (HCH), 4.4'-DDE and 4.4'-DDT in continental
air. Chemosphere 16(10-12): 2497-2513.
Wolfe GW & Ralph JA (1980) Acute oral toxicity study in B6C3F1 mice:
Lindane. Vienna, Virginia, Hazleton Laboratories America Inc.
(Unpublished report submitted to WHO by CIEL).
Wolff G (1986) Maternal influence on phenotypic differentiation of a
mutant mouse susceptible to neoplasia and obesity. In: Genetic
toxicology of environmental chemicals, Part B, Genetic effects and
applied mutagenesis. New York, Alan R. Liss Inc., pp. 33-38.
Wolff GL & Morrissey RL (1986) Increased responsiveness of lean
pseudoagouti Avy/a female mice to lindane enhancement of lung and
liver tumorigenesis. Proc Am Assoc Cancer Res 26: 138.
Wolff GL, Roberts DW, & Galbraith DB (1986) Prenatal determination
of obesity, tumour susceptibility and coat colour pattern in viable
yellow (Avy/a) mice. J Hered 77: 151-158.
Woodard F & Hagan EC (1947) Toxicological studies on the isomars and
mixtures of isomers of benzenehexachloride. Fed Proc 6: 386.
Yamaguchi I, Matsumura F, & Kadous AA (1979) Inhibition of synpatic
ATPase by heptachlor epoxide in rat brain. Pestic Biochem Physiol
11: 285-293.
Yamato Y, Kiyonaga M, & Watanabe T (1983) Comparative
bioaccumulation and elimination of HCH-isomars in short-necked clam
(Venerupis japonica) and guppy (Poecilia reticulata). Bull
Environ Contam Toxicol 31: 352-359.
Yoshida T & Castro TF (1970) Degradation of gamma-BHC in rice soils.
Soil Sci Soc Am Proc 34: 440-442.
Yule WN, Chiba M, & Morley HV (1967) Fate of insecticide residues.
Decomposition of lindane in soil. J Agric Food Chem 15(6): 1000-
1004.
Zeilmaker MJ & Yamasaki H (1986) Inhibition of junctional
intercellular communication as a possible short-term test to detect
tumour promoting agents. Results with nine chemicals tested by dye-
transfer assay in Chinese hamster V79 cells. Cancer Res 46: 6180-
6186.
Zesch A, Nitzsche K, & Lange M (1982) Demonstration of the
percutaneous resorption of a lipophilic pesticide and its possible
storage in the human body. Arch Dermatol Res 273: 43-49.
RESUME ET EVALUATION; CONCLUSIONS; RECOMMANDATIONS
1. Résumé et évaluation
1.1 Propriétés générales
L'hexachlorocyclohexane technique (HCH) est composé de 65-70%
d'alpha-HCH, de 7-10% de beta-HCH, de 14-15% de gamma-HCH, et
d'environ 10% d'autres isomères et composés. Le lindane contient
> 99% de gamma-HCH. C'est un solide, avec une tension de vapeur
faible, peu soluble dans l'eau mais très soluble dans les solvants
organiques comme l'acétone, et dans les solvants aromatiques et
chlorés. Le coefficient de partage n-octanol/eau (Log Poe) est
de 3,2-3,7.
Le lindane peut être dosé à part des autres isomères du HCH
après extraction par partage liquide/liquide, chromatographie sur
colonne et chromatographie en phase gazeuse avec détection par
capture d'électrons. Extrêmement sensibles, ces méthodes analytiques
permettent d'identifier des résidus de lindane de l'ordre du
nanogramme par kilogramme ou par litre.
Depuis le début des années 50, le lindane est utilisé comme
insecticide à large spectre; le secteur agricole y a recours
notamment pour traiter les semences et les sols; on l'utilise pour
traiter les arbres, le bois de construction et les matériaux
entreposés; on l'applique sur le pelage des animaux pour éliminer
les ectoparasites; on en fait également usage dans le domaine de la
santé publique.
1.2 Transport, réparation et transformation dans l'environnement
Le lindane est fortement adsorbé aux sols contenant une grande
quantité de matière organique; en outre, il peut s'imprégner dans le
sol à la faveur d'une chute de pluie ou d'une irrigation
artificielle. Sous l'effet des températures élevées des régions
tropicales, il se dissipe en grande partie par volatilisation.
Le lindane subit une dégradation rapide (déchloration) sous
l'action des rayons ultra-violets, pour former des
pentachlorocyclohexènes (PCCHS) et des tetrachlorocyclohexènes
(TCCHs). Lorsque le lindane se dégrade en sol humide ou inondé ou en
plein champ, sa demi-vie peut aller de quelques jours à trois ans,
selon le type de sol, le climat, la profondeur à laquelle il se
trouve, etc. Dans les sols consacrés aux cultures que l'on trouve
habituellement en Europe, sa demi-vie est de 40 à 70 jours. Les sols
non-stériles permettent une biodégradation plus rapide du lindane
que les sols stériles. Les conditions anaérobies sont les plus
propices à sa métabolisation microbienne. Le lindane présent dans
l'eau se dégrade sous l'action des microorganismes contenus dans les
sédiments, pour former les mêmes produits.
Le lindane et les gamma-PCCHs sont fixés en quantité limitée
par les plantes où ils subissent une translocation, surtout lorsque
les sols contiennent une forte proportion de matières organiques. On
trouve les résidus essentiellement dans les racines des plantes; la
translocation ne s'effectue que peu - ou pas du tout - dans les
tiges, les feuilles ou les fruits. La bioconcentration est rapide
chez les microorganismes, les invertébrés, les poissons, les oiseaux
et l'homme, mais la biotransformation et l'élimination le sont
également lorsque l'exposition est interrompue. Les organismes
aquatiques le fixent en plus grande quantité à partir de l'eau qu'à
partir de la nourriture. Les facteurs de bioconcentration dans les
organismes aquatiques vont de 10 à 6000 en laboratoire, et de 10 à
2600 sur le terrain.
1.3 Concentration dans l'environnement et exposition humaine
On a trouvé du lindane en suspension dans l'air au-dessus des
océans à des niveaux de concentration de 0,039-0,68 ng/m3; son
niveau de concentration a atteint 1 ng/m3 dans l'air de certains
pays. Les concentrations dans les eaux de surface de nombreux pays
européens étaient la plupart du temps inférieures à 0,1 µg/litre.
Dans le cas du Rhin et de ses affluents, le niveau de concentration
variait entre 0,01 et 0,4 µg/litre de 1969 à 1974; après 1974, il
était inférieur à 0,1 µg/litre. Dans l'eau de mer, des
concentrations de 0,001-0,002 µg/litre ont été enregistrées. Les
concentrations de lindane dans le sol sont généralement faibles -
l'ordre de 0,001-0,01 mg/kg, sauf dans les zones de décharge.
On a découvert du gamma-HCH chez les poissons et les crustacés,
à des niveaux de concentration allant de 'indécelable' à 2,5 mg/kg
(calculés par rapport aux matières grasses) selon qu'ils vivent en
eau douce ou dans l'eau de mer et en fonction de leur teneur - faible
ou élevée - en matières grasses. Des valeurs d'environ 330 et 440
µg/kg (poids humide) ont été trouvées dans le tissu adipeux des ours
polaires respectivement en 1982 et 1984. La concentration de lindane
dans le foie des oiseaux prédateurs variait entre 0,01 et 0,1 mg/kg.
En 1972-73, on a mesuré dans des oeufs d'épervier, en République
fédérale d'Allemagne, des valeurs allant de 0,6 mg/kg à 11,1 mg/kg
(calculées par rapport aux matières grasses).
Les niveaux de concentration de lindane dans l'eau potable sont
généralement inférieurs à 0,001 µg/litre, et dans les pays
industrialisés, c'est dans la nourriture que se trouve 90% du
lindane absorbé par l'homme. Au cours des 25 dernières années, on a
analysé dans bon nombre de pays certains produits alimentaires à la
recherche de leur teneur en lindane: dans les céréales, les fruits,
les légumes, les légumes à gousse, les huiles végétales, les
concentrations allaient de 'indécelable' à 5 mg/kg de produit. En ce
qui concerne le lait, les matières grasses animales, la viande et
les oeufs, les concentrations allaient de 'indécelable' à 5,1 mg/kg
de produit (calculés par rapport aux matières grasses). On a
rarement trouvé des concentrations plus élevées. Les niveaux de
concentration dans le poisson étaient généralement très inférieurs à
0,05 mg/kg de produit (calculés par rapport aux matières grasses).
Des études de ration totale et de panier de la ménagère pour estimer
l'apport quotidien de lindane chez l'homme, ont révélé d'importantes
variations en fonction des époques: vers 1970, l'apport quotidien
était égal ou inférieur à 0,05 µg/kg de poids corporel; en 1980, par
contre, cet apport a diminué pour n'être plus qu'égal ou inférieur à
0,003 µg/kg. Aux USA, l'apport journalier de gamma-HCH entre 1976 et
1979 a diminué, passant de 0,005 à 0,001 µg/kg pour les tout-petits
et de 0,01 à 0,006 µg/kg pour les enfants.
Dans un certain nombre de pays, on a déterminé la concentration
en lindane dans des tissus d'individus appartenant à la population
générale. Aux Pays-Bas, la teneur du sang était de l'ordre de
< 0,1-0,2 µg/litre, mais des concentrations bien supérieures ont été
trouvées dans le sang d'individus vivant dans des pays utilisant du
HCH technique. Dans différents pays, on a mesuré des niveaux moyens
de concentrations dans le tissu adipeux de l'homme allant de < 0,0
1 à 0,2 mg/kg (calculés par rapport aux matières grasses). Dans le
lait humain, les concentrations de lindane sont généralement plutôt
faibles, avec des moyennes allant de moins de 0,001 à 0,1 mg/kg,
rapportées aux matières grasses; toutefois, on a constaté que ces
valeurs s'abaissaient à la longue.
Ainsi, le lindane est présent partout dans le monde; on le
retrouve dans l'air, l'eau, le sol, les sédiments, les organismes
aquatiques et terrestres, ainsi que dans la nourriture, encore que
les concentrations dans ces compartiments du milieu soient
généralement faibles et diminuent peu à peu. L'homme est exposé
quotidiennement, par les aliments qu'il consomme; c'est ainsi que
l'on peut trouver du lindane dans le sang, les tissus adipeux, et
dans le lait de femme; toutefois, les doses ingérées sont également
en diminution.
1.4 Cinétique et métabolisme
Chez les rats, le lindane est absorbé rapidement au niveau des
voies digestives et se répartit dans l'ensemble des organes et
tissus en quelques heures. C'est dans les tissus adipeux et la peau
que l'on trouve les niveaux de concentration les plus élevés;
diverses études ont fait état d'un rapport teneur des
graisses/teneur du sang égal à 150-200, d'un rapport foie/sang de
5,3-9,6, et d'un rapport cerveau/sang de 4-6,5. Un rapport
graisses/sang identique a été trouvé chez des rats ayant inhalé du
lindane. Ces rapports, qui varient en fonction du sexe, sont
supérieurs chez les femelles. Au niveau de la peau, la résorption
s'effectue très lentement et dans une faible proportion, ce qui
explique la faible toxicité du lindane après exposition cutanée.
Le lindane est métabolisé essentiellement dans le foie selon
quatre réactions enzymatiques: la déshydrogénation en gamma-HCH, la
déshydrochloration en gamma-PCCH, la déchloration en gamma-TCCH et
l'hydroxylation en hexachlorocyclohexanol. Les produits finals après
biotransformation sont des dérivés di-, tri-, tetra-, penta-, et
hexachlorés. Ces métabolites sont excrétés essentiellement dans les
urines sous forme libre ou conjugués à l'acide glucuronique, à
l'acide sulfurique ou à la N-acétylcystéine. Le processus
d'élimination est relativement rapide, avec des demi-vies de 3-4
jours chez le rat. Les bactéries et les champignons métabolisent le
lindane en TCCH et en PCCH. La vitesse de la transformation
métabolique dans les plantes est faible, la voie de dégradation
principale passant par le PCCH pour aboutir au tetrachlorophenol, à
des conjugués avec le bêta-glucose et à d'autres composés inconnus.
Rien ne prouve que le lindane s'isomérise en alpha-HCH.
1.5 Effets sur les êtres vivants dans leur milieu naturel
Le lindane n'est pas très toxique pour les bactéries, les
algues, les protozoaires: la dose sans effet est généralement de
1 mg/litre. Son action sur les champignons est variable, avec des
doses sans effet variant entre 1 et 30 mg/litre, selon l'espèce. Il
est modérément toxique pour les invertébrés et les poissons, les
valeurs CL(E)50 pour ces organismes étant de 20-90 µg/litre. Les
études à court et à long terme portant sur trois espèces de poisson
ont révélé que la dose sans effet est de 9 µg/litre; des
concentrations de 2,1-23,4 µg/litre n'ont aucune conséquence sur la
reproduction. Les valeurs de la CL50 pour les crustacés d'eau
douce et marins varient entre 1 et 1100 µg/litre. Chez Daphnia
magna, il y a réduction, fonction de la dose, du taux de
reproduction; la dose sans effet se situe entre 11 et 19 µg/litre.
Une dose de 1 mg/litre n'a eu aucun effet néfaste sur la
reproduction des mollusques.
La DL50 pour les abeilles domestiques est de 0,56 µg/abeille.
Les DL50 orales aiguës pour un certain nombre d'espère
d'oiseaux se situent entre 100 et 1000 mg/kg de poids corporel. Des
études à courte terme sur les oiseaux ont permis d'établir que des
doses de 4-10 mg/kg de nourriture n'ont aucun effet, pas même sur la
qualité de la coquille des oeufs. On a néanmoins constaté une
moindre ponte chez les canes exposées à des doses de lindane allant
jusqu'à 20 mg/kg de poids corporel.
Des chauves-souris sont mortes dans les 17 jours qui ont suivi
leur exposition à des copeaux sur lesquels on avait appliqué du
lindane à la dose indiquée; les copeaux en contenaient initialement
10-866 mg/m2. On ne possède aucune donnée concernant les effets
sur les individus et les écosystèmes.
1.6 Effets sur les animaux de laboratoire et effets in vitro
La toxicité orale aiguë du lindane est modérée: la DL50 pour
les souris et les rats est de l'ordre de 60-250 mg/kg de poids
corporel, selon le véhicule utilisé. La LD50 dermique pour les
rats est d'environ 900 mg/kg de poids corporel. La toxicité est
révélée par des signes d'excitation au niveau du système nerveux
central.
Le lindane ne provoque aucune irritation ou sensibilisation de
la peau; il est légèrement irritant pour les yeux.
Une étude de 90 jours sur des rats a permis d'établir à 10
mg/kg de nourriture (soit 0,5 mg/kg de poids corporel) la dose sans
effet. A 50 et 250 mg/kg de nourriture, il y avait augmentation du
poids du foie, des reins et de la thyroïde; à 250 mg/kg de
nourriture, on constatait une augmentation de l'activité enzymatique
du foie. Cette augmentation de l'activité enzymatique accélère la
dégradation du lindane et autres dérivés. Une autre étude de 90
jours sur des rats a montré qu'une dose de 4 mg/kg de nourriture
(soit 0,2 mg/kg de poids corporel) pouvait être définie comme dose
sans effet nocif; on a observé que des concentrations égales ou
supérieures à 20 mg/kg de nourriture pouvaient avoir un effet
toxique au niveau des reins et du foie. Une étude toxicologique à
court-terme sur les souris n'a pas permis de définir la dose sans
effet.
On a constaté aucun effet toxique après administration de
lindane à des chiens, pendant 63 semaines, 15 mg/kg de nourriture
(soit 0,6 mg/kg de poids corporel). Une étude toxicologique menée
pendant 2 ans sur des chiens et au cours de laquelle un grand nombre
de paramètres ont été mesurés, a permis d'établir que
l'administration de doses égales ou supérieures à 50 mg/kg de
nourriture (soit 2 mg/kg de poids corporel) ne provoquait aucune
anomalie apparente liée au traitement. Chez les animaux auxquels on
avait administré des doses de 100 mg/kg de nourriture, on constatait
toutefois une hausse des niveaux de phosphatage alcaline; avec des
doses de 200 mg/kg, on observait des anomalies du tracé électro-
encéphalographique, indiquant une excitation neuronale aspécifique.
Chez des rats ayant inhalé du lindane à raison de 0,02-4,54
mg/m3, 6 heures par jour pendant 3 mois, la dose la plus forte a
entraîné une élévation des valeurs du cytochrome P450 hépatique; la
dose sans effet nocif a été fixée à 0,6 mg/m3. Lors de deux études
au long cours menées sur des rats de nombreuses années auparavant,
on a expérimenté des doses de 10-1600 mg/kg de nourriture. L'une de
ces études fixe à 50 mg/kg de nourriture la dose sans effet nocif. A
100 mg/kg de nourriture, le foie augmente, une hypertrophie
hépatocellulaire apparaît, ainsi qu'une dégénérescence graisseuse et
une nécrose. L'autre étude établit à 25 mg/kg de nourriture (soit
1,25 mg/kg de poids corporel) la dose sans effet; en doublant cette
dose, on note une hypertrophie hépatocellulaire et une
dégénérescence graisseuse.
Des recherches ont été entreprises pour vérifier les effets que
le lindane, après avoir été administré à des souris, des rats, des
chiens et des porcs par voie orale, sous-cutanée et
intrapéritonéale, pouvait avoir sur tous les aspects de la
reproduction (chez les rats, sur trois générations), et évaluer sa
toxicité pour l'embryon et sa tératogénicité. Le lindane administré
par voie orale et parentérale n'a eu aucun effet tératogène (les
côtes surnuméraires étant considérées comme des variations). Des
doses de 10 mg/kg de poids corporel et plus administrées oralement-
par gavage-se sont avérées toxiques pour le foetus et/ou la mère; on
considère qu'une dose de 5 mg/kg de poids corporel est sans effet
nocif. L'étude effectuée sur trois générations de rats, a montré que
des doses de lindane allant jusqu'à 100 mg/kg de nourriture
n'avaient d'influence ni sur la reproduction, ni sur la maturation;
avec 50 mg/kg de nourriture, des modifications morphologiques inter-
venaient au niveau du foie parmi la progéniture de la troisième
génération, preuve d'une induction enzymatique. La dose sans effet
était dans ce cas de 25 mg/kg (soit 1,25 mg/kg de poids corporel).
Dans une étude de 22 jours effectuée sur des rats, on a
déterminé que la dose sans effet neurotoxique était de 2,5 mg/kg de
poids corporel.
Des études bien conçues ont été effectuées pour déterminer la
mutagénicité du lindane. Selon les résultats des recherches très
larges entreprises, le lindane ne peut en aucun cas provoquer des
mutations génétiques chez les bactéries ni dans des cellules
mammaliennes; il n'entraîne pas non plus, chez Drosophila
melanogaster, des mutations récessives liées au sexe. D'autres
expériences - in vitro et in vivo - ont montré qu'en outre le
lindane ne provoque ni anomalies chromosomiques, ni échange de
chromatides soeurs dans des cellules mammaliennes. La recherche de
lésions de l'ADN bactérien et de liaisons covalentes avec l'ADN dans
le foie de rats et de souris in vivo, après administration par
voie orale, a également donné des résultats négatifs. Les rares
études où des résultats positifs ont été obtenus péchaient soit par
une mauvaise conception d'ensemble, soit par le fait que la pureté
du composé étudié n'avait pas été précisée. Quoi qu'il en soit, on
peut dire que le lindane n'a globalement aucun pouvoir mutagène.
Des études de cancérogénécité ont été effectuées chez la souris
et le rat, avec des doses allant respectivement jusqu'à 600 mg/kg de
nourriture et 1600 mg/kg de nourriture. Aux doses égales ou
supérieures à 160 mg/kg de nourriture, on a observé des nodules
hyperplasiques et/ou des adénomes hépatocellulaires chez les souris;
lors de certaines études, les doses administrées ont dépassé le
maximum toléré. Deux expériences ont montré qu'aucune élévation de
l'incidence des tumeurs n'intervient lorsque l'on donne à des souris
et à des rats des doses allant respectivement jusqu'à 160 et 640
mg/kg de nourriture.
Les résultats d'études sur l'initiation-promotion de la
cancérogénicité, sur le mode d'action du lindane et sur sa
mutagénicité indiquent que la réponse tumorigène observée avec le
gamma-HCH chez la souris est sous la dépendance d'un mécanisme non
génétique.
1.7 Effets sur l'homme
Le lindane a été à l'origine de plusieurs cas d'intoxications
mortelles et non mortelles; il s'agissait soit d'accidents, soit
d'absorption délibérée (suicide), soit d'une simple négligence
(absence de précautions) ou d'utilisation impropre de produits
médicaux contenant du lindane. Les symptômes consistaient en
nausées, agitation, maux de tête, vomissements, tremblements,
ataxie, convulsions toniques-cloniques et/ou modifications du tracé
électroencéphalographique. Ces effets sont réversibles après
interruption de l'exposition ou traitement symptomatique.
Depuis 40 ans, l'utilisation du lindane est très répandue;
pourtant on cite peu de cas d'intoxications survenant dans le
contexte professionnel. Chez les individus qui travaillent à la
fabrication du lindane ou à son épandage, donc soumis à une
exposition prolongée, on a seulement constaté une augmentation de
l'activité des enzymes métabolisantes au niveau du foie. Rien ne
prouve l'existence d'une quelconque relation - évoquée dans
certaines publications - entre exposition au lindane et apparition
d'anomalies hématologiques. Quelques études de toxicologie aiguë ou
à courte terme chez l'homme indiquent qu'une dose d'environ 1,0
mg/kg de poids corporel ne provoque pas d'intoxication; en revanche,
à la dose de 15-17 mg/kg de poids corporel, apparaissent de graves
symptômes d'intoxication.
Appliqué sur la peau, le lindane est absorbé à hauteur
d'environ 10%; la proportion est plus élevée s'il y a lésions.
2. Conclusions
2.1 Population générale
Le lindane circule dans l'environnement et il est présent dans
les chaînes alimentaires; de ce fait, l'homme ne peut échapper à
l'exposition. Toutefois, l'apport alimentaire quotidien et
l'exposition totale de la population dans son ensemble diminuent peu
à peu, et, nettement inférieurs à la dose journalière admissible
(DJA) conseillés, ne constituent pas une menace sérieuse pour la
santé publique.
2.2 Groupes de population particulièrement exposés
La présence de lindane dans le lait maternel expose les enfants
nourris au sein, à des doses généralement inférieures à la DJA, donc
non toxiques. Les niveaux d'exposition existants - que l'on les
souhaiterait tout de même inférieur - n'interdisent pas
l'allaitement au sein.
En ce qui concerne l'utilisation thérapeutique du lindane pour
traiter la gale et les poux de corps, il convient de se conformer
strictement aux doses prescrites.
2.3 Exposition professionnelle
Manipuler du lindane ne présente aucun danger, à condition de
prendre toutes les précautions indiquées pour éviter le plus
possible l'exposition.
2.4 Effets sur l'environnement
Les chauves-souris, qui s'accrochent au bois traité avec du
lindane aux doses indiquées, en subissent les effets toxiques.
Exception faite des résultats d'étude concernant les déversements
accidentels dans le milieu aquatique, rien ne permet d'affirmer que
la présence de lindane dans l'environnement constitue un danger
sérieux pour d'autres êtres vivants.
3. Recommandations
1. Afin de réduire au minimum la pollution de l'environnement par
d'autres isomères de HCH, il convient d'utiliser le lindane
(> 99% de gamma-HCH) au lieu du HCH technique.
2. Afin d'éviter la pollution de l'environnement, il faut adopter
des solutions adéquates pour se débarrasser des sous-produits
et des effluents provenant des usines de fabrication de
lindane.
3. Il faut prendre garde à ce que les déchets de lindane ne
polluent ni les sols, ni les eaux.
4. Il faut donner à ceux qui manipulent du lindane les indications
nécessaires sur les méthodes d'application et les précautions
d'utilisation.
5. Il faut effectuer des études de cancérogénécité au long cours,
qui soient conformes aux nonnes actuelles.
6. Il faut poursuivre la surveillance de la dose de lindane
quotidiennement absorbée par la population générale.
RESUMEN Y EVALUACIONES; CONCLUSIONES; RECOMENDACIONES
Resumen y evaluación
1.1 Propiedades generales
El hexaclorociclohexano (HCH) de calidad técnica está formado
por un 65-70% de alfa-HCH, un 7-10% de beta-HCH, un 14-15% de gamma-
HCH y aproximadamente un 10% de otros isómeros y compuestos. El
lindano contiene más del 99% de gamma-HCH. Es un compuesto sólido,
con baja presión de vapor y poco soluble en agua, pero muy soluble
en disolventes orgánicos, como la acetona, y en disolventes
aromáticos y clorados. El coeficiente de reparto n-octanol/agua
(log Poa) es de 3,2-3,7.
El lindano puede determinarse por separado de los demás
isómeros del HCH tras su extracción por reparto líquido/líquido,
cromatografía en columna y detección por cromatografía de gases con
captura de electrones. Como estos métodos analíticos son sumamente
sensibles, es posible identificar residuos de lindano del orden de
nanogramos por kilogramo o por litro.
El lindano lleva utilizándose desde el comienzo los años 50
como insecticida de amplio espectro con fines agrícolas y de otro
tipo, de los que cabe mencionar el tratamiento de semillas y de
suelos, las aplicaciones en árboles, madera y materiales
almacenados, el tratamiento de animales contra los ectoparásitos y
en la salud pública.
1.2 Transporte, distribución y transformación en el medio ambiente
En los suelos con un alto contenido de materia orgánica se
observa una intensa adsorción del lindano; además, puede penetrar en
el suelo con el agua de la lluvia o del riego artificial. La
volatilización parece ser una importante vía de dispersión en las
elevadas temperaturas de las regiones tropicales.
El lindano experimenta una rápida degradación (descloración)
por acción de los rayos ultravioleta, formando
pentaclorociclohexenos (PCCH) y tetraclorociclohexenos (TCCH).
Cuando el lindano se descompone en el medio ambiente en condiciones
de humedad o inmersión y en condiciones de campo, su semivida varía
de unos días a tres años, en función del tipo de suelo, del clima,
de la profundidad a la que se haya aplicado y de otros factores. En
los suelos agrícolas normales en Europa su semivida es de 40 a 70
días. La biodegradación del lindano es mucho más rápida en suelos no
esterilizados que en los esterilizados. Las condiciones anaerobias
son las más favorables para su metabolización microbiana. El lindano
presente en el agua es degradado principalmente por microorganismos
de los sedimentos para formar los mismos productos de degradación.
Las plantas absorben y translocan en su interior cantidades
limitadas de lindano y de gamma-PCCH, especialmente en suelos con un
elevado contenido de materia orgánica. Los residuos se depositan
sobre todo en las raíces de las plantas, y son pocos o ninguno los
que se desplazan a las ramas, las hojas o los frutos. En los
microorganismos, los invertebrados, los peces, las aves y el hombre
tiene lugar una bioconcentración rápida, pero cuando se interrumpe
la exposición se biotransforman y eliminan en un tiempo
relativamente breve. En los organismos acuáticos es más importante
su absorción a partir del agua que de los alimentos. Los factores de
bioconcentración de estos organismos en condiciones de laboratorio
variaron desde un valor aproximado de 10 hasta 6000; en condiciones
de campo oscilaron entre 10 y 2600.
1.3 Niveles medioambientales y exposición humana
En el aire oceánico se han encontrado concentraciones de
lindano de 0,039-0,68 ng/m3, y en el aire de algunos países se han
medido cantidades de hasta 11 ng/m3. Las concentraciones estimadas
en aguas de superficie de varios países europeos fueron en general
inferiores a 0,1 µ/litro. Su concentración en el río Rin y sus
afluentes en el período 1969-74 osciló entre 0,01 y 0,4 µg/litro;
después de 1974 se mantuvo por debajo de 0,1 µg/litro. En el agua
marina se han detectado niveles de 0,001-0,02 µg/litro. Las
concentraciones de lindano en el suelo son por lo general bajas, del
orden de 0,001-0,01 mg/kg, excepto en zonas de vertido de basuras.
En pescados y mariscos se han detectado concentraciones de
gamma-HCH que oscilan entre valores no detectables y 2,5 mg/kg
(valores referidos a las grasas), dependiendo de que vivan en agua
dulce o agua marina y de que su contenido en grasa sea alto o bajo.
En el tejido adiposo de los osos polares se encontraron en 1982 y
1984 niveles aproximados de 330 y 440 µg/kg (peso húmedo)
respectivamente. La concentración de lindano en el hígado de aves
predadoras oscilaba entre 0,01 y 0,1 mg/kg. Los huevos de gavilán
recogidos en 1972-73 en la República Federal de Alemania contenían
entre 0,6 y 11,1 mg/kg (cálculo referido a las grasas).
Las concentraciones de lindano en el agua potable generalmente
son inferiores a 0,001 µg/litro; en los países industrializados más
del 90% de la ingestión humana de lindano procede de los alimentos.
En los últimos 25 años se ha analizado el contenido de lindano de
determinados productos alimenticios de un gran número de países. Las
concentraciones halladas en cereales, frutas, hortalizas, legumbres
y aceites vegetales variaron entre valores no detectables y 5 mg/kg
de producto, y en la leche, las grasas, la carne y los huevos, entre
valores no detectables y 5,1 mg/kg (referido a las grasas). Sólo en
unos pocos casos se detectaron concentraciones más altas. Sus
niveles en el pescado eran, en general, muy inferiores a 0,05 mg/kg
de producto (referido a las grasas).
En estudios sobre dieta total y cesta de la compra para estimar
la ingestión humana diaria de lindano, se observó una clara
diferencia con el paso del tiempo: la ingestión en el período de
alrededor de 1970 llegaba a 0,05 µg/kg de peso corporal al día,
mientras que en 1980 esta cifra había descendido a 0,003 µg/kg de
peso corporal al día o menos. En los Estados Unidos, la ingestión de
gamma-HCH entre 1976 y 1979 disminuyó de 0,005 a 0,001 µg/kg de peso
corporal al día en los lactantes y de 0,01 a 0,006 µg/kg de peso
corporal al día en los niños de corta edad.
En algunos países se ha determinado el contenido de lindano en
los tejidos corporales de la población general. En los Países Bajos,
el contenido en la sangre era del orden de < 0,1-0, µg/litro, pero
se hallaron concentraciones mucho más altas en varios países en los
que se utilizaba HCH de calidad técnica. Las concentraciones medias
en el tejido adiposo humano en distintos países varió entre < 0,01
y 0,2 mg/kg (referido a las grasas). La concentración de lindano en
la leche humana suele ser bastante baja, con unos niveles medios que
van desde < 0,001 hasta 0,1 mg/kilo (referido a las grasas); sin
embargo, se ha producido una disminución manifiesta con el tiempo.
Así pues, el lindano se halla distribuido por todo el mundo y
se puede detectar en el aire, el agua, el suelo, los sedimentos, los
organismos acuáticos y terrestres y los alimentos, aunque las
concentraciones en estos distintos compartimentos ambientales son en
general bajas y están decreciendo progresivamente. El hombre está
expuesto a diario por conducto de los alimentos, habiéndose
detectado lindano en los tejidos sanguíneo y adiposo y en la leche
materna; sin embargo, los niveles de ingestión también están
disminuyendo.
1.4 Cinética y metabolismo
En las ratas, el lindano se absorbe rápidamente de¡ tracto
gastrointestinal y en unas horas se distribuye por todos los órganos
y tejidos. Las concentraciones más elevadas se dan en el tejido
adiposo y en la piel; en varios estudios, el cociente grasa:sangre
era de alrededor de 150-200, el cociente hígado:sangre, 5,3-9,6 y el
cociente cerebro:sangre, 4-6,5. El mismo cociente grasa:sangre se
encontró en ratas expuestas por inhalación. Estos cocientes varían
en función del sexo, siendo más elevados en las hembras. La
absorción por la piel tras la aplicación cutánea de lindano es lenta
y muy limitada; esto puede explicar la baja toxicidad del lindano
después de la exposición cutánea.
El lindano se metaboliza sobre todo en el hígado mediante
cuatro reacciones enzimáticas: deshidrogenación a gamma-HCH,
deshidrocloración a gamma-PCCH, descloración a gamma-TCCH e
hidroxilación a hexaclorociclohexanol. Los productos finales de la
biotransformación son compuestos di-, tri-, tetra-, penta- y
hexaclorados. Estos metabolitos se excretan fundamentalmente por la
orina, en forma libre o conjugada con ácido glucurónico, ácido
sulfúrico o N-acetilcisteína. La eliminación es relativamente
rápida, con una semivida en ratas de 3 a 4 días. Las bacterias y los
hongos metabolizan el lindano a TCCH y PCCH. La velocidad de
transformación metabólica en las plantas es baja, y la vía de
degradación más importante es a través del PCCH a tri- y
tetraclorofenol y productos conjugados con beta-glucosa y otros
compuestos desconocidos. No existen pruebas de la isomerización del
lindano a alfa HCH.
1.5 Efectos en los seres vivos del medio ambiente
El lindano no es muy tóxico para las bacterias, las algas ni
los protozoos: el nivel carente de efecto fue en general de 1
mg/litro. Su acción sobre los hongos es variable; los niveles sin
observación de efectos fueron de 1 a 30 mg/litro, según las
especies. Es moderadamente tóxico para los invertebrados y los
peces, siendo los valores de la C(E)L50 para esos organismos de
20-9 µg/litro. En estudios de corta y larga duración con tres
especies de peces, el nivel sin observación de efectos fue de 9
µg/litro; no se observaron efectos en la reproducción con niveles de
2,1-23,4 µg/litro. Los valores de la CL50 para crustáceos
dulceacuícolas y marinos variaron entre 1 y 1100 µg/litro. La
inhibición de la reproducción de Daphnia magna dependía de la
dosis; el nivel sin observación de efectos fue del orden de 11-19
µg/litro. No se observaron efectos adversos en la reproducción de
moluscos con dosis de 1 mg/litro.
La DL50 para la abeja de la miel fue de 0,56 µg/abeja.
Los valores de la DL50 aguda por vía oral para varias
especies de aves fueron de 100 a 1000 mg/kg de peso corporal. En
estudios de corta duración con aves, las dosis de 4-10 mg/kg en la
dieta no tuvieron efecto, ni siquiera sobre la calidad de la cáscara
de los huevos. Sin embargo, en patas ponedoras tratadas con dosis de
lindano de hasta 20 mg/kg de peso corporal disminuyó la producción
de huevos.
Todos los murciélagos expuestos a virutas de madera con un
contenido inicial de lindano de 10-866 mg/M2, resultado de la
aplicación de la dosis recomendada, murieron en un plazo de 17 días.
No se obtuvieron datos acerca de los efectos en poblaciones y
ecosistemas.
1.6 Efectos en los animales de experimentación e in vitro
La toxicidad aguda por via oral del lindano es moderada: la
DL50 para ratones y ratas oscila entre 60 y 250 mg/kg de peso
corporal, en función del vehículo utilizado. La DL50 por vía
cutánea en ratas es de aproximadamente 900 mg/kg de peso corporal.
La toxicidad se manifestó en forma de estimulación del sistema
nervioso central.
El lindano no irrita ni sensibiliza la piel; es ligeramente
irritante para los ojos.
En un estudio de 90 días en ratas, la concentración máxima sin
efecto fue de 10 mg/kg alimento (equivalente a 0,5 mg/kg de peso
corporal). Con niveles de 50 y 250 mg/kg de alimento aumentaron los
pesos del hígado, los riñones y el tiroides; con 250 mg/kg de
alimento, se observó un aumento en la actividad enzimática del
hígado. Este aumento acelera la degradación del lindano y de otros
compuestos. En otro estudio de 90 días en ratas, se consideró que el
nivel máximo sin efectos adversos era de 4 mg/kg de alimento
(equivalente a 0,2 mg/kg de peso corporal); se observó toxicidad
renal y hepática a concentraciones de 20 mg/kg y superiores. Un
estudio de toxicidad de corta duración en ratones se consideró
insuficiente para establecer la concentración sin efectos.
La administración de lindano a perros en dosis de 15 mg/kg de
alimento (equivalentes a 0,6 mg/kg de peso corporal) durante 63
semanas no tuvo efectos tóxicos. En un estudio de dos años de
duración sobre la toxicidad de este compuesto en perros, en el que
se midió un gran número de parámetros, no se observaron anomalías
relacionadas con el tratamiento con dosis de 50 mg/kg de alimento
(equivalentes a 2 mg/kg de peso corporal) e inferiores. Sin embargo,
en el grupo que recibió 100 mg/kg de alimento aumentó el nivel de
fosfatasa alcalina; y con 200 mg/kg de alimento aparecieron
anomalías electroencefalográficas indicativas de irritación neurona]
inespecífico.
En ratas expuestas por vía respiratoria a concentraciones de
lindano de 0,02-4,54 mg/m3, 6 horas al día durante 3 meses, la
dosis más alta indujo un incremento de los valores del citocromo
P450 hepático; el nivel sin observación de efectos adversos fue de
0,6 mg/m33. En dos estudios de larga duración en ratas, realizados
hace muchos años, se ensayaron dosis de 10-1600 mg/kg de alimentos.
En uno de estos estudios se determinó un nivel sin observación de
efectos adversos de 50 mg/kg de alimento (equivalente a 2,5 mg/kg de
peso corporal). Con 100 mg/kg de alimento se producía un aumento del
peso del hígado, hipertrofia hepatocelular, degeneración grasas y
necrosis. En el otro estudio, la dosis de 25 mg/kg de alimento
(equivalente a 1,25 mg/kg de peso corporal) no tenía efectos, pero
con 50 mg/kg de alimento se observaron signos de hipertrofia
hepatocelular y degeneración grasas.
Se han investigado los efectos del lindano en todos los
aspectos de la reproducción (en tres generaciones de ratas), y su
embriotoxicidad y teratogenia tras la administración oral,
subcutánea e intraperitoneal en ratones, ratas, perros y cerdos. No
se observaron efectos teratogénicos tras la administración oral o
parenteral (las costillas supernumerarias se consideraron
variaciones). Se pusieron de manifiesto fetoxicidad y/o efectos
tóxicos matemos con dosis de 10 mg/kg de peso corporal y superiores
administradas mediante sonda oral; se considera que el nivel sin
efectos adversos es de 5 mg/kg de peso corporal. En el estudio de
tres generaciones de ratas con dosis de hasta 100 mg/kg de alimentos
el lindano no ejerció efecto alguno en la reproducción ni la
maduración, pero con 50 mg/kg de alimento se produjeron cambios
morfológicos en el hígado, que demostraban la inducción enzimática
registrada en la descendencia de la tercera generación. El nivel sin
observación de efectos en este ensayo fue de 25 mg/kg de alimento
(equivalente a 1,25 mg/kg peso corporal).
En un estudio de 22 días en ratas se observó que la dosis sin
efecto neurotóxico era de 2,5 mg/kg de peso corporal.
Se han hecho estudios suficientes sobre la mutagenicidad del
lindano. En las amplias investigaciones realizadas sobre su
capacidad para inducir mutaciones génicas en bacterias y células de
mamiferos y para provocar mutaciones letales recesivas ligadas al
sexo en Drosophila melanogaster, se obtuvieron siempre resultados
negativos. El lindano también dio resultados negativos en los
ensayos in vitro e in vivo realizados con células de mamíferos
sobre lesiones cromosómicas e intercambio de cromátidas hermanas.
Tambien fueron negativos los resultados de los ensayos para
determinar las lesiones en el ADN de bacterias y los de las pruebas
in vivo para observar la formación de enlaces covalentes con el
ADN de hepatocitos de ratones y ratas tras su administración oral.
En los escasos ensayos en los que se obtuvieron resultados
positivos, el sistema de estudio no era adecuado o no se informó
sobre la pureza del compuesto ensayado. Sin embargo, en conjunto, el
lindano no parece tener potencial mutagénico.
Se han llevado a cabo estudios en ratones y ratas para
determinar el potencial carcinogénico del lindano con dosis de hasta
600 mg/kg de alimento en ratones y de hasta 1600 mg/kg de alimento
en ratas. En ratones que recibieron dosis de 160 mg/kg de alimento o
superiores se observaron nódulos hiperplásicos y/o adenomas
hepatocelulares; en algunos estudios, las dosis utilizadas superaban
la máxima tolerada. En dos estudios en ratones con dosis de hasta
160 mg/kg de alimentos como máximo y uno en ratas con 640 mg/kg de
alimentos no se vio ningún aumento en la incidencia de tumores.
Los resultados de los estudios sobre la iniciación y el
estímulo de la carcinogenicidad, sobre el mecanismo de acción y
sobre la mutagenicidad ponen de manifiesto que en la respuesta
tumorigénica observada con el gamma-HCH en ratones interviene un
mecanismo no genético.
1.7 Efectos en el ser humano
Se ha informado de varios casos de envenenamiento mortal y de
enfermedad no mortal por lindano, producidos de manera accidental,
intencionada (suicidio) o por una grave negligencia en las
precauciones de seguridad o la utilización inadecuada de productos
médicos con lindano. Los síntomas son náuseas, agitación, dolor de
cabeza, vómitos, temblor, ataxia, convulsiones tónico-clónicas y/o
cambios en el trazado electroencefalográfico. Estos efectos eran
reversibles tras la interrupción de la exposición o el tratamiento
sintomático.
A pesar de su uso generalizado durante 40 años, se ha informado
de muy pocos casos de envenenamiento en el trabajo. En los
trabajadores expuestos durante largos períodos, en la fabricación o
la aplicación del lindano, el único síntoma observado fue una mayor
actividad de las enzimas hepáticas metabolizadoras de fármacos. No
hay pruebas de la relación, sugerida en algunas publicaciones, entre
la exposición al lindano y la aparición de anomalías hematológicas.
Algunos estudios de toxicidad aguda y de corta duración en la
especie humana indican que una dosis aproximada de 1,0 mg/kg de peso
corporal no produce envenenamiento; sin embargo, con una dosis de
15-17 mg/kg de peso corporal se observaron síntomas de intoxicación
grave.
Se absorbe alrededor del 10% de la dosis de aplicación cutánea,
aunque a través de la piel lesionada pasa mayor cantidad.
2. Conclusiones
2.1 Población general
El lindano circula en el medio ambiente y está presente en las
cadenas troficas, de manera que la especie humana seguirá estando
expuesta. Sin embargo, la ingestión diaria y la exposición total de
la población general están disminuyendo gradualmente; se encuentran
claramente por debajo de la ingestión diaria admisible y no
constituyen un problema para la salud pública.
2.2 Subpoblaciones especialmente expuestas
La presencia de lindano en la leche materna determina la
exposición de los lactantes a niveles que generalmente son
inferiores a la ingesta diaria admisible y que, por consiguiente, no
son un problema para la salud. Aunque sería preferible que los
niveles de exposición fueran inferiores, los actuales no representan
un factor limitante de la práctica de la lactancia natural.
Se deben seguir rigurosamente las prescripciones en relación
con el uso terapéutico de¡ lindano contra la sarna y los piojos.
2.3 Exposición profesional
El lindano se puede manejar sin riesgo siempre que se observen
las precauciones recomendadas para reducir al mínimo la exposición.
2.4 Efectos en el medio ambiente
El lindano es tóxico para los murciélagos que reposan en
estrecho contacto con madera tratada de acuerdo con las
recomendaciones para la aplicación. Si se exceptúan los resultados
obtenidos en los estudios sobre derrames en el medio acuático, no
hay pruebas que indiquen que la presencia de lindano en el medio
ambiente plantee un riesgo importante para las poblaciones de otros
organismos.
3. Recomendaciones
1. A fin de reducir al mínimo la contaminación del medio ambiente
por otros isómeros del HCH, se debe utilizar lindano (> 99% de
gamma-HCH) en lugar de HCH de calidad técnica.
2. Con objeto de evitar la contaminación del medio ambiente, los
subproductos y efluentes de la fabricación del lindano se deben
eliminar de manera adecuada.
3. En la eliminación de lindano, hay que tomar precauciones para
evitar la contaminación de las aguas naturales y del suelo.
4. Como en el caso de otros plaguicidas, las personas encargadas
del manejo del lindano deben recibir instrucciones adecuadas
acerca de la manera de aplicarlo.
5. Se deben realizar ensayos de carcinogenicidad de larga duración
diseñados con arreglo a las normas actuales.
6. Se debe seguir vigilando la ingestión diaria de lindano por
parte de la población general.
Unspecified fungi were also able to metabolize lindane,
although at a lower rate than bacteria. The following metabolites
were identified: gamma-PCCH; hexachlorobenzene; pentachlorobenzene;
TCCH; 1,2,3,4-, 1,2,3,5-, and 1,2,4,5- tetrachlorobenzene; 1,2,3-,
1,2,4-, and 1,3,5-trichlorobenzene; 1,2- and 1,4- dichlorobenzene;
pentachlorophenol; 2,3,4,5-, 2,3,4,6-, and
2,3,5,6-tetrachlorophenol; 2,3,4- and 2,4,6-trichlorophenol, and
carbon dioxide. Metabolic intermediates such as PCCH were found at
up to 1% of the initial dose of lindane. About 1% of the intial dose
was converted to carbon dioxide after an incubation period of 52
days (Engst et al., 1974; Kujawa et al., 1976; Engst et al., 1977).
6.6.2.1 Anaerobic conditions
The influence of growth conditions on the metabolic route of
lindane in bacteria was demonstrated in the facultative anaerobe E.
coli as well as with mixed populations of soil microorganisms
(Mathur & Saha, 1977; Vonk & Quirijns, 1979). These reports and
others demonstrate the predominant formation of gamma-TCCH under
anaerobic growth conditions. Anaerobic metabolism consists of a
series of dechlorinating steps, leading to rapid formation of
chlorine-free, volatile hydrocarbons and chloride (Haider & Jagnow,
1975; Jagnow et al., 1977). Carbon dioxide is not formed under
anaerobic conditions.
A possible metabolic pathway under anaerobic conditions was
proposed by Ohisa et al. (1980). Lindane is dechlorinated by a
cytochrome P-450-dependent reaction to TCCH, followed by
dechlorination to the unstable dichlorocyclohexadiene and
dehydrochlorination to monochlorobenzene. The degradation of lindane
serves as an energy source for the cells (Ohisa et al., 1982). A
relationship between the metabolization of lindane and the Stickland
reaction (a coupled oxidation-reduction reaction between pairs of
amino acids) has been discussed (Ohisa & Yamaguchi, 1979; Ohisa et
al., 1980, 1982). The ability to degrade lindane is linked to a
bacterial enzyme system that catalyses the evolution of hydrogen
during fermentation (Jagnow et al., 1977).
6.6.2.2 Aerobic conditions
Under aerobic conditions, the metabolism of lindane in bacteria
is initiated predominantly by dehydrochlorination to gamma-PCCH
(Vonk & Quirijns, 1979). Further intermediates are chlorobenzenes,
and the end-product is carbon dioxide. No phenolic intermediate was
observed under submerged conditions (Mathur & Saha, 1975).
6.7 Isomerization
The detection of beta-HCH in the tissues of rats fed gamma-HCH
led to the conclusion that isomerization of lindane had occurred
(Kamada, 1971); however, the purity of the gamma-HCH used was not
given in this report, and it is possible that impurities were
measured.
Studies in rats by Copeland & Chadwick (1979) and Eichler et
al. (1983) demonstrated that lindane did not undergo
bioisomerization. Gopalaswamy & Aiyar (1984) reported
biotransformation of gamma-HCH to hexachlorobenzene in male rats.
The results of a study by Chadwick & Copeland (1985), however, using
six young female Fischer 344 rats administered lindane in arachis
oil at 20 mg/kg body weight daily for six days (control animals
received the vehicle only), indicated that no significant
biotransformation of lindane to hexachlorobenzene occurred in these
animals. The gamma-HCH contents of adipose tissue on a fat basis
were 0.04 ± 0.003 mg/kg in controls and 129 ± 6 mg/kg in
lindane-treated rats.
The possibility of isomerization of lindane to alpha- and
beta-HCH was also investigated in mixed populations of soil
microorganisms and other defined bacterial strains.
Newland et al. (1969) studied the degradation of lindane in
simulated lake impoundments and found traces of alpha-HCH under
anaerobic incubation conditions. Benezet & Matsumura (1973)
described the formation of small amounts of alpha-HCH from lindane
incubated either with aquatic sediments or with suspension cultures
of Pseudomonas putida supplemented with NAD. Matsumura et al.
(1976) described an NAD-dependent pathway in P. putida leading to
the formation of alpha-HCH under anaerobic conditions. Three percent
of the initial radioactivity of 14C-lindane was found at a
location with the same Rf value as alpha-HCH after separation of the
metabolites by thin-layer chromatography.
Engst et al. (1979b) indicated isomerization of lindane to
alpha- and beta-HCH in anaerobically grown cultures of P.
aeruginosa, in a study in which metabolites were analysed by gas
chromatography. Lindane was not reported to be isomerized to alpha-
or beta-HCH in fungi (Engst et al., 1977).
Vonk & Quirijns (1979) found a conversion rate of lindane to
alpha-HCH of 0.2% after anaerobic incubation of lindane for 4 or 8
weeks with either sandy or silt loam soil samples and E. coli. No
alpha-HCH was formed in control experiments in which sterile
nutrient medium was incubated with gamma-HCH for 28 days. Growing
mycelium of Aspergillus niger produced no alpha-HCH. In this
study, metabolites were identified by gas-liquid chromatography and
verified by mass spectrometry.
Haider (1979, 1983) tested anaerobic, semi-anaerobic, and
aerobic incubation of radioactive labelled lindane with
Citrobacter, Serratia, Clostridium, Klebsiella, Pseudomonas, and
E. coli. Incubation did not increase the level of alpha-HCH. The
results of the experiment with Pseudomonas under anaerobic
conditions were not reported.
Deo et al. (1981) studied the interconversion of gamma-HCH in a
sterile aquatic solution over periods of 1 day up to 4 weeks.
Gas-liquid chromatography indicated a slow interconversion of
gamma-HCH with time. Solvent extracts were tested for their toxicity
by topical application onto 2-day-old Drosophila melanogaster. The
observed decrease in toxicity of the gamma-HCH solution with time
may have been due to both degradation and isomerization to less
toxic isomers, such as alpha-HCH.
Taken together, the results of tests conducted under anaerobic
conditions show that only a very small amount of lindane, if any, is
converted to alpha-HCH, and there is no conclusive indication of
isomerization to beta-HCH.
7. EFFECTS ON LABORATORY MAMMALS AND IN IN-VITRO TEST SYSTEMS
Lindane has been tested for acute and for short- and long-term
toxicity in a number of animal species. Some of the earlier studies
were undertaken using material of unspecified purity; and in some
others, technical-grade HCH was used that contained various
quantities of alpha- and beta-HCH, in addition to lindane. Those
studies that are relevant to this review have been included.
7.1 Single exposure
The acute toxicity of lindane has been investigated in numerous
studies in a variety of species and strains of laboratory animals
via several routes of application. The reported LD50 values for
lindane given by different routes of administration are of the same
order of magnitude in the various species, and no sex-dependent
difference was seen. A marked difference in acute oral toxicity
results from the type of vehicle used: oily solutions of lindane
were more toxic than suspensions in water, and when mineral oils
were used as carriers fewer toxic effects were seen than with
vegetable oils (Muralidhara et al., 1979). Young animals were
generally more sensitive than adults. Lindane was more toxic to
animals suffering from protein deficiency than to rats with a normal
protein supply (Chen, 1968).
7.1.1 Oral
The LD50 values for the mouse, rat, guinea-pig, and rabbit
are summarized in Table 8.
The choice of vehicle used for administering lindane in studies
of its acute oral toxicity is important: the LD50 after
administration in an oily solution or of an emulsifiable concentrate
was 88 mg/kg body weight, but that for wettable powders, granules,
flowable concentrations and aqueous suspensions was in the order of
170 mg/kg or even higher.
Single oral doses of 40 mg/kg body weight dissolved in oil were
lethal to dogs; dogs that received 30 mg/kg survived but had
convulsions (Barke, 1950). McNamara and Krop (1948) found that a
dose of 100 mg/kg body weight was lethal to all of three treated
dogs, and 50 mg/kg caused death in four out of seven animals. These
data indicate that the lethal dose for dogs of lindane administered
as an oily solution is about 40-50 mg/kg body weight.
Table 8. Reported oral LD50 values for g-HCH in experimental
animals
Species LD50(mg/kg) References
Rats (male, female, or 90-270 Slade (1945); Woodard &
males and females) Hagan (1947);
Riemshneider (1949);
Antonovic (1958); Gaines
(1960); Edson et al.
(1966); Muacevic (1966,
1970, 1971a,b); Chen
(1968); Schafer (1972);
Frohberg et al. (1972b)
Mice (different strains) 55-250 Woodard & Hagan (1947);
Graeve & Herrnring
(1951); Nurmatov (1965);
Frohberg et al. (1972a);
Paul et al. (1980);
Wolfe & Ralph (1980)
Guinea-pig 100 Cameron (1945)
Rabbit 90-200 Cameron (1945); Nurmatov
(1965)
In an incident in which eight cows ingested a powder containing
19.1% gamma-HCH, those that ate 112 g or more of the powder died,
while those given 70 g survived. These findings indicate that, for
cows, the fatal dose was between 70 and 112 g or 140-225 mg/kg body
weight of the powder (equivalent to 28-45 mg/kg body weight
(McParland et al., 1973).
7.1.2 Intraperitoneal and intramuscular
Mice of the NMRI-EMD strain (SPF) were administered lindane as
a 0.5% suspension in 0.5% carboxymethylcellulose solution and were
observed for 14 days. The intraperitoneal and intramuscular LD50
values were found to be 97 and 152 mg/kg body weight, respectively
(Frohberg et al., 1972a). In rats, only the intraperitoneal LD50
has been determined: it was found to be 69 mg/kg body weight in
Wistar-AF/HAN-EMD administered the compound in
carboxymethylcellulose (Frohberg et al., 1972b).
7.1.3 Inhalation
Wistar (HAN/Boe) rats were exposed by whole-body exposure to
lindane at (analytical) concentrations of 0, 273, or 603 mg/m3 for
4 h. The average particle size was 0.4 µm, and the animals were
observed for 14 days. Neither deaths nor abnormalities were found
(Oldiges et al., 1980). The
The 4-h acute LC50 for a lindane (99.6%) aerosol was determined
by exposing four groups of five males and five female KFM-HAN Wistar
rats by inhalation to aerosols containing lindane at 0.1, 0.38,
0.64, or 2.1 mg/litre; 50% or more of the particles had a diameter
of less than 7 µm. The observation time was 22 days. At toxic doses,
signs of neurotoxicity (curved body posture, paddling movements and
spasms) were observed. The acute 4-h LC50 was found to be about
1600 mg/m3 for animals of each sex (Ullman et al., 1986d).
7.1.4 Dermal
The acute dermal toxicity for rabbits was 200-300 mg/kg
(Medvedev, 1974; see International Register for Potentially Toxic
Chemicals, 1983).
Sherman strain rats were given one dermal application of
lindane (99%) dissolved in xylene, and no attempt was made to remove
the compound during the observation time of 14 days. The LD50 was
1000 mg/kg body weight for males and 900 mg/kg body weight for
females (Gaines, 1960).
Male New Zealand rabbits, both young adult (2-3 kg) and just
weaned (1 kg) were shaved only, shaved and depilated or shaved,
depilated and 'stripped', and a commercial preparation of 1% lindane
and 99% inert material was applied once to the entire body except
the head, limbs, and perineal surface at a dose of 6 ml/kg body
weight (equivalent to a dose of lindane of 60 mg/kg body weight, a
dose reportedly used in infants). The lindane was allowed to remain
on the skin during the experiment. Two of four adult rabbits that
were treated after having been shaved, depilated, and 'stripped'
exhibited excitement after about 24 h. Adult rabbits that had been
shaved only showed no effect. Weanling rabbits exhibited severe
anorexia and convulsions, and death occurred in some cases. The
effects were more pronounced in weanlings with inflamed or damaged
skin. The concentrations of lindane in whole blood of weanlings when
convulsions occurred (about 24 h after treatment) were 0.7-2.5 µg/ml
(Hanig et al., 1976).
7.2 Short-term exposure
7.2.1 Oral
7.2.1.1 Mouse
In young dd mice fed diets containing lindane at 0, 2, 4, or 10
mg/kg diet for three months, no effect on growth and no
histopathological change in the main organs were seen at any dose
level (Chen & Liang, 1956). Similarly, Kitamura et al. (1970) saw no
difference in behaviour, food consumption or body weight gain from
that in controls in ICR mice fed diets containing lindane at 0.1, 1,
10, and 100 mg/kg diet for 36 days. No histopathological examination
was carried out.
7.2.1.2 Rat
Short-term studies carried out by Slade (1945) and Laug (1948)
were more or less inadequate for an evaluation.
Doisy & Bocklage (1949, 1950) fed lindane-containing diets to
weanling rats for four weeks; doses of 400, 600, and 800 mg/kg diet
caused high mortality rates. Food intake and weight gain were
markedly reduced, especially in the group receiving 800 mg/kg diet.
The animals showed irritability, hyperactivity, and convulsions. A
dose of 200 mg/kg diet was without effect. Young rats were more
susceptible than adults.
In a three-month toxicity study, groups of 15 male and 15
female Wistar KFM-Han (outbred) SPF rats were fed diets containing
lindane (99.85%) at 0, 0.2, 0.8, 4, 20, or 100 mg/kg diet. After 12
weeks of treatment, most of the animals were sacrificed; the
remaining rats were placed on a control diet for six weeks and then
sacrificed. Lindane had no effect on mortality, food consumption,
haematological parameters, the results of urinalysis, or clinical
symptoms, although rats fed 100 mg/kg diet gained 8.4-14.9% less
weight than controls. Liver cytochrome P-450 levels were increased
in females given diets containing lindane at 0.8 mg/kg diet or more
and in high-dose males at the termination of dosing; these values
returned to the control levels during the recovery period. Such
increases in cytochrome P-450 activity are regarded as an adaptation
phenomenon due to induction of the microsomal detoxifying enzymes.
Slight, dose-related, reversible increases in absolute and/or
relative weights of livers and kidneys were observed in male and
female rats fed lindane at 20 or 100 mg/kg diet, and
histopathological examination revealed changes in these animals.
Those in the liver included dose-dependent, minimal-to-slight
centrilobular hepatocellular hypertrophy at the end of the
application period. After the recovery period, liver weights were
found to be normal, and no centrilobular hypertrophy was seen. In
the kidneys, minimal-to-slight, unicellular and multicellular
necrosis of epithelial cells was observed in proximal convoluted
tubules, and basophilic tubules, interstitial nephritis and hyaline
droplets were seen in epithelial cells of the convoluted tubules.
After the recovery period, the tubular degeneration was no longer
present, but the nephritis and basophilic tubules were still present
in the animals that had received 100 mg/kg. No effects were observed
with doses of 4 mg/kg diet (equivalent to 0.3 mg/kg body weight) and
below (Suter et al., 1983).
In a 12-week study with groups of 10 male and 10 female Wistar
RIV:TOX (C-S) rats, four weeks old at the beginning of the
experiment, lindane (99.8%) was administered in the diet at
concentrations of 0, 2, 10, 50, and 250 mg/kg. At the highest dose,
increases were seen in the induction of enzymes, such as
aminopyrine- N-demethylase and ethoxyresorufine- O-deethylase, but
cytochrome P-450 and aryl hydroxylase activity were not increased.
At the two highest dose levels, the weights of livers, kidneys, and
thyroid were increased. The no-effect level of lindane in this study
was 10 mg/kg diet (equivalent to 0.75 mg/kg body weight) (van Velsen
et al., 1984).
Young male Wistar rats were fed gamma-HCH at a dose of 0 (five
rats) or 800 mg/kg diet (eight rats) for two weeks, and urinary
excretion of body constituents that reflect renal function was
measured. Glucosuria and increased excretion of creatinine and urea
were found, and hypertrophy and degeneration of the renal tubular
epithelia were observed histologically (Srinivasan et al., 1984). In
young male Wistar rats administered gamma-HCH at 800 mg/kg diet for
two weeks, liver weights were increased, but no difference was found
in moisture, nitrogen, protein, or glycogen levels. The fat and DNA
content of the liver were found to be increased, but the DNA content
per unit issue was decreased. The predominant change in the liver
was hypertrophy. Testicular weight was no different from that in
control animals, but the protein content was higher, and the DNA
content was lower. The histological changes observed were tubular
atrophy and spermatogenic arrest; the interstitial space was found
to be oedematous (Srinivasan et al., 1988).
Liver function was studied in male Wistar rats fed a control
diet (6-8 rats) or gamma-HCH at 800 mg/kg diet (8-12 rats).
Gamma-HCH produced noticeable hepatocellular effects, as indicated
by increased activity of serum aminotransferases, hepatic
glucose-6-phosphate dehydrogenase and aldolase and decreased
activity of liver glucose-6-phosphatase. Liver mitochondrial
dinitrophenol/Mg++/Ca++-activated ATPase activity was decreased,
and levels of microsomal Na+, K+-ATPases were lower in treated
than control animals (Srinivasan & Radhakrishnamurty, 1988).
In a preliminary study, 24 Fischer-344 weanling, female rats,
received daily oral doses of either arachis oil or lindane at 0.069
mmol/kg body weight for 189 days. Lindane induced a significant
increase in body weight after 112 days of treatment. In a subsequent
dose-response study, female Fischer-344 rats, 21 days of age, were
gavaged daily with arachis oil (six rats) or lindane at 5 (six
rats), 10(eight rats), 20 (12 rats), or 40 mg/kg body weight (12
rats). At 20 mg/kg, lindane induced an increase in body weight after
10 weeks of treatment. At 40 mg/kg, 7 out of the 12 rats died; the
other animals had increased body weight gain. Greater food
consumption was observed, and obesity was induced, as indicated by
the Lee index. In addition, lindane caused delay in vaginal opening,
disrupted oestrous cycling, reduced pituitary and uterine weights
and elevated food consumption during pro-oestrous. This response
suggests that, by inducing alterations in the reproductive function
of female rats and by interfering with hormonal regulation of the
energy balance, lindane may be anti-oestrogenic rather than
oestrogenic as previously proposed (Chadwick et al., 1988).
7.2.1.3 Dog
Lehman (1952) found a high mortality rate in dogs given lindane
at daily doses of 10 or 15 mg/kg body weight on five days per week
over a period of 2 to 221 days. (No details available). Lehman
(1965) reported a study initially conducted by Fitzhugh et al., in
which dogs (two males and two females per group) were exposed to
lindane at 0 or 15 ppm (equivalent to approximately 0.6 mg/kg body
weight) in the diet for a total of 63 weeks. No effect was observed
on mortality, organ weights, body weight gain, haematological
parameters, or histological appearance.
During a two-year toxicity study, groups of four male and four
female beagle dogs were fed lindane (99%) at 0, 25, 50, or 100 mg/kg
diet. The amounts of lindane that were actually ingested were 0.83,
1.60, and 2.92 mg/kg body weight per day. Convulsions seen
occasionally in control and low-dose animals were not related to the
treatment. No treatment-related change was observed in body weight,
food or water consumption, ophthalmological parameters,
electroencephalographic traces, results of haematological
examinations, urinalysis, and liver function tests, or organ
weights. At autopsy, somewhat darker colouration and a brittle
consistency of the liver were seen at 100 mg/kg. In addition,
alkaline phosphatase activity was increased in the highest dose
group. No treatment-related abnormality was apparent with 25 or 50
mg/kg diet.
A supplementary group of four male and four female dogs was
administered lindane at 200 mg/kg diet for 32 weeks. High-voltage
slow-wave activity changes, possibly indicative of nonspecific
neuronal irritation, were recorded in electroencephalographic
tracings at this dose level. No such effect was observed in the
two-year study at 100 mg/kg diet (Rivett et al., 1978).
7.2.1.4 Pig
Schnell (1965) fed diets containing lindane (99.5%) at 0, 5,
10, 20, 40, or 80 mg/kg diet to groups of five pigs over a period of
nine months. No clinical symptom was seen in any animal during the
test period. Food and water intake remained normal, and
haematological investigations and histopathological examination of
the liver, spleen, kidneys, adrenals, heart, and brain revealed no
substance-related change, even at the highest dose level tested.
7.2.2 Inhalation
7.2.2.1 Mouse
Balaschow (1964) exposed mice for 6 h/day to a lindane aerosol
containing a nominal concentration of 1 mg/m3 for 2.5 months.
During the first two weeks, white blood cell counts showed the
presence of leukocytosis, with a shift to the left; from the end of
the first month, leukopenia with a shift to the right was observed,
and toxic granulations and vacuoles appeared in the nuclei and
cytoplasm of some leukocytes. Later, a reduced mitotic rate was
observed. The relationship between the different cell types in the
bone marrow was undisturbed.
Four groups of 45 male and 45 female CD-1 (Charles River) mice
were exposed by whole-body inhalation to aerosols (geometric mean
particle diameter, about 3 µm) containing lindane (purity at least
99.6%) at 0, 0.3, 1.0, or 5-10 g/m3 for 6 h/day, on five days per
week for 14 weeks. The test dose levels were selected on the basis
of a preliminary range-finding study, which showed that males did
not develop major signs of toxicity after five exposures for 6 h/day
to lindane at 1.0 and 10.0 mg/m3. During the main study, however,
an unexpectedly high mortality rate was seen in females exposed to
10 mg/m3; after the first five exposures, therefore, the
concentration for the high-dose group was lowered to 5 mg/m3.
Subgroups of 15 mice of each sex in each group were sacrificed after
7, 14, and 20 weeks. The group sacrificed at 20 weeks was a recovery
group, which was not exposed to lindane after exposure week 14. Ten
of the 15 mice in each subset were used for pathological evaluation,
and the remaining five were examined for serum lindane levels. The
lindane aerosol was highly toxic to female mice at 5 mg/m3, and
probably also at 1 mg/m3. The no-observed-effect level was
concluded to be 0.3 mg/m3 (Klonne & Kintigh, 1988).
7.2.2.2 Rat
Groups of 12 male and 12 female Wistar Han/Boe SPF rats were
exposed by whole-body inhalation to lindane (99.9%) at nominal
concentrations of 0, 0.02, 0.12, 0.6, or 4.5 mg/m3 (average
particle size, 0.92 µm) for 6 h/day for three months. The two groups
that received 0 or 4.54 mg/m3 were used to investigate recovery.
Slight diarrhoea and ruffled fur were observed temporarily in the
high-dose animals only. Measurements of body and organ weights and
of food and water intake, clinical chemistry, and histopathology
showed no treatment-related change. Hepatic cytochrome P-450 values
were increased at the end of the exposure period in animals at the
highest dose, but all values returned to that of the control during
the six-week recovery period. Increased kidney weights and cloudy
swelling of the tubular epithelium were observed especially in males
at the two highest dose levels, but, again, all values were
comparable with those in controls at the end of the recovery period.
The no-effect-level was probably 0.6 mg/m3 (Oldiges et al., 1983).
7.2.3 Dermal
Four groups of 49 male and 49 female Charles River rats (Crt:
(WI)BR strain) were exposed dermally to lindane (purity at least
99.5%) at dose levels of 0, 10, 60, or 400 mg/kg per day, selected
on the basis of a preliminary range-finding study, on five
consecutive days per week. The test substance was applied as a
suspension in aqueous carboxymethyl cellulose at a constant volume
of 4 ml/kg to a clipped area of the skin on the dorsal area and was
retained for 6 h with a dressing consisting of a gauze pad
heat-welded to plastic-backed aluminium foil. In the main phase of
the study, 23 animals of each sex in each group were treated for 13
weeks before sacrifice; one group of 13 animals/sex were sacrificed
after 6 weeks of treatment; a third (recovery phase) group,
consisting of 13 animals of each sex, was retained in the study for
an additional six weeks. At sacrifice, three animals in each group
were selected for determination of tissue levels of lindane (Brown,
1988).
The toxic effects induced by subchronic dermal exposure to 60
and 400 mg/kg consisted of pathological lesions of the kidneys in
males (increased organ weight, hyaline droplet formation, tubular
degeneration with necrosis, basophilic tubules, casts) and
hypertrophy of the liver in males and females. Whereas the effects
on the liver were reversible, some of the histopathological changes
in the kidneys persisted (tubular degeneration with necrosis,
granular casts) after the recovery period. Although there was
evidence of increased intensity of hyaline droplet formation at the
lowest dose tested (10 mg/kg per day), this effect was very slight;
that level could therefore be considered to be the
no-observed-effect level. The use in this study of semi-quantitative
methods for determination of blood levels, protein, and turbidity
makes it difficult to come to any definite conclusion; however, the
results of the urinalysis did not provide evidence that lindane
adversely affects kidney function.
7.3 Skin and eye irritation; sensitization
7.3.1 Primary skin irritation
Application of 0.5 g of lindane to the intact skin of New
Zealand white rabbits, in a study performed in compliance with the
guidelines of the Organization for Economic Co-operation and
Development and the US Environmental Protection Agency, did not
cause irritation (Ullmann et al., 1986a).
7.3.2 Primary eye irritation
Lindane placed in the conjunctival sac of the left eye of New
Zealand white rabbits at 0.1 g was slightly irritating (Ullmann et
al., 1986b).
7.3.3 Sensitization
The allergic potential of lindane was tested in a
Magnusson-Kligman maximization test (according to the guidelines of
the Organization for Economic Co-operation and Development) on
Durkin-Hartley albino guinea-pigs. Ten males and ten females
received lindane (99.6%) and five males and five females received
the vehicle, ethanol. No difference was seen between the test group
and the controls after the first and second challenge application 24
and 48 h later, and it was concluded that lindane has no skin
sensitizing (contact allergenic) potential in these guinea-pigs
(Ullmann et al., 1986b).
Ullmann et al. (1987a) conducted a further maximization test
(following the guidelines of the Organization for Economic
Co-operation and Development) with Dunkin-Hartley albino guinea-pigs
to test the contact hypersensitization potential of a lindane
formulation. Ten males and ten females received intradermal
injections of 5% 'Nexit fluessig' (containing 25.9% lindane) in
saline, and five males and five females received the saline vehicle.
No sensitization reaction was observed after the first and second
challenge application, 24 and 48 h later.
Comparable experiments were carried out with two other
formulations, 'Nexit stark', a powder containing 78.9% lindane
(Ullmann et al., 1987b), and 'Agronex Saatgutpuder', a powder
containing 20.1% (Ullmann et al., 1987c). Each was administered as
intradermal injections of 0.1% in saline. No sensitization reaction
was observed after two challenge reactions 24 and 48 h later.
7.4 Long-term exposure
7.4.1 Oral
In two long-term studies, lindane powder was mixed into the
diet of Wistar rats (10 males and 10 females) at 10, 100, or 800
mg/kg diet and as an oily solution at 5, 10, 50, 100, 400, 800, or
1600 mg/kg diet, either as the gamma isomer or as technical HCH
(containing only 13% of the gamma isomer). Two control groups were
used. At 100 mg/kg diet, liver weight was increased, and
histopathological examination revealed hepatocellular hypertrophy,
fatty degeneration and necrosis as well as nephritic reactions
(granular degeneration and calcification in male rats). These
findings were more pronounced at the 400, 800, and 1600 mg/kg
dietary levels. At these concentrations, the life span of animals in
groups treated with the oily solution was shortened by 20-40%. The
no-effect level in this experiment was 50 mg/kg diet (Fitzhugh et
al., 1950; Lehman, 1952).
Similar results were obtained in another lifetime study, in
which groups of 10 male and 10 female rats received lindane at 25,
50, or 100 mg/kg diet. The dose of 25 mg/kg had no effect on the
liver, but hepatocellular hypertrophy was observed with 50 mg/kg and
slight fatty liver-cell degeneration was described in the group
receiving 100 mg/kg diet (Truhaut, 1954).
7.4.2 Appraisal of acute and short- and long-term studies
The acute oral toxicity (LD50) of lindane in different
species, depending on the vehicle used, ranges from 56 to 480 mg/kg
body weight. Preparations in oil were more toxic than aqueous
solutions or suspensions. The ranges for rats and mice were similar
(88-270 and 56-246 mg/kg, respectively). The dermal LD50 for rats
is approximately 900 mg/kg body weight, but smaller amounts (60
mg/kg as a 1% cream) caused convulsions, anorexia, and deaths in
weanling rabbits. No skin irritation or sensitization was observed,
and eye irritation was slight.
Although older, long-term studies in the rat suggest a
no-observed-adverse-effect level of 25 mg/kg diet, contemporary
short-term studies in rats indicate that this level is10 mg/kg diet,
equivalent to 0.75 mg/kg body weight on the basis of increased
hepatic, renal, and thyroid weights, increased cytochrome P-450
activity and histopathological findings in liver and kidneys.
7.5 Reproduction, embryotoxicity, and teratogenicity
7.5.1 Reproduction
Trifonova et al. (1970) found no reduction in the fertilization
rate of female rats after oral treatment for 90 days with lindane at
approximately 5 mg/kg body weight. When the dose was doubled over a
test period of 138 days, the fertilization rate was reduced.
(Details not given.)
A three-generation test was carried out in which 10 male and 10
female CD-rats were administered lindane at concentrations of 25,
50, or 100 mg/kg diet continuously. The treatment had no influence
on fertility, litter size, breeding rate, weight of newborn animals,
lactation, malformation rate, or maturation. The liver weights of
young animals of the F3b generation were increased, especially
among females. Histopathological examination of the liver showed
enlarged hepatocytes and vacuolization in animals treated with 50
and 100 mg/kg diet (Palmer et al., 1978a).
7.5.2 Embryotoxicity and teratogenicity
7.5.2.1 Oral administration
Mouse: Lindane was administered orally to seven groups of 25
pregnant NMRI-EMD (SPF) female mice at 0, 12, 30, or 60 mg/kg body
weight in 0.5% carboxymethyl cellulose, on either days 6-15 or days
11-12 of pregnancy. In the group receiving the highest dose, fetal
mortality was increased and fetal weights were decreased. A slight,
non-dose-related increase in malformation rate was found in the
mid-dose group (4.2%, as compared to 1.9% in controls). At the
highest dose, increased maternal mortality (48%) and reduced body
weight gain were observed. The treatment had no effect on the number
of implantations per dam, the percentages of early and late
resorptions, the number of runts or the malformation rate (Frohberg
& Bauer, 1972b).
Rat: Groups of 20 female CFY-rats received lindane at 5, 10,
or 20 mg/kg body weight by gavage during days 6-15 of pregnancy. In
the groups given 10 and 20 mg/kg, maternal toxicity (reduced food
intake and reduced weight gain) was observed, and two female rats
given 20 mg/kg died. At the same dose, there was a dose-related
increase in the incidence of offspring with extra (14th) ribs, which
was statistically significant. Other anomalies and litter parameters
were comparable to those in the controls, and there was no evidence
of embryo- or fetoxicity (Palmer et al., 1978b).
Khera et al. (1979) gave female Wistar rats (20 animals per
group) a lindane formulation (50% in corn oil) at 3.12, 6.25, or
12.5 mg/kg body weight (expressed as 100% lindane) by intubation on
days 6-15 of gestation. No effect was seen on the number of living
fetuses per litter, the number of dead plus resorbed fetuses or mean
fetal weight at 22 days of gestation. No malformation other than the
usual range of developmental variants was observed in any group. A
slight increase in the frequency of anomalies of the ribs and
reduced cranial ossification were seen in the fetuses exposed to
6.25 mg/kg; these effects were confined to two litters and were
probably not dose-related.
Female rats that received lindane orally at a dose of 25 mg/kg
body weight daily during pregnancy had higher post-implantation
embryonal mortality than aontrols, and at 12.5 mg/kg, no mortality
was found. Neither dose level induced teratological abnormalities
(Mametkuliev, 1976; see International Register for Potentially Toxic
Chemicals, 1983).
Rabbit: Lindane was administered by intragastric intubation
to New Zealand white rabbits (13 animals per group) on days 6-18 of
gestation at doses of 5, 10, and 20 mg/kg body weight. All treated
animals showed slight tachypnoea and lethargy during the treatment
period, and body weight gain and food intake were reduced.
Pre-implantation loss was significantly higher in the group given 20
mg/kg, but, as treatment did not start until day 6 of gestation,
this effect is unlikely to have been due to lindane.
Post-implantation loss and the incidence of resorptions were
increased at 5 and 20 mg/kg. The number of offspring with extra
(13th) ribs was significantly lower in animals given 5 mg/kg and
significantly higher in rabbits at 20 mg/kg than in controls. Fetal
and litter weights were unaffected, and the incidence of other
anomalies was similar to that in controls (Palmer et al., 1978b).
Dog: An increased frequency of stillbirths, unrelated to
dosage or period of administration, was seen in beagle dogs fed
lindane at 0 (five dogs), 7.5 (13 dogs) or 15 mg/kg body weight (14
dogs) from day 1 or 5 throughout gestation. No significant
teratogenic effect was observed. The number of living pups was
similar in control and test groups (Earl et al., 1973).
Pig: Groups of six female pigs received lindane at 0, 50, or
500 mg/kg diet from 30 days prior to mating until day 30 of
gestation. No treatment-related effect was found on number of
embryos, embryo weight, or rate of ovulation (Duee et al., 1975).
Cow: In an accidental poisoning incident, four pregnant cows
received lindane at 13.4 g (28 mg/kg body weight) 6-17 weeks
pre-partum. All had convulsions and muscular tremors in the ensuing
48 h but recovered with veterinary treatment. All calved on time and
produced normal, healthy calves. Four non-pregnant cows died after
receiving 21 g of lindane, which suggests that the minimum lethal
dose is 28-45 mg/kg body weight (McParland et al., 1973).
7.5.2.2 Subcutaneous injection
Mouse: Lindane was administered subcutaneously (in a 0.5%
carboxymethyl cellulose solution) to groups of 25 pregnant NMRI
miceat 6 mg/kg body weight on either days 11-13 or days 6-15 of
pregnancy. Except for a slight increase in the frequency of runts in
the latter group, no effect was seen on the number of implantations
or of living embryos per dam or on the percentage of absorptions or
resorptions; no treatment-related malformation was reported
(Frohberg & Bauer, 1972a).
Rat: Groups of 20 Sprague-Dawley rats received lindane at
doses of 0, 5, 15, or 30 mg/kg body weight by subcutaneous injection
on days 6-15 of gestation. Maternal toxicity was observed in the
mid- and high-dose groups. No effect attributable to the
administration of lindane was noted on pregnancy rates, maternal
gross pathology or reproduction, or offspring viability and
development. No teratogenic effect was found at necropsy or in
visceral and skeletal examinations (Reno, 1976a; Hazelton
Laboratories, 1976a).
Rabbit: Lindane was injected subcutaneously at 0, 5, 15, 30
or 45 mg/kg body weight into pregnant rabbits on days 6-18 of
pregnancy, except that the highest dose was given on days 6-9 and
then reduced to 30 mg/kg body weight. No embryotoxic or teratogenic
effect was found in fetuses exposed to the two lower doses. At the
two higher dose levels, increased maternal toxicity was found. At
the highest dose, the number of resorptions was increased, and 14
out of 15 animals died (Reno, 1976b; Hazelton Laboratories, 1976b).
7.5.3 Reproductive behaviour
Adult female Fischer (CDF-344) rats were injected
intraperitoneally on the morning of pro-oestrus with lindane at 25,
33, 50, or 75 mg/kg body weight in sesame oil, and in the evening,
they were examined for lordosis behaviour with a sexually
experienced male. A dose-dependent reduction in sexual receptivity
was seen with increasing doses of lindane: treated animals required
a greater number of mounts before the first lordosis response was
observed, and they may have required more sensory stimulation to
elicit the lordosis reflex. Most of the females also failed to
exhibit proceptive behaviour (darting and hopping) during the mating
test. This inhibition resembles the rapid effects of another
chlorinated pesticide, chlordecone, and does not appear to depend
upon disruption by lindane of the inhibition of the central nervous
system by gamma-aminobutyric acid (GABA). The results substantiate
previous suggestions that the ability of chlorinated pesticides to
interfere with intracellular oestradiol receptors cannot explain
their rapid attenuation of reproductive behaviour (Uphouse, 1987).
7.5.4 Appraisal of reproductive toxicology
Lindane was investigated in tests covering all aspects of
reproduction (three-generation studies in rats) and in tests for
embryotoxicity and teratogenicity by oral, subcutaneous and
intraperitoneal administration in mice, rats, dogs, and pigs).
Lindane did not exhibit teratogenic properties after oral or
parenteral application (extra ribs were regarded as variations).
Fetal and/or maternal toxic effects were observed in rats with doses
of 10 mg/kg body weight and higher given by oral gavage; 5 mg/kg is
therefore considered to be the NOAEL.
No effect on reproduction or maturation was seen in the
three-generation study at doses of lindane up to 100 mg/kg diet, but
morphological signs suggesting liver enzyme induction occurred with
doses from 50 mg/kg diet in the third generation. The no-effect
level in this test was 25 mg/kg diet (equivalent to approximately
1.25 mg/kg body weight).
7.6 Mutagenicity and related end-points
Lindane was tested in mutagenicity tests with a variety of
end-points. The relevant experiments are summarized in Tables 9, 10,
and 11. Those studies that were not performed according to protocols
which comply to the present international standards are considered
to be of limited relevance; some studies used lindane preparations
of less than 99% purity or of unknown purity. The results are so
consistent, however, that the limitations of some studies did not
vitiate a final assessment.
7.6.1 DNA damage
The ability of lindane to damage DNA was tested in Bacillus
subtilis and in Escherichia coli WP2 in the rec assay, and
tests for unscheduled DNA synthesis tests were performed in primary
rat hepatocytes and human fibroblasts. No mutagenic potential was
detected.
Sina et al. (1983) developed a sensitive alkaline elution assay
in non-radiolabelled rat hepatocytes to measure DNA single-strand
breaks induced by chemicals. This assay is used to predict
carcinogenic/mutagenic activity. Lindane at doses of 0.03 and 0.3
mmol/litre induced DNA damage, increasing with dose.
After oral administration of lindane to rats and mice, a very
low covalent binding index (0.02-0.01) was calculated, suggesting
that no significant binding to DNA had occurred.
The incorporation of orally admini stered radiolabelled
thymidine into liver DNA was determined in SIV-50-SD-rats 24 h after
a single oral dose by gavage of 0.01, 0.1, or 1.0 mmol/kg gamma-HCH.
No effect on liver DNA synthesis was seen (Büsser & Lutz, 1987).
7.6.2 Mutation
The ability of lindane to induce gene mutation has been
investigated extensively in S. typhimurium and E. coli, using an
adequate range of strains to cover both base-pair and frame-shift
mutations. Most of the tests were performed both with and without
metabolic activation by 9000 x g preparations from the livers of
induced rats or mice (Table 9).
Negative reults were obtained in the host-mediated assay using
mice and S. typhimurium or Serratia marescens. Furthermore, a
test for point mutations in V79 Chinese hamster cells, the hprt
test for forward mutations, indicated no mutagenic effect of
lindane. A test for sex-linked recessive lethal mutation in
Drosophila melanogaster also gave a negative result (Table 9).
D. melanogaster were also used to test for dominant lethal
mutation. Groups of 25 males and 25 females aged 6-24 h were
transferred to food containing HCH (Gammexane) at 20 mg/kg food
medium, and their progeny were raised on this food. Five males and
five females of the F1 generation (the 'toxic generation') were
raised on normal food and were allowed to mate with each other and
lay eggs for 24 h in 10 oviposition jars. From this generation of
flies, three successive mutation-generations were raised on normal
food, and the numbers of larvae hatched from eggs laid on each of
the first 10 days after enclosion were again recorded. The
percentage of larvae hatched from the total number of eggs laid,
cumulated over the entire period was significantly decreased in the
second and third generations. These results suggest that the
preparation tested is mutagenic (Sinha & Sinha, 1983).
A test for induction of reverse mutations in Saccharomyces
cerevisiae gave inconclusive results.
7.6.3 Chromosomal effects
Most of the cytogenetic tests performed with lindane both in
vivo and in vitro did not indicate mutagenic properties of
lindane. In only une study were there positive findings, but the
purity of the material tested was not given and the description of
the test was poor. Lindane therefore apparently does not induce
chromosomal breakage (Table 10).
Table 9. Result of mutagenicity tests of gamma-HCH
Test system Dose Type of test Metabolic Result Reference
(Organisms/Cells) activation
Bacillus subtilis
H17 rec+ 0.02 ml of solution plate none - Shirasu et al. (1976)
M45 rec- containing 1 mg/ml plate none -
in DMSO
Escherichia coli
WP2 try- approx. 1 mg plate none - Ashwood-Smith et al. (1972)
WP2 4 gradient plates, plate S9-mix - Probst et al. (1981)
WP2 uvr A- covering 10 000-fold plate S9-mix -
concentration range
WP2 urv A 6-7 dose levels up to plate none - Oesch (1980)
5000 µg S9-mix -
Salmonella typhimurium
TA98, TA100, TA1535, 1-1000 µg plate S9-mix - van Dijck & van de Voorde
TA1537, TA1538, (1976)
TA1950, TA1978
TA98, TA100, 93, 139, 208 µg plate none - Röhrborn (1977a)
TA1535, TA1538
TA100, TA1535, 6-7 dose levels up to plate none - Oesch (1980)
TA1537, TA98 5000 µg plate S9-mix -
TA100, TA1535, 8 dose levels, 0 up to plate none - Haworth et al. (1983)
TA1537, TA98 333 µg S9-mix -
Table 9 (contd)
Test system Dose Type of test Metabolic Result Reference
(Organisms/Cells) activation
Salmonella typhimurium contd)
TA100, TA1535, 4, 20, 100, 500, or plate S9 - Anderson & Styles (1978)
TA1538, TA98 2500 µg in DMSO
G46, TA98, TA100, concentration gradient plate S9-mix - Probst et al. (1981)
TA1537, TA1538,
C3076, TA1535, D3052
Host-mediated assay
Salmonella typhimurium G46 25 mg/kg bw mouse nr - Buselmaier et al. (1972)
(subcutaneous) (NMRI)
Serratia marescens 25 mg/kg bw mouse nr - Buselmaier et al. (1972)
a 21. leu- (subcutaneous) (NMRI)
Mammalian cells
hprt locus
V79 Chinese hamster cells 0.5-500 µg/ml plate S9-mix - Glatt & Oesch (1984);
0.5-250 µg/ml plate S9-mix - Oesch & Glatt (1984)
Sex-linked recessive
lethal test
Drosophila melanogaster 0.001% injected nr - Benes & Sram (1969)
(aqueous sol.) into
abdomen
(0.2 µl)
DMSO, dimethyl sulfoxide; nr, not relevant; bw, body weight
Table 10. Results of tests for other genetic effects
End-point Dose Effects Result Reference
Chromosomal aberrations in vitro
Chinese hamster fibroblast cell line 2.1 mg/mla (in ethanol) Chromatid gaps, chromatid Equivocal Ishidate & Odashima
(CHL) and chromosomal breaks (1977)
Lymphocytes from human peripheral 0.1, 0.5, 1.0, 5.0, or Chromosomal breakage Equivocal b Tzoneva-Maneva et al.
blood (different donors) 10 µg/ml only at toxic dosages (1971)
(5 and 10 µg/ml)
Chromosomal aberrations in vivo
Chinese hamster bone-marrow cells 0.125, 1.25, or 12.5 mg/kg Increase in chromosomal (-) Röhrborn (1976, 1977a)
body weight orally for gaps at highest dose
5 days level
Syrian hamster bone-marrow cells 64, 128, 280, or 640 mg/kg No chromosomal - Dzwonkowska & Hubner
body weight aberration (1986)
Rat bone-marrow cells 1.5, 7.0, or 15 mg/kg - - Gencik (1977)
body weight orally for
12 weeks
Human lymphocytes occupational exposure - Desi (1972)
(no further details)
Sister chromatid exchange in vivo
Mouse bone-marrow cells (strain CF1) Male/female: 2/1.6, 10/8, or
50/40 mg/kg body weight - - Guenard et al. (1984a)
as a single oral
application
Mouse bone-marrow cells Single intraperitoneal - - Guenard et al. (1984b)
(strain CF1) injection of 1.3, 6.4, or
32.1 mg/kg body weight
Table 10 (contd)
End-point Dose Effects Result Reference
Micronucleus test in vivo
Mouse erythroblasts (CBA male mice) 75 mg/kg body weight - - Jenssen & Ramel (1980)
Dominant lethal test
Rats (males; strain Chbb = THOM) 1.5, 7.0, or 15 mg/kg body Röhrborn (1977b)
weight daily for 8 weeks
orally (in olive oil)c
Rat (males; strain Wistar) 1.5, 7.0, or 15 mg/kg body Questionable Cerey et al. (1975)
weight in olive oil positiveb
Mouse (males; strain ICR/Ha Swiss) 15, 75, 200, or 1000 mg/kg - - Epstein et al. (1972)
body weight once
intraperitoneally
15 mg/kg body weight - Equivocal Epstein et al. (1972)
five times, orally
Mouse (males; strain NMRI-EMD) Single intraperitoneal - - Frohberg & Bauer (1972c)
injection of 12.5, 25, or
50 mg/kg body weight
a Maximal effective dose
b Inadequate study; protocol does not comply with international standards
c Males dosed continuously during the whole mating period (8 weeks)
Table 11. Results of tests for DNA damage
End-point Dose Type of test Metabolic Result Reference
activation
Unscheduled DNA synthesis
(transformed SV-40) human 1, 1000 µM (in acetone) Tissue culture None - Ahmed et al. (1977)
fibroblast (cell-line VA-4) fluid S9-mix -
Unscheduled DNA synthesis and 500 µg/ml Tissue culture - 50-70% Rocchi et al. (1980)
repair capacity after damage inhibition
by UV-rays (human lymphocytes)
Primary rat hepatocytes 100 nmol/ml Plate - - Probst et al. (1981)
(in DMSO)
Covalent DNA binding
Male mouse (strain NMRI, CF1 12-13 mg/kg body Liver DNA Not relevant - Sagelsdorff et al. (1983)
and C6B3F1) weight orally and 8.7 (covalent
up to 23 mg/kg body binding
weight orally index,
0.02-0.1)
Lindane was tested for its ability to induce sister chromatid
exchange in vivo (in mice by oral and intraperitoneal
administration) and in vitro (in Chinese hamster ovary cells); no
effect was seen. No mutagenic effect was observed in a test for
micronucleus formation in the bone marrow of mice treated in vivo,
and lindane did not induce chromosomal damage in vivo.
Two of three tests for induction of dominant lethal mutation in
rats gave clearly negative results, and the other gave a
questionably positive response. The significance of the latter test
must be regarded as low, because the purity of the material tested
was not given and the test was not performed in compliance with an
acceptable standard.
7.6.4 Miscellaneous tests
Lindane tested in a MO4 cell culture at doses of 1, 10, and
100 µg/ml induced no multinucleation or major toxicity (de Brabander
et al., 1976).
Lindane at a concentration of 101.8 µg/ml did not induce
6-thioguanine-resistant mutations in Chinese hamster V79 cells.
Concentrations of 100 and 200 µg/ml were significantly cytotoxic;
the concentration that allowed 10% survival was 120 µg/ml. At 11.6
µg/ml, lindane weakly inhibited metabolic cooperation between 6-TGs
and 6-TGr V79 cells. It was concluded from these studies that
lindane is not mutagenic in this test system; however, it inhibits
metabolic cooperation, mimicking the powerful tumour promotor
12- O-tetradecanoylphorbol 13-acetate in this assay system
(Tsushimoto et al., 1983).
The morphology of primary monkey kidney cells was examined 24 h
after addition of lindane (99.8% in 1% dimethylformamide) to the
growth medium, and readings were made daily for three days. Lindane
applied at concentrations above 10 mg/litre induced marked cellular
damage, and 250 mg/litre had cytotoxic effects (Desi et al., 1977).
7.6.5 Appraisal of mutagenicity and related end-points
The mutagenicity of lindane has been adequately studied. This
compound has been extensively investigated for its ability to induce
gene mutation in both bacteria and mammalian cells, and for its
activity in the assay for sex-linked recessive lethal mutation in
D. melanogaster. Negative results were obtained consistently. Its
ability to induce chromosomal damage and sister chromatid exchange
has been investigated in mammalian cells both in vitro and in
vivo, again with negative results. Both assays for DNA damage in
bacteria and studies in vivo to investigate covalent binding to
DNA in the liver of rats and mice following oral administration also
gave negative results. The few studies in which positive results
were obtained involved invalid study designs or lindane of unknown
purity.
Overall, lindane appears not to have mutagenic potential.
7.7 Carcinogenicity
7.7.1 Mouse
Gamma-HCH was fed to 20 male ICR/JCL mice (five weeks old) at
300 or 600 mg/kg diet for 26 weeks. Increased liver weights were
reported in the group receiving the higher dose. Five of 10 mice in
this dose group had type 0 or type I liver lesions. Type 0 lesions
were characterized as areas of atypical, small liver cells, uniform
in size and with a small nucleus, which normally forms round spots
and is readily distinguishable from the surrounding liver tissue.
Type I lesions were described as 'benign liver tumours' (Goto et
al., 1972).
Groups of 20 male mice (eight weeks old) were fed gamma-HCH at
100, 250, or 500 mg/kg diet for 24 weeks. The highest dose level
resulted in increased liver weight. No nodular hyperplasia or
hepatocellular tumour was observed (Ito et al., 1973b).
Hanada et al. (1973) treated 10-11 dd mice of each sex with
lindane at 100, 300, or 600 mg/kg diet for 32 weeks and killed the
survivors 5-6 weeks after the end of exposure. Hepatomas were found
in 1/3 females and 3/4 males that ingested 600 mg/kg diet and
survived for 36-38 weeks; none were found in animals fed 100 or 300
mg/kg diet. At the two higher doses, most animals had atypical
proliferations in the liver. alpha-Fetoprotein could not be
identified in the serum of the animals with hepatomas.
In an experiment lasting 110 weeks, 30 male and 30 female CF1
mice were fed lindane (> 99.5%) at 400 mg/kg diet. A group of
controls comprising 45 male and 44 female mice were fed a standard
diet. Benign and malignant liver tumours were diagnosed in 24% of
male controls and 23% of female controls and 93% of treated males
and 69% of treated females. Significant mortality (15%) occurred
during the early phase of the study in the treated group (Thorpe &
Walker, 1973). In a complete reexamination of all slides, the
reviewer concluded that lindane had not affected the incidence of
hepatocellular carcinomas inanimals of either sex but had enhanced
the incidence of hepatocellular adenoma (and hyperplastic nodules)
in male mice. In this strain, therefore, lindane had a tumorigenic
effect only in male mice (Vesselinovitch & Carlborg, 1983).
Herbst et al. (1975) and Weisse & Herbst (1977) studied the
carcinogenic potential of lindane at 12.5, 25, or 50 mg/kg diet
administered for 80 weeks to 50 male and 50 female Chbi:NMRI mice (a
strain with a low (2%) spontaneous rate of hepatomas). The control
group consisted of 100 males and 100 females. No evidence of
substance-related tumour formation was seen in animals of either sex
at any dose level. Electron microscopic examination showed no fine
structural hepatocellular alterations.
Groups of 50 B6C3F1 hybrid mice of each sex were fed lindane
at 80 or 160 mg/kg diet for 80 weeks and were killed 10-11 weeks
after the end of treatment. Hepatocellular carcinomas were found in
5/49 pooled male controls, 2/10 matched male controls, 19/49 males
fed 80 mg/kg diet and 9/46 males fed 160 mg/kg diet, and in 2/47
pooled female controls, no matched female controls, 2/47 females fed
80 mg/kg diet, and 3/46 females fed 160 mg/kg diet. Only the
incidence of hepatocellular carcinomas in the males at the lower
dose was significantly different from that in controls. It was
concluded that lindane is not carcinogenic in this test system (US
National Cancer Institute, 1977). In a reexamination of the slides,
the reviewer was in full agreement with the conclusions of the
original authors (Vesselinovitch & Carlborg, 1983).
Wolff & Morrissey (1986) administered diets containing lindane
at 160 mg/kg diet for 24 months to three phenotypes of (YS x VY)
F1 hybrid mice: obese yellow Avy/a, lean pseudoagouti Avy/a, and
lean black a/a. Hepatocellular adenomas were found in 35% of yellow
A vy/a mice (9% in controls) and in 12% of pseudoagouti Avy/a mice
(5% in controls); no increase in the incidence of liver tumours was
seen in the black a/a mice.
7.7.2 Rat
Groups of 10 male and 10 female Wistar rats were fed for life
on diets containing 10, 100, or 800 mg/kg diet of powdered lindane
or 5, 10, 50, 100, 400, 800, and 1600 mg/kg diet of lindane in corn
oil. The life span of the animals was shortened by 20-40% in a
dose-dependent manner with administration of 400, 800, and 1600
mg/kg diet, except in those given 800 mg/kg diet of powdered
lindane. No increase in tumour incidence was reported in the 200
treated rats (Fitzhugh et al., 1950) (see also section 7.4.1).
In a lifetime study, groups of 10 rats of each sex received
lindane at 0, 25, 50, or 100 mg/kg diet. No tumour formation was
found (Truhaut, 1954). (Details not given.)
Groups of 18-24 male W rats received lindane (99%) at 500 mg/kg
diet for 24 or 48 weeks. High mortality was seen; none of the six or
eight surviving animals had developed a liver tumour by 24 or 48
weeks, respectively (Ito et al., 1975).
Groups of 50 Osborne-Mendel rats of each sex were administered
lindane for 80 weeks and were then transferred to the control diet
for an additional 28-30 weeks; survivors were killed at 108-110
weeks. The males received 320 or 640 mg/kg diet for 38 weeks,
lowered thereafter to 160 and 320 mg/kg diet; the females received
320 and 640 mg/kg for two weeks, then 160 and 320 mg/kg for 49 weeks
followed by 80 and 160 mg/kg diet for 29 weeks. Matched controls
consisted of 10 animals per sex; these were combined for statistical
evaluation with 45 untreated male and female rats from other
bioassays. No increase in tumour rate was seen in treated groups of
either sex (US National Cancer Institute, 1977).
7.7.3 Initiation-promotion
The tumour-initiating activity of gamma-HCH was studied by
observing the appearance of phenotypically altered foci in female
Wistar rats (Schröter et al., 1987). Groups of 3-8 rats were
operated to remove the median and right liver lobes; they were then
administered gamma-HCH at 30 mg/kg body weight daily for two weeks,
followed by phenobarbital at 50 mg/kg body weight daily for 15
weeks. Liver foci were identified by means of the
gamma-glutamyltransferase reaction and morphological alterations. No
evidence of initiating activity was found.
Promoting activity was studied by administering
N-nitrosomorpholine as a single dose of 250 mg/kg body weight by
gavage, followed by 4, 15, and 20 weeks' administration of gamma-HCH
at 0.1, 0.5, 2.5, 10.0, or 30.0 mg/kg body weight per day. Both the
number and the size of altered foci were enhanced by doses of 2-3
mg/kg. The authors concluded that gamma-HCH could be classified as a
tumour promotor.
In an experiment using male dd mice (26-30 per group, eight
weeks old), administration of Kanechlor-500 at 500 mg/kg diet
induced nodular hyperplasia and hepatocellular carcinoma in the
livers of mice after 32 weeks' exposure. Administration of lindane
(99% pure) at 50, 100, or 250 mg/kg dietwith or without the
polychlorinated biphenyl at 250 mg/kg diet induced none of those
lesions after 24 weeks. Lindane was therefore neither tumorigenic a
promoter in this experiment (Ito et al., 1973a).
7.7.4 Mode of action
Considerable work has been done using mice generated
genetically from (C3H x VY)F1 or (YS x VY)F1 mice. The resulting
Avy/Avy, Avy/a and A/a crosses contain a genomic locus known as the
Agouti locus, which has been linked to tumorigenicity in these mice.
Treatment of Agouti mice with lindane at 160 mg/kg diet has been
found to saturate the lindane elimination pathways and thereby
result in an increased burden of lindane and its metabolites. This
excessive build-up could explain the tumorigenicity of lindane, at
least when given at 'excessive' levels (Wolff, 1986; Wolff et al.,
1986).
The tumour response to lindane has been characterized in (YS x
VY) F1 hybrid mice (Table 12). Lindane increased the incidence of
benign tumours only in the Avy/a genotype, while the 'normal' A/a
mice had no tumours. This finding indicates the existence of a
genetic predisposing factor, which may be of some importance in
evaluating the hazard of exposure to lindane. A phenotypic factor is
apparently involved, as mice of the obese yellow phenotype had a
greater tumour response in the liver than their isogenic siblings,
pseudoagouti mice; such factors may themselves result in more
tumours. Tumour incidence was not increased in normal black mice,
but the incidence of benign tumours of the liver and lung was
increased in Y genotype mice. The time of tumour onset was as early
as 18 months in pseudoagouti mice, but the normal black mice had no
tumours in the 24-month test period. Avy/a yellow mice thus have a
proclivity to form hepatocellular adenomas and lung tumours, which
is augmented (and not caused exclusively) by exposure to lindane.
The pseudoagouti and normal black mice have a low rate of
spontaneous tumours in the liver and lung, but only the pseudoagouti
respond to lindane. Thus, some genetically derived mice form benign
tumours, but the 'normal' A/a controls do not. Holder & Stöhrer
(1989) concluded that these findings are of limited applicability to
the situation in humans.
Table 12. Carcinogenic responses in normal (A/a), pseudoagouti (Avy/a), and
agouti (Avy/a) mice after 24 months of dietary exposure to lindane a
Tumour Phenotypeb No lindane 160 ppm lindane p value
No. % No. %
Liver adenoma B 6/96 6 3/96 3 -
PS 5/95 5 11/95 12* 0.11
Y 8/93 9 33/94 35* 8.2E-06
Hepatocellular B 3/96 3 1/96 1 -
carcinoma PS 2/95 2 5/95 5 -
Y 12/93 13** 16/94 27 -
Combined liver B 9/96 9 4/96 4 -
tumours PS 7/95 7 16/95 17* 0.036
Y 20/93 22** 49/94 52* 1.1E-05
Hyperplasia of lung B 10/96 10 79/96 82* <E-08
Clara cells PS 10/95 10 71/94 76* <E-08
Y 14/95 15 68/95 52* <E-08
Lung tumourc B 2/29 2 1/96 3 -
PS 2/95 6 5/94 14 0.0692
Y 12/95 4 16/95 19* 0.0012
Table 12 (continued)
* Statistically significant dose-related response compared to non-treated
comparable controls of the same phenotype
** Liver response increased in obese yellow (Avy/a) mice compared to A/a black
controls even in absence of treatment
a From Holder & Stöhrer (1989)
b B, black normal controls; PS, pseudoagouti; Y, yellow
c Not malignant; origin of cells uncertain
Tumour promotion was tested as a mechanism of action for
lindane in both the yellow and pseudoagouti variants of the (C3H x
VY) F1 mouse using phenobarbital, which increases the incidence of
benign tumours in the liver of yellow mice, as the tumour-promoter.
Lindane at a dose of 160 mg/kg diet may exceed saturation of the
metabolic mechanisms, especially in yellow mice with the (C3H x
VY)F1 genotype. Chadwick et al. (1987) found reduced elimination
of lindane in yellow and pseudoagouti mice and explained their
findings as follows: The yellow mouse carrying the Avy locus has a
propensity for tumorigenicity, which is enhanced by the yellow obese
phenotype. The lungs and livers of these animals therefore are very
likely to contain cells that are already transformed, whereas in
normal black mice there may be none or very few transformed cells.
Hence, Avy strain mice would be expected to respond to a tumour
promoter, whereas black mice would not; this was also the pattern of
tumour response observed. The authors concluded that, because
phenobarbital promotes tumours in this strain of mice, lindane is
also a promoter.
Oesch et al. (1982) studied the specific activities in CF1
and B6C3F1 mice and Osborne-Mendel rats of some of the enzymes
thought to be involved in lindane metabolism. Lindane was
administered at 51-360 mg/kg diet for three days or three months. No
clear change was seen in animals treated for three days, but changes
in enzyme activity were noted after three months' treatment. In the
CF1 strain (sensitive to liver tumour induction), a large increase
in liver weight was observed; this was not the case in B6C3F1
mice. In the Osborne-Mendel rats, a smaller increase was found.
Glutathione- S-transferase activity was increased in CF1 mice and
to a lesser extent in B6C3F1 mice and the rats. Increased
glutathione- S-transferase activity may lead to rapid conjugation
of glutathione with reactive metabolites, as, for example, epoxides
derived from lindane. Rat liver microsomes had more
UDP-glucuronosyltransferase activity than those from mouse liver.
This increased activity in rats could also lead to rapid conjugation
of phenols derived from lindane. The most striking difference,
however, was that CF1 mice had more monooxygenase activity and
less epoxide hydroxylase activity than rats; whether either of these
changes would result in an accumulation of reactive epoxides from
lindane remains to be elucidated.
Iverson et al. (1984) studied the ability of 14C-gamma-HCH to
bind to liver macromolecules of untreated and
phenobarbital-pretreated male HPB black mice in vivo and in
vitro. There was preferential binding of gamma-HCH to protein but
not to DNA.
These studies in mice indicate that lindane does not behave as
an initiator, in that it does not induce the preneoplastic foci seen
with known carcinogens, such as N-nitrosoporpholine and
N-methyl- N-nitrosourea (Holder & Stöhrer, 1989). Lindane can,
however, act as a tumour promoter, in that it caused outgrowth of
foci and increased the areas of the foci, indicative of
preneoplastic conditions. Whether these foci actually go on to form
tumours was not determined.
The notion that lindane has some characteristics in common with
tumour promoters is corroborated by the finding that it inhibits
cell-to-cell communication of the low-molecular-weight compound,
tritiated uridine. Trosko (1982) found that such inhibition occurred
when cells were pretreated with a variety of free-radical scavengers
and suggested that the inhibition might involve a free-radical
generating process. Some tumour promoters have been suggested to act
by a mechanism involving free radicals (Kensler & Trush, 1984; Rao &
Reddy, 1987).
If lindane acts by the tumour promotion mechanism suggested by
formation of gamma-glutamyltransferase-positive foci in the liver
and inhibition of cell-to-cell communication, it is likely to be a
dose-rate-limited process because of its known reversibility. That
is, the compound must be administered at above a certain amount and
rate or its carcinogenic effects are reversible and cease to be
manifested. Such a mechanism would therefore probably result in a
sigmoid response in models.
Zeilmaker & Yamasaki (1986) studied the effect of lindane on
gap-junctional intercellular communication in cultured Chinese
hamster V79 cells, grown as monolayer using a microinjection/dye
transfer technique. Intercellular communication via gap junctions is
thought to play a crucial role in cell proliferation and
differentiation and in tissue homeostasis, and consequently in
carcinogenesis. Lindane inhibited junctional communication in a
dose-response relationship (0-20 µg/ml) after a 60-h exposure, but
inhibition was seen after 24 h incubation only with the highest dose
level. In an earlier study, lindane strongly inhibited metabolic
cooperation between V79 cells at a non-toxic dose of 10 µg/ml
(Tsushimoto et al., 1983).
Another explanation for the finding that Avy/a yellow mice have
a predisposition for tumorigenicity but not necessarily for
carcinogenicity is their reduced immunocompetence, as evidenced by
decreased antibody response to T-cell-dependent immunogen tetanus
toxoid, enhanced antibody response to T-cell-independent immunogen
type III pneumococcal polysaccharide, decreased rates of carbon
clearance and increased rates of immunoglobulin A formation. The
pseudoagouti mice did not have reduced immunocompetence and had
reactions similar to those of normal black A/a mice in these
immunological tests (Holder & Stöhrer, 1989).
The lindane metabolite, 2,4,6-trichlorophenol (TCP),
constitutes a significant proportion of the urinary metabolites of
lindane and is considered to be a carcinogen. However, direct
measurements of comparative potency indicate that TCP contributes
only a small fraction of the 'lindane cancer potency' and therefore
may not add significantly to the quantitative impact of lindane. The
notion that TCP adds quantitatively to the carcinogenicity of
lindane per se remains a major element in the evaluation of the
carcinogenic hazard of lindane to humans (Holder & Stöhrer, 1989).
7.7.5 Appraisal of carcinogenicity
Studies to define the carcinogenic potential of lindane have
been conducted with mice and rats, at doses of up to 600 mg/kg diet
in mice and up to 1600 mg/kg diet in rats. In some studies, the dose
levels exceeded the maximum tolerated dose. Hyperplastic nodules
and/or hepatocellular adenomas were found in studies with mice at
doses from 160 mg/kg diet. Two studies using mice and one study
using rats, with dose levels of up to 160 mg/kg diet in mice and 640
mg/kg diet in rats, showed no increase in the incidence of tumours.
The results of studies on initiation-promotion, on mode of action,
and on mutagenicity indicate that the tumorigenic effect of
gamma-HCH in mice results from non-genetic mechanisms.
7.8 Special studies
7.8.1 Immunosuppression
Desi (1976) and Desi et al. (1978) reported the results of a
subacute study in which groups of 30-36 male rabbits were treated
orally, five times per week for 5-6 weeks, with doses of lindane
representing 0, 1/5, 1/10, 1/20, and 1/40 of the oral LD50, which
was 60 mg/kg body weight. Once a week, different doses of S.
typhimurium 'Ty 2' vaccine were injected intravenously. The
humoral immune response was determined by the tube agglutination
test. A linear regression was found between the dose of lindane and
reduction in antibody titres, in a time-dependent manner. The lowest
dose, 1.5 mg/kg body weight (1/40 of the oral LD50) caused no
immunosuppression.
7.8.2 Behavioural studies
The learning rate in a maze and responses to conditioning in a
Skinner box were studied after feeding lindane at daily doses of
2.5, 5, 10, or 50 mg/kg body weight to Wistar rats for 40 days. In
the maze, no effect was seen with 2.5 mg/kg, but at 5 mg/kg there
was stimulation accompanied by an increased error rate in maze
running activity; at 10 and 50 mg/kg, the animals became sedated and
committed more errors than the controls. In the Skinner box,
stimulation was seen with 2.5 and 5 mg/kg. At 10 mg/kg, no
difference was seen from the controls, whereas animals treated at 50
mg/kg were less active than the controls (Desi, 1974).
7.8.3 Neurotoxicity
Lindane produces a variety of neurological effects, both
central and peripheral, in mammals. The induced increase in neuronal
excitability and the underlying mechanisms of action have been
investigated both in vivo and in vitro.
7.8.3.1 Dose-response studies using intact animals
The effects of lindane on body temperature, food intake, and
body weight were studied in Wistar rats given single or repeated
non-convulsant oral doses. Groups of eight male and eight female
rats were given lindane as a single oral dose of 30 mg/kg in olive
oil. Controls received olive oil alone. Further groups of eight
males and eight females received 10 mg/kg and two groups of male
rats received 30 mg/kg once daily for seven days at either
thermoneutral ambient temperature or cold ambient temperature
(4 °C). The single dose of 30 mg/kg significantly decreased core
temperature 5 h later; this lindane-induced hypothermia was strongly
potentiated by cold stress in rats kept at 4 °C. A decrease in body
weight gain was also observed. No hypothermic effect was seen with
10 mg/kg (Camon et al., 1988a).
The relationship between the brain concentration of lindane and
its convulsant effect was studied in male Wistar rats administered
lindane (99.5%) dissolved in olive oil daily by gavage at doses of
5, 12, or 20 mg/kg body weight for 12 days. The mean plateau
concentration in brain was achieved within 5-8 days. There was a
strong correlation between the doses administered and the
concentration in brain at the plateau. A convulsant response was not
seen with 5 mg/kg, but tonic convulsions occurred at the two higher
doses. The rate of response (percentage of rats with convulsions)
was also correlated with the log of the concentration of lindane in
brain. The concentration in brain decreased after 12 days of daily
administration of doses of 5 and 12 mg/kg, but not with 20 mg/kg
(Tusell et al., 1988).
Camon et al. (1988b) investigated the effect of convulsant and
non-convulsant doses of lindane on regional glucose uptake in the
brain. Male Wistar rats received intraperitoneal injections of
3H-2-deoxyglucose, and the amount of label in different brain
structures was assayed by liquid-scintillation counting in 18
dissected brain regions. Lindane at a single convulsant dose (150
mg/kg orally) increased 2-deoxyglucose uptake in olfactory
tubercules, hypothalamus, hippocampus, paraflocculi, and the
post-medulla. With a single, non-convulsant dose of 30 mg/kg, the
uptake of 2-deoxyglucose was less affected; after treatment with 10
mg/kg per day for one week, 2-deoxyglucose uptake was observed in
superior colliculi but was decreased in the parietal cortex. The
increased uptake in limbic regions seen at the convulsive dose
correlates with the experimentally observed association between
signs of poisoning induced by lindane and damage to the limbic
system.
Intraperitoneal injections of gamma-HCH (99.0%) in corn oil at
80-480 mg/kg body weight increased the accumulation of cerebellar
cyclic GM in male CD-1 mice. Furthermore, it inhibited the binding
of 3H-tert-butylbicyclo- ortho-benzoate (a ligand for the
GABA-A receptor-linked chloride channel) in mouse cerebellum
(Fishman & Gianutsos, 1987).
Fishman & Gianutsos (1988) gave male CD-1 mice gamma-HCH at
single intraperitoneal doses of 80-400 mg/kg body weight in corn
oil. At the lowest dose, gamma-HCH increased the lethality and the
frequency of tonic/clonic seizures induced by intraperitoneal
injection of 50 mg/kg pentylenetetrazole or 20 mg/kg picrotoxin but
had no effect on locomotor activity.
Sunol et al. (1988) studied the effect of administering lindane
by gavage at 150 mg/kg body weight in olive oil on the GABAergic and
dopaminergic systems, by measuring the concentrations of GABA,
dopamine and its metabolites in seven brain areas at the onset of
seizures. All animals suffered tonic convulsions 18.3 ± 1.4 min
after lindane administration. The concentration of GABA was
decreased only in the colloculi and not in the other areas. Dopamine
concentrations were increased in the mesencephalon, and those of its
metabolite, DOPAC, were also increased in the mesencephalon and the
striatum (abstract only).
In studies by Desi (1983), adult female CFY rats were given a
daily dose of lindane (99.5%) at 2.5 mg/kg body weight. This dose
level had no functional, neurological, electroencephalographic, or
psychophysiological effect, used as early signs of disturbances of
the nervous system. A dose of 5.0 mg/kg body weight altered the
electrical activity of the brain, as indicated by changes in the
complex electroencephalograph, the number of changing
electroencepahlographic bands and the index number. In behavioural
experiments, running speed and number of errors indicated an
inhibitory effect of lindane at 5.0 mg/kg body weight on learning
capacity; this result was not seen with 2.5 mg/kg.
Müller et al. (1981) studied the electroneurophysiological
effects of various HCH isomers on groups of 15 male Wistar rats by
feeding them diets containing each isomer for 30 days. Conduction
velocity delay was observed in the animals fed the gamma isomer at a
daily dose of 25.4 mg/kg, but not at 12.3 or 1.3 mg/kg. The greatest
delay was induced by the lindane metabolite
gamma-pentachlorocyclohexene (38-783 mg/kg body weight).
Lindane was reported to lower the threshold for kindled
seizures (resulting from repetitive stimulation of the limbic system
within the brain) in rats (Joy et al., 1982, 1983). In these
experiments, stimulating electrodes were implanted in the amygdala
and other part of the limbic system, and the animals were stimulated
with a one-second train of pulses at 60 Hz on each day of the study.
In this procedure, no response is elicited initially; however, with
repeated stimulation, the procedure induces increasing levels of
electrical seizure, with clonic seizures resulting after many
trials. A developing after-discharge becomes progressively longer,
and the severity of the accompanying motor signs becomes more
pronounced, until kindling is completed. The subject then exhibits
stable convulsive responses for weeks or months afterwards. The
duration of the electrical seizure and the severity of the
behavioural response were found to increase much more rapidly when
lindane was administered at daily oral doses of 1, 3, or 10 mg/kg
body weight 3 h before each kindling trial. These effects were found
to be dose-dependent, and a threshold exposure of 0.5 mg/kg per day
was calculated. Rats administered lindane at this concentration were
found to develop brain levels of lindane which fluctuated between
0.2 and 0.4 µg/g.
Joy & Albertson (1987a,b) demonstrated that lindane alters
dentate gyrus granule response to perforant path input in the intact
rat in a manner indistinguishable from picrotoxin or
pentylenetetrazol, which are known GABA-mediated chloride channel
antagonists. In this study, 19 male Sprague-Dawley rats were
anaesthetized with urethane, electrodes for stimulating and
recording responses from the dentate gyrus of the hippocampus were
implanted, and the animals were placed in a stereotaxic device.
Lindane was then administered intraperitoneally in dimethylsulfoxide
to each animal at sequential doses of 5, 10, 20, and 40 mg/kg body
weight. Single or paired electrical stimuli were presented at
different intensities and at different intervals to evaluate the
effects of lindane on inhibition and facilitation. These studies
demonstrated a dose-dependent change in perforant path granule cell
function, manifested as an increase in the excitability of the
granule cell to other stimuli. Lindane was also found to induce a
small but statistically significant, dose-dependent increase in
presynaptic inhibition, as well as a significant increase in
postsynaptic inhibition. A dose-dependent effect on GABA-mediated
inhibition was measurable at exposures that were not convulsant in
unanaesthetized animals. The results of this in-vivo study
indicate that inhibition of GABA-mediated chloride channels in the
brain is probably an important mechanism by which lindane produces
neuronal hyperexcitability and convulsions.
7.8.3.2 Studies on mechanism
Although the precise mechanism by which lindane exerts its
neurotoxic action is not fully resolved, studies using preparations
of synaptosomes (pinched-off nerve endings) and of cholinergic
neuromuscular junctions, as well as studies using intact animals,
have provided insight into this issue. The results of representative
studies with each type of preparation are summarized in Table 13.
TABLE 13. Known effects of lindane on central and peripheral nerves or muscle tissue
Effect Species Type of preparation Reference
Inhibition of Rat in vivo Joy & Albertson (1987 a,b)
GABA-mediated chloride
ion uptake in CNS
Rat Brain synaptosomes/ Eldefrawi et al. (1985)
neuronal membrane Matsumura & Tanaka (1984)
Abalis et al. (1985, 1986)
Mouse Brain synaptosomes Fishman & Gianutsos (1988)
Increased availability Rat Brain synaptosomes Hawkinson et al. (1989)
of intracellular Ca++
Rat Neuroblastoma cells Joy et al. (1987)
Rat Neurohybridoma cells Joy et al. (1988)
Frog Neuromuscular junction Joy et al. (1987)
Vogel et al. (1985)
Publicover & Duncan (1979)
Decreased conduction Rat Tail nerve Müller et al. (1981)
velocity
Mitochondrial damage Frog Skeletal muscle Publicover et al. (1979)
Na+-K+-ATPase inhibition Frog Skeletal muscle Pandy et al. (1985)
Rat Liver mitochondria Srinivasan &
Radhakrishnamurty (1988)
TABLE 13. (Continued)
Effect Species Type of preparation Reference
Ca++-Mg++-ATPase Beef Brain Uchida et al. (1974)
inhibition
Rat Liver mitochondria Srinivasen &
Radhakrishnamurty (1988)
Lindane was shown to inhibit the uptake of chloride ions at
inhibitory synapses in the brain (Matsumura et al., 1984; Abalis et
al., 1985, 1986; Fishman & Gianutsos, 1988), and it is this mode of
action that is now widely considered to account primarily for the
convulsant activity of this insecticide. Because of its structural
similarity to picrotoxinin, lindane has a good geometric fit to the
picrotoxinin-binding site at the outer end of the chloride channel.
Once bound, the lindane is believed to block the action of the
neurotransmitter GABA, which mediates the entry of the Cl- necessary
for inhibitory neuronal function (Matsumura, 1985). More recent
studies by Joy & Albertson (1987a,b) provide evidence of such
inhibition of GABA in rats in vivo, demonstrating that: (a) this
mechanism operates at clinically relevant exposure levels (5-40
mg/kg); (b) the magnitude of this effect is dose-dependent; and (c)
this effect can be clearly measured at subconvulsant exposure levels
in unanaesthetized subjects.
Lindane has also been demonstrated to increase the excitability
of presynaptic cholinergic neurons at central (Hawkinson et al.,
1989) and peripheral (Vogel et al., 1985; Publicover & Duncan, 1979)
synapses. Using a nerve-muscle preparation, a universally accepted
model for a cholinergic synapse, Publicover & Duncan demonstrated
that concentrations of lindane as low as 5 x 10-5 mol/litre
increase the spontaneous release of acetylcholine. Similar results
were reported by Vogel et al. (1985), who observed increases up to
100 fold in spontaneous release of neurotransmitter in the presence
of lindane at 10-4 mol/litre. This increase was found in both
studies to be somewhat dependent upon the concentration of
extracellular calcium ions. In the presence of normal (approx. 1-2
mmol/litre) Ca++, the increase in spontaneous transmitter release
was 2.5-20 times greater than that measured when the extracellular
Ca++ level was buffered to 10-7 mol/litre, the homeostatically
maintained intracellular Ca++ concentration. An expanded report of
the study of Vogel et al. (1985) was made by Joy et al. (1987), who
additionally described a similar lindane-induced excitability in
neuroblastoma cells. Hutchison verified the involvement of Ca++ in
the enhanced release of acetylcholine using a rat brain synaptosome
preparation, and he postulated that the increased excitability of
cholinergic synapses (widely dispersed throughout the brain) may
contribute to the convulsant effect of lindane.
Pandy et al. (1985) reported that lindane weakly inhibits
Na+-K+-ATPase activity in skeletal muscle, and they suggested
that lindane can inhibit Ca++-Mg++, Ca++-, and mg++-ATPase.
Uchida et al. (1974) reported decreased Ca++-Mg++-ATPase
activity in beef brain. Since all of these ATPases are involved in
the maintenance of intracellular Ca++ concentration and have been
identified in a neuronal membrane preparation (Yamaguchi et al.,
1979), it is possible that the disruption of homeostatic calcium
regulation by lindane contributes to its excitatory action on the
central nervous system.
7.8.3.3 Summary
Chronic exposure to low levels of lindane can result in
proconvulsant activity, as demonstrated experimentally using the
kindling model of experimental epilepsy. Lindane has also been shown
to cause convulsions in rats administered oral doses of 12 mg/kg
daily for 12 days; a dose of 5 mg/kg body weight did not induce
convulsions within that period. The convulsive effect has been
suggested to be associated with inhibition of GABA-mediated chloride
channels in the brain, as demonstrated experimentally in mammals
both in vivo and in vitro. Changes in the electroencephalogram
and decrements in a variety of behavioural parameters have been
observed with lindane at a dose of 5 mg/kg/body weight per day for
40 days, but not at 2.5 mg/kg for 22 days. A delay in peripheral
nerve conduction velocity was observed in rats administered a
dietary concentrations of 25 mg/kg body weight, but not at 12 mg/kg.
7.9 Factors that modify toxicity; toxicity of metabolites
Pretreatment of rats with lindane minimized or inhibited the
convulsive effects of pentazole, picrotoxin, loramine, strychnine,
mintracol, cyclohexane sulfonamide and electro-shock (Herken,
1950a,b,c, 1951; Coper et al., 1951; Kewitz et al., 1952; Lange,
1965). It was demonstrated that premedication for two weeks with
daily doses of 2 mg lindane not only accelerated the metabolism of
other chemicals, but also caused an acceleration of its own
breakdown: in Fischer rats given two oral administrations of lindane
in oil at 2 mg/kg, there was increased excretion of glucuronic acid
conjugates (Chadwick et al., 1971).
The smallest single oral dose of lindane that reduced
pentobarbital sleeping time in FW-49 rats was 5 mg/kg body weight
(Schwabe & Wendling, 1967). The smallest single intraperitoneal dose
of lindane that shortened hexabarbital anaesthesia was 15 mg/kg body
weight. A similar result was obtained in Sprague-Dawley rats fed a
diet containing lindane at 0.5 mg/kg; the effect was more distinct
with a dose of 4 mg/kg diet (Kolmodin-Hedman et al., 1971).
8. EFFECTS ON HUMANS
8.1 Exposure of the general population
8.1.1 Acute toxicity, poisoning incidents
Several cases of fatal poisoning and numerous cases of
non-fatal illness caused by or ascribed to lindane have been
reported. These incidents were either accidental, intentional
(suicide) or due to gross neglect of safety precautions. In many of
these cases, the effects ascribed to lindane were more likely to
have been due, in total or in part, to other substances. A critical
review of these cases is provided by Hayes (1982).
The toxic or lethal dose appears to vary considerably with the
carrier and/or the degree of homogenization of the product. Under
certain conditions, 10-20 mg/kg body weight can present a lethal
hazard to humans, but higher concentrations can be tolerated when
followed by timely and appropriate medication. Starr & Clifford
(1972) described a case of acute lindane intoxication in a
2.5-year-old child who had severe epileptiform convulsions after
ingesting presumably two 0.78-g pellets containing 95% lindane. The
child recovered under medical care.
A suicide attempt was reported by Ohly (1973). The person
ingested about 100 ml of a 25.5% lindane emulsion concentrate, which
corresponds to a dose of about 309 mg/kg body weight. No vomiting or
diarrhoea was seen, but severe convulsions occurred within the first
24 h after lindane ingestion. Elevated serum levels of
glutamic-oxaloacetic transaminase, lactic dehydrogenase,
glutamic-pyruvate transaminase, and creatine phosphokinase activity,
in conjunction with the results of a liver biopsy, suggested fatty
degeneration and severe toxic damage to the liver. After five weeks
of clinical treatment, the patient recovered completely.
A number of cases of poisoning cases have been described after
medical treatment or abuse (Davies et al., 1983; Kurt et al., 1986;
Petring et al., 1986; Berry et al., 1987).
Clinical signs of intoxication can appear from a few minutes to
some hours after intake of lindane, depending on the route of
administration, the formulation, the concentration of lindane, and
the quantity involved. In mild cases, indisposition, nausea,
dizziness, restlessness, frontal headaches, and sometimes vomiting
occur. Muscular fasciculation, disturbances of equilibrium, ataxia,
and tremor may appear. Pains in the upper abdomen are frequently
coupled with diarrhoea and uncontrolled micturition. Clonic-tonic
convulsions of some minutes' duration may occur, and these may recur
after several hours or even days in response to optical, tactile,
and acoustic stimuli. In fatal cases, death follows several hours to
several days after intake. The cause of death is usually central
respiratory failure or acute circulatory collapse, often after
convulsions (Hayes, 1982; Jaeger et al., 1984).
8.1.2 Effects of short- and long-term exposures - controlled human
studies
8.1.2.1 Oral administration
Lindane given orally as a vermicide at a dose of 45 mg to an
adult patient (26 years old) in poor condition induced convulsions,
nausea, and vomiting. Recovery took place within 3-4 h. Of 15
patients in the same trial, who repeatedly received up to 30
mg/person for up to three days, six complained of nausea; no other
symptom was reported (Graeve & Herrnring, 1949).
Severe toxic symptoms were described in healthy volunteers
after oral intake of 15-17 mg/kg body weight of lindane in a liquid
carrier (Hofer, 1953; Schmiedeberg & Wasserburger, 1953). Reports of
toxic effects after administration of lindane against scabies
indicate that children are more sensitive to lindane than adults,
and rare cases of aplastic anaemia have been reported (G. Volans,
National Poison Unit, London, letter to IPCS, 1989).
8.1.2.2 Dermal application
Clinical reports after pharmaceutical use of lindane
cutaneously suggest that exposures somewhat higher than 5 mg/kg body
weight per day do not usually result in acute neurotoxic symptoms.
No cause-effect relationship was found between lindane and blood
dyscrasias (Ginsburg et al., 1977; Kramer et al., 1980; Morgan et
al., 1980).
Three groups, each consisting of one male and one female
volunteer, received an application of 30 g of a 0.3% commercial
lindane emulsion over the entire body (except the head and the angle
of the elbow) on three consecutive days. The first group removed the
emulsion by washing with soap and water 3 h after application; the
second removed it 10 h after application by washing with water only
at body temperature, and the third group by washing with soap and
water 10 h after application. A group of scabies patients were given
an application of 50 g of the emulsion over the entire body, except
the angles of the elbows. Blood samples were taken regularly from
all participants in the trial over a period of 1-144 h. The highest
average serum concentration found approximately 5.5 h after
application in the healthy volunteers was slightly less than 5
µg/litre, while in scabies patients an average of about 200 µg/litre
was found after 4 h. It was concluded that scabies patients deposit
greater concentrations of lindane in their bodies than do healthy
persons; the levels were higher in women than in men (Lange et al.,
1981; Zesch et al., 1982).
Feldmann & Maibach (1974) studied the absorption and excretion
of 14C-labelled lindane dissolved in acetone, applied at 4
µg/cm2 to the forearms of six human subjects. The usual dose of
radioactivity was 1, 2, or 5 µCi. Data obtained after intravenous
dosing was used to correct the results obtained on skin penetration
for incomplete urinary recovery. Total excretion of 14C after
topical application was 9.3% ± 3.7 of the dose in five days. Skin
absorption is incomplete because the chemical is lost from the skin
surface by washing, evaporation or gradual exfoliation of outer
layers of the stratum corneum. The amount absorbed into the body
depends on the relationship between the speed with which it
penetrates the skin and the speed with which it is lost from the
skin surface.
Serum concentrations of gamma-HCH were determined in nine
children (3.5-18 years old) following application of a 1% gamma-HCH
shampoo to treat pediculosis capitis (head lice). The shampoo was
applied vigorously to dry hair in a sufficient amount to saturate
the hair and scalp thoroughly. After 10 min, small quantities of
water were added until a lather formed, and shampooing was continued
for 4 min; thereafter, the hair was rinsed and blown dry. Gamma-HCH
was present in the serum of all children 2-24 h following the
application. A maximal concentration of 1.4 µg/litre was found after
2-4 h; this level decreased within 24 h to 0.41 µg/litre.
Re-treatment increased the maximal level to 3.6 µg/litre (Ginsburg &
Lowry, 1983).
Nitsche et al. (1985) applied emulsions of 0.3 and 1.0%
14C-lindane to defined areas of 2 cm2 intact or stripped skin.
They found that the flux of lindane in the skin was time-dependent:
Generally, the concentration increased with the depth of the layer.
Increased availability of lindane, induced by absence of the stratum
corneum and a long application period, resulted in preferential
accumulation in the epidermis, with none in the subcutaneous fat.
When intact skin was washed with soap and water 3 h after the
application, the concentration of lindane in the layer below the
stratum corneum 7 h later was higher than that in skin that had not
been washed. Washing of intact skin after a short penetration period
(3 h) resulted in introduction of lindane; this phenomenon was not
seen with stripped skin. Lindane could be more effectively removed
from the stratum corneum with soap and water than with water alone.
8.1.3 Epidemiological studies (general population)
The rate of mortality from liver cancer in the USA was related
to the 'domestic disappearance' of organochlorine pesticides. In
1962, 18 and 15 years after the introduction of DDT and technical
HCH, respectively, by which time any increase in the mortality rate
from primary liver cancer would be manifest, the number of cases of
primary liver cancer as a percentage of the total number of deaths
from liver cancer began a gradual, steady decline, from 61.3% in
1962 to 56.9% in 1972. The death rate (per 100 000 per year) from
primary liver cancer during this period declined from 3.46 to 3.18
(Deichmann & MacDonald, 1977).
A considerable number of case reports have been published in
which different blood dyscrasias were described in people who had
been exposed to lindane or to lindane and other chemicals (Mendeloff
& Smith, 1955; Albahary et al., 1957; Jedlicka et al., 1958;
Stieglitz et al., 1967; West, 1967; Hoshizaki et al., 1969;
Vodopick, 1975). The issuance by the US Environmental Protection
Agency (1977) of a 'rebuttable presumption against registration and
continued reregistration' of lindane in 1977 was triggered in part
by the problem of blood dyscrasias. Studies conducted over periods
of several weeks to several years, however, have given no indication
that there might be a cause-effect relationship between exposure to
lindane and blood dyscrasias (Milby & Samuels, 1971; Samuels &
Milby, 1971; Morgan et al., 1980; Wang & Grufferman, 1981), and this
was the final conclusion of the US Environmental Protection Agency
(1984a).
8.2 Occupational exposure
8.2.1 Toxic effects
Evaluation of the effects of gamma-HCH in occupationally
exposed workers is seriously hampered by the fact that most of the
studies are of workers who were exposed during manufacturing and
handling of lindane, or in the handling or spraying of
technical-grade HCH among other pesticides. All of these groups are
also potentially exposed to other HCH isomers, to impurities and to
other (process) chemicals. It is, therefore, difficult to relate the
effects found in these studies to any individual substance. The only
such studies mentioned here are those that were considered to be
useful for the evaluation.
Kolmodin-Hedman (1974) investigated blood levels of gamma-HCH
in 54 spraymen exposed to 4% lindane and other insecticides in the
form of aerosols and mists and who also occasionally diluted stock
solutions. Their exposure varied from once daily to once weekly, and
the length of exposure was 1-20 years; they did not always wear
protective gloves and respirators. Spraymen exposed to lindane had
mean plasma levels of gamma-HCH of 6.4, 7.5, and 9.9 µg/litre; a
maximal concentration of 87.0 µg/litre was found. Antipyrine
half-lives in exposed subjects and in controls were compared to
investigate whether lindane induces drug metabolism in humans. In 26
workers exposed mainly to lindane, the mean antipyrine plasma
half-life was significantly shorter than that in 33 controls: 7.7 ±
2.6 h compared with 13.1 ± 7.5 h, respectively. Induction occurred
with plasma levels above 10 g/litre; workers who were exposed to
lindane had shorter antipyrine and phenylbutazone half-lives when
their serum levels were above this value. Hyperlipoproteinaemia
(defined as a serum cholesterol level above 800 mg/litre and a
phospholipid level above 1400 mg/litre in the HDL fraction of the
serum lipoproteins) was found in 40% of the spraymen
(Kolmodin-Hedman, 1974, 1984).
Three workers mixed rapeseed manually with 75% lindane powder,
which also contained 10% thiram, and usually closed the sacks by
hand. This mixing procedure was repeated up to 80 times during a
workshift. The working period comprised the spring months of each
year, and the total exposure period varied between one and five
years. Gloves and masks were not always used, so dermal and
respiratory exposures were intensive. The plasma levels during
exposure in the three people who prepared rapeseed were 102, 100,
and 4.2 µg/litre; the last person had less frequent exposure than
the other two. The plasma level in the person who had 100 µg/litre
during exposure had decreased to the level before exposure within
five months (Kolmodin-Hedman, 1974, 1984).
Workers engaged in the production of lindane and exposed for at
least six months (8 h/day; wearing face masks in an air-ventilated
location) were tested for the presence of chromatid-type and labile
chromosome-type aberrations in their lymphocytes. The frequency of
stable chromosomal aberrations did not differ significantly from
that in normal controls (Kiraly et al., 1979).
Herbst (1976) examined workers engaged in the production of
lindane in three factories. The people were exposed not only to the
gamma isomer but also to the alpha, beta, delta, and epsilon
isomers. The average length of service was slightly more than 10
years. Of the 118 persons examined, 115 were men and three were
women, and the average age was 39 years. No abnormality was detected
in the haematopoietic system, the liver, the kidneys, or the nervous
system.
A series of reports were made on groups of 54-60 male workers
(24-62 years of age) in a lindane-producing factory, with a
geometric mean duration of exposure of 7.2 years (range, 1-30 years)
(Baumann et al., 1981; Brassow et al., 1981; Tomczak et al., 1981).
The lindane concentrations in serum were in the range 5-188
µg/litre; that of alpha-HCH was 10-273 µg/litre, and that of
beta-HCH, 17-760 µg/litre. None of the controls had a HCH
concentration in serum above the limit of detection (O.7µg/litre).
The time-weighted average threshold limit value of 0.5 mg/m3 was
not exceeded at any of the workplaces (range, 0.004-0.15 mg/m3);
the level of alpha-HCH was 0.002-1.99 mg/m3 and that of beta-HCH,
0.001-0.38 mg/m3. Only small deviations were found in some
laboratory tests: higher polymorphonuclear leukocyte count, lower
lymphocyte count, higher reticulocyte count, lower prothrombin
level, lower blood concentrations of creatinine and uric acid; these
findings were regarded as of no pathological significance. No
significant difference was observed in total red cell, white cell,
and platelet counts, in haemoglobin content, or in levels of
gamma-glutamine transferase, glutamic-oxaloacetic transaminase,
glutamic-pyruvic transaminase, lactic dehydrogenase, cholinesterase,
triglyceride, cholesterol or urea. No sign of health impairment was
observed. Examination of reflexes, sensitivity, amplitude and
frequency of fore-finger tremor and manual skills showed no
significant difference between the HCH-exposed and the control
groups. In addition, no pathological result was obtained in tests
for electromyography, maximal motor nerve conduction velocity in
ulnar nerves, or neuromuscular conduction. Furthermore,
electroencephalographic recordings showed no specific pathological
sign. The authors concluded that, even after decades of occupational
exposure to HCH, no sign of neurological impairment or perturbation
of neuromuscular function had occurred. Serum levels of luteinizing
hormone were higher in HCH-exposed workers than in controls (8.8 vs
5.7 mIU/ml). Levels of follicle stimulating hormone were
insignificantly higher and testosterone levels were insignificantly
lower in exposed men than in controls.
In malaria-control workers, who sprayed technical-grade HCH for
16 weeks, the serum level of gamma-HCH increased from a mean of
0.009 to 0.037 mg/litre in previously unexposed workers, and from
0.009 to 0.034 mg/litre in workers who had been exposed during three
previous spraying seasons (Gupta et al., 1982). In comparison with
the alpha and beta isomers, the gamma isomer was the least
cumulative and the least persistent in serum. These findings, as
well as those of Milby et al. (1968), suggest that gamma-HCH levels
in blood are mainly a reflection of recent exposure to lindane.
Nigam et al. (1986) studied 64 employees at a manufacturing
plant who were directly or indirectly associated with the production
of HCH. The exposed group comprised 19 workers who handled and
packaged the insecticide, 26 plant operators and supervisors who
were exposed indirectly to HCH, and 19 members of the maintenance
staff who visited the plant frequently. The control group consisted
of 14 workers who had no occupational contact with HCH. The length
of exposure varied from 0 to 30 years. The mean serum concentrations
of lindane in the four groups were: control, 0.0007, maintenance
staff, 0.0227, indirect exposure, 0.016, and handlers, 0.0571
mg/litre. Alpha-, beta-, and delta-HCH were also present; the total
HCH concentrations in serum were 0.0514 mg/litre in the controls,
0.1436 mg/litre in the maintenance staff, 0.2656 mg/litre in the
indirectly exposed workers, and 0.604 mg/litre in the handlers. Most
of the directly and indirectly exposed workers had paraesthesia of
the face and extremities, headache, and giddiness, and some had
symptoms of malaise, vomiting, tremors, apprehension, confusion,
loss of sleep, impaired memory and loss of libido. The same symptoms
were found in the group of maintenance workers but were less severe
and occurred in fewer cases.
Chattopadhyay et al. (1988) studied 45 male workers exposed to
HCH during its manufacture and compared them with 22 matched
controls. Paraesthesia of the face and extremities, headache,
giddiness, vomiting, apprehension, and loss of sleep, as well as
some changes in liver function were reported. These changes were
found to be more closely related to intensity of exposure (as
measured by HCH levels in blood serum) than to duration of exposure.
The measured exposures to total HCH were 13 to 20 times higher than
those in the control groups (details not given). Of the total HCH,
60-80% was gamma-HCH.
Plasma levels of gamma-HCH and urinary levels of three TCPs
were measured in 45 forestry workers who were engaged in dipping
conifer seedlings in a gamma-HCH solution, transporting the dipped
seedlings to planting sites or planting the seedlings. Protective
clothing was supplied. The work started in April, and until June the
workers had plasma concentrations below the detection limit (5
nmol/litre). From June onwards, there was an upward trend in the
number of workers who had gamma-HCH levels in plasma of up to 40
nmol/litre. In July, levels of 59, 75, and 123 nmol/litre were found
in three workers, and the latter two had symptoms of poisoning that
included feeling unwell with a flu-like illness, fatigue, sore
throat, and nausea. No sign of hepatotoxicity was observed. People
with levels greater than 70 nmol/litre were removed from further
exposure. When exposure ceased at the end of July and the people who
had had elevated levels were monitored in August, 80% of the workers
had no detectable gamma-HCH in their plasma; the group mean
concentration was 16 nmol/litre. By September, all plasma levels had
returned to the pre-exposure level, below the detection limit. The
mean half-life of gamma-HCH in plasma was calculated to be about
eight days. 2,4,6-, 2,3,5-, and 2,4,5-TCP were the major metabolites
of gamma-HCH (Drummond et al., 1988).
Neurological studies on 37 workers exposed to lindane over a
period of two years revealed three with serious
electroencephalographic disturbances; minor symptoms and signs were
seen in 14 of the workers. No change was observed in the
electroencephalographic patterns of 21 of the exposed individuals.
Blood levels of gamma-HCH were 0.002-0.34 mg/litre. The frequency of
clinical symptoms and electroencephalographic changes was higher
among individuals whose blood contained 0.02 mg/litre or more of
gamma-HCH (Czeglédi-Janko & Avar, 1970; American Conference of
Governmental Industrial Hygienists, Inc., 1986).
Tolot et al. (1969) and Schüttmann (1972) reported peripheral
neuropathies after contact with technical-grade HCH or lindane.
8.2.2 Irritation and sensitization
Behrbohm & Brandt (1959) described 26 cases of allergic and
toxic dermatitis in workers exposed during the manufacture of
technical-grade HCH. Patch testing with pure alpha-, beta-, gamma-,
and delta-HCH gave negative results, but positive reactions were
obtained with residual fractions. Baumgartner (1953) also described
skin sensitization in four workers involved in lindane manufacture.
Cases of allergic disease (rhinitis, conjunctivitis, and eczema)
were also reported among workers in the USSR exposed to lindane
(Krzhyzhanovskaya, 1973, and Bezuglikh et al., 1976; see Izmerov,
1983).
Patch tests were performed with 1% lindane dissolved in a
petroleum solvent on the upper back of 200 subjects, 105 men and 95
women, aged 18-76 years; 50 of them (34 men and 16 women) were
agricultural workers, 24 (18 men and six women) had worked on the
land in the past, and the other 53 men and 73 women had never used
pesticides. Results were read after 48 and 72 h. A positive reaction
was found in none of the 200 subjects tested (Lisi et al., 1986).
In a study to establish the optimal test concentration of lindane
and the frequency of irritant and sensitization reactions, Lisi et al.
(1987) tested 335 men and women, of whom 70 were employed in
agriculture, 25 had been employed in agriculture in the past and 240
had never been exposed to pesticides. The results of the patch test,
using 1% lindane in a petroleum solvent, were all negative, and no
sensitization reaction was observed.
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1 Microorganisms
9.1.1 Bacteria
The effect of lindane at concentrations up to 100 times the
recommended dose for field application (1 mg/kg of soil) was studied
on organic mineralization and on nitrification rates in alluvial
soil around Delhi, India. Lindane inhibited the evolution of CO2
from soil at a concentration of 100 mg/kg but not or only slightly
with 1 and 10 mg/kg over an incubation period of 120 days.
Nitrification by the bacterial species Nitrobacter and
Nitrosomonas was decreased with the dose of 100 mg/kg, and 10
mg/kg induced inhibition during the first three weeks after
application. Nitrification was restored within 35 days (Gaur &
Misra, 1977).
The influence of technical-grade HCH on the nitrifying
characteristics of Nitrosomonas and Nitrobacter species isolated
from an alluvial soil was studied in artificial growth media and in
a flooded, autoclaved soil. The concentrations tested were 0, 5, 20,
and 50 mg/kg soil or culture medium; the observation period was
10-40 days. The rate of nitrification was slower in autoclaved soil
than in culture media. Technical-grade HCH inhibited nitrification
at concentrations of 5 mg/kg and more (Ray, 1983).
Oxidation of substrate nitrogen was evaluated on the basis of
the appearance of nitrite in the medium in the presence of
Nitrosomonas europaea and on the disappearance of nitrite in the
presence of Nitrobacter agilis. N. agilis was sensitive to lindane
at concentrations as low as 1 µg/ml, and the highest concentration
tested, 1000 µg/litre, delayed nitrate production. In N. europaea,
lindane induced complete inhibition at a concentration of 10 µg/ml
within 6-7 days (Garretson & San Clemente, 1968).
9.1.2 Algae
9.1.2.1 Blue-green algae
Bringmann & Kühn (1978) saw no inhibition of growth of
Microcystis aeruginosa at concentrations of lindane up to 0.3
mg/litre, and Palmer & Maloney (1955) found no inhibition of the
growth of Microcystis aeruginosa or Cylindrospermum licheniforme
at 2 mg/litre. Both concentrations are higher than those that would
result in adverse effects in fish or crustaceans.
Concentrations of 50, 60, and 80 µg/ml of lindane were lowered
in the presence of 25 species of blue-green algae, thus reducing its
toxicity (Das & Singh, 1977).
9.1.2.2 Freshwater algae
Lindane was lethal to Scenedesmus acutus (age of culture 1,
3, or 5 days) at a concentration of 5 mg/litre after 5 days'
exposure. At concentrations of 1-5 mg/litre, 50% growth reduction
occurred (Krishnakumari, 1977). Jeanne-Levain (1974) also observed
about 50% inhibition of growth of Dunaliella bioculata with
lindane at 5 mg/litre; 10 mg/litre completely inhibited growth.
Similar findings were described by Jeanne (1979). No observable
inhibition of growth was found by Bringmann & Kühn (1978) in
Scenedesmus quadricauda at concentrations up to 1.9 mg/litre; and
Palmer & Maloney (1955) saw no inhibition in Scenedesmus obliquus,
Chlorella variegata, Gomphonema parvulum, or Nitzschia palea at
the only dose tested, 2 mg/litre.
9.1.2.3 Marine algae
Growth inhibition was studied in three species of marine algae
by Ukeles (1962), using concentrations of 1-9 mg/litre. No
inhibitory effect was found in Protococcus sp. at up to the
highest concentration tested or in Pheodactylum tricornutum at up
to 1 mg/litre. Chlorella sp. were slight inhibited at the lowest
concentration tested, but the dose-response relationship indicated
that the threshold was not far below this dose. In tests with green
algae and diatoms, growth was not inhibited by lindane at
concentrations below about 1 mg/litre.
9.1.3 Dinoflagellates, flagellates, and ciliates
Representative members of these three groups were exposed to
lindane at concentrations of 0.5-60 mg/litre (Jeanne-Levain, 1974).
Lethality was reached in Amphidinium carteri (dinoflagellate) at 2
mg/litre and in Tetrahymena pyriformis (ciliate) at > 10
mg/litre; no lethal effect was observed in Euglena gracilis
(flagellate) at up to the highest dose of 60 mg/litre. All doses had
inhibitory effects.
Ukeles (1962) observed inhibition of the growth of the
flagellates Monochrysis lutheri and Dunaliella euchlora with
concentrations of lindane > 5 mg/litre.
9.2 Aquatic organisms
9.2.1 Invertebrates
Green et al. (1986) investigated a range of 10 common European
freshwater macroinvertebrates in a continuous flow system to
establish their response to lindane at concentrations ranging from
25 ± 4.8 to 430 ± 32 µg/litre under similar test conditions: water
temperature, 11 ± 1 °C; pH, 7.5-8.0; dissolved oxygen, > 96%; and
hardness of water (as CaCO3), 92.9 ± 6.3 mg. The 96-h LC50 varied
from 4.5 up to > 430 µg/litre (Table 14). The ephemeropteran
Baetis rhodani and the plecopterans Leuctra moselyi and
Protonemura meyeri were the most sensitive species; Physa
fontinalis and Polycelis tenuis were the most tolerant.
Table 14. LC50 values for invertebrates
Taxon Species 96-h LC50 (µ/litre)
Insecta
Plecoptera Pteronarcys california 4.5
Leuctra moselyi < 130
Protonemura meyeri < 130
Ephemeroptera Baetis rhodani 54
Diptera Chironomus riparius 235
Trichoptera Hudropsyche angustipennis 330
Crustacea
Amphipoda Gammerus rhodani 48
Gammerus facia 10
Gammerus pulex 225
Isopoda Asellus aquaticus 375
Mollusca
Gastropoda Physa fontinalis > 430
Platyhelminthes
Tricladia Polycelis tenui s > 430
Annelida
Oligocheata Limnodrilus hoffmeisteri > 430
9.2.1.1 Crustaceans
The sensitivity of both freshwater and seawater crustaceans to
the acute toxicity of lindane is summarized in Table 15. In general,
freshwater crustaceans were much less sensitive to lindane than
those living in seawater. The LC50 values for freshwater
crustaceans are comparable to or about one order of magnitude higher
than the median values for fish, whereas seawater crustaceans are
generally an order of magnitude more sensitive than fish.
Daphnia magna were exposed to lindane at 11-210 µg/litre
continuously for 64 days, thus covering three succeeding generations
(Macek et al., 1976). A clear dose-dependent depression of
reproduction was found at higher concentrations. The authors
assessed the NOEL to be between 11 and 19 µg/litre. In the same
study, Gammarus fasciatus was exposed to lindane at concentrations
of 1.2-17.7 µg/litre. The NOEL for survival and reproductive success
was 4.3 µg/litre. Gammarus fasciatus is thus more sensitive to
both the acute and chronic effects of lindane.
9.2.1.2 Aquatic arthropods
The results of studies on the acute toxicity of lindane in
freshwater insects (mainly nymphs) are summarized in Table 15.
Aquatic insects can be seen to be sensitive to lindane, with
considerable differences between the tested species. The NOEL for
continuous exposure of two successive generations of Chironomus
tentans was 2.2 µg/litre (Macek et al., 1976).
The 48-h LC50 for the water mite Hydrachna trilobata Viets,
an aquatic arachnida, was 0.05 mg/litre (Nair, 1981).
9.2.1.3 Molluscs
The acute effects of lindane have been investigated in bivalves
and gastropods; no data were available on cephalopods. The LC50
values determined (Table 16) are all > 1 mg/litre, and therefore at
least one order of magnitude higher than those reported for fish,
indicating that molluscs are much less sensitive to the acute action
of lindane.
Butler (1963b) determined shell growth in oysters exposed to
various concentrations of lindane. When sufficiently irritated,
oysters close their shells, do not feed and therefore do not grow.
Even at the lowest dose tested, 1 mg/litre, a decreased shell growth
of 43% was observed.
The freshwater snail Lymnea stagnalis was exposed to lindane
at concentrations of 1 or 2 mg/litre for periods up to seven weeks,
and therefore at least two successive generations. No increase in
mortality over that in controls was observed at either
concentration. A slight decrease in shell growth was found at 2
mg/litre, and egg production was reduced at 1 and 2 mg/litre.
Embryonic development was also disturbed at both concentrations in a
dose-dependent manner. A NOEL for reproduction was not obtained in
this study, but the results suggest that it is close to 1 mg/litre
(Bluzat & Seuge, 1979; Seuge & Bluzat, 1979a,b, 1982).
In a study on the effects of lindane on egg production in the
mud snail Nassa obsoleta (Eisler, 1970a), the number of egg cases
deposited by day 33 after treatment following initial exposure for
96 h was measured. A clear NOEL was found at 1 mg/litre; the next
dose tested (10 mg/litre) induced a clear decrease.
Table 15. Acute toxicity of gamma-HCH to freshwater and marine crustaceans
Species Size (g, mm, Temperature Test 96-h LC50 Reference
cm, age) (°C) conditions (µg/litre)
Freshwater
Daphnia magna 24-h old 20 Static 516 Randall et al. (1979)
Static 485 Juhnkje & Lüdemann (1978)
18-h old 20 Static 1100 Sanders & Cope (1966)
Daphnia pulex 18-h old 21 Static 460 Sanders & Cope (1966)
Gammarus fasciatus Static 39 Juhnke & Lüdemann (1978)
Gammarus pulex 1.0-1.5 cm 15 Statica 34 Abel (1980)
Gammarus lacustris 2 months? 21 Static 48 Sanders (1969)
Simocephalus serrulatus 18-h old 21 Static 880 Sanders & Cope (1966)
Marine
Pink shrimp (Penaeus duorarum) 30-52 mm 24-26 Flow-through 0.17 Schimmel et al. (1977)
Brown shrimp (Penaeus aztecus) Adult 30 Flow-through 0.4 Butler (1963b)
Brown shrimp (Crangon crangon) - 15 Static 1-3.3 Portmann (1970)
Grass shrimp (Palaemonetes pugio) - 23-27 Flow-through 4.4 Schimmel et al. (1977)
Grass shrimp (Palaemonetes vulgaris) 0.47 g 20 Static 10 Eisler (1969)
Sand shrimp (Crangon septemspinosa) 0.25 g 20 Static 5 Eisler (1969)
Hermit crab (Pagurus longicarpus) 0.28 g 20 Static 5 Eisler (1969)
Mysid shrimp (Mysidopsis bahia) 8-9.5 mm 23-25 Flow-through 6.3 Schimmel et al. (1977)
a Water renewed regularly
Table 16. Acute toxicity of gamma-HCH to aquatic invertebrates
Species Freshwater/ Size (g, mm, Temperature Test 96-h LC50 Reference
Marine water cm, age) (°C) conditions (mg/litre)
Bivalves
Mytulis galtoprovincialis Marine 50-70 mm 17 Static 5.5 Rao (1981)
Mercenaria mercenaria Marine Eggs 20 Static > 10 Eister (1970a)
Marine Eggs 24 Static > 10a Davis & Hidu (1969)
Crassostrea virginica Marine Eggs 24 Static 9.1a Davis & Hidu (1969)
Cardium edule Marine - 15 Static 10a Portmann (1970)
Gastropods
Physa acuta Freshwater - 20-28 Static 8.1a Hashimoto & Nishiuchi (1981)
Semisulcospira libertina Freshwater - 20-28 Static 6.2a Hashimoto & Nishiuchi (1981)
Indoplanorbis exastus Freshwater - 20-28 Static 7.1a Hashimoto & Nishiuchi (1981)
Lymnea stagnalis Freshwater Staticb 7.3a Bluzat & Seuge (1979)
Insects
Cloeon diprerum Freshwater - 20-28 Static 0.15a Hashimoto & Nishiuchi (1981)
Chironomus tentans Freshwater Static 0.207a Juhnke & Lüdemann (1978)
Pteronarcys californica Freshwater 30-35 mm 15.5 Static 0.0045 Sanders & Cope (1968)
a 48-h LC50
b Water renewed regularly
Davis & Hidu (1969) found 67% survival of the larvae of the
hard clam Mercenaria mercenaria over 12 days after exposure to
lindane at a concentration of 10 mg/litre. For oysters (Crassostrea
virginica), 50% survival was seen after exposure at 9.1 mg/litre
over 48 h.
Overall, these studies show that reproduction of molluscs is
not adversely affected at concentrations just below 1 mg/litre, a
level much higher than the NOELs for fish and crustaceans.
9.2.2 Fish
9.2.2.1 Acute toxicity
The results of studies on the acute toxicity of lindane in fish
have been reported in the literature since 1959, although in most of
these no data were given on the purity of the lindane used. Since
there is no significant difference in the values reported around
1960 and those reported in 1975-80, however, data on the acute
toxicity of lindane are summarized here regardless of whether the
purity of the compound tested was reported.
LC50 values for lindane in several fish species are
summarized in Table 17. Most values fall within a range of 0.02-0.09
mg/litre, the majority being around 0.05 mg/litre. Only Macek &
McAllister (1970) found an extraordinarily low LC50 value for
brown trout of 0.002 mg/litre.
The symptoms of acute poisoning are mainly gross irritability,
loss of equilibrium, changes in pigmentation and localized
peripheral haemorrhage. Irritability appears within the first
minutes of exposure and is accompanied by loss of equilibrium and
disturbed swimming motion. Poisoned fish show signs of respiratory
distress. Haemorrhages appear at sub-lethal doses 2-4 days after the
beginning of exposure.
A clear temperature dependence of the LC50 was found in
bluegills (Cope, 1965; Macek et al., 1969), lindane being more toxic
at higher temperatures, although the range of LC50 values was the
same. Macek et al. (1969) found LC50 values of 0.054 mg/litre at
12.7 °C and 0.037 mg/litre at 23.8 °C.
In two studies, wild populations of mosquito fish were compared
to laboratory strains and to each other with respect to the toxic
action of lindane (Culley & Ferguson, 1969). Wild populations from
areas that had previously been treated with lindane tolerated higher
concentrations of the substance: one wild population had a LC50
value of 3.104 mg/litre, whereas a susceptible population from a
different region had a value of 0.074 mg/litre. These results
suggest that wild populations can adapt to lindane when exposed
repeatedly. Boyd & Ferguson (1964) found a similar effect.
9.2.2.2 Long-term toxicity
Macek et al. (1969) exposed bluegills to lindane at 0.6-9.1
µg/litre for 18 months. No adverse effect was observed at any of the
tested concentrations; the NOEL was therefore considered to be
> 9.1 µg/litre. In the same study, fathead minnows (Pimephales
promelas) were exposed to lindane at 1.4-23.5 µg/litre for 43
weeks. A statistically significant increase in mortality was
observed at the highest dose. Growth of the surviving fish was not
adversely affected, and spawning appeared to be normal in all test
groups. The NOEL in this experiment was considered to be 9.1
µg/litre.
Macek et al. (1969) exposed brook trout (Salvelinus
fontinalis) to lindane at concentrations of 1.0-16.6 µg/litre for
261 days. Only slight effects on growth were observed at the end of
the exposure period. Although no statistical assessment of spawning
was performed, fish exposed to the highest test level were adversely
affected in this respect, and the NOEL was set at 8.8 µg/litre.
The results of these studies indicate that concentrations not
far below those inducing 50% mortality are well tolerated for long
periods (up to 18 months). A 5-10-fold difference can be seen
between the maximum dose tolerated in long-term tests and the LC50
in the three species tested (Macek et al., 1976).
9.2.2.3 Reproduction
The reproductive effects of lindane were tested in bluegills,
fathead minnows, and brook trout (Macek et al., 1969). Spawning,
hatchability of eggs, and survival of the fry appeared not to be
adversely affected by concentrations of up to 9.1 µg/litre in
guppies, up to 23.4 µg/litre in fathead minnows, and up to 2.1
µg/litre in brook trout.
9.2.3 Amphibia
9.2.3.1 Acute toxicity
All of the published studies on the acute toxicity of lindane
in amphibia (Table 18) were undertaken with tadpoles. Tests on
tadpoles are considered to be the most reliable for assessing
possible adverse effects to aquatic organisms since these larvae
live exclusively in an aquatic environment, whereas adults spend a
major part of their lifetime outside the water. The results show
that larvae of amphibia are less sensitive to lindane than fish.
Table 17. Acute toxicity of gamma-HCH to fish
Species Freshwater/ Size (g, Temperature Test conditions 96-h LC50 Reference
Marine water mm, cm) (°C) (µg/litre)
Rainbow trout (Salmo gairdneri) Freshwater 0.69 g 12 Statica 32 McLeay (1976)
Freshwater 3.2 g 20 Static 38 Katz (1961)
0.6-1.7 g 13 Static 27 Macek & McAllister (1970)
0.7 g 13 Static 22 Cope (1965)
3-cm fry 12 Flow-through 22 Tooby & Durbin (1975)
Yearling 12 Flow-through 30 Tooby & Durbin (1975)
2.6 g 15 No detail 34b Dion (1984)
Brown trout (Salmo trutta) Freshwater 0.6-1.7 g 13 Static 2 Macek & McAllister (1970)
1.1 g 15 No detail 38b Dion (1984)
Coho salmon (Oncorhynchus kisutch) Freshwater 2.7-4.1 g 20 Static 50 Katz (1961)
0.6-1.7 g 13 Static 41 Macek & McAllister (1970)
Chinook salmon (Oncorhynchus Freshwater 1.5-5 g 20 Static 40 Katz (1961)
tschawytscha)
Bluegill (Lepomis macrochirus) Freshwater 0.6-1.7 g 18 Static 68 Macek & McAllister (1970)
1.0 g 18 Static 53 Cope (1965)
0.26 g 19 Static 57 Randall et al. (1979)
- 25 Static 77 Henderson et al. (1959)
0.6-1.5 g 18 Static 51 Macek et al. (1969)
Redear sunfish (Lepomis microlophus) Freshwater 0.6-1.7 g 18 Static 83 Macek & McAllister (1970)
Threespine stickleback (Gasterosteus Freshwater 0.38-0.77 g Room Static 44 and Katz (1961)
aculeatus) temperature 50c
Carp (Cyprimus carpio) Freshwater 0.6-1.7 g 18 Static 90 Macek & McAllister (1970)
6.8 cm 17-19 Static 280b Lüdemann & Neumann (1960a)
- - Static 310b Hashimoto & Nishiuchi (1981)
Table 18. Acute toxicity (48-h LC50) of lindane in tadpodes of freshwater amphibia under static conditions
Species Size (g, mm, cm, age) Temperature (°C) LC50 (mg/litre) Reference
Pseudacris triserata 7-days old 15.5 3.8 Sanders (1970)
Bufo woodhousii 4-5-weeks old 15.5 5.4 Sanders (1970)
Bufo bufo japonicus - - 24 Hashimoto & Nishiuchi (1981)
Bufo bufo L. 25-30 mm 18-21 0.3 Lüdemann & Neumann (1960b)
9.2.3.2 Effects on hatching and larval development
Marchal-Segault & Ramade (1981) exposed eggs and larvae of
Xenopus laevis to lindane at concentrations of 0.5-2 mg/litre in
tap water. Hatchability was reduced by the highest dose only;
however, development of the larvae was disturbed at all
concentrations testing, as seen by lowered body weights, longer
periods (four weeks) from hatching to metamorphosis, and
morphological abnormalities. Altered function of the
hypothalamo-hypophyseal axis, which regulates growth and
metamorphosis, and dysfunction of the intermediate lobe of the
hypophysis, which controls pigmentation, were suggested. A NOEL
could not be established.
9.3 Terrestrial organisms
9.3.1 Honey-bees
Atkins et al. (1973) estimated the LD50 in the honey-bee to
be 0.56 µg.
9.3.2 Birds
9.3.2.1 Acute toxicity
Some of the studies of the acute toxicity of lindane in birds
were undertaken soon after its introduction as an insecticide, and,
in these, the quality of lindane used is not specified.
Nevertheless, these studies are included in this review, as well as
studies in which no precise LD50 could be obtained.
The results are summarized in Table 19. The values obtained
cover a wide range, but most are in the order of 100 mg/kg body
weight. Common symptoms of poisoning are vomiting and loss of
appetite, loss of weight, and hyperexcitability; central nervous
system symptoms occur as incoordination, convulsions, and tremors
(Rosenberg et al.,1953; Dahlen & Haugen, 1954; Adamic, 1958; Turtle
et al., 1963; Dittrich, 1966).
In hens, lethal oral doses of 330-1440 mg/kg body weight caused
inflammation of the gastrointestinal tract, degeneration of the
liver and kidneys, and changes in ganglionic cells (Adamic, 1958).
Similar findings were obtained in bobwhite quails and mourning doves
(Rosenberg et al., 1953; Dahlen & Haugen, 1954) at 120-210 mg/kg
body weight. In doves, doses of > 300 mg/kg body weight caused
mainly liver atrophy, congested lungs and kidneys, and haemorrhage.
Table 19. Toxicity of lindane to birds
Species Parameter Concentration (mg/kg)a Reference
Bobwhite quail (Colinus virginianus) 5-d LC50 882 (755-1041) Hill et al. (1975)
acute LD50 120-130 (male) Dahlen & Haugen (1954)
acute LD50 190-210 (female) Dahlen & Haugen (1954)
Japanese quail (Cotumix coturnix japonica) 5-d LC50 490 (408-589) Hill & Camardese (1986)
205 and 425 (347-520) Clausing et al. (1980)
Ring-necked pheasant (Phasianus colchicus) 5-d LC50 561 (445-590) Hill et al. (1975)
Mallard (Anas platyrhynchos) acute LD50 > 2000 (male) Hudson et al. (1984)
5-d LC50 > 5000 Hill et al. (1975)
Starling (Stumus vulgaris) acute LD50 100 Schafer (1972)
Red-winged blackbird (Agelaius phoeniceus) acute LD50 75 Schafer (1972)
Common grackle (Quiscalus quiscula) acute LD50 > 100 Schafer (1972)
House sparrow (Passer domesticus) acute LD50 56 (320-100) Schafer (1972)
Common crow (Corvus brachyrhynchos) acute LD50 > 100 Schafer (1972)
Mourning dove (Streptopelia risoria) acute LD50 350-400 Dahlen & Haugen (1954)
Feral pigeon (Columba livia) acute LD50 > 600 Turtle et al. (1963)
a Acute LD50 expressed as milligrams per kilogram body weight in a single oral dose; otherwise,
concentration expressed as milligrams per kilogram food (i.e., birds were fed with a dosed diet
for 5 days followed by a "clean" diet for 3 days)
The concentrations of lindane in the diet that caused 50%
mortality in young and adult bobwhite quail, ring-necked pheasants
and mallard ducks within < 10 days and < 100 days (Dewitt et al.,
1963) are summarized in Table 20.
Table 20. Oral LD50 of lindane in birdsa
Species Lindane intake (mg/kg body weight)
< 10 days < 100 days
Young bobwhite 1070 930
Adult bobwhite - 1050
Young ring-necked pheasant 175 > 1800
Adult ring-necked pheasant - > 630
Young mallard 415 -
Adult mallard 1000 -
a From Dewitt et al. (1963)
9.3.2.2 Short-term toxicity
Chen & Liang (1956) fed white leghorn chickens and hybrid
native ducks diets containing lindane at 2, 4, or 10 mg/kg of diet
for three months. No adverse effect was observed in either species
at any dose.
When laying hens were fed diets containing lindane at 0.01,
0.1, 1, or 10 mg/kg of diet for 60 days, no effect was observed on
body weight gain, mortality, clinical symptoms, or egg production.
The authors concluded that lindane does not adversely affect
reproduction in hens at doses up to 10 mg/kg of diet (Ware & Naber,
1961).
Harrison et al. (1963) fed diets containing lindane at 4, 16,
or 64 mg/kg to white Leghorn x Australorp chickens for 27 days.
Increased mortality was seen in the highest dose group, and the two
higher doses resulted in enlarged livers. No pathological change was
found in the animals given the lowest dose, but in the two higher
dose groups dose-dependent liver hypertrophy was observed. Tissues
were not examined microscopically. The NOEL in this experiment was
concluded to be 4 mg/kg of diet.
The 30-day oral LD50 for male mallard ducks (12 animals) was
30 mg/kg body weight; as the acute LD50 was > 2000 mg/kg body
weight, the toxic action of lindane appears to be cumulative (Hudson
et al., 1984).
The LC50 values for lindane given in the diet for five days
were 882 mg/kg of diet in bobwhite quail (aged 9 days), 425 mg/kg of
diet in Japanese quail (aged 7 days), 561 mg/kg of diet in
ring-necked pheasant (aged 10 days), and > 5000 mg/kg of diet in
mallard ducks (aged 15 days) (Hill et al., 1975).
(a) Effect on egg-shell quality: Whitehead et al. (1972a,b,
1974) found that the shells of hens' eggs were not adversely
affected by administration of lindane in amounts up to 200 mg/kg of
diet; however, egg production was reduced at 100 and 200 mg/kg of
diet. Doses of up to 100 mg/kg of diet had no effect on
hatchability, egg weight, yolk weight, shell thickness, calcium
content, shearing strength or structure. The NOEL was 10 mg/kg of
diet. Similar findings were obtained in Japanese quail.
(b) Field experience: A population of Canada geese (Branta
canadensis) living in the Pacific Northwest of the USA was
observed from 1978 through 1981. Lowered reproductive success,
increased mortality among adults and a population decline in this
region were associated with the use of heptachlor for treating wheat
seed. This hypothesis was supported by the results of analyses of
eggs and tissues. In 1979, heptachlor was replaced by lindane for
use in this area; the reproductive success of the geese increased,
mortality decreased, and the population increased. There was no
evidence of either biomagnification of lindane from treated seed to
goose tissues or eggs or of induction of adverse effects by lindane
(Blus et al., 1984).
9.3.2.3 Reproduction
Lindane (99.8% in olive oil) was administered by stomach tube
to four groups of laying ducks (Anas platyrhynchus domesticus),
comprising one drake and four ducks, at doses of 0 or 20 mg/kg body
weight daily, three times per week, or twice a week for eight weeks.
Egg laying stopped immediately in the groups treated daily and three
times weekly and was irregular when it resumed, with drastically
reduced clutch sizes. The effect in the group treated twice weekly
was marginal. At the end of treatment, the laying frequencies for
the four groups were 50%, 8.3%, 11.7%, and 40%, respectively.
Hepatic, plasma, and ovarian vitellogenin levels were reduced in the
groups treated daily and three times per week; the ovaries of the
birds in these groups lacked mature vitellogenic follicles, and the
thecal layer of moderately differentiated oocytes became highly
atrophic. Levels of liver RNA were markedly reduced. A single
injection of stilboestrol at 50 mg/kg body weight restored egg
laying and the other parameters to normal within 24 h, suggesting
that lindane imposed its effects by inducing oestradiol
insufficiency (Chakravarty et al., 1986).
9.3.3 Mammals
The toxicity of lindane to bats has been studied because of its
use in timber treatment. Racey & Swift (1986) exposed pipistrelle
bats (Pipistrellus pipistrellus) to 1% lindane, both in
combination with 5% pentachlorophenol in an organic solvent and
alone. When it was applied in combination with pentachlorophenol, at
the rates recommended by the manufacturer, to wooden roosting boxes
six weeks before bats used them, the animals were killed within
seven days; when the combination was applied 14 months before use,
the animals died within 23 days. When lindane was applied alone two
weeks before exposure of bats in the boxes, all animals died within
four days. These results were significant at the 0.1% level.
Boyd et al. (1988) exposed pipistrelle bats to wood blocks
coated with lindane at 9.9 mg/m2 for 44 days and then for a further
44 days to blocks coated with lindane at 866 mg/m2, 24 h after
coating the blocks. Significant mortality ( p < 0.007) was
recorded. In a second experiment, all bats exposed to lindane at
either 147 or 211 mg/m2 died within 17 days, whereas no death
occurred among controls exposed to the solvent only.
Turner (1979) studied the distribution and concentration of
gamma-HCH in maternal and fetal tissues of a 6.5-year-old desert
bighorn (Ovis canadensis cremnobates). The maternal organs and
tissues and the tissues of the term ram fetus contained residue
levels ranging from none detected to 0.01 mg/kg on a fat basis.
Residues of 0.01 mg/kg were present in adipose tissues, muscle,
liver, gonads, and placenta. Placental transfer of gamma-HCH thus
appears to be very low.
9.4 Appraisal
The toxicity of lindane to organisms in the environment must be
assessed on the basis of the results of laboratory toxicity tests
and of the probable bioavailability of lindane to similar organisms
exposed in the wild. The strong adsorption of lindane to particles
might be expected to reduce its toxic effects below that seen in
laboratory studies of microorganisms in culture and of aquatic
organisms in water without sediment; however, there is insufficient
information to substantiate this hypothesis. No information was
available on the toxicity of lindane to organisms that feed on or
live in sediment.
Low levels of residues in birds in the wild, coupled with the
reported low toxicity of lindane in laboratory tests, make it
unlikely that it affects birds in the wild.
Bats are killed by applications of lindane to wood at normal
rates and are affected by residues of past wood treatment. Since
many bat species are declining in numbers or are extremely rare,
lindane must be regarded as a major hazard and its use avoided in
areas where bats might be found. Other mammals are unlikely to be
adversely affected by this compound.
10. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
The International Agency for Research on Cancer (1987)
evaluated the hexachlorocyclohexanes and concluded that there is
sufficient evidence for the carcinogenicity to experimental animals
of the technical grade and the alpha isomer; such evidence was
considered to be limited for the beta and gamma isomers. There is
considered to be inadequate evidence for their carcinogenicity in
humans. Hexachlorocyclohexanes were thus classified into group 2B,
possibly carcinogenic to humans.
WHO (1990) classified technical-grade lindane as 'moderately
hazardous' in normal use, on the basis of an LD50 of 88 mg/kg.
The WHO/FAO (1975) issued a data sheet on lindane (No. 12), dealing
with labelling, safe handling, transport, storage, disposal,
decontamination, training, and medical supervision of workers,
first-aid and medical treatment.
Lindane was evaluated by the FAO/WHO Joint Meeting on Pesticide
Residues in 1966, 1967, 1968, 1969, 1973, 1974, 1975, 1977, 1979,
and 1989 (FAO/WHO, 1967, 1968, 1969, 1970, 1974, 1975, 1976, 1978,
1980, 1990). A maximal acceptable daily intake of lindane in humans
was established at 0-0.008 mg/kg body weight by the 1989 Joint
Meeting (WHO, 1990). This value is based on a NOAEL of 10 mg/kg in
the diet, equivalent to 0.75 mg/kg body weight per day in rats and
1.6 mg/kg body weight per day in dogs. Maximum residue limits have
been recommended for more than 35 commodities, ranging from 0.01
mg/kg in milk to 3 mg/kg on strawberries; a limit of 0.5 mg/kg was
recommended for most fruit and vegetables (Codex Alimentarius
Commission, 1986; Table 21).
Table 21. Maximum residue limits (MRL) for gamma-HCH of the
Codex Alimentarius Commission (1986)
Crop/Commodity MRL (mg/kg)
Apples 0.5
Beans (dried) 1
Brussels sprouts 0.5
Cabbage 0.5
Cabbage, Savoy 0.5
Carrots 0.2 Eb
Cattle, carcase meat (in the carcase fat) 2
Cauliflower 0.5
Cereal grains (including rice) 0.5
Cherries 0.5
Cocoa beans 1
Cocoa butter 1
Cocoa mass 1
Cranberries 3
Currants (red) 0.5
Eggs (on a shell-free basis) 0.1 E
Endive 2
Grapes 0.5
Kohlrabi 1
Lettuce 2
Milk 0.01
Pears 0.5
Peas 0.1
Pigs, carcase meat (in the carcase fat) 2
Plums 0.5
Potatoes 0.05a
Poultry (in the carcase fat) 0.7 E
Radishes 1
Rapeseed 0.05a
Sheep, carcase meat (in the carcase fat) 2
Strawberries 3
Sugar beets (roots) 0.1
Sugar beets (tops) 0.1
Spinach 2
Tomatoes 2
a Level at or about the limit of determination
b E, Extraneous residue limit
APPENDIX I. CHEMICAL STRUCTURE
The basic structure of HCH is a closed chain of six carbon
atoms. The structure can have two spatial forms, a cis and a
trans configuration. Each carbon atom is bound to a hydrogen and a
chlorine atom, and one of these substituents forms a plane with the
two connecting carbon atoms. Since this plane is parallel to the
'equator' of the molecule, this atom is said to be in the equatorial
position. The bond with the other atom is parallel to the 'axis' of
the molecule and is thus in the axial position. Owing to the size of
the chlorine atom, the carbon atoms are not free to rotate, so the
positions of the chlorine and hydrogen atoms are fixed: one is
always in the equatorial position and the other in the axial
position.
The various combinations of the spatial orientations of the
hydrogen and chlorine atoms on each of the carbon atoms of
cyclohexane result in different isomeric compounds. Theoretically,
17 isomers of HCH are possible; but, owing to spatial
incompatibilities and thermodynamic instability, only nine isomers
have been detected. They all have the trans configuration. In the
beta isomer, all of the chlorine atoms are in the equatorial
position. The positions in the major isomers of HCH are shown in
Table 22 (Demozay & Marechal, 1972; Van Velsen, 1986).
Table 22. Positions of chlorine atoms in the major isomers of
HCHa
Isomer Chlorine positionb Physical structure
alphac AAEEEE Monoclinic prisms
ß EEEEEE Octahedral cubic
lamda AAAEEE Monoclinic crystals
delta AEEEEE Crystals/fine patelets
epsilon AEEAEE Monoclinic needles
or hexagonal
monoclinic crystals
a From van Velsen (1986)
b A, axial position; E, equatorial position
c Racemate of two optical isomers
REFERENCES
Abalis IM, Eldefrawi ME, & Eldefrawi AT (1985) High-affinity
stereospecific binding of cyclodiene insecticides and gamma-
hexachlorocyclohexane to gamma-aminobutyric acid receptors of rat
brain. Pestic Biochem Physiol 24: 95-102.
Abalis IM, Eldefrawi ME, & Eidefrawi AT (1986) Effects of
insecticides on GABA-induced chloride influx into rat brain
microsacs. J Toxicol Environ Health 18: 13-23.
Abel PD (1980) Toxicity of gamma-hexachlorocyclohexane (lindane) to
Gammarus pulex: Mortality in relation to concentration and
duration of exposure. Freshwater Biol 10: 251-259.
Adamic S (1958) [The influence of gamma isomers of
hexachlorocyclohexane on hen and duck.] Veterinarija (Sarajevo) 7:
329-334 (in Yugoslav).
Adams M, Coon FB, & Poling CE (1974) Insecticides in the tissues of
four generations of rats fed different dietary fats containing a
mixture of chlorinated hydrocarbon insecticides. J Agric Food Chem
22(1): 69-75.
Advisory Committee on Pesticides and Other Toxic Chemicals (1969)
Further review of certain persistent organochlorine pesticides used
in Groat Britain. London, Her Majesty's Stationery Office.
Agnihotri NP, Pandey SY, Jain HK, & Srivastava KP (1977) Persistence
of aldrin, dieldrin, lindane, heptachlor, and pp'-DDT in soil. J
Entomol Res 1(1): 89-91.
Ahmed FE, Hart RW, & Lewis NJ (1977) Pesticide induced DNA damage
and its repair in cultured human cells. Mutat Res 42: 161-174.
Aiyar AS (1980) Biological implications of pesticides: studies with
lindane. Manage Environ pp. 182-192.
Alabaster JS (1969) Survival of fish in 164 herbicides,
insecticides, fungicides, wetting agents, and miscellaneous
substances. Int Pest Control 11(2): 29-35.
Albahary C, Dubrisay J, & Guerin (1957) Pancytopenic rebelle au
lindane [isomère gamma de l'hexachlorocyclohexane]. Arch Mal Prof
Méd Trav Sécur Soc 18: 687-691.
Al-Omar MA, Al-Bassomy M, Al-Ogaily NV, & Al-Din Shebl D (1985)
Residue levels of organochlorine insecticides in lamb and beef from
Bagdad. Bull Environ Contam Toxicol 34: 509-512.
Al-Omar MA, AbduI-Jalil FH, Al-Ogaily NH, Tawfiq SJ, & Al-Bassomy MA
(1986) A follow-up study of maternal milk contamination with
organochlorine insecticide residues. Environ Pollut A42: 790-791.
American Conference of Governmental Industrial Hygienists Inc.
(1986) Documentation of the threshold limit values and biological
exposure indices, 5th ed. Cincinnati, Ohio, p. 348.
Anderson D & Styles JA (1978) The bacterial mutation test. Br J
Cancer 37: 924-930.
Angerer J & Barchet R (1983) [alpha, ß- and gamma-HCH in serum.] In:
Henschler D, ed. [Analytical methods for the testing of working
media prejudicial to health. Analyses in biological material, 7th
instalment.] Weinheim, Verlag Chemie, Vol. 2 (in German).
Angerer J, Maasz R, & Heinrich R (1983) Occupational exposure to
hexachlorocyclohexane. VI. Metabolism of gamma-hexachlorocyclohexane
in man. Int Arch Occup Environ Health 52: 59-67.
Anon (1984) [Organochlorine compounds including PCB: 1984 nitrition
report.] Frankfurt am Main, German Nutrition Association (in
German).
Anon (1989) FDA industry trying to cut pesticide contamination of
lanolin. Pest Toxic Chem News, 3 May, pp. 26-27.
Antonovic EA (1958) Evaluation of the toxicity of gamma-isomer of
hexachlorocyclohexane and its standardization in foodstuffs. Vopr
Pitan 17(6): 54-59.
Arbeitsgemeinschaft für die Reinhaltung der Elbe (1982) [Chlorinated
hydrocarbons; data for the River Elbe. Report on the result of the
programme for the measurement of chlorinated hydrocarbons in the
section of the Elbe between Schnackenburg and the North Sea, 1980-
1982]. Hamburg, Study Group for Keeping the Elbe Clean, pp. 64-65,
84, 86-94 (in German).
Arbeitsgemeinschaft für die Reinhaltung der Elbe (1988) [Water
quality data for the Elbe from Schnackenburg to the sea. Numerical
table, 1988.] Hamburg, Study Group for Keeping the Elbe Clean, p.
158 (in German).
Artigas F, Martinez E, Camon L, Gelpi E, & Rodriguez-Farre E (1988)
Brain metabolites of lindane and related isomers: Identification by
negative ion mass spectrometry. Toxicology 49: 57-63.
Ashwood-Smith MJ, Trevino J, & Ring R (1972) Mutagenicity of
dichlorvos. Nature 240: 418-419.
van Asperen K (1958) [Distribution and metabolism of
hexachlorocyclohexane in mammals.] In: [Proceedings of the IVth
International Congress on Crop Protection, Hamburg, 6-15 September
1957.] Braunschweig, ACO Druck GmbH, Vol. 2, pp. 1619-1623 (in
German).
Association of Official Government Chemists (1980) AOAC Methods: 29.
Pesticide residues. Multiresidue methods: general method for
organochlorine and organophosphorus pesticides (1), Washington, DC,
pp. 466-475.
Atkins EL, Greywood EA, & MacDonald RL (1973) Toxicity of pesticides
and other agricultural chemicals to honey bees. Riverside,
California, University of California, Agricultural Extension
(Unpublished report M-16).
Atlas E & Giam CS (1981) Global transport of organic pollutants:
Ambient concentrations in the remote marine atmosphere. Science 211:
163-165.
Bacci E, Calamari D, Gaggi C, Fanelli R, Focardi S, & Morosini M
(1986) Chlorinated hydrocarbons in lichen and moss samples from the
Antarctic Peninsula. Chemosphere 15(6): 747-754.
Balaschow WE (1964) [Haematopoietic disorders following the action
of the gamma-isomer of hexachlorocyclohexane.] Gig Tr Prof Zabol 8:
55-56 (in Russian).
Balba MH & Saha JG (1974) Metabolism of lindane-14C by wheat
plants grown from treated seed. Environ Lett 7(3): 181-194.
Baluja G, Murado MA, & Tejedor MC (1975) Adsorption-desorption of
lindane and aldrin by soils as affected by soil main components.
Environ Qual Saf Suppl. 3: 243-249.
Barke A (1950) [Toxicity of the gamma-isomers of
hexachlorocyclohexane.] Tierärtztl Umsch 5: 61-63 (in German).
Baron RL, Copeland F, & Walton MS (1975) Pharmacokinetics of
organochlorine pesticides in mammalian adipose tissue. Environ Qual
Saf Suppl. 3: 855-860.
Bauer U (1972) [Cncentration of insecticides, chlorinated
hydrocarbons and PCB in algae. Schr Reihe Verh Wasser- Boden-Lufthyg
37: 211-219 (in German).
Baumann K, Behling, K, Brassow HL, & Stapel K (1981) Occupational
exposure to hexachlorocyclohexane. III. Neurophysiological findings
and neuromuscular function in chronically exposed workers. Int Arch
Occup Environ Health 48: 165-172.
Baumgartner O (1953) [Sensitization to hexachlorocyclohexane.]
Schweiz Med Wochenschr 83(45): 1093-1094 (in German).
Bednarek W, Hansdorf W, Jörissen V, Schulte E, & Wegener H (1975)
[The effects of chemical pollution of the environment on birds of
prey in two test areas of Westphalia.] J Ornitol 116(2): 181-194 (in
German).
Behrbohm P & Brandt B (1959) [Allergic and toxic dermatitis in the
manufacturing and processing of hexachlorocyclohexane.] Arch
Gewerbepathol Gewerbehyg 17: 365-383 (in German).
Benes V & Sram R (1969) Mutagenic activity of some pesticides in
Drosophila melanogaster. Ind Med Surg 38(12): 442-444.
Benezet HJ & Matsumura F (1973) Isomerization of gamma-BHC to alpha-
BHC in the environment. Nature 243: 480-481.
Bercovici B, Wassermann M, Cucos S, Ron M, Wassermann D, & Pines A
(1983) Serum levels of polychlorinated biphenyls and some
organochlorine insecticides in women with recent and former missed
abortion. Environ Res 30: 169-174.
Bernhardt H & Ziemons E (1974) [Pesticide content of 19 German
reservoirs. German Association of Gas and Water Experts.] Wasser 5:
1-104 (in German).
Berry DH, Brewster MA, Watson R, & Neuberg RW (1987) Untoward
effects associated with lindane abuse. Am J Dis Child 141(2): 125-
126.
Bertram H., Kemper FH, & Zenzen C (1980) [Occurrence of HCH isomers
in man.] In: Deutsche Forschungsgemeinschaft, ed.
[Hexachlorcyclohexane as a harmful substance in foods. Papers from
two symposia of the Senate Committee on the Testing of Residues in
Foods, held on 28-29 November 1979 and 6 March 1980.] Weinheim,
Verlag Chemie, pp. 155-163 (in German).
Bidleman TF & Leonard R (1982) Aerial transport of pesticides over
the northern Indian Ocean and adjacent seas. Atmos Environ 16: 1099-
1107.
Blok SMG, Greve PA, Sangster B, Savelkoul TJF, & Wegman RCC (1984)
[Investigation of normally occurring values of a number of
organochlorine pesticides and related compounds and their
metabolites, of polychlorobiphenyls and of chlorophenols in the
blood or plasma of healthy volunteers.] Bilthoven, National
Institute of Public Health and Environmental Hygiene (Report No.
638101001) (in Dutch).
Blus LJ, Henny C J, Lenhart D J, & Kaiser TE (1984) Effects of
heptachlor- and lindane-treated seed on Canada geese. J Wildl Manage
48(4): 1097-1110.
Bluzat R & Seuge J (1979) Etude de la toxicité chronique de deux
insecticides (carbaryl et lindane) de la generation F1 de Lymnea
stagnalis L. (mollusque gasteropode pulmone). I. Croissance des
coquilles. Hydrobiologia 65(3): 245-255.
Bosch AL (1987a) Dermal absorption of 14C-lindane in male rabbits.
Madison, Wisconsin, Hazleton Laboratories America, Inc. (Unpublished
report No. 6188-103, submitted to WHO by CIEL).
Bosch AL (1987b) Dermal absorption of 14C-lindane in male rats.
Madison, Wisconsin, Hazleton Laboratories America, Inc. (Unpublished
report No. 6188-104, submitted to WHO by CIEL).
Boulekbache H (1980) Study of the toxicity of the alpha-, beta-, and
gamma isomers of hexachlorocyclohexane to fish, using the guppy as
the test species. Paris, Université de Paris VII, Laboratoire
d'Anatomie comparée (Unpublished report).
Boyd CE & Ferguson DE (1964) Susceptibility and resistance of
mosquito fish to several insecticides. J Econ Entomol 57: 430-431.
Boyd IL, Myhill DG, & Mitchell-Jones AJ (1988) Uptake of gamma-HCH
(lindane) by Pipistrelle bats and its effect on survival. Environ
Pollut 51: 95-111.
Bradbury FR (1963) The systemic action of benzene hexachloride seed
dressings. Ann Appl Biol 52: 361-370.
Bradbury FR & Whitaker WO (1956) The systemic action of
benzenehexachloride in plants: Quantitative measurements. J Sci Food
Agric 7: 248-253.
Brassow HL, Baumann K, & Lehnert G (1981) Occupational exposure to
hexachlorocyclohexane. II. Health conditions of chronically exposed
workers. Int Arch Occup Environ Health 48: 81-87.
Bringmann G & Kühn R (1978) [Threshold values for the harmful effect
of water-endangering substances on blue algae (Microcystis
aeruginosa) and green algae (Scenedesmus quadricauda) in the
cell reproduction inhibition test. Jahresber Wasser 50: 45-60 (in
German).
Brown D (1988) Lindane: 13-week dermal toxicity study (with interim
kill and recovery period) in the rat (HUK Project No. 580/2).
Harrogate, North Yorkshire, Hazleton, UH (Unpublished report
submitted to WHO by CIEL).
Buscher CA, Dougherty JH, & Skrinde RJ (1964) Chemical oxidation of
selected organic pesticides. J Water Pollut Control Fed 36: 1005.
Buselmaier W, Röhrborn G, & Propping P (1972) Mutagenicity
investigations with pesticides in the host-mediated assay and the
dominant lethal test in mice. Biol Zbl 91: 311-325 (in German).
Büsser MT & Lutz WK (1987) Stimulation of DNA synthesis in rat and
mouse liver by various tumor promoters. Carcinogenesis 8(10): 1433-
1437.
Butler PA (1963a) Commercial fisheries investigations. Washington,
DC, US Fish and Wildlife Service (Circular 199), pp. 5-28.
Butler PA (1963b) Commercial fisheries investigations. Washington,
DC, US Fish and Wildlife Service (Circular 167), pp. 11-25.
Butler PA (1971) Influence of pesticides on marine ecosystems. Proc
R Soc Lond B177: 321-329.
Cameron GR (1945) Risks to man and animals from the use of 2,2-
bis(p-chlorophenyl),1,1,1-trichlorethane (DDT): with a note on the
toxicology of gamma-benzenehexachloride (666, gammexane). Br Med
Bull 3: 783-785.
Camon L, Martinez E, Artigas F, Sola C, & Rodriguez-Farre E (1988a)
The effect of non-convulsant doses of lindane on temperature and
body weight. Toxicology 49: 389-394.
Camon L, Sola C, Martinez E, Sanfeliu C, & Rodriguez-Farre E (1988b)
Cerebral glucose uptake in lindane-treated rats. Toxicology 49: 381-
387.
Carey AE, Gowen JA, Tai H, Mitchell G, & Wiersma GB (1979) Pesticide
residue levels in soils and crops from 37 states, 1972-National
Soils Monitoring Program (IV). Pestic Monit J 12(4): 209-229.
Carrasco JM, Cunat P, Martinez M, & Primo E (1976) Pesticide
residues in total diet samples, Spain 1971-1972. Pestic Monit J
10(1): 18-23.
Cerey K, Szokolayova J, Rosival L, & Gencik A (1975) Detection of
mutagenic actions of lindane by means of the dominant lethal test.
In: VIII International Plant Protection Congress. Reports and
information. Section IV: Plant protection in relation to human
health and environmental pollution. Moscow, pp. 34-43.
Cetinkaya M, Gabel B, Podbielski A, & Thiemann W (1984) [On the
correlation of nutrition and living habits of feeding mothers and
contamination of human milk with nonvolatile chlorinated organic
chemicals.] Akt Ernähr 9(4): 157-162 (in German).
Chabert D & Vicente N (1978) Contamination de mollusques
méditerranéens par un biocide organo-chloré: le lindane. Rev Int
Océanog Méd 49: 45-48.
Chadwick RW & Copeland MF (1985) Investigation of HCB as a
metabolite from female rats treated daily for six days with lindane.
J Anal Toxicol 9: 262-266.
Chadwick RW & Freal JJ (1972a) The identification of five unreported
lindane metabolites recovered from rat urine. Bull Environ Contam
Toxicol 7(2/3): 137-146.
Chadwick RW & Freal JJ (1972b) Comparative acceleration of lindane
metabolism to chlorophenols by pretreatrnent of rats with lindane or
with DDT and lindane. Food Cosmet Toxicol 10: 789-795.
Chadwick RW, Cranmer MF, & Peoples AJ (1971) Comparative stimulation
of gamma-HCH metabolism by pretreatment of rats with gamma-HCH, DDT,
and DDT + gamma-HCH. Toxicol Appl Pharmacol 18: 685-695.
Chadwick RW, Chuang LT, & Williams K (1975) Dehydrogenation. A
previously unreported pathway of lindane metabolism in mammals.
Pestic Biochem Physiol 5: 575-586.
Chadwick RW, Freal JJ, Sovocool GW, Bryden CC, & Copeland MF (1978)
The identification of three previously unreported lindane
metabolites recovered from mammals. Chemosphere 8: 633-640.
Chadwick RW, Copeland MF, Mole ML, Nesnow S, & Cooke N (1981)
Comparative effect of pretreatment with phenobarbital, Arochlor
1254, and ß-naphthaflavone on the metabolism of lindane. Pestic
Biochem Physiol 15: 120-136.
Chadwick RW, Copeland MF, Wolff GL, Stead AG, Mole ML, & Whitehouse
DA (1987) Saturation of lindane metabolism in chronically treated
(YS x VY)F1 hybrid mice. J Toxicol Environ Health 20: 411-434,
Chadwick RW, Cooper RL, Chang J, Rehnberg GL, & McElroy WK (1988)
Possible antiestrogenic activity of lindane in female rats. J
Biochem Toxicol 3: 147-158.
Chakravarty S, Mandal A, & Lahiri P (1986) Effect of lindane on
clutch size and level of egg protein in domestic duck. Toxicology
39: 93-103.
Chametski WA & Lichtenstein EP (1973) Penetration and translocation
of 14C-lindane in pea plants. J Econ Entomol 66(2): 344-349.
Chattopadhyay P, Karnik AB, Thakore KN, Lakkad BC, Nigam SK &
Kashyap SK (1988) Health effects among workers involved in the
manufacture of hexachlorocyclohexane. J Soc Occup Med 38: 77--81.
Chen C-P (1968) The effect of a protein-deficient diet on the acute
oral toxicity of lindane. Kingston, Ontario, Queen's University
(Thesis).
Chen T & Liang C (1956) Oral toxicity of lindane and its tolerance
in poultry and mice. J Agric Assoc China 15: 78-90.
Chessells MJ, Hawker DW, Connell DW, & Papajcsik IA (1988) Factors
influencing the distribution of lindane and isomers in soil of an
agricultural environment. Chemosphere 17(9): 1741-1749.
Clausing P, Grün G, & Beitz H (1980) [Possibilities for
investigating and avoiding damage to bird life by pesticides.]
Nachtrichtenbl Pflanzenschutzdienst DDR 34(7): (in German).
Cliath MM & Spencer WF (1971) Division S-6-soil and water management
and conservation. Soil Sci Soc Am Proc 35: 791-795.
Cliath MM & Spencer WF (1972) Dissipation of pesticides from soil by
volatilization of degradation products. I. Lindane and DDT. Environ
Sci Technol 6(10): 910-914.
Codex Alimentarius Commission (1986) Guide to Codex recommendations
concerning pesticide residues, Part 2. Maximum limits for pesticide
residues, 3rd prelim. issue, Rome, Food and Agricultural
Organization of the United Nations (CAC/Vol. 8-Ed 2-1986).
Cope OB (1965) Effects of pesticides on fish and wildlife (1964
research finding). Washington, DC, US Fish and Wildlife Service
(Circular 226), pp. 51-63.
Copeland MF & Chadwick RW (1979) Bioisomerization of lindane in
rats. J Environ Pathol Toxicol 2: 737-749.
Coper H, Herken H, & Klempau I (1951) [Antagonism and synergism of
ß- and gamma-hexachlorocyclohexane.] Kiln Wochenschr 29(13/14): 264-
265 (in German).
Corneliussen PE (1969) Residues in food and feed. Pesticide residues
in total diet samples (IV). Pestic Monit J 2(4): 140-152.
Cosson Mannevy MA & Marchand MH (1980) Bioaccumulation des éléments
chimiques chez les organismes marins: limites des études in vitro.
Exposé fait au symposium de Valbonne, 1-2 October 1980. (Unpublished
report Celamerck C80 P007C submitted to WHO by CIEL).
Cowan AA (1981) Organochlorine compounds in mussels from Scottish
coastal waters. Environ Pollut B2: 129-143.
Culley DD Jr & Ferguson DE (1969) Patterns of insecticide resistance
in the mosquito fish, (Gambusia affinis). J Fish Res Board Can 26:
2395-2401.
Cummings JG, Zee KT, Turner V, & Quinn F (1966) Residues in eggs
from low level feeding of five chlorinated hydrocarbon insecticides
to hens. J Assoc Off Anal Chem 49(2): 354-364.
Czeglédi-Janko G & Avar P (1970) Occupational exposure to lindane;
clinical and laboratory findings. Br J Ind Med 27: 283-286.
Dahlen JH & Haugen AO (1954) Acute toxicity of certain insecticides
to the bobwhite quail and mourning dove. J Wildl Manage 18: 477-481.
Darskus R (1982) Water solubility and partition coefficient
n-octano/water for CM active ingredients. (Unpublished report
Celamerck 111AC-114-006 submitted to WHO by CIEL).
Das B & Singh PK (1977) Detoxication of the pesticide
benzenehexachloride by blue-green algae. Microbios Lett 4: 99-102.
Davies D & Mes J (1987) Comparison of the residue levels of some
organochlorine compounds in breast milk of the general and
indigenous Canadian populations. Bull Environ Contam Toxicol 39:
743-749.
Davies JE, Dedhia HV, Morgade C, Barquet A, & Maibach HI (1983)
Lindane poisoning. Arch Dermatol 119(2): 142-144.
Davis HC & Hidu H (1969) Effects of pesticides on embryonic
development of clams and oysters and on survival and growth of
larvae. Fish Bull 67; 393-404.
De Brabander M, van de Veire R, Aerts F, Geuens S, & Hoebeke J
(1976) New culture model facilitating rapid quantitative testing of
mitotic spindle inhibition in mammalian cells. J Natl Cancer Inst
56(2): 357-363.
De Bruijn J (1979) Reduction of chlordane, DDT, heptachlor,
hexachlorobenzene and hexachlorocyclohexane isomers contained in
effluents. Brussels, Commission of the European Communities (Report
ENV/223/74-E Rev.2).
Deichmann WB & MacDonald WE (1977) Organochlorine pesticides and
liver cancer deaths in the United States, 1930-1972. Ecotoxicol
Environ Saf 1: 89-110.
Demozay D & Marechal G (1972) Physical and chemical properties. In:
Ulmann, E., ed., Lindane: Monograph of an insecticide. Freiburg im
Breisgau, K. Schillinger Verlag, pp. 14-21.
Deo PG, Hasan SB, & Majumder SK (1981) Interconversions and toxicity
changes in hexachlorocyclohexane isomers on dispersion in water. J
Environ Sci Health B16(6): 691-701.
Desi I (1972) In: Proceedings of the International Symposium on
Lindane, Vienna, 9 June. Budapest, National Institute of Public
Health. (Unpublished report submitted to WHO by CIEL).
Desi I (1974) Neurotoxicological effect of small quantities of
lindane. Int Arch Arbeitsmed 33: 152-162.
Desi I (1976) Lindane: Toxicological studies. In: Proceedings of the
Symposium on Lindane, Lyon-Chazay, 29 April 1976. Brussels, Centre
International d'Etudes du Lindane, pp. 67-69.
Desi I (1983) Neurotoxicological investigation of pesticides in
animal experiments, Neurobehav Toxicol Teratol 5: 503-515.
Desi I, Dura G, & Szolobodnyik J (1977) Testing of pesticide
toxicity in tissue culture. J Toxicol Environ Health 2: 1053-1066.
Desi I, Varga L, & Farkas I (1978) Studies on the immunosuppressive
effect of organochlorine and organophosphoric pesticides in subacute
experiments. J Hyg Epidemiol Microbiol Immunol 22(1): 115-122.
Deutsche Forschungsgemeinschaft (1978) [Residues in breast milk-
situation and assessment]. Boppard, Harald Boldt Verlag (in German).
Deutsche Forschungsgemeinschaft (1979) [Residue analysis of
pesticides (5th instalment): Organochlorine pesticides]. Weinheim,
Verlag Chemie, pp. 12-18 (in German).
Deutsche Forschungsgemeinschaft (1983) [Hexachlorcyclohexane as a
harmful substance in foods. Papers from two symposia of the Senate
Committee on the Testing of Residues in Foods, held on 28-29
November 1979 and 6 March 1980]. Weinheim, Verlag Chemie (in
German).
Dewitt JB, Stickel WH, & Springer PF (1963) Wildlife studies,
Patuxent Wildlife Research Center, 1961-1962. US Fish and Wildlife
Service (Circular 167), pp. 74-96.
van Dijck P & van de Voorde H (1976) Mutagenicity versus
carcinogenicity of organochlorine insecticides. Meded Fac
Landbouwwet Rijksuniv Gent 41(2): 1491-1498.
Dion BM (1984) Toxicité aigue de l'isomère gamma de
l'hexachlorocyclohexane à l'égard des truites arc-en-ciel et fario.
(Unpublished report Celamerck 111AC-442-006 submitted to WHO by
CIEL).
Dittrich V (1966) Investigation on the acute oral toxicity of
different pesticides on nestlings of Parus ater and adults of
Passer domesticus. Z Angew Entomol 57: 430-437.
Doisy EA & Bocklage BC (1949) Chronic toxicity of gamma isomer of
hexachlorocyclohexane in the albino rat. Proc Soc Exp Biol Med 71:
490-493.
Doisy EA & Bocklage BC (1950) Inositol and the toxicity of four
isomers of benzenehexachloride for the rat. Proc Soc Exp Biol Med
74: 613-616.
Drummond L, Gillanders EM, & Wilson HK (1988) Plasma gamma-
hexachlorocyclohexane concentrations in forestry workers exposed to
lindane. Br J Ind Med 45: 493-497.
Duee PH, Bories G, Froc J, Hascoet M, Henry Y, Peleran JC, & Consell
G (1975) Influence d'une teneur élevée en pesticide (lindane) dans
l'aliment sur le taux d'ovulation et la mortalité embryonnaire chez
la truie. J Rech Porcine Fr: 331-335.
Dugast P (1980) Cheminement du lindane dans une châine trophique
terrestre de laboratoire. Meded Fac Landbouwwet Rijksuniv Gent
45(4): 905-914.
Duggan RE & Corneliussen PE (1972) Dietary intake of pesticide
chemicals in the United States (Ill), June 1968-April 1970. Pestic
Monit J 5(4): 331-341.
Dutch Chemical Industry Association (1980) 1alpha, 2alpha, 3ß,
4alpha, 5alpha, 6ß-hexachlorocyclohexane. In: Handling chemicals
safely, 2nd ed. Amsterdam, Dutch Association of Safety Experts and
Dutch Safety Institute, p. 529.
Dzwonkowska A & Hubner H (1986) Induction of chromosomal aberrations
in the Syrian hamster by insecticides tested in vivo.
Arch Toxicol 58: 152-156.
Earl FL, Miller E & van Loon EJ (1973) Reproductive, teratogenic and
neonatal effects of some pesticides and related compounds in beagle
dogs and miniature swine. In: Deichmann WB, ed., Pesticides and
environment: a continuing controversy. New York, Intercontinental
Medical Book Corporation, pp. 253-266.
Eckenhausen FW, Bennett D, Beynon KI, & Elgar KE (1981)
Organochlorine pesticide concentrations in perinatal samples from
mothers and babies. Arch Environ Health 36(2): 81-92.
Edelman T (1984) [Background values of a number of inorganic and
organic substances in the soil of the Netherlands; an initial
reconnaissance]. The Hague, State Publishing House (Soil protection,
report VROM 34) (in Dutch).
Eder G, Sturm R, & Ernst W (1987) Chlorinated hydrocarbons in
sediments of the Elbe River and the estuary. Chemosphere 16(10-12):
2487-2496.
Edson EF, Sanderson DM, & Noakes DN (1966) Acute toxicty data for
pesticides. World Rev Pestic Control 5(3): 143-151.
Edwards CA (1966) Insecticide residues in soils. Residue Rev 13: 83-
132.
Edwards CA (1973a) Persistent pesticides in the environment.
Cleveland, Ohio, CRC Press.
Edwards CA, ed. (1973b) Environmental pollution by pesticides. New
York, Plenum Press.
Edwards CA (1977) Environmental aspects of the usage of pesticides
in developing countries. Med. Fac. Landbouwwet Rijksuniv Gent 42/2:
853-868.
Edwards CA (1981) Lindane exposure analysis (August 1980). in:
Response of the Centre International d'Etudes du Lindane to EPA's
preliminary Notice of Determination and Position Document 2/3 on
lindane. Brussels, Centre International d'Etudes du Lindane, Vol. 1.
Egan H & Hubbard AW (1975) Analytical surveys of food. Br Med Bull
31(3): 201-208.
Ehlers W, Farmer WJ, Spencer WF, & Letey J (1969a) Lindane diffusion
in soils. II. Water content, bulk density and temperature effects.
Soil Sci Soc Am Proc 33: 505-508.
Ehlers W, Letey, J, Spencer WF, & Farmer WJ (1969b) Lindane
diffusion in soils. I. Theoretical consideration and mechanism of
movement. Soil Sci Soc Am Proc 33: 501-504.
Eichler D (1975) Investigations of residue samples for possible
metabolites of lindane. (Unpublished report Celamerck 111AA-641-003
submitted to WHO by CIEL).
Eichler D (1977) Experiments on the decomposition of lindane when
exposed to UV-light. (Unpublished report Celamerck 111AC-143-002
submitted to WHO by CIEL).
Eichler D (1980) [Biotic and abiotic breakdown and conversion
behaviour, including isomerization (plant).]. In: Deutsche
Forschungsgemeinschaft, ed. [Hexachlorcyclohexane as a harmful
substance in foods. Papers from two symposia of the Senate Committee
on the Testing of Residues in Foods, held on 28-29 November 1979 and
6 March 1980]. Weinheim, Verlag Chemie, pp. 65-72 (in German).
Eichler D, Heupt W, & Fischer O (1976) [Attempts at lindane
breakdown in biomes]. (Unpublished report Celamerck no. 111AX-912002
submitted to WHO by CIEL) (in German).
Eichler D, Heupt W, & Paul W (1983) Comparative study on the
distribution of alpha- and gamma-hexachlorocyclohexane in the rat
with particular reference to the problem of isomerization.
Xenobiotica 13(11): 639-647.
Eisler R (1969) Acute toxicities of insecticides to marine decapod
crustaceans. Crustaceana Int J Crustacean Res 16(3): 302-310.
Eisler R (1970a) Latent effects of insecticide intoxication to
marine molluscs. Hydrobiologia 36(3-4): 345-352.
Eisler R (1970b) Acute toxicities of organochlorine and
organophosphorus insecticides to estuarine fishes. Washington, DC,
US Department of Interior, Bureau of Sport Fisheries and Wildlife
(Tech. paper 45), pp. 3-12.
Eisler R (1970c) Factors affecting pesticide-induced toxicity in an
estuarine fish. Washington DC, US Department of Interior, Bureau of
Sport Fisheries and Wildlife (Technical paper 45), pp. :2-20.
Eldefrawi ME, Sherby SM, Abalis IM, & Eldefrawi AT (1985)
Interaction of pyrethroid and cyclodiene insecticides with nicotinic
acetyicholine and GABA receptors. Neurotoxicology 6: 47-62.
El-Dib MA & Badawy MI (1985) Organochlorine insecticides and PCBs in
water, sediment and fish from the Mediterranean Sea. Bull Environ
Contam Toxicol 34: 216-227.
Eisner E, Bieniek D, Klein W, & Korte F (1972) [Contributions to
ecological chemistry. LII. Distribution and conversion of aldrin-
14C, heptachlor-14C and lindane-14C in the green alga
Chlorella pyrenoidosa.] Chemosphere 6: 247-250 (in German).
Engst R, Blazovich M, & Knoll R (1967) [Occurrence of lindane in
carrots and its influence on the carotene content]. Nahrung 11(5):
389-399 (in German).
Engst R, Macholz RM, & Kujawa M (1970) Recent state of lindane
metabolism. Residue Rev 68: 59-84.
Engst R, Macholz RM, & Kujawa M (1974) [Lindane metabolism.
Degradation of lindane by mould cultures. Unconjugated metabolites].
Nahrung 18(8): 737-745 (in German).
Engst R, Macholz RM, Kujawa M, Lewerenz HJ, & Plass R (1976) The
metabolism of lindane and its metabolites gamma-2,3,4,5,6-
pentachlorocyclohexene, pentachlorobenzene and pentachlorophenol in
rats and the pathways of lindane-metabolism. J Environ Sci Health
Bl1(2): 95-117.
Engst R, Macholz RM, & Kujawa M (1977) The metabolism of lindane in
a culture of mould and the degradation scheme of lindane. Chemospere
7: 401-418.
Engst R, Macholz RM, & Kujawa M (1978a) [Confirmations of the
degradation scheme of gamma-hexachlorocyclohexane]. Nahrung 22(6):
K29-K32 (in German).
Engst R, Macholz RM, & Kujawa M (1978b) [Metabolites of
hexachlorocyclohexane (HCH) isomers in human blood]. Pharmazie
33(2/3): 109-111 (in German).
Engst R, Macholz RM, & Kujawa M (1979a) Recent state of lindane
metabolism, Part II. Residue Rev 75: 71-95.
Engst R, Fritsche W, Knoll R, Kujawa M, Macholz RM, & Straube G
(1979b) Interim results of studies of microbial isomerization of
gamma-hexachlorocyclohexane. Bull Environ Contam Toxicol 22: 699-
707.
Epstein SS, Arnold E, Andrea J, Bass W, & Bishop Y (1972) Detection
of chemical mutagens by the dominant lethal assay in the mouse.
Toxicol Appl Pharmacol 23: 288-325.
Ernst W (1975) [Uptake, excretion and conversion of lindane-14C by
Mytilus edulis (common mussel)]. Chemosphare 6: 375-380 (in
German).
Ernst W (1979) Factors affecting the evaluation of chemicals in
laboratory experiments using marine organisms. Ecotoxicol Environ
Saf 3: 90-98.
Faladysz J & Szeler P (1982) Chlorinated hydrocarbons in diving
ducks wintering in Gdansk Bay, Baltic Sea. Sci Total Environ 24:
119-127.
FAO (1973) Organochlorine insecticides 1973 (BHC, BHC + DDT, gamma-
BHC, campheclor, chlordane, DDT, endosuifan, endrin, heod,
heptachlor, HHDN). Rome, Food and Agricultural Organization of the
United Nations, pp. 29-37 (FAO Specifications for Pesticides).
FAO/WHO (1967) Evaluation of some pesticide residues in food.
Geneva, World Health Organization (FAO: PL/CP/15; WHO Food
Add./67.32).
FAO/WHO (1968) 1967 Evaluation of some pesticide residues in food.
Geneva, World Health Organization (FAO: PL: 1967/M/11/1; WHO Food
Add./68.30).
FAO/WHO (1969) 1968 Evaluation of some pesticide residues in food.
Geneva, World Health Organization (FAO: PL:1968/M/9/1; WHO Food
Add./69.35).
FAO/WHO (1970) 1969 Evaluations of some pesticide residues in food.
Geneva, World Health Organization (FAO: PL:1969/M/17/1; WHO Food
Add./70.38).
FAO/WHO (1974) 1973 Evaluations of some pesticide residues in food.
Geneva, World Health Organization (AGP: 1973/M/9/1; WHO Pesticide
Residues Series No. 3).
FAO/WHO (1975) 1974 Evaluations of some pesticide residues in food.
Geneva, World Health Organization (AGP: 1974/M/11; WHO Pesticide
Residues Series No. 4).
FAO/WHO (1976) 1975 Evaluations of some pesticide residues in food.
Geneva, World Health Organization (AGP: 1975/M/13; WHO Pesticide
Residues series No. 5).
FAO/WHO (1978) 1977 Evaluations of some pesticide residues in food.
Rome, Food and Agriculture Organization of the United Nations (FAO
Plant Production and Protection Paper 10 Sup).
FAO/WHO (1980) 1979 Evaluations of some pesticide residues in food.
Rome, Food and Agriculture Organization of the United Nations (FAO
Plant Production and Protection Paper 20 Sup).
FAO/WHO (1990) Pesticide residues in food. Report of the 1989 Joint
Meeting of the FAO Panel of Experts on Pesticide Residues in Food
and the Environment and the WHO Expert Group on Pesticide Residues.
Rome, Food and Agriculture Organization of the United Nations (FAO
Plant Production and Protection Paper 99).
Feldmann RJ & Maibach HI (1974) Percutaneous penetration of some
pesticides and herbicides in man. Toxicol Appl Pharmacol 28: 126-
132.
Fieggen W (1983) [Report on investigation of purification mud on
pesticides and PCBs in 1981]. The Hague, Union of Polder Boards (in
Dutch).
Fishmen BE & Gianutsos G (1987) Opposite effects of different
hexachlorocyclohexane (lindane) isomers on cerebellar cyclic GMP:
relation of cyclic GMP accumulation to seizure activity. Life Sci
41: 1703-1709.
Fishman BE & Gianutsos G (1988) CNS biochemical and pharmacological
effects of the isomers of hexachlorocyclohexane (lindane) in the
mouse. Toxicol Appl Pharmacol 93: 146-153.
Fitzhugh OG, Nelson AA, & Frawley JP (1950) The chronic toxicities
of technical benzenehexachloride and its alpha, beta- and gamma
isomers. J Pharm Exp Ther 100(1): 59-66.
Fitzloff JF & Pan JC (1984) Epoxidation of the lindane metabolite,
gamma-PCCH, by human and rat liver microsomes. Xenobiotica 14(7):
599-604.
Fitzloff J., Portig J, & Stein K (1982) Lindane metabolism by human
and rat liver microsomes. Xenobiotica 12(3): 197-202.
Fooken C & Butte W (1987) Organochlorine pesticides and
polychlorinated biphenyls in human milk during lactation.
Chemosphere 16(6): 1301-1309.
Foulquier L, Reynier B, Granby A, & Bovard P (1971) Toxicité du
lindane sur l'anguille, France. CR Acad Agric 57(12): 1060-1068.
Francis AJ, Spanggord RJ, & Ouchi GI (1975) Degradation of lindane
by Escherichia coli. Appl Microbiol 29(4): 567-568.
Frank R, Braun HE, Sirons GH, Rasper J, & Ward GG (1985)
Organochlorine and organophosphorus insecticides and industrial
pollutants in the milk supplies of Ontario, 1983. J Food Prod 48(6):
499-504.
Franklin A (1987) The concentration of metals, organochlorine
pesticide and PCB residues in marine fish and shell fish: results
from MAFF fish and shell fish monitoring programmes, 1977-1984.
Lowestoft, Ministry of Agriculture, Fisheries and Food, Directorate
of Fisheries Research (Aquatic Environment Monitoring Report No.
16).
Freal JJ & Chadwick RW (1973) Metabolism of hexachlorocyclohexane to
chlorophenols and effect of isomer pretreatment on lindane
metabolism in rat. J Agric Food Chem 21(3): 424-427.
Fricke G (1972) [Contamination of soils with chlorinated hydrocarbon
insecticides, comparison of 1969 with 1972 (Communication 1, field
vegetable growing). Gesunde Pfianz 24(11): 177-179 (in German).
Frisque GE, Galoux M, & Bernes A (1983) Accumulation de deux
micropollutants (les polychlorobiphenyles et le gamma-HCH) par des
bryophytes aquatiques de la Meuse. Meded Fac Landbouwwet Rijksuniv
Gent 48(4): 971-983.
Frohberg H & Bauer A (1972a) Lindane. Testing for teratogenic
effects in mice following subcutaneous injection. Darmstadt, E.
Merck (Unpublished report 4/24/72 submitted to WHO by CIEL).
Frohberg H & Bauer A (1972b) Lindane. Testing for teratogenic
effects in mice following oral administration. Darmstadt, E. Merck
(Unpublished report 4/107/72 submitted to WHO by CIEL).
Frohberg H & Bauer A (1972c) [Lindane: Testing for mutagenic effect.
Dominant lethality test on male mice]. Darmstadt, E. Merck
(Unpublished report 4/8/72 submitted to WHO by CIEL).
Frohberg H, Wolf HP, & von Eberstein M (1972a) [Testing for acute
toxicity in rats after oral administration, intraperitoneal
injection and intramuscular injection]. Darmstadt, E. Merck
(Unpublished report submitted to WHO by CIEL) (in German).
Frohberg H, von Eberstein M, Engemann J, & Weisse G (1972b) [Testing
for acute toxicity in rats after oral administration and
intraperitoneal injection]. Darmstadt, E. Merck (Unpublished report
submitted to WHO by CIEL) (in German).
Fuhremann TW & Lichtenstein EP (1980) A comparative study of the
persistence, movement, and metabolism of six carbon-14 insecticides
in soils and plants. J Agric Food Chem 28: 446-452.
Gaggi C, Bacci E, Calamari D, & Fanelli R (1986) Chlorinated
hydrocarbons in plant foliage: An indication of the tropospheric
contamination level. Chemosphere 14(11-12): 1673-1686.
Gaines TB (1960) The acute toxicity of pesticides to rats. Toxicol
Appl Pharmacol 2: 88-99.
Gardais M & Scherrer M (1979) Photodecomposition of lindane (studies
realized in Rhône-Poulenc (CRD)). Lyon, Rhône-Poulenc (Unpublished
report submitted to WHO by CIEL).
Gartrell MJ, Craun JC, Podrebarac DS, & Gunderson EL (1985a)
Pesticides, selected elements and other chemicals in adult total
diet samples, October 1978-September 1979. J Assoc Off Anal Chem
68(5): 862-875.
Gartrell MJ, Craun JC, Podrebarac DS, & Gunderson EL (1985b)
Pesticides, selected elements and other chemicals in infant and
toddler total diet samples, October 1978-September 1979. J.Assoc Off
Anal Chem 68(5): 842-861.
Garretson AL & San Clemente CL (1968) Inhibition of nitrifying
chemolithotrophic bacteria by several insecticides. J Econ Entomol
61(1): 285-288.
Gaur AC & Misra KC (1977) Effect of simazine, lindane and ceresan on
soil respiration and nitrification rates. Plant Soil 46: 5-15.
Gencik A (1977) [Cytogenic examination of the bone marrow of rats
after administration of lindane]. Bratislava Lek Listy 67(5): 579-
582 (in Slovak).
Geyer H, Sheehan P, Kotzias D, Freitag D, & Korte F (1982)
Prediction of ecotoxicological behaviour of chemicals. Relationship
between physico-chemical properties and bioaccumulation of organic
chemicals in the mussel Mytilus edulis. Chemosphere 11(11): 1121-
1134.
Geyer H, Politzki G, & Freitag D (1984) Prediction of
ecotoxicological behaviour of chemicals. Relationship between n-
octanol/water partition coefficient and bioaccumulation of organic
chemicals by alga Chlorella. Chemosphere 13(2): 269-284.
Geyer H, Scheunert I, & Korte F (1986) Bioconcentration potential of
organic environmental chemicals in humans. Regul Toxicol Pharmacol
6: 313-347.
Ginsburg CM & Lowry W (1983) Absorption of gamma benzene
hexachloride following application of Kwell shampoo. Pediatr
Dermatol 1(1): 74-76.
Ginsburg CM, Lowry W, & Reisch J (1977) Absorption of lindane (gamma
benzene hexachloride) in infants and children. J Pediatr 91(6): 998-
1000.
Glatt HR & Oesch F (1984) Mammalian cell (V79) mutagenicity test on
lindane. Brussels, CIEL (Unpublished report Celamerck No. 111AC-457-
019, submitted to WHO by CIEL).
Gopalswamy UV & Aiyar AS (1984) Biotransformation of lindane in the
rat. Bull Environ Contam Toxicol 32: 148-156.
Gorchev HG & Jelinek CF (1985) A review of the dietary intakes of
chemical contaminants. World Health Organ Bull 63(5): 945-962.
Goto M, Hattod M, & Miyagawa T (1972) [Contribution to ecological
chemistry. II. Hepatoma formation in mice after administration of
high doses of HCH isomers]. Chemosphere 6: 279-282 (in German).
Graeve K & Herrnring G (1949) [The application of the gamma isomers
of hexachlorocyclohexane as an anthelmintic]. Klin Wochenschr 27:
318 (in German).
Graeve K & Herrnring G (1951) [The toxicity of gamma-
hexachlorocyclohexane]. Arch int pharmacodyn 85(1/2): 64-72 (in
German).
Green DWJ, Kendall A, Williams A, & Pascoe D (1986) Studies on the
acute toxicity of pollutants to freshwater macroinvertebrates. 4.
Lindane (gamma-hexachlorocyclohexane). Arch Hydrobiol 106(2): 263-
273.
Greve PA (1972) Potentially hazardous substances in surface waters.
Part I. Pesticides in the River Rhine. Sci Total Environ 1: 173-180.
Greve PA & van Harten DC (1983) [Relationship between levels of
organochlorine pesticides and PCBs in fat and blood in human
subjects]. Bilthoven, National Institute of Public Health and
Environmental Hygiene (Unpublished report No. 638219001) (in Dutch).
Greve PA & van Hulst S (1977) [Organochlorine pesticides and PCBs in
total diets]. Bilthoven, National Institute of Public Health and
Environmental Hygiene (Unpublished report No. 192/77 Tox-ROB) (in
Dutch).
Greve PA & Wegman RCC (1985) Organochlorine compounds in human milk;
data from a recent investigation in the Netherlands. Working paper
for the WHO consultation on organochlorine compounds in human milk
and related hazards, Bilthoven, 9-11 January 1985. Bilthoven,
Institute of Public Health and Environmental Hygiene.
Grover PL & Sims P (1965) The metabolism of gamma-2, 3,4,5,6-
pentachlorocyclo-1-ene and gamma-hexachlorocyclohexane in rats.
Biochem J 96: 521-525.
Guenard J, Eichelberger HP, & Terrier C (1984a) In vivo sister
chromatid exchange assay in CF1-mouse bone marrow cells with
lindane (oral application). Itingen, Switzerland, Research and
Consulting Company AG. (Unpublished report No. 025705, Celamerck No.
111AC-457-020, submitted to WHO by CIEL).
Guenard J, Eichelberger HP, & Terrier C (1984b) In vivo sister
chromatid exchange assay in CF1 mouse bone marrow cells with
lindane (intraperitoneal injection). Itingen, Switzerland, Research
and Consulting Company AG. (Unpublished report No. 025716, Celamerck
No. 111AC-457-021, submitted to WHO by CIEL).
Guenzi WD & Beard WE (1970) Volatilization of lindane and DDT from
soils. Soil Sci Soc Am Proc 34: 443-447.
Guicherit R & Schulting FL (1985) The occurrence of organic
chemicals in the atmosphere of the Netherlands. Sci Total Environ
43: 193-219.
Gunderson EL (1988) FDA Total Diet Study, April 1982-April 1984,
dietary intakes of pesticides, selected elements and other
chemicals. J Assoc Off Anal Chem 71(6): 1200-1209.
Gupta SK, Parikh JR, Shah M, Chatterjee SK & Kashyap SK (1982)
Changes in serum HCH residues in malaria spraymen after short-term
occupational exposure. Arch Environ Health 37(1): 41-44.
Haider K (1979) Degradation and metabolization of lindane and other
hexachlorocyclohexane isomers by anaerobic and aerobic soil
microorganisms. Z Naturtorsch 34C: 1066-1069.
Haider K (1983) [Breakdown and conversion of gamma-HCH and other HCH
isomers by soil microorganisms]. In: [Hexachlorcyclohexane as a
harmful substance in foods]. Bonn, German Research Council, pp. 73-
78 (DFG research report) (in German).
Haider K & Jagnow G (1975) [Degradation of 14C-, 3H- and 36Cl-
labelled gamma-hexachlorocyclohexan by anaerobic soil
microorganisms]. Arch Microbiol 104(2): 113-121 (in German).
Haider K, Jagnow G, Kohnen R, & Lim SU (1974) [Degradation of
chlorinated benzenes, phenols and cyclohexane derivatives by benzene
and phenol using soil bacteria under aerobic conditions]. Arch
Microbiol 96: 183-200 (in German).
Haider K, Jagnow, G, & Rohr K (1975) [Anaerobic degradation of
gamma-hexachlorocyclohexane by a mixed bacterial flora from arable
soils and from the cow rumen]. Landwirtsch Forsch 32(2): 147-152 (in
German).
Haider K, Jagnow G, & Rohr R (1976) [Anaerobic breakdown of gamma-
hexachlorocyclohexane by mixed bacterial flora from soil and cow's
rumen]. Landwirtsch Forsch 32(11): 147-152 (in German).
Hamada M, Kawano E, Kawamura S, & Shiro M (1981) Radiation- and
photo-induced degradation of five isomers of 1,2,3,4,5,6-
hexachlorocyclohexane. Agric Biol Chem 45(3): 659-665.
Hamada M, Kawano E, Kawamura S, & Shiro M (1982) A new isomer of
1,2,3,4,5-pentachlorocyclohexane from UV-irradiation products of
(alpha-, ß-, and delta-isomers of 1,2,3,4,5,6-hexachlorocyclohexane.
Agric Biol Chem 46(1): 153-157.
Hanada M, Yutani C, & Miyaji T (1973) Induction of hepatoma in mice
by benzenehexachloride. Gann 64(5): 511-513.
Hanig JP, Yoder PD, & Krop S (1976) Convulsions in weanling rabbits
after a single topical application of 1% lindane. Toxicol Appl
Pharmacol 36: 463-469.
Hansen PD (1980) Uptake and transfer of the chlorinated hydrocarbon
lindane (gamma-BHC) in a laboratory freshwater food chain. Environ
Pollut (Ser A), 21: 97-108.
Harrison DL, Poole WSH, & Mol JCM (1963) Observations on feeding
lindane-fortified mash to chickens. N Z Vet J 11(6): 137-140.
Hartley DM & Johnston JB (1983) Use of the freshwater clam
Corbicula manilensis as a monitor for organochlorine pesticides.
Bull Environ Contam Toxicol 31: 33-40.
Hashimoto Y & Nishiuchi Y (1981) [Establishment of bioassay methods
for the evaluation of acute toxicity of pesticides to aquatic
organisms]. J Pestic Sci (Jpn), 6: 257-264 (in Japanese).
Hawkins GS & Reifenrath WG (1984) Development of an in vitro model
for determining the fate of chemicals applied to skin. Fundam Appl
Toxicol 4: 133-144.
Hawkinson JE, Shull LR, & Joy RM (1989) Effects of lindane on
calcium fluxes in synaptosomes. Neurotoxicology 10: 29-40.
Haworth S, Lawlor T, Mortelmans K, Speck W, & Zeiger E (1983)
Salmonella mutagenicity test results for 250 chemicals. Environ
Mutagen Suppl 1: 3-142.
Hayes WJ Jr (1982) Benzene hexachioride. In: Hayes WJ Jr (ed.)
Pesticides studied in man. Baltimore, Williams and Wilkins, pp. 211-
228.
Hazleton Laboratories America Inc (1976a) Teratology studies in
rats-lindane (gamma benzene hexachloride, USP). Madison, Wisconsin.
(Unpublished report submitted to WHO by CIEL).
Hazleton Laboratories America Inc (1976b) Teratology studies in
rabbits-lindane (gamma benzene hexachloride, USP). Madison,
Wisconsin. (Unpublished report submitted to WHO by CIEL).
Heeschen W, Nyhuis H & Blüthgen, A (1980) [Investigations on the
significance of the transformation and degradation of benzene
hexachloride (BHC) in the lactating cow and in the surroundings for
the BHC-contamination of milk]. Milchwissenschaft 35(4): 221-224 (in
German).
Henderson C, Pickering QH, & Tarzwell CM (1959) Relative toxicity of
ten chlorinated hydrocarbon insecticides to four carbon insecticides
to species of fish. Trans Am Fish Soc 88: 23-32.
Herbst M (1976) Toxicology of lindane. In: Proceedings of the
symposium on lindane, Lyon-Chazay, 29 April 1976. Brussels, CIEL,
pp. 43-69.
Herbst M, Weisse I, & Keel LMER (1975) A contribution to the
question of the possible hepato-carcinogenic effects of lindane.
Toxicology 4: 91-96.
Heritage AD & MacRae LC (1977a) Identification of intermediates
formed during the degradation of hexachlorocyclohexanes by
Clostridium sphenoïdes. Appl Environ Microbiol 33(6): 1295-1297.
Heritage AD & MacRae LC (1977b) Degradation of lindane by cell-free
preparations of Clostridium sphenoïdes. Appl Environ Microbiol
34(2): 222-224.
Heritage AD & MacRae LC (1979) Degradation of hexachlorocyclohexanes
and structurally related substances by Clostridium sphenoïdes.
Aust J Biol Sci 32: 493-500.
Herken H (1950a) [Functional disorders of the central nervous system
caused by hexachlorocyclohexane]. Naunyn-Schmiedeberger Arch Exp
Pathel Pharmacol 212: 158-159 (in German).
Herken H (1950b) [Functional disorclers of the central nervous
system following the action of hexachlorocyclohexane]. Arztliche
Wochenschr 5(13/14): 193-195 (in German).
Herken H (1950c) [Alterations in cell function in the nervous system
caused by hexachlorocyclohexane]. Naunyn-Schmiedeberger Arch Exp
Pathel Pharmacol 211: 143-152 (in German).
Herken H (1951) [Disruption of the nervous system with
hexachlorcyclohexane]. Arzneimittelforschung 1: 356-359 (in German).
Hermens J & Leeuwangh P (1982) Joint toxicity of mixtures of 8 and
24 chemicals to the guppy (Poecilia reticulata). Ecotoxicol
Environ Saf 6: 302-310.
Herrmann R, Thomas W, & Huebrier D (1984) [Behaviour of organic
micropollutants (PAH, PCB and BHC) and of a fecal sterol in the
Husum estuary and in the adjacent North Frisian Wadden Sea]. Dtsch
Gewässerkd Mitt 4: 101-107 (in German).
Herve S, Heinonen P, Paukku R, Knuutila M, Koistinen J, & Paasivirta
J (1988) Mussel incubation method for monitoring organochlorine
pollutants in water courses. Four-year application in Finland.
Chemosphere 17(10): 1945-1961.
Heupt W (1974) [Seepage behaviour of pesticides]. (Unpublished
report Celamerck-Ingelheim No. 11110-922-001-11110-922-009,
submitted to WHO by CIEL) (in German).
Heupt W (1979) [Behaviour of the active ingredients of pesticides in
the soil]. (Unpublished report Celamerck No. 111 AA-921-008/009,
submitted to WHO by CIEL) (in German).
Heupt W (1983) Determination of the hydrolytic stability of lindane.
(Unpublished report Celamerck Ingelheim, submitted to WHO by CIEL).
Hildebrandt G, Jeep H, Hurka H, Stuke T, Heitmann M, & Boiselle C
(1986) [Breast milk as a biological indicator: study on the organic
halogen burden in breast milk and foodstuffs]. Berlin, Schmidt E.
Verlag. (Report 5/86 of the Federal Office for the Environment) (in
German).
Hill EF & Camardese MB (1986) Lethal dietary toxicities of
environmental contaminants and pesticides to Coturnix. Washington,
DC, US Department of Interior, Fish and Wildlife Service, p. 89
(Fish and Wildlife Technical Report No. 2).
Hill EF, Heath RG, Spano JW, & Williams JD (1975) Lethal dietary
toxicities of environmental pollutants to birds. Washington, DC, US
Department of Interior Fish and Wildlife Service (Special Scientific
Report. Wildlife No. 191).
Hofer K (1953) [Investigations on the efficacy of lindane
preparations in cow-sheds and on the influence of gamma-HCH on the
ripening of Emmental cheese]. University of Munich, Institute o!
Food Science, Veterinary Faculty (Thesis) (in German).
Holder JW & Stöhrer G (1989) Review of studies relating to lindane
carcinogenicity. Washington, DC, US Environmental Protection Agency,
Office of Health and Environmental Assessment, Cancer Assessment
Group (Unpublished report).
Hoshizaki H, Niki Y, Tajima H, Terada Y, & Kasahara A (1969) A case
of leukemia following exposure to insecticide. Acta haematol jpn
32(14): 672-677.
Hudson RH, Tucker RK, & Haegele MA (1984) Handbook of toxicology of
pesticides to wildlife, 2nd ed. Washington DC, US Department of
Interior, Fish and Wildlife Service, p. 49 (Resource Publication No.
153).
International Agency for Research on Cancer (1987) Overall
evaluations of carcinogenicity: an updating of IARC Monographs
Volumes 1 to 42. Lyon, pp. 220-222 (IARC monographs on the
evaluation of carcinogenic risks to humans. Suppl 7).
International Atomic Energy Agency (1988) isotope techniques for
studying the fate of persistent pesticides in the tropics. Vienna
(IAEA-TELDOC-476).
International Register for Potentially Toxic Chemicals (1989) Data
profile on lindane. Geneva, United Nations Environment Programme.
Ishidate M, Jr & Odashima S (1977) Chromosome tests with 134
compounds on Chinese hamster cells in vitro-A screening for
chemical caminogens. Mutat Res 48: 337-354.
Ito N, Nagasaki H, Arai M, Makiura S, Sugihara S, & Hirao K (1973a)
Histopathologic studies on liver tumorigenesis induced in mice by
technical polychlorinated biphenyls and its promoting effect on
liver tumours induced by benzenehexachloride. J Natl Cancer Inst
51(5): 1637-1646.
Ito N, Nagasaki H, Arai M, Sugihara S, & Makiura S (1973b)
Histologic and ultrastructural studies on the hepatocarcinogenicity
of benzenehexachloride in mice. J Natl Cancer Inst 51(3): 817-826.
Ito N, Nagasaki H, Aoe H, Sugihara S, Miyata Y, Arai M, & Shirai T
(1975) Brief communication: Development of hepatocellular carcinomas
in rats treated with benzenehexachloride. J Natl Cancer Inst 54(3):
801-805.
Itokawa H, Schallah A, Weisgerber I, Klein W, & Korte F (1970)
[Contributions to ecological chemistry. XXII. Metabolism and residue
behaviour of lindane-14C in higher plants]. Tetrahedron 26: 763-
773 (in German).
Iverson F, Ryan JJ, Lizotte R, & Hierlihy SL (1984) In vivo and
in vitro binding of alpha and gamma-hexachlorocyclohexane to mouse
liver macromolecules. Toxicol Lett 20(3): 331-335.
Izmerov NF ed. (1983) Lindane. Moscow, Centre of International
Projects, GKNT (IRPTC Scientific reviews of Soviet literature on
toxicity and hazards of chemicals No. 40).
Jackson MD, Sheets TJ, & Moffett CL (1974) Persistence and movement
of BHC in a watershed. Mount Mitchell State Park, North Carolina,
1967-1972. Pestic Monit J 8(3): 202-208.
Jacobs A, Blangetti M, & Hellmund E (1974) [Storage of chlorinated
harmful substances found in Rhine water in the fatty tissue of
rats]. Wasser 43: 259-274 (in German).
Jaeger U, Podczek A, Haubenstock A, Pirich K, Donner A, & Hruby K
(1984) Acute oral poisoning with lindane-solvent mixtures. Vet Hum
Toxicol 26(1): 11-14.
Jagnow G, Haider K, & Ellwardt PC (1977) Anaerobic dechlorination
and degradation of hexachlorocyclohexane isomers by anaerobic and
facultative anaerobic bacteria. Arch Microbiol 115: 285-292.
Jeanne N (1979) Effets du lindane sur la division, le cycle
cellulaire et les biosynthèses de deux algues unicellulaires. Can J.
Bot 57: 1464-1472.
Jeanne-Levain N (1974) Etude des effets du lindane sur la croissance
et le developpement de quelques organismes unicellulaires. Bull Soc
Zool Fr 99(1): 105-109.
Jedlicka VL, Hermanska Z, Smida I, & Kouba A (1958) Paramyeloblastic
leukaemia appearing simultaneously in two blood cousins after
simultaneous contact with gammexane (hexachlorocyclohexane). Acta
med scand 161(6): 147-151.
Jenssen D & Ramel C (1980) The micronucleus test as part of a short-
term mutagenicity test program for the prediction of carcinogenicity
evaluated by 143 agents tested. Mutat Res 75: 191-202.
Johnson RD & Manske DD (1976) Residues in food and feed. Pesticide
residues in total diet samples (IX). Pestic Monit J 9(4): 157-169.
Johnson RD, Manske DD, New DH, & Podrebarac DS (1979) Pesticides and
other chemical residues in infant and toddler total diet samples,
August 1974-July 1975. Pestic Monit J 13(3): 87-98.
Joy RM & Albertson TE (1987a) Factors responsible for increased
excitability of dentate gyrus granule cells during exposure to
lindane. Neurotoxicology 8(4): 517-528.
Joy RM & Albertson TE (1987b) Interactions of lindane with
synaptically mediated inhibition and facilitation in the dentate
gyrus. Neurotoxicology 8(4): 529-542.
Joy RM, Stark LG, & Albertson TE (1982) Proconsulvant effects of
lindane: enhancement of amygdaloid kindling in the rat. Neurobehav
Toxicol Teratol 4: 347-354.
Joy RM, Stark LG, & Albertson TE (1983) Proconvulsant action of
lindane compared at two different kindling sites in the rat-amygdala
and hippocampus. Neurobehav Toxicol Teratol 5: 461-465.
Joy, RM, Burns VW, & Stark LG (1988) Increases in free intracellular
Ca++ accompany exposure of neurohybridoma cells to the insecticide
lindane. Toxicology 8(1): 44.
Juhnke J & Lüdemann D (1978) [Results of the testing of 200 chemical
compounds for acute toxicity to fish in the golden orfe test].
Wasser-Abwasser-Forsch 11(5): 161-164 (in German).
Kahunyo JM, Froslie A, & Maitai CK (1988) Organochlorine pesticide
residues in chicken eggs: a survey. J Toxicol Environ Health 24:
543-550.
Kamada T (1971) [Part I. Accumulation of BHC (alpha, ß-, gamma-, and
delta-) isomers in rat bodies and excretion into urine following
oral administration]. Nippon Eiseigaku Zasshi 26(4): 358-364 (in
Japanese).
Kampe W (1980) [Occurrence of hexachlorocyclohexane in the soil].
In: [Hexachlorocyclohexane as a harmful substance in foodstuffs].
Weinheim, Verlag Chemie, pp. 18-23 (Research Report) (in German).
Kampe W & Andre W (1980) [Heavy metals, nitrate and chlorinated
hydrocarbons in the vegetable components of total weekly food
consumption]. Ernähr Umsch 27(11): 356-364 (in German).
Kaphalia BS & Seth TD (1983) Chlorinated pesticide residues in blood
plasma and adipose tissue of normal and exposed human population.
Indian J Med Res 77: 245-247.
Karapally JC, Saha JG, & Lee YW (1973) Metabolism of lindane-14C
in the rabbit: Ether-soluble urinary metabolites. J Agric Food Chem
21(5): 811-818.
Kathpal TS, Yadav GS, Kushawa KS, & Singh G (1988) Research
communication: persistence behavior of HCH in rise soil and its
uptake by rice plants. Ecotoxicol Environ Saf 15: 336-338.
Katz M (1961) Acute toxicity of some organic insecticides to three
species of salmonids and to the three-spine stickleback. Trans Am
Fish Soc 90(3): 264-268.
Kawahara T (1972) [Studies on organochlorine pesticide residues in
crops and soils. Report XV. Absorption of BHC by stems and leaves of
rice plants]. Noyaku Kensasho Hokohu 12: 31-34 (in Japanese, with
English summary).
Kawahara T & Nakamura T (1972) [Studies on the residual
organochlorine pesticides in crops and soils. Report XXl. Absorption
and translocation of chlorinated hydrocarbons in potted turnips].
Bull Agric Chem Insp Stn 12: 52-56 (Abstract) (in Japanese).
Kawahara T, Konuma S, Wada T, Kureha Y, & Nakamura H (1971)
[Absorption and translocation of chlorinated hydrocarbons into crops
in the high-cooling region]. Noyaku Kensasho Hokohu 11: 67-72 (in
Japanese, with English summary).
Kay BD & Elrick DE (1967) Adsorption and movement of lindane in
soils. Soil Sci 104(5): 314-322.
Kensler TW & Trush MA (1984) The role of oxygen radicals in tumour
promoters. Environ Mutagen 6: 593-616.
Kewitz H & Reinert H (1952) [Demonstration of functional changes to
the central nervous system through electrically and chemically
induced spasma]. Naunyn-Schmiedebergs Arch Exp Pathol Pharmakol 215:
93-99 (in Germen).
Khera KS, Whalen C, Trivett G, & Angers G (1979) Teratogenicity
studies on pesticidal formulations of dimethoate, diuron, and
lindane in rats. Bull Environ Contam Toxicol 22: 522-529.
Kiraly J, Szentesi I, Ruzicska M, & Czeize A (1979) Chromosome
studies in workers producing organophosphate insecticides. Arch
Environ Contam Toxicol 8: 309-319.
Kitamura S, Sumino K, & Hayakawa K (1970) [Decomposition of gamma-
BHC in vivo]. Jpn J Public Health 17(11): 108-109 (in Japanese).
Klone PR & Kintigh WJ (1988) Lindane technical-fourteen week dust
aerosol inhalation study on mice. Bushy Run Research Center
(Unpublished report Met Path No. 14014; Laboratory Project No. ID
BRRC/51-524).
Koelling K (1978) [Residues in poultry and eggs. Report of the
symposium, 28 Mai 1975]. Boppard, Harald Boldt Verlag, pp. 9-159 (in
German).
Kohli J, Weisgerber I, Klein W, & Korte F (1976a) Contributions to
ecological chemistry: CVII. Fate of lindane-14C in lettuce,
endives, and soil under outdoor conditions. J Environ Sci Health
B11(1): 23-32.
Kohli J, Weisgerber I, & Klein W (1976b) Balance of conversion of
[14C]-lindane in lettuce in hydroponic culture. Pestic Biochem
Physiol 6: 91-97.
Kohnen R, Haider K, & Jagnow G (1975) Investigations on the
microbial degradation of lindane in submerged and aerated moist
soil. Environ Qual Saf 3: 222-225.
Kolmodin-Hedman B (1974) Exposure to lindane and DDT and its effects
on drug metabolism and serum lipoproteins. Stockholm, Departments of
Clinical Pharmacology (at Huddinge Hospital) and Pharmacology,
Karotinska Institute, Toxicology (Swedish Medical Research Council)
and Division of Occupational Medicine, National Board of
Occupational Safety and Health (Thesis).
Kolmodin-Hedman B (1984) Pesticides. In: Aitio A, Riihimaki V, &
Vainio H, ed. Biological monitoring and surveillance of workers
exposed to chemicals. Washington, DC, Hemisphere Publishing Corp.,
pp. 209-218.
Kolmodin-Hedman B, Alexanderson B, & Sjöqvist F (1971) Effect of
exposure to lindane on drug metabolism: Decreased hexobarbital
sleeping-times and increased antipyrine disappearance rate in rats.
Toxicol Appl Pharmacol 20(3): 299-307.
Kopf W & Schwoerbel J (1980) [Accumulation studies of the
insecticide lindane (gamma-HCH, BHC) by aquatic insects of the genus
Sigara (Hemiptera, Corixidae)]. Arch Hydrobiol Suppl 59(1): 32-
42 (in German).
Koransky W, Portig J, & Münch G (1963) [Absorption, distribution and
excretion of alpha- and gamma-hexachlorocyclohexane]. Naunyn-
Schmiedebergs Arch Exp Pathol Pharmakol 244: 564-575 (in German).
Koransky W, Portig J, Vohland HW, & Klempau I (1964) [Elimination of
alpha- and gamma-hexachlorocyclohexane and the way it is influenced
by liver microsomal enzymes]. Naunyn-Schmiedebergs Arch Exp Pathol
Pharmakol 247: 49-60 (in German).
Korn S & Earnest R (1974) Acute toxicity of twenty insecticides to
striped bass, Morone saxatilis. California Fish Game 60(3): 128-131.
Korte F (1980) [Occurrence of HCH-hexachlorcyclohexane as a harmful
substance in foods. Papers from two symposia of the Senate Committee
on the Testing of Residues in Foods, held on 28-29 November 1979 and
6 March 1980]. Weinhelm, Verlag Chemie, pp. 46-64 (in German).
Kotzias D, Nitz S, & Korte F (1981) [Light-induced total breakdown
of organic molecules adsorbed on silica gel]. Chemosphere 10(4):
415-422 (in German).
Kramer MS, Hutchinson TA, Rudnick SA, Leventhal JM, & Feinstein AR
(1980) Operational criteria for adverse drug reactions in evaluating
suspected toxicity of a popular scabicide. Clin Pharmacol Ther 27:
149-154.
Krauthacker B, Alebic-Kolbah T, Kralj M, Tjalcevic B, & Reiner E
(1980) Organochlorine pesticides in blood serum of the general
Yugoslav population and in occupationally exposed workers. Int Arch
Occup Environ Health 45: 217-220.
Krishnakumari MK (1977) Sensitivity of the alga Scenedesrnus acutus
to some pesticides. Life Sci 20: 1525-1532.
Kujawa M, Härtig M, Macholz RM, & Engst R (1976) [The degradation of
14C-lindane by a mould culture]. Nahrung 20(2): 181-183 (in
German).
Kujawa M, Engst R, & Macholz R (1977) On the metabolism of lindane.
In: Proceedings of International Symposium on Industrial Toxicology,
Environmental Pollution and Human Health, pp. 661-672.
Kumar, A. & Dwivedi PP (1988) Relative induction of molecular forms
of cytochrome P-450 in gamma-hexachlorocyclohexane exposed rat liver
microsomes. Arch Toxicol 62: 479-481.
Kurihara N & Nakajima M (1974) Studies on BHC isomers and related
compounds. VIII. Urinary metabolites produced from gamma- and ß-BHC
in the mouse: Chlorophenol conjugates. Pestic Biochem Physiol 4:
220-231.
Kurihara N, Uchido M, Fujita T, & Nakajima M (1973) Studies on BHC
isomers and related compounds. V. Some physicochemical properties of
BHC-isomers(1). Pestic Biochem Physiol 2: 383-390.
Kurihara N, Tanaka K, & Nakajima M (1979) Mercapturic acid formation
from lindane in rats. Pestic Biochem Physiol 10: 137-150.
Kurihara N, Suzuki T, & Nakajima M (1980) Deuterium isotope effects
on the formation of mercapturic acids from lindane in rats. Pestic
Biochem Physiol 14: 41-49.
Kurt TL, Dost R, Gilliland M, Reed G, & Petty C (1986) Accidental
Kwell (lindane) ingestions. Vet Hum Toxicol 28(6): 569-571.
(Published erratum appears in Vet Hum Toxicol 29(1): 24, 1987.)
Landesamt für Wasser und Abfall (1988) [Water quality report 1987].
Dusseldorf, North Rhine-Westphalia Office for Water and Refuse (in
German).
Lange P (1965) [Studies on the convulsive process and the effect of
convulsion-inhibiting substances thereon during progressive infusion
of convulsive poisons in mice]. Acta Biol Med Ger 15: 109-119 (in
German).
Lange M, Nitzsche K, & Zesch A (1981) Percutaneous absorption of
lindane in healthy volunteers and scabies patients. Dependency of
penetration kinetics in serum upon frequency of application, time
and mode of washing. Arch Dermatol Res 271: 387-399.
Laug EP (1948) Tissue distribution of a toxicant following oral
ingestion of the gamma isomer of benzene hexachloride by rats. J
Pharmacol Exp Ther 93: 277-281.
Laugel P (1981) [The residue situation in France and ways of
overcoming it]. Lebensmitteichem Gerichtl Chem 35: 29-32 (in
German).
Lehman AJ (1952) Chemicals in foods: A report to the association of
Food and Drug officials on current developments. Part II.
Pesticides. Section III. Subacute and chronic toxicity. Q Bull Assoc
Food Drug Off US 16(2): 47-53.
Lehman AJ (1965) Summaries of pesticide toxicity. Part I.
Chlorinated organic compounds. Topeka, Kansas, Associations with
Food and Drug Officials of the United States, pp. 27-29.
Lichtenstein EP & Polivka JB (1959) Persistence of some chlorinated
hydrocarbon insecticides in turf soils. J Econ Entomol 52(2): 289-
293.
Lichtenstein EP & Schulz KR (1958a) Persistence of some chlorinated
hydrocarbon insecticides as influenced by soil types, rate of
application and temperature. J Econ Entomol 52(1): 124-131.
Lichtenstein EP & Schulz KR (1958b) Breakdown of lindane and aldrin
in soils. J Econ Entomol 52(1): 118-124.
Lichtenstein EP & Schulz KR (1970) Volatilization of insecticides
from various substrates. J Agric Food Chem 18: 814.
Lichtenstein EP, de Peu LJ, Eshbaugh EL, & Sleesman JP (1960)
Persistence of DDT, aldrin, and lindane in some Midwestern soils. J
Econ Entomol 53(1): 136-142.
Lisi P, Caraffini S, & Assalve D (1986) A test series for pesticide
dermatitis. Contact Derm 15: 266-269.
Lisi P, Caraffini S, & Assalve D (1987) Irritation and sensitization
potential of pesticides. Contact Derm 17: 212-218.
Lowden GF, Saunders CL, & Edwards RW (1969) Organochlorine
insecticides in water (Part II). J Water Treat Exam 18: 275-287.
Lüdemann D & Neumann H (1960a) [Tests on the acute and toxic effects
of modern contact insecticides on one-year-old carp (Cyprinus
carpio L.)]. Z Angew Zool 47: 11-33 (in German).
Lüdemann D & Neumann H (1960b) [Tests on the acute and toxic effects
of modern contact insecticides on freshwater animals]. Z. Angew Zool
47: 303-321 (in German).
Macek KJ & McAllister WA (1970) Insecticide susceptibility of some
common fish family representatives. Trans Am Fish Soc 99: 20-27.
Macek KJ, Hutchinson C, & Cope OB (1969) The effects of temperature
on the susceptibility of bluegills and rainbow trout to selected
pesticides. Bull Environ Contam Toxicol 4(3): 174-183.
Macek KJ, Buxton KS, Derr SK, Dean JW, & Sauter S (1976) Chronic
toxicity of lindane to selected aquatic invertebrates and fish.
Washington, DC, US Environmental Protection Agency (Ecological
Research Series, EPA-600/3-76-046).
Macholz RM, Knoll R, Lewerenz H J, Plass R, & Schulze J (1983)
[Metabolism of gamma-hexachlorocyclohexane (HCH) in germ-free and
conventional rats]. Zentralbl Pharmakol 122(2): 221 (in German).
MacKay D & Walkoff AW (1973) Rate of evaporation of low-solubility
contaminants from water bodies to atmosphere. Environ Sci Technol 7:
611.
MacRae IC, Raghu K, & Castro TF (1967) Persistence and
biodegradation of four common isomers of benzenehexachloride in
submerged soils. J Agric Food Chem 15(5): 911-914.
MacRae JC, Yamaya Y, & Yoshida T (1984) Persistence of HCH-isomers
in soil suspensions. Soil Biol Biochem 16: 285-286.
Marcelle C & Thome JP (1983) Acute toxicity and bioaccumulation of
lindane in gudgeon, Gobio gobio (L.). Bull Environ Contam Toxicol
31: 453-458.
Marcelle C & Thome JP (1984) Relative importance of dietary and
environmental sources of lindane in fish. Bull Environ Contam
Toxicol 33: 423-429.
Marchal-Segault D & Ramade F (1981) The effects of lindane, an
insecticide, on hatching and post-embryonic development of Xenopus
laevis (Daudin). Anuran amphibian. Environ Res 24: 250-258.
Martin RJ & Duggan RE (1968) Pesticide residues in total diet
samples (III). Pestic Monit J 1(4): 1-20.
Martin DB & Hartmann WA (1985) Organochlorine pesticides and
polychlorinated biphenyls in sediment and fish from wetlands in the
North-Central United States. J Assoc Off Anal Chem 69(4): 712-717.
Mathur SP & Saha JG (1975) Microbial degradation of lindane-14C in
a flooded sandy loam soil. Soil Sci 120(4): 301-307.
Mathur SP & Saha JG (1977) Degradation of lindane-14C in a mineral
soil and in an organic soil. Bull Environ Contam Toxicol 17(4): 424-
430.
Matsumura F (1985) Toxicology of insecticides, 2nd ed. New York,
Plenum Press, pp. 128-143.
Matsumura F & Benezet HJ (1973) Studies on the bioaccumulation and
microbial degradation of 2,3,7,8,-tetrachlorodibenzo-p-dioxin.
Environ Health Perspect 5: 253-258.
Matsumura F & Tanaka K (1984) Molecular basis of neuroexcitability
actions of cyclodiene-type insecticides. In: Narahashi T, ed.,
Cellular and molecular neurotoxicology. New York, Raven Press, pp.
225-240.
Matsumura F, Benezet HJ, & Patil KC (1976) Factors affecting
microbial metabolism of gamma-BHC. J Pestic Sci 1: 3-8.
McLeay DJ (1976) A rapid method for measuring the acute toxicity of
pulpmill effluents and other toxicants to salmonid fish at ambient
room temperature. J Fish Res Board Canada 33; 1303-1311,
McNamara B & Krop S (1948) The treatment of acute poisoning produced
by gamma hexachlorocyclohexane. J Pharmacol 92: 147-152.
McParland PJ, McCracken RM, O'Hare MB, & Raven AM (1973)
Benzenehexachloride poisoning in cattle. Vet Rec 93: 369-371.
Meemken HA, Habersaat K, & Groebel W (1982) [Burden of breast milk
with chlorinated hydrocarbons from creams containing anhydrous
lanolin]. Lebensmittelchem Gerichtl Chem 36: 51-53 (in German).
Mendeloff AI & Smith DE (1955) Exposure to insecticides, bone marrow
failure, gastrointestinal bleeding, and uncontrollable infections.
Am J Med 19(8): 274-284.
Mes J, Davies DJ, & Turton D (1982) Polychlorinated biphenyl and
other chlorinated hydrocarbon residues in adipose tissue of
Canadians. Bull Environ Contam Toxicol 28: 97-104.
Mes J, Davies DJ, Turton D, & Sun WF (1986) Levels and trends of
chlorinated hydrocarbon contaminants in the breast milk of Canadian
women. Food Addit Contam 3(4): 313-322.
Mestres R (1974) Report of a group of experts on the content of
organohalogen compounds detected between 1968 and 1972 in water, air
and foodstuffs and the methods of analysis used in the nine Member
States of the European Community. Presented at the European
Colloquium on problems raised by the contamination of man and his
environment by persistent pesticides and organo-halogenated
compounds, Luxembourg, 14-16 May 1974. Luxembourg, Commission of the
European Communities (Paper No. 1).
Milby TH & Samuels AJ (1971) Human exposure to lindane-comparison of
an exposed and unexposed population. J Occup Med 13(5): 256-258.
Milby TH, Samuels AJ, & Ottoboni F (1968) Human exposure to lindane;
blood lindane levels as a function of exposure. J Occup Med 10(10):
584-587.
Mills AC & Biggar JW (1969) Solubility-temperature effect on the
adsorption of gamma- and beta-BHC from aqueous and hexane solutions
by soil materials. Soil Sci Soc Am Proc 33: 210-216.
Morgan DP, Stockdale EM, Roberts R J, & Walter AW (1980) Anemia
associated with exposure to lindane. Arch Environ Health 35(5): 307-
310.
Mosha RD, Gyrd-Hansen N, & Kraul I (1986) Distribution and
elimination of lindane in goats. Bull Environ Contam Toxicol 36:
516-522.
Mottram DS, Psomas IE, & Patterson RLS (1983) Chlorinated residues
in the adipose tissue of pigs treated with gamma-
hexachlorocyclohexane. J Sci Food Agric 34: 378-387.
Mouvet C (1985) Monitoring of polychlorinated biphenyls (PCBs) and
hexachlorocyclohexanes (HCH) in fresh water using the aquatic moss
Cinclidotus danubicus. Sci Total Environ 44: 253-267.
Moza P, Mostafa I, & Klein W (1974) [ntributions to ecological
chemistry. LXXXIX. Exploratory investigations of the metabolism of
gamma-pentachlorcyclohex-1-ene in higher plants grown
hydroponically]. Chemosphere 6: 255-258 (in German).
Muacevic G (1966) [Oral toxicity in male rats]. Ingelheim am Rhein,
Boehringer & Son (Unpublished report submitted to WHO by CIEL) (in
German).
Muacevic G (1970) [LD50 determination in female rats]. Ingelheim
am Rhein, Boehringer & Son (Unpublished report submitted to WHO by
CIEL) (in German).
Muacevic G (1971a) [LD50 determination in male rats]. Ingelheim am
Rhein, Boehringer & Son (Unpublished report submitted to WHO by
CIEL) (in German).
Muacevic G (1971b) [LD50 determination in the rat]. Ingelheim am
Rhein, Boehringer & Son (Unpublished report submitted to WHO by
CIEL) (in German).
Müller D, Klepel H, Macholz RM, Lewerenz HJ, & Engst R (1981)
Electroneurophysiological studies on neurotoxic effects of
hexachlorocyclohexane isomers and gamma-pentachlorocyclohexene. Bull
Environ Contam Toxicol 27: 704-706.
Muralidhara MK, Krishnakumari MK, & Majumder SK (1979) Effects of
carriers on the oral toxicity of lindane (gamma-BHC) to albino rats.
J Food Sci Technol 16: 105-107.
Nair GA (1981) Toxic effects of certain biocides on a freshwater
mite, Hydrachna trilobata Viets (Arachnida; Hydrachnoidea;
Hydrachnidae). J Environ Biol 2(2): 91-96.
Nash RG (1983) Determining environmental fate of pesticides with
microagroecosystems. Residue Rev 85: 199-215.
Nash RG, Harris WG, Ensor PD, & Woolson EA (1973) Comparative
extraction of chlorinated hydrocarbon insecticides from soils 20
years after treatment. J Assoc Off Anal Chem 56(3): 728-732.
Newland LW, Chesters G, & Lee GB (1969) Degradation of gamma-BHC in
simulated lake impoundments as affected by aeration. J Water Pollut
Control Fed 41: R174-R188.
Niessen KH, Ramolla J, Binder M, Brümann G, & Holmann U (1984)
Chlorinated hydrocarbons in adipose tissue of infants and toddlers:
inventory and studies on their association with intake of mothers'
milk. Eur J Pediatr 142: 238-243.
Nigam SK, Karnik AB, Majumder SK, Visweswarriah K, Suryanarayana
Raju G, Muktha Bai K, Lakkad BC, Thakore KN, & Chatterjee BB (1986)
Serum hexachlorocyclohexane residues in workers engaged at a HCH
manufacturing plant. Int Arch Occup Environ Health 57: 315-320.
Nitsche K, Lange M, Bauer E, & Zesch A (1985) Quantitative
distribution of locally applied lindane in human skin and
subcutaneous fat in vitro. Dermatosen 32(5): 161-165.
Noren K (1983) Levels of organochlorine contaminants in human milk
in relation to the dietary habits of the mothers. Acta Paediatr
Scand 72: 811-816.
Norstrom RJ, Simon M, Muir DCG, & Schweinsburg RE (1988)
Organochlorine contaminants in Arctic marine food chains:
identification, geographical distribution and temporal trends in
polar bears. Environ Sci Technol 22: 1063-1071.
Nurmatov RS (1965) [Effect of trichlormetaphos-3 and lindane on
animals]. Veterinarija 44(8): 85-87 (in Russian).
Ocker HD (1983) [Contaminants in cereal grains-state of knowledge
and list of deficiencies]. Getreide-Mehl-Brot 37: 3-7 (in German).
Oehme M & Mano S (1984) The long-range transport of organic
pollutants in the Arctic. Fresenius Z Anal Chem 319: 141-146.
Oehme M & Stray H (1982) Quantitative determination of ultra-traces
of chlorinated compounds in high-volume air samples from the Arctic
using polyurethane foam as collecting medium. Fresenius Z Anal Chem
311: 655-673.
Oesch F (1980) Bacterial mutagenicity tests of lindane with mouse
liver preparations as metabolizing systems. University of Mainz
(Unpublished report Celamerck No. 111AA-457-006, submitted to WHO by
CIEL).
Oesch F & Glatt HR (1984) Mammalian cell (V79) mutagenicity test on
lindane. University of Mainz (Unpublished report Celamerck No.
111AC-457-019/SP 540-VT21, submitted to WHO by CIEL).
Oesch F, Friedberg T, Herbst M, Paul W, Wilhelm N, & Bentley P
(1982) Effects of lindane treatment on drug metabolizing enzymes and
liver weight of CF1 mice in which it evoked hepatomas and in non-
susceptible rodents. Chem-biol Interactions 40: 1-14.
Ohisa N & Yamaguchi M (1978a) Degradation of gamma-BHC in flooded
soils enriched with peptone. Agric Biol Chem 42(11): 1983-1987.
Ohisa N & Yamaguchi M (1978b) Gamma BHC degradation accompanied by
the growth of Clostridium rectum isolated from paddy field soil.
Agric Biol Chem 42(10): 1819-1823.
Ohisa N & Yamaguchi M (1979) Clostridium species and gamma-BHC
degradation in paddy soil. Soil Biol Biochem 11: 645-649.
Ohisa N, Yamaguchi M, & Kurihara N (1980) Lindane degradation by
cell free extracts of Clostridium rectum. Arch Microbiol 125: 221-
225.
Ohisa N, Kurihara N, & Nakajima M (1982) ATP synthesis associated
with the conversion of hexachlorocyclohexane related compounds. Arch
Microbiol 131: 330-333.
Ohly A (1973) Toxicology of lindane. In: Ulmann, E. ed., Lindane,
Vol. I, Supplement 1974. Brussels, CIEL, p. 27.
Oldiges H, Takenaka S, & Hochrainer D (1980) [Inhalation test with
lindane (gamma-hexachlorocyclohexane) to determine the LC50.
(Unpublished report Celamerck No. 111AA-423-001 submitted to WHO by
CIEL) (in German).
Oldiges H, Hertel R, Kördel W, Hochrainer D, & Mohr U (1983) [90-Day
inhalation with lindane]. Schmallenberg, Fraunhofer Institut für
Toxikologie und Aerosolforschung (Unpublished report Celamerck No.
111AC-435-004, submitted to WHO by CIEL) (in German).
Oloffs PC, Albright LJ, Szeto SY, & Lau J (1973) Factors affecting
the behaviour of five chlorinated hydrocarbons in two natural waters
and their sediments. J Fish Res Board Canada 30(11): 161 9-1623.
Oshiba K (1972) Experimental studies on the fate of ß- and gamma-BHC
in vivo following daily administration. Osaka Shiritsu Daigaku
Igaku Zasshi 21(1-3): 1-19.
Paasivirta J, Palm H, Paukka R, Akhabuhaya J, & Lodenius M (1988)
Chlorinated insecticide residues in Tanzanian environment,
Tanzadrin. Chemosphere 17(10): 2055-2062.
Palmer CM & Maloney TE (1955) Preliminary screening for potential
algicides. Ohio J Sci 55(1): 1-8.
Palmer L & Kolmodin-Hedman B (1972) Improved quantitative gas
chromatographic method for the analysis of small amounts of
chlorinated hydrocarbon pesticides in human plasma. J Chromatogr 74:
21-30.
Palmer AK, Cozen DD, Spicer EJF, & Worden AN (1978a) Effects of
lindane upon reproductive function in a 3-generation study in rats.
Toxicology 10: 45-54.
Palmer AK, Bottomley AM, Worden AN, Frohberg H, & Bauer A (1978b)
Effect of lindane on pregnancy in the rabbit and rat. Toxicology 9:
239-247.
Pandy RN, Zaidi SIM, & Kidawl AM (1985) Effect of pesticides on frog
skeletal muscle sarcolemmal enzymes. J Environ Biol 6(1): 7-9.
Panwar RS, Gupta RA, Joshi HC, & Kapoor D (1982) Toxicity of some
chlorinated hydrocarbons and organophosphorus insecticides to
gastropod, Viviparus bengalensis Swainson. J Environ Biol 3(1):
31-36.
Pastor A, Hernandez F, Medina J, Melero R, Lopez FJ, & Conesa M
(1988) Organochlorine pesticides in marine organisms from the
Castelion and Valencia coasts of Spain. Mar Pollut Bull 19(5): 235-
238.
Paul W, Knappen F, & Stötzer H (1980) [Testing for acute toxicity
after oral administration in an oily solution to Chbi: NMRI (SPF)
mice]. Ingelheim am Rhein, Boehringer & Son (Unpublished report
submitted to WHO by CIEL) (in German).
Petring OU, Adelhoj B, & Jorgensen FR (1986) Acute poisoning after
oral intake of lindane. Ugeskr Laeg 148(50): 3377.
Platford RF (1981) The environmental significance of surface films.
II. Enhanced partitioning of lindane in thin films of octanol on the
surface of water. Chemosphere 10(7): 719-722.
Podlejski J & Dervieux AM (1978) Persistance de quatre produits
phytosanitaires dans les eaux de rivières en Camargue:
caractérisation de leurs effets sur l'écosystème. Trav Soc Pharm
Montpellier 38(2): 153-164.
Polishuk ZW, Wassermann M, Wassermann D, Groner Y, Lazarovici S, &
Tomatis L (1970) Effects of pregnancy on storage of organochlorine
insecticides. Arch Environ Health 20: 215-217.
Portig J, Kraus P, Stein K, Koransky W, Noack G, Gross B, & Sodomann
S (1979) Glutathione conjugate formation from hexachlorocyclohexane
and pentachlorocyclohexene by rat liver in vitro. Xenobiotica 9:
353-378.
Portmann JE (1970) Results of acute toxicity test with marine
organisms, using a standard method. In: Ruivo, M. ed., Marine
pollution and sea life. London, Fishing News (Books) Ltd, pp. 212-
217.
Portmann JE (1979) Evaluation of the impact on the aquatic
environment of hexachlorocyclohexane (HCH-isomers). Brussels,
Commission of the European Communities (Contract No. U/78/180-SEC
(78)792).
Probst GS, McMahon RE, Hill LE, Thompson CZ, Epp JK, & Neal SB
(1981) Chemically-induced unscheduled DNA synthesis in primary rat
hepatocyte cultures: A comparison with bacterial mutagenicity using
218 compounds. Environ Mutagen 3: 11-32.
Publicover SJ & Duncan CJ (1979) The action of lindane in
accelerating the spontaneous release of neurotransmitter at the frog
neuromuscular junction. Naunyn-Schmledebergs Arch Pharmacol 381:
179-182.
Publicover SJ, Duncan CJ, & Smith JL (1979) The action of lindane in
causing ultrastructural damage in frog skeletal muscle. Comp Biochem
Physiol C64: 237-241.
Racey PA & Swift SM (1986) The residual effects of remedial timber
treatments on bats. Biol Conserv 35: 205-214.
Ramamoorthy S (1985) Competition of fate processes in the
bioconcentration of lindane. Bull Environ Contam Toxicol 34: 349-
358.
Randall WF, Dennis WH, & Warner MC (1979) Acute toxicity of
dechlorinated DDT, chlordane, and lindane to bluegill (Lepomis
macrochirus) and Daphnia magna. Bull Environ Contam Toxicol 21:
849-854.
Rao MB (1981) Effect of gamma-hexachloran and Sevin on the survival
of the black sea mussel, Mytilus galloprovincialis Lam.
Hydrobiologia 78: 33-37.
Rao PSC & Davidson JM (1982) Retention and transformation of
selected pesticides and phosphorus in soil-water system: a critical
review. Springfield, Virginia, US Department of Commerce, National
Technical Information Services (EPA Report No. 600/3-82-060, PB 92-
256884).
Rao SM & Reddy JK (1987) Peroxisome proliferation and
hepatocarcinogenesis. Carcinogenesis 8(5): 631-636.
Rappe C, Nygren M, Lindström G, Buser HR, Blaser O, & Wüthrich C
(1987) Polychlorinated dibenzofurans and dibenzo-p-dioxins and other
chlorinated contaminants in cow milk from various locations in
Switzerland. Environ Sci Technol 21: 964-970.
Raw GR, ed. (1970) ClPAC Handbook. Vol. 1. Analysis of technical and
formulated pesticides. Collaborative International Pesticides
Analytical Council Ltd, New York, Academic Press, pp. 71-78, 129-
131.
Ray RC (1983) Toxicity of the pesticides hexachlorocyclohexane and
Benomyl to nitrifying bacteria in flooded autoclaved soil and in
culture media. Environ Pollut A32: 147-155.
Reltenrath WG, Chellquist EM, Shipwash EA, Jederberg WW, & Krueger
GG (1984) Percutaneous penetration in the hairless dog, weanling pig
and grafted athymic nude mouse: evaluation of models for predicting
skin penetration in man. Br J Dermatol 27 (Suppl. III): 123-135.
Reiner E, Krauthacker B, Stipcevic M, & Stefanac Z (1977) Blood
levels of chlorinated hydrocarbon residues in the population of a
continental town in Croatia (Yugoslavia). Pestic Monit J 11: 54-55.
Reno FE (1976a) Teratology study in rats: Lindane (gamma benzene
hexachloride, USP). Madison, Wisconsin, Hazelton Laboratories
America Inc. (Unpublished report submitted to WHO by CIEL).
Reno FE (1976b) Teratology study in rabbits: Lindane (gamma benzene
hexachloride, USP). Madison, Wisconsin, Hazelton Laboratories
America Inc. (Unpublished report submitted to WHO by CIEL).
Rhöne-Poulenc Agrochimie (1986) Lindane. Information kit. Lyon.
Riemschneider R (1949) [A contribution to the toxicity of contact
insecticides]. Anz Schädlingskd 22(1): 1-3 (in German).
Rivett KF, Chesterman H, Kellett DN, Newman AJ, & Worden AN (1978)
Effects of feeding lindane to dogs for periods of up to 2 years.
Toxicology 9: 273-289.
Rocohi P, Perocco P, Alberghini W, Fini A, & Prodi G (1980) Effect
of pesticides on scheduled and unscheduled DNA synthesis of rat
thymocytes and human lymphocytes. Arch Toxicol 45: 101-108.
Röhrborn G (1976) Cytogenetic analysis of bone marrow of Chinese
harnster (Cricetulus grisens) after sub-acute treatment with
lindane. (Unpublished report Celamerck No. 111AA-457-008, submitted
to WHO by CIEL).
Röhrborn G (1977a) Statement on the potential mutagenicity of
lindane (gamma-hexachlorocyclohexane). (Unpublished report Celamerck
No. 111AA-457-018, submitted to WHO by CIEL).
Röhrborn G (1977b) Dominant lethal test after treatment of male rats
with lindane. (Unpublished report Celamerck No. 111AA-457-004,
submitted to WHO by CIEL).
Rosenberg LE, Rudd RL, Devaul J, Lundberg DE, Recca JM, & Smith MB
(1953) The effects of economic poisons on wildlife. Quarterly
progress report of the Federal Aid in Wildlife Restoration Act pp.
2, 7, 11-12, 19, 43-45 (Project No. W-45R-1) (Unpublished report).
Sagelsdorff P, Lutz WK, & Schlatter C (1983) The relevance of
covalent binding to mouse liver DNA to the carcinogenic action of
hexachlorocyclohexane isomers. Carcinogenesis 4(10): 1267-1273.
Samuels AJ & Milby TH (1971) Human exposure to lindane. Clinical,
haematological, and biochemical effects. J Occup Med 13(3): 147-151.
San Antonio JP (1959) Demonstration of lindane and a lindane
metabolite in plants by paper chromatography. J Agric Food Chem
7(5): 322-325.
Sanders HO (1969) Toxicity of pesticides to the crustacean Gammarus
lacustrus. Washington, DC, US Department of Interior, Bureau of
Sport Fisheries and Wildlife (Technical Paper No. 25), pp. 2-18.
Sanders HO (1970) Pesticide toxicities to tadpoles of the western
chorus frog Pseudacris triseriata and Fowler's toad Bufo
woodhousii fowleri. Copeia 2: 246-251.
Sanders HO & Cope OB (1966) Toxicities of several pesticides to two
species of Cladocerans. Trans Am Fish Soc 95: 165-169.
Sanders HO & Cope OB (1968) The relative toxicities of several
pesticides to naiads of three species of stoneflies. Limnolog
Oceanogr 13: 112-117.
Sanfeliu C, Camon L, Martinez E, Sola C, Artigas F, & Rodriguez-
Farre E (1988) Regional distribution of lindane in rat brain.
Toxicology 49: 189-196.
Saxena MC, Siddique MKJ, Bhargava AK, Krishna Murti CR, & Kutty D
(1981) Placental transfer of pesticides in humans. Arch Toxicol 48:
127-134.
Schafer EW (1972) The acute oral toxicity of 369 pesticidal,
pharmaceutical and other chemicals to wild birds. Toxicol Appl
Pharmacol 21: 315-330
Schimmel SC, Patrick JM, Jr, & Forester J (1977) Toxicity and
bioconcentration of BHC and lindane in selected animals. Arch
Environ Contam Toxicol 6: 355-363.
Schmiedeberg J & Wasserberger HJ (1953) [Acute toxicity of
hexachlorocyclohexane for man]. Anz Schädlingskd 26(9): 129-133 (in
German).
Schmitt F (1956) [Experimental investigations on the behaviour of
hexachlorocyclohexane and aldrin in the soil, with special reference
to duration of activity, effects on small animal fauna, and
impairment of the taste of geocarpic fruits]. Hohenheim,
Agricultural College (Thesis) (in German).
Schneeweis JC, Greichus TA, & Linder RL (1974) Organochlorine
pesticide residue levels in North American timber wolves-1969-71.
Pestic Monit J 8(2): 142-143.
Schnell U (1965) [Investigations on the chronic toxicity of lindane
for pigs, with special reference to investigations for residues in
fat and liver]. Berlin, Humboldt University (Thesis) (in German).
Schröter C, Parzefall W, Schröter H, & Schulte-Hermann R (1987)
Dose-response studies on the effects of alph-, ß- and gamma-
hexachlorocyclohexane on putative preneoplastic foci,
monooxygenases, and growth in rat liver. Cancer Res 47: 80-88.
Schüttmann W (1972) Clinical observations on the long-term toxicity
of organochlorine pesticides. Z Gesamte Hyg 17: 12-18 (in German).
Schwabe U & Wendling I (1967) [Stimulation of drug metabolism by low
doses of DDT and other chlorlnated hydrocarbon insecticides].
Arzneimittelforschung 17(5): 614-618 (in German).
Seidler H, Machoiz RM, Härtig M, Kujawa M, & Engst R (1975) [Studies
on the metabolism of certain insecticides and fingicides in the rat.
Part IV. Distribution, degradation and excretion of 14C-labelled
lindane]. Nahrung 19(5/6): 473-482 (in German).
Selby LA, Newell KW, Hauser GA, & Junker G (1969) Comparison of
chlorinated hydrocarbon pesticides in maternal blood and placental
tissues. Environ Res 2: 247-255.
Seuge J & Bluzat R (1979a) Toxicité chronique du carbaryl et du
lindane chez le mollusque d'eau douce Lymnea stagnails L. Water
Res 13(3): 285-293.
Seuge J & Bluzat R (1979b) Etude de la toxicité chronique de deux
insecticides (carbaryl et lindane) à la génération F1 de Lymnea
stagnails L. (mollusque gastéropode pulmone). II. Conséquences sur
le potentiel reproducteur. Hydrobiologia 66(1): 25-31.
Seuge J & Bluzat R (1982) Influence d'une intoxication par le
lindane en fonction de la dureté de l'eau chez Lymnea stagnails
(Mollusca pulmonata). Malacologia 22(1-2): 1 5-18.
Sharom MS, Miles JRW, Harris CR, & McEwen FL (1980) Persistence of
12 insecticides in water. Water Res 14: 1089-1093.
Shearer RC, Letey J, Farmer WJ, & Klute A (1973) Lindane diffusion
in soil. Soil Sci Soc Am Proc 37: 189-193.
Shiota Y & Kanda S (1972) [Fate of benzene hexachlonde in paddy
soils]. Aichi-Ken Nogyo Sogo Shikenjho, Kenkyu Hokoku 1972A/4: 128-
133 (in Japanese).
Shirasu Y, Moriya M, Karo K, Furuhashi A, & Kada T (1976)
Mutagenicity screening of pesticides in the microbial system. Mutat
Res 40: 1 9-30.
Siddaramappa R & Sethunathan N (1975) Persistence of gamma-BHC and
beta-BHC in Indian rice soils under flooded conditions. Pestic Sci
6: 395-403.
Siddique MKJ, Saxena MC, Mishra UK, Krishnamurti CR, & Nag D (1981)
Long-term occupational exposure to DDT. Int Arch Occup Environ
Health 48: 301-308.
Sierra M & Santiago D (1987) Organochlorine pesticide levels in barn
owls collected in Leon, Spain. Bull Environ Contam Toxicol 38: 261-
265.
Sina JF, Bean CL, Dysart GR, Taylor VI, & Bradley MO (1983)
Evaluation of the alkaline elution/rat hepatocyte assay as a
predictor of carcinogenic/mutagenic potential. Mutat Res 113: 357-
391.
Sinha RRP & Sinha SP (1983) Induction of dominant lethal mutations
in Drosophila melanogaster by three pesticides: Gammexane, Sevin,
and Folidol. Comp Physiol Ecol 8(2): 87-89.
Skaare JU, Tuveng JM, & Sande HA (1988) Organochlorine pesticides
and polychlorinated biphenyls in maternal adipose tissue, blood,
milk, and cord blood from mothers and their infants living in
Norway. Arch Environ Toxicol 17: 55-63.
Skaftason JF & Johannesson T (1979) Organochlorine compounds (DDT,
hexachlorocylohexane, hexachlorobenzene) in Icelandic animal body
fat and butter fat: Local and global sources of contamination.
Letter to the Editor. Acta Pharmacol Toxicol 44: 156-157.
Slade RE (1945) The gamma-isomer of hexachlorocyclohexane
(gammexane). Chem Ind 65: 314-319.
Slooff W & Matthijsen AJCM (1988) Integrated criteria document:
hexachlorocyclohexanes. Bilthoven, National Institute of Public
Health and Environmental Protection (Report No. 758473011).
Srinivasan K & Radhakrishnamurty R (1988) Biochemical changes
produced by ß- and gamma-hexachlorocyclohexane isomers in albino
rats. J Environ Sci Health B23(4); 367-386.
Srinivasan K, Ramesh HP, & Radhakrishnamurty R (1984) Renal tubular
dysfunction caused by dietary hexachlorocyclohexane (HCH) isomers. J
Environ Sci Health B19(4/5): 453-466.
Srinivasan K, Ramesh HP, & Radhakrishnamurty R (1988) Changes
induced by hexachlorocyclohexane isomers in rat livers and testes.
Bull Environ Contam Toxicol 41: 531-539.
Starr HG & Clifford NJ (1972) Acute lindane intoxication. A case
study. Arch Environ Health 25: 374-375.
Starr RI & Johnson RE (1968) Laboratory method for determining the
rate of volatilization of insecticides from plants. J Agric Food
Chem 16(3): 411-414.
Steering Group on Food Surveillance (1982) Report of the Working
Party on Pesticide Residues (1977-1981). London, Her Majesty's
Stationery Office (Food Surveillance Paper No. 9).
Steering Group on Food Surveillance (1986) Report of the Working
Party on Pesticide Residues (1982-1985). London, Her Majesty's
Stationery Office (Food Surveillance Paper No. 16).
Steering Group on Food Surveillance (1989) Report of the Working
Party on Pesticide Residues (1985-1988). London, Her Majesty's
Stationery Office (Food Surveillance Paper No. 25).
Stein K, Portig J, & Koransky W (1977) Oxidative transformation of
hexachlorocyclohexane in rats and with rat liver microsomes. Naunyn-
Schmiedebergs Arch Pharmacol 298: 115-128.
Stein K, Portig J, Fuhrmann H, Koransky W, & Noack G (1980) Steric
factors in the pharmacokinetics of lindane and alpha-
hexachlorocyclohexane in rats. Xenobiotica 10(1): 65-77.
Steinwandter H (1976) [Lindane metabolism in plants. II. Formation
of alpha-HCH]. Chemosphere 4: 221-225 (in German).
Stewart DKR & Chisholm D (1971) Long-term persistence of BHC, DDT,
and chlordane in a sandy loam soil. Can J Soil Sci 51: 379-383.
Stewart DKR & Fox CJS (1971) Persistence of organochlorine
insecticides and their metabolites in Nova Sootian soils. J Econ
Entomol 64(2): 367-371.
Stieglitz R, Stobbe H, & Schüttmann W (1967) [Bone marrow damage
following exposure to the insecticide gamma-hexachlorocyclohexane
(lindane)]. Acta Haematol 38: 337-350 (in German).
Stöckigt J (1976) Investigations of the metabolism of gamma-HCH in
plant tissue. (Unpublished report Celamerck No. 111AA-641-004,
submitted to WHO by CIEL).
Stöckigt J & Ries B (1976) Catabolism of gamma-hexachlorocyclohexane
(lindane) by plants and cell cultures, a comparison. Bochum, Chair
of Plant Physiology, Ruhr University (Unpublished report Celamerck
No. 111AA-641-005, submitted to WHO by CIEL).
Strachan WMJ, Huneault H, Schertzer WM, & Elder FC (1980)
Organochlorines in precipitation in the Great Lakes region. In:
Afghan BK & MacKay D ed., Hydrocarbons and halogenated hydrocarbons
in the aquatic environment. New York, Plenum Press, pp. 387-396.
Sugiura K, Washino T, Hattori M, Sato E, & Goto M (1979)
Accumulation of organochlorine compounds in fishes. Difference of
accumulation factors by fishes. Chemosphere 6: 359-364.
Sunol C, Tusell JM, Gelp E, & Rodriguez-Farre E (1988) Convulsant
effect of lindane and regional brain concentration of GABA and
dopamine. Toxicology 49(2/3): 247-252.
Suter P, Horst K, Luetkemeier H, Herbst M, Terrier C, Lind H, &
Ellgehausen H (1983) Three months toxicity study in rats with
lindane: Part 1 & 2. Itingen, Research and Consulting Company AG
(Unpublished report Celamerck No. 111AA-433-007/009, submitted to
WHO by CIEL).
Suzuki M, Yamato Y, & Watanabe T (1975) Persistence of BHC
(1,2,3,4,5,6-hexachlorocyclohexane) and dieldrin residues in field
soils. Bull Environ Contam Towicol 14(5): 520-529.
Sweeney RA (1969) Metabolism of lindane by unicellular algae. In:
Proceedings of the 12th Conference on Great Lakes Research,
International Associationforf Great Lakes Research, pp. 98-102.
Szymczynski GA, Waliszewski SM, Tuszewski M, & Pyda P (1986)
Chlorinated pesticides levels in human adipose tissue in the
district of Poznan. J Environ Sci Health A21(1): 5-14.
Takabatake E (1978) Levels of organochlorine compounds and heavy
metals in tissues of the general population in Japan. Geogr Med 8:
1-23.
Tanabe S, Tatsukawa R, Kawano M, & Hidaka H (1982) Global
distribution and atmospheric transport of chlorinated hydrocarbons:
HCH(BHC) isomers and DDT compounds in the Western Pacific, Eastern
Indian, and Antarctic Oceans. J Oceanogr Soc Jpn 38(3): 137-148.
Tanabe S, Tanaka H, & Tatsukawa R (1984) Polychlorobiphenyls, Sigma-
DDT, and hexachlorocyclohexane isomars in the Western North Pacific
ecosystem. Arch Environ Contam Toxicol 13: 731-738.
Tanaka K, Kudhara N, & Nakajima M (1977) Pathways of chlorophenol
formation in oxidative biodegradation of BHC. Agric Biol Chem 41(4):
723-725.
Tanaka K, Kurihara N, & Nakajima M (1979) Oxidative metabolism of
lindane and its isomers with microsomes from rat liver and housefly
abdomen. Pestic Biochem Physiol 10: 96-103.
Thorpe E & Walker AIT (1973) The toxicology of dieldrin (HEOD). II.
Comparative long-term oral toxicity studies in mice with dieldrin,
DDT, phenobarbitone, ß-BHC and gamma-BHC. Food Cosmet Toxicol 11:
433-442.
Tolot F, Longlet JP, & Berthelon J (1969) Polyneurite due au
lindane. J Méd Lyon 50: 747-754.
Tomczak S, Baumann K, & Lehnert G (1981) Occupational exposure to
hexachlorocyclohexane. IV. Sex hormone alterations in HCH-exposed
workers. Int Arch Occup Environ Health 48: 283-287.
Tooby TE & Durbin FJ (1975) Lindane residue accumulation and
elimination in rainbow trout (Salmo gairdnerii Richardson) and
roach (Rutilus rutilus L.). Environ Pollut 8(2): 79-89.
Trautmann A & Streit B (1979) [Sorption of lindane (gamma-
hexachlorocyclohexane) in Nitzschia actinastroïdes (Lemm.) v. Goor
(dimethoate) at different growth conditions]. Arch Hydrobiol Suppl
55(3/4): 324-348 (in German).
Trifonova TK, Gladenko IN, & Schukla WD (1970) [Effect of gamma-BHC
and Sevin on reproduction]. Veterinarja 47: 91-93 (in Russian).
Trosko JE (1982) Inhibition of cell-to-cell communication by tumour
promoters. Carcinogenesis 7: 565-585.
Truhaut R (1954) Communication to the International Symposium on
Prevention of Cancer, São Paulo, 1954. In: Evaluation of the
toxicity of pesticide residues in food. Report of a Joint Meeting of
the FAO Committee on Pesticides in Agriculture and te WHO Expert
Committee on Pesticide Residues, Geneva, 30 September-7 October
1963. Rome, Food and Agriculture Organization of the United Nations,
p. 48 (FAO Meeting Report No. PL/1963/13. WHO/Food Add., 23 (1964)).
Tsukano Y (1973) Factors affecting disappearance o! BHC isomers from
rice field soil. Jpn Agric Res Q 7(2): 93-97.
Tsushimoto G, Chang CC, Trosko JE, & Matsumura F (1983) Cytotoxic,
mutagenic, and cell-cell communication inhibitory properties of DDT,
lindane, and chlordane on Chinese hamster cells in vitro. Arch
Environ Contam Toxicol 12: 721-730.
Tu CM (1975) Interaction between lindane and microbes in soils. Arch
Microbiol 105(2): 131-134.
Tu CM (1976) Utilization and degradation of lindane by soil
microorganisms. Arch Microbiol 108: 259-263.
Tuinstra LGMT (1971) Organochlorine insecticide residues in human
milk in the Leiden region. Neth Milk Dairy J 25: 24-32.
Turner JC (1979) Transplacental movement of organochlorine pesticide
residues in desert bighorn sheep. Bull Environ Contam Toxicol 21:
116-124.
Turner JC & Shanks V (1980) Absorption of some organochlorine
compounds by the rat small intestine- in vivo. Bull Environ Contam
Toxicol 24: 652-655.
Turtle EE, Taylor A, Wright EN, Thearle RJP, Egan H, Evans WH, &
Soutar NM (1963) The effects on birds of certain chlorinated
insecticides used as seed dressings. J Sci Food Agric 14: 567-577.
Tussel JM, Sunol C, Gelpi E, & Rodriguez-Farre E (1988) Effect of
lindane at repeated low doses. Toxicology 49: 375-379.
Tzoneva-Maneva MT, Kaloyaneva F, & Georgieva V (1971) Influence of
diazinon and lindane on the mitotic activity and the caryotype of
human lymphocytes, cultivated in vitro. Bibl Haematol 38(1): 344-
347.
Uchida M, Kurihare N, Fujita T, & Nakajima M (1974) BHC isomers and
related compounds. VII. Inhibitory effects of BHC isomers on sodium-
potassium ion-dependent ATPase, yeast growth, and nerve conduction.
Pestic Biochem Physiol 4(3): 260-265.
Ukeles R (1962) Growth of pure cultures of marine phytoplankton in
the presence of toxicants. Appl Microbiol 10: 532-537.
Ullmann L, Sacher R, & Porrizello T (1986a) Primary skin irritation
study with lindane in rabbits (4-hour occlusive application).
(Unpublished report Celamerck No 111AD-465-003, submitted to WHO by
CIEL).
Ullmann L, Claire R, & Bognar G (1986b) Test for delayed contact
hypersensitivity in the albino guinea pig with lindane (maximization
test). Itingen, Research and Consulting Company AG (Unpublished
report No. 061650, Celamerck No. 111AC-467-001, submitted to WHO by
CIEL).
Ullmann L, Mohler H, & Gembardt G (1986c) Four-hour acute aerosol
inhalation toxicity study with lindane in rats. Itingen, Research
and Consulting Company AG (Unpublished report No. 061637, submitted
to WHO by CIEL).
Ullmann L, Claire R, & Bognar G (1987a) Contact hypersensitivity to
Nexit Fluessig (CME 11129) in albino guinea pigs (maximization
test). Itingen, Research and Consulting Company AG (Unpublished
report Celamerck No. 11129-467-004, submitted to WHO by CIEL).
Ullmann J, Claire R, & Bognar G (1987b) Contact hypersensitivity to
Nexit Stark (CME 11125) in albino guinea pigs (maximization test).
Itingen, Research and Consulting Company AG (Unpublished report
Celamerck No. 11125-467-002, submitted to WHO by CIEL).
Ullmann J, Claire R, & Bognar G (1987c) Contact hypersensitivity to
Agronex Saatgutpuder (CME 11102) in albino guinea pigs (maximization
test). Itingen, Research and Consulting Company AG (Unpublished
report Celamerck No. 11102-467-003, submitted to WHO by CIEL).
Umweltbundesamt (1988-89) [Data on the environment 1988/89. Berlin,
Schmidt E. Verlag, pp. 403, 410-412, 416-417, 521-522, 524, 526 (in
German).
Uphouse L (1987) Decreased rodent sexual receptivity after lindane.
Toxicol Lett 39: 7-14.
US Environmental Protection Agency (1976) National Interim Primary
Drinking Water Regulations, Subpart B-Maximum contaminant levels.
Washington, DC, Office of Water Supply (Report No. EPA-570/9-76-
003).
US Environmental Protection Agency (1977) Notice of rebuttable
presumption against registration and continued registration of
pesticide products containing lindane. Fed Reg 42(33): 9816-9947.
US Environmental Protection Agency (1980) Lindane position document
2/3. Washington, DC, Office of Pesticide Programs.
US Environmental Protection Agency (1984) Health effects assessment
for lindane. Cincinnatti, Ohio (Report No. EPA/540/1-86/056).
US Environmental Protection Agency (1985) Guidance for the
reregistration of pesticide products containing lindane as the
active ingredient. Washington, DC (Report No. RS-85).
US National Cancer Institute (1977) Bioassay of lindane for possible
carcinogenicity. Washington, DC, US Department of Health, Education,
and Weltare (Technical Report Series No. 14).
van Velsen FL (1986) The oestrogenicity of the environmental
contaminant ß-hexachlorocyclohexane. University of Utrecht (Thesis).
van Velsen FL, Franken MAM, van Leeuwen FXR, & Loeber JG (1984)
[Study of semi-chronic oral toxicity of gamma-HCH in the rat].
Bilthoven, National Institute of Public Health and Environmental
Hygiene (Unpublished report No. 618209001) (in Dutch).
Vesselinovitch SD & Carlborg FW (1983) Lindane bioassay studies and
human cancer risk assessment. Toxicol Pathol 11(1): 12-22.
Vetter H, Kampe W, & Ranfft, K (1983) [Results of three years of
comparative tests on vegetables, fruit and bread from modern and
alternative producers]. Darmstadt, Association of German Institutes
for Agricultural Testing and Research, pp. 1-11,143 (VDLUFA Report
No. 7) (in German).
Vodopick H (1975) Erythropoietic hypoplasia after exposure to gamma-
benzene hexachloride. J Am Med Assoc 234(8): 850-851.
Voerman S & Besemer AFH (1970) Residues of dieldrin, lindane, DDT,
and parathion in a light sandy soil after repeated application
throughout a period of 15 years. J Agric Food Chem 18(4): 717-719.
Voerman S & Besemer AFH (1975) Persistence of dieldrin, lindane, and
DDT in a light sandy soil and their uptake by grass. Bull Environ
Contam. Toxicol 13(4): 501-505.
Vogel SM, Joy RM, & Narahashi T (1985) Lindane exerts both
presynaptic and postsynaptic actions at frog neuromuscular junction.
In: 15th Annual Meeting of the Society for Neuroscience, Part 2.
Dallas, Texas, 20-25 October 1985. Soc Neurosci Abstr 11: 1004.
Vohland HW, Portig J & Stein K (1981) Neuropharmacological effects
of isomers of hexachlorocyclohexane. Toxicol Appl Pharmacol 57(3):
425-438.
Vonk JW & Quirijns JK (1979) Anaerobic formation of (alpha-
hexachlorocyclohexane from gamma-hexachlorocyclohexane in soil and
by Escherichia coli. Pestic Biochem Physiol 12: 68-74.
de Vos RH, van Dokkum W, Olthof PDA, Quirijns JK, Muys T, & van der
Poll JM (1984) Pesticides and other chemical residues in Dutch total
diet samples (June 1976-July 1978). Food Chem Toxicol 22(1): 11-21.
Vukavic T, Pavkov S, Cusic S, Roncevic N, Vojinovic M, & Tokovic B
(1986) Pesticide residues in human colostrum: seasonal variations,
Yugoslavia. Arch Environ Contam Toxicol 15: 525-528.
Wahid PA & Sethunathan N (1979) Sorption and desorption of (alpha,
ß, and gamma isomers of hexachlorocyclohexane in soils. J Agric Food
Chem 27(5): 1050-1053.
Wahid PA & Sethunathan N (1980) Sorption and desorption of lindane
by anaerobic and aerobic soils. J Agric Food Chem 28: 623-625.
Wammes JY, Wegman RCC, Hofstee AWM, Janssens H, Rieffe DW, Marsman
JA, Groenemeijer GS, & van den Broek HH (1983) [Investigation into
the presence of pesticides and related compounds in surface waters].
Bilthoven, National Institute of Public Health and Environmental
Hygiene (Unpublished report No. 218102003) (in Dutch).
Wang HH & Grufferman S (1981) Aplastic anemia and occupational
pesticide exposure: a case-control study. J Occup Med 23(5): 364-
366.
Ware GW & Naber EC (1961) Lindane in eggs and chicken tissues. J
Econ Entomol 54(4): 675-677.
Wassermann M, Ron M, Bercovici B, Wassermann D, Cucos S, & Pines A
(1982) Premature delivery and organochlorine compounds.
Polychlorinated biphenyls and some organochlorine insecticides.
Environ Res 28: 106-112.
Wedberg JL, Moore S, III, Amore FJ, & McAvoy H (1978) Residues in
food and feed. Organochlorine insecticide residues in bovine milk
and manufactured milk products in Illinois, 1971-76. Pest Monit J
11(4): 161-164.
Wegman RCC & Greve PA (1980) Halogenated hydrocarbons in Dutch water
samples over the years 1969-1977. In: Afghan BK & MacKay D (eds.),
Hydrocarbons and halogenated hydrocarbons in the aquatic
environment. New York, Plenum Press, pp. 405-415.
Weigert P, MUller J, Hans R, & Zufelde KP (1983) [Pesticide residues
in foodstuffs. Communication I: Organochlorine compounds]. Berlin,
Dietrich Reimer Verlag (ZEBS-Berichte 3) (in German).
Weisse I & Herbst M (1977) Carcinogenicity study of lindane in the
mouse. Toxicology 7: 233-238.
Wellborn TL, Jr (1971) Toxicity of some compounds to striped bass
fingerlings. Progr Fish-Cult 33: 32-36.
West I (1967) Lindane and haematologic reactions. Arch Environ
Health 15: 97-101.
Wheatley GA (1965) The assessment and persistence of residues of
organochlorine insecticides in soils and their uptake by crops. Proc
Assoc Appl Biol 55: 325-329.
Whitehead CC, Downing AG, & Pettigrew RJ (1972a) The effects of
lindane on laying hens. Br Poult Sci 13: 293-299.
Whitehead CC, Downie JN, & Phillips JA (1972b) BHC not found to
reduce the shell quality of hen's eggs. Nature 239: 411-412.
Whitehead CC, Downie JN, & Phillips JA (1974) Some characteristics
of the egg shells of quail fed gamma-BHC. Pestic Sci 5: 275-279.
WHO/FAO (1975) Data sheets on pesticides: No. 1- Lindane. Geneva
(Unpublished document VBC/DS/75.12).
WHO (1985) Specifications for pesticides used in public health:
insecticides-molluscicides-repellents - methods, 6th ed., Geneva.
WHO (1990) The WHO recommended classification of pesticides by
hazard and guidelines to classification 1990-1991. Geneva
(Unpublished document WHO/PCS/90.1 Rev. 1).
Wilkes LC, Tapprich BL, & Mulkey NS (1987a) Determination of 14C-
residues following oral administration of 14C-lindane to lactating
goats (in vivo and in vitro studies). Monument, California,
Development Corporation (Unpublished report No. ADC 957a, submitted
to WHO by CIEL).
Wilkes LC, Mulkey NS, Hallenbeck SA, Piznik M, & Wargo JP (1987b)
Metabolism study of 14C-lindane fed or topically applied to
lactating goats. Monument, California, Development Corporation
(Unpublished report No. ADC 957b, submitted to WHO by CIEL).
Williams DT, Benoit, FM, McNeil EE, & Otson R (1978) Organochlorine
pesticide levels in Ottawa drinking water, 1976. Pestic Monit J
12(3): 163.
Wirth H (1985) The adsorbtion of 14C-labelled gamma-HCH (lindane)
at levels in the range of ng/litre to soil components. Geesthacht,
GKSS Research Institute (Report No. GKSS 85/E/30).
Wittlinger R & Ballschmiter K (1987) Global baseline pollution
studies. XI: Congener specific determination of polychlorinated
biphenyls (PCBs) and occurrence of alpha- and gamma-
hexachlorocyclohexane (HCH), 4.4'-DDE and 4.4'-DDT in continental
air. Chemosphere 16(10-12): 2497-2513.
Wolfe GW & Ralph JA (1980) Acute oral toxicity study in B6C3F1 mice:
Lindane. Vienna, Virginia, Hazleton Laboratories America Inc.
(Unpublished report submitted to WHO by CIEL).
Wolff G (1986) Maternal influence on phenotypic differentiation of a
mutant mouse susceptible to neoplasia and obesity. In: Genetic
toxicology of environmental chemicals, Part B, Genetic effects and
applied mutagenesis. New York, Alan R. Liss Inc., pp. 33-38.
Wolff GL & Morrissey RL (1986) Increased responsiveness of lean
pseudoagouti Avy/a female mice to lindane enhancement of lung and
liver tumorigenesis. Proc Am Assoc Cancer Res 26: 138.
Wolff GL, Roberts DW, & Galbraith DB (1986) Prenatal determination
of obesity, tumour susceptibility and coat colour pattern in viable
yellow (Avy/a) mice. J Hered 77: 151-158.
Woodard F & Hagan EC (1947) Toxicological studies on the isomars and
mixtures of isomers of benzenehexachloride. Fed Proc 6: 386.
Yamaguchi I, Matsumura F, & Kadous AA (1979) Inhibition of synpatic
ATPase by heptachlor epoxide in rat brain. Pestic Biochem Physiol
11: 285-293.
Yamato Y, Kiyonaga M, & Watanabe T (1983) Comparative
bioaccumulation and elimination of HCH-isomars in short-necked clam
(Venerupis japonica) and guppy (Poecilia reticulata). Bull
Environ Contam Toxicol 31: 352-359.
Yoshida T & Castro TF (1970) Degradation of gamma-BHC in rice soils.
Soil Sci Soc Am Proc 34: 440-442.
Yule WN, Chiba M, & Morley HV (1967) Fate of insecticide residues.
Decomposition of lindane in soil. J Agric Food Chem 15(6): 1000-
1004.
Zeilmaker MJ & Yamasaki H (1986) Inhibition of junctional
intercellular communication as a possible short-term test to detect
tumour promoting agents. Results with nine chemicals tested by dye-
transfer assay in Chinese hamster V79 cells. Cancer Res 46: 6180-
6186.
Zesch A, Nitzsche K, & Lange M (1982) Demonstration of the
percutaneous resorption of a lipophilic pesticide and its possible
storage in the human body. Arch Dermatol Res 273: 43-49.
RESUME ET EVALUATION; CONCLUSIONS; RECOMMANDATIONS
1. Résumé et évaluation
1.1 Propriétés générales
L'hexachlorocyclohexane technique (HCH) est composé de 65-70%
d'alpha-HCH, de 7-10% de beta-HCH, de 14-15% de gamma-HCH, et
d'environ 10% d'autres isomères et composés. Le lindane contient
> 99% de gamma-HCH. C'est un solide, avec une tension de vapeur
faible, peu soluble dans l'eau mais très soluble dans les solvants
organiques comme l'acétone, et dans les solvants aromatiques et
chlorés. Le coefficient de partage n-octanol/eau (Log Poe) est
de 3,2-3,7.
Le lindane peut être dosé à part des autres isomères du HCH
après extraction par partage liquide/liquide, chromatographie sur
colonne et chromatographie en phase gazeuse avec détection par
capture d'électrons. Extrêmement sensibles, ces méthodes analytiques
permettent d'identifier des résidus de lindane de l'ordre du
nanogramme par kilogramme ou par litre.
Depuis le début des années 50, le lindane est utilisé comme
insecticide à large spectre; le secteur agricole y a recours
notamment pour traiter les semences et les sols; on l'utilise pour
traiter les arbres, le bois de construction et les matériaux
entreposés; on l'applique sur le pelage des animaux pour éliminer
les ectoparasites; on en fait également usage dans le domaine de la
santé publique.
1.2 Transport, réparation et transformation dans l'environnement
Le lindane est fortement adsorbé aux sols contenant une grande
quantité de matière organique; en outre, il peut s'imprégner dans le
sol à la faveur d'une chute de pluie ou d'une irrigation
artificielle. Sous l'effet des températures élevées des régions
tropicales, il se dissipe en grande partie par volatilisation.
Le lindane subit une dégradation rapide (déchloration) sous
l'action des rayons ultra-violets, pour former des
pentachlorocyclohexènes (PCCHS) et des tetrachlorocyclohexènes
(TCCHs). Lorsque le lindane se dégrade en sol humide ou inondé ou en
plein champ, sa demi-vie peut aller de quelques jours à trois ans,
selon le type de sol, le climat, la profondeur à laquelle il se
trouve, etc. Dans les sols consacrés aux cultures que l'on trouve
habituellement en Europe, sa demi-vie est de 40 à 70 jours. Les sols
non-stériles permettent une biodégradation plus rapide du lindane
que les sols stériles. Les conditions anaérobies sont les plus
propices à sa métabolisation microbienne. Le lindane présent dans
l'eau se dégrade sous l'action des microorganismes contenus dans les
sédiments, pour former les mêmes produits.
Le lindane et les gamma-PCCHs sont fixés en quantité limitée
par les plantes où ils subissent une translocation, surtout lorsque
les sols contiennent une forte proportion de matières organiques. On
trouve les résidus essentiellement dans les racines des plantes; la
translocation ne s'effectue que peu - ou pas du tout - dans les
tiges, les feuilles ou les fruits. La bioconcentration est rapide
chez les microorganismes, les invertébrés, les poissons, les oiseaux
et l'homme, mais la biotransformation et l'élimination le sont
également lorsque l'exposition est interrompue. Les organismes
aquatiques le fixent en plus grande quantité à partir de l'eau qu'à
partir de la nourriture. Les facteurs de bioconcentration dans les
organismes aquatiques vont de 10 à 6000 en laboratoire, et de 10 à
2600 sur le terrain.
1.3 Concentration dans l'environnement et exposition humaine
On a trouvé du lindane en suspension dans l'air au-dessus des
océans à des niveaux de concentration de 0,039-0,68 ng/m3; son
niveau de concentration a atteint 1 ng/m3 dans l'air de certains
pays. Les concentrations dans les eaux de surface de nombreux pays
européens étaient la plupart du temps inférieures à 0,1 µg/litre.
Dans le cas du Rhin et de ses affluents, le niveau de concentration
variait entre 0,01 et 0,4 µg/litre de 1969 à 1974; après 1974, il
était inférieur à 0,1 µg/litre. Dans l'eau de mer, des
concentrations de 0,001-0,002 µg/litre ont été enregistrées. Les
concentrations de lindane dans le sol sont généralement faibles -
l'ordre de 0,001-0,01 mg/kg, sauf dans les zones de décharge.
On a découvert du gamma-HCH chez les poissons et les crustacés,
à des niveaux de concentration allant de 'indécelable' à 2,5 mg/kg
(calculés par rapport aux matières grasses) selon qu'ils vivent en
eau douce ou dans l'eau de mer et en fonction de leur teneur - faible
ou élevée - en matières grasses. Des valeurs d'environ 330 et 440
µg/kg (poids humide) ont été trouvées dans le tissu adipeux des ours
polaires respectivement en 1982 et 1984. La concentration de lindane
dans le foie des oiseaux prédateurs variait entre 0,01 et 0,1 mg/kg.
En 1972-73, on a mesuré dans des oeufs d'épervier, en République
fédérale d'Allemagne, des valeurs allant de 0,6 mg/kg à 11,1 mg/kg
(calculées par rapport aux matières grasses).
Les niveaux de concentration de lindane dans l'eau potable sont
généralement inférieurs à 0,001 µg/litre, et dans les pays
industrialisés, c'est dans la nourriture que se trouve 90% du
lindane absorbé par l'homme. Au cours des 25 dernières années, on a
analysé dans bon nombre de pays certains produits alimentaires à la
recherche de leur teneur en lindane: dans les céréales, les fruits,
les légumes, les légumes à gousse, les huiles végétales, les
concentrations allaient de 'indécelable' à 5 mg/kg de produit. En ce
qui concerne le lait, les matières grasses animales, la viande et
les oeufs, les concentrations allaient de 'indécelable' à 5,1 mg/kg
de produit (calculés par rapport aux matières grasses). On a
rarement trouvé des concentrations plus élevées. Les niveaux de
concentration dans le poisson étaient généralement très inférieurs à
0,05 mg/kg de produit (calculés par rapport aux matières grasses).
Des études de ration totale et de panier de la ménagère pour estimer
l'apport quotidien de lindane chez l'homme, ont révélé d'importantes
variations en fonction des époques: vers 1970, l'apport quotidien
était égal ou inférieur à 0,05 µg/kg de poids corporel; en 1980, par
contre, cet apport a diminué pour n'être plus qu'égal ou inférieur à
0,003 µg/kg. Aux USA, l'apport journalier de gamma-HCH entre 1976 et
1979 a diminué, passant de 0,005 à 0,001 µg/kg pour les tout-petits
et de 0,01 à 0,006 µg/kg pour les enfants.
Dans un certain nombre de pays, on a déterminé la concentration
en lindane dans des tissus d'individus appartenant à la population
générale. Aux Pays-Bas, la teneur du sang était de l'ordre de
< 0,1-0,2 µg/litre, mais des concentrations bien supérieures ont été
trouvées dans le sang d'individus vivant dans des pays utilisant du
HCH technique. Dans différents pays, on a mesuré des niveaux moyens
de concentrations dans le tissu adipeux de l'homme allant de < 0,0
1 à 0,2 mg/kg (calculés par rapport aux matières grasses). Dans le
lait humain, les concentrations de lindane sont généralement plutôt
faibles, avec des moyennes allant de moins de 0,001 à 0,1 mg/kg,
rapportées aux matières grasses; toutefois, on a constaté que ces
valeurs s'abaissaient à la longue.
Ainsi, le lindane est présent partout dans le monde; on le
retrouve dans l'air, l'eau, le sol, les sédiments, les organismes
aquatiques et terrestres, ainsi que dans la nourriture, encore que
les concentrations dans ces compartiments du milieu soient
généralement faibles et diminuent peu à peu. L'homme est exposé
quotidiennement, par les aliments qu'il consomme; c'est ainsi que
l'on peut trouver du lindane dans le sang, les tissus adipeux, et
dans le lait de femme; toutefois, les doses ingérées sont également
en diminution.
1.4 Cinétique et métabolisme
Chez les rats, le lindane est absorbé rapidement au niveau des
voies digestives et se répartit dans l'ensemble des organes et
tissus en quelques heures. C'est dans les tissus adipeux et la peau
que l'on trouve les niveaux de concentration les plus élevés;
diverses études ont fait état d'un rapport teneur des
graisses/teneur du sang égal à 150-200, d'un rapport foie/sang de
5,3-9,6, et d'un rapport cerveau/sang de 4-6,5. Un rapport
graisses/sang identique a été trouvé chez des rats ayant inhalé du
lindane. Ces rapports, qui varient en fonction du sexe, sont
supérieurs chez les femelles. Au niveau de la peau, la résorption
s'effectue très lentement et dans une faible proportion, ce qui
explique la faible toxicité du lindane après exposition cutanée.
Le lindane est métabolisé essentiellement dans le foie selon
quatre réactions enzymatiques: la déshydrogénation en gamma-HCH, la
déshydrochloration en gamma-PCCH, la déchloration en gamma-TCCH et
l'hydroxylation en hexachlorocyclohexanol. Les produits finals après
biotransformation sont des dérivés di-, tri-, tetra-, penta-, et
hexachlorés. Ces métabolites sont excrétés essentiellement dans les
urines sous forme libre ou conjugués à l'acide glucuronique, à
l'acide sulfurique ou à la N-acétylcystéine. Le processus
d'élimination est relativement rapide, avec des demi-vies de 3-4
jours chez le rat. Les bactéries et les champignons métabolisent le
lindane en TCCH et en PCCH. La vitesse de la transformation
métabolique dans les plantes est faible, la voie de dégradation
principale passant par le PCCH pour aboutir au tetrachlorophenol, à
des conjugués avec le bêta-glucose et à d'autres composés inconnus.
Rien ne prouve que le lindane s'isomérise en alpha-HCH.
1.5 Effets sur les êtres vivants dans leur milieu naturel
Le lindane n'est pas très toxique pour les bactéries, les
algues, les protozoaires: la dose sans effet est généralement de
1 mg/litre. Son action sur les champignons est variable, avec des
doses sans effet variant entre 1 et 30 mg/litre, selon l'espèce. Il
est modérément toxique pour les invertébrés et les poissons, les
valeurs CL(E)50 pour ces organismes étant de 20-90 µg/litre. Les
études à court et à long terme portant sur trois espèces de poisson
ont révélé que la dose sans effet est de 9 µg/litre; des
concentrations de 2,1-23,4 µg/litre n'ont aucune conséquence sur la
reproduction. Les valeurs de la CL50 pour les crustacés d'eau
douce et marins varient entre 1 et 1100 µg/litre. Chez Daphnia
magna, il y a réduction, fonction de la dose, du taux de
reproduction; la dose sans effet se situe entre 11 et 19 µg/litre.
Une dose de 1 mg/litre n'a eu aucun effet néfaste sur la
reproduction des mollusques.
La DL50 pour les abeilles domestiques est de 0,56 µg/abeille.
Les DL50 orales aiguës pour un certain nombre d'espère
d'oiseaux se situent entre 100 et 1000 mg/kg de poids corporel. Des
études à courte terme sur les oiseaux ont permis d'établir que des
doses de 4-10 mg/kg de nourriture n'ont aucun effet, pas même sur la
qualité de la coquille des oeufs. On a néanmoins constaté une
moindre ponte chez les canes exposées à des doses de lindane allant
jusqu'à 20 mg/kg de poids corporel.
Des chauves-souris sont mortes dans les 17 jours qui ont suivi
leur exposition à des copeaux sur lesquels on avait appliqué du
lindane à la dose indiquée; les copeaux en contenaient initialement
10-866 mg/m2. On ne possède aucune donnée concernant les effets
sur les individus et les écosystèmes.
1.6 Effets sur les animaux de laboratoire et effets in vitro
La toxicité orale aiguë du lindane est modérée: la DL50 pour
les souris et les rats est de l'ordre de 60-250 mg/kg de poids
corporel, selon le véhicule utilisé. La LD50 dermique pour les
rats est d'environ 900 mg/kg de poids corporel. La toxicité est
révélée par des signes d'excitation au niveau du système nerveux
central.
Le lindane ne provoque aucune irritation ou sensibilisation de
la peau; il est légèrement irritant pour les yeux.
Une étude de 90 jours sur des rats a permis d'établir à 10
mg/kg de nourriture (soit 0,5 mg/kg de poids corporel) la dose sans
effet. A 50 et 250 mg/kg de nourriture, il y avait augmentation du
poids du foie, des reins et de la thyroïde; à 250 mg/kg de
nourriture, on constatait une augmentation de l'activité enzymatique
du foie. Cette augmentation de l'activité enzymatique accélère la
dégradation du lindane et autres dérivés. Une autre étude de 90
jours sur des rats a montré qu'une dose de 4 mg/kg de nourriture
(soit 0,2 mg/kg de poids corporel) pouvait être définie comme dose
sans effet nocif; on a observé que des concentrations égales ou
supérieures à 20 mg/kg de nourriture pouvaient avoir un effet
toxique au niveau des reins et du foie. Une étude toxicologique à
court-terme sur les souris n'a pas permis de définir la dose sans
effet.
On a constaté aucun effet toxique après administration de
lindane à des chiens, pendant 63 semaines, 15 mg/kg de nourriture
(soit 0,6 mg/kg de poids corporel). Une étude toxicologique menée
pendant 2 ans sur des chiens et au cours de laquelle un grand nombre
de paramètres ont été mesurés, a permis d'établir que
l'administration de doses égales ou supérieures à 50 mg/kg de
nourriture (soit 2 mg/kg de poids corporel) ne provoquait aucune
anomalie apparente liée au traitement. Chez les animaux auxquels on
avait administré des doses de 100 mg/kg de nourriture, on constatait
toutefois une hausse des niveaux de phosphatage alcaline; avec des
doses de 200 mg/kg, on observait des anomalies du tracé électro-
encéphalographique, indiquant une excitation neuronale aspécifique.
Chez des rats ayant inhalé du lindane à raison de 0,02-4,54
mg/m3, 6 heures par jour pendant 3 mois, la dose la plus forte a
entraîné une élévation des valeurs du cytochrome P450 hépatique; la
dose sans effet nocif a été fixée à 0,6 mg/m3. Lors de deux études
au long cours menées sur des rats de nombreuses années auparavant,
on a expérimenté des doses de 10-1600 mg/kg de nourriture. L'une de
ces études fixe à 50 mg/kg de nourriture la dose sans effet nocif. A
100 mg/kg de nourriture, le foie augmente, une hypertrophie
hépatocellulaire apparaît, ainsi qu'une dégénérescence graisseuse et
une nécrose. L'autre étude établit à 25 mg/kg de nourriture (soit
1,25 mg/kg de poids corporel) la dose sans effet; en doublant cette
dose, on note une hypertrophie hépatocellulaire et une
dégénérescence graisseuse.
Des recherches ont été entreprises pour vérifier les effets que
le lindane, après avoir été administré à des souris, des rats, des
chiens et des porcs par voie orale, sous-cutanée et
intrapéritonéale, pouvait avoir sur tous les aspects de la
reproduction (chez les rats, sur trois générations), et évaluer sa
toxicité pour l'embryon et sa tératogénicité. Le lindane administré
par voie orale et parentérale n'a eu aucun effet tératogène (les
côtes surnuméraires étant considérées comme des variations). Des
doses de 10 mg/kg de poids corporel et plus administrées oralement-
par gavage-se sont avérées toxiques pour le foetus et/ou la mère; on
considère qu'une dose de 5 mg/kg de poids corporel est sans effet
nocif. L'étude effectuée sur trois générations de rats, a montré que
des doses de lindane allant jusqu'à 100 mg/kg de nourriture
n'avaient d'influence ni sur la reproduction, ni sur la maturation;
avec 50 mg/kg de nourriture, des modifications morphologiques inter-
venaient au niveau du foie parmi la progéniture de la troisième
génération, preuve d'une induction enzymatique. La dose sans effet
était dans ce cas de 25 mg/kg (soit 1,25 mg/kg de poids corporel).
Dans une étude de 22 jours effectuée sur des rats, on a
déterminé que la dose sans effet neurotoxique était de 2,5 mg/kg de
poids corporel.
Des études bien conçues ont été effectuées pour déterminer la
mutagénicité du lindane. Selon les résultats des recherches très
larges entreprises, le lindane ne peut en aucun cas provoquer des
mutations génétiques chez les bactéries ni dans des cellules
mammaliennes; il n'entraîne pas non plus, chez Drosophila
melanogaster, des mutations récessives liées au sexe. D'autres
expériences - in vitro et in vivo - ont montré qu'en outre le
lindane ne provoque ni anomalies chromosomiques, ni échange de
chromatides soeurs dans des cellules mammaliennes. La recherche de
lésions de l'ADN bactérien et de liaisons covalentes avec l'ADN dans
le foie de rats et de souris in vivo, après administration par
voie orale, a également donné des résultats négatifs. Les rares
études où des résultats positifs ont été obtenus péchaient soit par
une mauvaise conception d'ensemble, soit par le fait que la pureté
du composé étudié n'avait pas été précisée. Quoi qu'il en soit, on
peut dire que le lindane n'a globalement aucun pouvoir mutagène.
Des études de cancérogénécité ont été effectuées chez la souris
et le rat, avec des doses allant respectivement jusqu'à 600 mg/kg de
nourriture et 1600 mg/kg de nourriture. Aux doses égales ou
supérieures à 160 mg/kg de nourriture, on a observé des nodules
hyperplasiques et/ou des adénomes hépatocellulaires chez les souris;
lors de certaines études, les doses administrées ont dépassé le
maximum toléré. Deux expériences ont montré qu'aucune élévation de
l'incidence des tumeurs n'intervient lorsque l'on donne à des souris
et à des rats des doses allant respectivement jusqu'à 160 et 640
mg/kg de nourriture.
Les résultats d'études sur l'initiation-promotion de la
cancérogénicité, sur le mode d'action du lindane et sur sa
mutagénicité indiquent que la réponse tumorigène observée avec le
gamma-HCH chez la souris est sous la dépendance d'un mécanisme non
génétique.
1.7 Effets sur l'homme
Le lindane a été à l'origine de plusieurs cas d'intoxications
mortelles et non mortelles; il s'agissait soit d'accidents, soit
d'absorption délibérée (suicide), soit d'une simple négligence
(absence de précautions) ou d'utilisation impropre de produits
médicaux contenant du lindane. Les symptômes consistaient en
nausées, agitation, maux de tête, vomissements, tremblements,
ataxie, convulsions toniques-cloniques et/ou modifications du tracé
électroencéphalographique. Ces effets sont réversibles après
interruption de l'exposition ou traitement symptomatique.
Depuis 40 ans, l'utilisation du lindane est très répandue;
pourtant on cite peu de cas d'intoxications survenant dans le
contexte professionnel. Chez les individus qui travaillent à la
fabrication du lindane ou à son épandage, donc soumis à une
exposition prolongée, on a seulement constaté une augmentation de
l'activité des enzymes métabolisantes au niveau du foie. Rien ne
prouve l'existence d'une quelconque relation - évoquée dans
certaines publications - entre exposition au lindane et apparition
d'anomalies hématologiques. Quelques études de toxicologie aiguë ou
à courte terme chez l'homme indiquent qu'une dose d'environ 1,0
mg/kg de poids corporel ne provoque pas d'intoxication; en revanche,
à la dose de 15-17 mg/kg de poids corporel, apparaissent de graves
symptômes d'intoxication.
Appliqué sur la peau, le lindane est absorbé à hauteur
d'environ 10%; la proportion est plus élevée s'il y a lésions.
2. Conclusions
2.1 Population générale
Le lindane circule dans l'environnement et il est présent dans
les chaînes alimentaires; de ce fait, l'homme ne peut échapper à
l'exposition. Toutefois, l'apport alimentaire quotidien et
l'exposition totale de la population dans son ensemble diminuent peu
à peu, et, nettement inférieurs à la dose journalière admissible
(DJA) conseillés, ne constituent pas une menace sérieuse pour la
santé publique.
2.2 Groupes de population particulièrement exposés
La présence de lindane dans le lait maternel expose les enfants
nourris au sein, à des doses généralement inférieures à la DJA, donc
non toxiques. Les niveaux d'exposition existants - que l'on les
souhaiterait tout de même inférieur - n'interdisent pas
l'allaitement au sein.
En ce qui concerne l'utilisation thérapeutique du lindane pour
traiter la gale et les poux de corps, il convient de se conformer
strictement aux doses prescrites.
2.3 Exposition professionnelle
Manipuler du lindane ne présente aucun danger, à condition de
prendre toutes les précautions indiquées pour éviter le plus
possible l'exposition.
2.4 Effets sur l'environnement
Les chauves-souris, qui s'accrochent au bois traité avec du
lindane aux doses indiquées, en subissent les effets toxiques.
Exception faite des résultats d'étude concernant les déversements
accidentels dans le milieu aquatique, rien ne permet d'affirmer que
la présence de lindane dans l'environnement constitue un danger
sérieux pour d'autres êtres vivants.
3. Recommandations
1. Afin de réduire au minimum la pollution de l'environnement par
d'autres isomères de HCH, il convient d'utiliser le lindane
(> 99% de gamma-HCH) au lieu du HCH technique.
2. Afin d'éviter la pollution de l'environnement, il faut adopter
des solutions adéquates pour se débarrasser des sous-produits
et des effluents provenant des usines de fabrication de
lindane.
3. Il faut prendre garde à ce que les déchets de lindane ne
polluent ni les sols, ni les eaux.
4. Il faut donner à ceux qui manipulent du lindane les indications
nécessaires sur les méthodes d'application et les précautions
d'utilisation.
5. Il faut effectuer des études de cancérogénécité au long cours,
qui soient conformes aux nonnes actuelles.
6. Il faut poursuivre la surveillance de la dose de lindane
quotidiennement absorbée par la population générale.
RESUMEN Y EVALUACIONES; CONCLUSIONES; RECOMENDACIONES
Resumen y evaluación
1.1 Propiedades generales
El hexaclorociclohexano (HCH) de calidad técnica está formado
por un 65-70% de alfa-HCH, un 7-10% de beta-HCH, un 14-15% de gamma-
HCH y aproximadamente un 10% de otros isómeros y compuestos. El
lindano contiene más del 99% de gamma-HCH. Es un compuesto sólido,
con baja presión de vapor y poco soluble en agua, pero muy soluble
en disolventes orgánicos, como la acetona, y en disolventes
aromáticos y clorados. El coeficiente de reparto n-octanol/agua
(log Poa) es de 3,2-3,7.
El lindano puede determinarse por separado de los demás
isómeros del HCH tras su extracción por reparto líquido/líquido,
cromatografía en columna y detección por cromatografía de gases con
captura de electrones. Como estos métodos analíticos son sumamente
sensibles, es posible identificar residuos de lindano del orden de
nanogramos por kilogramo o por litro.
El lindano lleva utilizándose desde el comienzo los años 50
como insecticida de amplio espectro con fines agrícolas y de otro
tipo, de los que cabe mencionar el tratamiento de semillas y de
suelos, las aplicaciones en árboles, madera y materiales
almacenados, el tratamiento de animales contra los ectoparásitos y
en la salud pública.
1.2 Transporte, distribución y transformación en el medio ambiente
En los suelos con un alto contenido de materia orgánica se
observa una intensa adsorción del lindano; además, puede penetrar en
el suelo con el agua de la lluvia o del riego artificial. La
volatilización parece ser una importante vía de dispersión en las
elevadas temperaturas de las regiones tropicales.
El lindano experimenta una rápida degradación (descloración)
por acción de los rayos ultravioleta, formando
pentaclorociclohexenos (PCCH) y tetraclorociclohexenos (TCCH).
Cuando el lindano se descompone en el medio ambiente en condiciones
de humedad o inmersión y en condiciones de campo, su semivida varía
de unos días a tres años, en función del tipo de suelo, del clima,
de la profundidad a la que se haya aplicado y de otros factores. En
los suelos agrícolas normales en Europa su semivida es de 40 a 70
días. La biodegradación del lindano es mucho más rápida en suelos no
esterilizados que en los esterilizados. Las condiciones anaerobias
son las más favorables para su metabolización microbiana. El lindano
presente en el agua es degradado principalmente por microorganismos
de los sedimentos para formar los mismos productos de degradación.
Las plantas absorben y translocan en su interior cantidades
limitadas de lindano y de gamma-PCCH, especialmente en suelos con un
elevado contenido de materia orgánica. Los residuos se depositan
sobre todo en las raíces de las plantas, y son pocos o ninguno los
que se desplazan a las ramas, las hojas o los frutos. En los
microorganismos, los invertebrados, los peces, las aves y el hombre
tiene lugar una bioconcentración rápida, pero cuando se interrumpe
la exposición se biotransforman y eliminan en un tiempo
relativamente breve. En los organismos acuáticos es más importante
su absorción a partir del agua que de los alimentos. Los factores de
bioconcentración de estos organismos en condiciones de laboratorio
variaron desde un valor aproximado de 10 hasta 6000; en condiciones
de campo oscilaron entre 10 y 2600.
1.3 Niveles medioambientales y exposición humana
En el aire oceánico se han encontrado concentraciones de
lindano de 0,039-0,68 ng/m3, y en el aire de algunos países se han
medido cantidades de hasta 11 ng/m3. Las concentraciones estimadas
en aguas de superficie de varios países europeos fueron en general
inferiores a 0,1 µ/litro. Su concentración en el río Rin y sus
afluentes en el período 1969-74 osciló entre 0,01 y 0,4 µg/litro;
después de 1974 se mantuvo por debajo de 0,1 µg/litro. En el agua
marina se han detectado niveles de 0,001-0,02 µg/litro. Las
concentraciones de lindano en el suelo son por lo general bajas, del
orden de 0,001-0,01 mg/kg, excepto en zonas de vertido de basuras.
En pescados y mariscos se han detectado concentraciones de
gamma-HCH que oscilan entre valores no detectables y 2,5 mg/kg
(valores referidos a las grasas), dependiendo de que vivan en agua
dulce o agua marina y de que su contenido en grasa sea alto o bajo.
En el tejido adiposo de los osos polares se encontraron en 1982 y
1984 niveles aproximados de 330 y 440 µg/kg (peso húmedo)
respectivamente. La concentración de lindano en el hígado de aves
predadoras oscilaba entre 0,01 y 0,1 mg/kg. Los huevos de gavilán
recogidos en 1972-73 en la República Federal de Alemania contenían
entre 0,6 y 11,1 mg/kg (cálculo referido a las grasas).
Las concentraciones de lindano en el agua potable generalmente
son inferiores a 0,001 µg/litro; en los países industrializados más
del 90% de la ingestión humana de lindano procede de los alimentos.
En los últimos 25 años se ha analizado el contenido de lindano de
determinados productos alimenticios de un gran número de países. Las
concentraciones halladas en cereales, frutas, hortalizas, legumbres
y aceites vegetales variaron entre valores no detectables y 5 mg/kg
de producto, y en la leche, las grasas, la carne y los huevos, entre
valores no detectables y 5,1 mg/kg (referido a las grasas). Sólo en
unos pocos casos se detectaron concentraciones más altas. Sus
niveles en el pescado eran, en general, muy inferiores a 0,05 mg/kg
de producto (referido a las grasas).
En estudios sobre dieta total y cesta de la compra para estimar
la ingestión humana diaria de lindano, se observó una clara
diferencia con el paso del tiempo: la ingestión en el período de
alrededor de 1970 llegaba a 0,05 µg/kg de peso corporal al día,
mientras que en 1980 esta cifra había descendido a 0,003 µg/kg de
peso corporal al día o menos. En los Estados Unidos, la ingestión de
gamma-HCH entre 1976 y 1979 disminuyó de 0,005 a 0,001 µg/kg de peso
corporal al día en los lactantes y de 0,01 a 0,006 µg/kg de peso
corporal al día en los niños de corta edad.
En algunos países se ha determinado el contenido de lindano en
los tejidos corporales de la población general. En los Países Bajos,
el contenido en la sangre era del orden de < 0,1-0, µg/litro, pero
se hallaron concentraciones mucho más altas en varios países en los
que se utilizaba HCH de calidad técnica. Las concentraciones medias
en el tejido adiposo humano en distintos países varió entre < 0,01
y 0,2 mg/kg (referido a las grasas). La concentración de lindano en
la leche humana suele ser bastante baja, con unos niveles medios que
van desde < 0,001 hasta 0,1 mg/kilo (referido a las grasas); sin
embargo, se ha producido una disminución manifiesta con el tiempo.
Así pues, el lindano se halla distribuido por todo el mundo y
se puede detectar en el aire, el agua, el suelo, los sedimentos, los
organismos acuáticos y terrestres y los alimentos, aunque las
concentraciones en estos distintos compartimentos ambientales son en
general bajas y están decreciendo progresivamente. El hombre está
expuesto a diario por conducto de los alimentos, habiéndose
detectado lindano en los tejidos sanguíneo y adiposo y en la leche
materna; sin embargo, los niveles de ingestión también están
disminuyendo.
1.4 Cinética y metabolismo
En las ratas, el lindano se absorbe rápidamente de¡ tracto
gastrointestinal y en unas horas se distribuye por todos los órganos
y tejidos. Las concentraciones más elevadas se dan en el tejido
adiposo y en la piel; en varios estudios, el cociente grasa:sangre
era de alrededor de 150-200, el cociente hígado:sangre, 5,3-9,6 y el
cociente cerebro:sangre, 4-6,5. El mismo cociente grasa:sangre se
encontró en ratas expuestas por inhalación. Estos cocientes varían
en función del sexo, siendo más elevados en las hembras. La
absorción por la piel tras la aplicación cutánea de lindano es lenta
y muy limitada; esto puede explicar la baja toxicidad del lindano
después de la exposición cutánea.
El lindano se metaboliza sobre todo en el hígado mediante
cuatro reacciones enzimáticas: deshidrogenación a gamma-HCH,
deshidrocloración a gamma-PCCH, descloración a gamma-TCCH e
hidroxilación a hexaclorociclohexanol. Los productos finales de la
biotransformación son compuestos di-, tri-, tetra-, penta- y
hexaclorados. Estos metabolitos se excretan fundamentalmente por la
orina, en forma libre o conjugada con ácido glucurónico, ácido
sulfúrico o N-acetilcisteína. La eliminación es relativamente
rápida, con una semivida en ratas de 3 a 4 días. Las bacterias y los
hongos metabolizan el lindano a TCCH y PCCH. La velocidad de
transformación metabólica en las plantas es baja, y la vía de
degradación más importante es a través del PCCH a tri- y
tetraclorofenol y productos conjugados con beta-glucosa y otros
compuestos desconocidos. No existen pruebas de la isomerización del
lindano a alfa HCH.
1.5 Efectos en los seres vivos del medio ambiente
El lindano no es muy tóxico para las bacterias, las algas ni
los protozoos: el nivel carente de efecto fue en general de 1
mg/litro. Su acción sobre los hongos es variable; los niveles sin
observación de efectos fueron de 1 a 30 mg/litro, según las
especies. Es moderadamente tóxico para los invertebrados y los
peces, siendo los valores de la C(E)L50 para esos organismos de
20-9 µg/litro. En estudios de corta y larga duración con tres
especies de peces, el nivel sin observación de efectos fue de 9
µg/litro; no se observaron efectos en la reproducción con niveles de
2,1-23,4 µg/litro. Los valores de la CL50 para crustáceos
dulceacuícolas y marinos variaron entre 1 y 1100 µg/litro. La
inhibición de la reproducción de Daphnia magna dependía de la
dosis; el nivel sin observación de efectos fue del orden de 11-19
µg/litro. No se observaron efectos adversos en la reproducción de
moluscos con dosis de 1 mg/litro.
La DL50 para la abeja de la miel fue de 0,56 µg/abeja.
Los valores de la DL50 aguda por vía oral para varias
especies de aves fueron de 100 a 1000 mg/kg de peso corporal. En
estudios de corta duración con aves, las dosis de 4-10 mg/kg en la
dieta no tuvieron efecto, ni siquiera sobre la calidad de la cáscara
de los huevos. Sin embargo, en patas ponedoras tratadas con dosis de
lindano de hasta 20 mg/kg de peso corporal disminuyó la producción
de huevos.
Todos los murciélagos expuestos a virutas de madera con un
contenido inicial de lindano de 10-866 mg/M2, resultado de la
aplicación de la dosis recomendada, murieron en un plazo de 17 días.
No se obtuvieron datos acerca de los efectos en poblaciones y
ecosistemas.
1.6 Efectos en los animales de experimentación e in vitro
La toxicidad aguda por via oral del lindano es moderada: la
DL50 para ratones y ratas oscila entre 60 y 250 mg/kg de peso
corporal, en función del vehículo utilizado. La DL50 por vía
cutánea en ratas es de aproximadamente 900 mg/kg de peso corporal.
La toxicidad se manifestó en forma de estimulación del sistema
nervioso central.
El lindano no irrita ni sensibiliza la piel; es ligeramente
irritante para los ojos.
En un estudio de 90 días en ratas, la concentración máxima sin
efecto fue de 10 mg/kg alimento (equivalente a 0,5 mg/kg de peso
corporal). Con niveles de 50 y 250 mg/kg de alimento aumentaron los
pesos del hígado, los riñones y el tiroides; con 250 mg/kg de
alimento, se observó un aumento en la actividad enzimática del
hígado. Este aumento acelera la degradación del lindano y de otros
compuestos. En otro estudio de 90 días en ratas, se consideró que el
nivel máximo sin efectos adversos era de 4 mg/kg de alimento
(equivalente a 0,2 mg/kg de peso corporal); se observó toxicidad
renal y hepática a concentraciones de 20 mg/kg y superiores. Un
estudio de toxicidad de corta duración en ratones se consideró
insuficiente para establecer la concentración sin efectos.
La administración de lindano a perros en dosis de 15 mg/kg de
alimento (equivalentes a 0,6 mg/kg de peso corporal) durante 63
semanas no tuvo efectos tóxicos. En un estudio de dos años de
duración sobre la toxicidad de este compuesto en perros, en el que
se midió un gran número de parámetros, no se observaron anomalías
relacionadas con el tratamiento con dosis de 50 mg/kg de alimento
(equivalentes a 2 mg/kg de peso corporal) e inferiores. Sin embargo,
en el grupo que recibió 100 mg/kg de alimento aumentó el nivel de
fosfatasa alcalina; y con 200 mg/kg de alimento aparecieron
anomalías electroencefalográficas indicativas de irritación neurona]
inespecífico.
En ratas expuestas por vía respiratoria a concentraciones de
lindano de 0,02-4,54 mg/m3, 6 horas al día durante 3 meses, la
dosis más alta indujo un incremento de los valores del citocromo
P450 hepático; el nivel sin observación de efectos adversos fue de
0,6 mg/m33. En dos estudios de larga duración en ratas, realizados
hace muchos años, se ensayaron dosis de 10-1600 mg/kg de alimentos.
En uno de estos estudios se determinó un nivel sin observación de
efectos adversos de 50 mg/kg de alimento (equivalente a 2,5 mg/kg de
peso corporal). Con 100 mg/kg de alimento se producía un aumento del
peso del hígado, hipertrofia hepatocelular, degeneración grasas y
necrosis. En el otro estudio, la dosis de 25 mg/kg de alimento
(equivalente a 1,25 mg/kg de peso corporal) no tenía efectos, pero
con 50 mg/kg de alimento se observaron signos de hipertrofia
hepatocelular y degeneración grasas.
Se han investigado los efectos del lindano en todos los
aspectos de la reproducción (en tres generaciones de ratas), y su
embriotoxicidad y teratogenia tras la administración oral,
subcutánea e intraperitoneal en ratones, ratas, perros y cerdos. No
se observaron efectos teratogénicos tras la administración oral o
parenteral (las costillas supernumerarias se consideraron
variaciones). Se pusieron de manifiesto fetoxicidad y/o efectos
tóxicos matemos con dosis de 10 mg/kg de peso corporal y superiores
administradas mediante sonda oral; se considera que el nivel sin
efectos adversos es de 5 mg/kg de peso corporal. En el estudio de
tres generaciones de ratas con dosis de hasta 100 mg/kg de alimentos
el lindano no ejerció efecto alguno en la reproducción ni la
maduración, pero con 50 mg/kg de alimento se produjeron cambios
morfológicos en el hígado, que demostraban la inducción enzimática
registrada en la descendencia de la tercera generación. El nivel sin
observación de efectos en este ensayo fue de 25 mg/kg de alimento
(equivalente a 1,25 mg/kg peso corporal).
En un estudio de 22 días en ratas se observó que la dosis sin
efecto neurotóxico era de 2,5 mg/kg de peso corporal.
Se han hecho estudios suficientes sobre la mutagenicidad del
lindano. En las amplias investigaciones realizadas sobre su
capacidad para inducir mutaciones génicas en bacterias y células de
mamiferos y para provocar mutaciones letales recesivas ligadas al
sexo en Drosophila melanogaster, se obtuvieron siempre resultados
negativos. El lindano también dio resultados negativos en los
ensayos in vitro e in vivo realizados con células de mamíferos
sobre lesiones cromosómicas e intercambio de cromátidas hermanas.
Tambien fueron negativos los resultados de los ensayos para
determinar las lesiones en el ADN de bacterias y los de las pruebas
in vivo para observar la formación de enlaces covalentes con el
ADN de hepatocitos de ratones y ratas tras su administración oral.
En los escasos ensayos en los que se obtuvieron resultados
positivos, el sistema de estudio no era adecuado o no se informó
sobre la pureza del compuesto ensayado. Sin embargo, en conjunto, el
lindano no parece tener potencial mutagénico.
Se han llevado a cabo estudios en ratones y ratas para
determinar el potencial carcinogénico del lindano con dosis de hasta
600 mg/kg de alimento en ratones y de hasta 1600 mg/kg de alimento
en ratas. En ratones que recibieron dosis de 160 mg/kg de alimento o
superiores se observaron nódulos hiperplásicos y/o adenomas
hepatocelulares; en algunos estudios, las dosis utilizadas superaban
la máxima tolerada. En dos estudios en ratones con dosis de hasta
160 mg/kg de alimentos como máximo y uno en ratas con 640 mg/kg de
alimentos no se vio ningún aumento en la incidencia de tumores.
Los resultados de los estudios sobre la iniciación y el
estímulo de la carcinogenicidad, sobre el mecanismo de acción y
sobre la mutagenicidad ponen de manifiesto que en la respuesta
tumorigénica observada con el gamma-HCH en ratones interviene un
mecanismo no genético.
1.7 Efectos en el ser humano
Se ha informado de varios casos de envenenamiento mortal y de
enfermedad no mortal por lindano, producidos de manera accidental,
intencionada (suicidio) o por una grave negligencia en las
precauciones de seguridad o la utilización inadecuada de productos
médicos con lindano. Los síntomas son náuseas, agitación, dolor de
cabeza, vómitos, temblor, ataxia, convulsiones tónico-clónicas y/o
cambios en el trazado electroencefalográfico. Estos efectos eran
reversibles tras la interrupción de la exposición o el tratamiento
sintomático.
A pesar de su uso generalizado durante 40 años, se ha informado
de muy pocos casos de envenenamiento en el trabajo. En los
trabajadores expuestos durante largos períodos, en la fabricación o
la aplicación del lindano, el único síntoma observado fue una mayor
actividad de las enzimas hepáticas metabolizadoras de fármacos. No
hay pruebas de la relación, sugerida en algunas publicaciones, entre
la exposición al lindano y la aparición de anomalías hematológicas.
Algunos estudios de toxicidad aguda y de corta duración en la
especie humana indican que una dosis aproximada de 1,0 mg/kg de peso
corporal no produce envenenamiento; sin embargo, con una dosis de
15-17 mg/kg de peso corporal se observaron síntomas de intoxicación
grave.
Se absorbe alrededor del 10% de la dosis de aplicación cutánea,
aunque a través de la piel lesionada pasa mayor cantidad.
2. Conclusiones
2.1 Población general
El lindano circula en el medio ambiente y está presente en las
cadenas troficas, de manera que la especie humana seguirá estando
expuesta. Sin embargo, la ingestión diaria y la exposición total de
la población general están disminuyendo gradualmente; se encuentran
claramente por debajo de la ingestión diaria admisible y no
constituyen un problema para la salud pública.
2.2 Subpoblaciones especialmente expuestas
La presencia de lindano en la leche materna determina la
exposición de los lactantes a niveles que generalmente son
inferiores a la ingesta diaria admisible y que, por consiguiente, no
son un problema para la salud. Aunque sería preferible que los
niveles de exposición fueran inferiores, los actuales no representan
un factor limitante de la práctica de la lactancia natural.
Se deben seguir rigurosamente las prescripciones en relación
con el uso terapéutico de¡ lindano contra la sarna y los piojos.
2.3 Exposición profesional
El lindano se puede manejar sin riesgo siempre que se observen
las precauciones recomendadas para reducir al mínimo la exposición.
2.4 Efectos en el medio ambiente
El lindano es tóxico para los murciélagos que reposan en
estrecho contacto con madera tratada de acuerdo con las
recomendaciones para la aplicación. Si se exceptúan los resultados
obtenidos en los estudios sobre derrames en el medio acuático, no
hay pruebas que indiquen que la presencia de lindano en el medio
ambiente plantee un riesgo importante para las poblaciones de otros
organismos.
3. Recomendaciones
1. A fin de reducir al mínimo la contaminación del medio ambiente
por otros isómeros del HCH, se debe utilizar lindano (> 99% de
gamma-HCH) en lugar de HCH de calidad técnica.
2. Con objeto de evitar la contaminación del medio ambiente, los
subproductos y efluentes de la fabricación del lindano se deben
eliminar de manera adecuada.
3. En la eliminación de lindano, hay que tomar precauciones para
evitar la contaminación de las aguas naturales y del suelo.
4. Como en el caso de otros plaguicidas, las personas encargadas
del manejo del lindano deben recibir instrucciones adecuadas
acerca de la manera de aplicarlo.
5. Se deben realizar ensayos de carcinogenicidad de larga duración
diseñados con arreglo a las normas actuales.
6. Se debe seguir vigilando la ingestión diaria de lindano por
parte de la población general.