Nephrotoxicity associated with exposure to chemicals, principles and methods for the assessment of (EHC 119, 1991)

INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 119
PRINCIPLES AMD METHODS FOR THE ASSESSMENT OF NEPHROTOXICITY
ASSOCIATED WITH EXPOSURE TO CHEMICALS
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Neither the Commission of the European Communities nor any person
acting on behalf of the Commission is responsible for the use which
might be made of the information contained in this report.
(EUR 13222 EN)
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization, and on
behalf of the Commission of the
European Communities
World Health Orgnization
Geneva, 1991
The International Programme on Chemical Safety (IPCS) is a joint
venture of the United Nations Environment Programme, the International
Labour Organisation, and the World Health Organization. The main
objective of the IPCS is to carry out and disseminate evaluations of
the effects of chemicals on human health and the quality of the
environment. Supporting activities include the development of
epidemiological, experimental laboratory, and risk-assessment methods
that could produce internationally comparable results, and the
development of manpower in the field of toxicology. Other activities
carried out by the IPCS include the development of know-how for coping
with chemical accidents, coordination of laboratory testing and
epidemiological studies, and promotion of research on the mechanisms
of the biological action of chemicals.
WHO Library Cataloguing in Publication Data
Principles and methods for the assessment of nephrotoxicity
associated with exposure to chemicals.
(Environmental health criteria: 119) (EUR ; 13222)
1. Kidney diseases - chemically induced
2. Kidney neoplasms - chemically induced
3. Kidney - drug effects I. Series II. Series EUR; 13222
ISBN 92 4 157119 5 (NLM Classification WJ 300)
ISSN 0250-863X
(c) World Health Organization 1991
(c) ECSC-EEC-EAEC, Brussels-Luxembourg, 1991
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. For rights of reproduction or
translation of WHO publications, in part or in toto, application
should be made to the Office of Publications, World Health
Organization, Geneva, Switzerland. The World Health Organization
welcomes such applications.
The designations employed and the presentation of the material in
this publication do not imply the impression of any opinion whatsoever
on the part of the Secretariat of the World Health Organization
concerning the legal status of every country, territory, city, or area
or of its authorities, or concerning the delimitation of its frontiers
or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar nature
that are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
PRINCIPLES AND METHODS FOR THE ASSESSMENT OF NEPHROTOXICITY
ASSOCIATED WITH EXPOSURE TO CHEMICALS
1. SCOPE OF THE HEALTH SIGNIFICANCE OF NEPHROTOXICITY
2. NEPHROTOXICITY
2.1. Target selectivity
2.2. The dynamics of renal injury
2.3. Classification of renal disease
2.4. The epidemiology of nephrotoxicity
2.5. Risk factors for toxic nephropathies
2.5.1. Factors related to renal function
2.5.2. Clinical risk factors
2.5.3. Extrapolation of animal data to man
2.5.4. Risk assessment from nephrotoxicity
studies in animals
2.5.5. Special risk groups in humans
2.5.6. Multichemical exposure
2.5.7. Renal functional reserve
2.5.8. The effects of chemicals on kidneys
with pre-existing renal lesions
2.5.8.1 Nephrotoxicity in the presence
of renal and extrarenal disease
3. KIDNEY STRUCTURE AND FUNCTION
3.1. Renal anatomy
3.1.1. Histology
3.1.2. Enzyme histochemistry and quantification
3.1.3. Immunohistochemistry
3.2. The renal blood supply
3.2.1. Renal haemodynamics
3.3. The nephron
3.3.1. Cellular heterogeneity and cell-cell
interaction
3.3.2. The glomerulus
3.3.3. The proximal tubule
3.3.4. The medulla 58
3.3.4.1 The loops of Henle
3.3.4.2 Collecting ducts
3.3.4.3 The distal tubule
3.3.4.4 The countercurrent multiplier
system and urine concentration
3.3.4.5 The interstitial cells
3.4. Species, strain, and sex differences in renal
structure and function
3.5. Renal biochemistry
3.5.1. Biochemistry and metabolism in the cortex
3.5.2. Biochemistry and metabolism in the medulla
3.5.2.1 The biochemistry of renal
prostaglandins (PG)
3.5.2.2 Lipid metabolism
3.5.2.3 Carbohydrate metabolism in the
medulla
3.5.2.4 Medullary glycosaminoglycan (GAG)
3.6. The metabolism of xenobiotic molecules in the kidney
3.6.1. Oxidases
3.6.1.1 Cytochrome P-450-dependent mixed-
function oxidases (monooxygenases)
3.6.1.2 Prostaglandin peroxidase-
mediated metabolic activation
3.6.2. Conjugation
3.6.2.1 Glucuronide conjugation
3.6.2.2 Sulfate conjugation
3.6.2.3 Glutathione conjugation
3.6.2.4 Mercapturic acid synthesis
3.6.2.5 Amino acid conjugation
3.6.3. Other enzymes involved in xenobiotic
metabolism
4. THE MECHANISTIC BASIS OF CHEMICALLY INDUCED RENAL INJURY
4.1. Immunologically induced glomerular disease
4.2. Direct glomerular toxicity
4.3. Tubulointerstitial disease
4.3.1. Acute interstitial nephritis
4.3.2. Acute tubular toxicity
4.3.3. Chronic interstitial nephritis
4.4. Mechanisms of cellular toxicity
4.5. Factors that modify cellular injury by toxins
4.5.1. Cellular transport and accumulation
4.5.2. Metabolic degradation
4.5.3. Intracellular protein binding
4.5.4. Membrane reactions and pinocytosis
5. THERAPEUTIC AGENTS AND CHEMICALS THAT HAVE THE POTENTIAL TO
CAUSE NEPHROTOXICITY
5.1. Therapeutic agents
5.1.1. Analgesics and non-steroidal
anti-inflammatory drugs (NSAIDs)
5.1.2. Paracetamol and para-aminophenol
5.1.3. Antibiotics
5.1.3.1 Aminoglycosides
5.1.3.2 Cephalosporins
5.1.3.3 Amphotericin B
5.1.3.4 Tetracyclines
5.1.4. Penicillamine
5.1.5. Lithium
5.1.6. Urographic contrast media (UCM)
5.1.7. Anticancer drugs
5.1.7.1 Cisplatin
5.1.7.2 Adriamycin
5.1.8. Immunosuppressive agents
5.1.8.1 Cyclosporin A
5.1.9. Heroin
5.1.10. Puromycin aminonucleoside
5.2. Chemicals
5.2.1. Ethylene glycol
5.2.2. Organic chemicals and solvents
5.2.2.1 Volatile hydrocarbons
5.2.2.2 Chloroform
5.2.2.3 Halogenated alkenes
5.2.2.4 Hydrocarbon-induced
nephrotoxicity
5.2.2.5 Bipyridyl herbicides
5.3. Mycotoxins
5.4. Silicon
5.5. Metals
5.5.1. Lead
5.5.2. Cadmium
5.5.3. Mercury
5.5.4. Gold
5.5.5. Bismuth
5.5.6. Uranium
5.5.7. Chromium
5.5.8. Arsenic
5.5.9. Germanium
6. RENAL CANCER
6.1. Renal tumour classification
6.2. Renal adenocarcinoma
6.3. Upper urothelial carcinoma (transitional
cell carcinoma)
6.4. Experimentally induced renal adenomas and
adenocarcinomas
6.4.1. Background incidence of spontaneous
tumours in experimental animals
6.4.2. Inorganic compounds
6.4.3. Organic molecules
6.4.3.1 Nitrosamines and related
compounds
6.4.3.2 Morphological changes
6.4.3.3 Biochemical changes in cells
6.4.3.4 The mechanistic basis of renal
carcinoma
6.5. Experimentally induced upper urothelial
carcinomas (transitional cell carcinomas)
7. ASSESSMENT OF NEPHROTOXICITY
7.1. In vitro studies
7.1.1. Choice of chemical concentrations
for in vitro studies
7.1.1.1 Proximate and ultimate
nephrotoxicants in vitro
7.1.2. In vitro investigations of
nephrotoxicity
7.1.2.1 Perfusion and micropuncture
7.1.2.2 Renal cortical slice
7.1.2.3 Isolated nephron segments
7.1.2.4 Primary cell cultures
7.1.2.5 Established renal cell lines
7.1.2.6 Subcellular fractions
7.2. In vivo experimental studies
7.2.1. Methods for assessing chemically reactive
nephrotoxic metabolites in animals
7.2.2. Evaluation of glomerular function
7.2.3. Evaluation of tubular functions
7.2.4. Proteinuria
7.2.4.1 Total proteinuria and
electrophoretic pattern
7.2.4.2 Urinary excretion of single
plasma proteins
7.2.4.3 Enzymuria
7.2.4.4 Immunoreactive tissue
constituents
7.2.4.5 Urinary excretion of
prostaglandins
7.2.5. Clinical context
7.2.6. Radiological techniques
7.2.7. Other non-invasive renal assessment
8. DETECTION OF NEPHROTOXICITY IN HUMANS
8.1. Markers of nephrotoxicity
8.1.1. General requirements
8.1.2. Diagnostic value
8.1.3. Prognostic value
8.2. Screening for nephrotoxicity in humans
8.2.1. Glomerular filtration
8.2.2. Tests designed to assess selective
dysfunction
8.2.3. Tests designed to assess tissue damage
8.2.3.1 Enzymuria
8.2.3.2 Immunoreactive tissue
constituents
8.3. Clinical investigations
8.3.1. Invasive techniques
8.3.1.1 Biopsies from humans
8.3.1.2 Autopsy in humans
8.3.2. Tests designed to assess glomerular
filtration and renal blood flow
8.3.3. Proteinuria
8.3.4. Tests designed to assess selective
damage
9. SUMMARY AND CONCLUSIONS
10. RECOMMENDATIONS
REFERENCES
RESUME ET CONCLUSIONS
RECOMMANDATIONS
RESUMEN Y CONCLUSIONES
RECOMENDACIONES
WHO/CEC TASK GROUP ON PRINCIPLES AND METHODS FOR THE ASSESSMENT OF
NEPHROTOXICITY ASSOCIATED WITH EXPOSURE TO CHEMICALS
Members
Professor E.A. Bababunmi, Biomembrane Research Laboratories,
Department of Biochemistry, University of Ibadan, Ibadan, Nigeria
(Vice-Chairman)
Dr P. Bach, Nephrotoxicity Research Group, Robens Institute of Health
and Safety, University of Surrey, Guildford, Surrey, United Kingdom
Professor G. Baverel, Department of Pharmacology, Alexis Carrel
Faculty of Medicine, Lyon, France
Professor W.O. Berndt, University of Nebraska Medical Centre, Omaha,
Nebraska, USA (Chairman)
Dr G. Duggin, Toxicology Unit, Royal Prince Alfred Hospital,
Camperdown, New South Wales, Australia
Dr H. Endou, Department of Pharmacology, Faculty of Medicine,
University of Tokyo, Bunkyo-ku, Tokyo, Japan
Professor R. Goyer, Department of Pharmacology, University of Western
Ontario, Health Science Centre, London, Ontario, Canada
Dr M. Robbins, Tissue Radiobiology Research Unit, Churchill Hospital,
Headington, Oxford, United Kingdom (Rapporteur)
Observer
Dr C. Cojocel, European Chemical Industries Ecology and Toxicology
Centre, Brussels, Belgium
Secretariat
Dr J.C. Berger, Health and Safety Directorate, Commission of the
European Communities (CEC), Luxembourg
Dr E. Smith, International Programme on Chemical Safety, Division of
Environmental Health, World Health Organization, Geneva,
Switzerland
Consultants representing the CEC
Dr A. Bernard, Unit of Industrial Toxicology and Occupational
Medicine, Catholic University of Louvain, Brussels, Belgiuma
Secretariat (contd.)
Dr P. Druet, National Institute of Health and Medical Research
(INSERM), Broussais Hospital, Paris, Francea
Professor A. Mutti, Institute of Clinical Medicine and Nephrology,
University of Parma, Parma, Italya
a Attended 6 December 1989 only.
NOTE TO READERS OF THE CRITERIA DOCUMENTS
Every effort has been made to present information in the criteria
documents as accurately as possible without unduly delaying their
publication. In the interest of all users of the environmental health
criteria documents, readers are kindly requested to communicate any
errors that may have occurred to the Manager of the International
Programme on Chemical Safety, World Health Organization, Geneva,
Switzerland, in order that they may be included in corrigenda, which
will appear in subsequent volumes.
* * *
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Palais des
Nations, 1211 Geneva 10, Switzerland (Telephone No. 7988400 or
7985850).
PREFACE
The preparation of this monograph was undertaken jointly by the
International Programme on Chemical Safety (UNEP/ILO/WHO) and the
Commission of the European Communities.
A joint WHO/CEC Task Group on Principles and Methods for the
Assessment of Nephrotoxicity Associated with Exposure to Chemicals met
at the National Institute of Public Health and Environmental
Protection, Bilthoven, the Netherlands, from 4 to 8 December 1989. The
meeting was opened by Dr K.A. van der Heijden on behalf of the
Netherlands and the host institute. The Secretariat responded and
welcomed the participants on behalf of the three cooperating
organizations of the IPCS (UNEP/ILO/WHO) and the Commission of the
European Communities. The Task Group reviewed and revised the draft
criteria document and prepared a final text.
The drafts of this Monograph were prepared by DR P. BACH,
Guildford, United Kingdom, PROFESSOR W.O. BERNDT, Omaha, USA and
PROFESSOR R. GOYER, London, Ontario, Canada. During the preparation of
the monograph, many scientists made constructive suggestions and their
contributions are gratefully acknowledged. The photographs in Figure
2 were supplied by Dr N.J. Gregg, in Figure 4 by Professor W. Guder,
and in Figure 14 by the Department of Toxicology, Institute for
Medical Research and Occupational Health, University of Zagreb.
Following the Task Group, Dr P. Bach and Dr M. Robbins collated the
text for IPCS. Dr E. Smith and Dr P.G. Jenkins, both of the Central
Unit, IPCS, were responsible for the overall scientific content and
technical editing, respectively.
ABBREVIATIONS
ADH anti-diuretic hormone
AIN acute interstitial nephritis
ARF acute renal failure
BEA 2-bromoethalamine
BEN Balkan endemic nephropathy
BUN blood urea nitrogen
CIN chronic interstitial nephritis
DBCP 1,2-dibromo-3-chloropropane
DCVC S-(1,2-dichlorovinyl)-L-cysteine
DTPA diethylenetriamine pentaacetic acid
EDTA ethylenediaminetetraacetic acid
ESRD end-stage renal disease
GAG glycosaminoglycan
GBM glomerular basement membrane
GFR glomerular filtration rate
GSH glutathione-SH
H & E haematoxolin and eosin
HCBD hexachloro-1,3-butadiene
HPLC high-performance liquid chromatography
LDH lactate dehydrogenase
3MC 3-methylcholanthrene
NAC N-acetyl cysteine
NADPH reduced nicotinamide adenine dinucleotide phosphate
NNM N-nitrosomorpholine
NSAID non-steroidal anti-inflammatory drugs
PAH p-aminohippurate
PAS periodic acid Schiff stain
PCBD S-(1,2,3,4,4-pentachloro-1,3-butadienyl
PG prostaglandin
PoG proteoglycan
RPN renal papillary necrosis
TEA tetraethyl ammonium
UCM urographic contrast medium
UDP uridine diphosphate
1. SCOPE OF THE HEALTH SIGNIFICANCE OF NEPHROTOXICITY
Over the last 20 years it has become increasingly obvious that
the kidney is adversely affected by an array of chemicals. Man is
exposed to these as medicines, industrial and environmental chemicals,
and a variety of naturally occurring substances. The level of exposure
varies from minute quantities to very high doses. Exposure may be over
a long period of time or limited to a single event, and it may be due
to a single substance or to multiple chemicals. The circumstances of
exposure may be inadvertent, accidental, or intentional overdose or
therapeutic necessity. Some chemicals cause an acute injury and others
produce chronic renal changes that may lead to end-stage renal failure
and renal malignancies. The extent and cost of clinically relevant
nephrotoxicity has only started to become apparent during the last
decade. However, the full extent of the economic impact of chemically
induced or associated nephropathy is difficult to define because the
diagnosis of early injury and the documentation of the cascade of
secondary degenerative changes have not been adequately identified.
Instead most chemically associated renal disease is only identified as
an acute renal failure or as chronic renal failure at a very late
stage when therapeutic intervention is impossible. More importantly at
this stage, the etiology may be obscured by lack of reliable
information on the likely causative agents, the levels and duration of
exposure, and other possible contributing and exacerbating factors. At
present, epidemiological evidence indicates that nephrotoxicity
leading to acute and/or chronic renal failure represents a substantial
financial burden to society (Nuyts et al., 1989). Indeed, there is
some indication that chemical exposure could play a much greater
influence in the very high incidence of end-stage renal disease
encountered in nephrology and dialysis clinics than is currently
considered to be the case.
There are already several examples of this type of chemically
associated disease that went unrecognized for some time. These include
those nephropathies caused by cadmium, other environmental heavy
metals, and, more recently, the organo-metallic compounds used as
therapeutic agents, anti-cancer drugs, cyclosporin, analgesic abuse,
and antibiotics.
Owing to its diverse functions and small mass in relation to the
resting cardiac output that it handles, the kidney is a target both
for chemicals that are pharmacologically active and for toxic
material. The nephron and its related cells perform a diversity of
physiological functions. It is the major organ of excretion and
homeostasis for water-soluble molecules; because it is a metabolically
active organ, it can concentrate certain substances actively. In
addition, its cells have the potential to bioconvert chemicals and
metabolically activate a variety of compounds. There are a number of
other processes described below that establish the potential for
cellular injury. Specific physiological characteristics are localized
to specific cell types. This makes them susceptible to, and the target
for, chemicals. The effect of any chemical on a cell may be
pharmacological, in which case the effect is dose related and occurs
only as long as the concentration of the effector is high enough to be
active. Alternatively, the chemical may cause damage to the cell. The
cell responds to injury by repair and the kidney responds to cellular
lesion by renal and extrarenal adaptation to compensate for loss of
that cell function. Although there is a substantial capacity within
the kidney for repair, there are also several circumstances where
damage may be irreversible. In general, the proximal and distal
tubules and urothelia can be repaired, but the glomeruli and medulla
may have a significantly lower repair facility. It is, therefore,
possible to initiate a series of degenerative changes as a result of
interfering with one or more of the normal physiological processes.
The Environmental Health Criteria monographs normally focus on
industrial chemicals, but at present most of the experimental and
human information on nephrotoxicity is based on therapeutic
substances. These data are most useful because there are animal and
human comparisons for specific chemicals where the levels of exposure
and the nephrotoxicological consequences are well documented. From
these data it has been possible to glean some understanding of
mechanisms of primary injury and the long-term consequences and health
significance. Thus, these compounds are generally well studied, and
the more rational understanding of the mechanism of their
nephrotoxicity in animals and man provides the basis for validating
extrapolation between species and making rational risk assessment.
Most risk assessment decisions are currently based on information
concerning the aminoglycosides, halogenated anaesthetics, several
heavy metals, and lithium, where there is an excellent concordance
between animal data and findings in humans exposed to these agents
(Kluwe et al., 1984; Porter & Bennett, 1989). This has provided some
predictive indication of what will take place in humans exposed to
analogues of these compounds. On the other hand, the demonstration
that the occurrence of light hydrocarbon-related adenocarcinomas is
specific to male rats shows that there are examples where the
molecular understanding of a renal lesion in animals is irrelevant to
humans.
There are also therapeutic agents where attempts to extrapolate
from animals to man have not been as successful. These include
compounds such as cyclosporin, analgesics and non-steroidal
anti-inflammatory agents. It has, however, been possible to develop
some model lesions that parallel those in humans using these
compounds. Generally, different protocols have had to be used, such as
water deprivation and renal injury, but these have in turn provided
the basis for developing improved screening methods for such chemicals
and also for probing the molecular nature of the lesion. There are,
however, a number of chemicals, such as renal carcinogens, mycotoxins,
other natural toxins, and anti-cancer drugs, and some types of lesion,
such as the immunonephropathies, where it has been difficult to
establish good models in animals. A host of chemicals alter glomerular
filtration rate (GFR) or some other aspect of renal function, but the
long-term health significance is still not known and it is uncertain
how to extrapolate such data to man.
2. NEPHROTOXICITY
Nephrotoxicity can be defined as renal disease or dysfunction
that arises as a direct or indirect result of exposure to medicines,
and industrial or environmental chemicals. It is well established that
toxic nephropathies are not restricted to a single type of renal
injury. Some chemicals target one discrete anatomical region of the
kidney and may affect only one cell type. Chemical insult to the
kidney may result in a spectrum of nephropathies that are
indistinguishable from those that do not have a chemical etiology.
2.1 Target selectivity
It has become increasingly apparent that there are a number of
chemicals that may adversely affect one or more of the anatomical
elements of the kidney, such as the glomerulus, proximal,
intermediate, and distal tubules, and medullary, endothelial, and
urothelial cells. Although some of these cell types (such as the
proximal tubular cells) have a marked ability to repair damaged
regions, others, such as the glomerular epithelium and the "type 1"
medullary interstitial cells, do not. It is for this reason that the
dynamic process that follows any renal injury can affect the outcome
of the chemical insult.
2.2 The dynamics of renal injury
The renal response to injury is dynamic, and the kidney adapts to
maintain homeostasis during the cascade of repair and recovery that
follows the primary insult (Bach, 1989). Depending on the type and
frequency of the damage, and the region of the kidney that is damaged,
the organ can respond by a recovery, a reduced functional reserve, or
by a progressive degenerative change. A reduced functional reserve may
play a very important role in sensitizing the kidney to subsequent
renal injury, and an initiated degenerative cascade may either
stabilize or progress to acute or chronic renal failure. It is not
possible to differentiate between a kidney that has totally recovered,
one with a reduced functional reserve, and an organ with early
progressive degenerative change, except in animals where function and
morphology can be assessed under well controlled conditions.
2.3 Classification of renal disease
Classification of renal disease can be based on clinical
manifestations, pathological changes, or etiological agents. WHO has
prepared a number of detailed and illustrated publications in recent
years on the classification of renal disease (WHO 1982, l985, 1987,
1988). The general approach is to subdivide the kidney into major
anatomical components (i.e. glomeruli, tubules and interstitium, and
blood vessels) and to relate these to the major clinical syndromes
characteristic of renal diseases. Table 1 contains a modified
classification of renal disease that focuses on major disorders of the
kidney that may be associated with nephrotoxins. This classification
is consistent with previous WHO publications and textbooks of
nephrology and pathology and provides a framework for discussing the
mechanisms and pathology of nephrotoxicity. It must be appreciated
that nephrotoxic agents may have multiple anatomical targets and that
toxicity manifests itself in more than one clinical syndrome. Further
discussion of renal effects due to specific agents are discussed
below.
2.4 The epidemiology of nephrotoxicity
Putting the health significance of nephrotoxicity into
perspective is difficult because of the diverse array of chemicals
that target different parts of the kidney, the spectrum of disease
consequences, and the many interacting factors. There is also
uncertainty in assessing changes in renal function before they reach
the point where preventive medicine can no longer be practised and
therapeutic intervention may be appropriate.
Many industrial and environmental chemicals and therapeutic
agents have been shown in experimental studies and from acute toxic
exposures to be nephrotoxins, but the extent of their contributions to
the overall incidence of chronic renal failure is not known. Data
extracted from the European Dialysis and Transplant Association
Registry identified only about 4% of patients starting renal
replacement therapy in 1984 as having drug or chemical associated
renal disease. However, nearly 50% of these patients were considered
possible (but not diagnosed) cases of toxic nephropathy (Dieperink,
1989). Of those patients identified as having chemical-related renal
disease, analgesic nephropathy is the most important recognized
outcome. In an analysis of the European Dialysis and Transplant
Association Registry (1986), the prevalence of analgesic nephropathy
was found to vary greatly between countries. It is highest in
Switzerland (18.1%) and Belgium (11.8%) and accounts for over 4% of
patients in Denmark, Germany, Czechoslovakia, and Austria. In 20
countries the prevalence is lower than one patient in every hundred
(European Dialysis and Transport Association Registry, 1986; Wing et
al., 1989). Other specific drug nephropathies recorded less frequently
include those due to cisplatin ( cis-platinum) and cyclosporin A. A
small number of patients had other specific drug or chemical-related
nephropathies.
The role of the toxic agents that may contribute to the 50% of
cases of chronic renal failure of undiagnosed etiology is less
certain. There is opportunity for exposure to a number of chemicals in
the workplace or ambient environment (drugs included) that are
possible nephrotoxins. It has been estimated that there were nearly
four million workers in the USA with potential occupational exposure
to known or suspected nephrotoxins in the 1970s (Landrigan et al.,
1984). The major occupational exposure is to workplace solvents, but
Table 1. Classification of renal disease due to nephrotoxins in humans
1. Immunologically mediated
Antibody mediated
Membranous glomerulonephritis or immune complex type disease
metals (gold, mercury)
D-penicillamine
drugs responsible for a lupus-like syndrome
(hydralazine, procainamide, diphenylhydantoin)
Anti-glomerular basement membrane antibody mediated
organic solvents
hydrocarbons
2. T cell mediated (?)
Nephrotic syndrome with minimal glomerular changes
lithium salts
non-steroidal anti-inflammatory agents
toxic metals and organic compounds, including pesticides, are of great
concern. The well documented occurrence of subclinical nephropathies
in subjects occupationally exposed to nephrotoxins such as lead or
cadmium (see section 5.5), the excess of mortality for renal diseases
in cohorts of workers with previous exposure to these two heavy metals
(Bennett, 1985; Bernard & Lauwerys, 1986), and, more recently, the
suggestion that subclinical renal effects caused by cadmium are early
signs of an accelerated and irreversible decline of renal function
(Roels et al., 1989) should all be noted. The linkage of these risks
to the actual occurrence of chronic renal failure has not, however,
been possible. A review of the end-stage renal disease (ESRD)
population in the USA has shown that at least 19% have a renal disease
of unknown or non-specific etiology (Burton & Hirschman, 1979). If
other diagnostic groups that have uncertain etiologies, such as the
30% with glomerulo-nephritis, 5% with interstitial nephritis of
suspected etiology (lead, analgesics, etc.) and possibly the 8%
diagnosed as having pyelonephritis, are added, it becomes apparent
that the etiology of a large portion of patients with chronic renal
failure is unrecognized or undefined. Environmental factors may have
a previously unrecognized role in the etiology of these lesions.
There are many reasons for the failure to recognize toxic
etiologies (Sandler, 1987). A major reason is that chronic renal
failure develops slowly over a number of years, so that retrospective
identification of a toxic agent requires knowledge of lifestyles,
therapies, or workplace environments that might provide risk factors.
However, such data are generally not available. Unless the drug or
toxic chemical is persistent in tissues, it is usually not possible to
confirm or quantify exposure. The inability to recognize multiple
etiologies or confounding factors adds further complexity to the
problem. There is also lack of uniformity in clinical and pathological
diagnoses. A further complexity is that there is a tendency to
categorize chronic renal failure by a mixture of pathological and
etiological classifications. For example, in one instance a patient
may be classified as having chronic interstitial nephropathy on the
basis of a renal biopsy, whereas another patient with the same
pathology might be classified as having toxic nephropathy due to
exposure to a nephrotoxic chemical because of knowledge of exposure.
In a survey of patients requiring dialysis in Israel, Modan et al.
(1975) found diagnostic inconsistencies between hospital diagnosis,
autopsy reports, and diagnosis made by the study reviewers.
Disagreement was most often seen for chronic glomerulo-nephritis,
chronic pyelonephritis and nephrosclerosis. The stage of the
pathological process or severity influences classification.
Interstitial nephritis tends to be diagnosed more frequently in the
early stages of chronic renal failure, whereas glomerulonephritis is
a more common diagnosis for patients undergoing dialysis. There may
be a rational basis for this in that the scarring in persistent
interstitial nephritis does impede blood supply to the glomerulus.
This could lead to glomerular disease and interstitial nephritis
despite the fact that there are different etiologies or risk factors
for the two conditions.
The identification of chronic pyelonephritis is made more precise
by following established criteria. These include the presence of gross
irregular scarring, inflammation, fibrosis and deformity of calyces
underlying parenchymal scars, predominant tubulointerstitial
histological damage, and relative lack of glomeruli. There is evidence
that some of the chronic interstitial nephritis that is labelled
chronic pyelonephritis is due to something other than bacterial
infection.
Environmental Health Criteria 27: Guidelines on Studies in
Environmental Epidemiology (WHO, 1983) provides guidelines for
obtaining human data concerning the health effects of exposure to
chemical agents. For agents that produce acute renal failure,
long-term follow-up may identify those instances where chronic renal
disease has persisted. For agents that give rise to accidental
poisoning, clinical case reports can provide important information. In
the case of agents where exposure to larger population segments
occurs, information may be obtained by using statistical and
epidemiological methods to investigate possible nephrotoxicity from
such exposures (as compared to a non-exposed control group). Specific
segments of the population that might be at higher risk to a potential
nephrotoxic drug or workplace chemical should be particularly closely
monitored for renal effects.
There are marked differences between the incidence of
analgesic-associated ESRD in different countries and within the same
country (Table 2). This varies from up to 22% in Australia (in 1982)
Table 2. National prevalence of analgesic nephropathy
in patients with end-stage renal failure
% %
South Africa 22 Scandinavia (1979) 3
Switzerland (1980) 20 France (1979) 2
Belgium (1984) 18 USA 2
Australia (1985) 15 United Kingdom (1979) 1
Federal Republic of 13 Italy (1979) 1
Germany (1983) Spain (1979) 0.4
Canada (1976) 3
and in parts of some of the European countries to as low as 0.2% in
the USA. It is generally considered that the withdrawal of phenacetin
has lead to the disappearance of the high incidence of renal papillary
necrosis (RPN) in Scandinavia, Canada, and Australia, but a high
incidence remains in Switzerland, Belgium, and the Federal Republic of
Germany (Gregg et al., 1989). Specific geographical locations may have
analgesic abuse problems such as the Winston-Salem area (USA).
Worldwide variability in the prevalence of analgesic nephropathy has
long been recognized. The correlation of the incidence of this disease
with local analgesic consumption has been demonstrated. However, the
relation between both phenomena is not well established since
comparable consumption data, focussed on the sales of analgesic
mixtures, are not available in most countries. The high frequency
abuse area in Belgium is situated in the north (Fig. 1a), where up to
51% of dialysis patients are analgesic abusers, but this is markedly
lower in the south (Elseviers & De Broe, 1988). In Germany (Fig. 1b)
the highest prevalence is in West Berlin (up to 50%), Hamburg, and
Bremen (Pommer et al., 1986). These data indicate that the prevalence
of this disease has been underestimated on a national basis. There
are indications that the overall prevalence of this disease has also
been underestimated in several other countries. Local well-conducted
studies of the prevalence of analgesic nephropathy showed higher
prevalences than the European Dialysis and Transport Association
Registry. In the Federal Republic of Germany, a prevalence of 13% of
analgesic nephropathy in dialysis patients was found, while the
appropriate European Dialysis and Transport Association data was never
more than 6% (Pommer et al., 1986). In Belgium, the prevalence of
analgesic nephropathy was 18%, whereas the European Dialysis and
Transport Association registered a prevalence of 12% (Elseviers & De
Broe, 1988). The percentage of nephropathies of unknown etiology and
of pyelointerstitial nephritis may also indicate an underestimation of
analgesic nephropathy. These percentages are low in countries with a
high prevalence of analgesic nephropathy (Switzerland and Belgium) and
are high in countries such as Italy and Spain (Wing et al., 1989)
where analgesic nephropathy is considered to be rare. Analgesic
nephropathy progresses silently over a long period, and so the
diagnosis is difficult. In addition, most patients deny being
analgesic abusers, which further confounds diagnosis. Symptoms are
nonspecific until the degenerative cascade affects the cortex, when
renal failure occurs. Moreover, even when the renal failure is
recognized, the diagnosis of analgesic nephropathy remains difficult
unless diagnostic criteria are established.
2.5 Risk factors for toxic nephropathies
The risk of developing a clinically significant nephrotoxicity
depends on pre-existing clinical conditions and may be identified in
specific patient populations. Hypertension, diabetes, cardiovascular
disease, etc. are all thought to have the potential to exacerbate
nephrotoxicity, but many of these conditions have not been
systematically investigated for all types of nephrotoxicity. There are
examples where both chemicals and other disease factors cause a
lesion. For example, sickle cell disease and diabetes can cause renal
papillary necrosis, a condition that is also common in individuals who
abuse analgesics. The risk factors that predispose individuals to
renal papillary necrosis are not clear, and it is also unclear whether
diabetics are at greater risk of developing the lesion if they take
high doses of analgesics. This question cannot be resolved until
better diagnostic criteria are developed to identify the lesion before
it involves the cortex. Risk may also vary for different nephrotoxins.
While Bence-Jones protein excretion considerably increases the risk of
radiocontrast-induced renal injury, the effects of other types of
chemicals in patients with multiple myeloma are not clear. Therefore
recognition of the risk factors is necessary for the understanding and
prevention of renal damage.
There are also many examples of animal data that have not yet
been translated into risk terms in humans. For example, the immature
kidney may be resistant to amino-glycosides (Marre et al., 1980) and
cephalosporins (Tune, 1975), but the reverse is true for other
chemicals such as hexachlorobutadiene (Hook et al., 1983). Other
factors, such as electrolyte and volume changes or an alteration in
the renin-angiotensin system, may affect some types of drug-induced
acute renal failure (Bennett et al., 1983).
Risk factors as a measure of vulnerability to potential
nephrotoxicity that could be caused by drugs and chemicals are not
well defined at present. Clearly, wide variation exists among
individuals and even groups of people. Multiple exposure to toxic
agents and multiple drug usage are certainly factors, but the ability
to estimate risks to multiple exposure is limited. Factors intrinsic
to the nature of renal function and risk factors presented by clinical
disease have been reviewed (Porter, 1989) and are discussed below.

 2.5.1 Factors related to renal function
The vulnerability of the kidney to toxicity from exposure to a
particular drug or chemical is the product of several groups of risk
factors. In any one person, multiple factors may be operative. The
nature of normal renal function in itself contributes to the
vulnerability to toxins. The intimate association of the capillary
endothelial surface during the process of ultrafiltration provides
opportunity for direct toxicity. A further contribution to the
glomerular capillary vulnerability is the positive hydrostatic
pressure required for producing the plasma ultrafiltrate. Adding to
this vulnerability is the "hyperfiltration injury" hypothesis proposed
by Brenner (1983) and Brenner et al. (1978, 1982), who showed that
when a nephron ceases to function, the remaining nephrons hypertrophy
and the flow rate per functioning nephron is raised, thus increasing
the exposure to the drug or chemical. A further aspect is the concept
of renal reserve, i.e. fewer functioning nephrons further increase the
vulnerability of the kidney to toxicity.
Another aspect of the structure of the glomerular endothelial
cells that can lead to injury to the kidney is the negative charge of
the filtration membranes. Positively charged ligands can become
electrostatically attached and alter the permeability coefficient of
the glomerulus. In addition, cationic proteins can be sequestered in
the glomerulus and act as "planted antigens", and a circulating
antibody can attach to such antigens resulting in an in situ immune
complex formation; hydro-carbons from petroleum products can act in a
similar way (Ravnskov, 1985). This is but one of a wide variety of
immunologically mediated glomerular injury patterns that have been
identified. This variety is not surprising when one considers the
heterogeneity of the biochemical composition of the glomerulus and the
wide spectrum of antigenic compounds to which the body is exposed
(Glassock, 1986).
Tubular vulnerability to nephrotoxins is related to the nature of
normal tubular function. The medullary countercurrent multiplier
system provides a mechanism for eliminating body waste products while
minimizing body water loss. A consequence is the reabsorption and
recycling of compounds of low relative molecular mass, in particular
urea and neutral toxicants and/or their metabolites, which can
accumulate in the medullary interstitium. Depending on their chemical
properties, they may initiate an inflammatory response through
activation of mediators, a factor that may be relevant in the
pathogenesis of analgesic nephropathy (Mudge, 1982).
The organic acid and base transport systems of the proximal
tubule serve to excrete certain molecular species. Several commonly
used drugs, including the organic acid penicillin, utilize this
mechanism of transport. Toxicants that are involved in these systems
might induce renal injury directly because of high cellular
concentration or by acting as competitive inhibitors to block the
elimination of endogenously produced toxic metabolites. Tubular
mechanisms for acidification may play a part in tubular injury by
drugs or chemicals that induce an acidification defect, e.g., lithium
(Batelle et al., 1982). Drugs or chemicals that are absorbed by
pinocytosis become concentrated in lysosomes where they are subjected
to digestion by hydrolytic enzymes. Some toxicants, however, may
inhibit the hydrolytic process, resulting in drug accumulation and
tubular cell toxicity that may resemble lysosomal storage disease (as
occurs in aminoglycoside nephrotoxicity).
2.5.2 Clinical risk factors
The application of multivariate analysis for investigating
clinical risk factors in the onset of acute renal failure (Rasmussen
& Ibels, 1982) may provide some insight into factors that may increase
vulnerability to nephrotoxicity from drugs and chemicals. The risk
factors summarized in Table 3 show that multiple risk factors coexist
in the majority of patients with acute renal failure. Although age has
been recognized as a factor in a number of studies (Porter, 1989;
Porter & Bennett, 1989), it may simply be a convenient marker for the
change in renal vulnerability that relates to the decline in
glomerular filtration rate (GFR) occurring beyond the age of 50
(Davies & Shock, 1950). The pathological basis for this decline is not
certain but may be related to vascular changes that accompany aging
(Avendano & Lopez-Novoa, 1987). Another possible explanation is that
the kidneys of people over 50 years of age no longer respond to
hypertrophic growth factors. Renal donors aged 50 or more show little
or no functional increase after the loss of one kidney (Boner et al.,
1972). Indeed, renal function reserve declines linearly with time
after 30 years of age (Anderson & Brenner, 1986). Age may also reflect
a loss of the ability of the renal tissue to repair. In young
individuals nephrotoxicity in terms of tubular necrosis may be
compensated for by constant repair, while in older patients this
repair capacity may be diminished, resulting in the clinical
expression of renal injury (Laurent et al., 1988).
Pre-existing renal disease is an obvious risk factor predisposing
to abnormal accumulation and excess blood levels of many nephrotoxic
drugs and chemicals. It is not clear whether sex is a predisposing
risk factor in humans, but male rodents are considerably more
susceptible than females to nephrotoxicity and carcinogenicity from
many environmental toxins (NTP, 1983, 1986, 1987).
Factors such as short-term and high-dose exposure versus chronic
and/or low-dose exposure influence vulnerability via the mode of
metabolism, rate of excretion, etc. Long-term, low-dose exposure to
substances that have a long biological half-life, such as lead or
cadmium, increases risk from these nephrotoxins, but their role as
co-risk factors is not known.
Table 3. Frequency of combined risk factors in 143 patients
with acute renal failure (ARF)a
Age Hyper- Gout/ Diabetes Renal Diuretics
tension hyper- disease
uricaemia
Age (> 59 years) 30
Hypertension 29 4
Gout/hyperuricaemia 21 18 4
Diabetes 11 6 4 1
Renal disease 18 12 12 6 4
Diuretics 29 27 21 8 13 0
Multiple risksb 108 63 37 14 13 0
a Modified from: Rasmussen & Ibels (1982).
b Significant risk contribution to ARF based on discriminant multiple linear
regression analysis.
2.5.3 Extrapolation of animal data to man
The use of animals has been essential to help define the
molecular basis and the progression of model nephropathies, but it may
be inappropriate to extrapolate animal toxicology data directly to man
because of marked species, strain, dietary, and sex differences. In
addition, there may be differences in dosing levels and regimen and in
the absorption, distribution, metabolism, and excretion of potential
nephrotoxins. There are also very significant differences in renal
structural and functional characteristics in the common laboratory
species used for risk assessment.
Chemical safety assessment has generally been undertaken in
relatively few strains of animals, such as the Sprague-Dawley, Wistar
and Fisher-344 rats. There is limited information on inter-species
comparisons. In addition to assessing the renal differences in each
of the species or strains used, it is necessary to examine extrarenal
differences. Thus, for example, there are marked species differences
in the hepatic handling of chemicals (Smith 1974; Testa & Jenner,
1976) and the metabolic capacity of each of the major organ systems
(Litterst et al., 1975a; Kluwe, 1983). This will have profound
consequences on the amount of a parent chemical and the pattern of
metabolites that reach the kidney. Dietary factors such as
carbohydrate, lipid, and protein intake alter renal function, and the
presence of contaminants and natural toxicants may add to the toxic
burden of the kidney (Bridges et al., 1982).
If the mechanistic basis of a renal injury is clearly
established, it is easier to assess the risk of chemical injury in
man, but such data are at present only available for a few chemicals.
There are relatively few examples of nephrotoxic chemicals where there
is a full profile of information from experimental animals and man,
and in the vast majority of cases data are available only in rats.
In order to extrapolate animal data for risk assessment, each
screening procedure should cover a sensible level of exposure and a
comparable condition to that found in man. The experience gained
should provide a foundation from which a rational basis can be
developed to identify potentially exacerbating risk factors and from
which nephrotoxicity can be reduced. Some lesions can only be induced
in rats with difficulty, and there may be a need to use sensitive
species or strains and/or to adapt certain experimental manoeuvres to
produce a lesion similar to that which occurs in man.
2.5.4 Risk assessment from nephrotoxicity studies in animals
Risk assessment from nephrotoxicity studies in animals has been
best defined for therapeutic agents. Many have been widely tested in
animals as a pre-clinical safety evaluation or used to study the
mechanism of renal injury where there are adverse reactions caused by
these compounds in clinical usage. The risk assessment for a number of
workplace or environmental chemicals has been developed from animal
models that have been used to study the mechanisms of these effects,
especially those of the heavy metals and some of the industrial
organic chemicals.
2.5.5 Special risk groups in humans
The marked variability in the response of any study population to
potentially nephrotoxic compounds establishes clearly that there are
groups at risk. There are a number of factors that could be
responsible for increasing the risk of nephrotoxicity. These include
existing renal disease, loss of renal parenchyma, high protein diet,
chemical exposure, predisposing factors, multiple myeloma, and other
conditions where there is an added level of protein excretion when the
kidney is under an additional work-load.
So far there has been relatively little interest in the
individuals that do not appear to be at risk from exposure to
potential nephrotoxins. While this is generally assumed to be the
result of an absence of predisposing factors, there may well be other
chemical, dietary, or disease considerations that provide a protective
effect. There is experimental evidence to suggest that a pre-existing
streptozotocin-induced diabetes and also poly-aspartic acid protect
against aminoglycoside-induced renal injury, and that fish oil diets
(high in omega-3 polyunsaturated fatty acids) reduce cyclosporin-A
nephrotoxicity. These factors could well be used to reduce the health
impact of nephrotoxicity.
2.5.6 Multichemical exposure
At present, there is virtually no information on the effects of
multichemical exposure in man and very little data on the effects of
more than one chemical administered simultaneously in animals.
Simultaneous exposure to several chemicals represents a major
toxicological problem, as man is generally exposed to more than one
substance in medicines, in food, and from environmental factors.
However, most experimental studies have investigated only single
chemicals. Interactions have been studied between mercuric chloride,
potassium dichromate, citrinin, and hexachloro-1,3-butadiene (HCBD)
in vivo and in vitro using a rat model (Baggett & Berndt 1984a,b;
1985). Dichromate potentiates the mercuric chloride effect, i.e. the
effects produced by the combination of metals are always greater than
the sum of the individual effects. There appears to be no simple
kinetic explanation, i.e. no enhanced renal accumulation of the
mercuric ion. The plasma membrane may be a site for the interaction of
these metal ions and could be the preliminary step that leads to
overall renal dysfunction and an ultimately enhanced acute renal
failure. Dichromate-citrinin and dichromate-HCBD interactions have
been demonstrated by alterations in urine flow, glucose excretion, and
transport processes. Some experimental data suggest that a
synergistic interaction may occur in analgesic nephropathy. The
mechanisms that underlie these interactions are not understood, and at
present there is no rational basis to predict them. Experimental
studies have shown that tubular cell injury, induced by
trichloroethylene and carbon tetrachloride, is potentiated by exposure
to polyhalogenated biphenyls, e.g., polychlorinated biphenyls (Kluwe
et al., 1979).
2.5.7 Renal functional reserve
The concept of renal functional reserve is a simple one in which
not all of the nephrons nor all of the cellular functions in a single
nephron are available or used at any one time (Friedlander et al.,
1989). Thus there is a buffering capacity in the kidney that can cope
with short-lived or protracted demands on function that exceed the
normal level. Part of this functional reserve is used to meet the
response to perturbation of the homeostatic system by water or
electrolyte loading. Most of the studies on and understanding of renal
functional reserve relate to changes in glomerular filtration rate and
renal blood flow. It is likely that additional approaches are needed
to test for other types of functional reserve.
2.5.8 The effects of chemicals on kidneys with pre-existing renal lesions
Although it is generally acknowledged that there are several
types of renal lesions that exist as a nephropathy in the general
population at a low but significant level (e.g., nephrotic syndrome),
little is known about how these pre-existing lesions affect the
response of the kidney to subsequent nephrotoxic insults.
2.5.8.1 Nephrotoxicity in the presence of renal and extrarenal disease
Safety screening is conducted on young, disease-free animals,
housed under optimal conditions, fed contamination-free, high-protein
food. By contrast, man is exposed to a variety of dietary and
environmental chemicals and to a poly-pharmacy of both prescribed and
self-administered medications over many years. In addition, screening
is generally undertaken using normal experimental animals. This may be
inappropriate because, with the exception of occupational and
environmental exposure, man is exposed to potentially nephrotoxic
therapeutic substances to treat disease. Pre-existing diseases can
have a profound effect on the direct or indirect response of the
kidney to handling chemicals (Bennett, 1986). There is an increasing
wealth of animal data to demonstrate that common clinical conditions
in man, such as hypertension, renal compromise, and renal ischaemic
injury, exacerbate cyclosporin nephrotoxicity and bacterial endotoxins
in animals and that systemic infection increases the sensitivity of
the kidney to aminoglycoside toxicity (Bergeron et al., 1982).
The role of pre-existing renal lesions on nephrotoxicity is
important, but there are few clear indications of what can be
predicted from existing clinical data and from animal studies.
Diabetes is generally associated with reduced renal function, diabetic
nephropathy, and renal papillary necrosis, and might be expected to
exacerbate chemical-associated nephrotoxicity. Untreated
streptozotocin-induced diabetes, however, protects rats against
gentamicin, low-dose cisplatin, and uranyl nitrate nephrotoxicity
(Teixeira et al., 1982; Vaamonde et al., 1984). Recent studies on the
acute effects of intravenous radio-contrast media on anaesthetized
diabetic rats were inconclusive (Reed et al., 1983, Golman & Almen,
1985, Leeming et al., 1985).
3. KIDNEY STRUCTURE AND FUNCTION
An in-depth review of kidney structure and function is beyond the
scope of this monograph. Only sufficient information will be given to
provide a general background against which nephrotoxicity can be
framed. A fuller insight into the complexities of the kidney in
health, disease, and nephrotoxicity has been described by Valtin
(1973), Orloff & Berliner (1973), Hook (1981), Porter (1982), Bach et
al. (1982, 1989), Bach & Lock (1982, 1985, 1987, 1989), Seldin &
Giebisch (1985), Brenner & Rector (1986).
3.1 Renal anatomy
The two kidneys are situated retro-peritoneally, on either side
of the vertebral column, and process 25% of the resting cardiac output
via an arterial blood supply. Much of the fluid and most of the
solutes in blood are filtered through the glomeruli into the proximal
part of the nephron (the functional unit of the kidney) from which
essential small molecules are reabsorbed. Numerous macro-molecules are
reabsorbed into the tubular cells by an endocytotic process and are
digested in tubular lysosomes. Many organic acids and bases (including
many drugs) are secreted (and reabsorbed) by carrier-mediated
processes located principally in the proximal tubule. There is some
secretion, mainly of waste solutes, from the blood into the distal
part of the nephron, and much of the water in which they are dissolved
is subsequently reabsorbed.
Each kidney is made up of a large number of nephrons, groups of
which unite to continue as collecting ducts or tubules, and these in
turn combine to make up the ducts of Bellini, which exit around the
papilla tip. The papilla opens into the calix, which is in continuity
with the renal pelvis, a funnel-shaped area that narrows to the
ureter. The continued production of urine, together with peristalsis
of the ureter, carries excreted waste to the bladder. The
morphophysiology of the kidney varies markedly between species.
Therefore, a generalized description will be provided, and only the
important differences between the rat (and other common laboratory
animals) and man will be described (Moffat, 1979).
3.1.1 Histology
Renal lesions occur in discrete anatomical regions. This
highlights the need to understand changes in terms of the biochemical
properties of the specifically affected region and its adjacent cells.
While haematoxylin and eosin staining and a number of other routinely
used staining procedures identify nephropathies and renal
degeneration, these are generally based on a relatively non-specific
assessment. The non-specificity of routine histopathology has, in
fact, been the strength of these methods in the preliminary
assessment of chemically induced nephropathies. It may, however, miss
some types of lesions and generally gives little information that can
help identify the mechanistic basis of a lesion.
Histochemical techniques can provide insight into primary and
secondary cellular mechanisms. One aspect of "histochemistry" is the
use of frozen segments of the nephron (Bach et al., 1987) and the
application of fluori-metric or radiochemical assays to measure the
activities and distribution of specific biochemical characteristics.
Microdissection generally fails to give a detailed localization of
these properties in relation to specific or individual cells. In
addition, the technique is difficult to apply to injured renal cells.
The most widely used histochemical approach is based on obtaining
frozen, fixed frozen, or fixed embedded sections of the kidney that
are then used with chromo- or fluoro-phores. These react with a
selected type of material. The types of materials that can be
visualized in section depends on their chemical structure (e.g.,
carbohydrate), enzymic activity (e.g., lactate dehydrogenase),
antigenicity (e.g., specific molecules), or physicochemical
properties (e.g., lipophilicity), some of which are shown in Fig. 2.
This approach also includes the distribution or incorporation of
radiolabelled molecules by their interaction with a photographic film
laid over the section (Bach et al., 1987). Immunohistochemical
techniques using labelled antibodies permit antigens to be localized
at the light microscopical and ultrastructural levels.
These microscopic histochemical techniques provide information on
the distribution at, or within, specific cells and their relative
activities, and have been used to define a variety of characteristics
of the kidney. It is important to stress that each method has its own
inherent strength and weakness, and that it may be difficult to relate
data from tissue sections to absolute biochemical measurements. This
is a consequence of the complex mixture of materials that are present
in tissue sections and the chemical changes that may take place in
these sections, particularly once they have been fixed. While the
biochemical characteristics of tissue in frozen sections are least
likely to be adversely affected, subtle and misleading alterations in
chemical properties can still occur. In addition, treatments to
conserve morphological features (fixation and embedding) alter these
values. It may also be difficult to be certain whether an increased
intensity of staining for a certain substance represents de novo
synthesis, unmasking, or the loss of factors that suppress staining.
These two techniques have proved to be very powerful in providing
information on the biochemical characteristics of cells and the
changes associated with renal injury. The in situ hybridization
techniques, using specific labelled nucleotidic probes, permit the
detection of protein synthesis at a subcellular level.
3.1.2 Enzyme histochemistry and quantification
Histochemical heterogeneity is evident in each part of the
nephron and varies between different species. In addition, the profile
of characteristics within any region of the kidney and nephron may be
related to sex and age. Antibodies are directed against unique or
novel characteristics along the nephron that are present on enzymes,
2.5.1 Factors related to renal function
The vulnerability of the kidney to toxicity from exposure to a
particular drug or chemical is the product of several groups of risk
factors. In any one person, multiple factors may be operative. The
nature of normal renal function in itself contributes to the
vulnerability to toxins. The intimate association of the capillary
endothelial surface during the process of ultrafiltration provides
opportunity for direct toxicity. A further contribution to the
glomerular capillary vulnerability is the positive hydrostatic
pressure required for producing the plasma ultrafiltrate. Adding to
this vulnerability is the "hyperfiltration injury" hypothesis proposed
by Brenner (1983) and Brenner et al. (1978, 1982), who showed that
when a nephron ceases to function, the remaining nephrons hypertrophy
and the flow rate per functioning nephron is raised, thus increasing
the exposure to the drug or chemical. A further aspect is the concept
of renal reserve, i.e. fewer functioning nephrons further increase the
vulnerability of the kidney to toxicity.
Another aspect of the structure of the glomerular endothelial
cells that can lead to injury to the kidney is the negative charge of
the filtration membranes. Positively charged ligands can become
electrostatically attached and alter the permeability coefficient of
the glomerulus. In addition, cationic proteins can be sequestered in
the glomerulus and act as "planted antigens", and a circulating
antibody can attach to such antigens resulting in an in situ immune
complex formation; hydro-carbons from petroleum products can act in a
similar way (Ravnskov, 1985). This is but one of a wide variety of
immunologically mediated glomerular injury patterns that have been
identified. This variety is not surprising when one considers the
heterogeneity of the biochemical composition of the glomerulus and the
wide spectrum of antigenic compounds to which the body is exposed
(Glassock, 1986).
Tubular vulnerability to nephrotoxins is related to the nature of
normal tubular function. The medullary countercurrent multiplier
system provides a mechanism for eliminating body waste products while
minimizing body water loss. A consequence is the reabsorption and
recycling of compounds of low relative molecular mass, in particular
urea and neutral toxicants and/or their metabolites, which can
accumulate in the medullary interstitium. Depending on their chemical
properties, they may initiate an inflammatory response through
activation of mediators, a factor that may be relevant in the
pathogenesis of analgesic nephropathy (Mudge, 1982).
The organic acid and base transport systems of the proximal
tubule serve to excrete certain molecular species. Several commonly
used drugs, including the organic acid penicillin, utilize this
mechanism of transport. Toxicants that are involved in these systems
might induce renal injury directly because of high cellular
concentration or by acting as competitive inhibitors to block the
elimination of endogenously produced toxic metabolites. Tubular
mechanisms for acidification may play a part in tubular injury by
drugs or chemicals that induce an acidification defect, e.g., lithium
(Batelle et al., 1982). Drugs or chemicals that are absorbed by
pinocytosis become concentrated in lysosomes where they are subjected
to digestion by hydrolytic enzymes. Some toxicants, however, may
inhibit the hydrolytic process, resulting in drug accumulation and
tubular cell toxicity that may resemble lysosomal storage disease (as
occurs in aminoglycoside nephrotoxicity).
2.5.2 Clinical risk factors
The application of multivariate analysis for investigating
clinical risk factors in the onset of acute renal failure (Rasmussen
& Ibels, 1982) may provide some insight into factors that may increase
vulnerability to nephrotoxicity from drugs and chemicals. The risk
factors summarized in Table 3 show that multiple risk factors coexist
in the majority of patients with acute renal failure. Although age has
been recognized as a factor in a number of studies (Porter, 1989;
Porter & Bennett, 1989), it may simply be a convenient marker for the
change in renal vulnerability that relates to the decline in
glomerular filtration rate (GFR) occurring beyond the age of 50
(Davies & Shock, 1950). The pathological basis for this decline is not
certain but may be related to vascular changes that accompany aging
(Avendano & Lopez-Novoa, 1987). Another possible explanation is that
the kidneys of people over 50 years of age no longer respond to
hypertrophic growth factors. Renal donors aged 50 or more show little
or no functional increase after the loss of one kidney (Boner et al.,
1972). Indeed, renal function reserve declines linearly with time
after 30 years of age (Anderson & Brenner, 1986). Age may also reflect
a loss of the ability of the renal tissue to repair. In young
individuals nephrotoxicity in terms of tubular necrosis may be
compensated for by constant repair, while in older patients this
repair capacity may be diminished, resulting in the clinical
expression of renal injury (Laurent et al., 1988).
Pre-existing renal disease is an obvious risk factor predisposing
to abnormal accumulation and excess blood levels of many nephrotoxic
drugs and chemicals. It is not clear whether sex is a predisposing
risk factor in humans, but male rodents are considerably more
susceptible than females to nephrotoxicity and carcinogenicity from
many environmental toxins (NTP, 1983, 1986, 1987).
Factors such as short-term and high-dose exposure versus chronic
and/or low-dose exposure influence vulnerability via the mode of
metabolism, rate of excretion, etc. Long-term, low-dose exposure to
substances that have a long biological half-life, such as lead or
cadmium, increases risk from these nephrotoxins, but their role as
co-risk factors is not known.
Table 3. Frequency of combined risk factors in 143 patients
with acute renal failure (ARF)a
Age Hyper- Gout/ Diabetes Renal Diuretics
tension hyper- disease
uricaemia
Age (> 59 years) 30
Hypertension 29 4
Gout/hyperuricaemia 21 18 4
Diabetes 11 6 4 1
Renal disease 18 12 12 6 4
Diuretics 29 27 21 8 13 0
Multiple risksb 108 63 37 14 13 0
a Modified from: Rasmussen & Ibels (1982).
b Significant risk contribution to ARF based on discriminant multiple linear
regression analysis.
2.5.3 Extrapolation of animal data to man
The use of animals has been essential to help define the
molecular basis and the progression of model nephropathies, but it may
be inappropriate to extrapolate animal toxicology data directly to man
because of marked species, strain, dietary, and sex differences. In
addition, there may be differences in dosing levels and regimen and in
the absorption, distribution, metabolism, and excretion of potential
nephrotoxins. There are also very significant differences in renal
structural and functional characteristics in the common laboratory
species used for risk assessment.
Chemical safety assessment has generally been undertaken in
relatively few strains of animals, such as the Sprague-Dawley, Wistar
and Fisher-344 rats. There is limited information on inter-species
comparisons. In addition to assessing the renal differences in each
of the species or strains used, it is necessary to examine extrarenal
differences. Thus, for example, there are marked species differences
in the hepatic handling of chemicals (Smith 1974; Testa & Jenner,
1976) and the metabolic capacity of each of the major organ systems
(Litterst et al., 1975a; Kluwe, 1983). This will have profound
consequences on the amount of a parent chemical and the pattern of
metabolites that reach the kidney. Dietary factors such as
carbohydrate, lipid, and protein intake alter renal function, and the
presence of contaminants and natural toxicants may add to the toxic
burden of the kidney (Bridges et al., 1982).
If the mechanistic basis of a renal injury is clearly
established, it is easier to assess the risk of chemical injury in
man, but such data are at present only available for a few chemicals.
There are relatively few examples of nephrotoxic chemicals where there
is a full profile of information from experimental animals and man,
and in the vast majority of cases data are available only in rats.
In order to extrapolate animal data for risk assessment, each
screening procedure should cover a sensible level of exposure and a
comparable condition to that found in man. The experience gained
should provide a foundation from which a rational basis can be
developed to identify potentially exacerbating risk factors and from
which nephrotoxicity can be reduced. Some lesions can only be induced
in rats with difficulty, and there may be a need to use sensitive
species or strains and/or to adapt certain experimental manoeuvres to
produce a lesion similar to that which occurs in man.
2.5.4 Risk assessment from nephrotoxicity studies in animals
Risk assessment from nephrotoxicity studies in animals has been
best defined for therapeutic agents. Many have been widely tested in
animals as a pre-clinical safety evaluation or used to study the
mechanism of renal injury where there are adverse reactions caused by
these compounds in clinical usage. The risk assessment for a number of
workplace or environmental chemicals has been developed from animal
models that have been used to study the mechanisms of these effects,
especially those of the heavy metals and some of the industrial
organic chemicals.
2.5.5 Special risk groups in humans
The marked variability in the response of any study population to
potentially nephrotoxic compounds establishes clearly that there are
groups at risk. There are a number of factors that could be
responsible for increasing the risk of nephrotoxicity. These include
existing renal disease, loss of renal parenchyma, high protein diet,
chemical exposure, predisposing factors, multiple myeloma, and other
conditions where there is an added level of protein excretion when the
kidney is under an additional work-load.
So far there has been relatively little interest in the
individuals that do not appear to be at risk from exposure to
potential nephrotoxins. While this is generally assumed to be the
result of an absence of predisposing factors, there may well be other
chemical, dietary, or disease considerations that provide a protective
effect. There is experimental evidence to suggest that a pre-existing
streptozotocin-induced diabetes and also poly-aspartic acid protect
against aminoglycoside-induced renal injury, and that fish oil diets
(high in omega-3 polyunsaturated fatty acids) reduce cyclosporin-A
nephrotoxicity. These factors could well be used to reduce the health
impact of nephrotoxicity.
2.5.6 Multichemical exposure
At present, there is virtually no information on the effects of
multichemical exposure in man and very little data on the effects of
more than one chemical administered simultaneously in animals.
Simultaneous exposure to several chemicals represents a major
toxicological problem, as man is generally exposed to more than one
substance in medicines, in food, and from environmental factors.
However, most experimental studies have investigated only single
chemicals. Interactions have been studied between mercuric chloride,
potassium dichromate, citrinin, and hexachloro-1,3-butadiene (HCBD)
in vivo and in vitro using a rat model (Baggett & Berndt 1984a,b;
1985). Dichromate potentiates the mercuric chloride effect, i.e. the
effects produced by the combination of metals are always greater than
the sum of the individual effects. There appears to be no simple
kinetic explanation, i.e. no enhanced renal accumulation of the
mercuric ion. The plasma membrane may be a site for the interaction of
these metal ions and could be the preliminary step that leads to
overall renal dysfunction and an ultimately enhanced acute renal
failure. Dichromate-citrinin and dichromate-HCBD interactions have
been demonstrated by alterations in urine flow, glucose excretion, and
transport processes. Some experimental data suggest that a
synergistic interaction may occur in analgesic nephropathy. The
mechanisms that underlie these interactions are not understood, and at
present there is no rational basis to predict them. Experimental
studies have shown that tubular cell injury, induced by
trichloroethylene and carbon tetrachloride, is potentiated by exposure
to polyhalogenated biphenyls, e.g., polychlorinated biphenyls (Kluwe
et al., 1979).
2.5.7 Renal functional reserve
The concept of renal functional reserve is a simple one in which
not all of the nephrons nor all of the cellular functions in a single
nephron are available or used at any one time (Friedlander et al.,
1989). Thus there is a buffering capacity in the kidney that can cope
with short-lived or protracted demands on function that exceed the
normal level. Part of this functional reserve is used to meet the
response to perturbation of the homeostatic system by water or
electrolyte loading. Most of the studies on and understanding of renal
functional reserve relate to changes in glomerular filtration rate and
renal blood flow. It is likely that additional approaches are needed
to test for other types of functional reserve.
2.5.8 The effects of chemicals on kidneys with pre-existing renal lesions
Although it is generally acknowledged that there are several
types of renal lesions that exist as a nephropathy in the general
population at a low but significant level (e.g., nephrotic syndrome),
little is known about how these pre-existing lesions affect the
response of the kidney to subsequent nephrotoxic insults.
2.5.8.1 Nephrotoxicity in the presence of renal and extrarenal disease
Safety screening is conducted on young, disease-free animals,
housed under optimal conditions, fed contamination-free, high-protein
food. By contrast, man is exposed to a variety of dietary and
environmental chemicals and to a poly-pharmacy of both prescribed and
self-administered medications over many years. In addition, screening
is generally undertaken using normal experimental animals. This may be
inappropriate because, with the exception of occupational and
environmental exposure, man is exposed to potentially nephrotoxic
therapeutic substances to treat disease. Pre-existing diseases can
have a profound effect on the direct or indirect response of the
kidney to handling chemicals (Bennett, 1986). There is an increasing
wealth of animal data to demonstrate that common clinical conditions
in man, such as hypertension, renal compromise, and renal ischaemic
injury, exacerbate cyclosporin nephrotoxicity and bacterial endotoxins
in animals and that systemic infection increases the sensitivity of
the kidney to aminoglycoside toxicity (Bergeron et al., 1982).
The role of pre-existing renal lesions on nephrotoxicity is
important, but there are few clear indications of what can be
predicted from existing clinical data and from animal studies.
Diabetes is generally associated with reduced renal function, diabetic
nephropathy, and renal papillary necrosis, and might be expected to
exacerbate chemical-associated nephrotoxicity. Untreated
streptozotocin-induced diabetes, however, protects rats against
gentamicin, low-dose cisplatin, and uranyl nitrate nephrotoxicity
(Teixeira et al., 1982; Vaamonde et al., 1984). Recent studies on the
acute effects of intravenous radio-contrast media on anaesthetized
diabetic rats were inconclusive (Reed et al., 1983, Golman & Almen,
1985, Leeming et al., 1985).
3. KIDNEY STRUCTURE AND FUNCTION
An in-depth review of kidney structure and function is beyond the
scope of this monograph. Only sufficient information will be given to
provide a general background against which nephrotoxicity can be
framed. A fuller insight into the complexities of the kidney in
health, disease, and nephrotoxicity has been described by Valtin
(1973), Orloff & Berliner (1973), Hook (1981), Porter (1982), Bach et
al. (1982, 1989), Bach & Lock (1982, 1985, 1987, 1989), Seldin &
Giebisch (1985), Brenner & Rector (1986).
3.1 Renal anatomy
The two kidneys are situated retro-peritoneally, on either side
of the vertebral column, and process 25% of the resting cardiac output
via an arterial blood supply. Much of the fluid and most of the
solutes in blood are filtered through the glomeruli into the proximal
part of the nephron (the functional unit of the kidney) from which
essential small molecules are reabsorbed. Numerous macro-molecules are
reabsorbed into the tubular cells by an endocytotic process and are
digested in tubular lysosomes. Many organic acids and bases (including
many drugs) are secreted (and reabsorbed) by carrier-mediated
processes located principally in the proximal tubule. There is some
secretion, mainly of waste solutes, from the blood into the distal
part of the nephron, and much of the water in which they are dissolved
is subsequently reabsorbed.
Each kidney is made up of a large number of nephrons, groups of
which unite to continue as collecting ducts or tubules, and these in
turn combine to make up the ducts of Bellini, which exit around the
papilla tip. The papilla opens into the calix, which is in continuity
with the renal pelvis, a funnel-shaped area that narrows to the
ureter. The continued production of urine, together with peristalsis
of the ureter, carries excreted waste to the bladder. The
morphophysiology of the kidney varies markedly between species.
Therefore, a generalized description will be provided, and only the
important differences between the rat (and other common laboratory
animals) and man will be described (Moffat, 1979).
3.1.1 Histology
Renal lesions occur in discrete anatomical regions. This
highlights the need to understand changes in terms of the biochemical
properties of the specifically affected region and its adjacent cells.
While haematoxylin and eosin staining and a number of other routinely
used staining procedures identify nephropathies and renal
degeneration, these are generally based on a relatively non-specific
assessment. The non-specificity of routine histopathology has, in
fact, been the strength of these methods in the preliminary
assessment of chemically induced nephropathies. It may, however, miss
some types of lesions and generally gives little information that can
help identify the mechanistic basis of a lesion.
Histochemical techniques can provide insight into primary and
secondary cellular mechanisms. One aspect of "histochemistry" is the
use of frozen segments of the nephron (Bach et al., 1987) and the
application of fluori-metric or radiochemical assays to measure the
activities and distribution of specific biochemical characteristics.
Microdissection generally fails to give a detailed localization of
these properties in relation to specific or individual cells. In
addition, the technique is difficult to apply to injured renal cells.
The most widely used histochemical approach is based on obtaining
frozen, fixed frozen, or fixed embedded sections of the kidney that
are then used with chromo- or fluoro-phores. These react with a
selected type of material. The types of materials that can be
visualized in section depends on their chemical structure (e.g.,
carbohydrate), enzymic activity (e.g., lactate dehydrogenase),
antigenicity (e.g., specific molecules), or physicochemical
properties (e.g., lipophilicity), some of which are shown in Fig. 2.
This approach also includes the distribution or incorporation of
radiolabelled molecules by their interaction with a photographic film
laid over the section (Bach et al., 1987). Immunohistochemical
techniques using labelled antibodies permit antigens to be localized
at the light microscopical and ultrastructural levels.
These microscopic histochemical techniques provide information on
the distribution at, or within, specific cells and their relative
activities, and have been used to define a variety of characteristics
of the kidney. It is important to stress that each method has its own
inherent strength and weakness, and that it may be difficult to relate
data from tissue sections to absolute biochemical measurements. This
is a consequence of the complex mixture of materials that are present
in tissue sections and the chemical changes that may take place in
these sections, particularly once they have been fixed. While the
biochemical characteristics of tissue in frozen sections are least
likely to be adversely affected, subtle and misleading alterations in
chemical properties can still occur. In addition, treatments to
conserve morphological features (fixation and embedding) alter these
values. It may also be difficult to be certain whether an increased
intensity of staining for a certain substance represents de novo
synthesis, unmasking, or the loss of factors that suppress staining.
These two techniques have proved to be very powerful in providing
information on the biochemical characteristics of cells and the
changes associated with renal injury. The in situ hybridization
techniques, using specific labelled nucleotidic probes, permit the
detection of protein synthesis at a subcellular level.
3.1.2 Enzyme histochemistry and quantification
Histochemical heterogeneity is evident in each part of the
nephron and varies between different species. In addition, the profile
of characteristics within any region of the kidney and nephron may be
related to sex and age. Antibodies are directed against unique or
novel characteristics along the nephron that are present on enzymes,








 glycoproteins, or other molecules associated with membranes or soluble
cytosolic constituents. The biochemical characteristic can be
visualized using enzyme, fluoro-phore, or radioactive labels (Bach et
al., 1987). Individual microdissected nephron segments have been
studied to determine the quantitative distribution of selected enzymes
(Table 4). Table 5 lists biochemical and function parameters for the
different nephron segments, showing their individual receptor
sensitivity.
3.1.3 Immunohistochemistry
The use of antibodies raised towards enzymes can help in the
study of isoenzyme distribution and factors affecting changes in their
distribution. For instance, aldolase-B monomers increase in the
proximal tubules of rats during renal maturation, but not in the
distal tubules. In contrast, aldolase-A monomers increase in the
distal tubules but not in the proximal tubules.
Immunohistochemical techniques demonstrate many functional
proteins at discrete locations in the kidney, including the glomeruli
and the tubular basement membrane, but there are few data on changes
in these proteins as a result of nephrotoxicity. A variety of
immunodeposits are associated with glomerulopathies including those
caused by heavy metals, but these appear to be T-cell mediated. They
are assessed by immunofluorescent monitoring in the glomerulus, but
this acts as a passive sieve and may be involved as a secondary
consequence of immunodeposition.
Histochemistry can also be used to show the distribution of a
number of oxidative enzymes. Cytochrome P-450 mixed-function oxidase
activities have been shown immuno-histochemically to be localized in
the proximal tubule, particularly in the S2 and S3 segments in the
Table 4. Distribution of enzymes in individual nephron segments
in various animal species
Enzyme Relative activitya Animal
Specific to the glomerulus
Adenosine deaminase rat
Specific to the proximal tubule
Glucose-6-phosphatase S1>S2>S3 rat
Fructose-1,6-bisphosphatase S1<S2>S3 rat
Phosphoenolpyruvate carboxykinase S1>S2>S3 rat, rabbit
Fructokinase S1=S2<S3 rat
Fructose-1-phosphate aldolase S1=S2>S3 rat
Glycerokinase S1=S2>S3 rat, rabbit
Glycerol-3-phosphate dehydrogenase S1=S2<S3 rat
Glutamine synthetase S3 rat
Alanine aminotransferase S1=S2<S3 rat
Gamma-glutamyltraspeptidase S1<S2<S3 rabbit
Gamma-glutamyl-cysteine synthetase S3 rat
Glutathione-S-transferase S1<S2<S3 rabbit
Cytochrome P-450 S1=S2>S3 rat, rabbit
Alanine aminopeptidase S1<S2<S3 rat
Alkaline phosphatase S1=S2=S3 rat
Leucine aminopeptidase S1<S2<S3 rat
D-Amino acid oxidase S1=S2<S3 rat
L-Hydroxy acid oxidase S1=S2<S3 rat
Fatty-acyl-CoA oxidase S1=S2<S3 rat
Choline oxidase S1=S2<S3 rat
25(OH)-D3-1alpha-hydroxylase S1<S2=S3 rat
(D3 deficient)
rabbit (fetus)
Table 4 (contd).
Enzyme Relative activitya Animal
Relatively specific to the proximal tubule
Glutamate dehydrogenase S1=S2>S3 rat, rabbit
Malic enzyme S1=S2<S3 rat
Trypsin-type protease S1>S2>S3 rat
ß-D-Galactosidase S1=S2<S3 rat
N-Acetyl-ß-D-glucosaminidase S1=S2>S3 rat
Xanthine oxidase S1>S2>S3 rat
Superoxide dismutase S1>S2>S3 rat
Specific to the lower nephron
Hexokinase MTAL=CTAL>DCT>CCD=MCD rat, rabbit
Phosphofructokinase MTAL=CTAL=CCD=MCD rat, rabbit
Pyruvate kinase MTAL=CTAL<DCT<CCD=MCD rat, rabbit
Kallikrein CNT rabbit
Relatively specific to the lower nephron
Fructose 1,6-bisphosphate aldolase MTAL=CTAL>DCT rat
Glycerol dehydrophosphate CCD<MCD rabbit
dehydrogenase
Table 4 (contd).
Enzyme Relative activitya Animal
Relatively specific to the
lower nephron (contd.)
Lactate dehydrogenase MTAL=CTAL-DCT=CCD rat, rabbit
Aspartate aminotransferase MTAL=CTAL>DCT rat
Citrate synthase MTAL=CTAL=DCT>CCD rat
Isocitrate dehydrogenase (NAD+) MTAL=CTAL=DCT=CCD rat
Na,K-ATPase MTAL<CTAL<DCT>CCD rat
a CCD = cortical collecting duct; CNT = connecting tubule; CTAL = cortical thick ascending
limb of Henle's loop; DCT = distal convoluted tubule; MCD = medullary collecting duct;
MTAL = medullary thick ascending limb of Henle's loop;
S1 = early proximal tubule; S2 = middle proximal tubule; S3 = late proximal tubule.
rat and rabbit. Large numbers of peroxisomes containing D-amino acid
oxidase and catalase are localized in the S3 portion of the proximal
tubule, but they are absent from the glomerulus and the distal
nephron. There is little immuno-histochemical data on the distribution
of molecules that are likely to protect renal cells from the effects
of reactive intermediates. Ligandin or glutathione- S-transferase B
is located in the proximal tubule of both animals and man and in the
thick limb of the loop of Henle in man. Catalase activity is greatest
in the proximal tubule (where it is localized in the peroxisomes),
less in the distal tubule, and very low in the glomerulus. Glutathione
has been shown by histochemistry to be localized in the proximal
convoluted tubule. However, there are some uncertainties as to what is
being assessed, since the reaction measures sulfhydryl groups and not
only glutathione. The distribution of at least one superoxide
dismutase isoenzyme shows a marked species difference between the dog
and rat, but is localized in the proximal tubules in both (Bach et
al., 1987).
3.2 The renal blood supply
Each kidney is supplied by a renal artery (a branch of the
abdominal aorta), which divides to form several interlobar arteries
(Fig. 3). These in turn give rise to the arcuate arteries, which run
between the cortex and medulla parallel to the kidney surface. Many
cortical radial arteries arise from the arcuate vessel and pass
through the cortex. Here a small amount of blood reaches the surface
to supply the kidney capsule, but most of the blood flow is directed
through branches that form the afferent arterioles to the glomeruli.
Each afferent arteriole breaks up to form the capillary plexus of the
glomerulus; this is drained into the efferent arteriole. The efferent
arterioles form two types of capillary networks
* In the "mid" and "superficial" cortical regions, they form the
peritubular capillaries surrounding proximal and distal tubules
(in the superficial regions some peritubular capillary networks
interlace the nephron from which they were derived, but such an
association appears to be the exception rather than the rule).
* In the juxtamedullary region (and some mid-cortical areas in man)
each efferent arteriole is directed into the medulla, where it
branches into the vasa recta bundles. Each bundle consists of up
to 30 descending vessels, the peripheral vessels of which give
rise to a highly branched capillary network in the outer medulla.
The core of the vasa recta bundle continues to the inner medulla
where it terminates in a capillary network (Beeuwkes, 1980).
The walls of all peritubular capillaries in the kidney are made
up of a thin fenestrated endothelium resting on a basal lamina. The
capillaries in the cortex generally open into the cortical radial
vein, from which blood flows via the arcuate vein to the renal vein
and finally to the inferior vena cava. The capillary plexuses in the
Table 5. Summary of nephron heterogeneitya
Anatomical regionb Biochemical featuresc Functional features
Glomerulus SF<JM Renin:(SF>JM) GFR:(SF<JM)
epithelium Adenosine-AC Mesangial contraction
(AII, histamine)
endothelium Histamine-AC (intra-mesangium)
mesangium Serotonin-AC (extra-mesangium)
ANP-GC ROM generation (intra-
and extra-mesangium)
ET-PGE2 (intra-mesangium)
AVP-PGE2 (intra- and extra-mesangium)
AII-PGE2 (intra- and extra-mesangium)
Proximal tubule Glucose carrier: J-glucose:
S1, S2, S3 (brush border) (S1>S2,S3)
Gluconeogenesis
(S1>S2>S3) J-V:(S1>S2>S3)
Cytochrome P-450: 1,25(OH)2D3 synthesis
(S1<S2>S3)
NADPH-cytochrome c reductase: J-V decreased by ANP
(S1<S2>S3)
Ammoniagenesis P-Cl/P-Na:(SF>JM)
(S1>S2>S3)
PTH-AC(S1>S3) PAHsecretion:
(S1<S2>S3)
Adenosine-AC J-aminoacid:
(S1>S2,S3)
Henle's loop
Thin: DTL<ATL AVP-AC(+ATL,-DTL) P-water:(DTL>ATL)
P-NaCl:(DTL<ATL)
P-urea:(DTL<ATL)
Table 5 (contd).
Anatomical regionb Biochemical featuresc Functional features
Thick: MTAL>CTAL AVP-AC:(MTAL>CTAL) AVP stimulation of
J-Cl: (MTAL>CTAL)
PTH-AC:(MTAL<CTAL) PTH stimulation of
J-Ca: (MTAL<CTAL)
SCT-AC:(MTAL>CTAL)
Tamm-Horsfall glycoprotein
PGE2 synthesis J-NaCl:(MTAL>CTAL)
(MTAL>CTAL) PG inhibition of J-Na:
(+MTAL, -CTAL)
EGF synthesis
J-Na decreased by ANP
Distal tubule
DCT:single cell type SCT-AC:(+DCT) SCT suppression of
Vt:(DCT)
CNT:multiple cell PTH-AC:(+CNT) PTH stimulation of
types J-Ca: (CNT)
AVP-AC:(+CNT) AVP suppression of
Vt:(CNT)
ISO-AC:(+CNT) ISO suppression of
Vt:(CNT)
Kallikrein:(+CNT)
Aldosterone binding Vt:(DCT < CNT)
K-secretion:(DCT<CNT)?
Collecting duct system
two cell types AVP-AC:(CCD > OMCD)
CCD (P.cell>I.cell) ISO-AC:(CCD > OMCD) Vt:(CCD > OMCD)
J-Na and J-V decreased
by ANP
Table 5 (contd).
Anatomical regionb Biochemical featuresc Functional features
OMCD (P.cell>I.cell) PG-AC:(CCD < OMCD) P-urea:(CCD,OMCD<IMCD)
PGE2 synthesis
(CCD<OMCD<IMCD)
Aldosterone binding
Adenosine-AC (CCD>OMCD)
ANP-GC (CCD<OMCD<IMCD)
IMCD J-V decreased by ANP
a Based on data obtained from the rabbit and rat kidney.
b Parts of the nephron: ATL = ascending thin limb of Henle's loop; CCD = cortical collecting
duct; CNT = connecting tubule; CTAL = cortical thick ascending limb of Henle's loop; DCT =
distal convoluted tubule; DTL = descending thin limb of Henle's loop; I.cell = intercalated
cell; IMCD = inner medullary collecting duct; JM = juxtamedullary nephron; MCT = medullary
collecting duct; MTAL = medullary thick ascending limb of Henle's loop; OMCD = outer
medullary collecting duct; P.cell = principal cell; S1 = early proximal tubule; S2 = middle
proximal tubule; S3 = late proximal tubule; SF = superficial nephrons.
c Hormone effects on nephron receptors: AII = angiotensin II; AC = adenylate cyclase; ANP =
atrial natriuretic peptide; AVP = arginine vasopressin; GC = guanylate cyclase; EGF =
epidermal growth factor; ET = endothelin; ISO = isoproterenol; PG = prostaglandin; PTH =
parathyroid hormone; ROM = reactive oxygen metabolites; SCT = salmon calcitonin.
Transport substances: PAH = para-aminohippurate; J-x = flux of substance x; J-V = flux of
fluid volume; P-x = permeability for substance x; Vt = transcellular voltage.
glycoproteins, or other molecules associated with membranes or soluble
cytosolic constituents. The biochemical characteristic can be
visualized using enzyme, fluoro-phore, or radioactive labels (Bach et
al., 1987). Individual microdissected nephron segments have been
studied to determine the quantitative distribution of selected enzymes
(Table 4). Table 5 lists biochemical and function parameters for the
different nephron segments, showing their individual receptor
sensitivity.
3.1.3 Immunohistochemistry
The use of antibodies raised towards enzymes can help in the
study of isoenzyme distribution and factors affecting changes in their
distribution. For instance, aldolase-B monomers increase in the
proximal tubules of rats during renal maturation, but not in the
distal tubules. In contrast, aldolase-A monomers increase in the
distal tubules but not in the proximal tubules.
Immunohistochemical techniques demonstrate many functional
proteins at discrete locations in the kidney, including the glomeruli
and the tubular basement membrane, but there are few data on changes
in these proteins as a result of nephrotoxicity. A variety of
immunodeposits are associated with glomerulopathies including those
caused by heavy metals, but these appear to be T-cell mediated. They
are assessed by immunofluorescent monitoring in the glomerulus, but
this acts as a passive sieve and may be involved as a secondary
consequence of immunodeposition.
Histochemistry can also be used to show the distribution of a
number of oxidative enzymes. Cytochrome P-450 mixed-function oxidase
activities have been shown immuno-histochemically to be localized in
the proximal tubule, particularly in the S2 and S3 segments in the
Table 4. Distribution of enzymes in individual nephron segments
in various animal species
Enzyme Relative activitya Animal
Specific to the glomerulus
Adenosine deaminase rat
Specific to the proximal tubule
Glucose-6-phosphatase S1>S2>S3 rat
Fructose-1,6-bisphosphatase S1<S2>S3 rat
Phosphoenolpyruvate carboxykinase S1>S2>S3 rat, rabbit
Fructokinase S1=S2<S3 rat
Fructose-1-phosphate aldolase S1=S2>S3 rat
Glycerokinase S1=S2>S3 rat, rabbit
Glycerol-3-phosphate dehydrogenase S1=S2<S3 rat
Glutamine synthetase S3 rat
Alanine aminotransferase S1=S2<S3 rat
Gamma-glutamyltraspeptidase S1<S2<S3 rabbit
Gamma-glutamyl-cysteine synthetase S3 rat
Glutathione-S-transferase S1<S2<S3 rabbit
Cytochrome P-450 S1=S2>S3 rat, rabbit
Alanine aminopeptidase S1<S2<S3 rat
Alkaline phosphatase S1=S2=S3 rat
Leucine aminopeptidase S1<S2<S3 rat
D-Amino acid oxidase S1=S2<S3 rat
L-Hydroxy acid oxidase S1=S2<S3 rat
Fatty-acyl-CoA oxidase S1=S2<S3 rat
Choline oxidase S1=S2<S3 rat
25(OH)-D3-1alpha-hydroxylase S1<S2=S3 rat
(D3 deficient)
rabbit (fetus)
Table 4 (contd).
Enzyme Relative activitya Animal
Relatively specific to the proximal tubule
Glutamate dehydrogenase S1=S2>S3 rat, rabbit
Malic enzyme S1=S2<S3 rat
Trypsin-type protease S1>S2>S3 rat
ß-D-Galactosidase S1=S2<S3 rat
N-Acetyl-ß-D-glucosaminidase S1=S2>S3 rat
Xanthine oxidase S1>S2>S3 rat
Superoxide dismutase S1>S2>S3 rat
Specific to the lower nephron
Hexokinase MTAL=CTAL>DCT>CCD=MCD rat, rabbit
Phosphofructokinase MTAL=CTAL=CCD=MCD rat, rabbit
Pyruvate kinase MTAL=CTAL<DCT<CCD=MCD rat, rabbit
Kallikrein CNT rabbit
Relatively specific to the lower nephron
Fructose 1,6-bisphosphate aldolase MTAL=CTAL>DCT rat
Glycerol dehydrophosphate CCD<MCD rabbit
dehydrogenase
Table 4 (contd).
Enzyme Relative activitya Animal
Relatively specific to the
lower nephron (contd.)
Lactate dehydrogenase MTAL=CTAL-DCT=CCD rat, rabbit
Aspartate aminotransferase MTAL=CTAL>DCT rat
Citrate synthase MTAL=CTAL=DCT>CCD rat
Isocitrate dehydrogenase (NAD+) MTAL=CTAL=DCT=CCD rat
Na,K-ATPase MTAL<CTAL<DCT>CCD rat
a CCD = cortical collecting duct; CNT = connecting tubule; CTAL = cortical thick ascending
limb of Henle's loop; DCT = distal convoluted tubule; MCD = medullary collecting duct;
MTAL = medullary thick ascending limb of Henle's loop;
S1 = early proximal tubule; S2 = middle proximal tubule; S3 = late proximal tubule.
rat and rabbit. Large numbers of peroxisomes containing D-amino acid
oxidase and catalase are localized in the S3 portion of the proximal
tubule, but they are absent from the glomerulus and the distal
nephron. There is little immuno-histochemical data on the distribution
of molecules that are likely to protect renal cells from the effects
of reactive intermediates. Ligandin or glutathione- S-transferase B
is located in the proximal tubule of both animals and man and in the
thick limb of the loop of Henle in man. Catalase activity is greatest
in the proximal tubule (where it is localized in the peroxisomes),
less in the distal tubule, and very low in the glomerulus. Glutathione
has been shown by histochemistry to be localized in the proximal
convoluted tubule. However, there are some uncertainties as to what is
being assessed, since the reaction measures sulfhydryl groups and not
only glutathione. The distribution of at least one superoxide
dismutase isoenzyme shows a marked species difference between the dog
and rat, but is localized in the proximal tubules in both (Bach et
al., 1987).
3.2 The renal blood supply
Each kidney is supplied by a renal artery (a branch of the
abdominal aorta), which divides to form several interlobar arteries
(Fig. 3). These in turn give rise to the arcuate arteries, which run
between the cortex and medulla parallel to the kidney surface. Many
cortical radial arteries arise from the arcuate vessel and pass
through the cortex. Here a small amount of blood reaches the surface
to supply the kidney capsule, but most of the blood flow is directed
through branches that form the afferent arterioles to the glomeruli.
Each afferent arteriole breaks up to form the capillary plexus of the
glomerulus; this is drained into the efferent arteriole. The efferent
arterioles form two types of capillary networks
* In the "mid" and "superficial" cortical regions, they form the
peritubular capillaries surrounding proximal and distal tubules
(in the superficial regions some peritubular capillary networks
interlace the nephron from which they were derived, but such an
association appears to be the exception rather than the rule).
* In the juxtamedullary region (and some mid-cortical areas in man)
each efferent arteriole is directed into the medulla, where it
branches into the vasa recta bundles. Each bundle consists of up
to 30 descending vessels, the peripheral vessels of which give
rise to a highly branched capillary network in the outer medulla.
The core of the vasa recta bundle continues to the inner medulla
where it terminates in a capillary network (Beeuwkes, 1980).
The walls of all peritubular capillaries in the kidney are made
up of a thin fenestrated endothelium resting on a basal lamina. The
capillaries in the cortex generally open into the cortical radial
vein, from which blood flows via the arcuate vein to the renal vein
and finally to the inferior vena cava. The capillary plexuses in the
Table 5. Summary of nephron heterogeneitya
Anatomical regionb Biochemical featuresc Functional features
Glomerulus SF<JM Renin:(SF>JM) GFR:(SF<JM)
epithelium Adenosine-AC Mesangial contraction
(AII, histamine)
endothelium Histamine-AC (intra-mesangium)
mesangium Serotonin-AC (extra-mesangium)
ANP-GC ROM generation (intra-
and extra-mesangium)
ET-PGE2 (intra-mesangium)
AVP-PGE2 (intra- and extra-mesangium)
AII-PGE2 (intra- and extra-mesangium)
Proximal tubule Glucose carrier: J-glucose:
S1, S2, S3 (brush border) (S1>S2,S3)
Gluconeogenesis
(S1>S2>S3) J-V:(S1>S2>S3)
Cytochrome P-450: 1,25(OH)2D3 synthesis
(S1<S2>S3)
NADPH-cytochrome c reductase: J-V decreased by ANP
(S1<S2>S3)
Ammoniagenesis P-Cl/P-Na:(SF>JM)
(S1>S2>S3)
PTH-AC(S1>S3) PAHsecretion:
(S1<S2>S3)
Adenosine-AC J-aminoacid:
(S1>S2,S3)
Henle's loop
Thin: DTL<ATL AVP-AC(+ATL,-DTL) P-water:(DTL>ATL)
P-NaCl:(DTL<ATL)
P-urea:(DTL<ATL)
Table 5 (contd).
Anatomical regionb Biochemical featuresc Functional features
Thick: MTAL>CTAL AVP-AC:(MTAL>CTAL) AVP stimulation of
J-Cl: (MTAL>CTAL)
PTH-AC:(MTAL<CTAL) PTH stimulation of
J-Ca: (MTAL<CTAL)
SCT-AC:(MTAL>CTAL)
Tamm-Horsfall glycoprotein
PGE2 synthesis J-NaCl:(MTAL>CTAL)
(MTAL>CTAL) PG inhibition of J-Na:
(+MTAL, -CTAL)
EGF synthesis
J-Na decreased by ANP
Distal tubule
DCT:single cell type SCT-AC:(+DCT) SCT suppression of
Vt:(DCT)
CNT:multiple cell PTH-AC:(+CNT) PTH stimulation of
types J-Ca: (CNT)
AVP-AC:(+CNT) AVP suppression of
Vt:(CNT)
ISO-AC:(+CNT) ISO suppression of
Vt:(CNT)
Kallikrein:(+CNT)
Aldosterone binding Vt:(DCT < CNT)
K-secretion:(DCT<CNT)?
Collecting duct system
two cell types AVP-AC:(CCD > OMCD)
CCD (P.cell>I.cell) ISO-AC:(CCD > OMCD) Vt:(CCD > OMCD)
J-Na and J-V decreased
by ANP
Table 5 (contd).
Anatomical regionb Biochemical featuresc Functional features
OMCD (P.cell>I.cell) PG-AC:(CCD < OMCD) P-urea:(CCD,OMCD<IMCD)
PGE2 synthesis
(CCD<OMCD<IMCD)
Aldosterone binding
Adenosine-AC (CCD>OMCD)
ANP-GC (CCD<OMCD<IMCD)
IMCD J-V decreased by ANP
a Based on data obtained from the rabbit and rat kidney.
b Parts of the nephron: ATL = ascending thin limb of Henle's loop; CCD = cortical collecting
duct; CNT = connecting tubule; CTAL = cortical thick ascending limb of Henle's loop; DCT =
distal convoluted tubule; DTL = descending thin limb of Henle's loop; I.cell = intercalated
cell; IMCD = inner medullary collecting duct; JM = juxtamedullary nephron; MCT = medullary
collecting duct; MTAL = medullary thick ascending limb of Henle's loop; OMCD = outer
medullary collecting duct; P.cell = principal cell; S1 = early proximal tubule; S2 = middle
proximal tubule; S3 = late proximal tubule; SF = superficial nephrons.
c Hormone effects on nephron receptors: AII = angiotensin II; AC = adenylate cyclase; ANP =
atrial natriuretic peptide; AVP = arginine vasopressin; GC = guanylate cyclase; EGF =
epidermal growth factor; ET = endothelin; ISO = isoproterenol; PG = prostaglandin; PTH =
parathyroid hormone; ROM = reactive oxygen metabolites; SCT = salmon calcitonin.
Transport substances: PAH = para-aminohippurate; J-x = flux of substance x; J-V = flux of
fluid volume; P-x = permeability for substance x; Vt = transcellular voltage.
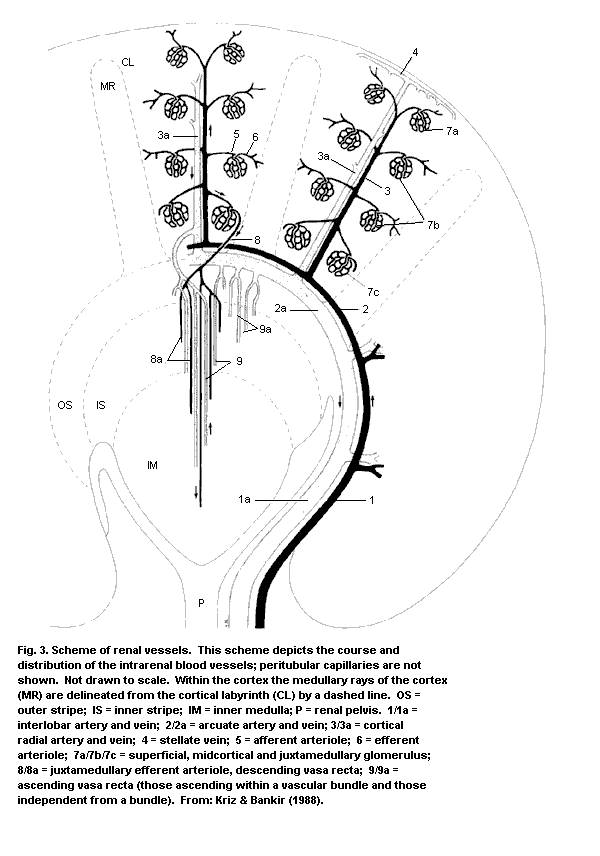 medulla drain into the ascending vasa recta, which join the arcuate
veins. The arterial branches are terminal, without anastomoses. In
contrast, the veins are richly anastomosed.
There is a well-defined structural relationship between the vasa
recta bundles and the nephrons in the outer medulla, at least in the
animals that have been studied. A central core (consisting of
descending and ascending vasa recta) is surrounded by a peripheral
layer consisting of a closely intermingled ascending vasa recta and
the descending thin limbs of the loops of Henle. Between these bundles
are the thick ascending limbs of the loops of Henle, some descending
limbs, and the collecting ducts. Within the bundles both ascending
and descending vasa recta are in intimate contact with each other
(rather than with the same type of vessel). There are more ascending
vessels, all of larger diameter, than descending ones, and this
increased volume capacity relates to the removal of excess water from
the interstitium and the maintenance of the medullary osmotic gradient
shown in Fig. 4.
Many of the "major" and "minor" blood vessels in the kidney have
either smooth muscle cells as an integral part of their structure, or
other cells that may have a contractile function. Thus most of the
intrarenal vascular system has both adrenergic and cholinergic
innervation. Intrarenal blood flow, the factors which alter it, and
its effects on renal function are poorly understood. Although there
appear to be the facilities for a direct and effective perfusion of
the medulla from the arcuate artery, this does not seem to occur
(Moffat, 1979; Beeuwkes, 1980).
3.2.1 Renal haemodynamics
The measurement of total blood flow through the kidneys can be
measured relatively easily using modern techniques (Grunfeld et al.,
1971; Pearson, 1979). Defining the zonal blood flow has given some
conflicting results, but assessing regional blood flow within the
kidney is fraught with difficulties and is subject to varied
interpretations (Grandchamp et al., 1971; Grunfeld et al., 1971;
Pearson, 1979; Aukland, 1980; Knox et al., 1984). There are, however,
consistent data (derived from a number of fundamentally different
techniques) to show that intrarenal blood flow is greatest in the
cortex (80-85% of total renal flow) and that it decreases through the
juxtamedullary region to less than 10% of the total renal flow in the
medulla. It must be stressed that, although the medulla is poorly
perfused in comparison to the rest of the kidney, it is, nonetheless
(because of the 25% resting cardiac output and therefore abundant
renal blood flow), a well-perfused tissue. According to Thurau (1964),
the medullary blood flow is about 15 times that of resting muscle and
the same as that of the brain. In addition, the capillary volume
fraction of the medulla is more than twice that of the renal cortex
(Beeuwkes, 1980). Despite this, there is considerable variation in
tissue pO2 in the kidney; a marked decrease in pO2 levels is seen
with increasing tissue depth (Brezis et al., 1984).
3.3 The nephron
The kidney is divided into three main regions, cortex (outer),
medulla (inner), and pelvis (Fig. 5). Within the cortex arise the
renal corpuscles, defined as superficial, midcortical or
juxtamedullary depending on the anatomical location of the renal
corpuscle in the cortex. The nephron is the functional unit of the
kidney and consists of a continuous tube of highly specialized
heterogeneous cells, which show sub-specialization along the length of
nephrons and between them. There are marked structural and functional
differences between the nephrons arising in the cortex and those
arising in the juxtamedullary regions. The total number of nephrons
varies between different species and within any one species as a
function of age. The macroscopic differentiation of the kidney into
distinct zones arises not only from the regional vascularity but also
from the way different functional parts of the nephron are arranged
within the kidney. A more detailed account of the ultrastructure of
the morphologically definable regions of the nephron and their
functional inter-relationship has been provided by Moffat (1981,
1982), Bohman (1980), and Maunsbach et al. (1980). Recently the
nephron nomenclature has been standardized by the Renal Commission of
the International Union of Physical Sciences (Kriz & Bankir, 1988).
This is summarized in Figures 3, 5, and 6, and Table 6.
3.3.1 Cellular heterogeneity and cell-cell interaction
There are well over 20 morphologically different cell types
(based on light microscopy alone) in the kidney, and when
histochemical and immunohistochemical methods are applied to renal
tissue sections the diversity of cell types is even more apparent. The
spectrum of biochemical (and structural and functional)
characteristics in these cells demonstrates the very marked
heterogeneity that is the hallmark of the kidney. It is well
established that the expression of many of these biochemical
characteristics is an integral of the functions of that particular
region of the kidney, and there is the potential to change the
expression of these characteristics in terms of the demands on the
kidney. These include both water and electrolytes, dietary factors,
and chemicals with pharmacological and toxic effects, or may be as a
result of chemical and other types of injury. More importantly, the
characteristics of a cell may make it either resistant or sensitive to
the target selective toxicity of a chemical.
medulla drain into the ascending vasa recta, which join the arcuate
veins. The arterial branches are terminal, without anastomoses. In
contrast, the veins are richly anastomosed.
There is a well-defined structural relationship between the vasa
recta bundles and the nephrons in the outer medulla, at least in the
animals that have been studied. A central core (consisting of
descending and ascending vasa recta) is surrounded by a peripheral
layer consisting of a closely intermingled ascending vasa recta and
the descending thin limbs of the loops of Henle. Between these bundles
are the thick ascending limbs of the loops of Henle, some descending
limbs, and the collecting ducts. Within the bundles both ascending
and descending vasa recta are in intimate contact with each other
(rather than with the same type of vessel). There are more ascending
vessels, all of larger diameter, than descending ones, and this
increased volume capacity relates to the removal of excess water from
the interstitium and the maintenance of the medullary osmotic gradient
shown in Fig. 4.
Many of the "major" and "minor" blood vessels in the kidney have
either smooth muscle cells as an integral part of their structure, or
other cells that may have a contractile function. Thus most of the
intrarenal vascular system has both adrenergic and cholinergic
innervation. Intrarenal blood flow, the factors which alter it, and
its effects on renal function are poorly understood. Although there
appear to be the facilities for a direct and effective perfusion of
the medulla from the arcuate artery, this does not seem to occur
(Moffat, 1979; Beeuwkes, 1980).
3.2.1 Renal haemodynamics
The measurement of total blood flow through the kidneys can be
measured relatively easily using modern techniques (Grunfeld et al.,
1971; Pearson, 1979). Defining the zonal blood flow has given some
conflicting results, but assessing regional blood flow within the
kidney is fraught with difficulties and is subject to varied
interpretations (Grandchamp et al., 1971; Grunfeld et al., 1971;
Pearson, 1979; Aukland, 1980; Knox et al., 1984). There are, however,
consistent data (derived from a number of fundamentally different
techniques) to show that intrarenal blood flow is greatest in the
cortex (80-85% of total renal flow) and that it decreases through the
juxtamedullary region to less than 10% of the total renal flow in the
medulla. It must be stressed that, although the medulla is poorly
perfused in comparison to the rest of the kidney, it is, nonetheless
(because of the 25% resting cardiac output and therefore abundant
renal blood flow), a well-perfused tissue. According to Thurau (1964),
the medullary blood flow is about 15 times that of resting muscle and
the same as that of the brain. In addition, the capillary volume
fraction of the medulla is more than twice that of the renal cortex
(Beeuwkes, 1980). Despite this, there is considerable variation in
tissue pO2 in the kidney; a marked decrease in pO2 levels is seen
with increasing tissue depth (Brezis et al., 1984).
3.3 The nephron
The kidney is divided into three main regions, cortex (outer),
medulla (inner), and pelvis (Fig. 5). Within the cortex arise the
renal corpuscles, defined as superficial, midcortical or
juxtamedullary depending on the anatomical location of the renal
corpuscle in the cortex. The nephron is the functional unit of the
kidney and consists of a continuous tube of highly specialized
heterogeneous cells, which show sub-specialization along the length of
nephrons and between them. There are marked structural and functional
differences between the nephrons arising in the cortex and those
arising in the juxtamedullary regions. The total number of nephrons
varies between different species and within any one species as a
function of age. The macroscopic differentiation of the kidney into
distinct zones arises not only from the regional vascularity but also
from the way different functional parts of the nephron are arranged
within the kidney. A more detailed account of the ultrastructure of
the morphologically definable regions of the nephron and their
functional inter-relationship has been provided by Moffat (1981,
1982), Bohman (1980), and Maunsbach et al. (1980). Recently the
nephron nomenclature has been standardized by the Renal Commission of
the International Union of Physical Sciences (Kriz & Bankir, 1988).
This is summarized in Figures 3, 5, and 6, and Table 6.
3.3.1 Cellular heterogeneity and cell-cell interaction
There are well over 20 morphologically different cell types
(based on light microscopy alone) in the kidney, and when
histochemical and immunohistochemical methods are applied to renal
tissue sections the diversity of cell types is even more apparent. The
spectrum of biochemical (and structural and functional)
characteristics in these cells demonstrates the very marked
heterogeneity that is the hallmark of the kidney. It is well
established that the expression of many of these biochemical
characteristics is an integral of the functions of that particular
region of the kidney, and there is the potential to change the
expression of these characteristics in terms of the demands on the
kidney. These include both water and electrolytes, dietary factors,
and chemicals with pharmacological and toxic effects, or may be as a
result of chemical and other types of injury. More importantly, the
characteristics of a cell may make it either resistant or sensitive to
the target selective toxicity of a chemical.
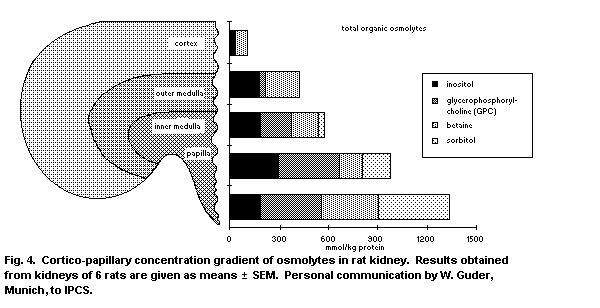 3.3.2 The glomerulus
The glomerulus forms the initial part of the nephron and
functions as a relatively poorly selective macro-molecular exclusion
filter to the hydrostatic pressure of the blood. The number of
glomeruli is, in general, related to the mass of the species, and the
size of each glomerulus depends, among other factors, on the
environmental water balance. Three anatomically distinct types of
glomeruli can be identified: those in the superficial cortex, which
are part of the superficial nephrons; those arising in the midcortical
area; and those ofjuxtamedullary origin, which continue as nephrons
that loop down into the medulla. The structure of the glomerulus is
complex (Fig. 7) and has only been defined using scanning and
transmission electron microscopy (Maunsbach et al., 1980; Moffat 1981,
1982).
The glomerular "tuft" is made up of a number of capillary
branches that arise from the afferent arteriole, anastomose, and drain
to the efferent arteriole. There are also communicating vessels
between the branch capillaries. The fenestrated endothelium cannot
prevent plasma molecules from leaving the lumen, but a negatively
charged cell coat imparts some selective permeability. The capillaries
are in direct contact with the glomerular basement membrane (or basal
lamina), which, when viewed under the electron microscope, can be
divided into three layers: the lamina rara interna on the endothelial
side; the central lamina densa; and the lamina rara externa, which is
in direct contact with the epithelial cells (the podocytes). The basal
lamina contains collagen (mostly Type IV) and sialic acid and is rich
in glycosaminoglycans, mainly heparan sulfate (Kanwar & Farquhar,
1979), which provides a strongly anionic macromolecular filtration
barrier.
The capillary tuft (ensheathed in its basal lamina) is surrounded
by a number of podocytes, each of which gives rise to several primary
processes (trabeculae). These in turn give rise to secondary
processes, and, finally, to numerous tertiary foot processes that are
embedded in the lamina rara externa.
The foot processes of one podocyte interdigitate with those of an
adjacent epithelial cell for adjacent trabeculae. The surfaces of the
podocytes are covered by a strongly anionic cell coat that extends to
the spaces between the foot processes. It is through these spaces that
the glomerular filtrate reaches the lumen of Bowman's space. Thus, the
podocyte provides a structural support for the basal lamina and may
also serve to provide additional anionic forces for the process of
biological ultrafiltration. It has been suggested that podocytes may
have phagocytic properties and undergo contraction (Moffat, 1981).
3.3.2 The glomerulus
The glomerulus forms the initial part of the nephron and
functions as a relatively poorly selective macro-molecular exclusion
filter to the hydrostatic pressure of the blood. The number of
glomeruli is, in general, related to the mass of the species, and the
size of each glomerulus depends, among other factors, on the
environmental water balance. Three anatomically distinct types of
glomeruli can be identified: those in the superficial cortex, which
are part of the superficial nephrons; those arising in the midcortical
area; and those ofjuxtamedullary origin, which continue as nephrons
that loop down into the medulla. The structure of the glomerulus is
complex (Fig. 7) and has only been defined using scanning and
transmission electron microscopy (Maunsbach et al., 1980; Moffat 1981,
1982).
The glomerular "tuft" is made up of a number of capillary
branches that arise from the afferent arteriole, anastomose, and drain
to the efferent arteriole. There are also communicating vessels
between the branch capillaries. The fenestrated endothelium cannot
prevent plasma molecules from leaving the lumen, but a negatively
charged cell coat imparts some selective permeability. The capillaries
are in direct contact with the glomerular basement membrane (or basal
lamina), which, when viewed under the electron microscope, can be
divided into three layers: the lamina rara interna on the endothelial
side; the central lamina densa; and the lamina rara externa, which is
in direct contact with the epithelial cells (the podocytes). The basal
lamina contains collagen (mostly Type IV) and sialic acid and is rich
in glycosaminoglycans, mainly heparan sulfate (Kanwar & Farquhar,
1979), which provides a strongly anionic macromolecular filtration
barrier.
The capillary tuft (ensheathed in its basal lamina) is surrounded
by a number of podocytes, each of which gives rise to several primary
processes (trabeculae). These in turn give rise to secondary
processes, and, finally, to numerous tertiary foot processes that are
embedded in the lamina rara externa.
The foot processes of one podocyte interdigitate with those of an
adjacent epithelial cell for adjacent trabeculae. The surfaces of the
podocytes are covered by a strongly anionic cell coat that extends to
the spaces between the foot processes. It is through these spaces that
the glomerular filtrate reaches the lumen of Bowman's space. Thus, the
podocyte provides a structural support for the basal lamina and may
also serve to provide additional anionic forces for the process of
biological ultrafiltration. It has been suggested that podocytes may
have phagocytic properties and undergo contraction (Moffat, 1981).
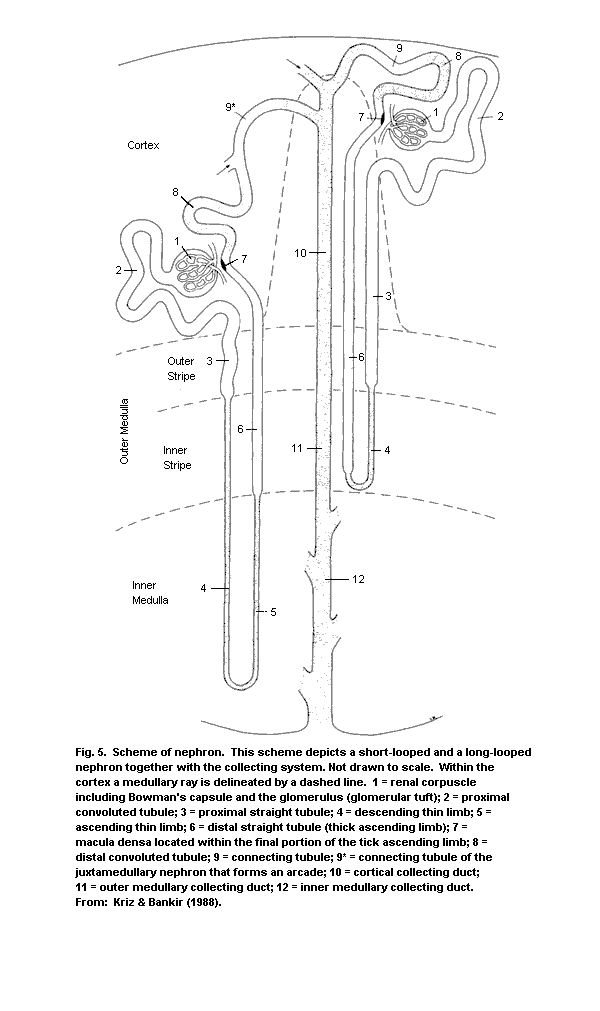
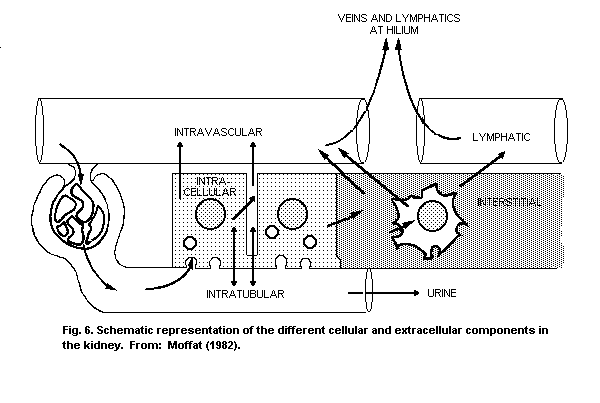 Table 6. Summary of nomenclature of segments and cells of the renal tubule (From: Kriz & Bankir, 1988). A continuous serpentine
arrow ( ) means that the transition between the two structures is gradual. An interrupted serpentine arrow ( ) means that the
transition is gradual in some species, abrupt in others. Abbreviations marked by a star were introduced by Morel and coworkers
(Kidney Int 9: 264, 1976). They mean: DCTa = Distal convoluted tubule, initial portion; DCTb = Distal convoluted tubule, bright
portion; DCTg = Distal convoluted tubule, granular portion; DCTI = Distal convoluted tubule, light portion; CCTg = Cortical
collecting tubule, granular portion; CCTI = Cortical collecting tubule, light portion
Micro-
Anatomical Main division Subdivisions Segmentation Abbre- Cell types Other frequently used
terms viation denominations
Proximal PROXIMAL pars convoluta Proximal S 1 cells
convolution TUBLULE or Convoluted S 1 - segment PCT P 1 segment PT
convulated part Tubule
S 2 - segment S 2 cells P 2 segment
pars recta Proximal
or Straight S 3 - segment PST P 3 segment PR
straight part Tubule S 3 cells
Loop of INTERMEDIATE pars descendens Descending DTL DTL cells
Henle TUBULE or Thin of short loops Type 1 Short Descending Thin Limb of
descending part Limb of long loops Henle's loop (SDL)
upper part Type 2 Long Descending Thin Limb,
lower part upper part (LDLu)
Type 3 Long Descending Thin Limb,
lower part (LDLl)
pre-bend segment ATL cells
pars of ascendens
or Ascending Thin Limb ATL Type 4 Tal Thin Ascending Limb
ascending part (of long loops only)
ascending part
Table 6 (contd.)
Micro-
Anatomical Main division Subdivisions Segmentation Abbre- Cell types Other frequently used
terms viation denominations
DISTAL pars recta Distal Medullary D MTAL DST or Tal MAL Thick Ascending Limb of
TUBULE or Straight straight S cells Henle's Loop
straight part Tubule part T mTALH Medullary Thick Limb
or Cortical o CAL Cortical Thick Limb
Thick straight r CTAL cTALH (incl. Macula Densa)
Ascending part Macula Densa T MD MD MD
Limb postmacular A cells
segment L DCTa* early
Distal pars convoluta DCT cells distal Distal Tubule
convolution or Distal Convoluted Tubule DCT (+ IC cells) DCTb* tubule
convoluted part
COLLECTING CONNECTING TUBULE CNT cells
SYSTEM CNT + DCTg* late Connecting
IC cells CCTg* distal Segment
Collecting COLLECTING CD cells tubule
duct DUCT Cortical Collecting Duct CCD = principal
cells DCTl* Initial
Collecting
Tubule
= light cells CCTl* Cortical
+ Collecting
IC cells Tubule (CCT)
= intercalated
cells
= mitochondria-
rich cells Outer Medullary Collecting
Outer Medullary OMCD = carboanhyd- Tubule (OMCT)
Collecting Duct rase-rich cells
= dark cells
CD cells Inner Medullary Collecting
Inner Medullary IMCD = prinicpal Tubule (IMCT)
Collecting Duct cells Papillary Collecting Duct
(PCD) or ducts of Bellini
The axial regions of each glomerulus contain mesangial cells.
Information on the structure and possible functions of mesangial cells
has been reviewed by Moffat (1981). In brief, they undergo contraction
and may thus control glomerular blood flow via biogenic amine or
hormonal control. Of equal importance is the observation that these
cells take up large molecules (such as colloids, immune complexes, and
protein aggregates), which may eventually be disposed of via the renal
lymphatic system.
The driving force for filtration is provided by the glomerular
capillary hydrostatic pressure (which is controlled mainly by the
vascular tone of the afferent and efferent arterioles), minus both the
plasma osmotic pressure and the hydrostatic pressure in the Bowman's
space. The resulting "effective filtration pressure" across the basal
lamina is about 1.2-2.0 kPa (10-15 mmHg). Selective filtration is
achieved primarily on the basis of size restriction by the basement
membrane, which impedes the passage of macromolecules with an
effective radius greater than 1.8 nm and completely prevents the
filtration of macromolecules with an effective radius greater than 4.5
nm. In addition the presence of fixed negative charges on the
endothelial, epithelial, and basement membranes hinders the filtration
of anionic macromolecules while facilitating the passage of cationic
macromolecules. The selectivity of filtration is, in part, a
consequence of the anionic nature of the basement membrane, which
blocks or slows the passage of negatively charged or neutral
macromolecules and leaves those carrying a cationic charge and small
molecules (irrespective of charge) to pass unimpeded.
3.3.3 The proximal tubule
The proximal tubule is found only in the cortex or subcortical
zones of the kidney. Anatomically each proximal tubule can be divided
into the convoluted portion (pars convoluta) and the shorter straight
descending portion (the pars recta), which then continues to become
the descending limb of the loop of Henle. It may be sub-divided, by a
number of morphological and functional features, into three segments,
S1, S2, and S3.
The proximal tubule plays a decisive role in maintaining
homeostasis. This is achieved when sodium and chloride ions flux from
the tubule lumen to the peritubular capillaries under the control of
a number of processes such as nonspecific electrophysiological
gradients and selective active transport mechanisms. Water follows the
ions by osmotic effects. In addition, hydrostatic pressure,
attributable to the presence of both proteins and glycosamino-glycans
(Wolgast et al., 1973), contributes to water movement from the
epithelial cell to the interstitium and thence, by an osmotic
gradient, into the capillaries (Valtin, 1973). The flux of ions within
the proximal tubule, including the absorption and secretion of
Table 6. Summary of nomenclature of segments and cells of the renal tubule (From: Kriz & Bankir, 1988). A continuous serpentine
arrow ( ) means that the transition between the two structures is gradual. An interrupted serpentine arrow ( ) means that the
transition is gradual in some species, abrupt in others. Abbreviations marked by a star were introduced by Morel and coworkers
(Kidney Int 9: 264, 1976). They mean: DCTa = Distal convoluted tubule, initial portion; DCTb = Distal convoluted tubule, bright
portion; DCTg = Distal convoluted tubule, granular portion; DCTI = Distal convoluted tubule, light portion; CCTg = Cortical
collecting tubule, granular portion; CCTI = Cortical collecting tubule, light portion
Micro-
Anatomical Main division Subdivisions Segmentation Abbre- Cell types Other frequently used
terms viation denominations
Proximal PROXIMAL pars convoluta Proximal S 1 cells
convolution TUBLULE or Convoluted S 1 - segment PCT P 1 segment PT
convulated part Tubule
S 2 - segment S 2 cells P 2 segment
pars recta Proximal
or Straight S 3 - segment PST P 3 segment PR
straight part Tubule S 3 cells
Loop of INTERMEDIATE pars descendens Descending DTL DTL cells
Henle TUBULE or Thin of short loops Type 1 Short Descending Thin Limb of
descending part Limb of long loops Henle's loop (SDL)
upper part Type 2 Long Descending Thin Limb,
lower part upper part (LDLu)
Type 3 Long Descending Thin Limb,
lower part (LDLl)
pre-bend segment ATL cells
pars of ascendens
or Ascending Thin Limb ATL Type 4 Tal Thin Ascending Limb
ascending part (of long loops only)
ascending part
Table 6 (contd.)
Micro-
Anatomical Main division Subdivisions Segmentation Abbre- Cell types Other frequently used
terms viation denominations
DISTAL pars recta Distal Medullary D MTAL DST or Tal MAL Thick Ascending Limb of
TUBULE or Straight straight S cells Henle's Loop
straight part Tubule part T mTALH Medullary Thick Limb
or Cortical o CAL Cortical Thick Limb
Thick straight r CTAL cTALH (incl. Macula Densa)
Ascending part Macula Densa T MD MD MD
Limb postmacular A cells
segment L DCTa* early
Distal pars convoluta DCT cells distal Distal Tubule
convolution or Distal Convoluted Tubule DCT (+ IC cells) DCTb* tubule
convoluted part
COLLECTING CONNECTING TUBULE CNT cells
SYSTEM CNT + DCTg* late Connecting
IC cells CCTg* distal Segment
Collecting COLLECTING CD cells tubule
duct DUCT Cortical Collecting Duct CCD = principal
cells DCTl* Initial
Collecting
Tubule
= light cells CCTl* Cortical
+ Collecting
IC cells Tubule (CCT)
= intercalated
cells
= mitochondria-
rich cells Outer Medullary Collecting
Outer Medullary OMCD = carboanhyd- Tubule (OMCT)
Collecting Duct rase-rich cells
= dark cells
CD cells Inner Medullary Collecting
Inner Medullary IMCD = prinicpal Tubule (IMCT)
Collecting Duct cells Papillary Collecting Duct
(PCD) or ducts of Bellini
The axial regions of each glomerulus contain mesangial cells.
Information on the structure and possible functions of mesangial cells
has been reviewed by Moffat (1981). In brief, they undergo contraction
and may thus control glomerular blood flow via biogenic amine or
hormonal control. Of equal importance is the observation that these
cells take up large molecules (such as colloids, immune complexes, and
protein aggregates), which may eventually be disposed of via the renal
lymphatic system.
The driving force for filtration is provided by the glomerular
capillary hydrostatic pressure (which is controlled mainly by the
vascular tone of the afferent and efferent arterioles), minus both the
plasma osmotic pressure and the hydrostatic pressure in the Bowman's
space. The resulting "effective filtration pressure" across the basal
lamina is about 1.2-2.0 kPa (10-15 mmHg). Selective filtration is
achieved primarily on the basis of size restriction by the basement
membrane, which impedes the passage of macromolecules with an
effective radius greater than 1.8 nm and completely prevents the
filtration of macromolecules with an effective radius greater than 4.5
nm. In addition the presence of fixed negative charges on the
endothelial, epithelial, and basement membranes hinders the filtration
of anionic macromolecules while facilitating the passage of cationic
macromolecules. The selectivity of filtration is, in part, a
consequence of the anionic nature of the basement membrane, which
blocks or slows the passage of negatively charged or neutral
macromolecules and leaves those carrying a cationic charge and small
molecules (irrespective of charge) to pass unimpeded.
3.3.3 The proximal tubule
The proximal tubule is found only in the cortex or subcortical
zones of the kidney. Anatomically each proximal tubule can be divided
into the convoluted portion (pars convoluta) and the shorter straight
descending portion (the pars recta), which then continues to become
the descending limb of the loop of Henle. It may be sub-divided, by a
number of morphological and functional features, into three segments,
S1, S2, and S3.
The proximal tubule plays a decisive role in maintaining
homeostasis. This is achieved when sodium and chloride ions flux from
the tubule lumen to the peritubular capillaries under the control of
a number of processes such as nonspecific electrophysiological
gradients and selective active transport mechanisms. Water follows the
ions by osmotic effects. In addition, hydrostatic pressure,
attributable to the presence of both proteins and glycosamino-glycans
(Wolgast et al., 1973), contributes to water movement from the
epithelial cell to the interstitium and thence, by an osmotic
gradient, into the capillaries (Valtin, 1973). The flux of ions within
the proximal tubule, including the absorption and secretion of
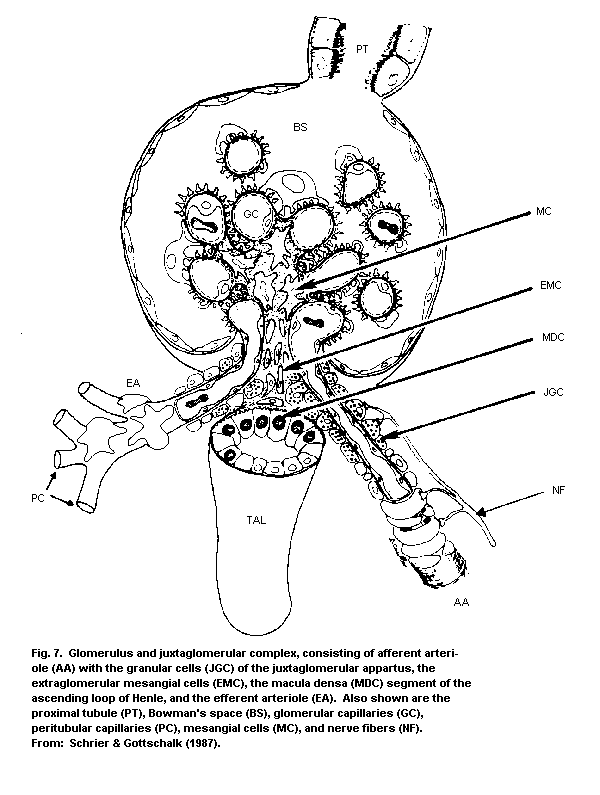 HCO3- and H+ and the "lumen trapping" of ammonium ions, controls
renal acid-base regulation (Valtin, 1973).
Those proteins that have passed from Bowman's capsule (a
significant amount of albumin in the case of normal rats) are
reabsorbed in the proximal tubule by pinocytotic removal from the base
of the microvillous brush border into the epithelial cells. The
vesicles thus formed combine, form protein-filled vacuoles, and fuse
with lysosomes, from which the digestion products of the protein
diffuse, eventually, to the capillary system or are used in the
metabolic processes of the cell.
There are, in addition, other absorptive and secretory
mechanisms. These include the co-transport process that reabsorbs
glucose and the secretion of both acidic and basic organic compounds
(Valtin, 1973; Orloff & Berliner, 1973; Brenner & Rector, 1986;
Berndt, 1989).
3.3.4 The medulla
The medulla differs from the cortex (Fig. 5 and Fig. 8) both at
the macroscopic and at the microscopic levels. This region can be
divided into the outer medulla (which is made up of the thin
descending and the thick ascending limbs of the loops of Henle,
collecting ducts, the vasa recta, and a dense capillary network) and
the inner medulla, the free part of which is referred to as the
"papilla" (although some researchers apply that name only to the apex
of this region). The inner medulla contains the thin limbs of the
loops of Henle, collecting ducts, the vasa recta, and a diffuse
network of capillaries. Packed into the spaces between these
structures are interstitial cells embedded in a matrix rich in
glycosaminoglycans.
The collecting ducts terminate as the ducts of Bellini around the
tip of the papilla. Whereas the mouse, gerbil, rat, guinea-pig,
rabbit, dog, cat, and primate kidneys have only a single papilla, the
pig and man have multi-papillate kidneys. There are between 9 and 20
papillae in each human kidney (Burry et al., 1977), of which there are
two anatomically distinguishable types. The conical non-refluxing
papillae, where the surface orifices of the ducts of Bellini are
slit-like, close when there is an increase in the "back-pressure" of
urine from the bladder and so prevent intrarenal reflux when reflux
occurs from the bladder. These papillae occur predominantly in the
mid zone. The refluxing papillae occur predominantly in the polar
regions, and, as they have flattened tips, the collecting duct
orifices are wide and prone to retrograde flow of urine into the
tubules during vesico-ureteric reflux (Ransley & Risdon, 1979). The
microscopic and ultrastructural features of the medulla have been
described by several researchers (Moffat, 1979, 1981, 1982; Bohman,
1980; Maunsbach et al., 1980).
HCO3- and H+ and the "lumen trapping" of ammonium ions, controls
renal acid-base regulation (Valtin, 1973).
Those proteins that have passed from Bowman's capsule (a
significant amount of albumin in the case of normal rats) are
reabsorbed in the proximal tubule by pinocytotic removal from the base
of the microvillous brush border into the epithelial cells. The
vesicles thus formed combine, form protein-filled vacuoles, and fuse
with lysosomes, from which the digestion products of the protein
diffuse, eventually, to the capillary system or are used in the
metabolic processes of the cell.
There are, in addition, other absorptive and secretory
mechanisms. These include the co-transport process that reabsorbs
glucose and the secretion of both acidic and basic organic compounds
(Valtin, 1973; Orloff & Berliner, 1973; Brenner & Rector, 1986;
Berndt, 1989).
3.3.4 The medulla
The medulla differs from the cortex (Fig. 5 and Fig. 8) both at
the macroscopic and at the microscopic levels. This region can be
divided into the outer medulla (which is made up of the thin
descending and the thick ascending limbs of the loops of Henle,
collecting ducts, the vasa recta, and a dense capillary network) and
the inner medulla, the free part of which is referred to as the
"papilla" (although some researchers apply that name only to the apex
of this region). The inner medulla contains the thin limbs of the
loops of Henle, collecting ducts, the vasa recta, and a diffuse
network of capillaries. Packed into the spaces between these
structures are interstitial cells embedded in a matrix rich in
glycosaminoglycans.
The collecting ducts terminate as the ducts of Bellini around the
tip of the papilla. Whereas the mouse, gerbil, rat, guinea-pig,
rabbit, dog, cat, and primate kidneys have only a single papilla, the
pig and man have multi-papillate kidneys. There are between 9 and 20
papillae in each human kidney (Burry et al., 1977), of which there are
two anatomically distinguishable types. The conical non-refluxing
papillae, where the surface orifices of the ducts of Bellini are
slit-like, close when there is an increase in the "back-pressure" of
urine from the bladder and so prevent intrarenal reflux when reflux
occurs from the bladder. These papillae occur predominantly in the
mid zone. The refluxing papillae occur predominantly in the polar
regions, and, as they have flattened tips, the collecting duct
orifices are wide and prone to retrograde flow of urine into the
tubules during vesico-ureteric reflux (Ransley & Risdon, 1979). The
microscopic and ultrastructural features of the medulla have been
described by several researchers (Moffat, 1979, 1981, 1982; Bohman,
1980; Maunsbach et al., 1980).
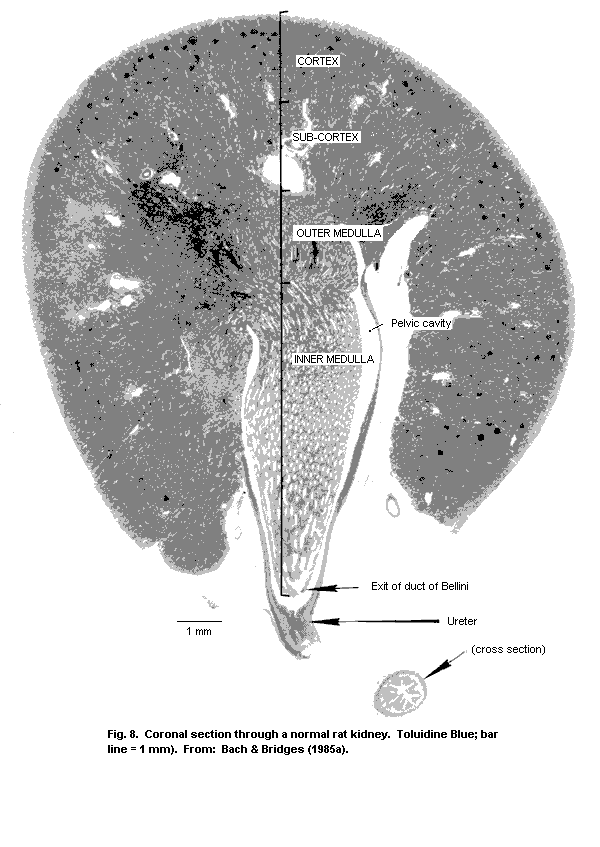 3.3.4.1 The loops of Henle
The loops of Henle may be divided into two populations on
anatomical grounds. Short loops penetrate no further than the outer
medulla. The proximal tubule and thick ascending limb are closely
associated in the cortex, but in the medulla the descending limb is
intimately related to the ascending vasa recta, and the ascending limb
to the collecting duct. The association of the ascending and
descending limbs of the loop of Henle with the vascular system or with
the collecting ducts provides a multi-dimensional network in which
solutes or water may undergo countercurrent exchange. These exchanges
may either provide a shunt that excludes selected solutes (and water)
from the inner medulla or, alternatively, solutes (e.g., sodium
chloride and urea) may be trapped in this zone. This exclusion of
water and trapping of sodium chloride, urea, and osmolytes helps
maintain the osmotic gradient along the inner medulla. In long loops
(the length is proportional to the renal concentrating potential), the
loop of Henle penetrates the inner medulla. Only about a third of the
ascending and descending limbs of long loops lie together; in the
other instances the ascending limbs are nearer to collecting ducts
than to descending limbs.
3.3.4.2 Collecting ducts
Collecting ducts consists of three identifiable segments, which
lie, respectively, in the cortex, the outer medulla, and the inner
medulla. These segments demonstrate different permeabilities to water
and osmolytes. The difference in permeability may be related to the
presence of two cell types, the intercalated and collecting duct (or
principal) cells.
3.3.4.3 The distal tubule
The distal tubule connects the thick ascending limb of the loop
of Henle to that part of the collecting duct which originates in the
cortex. The distal tubules are in-involved in both ion and water
reabsorption, but play a much less significant role than the proximal
tubules. The underlying mechanisms responsible for reabsorption
appear, in essence, to be similar to those already outlined. The major
differences include a stronger Na+ gradient against which to "pump",
the ability to reabsorb sodium without reabsorbing water, the
controlling effects of anti-diuretic hormone (ADH) and aldosterone
(among other mediators), and the very limited (or lack of) protein
reabsorption. The secretion of potassium ions appears to be under the
control of an active transport mechanism, the regulating factors of
which are many and complex (Valtin, 1973; Orloff & Berliner, 1973;
Brenner & Rector, 1986).
3.3.4.4 The countercurrent multiplier system and urine concentration
Less than 1% of the glomerular filtrate leaves the kidney as
urine (unless there is a state of diuresis), the remainder having been
reabsorbed. The process of urine concentration is complex and depends
(at least in part) on the countercurrent multiplier system, which
establishes a steep osmotic gradient along the inner medulla. The high
osmolality is a consequence of the differential permeability of the
limbs of the loops of Henle and the collecting ducts to water and
ions. The thick ascending limb is thought to have an active mechanism
which transports chloride and sodium out of the lumen and into the
interstitium, but the limb remains impermeable to water. As a
consequence the osmolality decreases in this part of the tubule (the
diluting segment). The descending limb, on the other hand, is freely
permeable to water, but probably not to sodium ions. The high ion
concentration in the interstitium would draw water out of the
descending limb, increasing the osmolality towards the turn of the
U-loop. This is augmented by urea and other osmolytes that leave the
collecting ducts and enter the ascending limb via the interstitium,
thus being recirculated to the medulla.
The collecting ducts regulate the final urine concentration by
controlling the amount of water that is reabsorbed. The passage of
water out of the ducts is thought to be mediated largely by cyclic
adenosine-monophosphate (cAMP), the synthesis of which is stimulated
by ADH, which increases the permeability of the luminal cell membrane
to water. Osmotic effects draw the water out of the cell (through the
basement membrane) into the hyperosmotic interstitium. In the absence
of ADH the collecting duct is thought to be impermeable and relatively
little water is reabsorbed from it. The interstitial osmotic gradient
is assumed to be maintained by the effective removal of water via the
ascending vasa recta, which have both a greater radius than the
descending vasa recta and are about twice as numerous. The
countercurrent exchange associated with the loops of Henle arising
from cortical nephrons offers an important "barrier" zone, which is
thought to facilitate solute trapping in and solvent exclusion from
the inner medulla, and thus helps to maintain the hyperosmolality in
this "compartment".
There are a number of other factors that control, alter, or
contribute to urine concentration. Medullary blood flow is complex, as
are the factors controlling it. Increased blood flow rates will
decrease the efficiency of countercurrent exchange in the outer
medulla, as a consequence of which the high osmotic gradient in the
inner medullary compartment will be "washed out", and urine will not
be concentrated. Diuresis is associated with increased blood flow
rates (Earley & Friedler, 1964, 1965; Chuang et al., 1978).
A unique feature of the vasa recta is their permeability to
macromolecules, a consequence of which is that the medulla contains a
large pool of albumin. The factors controlling the rapid turnover of
this milieu are poorly understood. It is generally assumed that
(together with the glycosaminoglycans) these proteins provide an
interstitial osmotic pressure that facilitates water reabsorption (see
Brenner & Rector (1986) for a fuller discussion and list of
references).
3.3.4.5 The interstitial cells
Interstitial cells occur in most organs. Three types of
interstitial cells have been described in the medulla of the rat
kidney (Bohman, 1980). Type I cells are the most abundant and
represent the typical renal medullary cells. Type 2 medullary
interstitial cells are generally round and lack lipid droplets, while
Type 3 cells correspond to the pericytes. Types 2 and 3 are sparsely
distributed and are often overlooked between the tubules, ducts, and
blood vessels. In the inner medulla, however, Type I cells are
numerous and especially prominent because they are set in a dense
matrix of glycosaminoglycans (previously referred to as
mucopolysaccharides or acidic mucopolysaccharides).
The medullary interstitial cells have been described by Moffat
(1979, 1981, 1982), Bohman (1980), and Maunsbach et al. (1980). The
number of cells and the amount of matrix substance occupies 10-20% of
the tissue volume in the outer medulla, and 40% near the apex of the
inner medulla (Bohman, 1980). The cells, which are arranged in a
regular pattern perpendicular to the tubules and vessels, are
irregular in shape and have many long slender processes. These come
into close contact with adjacent interstitial cells, capillaries, and
the limbs of the loop of Henle, but there is no such relationship with
the collecting ducts.
One of the most characteristic features associated with the Type
I cells is the presence of lipid inclusion droplets, which occupy at
least 2-4% of the total cell volume. The lipid content is largely
triglycerides, with variable amounts of cholesterol esters and
phospholipids. A number of conditions have been described where there
are marked changes in the size and number of lipid droplets. The
pathophysiological significance of these changes is difficult to
interpret because of varied experimental approaches, species
variation, and contradictory reports (Bohman, 1980).
3.4 Species, strain, and sex differences in renal structure and function
There are important differences between the renal structure of
animals and man that may have a direct effect on the interpretation of
toxicological data (Stolte & Alt 1980, 1982; Mudge 1985). The kidney
varies greatly in structure and function between different species and
strains, and there are also more subtle differences between the sexes
of several animals. In general, the kidneys can be classified into
those that are multipapillate, such as those of man and the pig, those
that have more than one papilla (e.g., spider monkey), and those of
the vast majority of animal species, which are unipapillate. The
papillae may either be present as a well defined pyramidal structure,
as in rodents, man, pigs, and dogs, or represent only a ridge as in
the non-human primates. Furthermore the kidney may be unilobar (and
have a compact structure) or consist of a multilobar structure, as in
bovines and elephants.
Table 7 compares some of the structural and functional features
of the most species most commonly used in toxicity studies. It
illustrates clearly that there are a number of differences between man
and the rodents, which are the most commonly used species to assess
nephrotoxic potential and study mechanisms of injury. There are also
major strain differences between Sprague-Dawley, Wistar, and Fischer
344 rats, which probably account for the vast majority of animals
studied. The Brown Norway rat is a most useful model for studying
mercuric chloride immunomediated nephropathy (Druet et al., 1987), and
the differences between the metabolism of methoxyflurane anaesthetic
in Fischer 344, Buffalo, Wistar, Long Evans, and Sprague-Dawley rat
was used as the basis for demonstrating the toxicity of the fluoride
ion released by hepatic mixed-function oxidase activity (Mazze, 1976,
1981). In addition there is evidence that there are consistent
differences between the renal structure and function of male and
female mice. Other sex differences have been reported in other
species.
Species or strains with unique anatomical and functional
attributes can offer an important way of helping to understand toxic
mechanisms, but at present there are few published data on non-human
primates, marmosets, or the pig.
3.5 Renal biochemistry
There is little doubt that renal metabolism is coupled tightly to
specific functions in the kidney. In particular, reabsorption of
sodium chloride can be correlated directly with oxygen consumption and
is probably the most energy-demanding transport function of this
organ. The regions of the kidney vary in their ability to metabolize
and produce various substrates.
3.5.1 Biochemistry and metabolism in the cortex
The movement of the sodium ions from the tubular fluid to the
blood is quantitatively one of the the most important functions that
the kidney performs. This is accomplished by aerobic metabolism linked
to adenosinetriphosphate (ATP) production and utilization. The exact
mechanisms are not fully understood.
Renal cortical nephrons are capable of utilizing a variety of
substrates, and the substrate utilization varies from nephron segment
to nephron segment. For example, Klein et al. (1981) demonstrated
that the convoluted portion of the proximal tubule utilized succinate,
glutamate, glutamine and other substrates quite extensively. This same
nephron segment, however, utilized glucose, lactate, and palmitate
only minimally. The hexosemonophosphate shunt is present at highest
activity in the distal segment of the nephron and in the thick
ascending limb. Although this pathway may account for relatively
little glucose oxidation, it would appear important as a source of
reduced nicotinamide adenine dinucleotide phosphate (NADPH).
The kidney cortex is also capable of producing glucose from
non-carbohydrate precursors. Although different substrates are
utilized for gluconeogenesis in the kidney, compared with the liver,
the gluconeogenic pathways are similar. Changes in hydrogen ion
activity do not alter hepatic gluconeogenesis, but markedly effect
that in the kidney.
Additionally, renal gluconeogenesis is influenced by the
concentration of substrate in the renal arterial blood. Guder &
Schmidt (1974) and Schmidt & Guder (1976) have demonstrated that the
rate-limiting enzymes for gluconeogenesis are not uniformly
distributed throughout the nephron. For example, the highest activity
is found in the proximal convoluted tubule, there being relatively
little activity in the thick ascending limb. The glycolytic enzymes,
on the other hand, are present in the thick ascending limb, the distal
tubule, and the collecting duct. A specific role for glucose in
supporting the various renal transport processes has not been
adequately described. Renal phospholipid metabolism, however, may be
important in support of transport and may play a direct role in sodium
movement.
Table 7. Comparison between the renal structure and function in man
and in commonly investigated speciesa
Man Rat Dog Pig
Cortical structure
Nephrons per g body weight 16 128 45 26
Glomerular radius (µm) 100 61 90 83
Proximal tubular length (mm) 16 12 20 30
Tubular radius (µm) 36 29 33 35
Cortical function
Glomerular filtration rate (ml/min per m2) 75 35 104 72
Inulin clearance (ml/min per kg body weight) 2.0 6.0 4.3 2.1
p-Aminohippurate transport maxima
(mg/min per kg body weight) 1.3 3.0 1.0 -
Drug-metabolizing enzymesb
Mixed-function oxidase 4 5 - -
NADPH-cytochrome c reductase 15 48 - -
Medullary structure
Number of papilla 15-20 1 1 6-10
Percent long loops 14 28 100 3
Relative medullary thickness 3.0 5.8 4.3 1.6
Medullary function
Maximum urine osmolality
(mOsmol/kg) 1400 2610 2610 1080
a Data from Mudge, 1985; Stolte & Alt, 1980, 1982; Gyrd-Hansen, 1968
b Renal enzyme activity expressed as a percentage of liver activity
- No data published on this parameter
3.5.2 Biochemistry and metabolism in the medulla
Guder & Ross (1984) have highlighted the biochemical aspects of
heterogeneity along the nephrons. Much of the published information
has been derived from whole medulla or medullary slices and fails to
differentiate between the metabolic contribution from the nephrons
(loops of Henle), as opposed to the collecting duct epithelia, versus
the interstitial cells.
3.5.2.1 The biochemistry of renal prostaglandins (PG)
The PGs and endoperoxides are a group of ubiquitously distributed
hormones with a broad spectrum of potent biological activity that
shows marked receptor specificity. They are synthesized from the C20:4
fatty acid arachidonic acid by an enzyme system (which includes
cyclo-oxygenases, peroxidases, isomerases, and reductases)
collectively called PG synthetase.
The PGs (Fig. 9) are structurally similar and are only present in
minute concentrations. Several are labile and undergo spontaneous
chemical changes. Thus, most of the methods (both qualitative and
quantitative) needed for their biochemical investigations are fraught
with subtle pitfalls (Frolich & Walker, 1980). The literature on renal
PG biology is large, complex, contradictory, and difficult to
interpret. The subject has been reviewed recently by Dunn & Hood
(1977), Dunn & Zambraski (1980), Horrobin (1980), Morrison (1980),
Zusman (1980), Dunn (1981), Frolich et al. (1981), and Levenson et al.
(1982).
PGs are not stored in renal tissue but are synthesized de novo
from arachidonic acid, which is released from stored phospholipid or
triglyceride pools by the action of phospholipase A2. The factors
that regulate the release of arachidonic acid include both
receptor-mediated responses (such as vasoactive peptides and biogenic
amines) and non-specific stimuli (ischaemia). The prostaglandin
precursor may be drawn from different lipid pools. Any arachidonate
that is not channelled into prostaglandin synthesis may be re-acylated
(as are the de novo synthesized molecules) or disposed of via
several other metabolic routes. Arachidonic acid (the availability of
which is rate limiting) is converted to PGG2 and thence to other
PG-related substances.
The anatomically identifiable areas of the kidney each synthesize
a different pattern of PGs in vitro. The in vivo contributions of
each area to PG synthesis and the function of each PG remains largely
a matter of speculation at present. Total PG synthesis is several
times higher in the medulla (where typically it is greatest in the
papilla) than in the cortex (Dunn & Hood, 1977). However, the
distribution of, for example, PGE2 synthesis reflects a more complex
picture, its concentration being lower in the papilla than the rest of
the inner medulla (van Dorp, 1971). A recent study using isolated
nephrons showed that the highest PGE2 production is in the medullary
3.3.4.1 The loops of Henle
The loops of Henle may be divided into two populations on
anatomical grounds. Short loops penetrate no further than the outer
medulla. The proximal tubule and thick ascending limb are closely
associated in the cortex, but in the medulla the descending limb is
intimately related to the ascending vasa recta, and the ascending limb
to the collecting duct. The association of the ascending and
descending limbs of the loop of Henle with the vascular system or with
the collecting ducts provides a multi-dimensional network in which
solutes or water may undergo countercurrent exchange. These exchanges
may either provide a shunt that excludes selected solutes (and water)
from the inner medulla or, alternatively, solutes (e.g., sodium
chloride and urea) may be trapped in this zone. This exclusion of
water and trapping of sodium chloride, urea, and osmolytes helps
maintain the osmotic gradient along the inner medulla. In long loops
(the length is proportional to the renal concentrating potential), the
loop of Henle penetrates the inner medulla. Only about a third of the
ascending and descending limbs of long loops lie together; in the
other instances the ascending limbs are nearer to collecting ducts
than to descending limbs.
3.3.4.2 Collecting ducts
Collecting ducts consists of three identifiable segments, which
lie, respectively, in the cortex, the outer medulla, and the inner
medulla. These segments demonstrate different permeabilities to water
and osmolytes. The difference in permeability may be related to the
presence of two cell types, the intercalated and collecting duct (or
principal) cells.
3.3.4.3 The distal tubule
The distal tubule connects the thick ascending limb of the loop
of Henle to that part of the collecting duct which originates in the
cortex. The distal tubules are in-involved in both ion and water
reabsorption, but play a much less significant role than the proximal
tubules. The underlying mechanisms responsible for reabsorption
appear, in essence, to be similar to those already outlined. The major
differences include a stronger Na+ gradient against which to "pump",
the ability to reabsorb sodium without reabsorbing water, the
controlling effects of anti-diuretic hormone (ADH) and aldosterone
(among other mediators), and the very limited (or lack of) protein
reabsorption. The secretion of potassium ions appears to be under the
control of an active transport mechanism, the regulating factors of
which are many and complex (Valtin, 1973; Orloff & Berliner, 1973;
Brenner & Rector, 1986).
3.3.4.4 The countercurrent multiplier system and urine concentration
Less than 1% of the glomerular filtrate leaves the kidney as
urine (unless there is a state of diuresis), the remainder having been
reabsorbed. The process of urine concentration is complex and depends
(at least in part) on the countercurrent multiplier system, which
establishes a steep osmotic gradient along the inner medulla. The high
osmolality is a consequence of the differential permeability of the
limbs of the loops of Henle and the collecting ducts to water and
ions. The thick ascending limb is thought to have an active mechanism
which transports chloride and sodium out of the lumen and into the
interstitium, but the limb remains impermeable to water. As a
consequence the osmolality decreases in this part of the tubule (the
diluting segment). The descending limb, on the other hand, is freely
permeable to water, but probably not to sodium ions. The high ion
concentration in the interstitium would draw water out of the
descending limb, increasing the osmolality towards the turn of the
U-loop. This is augmented by urea and other osmolytes that leave the
collecting ducts and enter the ascending limb via the interstitium,
thus being recirculated to the medulla.
The collecting ducts regulate the final urine concentration by
controlling the amount of water that is reabsorbed. The passage of
water out of the ducts is thought to be mediated largely by cyclic
adenosine-monophosphate (cAMP), the synthesis of which is stimulated
by ADH, which increases the permeability of the luminal cell membrane
to water. Osmotic effects draw the water out of the cell (through the
basement membrane) into the hyperosmotic interstitium. In the absence
of ADH the collecting duct is thought to be impermeable and relatively
little water is reabsorbed from it. The interstitial osmotic gradient
is assumed to be maintained by the effective removal of water via the
ascending vasa recta, which have both a greater radius than the
descending vasa recta and are about twice as numerous. The
countercurrent exchange associated with the loops of Henle arising
from cortical nephrons offers an important "barrier" zone, which is
thought to facilitate solute trapping in and solvent exclusion from
the inner medulla, and thus helps to maintain the hyperosmolality in
this "compartment".
There are a number of other factors that control, alter, or
contribute to urine concentration. Medullary blood flow is complex, as
are the factors controlling it. Increased blood flow rates will
decrease the efficiency of countercurrent exchange in the outer
medulla, as a consequence of which the high osmotic gradient in the
inner medullary compartment will be "washed out", and urine will not
be concentrated. Diuresis is associated with increased blood flow
rates (Earley & Friedler, 1964, 1965; Chuang et al., 1978).
A unique feature of the vasa recta is their permeability to
macromolecules, a consequence of which is that the medulla contains a
large pool of albumin. The factors controlling the rapid turnover of
this milieu are poorly understood. It is generally assumed that
(together with the glycosaminoglycans) these proteins provide an
interstitial osmotic pressure that facilitates water reabsorption (see
Brenner & Rector (1986) for a fuller discussion and list of
references).
3.3.4.5 The interstitial cells
Interstitial cells occur in most organs. Three types of
interstitial cells have been described in the medulla of the rat
kidney (Bohman, 1980). Type I cells are the most abundant and
represent the typical renal medullary cells. Type 2 medullary
interstitial cells are generally round and lack lipid droplets, while
Type 3 cells correspond to the pericytes. Types 2 and 3 are sparsely
distributed and are often overlooked between the tubules, ducts, and
blood vessels. In the inner medulla, however, Type I cells are
numerous and especially prominent because they are set in a dense
matrix of glycosaminoglycans (previously referred to as
mucopolysaccharides or acidic mucopolysaccharides).
The medullary interstitial cells have been described by Moffat
(1979, 1981, 1982), Bohman (1980), and Maunsbach et al. (1980). The
number of cells and the amount of matrix substance occupies 10-20% of
the tissue volume in the outer medulla, and 40% near the apex of the
inner medulla (Bohman, 1980). The cells, which are arranged in a
regular pattern perpendicular to the tubules and vessels, are
irregular in shape and have many long slender processes. These come
into close contact with adjacent interstitial cells, capillaries, and
the limbs of the loop of Henle, but there is no such relationship with
the collecting ducts.
One of the most characteristic features associated with the Type
I cells is the presence of lipid inclusion droplets, which occupy at
least 2-4% of the total cell volume. The lipid content is largely
triglycerides, with variable amounts of cholesterol esters and
phospholipids. A number of conditions have been described where there
are marked changes in the size and number of lipid droplets. The
pathophysiological significance of these changes is difficult to
interpret because of varied experimental approaches, species
variation, and contradictory reports (Bohman, 1980).
3.4 Species, strain, and sex differences in renal structure and function
There are important differences between the renal structure of
animals and man that may have a direct effect on the interpretation of
toxicological data (Stolte & Alt 1980, 1982; Mudge 1985). The kidney
varies greatly in structure and function between different species and
strains, and there are also more subtle differences between the sexes
of several animals. In general, the kidneys can be classified into
those that are multipapillate, such as those of man and the pig, those
that have more than one papilla (e.g., spider monkey), and those of
the vast majority of animal species, which are unipapillate. The
papillae may either be present as a well defined pyramidal structure,
as in rodents, man, pigs, and dogs, or represent only a ridge as in
the non-human primates. Furthermore the kidney may be unilobar (and
have a compact structure) or consist of a multilobar structure, as in
bovines and elephants.
Table 7 compares some of the structural and functional features
of the most species most commonly used in toxicity studies. It
illustrates clearly that there are a number of differences between man
and the rodents, which are the most commonly used species to assess
nephrotoxic potential and study mechanisms of injury. There are also
major strain differences between Sprague-Dawley, Wistar, and Fischer
344 rats, which probably account for the vast majority of animals
studied. The Brown Norway rat is a most useful model for studying
mercuric chloride immunomediated nephropathy (Druet et al., 1987), and
the differences between the metabolism of methoxyflurane anaesthetic
in Fischer 344, Buffalo, Wistar, Long Evans, and Sprague-Dawley rat
was used as the basis for demonstrating the toxicity of the fluoride
ion released by hepatic mixed-function oxidase activity (Mazze, 1976,
1981). In addition there is evidence that there are consistent
differences between the renal structure and function of male and
female mice. Other sex differences have been reported in other
species.
Species or strains with unique anatomical and functional
attributes can offer an important way of helping to understand toxic
mechanisms, but at present there are few published data on non-human
primates, marmosets, or the pig.
3.5 Renal biochemistry
There is little doubt that renal metabolism is coupled tightly to
specific functions in the kidney. In particular, reabsorption of
sodium chloride can be correlated directly with oxygen consumption and
is probably the most energy-demanding transport function of this
organ. The regions of the kidney vary in their ability to metabolize
and produce various substrates.
3.5.1 Biochemistry and metabolism in the cortex
The movement of the sodium ions from the tubular fluid to the
blood is quantitatively one of the the most important functions that
the kidney performs. This is accomplished by aerobic metabolism linked
to adenosinetriphosphate (ATP) production and utilization. The exact
mechanisms are not fully understood.
Renal cortical nephrons are capable of utilizing a variety of
substrates, and the substrate utilization varies from nephron segment
to nephron segment. For example, Klein et al. (1981) demonstrated
that the convoluted portion of the proximal tubule utilized succinate,
glutamate, glutamine and other substrates quite extensively. This same
nephron segment, however, utilized glucose, lactate, and palmitate
only minimally. The hexosemonophosphate shunt is present at highest
activity in the distal segment of the nephron and in the thick
ascending limb. Although this pathway may account for relatively
little glucose oxidation, it would appear important as a source of
reduced nicotinamide adenine dinucleotide phosphate (NADPH).
The kidney cortex is also capable of producing glucose from
non-carbohydrate precursors. Although different substrates are
utilized for gluconeogenesis in the kidney, compared with the liver,
the gluconeogenic pathways are similar. Changes in hydrogen ion
activity do not alter hepatic gluconeogenesis, but markedly effect
that in the kidney.
Additionally, renal gluconeogenesis is influenced by the
concentration of substrate in the renal arterial blood. Guder &
Schmidt (1974) and Schmidt & Guder (1976) have demonstrated that the
rate-limiting enzymes for gluconeogenesis are not uniformly
distributed throughout the nephron. For example, the highest activity
is found in the proximal convoluted tubule, there being relatively
little activity in the thick ascending limb. The glycolytic enzymes,
on the other hand, are present in the thick ascending limb, the distal
tubule, and the collecting duct. A specific role for glucose in
supporting the various renal transport processes has not been
adequately described. Renal phospholipid metabolism, however, may be
important in support of transport and may play a direct role in sodium
movement.
Table 7. Comparison between the renal structure and function in man
and in commonly investigated speciesa
Man Rat Dog Pig
Cortical structure
Nephrons per g body weight 16 128 45 26
Glomerular radius (µm) 100 61 90 83
Proximal tubular length (mm) 16 12 20 30
Tubular radius (µm) 36 29 33 35
Cortical function
Glomerular filtration rate (ml/min per m2) 75 35 104 72
Inulin clearance (ml/min per kg body weight) 2.0 6.0 4.3 2.1
p-Aminohippurate transport maxima
(mg/min per kg body weight) 1.3 3.0 1.0 -
Drug-metabolizing enzymesb
Mixed-function oxidase 4 5 - -
NADPH-cytochrome c reductase 15 48 - -
Medullary structure
Number of papilla 15-20 1 1 6-10
Percent long loops 14 28 100 3
Relative medullary thickness 3.0 5.8 4.3 1.6
Medullary function
Maximum urine osmolality
(mOsmol/kg) 1400 2610 2610 1080
a Data from Mudge, 1985; Stolte & Alt, 1980, 1982; Gyrd-Hansen, 1968
b Renal enzyme activity expressed as a percentage of liver activity
- No data published on this parameter
3.5.2 Biochemistry and metabolism in the medulla
Guder & Ross (1984) have highlighted the biochemical aspects of
heterogeneity along the nephrons. Much of the published information
has been derived from whole medulla or medullary slices and fails to
differentiate between the metabolic contribution from the nephrons
(loops of Henle), as opposed to the collecting duct epithelia, versus
the interstitial cells.
3.5.2.1 The biochemistry of renal prostaglandins (PG)
The PGs and endoperoxides are a group of ubiquitously distributed
hormones with a broad spectrum of potent biological activity that
shows marked receptor specificity. They are synthesized from the C20:4
fatty acid arachidonic acid by an enzyme system (which includes
cyclo-oxygenases, peroxidases, isomerases, and reductases)
collectively called PG synthetase.
The PGs (Fig. 9) are structurally similar and are only present in
minute concentrations. Several are labile and undergo spontaneous
chemical changes. Thus, most of the methods (both qualitative and
quantitative) needed for their biochemical investigations are fraught
with subtle pitfalls (Frolich & Walker, 1980). The literature on renal
PG biology is large, complex, contradictory, and difficult to
interpret. The subject has been reviewed recently by Dunn & Hood
(1977), Dunn & Zambraski (1980), Horrobin (1980), Morrison (1980),
Zusman (1980), Dunn (1981), Frolich et al. (1981), and Levenson et al.
(1982).
PGs are not stored in renal tissue but are synthesized de novo
from arachidonic acid, which is released from stored phospholipid or
triglyceride pools by the action of phospholipase A2. The factors
that regulate the release of arachidonic acid include both
receptor-mediated responses (such as vasoactive peptides and biogenic
amines) and non-specific stimuli (ischaemia). The prostaglandin
precursor may be drawn from different lipid pools. Any arachidonate
that is not channelled into prostaglandin synthesis may be re-acylated
(as are the de novo synthesized molecules) or disposed of via
several other metabolic routes. Arachidonic acid (the availability of
which is rate limiting) is converted to PGG2 and thence to other
PG-related substances.
The anatomically identifiable areas of the kidney each synthesize
a different pattern of PGs in vitro. The in vivo contributions of
each area to PG synthesis and the function of each PG remains largely
a matter of speculation at present. Total PG synthesis is several
times higher in the medulla (where typically it is greatest in the
papilla) than in the cortex (Dunn & Hood, 1977). However, the
distribution of, for example, PGE2 synthesis reflects a more complex
picture, its concentration being lower in the papilla than the rest of
the inner medulla (van Dorp, 1971). A recent study using isolated
nephrons showed that the highest PGE2 production is in the medullary
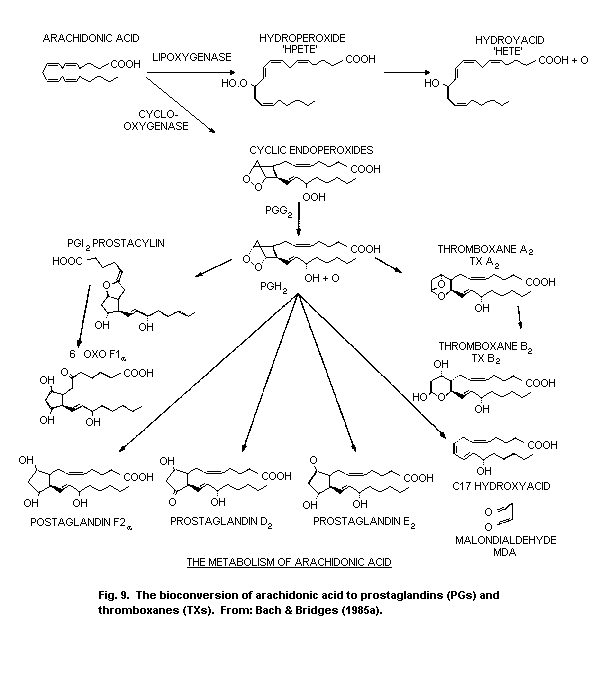 collecting tubules, followed by cortical collecting ducts and
glomeruli. Furthermore, there are marked sex-related differences in
the effects of cofactors on medullary PG synthetase activity (Hirafuji
et al., 1980). Some PGs break down spontaneously (e.g., PGI2 to
alpha-6-keto-PGF), but the majority are metabolically degraded
(Morrison, 1980). The enzymic conversions are mediated by a number of
enzymes, including dehydrogenases, reductases, and ß- and
omega-oxidases. The enzymes that degrade PGs are located mainly in the
cortex, but there are species differences in the corticomedullary
ratio of these enzymic activities (Powell, 1980).
The factors regulating the biosynthesis of each type of PG are
only poorly defined (Horrobin, 1980). A large number of endogenous and
exogenous substances have been reported to alter renal PG synthesis,
and several patho-physiological conditions have been described in
which renal PG synthesis is increased. Most attention has been
focussed on the inhibitory effects of the anti-inflammatory drugs. The
steroidal compounds (e.g., corticosteroids) prevent the release of
arachidonic acid from its lipid pools, and the non-steroidal
products (e.g., indomethacin) inhibit cyclo-oxygenase. It is, however,
essential to be aware that any factor that perturbs PG synthesis may
act differently at different sites in the synthetic (or degradative)
pathway.
Indomethacin (one of the most extensively studied cyclo-oxygenase
inhibitors) produces several alterations in renal PG dynamics.
Uncertainties in defining PG-"related" pathophysiological changes are
compounded by the observation (Attallah & Stahl, 1980) that PGE2
synthesis in slices from each zone of the kidney has a different dose
response to indomethacin inhibition: the cortex is most sensitive and
the papilla least sensitive. The cyclo-oxygenase inhibitors are
generally classified as either reversible or irreversible, but the
multiplicity of effects and the possibility of enzymic polymorphism in
different regions of the kidney suggest that such a classification may
be an over-simplification.
The exact physiological roles of the PGs in normal renal function
and the way in which these are altered in the development of
nephropathies are not clear. Firstly, indomethacin, for example, has
been shown to cause biochemical changes that may be classified as
either related or unrelated to altering PG dynamics. Secondly, many
attempts to define renal PG function have been based on the hypothesis
that urinary PG excretion reflects de novo renal synthesis (Dunn &
Hood, 1977; Dunn & Zambraski, 1980; Dunn 1981), notwithstanding
analytical difficulties of measuring very low levels of various PGs
and apparently ignoring the fact that de novo synthesized PGs may
have undergone extensive degradation. The measurement of urinary PGs,
as an estimate of their de novo renal synthesis, remains equivocal
because, firstly, seminal PGE2 is an unavoidable and variable
contaminant in the urine of males (Suzuki et al., 1980) and, secondly,
Brown et al. (1980) have demonstrated that both rabbit and rat urinary
bladders can synthesize PGE from arachidonic acid. Finally, the
physiology of renal function is controlled by several hormonal
systems, the detailed functioning of which has not been clearly
established. It is known that renal PGs may be altered by (or may
alter) the renin/angiotensin II/aldosterone system (Hackenthal et
al., 1980; Lee, 1980; Weber, 1980; Baer, 1981), the kallikrein-kinin
system (Margolius, 1980; Rockel & Heidland, 1980), and the regulation
of fluid balance and water reabsorption via ADH (Blair-West et al.,
1980). Furthermore, each of these hormonal systems may interact with
the others via direct or indirect mechanisms. It seems likely that a
full understanding of the pathophysiology of the renal hormonal
systems will take some time to crystallize.
In spite of the rather abstruse biology, there is general
consensus (Dunn & Hood 1977; Dunn & Zambraski, 1980; Morrison, 1980)
that PGs have a central role in renal function. It seems, however,
that renal PGs play little, if any, major regulatory role in basal
renal blood flow in normal conscious animals. There is evidence that
PGs are released in response to ischaemic and vasoconstrictive stress,
where their role seems to be to provide a protective effect by
maintaining glomerular dynamics. The role of PGs (especially PGE2)
in preventing experimentally induced acute renal failure is
conflicting. Arachidonic acid does stimulate renin release, a
response that is blocked by cyclo-oxygenase inhibitors, but it remains
uncertain which of the PGs mediate this effect in vivo. It is also
unclear from which renal zone such mediators are synthesized and
released. Renin release may, in turn, affect PG synthesis and the
kallikrein-kinin system (which in turn may modulate PG synthesis and
the renin system). ADH is assumed to stimulate PGE2, but published
data on the controlling effects of PGs on salt and water balance are
very difficult to interpret. Similarly, the mass of literature on
hypertension and PGs favours the concept that the two are related, but
fails to propound a unifying hypothesis.
3.5.2.2 Lipid metabolism
The contents of the interstitial lipid droplets are too
specialized to be used as a metabolic energy store (Bohman, 1980),
although this droplet population undergoes marked and rapid change
during the short periods that precede various pathophysiological
conditions. The interstitial cells produce the ground substance matrix
that surrounds them. Early evidence that the interstitial cells of the
renal medulla are only a highly specialized PG-producing cell type has
become equivocal. The interstitial lipid droplets do not, in fact,
provide the sole source of arachidonic acid for PG synthesis. Only 50%
of the medullary capacity for synthesizing PG is confined to the
interstitial cells; the rest is in the collecting ducts (Bohman,
1980). The significance of PG synthesis in the medullary cells cannot,
however, be overlooked, as it may play an important role in regulating
blood pressure and other renal functions. It has also been suggested
to occupy a central position in the pathogenesis of renal papillary
necrosis.
In recent years the importance of the endocrine function of the
medullary interstitial cells in regulating blood pressure has been
highlighted by several workers (Mandal & Bohman, 1980). Three groups
of vaso-active compounds have been isolated from the medulla or
cultured interstitial cells. Experimentally induced, spontaneously
occurring, and pathologically precipitated hypertensive states have
been reversed by subcutaneous transplants of renal papillary fragments
and by cultured interstitial cells. In addition, the systemic
administration of both the polar and neutral reno-medullary lipids
reduces arterial blood pressure (Muirhead & Pitcock, 1980).
The numerous lipid droplets in the interstitial cells have been
found to contain traces of cholesterol esters, a few percent of
phospholipids, mainly phosphatidylcholine, and, rarely, trace amounts
of phosphatidyl-ethanolamine. A few percent of free fatty acids and
tri-acylglycerols make up the remaining 80-90%, the composition of
which varies in different species. The most striking features are the
varied types and large amounts of unsaturated fatty acids; most
notably those of 20 or more carbon atoms such as arachidonic acid and
especially adrenic acid. The large amount of arachidonic acid suggests
that the interstitial lipid droplets may be an important pool for PG
synthesis in the kidney (Bojesen, 1974).
3.5.2.3 Carbohydrate metabolism in the medulla
The metabolism of carbohydrate in the renal medulla has been
reviewed by Cohen (1979). Some early observations suggested that the
low oxygen tension in the inner medulla (a pO2 as low as 0.67-2.0
kPa (5-l5 mmHg) compared with 10 kPa (75 mmHg) in the cortex) would
necessitate anaerobic metabolism. However, aerobic metabolism is only
limited at an O2 availability of less than 0.13 kPa (1 mmHg).
Many metabolic investigations have used medullary slices or
homogenates. Thus, the exact contribution of the different cell types
in the inner medulla to functional energy dynamics and to the changes
that underlie, for example, diuresis or anti-diuresis have yet to be
related to the phosphorylation and redox states within these
individual cell types.
Carbohydrates are stored in the medulla as either glycogen or as
glycosaminoglycan (GAG), the former in collecting ducts and epithelia
and the latter as an important constituent of the interstitial ground
substance. There is evidence to suggest that either can be mobilized
to provide an energy source or the glucose units for the synthesis of
the other macromolecular carbohydrates (Darnton, 1967, 1969).
3.5.2.4 Medullary glycosaminoglycan (GAG)
The biology of GAGs has been described by Kennedy (1979). GAGs
are linear polysaccharides that are made up of repeating disaccharide
units, one carbohydrate moiety of which is a hexuronic acid (or a
neutral sugar in one case) and the other a hexosamine. The nature of
the disaccharide units and the occurrence of N-acetyl groups,
together with the position of O-sulfate groups, define the species
of macromolecule. There are seven basic types of GAG. These molecules
also show heterogeneity of relative molecular mass (when isolated from
the same or different organs; Toledo & Dietrich, 1977), and the molar
ratio of sulfate to hexosamine varies by up to 2-fold for the same
type of GAG (Suzuki et al., 1976). Most of these substances probably
occur in vivo as proteoglycans (PoGs). These supramolecular
structures are composed of a linear protein backbone that carries GAGs
covalently bound at intervals along its length. In theory, any
combination and ratio of GAGs may occur. It is only recently that the
concept of PoGs has been accepted; before this the presence of protein
was assumed to be a contamination and vigorous steps were taken to
remove it.
In spite of the ubiquity of PoGs and their composite GAGs,
relatively little is known about their physiological functions, with
the exception of their anti-coagulant and anti-lipaemic properties,
which are best studied in heparin. These molecules are bound to cell
surfaces (Kjellen et al., 1977), where they may control the access of
endogenous and exogenous molecules to cell membrane receptors.
Similarly, the functions of this intercellular polyanionic matrix most
probably extend beyond that of "immobilized anti-coagulants" or "space
filling", and include controlling the micro-environment of cells (by
binding either inorganic or organic cations and by their immense
water-holding capacity) and regulating cell-cell communications. GAGs
may also control cell recognition and adhesion, and contribute to the
control of cell movement, growth, differentiation, and proliferation
(Long & Williamson, 1979). The association of GAGs with mitochondria
and nuclear membranes suggests that these macromolecules may also play
a direct role in controlling some intracellular functions.
The distribution of GAGs has been assessed in tissue either by
the autoradiographic distribution of precursor carbohydrates or
35SO2 or by histochemical staining. It is generally assumed that
sulfate radiolabel distribution is relatively specific for GAGs.
However, most of the staining procedures are nonspecific (e.g.,
toluidine blue interacts with any polyanion to give a metachromatic
colour shift) and depend either on a priori knowledge of
distribution or, for example, the use of control sections that have
been exposed to selective enzymic digestion.
The amount and types of GAGs in the kidneys of various species
have been reported. The quantity of polyanionic macromolecule has been
found to be greater in the medulla than in the cortex for the rat
(Jacobsen et al., 1964; Kresse & Grossmann, 1970), pig (Kresse &
Grossmann 1970), dog (Castor & Green, 1968; Kresse & Grossmann, 1970),
and normal human kidney (Inoue et al., 1973; Constantopoulos et al.,
1973). The medulla:cortex ratio in the human kidney was found to be
age related, increasing rapidly to a maximum in the fourth decade and
then declining slowly (Inone et al., 1973). The heterogeneous
distribution of the types of GAG in the kidney is supported by the
data of Constantopoulos et al. (1973) for the human kidney and Castor
& Green (1968), who reported that hyaluronic acid had a high relative
molecular mass in the medulla but a low one in the cortex of dogs.
However, other data on the dog, pig, and sheep (Dicker & Franklin,
1966) and the rat (Barry & Bowness, 1975) suggest that the types and
quantities of GAG are the same in both the cortex and medulla.
The processes underlying and controlling the biosynthesis of PoGs
are complex and incompletely documented (Kennedy, 1979). Muirhead &
Pitcock (1980) reported that medullary interstitial cells synthesize
PoGs (both in situ and in culture) and that these macromolecules are
associated with the cellular cisternae (dilated rough endoplasmic
reticulum). Darnton (1967, 1969) presented data to show that glycogen
associated with the epithelial cells of the collecting duct in the
rabbit is mobilized and incorporated into GAGs.
The functions of the medullary GAGs have been the centre of
controversy since Ginetzinsky (1958) suggested that the action of ADH
was mediated by the release of hyaluronidase. This would depolymerize
medullary GAG and (so it was argued) allow greater water reabsorbtion
from the tubules into the interstitium and thence to the blood supply.
This hypothesis has been supported by some workers (Jacobson et al.,
1964; Farber et al., 1971) but refuted by others (Sun et al., 1972;
McAuliffe 1978, 1980; Sun, 1980) in animals with spontaneous diabetes
insipidus. These conflicting data are difficult to resolve into a
single unifying theory relating the physiological function of GAGs to
the urine-concentrating process.
3.6 The metabolism of xenobiotic molecules in the kidney
Chemically induced lesions may depend to varying extents on the
metabolic capacity of tissues to deal with "insults". The metabolism
of xenobiotic molecules may either prevent lesions (by deactivation),
or be directly responsible for damage (by bio-activation). The renal
metabolism of chemicals (and its consequences) has been reviewed by
Hook et al. (1979), Anders (1980), Connelly & Bridges (1980), Kluwe &
Hook (1980), Davis et al. (1981), Rush et al., (1984) and Tarloff et
al. (1987).
It is likely that the liver meets the challenge of metabolizing
a major proportion of exogenous compounds in vivo before they reach
the systemic circulation. Most fundamental types of bioconversion have
been described for the perfused kidney of several species (Szefler &
Acara, 1979; Elbers et al., 1980; Ross et al., 1980; Emslie et al.,
1981) and for isolated renal cells and tubular fragments (Fry et al.,
1978; Jones et al., 1979; Ormstad, 1982). Similarly, kidney
microsomes have been shown to have most of the enzymic and
cytochrome-mediated metabolic activities that have been described in
other tissues. The xenobiotic-transformating capacity of the kidney is
about 3-50% (depending on the system, species, and source of data) of
that found in the liver (Litterst et al., 1975a, Navran &
Louis-Ferdinand 1975; Fry et al., 1978), but it may be much higher
than that of the liver under certain circumstances (Anders, 1980).
There are marked qualitative differences between hepatic and renal
xenobiotic metabolism. Renal enzymes are stable during the Ca2+
method of preparing microsomes (Litterst et al., 1975b). Enzymic
kinetic constants vary between microsomes isolated from the two organs
(Navran & Louis-Ferdinand, 1975). Whereas there are marked sex-related
differences in hepatic metabolism, there are few in the kidney
(Litterst et al., 1977). There is evidence to suggest that liver
cytochrome P-450 is similar to that of the kidney, based on
electro-phoretic and electron paramagnetic resonance studies
(Armbrecht et al., 1979), immunological criteria (Guengerich & Mason,
1979), and on immunometabolic studies (Kaminsky et al., 1979).
However, these data are most difficult to interpret in "absolute"
terms, because the samples of cytochrome P-450 were from organs
exposed to different inducing agents. There is now substantial
evidence that hepatic and renal tissue respond differently, both
quantitatively and qualitatively, to the various inducers of
cytochrome P-450 (Litterst et al., 1977; Zenser et al., 1978a;
Kaminsky et al., 1979). Ascorbic acid deficiency (Sikic et al., 1977)
and carbon tetrachloride pretreatment (Litterst et al., 1977) alter
the metabolism of xenobiotics in a different way in the liver and
kidney. In addition, the inhibitory effects of
2-diethylaminoethyl-2,2-diphenylvalerate (SKF-525A) on renal and
hepatic microsomes studied in vitro are similar but not identical
(Litterst et al., 1977).
There are several enzymes involved in renal xenobiotic
metabolism. It is not possible to comment on all of those that may be
relevant to nephrotoxicity nor, indeed, is it clearly established what
role each renal enzyme plays in the realization of the potential
toxicity of a chemical. Many of the enzymes that metabolize
xenobiotics are compartmentalized in specific regions of the kidney.
The anatomical localization of these characteristics may play a key
role in the toxicological consequence that follows the entry of a
xenobiotic into the kidney. The distribution and regulation of these
enzymes may predispose to the toxic effects of chemicals. Thus,
although intrarenal metabolism may be a prerequisite for the target
selective effects of some chemicals, the final outcome of the toxic
response relates to the sum of a number of factors. These include the
localization of those renal enzymes involved in xenobiotic metabolism
(this may be metabolic activation or other processes, perhaps in an
adjacent cell) and the processes controlling the intracellular
concentration of toxic chemicals. The intracellular concentration of
a chemical can be influenced by xenobiotic metabolism per se and by
many of the inherent processes in the kidney, such as transport, pH,
and solute gradients on either side of a membrane. The outcome of a
chemical exposure may also be affected by the numbers and types of
organelles in a specific cell type that have critical properties
relevant to the functions of that cell and by the presence of a
protective mechanism in a particular cell type (such as antioxidants,
free radical scavengers). Most of the processes that underlie
nephrotoxicity are probably multi-step events that are affected by
more than one metabolic pathway and occur via competing and sequential
pathways. Little is known about the control of these pathways, so that
it is difficult to predict from the structure of a chemical alone what
effects it will have on the kidney. In addition, a major role is
obviously played by the extra-renal metabolism (in the liver, lung,
gut, etc.) of the parent chemical and by a variety of other organ
functions. These include the lung (exhalation of volatile
metabolites), liver (biliary excretion), and gastrointestinal
microflora (enterohepatic circulation, serum protein binding), and
they determine the types of chemicals, the concentration that reaches
the kidney, and their renal pharmacokinetics.
3.6.1 Oxidases
Oxidases can convert chemicals into active intermediates or
generate reactive species by redox cycling. This is potentially
important for compounds that contain arylamine, quaternary bipyridyl
(paraquat), quinone (adriamycin), or nitro (nitrofurantoin)
structures. It is generally accepted that, in the liver, biologically
reactive intermediates mediate their toxic effects by binding to
cellular macromolecules and blocking normal functional processes
(Jollow et al., 1976; Snyder et al., 1981). Similar mechanisms have
been proposed to explain various types of chemically induced renal
lesions, including papillary necrosis. The metabolically generated
reactive intermediates have a relatively short life and are most
likely formed in the organ or anatomical area in which they induce
damage.
3.6.1.1 Cytochrome-P-450-dependent mixed-function oxidases (monooxygenases)
This enzyme system carries out a two-electron flow pathway, and
the flavoprotein component can catalyse single electron reductions
such as the reduction of quinones to semiquinone radicals (Bachur et
al., 1979). Multiple forms of cytochrome P-450 have been identified in
the kidney. These include phenobarbital-, 3-methylcholanthrene-, and
ß-naphthoflavone-inducible cytochrome P-450. Renal cytochrome P-450
induction varies in different species. Polycyclic aromatic
hydrocarbons induce cytochrome P-450 in most species, whereas
phenobarbital is effective in hamsters and rabbits but not in
guinea-pigs, rats, and mice (Smith et al., 1986). Similarly, renal
P-450 responds differently to inhibitors of mixed-function oxidases.
The effects of inhibitors such as SKF-525A are further
complicated by multiple actions on renal transport, intracellular
binding of chemicals at noncatalytic sites, and on cytochrome
P-450-dependent metabolism. Other inhibitors do not appear to have
been as fully studied, and the paucity of data in this area makes
other studies on the effects of inhibitors most difficult to
interpret. Some species also have sex-related differences, e.g., male
mice have higher concentrations and activities of P-450 than female
mice (Krijsheld & Gram, 1984; Smith et al., 1984; Hawke & Welch,
1985), but this is not the case for rats (Litterst et al., 1977; Hook
et al., 1982) or rabbits (Litterst et al., 1977). There is no clear
data on other species, such as man, nor on how different types of
renal disease affect the concentration of renal P-450 or its induction
or inhibition.
The specific activity of the renal mixed-function oxidases varies
widely between species and is about 10% of the hepatic activity
(Zenser et al., 1978a,b; Endou, 1983). This suggests a role for renal
P-450 that is quantitatively less important than that of the liver.
However, this is not the case for all chemicals, since the renal
metabolism of chloroform is about 2-fold higher than the hepatic
activity (Smith & Hook 1984). More importantly, mixed-function
oxidase activities are intra-renally localized to discrete areas
where their significance in metabolism may be far greater than in the
liver. The S2 proximal segment has a cytochrome P-450 concentration
that is 2-3 times higher than the S1 or S3 segments. The distal
tubules, cortical collecting ducts, and the medulla contain no
measurable cytochrome P-450 activity (Endou, 1983). By contrast
NADPH-cytochrome-P-450 reductase activity is highest in the S2 and
S3 segments, but it also extends to the distal tubule and medullary
structures.
3.6.1.2 Prostaglandin peroxidase-mediated metabolic activation
Recently, it has been shown that there are marked quantitative
and qualitative differences in the regional distribution of microsomal
mixed-function oxidase activity within the rabbit kidney (Zenser et
al., 1978a,b; Armbrecht et al., 1979). Most mixed-function oxidase
activity is located in the cortex and least in the inner medulla in
control tissue and in that taken from animals induced with
3-methylcholanthrene. Cytochrome P-450 is not detected in the medulla
of controls or even those of induced animals. In addition, laurate
hydroxylase activity (the only mixed-function oxidase activity found
in the inner medulla) shows marked differences in the pattern of
inhibition by carbon monoxide, alpha-naphthoflavone, and metyrapone in
the cortex and the outer and inner medulla. This suggests differences
in the genetic expression of the same type of enzymic activity in
different zones of the kidney.
Davis et al. (1981) showed that oxidative metabolism in the
medulla is mediated in the absence of spectrophotometrically
measurable cytochrome P-450. Zenser et al. (1979a) reported that
cortex microsomes metabolized 1,3diphenylisobenzofuran to
O-dibenzoylbenzene largely via a cytochrome P-450-like system (it
was NADPH dependent and inhibited by carbon monoxide and metyrapone).
The inner medulla microsomes had the same metabolic capacity in the
presence of arachidonic acid (the system was independent of NADPH, and
it was inhibited by non-steroidal antiinflammatory compounds such as
indomethacin and not by carbon monoxide or metyrapone). The outer
medulla microsomes had both types of activity. The antioxidant
ethoxyquin inhibited the arachidonic acid and the NADPH-dependent
metabolic processes.
The specific arachidonic-acid-dependent PG
cyclooxygenase-mediated metabolism of benzidine has been shown to be
absent from hepatic and renal cortical microsomes but active in
medullary microsomes, especially those from the inner medulla. The
metabolism was inhibited by nonsteroidal anti-inflammatory drugs,
ethoxyquin, and arachidonic acid analogues. Approximately 75% of
metabolized benzidine was covalently bound to macromolecules,
presumably via a reactive intermediate. Addition of sulfhydryl
protectors, such as glutathione, reduced the amount of covalently
bound metabolite to 25% (Zenser et al., 1979b,c). Using rabbit renal
inner medullary slices, Rapp et al. (1980) have confirmed the
arachidonic-acid-dependent co-oxidative activation of low
concentrations of benzidine, and its covalent binding to tissue.
Mohandas et al. (1981a,b) examined the activation of paracetamol
(acetaminophen) and the covalent binding to protein in the inner
medulla, outer medulla, and renal cortex, and compared this to the
situation in the liver. The arachidonicacid-dependent pathway showed
a ten times greater degree of activity in the inner medulla compared
to the renal cortex, intermediate activity being found in the outer
medulla. In the liver the arachadonic-acid-dependent pathway activity
was approximately 50% of that of the renal cortex. The total
activation of paracetamol (acetaminophen) by arachadonic-acid- and
NADPH-dependent pathways in the renal cortex and the liver was
essentially the same.
3.6.2 Conjugation
Conjugation takes place on existing groups or those produced by
oxidation, and greatly increases the polarity of compounds. This
facilitates their elimination and generally terminates any
pharmacological activity. There are several examples, however, where
conjugation may give rise to reactive compounds (e.g., the
glucuronides of N-hydroxy-2-acetylaminofluorene and
N-hydroxyphenacetin are potently toxic). Similarly, glutathione
conjugates may be toxic (section 6.3.2.1).
3.6.2.1 Glucuronide conjugation
Glucuronide conjugates are formed by the action of uridine
diphosphate (UDP) glucuronyl transferase. This enzyme has at least
three isozymes, each of which preferentially conjugates different
types of molecules. Only UDP-GT1 occurs in the rat kidney, where the
substrates include planar compounds such as 1-naphthol and
4-nitrophenol. UDP-GT1 activity is increased by 3-methylcholanthrene.
Human kidneys have UDP-GT1 and high GT2 activities, while rabbit
kidneys have all three isoenzymes. UDPglucuronyl transferase activity
is highest in the cortex, and the distal tubule activity is about 50%
of that found in the proximal tubule (Cojocel et al., 1983). The
enzyme is also measurable in rat kidney medulla, but here its activity
may be limited by the availability of the cosubstrate, UDP-glucuronic
acid. Renal glucuronidation capacity may be comparable to or greater
than that of the liver, depending on the substrate, and microsomes
from female rats form considerably more glucuronide conjugates than
those from male rat kidneys.
3.6.2.2 Sulfate conjugation
Sulfotransferases form highly polar and, therefore, rapidly
excreted sulfate esters. The concentrations of both sulfotransferase
and activated sulfate are higher in the renal cortex than in the
medulla, and renal sulfotransferase activity is markedly lower than
that of the liver. The capacity to synthesize sulfate conjugates is
not increased by standard inducers.
3.6.2.3 Glutathione conjugation
Glutathione is the most abundant thiol-containing peptide in the
kidney, where it is synthesized in the proximal tubule and provides a
scavenger for detoxifying electrophilic radicals formed from alkyl and
aryl halides, epoxides, and alkenes (Ormstad, 1987). These compounds
are degraded to the cysteine conjugate and are generally excreted as
the N-acetyl-cysteine conjugate. Glutathione may also have ligand
binding and transport properties, and it has been suggested that it is
an important carrier in the transfer of amino acids from the extra- to
the intracellular space. Glutathione S-transferase plays an active
role in metabolism, where it catalyses the initial step in glutathione
conjugation of halogenated aromatics, epoxides, halogenated alkyls and
aralkyls, and alpha,ß-unsaturated compounds (Reed & Beatty, 1980),
drugs such as paracetamol (acetaminophen), and endogenous substrates
such as estrogen and PGs (Moldeus et al., 1978; Jones et al., 1979;
Kaplowitz, 1980).
The total renal GSH S-transferase activity per g wet tissue is
considerably less than the corresponding hepatic activity (Hales et
al., 1978). There are sex differences in the renal GSH S-transferase
activities in rats, aralkyl, epoxide, and alkyl transferase activities
being lower in males than in females. Rat kidney glutathione
S-transferases consist of three distinct proteins. One is identical
to hepatic transferase B (ligandin), a second conjugates
alpha,ß-unsaturated substrates similarly to the hepatic enzyme, and
the third, renal transferase, is unique to the kidney and active with
p-nitrobenzyl chloride (Hales et al., 1978).
Renal GSH S-transferases are under complex hormonal control.
Hypophysectomy in male rats significantly increases GSH S-aryl,
aralkyl, and epoxide transferase activities without altering GSH
S-alkyl and alkene activities. GSH S-transferase can be induced by
a number of chemicals, but the profile of activities affected is
complex. Phenobarbital fails to induce cytochrome P-450 activity in
rat kidney, but increases GSH S-aralkyl transferase activity without
affecting GSH S-alkyl, aryl, and epoxide transferases.
3-Methylcholanthrene (3MC) induces renal GSH S-aryl and aralkyl
transferase activities but not GSH S-alkyl or epoxide transferases
(Clifton et al., 1975; Chasseaud, 1980).
3.6.2.4 Mercapturic acid synthesis
The formation of glutathione conjugates is the first in the
pathway of renal metabolism to mercapturic acid. The enzyme
gamma-glutamyl transpeptidase is localized on the brush border of
the proximal tubule, where it cleaves the gamma-glutamyl linkage of
glutathione to produce the cysteinyl-glycine conjugate in the tubule
lumen. This metabolite is a substrate for a number of peptidases that
produce the cysteinyl conjugate, which in turn is converted to the
mercapturic acid by microsomal N-acetyltransferase (Green & Elce,
1975). The cells of the proximal tubule in the outer medulla produce
the N-acetyl-cysteine conjugate of paracetamol (Jones et al., 1979).
Glutathione conjugation is generally a detoxification pathway,
but some compounds may undergo bioactivation by ß-lyase (localized in
the outer mitochondrial membrane and the cytoplasm of the proximal
tubule). This enzyme is now known to be capable of cleaving the C-S
bond, leaving a reactive intermediate.
Extrarenal biotransformations are now known to produce substrates
for renal enzymes, which convert these metabolites into reactive
intermediates that cause target selective toxicity.
Hexachloro-1,3-butadiene (HCBD) is metabolized in the liver probably
by GSH S-transferasecatalysed halogen substitution rather than a
cytochrome P-450-mediated reaction to the GSH conjugate. In the
process, hepatic but not renal GSH is depleted in the male rat,
whereas GSH decreases in the female rat kidney. The HCBD-GSH conjugate
may be transported to the bile, returned to the bloodstream via
intestinal reabsorption, and excreted via the kidneys (Nash et al.,
1984). The cysteine conjugate of HCBD, S-pentachlorobuta-1,3-dienyl
cysteine, causes the same lesion. The enzyme ß-lyase is present in
both the liver and kidney. The unique renal susceptibility to the
HCBD-GSH metabolite appears to be related to accumulation via the
organic anion transport, as this is inhibited by probenecid both
in vivo and in vitro (Fig. 10).
Some alkylhalides, such as 1,2-dibromoethane (Lock, 1987) and
1,2-dibromo-3-chloropropane (DBCP) (Dybing et al., 1989), may form
reactive, nephrotoxic intermediates (presumably episulfonium ions)
following conjugation with glutathione without further metabolism by
ß-lyase. 1,2-Dibromoethane and DBCP are metabolized in the liver by
both cytochrome P-450 oxidative dehalogenation and by
glutathione- S-transferase-mediated substitution. The renal cortex
of the rat contains substantial quantities of glutathione
S-transferase with a high activity towards 1,2-dibromoethane (Lock,
1987). In the male rat kidney, studies with perdeutero-DBCP indicate
that DBCP is not metabolized by cytochrome P-450, but presumably by
glutathione S-transferase. Furthermore, inhibitors of
gamma-glutamyl transpeptidase and ß-lyase do not affect DBCP-induced
renal tubular necrosis (Omichinski et al., 1987; Dybing et al., 1989).
3.6.2.5 Amino acid conjugation
The amino acid glycine forms a glyco-conjugate via the activation
of a carboxylic acid (e.g., salicylic acid) by coenzyme A. Salicyl-CoA
and glycine are co-substrates for acyl-CoA-glycine-
N-acetyltransferase, which catalyses the condensation to
salicyluric acid. Glycine N-acetyltransferase activity is present in
the kidneys of rabbits, monkeys, and humans (Bekersky et al., 1980).
The kidney may be a major site for the metabolism of benzoic acid to
hippuric acid, p-aminobenzoic acid to p-aminohippuric acid, and
salicylate to salicyluric acid (Wan & Riegelman, 1972a,b; Wan et al.,
1972). The isolated rat kidney perfused with glycine and salicylic
acid excretes 3-4% of the salicylic acid as the glycine conjugate (Wan
& Riegelman, 1972a,b; Wan et al., 1972; Bekersky et al., 1980). These
reactions are reversible in the kidney. About 20% of salicyluric acid
is converted to salicylic acid by the isolated perfused rat kidney,
whereas the liver does not deconjugate salicyluric acid to salicylate.
The freshly deconjugated salicylate is more rapidly excreted than the
parent salicylate. Possibly the diffusion of salicylate into the renal
cell is the rate-limiting step of elimination. This is not the case
for salicyluric acid, which is converted to salicylate within tubular
cells (Bekersky et al., 1980).
collecting tubules, followed by cortical collecting ducts and
glomeruli. Furthermore, there are marked sex-related differences in
the effects of cofactors on medullary PG synthetase activity (Hirafuji
et al., 1980). Some PGs break down spontaneously (e.g., PGI2 to
alpha-6-keto-PGF), but the majority are metabolically degraded
(Morrison, 1980). The enzymic conversions are mediated by a number of
enzymes, including dehydrogenases, reductases, and ß- and
omega-oxidases. The enzymes that degrade PGs are located mainly in the
cortex, but there are species differences in the corticomedullary
ratio of these enzymic activities (Powell, 1980).
The factors regulating the biosynthesis of each type of PG are
only poorly defined (Horrobin, 1980). A large number of endogenous and
exogenous substances have been reported to alter renal PG synthesis,
and several patho-physiological conditions have been described in
which renal PG synthesis is increased. Most attention has been
focussed on the inhibitory effects of the anti-inflammatory drugs. The
steroidal compounds (e.g., corticosteroids) prevent the release of
arachidonic acid from its lipid pools, and the non-steroidal
products (e.g., indomethacin) inhibit cyclo-oxygenase. It is, however,
essential to be aware that any factor that perturbs PG synthesis may
act differently at different sites in the synthetic (or degradative)
pathway.
Indomethacin (one of the most extensively studied cyclo-oxygenase
inhibitors) produces several alterations in renal PG dynamics.
Uncertainties in defining PG-"related" pathophysiological changes are
compounded by the observation (Attallah & Stahl, 1980) that PGE2
synthesis in slices from each zone of the kidney has a different dose
response to indomethacin inhibition: the cortex is most sensitive and
the papilla least sensitive. The cyclo-oxygenase inhibitors are
generally classified as either reversible or irreversible, but the
multiplicity of effects and the possibility of enzymic polymorphism in
different regions of the kidney suggest that such a classification may
be an over-simplification.
The exact physiological roles of the PGs in normal renal function
and the way in which these are altered in the development of
nephropathies are not clear. Firstly, indomethacin, for example, has
been shown to cause biochemical changes that may be classified as
either related or unrelated to altering PG dynamics. Secondly, many
attempts to define renal PG function have been based on the hypothesis
that urinary PG excretion reflects de novo renal synthesis (Dunn &
Hood, 1977; Dunn & Zambraski, 1980; Dunn 1981), notwithstanding
analytical difficulties of measuring very low levels of various PGs
and apparently ignoring the fact that de novo synthesized PGs may
have undergone extensive degradation. The measurement of urinary PGs,
as an estimate of their de novo renal synthesis, remains equivocal
because, firstly, seminal PGE2 is an unavoidable and variable
contaminant in the urine of males (Suzuki et al., 1980) and, secondly,
Brown et al. (1980) have demonstrated that both rabbit and rat urinary
bladders can synthesize PGE from arachidonic acid. Finally, the
physiology of renal function is controlled by several hormonal
systems, the detailed functioning of which has not been clearly
established. It is known that renal PGs may be altered by (or may
alter) the renin/angiotensin II/aldosterone system (Hackenthal et
al., 1980; Lee, 1980; Weber, 1980; Baer, 1981), the kallikrein-kinin
system (Margolius, 1980; Rockel & Heidland, 1980), and the regulation
of fluid balance and water reabsorption via ADH (Blair-West et al.,
1980). Furthermore, each of these hormonal systems may interact with
the others via direct or indirect mechanisms. It seems likely that a
full understanding of the pathophysiology of the renal hormonal
systems will take some time to crystallize.
In spite of the rather abstruse biology, there is general
consensus (Dunn & Hood 1977; Dunn & Zambraski, 1980; Morrison, 1980)
that PGs have a central role in renal function. It seems, however,
that renal PGs play little, if any, major regulatory role in basal
renal blood flow in normal conscious animals. There is evidence that
PGs are released in response to ischaemic and vasoconstrictive stress,
where their role seems to be to provide a protective effect by
maintaining glomerular dynamics. The role of PGs (especially PGE2)
in preventing experimentally induced acute renal failure is
conflicting. Arachidonic acid does stimulate renin release, a
response that is blocked by cyclo-oxygenase inhibitors, but it remains
uncertain which of the PGs mediate this effect in vivo. It is also
unclear from which renal zone such mediators are synthesized and
released. Renin release may, in turn, affect PG synthesis and the
kallikrein-kinin system (which in turn may modulate PG synthesis and
the renin system). ADH is assumed to stimulate PGE2, but published
data on the controlling effects of PGs on salt and water balance are
very difficult to interpret. Similarly, the mass of literature on
hypertension and PGs favours the concept that the two are related, but
fails to propound a unifying hypothesis.
3.5.2.2 Lipid metabolism
The contents of the interstitial lipid droplets are too
specialized to be used as a metabolic energy store (Bohman, 1980),
although this droplet population undergoes marked and rapid change
during the short periods that precede various pathophysiological
conditions. The interstitial cells produce the ground substance matrix
that surrounds them. Early evidence that the interstitial cells of the
renal medulla are only a highly specialized PG-producing cell type has
become equivocal. The interstitial lipid droplets do not, in fact,
provide the sole source of arachidonic acid for PG synthesis. Only 50%
of the medullary capacity for synthesizing PG is confined to the
interstitial cells; the rest is in the collecting ducts (Bohman,
1980). The significance of PG synthesis in the medullary cells cannot,
however, be overlooked, as it may play an important role in regulating
blood pressure and other renal functions. It has also been suggested
to occupy a central position in the pathogenesis of renal papillary
necrosis.
In recent years the importance of the endocrine function of the
medullary interstitial cells in regulating blood pressure has been
highlighted by several workers (Mandal & Bohman, 1980). Three groups
of vaso-active compounds have been isolated from the medulla or
cultured interstitial cells. Experimentally induced, spontaneously
occurring, and pathologically precipitated hypertensive states have
been reversed by subcutaneous transplants of renal papillary fragments
and by cultured interstitial cells. In addition, the systemic
administration of both the polar and neutral reno-medullary lipids
reduces arterial blood pressure (Muirhead & Pitcock, 1980).
The numerous lipid droplets in the interstitial cells have been
found to contain traces of cholesterol esters, a few percent of
phospholipids, mainly phosphatidylcholine, and, rarely, trace amounts
of phosphatidyl-ethanolamine. A few percent of free fatty acids and
tri-acylglycerols make up the remaining 80-90%, the composition of
which varies in different species. The most striking features are the
varied types and large amounts of unsaturated fatty acids; most
notably those of 20 or more carbon atoms such as arachidonic acid and
especially adrenic acid. The large amount of arachidonic acid suggests
that the interstitial lipid droplets may be an important pool for PG
synthesis in the kidney (Bojesen, 1974).
3.5.2.3 Carbohydrate metabolism in the medulla
The metabolism of carbohydrate in the renal medulla has been
reviewed by Cohen (1979). Some early observations suggested that the
low oxygen tension in the inner medulla (a pO2 as low as 0.67-2.0
kPa (5-l5 mmHg) compared with 10 kPa (75 mmHg) in the cortex) would
necessitate anaerobic metabolism. However, aerobic metabolism is only
limited at an O2 availability of less than 0.13 kPa (1 mmHg).
Many metabolic investigations have used medullary slices or
homogenates. Thus, the exact contribution of the different cell types
in the inner medulla to functional energy dynamics and to the changes
that underlie, for example, diuresis or anti-diuresis have yet to be
related to the phosphorylation and redox states within these
individual cell types.
Carbohydrates are stored in the medulla as either glycogen or as
glycosaminoglycan (GAG), the former in collecting ducts and epithelia
and the latter as an important constituent of the interstitial ground
substance. There is evidence to suggest that either can be mobilized
to provide an energy source or the glucose units for the synthesis of
the other macromolecular carbohydrates (Darnton, 1967, 1969).
3.5.2.4 Medullary glycosaminoglycan (GAG)
The biology of GAGs has been described by Kennedy (1979). GAGs
are linear polysaccharides that are made up of repeating disaccharide
units, one carbohydrate moiety of which is a hexuronic acid (or a
neutral sugar in one case) and the other a hexosamine. The nature of
the disaccharide units and the occurrence of N-acetyl groups,
together with the position of O-sulfate groups, define the species
of macromolecule. There are seven basic types of GAG. These molecules
also show heterogeneity of relative molecular mass (when isolated from
the same or different organs; Toledo & Dietrich, 1977), and the molar
ratio of sulfate to hexosamine varies by up to 2-fold for the same
type of GAG (Suzuki et al., 1976). Most of these substances probably
occur in vivo as proteoglycans (PoGs). These supramolecular
structures are composed of a linear protein backbone that carries GAGs
covalently bound at intervals along its length. In theory, any
combination and ratio of GAGs may occur. It is only recently that the
concept of PoGs has been accepted; before this the presence of protein
was assumed to be a contamination and vigorous steps were taken to
remove it.
In spite of the ubiquity of PoGs and their composite GAGs,
relatively little is known about their physiological functions, with
the exception of their anti-coagulant and anti-lipaemic properties,
which are best studied in heparin. These molecules are bound to cell
surfaces (Kjellen et al., 1977), where they may control the access of
endogenous and exogenous molecules to cell membrane receptors.
Similarly, the functions of this intercellular polyanionic matrix most
probably extend beyond that of "immobilized anti-coagulants" or "space
filling", and include controlling the micro-environment of cells (by
binding either inorganic or organic cations and by their immense
water-holding capacity) and regulating cell-cell communications. GAGs
may also control cell recognition and adhesion, and contribute to the
control of cell movement, growth, differentiation, and proliferation
(Long & Williamson, 1979). The association of GAGs with mitochondria
and nuclear membranes suggests that these macromolecules may also play
a direct role in controlling some intracellular functions.
The distribution of GAGs has been assessed in tissue either by
the autoradiographic distribution of precursor carbohydrates or
35SO2 or by histochemical staining. It is generally assumed that
sulfate radiolabel distribution is relatively specific for GAGs.
However, most of the staining procedures are nonspecific (e.g.,
toluidine blue interacts with any polyanion to give a metachromatic
colour shift) and depend either on a priori knowledge of
distribution or, for example, the use of control sections that have
been exposed to selective enzymic digestion.
The amount and types of GAGs in the kidneys of various species
have been reported. The quantity of polyanionic macromolecule has been
found to be greater in the medulla than in the cortex for the rat
(Jacobsen et al., 1964; Kresse & Grossmann, 1970), pig (Kresse &
Grossmann 1970), dog (Castor & Green, 1968; Kresse & Grossmann, 1970),
and normal human kidney (Inoue et al., 1973; Constantopoulos et al.,
1973). The medulla:cortex ratio in the human kidney was found to be
age related, increasing rapidly to a maximum in the fourth decade and
then declining slowly (Inone et al., 1973). The heterogeneous
distribution of the types of GAG in the kidney is supported by the
data of Constantopoulos et al. (1973) for the human kidney and Castor
& Green (1968), who reported that hyaluronic acid had a high relative
molecular mass in the medulla but a low one in the cortex of dogs.
However, other data on the dog, pig, and sheep (Dicker & Franklin,
1966) and the rat (Barry & Bowness, 1975) suggest that the types and
quantities of GAG are the same in both the cortex and medulla.
The processes underlying and controlling the biosynthesis of PoGs
are complex and incompletely documented (Kennedy, 1979). Muirhead &
Pitcock (1980) reported that medullary interstitial cells synthesize
PoGs (both in situ and in culture) and that these macromolecules are
associated with the cellular cisternae (dilated rough endoplasmic
reticulum). Darnton (1967, 1969) presented data to show that glycogen
associated with the epithelial cells of the collecting duct in the
rabbit is mobilized and incorporated into GAGs.
The functions of the medullary GAGs have been the centre of
controversy since Ginetzinsky (1958) suggested that the action of ADH
was mediated by the release of hyaluronidase. This would depolymerize
medullary GAG and (so it was argued) allow greater water reabsorbtion
from the tubules into the interstitium and thence to the blood supply.
This hypothesis has been supported by some workers (Jacobson et al.,
1964; Farber et al., 1971) but refuted by others (Sun et al., 1972;
McAuliffe 1978, 1980; Sun, 1980) in animals with spontaneous diabetes
insipidus. These conflicting data are difficult to resolve into a
single unifying theory relating the physiological function of GAGs to
the urine-concentrating process.
3.6 The metabolism of xenobiotic molecules in the kidney
Chemically induced lesions may depend to varying extents on the
metabolic capacity of tissues to deal with "insults". The metabolism
of xenobiotic molecules may either prevent lesions (by deactivation),
or be directly responsible for damage (by bio-activation). The renal
metabolism of chemicals (and its consequences) has been reviewed by
Hook et al. (1979), Anders (1980), Connelly & Bridges (1980), Kluwe &
Hook (1980), Davis et al. (1981), Rush et al., (1984) and Tarloff et
al. (1987).
It is likely that the liver meets the challenge of metabolizing
a major proportion of exogenous compounds in vivo before they reach
the systemic circulation. Most fundamental types of bioconversion have
been described for the perfused kidney of several species (Szefler &
Acara, 1979; Elbers et al., 1980; Ross et al., 1980; Emslie et al.,
1981) and for isolated renal cells and tubular fragments (Fry et al.,
1978; Jones et al., 1979; Ormstad, 1982). Similarly, kidney
microsomes have been shown to have most of the enzymic and
cytochrome-mediated metabolic activities that have been described in
other tissues. The xenobiotic-transformating capacity of the kidney is
about 3-50% (depending on the system, species, and source of data) of
that found in the liver (Litterst et al., 1975a, Navran &
Louis-Ferdinand 1975; Fry et al., 1978), but it may be much higher
than that of the liver under certain circumstances (Anders, 1980).
There are marked qualitative differences between hepatic and renal
xenobiotic metabolism. Renal enzymes are stable during the Ca2+
method of preparing microsomes (Litterst et al., 1975b). Enzymic
kinetic constants vary between microsomes isolated from the two organs
(Navran & Louis-Ferdinand, 1975). Whereas there are marked sex-related
differences in hepatic metabolism, there are few in the kidney
(Litterst et al., 1977). There is evidence to suggest that liver
cytochrome P-450 is similar to that of the kidney, based on
electro-phoretic and electron paramagnetic resonance studies
(Armbrecht et al., 1979), immunological criteria (Guengerich & Mason,
1979), and on immunometabolic studies (Kaminsky et al., 1979).
However, these data are most difficult to interpret in "absolute"
terms, because the samples of cytochrome P-450 were from organs
exposed to different inducing agents. There is now substantial
evidence that hepatic and renal tissue respond differently, both
quantitatively and qualitatively, to the various inducers of
cytochrome P-450 (Litterst et al., 1977; Zenser et al., 1978a;
Kaminsky et al., 1979). Ascorbic acid deficiency (Sikic et al., 1977)
and carbon tetrachloride pretreatment (Litterst et al., 1977) alter
the metabolism of xenobiotics in a different way in the liver and
kidney. In addition, the inhibitory effects of
2-diethylaminoethyl-2,2-diphenylvalerate (SKF-525A) on renal and
hepatic microsomes studied in vitro are similar but not identical
(Litterst et al., 1977).
There are several enzymes involved in renal xenobiotic
metabolism. It is not possible to comment on all of those that may be
relevant to nephrotoxicity nor, indeed, is it clearly established what
role each renal enzyme plays in the realization of the potential
toxicity of a chemical. Many of the enzymes that metabolize
xenobiotics are compartmentalized in specific regions of the kidney.
The anatomical localization of these characteristics may play a key
role in the toxicological consequence that follows the entry of a
xenobiotic into the kidney. The distribution and regulation of these
enzymes may predispose to the toxic effects of chemicals. Thus,
although intrarenal metabolism may be a prerequisite for the target
selective effects of some chemicals, the final outcome of the toxic
response relates to the sum of a number of factors. These include the
localization of those renal enzymes involved in xenobiotic metabolism
(this may be metabolic activation or other processes, perhaps in an
adjacent cell) and the processes controlling the intracellular
concentration of toxic chemicals. The intracellular concentration of
a chemical can be influenced by xenobiotic metabolism per se and by
many of the inherent processes in the kidney, such as transport, pH,
and solute gradients on either side of a membrane. The outcome of a
chemical exposure may also be affected by the numbers and types of
organelles in a specific cell type that have critical properties
relevant to the functions of that cell and by the presence of a
protective mechanism in a particular cell type (such as antioxidants,
free radical scavengers). Most of the processes that underlie
nephrotoxicity are probably multi-step events that are affected by
more than one metabolic pathway and occur via competing and sequential
pathways. Little is known about the control of these pathways, so that
it is difficult to predict from the structure of a chemical alone what
effects it will have on the kidney. In addition, a major role is
obviously played by the extra-renal metabolism (in the liver, lung,
gut, etc.) of the parent chemical and by a variety of other organ
functions. These include the lung (exhalation of volatile
metabolites), liver (biliary excretion), and gastrointestinal
microflora (enterohepatic circulation, serum protein binding), and
they determine the types of chemicals, the concentration that reaches
the kidney, and their renal pharmacokinetics.
3.6.1 Oxidases
Oxidases can convert chemicals into active intermediates or
generate reactive species by redox cycling. This is potentially
important for compounds that contain arylamine, quaternary bipyridyl
(paraquat), quinone (adriamycin), or nitro (nitrofurantoin)
structures. It is generally accepted that, in the liver, biologically
reactive intermediates mediate their toxic effects by binding to
cellular macromolecules and blocking normal functional processes
(Jollow et al., 1976; Snyder et al., 1981). Similar mechanisms have
been proposed to explain various types of chemically induced renal
lesions, including papillary necrosis. The metabolically generated
reactive intermediates have a relatively short life and are most
likely formed in the organ or anatomical area in which they induce
damage.
3.6.1.1 Cytochrome-P-450-dependent mixed-function oxidases (monooxygenases)
This enzyme system carries out a two-electron flow pathway, and
the flavoprotein component can catalyse single electron reductions
such as the reduction of quinones to semiquinone radicals (Bachur et
al., 1979). Multiple forms of cytochrome P-450 have been identified in
the kidney. These include phenobarbital-, 3-methylcholanthrene-, and
ß-naphthoflavone-inducible cytochrome P-450. Renal cytochrome P-450
induction varies in different species. Polycyclic aromatic
hydrocarbons induce cytochrome P-450 in most species, whereas
phenobarbital is effective in hamsters and rabbits but not in
guinea-pigs, rats, and mice (Smith et al., 1986). Similarly, renal
P-450 responds differently to inhibitors of mixed-function oxidases.
The effects of inhibitors such as SKF-525A are further
complicated by multiple actions on renal transport, intracellular
binding of chemicals at noncatalytic sites, and on cytochrome
P-450-dependent metabolism. Other inhibitors do not appear to have
been as fully studied, and the paucity of data in this area makes
other studies on the effects of inhibitors most difficult to
interpret. Some species also have sex-related differences, e.g., male
mice have higher concentrations and activities of P-450 than female
mice (Krijsheld & Gram, 1984; Smith et al., 1984; Hawke & Welch,
1985), but this is not the case for rats (Litterst et al., 1977; Hook
et al., 1982) or rabbits (Litterst et al., 1977). There is no clear
data on other species, such as man, nor on how different types of
renal disease affect the concentration of renal P-450 or its induction
or inhibition.
The specific activity of the renal mixed-function oxidases varies
widely between species and is about 10% of the hepatic activity
(Zenser et al., 1978a,b; Endou, 1983). This suggests a role for renal
P-450 that is quantitatively less important than that of the liver.
However, this is not the case for all chemicals, since the renal
metabolism of chloroform is about 2-fold higher than the hepatic
activity (Smith & Hook 1984). More importantly, mixed-function
oxidase activities are intra-renally localized to discrete areas
where their significance in metabolism may be far greater than in the
liver. The S2 proximal segment has a cytochrome P-450 concentration
that is 2-3 times higher than the S1 or S3 segments. The distal
tubules, cortical collecting ducts, and the medulla contain no
measurable cytochrome P-450 activity (Endou, 1983). By contrast
NADPH-cytochrome-P-450 reductase activity is highest in the S2 and
S3 segments, but it also extends to the distal tubule and medullary
structures.
3.6.1.2 Prostaglandin peroxidase-mediated metabolic activation
Recently, it has been shown that there are marked quantitative
and qualitative differences in the regional distribution of microsomal
mixed-function oxidase activity within the rabbit kidney (Zenser et
al., 1978a,b; Armbrecht et al., 1979). Most mixed-function oxidase
activity is located in the cortex and least in the inner medulla in
control tissue and in that taken from animals induced with
3-methylcholanthrene. Cytochrome P-450 is not detected in the medulla
of controls or even those of induced animals. In addition, laurate
hydroxylase activity (the only mixed-function oxidase activity found
in the inner medulla) shows marked differences in the pattern of
inhibition by carbon monoxide, alpha-naphthoflavone, and metyrapone in
the cortex and the outer and inner medulla. This suggests differences
in the genetic expression of the same type of enzymic activity in
different zones of the kidney.
Davis et al. (1981) showed that oxidative metabolism in the
medulla is mediated in the absence of spectrophotometrically
measurable cytochrome P-450. Zenser et al. (1979a) reported that
cortex microsomes metabolized 1,3diphenylisobenzofuran to
O-dibenzoylbenzene largely via a cytochrome P-450-like system (it
was NADPH dependent and inhibited by carbon monoxide and metyrapone).
The inner medulla microsomes had the same metabolic capacity in the
presence of arachidonic acid (the system was independent of NADPH, and
it was inhibited by non-steroidal antiinflammatory compounds such as
indomethacin and not by carbon monoxide or metyrapone). The outer
medulla microsomes had both types of activity. The antioxidant
ethoxyquin inhibited the arachidonic acid and the NADPH-dependent
metabolic processes.
The specific arachidonic-acid-dependent PG
cyclooxygenase-mediated metabolism of benzidine has been shown to be
absent from hepatic and renal cortical microsomes but active in
medullary microsomes, especially those from the inner medulla. The
metabolism was inhibited by nonsteroidal anti-inflammatory drugs,
ethoxyquin, and arachidonic acid analogues. Approximately 75% of
metabolized benzidine was covalently bound to macromolecules,
presumably via a reactive intermediate. Addition of sulfhydryl
protectors, such as glutathione, reduced the amount of covalently
bound metabolite to 25% (Zenser et al., 1979b,c). Using rabbit renal
inner medullary slices, Rapp et al. (1980) have confirmed the
arachidonic-acid-dependent co-oxidative activation of low
concentrations of benzidine, and its covalent binding to tissue.
Mohandas et al. (1981a,b) examined the activation of paracetamol
(acetaminophen) and the covalent binding to protein in the inner
medulla, outer medulla, and renal cortex, and compared this to the
situation in the liver. The arachidonicacid-dependent pathway showed
a ten times greater degree of activity in the inner medulla compared
to the renal cortex, intermediate activity being found in the outer
medulla. In the liver the arachadonic-acid-dependent pathway activity
was approximately 50% of that of the renal cortex. The total
activation of paracetamol (acetaminophen) by arachadonic-acid- and
NADPH-dependent pathways in the renal cortex and the liver was
essentially the same.
3.6.2 Conjugation
Conjugation takes place on existing groups or those produced by
oxidation, and greatly increases the polarity of compounds. This
facilitates their elimination and generally terminates any
pharmacological activity. There are several examples, however, where
conjugation may give rise to reactive compounds (e.g., the
glucuronides of N-hydroxy-2-acetylaminofluorene and
N-hydroxyphenacetin are potently toxic). Similarly, glutathione
conjugates may be toxic (section 6.3.2.1).
3.6.2.1 Glucuronide conjugation
Glucuronide conjugates are formed by the action of uridine
diphosphate (UDP) glucuronyl transferase. This enzyme has at least
three isozymes, each of which preferentially conjugates different
types of molecules. Only UDP-GT1 occurs in the rat kidney, where the
substrates include planar compounds such as 1-naphthol and
4-nitrophenol. UDP-GT1 activity is increased by 3-methylcholanthrene.
Human kidneys have UDP-GT1 and high GT2 activities, while rabbit
kidneys have all three isoenzymes. UDPglucuronyl transferase activity
is highest in the cortex, and the distal tubule activity is about 50%
of that found in the proximal tubule (Cojocel et al., 1983). The
enzyme is also measurable in rat kidney medulla, but here its activity
may be limited by the availability of the cosubstrate, UDP-glucuronic
acid. Renal glucuronidation capacity may be comparable to or greater
than that of the liver, depending on the substrate, and microsomes
from female rats form considerably more glucuronide conjugates than
those from male rat kidneys.
3.6.2.2 Sulfate conjugation
Sulfotransferases form highly polar and, therefore, rapidly
excreted sulfate esters. The concentrations of both sulfotransferase
and activated sulfate are higher in the renal cortex than in the
medulla, and renal sulfotransferase activity is markedly lower than
that of the liver. The capacity to synthesize sulfate conjugates is
not increased by standard inducers.
3.6.2.3 Glutathione conjugation
Glutathione is the most abundant thiol-containing peptide in the
kidney, where it is synthesized in the proximal tubule and provides a
scavenger for detoxifying electrophilic radicals formed from alkyl and
aryl halides, epoxides, and alkenes (Ormstad, 1987). These compounds
are degraded to the cysteine conjugate and are generally excreted as
the N-acetyl-cysteine conjugate. Glutathione may also have ligand
binding and transport properties, and it has been suggested that it is
an important carrier in the transfer of amino acids from the extra- to
the intracellular space. Glutathione S-transferase plays an active
role in metabolism, where it catalyses the initial step in glutathione
conjugation of halogenated aromatics, epoxides, halogenated alkyls and
aralkyls, and alpha,ß-unsaturated compounds (Reed & Beatty, 1980),
drugs such as paracetamol (acetaminophen), and endogenous substrates
such as estrogen and PGs (Moldeus et al., 1978; Jones et al., 1979;
Kaplowitz, 1980).
The total renal GSH S-transferase activity per g wet tissue is
considerably less than the corresponding hepatic activity (Hales et
al., 1978). There are sex differences in the renal GSH S-transferase
activities in rats, aralkyl, epoxide, and alkyl transferase activities
being lower in males than in females. Rat kidney glutathione
S-transferases consist of three distinct proteins. One is identical
to hepatic transferase B (ligandin), a second conjugates
alpha,ß-unsaturated substrates similarly to the hepatic enzyme, and
the third, renal transferase, is unique to the kidney and active with
p-nitrobenzyl chloride (Hales et al., 1978).
Renal GSH S-transferases are under complex hormonal control.
Hypophysectomy in male rats significantly increases GSH S-aryl,
aralkyl, and epoxide transferase activities without altering GSH
S-alkyl and alkene activities. GSH S-transferase can be induced by
a number of chemicals, but the profile of activities affected is
complex. Phenobarbital fails to induce cytochrome P-450 activity in
rat kidney, but increases GSH S-aralkyl transferase activity without
affecting GSH S-alkyl, aryl, and epoxide transferases.
3-Methylcholanthrene (3MC) induces renal GSH S-aryl and aralkyl
transferase activities but not GSH S-alkyl or epoxide transferases
(Clifton et al., 1975; Chasseaud, 1980).
3.6.2.4 Mercapturic acid synthesis
The formation of glutathione conjugates is the first in the
pathway of renal metabolism to mercapturic acid. The enzyme
gamma-glutamyl transpeptidase is localized on the brush border of
the proximal tubule, where it cleaves the gamma-glutamyl linkage of
glutathione to produce the cysteinyl-glycine conjugate in the tubule
lumen. This metabolite is a substrate for a number of peptidases that
produce the cysteinyl conjugate, which in turn is converted to the
mercapturic acid by microsomal N-acetyltransferase (Green & Elce,
1975). The cells of the proximal tubule in the outer medulla produce
the N-acetyl-cysteine conjugate of paracetamol (Jones et al., 1979).
Glutathione conjugation is generally a detoxification pathway,
but some compounds may undergo bioactivation by ß-lyase (localized in
the outer mitochondrial membrane and the cytoplasm of the proximal
tubule). This enzyme is now known to be capable of cleaving the C-S
bond, leaving a reactive intermediate.
Extrarenal biotransformations are now known to produce substrates
for renal enzymes, which convert these metabolites into reactive
intermediates that cause target selective toxicity.
Hexachloro-1,3-butadiene (HCBD) is metabolized in the liver probably
by GSH S-transferasecatalysed halogen substitution rather than a
cytochrome P-450-mediated reaction to the GSH conjugate. In the
process, hepatic but not renal GSH is depleted in the male rat,
whereas GSH decreases in the female rat kidney. The HCBD-GSH conjugate
may be transported to the bile, returned to the bloodstream via
intestinal reabsorption, and excreted via the kidneys (Nash et al.,
1984). The cysteine conjugate of HCBD, S-pentachlorobuta-1,3-dienyl
cysteine, causes the same lesion. The enzyme ß-lyase is present in
both the liver and kidney. The unique renal susceptibility to the
HCBD-GSH metabolite appears to be related to accumulation via the
organic anion transport, as this is inhibited by probenecid both
in vivo and in vitro (Fig. 10).
Some alkylhalides, such as 1,2-dibromoethane (Lock, 1987) and
1,2-dibromo-3-chloropropane (DBCP) (Dybing et al., 1989), may form
reactive, nephrotoxic intermediates (presumably episulfonium ions)
following conjugation with glutathione without further metabolism by
ß-lyase. 1,2-Dibromoethane and DBCP are metabolized in the liver by
both cytochrome P-450 oxidative dehalogenation and by
glutathione- S-transferase-mediated substitution. The renal cortex
of the rat contains substantial quantities of glutathione
S-transferase with a high activity towards 1,2-dibromoethane (Lock,
1987). In the male rat kidney, studies with perdeutero-DBCP indicate
that DBCP is not metabolized by cytochrome P-450, but presumably by
glutathione S-transferase. Furthermore, inhibitors of
gamma-glutamyl transpeptidase and ß-lyase do not affect DBCP-induced
renal tubular necrosis (Omichinski et al., 1987; Dybing et al., 1989).
3.6.2.5 Amino acid conjugation
The amino acid glycine forms a glyco-conjugate via the activation
of a carboxylic acid (e.g., salicylic acid) by coenzyme A. Salicyl-CoA
and glycine are co-substrates for acyl-CoA-glycine-
N-acetyltransferase, which catalyses the condensation to
salicyluric acid. Glycine N-acetyltransferase activity is present in
the kidneys of rabbits, monkeys, and humans (Bekersky et al., 1980).
The kidney may be a major site for the metabolism of benzoic acid to
hippuric acid, p-aminobenzoic acid to p-aminohippuric acid, and
salicylate to salicyluric acid (Wan & Riegelman, 1972a,b; Wan et al.,
1972). The isolated rat kidney perfused with glycine and salicylic
acid excretes 3-4% of the salicylic acid as the glycine conjugate (Wan
& Riegelman, 1972a,b; Wan et al., 1972; Bekersky et al., 1980). These
reactions are reversible in the kidney. About 20% of salicyluric acid
is converted to salicylic acid by the isolated perfused rat kidney,
whereas the liver does not deconjugate salicyluric acid to salicylate.
The freshly deconjugated salicylate is more rapidly excreted than the
parent salicylate. Possibly the diffusion of salicylate into the renal
cell is the rate-limiting step of elimination. This is not the case
for salicyluric acid, which is converted to salicylate within tubular
cells (Bekersky et al., 1980).
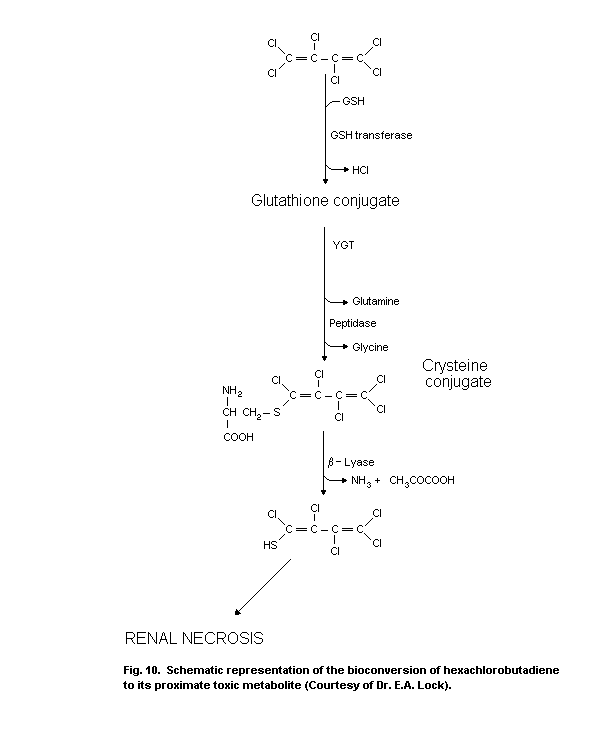 Other aromatic acids (such as benzoic acid or p-aminobenzoic
acid) competitively inhibited glycine conjugation. Cellular glycine is
limited, and so its use in conjugation would be saturated with
increasing concentrations of substrate.
3.6.3 Other enzymes involved in xenobiotic metabolism
Highly electrophilic epoxides play a key role in tissue
alkylation of macromolecules, especially nucleic acids, leading to
mutagenic and carcinogenic effects. Epoxide hydrase (hydrolase)
converts aliphatic and aromatic epoxides to their trans-hydrodiols, a
process that may generate both toxic and non-toxic products (Anders,
1980).
The oxidation of aldehydes to ketones or carboxylic acids in the
kidney is mediated by aldehyde oxidase and aldehyde dehydrogenase
(Goldberg & Anderson, 1985). Renal aldehyde oxidase activity is
localized in the proximal tubule but represents only 40% of the liver
activity (Anders, 1980), but aldehyde oxidase represents only 10% of
renal aldehyde dehydrogenase activity (Anders, 1980). At least two
aldehyde dehydrogenase isozymes occur in proximal tubule mitochondria
and cytosol. Substrates include formaldehyde, acetaldehyde, acrolein,
and malondialdehyde (Hjelle et al., 1983).
DT-diaphorase (NADPH-quinone oxidoreductase) is a cytosolic
enzyme that is present at highest concentration in the medulla and
catalyses a two-electron reduction of quinones to less reactive
hydroquinones. It therefore blocks redox cycling and the generation
of superoxide anion radicals (Lind et al., 1978).
4. THE MECHANISTIC BASIS OF CHEMICALLY INDUCED RENAL INJURY
Over the past 20-30 years there has been a growing understanding
of the molecular basis of disease and the biochemical mechanisms that
are associated with chemically induced cellular degeneration and
lesions in target organ systems. The application of this understanding
provides a foundation upon which to study chemically induced renal
injury and, in particular, a rational basis for the extrapolation of
animal toxicity data to man and risk assessment.
4.1 Immunologically induced glomerular disease
Immunologically mediated glomerulonephritis can result from the
deposition of free circulating antibodies interacting with a
structural glomerular antigen or with a "planted", non-glomerular
antigen (such as cationic proteins that fix on anionic sites of the
glomerular basement membrane). Alternatively, glomerular injury may
be the consequence of the deposition of circulating immune complexes.
The two main forms of antibody-mediated glomerulonephritis are the
anti-GBM-mediated disease and membranous glomerulonephritis (or
immune complex-type mediated glomerulonephritis). The former disease
is characterized by the presence of linear IgG deposits along the
glomerular basement membrane. The latter form may result from either
of the mechanisms mentioned, i.e. deposition of free circulating
antibodies against an irregularly distributed antigen, which may be of
glomerular origin, or deposition of circulating immune complexes. A
membranous glomerulonephritis is characterized by the presence of
granular IgG deposits along the glomerular basement membrane.
Besides antibody-mediated glomerulonephritis, it is more and more
apparent that there are cell-mediated glomerulonephritides without Ig
deposition. An example of such a disease is probably the nephrotic
syndrome with minimal glomerular changes at the light microscope
level.
The role of genetic factors, which has been well demonstrated in
the mercury model in the rat, is also clear in the human situation.
Membranous glomerulonephritis induced by gold and D-penicillamine is
much more frequent in DR3-positive rheumatoid arthritis patients and
in poor sulfoxydators. The lupus-like disease observed in
hydralazine-treated patients is more frequent in those with the DR4
antigen and "slow-acetylators".
Other drugs such as non-steroidal anti-inflammatory agents or
lithium salts may induce the nephrotic syndrome with minimal
glomerular changes. Immunofluorescence is negative in these cases, and
there is indirect evidence that such disease could be due to T
cell-mediated immunity.
There are many potential environmental agents, drugs, and toxic
chemicals that have been related to this form of glomerulonephritis.
Drugs and toxic chemicals that may induce glomerulonephritis in humans
include gold and mercury, d-penicillamine, non-steroidal
anti-inflammatory agents, and heroin. Other drugs and chemicals, in
which association is suspected but not certain, include silica
exposure, toxic-oil syndrome, hydrocarbon exposure, and interferon.
Other drugs may induce an immune complex type of glomerulonephritis in
the context of a lupus-like syndrome (e.g., hydralazine, procanamide,
and diphenylhydantoin. Furthermore, there are many other substances
that have been implicated by case reports or unconfirmed experiments.
Anti-GBM-mediated glomerulonephritis is not the usual mechanism
responsible for toxic glomerular nephropathy. However, exposure to
organic solvents is thought to be a factor in some cases of
Goodpasture's syndrome, which consists of the simultaneous occurrence
of necrotizing haemorrhagic interstitial pneumonitis and
proliferative usually rapidly progressive glomerulonephritis. This is
an auto-immune disease resulting from antiGBM antibodies that
cross-react with basement membranes in lung alveoli.
The mechanisms by which drugs and chemicals induce
immunologically related renal disease are both complex and not
entirely understood. It is unlikely that drugs or toxins induce renal
damage by modifying self antigens or by acting as haptens. On the
other hand, many agents such as gold, d-penicillamine, and mercury
may have an immunomodulatory effect. This mechanism of action is also
not understood, but Druet et al. (1987) and Druet (1989) have
summarized the evidence that this effect is dependent on genetic
factors and may be related in the rat to the appearance of anti-class
II T cells.
4.2 Direct glomerular toxicity
Glomerular lesions may be caused by the direct toxicity of a drug
or chemical. Direct toxicity to components of the glomerular
apparatus, apart from immunologically induced injury, is relatively
uncommon. However, it has been described following exposure to
puromycin or to materials that may be deposited in the basement
membrane (Caulfield et al., 1976). Particulate substances such as gold
and silica may become deposited in mesangial cells (Burkholder, 1982).
Whether material within mesangial cells is actually phagocytosed is
unclear, but the reaction to such deposits may be a proliferation of
glomerular cells and inflammatory cell response. Injury to the
mesangium may alter glomerular permeability. Solutes and water move
across glomerular capillary walls through an extracellular pathway
that consists sequentially of endothelial fenestrae, the glomerular
basement membrane, the pores of slit diaphragms, and the filtration
slits. Water permeability is determined by the total area of
epithelial slit pores. Contraction of the glomerular mesangium
shortens and narrows glomerular capillaries, which in turn narrows
epithelial slit pores and reduces glomerular filtration.
Damage to the glomeruli may also occur as a result of fibrin
deposition due to local or systemic activation of the coagulation
system. This may be induced by a variety of renal diseases including
toxic or immunotoxic disorders. Fibrin deposits per se may damage
glomeruli in several ways, including occlusion of glomerular
capillaries, involvement in an inflammatory reaction, or direct
toxicity to glomerular mesangial cells (Kanfer, 1989).
4.3 Tubulointerstitial disease
Tubulointerstitial disease may result from hypersensitivity to
specific drugs or chemicals or from direct toxicity to tubular
epithelial cells. Most forms of tubular injury also involve the
interstitium, and so these forms of renal injury are considered
together. However, they may be subdivided, on the basis of
clinico-pathological features, into three groups of disorders.
4.3.1 Acute interstitial nephritis
Acute interstitial nephritis (AIN) occurs as an immunoallergic or
cell-mediated immune response to a variety of drugs, particularly
penicillin and its derivatives (e.g., methicillin), but is also
reported after therapy with thiazdes, non-steroidal anti-inflammatory
drugs, gold salts, and occupational exposure to mercury (Kleinknecht
et al., 1978; Clive & Stoff, 1984).
Both humoral and cellular immunity are involved. Anti-tubular
basement membrane antibodies are probably involved in some cases of
methicillin- or diphenyhydantoin-induced immunological nephritis. In
the majority of cases of acute interstitial nephritis no immune
reactants are found. The most striking feature is the presence of
cells infiltrating the interstitium with mononuclear cells and
eosinophiles. Most lymphocytes have been shown to be T cells; most of
these are T4 cells (helper/inducer cells) and a lesser fraction
composed of T8 cells (suppressor/ cytotoxic cells). It has been
suggested that the T cells may be activated by drug exposure (Druet et
al., 1987; Druet, 1989).
When extrarenal signs and clinical symptoms are present, they may
reflect a systemic hypersensitivity reaction that includes fever, skin
rash, and eosinophilia. Renal involvement is manifested by mild
proteinuria and haematuria. AIN tends to be more severe, with a high
incidence of renal failure, in adult patients, but is usually milder
in patients under 15 years of age. It has also been pointed out that
absence of prior allergic reaction to a drug (e.g., penicillin) does
not alter the risk of AIN. The kidneys are usually swollen (as
visualized radio-graphically) because of oedema fluid and cellular
infiltration, composed most commonly of lymphocytes and plasma cells
as well as eosinophilic and polymorphonuclear neutrophils. In some
instances the histological appearances may suggest chronic
inflammation, and macrophages and giant cells may be present. Renal
tubular cell damage is always present, but there is no fibrosis in the
acute stages. Glomerular and vascular lesions are uncommon, and the
lesions are usually reversible. However, persistent loss of renal
function indicates progression to fibrosis and chronic interstitial
disease in an undetermined number of cases. Anti-tubular basement
membrane antibodies are probably involved in some cases of
methicillin- or diphenylhydantoin-induced nephritis. Deposits of IgG
and C3 may be detectable along tubules in biopsies during the acute
phase. IgE may be elevated in the serum, confirming the allergic or
hypersensitivity process. The reaction can be further indicated by
tests of cell-mediated hypersensitivity (lymphocyte transformation
test) or antibodymediated hypersensitivity (circulating antibodies
reacting with the drug).
4.3.2 Acute tubular toxicity
Acute tubular effects of drugs and toxins are the result of
direct cellular toxicity. They may vary from necrosis of tubular
cells, leading to acute renal failure, to subtle subcellular lesions
and functional effects. The major groups of agents causing acute
tubular toxicity are antibiotics, particularly aminoglycosides,
contrast agents, non-steroidal anti-inflammatory drugs, and
chemotherapeutic agents including cyclosporin A and cis-platinum.
Injury to proximal tubular lining cells is manifest by increased
excretion of substances normally resorbed by these cells, such as
glucose, amino acids, phosphate, and sodium. Extension of the lesion
to distal portions of the tubule is accompanied by loss of the ability
to acidify the urine and to maintain water and electrolyte balance.
Tubular toxicity may be accompanied by glomerular effects, and, if the
process is persistent, may lead to a chronic interstitial nephropathy,
as in lead and cadmium toxicity.
4.3.3 Chronic interstitial nephritis
Chronic interstitial nephritis (CIN) generally has fewer
distinguishing morphological features than most forms of acute renal
disease. It is characterized morphologically by infiltration with
mononuclear cells, prominent interstitial fibrosis, and tubular
atrophy. The best documented cause of CIN is analgesic nephropathy
which is often accompanied by acute papillary necrosis. CIN may also
occur as a sequela to severe acute tubular disease or acute
interstitial nephritis, or as an expression of chronic low-dose
exposure to specific nephrotoxins (lead or cadmium nephropathy). The
term "tubulointerstitial disease" may be preferable, because it better
identifies the primary sites of the pathological process. Progressive
fibrosis of the interstitial tissue results in a decreasing number of
functional nephrons with eventual reduction in glomerular filtration
rate and azotaemia. There may be few symptoms preceding the onset of
renal failure.
4.4 Mechanisms of cellular toxicity
There are several mechanisms that are thought to be central to
toxicological injury, including impaired lysosmal function, membrane
changes, and oxidative stress. It is now widely accepted that
Ca2+-homeostasis in the cell and Ca2+-mediated cell functions are
critical targets for numerous pathophysiological processes including
toxicant-induced cell death (Recknagel, 1983; Pounds & Rosen, 1988).
Many classes of pharmaceuticals and other chemicals (e.g., metals,
pesticides, and solvents) impair the calcium messenger system (Pounds,
1984; Olorunsogo et al., 1985; Moore et al., 1986). Disturbances in
intracellular Ca2+ homeostatis and sustained increase in cytosolic
Ca2+ cause cell death by the disruption of the plasma membrane,
cytoskeleton, endoplasmic reticulum, and mitochondria. In addition,
chemicals (alkylating or arylating agents) can be toxic and may induce
cell death through an initial DNA damage or by apoptosis
(receptor-mediated programmed cell death). In cell injury caused by
chemical toxicants, cellular accumulation of Ca2+ and the generation
of oxygen free radicals damage cellular components, particularly
mitochondrial membranes. Indeed Ca2+ potentiates oxygen free radical
injury to renal mitochondria (Malis & Bonventre, 1986), and the result
of this detrimental interaction could be due, in part, to the
activation of phospholipase A2.
Lipid peroxidation has been suggested as one of the possible
mechanisms whereby chemicals may produce membrane damage and cell
death. Free radicals, generated either directly by the metabolism of
a chemical or from the reduction of oxygen (forming O2-, H2O2
and OH*), can initiate lipid peroxidation via hydrogen abstraction
from polyunsaturated fatty acids. This interaction will form lipid
peroxyradicals and lipid hydroxyperoxides, propagating the chain
reaction. Such a chain reaction may destroy cellular membranes and
thereby result in increased plasma membrane permeability or altered
fluidity and cell death. Lipid peroxidation may also cause cell death
through the formation of potent toxic lipid metabolites (such as
hydroxyalkenals). However, several lines of evidence indicate that
lipid peroxidation is most often independent (or is a consequence
rather than the cause) of cell death (Witschi et al., 1987). One or
more of these mechanisms of cellular injury could closely interact.
Proximal renal tubular cells are particularly vulnerable to the
toxic action of chemicals, owing to their high energy demand (such as
reabsorptive and secretory functions). Redox-active agents may cause
extensive oxidation of GSH to oxidized glutathione (GSSG). Under such
conditions, often referred to as "oxidative stress", reduction of
GSSG back to GSH by the NADPH-dependent GSSG reductase is lower than
the rate of GSH oxidation. This may lead to gluthathione depletion and
cause oxidation of cellular enzymes, depletion of cellular ATP, and
loss of mitochondrial function (Trump et al., 1989).
Reactive electrophilic metabolites are known to bind covalently
to tissue proteins, and it has been suggested that cell injury and
death are a consequence of the interaction of such reactive
intermediates with critical cellular molecules. Free sulfhydryl groups
are involved in the catatylic activity of many proteins. Modification
of such sulfhydryl groups by covalent binding or by oxidation may
inactivate critical enzymes and lead to cell death. For some chemicals
the loss of protein sulfhydryl groups results mainly from a reversible
oxidative process, which leads to the formation of disulfide
cross-links or mixed disulfides with another protein or GSH. Enzymes
involved in Ca2+ homeostasis may be one example of such critical
cellular target molecules for alkylating/arylating or oxidizing
metabolites.
Studies on isolated cells have shown that exposure to lethal
concentrations of some cytotoxic chemicals leads to a rapid and
sustained rise in cytosolic Ca2+. Ca2+-mediated cytotoxicity may
at least in part be related to effects on cytoskeletal organization.
High Ca2+ concentrations affect the regulation of the formation of
actin bundles and tubulin polymerization. Activation, by high
intracellular Ca2+, of Ca2+-dependent proteases that cleave
cytoskeletal proteins has been proposed as one mechanism of cell death
(Fig. 11). Activation of other enzymes, such as phospholipase A2
(causing disruption of the plasma membrane and formation of toxic
membrane breakdown products) and endonucleases (extensive DNA
fragmentation), has also been associated with cell death (Trump et
al., 1989).
Certain alkyl halides such as DBCP (Omichinski et al., 1987;
Dybing et al., 1989) and reactive oxygen species cause single-strand
breaks in DNA. Extensive DNA damage activates poly
ADP-ribosyltransferase, which leads to a critical reduction in
cellular NAD+ levels, followed by depletion of ATP and eventual
cell death.
The process by which glucocorticoids induce killing of immature
thymocytes has been termed programmed cell death (apoptosis). Recent
studies indicates that the environmental contaminant
2,3,7,8-tetrachlorinated dibenzo- p-dioxin (TCDD) causes a
receptor-mediated influx of Ca2+ in thymic cells, which in turn
activates endonucleases and thereby causes programmed cell death. The
role of such a process in the chemically mediated killing of kidney
cells has yet to be determined (McConkey et al., 1988).
4.5 Factors that modify cellular injury by toxins
4.5.1 Cellular transport and accumulation
Drugs and other chemicals including metals may be transported
across proximal tubular cells, i.e., from renal capillaries across
tubular cells to be excreted in the tubular lumen or vice versa
(absorption). Many organic anions are excreted against concentration
gradients at rates that exceed glomerular filtration. This implies an
active carrier-mediated transport process. Such a process requires
energy obtained from oxidative metabolism located in mitochondria. An
active process for transporting solutes in renal tubular cells has
certain implications concerning the susceptibility of tubular cells to
effects of toxins (Berndt, 1989). If cationic drugs or chemicals are
actively transported, there is the immediate problem of competition
with the transport of essential cations. Active transport with the
capability of concentrating absorbed material may concentrate
potential nephrotoxins as well as essential solutes in the renal
cortex. The same toxins that impair energy metabolism will impede the
cellular transport of essential solutes (Rennick, 1978). Other toxic
substances may be concentrated in the medulla or the papillae,
probably as a consequence of the physiological mechanism that
concentrates urine. The renal accumulation of chemicals such as
gentamicin, cephaloridine, or cadmium is well documented.
4.5.2 Metabolic degradation
Metabolic degradation or transformation most often occurs in the
liver, but many of the same enzyme systems are present in the kidney
as well. The metabolism of drugs and chemicals within the kidney may
result in substances that are either more or less toxic. Those drugs
and chemicals that are metabolized by the mixed-function oxidase
system have received the most attention. For example, several
chlorinated alkyl hydrocarbons of low relative molecular mass, such as
carbon tetrachloride and trichloromethane, may be transformed into
reactive, toxic products that bind covalently to renal tissue,
producing membrane injury. In addition, low-level exposure to other
substances, such as polychlorinated biphenyls (PCBs), that activate
the enzyme systems may enhance the production of toxic products (Kluwe
et al., 1979). Similarly, pretreatment with phenobarbital enhances the
activity of mixedfunction oxidase enzymes and, hence, the toxicity of
compounds like methoxyflurane whose metabolic products are fluoride
and oxalate, two substances potentially toxic to the kidney. The
fluoride ion is toxic to cell membranes, whereas oxalate may
precipitate within the lumen of nephrons (Mazze, 1976). On the other
hand, phenobarbital reduces the renal toxicity of DBCP due to an
increase in its detoxication in the liver (Kluwe, 1983).
4.5.3 Intracellular protein binding
The intracellular concentration of toxins may be influenced by
protein binding. The soluble cytoplasmic protein, metallothionein, and
insoluble acidic protein complexes forming nuclear inclusion bodies
are examples of a phenomenon that concentrates two different groups of
metals.
Metallothioneins are proteins of low relative molecular mass
(6000-7000 Daltons) characterized by a high cysteine content (23-33%),
a complete lack of aromatic acids, and a high content of heavy metals
(7-12 metal atoms/molecule of protein). Metallothioneins can bind
Other aromatic acids (such as benzoic acid or p-aminobenzoic
acid) competitively inhibited glycine conjugation. Cellular glycine is
limited, and so its use in conjugation would be saturated with
increasing concentrations of substrate.
3.6.3 Other enzymes involved in xenobiotic metabolism
Highly electrophilic epoxides play a key role in tissue
alkylation of macromolecules, especially nucleic acids, leading to
mutagenic and carcinogenic effects. Epoxide hydrase (hydrolase)
converts aliphatic and aromatic epoxides to their trans-hydrodiols, a
process that may generate both toxic and non-toxic products (Anders,
1980).
The oxidation of aldehydes to ketones or carboxylic acids in the
kidney is mediated by aldehyde oxidase and aldehyde dehydrogenase
(Goldberg & Anderson, 1985). Renal aldehyde oxidase activity is
localized in the proximal tubule but represents only 40% of the liver
activity (Anders, 1980), but aldehyde oxidase represents only 10% of
renal aldehyde dehydrogenase activity (Anders, 1980). At least two
aldehyde dehydrogenase isozymes occur in proximal tubule mitochondria
and cytosol. Substrates include formaldehyde, acetaldehyde, acrolein,
and malondialdehyde (Hjelle et al., 1983).
DT-diaphorase (NADPH-quinone oxidoreductase) is a cytosolic
enzyme that is present at highest concentration in the medulla and
catalyses a two-electron reduction of quinones to less reactive
hydroquinones. It therefore blocks redox cycling and the generation
of superoxide anion radicals (Lind et al., 1978).
4. THE MECHANISTIC BASIS OF CHEMICALLY INDUCED RENAL INJURY
Over the past 20-30 years there has been a growing understanding
of the molecular basis of disease and the biochemical mechanisms that
are associated with chemically induced cellular degeneration and
lesions in target organ systems. The application of this understanding
provides a foundation upon which to study chemically induced renal
injury and, in particular, a rational basis for the extrapolation of
animal toxicity data to man and risk assessment.
4.1 Immunologically induced glomerular disease
Immunologically mediated glomerulonephritis can result from the
deposition of free circulating antibodies interacting with a
structural glomerular antigen or with a "planted", non-glomerular
antigen (such as cationic proteins that fix on anionic sites of the
glomerular basement membrane). Alternatively, glomerular injury may
be the consequence of the deposition of circulating immune complexes.
The two main forms of antibody-mediated glomerulonephritis are the
anti-GBM-mediated disease and membranous glomerulonephritis (or
immune complex-type mediated glomerulonephritis). The former disease
is characterized by the presence of linear IgG deposits along the
glomerular basement membrane. The latter form may result from either
of the mechanisms mentioned, i.e. deposition of free circulating
antibodies against an irregularly distributed antigen, which may be of
glomerular origin, or deposition of circulating immune complexes. A
membranous glomerulonephritis is characterized by the presence of
granular IgG deposits along the glomerular basement membrane.
Besides antibody-mediated glomerulonephritis, it is more and more
apparent that there are cell-mediated glomerulonephritides without Ig
deposition. An example of such a disease is probably the nephrotic
syndrome with minimal glomerular changes at the light microscope
level.
The role of genetic factors, which has been well demonstrated in
the mercury model in the rat, is also clear in the human situation.
Membranous glomerulonephritis induced by gold and D-penicillamine is
much more frequent in DR3-positive rheumatoid arthritis patients and
in poor sulfoxydators. The lupus-like disease observed in
hydralazine-treated patients is more frequent in those with the DR4
antigen and "slow-acetylators".
Other drugs such as non-steroidal anti-inflammatory agents or
lithium salts may induce the nephrotic syndrome with minimal
glomerular changes. Immunofluorescence is negative in these cases, and
there is indirect evidence that such disease could be due to T
cell-mediated immunity.
There are many potential environmental agents, drugs, and toxic
chemicals that have been related to this form of glomerulonephritis.
Drugs and toxic chemicals that may induce glomerulonephritis in humans
include gold and mercury, d-penicillamine, non-steroidal
anti-inflammatory agents, and heroin. Other drugs and chemicals, in
which association is suspected but not certain, include silica
exposure, toxic-oil syndrome, hydrocarbon exposure, and interferon.
Other drugs may induce an immune complex type of glomerulonephritis in
the context of a lupus-like syndrome (e.g., hydralazine, procanamide,
and diphenylhydantoin. Furthermore, there are many other substances
that have been implicated by case reports or unconfirmed experiments.
Anti-GBM-mediated glomerulonephritis is not the usual mechanism
responsible for toxic glomerular nephropathy. However, exposure to
organic solvents is thought to be a factor in some cases of
Goodpasture's syndrome, which consists of the simultaneous occurrence
of necrotizing haemorrhagic interstitial pneumonitis and
proliferative usually rapidly progressive glomerulonephritis. This is
an auto-immune disease resulting from antiGBM antibodies that
cross-react with basement membranes in lung alveoli.
The mechanisms by which drugs and chemicals induce
immunologically related renal disease are both complex and not
entirely understood. It is unlikely that drugs or toxins induce renal
damage by modifying self antigens or by acting as haptens. On the
other hand, many agents such as gold, d-penicillamine, and mercury
may have an immunomodulatory effect. This mechanism of action is also
not understood, but Druet et al. (1987) and Druet (1989) have
summarized the evidence that this effect is dependent on genetic
factors and may be related in the rat to the appearance of anti-class
II T cells.
4.2 Direct glomerular toxicity
Glomerular lesions may be caused by the direct toxicity of a drug
or chemical. Direct toxicity to components of the glomerular
apparatus, apart from immunologically induced injury, is relatively
uncommon. However, it has been described following exposure to
puromycin or to materials that may be deposited in the basement
membrane (Caulfield et al., 1976). Particulate substances such as gold
and silica may become deposited in mesangial cells (Burkholder, 1982).
Whether material within mesangial cells is actually phagocytosed is
unclear, but the reaction to such deposits may be a proliferation of
glomerular cells and inflammatory cell response. Injury to the
mesangium may alter glomerular permeability. Solutes and water move
across glomerular capillary walls through an extracellular pathway
that consists sequentially of endothelial fenestrae, the glomerular
basement membrane, the pores of slit diaphragms, and the filtration
slits. Water permeability is determined by the total area of
epithelial slit pores. Contraction of the glomerular mesangium
shortens and narrows glomerular capillaries, which in turn narrows
epithelial slit pores and reduces glomerular filtration.
Damage to the glomeruli may also occur as a result of fibrin
deposition due to local or systemic activation of the coagulation
system. This may be induced by a variety of renal diseases including
toxic or immunotoxic disorders. Fibrin deposits per se may damage
glomeruli in several ways, including occlusion of glomerular
capillaries, involvement in an inflammatory reaction, or direct
toxicity to glomerular mesangial cells (Kanfer, 1989).
4.3 Tubulointerstitial disease
Tubulointerstitial disease may result from hypersensitivity to
specific drugs or chemicals or from direct toxicity to tubular
epithelial cells. Most forms of tubular injury also involve the
interstitium, and so these forms of renal injury are considered
together. However, they may be subdivided, on the basis of
clinico-pathological features, into three groups of disorders.
4.3.1 Acute interstitial nephritis
Acute interstitial nephritis (AIN) occurs as an immunoallergic or
cell-mediated immune response to a variety of drugs, particularly
penicillin and its derivatives (e.g., methicillin), but is also
reported after therapy with thiazdes, non-steroidal anti-inflammatory
drugs, gold salts, and occupational exposure to mercury (Kleinknecht
et al., 1978; Clive & Stoff, 1984).
Both humoral and cellular immunity are involved. Anti-tubular
basement membrane antibodies are probably involved in some cases of
methicillin- or diphenyhydantoin-induced immunological nephritis. In
the majority of cases of acute interstitial nephritis no immune
reactants are found. The most striking feature is the presence of
cells infiltrating the interstitium with mononuclear cells and
eosinophiles. Most lymphocytes have been shown to be T cells; most of
these are T4 cells (helper/inducer cells) and a lesser fraction
composed of T8 cells (suppressor/ cytotoxic cells). It has been
suggested that the T cells may be activated by drug exposure (Druet et
al., 1987; Druet, 1989).
When extrarenal signs and clinical symptoms are present, they may
reflect a systemic hypersensitivity reaction that includes fever, skin
rash, and eosinophilia. Renal involvement is manifested by mild
proteinuria and haematuria. AIN tends to be more severe, with a high
incidence of renal failure, in adult patients, but is usually milder
in patients under 15 years of age. It has also been pointed out that
absence of prior allergic reaction to a drug (e.g., penicillin) does
not alter the risk of AIN. The kidneys are usually swollen (as
visualized radio-graphically) because of oedema fluid and cellular
infiltration, composed most commonly of lymphocytes and plasma cells
as well as eosinophilic and polymorphonuclear neutrophils. In some
instances the histological appearances may suggest chronic
inflammation, and macrophages and giant cells may be present. Renal
tubular cell damage is always present, but there is no fibrosis in the
acute stages. Glomerular and vascular lesions are uncommon, and the
lesions are usually reversible. However, persistent loss of renal
function indicates progression to fibrosis and chronic interstitial
disease in an undetermined number of cases. Anti-tubular basement
membrane antibodies are probably involved in some cases of
methicillin- or diphenylhydantoin-induced nephritis. Deposits of IgG
and C3 may be detectable along tubules in biopsies during the acute
phase. IgE may be elevated in the serum, confirming the allergic or
hypersensitivity process. The reaction can be further indicated by
tests of cell-mediated hypersensitivity (lymphocyte transformation
test) or antibodymediated hypersensitivity (circulating antibodies
reacting with the drug).
4.3.2 Acute tubular toxicity
Acute tubular effects of drugs and toxins are the result of
direct cellular toxicity. They may vary from necrosis of tubular
cells, leading to acute renal failure, to subtle subcellular lesions
and functional effects. The major groups of agents causing acute
tubular toxicity are antibiotics, particularly aminoglycosides,
contrast agents, non-steroidal anti-inflammatory drugs, and
chemotherapeutic agents including cyclosporin A and cis-platinum.
Injury to proximal tubular lining cells is manifest by increased
excretion of substances normally resorbed by these cells, such as
glucose, amino acids, phosphate, and sodium. Extension of the lesion
to distal portions of the tubule is accompanied by loss of the ability
to acidify the urine and to maintain water and electrolyte balance.
Tubular toxicity may be accompanied by glomerular effects, and, if the
process is persistent, may lead to a chronic interstitial nephropathy,
as in lead and cadmium toxicity.
4.3.3 Chronic interstitial nephritis
Chronic interstitial nephritis (CIN) generally has fewer
distinguishing morphological features than most forms of acute renal
disease. It is characterized morphologically by infiltration with
mononuclear cells, prominent interstitial fibrosis, and tubular
atrophy. The best documented cause of CIN is analgesic nephropathy
which is often accompanied by acute papillary necrosis. CIN may also
occur as a sequela to severe acute tubular disease or acute
interstitial nephritis, or as an expression of chronic low-dose
exposure to specific nephrotoxins (lead or cadmium nephropathy). The
term "tubulointerstitial disease" may be preferable, because it better
identifies the primary sites of the pathological process. Progressive
fibrosis of the interstitial tissue results in a decreasing number of
functional nephrons with eventual reduction in glomerular filtration
rate and azotaemia. There may be few symptoms preceding the onset of
renal failure.
4.4 Mechanisms of cellular toxicity
There are several mechanisms that are thought to be central to
toxicological injury, including impaired lysosmal function, membrane
changes, and oxidative stress. It is now widely accepted that
Ca2+-homeostasis in the cell and Ca2+-mediated cell functions are
critical targets for numerous pathophysiological processes including
toxicant-induced cell death (Recknagel, 1983; Pounds & Rosen, 1988).
Many classes of pharmaceuticals and other chemicals (e.g., metals,
pesticides, and solvents) impair the calcium messenger system (Pounds,
1984; Olorunsogo et al., 1985; Moore et al., 1986). Disturbances in
intracellular Ca2+ homeostatis and sustained increase in cytosolic
Ca2+ cause cell death by the disruption of the plasma membrane,
cytoskeleton, endoplasmic reticulum, and mitochondria. In addition,
chemicals (alkylating or arylating agents) can be toxic and may induce
cell death through an initial DNA damage or by apoptosis
(receptor-mediated programmed cell death). In cell injury caused by
chemical toxicants, cellular accumulation of Ca2+ and the generation
of oxygen free radicals damage cellular components, particularly
mitochondrial membranes. Indeed Ca2+ potentiates oxygen free radical
injury to renal mitochondria (Malis & Bonventre, 1986), and the result
of this detrimental interaction could be due, in part, to the
activation of phospholipase A2.
Lipid peroxidation has been suggested as one of the possible
mechanisms whereby chemicals may produce membrane damage and cell
death. Free radicals, generated either directly by the metabolism of
a chemical or from the reduction of oxygen (forming O2-, H2O2
and OH*), can initiate lipid peroxidation via hydrogen abstraction
from polyunsaturated fatty acids. This interaction will form lipid
peroxyradicals and lipid hydroxyperoxides, propagating the chain
reaction. Such a chain reaction may destroy cellular membranes and
thereby result in increased plasma membrane permeability or altered
fluidity and cell death. Lipid peroxidation may also cause cell death
through the formation of potent toxic lipid metabolites (such as
hydroxyalkenals). However, several lines of evidence indicate that
lipid peroxidation is most often independent (or is a consequence
rather than the cause) of cell death (Witschi et al., 1987). One or
more of these mechanisms of cellular injury could closely interact.
Proximal renal tubular cells are particularly vulnerable to the
toxic action of chemicals, owing to their high energy demand (such as
reabsorptive and secretory functions). Redox-active agents may cause
extensive oxidation of GSH to oxidized glutathione (GSSG). Under such
conditions, often referred to as "oxidative stress", reduction of
GSSG back to GSH by the NADPH-dependent GSSG reductase is lower than
the rate of GSH oxidation. This may lead to gluthathione depletion and
cause oxidation of cellular enzymes, depletion of cellular ATP, and
loss of mitochondrial function (Trump et al., 1989).
Reactive electrophilic metabolites are known to bind covalently
to tissue proteins, and it has been suggested that cell injury and
death are a consequence of the interaction of such reactive
intermediates with critical cellular molecules. Free sulfhydryl groups
are involved in the catatylic activity of many proteins. Modification
of such sulfhydryl groups by covalent binding or by oxidation may
inactivate critical enzymes and lead to cell death. For some chemicals
the loss of protein sulfhydryl groups results mainly from a reversible
oxidative process, which leads to the formation of disulfide
cross-links or mixed disulfides with another protein or GSH. Enzymes
involved in Ca2+ homeostasis may be one example of such critical
cellular target molecules for alkylating/arylating or oxidizing
metabolites.
Studies on isolated cells have shown that exposure to lethal
concentrations of some cytotoxic chemicals leads to a rapid and
sustained rise in cytosolic Ca2+. Ca2+-mediated cytotoxicity may
at least in part be related to effects on cytoskeletal organization.
High Ca2+ concentrations affect the regulation of the formation of
actin bundles and tubulin polymerization. Activation, by high
intracellular Ca2+, of Ca2+-dependent proteases that cleave
cytoskeletal proteins has been proposed as one mechanism of cell death
(Fig. 11). Activation of other enzymes, such as phospholipase A2
(causing disruption of the plasma membrane and formation of toxic
membrane breakdown products) and endonucleases (extensive DNA
fragmentation), has also been associated with cell death (Trump et
al., 1989).
Certain alkyl halides such as DBCP (Omichinski et al., 1987;
Dybing et al., 1989) and reactive oxygen species cause single-strand
breaks in DNA. Extensive DNA damage activates poly
ADP-ribosyltransferase, which leads to a critical reduction in
cellular NAD+ levels, followed by depletion of ATP and eventual
cell death.
The process by which glucocorticoids induce killing of immature
thymocytes has been termed programmed cell death (apoptosis). Recent
studies indicates that the environmental contaminant
2,3,7,8-tetrachlorinated dibenzo- p-dioxin (TCDD) causes a
receptor-mediated influx of Ca2+ in thymic cells, which in turn
activates endonucleases and thereby causes programmed cell death. The
role of such a process in the chemically mediated killing of kidney
cells has yet to be determined (McConkey et al., 1988).
4.5 Factors that modify cellular injury by toxins
4.5.1 Cellular transport and accumulation
Drugs and other chemicals including metals may be transported
across proximal tubular cells, i.e., from renal capillaries across
tubular cells to be excreted in the tubular lumen or vice versa
(absorption). Many organic anions are excreted against concentration
gradients at rates that exceed glomerular filtration. This implies an
active carrier-mediated transport process. Such a process requires
energy obtained from oxidative metabolism located in mitochondria. An
active process for transporting solutes in renal tubular cells has
certain implications concerning the susceptibility of tubular cells to
effects of toxins (Berndt, 1989). If cationic drugs or chemicals are
actively transported, there is the immediate problem of competition
with the transport of essential cations. Active transport with the
capability of concentrating absorbed material may concentrate
potential nephrotoxins as well as essential solutes in the renal
cortex. The same toxins that impair energy metabolism will impede the
cellular transport of essential solutes (Rennick, 1978). Other toxic
substances may be concentrated in the medulla or the papillae,
probably as a consequence of the physiological mechanism that
concentrates urine. The renal accumulation of chemicals such as
gentamicin, cephaloridine, or cadmium is well documented.
4.5.2 Metabolic degradation
Metabolic degradation or transformation most often occurs in the
liver, but many of the same enzyme systems are present in the kidney
as well. The metabolism of drugs and chemicals within the kidney may
result in substances that are either more or less toxic. Those drugs
and chemicals that are metabolized by the mixed-function oxidase
system have received the most attention. For example, several
chlorinated alkyl hydrocarbons of low relative molecular mass, such as
carbon tetrachloride and trichloromethane, may be transformed into
reactive, toxic products that bind covalently to renal tissue,
producing membrane injury. In addition, low-level exposure to other
substances, such as polychlorinated biphenyls (PCBs), that activate
the enzyme systems may enhance the production of toxic products (Kluwe
et al., 1979). Similarly, pretreatment with phenobarbital enhances the
activity of mixedfunction oxidase enzymes and, hence, the toxicity of
compounds like methoxyflurane whose metabolic products are fluoride
and oxalate, two substances potentially toxic to the kidney. The
fluoride ion is toxic to cell membranes, whereas oxalate may
precipitate within the lumen of nephrons (Mazze, 1976). On the other
hand, phenobarbital reduces the renal toxicity of DBCP due to an
increase in its detoxication in the liver (Kluwe, 1983).
4.5.3 Intracellular protein binding
The intracellular concentration of toxins may be influenced by
protein binding. The soluble cytoplasmic protein, metallothionein, and
insoluble acidic protein complexes forming nuclear inclusion bodies
are examples of a phenomenon that concentrates two different groups of
metals.
Metallothioneins are proteins of low relative molecular mass
(6000-7000 Daltons) characterized by a high cysteine content (23-33%),
a complete lack of aromatic acids, and a high content of heavy metals
(7-12 metal atoms/molecule of protein). Metallothioneins can bind
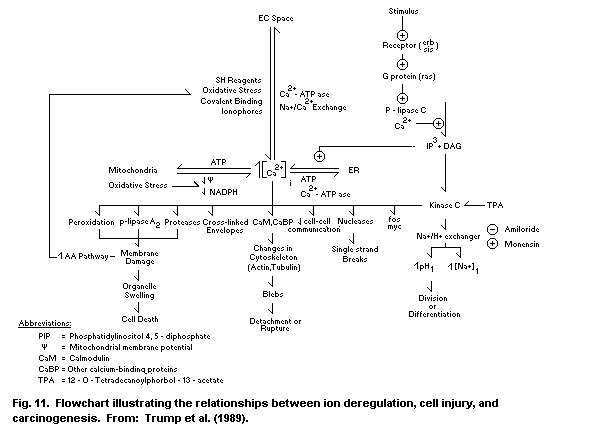 several essential or non-essential heavy metal ions including zinc,
copper, cadmium, mercury, silver, gold, and cobalt (Goyer & Cherian,
1977). The metal ions are bound exclusively through thiolate
coordination complexes, which involve all the cysteine residues (20 in
rat liver metallothionein) located in two domains (alpha and ß
domains). Metal ions that can bind to metallothioneins can also, to
variable extents, promote the transcriptional activity of
metallothionein genes. In the kidney, Cd2+ and Hg2+ are the best
inducers of metallothionein synthesis. Metallothionein synthesis can
also be induced by various stresses (e.g., tissue injury, food
restriction, infections). Not all the biological functions of
metallothionein have been fully elucidated. They probably include
protection against and detoxification of heavy metals, regulation of
the metabolism and possibly the function of essential elements such as
copper and zinc, and a protective response to various stresses by
altering zinc distributions between tissues and within cells (e.g.,
macrophages) and by acting as a free radical scavenger (Dunn et al.,
1987).
Lead and bismuth accumulate in renal tubular cells bound to a
complex of acidic proteins that form morphologically discernible
inclusion bodies (Goyer & Cherian, 1977). As with metallothionein, the
sequestering of toxic metals by the protein complex is thought to
reduce the intracellular toxicity of these metals.
4.5.4 Membrane reactions and pinocytosis
Macromolecular substances are transported by pinocytosis and
included in intracellular vacuoles. Proteins that are normally in the
glomerular filtrate are taken up by the cell membrane by pinocytosis.
Such pinocytotic vesicles fuse with primary lysosomes, which contain
lytic enzymes. Secondary lysosomes are formed, and the macromolecular
material is degraded or broken down. The products of low relative
molecular mass then leave the lysosomes in order to prevent an
increase in osmolality and lysosome swelling (Jacques, 1975).
Potential nephrotoxins that may be taken into renal tubular cells
in this manner include chelating agents such as nitrilotriacetic acid,
ethylenediaminetetraacetic acid (EDTA), and metallothionein. Membrane
binding of EDTA administered as the calcium-EDTA chelate persists, the
calcium but not the EDTA being dislocated to other cellular
components. This suggests the manner in which EDTA may sequester
cellular lead or other metals for excretion (Schwartz et al., 1970).
5. THERAPEUTIC AGENTS AND CHEMICALS THAT HAVE THE POTENTIAL TO CAUSE
NEPHROTOXICITY
5.1 Therapeutic agents
Many therapeutic agents have been linked to clinically
significant nephrotoxicity. At present much is known and understood
about some of these agents, as there is a substantial amount of
relevant animal toxicity and human data for comparison.
5.1.1 Analgesics and non-steroidal anti-inflammatory drugs (NSAIDs)
Analgesic nephropathy may be a consequence of the excessive
consumption of mixed analgesics. Originally, phenacetin was common to
all of these mixtures, which led to the conclusion that this drug was
the only cause of "phenacetin kidney". Subsequently, a variety of
analgesics, NSAIDs, and a number of industrial and environmental
chemicals have been shown to have the potential to cause RPN and
interstitial nephritis (Burry et al., 1977; Schwarz, 1987).
The prevalence of analgesic-associated nephropathy varies
worldwide and is probably related to patterns of analgesic use. It has
been found most often in women aged 45-55 as a result of a high
incidence of analgesic abuse, and has been reported more frequently in
Australia and Switzerland and less frequently in the USA, Canada, and
Germany. It is estimated that more than 37 million people in the USA
have arthritis and use these drugs. This is indeed a very large
population at risk. Analgesic effects are said to be a factor in as
many as 20-30% of cases of interstitial nephritis in the south-east of
the USA (Murray & Goldberg, 1975).
Diagnosis is often made by coupling the history of analgesic
abuse with morphological evidence of renal papillary necrosis and
chronic interstitial nephritis. The resultant lesions can be
recognized by radiological examinations or ultrasonography and consist
of calcifications along the line of Hodson, shrinking of the kidneys
resulting in irregular contours, and decreased length of both kidneys.
Necrotic papillae may be voided in the urine and this is occasionally
observed.
At least two countries have legislated to prevent phenacetin from
being sold over the counter. This legislation has had the effect of a
change in analgesic formulation, usually towards that of a single drug
such as aspirin or paracetamol (acetaminophen). In Sweden, the removal
of phenacetin resulted in the progressive decline in the incidence of
analgesic nephropathy. This began approximately 6 years after the
removal of phenacetin and the major impact was seen after 12 years. In
Australia, phenacetin was removed from combination analgesics by 1976
and replaced with acetaminophen (the principle metabolite of
phenacetin); by 1979 legislation prohibited the overthe-counter sale
of combination analgesics. According to the Australia-New-Zealand
Dialysis and Transplant Registry, this has resulted in a progressive
decline in the incidence of analgesic nephropathy in patients
presenting with end-stage chronic renal failure in dialysis and
transplant programmes. In 1982, 22% of patients in dialyses and
transplant programmes had analgesic nephropathy; by 1988, the
incidence had declined to approximately 13%. By contrast, the sale of
phenacetin-containing analgesics declined to 21% (in 1976) and 9% (in
1983) of the total volume of analgesics sold in Belgium, but the
prevalence of analgesic nephropathy remained unchanged over this
period. Retrospective and prospective studies in Belgium have shown
decreased renal function in analgesic abusers who never took analgesic
mixtures containing phenacetin (Elseviers & De Broe, 1988).
Paracetamol, which largely replaced phenacetin in analgesic mixtures),
is assumed to be involved in the pathogenesis of RPN. Recently, an
increased risk of renal disease was found in daily users of
paracetamol in North Carolina, USA (Sandler et al., 1989). Other
therapeutic agents (mostly analgesics and NSAIDs) have been
implicated in RPN, and a number of industrial and environmental
chemicals also have the potential to cause this lesion (Bach &
Bridges, 1985a).
Most patients deny analgesic abuse, thus making epidemiological
studies that attempt to identify the causative agent difficult. Most
of the epidemiological data that has been reported over-reflected the
intake of phenacetin at the expense of other agents that could equally
be implicated. The estimate of the total lifetime dose of analgesic
that produces papillary necrosis varies from < 1 to 35 kg (but this
was based only on phenacetin). No data was given in the
epidemiological studies on what quantities of other analgesics were
also taken or on exposure to papillotoxic chemicals. Thus the etiology
of RPN has been complicated by poly-pharmacy, lack of documentation on
the intake of other analgesics and NSAIDs, exposure to other
chemicals, and the difficulty in diagnosing RPN.
Analgesic abuse (Nanra et al., 1978; Nanra, 1980; Prescott, 1982;
Bach & Bridges, 1985a; Schwarz, 1987) and addiction have been linked
to co-formulation with caffeine, but there is no firm supporting
evidence. Few analgesic abusers take the drugs for appropriate
indications. The origins of abuse are usually psycho-social and
represent neurotic, dependent, immature, introverted, anxious, or
depressed individuals, up to 20% of whom also smoke and abuse alcohol,
psychotropic drugs, and sleeping tablets. Most analgesic abusers are
women (over 30 years of age) from lower socioeconomic/education
groups, who have taken these mixtures for 5-30 years. Several factors
such as dehydration, secondary to high ambient temperatures and
bacterial infection, have been implicated in the development of renal
failure (Kincaid-Smith, 1979). Renal function may be preserved and the
progression to ESRD may be prevented by stopping analgesic exposure,
but patients who continue to abuse analgesics have a poor prognosis
(Schwarz, 1987). Analgesic abusers also have an increased incidence of
anaemia, gastric ulcers, and cardiovascular heart disease (Dubach et
al., 1978).
Clinical features of analgesic nephropathy include loss of
urine-concentrating capacity (Bengtsson, 1962), electrolyte
disturbances, (sodium wastage and hypocalcaemia), and defective
urinary acidification after ammonium chloride loading (Bengtsson,
1962; Nanra et al., 1978). An increase in BUN or creatinine
identifies incipient renal failure; at this stage papillary necrosis
is well advanced and includes secondary degenerative changes.
Radiology and ultrasound may identify irregular shrinking of the renal
tissue and medullary calcifications, but these are advanced changes.
Very early in the course of the injury histological changes are
confined to the medulla (a region of the kidney that is not always
assessed at autopsy). This situation progresses to include other
changes, especially in the cortex, that are easily biopsied to show
interstitial nephritis, but they do not define the underlying cause.
The earliest degenerative changes begin at the papilla tip and
affect interstitial cells, loops of Henle, capillaries, and the
proteoglycan ground substance, and result in lipidosis. More advanced
or intermediate RPN affects the outer medulla, as seen by atrophy,
sclerosis, and inflammatory response and calcification of the necrosed
papilla tip (Burry et al., 1977; Gloor, 1978). Total RPN affects the
corticomedullary and cortical regions and is characterized by chronic
interstitial nephritis, tubular dilatation, atrophy, basement membrane
thickening, fibrosis, sclerosis, and inflammatory cell infiltration.
Vascular degeneration such as suburothelial capillary sclerosis is
pathognomonic for RPN (Mihatsch et al., 1984). Pelvic, ureteric, and
bladder urothelia show thickening of capillary walls, sclerosis of
lamina propria, altered fat and collagen deposition, and epithelial
hyperplasia advancing to malignancy and tumours (Burry et al., 1977;
Mihatsch et al., 1984).
The pathological changes in humans with RPN have been obtained
from autopsy or postmortem tissues, where autolytic degradation may
alter the appearance and make interpretation of the stage of the
lesion difficult. Animal models of RPN in a laboratory situation have
provided more detail on the focus of primary injury and the course of
degenerative changes and also allow mechanisms to be studied, but they
do not necessarily reflect the situation in humans.
Analgesics (e.g., aspirin, phenacetin, and paracetamol) do not
always cause RPN in rats. Thus inappropriately high doses have
sometimes been given for prolonged periods (Prescott, 1982; Bach &
Bridges, 1985a). Biological variation within such groups is very high
and experimental use of NSAIDs has caused fatalities due to
extra-renal toxicity (gastric ulceration and perforation; see Kaump,
1966). This has produced experimental models that are of limited
value for studying the course of the lesion or mechanism of RPN. The
renal functional changes and the pathomorphological progression of the
lesion in several acute model systems show marked similarities with
those reported for the analgesic-associated lesion in both
experimental animals and man (Bach & Hardy, 1985; Bach & Bridges,
1985a).
The histological changes induced by experimental RPN follow a
similar pattern of early, intermediate and total RPN to that described
in man (Fig. 12), and are dose and time dependent. The earliest
morphological changes induced by papillotoxins occur in the renal
medullary interstitial cells. The medullary glycosaminoglycan matrix
also undergoes changes, showing an increase and then a decrease in
staining intensity. It is only subsequently that there are platelet
adhesions, blocking of blood vessels, degenerative changes in the
collecting ducts and proximal tubules, and the accumulation of lipid
material in capillaries and epithelial cells. At the same time as
repair and reepithelization are taking place, there is an increase in
the presence of tubular casts, proximal and distal tubular dilatation,
and hyperplasia of the collecting ducts, and pelvic urothelia, and
the suburothelial capillaries undergo sclerotic changes. When the
repair phase is advanced or complete, there are also degenerative
changes in the cortex, including fibrosis, glomerular sclerosis, and
cystic dilatation. The histological and functional changes produced by
model RPN are remarkably similar to those observed in human analgesic
abusers. The use of high-resolution light microscopy and
ultrastructural studies (in conjunction with histochemistry and
immuno-histochemistry) can help establish the changes in adjacent
cells and link the "cause-and-effect" relationships in the sequence of
degenerative events.
The mechanism of analgesic-induced renal papillary necrosis is
still not fully understood. Progress in our understanding of the
pathogenesis of the model lesions (Bach & Bridges, 1984, 1985a) has
enabled some factors to be identified that may be involved in the
molecular changes. There is no evidence to suggest that the model
lesion has an early immunological basis, nor that it is a consequence
of renal hypoxia or vasoconstriction, and there is no experimental
basis to suggest that the altered intermediary metabolism is a
critical factor (Bach et al., 1983). The concept that altered PG
metabolism gives rise to vascular (or other) changes is an attractive
one, but the exceptionally low levels of these hormones, combined with
their instability, have made it very difficult to test this
hypothesis. The countercurrent concentration mechanism is an important
normal renal function and is thought to play an important role in
concentrating chemicals to toxic levels within the medulla. One of
the earliest changes in the development of RPN is the loss of
concentrating processes, which detracts from this hypothesis.
Furthermore, the concentrating of a compound in the medulla does not
explain the molecular mechanism by which it causes RPN (Bach &
Bridges, 1985a).
At present the most attractive explanation for the development of
RPN relates to a metabolic activation within the kidney. There are two
major oxidative systems for xenobiotic metabolism in the kidney. The
cytochrome P-450 system is localized to the cortex, whereas the PG
hydroperoxidase system (PGH) is located almost exclusively in the
medulla. The reasons for the selective targeting of particular
chemicals for the renal medulla interstitial cells are uncertain, but
several essential or non-essential heavy metal ions including zinc,
copper, cadmium, mercury, silver, gold, and cobalt (Goyer & Cherian,
1977). The metal ions are bound exclusively through thiolate
coordination complexes, which involve all the cysteine residues (20 in
rat liver metallothionein) located in two domains (alpha and ß
domains). Metal ions that can bind to metallothioneins can also, to
variable extents, promote the transcriptional activity of
metallothionein genes. In the kidney, Cd2+ and Hg2+ are the best
inducers of metallothionein synthesis. Metallothionein synthesis can
also be induced by various stresses (e.g., tissue injury, food
restriction, infections). Not all the biological functions of
metallothionein have been fully elucidated. They probably include
protection against and detoxification of heavy metals, regulation of
the metabolism and possibly the function of essential elements such as
copper and zinc, and a protective response to various stresses by
altering zinc distributions between tissues and within cells (e.g.,
macrophages) and by acting as a free radical scavenger (Dunn et al.,
1987).
Lead and bismuth accumulate in renal tubular cells bound to a
complex of acidic proteins that form morphologically discernible
inclusion bodies (Goyer & Cherian, 1977). As with metallothionein, the
sequestering of toxic metals by the protein complex is thought to
reduce the intracellular toxicity of these metals.
4.5.4 Membrane reactions and pinocytosis
Macromolecular substances are transported by pinocytosis and
included in intracellular vacuoles. Proteins that are normally in the
glomerular filtrate are taken up by the cell membrane by pinocytosis.
Such pinocytotic vesicles fuse with primary lysosomes, which contain
lytic enzymes. Secondary lysosomes are formed, and the macromolecular
material is degraded or broken down. The products of low relative
molecular mass then leave the lysosomes in order to prevent an
increase in osmolality and lysosome swelling (Jacques, 1975).
Potential nephrotoxins that may be taken into renal tubular cells
in this manner include chelating agents such as nitrilotriacetic acid,
ethylenediaminetetraacetic acid (EDTA), and metallothionein. Membrane
binding of EDTA administered as the calcium-EDTA chelate persists, the
calcium but not the EDTA being dislocated to other cellular
components. This suggests the manner in which EDTA may sequester
cellular lead or other metals for excretion (Schwartz et al., 1970).
5. THERAPEUTIC AGENTS AND CHEMICALS THAT HAVE THE POTENTIAL TO CAUSE
NEPHROTOXICITY
5.1 Therapeutic agents
Many therapeutic agents have been linked to clinically
significant nephrotoxicity. At present much is known and understood
about some of these agents, as there is a substantial amount of
relevant animal toxicity and human data for comparison.
5.1.1 Analgesics and non-steroidal anti-inflammatory drugs (NSAIDs)
Analgesic nephropathy may be a consequence of the excessive
consumption of mixed analgesics. Originally, phenacetin was common to
all of these mixtures, which led to the conclusion that this drug was
the only cause of "phenacetin kidney". Subsequently, a variety of
analgesics, NSAIDs, and a number of industrial and environmental
chemicals have been shown to have the potential to cause RPN and
interstitial nephritis (Burry et al., 1977; Schwarz, 1987).
The prevalence of analgesic-associated nephropathy varies
worldwide and is probably related to patterns of analgesic use. It has
been found most often in women aged 45-55 as a result of a high
incidence of analgesic abuse, and has been reported more frequently in
Australia and Switzerland and less frequently in the USA, Canada, and
Germany. It is estimated that more than 37 million people in the USA
have arthritis and use these drugs. This is indeed a very large
population at risk. Analgesic effects are said to be a factor in as
many as 20-30% of cases of interstitial nephritis in the south-east of
the USA (Murray & Goldberg, 1975).
Diagnosis is often made by coupling the history of analgesic
abuse with morphological evidence of renal papillary necrosis and
chronic interstitial nephritis. The resultant lesions can be
recognized by radiological examinations or ultrasonography and consist
of calcifications along the line of Hodson, shrinking of the kidneys
resulting in irregular contours, and decreased length of both kidneys.
Necrotic papillae may be voided in the urine and this is occasionally
observed.
At least two countries have legislated to prevent phenacetin from
being sold over the counter. This legislation has had the effect of a
change in analgesic formulation, usually towards that of a single drug
such as aspirin or paracetamol (acetaminophen). In Sweden, the removal
of phenacetin resulted in the progressive decline in the incidence of
analgesic nephropathy. This began approximately 6 years after the
removal of phenacetin and the major impact was seen after 12 years. In
Australia, phenacetin was removed from combination analgesics by 1976
and replaced with acetaminophen (the principle metabolite of
phenacetin); by 1979 legislation prohibited the overthe-counter sale
of combination analgesics. According to the Australia-New-Zealand
Dialysis and Transplant Registry, this has resulted in a progressive
decline in the incidence of analgesic nephropathy in patients
presenting with end-stage chronic renal failure in dialysis and
transplant programmes. In 1982, 22% of patients in dialyses and
transplant programmes had analgesic nephropathy; by 1988, the
incidence had declined to approximately 13%. By contrast, the sale of
phenacetin-containing analgesics declined to 21% (in 1976) and 9% (in
1983) of the total volume of analgesics sold in Belgium, but the
prevalence of analgesic nephropathy remained unchanged over this
period. Retrospective and prospective studies in Belgium have shown
decreased renal function in analgesic abusers who never took analgesic
mixtures containing phenacetin (Elseviers & De Broe, 1988).
Paracetamol, which largely replaced phenacetin in analgesic mixtures),
is assumed to be involved in the pathogenesis of RPN. Recently, an
increased risk of renal disease was found in daily users of
paracetamol in North Carolina, USA (Sandler et al., 1989). Other
therapeutic agents (mostly analgesics and NSAIDs) have been
implicated in RPN, and a number of industrial and environmental
chemicals also have the potential to cause this lesion (Bach &
Bridges, 1985a).
Most patients deny analgesic abuse, thus making epidemiological
studies that attempt to identify the causative agent difficult. Most
of the epidemiological data that has been reported over-reflected the
intake of phenacetin at the expense of other agents that could equally
be implicated. The estimate of the total lifetime dose of analgesic
that produces papillary necrosis varies from < 1 to 35 kg (but this
was based only on phenacetin). No data was given in the
epidemiological studies on what quantities of other analgesics were
also taken or on exposure to papillotoxic chemicals. Thus the etiology
of RPN has been complicated by poly-pharmacy, lack of documentation on
the intake of other analgesics and NSAIDs, exposure to other
chemicals, and the difficulty in diagnosing RPN.
Analgesic abuse (Nanra et al., 1978; Nanra, 1980; Prescott, 1982;
Bach & Bridges, 1985a; Schwarz, 1987) and addiction have been linked
to co-formulation with caffeine, but there is no firm supporting
evidence. Few analgesic abusers take the drugs for appropriate
indications. The origins of abuse are usually psycho-social and
represent neurotic, dependent, immature, introverted, anxious, or
depressed individuals, up to 20% of whom also smoke and abuse alcohol,
psychotropic drugs, and sleeping tablets. Most analgesic abusers are
women (over 30 years of age) from lower socioeconomic/education
groups, who have taken these mixtures for 5-30 years. Several factors
such as dehydration, secondary to high ambient temperatures and
bacterial infection, have been implicated in the development of renal
failure (Kincaid-Smith, 1979). Renal function may be preserved and the
progression to ESRD may be prevented by stopping analgesic exposure,
but patients who continue to abuse analgesics have a poor prognosis
(Schwarz, 1987). Analgesic abusers also have an increased incidence of
anaemia, gastric ulcers, and cardiovascular heart disease (Dubach et
al., 1978).
Clinical features of analgesic nephropathy include loss of
urine-concentrating capacity (Bengtsson, 1962), electrolyte
disturbances, (sodium wastage and hypocalcaemia), and defective
urinary acidification after ammonium chloride loading (Bengtsson,
1962; Nanra et al., 1978). An increase in BUN or creatinine
identifies incipient renal failure; at this stage papillary necrosis
is well advanced and includes secondary degenerative changes.
Radiology and ultrasound may identify irregular shrinking of the renal
tissue and medullary calcifications, but these are advanced changes.
Very early in the course of the injury histological changes are
confined to the medulla (a region of the kidney that is not always
assessed at autopsy). This situation progresses to include other
changes, especially in the cortex, that are easily biopsied to show
interstitial nephritis, but they do not define the underlying cause.
The earliest degenerative changes begin at the papilla tip and
affect interstitial cells, loops of Henle, capillaries, and the
proteoglycan ground substance, and result in lipidosis. More advanced
or intermediate RPN affects the outer medulla, as seen by atrophy,
sclerosis, and inflammatory response and calcification of the necrosed
papilla tip (Burry et al., 1977; Gloor, 1978). Total RPN affects the
corticomedullary and cortical regions and is characterized by chronic
interstitial nephritis, tubular dilatation, atrophy, basement membrane
thickening, fibrosis, sclerosis, and inflammatory cell infiltration.
Vascular degeneration such as suburothelial capillary sclerosis is
pathognomonic for RPN (Mihatsch et al., 1984). Pelvic, ureteric, and
bladder urothelia show thickening of capillary walls, sclerosis of
lamina propria, altered fat and collagen deposition, and epithelial
hyperplasia advancing to malignancy and tumours (Burry et al., 1977;
Mihatsch et al., 1984).
The pathological changes in humans with RPN have been obtained
from autopsy or postmortem tissues, where autolytic degradation may
alter the appearance and make interpretation of the stage of the
lesion difficult. Animal models of RPN in a laboratory situation have
provided more detail on the focus of primary injury and the course of
degenerative changes and also allow mechanisms to be studied, but they
do not necessarily reflect the situation in humans.
Analgesics (e.g., aspirin, phenacetin, and paracetamol) do not
always cause RPN in rats. Thus inappropriately high doses have
sometimes been given for prolonged periods (Prescott, 1982; Bach &
Bridges, 1985a). Biological variation within such groups is very high
and experimental use of NSAIDs has caused fatalities due to
extra-renal toxicity (gastric ulceration and perforation; see Kaump,
1966). This has produced experimental models that are of limited
value for studying the course of the lesion or mechanism of RPN. The
renal functional changes and the pathomorphological progression of the
lesion in several acute model systems show marked similarities with
those reported for the analgesic-associated lesion in both
experimental animals and man (Bach & Hardy, 1985; Bach & Bridges,
1985a).
The histological changes induced by experimental RPN follow a
similar pattern of early, intermediate and total RPN to that described
in man (Fig. 12), and are dose and time dependent. The earliest
morphological changes induced by papillotoxins occur in the renal
medullary interstitial cells. The medullary glycosaminoglycan matrix
also undergoes changes, showing an increase and then a decrease in
staining intensity. It is only subsequently that there are platelet
adhesions, blocking of blood vessels, degenerative changes in the
collecting ducts and proximal tubules, and the accumulation of lipid
material in capillaries and epithelial cells. At the same time as
repair and reepithelization are taking place, there is an increase in
the presence of tubular casts, proximal and distal tubular dilatation,
and hyperplasia of the collecting ducts, and pelvic urothelia, and
the suburothelial capillaries undergo sclerotic changes. When the
repair phase is advanced or complete, there are also degenerative
changes in the cortex, including fibrosis, glomerular sclerosis, and
cystic dilatation. The histological and functional changes produced by
model RPN are remarkably similar to those observed in human analgesic
abusers. The use of high-resolution light microscopy and
ultrastructural studies (in conjunction with histochemistry and
immuno-histochemistry) can help establish the changes in adjacent
cells and link the "cause-and-effect" relationships in the sequence of
degenerative events.
The mechanism of analgesic-induced renal papillary necrosis is
still not fully understood. Progress in our understanding of the
pathogenesis of the model lesions (Bach & Bridges, 1984, 1985a) has
enabled some factors to be identified that may be involved in the
molecular changes. There is no evidence to suggest that the model
lesion has an early immunological basis, nor that it is a consequence
of renal hypoxia or vasoconstriction, and there is no experimental
basis to suggest that the altered intermediary metabolism is a
critical factor (Bach et al., 1983). The concept that altered PG
metabolism gives rise to vascular (or other) changes is an attractive
one, but the exceptionally low levels of these hormones, combined with
their instability, have made it very difficult to test this
hypothesis. The countercurrent concentration mechanism is an important
normal renal function and is thought to play an important role in
concentrating chemicals to toxic levels within the medulla. One of
the earliest changes in the development of RPN is the loss of
concentrating processes, which detracts from this hypothesis.
Furthermore, the concentrating of a compound in the medulla does not
explain the molecular mechanism by which it causes RPN (Bach &
Bridges, 1985a).
At present the most attractive explanation for the development of
RPN relates to a metabolic activation within the kidney. There are two
major oxidative systems for xenobiotic metabolism in the kidney. The
cytochrome P-450 system is localized to the cortex, whereas the PG
hydroperoxidase system (PGH) is located almost exclusively in the
medulla. The reasons for the selective targeting of particular
chemicals for the renal medulla interstitial cells are uncertain, but
 may relate to the absence of free radical scavengers or nucleophiles
and/or to the presence of extensive numbers of lipid droplets
containing polyunsaturated fatty acids within these cells. These would
form an ideal substrate for extensive lipid peroxidation (Porter et
al., 1980) within the renal medullary interstitial cells once a
reactive species had been generated within these cells. The role of
co-oxidation of substrates as a consequence of PG synthesis has become
an attractive mechanistic basis on which to explain papillary damage
(Fig. 13) and the activation of bladder carcinogens, and it may also
be pertinent to other types of renal toxicity that are not associated
with mixed-function oxidase activity (Bach & Bridges, 1984).
Prostaglandin endoperoxide synthetase consists of two inseparable
activities. Fatty acid cyclo-oxygenase catalyses the oxidation of
arachidonic acid to PG hydroperoxy-endoperoxide (PGG2), while the
other activity, PG hydroperoxidase, reduces PGG2 to PGH2 and
co-oxidizes another molecule. PGH2 is the precursor for both PGs and
thromboxanes (Davis et al., 1981). PG hydroperoxidase is inhibited by
antioxidants and will reduce many different fatty acid peroxides,
other organic peroxides (cumene hydroperoxide and tert-butyl
hydroperoxide), and inorganic peroxides (hydrogen peroxide) and, in
the process, co-oxidize a range of substrates including several
bladder carcinogens that produce free radicals.
Paracetamol forms a reactive intermediate that co-valently binds
to trichloroacetic-acid-precipitable macro-molecules but is inhibited
by aspirin and other inhibitors of fatty acid cyclo-oxygenase.
Ethoxyquin, ascorbic acid, and glutathione also inhibit covalent
binding of paracetamol during peroxidative activation by reacting with
electrophilic intermediates generated by co-oxidation (Zenser et al.,
1983).
Other non-steroidal anti-inflammatory agents may produce
hypersensitivity reactions, lipoid nephrosis, and interstitial
nephritis (Finkelstein et al., 1982).
5.1.2 Paracetamol (acetaminophen) and para-aminophenol
Large doses of paracetamol can produce acute proximal tubular
necrosis, especially in male Fischer-344 rats (Mitchell et al., 1977;
McMurtry et al., 1978; Hennis et al., 1981; Newton et al., 1983a,b).
Microsomal cytochrome P-450 activation to a reactive arylating
intermediate is thought to be an obligatory biochemical event in
paracetamol-induced hepatic necrosis (Mitchell et al., 1973; Nelson,
1982). Nephrotoxic dosages of paracetamol bind covalently to renal
protein (Mitchell et al., 1977; McMurtry et al., 1978; Nelson, 1982)
by an NADPH-dependent, cytochrome-P-450-mediated process (McMurtry et
al., 1978; Newton et al., 1983a,b). Alternatively, paracetamol is
enzymically deacetylated to para-aminophenol, a potent selective
nephrotoxin that damages the proximal tubule (Calder et al., 1979).
Para-aminophenol produces acute necrosis of the proximal convoluted
tubules in rats after a single injection (Green et al., 1969), and has
may relate to the absence of free radical scavengers or nucleophiles
and/or to the presence of extensive numbers of lipid droplets
containing polyunsaturated fatty acids within these cells. These would
form an ideal substrate for extensive lipid peroxidation (Porter et
al., 1980) within the renal medullary interstitial cells once a
reactive species had been generated within these cells. The role of
co-oxidation of substrates as a consequence of PG synthesis has become
an attractive mechanistic basis on which to explain papillary damage
(Fig. 13) and the activation of bladder carcinogens, and it may also
be pertinent to other types of renal toxicity that are not associated
with mixed-function oxidase activity (Bach & Bridges, 1984).
Prostaglandin endoperoxide synthetase consists of two inseparable
activities. Fatty acid cyclo-oxygenase catalyses the oxidation of
arachidonic acid to PG hydroperoxy-endoperoxide (PGG2), while the
other activity, PG hydroperoxidase, reduces PGG2 to PGH2 and
co-oxidizes another molecule. PGH2 is the precursor for both PGs and
thromboxanes (Davis et al., 1981). PG hydroperoxidase is inhibited by
antioxidants and will reduce many different fatty acid peroxides,
other organic peroxides (cumene hydroperoxide and tert-butyl
hydroperoxide), and inorganic peroxides (hydrogen peroxide) and, in
the process, co-oxidize a range of substrates including several
bladder carcinogens that produce free radicals.
Paracetamol forms a reactive intermediate that co-valently binds
to trichloroacetic-acid-precipitable macro-molecules but is inhibited
by aspirin and other inhibitors of fatty acid cyclo-oxygenase.
Ethoxyquin, ascorbic acid, and glutathione also inhibit covalent
binding of paracetamol during peroxidative activation by reacting with
electrophilic intermediates generated by co-oxidation (Zenser et al.,
1983).
Other non-steroidal anti-inflammatory agents may produce
hypersensitivity reactions, lipoid nephrosis, and interstitial
nephritis (Finkelstein et al., 1982).
5.1.2 Paracetamol (acetaminophen) and para-aminophenol
Large doses of paracetamol can produce acute proximal tubular
necrosis, especially in male Fischer-344 rats (Mitchell et al., 1977;
McMurtry et al., 1978; Hennis et al., 1981; Newton et al., 1983a,b).
Microsomal cytochrome P-450 activation to a reactive arylating
intermediate is thought to be an obligatory biochemical event in
paracetamol-induced hepatic necrosis (Mitchell et al., 1973; Nelson,
1982). Nephrotoxic dosages of paracetamol bind covalently to renal
protein (Mitchell et al., 1977; McMurtry et al., 1978; Nelson, 1982)
by an NADPH-dependent, cytochrome-P-450-mediated process (McMurtry et
al., 1978; Newton et al., 1983a,b). Alternatively, paracetamol is
enzymically deacetylated to para-aminophenol, a potent selective
nephrotoxin that damages the proximal tubule (Calder et al., 1979).
Para-aminophenol produces acute necrosis of the proximal convoluted
tubules in rats after a single injection (Green et al., 1969), and has
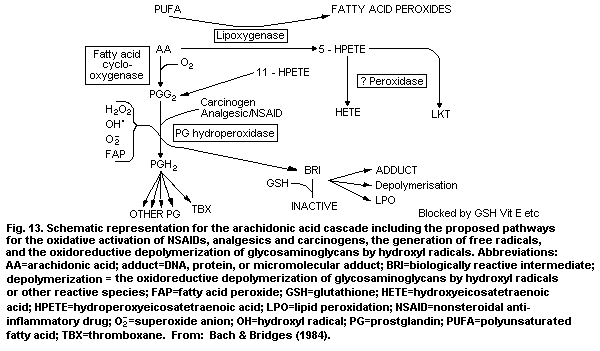 been demonstrated to be a minor metabolite of paracetamol in the
Fischer-344 rat and its isolated perfused kidney (Newton et al.,
1982). Paracetamol ( N-acetyl- p-aminophenol) is structurally
closely related to para-aminophenol, and metabolites have been shown
to be excreted by the biliary route in rats (Siegers & Klaassen, 1984)
and mice (Fischer et al., 1985). These metabolites are the glucuronic
acid and sulfate conjugates (Siegers & Klaassen, 1984) and the
glutathione conjugate (Hinson et al., 1982). Toxicity arising from
para-aminophenol has been previously suggested to result from a
dose-related depletion of kidney reduced glutathione and covalent
binding to essential renal macromolecules (Crowe et al., 1977; 1979).
Both paracetamol and para-aminophenol deplete renal cortical
reduced glutathione concentrations and arylate renal macromolecules
(McMurtry, et al., 1978; Crowe et al., 1979). The changes produced by
para-aminophenol are indistinguishable from those caused by
paracetamol (Newton et al., 1983a,b). Mouse renal cortical slices and
homogenates are capable of deacetylating paracetamol to
para-aminophenol (Carpenter & Mudge, 1981), which has also been
identified as a urinary metabolite of paracetamol in both hamsters
(Gemborys & Mudge, 1981) and Fischer-344 rats (Newton et al.,
1983a,b). Thus the rat is capable of deacetylating paracetamol to
para-aminophenol. In the renal cortex, paracetamol deacetylation
occurs primarily in the cytosolic fraction (Newton et al., 1983a).
Similarly, metabolic activation of paracetamol to an arylating
intermediate is dependent on the presence of a cytosolic deacetylase
(Newton et al., 1983b).
Both para-aminophenol and bis-( p-nitro-phenyl)-phosphate (a
carboxylesterase-amidase inhibitor) inhibit the covalent binding of
paracetamol to renal macromolecules (Newton et al., 1983b). Conclusive
evidence that para-cetamol binds to renal macromolecules after
deacetylation and metabolic activation to para-aminophenol has been
provided by the demonstration of covalent binding of [ring-
14C]-paracetamol, but not [acetyl-14C]-paracetamol, to renal
protein (Newton et al., 1983b).
Thus, paracetamol activation by renal cortical tissue can occur
in two different ways, i.e. either a microsomal
cytochrome-P-450-dependent pathway or deacetylation to
para-aminophenol and subsequent metabolic activation. The reactive
intermediates formed by each pathway suggest that both mechanisms may
play a role in paracetamolinduced renal cortical necrosis.
5.1.3 Antibiotics
Nephrotoxicity related to antibiotics is most often due to
effects on transport, concentration, and excretory functions. All
parts of the nephron or kidney may be affected. However, there is
usually some specificity in the site of action, particular toxins
affecting specific portions of the nephron (Curtis, 1979).
Mechanisms of injury span a broad spectrum of potential lesions.
The most common effect is direct toxicity to renal tubular cells
manifested by cell injury and necrosis. Direct toxicity to glomeruli
is not as conspicuous but does occur. Immunologically induced lesions
in glomeruli and interstitial tissue may also occur.
5.1.3.1 Aminoglycosides
Nephrotoxicity is a common complication of aminoglycoside
antibiotic therapy in man (Bennett, 1983; Matzke et al., 1983;
Kahlmeter & Dahlager, 1984). Early signs of nephrotoxicity include
increased urinary excretion of proximal tubular cell brush-border
membrane enzymes such as alanine aminopeptidase, proteins of relative
low molecular mass such as lysozyme and ß2-microglobulin, and granular
casts (Schentag, 1983). A urine-concentrating defect is usually
evident and may explain the non-oliguric acute renal failure typically
observed in these patients. Less common manifestations of tubular
dysfunction include potassium, magnesium, calcium, and glucose loss in
the urine. Azotaemia and elevation of the serum creatinine
concentration are relatively late manifestations of nephrotoxicity
and reflect depression of glomerular filtration rate consequent to
extensive proximal tubular cell necrosis. Patients receiving standard
doses of amino-glycoside antibiotics usually do not manifest
depression of glomerular filtration until after seven or more days of
drug therapy. However, pathological changes confined to the proximal
tubule can be seen in renal biopsy material obtained before this time
(DeBroe et al., 1984). At the light microscope level, these changes
range from loss of brush-border membrane, apical blebbing, and
prominent vacuoles to cloudy swelling, patchy cell necrosis, and
sloughing of necrotic cells with cast formation in the lumen. Electron
microscopy reveals the presence of multicentric multilamellar membrane
structures known as myeloid bodies within distended lysosomes (Kosek
et al., 1974). These lysosomal lesions can be seen within 1-2 days of
drug treatment and they increase in size and number as therapy is
prolonged.
The nephrotoxicity potential of aminoglycosides has been ranked
as neomycin > gentamicin > sisomicin = kanamycin > tobramycin >
netilmicin > streptomycin (Parker et al., 1982). The situation with
amikacin has been somewhat controversial, but recent studies have
suggested that it is less nephrotoxic, even in experimental animals,
than the other clinically available aminoglycosides, except for
streptomycin. Clear-cut therapeutic advantages of any particular
aminoglycoside are not readily apparent in patients because of the
serious nature of their underlying illness and concurrent therapy with
multiple drugs. Furthermore, the relative nephrotoxicity is usually
assessed by insensitive techniques, such as blood urea nitrogen, serum
creatinine, and enzymuria, that do not give a quantitative
representation of the extent of renal injury. In humans, few would
argue that neomycin and gentamicin are much more nephrotoxic in
therapeutic use than streptomycin, but there are also other important
risk factors that relate to the clinical condition of the patient:
* dehydration, volume depletion, diuretic-induced volume depletion;
* advanced age;
* pre-existing renal disease;
* electrolyte imbalance (acidosis, hypomagnesaemia, hypokalaemia,
hypocalcaemia);
* hypotension/renal ischaemia;
* extrarenal target organ disease such as cirrhosis of the liver;
* exposure to multiple nephrotoxins;
* frequent dose regimens as opposed to larger doses given less
frequently;
* elevated aminoglycoside trough concentrations.
Current understanding of the pathogenesis of amino-glycoside
nephrotoxicity has been derived primarily from studies in rats, which
exhibit a pattern of renal injury indistinguishable from that observed
in man (Kaloyanides & Pastoriza-Munoz, 1980; Humes et al., 1982;
Bennett, 1983; Tulkens, 1989). The drug dose, in relation to body
weight, required to induce injury in the rat is considerably larger
than that required in man, whereas the dose is approximately the same
when expressed in relation to body surface area. From such studies has
emerged unequivocal evidence that aminoglycoside nephrotoxicity is
causally linked to the transport and accumulation of drugs by renal
proximal tubular cells. Following parenteral administration,
aminoglycosides are eliminated unchanged in the urine by glomerular
filtration. A small fraction of the filtered drug is taken up by the
renal proximal tubular cells via a low affinity, high capacity
transport mechanism that exhibits saturation kinetics (Kaloyanides,
1984a; Giuliano et al., 1986). The first step in this transport
process involves binding of the cationic aminoglycoside to apical
membrane receptors, thought to be anionic phospholipids such as
phosphatidylinositol (Sastrasinh et al., 1982). This is followed by
uptake into the cell by adsorptive endocytosis (Silverblatt & Kuehn,
1979) with subsequent translocation and sequestration of the drug in
high concentration within lysosomes (Morin et al., 1980; Josepovitz et
al., 1985). In addition a small quantity of drug appears to gain
access into the cell across the basolateral membrane (Collier et al.,
1979). Following uptake into proximal tubular cells, aminoglycosides
express their nephrotoxicity potential by disrupting one or more
critical intracellular metabolic pathways.
Although these drugs have been shown to effect a variety of
biochemical processes at several sites within proximal tubular cells
(Kaloyanides, 1984b), it remains to be established which if any of
these actions are causally linked to the cascade that eventuates in
cell injury and necrosis. Prominent among the biochemical derangements
is a disturbance of phospholipid metabolism reflected by an increase
in renal cortical phospholipid enriched in phosphatidylinositol
(Kaloyanides, 1984b). The phospholipidosis has been shown to be due
primarily to the accumulation of lysosomal myeloid bodies (Josepovitz
et al., 1985), which form as a consequence of the inhibition of
lysosomal phospholipases (Laurent et al., 1982; Carlier et al., 1983)
by the high concentration of drug within the lysosomal compartment
(Ramsammy et al., 1989a). The mechanism of inhibition is thought to be
related to an electrostatic interaction between the cationic
aminoglycoside and anionic phospholipid. Another example of an adverse
interaction between aminoglycosides and phospholipid is the
observation that gentamicin inhibits agonist activation of the
phosphatidylinositol cascade (Ramsammy et al., 1988a), an effect that
localizes the site of interaction at the cytoplasmic surface of the
plasma membrane and most likely reflects binding of the polycationic
gentamicin to the polyanionic phospholipid,
phosphatidylinositol-4,5-bis-phosphate. This effect may also explain
the observation that aminoglycosides inhibit phosphatidylinositol-
specific phospholipase C in renal brush-border membranes (Schwertz et
al., 1984). Alterations of other biochemical processes associated with
plasma membranes have been described, including depressions of
Na+-K+-ATPase, adenylate cyclase, alkaline phosphatase, and
calcium binding (Morin et al., 1980; Williams et al., 1981). Impaired
mitochondrial respiration (Weinberg & Humes, 1980) and decreased
incorporation of leucine into microsomal protein (Bennett et al.,
1988) have also been observed prior to the onset of obvious
irreversible cell injury. These findings emphasize that multiple
sites serve as targets for drug-cell interaction. However, it remains
uncertain which of these biochemical abnormalities are proximal
events causally linked to toxicity.
One theory that attempts to integrate these diverse observations
focuses on the lysosomal accumulation of aminoglycosides, with
induction of a lysosomal phospholipidosis as the critical first step
(Tulkens, 1989). If the injury threshold concentration of
aminoglycoside is not reached, the lysosomal phospholipidosis
regresses without any biochemical or morphological evidence of
cellular necrosis and regeneration (Giuliano et al., 1984). However,
if the injury threshold concentration is exceeded, the lysosomal
phospholipidosis progresses and the overloaded lysosomes swell,
resulting in the loss of integrity of the lysosomal membrane and the
release of lysosomal enzymes, toxins, and large quantities of
aminoglycosides into the cytosol. The extralysosomal aminoglycoside
interacts with and disrupts the functional integrity of other
subcellular membranes, thereby initiating the injury cascade that
eventuates in cell death.
It should be emphasized that aminoglycoside-induced proximal
tubular cell necrosis is accompanied by a conspicuous regenerative
response (Parker et al., 1982; Toubeau et al., 1986). Thus, the
clinical threshold for nephrotoxicity is determined by the balance
between the rate of necrosis and the rate of regeneration of proximal
tubular cells (Soberon et al., 1979). If necrosis dominates, overt
renal failure ensues.
Aminoglycoside nephrotoxicity is accompanied by increased
generation of free radicals. Furthermore, nephrotoxicity is blocked
with free radical scavengers/antioxidants such as dimethylthiourea,
dimethyl sufoxide, sodium benzoate, or deferoxamine (Walker & Shah,
1987). However other studies have demonstrated that antioxidants such
as vitamin E do not protect against aminoglycoside-induced injury
(Ramsammy et al., 1986, 1987, 1988b). The reasons for this apparent
discrepancy are not known, and the exact role of lipid peroxidation in
gentamicin nephrotoxicity therefore remains unclear.
Polyaspartic acid has recently been shown to protect rats
completely from developing aminoglycoside nephrotoxicity without
inhibiting proximal tubular cell drug uptake (Williams et al., 1986;
Gilbert et al., 1989; Ramsammy et al., 1989b). In vitro studies
suggest that the protective effect of polyaspartic acid is due to the
ability of this polyanionic peptide to bind the cationic
aminoglycosides, thereby preventing these drugs from interacting
electrostatically with various targets, presumably anionic
phospholipids, within the cell.
5.1.3.2 Cephalosporins
The nephrotoxicity of cephalosporins was first noted when the
drugs were used in combination with aminoglycosides, but it is now
recognized that cephalosporins, particularly cephaloridine, may
produce degeneration and necrosis of proximal tubular lining cells and
acute renal failure. It has been suggested that the cellular toxicity
is the result of metabolic activation of the five-member thiophene
ring present in cephalothin and cephaloridine, the only two
cephalosporins that seem capable of producing dose-dependent direct
nephrotoxicity (Mitchell et al., 1977). More recently, it has been
suggested that lipid peroxidation (Goldstein et al., 1989) and direct
mitochondrial toxicity may be involved in the mechanisms of
cephaloridine nephrotoxicity. Necrosis occurs when the concentration
exceeds 1000 mg/kg wet tissue. The correlation between dose and
response, as well as the localization of the lesion in the proximal
portion of the nephron, may be explained by a striking
corticomedullary gradient in tissue concentration. The cellular uptake
of cephaloridine and nephrotoxicity have been modified or eliminated
in experimental animals by pretreatment with either probenecid or
p-aminohippuric acid. The mechanisms of toxicity are complex (Wold
et al., 1979; Tune & Fravert, 1980; Tune, 1986; Goldstein et al.,
1987; Tune et al., 1988).
5.1.3.3 Amphotericin B
The increasing use of immunosuppressive therapy and the attendant
systemic mycotic infections have resulted in an increase in the
administration of amphotericin B. This drug is almost always
associated with some degree of toxicity to the distal renal tubule and
accompanying acidosis, hypokalaemia, and polyuria (Butler, 1966;
Douglas & Healy, 1969). Reduced renal blood flow and glomerular
filtration rate may also occur. Pretreatment of experimental animals
with furosemide or sodium protects against decreases in renal plasma
flow and glomerular filtration rate immediately following amphotericin
B treatment. Salt loading also protects against
amphotericin-induced decreases in renal plasma flow and glomerular
filtration rate upon chronic drug administration in rats. However, it
is important to note that tubular toxicity in these studies was not
ameliorated by salt loading (Tolins & Raij, 1988). These data suggest
that the tubular toxicity of amphotericin B is not secondary to renal
vasoconstriction and ischaemia.
5.1.3.4 Tetracyclines
The nephrotoxicity of tetracycline incited considerable interest
in the early 1960s, shortly after its introduction. People,
particularly children, developed a reversible proximal tubular
dysfunction after receiving outdated drugs. The nephrotoxicity was
found to be due to a degradation product, anhydro-4-epitetracycline.
The problem has disappeared with the substitution of citric acid for
lactose as a vehicle (Curtis, 1979).
Other rare effects of tetracycline that have been reported are
impairment of renal-concentrating ability by
demethylchlorotetracycline and occurrences of acute interstitial
nephritis after minocycline treatment. More important to current usage
is the awareness that the serum half-life of the two most commonly
used drugs, tetracycline and oxytetracycline, is greatly prolonged in
renal failure, and that the anti-anabolic effect of the tetracyclines,
which inhibit the incorporation of amino acids into protein, may
further contribute to negative nitrogen balance and uraemia by raising
blood urea nitrogen (Curtis, 1979).
5.1.4 Penicillamine
Penicillamine (3,3-dimethylcysteine) was first used clinically as
a copper-chelating agent to treat Wilson's disease. Because of the
drug's potential for decreasing collagen formation, its use has been
extended to a number of clinical disorders in which fibrosis is a
major component, such as rheumatoid arthritis, pulmonary fibrosis,
and liver disorders.
It has been suggested that the drug acts by reducing disulfide
linkages. This inhibits polymerization of macromolecules and leads
to impairment of collagen formation. Use of the drug has been tempered
by the occurrence of side effects in as many as 30% of patients. The
most important side effect (20% of cases) is proteinuria. The
morphological appearance of the glomerular lesions is typically that
of perimembranous glomerulonephritis with segmental subepithelial
immune-complex deposits. These changes are best demonstrated as
granular immunofluorescent deposits of IgG and C3. Withdrawal of
penicillamine therapy results in disappearance of the proteinuria and
repair of the basement membrane changes in 60% of cases (Gartner,
1980). Immune-complex glomerulonephritis with granular deposits along
basement membrane and in the mesangium can be produced experimentally,
confirming the role of an immunological mechanism in the pathogenesis
of the nephropathy. In addition the drug is associated with the
development of Goodpasture's syndrome with linear glomerular basement
membrane deposits.
5.1.5 Lithium
Lithium salts, mainly lithium carbonate, have been used for 40
years to prevent relapses of maniac-depressive illness. Impaired renal
ability to acidify and concentrate urine is a common finding among
patients given lithium (Batelle et al., 1982). It is usually regarded
as a minor side-effect of the drug, i.e. a pharmacologically induced
physiological impairment of distal tubules and collecting ducts, such
a target-selective effect usually being reversible after
discontinuation of the therapy. Since the polyurea is resistant to
ADH, the effect has been characterized as resembling nephrogenic
diabetes insipidus (Bendz, 1983).
Although several case reports of lithium-induced chronic renal
insufficiency have been published (Hestbech et al., 1977; Hansen et
al., 1979, 1981; Kincaid-Smith et al., 1979; Cohen et al., 1981;
Walker et al., 1982,1986; Ottosen et al., 1984), the overall evidence
suggesting progressive renal damage in patients taking lithium is
rather limited because of methodological weaknesses in human studies
(Lippmann, 1982). However, animal studies support the view that
long-term treatment with lithium salts may lead to tubulo-interstitial
nephropathies. Experimentally induced focal fibrosis, tubular atrophy,
and cystic dilatation of distal tubules were obtained by exposing
animals to toxic doses (Ottosen et al., 1984; Walker et al., 1986).
In addition to tubular effects, the occurrence of nephrotic
syndrome in psychiatric patients has been attributed to long-term
treatment with lithium salts (Richman et al., 1980; Depner, 1982).
Thus, although case reports do not constitute evidence, there is some
indication that lithium may adversely affect other segments along the
nephron. A proportion of cases, ranging from 0 to 50% (median 8%) of
patients on lithium, may eventually develop chronic renal
insufficiency, evolving towards end-stage renal disease (Cohen et al.,
1981). Such an increased risk may still be regarded as acceptable,
especially when compared to the benefits of such a therapeutic
approach to serious psychiatric problems. Thus, fear of renal disease
may not require the therapy to be stopped. However, a close monitoring
of renal function is strongly recommended. Once-daily dosing to
maintain serum lithium levels at the lower therapeutic range, i.e.
0.4-0.7 mEq/litre, is advisable. Furthermore, it is critical to avoid
salt depletion, which can disturb the equilibrium of serum lithium and
induce acute intoxication.
5.1.6 Urographic contrast media (UCM)
Radiographic procedures are normally safe, but a small proportion
of patients subsequently experience a transient or, very rarely, a
permanent decline in renal function (Cedgard et al., 1986). There are
various predisposition factors such as dose, age, multiple utilization
of UCM, dehydration, diabetes (Taliercio et al., 1986), multiple
myeloma (Harkonen & Kjellstrand, 1981), hypertension, atherosclerosis,
prior kidney or liver diseases, the co-administration of nephrotoxic
drugs, and kidney transplantation. Prospective studies suggest that
diabetes per se is not a risk factor when matched for pre-existing
renal disease (Teruel et al., 1981; D'Elia et al., 1982). This
highlights pre-existing renal disease as a major risk factor. In view
of the fact that there are several million radiological procedures
each year, the number of patients at risk of developing adverse
effects from the administration of contrast media is significant. Up
to 10% of cases of acute renal failure in hospitalized patients may be
due to intravascular urographic contrast medium administration (Hou
et al., 1983).
The cause of such renal injury is not well understood, but
hyperosmolality (e.g., with meglumine diatrizoate) has been claimed to
be an important factor in renal damage (Forrest et al., 1981). New
low-osmolar urographic contrast media (such as iopamidol) are being
introduced, some of which are isotonic with plasma, but a progressive
increase in the incidence of ARF from 0-12% up to 100% in high-risk
patients has been reported (Eisenberg et al., 1980). About 65% of ARF
follow intravenous urography, and 30% follow arteriography. The rest
are associated with computerized tomography (Hanaway & Black, 1977;
Harkonen & Kjellstrand, 1979; Fang et al., 1980). The reported
increase in UCM-induced ARF could be due to better monitoring and
awareness, higher health standards, or a prolonged survival of
patients with critical illnesses (who would then be more prone to
multiple X-ray contrast media examinations), or could represent other
types of nephrotoxicity.
The pathophysiology of UCM-induced ARF is unclear, but may
involve renal ischaemia and haemodynamic effects on glomerular
function and/or intrarenal flow distribution. Several hormonal systems
may be activated prior to and/or during ARF (Caldicott et al., 1970;
Chou et al., 1974; Katzberg et al., 1977). Thus, the effects of
contrast media on kidney function continue to be conflicting and
represent both glomerular and tubular dysfunctions (Milman & Gottlieb,
1977; Rahimi et al., 1981; Teruel et al., 1981; Khoury et al., 1983).
It has been suggested, but not confirmed, that non-ionic low-osmolal
contrast media have reduced nephrotoxicity (Gale et al., 1984;
Spataro, 1984; Smith et al., 1985; Cedgard et al., 1986; Cavaliere et
al., 1987).
Attempts to induce radiocontrast nephrotoxicity in animals have
led to inconclusive or contradictory results. Intact hydrated rats
with or without experimentally induced acute renal failure do not
develop radiocontrast nephrotoxicity (McIntosh, et al., 1975; Moreau
et al., 1980). Transient reductions in glomerular filtration rate and
renal blood flow have been reported immediately following
radiocontrast injection in rats and dogs (Norby & DiBona, 1975;
Cunningham et al., 1986; Katzberg et al., 1986), but rarely has acute
renal failure been studied or documented in the intact animal
following these acute measurements. Nephrotoxicity may occur, however,
when the radiocontrast agent is given in association with experimental
manoeuvres designed to reduce renal function. These include repeated
dehydration with furosemide injections, renal ischaemia (Schultz et
al., 1982), and acute renal failure induced by mercuric chloride or
glycerol (McLachlan et al., 1972).
5.1.7 Anticancer drugs
5.1.7.1 Cisplatin
Cisplatin ( cis-diamminedichloroplatinum II) has become the
chemotherapeutic agent of choice in the treatment of several solid
tumours, particularly testicular and ovarian cancers (Einhorn &
Donohue, 1977). Unfortunately cisplatin is also one of the most toxic
anticancer drugs, its dose-limiting toxicity being nephrotoxicity
(Madias & Harrington, 1978; Goldstein & Mayor, 1983; Safirstein et
al., 1986). Despite the use of optimal methods for administering
cisplatin, such as the use of active hydration (Cvitkovic et al.,
1977) or sodium chloride as the vehicle (Ozols et al., 1984),
approximately 30% of patients will manifest nephrotoxicity.
Early clinical trials of cisplatin in cancer patients showed a
striking incidence of persistent azotaemia and acute renal failure
(Rossof et al., 1972; Lippman et al., 1973). In later studies serum
creatinine levels increased within 6-7 days of treatment, and then
apparently returned to pre-treatment levels by approximately 3 weeks
(Hayes et al., 1977). Similar results were seen following the
injection of cisplatin into rats (Ward & Fauvie, 1976; Chopra et al.,
1982). Thus cisplatin-induced nephrotoxicity initially appeared to be
an acute reversible condition. However, more recent findings suggest
that cisplatin causes a permanent reduction in GFR (Dentino et al.,
1978; Meijer et al., 1983; Fjeldborg et al., 1986), which may indeed
be progressive in nature (Groth et al., 1986; Jaffe et al., 1987).
Hypomagnesaemia is frequently noted in patients receiving
cisplatin (Buckley et al., 1984; Vogelzang et al., 1985), and is
associated with inappropriately high levels of urinary excretion of
magnesium. This deficiency in magnesium leads to hypokalaemia and
hypocalcaemia. This selective renal loss of magnesium is not unusual
and may be even more common than other renal abnormalities as an
expression of cisplatin nephrotoxicity.
Light microscope studies of human kidneys have revealed focal
acute tubular necrosis, affecting primarily the distal and collecting
tubules, with dilatation of convoluted tubules and cast formation
(Gonzalez-Vitale et al., 1977). More recently, Tanaka et al. (1986)
reported sporadic degenerative lesions, necrosis, and regenerative
changes in the S2 and S3 regions of the proximal tubule and also in
the distal tubule and collecting duct. The glomeruli and vasculature
appeared uninvolved. These observations are somewhat different to
those seen in the rat, where cisplatin-induced damage is largely
confined to the S3 segment of the proximal tubule, located in the
outer stripe of the outer medulla (Chopra et al., 1982). With
increasing time cystic tubules develop in this region (Dobyan, 1985).
However, these cysts have not been reported clinically.
The activity of a number of urinary enzymes, including alanine
aminopeptidase, N-acetyl-ß-D-glucosaminidase, leucine aminopeptidase
and ß-glucurinidase, has been shown to be elevated as early as 36-48
h after cisplatin treatment (Kuhn et al., 1978; Jones et al., 1980).
ß2-micro-globulin excretion has also been shown to be transiently
increased after cisplatin treatment (Daugaard et al., 1988a,b). It is
of interest to note that this proteinuria (involving proteins of low
relative molecular mass), predominantly tubular in origin, was
transient, whereas a persistent proteinuria consisting of proteins of
high relative molecular mass, such as albumin and IGg, and glomerular
in origin was seen after the completion of cisplatin treatment.
The pathophysiology of the GFR reduction remains ill defined. It
is clear that cisplatin produces an acute, mainly proximal, tubular
functional impairment within hours of administration (Daugaard et al.,
1988a,b). It has been suggested that the former is a consequence of
the latter. Thus, Groth et al. (1986) attributed the chronic reduction
in GFR to increased intratubular pressure within damaged tubules. The
glomerular proteinuria reported by Daugaard et al. (1988a) suggests
that cisplatin may directly damage glomeruli. Cisplatin-induced
glomerular lesions have been reported in the pig (Robbins et al.,
1990).
5.1.7.2 Adriamycin
The anthracycline antibiotic Adriamycin is widely used in
clinical oncology to treat several cancers, including breast
carcinoma, malignant lymphomas, and sarcomas (Blum & Carter, 1974).
Its clinical use is limited by its cardiotoxicity. Experimentally
adriamycin has been shown to produce a nephrotic syndrome in rats
(Young, 1975), rabbits (Fajardo et al., 1980), and pigs (van Fleet et
al., 1979).
Rats treated with a single dose of Adriamycin exhibited a marked
proteinuria evident within several days of treatment (Bertani et al.,
1982). Maximal levels were seen after approximately 2 weeks, after
which levels declined but remained significantly above control levels
10 weeks after treatment. Serum albumin levels were also significantly
reduced, whereas there was a concomitant hyperlipidaemia (Bertani et
al., 1986). Adriamycin-induced changes in renal functional parameters
are less well defined. Litterst & Weiss (1987) reported that BUN and
serum creatinine values were either unaffected or only minimally
increased. However, more recent studies indicate significant and
progressive reductions in GFR (Hall et al., 1986). Single nephron
glomerular filtration rate is reduced due to a decreased
ultrafiltration coefficient (Michels et al., 1983).
Morphological damage is first seen in the glomerulus;
ultrastructural examination reveals extensive damage to the glomerular
epithelial cells occurring within 36-48 h of injection (Bertani et
al., 1982). This leads to the eventual loss of the foot processes.
Light microscope studies reveal the characteristic presence of
vacuoles in the glomeruli; with time progressive glomerulosclerosis is
seen. Associated with these glomerular changes are tubular changes;
these consist of dilated tubules filled with casts, predominantly in
the outer stripe of the outer medulla, and atrophic tubules associated
with areas of interstitial fibrosis. It appears that Adriamycin
primarily damages the glomeruli and that the tubulo-interstitial
damage results from the proteinuria, which induces cast formation and
interstitial inflammatory reaction. Fajardo et al. (1980) reported
that juxtamedullary glomeruli were more sensitive than cortical
glomeruli; micropuncture studies in the Munich Wistar Fromter rat
confirm this observation (Soose et al., 1988).
Renal toxicity in patients appears rare, although there has been
a report of acute renal failure following Adriamycin-treatment (Burke
et al., 1977). This may reflect species differences in sensitivity or
may reflect the use of inappropriate test protocols for detecting
renal damage.
5.1.8 Immunosuppressive agents
5.1.8.1 Cyclosporin A
Cyclosporin A has been widely used for preventing organ rejection
after transplantation and in auto-immune diseases, but it is highly
nephrotoxic in the clinical situation (Kostakis et al., 1977; Calne et
al., 1978; Powles et al., 1978). Clinically, cyclosporin
nephrotoxicity has been reported as an acute reversible renal
dysfunction, an acute vasculopathy (thrombotic microangiopathy),
and/or a chronic nephropathy with interstitial fibrosis (Mihatsch et
al., 1985; Palestine et al., 1986). After cyclosporin A treatment
there is an inverse linear relation between GFR and the severity of
the lesions. During the first 6 months of treatment, renal fibrosis in
patients given high doses of cyclosporin A shows a dosedependent
progression of increased severity (Klintmalm et al., 1981). The rat
model of cyclosporin A nephrotoxicity may adequately represent the
acute condition in man (Mihatsch et al., 1985, 1986) but does not
represent that seen in the clinical situation. Although rats dosed
with cyclosporin also develop renal surface changes that correspond to
focal areas of collapsed proximal tubular regions with subcapsular
fibrosis, degenerating tubular epithelium and thickening of the
basement membrane, the chronic striped fibrosis and arteriolar lesions
have not been reproduced experimentally.
Some animal data have shown increased blood urea nitrogen and
creatinine, brush-border and lysosomal enzyme leakage, and
vacuolation, necrosis, and regeneration of P3 cells in rats (Dieperink
et al., 1983, 1985; Ryffel et al., 1983, 1986; Murray et al., 1985;
Dieperink, 1989). However, most studies find no necrosis or enzymuria
despite profound reductions in GFR. Functional changes in animals and
man given cyclosporin A are similar. They represent haemodynamic
changes such as an increased renal vascular resistance (Murray et al.,
1985), proximal fractional reabsorption (Dieperink et al., 1983,
1985; Dieperink, 1989), and renal blood flow, plasma flow, and GFR
decrease. The reduction in renal perfusion and filtration has been
prevented experimentally by vasoactive alpha-adrenergic antagonists
and renal denervation (Murray et al., 1985), but this has not been
established in humans. Cyclosporin A appears to have a direct
preglomerular vasoconstriction effect, decreasing the ultrafiltration
pressure and increasing proximal fractional reabsorption. Presumably,
tubular flow rates and end proximal tubular delivery decrease, and,
due to varying tubular hypoperfusion, there is a focal tubular
collapse, and degeneration and peritubular interstitial fibrosis
develop. The precise relationship between renal vasoconstriction and
chronic tubulo-interstitial pathology is poorly understood.
5.1.9 Heroin
About 1% of heroin addicts develop haematuria, proteinuria, or
the nephrotic syndrome (Cunningham et al., 1980). Morphologically,
the renal lesion has been described as focal sclerosing
glomerulonephritis. In the absence of proliferative lesions or immune
deposits, a direct toxic effect of heroin or even a contaminant or
solvent employed in the administration of heroin has been suggested as
the major pathogenetic mechanism. However, heroin-related increases in
IgM titres have been regarded as evidence that an immunological
mechanism may play some role in this disorder.
5.1.10 Puromycin aminonucleoside
Direct toxicity of the puromycin aminonucleoside to glomerular
components may also occur and may be a factor in renal failure.
Experimental studies of the effects of puromycin have provided
considerable basic information regarding the pathogenesis of direct
chemical injury to glomerular structures. The effects are limited to
rats and monkeys. Epithelial cells become swollen, and there is an
increase in lysosome and pinocytotic activity, fusion and loss of foot
processes, and a reduction in the number of filtration slits. As the
lesion progresses, epithelial cells become detached, leaving "naked"
basement membrane in direct contact with Bowman's capsule, which may
account for the severe proteinuria (Caulfield et al., 1976). It is
suggested that as the lesion progresses there is an increase in
basement membrane synthesis, mesangial cell proliferation and fusion,
and crescent formation leading to the light microscope appearance of
focal glomerular sclerosis (Gartner, 1980). The glomerular lesions
produced by puromycin do not appear to invoke an immunological
response, so that the resulting alterations are entirely related to
the direct toxicity of the drug.
5.2 Chemicals
The diversity of organic molecules is such that there are
chemicals that are now known to adversely affect each part of the
kidney. This section will examine those chemicals that do not fit into
any conventional section on therapeutically used agents.
5.2.1 Ethylene glycol
Ethylene glycol, a constituent of antifreeze, is occasionally
ingested and causes severe acute toxicity to the brain and kidney.
Acute tubular necrosis is followed by renal failure. Exposure often
leads to permanent renal damage. A morphological feature of mild
ethylene glycol toxicity is cytoplasmic vacuolation, which may suggest
hypokalaemic nephropathy or osmotic nephrosis due to mannitol. Most
ethylene glycol is excreted unmetabolized, but a small percentage is
metabolized to oxalic acid. This is accompanied by deposition of
calcium oxalate crystals in the kidney, which may contribute to a
persistent inflammatory reaction and interstitial fibrosis. Excessive
urinary excretion of oxalate and crystal formation may also be seen
following administration of halogen-containing anaesthetic agents,
particularly methoxyflurane and halothane (Roxe, 1980). Acute ethylene
glycol toxicity is treated with ethanol, which competes as a substrate
for alcohol dehydrogenase (Peterson et al., 1981).
5.2.2 Organic chemicals and solvents
5.2.2.1 Volatile hydrocarbons
Volatile hydrocarbons, particularly chlorinated compounds such as
carbon tetrachloride and trichloroethylene, may produce glomerular
lesions leading to nephrotic syndrome and renal failure. The
relationship of volatile hydrocarbon exposure to the development of
glomerulo-nephritis in populations is not clear. It has been found
that among patients with glomerulonephritis there are more with a
history of exposure to hydrocarbon solvents than would be expected.
Attempts to reproduce in rats the glomerular lesions observed in
patients have only been partially successful. Solvent-exposed rats had
increased proteinuria and glomerular sclerosis, but proliferative
lesions and significant immune deposits were not observed (Zimmerman
& Norbach, 1980). Of 15 patients studied in Sweden with
post-streptococcal glomerulonephritis, 6 had a history of brief
exposure to organic solvents before the development of their disease.
This suggested to these investigators that solvent exposure may
influence the outcome of an infection with streptococci. Prior
exposure to hydrocarbon-containing solvents has been identified in a
number of patients with Goodpasture's syndrome (Gartner, 1980). Apart
from the pulmonary manifestation of cough, shortness of breath, and
haemoptysis, there may be haematuria and proteinuria. Renal morphology
consists of a proliferative glomerulonephritis with IgG and C3 in the
glomerular basement membrane.
Studies conducted in Sweden (Askergren 1981; Askergren et al.,
1981) and in Belgium (Viau et al., 1987) have reported a slight
increase in the urinary excretion of albumin in groups of workers
exposed to industrial solvents, particularly styrene. This effect
probably reflects an enhanced glomerular permeability since the
urinary output of markers of proximal tubular function
(ß2-micro-globulin, retinol-binding protein) was not affected. In
Italy, Franchini et al. (1983) also reported slight renal disturbances
in workers occupationally exposed to solvents. These effects
consisted of enhanced urinary excretion of total proteins, lysozyme,
and ß-glucuronidase and pointed to a tubular lesion, since they were
not accompanied by a rise in albuminuria. It is at present impossible
to relate nephrotoxic effects reported in these studies to exposure to
one solvent or one class of solvents, although styrene has been
incriminated by Askergren et al. (1981). One must also recognize that
the renal effects reported in these studies are mild and do not appear
to correlate with indices of exposure to solvents.
An auto-immune mechanism following chronic exposure is probably
responsible for the glomerular lesions. In patients who have
Goodpasture's syndrome, the primary site of damage may be the alveolar
basement membrane of the lung, which is damaged by inhalation of the
solvents, and antibodies to altered alveolar basement membrane may
cross-react with glomerular basement membrane. Alternatively,
auto-immunity may follow direct toxic injury to renal tubular or
glomerular structures. Acute exposure to these solvents does produce
acute tubular necrosis, and it is likely that prolonged exposure to
low levels that do not result in cell necrosis produces cell injury
sufficient to damage renal cell membranes and provide the antigen for
the immune reaction (Gartner, 1980).
5.2.2.2 Chloroform
In vitro exposure to chloroform has been shown to produce
toxicity in kidney slices from male but not from female mice (Smith &
Hook, 1984). Furthermore, 14C-labelled chloroform was metabolized
to 14CO2, and the radioactivity was covalently bound by cortical
microsomes from male but not female mice. The in vitro metabolism of
chloroform by male, but not female, renal slices is consistent with
reduced susceptibility of female mice to in vivo chloroform
nephrotoxicity (Smith et al., 1983; Smith et al., 1984). Metabolism is
dependent on oxygen, a NADPH-regenerating system, incubation time,
microsomal protein concentration, and substrate concentration, and is
inhibited by carbon monoxide (Smith & Hook, 1984). The negligible
degree of chloroform metabolism and toxicity in female mice is
consistent with a lower renal cytochrome P-450 concentration and
activity in female mice than in males (Smith & Hook, 1984).
Pretreatment of rabbits with phenobarbital, a renal cytochrome P-450
inducer in this species, enhances the toxic response of renal cortical
slices to chloroform in vitro (Bailie et al., 1984). The rate at
which deuterated chloroform is metabolized by the liver to phosgene is
approximately half that of chloroform. Deuterated chloroform is also
less hepatotoxic that chloroform since the C-D bond is stronger than
the C-H bond. These data suggest that cleavage of the C-H bond is the
rate-limiting step in the activation of chloroform. Deuterated
chloroform is also less toxic to the kidney than chloroform
(Ahmadizadeh et al., 1981; Branchflower et al., 1984). This deuterium
isotope effect on chloroforminduced nephrotoxicity suggests that the
kidney metabolizes chloroform in the same manner as the liver, e.g.,
by oxidation to phosgene. Indeed, rabbit renal cortical microsomes
incubated in media supplemented with L-cysteine metabolize
14C-labelled chloroform to radioactive phosgene-cysteine
2-oxothiazolidine-4-carboxylic acid (Bailie et al., 1984). These
in vitro data collectively support the hypothesis that mouse and
rabbit kidneys biotransform chloroform to a metabolite (phosgene)
that mediates nephrotoxicity.
5.2.2.3 Halogenated alkenes
The nephrotoxin S-(1,2-dichlorovinyl)-L-cysteine (DCVC) is
formed by trichloroethylene extraction of proteinaceous substances and
was first identified in extracted animal food (McKinney et al., 1957,
1959). It has been widely used as a model compound in nephrotoxicity
studies. DCVC is accumulated in the proximal tubules by an active
carrier system for organic anions (Elfarra et al., 1986a,b). It is
then activated by cysteine conjugate ß-lyase to a reactive thiol
(Bhattacharya & Schultze, 1967) and causes tubular damage (Terracini
& Parker, 1965). DCVC is a potent specific nephrotoxin, which produces
proximal tubular damage in vivo and in vitro (Elfarra et al.,
1986a,b; Lash & Anders, 1986; Lash et al., 1986). In vivo DCVC causes
its primary lesion in the straight segment (S-3) of the proximal
tubule, and the molecule is also cytotoxic both for primary cultures
of proximal tubular cells and for cell lines derived from this region
of the nephron. There is a close correlation between the in vivo and
in vitro effects of this compound with regards to its metabolism and
effects on cells (Hassall et al., 1983).
Cysteine conjugates such as DCVC are metabolized by cysteine
conjugate ß-lyase to their ultimate toxic species i.e. pyruvate,
ammonia, and a reactive thiol (Anderson & Schultze, 1965). This
reaction plays a role in the nephrotoxicity of DCVC (Lash et al.,
1986). ß-lyase has been found to predominate in cytosolic and
mitochondrial fractions (Lash et al., 1986) and has a requirement for
pyridoxal phosphate. The enzyme activity can be inhibited by pyridoxal
phosphate inhibitors such as amino-oxyacetic acid and propargylglycine
(Elfarra et al., 1986a). In addition to monitoring enzyme activity,
renal cortical slices can be utilized to assess the regulation of
enzyme activity and the resultant effects on toxicity.
There is a greater sensitivity to DCVC-induced kidney damages in
the adult mouse than there is in the newborn. Similar findings have
been reported using cephaloridine, where the newborn animal is more
resistant to nephrotoxicity than the adult rabbit (Tune, 1975). These
finding for DCVC differ from those for hexachlorobutadiene (Kuo &
Hook, 1983; Lock et al., 1984), where nephrotoxicity is greater in the
young rat and mouse than in the adults.
Chlorotrifluoroethylene is a potent nephrotoxin (Potter et al.,
1981) and is metabolized by hepatic cytosolic and microsomal
glutathione S-transferases to
S-(2-chloro-1,1,2-trifluoroethyl)glutathione (Dohn et al., 1985a),
which is nephrotoxic in rats and cytotoxic in isolated rat kidney
proximal tubular cells (Dohn et al., 1985b). The corresponding
cysteine S-conjugate, S-(2-chloro-1,1,2-trifluoroethyl)-L-cysteine
(CTFC) is also nephrotoxic in rats and cytotoxic in isolated kidney
cells, and its bioactivation is dependent on metabolism by renal
ß-lyase (Dohn et al., 1985b). Pyruvate and hydrogen sulfide have been
identified as metabolites of CTFC (Banki et al., 1986; Lash et al.,
1986).
5.2.2.4 Hydrocarbon-induced nephrotoxicity
Inhalation of unleaded gasoline for 2 years produced renal
tumours (adenomas and adenocarcinomas) in male Fischer-344 rats but
not in female rats or mice of either sex (Kitchen, 1984; MacFarland,
1984; Mehlmann et al., 1984). Subchronic inhalation exposure
increased protein (hyaline) droplets in proximal convoluted tubules
(Halder et al., 1984), accumulated casts at the cortico-medullary
junction and single cell necrosis and regeneration of the nephron
(Short et al., 1986) in male rats. When different fractions of
unleaded gasoline were screened for their specific effect on the male
rat kidney, it was found that the branched-chain saturated hydrocarbon
components (used as anti-knocking agents) caused hyaline droplet
formation. A number of chemicals, such as 2,2,4-trimethyl-pentane
(Phillips & Egan, 1984a,b; Halder et al., 1985; Viau et al., 1986a),
decalin (Alden et al., 1984), 1,4-dichlorobenzene (NTP, 1987), and
p-dichlorobenzene (NTP, 1986; Bomhard et al., 1988), have now been
shown to cause such hyaline droplets in male rats. Although these
chemicals cause minimal renal functional impairment, a protein droplet
nephrosis develops, progressing to mild tubular degeneration,
necrosis, and regeneration after several weeks of treatment (Phillips
& Cockrell, 1984a,b).
Male mice excrete a sex-related protein, which results in a
urinary protein level 2.5 to 3 times that of female mice. However, the
male mouse sex-associated urinary protein hydrolyses readily and thus
does not accumulate in the proximal tubule (Alden et al., 1984; Alden,
1989). In humans, it has recently been reported that protein 1 (an
alpha2-microglobulin of about 20 000 Daltons) has a sex-linked
behaviour just like the androgen-dependent alpha2u-globulin. Protein
1 is excreted in greater amounts in the urine of males after puberty.
In the age group 15 to 20 years, its concentration in the urine of
males is on average fifty times higher than that in the urine of
females (Bernard et al., 1989). The relevance of this observation in
humans is unknown.
The basis for the marked sex dependence and species difference in
the development of hyaline droplet deposition in male rats relates to
the fact that they excrete the sex-hormone-related and therefore
male-specific protein alpha2u-globulin (Stonard et al., 1986; Loury
et al., 1987; Olson et al., 1987). This is also species specific to
the rat and has not been reported in any other commonly used animals
or man. alpha2u-Globulin is synthesized by the male rat liver and is
an important constituent of the physiological proteinuria in adult
male rats. At maturity the total urinary protein is 20-30%
alpha2u-globulin and 10% albumin. At 160 days of age the excretion of
albumin and total urinary proteins is markedly increased. By one
year, albumin represents nearly 60% of the total protein while
alpha2u-globulin is less than 10%. This reversal in relative content
may be the consequence of a progressive glomerulonephrosis, associated
with an apparent spontaneous accumulation of hyaline droplets. The
nephrotic condition may be the consequence of the burden of excreting
alpha2u-globulin. Its early onset is sex dependent; female rats do not
exhibit the proteinuria until a later age. In both aging and the
response to hydrocarbons, the common pathological factor may be the
accumulation of alpha2u-globulin, via a susceptible pathway not shared
with other proteins (Neuhaus, 1986; Stonard et al., 1986).
The chronic regenerative response subsequent to moderate proximal
tubular damage in the kidney of male rats exposed to petroleum
hydrocarbons may be an important stimulus in renal tumour formation
caused by this group of chemicals (Short et al., 1986). Loury et al.
(1987) have shown a 5- to 8-fold increase in S phase of renal cells
induced by unleaded gasoline, and the renal proliferative effects of
2,2,4-trimethylpentane are localized to the S2 segment of the
proximal tubule of the male rat (Short et al., 1986).
Administration of 2,2,4-trimethylpentane to male rats produces a
dose-related increase in the concentration of
2,2,4-trimethylpentane-derived radiolabel in the kidney, which appears
to parallel the dose-related accumulation of 2u-globulin (Stonard et
al., 1986; Charbonneau et al., 1987). The reversible binding of a
metabolite of 2,2,4trimethylpentane to alpha2u-globulin in the male
rat kidney (Lock et al., 1987) is thought to alter endocytosis or
lysosomal handling of the alpha2u-globulin-2,2,4-trimethylpentane
metabolite complex. This may increase cell turnover of the S2 cells
via lysosomal enlargement and/or instability, leading to cell death.
It appears that lysosomal catabolism of the
2,4,4-trimethyl-2-pentanol-alpha2u-globulin complex compound to
alpha2u-globulin causes lysosomal protein overload, resulting in
cell necrosis (Swenberg et al., 1989).
There are other sex-related differences in the handling of
2,2,4-trimethylpentane by rats. Female rats rapidly metabolize
2,2,4-trimethylpentane and excrete it in urine, while in male rats the
compound is eliminated more slowly and is retained in the kidneys
(Kloss et al., 1985). Recent studies have also shown that
2,2,4-trimethylpentane is metabolized in male and female rats to
trimethyl-pentanols, pentanoic acids, and hydroxypentanoic acids
(Olson et al., 1986; Charbonneau et al., 1987).
5.2.2.5 Bipyridyl herbicides
Paraquat is a potent bipyridyl herbicide that has multiple organ
effects. The kidney is frequently involved in serious cases of
paraquat poisoning (WHO, 1984). The compound is actively secreted by
the organic cation transport in the proximal tubule. Renal
histological examinations in a variety of animals exposed to paraquat
show vacuolation of the proximal convoluted tubules and proximal
tubular cell necroses (Lock, 1979; Lock & Ishmael, 1979). Acute
oliguric renal failure is common in severely poisoned patients. Less
severe manifestations include impaired glomerular filtration, which
often recovers after several days and before the paraquat induces
severe pulmonary fibrosis. Other renal functional abnormalities
include proteinuria and haematuria. Tubular damage may be shown by the
presence of glucosuria or all of the features of the Fanconi syndrome.
The severity of the acute renal failure is a major determinant of the
outcome of the poisoning (WHO, 1984).
The biochemical mechanism of nephrotoxicity has not been fully
elucidated, but it is assumed to be identical to that seen in other
tissues. Paraquat undergoes redox cycling in the presence of NADPH and
oxygen with the generation of superoxide and subsequent development of
lipid peroxidation and membrane damage. The development of hydroxyl
radicals results in oxidative damage to nucleic acids, proteins, and
polysaccharides (Autor, 1977).
Diquat is another bipyridyl herbicide that produces multiple
organ toxicity. It undergoes active tubular secretion by the organic
cation system in the proximal tubule (Lock, 1979; Lock & Ishmael,
1979). The histological lesion produced by diquat is necrosis of the
proximal tubular cells and some distal tubular cells (Lock & Ishmael,
1979). Human cases of diquat poisoning result in acute renal failure.
The mechanism of toxicity of diquat appears to be identical to that of
paraquat (Autor, 1977; WHO, 1984).
5.3 Mycotoxins
A high frequency of endemic chronic nephropathy has been
recognized in localized areas of Bulgaria, Rumania, and Yugoslavia
since the 1920s. The affected people live in villages in valleys near
the Danube (Hall & Dammin, 1978; WHO, 1979; Hall, 1982). The
condition, known as Balkan endemic nephropathy (Fig. 14), is an
interesting case study of an environmentally related chronic renal
disease. The etiology is unknown at present. Mycotoxins, particularly
ochratoxin A, have been implicated because of similarities with
disease in animals and identification of the mycotoxin in food (Krogh
et al., 1977; Pepeljnjak & Cvetnic, 1985; Petkova-Bocharova &
Castegnaro, 1985) and in human tissues (Hult & Fuchs, 1986) where
nephrotoxicity is most frequent. Silicates have been suggested because
of the proximity of villages with affected families to streams and
rivers containing silicon.
Although not clearly implicated in BEN, the fungal toxin citrinin
has been suggested as a causative agent in porcine citrinin
nephropathy and clearly has nephrotoxic effects in a number of species
(Berndt & Hayes, 1977; Phillips et al., 1979; Phillips et al.,
1980a,b; Lockard et al., 1980). Citrinin produces acute tubular
necrosis primarily of the S1 section of the proximal tubule. It is
eliminated rapidly by the kidney and only metabolized to the extent of
10-15%, which suggests that effects are due to the parent compound.
Little information is available concerning the cascade leading from
the initial insult to the production of acute tubular necrosis 2-4
days after its administration. Citrinin has been shown to exert a
synergistic effect on ochratoxin A toxicity in animal models. This is
important because the same fungal species that synthesize ochratoxin
A also produce citrinin. This was clearly demonstrated by the presence
of citrinin in 19 out of 21 food samples contaminated with ochratoxin
A. Both genetic and environmental factors, such as exposure to
ochratoxin A, appear to be involved in BEN and the associated renal
tract tumours (Castegnaro & Chernozemsky, 1987). In one endemic area
in Bulgaria the relative risk of patients with BEN developing urinary
tract tumours is 90-fold greater than in people from non-endemic areas
(Castegnaro & Chernozemsky, 1987). Inhabitants of the 15 villages in
the Vratza region of northern Bulgaria have a 30-40% mortality rate
from chronic nephropathy, while urinary tract tumours comprise 25-30%
of all neoplasms in males and females in these geographic areas
(Markovic, 1972). In recent studies ochratoxin A has been found to
induce renal adenomas and carcinomas both in mice (Kanisawa &
Suzuki, 1978; Bendele et al., 1985) and in rats (NTP, 1988). Frequent
metastases, mainly to the lung, were found in the rat study. The
target of ochratoxin nephrotoxicity has been reported to be the S2
and S3 nephron segments (Jung et al., 1989).
Thus the animal and human data indicate that ochratoxin A is a
risk factor for toxic nephropathies and in the etiology of human
nephropathy and associated renal tumours.
5.4 Silicon
An association between occupational exposure to free silica
(SiO2) and chronic nephropathy has been suspected for several years,
but the number of reported cases is few. Clinically, lung fibrosis is
been demonstrated to be a minor metabolite of paracetamol in the
Fischer-344 rat and its isolated perfused kidney (Newton et al.,
1982). Paracetamol ( N-acetyl- p-aminophenol) is structurally
closely related to para-aminophenol, and metabolites have been shown
to be excreted by the biliary route in rats (Siegers & Klaassen, 1984)
and mice (Fischer et al., 1985). These metabolites are the glucuronic
acid and sulfate conjugates (Siegers & Klaassen, 1984) and the
glutathione conjugate (Hinson et al., 1982). Toxicity arising from
para-aminophenol has been previously suggested to result from a
dose-related depletion of kidney reduced glutathione and covalent
binding to essential renal macromolecules (Crowe et al., 1977; 1979).
Both paracetamol and para-aminophenol deplete renal cortical
reduced glutathione concentrations and arylate renal macromolecules
(McMurtry, et al., 1978; Crowe et al., 1979). The changes produced by
para-aminophenol are indistinguishable from those caused by
paracetamol (Newton et al., 1983a,b). Mouse renal cortical slices and
homogenates are capable of deacetylating paracetamol to
para-aminophenol (Carpenter & Mudge, 1981), which has also been
identified as a urinary metabolite of paracetamol in both hamsters
(Gemborys & Mudge, 1981) and Fischer-344 rats (Newton et al.,
1983a,b). Thus the rat is capable of deacetylating paracetamol to
para-aminophenol. In the renal cortex, paracetamol deacetylation
occurs primarily in the cytosolic fraction (Newton et al., 1983a).
Similarly, metabolic activation of paracetamol to an arylating
intermediate is dependent on the presence of a cytosolic deacetylase
(Newton et al., 1983b).
Both para-aminophenol and bis-( p-nitro-phenyl)-phosphate (a
carboxylesterase-amidase inhibitor) inhibit the covalent binding of
paracetamol to renal macromolecules (Newton et al., 1983b). Conclusive
evidence that para-cetamol binds to renal macromolecules after
deacetylation and metabolic activation to para-aminophenol has been
provided by the demonstration of covalent binding of [ring-
14C]-paracetamol, but not [acetyl-14C]-paracetamol, to renal
protein (Newton et al., 1983b).
Thus, paracetamol activation by renal cortical tissue can occur
in two different ways, i.e. either a microsomal
cytochrome-P-450-dependent pathway or deacetylation to
para-aminophenol and subsequent metabolic activation. The reactive
intermediates formed by each pathway suggest that both mechanisms may
play a role in paracetamolinduced renal cortical necrosis.
5.1.3 Antibiotics
Nephrotoxicity related to antibiotics is most often due to
effects on transport, concentration, and excretory functions. All
parts of the nephron or kidney may be affected. However, there is
usually some specificity in the site of action, particular toxins
affecting specific portions of the nephron (Curtis, 1979).
Mechanisms of injury span a broad spectrum of potential lesions.
The most common effect is direct toxicity to renal tubular cells
manifested by cell injury and necrosis. Direct toxicity to glomeruli
is not as conspicuous but does occur. Immunologically induced lesions
in glomeruli and interstitial tissue may also occur.
5.1.3.1 Aminoglycosides
Nephrotoxicity is a common complication of aminoglycoside
antibiotic therapy in man (Bennett, 1983; Matzke et al., 1983;
Kahlmeter & Dahlager, 1984). Early signs of nephrotoxicity include
increased urinary excretion of proximal tubular cell brush-border
membrane enzymes such as alanine aminopeptidase, proteins of relative
low molecular mass such as lysozyme and ß2-microglobulin, and granular
casts (Schentag, 1983). A urine-concentrating defect is usually
evident and may explain the non-oliguric acute renal failure typically
observed in these patients. Less common manifestations of tubular
dysfunction include potassium, magnesium, calcium, and glucose loss in
the urine. Azotaemia and elevation of the serum creatinine
concentration are relatively late manifestations of nephrotoxicity
and reflect depression of glomerular filtration rate consequent to
extensive proximal tubular cell necrosis. Patients receiving standard
doses of amino-glycoside antibiotics usually do not manifest
depression of glomerular filtration until after seven or more days of
drug therapy. However, pathological changes confined to the proximal
tubule can be seen in renal biopsy material obtained before this time
(DeBroe et al., 1984). At the light microscope level, these changes
range from loss of brush-border membrane, apical blebbing, and
prominent vacuoles to cloudy swelling, patchy cell necrosis, and
sloughing of necrotic cells with cast formation in the lumen. Electron
microscopy reveals the presence of multicentric multilamellar membrane
structures known as myeloid bodies within distended lysosomes (Kosek
et al., 1974). These lysosomal lesions can be seen within 1-2 days of
drug treatment and they increase in size and number as therapy is
prolonged.
The nephrotoxicity potential of aminoglycosides has been ranked
as neomycin > gentamicin > sisomicin = kanamycin > tobramycin >
netilmicin > streptomycin (Parker et al., 1982). The situation with
amikacin has been somewhat controversial, but recent studies have
suggested that it is less nephrotoxic, even in experimental animals,
than the other clinically available aminoglycosides, except for
streptomycin. Clear-cut therapeutic advantages of any particular
aminoglycoside are not readily apparent in patients because of the
serious nature of their underlying illness and concurrent therapy with
multiple drugs. Furthermore, the relative nephrotoxicity is usually
assessed by insensitive techniques, such as blood urea nitrogen, serum
creatinine, and enzymuria, that do not give a quantitative
representation of the extent of renal injury. In humans, few would
argue that neomycin and gentamicin are much more nephrotoxic in
therapeutic use than streptomycin, but there are also other important
risk factors that relate to the clinical condition of the patient:
* dehydration, volume depletion, diuretic-induced volume depletion;
* advanced age;
* pre-existing renal disease;
* electrolyte imbalance (acidosis, hypomagnesaemia, hypokalaemia,
hypocalcaemia);
* hypotension/renal ischaemia;
* extrarenal target organ disease such as cirrhosis of the liver;
* exposure to multiple nephrotoxins;
* frequent dose regimens as opposed to larger doses given less
frequently;
* elevated aminoglycoside trough concentrations.
Current understanding of the pathogenesis of amino-glycoside
nephrotoxicity has been derived primarily from studies in rats, which
exhibit a pattern of renal injury indistinguishable from that observed
in man (Kaloyanides & Pastoriza-Munoz, 1980; Humes et al., 1982;
Bennett, 1983; Tulkens, 1989). The drug dose, in relation to body
weight, required to induce injury in the rat is considerably larger
than that required in man, whereas the dose is approximately the same
when expressed in relation to body surface area. From such studies has
emerged unequivocal evidence that aminoglycoside nephrotoxicity is
causally linked to the transport and accumulation of drugs by renal
proximal tubular cells. Following parenteral administration,
aminoglycosides are eliminated unchanged in the urine by glomerular
filtration. A small fraction of the filtered drug is taken up by the
renal proximal tubular cells via a low affinity, high capacity
transport mechanism that exhibits saturation kinetics (Kaloyanides,
1984a; Giuliano et al., 1986). The first step in this transport
process involves binding of the cationic aminoglycoside to apical
membrane receptors, thought to be anionic phospholipids such as
phosphatidylinositol (Sastrasinh et al., 1982). This is followed by
uptake into the cell by adsorptive endocytosis (Silverblatt & Kuehn,
1979) with subsequent translocation and sequestration of the drug in
high concentration within lysosomes (Morin et al., 1980; Josepovitz et
al., 1985). In addition a small quantity of drug appears to gain
access into the cell across the basolateral membrane (Collier et al.,
1979). Following uptake into proximal tubular cells, aminoglycosides
express their nephrotoxicity potential by disrupting one or more
critical intracellular metabolic pathways.
Although these drugs have been shown to effect a variety of
biochemical processes at several sites within proximal tubular cells
(Kaloyanides, 1984b), it remains to be established which if any of
these actions are causally linked to the cascade that eventuates in
cell injury and necrosis. Prominent among the biochemical derangements
is a disturbance of phospholipid metabolism reflected by an increase
in renal cortical phospholipid enriched in phosphatidylinositol
(Kaloyanides, 1984b). The phospholipidosis has been shown to be due
primarily to the accumulation of lysosomal myeloid bodies (Josepovitz
et al., 1985), which form as a consequence of the inhibition of
lysosomal phospholipases (Laurent et al., 1982; Carlier et al., 1983)
by the high concentration of drug within the lysosomal compartment
(Ramsammy et al., 1989a). The mechanism of inhibition is thought to be
related to an electrostatic interaction between the cationic
aminoglycoside and anionic phospholipid. Another example of an adverse
interaction between aminoglycosides and phospholipid is the
observation that gentamicin inhibits agonist activation of the
phosphatidylinositol cascade (Ramsammy et al., 1988a), an effect that
localizes the site of interaction at the cytoplasmic surface of the
plasma membrane and most likely reflects binding of the polycationic
gentamicin to the polyanionic phospholipid,
phosphatidylinositol-4,5-bis-phosphate. This effect may also explain
the observation that aminoglycosides inhibit phosphatidylinositol-
specific phospholipase C in renal brush-border membranes (Schwertz et
al., 1984). Alterations of other biochemical processes associated with
plasma membranes have been described, including depressions of
Na+-K+-ATPase, adenylate cyclase, alkaline phosphatase, and
calcium binding (Morin et al., 1980; Williams et al., 1981). Impaired
mitochondrial respiration (Weinberg & Humes, 1980) and decreased
incorporation of leucine into microsomal protein (Bennett et al.,
1988) have also been observed prior to the onset of obvious
irreversible cell injury. These findings emphasize that multiple
sites serve as targets for drug-cell interaction. However, it remains
uncertain which of these biochemical abnormalities are proximal
events causally linked to toxicity.
One theory that attempts to integrate these diverse observations
focuses on the lysosomal accumulation of aminoglycosides, with
induction of a lysosomal phospholipidosis as the critical first step
(Tulkens, 1989). If the injury threshold concentration of
aminoglycoside is not reached, the lysosomal phospholipidosis
regresses without any biochemical or morphological evidence of
cellular necrosis and regeneration (Giuliano et al., 1984). However,
if the injury threshold concentration is exceeded, the lysosomal
phospholipidosis progresses and the overloaded lysosomes swell,
resulting in the loss of integrity of the lysosomal membrane and the
release of lysosomal enzymes, toxins, and large quantities of
aminoglycosides into the cytosol. The extralysosomal aminoglycoside
interacts with and disrupts the functional integrity of other
subcellular membranes, thereby initiating the injury cascade that
eventuates in cell death.
It should be emphasized that aminoglycoside-induced proximal
tubular cell necrosis is accompanied by a conspicuous regenerative
response (Parker et al., 1982; Toubeau et al., 1986). Thus, the
clinical threshold for nephrotoxicity is determined by the balance
between the rate of necrosis and the rate of regeneration of proximal
tubular cells (Soberon et al., 1979). If necrosis dominates, overt
renal failure ensues.
Aminoglycoside nephrotoxicity is accompanied by increased
generation of free radicals. Furthermore, nephrotoxicity is blocked
with free radical scavengers/antioxidants such as dimethylthiourea,
dimethyl sufoxide, sodium benzoate, or deferoxamine (Walker & Shah,
1987). However other studies have demonstrated that antioxidants such
as vitamin E do not protect against aminoglycoside-induced injury
(Ramsammy et al., 1986, 1987, 1988b). The reasons for this apparent
discrepancy are not known, and the exact role of lipid peroxidation in
gentamicin nephrotoxicity therefore remains unclear.
Polyaspartic acid has recently been shown to protect rats
completely from developing aminoglycoside nephrotoxicity without
inhibiting proximal tubular cell drug uptake (Williams et al., 1986;
Gilbert et al., 1989; Ramsammy et al., 1989b). In vitro studies
suggest that the protective effect of polyaspartic acid is due to the
ability of this polyanionic peptide to bind the cationic
aminoglycosides, thereby preventing these drugs from interacting
electrostatically with various targets, presumably anionic
phospholipids, within the cell.
5.1.3.2 Cephalosporins
The nephrotoxicity of cephalosporins was first noted when the
drugs were used in combination with aminoglycosides, but it is now
recognized that cephalosporins, particularly cephaloridine, may
produce degeneration and necrosis of proximal tubular lining cells and
acute renal failure. It has been suggested that the cellular toxicity
is the result of metabolic activation of the five-member thiophene
ring present in cephalothin and cephaloridine, the only two
cephalosporins that seem capable of producing dose-dependent direct
nephrotoxicity (Mitchell et al., 1977). More recently, it has been
suggested that lipid peroxidation (Goldstein et al., 1989) and direct
mitochondrial toxicity may be involved in the mechanisms of
cephaloridine nephrotoxicity. Necrosis occurs when the concentration
exceeds 1000 mg/kg wet tissue. The correlation between dose and
response, as well as the localization of the lesion in the proximal
portion of the nephron, may be explained by a striking
corticomedullary gradient in tissue concentration. The cellular uptake
of cephaloridine and nephrotoxicity have been modified or eliminated
in experimental animals by pretreatment with either probenecid or
p-aminohippuric acid. The mechanisms of toxicity are complex (Wold
et al., 1979; Tune & Fravert, 1980; Tune, 1986; Goldstein et al.,
1987; Tune et al., 1988).
5.1.3.3 Amphotericin B
The increasing use of immunosuppressive therapy and the attendant
systemic mycotic infections have resulted in an increase in the
administration of amphotericin B. This drug is almost always
associated with some degree of toxicity to the distal renal tubule and
accompanying acidosis, hypokalaemia, and polyuria (Butler, 1966;
Douglas & Healy, 1969). Reduced renal blood flow and glomerular
filtration rate may also occur. Pretreatment of experimental animals
with furosemide or sodium protects against decreases in renal plasma
flow and glomerular filtration rate immediately following amphotericin
B treatment. Salt loading also protects against
amphotericin-induced decreases in renal plasma flow and glomerular
filtration rate upon chronic drug administration in rats. However, it
is important to note that tubular toxicity in these studies was not
ameliorated by salt loading (Tolins & Raij, 1988). These data suggest
that the tubular toxicity of amphotericin B is not secondary to renal
vasoconstriction and ischaemia.
5.1.3.4 Tetracyclines
The nephrotoxicity of tetracycline incited considerable interest
in the early 1960s, shortly after its introduction. People,
particularly children, developed a reversible proximal tubular
dysfunction after receiving outdated drugs. The nephrotoxicity was
found to be due to a degradation product, anhydro-4-epitetracycline.
The problem has disappeared with the substitution of citric acid for
lactose as a vehicle (Curtis, 1979).
Other rare effects of tetracycline that have been reported are
impairment of renal-concentrating ability by
demethylchlorotetracycline and occurrences of acute interstitial
nephritis after minocycline treatment. More important to current usage
is the awareness that the serum half-life of the two most commonly
used drugs, tetracycline and oxytetracycline, is greatly prolonged in
renal failure, and that the anti-anabolic effect of the tetracyclines,
which inhibit the incorporation of amino acids into protein, may
further contribute to negative nitrogen balance and uraemia by raising
blood urea nitrogen (Curtis, 1979).
5.1.4 Penicillamine
Penicillamine (3,3-dimethylcysteine) was first used clinically as
a copper-chelating agent to treat Wilson's disease. Because of the
drug's potential for decreasing collagen formation, its use has been
extended to a number of clinical disorders in which fibrosis is a
major component, such as rheumatoid arthritis, pulmonary fibrosis,
and liver disorders.
It has been suggested that the drug acts by reducing disulfide
linkages. This inhibits polymerization of macromolecules and leads
to impairment of collagen formation. Use of the drug has been tempered
by the occurrence of side effects in as many as 30% of patients. The
most important side effect (20% of cases) is proteinuria. The
morphological appearance of the glomerular lesions is typically that
of perimembranous glomerulonephritis with segmental subepithelial
immune-complex deposits. These changes are best demonstrated as
granular immunofluorescent deposits of IgG and C3. Withdrawal of
penicillamine therapy results in disappearance of the proteinuria and
repair of the basement membrane changes in 60% of cases (Gartner,
1980). Immune-complex glomerulonephritis with granular deposits along
basement membrane and in the mesangium can be produced experimentally,
confirming the role of an immunological mechanism in the pathogenesis
of the nephropathy. In addition the drug is associated with the
development of Goodpasture's syndrome with linear glomerular basement
membrane deposits.
5.1.5 Lithium
Lithium salts, mainly lithium carbonate, have been used for 40
years to prevent relapses of maniac-depressive illness. Impaired renal
ability to acidify and concentrate urine is a common finding among
patients given lithium (Batelle et al., 1982). It is usually regarded
as a minor side-effect of the drug, i.e. a pharmacologically induced
physiological impairment of distal tubules and collecting ducts, such
a target-selective effect usually being reversible after
discontinuation of the therapy. Since the polyurea is resistant to
ADH, the effect has been characterized as resembling nephrogenic
diabetes insipidus (Bendz, 1983).
Although several case reports of lithium-induced chronic renal
insufficiency have been published (Hestbech et al., 1977; Hansen et
al., 1979, 1981; Kincaid-Smith et al., 1979; Cohen et al., 1981;
Walker et al., 1982,1986; Ottosen et al., 1984), the overall evidence
suggesting progressive renal damage in patients taking lithium is
rather limited because of methodological weaknesses in human studies
(Lippmann, 1982). However, animal studies support the view that
long-term treatment with lithium salts may lead to tubulo-interstitial
nephropathies. Experimentally induced focal fibrosis, tubular atrophy,
and cystic dilatation of distal tubules were obtained by exposing
animals to toxic doses (Ottosen et al., 1984; Walker et al., 1986).
In addition to tubular effects, the occurrence of nephrotic
syndrome in psychiatric patients has been attributed to long-term
treatment with lithium salts (Richman et al., 1980; Depner, 1982).
Thus, although case reports do not constitute evidence, there is some
indication that lithium may adversely affect other segments along the
nephron. A proportion of cases, ranging from 0 to 50% (median 8%) of
patients on lithium, may eventually develop chronic renal
insufficiency, evolving towards end-stage renal disease (Cohen et al.,
1981). Such an increased risk may still be regarded as acceptable,
especially when compared to the benefits of such a therapeutic
approach to serious psychiatric problems. Thus, fear of renal disease
may not require the therapy to be stopped. However, a close monitoring
of renal function is strongly recommended. Once-daily dosing to
maintain serum lithium levels at the lower therapeutic range, i.e.
0.4-0.7 mEq/litre, is advisable. Furthermore, it is critical to avoid
salt depletion, which can disturb the equilibrium of serum lithium and
induce acute intoxication.
5.1.6 Urographic contrast media (UCM)
Radiographic procedures are normally safe, but a small proportion
of patients subsequently experience a transient or, very rarely, a
permanent decline in renal function (Cedgard et al., 1986). There are
various predisposition factors such as dose, age, multiple utilization
of UCM, dehydration, diabetes (Taliercio et al., 1986), multiple
myeloma (Harkonen & Kjellstrand, 1981), hypertension, atherosclerosis,
prior kidney or liver diseases, the co-administration of nephrotoxic
drugs, and kidney transplantation. Prospective studies suggest that
diabetes per se is not a risk factor when matched for pre-existing
renal disease (Teruel et al., 1981; D'Elia et al., 1982). This
highlights pre-existing renal disease as a major risk factor. In view
of the fact that there are several million radiological procedures
each year, the number of patients at risk of developing adverse
effects from the administration of contrast media is significant. Up
to 10% of cases of acute renal failure in hospitalized patients may be
due to intravascular urographic contrast medium administration (Hou
et al., 1983).
The cause of such renal injury is not well understood, but
hyperosmolality (e.g., with meglumine diatrizoate) has been claimed to
be an important factor in renal damage (Forrest et al., 1981). New
low-osmolar urographic contrast media (such as iopamidol) are being
introduced, some of which are isotonic with plasma, but a progressive
increase in the incidence of ARF from 0-12% up to 100% in high-risk
patients has been reported (Eisenberg et al., 1980). About 65% of ARF
follow intravenous urography, and 30% follow arteriography. The rest
are associated with computerized tomography (Hanaway & Black, 1977;
Harkonen & Kjellstrand, 1979; Fang et al., 1980). The reported
increase in UCM-induced ARF could be due to better monitoring and
awareness, higher health standards, or a prolonged survival of
patients with critical illnesses (who would then be more prone to
multiple X-ray contrast media examinations), or could represent other
types of nephrotoxicity.
The pathophysiology of UCM-induced ARF is unclear, but may
involve renal ischaemia and haemodynamic effects on glomerular
function and/or intrarenal flow distribution. Several hormonal systems
may be activated prior to and/or during ARF (Caldicott et al., 1970;
Chou et al., 1974; Katzberg et al., 1977). Thus, the effects of
contrast media on kidney function continue to be conflicting and
represent both glomerular and tubular dysfunctions (Milman & Gottlieb,
1977; Rahimi et al., 1981; Teruel et al., 1981; Khoury et al., 1983).
It has been suggested, but not confirmed, that non-ionic low-osmolal
contrast media have reduced nephrotoxicity (Gale et al., 1984;
Spataro, 1984; Smith et al., 1985; Cedgard et al., 1986; Cavaliere et
al., 1987).
Attempts to induce radiocontrast nephrotoxicity in animals have
led to inconclusive or contradictory results. Intact hydrated rats
with or without experimentally induced acute renal failure do not
develop radiocontrast nephrotoxicity (McIntosh, et al., 1975; Moreau
et al., 1980). Transient reductions in glomerular filtration rate and
renal blood flow have been reported immediately following
radiocontrast injection in rats and dogs (Norby & DiBona, 1975;
Cunningham et al., 1986; Katzberg et al., 1986), but rarely has acute
renal failure been studied or documented in the intact animal
following these acute measurements. Nephrotoxicity may occur, however,
when the radiocontrast agent is given in association with experimental
manoeuvres designed to reduce renal function. These include repeated
dehydration with furosemide injections, renal ischaemia (Schultz et
al., 1982), and acute renal failure induced by mercuric chloride or
glycerol (McLachlan et al., 1972).
5.1.7 Anticancer drugs
5.1.7.1 Cisplatin
Cisplatin ( cis-diamminedichloroplatinum II) has become the
chemotherapeutic agent of choice in the treatment of several solid
tumours, particularly testicular and ovarian cancers (Einhorn &
Donohue, 1977). Unfortunately cisplatin is also one of the most toxic
anticancer drugs, its dose-limiting toxicity being nephrotoxicity
(Madias & Harrington, 1978; Goldstein & Mayor, 1983; Safirstein et
al., 1986). Despite the use of optimal methods for administering
cisplatin, such as the use of active hydration (Cvitkovic et al.,
1977) or sodium chloride as the vehicle (Ozols et al., 1984),
approximately 30% of patients will manifest nephrotoxicity.
Early clinical trials of cisplatin in cancer patients showed a
striking incidence of persistent azotaemia and acute renal failure
(Rossof et al., 1972; Lippman et al., 1973). In later studies serum
creatinine levels increased within 6-7 days of treatment, and then
apparently returned to pre-treatment levels by approximately 3 weeks
(Hayes et al., 1977). Similar results were seen following the
injection of cisplatin into rats (Ward & Fauvie, 1976; Chopra et al.,
1982). Thus cisplatin-induced nephrotoxicity initially appeared to be
an acute reversible condition. However, more recent findings suggest
that cisplatin causes a permanent reduction in GFR (Dentino et al.,
1978; Meijer et al., 1983; Fjeldborg et al., 1986), which may indeed
be progressive in nature (Groth et al., 1986; Jaffe et al., 1987).
Hypomagnesaemia is frequently noted in patients receiving
cisplatin (Buckley et al., 1984; Vogelzang et al., 1985), and is
associated with inappropriately high levels of urinary excretion of
magnesium. This deficiency in magnesium leads to hypokalaemia and
hypocalcaemia. This selective renal loss of magnesium is not unusual
and may be even more common than other renal abnormalities as an
expression of cisplatin nephrotoxicity.
Light microscope studies of human kidneys have revealed focal
acute tubular necrosis, affecting primarily the distal and collecting
tubules, with dilatation of convoluted tubules and cast formation
(Gonzalez-Vitale et al., 1977). More recently, Tanaka et al. (1986)
reported sporadic degenerative lesions, necrosis, and regenerative
changes in the S2 and S3 regions of the proximal tubule and also in
the distal tubule and collecting duct. The glomeruli and vasculature
appeared uninvolved. These observations are somewhat different to
those seen in the rat, where cisplatin-induced damage is largely
confined to the S3 segment of the proximal tubule, located in the
outer stripe of the outer medulla (Chopra et al., 1982). With
increasing time cystic tubules develop in this region (Dobyan, 1985).
However, these cysts have not been reported clinically.
The activity of a number of urinary enzymes, including alanine
aminopeptidase, N-acetyl-ß-D-glucosaminidase, leucine aminopeptidase
and ß-glucurinidase, has been shown to be elevated as early as 36-48
h after cisplatin treatment (Kuhn et al., 1978; Jones et al., 1980).
ß2-micro-globulin excretion has also been shown to be transiently
increased after cisplatin treatment (Daugaard et al., 1988a,b). It is
of interest to note that this proteinuria (involving proteins of low
relative molecular mass), predominantly tubular in origin, was
transient, whereas a persistent proteinuria consisting of proteins of
high relative molecular mass, such as albumin and IGg, and glomerular
in origin was seen after the completion of cisplatin treatment.
The pathophysiology of the GFR reduction remains ill defined. It
is clear that cisplatin produces an acute, mainly proximal, tubular
functional impairment within hours of administration (Daugaard et al.,
1988a,b). It has been suggested that the former is a consequence of
the latter. Thus, Groth et al. (1986) attributed the chronic reduction
in GFR to increased intratubular pressure within damaged tubules. The
glomerular proteinuria reported by Daugaard et al. (1988a) suggests
that cisplatin may directly damage glomeruli. Cisplatin-induced
glomerular lesions have been reported in the pig (Robbins et al.,
1990).
5.1.7.2 Adriamycin
The anthracycline antibiotic Adriamycin is widely used in
clinical oncology to treat several cancers, including breast
carcinoma, malignant lymphomas, and sarcomas (Blum & Carter, 1974).
Its clinical use is limited by its cardiotoxicity. Experimentally
adriamycin has been shown to produce a nephrotic syndrome in rats
(Young, 1975), rabbits (Fajardo et al., 1980), and pigs (van Fleet et
al., 1979).
Rats treated with a single dose of Adriamycin exhibited a marked
proteinuria evident within several days of treatment (Bertani et al.,
1982). Maximal levels were seen after approximately 2 weeks, after
which levels declined but remained significantly above control levels
10 weeks after treatment. Serum albumin levels were also significantly
reduced, whereas there was a concomitant hyperlipidaemia (Bertani et
al., 1986). Adriamycin-induced changes in renal functional parameters
are less well defined. Litterst & Weiss (1987) reported that BUN and
serum creatinine values were either unaffected or only minimally
increased. However, more recent studies indicate significant and
progressive reductions in GFR (Hall et al., 1986). Single nephron
glomerular filtration rate is reduced due to a decreased
ultrafiltration coefficient (Michels et al., 1983).
Morphological damage is first seen in the glomerulus;
ultrastructural examination reveals extensive damage to the glomerular
epithelial cells occurring within 36-48 h of injection (Bertani et
al., 1982). This leads to the eventual loss of the foot processes.
Light microscope studies reveal the characteristic presence of
vacuoles in the glomeruli; with time progressive glomerulosclerosis is
seen. Associated with these glomerular changes are tubular changes;
these consist of dilated tubules filled with casts, predominantly in
the outer stripe of the outer medulla, and atrophic tubules associated
with areas of interstitial fibrosis. It appears that Adriamycin
primarily damages the glomeruli and that the tubulo-interstitial
damage results from the proteinuria, which induces cast formation and
interstitial inflammatory reaction. Fajardo et al. (1980) reported
that juxtamedullary glomeruli were more sensitive than cortical
glomeruli; micropuncture studies in the Munich Wistar Fromter rat
confirm this observation (Soose et al., 1988).
Renal toxicity in patients appears rare, although there has been
a report of acute renal failure following Adriamycin-treatment (Burke
et al., 1977). This may reflect species differences in sensitivity or
may reflect the use of inappropriate test protocols for detecting
renal damage.
5.1.8 Immunosuppressive agents
5.1.8.1 Cyclosporin A
Cyclosporin A has been widely used for preventing organ rejection
after transplantation and in auto-immune diseases, but it is highly
nephrotoxic in the clinical situation (Kostakis et al., 1977; Calne et
al., 1978; Powles et al., 1978). Clinically, cyclosporin
nephrotoxicity has been reported as an acute reversible renal
dysfunction, an acute vasculopathy (thrombotic microangiopathy),
and/or a chronic nephropathy with interstitial fibrosis (Mihatsch et
al., 1985; Palestine et al., 1986). After cyclosporin A treatment
there is an inverse linear relation between GFR and the severity of
the lesions. During the first 6 months of treatment, renal fibrosis in
patients given high doses of cyclosporin A shows a dosedependent
progression of increased severity (Klintmalm et al., 1981). The rat
model of cyclosporin A nephrotoxicity may adequately represent the
acute condition in man (Mihatsch et al., 1985, 1986) but does not
represent that seen in the clinical situation. Although rats dosed
with cyclosporin also develop renal surface changes that correspond to
focal areas of collapsed proximal tubular regions with subcapsular
fibrosis, degenerating tubular epithelium and thickening of the
basement membrane, the chronic striped fibrosis and arteriolar lesions
have not been reproduced experimentally.
Some animal data have shown increased blood urea nitrogen and
creatinine, brush-border and lysosomal enzyme leakage, and
vacuolation, necrosis, and regeneration of P3 cells in rats (Dieperink
et al., 1983, 1985; Ryffel et al., 1983, 1986; Murray et al., 1985;
Dieperink, 1989). However, most studies find no necrosis or enzymuria
despite profound reductions in GFR. Functional changes in animals and
man given cyclosporin A are similar. They represent haemodynamic
changes such as an increased renal vascular resistance (Murray et al.,
1985), proximal fractional reabsorption (Dieperink et al., 1983,
1985; Dieperink, 1989), and renal blood flow, plasma flow, and GFR
decrease. The reduction in renal perfusion and filtration has been
prevented experimentally by vasoactive alpha-adrenergic antagonists
and renal denervation (Murray et al., 1985), but this has not been
established in humans. Cyclosporin A appears to have a direct
preglomerular vasoconstriction effect, decreasing the ultrafiltration
pressure and increasing proximal fractional reabsorption. Presumably,
tubular flow rates and end proximal tubular delivery decrease, and,
due to varying tubular hypoperfusion, there is a focal tubular
collapse, and degeneration and peritubular interstitial fibrosis
develop. The precise relationship between renal vasoconstriction and
chronic tubulo-interstitial pathology is poorly understood.
5.1.9 Heroin
About 1% of heroin addicts develop haematuria, proteinuria, or
the nephrotic syndrome (Cunningham et al., 1980). Morphologically,
the renal lesion has been described as focal sclerosing
glomerulonephritis. In the absence of proliferative lesions or immune
deposits, a direct toxic effect of heroin or even a contaminant or
solvent employed in the administration of heroin has been suggested as
the major pathogenetic mechanism. However, heroin-related increases in
IgM titres have been regarded as evidence that an immunological
mechanism may play some role in this disorder.
5.1.10 Puromycin aminonucleoside
Direct toxicity of the puromycin aminonucleoside to glomerular
components may also occur and may be a factor in renal failure.
Experimental studies of the effects of puromycin have provided
considerable basic information regarding the pathogenesis of direct
chemical injury to glomerular structures. The effects are limited to
rats and monkeys. Epithelial cells become swollen, and there is an
increase in lysosome and pinocytotic activity, fusion and loss of foot
processes, and a reduction in the number of filtration slits. As the
lesion progresses, epithelial cells become detached, leaving "naked"
basement membrane in direct contact with Bowman's capsule, which may
account for the severe proteinuria (Caulfield et al., 1976). It is
suggested that as the lesion progresses there is an increase in
basement membrane synthesis, mesangial cell proliferation and fusion,
and crescent formation leading to the light microscope appearance of
focal glomerular sclerosis (Gartner, 1980). The glomerular lesions
produced by puromycin do not appear to invoke an immunological
response, so that the resulting alterations are entirely related to
the direct toxicity of the drug.
5.2 Chemicals
The diversity of organic molecules is such that there are
chemicals that are now known to adversely affect each part of the
kidney. This section will examine those chemicals that do not fit into
any conventional section on therapeutically used agents.
5.2.1 Ethylene glycol
Ethylene glycol, a constituent of antifreeze, is occasionally
ingested and causes severe acute toxicity to the brain and kidney.
Acute tubular necrosis is followed by renal failure. Exposure often
leads to permanent renal damage. A morphological feature of mild
ethylene glycol toxicity is cytoplasmic vacuolation, which may suggest
hypokalaemic nephropathy or osmotic nephrosis due to mannitol. Most
ethylene glycol is excreted unmetabolized, but a small percentage is
metabolized to oxalic acid. This is accompanied by deposition of
calcium oxalate crystals in the kidney, which may contribute to a
persistent inflammatory reaction and interstitial fibrosis. Excessive
urinary excretion of oxalate and crystal formation may also be seen
following administration of halogen-containing anaesthetic agents,
particularly methoxyflurane and halothane (Roxe, 1980). Acute ethylene
glycol toxicity is treated with ethanol, which competes as a substrate
for alcohol dehydrogenase (Peterson et al., 1981).
5.2.2 Organic chemicals and solvents
5.2.2.1 Volatile hydrocarbons
Volatile hydrocarbons, particularly chlorinated compounds such as
carbon tetrachloride and trichloroethylene, may produce glomerular
lesions leading to nephrotic syndrome and renal failure. The
relationship of volatile hydrocarbon exposure to the development of
glomerulo-nephritis in populations is not clear. It has been found
that among patients with glomerulonephritis there are more with a
history of exposure to hydrocarbon solvents than would be expected.
Attempts to reproduce in rats the glomerular lesions observed in
patients have only been partially successful. Solvent-exposed rats had
increased proteinuria and glomerular sclerosis, but proliferative
lesions and significant immune deposits were not observed (Zimmerman
& Norbach, 1980). Of 15 patients studied in Sweden with
post-streptococcal glomerulonephritis, 6 had a history of brief
exposure to organic solvents before the development of their disease.
This suggested to these investigators that solvent exposure may
influence the outcome of an infection with streptococci. Prior
exposure to hydrocarbon-containing solvents has been identified in a
number of patients with Goodpasture's syndrome (Gartner, 1980). Apart
from the pulmonary manifestation of cough, shortness of breath, and
haemoptysis, there may be haematuria and proteinuria. Renal morphology
consists of a proliferative glomerulonephritis with IgG and C3 in the
glomerular basement membrane.
Studies conducted in Sweden (Askergren 1981; Askergren et al.,
1981) and in Belgium (Viau et al., 1987) have reported a slight
increase in the urinary excretion of albumin in groups of workers
exposed to industrial solvents, particularly styrene. This effect
probably reflects an enhanced glomerular permeability since the
urinary output of markers of proximal tubular function
(ß2-micro-globulin, retinol-binding protein) was not affected. In
Italy, Franchini et al. (1983) also reported slight renal disturbances
in workers occupationally exposed to solvents. These effects
consisted of enhanced urinary excretion of total proteins, lysozyme,
and ß-glucuronidase and pointed to a tubular lesion, since they were
not accompanied by a rise in albuminuria. It is at present impossible
to relate nephrotoxic effects reported in these studies to exposure to
one solvent or one class of solvents, although styrene has been
incriminated by Askergren et al. (1981). One must also recognize that
the renal effects reported in these studies are mild and do not appear
to correlate with indices of exposure to solvents.
An auto-immune mechanism following chronic exposure is probably
responsible for the glomerular lesions. In patients who have
Goodpasture's syndrome, the primary site of damage may be the alveolar
basement membrane of the lung, which is damaged by inhalation of the
solvents, and antibodies to altered alveolar basement membrane may
cross-react with glomerular basement membrane. Alternatively,
auto-immunity may follow direct toxic injury to renal tubular or
glomerular structures. Acute exposure to these solvents does produce
acute tubular necrosis, and it is likely that prolonged exposure to
low levels that do not result in cell necrosis produces cell injury
sufficient to damage renal cell membranes and provide the antigen for
the immune reaction (Gartner, 1980).
5.2.2.2 Chloroform
In vitro exposure to chloroform has been shown to produce
toxicity in kidney slices from male but not from female mice (Smith &
Hook, 1984). Furthermore, 14C-labelled chloroform was metabolized
to 14CO2, and the radioactivity was covalently bound by cortical
microsomes from male but not female mice. The in vitro metabolism of
chloroform by male, but not female, renal slices is consistent with
reduced susceptibility of female mice to in vivo chloroform
nephrotoxicity (Smith et al., 1983; Smith et al., 1984). Metabolism is
dependent on oxygen, a NADPH-regenerating system, incubation time,
microsomal protein concentration, and substrate concentration, and is
inhibited by carbon monoxide (Smith & Hook, 1984). The negligible
degree of chloroform metabolism and toxicity in female mice is
consistent with a lower renal cytochrome P-450 concentration and
activity in female mice than in males (Smith & Hook, 1984).
Pretreatment of rabbits with phenobarbital, a renal cytochrome P-450
inducer in this species, enhances the toxic response of renal cortical
slices to chloroform in vitro (Bailie et al., 1984). The rate at
which deuterated chloroform is metabolized by the liver to phosgene is
approximately half that of chloroform. Deuterated chloroform is also
less hepatotoxic that chloroform since the C-D bond is stronger than
the C-H bond. These data suggest that cleavage of the C-H bond is the
rate-limiting step in the activation of chloroform. Deuterated
chloroform is also less toxic to the kidney than chloroform
(Ahmadizadeh et al., 1981; Branchflower et al., 1984). This deuterium
isotope effect on chloroforminduced nephrotoxicity suggests that the
kidney metabolizes chloroform in the same manner as the liver, e.g.,
by oxidation to phosgene. Indeed, rabbit renal cortical microsomes
incubated in media supplemented with L-cysteine metabolize
14C-labelled chloroform to radioactive phosgene-cysteine
2-oxothiazolidine-4-carboxylic acid (Bailie et al., 1984). These
in vitro data collectively support the hypothesis that mouse and
rabbit kidneys biotransform chloroform to a metabolite (phosgene)
that mediates nephrotoxicity.
5.2.2.3 Halogenated alkenes
The nephrotoxin S-(1,2-dichlorovinyl)-L-cysteine (DCVC) is
formed by trichloroethylene extraction of proteinaceous substances and
was first identified in extracted animal food (McKinney et al., 1957,
1959). It has been widely used as a model compound in nephrotoxicity
studies. DCVC is accumulated in the proximal tubules by an active
carrier system for organic anions (Elfarra et al., 1986a,b). It is
then activated by cysteine conjugate ß-lyase to a reactive thiol
(Bhattacharya & Schultze, 1967) and causes tubular damage (Terracini
& Parker, 1965). DCVC is a potent specific nephrotoxin, which produces
proximal tubular damage in vivo and in vitro (Elfarra et al.,
1986a,b; Lash & Anders, 1986; Lash et al., 1986). In vivo DCVC causes
its primary lesion in the straight segment (S-3) of the proximal
tubule, and the molecule is also cytotoxic both for primary cultures
of proximal tubular cells and for cell lines derived from this region
of the nephron. There is a close correlation between the in vivo and
in vitro effects of this compound with regards to its metabolism and
effects on cells (Hassall et al., 1983).
Cysteine conjugates such as DCVC are metabolized by cysteine
conjugate ß-lyase to their ultimate toxic species i.e. pyruvate,
ammonia, and a reactive thiol (Anderson & Schultze, 1965). This
reaction plays a role in the nephrotoxicity of DCVC (Lash et al.,
1986). ß-lyase has been found to predominate in cytosolic and
mitochondrial fractions (Lash et al., 1986) and has a requirement for
pyridoxal phosphate. The enzyme activity can be inhibited by pyridoxal
phosphate inhibitors such as amino-oxyacetic acid and propargylglycine
(Elfarra et al., 1986a). In addition to monitoring enzyme activity,
renal cortical slices can be utilized to assess the regulation of
enzyme activity and the resultant effects on toxicity.
There is a greater sensitivity to DCVC-induced kidney damages in
the adult mouse than there is in the newborn. Similar findings have
been reported using cephaloridine, where the newborn animal is more
resistant to nephrotoxicity than the adult rabbit (Tune, 1975). These
finding for DCVC differ from those for hexachlorobutadiene (Kuo &
Hook, 1983; Lock et al., 1984), where nephrotoxicity is greater in the
young rat and mouse than in the adults.
Chlorotrifluoroethylene is a potent nephrotoxin (Potter et al.,
1981) and is metabolized by hepatic cytosolic and microsomal
glutathione S-transferases to
S-(2-chloro-1,1,2-trifluoroethyl)glutathione (Dohn et al., 1985a),
which is nephrotoxic in rats and cytotoxic in isolated rat kidney
proximal tubular cells (Dohn et al., 1985b). The corresponding
cysteine S-conjugate, S-(2-chloro-1,1,2-trifluoroethyl)-L-cysteine
(CTFC) is also nephrotoxic in rats and cytotoxic in isolated kidney
cells, and its bioactivation is dependent on metabolism by renal
ß-lyase (Dohn et al., 1985b). Pyruvate and hydrogen sulfide have been
identified as metabolites of CTFC (Banki et al., 1986; Lash et al.,
1986).
5.2.2.4 Hydrocarbon-induced nephrotoxicity
Inhalation of unleaded gasoline for 2 years produced renal
tumours (adenomas and adenocarcinomas) in male Fischer-344 rats but
not in female rats or mice of either sex (Kitchen, 1984; MacFarland,
1984; Mehlmann et al., 1984). Subchronic inhalation exposure
increased protein (hyaline) droplets in proximal convoluted tubules
(Halder et al., 1984), accumulated casts at the cortico-medullary
junction and single cell necrosis and regeneration of the nephron
(Short et al., 1986) in male rats. When different fractions of
unleaded gasoline were screened for their specific effect on the male
rat kidney, it was found that the branched-chain saturated hydrocarbon
components (used as anti-knocking agents) caused hyaline droplet
formation. A number of chemicals, such as 2,2,4-trimethyl-pentane
(Phillips & Egan, 1984a,b; Halder et al., 1985; Viau et al., 1986a),
decalin (Alden et al., 1984), 1,4-dichlorobenzene (NTP, 1987), and
p-dichlorobenzene (NTP, 1986; Bomhard et al., 1988), have now been
shown to cause such hyaline droplets in male rats. Although these
chemicals cause minimal renal functional impairment, a protein droplet
nephrosis develops, progressing to mild tubular degeneration,
necrosis, and regeneration after several weeks of treatment (Phillips
& Cockrell, 1984a,b).
Male mice excrete a sex-related protein, which results in a
urinary protein level 2.5 to 3 times that of female mice. However, the
male mouse sex-associated urinary protein hydrolyses readily and thus
does not accumulate in the proximal tubule (Alden et al., 1984; Alden,
1989). In humans, it has recently been reported that protein 1 (an
alpha2-microglobulin of about 20 000 Daltons) has a sex-linked
behaviour just like the androgen-dependent alpha2u-globulin. Protein
1 is excreted in greater amounts in the urine of males after puberty.
In the age group 15 to 20 years, its concentration in the urine of
males is on average fifty times higher than that in the urine of
females (Bernard et al., 1989). The relevance of this observation in
humans is unknown.
The basis for the marked sex dependence and species difference in
the development of hyaline droplet deposition in male rats relates to
the fact that they excrete the sex-hormone-related and therefore
male-specific protein alpha2u-globulin (Stonard et al., 1986; Loury
et al., 1987; Olson et al., 1987). This is also species specific to
the rat and has not been reported in any other commonly used animals
or man. alpha2u-Globulin is synthesized by the male rat liver and is
an important constituent of the physiological proteinuria in adult
male rats. At maturity the total urinary protein is 20-30%
alpha2u-globulin and 10% albumin. At 160 days of age the excretion of
albumin and total urinary proteins is markedly increased. By one
year, albumin represents nearly 60% of the total protein while
alpha2u-globulin is less than 10%. This reversal in relative content
may be the consequence of a progressive glomerulonephrosis, associated
with an apparent spontaneous accumulation of hyaline droplets. The
nephrotic condition may be the consequence of the burden of excreting
alpha2u-globulin. Its early onset is sex dependent; female rats do not
exhibit the proteinuria until a later age. In both aging and the
response to hydrocarbons, the common pathological factor may be the
accumulation of alpha2u-globulin, via a susceptible pathway not shared
with other proteins (Neuhaus, 1986; Stonard et al., 1986).
The chronic regenerative response subsequent to moderate proximal
tubular damage in the kidney of male rats exposed to petroleum
hydrocarbons may be an important stimulus in renal tumour formation
caused by this group of chemicals (Short et al., 1986). Loury et al.
(1987) have shown a 5- to 8-fold increase in S phase of renal cells
induced by unleaded gasoline, and the renal proliferative effects of
2,2,4-trimethylpentane are localized to the S2 segment of the
proximal tubule of the male rat (Short et al., 1986).
Administration of 2,2,4-trimethylpentane to male rats produces a
dose-related increase in the concentration of
2,2,4-trimethylpentane-derived radiolabel in the kidney, which appears
to parallel the dose-related accumulation of 2u-globulin (Stonard et
al., 1986; Charbonneau et al., 1987). The reversible binding of a
metabolite of 2,2,4trimethylpentane to alpha2u-globulin in the male
rat kidney (Lock et al., 1987) is thought to alter endocytosis or
lysosomal handling of the alpha2u-globulin-2,2,4-trimethylpentane
metabolite complex. This may increase cell turnover of the S2 cells
via lysosomal enlargement and/or instability, leading to cell death.
It appears that lysosomal catabolism of the
2,4,4-trimethyl-2-pentanol-alpha2u-globulin complex compound to
alpha2u-globulin causes lysosomal protein overload, resulting in
cell necrosis (Swenberg et al., 1989).
There are other sex-related differences in the handling of
2,2,4-trimethylpentane by rats. Female rats rapidly metabolize
2,2,4-trimethylpentane and excrete it in urine, while in male rats the
compound is eliminated more slowly and is retained in the kidneys
(Kloss et al., 1985). Recent studies have also shown that
2,2,4-trimethylpentane is metabolized in male and female rats to
trimethyl-pentanols, pentanoic acids, and hydroxypentanoic acids
(Olson et al., 1986; Charbonneau et al., 1987).
5.2.2.5 Bipyridyl herbicides
Paraquat is a potent bipyridyl herbicide that has multiple organ
effects. The kidney is frequently involved in serious cases of
paraquat poisoning (WHO, 1984). The compound is actively secreted by
the organic cation transport in the proximal tubule. Renal
histological examinations in a variety of animals exposed to paraquat
show vacuolation of the proximal convoluted tubules and proximal
tubular cell necroses (Lock, 1979; Lock & Ishmael, 1979). Acute
oliguric renal failure is common in severely poisoned patients. Less
severe manifestations include impaired glomerular filtration, which
often recovers after several days and before the paraquat induces
severe pulmonary fibrosis. Other renal functional abnormalities
include proteinuria and haematuria. Tubular damage may be shown by the
presence of glucosuria or all of the features of the Fanconi syndrome.
The severity of the acute renal failure is a major determinant of the
outcome of the poisoning (WHO, 1984).
The biochemical mechanism of nephrotoxicity has not been fully
elucidated, but it is assumed to be identical to that seen in other
tissues. Paraquat undergoes redox cycling in the presence of NADPH and
oxygen with the generation of superoxide and subsequent development of
lipid peroxidation and membrane damage. The development of hydroxyl
radicals results in oxidative damage to nucleic acids, proteins, and
polysaccharides (Autor, 1977).
Diquat is another bipyridyl herbicide that produces multiple
organ toxicity. It undergoes active tubular secretion by the organic
cation system in the proximal tubule (Lock, 1979; Lock & Ishmael,
1979). The histological lesion produced by diquat is necrosis of the
proximal tubular cells and some distal tubular cells (Lock & Ishmael,
1979). Human cases of diquat poisoning result in acute renal failure.
The mechanism of toxicity of diquat appears to be identical to that of
paraquat (Autor, 1977; WHO, 1984).
5.3 Mycotoxins
A high frequency of endemic chronic nephropathy has been
recognized in localized areas of Bulgaria, Rumania, and Yugoslavia
since the 1920s. The affected people live in villages in valleys near
the Danube (Hall & Dammin, 1978; WHO, 1979; Hall, 1982). The
condition, known as Balkan endemic nephropathy (Fig. 14), is an
interesting case study of an environmentally related chronic renal
disease. The etiology is unknown at present. Mycotoxins, particularly
ochratoxin A, have been implicated because of similarities with
disease in animals and identification of the mycotoxin in food (Krogh
et al., 1977; Pepeljnjak & Cvetnic, 1985; Petkova-Bocharova &
Castegnaro, 1985) and in human tissues (Hult & Fuchs, 1986) where
nephrotoxicity is most frequent. Silicates have been suggested because
of the proximity of villages with affected families to streams and
rivers containing silicon.
Although not clearly implicated in BEN, the fungal toxin citrinin
has been suggested as a causative agent in porcine citrinin
nephropathy and clearly has nephrotoxic effects in a number of species
(Berndt & Hayes, 1977; Phillips et al., 1979; Phillips et al.,
1980a,b; Lockard et al., 1980). Citrinin produces acute tubular
necrosis primarily of the S1 section of the proximal tubule. It is
eliminated rapidly by the kidney and only metabolized to the extent of
10-15%, which suggests that effects are due to the parent compound.
Little information is available concerning the cascade leading from
the initial insult to the production of acute tubular necrosis 2-4
days after its administration. Citrinin has been shown to exert a
synergistic effect on ochratoxin A toxicity in animal models. This is
important because the same fungal species that synthesize ochratoxin
A also produce citrinin. This was clearly demonstrated by the presence
of citrinin in 19 out of 21 food samples contaminated with ochratoxin
A. Both genetic and environmental factors, such as exposure to
ochratoxin A, appear to be involved in BEN and the associated renal
tract tumours (Castegnaro & Chernozemsky, 1987). In one endemic area
in Bulgaria the relative risk of patients with BEN developing urinary
tract tumours is 90-fold greater than in people from non-endemic areas
(Castegnaro & Chernozemsky, 1987). Inhabitants of the 15 villages in
the Vratza region of northern Bulgaria have a 30-40% mortality rate
from chronic nephropathy, while urinary tract tumours comprise 25-30%
of all neoplasms in males and females in these geographic areas
(Markovic, 1972). In recent studies ochratoxin A has been found to
induce renal adenomas and carcinomas both in mice (Kanisawa &
Suzuki, 1978; Bendele et al., 1985) and in rats (NTP, 1988). Frequent
metastases, mainly to the lung, were found in the rat study. The
target of ochratoxin nephrotoxicity has been reported to be the S2
and S3 nephron segments (Jung et al., 1989).
Thus the animal and human data indicate that ochratoxin A is a
risk factor for toxic nephropathies and in the etiology of human
nephropathy and associated renal tumours.
5.4 Silicon
An association between occupational exposure to free silica
(SiO2) and chronic nephropathy has been suspected for several years,
but the number of reported cases is few. Clinically, lung fibrosis is

 the primary problem, but in an early study from Italy chronic renal
failure was found in 40% and proteinuria in 20% of 20 patients with
chronic silicosis. The renal silicon content of patients with
proteinuria and chronic silicon exposure has been shown to be much
higher than the normal level, and there appears to be a direct
relationship between level of exposure and probability of renal
disease. Animal studies have demonstrated that silicon is excreted by
glomerular filtration, and a morphological study of experimental
animals and human biopsy material has demonstrated silicon deposits in
subepithelial and subendothelial areas of the basement membrane and in
epithelial cells. Human biopsy materials show a mild focal or
segmental proliferative glomerulonephritis and the absence of
significant immune-complex deposits. These findings suggest a direct
toxic effect on the glomerulus. These cases also have varying degrees
of tubular cell degeneration. Animal studies demonstrate a
dose-related nephropathy that is primarily tubular, with an
interstitial inflammatory reaction and fibrosis. The proliferative
glomerular lesions observed in humans are not seen in animals, but
this difference in response may be related to dose or species
(Hauglustaine et al., 1980).
5.5 Metals
Metals constitute some of the earliest recognized and the best
investigated nephrotoxins. X-ray fluorescence gives a clear
indication of the metal burden that an individual carries.
5.5.1 Lead
Lead has been a very common cause of acute or chronic renal
failure in the past. Acute tubular necrosis has been described
following accidental or intentional absorption of high doses of lead.
Cases of chronic renal failure have been reported in adults who
ingested large amounts of leaded paint during childhood (Queensland,
Australia), in people who consumed alcohol illicitly distilled in
lead-containing stills, and in workers with a long history of
occupational lead exposure (Emmerson, 1973; Bennett, 1985).
Several epidemiological studies have consistently reported that
workers with a heavy industrial exposure to lead experience an
increased risk of death through chronic renal failure (Cooper &
Gaffey, 1975; Malcolm & Barnett, 1982; McMichael & Johnson, 1982;
Davies 1984; Selevan et al., 1985; Cooper et al., 1985). There is also
some evidence that occult lead poisoning may contribute to renal
insufficiency in patients with gout and essential hypertension
(Batuman et al., 1981, 1983; Colleoni & D'Amico, 1986).
In adults, lead nephropathy occurs as an insidious progressive
disease characterized by the absence of proteinuria, albuminuria, and
urinary concentration deficit in its early phases (Wedeen et al.,
1979). This renal disease can be diagnosed only by functional tests
(e.g., estimation of GFR on the basis of blood urea nitrogen or
creatinine clearance). Several cross-sectional studies have attempted
to detect early renal effects in workers exposed to lead (Hammond et
al., 1980; Buchet et al., 1980; Verschoor et al., 1987). These studies
confirm that lead nephropathy in adults, even at an advanced stage
(i.e. with decreased GFR), cannot be detected by the determination of
urinary proteins of low or high relative molecular mass (e.g.,
ß2-microglobulin, albumin). The only marker that seems to respond at
an early stage of lead nephropathy is the urinary excretion of the
lysosomal enzyme, N-acetyl-ß-D-glucosaminidase (NAG) (Verschoor et
al., 1987). However, the underlying mechanism of this renal effect
remains to be elucidated. Increased urinary leakage of NAG might
result from cell damage and exfoliation, but also from a stimulation
by lead of exocytosis or of the renal activity of the enzyme.
The renal effects of lead are primarily tubular or
tubulo-interstitial and they may be both acute and chronic. However,
the acute effects of lead differ from those of most of the other
metals in that cell injury is for the most part reversible and
necrosis is uncommon. Cells of the proximal tubule are most severely
affected, and this effect is characterized by a reduction in
resorptive function leading to a generalized amino-aciduria,
glycosuria, and hyperphosphaturia. These components of the Fanconi
syndrome have been observed in children with acute lead toxicity and
who also have overt symptoms of central nervous system toxicity, and
in rats exposed to lead. Proximal tubular dysfunction has been more
difficult to demonstrate in workers with chronic lead nephropathy
(Goyer & Rhyne, 1973).
The effects of lead on renal tubular cells and sodium
reabsorption are less clear. Increase in plasma renin and aldosterone
while a low-sodium diet is consumed has been observed in a group of
men with a history of "moonshine" ingestion and occult lead toxicity
(Sandstead et al., 1970). In contrast, studies on the effects of
minimally toxic levels of lead exposure in rats showed a reduction in
plasma renin activity in spite of a significant increase in blood
pressure (Victery et al., 1982). These differences may reflect a
difference in time-dose relationship.
The renal effects of lead may also be influenced by interactions
with calcium. Decreasing dietary calcium increases lead retention,
possibly because of a decrease in lead excretion. Increased blood lead
in children is associated with decreased 1,2,5-dihydroxyvitamin D
(synthesized in the kidney) and may reflect impaired synthesis
(Mahaffey, 1980).
The renal proximal tubular cells of people and experimental
animals with lead poisoning are characterized morphologically by the
presence of intranuclear inclusion bodies. In conventional
paraffin-embedded haematoxylin and eosin-stained sections of renal
tissue, the inclusions appear as dense, homogeneous, and eosinophilic
bodies, and at the electon microscope level they have a characteristic
fibrillary margin around a dense central core. Morphologically they
are always separate and distinct from the nucleoli and several may be
found in the same nucleus (Fig. 15). The inclusion bodies contain a
protein-lead complex, and they may be isolated by differential
centrifugation. The protein is a non-histone protein rich in glutamic
and aspartic acids and glycine, and may be a mixture of acidic
proteins with similar physicochemical properties (Moore et al., 1973).
The origin and nature of the protein has not yet been determined, but
recent studies of formation of inclusion bodies in renal cell cultures
suggest that they form initially in the cytoplasm and then migrate
into the nucleus (McLachlan et al., 1980). The major fraction of lead
in the kidney during the acute phase of lead toxicity is bound in the
inclusion bodies. For this reason, the inclusion bodies have been
interpreted as serving as an intracellular depot for lead.
Nevertheless, proximal renal tubular cells during the acute phase of
lead toxicity are usually swollen, and the mitochondria show a
decrease in matrical granules and altered cristae. Functional studies
of mitochondria show reduced respiration and oxidative
phosphorylation. Lysosomes do not seem to have a role in sequestering
intracellular lead.
Chelation therapy following lead toxicity produces a marked
increase in lead excretion. This is accompanied by reversal of the
acute morphological effects of lead on proximal renal tubular cells,
loss of inclusion bodies from nuclei, and restoration of normal renal
cell morphology and function (Goyer & Wilson, 1975).
Both experimental animals and people with chronic exposure to
lead may develop a progressive interstitial nephropathy. In laboratory
animals, progression from acute tubular to chronic tubulo-interstitial
disease may be followed as a continuum. An increase in chronic
interstitial renal disease has been reported in workers with long
histories of occupational exposure, but the nonspecific nature of the
morphological changes makes it difficult to identify lead as the
etiological agent except by association. There is a progressive
increase in fibrosis, beginning in peritubular areas extending into
the interstitium (Cramer et al., 1974). Inflammatory cells are
uncommon and are probably not a primary component of the process.
There is eventual tubule atrophy and hyperplasia of surviving tubules.
There is little evidence that the glomerulus is directly affected by
excessive exposure to lead, except for some nonspecific swelling of
mesangial and epithelial cells. In the terminal stage, glomeruli
become sclerotic. An immunological basis for the progression of
lead-induced nephropathy, as suggested following gold and mercury
exposures, might be suspected. However, there is at present no
published documentation of antirenal antibodies or immune-complex
formation in the pathogenesis of lead nephropathy. One study suggested
that lowered glomerular filtration rate occurs in occupational
exposure to lead that does not produce clinical toxicity (Wedeen et
al., 1979). The pathophysiological basis for this observation has not
yet been determined but may be a consequence of direct toxicity to
epithelial cells of the glomerular apparatus.
the primary problem, but in an early study from Italy chronic renal
failure was found in 40% and proteinuria in 20% of 20 patients with
chronic silicosis. The renal silicon content of patients with
proteinuria and chronic silicon exposure has been shown to be much
higher than the normal level, and there appears to be a direct
relationship between level of exposure and probability of renal
disease. Animal studies have demonstrated that silicon is excreted by
glomerular filtration, and a morphological study of experimental
animals and human biopsy material has demonstrated silicon deposits in
subepithelial and subendothelial areas of the basement membrane and in
epithelial cells. Human biopsy materials show a mild focal or
segmental proliferative glomerulonephritis and the absence of
significant immune-complex deposits. These findings suggest a direct
toxic effect on the glomerulus. These cases also have varying degrees
of tubular cell degeneration. Animal studies demonstrate a
dose-related nephropathy that is primarily tubular, with an
interstitial inflammatory reaction and fibrosis. The proliferative
glomerular lesions observed in humans are not seen in animals, but
this difference in response may be related to dose or species
(Hauglustaine et al., 1980).
5.5 Metals
Metals constitute some of the earliest recognized and the best
investigated nephrotoxins. X-ray fluorescence gives a clear
indication of the metal burden that an individual carries.
5.5.1 Lead
Lead has been a very common cause of acute or chronic renal
failure in the past. Acute tubular necrosis has been described
following accidental or intentional absorption of high doses of lead.
Cases of chronic renal failure have been reported in adults who
ingested large amounts of leaded paint during childhood (Queensland,
Australia), in people who consumed alcohol illicitly distilled in
lead-containing stills, and in workers with a long history of
occupational lead exposure (Emmerson, 1973; Bennett, 1985).
Several epidemiological studies have consistently reported that
workers with a heavy industrial exposure to lead experience an
increased risk of death through chronic renal failure (Cooper &
Gaffey, 1975; Malcolm & Barnett, 1982; McMichael & Johnson, 1982;
Davies 1984; Selevan et al., 1985; Cooper et al., 1985). There is also
some evidence that occult lead poisoning may contribute to renal
insufficiency in patients with gout and essential hypertension
(Batuman et al., 1981, 1983; Colleoni & D'Amico, 1986).
In adults, lead nephropathy occurs as an insidious progressive
disease characterized by the absence of proteinuria, albuminuria, and
urinary concentration deficit in its early phases (Wedeen et al.,
1979). This renal disease can be diagnosed only by functional tests
(e.g., estimation of GFR on the basis of blood urea nitrogen or
creatinine clearance). Several cross-sectional studies have attempted
to detect early renal effects in workers exposed to lead (Hammond et
al., 1980; Buchet et al., 1980; Verschoor et al., 1987). These studies
confirm that lead nephropathy in adults, even at an advanced stage
(i.e. with decreased GFR), cannot be detected by the determination of
urinary proteins of low or high relative molecular mass (e.g.,
ß2-microglobulin, albumin). The only marker that seems to respond at
an early stage of lead nephropathy is the urinary excretion of the
lysosomal enzyme, N-acetyl-ß-D-glucosaminidase (NAG) (Verschoor et
al., 1987). However, the underlying mechanism of this renal effect
remains to be elucidated. Increased urinary leakage of NAG might
result from cell damage and exfoliation, but also from a stimulation
by lead of exocytosis or of the renal activity of the enzyme.
The renal effects of lead are primarily tubular or
tubulo-interstitial and they may be both acute and chronic. However,
the acute effects of lead differ from those of most of the other
metals in that cell injury is for the most part reversible and
necrosis is uncommon. Cells of the proximal tubule are most severely
affected, and this effect is characterized by a reduction in
resorptive function leading to a generalized amino-aciduria,
glycosuria, and hyperphosphaturia. These components of the Fanconi
syndrome have been observed in children with acute lead toxicity and
who also have overt symptoms of central nervous system toxicity, and
in rats exposed to lead. Proximal tubular dysfunction has been more
difficult to demonstrate in workers with chronic lead nephropathy
(Goyer & Rhyne, 1973).
The effects of lead on renal tubular cells and sodium
reabsorption are less clear. Increase in plasma renin and aldosterone
while a low-sodium diet is consumed has been observed in a group of
men with a history of "moonshine" ingestion and occult lead toxicity
(Sandstead et al., 1970). In contrast, studies on the effects of
minimally toxic levels of lead exposure in rats showed a reduction in
plasma renin activity in spite of a significant increase in blood
pressure (Victery et al., 1982). These differences may reflect a
difference in time-dose relationship.
The renal effects of lead may also be influenced by interactions
with calcium. Decreasing dietary calcium increases lead retention,
possibly because of a decrease in lead excretion. Increased blood lead
in children is associated with decreased 1,2,5-dihydroxyvitamin D
(synthesized in the kidney) and may reflect impaired synthesis
(Mahaffey, 1980).
The renal proximal tubular cells of people and experimental
animals with lead poisoning are characterized morphologically by the
presence of intranuclear inclusion bodies. In conventional
paraffin-embedded haematoxylin and eosin-stained sections of renal
tissue, the inclusions appear as dense, homogeneous, and eosinophilic
bodies, and at the electon microscope level they have a characteristic
fibrillary margin around a dense central core. Morphologically they
are always separate and distinct from the nucleoli and several may be
found in the same nucleus (Fig. 15). The inclusion bodies contain a
protein-lead complex, and they may be isolated by differential
centrifugation. The protein is a non-histone protein rich in glutamic
and aspartic acids and glycine, and may be a mixture of acidic
proteins with similar physicochemical properties (Moore et al., 1973).
The origin and nature of the protein has not yet been determined, but
recent studies of formation of inclusion bodies in renal cell cultures
suggest that they form initially in the cytoplasm and then migrate
into the nucleus (McLachlan et al., 1980). The major fraction of lead
in the kidney during the acute phase of lead toxicity is bound in the
inclusion bodies. For this reason, the inclusion bodies have been
interpreted as serving as an intracellular depot for lead.
Nevertheless, proximal renal tubular cells during the acute phase of
lead toxicity are usually swollen, and the mitochondria show a
decrease in matrical granules and altered cristae. Functional studies
of mitochondria show reduced respiration and oxidative
phosphorylation. Lysosomes do not seem to have a role in sequestering
intracellular lead.
Chelation therapy following lead toxicity produces a marked
increase in lead excretion. This is accompanied by reversal of the
acute morphological effects of lead on proximal renal tubular cells,
loss of inclusion bodies from nuclei, and restoration of normal renal
cell morphology and function (Goyer & Wilson, 1975).
Both experimental animals and people with chronic exposure to
lead may develop a progressive interstitial nephropathy. In laboratory
animals, progression from acute tubular to chronic tubulo-interstitial
disease may be followed as a continuum. An increase in chronic
interstitial renal disease has been reported in workers with long
histories of occupational exposure, but the nonspecific nature of the
morphological changes makes it difficult to identify lead as the
etiological agent except by association. There is a progressive
increase in fibrosis, beginning in peritubular areas extending into
the interstitium (Cramer et al., 1974). Inflammatory cells are
uncommon and are probably not a primary component of the process.
There is eventual tubule atrophy and hyperplasia of surviving tubules.
There is little evidence that the glomerulus is directly affected by
excessive exposure to lead, except for some nonspecific swelling of
mesangial and epithelial cells. In the terminal stage, glomeruli
become sclerotic. An immunological basis for the progression of
lead-induced nephropathy, as suggested following gold and mercury
exposures, might be suspected. However, there is at present no
published documentation of antirenal antibodies or immune-complex
formation in the pathogenesis of lead nephropathy. One study suggested
that lowered glomerular filtration rate occurs in occupational
exposure to lead that does not produce clinical toxicity (Wedeen et
al., 1979). The pathophysiological basis for this observation has not
yet been determined but may be a consequence of direct toxicity to
epithelial cells of the glomerular apparatus.
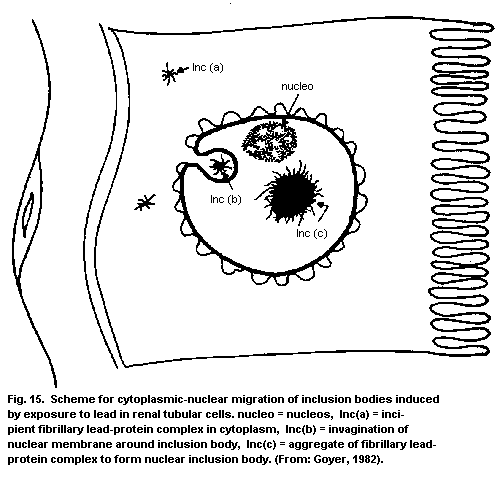 Intranuclear inclusion bodies are uncommon in the late stages of
lead nephropathy, although they may be seen in renal biopsy or autopsy
as a manifestation of a super-imposed severe acute exposure. It has
been shown that inclusion bodies may be found in the urine of workers
with occupational exposure to lead, but their presence or absence in
urine has not been related to the severity of lead nephropathy
(Schumann et al., 1980).
5.5.2 Cadmium
Cadmium is an occupational and environmental contaminant that has
received a great deal of attention. An important toxicological
feature of cadmium is its exceptionally long biological half-life in
the human organism (10-30 years). Once absorbed, cadmium is
efficiently retained in the organism and accumulates throughout life.
In the newborn baby, cadmium is present only at very low levels, but
by the age of 50 the cadmium body burden may have reached up to 20-30
mg and, in people occupationally exposed, it may reach values as high
as 200-300 mg. Furthermore, cadmium concentrates in vital organs,
particularly in the kidneys. At low levels of exposure, such as those
prevailing in the general environment, 30-50% of the cadmium body
burden is found in the kidneys alone (Nomiyama, 1980; Bernard &
Lauwerys 1986; Friberg et al., 1986).
The accumulation of cadmium in the kidney, may give rise to a
progressive form of tubulo-interstitial nephritis. In contrast to the
situation with many nephrotoxins, including other heavy metals such as
lead and mercury, there are virtually no acute effects of inorganic
cadmium salts on the kidney, except perhaps for some nonspecific
effects that have been seen in animals given near-lethal doses. One of
the most challenging questions regarding the metabolism of cadmium has
been the role of metallothionein in cellular metabolism and its
potential toxicity. Metallothionein synthesized within the kidney
protects from cadmium toxicity, but intravenously injected
cadmiummetallothionein is more nephrotoxic than inorganic cadmium (see
section 4.5.3.).
Cadmium nephropathy was first described by Friberg (1948, 1950)
who studied a group of alkaline battery workers in Sweden during the
late 1940s. Since these reports, a number of epidemiological studies
have shown the occurrence of nephrotoxic effects in populations
exposed to cadmium at work and in the general environment. These
studies have demonstrated that the most prominent feature and probably
the earliest sign of cadmium nephropathy is increased proteinuria.
Studies performed in the 1950s and 1960s (reviewed by Friberg et
al., 1986 and Bernard & Lauwerys, 1986) showed that cadmium
proteinuria is similar to the tubular-type proteinuria described by
Butler & Flynn (1958) in patients with tubular disorders, and consists
of unidentified proteins of low relative molecular mass derived from
plasma. Characterization of these proteins led to the discovery of
ß2-microglobulin, retinol-binding protein, and alpha1-microglobulin.
Subsequent studies demonstrated that the increased urinary excretion
of proteins of low relative molecular mass observed in cadmium
nephropathy and other renal disease was due to the failure of the
proximal tubules to reabsorb proteins filtered through the glomeruli.
The effects of cadmium on the excretion of ß2-microglobulin have
been extensively documented (Bernard et al., 1976, 1979a,b, 1982,
1987). Measurement of retinol-binding protein is much more reliable in
acidic urine and detects tubular proteinuria with equal sensitivity
(Bernard et al., 1982).
As cadmium nephropathy progresses, it increasingly presents the
signs of a complete Fanconi's syndrome, i.e. aminoaciduria,
glucosuria, increased urinary excretion of calcium, phosphorus, and
uric acid, and decreased concentrating ability of the kidneys. In the
most severe cases, the GRF decreases. The disturbances in calcium and
phosphorus metabolism may lead to a demineralization of the bones and
the formation of kidney stones (Friberg et al., 1974).
In cadmium-polluted areas of Japan, signs of renal dysfunction
very similar to those observed in cadmium workers have been frequently
found. A higher incidence of proteinuria, glucosuria, and
aminoaciduria, and increased excretion of ß2-microglobulin have been
observed in the Zinzu river basin in Toyama where Itai-Itai disease
was first seen (Fukushima et al., 1974; Kjellström et al., 1977;
Shiroishi et al., 1977; Kjellström & Nordberg, 1978). In the endemic
area of Toyama, the increased urinary excretion of ß2-microglobulin
was strongly related to the residence time in that area as well as to
the purposes for which contaminated river water was used (Kjellström
et al., 1977). In urine, ß2-microglobulin concentration correlated
with the cadmium level (Nogawa et al., 1979a,b).
Further investigations of the renal function of the inhabitants
in this area revealed a significant decrease in both creatinine
clearance and renal phosphorus reabsorption. Renal dysfunction due to
chronic cadmium poisoning was also found in other areas of Japan where
the rice was contaminated by cadmium (Saito et al., 1977; Kojima et
al., 1977). Studies carried out in Belgium suggested that
environmental exposure to cadmium in an industrialized area polluted
by this metal may exacerbate the age-related decline of renal function
in elderly residents (Lauwerys et al., 1980; Roels et al., 1981a,b).
Since cadmium-induced nephropathy may occur within the general
population, it is of major public health importance to know what level
of cadmium exposure carries a risk of renal tubular dysfunction and
cadmium nephropathy.
The concept of a critical concentration of cadmium has very
important implications with regard to establishing maximum levels of
cadmium that human populations may be exposed to with some margin of
safety. From a comparison of the cadmium concentrations in the renal
cortex of cadmium-exposed people with and without signs of kidney
damage, Friberg et al. (1974) suggested that the critical level of
cadmium in the renal cortex for the appearance of tubular proteinuria
is around 200 mg/kg. With the development of neutron activation
techniques allowing the in vivo determination of cadmium in
tissues, the critical level of cadmium in the human kidney has been
more precisely assessed.
Investigations conducted by Roels et al. (1981a) in Belgium and
Ellis et al. (1981) in the USA have shown that when the concentration
of cadmium in the kidney cortex reaches about 200 mg/kg, signs of
renal dysfunction (e.g., increased urinary excretion of albumin and
ß2-microglobulin) develop in about 10% of male workers exposed to this
metal. On the basis of the relationship between the concentrations of
cadmium in the urine and renal cortex and the prevalence of renal
anomalies, the critical concentration of cadmium in urine has been
estimated to be 10 µg/g creatinine (Bernard et al., 1979a,b; Buchet et
al., 1980; Roels et al., 1981a,b). Epidemiological studies of people
living in cadmium-polluted areas of Japan have shown that
ß2-microglobulinuria occurs after a lifetime accumulation of 2000 mg
cadmium or more (Nogawa et al., 1989).
Since several studies have shown that, in most cases, once
cadmium proteinuria has developed, it is irreversible, the progression
of renal dysfunction after cessation of exposure is very slow (Roels
et al., 1982; Elinder et al., 1985a,b). Persistent proteinuria is
found frequently among retired cadmium workers with no evidence of
renal insufficiency. In a group of workers removed from exposure after
the finding of microproteinuria (low or high relative molecular mass),
the reduction in GFR during a 5-year follow-up was about five times
greater than that accounted for by aging (Roels et al., 1989).
5.5.3 Mercury
It has been known for a long time that patients treated with
mercurial compounds can develop a glomerulonephritis that is usually
of the immune complex type (Becker et al., 1962; Druet et al., 1982).
Cases of mercury glomerulonephritis have also been reported as a
result of chronic exposure to high levels of mercury in industry
(Tubbs et al., 1982). Patients with mercurial nephropathy usually
present with a proteinuria and occasionally a nephrotic syndrome, but
no renal insufficiency (Druet et al., 1982).
Mercury may produce different effects on the kidney depending on
the biochemical form of the metal and nature of exposure. Inorganic
mercury compounds are classic examples of agents that cause acute
tubular necrosis. Mercuric chloride was used as a suicidal agent
during the nineteenth and early part of the twentieth centuries but
was unpopular for this purpose because of the painful accompanying
corrosive injuries it produced.
Regardless of the route of administration, mercuric chloride
produces acute tubular necrosis within hours of administration,
resulting in anuria and death. If the patient can be maintained by
dialysis, regeneration of tubular lining cells is possible. These may
be followed by ultrastructural changes consistent with irreversible
cell injury, including actual disruption of mitochondria, release of
lysosomal enzymes, and rupture of cell membranes.
The necrosis of the epithelium of the pars recta following
injection of mercuric chloride has been described in detail in the
rat. Cellular changes include fragmentation and disruption of the
plasma membrane and its appendages, vesiculation and disruption of the
endoplasmic reticulum and other cytoplasmic membranes, dissociation of
polysomes and loss of ribosomes, mitochondrial swelling with
appearance of amorphous intramatrical deposits, and condensation of
nuclear chromatin. These changes are common to renal cell necrosis
resulting from a variety of causes (Gritzka & Trump, 1968).
Mercury and its compounds are used widely, not only in various
industrial processes but also in a number of other applications such
as fungicides, contraceptive spermicides, and disinfectants. Several
studies have been carried out to determine the extent to which current
exposure of human populations to mercury can cause adverse renal
effects. Foa' et al., (1976) reported an increased prevalence of
glomerular proteinuria in workers exposed to mercury vapour in a
chloralkali plant.
Studies carried out between 1979 and 1984 (Buchet et al., 1980;
Roels et al., 1985) provided further evidence that occupational
exposure to mercury vapour can lead to subclinical renal
disturbances. These consisted of increased urinary excretion of
proteins of high relative molecular mass (albumin, transferrin, and
immunoglobulin G), lysosomal enzymes, and retinol-binding protein,
which occurred at a higher prevalence in subjects who excreted more
than 50 µg mercury/g creatinine. These observations were not confirmed
by Stonard et al. (1983), who found only a slight increase in the
prevalence of NAG and gamma-glutamyltranspeptidase (an enzyme of the
brush-border) in workers with urinary mercury levels higher than 100
µg/g creatinine.
Increased urinary excretion of NAG has also been found in workers
involved in the production of various mercuric salts (Rosenman et al.,
1986). Studies on patients with Minamata disease have provided
inconsistent results regarding the induction of proximal tubular
injury by methylmercury (Iesato et al., 1977; Ohi et al., 1982). By
contrast, in a study of 509 infants exposed to phenylmercury fungicide
on cloth diapers, Gotelli et al. (1985) clearly demonstrated that the
kidney is a target organ during prolonged exposure to this compound.
They showed that the urinary excretion of
gamma-glutamyl-transpeptidase increased in a dose-dependent manner
when urinary mercury exceeded approximately 220 µg/litre. This effect
was, however, completely reversible and had disappeared when the
infants were re-examined two years later.
Although exposure to a high dose of mercuric chloride is directly
toxic to renal tubular lining cells, chronic low-dose exposure to
mercuric salts or even elemental mercury vapour may induce an
immunological glomerular disease. This form of mercury injury to the
kidney is clinically the most common form of mercury-induced
nephropathy. Exposed workers may develop a proteinuria that is
reversible after they are removed from exposure. It has been stated
that mercury-induced nephropathy seldom occurs without sufficient
exposure to produce detectable mercury neuropathy as well.
Experimental studies have shown that the pathogenesis of mercury
nephropathy has two phases: an early phase characterized by an
anti-basement-membrane glomerulonephritis followed by a superimposed
immune-complex glomerulonephritis (Roman-Franco et al., 1978). The
pathogenesis of the nephropathy in humans appears similar, although
antigens have not been characterized. Also, the early
glomerulonephritis may progress in humans to an interstitial
immune-complex nephritis (Tubbs et al., 1982).
5.5.4 Gold
The use of gold in the form of organic salts to treat rheumatoid
arthritis may be complicated by development of proteinuria and the
nephrotic syndrome (Hall et al., 1987). Morphologically, the kidney
shows an immune-complex glomerulonephritis with granular deposits
along the glomerular basement membrane and in the mesangium. The
pathogenesis of the immune-complex disease is not known for certain,
but gold may behave as a hapten and generate the production of
antibodies with subsequent deposition of gold protein-antibody
complexes in the glomerular subepithelium. Another hypothesis is that
antibodies are formed against damaged tubular structures, particularly
mitochondria, providing immune complexes for the glomerular deposits
(Viol et al., 1977).
The pathogenesis of the tubular cell lesions induced by gold
therapy is probably initiated by the direct toxicity of gold to
tubular cell components. From experimental studies it appears that
gold salts have an affinity for the mitochondria of proximal tubular
lining cells. This is followed by autophagocytosis and accumulation of
gold in amorphous phagolysosomes (Stuve & Galle, 1970). Gold particles
can be identified in degenerating mitochondria, in tubular lining
cells, and in glomerular epithelial cells by X-ray microanalysis
(Ainsworth et al., 1981).
5.5.5 Bismuth
The effects of bismuth on the kidney are similar to those of
lead, but it is a less frequent cause of renal disease. This is
because bismuth is not present in such large amounts in the ambient
environment, nor is it as important industrially. However, bismuth has
been used therapeutically to treat a variety of ailments, most
particularly syphilis. Bismuth administration results in the formation
in proximal renal tubular lining cells of characteristic nuclear
inclusion bodies that are similar to the lead-induced bodies and are
composed of a bismuth-protein complex. The protein is acidic and has
an amino acid composition similar to that forming the lead inclusion
bodies. However, there is a slight difference in morphology between
the lead- and bismuth-induced inclusion bodies. The bismuth-protein
complexes are also observed in the mitochondria of proximal tubular
lining cells (Fowler & Goyer, 1975). The bismuth content of the
inclusion bodies has been confirmed by X-ray microanalysis of tissue
sections. Whether bismuth produces a chronic interstitial nephropathy
like lead has not yet been documented. However, bismuth inclusions
have been found at autopsy more than 30 years after a course of
bismuth therapy.
5.5.6 Uranium
Exposure of humans or experimental animals to compounds of
uranium results in injury and necrosis of proximal renal tubules. The
most sensitive site is the pars recta (as in the case of mercury),
but, depending on the dose, injury and necrosis may extend to other
parts of the proximal tubule. Acute injury is followed by regeneration
of tubular epithelial cells. Chronic effects have not been reported.
An increase in the urinary excretion of ß2-microglobulin and of
specific amino acids has been reported by Thun et al. (1985) in
uranium mill workers.
5.5.7 Chromium
The acute and chronic effects of chromium (mainly on the
respiratory tract and skin) are due largely to hexavalent compounds.
The acute tubular toxicity of chromate and dichromates salts in
animals is well documented, and renal tubular necrosis has also been
described in humans following acute poisoning (Langard & Norseth,
1986). Epidemiological studies have shown that chromium(VI) can
produce slight tubular dysfunction in chronically exposed workers.
Mutti et al. (1979) reported an increased prevalence of elevated
ß-glucuronidase and total protein levels in the urine of welders
exposed to chromium. These observations have been confirmed by recent
studies using more sensitive, reliable markers of tubular injury, such
as ß2-microglobulin (Lindberg & Vesterberg, 1983), retinol-binding
protein, and the BB-50 renal antigen (Mutti et al., 1985).
Franchini & Mutti (1988) have studied dose-effect/response
relationships between the urinary excretion of chromium and that of
retinol-binding protein or the renal antigen BB-50. Most of the
abnormal values were observed in subjects with urinary excretion of
chromium greater than 15 µg/g creatinine; however, above this
threshold the degree of tubular impairment was not related to urinary
excretion of chromium. Franchini & Mutti (1988) explained this
phenomenon by postulating that the tubular damage observed in
chromium(VI)-exposed workers is transient and due mainly to acute
exposure, and that workers become progressively resistant to the
effects of more severe or prolonged exposure.
5.5.8 Arsenic
Acute arsenic poisoning may cause tubular necrosis. Acute or
severe chronic poisoning is usually treated with the chelating agent
BAL (2,3-dimercaptopropanol). Inhalation of arsine may also produce an
acute tubular necrosis as a result of intravascular haemolysis.
In a cross-sectional study, Foa' et al. (1987) failed to show
significant differences between occupationally exposed workers and
matched controls, with the exception of a slight increase in the
urinary excretion of retinolbinding protein. However, owing to the
small sample size and the low power of the study, no definite
conclusion could be drawn from the slight increases in albuminuria,
ß2-microglobulin, and the brush-border antigen BB50. The authors
concluded that extended population surveys would be desirable for a
complete definition of such subtle effects.
5.5.9 Germanium
Germanium is naturally present in the diet, normal intake being
about 1 mg per day. It is being used increasingly in the semiconductor
industry. A recent report from Japan documented renal failure in ten
individuals (including two deaths) among previously healthy
individuals taking large doses of germanium (of the order of 50-250 mg
per day) over periods of 4-18 months (Matsusaka et al., 1989). Renal
biopsy or autopsy in seven cases showed degeneration of the renal
tubular epithelium in all cases with or without interstitial fibroses
or oedema. The glomeruli were only minimally effected in two cases.
6. RENAL CANCER
Tumours of the renal parenchyma, pelvis, and ureters are
uncommon, accounting for less than 2-3% of all human cancers
(DeKernion & Berry, 1980; Dayal & Kinman, 1983). The role of drugs,
chemicals, and other environmental factors in the etiology of
parenchymal and urinary tract tumours is unclear, but cancer of these
sites is most common in certain industrialized nations (Sweden) and in
people in the higher socio-economic groups (Rimpela & Pukkala, 1987).
Other risk factors have been stratified (Selli et al., 1983). The
ratio between tumours of the renal parenchyma and pelvis is fairly
constant (about 5:1), and the parallel trends in increasing incidence
argue for some commonality in the etiology of tumours at both sites,
although some factors may be site specific. Tumours are nearly twice
as common in males as in females. A compilation of trends in cancer
rates in the USA indicates that the incidence of both kidney and
bladder cancer is increasing (Pollack & Horm, 1980). A similar
observation has been made with regard to renal parenchymal tumours in
males in Scotland (Ritchie et al., 1984).
6.1 Renal tumour classification
The International Classification of Diseases for Oncology (code
189) divides tumours of the urinary system into five groups according
to their size, i.e. parenchyma of the kidney (189.0), renal pelvis
(189.1), ureter (189.2), urethra (189.3), and paraurethral gland
(189.4) (Mostofi et al., 1981; WHO, 1990). These distinctions have
only been made in recent years, so that many mortality studies of
renal tumours have included this whole category. Of the five types of
urinary tract tumours, about 90-95% of renal tumours in adults are
adenocarcinomas arising from the renal parenchyma. Nephroblastoma
(Wilm's tumour) is the second most common histological type of renal
tumour and accounts for 2-4% of kidney cancer in Sweden and the USA.
It is easily distinguished morphologically from renal adenocarcinoma
and usually appears in the first five years of life; 95% of cases
occur before the age of 15 years. Although nephroblastomas are the
fourth most common tumour in childhood, they are relatively rare in
adults.
6.2 Renal adenocarcinoma
Renal adenocarcinoma has been known under several synonyms (clear
cell carcinoma, hypernephroma, Grawitz tumour) reflecting uncertainty
about its origin, but immunological studies have established that
renal adenocarcinomas arise from the proximal convoluted tubule
(Wallace & Nairn, 1972). They tend to be circumscribed, ranging in
size from microscopic lesions to large neoplasms (Hamilton, 1975). The
spectrum from small benign lesions to clearly malignant lesions
suggests a continuous pathological process, so that it is often
difficult to label smaller tumours as benign or malignant. Hellsten et
al. (l983) have defined all tumours of 2 cm or more in diameter as
adenocarcinomas. Postmortem studies have shown that adenomas are
present in approximately 25% of all males over 50 years of age, and it
was found that 34% of 235 clinically unrecognized tumours present at
autopsy were less than 3 cm in diameter. In the absence of invasion of
surrounding tissue, features such as frequent mitotic figures,
cellular pleomorphism, and haemorrhage and necrosis generally indicate
a malignant potential regardless of size. Calcification may be
detected by X-ray examination in about 15% of cases. Although 2-3%
may be cystic, the commonest form is a solid tumour that is usually
composed of clear cells rich in lipid, glycogen, or both, but may
contain granular cells or even tightly packed eosinophilic cells
referred to as oncocytes. The cell pattern may be trabecular, solid,
or mixed, but it is doubtful that cell type or structural pattern has
any clinical significance. Grading is difficult and has not been shown
to be clinically useful. These tumours usually grow slowly, and
overall survival is 20-25% after nephrectomy. The presence of
multiple tumours, renal vein invasion, or regional lymph node
metastases indicates a poorer prognosis. Hellsten et al. (1983)
recorded metastasizing renal carcinoma as cause of death in 21% of a
postmortem series, and in 33% a second malignant tumour was observed
causing the death of 20%.
Immunological mechanisms are thought to determine the natural
history of the disease. The development of monoclonal antibodies and
flow cytometry have provided new methods for investigating
immunological responses. Total T lymphocyte counts were found in a
study of 32 patients to be lower than in controls (Ritchie et al.,
1984), due largely to a deficit of T helper cells but not T
suppressor-cytotoxic cells. This effect of the tumour is reversed by
removal of the primary tumour, and recurs with return of the tumour.
These findings are believed to suggest that there is a systemic effect
of the tumour acting at the level of the bone marrow or thymus to
affect the production or maturation of T helper cells.
The role of specific environmental factors in the etiology of
urinary tract tumours has been difficult to define (Newson & Vugrin,
1987). Apart from increases in renal tumours in asbestos workers and
the identification of some occupationally related bladder tumours,
there does not appear to be a clearly defined association with
specific chemicals or environmental factors. However, there is an
association with a combination of exposure to substances in the
environment and life-style practices such as tobacco use. This
suggests that there may be interactions between substances or that the
urinary tract, like the lung, has to deal with a number of substances
with promoter activity. Cigarette smokers have a 2-fold increase in
risk of urinary tract tumours (Goodman et al., 1986), and an increase
associated with alcohol and coffee usage has been suggested (Jacobsen
et al., 1986). In addition, chronic interstitial nephritis may
predispose to urinary tract tumours. People with endemic (Balkan)
nephropathy have an increase in renal tumours and a possible
relationship between chronic interstitial nephritis and renal
neoplasia (see section 5.3). It is also noteworthy that human
populations with excessive exposure to some known carcinogens (e.g.,
cycasin in Guam and aflatoxins in Africa and Asia) have not yet been
shown to have an increase in kidney cancer (Sufrin & Beckley, 1980).
Renal adenocarcinoma has been diagnosed with increasing frequency in
patients with chronic renal failure, particularly in those patients
treated with long-term dialysis (Dunhill et al., 1977).
An association between renal cancer and excess exposure to lead
has not been clearly established, but a study of lead smelter and
battery workers found a significant excess of malignancies at all
sites, these being mostly lung tumours (Cooper & Gaffey, 1975). Case
reports of renal tumours in workers with lead nephropathy have
appeared (Baker et al., 1980; Lilis, 1981).
6.3 Upper urothelial carcinoma (transitional cell carcinoma)
Tumours of the renal pelvis form a spectrum from benign
papillomas to frank papillary carcinomas and, like bladder tumours,
are generally low-grade cancers. However, they tend to recur
regardless of their morphology. Upper urothelial carcinoma has been
associated with RPN and analgesic abuse, but a cause-and-effect
relationship between the two has not been proven (Bach & Bridges,
1985a). The incidence of upper urothelial carcinoma among analgesic
abusers is very high, and females predominate in analgesic-associated
upper urothelial carcinoma. The female:male ratio is 2.5 to 1
(Bengtsson et al., 1978), which is in keeping with the ratio for
analgesic abusers. Analgesic abusers also develop upper urothelial
carcinoma at a younger age than non-analgesic abusers (Mihatsch et
al., 1980a,b,c). The distribution of urothelial carcinomas in
analgesic abusers has a distinct pattern; tumours of the renal pelvis,
ureter, and bladder are found 80 times, 90 times, and 7 times,
respectively, more frequently than in non-analgesic abusers. The
tumours are typically multiple, diffuse, and poorly differentiated,
and spread rapidly (Mihatsch et al., 1980c).
Patients who discontinue abuse of the drugs are at a greater risk
of developing upper urothelial carcinoma, often after a latent
period of 10-20 years after initiating analgesic abuse. Greatly
improved dialysis techniques may result in the survival of
analgesic-abusing patients who would otherwise have developed
end-stage renal disease and subsequently died (Mihatsch et al.,
1980a). It has therefore been suggested that the incidence of upper
urothelial carcinoma will increase.
The diagnosis of upper urothelial carcinoma is difficult in the
clinical situation because of few specific clinical symptoms to
indicate the malignant changes (Johansson et al., 1976; Bengtsson et
al., 1978; Mihatsch et al., 1980a,b,c; Mihatsch & Knusli, 1982; Bach
& Bridges, 1985a; Pommer et al., 1986). The prognosis is poor, and
patients with upper urothelial carcinoma only have a mean survival
time of 22 months (Mihatsch et al., 1980a) owing to the difficulty of
diagnosis, compromised renal function of patients with RPN, and
multifocal sites of rapidly developing and widespread invasion and
metastases (Johansson et al., 1976; Mihatsch & Knusli, 1982).
6.4 Experimentally induced renal adenomas and adenocarcinomas
Renal adenomas and adenocarcinomas may be induced in laboratory
animals by various natural products and biological and chemical
agents. However, linkage of exposure of these substances to renal
cancer in humans is lacking in most instances, or, at best, is only
suspected.
6.4.1 Background incidence of spontaneous tumours in experimental animals
The incidence of spontaneous renal parenchymal tumours in most
commonly used strains of male rats and mice is in the region of 0.2%,
whereas it is < 0.1% for females (Crain, 1958; Goodman et al., 1979,
1980; Ward et al., 1979; Maekawa et al., 1983). The incidence may be
up to 2.7% (Pour et al., 1979) in hamsters, which is generally low
enough for investigative studies.
Renal tumours have been induced experimentally by a large number
of compounds including lead salts (Kilham et al., 1962; van Esch &
Kroes, 1969; Goyer & Moore, 1974), nickel sulfides (Jasmin & Riopelle,
1976; Sunderman et al., 1984), methylmercury chloride (Mitsumori et
al., 1981), N-(4 -fluoro-4-biphenylyl) acetamide (Hinton et al.,
1980), trisodium nitrilotriacetic acid (Goyer et al., 1981), potassium
bromate (Kurokawa et al., 1983), halogenated alkenes (Kociba et al.,
1977; Reichert et al., 1984), and tris(2,3-dibromopropyl)phosphate
(Reznik et al., 1979). Natural products include cycasin (Laqueur &
Spatz, 1968), aflatoxins B1 (Butler et al., 1969; Epstein et al.,
1969), ochratoxin A (Kanisawa & Suzuki, 1978), citrinin (Arai &
Hibino, 1983), the fermentation-derived anti-neoplastic agent
daunomycin (Sternberg et al., 1972), and streptozotocin (Rakieten et
al., 1968; Hard, 1985). Diethylstilbestrol (Horning & Whittick, 1954)
and related estrogens (Li et al., 1983) produce a high incidence of
parenchymal tumours in hamsters.
The classical two-stage model of carcinogenesis also applies to
several of the nitrosamines, where promoters include sodium arsenite
(Shirachi et al., 1983), DL-serine (Hiasa et al., 1984a), folic acid
(Shirai et al., 1984), lead acetate (Hiasa et al., 1983), nicotinamide
(Rosenberg et al., 1985), trisodium nitrilotriacetate (Hiasa et al.,
1984b), and citrinin (Shinohara et al., 1976). There are several
factors that may affect the development of carcinomas, including diet
(Hard & Butler, 1970; McLean & Magee, 1970; Hard, 1980, 1984; Swann et
al., 1980), partial hepatectomy (Evarts et al., 1982), unilateral
nephrectomy (Ito et al., 1969), and unilateral hydronephrosis (Ohmori
& Tabei, 1983).
6.4.2 Inorganic compounds
Various inorganic compounds of lead have been investigated over
the last 30 years (Van Esch & Kroes, 1969). The tumours arise from
kidney tubular epithelial cells in kidneys and are similar to renal
cortical tumours found in humans. Production of tumours requires
continuous exposure to relatively high concentrations of lead in the
diet or drinking-water for 1 to 2 years. The tumours occur in a
background of severe interstitial nephritis characterized by tubular
atrophy as well as focal areas of hyperplasia. They are usually
multifocal and vary from microscopic adenomas to large renal
adenocarcinomas that may invade contiguous structures or metastasize
to the lungs. Intra-nuclear inclusions, which are usually present in
proximal tubular epithelial cells in lead toxicity, are absent in
neoplastic cells, and the tumors contain much less lead than adjacent
renal parenchyma (Mao & Molnar, 1967). Tumor cells are pleomorphic,
and ultrastructural studies have shown marked morphological
alterations in mitochondria.
Renal cancer occurs after injection of crystalline nickel
subsulfide (Ni3S2) into the kidney of rats, but not after
treatment with amorphous nickel sulfide (NiS). No evidence indicates
that nickel compounds are carcinogenic in experimental animals when
administered by oral or subcutaneous routes (Sunderman, 1981).
6.4.3 Organic molecules
Nitrilotriacetic acid, a polyamino polycarboxylic acid with
chelating properties similar to EDTA (used to treat lead poisoning),
produces chronic interstitial nephropathy in rodents. A spectrum of
tubular cell histological changes occurs from hyperplasia to small
adenomas to adenocarcinomas (Goyer et al., 1981). Renal adenocarcinoma
has been induced in male rats by the chronic inhalation of unleaded
gasoline vapour (MacFarland et al., 1984), but this relationship has
not been supported by epidemiological studies on workers in the
petroleum industry (Enterline & Viren, 1985).
6.4.3.1 Nitrosamines and related compounds
The nitrosamines represent one of the most widely investigated
groups of model compounds. They include dimethylnitrosamine (Murphy
et al., 1966; Mohr et al., 1974; Hard, 1984), which produces
mesenchymal (connective tissue) tumours in young animals but adenomas
and adenocarcinomas in mature animals (Hard, 1979). The
co-administration of putrescine (Ohmori & Tabei, 1983) or
N-3,5-dichlorophenyl-succinimide (Ito et al., 1974) with
dimethylnitrosamine caused a dose-related incidence of up to 100%
renal tumours after 100 weeks. N-ethyl- N-hydroxyethylnitrosamine
(Hiasa et al., 1979) on its own, or, especially, when administered
with basic lead acetate (Hiasa et al., 1983) or serine (Hiasa et al.,
1984a) causes tumours in up to 95% of animals by 32-38 weeks.
N-Nitrosomorpholine causes oncocytomas (Bannasch et al., 1978a,b,
1980). There are interesting differences between several of the model
compounds, interspecies responses, effects of dose regimens, etc.
However, the investigation of these models has generally permitted the
progression of cellular injury to be described in terms of the acute
effects, early hyperplasia, dysplasia, and different types of renal
tumours.
6.4.3.2 Morphological changes
Many of these model compounds have been used to study the early,
intermediate, and late changes at the light microscope and
ultrastructural levels (Horning & Whittick, 1954; Butler, 1964; Butler
& Lijinsky, 1970; Ertürk et al., 1970; Hard & Butler, 1971; Sternberg
et al., 1972; Bennington, 1973; Hard, 1975, 1984, 1985; Bannasch et
al., 1978a,b, 1980; Dees et al., 1980a,b; Ohmori et al., 1982; Tsuda
et al., 1983; Eble & Hull, 1984; Hard et al., 1984; Hiasa et al.,
1984a,b,c). The phenotypic changes associated with loss of normal
growth control have concentrated on the focal preneoplastic changes in
heterogeneous cells. These undergo slow changes (where it is highly
desirable to define the origins of the neoplasm) in a limited number
of enzyme markers and in lipids, and carbohydrates. In addition, an
increase in cytoplasmic RNA (shown by enhanced cytoplasmic basophilia
or numerous ribosomes at the ultrastructural level) has been observed
as a marker for hyperbasophilic and basophilic preneoplastic foci in
the epithelium of the renal tubular system (Hard, 1986; Bannasch &
Zerban, 1986).
The nomenclature of renal parenchymal tumours is based on several
criteria, including the size of the tumour and the
morphological-histochemical characteristics of cells and their
organization. The progression in tissue mass from hyperplasia, through
dysplasia, to adenoma, adenocarcinoma, and carcinoma is a continuum,
although the earliest changes, particularly hyperplasia, may be
reversible. Cells may have no cytoplasmic staining (clear cells) or
granular acidophilic or basophilic cytoplasm staining, and tumours may
have a mixture of cells with different staining characteristics. Where
adenomas contain a uniform population of finely granulated
eosinophilic cells, they are termed renal oncocytomas. Tumours can
also be classified as tubular, solid, lobular, disorganized, invasive,
papillary or cystadenoma, or by a composite of such terms based on
their appearance (Hard, 1987).
Clear and acidophilic (granular) cell kidney tumours induced by
limited exposure of rats to N-nitrosomorpholine are associated with
a transient storage of glycogen (Bannasch et al., 1978a) and closely
parallel the most common malignant renal neoplasm in man. The tumours
originate from segments of the collecting duct system storing large
amounts of glycogen (Nogueira et al., 1989). However, when
microadenomas develop, the clear (glycogenotic) cells loose glycogen
and acquire an acidophilic (granular) cytoplasm, although both cell
types can coexist in large tumours. Lipid-storing cells are often
found in the clear cell tumours, but the significance is not known. By
contrast, there are no such relationships between cells storing
glycogen and the so-called renal oncocytomas also found in NNM-induced
rats (Bannasch et al., 1978b; Nogueira et al., 1989), although rat
renal oncocytomas also originate from the collecting duct system
(Nogueira & Bannasch, 1988). They are, however, benign end-stage
lesions where the cytoplasm is crowded with pathologically altered
mitochondria (Krech et al., 1981). A temporary focal storage of
glycosamino-glycans has been reported in chromophobic rat renal cell
tubules and tumours (Bannasch et al., 1980, 1981) and in the
corresponding type of human tumour (Thoenes et al., 1985).
6.4.3.3 Biochemical changes in cells
An immunohistochemical increase in glucose-6-phosphate
dehydrogenase is associated with basophilic renal cell tumours (Tsuda
et al., 1986) and nephroblastomas (Moore et al., 1986). The pentose
phosphate shunt provides sugars for RNA and DNA synthesis, and the
activation of this pathway is probably closely related to certain
phenotypic changes, such as an increase in ribosomes and an enhanced
cell proliferation in preneoplastic and neoplastic lesions. In line
with this interpretation, rat renal oncocytic tubules and tumours,
which are poor in ribosomes and grow very slowly, usually show a
normal or even decreased activity of glucose-6-phosphate dehydrogenase
(Tsuda et al., 1986). However, in some experimental models a reduced
amount or activity of glucose-6-phosphate dehydrogenase was found in
the more malignant populations, suggesting involvement of the enzyme
in other metabolic aberrations relevant to tumorigenesis (Moore et
al., 1986).
Alterations in drug-metabolizing enzymes (see below) during
carcinogenesis have been detected by immuno-histochemical methods in
various tissues, especially in the renal tubular system, but they have
not been correlated to the same extent with the respective enzyme
activities and with other changes in the cellular phenotype as have
those of carbohydrate metabolism. By contrast,
N-ethyl- N-hydroxyethylnitrosamine-induced renal carcinomas show
opposite alterations in drug-metabolizing enzymes in preneoplastic and
neoplastic lesions of these tissues (Tsuda et al., 1987). Reduced
activities of gamma-glutamyl-transpeptidase (Ohmori et al., 1982;
Tsuda et al., 1986), succinate dehydrogenase (Tsuda et al., 1986), and
alkaline phosphatase (Tsuda et al., 1986) are seen as early changes
during the development of basophilic cell tumours from hyperbasophilic
segments of the proximal nephron. In contrast, however, there are no
similar changes in gamma-glutamyltranspeptidase or alkaline
phosphatase activity (but there is an increase in succinate
dehydrogenase activity) in oncocytic tubular lesions seen in these
animals (Tsuda et al., 1986). The increased binding of anti-cytochrome
c oxidase to the oncocytic lesions in both man (Ortmann et al., 1988)
and rat (Mayer et al., 1989) may be a useful marker for preneoplastic
renal changes.
6.4.3.4 The mechanistic basis of renal carcinoma
The mechanistic basis for the development of renal carcinoma may
be genotoxic or non-genotoxic. The common feature to all genotoxic
agents is the generation of a reactive electrophilic
(electron-deficient) species which is capable of binding to
nucleophilic (electron-rich) sites on cellular macromolecules
including proteins, lipids, RNA, and especially DNA (Miller & Miller,
1981). For example nitroso-compounds alkylate DNA in the N-7 and,
especially (due to its prolonged stability), O-6 positions (Nicoll et
al., 1975).
Genotoxic compounds are either direct alkylating agents
(requiring no activation) or they require one or more
biotransformation steps by the P-450-dependent mono-oxygenases (e.g.,
chloroform) (Bailie et al., 1984; Smith & Hook, 1984). However,
hexachloro-1,3-butadiene is transformed by ß-lyase (Elfarra & Anders,
1984) or prostaglandin hydroperoxidase-mediated co-oxidation (Davis et
al., 1981), which may also be involved in
N-[4-(5-nitro-2-furyl)-2-thiazolyl]formamide transformation (Zenser
& Davis, 1984). In addition, there may be several other renal and
extra-renal metabolic steps. There are also some molecules that cannot
be reliably classified into either group. Diethystilbestol is a very
weak alkylating agent (Lutz et al., 1982), and its mechanism of action
is thought to be mediated by renal estrogen receptors (Li & Li, 1984).
However, some metabolic component may be involved, e.g., hepatic
mixed-function oxidase (Metzler, 1981) and peroxidative activation
(Metzler & McLachlan, 1978).
6.5 Experimentally induced upper urothelial carcinomas
(transitional cell carcinomas)
There is experimental evidence to connect analgesic exposure to
the development of urothelial tumours (Johansson & Angervall, 1976;
Bengtsson et al., 1978; Bach & Bridges, 1985a). While bladder tumours
have been studied extensively in animal models, the practical
difficulties of looking for malignancies in the ureter or pelvis has
limited studies in this area. Based on long-term carcinogenicity
studies, there are few data to establish a clear experimental
relationship between analgesic exposure and upper urothelial
carcinoma. There is, however, experimental evidence to suggest that
upper urothelial carcinomas can be induced using a classical two-stage
initiation/promotion regimen (Bach & Gregg, 1988; Gregg et al., 1989).
These data suggest that localized injury associated with papillary
necrosis adjacent to urothelium that has already been initiated will
result in a proliferation of changes that lead to malignancy. At
present the full significance of these findings in terms of the human
analgesic problem is not clear.
7. ASSESSMENT OF NEPHROTOXICITY
No single in vivo or in vitro method of studying the
nephrotoxicity of chemicals can address all of the questions that must
be asked. It is therefore inadvisable to separate mechanistic research
into target cell toxicity from the screening of novel compounds for
their potential nephrotoxicity. The holistic approach to
nephrotoxicity assessment also demands that in vivo investigations
are not separated from in vitro studies, and that data continue to be
derived from several different animal species and related to
accurately conducted epidemiological and clinical studies (where these
data are available).
Present data suggest that most in vitro methods can provide
information on the mechanism of primary insult and the effect on cell
viability. However, there appears at present to be little place for
in vitro techniques in the assessment of secondary renal changes, as
the factors that contribute to the cascade of degenerative changes
that follows renal insult are largely obscure. Thus there is inherent
uncertainty as to what should be studied in in vitro systems. There
are, however, several approaches that can be used to provide a better
understanding of the contribution to degenerative changes in vivo.
These include harvesting tissue at different time points following an
insult and using this tissue to study function in different cell
types, or studying the effects of chemicals on target and non-target
cells using pure and mixed cell cultures in the presence and absence
of cells that are related anatomically. It is also possible to
exchange culture media between cells that have been insulted, and
those that have not, in order to assess the release of factors that
may be toxic to other cells. Different cells can be studied in the
same media to define cell-cell interactions and how chemical insult
affects this process.
7.1 In vitro studies
Despite the complexity of the kidney and nephrotoxicity, and the
difficulty in defining what any one in vitro system achieves, there
are several ways to progress in the use of screening methods.
One such rational approach could include:
* the careful identification of compounds with well-documented in
vivo nephrotoxicity, including the sequence of pathological and
functional changes, the metabolites formed, quantities excreted,
and the cellular pharmacodynamic effects;
* the choice of chemicals that target specifically for one
anatomically discrete cell type in vivo;
* the use of both more and less nephrotoxic analogues of the
chemical for determining structure-activity relationships and
computer-based simulations;
* the systematic study of these compounds by several different in
vitro methods, and the use of several criteria for assessing in
vitro nephrotoxicity for each.
7.1.1 Choice of chemical concentrations for in vitro studies
The validity of using the cytotoxicity (in vitro) for a given
concentration of a chemical as a likely indicator of a toxicological
effect in vivo can be very difficult to establish. It may be
impossible to assess this in the kidney, because this organ is highly
compartmentalized. In the intact and functioning organ, it is
currently not possible to establish the concentrations of a xenobiotic
(or its metabolites) associated with any one cell type. This
uncertainty relates, for instance, to the different transport systems
that are distributed in discrete parts of the nephron, transcellular
pH gradients, and the selective accumulation of certain chemicals in
cell organelles (which have heterogeneous distribution in different
types of cells). Thus some chemicals can be selectively concentrated
in a discrete area of the kidney to several times the plasma
concentration. Alternatively, certain chemicals may be actively or
selectively excluded from some cell types (Mudge, 1985).
Drug metabolism systems that alter the physicochemical
characteristics of xenobiotics and their metabolites (that will
facilitate the redistribution of chemicals within and between cells)
are also heterogeneously distributed. Thus, certain xenobiotic
products may be selectively concentrated in (while others are excluded
from) specific cell types. Therefore, even though arterial and venous
blood and urinary concentrations of chemicals can be measured, there
is no certainty that such data relate to the concentrations of any
specific metabolite that reaches a target cell. Autoradiography may
be very valuable in providing some idea on how chemicals are
distributed, but it only shows the distribution of radiolabel-derived
material, not its chemical nature. Once the anatomical integrity of
the kidney has been altered, it may be difficult to relate the
concentration of any chemical to the same cells in vivo. While
structure is maintained in the perfused kidney, the functional changes
impose a similar constraint. The uncertainty as to what concentrations
of chemicals to use in in vitro systems is exacerbated by the
undefined influence of extrarenal and renal metabolism on the delivery
of the proximate and ultimate toxins to the target sites of injury in
vivo.
7.1.1.1 Proximate and ultimate nephrotoxicants in vitro
In addition to the need to consider carefully the consequences of
the changes in the route of chemical delivery when the anatomical
integrity of the kidney has been disrupted (i.e. in all systems other
than the isolated perfused kidney), it is important to consider how a
chemical is delivered to the cells. This consideration should be
expressed in terms of the chemical being free or bound (should the
media contain protein or not?), the physicochemical characteristics of
the solution in which the chemical is delivered (such as its pH, ionic
concentration, and endogenous and exogenous micro- or
macro-molecules), and the kinetics of delivery (this is generally
zero-order in most in vitro systems but follows first or second
order kinetics in vivo).
Chemically induced nephrotoxicity may be the result of a direct
action of the parent chemical in the kidney or may be due to an
extrarenally formed metabolite. However, in some instances the
chemical/metabolite has to be further metabolized in situ to form the
ultimate nephro-toxic species. Assessing the role of in situ
metabolism in nephrotoxicity from in vivo data may be difficult,
since most nephrotoxic chemicals are also extensively metabolized in
extrarenal tissue such as the liver. Experimental approaches using
different species and strains of animals, inducers and inhibitors of
drug metabolizing enzymes, and candidate proximate and ultimate
nephrotoxic metabolites may give some evidence for the involvement of
intrarenal metabolic activation (Rush et al., 1984). More direct
evidence for a direct renal activation of a chemical has to come from
in vitro studies or studies with perfused kidneys where extrarenal
metabolism/activation can be excluded. In vitro studies may include
experiments with isolated tissue preparation (kidney slices and
tubules), cells (primary cells or cell lines) or subcellular fractions
to assess chemically induced toxicity. In order to determine the role
of extrarenal metabolism in the formation of nephrotoxic metabolites,
co-culture systems using liver cells (as an activation system) and
kidney cells (as target cells) may be very useful (Moldeus et al.,
1978). The stability of a reactive metabolite, generated by liver
cells, may be measured by transferring, after various time intervals,
the incubation medium to the target cell population.
7.1.2 In vitro investigations of nephrotoxicity
In vitro techniques can be divided into those where the
anatomical relationship between cells is maintained (perfusion,
micropuncture, and slices), those where glomeruli and tubular
fragments are isolated, and those where cells are isolated. The
different techniques for assessing nephrotoxicity in vitro have been
reviewed and the strengths and weaknesses of each presented in broad
terms (Bach et al., 1985, 1986; Bach & Kwizera, 1988). Some methods
are technically difficult, depend on sophisticated equipment, are
subject to artefacts in inexperienced hands (perfusion and
microperfusion), and are difficult to interpret.
7.1.2.1 Perfusion and micropuncture
Micropuncture methodology has not been widely used to assess the
toxicological effects of chemicals (Bank et al., 1967; Biber et al.,
1968) because the methodology is extremely complex. However, a few of
the problem areas should be mentioned. Micro-puncture procedures
succeed only when the experimentalists can collect measured small
samples of tubular fluid and subject these to appropriate chemical
analyses. These procedures are extremely complex, and, because of the
small volumes involved, subject to considerable error. To assure that
the micropuncture collections reflect the "physiological state", most
workers attempt to collect only very small volumes at the
"physiological" flow rate past the point of micropuncture. Stationary
microperfusion has been used to assess tubular function in a
restricted area of the nephron.
Micro-injection into a tubular segment has been used by many
physiologists and may be of particular toxicological interest
(Gottschalk & Lassiter, 1973; Roch-Ramel & Peters, 1979; Diezi &
Roch-Ramel, 1987). With this technique, various renal function
markers, as well as potential nephrotoxicants, may be injected into a
segment of the proximal tubule during a free-flow situation. The urine
from the injected kidney may then be collected to assess
nephrotoxicant effects on the injected renal function markers. For
micropuncture specialists, this is a relatively straight-forward
technique and does not involve the problems associated with removal of
tubular fluid by micropuncture. This procedure might permit the
assessment of nephrotoxicant effects on membrane permeability by
examining, for example, inulin excretion from the injected kidney.
The isolated perfused tubule technique represents the development
of an in vitro procedure that permits the assessment of intact
tubular function under carefully controlled in vitro conditions.
Hence all of the advantages of in vitro methodology are available
while intact tubular function is being studied (Diezi & Roch-Ramel,
1987).
Tubule segments can also be isolated by manual dissection for
microperfusion, where they are attached to micropipettes suspended in
a bathing solution and perfused with an artificial tubular fluid
(Ullrich & Greger, 1985). Relatively few attempts have been made to
apply this sophisticated methodology to the study of nephrotoxicants,
where it would be possible to add chemicals to either the perfusate or
the bathing solution and examine effects on either the tubular or
basolateral side of the cell. This technique has been used to
demonstrate that organic anion transport across the tubular cell is
active on the basolateral side but not on the luminal side (Tune et
al., 1969).
The major advantage of this procedure is that it permits an in
vitro assessment of renal function with a tissue segment that is
essentially intact. The situation in an isolated nephron segment is
obviously not identical to that in the intact kidney, but by carefully
regulating the perfusion solution and the bathing solution one can
approximate in vivo physiology. This is a potentially important
procedure that needs to be assessed for its utility.
7.1.2.2 Renal cortical slice
These techniques have been reviewed extensively (Berndt,
1976,1987; Bach & Lock, 1982; Kacew, 1987) and used to show
deleterious effects of chemicals and drugs on the kidney. Much of the
published information has focused on renal tubular transport as the
criterion for establishing the nephrotoxic potential of chemicals. The
tests are based on measuring the accumulation of the organic ions
p-aminohippurate (PAH) and tetraethyl-ammonium (TEA) (Hirsch, 1976)
by renal slices. The organic anion transport is a sensitive indicator
of aminoglycoside (Kluwe & Hook, 1978; Kaloyanides & Pastoriza-Munoz,
1980) and cephaloridine toxicity (Kuo & Hook, 1982; Kuo et al., 1982).
Similarly, the effects of mercuric ions, chromate ions,
hexachlorobutadiene conjugates, and other nephrotoxins appear readily
detectable by this approach. Organic ion accumulation and
gluconeogenesis in renal cortical slices may be poor indicators of
early toxic effects to the kidney resulting from cisplatin
administration because these parameters are not affected except at
high doses. The poor sensitivity and delayed response of renal slice
parameters indicate that membrane function and cell metabolism may not
be early targets of cisplatin at the cellular level. Slices maintain
ß-lyase activity for up 12 h and have been used to study halo-alkene
toxicity. Aminooxyacetic acid inhibits ß-lyase activity almost
completely. DCVC decreases PAH accumulation, but does not appear to
use the same transport process.
7.1.2.3 Isolated nephron segments
Isolated tubules overcome many of the disadvantages of cortical
slices, in that they remain in contact with substrates and toxins in
the medium, whereas cortical slices may show lumen collapse within a
short period (Chahwala & Harpur, 1986).
Freshly isolated tubule or cell suspensions offer an important
way of studying the mechanisms of nephrotoxicity and screening novel
compounds for their potential acute effects on the kidney. A
limitation, however, is the short in vitro lifespan of isolated
tubules prepared, by any technique, for studying early toxic effects.
In general, most investigators limit incubations to no more than 2-4
h because of loss of viability and functional capabilities. Ormstad
(1982) reported rapid loss of viability of isolated renal tubules and
cells, more than 25% of the cellular lactate dehydrogenase (LDH)
leaking to the medium during 1.5 to 2 h of incubation. Obatomi &
Plummer (1986) observed a 40% loss in tubule cell viability during 3-h
incubations of rat proximal tubules. Loss of renal function, such as
O2 consumption, has also been reported for isolated tubules (Harris et
al., 1981).
A number of fresh tubular systems exhibiting high initial
viability (> 90% by trypan blue exclusion) have been prepared from
collagenase digests of rat cortical tissue (Cunnaro & Weiner, 1978;
Belleman, 1980; Cojocel et al., 1983; Gstraunthaler et al., 1985;
Obatomi & Plummer, 1986) using tubular fragments of proximal origin.
The tubules can be used to study the metabolism of xenobiotics liable
to be converted into compounds responsible for the alteration of
normal renal metabolism (Jones et al., 1979). Tubular fragments
obtained by collagenase treatment of dog (Baverel et al., 1978,
1980a), human (Baverel et al., 1979), baboon (Michoudet & Baverel,
1987), and guinea-pig (Baverel et al., 1980b) renal cortex have also
been used for metabolic studies. These fragments retain the
gluconeogenic capacity that is specific to the proximal convoluted
tubule (Guder & Ross, 1984). Human, dog, and rat renal cortex tubules
also release ammonia from glutamine, and any drug-induced disturbance
of renal ammoniagenesis can be studied with these models (Martin et
al., 1987, 1989, 1990).
Proximal tubules have been purified from the above preparations
by centrifugation on a Percoll density gradient. The samples obtained
exhibit enrichment in proximal tubule cells relative to cells from
other areas of the nephron, as indicated by the distribution of
alkaline phosphatase and hexokinase (Vinay et al., 1981).
Suspensions of thick ascending limb fragments have also been
prepared from dog (Baverel et al., 1980a,b; Anand-Srivastava et al.,
1986), rat (Trinh-Trang-Tan et al., 1986), and rabbit (Chamberlin et
al., 1984) outer medulla. Suspensions of collecting tubules obtained
from the inner medulla (Anand-Srivastava et al., 1986; Wirthensohn
et al., 1987, 1989) may prove very useful in studies of the effect of
nephrotoxic substances that interfere with the function of this renal
zone.
Direct addition of different concentrations of nephrotoxic agents
such as ochratoxin A, citrinin, furosemide, and potassium chromate to
the suspension releases enzymes specific to the proximal tubule (Table
4), such as alanine aminopeptidase, leucine aminopeptidase and
alkaline phosphatase, to the incubation medium in a dosedependent
manner (Endou et al., 1985). To characterize further the intrarenal
site(s) and mode of nephrotoxicity, definite portions of a single
nephron can be microdissected from the collagenase-treated kidney.
There are two different methods available for the microdissection of
individual nephron segments; one is from lyophilized kidney sections
and the other is from collagenase-treated fresh kidneys (Morel et al.,
1976). In nephrotoxicity studies, fresh individual nephron segments
are used. The following segments can be isolated: the glomerulus, the
proximal tubule (S1, S2, S3), the thin descending limb of
Henle's loop, the medullary and cortical thick ascending limb of
Henle's loop, the distal convoluted tubule, the connecting tubule, and
the cortical and medullary collecting tubules.
Several functional parameters can be studied in nephron segments.
Gluconeogenesis is a unique function of the proximal tubule, within
which the S1 segment is most active (Maleque et al., 1980; Endou et
al., 1985). Gluconeogenesis is strongly induced by metabolic acidosis
or by alpha1-adrenergic stimulation (Nakada et al., 1986a).
First-generation cephalosporins, cephaloridine and cephalothin cause
a time- and concentration-dependent decrease in gluconeogenesis, and
it is clearly indicated that the site of nephrotoxicity of these
antibiotics is the proximal tubule (S1, S2, S3). Ammoniagenic
activity is distributed in all the nephron segments, but the highest
production rate of ammonia from glutamine is observed in the proximal
tubule (Nonoguchi et al., 1985). Ammoniagenesis via the purine
nucleotide cycle from asparatate as a substrate is also high in the
proximal tubule (Tamura & Endou, 1988). Ammonia production is
increased in a similar way by metabolic acidosis or potassium
depletion (Nonoguchi et al., 1986). Cisplatin nephrotoxicity is
morphologically known to be focussed on the S3 segment. However, this
drug decreases ammoniagenesis from glutamine not only in S3, but
also in S2, suggesting a discrepancy between morphological and
biochemical evaluations, although cisplatin does not affect
gluconeogenesis in isolated nephron segments (Nakada et al., 1986b).
The kidney possesses various active transport processes that
consume ATP at a high rate. Individual nephron segments require their
own particular substrates for synthesizing the necessary ATP: this has
been shown in both mice (Uchida & Endou, 1988) and rats (Jung et al.,
1989). The proximal tubule cannot use glucose to produce ATP, whereas
the other nephron segments can use it. In general, pyruvate or lactate
is the preferred substrate in all segments. Nephrotoxicity assessment
by measuring cellular ATP content shows clearly that mercuric chloride
decreases ATP content only in S2 (Jung et al., 1989) and that
ochratoxin A nephrotoxicity localizes in S2 and S3 (Jung & Endou,
1989). Thus, measurement of cellular ATP in specific nephron segments
enables possible nephrotoxicants to be evaluated. A similar principle
can be applied by measuring intracellular free calcium (Jung & Endou,
1990). From the biological point of view, it is essential to keep
cellular ATP at a high level and to maintain a low concentration of
intracellular free calcium for all living cells. It should,
therefore, be reasonable and useful to introduce these sensitive
parameters to nephrotoxicity assessment, although the methods require
special techniques for microdissecting nephron segments or special
instruments.
An advantage of the use of isolated tubules, as compared to in
vivo experiments, is that it permits a cellular environment that is
defined both quantitatively and qualitatively. This allows the
relationship between the concentration of a nephrotoxin, exposure
time, and effect to be studied. Extrarenal effects can be avoided, and
so isolated tubules are very suitable for studying the effects of
nephrotoxins that act directly at the tubular site. Owing to a lack
of polarity (Koseki et al., 1988), isolated renal cell suspension may
have limited usefulness.
There are several limitations associated with the use of freshly
isolated or cultured renal cells. Cells released by enzymic digestion
or fresh fragments can be cultured in the presence of serum-free,
hormonally defined culture media. This prevents fibroblast
proliferation and encourages epithelial cell growth (Chuman et al.,
1982), but both cell preparations lack a brush border, which may be
critical for the active uptake of drugs and chemicals.
Alternative approaches to obtain a preparation with an intact
brush border include the use of different sized sieves to separate
glomeruli and tubules (Bach et al., 1986), which may not result in a
pure preparation of proximal tubular cells. The choice of method for
monitoring cell viability may circumvent this, e.g., the use of
prostaglandin synthesis (Sraer et al., 1980) to assess effects of
chemicals selectively on glomeruli. The density gradient technique
(Vinay et al., 1981) uses Percoll centrifugation to separate the
different cell types to obtain a > 90% pure preparation of proximal
tubular cells. These cells retain their viability and their GSH levels
at > 50% for 2 h at 37 °C, and demonstrate cytochrome-P450-dependent
mono-oxygenase activity profiles that are inducible only by
3-methylcholanthrene (3MC). This may be an appropriate preparation to
study the effects of various drugs and chemicals, in both rats and
man, that demonstrate nephrotoxicity to either the S1, S2, or S3
regions after administration (Smith et al., 1986; Rosenberg &
Michalopoulos, 1987).
7.1.2.4 Primary cell cultures
Cell to be cultured should be of well defined origin. For this,
purification of a homogeneous glomerular or tubular cell population
can be achieved by several methods (Jakoby & Pastan, 1979), including
sieving techniques (Striker et al., 1980), magnetic and mechanical
techniques (Meezan & Brendel, 1973), density gradient centrifugation
(Scholer & Edelman, 1979; Vinay et al., 1981), and collagenase
digestion (Curthoys & Bellemann, 1979; Belleman, 1980; Ormstad et al.,
1981). More recently, techniques such as immunodissection, cell
sorting, free-flow electrophoresis, and microdissection have been used
(Pretlow & Pretlow, 1982, 1983, 1984). The advantage of using primary
cell cultures is that it allows long exposure to xenobiotics and the
choice of appropriate metabolites. In addition, it is possible to
monitor a variety of cell functional, biomedical, or morphological
responses in a dose- and time-related manner (Fry et al., 1978;
Belleman, 1980; Fry & Perry, 1981; Bach et al., 1986).
Mechanical or enzymic dispersal may damage cells, and once cells
are dispersed it is generally difficult to establish their anatomical
identity unless suitable markers are used. These markers include both
the presence and absence of a range of functional and biochemical
characteristics, such as transport systems, and an array of structural
and functional molecules. These can best be assessed by a variety of
histochemical and immunocytochemical methods (Bach et al., 1985,
1987). At present, isolated cells are generally mixtures (although
they may be enriched) and must be used within a few hours. Primary
cell cultures may rapidly dedifferentiate (Curthoys & Bellemann, 1979)
or adapt to a new environment and change their characteristics as a
result of the presence or absence of factors in the culture media,
which may obfuscate their anatomic origins. More importantly, loss of
a biochemical characteristic that is part of the molecular basis for
target cell toxicity may invalidate in vitro studies. Changes in
other aspects of cellular integrity can increase or decrease both the
sensitivity and selectivity of screening methods used for cytotoxicity
studies.
Two approaches have been used to modulate the expression of cell
characteristics. The polarity of epithelial cells is better expressed
when cell are grown on permeable supports such as collagen/filters
(Jakoby & Pastan, 1979). Similarly, the appropriate modulation of
culture media has been used to alter rabbit proximal tubule cell
metabolism to the gluconeogenic pathway and these cells then develop
brush-border characteristics. Thus, media can be an important
variable, especially because of the diverse combination of buffers and
growth supplements used. There are major advantages in using fully
defined culture media (Sato & Reid, 1978), but these have not been
widely adopted.
Human proximal tubules have been shown to been sensitive to
cyclosporin A (Trifillis et al., 1986), but there are no data on the
mechanistic bases of these changes. Rat, rabbit, dog, and human
glomerular mesangial and epithelial cells may be co-cultured or each
type derived separately (Kreisberg et al., 1977, 1978; Foidart et al.,
1979, 1980, 1981; Morita et al., 1980; Striker et al., 1980; Kreisberg
& Karnovsky, 1983). Rat epithelial cells are more sensitive to
puromycin aminonucleoside and Adriamycin than are mesangial cells, as
is the case in vivo, but there is little mechanistic information.
Rat medullary interstitial cells can be cultured at high osmolality
and have been shown to be sensitive to a number of compounds that
cause renal papillary necrosis.
7.1.2.5 Established renal cell lines
Several established renal cell lines have been studied that have
properties reminiscent of specific parts of the nephron, such as
LLC-PK1 (of proximal tubule type) and MDCK (of distal tubule type).
The major disadvantage is that the exact site of origin, within the
nephron, of each, is not known, and it may not totally represent the
normal physiological state. However, these lines are often
heterogeneous and there is a need to characterize them more
systematically so as to establish where they may be useful in
screening chemicals for toxicity or in understanding the mechanisms of
target cell toxicity.
Differences exist between the apical and basolateral membrane
transport of substances into cells, which may be central to the
mechanism of nephrotoxicity. When cells are cultured on solid
surfaces, only apical exposure to chemicals occurs, whereas in vivo,
proximal tubule cells are exposed from the apical or basolateral sides
or both. This disadvantage can be overcome by culturing renal cells on
microporous membranes suspended in culture wells. These cells, which
form a confluent single-cell monolayer covering the membrane within
some days, more closely mimic the in vivo state than those grown on
plastic plates. They show anatomical and functional polarization. This
culture technique allows access to the cell monolayer from both the
apical and the basolateral sides, and apical and basolateral fluid may
be studied simultaneously. This new experimental tool allows the study
of transport and epithelial resistance across the cell monolayer and
polarized uptake of various molecules, including potentially
nephrotoxic drugs, as well as to perform a variety of analytical
techniques.
The various cell lines used in nephrotoxicity studies have been
reviewed by Wilson (1986). The LLC-PK1 cell lines have a typical
epithelial polarity and have features similar to proximal tubular
epithelium, such as transport systems (Handler, 1983) and the enzyme
marker gamma-glutamyltranspeptidase (Perantoni & Berman, 1979).
Confluent LLC-PK1 cells cultured on a solid support form domes (due to
transcellular transport), but monolayers grown on a porous membrane do
not. More importantly these cells have polarity and have a
well-developed brush border. Confluent LLC-PK1 monolayers exposed to
PCBD-GSH from the apical side are more sensitive than when exposed
from the basolateral side. This is due to the brush border
localization of gamma-glutamyltranspeptidase, which catalyses the
first step of the breakdown of the conjugate to the ultimate reactive
intermediate. Neither apical nor basolateral treatment with PCBD-NAC
elicits any toxicity. It is assumed that the absence of an organic
anion transporter from these cells could explain this finding, since
it has been established that haloalkene conjugates enter cells via the
basolaterally located anion transporters (Lock et al., 1986). The
absence of an organic anion transport system limits the usefulness of
LLC-PK1 cell lines for studying nephrotoxic compounds, such as
PCBD-NAC, that need active transport to enter the cells. However, an
active basolateral organic cation transport system
(gamma-glutamyltranspeptidase and dipeptidase) makes these cells
especially useful for testing compounds that have a toxic action on
these transport systems.
7.1.2.6 Subcellular fractions
It is also possible (and sometimes desirable) to use homogeneous
or fractionated organelles, membranes, or cytoplasm from defined
cells for specific cell-free investigations. The constraints on the
preparation of these systems should be apparent from the foregoing
discussion. Subcellular fractions, such as vesicles, nuclei,
lysosomes, and microsomes, can be used to study subcellular
distribution, the interaction between a cellular compartment and a
chemical, and the kinetics of binding or release of substances. It is
also possible to study specific effects, such as enzyme inhibition,
metabolic activation, covalent binding, or the modulation of lipid
peroxidation, using purified or commercially available biochemicals
with appropriate cofactors and suitable techniques for monitoring
these interactions (Bach & Bridges, 1985b, 1987).
Many nephrotoxic agents interact with cell membranes, where they
bind with receptors, effect transport systems, or disrupt structure
and function per se. Thus, membrane vesicles may be useful for
studying these interactions and the mechanisms of cell injury. It is
possible to isolate vesicles from the brush border and basement
membranes to study transport systems at each site in vitro. Williams
et al. (1986) showed a very good correlation between the in vitro
binding of aminoglycosides to brush-border membrane vesicles and their
in vivo nephrotoxicity. Inhibition of aminoglycoside membrane
binding by polyaspartate reduces nephrotoxicity and suggests that
binding of these antibiotics to brush-border phospholipid may be a
crucial event in nephrotoxicity.
7.2 in vivo experimental studies
Current methods for diagnosing renal injury and predicting the
health significance are not sufficient to deal with the diversity of
possible chemical injuries (for full discussion, see Bach et al.,
1989). This is because the kidney can undergo substantial chemically
induced injury without any clinical indication, since subtle injury
may be buffered within the considerable functional reserve. This masks
a substantial amount of renal degeneration (Friedlander et al., 1989).
Thus, for example, the single cross-sectional measurement of GFR may
only show incipient acute or chronic renal failure. Quantitative
urinary enzyme excretion patterns cannot identify either the type or
severity of renal injury, and often they do not correlate with
morphological and functional changes (Schentag et al., 1978).
There are a number of inherent difficulties in diagnostic
procedures for nephropathy, which include the absence of standard
diagnostic criteria and the inability to relate exposure to a given
agent and the observed effect. In addition, renal functional reserve
is a major factor that masks renal degeneration, as assessed by GFR,
blood urea nitrogen, and creatinine, up to the point where over 75% of
the functioning nephrons have been lost. Thus, it should be stressed
that these factors measure incipient renal failure and that the fact
that values are normal (something that is subject to age-related
change and varies between the two sexes) does not signify the absence
of renal dysfunction or even, in some cases, gross renal
insufficiency. Therefore, cause and effect cannot be clearly
established on the basis of available knowledge when the renal lesion
results from a multifactorial process with a long latency. Part of
this uncertainty can be addressed by studies on experimental animals.
7.2.1 Methods for assessing chemically reactive nephrotoxic
metabolites in animals
It is known that many nephrotoxicities that follow the
administration of inert, relatively nontoxic chemicals are related to
the formation of reactive electrophiles during the metabolism of these
chemicals (Ford & Hook, 1984). These electrophilic products can react
covalently with nucleophilic sites on renal macromolecules such as
protein, lipid, and DNA. The covalent binding may be measured by the
use of radiolabelled forms of the chemical, by immunological detection
of DNA/protein adducts (Harris et al., 1987), or by the 32P-DNA
postlabelling method (Reddy et al., 1984). Furthermore, chemicals that
are metabolized to DNA-damaging intermediates may be detected in vivo
by measuring the alkaline elution of isolated kidney nuclei
(Omichinski et al., 1987; Brunborg et al., 1988) or unscheduled DNA
synthesis in isolated kidney cells (Tyson & Mirsalis, 1985) after in
vivo exposure of animals.
7.2.2 Evaluation of glomerular function
Evaluation of blood urea nitrogen is probably the most common
procedure for indirectly evaluating GFR in experimental animals.
Although insensitive, this test may be sufficient to establish the
time course of chronically developing renal failure in the
experimental setting. Serum creatinine is also used for the same
purpose. However, owing to interferences from nonspecific chromogens,
this test is unreliable in most experimental animals and especially in
the rat. Although this problem may be overcome, it has not been dealt
with adequately in most available studies, thus generating wide
scatter in "normal" ranges.
In animals, more subtle changes in GFR occurring during
subchronic and chronic studies should be assessed by evaluating the
clearance of exogenous substances such as inulin, EDTA, or
iothalamate. The latter may be determined either by measuring the
radioactivity of labelled material or by means of reliable HPLC
methods (Prueksaritanont et al., 1984). Furthermore, the same HPLC
method may be used to measure PAH and to assess other haemodynamic
parameters. Two (or more) clearance periods should be calculated and
averaged in order to ensure greater accuracy.
There is a growing body of evidence to suggest that reduced renal
reserve due to hypertension/hyperfunction/hyperfiltration of remnant
nephrons is important in the course towards end-stage renal disease.
This can in part be lowered by reducing protein intake and blood
pressure. The concept of renal functional reserve includes the
evaluation of renal blood flow and GFR by measuring their increase
after protein load or the administration of aminoacids, glucagon, or
vasodilatory drugs. At present such tests do not have defined
standardized stimuli, and there are no data on their use for detecting
nephrotoxicity.
7.2.3 Evaluation of tubular functions
In experimental animals, tubular dysfunctions are usually
detected through simple and inexpensive tests, such as those of
glycosuria, enzymuria, and osmolality, which may provide other useful
information. Some of these tests are sensitive enough to detect acute
tubular damage, although caution must be exercised in predicting
specific effects on transport processes or cell viability on the basis
of data obtained from in vivo experiments (Berndt, 1981). More
subtle renal changes occurring during chronic studies may be evaluated
by measuring the renal clearance of lithium (Dieperink et al., 1983;
Daugaard et al., 1988b). This non-invasive method is applicable both
to human (Thompson et al., 1984) and animal studies. The loss of renal
tubular functions can be assessed by test procedures that impose one
or more stressing conditions to force compensatory changes, e.g.,
maximal urinary dilution/concentration or
acidification/alkalinization, and maximal tubular reabsorption of
glucose and phosphate. The value of these tests is limited for group
studies by practical considerations but may be useful for individuals.
7.2.4 Proteinuria
Proteinuria is the loss of proteins following increased
permeability of the glomerular barrier, reduced tubular reabsorption
of filtered proteins, or shedding of specific constituents into the
urine as a consequence of cellular turnover or selective tissue
damage. Since pathological changes either at the glomerular or
tubulo-interstitial level may occur even in the absence of a
substantial reduction in GFR, the evaluation of proteinuria may also
be useful in some circumstances to detect renal dysfunction occurring
either at the glomerular or at the tubular level. Proteins may be
measured by nonspecific assays, by immunochemical methods or by their
enzymic activity. Sensitive methods have been developed to detect
small amounts of proteins in microlitre quantities of unconcentrated
urine.
7.2.4.1 Total proteinuria and electrophoretic pattern
Measurement of total proteinuria and electrophoretic separation
of single proteins provide a comprehensive approach to chemically
induced renal dysfunction.
The rationale for such an approach relies on the
pathophysiological mechanisms controlling the renal handling of
plasmaproteins. Proteins of high relative molecular mass (> 45 000
Daltons) are usually confined to the vascular compartment by basal
membranes. Furthermore, the glomerular polyanion acts as a selective
filter that retains negatively charged proteins, such as albumin,
because of electrostatic interactions. The glomerular pore size is
thought to have an important role in retaining proteins of higher
relative molecular mass (e.g., immunoglobulins). Proteins with lower
relative molecular mass (< 45 000 Daltons) pass the glomerular
barrier with sieving coefficients inversely related to their mass.
Filtered proteins of low relative molecular mass are efficiently taken
up by the proximal tubules (more than 99%). Even slight decreases in
tubular fractional reabsorption due to tissue damage or dysfunction
will result in increased low relative molecular mass proteinuria (Fig.
16).
Intranuclear inclusion bodies are uncommon in the late stages of
lead nephropathy, although they may be seen in renal biopsy or autopsy
as a manifestation of a super-imposed severe acute exposure. It has
been shown that inclusion bodies may be found in the urine of workers
with occupational exposure to lead, but their presence or absence in
urine has not been related to the severity of lead nephropathy
(Schumann et al., 1980).
5.5.2 Cadmium
Cadmium is an occupational and environmental contaminant that has
received a great deal of attention. An important toxicological
feature of cadmium is its exceptionally long biological half-life in
the human organism (10-30 years). Once absorbed, cadmium is
efficiently retained in the organism and accumulates throughout life.
In the newborn baby, cadmium is present only at very low levels, but
by the age of 50 the cadmium body burden may have reached up to 20-30
mg and, in people occupationally exposed, it may reach values as high
as 200-300 mg. Furthermore, cadmium concentrates in vital organs,
particularly in the kidneys. At low levels of exposure, such as those
prevailing in the general environment, 30-50% of the cadmium body
burden is found in the kidneys alone (Nomiyama, 1980; Bernard &
Lauwerys 1986; Friberg et al., 1986).
The accumulation of cadmium in the kidney, may give rise to a
progressive form of tubulo-interstitial nephritis. In contrast to the
situation with many nephrotoxins, including other heavy metals such as
lead and mercury, there are virtually no acute effects of inorganic
cadmium salts on the kidney, except perhaps for some nonspecific
effects that have been seen in animals given near-lethal doses. One of
the most challenging questions regarding the metabolism of cadmium has
been the role of metallothionein in cellular metabolism and its
potential toxicity. Metallothionein synthesized within the kidney
protects from cadmium toxicity, but intravenously injected
cadmiummetallothionein is more nephrotoxic than inorganic cadmium (see
section 4.5.3.).
Cadmium nephropathy was first described by Friberg (1948, 1950)
who studied a group of alkaline battery workers in Sweden during the
late 1940s. Since these reports, a number of epidemiological studies
have shown the occurrence of nephrotoxic effects in populations
exposed to cadmium at work and in the general environment. These
studies have demonstrated that the most prominent feature and probably
the earliest sign of cadmium nephropathy is increased proteinuria.
Studies performed in the 1950s and 1960s (reviewed by Friberg et
al., 1986 and Bernard & Lauwerys, 1986) showed that cadmium
proteinuria is similar to the tubular-type proteinuria described by
Butler & Flynn (1958) in patients with tubular disorders, and consists
of unidentified proteins of low relative molecular mass derived from
plasma. Characterization of these proteins led to the discovery of
ß2-microglobulin, retinol-binding protein, and alpha1-microglobulin.
Subsequent studies demonstrated that the increased urinary excretion
of proteins of low relative molecular mass observed in cadmium
nephropathy and other renal disease was due to the failure of the
proximal tubules to reabsorb proteins filtered through the glomeruli.
The effects of cadmium on the excretion of ß2-microglobulin have
been extensively documented (Bernard et al., 1976, 1979a,b, 1982,
1987). Measurement of retinol-binding protein is much more reliable in
acidic urine and detects tubular proteinuria with equal sensitivity
(Bernard et al., 1982).
As cadmium nephropathy progresses, it increasingly presents the
signs of a complete Fanconi's syndrome, i.e. aminoaciduria,
glucosuria, increased urinary excretion of calcium, phosphorus, and
uric acid, and decreased concentrating ability of the kidneys. In the
most severe cases, the GRF decreases. The disturbances in calcium and
phosphorus metabolism may lead to a demineralization of the bones and
the formation of kidney stones (Friberg et al., 1974).
In cadmium-polluted areas of Japan, signs of renal dysfunction
very similar to those observed in cadmium workers have been frequently
found. A higher incidence of proteinuria, glucosuria, and
aminoaciduria, and increased excretion of ß2-microglobulin have been
observed in the Zinzu river basin in Toyama where Itai-Itai disease
was first seen (Fukushima et al., 1974; Kjellström et al., 1977;
Shiroishi et al., 1977; Kjellström & Nordberg, 1978). In the endemic
area of Toyama, the increased urinary excretion of ß2-microglobulin
was strongly related to the residence time in that area as well as to
the purposes for which contaminated river water was used (Kjellström
et al., 1977). In urine, ß2-microglobulin concentration correlated
with the cadmium level (Nogawa et al., 1979a,b).
Further investigations of the renal function of the inhabitants
in this area revealed a significant decrease in both creatinine
clearance and renal phosphorus reabsorption. Renal dysfunction due to
chronic cadmium poisoning was also found in other areas of Japan where
the rice was contaminated by cadmium (Saito et al., 1977; Kojima et
al., 1977). Studies carried out in Belgium suggested that
environmental exposure to cadmium in an industrialized area polluted
by this metal may exacerbate the age-related decline of renal function
in elderly residents (Lauwerys et al., 1980; Roels et al., 1981a,b).
Since cadmium-induced nephropathy may occur within the general
population, it is of major public health importance to know what level
of cadmium exposure carries a risk of renal tubular dysfunction and
cadmium nephropathy.
The concept of a critical concentration of cadmium has very
important implications with regard to establishing maximum levels of
cadmium that human populations may be exposed to with some margin of
safety. From a comparison of the cadmium concentrations in the renal
cortex of cadmium-exposed people with and without signs of kidney
damage, Friberg et al. (1974) suggested that the critical level of
cadmium in the renal cortex for the appearance of tubular proteinuria
is around 200 mg/kg. With the development of neutron activation
techniques allowing the in vivo determination of cadmium in
tissues, the critical level of cadmium in the human kidney has been
more precisely assessed.
Investigations conducted by Roels et al. (1981a) in Belgium and
Ellis et al. (1981) in the USA have shown that when the concentration
of cadmium in the kidney cortex reaches about 200 mg/kg, signs of
renal dysfunction (e.g., increased urinary excretion of albumin and
ß2-microglobulin) develop in about 10% of male workers exposed to this
metal. On the basis of the relationship between the concentrations of
cadmium in the urine and renal cortex and the prevalence of renal
anomalies, the critical concentration of cadmium in urine has been
estimated to be 10 µg/g creatinine (Bernard et al., 1979a,b; Buchet et
al., 1980; Roels et al., 1981a,b). Epidemiological studies of people
living in cadmium-polluted areas of Japan have shown that
ß2-microglobulinuria occurs after a lifetime accumulation of 2000 mg
cadmium or more (Nogawa et al., 1989).
Since several studies have shown that, in most cases, once
cadmium proteinuria has developed, it is irreversible, the progression
of renal dysfunction after cessation of exposure is very slow (Roels
et al., 1982; Elinder et al., 1985a,b). Persistent proteinuria is
found frequently among retired cadmium workers with no evidence of
renal insufficiency. In a group of workers removed from exposure after
the finding of microproteinuria (low or high relative molecular mass),
the reduction in GFR during a 5-year follow-up was about five times
greater than that accounted for by aging (Roels et al., 1989).
5.5.3 Mercury
It has been known for a long time that patients treated with
mercurial compounds can develop a glomerulonephritis that is usually
of the immune complex type (Becker et al., 1962; Druet et al., 1982).
Cases of mercury glomerulonephritis have also been reported as a
result of chronic exposure to high levels of mercury in industry
(Tubbs et al., 1982). Patients with mercurial nephropathy usually
present with a proteinuria and occasionally a nephrotic syndrome, but
no renal insufficiency (Druet et al., 1982).
Mercury may produce different effects on the kidney depending on
the biochemical form of the metal and nature of exposure. Inorganic
mercury compounds are classic examples of agents that cause acute
tubular necrosis. Mercuric chloride was used as a suicidal agent
during the nineteenth and early part of the twentieth centuries but
was unpopular for this purpose because of the painful accompanying
corrosive injuries it produced.
Regardless of the route of administration, mercuric chloride
produces acute tubular necrosis within hours of administration,
resulting in anuria and death. If the patient can be maintained by
dialysis, regeneration of tubular lining cells is possible. These may
be followed by ultrastructural changes consistent with irreversible
cell injury, including actual disruption of mitochondria, release of
lysosomal enzymes, and rupture of cell membranes.
The necrosis of the epithelium of the pars recta following
injection of mercuric chloride has been described in detail in the
rat. Cellular changes include fragmentation and disruption of the
plasma membrane and its appendages, vesiculation and disruption of the
endoplasmic reticulum and other cytoplasmic membranes, dissociation of
polysomes and loss of ribosomes, mitochondrial swelling with
appearance of amorphous intramatrical deposits, and condensation of
nuclear chromatin. These changes are common to renal cell necrosis
resulting from a variety of causes (Gritzka & Trump, 1968).
Mercury and its compounds are used widely, not only in various
industrial processes but also in a number of other applications such
as fungicides, contraceptive spermicides, and disinfectants. Several
studies have been carried out to determine the extent to which current
exposure of human populations to mercury can cause adverse renal
effects. Foa' et al., (1976) reported an increased prevalence of
glomerular proteinuria in workers exposed to mercury vapour in a
chloralkali plant.
Studies carried out between 1979 and 1984 (Buchet et al., 1980;
Roels et al., 1985) provided further evidence that occupational
exposure to mercury vapour can lead to subclinical renal
disturbances. These consisted of increased urinary excretion of
proteins of high relative molecular mass (albumin, transferrin, and
immunoglobulin G), lysosomal enzymes, and retinol-binding protein,
which occurred at a higher prevalence in subjects who excreted more
than 50 µg mercury/g creatinine. These observations were not confirmed
by Stonard et al. (1983), who found only a slight increase in the
prevalence of NAG and gamma-glutamyltranspeptidase (an enzyme of the
brush-border) in workers with urinary mercury levels higher than 100
µg/g creatinine.
Increased urinary excretion of NAG has also been found in workers
involved in the production of various mercuric salts (Rosenman et al.,
1986). Studies on patients with Minamata disease have provided
inconsistent results regarding the induction of proximal tubular
injury by methylmercury (Iesato et al., 1977; Ohi et al., 1982). By
contrast, in a study of 509 infants exposed to phenylmercury fungicide
on cloth diapers, Gotelli et al. (1985) clearly demonstrated that the
kidney is a target organ during prolonged exposure to this compound.
They showed that the urinary excretion of
gamma-glutamyl-transpeptidase increased in a dose-dependent manner
when urinary mercury exceeded approximately 220 µg/litre. This effect
was, however, completely reversible and had disappeared when the
infants were re-examined two years later.
Although exposure to a high dose of mercuric chloride is directly
toxic to renal tubular lining cells, chronic low-dose exposure to
mercuric salts or even elemental mercury vapour may induce an
immunological glomerular disease. This form of mercury injury to the
kidney is clinically the most common form of mercury-induced
nephropathy. Exposed workers may develop a proteinuria that is
reversible after they are removed from exposure. It has been stated
that mercury-induced nephropathy seldom occurs without sufficient
exposure to produce detectable mercury neuropathy as well.
Experimental studies have shown that the pathogenesis of mercury
nephropathy has two phases: an early phase characterized by an
anti-basement-membrane glomerulonephritis followed by a superimposed
immune-complex glomerulonephritis (Roman-Franco et al., 1978). The
pathogenesis of the nephropathy in humans appears similar, although
antigens have not been characterized. Also, the early
glomerulonephritis may progress in humans to an interstitial
immune-complex nephritis (Tubbs et al., 1982).
5.5.4 Gold
The use of gold in the form of organic salts to treat rheumatoid
arthritis may be complicated by development of proteinuria and the
nephrotic syndrome (Hall et al., 1987). Morphologically, the kidney
shows an immune-complex glomerulonephritis with granular deposits
along the glomerular basement membrane and in the mesangium. The
pathogenesis of the immune-complex disease is not known for certain,
but gold may behave as a hapten and generate the production of
antibodies with subsequent deposition of gold protein-antibody
complexes in the glomerular subepithelium. Another hypothesis is that
antibodies are formed against damaged tubular structures, particularly
mitochondria, providing immune complexes for the glomerular deposits
(Viol et al., 1977).
The pathogenesis of the tubular cell lesions induced by gold
therapy is probably initiated by the direct toxicity of gold to
tubular cell components. From experimental studies it appears that
gold salts have an affinity for the mitochondria of proximal tubular
lining cells. This is followed by autophagocytosis and accumulation of
gold in amorphous phagolysosomes (Stuve & Galle, 1970). Gold particles
can be identified in degenerating mitochondria, in tubular lining
cells, and in glomerular epithelial cells by X-ray microanalysis
(Ainsworth et al., 1981).
5.5.5 Bismuth
The effects of bismuth on the kidney are similar to those of
lead, but it is a less frequent cause of renal disease. This is
because bismuth is not present in such large amounts in the ambient
environment, nor is it as important industrially. However, bismuth has
been used therapeutically to treat a variety of ailments, most
particularly syphilis. Bismuth administration results in the formation
in proximal renal tubular lining cells of characteristic nuclear
inclusion bodies that are similar to the lead-induced bodies and are
composed of a bismuth-protein complex. The protein is acidic and has
an amino acid composition similar to that forming the lead inclusion
bodies. However, there is a slight difference in morphology between
the lead- and bismuth-induced inclusion bodies. The bismuth-protein
complexes are also observed in the mitochondria of proximal tubular
lining cells (Fowler & Goyer, 1975). The bismuth content of the
inclusion bodies has been confirmed by X-ray microanalysis of tissue
sections. Whether bismuth produces a chronic interstitial nephropathy
like lead has not yet been documented. However, bismuth inclusions
have been found at autopsy more than 30 years after a course of
bismuth therapy.
5.5.6 Uranium
Exposure of humans or experimental animals to compounds of
uranium results in injury and necrosis of proximal renal tubules. The
most sensitive site is the pars recta (as in the case of mercury),
but, depending on the dose, injury and necrosis may extend to other
parts of the proximal tubule. Acute injury is followed by regeneration
of tubular epithelial cells. Chronic effects have not been reported.
An increase in the urinary excretion of ß2-microglobulin and of
specific amino acids has been reported by Thun et al. (1985) in
uranium mill workers.
5.5.7 Chromium
The acute and chronic effects of chromium (mainly on the
respiratory tract and skin) are due largely to hexavalent compounds.
The acute tubular toxicity of chromate and dichromates salts in
animals is well documented, and renal tubular necrosis has also been
described in humans following acute poisoning (Langard & Norseth,
1986). Epidemiological studies have shown that chromium(VI) can
produce slight tubular dysfunction in chronically exposed workers.
Mutti et al. (1979) reported an increased prevalence of elevated
ß-glucuronidase and total protein levels in the urine of welders
exposed to chromium. These observations have been confirmed by recent
studies using more sensitive, reliable markers of tubular injury, such
as ß2-microglobulin (Lindberg & Vesterberg, 1983), retinol-binding
protein, and the BB-50 renal antigen (Mutti et al., 1985).
Franchini & Mutti (1988) have studied dose-effect/response
relationships between the urinary excretion of chromium and that of
retinol-binding protein or the renal antigen BB-50. Most of the
abnormal values were observed in subjects with urinary excretion of
chromium greater than 15 µg/g creatinine; however, above this
threshold the degree of tubular impairment was not related to urinary
excretion of chromium. Franchini & Mutti (1988) explained this
phenomenon by postulating that the tubular damage observed in
chromium(VI)-exposed workers is transient and due mainly to acute
exposure, and that workers become progressively resistant to the
effects of more severe or prolonged exposure.
5.5.8 Arsenic
Acute arsenic poisoning may cause tubular necrosis. Acute or
severe chronic poisoning is usually treated with the chelating agent
BAL (2,3-dimercaptopropanol). Inhalation of arsine may also produce an
acute tubular necrosis as a result of intravascular haemolysis.
In a cross-sectional study, Foa' et al. (1987) failed to show
significant differences between occupationally exposed workers and
matched controls, with the exception of a slight increase in the
urinary excretion of retinolbinding protein. However, owing to the
small sample size and the low power of the study, no definite
conclusion could be drawn from the slight increases in albuminuria,
ß2-microglobulin, and the brush-border antigen BB50. The authors
concluded that extended population surveys would be desirable for a
complete definition of such subtle effects.
5.5.9 Germanium
Germanium is naturally present in the diet, normal intake being
about 1 mg per day. It is being used increasingly in the semiconductor
industry. A recent report from Japan documented renal failure in ten
individuals (including two deaths) among previously healthy
individuals taking large doses of germanium (of the order of 50-250 mg
per day) over periods of 4-18 months (Matsusaka et al., 1989). Renal
biopsy or autopsy in seven cases showed degeneration of the renal
tubular epithelium in all cases with or without interstitial fibroses
or oedema. The glomeruli were only minimally effected in two cases.
6. RENAL CANCER
Tumours of the renal parenchyma, pelvis, and ureters are
uncommon, accounting for less than 2-3% of all human cancers
(DeKernion & Berry, 1980; Dayal & Kinman, 1983). The role of drugs,
chemicals, and other environmental factors in the etiology of
parenchymal and urinary tract tumours is unclear, but cancer of these
sites is most common in certain industrialized nations (Sweden) and in
people in the higher socio-economic groups (Rimpela & Pukkala, 1987).
Other risk factors have been stratified (Selli et al., 1983). The
ratio between tumours of the renal parenchyma and pelvis is fairly
constant (about 5:1), and the parallel trends in increasing incidence
argue for some commonality in the etiology of tumours at both sites,
although some factors may be site specific. Tumours are nearly twice
as common in males as in females. A compilation of trends in cancer
rates in the USA indicates that the incidence of both kidney and
bladder cancer is increasing (Pollack & Horm, 1980). A similar
observation has been made with regard to renal parenchymal tumours in
males in Scotland (Ritchie et al., 1984).
6.1 Renal tumour classification
The International Classification of Diseases for Oncology (code
189) divides tumours of the urinary system into five groups according
to their size, i.e. parenchyma of the kidney (189.0), renal pelvis
(189.1), ureter (189.2), urethra (189.3), and paraurethral gland
(189.4) (Mostofi et al., 1981; WHO, 1990). These distinctions have
only been made in recent years, so that many mortality studies of
renal tumours have included this whole category. Of the five types of
urinary tract tumours, about 90-95% of renal tumours in adults are
adenocarcinomas arising from the renal parenchyma. Nephroblastoma
(Wilm's tumour) is the second most common histological type of renal
tumour and accounts for 2-4% of kidney cancer in Sweden and the USA.
It is easily distinguished morphologically from renal adenocarcinoma
and usually appears in the first five years of life; 95% of cases
occur before the age of 15 years. Although nephroblastomas are the
fourth most common tumour in childhood, they are relatively rare in
adults.
6.2 Renal adenocarcinoma
Renal adenocarcinoma has been known under several synonyms (clear
cell carcinoma, hypernephroma, Grawitz tumour) reflecting uncertainty
about its origin, but immunological studies have established that
renal adenocarcinomas arise from the proximal convoluted tubule
(Wallace & Nairn, 1972). They tend to be circumscribed, ranging in
size from microscopic lesions to large neoplasms (Hamilton, 1975). The
spectrum from small benign lesions to clearly malignant lesions
suggests a continuous pathological process, so that it is often
difficult to label smaller tumours as benign or malignant. Hellsten et
al. (l983) have defined all tumours of 2 cm or more in diameter as
adenocarcinomas. Postmortem studies have shown that adenomas are
present in approximately 25% of all males over 50 years of age, and it
was found that 34% of 235 clinically unrecognized tumours present at
autopsy were less than 3 cm in diameter. In the absence of invasion of
surrounding tissue, features such as frequent mitotic figures,
cellular pleomorphism, and haemorrhage and necrosis generally indicate
a malignant potential regardless of size. Calcification may be
detected by X-ray examination in about 15% of cases. Although 2-3%
may be cystic, the commonest form is a solid tumour that is usually
composed of clear cells rich in lipid, glycogen, or both, but may
contain granular cells or even tightly packed eosinophilic cells
referred to as oncocytes. The cell pattern may be trabecular, solid,
or mixed, but it is doubtful that cell type or structural pattern has
any clinical significance. Grading is difficult and has not been shown
to be clinically useful. These tumours usually grow slowly, and
overall survival is 20-25% after nephrectomy. The presence of
multiple tumours, renal vein invasion, or regional lymph node
metastases indicates a poorer prognosis. Hellsten et al. (1983)
recorded metastasizing renal carcinoma as cause of death in 21% of a
postmortem series, and in 33% a second malignant tumour was observed
causing the death of 20%.
Immunological mechanisms are thought to determine the natural
history of the disease. The development of monoclonal antibodies and
flow cytometry have provided new methods for investigating
immunological responses. Total T lymphocyte counts were found in a
study of 32 patients to be lower than in controls (Ritchie et al.,
1984), due largely to a deficit of T helper cells but not T
suppressor-cytotoxic cells. This effect of the tumour is reversed by
removal of the primary tumour, and recurs with return of the tumour.
These findings are believed to suggest that there is a systemic effect
of the tumour acting at the level of the bone marrow or thymus to
affect the production or maturation of T helper cells.
The role of specific environmental factors in the etiology of
urinary tract tumours has been difficult to define (Newson & Vugrin,
1987). Apart from increases in renal tumours in asbestos workers and
the identification of some occupationally related bladder tumours,
there does not appear to be a clearly defined association with
specific chemicals or environmental factors. However, there is an
association with a combination of exposure to substances in the
environment and life-style practices such as tobacco use. This
suggests that there may be interactions between substances or that the
urinary tract, like the lung, has to deal with a number of substances
with promoter activity. Cigarette smokers have a 2-fold increase in
risk of urinary tract tumours (Goodman et al., 1986), and an increase
associated with alcohol and coffee usage has been suggested (Jacobsen
et al., 1986). In addition, chronic interstitial nephritis may
predispose to urinary tract tumours. People with endemic (Balkan)
nephropathy have an increase in renal tumours and a possible
relationship between chronic interstitial nephritis and renal
neoplasia (see section 5.3). It is also noteworthy that human
populations with excessive exposure to some known carcinogens (e.g.,
cycasin in Guam and aflatoxins in Africa and Asia) have not yet been
shown to have an increase in kidney cancer (Sufrin & Beckley, 1980).
Renal adenocarcinoma has been diagnosed with increasing frequency in
patients with chronic renal failure, particularly in those patients
treated with long-term dialysis (Dunhill et al., 1977).
An association between renal cancer and excess exposure to lead
has not been clearly established, but a study of lead smelter and
battery workers found a significant excess of malignancies at all
sites, these being mostly lung tumours (Cooper & Gaffey, 1975). Case
reports of renal tumours in workers with lead nephropathy have
appeared (Baker et al., 1980; Lilis, 1981).
6.3 Upper urothelial carcinoma (transitional cell carcinoma)
Tumours of the renal pelvis form a spectrum from benign
papillomas to frank papillary carcinomas and, like bladder tumours,
are generally low-grade cancers. However, they tend to recur
regardless of their morphology. Upper urothelial carcinoma has been
associated with RPN and analgesic abuse, but a cause-and-effect
relationship between the two has not been proven (Bach & Bridges,
1985a). The incidence of upper urothelial carcinoma among analgesic
abusers is very high, and females predominate in analgesic-associated
upper urothelial carcinoma. The female:male ratio is 2.5 to 1
(Bengtsson et al., 1978), which is in keeping with the ratio for
analgesic abusers. Analgesic abusers also develop upper urothelial
carcinoma at a younger age than non-analgesic abusers (Mihatsch et
al., 1980a,b,c). The distribution of urothelial carcinomas in
analgesic abusers has a distinct pattern; tumours of the renal pelvis,
ureter, and bladder are found 80 times, 90 times, and 7 times,
respectively, more frequently than in non-analgesic abusers. The
tumours are typically multiple, diffuse, and poorly differentiated,
and spread rapidly (Mihatsch et al., 1980c).
Patients who discontinue abuse of the drugs are at a greater risk
of developing upper urothelial carcinoma, often after a latent
period of 10-20 years after initiating analgesic abuse. Greatly
improved dialysis techniques may result in the survival of
analgesic-abusing patients who would otherwise have developed
end-stage renal disease and subsequently died (Mihatsch et al.,
1980a). It has therefore been suggested that the incidence of upper
urothelial carcinoma will increase.
The diagnosis of upper urothelial carcinoma is difficult in the
clinical situation because of few specific clinical symptoms to
indicate the malignant changes (Johansson et al., 1976; Bengtsson et
al., 1978; Mihatsch et al., 1980a,b,c; Mihatsch & Knusli, 1982; Bach
& Bridges, 1985a; Pommer et al., 1986). The prognosis is poor, and
patients with upper urothelial carcinoma only have a mean survival
time of 22 months (Mihatsch et al., 1980a) owing to the difficulty of
diagnosis, compromised renal function of patients with RPN, and
multifocal sites of rapidly developing and widespread invasion and
metastases (Johansson et al., 1976; Mihatsch & Knusli, 1982).
6.4 Experimentally induced renal adenomas and adenocarcinomas
Renal adenomas and adenocarcinomas may be induced in laboratory
animals by various natural products and biological and chemical
agents. However, linkage of exposure of these substances to renal
cancer in humans is lacking in most instances, or, at best, is only
suspected.
6.4.1 Background incidence of spontaneous tumours in experimental animals
The incidence of spontaneous renal parenchymal tumours in most
commonly used strains of male rats and mice is in the region of 0.2%,
whereas it is < 0.1% for females (Crain, 1958; Goodman et al., 1979,
1980; Ward et al., 1979; Maekawa et al., 1983). The incidence may be
up to 2.7% (Pour et al., 1979) in hamsters, which is generally low
enough for investigative studies.
Renal tumours have been induced experimentally by a large number
of compounds including lead salts (Kilham et al., 1962; van Esch &
Kroes, 1969; Goyer & Moore, 1974), nickel sulfides (Jasmin & Riopelle,
1976; Sunderman et al., 1984), methylmercury chloride (Mitsumori et
al., 1981), N-(4 -fluoro-4-biphenylyl) acetamide (Hinton et al.,
1980), trisodium nitrilotriacetic acid (Goyer et al., 1981), potassium
bromate (Kurokawa et al., 1983), halogenated alkenes (Kociba et al.,
1977; Reichert et al., 1984), and tris(2,3-dibromopropyl)phosphate
(Reznik et al., 1979). Natural products include cycasin (Laqueur &
Spatz, 1968), aflatoxins B1 (Butler et al., 1969; Epstein et al.,
1969), ochratoxin A (Kanisawa & Suzuki, 1978), citrinin (Arai &
Hibino, 1983), the fermentation-derived anti-neoplastic agent
daunomycin (Sternberg et al., 1972), and streptozotocin (Rakieten et
al., 1968; Hard, 1985). Diethylstilbestrol (Horning & Whittick, 1954)
and related estrogens (Li et al., 1983) produce a high incidence of
parenchymal tumours in hamsters.
The classical two-stage model of carcinogenesis also applies to
several of the nitrosamines, where promoters include sodium arsenite
(Shirachi et al., 1983), DL-serine (Hiasa et al., 1984a), folic acid
(Shirai et al., 1984), lead acetate (Hiasa et al., 1983), nicotinamide
(Rosenberg et al., 1985), trisodium nitrilotriacetate (Hiasa et al.,
1984b), and citrinin (Shinohara et al., 1976). There are several
factors that may affect the development of carcinomas, including diet
(Hard & Butler, 1970; McLean & Magee, 1970; Hard, 1980, 1984; Swann et
al., 1980), partial hepatectomy (Evarts et al., 1982), unilateral
nephrectomy (Ito et al., 1969), and unilateral hydronephrosis (Ohmori
& Tabei, 1983).
6.4.2 Inorganic compounds
Various inorganic compounds of lead have been investigated over
the last 30 years (Van Esch & Kroes, 1969). The tumours arise from
kidney tubular epithelial cells in kidneys and are similar to renal
cortical tumours found in humans. Production of tumours requires
continuous exposure to relatively high concentrations of lead in the
diet or drinking-water for 1 to 2 years. The tumours occur in a
background of severe interstitial nephritis characterized by tubular
atrophy as well as focal areas of hyperplasia. They are usually
multifocal and vary from microscopic adenomas to large renal
adenocarcinomas that may invade contiguous structures or metastasize
to the lungs. Intra-nuclear inclusions, which are usually present in
proximal tubular epithelial cells in lead toxicity, are absent in
neoplastic cells, and the tumors contain much less lead than adjacent
renal parenchyma (Mao & Molnar, 1967). Tumor cells are pleomorphic,
and ultrastructural studies have shown marked morphological
alterations in mitochondria.
Renal cancer occurs after injection of crystalline nickel
subsulfide (Ni3S2) into the kidney of rats, but not after
treatment with amorphous nickel sulfide (NiS). No evidence indicates
that nickel compounds are carcinogenic in experimental animals when
administered by oral or subcutaneous routes (Sunderman, 1981).
6.4.3 Organic molecules
Nitrilotriacetic acid, a polyamino polycarboxylic acid with
chelating properties similar to EDTA (used to treat lead poisoning),
produces chronic interstitial nephropathy in rodents. A spectrum of
tubular cell histological changes occurs from hyperplasia to small
adenomas to adenocarcinomas (Goyer et al., 1981). Renal adenocarcinoma
has been induced in male rats by the chronic inhalation of unleaded
gasoline vapour (MacFarland et al., 1984), but this relationship has
not been supported by epidemiological studies on workers in the
petroleum industry (Enterline & Viren, 1985).
6.4.3.1 Nitrosamines and related compounds
The nitrosamines represent one of the most widely investigated
groups of model compounds. They include dimethylnitrosamine (Murphy
et al., 1966; Mohr et al., 1974; Hard, 1984), which produces
mesenchymal (connective tissue) tumours in young animals but adenomas
and adenocarcinomas in mature animals (Hard, 1979). The
co-administration of putrescine (Ohmori & Tabei, 1983) or
N-3,5-dichlorophenyl-succinimide (Ito et al., 1974) with
dimethylnitrosamine caused a dose-related incidence of up to 100%
renal tumours after 100 weeks. N-ethyl- N-hydroxyethylnitrosamine
(Hiasa et al., 1979) on its own, or, especially, when administered
with basic lead acetate (Hiasa et al., 1983) or serine (Hiasa et al.,
1984a) causes tumours in up to 95% of animals by 32-38 weeks.
N-Nitrosomorpholine causes oncocytomas (Bannasch et al., 1978a,b,
1980). There are interesting differences between several of the model
compounds, interspecies responses, effects of dose regimens, etc.
However, the investigation of these models has generally permitted the
progression of cellular injury to be described in terms of the acute
effects, early hyperplasia, dysplasia, and different types of renal
tumours.
6.4.3.2 Morphological changes
Many of these model compounds have been used to study the early,
intermediate, and late changes at the light microscope and
ultrastructural levels (Horning & Whittick, 1954; Butler, 1964; Butler
& Lijinsky, 1970; Ertürk et al., 1970; Hard & Butler, 1971; Sternberg
et al., 1972; Bennington, 1973; Hard, 1975, 1984, 1985; Bannasch et
al., 1978a,b, 1980; Dees et al., 1980a,b; Ohmori et al., 1982; Tsuda
et al., 1983; Eble & Hull, 1984; Hard et al., 1984; Hiasa et al.,
1984a,b,c). The phenotypic changes associated with loss of normal
growth control have concentrated on the focal preneoplastic changes in
heterogeneous cells. These undergo slow changes (where it is highly
desirable to define the origins of the neoplasm) in a limited number
of enzyme markers and in lipids, and carbohydrates. In addition, an
increase in cytoplasmic RNA (shown by enhanced cytoplasmic basophilia
or numerous ribosomes at the ultrastructural level) has been observed
as a marker for hyperbasophilic and basophilic preneoplastic foci in
the epithelium of the renal tubular system (Hard, 1986; Bannasch &
Zerban, 1986).
The nomenclature of renal parenchymal tumours is based on several
criteria, including the size of the tumour and the
morphological-histochemical characteristics of cells and their
organization. The progression in tissue mass from hyperplasia, through
dysplasia, to adenoma, adenocarcinoma, and carcinoma is a continuum,
although the earliest changes, particularly hyperplasia, may be
reversible. Cells may have no cytoplasmic staining (clear cells) or
granular acidophilic or basophilic cytoplasm staining, and tumours may
have a mixture of cells with different staining characteristics. Where
adenomas contain a uniform population of finely granulated
eosinophilic cells, they are termed renal oncocytomas. Tumours can
also be classified as tubular, solid, lobular, disorganized, invasive,
papillary or cystadenoma, or by a composite of such terms based on
their appearance (Hard, 1987).
Clear and acidophilic (granular) cell kidney tumours induced by
limited exposure of rats to N-nitrosomorpholine are associated with
a transient storage of glycogen (Bannasch et al., 1978a) and closely
parallel the most common malignant renal neoplasm in man. The tumours
originate from segments of the collecting duct system storing large
amounts of glycogen (Nogueira et al., 1989). However, when
microadenomas develop, the clear (glycogenotic) cells loose glycogen
and acquire an acidophilic (granular) cytoplasm, although both cell
types can coexist in large tumours. Lipid-storing cells are often
found in the clear cell tumours, but the significance is not known. By
contrast, there are no such relationships between cells storing
glycogen and the so-called renal oncocytomas also found in NNM-induced
rats (Bannasch et al., 1978b; Nogueira et al., 1989), although rat
renal oncocytomas also originate from the collecting duct system
(Nogueira & Bannasch, 1988). They are, however, benign end-stage
lesions where the cytoplasm is crowded with pathologically altered
mitochondria (Krech et al., 1981). A temporary focal storage of
glycosamino-glycans has been reported in chromophobic rat renal cell
tubules and tumours (Bannasch et al., 1980, 1981) and in the
corresponding type of human tumour (Thoenes et al., 1985).
6.4.3.3 Biochemical changes in cells
An immunohistochemical increase in glucose-6-phosphate
dehydrogenase is associated with basophilic renal cell tumours (Tsuda
et al., 1986) and nephroblastomas (Moore et al., 1986). The pentose
phosphate shunt provides sugars for RNA and DNA synthesis, and the
activation of this pathway is probably closely related to certain
phenotypic changes, such as an increase in ribosomes and an enhanced
cell proliferation in preneoplastic and neoplastic lesions. In line
with this interpretation, rat renal oncocytic tubules and tumours,
which are poor in ribosomes and grow very slowly, usually show a
normal or even decreased activity of glucose-6-phosphate dehydrogenase
(Tsuda et al., 1986). However, in some experimental models a reduced
amount or activity of glucose-6-phosphate dehydrogenase was found in
the more malignant populations, suggesting involvement of the enzyme
in other metabolic aberrations relevant to tumorigenesis (Moore et
al., 1986).
Alterations in drug-metabolizing enzymes (see below) during
carcinogenesis have been detected by immuno-histochemical methods in
various tissues, especially in the renal tubular system, but they have
not been correlated to the same extent with the respective enzyme
activities and with other changes in the cellular phenotype as have
those of carbohydrate metabolism. By contrast,
N-ethyl- N-hydroxyethylnitrosamine-induced renal carcinomas show
opposite alterations in drug-metabolizing enzymes in preneoplastic and
neoplastic lesions of these tissues (Tsuda et al., 1987). Reduced
activities of gamma-glutamyl-transpeptidase (Ohmori et al., 1982;
Tsuda et al., 1986), succinate dehydrogenase (Tsuda et al., 1986), and
alkaline phosphatase (Tsuda et al., 1986) are seen as early changes
during the development of basophilic cell tumours from hyperbasophilic
segments of the proximal nephron. In contrast, however, there are no
similar changes in gamma-glutamyltranspeptidase or alkaline
phosphatase activity (but there is an increase in succinate
dehydrogenase activity) in oncocytic tubular lesions seen in these
animals (Tsuda et al., 1986). The increased binding of anti-cytochrome
c oxidase to the oncocytic lesions in both man (Ortmann et al., 1988)
and rat (Mayer et al., 1989) may be a useful marker for preneoplastic
renal changes.
6.4.3.4 The mechanistic basis of renal carcinoma
The mechanistic basis for the development of renal carcinoma may
be genotoxic or non-genotoxic. The common feature to all genotoxic
agents is the generation of a reactive electrophilic
(electron-deficient) species which is capable of binding to
nucleophilic (electron-rich) sites on cellular macromolecules
including proteins, lipids, RNA, and especially DNA (Miller & Miller,
1981). For example nitroso-compounds alkylate DNA in the N-7 and,
especially (due to its prolonged stability), O-6 positions (Nicoll et
al., 1975).
Genotoxic compounds are either direct alkylating agents
(requiring no activation) or they require one or more
biotransformation steps by the P-450-dependent mono-oxygenases (e.g.,
chloroform) (Bailie et al., 1984; Smith & Hook, 1984). However,
hexachloro-1,3-butadiene is transformed by ß-lyase (Elfarra & Anders,
1984) or prostaglandin hydroperoxidase-mediated co-oxidation (Davis et
al., 1981), which may also be involved in
N-[4-(5-nitro-2-furyl)-2-thiazolyl]formamide transformation (Zenser
& Davis, 1984). In addition, there may be several other renal and
extra-renal metabolic steps. There are also some molecules that cannot
be reliably classified into either group. Diethystilbestol is a very
weak alkylating agent (Lutz et al., 1982), and its mechanism of action
is thought to be mediated by renal estrogen receptors (Li & Li, 1984).
However, some metabolic component may be involved, e.g., hepatic
mixed-function oxidase (Metzler, 1981) and peroxidative activation
(Metzler & McLachlan, 1978).
6.5 Experimentally induced upper urothelial carcinomas
(transitional cell carcinomas)
There is experimental evidence to connect analgesic exposure to
the development of urothelial tumours (Johansson & Angervall, 1976;
Bengtsson et al., 1978; Bach & Bridges, 1985a). While bladder tumours
have been studied extensively in animal models, the practical
difficulties of looking for malignancies in the ureter or pelvis has
limited studies in this area. Based on long-term carcinogenicity
studies, there are few data to establish a clear experimental
relationship between analgesic exposure and upper urothelial
carcinoma. There is, however, experimental evidence to suggest that
upper urothelial carcinomas can be induced using a classical two-stage
initiation/promotion regimen (Bach & Gregg, 1988; Gregg et al., 1989).
These data suggest that localized injury associated with papillary
necrosis adjacent to urothelium that has already been initiated will
result in a proliferation of changes that lead to malignancy. At
present the full significance of these findings in terms of the human
analgesic problem is not clear.
7. ASSESSMENT OF NEPHROTOXICITY
No single in vivo or in vitro method of studying the
nephrotoxicity of chemicals can address all of the questions that must
be asked. It is therefore inadvisable to separate mechanistic research
into target cell toxicity from the screening of novel compounds for
their potential nephrotoxicity. The holistic approach to
nephrotoxicity assessment also demands that in vivo investigations
are not separated from in vitro studies, and that data continue to be
derived from several different animal species and related to
accurately conducted epidemiological and clinical studies (where these
data are available).
Present data suggest that most in vitro methods can provide
information on the mechanism of primary insult and the effect on cell
viability. However, there appears at present to be little place for
in vitro techniques in the assessment of secondary renal changes, as
the factors that contribute to the cascade of degenerative changes
that follows renal insult are largely obscure. Thus there is inherent
uncertainty as to what should be studied in in vitro systems. There
are, however, several approaches that can be used to provide a better
understanding of the contribution to degenerative changes in vivo.
These include harvesting tissue at different time points following an
insult and using this tissue to study function in different cell
types, or studying the effects of chemicals on target and non-target
cells using pure and mixed cell cultures in the presence and absence
of cells that are related anatomically. It is also possible to
exchange culture media between cells that have been insulted, and
those that have not, in order to assess the release of factors that
may be toxic to other cells. Different cells can be studied in the
same media to define cell-cell interactions and how chemical insult
affects this process.
7.1 In vitro studies
Despite the complexity of the kidney and nephrotoxicity, and the
difficulty in defining what any one in vitro system achieves, there
are several ways to progress in the use of screening methods.
One such rational approach could include:
* the careful identification of compounds with well-documented in
vivo nephrotoxicity, including the sequence of pathological and
functional changes, the metabolites formed, quantities excreted,
and the cellular pharmacodynamic effects;
* the choice of chemicals that target specifically for one
anatomically discrete cell type in vivo;
* the use of both more and less nephrotoxic analogues of the
chemical for determining structure-activity relationships and
computer-based simulations;
* the systematic study of these compounds by several different in
vitro methods, and the use of several criteria for assessing in
vitro nephrotoxicity for each.
7.1.1 Choice of chemical concentrations for in vitro studies
The validity of using the cytotoxicity (in vitro) for a given
concentration of a chemical as a likely indicator of a toxicological
effect in vivo can be very difficult to establish. It may be
impossible to assess this in the kidney, because this organ is highly
compartmentalized. In the intact and functioning organ, it is
currently not possible to establish the concentrations of a xenobiotic
(or its metabolites) associated with any one cell type. This
uncertainty relates, for instance, to the different transport systems
that are distributed in discrete parts of the nephron, transcellular
pH gradients, and the selective accumulation of certain chemicals in
cell organelles (which have heterogeneous distribution in different
types of cells). Thus some chemicals can be selectively concentrated
in a discrete area of the kidney to several times the plasma
concentration. Alternatively, certain chemicals may be actively or
selectively excluded from some cell types (Mudge, 1985).
Drug metabolism systems that alter the physicochemical
characteristics of xenobiotics and their metabolites (that will
facilitate the redistribution of chemicals within and between cells)
are also heterogeneously distributed. Thus, certain xenobiotic
products may be selectively concentrated in (while others are excluded
from) specific cell types. Therefore, even though arterial and venous
blood and urinary concentrations of chemicals can be measured, there
is no certainty that such data relate to the concentrations of any
specific metabolite that reaches a target cell. Autoradiography may
be very valuable in providing some idea on how chemicals are
distributed, but it only shows the distribution of radiolabel-derived
material, not its chemical nature. Once the anatomical integrity of
the kidney has been altered, it may be difficult to relate the
concentration of any chemical to the same cells in vivo. While
structure is maintained in the perfused kidney, the functional changes
impose a similar constraint. The uncertainty as to what concentrations
of chemicals to use in in vitro systems is exacerbated by the
undefined influence of extrarenal and renal metabolism on the delivery
of the proximate and ultimate toxins to the target sites of injury in
vivo.
7.1.1.1 Proximate and ultimate nephrotoxicants in vitro
In addition to the need to consider carefully the consequences of
the changes in the route of chemical delivery when the anatomical
integrity of the kidney has been disrupted (i.e. in all systems other
than the isolated perfused kidney), it is important to consider how a
chemical is delivered to the cells. This consideration should be
expressed in terms of the chemical being free or bound (should the
media contain protein or not?), the physicochemical characteristics of
the solution in which the chemical is delivered (such as its pH, ionic
concentration, and endogenous and exogenous micro- or
macro-molecules), and the kinetics of delivery (this is generally
zero-order in most in vitro systems but follows first or second
order kinetics in vivo).
Chemically induced nephrotoxicity may be the result of a direct
action of the parent chemical in the kidney or may be due to an
extrarenally formed metabolite. However, in some instances the
chemical/metabolite has to be further metabolized in situ to form the
ultimate nephro-toxic species. Assessing the role of in situ
metabolism in nephrotoxicity from in vivo data may be difficult,
since most nephrotoxic chemicals are also extensively metabolized in
extrarenal tissue such as the liver. Experimental approaches using
different species and strains of animals, inducers and inhibitors of
drug metabolizing enzymes, and candidate proximate and ultimate
nephrotoxic metabolites may give some evidence for the involvement of
intrarenal metabolic activation (Rush et al., 1984). More direct
evidence for a direct renal activation of a chemical has to come from
in vitro studies or studies with perfused kidneys where extrarenal
metabolism/activation can be excluded. In vitro studies may include
experiments with isolated tissue preparation (kidney slices and
tubules), cells (primary cells or cell lines) or subcellular fractions
to assess chemically induced toxicity. In order to determine the role
of extrarenal metabolism in the formation of nephrotoxic metabolites,
co-culture systems using liver cells (as an activation system) and
kidney cells (as target cells) may be very useful (Moldeus et al.,
1978). The stability of a reactive metabolite, generated by liver
cells, may be measured by transferring, after various time intervals,
the incubation medium to the target cell population.
7.1.2 In vitro investigations of nephrotoxicity
In vitro techniques can be divided into those where the
anatomical relationship between cells is maintained (perfusion,
micropuncture, and slices), those where glomeruli and tubular
fragments are isolated, and those where cells are isolated. The
different techniques for assessing nephrotoxicity in vitro have been
reviewed and the strengths and weaknesses of each presented in broad
terms (Bach et al., 1985, 1986; Bach & Kwizera, 1988). Some methods
are technically difficult, depend on sophisticated equipment, are
subject to artefacts in inexperienced hands (perfusion and
microperfusion), and are difficult to interpret.
7.1.2.1 Perfusion and micropuncture
Micropuncture methodology has not been widely used to assess the
toxicological effects of chemicals (Bank et al., 1967; Biber et al.,
1968) because the methodology is extremely complex. However, a few of
the problem areas should be mentioned. Micro-puncture procedures
succeed only when the experimentalists can collect measured small
samples of tubular fluid and subject these to appropriate chemical
analyses. These procedures are extremely complex, and, because of the
small volumes involved, subject to considerable error. To assure that
the micropuncture collections reflect the "physiological state", most
workers attempt to collect only very small volumes at the
"physiological" flow rate past the point of micropuncture. Stationary
microperfusion has been used to assess tubular function in a
restricted area of the nephron.
Micro-injection into a tubular segment has been used by many
physiologists and may be of particular toxicological interest
(Gottschalk & Lassiter, 1973; Roch-Ramel & Peters, 1979; Diezi &
Roch-Ramel, 1987). With this technique, various renal function
markers, as well as potential nephrotoxicants, may be injected into a
segment of the proximal tubule during a free-flow situation. The urine
from the injected kidney may then be collected to assess
nephrotoxicant effects on the injected renal function markers. For
micropuncture specialists, this is a relatively straight-forward
technique and does not involve the problems associated with removal of
tubular fluid by micropuncture. This procedure might permit the
assessment of nephrotoxicant effects on membrane permeability by
examining, for example, inulin excretion from the injected kidney.
The isolated perfused tubule technique represents the development
of an in vitro procedure that permits the assessment of intact
tubular function under carefully controlled in vitro conditions.
Hence all of the advantages of in vitro methodology are available
while intact tubular function is being studied (Diezi & Roch-Ramel,
1987).
Tubule segments can also be isolated by manual dissection for
microperfusion, where they are attached to micropipettes suspended in
a bathing solution and perfused with an artificial tubular fluid
(Ullrich & Greger, 1985). Relatively few attempts have been made to
apply this sophisticated methodology to the study of nephrotoxicants,
where it would be possible to add chemicals to either the perfusate or
the bathing solution and examine effects on either the tubular or
basolateral side of the cell. This technique has been used to
demonstrate that organic anion transport across the tubular cell is
active on the basolateral side but not on the luminal side (Tune et
al., 1969).
The major advantage of this procedure is that it permits an in
vitro assessment of renal function with a tissue segment that is
essentially intact. The situation in an isolated nephron segment is
obviously not identical to that in the intact kidney, but by carefully
regulating the perfusion solution and the bathing solution one can
approximate in vivo physiology. This is a potentially important
procedure that needs to be assessed for its utility.
7.1.2.2 Renal cortical slice
These techniques have been reviewed extensively (Berndt,
1976,1987; Bach & Lock, 1982; Kacew, 1987) and used to show
deleterious effects of chemicals and drugs on the kidney. Much of the
published information has focused on renal tubular transport as the
criterion for establishing the nephrotoxic potential of chemicals. The
tests are based on measuring the accumulation of the organic ions
p-aminohippurate (PAH) and tetraethyl-ammonium (TEA) (Hirsch, 1976)
by renal slices. The organic anion transport is a sensitive indicator
of aminoglycoside (Kluwe & Hook, 1978; Kaloyanides & Pastoriza-Munoz,
1980) and cephaloridine toxicity (Kuo & Hook, 1982; Kuo et al., 1982).
Similarly, the effects of mercuric ions, chromate ions,
hexachlorobutadiene conjugates, and other nephrotoxins appear readily
detectable by this approach. Organic ion accumulation and
gluconeogenesis in renal cortical slices may be poor indicators of
early toxic effects to the kidney resulting from cisplatin
administration because these parameters are not affected except at
high doses. The poor sensitivity and delayed response of renal slice
parameters indicate that membrane function and cell metabolism may not
be early targets of cisplatin at the cellular level. Slices maintain
ß-lyase activity for up 12 h and have been used to study halo-alkene
toxicity. Aminooxyacetic acid inhibits ß-lyase activity almost
completely. DCVC decreases PAH accumulation, but does not appear to
use the same transport process.
7.1.2.3 Isolated nephron segments
Isolated tubules overcome many of the disadvantages of cortical
slices, in that they remain in contact with substrates and toxins in
the medium, whereas cortical slices may show lumen collapse within a
short period (Chahwala & Harpur, 1986).
Freshly isolated tubule or cell suspensions offer an important
way of studying the mechanisms of nephrotoxicity and screening novel
compounds for their potential acute effects on the kidney. A
limitation, however, is the short in vitro lifespan of isolated
tubules prepared, by any technique, for studying early toxic effects.
In general, most investigators limit incubations to no more than 2-4
h because of loss of viability and functional capabilities. Ormstad
(1982) reported rapid loss of viability of isolated renal tubules and
cells, more than 25% of the cellular lactate dehydrogenase (LDH)
leaking to the medium during 1.5 to 2 h of incubation. Obatomi &
Plummer (1986) observed a 40% loss in tubule cell viability during 3-h
incubations of rat proximal tubules. Loss of renal function, such as
O2 consumption, has also been reported for isolated tubules (Harris et
al., 1981).
A number of fresh tubular systems exhibiting high initial
viability (> 90% by trypan blue exclusion) have been prepared from
collagenase digests of rat cortical tissue (Cunnaro & Weiner, 1978;
Belleman, 1980; Cojocel et al., 1983; Gstraunthaler et al., 1985;
Obatomi & Plummer, 1986) using tubular fragments of proximal origin.
The tubules can be used to study the metabolism of xenobiotics liable
to be converted into compounds responsible for the alteration of
normal renal metabolism (Jones et al., 1979). Tubular fragments
obtained by collagenase treatment of dog (Baverel et al., 1978,
1980a), human (Baverel et al., 1979), baboon (Michoudet & Baverel,
1987), and guinea-pig (Baverel et al., 1980b) renal cortex have also
been used for metabolic studies. These fragments retain the
gluconeogenic capacity that is specific to the proximal convoluted
tubule (Guder & Ross, 1984). Human, dog, and rat renal cortex tubules
also release ammonia from glutamine, and any drug-induced disturbance
of renal ammoniagenesis can be studied with these models (Martin et
al., 1987, 1989, 1990).
Proximal tubules have been purified from the above preparations
by centrifugation on a Percoll density gradient. The samples obtained
exhibit enrichment in proximal tubule cells relative to cells from
other areas of the nephron, as indicated by the distribution of
alkaline phosphatase and hexokinase (Vinay et al., 1981).
Suspensions of thick ascending limb fragments have also been
prepared from dog (Baverel et al., 1980a,b; Anand-Srivastava et al.,
1986), rat (Trinh-Trang-Tan et al., 1986), and rabbit (Chamberlin et
al., 1984) outer medulla. Suspensions of collecting tubules obtained
from the inner medulla (Anand-Srivastava et al., 1986; Wirthensohn
et al., 1987, 1989) may prove very useful in studies of the effect of
nephrotoxic substances that interfere with the function of this renal
zone.
Direct addition of different concentrations of nephrotoxic agents
such as ochratoxin A, citrinin, furosemide, and potassium chromate to
the suspension releases enzymes specific to the proximal tubule (Table
4), such as alanine aminopeptidase, leucine aminopeptidase and
alkaline phosphatase, to the incubation medium in a dosedependent
manner (Endou et al., 1985). To characterize further the intrarenal
site(s) and mode of nephrotoxicity, definite portions of a single
nephron can be microdissected from the collagenase-treated kidney.
There are two different methods available for the microdissection of
individual nephron segments; one is from lyophilized kidney sections
and the other is from collagenase-treated fresh kidneys (Morel et al.,
1976). In nephrotoxicity studies, fresh individual nephron segments
are used. The following segments can be isolated: the glomerulus, the
proximal tubule (S1, S2, S3), the thin descending limb of
Henle's loop, the medullary and cortical thick ascending limb of
Henle's loop, the distal convoluted tubule, the connecting tubule, and
the cortical and medullary collecting tubules.
Several functional parameters can be studied in nephron segments.
Gluconeogenesis is a unique function of the proximal tubule, within
which the S1 segment is most active (Maleque et al., 1980; Endou et
al., 1985). Gluconeogenesis is strongly induced by metabolic acidosis
or by alpha1-adrenergic stimulation (Nakada et al., 1986a).
First-generation cephalosporins, cephaloridine and cephalothin cause
a time- and concentration-dependent decrease in gluconeogenesis, and
it is clearly indicated that the site of nephrotoxicity of these
antibiotics is the proximal tubule (S1, S2, S3). Ammoniagenic
activity is distributed in all the nephron segments, but the highest
production rate of ammonia from glutamine is observed in the proximal
tubule (Nonoguchi et al., 1985). Ammoniagenesis via the purine
nucleotide cycle from asparatate as a substrate is also high in the
proximal tubule (Tamura & Endou, 1988). Ammonia production is
increased in a similar way by metabolic acidosis or potassium
depletion (Nonoguchi et al., 1986). Cisplatin nephrotoxicity is
morphologically known to be focussed on the S3 segment. However, this
drug decreases ammoniagenesis from glutamine not only in S3, but
also in S2, suggesting a discrepancy between morphological and
biochemical evaluations, although cisplatin does not affect
gluconeogenesis in isolated nephron segments (Nakada et al., 1986b).
The kidney possesses various active transport processes that
consume ATP at a high rate. Individual nephron segments require their
own particular substrates for synthesizing the necessary ATP: this has
been shown in both mice (Uchida & Endou, 1988) and rats (Jung et al.,
1989). The proximal tubule cannot use glucose to produce ATP, whereas
the other nephron segments can use it. In general, pyruvate or lactate
is the preferred substrate in all segments. Nephrotoxicity assessment
by measuring cellular ATP content shows clearly that mercuric chloride
decreases ATP content only in S2 (Jung et al., 1989) and that
ochratoxin A nephrotoxicity localizes in S2 and S3 (Jung & Endou,
1989). Thus, measurement of cellular ATP in specific nephron segments
enables possible nephrotoxicants to be evaluated. A similar principle
can be applied by measuring intracellular free calcium (Jung & Endou,
1990). From the biological point of view, it is essential to keep
cellular ATP at a high level and to maintain a low concentration of
intracellular free calcium for all living cells. It should,
therefore, be reasonable and useful to introduce these sensitive
parameters to nephrotoxicity assessment, although the methods require
special techniques for microdissecting nephron segments or special
instruments.
An advantage of the use of isolated tubules, as compared to in
vivo experiments, is that it permits a cellular environment that is
defined both quantitatively and qualitatively. This allows the
relationship between the concentration of a nephrotoxin, exposure
time, and effect to be studied. Extrarenal effects can be avoided, and
so isolated tubules are very suitable for studying the effects of
nephrotoxins that act directly at the tubular site. Owing to a lack
of polarity (Koseki et al., 1988), isolated renal cell suspension may
have limited usefulness.
There are several limitations associated with the use of freshly
isolated or cultured renal cells. Cells released by enzymic digestion
or fresh fragments can be cultured in the presence of serum-free,
hormonally defined culture media. This prevents fibroblast
proliferation and encourages epithelial cell growth (Chuman et al.,
1982), but both cell preparations lack a brush border, which may be
critical for the active uptake of drugs and chemicals.
Alternative approaches to obtain a preparation with an intact
brush border include the use of different sized sieves to separate
glomeruli and tubules (Bach et al., 1986), which may not result in a
pure preparation of proximal tubular cells. The choice of method for
monitoring cell viability may circumvent this, e.g., the use of
prostaglandin synthesis (Sraer et al., 1980) to assess effects of
chemicals selectively on glomeruli. The density gradient technique
(Vinay et al., 1981) uses Percoll centrifugation to separate the
different cell types to obtain a > 90% pure preparation of proximal
tubular cells. These cells retain their viability and their GSH levels
at > 50% for 2 h at 37 °C, and demonstrate cytochrome-P450-dependent
mono-oxygenase activity profiles that are inducible only by
3-methylcholanthrene (3MC). This may be an appropriate preparation to
study the effects of various drugs and chemicals, in both rats and
man, that demonstrate nephrotoxicity to either the S1, S2, or S3
regions after administration (Smith et al., 1986; Rosenberg &
Michalopoulos, 1987).
7.1.2.4 Primary cell cultures
Cell to be cultured should be of well defined origin. For this,
purification of a homogeneous glomerular or tubular cell population
can be achieved by several methods (Jakoby & Pastan, 1979), including
sieving techniques (Striker et al., 1980), magnetic and mechanical
techniques (Meezan & Brendel, 1973), density gradient centrifugation
(Scholer & Edelman, 1979; Vinay et al., 1981), and collagenase
digestion (Curthoys & Bellemann, 1979; Belleman, 1980; Ormstad et al.,
1981). More recently, techniques such as immunodissection, cell
sorting, free-flow electrophoresis, and microdissection have been used
(Pretlow & Pretlow, 1982, 1983, 1984). The advantage of using primary
cell cultures is that it allows long exposure to xenobiotics and the
choice of appropriate metabolites. In addition, it is possible to
monitor a variety of cell functional, biomedical, or morphological
responses in a dose- and time-related manner (Fry et al., 1978;
Belleman, 1980; Fry & Perry, 1981; Bach et al., 1986).
Mechanical or enzymic dispersal may damage cells, and once cells
are dispersed it is generally difficult to establish their anatomical
identity unless suitable markers are used. These markers include both
the presence and absence of a range of functional and biochemical
characteristics, such as transport systems, and an array of structural
and functional molecules. These can best be assessed by a variety of
histochemical and immunocytochemical methods (Bach et al., 1985,
1987). At present, isolated cells are generally mixtures (although
they may be enriched) and must be used within a few hours. Primary
cell cultures may rapidly dedifferentiate (Curthoys & Bellemann, 1979)
or adapt to a new environment and change their characteristics as a
result of the presence or absence of factors in the culture media,
which may obfuscate their anatomic origins. More importantly, loss of
a biochemical characteristic that is part of the molecular basis for
target cell toxicity may invalidate in vitro studies. Changes in
other aspects of cellular integrity can increase or decrease both the
sensitivity and selectivity of screening methods used for cytotoxicity
studies.
Two approaches have been used to modulate the expression of cell
characteristics. The polarity of epithelial cells is better expressed
when cell are grown on permeable supports such as collagen/filters
(Jakoby & Pastan, 1979). Similarly, the appropriate modulation of
culture media has been used to alter rabbit proximal tubule cell
metabolism to the gluconeogenic pathway and these cells then develop
brush-border characteristics. Thus, media can be an important
variable, especially because of the diverse combination of buffers and
growth supplements used. There are major advantages in using fully
defined culture media (Sato & Reid, 1978), but these have not been
widely adopted.
Human proximal tubules have been shown to been sensitive to
cyclosporin A (Trifillis et al., 1986), but there are no data on the
mechanistic bases of these changes. Rat, rabbit, dog, and human
glomerular mesangial and epithelial cells may be co-cultured or each
type derived separately (Kreisberg et al., 1977, 1978; Foidart et al.,
1979, 1980, 1981; Morita et al., 1980; Striker et al., 1980; Kreisberg
& Karnovsky, 1983). Rat epithelial cells are more sensitive to
puromycin aminonucleoside and Adriamycin than are mesangial cells, as
is the case in vivo, but there is little mechanistic information.
Rat medullary interstitial cells can be cultured at high osmolality
and have been shown to be sensitive to a number of compounds that
cause renal papillary necrosis.
7.1.2.5 Established renal cell lines
Several established renal cell lines have been studied that have
properties reminiscent of specific parts of the nephron, such as
LLC-PK1 (of proximal tubule type) and MDCK (of distal tubule type).
The major disadvantage is that the exact site of origin, within the
nephron, of each, is not known, and it may not totally represent the
normal physiological state. However, these lines are often
heterogeneous and there is a need to characterize them more
systematically so as to establish where they may be useful in
screening chemicals for toxicity or in understanding the mechanisms of
target cell toxicity.
Differences exist between the apical and basolateral membrane
transport of substances into cells, which may be central to the
mechanism of nephrotoxicity. When cells are cultured on solid
surfaces, only apical exposure to chemicals occurs, whereas in vivo,
proximal tubule cells are exposed from the apical or basolateral sides
or both. This disadvantage can be overcome by culturing renal cells on
microporous membranes suspended in culture wells. These cells, which
form a confluent single-cell monolayer covering the membrane within
some days, more closely mimic the in vivo state than those grown on
plastic plates. They show anatomical and functional polarization. This
culture technique allows access to the cell monolayer from both the
apical and the basolateral sides, and apical and basolateral fluid may
be studied simultaneously. This new experimental tool allows the study
of transport and epithelial resistance across the cell monolayer and
polarized uptake of various molecules, including potentially
nephrotoxic drugs, as well as to perform a variety of analytical
techniques.
The various cell lines used in nephrotoxicity studies have been
reviewed by Wilson (1986). The LLC-PK1 cell lines have a typical
epithelial polarity and have features similar to proximal tubular
epithelium, such as transport systems (Handler, 1983) and the enzyme
marker gamma-glutamyltranspeptidase (Perantoni & Berman, 1979).
Confluent LLC-PK1 cells cultured on a solid support form domes (due to
transcellular transport), but monolayers grown on a porous membrane do
not. More importantly these cells have polarity and have a
well-developed brush border. Confluent LLC-PK1 monolayers exposed to
PCBD-GSH from the apical side are more sensitive than when exposed
from the basolateral side. This is due to the brush border
localization of gamma-glutamyltranspeptidase, which catalyses the
first step of the breakdown of the conjugate to the ultimate reactive
intermediate. Neither apical nor basolateral treatment with PCBD-NAC
elicits any toxicity. It is assumed that the absence of an organic
anion transporter from these cells could explain this finding, since
it has been established that haloalkene conjugates enter cells via the
basolaterally located anion transporters (Lock et al., 1986). The
absence of an organic anion transport system limits the usefulness of
LLC-PK1 cell lines for studying nephrotoxic compounds, such as
PCBD-NAC, that need active transport to enter the cells. However, an
active basolateral organic cation transport system
(gamma-glutamyltranspeptidase and dipeptidase) makes these cells
especially useful for testing compounds that have a toxic action on
these transport systems.
7.1.2.6 Subcellular fractions
It is also possible (and sometimes desirable) to use homogeneous
or fractionated organelles, membranes, or cytoplasm from defined
cells for specific cell-free investigations. The constraints on the
preparation of these systems should be apparent from the foregoing
discussion. Subcellular fractions, such as vesicles, nuclei,
lysosomes, and microsomes, can be used to study subcellular
distribution, the interaction between a cellular compartment and a
chemical, and the kinetics of binding or release of substances. It is
also possible to study specific effects, such as enzyme inhibition,
metabolic activation, covalent binding, or the modulation of lipid
peroxidation, using purified or commercially available biochemicals
with appropriate cofactors and suitable techniques for monitoring
these interactions (Bach & Bridges, 1985b, 1987).
Many nephrotoxic agents interact with cell membranes, where they
bind with receptors, effect transport systems, or disrupt structure
and function per se. Thus, membrane vesicles may be useful for
studying these interactions and the mechanisms of cell injury. It is
possible to isolate vesicles from the brush border and basement
membranes to study transport systems at each site in vitro. Williams
et al. (1986) showed a very good correlation between the in vitro
binding of aminoglycosides to brush-border membrane vesicles and their
in vivo nephrotoxicity. Inhibition of aminoglycoside membrane
binding by polyaspartate reduces nephrotoxicity and suggests that
binding of these antibiotics to brush-border phospholipid may be a
crucial event in nephrotoxicity.
7.2 in vivo experimental studies
Current methods for diagnosing renal injury and predicting the
health significance are not sufficient to deal with the diversity of
possible chemical injuries (for full discussion, see Bach et al.,
1989). This is because the kidney can undergo substantial chemically
induced injury without any clinical indication, since subtle injury
may be buffered within the considerable functional reserve. This masks
a substantial amount of renal degeneration (Friedlander et al., 1989).
Thus, for example, the single cross-sectional measurement of GFR may
only show incipient acute or chronic renal failure. Quantitative
urinary enzyme excretion patterns cannot identify either the type or
severity of renal injury, and often they do not correlate with
morphological and functional changes (Schentag et al., 1978).
There are a number of inherent difficulties in diagnostic
procedures for nephropathy, which include the absence of standard
diagnostic criteria and the inability to relate exposure to a given
agent and the observed effect. In addition, renal functional reserve
is a major factor that masks renal degeneration, as assessed by GFR,
blood urea nitrogen, and creatinine, up to the point where over 75% of
the functioning nephrons have been lost. Thus, it should be stressed
that these factors measure incipient renal failure and that the fact
that values are normal (something that is subject to age-related
change and varies between the two sexes) does not signify the absence
of renal dysfunction or even, in some cases, gross renal
insufficiency. Therefore, cause and effect cannot be clearly
established on the basis of available knowledge when the renal lesion
results from a multifactorial process with a long latency. Part of
this uncertainty can be addressed by studies on experimental animals.
7.2.1 Methods for assessing chemically reactive nephrotoxic
metabolites in animals
It is known that many nephrotoxicities that follow the
administration of inert, relatively nontoxic chemicals are related to
the formation of reactive electrophiles during the metabolism of these
chemicals (Ford & Hook, 1984). These electrophilic products can react
covalently with nucleophilic sites on renal macromolecules such as
protein, lipid, and DNA. The covalent binding may be measured by the
use of radiolabelled forms of the chemical, by immunological detection
of DNA/protein adducts (Harris et al., 1987), or by the 32P-DNA
postlabelling method (Reddy et al., 1984). Furthermore, chemicals that
are metabolized to DNA-damaging intermediates may be detected in vivo
by measuring the alkaline elution of isolated kidney nuclei
(Omichinski et al., 1987; Brunborg et al., 1988) or unscheduled DNA
synthesis in isolated kidney cells (Tyson & Mirsalis, 1985) after in
vivo exposure of animals.
7.2.2 Evaluation of glomerular function
Evaluation of blood urea nitrogen is probably the most common
procedure for indirectly evaluating GFR in experimental animals.
Although insensitive, this test may be sufficient to establish the
time course of chronically developing renal failure in the
experimental setting. Serum creatinine is also used for the same
purpose. However, owing to interferences from nonspecific chromogens,
this test is unreliable in most experimental animals and especially in
the rat. Although this problem may be overcome, it has not been dealt
with adequately in most available studies, thus generating wide
scatter in "normal" ranges.
In animals, more subtle changes in GFR occurring during
subchronic and chronic studies should be assessed by evaluating the
clearance of exogenous substances such as inulin, EDTA, or
iothalamate. The latter may be determined either by measuring the
radioactivity of labelled material or by means of reliable HPLC
methods (Prueksaritanont et al., 1984). Furthermore, the same HPLC
method may be used to measure PAH and to assess other haemodynamic
parameters. Two (or more) clearance periods should be calculated and
averaged in order to ensure greater accuracy.
There is a growing body of evidence to suggest that reduced renal
reserve due to hypertension/hyperfunction/hyperfiltration of remnant
nephrons is important in the course towards end-stage renal disease.
This can in part be lowered by reducing protein intake and blood
pressure. The concept of renal functional reserve includes the
evaluation of renal blood flow and GFR by measuring their increase
after protein load or the administration of aminoacids, glucagon, or
vasodilatory drugs. At present such tests do not have defined
standardized stimuli, and there are no data on their use for detecting
nephrotoxicity.
7.2.3 Evaluation of tubular functions
In experimental animals, tubular dysfunctions are usually
detected through simple and inexpensive tests, such as those of
glycosuria, enzymuria, and osmolality, which may provide other useful
information. Some of these tests are sensitive enough to detect acute
tubular damage, although caution must be exercised in predicting
specific effects on transport processes or cell viability on the basis
of data obtained from in vivo experiments (Berndt, 1981). More
subtle renal changes occurring during chronic studies may be evaluated
by measuring the renal clearance of lithium (Dieperink et al., 1983;
Daugaard et al., 1988b). This non-invasive method is applicable both
to human (Thompson et al., 1984) and animal studies. The loss of renal
tubular functions can be assessed by test procedures that impose one
or more stressing conditions to force compensatory changes, e.g.,
maximal urinary dilution/concentration or
acidification/alkalinization, and maximal tubular reabsorption of
glucose and phosphate. The value of these tests is limited for group
studies by practical considerations but may be useful for individuals.
7.2.4 Proteinuria
Proteinuria is the loss of proteins following increased
permeability of the glomerular barrier, reduced tubular reabsorption
of filtered proteins, or shedding of specific constituents into the
urine as a consequence of cellular turnover or selective tissue
damage. Since pathological changes either at the glomerular or
tubulo-interstitial level may occur even in the absence of a
substantial reduction in GFR, the evaluation of proteinuria may also
be useful in some circumstances to detect renal dysfunction occurring
either at the glomerular or at the tubular level. Proteins may be
measured by nonspecific assays, by immunochemical methods or by their
enzymic activity. Sensitive methods have been developed to detect
small amounts of proteins in microlitre quantities of unconcentrated
urine.
7.2.4.1 Total proteinuria and electrophoretic pattern
Measurement of total proteinuria and electrophoretic separation
of single proteins provide a comprehensive approach to chemically
induced renal dysfunction.
The rationale for such an approach relies on the
pathophysiological mechanisms controlling the renal handling of
plasmaproteins. Proteins of high relative molecular mass (> 45 000
Daltons) are usually confined to the vascular compartment by basal
membranes. Furthermore, the glomerular polyanion acts as a selective
filter that retains negatively charged proteins, such as albumin,
because of electrostatic interactions. The glomerular pore size is
thought to have an important role in retaining proteins of higher
relative molecular mass (e.g., immunoglobulins). Proteins with lower
relative molecular mass (< 45 000 Daltons) pass the glomerular
barrier with sieving coefficients inversely related to their mass.
Filtered proteins of low relative molecular mass are efficiently taken
up by the proximal tubules (more than 99%). Even slight decreases in
tubular fractional reabsorption due to tissue damage or dysfunction
will result in increased low relative molecular mass proteinuria (Fig.
16).
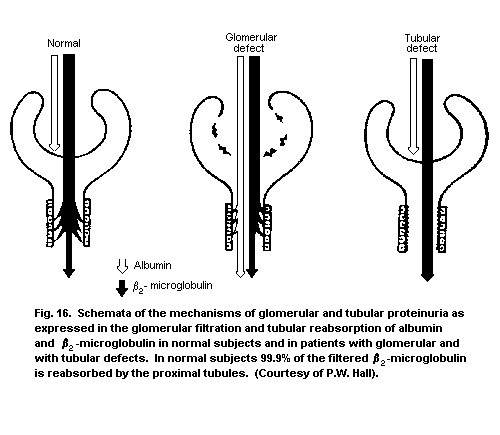 On the basis of the electrophoretic pattern, proteinuria may
reveal glomerular damage, tubular dysfunction or mixed patterns. The
glomerular damage may be selective (mostly due to a loss of glomerular
polyanion) or unselective (involving more extensive damage, and
glomerular hyperfiltration and hypertension). However, it should be
recognized that some features unique to experimental animals may
account for large variations, which could lead to wrong conclusions.
For instance, marked sex-, age-, and diet-related changes may occur,
especially in the male rat (Neuhaus et al., 1981). Even if most of
these changes have a counterpart in man, they are amplified by the
lack of variations in housing conditions. As a result, the young male
rat may physiologically show "tubular" proteinuria, whereas the aging
rat displays "glomerular" patterns, owing to a spontaneous
nephropathy, which can be prevented in part by reducing dietary
protein content. Depending on other factors, especially diet and
concomitant treatments, such electrophoretic patterns may be accounted
for by underlying mechanisms and related morphological changes.
Owing to their limited affinity for most dyes, proteins of low
relative molecular mass are better identified by the quantitative
measurement of single components such as ß2-microglobulin (Viau et
al., 1986b). Because of its peculiar metabolism, the urinary excretion
of alpha2u-globulin, a sex-related protein of low relative molecular
mass, cannot be recommended as a marker of tubular damage.
Furthermore, its renal handling and disposition may interfere with
those of other proteins, accounting for some of the spontaneous
changes occurring in the pattern of proteinuria from the male rat.
Thus, when evaluating proteinuria, preference should be given to
female rats, since in this case extrapolation to man seems to be less
affected by species-related problems.
7.2.4.2 Urinary excretion of single plasma proteins
Very sensitive immunochemical methods are available for measuring
the urinary excretion of single plasma proteins, such as IgG, albumin,
ß2-microglobulin and alpha2u-globulin in the rat (Bernard et al.,
1988). In addition to better analytical features, in terms of
sensitivity, specificity, accuracy, and reproducibility, the
quantification of single urinary proteins has two other inherent
advantages. Firstly, single proteins may be significantly increased
without giving rise to pathological values in total proteinuria
(Barratt, 1983). Secondly, the power of experimental studies is
greatly increased by quantitative data making it possible to use
parametric tests.
7.2.4.3 Enzymuria
Several different enzymes have been studied (Table 8), but none
satisfies all the criteria for nephrotoxicity (Dubach et al., 1989).
Enzymes are not uniformly distributed along or between nephrons.
Although it should be possible to localize the area of kidney damage
on the basis of the pattern of enzymuria, the site-selectivity of
single enzymes is questionable. The failure to recognize selective
damage by measuring enzyme activity may be accounted for by two
factors. Firstly, it is possible that chemically induced early renal
changes are less selective than advanced lesions preceding end-stage
renal disease. Secondly, the poor analytical features of most enzyme
measurements in urine may give rise to aspecific patterns. This
important question may be addressed by measuring immunoreactive
antigens, including enzymes, since the use of reliable immunochemical
techniques would limit the effects of analytical problems.
Most enzymes are stable over a narrow range of pH, and their
activity may be affected by the presence of inhibitors such as urea
(Price, 1982). It should be stressed that all rat enzyme studies must
be carried out under carefully controlled conditions, after an
adequate period of acclimatization, and after changing to day instead
of night feeding. Urine must be minimally contaminated with
microorganisms. This is achieved by surrounding the urine collecting
vessel with ice, so that bacteria cannot multiply as readily as in
normal metabolic cages (Berlyne, 1984).
7.2.4.4 Immunoreactive tissue constituents
Tissue constituents may be released into urine due to increased
cellular turnover or cell death and may be detected by immunochemical
methods. Monoclonal antibodies have been produced against both rat
(Tokoff-Rubin, 1986) and human brush-border antigens (Mutti et al.,
1985; Mutti, 1989). In both cases, cross-reactivity between species
has been shown (Mutti, 1987, 1989). The specificity of such an
approach relies on the site-selectivity of target proteins and on the
advantages of monoclonal antibodies, including monospecificity and
reproducibility of reagents. Its sensitivity has been proved by
Table 8. Some enzymes used as an index of nephrotoxicity
Enzymes Cellular location
Alanine aminopeptidase brush border
Alkaline phosphatase
gamma-Glutamyltransferase
Maltase
Trehalase
Glutamic oxaloacetic transaminase cytosol
Glutamic pyruvic transaminase
Lactate dehydrogenase
Malate dehydrogenase
N-Acetyl-ß-D-glucosaminidase lysosome
Acid phosphatase
ß-galactosidase
ß-glucosidase
ß-glucuronidase
Glutamate dehydrogenase mitochondria
comparison with other markers in various situations, but it needs
further validation in carefully designed chronic studies, since the
prognostic value of slight changes in such a sensitive test is
currently unknown.
In general terms, it has been through the use of
histopathological studies on kidney tissue that advanced renal lesions
have been identified. This approach has many inherent advantages, and
represents the method of choice, particularly if it can be used for
the early diagnosis of renal lesions or dysfunction in experimental
animals being used for nephrotoxicity screening studies. For clinical
investigations in human and population studies, it is obviously the
least desirable of the techniques that are available.
7.2.4.5 Urinary excretion of prostaglandins
The urinary excretion of PGs (mainly PGE2) may reflect the rate
of renal synthesis. This is modified in several nephropathies and may
be affected by a number of nephrotoxic chemicals.
7.2.5 Clinical context
At present, the clinical diagnosis of toxic nephropathy still
relies heavily on the case history of patients showing symptoms and/or
laboratory abnormalities suggesting chronic renal failure without any
obvious recent cause.
In such circumstances, history is the cornerstone of diagnosis.
It can only be made by excluding other known conditions or risk
factors that lead to renal insufficiency and assessing exposure to
nephrotoxins, which has to be consistent with known
dose-effect/response relationships and with the temporal sequence of
events leading to the observed effect. For immune reactions, the role
of individual susceptibility should also be considered. In some
circumstances, kidney toxicity may be considered as a factor
contributing to the clinical outcome in a multifactorial process. In
this case, it is difficult to distinguish determinants from
predisposing and/or aggravating factors.
There are two well-recognized clinical syndromes that result from
immunologically mediated glomerular disease. These are the nephrotic
syndrome and the nephritic syndrome. The nephrotic syndrome is
characterized by massive proteinuria, hypoalbuminaemia (from
proteinuria) and generalized oedema. The syndrome is the result of
pathology that affects the integrity of the GBM. Although the cause of
85% of nephrotic syndrome is not known, it is the syndrome most
frequently occurring with toxic nephropathies, it is generally dose
related, and it is reversible. Glomerular pathology is most often
membranous glomerulonephritis or circulating immune-complex disease.
The nephritic syndrome is characterized by haematuria and a decreasing
GFR and is likely to be accompanied by some degree of hypertension.
The pathological changes in the glomeruli are usually caused by those
diseases or toxic agents that produce an inflammatory proliferative
response within the glomeruli. The proliferation may involve
endothelial mesangial or epithelial cells and may be associated with
an inflammatory cell infiltration.
7.2.6 Radiological techniques
Radiocontrast media can be used to study the kidney either by
conventional or by retrograde pyelography. There are a number of
limitations to this technique, such as the need for adequate renal
function by which to image the organ. There is adequate evidence that
radiocontrast agents have a nephrotoxic potential. Media of high
osmolality are especially likely to precipitate renal failure. In
addition, those patients who are dehydrated or with reduced blood
volume are at special risk. There is also evidence to show that
patients who have multiple myeloma represent a special risk group.
7.2.7 Other non-invasive renal assessment
Whereas gamma camera renography and ultrasound are
well-established techniques for the assessment of renal function,
increasing use is being made of ultrasound linked with Doppler flow,
nuclear magnetic resonance imaging, and spectoscopy; ureteral
fibroscopy may be used to view the pelvis directly. Ultrasound
evaluation of the kidneys provides a means of excluding obstruction as
a cause of anuria or oliguria in ARF. It may also reveal papillary
necrosis or perirenal haematomas and obviate the intrinsic dangers of
pyelography. Radionuclide scans can be used to identify major
atherothrombotic events and cortical necrosis. They can also be used
to show reduced renal blood flow, which is usually preserved (some 50%
of normal) in acute tubular necrosis, but is severely compromised in
acute glomerulonephritis and vasculitis. Renal biopsy should not be
used for ARF unless the duration exceeds three weeks and there is no
obvious cause. It can then help diagnose glomerulonephritis,
vasculitis, and AIN.
8. DETECTION OF NEPHROTOXICITY IN HUMANS
Traditional methods of diagnosing renal damage and predicting
their health significance are not sufficient to deal with the
diversity of chemically induced lesions (Bach et al., 1989). This is
because the kidney can undergo substantial chemically induced injury
without any clinical indication, owing to its considerable functional
reserve. This masks a substantial amount of renal degeneration
(Friedlander et al., 1989). Thus, for example, the single common
measurement of GFR may only show incipient acute or chronic renal
failure.
Most nephropathies in humans are of unknown origin. There is,
however, some indirect evidence that toxicant exposure could be
involved. For example, 80% of the cases of membranous nephropathy are
of unknown origin. Among the remaining 20%, half of them have been
found to be associated with drug exposure. It is likely, therefore,
that a percentage of those cases of unknown origin are also related to
toxic exposure.
8.1 Markers of nephrotoxicity
Over the last ten years, new biochemical and immunochemical
methods have been developed for detecting early renal changes in
humans. Their application has given rise to a number of markers, some
of which also are available for studies on experimental animals and
have been reviewed in section 7.2. Although some tests may open new
perspectives in terms of selectivity, sensitivity, and specificity,
most of the recently developed markers need further validation. Hence,
some conceptual and methodological problems must be addressed and new
methods critically evaluated. This is especially important when
implementing screening programmes for the early detection of kidney
damage and/or dysfunction in humans.
8.1.1 General requirements
Markers to be used for screening purposes in population groups
should fulfill certain general criteria, including specificity,
non-invasiveness, sensitivity in detecting early renal functional
changes, and predictive value for development of renal insufficiency.
Analytical methods must be reproducible, easy to perform, and
applicable to a large number of samples, and the samples must be
stored appropriately to ensure stability.
The prevalence of true positive results among subjects who are
actually ill indicates the sensitivity of the marker, whereas the
prevalence of true negative findings among healthy individuals is a
measure of its specificity.
Some markers meet the requirement of specificity, although it may
be difficult to establish clear-cut distinctions in individual cases.
For instance, whereas gross changes in proteinuria involving proteins
of low and high relative molecular mass indicate tubular and
glomerular damage, respectively, marginal increases over reference
limits may be due to either condition (Mutti, 1989).
The predictive value of markers of nephrotoxicity has not been
systematically tested, but it is usually assumed that such markers as
the urinary excretion of single plasma proteins are highly
sensitive but somewhat aspecific. This is due to the fact that they
may be increased by a number of physiological conditions (e.g.,
physical workload, posture, pharmacological effects of exogenous
substances, or even meat meals). On the contrary, other markers such
as serum creatinine are considered to be relatively specific, but
relatively insensitive, since they reflect late changes, occurring
when more than 50% of the renal reserve has been lost. Within these
extremes, there are various possibilities which should be carefully
weighed according to the methodological context dealt with.
The predictive value of markers, in terms of sensitivity and
specificity, should be taken into account when evaluating individual
results. For some parameters (e.g., proteinuria involving proteins of
low relative molecular mass) minor dysfunctions at the tubular level
will result in deviations of several orders of magnitude from
reference values, whereas the same relative change in other parameters
(e.g., brush-border antigens, enzyme activities) may indicate severe
tubular injury.
In the clinical context, it is often difficult to establish
correct etiological diagnoses because of the long latency between
exposure and the development of overt disease. For the same reason, it
is sometimes difficult in epidemiological work to assess the
prognostic value of early changes. Nonetheless, it is clear that
markers used in the clinical context must be specific enough to avoid
further undue and sometimes invasive procedures, which tend to
accumulate once a subject enters a diagnostic decision tree (Mold &
Stein, 1986).
The quantification of any constituents in urine may only be
obtained by assessing urine flow rate. The accuracy of timed urine
samples (especially when obtained during epidemiological surveys) is
unreliable and most analytes may be unstable. There are still a number
of factors that can confound or modify the renal response, such as
time of sampling (owing to spontaneous rhythms), posture, physical
work load, and diet. Thus, there is a need to use standardized
procedures to limit possible interferences that can increase the
variability between and within subjects. To overcome these
methodological problems, there is an increasing tendency to use spot
samples, generally the second sample of the day, and to express
results as a function of creatinine.
8.1.2 Diagnostic value
The predictive value of a marker only in part contributes to its
diagnostic validity, which is defined in terms of the probability that
the classification based on test values actually corresponds to the
subject's condition. The diagnostic value may be both positive and
negative, the positive diagnostic value being the probability that a
subject classified as positive by the test is actually ill, and the
negative diagnostic value being the probability that subjects negative
at the test are actually healthy individuals. It must be stressed that
the diagnostic validity is only marginally affected by the predictive
value of the markers, since in most screening programmes a low
prevalence of disease is the major determinant of pitfalls.
Thus, markers must be selected according to the condition under
evaluation. When studying groups at risk, emphasis should be given to
sensitivity, whereas specificity should receive adequate attention in
the clinical setting.
8.1.3 Prognostic value
Another important property of markers, which has so far only been
addressed in a few studies, is their prognostic value, i.e. their
ability to predict the "natural" course of the disease. In most cases
early functional changes may be compensated and structural damage may
be repaired. However, both conditions may also trigger a cascade of
events leading to end-stage renal disease. The ability to target
groups actually at risk and to predict the "natural" evolution of the
disease is part of effective prevention. This can be achieved by
reducing exposure (primary prevention) or by establishing an
individual diagnosis at reversible stages (secondary prevention).
Microscopic examination of urine sediment, although impractical
in population studies, may help in establishing individual
differential diagnosis. Even if it is insensitive for detecting
nephrotoxicity, this test is suitable at the individual level to
exclude confounding factors accounting for increased excretion of
plasma proteins (e.g., leukocyturia, haematuria, or bacteriuria).
8.2 Screening for nephrotoxicity in humans
In the epidemiological context, it can be assumed that negative
results are recorded in actually healthy subjects, whereas the
probability of false positive values is rather high because of the low
prevalence of the disease. Thus, health surveillance programmes should
be mainly aimed at excluding harmful situations rather than at
identifying ill people. This goal may be achieved using very sensitive
markers of subclinical renal impairment. A clinical diagnosis in
individuals showing abnormal test values should then be established
only after repeated measurements and complementary confirmatory tests.
Practical considerations are also important when planning population
studies, where sampling procedures that are invasive or too elaborate
cannot be included in the protocol.
8.2.1 Glomerular filtration
Clearance procedures can seldom be adopted when screening
population groups, since only single blood and spot urine samples are
usually available. However, it must be stressed that some tests
fulfilling the above objective have proved their diagnostic and
prognostic validity and for this reason they are now also being
employed in clinical practice.
An indirect method of assessing GFR by using a single blood
sample is the measurement of serum creatinine and the expression of
its reciprocal value adjusted to constant body surface area. This has
been correlated with GFR (Siersbaek-Nielson et al., 1971). The same
concept applied to ß2-microglobulin would increase the sensitivity of
such an approach, owing to the higher relative molecular mass and the
use of more accurate and reproducible analytical methods (Wibell et
al., 1973).
8.2.2 Tests designed to assess selective dysfunction
Serious renal diseases (e.g., nephrotic or Fanconi's syndromes)
may occur in the absence of haemodynamic changes. In such
circumstances, renal insufficiency is a late event that is preceded by
earlier, though serious, selective dysfunctions occurring at the
glomerular and proximal tubular level. These may be revealed by
measuring single plasma proteins in urine (see also section 8.3.3.).
The excretion in urine of proteins of high relative molecular mass
generally reflects glomerular dysfunction, whereas urinary excretion
of proteins of low relative molecular mass may be indicative of
tubular dysfunction. The assessment of protein electrophoretic
patterns has been discussed in section 7.2.4.
8.2.3 Tests designed to assess tissue damage
Tubular dysfunction is not necessarily associated with
histopathological changes. It has been shown that pharmacological
inhibition of tubular uptake may account for proteinuria (involving
proteins of low relative molecular mass) observed after protein load
(Buzio et al., 1989). Furthermore, highly selective damage to a
tubular segment may be functionally compensated by other segments of
the nephron. Methods aimed at detecting cellular damage may thus help
both to show subclinical lesions and to interpret associated
dysfunctions. The urinary excretion of tissue constituents may be
measured through the enzymic or immunochemical characteristics of
material shed into the urine.
Renal functional changes may be reversible, depending on the
efficiency of repair mechanisms and the cessation of exposure to the
offending agent. Repeated monitoring may help to distinguish
progressive renal disease from transient lesions (Thornley et al.,
1985).
8.2.3.1 Enzymuria
The general principles describe in section 7.2.4.3. also apply to
humans. Even when using freshly voided spot samples, urine is a
hostile environment for most enzymes. Only a few enzymes (e.g.,
N-acetly-ß-D-glycosaminidase) show acceptable stability and
analytical precision (Price, 1982). Table 8 lists some human urinary
enzymes that have been used in nephrotoxic studies.
8.2.3.2 Immunoreactive tissue constituents
Tissue constituents (including enzymes) are physiologically shed
into the urine as a consequence of cell turnover and metabolism. When
they are detected by immunochemical methods (e.g., immunofluorescence,
enzyme-linked immunosorbent assay), they are referred to as antigens.
Tamm-Horsfall glycoprotein is a specific renal protein, localized on
the membrane of cells of the thick ascending limb of the loop of
Henle, which is excreted in the urine at a relatively constant rate.
This can increase following injury to the distal part of the tubule
and is depressed when the renal mass is reduced (Thornley et al.,
1985). Although increased excretion of proximal tubular antigens was
reported twenty years ago in various clinical situations such as
tubular necrosis, allograft rejection, and
cephalotin/gentamicin-induced nephrotoxicity (Antoine et al., 1969;
Rosenmann et al., 1971; Scherberich et al., 1976, 1984), only recently
has it been possible to improve significantly the specificity and
reproducibility of such an approach, relying on the properties of
monoclonal antibodies.
Monoclonal antibodies to human brush-border antigens have been
produced by Mutti et al. (1985). Monoclonal antibodies may be
conveniently employed in ELISA procedures set up to detect trace
amounts of antigens in biological samples. Although the BB-50
brush-border antigen was also localized in peritubular capillaries
(Mutti et al., 1985), subsequent work lead to the identification of a
monoclonal antibody reacting with an antigen located specifically in
the brush border and thus called BBA or brush-border antigen (Mutti et
al., 1988). Promising results have been obtained in several
cross-sectional investigations on groups at risk of chemically induced
renal damage (for a review, see Mutti, 1989). Similar results were
obtained with monoclonal antibodies to rat brush-border cross-reacting
with the human kidney (Tokoff-Rubin et al., 1986).
Monoclonal antibodies have also been produced that react
specifically with other segments along the nephron (namely S3) where
the intestinal isoform of alkaline phosphatase is located (Verpooten
et al., 1989). They could be used to monitor the effects of chemicals
(e.g., mercury) acting selectively on the straight part of proximal
tubules. All of these recently developed tests need further validation
in well-designed longitudinal studies, since their prognostic value is
currently unknown.
8.3 Clinical investigations
The clinical diagnosis of toxic nephropathy frequently relies
heavily on the history of patients who occasionally showed symptoms
and/or laboratory abnormalities suggesting chronic failure without any
obvious recent cause.
Diagnosis can only be made by excluding other known conditions or
risk factors. This has to be assessed by estimating exposure to
nephrotoxins in relation to the known dose-effect/response
relationship and the temporal sequence of events that follow such
exposure. The role of individual susceptibility should also be
considered. In some circumstances, nephrotoxicity may be one factor in
a multifactorial process leading to clinical renal disease.
Retrospective data about exposure and early changes in renal function
are usually not available.
Most progressive kidney diseases have a subtle onset. This is the
reason why the markers designed for use in epidemiological studies are
becoming part of clinical investigations.
Owing to the short latency between exposure and the development
of severe symptoms, acute renal failure should be accurately diagnosed
in all cases. The simple evaluation of serum creatinine (increases of
3-5 mg/litre or 50% above baseline values) makes it possible to detect
nephrotoxic reactions.
8.3.1 Invasive techniques
The use of invasive techniques is limited by ethical constraints.
8.3.1.1 Biopsies from humans
Renal biopsy is the only way to identify glomerular, tubular, or
interstitial renal diseases. However, the risk-to-benefit ratio must
be considered carefully in each individual patient being evaluated,
and there is no universal agreement on the conditions in which
percutaneous renal biopsy may be useful.
8.3.1.2 Autopsy in humans
Autopsies performed on patients with end-stage renal failure only
reveal the etiology of the renal disease in a small percentage of
cases.
8.3.2 Tests designed to assess glomerular filtration and renal blood flow
Traditional methods based on inulin and PAH clearances are
progressively being substituted by more convenient methods such as the
clearance of 51Cr-EDTA and 99Tc-DTPA to assess GFR or the
clearance of 125I-hippuran to measure renal plasma flow. These
techniques are thought to be more sensitive than creatinine clearance
and more accurate than colorimetric methods.
8.3.3 Proteinuria
Section 7.2.4. contains a detailed discussion of experimental
studies involving proteinuria. Table 9 gives information on some
proteins excreted in human urine. Values that are in excess of the
normal range may be indicative of renal dysfunction.
Table 9. Excretion rates of common urine proteins
Protein Relative molecular mass Normal range
(Daltons)
Albumin 68 000 < 30 mg/day
ß2-microglobulin 12 800 < 0.3 mg/day
Retinol-binding Protein 21 400 < 0.3 mg/day
IgG 160 000 2-3 mg/day
alpha2-microglobulin ± 30 000 ?
8.3.4 Tests designed to assess selective damage
Tissue constituents may be shed into the urine following toxic
damage to specific structures. All of these specific antigens may be
detected by using immunochemical or biochemical methods designed to
measure their concentration or enzymic activity, respectively.
Although they have been designed expressly for evaluating population
groups, these tests may also be useful in the clinical setting to
monitor patients at risk. Even if they are very sensitive and
relatively specific, they need further validation under carefully
controlled clinical conditions, particularly in longitudinal studies.
9. SUMMARY AND CONCLUSIONS
The kidneys are the main organs of excretion and homeostasis for
water-soluble molecules. The functional unit of the kidney is the
nephron, essentially a continuous tube of highly specialized
heterogeneous cells, which exhibits marked structural, functional, and
biochemical organization. There are pronounced differences between the
nephrons themselves, depending on the cortical localization of the
individual glomeruli. This complex structural organization, combined
with differences in regional vascularity arising from the specialized
vascular arrangement, produces a highly complex heterogeneous organ.
Much of the anatomical and functional understanding developed
from animals is directly applicable to the human kidney. However, the
biomedical and metabolic processes in the human kidney, as well as the
differences among animal species, have not been as thoroughly
elucidated. Thus, the ability to extrapolate the effects of chemicals
among species is limited.
Several chemicals (both therapeutic and non-therapeutic) have
toxic effects on one or more anatomical elements of the kidney. Toxic
effects may be acute or chronic, and they may be direct or mediated
indirectly through immunological mechanisms. The health impact of
nephrotoxic chemicals is related to risk factors, which include the
intergrade of the renal functional reserve and factors such as
pre-existing renal damage, disease, age, sex, and diet.
The epidemiology of chemically induced nephrotoxicity by
individual chemicals or in mixed exposures has been inadequately
studied. The contribution of chemicals to the overall incidence of
nephropathy and of chronic renal failure is, with few exceptions,
undefined. In the case of some occupationally exposed groups and
analgesic associated renal disease, there has been extensive research
that has shown variations in incidence between groups and countries.
However, it is estimated that up to 18% of end-stage renal disease may
be due to analgesic nephropathy and up to 5% to other toxic
nephropathies. About 50% of end-stage renal disease is of unknown
etiology. A major problem in assigning a cause for end-stage renal
disease is the long latency and/or slow development of chronic renal
failure, which makes retrospective identification of the causative
agent difficult. In only a few cases is measurement of tissue (body,
kidney) levels of chemicals relevant to the diagnosis. A further
problem has been the lack of consistency in diagnostic and
pathological criteria and terminology.
Renal anatomical and physiological differences make direct
extrapolation from experimental systems (in vivo and in vitro) to
humans difficult. There are very few examples of nephrotoxic chemicals
where there are adequate comparable data from animals and humans to
form a firm basis for the assessment of potential human health risk.
Chemicals may damage selectively vulnerable kidney structures or
activate immunological mechanisms. Mechanisms of renal injury fall
generally into two categories: (a) immunologically induced disease of
acute interstitial nephritis; (b) those that primarily affect the
glomerulus by either anti-GBM-mediated antibodies or immune complexes.
Another major group is composed of diseases initiated by chemicals or
their metabolites that interfere with cellular biochemical and
haemodynamic effects, etc. Factors that can modify cellular injury by
toxicants include cellular transport systems, pinocytosis, metabolic
degradation, and interaction with cellular proteins, lipids,
membranes, DNA, and perhaps other cellular constituents.
The increasing use of therapeutic agents and chemicals increases
the possibility of nephrotoxicity. Nephrotoxicity induced by
therapeutic agents depends on the dose and duration of exposure (e.g.,
combination analgesics leading to renal papillary necrosis).
Nephrotoxic effects of analgesics, antibiotics (such as the
aminoglycosides), anticancer agents (such as cis-platinum), and a
variety of other agents have been investigated extensively. Chemicals
frequently used in industry or the home, e.g., chlorinated
hydrocarbons and ethylene glycol, also have the potential to produce
renal damage. Environmental chemicals such as lead and cadmium are
capable of inducing nephrotoxicity. These agents act as toxicants
after intracellular accumulation of the parent compound or after renal
or extrarenal hiptransformation. Multichemical exposure may result in
antagonistic or synergistic responses.
Turnours of the kidney and urinary tract account for less than
2-3% of all human cancers, but the frequency of these tumours is
increasing, suggesting a role for environmental factors. Although many
drugs and chemicals have been shown to be carcinogenic in experimental
models, only a few specific substances have been related to turnouts
in man. These include asbestos (renal adenocarcinoma), analgesic abuse
(transitional carcinomas), and lifestyle factors (tobacco use,
alcohol, coffee). There are population groups with urinary tract
cancer of as yet undetermined etiology. Occupational chemicals have
been related to the etiology of cancer of the urinary bladder. Many
drugs and chemicals cause interstitial nephritis, which may in itself
be a factor in developing urinary tract cancers.
No single in vivo or in vitro method is sufficient to assess
chemically induced renal dysfunction. Therefore, it is advisable not
only to screen compounds for nephrotoxic potential, but to incorporate
mechanistic studies of target cell toxicity into the experimental
protocols. To accomplish this, both in vivo and in vitro
investigations should be utilized in concert. In vitro studies
involve those where anatomical cellular relationships are maintained
(e.g., isolated perfused kidney and tubules, renal cortical slices,
and isolated nephron segments) and those where renal cells are used
(e.g., cell suspension, primary cell cultures, established cell lines,
and subcellular fractions). In vivo studies utilize both invasive
and non-invasive techniques. Invasive procedures include
histopathological and routine renal function measurements.
Non-invasive procedures permit repeated assessment of renal function
in animals through the measurement of an array of renal function tests
(glomerular filtration, tubular function, proteinuria, enzymuria,
etc.). Specialized biochemical tests should be used where relevant.
The appropriate mixture of in vivo and in vitro experiments will
reveal whether or not chemicals are nephrotoxic and give insights into
potencies, sites, and mechanisms of toxicity.
Traditional methods for the assessment of renal function in
humans are inadequate for the timely diagnosis of chemically induced
renal dysfunctionand prediction of its health significance. The lack
of specific, early markers for nephrotoxicity is particularly
troublesome. Non-invasive assessment of nephrotoxity should employ
markers of high specificity, sensitivity for detection of early renal
changes, and predictive value for the development of renal
insufficiency. Present techniques for monitoring glomerular or tubular
function are useful only when severe renal damage has developed.
Although immuno-reactive tissue constituents are being suggested as
appropriate markers, their suitability needs to be validated in
well-designed longitudinal studies.
10. RECOMMENDATIONS
1. A continued effort should be made to develop and validate more
selective and specific markers, including monoclonal antibodies, for
assessment of renal dysfunction in animals. These markers may be
applicable to humans.
2. The data base for predicting the potential of chemicals for human
nephrotoxicity should be extended. This includes development and
validation of experimental animal approaches ( in vivo and in
vitro), alternative methods for studying nephrotoxicity, information
on interspecies differences, and experience from the preclinical
evaluation of new therapeutic agents in humans.
3. Epidemiological studies (i.e. prospective studies in occupational
and general population groups exposed to nephrotoxic chemicals or
involved with analgesic abuse) should be reinforced.
4. More effort should be made to establish the role of chemicals in
the etiology of renal disease at the earliest diagnostic stage (e.g.,
work history, tissue monitoring for nephrotoxins).
5. Understanding of the mechanisms of action of nephro-toxicants
will be helpful in the prevention and clinical management of unwanted
renal effects, and may help in predicting the nephrotoxic potential
for new drugs and chemicals. Areas of particular importance for
further research are:
* immunological mechanisms;
* direct effects of chemicals on membranes, including mechanisms of
lipid peroxidation, membrane/chemical interaction, ion shifts,
and receptor-mediated events;
* activation of proto-oncogenes and cell differentiation;
* regulation of cellular metabolism.
6. There is a need to identify and correlate specific functions with
discrete anatomical locations within the kidney.
7. The role of genetic variation and susceptibility to the toxic
effects of drugs and chemicals should be studied further.
8. The relationship between nephrotoxicity and renal carcinogenesis
(e.g., mycotoxins and Balkan nephropathy) needs further study.
REFERENCES
AHMADIZADEH, M., KUO, C.-H., & HOOK, J.B. (1981) Nephrotoxicity and
hepatotoxicity of chloroform in mice: effect of deuterium
substitution. J. Toxicol. environ. Health, 8: 105-111.
AINSWORTH, S.K., SWAIN, R.P., WATABE, N., BRACKETT, N.C., PILIA, P.,
& HENNIGAR, G.R. (1981) Gold nephropathy, ultrastructural fluorescent
and energy-dispersive X-ray microanalysis study. Arch. Pathol. lab.
Med., 105: 373-378.
ALDEN, C.L. (1989) Male rat specific alpha2u-globulin nephropathy and
tumourigenesis. In: Bach, P.H. & Lock, E.A., ed. Nephrotoxicity:
Extrapolations from in vitro to in vivo, and animals to man, New
York, London, Plenum Press, pp. 535-542.
ALDEN, C.L., KANERVA, R.L., RIDDER, G., & STONE, L.C. (1984) The
pathogenesis of the nephrotoxicity of volatile hydrocarbons in the
male rat. In: Mehlman, M.A., Hemstreet, C.P., Thorpe, J.J., & Weaver,
N.K., ed. Renal effects of petroleum hydrocarbons, Princeton, New
Jersey, Princeton Scientific Publishers, pp. 107-120 (Advances in
Modern Environmental Toxicology, Vol. VII).
ANAND-SRIVASTAVA, M.B., VINAY, P., GENEST, J., & CANTIN, M. (1986)
Effect of atrial natriuretic factor on adenylate cyclase in various
nephron segments. Am. J. Physiol., 251: F417-F423.
ANDERS, M.W. (1980) Metabolism of drugs by the kidney. Kidney Int., 8:
636-647.
ANDERSON, P.M. & SCHULTZE, M.O. (1965) Cleavage of
S-(1,2-dichlorovinyl)-L-cysteine by an enzyme of bovine origin. Arch.
Biochem. Biophys., 111: 593-602.
ANDERSON, S. & BRENNER, B.M. (1986) Effects of aging on the renal
glomerulus. Am. J. med., 80: 435-442.
ANTOINE, B., NEVEU, T., LESKI, M., & BACH J.F. (1969) Histuria during
renal transplantation. Transplantation, 8: 110-120.
ARMBRECHT, H.J., BIRNBAUM, L.S., ZENSER, T.V., MATTAMMAL, M.B., &
DAVIS, B.B. (1979) Renal cytochrome P-450's - Electrophoretic and
electron paramagnetic resonance studies. Arch. Biochem. Biophys., 197:
277-284.
ASKERGREN, A. (1981) Studies on kidney function in subjects exposed to
organic solvents. III. Excretion of cells in the urine. Acta med.
Scand., 210: 103-106.
ASKERGREN, A., ALLGEN, L.G., KARLSSON, C., LUNDBERG, I., & NYBERG, E.
(1981) Studies on kidney function in subjects exposed to organic
solvents. I. Excretion of albumin and ß2-microglobulin in the urine.
Acta med. Scand., 209: 479-483.
ATTALLAH, A. & STAHL, R. (1980) Inhibition of renal PGE2
biosynthesis in vivo: regional differences. Prostaglandins, 19:
649-650.
AUKLAND, K. (1980) Methods for measuring renal blood flow: Total flow
and regional distribution. Annu. Rev. Physiol., 42: 543-555.
AUTOR, A.P., ed. (1977) Biochemical mechanisms of paraquat toxicity,
New York, London, San Francisco, Academic Press.
AVENDANO, L.H. & LOPEZ-NOVOA, J.M. (1987) Glomerular filtration and
renal blood flow in the aged. In: Macias-Nunez, I.F. & Cameron, J.S.,
ed. Renal function and diseases in the elderly, London, Boston,
Toronto, Butterworth, pp. 27-48.
BACH, P.H. (1989) The detection of chemically induced renal injury,
the cascade of degenerative morphological and functional changes that
follow the primary nephrotoxic insult and the evaluation of these
changes by in vitro methods. Toxicol. Lett., 46: 237-250.
BACH, P.H. & BRIDGES, J.W. (1984) The role of prostaglandin synthase
mediated metabolic activation of analgesics and non-steroidal
anti-inflammatory drugs in the development of renal papillary necrosis
and upper urothelial carcinoma. Prostaglandins Leukotrienes Med., 15:
251-274.
BACH, P.H. & BRIDGES, J.W. (1985a) Chemically induced renal papillary
necrosis and upper urothelial carcinoma. CRC crit. Rev. Toxicol., 15:
217-439.
BACH, P.H. & BRIDGES, J.W. (1985b) A decision tree approach for the
application of drug metabolism and kinetics to in vivo and in vitro
toxicological and pharmacological testing. Arch. Toxicol., Suppl. 8:
173-188.
BACH, P.H. & GREGG, N. (1988) Experimentally induced renal papillary
necrosis and upper urothelial carcinoma. Inter. Rev. exp. Pathol., 30:
1-54.
BACH, P.H. & HARDY, T.L. (1985) The relevance of animal models to the
study of analgesic associated renal papillary necrosis in man. Kidney
Int., 28: 605-613.
BACH, P.H. & KWIZERA, E.N. (1988) Nephrotoxicity: A rational approach
to in vitro target cell injury in the kidney. Xenobiotica, 16:
685-698.
BACH, P.H. & LOCK, E.A. (1982) The use of renal tissue slices,
perfusion and infusion techniques to assess renal function and
malfunction. In: Bach, P.H., Bonner, F.W., Bridges, J.W., & Lock,
E.A., ed. Nephrotoxicity: Assessment and pathogenesis, New York,
Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 128-143.
BACH, P.H. & LOCK, E.A., ed. (1985) Renal heterogeneity and target
cell toxicity, New York, Chichester, Brisbane, Toronto, John Wiley and
Sons.
BACH, P.H. & LOCK, E.A., ed. (1987) Nephrotoxicity in the experimental
and the clinical situation, Dordrecht, Boston, Lancaster, Martinus
Nijhoff Publishers.
BACH, P.H. & LOCK, E.A., ed. (1989) Nephrotoxicity: Extrapolation from
in vitro to in vivo, and animals to man, New York, London, Plenum
Press.
BACH, P.H., BONNER, F.W., BRIDGES, J.W., & LOCK, E.A., ed. (1982)
Nephrotoxicity: Assessment and pathogenesis, New York, Chichester,
Brisbane, Toronto, John Wiley and Sons.
BACH, P.H., GRASSO, P., MOLLAND, E.A., & BRIDGES, J.W. (1983) Changes
in the medullary glycosaminoglycan histochemistry and microvascular
filling during the development of 2-bromoethanamine
hydrobromide-induced renal papillary necrosis. Toxicol. appl.
Pharmacol., 69: 333-344.
BACH, P.H., KETLEY, C.P., BENNS, S.E., AHMED, I., & DIXIT., M. (1985)
The use of isolated and cultured renal cells in nephrotoxicity -
practice, potential and problems. In: Bach, P.H. & Lock, E.A., ed.
Renal heterogeneity and target cell toxicity, New York, Chichester,
Brisbane, Toronto, John Wiley and Sons, pp. 505-518.
BACH, P.H., KETLEY, C.P., DIXIT, M., & AHMED, I. (1986) The mechanisms
of target cell injury in nephrotoxicity. Food chem. Toxicol., 24:
775-779.
BACH, P.H., GREGG, N.J., & WACHSMUTH, E.D. (1987) The application of
histochemistry at the light microscopic level to the study of
nephrotoxicity. In: Bach, P.H. & Lock, E.A., ed. Nephrotoxicity in the
experimental and the clinical situation, Dordrecht, Boston, Lancaster,
Martinus Nijhoff Publishers, pp. 19-84.
BACH, P.H., BERLIN, A., HESELTINE, E., KRUG, E., LAUWERYS, R., SMITH,
E., & VAN DER VENNE, M.-T., ed. (1989) Proceedings of the
International Workshop on the Health Significance of Nephrotoxicity.
Toxicol. Lett., 46: 1-306.
BACHUR, N.R., GORDON, S.L., GEE, M.V., & KON, H. (1979) NADPH
cytochrome P-450 reductase activation of quinone anticancer agents to
free radicals. Proc. Natl Acad. Sci., 76: 954-957.
BAER, P.G. (1981) The contribution of prostaglandins to renal blood
flow maintenance is determined by the level of activity of the
renin-angiotensin system. Life Sci., 28: 587-593.
BAGGETT, J.M. & BERNDT, W.O. (1984a) Renal and hepatic glutathione
concentrations in rats after treatment with hexachloro-1,3-butadiene
and citrinin. Arch. Toxicol., 56: 46-49.
BAGGETT, J.M. & BERNDT, W.O. (1984b) Interaction of potassium
dichromate with the nephrotoxins, mercuric chloride and citrinin.
Toxicology, 33: 157-169.
BAGGETT, J.M. & BERNDT, W.O. (1985) The effect of potassium dichromate
on the urinary excretion, organ and subcellular distribution of
203Hg-mercuric chloride. Toxicol. Lett., 29: 115-121.
BAILIE, M.B., SMITH, J.H., NEWTON, J.F., & HOOK, J.B. (1984) Mechanism
of chloroform nephrotoxicity. IV. Phenobarbital potentiation of in
vitro chloroform metabolism and toxicity in rabbit kidneys. Toxicol.
appl. Pharmacol., 74: 285-292.
BAKER, E.L., GOYER, R.A., FOWLER, B.A., KHETTERY, U., BERNARD, D.B.,
ADLER, S., DEVERE WHITE, R., BABAYAN, R., & FELDMAN, R.G. (1980)
Occupational lead exposure, nephropathy, and renal cancer. Am. J. ind.
Med., 1: 139-148.
BANK, N., MUTZ, E.F., & AYNEDIJIAN, H.S. (1967) The role of "leakage"
of tubular fluid in anuria due to mercury poisoning. J. clin. Invest.,
46: 695-704.
BANKI, K., ELFARRA, A.A., LASH, L.H., & ANDERS, M.W. (1986) Metabolism
of S-(2-chloro-1,1,2-trifluoroethyl)-L-cysteine to hydrogen sulfide
and the role of hydrogen sulfide in
S-(2-chloro-1,1,2-trifluoroethyl)-L-cysteine-induced mitochondrial
toxicity. Biochem. biophys. Res. Commun., 138: 707-713.
BANNASCH, P. & ZERBAN, H. (1986) Renal cell adenoma and carcinoma. In:
Jones, T.C., Mohr, U., Hunt, R.D., ed. Monographs on pathology of
laboratory animals, urinary system, Berlin, Heidelberg, New York,
Springer-Verlag, pp. 112-139.
BANNASCH, P., KRECH, R., & ZERBAN, H. (1978a) [Morphogenesis and
micro-morphology of epithelial kidney tumours in rats poisoned with
nitrosomorpho-line. II. Tubular glycogenesis and the formation of
clear-cell or acidophilic-cell tumours.] Z. Krebsforsch., 92: 63-86
(in German).
BANNASCH, P., KRECH, R., & ZERBAN, H. (1978b) [Morphogenesis and
micro-morphology of epithelial kidney tumours in rats poisoned with
nitrosomorpho-line. III. Oncocyte tubules and oncocytomas.] Z.
Krebsforsch., 92: 87-104 (in German).
BANNASCH, P., KRECH, R., & ZERBAN, H. (1980) [Morphogenesis and
micro-morphology of epithelial kidney tumours in rats poisoned with
nitrosomorpho-line. IV. Tubular lesions and basophilic tumours.] J.
Cancer Res. clin. Oncol., 98: 243-265 (in German).
BANNASCH, P., BENNER, U., HACKER, H.J., KLIMEK, F., MAYER, D., MOORE,
M., & ZERBAN, H. (1981) Cytochemical and biochemical analysis of
carcinogenesis. Histochem. J., 13: 799-820.
BARRATT, M. (1983) Proteinuria. Br. med. J., 287: 1489-1490.
BARRY, D.N. & BOWNESS, J.M. (1975) Identification and turnover of
glyco-saminoglycans in rat kidneys. Can. J. Biochem., 53: 713-720.
BATELLE, D., GAVIRIA, M., GRUPP, M., ARRUDA, J.A., WYNN, J., &
KURTZMAN, N.A. (1982) Distal nephron function in patients receiving
lithium therapy. Kidney Int., 21: 477-486.
BATUMAN, V., MAESAKA, J.K., HADDAD, B., TEPPER, E., LANDY, E., &
WEDEEN, R.P. (1981) The role of lead in gout nephropathy. New Engl. J.
Med., 304: 520-523.
BATUMAN, V., LANDY, E., MAESAKA, J.K., & WEDEEN, R.P. (1983)
Contribution of lead to hypertension with renal impairment. New Engl.
J. Med., 309: 17-21.
BAVEREL, G., BONNARD, M., D'ARMAGNAC DE CASTANET, E., & PELLET, M.
(1978) Lactate and pyruvate metabolism in isolated renal tubules of
normal dogs. Kidney Int., 14: 567-575.
BAVEREL, G., BONNARD, M., & PELLET, M. (1979) Lactate and pyruvate
metabolism in isolated human kidney tubules. FEBS Lett., 101: 282-286.
BAVEREL, G., FORISSIER, M., & PELLET, M. (1980a) Lactate and pyruvate
metabolism in dog renal outer medulla. Effects of oleate and ketone
bodies. Int. J. Biochem., 12: 163-168.
BAVEREL, G., GENOUX, C., FORISSIER, M., & PELLET, M. (1980b) Fate of
glutamate carbon and nitrogen in isolated guinea-pig kidney cortex
tubules. Biochem. J., 188: 873-880.
BECKER, C.G., BECKER, E.L., MAHER, J.F., & SCHREINER, G.E. (1962)
Nephrotic syndrome after contact with mercury. A report of five cases,
three after use of ammoniated mercury. Arch. intern. Med., 110:
178-186.
BEEUWKES, R. (1980) The vascular organisation of the kidney. Annu.
Rev. Physiol., 42: 531-542.
BEKERSKY, I., COLBURN, W.A., FISHMAN, L., & KAPLAN, S.A. (1980)
Metabolism of salicylic acid in the isolated perfused rat kidney.
Intercon-version of salicyluric and salicylic acids. Drug Metab.
Disp., 8: 319-324.
BELLEMAN, P. (1980) Primary monolayer cultures of liver parenchymal
cells and kidney tubules as a useful new model for biochemical
pharmacology and experimental toxicology. Studies in vitro on
hepatic membrane transport, induction of liver enzymes and adaptive
changes in renal cortical enzymes. Arch. Toxicol., 44: 63-84.
BENDELE, A.M., CARLTON, W.W., KROGH, P., & LILLEHOJ, E.B. (1985)
Ochratoxin A carcinogenesis in the (C57BL/6J x C3H)F1 mouse. J. Natl
Cancer Inst., 75: 733-742.
BENDZ, H. (1983) Kidney function in lithium-treated patients: a
literature survey. Acta psychiatr. Scand., 68: 303-24.
BENGTSSON, U. (1962) A comparative study of chronic non-obstructive
pyelonephritis and renal papillary necrosis. Acta med. Scand.,
388(Suppl.): 1-71.
BENGTSSON, U., JOHANSSON, S., & ANGERVALL, L. (1978) Malignancies of
the urinary tract and their relation to analgesic abuse. Kidney Int.,
13: 107-113.
BENNETT, W.M. (1983) Aminoglycoside nephrotoxicity. Nephron, 35:
73-77.
BENNETT, W.M. (1985) Lead nephropathy. Kidney Int., 28: 212-220.
BENNETT, W.M. (1986) Drugs and renal disease, New York, Churchill
Livingstone.
BENNETT, W.M., HOUGHTON, D.C., MCCARRON, D.A., ELLIOTT, W.C., PORTER,
G.A., & GILBERT, D.N. (1983) Interventions to modify
aminoglycoside-induced acute renal failure. In: Solez, K. & Whelton,
A., ed. Acute renal failure: Correlations between morphology and
function, New York, Basel, Marcel Dekker, pp. 331-358.
BENNETT, W.M., MELA-RIKER, L., HOUGHTON, D.C., GILBERT, D.N., & BUSS,
W.C. (1988) Microsomal protein synthesis inhibition: an early
mani-festation of gentamicin nephrotoxicity. Am. J. Physiol., 24:
F265-F269.
BENNINGTON, J.L. (1973) Cancer of the kidney - etiology, epidemiology,
and pathology. Cancer, 32: 1017-1029.
BERGERON, M.G., TROTTIER, S., LESSARD, C., BEAUCHAMP, D., & GAGNON, P.
(1982) Disturbed intrarenal distribution of gentamicin in experimental
pyelonephritis due to Escherichia coli. J. infect. Dis., 146:
436-439.
BERLYNE, G.M. (1984) Toxic nephropathies and current methods for early
detection of the toxicity of the kidney. In: Mehlman, M.A., Hemstreet,
C.P., Thorpe, J.J., & Weaver, N.K., ed. Renal effects of petroleum
hydrocarbons, Princeton, New Jersey, Princeton Scientific Publishers,
pp. 173-184 (Advances in Modern Environmental Toxicology, Vol. VII).
BERNARD, A. & LAUWERYS, R. (1986) Effects of cadmium exposure in man.
In: Foulkes, E.C., ed. Cadmium toxicology. Handbook of experimental
pharmacology, Berlin, Heidelberg, New York, Springer-Verlag, Vol. 80,
pp. 136-177.
BERNARD, A., ROELS, H., HUBERMONT, G., BUCHET, J.P., MASSON, P.L., &
LAUWERYS, R. (1976) Characterization of the proteinuria in cadmium
exposed workers. Int. Arch. occup. environ. Health, 38: 19-30.
BERNARD, A., BUCHET, J.P., ROELS, H., MASSON, P., & LAUWERYS, R.
(1979a) Renal excretion of proteins and enzymes in workers exposed to
cadmium. Eur. J. clin. Invest., 9: 11-22.
BERNARD, A., GORET, A., BUCHET, J.P., ROELS, H., & LAUWERYS, R.
(1979b) Comparison of sodium dodecyl sulfate polyacrylamide gel
electro-phoresis with quantitative methods for the analysis of
cadmium-induced proteinuria. Int. Arch. occup. environ. Health, 44:
139-148.
BERNARD, A., MOREAU, D., & LAUWERYS, R. (1982) Comparison of
retinol-binding protein and beta2-microglobulin determination in urine
for the early detection of tubular proteinuria. Clin. Chim. Acta, 126:
1-7.
BERNARD, A., OULED AMOR, A., & LAUWERYS, R. (1987) The effects of low
doses of cadmium-metallothionein on the renal uptake of
beta2-microglobulin in rats. Toxicol. appl. Pharmacol., 15: 440-445.
BERNARD, A., OULED AMOR, A., VIAU, C., & LAUWERYS, R. (1988) The renal
uptake of proteins: a non selective process in conscious rats. Kidney
Int., 34: 175-185.
BERNARD, A., LAUWERYS, R., NOEL, A., VANDELEENE, B., & LAMBERT, A.
(1989) Urine protein 1: a sex dependent marker of tubular or
glomerular dysfunction. Clin. Chem., 35: 2141-2142.
BERNDT, W.O. (1976) Use of renal slice technique for evaluation of
renal tubular transport process. Environ Health Perspect., 15: 73-88.
BERNDT, W.O. (1981) Use of renal function tests in the evaluation of
nephrotoxic effects. In: Hook, J.B., ed. Toxicology of the kidney, New
York, Raven Press, pp. 1-29 (Target Organ Toxicology Series).
BERNDT, W.O. (1987) Renal slices and perfusion. In: Bach, P.H. & Lock,
E.A., ed. Nephrotoxicity in the experimental and the clinical
situation, Dordrecht, Boston, Lancaster, Martinus Nijhoff Publishers,
pp. 301-316.
BERNDT, W.O. (1989) Potential involvement of renal transport
mechanisms in nephrotoxicity. Toxicol. Lett., 46: 77-82.
BERNDT, W.O. & HAYES, A.W. (1977) Effects of citrinin on renal
transport processes. J. environ. Pathol. Toxicol., 1: 93-103.
BERTANI, T., POGGI, A., POZZONI, R., DELAINI, F., SACCHI, G., THOUA,
Y., MECCA, G., REMUZZI, G., & DONATI, M.D. (1982) Adriamycin-induced
nephrotic syndrome in rats: sequence of pathologic events. Lab.
Invest., 46: 16-23.
BERTANI, T., ROCCHI, G., SACCHI, G., MECCA, G., & REMUZZI, G. (1986)
Adriamycin-induced glomerulosclerosis in the rat. Am. J. Kidney Dis.,
7: 12-19.
BHATTACHARYA, R.K. & SCHULTZE, M.O. (1967) Enzymes from bovine and
turkey kidneys which cleave S-(1,2-dichlorovinyl)-L-cysteine. Comp.
Biochem. Physiol., 22: 723-735.
BIBER, T.U.L., MYLLE, M., BAINES, A.D., & GOTTSCHALK, C.W. (1968) A
study by micropuncture and microdissection of acute renal failure in
rats. Am. J. Med., 44: 664-705.
BLAIR-WEST, J.R., BOBIK, A., BROOK, A.H., ESLER, M.D., GIBSON, A.,
MORRIS, M., MCKINLEY, M.J., & PULLAN, P.T. (1980) Renin, ADH and the
kidney: a congeries of conundrums. Prog. biochem. Pharmacol., 17:
20-28.
BLUM, R.H. & CARTER, S.K. (1974) Adriamycin: a new anticancer drug
with significant clinical activity. Ann. intern. Med., 80: 249-259.
BOHMAN, S.-O. (1980) The ultrastructure of the renal medulla and the
interstitial cells. In: Mandal, A.K. & Bohman, S.-O., ed. The renal
papilla and hypertension, New York, London, Plenum Press, pp. 7-33.
BOJESEN, I. (1974) Quantitative and qualitative analyses of isolated
lipid droplets from interstitial cells in renal papillae from various
species. Lipids, 9: 835-843.
BOMHARD, E., LUCKHAUS, G., VOIGT, W.-H., & LOESER, E. (1988) Induction
of light hydrocarbons nephropathy by p-dichlorobenzene. Arch.
Toxicol., 61: 433-439.
BONER, G., SHERRY, J., & RIESELBACH, R.E. (1972) Hypertrophy of the
normal human kidney following contralateral nephrectomy. Nephron, 9:
364-372.
BRENNER, B.M. (1983) Hemodynamically mediated glomerular injury and
the progressive nature of kidney disease. Kidney Int., 23: 647-655.
BRENNER, B.M. & RECTOR, F.C., ed. (1986) The kidney, Philadelphia,
W.B. Saunders.
BRENNER, B.M., HOSTETTER, T.H., & HUMAS, H.D. (1978) Glomerular
permselectivity: barrier function based on discrimination of molecular
size and charge. Am. J. Physiol., 234: F455-F460.
BRENNER, B.M., MEYER, T.W., & HOSTETTER, T.H. (1982) Dietary protein
intake and the progressive nature of kidney disease: the role of
hemodynami-cally mediated glomerular injury in the pathogenesis of
progressive glomerular sclerosis in aging, renal ablation and
intrinsic renal disease. New Engl. J. Med., 307: 652-659.
BREZIS, M., ROSEN, D., SILVA, P., & EPSTEIN, F.H. (1984) Renal
ischaemia: a new perspective. Kidney Int., 26: 375-383.
BRIDGES, J.W., BACH, P.H., BONNER, F.W., & BEATON, E.M. (1982) Effects
of nutritional factors on renal response to toxins. In: Bach, P.H.,
Bonner, F.W., Bridges, J.W., & Lock, E.A., ed. Nephrotoxicity:
Assessment and pathogenesis, New York, Chichester, Brisbane, Toronto,
John Wiley and Sons, pp. 182-198.
BROWN, W.W., ZENSER, T.V., & DAVIS, B.B. (1980) Prostaglandin E2
production by rabbit urinary bladder. Am. J. Physiol., 239: F452-F458.
BRUNBORG, G., HOLME, J.A., SODERLUND, E.J., OMICHINSKI, J.G., &
DYBING, E. (1988) An automated alkaline elution system: DNA damage
induced by 1,2-dibromo-3-chloropropane in vivo and in vitro. Anal.
Biochem., 174: 522-536.
BUCHET, J.-P., ROELS, H., BERNARD, A., & LAUWERYS, R. (1980)
Assessment of renal function of workers simultaneously exposed to
inorganic lead, cadmium or mercury vapour. J. occup. Med., 22:
741-750.
BUCKLEY, J.E., CLARK, V.L., MEYER, T.H., & PEARLMAN, N.W. (1984)
Hypomagnesemia after cisplatin combination chemotherapy. Arch. intern.
Med., 144: 2347-2348.
BURKE, J.F., LAUCIUS, J.F., BRODOVSKY, H.S., & SORIANO, R.Z. (1977)
Doxorubicin hydrochloride-associated renal failure. Arch. intern.
Med., 137: 385-388.
BURKHOLDER, P.M. (1982) Functions and pathophysiology of the
glomerular mesangium (editorial). Lab. Invest., 46: 239-241.
BURRY, A., CROSS, R., & AXELSEN, R. (1977) Analgesic nephropathy and
the renal concentrating mechanism. Pathol. Annu., 12: 1-31.
BURTON, B.T. & HIRSCHMAN, G.H. (1979) Demographic analysis: end-stage
renal disease and its treatment in the United States. Clin. Nephrol.,
11: 47-51.
BUTLER, E.A. & FLYNN, F.V. (1958) The proteinuria of renal tubular
disorders. Lancet, 2: 978-980.
BUTLER, W.H. (1964) Acute toxicity of aflatoxin B1 in rats. Br. J.
Cancer, 18: 756-762.
BUTLER, W.H. & LIJINSKY, W. (1970) Acute toxicity of aflatoxin G1 to
the rat. J. Pathol., 102: 209-212.
BUTLER, W.H., GREENBLATT, M., & LIJINSKY, W. (1969) Carcinogenesis in
rats by aflatoxins B1, G1, and B2. Cancer Res., 29: 2206-2211.
BUTLER, W.T. (1966) Pharmacology, toxicity, and therapeutic usefulness
of amphotericin B. J. Am. Med. Assoc, 195: 371-375.
BUZIO, C., MUTTI, A., CAPANI, F., ANDRULLI, S., PERAZZOLI, F.,
ALINOVI, R., NEGRO, A., & RUSTICHELLI, R. (1989) Circadian rhythm of
proteinuria: effects of an evening meat meal. Nephrol. Dialysis
Transplant., 4: 266-270.
CALDER, I.C., YONG, A.C., WOODS, R.A., CROWN, C.A., HAM, K.N., &
TANGE, J.D. (1979) The nephrotoxicity of p-aminophenol. II. The effect
of metabolic inhibitors and inducers. Chem.-biol. Interact., 27:
245-254.
CALDICOTT, W.J.H., HOLLENBERG, N.K., & ABRAMS, H.L. (1970)
Characteristics of response of renal vascular bed to contrast media.
Evidence of vasoconstriction induced by renin-angiotensin system.
Invest. Radiol., 5: 539-547.
CALNE, R.Y., WHITE, D.J.G., THIRU, S., EVANS, D.B., MCMASTER, P.,
DUNN, D.C., CRADDOCK, G.N., PENTLOW, B.D., & ROLLES, K. (1978)
Cyclosporin-A in patients receiving renal allografts from cadaver
donors. Lancet, 2: 1323-1327.
CARLIER, M.-B., LAURENT, G., CLAES, P.J., VANDERHAEGHE, H.J., &
TULKENS, P.M. (1983) Inhibition of lysosomal phospholipases by
aminoglycoside antibiotics: in vitro comparative studies.
Antimicrob. Agents Chemother., 23: 440-449.
CARPENTER, H.M. & MUDGE, G.H. (1981) Acetaminophen nephrotoxicity.
Studies on renal acetylation and deacetylation. J. Pharmacol. exp.
Ther., 218: 161-167.
CASTEGNARO, M. & CHERNOZEMSKY, I. (1987) Endemic nephropathy and
urinary tract tumors in the Balkans. Cancer Res., 47: 3608-3609.
CASTOR, C.W. & GREEN, J.A. (1968) Regional distribution of acid
mucopolysaccharides in the kidney. J. clin. Invest., 47: 2125-2132.
CAULFIELD, J.P., REID, J.J., & FARQUBAR, M.G. (1976) Alterations of
the glomerular epithelium in aminonucleoside nephrosis. Lab. Invest.,
34: 43-59.
CAVALIERE, G., ARRIGO, G., D'AMICO, G., BERNASCONI, P., SCHIAVINA,
G., DELLAFIORE, L., & VERGNAGHI, D. (1987) Tubular nephro-toxicity
after intravenous urography with ionic high-osmolal and nonionic
low-osmolal contrast media in patients with chronic renal
insufficiency. Nephron, 46: 128-133.
CEDGARD, S., HERLITZ, H., GETERUD, K., ALTMAN, P.O., & AURELL, M.
(1986) Acute renal insufficiency after administration of low-osmolar
contrast media. Lancet, 2: 1281.
CHAHWALA, S.B. & HARPUR, E.S. (1986) The use of renal tubule fragments
isolated from the rat to investigate aspects of gentamicin
nephrotoxicity. J. Pharmacol. Methods, 15: 21-34.
CHAMBERLIN, M.E., LEFURGEY, A., & MANDEL, L.J. (1984) Suspension of
medullary thick ascending limb tubules from the rabbit kidney. Am. J.
Physiol., 247: F955-F964.
CHARBONNEAU, M., LOCK, E.A., STRASSER, J., COX, M.G., TURNER, M.J., &
BUS, J.S. (1987) 2,2,4-Trimethylpentane-induced nephrotoxicity. I.
Metabolic disposition of TMP in male and female Fischer-344 rats.
Toxicol. appl. Pharmacol., 91: 171-181.
CHASSEAUD, L.F. (1980) Extrahepatic conjugation with glutathione. In:
Gram, T.E., ed. Extrahepatic metabolism of drugs and other foreign
compounds, New York, Spectrum Publications, pp. 427-443.
CHOPRA, S., KAUFMAN, J.S., JONES, T.W., HONG, W.K., GEHR, M.K.,
HAMBERGER, R.J., FLAMENBAUM, W., & TRUMP, B.F. (1982)
Cis-diamminedichloro-platinum-induced acute renal failure in the rat.
Kidney. Int., 21: 54-64.
CHOU, C.C., HOOK, J.B., & HSIEH, C.P. (1974) Effects of radiopaque
dyes on renal vascular resistance. J. lab. clin. Med., 78: 705-712.
CHUANG, E.L., REINECK, H.J., OSGOOD, R.W., LIMAI, R.T., & STEIN, J.H.
(1978) Studies on the mechanism of reduced urinary osmolality after
exposure of renal papilla. J. clin. Invest., 61: 633-639.
CHUMAN, L., FINE, L.G., COHEN, A.H., & SAIER, M.H. Jr. (1982)
Continuous growth of proximal tubular kidney epithelial cells in
hormone-supplemented serum-free medium. J. Cell Biol., 94: 506-510.
CLIFTON, G., KAPLOWITZ, N., WALLIN, J.D., & KUHLENKAMP, J. (1975) Drug
induction and sex differences of renal glutathione S-transferases in
the rat. Biochem. J., 150: 259-262.
CLIVE, D.M. & STOFF, J.S. (1984) Renal syndrome associated with
nonsteroidal anti-inflammatory drugs. New Engl. J. Med., 310: 563-572.
COHEN, J.J. (1979) Is the function of the renal papilla coupled
exclusively to an anaerobic pattern of metabolism? Am. J. Physiol.,
236: F423-F433.
COHEN, J.J., HARRINGTON, J.T., & KASSIREV, J.P. (1981) Lithium and the
kidney. Kidney Int., 19: 374-387.
COJOCEL, C., MAITA, K., PASINO, D.A., KUO, C., & HOOK, J.B. (1983)
Metabolic heterogeneity of the proximal and distal kidney tubules.
Life Sci., 33: 855-861.
COLLEONI, N. & D'AMICO, G. (1986) Chronic lead accumulation as a
possible cause of renal failure in gouty patients. Nephron, 44: 32-35.
COLLIER, V., LIETMAN, P.S., & MITCH, W.E. (1979) Evidence for luminal
uptake of gentamicin in the perfused rat kidney. J. Pharmacol. exp.
Ther., 210: 247-251.
CONNELLY, J.C. & BRIDGES, J.W. (1980) The distribution and role of
cytochrome P-450 in extrahepatic organs. Prog. Drug. Metab., 5: 1-111.
CONSTANTOPOULOS, G., LOUIE, M., & DEKABAN, A.S. (1973) Acid
mucopolysaccharides (glycosaminoglycans) in normal human kidneys and
in kidneys of patients with mucopolysaccaridoses. Biochem. Med., 7:
376-388.
COOPER, W.C. & GAFFEY, W.R. (1975) Mortality of lead workers. J.
occup. Med., 17: 100-109.
COOPER, W.C., WONG, O., & KHEIFETS, L. (1985) Mortality among
employees of lead battery plants and lead producing plants. Scand. J.
Work Environ. Health., 11: 331-345.
CRAIN, R.C. (1958) Spontaneous tumors in the Rochester strain of the
Wistar rat. Am. J. Pathol., 34: 311-336.
CRAMER, K., GOYER, R.A., JAGENBURG, R., & WILSON, M. (1974) Renal
ultrastructure, renal function and parameters of lead toxicity in
workers with different periods of lead exposure. Br. J. ind. Med., 31:
113-127.
CROWE, C.A., CALDER, I.C., MADSEN, N.P., FUNDER, C.C., GREEN, C.R.,
HAM, K.N., & TANGE, J.D. (1977) An experimental model of
analgesic-induced renal damage. Some effects of p-aminophenol on rat
kidney mitochondria. Xenobiotica, 7: 345-356.
CROWE, C.A., YONG, A.C., CALDER, I.C., HAM, K.N., & TANGE, J.D. (1979)
The nephrotoxicity of p-aminophenol. I. The effect of microsomal
cyto-chromes, glutathione and covalent binding in kidney and liver.
Chem.-biol. Interact., 27: 235-243.
CUNARRO, J.A. & WEINER, M.W. (1978) Effects of ethacrynic acid and
furosemide on respiration of isolated kidney tubules: the role of ion
transport and the source of metabolic energy. J. Pharmacol. exp.
Ther., 206: 198-206.
CUNNINGHAM, E.E., BRENTJENS, J.R., ZIELEZREY, M.A., ANDRES, G.A., &
VERUTO, R.C. (1980) Heroin nephropathy: a clinicopathologic and
epidemiologic study. Am. J. Med., 68: 47-53.
CUNNINGHAM, E.E., BARONE, P.P., NASCIMENTO, L., & VENUTO, R.C. (1986)
Effect of a radiographic contrast agent on renal function in the rat.
Comparison with equiosmolar mannitol. Miner. electrolyte Metab., 12:
157-160.
CURTHOYS, N.P. & BELLEMANN, P. (1979) Renal cortical cells in primary
monolayer culture. Enzymatic changes and morphological observations.
Exp. cell Res., 121: 31-45.
CURTIS, J.R. (1979) Drug-induced renal disease. Drugs, 18: 377-391.
CVITKOVIC, E., SPAULDING, J., BETHUNE, V., MARTIN, J., & WHITMORE,
W.F. (1977) Improvement of cis-dichlorodiammineplatinum (NSC-119875):
Therapeutic index in an animal model. Cancer, 39: 1357-1361.
DARNTON, S.J. (1967) Glycogen metabolism in rabbit kidney under
differing physiological states. Q. J. exp. Physiol., 52: 392-400.
DARNTON, S.J. (1969) The conversion of injected glucose into renal
glycogen and mucopolysaccharides. An autoradiographic study of rabbits
in various states of hydration. Z. Zellforsch., 102: 273-282.
DAUGAARD, G., ROSSING, N., & RORTH, M. (1988a) Effects of cisplatin on
different measures of glomerular function in the human kidney with
special emphasis on high-dose. Cancer Chemother. Pharmacol., 21:
163-167.
DAUGAARD, G., HOLSTEIN-RATHLOU, N.-H., & LEYSSAC, P.P. (1988b) Effects
of cisplatin on proximal convoluted and straight segments of the rat
kidney. J. Pharmacol. exp. Ther., 244: 1081-1085.
DAVIES, D.F. & SHOCK, N.W. (1950) Age changes in glomerular filtration
rate, effective renal plasma flow and tubular excretory capacity in
adult males. J. clin. Invest., 29: 496-507.
DAVIES, J.M. (1984) Long term mortality study of chromate pigment
workers who suffered lead poisoning. Br. J. ind. Med., 41: 170-178.
DAVIS, B.B., MATTAMMAL, M.B., & ZENSER, T.V. (1981) Renal metabolism
of drugs and xenobiotics. Nephron, 27: 187-196.
DAYAL, H. & KINMAN, J. (1983) Epidemiology of kidney cancer. Semin.
Oncol., 10: 366-377.
DEBROE, M.E., PAULUS, G.J., VERPOOTEN, G.A., ROELS, F., BUYSSENS, N.,
WEDEEN, R., VANHOOF, F., & TULKENS, P.M. (1984) Early effects of
gentamicin, tobramycin and amikacin on the human kidney. Kidney Int.,
25: 643-652.
DEES, J.H., HEATFIELD, B.M., REUBER, M.D., & TRUMP, B.F. (1980a)
Adenocarcinoma of the kidney. III. Histogenesis of renal
adenocarcinomas induced in rats by
N-(4'-fluoro-4-biphenylyl)acetamide, J. Natl Cancer Inst., 64:
1537-1545.
DEES, J.H., HEATFIELD, B.M., & TRUMP, B.F. (1980b) Adenocarcinoma of
the kidney. IV. Electron microscopic study of the development of renal
adenocar-cinomas induced in rats by
N-(4'-fluoro-4-biphenylyl)acetamide. J. Natl Cancer Inst., 64:
1547-1562.
DEKERNION, J.B. & BERRY, D. (1980) The diagnosis and treatment of
renal carcinoma. Cancer, 45(Suppl.): 1947-1982.
D'ELIA, J.A., GLEASON, K.E., ALDAY, M., MALARICK, C., GODLEY, K.,
WARRAM, J., KALDANY, A., & WEINRAUCH, L.A. (1982) Nephrotoxicity from
angiographic contrast material. A prospective study. Am. J. Med., 72:
719-725.
DENTINO, M., LUFT, F.C., YUM, M.W., WILLIAMS, S.D., & EINHORN, L.H.
(1978) Long term effect of cis-diamminedichloride platinum (CDDP) on
renal function and structure in man. Cancer, 41: 1274-1281.
DEPNER, T.A. (1982) Nephrotic syndrome secondary to lithium therapy.
Nephron, 30: 286-289.
DICKER, S.E. & FRANKLIN, C.S. (1966) The isolation of hyaluronic acid
and chondroitin sulphate from kidneys and their reaction with urinary
hyaluronidase. J. Physiol., 186: 110-120.
DIEPERINK, H.H. (1989) Identification of groups at risk for renal
diseases (including nephrotoxicity). Toxicol. Lett., 46: 257-268.
DIEPERINK, H., STARKLINT, H., & LEYSSAC, P.P. (1983) Nephrotoxicity of
cyclosporin A - an animal model. A study of the nephrotoxic effect of
cyclosporin on overall renal and tubular function in conscious rats.
Transplant Proc., 15(Suppl. 1): 2736-2741.
DIEPERINK, H., LEYSSAC, P.P., STARKLINT, H., & KEMP, E. (1985)
Glomerulotubular function in cyclosporin A treated rats. A lithium
clearance, occlusion time/transit time and micropuncture study. Proc.
Eur. Dialysis Transplant Assoc., 21: 853-859.
DIEZI, J. & ROCH-RAMEL, F. (1987) The use of single nephron techniques
in renal toxicity studies. In: Bach, P.H. & Lock, E.A., ed.
Nephrotoxicity in the experimental and the clinical situation,
Dordrecht, Boston, Lancaster, Martinus Nijhoff Publishers, pp.
317-358.
DOBYAN, D.C. (1985) Long-term consequences of cis-platinum-induced
renal injury: a structural and functional study. Anat. Rec., 212:
239-245.
DOHN, D.R., QUEBBEMANN, A.J., BORCH, R.F., & ANDERS, M.W. (1985a)
Enzymatic reaction of chlorotrifluoroethene with glutathione: 19F NMR
evidence for stereochemical control of the reaction. Biochemistry, 24:
5137-5143.
DOHN, D.R., LEININGER, J.R., LASH, L.H., QUEBBEMANN, A.J., & ANDERS,
M.W. (1985b) S-(2-chloro-1,1,2-trifluoroethyl)glutathione and
S-(2-chloro-1,1,2-trifluoroethyl)-L-cysteine, the glutathione and
cysteine conju-gates of chlorotrifluoroethene. J. Pharmacol. exp.
Ther., 235: 851-857.
DOUGLAS, J.B. & HEALY, J.K. (1969) Nephrotoxic effects of amphotericin
B, including renal tubular acidosis. Am. J. Med., 46: 154-162.
DRUET, P. (1989) Contribution of immunological reactions to
nephrotoxicity. Toxicol. Lett., 46: 55-64.
DRUET, P., BERNARD, A., HIRSCH, F., WEENING, J.J., GENOUX, P., MAHIEU,
P., & BIRKELAND, S. (1982) Immunologically mediated glomerulonephritis
induced by heavy metals. Arch. Toxicol., 50: 187-194.
DRUET, P., JACQUOT, C., BARAN, D., KLEINKNECHT, D., FILLASTRE, J.P.,
& MERY, J.Ph. (1987) Immunologically mediated nephritis induced by
toxins and drugs. In: Bach, P.H. & Lock, E.A., ed. Nephrotoxicity in
the experimental and the clinical situation, Dordrecht, Boston,
Lancaster, Martinus Nijhoff Publishers, pp. 727-770.
DUBACH, U.C., LE HIR, M., & GANDHI, R. (1989) Use of urinary enzymes
as markers of nephrotoxicity. Toxicol. Lett., 46: 193-196.
DUNHILL, M.S., MILLARD, P.R., & OLIVER, D. (1977) Acquired cystic
disease of the kidney: a hazard of long-term intermittent maintenance
dialysis. J. clin. Pathol., 30: 868-877.
DUNN, M.A., BLALOCK, T.L., & COUSIN, R.J. (1987) Minireview of
metallothionein. Proc. Soc. Exp. Biol. Med., 185: 107-119.
DUNN, M.J. (1981) Prostaglandins and Bartter's syndrome. Kidney Int.,
19: 86-102.
DUNN, M.J. & HOOD, V.L. (1977) Prostaglandins and the kidney. Am. J.
Physiol. 223: F169-F184.
DUNN, M.J. & ZAMBRASKI, E.J. (1980) Renal effects of drugs that
inhibit prostaglandin synthesis. Kidney Int., 18: 609-622.
DYBING, E., OMICHINSKI, J.G., SODERLUND, E.J., BRUNBORG, G., LAG, M.,
HOLME, J.A., & NELSON, S.D. (1989) Mutagenicity and organ damage of
1,2-dibromo-3-chloropropane (DBCP) and
tris(2,3-dibromopropyl)phosphate (tris-BP): role of metabolic
activation. In: Hodgson, E., Bend, J.R., & Philpot, R.M., ed. Reviews
in biochemical toxicology, Amsterdam, Oxford, New York, Elsevier
Science Publishers, Vol. 10, pp. 139-186.
EARLEY, L.E. & FRIEDLER, R.M. (1964) Observations on the mechanism of
decreased tubular reabsorption of sodium and water during saline
loading. J. clin. Invest., 43: 1928-1936.
EARLEY, L.E. & FRIEDLER, R.M. (1965) Changes in renal blood flow and
possibly the intra-renal distribution of blood during natriuresis
accompanying saline loading in the dog. J. clin. Invest., 44: 929-941.
EBLE, J.N. & HULL, M.T. (1984) Morphologic features of renal
oncocytoma. A light and electron microscopic study. Hum. Pathol., 15:
1054-1061.
EINHORN, L.H. & DONOHUE, J. (1977) Cis-diamminedichloroplatinum,
vinblastin and bleomycin combination chemotherapy in disseminated
testicular cancer. Ann. intern. Med., 87: 293-298.
EISENBERG, R.L., BANK, W.O., & HEDGCOCK, M.W. (1980) Renal failure
after major angiography. Am. J. Med., 68: 43-46.
ELBERS, R., KAMPFFMEYER, H.G., & RABES, H. (1980) Effects and
metabolic pathway of 4-dimethylaminophenol during kidney perfusion.
Xenobiotica, 10: 621-632.
ELFARRA, A.A. & ANDERS, M.W. (1984) Renal processing of glutathione
conjugates. Role in nephrotoxicity. Biochem. Pharmacol., 33:
3729-3732.
ELFARRA, A.A., JACOBSON, I., & ANDERS, M.W. (1986a) Mechanism of
S-(1,2-dichlorovinyl) glutathione-induced nephrotoxicity. Biochem.
Pharmacol., 35: 283-288.
ELFARRA, A.A., LASH, L.H., & ANDERS, M.W. (1986b) Metabolic activation
and detoxification of nephrotoxic cysteine and homocysteine
S-conjugates. Proc. Natl Acad. Sci., 83: 2667-2671.
ELINDER, C.G., EDLING, C., LINDBERG, E., BERTIL, K., & VESTERBERG, O.
(1985a) Beta-2-microglobulinuria among workers previously exposed to
cadmium: follow-up and dose-response analyses. Am. J. ind. Med., 8:
553-564.
ELINDER, C.G., EDLING, C., LINDBERG, E., KAGEDAL, B., & VESTERBERG, O.
(1985b) Assessment of renal function in workers previously exposed to
cadmium. Br. J. ind. Med., 42: 754-760.
ELLIS, K.J., MORGAN, W.C., ZANZI, I., YASUMURA, S., VARTSKY, D., &
COHN, S.H. (1981) Critical concentration of Cd in human renal cortex:
dose-effect studies in Cd smelter workers. J. Toxicol. environ.
Health, 7: 691-703.
ELSEVIERS, M.M. & DE BROE, M.E. (1988) Is analgesic nephropathy still
a problem in Belgium? Nephrol. Dialysis Transplant., 2: 143-149.
EMMERSON, B.T. (1973) Chronic lead nephropathy. Kidney Int., 4: 1-5.
EMSLIE, K.R., CALDER, I.C., HART, S.J., & TANGE, J.D. (1981) Induction
of paracetamol metabolism in the isolated perfused kidney.
Xenobiotica, 11: 43-50.
ENDOU, H. (1983) Cytochrome P-450 monooxygenase system in the rabbit
kidney: its intranephron localization and its induction. Jpn. J.
Pharmacol., 33: 423-433.
ENDOU, H., NONOGUCHI, Y., TAKEHARA, H., YAMADA, H., & NAKADA, J.
(1985) Intranephron heterogeneity of ammoniagenesis and
gluconeogenesis in rats. Contrib. Nephrol., 47: 98-104.
ENTERLINE, P.E. & VIREN, J. (1985) Epidemiologic evidence for an
association between gasoline and kidney cancer. Environ. Health
Perspect., 62: 303-312.
EPSTEIN, S.M., BARTUS, B., & FARBER, E. (1969) Renal epithelial
neoplasms induced in male Wistar rats by oral aflatoxin B1. Cancer
Res., 29: 1045-1050.
ERTÜRK, E., COHEN, S.M., & BRYAN, G.T. (1970) Induction, histogenesis,
and isotransplantability of renal tumors induced by formic acid
2-[4-(5-nitro-2-furyl)-2-thiazolyl]hydrazide in rats. Cancer Res., 30:
2098-2106.
EUROPEAN DIALYSIS AND TRANSPLANT ASSOCIATION REGISTRY (l986)
Demography of dialysis and transplantation in Europe, 1984. Nephrol.
Dialysis Transplant., 1: 1.
EVARTS, R.P., BROWN, C.A., & MOSTAFA, H. (1982) Production of kidney
tumors in rats with low dose of dimethylnitrosamine after partial
hepatectomy. J. Natl Cancer Inst., 68: 293-298.
FAJARDO, L.F., ELTRINGHAM, J.R., STEWART, J.R., & KLAUBER, M.R. (1980)
Adriamycin nephrotoxicity. Lab. Invest., 43: 242-253.
FANG, L.S., SIROTA, R.A., EBERT, T.H., & LICHTENSTEIN, N.S. (1980) Low
fractional excretion of sodium with contrast media-induced acute renal
failure. Arch. intern. Med., 140: 531-533.
FARBER, S.J., WALAT, R.J., BENJAMIN, R., & VAN PRAAG, D. (1971) Effect
of increased osmolality on glycosaminoglycan metabolism of rabbit
renal papilla. Am. J. Physiol., 220: 880-885.
FINKELSTEIN, A., FRALEY, D.S., STACHURA, I., FELDMAN, H.A., GANDY,
D.R., & BOURKE, E. (1982) Fenoprofen nephropathy: Lipoid nephrosis and
interstitial nephritis. Am. J. Med., 72: 81-86.
FISCHER, L.J., GREEN, M.D., & HARMAN, A.W. (1985) Studies on the fate
of the glutathione and cysteine conjugates of acetaminophen in mice.
Drug Metab. Disp., 13: 121-126.
FJELDBORG, P., SORENSEN, J., & HELKJAER, P.E. (1986) The long-term
effect of cisplatin on renal function. Cancer, 58: 2214-2217.
FOA', V., CAIMI, L., AMANTI, L., ANTONINI, C., GATTINONI, A.,
TETTAMANTI, G., LOMBARDO, A., & GIULIANI, A. (1976) Patterns of some
lysosomal enzymes in the plasma and of proteins in urine of workers
exposed to inorganic mercury. Int. Arch. occup. environ. Health, 37:
115-124.
FOA', V., COLOMBI, A., MARONI, M., BARBIERI, F., FRANCHINI, I., MUTTI,
A., DE ROSA, E., & BARTOLUCCI, G.B. (1987) Study of kidney function of
workers with low level exposure to inorganic arsenic. In: Foa', V.,
Emmett, E.A., Maroni, M., & Colombi, A., ed. Occupational and
environmental chemical hazards, Chichester, Ellis Horwood Limited, pp.
362-367.
FOIDART, J.B., DECKANNE, C.A., MAHIEU, P., CREUTZ, C.E., & DE MEY, J.
(1979) Tissue culture of normal rat glomeruli: Isolation and
morphological characterization of two homogeneous cell lines. Invest.
cell Pathol., 2: 15-26.
FOIDART, J.B., DUBOIS, C.H., & FOIDART, J.-M. (1980) Tissue culture of
normal rat glomeruli: Basement membrane biosynthesis by homogenous
epithelial and mesangial cell lines. Int. J. Biochem., 12: 197-202.
FOIDART, J.B., DECHENNE, C.A., & MAHIEU, P. (1981) Tissue culture of
normal rat glomeruli: Characterization of collagenous and
non-collagenous basement membrane antigens on the epithelial and
mesangial cells. Diagn. Histopathol., 4: 71-77.
FORD, S.M. & HOOK, J.B. (1984) Biochemical mechanisms of toxic
nephropathies. Semin. Nephrol., 4: 88-106.
FORREST, J.B., HOWARDS, S.S., & GILLENWATER, I.Y. (1981) Osmotic
effects of intravenous contrast agents on renal function. J. Urol.,
125: 147-150.
FOWLER, B.A. & GOYER, R.A. (1975) Bismuth localization within nuclear
inclusions by X-ray microanalysis: Effects of accelerating voltage.
Histochem. Cytochem., 23: 722-727.
FRANCHINI, I. & MUTTI, A. (1988) Selected toxicological aspects of
chromium compounds. Sci. total Environ., 71: 379-387.
FRANCHINI, I., CAVATORTA, A., FALZOI, M., LUCERTINI, S., & MUTTI, A.
(1983) Early indicator of renal damage in workers exposed to organic
solvents. Int. Arch. occup. environ. Health, 51: 1-9.
FRIBERG, L. (1948) Proteinuria and kidney injury among workers exposed
to cadmium and nickel dust. J. ind. Toxicol., 30: 32-36.
FRIBERG, L. (1950) Health hazards in the manufacture of alkaline
accumulators with special reference to chronic cadmium poisoning. Acta
med. Scand., 138 (Suppl. 240): 1-154.
FRIBERG, L., PISCATOR, M., NORDBERG, G.F., & KJELLSTROM, T. (1974)
Cadmium in the environment, 2nd ed., Cleveland, Ohio, Chemical Rubber
Co. Press.
FRIBERG, L., ELINDER, C.G., KJELLSTROM, T., & NORDBERG, G.F. (1986)
Cadmium and health: a toxicological and epidemiological appraisal,
Boca Raton, Florida, CRC Press, pp. 1-307.
FRIEDLANDER, G., BLANCHET, F., & AMIEL, C. (1989) Renal functional
reserve. Toxicol. Lett., 46: 227-235.
FROLICH, J.C. & WALKER, L.A. (1980) Determination, source, metabolism
and functional role of renal prostaglandins. Clin. exp. Hypertension,
2: 709-728.
FROLICH, J.C., NIES, A.S., & SCHRIER, R.W., ed. (1981) Prostaglandins
and the kidney. Kidney Int., 19: 755-868.
FRY, J.R. & PERRY, N.K. (1981) The effect of Aroclor-1254 pretreatment
on the phase I and phase II metabolism of 7-ethoxycoumarin in isolated
viable rat kidney cells. Biochem. Pharmacol., 30: 1197-1201.
FRY, J.R., WIEBKIN, P., KAO, J., JONES, C.A., GWYNN, J., & BRIDGES,
J.W. (1978) A comparison of drug-metabolising capability in isolated
viable rat hepatocytes and renal tubule fragments. Xenobiotica, 8:
113-120.
FUKUSHIMA, M., ISHIZAKI, A., NOGAWA, K., SAKAMOTO, M., & KOBAYASHI, E.
(1974) Epidemiological studies on renal failure of inhabitants in
Itai-Itai disease endemic district. I. Some findings of inhabitants
living in and around the endemic district of the Jinzu River basin.
Jpn. J. pub. Health, 21: 67-73.
GALE, M.E., ROBBINS, A.H., HAMBURGER, R.J., & WIDRICH, W.C. (1984)
Renal toxicity of contrast agents: iopamidol, iothalamate, and
diatrizoate. Am. J. Roentgenol., 142: 333-335.
GARTNER, H.V. (1980) Drug-associated nephropathy. In: Berry, C.L.,
Grundmann, E., & Kirsten, W.H., ed. Drug-induced pathology, Berlin,
Heidelberg, New York, Springer-Verlag, pp. 144-174 (Current Topics in
Pathology, Vol. 69).
GEMBORYS, M.W. & MUDGE, G.H. (1981) Formation and disposition of the
minor metabolites of acetaminophen in the hamster. Drug Metab. Disp.,
9: 340-351.
GILBERT, D.N., WOOD, C.A., KOHLHEPP, S.J., KOHNEN, P.W., HOUGHTON,
D.B., FINKBEINER, H.C., LINDSLEY, J., & BENNETT, W.M. (1989)
Polyaspartic acid prevents experimental aminoglycoside nephrotoxicity.
J. infect. Dis., 159: 945-953.
GINETZINSKY, A.G. (1958) Role of hyaluronidase in the reabsorption of
water in renal tubules. The mechanism of action of the anti-diuretic
hormone. Nature (Lond.), 182: 1218-1219.
GIULIANO, R.A., PAULUS, G.J., VERPOOTEN, R.A., PATTYN, V., POLLET,
D.E., TULKENS, P.M., & DEBROE, M.E. (1984) Recovery of cortical
phospholipidosis and necrosis after acute gentamicin loading in rats.
Kidney Int., 26: 838-847.
GIULIANO, R.A., VERPOOTEN, G.A., VERBIST, L., WEDEEN, R., & DEBROE,
M.E. (1986) In vivo uptake kinetics of aminoglycosides in the kidney
cortex of rats. J. Pharmacol. exp. Ther., 236: 470-475.
GLOOR, F.J. (1978) Changing concept in the pathogenesis and morphology
of analgesic nephropathy as seen in Europe. Kidney Int., 13: 27-33.
GOLDBERG, J.P. & ANDERSON, R.J. (1985) Renal metabolism and excretion
of drugs. In: Seldin, D.W. & Giebisch, G., ed. The kidney: Physiology
and pathophysiology, New York, Raven Press, pp. 2097-2110.
GOLDSTEIN, R.S. & MAYOR, G.H. (1983) Minireview: The nephrotoxicity of
cis-platin. Life Sci., 32: 685-690.
GOLDSTEIN, R.S., CONTARDI, L.R., PASINO, D.A., & HOOK, J.B. (1987)
Mechanisms mediating cephaloridine inhibition of gluconeogenesis.
Toxicol. appl. Pharmacol., 87: 297-305.
GOLDSTEIN, R.S., SOZIO, R.S., TARLOFF, J.B., & HOOK, J.B. (1989) The
role of oxidative stress in cephaloridine nephrotoxicity. In: Bach,
P.H. & Lock, E.A., ed. Nephrotoxicity: Extrapolation from in vitro
to in vivo, and animals to man, New York, London, Plenum Press, pp.
457-461.
GOLMAN, K. & ALMEN, T. (1985) Contrast media-induced nephrotoxicity.
Survey and present state. Invest. Radiol., 20: S92-S97.
GONZALEZ-VITALE, J.C., HAYES, D.M., CVITKOVIC, E., & STERNBERG, S.
(1977) The renal pathology in clinical trials of cis-platinum (II)
diammine-dichloride. Cancer, 39: 1362-1371.
GOODMAN, D.G., WARD, J.M., SQUIRE, R.A., CHU, K.C., & LINHART, M.S.
(1979) Neoplastic and non-neoplastic lesions in aging F344 rats,
Toxicol. appl. Pharmacol., 48: 237-248.
GOODMAN, D.G., WARD, J.M., SQUIRE, R.A., PAXTON, M.B., REICHARDT,
W.D., CHU, K.C., & LINHART, M.S. (1980) Neoplastic and non-neoplastic
lesions in aging Osborne-Mendel rats. Toxicol. appl. Pharmacol., 55:
433-447.
GOODMAN, M.T., MORGENSTERN, H., & WYNDER, E.L. (1986) A case-control
study of factors affecting the development of renal cell cancer. Am.
J. Epidem., 124: 926-941.
GOTELLI, C.A., ASTOLFI, E., COX, C., CERNICHIARI, E., & CLARKSON, T.W.
(1985) Early biochemical effects of an organic mercury fungicide on
infants: dose makes the poison. Science, 227: 638-640.
GOTTSCHALK, C.W. & LASSITER, W.E. (1973) Micropuncture methodology.
In: Orloff, J. & Berliner, W.R., ed. Handbook of physiology. Section
8: Renal physiology, Washington, DC, American Physiological Society.
GOYER, R.A. (1982) Nephrotoxic effects of lead. In: Bach, P.H.,
Bonner, F.W., Bridges, J.W., & Lock, E.A., ed. Nephrotoxicity:
Assessment and pathogenesis, New York, Chichester, Brisbane, Toronto,
John Wiley and Sons.
GOYER, R.A. & CHERIAN, M.G. (1977) Tissue and cellular toxicology of
metals. In: Brown, S.S., ed. Clinical chemistry and chemical
toxicology of metals, Amsterdam, Elsevier-North Holland Biomedical
Press, pp. 89-103.
GOYER, R.A. & MOORE, J.F. (1974) Cellular effects of lead. Adv. exp.
Med. Biol., 48: 447-462.
GOYER, R.A. & RHYNE, B.C. (1973) Pathological effects of lead. Int.
Rev. exp. Pathol., 12: 1-77.
GOYER, R.A. & WILSON, M.H. (1975) Lead-induced inclusion bodies:
Results of EDTA treatment. Lab. Invest., 32: 149-156.
GOYER, R.A., FALK, H.L., HOGAN, M., FELDMAN, D.D., & RICHTER, W.
(1981) Renal tumors in rats given trisodium nitrilotriacetic acid in
drinking water for 2 years. J. Natl Cancer Inst., 66: 869-880.
GRANDCHAMP, A., AYER, G., SCHERRER, J.R., & TRUNIGER, B. (1971)
Intrarenal haemodynamics of the rat kidney determined by the Xenon
washout technique. Nephron, 8: 33-45.
GREEN, C.R., HAM, K.N., & TANGE, J.D. (1969) Kidney lesions induced in
rats by p-aminophenol. Br. med. J., 1: 162-164.
GREEN, R.M. & ELCE, J.S. (1975) Acetylation of S-substituted cysteines
by a rat liver and kidney microsomal N-acetyltransferase. Biochem. J.,
147: 283-289.
GREGG, N., ELSEVIERS, M.M., DEBROE, M.E., & BACH, P.H. (1989) The
epidemiological and mechanistic basis of analgesic associated
nephropathy. Toxicol. Lett., 46: 141-151.
GRITZKA, T.L. & TRUMP, B.F. (1968) Renal tubular lesions caused by
mercuric chloride. Am. J. Pathol., 52: 1225-1227.
GROTH, S., NIELSEN, H., SORENSEN, J.B., CHRISTENSEN, A.B., PEDERSEN,
A.G., & RORTH, M. (1986) Acute and long-term nephrotoxicity of
cisplatinum in man. Cancer Chemother. Pharmacol., 17: 191-196.
GRUNFELD, J.P., RAPHAEL, J.C., BANKIR, W., KLEINKNECHT, D., & BARGER,
A.C. (1971) Intrarenal distribution of blood flow. Adv. Nephrol., 1:
125-143.
GSTRAUNTHALER, G., PFALLER, W., & KOTANKO, P. (1985) Interrelation
between oxygen consumption and Na-K-ATPase activity in rat renal
proximal tubule suspension. Renal Physiol., 8: 38-44.
GUDER, W.G. & ROSS, B.D. (1984) Enzyme distribution along the nephron.
Kidney Int., 26: 101-111.
GUDER, W.G. & SCHMIDT, U. (1974) The localization of gluconeogenesis
in the rat nephron: determination of PEPCK in microdissected tubules.
Hoppe Seylers Z. Physiol. Chem., 355: 273-278.
GUENGERICH, F.P. & MASON, P.S. (1979) Immunological comparison of
hepatic and extrahepatic cytochromes P-450. Mol. Pharmacol., 15:
154-64.
GYRD-HANSEN, N. (1968) Renal clearances in pigs. Acta vet. Scand., 9:
183-198.
HACKENTHAL, E., SCHWERTSCHLAG, U., & SEYBERTH, H.W. (1980)
Prostaglandins and renin release studies in the isolated perfused rat
kidney. Prog. Biochem. Pharmacol., 17: 98-107.
HALDER, C.A., WARNE, T.M., & HATOUM, N.S. (1984) Renal toxicity of
gasoline and related petroleum naphthas in male rats. In: Mehlman,
M.A., Hemstreet, C.P., Thorpe, J.J., & Weaver, N.K., ed. Renal effects
of petroleum hydrocarbons, Princeton, New Jersey, Princeton Scientific
Publishers, pp. 73-88.
HALDER, C.A., HOLDSWORTH, C.E., & COOKRELL, B.Y. (1985) Hydrocarbon
nephropathy in male rats. Identification of the nephrotoxic components
of gasoline. In: Bach, P.H. & Lock, E.A., ed. Renal heterogeneity and
target cell toxicity, New York, Chichester, Brisbane, Toronto, John
Wiley and Sons, pp. 481-484.
HALES, B.F., JAEGER, V., & NEIMS, A.H. (1978) Isoelectric focusing of
glutathione S-transferases from rat liver and kidney. Biochem. J.,
175: 937-943.
HALL, C.L., FOTHERGILL, N.J., BLACKWELL, M.M., HARRISON, P.R.,
MACKENZIE, J.C., & MACIVER, A.G. (1987) The natural course of gold
nephropathy: long-term study of 21 patients. Br. med. J., 295:
745-748.
HALL, P.W., III (1982) Endemic Balkan nephropathy. In: Porter, G., ed.
Nephrotoxic mechanisms and environmental toxins, New York, London,
Plenum Press, pp. 227-240.
HALL, P.W., III & DAMMIN, G.J. (1978) Balkan nephropathy. Nephron, 22:
281-300.
HALL, R.L., WILKE, W.L., & FETTMAN, M.J. (1986) The progression of
adriamycin-induced nephrotic syndrome in rats and the effect of
captopril. Toxicol. appl. Pharmacol., 82: 164-174.
HAMILTON, J.M. (1975) Renal carcinogenesis. Adv. Cancer Res., 22:
1-56.
HAMMOND, P.B., LERNER, S.I., GARTSIDE, P.S., HANENSON, I.B., RODA,
S.B., FOULKES, E.C., JOHNSON, D.R., & PESCI, A.J. (1980) The
relationship of biological indices of lead exposure to the health
status of workers in a secondary lead smelter. J. occup. Med., 22:
475-484.
HANDLER, J.S. (1983) Use of cultured epithelia to study transport and
its regulation. J. exp. Biol., 106: 55-69.
HANSEN, H.E., HESTBECH, J., SORENSEN, J.L., NORGAARD, K., HEILSKOV,
J., & AMDISEN, A. (1979) Chronic interstitial nephritis in patients on
long-term lithium treatment. Q. J. Med., 48: 577-591.
HANSEN, H.E., MOGENSEN, C.E., SORENSEN, J.L., NORGAARD, K., HEILSKOV,
J., & ANDERESEN, A. (1981) Albumin and beta-2-microglobulin excretion
in patients on long-term lithium treatment. Nephron, 29: 229-232.
HARD, G.C. (1975) Autoradiographic analysis of proliferative activity
in rat kidney epithelial and mesenchymal cell subpopulations following
a carcinogenic dose of dimethylnitrosamine. Cancer Res., 35:
3762-3773.
HARD, G.C. (1979) Effect of age at treatment on incidence and type of
renal neoplasm induced in the rat by a single dose of
dimethylnitrosamine. Cancer Res., 39: 4965-4970.
HARD, G.C. (1980) Morphological correlates of irreversible tumour
formation. In: Holmstedt, B., Lauwerys, R., Mercier, M., & Roberfroid,
M., ed. Mechanisms of toxicity and hazard evaluation, Amsterdam,
Elsevier/North Holland Biomedical Press, pp. 231-240.
HARD, G.C., (1984) High frequency, single-dose model of renal
adenoma/carcinoma induction using dimethylnitrosamine in Crl:(W)BR
rats. Carcinogenesis, 5: 1047-1050.
HARD, G.C. (1985) Identification of a high frequency model for renal
carcinoma by the induction of renal tumors in the mouse with a single
dose of streptozotocin. Cancer Res., 45: 703-708.
HARD, G.C. (1986) Renal carcinogenesis, rat. In: Jones, T.C., Mohr,
U., & Hunt, R.D., ed. Monographs on pathology of laboratory animals,
urinary system, Berlin, Heidelberg, New York, Springer-Verlag, pp.
45-49.
HARD, G.C. (1987) Chemically induced epithelial tumours and
carcinogenesis of the renal parenchyma. In: Bach, P.H. & Lock, E.A.,
ed. Nephrotoxicity in the experimental and the clinical situation,
Dordrecht, Boston, Lancaster, Martinus Nijhoff Publishers, pp.
211-251.
HARD, G.C. & BUTLER, W.H. (1970) Cellular analysis of renal neoplasia:
induction of renal tumors in dietary-conditioned rats by
dimethylnitrosamine with a reappraisal of morphological
characteristics. Cancer Res., 30: 2796-2805.
HARD, G.C. & BUTLER, W.H. (1971) Morphogenesis of epithelial neoplasms
induced in the rat kidney by dimethylnitrosamine. Cancer Res., 31:
1496-1505.
HARD, G.C., MACKAY, R.L., & KOCHHAR, O.S. (1984) Electron microscopic
determination of the sequence of acute tubular and vascular injury
induced in the rat kidney by a carcinogenic dose of
dimethylnitrosamine. Lab. Invest., 50: 659-672.
HARKONEN, S. & KJELLSTRAND, C.M. (1979) Intravenous pyelography in
non-uremic diabetic patients. Nephron, 24: 268-270.
HARKONEN, S. & KJELLSTRAND, C.M. (1981) Contrast nephropathy. Am. J.
Nephrol., 1: 69-77.
HARRIS, C.C., WESTON, A., WILLEY, J.C., TRIVERS, G.E., & MANN, D.L.
(1987) Biochemical and molecular epidemiology of human cancer:
Indicators of carcinogen exposure, DNA damage, and genetic
predisposition. Environ. Health Perspect., 75: 109-119.
HARRIS, S.I., BALABAN, R.S., BARRETT, L., & MANDEL, L.J. (1981)
Mitochondrial respiratory capacity and Na+- and K+-dependent
adenosine triphosphatase-mediated ion transport in the intact renal
cell. J. biol. Chem., 256: 10319-10328.
HASSALL, C.D., GANDOLFI A.J., & BRENDEL K. (1983) Correlation of the
in vivo and in vitro renal toxicity of
S-(1,2-dichlorovinyl)-L-cysteine. Drug chem. Toxicol., 6: 507-520.
HAUGLUSTAINE, D., VAN DAMME, B., DAENENS, P., & MICHIELSEN, P. (1980)
Silicon nephropathy, a possible occupational hazard. Nephron, 26:
219-224.
HAWKE, R.L. & WELCH, R.M. (1985) Major differences in the specificity
and regulation of mouse renal cytochrome P-450 dependent
monooxygenases. Mol. Pharmacol., 28: 283-289.
HAYES, D.M., CVITKOVIC, E., GOLBERG, R.B., SCHEINER, E., HELSON, L.,
& KRAKOFF, I.H. (1977) High dose cis-platinum diammine chloride:
amelioration of renal toxicity by mannitol diuresis. Cancer, 39:
1372-1381.
HELLSTEN, S., BERGE, T., & LINELL, F. (1983) Clinically unrecognized
renal carcinoma. Aspects of tumor morphology, lymphatic and
haematogenous metastatic spread. Br. J. Urol., 55: 166-170.
HENNIS, H.L., ALLEN, R.C., HENNIGAR, G.R., & SIMMONS, M.A. (1981) A
sensitive method for determining the nephrotoxic effects of the
analgesic acetaminophen upon esterases using isoelectric focusing.
Electrophoresis, 2: 187-190.
HESTBECH, J., HANSEN, H.E., AMDISEN, A., & OLSEN, S. (1977) Chronic
renal lesions following long-term treatment with lithium. Kidney Int.,
12: 205-213.
HIASA, Y., OHSHIMA, M., IWATA, C., & TANIKAKE, T. (1979)
Histopathological studies on renal tubular cell tumours in rats
treated with N-ethyl-N-hydroxyethylnitrosamine. Gann, 70: 817-820.
HIASA, Y., OHSHIMA, M., KITAHORI, Y., FUJIYATA, T., YUASA, T., &
MIYASHIRO, A. (1983) Basic lead acetate: Promoting effect on the
development of renal tubular cell tumours in rats treated with
N-ethyl-N-hydroxyethyl-nitrosamine. J. Natl Cancer Inst., 70: 761-765.
HIASA, Y., ENOKI, N., KITAHORI, Y., KONISHI, N., & SHIMOYAMA, T.
(1984a) DL-serine: Promoting activity on renal tumorigenesis by
N-ethyl-N-hydroxyethylnitrosamine in rats. J. Natl Cancer Inst., 73:
297-299.
HIASA, Y., KITAHORI, Y., KONISHI, N., ENOKI, N., SHIMOYAMA, T., &
MIYASHIRO, A. (1984b) Trisodium nitrilotriacetate monohydrate:
promoting effects on the development of renal tubular cell tumors in
rats treated with N-ethyl-N-hydroxyethylnitrosamine. J. Natl Cancer
Inst., 72: 483-489.
HIASA, Y., LIN, J.-C., KONISHI, N., KITAHORI, Y., ENOKI, N., &
SHIMOYAMA, T. (1984c) Histopathological and biochemical analyses of
transplantable renal adenocarcinoma in rats induced by
N-ethyl-N-hydroxyethyl-nitrosamine. Cancer Res., 44: 1664-1670.
HINSON, J.A., MONKS, T.T., HONG, M., HIGHET, R.J., & POHL, L.R. (1982)
3-(Glutathion-S-yl)acetaminophen: a biliary metabolite of
acetamino-phen. Drug Metab. Disp., 10: 47-50.
HINTON, D.E., HEATFIELD, B.M., LIPSKY, M.M., & TRUMP, B.F. (1980)
Animal model: chemically induced renal tubular carcinoma in rats. Am.
J. Pathol., 100: 317-320.
HIRAFUJI, M., SATOH, S., & OGURA, Y. (1980) Sex difference in
stimulatory actions of cofactors on prostaglandin synthetase in
microsomes from rat kidney medulla. Biochem. Pharmacol., 29:
2635-2637.
HIRSCH, G.H. (1976) Differential effects of nephrotoxic agents on
renal transport and metabolism by use of in vitro techniques.
Environ. Health Perspect., 15: 89-99.
HJELLE, J.T., PETERSON, D.R., & HJELLE, J.J. (1983) Drug metabolism in
isolated proximal tubule cells: aldehyde dehydrogenase. J. Pharmacol.
exp. Ther., 224: 699-706.
HOOK, J.B., ed. (1981) Toxicology of the kidney, New York, Raven
Press.
HOOK, J.B., MCCORMACK, K.M., & KLUWE, W.M. (1979) Biochemical
mechanisms of nephrotoxicity. In: Hodgson, E., Bend, J.R., & Philpot,
R.M., ed. Reviews in biochemical toxicology, Amsterdam, Oxford, New
York, Elsevier Science Publishers, pp. 53-78.
HOOK, J.B., ELCOMBE, C.R., ROSE, M.S., & LOCK, E.A. (1982)
Characterization of the effects of known hepatic monooxygenase
inducers on male and female rat and mouse kidneys. Life Sci., 31:
1077-1084.
HOOK, J.B., ISHMAEL, J., & LOCK, E.A. (1983) Nephrotoxicity of
hexachloro-1:3-butadiene in the rat: the effect of age, sex and
strain. Toxicol. appl. Pharmacol., 67: 121-131.
HORNING, E.S. & WHITTICK, J.W. (1954) The histogenesis of
stilboestrol-induced renal tumours in the male golden hamster. Br. J.
Cancer, 8: 451-457.
HORROBIN, D.F. (1980) The regulation of prostaglandin biosynthesis:
negative feedback mechanisms and the selective control of formulation
of 1 and 2 series prostaglandin: relevance to inflammation and
immunity. Med. Hypotheses, 6: 687-709.
HOU, S.H., BUSHINSKY, D.A., WISH, J.B., COHEN, J.J., & HARRINGTON,
J.T. (1983) Harrington, Hospital-acquired renal insufficiency: a
prospective study. Am. J. Med., 74: 243-248.
HULT, K. & FUCHS, R. (1986) Analysis and dynamics of ochratoxin A in
biological systems. In: Steyn, P.S. & Vleggaar, R., ed. Mycotoxins and
phycotoxins. Sixth International IUPAC Symposium on Mycotoxins and
Phycotoxins, Pretoria, Republic of South Africa, 22-25 July, 1985,
Amsterdam, Oxford, New York, Elsevier Science Publishers, pp. 365-376.
HUMES, H.D., WEINBERG, J.M., & KNAUSS, T.C. (1982) Clinical and
pathophysiological aspects of aminoglycoside nephrotoxicity. Am. J.
kidney Dis., 2: 5-29.
IESATO, K., WAKASHIN, M., WAKASHIN, Y., & TOJO, S. (1977) Renal
tubular dysfunction in Minamata disease. Ann. intern. Med., 86:
731-737.
INOUE, G., SAWADA, T., & YOSHIKAWA, M. (1973) Age-related change in
acid mucopolysaccharides level and water content in papillae.
Gerontologia, 19: 73-78.
ITO, N., HIASA, Y., TAMAI, A., & YOSHIDA, K. (1969) Effect of
unilateral nephrectomy on the development of kidney tumor in rats
treated with N-nitro-sodimethylamine. Gann, 60: 319-327.
ITO, N., SUGIHARA, S., MAKIURA, S., ARAI, M., HIRAO, K., DENDA, A., &
NISHIO, O. (1974) Effect of N-(3,5-dichlorophenyl)succinimide on the
histological pattern and incidence of kidney tumors in rats induced by
dimethylnitrosoamine. Gann, 65: 131-138.
JACOBSON, A., GRIECO, A.J., & FARBER, S.J. (1964) Hexosamine analysis
of renal papillae in diuretic and antiduiretic rats. Proc. Soc. Exp.
Biol. Med., 115: 1153-1156.
JACOBSEN, B.K., BJELKE, E., KVALE, G., & HEUCH, I. (1986) Coffee
drinking, mortality and cancer incidence: results from a Norwegian
prospective study. J. Natl Cancer Inst., 76: 823-831.
JACQUES, P.J. (1975) The endocytic uptake of macromolecules. In:
Trump, B.F. & Arstilla, U.A., ed. Pathology of cell membranes, New
York, London, San Franciso, Academic Press, Vol. 1, pp. 225-276.
JAFFE, N., KEIFER, R., ROBERTSON, R., CANGIR, A., & WANG, A. (1987)
Renal toxicity with cumulative doses of
cis-diamminedichloroplatinum-II in pediatric patients with
osteosarcoma. Cancer, 59: 1577-1581.
JAKOBY, W.B. & PASTAN, I.H., ed. (1979) Cell culture, methods in
enzymology, New York, London, San Franciso, Academic Press, Vol. 58,
642 pp.
JASMIN, G. & RIOPELLE, J.L. (1976) Renal carcinomas and erythrocytosis
in rats following intrarenal injection of nickel subsulfide. Lab.
Invest., 35: 71-78.
JOHANSSON, S. & ANGERVALL, L. (1976) Urothelial hyperplasia of the
renal papillae in female Sprague-Dawley rats induced by long term
feeding of phenacetin. Acta pathol. microbiol. Scand. Sect. A., 84:
353-354.
JOHANSSON, S., ANGERVALL, L., BENGTSSON, U., & WAHLQVIST, L. (1976) A
clinicopathologic and prognostic study of epithelial tumors of the
renal pelvis. Cancer, 37: 1376-1383.
JOLLOW, D.J., KOCSIS, J.J., SNYDER, R., & VAINIO, H., ed. (1976)
Biological reactive intermediates. Formation, toxicity and
inactivation, New York, London, Plenum Press.
JONES, B.R., BHALLA, R.B., MLADEK, J., KALEYA, R.N., GRALLA, R.J.,
ALLOCK, N.N., SCHWARTZ, M.K., YOUNG, C.W., & KEIDENBERG, M.M. (1980)
Comparison of methods of evaluating nephrotoxicity of cis-platinum.
Clin. Pharmacol. Ther., 24: 557-562.
JONES, D.P., SUNDBY, G.-B., ORMSTAD, K., & ORRENIUS, S. (1979) Use of
isolated kidney cells for study of drug metabolism. Biochem.
Pharmacol., 28: 929-935.
JOSEPOVITZ, C., FARRUGGELLA, T., LEVINE, R., LANE, B., & KALOYANIDES,
G.J. (1985) Effect of netilmicin on the phospholipid composition of
subcellular fractions of rat renal cortex. J. Pharmacol. exp. Ther.,
235: 810-819.
JUNG, K.Y. & ENDOU, H. (1989) Nephrotoxicity assessment by measuring
cellular ATP content II. Intranephron site of ochratoxin A
nephrotoxicity. Toxicol. appl. Pharmacol., 100: 383-390.
JUNG, K.Y. & ENDOU, H. (1989) Biphasic increasing effect of
angiotensin II on intracellular free calcium in isolated rat early
proximal tubule. Biochem. Biophys. Res. Commun., 165: 1221-1228.
JUNG, K.Y., UCHIDA, S., & ENDOU. H. (1989) Nephrotoxicity assessment
by measuring cellular ATP content I. Substrate specificities in the
maintenance of ATP content in isolated rat nephron segments. Toxicol.
appl. Pharmacol., 100: 369-382.
KACEW, S. (1987) Detection of nephrotoxicity of foreign compounds with
the use of in vitro and in vivo techniques. In: Bach, P.H. & Lock,
E.A., ed. Nephrotoxicity in the experimental and the clinical
situation, Dordrecht, Boston, Lancaster, Martinus Nijhoff Publishers,
pp. 533-562.
KAHLMETER, G. & DAHLAGER, J.I. (1984) Aminoglycoside toxicity - a
review of clinical studies published between 1975 and 1982. J.
antimicrob. Chemother., 13(Suppl. A): 9-22.
KALOYANIDES, G.J. (1984a) Renal pharmacology of aminoglycoside
nephrotoxicity. Contrib. Nephrol., 42: 148-167.
KALOYANIDES, G.J. (1984b) Aminoglycoside induced functional and
biochemical defects in the renal cortex. Fundam. appl. Toxicol., 4:
930-943.
KALOYANIDES, G.G. & PASTORIZA-MUNOZ, E. (1980) Aminoglycoside
nephro-toxicity. Kidney Int., 18: 571-582.
KAMINSKY, L.S., FASCO, M.J., & GUENGERICH, F.P. (1979) Comparison of
different forms of liver, kidney and lung microsomal cytochrome P-450
by immunological inhibition of regio- and stereoselective metabolism
of warfarin. J. biol. Chem., 254: 9657-9662.
KANFER, A. (1989) The role of coagulation in glomerular injury.
Toxicol. Lett., 46: 83-92.
KANISAWA, M. & SUZUKI, S. (1978) Induction of renal and hepatic tumors
in mice by ochratoxin A, a mycotoxin. Gann, 69: 599-600.
KANWAR, Y.S. & FARQUHAR, M.G. (1979) Presence of heparan sulfate in
the glomerular basement membrane. Proc. Natl Acad. Sci., 76:
1303-1307.
KAPLOWITZ, N. (1980) Physiological significance of glutathione
S-transferases. Am. J. Physiol., 239: G439-G444.
KATZBERG, R.W., MORRIS T.W., & BURGENER, F.A. (1977) Renal renin and
hemodynamic responses to selective renal artery catheterization and
angiography. Invest. Radiol., 12: 381-388.
KATZBERG, R.W., PABICO, R.C., MORRIS, T.W., HAYAKAWA, K., MCKENNA,
B.A., PANNER, B.J., VENTURA, J.A., & FISHER, H.W. (1986) Effects of
contrast media on renal function and subcellular morphology in the
dog. Invest. Radiol., 21: 64-70.
KAUMP, D.H. (1966) Pharmacology of the fenamates: II. Toxicology in
animals. Ann. physiol. Med., Suppl. 1: 16-23.
KENNEDY, J.F. (1979) Proteoglycans: biological and chemical aspects in
human life, Amsterdam, Oxford, New York, Elsevier Scientific
Publishers.
KHOURY, G.A., HOPPER, J.C., VARGHESE, Z., FARRINGTON, K., DICK, R.,
IRVING, J.D., SWENY, P., FERNANDO, O.N., & MOORHEAD, J.F. (1983)
Nephrotoxicity of ionic and non-ionic contrast material in digital
vascular imaging and selective renal arteriography. Br. J. Radiol.,
56: 631-635.
KILHAM, L., LOW, R.J., CONTI, S.F., & DALLENBACH, F.D. (1962)
Intranuclear inclusions and neoplasms in the kidneys of wild rats. J.
Natl Cancer Inst., 29: 863-885.
KINCAID-SMITH, P. (1979) Analgesis nephropathy in Australia. Contrib.
Nephrol., 16: 57-64.
KINCAID-SMITH, P., BURROWS, G.D., DAVIES, B.M., HOLWILL, B., WALTER,
M., & WALKER, R.G. (1979) Renal biopsy findings in lithium and
prelithium patients. Lancet, 2(8144): 700-701.
KITCHEN, D.N. (1984) Neoplastic renal effects of unleaded gasoline in
Fischer 344 rats. In: Mehlman, M.A., Hemstreet, C.P., Thorpe, J.J., &
Weaver, N.K., ed. Renal effects of petroleum hydrocarbons, Princeton,
New Jersey, Princeton Scientific Publishers, pp. 65-71.
KJELLEN, P., OLDBERG, A., RUBIN, K., & HOOK, M. (1977) Binding of
heparin and heparan sulphate to rat liver cells. Biochem. biophys.
Res. Commun., 74: 126-133.
KJELLSTROM, T. & NORDBERG, G.F. (1978) The kinetic model of cadmium
metabolism in the human being. Environ. Res., 16: 248-269.
KJELLSTROM, T., SHIROISHI, K., & EVRIN, P. (1977) Urinary
beta2-microglobulin excretion among people exposed to cadmium in the
general environment. An epidemiological study in cooperation between
Japan and Sweden. Environ. Res., 13: 318-344.
KLEIN, K.L., WANG, M.S., TORIKAI, S., DAVISDON, W.D., & KUROKAWA, K.
(1981) Substrate oxidation by isolated single nephron segments of the
rat. Kidney Int., 20: 29-35.
KLEINKNECHT, D., KANFER, A., MOREL-MAROGER, L., & MERY, J.P. (1978)
Immunologically mediated drug-induced acute renal failure. Contrib.
Nephrol., 10: 42-52.
KLINTMALM, G.B.M., IWATSUKI, S., & STARZL, T.E. (1981) Nephro-toxicity
of cyclosporine in liver and kidney transplant recipients. Lancet,
1(8218): 470-471.
KLOSS, M.W., COX, M.G., NORTON, R.M., SWENBERG, J.A., & BUS, J.S.
(1985) Sex-dependent differences in the disposition of
14C-5-2,2,4-trimethylpentane in Fischer 344 rats. In: Bach, P.H. &
Lock, E.A., ed. Renal heterogeneity and target cell toxicity, New
York, Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 489-492.
KLUWE, W.M. (1983) Chemical modulation of 1,2-dibromo-3-chloropropane.
Toxicology, 27: 287-299.
KLUWE, W.M. & HOOK, J.B. (1978) Analysis of gentamicin uptake by rat
renal cortical slices. Toxicol. appl. Pharmacol., 45: 531-539.
KLUWE, W.M. & HOOK, J.B. (1980) Effects of environmental chemicals on
kidney metabolism and function. Kidney Int., 18: 648-655.
KLUWE, W.M., HERRMANN, C.L., & HOOK, J.B. (1979) Effects of dietary
polychlorinated biphenyls and polybrominated biphenyls on the renal
and hepatic toxicities of several chlorinated hydrocarbon solvents in
mice. J. Toxicol. environ. Health, 5: 605-615.
KLUWE, W.M., ABDO, K.M., & HUFF, J. (1984) Chronic kidney disease and
organic chemical exposures: evaluations of causal relationships in
humans and experimental animals. Fundam. appl. Toxicol., 4: 889-901.
KOCIBA, R.J., KEYES, D.G., JERSEY, G.C., BALLARD, J.J., DITTENBER,
D.A., QUAST, J.F., WADE, C.E., HUMISTON, C.G., & SCHWARTZ, B.A. (1977)
Results of a two year chronic toxicity study with hexachlorobutadiene
in rats. Am. Ind. Hyg. Assoc. J., 38: 589-602.
KOJIMA, S., HAGA, Y., KURIHARA, T., & YAMAWAKI, T. (1977) A comparison
between fecal cadmium and urinary beta2-microglobulin, total protein
and cadmium among Japanese farmers. Environ. Res., 14: 436-451.
KOSEK, J.D., MAZZE, R.I., & COUSINS, M.J. (1974) Nephrotoxicity of
gentamicin. Lab. Invest., 30: 48-57.
KOSEKI, C., YAMAGUCHI, Y., FURUSAWA, M., & ENDOU, H. (1988) Isolation
by monoclonal antibody of intercalated cells of rabbit kidney. Kidney
Int., 33: 543-554.
KOSTAKIS, A.J., WHITE, D.J.G., & CALNE, R.Y. (1977) Toxic effects in
the use of cyclosporin A in alcoholic solution as an immunosuppressant
of rat heart allografts. IRCS Surg. Transplant., 5: 243.
KRECH, R., ZERBAN, H., & BANNASCH, P. (1981) Mitochondrial anomalies
in renal oncocytes induced in rats by N-nitrosomorpholine. Eur. J.
cell Biol., 25: 331-339.
KREISBERG, J.I. & KARNOVSKY, M.J. (1983) Glomerular cells in culture.
Kidney Int., 23: 439-447
KREISBERG, J.I., PITTS, A.M., & PRETLOW, T.G. (1977) Separation of
proximal tubule cells from suspensions of rat kidney cells in density
gradients of ficoll in tissue culture medium. Am. J. Pathol., 86:
591-600.
KREISBERG, J.I., HOOVER, R.L., & KARNOVSKY, M.J. (1978) Isolation and
characterization of rat glomerular epithelial cells in vitro. Kidney
Int., 14: 21-30.
KRESSE, H. & GROSSMANN, A. (1970) [Comparative study of the
mucopolysaccharide and collagen content in various topographical areas
of the kidney in the rat, dog and pig.] Z. Klin. Chem., 8: 420-424 (in
German).
KRIJSHELD, K.R. & GRAM, T.E. (1984) Selective induction of renal
microsomal cytochrome P-450-linked monooxygenases by
1,1-dichloroethylene in mice. Biochem. Pharmacol., 33: 1951-1956.
KRIZ, W. & BANKIR, L. (1988) A standard nomenclature for structures of
the kidney, Am. J. Physiol., 254: F1-F8.
KROGH, P., HALD, B., PLESTINA, R., & CEOVIC, S. (1977) Balkan
(endemic) nephropathy and foodborne ochratoxin A: preliminary results
of a survey of foodstuffs. Acta pathol. microbiol. Scand. Sect. B, 85:
238-240.
KUHN, J.A., ARGY, W.R., HAKOWSKI, T.A., SCHREINER, G.E., & SCHEIN,
P.S. (1978) Nephrotoxicity of cis-diamminedichloroplatinum as measured
by urinary beta glucuronidase. Clin. Res., 26: 776.
KUO, C.-H. & HOOK, J.B. (1982) Depletion of renal glutathione content
and nephrotoxicity of cephaloridine in rabbits, rats and mice.
Toxicol. appl. Pharmacol., 63: 292-302.
KUO, C.-H. & HOOK, J.B. (1983) Effects of age and sex on
hexachloro-1,3-butadiene toxicity in the Fisher 344 rat. Life Sci.,
33: 517-523.
KUO, C.-H., BRASELTON, W.E., & HOOK, J.B. (1982) Effect of
phenobarbital on cephaloridine toxicity and accumulation in rabbit and
rat kidneys. Toxicol. appl. Pharmacol., 64: 244-254.
KUROKAWA, Y., HAYASHI, Y., MAEKAWA, A., TAKAHASHI, M., KOKUBO, T., &
ODASHIMA, S. (1983) Carcinogenicity of potassium bromate administered
orally to F344 rats. J. Natl Cancer Inst., 71: 965-972.
LANDRIGAN, P.J., GOYER, R.A., CLARKSON, T.W., SANDLER, D.P., SMITH,
J.H., THUN, M.J., & WEDEEN, R.P. (l984) The work-relatedness of renal
disease. Arch. environ. Health, 39: 225-230.
LANGARD, S. & NORSETH, T. (1986) Chromium. In: Friberg, L., Nordberg,
G., & Vouk, V.B., ed. Handbook on the toxicology of metals, Amsterdam,
Oxford, New York, Elsevier Science Publishers, Vol. II, pp. 185-210.
LAQUEUR, G.L. & SPATZ, M. (1968) Toxicology of cycasin. Cancer Res.,
28: 2262-2267.
LASH L.H. & ANDERS M.W. (1986) Cytotoxicity of
S-(1,2-dichlorovinyl)-glutathione and S-(1,2-dichlorovinyl)-L-cysteine
in isolated rat kidney cells. J. biol. Chem., 261: 13076-13081.
LASH, L.H., ELFARRA, A.A., & ANDERS, M.W. (1986) Renal cysteine
conjugate betalyase. Bioactivation of nephrotoxic cysteine
S-conjugates in mitochondrial outer membrane. J. biol. Chem., 261:
5930-5935.
LAURENT, G., CARLIER, M.-B., ROLLMAN, B., VANHOOF, F., & TULKENS, P.M.
1982) Mechanism of aminoglycoside-induced lysosomal phospholipidosis:
in vitro and in vivo studies with gentamicin and amikacin.
Biochem. Pharmacol., 31: 3861-3870.
LAURENT, G., TOUBEAU, G., HEUSON-STEINNON, J.A., TULKENS, P., &
MALDAUGE, P. (1988) Kidney tissue repair after nephrotoxic injury:
biochemical and morphologic characterization. CRC crit. Rev. Toxicol.,
19: 147-183.
LAUWERYS, R., ROELS, H., BERNARD, A., & BUCHET, J.-P. (1980) Renal
response to cadmium in a population living in a non-ferrous smelter
area in Belgium. Int. Arch. occup. environ. Health, 45: 271-274.
LEE, J.B. (1980) Prostaglandins and the renin-angiotensin axis. Clin.
Nephrol., 14: 159-163.
LEEMING, B.W.A., SPOKES, K.C., & SILVA, P. (1985) Effect of meglumine
iothalamate on renal hemodynamics and function in the diabetic rat.
Invest. Radiol., 20: 971-977.
LEVENSON, D.J., SIMMONS, C.E., & BRENNER, B.H. (1982) Arachadonic acid
metabolism, prostaglandins and the kidney. Am. J. Med., 72: 354-374.
LI, J.J. & LI, S.A. (1984) Estrogen-induced tumorigenesis in hamsters:
roles for hormonal and carcinogenic activities. Arch. Toxicol., 55:
110-118.
LI, J.J., LI, S.A., KLICKA, J.K., PARSONS, J.A., & LAM, L.K.T. (1983)
Relative carcinogenic activity of various synthetic and natural
estrogens in the Syrian hamster kidney. Cancer Res., 43: 5200-5204.
LILIS, R. (1981) Long-term lead exposure, chronic nephropathy, and
renal cancer: a case report. Am. J. ind. Med., 2: 293-297.
LIND, C., VADI, H., & ERNSTER, L. (1978) Metabolism of
benzo(a)pyrene-3,6-quinone and 3-hydroxybenzo(a)pyrene in liver
microsomes from 3-methyl-cholanthrene-treated rats. Arch. Biochem.
Biophys., 190: 97-108.
LINDBERG, E. & VESTERBERG, O. (1983) Urinary excretion of proteins in
chromeplaters, ex-chromeplaters and referents. Scand. J. Work Environ.
Health, 9: 505-510.
LIPPMAN, A.J., HELSON, C., HELSON, L., & KRAKOFF, I.H. (1973) Clinical
trials of cis-diamminedichloroplatinum (NSC-119875). Cancer Chemother.
Rep., 57: 191-200.
LIPPMANN, S. (1982) Lithium's effects on the kidney. Postgrad. Med.,
71: 99-104, 107-108.
LITTERST, C.L. & WEISS, R.B. (1987) Clinical and experimental
nephrotoxicity of cancer chemotherapeutic agents. In: Bach, P.H. &
Lock, E.A., ed. Nephrotoxicity in the experimental and clinical
situation, Dordrecht, Boston, Lancaster, Martinus Nijhoff Publishers,
pp. 771-816.
LITTERST, C.L., MIMNAUGH, E.G., REAGAN, R.L., & GRAM, T.E. (1975a)
Comparison of in vitro drug metabolism by lung, liver and kidney of
several common laboratory species. Drug Metab. Disp., 3: 259-265.
LITTERST, C.L., MIMNAUGH, E.G., REAGAN, R.L., & GRAM, T.E. (1975b)
Drug metabolism by microsomes from extrahepatic organs of rat and
rabbit prepared by calcium aggregation. Life Sci., 17: 813-818.
LITTERST, C.L., MIMNAUGH, E.G., & GRAM, T.E. (1977) Alterations in
extrahepatic drug metabolism by factors known to affect hepatic
activity. Biochem. Pharmacol., 26: 749-755.
LOCK, E.A. (1979) The effect of paraquat and diquat on renal function
in the rat. Toxicol. appl. Pharmacol., 48: 327-336.
LOCK, E.A. (1987) Metabolic activation of halogenated chemicals and
its relevance to nephrotoxicity. In: Bach, P.H. & Lock, E.A., ed.
Nephrotoxicity in the experimental and clinical situation, Dordrecht,
Martinus Nijhoff, pp. 429-461.
LOCK, E.A. & ISHMAEL, J. (1979) The acute toxic effects of paraquat
and diquat on the rat kidney. Toxicol. appl. Pharmacol., 50: 67-76.
LOCK, E.A., ISHMAEL, J., & HOOK, J.B. (1984) Nephrotoxicity of
hexachloro-1,3-butadiene in the mouse: the effect of age, sex, strain,
monooxygenase modifiers, and the role of glutathione. Toxicol. appl.
Pharmacol., 72: 484-494.
LOCK, E.A., ODUM, J., & ORMOND, P. (1986) Transport of
N-acetyl-S-pentachloro-1,3-butadienylcysteine by the renal cortex.
Arch. Toxicol., 59: 12-15.
LOCKARD, W.V., PHILLIPS, R.D., HAYES, A.W., BERNDT, W.O., & O'NEAL,
R.M. (1980) Citrinin nephrotoxicity in rats: A light electron
microscopic study. Exp. mol. Pathol., 32: 266-340.
LONG, W.F. & WILLIAMSON, F.B. (1979) Glycosaminoglycans, calcium ions
and the control of cell proliferation. IRCS med. Sci., 7: 429-434.
LOURY, D., SMITH-OLIVER, T., & BUTTERWORTH, B.E. (1987) Assessment of
the binding potential of 2,2,4-trimethylpentane to the rat
alpha-2u-globulin. Toxicol. appl. Pharmacol., 88: 44-56.
LUTZ, W.K, JAGGI, W., & SCHLATTER, C. (1982) Covalent binding of
diethylstilbestrol to DNA in rat and hamster liver and kidney.
Chem.-biol. Interact., 42: 251-257.
MACFARLAND, H.N., ULRICH, C.E., HOLDSWORTH, C.E., KITCHEN, D.N.,
HALLIWELL, W.H., & BLUM, S.C. (1984) A chronic inhalation study with
unleaded gasoline vapor. J. Am. Coll. Toxicol., 3: 231-248.
MCAULIFFE, W.G. (1978) The effects of anti-diuretic hormone on the
morphology and histochemistry of the renal medullary interstitium of
rats with hereditary hypothalmic diabetes insipidus (Brattleboro
strain). Anat. Rec., 190: 474.
MCAULIFFE, W.G. (1980) Histochemistry and ultrastructure of the
interstitium of the renal papilla in rats with hereditary diabetes
insipidus (Brattleboro strain). Am. J. Anat., 157: 17-26.
MCCONKEY, D.J., HARTZELL, P., DUDDY, S.K., HÅKANSSON, H., & ORRENIUS,
S. (1988) 2,3,7,8-Tetrachlorodibenzo-p-dioxin kills immature
thymocytes by Ca2+- mediated endonuclease activation. Science, 242:
256-259.
MCINTOSH, C.S., MOSELEY, I.F., FRY, I.K., & CATTELL, W.R. (1975)
Excretion urography toxicity studies in experimental acute renal
failure. Nephron, 14: 373-377.
MCKINNEY, L.L., WEAHLY, F.B., ELDRIDGE, A.C., CAMPBELL, R.E., COWAN,
J.C., PICKEN, J.C., Jr, & BIESTER, H.E. (1957)
S-(Dichlorovinyl)-L-cysteine: an agent causing total aplastic anemia
in calves. J. Am. Chem. Soc., 79: 3932-3933.
MCKINNEY, L.L., PICKEN J.C., Jr, WEAKLEY, F.B., ELDRIDGE, A.C.,
CAMPBELL, R.E., COWAN, J.C., & BIESTER, H.E. (1959) Possible toxic
factor of trichloroethylene-extracted soybean oil meal. J. Am. Chem.
Soc., 81: 909-915.
MCLACHLAN, J.R., GOYER, R.A., & CHERIAN, M.G. (1980) Formation of
lead-induced inclusion bodies in primary rat kidney epithelial cell
cultures. Toxicol. appl. Pharmacol., 56: 418-431.
MCLACHLAN, M.S.F., CHICK, S., ROBERTS, E.E., & ASSCHER, A.W. (1972)
Intravenous urography in experimental acute renal failure in the rat.
Invest. Radiol., 7: 466-473.
MCLEAN, A.E.M. & MAGEE, P.N. (1970) Increased renal carcinogenesis by
dimethylnitrosamine in protein deficient rats. Br. J. exp. Pathol.,
51: 587-590.
MCMICHAEL, A.J. & JOHNSON, H.M. (1982) Long term mortality profile of
heavily-exposed lead smelter workers. J. occup. Med., 24: 375-378.
MCMURTRY, R.J., SNODGRASS, W.R., & MITCHELL, J.R. (1978) Renal
necrosis, glutathione depletion and covalent binding after
acetaminophen. Toxicol. appl. Pharmacol., 46: 87-100.
MADIAS, N.E. & HARRINGTON, J.T. (1978) Platinum nephrotoxicity
(review). Am. J. Med., 65: 307-314.
MAEKAWA, A., KUROKAWA, Y., TAKAHASHI, M., KOKUBO, T., OGIU, T.,
ONODERA, H., TANIGAWA, H., OHNO, Y., FURUKAWA, F., & HAYASHI, Y.
(1983) Spontaneous tumors in F-344/DuCrj rats. Gann, 74: 365-372.
MAHAFFEY, J.R. (1980) Nutrient-lead interactions. In: Singhal, R.L. &
Thomas, J.A., ed. Lead toxicity, Baltimore, Maryland, Urban and
Schwarzenberg, pp. 425-460.
MALCOLM, D. & BARNETT, H.A.R. (1982) A mortality study of lead workers
1925-76. Br. J. ind. Med., 39: 404-410.
MALEQUE, A., ENDOU, H., KOSEKI, C., & SAKAI, F. (1980) Nephron
hetero-geneity: gluconeogenesis from pyruvate in rabbit nephron. FEBS
Lett., 116: 154-156.
MALIS, C.D. & BONVENTRE, J.V. (1986) Mechanism of calcium potentiation
of oxygen free radical injury to renal mitochondria. J. biol. Chem.,
261: 14201-14208.
MANDAL, A.K. & BOHMAN, S.O., ed. (1980) The renal papilla and
hypertension, New York, London, Plenum Press.
MAO, P. & MOLNAR, J.J. (1967) The fine structure and histochemistry of
lead induced renal tumors in rats. Am. J. Pathol., 50: 571-603.
MARKOVIC, B. (1972) Endemic nephritis and urinary tract cancer in
Yugoslavia, Bulgaria and Rumania. J. Urol., 107: 212-219.
MARRE, R., TARARA, N., & LOUTON, T. (1980) Age dependent
nephrotoxicity and the pharmacokinetics of gentamicin in rats. Eur. J.
Pediatr., 133: 25-29.
MARTIN, G., DUROZARD, D., & BAVEREL, G. (1987) Acceleration of
ammonia-genesis in isolated rat kidney tubules by the antiepileptic
drug: valproic acid. In: Kovacevic, Z. & Guder, W.G., ed. Molecular
nephrology. Biochemical aspects of kidney function, Berlin, New York,
Walter de Gruyter & Co., pp. 287-292.
MARTIN, G., MICHOUDET, C., & BAVEREL, G. (1989) Stimulation of
glutamine metabolism by the antiepileptic drug, sodium valproate, in
isolated dog kidney tubules. Biochem. Pharmacol., 38: 3947-3952.
MARTIN, G., DUROZARD, D., BESSON, J., & BAVEREL, G. (1990) Effect of
the antiepileptic drug, sodium valproate, on glutamine and glutamate
metabolism in isolated human kidney tubules. Biochim. Biophys. Acta,
1033: 261-266.
MATSUSAKA, T., FUJII, M., NAKANO, T., TERAI., T., KURATA, A.,
IMAIZUMI, M., & ABE, H. (1989) Germanium-induced nephropathy: Report
of two cases and review of the literature. Clin. Nephrol., 30:
341-345.
MATZKE, G., LUCAROTTI, R., & SHARPIRO, H.S. (1983) Controlled
comparison of gentamicin and tobramycin nephrotoxicity. Am. J.
Nephrol., 3: 11-17.
MAUNSBACH, A.N., OLSEN, T.S., & CHRISTENSEN, E.I., ed. (1980)
Functional ultrastructure of the kidney, New York, London, Academic
Press.
MAYER, D., WEBER, E., KADENBACH, B., & BANNASCH, P. (1989)
Immunocyto-chemical demonstration of cytochrome c oxidase as a marker
for renal oncocytes and oncocytomas. Toxicol. Pathol., 17: 46-49.
MAZZE, R.I. (1976) Methoxyflurane nephropathy. Environ. Health
Perspect., 15: 111-119.
MAZZE, R.I. (1981) Methoxyflurane nephropathy. In: Hook, J.B., ed.
Toxicology of the kidney, New York, Raven Press, pp. 135-149.
MEHLMANN, M.A., HEMSTREET, C.P., THORPE, J.J., & WEAVER, N.K., ed.
(1984) Renal effects of petroleum hydrocarbons, Princeton, New Jersey,
Princeton Scientific Publishers.
MEEZAN, E. & BRENDEL, K. (1973) Effect of ethacrynic acid on oxidative
metabolism in isolated glomeruli. J. Pharmacol. exp. Ther., 187:
352-364.
MEIJER, S., SLEIJFER, D., MULDER, N., SLUITER, W.J., MARRINK, J.,
SCHRAFFORD KOOPS, H., BROUWERS, T.M., OLDHOFF, J., VAN DER HEM, G.K.,
& MANDEMA, E. (1983) Some effects of combination chemotherapy with
cisplatin on renal function in patients with nonseminomatous
testicular carcinoma. Cancer, 51: 2035-2040.
METZLER, M. (1981) Studies on the mechanism of carcinogenicity of
diethylstilboestrol: role of metabolic activation. Food Cosmet.
Toxicol., 19: 611-615.
METZLER, M. & MCLACHLAN, J.A. (1978) Peroxidase-mediated oxidation, a
possible pathway for metabolic activation of diethylstilbestrol.
Biochem. biophys. Res. Commun., 85: 874-884.
MICHELS, L.D., DAVIDMAN, M., & KEANE, W.F. (1983) Adriamycin nephrotic
syndrome: glomerular haemodynamics and permselectivity. Kidney Int.,
23: 246.
MICHOUDET, C. & BAVEREL, G. (1987) Metabolism of acetaldehyde in human
and baboon renal cortex. FEBS Lett., 216: 113-117.
MIHATSCH, M.J. & KNUSLI, K. (1982) Phenacetin abuse and malignant
tumours. An autopsy study covering 25 years (1953-1977). Klin.
Wochenschr., 60: 1339-1349.
MIHATSCH, M.J., HOFER, H.O., GUTZWILER, F., BRUNNER, F.P., &
ZOLLINGER, H.U. (1980a) [Phenacetin abuse. I. Frequency, per capita
consumption and consequential costs.] Schweiz. med. Wochenschr., 110:
108-115 (in German).
MIHATSCH, M.J., SCHMIDLIN, P., BRUNNER, F.P. HOFER, H.O., SIX, P., &
ZOLLINGER, H.U. (1980b) [Phenacetin abuse. II. Chronic renal
insufficiency and autopsy material in Basel.] Schweiz. med.
Wochenschr., 110: 116-124 (in German).
MIHATSCH, M.J., MANZ, T., KNUSLI, C., HOFER, H.O., RIST, M., GUETG,
R., RUTISHAUSER, G., & ZOLLINGERS, H.U. (1980c) [Phenacetin abuse.
III. Malignant tumours of the urinary tract associated with phenacetin
abuse in Basel, 1963-1977.] Schweiz. med. Wochenschr., 110: 255-264
(in German).
MIHATSCH, M.J., HOFER, H.O., GUDAT, F., KNUSLI, C., TORHORST, J., &
ZOLLINGER, U. (1984) Capillary sclerosis of the lower urinary tract
and analgesic nephropathy. Clin. Nephrol., 20: 285-301.
MIHATSCH, M.J., THIEL, G., BASLER, V., RYFFEL, B., LANDSMANN, J., VON
OVERBECK, J., & ZOLLINGER, H.U. (1985) Morphologic patterns in
cyclosporin-treated renal transplant recipients. Transplant. Proc.,
17(Suppl. 1): 101-116.
MIHATSCH, M.J., RYFFEL, B., HERMLE, M., BRUNNER, F.P., & THIEL, G.
(1986) Morphology of cyclosporine nephrotoxicity in the rat. Clin.
Nephrol., 25(Suppl. 1): S2-S8.
MILLER, E.C. & MILLER, J.A. (1981) Searches for ultimate chemical
carcinogens and their reactions with cellular macromolecules. Cancer,
47: 2327-2345.
MILMAN, N. & GOTTLIEB, P. (1977) Renal function after high-dose
urography in patients with chronic renal insufficiency. Clin.
Nephrol., 7: 250-254.
MITCHELL, J.R., JOLLOW, D.J., POTTER, W.Z., DAVIS, D.C., GILLETTE,
J.R., & BRODIE, B.B. (1973) Acetaminophen-induced hepatic necrosis. I.
Role of drug metabolism. J. Pharmacol. exp. Ther., 187: 185-194.
MITCHELL, J.R., MCMURTRY, R.J., STATHAM, C.N., & NELSON, S.D. (1977)
Molecular basis for several drug-induced nephropathies. Am. J. Med.,
62: 518-526.
MITSUMORI, K., MAITA, K., SAITO, T., TSUDA, S., & SHIRASU, Y. (1981)
Carcinogenicity of methylmercury chloride in ICR mice: preliminary
note on renal carcinogenesis. Cancer Lett., 12: 305-310.
MODAN, B., BOICHIS, H., BOTT-KANNER, G., BARELL, V., BAR-HOACH, N., &
ELIAHOU, H.E. (l975) An epidemiological study of renal failure. l. The
need for maintenance dialysis. Am. J. Epidemiol., 101: 276-280.
MOFFAT, D.B. (1979) The mammalian kidney, Cambridge, New York,
Cambridge University Press.
MOFFAT, D.B. (1981) New ideas on the anatomy of the kidney. J. clin.
Pathol., 34: 1197-1206.
MOFFAT, D.B. (1982) Morphology of the kidney in relation to
nephrotoxicity -portae renales. In: Bach, P.H., Bonner, F.W., & Lock,
E.A., ed. Nephrotoxicity: Assessment and pathogenesis, New York,
Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 10-26.
MOHANDAS, J., DUGGIN, G.G., HORVATH, J.S., & TILLER, D.J. (1981a)
Regional differences in peroxidatic activation of paracetamol
(acetaminophen) mediated by cytochrome P-450 and prostaglandin
endoperoxide synthetase in rabbit kidney. Res. Commun. chem. Pathol.
Pharmacol., 34: 69-80.
MOHANDAS, J., DUGGIN, G.G., HORVATH, J.S., & TILLER, D.J. (1981b)
Metabolic oxidation of acetaminophen (paracetamol) mediated by
cytochrome P-450 mixed function oxidase and prostaglandin endoperoxide
synthetase in rabbit kidney. Toxicol. appl. Pharmacol., 61: 252-259.
MOHR, U., HAAS, H., & HILFRICH, J. (1974) The carcinogenic effects of
dimethylnitrosamine and nitrosomethylurea in European hamsters
( Cricetus cricetus L.). Br. J. Cancer, 29: 359-364.
MOLD, J.W. & STEIN, H.F. (1986) Sounding board: the cascade effect in
the clinical care of patients. New Engl. J. Med., 314: 512-514.
MOLDEUS, P., JONES, D.P., ORMSTAD, K., & ORRENIUS, S. (1978) Formation
and metabolism of a glutathione S-conjugate in isolated rat liver and
kidney cells. Biochem. biophys. Res. Commun., 83: 195-200.
MOORE, J.R., GOYER, R.A., & WILSON, M.H. (1973) Lead-induced inclusion
bodies: Solubility, amino acid content, and relationship to residual
acidic proteins. Lab. Invest., 29: 488-494.
MOORE, M.A., NAKAMURA, T., SHIRAI, T., & ITO, N. (1986)
Immuno-histochemical demonstration of increased glucose-6-phosphate
dehydrogenase in preneoplastic and neoplastic lesions induced by
propylnitrosamines in F 344 rats and Syrian hamsters. Gann, 77:
131-138.
MOREAU, J.F., DROZ, D., NOEL, L.H., LEIBOWITCH, J., JUNGERS, P., &
MICHEL, J.R. (1980) Tubular nephrotoxicity of water soluble iodinated
contrast media. Invest. Radiol., 15: S54-S60.
MOREL, F., CHABARDES, D., & IMBERT, M. (1976) Functional segmentation
of the rabbit distal tubule by microdetermination of hormone-dependent
adenylate cyclase activity. Kidney Int., 9: 264.
MORIN, J.P., VIOTTE, G., VANDERWALLE, A., VAN HOOF, F., TULKENS, P.,
& FILLASTRE, J.P. (1980) Gentamicin-induced nephrotoxicity: a cell
biology approach. Kidney Int., 18: 583-590.
MORITA, T., OITE, T., KIHARA, I., YAMAMOTO, T., HARA, M., NAKA, A., &
OHNO, S. (1980) Culture of isolated glomeruli from normal and
nephritic rabbits. I. Characterization of outgrowing cells. Acta
pathol. Jpn., 30: 917-926.
MORRISON, A.R. (1980) Prostaglandins and the kidney. Am. J. Med., 69:
171-173.
MOSTOFI, F.K., SESTERHENN, I.A., & SOBIN, L.H. (1981) Histological
typing of kidney tumours, Geneva, World Health Organization, 26 pp.
MUDGE, G.H. (1982) Analgesic nephropathy: renal drug distribution and
metabolism. In: Porter, G.A., ed. Nephrotoxic mechanisms of drugs and
environmental toxins, New York, London, Plenum Press, pp. 209-225.
MUDGE, G.H. (1985) Pathogenesis of nephrotoxicity: Pharmacological
principles. In: Bach, P.H. & Lock, E.A., ed. Renal heterogeneity and
target cell toxicity, New York, Chichester, Brisbane, Toronto, John
Wiley and Sons, pp. 1-12.
MUIRHEAD, E.E. & PITCOCK, J.A. (1980) Evidence on involvement of the
renal papilla in hypertension. In: Mandal, A.K. & Bohman, S.-O., ed.
The renal papilla and hypertension, New York, London, Plenum Press,
pp. 35-61.
MURPHY, G.P., MIRAND, E.A., JOHNSTON, G.S., SCHMIDT, J.D., & SCOTT,
W.W. (1966) Renal tumors induced by a single dose of
dimethylnitrosamine: morphologic, functional, enzymatic and hormonal
characterizations. Invest. Urol., 4: 39-56.
MURRAY, B.M., PALLER, M.S., & FERRIS, T.F. (1985) Effect of
cyclosporine administration on renal hemodynamics in conscious rats.
Kidney Int., 28: 767-774.
MURRAY, T. & GOLDBERG, M. (1975) Chronic interstitial nephritis:
Etiologic factors. Ann. intern. Med., 82: 453-459.
MUTTI, A. (1987) The study of urinary excretion of kidney antigens to
reveal early effects of exposure to exogenous chemicals. In: Foa', V.,
Emmett, E.A., Maroni, M., & Colombi, A., ed. Occupational and
environmental chemical hazards, Chichester, Ellis Horwood Limited, pp.
315-322.
MUTTI, A. (1989) Detection of renal diseases in humans: developing
markers and methods. Toxicol. Lett., 46: 177-191.
MUTTI, A., CAVATORTA, A., PEDRONI, C., BORGHI, A., GIAROLI, C., &
FRANCHINI, I. (1979) The role of chromium accumulation in the
relationship between airborne and urinary chromium in welders. Int.
Arch. occup. environ. Health, 43: 123-133.
MUTTI, A., LUCERTINI, S., VALCAVI, P.P., NERI, T.M., FORNARI, M.,
ALINOVI, R., & FRANCHINI, I. (1985) Urinary excretion of brush-border
antigen revealed by monoclonal antibody: early indicator of toxic
nephropathy. Lancet, 2(8461): 914-917.
MUTTI, A., ALINOVI, R., BERGAMASCHI, E., FORNARI, M., & FRANCHINI, I.
(1988) Monoclonal antibodies to brush-border antigens for the early
diagnosis of nephrotoxicity. Arch. Toxicol., Suppl. 12: 162-165.
NAKADA, J., YAMADA, H., & ENDOU, H. (1986a) Evidence that
alpha-1-adrenergic stimuli specifically increase gluconeogenesis of
the isolated proximal convoluted tubule in the rat. Renal Physiol., 9:
213-222.
NAKADA, J., MACHIDA, T., & ENDOU, H. (1986b) Nephrotoxicity of
cisplatin in rats. In: Tanabe, T., Hook, J.B., & Endou, H., ed.
Nephrotoxicity of antibiotics and immunosuppressants, Amsterdam,
Oxford, New York, Elsevier Science Publishers, pp. 179-182.
NANRA, R.S. (1980) Clinical and pathological aspects of analgesic
nephropathy. Br. J. clin. Pharmacol., 10: 359S-368S.
NANRA, R.S., STUART-TAYLOR, J., DELEON, A.H., & WHITE, K.H. (1978)
Analgesic nephropathy: etiology, clinical syndrome, and
clinicopatho-logic correlations in Australia. Kidney Int., 13: 79-92.
NASH, J.A., KING, L.J., LOCK, E.A., & GREEN, T. (1984) The metabolism
and disposition of hexachloro-1:3-butadiene in the rat and its
relevance to nephrotoxicity. Toxicol. appl. Pharmacol., 73: 124-137.
NAVRAN, S.S. & LOUIS-FERDINAND, R.T. (1975) P-Chloro-N-methylaniline
demethylation by rat kidney subcellular fractions. Res. Commun. chem.
Pathol. Pharmacol., 12: 713-721.
NELSON, S.D. (1982) Metabolic activation and drug toxicity. J. med.
Chem., 25: 753-765.
NEUHAUS, O.W. (1986) Renal reabsorption of low-molecular weight
proteins in adult male rats: alpha-2u globulin. Proc. Soc. Exp. Biol.
Med., 182: 531-539.
NEUHAUS, O.W., FLORY, W., BISWAS, N., & HOLLERMAN, C.E. (1981) Urinary
excretion of alpha2u-globulin and albumin by adult male rats following
treatment with nephrotoxic agents. Nephron, 28: 133-140.
NEWSON, G.D. & VUGRIN, D. (1987) Etiologic factors in renal cell
adeno-carcinoma. Semin. Nephrol., 7: 108-116.
NEWTON, J.F., KUO, C.H., GEMBORYS, M.W., MUDGE, G.H., & HOOK, J.B.
(1982) Nephrotoxicity of p-aminophenol: a metabolite of acetaminophen
in the Fischer 344 rat. Toxicol. appl. Pharmacol., 65: 336-344.
NEWTON, J.F., BAILIE, M.B., & HOOK, J.B. (1983a) Acetaminophen
nephro-toxicity in the rat. Renal metabolic activation in vitro.
Toxicol. appl. Pharmacol., 70: 433-444.
NEWTON, J.F., YOSHIMOTO, J., BERNSTEIN, J., RUSH, G.F., & HOOK, J.B.
(1983b) Acetaminophen nephrotoxicity in the rat. I. Strain differences
in nephrotoxicity and metabolism. Toxicol. appl. Pharmacol., 69:
291-306.
NICOLL, J.W., SWANN, P.F., & PEGG, A.E. (1975) Effect of
dimethylnitro-samine on persistence of methylated guanines in rat
liver and kidney DNA. Nature (Lond)., 254: 261-262.
NOGAWA, K., KOBAYASHI, E., & HONDA, R. (1979a) A study of the
relationship between cadmium concentrations in urine and renal effects
of cadmium. Environ. Health Perspect., 28: 161-168.
NOGAWA, K., ISHIZAKI, A., & KOBAYASHI, E. (1979b) A comparison between
health effects of cadmium and cadmium concentration in urine among
inhabitants of the Itai-Itai disease endemic districts. Environ. Res.,
18: 397-409.
NOGAWA, K., HONDA, R., KIDO, T., TSURITANI, I., YAMADA, Y., ISHIZAKI,
M., & YAMAYA, H. (1989) A dose-response analysis of cadmium in the
general environment with special reference to total cadmium intake
limit. Environ. Res., 48: 7-16.
NOGUEIRA, E. & BANNASCH, P. (1988) Cellular origin of rat renal
oncocytoma. Lab. Invest., 59: 337-343.
NOGUEIRA, E., KLIMEK, F., WEBER, E., & BANNASCH, P. (1989) Collecting
duct: origin of rat renal clear cell tumours. Virchows Arch. B Cell
Pathol., 57(5): 275-283.
NOMIYAMA, K. (1980) Recent progress and perspectives in cadmium health
effects studies. Sci. total Environ., 14: 199-232.
NONOGUCHI, H., UCHIDA, S., SHIIGAI, T., & ENDOU, H. (1985) Effect of
chronic metabolic acidosis on ammonia production from L-glutamine in
microdissected rat nephron segments. Pflugers Arch., 403: 229-235.
NONOGUCHI, H., TAKEHARA, Y., & ENDOU, H. (1986) Intra- and
inter-nephron heterogeneity of ammoniagenesis in rats: Effect of
chronic metabolic acidosis and potassium depletion. Pflugers Arch.,
407: 245-251.
NORBY L.H. & DIBONA, G.F. (1975) The renal vascular effects of
meglumine diatrizoate. J. Pharmacol. exp. Ther., 193: 932-940.
NTP (1983) Technical report on the carcinogenesis bioassay of
penta-chloroethane (CAS No. 76-01-7) in F344/N rats and B6c3F1 mice
(gavage studies), Research Triangle Park, North Carolina, US
Department of Health and Human Services, National Toxicology Program
(NIH Publication No. 83-1788).
NTP (1986) Technical report on the toxicology and carcinogenesis
studies of 1,4-dichlorobenzene (CAS No. 106-46-7) in F344/N rats and
B6c3F1 mice (gavage studies), Research Triangle Park, North Carolina,
US Department of Health and Human Services, National Toxicology
Program (NIH Publication No. 86-2575).
NTP (1987) Toxicology and carcinogenesis studies of
1,4-dichlorobenzene in F344/N rats and B6C3F1 mice, Research Triangle
Park, North Carolina, US Department of Health and Human Services,
National Toxicology Program (NTP TR 319) (NIH Publication No.
87-2575).
NTP (1988) NTP draft technical report on the toxicology and
carcinogenesis studies of ochratoxin A (CAS No. 303-47-9) in F344/N
rats (gavage studies), Research Triangle Park, North Carolina, US
Department of Health and Human Services, National Toxicology Program
(NIH Publication No. 88-2813).
NUYTS, G.D., ELSEVIERS, M.M., & DE BROE, M.E. (1989) Health impact of
renal disease due to nephrotoxicity. Toxicol. Lett., 46: 31-44.
OBATOMI, D.K. & PLUMMER, D.T. (1986) Oxygen uptake and glutamate
dehydro-genase release in isolated renal tubules from rat kidney.
Biochem. Soc. Trans., 14: 1281-1282.
OHI, G., YAMAMOTO, S., FUJINO, T., ITAI, Y., UENO, K., SAITO, H.,
INOBA, Y., NAKAJIMA, K., & SHIROISHI, K. (1982) Urinary
beta2-microglobulin does not serve as diagnostic tool for Minamata
disease. Arch. environ. Health, 37: 336-341.
OHMORI, T., HIASA, Y., MURATA, Y., & WILLIAMS, G.M. (1982) Gamma
glutamyl transpeptidase activity in carcinogen-induced epithelial
lesions of rat kidney. Gann, 73: 543-548.
OHMORI, T. & TABEI, R. (1983) Modulation of N-nitrosodimethylamine
kidney tumorigenesis by unilateral hydronephrosis and multiple
putrescine adminis-trations. J. Natl Cancer Inst., 71: 787-793.
OLORUNSOGO, O.O., MALOMO, S.O., & BABABUNMI, E.A. (1985)
Proto-nophoric properties of fluorinated arylalkylsulphonamides:
Observations with perfluidone. Biochem. Pharmacol., 34: 2945-2952.
OLSON, C.T., YU, K.O., & SERVE, M.P. (1986) Metabolism of nephrotoxic
cis-and trans-decalin in Fischer-344 rats. J. Toxicol. environ.
Health, 18: 285-292.
OLSON, M.J., GARG, B.D., MURTY, C.V.R., & ROY, A.K. (1987)
Accumulation of alpha2u-globulin in the renal proximal tubules of male
rats exposed to unleaded gasoline. Toxicol. appl. Pharmacol., 90:
43-51.
OMICHINSKI, J.G., BRUNBORG, G., SODERLUND, E.J., DAHL, J.E., BAUSANO,
J.A., HOLME, J.A., NELSON, S.D., & DYBING, E. (1987) Renal necrosis
and DNA damage caused by selectively deuterated and methylated analogs
of 1,2-dibromo-3-chloropropane in the rat. Toxicol. appl. Pharmacol.,
91: 358-370.
ORLOFF, J. & BERLINER, R.W., ed. (1973) Handbook of physiology.
Section 8: Renal physiology, Washington, DC, American Physiological
Society.
ORMSTAD, K. (1982) The use of isolated kidney cells, glomeruli and
subcellular fractions for assessing renal function. In: Bach, P.H.,
Bonner, F.W., Bridges, J.W., & Lock, E.A., ed. Nephrotoxicity:
Assessment and pathogenesis, New York, Chichester, Brisbane, Toronto,
John Wiley and Sons, pp. 161-168.
ORMSTAD, K. (1987) Metabolism of glutathione in the kidney. In: Bach,
P.H. & Lock, E.A., ed. Nephrotoxicity in the experimental and the
clinical situation, Dordrecht, Boston, Lancaster, Martinus Nijhoff
Publishers, pp. 405-428.
ORMSTAD, K., JONES, D.P., & ORRENIUS, S. (1981) Preparation and
characterisation of isolated kidney cells. Methods Enzymol., 77:
137-146.
ORTMANN, M., VIERBUCHEN, M., KOLLER, G., & FISCHER, R. (1988) Renal
oncocytoma. I. Cytochrome c oxidase in normal and neoplastic renal
tissue as detected by immunohistochemistry - a valuable aid to
distinguish oncocytomas from renal cell carcinomas. Virchows Arch. B
Cell Pathol., 56: 165-173.
OTTOSEN, P.D., SIGH, B., CHRISTENSEN, J., OLSEN, S., & CHRISTENSEN,
S. (1984) Lithium induced interstitial nephropathy associated with
chronic renal failure. Acta pathol. microbiol. immunol. Scand., 92(6):
447-54.
OZOLS, R.F., CORDEN, B.J., JACOB, J., WESLEY, M.N., OSTCHAGA, Y., &
YOUNG, R.C. (1984) High dose cisplatin in hypertonic saline. Ann.
intern. Med., 100: 19-24.
PALESTINE, A.G., AUSTIN, H.A., BALOW, J.E., ANTONOVYCH, T.T., SABNIS,
S.G., PREUSS, H.G., & NUSSENBLATT, R.B. (1986) Renal histo-pathologic
alterations in patients treated with cyclosporine for uveitis. New
Engl. J. Med., 314: 1293-1298.
PARKER, R.A., BENNETT, W.M., & PORTER, G.A. (1982) Animal models in
the study of aminoglycoside nephrotoxicity. In: Whelton, A. & Neu,
H.S., ed. The aminoglycosides: Microbiology, clinical use and
toxicology, New York, Basel, Marcel Dekker, pp. 235-267.
PEARSON, R.M. (1979) Methods for the assessment of the effects of
drugs on renal blood flow. Br. J. clin. Pharmacol., 7: 129-138.
PEPELJNJAK, S. & CVETNIC, Z. (1985) The mycotoxicological chain and
contamination of food by ochratoxin A in the nephropathic and
non-nephropathic areas in Yugoslavia. Mycopathologia, 90: 147-153.
PERANTONI, A. & BERMAN, J.J. (1979) Properties of Wilms' tumor line
(TuWi) and pig kidney line (LLC-PK1) typical of normal kidney tubular
epithelium. In Vitro, 15: 446-454.
PETERSON, C.D., COLLINS, A.J., HINES, J. M., BULLOCK, M.L., & KEANE,
W.F. (1981) Ethylene glycol poisoning: pharmacokinetics during therapy
with ethanol and hemodialysis. New Engl. J. Med., 304: 21-23.
PETKOVA-BOCHAROVA, T. & CASTEGNARO, M. (1985) Ochratoxin A
con-tamination of cereals in an area of high incidence of Balkan
endemic nephropathy in Bulgaria. Food Addit. Contam., 2: 267-270.
PHILLIPS R.D. & COCKRELL, B.Y. (1984a) Kidney structural changes in
rats following inhalation exposure to C10-C11 isoparaffinic solvent.
Toxicology, 33: 261-273.
PHILLIPS, R.D. & COCKRELL, B.Y. (1984b) Effect of certain light
hydrocarbons on kidney function and structure in male rats. In:
Mehlman, M.D., Hemstreet, C.O., Thrope, J.J., & Weaver, N.K., ed.
Renal effects of petroleum hydro-carbons, Princeton, New Jersey,
Princeton Scientific Publishers, pp. 89-105.
PHILLIPS, R.D. & EGAN, G.F. (1984a) Effect of C10-C11 isoparaffinic
solvent on kidney function in Fischer 344 rats during eight weeks of
inhalation. Toxicol. appl. Pharmacol., 73: 500-510.
PHILLIPS, R.D. & EGAN, G.F. (1984b) Subchronic inhalation exposure of
dearomatized white spirit and C10-C11 isoparaffinic hydrocarbon in
Sprague-Dawley rats. Fundam. appl. Toxicol., 4: 808-818.
PHILLIPS, R.D., HAYES, A.W., & BERNDT W.O. (1979) Disposition of
14C-citrinin in the rat. Toxicology, 12: 285-298.
PHILLIPS, R.D., HAYES, A.W., & BERNDT, W.O. (1980a) High pressure
liquid chromatographic analysis of citrinin and its application to
biological systems. J. Chromatogr., 190: 419-425.
PHILLIPS, R.D., HAYES, A.W., BERNDT, W.O., & WILLIAMS. W.L. (1980b)
Effects of citrinin on renal function and structure. Toxicology, 16:
123-137.
POLLACK, E.S. & HORM, J.W. (1980) Trends in cancer incidence and
mortality in the United States, 1969-76. J. Natl Cancer Inst., 64:
1091-1103.
POMMER, W., GLAESKE, G., & MOLZAHN, M. (1986) The analgesic problem in
the Federal Republic of Germany: analgesic consumption, frequency of
analgesic nephropathy and regional differences. Clin. Nephrol., 26:
273-278.
PORTER, G.A., ed. (1982) Nephrotoxic mechanisms of drugs and
environmental toxins, New York, London, Plenum Press.
PORTER, G.A. (1989) Risk factors for toxic nephropathies. Toxicol.
Lett., 46: 269-279.
PORTER, G.A. & BENNETT, W.M. (1989) Drug-induced renal effects of
cyclosporine, aminoglycoside antibiotics and lithium: extrapolation of
animal data to man. In: Bach, P.H. & Lock, E.A., ed. Nephrotoxicity:
Extrapolation from in vitro to in vivo, and animals to man, New
York, London, Plenum Press, pp. 147-170.
PORTER, N.A., WOLF, R.A., & WEENEN, H. (1980) The free radical
oxidation of polyunsaturated lecithins. Lipids, 15: 163-167.
POTTER, C.L., GANDOLFI, A.J., NAGLE, R.B., & CLAYTON, J.W. (1981)
Effects of inhaled chlorotrifluoroethylene and and hexafluoropropene
on the rat kidney. Toxicol. appl. Pharmacol., 59: 431-440.
POUNDS, J.G. (1984) Effect of lead intoxication on calcium-mediated
cell function: a review. Neurotoxicology, 5: 295-332.
POUNDS, J.G. & ROSEN, J.F. (1988) Cellular Ca2+ Homeostasis and
Ca2+-mediated cell processes as critical targets for toxicant
action: con-ceptual and methodological pitfalls. Toxicol. appl.
Pharmacol., 94: 331-341.
POUR, P., ALTHOFF, J., SALMASI, S.Z., & STEPAN, K. (1979) Spontaneous
tumors and common diseases in three types of hamsters. J. Natl Cancer
Inst., 63: 797-811.
POWELL, W.S. (1980) Distribution of prostaglandin - hydroxylases in
different tissues. Prostaglandins, 19: 701-710.
POWLES, R.L., BARRETT, A.J., CLINK, H., KAY, H.E.M., SLOANE, J., &
MCELWAIN, T.J. (1978) Cyclosporin A for the treatment of
graft-versus-host disease in man. Lancet, 2: 1327-1331.
PRESCOTT, L.F. (1982) Analgesic nephropathy: a reassessment of the
role of phenacetin and other analgesics. Drugs, 23: 75-149.
PRETLOW, T.G., II & PRETLOW, T.P., ed. (1982) Cell separation: Methods
and selected applications, New York, London, San Francisco, Academic
Press, Vol. 1.
PRETLOW, T.G., II & PRETLOW, T.P., ed. (1983) Cell separation: Methods
and selected applications, New York, London, San Francisco, Academic
Press, Vol. 2.
PRETLOW, T.G., II & PRETLOW, T.P., ed. (1984) Cell separation: methods
and selected applications, New York, London, San Francisco, Academic
Press, Vol. 3.
PRICE, R.G. (1982) Urinary enzymes, nephrotoxicity and renal disease.
Toxicology, 23: 99-134.
PRUEKSARITANONT, T., CHEN, M.-L., & CHIOU, W.L. (1984) Simple and
micro high-performance liquid chromatographic method for simultaneous
analysis of p-aminohippuric acid and iothalamate in biological fluids.
J. Chromatogr., 306: 89-97.
RAHIMI, A., EDMONDSON, R.P.S., & JONES, N.F. (1981) Effect of
radiocontrast media on kidney of patients with renal disease. Br. Med.
J., 282: 1194-1195.
RAKIETEN, N., GORDON, B.S., COONEY, D.A., DAVIS, R.D., & SCHEIN, P.S.
(1968) Renal tumorigenic action of streptozotocin (NSC-85998) in rats.
Cancer Chemother. Rep., 52: 563-567.
RAMSAMMY, L.S., JOSEPOVITZ, C., LING, K.-Y., LANE, B.P., &
KALOYANIDES, G.J. (1986) Effects of diphenyl-phenylenediamine on
gentamicin-induced lipid peroxidation and toxicity in rat renal
cortex. J. Pharmacol. exp. Ther., 238: 83-88.
RAMSAMMY, L.S., JOSEPOVITZ, C., LING, K.-Y., LANE, B.P., &
KALOYANIDES, G.J. (1987) Failure of inhibition of lipid peroxidation
by vitamin E to protect against gentamicin nephrotoxicity in the rat.
Biochem. Pharmacol., 36: 2125-2132.
RAMSAMMY, L.S., JOSEPOVITZ, C., & KALOYANIDES, G.J. (1988a) Gentamicin
inhibits agonist stimulation of the phosphatidylinositol cascade in
primary cultures of rabbit proximal tubular cells and in rat renal
cortex. J. Pharmacol. exp. Ther., 247: 989-996.
RAMSAMMY, L.S., JOSEPOVITZ, C., LANE, B., & KALOYANIDES, G.J. (1988b)
Failure of hydroxyl radical scavengers to protect against
gentamicin-induced acute renal failure in the rat. Fed. Am. Soc. Exp.
Biol. J., 2: 407A.
RAMSAMMY, L.S., JOSEPOVITZ, C., LANE, B., & KALOYANIDES, G.J. (1989a)
Effect of gentamicin on phospholipid metabolism in cultured rabbit
proximal tubular cells. Am. J. Physiol., 256: C204-C213.
RAMSAMMY, L.S., JOSEPOVITZ, C., LANE, B.P., & KALOYANIDES, G.J.
(1989b) Polyaspartic acid protects against gentamicin nephrotoxicity
in the rat. J. Pharmacol. exp. Ther., 250: 149-153.
RANSLEY, P.G. & RISDON, R.A. (1979) The pathogenesis of reflux
nephropathy. Contrib. Nephrol., 16: 90-97.
RAPP, N.S., ZENSER, T.V., BROWN, W.W., & DAVIS, B.B. (1980) Metabolism
of benzidine by a prostaglandin-mediated process in renal inner
medullary slices. J. Pharmacol. exp. Ther., 215: 401-406.
RASMUSSEN, H.H. & IBELS, L.S. (1982) Acute renal failure: multivarite
analysis of causes and risk factors. Am. J. Med., 73: 211-218.
RAVNSKOV, U. (1985) Possible mechanism of hydrocarbon-associated
glomerulo-nephritis. Clin. Nephrol., 23: 294-298.
RECKNAGEL, R.O. (1983) Carbon tetrachloride hepatotoxicity: status quo
and future prospects. Trends Pharmacol. Sci., March: 129-131.
REDDY, M.V., GUPTA, R.C., RANDERATH, E., & RANDERATH, K. (1984)
32P-Postlabeling test for covalent DNA binding of chemicals in
vivo: Application to a variety of aromatic carcinogens and methylating
agents. Carcinogenesis, 5: 231-243.
REED, D.J. & BEATTY, P.W. (1980) Biosynthesis and regulation of
glutathione: toxicological implications. In: Hodgson, E., Bend, J.R.,
& Philpot, R.M., ed. Reviews in biochemical toxicology, Amsterdam,
Oxford, New York, Elsevier Science Publishers, Vol. 2, pp. 213-241.
REED, J.R., WILLIAMS R.H., & LUKE, R.G. (1983) The renal hemodynamic
response to diatrizoate in normal and diabetic rats. Invest. Radiol.,
18: 536-540.
REICHERT, D., SPENGLER, V., ROMEN, W., & HENSCHLER, D. (1984)
Carcinogenicity of dichloroacetylene: an inhalation study.
Carcinogenesis, 5: 1411-1420.
RENNICK, G. (1978) Renal excretion of cationic drugs - Tubule
transport and metabolism. In: Fillastre, J.P., ed. Nephrotoxicity
interaction of drugs with membrane systems, mitochondria-lysosomes,
New York, Masson Publishing Co., pp. 1-41.
REZNIK, G., WARD, J.M., HARDISTY, J.F., & RUSSFIELD, A. (1979) Renal
carcinogenic and nephrotoxic effects of the flame retardant
tris(2,3-dibromopropyl) phosphate in F344 rats and (C57BL/6N X
C3H/HeN)F1 mice. J. Natl Cancer Inst., 63: 205-212.
RICHMAN, A.V., MASCO, H.L., RIFKIN, S.I., & ACHARYA, M.K. (1980)
Minimal-change disease and the nephrotic syndrome associated with
lithium therapy. Ann. intern. Med., 92: 70-72.
RIMPELA, A.H. & PUKKALA, E.I. (1987) Cancers of affluence: positive
social class gradient and rising incidence trend in some cancer forms.
Soc. Sci. Med., 24: 601-606.
RITCHIE, A.W.S., KEMP, I.W., & CHISHOLM, G.D. (1984) Is the incidence
of renal carcinoma increasing? Br. J. Urol., 56: 571-573.
ROBBINS, M.E.C., CAMPLING, D., WHITEHOUSE, E., HOPEWELL, J.W., &
MICHALOWSKI, A. (1990) Cisplatin-induced reductions in renal reserve
uncovered by unilateral nephrectomy: an experimental study in the pig.
Cancer Chemother. Pharmacol., 27: 211-218.
ROCH-RAMEL, F. & PETERS, G. (1979) Micropuncture techniques as a tool
in renal pharmacology. Annu. Rev. Pharmacol. Toxicol., 19: 323-345.
ROELS, H., LAUWERYS, R., BUCHET, J.P., BERNARD, A., CHETTLE, D.R.,
HARVEY, T.C., & AL HADDAD, I.K. (1981a) In vivo measurement of liver
and kidney cadmium in workers exposed to this metal. Environ. Res.,
26: 217-240.
ROELS, H., LAUWERYS, R., BUCHET, J.P., & BERNARD, A. (1981b)
Environmental exposure to cadmium and renal function of aged women in
three areas of Belgium. Environ. Res., 24: 117-130.
ROELS, H., DJUBGANG, J., BUCHET, J.P., BERNARD, A., & LAUWERYS, R.
(1982) Evolution of cadmium-induced renal dysfunction in workers
removed from exposure. Scand. J. Work Environ. Health, 8: 191-200.
ROELS, H., GENNART, J.P., LAUWERYS, R., BUCHET, J.P., MALCHAIRE, J.,
& BERNARD, A. (1985) Surveillance of workers exposed to mercury
vapour: validation of a previously proposed biological threshold limit
value for mercury concentration in urine. Am. J. ind. Med., 7: 45-71.
ROELS, H., LAUWERYS, R., BUCHET, J.P., BERNARD, A., VOS, A., &
OVERSTEYNS, M. (1989) A prospective study of proteinuria in cadmium
workers. In: Bach, P.H. & Lock, E.A., ed. Nephrotoxicity:
Extrapolation from in vitro to in vivo, and animals to man, New
York, London, Plenum Press, pp. 33-36.
ROMAN-FRANCO, A.A., TWIRELLO, M., ALBINI, B., OSSI, E., MILGROM,
F., & ANDRES, G.A. (1978) Anti-basement membrane antibodies and
antigen-antibody complexes in rabbits injected with mercuric chloride.
Clin. Immunol. Immunopathol., 9: 404-481.
ROSENBERG, M.R. & MICHALOPOULOS, G. (1987) Kidney proximal tubular
cells isolated by collagenase perfusion grow in defined media in the
absence of growth factors. J. cell Physiol., 131: 107-113.
ROSENBERG, M.R., NOVICKI, D.L., JIRTLE, R.L., NOVOTNY, A., &
MICHALOPOULOS, G. (1985) Promoting effect of nicotinamide on the
development of renal tubular cell tumours in rats initiated with
diethylnitrosamine. Cancer Res., 45: 809-814.
ROSENMAN, K.D., VALCIUKAS, J.A., GLICKMAN, L., MYERS, B.R., & CINOTTI,
A. (1986) Sensitive indicators of inorganic mercury toxicity. Arch.
environ. Health, 41: 208-215.
ROSENMANN, E., DISHON, T., DURST, A., & BOSS, J.H. (1971) Urinary
excretion of kidney antigens in experimental renal disease of the rat.
Br. J. exp. Pathol., 52: 388-394.
ROSS, B., TANGE, J., EMSLIE, K., HART, S., SMAIL, M., & CALDER, I.
(1980) Paracetamol metabolism by the isolated perfused rat kidney.
Kidney Int., 18: 562-570.
ROSSOF, A.H., SLAYTON, R.E., & PERLIA, C.P. (1972) Preliminary
clinical experience with cis-diamminedichloroplatinum (NSC-119875
CACP). Cancer, 30: 1451-1456.
ROXE, D.M. (1980) Toxic nephropathy from diagnostic and therapeutic
agents. Am. J. Med., 69: 759-766.
RUSH, G.F., SMITH, J.H., NEWTON, J.F., & HOOK, J.B. (1984) Chemically
induced nephrotoxicity: role of metabolic activation. CRC crit. Rev.
Toxicol., 13: 99-160.
RYFFEL, B., DONATSCH, P., MADORIN, M., MATTER, B.E., RUTTIMAN, G.,
SCHON, H., STOLL, R., & WILSON, J. (1983) Toxicological evaluation of
cyclosporin A. Arch. Toxicol., 53: 107-141.
RYFFEL, B., HIESTAND, P., FOXWELL, B., DONATSCH, P., BOELSTERLI, H.J.,
MAURER, G., & MIHATSCH, M.J. (1986) Nephrotoxic and immunosuppressive
potentials of cyclosporine metabolites in rats. Transplant. Proc.,
18(Suppl. 5): 41-45.
SAFIRSTEIN, R., WINSTON, J., GOLDSTEIN, M., MOEL, D., DICKMAN, S., &
GUTTENPLAN, J. (1986) Cisplatin nephrotoxicity. Am. J. kidney Dis., 8:
356-367.
SAITO, H., SHIOJI, R., HURUKAWA, Y., NAGAI, K., ARIKEWA, T., SAITO,
T., SASAKI, Y., FURUYAMA, T., & YOSHINAGA, K. (1977) Cadmium-induced
proximal tubular dysfunction in a cadmium polluted area. Contrib.
Nephrol., 6: 1-12.
SANDLER, D.P. (1987) Epidemiology in the assessment of nephrotoxicity
In: Bach, P.H. & Lock, E.A., ed. Nephrotoxicity in the experimental
and clinical situation. Part 2, Dordrecht, Boston, Lancaster, Martinus
Nijhoff Publishers, pp. 847-883.
SANDLER, D.P., SMITH, J.C., WEINBERG, C.R., BUCKALEW, V.M., DENNIS,
V.W., BLYTHE, W.B., & BURGESS, W.P. (1989) Analgesic use and chronic
renal disease. New Engl. J. Med., 320: 1238-1243.
SANDSTEAD, H.H., MICHELAKIS, A.M., & TEMPLE, T.E. (1970) Lead
intoxication. Its effect on the renin-aldosterone response to sodium
deprivation. Arch. environ. Health, 20: 356-363.
SASTRASINH, M., KNAUSS, T.C., WEINBERG, J.M., & HUMES, H.D. (1982)
Identification of the aminoglycoside binding site in rat renal brush
border membranes. J. Pharmacol. exp. Ther., 222: 350-358.
SATO, G. & REID, L. (1978) Replacement of serum in cell culture by
hormones. Int. Rev. Biochem., 20: 219-251.
SCHENTAG, J.J. (1983) Specificity of renal tubular damage criteria for
aminoglycoside nephrotoxicity in critically ill patients. J. clin.
Pharmacol., 23: 473-483.
SCHENTAG, J.J., SUFTEN, T.A., & PLAUT, M.E. (1978) Early detection of
aminoglycoside nephrotoxicity with urinary beta-2-microglobulin. J.
Med., 9: 201-210.
SCHERBERICH, J.E., FALKENBERG, F., MONDORF, W., PFLEIDERER, G., &
SCHEOPPE, W. (1976) Isolation of kidney tissue antigens from urine by
biospecific immunosorbents. Verh. Dtsch. Ges. Inn. Med., 82:
1469-1473.
SCHERBERICH, J.E., MONDORF, W., FALKENBERG, F.W., PIERARD, D., &
SCHOEPPE, W. (1984) Monitoring drug nephrotoxicity. Quantitative
estimation of human kidney brush border antigens in urine as a
specific marker of tubular damage. Contrib. Nephrol., 42: 81-92.
SCHMIDT, U. & GUDER, W.G. (1976) Sites of enzyme activity along the
nephron. Kidney Int., 9: 233-242.
SCHOLER, D.W. & EDELMAN, I.S. (1979) Isolation of rat kidney cortical
tubules enriched in proximal and distal segments. Am. J. Physiol.,
237: F350-F359.
SCHRIER, R.W. & GOTTSCHALK, C.W. (1987) Diseases of the kidney,
Boston, Little, Brown and Co.
SCHULTZ, S.G., LAVELLE K.J., & SWAIN, R. (1982) Nephrotoxicity of
radiocontrast media in ischemia renal failure in rabbits. Nephron, 32:
113-117.
SCHUMANN, G.B., LERNER, S.I., WEISS, M.A., GAWRONSKI, L., & LOHIYA,
G.K. (1980) Inclusion-bearing cells in industrial workers exposed to
lead. Am. J. clin. Med., 74: 192-196.
SCHWARTZ, S.L., JOHNSON, C.B., & DOOLAN, P.D. (1970) Study of the
mechanism of renal vasculogenesis induced in the rat by EDTA.
Comparison of the cellular activities of calcium and chromium. Mol.
Pharmacol., 6: 54-59.
SCHWARZ, A. (1987) Analgesic-associated nephropathy. Klin.
Wochenschr., 65: 1-16.
SCHWERTZ, D.W., KREISBERG, J.I., & VENKATACHALEM, M.A. (1984) Effects
of aminoglycosides on proximal tubule brush border membrane
phosphatidylinositol-specific phospholipase C. J. Pharmacol. exp.
Ther., 231: 48-55.
SELDIN, D.D. & GIEBISCH, G. (1985) The kidney, physiology and
pathophysiology, New York, Raven Press, Vol. 1.
SELEVAN, S.G., LANDRIGAN, P.J., STERN, F.B., & JONES, J.H. (1985)
Mortality of lead smelter workers. Am. J. Epidemiol., 122: 673-683.
SELLI, C., HINSHAW, W.M., WOODARD, B.H., & PAULSON, D.F. (1983)
Stratification of risk factors in renal cell carcinoma. Cancer, 52:
899-903.
SHINOHARA, Y., ARAI, M., HIRAO, K., SUGIHARA, S., NAKANISHI, K.,
TSUNODA, H., & ITO, N. (1976) Combination effect of citrinin and other
chemicals on rat kidney tumorigenesis. Gann, 67: 147-155.
SHIRACHI, D.Y., JOHANSEN, M.G., MCGOWAN, J.P., & TU, S.-H. (1983)
Tumorigenic effect of sodium arsenite in rat kidney. Proc. West.
Pharmacol. Soc., 26: 413-415.
SHIRAI, T., OHSHIMA, M., MASUDA, A., TAMANO, S., & ITO, N. (1984)
Promotion of 2-(ethylnitrosamino)ethanol-induced renal carcinogenesis
in rats by nephrotoxic compounds: positive responses with folic acid,
basic lead acetate, and N-(3,5-dichlorophenyl)succinimide but not with
2,3-dibromo-1-propanol phosphate. J. Natl Cancer Inst., 72: 477-482.
SHIROISHI, K., KJELLSTROM, T., KUBOTA, K., EVRIN, P.E., ANAYAMA, M.,
VESTERBERG, O., SHIMADA, T., PISCATOR, M., IWATA, T., & NISHINO, H.
(1977) Urine analysis for detection of cadmium-induced renal changes,
with special reference to beta2-microglobulin. Environ. Res., 13:
407-424.
SHORT, B.G., BURNETT, V.L., & SWENBERG, J.A. (1986) Histopathology and
cell proliferation induced by 2,2,4-trimethylpentane in the male rat
kidney. Toxicol. Pathol., 14: 194-203.
SIEGERS C.-P. & KLAASSEN, C.D. (1984) Biliary excretion of
acetaminophen in ureter-ligated rats. Pharmacology, 28: 177-180.
SIERSBAEK-NIELSON, K., MOLHOLM HANSEN, J.M., KAMPMANN, J., &
KRISTENSEN, M. (1971) Rapid evaluation of creatinine clearance.
Lancet, 1: 1133-1134.
SIKIC, B.I., MIMNAUGH, E.G., LITTERST, C.L., & GRAM, T.E. (1977) The
effects of ascorbic acid deficiency and repletion on pulmonary, renal
and hepatic drug metabolism in the guinea-pig. Arch. Biochem.
Biophys., 179: 663-671.
SILVERBLATT, F.J. & KUEHN, C. (1979) Autoradiography of gentamicin
uptake by the rat proximal tubule cell. Kidney Int., 15: 335-345.
SMITH, H.J., LEVORSTAD, K., BERG, K.J., ROOTWELT, K., & SVEEN, K.
(1985) High dose urography in patients with renal failure. A double
blind investigation of iohexol and metrizoate. Acta radiol. Diagn.,
26: 213-220.
SMITH, J.H. & HOOK, J.B. (1984) Mechanism of chloroform
nephrotoxicity. III. Renal and hepatic microsomal metabolism of
chloroform in mice. Toxicol. appl. Pharmacol., 73: 511-524.
SMITH, J.H., MAITA, K., SLEIGHT, S.D., & HOOK, J.B. (1983) Mechanism
of chloroform nephrotoxicity. I. Time course of chloroform toxicity in
male and female mice. Toxicol. appl. Pharmacol., 70: 467-479.
SMITH, J.H., MAITA, K., SLEIGHT, S.D., & HOOK, J.B. (1984) Effect of
sex hormone status on chloroform nephrotoxicity and renal mixed
function oxidases in mice. Toxicology, 30: 305-316.
SMITH, J.H., RUSH, G.F., & HOOK, J.B. (1986) Induction of renal and
hepatic mixed function oxidases in the hamster and guinea-pig.
Toxicology, 38: 209-218.
SMITH, R.L. (1974) The excretory function of bile: the elimination of
drugs and toxic substances in bile - factors affecting biliary
excretion, London, Chapman & Hall, pp. 16-34.
SNYDER, R., PARKE, D., KOCSIS, J.J., JOLLOW, D.J., & GIBSON, G.G., ed.
(1981) Biological reactive intermediates. Part II, New York, London,
Plenum Press.
SOBERON, L., BOWMAN, R.L., PASTORIZA-MUNOZ, E., & KALOYANIDES, G.J.
(1979) Comparative nephrotoxicities of gentamicin, netilmicin and
tobramycin in the rat. J. Pharmacol. exp. Ther., 210: 334-343.
SOOSE, M., HABERSTROH, U., ROVIRA-HOLBACH, G., BRUNKHORST, R., &
STOLTE, H. (1988) Heterogeneity of glomerular barrier function in
early adriamycin nephrosis of MWF rats. Clin. Physiol. Biochem., 6:
310-315.
SPATARO, R. (1984) New contrast agents for urography. Radiol. Clin.
North Am., 22: 365-380.
SRAER, J., FOIDART, J., CHANSEL, D., MAHIEU, P., & ARDAILLOU, R.
(1980) Prostaglandin synthesis by rat isolated glomeruli and
glomerular cultured cells. Int. J. Biochem., 12: 203-207.
STERNBERG, S.S., PHILIPS, F.S., & CRONIN, A.P. (1972) Renal tumors and
other lesions in rats following a single intravenous injection of
Daunomycin. Cancer Res., 32: 1029-1036.
STOLTE, H. & ALT, J., ed. (1980) Research animals and experimental
design in nephrology. Contrib. Nephrol., 19: 1-249.
STOLTE, H. & ALT, J. (1982) The choice of animals for nephrotoxic
investigations. In: Bach, P.H., Bonner, F.W., Bridges, J.W., & Lock,
E.A., ed. Nephrotoxicity: Assessment and pathogenesis, New York,
Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 102-112.
STONARD, M.D., CHATER, B.V., DUFFIELD, D.P., NEVITT, A.L., O'SULLIVAN,
J.J., & STEEL, G.T. (1983) An evaluation of renal function in workers
occupationally exposed to mercury vapour. Int. Arch. occup. environ.
Health, 52: 177-189.
STONARD, M.D., PHILLIPS, P.G.N., FOSTER, J.R., SIMPSON, M.G. & LOCK,
E.A. (1986) Alpha-2u-globulin: Measurement in rat kidney following
administration of 2,2,4-trimethylpentane. Toxicology, 41: 161-168.
STRIKER, G.E., KILLEN, P.D., & FARIN, F.M. (1980) Human glomerular
cells in vitro: Isolation and characterization. Transplant. Proc.,
12: 88-99.
STUVE, J. & GALLE, P. (1970) Role of mitochondria in the handling of
gold by the kidney. J. cell. Biol., 44: 667-676.
SUFRIN, G. & BECKLEY, S.A. (1980) Renal adenocarcinoma, Geneva,
International Union Against Cancer, pp. 133-155 (Technical Report No.
49).
SUN, C.N. (1980) Effect of anti-diuretic hormone to the connective
tissue of rat renal papilla. Exp. Pathol., 18: 469-473.
SUN, C.N., WHITE, H.J., & TOWBIN, E.J. (1972) Histochemistry and
electron microscopy of renal papilla in a genetic strain of rats with
diabetic insipidus. Nephron, 9: 308-317.
SUNDERMAN, F.W., Jr (1981) Present research on nickel carcinogenesis.
Environ. Health Perspect., 40: 131-142.
SUNDERMAN, F.W., MCCULLY, R.S., & HOPFER, S.M. (1984) Association
between erythrocytosis and renal cancers in rats following intrarenal
injection of nickel compounds. Carcinogenesis, 5: 1511-1517.
SUZUKI, S., SUZUKI, S., NAKAMURA, N., & KOIZUMI, T. (1976) The
heterogeneity of dermatan sulfate and heparan sulfate in rat liver and
a shift in the glycosaminoglycan contents in carbon tetrachloride
damaged liver. Biochim. Biophys. Acta, 428: 166-181.
SUZUKI, S., SHAPIRO, R., MULROW, P.J., & TAN, S.Y. (1980) Urinary
prostaglandin E2 excretion in chronic renal disease. Prostaglandins
Med., 4: 377-382.
SWANN, P.F., KAUFMAN, D.G., MAGEE, P.N., & MACE, R. (1980) Induction
of kidney tumours by a single dose of dimethylnitrosamine: dose
response and influence of diet and benzo(a)pyrene pretreatment. Br. J.
Cancer, 41: 285-294.
SWENBERG, J.A., SHORT, B., BORGHOFF, S., STRASSER, J., & CHARBONNEAU,
J. (1989) The comparative pathobiology of alpha-2u-globulin
nephropathy. Toxicol. appl. Pharmacol., 97: 35-46.
SZEFLER, S.J. & ACARA, M. (1979) Isoproterenol excretion and
metabolism in the isolated perfused rat kidney. J. Pharmacol. exp.
Ther., 210: 295-300.
TALIERCIO, C.P., VLIETSTRA, R.E., FISHER, L.D., & BURNETT, J.C. (1986)
Risks for renal dysfunction with cardiac angiography. Ann. intern.
Med., 104: 501-504.
TAMURA, T. & ENDOU, H. (1988) Contribution of purine nucleotide cycle
to intranephron ammoniagenesis in rats. Am. J. Physiol., 255:
F1122-F1127.
TANAKA, H., ISHIKAWA, E., TESHIMA, S., & SHIMUZI, E. (1986)
Histopathological study of human cisplatin nephropathy. Toxicol.
Pathol., 14: 247-257.
TARLOFF, J.B., GOLSTEIN, R.S., & HOOK, J.B. (1987) Xenobiotic
metabolism in the mammalian kidney. In: Bach, P.H. & Lock, E.A., ed.
Nephro-toxicity in the experimental and the clinical situation,
Dordrecht, Boston, Lancaster, Martinus Nijhoff Publishers, pp.
371-404.
TEIXEIRA, R.B., KELLEY, F., ALPERT, H., PARDOV, V., & VAAMONDE, C.A.
(1982) Complete protection from gentamicin-induced acute renal failure
in the untreated streptozotocin diabetes mellitus rat. Kidney Int.,
21: 600-612.
TERRACINI, B. & PARKER, V.H. (1965) A pathological study on the
toxicity of S-(dichlorovinyl)-L-cysteine. Food Cosmet. Toxicol., 3:
67-74.
TERUEL, J.L., MARCEN, R., ONAINDIA, J.M., SERRANO, A., QUEREDA, C., &
ORTUNO, J. (1981) Renal function impairment caused by intravenous
urography, a prospective study. Arch. intern. Med., 141: 1271-1274.
TESTA, B. & JENNER, P. (1976) Drug metabolism: chemical and
biochemical aspects, New York, Basel, Marcel Dekker.
THOENES, W., STÖRKEL, S., & RUMPELT, H.-J. (1985) Human chromophobe
cell renal carcinoma. Virchows Arch. B Cell Pathol., 48: 207-217.
THOMPSON, W.M., FOSTER, W.L., Jr, HALVORSEN, R.A., DUNNICK, N.R.,
ROMMEL, A., & BATES, M. (1984) Iopamidol: new, nonionic contrast agent
for excretory urography. Am. J. Roentgenol., 142: 329-332.
THORNLEY, C., DAWNAY, A., & CATTEL, W.R. (1985) Human Tamm-Horsfall
glycoprotein: urinary and plasma levels in normal subjects and
patients with renal disease determined by a fully validated
radioimmunoassay. Clin. Sci., 68: 529-535.
THUN, M.J., BAKER, D.B., STEENLAND, K., SMITH, A.B., HALPERIN, W., &
BERL, T.B. (1985) Renal toxicity in uranium mill workers. Scand. J.
Work Environ. Health, 11: 83-90.
THURAU, K. (1964) Renal hemodynamics. Am. J. Med., 36: 698-719.
TOKOFF-RUBIN, N.E. (1986) Diagnosis of tubular injury in renal
transplant patients by urinary assay for proximal tubular antigen, the
adenosine-deaminase-binding protein. Transplantation, 41: 593-599.
TOLEDO, O.M.S. & DIETRICH, C.P. (1977) Tissue specific distribution of
sulfated mucopolysaccharides in mammals. Biochim. Biophys. Acta, 498:
114-122.
TOLINS, J.P. & RAIJ, L. (1988) Chronic amphotericin B nephrotoxicity
in the rat; protective effect of prophylactic salt loading. Am. J.
kidney Dis., 11: 313-317.
TOUBEAU, G., LAURENT, G., CARLIER, M.-B., ABID, S., MALDAGUE, P.,
HEUSON-STIENNON, J.A., & TULKENS, P.M. (1986) Tissue repair in rat
kidney cortex after short treatment with aminoglycosides at low doses.
Lab. Invest., 54: 385-393.
TRIFILLIS, A.L., REGEC, A., HALL-CRAGGS, M., & TRUMP, B.F. (1986)
Effects of cyclosporine on cultured human renal tubular cells.
Toxicol. Pathol., 14: 210-212.
TRINH-TRANG-TAN, M.M., BOUBY, N., COUTAUD, C., & BANKIR, L. (1986)
Quick isolation of rat medullary thick ascending limbs. Enzymatic and
metabolic characterization. Pflügers Arch., 407: 228-234.
TRUMP, B.F., BEREZESKY, I.K., SMITH, M.W., PHELPS, P.C., & ELLIGET,
K.A. (1989) The relationship between cellular ion deregulation and
acute and chronic toxicity. Toxicol. appl. Pharmacol., 97: 6-22.
TSUDA, H., SAKATA, T., TAMANO, S., OKUMURA, M., & ITO, N. (1983)
Sequential observations on the appearance of neoplastic lesions in the
liver and kidney after treatment with
N-ethyl-N-hydroxyethylnitrosamine followed by partial hepatectomy and
unilateral nephrectomy. Carcinogenesis, 4: 523-528.
TSUDA, H., HACKER, H.J., KATAYAMA, H., MASUI, T., ITO, N., & BANNASCH,
P. (1986) Correlative histochemical studies on preneoplastic and
neoplastic lesions in the kidney of rats treated with nitrosamines.
Virchows Arch. B Cell Pathol., 51: 385-404.
TSUDA, H., MOORE, M.A., ASAMOTO, M., INOUE, T., FUKUSHIMA, S., ITO,
N., SATOH, K., AMELIZAD, Z., & OESCH, F. (1987) Immunohistochemically
demonstrated altered expression of cytochrome P-450 molecular forms
and epoxide hydrolase in N-ethyl-N-hydroxyethylnitrosamine-induced rat
kidney and liver lesions. Carcinogenesis, 8: 711-717.
TUBBS, R.R., GEPHARDT, G.N., MCMAHON, J.T., POHL, M.C., VIDT, D.G.,
BARENBERG, S.A., & VALENZUELA, R. (1982) Membranous
glomerulo-nephritis associated with industrial mercury exposure. Am.
J. clin. Pathol., 77: 409-413.
TULKENS, P.M. (1989) Nephrotoxicity of aminoglycoside antibiotics.
Toxicol. Lett., 46: 107-123.
TUNE, B.M. (1975) Relationship between the transport and toxicity of
cephalosporine in the kidney. J. infect. Dis., 132: 189-194.
TUNE, B.M. (1986) The nephrotoxicity of cephalosporin antibiotics -
structure-activity relationships. Comments Toxicol., 1: 145-170.
TUNE, B.M. & FRAVERT, D. (1980) Mechanisms of cephalosporin
nephrotoxicity. A comparison of cephaloridine and cephaloglycin.
Kidney Int., 18: 591-600.
TUNE, B.M., BURG, M.D., & PATLAK, C.S. (1969) Characteristics of
p-aminohippurate transport in proximal renal tubules. Am. J. Physiol.,
217: 1057-1063.
TUNE, B.M., SIBLEY, R.K., & HSU, C.-Y. (1988) The mitochondrial
respiratory toxicity of cephalosporin antibiotics. An inhibitory
effect on substrate uptake. J. Pharmacol. exp. Therap., 245:
1054-1059.
TYSON, C.K. & MIRSALIS, J.C. (1985) Measurement of unscheduled DNA
synthesis in rat kidney cells following in vivo treatment with
genotoxic agents. Environ. Mutagen., 7: 889-899.
UCHIDA, S. & ENDOU, H. (1988) Substrate specificity to maintain
cellular ATP along the mouse nephron. Am. J. Physiol., 255: F977-F983.
ULLRICH, K.J. & GREGER, R. (1985) Approaches to the study of tubule
transport function. In: Seldin, D.G. & Giebisch, G., ed. The kidney,
physiology and pathology, New York, Raven Press, Chapter 20, pp.
427-455.
VAAMONDE, C.A., BIER, R., GOUVEA, W., ALPERT, H., KELLY, J., & PARDO,
V. (1984) Effect of duration of diabetes on the protection observed in
the diabetic rat against gentamicin-induced acute renal failure.
Miner. Electrolyte Metab., 10: 209-216.
VALTIN, H., ed. (1973) Renal functions: mechanisms preserving fluid
and solute balance in health, Boston, Toronto, Little Brown and Co.
VAN DORP, D. (1971) Recent developments in the biosynthesis and the
analyses of prostaglandins. Ann. NY Acad. Sci., 180: 181-199.
VAN ESCH, G.J. & KROES, R. (1969) The induction of renal tumours by
feeding basic lead acetate to mice and hamsters. Br. J. Cancer, 23:
765-771.
VAN FLEET, J.F., GREENWOOD, L.A., & FERRANO, F.J. (1979) Pathologic
features of adriamycin toxicosis in young pigs: non skeletal lesions.
Am. J. vet. Res., 40: 1537-1552.
VERPOOTEN, G.F., NOUWEN, E.J., HOYLAERTS, M.F., HENDRIX, P.G., & DE
BROE, M. (1989) Segment-specific localization of intestinal-type
alkaline phosphatase in human kidney. Kidney Int., 36: 617-625.
VERSCHOOR, M., WIBOWO, A., HERBER, R., VAN HEMMEN, J., & ZIELHUIS, R.
(1987) Influence of occupational low-level lead exposure on renal
parameters. Am. J. ind. Med., 12: 341-351.
VIAU, C., BERNARD, A., GUERET, F., MALDAGUE, P., GENGOUX, P., &
LAUWERYS, R. (1986a) Isoparaffinic solvent-induced nephrotoxicity in
the rat. Toxicology, 38: 227-240.
VIAU, C., BERNARD, A., & LAUWERYS, R. (1986b) Determination of rat
beta2-microglobulin in urine and in serum. I. Development of an
immunoassay based on latex particles agglutination. J. appl. Toxicol.,
6: 185-189.
VIAU, C., BERNARD, C., LAUWERYS, R., QUAGHEBEUR, L., CORNU, M.E.,
PHILLIPS, S.C., MUTTI, A., LUCERTINI, S., & FRANCHINI, I. (1987) A
cross-sectional survey of kidney function in refinery employees. Am.
J. ind. Med., 11: 177-187.
VICTERY, W., VANER, A.J., SHULAK, J.M., SCHOEPS, P., & JULIUS, S.
(1982) Lead, hypertension and the renin-angiotension system in rats.
J. lab. clin. Med., 99: 354-362.
VIOL, G.W., MINIELLY, J.A., & BISTRICKI, T. (1977) Gold nephropathy.
Tissue analysis by X-ray fluorescent spectroscopy. Arch. Pathol. lab.
Med., 101: 635-640.
VINAY, P., GOUGOUX, A., & LEMIEUX, G. (1981) Isolation of a pure
suspension of rat proximal tubules. Am. J. Physiol., 241: F403-F411.
VOGELZANG, N.J., TORKELSON, J.L., & KENNEDY, B.J. (1985)
Hypo-magnesemia, renal dysfunction and Raynaud's phenomenon in
patients treated with cisplatin, vinblastine and bleomycin. Cancer,
56: 2765-2770.
WALKER, P.D. & SHAH, S.V. (1987) Evidence suggesting a role for
hydroxyl radical in gentamicin-induced acute renal failure in rats. J.
clin. Invest., 81: 334-341.
WALKER, R.G., DAVIES, B.M., HOLWILL, B.J., DOWLING, J.P., &
KINCAID-SMITH, P. (1982) A clinicopathological study of lithium
nephrotoxicity. J. chron. Dis., 35: 685-695.
WALKER, R.G., ESCOTT, M., BIRCHALL, I., DOWLING, J.P., &
KINCAID-SMITH, P. (1986) Chronic progressive renal lesions induced by
lithium. Kidney Int., 29: 875-881.
WALLACE, A.C. & NAIRN, R.C. (1972) Renal tubular antigens in kidney
tumors. Cancer, 29: 977-981.
WAN, S.H. & RIEGELMAN, S. (1972a) Renal contribution to overall
metabolism of drug. I: Conversion of benzoic acid to hippuric acid. J.
pharm. Sci., 61: 1278-1284.
WAN, S.H. & RIEGELMAN, S. (1972b) Renal contribution to overall
metabolism of drugs. II: Biotransformation of salicylic acid to
salicyluric acid. J. pharm. Sci., 61: 1284-1287.
WAN, S.H., VON LEHMANN, B., & RIEGELMAN, S. (1972) Renal contribution
to overall metabolism of drugs. III: Metabolism of p-aminobenzoic
acid. J. pharm. Sci., 61: 1288-1292.
WARD, J.M. & FAUVIE, K.A. (1976) The nephrotoxic effects of
cis-diammine-dichloroplatinum (II) (NSC-119875) in male F344 rats.
Toxicol. appl. Pharmacol., 38: 535-547.
WARD, J.M., GOODMAN, D.G., SQUIRE, R.A., CHU, R.C., & LINHART, M.S.
(1979) Neoplastic and non-neoplastic lesions in aging (C57BL/6N X
CH3/HeN)F1 (B6C3F11) mice. J. Natl Cancer Inst., 63: 849-854.
WEBER, P.C. (1980) Renal prostaglandins, kidney function and effect of
aspirin on metabolism of acetaminophen and benzidine by renal inner
medulla prostaglandin hydroperoxidase. J. lab. clin. Med., 101: 58-65.
WEDEEN, R.P., MALLIK, D.K., & BATUMAN, V. (1979) Detection and
treatment of occupational lead nephropathy. Arch. intern. Med., 139:
53-57.
WEINBERG, J.M. & HUMES, H.D. (1980) Mechanisms of gentamicin-induced
dysfunction of renal cortical mitochondria. I. Effects on
mitochondrial respiration. Arch. Biochem. Biophys., 205: 222-231.
WHO (1979) IPCS Environmental Health Criteria 11: Mycotoxins, Geneva,
World Health Organization, 127 pp.
WHO COLLABORATING CENTRE FOR THE HISTOLOGICAL CLASSIFICATION OF RENAL
DISEASES (1982) Renal disease: Classification and atlas of glomerular
diseases, Tokyo, Igaku-Shoin, 359 pp.
WHO (1983) IPCS Environmental Health Criteria 27: Guidelines on
studies in environmental epidemiology, Geneva, World Health
Organization, pp. 184-185.
WHO (1984) IPCS Environmental Health Criteria 39: Paraquat and Diquat,
Geneva, World Health Organization, 181 pp.
WHO (1990) International classification of diseases for oncology, 2nd
ed., Geneva, World Health Organization, 144 pp.
WHO COLLABORATING CENTRE FOR THE HISTOLOGICAL CLASSIFICATION OF RENAL
DISEASES (1985) Renal disease: Classification and atlas of
tubulo-interstitial diseases, New York, Igaku-Shoin, 221 pp.
WHO COLLABORATING CENTRE FOR THE HISTOLOGICAL CLASSIFICATION OF RENAL
DISEASES (1987) Renal disease: Classification and atlas, New York,
Igaku-Shoin, 291 pp.
WHO COLLABORATING CENTRE FOR THE HISTOLOGICAL CLASSIFICATION OF RENAL
DISEASES (1988) Classification and atlas of infectious and tropical
diseases, Chicago, American Society of Clinical Pathologists, 273 pp.
WIBELL, L., EVRIN, P.E., & BERGGARD, I. (1973) Serum
beta2-microglobulin in renal disease. Nephron, 10: 320-331.
WILLIAMS, P.D., HOLOHAN, P.D., & ROSS, C.R. (1981) Gentamicin
nephro-toxicity. II. Plasma membrane changes. Toxicol. appl.
Pharmacol., 61: 243-251.
WILLIAMS, P.D., HOTTENDORF, G.H., & BENNETT, D.B. (1986) Inhibition of
renal membrane binding and nephrotoxicity of aminoglycosides. J.
Pharmacol. exp. Ther., 237: 919-925.
WILSON, P.D. (1986) Use of cultured renal tubular cells in the study
of cell injury. Miner. Electrolyte Metab., 12: 71-83.
WING, A.J., BRUNNER, F.P., GEERLINGS, W., BROYER, M., BRYNGER, H.,
FASSBINDER, W., RISSONI, G., SELWOOD, N.H., & TUFESON, G. (1989)
Contribution of toxic nephropathies to end-stage renal failure in
Europe: a report form the EDTA-ERA registry. Toxicol. Lett., 46:
281-292.
WIRTHENSOHN, G., BECK, F.X., & GUDER, W.G. (1987) Role and regulation
of glycerophosphorylcholine in rat renal papilla. Pflügers Arch., 409:
411-415.
WIRTHENSOHN, G., LEFRANK, S., SCHMOLKE, M., & GUDER, W.G. (1989)
Regulation of organic osmolyte concentrations in tubules from rat
renal inner medulla. Am. J. Physiol., 256: F128-F135.
WITSCHI, H.P., ALDRIDGE, W.N., AUST, S.D., AUTOR, A.P., DE MATTEIS,
F., DRUET, P., ELBERS, R., FOWLER, B.A., GRONIOWSKI, J.A., HAGMAN, W.,
ORRENIUS, S., PETERING, D.H., SEINEN, W., SUMMER, K.H., & TRUMP, B.F.
(1987) Mechanisms and target cell injury. In: Fowler, B.A., ed.
Mehanisms of cell injury: Implications for human health, New York,
Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 385-403.
WOLD, J.S., TURNIPSEED, S.A., & MILLER, B.L. (1979) The effect of
renal cation transport inhibition on cephaloridine nephrotoxicity.
Toxicol. appl. Pharmacol., 47: 115-122.
WOLGAST, M., PERSSON, E., SCHNERMANN, J., ULFENDAHL, H., & WUNDERLICH,
P. (1973) Colloid osmotic pressure of the subcapsular interstitial
fluid of rat kidneys during hydropenia and volume expansion. Pflügers
Arch., 340: 123-131.
YOUNG, D.M. (1975) Pathologic effects of adriamycin in experimental
systems. Cancer Chemother. Rep., 6: 159-175.
ZENSER, T.V. & DAVIS, B.B. (1984) Enzyme systems involved in the
formation of reactive metabolites in the renal medulla: cooxidation
via prostaglandin H synthase. Fundam. appl. Toxicol., 4: 922-929.
ZENSER, T.V., MATTAMMAL, M.B., & DAVIS, B.B. (1978a) Differential
distribution of the mixed function oxidase in rabbit kidney. J.
Pharmacol. exp. Ther., 207: 719-725.
ZENSER, T.V., MATTAMMAL, M.B., HERMAN, C.A., JOSHI, S., & DAVIS, B.B.
(1978b) Effect of acetaminophen on prostaglandin E2 and
prostaglandin F2 alpha synthesis in the renal inner medulla of rat.
Biochim. Biophys. Acta, 542: 486-495.
ZENSER, T.V., MATTAMMAL, M.B., & DAVIS, B.B. (1979a) Demonstration of
separate pathways for the metabolism of organic compounds in rabbit
kidney. J. Pharmacol. exp. Ther., 208: 418-421.
ZENSER, T.V., MATTAMMAL, M.B., & DAVIS, B.B. (1979b) Cooxidation of
benzidine by renal medullary prostaglandin cyclooxygenase. J.
Pharmacol. exp. Ther., 211: 460-464.
ZENSER, T.V., MATTAMMAL, M.B., BROWN, W.W., & DAVIS, B.B. (1979c)
Cooxygenation by prostaglandin cyclooxygenase from rabbit inner
medulla. Kidney Int., 16: 688-694.
ZENSER, T.V., MATTAMMAL, M.B., RAPP, N.S., & DAVIS, B.B. (1983) Effect
of aspirin on metabolism of acetaminophen and benzidine by renal inner
medulla prostaglandin hydroperoxidase. J. lab. clin. Med., 101: 58-65.
ZIMMERMAN, S.W. & NORBACH, D.H. (1980) Nephrotoxic effects of
long-term carbon tetrachloride administration in rats. Arch. Pathol.
lab. Med., 104: 94-99.
ZUSMAN, R.M. (1980) Prostaglandin E2 biosynthesis by renomedullary
inter-stitial cells: In vitro studies and pathophysiological
correlations. In: Mandal, A.K. & Bohman, S.-O., ed. The renal papilla
and hypertension, New York, London, Plenum Press, pp. 187-207.
RESUME ET CONCLUSIONS
L'excrétion et l'homéostase des molécules hydrosolubles sont
principalement assurées par les reins. Ceuxci sont constitués
d'unités fonctionnelles appelées néphrons, qui consistent
essentiellement en un tube continu formé de cellules hétérogènes
hautement spécialisées qui présentent une organisation structurale,
fonctionnelle et biochimique poussée. Il existe des différences
marquées entre les néphrons eux-mêmes, qui dépendent de la
localisation plus ou moins profonde du glomérule dans le cortex.
C'est l'agencement complexe de cette structure qui, joint à des
différences régionales de vascularisation tenant à la disposition
particulière des vaisseaux, constitue cet organe hétérogène et
hautement organisé qu'est le rein.
Une grande partie des connaissances sur l'anatomie et le
fonctionnement du rein, obtenues grâce à l'expérimentation animale,
est directement transposable à l'homme. Toutefois, les processus
biologiques et métaboliques qui se déroulent dans le rein humain ne
sont pas encore complètement élucidés, non plus que les différences
constatées d'une espèce animale à une autre. Dans ces conditions, on
comprend qu'on ne puisse pas toujours extrapoler d'une espèce à une
autre les effets toxicologiques observés.
Plusieurs substances (qu'elles aient ou non un effet
thérapeutique) exercent des effets toxiques sur les divers éléments
anatomiques du rein. Les effets toxiques peuvent être aigus ou
chroniques; ils peuvent être directs ou se produire par
l'intermédiaire de mécanismes immunologiques. L'impact qu'une
substance néphrotoxique peut avoir sur la santé dépend de plusieurs
facteurs de risque: état fonctionnel du rein, lésion préexistante,
maladie, âge, sexe, régime alimentaire, etc..
On n'a pas suffisamment étudié l'épidémiologie des cas de
néphrotoxicité imputables à des diverses substances chimiques agissant
seules ou en association. A quelques exceptions près, la contribution
des produits chimiques à l'incidence globale des néphropathies et de
l'insuffisance rénale chronique reste mal définie. D'importantes
recherches ont été consacrées à certains groupes exposés de par leur
profession ainsi qu'aux néphropathies résultant de la consommation
d'analgésiques : elles ont montré l'existence de variations entre les
différents groupes et les différents pays. Toutefois on estime que
jusqu'à 18% des néphropathies terminales pourraient être dues à la
prise d'analgésiques et jusqu'à 5% avoir une origine toxique
quelconque. Environ 50% des néphropathies terminales sont d'origine
inconnue. L'un des principaux problèmes qui se posent dans la
recherche de l'étiologie des néphropathies terminales tient à la
longue période de latence ou à l'évolution lente de l'insuffisance
rénale chronique, qui rendent difficile l'identification rétrospective
de l'agent causal. Ce n'est que dans quelques cas que le dosage de
certaines substances dans les tissus (organisme, rein) peut conduire
au diagnostic. Par ailleurs on se heurte également à un certain
manque de cohérence dans les critères et la terminologie utilisés sur
le plan diagnostique et anatomopathologique.
Les différences anatomiques et physiologiques rendent difficile
une extrapolation directe à l'homme des résultats obtenus sur des
systèmes expérimentaux ( in vivo ou in vitro). On ne connaît que
très peu de cas où les données de néphrotoxicité sont comparables chez
l'homme et chez l'animal et permettent d'évaluer de façon fiable le
risque pour la santé humaine.
Certaines substances chimiques peuvent provoquer des lésions
sélectives au niveau des structures rénales vulnérables ou déclencher
des mécanismes immunologiques. On considère qu'il existe en général
deux types de mécanismes à la base des lésions rénales : a) les
mécanismes immunologiques qui conduisent à la néphrite aiguë
interstitielle et b) les mécanismes qui affectent principalement le
glomérule par l'intermédiaire d'anticorps anti-GBM ou
d'immunocomplexes. Il existe un autre grand groupe de maladies dues
à des substances chimiques ou à leurs métabolites qui perturbent
l'activité biochimique cellulaire ou modifient les propriétés
hémodynamiques, etc. Parmi les facteurs susceptibles d'influer sur
les lésions cellulaires provoquées par les produits toxiques figurent
les systèmes de transport cellulaire, la pinocytose, la dégradation
métabolique, l'interaction avec les protéines, les lipides, les
membranes et l'ADN cellulaires et éventuellement avec d'autres
constituants de la cellule.
Plus on utilise de produits pharmaceutiques et de substances
chimiques, plus on accroît les risques de néphrotoxicité. La
néphrotoxicité des médicaments dépend de la dose et de la durée du
traitement (par exemple dans le cas des associations d'analgésiques
qui conduisent à une nécrose papillaire). On a beaucoup étudié les
effets néphrotoxiques des analgésiques, des antibiotiques (comme les
aminoglycosides), des agents anticancéreux (comme le cis-platine) et
de divers autres agents. Nombre de substances fréquemment utilisées
dans l'industrie ou à la maison comme les hydrocarbures chlorés ou
l'éthylèneglycol peuvent également avoir une action toxique sur le
rein. D'autres substances, présentes dans l'environnement comme le
plomb ou le cadmium sont effectivement néphrotoxiques. L'action
toxique de ces substances intervient après accumulation
intracellulaire du produit initial ou métabolisation intra- ou
extrarénale. L'exposition à plusieurs substances chimiques peut
entraîner des réactions antagonistes ou synergistiques.
Les tumeurs du rein et des voies urinaires représentent moins de
2 à 3 % de l'ensemble des cancers humains, mais leur fréquence est en
augmentation, ce qui incite à penser que des facteurs environnementaux
pourraient être à l'oeuvre. Nombre de produits pharmaceutiques et de
substances chimiques se sont révélés cancérogènes sur des modèles
expérimentaux mais seuls quelques-uns d'entre eux sont responsables de
l'apparition de cancers chez l'homme. Il s'agit de l'amiante
(adénocarcinome rénal), de l'abus d'analgésiques (carcinomes de type
transitionnel) et de facteurs tenant au mode de vie (tabagisme,
consommation d'alcool et de café). Il existe des groupes de
population chez lesquels certains cancers des voies urinaires ont une
étiologie encore inconnue. Certaines substances chimiques utilisées
dans le cadre professionnel sont à l'origine de cancers de la vessie.
De nombreuses substances chimiques ou pharmaceutiques peuvent
entraîner une néphrite interstitielle, affection qui serait
susceptible de favoriser l'apparition de cancers des voies urinaires.
Il n'existe pas de méthode qui permette d'évaluer in vivo ou in
vitro dans des conditions satisfaisantes une insuffisance rénale
provoquée par des substances chimiques. Aussi faut-il non seulement
contrôler l'activité néphrotoxique des produits chimiques, mais
également inclure dans les protocoles expérimentaux l'étude du
mécanisme toxique au niveau des cellules cibles. Pour y parvenir il
faut faire appel tant à l'expérimentation in vivo qu'à des études in
vitro. Les études in vitro doivent porter sur des éléments où les
relations anatomiques et cellulaires sont maintenues (par
microperfusion de reins et de tubules isolés, coupes de cortex,
fragments isolés de néphrons, etc.) et sur des cellules rénales (par
exemple suspensions cellulaires, cultures cellulaires primaires,
lignées cellulaires et fractions infracellulaires). Pour les études
in vivo, on peut faire appel à des techniques effractives ou non
effractives. Parmi les techniques effractives figurent les examens
histopathologiques ainsi que les techniques classiques de bilan de la
fonction rénale. Les techniques non effractives permettent un bilan
répété de la fonction rénale chez l'animal par la mesure d'une série
de paramètres fonctionnels (filtration glomérulaire, fonction
tubulaire, protéinurie, enzymurie, etc.). Des épreuves biochimiques
spécialisées devront être effectuées le cas échéant. En associant de
façon convenable les épreuves in vivo et les épreuves in vitro on
pourra déterminer si telle ou telle substance chimique est
néphrotoxique et avoir une idée de son activité, de son site d'action
et du mécanisme de son action toxique.
Les méthodes habituellement utilisées pour l'exploration
fonctionnelle du rein chez l'homme ne permettent pas un diagnostic
suffisamment rapide d'une insuffisance rénale due à des substances
chimiques ni d'en apprécier le retentissement sur la santé. L'absence
de marqueurs spécifiques précoces de la néphrotoxicité est
particulièrement gênante. Les méthodes non effractives devront faire
appel à des marqueurs très spécifiques et sensibles, qui permettent de
déceler rapidement les modifications qui se produisent au niveau rénal
afin de prévoir l'apparition éventuelle d'une insuffisance
fonctionnelle. Les techniques actuelles de contrôle de la fonction
glomérulaire ou tubulaire ne sont utilisables que lorsque les lésions
sont déjà très importantes. On a proposé comme marqueurs
l'utilisation de constituants tissulaires immunoréactifs mais encore
faut-il en confirmer la valeur au moyen d'études longitudinales bien
conçues.
RECOMMANDATIONS
1. Il faut faire un effort soutenu pour développer et valider des
marqueurs plus sélectifs et plus spécifiques et notamment des
anticorps monoclonaux en vue d'étudier l'insuffisance rénale chez
l'animal. Ces marqueurs pourraient être utilisables chez l'homme.
2. Il faudrait compléter la base de données utilisables pour
déterminer le potentiel néphrotoxique des substances chimiques pour
l'homme. A cet effet, on développera et on validera diverses méthodes
d'expérimentation animale ( in vivo et in vitro), on mettra au
point des méthodes nouvelles d'étude de la néphrotoxicité, on étudiera
les différences interspécifiques et on prendra en compte l'expérience
tirée des essais précliniques de nouveaux médicaments chez l'homme.
3. Il faudrait renforcer les études épidémiologiques (c'est-à-dire
les études prospectives sur des groupes professionnels ou des groupes
de la population générale exposés à des substances néphrotoxiques ou
qui font une consommation excessive d'analgésiques).
4. Des efforts plus importants devront être consentis afin d'établir
le rôle de certaines substances chimiques dans l'étiologie des
maladies rénales au stade le plus précoce possible (antécédents
professionnels, recherches de néphrotoxines dans les tissus).
5. L'élucidation du mode d'action des substances néphrotoxiques peut
être utile à la prévention et au traitement des effets rénaux
indésirables et pourrait contribuer à la prévision du pouvoir
néphrotoxique des nouveaux médicaments et des nouveaux produits.
Parmi les secteurs de recherche particulièrement importants on peut
citer :
* les mécanismes immunologiques
* les effets directs des substances chimiques sur les membranes et
notamment les mécanismes de peroxydation des lipides, les
interactions membranes/substances chimiques, les déplacements
d'ions, les événements au niveau des récepteurs
* l'activation des proto-oncogènes et la différenciation cellulaire
* la régulation du métabolisme cellulaire.
6. Il faut également préciser la localisation anatomique de
certaines fonctions rénales.
7. Il faudrait étudier de façon plus approfondie le rôle des
variations et de la prédisposition génétiques aux effets toxiques des
médicaments et des produits chimiques.
8. La relation entre néphrotoxicité et cancer (mycotoxines et
néphropathie des Balkans par exemple) doit être étudiée plus à fond.
RESUMEN Y CONCLUSIONES
Los riñones son los principales órganos de excreción y
homeostasis de las moléculas hidrosolubles. La unidad funcional del
riñón es el nefrón, que consiste esencialmente en un tubo continuo de
células heterogéneas sumamente especializadas, y que exhibe una
notable organización estructural, funcional y bioquímica. Existen
diferencias pronunciadas entre unos nefrones y otros, atendiendo a la
localización de los glomérulos individuales correspondientes en la
corteza. Esta compleja organización estructural, combinada con las
diferencias en la vascularidad regional debida a lo especializado de
la disposición vascular, da lugar a un órgano sumamente complejo y
heterogéneo.
Gran parte de los conocimientos anatómicos y funcionales
obtenidos en los animales pueden aplicarse directamente al riñón
humano. No obstante, los procesos biomédicos y metabólicos del riñón
humano, así como las diferencias entre las especies animales, no se
han elucidado de forma tan detallada. Así, los efectos de las
sustancias químicas sólo se pueden extrapolar hasta cierto punto de
unas especies a otras.
Varias sustancias químicas (tanto terapéuticas como no
terapéuticas) tienen efectos tóxicos en uno o más elementos anatómicos
del riñón. Los efectos tóxicos pueden ser agudos o crónicos, y pueden
ser directos o estar indirectamente mediados por mecanismos
inmunológicos. El impacto que tienen en la salud las sustancias
químicas nefrotóxicas está relacionado con los factores de riesgo,
entre los que figuran el estado de la reserva funcional del riñón y
factores como las lesiones renales ya existentes, las enfermedades, la
edad, el sexo y la dieta.
No se ha estudiado bastante la epidemiología de la nefrotoxicidad
inducida con sustancias químicas aisladas o en exposiciones mixtas.
La contribución de las sustancias químicas a la incidencia global de
la nefropatía y del fallo renal crónico está, salvo raras excepciones,
sin definir. En el caso de algunos grupos expuestos por su profesión
y de enfermedad renal asociada a los analgésicos, se han hecho amplios
estudios que han demostrado la existencia de la incidencia entre
grupos y países. Sin embargo, se estima que hasta el 18% de las
enfermedades renales en fase terminal pueden deberse a nefropatía por
analgésicos y hasta el 5% a otras nefropatías tóxicas. Alrededor del
50% de las enfermedades renales en fase terminal son de etiología
desconocida. Uno de los principales problemas para atribuir una causa
a la enfermedad renal en fase terminal es la larga latencia y/o la
lenta evolución del fallo renal crónico, lo que dificulta la
identificación retrospectiva del agente causal. Sólo en algunos
casos, la medida de los niveles tisulares (organismo, riñón) de
sustancias químicas es útil para el diagnóstico. Otro problema ha
sido la falta de coherencia en la terminología y los criterios
patológicos y de diagnóstico.
Las diferencias anatómicas y fisiológicas del riñón dificultan la
extrapolación directa al ser humano a partir de los sistemas
experimentales ( in vivo e in vitro). Existen muy pocos ejemplos
de sustancias químicas nefrotóxicas para las que se disponga de datos
adecuados y comparables entre los animales y el hombre como para
formar una base sólida sobre la que evaluar el riesgo potencial para
la salud humana.
Las sustancias químicas pueden dañar de modo selectivo las
estructuras vulnerables del riñón o activar mecanismos inmunológicos.
Los mecanismos de lesión renal pueden dividirse generalmente en dos
categorías: a) nefritis intersticiales agudas inmunológicamente
inducidas; b) aquellos que afectan primordialmente al glomérulo por
conducto de anticuerpos mediados por la anti-GBM o por conducto de
complejos inmunes. Otro grupo principal está compuesto por las
enfermedades desencadenadas por sustancias químicas o sus metabolitos
que interfieren con los efectos bioquímicos y hemodinámicos en la
célula, entre otros. Entre los factores que pueden modificar la
lesión celular producida por sustancias tóxicas figuran los sistemas
de transporte celular, la pinocitosis, la degradación metabólica y la
interacción con las proteínas, los lípidos, las membranas y el ADN
celulares, y tal vez otros constituyentes de la célula.
El uso cada vez más extendido de agentes y sustancias químicas
terapéuticas aumenta las posibilidades de aparición de nefrotoxicidad.
La nefrotoxicidad inducida por agentes terapéuticos depende de la
dosis y del tiempo de exposición (por ejemplo, analgésicos de
combinación que producen necrosis papilar del riñón). Los efectos
nefrotóxicos de los analgésicos, los antibióticos (como los
aminoglucósidos), los agentes anticancerosos (como el cis-platino), y
varios agentes más han sido objeto de amplios estudios. Las
sustancias químicas de uso frecuente en la industria o el hogar, como
los hidrocarburos clorurados y el etilenglicol, también tienen
potencial para producir lesiones renales. Las sustancias químicas del
medio ambiente como el plomo y el cadmio son capaces de inducir
nefrotoxicidad. Estos agentes tienen efectos tóxicos tras la
acumulación intracelular del compuesto original o tras la
biotransformación renal o extrarrenal. La exposición multiquímica
puede dar lugar a respuestas antagonistas o sinérgicas.
Aunque los tumores del riñón y del tracto urinario representan
menos del 2-3% de todos los cánceres humanos, la frecuencia de esos
tumores está aumentando, lo que indica que los factores ambientales
ejercen cierta influencia. Si bien se ha demostrado que muchos
fármacos y sustancias químicas son carcinogénicos en modelos
experimentales, sólo algunas sustancias concretas se han relacionado
con tumores en el hombre. Entre ellos figuran el amianto
(adenocarcinoma renal), el uso indebido de analgésicos (carcinomas de
transición), y los factores relacionados con el modo de vida (uso de
tabaco, alcohol, café). Existen grupos de población con cáncer del
tracto urinario de etiología aún por determinar. Las sustancias
químicas presentes en el medio profesional se han relacionado con la
etiología del cáncer de la vejiga urinaria. Muchos fármacos y
sustancias químicas provocan nefritis intersticial, que en sí misma
puede ser un factor desencadenante de cánceres en el tracto urinario.
No hay ningún método in vivo o in vitro que por sí solo baste
para evaluar la disfunción renal químicamente inducida. Así pues, es
aconsejable no sólo estudiar compuestos en busca de su potencial
nefrotóxico, sino incorporar a los protocolos experimentales estudios
mecanicistas sobre la toxicidad para células diana. Para conseguirlo,
deben realizarse investigaciones concertadas in vivo e in vitro.
Los estudios in vitro son aquellos en los que se mantienen las
relaciones anatómicas entre células (por ejemplo, riñón y túbulos
aislados y perfundidos, secciones de la corteza renal y segmentos
aislados de nefrones) y aquellos en los que se utilizan células
renales (por ejemplo, suspensión de células, cultivos de células
primarias, líneas celulares establecidas y fracciones subcelulares).
En los estudios in vivo se utilizan técnicas tanto invasivas como no
invasivas. Los procedimientos invasivos comprenden las mediciones
histopatológicas y ordinarias de la función renal. Los procedimientos
no invasivos permiten evaluar de modo repetido la función renal en los
animales midiendo una serie de pruebas de la función renal (filtración
glomerular, función tubular, proteinuria, enzimuria, etc). Cuando
convenga, deben realizarse ensayos bioquímicos especializados. La
combinación adecuada de experimentos in vivo e in vitro revelará
si las sustancias químicas son nefrotóxicas o no y dará idea de la
potencia, la localización y los mecanismos de la toxicidad.
Los métodos tradicionales de evaluación de la función renal en el
hombre no bastan para diagnosticar de modo oportuno la disfunción
renal inducida por sustancias químicas y la predicción de su
importancia para la salud. La falta de marcadores específicos y
precoces en la nefrotoxicidad resulta particularmente problemática.
La evaluación no invasiva de la nefrotoxicidad debe hacer uso de
marcadores de gran especificidad, sensibilidad para la detección de
las alteraciones renales precoces y valor predictivo para la evolución
de la insuficiencia renal. Las técnicas actuales de seguimiento de la
función glomerular o tubular sólo son útiles en los casos en que se ha
producido una lesión renal grave. Aunque actualmente se señalan los
constituyentes tisulares inmunorreactivos como marcadores apropiados,
es preciso validar su idoneidad en estudios longitudinales bien
diseñados.
RECOMENDACIONES
1. Debe hacerse un esfuerzo continuado por desarrollar y validar
marcadores más selectivos y específicos, inclusive anticuerpos
monoclonales, para evaluar la disfunción renal en animales. Es
posible que esos marcadores sean aplicables al hombre.
2. Debe ampliarse la base de datos para predecir el potencial
nefrotóxico de las sustancias químicas para el hombre. Ello comprende
el desarrollo y la validación de criterios experimentales en animales
( in vivo e in vitro), métodos alternativos para estudiar la
nefrotoxicidad, información sobre diferencias interespecíficas, y
experiencia de la evaluación preclínica de nuevos agentes terapéuticos
en el hombre.
3. Deben reforzarse los estudios epidemiológicos (es decir, estudios
prospectivos en grupos profesionales y de la población general
expuestos a sustancias químicas nefrotóxicas o que hagan uso indebido
de analgésicos).
4. Deben intensificarse los esfuerzos por establecer el papel de las
sustancias químicas en la etiología de las enfermedades renales en la
etapa más temprana de diagnóstico (por ejemplo, pasado laboral,
vigilancia de los tejidos en busca de nefrotoxinas).
5. La comprensión de los mecanismos de acción de los nefrotóxicos
ayudará a prevenir y gestionar desde el punto de vista clínico los
efectos renales no deseados, y puede ayudar a predecir el potencial
nefrotóxico de nuevos fármacos y sustancias químicas. Los aspectos de
particular importancia para las investigaciones futuras son:
* los mecanismos inmunológicos;
* los efectos directos de las sustancias químicas en las membranas,
inclusive los mecanismos de peroxidación lipídica, la interacción
membrana/sustancias químicas, los cambios de iones, y los
procesos mediados por receptores;
* la activación de los proto-oncogenes y de la diferen ciación
celular;
* la regulación del metabolismo celular.
6. Es necesario determinar y correlacionar las funciones específicas
con localizaciones anatómicas discretas dentro del riñón.
7. Debe estudiarse más a fondo el papel de la variación genética y
la susceptibilidad a los efectos tóxicos de fármacos y sustancias
químicas.
8. Debe estudiarse con más detalle la relación entre la
nefrotoxicidad y la carcinogénesis renal (por ejemplo micotoxinas y
nefropatía de los Balcanes).
On the basis of the electrophoretic pattern, proteinuria may
reveal glomerular damage, tubular dysfunction or mixed patterns. The
glomerular damage may be selective (mostly due to a loss of glomerular
polyanion) or unselective (involving more extensive damage, and
glomerular hyperfiltration and hypertension). However, it should be
recognized that some features unique to experimental animals may
account for large variations, which could lead to wrong conclusions.
For instance, marked sex-, age-, and diet-related changes may occur,
especially in the male rat (Neuhaus et al., 1981). Even if most of
these changes have a counterpart in man, they are amplified by the
lack of variations in housing conditions. As a result, the young male
rat may physiologically show "tubular" proteinuria, whereas the aging
rat displays "glomerular" patterns, owing to a spontaneous
nephropathy, which can be prevented in part by reducing dietary
protein content. Depending on other factors, especially diet and
concomitant treatments, such electrophoretic patterns may be accounted
for by underlying mechanisms and related morphological changes.
Owing to their limited affinity for most dyes, proteins of low
relative molecular mass are better identified by the quantitative
measurement of single components such as ß2-microglobulin (Viau et
al., 1986b). Because of its peculiar metabolism, the urinary excretion
of alpha2u-globulin, a sex-related protein of low relative molecular
mass, cannot be recommended as a marker of tubular damage.
Furthermore, its renal handling and disposition may interfere with
those of other proteins, accounting for some of the spontaneous
changes occurring in the pattern of proteinuria from the male rat.
Thus, when evaluating proteinuria, preference should be given to
female rats, since in this case extrapolation to man seems to be less
affected by species-related problems.
7.2.4.2 Urinary excretion of single plasma proteins
Very sensitive immunochemical methods are available for measuring
the urinary excretion of single plasma proteins, such as IgG, albumin,
ß2-microglobulin and alpha2u-globulin in the rat (Bernard et al.,
1988). In addition to better analytical features, in terms of
sensitivity, specificity, accuracy, and reproducibility, the
quantification of single urinary proteins has two other inherent
advantages. Firstly, single proteins may be significantly increased
without giving rise to pathological values in total proteinuria
(Barratt, 1983). Secondly, the power of experimental studies is
greatly increased by quantitative data making it possible to use
parametric tests.
7.2.4.3 Enzymuria
Several different enzymes have been studied (Table 8), but none
satisfies all the criteria for nephrotoxicity (Dubach et al., 1989).
Enzymes are not uniformly distributed along or between nephrons.
Although it should be possible to localize the area of kidney damage
on the basis of the pattern of enzymuria, the site-selectivity of
single enzymes is questionable. The failure to recognize selective
damage by measuring enzyme activity may be accounted for by two
factors. Firstly, it is possible that chemically induced early renal
changes are less selective than advanced lesions preceding end-stage
renal disease. Secondly, the poor analytical features of most enzyme
measurements in urine may give rise to aspecific patterns. This
important question may be addressed by measuring immunoreactive
antigens, including enzymes, since the use of reliable immunochemical
techniques would limit the effects of analytical problems.
Most enzymes are stable over a narrow range of pH, and their
activity may be affected by the presence of inhibitors such as urea
(Price, 1982). It should be stressed that all rat enzyme studies must
be carried out under carefully controlled conditions, after an
adequate period of acclimatization, and after changing to day instead
of night feeding. Urine must be minimally contaminated with
microorganisms. This is achieved by surrounding the urine collecting
vessel with ice, so that bacteria cannot multiply as readily as in
normal metabolic cages (Berlyne, 1984).
7.2.4.4 Immunoreactive tissue constituents
Tissue constituents may be released into urine due to increased
cellular turnover or cell death and may be detected by immunochemical
methods. Monoclonal antibodies have been produced against both rat
(Tokoff-Rubin, 1986) and human brush-border antigens (Mutti et al.,
1985; Mutti, 1989). In both cases, cross-reactivity between species
has been shown (Mutti, 1987, 1989). The specificity of such an
approach relies on the site-selectivity of target proteins and on the
advantages of monoclonal antibodies, including monospecificity and
reproducibility of reagents. Its sensitivity has been proved by
Table 8. Some enzymes used as an index of nephrotoxicity
Enzymes Cellular location
Alanine aminopeptidase brush border
Alkaline phosphatase
gamma-Glutamyltransferase
Maltase
Trehalase
Glutamic oxaloacetic transaminase cytosol
Glutamic pyruvic transaminase
Lactate dehydrogenase
Malate dehydrogenase
N-Acetyl-ß-D-glucosaminidase lysosome
Acid phosphatase
ß-galactosidase
ß-glucosidase
ß-glucuronidase
Glutamate dehydrogenase mitochondria
comparison with other markers in various situations, but it needs
further validation in carefully designed chronic studies, since the
prognostic value of slight changes in such a sensitive test is
currently unknown.
In general terms, it has been through the use of
histopathological studies on kidney tissue that advanced renal lesions
have been identified. This approach has many inherent advantages, and
represents the method of choice, particularly if it can be used for
the early diagnosis of renal lesions or dysfunction in experimental
animals being used for nephrotoxicity screening studies. For clinical
investigations in human and population studies, it is obviously the
least desirable of the techniques that are available.
7.2.4.5 Urinary excretion of prostaglandins
The urinary excretion of PGs (mainly PGE2) may reflect the rate
of renal synthesis. This is modified in several nephropathies and may
be affected by a number of nephrotoxic chemicals.
7.2.5 Clinical context
At present, the clinical diagnosis of toxic nephropathy still
relies heavily on the case history of patients showing symptoms and/or
laboratory abnormalities suggesting chronic renal failure without any
obvious recent cause.
In such circumstances, history is the cornerstone of diagnosis.
It can only be made by excluding other known conditions or risk
factors that lead to renal insufficiency and assessing exposure to
nephrotoxins, which has to be consistent with known
dose-effect/response relationships and with the temporal sequence of
events leading to the observed effect. For immune reactions, the role
of individual susceptibility should also be considered. In some
circumstances, kidney toxicity may be considered as a factor
contributing to the clinical outcome in a multifactorial process. In
this case, it is difficult to distinguish determinants from
predisposing and/or aggravating factors.
There are two well-recognized clinical syndromes that result from
immunologically mediated glomerular disease. These are the nephrotic
syndrome and the nephritic syndrome. The nephrotic syndrome is
characterized by massive proteinuria, hypoalbuminaemia (from
proteinuria) and generalized oedema. The syndrome is the result of
pathology that affects the integrity of the GBM. Although the cause of
85% of nephrotic syndrome is not known, it is the syndrome most
frequently occurring with toxic nephropathies, it is generally dose
related, and it is reversible. Glomerular pathology is most often
membranous glomerulonephritis or circulating immune-complex disease.
The nephritic syndrome is characterized by haematuria and a decreasing
GFR and is likely to be accompanied by some degree of hypertension.
The pathological changes in the glomeruli are usually caused by those
diseases or toxic agents that produce an inflammatory proliferative
response within the glomeruli. The proliferation may involve
endothelial mesangial or epithelial cells and may be associated with
an inflammatory cell infiltration.
7.2.6 Radiological techniques
Radiocontrast media can be used to study the kidney either by
conventional or by retrograde pyelography. There are a number of
limitations to this technique, such as the need for adequate renal
function by which to image the organ. There is adequate evidence that
radiocontrast agents have a nephrotoxic potential. Media of high
osmolality are especially likely to precipitate renal failure. In
addition, those patients who are dehydrated or with reduced blood
volume are at special risk. There is also evidence to show that
patients who have multiple myeloma represent a special risk group.
7.2.7 Other non-invasive renal assessment
Whereas gamma camera renography and ultrasound are
well-established techniques for the assessment of renal function,
increasing use is being made of ultrasound linked with Doppler flow,
nuclear magnetic resonance imaging, and spectoscopy; ureteral
fibroscopy may be used to view the pelvis directly. Ultrasound
evaluation of the kidneys provides a means of excluding obstruction as
a cause of anuria or oliguria in ARF. It may also reveal papillary
necrosis or perirenal haematomas and obviate the intrinsic dangers of
pyelography. Radionuclide scans can be used to identify major
atherothrombotic events and cortical necrosis. They can also be used
to show reduced renal blood flow, which is usually preserved (some 50%
of normal) in acute tubular necrosis, but is severely compromised in
acute glomerulonephritis and vasculitis. Renal biopsy should not be
used for ARF unless the duration exceeds three weeks and there is no
obvious cause. It can then help diagnose glomerulonephritis,
vasculitis, and AIN.
8. DETECTION OF NEPHROTOXICITY IN HUMANS
Traditional methods of diagnosing renal damage and predicting
their health significance are not sufficient to deal with the
diversity of chemically induced lesions (Bach et al., 1989). This is
because the kidney can undergo substantial chemically induced injury
without any clinical indication, owing to its considerable functional
reserve. This masks a substantial amount of renal degeneration
(Friedlander et al., 1989). Thus, for example, the single common
measurement of GFR may only show incipient acute or chronic renal
failure.
Most nephropathies in humans are of unknown origin. There is,
however, some indirect evidence that toxicant exposure could be
involved. For example, 80% of the cases of membranous nephropathy are
of unknown origin. Among the remaining 20%, half of them have been
found to be associated with drug exposure. It is likely, therefore,
that a percentage of those cases of unknown origin are also related to
toxic exposure.
8.1 Markers of nephrotoxicity
Over the last ten years, new biochemical and immunochemical
methods have been developed for detecting early renal changes in
humans. Their application has given rise to a number of markers, some
of which also are available for studies on experimental animals and
have been reviewed in section 7.2. Although some tests may open new
perspectives in terms of selectivity, sensitivity, and specificity,
most of the recently developed markers need further validation. Hence,
some conceptual and methodological problems must be addressed and new
methods critically evaluated. This is especially important when
implementing screening programmes for the early detection of kidney
damage and/or dysfunction in humans.
8.1.1 General requirements
Markers to be used for screening purposes in population groups
should fulfill certain general criteria, including specificity,
non-invasiveness, sensitivity in detecting early renal functional
changes, and predictive value for development of renal insufficiency.
Analytical methods must be reproducible, easy to perform, and
applicable to a large number of samples, and the samples must be
stored appropriately to ensure stability.
The prevalence of true positive results among subjects who are
actually ill indicates the sensitivity of the marker, whereas the
prevalence of true negative findings among healthy individuals is a
measure of its specificity.
Some markers meet the requirement of specificity, although it may
be difficult to establish clear-cut distinctions in individual cases.
For instance, whereas gross changes in proteinuria involving proteins
of low and high relative molecular mass indicate tubular and
glomerular damage, respectively, marginal increases over reference
limits may be due to either condition (Mutti, 1989).
The predictive value of markers of nephrotoxicity has not been
systematically tested, but it is usually assumed that such markers as
the urinary excretion of single plasma proteins are highly
sensitive but somewhat aspecific. This is due to the fact that they
may be increased by a number of physiological conditions (e.g.,
physical workload, posture, pharmacological effects of exogenous
substances, or even meat meals). On the contrary, other markers such
as serum creatinine are considered to be relatively specific, but
relatively insensitive, since they reflect late changes, occurring
when more than 50% of the renal reserve has been lost. Within these
extremes, there are various possibilities which should be carefully
weighed according to the methodological context dealt with.
The predictive value of markers, in terms of sensitivity and
specificity, should be taken into account when evaluating individual
results. For some parameters (e.g., proteinuria involving proteins of
low relative molecular mass) minor dysfunctions at the tubular level
will result in deviations of several orders of magnitude from
reference values, whereas the same relative change in other parameters
(e.g., brush-border antigens, enzyme activities) may indicate severe
tubular injury.
In the clinical context, it is often difficult to establish
correct etiological diagnoses because of the long latency between
exposure and the development of overt disease. For the same reason, it
is sometimes difficult in epidemiological work to assess the
prognostic value of early changes. Nonetheless, it is clear that
markers used in the clinical context must be specific enough to avoid
further undue and sometimes invasive procedures, which tend to
accumulate once a subject enters a diagnostic decision tree (Mold &
Stein, 1986).
The quantification of any constituents in urine may only be
obtained by assessing urine flow rate. The accuracy of timed urine
samples (especially when obtained during epidemiological surveys) is
unreliable and most analytes may be unstable. There are still a number
of factors that can confound or modify the renal response, such as
time of sampling (owing to spontaneous rhythms), posture, physical
work load, and diet. Thus, there is a need to use standardized
procedures to limit possible interferences that can increase the
variability between and within subjects. To overcome these
methodological problems, there is an increasing tendency to use spot
samples, generally the second sample of the day, and to express
results as a function of creatinine.
8.1.2 Diagnostic value
The predictive value of a marker only in part contributes to its
diagnostic validity, which is defined in terms of the probability that
the classification based on test values actually corresponds to the
subject's condition. The diagnostic value may be both positive and
negative, the positive diagnostic value being the probability that a
subject classified as positive by the test is actually ill, and the
negative diagnostic value being the probability that subjects negative
at the test are actually healthy individuals. It must be stressed that
the diagnostic validity is only marginally affected by the predictive
value of the markers, since in most screening programmes a low
prevalence of disease is the major determinant of pitfalls.
Thus, markers must be selected according to the condition under
evaluation. When studying groups at risk, emphasis should be given to
sensitivity, whereas specificity should receive adequate attention in
the clinical setting.
8.1.3 Prognostic value
Another important property of markers, which has so far only been
addressed in a few studies, is their prognostic value, i.e. their
ability to predict the "natural" course of the disease. In most cases
early functional changes may be compensated and structural damage may
be repaired. However, both conditions may also trigger a cascade of
events leading to end-stage renal disease. The ability to target
groups actually at risk and to predict the "natural" evolution of the
disease is part of effective prevention. This can be achieved by
reducing exposure (primary prevention) or by establishing an
individual diagnosis at reversible stages (secondary prevention).
Microscopic examination of urine sediment, although impractical
in population studies, may help in establishing individual
differential diagnosis. Even if it is insensitive for detecting
nephrotoxicity, this test is suitable at the individual level to
exclude confounding factors accounting for increased excretion of
plasma proteins (e.g., leukocyturia, haematuria, or bacteriuria).
8.2 Screening for nephrotoxicity in humans
In the epidemiological context, it can be assumed that negative
results are recorded in actually healthy subjects, whereas the
probability of false positive values is rather high because of the low
prevalence of the disease. Thus, health surveillance programmes should
be mainly aimed at excluding harmful situations rather than at
identifying ill people. This goal may be achieved using very sensitive
markers of subclinical renal impairment. A clinical diagnosis in
individuals showing abnormal test values should then be established
only after repeated measurements and complementary confirmatory tests.
Practical considerations are also important when planning population
studies, where sampling procedures that are invasive or too elaborate
cannot be included in the protocol.
8.2.1 Glomerular filtration
Clearance procedures can seldom be adopted when screening
population groups, since only single blood and spot urine samples are
usually available. However, it must be stressed that some tests
fulfilling the above objective have proved their diagnostic and
prognostic validity and for this reason they are now also being
employed in clinical practice.
An indirect method of assessing GFR by using a single blood
sample is the measurement of serum creatinine and the expression of
its reciprocal value adjusted to constant body surface area. This has
been correlated with GFR (Siersbaek-Nielson et al., 1971). The same
concept applied to ß2-microglobulin would increase the sensitivity of
such an approach, owing to the higher relative molecular mass and the
use of more accurate and reproducible analytical methods (Wibell et
al., 1973).
8.2.2 Tests designed to assess selective dysfunction
Serious renal diseases (e.g., nephrotic or Fanconi's syndromes)
may occur in the absence of haemodynamic changes. In such
circumstances, renal insufficiency is a late event that is preceded by
earlier, though serious, selective dysfunctions occurring at the
glomerular and proximal tubular level. These may be revealed by
measuring single plasma proteins in urine (see also section 8.3.3.).
The excretion in urine of proteins of high relative molecular mass
generally reflects glomerular dysfunction, whereas urinary excretion
of proteins of low relative molecular mass may be indicative of
tubular dysfunction. The assessment of protein electrophoretic
patterns has been discussed in section 7.2.4.
8.2.3 Tests designed to assess tissue damage
Tubular dysfunction is not necessarily associated with
histopathological changes. It has been shown that pharmacological
inhibition of tubular uptake may account for proteinuria (involving
proteins of low relative molecular mass) observed after protein load
(Buzio et al., 1989). Furthermore, highly selective damage to a
tubular segment may be functionally compensated by other segments of
the nephron. Methods aimed at detecting cellular damage may thus help
both to show subclinical lesions and to interpret associated
dysfunctions. The urinary excretion of tissue constituents may be
measured through the enzymic or immunochemical characteristics of
material shed into the urine.
Renal functional changes may be reversible, depending on the
efficiency of repair mechanisms and the cessation of exposure to the
offending agent. Repeated monitoring may help to distinguish
progressive renal disease from transient lesions (Thornley et al.,
1985).
8.2.3.1 Enzymuria
The general principles describe in section 7.2.4.3. also apply to
humans. Even when using freshly voided spot samples, urine is a
hostile environment for most enzymes. Only a few enzymes (e.g.,
N-acetly-ß-D-glycosaminidase) show acceptable stability and
analytical precision (Price, 1982). Table 8 lists some human urinary
enzymes that have been used in nephrotoxic studies.
8.2.3.2 Immunoreactive tissue constituents
Tissue constituents (including enzymes) are physiologically shed
into the urine as a consequence of cell turnover and metabolism. When
they are detected by immunochemical methods (e.g., immunofluorescence,
enzyme-linked immunosorbent assay), they are referred to as antigens.
Tamm-Horsfall glycoprotein is a specific renal protein, localized on
the membrane of cells of the thick ascending limb of the loop of
Henle, which is excreted in the urine at a relatively constant rate.
This can increase following injury to the distal part of the tubule
and is depressed when the renal mass is reduced (Thornley et al.,
1985). Although increased excretion of proximal tubular antigens was
reported twenty years ago in various clinical situations such as
tubular necrosis, allograft rejection, and
cephalotin/gentamicin-induced nephrotoxicity (Antoine et al., 1969;
Rosenmann et al., 1971; Scherberich et al., 1976, 1984), only recently
has it been possible to improve significantly the specificity and
reproducibility of such an approach, relying on the properties of
monoclonal antibodies.
Monoclonal antibodies to human brush-border antigens have been
produced by Mutti et al. (1985). Monoclonal antibodies may be
conveniently employed in ELISA procedures set up to detect trace
amounts of antigens in biological samples. Although the BB-50
brush-border antigen was also localized in peritubular capillaries
(Mutti et al., 1985), subsequent work lead to the identification of a
monoclonal antibody reacting with an antigen located specifically in
the brush border and thus called BBA or brush-border antigen (Mutti et
al., 1988). Promising results have been obtained in several
cross-sectional investigations on groups at risk of chemically induced
renal damage (for a review, see Mutti, 1989). Similar results were
obtained with monoclonal antibodies to rat brush-border cross-reacting
with the human kidney (Tokoff-Rubin et al., 1986).
Monoclonal antibodies have also been produced that react
specifically with other segments along the nephron (namely S3) where
the intestinal isoform of alkaline phosphatase is located (Verpooten
et al., 1989). They could be used to monitor the effects of chemicals
(e.g., mercury) acting selectively on the straight part of proximal
tubules. All of these recently developed tests need further validation
in well-designed longitudinal studies, since their prognostic value is
currently unknown.
8.3 Clinical investigations
The clinical diagnosis of toxic nephropathy frequently relies
heavily on the history of patients who occasionally showed symptoms
and/or laboratory abnormalities suggesting chronic failure without any
obvious recent cause.
Diagnosis can only be made by excluding other known conditions or
risk factors. This has to be assessed by estimating exposure to
nephrotoxins in relation to the known dose-effect/response
relationship and the temporal sequence of events that follow such
exposure. The role of individual susceptibility should also be
considered. In some circumstances, nephrotoxicity may be one factor in
a multifactorial process leading to clinical renal disease.
Retrospective data about exposure and early changes in renal function
are usually not available.
Most progressive kidney diseases have a subtle onset. This is the
reason why the markers designed for use in epidemiological studies are
becoming part of clinical investigations.
Owing to the short latency between exposure and the development
of severe symptoms, acute renal failure should be accurately diagnosed
in all cases. The simple evaluation of serum creatinine (increases of
3-5 mg/litre or 50% above baseline values) makes it possible to detect
nephrotoxic reactions.
8.3.1 Invasive techniques
The use of invasive techniques is limited by ethical constraints.
8.3.1.1 Biopsies from humans
Renal biopsy is the only way to identify glomerular, tubular, or
interstitial renal diseases. However, the risk-to-benefit ratio must
be considered carefully in each individual patient being evaluated,
and there is no universal agreement on the conditions in which
percutaneous renal biopsy may be useful.
8.3.1.2 Autopsy in humans
Autopsies performed on patients with end-stage renal failure only
reveal the etiology of the renal disease in a small percentage of
cases.
8.3.2 Tests designed to assess glomerular filtration and renal blood flow
Traditional methods based on inulin and PAH clearances are
progressively being substituted by more convenient methods such as the
clearance of 51Cr-EDTA and 99Tc-DTPA to assess GFR or the
clearance of 125I-hippuran to measure renal plasma flow. These
techniques are thought to be more sensitive than creatinine clearance
and more accurate than colorimetric methods.
8.3.3 Proteinuria
Section 7.2.4. contains a detailed discussion of experimental
studies involving proteinuria. Table 9 gives information on some
proteins excreted in human urine. Values that are in excess of the
normal range may be indicative of renal dysfunction.
Table 9. Excretion rates of common urine proteins
Protein Relative molecular mass Normal range
(Daltons)
Albumin 68 000 < 30 mg/day
ß2-microglobulin 12 800 < 0.3 mg/day
Retinol-binding Protein 21 400 < 0.3 mg/day
IgG 160 000 2-3 mg/day
alpha2-microglobulin ± 30 000 ?
8.3.4 Tests designed to assess selective damage
Tissue constituents may be shed into the urine following toxic
damage to specific structures. All of these specific antigens may be
detected by using immunochemical or biochemical methods designed to
measure their concentration or enzymic activity, respectively.
Although they have been designed expressly for evaluating population
groups, these tests may also be useful in the clinical setting to
monitor patients at risk. Even if they are very sensitive and
relatively specific, they need further validation under carefully
controlled clinical conditions, particularly in longitudinal studies.
9. SUMMARY AND CONCLUSIONS
The kidneys are the main organs of excretion and homeostasis for
water-soluble molecules. The functional unit of the kidney is the
nephron, essentially a continuous tube of highly specialized
heterogeneous cells, which exhibits marked structural, functional, and
biochemical organization. There are pronounced differences between the
nephrons themselves, depending on the cortical localization of the
individual glomeruli. This complex structural organization, combined
with differences in regional vascularity arising from the specialized
vascular arrangement, produces a highly complex heterogeneous organ.
Much of the anatomical and functional understanding developed
from animals is directly applicable to the human kidney. However, the
biomedical and metabolic processes in the human kidney, as well as the
differences among animal species, have not been as thoroughly
elucidated. Thus, the ability to extrapolate the effects of chemicals
among species is limited.
Several chemicals (both therapeutic and non-therapeutic) have
toxic effects on one or more anatomical elements of the kidney. Toxic
effects may be acute or chronic, and they may be direct or mediated
indirectly through immunological mechanisms. The health impact of
nephrotoxic chemicals is related to risk factors, which include the
intergrade of the renal functional reserve and factors such as
pre-existing renal damage, disease, age, sex, and diet.
The epidemiology of chemically induced nephrotoxicity by
individual chemicals or in mixed exposures has been inadequately
studied. The contribution of chemicals to the overall incidence of
nephropathy and of chronic renal failure is, with few exceptions,
undefined. In the case of some occupationally exposed groups and
analgesic associated renal disease, there has been extensive research
that has shown variations in incidence between groups and countries.
However, it is estimated that up to 18% of end-stage renal disease may
be due to analgesic nephropathy and up to 5% to other toxic
nephropathies. About 50% of end-stage renal disease is of unknown
etiology. A major problem in assigning a cause for end-stage renal
disease is the long latency and/or slow development of chronic renal
failure, which makes retrospective identification of the causative
agent difficult. In only a few cases is measurement of tissue (body,
kidney) levels of chemicals relevant to the diagnosis. A further
problem has been the lack of consistency in diagnostic and
pathological criteria and terminology.
Renal anatomical and physiological differences make direct
extrapolation from experimental systems (in vivo and in vitro) to
humans difficult. There are very few examples of nephrotoxic chemicals
where there are adequate comparable data from animals and humans to
form a firm basis for the assessment of potential human health risk.
Chemicals may damage selectively vulnerable kidney structures or
activate immunological mechanisms. Mechanisms of renal injury fall
generally into two categories: (a) immunologically induced disease of
acute interstitial nephritis; (b) those that primarily affect the
glomerulus by either anti-GBM-mediated antibodies or immune complexes.
Another major group is composed of diseases initiated by chemicals or
their metabolites that interfere with cellular biochemical and
haemodynamic effects, etc. Factors that can modify cellular injury by
toxicants include cellular transport systems, pinocytosis, metabolic
degradation, and interaction with cellular proteins, lipids,
membranes, DNA, and perhaps other cellular constituents.
The increasing use of therapeutic agents and chemicals increases
the possibility of nephrotoxicity. Nephrotoxicity induced by
therapeutic agents depends on the dose and duration of exposure (e.g.,
combination analgesics leading to renal papillary necrosis).
Nephrotoxic effects of analgesics, antibiotics (such as the
aminoglycosides), anticancer agents (such as cis-platinum), and a
variety of other agents have been investigated extensively. Chemicals
frequently used in industry or the home, e.g., chlorinated
hydrocarbons and ethylene glycol, also have the potential to produce
renal damage. Environmental chemicals such as lead and cadmium are
capable of inducing nephrotoxicity. These agents act as toxicants
after intracellular accumulation of the parent compound or after renal
or extrarenal hiptransformation. Multichemical exposure may result in
antagonistic or synergistic responses.
Turnours of the kidney and urinary tract account for less than
2-3% of all human cancers, but the frequency of these tumours is
increasing, suggesting a role for environmental factors. Although many
drugs and chemicals have been shown to be carcinogenic in experimental
models, only a few specific substances have been related to turnouts
in man. These include asbestos (renal adenocarcinoma), analgesic abuse
(transitional carcinomas), and lifestyle factors (tobacco use,
alcohol, coffee). There are population groups with urinary tract
cancer of as yet undetermined etiology. Occupational chemicals have
been related to the etiology of cancer of the urinary bladder. Many
drugs and chemicals cause interstitial nephritis, which may in itself
be a factor in developing urinary tract cancers.
No single in vivo or in vitro method is sufficient to assess
chemically induced renal dysfunction. Therefore, it is advisable not
only to screen compounds for nephrotoxic potential, but to incorporate
mechanistic studies of target cell toxicity into the experimental
protocols. To accomplish this, both in vivo and in vitro
investigations should be utilized in concert. In vitro studies
involve those where anatomical cellular relationships are maintained
(e.g., isolated perfused kidney and tubules, renal cortical slices,
and isolated nephron segments) and those where renal cells are used
(e.g., cell suspension, primary cell cultures, established cell lines,
and subcellular fractions). In vivo studies utilize both invasive
and non-invasive techniques. Invasive procedures include
histopathological and routine renal function measurements.
Non-invasive procedures permit repeated assessment of renal function
in animals through the measurement of an array of renal function tests
(glomerular filtration, tubular function, proteinuria, enzymuria,
etc.). Specialized biochemical tests should be used where relevant.
The appropriate mixture of in vivo and in vitro experiments will
reveal whether or not chemicals are nephrotoxic and give insights into
potencies, sites, and mechanisms of toxicity.
Traditional methods for the assessment of renal function in
humans are inadequate for the timely diagnosis of chemically induced
renal dysfunctionand prediction of its health significance. The lack
of specific, early markers for nephrotoxicity is particularly
troublesome. Non-invasive assessment of nephrotoxity should employ
markers of high specificity, sensitivity for detection of early renal
changes, and predictive value for the development of renal
insufficiency. Present techniques for monitoring glomerular or tubular
function are useful only when severe renal damage has developed.
Although immuno-reactive tissue constituents are being suggested as
appropriate markers, their suitability needs to be validated in
well-designed longitudinal studies.
10. RECOMMENDATIONS
1. A continued effort should be made to develop and validate more
selective and specific markers, including monoclonal antibodies, for
assessment of renal dysfunction in animals. These markers may be
applicable to humans.
2. The data base for predicting the potential of chemicals for human
nephrotoxicity should be extended. This includes development and
validation of experimental animal approaches ( in vivo and in
vitro), alternative methods for studying nephrotoxicity, information
on interspecies differences, and experience from the preclinical
evaluation of new therapeutic agents in humans.
3. Epidemiological studies (i.e. prospective studies in occupational
and general population groups exposed to nephrotoxic chemicals or
involved with analgesic abuse) should be reinforced.
4. More effort should be made to establish the role of chemicals in
the etiology of renal disease at the earliest diagnostic stage (e.g.,
work history, tissue monitoring for nephrotoxins).
5. Understanding of the mechanisms of action of nephro-toxicants
will be helpful in the prevention and clinical management of unwanted
renal effects, and may help in predicting the nephrotoxic potential
for new drugs and chemicals. Areas of particular importance for
further research are:
* immunological mechanisms;
* direct effects of chemicals on membranes, including mechanisms of
lipid peroxidation, membrane/chemical interaction, ion shifts,
and receptor-mediated events;
* activation of proto-oncogenes and cell differentiation;
* regulation of cellular metabolism.
6. There is a need to identify and correlate specific functions with
discrete anatomical locations within the kidney.
7. The role of genetic variation and susceptibility to the toxic
effects of drugs and chemicals should be studied further.
8. The relationship between nephrotoxicity and renal carcinogenesis
(e.g., mycotoxins and Balkan nephropathy) needs further study.
REFERENCES
AHMADIZADEH, M., KUO, C.-H., & HOOK, J.B. (1981) Nephrotoxicity and
hepatotoxicity of chloroform in mice: effect of deuterium
substitution. J. Toxicol. environ. Health, 8: 105-111.
AINSWORTH, S.K., SWAIN, R.P., WATABE, N., BRACKETT, N.C., PILIA, P.,
& HENNIGAR, G.R. (1981) Gold nephropathy, ultrastructural fluorescent
and energy-dispersive X-ray microanalysis study. Arch. Pathol. lab.
Med., 105: 373-378.
ALDEN, C.L. (1989) Male rat specific alpha2u-globulin nephropathy and
tumourigenesis. In: Bach, P.H. & Lock, E.A., ed. Nephrotoxicity:
Extrapolations from in vitro to in vivo, and animals to man, New
York, London, Plenum Press, pp. 535-542.
ALDEN, C.L., KANERVA, R.L., RIDDER, G., & STONE, L.C. (1984) The
pathogenesis of the nephrotoxicity of volatile hydrocarbons in the
male rat. In: Mehlman, M.A., Hemstreet, C.P., Thorpe, J.J., & Weaver,
N.K., ed. Renal effects of petroleum hydrocarbons, Princeton, New
Jersey, Princeton Scientific Publishers, pp. 107-120 (Advances in
Modern Environmental Toxicology, Vol. VII).
ANAND-SRIVASTAVA, M.B., VINAY, P., GENEST, J., & CANTIN, M. (1986)
Effect of atrial natriuretic factor on adenylate cyclase in various
nephron segments. Am. J. Physiol., 251: F417-F423.
ANDERS, M.W. (1980) Metabolism of drugs by the kidney. Kidney Int., 8:
636-647.
ANDERSON, P.M. & SCHULTZE, M.O. (1965) Cleavage of
S-(1,2-dichlorovinyl)-L-cysteine by an enzyme of bovine origin. Arch.
Biochem. Biophys., 111: 593-602.
ANDERSON, S. & BRENNER, B.M. (1986) Effects of aging on the renal
glomerulus. Am. J. med., 80: 435-442.
ANTOINE, B., NEVEU, T., LESKI, M., & BACH J.F. (1969) Histuria during
renal transplantation. Transplantation, 8: 110-120.
ARMBRECHT, H.J., BIRNBAUM, L.S., ZENSER, T.V., MATTAMMAL, M.B., &
DAVIS, B.B. (1979) Renal cytochrome P-450's - Electrophoretic and
electron paramagnetic resonance studies. Arch. Biochem. Biophys., 197:
277-284.
ASKERGREN, A. (1981) Studies on kidney function in subjects exposed to
organic solvents. III. Excretion of cells in the urine. Acta med.
Scand., 210: 103-106.
ASKERGREN, A., ALLGEN, L.G., KARLSSON, C., LUNDBERG, I., & NYBERG, E.
(1981) Studies on kidney function in subjects exposed to organic
solvents. I. Excretion of albumin and ß2-microglobulin in the urine.
Acta med. Scand., 209: 479-483.
ATTALLAH, A. & STAHL, R. (1980) Inhibition of renal PGE2
biosynthesis in vivo: regional differences. Prostaglandins, 19:
649-650.
AUKLAND, K. (1980) Methods for measuring renal blood flow: Total flow
and regional distribution. Annu. Rev. Physiol., 42: 543-555.
AUTOR, A.P., ed. (1977) Biochemical mechanisms of paraquat toxicity,
New York, London, San Francisco, Academic Press.
AVENDANO, L.H. & LOPEZ-NOVOA, J.M. (1987) Glomerular filtration and
renal blood flow in the aged. In: Macias-Nunez, I.F. & Cameron, J.S.,
ed. Renal function and diseases in the elderly, London, Boston,
Toronto, Butterworth, pp. 27-48.
BACH, P.H. (1989) The detection of chemically induced renal injury,
the cascade of degenerative morphological and functional changes that
follow the primary nephrotoxic insult and the evaluation of these
changes by in vitro methods. Toxicol. Lett., 46: 237-250.
BACH, P.H. & BRIDGES, J.W. (1984) The role of prostaglandin synthase
mediated metabolic activation of analgesics and non-steroidal
anti-inflammatory drugs in the development of renal papillary necrosis
and upper urothelial carcinoma. Prostaglandins Leukotrienes Med., 15:
251-274.
BACH, P.H. & BRIDGES, J.W. (1985a) Chemically induced renal papillary
necrosis and upper urothelial carcinoma. CRC crit. Rev. Toxicol., 15:
217-439.
BACH, P.H. & BRIDGES, J.W. (1985b) A decision tree approach for the
application of drug metabolism and kinetics to in vivo and in vitro
toxicological and pharmacological testing. Arch. Toxicol., Suppl. 8:
173-188.
BACH, P.H. & GREGG, N. (1988) Experimentally induced renal papillary
necrosis and upper urothelial carcinoma. Inter. Rev. exp. Pathol., 30:
1-54.
BACH, P.H. & HARDY, T.L. (1985) The relevance of animal models to the
study of analgesic associated renal papillary necrosis in man. Kidney
Int., 28: 605-613.
BACH, P.H. & KWIZERA, E.N. (1988) Nephrotoxicity: A rational approach
to in vitro target cell injury in the kidney. Xenobiotica, 16:
685-698.
BACH, P.H. & LOCK, E.A. (1982) The use of renal tissue slices,
perfusion and infusion techniques to assess renal function and
malfunction. In: Bach, P.H., Bonner, F.W., Bridges, J.W., & Lock,
E.A., ed. Nephrotoxicity: Assessment and pathogenesis, New York,
Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 128-143.
BACH, P.H. & LOCK, E.A., ed. (1985) Renal heterogeneity and target
cell toxicity, New York, Chichester, Brisbane, Toronto, John Wiley and
Sons.
BACH, P.H. & LOCK, E.A., ed. (1987) Nephrotoxicity in the experimental
and the clinical situation, Dordrecht, Boston, Lancaster, Martinus
Nijhoff Publishers.
BACH, P.H. & LOCK, E.A., ed. (1989) Nephrotoxicity: Extrapolation from
in vitro to in vivo, and animals to man, New York, London, Plenum
Press.
BACH, P.H., BONNER, F.W., BRIDGES, J.W., & LOCK, E.A., ed. (1982)
Nephrotoxicity: Assessment and pathogenesis, New York, Chichester,
Brisbane, Toronto, John Wiley and Sons.
BACH, P.H., GRASSO, P., MOLLAND, E.A., & BRIDGES, J.W. (1983) Changes
in the medullary glycosaminoglycan histochemistry and microvascular
filling during the development of 2-bromoethanamine
hydrobromide-induced renal papillary necrosis. Toxicol. appl.
Pharmacol., 69: 333-344.
BACH, P.H., KETLEY, C.P., BENNS, S.E., AHMED, I., & DIXIT., M. (1985)
The use of isolated and cultured renal cells in nephrotoxicity -
practice, potential and problems. In: Bach, P.H. & Lock, E.A., ed.
Renal heterogeneity and target cell toxicity, New York, Chichester,
Brisbane, Toronto, John Wiley and Sons, pp. 505-518.
BACH, P.H., KETLEY, C.P., DIXIT, M., & AHMED, I. (1986) The mechanisms
of target cell injury in nephrotoxicity. Food chem. Toxicol., 24:
775-779.
BACH, P.H., GREGG, N.J., & WACHSMUTH, E.D. (1987) The application of
histochemistry at the light microscopic level to the study of
nephrotoxicity. In: Bach, P.H. & Lock, E.A., ed. Nephrotoxicity in the
experimental and the clinical situation, Dordrecht, Boston, Lancaster,
Martinus Nijhoff Publishers, pp. 19-84.
BACH, P.H., BERLIN, A., HESELTINE, E., KRUG, E., LAUWERYS, R., SMITH,
E., & VAN DER VENNE, M.-T., ed. (1989) Proceedings of the
International Workshop on the Health Significance of Nephrotoxicity.
Toxicol. Lett., 46: 1-306.
BACHUR, N.R., GORDON, S.L., GEE, M.V., & KON, H. (1979) NADPH
cytochrome P-450 reductase activation of quinone anticancer agents to
free radicals. Proc. Natl Acad. Sci., 76: 954-957.
BAER, P.G. (1981) The contribution of prostaglandins to renal blood
flow maintenance is determined by the level of activity of the
renin-angiotensin system. Life Sci., 28: 587-593.
BAGGETT, J.M. & BERNDT, W.O. (1984a) Renal and hepatic glutathione
concentrations in rats after treatment with hexachloro-1,3-butadiene
and citrinin. Arch. Toxicol., 56: 46-49.
BAGGETT, J.M. & BERNDT, W.O. (1984b) Interaction of potassium
dichromate with the nephrotoxins, mercuric chloride and citrinin.
Toxicology, 33: 157-169.
BAGGETT, J.M. & BERNDT, W.O. (1985) The effect of potassium dichromate
on the urinary excretion, organ and subcellular distribution of
203Hg-mercuric chloride. Toxicol. Lett., 29: 115-121.
BAILIE, M.B., SMITH, J.H., NEWTON, J.F., & HOOK, J.B. (1984) Mechanism
of chloroform nephrotoxicity. IV. Phenobarbital potentiation of in
vitro chloroform metabolism and toxicity in rabbit kidneys. Toxicol.
appl. Pharmacol., 74: 285-292.
BAKER, E.L., GOYER, R.A., FOWLER, B.A., KHETTERY, U., BERNARD, D.B.,
ADLER, S., DEVERE WHITE, R., BABAYAN, R., & FELDMAN, R.G. (1980)
Occupational lead exposure, nephropathy, and renal cancer. Am. J. ind.
Med., 1: 139-148.
BANK, N., MUTZ, E.F., & AYNEDIJIAN, H.S. (1967) The role of "leakage"
of tubular fluid in anuria due to mercury poisoning. J. clin. Invest.,
46: 695-704.
BANKI, K., ELFARRA, A.A., LASH, L.H., & ANDERS, M.W. (1986) Metabolism
of S-(2-chloro-1,1,2-trifluoroethyl)-L-cysteine to hydrogen sulfide
and the role of hydrogen sulfide in
S-(2-chloro-1,1,2-trifluoroethyl)-L-cysteine-induced mitochondrial
toxicity. Biochem. biophys. Res. Commun., 138: 707-713.
BANNASCH, P. & ZERBAN, H. (1986) Renal cell adenoma and carcinoma. In:
Jones, T.C., Mohr, U., Hunt, R.D., ed. Monographs on pathology of
laboratory animals, urinary system, Berlin, Heidelberg, New York,
Springer-Verlag, pp. 112-139.
BANNASCH, P., KRECH, R., & ZERBAN, H. (1978a) [Morphogenesis and
micro-morphology of epithelial kidney tumours in rats poisoned with
nitrosomorpho-line. II. Tubular glycogenesis and the formation of
clear-cell or acidophilic-cell tumours.] Z. Krebsforsch., 92: 63-86
(in German).
BANNASCH, P., KRECH, R., & ZERBAN, H. (1978b) [Morphogenesis and
micro-morphology of epithelial kidney tumours in rats poisoned with
nitrosomorpho-line. III. Oncocyte tubules and oncocytomas.] Z.
Krebsforsch., 92: 87-104 (in German).
BANNASCH, P., KRECH, R., & ZERBAN, H. (1980) [Morphogenesis and
micro-morphology of epithelial kidney tumours in rats poisoned with
nitrosomorpho-line. IV. Tubular lesions and basophilic tumours.] J.
Cancer Res. clin. Oncol., 98: 243-265 (in German).
BANNASCH, P., BENNER, U., HACKER, H.J., KLIMEK, F., MAYER, D., MOORE,
M., & ZERBAN, H. (1981) Cytochemical and biochemical analysis of
carcinogenesis. Histochem. J., 13: 799-820.
BARRATT, M. (1983) Proteinuria. Br. med. J., 287: 1489-1490.
BARRY, D.N. & BOWNESS, J.M. (1975) Identification and turnover of
glyco-saminoglycans in rat kidneys. Can. J. Biochem., 53: 713-720.
BATELLE, D., GAVIRIA, M., GRUPP, M., ARRUDA, J.A., WYNN, J., &
KURTZMAN, N.A. (1982) Distal nephron function in patients receiving
lithium therapy. Kidney Int., 21: 477-486.
BATUMAN, V., MAESAKA, J.K., HADDAD, B., TEPPER, E., LANDY, E., &
WEDEEN, R.P. (1981) The role of lead in gout nephropathy. New Engl. J.
Med., 304: 520-523.
BATUMAN, V., LANDY, E., MAESAKA, J.K., & WEDEEN, R.P. (1983)
Contribution of lead to hypertension with renal impairment. New Engl.
J. Med., 309: 17-21.
BAVEREL, G., BONNARD, M., D'ARMAGNAC DE CASTANET, E., & PELLET, M.
(1978) Lactate and pyruvate metabolism in isolated renal tubules of
normal dogs. Kidney Int., 14: 567-575.
BAVEREL, G., BONNARD, M., & PELLET, M. (1979) Lactate and pyruvate
metabolism in isolated human kidney tubules. FEBS Lett., 101: 282-286.
BAVEREL, G., FORISSIER, M., & PELLET, M. (1980a) Lactate and pyruvate
metabolism in dog renal outer medulla. Effects of oleate and ketone
bodies. Int. J. Biochem., 12: 163-168.
BAVEREL, G., GENOUX, C., FORISSIER, M., & PELLET, M. (1980b) Fate of
glutamate carbon and nitrogen in isolated guinea-pig kidney cortex
tubules. Biochem. J., 188: 873-880.
BECKER, C.G., BECKER, E.L., MAHER, J.F., & SCHREINER, G.E. (1962)
Nephrotic syndrome after contact with mercury. A report of five cases,
three after use of ammoniated mercury. Arch. intern. Med., 110:
178-186.
BEEUWKES, R. (1980) The vascular organisation of the kidney. Annu.
Rev. Physiol., 42: 531-542.
BEKERSKY, I., COLBURN, W.A., FISHMAN, L., & KAPLAN, S.A. (1980)
Metabolism of salicylic acid in the isolated perfused rat kidney.
Intercon-version of salicyluric and salicylic acids. Drug Metab.
Disp., 8: 319-324.
BELLEMAN, P. (1980) Primary monolayer cultures of liver parenchymal
cells and kidney tubules as a useful new model for biochemical
pharmacology and experimental toxicology. Studies in vitro on
hepatic membrane transport, induction of liver enzymes and adaptive
changes in renal cortical enzymes. Arch. Toxicol., 44: 63-84.
BENDELE, A.M., CARLTON, W.W., KROGH, P., & LILLEHOJ, E.B. (1985)
Ochratoxin A carcinogenesis in the (C57BL/6J x C3H)F1 mouse. J. Natl
Cancer Inst., 75: 733-742.
BENDZ, H. (1983) Kidney function in lithium-treated patients: a
literature survey. Acta psychiatr. Scand., 68: 303-24.
BENGTSSON, U. (1962) A comparative study of chronic non-obstructive
pyelonephritis and renal papillary necrosis. Acta med. Scand.,
388(Suppl.): 1-71.
BENGTSSON, U., JOHANSSON, S., & ANGERVALL, L. (1978) Malignancies of
the urinary tract and their relation to analgesic abuse. Kidney Int.,
13: 107-113.
BENNETT, W.M. (1983) Aminoglycoside nephrotoxicity. Nephron, 35:
73-77.
BENNETT, W.M. (1985) Lead nephropathy. Kidney Int., 28: 212-220.
BENNETT, W.M. (1986) Drugs and renal disease, New York, Churchill
Livingstone.
BENNETT, W.M., HOUGHTON, D.C., MCCARRON, D.A., ELLIOTT, W.C., PORTER,
G.A., & GILBERT, D.N. (1983) Interventions to modify
aminoglycoside-induced acute renal failure. In: Solez, K. & Whelton,
A., ed. Acute renal failure: Correlations between morphology and
function, New York, Basel, Marcel Dekker, pp. 331-358.
BENNETT, W.M., MELA-RIKER, L., HOUGHTON, D.C., GILBERT, D.N., & BUSS,
W.C. (1988) Microsomal protein synthesis inhibition: an early
mani-festation of gentamicin nephrotoxicity. Am. J. Physiol., 24:
F265-F269.
BENNINGTON, J.L. (1973) Cancer of the kidney - etiology, epidemiology,
and pathology. Cancer, 32: 1017-1029.
BERGERON, M.G., TROTTIER, S., LESSARD, C., BEAUCHAMP, D., & GAGNON, P.
(1982) Disturbed intrarenal distribution of gentamicin in experimental
pyelonephritis due to Escherichia coli. J. infect. Dis., 146:
436-439.
BERLYNE, G.M. (1984) Toxic nephropathies and current methods for early
detection of the toxicity of the kidney. In: Mehlman, M.A., Hemstreet,
C.P., Thorpe, J.J., & Weaver, N.K., ed. Renal effects of petroleum
hydrocarbons, Princeton, New Jersey, Princeton Scientific Publishers,
pp. 173-184 (Advances in Modern Environmental Toxicology, Vol. VII).
BERNARD, A. & LAUWERYS, R. (1986) Effects of cadmium exposure in man.
In: Foulkes, E.C., ed. Cadmium toxicology. Handbook of experimental
pharmacology, Berlin, Heidelberg, New York, Springer-Verlag, Vol. 80,
pp. 136-177.
BERNARD, A., ROELS, H., HUBERMONT, G., BUCHET, J.P., MASSON, P.L., &
LAUWERYS, R. (1976) Characterization of the proteinuria in cadmium
exposed workers. Int. Arch. occup. environ. Health, 38: 19-30.
BERNARD, A., BUCHET, J.P., ROELS, H., MASSON, P., & LAUWERYS, R.
(1979a) Renal excretion of proteins and enzymes in workers exposed to
cadmium. Eur. J. clin. Invest., 9: 11-22.
BERNARD, A., GORET, A., BUCHET, J.P., ROELS, H., & LAUWERYS, R.
(1979b) Comparison of sodium dodecyl sulfate polyacrylamide gel
electro-phoresis with quantitative methods for the analysis of
cadmium-induced proteinuria. Int. Arch. occup. environ. Health, 44:
139-148.
BERNARD, A., MOREAU, D., & LAUWERYS, R. (1982) Comparison of
retinol-binding protein and beta2-microglobulin determination in urine
for the early detection of tubular proteinuria. Clin. Chim. Acta, 126:
1-7.
BERNARD, A., OULED AMOR, A., & LAUWERYS, R. (1987) The effects of low
doses of cadmium-metallothionein on the renal uptake of
beta2-microglobulin in rats. Toxicol. appl. Pharmacol., 15: 440-445.
BERNARD, A., OULED AMOR, A., VIAU, C., & LAUWERYS, R. (1988) The renal
uptake of proteins: a non selective process in conscious rats. Kidney
Int., 34: 175-185.
BERNARD, A., LAUWERYS, R., NOEL, A., VANDELEENE, B., & LAMBERT, A.
(1989) Urine protein 1: a sex dependent marker of tubular or
glomerular dysfunction. Clin. Chem., 35: 2141-2142.
BERNDT, W.O. (1976) Use of renal slice technique for evaluation of
renal tubular transport process. Environ Health Perspect., 15: 73-88.
BERNDT, W.O. (1981) Use of renal function tests in the evaluation of
nephrotoxic effects. In: Hook, J.B., ed. Toxicology of the kidney, New
York, Raven Press, pp. 1-29 (Target Organ Toxicology Series).
BERNDT, W.O. (1987) Renal slices and perfusion. In: Bach, P.H. & Lock,
E.A., ed. Nephrotoxicity in the experimental and the clinical
situation, Dordrecht, Boston, Lancaster, Martinus Nijhoff Publishers,
pp. 301-316.
BERNDT, W.O. (1989) Potential involvement of renal transport
mechanisms in nephrotoxicity. Toxicol. Lett., 46: 77-82.
BERNDT, W.O. & HAYES, A.W. (1977) Effects of citrinin on renal
transport processes. J. environ. Pathol. Toxicol., 1: 93-103.
BERTANI, T., POGGI, A., POZZONI, R., DELAINI, F., SACCHI, G., THOUA,
Y., MECCA, G., REMUZZI, G., & DONATI, M.D. (1982) Adriamycin-induced
nephrotic syndrome in rats: sequence of pathologic events. Lab.
Invest., 46: 16-23.
BERTANI, T., ROCCHI, G., SACCHI, G., MECCA, G., & REMUZZI, G. (1986)
Adriamycin-induced glomerulosclerosis in the rat. Am. J. Kidney Dis.,
7: 12-19.
BHATTACHARYA, R.K. & SCHULTZE, M.O. (1967) Enzymes from bovine and
turkey kidneys which cleave S-(1,2-dichlorovinyl)-L-cysteine. Comp.
Biochem. Physiol., 22: 723-735.
BIBER, T.U.L., MYLLE, M., BAINES, A.D., & GOTTSCHALK, C.W. (1968) A
study by micropuncture and microdissection of acute renal failure in
rats. Am. J. Med., 44: 664-705.
BLAIR-WEST, J.R., BOBIK, A., BROOK, A.H., ESLER, M.D., GIBSON, A.,
MORRIS, M., MCKINLEY, M.J., & PULLAN, P.T. (1980) Renin, ADH and the
kidney: a congeries of conundrums. Prog. biochem. Pharmacol., 17:
20-28.
BLUM, R.H. & CARTER, S.K. (1974) Adriamycin: a new anticancer drug
with significant clinical activity. Ann. intern. Med., 80: 249-259.
BOHMAN, S.-O. (1980) The ultrastructure of the renal medulla and the
interstitial cells. In: Mandal, A.K. & Bohman, S.-O., ed. The renal
papilla and hypertension, New York, London, Plenum Press, pp. 7-33.
BOJESEN, I. (1974) Quantitative and qualitative analyses of isolated
lipid droplets from interstitial cells in renal papillae from various
species. Lipids, 9: 835-843.
BOMHARD, E., LUCKHAUS, G., VOIGT, W.-H., & LOESER, E. (1988) Induction
of light hydrocarbons nephropathy by p-dichlorobenzene. Arch.
Toxicol., 61: 433-439.
BONER, G., SHERRY, J., & RIESELBACH, R.E. (1972) Hypertrophy of the
normal human kidney following contralateral nephrectomy. Nephron, 9:
364-372.
BRENNER, B.M. (1983) Hemodynamically mediated glomerular injury and
the progressive nature of kidney disease. Kidney Int., 23: 647-655.
BRENNER, B.M. & RECTOR, F.C., ed. (1986) The kidney, Philadelphia,
W.B. Saunders.
BRENNER, B.M., HOSTETTER, T.H., & HUMAS, H.D. (1978) Glomerular
permselectivity: barrier function based on discrimination of molecular
size and charge. Am. J. Physiol., 234: F455-F460.
BRENNER, B.M., MEYER, T.W., & HOSTETTER, T.H. (1982) Dietary protein
intake and the progressive nature of kidney disease: the role of
hemodynami-cally mediated glomerular injury in the pathogenesis of
progressive glomerular sclerosis in aging, renal ablation and
intrinsic renal disease. New Engl. J. Med., 307: 652-659.
BREZIS, M., ROSEN, D., SILVA, P., & EPSTEIN, F.H. (1984) Renal
ischaemia: a new perspective. Kidney Int., 26: 375-383.
BRIDGES, J.W., BACH, P.H., BONNER, F.W., & BEATON, E.M. (1982) Effects
of nutritional factors on renal response to toxins. In: Bach, P.H.,
Bonner, F.W., Bridges, J.W., & Lock, E.A., ed. Nephrotoxicity:
Assessment and pathogenesis, New York, Chichester, Brisbane, Toronto,
John Wiley and Sons, pp. 182-198.
BROWN, W.W., ZENSER, T.V., & DAVIS, B.B. (1980) Prostaglandin E2
production by rabbit urinary bladder. Am. J. Physiol., 239: F452-F458.
BRUNBORG, G., HOLME, J.A., SODERLUND, E.J., OMICHINSKI, J.G., &
DYBING, E. (1988) An automated alkaline elution system: DNA damage
induced by 1,2-dibromo-3-chloropropane in vivo and in vitro. Anal.
Biochem., 174: 522-536.
BUCHET, J.-P., ROELS, H., BERNARD, A., & LAUWERYS, R. (1980)
Assessment of renal function of workers simultaneously exposed to
inorganic lead, cadmium or mercury vapour. J. occup. Med., 22:
741-750.
BUCKLEY, J.E., CLARK, V.L., MEYER, T.H., & PEARLMAN, N.W. (1984)
Hypomagnesemia after cisplatin combination chemotherapy. Arch. intern.
Med., 144: 2347-2348.
BURKE, J.F., LAUCIUS, J.F., BRODOVSKY, H.S., & SORIANO, R.Z. (1977)
Doxorubicin hydrochloride-associated renal failure. Arch. intern.
Med., 137: 385-388.
BURKHOLDER, P.M. (1982) Functions and pathophysiology of the
glomerular mesangium (editorial). Lab. Invest., 46: 239-241.
BURRY, A., CROSS, R., & AXELSEN, R. (1977) Analgesic nephropathy and
the renal concentrating mechanism. Pathol. Annu., 12: 1-31.
BURTON, B.T. & HIRSCHMAN, G.H. (1979) Demographic analysis: end-stage
renal disease and its treatment in the United States. Clin. Nephrol.,
11: 47-51.
BUTLER, E.A. & FLYNN, F.V. (1958) The proteinuria of renal tubular
disorders. Lancet, 2: 978-980.
BUTLER, W.H. (1964) Acute toxicity of aflatoxin B1 in rats. Br. J.
Cancer, 18: 756-762.
BUTLER, W.H. & LIJINSKY, W. (1970) Acute toxicity of aflatoxin G1 to
the rat. J. Pathol., 102: 209-212.
BUTLER, W.H., GREENBLATT, M., & LIJINSKY, W. (1969) Carcinogenesis in
rats by aflatoxins B1, G1, and B2. Cancer Res., 29: 2206-2211.
BUTLER, W.T. (1966) Pharmacology, toxicity, and therapeutic usefulness
of amphotericin B. J. Am. Med. Assoc, 195: 371-375.
BUZIO, C., MUTTI, A., CAPANI, F., ANDRULLI, S., PERAZZOLI, F.,
ALINOVI, R., NEGRO, A., & RUSTICHELLI, R. (1989) Circadian rhythm of
proteinuria: effects of an evening meat meal. Nephrol. Dialysis
Transplant., 4: 266-270.
CALDER, I.C., YONG, A.C., WOODS, R.A., CROWN, C.A., HAM, K.N., &
TANGE, J.D. (1979) The nephrotoxicity of p-aminophenol. II. The effect
of metabolic inhibitors and inducers. Chem.-biol. Interact., 27:
245-254.
CALDICOTT, W.J.H., HOLLENBERG, N.K., & ABRAMS, H.L. (1970)
Characteristics of response of renal vascular bed to contrast media.
Evidence of vasoconstriction induced by renin-angiotensin system.
Invest. Radiol., 5: 539-547.
CALNE, R.Y., WHITE, D.J.G., THIRU, S., EVANS, D.B., MCMASTER, P.,
DUNN, D.C., CRADDOCK, G.N., PENTLOW, B.D., & ROLLES, K. (1978)
Cyclosporin-A in patients receiving renal allografts from cadaver
donors. Lancet, 2: 1323-1327.
CARLIER, M.-B., LAURENT, G., CLAES, P.J., VANDERHAEGHE, H.J., &
TULKENS, P.M. (1983) Inhibition of lysosomal phospholipases by
aminoglycoside antibiotics: in vitro comparative studies.
Antimicrob. Agents Chemother., 23: 440-449.
CARPENTER, H.M. & MUDGE, G.H. (1981) Acetaminophen nephrotoxicity.
Studies on renal acetylation and deacetylation. J. Pharmacol. exp.
Ther., 218: 161-167.
CASTEGNARO, M. & CHERNOZEMSKY, I. (1987) Endemic nephropathy and
urinary tract tumors in the Balkans. Cancer Res., 47: 3608-3609.
CASTOR, C.W. & GREEN, J.A. (1968) Regional distribution of acid
mucopolysaccharides in the kidney. J. clin. Invest., 47: 2125-2132.
CAULFIELD, J.P., REID, J.J., & FARQUBAR, M.G. (1976) Alterations of
the glomerular epithelium in aminonucleoside nephrosis. Lab. Invest.,
34: 43-59.
CAVALIERE, G., ARRIGO, G., D'AMICO, G., BERNASCONI, P., SCHIAVINA,
G., DELLAFIORE, L., & VERGNAGHI, D. (1987) Tubular nephro-toxicity
after intravenous urography with ionic high-osmolal and nonionic
low-osmolal contrast media in patients with chronic renal
insufficiency. Nephron, 46: 128-133.
CEDGARD, S., HERLITZ, H., GETERUD, K., ALTMAN, P.O., & AURELL, M.
(1986) Acute renal insufficiency after administration of low-osmolar
contrast media. Lancet, 2: 1281.
CHAHWALA, S.B. & HARPUR, E.S. (1986) The use of renal tubule fragments
isolated from the rat to investigate aspects of gentamicin
nephrotoxicity. J. Pharmacol. Methods, 15: 21-34.
CHAMBERLIN, M.E., LEFURGEY, A., & MANDEL, L.J. (1984) Suspension of
medullary thick ascending limb tubules from the rabbit kidney. Am. J.
Physiol., 247: F955-F964.
CHARBONNEAU, M., LOCK, E.A., STRASSER, J., COX, M.G., TURNER, M.J., &
BUS, J.S. (1987) 2,2,4-Trimethylpentane-induced nephrotoxicity. I.
Metabolic disposition of TMP in male and female Fischer-344 rats.
Toxicol. appl. Pharmacol., 91: 171-181.
CHASSEAUD, L.F. (1980) Extrahepatic conjugation with glutathione. In:
Gram, T.E., ed. Extrahepatic metabolism of drugs and other foreign
compounds, New York, Spectrum Publications, pp. 427-443.
CHOPRA, S., KAUFMAN, J.S., JONES, T.W., HONG, W.K., GEHR, M.K.,
HAMBERGER, R.J., FLAMENBAUM, W., & TRUMP, B.F. (1982)
Cis-diamminedichloro-platinum-induced acute renal failure in the rat.
Kidney. Int., 21: 54-64.
CHOU, C.C., HOOK, J.B., & HSIEH, C.P. (1974) Effects of radiopaque
dyes on renal vascular resistance. J. lab. clin. Med., 78: 705-712.
CHUANG, E.L., REINECK, H.J., OSGOOD, R.W., LIMAI, R.T., & STEIN, J.H.
(1978) Studies on the mechanism of reduced urinary osmolality after
exposure of renal papilla. J. clin. Invest., 61: 633-639.
CHUMAN, L., FINE, L.G., COHEN, A.H., & SAIER, M.H. Jr. (1982)
Continuous growth of proximal tubular kidney epithelial cells in
hormone-supplemented serum-free medium. J. Cell Biol., 94: 506-510.
CLIFTON, G., KAPLOWITZ, N., WALLIN, J.D., & KUHLENKAMP, J. (1975) Drug
induction and sex differences of renal glutathione S-transferases in
the rat. Biochem. J., 150: 259-262.
CLIVE, D.M. & STOFF, J.S. (1984) Renal syndrome associated with
nonsteroidal anti-inflammatory drugs. New Engl. J. Med., 310: 563-572.
COHEN, J.J. (1979) Is the function of the renal papilla coupled
exclusively to an anaerobic pattern of metabolism? Am. J. Physiol.,
236: F423-F433.
COHEN, J.J., HARRINGTON, J.T., & KASSIREV, J.P. (1981) Lithium and the
kidney. Kidney Int., 19: 374-387.
COJOCEL, C., MAITA, K., PASINO, D.A., KUO, C., & HOOK, J.B. (1983)
Metabolic heterogeneity of the proximal and distal kidney tubules.
Life Sci., 33: 855-861.
COLLEONI, N. & D'AMICO, G. (1986) Chronic lead accumulation as a
possible cause of renal failure in gouty patients. Nephron, 44: 32-35.
COLLIER, V., LIETMAN, P.S., & MITCH, W.E. (1979) Evidence for luminal
uptake of gentamicin in the perfused rat kidney. J. Pharmacol. exp.
Ther., 210: 247-251.
CONNELLY, J.C. & BRIDGES, J.W. (1980) The distribution and role of
cytochrome P-450 in extrahepatic organs. Prog. Drug. Metab., 5: 1-111.
CONSTANTOPOULOS, G., LOUIE, M., & DEKABAN, A.S. (1973) Acid
mucopolysaccharides (glycosaminoglycans) in normal human kidneys and
in kidneys of patients with mucopolysaccaridoses. Biochem. Med., 7:
376-388.
COOPER, W.C. & GAFFEY, W.R. (1975) Mortality of lead workers. J.
occup. Med., 17: 100-109.
COOPER, W.C., WONG, O., & KHEIFETS, L. (1985) Mortality among
employees of lead battery plants and lead producing plants. Scand. J.
Work Environ. Health., 11: 331-345.
CRAIN, R.C. (1958) Spontaneous tumors in the Rochester strain of the
Wistar rat. Am. J. Pathol., 34: 311-336.
CRAMER, K., GOYER, R.A., JAGENBURG, R., & WILSON, M. (1974) Renal
ultrastructure, renal function and parameters of lead toxicity in
workers with different periods of lead exposure. Br. J. ind. Med., 31:
113-127.
CROWE, C.A., CALDER, I.C., MADSEN, N.P., FUNDER, C.C., GREEN, C.R.,
HAM, K.N., & TANGE, J.D. (1977) An experimental model of
analgesic-induced renal damage. Some effects of p-aminophenol on rat
kidney mitochondria. Xenobiotica, 7: 345-356.
CROWE, C.A., YONG, A.C., CALDER, I.C., HAM, K.N., & TANGE, J.D. (1979)
The nephrotoxicity of p-aminophenol. I. The effect of microsomal
cyto-chromes, glutathione and covalent binding in kidney and liver.
Chem.-biol. Interact., 27: 235-243.
CUNARRO, J.A. & WEINER, M.W. (1978) Effects of ethacrynic acid and
furosemide on respiration of isolated kidney tubules: the role of ion
transport and the source of metabolic energy. J. Pharmacol. exp.
Ther., 206: 198-206.
CUNNINGHAM, E.E., BRENTJENS, J.R., ZIELEZREY, M.A., ANDRES, G.A., &
VERUTO, R.C. (1980) Heroin nephropathy: a clinicopathologic and
epidemiologic study. Am. J. Med., 68: 47-53.
CUNNINGHAM, E.E., BARONE, P.P., NASCIMENTO, L., & VENUTO, R.C. (1986)
Effect of a radiographic contrast agent on renal function in the rat.
Comparison with equiosmolar mannitol. Miner. electrolyte Metab., 12:
157-160.
CURTHOYS, N.P. & BELLEMANN, P. (1979) Renal cortical cells in primary
monolayer culture. Enzymatic changes and morphological observations.
Exp. cell Res., 121: 31-45.
CURTIS, J.R. (1979) Drug-induced renal disease. Drugs, 18: 377-391.
CVITKOVIC, E., SPAULDING, J., BETHUNE, V., MARTIN, J., & WHITMORE,
W.F. (1977) Improvement of cis-dichlorodiammineplatinum (NSC-119875):
Therapeutic index in an animal model. Cancer, 39: 1357-1361.
DARNTON, S.J. (1967) Glycogen metabolism in rabbit kidney under
differing physiological states. Q. J. exp. Physiol., 52: 392-400.
DARNTON, S.J. (1969) The conversion of injected glucose into renal
glycogen and mucopolysaccharides. An autoradiographic study of rabbits
in various states of hydration. Z. Zellforsch., 102: 273-282.
DAUGAARD, G., ROSSING, N., & RORTH, M. (1988a) Effects of cisplatin on
different measures of glomerular function in the human kidney with
special emphasis on high-dose. Cancer Chemother. Pharmacol., 21:
163-167.
DAUGAARD, G., HOLSTEIN-RATHLOU, N.-H., & LEYSSAC, P.P. (1988b) Effects
of cisplatin on proximal convoluted and straight segments of the rat
kidney. J. Pharmacol. exp. Ther., 244: 1081-1085.
DAVIES, D.F. & SHOCK, N.W. (1950) Age changes in glomerular filtration
rate, effective renal plasma flow and tubular excretory capacity in
adult males. J. clin. Invest., 29: 496-507.
DAVIES, J.M. (1984) Long term mortality study of chromate pigment
workers who suffered lead poisoning. Br. J. ind. Med., 41: 170-178.
DAVIS, B.B., MATTAMMAL, M.B., & ZENSER, T.V. (1981) Renal metabolism
of drugs and xenobiotics. Nephron, 27: 187-196.
DAYAL, H. & KINMAN, J. (1983) Epidemiology of kidney cancer. Semin.
Oncol., 10: 366-377.
DEBROE, M.E., PAULUS, G.J., VERPOOTEN, G.A., ROELS, F., BUYSSENS, N.,
WEDEEN, R., VANHOOF, F., & TULKENS, P.M. (1984) Early effects of
gentamicin, tobramycin and amikacin on the human kidney. Kidney Int.,
25: 643-652.
DEES, J.H., HEATFIELD, B.M., REUBER, M.D., & TRUMP, B.F. (1980a)
Adenocarcinoma of the kidney. III. Histogenesis of renal
adenocarcinomas induced in rats by
N-(4'-fluoro-4-biphenylyl)acetamide, J. Natl Cancer Inst., 64:
1537-1545.
DEES, J.H., HEATFIELD, B.M., & TRUMP, B.F. (1980b) Adenocarcinoma of
the kidney. IV. Electron microscopic study of the development of renal
adenocar-cinomas induced in rats by
N-(4'-fluoro-4-biphenylyl)acetamide. J. Natl Cancer Inst., 64:
1547-1562.
DEKERNION, J.B. & BERRY, D. (1980) The diagnosis and treatment of
renal carcinoma. Cancer, 45(Suppl.): 1947-1982.
D'ELIA, J.A., GLEASON, K.E., ALDAY, M., MALARICK, C., GODLEY, K.,
WARRAM, J., KALDANY, A., & WEINRAUCH, L.A. (1982) Nephrotoxicity from
angiographic contrast material. A prospective study. Am. J. Med., 72:
719-725.
DENTINO, M., LUFT, F.C., YUM, M.W., WILLIAMS, S.D., & EINHORN, L.H.
(1978) Long term effect of cis-diamminedichloride platinum (CDDP) on
renal function and structure in man. Cancer, 41: 1274-1281.
DEPNER, T.A. (1982) Nephrotic syndrome secondary to lithium therapy.
Nephron, 30: 286-289.
DICKER, S.E. & FRANKLIN, C.S. (1966) The isolation of hyaluronic acid
and chondroitin sulphate from kidneys and their reaction with urinary
hyaluronidase. J. Physiol., 186: 110-120.
DIEPERINK, H.H. (1989) Identification of groups at risk for renal
diseases (including nephrotoxicity). Toxicol. Lett., 46: 257-268.
DIEPERINK, H., STARKLINT, H., & LEYSSAC, P.P. (1983) Nephrotoxicity of
cyclosporin A - an animal model. A study of the nephrotoxic effect of
cyclosporin on overall renal and tubular function in conscious rats.
Transplant Proc., 15(Suppl. 1): 2736-2741.
DIEPERINK, H., LEYSSAC, P.P., STARKLINT, H., & KEMP, E. (1985)
Glomerulotubular function in cyclosporin A treated rats. A lithium
clearance, occlusion time/transit time and micropuncture study. Proc.
Eur. Dialysis Transplant Assoc., 21: 853-859.
DIEZI, J. & ROCH-RAMEL, F. (1987) The use of single nephron techniques
in renal toxicity studies. In: Bach, P.H. & Lock, E.A., ed.
Nephrotoxicity in the experimental and the clinical situation,
Dordrecht, Boston, Lancaster, Martinus Nijhoff Publishers, pp.
317-358.
DOBYAN, D.C. (1985) Long-term consequences of cis-platinum-induced
renal injury: a structural and functional study. Anat. Rec., 212:
239-245.
DOHN, D.R., QUEBBEMANN, A.J., BORCH, R.F., & ANDERS, M.W. (1985a)
Enzymatic reaction of chlorotrifluoroethene with glutathione: 19F NMR
evidence for stereochemical control of the reaction. Biochemistry, 24:
5137-5143.
DOHN, D.R., LEININGER, J.R., LASH, L.H., QUEBBEMANN, A.J., & ANDERS,
M.W. (1985b) S-(2-chloro-1,1,2-trifluoroethyl)glutathione and
S-(2-chloro-1,1,2-trifluoroethyl)-L-cysteine, the glutathione and
cysteine conju-gates of chlorotrifluoroethene. J. Pharmacol. exp.
Ther., 235: 851-857.
DOUGLAS, J.B. & HEALY, J.K. (1969) Nephrotoxic effects of amphotericin
B, including renal tubular acidosis. Am. J. Med., 46: 154-162.
DRUET, P. (1989) Contribution of immunological reactions to
nephrotoxicity. Toxicol. Lett., 46: 55-64.
DRUET, P., BERNARD, A., HIRSCH, F., WEENING, J.J., GENOUX, P., MAHIEU,
P., & BIRKELAND, S. (1982) Immunologically mediated glomerulonephritis
induced by heavy metals. Arch. Toxicol., 50: 187-194.
DRUET, P., JACQUOT, C., BARAN, D., KLEINKNECHT, D., FILLASTRE, J.P.,
& MERY, J.Ph. (1987) Immunologically mediated nephritis induced by
toxins and drugs. In: Bach, P.H. & Lock, E.A., ed. Nephrotoxicity in
the experimental and the clinical situation, Dordrecht, Boston,
Lancaster, Martinus Nijhoff Publishers, pp. 727-770.
DUBACH, U.C., LE HIR, M., & GANDHI, R. (1989) Use of urinary enzymes
as markers of nephrotoxicity. Toxicol. Lett., 46: 193-196.
DUNHILL, M.S., MILLARD, P.R., & OLIVER, D. (1977) Acquired cystic
disease of the kidney: a hazard of long-term intermittent maintenance
dialysis. J. clin. Pathol., 30: 868-877.
DUNN, M.A., BLALOCK, T.L., & COUSIN, R.J. (1987) Minireview of
metallothionein. Proc. Soc. Exp. Biol. Med., 185: 107-119.
DUNN, M.J. (1981) Prostaglandins and Bartter's syndrome. Kidney Int.,
19: 86-102.
DUNN, M.J. & HOOD, V.L. (1977) Prostaglandins and the kidney. Am. J.
Physiol. 223: F169-F184.
DUNN, M.J. & ZAMBRASKI, E.J. (1980) Renal effects of drugs that
inhibit prostaglandin synthesis. Kidney Int., 18: 609-622.
DYBING, E., OMICHINSKI, J.G., SODERLUND, E.J., BRUNBORG, G., LAG, M.,
HOLME, J.A., & NELSON, S.D. (1989) Mutagenicity and organ damage of
1,2-dibromo-3-chloropropane (DBCP) and
tris(2,3-dibromopropyl)phosphate (tris-BP): role of metabolic
activation. In: Hodgson, E., Bend, J.R., & Philpot, R.M., ed. Reviews
in biochemical toxicology, Amsterdam, Oxford, New York, Elsevier
Science Publishers, Vol. 10, pp. 139-186.
EARLEY, L.E. & FRIEDLER, R.M. (1964) Observations on the mechanism of
decreased tubular reabsorption of sodium and water during saline
loading. J. clin. Invest., 43: 1928-1936.
EARLEY, L.E. & FRIEDLER, R.M. (1965) Changes in renal blood flow and
possibly the intra-renal distribution of blood during natriuresis
accompanying saline loading in the dog. J. clin. Invest., 44: 929-941.
EBLE, J.N. & HULL, M.T. (1984) Morphologic features of renal
oncocytoma. A light and electron microscopic study. Hum. Pathol., 15:
1054-1061.
EINHORN, L.H. & DONOHUE, J. (1977) Cis-diamminedichloroplatinum,
vinblastin and bleomycin combination chemotherapy in disseminated
testicular cancer. Ann. intern. Med., 87: 293-298.
EISENBERG, R.L., BANK, W.O., & HEDGCOCK, M.W. (1980) Renal failure
after major angiography. Am. J. Med., 68: 43-46.
ELBERS, R., KAMPFFMEYER, H.G., & RABES, H. (1980) Effects and
metabolic pathway of 4-dimethylaminophenol during kidney perfusion.
Xenobiotica, 10: 621-632.
ELFARRA, A.A. & ANDERS, M.W. (1984) Renal processing of glutathione
conjugates. Role in nephrotoxicity. Biochem. Pharmacol., 33:
3729-3732.
ELFARRA, A.A., JACOBSON, I., & ANDERS, M.W. (1986a) Mechanism of
S-(1,2-dichlorovinyl) glutathione-induced nephrotoxicity. Biochem.
Pharmacol., 35: 283-288.
ELFARRA, A.A., LASH, L.H., & ANDERS, M.W. (1986b) Metabolic activation
and detoxification of nephrotoxic cysteine and homocysteine
S-conjugates. Proc. Natl Acad. Sci., 83: 2667-2671.
ELINDER, C.G., EDLING, C., LINDBERG, E., BERTIL, K., & VESTERBERG, O.
(1985a) Beta-2-microglobulinuria among workers previously exposed to
cadmium: follow-up and dose-response analyses. Am. J. ind. Med., 8:
553-564.
ELINDER, C.G., EDLING, C., LINDBERG, E., KAGEDAL, B., & VESTERBERG, O.
(1985b) Assessment of renal function in workers previously exposed to
cadmium. Br. J. ind. Med., 42: 754-760.
ELLIS, K.J., MORGAN, W.C., ZANZI, I., YASUMURA, S., VARTSKY, D., &
COHN, S.H. (1981) Critical concentration of Cd in human renal cortex:
dose-effect studies in Cd smelter workers. J. Toxicol. environ.
Health, 7: 691-703.
ELSEVIERS, M.M. & DE BROE, M.E. (1988) Is analgesic nephropathy still
a problem in Belgium? Nephrol. Dialysis Transplant., 2: 143-149.
EMMERSON, B.T. (1973) Chronic lead nephropathy. Kidney Int., 4: 1-5.
EMSLIE, K.R., CALDER, I.C., HART, S.J., & TANGE, J.D. (1981) Induction
of paracetamol metabolism in the isolated perfused kidney.
Xenobiotica, 11: 43-50.
ENDOU, H. (1983) Cytochrome P-450 monooxygenase system in the rabbit
kidney: its intranephron localization and its induction. Jpn. J.
Pharmacol., 33: 423-433.
ENDOU, H., NONOGUCHI, Y., TAKEHARA, H., YAMADA, H., & NAKADA, J.
(1985) Intranephron heterogeneity of ammoniagenesis and
gluconeogenesis in rats. Contrib. Nephrol., 47: 98-104.
ENTERLINE, P.E. & VIREN, J. (1985) Epidemiologic evidence for an
association between gasoline and kidney cancer. Environ. Health
Perspect., 62: 303-312.
EPSTEIN, S.M., BARTUS, B., & FARBER, E. (1969) Renal epithelial
neoplasms induced in male Wistar rats by oral aflatoxin B1. Cancer
Res., 29: 1045-1050.
ERTÜRK, E., COHEN, S.M., & BRYAN, G.T. (1970) Induction, histogenesis,
and isotransplantability of renal tumors induced by formic acid
2-[4-(5-nitro-2-furyl)-2-thiazolyl]hydrazide in rats. Cancer Res., 30:
2098-2106.
EUROPEAN DIALYSIS AND TRANSPLANT ASSOCIATION REGISTRY (l986)
Demography of dialysis and transplantation in Europe, 1984. Nephrol.
Dialysis Transplant., 1: 1.
EVARTS, R.P., BROWN, C.A., & MOSTAFA, H. (1982) Production of kidney
tumors in rats with low dose of dimethylnitrosamine after partial
hepatectomy. J. Natl Cancer Inst., 68: 293-298.
FAJARDO, L.F., ELTRINGHAM, J.R., STEWART, J.R., & KLAUBER, M.R. (1980)
Adriamycin nephrotoxicity. Lab. Invest., 43: 242-253.
FANG, L.S., SIROTA, R.A., EBERT, T.H., & LICHTENSTEIN, N.S. (1980) Low
fractional excretion of sodium with contrast media-induced acute renal
failure. Arch. intern. Med., 140: 531-533.
FARBER, S.J., WALAT, R.J., BENJAMIN, R., & VAN PRAAG, D. (1971) Effect
of increased osmolality on glycosaminoglycan metabolism of rabbit
renal papilla. Am. J. Physiol., 220: 880-885.
FINKELSTEIN, A., FRALEY, D.S., STACHURA, I., FELDMAN, H.A., GANDY,
D.R., & BOURKE, E. (1982) Fenoprofen nephropathy: Lipoid nephrosis and
interstitial nephritis. Am. J. Med., 72: 81-86.
FISCHER, L.J., GREEN, M.D., & HARMAN, A.W. (1985) Studies on the fate
of the glutathione and cysteine conjugates of acetaminophen in mice.
Drug Metab. Disp., 13: 121-126.
FJELDBORG, P., SORENSEN, J., & HELKJAER, P.E. (1986) The long-term
effect of cisplatin on renal function. Cancer, 58: 2214-2217.
FOA', V., CAIMI, L., AMANTI, L., ANTONINI, C., GATTINONI, A.,
TETTAMANTI, G., LOMBARDO, A., & GIULIANI, A. (1976) Patterns of some
lysosomal enzymes in the plasma and of proteins in urine of workers
exposed to inorganic mercury. Int. Arch. occup. environ. Health, 37:
115-124.
FOA', V., COLOMBI, A., MARONI, M., BARBIERI, F., FRANCHINI, I., MUTTI,
A., DE ROSA, E., & BARTOLUCCI, G.B. (1987) Study of kidney function of
workers with low level exposure to inorganic arsenic. In: Foa', V.,
Emmett, E.A., Maroni, M., & Colombi, A., ed. Occupational and
environmental chemical hazards, Chichester, Ellis Horwood Limited, pp.
362-367.
FOIDART, J.B., DECKANNE, C.A., MAHIEU, P., CREUTZ, C.E., & DE MEY, J.
(1979) Tissue culture of normal rat glomeruli: Isolation and
morphological characterization of two homogeneous cell lines. Invest.
cell Pathol., 2: 15-26.
FOIDART, J.B., DUBOIS, C.H., & FOIDART, J.-M. (1980) Tissue culture of
normal rat glomeruli: Basement membrane biosynthesis by homogenous
epithelial and mesangial cell lines. Int. J. Biochem., 12: 197-202.
FOIDART, J.B., DECHENNE, C.A., & MAHIEU, P. (1981) Tissue culture of
normal rat glomeruli: Characterization of collagenous and
non-collagenous basement membrane antigens on the epithelial and
mesangial cells. Diagn. Histopathol., 4: 71-77.
FORD, S.M. & HOOK, J.B. (1984) Biochemical mechanisms of toxic
nephropathies. Semin. Nephrol., 4: 88-106.
FORREST, J.B., HOWARDS, S.S., & GILLENWATER, I.Y. (1981) Osmotic
effects of intravenous contrast agents on renal function. J. Urol.,
125: 147-150.
FOWLER, B.A. & GOYER, R.A. (1975) Bismuth localization within nuclear
inclusions by X-ray microanalysis: Effects of accelerating voltage.
Histochem. Cytochem., 23: 722-727.
FRANCHINI, I. & MUTTI, A. (1988) Selected toxicological aspects of
chromium compounds. Sci. total Environ., 71: 379-387.
FRANCHINI, I., CAVATORTA, A., FALZOI, M., LUCERTINI, S., & MUTTI, A.
(1983) Early indicator of renal damage in workers exposed to organic
solvents. Int. Arch. occup. environ. Health, 51: 1-9.
FRIBERG, L. (1948) Proteinuria and kidney injury among workers exposed
to cadmium and nickel dust. J. ind. Toxicol., 30: 32-36.
FRIBERG, L. (1950) Health hazards in the manufacture of alkaline
accumulators with special reference to chronic cadmium poisoning. Acta
med. Scand., 138 (Suppl. 240): 1-154.
FRIBERG, L., PISCATOR, M., NORDBERG, G.F., & KJELLSTROM, T. (1974)
Cadmium in the environment, 2nd ed., Cleveland, Ohio, Chemical Rubber
Co. Press.
FRIBERG, L., ELINDER, C.G., KJELLSTROM, T., & NORDBERG, G.F. (1986)
Cadmium and health: a toxicological and epidemiological appraisal,
Boca Raton, Florida, CRC Press, pp. 1-307.
FRIEDLANDER, G., BLANCHET, F., & AMIEL, C. (1989) Renal functional
reserve. Toxicol. Lett., 46: 227-235.
FROLICH, J.C. & WALKER, L.A. (1980) Determination, source, metabolism
and functional role of renal prostaglandins. Clin. exp. Hypertension,
2: 709-728.
FROLICH, J.C., NIES, A.S., & SCHRIER, R.W., ed. (1981) Prostaglandins
and the kidney. Kidney Int., 19: 755-868.
FRY, J.R. & PERRY, N.K. (1981) The effect of Aroclor-1254 pretreatment
on the phase I and phase II metabolism of 7-ethoxycoumarin in isolated
viable rat kidney cells. Biochem. Pharmacol., 30: 1197-1201.
FRY, J.R., WIEBKIN, P., KAO, J., JONES, C.A., GWYNN, J., & BRIDGES,
J.W. (1978) A comparison of drug-metabolising capability in isolated
viable rat hepatocytes and renal tubule fragments. Xenobiotica, 8:
113-120.
FUKUSHIMA, M., ISHIZAKI, A., NOGAWA, K., SAKAMOTO, M., & KOBAYASHI, E.
(1974) Epidemiological studies on renal failure of inhabitants in
Itai-Itai disease endemic district. I. Some findings of inhabitants
living in and around the endemic district of the Jinzu River basin.
Jpn. J. pub. Health, 21: 67-73.
GALE, M.E., ROBBINS, A.H., HAMBURGER, R.J., & WIDRICH, W.C. (1984)
Renal toxicity of contrast agents: iopamidol, iothalamate, and
diatrizoate. Am. J. Roentgenol., 142: 333-335.
GARTNER, H.V. (1980) Drug-associated nephropathy. In: Berry, C.L.,
Grundmann, E., & Kirsten, W.H., ed. Drug-induced pathology, Berlin,
Heidelberg, New York, Springer-Verlag, pp. 144-174 (Current Topics in
Pathology, Vol. 69).
GEMBORYS, M.W. & MUDGE, G.H. (1981) Formation and disposition of the
minor metabolites of acetaminophen in the hamster. Drug Metab. Disp.,
9: 340-351.
GILBERT, D.N., WOOD, C.A., KOHLHEPP, S.J., KOHNEN, P.W., HOUGHTON,
D.B., FINKBEINER, H.C., LINDSLEY, J., & BENNETT, W.M. (1989)
Polyaspartic acid prevents experimental aminoglycoside nephrotoxicity.
J. infect. Dis., 159: 945-953.
GINETZINSKY, A.G. (1958) Role of hyaluronidase in the reabsorption of
water in renal tubules. The mechanism of action of the anti-diuretic
hormone. Nature (Lond.), 182: 1218-1219.
GIULIANO, R.A., PAULUS, G.J., VERPOOTEN, R.A., PATTYN, V., POLLET,
D.E., TULKENS, P.M., & DEBROE, M.E. (1984) Recovery of cortical
phospholipidosis and necrosis after acute gentamicin loading in rats.
Kidney Int., 26: 838-847.
GIULIANO, R.A., VERPOOTEN, G.A., VERBIST, L., WEDEEN, R., & DEBROE,
M.E. (1986) In vivo uptake kinetics of aminoglycosides in the kidney
cortex of rats. J. Pharmacol. exp. Ther., 236: 470-475.
GLOOR, F.J. (1978) Changing concept in the pathogenesis and morphology
of analgesic nephropathy as seen in Europe. Kidney Int., 13: 27-33.
GOLDBERG, J.P. & ANDERSON, R.J. (1985) Renal metabolism and excretion
of drugs. In: Seldin, D.W. & Giebisch, G., ed. The kidney: Physiology
and pathophysiology, New York, Raven Press, pp. 2097-2110.
GOLDSTEIN, R.S. & MAYOR, G.H. (1983) Minireview: The nephrotoxicity of
cis-platin. Life Sci., 32: 685-690.
GOLDSTEIN, R.S., CONTARDI, L.R., PASINO, D.A., & HOOK, J.B. (1987)
Mechanisms mediating cephaloridine inhibition of gluconeogenesis.
Toxicol. appl. Pharmacol., 87: 297-305.
GOLDSTEIN, R.S., SOZIO, R.S., TARLOFF, J.B., & HOOK, J.B. (1989) The
role of oxidative stress in cephaloridine nephrotoxicity. In: Bach,
P.H. & Lock, E.A., ed. Nephrotoxicity: Extrapolation from in vitro
to in vivo, and animals to man, New York, London, Plenum Press, pp.
457-461.
GOLMAN, K. & ALMEN, T. (1985) Contrast media-induced nephrotoxicity.
Survey and present state. Invest. Radiol., 20: S92-S97.
GONZALEZ-VITALE, J.C., HAYES, D.M., CVITKOVIC, E., & STERNBERG, S.
(1977) The renal pathology in clinical trials of cis-platinum (II)
diammine-dichloride. Cancer, 39: 1362-1371.
GOODMAN, D.G., WARD, J.M., SQUIRE, R.A., CHU, K.C., & LINHART, M.S.
(1979) Neoplastic and non-neoplastic lesions in aging F344 rats,
Toxicol. appl. Pharmacol., 48: 237-248.
GOODMAN, D.G., WARD, J.M., SQUIRE, R.A., PAXTON, M.B., REICHARDT,
W.D., CHU, K.C., & LINHART, M.S. (1980) Neoplastic and non-neoplastic
lesions in aging Osborne-Mendel rats. Toxicol. appl. Pharmacol., 55:
433-447.
GOODMAN, M.T., MORGENSTERN, H., & WYNDER, E.L. (1986) A case-control
study of factors affecting the development of renal cell cancer. Am.
J. Epidem., 124: 926-941.
GOTELLI, C.A., ASTOLFI, E., COX, C., CERNICHIARI, E., & CLARKSON, T.W.
(1985) Early biochemical effects of an organic mercury fungicide on
infants: dose makes the poison. Science, 227: 638-640.
GOTTSCHALK, C.W. & LASSITER, W.E. (1973) Micropuncture methodology.
In: Orloff, J. & Berliner, W.R., ed. Handbook of physiology. Section
8: Renal physiology, Washington, DC, American Physiological Society.
GOYER, R.A. (1982) Nephrotoxic effects of lead. In: Bach, P.H.,
Bonner, F.W., Bridges, J.W., & Lock, E.A., ed. Nephrotoxicity:
Assessment and pathogenesis, New York, Chichester, Brisbane, Toronto,
John Wiley and Sons.
GOYER, R.A. & CHERIAN, M.G. (1977) Tissue and cellular toxicology of
metals. In: Brown, S.S., ed. Clinical chemistry and chemical
toxicology of metals, Amsterdam, Elsevier-North Holland Biomedical
Press, pp. 89-103.
GOYER, R.A. & MOORE, J.F. (1974) Cellular effects of lead. Adv. exp.
Med. Biol., 48: 447-462.
GOYER, R.A. & RHYNE, B.C. (1973) Pathological effects of lead. Int.
Rev. exp. Pathol., 12: 1-77.
GOYER, R.A. & WILSON, M.H. (1975) Lead-induced inclusion bodies:
Results of EDTA treatment. Lab. Invest., 32: 149-156.
GOYER, R.A., FALK, H.L., HOGAN, M., FELDMAN, D.D., & RICHTER, W.
(1981) Renal tumors in rats given trisodium nitrilotriacetic acid in
drinking water for 2 years. J. Natl Cancer Inst., 66: 869-880.
GRANDCHAMP, A., AYER, G., SCHERRER, J.R., & TRUNIGER, B. (1971)
Intrarenal haemodynamics of the rat kidney determined by the Xenon
washout technique. Nephron, 8: 33-45.
GREEN, C.R., HAM, K.N., & TANGE, J.D. (1969) Kidney lesions induced in
rats by p-aminophenol. Br. med. J., 1: 162-164.
GREEN, R.M. & ELCE, J.S. (1975) Acetylation of S-substituted cysteines
by a rat liver and kidney microsomal N-acetyltransferase. Biochem. J.,
147: 283-289.
GREGG, N., ELSEVIERS, M.M., DEBROE, M.E., & BACH, P.H. (1989) The
epidemiological and mechanistic basis of analgesic associated
nephropathy. Toxicol. Lett., 46: 141-151.
GRITZKA, T.L. & TRUMP, B.F. (1968) Renal tubular lesions caused by
mercuric chloride. Am. J. Pathol., 52: 1225-1227.
GROTH, S., NIELSEN, H., SORENSEN, J.B., CHRISTENSEN, A.B., PEDERSEN,
A.G., & RORTH, M. (1986) Acute and long-term nephrotoxicity of
cisplatinum in man. Cancer Chemother. Pharmacol., 17: 191-196.
GRUNFELD, J.P., RAPHAEL, J.C., BANKIR, W., KLEINKNECHT, D., & BARGER,
A.C. (1971) Intrarenal distribution of blood flow. Adv. Nephrol., 1:
125-143.
GSTRAUNTHALER, G., PFALLER, W., & KOTANKO, P. (1985) Interrelation
between oxygen consumption and Na-K-ATPase activity in rat renal
proximal tubule suspension. Renal Physiol., 8: 38-44.
GUDER, W.G. & ROSS, B.D. (1984) Enzyme distribution along the nephron.
Kidney Int., 26: 101-111.
GUDER, W.G. & SCHMIDT, U. (1974) The localization of gluconeogenesis
in the rat nephron: determination of PEPCK in microdissected tubules.
Hoppe Seylers Z. Physiol. Chem., 355: 273-278.
GUENGERICH, F.P. & MASON, P.S. (1979) Immunological comparison of
hepatic and extrahepatic cytochromes P-450. Mol. Pharmacol., 15:
154-64.
GYRD-HANSEN, N. (1968) Renal clearances in pigs. Acta vet. Scand., 9:
183-198.
HACKENTHAL, E., SCHWERTSCHLAG, U., & SEYBERTH, H.W. (1980)
Prostaglandins and renin release studies in the isolated perfused rat
kidney. Prog. Biochem. Pharmacol., 17: 98-107.
HALDER, C.A., WARNE, T.M., & HATOUM, N.S. (1984) Renal toxicity of
gasoline and related petroleum naphthas in male rats. In: Mehlman,
M.A., Hemstreet, C.P., Thorpe, J.J., & Weaver, N.K., ed. Renal effects
of petroleum hydrocarbons, Princeton, New Jersey, Princeton Scientific
Publishers, pp. 73-88.
HALDER, C.A., HOLDSWORTH, C.E., & COOKRELL, B.Y. (1985) Hydrocarbon
nephropathy in male rats. Identification of the nephrotoxic components
of gasoline. In: Bach, P.H. & Lock, E.A., ed. Renal heterogeneity and
target cell toxicity, New York, Chichester, Brisbane, Toronto, John
Wiley and Sons, pp. 481-484.
HALES, B.F., JAEGER, V., & NEIMS, A.H. (1978) Isoelectric focusing of
glutathione S-transferases from rat liver and kidney. Biochem. J.,
175: 937-943.
HALL, C.L., FOTHERGILL, N.J., BLACKWELL, M.M., HARRISON, P.R.,
MACKENZIE, J.C., & MACIVER, A.G. (1987) The natural course of gold
nephropathy: long-term study of 21 patients. Br. med. J., 295:
745-748.
HALL, P.W., III (1982) Endemic Balkan nephropathy. In: Porter, G., ed.
Nephrotoxic mechanisms and environmental toxins, New York, London,
Plenum Press, pp. 227-240.
HALL, P.W., III & DAMMIN, G.J. (1978) Balkan nephropathy. Nephron, 22:
281-300.
HALL, R.L., WILKE, W.L., & FETTMAN, M.J. (1986) The progression of
adriamycin-induced nephrotic syndrome in rats and the effect of
captopril. Toxicol. appl. Pharmacol., 82: 164-174.
HAMILTON, J.M. (1975) Renal carcinogenesis. Adv. Cancer Res., 22:
1-56.
HAMMOND, P.B., LERNER, S.I., GARTSIDE, P.S., HANENSON, I.B., RODA,
S.B., FOULKES, E.C., JOHNSON, D.R., & PESCI, A.J. (1980) The
relationship of biological indices of lead exposure to the health
status of workers in a secondary lead smelter. J. occup. Med., 22:
475-484.
HANDLER, J.S. (1983) Use of cultured epithelia to study transport and
its regulation. J. exp. Biol., 106: 55-69.
HANSEN, H.E., HESTBECH, J., SORENSEN, J.L., NORGAARD, K., HEILSKOV,
J., & AMDISEN, A. (1979) Chronic interstitial nephritis in patients on
long-term lithium treatment. Q. J. Med., 48: 577-591.
HANSEN, H.E., MOGENSEN, C.E., SORENSEN, J.L., NORGAARD, K., HEILSKOV,
J., & ANDERESEN, A. (1981) Albumin and beta-2-microglobulin excretion
in patients on long-term lithium treatment. Nephron, 29: 229-232.
HARD, G.C. (1975) Autoradiographic analysis of proliferative activity
in rat kidney epithelial and mesenchymal cell subpopulations following
a carcinogenic dose of dimethylnitrosamine. Cancer Res., 35:
3762-3773.
HARD, G.C. (1979) Effect of age at treatment on incidence and type of
renal neoplasm induced in the rat by a single dose of
dimethylnitrosamine. Cancer Res., 39: 4965-4970.
HARD, G.C. (1980) Morphological correlates of irreversible tumour
formation. In: Holmstedt, B., Lauwerys, R., Mercier, M., & Roberfroid,
M., ed. Mechanisms of toxicity and hazard evaluation, Amsterdam,
Elsevier/North Holland Biomedical Press, pp. 231-240.
HARD, G.C., (1984) High frequency, single-dose model of renal
adenoma/carcinoma induction using dimethylnitrosamine in Crl:(W)BR
rats. Carcinogenesis, 5: 1047-1050.
HARD, G.C. (1985) Identification of a high frequency model for renal
carcinoma by the induction of renal tumors in the mouse with a single
dose of streptozotocin. Cancer Res., 45: 703-708.
HARD, G.C. (1986) Renal carcinogenesis, rat. In: Jones, T.C., Mohr,
U., & Hunt, R.D., ed. Monographs on pathology of laboratory animals,
urinary system, Berlin, Heidelberg, New York, Springer-Verlag, pp.
45-49.
HARD, G.C. (1987) Chemically induced epithelial tumours and
carcinogenesis of the renal parenchyma. In: Bach, P.H. & Lock, E.A.,
ed. Nephrotoxicity in the experimental and the clinical situation,
Dordrecht, Boston, Lancaster, Martinus Nijhoff Publishers, pp.
211-251.
HARD, G.C. & BUTLER, W.H. (1970) Cellular analysis of renal neoplasia:
induction of renal tumors in dietary-conditioned rats by
dimethylnitrosamine with a reappraisal of morphological
characteristics. Cancer Res., 30: 2796-2805.
HARD, G.C. & BUTLER, W.H. (1971) Morphogenesis of epithelial neoplasms
induced in the rat kidney by dimethylnitrosamine. Cancer Res., 31:
1496-1505.
HARD, G.C., MACKAY, R.L., & KOCHHAR, O.S. (1984) Electron microscopic
determination of the sequence of acute tubular and vascular injury
induced in the rat kidney by a carcinogenic dose of
dimethylnitrosamine. Lab. Invest., 50: 659-672.
HARKONEN, S. & KJELLSTRAND, C.M. (1979) Intravenous pyelography in
non-uremic diabetic patients. Nephron, 24: 268-270.
HARKONEN, S. & KJELLSTRAND, C.M. (1981) Contrast nephropathy. Am. J.
Nephrol., 1: 69-77.
HARRIS, C.C., WESTON, A., WILLEY, J.C., TRIVERS, G.E., & MANN, D.L.
(1987) Biochemical and molecular epidemiology of human cancer:
Indicators of carcinogen exposure, DNA damage, and genetic
predisposition. Environ. Health Perspect., 75: 109-119.
HARRIS, S.I., BALABAN, R.S., BARRETT, L., & MANDEL, L.J. (1981)
Mitochondrial respiratory capacity and Na+- and K+-dependent
adenosine triphosphatase-mediated ion transport in the intact renal
cell. J. biol. Chem., 256: 10319-10328.
HASSALL, C.D., GANDOLFI A.J., & BRENDEL K. (1983) Correlation of the
in vivo and in vitro renal toxicity of
S-(1,2-dichlorovinyl)-L-cysteine. Drug chem. Toxicol., 6: 507-520.
HAUGLUSTAINE, D., VAN DAMME, B., DAENENS, P., & MICHIELSEN, P. (1980)
Silicon nephropathy, a possible occupational hazard. Nephron, 26:
219-224.
HAWKE, R.L. & WELCH, R.M. (1985) Major differences in the specificity
and regulation of mouse renal cytochrome P-450 dependent
monooxygenases. Mol. Pharmacol., 28: 283-289.
HAYES, D.M., CVITKOVIC, E., GOLBERG, R.B., SCHEINER, E., HELSON, L.,
& KRAKOFF, I.H. (1977) High dose cis-platinum diammine chloride:
amelioration of renal toxicity by mannitol diuresis. Cancer, 39:
1372-1381.
HELLSTEN, S., BERGE, T., & LINELL, F. (1983) Clinically unrecognized
renal carcinoma. Aspects of tumor morphology, lymphatic and
haematogenous metastatic spread. Br. J. Urol., 55: 166-170.
HENNIS, H.L., ALLEN, R.C., HENNIGAR, G.R., & SIMMONS, M.A. (1981) A
sensitive method for determining the nephrotoxic effects of the
analgesic acetaminophen upon esterases using isoelectric focusing.
Electrophoresis, 2: 187-190.
HESTBECH, J., HANSEN, H.E., AMDISEN, A., & OLSEN, S. (1977) Chronic
renal lesions following long-term treatment with lithium. Kidney Int.,
12: 205-213.
HIASA, Y., OHSHIMA, M., IWATA, C., & TANIKAKE, T. (1979)
Histopathological studies on renal tubular cell tumours in rats
treated with N-ethyl-N-hydroxyethylnitrosamine. Gann, 70: 817-820.
HIASA, Y., OHSHIMA, M., KITAHORI, Y., FUJIYATA, T., YUASA, T., &
MIYASHIRO, A. (1983) Basic lead acetate: Promoting effect on the
development of renal tubular cell tumours in rats treated with
N-ethyl-N-hydroxyethyl-nitrosamine. J. Natl Cancer Inst., 70: 761-765.
HIASA, Y., ENOKI, N., KITAHORI, Y., KONISHI, N., & SHIMOYAMA, T.
(1984a) DL-serine: Promoting activity on renal tumorigenesis by
N-ethyl-N-hydroxyethylnitrosamine in rats. J. Natl Cancer Inst., 73:
297-299.
HIASA, Y., KITAHORI, Y., KONISHI, N., ENOKI, N., SHIMOYAMA, T., &
MIYASHIRO, A. (1984b) Trisodium nitrilotriacetate monohydrate:
promoting effects on the development of renal tubular cell tumors in
rats treated with N-ethyl-N-hydroxyethylnitrosamine. J. Natl Cancer
Inst., 72: 483-489.
HIASA, Y., LIN, J.-C., KONISHI, N., KITAHORI, Y., ENOKI, N., &
SHIMOYAMA, T. (1984c) Histopathological and biochemical analyses of
transplantable renal adenocarcinoma in rats induced by
N-ethyl-N-hydroxyethyl-nitrosamine. Cancer Res., 44: 1664-1670.
HINSON, J.A., MONKS, T.T., HONG, M., HIGHET, R.J., & POHL, L.R. (1982)
3-(Glutathion-S-yl)acetaminophen: a biliary metabolite of
acetamino-phen. Drug Metab. Disp., 10: 47-50.
HINTON, D.E., HEATFIELD, B.M., LIPSKY, M.M., & TRUMP, B.F. (1980)
Animal model: chemically induced renal tubular carcinoma in rats. Am.
J. Pathol., 100: 317-320.
HIRAFUJI, M., SATOH, S., & OGURA, Y. (1980) Sex difference in
stimulatory actions of cofactors on prostaglandin synthetase in
microsomes from rat kidney medulla. Biochem. Pharmacol., 29:
2635-2637.
HIRSCH, G.H. (1976) Differential effects of nephrotoxic agents on
renal transport and metabolism by use of in vitro techniques.
Environ. Health Perspect., 15: 89-99.
HJELLE, J.T., PETERSON, D.R., & HJELLE, J.J. (1983) Drug metabolism in
isolated proximal tubule cells: aldehyde dehydrogenase. J. Pharmacol.
exp. Ther., 224: 699-706.
HOOK, J.B., ed. (1981) Toxicology of the kidney, New York, Raven
Press.
HOOK, J.B., MCCORMACK, K.M., & KLUWE, W.M. (1979) Biochemical
mechanisms of nephrotoxicity. In: Hodgson, E., Bend, J.R., & Philpot,
R.M., ed. Reviews in biochemical toxicology, Amsterdam, Oxford, New
York, Elsevier Science Publishers, pp. 53-78.
HOOK, J.B., ELCOMBE, C.R., ROSE, M.S., & LOCK, E.A. (1982)
Characterization of the effects of known hepatic monooxygenase
inducers on male and female rat and mouse kidneys. Life Sci., 31:
1077-1084.
HOOK, J.B., ISHMAEL, J., & LOCK, E.A. (1983) Nephrotoxicity of
hexachloro-1:3-butadiene in the rat: the effect of age, sex and
strain. Toxicol. appl. Pharmacol., 67: 121-131.
HORNING, E.S. & WHITTICK, J.W. (1954) The histogenesis of
stilboestrol-induced renal tumours in the male golden hamster. Br. J.
Cancer, 8: 451-457.
HORROBIN, D.F. (1980) The regulation of prostaglandin biosynthesis:
negative feedback mechanisms and the selective control of formulation
of 1 and 2 series prostaglandin: relevance to inflammation and
immunity. Med. Hypotheses, 6: 687-709.
HOU, S.H., BUSHINSKY, D.A., WISH, J.B., COHEN, J.J., & HARRINGTON,
J.T. (1983) Harrington, Hospital-acquired renal insufficiency: a
prospective study. Am. J. Med., 74: 243-248.
HULT, K. & FUCHS, R. (1986) Analysis and dynamics of ochratoxin A in
biological systems. In: Steyn, P.S. & Vleggaar, R., ed. Mycotoxins and
phycotoxins. Sixth International IUPAC Symposium on Mycotoxins and
Phycotoxins, Pretoria, Republic of South Africa, 22-25 July, 1985,
Amsterdam, Oxford, New York, Elsevier Science Publishers, pp. 365-376.
HUMES, H.D., WEINBERG, J.M., & KNAUSS, T.C. (1982) Clinical and
pathophysiological aspects of aminoglycoside nephrotoxicity. Am. J.
kidney Dis., 2: 5-29.
IESATO, K., WAKASHIN, M., WAKASHIN, Y., & TOJO, S. (1977) Renal
tubular dysfunction in Minamata disease. Ann. intern. Med., 86:
731-737.
INOUE, G., SAWADA, T., & YOSHIKAWA, M. (1973) Age-related change in
acid mucopolysaccharides level and water content in papillae.
Gerontologia, 19: 73-78.
ITO, N., HIASA, Y., TAMAI, A., & YOSHIDA, K. (1969) Effect of
unilateral nephrectomy on the development of kidney tumor in rats
treated with N-nitro-sodimethylamine. Gann, 60: 319-327.
ITO, N., SUGIHARA, S., MAKIURA, S., ARAI, M., HIRAO, K., DENDA, A., &
NISHIO, O. (1974) Effect of N-(3,5-dichlorophenyl)succinimide on the
histological pattern and incidence of kidney tumors in rats induced by
dimethylnitrosoamine. Gann, 65: 131-138.
JACOBSON, A., GRIECO, A.J., & FARBER, S.J. (1964) Hexosamine analysis
of renal papillae in diuretic and antiduiretic rats. Proc. Soc. Exp.
Biol. Med., 115: 1153-1156.
JACOBSEN, B.K., BJELKE, E., KVALE, G., & HEUCH, I. (1986) Coffee
drinking, mortality and cancer incidence: results from a Norwegian
prospective study. J. Natl Cancer Inst., 76: 823-831.
JACQUES, P.J. (1975) The endocytic uptake of macromolecules. In:
Trump, B.F. & Arstilla, U.A., ed. Pathology of cell membranes, New
York, London, San Franciso, Academic Press, Vol. 1, pp. 225-276.
JAFFE, N., KEIFER, R., ROBERTSON, R., CANGIR, A., & WANG, A. (1987)
Renal toxicity with cumulative doses of
cis-diamminedichloroplatinum-II in pediatric patients with
osteosarcoma. Cancer, 59: 1577-1581.
JAKOBY, W.B. & PASTAN, I.H., ed. (1979) Cell culture, methods in
enzymology, New York, London, San Franciso, Academic Press, Vol. 58,
642 pp.
JASMIN, G. & RIOPELLE, J.L. (1976) Renal carcinomas and erythrocytosis
in rats following intrarenal injection of nickel subsulfide. Lab.
Invest., 35: 71-78.
JOHANSSON, S. & ANGERVALL, L. (1976) Urothelial hyperplasia of the
renal papillae in female Sprague-Dawley rats induced by long term
feeding of phenacetin. Acta pathol. microbiol. Scand. Sect. A., 84:
353-354.
JOHANSSON, S., ANGERVALL, L., BENGTSSON, U., & WAHLQVIST, L. (1976) A
clinicopathologic and prognostic study of epithelial tumors of the
renal pelvis. Cancer, 37: 1376-1383.
JOLLOW, D.J., KOCSIS, J.J., SNYDER, R., & VAINIO, H., ed. (1976)
Biological reactive intermediates. Formation, toxicity and
inactivation, New York, London, Plenum Press.
JONES, B.R., BHALLA, R.B., MLADEK, J., KALEYA, R.N., GRALLA, R.J.,
ALLOCK, N.N., SCHWARTZ, M.K., YOUNG, C.W., & KEIDENBERG, M.M. (1980)
Comparison of methods of evaluating nephrotoxicity of cis-platinum.
Clin. Pharmacol. Ther., 24: 557-562.
JONES, D.P., SUNDBY, G.-B., ORMSTAD, K., & ORRENIUS, S. (1979) Use of
isolated kidney cells for study of drug metabolism. Biochem.
Pharmacol., 28: 929-935.
JOSEPOVITZ, C., FARRUGGELLA, T., LEVINE, R., LANE, B., & KALOYANIDES,
G.J. (1985) Effect of netilmicin on the phospholipid composition of
subcellular fractions of rat renal cortex. J. Pharmacol. exp. Ther.,
235: 810-819.
JUNG, K.Y. & ENDOU, H. (1989) Nephrotoxicity assessment by measuring
cellular ATP content II. Intranephron site of ochratoxin A
nephrotoxicity. Toxicol. appl. Pharmacol., 100: 383-390.
JUNG, K.Y. & ENDOU, H. (1989) Biphasic increasing effect of
angiotensin II on intracellular free calcium in isolated rat early
proximal tubule. Biochem. Biophys. Res. Commun., 165: 1221-1228.
JUNG, K.Y., UCHIDA, S., & ENDOU. H. (1989) Nephrotoxicity assessment
by measuring cellular ATP content I. Substrate specificities in the
maintenance of ATP content in isolated rat nephron segments. Toxicol.
appl. Pharmacol., 100: 369-382.
KACEW, S. (1987) Detection of nephrotoxicity of foreign compounds with
the use of in vitro and in vivo techniques. In: Bach, P.H. & Lock,
E.A., ed. Nephrotoxicity in the experimental and the clinical
situation, Dordrecht, Boston, Lancaster, Martinus Nijhoff Publishers,
pp. 533-562.
KAHLMETER, G. & DAHLAGER, J.I. (1984) Aminoglycoside toxicity - a
review of clinical studies published between 1975 and 1982. J.
antimicrob. Chemother., 13(Suppl. A): 9-22.
KALOYANIDES, G.J. (1984a) Renal pharmacology of aminoglycoside
nephrotoxicity. Contrib. Nephrol., 42: 148-167.
KALOYANIDES, G.J. (1984b) Aminoglycoside induced functional and
biochemical defects in the renal cortex. Fundam. appl. Toxicol., 4:
930-943.
KALOYANIDES, G.G. & PASTORIZA-MUNOZ, E. (1980) Aminoglycoside
nephro-toxicity. Kidney Int., 18: 571-582.
KAMINSKY, L.S., FASCO, M.J., & GUENGERICH, F.P. (1979) Comparison of
different forms of liver, kidney and lung microsomal cytochrome P-450
by immunological inhibition of regio- and stereoselective metabolism
of warfarin. J. biol. Chem., 254: 9657-9662.
KANFER, A. (1989) The role of coagulation in glomerular injury.
Toxicol. Lett., 46: 83-92.
KANISAWA, M. & SUZUKI, S. (1978) Induction of renal and hepatic tumors
in mice by ochratoxin A, a mycotoxin. Gann, 69: 599-600.
KANWAR, Y.S. & FARQUHAR, M.G. (1979) Presence of heparan sulfate in
the glomerular basement membrane. Proc. Natl Acad. Sci., 76:
1303-1307.
KAPLOWITZ, N. (1980) Physiological significance of glutathione
S-transferases. Am. J. Physiol., 239: G439-G444.
KATZBERG, R.W., MORRIS T.W., & BURGENER, F.A. (1977) Renal renin and
hemodynamic responses to selective renal artery catheterization and
angiography. Invest. Radiol., 12: 381-388.
KATZBERG, R.W., PABICO, R.C., MORRIS, T.W., HAYAKAWA, K., MCKENNA,
B.A., PANNER, B.J., VENTURA, J.A., & FISHER, H.W. (1986) Effects of
contrast media on renal function and subcellular morphology in the
dog. Invest. Radiol., 21: 64-70.
KAUMP, D.H. (1966) Pharmacology of the fenamates: II. Toxicology in
animals. Ann. physiol. Med., Suppl. 1: 16-23.
KENNEDY, J.F. (1979) Proteoglycans: biological and chemical aspects in
human life, Amsterdam, Oxford, New York, Elsevier Scientific
Publishers.
KHOURY, G.A., HOPPER, J.C., VARGHESE, Z., FARRINGTON, K., DICK, R.,
IRVING, J.D., SWENY, P., FERNANDO, O.N., & MOORHEAD, J.F. (1983)
Nephrotoxicity of ionic and non-ionic contrast material in digital
vascular imaging and selective renal arteriography. Br. J. Radiol.,
56: 631-635.
KILHAM, L., LOW, R.J., CONTI, S.F., & DALLENBACH, F.D. (1962)
Intranuclear inclusions and neoplasms in the kidneys of wild rats. J.
Natl Cancer Inst., 29: 863-885.
KINCAID-SMITH, P. (1979) Analgesis nephropathy in Australia. Contrib.
Nephrol., 16: 57-64.
KINCAID-SMITH, P., BURROWS, G.D., DAVIES, B.M., HOLWILL, B., WALTER,
M., & WALKER, R.G. (1979) Renal biopsy findings in lithium and
prelithium patients. Lancet, 2(8144): 700-701.
KITCHEN, D.N. (1984) Neoplastic renal effects of unleaded gasoline in
Fischer 344 rats. In: Mehlman, M.A., Hemstreet, C.P., Thorpe, J.J., &
Weaver, N.K., ed. Renal effects of petroleum hydrocarbons, Princeton,
New Jersey, Princeton Scientific Publishers, pp. 65-71.
KJELLEN, P., OLDBERG, A., RUBIN, K., & HOOK, M. (1977) Binding of
heparin and heparan sulphate to rat liver cells. Biochem. biophys.
Res. Commun., 74: 126-133.
KJELLSTROM, T. & NORDBERG, G.F. (1978) The kinetic model of cadmium
metabolism in the human being. Environ. Res., 16: 248-269.
KJELLSTROM, T., SHIROISHI, K., & EVRIN, P. (1977) Urinary
beta2-microglobulin excretion among people exposed to cadmium in the
general environment. An epidemiological study in cooperation between
Japan and Sweden. Environ. Res., 13: 318-344.
KLEIN, K.L., WANG, M.S., TORIKAI, S., DAVISDON, W.D., & KUROKAWA, K.
(1981) Substrate oxidation by isolated single nephron segments of the
rat. Kidney Int., 20: 29-35.
KLEINKNECHT, D., KANFER, A., MOREL-MAROGER, L., & MERY, J.P. (1978)
Immunologically mediated drug-induced acute renal failure. Contrib.
Nephrol., 10: 42-52.
KLINTMALM, G.B.M., IWATSUKI, S., & STARZL, T.E. (1981) Nephro-toxicity
of cyclosporine in liver and kidney transplant recipients. Lancet,
1(8218): 470-471.
KLOSS, M.W., COX, M.G., NORTON, R.M., SWENBERG, J.A., & BUS, J.S.
(1985) Sex-dependent differences in the disposition of
14C-5-2,2,4-trimethylpentane in Fischer 344 rats. In: Bach, P.H. &
Lock, E.A., ed. Renal heterogeneity and target cell toxicity, New
York, Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 489-492.
KLUWE, W.M. (1983) Chemical modulation of 1,2-dibromo-3-chloropropane.
Toxicology, 27: 287-299.
KLUWE, W.M. & HOOK, J.B. (1978) Analysis of gentamicin uptake by rat
renal cortical slices. Toxicol. appl. Pharmacol., 45: 531-539.
KLUWE, W.M. & HOOK, J.B. (1980) Effects of environmental chemicals on
kidney metabolism and function. Kidney Int., 18: 648-655.
KLUWE, W.M., HERRMANN, C.L., & HOOK, J.B. (1979) Effects of dietary
polychlorinated biphenyls and polybrominated biphenyls on the renal
and hepatic toxicities of several chlorinated hydrocarbon solvents in
mice. J. Toxicol. environ. Health, 5: 605-615.
KLUWE, W.M., ABDO, K.M., & HUFF, J. (1984) Chronic kidney disease and
organic chemical exposures: evaluations of causal relationships in
humans and experimental animals. Fundam. appl. Toxicol., 4: 889-901.
KOCIBA, R.J., KEYES, D.G., JERSEY, G.C., BALLARD, J.J., DITTENBER,
D.A., QUAST, J.F., WADE, C.E., HUMISTON, C.G., & SCHWARTZ, B.A. (1977)
Results of a two year chronic toxicity study with hexachlorobutadiene
in rats. Am. Ind. Hyg. Assoc. J., 38: 589-602.
KOJIMA, S., HAGA, Y., KURIHARA, T., & YAMAWAKI, T. (1977) A comparison
between fecal cadmium and urinary beta2-microglobulin, total protein
and cadmium among Japanese farmers. Environ. Res., 14: 436-451.
KOSEK, J.D., MAZZE, R.I., & COUSINS, M.J. (1974) Nephrotoxicity of
gentamicin. Lab. Invest., 30: 48-57.
KOSEKI, C., YAMAGUCHI, Y., FURUSAWA, M., & ENDOU, H. (1988) Isolation
by monoclonal antibody of intercalated cells of rabbit kidney. Kidney
Int., 33: 543-554.
KOSTAKIS, A.J., WHITE, D.J.G., & CALNE, R.Y. (1977) Toxic effects in
the use of cyclosporin A in alcoholic solution as an immunosuppressant
of rat heart allografts. IRCS Surg. Transplant., 5: 243.
KRECH, R., ZERBAN, H., & BANNASCH, P. (1981) Mitochondrial anomalies
in renal oncocytes induced in rats by N-nitrosomorpholine. Eur. J.
cell Biol., 25: 331-339.
KREISBERG, J.I. & KARNOVSKY, M.J. (1983) Glomerular cells in culture.
Kidney Int., 23: 439-447
KREISBERG, J.I., PITTS, A.M., & PRETLOW, T.G. (1977) Separation of
proximal tubule cells from suspensions of rat kidney cells in density
gradients of ficoll in tissue culture medium. Am. J. Pathol., 86:
591-600.
KREISBERG, J.I., HOOVER, R.L., & KARNOVSKY, M.J. (1978) Isolation and
characterization of rat glomerular epithelial cells in vitro. Kidney
Int., 14: 21-30.
KRESSE, H. & GROSSMANN, A. (1970) [Comparative study of the
mucopolysaccharide and collagen content in various topographical areas
of the kidney in the rat, dog and pig.] Z. Klin. Chem., 8: 420-424 (in
German).
KRIJSHELD, K.R. & GRAM, T.E. (1984) Selective induction of renal
microsomal cytochrome P-450-linked monooxygenases by
1,1-dichloroethylene in mice. Biochem. Pharmacol., 33: 1951-1956.
KRIZ, W. & BANKIR, L. (1988) A standard nomenclature for structures of
the kidney, Am. J. Physiol., 254: F1-F8.
KROGH, P., HALD, B., PLESTINA, R., & CEOVIC, S. (1977) Balkan
(endemic) nephropathy and foodborne ochratoxin A: preliminary results
of a survey of foodstuffs. Acta pathol. microbiol. Scand. Sect. B, 85:
238-240.
KUHN, J.A., ARGY, W.R., HAKOWSKI, T.A., SCHREINER, G.E., & SCHEIN,
P.S. (1978) Nephrotoxicity of cis-diamminedichloroplatinum as measured
by urinary beta glucuronidase. Clin. Res., 26: 776.
KUO, C.-H. & HOOK, J.B. (1982) Depletion of renal glutathione content
and nephrotoxicity of cephaloridine in rabbits, rats and mice.
Toxicol. appl. Pharmacol., 63: 292-302.
KUO, C.-H. & HOOK, J.B. (1983) Effects of age and sex on
hexachloro-1,3-butadiene toxicity in the Fisher 344 rat. Life Sci.,
33: 517-523.
KUO, C.-H., BRASELTON, W.E., & HOOK, J.B. (1982) Effect of
phenobarbital on cephaloridine toxicity and accumulation in rabbit and
rat kidneys. Toxicol. appl. Pharmacol., 64: 244-254.
KUROKAWA, Y., HAYASHI, Y., MAEKAWA, A., TAKAHASHI, M., KOKUBO, T., &
ODASHIMA, S. (1983) Carcinogenicity of potassium bromate administered
orally to F344 rats. J. Natl Cancer Inst., 71: 965-972.
LANDRIGAN, P.J., GOYER, R.A., CLARKSON, T.W., SANDLER, D.P., SMITH,
J.H., THUN, M.J., & WEDEEN, R.P. (l984) The work-relatedness of renal
disease. Arch. environ. Health, 39: 225-230.
LANGARD, S. & NORSETH, T. (1986) Chromium. In: Friberg, L., Nordberg,
G., & Vouk, V.B., ed. Handbook on the toxicology of metals, Amsterdam,
Oxford, New York, Elsevier Science Publishers, Vol. II, pp. 185-210.
LAQUEUR, G.L. & SPATZ, M. (1968) Toxicology of cycasin. Cancer Res.,
28: 2262-2267.
LASH L.H. & ANDERS M.W. (1986) Cytotoxicity of
S-(1,2-dichlorovinyl)-glutathione and S-(1,2-dichlorovinyl)-L-cysteine
in isolated rat kidney cells. J. biol. Chem., 261: 13076-13081.
LASH, L.H., ELFARRA, A.A., & ANDERS, M.W. (1986) Renal cysteine
conjugate betalyase. Bioactivation of nephrotoxic cysteine
S-conjugates in mitochondrial outer membrane. J. biol. Chem., 261:
5930-5935.
LAURENT, G., CARLIER, M.-B., ROLLMAN, B., VANHOOF, F., & TULKENS, P.M.
1982) Mechanism of aminoglycoside-induced lysosomal phospholipidosis:
in vitro and in vivo studies with gentamicin and amikacin.
Biochem. Pharmacol., 31: 3861-3870.
LAURENT, G., TOUBEAU, G., HEUSON-STEINNON, J.A., TULKENS, P., &
MALDAUGE, P. (1988) Kidney tissue repair after nephrotoxic injury:
biochemical and morphologic characterization. CRC crit. Rev. Toxicol.,
19: 147-183.
LAUWERYS, R., ROELS, H., BERNARD, A., & BUCHET, J.-P. (1980) Renal
response to cadmium in a population living in a non-ferrous smelter
area in Belgium. Int. Arch. occup. environ. Health, 45: 271-274.
LEE, J.B. (1980) Prostaglandins and the renin-angiotensin axis. Clin.
Nephrol., 14: 159-163.
LEEMING, B.W.A., SPOKES, K.C., & SILVA, P. (1985) Effect of meglumine
iothalamate on renal hemodynamics and function in the diabetic rat.
Invest. Radiol., 20: 971-977.
LEVENSON, D.J., SIMMONS, C.E., & BRENNER, B.H. (1982) Arachadonic acid
metabolism, prostaglandins and the kidney. Am. J. Med., 72: 354-374.
LI, J.J. & LI, S.A. (1984) Estrogen-induced tumorigenesis in hamsters:
roles for hormonal and carcinogenic activities. Arch. Toxicol., 55:
110-118.
LI, J.J., LI, S.A., KLICKA, J.K., PARSONS, J.A., & LAM, L.K.T. (1983)
Relative carcinogenic activity of various synthetic and natural
estrogens in the Syrian hamster kidney. Cancer Res., 43: 5200-5204.
LILIS, R. (1981) Long-term lead exposure, chronic nephropathy, and
renal cancer: a case report. Am. J. ind. Med., 2: 293-297.
LIND, C., VADI, H., & ERNSTER, L. (1978) Metabolism of
benzo(a)pyrene-3,6-quinone and 3-hydroxybenzo(a)pyrene in liver
microsomes from 3-methyl-cholanthrene-treated rats. Arch. Biochem.
Biophys., 190: 97-108.
LINDBERG, E. & VESTERBERG, O. (1983) Urinary excretion of proteins in
chromeplaters, ex-chromeplaters and referents. Scand. J. Work Environ.
Health, 9: 505-510.
LIPPMAN, A.J., HELSON, C., HELSON, L., & KRAKOFF, I.H. (1973) Clinical
trials of cis-diamminedichloroplatinum (NSC-119875). Cancer Chemother.
Rep., 57: 191-200.
LIPPMANN, S. (1982) Lithium's effects on the kidney. Postgrad. Med.,
71: 99-104, 107-108.
LITTERST, C.L. & WEISS, R.B. (1987) Clinical and experimental
nephrotoxicity of cancer chemotherapeutic agents. In: Bach, P.H. &
Lock, E.A., ed. Nephrotoxicity in the experimental and clinical
situation, Dordrecht, Boston, Lancaster, Martinus Nijhoff Publishers,
pp. 771-816.
LITTERST, C.L., MIMNAUGH, E.G., REAGAN, R.L., & GRAM, T.E. (1975a)
Comparison of in vitro drug metabolism by lung, liver and kidney of
several common laboratory species. Drug Metab. Disp., 3: 259-265.
LITTERST, C.L., MIMNAUGH, E.G., REAGAN, R.L., & GRAM, T.E. (1975b)
Drug metabolism by microsomes from extrahepatic organs of rat and
rabbit prepared by calcium aggregation. Life Sci., 17: 813-818.
LITTERST, C.L., MIMNAUGH, E.G., & GRAM, T.E. (1977) Alterations in
extrahepatic drug metabolism by factors known to affect hepatic
activity. Biochem. Pharmacol., 26: 749-755.
LOCK, E.A. (1979) The effect of paraquat and diquat on renal function
in the rat. Toxicol. appl. Pharmacol., 48: 327-336.
LOCK, E.A. (1987) Metabolic activation of halogenated chemicals and
its relevance to nephrotoxicity. In: Bach, P.H. & Lock, E.A., ed.
Nephrotoxicity in the experimental and clinical situation, Dordrecht,
Martinus Nijhoff, pp. 429-461.
LOCK, E.A. & ISHMAEL, J. (1979) The acute toxic effects of paraquat
and diquat on the rat kidney. Toxicol. appl. Pharmacol., 50: 67-76.
LOCK, E.A., ISHMAEL, J., & HOOK, J.B. (1984) Nephrotoxicity of
hexachloro-1,3-butadiene in the mouse: the effect of age, sex, strain,
monooxygenase modifiers, and the role of glutathione. Toxicol. appl.
Pharmacol., 72: 484-494.
LOCK, E.A., ODUM, J., & ORMOND, P. (1986) Transport of
N-acetyl-S-pentachloro-1,3-butadienylcysteine by the renal cortex.
Arch. Toxicol., 59: 12-15.
LOCKARD, W.V., PHILLIPS, R.D., HAYES, A.W., BERNDT, W.O., & O'NEAL,
R.M. (1980) Citrinin nephrotoxicity in rats: A light electron
microscopic study. Exp. mol. Pathol., 32: 266-340.
LONG, W.F. & WILLIAMSON, F.B. (1979) Glycosaminoglycans, calcium ions
and the control of cell proliferation. IRCS med. Sci., 7: 429-434.
LOURY, D., SMITH-OLIVER, T., & BUTTERWORTH, B.E. (1987) Assessment of
the binding potential of 2,2,4-trimethylpentane to the rat
alpha-2u-globulin. Toxicol. appl. Pharmacol., 88: 44-56.
LUTZ, W.K, JAGGI, W., & SCHLATTER, C. (1982) Covalent binding of
diethylstilbestrol to DNA in rat and hamster liver and kidney.
Chem.-biol. Interact., 42: 251-257.
MACFARLAND, H.N., ULRICH, C.E., HOLDSWORTH, C.E., KITCHEN, D.N.,
HALLIWELL, W.H., & BLUM, S.C. (1984) A chronic inhalation study with
unleaded gasoline vapor. J. Am. Coll. Toxicol., 3: 231-248.
MCAULIFFE, W.G. (1978) The effects of anti-diuretic hormone on the
morphology and histochemistry of the renal medullary interstitium of
rats with hereditary hypothalmic diabetes insipidus (Brattleboro
strain). Anat. Rec., 190: 474.
MCAULIFFE, W.G. (1980) Histochemistry and ultrastructure of the
interstitium of the renal papilla in rats with hereditary diabetes
insipidus (Brattleboro strain). Am. J. Anat., 157: 17-26.
MCCONKEY, D.J., HARTZELL, P., DUDDY, S.K., HÅKANSSON, H., & ORRENIUS,
S. (1988) 2,3,7,8-Tetrachlorodibenzo-p-dioxin kills immature
thymocytes by Ca2+- mediated endonuclease activation. Science, 242:
256-259.
MCINTOSH, C.S., MOSELEY, I.F., FRY, I.K., & CATTELL, W.R. (1975)
Excretion urography toxicity studies in experimental acute renal
failure. Nephron, 14: 373-377.
MCKINNEY, L.L., WEAHLY, F.B., ELDRIDGE, A.C., CAMPBELL, R.E., COWAN,
J.C., PICKEN, J.C., Jr, & BIESTER, H.E. (1957)
S-(Dichlorovinyl)-L-cysteine: an agent causing total aplastic anemia
in calves. J. Am. Chem. Soc., 79: 3932-3933.
MCKINNEY, L.L., PICKEN J.C., Jr, WEAKLEY, F.B., ELDRIDGE, A.C.,
CAMPBELL, R.E., COWAN, J.C., & BIESTER, H.E. (1959) Possible toxic
factor of trichloroethylene-extracted soybean oil meal. J. Am. Chem.
Soc., 81: 909-915.
MCLACHLAN, J.R., GOYER, R.A., & CHERIAN, M.G. (1980) Formation of
lead-induced inclusion bodies in primary rat kidney epithelial cell
cultures. Toxicol. appl. Pharmacol., 56: 418-431.
MCLACHLAN, M.S.F., CHICK, S., ROBERTS, E.E., & ASSCHER, A.W. (1972)
Intravenous urography in experimental acute renal failure in the rat.
Invest. Radiol., 7: 466-473.
MCLEAN, A.E.M. & MAGEE, P.N. (1970) Increased renal carcinogenesis by
dimethylnitrosamine in protein deficient rats. Br. J. exp. Pathol.,
51: 587-590.
MCMICHAEL, A.J. & JOHNSON, H.M. (1982) Long term mortality profile of
heavily-exposed lead smelter workers. J. occup. Med., 24: 375-378.
MCMURTRY, R.J., SNODGRASS, W.R., & MITCHELL, J.R. (1978) Renal
necrosis, glutathione depletion and covalent binding after
acetaminophen. Toxicol. appl. Pharmacol., 46: 87-100.
MADIAS, N.E. & HARRINGTON, J.T. (1978) Platinum nephrotoxicity
(review). Am. J. Med., 65: 307-314.
MAEKAWA, A., KUROKAWA, Y., TAKAHASHI, M., KOKUBO, T., OGIU, T.,
ONODERA, H., TANIGAWA, H., OHNO, Y., FURUKAWA, F., & HAYASHI, Y.
(1983) Spontaneous tumors in F-344/DuCrj rats. Gann, 74: 365-372.
MAHAFFEY, J.R. (1980) Nutrient-lead interactions. In: Singhal, R.L. &
Thomas, J.A., ed. Lead toxicity, Baltimore, Maryland, Urban and
Schwarzenberg, pp. 425-460.
MALCOLM, D. & BARNETT, H.A.R. (1982) A mortality study of lead workers
1925-76. Br. J. ind. Med., 39: 404-410.
MALEQUE, A., ENDOU, H., KOSEKI, C., & SAKAI, F. (1980) Nephron
hetero-geneity: gluconeogenesis from pyruvate in rabbit nephron. FEBS
Lett., 116: 154-156.
MALIS, C.D. & BONVENTRE, J.V. (1986) Mechanism of calcium potentiation
of oxygen free radical injury to renal mitochondria. J. biol. Chem.,
261: 14201-14208.
MANDAL, A.K. & BOHMAN, S.O., ed. (1980) The renal papilla and
hypertension, New York, London, Plenum Press.
MAO, P. & MOLNAR, J.J. (1967) The fine structure and histochemistry of
lead induced renal tumors in rats. Am. J. Pathol., 50: 571-603.
MARKOVIC, B. (1972) Endemic nephritis and urinary tract cancer in
Yugoslavia, Bulgaria and Rumania. J. Urol., 107: 212-219.
MARRE, R., TARARA, N., & LOUTON, T. (1980) Age dependent
nephrotoxicity and the pharmacokinetics of gentamicin in rats. Eur. J.
Pediatr., 133: 25-29.
MARTIN, G., DUROZARD, D., & BAVEREL, G. (1987) Acceleration of
ammonia-genesis in isolated rat kidney tubules by the antiepileptic
drug: valproic acid. In: Kovacevic, Z. & Guder, W.G., ed. Molecular
nephrology. Biochemical aspects of kidney function, Berlin, New York,
Walter de Gruyter & Co., pp. 287-292.
MARTIN, G., MICHOUDET, C., & BAVEREL, G. (1989) Stimulation of
glutamine metabolism by the antiepileptic drug, sodium valproate, in
isolated dog kidney tubules. Biochem. Pharmacol., 38: 3947-3952.
MARTIN, G., DUROZARD, D., BESSON, J., & BAVEREL, G. (1990) Effect of
the antiepileptic drug, sodium valproate, on glutamine and glutamate
metabolism in isolated human kidney tubules. Biochim. Biophys. Acta,
1033: 261-266.
MATSUSAKA, T., FUJII, M., NAKANO, T., TERAI., T., KURATA, A.,
IMAIZUMI, M., & ABE, H. (1989) Germanium-induced nephropathy: Report
of two cases and review of the literature. Clin. Nephrol., 30:
341-345.
MATZKE, G., LUCAROTTI, R., & SHARPIRO, H.S. (1983) Controlled
comparison of gentamicin and tobramycin nephrotoxicity. Am. J.
Nephrol., 3: 11-17.
MAUNSBACH, A.N., OLSEN, T.S., & CHRISTENSEN, E.I., ed. (1980)
Functional ultrastructure of the kidney, New York, London, Academic
Press.
MAYER, D., WEBER, E., KADENBACH, B., & BANNASCH, P. (1989)
Immunocyto-chemical demonstration of cytochrome c oxidase as a marker
for renal oncocytes and oncocytomas. Toxicol. Pathol., 17: 46-49.
MAZZE, R.I. (1976) Methoxyflurane nephropathy. Environ. Health
Perspect., 15: 111-119.
MAZZE, R.I. (1981) Methoxyflurane nephropathy. In: Hook, J.B., ed.
Toxicology of the kidney, New York, Raven Press, pp. 135-149.
MEHLMANN, M.A., HEMSTREET, C.P., THORPE, J.J., & WEAVER, N.K., ed.
(1984) Renal effects of petroleum hydrocarbons, Princeton, New Jersey,
Princeton Scientific Publishers.
MEEZAN, E. & BRENDEL, K. (1973) Effect of ethacrynic acid on oxidative
metabolism in isolated glomeruli. J. Pharmacol. exp. Ther., 187:
352-364.
MEIJER, S., SLEIJFER, D., MULDER, N., SLUITER, W.J., MARRINK, J.,
SCHRAFFORD KOOPS, H., BROUWERS, T.M., OLDHOFF, J., VAN DER HEM, G.K.,
& MANDEMA, E. (1983) Some effects of combination chemotherapy with
cisplatin on renal function in patients with nonseminomatous
testicular carcinoma. Cancer, 51: 2035-2040.
METZLER, M. (1981) Studies on the mechanism of carcinogenicity of
diethylstilboestrol: role of metabolic activation. Food Cosmet.
Toxicol., 19: 611-615.
METZLER, M. & MCLACHLAN, J.A. (1978) Peroxidase-mediated oxidation, a
possible pathway for metabolic activation of diethylstilbestrol.
Biochem. biophys. Res. Commun., 85: 874-884.
MICHELS, L.D., DAVIDMAN, M., & KEANE, W.F. (1983) Adriamycin nephrotic
syndrome: glomerular haemodynamics and permselectivity. Kidney Int.,
23: 246.
MICHOUDET, C. & BAVEREL, G. (1987) Metabolism of acetaldehyde in human
and baboon renal cortex. FEBS Lett., 216: 113-117.
MIHATSCH, M.J. & KNUSLI, K. (1982) Phenacetin abuse and malignant
tumours. An autopsy study covering 25 years (1953-1977). Klin.
Wochenschr., 60: 1339-1349.
MIHATSCH, M.J., HOFER, H.O., GUTZWILER, F., BRUNNER, F.P., &
ZOLLINGER, H.U. (1980a) [Phenacetin abuse. I. Frequency, per capita
consumption and consequential costs.] Schweiz. med. Wochenschr., 110:
108-115 (in German).
MIHATSCH, M.J., SCHMIDLIN, P., BRUNNER, F.P. HOFER, H.O., SIX, P., &
ZOLLINGER, H.U. (1980b) [Phenacetin abuse. II. Chronic renal
insufficiency and autopsy material in Basel.] Schweiz. med.
Wochenschr., 110: 116-124 (in German).
MIHATSCH, M.J., MANZ, T., KNUSLI, C., HOFER, H.O., RIST, M., GUETG,
R., RUTISHAUSER, G., & ZOLLINGERS, H.U. (1980c) [Phenacetin abuse.
III. Malignant tumours of the urinary tract associated with phenacetin
abuse in Basel, 1963-1977.] Schweiz. med. Wochenschr., 110: 255-264
(in German).
MIHATSCH, M.J., HOFER, H.O., GUDAT, F., KNUSLI, C., TORHORST, J., &
ZOLLINGER, U. (1984) Capillary sclerosis of the lower urinary tract
and analgesic nephropathy. Clin. Nephrol., 20: 285-301.
MIHATSCH, M.J., THIEL, G., BASLER, V., RYFFEL, B., LANDSMANN, J., VON
OVERBECK, J., & ZOLLINGER, H.U. (1985) Morphologic patterns in
cyclosporin-treated renal transplant recipients. Transplant. Proc.,
17(Suppl. 1): 101-116.
MIHATSCH, M.J., RYFFEL, B., HERMLE, M., BRUNNER, F.P., & THIEL, G.
(1986) Morphology of cyclosporine nephrotoxicity in the rat. Clin.
Nephrol., 25(Suppl. 1): S2-S8.
MILLER, E.C. & MILLER, J.A. (1981) Searches for ultimate chemical
carcinogens and their reactions with cellular macromolecules. Cancer,
47: 2327-2345.
MILMAN, N. & GOTTLIEB, P. (1977) Renal function after high-dose
urography in patients with chronic renal insufficiency. Clin.
Nephrol., 7: 250-254.
MITCHELL, J.R., JOLLOW, D.J., POTTER, W.Z., DAVIS, D.C., GILLETTE,
J.R., & BRODIE, B.B. (1973) Acetaminophen-induced hepatic necrosis. I.
Role of drug metabolism. J. Pharmacol. exp. Ther., 187: 185-194.
MITCHELL, J.R., MCMURTRY, R.J., STATHAM, C.N., & NELSON, S.D. (1977)
Molecular basis for several drug-induced nephropathies. Am. J. Med.,
62: 518-526.
MITSUMORI, K., MAITA, K., SAITO, T., TSUDA, S., & SHIRASU, Y. (1981)
Carcinogenicity of methylmercury chloride in ICR mice: preliminary
note on renal carcinogenesis. Cancer Lett., 12: 305-310.
MODAN, B., BOICHIS, H., BOTT-KANNER, G., BARELL, V., BAR-HOACH, N., &
ELIAHOU, H.E. (l975) An epidemiological study of renal failure. l. The
need for maintenance dialysis. Am. J. Epidemiol., 101: 276-280.
MOFFAT, D.B. (1979) The mammalian kidney, Cambridge, New York,
Cambridge University Press.
MOFFAT, D.B. (1981) New ideas on the anatomy of the kidney. J. clin.
Pathol., 34: 1197-1206.
MOFFAT, D.B. (1982) Morphology of the kidney in relation to
nephrotoxicity -portae renales. In: Bach, P.H., Bonner, F.W., & Lock,
E.A., ed. Nephrotoxicity: Assessment and pathogenesis, New York,
Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 10-26.
MOHANDAS, J., DUGGIN, G.G., HORVATH, J.S., & TILLER, D.J. (1981a)
Regional differences in peroxidatic activation of paracetamol
(acetaminophen) mediated by cytochrome P-450 and prostaglandin
endoperoxide synthetase in rabbit kidney. Res. Commun. chem. Pathol.
Pharmacol., 34: 69-80.
MOHANDAS, J., DUGGIN, G.G., HORVATH, J.S., & TILLER, D.J. (1981b)
Metabolic oxidation of acetaminophen (paracetamol) mediated by
cytochrome P-450 mixed function oxidase and prostaglandin endoperoxide
synthetase in rabbit kidney. Toxicol. appl. Pharmacol., 61: 252-259.
MOHR, U., HAAS, H., & HILFRICH, J. (1974) The carcinogenic effects of
dimethylnitrosamine and nitrosomethylurea in European hamsters
( Cricetus cricetus L.). Br. J. Cancer, 29: 359-364.
MOLD, J.W. & STEIN, H.F. (1986) Sounding board: the cascade effect in
the clinical care of patients. New Engl. J. Med., 314: 512-514.
MOLDEUS, P., JONES, D.P., ORMSTAD, K., & ORRENIUS, S. (1978) Formation
and metabolism of a glutathione S-conjugate in isolated rat liver and
kidney cells. Biochem. biophys. Res. Commun., 83: 195-200.
MOORE, J.R., GOYER, R.A., & WILSON, M.H. (1973) Lead-induced inclusion
bodies: Solubility, amino acid content, and relationship to residual
acidic proteins. Lab. Invest., 29: 488-494.
MOORE, M.A., NAKAMURA, T., SHIRAI, T., & ITO, N. (1986)
Immuno-histochemical demonstration of increased glucose-6-phosphate
dehydrogenase in preneoplastic and neoplastic lesions induced by
propylnitrosamines in F 344 rats and Syrian hamsters. Gann, 77:
131-138.
MOREAU, J.F., DROZ, D., NOEL, L.H., LEIBOWITCH, J., JUNGERS, P., &
MICHEL, J.R. (1980) Tubular nephrotoxicity of water soluble iodinated
contrast media. Invest. Radiol., 15: S54-S60.
MOREL, F., CHABARDES, D., & IMBERT, M. (1976) Functional segmentation
of the rabbit distal tubule by microdetermination of hormone-dependent
adenylate cyclase activity. Kidney Int., 9: 264.
MORIN, J.P., VIOTTE, G., VANDERWALLE, A., VAN HOOF, F., TULKENS, P.,
& FILLASTRE, J.P. (1980) Gentamicin-induced nephrotoxicity: a cell
biology approach. Kidney Int., 18: 583-590.
MORITA, T., OITE, T., KIHARA, I., YAMAMOTO, T., HARA, M., NAKA, A., &
OHNO, S. (1980) Culture of isolated glomeruli from normal and
nephritic rabbits. I. Characterization of outgrowing cells. Acta
pathol. Jpn., 30: 917-926.
MORRISON, A.R. (1980) Prostaglandins and the kidney. Am. J. Med., 69:
171-173.
MOSTOFI, F.K., SESTERHENN, I.A., & SOBIN, L.H. (1981) Histological
typing of kidney tumours, Geneva, World Health Organization, 26 pp.
MUDGE, G.H. (1982) Analgesic nephropathy: renal drug distribution and
metabolism. In: Porter, G.A., ed. Nephrotoxic mechanisms of drugs and
environmental toxins, New York, London, Plenum Press, pp. 209-225.
MUDGE, G.H. (1985) Pathogenesis of nephrotoxicity: Pharmacological
principles. In: Bach, P.H. & Lock, E.A., ed. Renal heterogeneity and
target cell toxicity, New York, Chichester, Brisbane, Toronto, John
Wiley and Sons, pp. 1-12.
MUIRHEAD, E.E. & PITCOCK, J.A. (1980) Evidence on involvement of the
renal papilla in hypertension. In: Mandal, A.K. & Bohman, S.-O., ed.
The renal papilla and hypertension, New York, London, Plenum Press,
pp. 35-61.
MURPHY, G.P., MIRAND, E.A., JOHNSTON, G.S., SCHMIDT, J.D., & SCOTT,
W.W. (1966) Renal tumors induced by a single dose of
dimethylnitrosamine: morphologic, functional, enzymatic and hormonal
characterizations. Invest. Urol., 4: 39-56.
MURRAY, B.M., PALLER, M.S., & FERRIS, T.F. (1985) Effect of
cyclosporine administration on renal hemodynamics in conscious rats.
Kidney Int., 28: 767-774.
MURRAY, T. & GOLDBERG, M. (1975) Chronic interstitial nephritis:
Etiologic factors. Ann. intern. Med., 82: 453-459.
MUTTI, A. (1987) The study of urinary excretion of kidney antigens to
reveal early effects of exposure to exogenous chemicals. In: Foa', V.,
Emmett, E.A., Maroni, M., & Colombi, A., ed. Occupational and
environmental chemical hazards, Chichester, Ellis Horwood Limited, pp.
315-322.
MUTTI, A. (1989) Detection of renal diseases in humans: developing
markers and methods. Toxicol. Lett., 46: 177-191.
MUTTI, A., CAVATORTA, A., PEDRONI, C., BORGHI, A., GIAROLI, C., &
FRANCHINI, I. (1979) The role of chromium accumulation in the
relationship between airborne and urinary chromium in welders. Int.
Arch. occup. environ. Health, 43: 123-133.
MUTTI, A., LUCERTINI, S., VALCAVI, P.P., NERI, T.M., FORNARI, M.,
ALINOVI, R., & FRANCHINI, I. (1985) Urinary excretion of brush-border
antigen revealed by monoclonal antibody: early indicator of toxic
nephropathy. Lancet, 2(8461): 914-917.
MUTTI, A., ALINOVI, R., BERGAMASCHI, E., FORNARI, M., & FRANCHINI, I.
(1988) Monoclonal antibodies to brush-border antigens for the early
diagnosis of nephrotoxicity. Arch. Toxicol., Suppl. 12: 162-165.
NAKADA, J., YAMADA, H., & ENDOU, H. (1986a) Evidence that
alpha-1-adrenergic stimuli specifically increase gluconeogenesis of
the isolated proximal convoluted tubule in the rat. Renal Physiol., 9:
213-222.
NAKADA, J., MACHIDA, T., & ENDOU, H. (1986b) Nephrotoxicity of
cisplatin in rats. In: Tanabe, T., Hook, J.B., & Endou, H., ed.
Nephrotoxicity of antibiotics and immunosuppressants, Amsterdam,
Oxford, New York, Elsevier Science Publishers, pp. 179-182.
NANRA, R.S. (1980) Clinical and pathological aspects of analgesic
nephropathy. Br. J. clin. Pharmacol., 10: 359S-368S.
NANRA, R.S., STUART-TAYLOR, J., DELEON, A.H., & WHITE, K.H. (1978)
Analgesic nephropathy: etiology, clinical syndrome, and
clinicopatho-logic correlations in Australia. Kidney Int., 13: 79-92.
NASH, J.A., KING, L.J., LOCK, E.A., & GREEN, T. (1984) The metabolism
and disposition of hexachloro-1:3-butadiene in the rat and its
relevance to nephrotoxicity. Toxicol. appl. Pharmacol., 73: 124-137.
NAVRAN, S.S. & LOUIS-FERDINAND, R.T. (1975) P-Chloro-N-methylaniline
demethylation by rat kidney subcellular fractions. Res. Commun. chem.
Pathol. Pharmacol., 12: 713-721.
NELSON, S.D. (1982) Metabolic activation and drug toxicity. J. med.
Chem., 25: 753-765.
NEUHAUS, O.W. (1986) Renal reabsorption of low-molecular weight
proteins in adult male rats: alpha-2u globulin. Proc. Soc. Exp. Biol.
Med., 182: 531-539.
NEUHAUS, O.W., FLORY, W., BISWAS, N., & HOLLERMAN, C.E. (1981) Urinary
excretion of alpha2u-globulin and albumin by adult male rats following
treatment with nephrotoxic agents. Nephron, 28: 133-140.
NEWSON, G.D. & VUGRIN, D. (1987) Etiologic factors in renal cell
adeno-carcinoma. Semin. Nephrol., 7: 108-116.
NEWTON, J.F., KUO, C.H., GEMBORYS, M.W., MUDGE, G.H., & HOOK, J.B.
(1982) Nephrotoxicity of p-aminophenol: a metabolite of acetaminophen
in the Fischer 344 rat. Toxicol. appl. Pharmacol., 65: 336-344.
NEWTON, J.F., BAILIE, M.B., & HOOK, J.B. (1983a) Acetaminophen
nephro-toxicity in the rat. Renal metabolic activation in vitro.
Toxicol. appl. Pharmacol., 70: 433-444.
NEWTON, J.F., YOSHIMOTO, J., BERNSTEIN, J., RUSH, G.F., & HOOK, J.B.
(1983b) Acetaminophen nephrotoxicity in the rat. I. Strain differences
in nephrotoxicity and metabolism. Toxicol. appl. Pharmacol., 69:
291-306.
NICOLL, J.W., SWANN, P.F., & PEGG, A.E. (1975) Effect of
dimethylnitro-samine on persistence of methylated guanines in rat
liver and kidney DNA. Nature (Lond)., 254: 261-262.
NOGAWA, K., KOBAYASHI, E., & HONDA, R. (1979a) A study of the
relationship between cadmium concentrations in urine and renal effects
of cadmium. Environ. Health Perspect., 28: 161-168.
NOGAWA, K., ISHIZAKI, A., & KOBAYASHI, E. (1979b) A comparison between
health effects of cadmium and cadmium concentration in urine among
inhabitants of the Itai-Itai disease endemic districts. Environ. Res.,
18: 397-409.
NOGAWA, K., HONDA, R., KIDO, T., TSURITANI, I., YAMADA, Y., ISHIZAKI,
M., & YAMAYA, H. (1989) A dose-response analysis of cadmium in the
general environment with special reference to total cadmium intake
limit. Environ. Res., 48: 7-16.
NOGUEIRA, E. & BANNASCH, P. (1988) Cellular origin of rat renal
oncocytoma. Lab. Invest., 59: 337-343.
NOGUEIRA, E., KLIMEK, F., WEBER, E., & BANNASCH, P. (1989) Collecting
duct: origin of rat renal clear cell tumours. Virchows Arch. B Cell
Pathol., 57(5): 275-283.
NOMIYAMA, K. (1980) Recent progress and perspectives in cadmium health
effects studies. Sci. total Environ., 14: 199-232.
NONOGUCHI, H., UCHIDA, S., SHIIGAI, T., & ENDOU, H. (1985) Effect of
chronic metabolic acidosis on ammonia production from L-glutamine in
microdissected rat nephron segments. Pflugers Arch., 403: 229-235.
NONOGUCHI, H., TAKEHARA, Y., & ENDOU, H. (1986) Intra- and
inter-nephron heterogeneity of ammoniagenesis in rats: Effect of
chronic metabolic acidosis and potassium depletion. Pflugers Arch.,
407: 245-251.
NORBY L.H. & DIBONA, G.F. (1975) The renal vascular effects of
meglumine diatrizoate. J. Pharmacol. exp. Ther., 193: 932-940.
NTP (1983) Technical report on the carcinogenesis bioassay of
penta-chloroethane (CAS No. 76-01-7) in F344/N rats and B6c3F1 mice
(gavage studies), Research Triangle Park, North Carolina, US
Department of Health and Human Services, National Toxicology Program
(NIH Publication No. 83-1788).
NTP (1986) Technical report on the toxicology and carcinogenesis
studies of 1,4-dichlorobenzene (CAS No. 106-46-7) in F344/N rats and
B6c3F1 mice (gavage studies), Research Triangle Park, North Carolina,
US Department of Health and Human Services, National Toxicology
Program (NIH Publication No. 86-2575).
NTP (1987) Toxicology and carcinogenesis studies of
1,4-dichlorobenzene in F344/N rats and B6C3F1 mice, Research Triangle
Park, North Carolina, US Department of Health and Human Services,
National Toxicology Program (NTP TR 319) (NIH Publication No.
87-2575).
NTP (1988) NTP draft technical report on the toxicology and
carcinogenesis studies of ochratoxin A (CAS No. 303-47-9) in F344/N
rats (gavage studies), Research Triangle Park, North Carolina, US
Department of Health and Human Services, National Toxicology Program
(NIH Publication No. 88-2813).
NUYTS, G.D., ELSEVIERS, M.M., & DE BROE, M.E. (1989) Health impact of
renal disease due to nephrotoxicity. Toxicol. Lett., 46: 31-44.
OBATOMI, D.K. & PLUMMER, D.T. (1986) Oxygen uptake and glutamate
dehydro-genase release in isolated renal tubules from rat kidney.
Biochem. Soc. Trans., 14: 1281-1282.
OHI, G., YAMAMOTO, S., FUJINO, T., ITAI, Y., UENO, K., SAITO, H.,
INOBA, Y., NAKAJIMA, K., & SHIROISHI, K. (1982) Urinary
beta2-microglobulin does not serve as diagnostic tool for Minamata
disease. Arch. environ. Health, 37: 336-341.
OHMORI, T., HIASA, Y., MURATA, Y., & WILLIAMS, G.M. (1982) Gamma
glutamyl transpeptidase activity in carcinogen-induced epithelial
lesions of rat kidney. Gann, 73: 543-548.
OHMORI, T. & TABEI, R. (1983) Modulation of N-nitrosodimethylamine
kidney tumorigenesis by unilateral hydronephrosis and multiple
putrescine adminis-trations. J. Natl Cancer Inst., 71: 787-793.
OLORUNSOGO, O.O., MALOMO, S.O., & BABABUNMI, E.A. (1985)
Proto-nophoric properties of fluorinated arylalkylsulphonamides:
Observations with perfluidone. Biochem. Pharmacol., 34: 2945-2952.
OLSON, C.T., YU, K.O., & SERVE, M.P. (1986) Metabolism of nephrotoxic
cis-and trans-decalin in Fischer-344 rats. J. Toxicol. environ.
Health, 18: 285-292.
OLSON, M.J., GARG, B.D., MURTY, C.V.R., & ROY, A.K. (1987)
Accumulation of alpha2u-globulin in the renal proximal tubules of male
rats exposed to unleaded gasoline. Toxicol. appl. Pharmacol., 90:
43-51.
OMICHINSKI, J.G., BRUNBORG, G., SODERLUND, E.J., DAHL, J.E., BAUSANO,
J.A., HOLME, J.A., NELSON, S.D., & DYBING, E. (1987) Renal necrosis
and DNA damage caused by selectively deuterated and methylated analogs
of 1,2-dibromo-3-chloropropane in the rat. Toxicol. appl. Pharmacol.,
91: 358-370.
ORLOFF, J. & BERLINER, R.W., ed. (1973) Handbook of physiology.
Section 8: Renal physiology, Washington, DC, American Physiological
Society.
ORMSTAD, K. (1982) The use of isolated kidney cells, glomeruli and
subcellular fractions for assessing renal function. In: Bach, P.H.,
Bonner, F.W., Bridges, J.W., & Lock, E.A., ed. Nephrotoxicity:
Assessment and pathogenesis, New York, Chichester, Brisbane, Toronto,
John Wiley and Sons, pp. 161-168.
ORMSTAD, K. (1987) Metabolism of glutathione in the kidney. In: Bach,
P.H. & Lock, E.A., ed. Nephrotoxicity in the experimental and the
clinical situation, Dordrecht, Boston, Lancaster, Martinus Nijhoff
Publishers, pp. 405-428.
ORMSTAD, K., JONES, D.P., & ORRENIUS, S. (1981) Preparation and
characterisation of isolated kidney cells. Methods Enzymol., 77:
137-146.
ORTMANN, M., VIERBUCHEN, M., KOLLER, G., & FISCHER, R. (1988) Renal
oncocytoma. I. Cytochrome c oxidase in normal and neoplastic renal
tissue as detected by immunohistochemistry - a valuable aid to
distinguish oncocytomas from renal cell carcinomas. Virchows Arch. B
Cell Pathol., 56: 165-173.
OTTOSEN, P.D., SIGH, B., CHRISTENSEN, J., OLSEN, S., & CHRISTENSEN,
S. (1984) Lithium induced interstitial nephropathy associated with
chronic renal failure. Acta pathol. microbiol. immunol. Scand., 92(6):
447-54.
OZOLS, R.F., CORDEN, B.J., JACOB, J., WESLEY, M.N., OSTCHAGA, Y., &
YOUNG, R.C. (1984) High dose cisplatin in hypertonic saline. Ann.
intern. Med., 100: 19-24.
PALESTINE, A.G., AUSTIN, H.A., BALOW, J.E., ANTONOVYCH, T.T., SABNIS,
S.G., PREUSS, H.G., & NUSSENBLATT, R.B. (1986) Renal histo-pathologic
alterations in patients treated with cyclosporine for uveitis. New
Engl. J. Med., 314: 1293-1298.
PARKER, R.A., BENNETT, W.M., & PORTER, G.A. (1982) Animal models in
the study of aminoglycoside nephrotoxicity. In: Whelton, A. & Neu,
H.S., ed. The aminoglycosides: Microbiology, clinical use and
toxicology, New York, Basel, Marcel Dekker, pp. 235-267.
PEARSON, R.M. (1979) Methods for the assessment of the effects of
drugs on renal blood flow. Br. J. clin. Pharmacol., 7: 129-138.
PEPELJNJAK, S. & CVETNIC, Z. (1985) The mycotoxicological chain and
contamination of food by ochratoxin A in the nephropathic and
non-nephropathic areas in Yugoslavia. Mycopathologia, 90: 147-153.
PERANTONI, A. & BERMAN, J.J. (1979) Properties of Wilms' tumor line
(TuWi) and pig kidney line (LLC-PK1) typical of normal kidney tubular
epithelium. In Vitro, 15: 446-454.
PETERSON, C.D., COLLINS, A.J., HINES, J. M., BULLOCK, M.L., & KEANE,
W.F. (1981) Ethylene glycol poisoning: pharmacokinetics during therapy
with ethanol and hemodialysis. New Engl. J. Med., 304: 21-23.
PETKOVA-BOCHAROVA, T. & CASTEGNARO, M. (1985) Ochratoxin A
con-tamination of cereals in an area of high incidence of Balkan
endemic nephropathy in Bulgaria. Food Addit. Contam., 2: 267-270.
PHILLIPS R.D. & COCKRELL, B.Y. (1984a) Kidney structural changes in
rats following inhalation exposure to C10-C11 isoparaffinic solvent.
Toxicology, 33: 261-273.
PHILLIPS, R.D. & COCKRELL, B.Y. (1984b) Effect of certain light
hydrocarbons on kidney function and structure in male rats. In:
Mehlman, M.D., Hemstreet, C.O., Thrope, J.J., & Weaver, N.K., ed.
Renal effects of petroleum hydro-carbons, Princeton, New Jersey,
Princeton Scientific Publishers, pp. 89-105.
PHILLIPS, R.D. & EGAN, G.F. (1984a) Effect of C10-C11 isoparaffinic
solvent on kidney function in Fischer 344 rats during eight weeks of
inhalation. Toxicol. appl. Pharmacol., 73: 500-510.
PHILLIPS, R.D. & EGAN, G.F. (1984b) Subchronic inhalation exposure of
dearomatized white spirit and C10-C11 isoparaffinic hydrocarbon in
Sprague-Dawley rats. Fundam. appl. Toxicol., 4: 808-818.
PHILLIPS, R.D., HAYES, A.W., & BERNDT W.O. (1979) Disposition of
14C-citrinin in the rat. Toxicology, 12: 285-298.
PHILLIPS, R.D., HAYES, A.W., & BERNDT, W.O. (1980a) High pressure
liquid chromatographic analysis of citrinin and its application to
biological systems. J. Chromatogr., 190: 419-425.
PHILLIPS, R.D., HAYES, A.W., BERNDT, W.O., & WILLIAMS. W.L. (1980b)
Effects of citrinin on renal function and structure. Toxicology, 16:
123-137.
POLLACK, E.S. & HORM, J.W. (1980) Trends in cancer incidence and
mortality in the United States, 1969-76. J. Natl Cancer Inst., 64:
1091-1103.
POMMER, W., GLAESKE, G., & MOLZAHN, M. (1986) The analgesic problem in
the Federal Republic of Germany: analgesic consumption, frequency of
analgesic nephropathy and regional differences. Clin. Nephrol., 26:
273-278.
PORTER, G.A., ed. (1982) Nephrotoxic mechanisms of drugs and
environmental toxins, New York, London, Plenum Press.
PORTER, G.A. (1989) Risk factors for toxic nephropathies. Toxicol.
Lett., 46: 269-279.
PORTER, G.A. & BENNETT, W.M. (1989) Drug-induced renal effects of
cyclosporine, aminoglycoside antibiotics and lithium: extrapolation of
animal data to man. In: Bach, P.H. & Lock, E.A., ed. Nephrotoxicity:
Extrapolation from in vitro to in vivo, and animals to man, New
York, London, Plenum Press, pp. 147-170.
PORTER, N.A., WOLF, R.A., & WEENEN, H. (1980) The free radical
oxidation of polyunsaturated lecithins. Lipids, 15: 163-167.
POTTER, C.L., GANDOLFI, A.J., NAGLE, R.B., & CLAYTON, J.W. (1981)
Effects of inhaled chlorotrifluoroethylene and and hexafluoropropene
on the rat kidney. Toxicol. appl. Pharmacol., 59: 431-440.
POUNDS, J.G. (1984) Effect of lead intoxication on calcium-mediated
cell function: a review. Neurotoxicology, 5: 295-332.
POUNDS, J.G. & ROSEN, J.F. (1988) Cellular Ca2+ Homeostasis and
Ca2+-mediated cell processes as critical targets for toxicant
action: con-ceptual and methodological pitfalls. Toxicol. appl.
Pharmacol., 94: 331-341.
POUR, P., ALTHOFF, J., SALMASI, S.Z., & STEPAN, K. (1979) Spontaneous
tumors and common diseases in three types of hamsters. J. Natl Cancer
Inst., 63: 797-811.
POWELL, W.S. (1980) Distribution of prostaglandin - hydroxylases in
different tissues. Prostaglandins, 19: 701-710.
POWLES, R.L., BARRETT, A.J., CLINK, H., KAY, H.E.M., SLOANE, J., &
MCELWAIN, T.J. (1978) Cyclosporin A for the treatment of
graft-versus-host disease in man. Lancet, 2: 1327-1331.
PRESCOTT, L.F. (1982) Analgesic nephropathy: a reassessment of the
role of phenacetin and other analgesics. Drugs, 23: 75-149.
PRETLOW, T.G., II & PRETLOW, T.P., ed. (1982) Cell separation: Methods
and selected applications, New York, London, San Francisco, Academic
Press, Vol. 1.
PRETLOW, T.G., II & PRETLOW, T.P., ed. (1983) Cell separation: Methods
and selected applications, New York, London, San Francisco, Academic
Press, Vol. 2.
PRETLOW, T.G., II & PRETLOW, T.P., ed. (1984) Cell separation: methods
and selected applications, New York, London, San Francisco, Academic
Press, Vol. 3.
PRICE, R.G. (1982) Urinary enzymes, nephrotoxicity and renal disease.
Toxicology, 23: 99-134.
PRUEKSARITANONT, T., CHEN, M.-L., & CHIOU, W.L. (1984) Simple and
micro high-performance liquid chromatographic method for simultaneous
analysis of p-aminohippuric acid and iothalamate in biological fluids.
J. Chromatogr., 306: 89-97.
RAHIMI, A., EDMONDSON, R.P.S., & JONES, N.F. (1981) Effect of
radiocontrast media on kidney of patients with renal disease. Br. Med.
J., 282: 1194-1195.
RAKIETEN, N., GORDON, B.S., COONEY, D.A., DAVIS, R.D., & SCHEIN, P.S.
(1968) Renal tumorigenic action of streptozotocin (NSC-85998) in rats.
Cancer Chemother. Rep., 52: 563-567.
RAMSAMMY, L.S., JOSEPOVITZ, C., LING, K.-Y., LANE, B.P., &
KALOYANIDES, G.J. (1986) Effects of diphenyl-phenylenediamine on
gentamicin-induced lipid peroxidation and toxicity in rat renal
cortex. J. Pharmacol. exp. Ther., 238: 83-88.
RAMSAMMY, L.S., JOSEPOVITZ, C., LING, K.-Y., LANE, B.P., &
KALOYANIDES, G.J. (1987) Failure of inhibition of lipid peroxidation
by vitamin E to protect against gentamicin nephrotoxicity in the rat.
Biochem. Pharmacol., 36: 2125-2132.
RAMSAMMY, L.S., JOSEPOVITZ, C., & KALOYANIDES, G.J. (1988a) Gentamicin
inhibits agonist stimulation of the phosphatidylinositol cascade in
primary cultures of rabbit proximal tubular cells and in rat renal
cortex. J. Pharmacol. exp. Ther., 247: 989-996.
RAMSAMMY, L.S., JOSEPOVITZ, C., LANE, B., & KALOYANIDES, G.J. (1988b)
Failure of hydroxyl radical scavengers to protect against
gentamicin-induced acute renal failure in the rat. Fed. Am. Soc. Exp.
Biol. J., 2: 407A.
RAMSAMMY, L.S., JOSEPOVITZ, C., LANE, B., & KALOYANIDES, G.J. (1989a)
Effect of gentamicin on phospholipid metabolism in cultured rabbit
proximal tubular cells. Am. J. Physiol., 256: C204-C213.
RAMSAMMY, L.S., JOSEPOVITZ, C., LANE, B.P., & KALOYANIDES, G.J.
(1989b) Polyaspartic acid protects against gentamicin nephrotoxicity
in the rat. J. Pharmacol. exp. Ther., 250: 149-153.
RANSLEY, P.G. & RISDON, R.A. (1979) The pathogenesis of reflux
nephropathy. Contrib. Nephrol., 16: 90-97.
RAPP, N.S., ZENSER, T.V., BROWN, W.W., & DAVIS, B.B. (1980) Metabolism
of benzidine by a prostaglandin-mediated process in renal inner
medullary slices. J. Pharmacol. exp. Ther., 215: 401-406.
RASMUSSEN, H.H. & IBELS, L.S. (1982) Acute renal failure: multivarite
analysis of causes and risk factors. Am. J. Med., 73: 211-218.
RAVNSKOV, U. (1985) Possible mechanism of hydrocarbon-associated
glomerulo-nephritis. Clin. Nephrol., 23: 294-298.
RECKNAGEL, R.O. (1983) Carbon tetrachloride hepatotoxicity: status quo
and future prospects. Trends Pharmacol. Sci., March: 129-131.
REDDY, M.V., GUPTA, R.C., RANDERATH, E., & RANDERATH, K. (1984)
32P-Postlabeling test for covalent DNA binding of chemicals in
vivo: Application to a variety of aromatic carcinogens and methylating
agents. Carcinogenesis, 5: 231-243.
REED, D.J. & BEATTY, P.W. (1980) Biosynthesis and regulation of
glutathione: toxicological implications. In: Hodgson, E., Bend, J.R.,
& Philpot, R.M., ed. Reviews in biochemical toxicology, Amsterdam,
Oxford, New York, Elsevier Science Publishers, Vol. 2, pp. 213-241.
REED, J.R., WILLIAMS R.H., & LUKE, R.G. (1983) The renal hemodynamic
response to diatrizoate in normal and diabetic rats. Invest. Radiol.,
18: 536-540.
REICHERT, D., SPENGLER, V., ROMEN, W., & HENSCHLER, D. (1984)
Carcinogenicity of dichloroacetylene: an inhalation study.
Carcinogenesis, 5: 1411-1420.
RENNICK, G. (1978) Renal excretion of cationic drugs - Tubule
transport and metabolism. In: Fillastre, J.P., ed. Nephrotoxicity
interaction of drugs with membrane systems, mitochondria-lysosomes,
New York, Masson Publishing Co., pp. 1-41.
REZNIK, G., WARD, J.M., HARDISTY, J.F., & RUSSFIELD, A. (1979) Renal
carcinogenic and nephrotoxic effects of the flame retardant
tris(2,3-dibromopropyl) phosphate in F344 rats and (C57BL/6N X
C3H/HeN)F1 mice. J. Natl Cancer Inst., 63: 205-212.
RICHMAN, A.V., MASCO, H.L., RIFKIN, S.I., & ACHARYA, M.K. (1980)
Minimal-change disease and the nephrotic syndrome associated with
lithium therapy. Ann. intern. Med., 92: 70-72.
RIMPELA, A.H. & PUKKALA, E.I. (1987) Cancers of affluence: positive
social class gradient and rising incidence trend in some cancer forms.
Soc. Sci. Med., 24: 601-606.
RITCHIE, A.W.S., KEMP, I.W., & CHISHOLM, G.D. (1984) Is the incidence
of renal carcinoma increasing? Br. J. Urol., 56: 571-573.
ROBBINS, M.E.C., CAMPLING, D., WHITEHOUSE, E., HOPEWELL, J.W., &
MICHALOWSKI, A. (1990) Cisplatin-induced reductions in renal reserve
uncovered by unilateral nephrectomy: an experimental study in the pig.
Cancer Chemother. Pharmacol., 27: 211-218.
ROCH-RAMEL, F. & PETERS, G. (1979) Micropuncture techniques as a tool
in renal pharmacology. Annu. Rev. Pharmacol. Toxicol., 19: 323-345.
ROELS, H., LAUWERYS, R., BUCHET, J.P., BERNARD, A., CHETTLE, D.R.,
HARVEY, T.C., & AL HADDAD, I.K. (1981a) In vivo measurement of liver
and kidney cadmium in workers exposed to this metal. Environ. Res.,
26: 217-240.
ROELS, H., LAUWERYS, R., BUCHET, J.P., & BERNARD, A. (1981b)
Environmental exposure to cadmium and renal function of aged women in
three areas of Belgium. Environ. Res., 24: 117-130.
ROELS, H., DJUBGANG, J., BUCHET, J.P., BERNARD, A., & LAUWERYS, R.
(1982) Evolution of cadmium-induced renal dysfunction in workers
removed from exposure. Scand. J. Work Environ. Health, 8: 191-200.
ROELS, H., GENNART, J.P., LAUWERYS, R., BUCHET, J.P., MALCHAIRE, J.,
& BERNARD, A. (1985) Surveillance of workers exposed to mercury
vapour: validation of a previously proposed biological threshold limit
value for mercury concentration in urine. Am. J. ind. Med., 7: 45-71.
ROELS, H., LAUWERYS, R., BUCHET, J.P., BERNARD, A., VOS, A., &
OVERSTEYNS, M. (1989) A prospective study of proteinuria in cadmium
workers. In: Bach, P.H. & Lock, E.A., ed. Nephrotoxicity:
Extrapolation from in vitro to in vivo, and animals to man, New
York, London, Plenum Press, pp. 33-36.
ROMAN-FRANCO, A.A., TWIRELLO, M., ALBINI, B., OSSI, E., MILGROM,
F., & ANDRES, G.A. (1978) Anti-basement membrane antibodies and
antigen-antibody complexes in rabbits injected with mercuric chloride.
Clin. Immunol. Immunopathol., 9: 404-481.
ROSENBERG, M.R. & MICHALOPOULOS, G. (1987) Kidney proximal tubular
cells isolated by collagenase perfusion grow in defined media in the
absence of growth factors. J. cell Physiol., 131: 107-113.
ROSENBERG, M.R., NOVICKI, D.L., JIRTLE, R.L., NOVOTNY, A., &
MICHALOPOULOS, G. (1985) Promoting effect of nicotinamide on the
development of renal tubular cell tumours in rats initiated with
diethylnitrosamine. Cancer Res., 45: 809-814.
ROSENMAN, K.D., VALCIUKAS, J.A., GLICKMAN, L., MYERS, B.R., & CINOTTI,
A. (1986) Sensitive indicators of inorganic mercury toxicity. Arch.
environ. Health, 41: 208-215.
ROSENMANN, E., DISHON, T., DURST, A., & BOSS, J.H. (1971) Urinary
excretion of kidney antigens in experimental renal disease of the rat.
Br. J. exp. Pathol., 52: 388-394.
ROSS, B., TANGE, J., EMSLIE, K., HART, S., SMAIL, M., & CALDER, I.
(1980) Paracetamol metabolism by the isolated perfused rat kidney.
Kidney Int., 18: 562-570.
ROSSOF, A.H., SLAYTON, R.E., & PERLIA, C.P. (1972) Preliminary
clinical experience with cis-diamminedichloroplatinum (NSC-119875
CACP). Cancer, 30: 1451-1456.
ROXE, D.M. (1980) Toxic nephropathy from diagnostic and therapeutic
agents. Am. J. Med., 69: 759-766.
RUSH, G.F., SMITH, J.H., NEWTON, J.F., & HOOK, J.B. (1984) Chemically
induced nephrotoxicity: role of metabolic activation. CRC crit. Rev.
Toxicol., 13: 99-160.
RYFFEL, B., DONATSCH, P., MADORIN, M., MATTER, B.E., RUTTIMAN, G.,
SCHON, H., STOLL, R., & WILSON, J. (1983) Toxicological evaluation of
cyclosporin A. Arch. Toxicol., 53: 107-141.
RYFFEL, B., HIESTAND, P., FOXWELL, B., DONATSCH, P., BOELSTERLI, H.J.,
MAURER, G., & MIHATSCH, M.J. (1986) Nephrotoxic and immunosuppressive
potentials of cyclosporine metabolites in rats. Transplant. Proc.,
18(Suppl. 5): 41-45.
SAFIRSTEIN, R., WINSTON, J., GOLDSTEIN, M., MOEL, D., DICKMAN, S., &
GUTTENPLAN, J. (1986) Cisplatin nephrotoxicity. Am. J. kidney Dis., 8:
356-367.
SAITO, H., SHIOJI, R., HURUKAWA, Y., NAGAI, K., ARIKEWA, T., SAITO,
T., SASAKI, Y., FURUYAMA, T., & YOSHINAGA, K. (1977) Cadmium-induced
proximal tubular dysfunction in a cadmium polluted area. Contrib.
Nephrol., 6: 1-12.
SANDLER, D.P. (1987) Epidemiology in the assessment of nephrotoxicity
In: Bach, P.H. & Lock, E.A., ed. Nephrotoxicity in the experimental
and clinical situation. Part 2, Dordrecht, Boston, Lancaster, Martinus
Nijhoff Publishers, pp. 847-883.
SANDLER, D.P., SMITH, J.C., WEINBERG, C.R., BUCKALEW, V.M., DENNIS,
V.W., BLYTHE, W.B., & BURGESS, W.P. (1989) Analgesic use and chronic
renal disease. New Engl. J. Med., 320: 1238-1243.
SANDSTEAD, H.H., MICHELAKIS, A.M., & TEMPLE, T.E. (1970) Lead
intoxication. Its effect on the renin-aldosterone response to sodium
deprivation. Arch. environ. Health, 20: 356-363.
SASTRASINH, M., KNAUSS, T.C., WEINBERG, J.M., & HUMES, H.D. (1982)
Identification of the aminoglycoside binding site in rat renal brush
border membranes. J. Pharmacol. exp. Ther., 222: 350-358.
SATO, G. & REID, L. (1978) Replacement of serum in cell culture by
hormones. Int. Rev. Biochem., 20: 219-251.
SCHENTAG, J.J. (1983) Specificity of renal tubular damage criteria for
aminoglycoside nephrotoxicity in critically ill patients. J. clin.
Pharmacol., 23: 473-483.
SCHENTAG, J.J., SUFTEN, T.A., & PLAUT, M.E. (1978) Early detection of
aminoglycoside nephrotoxicity with urinary beta-2-microglobulin. J.
Med., 9: 201-210.
SCHERBERICH, J.E., FALKENBERG, F., MONDORF, W., PFLEIDERER, G., &
SCHEOPPE, W. (1976) Isolation of kidney tissue antigens from urine by
biospecific immunosorbents. Verh. Dtsch. Ges. Inn. Med., 82:
1469-1473.
SCHERBERICH, J.E., MONDORF, W., FALKENBERG, F.W., PIERARD, D., &
SCHOEPPE, W. (1984) Monitoring drug nephrotoxicity. Quantitative
estimation of human kidney brush border antigens in urine as a
specific marker of tubular damage. Contrib. Nephrol., 42: 81-92.
SCHMIDT, U. & GUDER, W.G. (1976) Sites of enzyme activity along the
nephron. Kidney Int., 9: 233-242.
SCHOLER, D.W. & EDELMAN, I.S. (1979) Isolation of rat kidney cortical
tubules enriched in proximal and distal segments. Am. J. Physiol.,
237: F350-F359.
SCHRIER, R.W. & GOTTSCHALK, C.W. (1987) Diseases of the kidney,
Boston, Little, Brown and Co.
SCHULTZ, S.G., LAVELLE K.J., & SWAIN, R. (1982) Nephrotoxicity of
radiocontrast media in ischemia renal failure in rabbits. Nephron, 32:
113-117.
SCHUMANN, G.B., LERNER, S.I., WEISS, M.A., GAWRONSKI, L., & LOHIYA,
G.K. (1980) Inclusion-bearing cells in industrial workers exposed to
lead. Am. J. clin. Med., 74: 192-196.
SCHWARTZ, S.L., JOHNSON, C.B., & DOOLAN, P.D. (1970) Study of the
mechanism of renal vasculogenesis induced in the rat by EDTA.
Comparison of the cellular activities of calcium and chromium. Mol.
Pharmacol., 6: 54-59.
SCHWARZ, A. (1987) Analgesic-associated nephropathy. Klin.
Wochenschr., 65: 1-16.
SCHWERTZ, D.W., KREISBERG, J.I., & VENKATACHALEM, M.A. (1984) Effects
of aminoglycosides on proximal tubule brush border membrane
phosphatidylinositol-specific phospholipase C. J. Pharmacol. exp.
Ther., 231: 48-55.
SELDIN, D.D. & GIEBISCH, G. (1985) The kidney, physiology and
pathophysiology, New York, Raven Press, Vol. 1.
SELEVAN, S.G., LANDRIGAN, P.J., STERN, F.B., & JONES, J.H. (1985)
Mortality of lead smelter workers. Am. J. Epidemiol., 122: 673-683.
SELLI, C., HINSHAW, W.M., WOODARD, B.H., & PAULSON, D.F. (1983)
Stratification of risk factors in renal cell carcinoma. Cancer, 52:
899-903.
SHINOHARA, Y., ARAI, M., HIRAO, K., SUGIHARA, S., NAKANISHI, K.,
TSUNODA, H., & ITO, N. (1976) Combination effect of citrinin and other
chemicals on rat kidney tumorigenesis. Gann, 67: 147-155.
SHIRACHI, D.Y., JOHANSEN, M.G., MCGOWAN, J.P., & TU, S.-H. (1983)
Tumorigenic effect of sodium arsenite in rat kidney. Proc. West.
Pharmacol. Soc., 26: 413-415.
SHIRAI, T., OHSHIMA, M., MASUDA, A., TAMANO, S., & ITO, N. (1984)
Promotion of 2-(ethylnitrosamino)ethanol-induced renal carcinogenesis
in rats by nephrotoxic compounds: positive responses with folic acid,
basic lead acetate, and N-(3,5-dichlorophenyl)succinimide but not with
2,3-dibromo-1-propanol phosphate. J. Natl Cancer Inst., 72: 477-482.
SHIROISHI, K., KJELLSTROM, T., KUBOTA, K., EVRIN, P.E., ANAYAMA, M.,
VESTERBERG, O., SHIMADA, T., PISCATOR, M., IWATA, T., & NISHINO, H.
(1977) Urine analysis for detection of cadmium-induced renal changes,
with special reference to beta2-microglobulin. Environ. Res., 13:
407-424.
SHORT, B.G., BURNETT, V.L., & SWENBERG, J.A. (1986) Histopathology and
cell proliferation induced by 2,2,4-trimethylpentane in the male rat
kidney. Toxicol. Pathol., 14: 194-203.
SIEGERS C.-P. & KLAASSEN, C.D. (1984) Biliary excretion of
acetaminophen in ureter-ligated rats. Pharmacology, 28: 177-180.
SIERSBAEK-NIELSON, K., MOLHOLM HANSEN, J.M., KAMPMANN, J., &
KRISTENSEN, M. (1971) Rapid evaluation of creatinine clearance.
Lancet, 1: 1133-1134.
SIKIC, B.I., MIMNAUGH, E.G., LITTERST, C.L., & GRAM, T.E. (1977) The
effects of ascorbic acid deficiency and repletion on pulmonary, renal
and hepatic drug metabolism in the guinea-pig. Arch. Biochem.
Biophys., 179: 663-671.
SILVERBLATT, F.J. & KUEHN, C. (1979) Autoradiography of gentamicin
uptake by the rat proximal tubule cell. Kidney Int., 15: 335-345.
SMITH, H.J., LEVORSTAD, K., BERG, K.J., ROOTWELT, K., & SVEEN, K.
(1985) High dose urography in patients with renal failure. A double
blind investigation of iohexol and metrizoate. Acta radiol. Diagn.,
26: 213-220.
SMITH, J.H. & HOOK, J.B. (1984) Mechanism of chloroform
nephrotoxicity. III. Renal and hepatic microsomal metabolism of
chloroform in mice. Toxicol. appl. Pharmacol., 73: 511-524.
SMITH, J.H., MAITA, K., SLEIGHT, S.D., & HOOK, J.B. (1983) Mechanism
of chloroform nephrotoxicity. I. Time course of chloroform toxicity in
male and female mice. Toxicol. appl. Pharmacol., 70: 467-479.
SMITH, J.H., MAITA, K., SLEIGHT, S.D., & HOOK, J.B. (1984) Effect of
sex hormone status on chloroform nephrotoxicity and renal mixed
function oxidases in mice. Toxicology, 30: 305-316.
SMITH, J.H., RUSH, G.F., & HOOK, J.B. (1986) Induction of renal and
hepatic mixed function oxidases in the hamster and guinea-pig.
Toxicology, 38: 209-218.
SMITH, R.L. (1974) The excretory function of bile: the elimination of
drugs and toxic substances in bile - factors affecting biliary
excretion, London, Chapman & Hall, pp. 16-34.
SNYDER, R., PARKE, D., KOCSIS, J.J., JOLLOW, D.J., & GIBSON, G.G., ed.
(1981) Biological reactive intermediates. Part II, New York, London,
Plenum Press.
SOBERON, L., BOWMAN, R.L., PASTORIZA-MUNOZ, E., & KALOYANIDES, G.J.
(1979) Comparative nephrotoxicities of gentamicin, netilmicin and
tobramycin in the rat. J. Pharmacol. exp. Ther., 210: 334-343.
SOOSE, M., HABERSTROH, U., ROVIRA-HOLBACH, G., BRUNKHORST, R., &
STOLTE, H. (1988) Heterogeneity of glomerular barrier function in
early adriamycin nephrosis of MWF rats. Clin. Physiol. Biochem., 6:
310-315.
SPATARO, R. (1984) New contrast agents for urography. Radiol. Clin.
North Am., 22: 365-380.
SRAER, J., FOIDART, J., CHANSEL, D., MAHIEU, P., & ARDAILLOU, R.
(1980) Prostaglandin synthesis by rat isolated glomeruli and
glomerular cultured cells. Int. J. Biochem., 12: 203-207.
STERNBERG, S.S., PHILIPS, F.S., & CRONIN, A.P. (1972) Renal tumors and
other lesions in rats following a single intravenous injection of
Daunomycin. Cancer Res., 32: 1029-1036.
STOLTE, H. & ALT, J., ed. (1980) Research animals and experimental
design in nephrology. Contrib. Nephrol., 19: 1-249.
STOLTE, H. & ALT, J. (1982) The choice of animals for nephrotoxic
investigations. In: Bach, P.H., Bonner, F.W., Bridges, J.W., & Lock,
E.A., ed. Nephrotoxicity: Assessment and pathogenesis, New York,
Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 102-112.
STONARD, M.D., CHATER, B.V., DUFFIELD, D.P., NEVITT, A.L., O'SULLIVAN,
J.J., & STEEL, G.T. (1983) An evaluation of renal function in workers
occupationally exposed to mercury vapour. Int. Arch. occup. environ.
Health, 52: 177-189.
STONARD, M.D., PHILLIPS, P.G.N., FOSTER, J.R., SIMPSON, M.G. & LOCK,
E.A. (1986) Alpha-2u-globulin: Measurement in rat kidney following
administration of 2,2,4-trimethylpentane. Toxicology, 41: 161-168.
STRIKER, G.E., KILLEN, P.D., & FARIN, F.M. (1980) Human glomerular
cells in vitro: Isolation and characterization. Transplant. Proc.,
12: 88-99.
STUVE, J. & GALLE, P. (1970) Role of mitochondria in the handling of
gold by the kidney. J. cell. Biol., 44: 667-676.
SUFRIN, G. & BECKLEY, S.A. (1980) Renal adenocarcinoma, Geneva,
International Union Against Cancer, pp. 133-155 (Technical Report No.
49).
SUN, C.N. (1980) Effect of anti-diuretic hormone to the connective
tissue of rat renal papilla. Exp. Pathol., 18: 469-473.
SUN, C.N., WHITE, H.J., & TOWBIN, E.J. (1972) Histochemistry and
electron microscopy of renal papilla in a genetic strain of rats with
diabetic insipidus. Nephron, 9: 308-317.
SUNDERMAN, F.W., Jr (1981) Present research on nickel carcinogenesis.
Environ. Health Perspect., 40: 131-142.
SUNDERMAN, F.W., MCCULLY, R.S., & HOPFER, S.M. (1984) Association
between erythrocytosis and renal cancers in rats following intrarenal
injection of nickel compounds. Carcinogenesis, 5: 1511-1517.
SUZUKI, S., SUZUKI, S., NAKAMURA, N., & KOIZUMI, T. (1976) The
heterogeneity of dermatan sulfate and heparan sulfate in rat liver and
a shift in the glycosaminoglycan contents in carbon tetrachloride
damaged liver. Biochim. Biophys. Acta, 428: 166-181.
SUZUKI, S., SHAPIRO, R., MULROW, P.J., & TAN, S.Y. (1980) Urinary
prostaglandin E2 excretion in chronic renal disease. Prostaglandins
Med., 4: 377-382.
SWANN, P.F., KAUFMAN, D.G., MAGEE, P.N., & MACE, R. (1980) Induction
of kidney tumours by a single dose of dimethylnitrosamine: dose
response and influence of diet and benzo(a)pyrene pretreatment. Br. J.
Cancer, 41: 285-294.
SWENBERG, J.A., SHORT, B., BORGHOFF, S., STRASSER, J., & CHARBONNEAU,
J. (1989) The comparative pathobiology of alpha-2u-globulin
nephropathy. Toxicol. appl. Pharmacol., 97: 35-46.
SZEFLER, S.J. & ACARA, M. (1979) Isoproterenol excretion and
metabolism in the isolated perfused rat kidney. J. Pharmacol. exp.
Ther., 210: 295-300.
TALIERCIO, C.P., VLIETSTRA, R.E., FISHER, L.D., & BURNETT, J.C. (1986)
Risks for renal dysfunction with cardiac angiography. Ann. intern.
Med., 104: 501-504.
TAMURA, T. & ENDOU, H. (1988) Contribution of purine nucleotide cycle
to intranephron ammoniagenesis in rats. Am. J. Physiol., 255:
F1122-F1127.
TANAKA, H., ISHIKAWA, E., TESHIMA, S., & SHIMUZI, E. (1986)
Histopathological study of human cisplatin nephropathy. Toxicol.
Pathol., 14: 247-257.
TARLOFF, J.B., GOLSTEIN, R.S., & HOOK, J.B. (1987) Xenobiotic
metabolism in the mammalian kidney. In: Bach, P.H. & Lock, E.A., ed.
Nephro-toxicity in the experimental and the clinical situation,
Dordrecht, Boston, Lancaster, Martinus Nijhoff Publishers, pp.
371-404.
TEIXEIRA, R.B., KELLEY, F., ALPERT, H., PARDOV, V., & VAAMONDE, C.A.
(1982) Complete protection from gentamicin-induced acute renal failure
in the untreated streptozotocin diabetes mellitus rat. Kidney Int.,
21: 600-612.
TERRACINI, B. & PARKER, V.H. (1965) A pathological study on the
toxicity of S-(dichlorovinyl)-L-cysteine. Food Cosmet. Toxicol., 3:
67-74.
TERUEL, J.L., MARCEN, R., ONAINDIA, J.M., SERRANO, A., QUEREDA, C., &
ORTUNO, J. (1981) Renal function impairment caused by intravenous
urography, a prospective study. Arch. intern. Med., 141: 1271-1274.
TESTA, B. & JENNER, P. (1976) Drug metabolism: chemical and
biochemical aspects, New York, Basel, Marcel Dekker.
THOENES, W., STÖRKEL, S., & RUMPELT, H.-J. (1985) Human chromophobe
cell renal carcinoma. Virchows Arch. B Cell Pathol., 48: 207-217.
THOMPSON, W.M., FOSTER, W.L., Jr, HALVORSEN, R.A., DUNNICK, N.R.,
ROMMEL, A., & BATES, M. (1984) Iopamidol: new, nonionic contrast agent
for excretory urography. Am. J. Roentgenol., 142: 329-332.
THORNLEY, C., DAWNAY, A., & CATTEL, W.R. (1985) Human Tamm-Horsfall
glycoprotein: urinary and plasma levels in normal subjects and
patients with renal disease determined by a fully validated
radioimmunoassay. Clin. Sci., 68: 529-535.
THUN, M.J., BAKER, D.B., STEENLAND, K., SMITH, A.B., HALPERIN, W., &
BERL, T.B. (1985) Renal toxicity in uranium mill workers. Scand. J.
Work Environ. Health, 11: 83-90.
THURAU, K. (1964) Renal hemodynamics. Am. J. Med., 36: 698-719.
TOKOFF-RUBIN, N.E. (1986) Diagnosis of tubular injury in renal
transplant patients by urinary assay for proximal tubular antigen, the
adenosine-deaminase-binding protein. Transplantation, 41: 593-599.
TOLEDO, O.M.S. & DIETRICH, C.P. (1977) Tissue specific distribution of
sulfated mucopolysaccharides in mammals. Biochim. Biophys. Acta, 498:
114-122.
TOLINS, J.P. & RAIJ, L. (1988) Chronic amphotericin B nephrotoxicity
in the rat; protective effect of prophylactic salt loading. Am. J.
kidney Dis., 11: 313-317.
TOUBEAU, G., LAURENT, G., CARLIER, M.-B., ABID, S., MALDAGUE, P.,
HEUSON-STIENNON, J.A., & TULKENS, P.M. (1986) Tissue repair in rat
kidney cortex after short treatment with aminoglycosides at low doses.
Lab. Invest., 54: 385-393.
TRIFILLIS, A.L., REGEC, A., HALL-CRAGGS, M., & TRUMP, B.F. (1986)
Effects of cyclosporine on cultured human renal tubular cells.
Toxicol. Pathol., 14: 210-212.
TRINH-TRANG-TAN, M.M., BOUBY, N., COUTAUD, C., & BANKIR, L. (1986)
Quick isolation of rat medullary thick ascending limbs. Enzymatic and
metabolic characterization. Pflügers Arch., 407: 228-234.
TRUMP, B.F., BEREZESKY, I.K., SMITH, M.W., PHELPS, P.C., & ELLIGET,
K.A. (1989) The relationship between cellular ion deregulation and
acute and chronic toxicity. Toxicol. appl. Pharmacol., 97: 6-22.
TSUDA, H., SAKATA, T., TAMANO, S., OKUMURA, M., & ITO, N. (1983)
Sequential observations on the appearance of neoplastic lesions in the
liver and kidney after treatment with
N-ethyl-N-hydroxyethylnitrosamine followed by partial hepatectomy and
unilateral nephrectomy. Carcinogenesis, 4: 523-528.
TSUDA, H., HACKER, H.J., KATAYAMA, H., MASUI, T., ITO, N., & BANNASCH,
P. (1986) Correlative histochemical studies on preneoplastic and
neoplastic lesions in the kidney of rats treated with nitrosamines.
Virchows Arch. B Cell Pathol., 51: 385-404.
TSUDA, H., MOORE, M.A., ASAMOTO, M., INOUE, T., FUKUSHIMA, S., ITO,
N., SATOH, K., AMELIZAD, Z., & OESCH, F. (1987) Immunohistochemically
demonstrated altered expression of cytochrome P-450 molecular forms
and epoxide hydrolase in N-ethyl-N-hydroxyethylnitrosamine-induced rat
kidney and liver lesions. Carcinogenesis, 8: 711-717.
TUBBS, R.R., GEPHARDT, G.N., MCMAHON, J.T., POHL, M.C., VIDT, D.G.,
BARENBERG, S.A., & VALENZUELA, R. (1982) Membranous
glomerulo-nephritis associated with industrial mercury exposure. Am.
J. clin. Pathol., 77: 409-413.
TULKENS, P.M. (1989) Nephrotoxicity of aminoglycoside antibiotics.
Toxicol. Lett., 46: 107-123.
TUNE, B.M. (1975) Relationship between the transport and toxicity of
cephalosporine in the kidney. J. infect. Dis., 132: 189-194.
TUNE, B.M. (1986) The nephrotoxicity of cephalosporin antibiotics -
structure-activity relationships. Comments Toxicol., 1: 145-170.
TUNE, B.M. & FRAVERT, D. (1980) Mechanisms of cephalosporin
nephrotoxicity. A comparison of cephaloridine and cephaloglycin.
Kidney Int., 18: 591-600.
TUNE, B.M., BURG, M.D., & PATLAK, C.S. (1969) Characteristics of
p-aminohippurate transport in proximal renal tubules. Am. J. Physiol.,
217: 1057-1063.
TUNE, B.M., SIBLEY, R.K., & HSU, C.-Y. (1988) The mitochondrial
respiratory toxicity of cephalosporin antibiotics. An inhibitory
effect on substrate uptake. J. Pharmacol. exp. Therap., 245:
1054-1059.
TYSON, C.K. & MIRSALIS, J.C. (1985) Measurement of unscheduled DNA
synthesis in rat kidney cells following in vivo treatment with
genotoxic agents. Environ. Mutagen., 7: 889-899.
UCHIDA, S. & ENDOU, H. (1988) Substrate specificity to maintain
cellular ATP along the mouse nephron. Am. J. Physiol., 255: F977-F983.
ULLRICH, K.J. & GREGER, R. (1985) Approaches to the study of tubule
transport function. In: Seldin, D.G. & Giebisch, G., ed. The kidney,
physiology and pathology, New York, Raven Press, Chapter 20, pp.
427-455.
VAAMONDE, C.A., BIER, R., GOUVEA, W., ALPERT, H., KELLY, J., & PARDO,
V. (1984) Effect of duration of diabetes on the protection observed in
the diabetic rat against gentamicin-induced acute renal failure.
Miner. Electrolyte Metab., 10: 209-216.
VALTIN, H., ed. (1973) Renal functions: mechanisms preserving fluid
and solute balance in health, Boston, Toronto, Little Brown and Co.
VAN DORP, D. (1971) Recent developments in the biosynthesis and the
analyses of prostaglandins. Ann. NY Acad. Sci., 180: 181-199.
VAN ESCH, G.J. & KROES, R. (1969) The induction of renal tumours by
feeding basic lead acetate to mice and hamsters. Br. J. Cancer, 23:
765-771.
VAN FLEET, J.F., GREENWOOD, L.A., & FERRANO, F.J. (1979) Pathologic
features of adriamycin toxicosis in young pigs: non skeletal lesions.
Am. J. vet. Res., 40: 1537-1552.
VERPOOTEN, G.F., NOUWEN, E.J., HOYLAERTS, M.F., HENDRIX, P.G., & DE
BROE, M. (1989) Segment-specific localization of intestinal-type
alkaline phosphatase in human kidney. Kidney Int., 36: 617-625.
VERSCHOOR, M., WIBOWO, A., HERBER, R., VAN HEMMEN, J., & ZIELHUIS, R.
(1987) Influence of occupational low-level lead exposure on renal
parameters. Am. J. ind. Med., 12: 341-351.
VIAU, C., BERNARD, A., GUERET, F., MALDAGUE, P., GENGOUX, P., &
LAUWERYS, R. (1986a) Isoparaffinic solvent-induced nephrotoxicity in
the rat. Toxicology, 38: 227-240.
VIAU, C., BERNARD, A., & LAUWERYS, R. (1986b) Determination of rat
beta2-microglobulin in urine and in serum. I. Development of an
immunoassay based on latex particles agglutination. J. appl. Toxicol.,
6: 185-189.
VIAU, C., BERNARD, C., LAUWERYS, R., QUAGHEBEUR, L., CORNU, M.E.,
PHILLIPS, S.C., MUTTI, A., LUCERTINI, S., & FRANCHINI, I. (1987) A
cross-sectional survey of kidney function in refinery employees. Am.
J. ind. Med., 11: 177-187.
VICTERY, W., VANER, A.J., SHULAK, J.M., SCHOEPS, P., & JULIUS, S.
(1982) Lead, hypertension and the renin-angiotension system in rats.
J. lab. clin. Med., 99: 354-362.
VIOL, G.W., MINIELLY, J.A., & BISTRICKI, T. (1977) Gold nephropathy.
Tissue analysis by X-ray fluorescent spectroscopy. Arch. Pathol. lab.
Med., 101: 635-640.
VINAY, P., GOUGOUX, A., & LEMIEUX, G. (1981) Isolation of a pure
suspension of rat proximal tubules. Am. J. Physiol., 241: F403-F411.
VOGELZANG, N.J., TORKELSON, J.L., & KENNEDY, B.J. (1985)
Hypo-magnesemia, renal dysfunction and Raynaud's phenomenon in
patients treated with cisplatin, vinblastine and bleomycin. Cancer,
56: 2765-2770.
WALKER, P.D. & SHAH, S.V. (1987) Evidence suggesting a role for
hydroxyl radical in gentamicin-induced acute renal failure in rats. J.
clin. Invest., 81: 334-341.
WALKER, R.G., DAVIES, B.M., HOLWILL, B.J., DOWLING, J.P., &
KINCAID-SMITH, P. (1982) A clinicopathological study of lithium
nephrotoxicity. J. chron. Dis., 35: 685-695.
WALKER, R.G., ESCOTT, M., BIRCHALL, I., DOWLING, J.P., &
KINCAID-SMITH, P. (1986) Chronic progressive renal lesions induced by
lithium. Kidney Int., 29: 875-881.
WALLACE, A.C. & NAIRN, R.C. (1972) Renal tubular antigens in kidney
tumors. Cancer, 29: 977-981.
WAN, S.H. & RIEGELMAN, S. (1972a) Renal contribution to overall
metabolism of drug. I: Conversion of benzoic acid to hippuric acid. J.
pharm. Sci., 61: 1278-1284.
WAN, S.H. & RIEGELMAN, S. (1972b) Renal contribution to overall
metabolism of drugs. II: Biotransformation of salicylic acid to
salicyluric acid. J. pharm. Sci., 61: 1284-1287.
WAN, S.H., VON LEHMANN, B., & RIEGELMAN, S. (1972) Renal contribution
to overall metabolism of drugs. III: Metabolism of p-aminobenzoic
acid. J. pharm. Sci., 61: 1288-1292.
WARD, J.M. & FAUVIE, K.A. (1976) The nephrotoxic effects of
cis-diammine-dichloroplatinum (II) (NSC-119875) in male F344 rats.
Toxicol. appl. Pharmacol., 38: 535-547.
WARD, J.M., GOODMAN, D.G., SQUIRE, R.A., CHU, R.C., & LINHART, M.S.
(1979) Neoplastic and non-neoplastic lesions in aging (C57BL/6N X
CH3/HeN)F1 (B6C3F11) mice. J. Natl Cancer Inst., 63: 849-854.
WEBER, P.C. (1980) Renal prostaglandins, kidney function and effect of
aspirin on metabolism of acetaminophen and benzidine by renal inner
medulla prostaglandin hydroperoxidase. J. lab. clin. Med., 101: 58-65.
WEDEEN, R.P., MALLIK, D.K., & BATUMAN, V. (1979) Detection and
treatment of occupational lead nephropathy. Arch. intern. Med., 139:
53-57.
WEINBERG, J.M. & HUMES, H.D. (1980) Mechanisms of gentamicin-induced
dysfunction of renal cortical mitochondria. I. Effects on
mitochondrial respiration. Arch. Biochem. Biophys., 205: 222-231.
WHO (1979) IPCS Environmental Health Criteria 11: Mycotoxins, Geneva,
World Health Organization, 127 pp.
WHO COLLABORATING CENTRE FOR THE HISTOLOGICAL CLASSIFICATION OF RENAL
DISEASES (1982) Renal disease: Classification and atlas of glomerular
diseases, Tokyo, Igaku-Shoin, 359 pp.
WHO (1983) IPCS Environmental Health Criteria 27: Guidelines on
studies in environmental epidemiology, Geneva, World Health
Organization, pp. 184-185.
WHO (1984) IPCS Environmental Health Criteria 39: Paraquat and Diquat,
Geneva, World Health Organization, 181 pp.
WHO (1990) International classification of diseases for oncology, 2nd
ed., Geneva, World Health Organization, 144 pp.
WHO COLLABORATING CENTRE FOR THE HISTOLOGICAL CLASSIFICATION OF RENAL
DISEASES (1985) Renal disease: Classification and atlas of
tubulo-interstitial diseases, New York, Igaku-Shoin, 221 pp.
WHO COLLABORATING CENTRE FOR THE HISTOLOGICAL CLASSIFICATION OF RENAL
DISEASES (1987) Renal disease: Classification and atlas, New York,
Igaku-Shoin, 291 pp.
WHO COLLABORATING CENTRE FOR THE HISTOLOGICAL CLASSIFICATION OF RENAL
DISEASES (1988) Classification and atlas of infectious and tropical
diseases, Chicago, American Society of Clinical Pathologists, 273 pp.
WIBELL, L., EVRIN, P.E., & BERGGARD, I. (1973) Serum
beta2-microglobulin in renal disease. Nephron, 10: 320-331.
WILLIAMS, P.D., HOLOHAN, P.D., & ROSS, C.R. (1981) Gentamicin
nephro-toxicity. II. Plasma membrane changes. Toxicol. appl.
Pharmacol., 61: 243-251.
WILLIAMS, P.D., HOTTENDORF, G.H., & BENNETT, D.B. (1986) Inhibition of
renal membrane binding and nephrotoxicity of aminoglycosides. J.
Pharmacol. exp. Ther., 237: 919-925.
WILSON, P.D. (1986) Use of cultured renal tubular cells in the study
of cell injury. Miner. Electrolyte Metab., 12: 71-83.
WING, A.J., BRUNNER, F.P., GEERLINGS, W., BROYER, M., BRYNGER, H.,
FASSBINDER, W., RISSONI, G., SELWOOD, N.H., & TUFESON, G. (1989)
Contribution of toxic nephropathies to end-stage renal failure in
Europe: a report form the EDTA-ERA registry. Toxicol. Lett., 46:
281-292.
WIRTHENSOHN, G., BECK, F.X., & GUDER, W.G. (1987) Role and regulation
of glycerophosphorylcholine in rat renal papilla. Pflügers Arch., 409:
411-415.
WIRTHENSOHN, G., LEFRANK, S., SCHMOLKE, M., & GUDER, W.G. (1989)
Regulation of organic osmolyte concentrations in tubules from rat
renal inner medulla. Am. J. Physiol., 256: F128-F135.
WITSCHI, H.P., ALDRIDGE, W.N., AUST, S.D., AUTOR, A.P., DE MATTEIS,
F., DRUET, P., ELBERS, R., FOWLER, B.A., GRONIOWSKI, J.A., HAGMAN, W.,
ORRENIUS, S., PETERING, D.H., SEINEN, W., SUMMER, K.H., & TRUMP, B.F.
(1987) Mechanisms and target cell injury. In: Fowler, B.A., ed.
Mehanisms of cell injury: Implications for human health, New York,
Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 385-403.
WOLD, J.S., TURNIPSEED, S.A., & MILLER, B.L. (1979) The effect of
renal cation transport inhibition on cephaloridine nephrotoxicity.
Toxicol. appl. Pharmacol., 47: 115-122.
WOLGAST, M., PERSSON, E., SCHNERMANN, J., ULFENDAHL, H., & WUNDERLICH,
P. (1973) Colloid osmotic pressure of the subcapsular interstitial
fluid of rat kidneys during hydropenia and volume expansion. Pflügers
Arch., 340: 123-131.
YOUNG, D.M. (1975) Pathologic effects of adriamycin in experimental
systems. Cancer Chemother. Rep., 6: 159-175.
ZENSER, T.V. & DAVIS, B.B. (1984) Enzyme systems involved in the
formation of reactive metabolites in the renal medulla: cooxidation
via prostaglandin H synthase. Fundam. appl. Toxicol., 4: 922-929.
ZENSER, T.V., MATTAMMAL, M.B., & DAVIS, B.B. (1978a) Differential
distribution of the mixed function oxidase in rabbit kidney. J.
Pharmacol. exp. Ther., 207: 719-725.
ZENSER, T.V., MATTAMMAL, M.B., HERMAN, C.A., JOSHI, S., & DAVIS, B.B.
(1978b) Effect of acetaminophen on prostaglandin E2 and
prostaglandin F2 alpha synthesis in the renal inner medulla of rat.
Biochim. Biophys. Acta, 542: 486-495.
ZENSER, T.V., MATTAMMAL, M.B., & DAVIS, B.B. (1979a) Demonstration of
separate pathways for the metabolism of organic compounds in rabbit
kidney. J. Pharmacol. exp. Ther., 208: 418-421.
ZENSER, T.V., MATTAMMAL, M.B., & DAVIS, B.B. (1979b) Cooxidation of
benzidine by renal medullary prostaglandin cyclooxygenase. J.
Pharmacol. exp. Ther., 211: 460-464.
ZENSER, T.V., MATTAMMAL, M.B., BROWN, W.W., & DAVIS, B.B. (1979c)
Cooxygenation by prostaglandin cyclooxygenase from rabbit inner
medulla. Kidney Int., 16: 688-694.
ZENSER, T.V., MATTAMMAL, M.B., RAPP, N.S., & DAVIS, B.B. (1983) Effect
of aspirin on metabolism of acetaminophen and benzidine by renal inner
medulla prostaglandin hydroperoxidase. J. lab. clin. Med., 101: 58-65.
ZIMMERMAN, S.W. & NORBACH, D.H. (1980) Nephrotoxic effects of
long-term carbon tetrachloride administration in rats. Arch. Pathol.
lab. Med., 104: 94-99.
ZUSMAN, R.M. (1980) Prostaglandin E2 biosynthesis by renomedullary
inter-stitial cells: In vitro studies and pathophysiological
correlations. In: Mandal, A.K. & Bohman, S.-O., ed. The renal papilla
and hypertension, New York, London, Plenum Press, pp. 187-207.
RESUME ET CONCLUSIONS
L'excrétion et l'homéostase des molécules hydrosolubles sont
principalement assurées par les reins. Ceuxci sont constitués
d'unités fonctionnelles appelées néphrons, qui consistent
essentiellement en un tube continu formé de cellules hétérogènes
hautement spécialisées qui présentent une organisation structurale,
fonctionnelle et biochimique poussée. Il existe des différences
marquées entre les néphrons eux-mêmes, qui dépendent de la
localisation plus ou moins profonde du glomérule dans le cortex.
C'est l'agencement complexe de cette structure qui, joint à des
différences régionales de vascularisation tenant à la disposition
particulière des vaisseaux, constitue cet organe hétérogène et
hautement organisé qu'est le rein.
Une grande partie des connaissances sur l'anatomie et le
fonctionnement du rein, obtenues grâce à l'expérimentation animale,
est directement transposable à l'homme. Toutefois, les processus
biologiques et métaboliques qui se déroulent dans le rein humain ne
sont pas encore complètement élucidés, non plus que les différences
constatées d'une espèce animale à une autre. Dans ces conditions, on
comprend qu'on ne puisse pas toujours extrapoler d'une espèce à une
autre les effets toxicologiques observés.
Plusieurs substances (qu'elles aient ou non un effet
thérapeutique) exercent des effets toxiques sur les divers éléments
anatomiques du rein. Les effets toxiques peuvent être aigus ou
chroniques; ils peuvent être directs ou se produire par
l'intermédiaire de mécanismes immunologiques. L'impact qu'une
substance néphrotoxique peut avoir sur la santé dépend de plusieurs
facteurs de risque: état fonctionnel du rein, lésion préexistante,
maladie, âge, sexe, régime alimentaire, etc..
On n'a pas suffisamment étudié l'épidémiologie des cas de
néphrotoxicité imputables à des diverses substances chimiques agissant
seules ou en association. A quelques exceptions près, la contribution
des produits chimiques à l'incidence globale des néphropathies et de
l'insuffisance rénale chronique reste mal définie. D'importantes
recherches ont été consacrées à certains groupes exposés de par leur
profession ainsi qu'aux néphropathies résultant de la consommation
d'analgésiques : elles ont montré l'existence de variations entre les
différents groupes et les différents pays. Toutefois on estime que
jusqu'à 18% des néphropathies terminales pourraient être dues à la
prise d'analgésiques et jusqu'à 5% avoir une origine toxique
quelconque. Environ 50% des néphropathies terminales sont d'origine
inconnue. L'un des principaux problèmes qui se posent dans la
recherche de l'étiologie des néphropathies terminales tient à la
longue période de latence ou à l'évolution lente de l'insuffisance
rénale chronique, qui rendent difficile l'identification rétrospective
de l'agent causal. Ce n'est que dans quelques cas que le dosage de
certaines substances dans les tissus (organisme, rein) peut conduire
au diagnostic. Par ailleurs on se heurte également à un certain
manque de cohérence dans les critères et la terminologie utilisés sur
le plan diagnostique et anatomopathologique.
Les différences anatomiques et physiologiques rendent difficile
une extrapolation directe à l'homme des résultats obtenus sur des
systèmes expérimentaux ( in vivo ou in vitro). On ne connaît que
très peu de cas où les données de néphrotoxicité sont comparables chez
l'homme et chez l'animal et permettent d'évaluer de façon fiable le
risque pour la santé humaine.
Certaines substances chimiques peuvent provoquer des lésions
sélectives au niveau des structures rénales vulnérables ou déclencher
des mécanismes immunologiques. On considère qu'il existe en général
deux types de mécanismes à la base des lésions rénales : a) les
mécanismes immunologiques qui conduisent à la néphrite aiguë
interstitielle et b) les mécanismes qui affectent principalement le
glomérule par l'intermédiaire d'anticorps anti-GBM ou
d'immunocomplexes. Il existe un autre grand groupe de maladies dues
à des substances chimiques ou à leurs métabolites qui perturbent
l'activité biochimique cellulaire ou modifient les propriétés
hémodynamiques, etc. Parmi les facteurs susceptibles d'influer sur
les lésions cellulaires provoquées par les produits toxiques figurent
les systèmes de transport cellulaire, la pinocytose, la dégradation
métabolique, l'interaction avec les protéines, les lipides, les
membranes et l'ADN cellulaires et éventuellement avec d'autres
constituants de la cellule.
Plus on utilise de produits pharmaceutiques et de substances
chimiques, plus on accroît les risques de néphrotoxicité. La
néphrotoxicité des médicaments dépend de la dose et de la durée du
traitement (par exemple dans le cas des associations d'analgésiques
qui conduisent à une nécrose papillaire). On a beaucoup étudié les
effets néphrotoxiques des analgésiques, des antibiotiques (comme les
aminoglycosides), des agents anticancéreux (comme le cis-platine) et
de divers autres agents. Nombre de substances fréquemment utilisées
dans l'industrie ou à la maison comme les hydrocarbures chlorés ou
l'éthylèneglycol peuvent également avoir une action toxique sur le
rein. D'autres substances, présentes dans l'environnement comme le
plomb ou le cadmium sont effectivement néphrotoxiques. L'action
toxique de ces substances intervient après accumulation
intracellulaire du produit initial ou métabolisation intra- ou
extrarénale. L'exposition à plusieurs substances chimiques peut
entraîner des réactions antagonistes ou synergistiques.
Les tumeurs du rein et des voies urinaires représentent moins de
2 à 3 % de l'ensemble des cancers humains, mais leur fréquence est en
augmentation, ce qui incite à penser que des facteurs environnementaux
pourraient être à l'oeuvre. Nombre de produits pharmaceutiques et de
substances chimiques se sont révélés cancérogènes sur des modèles
expérimentaux mais seuls quelques-uns d'entre eux sont responsables de
l'apparition de cancers chez l'homme. Il s'agit de l'amiante
(adénocarcinome rénal), de l'abus d'analgésiques (carcinomes de type
transitionnel) et de facteurs tenant au mode de vie (tabagisme,
consommation d'alcool et de café). Il existe des groupes de
population chez lesquels certains cancers des voies urinaires ont une
étiologie encore inconnue. Certaines substances chimiques utilisées
dans le cadre professionnel sont à l'origine de cancers de la vessie.
De nombreuses substances chimiques ou pharmaceutiques peuvent
entraîner une néphrite interstitielle, affection qui serait
susceptible de favoriser l'apparition de cancers des voies urinaires.
Il n'existe pas de méthode qui permette d'évaluer in vivo ou in
vitro dans des conditions satisfaisantes une insuffisance rénale
provoquée par des substances chimiques. Aussi faut-il non seulement
contrôler l'activité néphrotoxique des produits chimiques, mais
également inclure dans les protocoles expérimentaux l'étude du
mécanisme toxique au niveau des cellules cibles. Pour y parvenir il
faut faire appel tant à l'expérimentation in vivo qu'à des études in
vitro. Les études in vitro doivent porter sur des éléments où les
relations anatomiques et cellulaires sont maintenues (par
microperfusion de reins et de tubules isolés, coupes de cortex,
fragments isolés de néphrons, etc.) et sur des cellules rénales (par
exemple suspensions cellulaires, cultures cellulaires primaires,
lignées cellulaires et fractions infracellulaires). Pour les études
in vivo, on peut faire appel à des techniques effractives ou non
effractives. Parmi les techniques effractives figurent les examens
histopathologiques ainsi que les techniques classiques de bilan de la
fonction rénale. Les techniques non effractives permettent un bilan
répété de la fonction rénale chez l'animal par la mesure d'une série
de paramètres fonctionnels (filtration glomérulaire, fonction
tubulaire, protéinurie, enzymurie, etc.). Des épreuves biochimiques
spécialisées devront être effectuées le cas échéant. En associant de
façon convenable les épreuves in vivo et les épreuves in vitro on
pourra déterminer si telle ou telle substance chimique est
néphrotoxique et avoir une idée de son activité, de son site d'action
et du mécanisme de son action toxique.
Les méthodes habituellement utilisées pour l'exploration
fonctionnelle du rein chez l'homme ne permettent pas un diagnostic
suffisamment rapide d'une insuffisance rénale due à des substances
chimiques ni d'en apprécier le retentissement sur la santé. L'absence
de marqueurs spécifiques précoces de la néphrotoxicité est
particulièrement gênante. Les méthodes non effractives devront faire
appel à des marqueurs très spécifiques et sensibles, qui permettent de
déceler rapidement les modifications qui se produisent au niveau rénal
afin de prévoir l'apparition éventuelle d'une insuffisance
fonctionnelle. Les techniques actuelles de contrôle de la fonction
glomérulaire ou tubulaire ne sont utilisables que lorsque les lésions
sont déjà très importantes. On a proposé comme marqueurs
l'utilisation de constituants tissulaires immunoréactifs mais encore
faut-il en confirmer la valeur au moyen d'études longitudinales bien
conçues.
RECOMMANDATIONS
1. Il faut faire un effort soutenu pour développer et valider des
marqueurs plus sélectifs et plus spécifiques et notamment des
anticorps monoclonaux en vue d'étudier l'insuffisance rénale chez
l'animal. Ces marqueurs pourraient être utilisables chez l'homme.
2. Il faudrait compléter la base de données utilisables pour
déterminer le potentiel néphrotoxique des substances chimiques pour
l'homme. A cet effet, on développera et on validera diverses méthodes
d'expérimentation animale ( in vivo et in vitro), on mettra au
point des méthodes nouvelles d'étude de la néphrotoxicité, on étudiera
les différences interspécifiques et on prendra en compte l'expérience
tirée des essais précliniques de nouveaux médicaments chez l'homme.
3. Il faudrait renforcer les études épidémiologiques (c'est-à-dire
les études prospectives sur des groupes professionnels ou des groupes
de la population générale exposés à des substances néphrotoxiques ou
qui font une consommation excessive d'analgésiques).
4. Des efforts plus importants devront être consentis afin d'établir
le rôle de certaines substances chimiques dans l'étiologie des
maladies rénales au stade le plus précoce possible (antécédents
professionnels, recherches de néphrotoxines dans les tissus).
5. L'élucidation du mode d'action des substances néphrotoxiques peut
être utile à la prévention et au traitement des effets rénaux
indésirables et pourrait contribuer à la prévision du pouvoir
néphrotoxique des nouveaux médicaments et des nouveaux produits.
Parmi les secteurs de recherche particulièrement importants on peut
citer :
* les mécanismes immunologiques
* les effets directs des substances chimiques sur les membranes et
notamment les mécanismes de peroxydation des lipides, les
interactions membranes/substances chimiques, les déplacements
d'ions, les événements au niveau des récepteurs
* l'activation des proto-oncogènes et la différenciation cellulaire
* la régulation du métabolisme cellulaire.
6. Il faut également préciser la localisation anatomique de
certaines fonctions rénales.
7. Il faudrait étudier de façon plus approfondie le rôle des
variations et de la prédisposition génétiques aux effets toxiques des
médicaments et des produits chimiques.
8. La relation entre néphrotoxicité et cancer (mycotoxines et
néphropathie des Balkans par exemple) doit être étudiée plus à fond.
RESUMEN Y CONCLUSIONES
Los riñones son los principales órganos de excreción y
homeostasis de las moléculas hidrosolubles. La unidad funcional del
riñón es el nefrón, que consiste esencialmente en un tubo continuo de
células heterogéneas sumamente especializadas, y que exhibe una
notable organización estructural, funcional y bioquímica. Existen
diferencias pronunciadas entre unos nefrones y otros, atendiendo a la
localización de los glomérulos individuales correspondientes en la
corteza. Esta compleja organización estructural, combinada con las
diferencias en la vascularidad regional debida a lo especializado de
la disposición vascular, da lugar a un órgano sumamente complejo y
heterogéneo.
Gran parte de los conocimientos anatómicos y funcionales
obtenidos en los animales pueden aplicarse directamente al riñón
humano. No obstante, los procesos biomédicos y metabólicos del riñón
humano, así como las diferencias entre las especies animales, no se
han elucidado de forma tan detallada. Así, los efectos de las
sustancias químicas sólo se pueden extrapolar hasta cierto punto de
unas especies a otras.
Varias sustancias químicas (tanto terapéuticas como no
terapéuticas) tienen efectos tóxicos en uno o más elementos anatómicos
del riñón. Los efectos tóxicos pueden ser agudos o crónicos, y pueden
ser directos o estar indirectamente mediados por mecanismos
inmunológicos. El impacto que tienen en la salud las sustancias
químicas nefrotóxicas está relacionado con los factores de riesgo,
entre los que figuran el estado de la reserva funcional del riñón y
factores como las lesiones renales ya existentes, las enfermedades, la
edad, el sexo y la dieta.
No se ha estudiado bastante la epidemiología de la nefrotoxicidad
inducida con sustancias químicas aisladas o en exposiciones mixtas.
La contribución de las sustancias químicas a la incidencia global de
la nefropatía y del fallo renal crónico está, salvo raras excepciones,
sin definir. En el caso de algunos grupos expuestos por su profesión
y de enfermedad renal asociada a los analgésicos, se han hecho amplios
estudios que han demostrado la existencia de la incidencia entre
grupos y países. Sin embargo, se estima que hasta el 18% de las
enfermedades renales en fase terminal pueden deberse a nefropatía por
analgésicos y hasta el 5% a otras nefropatías tóxicas. Alrededor del
50% de las enfermedades renales en fase terminal son de etiología
desconocida. Uno de los principales problemas para atribuir una causa
a la enfermedad renal en fase terminal es la larga latencia y/o la
lenta evolución del fallo renal crónico, lo que dificulta la
identificación retrospectiva del agente causal. Sólo en algunos
casos, la medida de los niveles tisulares (organismo, riñón) de
sustancias químicas es útil para el diagnóstico. Otro problema ha
sido la falta de coherencia en la terminología y los criterios
patológicos y de diagnóstico.
Las diferencias anatómicas y fisiológicas del riñón dificultan la
extrapolación directa al ser humano a partir de los sistemas
experimentales ( in vivo e in vitro). Existen muy pocos ejemplos
de sustancias químicas nefrotóxicas para las que se disponga de datos
adecuados y comparables entre los animales y el hombre como para
formar una base sólida sobre la que evaluar el riesgo potencial para
la salud humana.
Las sustancias químicas pueden dañar de modo selectivo las
estructuras vulnerables del riñón o activar mecanismos inmunológicos.
Los mecanismos de lesión renal pueden dividirse generalmente en dos
categorías: a) nefritis intersticiales agudas inmunológicamente
inducidas; b) aquellos que afectan primordialmente al glomérulo por
conducto de anticuerpos mediados por la anti-GBM o por conducto de
complejos inmunes. Otro grupo principal está compuesto por las
enfermedades desencadenadas por sustancias químicas o sus metabolitos
que interfieren con los efectos bioquímicos y hemodinámicos en la
célula, entre otros. Entre los factores que pueden modificar la
lesión celular producida por sustancias tóxicas figuran los sistemas
de transporte celular, la pinocitosis, la degradación metabólica y la
interacción con las proteínas, los lípidos, las membranas y el ADN
celulares, y tal vez otros constituyentes de la célula.
El uso cada vez más extendido de agentes y sustancias químicas
terapéuticas aumenta las posibilidades de aparición de nefrotoxicidad.
La nefrotoxicidad inducida por agentes terapéuticos depende de la
dosis y del tiempo de exposición (por ejemplo, analgésicos de
combinación que producen necrosis papilar del riñón). Los efectos
nefrotóxicos de los analgésicos, los antibióticos (como los
aminoglucósidos), los agentes anticancerosos (como el cis-platino), y
varios agentes más han sido objeto de amplios estudios. Las
sustancias químicas de uso frecuente en la industria o el hogar, como
los hidrocarburos clorurados y el etilenglicol, también tienen
potencial para producir lesiones renales. Las sustancias químicas del
medio ambiente como el plomo y el cadmio son capaces de inducir
nefrotoxicidad. Estos agentes tienen efectos tóxicos tras la
acumulación intracelular del compuesto original o tras la
biotransformación renal o extrarrenal. La exposición multiquímica
puede dar lugar a respuestas antagonistas o sinérgicas.
Aunque los tumores del riñón y del tracto urinario representan
menos del 2-3% de todos los cánceres humanos, la frecuencia de esos
tumores está aumentando, lo que indica que los factores ambientales
ejercen cierta influencia. Si bien se ha demostrado que muchos
fármacos y sustancias químicas son carcinogénicos en modelos
experimentales, sólo algunas sustancias concretas se han relacionado
con tumores en el hombre. Entre ellos figuran el amianto
(adenocarcinoma renal), el uso indebido de analgésicos (carcinomas de
transición), y los factores relacionados con el modo de vida (uso de
tabaco, alcohol, café). Existen grupos de población con cáncer del
tracto urinario de etiología aún por determinar. Las sustancias
químicas presentes en el medio profesional se han relacionado con la
etiología del cáncer de la vejiga urinaria. Muchos fármacos y
sustancias químicas provocan nefritis intersticial, que en sí misma
puede ser un factor desencadenante de cánceres en el tracto urinario.
No hay ningún método in vivo o in vitro que por sí solo baste
para evaluar la disfunción renal químicamente inducida. Así pues, es
aconsejable no sólo estudiar compuestos en busca de su potencial
nefrotóxico, sino incorporar a los protocolos experimentales estudios
mecanicistas sobre la toxicidad para células diana. Para conseguirlo,
deben realizarse investigaciones concertadas in vivo e in vitro.
Los estudios in vitro son aquellos en los que se mantienen las
relaciones anatómicas entre células (por ejemplo, riñón y túbulos
aislados y perfundidos, secciones de la corteza renal y segmentos
aislados de nefrones) y aquellos en los que se utilizan células
renales (por ejemplo, suspensión de células, cultivos de células
primarias, líneas celulares establecidas y fracciones subcelulares).
En los estudios in vivo se utilizan técnicas tanto invasivas como no
invasivas. Los procedimientos invasivos comprenden las mediciones
histopatológicas y ordinarias de la función renal. Los procedimientos
no invasivos permiten evaluar de modo repetido la función renal en los
animales midiendo una serie de pruebas de la función renal (filtración
glomerular, función tubular, proteinuria, enzimuria, etc). Cuando
convenga, deben realizarse ensayos bioquímicos especializados. La
combinación adecuada de experimentos in vivo e in vitro revelará
si las sustancias químicas son nefrotóxicas o no y dará idea de la
potencia, la localización y los mecanismos de la toxicidad.
Los métodos tradicionales de evaluación de la función renal en el
hombre no bastan para diagnosticar de modo oportuno la disfunción
renal inducida por sustancias químicas y la predicción de su
importancia para la salud. La falta de marcadores específicos y
precoces en la nefrotoxicidad resulta particularmente problemática.
La evaluación no invasiva de la nefrotoxicidad debe hacer uso de
marcadores de gran especificidad, sensibilidad para la detección de
las alteraciones renales precoces y valor predictivo para la evolución
de la insuficiencia renal. Las técnicas actuales de seguimiento de la
función glomerular o tubular sólo son útiles en los casos en que se ha
producido una lesión renal grave. Aunque actualmente se señalan los
constituyentes tisulares inmunorreactivos como marcadores apropiados,
es preciso validar su idoneidad en estudios longitudinales bien
diseñados.
RECOMENDACIONES
1. Debe hacerse un esfuerzo continuado por desarrollar y validar
marcadores más selectivos y específicos, inclusive anticuerpos
monoclonales, para evaluar la disfunción renal en animales. Es
posible que esos marcadores sean aplicables al hombre.
2. Debe ampliarse la base de datos para predecir el potencial
nefrotóxico de las sustancias químicas para el hombre. Ello comprende
el desarrollo y la validación de criterios experimentales en animales
( in vivo e in vitro), métodos alternativos para estudiar la
nefrotoxicidad, información sobre diferencias interespecíficas, y
experiencia de la evaluación preclínica de nuevos agentes terapéuticos
en el hombre.
3. Deben reforzarse los estudios epidemiológicos (es decir, estudios
prospectivos en grupos profesionales y de la población general
expuestos a sustancias químicas nefrotóxicas o que hagan uso indebido
de analgésicos).
4. Deben intensificarse los esfuerzos por establecer el papel de las
sustancias químicas en la etiología de las enfermedades renales en la
etapa más temprana de diagnóstico (por ejemplo, pasado laboral,
vigilancia de los tejidos en busca de nefrotoxinas).
5. La comprensión de los mecanismos de acción de los nefrotóxicos
ayudará a prevenir y gestionar desde el punto de vista clínico los
efectos renales no deseados, y puede ayudar a predecir el potencial
nefrotóxico de nuevos fármacos y sustancias químicas. Los aspectos de
particular importancia para las investigaciones futuras son:
* los mecanismos inmunológicos;
* los efectos directos de las sustancias químicas en las membranas,
inclusive los mecanismos de peroxidación lipídica, la interacción
membrana/sustancias químicas, los cambios de iones, y los
procesos mediados por receptores;
* la activación de los proto-oncogenes y de la diferen ciación
celular;
* la regulación del metabolismo celular.
6. Es necesario determinar y correlacionar las funciones específicas
con localizaciones anatómicas discretas dentro del riñón.
7. Debe estudiarse más a fondo el papel de la variación genética y
la susceptibilidad a los efectos tóxicos de fármacos y sustancias
químicas.
8. Debe estudiarse con más detalle la relación entre la
nefrotoxicidad y la carcinogénesis renal (por ejemplo micotoxinas y
nefropatía de los Balcanes).
See Also:
Toxicological Abbreviations