
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 118
INORGANIC MERCURY
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
First draft prepared by Dr. L. Friberg,
Karolinska Institute, Sweden
World Health Orgnization
Geneva, 1991
The International Programme on Chemical Safety (IPCS) is a
joint venture of the United Nations Environment Programme, the
International Labour Organisation, and the World Health
Organization. The main objective of the IPCS is to carry out and
disseminate evaluations of the effects of chemicals on human health
and the quality of the environment. Supporting activities include
the development of epidemiological, experimental laboratory, and
risk-assessment methods that could produce internationally
comparable results, and the development of manpower in the field of
toxicology. Other activities carried out by the IPCS include the
development of know-how for coping with chemical accidents,
coordination of laboratory testing and epidemiological studies, and
promotion of research on the mechanisms of the biological action of
chemicals.
WHO Library Cataloguing in Publication Data
Inorganic mercury.
(Environmental health criteria ; 118)
1.Mercury poisoning 2.Environmental pollutants
I.Series
ISBN 92 4 157118 7 (NLM Classification: QV 293)
ISSN 0250-863X
The World Health Organization welcomes requests for permission
to reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made
to the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1991
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material
in this publication do not imply the expression of any opinion
whatsoever on the part of the Secretariat of the World Health
Organization concerning the legal status of any country, territory,
city or area or of its authorities, or concerning the delimitation
of its frontiers or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar
nature that are not mentioned. Errors and omissions excepted, the
names of proprietary products are distinguished by initial capital
letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR INORGANIC MERCURY
1. SUMMARY AND CONCLUSIONS
1.1. Identity
1.2. Physical and chemical properties
1.3. Analytical methods
1.3.1. Analysis, sampling, and storage of urine
1.3.2. Analysis and sampling of air
1.4. Sources of human and environmental exposure
1.4.1. Natural occurrence
1.4.2. Sources due to human activities
1.5. Uses
1.6. Environmental transport, distribution, and transformation
1.7. Human exposure
1.8. Kinetics and metabolism
1.8.1. Reference and normal values
1.9. Effects in humans
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1. Identity
2.2. Physical and chemical properties
2.3. Conversion factors
2.4. Analytical methods
2.4.1. Analysis, sampling, and storage of urine
2.4.2. Analysis and sampling of air
2.4.3. Quality control and quality assurance
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Man-made sources
3.3. Uses
3.4. Dental amalgam in dentistry
3.5. Mercury-containing cream and soap
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. General population exposure
5.1.1. Exposure from dental amalgam
5.1.1.1 Human studies
5.1.1.2 Animal experiments
5.1.2. Skin-lightening soaps and creams
5.1.3. Mercury in paint
5.2. Occupational exposure during manufacture, formulation, and
use
6. KINETICS AND METABOLISM
6.1. Absorption
6.1.1. Absorption by inhalation
6.1.2. Absorption by ingestion
6.1.3. Absorption through skin
6.1.4. Absorption by axonal transport
6.2. Distribution
6.3. Metabolic transformation
6.4. Elimination and excretion
6.5. Retention and turnover
6.5.1. Biological half-time
6.5.2. Reference or normal values in indicator media
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
7.1. Uptake, elimination, and accumulation in organisms
7.2. Toxicity to microorganisms
7.3. Toxicity to aquatic organisms
7.4. Toxicity to terrestrial organisms
7.5. Effects of mercury in the field
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
8.1. Single and short-term exposure
8.2. Long-term exposure
8.2.1. General effects
8.2.2. Immunological effects
8.2.2.1 Auto-immunity
8.2.2.2 Genetics
8.2.2.3 Mechanisms of induction
8.2.2.4 Autoregulation
8.2.2.5 Immunosuppression
8.2.2.6 Conclusions
8.3. Reproduction, embryotoxicity, and teratogenicity
8.3.1. Males
8.3.2. Females
8.4. Mutagenicity and related end-points
8.5. Carcinogenicity
8.6. Factors modifying toxicity
8.7. Mechanisms of toxicity - mode of action
9. EFFECTS ON HUMANS
9.1. Acute toxicity
9.2. Effects on the nervous system
9.2.1. Relations between mercury in central nervous system
and effects/response
9.2.2. Relations between mercury in air, urine or blood
and effects/response
9.2.2.1 Occupational exposure
9.2.2.2 General population exposure
9.3. Effects on the kidney
9.3.1. Immunological effects
9.3.2. Relations between mercury in organs and effects/response
9.3.3. Relations between mercury in air, urine and/or blood and
effect/response
9.4. Skin reactions
9.4.1. Contact dermatitis
9.4.2. Pink disease and other skin manifestations
9.5. Carcinogenicity
9.6. Mutagenicity and related end-points
9.7. Dental amalgam and general health
9.8. Reproduction, embryotoxicity, and teratogenicity
9.8.1. Occupational exposure
9.8.1.1 In males
9.8.1.2 In females
10. EVALUATION OF HUMAN HEALTH RISKS
10.1. Exposure levels and routes
10.1.1. Mercury vapour
10.1.2. Inorganic mercury compounds
10.2. Toxic effects
10.2.1. Mercury vapour
10.2.2. Inorganic mercury compounds
10.3. Dose-response relationships
10.3.1. Mercury vapour
10.3.2. Inorganic mercury compounds
11. RECOMMENDATIONS FOR FURTHER RESEARCH
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
REFERENCES
RESUME ET CONCLUSIONS
RESUMEN Y CONCLUSIONES
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR INORGANIC
MERCURY
Members
Professor M. Berlin, Institute of Environmental Medicine,
University of Lund, Lund, Sweden (Chairman)
Professor P. Druet, Broussais Hospital, Paris, France
Professor V. Foà, Institute of Occupational Health, Uni-
versity of Milan, Milan, Italy
Professor L. Friberg, Karolinska Institute, Department of
Environmental Hygiene, Stockholm, Sweden
Professor P. Glantz, Prosthetic Dentistry, Faculty of
Odontology, University of Lund, Tandlakarhogskolan,
Malmö, Sweden
Professor C.A. Gotelli, Centre for Toxicological Research,
Buenos Aires, Argentina
Professor G. Kazantzis, Institute of Occupational Health,
London School of Hygiene and Tropical Medicine, London,
United Kingdom (Rapporteur)
Dr L. Magos, Toxicological Unit, Medical Research Council,
Carshalton, Surrey, United Kingdom
Dr W.B. Peirano, Environmental Criteria and Assessment
Office, Office of Research and Development, US Environ-
mental Protection Agency, Cincinnati, USA
Professor B.S. Sridhara Rama Rao, Department of Neurochem-
istry, National Institute of Mental Health and Neuro-
sciences, Bangalore, India
Professor M. Riolfatti, Institute of Hygiene, Faculty of
Pharmaceutical Science, Padova, Italy
Dr M.J. Vimy, Health Science Centre, Department of Medi-
cine and Medical Physiology, Faculty of Medicine, Uni-
versity of Calgary, Calgary, Alberta, Canada
Observers
Dr M. Ancora, Centro Italiano Studi e Indagini, Rome,
Italy
Professor K.S. Larsson, Institute for Odontological Toxi-
cology, Faculty of Dentistry, Karolinska Institute,
Huddinge, Sweden
Observers (contd.)
Professor C. Maltoni, Institute of Oncology, Bologna,
Italy
Dr A. Mochi, Centro Italiano Studi e Indagini, Rome, Italy
Professor A.A.G. Tomlinson, Centro Italiano Studi e
Indagini, Rome, Italy
Secretariat
Dr D. Kello, Toxicology and Food Safety, World Health
Organization Regional Office for Europe, Copenhagen,
Denmark
Dr T. Kjellström, Prevention of Environmental Pollution,
Division of Environmental Health, World Health Organiz-
ation, Geneva, Switzerland (Secretary)
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in
the criteria monographs as accurately as possible without
unduly delaying their publication. In the interest of all
users of the environmental health criteria monographs,
readers are kindly requested to communicate any errors
that may have occurred to the Manager of the International
Programme on Chemical Safety, World Health Organization,
Geneva, Switzerland, in order that they may be included in
corrigenda, which will appear in subsequent volumes.
* * *
A detailed data profile and a legal file can be
obtained from the International Register of Potentially
Toxic Chemicals, Palais des Nations, 1211 Geneva 10,
Switzerland (Telephone No. 7988400 or 7985850).
ENVIRONMENTAL HEALTH CRITERIA FOR INORGANIC MERCURY
A WHO Task Group on Environmental Health Criteria for
Inorganic Mercury met in Bologna, Italy, at the County
Council Headquarters (Provincia) from 25 to 30 September
1989. The meeting was sponsored by the Italian Ministry
of the Environment and organized locally by the Institute
of Oncology and Environmental Sciences with the assistance
of the County Council. Professor C. Maltoni, Director of
the Bologna Institute of Oncology, opened the meeting and
welcomed the participants on behalf of the host insti-
tution. Mr A. Vecchi, Dr M. Moruzzi, and Dr A. Lolli, wel-
comed the participants on behalf of the local authorities.
Dr A. Mochi, Centro Italiano Studi e Indagini, greeted the
participants on behalf of the Ministry of the Environment,
and Dr D. Kello, WHO Regional Office for Europe, addressed
the meeting on behalf of the cooperating organizations of
the IPCS (ILO/UNEP/WHO).
The Task Group reviewed and revised the draft document
and made an evaluation of the human health risks from
exposure to inorganic mercury.
The draft of this report was prepared by Dr L.
Friberg, Karolinska Institute, Stockholm, Sweden. Dr T.
Kjellström, WHO, Geneva, was responsible for the overall
scientific content and Dr P.G. Jenkins, WHO, Geneva, for
the technical editing.
* * *
Partial financial support for the publication of this
report was kindly provided by the National Institute of
Environmental Medicine, Stockholm, Sweden, and the Minis-
try of the Environment of Italy. The Centro Italiano Studi
e Indagini and the Institute of Oncology, Bologna, con-
tributed to the organization and provision of meeting
facilities.
ABBREVIATIONS
AAS atomic absorption spectrophotometry
CNS central nervous system
CVAA cold vapour atomic absorption
EEC European Economic Community
EEG electroencephalogram
GBM glomerular basement membrane
GC gas chromatography
GEMS Global Environment Monitoring System
GLC gas-liquid chromatography
LOAEL lowest-observed-adverse-effect level
MGP membranous glomerulopathy
NOAEL no-observed-adverse-effect level
SD standard deviation
SMR standardized mortality ratio
TWA time-weighted average
1. SUMMARY AND CONCLUSIONS
This monograph concentrates primarily on the risk to
human health from inorganic mercury, and examines research
reports that have appeared since the publication of
Environmental Health Criteria 1: Mercury (WHO, 1976). In
the period since 1976, new research data has become avail-
able for two main health concerns related to inorganic
mercury, i.e. mercury in dental amalgam and in skin-
lightening soaps. The emphasis in this monograph is on
exposure from these two sources, but the basic kinetics
and toxicology are reviewed with all aspects of inorganic
mercury in mind.
Human health concerns related to the global transport,
bioaccumulation, and transformation of inorganic mercury
almost exclusively arise from the conversion of mercury
compounds into methylmercury and exposure to methylmercury
in sea-food and other food. The global environmental and
ecological aspects of inorganic mercury have been summar-
ized in this monograph. More detailed descriptions may be
found in Environmental Health Criteria 86: Mercury -
Environmental Aspects (WHO, 1989) and Environmental Health
Criteria 101: Methylmercury (WHO, 1990).
1.1. Identity
Mercury exists in three states: Hg0 (metallic);
Hg2++ (mercurous); and Hg++ (mercuric). It can form
organometallic compounds, some of which have found
industrial and agricultural uses.
1.2. Physical and chemical properties
Elemental mercury has a very high vapour pressure.
The saturated atmosphere at 20 °C has a concentration over
200 times greater than the currently accepted concen-
tration for occupational exposure.
Solubility in water increases in the order: elemental
mercury < mercurous chloride < methylmercury chloride <
mercuric chloride. Elemental mercury and the halide com-
pounds of alkylmercurials are soluble in non-polar
solvents.
Mercury vapour is more soluble in plasma, whole blood,
and haemoglobin than in distilled water, where it dis-
solves only slightly. The organometallic compounds are
stable, although some are readily broken down by living
organisms.
1.3. Analytical methods
The most commonly used analytical methods for the
quantification of total and inorganic mercury compounds
are atomic absorption of cold vapour (CVAA) and neutron
activation. Detailed information relating to analytical
methods are given in Environmental Health Criteria 1:
Mercury (WHO, 1976) and Environmental Health Criteria 101:
Methylmercury (WHO, 1990).
All analytical procedures for mercury require careful
quality control and quality assurance.
1.3.1. Analysis, sampling, and storage of urine
Flameless atomic absorption spectrophotometry is used
in routine analysis for various media. Particular care
must be taken when choosing the anticoagulant used for
blood sampling in order to avoid contamination by mercury
compounds. Special care must also be taken in the sampling
and storage of urine, since bacterial growth can change
the concentration of the numerous forms of mercury that
may be present. Addition of hydrochloric acid or bacteri-
cidal substances and freezing the sample are the best
methods to prevent alteration of urine samples. Correc-
tion of concentration by reference to urine density or
creatinine content are recommended.
1.3.2. Analysis and sampling of air
Analytical methods for mercury in air may be divided
into instant reading methods and methods with separate
sampling and analysis stages. Instant reading methods can
be used for the quantification of elemental mercury
vapour. Sampling in acid-oxidizing media or on hopcalite
is used for the quantification of total mercury.
The cold vapour atomic absorption (CVAA) technique is
the most frequently used analytical method.
1.4. Sources of human and environmental exposure
1.4.1. Natural occurrence
The major natural sources of mercury are degassing of
the earth's crust, emissions from volcanoes, and evapor-
ation from natural bodies of water.
The natural emissions are of the order of 2700-6000
tonnes per year.
1.4.2. Sources due to human activities
The world-wide mining of mercury is estimated to yield
about 10 000 tonnes/year. These activities lead to some
losses of mercury and direct discharges to the atmos-
phere. Other important sources are fossil fuel combustion,
metal sulfide ore smelting, gold refining, cement pro-
duction, refuse incineration, and industrial applications
of metals.
The specific normal emission from a chloralkali plant
is about 450 g of mercury per ton of caustic soda
produced.
The total global amount and release of mercury, due to
human activities, to the atmosphere has been estimated to
be up to 3000 tonnes/year.
1.5. Uses
A major use of mercury is as a cathode in the elec-
trolysis of sodium chloride. Since the resultant chemicals
are contaminated with mercury, their use in other indus-
trial activities leads to a contamination of other
products. Mercury is used in the electrical industry, in
control instruments in the home and industry, and in lab-
oratory and medical instruments. Some therapeutic agents
contain inorganic mercury. A very large amount of mercury
is used for the extraction of gold.
Dental silver amalgam for tooth filling contains large
amounts of mercury, mixed (in the proportion of 1:1) with
alloy powder (silver, tin, copper, zinc). Copper amalgam,
used mostly in paediatric dentistry, contains up to 70%
mercury and up to 30% copper. These uses can cause ex-
posure of the dentist, dental assistants, and also of the
patients.
Some dark-skinned people use mercury-containing creams
and soap to achieve a lighter skin tone. The distribution
of these products is now banned in the EEC, in North
America, and in many African countries, but mercury-
containing soap is still manufactured in several European
countries. The soaps contain up to 3% of mercuric iodine
and the creams contain ammoniated mercury (up to 10%).
1.6. Environmental transport, distribution, and transformation
Emitted mercury vapour is converted to soluble forms
and deposited by rain onto soil and water. The atmospheric
residence time for mercury vapour is up to 3 years,
whereas soluble forms have a residence time of only a few
weeks.
The change in speciation of mercury from inorganic to
methylated forms is the first step in the aquatic bioac-
cumulation process. This can occur non-enzymically or
through microbial action. Methylmercury enters the food-
chain of predatory species where biomagnification occurs.
1.7. Human exposure
The general population is primarily exposed to mercury
through the diet and dental amalgam. Depending on the con-
centrations in air and water, significant contributions to
the daily intake of total mercury can occur. Fish is a
dominant source of human exposure to methylmercury.
Recent experimental studies have shown that mercury is
released from amalgam restorations in the mouth as vapour.
The release rate of this mercury vapour is increased, for
example, by chewing. Several studies have correlated the
number of dental amalgam fillings or amalgam surfaces with
the mercury content in tissues from human autopsy, as well
as in samples of blood, urine, and plasma. Both the pre-
dicted mercury uptake from amalgam and the observed ac-
cumulation of mercury show substantial individual vari-
ation. It is, therefore, difficult to make accurate
quantitative estimations of the mercury release and uptake
by the human body from dental amalgam tooth restorations.
Experimental studies in sheep have examined in greater
detail the distribution of mercury released from amalgam
restorations.
Use of skin-lightening soap and creams can give rise
to substantial mercury exposure.
Occupational exposure to inorganic mercury has been
investigated in chloralkali plants, mercury mines, ther-
mometer factories, refineries, and in dental clinics.
High mercury levels have been reported for all these
occupational exposure situations, although levels vary
according to work environment conditions.
1.8. Kinetics and metabolism
Results of both human and animal studies indicate that
about 80% of inhaled metallic mercury vapour is retained
by the body, whereas liquid metallic mercury is poorly
absorbed via the gastrointestinal tract (less than 1%).
Inhaled inorganic mercury aerosols are deposited in the
respiratory tract and absorbed, the rate depending on
particle size. Inorganic mercury compounds are probably
absorbed from the human gastrointestinal tract to a level
of less than 10% on average, but there is considerable
individual variation. Absorption is much higher in newborn
rats.
The kidney is the main depository of mercury after the
administration of elemental mercury vapour or inorganic
mercury compounds (50-90% of the body burden of animals).
Significantly more mercury is transported to the brain
of mice and monkeys after the inhalation of elemental
mercury than after the intravenous injection of equivalent
doses of the mercuric form. The red blood cell to plasma
ratio in humans is higher (> 1) after administration of
elemental mercury than mercuric mercury and more mercury
crosses the placental barrier. Only a small fraction of
the administered divalent mercury enters the rat fetus.
Several forms of metabolic transformation can occur:
* oxidation of metallic mercury to divalent mercury;
* reduction of divalent mercury to metallic mercury;
* methylation of inorganic mercury;
* conversion of methylmercury to divalent inorganic
mercury.
The oxidation of metallic mercury vapour to divalent
ionic mercury (section 6.1.1) is not fast enough to pre-
vent the passage of elemental mercury through the blood-
brain barrier, the placenta, and other tissues. Oxidation
in these tissues serves as a trap to hold the mercury and
leads to accumulation in brain and fetal tissues.
The reduction of divalent mercury to Hg0 has been
demonstrated both in animals (mice and rats) and humans.
The decomposition of organomercurials, including methyl-
mercury, is also a source of mercuric mercury.
The faecal and urinary routes are the main pathways
for the elimination of inorganic mercury in humans,
although some elemental mercury is exhaled. One form of
depletion is the transfer of maternal mercury to the
fetus.
The biological half-time, which only lasts a few days
or weeks for most of the absorbed mercury, is very long,
probably years, for a fraction of the mercury. Such long
half-times have been observed in animal experiments as
well as in humans. A complicated interplay exists between
mercury and some other elements, including selenium. The
formation of a selenium complex may be responsible for the
long half-time of a fraction of the mercury.
1.8.1. Reference and normal values
Limited information from deceased miners shows mercury
concentrations in the brain, years after cessation of
exposure, of several mg/kg, with still higher values in
some parts of the brain. However, lack of quality control
of the analysis makes these data uncertain. Among a small
number of deceased dentists, without known symptoms of
mercury intoxication, mercury levels varied from very low
concentrations up to a few hundred µg/kg in the occipital
lobe cortex and from about 100 µg/kg to a few mg/kg in
the pituitary gland.
From autopsies on subjects not occupationally exposed
but with a varying number of amalgam fillings, it seems
that a moderate number (about 25) of amalgam surfaces may
on average increase the brain mercury concentration by
about 10 µg/kg. The corresponding increase in the kid-
neys, based on a very limited number of analyses, is
probably 300-400 µg/kg. However, the individual vari-
ation is considerable.
Mercury levels in urine and blood can be used as indi-
cators of exposure provided that the exposure is recent
and relatively constant, is long-term, and is evaluated on
a group basis. Recent exposure data are more reliable
than those quoted in Environmental Health Criteria 1:
Mercury (WHO, 1976). Urinary levels of about 50 µg per g
creatinine are seen after occupational exposure to about
40 µg mercury/m3 of air. This relationship (5:4) between
urine and air levels is much lower that the 3:1 estimated
by WHO (1976). The difference may in part be explained by
different sampling technique for evaluating air exposure.
An exposure of 40 µg mercury/m3 of air will correspond
to about 15-20 µg mercury/litre of blood. However, inter-
ference from methylmercury exposure can make it difficult
to evaluate exposure to low concentrations of inorganic
mercury by means of blood analysis. A way to overcome the
problems is to analyse mercury in plasma or analyse both
inorganic mercury and methylmercury. The problem of inter-
ference from methylmercury is much smaller when analysing
urine, as methylmercury is excreted in the urine to only a
very limited extent.
1.9. Effects in humans
Acute inhalation exposure to mercury vapour may be
followed by chest pains, dyspnoea, coughing, haemoptysis,
and sometimes interstitial pneumonitis leading to death.
The ingestion of mercuric compounds, in particular
mercuric chloride, has caused ulcerative gastroenteritis
and acute tubular necrosis causing death from anuria where
dialysis was not available.
The central nervous system is the critical organ for
mercury vapour exposure. Subacute exposure has given rise
to psychotic reactions characterized by delirium, halluci-
nations, and suicidal tendency. Occupational exposure has
resulted in erethism as the principal feature of a broad
ranging functional disturbance. With continuing exposure a
fine tremor develops, initially involving the hands. In
the milder cases erethism and tremor regress slowly over a
period of years following removal from exposure. De-
creased nerve conduction velocity has been demonstrated in
mercury-exposed workers. Long-term, low-level exposure
has been associated with less pronounced symptoms of
erethism.
There is very little information available on brain
mercury levels in cases of mercury poisoning, and nothing
that makes it possible to estimate a no-observed-effect
level or a dose-response curve.
At a urinary mercury excretion level of 100 µg per g
creatinine, the probability of developing the classical
neurological signs of mercurial intoxication (tremor,
erethism) and proteinuria is high. An exposure correspond-
ing to 30 to 100 µg mercury/g creatinine increases the
incidence of some less severe toxic effects that do not
lead to overt clinical impairment. In a few studies
tremor, recorded electrophysiologically, has been observed
at low urine concentrations (down to 25-35 µg/g creati-
nine). Other studies did not show such an effect. Some of
the exposed people develop proteinuria (proteins of low
relative molecular mass and microalbuminuria). Appropriate
epidemiological data covering exposure levels correspond-
ing to less than 30-50 µg mercury/g creatinine are not
available.
The exposure of the general population is generally
low, but may occasionally be raised to the level of occu-
pational exposure and can even be toxic. Thus, the
mishandling of liquid mercury has resulted in severe
intoxication.
The kidney is the critical organ following the
ingestion of inorganic divalent mercury salts. Occu-
pational exposure to metallic mercury has long been
associated with the development of proteinuria, both in
workers with other evidence of mercury poisoning and in
those without such evidence. Less commonly, occupational
exposure has been followed by the nephrotic syndrome,
which has also occurred after the use of skin-lightening
creams containing inorganic mercury, and even after acci-
dental exposure. The current evidence suggests that this
nephrotic syndrome results from an immunotoxic response.
Until recently, effects of elemental mercury vapour on the
kidney had been reported only at doses higher than those
associated with the onset of signs and symptoms from the
central nervous system. New studies have, however, re-
ported kidney effects at lower exposure levels. Experi-
mental studies on animals have shown that inorganic
mercury may induce auto-immune glomerulonephritis in all
species tested, but not in all strains, indicating a
genetic predisposition. A consequence of an immunological
etiology is that, in the absence of dose-response studies
for groups of immunologically sensitive individuals, it is
not scientifically possible to set a level for mercury
(e.g., in blood or urine) below which (in individual
cases) mercury-related symptoms will not occur.
Both metallic mercury vapour and mercury compounds
have given rise to contact dermatitis. Mercurial pharma-
ceuticals have been responsible for Pink disease in
children, and mercury vapour exposure may be a cause of
"Kawasaki" disease. In some studies, but not in others,
effects on the menstrual cycle and/or fetal development
have been reported. The standard of published epidemio-
logical studies is such that it remains an open question
whether mercury vapour can adversely affect the menstrual
cycle or fetal development in the absence of the well-
known signs of mercury intoxication.
Recently, there has been an intense debate on the
safety of dental amalgams and claims have been made that
mercury from amalgam may cause severe health hazards.
Reports describing different types of symptoms and signs
and the results of the few epidemiological studies
produced are inconclusive.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1. Identity
This monograph focuses on the risk to human health
from the compounds of inorganic mercury. Other forms of
mercury are discussed where they are relevant to the full
evaluation of human health risks, e.g., the metabolic
transformation of methylmercury to inorganic mercury.
Elemental mercury has the CAS registry number
7439-97-6 and a relative atomic mass of 200.59. There
are three states of inorganic mercury: Hg0 (metallic),
Hg2++ (mercurous), and Hg++ (mercuric) mercury. The
mercurous and mercuric states form numerous inorganic and
organic chemical compounds. Organic forms are those in
which mercury is attached covalently to at least one
carbon atom.
2.2. Physical and chemical properties
In its elemental form, mercury is a heavy silvery
liquid at room temperature. At 20 °C the specific gravity
of the metal is 13.456 and the vapour pressure is 0.16 Pa
(0.0012 mmHg). Thus, a saturated atmosphere at 20 °C con-
tains approximately 15 mg/m3. This concentration is
300 times greater than the recommended health-based occu-
pational exposure limit of 0.05 mg/m3 (WHO, 1980).
Mercurials differ greatly in their solubilities.
Solubility values in water are: elemental mercury (30 °C),
2 µg/litre; mercurous chloride (25 °C), 2 mg/litre; mer-
curic chloride (20 °C), 69 g/litre (Linke, 1958; CRC,
1972). The solubility of methylmercury chloride in water
is higher than that of mercurous chloride by about three
orders of magnitude, this being related to the very high
solubility of the methylmercury cation in water (Linke,
1958; Clarkson et al., 1988b). Certain species of mercury
are soluble in non-polar solvents. These include elemental
mercury and the halide compounds of alkylmercurials
(Clarkson et al., 1988b).
From the biochemical point of view the most important
chemical property of mercuric mercury and alkylmercurials
is their high affinity for sulfhydryl groups.
Hursh (1985) showed that mercury vapour is more sol-
uble in plasma, whole blood, and haemoglobin than in dis-
tilled water or isotonic saline.
The following speciation among mercury compounds has
been proposed by Lindqvist et al. (1984), where V indi-
cates volatile species, R water-soluble particle-borne
reactive species, and NR non-reactive species:
V: Hg0 (elemental mercury), (CH3)2Hg
R: Hg2+, HgX2, HgX3-, and HgX42- (where X = OH-,
Cl-, or Br-), Hg0 on aerosol particles, Hg2+ com-
plexes with organic acids.
NR: CH3Hg+, CH3HgCl, CH3HgOH, and other organomer-
curic compounds, Hg(CN)2, HgS, and Hg2+ bound to
sulfur in fragments of humic matter.
The main volatile form in air is elemental mercury,
but dimethylmercury may also occur (Slemr et al., 1981).
Uncharged complexes, such as HgCl2 and CH3HgOH, oc-
cur in the gaseous phase, but are also relatively stable
in fresh water (snow and rain as well as standing or flow-
ing water). HgCl42- is the dominant form in sea water.
2.3. Conversion factors
1 ppm = 1 mg/kg = 5 µmol/kg
1 mol creatinine = 113.1 g creatinine
2.4. Analytical methods
Detailed information relating to analytical methods
was given in Environmental Health Criteria 1: Mercury
(WHO, 1976) and in Environmental Health Criteria 101:
Methylmercury (WHO, 1990). This monograph contains further
information concerning the sampling and analysis of urine
and air, the most frequently studied media for evaluation
of exposure to inorganic mercury. A summary of the com-
monly used analytical methods is given in Table 1. More
advanced methods, such as inductively coupled plasma
atomic emission spectrometry and spark source mass spec-
trometry, are described in Kneip & Friberg (1986).
2.4.1. Analysis, sampling, and storage of urine
For routine analysis, various forms of flameless
atomic absorption spectrophotometry (AAS) are used. The
"Magos" selective atomic absorption method determines
both total and inorganic mercury and, by difference,
organic mercury. The neutron activation procedure is
regarded as the most accurate and sensitive procedure and
is usually used as the reference method.
Table 1. Analytical methods for the determination of mercury
--------------------------------------------------------------------------------------------------------------------
Media Speciation Analytical Detection Comments References
method limit
(ng Hg/g)
--------------------------------------------------------------------------------------------------------------------
Food, tissues total mercury atomic 2.0 method has many adaptations Hatch & Ott (1968)
absorption (see Peter & Strunc, 1984)
Blood, urine total mercury atomic 0.5 also estimates organic mercury Magos (1971); Magos &
inorganic mercury absorption as difference between total Clarkson (1972)
and inorganic
Blood, urine total mercury atomic 2.5 automated form of the method Farant et al. (1981)
hair, tissues inorganic mercury absorption of Magos (1971)
Blood, urine total mercury atomic 4.0 automated form of the method Coyle & Hartley (1981)
hair, tissues inorganic mercury absorption of Magos (1971)
All media total mercury neutron 0.1 reference method (review) WHO (1976)
activation
--------------------------------------------------------------------------------------------------------------------
Blood samples are best collected in "vacutainers"
containing heparin (without mercury compounds as preserv-
ative) (WHO, 1980) and stored at 4 °C prior to analysis.
This method of collection is especially important if mer-
cury levels in plasma and red blood cells are to be
measured. Blood samples can usually be stored for one or
two days before haemolysis becomes significant (Clarkson
et al., 1988c).
The sampling and storage of urine have been discussed
in detail by Clarkson et al. (1988c). It is important to
avoid contamination of urine samples; special cleaning
procedures and the use of metal-free polyethylene con-
tainers have been recommended.
As a rule, urine is saturated with several inorganic
salts. Precipitates are sometimes seen in freshly voided
samples and are normally present in urine samples that
have been stored at low temperature (1-4 °C). To lessen
problems of precipitates, urine samples should be homogen-
ized by shaking before analysis. Alternatively, a strong
acid, preferably hydrochloric acid, can be added to the
urine sample to lower pH and increase the solubility of
the salts.
Bacterial growth is rapid in urine at room tempera-
ture. Even urine samples from healthy people become over-
grown with bacteria after only a few hours. If urine
samples are frozen (to below -20 °C), bacterial growth is
reduced substantially. Bacteria may reduce some mercury
compounds to elemental mercury, which might give rise to
significant losses of mercury by volatilization (WHO,
1976). Bactericidal substances, such as sodium azide, may
be added to urine samples. However, sodium azide is a
strong reducing agent and may form Hg0 from Hg2+. The
addition of 1 g sulfamic acid and 0.5 ml of a detergent
(Triton X-100) to 500 ml of urine produces stable urine
samples at room temperature for at least one month (Skare,
1972).
Even when the rate of metal excretion is constant,
metal concentration in urine varies according to the urine
flow rate (Diamond, 1988). It is therefore necessary to
adjust the measured concentrations of metals in spot urine
samples for variations in the urine flow rate. This can be
done by correcting for urine relative density or osmo-
lality or by dividing by the concentration of creatinine
in the urine sample. Another alternative is the use of
timed urine specimens (e.g., 4 h or 8 h). If the concen-
tration of a substance is standardized to a constant rela-
tive density (usually 1.018 or 1.024), the basis of cor-
rection chosen profoundly changes the figures obtained.
Correction to 1.024 gives values 33% higher than correc-
tion to 1.018 (Aitio, 1988). Furthermore, many chemicals,
including mercury, exhibit diurnal variation in concen-
tration (Piotrowski et al., 1975). Correction using cre-
atinine values has the advantage that the mercury concen-
tration will be independent of hydration status.
2.4.2. Analysis and sampling of air
Analytical methods for mercury in air may be divided
into instant reading methods and methods with separate
sampling and analysis stages (WHO, 1976).
One instant reading method is based on the "cold
vapour atomic absorption" (CVAA) technique, which
measures the absorption of mercury vapour by ultra-violet
light using a wave length of 253.7 nm. Most of the AAS
procedures have a detection limit in the range of 2 to
5 µg mercury/m3.
Another instant reading method that has been used
increasingly in recent years is a special type of gold
amalgamation technique. This method has been used in a
number of studies for evaluating the release of elemental
mercury vapour in the oral cavity from amalgam fillings
(Svare et al., 1981; Vimy & Lorscheider, 1985a,b).
McNerney et al. (1972) gave a detailed description of the
method, which is based on an increase in the electrical
resistance of a thin gold film after adsorption of mercury
vapour. The detection limit is 0.05 ng mercury. Within the
range of 0.5 to 25 ng, the relative standard deviation was
found to vary between 3 and 10% when 15 samples from each
of 6 mercury vapour standards were examined. At higher
mercury concentrations, the films become saturated with
mercury and precision decreases. It is possible to correct
for this saturation with a calibration curve. However,
there are no data on the accuracy of the method when used
in actual field studies, such as the ones by Svare et al.
(1981) or Vimy & Lorscheider (1985a,b).
In an analytical method based on separate sampling and
analysis, the air is sampled in two bubblers in series,
containing sulfuric acid and potassium permanganate (WHO,
1976). The mercury is subsequently determined by CVAA.
With this method the total mercury in the air is measured,
not just mercury vapour. Another sampling technique uses
solid absorbants. Different amalgamation techniques using
gold have been shown to have good collection efficiency
for mercury vapour (McCammon et al., 1980; Dumarey et al.,
1985; Skare & Engqvist, 1986). Roels et al. (1987) used a
filter with two layers of hopcalite (a mixture of metal
oxides that can absorb metals) to collect the mercury.
After solubilization, the mercury was analysed by a CVAA
technique. It was necessary also to measure blanks of hop-
calite and scrubbing solution. Large variations were found
for background mercury contamination of hopcalite from
batch to batch (6-93 ng mercury per 200 mg hopcalite).
Sampling of air for mercury analysis can be made by
static samplers or by personal monitoring. Personal
samplers are recommended. A study by Roels et al. (1987)
compared results obtained with the use of static samplers
with results from personal samplers. In most of the
workplaces, personal samplers yielded higher exposure
levels (time-weighted averages) than did static samplers
(see section 6.5.2).
2.4.3. Quality control and quality assurance
General considerations of quality control and quality
assurance have been recommended by WHO (UNEP/WHO, 1984;
WHO, 1986; Aitio, 1988). At a recent conference on "Bio-
logical Monitoring of Toxic Metals" (Friberg, 1988), a
WHO approach based on a GEMS programme (Vahter, 1982) was
described in detail. Specific quality control programmes
for mercury in hair using the GEMS approach have been
described (Lind et al., 1988). Roels et al. (1987) suc-
cessfully used another regression method when analysing
mercury in urine.
In almost any quality control programme, there is a
need for reference materials containing the metal in con-
centrations covering the expected working range of moni-
toring samples. Several reference materials are commer-
cially available for both environmental samples and for
urine and blood (Muramatsu & Parr, 1985; Parr et al.,
1987; Rasberry, 1987; Parr et al., 1988; Okamoto, 1988).
The following are suppliers of reference materials: NIST
(Office of Standard Reference Materials, National Insti-
tute of Standards and Technology, Rm. B311, Chemistry
Bldg., Gaithersburg, MD 20899, USA), IAEA (International
Atomic Energy Agency, Analytical Quality Control Services,
Laboratory Seibersdorf, A-1400 Vienna), BCR (Community
Bureau of Reference, Commission of the European Communi-
ties, 200 Rue de la Loi, B-1049 Brussels, Belgium); NIES
(National Institute for Environmental Studies, Japan
Environment Agency, P.O. Yatabe, Tsukuba Ibaraki 300-21,
Japan), NRCC (National Research Council Canada, Division
of Chemistry, Ottawa, K1A OR6, Canada), Nycomed AS Diag-
nostics (P.O. Box 4220, Torshov, 0401 Oslo 4, Norway),
Behring Institute (P.O. Box 1140, D-3550 Marburg 1,
Germany), Kaulson Laboratories Inc. (691 Bloomfield
Avenue, Caldwell, New Jersey 07006, USA). However, the
available reference materials do not cover the demand for
different mercury species, biological media or for differ-
ent concentrations. Only NRCC has a reference material
(fish) for total mercury and for methylmercury.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
The major natural sources of mercury are the degassing
of the earth's crust, emissions from volcanoes, and evap-
oration from natural bodies of water (National Academy of
Sciences, 1978; Nriagu, 1979; Lindqvist et al., 1984). The
most recent estimates indicate that natural emissions are
of the order of 2700-6000 tonnes per year (Lindberg et
al., 1987).
The earth's crust is also an important source of mer-
cury for bodies of natural water. Some of this mercury is
undoubtedly of natural origin, but some may have been
deposited from the atmosphere and may ultimately have been
generated by human activities (Lindqvist et al., 1984).
Thus, it is difficult to assess quantitatively the rela-
tive contributions of natural and anthropogenic mercury to
run-off from land to natural bodies of water. Data con-
cerning mercury in the general environment and in food
have been reviewed in Environmental Health Criteria 101:
Methylmercury (WHO, 1990).
3.2. Man-made sources
The worldwide mining of mercury is estimated to yield
about 10 000 tonnes/year. Mining activities result in
losses of mercury through the dumping of mine tailings and
direct discharges to the atmosphere. The Almaden mercury
mine in Spain, which accounts for 90% of the total output
of the European Community, was expected to produce 1380
tonnes in 1987 (Seco, 1987). Other important sources are
the combustion of fossil fuel, the smelting of metal sul-
fide ores, the refining of gold (sometimes under very
primitive conditions), the production of cement, refuse
incineration, and industrial metal applications. The
emissions of mercury to the atmosphere in Sweden in 1984
were estimated to be as follows (in kg/year): incineration
of household waste (3300), smelting (900), chloralkali
industry (400), crematories (300), mining (200), combus-
tion of coal and peat (200), other sources (200) (Swedish
Environmental Protection Board, 1986). Analogous data for
the estimated atmospheric emissions of mercury in the
United Kingdom were (in kg/year): fossil fuel combustion
(25 500), production and use of articles containing mer-
cury (10 100), municipal waste incineration (5900), non-
ferrous metal production (5000), cement manufacture
(2500), iron and steel production (1800), sewage sludge
incineration (500) (Dean & Suess, 1985). In developing
countries the emissions from industry and mining may be
much greater. For example, the emission to water from one
single chloralkali plant in Nicaragua in 1980 was 24 kg
per day (9 tonnes/year) (Velasquez et al., 1980). It was
estimated that 450 g of mercury was emitted per tonne of
soda produced in six chloroalkali plants in Argentina, and
the quantity of mercury released in the environment was
about 86 tonnes/year (Gotelli, 1989).
The total global release of mercury to the atmosphere
due to human activities has been estimated to be of the
order of 2000-3000 tonnes/year (Lindberg et al., 1987;
Pacyna, 1987). It should be stressed that there are con-
siderable uncertainties in the estimated fluxes of mercury
in the environment and in its speciation. Concentrations
in the unpolluted atmosphere and in natural bodies of
water are so low that they are near the limit of detection
of current analytical methods, even for the determination
of total mercury.
Although amounts of mercury resulting from human ac-
tivities may be quite small relative to global emissions,
the anthropogenic release of elemental metal mercury into
confined areas was the source of the poisoning outbreaks
in Minamata and Niigata (WHO, 1976).
3.3. Uses
A major use of mercury is as a cathode in the elec-
trolysis of sodium chloride solution to produce caustic
soda and chlorine gas, which has important uses in the
paper-pulp industry. It should be noted that all the elec-
trolytic products (hydrogen, chlorine, sodium hydroxide,
sodium hypochlorite, and hydrochloric acid) are contami-
nated with mercury (Gotelli, 1989). These substances are
important in the economy of other industrial activities
and the presence of mercury can contaminate other prod-
ucts. About 50 tonnes of liquid metal are used in each
manufacturing plant. In most industrialized countries,
stringent procedures have been taken to reduce losses of
mercury. Mercury is widely used in the electrical industry
(lamps, arc rectifiers, and mercury battery cells), in
control instruments in the home and industry (switches,
thermostats, barometers), and in other laboratory and
medical instruments. It is also widely used in the dental
profession for tooth amalgam fillings. Other therapeutic
agents, such as teething powders, ointments, and laxa-
tives, contain inorganic mercury (ATSDR, 1989), as do some
antihistaminic preparations sold in Italy (EDIMED, 1989).
Organic mercury compounds continue to be used in anti-
fouling and mildew-proofing latex paints and to control
fungus infections of seeds, bulb plants, and vegetation.
The World Health Organization has warned against the use
of alkylmercury compounds in seed dressing (WHO, 1976).
One of the uses of liquid metallic mercury that may
have a serious impact on health is the extraction of gold
from ore concentrates or from recycled gold articles.
Reports from China (Wu et al., 1989) indicate high ex-
posure in the vicinity of "cottage industry" operations
of this type, and Villaluz (1988) reported that 50 000
people may be exposed around small scale gold mining oper-
ations in Indonesia, Kampuchea, the Philippines, and
Viet Nam. The same problem also occurs in Brazil and
Colombia. The release of elemental mercury from these
activities is about 120 tonnes/year in Brazil (Gotelli,
1989).
3.4. Dental amalgam in dentistry
WHO (1976) estimated that in industrial countries
about 3% of the total consumption of mercury was used for
dental amalgam. Amalgam has been used extensively as a
tooth-filling material for more than 150 years and
accounts for 75-80% of all single tooth restorations
(Bauer & First, 1982; Wolff et al., 1983). It has been
estimated that each American dentist in private practice
uses on average 0.9-1.4 kg of amalgam per year (Naleway et
al., 1985).
Most conventional silver amalgams consist of a 1:1
mixture of metallic mercury and an alloy powder consisting
of silver (about 70% by weight), tin (about 25%), and
smaller amounts of copper (1-6%) and zinc (0-2%). A modern
type of silver amalgam is also available, containing
higher amounts of copper (up to about 25%). At the time of
trituration (mixing), the amalgam generally contains simi-
lar weights of alloy powders and mercury. Excess mercury
(< 5%) is removed immediately before or at the conden-
sation of the plastic amalgam mix in the prepared tooth
cavity. The amalgam begins to set within minutes of inser-
tion and therefore needs to be carved to satisfactory
anatomic form within this period of time. Finishing (e.g.,
polishing) with rotating instruments can take place after
setting for 24 h, but continuing hardening of amalgam
restorations takes place over many months (ADA, 1985;
Enwonwu, 1987; SOS, 1987).
Previously, amalgam was usually prepared with mortar
and pestle. The amalgam mixture was thereafter placed on
a cloth filter and squeezed to expel excess mercury. This
method of handling amalgam easily vapourizes mercury and
there is also a risk of spillage. The technique is still
in use in some countries (section 9.5.2.2). The modern,
safer method for the preparation of amalgam involves
mixing the alloy with mercury in a sealed capsule. This
decreases the occupational exposure substantially (Harris
et al., 1978; Skuba, 1984).
A second type of dental amalgam is the so-called
"copper amalgam" used mostly in paediatric dentistry
until a few decades ago. This material contained 60-70%
mercury and 30-40% copper, and was prepared by open
heating in the dental surgery. This process naturally gave
rise to considerable occupational mercury vapour exposure.
Copper amalgams were easier to retain in dental cavities
because of their higher initial plasticity than silver
amalgams. Contrary to silver amalgam fillings, copper
amalgam undergoes easily detectable dissolution with time.
This solubilization was, for some time, actually con-
sidered an advantage because of the associated bacteri-
cidal effects (SOS, 1987).
A source of mercury loss to the atmosphere is the
release of metallic mercury vapour during the cremation of
cadavers. Crematories are often located in densely popu-
lated areas and do not have high chimneys. All the mercury
from amalgam fillings vapourizes during the cremation, as
the temperature is above 800 °C. In a Swedish study, it
has been estimated that 170-180 kg of metallic mercury is
released annually from a total of about 50 000 cremations
per year (Mörner & Nilsson, 1986). The use of amalgam in
Sweden is estimated to be 5-7.5 tonnes per year (SOS,
1987), compared with 90-100 tonnes in the USA (Wolff et
al., 1983; Naleway et al., 1985). It is difficult to esti-
mate the global release of mercury vapour from cremation
due to uncertainties about dental status at the time of
death in relation to frequency of cremations.
3.5. Mercury-containing cream and soap
Mercury-containing cream and soap has for a long time
been used by dark-skinned people to obtain a lighter skin
tone, probably due to inhibition of pigment formation.
There are mainly two types of products distributed for
this purpose: skin-lightening creams and skin-lightening
soaps. This subject has recently been reviewed by Berlin
(personal communication to the IPCS by M. Berlin).
The distribution of the two products is now banned in
the European Economic Community, in North America, and in
many African states. Mercury-containing soap is, however,
manufactured in several European countries and sold as
germicidal soap to the Third World, and it has frequently
been found in European cities with a substantial black
population, such as London and Brussels. This implies that
the mercury-containing soap manufactured in Europe has
been re-imported illegally from African countries.
English community health authorities (Lambeth, 1988)
have identified several brands of soap containing mercury.
The soaps have been analysed and contain typically 1-3% of
mercuric iodide. There are also skin-lightening creams
containing ammoniated mercury from 1-5% (Marzulli & Brown,
1972) or 5-10% (Barr et al., 1973). Both the soap and the
cream are applied on the skin, allowed to dry on the skin
surface, and left overnight.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
There is a well-recognized global cycle for mercury,
whereby emitted mercury vapour is converted to soluble
forms (e.g., Hg++) and deposited by rain onto soil and
water. Mercury vapour has an atmospheric residence time of
between 0.4 and 3 years, whereas soluble forms have resi-
dence times of a few weeks. Transport in soil and water is
thus limited and deposition within a short distance is
highly likely.
The change in mercury speciation from inorganic to
methylated forms is the first step in the aquatic bioac-
cumulation process. Methylation can occur non-enzymati-
cally or through microbial action. Once methylmercury is
released, it enters the food chain by rapid diffusion and
tight binding to proteins. It attains its highest levels,
through food-chain biomagnification, in the tissues of
such predatory species as freshwater trout, pike, and bass
and marine tuna, swordfish, and shark. The ratio of the
methylmercury concentration in fish tissue to the concen-
tration of inorganic mercury in water is usually between
10 000 and 100 000 to one. Levels of selenium in the
water may affect the availability of mercury for uptake
into aquatic biota. Reports from Sweden and Canada point
to the likelihood of increased methylmercury concentration
in fish after the construction of artificial water reser-
voirs (WHO, 1990).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
The general population is primarily exposed to mercury
from dental amalgam and the diet. However, depending upon
the level of contamination, air and water can contribute
significantly to the daily intake of total mercury. In
most foodstuffs, mercury is usually in the inorganic form
and below the limit of detection (20 ng mercury/g fresh
weight). The exceptions are fish and fish products, which
are the main source of methylmercury in the diet. Levels
greater than 1.2 mg/kg are often found in the edible
portion of shark, swordfish, and Mediterranean tuna. Simi-
lar levels in pike, walleye, and bass taken from polluted
fresh water have been identified. Table 2 indicates the
average daily intake and retention of total mercury and
mercury compounds in the general population not occu-
pationally exposed to mercury.
The level of mercury in fish, even for humans consum-
ing small amounts (10-30 g of fish/day), can markedly
affect the intake of methylmercury and, thus, of total
mercury. The weekly consumption of 200 g of fish having
500 µg mercury/kg will result in the intake of 100 µg
mercury (predominantly methylmercury). This amount is one-
half of the tolerable recommended weekly intake (WHO,
1989).
The subject of human mercury dietary exposure has been
discussed in previous Environmental Health Criteria mono-
graphs (WHO, 1976, 1990). This section emphasizes human
exposure to inorganic mercury from dental amalgam and
skin-lightening creams and soaps among the general popu-
lation, and occupational exposure due to the use of
amalgam in dentistry. Industrial exposure was described in
detail in WHO (1976); more recent information is discussed
in section 9.
5.1. General population exposure
5.1.1. Exposure from dental amalgam
5.1.1.1 Human studies
The release of mercury vapour from dental amalgam
fillings has been known for a very long time (Stock,
1939). The next major contribution to this field was that
of Frykholm (1957). Using a radioactive mercury tracer, he
showed that the insertion of amalgam in both humans and
dogs resulted in significant concentrations of mercury in
urine and faeces. In humans, the concentration of urinary
mercury increased during a 5-day period following the
insertion of 4-5 small occlusal fillings. A new higher
peak occurred a couple of days after removal of these
fillings. Faecal elimination showed a similar pattern,
appearing on the second day after amalgam insertion.
Another maximum appeared 1-2 days after amalgam removal.
Frykholm (1957) also measured the concentration of mercury
in the oral cavity during amalgam placement in teeth.
Recently, concern over amalgam usage has been revived by
the publication of a number of experimental studies
showing that, among other elements, inorganic mercury is
released from amalgam in vitro (Brune, 1981; Brune & Evje,
1985). More importantly, mercury vapour released in the
mouth in vivo leads to an increased uptake of mercury in
body tissues (Gay et al., 1979; Svare et al., 1981;
Abraham et al., 1984; Ott et al., 1984; Patterson et al.,
1985; Vimy & Lorscheider, 1985a,b; Vimy et al., 1986;
Langworth et al., 1988; Nylander et al., 1987, 1989;
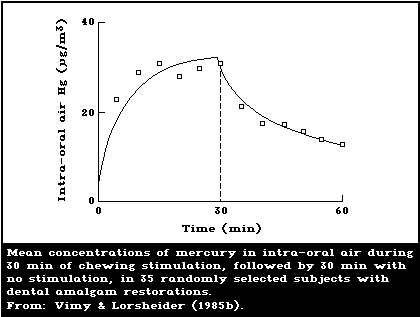
Berglund et al., 1988; Aronsson et al., 1989). Vimy &
Lorscheider (1985b) showed that the release rate of mer-
cury vapour increases dramatically when the amalgam is
stimulated by continuous chewing, reaching a plateau
within 10 min. After the cessation of chewing, it takes
approximately 90 min for the mercury release rate to
decline to the basal pre-chewing value (Fig. 1). A con-
firmatory study has recently been published by Aronsson et
al. (1989), who also made daily dose estimates.
Table 2. Estimated average daily intake and retention (µg/day) of
total mercury and mercury compounds in the general population not
occupationally exposed to mercurya
---------------------------------------------------------------------
Exposure Elemental Inorganic mercury Methylmercury
mercury vapour compounds
---------------------------------------------------------------------
Air 0.030 (0.024) 0.002 (0.001) 0.008 (0.0064)
Food
Fish 0 0.600 (0.042) 2.4 (2.3)
Non-fish 0 3.6 (0.25) 0
Drinking-water 0 0.050 (0.0035) 0
Dental amalgams 3.8-21 (3-17) 0 0
Total 3.9-21 (3.1-17) 4.3 (0.3) 2.41 (2.31)
---------------------------------------------------------------------
a From: Environmental Health Criteria 101: Methylmercury (WHO, 1990).
Values given are the estimated average daily intake; the figures
in parentheses represent the estimated amount retained in the body
of an adult.
Values are quoted to 2 significant figures.
Critical reviews have been made of published infor-
mation on mercury release and exposure from amalgam
(Enwonwu, 1987; Friberg & Nylander, 1987; Langan et al.,
1987; Mackert, 1987; Olsson & Bergman, 1987; Clarkson et
al., 1988a). From these reviews it can be concluded that
it is difficult to make accurate quantitative estimations
of the mercury release from amalgam and the uptake of
mercury by the human body. Problems include uncertainty
about analytical quality control, differences in sampling
methodology, breathing pattern, dilution with inhaled air,
and uncertainty about time since previous meals. Due to
these factors, some studies may have overestimated and
others underestimated the daily dose of mercury, while
others may have underestimated or overestimated the mer-
cury uptake.
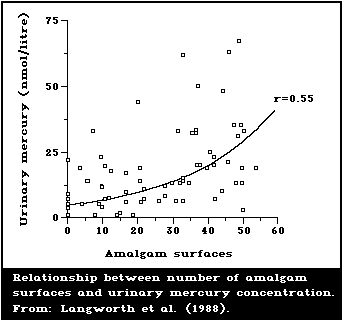
 Several studies have correlated the number of dental
amalgam fillings or amalgam surfaces with the mercury
content in brain and kidney tissue from human autopsy.
Subjects with no dental amalgam had a mean mercury level
of 6.7 ng/g (2.4-12.2) in the occipital cortex; whereas,
subjects with amalgams had a mean level of 12.3 ng/g
(4.8-28.7) (Friberg & Nylander, 1987; Nylander et al.,
1987). Amalgam-free subjects had a mean mercury level in
kidneys of 49 ng/g (21-105), whereas subjects with amalgam
fillings had a corresponding level of 433 ng/g (48-810).
In a similar investigation, Eggleston & Nylander (1987)
showed mean mercury levels of 6.7 ng/g (1.9-22.1) and 3.8
ng/g (1.4-7.1) in grey and white brain matter, respect-
ively, in subjects with no amalgam fillings. In subjects
with amalgam fillings, mercury levels were 15.2 ng/g
(3.0-121.4) and 11.2 ng/g (1.7-110.1) for grey and white
matter, respectively. In a more recent extensive study,
Schiele (1988) showed a mean brain occipital mercury con-
centration of 10 ng/g for 44 subjects with an average of
14 amalgam surfaces each. Kidneys from the same subjects
showed a sex difference in the mercury concentrations,
mean values being 484 ng/g for the 16 females and 263 for
the 28 males. Amalgam-free subjects were not included in
this study.
Using published experimental data (Svare et al., 1981;
Abraham et al., 1984; Patterson et al., 1985; Vimy &
Lorsheider, 1985b), the amalgam mercury release rate,
average daily mercury uptake, and its steady-state contri-
bution to blood, urine, brain, and kidney were estimated
by Clarkson et al. (1988a). These estimations gave brain,
kidney, and urine values that are similar to data reported
from human studies (brain and kidney autopsy samples:
Friberg et al., 1986; Nylander et al., 1987; Schiele,
1988; urine: Nilsson & Nilsson, 1986b; Olstad et al.,
1987; Langworth, 1987). A representative illustration of
the type of relationship found is given in Fig. 2. Esti-
mates of daily dosages of mercury attributed to amalgam
have also been reported by Mackert (1987) and Olsson &
Bergman (1987), although they are somewhat lower than
those of Clarkson et al. (1988a).
Snapp et al. (1989) studied the blood mercury level
before and 18 weeks after the removal of amalgam fillings.
After the removal, nine of the ten subjects examined
exhibited a statistically significant mean decrease of
1.13 ng (± 0.6) mercury/ml in the blood mercury level.
Recently, Molin et al. (1990) studied mercury concen-
trations in human plasma, erythrocytes, and urine before
and up to 12 months after removal of amalgam fillings and
replacements with gold alloy restorations. They noted an
initial increase in all recorded mercury levels after
amalgam removal. About three months thereafter, plasma and
erythrocyte levels decreased markedly. A continuous
reduction in urine mercury levels took place, reaching a
plateau of approximately 25% of the pre-removal mercury
level within 9 months.
It is important to note that, in the studies cited,
both the predicted mercury uptake from amalgam and the
observed accumulation of mercury in the body are average
values. It is also clear from the original reports that
substantial individual variations exist.
Several studies have correlated the number of dental
amalgam fillings or amalgam surfaces with the mercury
content in brain and kidney tissue from human autopsy.
Subjects with no dental amalgam had a mean mercury level
of 6.7 ng/g (2.4-12.2) in the occipital cortex; whereas,
subjects with amalgams had a mean level of 12.3 ng/g
(4.8-28.7) (Friberg & Nylander, 1987; Nylander et al.,
1987). Amalgam-free subjects had a mean mercury level in
kidneys of 49 ng/g (21-105), whereas subjects with amalgam
fillings had a corresponding level of 433 ng/g (48-810).
In a similar investigation, Eggleston & Nylander (1987)
showed mean mercury levels of 6.7 ng/g (1.9-22.1) and 3.8
ng/g (1.4-7.1) in grey and white brain matter, respect-
ively, in subjects with no amalgam fillings. In subjects
with amalgam fillings, mercury levels were 15.2 ng/g
(3.0-121.4) and 11.2 ng/g (1.7-110.1) for grey and white
matter, respectively. In a more recent extensive study,
Schiele (1988) showed a mean brain occipital mercury con-
centration of 10 ng/g for 44 subjects with an average of
14 amalgam surfaces each. Kidneys from the same subjects
showed a sex difference in the mercury concentrations,
mean values being 484 ng/g for the 16 females and 263 for
the 28 males. Amalgam-free subjects were not included in
this study.
Using published experimental data (Svare et al., 1981;
Abraham et al., 1984; Patterson et al., 1985; Vimy &
Lorsheider, 1985b), the amalgam mercury release rate,
average daily mercury uptake, and its steady-state contri-
bution to blood, urine, brain, and kidney were estimated
by Clarkson et al. (1988a). These estimations gave brain,
kidney, and urine values that are similar to data reported
from human studies (brain and kidney autopsy samples:
Friberg et al., 1986; Nylander et al., 1987; Schiele,
1988; urine: Nilsson & Nilsson, 1986b; Olstad et al.,
1987; Langworth, 1987). A representative illustration of
the type of relationship found is given in Fig. 2. Esti-
mates of daily dosages of mercury attributed to amalgam
have also been reported by Mackert (1987) and Olsson &
Bergman (1987), although they are somewhat lower than
those of Clarkson et al. (1988a).
Snapp et al. (1989) studied the blood mercury level
before and 18 weeks after the removal of amalgam fillings.
After the removal, nine of the ten subjects examined
exhibited a statistically significant mean decrease of
1.13 ng (± 0.6) mercury/ml in the blood mercury level.
Recently, Molin et al. (1990) studied mercury concen-
trations in human plasma, erythrocytes, and urine before
and up to 12 months after removal of amalgam fillings and
replacements with gold alloy restorations. They noted an
initial increase in all recorded mercury levels after
amalgam removal. About three months thereafter, plasma and
erythrocyte levels decreased markedly. A continuous
reduction in urine mercury levels took place, reaching a
plateau of approximately 25% of the pre-removal mercury
level within 9 months.
It is important to note that, in the studies cited,
both the predicted mercury uptake from amalgam and the
observed accumulation of mercury in the body are average
values. It is also clear from the original reports that
substantial individual variations exist.
 5.1.1.2 Animal experiments
Frykholm (1957), using radioactive mercury in amalgam,
studied the release and uptake of mercury in dogs and
monkeys. He concluded that the mercury exposure from
amalgam was essentially limited to the immediate placement
procedures. This is in contrast to more recent studies
that examined the disposition of radioactive mercury
released from amalgam restorations in sheep (Hahn et al.,
1989; Vimy et al., 1990a).
Hahn et al. (1989) demonstrated by whole-body image
scan that amalgam mercury could be readily visualized in
the kidney, liver, jawbone, and gastrointestinal tract
after only 29 days of chewing with amalgam. Vimy et al.
(1990a) demonstrated that the mercury levels in maternal
blood, fetal blood, and amniotic fluid reached a peak
within 48 h after amalgam placement and remained at that
level for the duration of the studies (140 days). Mercury
levels of 4 ng/g in maternal blood and amniotic fluid and
of 10 ng/g in fetal blood were found. The erythro-
cyte/plasma ratios of mercury from amalgam in both the
ewe and fetal lamb were less than unity. The maternal
urine mercury concentration ranged from 1-10 ng/g during a
16-day period. Approximately 7.7 mg of mercury could be
eliminated per day in the faeces.
All tissues examined displayed mercury accumulation.
By 29 days, kidney mercury levels rose to approximately
9000 ng/g, and these levels were maintained throughout the
duration of the study. A similar pattern was observed in
the liver, but the levels remained at approximately 1000
ng/g. The fetal kidney contained mercury levels of 10-14
ng/g, whereas fetal liver had levels of 100-130 ng/g.
The maternal brain (cerebrum, occipital lobe, and
thalamus) showed a mercury accumulation ranging from 3-13
ng/g. In the pituitary, thyroid, and adrenal glands, con-
centrations ranged from approximately 10-100 ng/g. In the
fetal cerebrum, occipital cortex, and thalamus the highest
levels were approximately 10 ng/g. The fetal pituitary
gland had mercury concentrations of more than 100 ng/g,
whereas the thyroid and adrenal glands contained less than
10 ng/g.
Milk obtained at lamb parturition or within several
days following birth (25-41 days after amalgam placement)
contained levels of mercury from dental amalgam that
reached as high as 60 ng/g.
Other recent reports indicate that both kidney func-
tion (Vimy et al., 1990b) and intestinal bacterial popu-
lation (Summers et al., 1990) may be affected when animals
are exposed to dental amalgam mercury.
5.1.2. Skin-lightening soaps and creams
Elemental mercury and soluble inorganic mercury com-
pounds can penetrate the human skin. Mercury-containing
skin-lightening soaps and creams are left on the skin
overnight. Therefore, the possibility of substantial mer-
cury exposure exists both via the skin and through inha-
lation. There are no empirical data showing the relative
importance of the different exposure routes, but the evi-
dence indicates that the total exposure to mercury is sub-
stantial from these sources. Barr et al. (1973) reported
that in a group of 60 African women using skin-lightening
creams (5-10% ammoniated mercury), the mean urinary mer-
cury excretion was 109 µg/litre (range: 0-220 µg per
litre). A subgroup of 26 women with a nephrotic syndrome
had a mean urinary mercury level of 150 µg/litre (range:
90-250 µg/litre). Marzulli & Brown (1972) reported uri-
nary mercury levels from 28 to 600 µg/litre among a group
of 6 women who had used skin-lightening cream containing
1-3% ammoniated mercury for two years.
Lauwerys et al. (1987) reported the case of a woman
who had recently given birth and who had used during preg-
nancy and lactation a soap containing 1% mercury as mer-
curic iodide and a mercury-containing cream. The urinary
mercury content of the mother was 784 µg/g creatinine
4 months after the birth at a time when she was still
using the soap and cream. Although no mercury-containing
cream or soap was used on her baby's skin and the lac-
tation period lasted only one month, the baby's blood (at
the age of three months) contained 19 µg/litre and the
urine 274 µg/g creatinine.
5.1.3. Mercury in paint
Mercury compounds are added to water-based latex
paints to inhibit the growth of bacteria and mould.
Several reports have highlighted that mercury vapour can
be released from the paint on interior house walls
(Hirschman et al., 1963; Jacobs & Goldwater, 1965; Foote,
1972; Sibbett et al., 1972).
A recent study by Agocs et al. (in press) compared
homes recently coated with a paint containing a median
concentration of 754 mg mercury/litre with homes not
coated with a mercury-containing paint to determine
whether the recent application of such a paint is associ-
ated with elevated concentrations of mercury in air and
urine. Air samples from the 19 homes of exposed people
contained a median level of 2 µg/m3 (range, undetectable
to 10 µg/m3), while concentrations of mercury in air
from 9 homes of unexposed people were below the detection
limit of 0.1 µg/m3 (p < 0.001). The median urine mercury
concentration was higher for the 65 exposed people
(8.4 µg/g creatinine; range, 2.5-118) than for the 28
unexposed people (1.9 µg/g creatinine; range, 0.04-7)
(p < 0.001).
5.2. Occupational exposure during manufacture, formulation,
and use
Occupational exposure to mercury in chloralkali plants
and in mercury mining was reviewed in WHO (1976). In more
recent studies, average urine mercury levels of 50-100 µg
per litre have been reported (see sections 9.1.2 and
9.2.2).
A NIOSH survey in 1983 of 84 workers in a thermometer
factory showed that five workers had urinary mercury
levels above 150 µg/g creatinine and three workers had
levels above 300 µg/g creatinine. Personal air sampling
showed exposure levels of 26-271 µg/m3 (Ehrenberg et
al., 1986). Other studies of instrument and thermometer
factories in the USA yielded similar results (Price &
Wisseman, 1977; Wallingford, 1982; Lee, 1984). In gold and
silver refineries in the USA, the mean urinary mercury
concentration was 108 µg/litre for four regularly exposed
workers (Handke & Pryor, 1981).
Recently, particular interest has focused on occu-
pational exposure to mercury in dentistry (see also
section 3.2). Several studies made during the period 1960-
1980 have reported average levels of mercury vapour in
dental clinics ranging between 20 and 30 µg/m3 air,
and certain clinics have been found to have levels of 150-
170 µg/m3 (Joselow et al., 1968; Gronka et al., 1970;
Buchwald, 1972; Schneider, 1974). Some of these studies
also reported the urine mercury levels of dental person-
nel. Joselow et al. (1968) found an average urinary
mercury concentration of 40 µg/litre among 50 dentists,
some values exceeding 100 µg/litre. These levels are
similar to the urinary mercury concentrations reported by
Gronka et al. (1970) and Buchwald (1972).
Kelman (1978) reported statistically significantly
higher urine mercury levels among dental assistants
(38 µg/litre) than among dentists (22 µg/litre). On the
other hand, Nixon et al. (1981) found only small differ-
ences between dentists and dental assistants. The average
environmental mercury exposure in 200 clinics studied was
11 µg/m3 (with a range from 0 to 82 µg/m3), while
the mean urine mercury concentration was 26 µg/litre
(2-149 µg/litre).
In a nationwide American study by Naleway et al.
(1985), the average mercury level in urine sampled between
1975 and 1983 from 4272 dentists was 14.2 µg/litre (SD
± 25.4 µg/litre; the frequency distribution did not re-
semble a normal distribution), the range being 0-556 µg
per litre. In 4.9% of the samples, levels were above
50 µg/litre, and above 100 µg/litre in 1.3% of
samples. The wide range of values was probably due to the
sampling techniques, methodological problems, and vari-
ations in occupational exposures to amalgam.
In a similar Norwegian study, Jokstad (1987) reported
that 2% of a group of 672 dentists had urine mercury
levels greater than 20 µg/litre. The highest recorded
value in this group was 50 µg/litre.
Recently Nilsson & Nilsson (1986a,b) reported a com-
paratively low mercury level (4 µg/m3) in the air of
private dental clinics. The median urine mercury concen-
tration was 6 µg/litre (range: 1-21 µg/litre) for den-
tists and 7 µg/litre (range: 1-70 µg/litre) for dental
assistants. In a Belgian study of dentists by Huberlant
et al. (1983), the mean urine mercury concentration was
also relatively low (11.5 µg/g creatinine).
Dentists and dental assistants may be momentarily
exposed to high local peaks of mercury vapour during
insertion, polishing, and removal of amalgam fillings,
especially if adequate protective measures are not taken
(Frykholm, 1957; Buchwald, 1972; Cooley & Barkmeier, 1978;
Reinhardt et al., 1983; Richards & Warren, 1985). Richards
& Warren (1985) reported mercury vapour concentrations
approaching 1000 µg/m3 in the breathing zone of dentists
not using coolants or adequate aspiration techniques
during operative procedures. The corresponding concen-
trations when proper measures were used were approxi-
matively ten times lower (110 µg/m3).
When Battistone et al. (1976) analysed the blood
mercury level of 1389 American dentists, the mean value
was 9.8 µg/litre (18 dentists having levels above 30 µg
per litre). In a study of 380 American dentists, Brady et
al. (1980) reported a mean concentration of 8.5 µg per
litre, 7.4% of the participants having blood mercury
levels greater than 15 µg/litre. These levels were found
to decrease within 16 h after termination of exposure.
This finding agrees with the documented short biological
half-time in blood for the majority of the mercury (see
section 6.5).
These studies suffered from variations in the sampling
techniques, the analytical techniques, and the occu-
pational exposure of the participants. Although the extent
of occupational exposure could be evaluated from mercury
concentrations found in critical organs, few data are
available in the literature. Kosta et al. (1975) reported
levels of mercury in the central nervous system and the
kidneys of deceased mercury miners several years after
cessation of exposure. Average levels of 700 µg/kg wet
weight of brain (SD ± 640 µg/kg) were, for example, re-
ported in six cases. In the same group plus an additional
miner, pituitary mercury levels were reported to be as
high as 27 100 µg/kg (SD ± 14 900 µg/kg). Non-exposed
controls showed mean brain levels of 4.2 µg per kg (SD
± 2.6 µg/kg, n = 5), mean pituitary levels of 40 µg per
kg (SD ± 26 µg/kg, n = 6), and mean kidney levels of
140 µg/kg (SD ± 160 µg/kg, n = 7) (see also sections
9.1.1 and 9.2.1).
A Swedish study of seven former dentists and one
dental nurse reported elevated concentrations of mercury
in the pituitary gland and occipital lobe cortex (Nylander
et al., 1989). Values of up to 4000 µg/kg wet weight
were observed in the pituitary gland, and of up to
300 µg/kg in the occipital lobe cortex. Two of the sub-
jects were 80 years old and had been retired for several
years. High mercury levels were also noted in the kidneys
and thyroid. In one subject, the thyroid concentration was
28 000 µg/kg despite several years retirement.
6. KINETICS AND METABOLISM
There are major differences in the kinetics and metab-
olism of the various mercury species. Metallic mercury is
rapidly oxidized to inorganic mercury compounds in the
body. However, its kinetics and membrane permeability are
different from those of mercuric mercury. Also methylmer-
cury can be converted to inorganic mercury in vivo (WHO,
1990). Thus, the ultimate fate of absorbed mercury com-
pounds will depend on their chemical transformation in the
body as well as the kinetics. The details of the kinetics
and metabolism of methylmercury have been described in WHO
(1990).
6.1. Absorption
6.1.1. Absorption by inhalation
Inhalation of mercury vapour is the most important
route of uptake for elemental mercury. Approximately 80%
of inhaled mercury vapour is retained. The retention
occurs almost entirely in the alveoli, where it is almost
100%. The retained amount is the same whether inhalation
takes place through the nose or the mouth (WHO, 1976;
Hursh et al., 1976).
The uptake of metallic mercury vapour from inspired
air into the blood depends on the dissolution of mercury
vapour in the blood as it passes through the pulmonary
circulation. The dissolved vapour is then very soon oxi-
dized to Hg++, partly in the red blood cells and partly
after diffusion into other tissues. This oxidation occurs
under the influence of the enzyme catalase. The oxidation,
and in consequence the absorption, of mercury vapour in
humans can be reduced considerably by alcohol or the
herbicide aminotriazole (WHO, 1976; Halbach & Clarkson,
1978; Magos et al., 1978; Hursh et al., 1980).
WHO (1976) concluded that information on pulmonary
retention of inorganic mercury compounds was lacking.
Deposition should follow the physical laws governing depo-
sition of aerosols in the respiratory system. Particulates
with a high probability of deposition in the upper respir-
atory tract should be cleared quickly. For particulates
deposited in the lower respiratory tract, a longer reten-
tion period would be expected, the length depending on
solubility, among other factors. In experiments on dogs,
approximately 45% of a radioactive mercury(II) oxide aero-
sol, with a median droplet diameter of 0.16 (± 0.06) µm,
was cleared in less than 24 h and the remainder with a
half-time of 33 days (Morrow et al., 1964). Radioactivity
was detected in blood as well as in urine. The concen-
tration in blood followed the curve of its disappearance
from the lungs. The in vivo solubility of the particles
was found to be of great importance for the clearance
during the slow phase. Recent evidence has shown that lung
macrophages are able to increase the solubility of only
slightly soluble metals (Lundborg et al., 1984; Marafante
et al., 1987) and that this is due to a low pH in the
phagolysosomes (Nilsen et al., 1988).
Although there are still no data to allow a quantitat-
ive evaluation of the absorption of different inorganic
mercury compounds, significant absorption must take place
directly from the lung and, probably, to some extent from
the gastrointestinal tract after mucociliary clearance of
non-absorbed mercury.
6.1.2. Absorption by ingestion
Liquid metallic mercury is poorly absorbed. Some data
indicate an absorption of less than 0.01% in rats. How-
ever, humans who accidently ingested several grams of
metallic mercury showed increased blood levels of mercury
(WHO, 1976). Metallic mercury has been incorporated into
tissues after accidental breakage of intestinal tubes,
containers, and thermometers. This has sometimes caused
local tissue reactions with or without signs of systemic
poisoning (Geller, 1976). The reason for the different
types of reactions is not known.
The absorption in humans of inorganic mercuric mercury
compounds from foods was estimated by WHO (1976) to be
about 7% on average and by Elinder et al. (1988) to be
less than 10% (probably about 5%). The data were mainly
obtained from tracer studies on human volunteers (Rahola
et al., 1973), who received single oral doses of protein-
bound inorganic mercuric mercury. Although individual
variation was considerable, the proportion of the dose
excreted in the faeces during the first 4-5 days was 75-92%.
Absorption in young children may be considerably
greater. Kostial et al. (1978, 1983) observed an average
absorption in newborn rats of 38% six days after an oral
dose of mercuric chloride. The absorption in older animals
was only about 1%. As breast milk may contain significant
amounts of inorganic as well as organic mercury, this
route of exposure should not be overlooked (section 6.4).
The low solubility of mercurous chloride limits absorp-
tion. However, after prolonged intake the accumulation of
mercury in tissues, urinary mercury excretion, and adverse
effects indicate that some absorption takes place.
6.1.3. Absorption through skin
Little information was available on skin absorption
when WHO (1976) was published, although some animal exper-
iments revealed a certain degree of skin penetration (a
few per cent of an aqueous solution of mercuric salts
during the first hours of skin application) (Friberg et
al., 1961; Skog & Wahlberg, 1964; Wahlberg, 1965). Recent
studies on human volunteers (Hursh et al., 1989) indicate
that uptake via the skin of metallic mercury vapour is
only about 1% of uptake by inhalation. However, it is
obvious that the use of skin-lightening creams containing
inorganic mercury salts causes substantial absorption and
accumulation into the body (section 5.1.2), although there
is no information on how much of the mercury is absorbed
through the skin and how much is absorbed via other
routes.
6.1.4. Absorption by axonal transport
Arvidson (1987) reported an accumulation of mercury
from a tracer dose of 203HgCl2 in the hypoglossal nuclei
of the brain stem of rats after a single injection into
the tongue. A similar accumulation was not seen in con-
trols after a similar injection into the gluteus maximus
muscle. The author concluded that the results provided
evidence of retrograde axonal transport of mercury in the
hypoglossal nerve.
6.2. Distribution
From studies on animals and humans (WHO, 1976; Khayat
& Dencker, 1983a, 1984; see also sections 8 and 9), it is
known that mercury has an affinity for ectodermal and
endodermal epithelial cells and glands. It accumulates in,
for instance, the thyroid, pituitary, brain, kidney,
liver, pancreas, testes, ovaries, and prostate. Within
the organs the distribution is not uniform. This explains
why biological half-times may differ not only between
organs but also within an organ. The kidney is the chief
depository of mercury after the administration of elemen-
tal mercury vapour or inorganic salts. Based on animal
data, 50-90% of the body burden is found in the kidneys.
Significant amounts were transported to the brain after
exposure of mice and monkeys to elemental mercury vapour.
The brain mercury levels were ten times higher than after
equal doses of mercuric mercury given intravenously
(Berlin & Johansson, 1964; Berlin et al., 1969; WHO,
1976). In rats given daily subcutaneous doses of mercuric
chloride for six weeks, only 0.01% of the total dose of
mercury was found in the brain, while about 3% of the dose
was retained in the kidneys (Friberg, 1956).
The red cell to plasma ratio in humans was approxi-
mately 1.0 after exposure to Hg0 vapour, but was 0.4
after exposure to inorganic mercury salts (WHO, 1976). The
ratio may vary, however. Suzuki et al. (1976) observed a
red cell to plasma ratio of about 1.5-2 for workers
exposed only to mercury vapour, while the corresponding
ratio for 6 chloralkali workers (where the exposure may
have been to both vapour and inorganic salts) averaged
only 0.02. The reason for this extremely low ratio is
unknown. In a report by Cherian et al. (1978), a ratio of
about 2 was observed during the first few days after
exposure of volunteers to metallic mercury vapour.
Jugo (1976) compared the retention of mercuric chlor-
ide after a single injection in adult and 2-week-old suck-
ling rats. The whole-body retention 6 days after treatment
was significantly higher in the suckling animals, and the
accumulation of mercury was 13- and 19-fold higher in the
brain and liver, respectively, compared to adult rats. On
the other hand, the mercury concentrations in the kidneys
were markedly higher in the adult group.
In two pregnant women who had been accidentally
exposed to metallic mercury vapour, the concentration of
mercury in the infant blood was similar to that in the
maternal blood at the time of delivery (Clarkson &
Kilpper, 1978). There are no other data on the transfer of
inhaled mercury vapour to the fetus in humans.
Based on studies in rodents, elemental mercury vapour
easily penetrates the placental barrier and, after oxi-
dation, accumulates in the fetal tissue. Only a fraction
of divalent mercury enters the fetus, but it can accumu-
late in the placenta. Clarkson et al. (1972) found that
mercury levels in the fetuses of rats exposed to mercury
vapour were 10-40 times higher than in animals exposed to
equivalent doses of mercuric chloride. Differences in the
penetration of the placental barrier have been confirmed
in mice by Khayat & Dencker (1982), who found a 4-fold
higher fetal mercury concentration after exposure to met-
allic mercury vapour than after exposure to mercuric
chloride. The uptake of mercury vapour increased with
gestational age. Only traces of radioactive mercury were
found in embryos at 8 and 10 days of gestation. A distinct
accumulation of mercury was seen in the fetal tissue from
day 12 of gestation with a pronounced uptake in the fetal
liver and heart. The mercury concentration in the CNS was
rather low in early and mid gestation but increased just
prior to birth (Ogata & Meguro, 1986).
Yoshida et al. (1986, 1987) studied the uptake and
distribution of mercury in the fetus of guinea-pigs during
late gestation after repeated exposure to 200-300 µg mer-
cury vapour/m3 2 h/day and after a single exposure for
150 min to 8-11 mg/m3. Mercury concentrations in fetal
brain, lungs, heart, kidneys, and blood were much lower
than those in maternal tissues, the concentrations dif-
fering by a factor of about 5 in the brain and a factor of
up to 100 in the kidneys. Mercury concentrations in fetal
liver were up to two times higher than those found in
maternal liver. In the fetal liver, more than 50% of the
mercury was bound to a metallothionein-like protein with a
relative molecular mass of about 10 000 to 12 000. The
bulk of the eluted mercury in the maternal liver was
associated with a protein of high relative molecular mass.
The authors suggested that the fetal metallothionein-like
protein plays a role in preventing further distribution of
mercury from the liver after in utero exposure to mercury
vapour.
Mercury distribution in the neonate differs from that
in the fetus (Yoshida et al., 1989). A significantly
increased level was found in kidney, lung, and brain in
neonate guinea-pigs, compared with fetuses, and there was
a progressive decrease in liver concentration, with dimin-
ishing hepatic metallothionein levels, in the neonates.
These results suggest a redistribution of mercury to other
tissues in the neonate.
The oxidation of elemental mercury vapour in the body
(section 6.1.1) can be reduced considerably (to about 50%
of normal values) by moderate amounts of alcohol. In an
in vivo study, the uptake of labelled mercury into human
red cells was reduced by almost a factor of ten by etha-
nol, while there was an increase in liver mercury concen-
trations (Hursh et al., 1980). Observations on rats, mice,
and monkeys confirm these results (Khayat & Dencker,
1983a,b, 1984). They also show a marked decrease in mer-
cury concentrations in several organs, including the
brain. However, somewhat higher concentrations of mercury
were observed in the brain and liver of pregnant mice with
a congenital catalase deficiency that were exposed for 1 h
to metallic mercury vapour during day 18 of gestation
(Ogata & Meguro, 1986). The blood mercury concentration
in the catalase-deficient mice was only about half of that
in the control mice. The uptake in the fetus was 2% of the
dose compared to 1.2% for the controls.
Lower mercury levels have been observed in the brain
tissue of humans classified as chronic alcohol abusers
than in controls (Fig. 3).
6.3. Metabolic transformation
Several forms of metabolic transformation occur:
* oxidation of metallic mercury vapour to divalent mer-
cury;
* reduction of divalent mercury to metallic mercury;
* methylation of inorganic mercury;
* conversion of methylmercury to divalent inorganic mer-
cury.
The oxidation of metallic mercury vapour to divalent
ionic mercury (section 6.1.1) takes place very soon after
absorption, but some elemental mercury remains dissolved
in the blood long enough (a few minutes) for it to be
carried to the blood-brain barrier and the placenta (WHO,
1976). Recent in vitro studies on the oxidation of mercury
by the blood (Hursh et al., 1988) indicate that because of
the short transit time from the lung to the brain almost
all the mercury vapour (97%) arrives at the brain unoxi-
dized. Its lipid solubility and high diffusibility allow
rapid transit across these barriers. Oxidation of the
mercury vapour in brain and fetal tissues converts it to
the ionic form, which is much less likely to cross the
blood-brain and placental barriers. Thus, oxidation in
these tissues serves as a trap to hold the mercury and
leads to accumulation in brain and fetal tissues (WHO,
1976).
5.1.1.2 Animal experiments
Frykholm (1957), using radioactive mercury in amalgam,
studied the release and uptake of mercury in dogs and
monkeys. He concluded that the mercury exposure from
amalgam was essentially limited to the immediate placement
procedures. This is in contrast to more recent studies
that examined the disposition of radioactive mercury
released from amalgam restorations in sheep (Hahn et al.,
1989; Vimy et al., 1990a).
Hahn et al. (1989) demonstrated by whole-body image
scan that amalgam mercury could be readily visualized in
the kidney, liver, jawbone, and gastrointestinal tract
after only 29 days of chewing with amalgam. Vimy et al.
(1990a) demonstrated that the mercury levels in maternal
blood, fetal blood, and amniotic fluid reached a peak
within 48 h after amalgam placement and remained at that
level for the duration of the studies (140 days). Mercury
levels of 4 ng/g in maternal blood and amniotic fluid and
of 10 ng/g in fetal blood were found. The erythro-
cyte/plasma ratios of mercury from amalgam in both the
ewe and fetal lamb were less than unity. The maternal
urine mercury concentration ranged from 1-10 ng/g during a
16-day period. Approximately 7.7 mg of mercury could be
eliminated per day in the faeces.
All tissues examined displayed mercury accumulation.
By 29 days, kidney mercury levels rose to approximately
9000 ng/g, and these levels were maintained throughout the
duration of the study. A similar pattern was observed in
the liver, but the levels remained at approximately 1000
ng/g. The fetal kidney contained mercury levels of 10-14
ng/g, whereas fetal liver had levels of 100-130 ng/g.
The maternal brain (cerebrum, occipital lobe, and
thalamus) showed a mercury accumulation ranging from 3-13
ng/g. In the pituitary, thyroid, and adrenal glands, con-
centrations ranged from approximately 10-100 ng/g. In the
fetal cerebrum, occipital cortex, and thalamus the highest
levels were approximately 10 ng/g. The fetal pituitary
gland had mercury concentrations of more than 100 ng/g,
whereas the thyroid and adrenal glands contained less than
10 ng/g.
Milk obtained at lamb parturition or within several
days following birth (25-41 days after amalgam placement)
contained levels of mercury from dental amalgam that
reached as high as 60 ng/g.
Other recent reports indicate that both kidney func-
tion (Vimy et al., 1990b) and intestinal bacterial popu-
lation (Summers et al., 1990) may be affected when animals
are exposed to dental amalgam mercury.
5.1.2. Skin-lightening soaps and creams
Elemental mercury and soluble inorganic mercury com-
pounds can penetrate the human skin. Mercury-containing
skin-lightening soaps and creams are left on the skin
overnight. Therefore, the possibility of substantial mer-
cury exposure exists both via the skin and through inha-
lation. There are no empirical data showing the relative
importance of the different exposure routes, but the evi-
dence indicates that the total exposure to mercury is sub-
stantial from these sources. Barr et al. (1973) reported
that in a group of 60 African women using skin-lightening
creams (5-10% ammoniated mercury), the mean urinary mer-
cury excretion was 109 µg/litre (range: 0-220 µg per
litre). A subgroup of 26 women with a nephrotic syndrome
had a mean urinary mercury level of 150 µg/litre (range:
90-250 µg/litre). Marzulli & Brown (1972) reported uri-
nary mercury levels from 28 to 600 µg/litre among a group
of 6 women who had used skin-lightening cream containing
1-3% ammoniated mercury for two years.
Lauwerys et al. (1987) reported the case of a woman
who had recently given birth and who had used during preg-
nancy and lactation a soap containing 1% mercury as mer-
curic iodide and a mercury-containing cream. The urinary
mercury content of the mother was 784 µg/g creatinine
4 months after the birth at a time when she was still
using the soap and cream. Although no mercury-containing
cream or soap was used on her baby's skin and the lac-
tation period lasted only one month, the baby's blood (at
the age of three months) contained 19 µg/litre and the
urine 274 µg/g creatinine.
5.1.3. Mercury in paint
Mercury compounds are added to water-based latex
paints to inhibit the growth of bacteria and mould.
Several reports have highlighted that mercury vapour can
be released from the paint on interior house walls
(Hirschman et al., 1963; Jacobs & Goldwater, 1965; Foote,
1972; Sibbett et al., 1972).
A recent study by Agocs et al. (in press) compared
homes recently coated with a paint containing a median
concentration of 754 mg mercury/litre with homes not
coated with a mercury-containing paint to determine
whether the recent application of such a paint is associ-
ated with elevated concentrations of mercury in air and
urine. Air samples from the 19 homes of exposed people
contained a median level of 2 µg/m3 (range, undetectable
to 10 µg/m3), while concentrations of mercury in air
from 9 homes of unexposed people were below the detection
limit of 0.1 µg/m3 (p < 0.001). The median urine mercury
concentration was higher for the 65 exposed people
(8.4 µg/g creatinine; range, 2.5-118) than for the 28
unexposed people (1.9 µg/g creatinine; range, 0.04-7)
(p < 0.001).
5.2. Occupational exposure during manufacture, formulation,
and use
Occupational exposure to mercury in chloralkali plants
and in mercury mining was reviewed in WHO (1976). In more
recent studies, average urine mercury levels of 50-100 µg
per litre have been reported (see sections 9.1.2 and
9.2.2).
A NIOSH survey in 1983 of 84 workers in a thermometer
factory showed that five workers had urinary mercury
levels above 150 µg/g creatinine and three workers had
levels above 300 µg/g creatinine. Personal air sampling
showed exposure levels of 26-271 µg/m3 (Ehrenberg et
al., 1986). Other studies of instrument and thermometer
factories in the USA yielded similar results (Price &
Wisseman, 1977; Wallingford, 1982; Lee, 1984). In gold and
silver refineries in the USA, the mean urinary mercury
concentration was 108 µg/litre for four regularly exposed
workers (Handke & Pryor, 1981).
Recently, particular interest has focused on occu-
pational exposure to mercury in dentistry (see also
section 3.2). Several studies made during the period 1960-
1980 have reported average levels of mercury vapour in
dental clinics ranging between 20 and 30 µg/m3 air,
and certain clinics have been found to have levels of 150-
170 µg/m3 (Joselow et al., 1968; Gronka et al., 1970;
Buchwald, 1972; Schneider, 1974). Some of these studies
also reported the urine mercury levels of dental person-
nel. Joselow et al. (1968) found an average urinary
mercury concentration of 40 µg/litre among 50 dentists,
some values exceeding 100 µg/litre. These levels are
similar to the urinary mercury concentrations reported by
Gronka et al. (1970) and Buchwald (1972).
Kelman (1978) reported statistically significantly
higher urine mercury levels among dental assistants
(38 µg/litre) than among dentists (22 µg/litre). On the
other hand, Nixon et al. (1981) found only small differ-
ences between dentists and dental assistants. The average
environmental mercury exposure in 200 clinics studied was
11 µg/m3 (with a range from 0 to 82 µg/m3), while
the mean urine mercury concentration was 26 µg/litre
(2-149 µg/litre).
In a nationwide American study by Naleway et al.
(1985), the average mercury level in urine sampled between
1975 and 1983 from 4272 dentists was 14.2 µg/litre (SD
± 25.4 µg/litre; the frequency distribution did not re-
semble a normal distribution), the range being 0-556 µg
per litre. In 4.9% of the samples, levels were above
50 µg/litre, and above 100 µg/litre in 1.3% of
samples. The wide range of values was probably due to the
sampling techniques, methodological problems, and vari-
ations in occupational exposures to amalgam.
In a similar Norwegian study, Jokstad (1987) reported
that 2% of a group of 672 dentists had urine mercury
levels greater than 20 µg/litre. The highest recorded
value in this group was 50 µg/litre.
Recently Nilsson & Nilsson (1986a,b) reported a com-
paratively low mercury level (4 µg/m3) in the air of
private dental clinics. The median urine mercury concen-
tration was 6 µg/litre (range: 1-21 µg/litre) for den-
tists and 7 µg/litre (range: 1-70 µg/litre) for dental
assistants. In a Belgian study of dentists by Huberlant
et al. (1983), the mean urine mercury concentration was
also relatively low (11.5 µg/g creatinine).
Dentists and dental assistants may be momentarily
exposed to high local peaks of mercury vapour during
insertion, polishing, and removal of amalgam fillings,
especially if adequate protective measures are not taken
(Frykholm, 1957; Buchwald, 1972; Cooley & Barkmeier, 1978;
Reinhardt et al., 1983; Richards & Warren, 1985). Richards
& Warren (1985) reported mercury vapour concentrations
approaching 1000 µg/m3 in the breathing zone of dentists
not using coolants or adequate aspiration techniques
during operative procedures. The corresponding concen-
trations when proper measures were used were approxi-
matively ten times lower (110 µg/m3).
When Battistone et al. (1976) analysed the blood
mercury level of 1389 American dentists, the mean value
was 9.8 µg/litre (18 dentists having levels above 30 µg
per litre). In a study of 380 American dentists, Brady et
al. (1980) reported a mean concentration of 8.5 µg per
litre, 7.4% of the participants having blood mercury
levels greater than 15 µg/litre. These levels were found
to decrease within 16 h after termination of exposure.
This finding agrees with the documented short biological
half-time in blood for the majority of the mercury (see
section 6.5).
These studies suffered from variations in the sampling
techniques, the analytical techniques, and the occu-
pational exposure of the participants. Although the extent
of occupational exposure could be evaluated from mercury
concentrations found in critical organs, few data are
available in the literature. Kosta et al. (1975) reported
levels of mercury in the central nervous system and the
kidneys of deceased mercury miners several years after
cessation of exposure. Average levels of 700 µg/kg wet
weight of brain (SD ± 640 µg/kg) were, for example, re-
ported in six cases. In the same group plus an additional
miner, pituitary mercury levels were reported to be as
high as 27 100 µg/kg (SD ± 14 900 µg/kg). Non-exposed
controls showed mean brain levels of 4.2 µg per kg (SD
± 2.6 µg/kg, n = 5), mean pituitary levels of 40 µg per
kg (SD ± 26 µg/kg, n = 6), and mean kidney levels of
140 µg/kg (SD ± 160 µg/kg, n = 7) (see also sections
9.1.1 and 9.2.1).
A Swedish study of seven former dentists and one
dental nurse reported elevated concentrations of mercury
in the pituitary gland and occipital lobe cortex (Nylander
et al., 1989). Values of up to 4000 µg/kg wet weight
were observed in the pituitary gland, and of up to
300 µg/kg in the occipital lobe cortex. Two of the sub-
jects were 80 years old and had been retired for several
years. High mercury levels were also noted in the kidneys
and thyroid. In one subject, the thyroid concentration was
28 000 µg/kg despite several years retirement.
6. KINETICS AND METABOLISM
There are major differences in the kinetics and metab-
olism of the various mercury species. Metallic mercury is
rapidly oxidized to inorganic mercury compounds in the
body. However, its kinetics and membrane permeability are
different from those of mercuric mercury. Also methylmer-
cury can be converted to inorganic mercury in vivo (WHO,
1990). Thus, the ultimate fate of absorbed mercury com-
pounds will depend on their chemical transformation in the
body as well as the kinetics. The details of the kinetics
and metabolism of methylmercury have been described in WHO
(1990).
6.1. Absorption
6.1.1. Absorption by inhalation
Inhalation of mercury vapour is the most important
route of uptake for elemental mercury. Approximately 80%
of inhaled mercury vapour is retained. The retention
occurs almost entirely in the alveoli, where it is almost
100%. The retained amount is the same whether inhalation
takes place through the nose or the mouth (WHO, 1976;
Hursh et al., 1976).
The uptake of metallic mercury vapour from inspired
air into the blood depends on the dissolution of mercury
vapour in the blood as it passes through the pulmonary
circulation. The dissolved vapour is then very soon oxi-
dized to Hg++, partly in the red blood cells and partly
after diffusion into other tissues. This oxidation occurs
under the influence of the enzyme catalase. The oxidation,
and in consequence the absorption, of mercury vapour in
humans can be reduced considerably by alcohol or the
herbicide aminotriazole (WHO, 1976; Halbach & Clarkson,
1978; Magos et al., 1978; Hursh et al., 1980).
WHO (1976) concluded that information on pulmonary
retention of inorganic mercury compounds was lacking.
Deposition should follow the physical laws governing depo-
sition of aerosols in the respiratory system. Particulates
with a high probability of deposition in the upper respir-
atory tract should be cleared quickly. For particulates
deposited in the lower respiratory tract, a longer reten-
tion period would be expected, the length depending on
solubility, among other factors. In experiments on dogs,
approximately 45% of a radioactive mercury(II) oxide aero-
sol, with a median droplet diameter of 0.16 (± 0.06) µm,
was cleared in less than 24 h and the remainder with a
half-time of 33 days (Morrow et al., 1964). Radioactivity
was detected in blood as well as in urine. The concen-
tration in blood followed the curve of its disappearance
from the lungs. The in vivo solubility of the particles
was found to be of great importance for the clearance
during the slow phase. Recent evidence has shown that lung
macrophages are able to increase the solubility of only
slightly soluble metals (Lundborg et al., 1984; Marafante
et al., 1987) and that this is due to a low pH in the
phagolysosomes (Nilsen et al., 1988).
Although there are still no data to allow a quantitat-
ive evaluation of the absorption of different inorganic
mercury compounds, significant absorption must take place
directly from the lung and, probably, to some extent from
the gastrointestinal tract after mucociliary clearance of
non-absorbed mercury.
6.1.2. Absorption by ingestion
Liquid metallic mercury is poorly absorbed. Some data
indicate an absorption of less than 0.01% in rats. How-
ever, humans who accidently ingested several grams of
metallic mercury showed increased blood levels of mercury
(WHO, 1976). Metallic mercury has been incorporated into
tissues after accidental breakage of intestinal tubes,
containers, and thermometers. This has sometimes caused
local tissue reactions with or without signs of systemic
poisoning (Geller, 1976). The reason for the different
types of reactions is not known.
The absorption in humans of inorganic mercuric mercury
compounds from foods was estimated by WHO (1976) to be
about 7% on average and by Elinder et al. (1988) to be
less than 10% (probably about 5%). The data were mainly
obtained from tracer studies on human volunteers (Rahola
et al., 1973), who received single oral doses of protein-
bound inorganic mercuric mercury. Although individual
variation was considerable, the proportion of the dose
excreted in the faeces during the first 4-5 days was 75-92%.
Absorption in young children may be considerably
greater. Kostial et al. (1978, 1983) observed an average
absorption in newborn rats of 38% six days after an oral
dose of mercuric chloride. The absorption in older animals
was only about 1%. As breast milk may contain significant
amounts of inorganic as well as organic mercury, this
route of exposure should not be overlooked (section 6.4).
The low solubility of mercurous chloride limits absorp-
tion. However, after prolonged intake the accumulation of
mercury in tissues, urinary mercury excretion, and adverse
effects indicate that some absorption takes place.
6.1.3. Absorption through skin
Little information was available on skin absorption
when WHO (1976) was published, although some animal exper-
iments revealed a certain degree of skin penetration (a
few per cent of an aqueous solution of mercuric salts
during the first hours of skin application) (Friberg et
al., 1961; Skog & Wahlberg, 1964; Wahlberg, 1965). Recent
studies on human volunteers (Hursh et al., 1989) indicate
that uptake via the skin of metallic mercury vapour is
only about 1% of uptake by inhalation. However, it is
obvious that the use of skin-lightening creams containing
inorganic mercury salts causes substantial absorption and
accumulation into the body (section 5.1.2), although there
is no information on how much of the mercury is absorbed
through the skin and how much is absorbed via other
routes.
6.1.4. Absorption by axonal transport
Arvidson (1987) reported an accumulation of mercury
from a tracer dose of 203HgCl2 in the hypoglossal nuclei
of the brain stem of rats after a single injection into
the tongue. A similar accumulation was not seen in con-
trols after a similar injection into the gluteus maximus
muscle. The author concluded that the results provided
evidence of retrograde axonal transport of mercury in the
hypoglossal nerve.
6.2. Distribution
From studies on animals and humans (WHO, 1976; Khayat
& Dencker, 1983a, 1984; see also sections 8 and 9), it is
known that mercury has an affinity for ectodermal and
endodermal epithelial cells and glands. It accumulates in,
for instance, the thyroid, pituitary, brain, kidney,
liver, pancreas, testes, ovaries, and prostate. Within
the organs the distribution is not uniform. This explains
why biological half-times may differ not only between
organs but also within an organ. The kidney is the chief
depository of mercury after the administration of elemen-
tal mercury vapour or inorganic salts. Based on animal
data, 50-90% of the body burden is found in the kidneys.
Significant amounts were transported to the brain after
exposure of mice and monkeys to elemental mercury vapour.
The brain mercury levels were ten times higher than after
equal doses of mercuric mercury given intravenously
(Berlin & Johansson, 1964; Berlin et al., 1969; WHO,
1976). In rats given daily subcutaneous doses of mercuric
chloride for six weeks, only 0.01% of the total dose of
mercury was found in the brain, while about 3% of the dose
was retained in the kidneys (Friberg, 1956).
The red cell to plasma ratio in humans was approxi-
mately 1.0 after exposure to Hg0 vapour, but was 0.4
after exposure to inorganic mercury salts (WHO, 1976). The
ratio may vary, however. Suzuki et al. (1976) observed a
red cell to plasma ratio of about 1.5-2 for workers
exposed only to mercury vapour, while the corresponding
ratio for 6 chloralkali workers (where the exposure may
have been to both vapour and inorganic salts) averaged
only 0.02. The reason for this extremely low ratio is
unknown. In a report by Cherian et al. (1978), a ratio of
about 2 was observed during the first few days after
exposure of volunteers to metallic mercury vapour.
Jugo (1976) compared the retention of mercuric chlor-
ide after a single injection in adult and 2-week-old suck-
ling rats. The whole-body retention 6 days after treatment
was significantly higher in the suckling animals, and the
accumulation of mercury was 13- and 19-fold higher in the
brain and liver, respectively, compared to adult rats. On
the other hand, the mercury concentrations in the kidneys
were markedly higher in the adult group.
In two pregnant women who had been accidentally
exposed to metallic mercury vapour, the concentration of
mercury in the infant blood was similar to that in the
maternal blood at the time of delivery (Clarkson &
Kilpper, 1978). There are no other data on the transfer of
inhaled mercury vapour to the fetus in humans.
Based on studies in rodents, elemental mercury vapour
easily penetrates the placental barrier and, after oxi-
dation, accumulates in the fetal tissue. Only a fraction
of divalent mercury enters the fetus, but it can accumu-
late in the placenta. Clarkson et al. (1972) found that
mercury levels in the fetuses of rats exposed to mercury
vapour were 10-40 times higher than in animals exposed to
equivalent doses of mercuric chloride. Differences in the
penetration of the placental barrier have been confirmed
in mice by Khayat & Dencker (1982), who found a 4-fold
higher fetal mercury concentration after exposure to met-
allic mercury vapour than after exposure to mercuric
chloride. The uptake of mercury vapour increased with
gestational age. Only traces of radioactive mercury were
found in embryos at 8 and 10 days of gestation. A distinct
accumulation of mercury was seen in the fetal tissue from
day 12 of gestation with a pronounced uptake in the fetal
liver and heart. The mercury concentration in the CNS was
rather low in early and mid gestation but increased just
prior to birth (Ogata & Meguro, 1986).
Yoshida et al. (1986, 1987) studied the uptake and
distribution of mercury in the fetus of guinea-pigs during
late gestation after repeated exposure to 200-300 µg mer-
cury vapour/m3 2 h/day and after a single exposure for
150 min to 8-11 mg/m3. Mercury concentrations in fetal
brain, lungs, heart, kidneys, and blood were much lower
than those in maternal tissues, the concentrations dif-
fering by a factor of about 5 in the brain and a factor of
up to 100 in the kidneys. Mercury concentrations in fetal
liver were up to two times higher than those found in
maternal liver. In the fetal liver, more than 50% of the
mercury was bound to a metallothionein-like protein with a
relative molecular mass of about 10 000 to 12 000. The
bulk of the eluted mercury in the maternal liver was
associated with a protein of high relative molecular mass.
The authors suggested that the fetal metallothionein-like
protein plays a role in preventing further distribution of
mercury from the liver after in utero exposure to mercury
vapour.
Mercury distribution in the neonate differs from that
in the fetus (Yoshida et al., 1989). A significantly
increased level was found in kidney, lung, and brain in
neonate guinea-pigs, compared with fetuses, and there was
a progressive decrease in liver concentration, with dimin-
ishing hepatic metallothionein levels, in the neonates.
These results suggest a redistribution of mercury to other
tissues in the neonate.
The oxidation of elemental mercury vapour in the body
(section 6.1.1) can be reduced considerably (to about 50%
of normal values) by moderate amounts of alcohol. In an
in vivo study, the uptake of labelled mercury into human
red cells was reduced by almost a factor of ten by etha-
nol, while there was an increase in liver mercury concen-
trations (Hursh et al., 1980). Observations on rats, mice,
and monkeys confirm these results (Khayat & Dencker,
1983a,b, 1984). They also show a marked decrease in mer-
cury concentrations in several organs, including the
brain. However, somewhat higher concentrations of mercury
were observed in the brain and liver of pregnant mice with
a congenital catalase deficiency that were exposed for 1 h
to metallic mercury vapour during day 18 of gestation
(Ogata & Meguro, 1986). The blood mercury concentration
in the catalase-deficient mice was only about half of that
in the control mice. The uptake in the fetus was 2% of the
dose compared to 1.2% for the controls.
Lower mercury levels have been observed in the brain
tissue of humans classified as chronic alcohol abusers
than in controls (Fig. 3).
6.3. Metabolic transformation
Several forms of metabolic transformation occur:
* oxidation of metallic mercury vapour to divalent mer-
cury;
* reduction of divalent mercury to metallic mercury;
* methylation of inorganic mercury;
* conversion of methylmercury to divalent inorganic mer-
cury.
The oxidation of metallic mercury vapour to divalent
ionic mercury (section 6.1.1) takes place very soon after
absorption, but some elemental mercury remains dissolved
in the blood long enough (a few minutes) for it to be
carried to the blood-brain barrier and the placenta (WHO,
1976). Recent in vitro studies on the oxidation of mercury
by the blood (Hursh et al., 1988) indicate that because of
the short transit time from the lung to the brain almost
all the mercury vapour (97%) arrives at the brain unoxi-
dized. Its lipid solubility and high diffusibility allow
rapid transit across these barriers. Oxidation of the
mercury vapour in brain and fetal tissues converts it to
the ionic form, which is much less likely to cross the
blood-brain and placental barriers. Thus, oxidation in
these tissues serves as a trap to hold the mercury and
leads to accumulation in brain and fetal tissues (WHO,
1976).
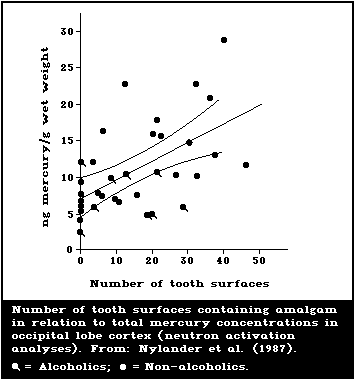 The reduction of divalent mercury to Hg0 has been
demonstrated both in animals (mice and rats) and humans
(WHO, 1976; Dunn et al., 1978, 1981a,b; Sugata & Clarkson,
1979). A small amount of exhaled mercury vapour is the
result of this reduction. It is increased in catalase-
deficient mice (Ogata et al., 1987) and by alcohol (both
in vitro and in vivo ) in both mice and humans (Dunn et.
al., 1981a,b). The increased exhalation of mercury vapour
in the latter case may be explained by assuming that the
oxidation by catalase is less than normal.
It was stated in WHO (1976) that there is no evidence
in the literature for the synthesis of organomercury com-
pounds in human or mammalian tissues. Minor methylation
may occur in vitro by intestinal or oral bacteria (Rowland
et al., 1975; Heintze et al., 1983). A slight increase in
the concentration of methylmercury in blood and/or urine
has been reported among dentists and workers in the
chloralkali industry (Cross et al., 1978; Pan et al.,
1980; Aitio et al., 1983). These data cannot be taken as
evidence of methylation, however, due to lack of analyti-
cal quality control and possible confounding by exposure
to methylmercury. Chang et al. (1987) did not observe any
methylation in a study of dentists.
The conversion of methylmercury to inorganic mercury
is considered a key step in the process of excretion of
mercury after exposure to methylmercury (WHO, 1990). If
the intact molecule of an organomercurial in an organ is
more rapidly excreted than inorganic mercury, biotrans-
formation will decrease the overall excretion rate, and
the ratio of inorganic to organic mercury in that particu-
lar organ will increase with time. The fraction of total
mercury present as Hg++ will depend on the duration of
exposure to methylmercury and/or the time elapsed since
cessation of exposure. Even if the demethylation rate is
very slow, this process may in the long run give rise to
considerable accumulation of inorganic mercury. The ratio
of methylmercury to inorganic mercury depends on the rate
of demethylation and the clearance half-times of methyl-
mercury and inorganic mercury.
After short-term exposure of experimental animals to
methylmercury the kidneys usually contain the highest
fraction of Hg++ in relation to total mercury, while the
relative concentration in the brain is low (WHO, 1976).
In studies on squirrel monkeys (Berlin et al., 1975), the
short-term biotransformation to inorganic mercury was as
follows: of the total mercury, about 20% was inorganic in
the liver; 50% in the kidney; 30%-85% in the bile; and
less than 5% in the brain.
More recent data from long-term studies on monkeys
show a different pattern. Mottet & Burbacher (1988) sum-
marized a long series of studies on the metabolism and
toxicity of methylmercury in monkeys (Macaca fascicu-
laris). The monkeys had been orally exposed to high levels
of methylmercury for a period of years and sacrificed
during the ongoing exposure. At the end of the exposure
period, 10-33% of the mercury in the brain was present in
the inorganic form (Lind et al., 1988). In monkeys that
had been without mercury exposure for 6 months to almost
two years after the same treatment, the relative concen-
tration of inorganic mercury was much higher, i.e. about
90%. Exact half-times for the different compounds could
not be established in the absence of data on the concen-
trations of inorganic and organic mercury in the brain at
different time intervals during the accumulation and
clearance phases. Recent data by Rice (1989) also demon-
strate demethylation in the brain. Female monkeys (Macaca
fascicularis) were dosed for at least 1.7 years with mer-
cury as methylmercury chloride (10-50 µg/kg per day).
After dosing ceased, the blood mercury half-time was about
14 days. Approximately 230 days after cessation of dosing,
the monkeys were sacrificed and brain total mercury levels
determined. These levels were considered to be at least
three orders of magnitude higher than those predicted by
assuming the half-time in brain to be the same as that in
blood. The author considered the most likely explanation
to be demethylation of methylmercury and subsequent bind-
ing of inorganic mercury to tissue.
Similar results were recently reported by Hansen et
al. (1989) who fed fish contaminated with methylmercury to
one Alsatian dog for 7 years. The dog was examined after
its death at the age of 12 years, 4 years after the
exposure to methylmercury had ceased. Two dogs of the same
age and breed served as controls. In the CNS, the mercury
was fairly uniformly distributed and 93% was in the inor-
ganic state, whereas the skeletal muscles contained
approximately 30% inorganic mercury. The authors concluded
that the results demonstrated time-dependent demethylation
and suggested a variation in the rate from one type of
tissue to another. High levels of mercury were demon-
strated by a histochemical method in the liver, thyroid
gland, and kidney, whereas practically no mercury was
found in any of the organs examined in the control dogs.
The distribution of inorganic mercury was determined by a
histochemical method for locating mercury in tissue
sections. Total mercury was analysed by flameless atomic
absorption and organic mercury by GC.
A considerable fraction of the mercury in human
brains is reported to be in the form of inorganic mercury.
Kitamura et al. (1976) analysed autopsy material from 20
Japanese subjects for total mercury using flameless atomic
absorption and for methylmercury using GC. The median
concentration of total mercury in the cerebrum was 0.097
mg/kg wet weight and of methylmercury 0.012 mg/kg wet
weight. The values for the cerebellum were similar. No
analytical quality control data were reported.
In a Swedish autopsy study covering six cases (Friberg
et al., 1986; Nylander et al., 1987), about 80% of the
mercury in the occipital lobe cortex was inorganic. The
concentration of inorganic mercury varied between 3 and
22 µg/kg wet weight. Both total mercury and inorganic
mercury were determined by the method of Magos (Magos,
1971; Magos & Clarkson, 1972). For quality control pur-
poses total mercury was also analysed by neutron acti-
vation analysis. In this study, however, the concen-
trations of mercury in the brain were considerably lower
than in the Japanese study. As has been discussed in sec-
tion 5.1.1, an association between the number of amalgam
fillings and total mercury concentration in the occipital
lobe has been found. Exposure to inorganic mercury from
dental fillings could explain the high proportion of inor-
ganic mercury in the Swedish study but not in the Japanese
study, as it seems reasonable to assume that the mercury
exposure from amalgam should be approximately the same in
the two countries. The exposure to methylmercury could,
however, easily differ considerably.
Takizawa (1986) reported the total mercury and methyl-
mercury brain concentrations in about 30 humans who had
died from 20 days to 18 years after the onset of symptoms
of methylmercury poisoning. The total mercury content was
measured by flameless atomic absorption spectrophotometry,
while methylmercury was analysed by electron capture GLC
(Minagawa et al., 1979; Takizawa, 1986). The total mercury
content in "acute" cases (autopsy < 100 days after onset
of symptoms) was 8.8-21.4 mg/kg and the concentration of
methylmercury was 1.85-8.42 mg mercury/kg. The concen-
trations for the "chronic" cases were 0.35-5.29 mg/kg
for total mercury and 0.31-1.02 mg mercury/g for methyl-
mercury. On average, only 28% of the mercury was present
as methylmercury in the acute cases and 17% in the chronic
cases. Takizawa (1986) also presented data for residents
near Minamata Bay and for a non-polluted area. The best
estimate from these data is that only 16% and 12%,
respectively, of the total mercury was present as methyl-
mercury. Unfortunately, in these reports quality control
data were not presented. The authors measured total mer-
cury and methylmercury and assumed that the difference
between these analyses was due to inorganic mercury. It
could in principle, in whole or in part, also have been
methylmercury that was not extracted in the gas chromato-
graphic procedure. Ideally, analyses should be carried out
using, for instance, the method by Magos (1971), which
measures total mercury and inorganic mercury.
The tissues in the studies by Takizawa (1986) were
stored for long periods after fixation with a 10% neutral
formalin solution. Miyama & Suzuki (1971) found that the
ratio of inorganic to total mercury in the cerebral cortex
increased from about 35% (tissues stored frozen) to about
50% after storage in 10% formalin for one year. However,
there was no loss of inorganic mercury. Eto et al. (1988)
compared results from a small number of analyses of
formalin-fixed tissues with results from analyses of
frozen tissues. There was no systematic loss related to
storage in formaldehyde.
The concentrations of inorganic mercury in the brain,
reported in overt cases of methylmercury poisoning, are
very high, similar to those observed after toxic exposure
to metallic mercury vapour. Whether or not an accumulation
of inorganic mercury actually contributed to the toxic
effects is not known, but seems unlikely. Even assuming
no analytical problems, it should be borne in mind that
the methylmercury poisoning usually occurred after rela-
tively short exposure to methylmercury when no significant
biotransformation should yet have taken place. However, a
comparison of the toxicology of methylmercury with that of
ethylmercury, which decomposes significantly more quickly,
indicated that cerebellar damage could not be related to
inorganic mercury. The higher concentration of inorganic
mercury in the brain of ethylmercury-treated rats, com-
pared with methylmercury-treated rats, was associated with
less cerebellar damage (Magos et al., 1985). It is more
difficult to evaluate the possible long-term effects of
inorganic mercury, which slowly accumulates in the brain.
The distribution of ionic mercury in the brain will
depend on whether Hg++ enters the brain in the ionic form
or as a result of in situ biotransformation following
penetration of the brain barrier by elemental mercury or
methylmercury. The toxicological aspects of such possible
differences in distribution are not known.
6.4. Elimination and excretion
A small portion of absorbed inorganic mercury is ex-
haled as metallic mercury vapour, formed by the reduction
of Hg++ in the tissues (Dunn et al., 1978), but urine and
faeces are the principal routes of elimination (WHO,
1976). The urinary route dominates when exposure is high.
After exposure to metallic mercury vapour, a small frac-
tion of the mercury in the urine may be present as elemen-
tal mercury (Stopford et al., 1978; Yoshida & Yamamura,
1982). One form of depletion is the transfer of maternal
mercury to the fetal unit. Thus, inorganic mercury was
detected in the amniotic fluid in all but two out of 57
Japanese pregnant women, while organic mercury was found
in only 30 women (Suzuki et al., 1977). In a study by
Skerfving (1988), it was reported that the concentrations
of total mercury in breast milk and in the blood plasma of
breast-fed infants were similar to those in the maternal
plasma of Swedish fishermen's wives. Although the women
were exposed to methylmercury, 80% of mercury excreted in
breast milk was in the inorganic form. No formal analyti-
cal quality control procedures were applied in the studies
where mercury was speciated.
6.5. Retention and turnover
6.5.1. Biological half-time
Only very limited data were available on the biologi-
cal half-time of inorganic mercury when WHO (1976) was
published. Studies on a small number of volunteers had
shown that the elimination of mercury, after a single
exposure to metallic mercury vapour, followed a single
exponential process with an average half-time of 58 days
during the first few months after the exposure. Similar
data were available from studies involving oral exposure
to mercuric mercury. It was pointed out that there had
been a few reports of high brain mercury concentrations
in workers several years after cessation of exposure to
mercury vapour. This indicated that the half-time in
brain is longer than that in other organs, although no
quantitative estimations were made.
As a result of tracer studies on human volunteers
(Nakaaki et al., 1975, 1978; Cherian et al., 1978; Newton
& Fry, 1978; Hursh et al., 1980) and animals (Berlin et
al., 1975), more data are now available on the kinetics
during the first few months after exposure. The elimin-
ation of inorganic mercury follows a complicated pattern
with biological half-times that differ according to the
tissue and the time after exposure. The best estimate is
that after short-term exposure to mercury vapour, the
first phase of elimination from blood has a half-time of
approximately 2-4 days and accounts for about 90% of the
mercury. This is followed by a second phase with a half-
time of 15-30 days.
In tracer studies on nine human volunteers (Hursh et
al., 1976, 1980; Clarkson et al., 1988a) the half-time for
most of the mercury in the brain was 19 (± 1.7) days
during the first 35 to 45 days. Newton & Fry (1978) found
half-times of 23 and 26 days in the head of two subjects
accidentally exposed to radioactive mercuric oxide. In a
study by Berlin et al. (1975), a steady state was not
reached in the brains of squirrel monkeys exposed for two
months to mercury vapour. In one study on monkeys (Macaca
fascicularis) (section 6.3) lasting several years where
inorganic mercury accumulated in the brain (probably as a
result of demethylation of methylmercury), there was still
considerable inorganic mercury in the brain 1-2 years
after cessation of exposure (Lind et al., 1988). These
results indicate a very long half-time for a fraction of
the inorganic mercury in the brain. This is in accordance
with data from deceased miners and dentists (section
5.2).
The half-time in the kidneys for inorganic mercury in
the studies by Hursh et al. (1976, 1980) was 64 days,
about the same as that for the body as a whole. As in the
case of the brain, a fraction of the mercury probably has
a long biological half-time (section 5.2).
A few attempts to perform a quantitative evaluation of
the half-time for inorganic mercury have been made using
multicompartment models (Sugita, 1978; Bernard & Purdue,
1984). According to the recommendation of ICRP (1980), the
four-compartment model of Bernard & Purdue (1984) included
one compartment with a half-time of 27 years. As the basic
assumptions are uncertain, the models are uncertain, but
may be of value for a possible "worst case" estimation
of the retention of inorganic mercury in the brain. The
model of Bernard & Purdue (1984) has been used by Vimy et
al. (1986) for calculating mercury accumulation in the
brain from amalgam fillings (section 5.1). The form of
mercury that is responsible for the long biological half-
time may be biochemically inactive mercury selenide.
6.5.2. Reference or normal values in indicator media
A considerable amount of information is given in
Environmental Health Criteria 101: Methylmercury (WHO,
1990). The mean concentration of total mercury in whole
blood (in the absence of consumption of fish with high
concentrations of methylmercury) is probably of the order
of 5-10 µg/litre, and in hair about 1-2 mg/kg. The aver-
age mercury concentration in urine is about 4 µg per
litre and in the placenta about 10 mg/kg wet weight,
although the individual variation is substantial. One
source of the variation in urine levels seems to be
exposure from dental amalgam (Fig. 4), while for blood and
hair levels fish consumption is the major source of ex-
posure. Increased hair levels may also be due to external
contamination.
The reduction of divalent mercury to Hg0 has been
demonstrated both in animals (mice and rats) and humans
(WHO, 1976; Dunn et al., 1978, 1981a,b; Sugata & Clarkson,
1979). A small amount of exhaled mercury vapour is the
result of this reduction. It is increased in catalase-
deficient mice (Ogata et al., 1987) and by alcohol (both
in vitro and in vivo ) in both mice and humans (Dunn et.
al., 1981a,b). The increased exhalation of mercury vapour
in the latter case may be explained by assuming that the
oxidation by catalase is less than normal.
It was stated in WHO (1976) that there is no evidence
in the literature for the synthesis of organomercury com-
pounds in human or mammalian tissues. Minor methylation
may occur in vitro by intestinal or oral bacteria (Rowland
et al., 1975; Heintze et al., 1983). A slight increase in
the concentration of methylmercury in blood and/or urine
has been reported among dentists and workers in the
chloralkali industry (Cross et al., 1978; Pan et al.,
1980; Aitio et al., 1983). These data cannot be taken as
evidence of methylation, however, due to lack of analyti-
cal quality control and possible confounding by exposure
to methylmercury. Chang et al. (1987) did not observe any
methylation in a study of dentists.
The conversion of methylmercury to inorganic mercury
is considered a key step in the process of excretion of
mercury after exposure to methylmercury (WHO, 1990). If
the intact molecule of an organomercurial in an organ is
more rapidly excreted than inorganic mercury, biotrans-
formation will decrease the overall excretion rate, and
the ratio of inorganic to organic mercury in that particu-
lar organ will increase with time. The fraction of total
mercury present as Hg++ will depend on the duration of
exposure to methylmercury and/or the time elapsed since
cessation of exposure. Even if the demethylation rate is
very slow, this process may in the long run give rise to
considerable accumulation of inorganic mercury. The ratio
of methylmercury to inorganic mercury depends on the rate
of demethylation and the clearance half-times of methyl-
mercury and inorganic mercury.
After short-term exposure of experimental animals to
methylmercury the kidneys usually contain the highest
fraction of Hg++ in relation to total mercury, while the
relative concentration in the brain is low (WHO, 1976).
In studies on squirrel monkeys (Berlin et al., 1975), the
short-term biotransformation to inorganic mercury was as
follows: of the total mercury, about 20% was inorganic in
the liver; 50% in the kidney; 30%-85% in the bile; and
less than 5% in the brain.
More recent data from long-term studies on monkeys
show a different pattern. Mottet & Burbacher (1988) sum-
marized a long series of studies on the metabolism and
toxicity of methylmercury in monkeys (Macaca fascicu-
laris). The monkeys had been orally exposed to high levels
of methylmercury for a period of years and sacrificed
during the ongoing exposure. At the end of the exposure
period, 10-33% of the mercury in the brain was present in
the inorganic form (Lind et al., 1988). In monkeys that
had been without mercury exposure for 6 months to almost
two years after the same treatment, the relative concen-
tration of inorganic mercury was much higher, i.e. about
90%. Exact half-times for the different compounds could
not be established in the absence of data on the concen-
trations of inorganic and organic mercury in the brain at
different time intervals during the accumulation and
clearance phases. Recent data by Rice (1989) also demon-
strate demethylation in the brain. Female monkeys (Macaca
fascicularis) were dosed for at least 1.7 years with mer-
cury as methylmercury chloride (10-50 µg/kg per day).
After dosing ceased, the blood mercury half-time was about
14 days. Approximately 230 days after cessation of dosing,
the monkeys were sacrificed and brain total mercury levels
determined. These levels were considered to be at least
three orders of magnitude higher than those predicted by
assuming the half-time in brain to be the same as that in
blood. The author considered the most likely explanation
to be demethylation of methylmercury and subsequent bind-
ing of inorganic mercury to tissue.
Similar results were recently reported by Hansen et
al. (1989) who fed fish contaminated with methylmercury to
one Alsatian dog for 7 years. The dog was examined after
its death at the age of 12 years, 4 years after the
exposure to methylmercury had ceased. Two dogs of the same
age and breed served as controls. In the CNS, the mercury
was fairly uniformly distributed and 93% was in the inor-
ganic state, whereas the skeletal muscles contained
approximately 30% inorganic mercury. The authors concluded
that the results demonstrated time-dependent demethylation
and suggested a variation in the rate from one type of
tissue to another. High levels of mercury were demon-
strated by a histochemical method in the liver, thyroid
gland, and kidney, whereas practically no mercury was
found in any of the organs examined in the control dogs.
The distribution of inorganic mercury was determined by a
histochemical method for locating mercury in tissue
sections. Total mercury was analysed by flameless atomic
absorption and organic mercury by GC.
A considerable fraction of the mercury in human
brains is reported to be in the form of inorganic mercury.
Kitamura et al. (1976) analysed autopsy material from 20
Japanese subjects for total mercury using flameless atomic
absorption and for methylmercury using GC. The median
concentration of total mercury in the cerebrum was 0.097
mg/kg wet weight and of methylmercury 0.012 mg/kg wet
weight. The values for the cerebellum were similar. No
analytical quality control data were reported.
In a Swedish autopsy study covering six cases (Friberg
et al., 1986; Nylander et al., 1987), about 80% of the
mercury in the occipital lobe cortex was inorganic. The
concentration of inorganic mercury varied between 3 and
22 µg/kg wet weight. Both total mercury and inorganic
mercury were determined by the method of Magos (Magos,
1971; Magos & Clarkson, 1972). For quality control pur-
poses total mercury was also analysed by neutron acti-
vation analysis. In this study, however, the concen-
trations of mercury in the brain were considerably lower
than in the Japanese study. As has been discussed in sec-
tion 5.1.1, an association between the number of amalgam
fillings and total mercury concentration in the occipital
lobe has been found. Exposure to inorganic mercury from
dental fillings could explain the high proportion of inor-
ganic mercury in the Swedish study but not in the Japanese
study, as it seems reasonable to assume that the mercury
exposure from amalgam should be approximately the same in
the two countries. The exposure to methylmercury could,
however, easily differ considerably.
Takizawa (1986) reported the total mercury and methyl-
mercury brain concentrations in about 30 humans who had
died from 20 days to 18 years after the onset of symptoms
of methylmercury poisoning. The total mercury content was
measured by flameless atomic absorption spectrophotometry,
while methylmercury was analysed by electron capture GLC
(Minagawa et al., 1979; Takizawa, 1986). The total mercury
content in "acute" cases (autopsy < 100 days after onset
of symptoms) was 8.8-21.4 mg/kg and the concentration of
methylmercury was 1.85-8.42 mg mercury/kg. The concen-
trations for the "chronic" cases were 0.35-5.29 mg/kg
for total mercury and 0.31-1.02 mg mercury/g for methyl-
mercury. On average, only 28% of the mercury was present
as methylmercury in the acute cases and 17% in the chronic
cases. Takizawa (1986) also presented data for residents
near Minamata Bay and for a non-polluted area. The best
estimate from these data is that only 16% and 12%,
respectively, of the total mercury was present as methyl-
mercury. Unfortunately, in these reports quality control
data were not presented. The authors measured total mer-
cury and methylmercury and assumed that the difference
between these analyses was due to inorganic mercury. It
could in principle, in whole or in part, also have been
methylmercury that was not extracted in the gas chromato-
graphic procedure. Ideally, analyses should be carried out
using, for instance, the method by Magos (1971), which
measures total mercury and inorganic mercury.
The tissues in the studies by Takizawa (1986) were
stored for long periods after fixation with a 10% neutral
formalin solution. Miyama & Suzuki (1971) found that the
ratio of inorganic to total mercury in the cerebral cortex
increased from about 35% (tissues stored frozen) to about
50% after storage in 10% formalin for one year. However,
there was no loss of inorganic mercury. Eto et al. (1988)
compared results from a small number of analyses of
formalin-fixed tissues with results from analyses of
frozen tissues. There was no systematic loss related to
storage in formaldehyde.
The concentrations of inorganic mercury in the brain,
reported in overt cases of methylmercury poisoning, are
very high, similar to those observed after toxic exposure
to metallic mercury vapour. Whether or not an accumulation
of inorganic mercury actually contributed to the toxic
effects is not known, but seems unlikely. Even assuming
no analytical problems, it should be borne in mind that
the methylmercury poisoning usually occurred after rela-
tively short exposure to methylmercury when no significant
biotransformation should yet have taken place. However, a
comparison of the toxicology of methylmercury with that of
ethylmercury, which decomposes significantly more quickly,
indicated that cerebellar damage could not be related to
inorganic mercury. The higher concentration of inorganic
mercury in the brain of ethylmercury-treated rats, com-
pared with methylmercury-treated rats, was associated with
less cerebellar damage (Magos et al., 1985). It is more
difficult to evaluate the possible long-term effects of
inorganic mercury, which slowly accumulates in the brain.
The distribution of ionic mercury in the brain will
depend on whether Hg++ enters the brain in the ionic form
or as a result of in situ biotransformation following
penetration of the brain barrier by elemental mercury or
methylmercury. The toxicological aspects of such possible
differences in distribution are not known.
6.4. Elimination and excretion
A small portion of absorbed inorganic mercury is ex-
haled as metallic mercury vapour, formed by the reduction
of Hg++ in the tissues (Dunn et al., 1978), but urine and
faeces are the principal routes of elimination (WHO,
1976). The urinary route dominates when exposure is high.
After exposure to metallic mercury vapour, a small frac-
tion of the mercury in the urine may be present as elemen-
tal mercury (Stopford et al., 1978; Yoshida & Yamamura,
1982). One form of depletion is the transfer of maternal
mercury to the fetal unit. Thus, inorganic mercury was
detected in the amniotic fluid in all but two out of 57
Japanese pregnant women, while organic mercury was found
in only 30 women (Suzuki et al., 1977). In a study by
Skerfving (1988), it was reported that the concentrations
of total mercury in breast milk and in the blood plasma of
breast-fed infants were similar to those in the maternal
plasma of Swedish fishermen's wives. Although the women
were exposed to methylmercury, 80% of mercury excreted in
breast milk was in the inorganic form. No formal analyti-
cal quality control procedures were applied in the studies
where mercury was speciated.
6.5. Retention and turnover
6.5.1. Biological half-time
Only very limited data were available on the biologi-
cal half-time of inorganic mercury when WHO (1976) was
published. Studies on a small number of volunteers had
shown that the elimination of mercury, after a single
exposure to metallic mercury vapour, followed a single
exponential process with an average half-time of 58 days
during the first few months after the exposure. Similar
data were available from studies involving oral exposure
to mercuric mercury. It was pointed out that there had
been a few reports of high brain mercury concentrations
in workers several years after cessation of exposure to
mercury vapour. This indicated that the half-time in
brain is longer than that in other organs, although no
quantitative estimations were made.
As a result of tracer studies on human volunteers
(Nakaaki et al., 1975, 1978; Cherian et al., 1978; Newton
& Fry, 1978; Hursh et al., 1980) and animals (Berlin et
al., 1975), more data are now available on the kinetics
during the first few months after exposure. The elimin-
ation of inorganic mercury follows a complicated pattern
with biological half-times that differ according to the
tissue and the time after exposure. The best estimate is
that after short-term exposure to mercury vapour, the
first phase of elimination from blood has a half-time of
approximately 2-4 days and accounts for about 90% of the
mercury. This is followed by a second phase with a half-
time of 15-30 days.
In tracer studies on nine human volunteers (Hursh et
al., 1976, 1980; Clarkson et al., 1988a) the half-time for
most of the mercury in the brain was 19 (± 1.7) days
during the first 35 to 45 days. Newton & Fry (1978) found
half-times of 23 and 26 days in the head of two subjects
accidentally exposed to radioactive mercuric oxide. In a
study by Berlin et al. (1975), a steady state was not
reached in the brains of squirrel monkeys exposed for two
months to mercury vapour. In one study on monkeys (Macaca
fascicularis) (section 6.3) lasting several years where
inorganic mercury accumulated in the brain (probably as a
result of demethylation of methylmercury), there was still
considerable inorganic mercury in the brain 1-2 years
after cessation of exposure (Lind et al., 1988). These
results indicate a very long half-time for a fraction of
the inorganic mercury in the brain. This is in accordance
with data from deceased miners and dentists (section
5.2).
The half-time in the kidneys for inorganic mercury in
the studies by Hursh et al. (1976, 1980) was 64 days,
about the same as that for the body as a whole. As in the
case of the brain, a fraction of the mercury probably has
a long biological half-time (section 5.2).
A few attempts to perform a quantitative evaluation of
the half-time for inorganic mercury have been made using
multicompartment models (Sugita, 1978; Bernard & Purdue,
1984). According to the recommendation of ICRP (1980), the
four-compartment model of Bernard & Purdue (1984) included
one compartment with a half-time of 27 years. As the basic
assumptions are uncertain, the models are uncertain, but
may be of value for a possible "worst case" estimation
of the retention of inorganic mercury in the brain. The
model of Bernard & Purdue (1984) has been used by Vimy et
al. (1986) for calculating mercury accumulation in the
brain from amalgam fillings (section 5.1). The form of
mercury that is responsible for the long biological half-
time may be biochemically inactive mercury selenide.
6.5.2. Reference or normal values in indicator media
A considerable amount of information is given in
Environmental Health Criteria 101: Methylmercury (WHO,
1990). The mean concentration of total mercury in whole
blood (in the absence of consumption of fish with high
concentrations of methylmercury) is probably of the order
of 5-10 µg/litre, and in hair about 1-2 mg/kg. The aver-
age mercury concentration in urine is about 4 µg per
litre and in the placenta about 10 mg/kg wet weight,
although the individual variation is substantial. One
source of the variation in urine levels seems to be
exposure from dental amalgam (Fig. 4), while for blood and
hair levels fish consumption is the major source of ex-
posure. Increased hair levels may also be due to external
contamination.
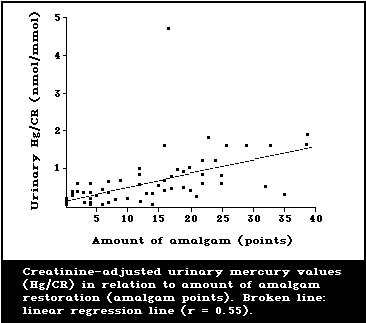 There are at present no suitable indicator media that
will reflect concentrations of inorganic mercury in the
critical organs, the brain or kidney, under different
exposure situations. This is to be expected in view of the
complicated pattern of metabolism for different mercury
compounds. One important consequence is that concen-
trations of mercury in urine or blood may be low quite
soon after exposure has ceased, despite the fact that
concentrations in the critical organs may still be high.
There is some information, obtained from subjects not
occupationally exposed and with only a moderate fish
consumption, on the relationship between exposure to
metallic mercury vapour and concentrations of mercury in
urine and brain tissue. This relationship (section 5.1)
indicates that ongoing long-term exposure to elemental
mercury vapour, leading to a mercury absorption of
5-10 µg/day, will result in a mercury excretion in urine
of about 5 µg/litre and average mercury concentrations
in the occipital lobe cortex and kidney of approximately
10 µg/kg and 500 µg/kg, respectively.
The distribution between blood and hair is well known
for different exposure levels of methylmercury, which
forms the basis for the use of hair as an indicator media
for this compound. There is no corresponding information
for inorganic mercury. When high levels of total mercury
in hair have been reported, for instance, among dentists
exposed to metallic mercury vapour (see e.g. Sinclair et
al., 1980; Pritchard et al., 1982; Sikorski et al., 1987),
it was not known how much was due to external contami-
nation. In a report on biological monitoring of toxic
metals, Elinder et al. (1988) concluded that hair is not a
suitable indicator medium for monitoring exposure to inor-
ganic mercury.
There is good epidemiological evidence from occu-
pational exposure that, on a group basis, recent exposure
is reflected in the mercury levels in blood and urine.
When exposure is low (e.g., from amalgam), it is difficult
to find an association between exposure levels and blood
concentrations due to confounding exposures to methylmer-
cury in fish. A way to overcome the problem may be to
analyse mercury in plasma or speciate the analysis for
inorganic mercury (Elinder et al., 1988). The problem of
confounding exposures is not so important when analysing
urine, as only a very small fraction of absorbed methyl-
mercury is excreted in urine.
Data amassed by Smith et al. (1970) from the chlorine
industry were used by WHO (1976) to evaluate the relation-
ship between concentrations of metallic mercury vapour in
air and concentrations of mercury in blood and urine.
Long-term time-weighted occupational exposure to an aver-
age air mercury concentration of 50 µg/m3 was considered
to be associated, on a group basis, with blood mercury
levels of approximately 35 µg/litre, and with urinary
concentrations of 150 µg/litre. The ratio of urine to air
concentrations was re-evaluated by WHO (1980) to be closer
to 2.0-2.5 instead of 3.0. The mercury concentrations in
air were measured with static samplers. Results from a
number of more recent studies have been reported where
both static samplers and personal samplers have been used
(Ishihara et al., 1977; Lindstedt et al., 1979; Müller et
al., 1980; Mattiussi et al., 1982; Roels et al., 1987).
Where personal samplers have been used, the ratio between
urinary mercury (µg/litre or per g creatinine) and
mercury in air (µg/m3) has as a rule been 1-2. When
blood values were reported they were either similar to
those given in WHO (1976) or somewhat lower.
In the study by Roels et al. (1987), personal monitor-
ing was used, detailed quality control procedures were
implemented and reported, and the examined subjects had
been exposed to defined concentrations for at least one
year. A good relationship could be established between the
daily time-weighted exposure to mercury vapour and the
daily level of mercury in blood and urine (Fig. 5A and B).
Urinary levels of about 50 µg/g creatinine were seen after
occupational exposure to about 40 µg/m3 of air. Such an
exposure would correspond to about 17 µg/litre of blood.
Several studies have reported a correlation between
mercury in blood and urine. The results vary considerably
and it is not known whether the ratio between concen-
trations in urine and blood is constant at different
exposure levels. At low exposure levels the possibilities
of a significant confounding effect on blood levels should
always be borne in mind.
There are at present no suitable indicator media that
will reflect concentrations of inorganic mercury in the
critical organs, the brain or kidney, under different
exposure situations. This is to be expected in view of the
complicated pattern of metabolism for different mercury
compounds. One important consequence is that concen-
trations of mercury in urine or blood may be low quite
soon after exposure has ceased, despite the fact that
concentrations in the critical organs may still be high.
There is some information, obtained from subjects not
occupationally exposed and with only a moderate fish
consumption, on the relationship between exposure to
metallic mercury vapour and concentrations of mercury in
urine and brain tissue. This relationship (section 5.1)
indicates that ongoing long-term exposure to elemental
mercury vapour, leading to a mercury absorption of
5-10 µg/day, will result in a mercury excretion in urine
of about 5 µg/litre and average mercury concentrations
in the occipital lobe cortex and kidney of approximately
10 µg/kg and 500 µg/kg, respectively.
The distribution between blood and hair is well known
for different exposure levels of methylmercury, which
forms the basis for the use of hair as an indicator media
for this compound. There is no corresponding information
for inorganic mercury. When high levels of total mercury
in hair have been reported, for instance, among dentists
exposed to metallic mercury vapour (see e.g. Sinclair et
al., 1980; Pritchard et al., 1982; Sikorski et al., 1987),
it was not known how much was due to external contami-
nation. In a report on biological monitoring of toxic
metals, Elinder et al. (1988) concluded that hair is not a
suitable indicator medium for monitoring exposure to inor-
ganic mercury.
There is good epidemiological evidence from occu-
pational exposure that, on a group basis, recent exposure
is reflected in the mercury levels in blood and urine.
When exposure is low (e.g., from amalgam), it is difficult
to find an association between exposure levels and blood
concentrations due to confounding exposures to methylmer-
cury in fish. A way to overcome the problem may be to
analyse mercury in plasma or speciate the analysis for
inorganic mercury (Elinder et al., 1988). The problem of
confounding exposures is not so important when analysing
urine, as only a very small fraction of absorbed methyl-
mercury is excreted in urine.
Data amassed by Smith et al. (1970) from the chlorine
industry were used by WHO (1976) to evaluate the relation-
ship between concentrations of metallic mercury vapour in
air and concentrations of mercury in blood and urine.
Long-term time-weighted occupational exposure to an aver-
age air mercury concentration of 50 µg/m3 was considered
to be associated, on a group basis, with blood mercury
levels of approximately 35 µg/litre, and with urinary
concentrations of 150 µg/litre. The ratio of urine to air
concentrations was re-evaluated by WHO (1980) to be closer
to 2.0-2.5 instead of 3.0. The mercury concentrations in
air were measured with static samplers. Results from a
number of more recent studies have been reported where
both static samplers and personal samplers have been used
(Ishihara et al., 1977; Lindstedt et al., 1979; Müller et
al., 1980; Mattiussi et al., 1982; Roels et al., 1987).
Where personal samplers have been used, the ratio between
urinary mercury (µg/litre or per g creatinine) and
mercury in air (µg/m3) has as a rule been 1-2. When
blood values were reported they were either similar to
those given in WHO (1976) or somewhat lower.
In the study by Roels et al. (1987), personal monitor-
ing was used, detailed quality control procedures were
implemented and reported, and the examined subjects had
been exposed to defined concentrations for at least one
year. A good relationship could be established between the
daily time-weighted exposure to mercury vapour and the
daily level of mercury in blood and urine (Fig. 5A and B).
Urinary levels of about 50 µg/g creatinine were seen after
occupational exposure to about 40 µg/m3 of air. Such an
exposure would correspond to about 17 µg/litre of blood.
Several studies have reported a correlation between
mercury in blood and urine. The results vary considerably
and it is not known whether the ratio between concen-
trations in urine and blood is constant at different
exposure levels. At low exposure levels the possibilities
of a significant confounding effect on blood levels should
always be borne in mind.
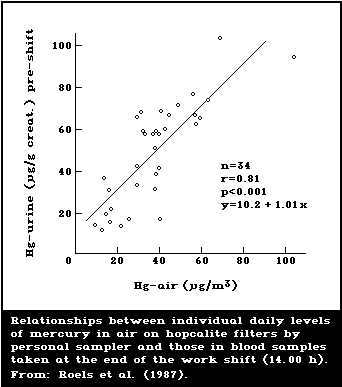
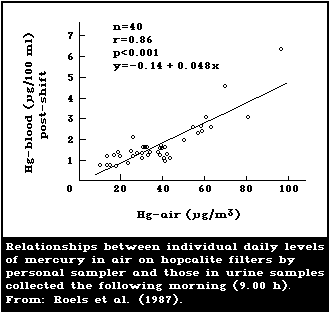 On the basis of studies by Smith et al. (1970) and
Lindstedt et al. (1979), Skerfving & Berlin (1985)
suggested that a urine mercury level of 50 µg/g creati-
nine is associated with a blood mercury level of 20 µg
per litre. Roels et al. (1987) reported a regression
equation, where a urine mercury level of 50 µg/g creati-
nine leads to a blood mercury level of 16 µg/litre.
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
This chapter is extracted from the summary of Environ-
mental Health Criteria 86: Mercury - Environmental Aspects
(WHO, 1989).
7.1. Uptake, elimination, and accumulation in organisms
Mercuric salts, and, to a much greater extent, organic
mercury, are readily taken up by organisms in water.
Aquatic invertebrates, and most particularly aquatic
insects, accumulate mercury to high concentrations. Fish
also take up the metal and retain it in tissues, princi-
pally as methylmercury, although most of the environmental
mercury to which they are exposed is inorganic. The source
of the methylation is uncertain, but there is strong indi-
cation that bacterial action leads to methylation in
aquatic systems. Environmental levels of methylmercury
depend upon the balance between bacterial methylation and
demethylation. The indications are that methylmercury in
fish arises from this bacterial methylation of inorganic
mercury, either in the environment or in bacteria associ-
ated with fish gills, surface, or gut. There is little
indication that fish themselves either methylate or
demethylate mercury. Elimination of methylmercury is slow
from fish (with half times in the order of months or
years) and from other aquatic organisms. Loss of inorganic
mercury is more rapid and so most of the mercury in fish
is retained in the form of methylmercury. Terrestrial
organisms are also contaminated by mercury, with birds
being the best studied. Sea birds and those feeding in
estuaries are most contaminated. The form of retained
mercury in birds is more variable and depends on species,
organ, and geographical site.
7.2. Toxicity to microorganisms
The metal is toxic to microorganisms. Inorganic mer-
cury has been reported to have effects at concentrations
of the metal in the culture medium of 5 µg/litre, and
organomercury compounds at concentrations at least 10
times lower than this. Organomercury compounds have been
used as fungicides. One factor affecting the toxicity of
the organometal is the rate of uptake of the metal by
cells. Mercury is bound to the cell walls or cell mem-
branes of microorganisms, apparently to a limited number
of binding sites. This means that effects are related to
cell density as well as to the concentration of mercury in
the substrate. These effects are often irreversible, and
mercury at low concentrations represents a major hazard to
microorganisms.
7.3. Toxicity to aquatic organisms
The organic forms of mercury are generally more toxic
to aquatic organisms than the inorganic forms. Aquatic
plants are affected by mercury in the water at concen-
trations approaching 1 mg/litre for inorganic mercury but
at much lower concentrations of organic mercury. Aquatic
invertebrates vary greatly in their susceptibility to
mercury. Generally, larval stages are more sensitive than
adults. The 96-h LC50s vary between 33 and 400 µg per
litre for freshwater fish and are higher for sea-water
fish. However, organic mercury compounds are more toxic.
Toxicity is affected by temperature, salinity, dissolved
oxygen, and water hardness. A wide variety of physiologi-
cal and biochemical abnormalities has been reported after
fish have been exposed to sublethal concentrations of
mercury, although the environmental significance of these
effects is difficult to assess. Reproduction is also
affected adversely by mercury.
7.4. Toxicity to terrestrial organisms
Plants are generally insensitive to the toxic effects
of mercury compounds. Birds fed inorganic mercury show a
reduction in food intake and consequent poor growth.
Other, more subtle, effects on enzyme systems, cardio-
vascular function, blood parameters, the immune response,
kidney function and structure, and behaviour have been
reported. Organomercury compounds are more toxic for birds
than are inorganic.
7.5. Effects of mercury in the field
Pollution of the sea with organomercury led to the
death of fish and fish-eating birds in Japan. Except for
this incident at Minamata, few follow-up studies of the
effects of localized release have been conducted. The use
of organomercury fungicides as seed dressings in Europe
led to the deaths of large numbers of granivorous birds,
together with birds of prey feeding on the corpses. Resi-
dues of mercury in birds' eggs have been associated with
deaths of embryos in shell. The presence of organochlorine
residues in the same birds and their eggs makes an accu-
rate assessment of the effects of mercury difficult. It
is, however, thought to be a contributing factor in the
population decline of some species of raptors.
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
8.1. Single and short-term exposure
Ashe et al. (1953) reported evidence of damage to
brain, kidney, heart, and lungs in rabbits exposed acutely
to metallic mercury vapour at a mercury concentration of
29 mg/m3 of air.
The LD50 for inorganic mercury, as well as for a num-
ber of organomercurials (e.g., arylmercury, alkoxyalkyl-
and alkylmercury compounds), lies between 10 and 40 mg/kg
body weight for all compounds tested. For mercuric chlor-
ide, a value of about 10 mg/kg body weight has been
observed after parenteral administration to mice. The
similarity in LD50 values for these various types of mer-
cury compounds is considered to result from the fact that,
when given in acute massive doses, mercury in any chemical
form will denature proteins, inactivate enzymes, and cause
severe disruption of any tissue with which it comes into
contact in sufficient concentrations. The features of
acute toxicity usually consist of shock, cardiovascular
collapse, acute renal failure, and severe gastrointestinal
damage.
8.2. Long-term exposure
8.2.1. General effects
WHO (1976), in evaluating a number of experimental
studies on animals, concluded that both reversible and
irreversible toxic effects may be caused by mercury and
its compounds. Microscopically detectable changes have
been seen in the organs of dogs, rabbits, and rats exposed
to concentrations of elemental mercury vapour ranging from
about 100 to 30 000 µg/m3 for different periods of time.
Severe damage was noted in kidneys and brains at mercury
levels in air of about 900 µg/m3 after an exposure
period of about 12 weeks. After exposure of dogs to
100 µg mercury/m3, for 7 h/day, 5 days/week over a
period of 83 weeks, no microscopically detectable effects
were seen, and tests revealed no abnormalities in kidney
function.
In two studies (Fukuda, 1971; Kishi et al., 1978),
tremor and behavioural effects were observed in rabbits
and rats after several weeks of exposure to metallic mer-
cury vapour at levels of several mg/m3, although there
were no morphological changes in the brain. The symptoms
were associated with brain mercury concentrations of about
1 and 20 mg/kg wet weight in the two studies.
8.2.2. Immunological effects
During the last 10-20 years, great attention has been
paid to effects of inorganic mercury on the immune system.
An important conclusion is that, depending upon the animal
strain tested, either auto-immunity or immunosuppression
is observed.
8.2.2.1 Auto-immunity
Bariety et al. (1971) showed that about 30% of outbred
Wistar rats exposed to mercuric chloride (1.5 or 2.5 mg/kg
body weight) 3 times per week for periods of several
months developed a membranous glomerulopathy characterized
by granular, subepithelial deposits of IgG and C3.
In Brown Norway rats (Sapin et al., 1977; Druet et
al., 1978) and in New Zealand rabbits (Roman-Franco et
al., 1978) injected with 1 or 2 mg/kg body weight, a sys-
temic auto-immune disease was observed. It appeared in
100% of the rats tested. This disease is characterized by
the production of auto-antibodies to renal and extra-renal
basement membranes. These antibodies are specific for
laminin, type IV collagen, and entactin (Bellon et al.,
1982; Fukatsu et al., 1987) and are found deposited along
the glomerular basement membrane in a linear pattern.
After 3 to 4 weeks, a typical membranous glomerulopathy
with granular, subepithelial IgG deposits is observed. The
majority of rats develop proteinuria, which progresses in
some animals to the nephrotic syndrome (Druet et al.,
1978). About half of those with this syndrome die. How-
ever, the remainder recover since the disease is transi-
ent. Dermatitis and Sjogren's syndrome have been recently
detected in Brown Norway rats injected with mercuric
chloride (Aten et al., 1988). Contact sensitization to
mercury has also been reported to occur in susceptible
strains of guinea-pigs (Polak et al., 1968). Auto-immunity
in Brown Norway rats appears in the context of a poly-
clonal activation of B cells. There is a lymphoprolifer-
ation (increase in the number of CD4+ T cells and B
cells) and hyperimmunoglobulinaemia affecting mainly IgE
(Prouvost-Danon et al., 1981; Hirsch et al., 1982;
Pelletier et al., 1988a), and several auto-antibodies such
as antinuclear antibodies (Hirsch et al., 1982) are
produced. All these manifestations are transient. They
appear from day 8 following the injection, peak during the
third week, and then progressively decline.
A number of other studies have been carried out using
Brown Norway rats. Low doses of mercuric chloride
(50 µg/kg body weight given 3 times per week) induced
auto-immune glomerulopathy, while 100 µg/kg body weight
(also 3 times per week) induced both auto-immune glomer-
ulopathy and proteinuria. Mercuric chloride was also
effective when given by inhalation (aerosol or intratra-
cheal instillation) or ingestion (Bernaudin et al., 1981;
Andres, 1984; Knoflach et al., 1986). Other inorganic
mercury compounds, e.g., mercurous chloride given orally
or HgNH2Cl-containing ointments, also induce auto-
immunity (Druet et al., 1981).
Auto-immune disorders including auto-immune glomer-
ulopathy have been described in other strains of rats
(Weening et al., 1978; Druet et al., 1982). In suscep-
tible strains of mice, especially those mice carrying the
H-2s haplotype, long-term exposure to mercuric chloride
induces extremely high titres of antinucleolar auto-anti-
bodies (Robinson et al., 1986; Mirtscheva et al., 1987).
These auto-antibodies and circulating immune complexes
(Hultman & Eneström, 1987) are involved in the glomerular
IgG deposits found in the mesangium and in the vessel
walls of H-2s mice treated with mercuric chloride
(Hultman & Eneström, 1988). Analysis of the fine speci-
ficity of the antinucleolar auto-antibodies revealed that
at least some of them react with fibrillarin, a component
of U3 small nuclear ribonucleoprotein. Sera from human
patients with idiopathic scleroderma contain auto-anti-
bodies with exactly the same specificity (Reuter et al.,
1989).
8.2.2.2 Genetics
Rats with certain major histocompatibility haplotypes,
such as Lewis rats, are resistant whatever the dose used,
while other strains are susceptible (Table 3). The suscep-
tibility of segregants obtained by crossing Brown Norway
and Lewis rats has been extensively studied (Druet et al.,
1977, 1982; Sapin et al., 1984). It has been demonstrated
that susceptibility depends upon 3 or 4 genes. One of them
is located within the major histocompatibility complex.
Both the major histocompatibility complex-linked and
-unlinked genes are required for these auto-immune abnor-
malities to occur.
Table 3. Susceptibility of various strains of rats to
auto-immune glomerulonephritis induced by mercuric chloride
-----------------------------------------------------------
Strain RT-1 Auto-immune
haplotypea glomerulonephritis
-----------------------------------------------------------
BN n anti-GBMb; MGPc
LEW, F/344 l none
BS, AS, BD IX l none
BN-1Ld l none
LEW-1Nd n none
PVG/c, AUG c glomerular granular deposits
DA, AVN a glomerular granular deposits
AS2 f glomerular granular deposits
OKA k glomerular granular deposits
___________________________________________________________
Table 3 (contd.)
___________________________________________________________
BUF b glomerular granular deposits
BD V d glomerular granular deposits
WAG, LOU u none
Wistar Furth u MGP
-----------------------------------------------------------
a Major histocompatibility complex in the rat.
b Antiglomerular basement membrane antibodies.
c Membranous glomerulopathy.
d Congenic rats with the l RT-1 haplotype on the
BN background (BN-1L) or with the n haplotype on the
LEW background (LEW-1N).
8.2.2.3 Mechanisms of induction
Mechanisms of induction have been thoroughly studied
in rats. Mercuric chloride induces in Brown Norway rats a
polyclonal activation of B cells (Hirsch et al., 1982;
Pelletier et al., 1988a). T cells are required, since
Brown Norway rats with the nude mutation or depleted of
T cells are resistant (Pelletier et al., 1987a). It
appears that mercuric chloride induces in this rat strain
the appearance of T cells able to stimulate class II de-
terminants (also called Ia antigens), which are expressed
on the cell membrane of all B cells (Pelletier et al.,
1986). The role of such T cells is strongly supported by
the fact that T cells from Brown Norway rats injected with
mercuric chloride are able to transfer auto-immune mani-
festations to normal Brown Norway recipients and also to
Brown Norway rats depleted of T cells (Pelletier et al.,
1988b). This strongly suggests that T cells from rats
injected with mercuric chloride are able to stimulate
B cells directly. The autoreactive, anticlass II, T cells
which recognize normal B cells as well as B cells from
rats injected with mercuric chloride may have initially
been induced following a modification of class II deter-
minants by mercury, as suggested by Gleichmann et al.
(1984). It is also possible that mercuric chloride affects
CD8+ (suppressor/cytotoxic) T cells, as suggested by
Weening et al. (1981).
The fine mechanism of action at the cellular level
(see section 8.7) remains to be elucidated.
8.2.2.4 Autoregulation
The auto-immune disease observed in Brown Norway rats
is self-regulated. Abnormalities progressively disappear
after the third week. It has been shown that CD8+ (sup-
pressor/cytotoxic) T cells are responsible for this effect
(Bowman et al., 1984), together with the appearance of
auto-anti-idiotypic antibodies (Chalopin & Lockwood,
1984).
8.2.2.5 Immunosuppression
Lewis rats do not develop auto-immune disorders when
injected with mercuric chloride. In contrast, CD8+
(suppressor/cytotoxic) T cells proliferate in the spleen
and in the lymph nodes of such animals. As a consequence
they develop a non-antigen-specific immunosuppression
(Pelletier et al., 1987c). They do not respond either to
classical mitogens or to allo-antigens. More interest-
ingly, mercuric chloride is able to inhibit the develop-
ment of organ-specific auto-immune disorders such as
Heymann's nephritis (Pelletier et al., 1987b) and exper-
imental allergic encephalomyelitis (Pelletier et al.,
1988c). The mechanisms are not yet understood.
The mercury model represents a unique tool for evalu-
ating the relationship between genetic and chemically
induced immune disregulation.
8.2.2.6 Conclusions
It may be concluded that the most sensitive adverse
effect caused by mercuric mercury is the formation of
mercuric-mercury-induced auto-immune glomerulonephritis,
the first step being the production and deposition of IgG
antibodies to the glomerular basement membrane. The Brown
Norway rat is a good test species for the study of
mercuric-mercury-induced auto-immune glomerulonephritis,
although this effect has also been observed in rabbits.
Table 4 presents the available studies on auto-immune
glomerulonephritis. The lowest-observed-adverse-effect
level found in these studies was 16 mg/kg per day via the
subcutaneous route of exposure.
8.3. Reproduction, embryotoxicity, and teratogenicity
8.3.1. Males
Very little information on male reproductive effects
is available. Lee & Dixon (1975) injected male mice with
single doses of mercuric chloride (1 mg mercury/kg body
weight) and found a significant decrease in fertility,
compared with controls, in controlled mating tests. Normal
fertility was restored after about 2 months. In studies by
Chowdhury et al. (1986), gradual alterations in testicular
tissues were noted in rats treated with mercuric chloride
at intraperitoneal dosages of 0.05 mg/kg and 0.1 mg/kg
body weight over a period of 90 days. There was a decrease
in seminiferous tubular diameter, spermatogenic cell
counts, and Leydig cell nuclear diameter, compared with
controls.
Table 4. Auto-immune effects of mercuric mercury on the glomerular
basement membrane
-----------------------------------------------------------------------
Animal Route Duration Adverse Reference
effect
level
(mg/kg
per day)
-----------------------------------------------------------------------
Brown Norway rat oral 60 days 320 Bernaudin
et al. (1981)
Brown Norway rat oral 60 days 630 Andres (1984)
Brown Norway rat subcutaneous 12 weeks 16 Druet et al.
(1977)
Brown Norway rat subcutaneous 8 weeks 32a Druet et al.
(1978)
Rabbit intramuscular 1-17 weeks 633 Roman-Franco
et al. (1978)
-----------------------------------------------------------------------
a Proteinuria was observed, in addition to the auto-immune
glomerulonephritis, in these rats.
8.3.2. Females
Lamperti & Printz (1973) injected female hamsters with
daily doses of 1 mg mercuric chloride (8-11 mg mercury/kg)
throughout one 4-day estrous cycle (the LD50 being 18 mg
mercury/kg body weight). There were effects on the repro-
ductive system, including morphological changes of corpora
lutea and inhibition of follicular maturation. In further
studies (Lamperti & Printz, 1974), it was reported that
60% of female hamsters did not ovulate by day one of the
third estrous cycle after having been given a total of
3-4 mg mercuric chloride during the first estrous cycle.
Watanabe et al. (1982) injected female hamsters with mer-
curic chloride at high doses (6.4 or 12.8 mg mercury/kg
body weight) during day one of the estrous cycle and
observed an inhibition of ovulation. Lamperti & Niewenhuis
(1976) injected female hamsters with 1 mg mercuric chlor-
ide per day during one estrous cycle and found signifi-
cantly higher levels of follicle-stimulating hormone in
the pituitary gland, compared with controls.
Several investigators have reported abortions follow-
ing exposure to elemental mercury vapour or inorganic
mercury compounds several days after implantation. There
are also reports of decreased fetal weight and malfor-
mations. Gale & Ferm (1971) injected three groups of
female hamsters with a single dose of 2, 3, or 4 mg mer-
curic acetate (about 1.3-2.5 mg mercury) intravenously on
day 8 of gestation. The exposed groups showed resorption
frequencies of 12, 34, and 52%, respectively, compared to
4% in the controls. The mothers showed signs of mercury
intoxication in the form of weight loss, kidney lesions,
and diarrhoea. In later studies (Gale, 1980, 1981), a
single injection of mercuric acetate (15 mg/kg body
weight) to hamsters produced a cluster of cardiac and non-
cardiac abnormalities. The most important aspects of
embryotoxicity were resorptions, retardation, and abnormal
heart. Significant but varied interstrain differences were
observed. Holt & Webb (1986) exposed pregnant Wistar rats
intravenously to mercuric chloride at different periods of
gestation. During mid-gestation the minimum effective
teratogenic dose of mercury (0.79 mg/kg total body weight)
was high in relation to the maternal LD50 and the inci-
dence of fetal malformations, mainly brain defects, was
23% in all live fetuses. In rats of different gestational
ages, uptake of Hg2+ by the fetus at this dose level
decreased sharply between day 12 and day 13. The terato-
genic effects on the fetus and damage to the maternal
kidneys, however, were essentially the same in animals
dosed with Hg2+ either immediately before or immediately
after these gestational ages. The authors considered it
probable, therefore, that fetal defects resulted not from
any direct action of Hg2+ on the conceptuses but either
from the inhibition of the transport of essential metab-
olites from the mother or from maternal kidney dysfunc-
tion.
In a study by Rizzo & Furst (1972), three groups of
female rats were exposed to single oral doses of mercuric
oxide equivalent to 2 mg mercury/dose. Each dose of mer-
cury was given suspended in 2 ml peanut oil. The three
groups received the mercury on day 5, 12, or 19 after con-
ception. External malformations were observed in 29.7%,
6.8%, and 3.4% of cases, respectively, while the three
control groups had values between 0 and 2%. The two
observable effects that mercury had on rat fetuses were
arrest of general growth, as indicated by the number of
runts, and inhibition of eye formation. The mothers showed
no effects from the treatment. The data, if confirmed, are
of particular interest, as mercuric oxide is fairly insol-
uble.
Steffek et al. (1987) reported the effects of elemen-
tal mercury vapour exposure on pregnant Sprague-Dawley
rats. The rats were exposed to elemental mercury vapour at
concentrations of 100, 500, or 1000 µg/m3 during the
entire gestational period (chronic exposure) or during the
period of organogenesis (days 10-15, acute exposure).
Macroscopic examination of fetuses obtained from pregnant
rats exposed acutely or chronically to 100 µg/m3 re-
vealed no increased incidence of congenital malformations
or resorptions when compared to room or chamber controls.
However, acute exposure to 500 µg/m3 resulted in an
increase in the number of resorptions (5/41), and chronic
exposure at this concentration resulted in two fetuses
(out of 84 that were examined) with cranial defects. There
were also single cases of encephalomeningocoele, dome-
shaped cranial configuration, and cleft palate (Steffek,
A.J., written personal communication to the American
Dental Association). Acute exposure at 1000 µg/m3 re-
sulted in an increase in the rate of resorptions (8/71),
and chronic exposure at this dose level produced a
decrease in maternal and fetal weights, relative to the
control groups, and an increase in the number of resorp-
tions (7/28).
8.4. Mutagenicity and related end-points
Mercuric mercury affects the mitotic spindle in
plants, which may lead to an abnormal distribution of
chromosomes (Ramel, 1972; Leonard et al., 1983). It is not
a potent inducer of dominant lethal mutations in mice
(Suter, 1975). Zasukhina et al. (1983) reported the induc-
tion of single-stranded DNA breaks after exposure of
cultures of mice embryo cells to mercury chloride. The
mercury did not induce mutations but had a strong lethal
effect in a survival test of vaccinia virus. A shortening
of the chromosome length in human lymphocytes exposed in
vitro has been observed (Andersen et al., 1983). Mercuric
mercury did not induce chromosomal aberrations in human
lymphocytes and in mammalian cells in vitro (Paton &
Allison 1972; Umeda & Nishimura, 1979). Positive results
in the recombination assay with mercuric chloride have
been reported by Kanematsu et al. (1980). Effects have
been reported on DNA repair in mammalian cells (Robison et
al., 1984). There was an increase in C-mitotic figures and
segregational errors in human lymphocytes and in Indian
muntjac fibroblasts (Verschaeve et al., 1984, 1985). Based
on studies of Drosophila melanogaster, Magnusson & Ramel
(1986) found a pronounced variation in tolerance between
12 wild type strains when testing a number of metal com-
pounds, including mercuric chloride. Morimoto et al.
(1982) reported that in human whole blood cultures sele-
nite prevents the induction of sister-chromatid exchanges
by mercuric chloride.
The US Agency for Toxic Substances and Disease Regis-
try (ATSDR, 1989) has reviewed several in vitro genotox-
icity studies on mercury compounds. Mercuric chloride was
found to induce gene mutations in mouse lymphoma cells
(Oberly et al., 1982) and DNA damage in rat and mouse
fibroblasts (Zasukhina et al., 1983). It was observed to
1983; Christie et al., 1986). Using the alkaline elution
assay in intact Chinese hamster ovary cells, several
studies have shown that mercuric chloride can cause
single-strand breaks in DNA (Cantoni et al., 1982, 1984a,
1984b; Cantoni & Costa, 1983; Christie et al., 1984,
1986). Furthermore, Cantoni & Costa (1983) found that the
DNA-damaging effect of mercuric chloride is enhanced by a
concurrent inhibitory effect that mercury has on DNA
repair mechanisms.
8.5. Carcinogenicity
WHO (1976) reported no evidence that inorganic mercury
is carcinogenic. In a study by Schroeder & Mitchener
(1975), groups of mice were exposed to various metals,
including mercuric chloride. After a lifetime of exposure
to 5 µg mercury/litre in basal drinking-water, 51.2%
(21 out of 41) of the mice revealed tumours compared to
29.8% (14 out of 47) among controls. The incidence was
higher than for any of the other metals tested, but the
authors concluded that no element was significantly
tumorigenic. Mercury has not been reviewed by IARC (IARC,
1987). The US EPA (1989) has classified inorganic mercury
as a group O compound, i.e. it is not classifiable as to
human carcinogenicity.
8.6. Factors modifying toxicity
Factors such as age, sex, nutritional state, and oral
exposure giving rise to sensitization are likely to affect
the relationship between dose and effect or response. The
type of chemical exposure (whether to elemental mercury or
to mercuric mercury salts) is an important determinant for
the toxic effect and to differences in distribution.
As described in section 6.1.2, Kostial et al. (1978)
observed a high degree of absorption of inorganic mercury
in newborn rats after oral exposure to mercuric chloride.
The immature rodent kidney is, on the other hand, less
sensitive to mercury exposure than the adult kidney, as
less accumulation takes place in the kidney of the newborn
pups (Daston et al., 1983). No information is available
concerning age effects in humans.
Exposure of rats to high concentrations of mercury
vapour induced metallothionein in kidney tissue that
resulted in the binding of divalent mercury (Sapota et
al., 1974). Female rats are less susceptible than male
rats to the nephrotoxic effect of mercuric mercury (Magos
et al., 1974). This seems to be related to the metallo-
thionein content of the kidney, which is higher in females
and is increased by estradiol treatment (Nishiyama et al.,
1987). Administration of zinc to rats reduced the renal
toxic effects of mercuric mercury and induced an increase
in the glutathione (Fukino et al., 1986) and metallo-
thionein content of renal tissue (Yoshikawa & Ohta,
1982). Zinc treatment in hamsters injected with mercuric
salt reduced the embryotoxic and teratogenic effects
produced by treatment with the mercuric salt alone (Gale,
1984).
Selenium has been found to affect the distribution of
mercuric mercury in rats (Parizek & Ostadalova, 1967;
Nygaard & Hansen, 1978), mice (Eybl et al., 1969), rabbits
(Imura & Naganuma, 1978; Naganuma & Imura, 1980), and pigs
(Hansen et al., 1981). As a consequence of this redistri-
bution, a decrease in toxicity has also been observed
(Parizek & Ostadalova, 1967; Johnson & Pond, 1974). Mer-
cury forms a mercury-protein complex with selenium (Burk
et al., 1974), which can be identified in plasma and blood
cells (Chen et al., 1974; Imura & Naganuma, 1978). When
given with selenium, mercury is retained longer in blood
and, as a consequence, accumulation in the kidney is
decreased. Mercury taken up by the kidney is bound to a
protein-selenium complex, and, on administration of equi-
valent amounts of selenium, the binding to metallothionein
is diminished and may be negligible (Komsta-Szumska &
Chmielnicka, 1977; Mengel & Karlog, 1980). A consequence
of the changed binding of mercury in blood brought about
by selenium is that transport of selenium and mercury
through the placenta membranes is inhibited (Parizek et
al., 1971).
So far only selenate or selenite compounds, and not
the naturally occurring selenium compounds in food, have
been studied in detail. However, Magos et al. (1984, 1987)
compared the distribution and form of mercury and protec-
tion against the nephrotoxic effects of mercury after
exposure to different forms or compounds of selenium. It
was concluded that dietary selenium is less efficient than
selenite as an antidote against mercurial nephrotoxicity.
Studies of selenium interaction with mercuric mercury
have mainly been carried out in rodents. Selenium metab-
olism in humans is different from that in most animals,
and selenium dependency in humans is comparatively less
than that in rodents. However, observations in workers
exposed to mercury vapour indicate that there is also a
selenium-mercury interaction in humans. Selenium and
mercury concentrations with a molar ratio of 1:1 have been
found in organs such as the brain, thyroid, and pituitary
(Kosta et al., 1975; Rossi et al., 1976). In renal
biopsies from two mercury-intoxicated patients, inclusion
bodies were seen in lysosomes of renal tubules, and it was
demonstrated that these inclusion bodies contained
selenium and mercury (Aoi et al., 1985). In 28 workers
exposed to mercury vapour, the selenium excretion in urine
was high compared to non-exposed workers (Alexander et
al., 1983). However, this was not confirmed by Suzuki et
al. (1986), who studied 57 workers exposed to mercury
vapour. They found a decrease in selenium excretion as
mercury excretion in the urine increased.
As discussed in section 6.2, ethanol inhibits the
enzyme catalase, which is the main enzyme responsible for
the oxidation of mercury in blood and tissues. Ethanol
consumption thus modifies the balance between oxidation
and reduction of mercury in tissues. As a consequence,
less mercury vapour is absorbed from the lungs, more
mercury exists unoxidized in the blood, and more mercury
is transported to the brain. It is unclear whether the
observed decrease of mercury in the brain is due to the
fact that less mercury is oxidized and retained in the
brain or that more retained mercury is lost from the brain
as a result of reduction following the intake of ethanol.
The mercury content of the brain following acute exposure
to mercury in acatalasaemic mice is greater than that of
mercury-exposed control mice (Ogata et al., 1987). This is
also the case with the fetal brain after exposure of preg-
nant female acatalasaemic mice (Ogata & Meguro, 1986).
8.7. Mechanisms of toxicity - mode of action
The neurotoxic effect seen after exposure to metallic
mercury vapour is attributable to the divalent mercury ion
formed through oxidation in the brain tissue. Interference
with enzyme function by binding to sulfhydryl groups is
one possible mechanism. Kark (1979) reviewed the available
evidence regarding the inhibitory effect of mercuric ions
on different enzyme systems ( in vitro and in vivo). Mer-
cury concentrations at which enzyme inhibition appears are
consistent with concentrations at which toxic effects on
the central nervous system are observed. The in vivo con-
ditions are, however, complicated. Ligands capable of
binding mercuric ions, e.g., sulfhydryl groups and seleno-
hydrol groups, are ubiquitous and associated with
proteins. These ligands may have a protective or scaveng-
ing effect, thereby preventing interference with important
receptors. Mercuric ions penetrate cell membranes to a
very limited degree. In contrast mercury vapour penetrates
more readily due to its lipophilicity. Miyamoto (1983)
demonstrated with frog nerve-muscle preparations that mer-
curic mercury penetrates the nerve cell membrane through
sodium and calcium channels, causing an irreversible
depolarization and an increase in transmitter release.
There is a subsequent irreversible block of transmitter
release. Transport through the cell membrane via the for-
mation of carrier complexes would also be a possibility,
although this has not been demonstrated. Mercury has been
found intracellularly in nerve cells after exposure to
mercury vapour (Cassano et al., 1966) and also after pro-
longed exposure to mercuric chloride in the rat (Moller-
Madsen & Danscher, 1986).
Mercuric ions react with DNA and RNA in vitro and may
change the tertiary structure of these molecules (Eichhorn
& Clark, 1963; Gruenwedel & Davidson, 1966). Inhibition
of protein synthesis has been observed in cell systems as
well as in cell-free systems at a mercury concentration
equivalent to 2 x 10-5 mol/litre (Nakada et al., 1980).
Similar mercury concentrations have been observed to
increase the pre-synaptic release of the transmitter sub-
stance acetylcholine (Kostial & Landeka, 1975; Manalis &
Cooper, 1975). An increase in the release of dopamine in
the rat brain after treatment with mercury (10-5 mol per
litre) at a level of 2 µg/g was observed by Bondy et al.
(1979).
Mercury may interfere with membrane structure in vitro
by hydrolysing specific lipids (Ganser & Kirschner,
1985), causing membrane lesions, and by reducing lipid
synthesis in nerve cells (Cloez et al., 1987). Further-
more, an irreversible interference with the post-synaptic
membrane has been observed (Manalis & Cooper, 1975; Juang,
1976).
Knowledge regarding the mechanism of mercury neurotox-
icity, following exposure to mercury vapour, is still
fragmentary. Little data has so far emerged concerning
morphological changes in the human or primate brain. Mer-
cury has been demonstrated in rats within the motor nuclei
of the rhombencephalon and the cerebral cortex, the
highest concentrations occurring in striated areas and
within the deep nuclei of the cerebellum. A proportion-
ately high level was seen in the anterior horn motor
neurones of the spinal cord. The localization was gener-
ally interneuronal, but was also seen in the cytoplasm of
glial and ependymal cells (Moller-Madsen & Danscher,
1986). In human cases of mercury poisoning, chromatolysis
in scattered neurones was observed in the occipital lobe
of the brain and there was a loss of Purkinje cells
(Takahata et al., 1970). Davis et al. (1974) demonstrated,
in two human cases, mercury in the cytoplasm of neurones
in the nuclei olivaris and dentatus, in Purkinje cells,
in anterior horn cells of the spinal cord, and in the
neurones of the substantia nigra. In both cases a decrease
in the number of neurones of the granular cell layer in
the cerebellum and, possibly, also of Purkinje cells was
seen.
The mechanism behind the nephrotoxic effect of inor-
ganic mercury is discussed in sections 8.2 and 9.3. Here
it can be summarized that two types of renal injury have
been observed. The first is a glomerular injury caused by
an auto-immune reaction induced by mercury and resulting
in antibody formation against the glomerular tissue, depo-
sition of immune complex, glomerular nephritis, protein-
uria, and nephrotic syndrome. Alternatively, immune com-
plexes containing other mercury-induced anti-bodies may be
deposited in the glomeruli. The second is a renal tubular
damage affecting the proximal tubules and developing in
parallel with the accumulation of mercury in the renal
tubular cells. This damage results in a loss of renal
tubular enzymes, such as gamma-glutamyl transferase, and
lysosome enzymes, such as beta-galactosidase, beta-glucur-
onidase and N-acetyl-beta-glucosaminidase (Foa et al.,
1976), and in decreased reabsorption leading to an
increased secretion of endogenous trace elements such as
zinc and copper (Chmielnicka et al., 1986). An early
effect is an inhibition of protein synthesis. A swel-
ling of the endoplasmic reticulum with disaggregation
of polyribosomes is observed in electron microscopy.
Eventually, renal tubular necrosis and renal failure
develop (Wessel, 1967; Gritzka & Trump, 1968; Barnes
et al., 1980; Pezerovic et al., 1981).
The immunotoxic effect of inorganic mercury is prob-
ably the least understood effect of exposure to inorganic
mercury. Mercuric mercury has been observed as a potent
stimulator of human T lymphocytes in vitro (Schöpf et al.,
1969; Nordlind & Henze, 1984; Nordlind, 1985). Mercury is
initially bound to lymphocyte membranes, but it has also
been demonstrated that there is an uptake of mercury by
the nuclei (Nordlind, 1985) at levels likely to occur in
blood following exposure to mercury vapour. It can be
speculated that this phenomenon may be related to the
rather generalized syndromes observed in children, such as
acrodynia or "Pink disease" (Warkany, 1966; Skerfving &
Vostal, 1972) and the rather generalized and unspecific
syndromes reported to be related to dental amalgam fill-
ings, but with an unproven relation to mercury exposure
(section 9.7).
9. EFFECTS ON HUMANS
WHO (1976) dealt primarily with effects of occu-
pational exposure to mercury vapour. Apart from accidental
exposure, there was little information on exposure to in-
organic mercury among the general population. Recent data
indicate that the release of mercury vapour from amalgam
fillings may dominate exposure to inorganic mercury among
the general population (Elinder et al., 1988; WHO, 1990).
Other sources are a fish-rich diet (biotransformation of
alkyl mercury present in some species of fish resulting in
the accumulation of inorganic mercury), environmental pol-
lution in the vicinity of industrial sources, toxic waste
sites, and accidental spillage. Mercury-containing phar-
maceuticals may also be significant sources of exposure
for some populations.
The present review will focus on possible chronic
effects of long-term, low-level occupational exposure and,
for the general population, of mercury released from
amalgam. However, a brief review of acute effects will be
given. In general, the available information is presented
according to the effects on organs or organ systems. Some-
times this is not possible, as is the case for the combi-
nation of a number of unspecific symptoms that have been
associated by some with exposure from dental amalgams.
Whenever dose-response relationships are discussed, it
should be borne in mind that data on exposure levels in
the past are scarce. Quality assurance data are with few
exceptions not reported. In most studies personal samplers
were not used but only static samplers, which can be a
source of considerable error in the estimated absorbed
dose, as demonstrated by Stopford et al. (1978) and Roels
et al. (1987).
Data related to response rates are sometimes difficult
to interpret. The studies are as a rule cross-sectional,
and so selection bias, which may lead to either an under-
estimation or an overestimation of risks, can occur. The
numbers of exposed subjects and controls are often small.
For quite a few of the parameters studied, the results may
be influenced by the investigator and "interviewer bias"
is thus possible.
9.1. Acute toxicity
Workers acutely exposed (4-8 h) to calculated elemen-
tal mercury levels of 1.1 to 44 mg/m3 due to an accident
exhibited chest pains, dyspnoea, cough, haemoptysis,
impairment of pulmonary function, and evidence of inter-
stitial pneumonitis (McFarland & Reigel, 1978). Acute mer-
cury poisoning with mercurial pneumonitis was reported
among four men after they were exposed to mercury vapour
while attempting home gold ore purification using a gold-
mercury amalgam and sulfuric acid (Levin et al., 1988).
Urine mercury levels were 169 to 520 µg/litre in two
cases. Probably there was also substantial exposure to
sulfur dioxide and sulfuric acid.
Troen et al. (1951) reported 18 cases of human poison-
ing following oral ingestion of single doses of mercuric
chloride, nine of which resulted in death. The lethal
doses ranged from 29 mg/kg body weight to at least
50 mg/kg. The most common autopsy findings in these cases
were gastrointestinal lesions (ranging from mild gastritis
to severe necrotizing ulceration of the mucosa) and renal
lesions that had resulted in renal failure.
9.2. Effects on the nervous system
Most information focuses on effects on the central
nervous system following occupational exposure. The
central nervous system is the critical organ for mercury
vapour exposure (WHO, 1976). Acute exposure has given rise
to psychotic reactions characterized by delirium, halluci-
nations, and suicidal tendency. Occupational exposure has
resulted in erethism, with irritability, excitability,
excessive shyness, and insomnia as the principal features
of a broad-ranging functional disturbance. With continuing
exposure, a fine tremor develops, initially involving the
hands and later spreading to the eyelids, lips, and
tongue, causing violent muscular spasms in the most severe
cases. The tremor is reflected in the handwriting which
has a characteristic appearance. In milder cases, erethism
and tremor regress slowly over a period of years following
removal from exposure. Decreased nerve conduction velocity
in mercury-exposed workers has been demonstrated (Bidstrup
et al., 1951). Long-term, low-level exposure has been
found to be associated with less pronounced symptoms of
erethism, characterized by fatigue, irritability, loss of
memory, vivid dreams, and depression (WHO, 1976).
9.2.1. Relations between mercury in the central nervous
system and effects/response
There is very little information on brain mercury
levels in cases of mercury poisoning, and nothing that
makes it possible to estimate a no-observed-effect level
or a dose-response curve. Furthermore, the available data
are uncertain as they are not accompanied by analytical
quality control information.
Brigatti (1949) reported mercury concentrations of
6-9 mg/kg in the brain of two workers exposed to mercury
vapour and with signs of mercury poisoning 2 years before
they died. In two similar cases, Takahata et al. (1970)
reported 5-34 mg mercury/kg in different parts of the
brain. In two fatal cases of poisoning, which followed
many years of exposure to high doses of calomel as a
laxative, mercury concentrations of about 4 mg/kg in the
frontal lobe cortex were reported, and in one of the cases
a concentration of 106 mg/kg was found in the inferior
olive (Davis et al., 1974; Wands et al., 1974). The symp-
toms were dementia, erethism, colitis, and renal failure.
A dose of two tablet laxatives, each containing 120 mg of
USP-grade mercurous chloride, had been taken daily for 25
years in one case and for 6 years in the other.
9.2.2. Relations between mercury in air, urine or blood and
effects/response
9.2.2.1 Occupational exposure
WHO (1976) found no evidence of the classical symptoms
of mercurialism, erethism, intentional tremor, or gingi-
vitis below a time-weighted occupational exposure to
mercury in air of 100 µg/m3. A report on cottage
industry mercury smelting in China (Wu et al., 1989) has
confirmed the classical symptoms at exposure levels of up
to 600 µg/m3. Symptoms such as loss of appetite and
psychological disturbance have also been found to occur at
mercury levels below 100 µg/m3 (WHO, 1976).
Zeglio (1958) observed that following cessation of
exposure, symptoms and signs of neurological impairment
regress slowly in the milder cases of poisoning affecting
the nervous system. However, in the more severe cases,
neurological impairment persists and may become exacer-
bated.
One of the studies on which WHO (1976) based its con-
clusions was carried out by Smith et al. (1970). This
study covered the prevalence of several medical findings
in 567 workers in a number of chloralkali factories in the
USA in relation to mercury vapour exposure (Fig. 6). The
authors concluded that the data showed no significant
signs or symptoms in persons exposed to mercury vapour at
or below a level of 0.1 mg/m3. Subjective symptoms
appeared to increase at lower exposure levels, but the
authors questioned this finding because of the confounding
effect of alcohol. In a follow-up of part of this study,
Bunn et al. (1986) did not find significant differences in
the frequency of objective or subjective findings related
to mercury exposure, which generally was said to range
from 50 to 100 µg/m3 (time-weighted average). The
report, however, did not give sufficient information about
several methodological questions, including quality assur-
ance aspects and possible confounding variables.
Later studies, covering industries where exposure has
been high, have pointed towards the importance of urine
mercury peaks in excess of 500 µg/litre for the develop-
ment of neurological signs and symptoms (Langolf et al.,
1978, 1981). Urine mercury peaks in excess of 100 µg per
litre have been associated with impaired performance in
mechanical and visual memory tasks and psychomotor ability
tests (Forzi et al., 1976).
There is also a report (Albers et al., 1988) that, as
long as 20 to 35 years after exposure, subjects who had
experienced urine mercury peak levels above 600 µg per
litre demonstrated significantly decreased strength,
decreased coordination, increased tremor, decreased sen-
sation, and increased prevalence of Babinski and snout
reflexes when compared with control subjects. Furthermore,
subjects with reported clinical polyneuropathy had sig-
nificantly higher peak levels of mercury in urine than the
subjects without those signs. The reported signs of the
presence of an upper motor neuron lesion will require
further investigation. Many measures demonstrating sig-
nificant differences between exposed and unexposed
subjects were age dependant, but a multiple regression
analysis showed that the association between neurological
signs and mercury exposure remained after allowing for age
(Albers et al., 1988).
On the basis of studies by Smith et al. (1970) and
Lindstedt et al. (1979), Skerfving & Berlin (1985)
suggested that a urine mercury level of 50 µg/g creati-
nine is associated with a blood mercury level of 20 µg
per litre. Roels et al. (1987) reported a regression
equation, where a urine mercury level of 50 µg/g creati-
nine leads to a blood mercury level of 16 µg/litre.
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
This chapter is extracted from the summary of Environ-
mental Health Criteria 86: Mercury - Environmental Aspects
(WHO, 1989).
7.1. Uptake, elimination, and accumulation in organisms
Mercuric salts, and, to a much greater extent, organic
mercury, are readily taken up by organisms in water.
Aquatic invertebrates, and most particularly aquatic
insects, accumulate mercury to high concentrations. Fish
also take up the metal and retain it in tissues, princi-
pally as methylmercury, although most of the environmental
mercury to which they are exposed is inorganic. The source
of the methylation is uncertain, but there is strong indi-
cation that bacterial action leads to methylation in
aquatic systems. Environmental levels of methylmercury
depend upon the balance between bacterial methylation and
demethylation. The indications are that methylmercury in
fish arises from this bacterial methylation of inorganic
mercury, either in the environment or in bacteria associ-
ated with fish gills, surface, or gut. There is little
indication that fish themselves either methylate or
demethylate mercury. Elimination of methylmercury is slow
from fish (with half times in the order of months or
years) and from other aquatic organisms. Loss of inorganic
mercury is more rapid and so most of the mercury in fish
is retained in the form of methylmercury. Terrestrial
organisms are also contaminated by mercury, with birds
being the best studied. Sea birds and those feeding in
estuaries are most contaminated. The form of retained
mercury in birds is more variable and depends on species,
organ, and geographical site.
7.2. Toxicity to microorganisms
The metal is toxic to microorganisms. Inorganic mer-
cury has been reported to have effects at concentrations
of the metal in the culture medium of 5 µg/litre, and
organomercury compounds at concentrations at least 10
times lower than this. Organomercury compounds have been
used as fungicides. One factor affecting the toxicity of
the organometal is the rate of uptake of the metal by
cells. Mercury is bound to the cell walls or cell mem-
branes of microorganisms, apparently to a limited number
of binding sites. This means that effects are related to
cell density as well as to the concentration of mercury in
the substrate. These effects are often irreversible, and
mercury at low concentrations represents a major hazard to
microorganisms.
7.3. Toxicity to aquatic organisms
The organic forms of mercury are generally more toxic
to aquatic organisms than the inorganic forms. Aquatic
plants are affected by mercury in the water at concen-
trations approaching 1 mg/litre for inorganic mercury but
at much lower concentrations of organic mercury. Aquatic
invertebrates vary greatly in their susceptibility to
mercury. Generally, larval stages are more sensitive than
adults. The 96-h LC50s vary between 33 and 400 µg per
litre for freshwater fish and are higher for sea-water
fish. However, organic mercury compounds are more toxic.
Toxicity is affected by temperature, salinity, dissolved
oxygen, and water hardness. A wide variety of physiologi-
cal and biochemical abnormalities has been reported after
fish have been exposed to sublethal concentrations of
mercury, although the environmental significance of these
effects is difficult to assess. Reproduction is also
affected adversely by mercury.
7.4. Toxicity to terrestrial organisms
Plants are generally insensitive to the toxic effects
of mercury compounds. Birds fed inorganic mercury show a
reduction in food intake and consequent poor growth.
Other, more subtle, effects on enzyme systems, cardio-
vascular function, blood parameters, the immune response,
kidney function and structure, and behaviour have been
reported. Organomercury compounds are more toxic for birds
than are inorganic.
7.5. Effects of mercury in the field
Pollution of the sea with organomercury led to the
death of fish and fish-eating birds in Japan. Except for
this incident at Minamata, few follow-up studies of the
effects of localized release have been conducted. The use
of organomercury fungicides as seed dressings in Europe
led to the deaths of large numbers of granivorous birds,
together with birds of prey feeding on the corpses. Resi-
dues of mercury in birds' eggs have been associated with
deaths of embryos in shell. The presence of organochlorine
residues in the same birds and their eggs makes an accu-
rate assessment of the effects of mercury difficult. It
is, however, thought to be a contributing factor in the
population decline of some species of raptors.
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
8.1. Single and short-term exposure
Ashe et al. (1953) reported evidence of damage to
brain, kidney, heart, and lungs in rabbits exposed acutely
to metallic mercury vapour at a mercury concentration of
29 mg/m3 of air.
The LD50 for inorganic mercury, as well as for a num-
ber of organomercurials (e.g., arylmercury, alkoxyalkyl-
and alkylmercury compounds), lies between 10 and 40 mg/kg
body weight for all compounds tested. For mercuric chlor-
ide, a value of about 10 mg/kg body weight has been
observed after parenteral administration to mice. The
similarity in LD50 values for these various types of mer-
cury compounds is considered to result from the fact that,
when given in acute massive doses, mercury in any chemical
form will denature proteins, inactivate enzymes, and cause
severe disruption of any tissue with which it comes into
contact in sufficient concentrations. The features of
acute toxicity usually consist of shock, cardiovascular
collapse, acute renal failure, and severe gastrointestinal
damage.
8.2. Long-term exposure
8.2.1. General effects
WHO (1976), in evaluating a number of experimental
studies on animals, concluded that both reversible and
irreversible toxic effects may be caused by mercury and
its compounds. Microscopically detectable changes have
been seen in the organs of dogs, rabbits, and rats exposed
to concentrations of elemental mercury vapour ranging from
about 100 to 30 000 µg/m3 for different periods of time.
Severe damage was noted in kidneys and brains at mercury
levels in air of about 900 µg/m3 after an exposure
period of about 12 weeks. After exposure of dogs to
100 µg mercury/m3, for 7 h/day, 5 days/week over a
period of 83 weeks, no microscopically detectable effects
were seen, and tests revealed no abnormalities in kidney
function.
In two studies (Fukuda, 1971; Kishi et al., 1978),
tremor and behavioural effects were observed in rabbits
and rats after several weeks of exposure to metallic mer-
cury vapour at levels of several mg/m3, although there
were no morphological changes in the brain. The symptoms
were associated with brain mercury concentrations of about
1 and 20 mg/kg wet weight in the two studies.
8.2.2. Immunological effects
During the last 10-20 years, great attention has been
paid to effects of inorganic mercury on the immune system.
An important conclusion is that, depending upon the animal
strain tested, either auto-immunity or immunosuppression
is observed.
8.2.2.1 Auto-immunity
Bariety et al. (1971) showed that about 30% of outbred
Wistar rats exposed to mercuric chloride (1.5 or 2.5 mg/kg
body weight) 3 times per week for periods of several
months developed a membranous glomerulopathy characterized
by granular, subepithelial deposits of IgG and C3.
In Brown Norway rats (Sapin et al., 1977; Druet et
al., 1978) and in New Zealand rabbits (Roman-Franco et
al., 1978) injected with 1 or 2 mg/kg body weight, a sys-
temic auto-immune disease was observed. It appeared in
100% of the rats tested. This disease is characterized by
the production of auto-antibodies to renal and extra-renal
basement membranes. These antibodies are specific for
laminin, type IV collagen, and entactin (Bellon et al.,
1982; Fukatsu et al., 1987) and are found deposited along
the glomerular basement membrane in a linear pattern.
After 3 to 4 weeks, a typical membranous glomerulopathy
with granular, subepithelial IgG deposits is observed. The
majority of rats develop proteinuria, which progresses in
some animals to the nephrotic syndrome (Druet et al.,
1978). About half of those with this syndrome die. How-
ever, the remainder recover since the disease is transi-
ent. Dermatitis and Sjogren's syndrome have been recently
detected in Brown Norway rats injected with mercuric
chloride (Aten et al., 1988). Contact sensitization to
mercury has also been reported to occur in susceptible
strains of guinea-pigs (Polak et al., 1968). Auto-immunity
in Brown Norway rats appears in the context of a poly-
clonal activation of B cells. There is a lymphoprolifer-
ation (increase in the number of CD4+ T cells and B
cells) and hyperimmunoglobulinaemia affecting mainly IgE
(Prouvost-Danon et al., 1981; Hirsch et al., 1982;
Pelletier et al., 1988a), and several auto-antibodies such
as antinuclear antibodies (Hirsch et al., 1982) are
produced. All these manifestations are transient. They
appear from day 8 following the injection, peak during the
third week, and then progressively decline.
A number of other studies have been carried out using
Brown Norway rats. Low doses of mercuric chloride
(50 µg/kg body weight given 3 times per week) induced
auto-immune glomerulopathy, while 100 µg/kg body weight
(also 3 times per week) induced both auto-immune glomer-
ulopathy and proteinuria. Mercuric chloride was also
effective when given by inhalation (aerosol or intratra-
cheal instillation) or ingestion (Bernaudin et al., 1981;
Andres, 1984; Knoflach et al., 1986). Other inorganic
mercury compounds, e.g., mercurous chloride given orally
or HgNH2Cl-containing ointments, also induce auto-
immunity (Druet et al., 1981).
Auto-immune disorders including auto-immune glomer-
ulopathy have been described in other strains of rats
(Weening et al., 1978; Druet et al., 1982). In suscep-
tible strains of mice, especially those mice carrying the
H-2s haplotype, long-term exposure to mercuric chloride
induces extremely high titres of antinucleolar auto-anti-
bodies (Robinson et al., 1986; Mirtscheva et al., 1987).
These auto-antibodies and circulating immune complexes
(Hultman & Eneström, 1987) are involved in the glomerular
IgG deposits found in the mesangium and in the vessel
walls of H-2s mice treated with mercuric chloride
(Hultman & Eneström, 1988). Analysis of the fine speci-
ficity of the antinucleolar auto-antibodies revealed that
at least some of them react with fibrillarin, a component
of U3 small nuclear ribonucleoprotein. Sera from human
patients with idiopathic scleroderma contain auto-anti-
bodies with exactly the same specificity (Reuter et al.,
1989).
8.2.2.2 Genetics
Rats with certain major histocompatibility haplotypes,
such as Lewis rats, are resistant whatever the dose used,
while other strains are susceptible (Table 3). The suscep-
tibility of segregants obtained by crossing Brown Norway
and Lewis rats has been extensively studied (Druet et al.,
1977, 1982; Sapin et al., 1984). It has been demonstrated
that susceptibility depends upon 3 or 4 genes. One of them
is located within the major histocompatibility complex.
Both the major histocompatibility complex-linked and
-unlinked genes are required for these auto-immune abnor-
malities to occur.
Table 3. Susceptibility of various strains of rats to
auto-immune glomerulonephritis induced by mercuric chloride
-----------------------------------------------------------
Strain RT-1 Auto-immune
haplotypea glomerulonephritis
-----------------------------------------------------------
BN n anti-GBMb; MGPc
LEW, F/344 l none
BS, AS, BD IX l none
BN-1Ld l none
LEW-1Nd n none
PVG/c, AUG c glomerular granular deposits
DA, AVN a glomerular granular deposits
AS2 f glomerular granular deposits
OKA k glomerular granular deposits
___________________________________________________________
Table 3 (contd.)
___________________________________________________________
BUF b glomerular granular deposits
BD V d glomerular granular deposits
WAG, LOU u none
Wistar Furth u MGP
-----------------------------------------------------------
a Major histocompatibility complex in the rat.
b Antiglomerular basement membrane antibodies.
c Membranous glomerulopathy.
d Congenic rats with the l RT-1 haplotype on the
BN background (BN-1L) or with the n haplotype on the
LEW background (LEW-1N).
8.2.2.3 Mechanisms of induction
Mechanisms of induction have been thoroughly studied
in rats. Mercuric chloride induces in Brown Norway rats a
polyclonal activation of B cells (Hirsch et al., 1982;
Pelletier et al., 1988a). T cells are required, since
Brown Norway rats with the nude mutation or depleted of
T cells are resistant (Pelletier et al., 1987a). It
appears that mercuric chloride induces in this rat strain
the appearance of T cells able to stimulate class II de-
terminants (also called Ia antigens), which are expressed
on the cell membrane of all B cells (Pelletier et al.,
1986). The role of such T cells is strongly supported by
the fact that T cells from Brown Norway rats injected with
mercuric chloride are able to transfer auto-immune mani-
festations to normal Brown Norway recipients and also to
Brown Norway rats depleted of T cells (Pelletier et al.,
1988b). This strongly suggests that T cells from rats
injected with mercuric chloride are able to stimulate
B cells directly. The autoreactive, anticlass II, T cells
which recognize normal B cells as well as B cells from
rats injected with mercuric chloride may have initially
been induced following a modification of class II deter-
minants by mercury, as suggested by Gleichmann et al.
(1984). It is also possible that mercuric chloride affects
CD8+ (suppressor/cytotoxic) T cells, as suggested by
Weening et al. (1981).
The fine mechanism of action at the cellular level
(see section 8.7) remains to be elucidated.
8.2.2.4 Autoregulation
The auto-immune disease observed in Brown Norway rats
is self-regulated. Abnormalities progressively disappear
after the third week. It has been shown that CD8+ (sup-
pressor/cytotoxic) T cells are responsible for this effect
(Bowman et al., 1984), together with the appearance of
auto-anti-idiotypic antibodies (Chalopin & Lockwood,
1984).
8.2.2.5 Immunosuppression
Lewis rats do not develop auto-immune disorders when
injected with mercuric chloride. In contrast, CD8+
(suppressor/cytotoxic) T cells proliferate in the spleen
and in the lymph nodes of such animals. As a consequence
they develop a non-antigen-specific immunosuppression
(Pelletier et al., 1987c). They do not respond either to
classical mitogens or to allo-antigens. More interest-
ingly, mercuric chloride is able to inhibit the develop-
ment of organ-specific auto-immune disorders such as
Heymann's nephritis (Pelletier et al., 1987b) and exper-
imental allergic encephalomyelitis (Pelletier et al.,
1988c). The mechanisms are not yet understood.
The mercury model represents a unique tool for evalu-
ating the relationship between genetic and chemically
induced immune disregulation.
8.2.2.6 Conclusions
It may be concluded that the most sensitive adverse
effect caused by mercuric mercury is the formation of
mercuric-mercury-induced auto-immune glomerulonephritis,
the first step being the production and deposition of IgG
antibodies to the glomerular basement membrane. The Brown
Norway rat is a good test species for the study of
mercuric-mercury-induced auto-immune glomerulonephritis,
although this effect has also been observed in rabbits.
Table 4 presents the available studies on auto-immune
glomerulonephritis. The lowest-observed-adverse-effect
level found in these studies was 16 mg/kg per day via the
subcutaneous route of exposure.
8.3. Reproduction, embryotoxicity, and teratogenicity
8.3.1. Males
Very little information on male reproductive effects
is available. Lee & Dixon (1975) injected male mice with
single doses of mercuric chloride (1 mg mercury/kg body
weight) and found a significant decrease in fertility,
compared with controls, in controlled mating tests. Normal
fertility was restored after about 2 months. In studies by
Chowdhury et al. (1986), gradual alterations in testicular
tissues were noted in rats treated with mercuric chloride
at intraperitoneal dosages of 0.05 mg/kg and 0.1 mg/kg
body weight over a period of 90 days. There was a decrease
in seminiferous tubular diameter, spermatogenic cell
counts, and Leydig cell nuclear diameter, compared with
controls.
Table 4. Auto-immune effects of mercuric mercury on the glomerular
basement membrane
-----------------------------------------------------------------------
Animal Route Duration Adverse Reference
effect
level
(mg/kg
per day)
-----------------------------------------------------------------------
Brown Norway rat oral 60 days 320 Bernaudin
et al. (1981)
Brown Norway rat oral 60 days 630 Andres (1984)
Brown Norway rat subcutaneous 12 weeks 16 Druet et al.
(1977)
Brown Norway rat subcutaneous 8 weeks 32a Druet et al.
(1978)
Rabbit intramuscular 1-17 weeks 633 Roman-Franco
et al. (1978)
-----------------------------------------------------------------------
a Proteinuria was observed, in addition to the auto-immune
glomerulonephritis, in these rats.
8.3.2. Females
Lamperti & Printz (1973) injected female hamsters with
daily doses of 1 mg mercuric chloride (8-11 mg mercury/kg)
throughout one 4-day estrous cycle (the LD50 being 18 mg
mercury/kg body weight). There were effects on the repro-
ductive system, including morphological changes of corpora
lutea and inhibition of follicular maturation. In further
studies (Lamperti & Printz, 1974), it was reported that
60% of female hamsters did not ovulate by day one of the
third estrous cycle after having been given a total of
3-4 mg mercuric chloride during the first estrous cycle.
Watanabe et al. (1982) injected female hamsters with mer-
curic chloride at high doses (6.4 or 12.8 mg mercury/kg
body weight) during day one of the estrous cycle and
observed an inhibition of ovulation. Lamperti & Niewenhuis
(1976) injected female hamsters with 1 mg mercuric chlor-
ide per day during one estrous cycle and found signifi-
cantly higher levels of follicle-stimulating hormone in
the pituitary gland, compared with controls.
Several investigators have reported abortions follow-
ing exposure to elemental mercury vapour or inorganic
mercury compounds several days after implantation. There
are also reports of decreased fetal weight and malfor-
mations. Gale & Ferm (1971) injected three groups of
female hamsters with a single dose of 2, 3, or 4 mg mer-
curic acetate (about 1.3-2.5 mg mercury) intravenously on
day 8 of gestation. The exposed groups showed resorption
frequencies of 12, 34, and 52%, respectively, compared to
4% in the controls. The mothers showed signs of mercury
intoxication in the form of weight loss, kidney lesions,
and diarrhoea. In later studies (Gale, 1980, 1981), a
single injection of mercuric acetate (15 mg/kg body
weight) to hamsters produced a cluster of cardiac and non-
cardiac abnormalities. The most important aspects of
embryotoxicity were resorptions, retardation, and abnormal
heart. Significant but varied interstrain differences were
observed. Holt & Webb (1986) exposed pregnant Wistar rats
intravenously to mercuric chloride at different periods of
gestation. During mid-gestation the minimum effective
teratogenic dose of mercury (0.79 mg/kg total body weight)
was high in relation to the maternal LD50 and the inci-
dence of fetal malformations, mainly brain defects, was
23% in all live fetuses. In rats of different gestational
ages, uptake of Hg2+ by the fetus at this dose level
decreased sharply between day 12 and day 13. The terato-
genic effects on the fetus and damage to the maternal
kidneys, however, were essentially the same in animals
dosed with Hg2+ either immediately before or immediately
after these gestational ages. The authors considered it
probable, therefore, that fetal defects resulted not from
any direct action of Hg2+ on the conceptuses but either
from the inhibition of the transport of essential metab-
olites from the mother or from maternal kidney dysfunc-
tion.
In a study by Rizzo & Furst (1972), three groups of
female rats were exposed to single oral doses of mercuric
oxide equivalent to 2 mg mercury/dose. Each dose of mer-
cury was given suspended in 2 ml peanut oil. The three
groups received the mercury on day 5, 12, or 19 after con-
ception. External malformations were observed in 29.7%,
6.8%, and 3.4% of cases, respectively, while the three
control groups had values between 0 and 2%. The two
observable effects that mercury had on rat fetuses were
arrest of general growth, as indicated by the number of
runts, and inhibition of eye formation. The mothers showed
no effects from the treatment. The data, if confirmed, are
of particular interest, as mercuric oxide is fairly insol-
uble.
Steffek et al. (1987) reported the effects of elemen-
tal mercury vapour exposure on pregnant Sprague-Dawley
rats. The rats were exposed to elemental mercury vapour at
concentrations of 100, 500, or 1000 µg/m3 during the
entire gestational period (chronic exposure) or during the
period of organogenesis (days 10-15, acute exposure).
Macroscopic examination of fetuses obtained from pregnant
rats exposed acutely or chronically to 100 µg/m3 re-
vealed no increased incidence of congenital malformations
or resorptions when compared to room or chamber controls.
However, acute exposure to 500 µg/m3 resulted in an
increase in the number of resorptions (5/41), and chronic
exposure at this concentration resulted in two fetuses
(out of 84 that were examined) with cranial defects. There
were also single cases of encephalomeningocoele, dome-
shaped cranial configuration, and cleft palate (Steffek,
A.J., written personal communication to the American
Dental Association). Acute exposure at 1000 µg/m3 re-
sulted in an increase in the rate of resorptions (8/71),
and chronic exposure at this dose level produced a
decrease in maternal and fetal weights, relative to the
control groups, and an increase in the number of resorp-
tions (7/28).
8.4. Mutagenicity and related end-points
Mercuric mercury affects the mitotic spindle in
plants, which may lead to an abnormal distribution of
chromosomes (Ramel, 1972; Leonard et al., 1983). It is not
a potent inducer of dominant lethal mutations in mice
(Suter, 1975). Zasukhina et al. (1983) reported the induc-
tion of single-stranded DNA breaks after exposure of
cultures of mice embryo cells to mercury chloride. The
mercury did not induce mutations but had a strong lethal
effect in a survival test of vaccinia virus. A shortening
of the chromosome length in human lymphocytes exposed in
vitro has been observed (Andersen et al., 1983). Mercuric
mercury did not induce chromosomal aberrations in human
lymphocytes and in mammalian cells in vitro (Paton &
Allison 1972; Umeda & Nishimura, 1979). Positive results
in the recombination assay with mercuric chloride have
been reported by Kanematsu et al. (1980). Effects have
been reported on DNA repair in mammalian cells (Robison et
al., 1984). There was an increase in C-mitotic figures and
segregational errors in human lymphocytes and in Indian
muntjac fibroblasts (Verschaeve et al., 1984, 1985). Based
on studies of Drosophila melanogaster, Magnusson & Ramel
(1986) found a pronounced variation in tolerance between
12 wild type strains when testing a number of metal com-
pounds, including mercuric chloride. Morimoto et al.
(1982) reported that in human whole blood cultures sele-
nite prevents the induction of sister-chromatid exchanges
by mercuric chloride.
The US Agency for Toxic Substances and Disease Regis-
try (ATSDR, 1989) has reviewed several in vitro genotox-
icity studies on mercury compounds. Mercuric chloride was
found to induce gene mutations in mouse lymphoma cells
(Oberly et al., 1982) and DNA damage in rat and mouse
fibroblasts (Zasukhina et al., 1983). It was observed to
1983; Christie et al., 1986). Using the alkaline elution
assay in intact Chinese hamster ovary cells, several
studies have shown that mercuric chloride can cause
single-strand breaks in DNA (Cantoni et al., 1982, 1984a,
1984b; Cantoni & Costa, 1983; Christie et al., 1984,
1986). Furthermore, Cantoni & Costa (1983) found that the
DNA-damaging effect of mercuric chloride is enhanced by a
concurrent inhibitory effect that mercury has on DNA
repair mechanisms.
8.5. Carcinogenicity
WHO (1976) reported no evidence that inorganic mercury
is carcinogenic. In a study by Schroeder & Mitchener
(1975), groups of mice were exposed to various metals,
including mercuric chloride. After a lifetime of exposure
to 5 µg mercury/litre in basal drinking-water, 51.2%
(21 out of 41) of the mice revealed tumours compared to
29.8% (14 out of 47) among controls. The incidence was
higher than for any of the other metals tested, but the
authors concluded that no element was significantly
tumorigenic. Mercury has not been reviewed by IARC (IARC,
1987). The US EPA (1989) has classified inorganic mercury
as a group O compound, i.e. it is not classifiable as to
human carcinogenicity.
8.6. Factors modifying toxicity
Factors such as age, sex, nutritional state, and oral
exposure giving rise to sensitization are likely to affect
the relationship between dose and effect or response. The
type of chemical exposure (whether to elemental mercury or
to mercuric mercury salts) is an important determinant for
the toxic effect and to differences in distribution.
As described in section 6.1.2, Kostial et al. (1978)
observed a high degree of absorption of inorganic mercury
in newborn rats after oral exposure to mercuric chloride.
The immature rodent kidney is, on the other hand, less
sensitive to mercury exposure than the adult kidney, as
less accumulation takes place in the kidney of the newborn
pups (Daston et al., 1983). No information is available
concerning age effects in humans.
Exposure of rats to high concentrations of mercury
vapour induced metallothionein in kidney tissue that
resulted in the binding of divalent mercury (Sapota et
al., 1974). Female rats are less susceptible than male
rats to the nephrotoxic effect of mercuric mercury (Magos
et al., 1974). This seems to be related to the metallo-
thionein content of the kidney, which is higher in females
and is increased by estradiol treatment (Nishiyama et al.,
1987). Administration of zinc to rats reduced the renal
toxic effects of mercuric mercury and induced an increase
in the glutathione (Fukino et al., 1986) and metallo-
thionein content of renal tissue (Yoshikawa & Ohta,
1982). Zinc treatment in hamsters injected with mercuric
salt reduced the embryotoxic and teratogenic effects
produced by treatment with the mercuric salt alone (Gale,
1984).
Selenium has been found to affect the distribution of
mercuric mercury in rats (Parizek & Ostadalova, 1967;
Nygaard & Hansen, 1978), mice (Eybl et al., 1969), rabbits
(Imura & Naganuma, 1978; Naganuma & Imura, 1980), and pigs
(Hansen et al., 1981). As a consequence of this redistri-
bution, a decrease in toxicity has also been observed
(Parizek & Ostadalova, 1967; Johnson & Pond, 1974). Mer-
cury forms a mercury-protein complex with selenium (Burk
et al., 1974), which can be identified in plasma and blood
cells (Chen et al., 1974; Imura & Naganuma, 1978). When
given with selenium, mercury is retained longer in blood
and, as a consequence, accumulation in the kidney is
decreased. Mercury taken up by the kidney is bound to a
protein-selenium complex, and, on administration of equi-
valent amounts of selenium, the binding to metallothionein
is diminished and may be negligible (Komsta-Szumska &
Chmielnicka, 1977; Mengel & Karlog, 1980). A consequence
of the changed binding of mercury in blood brought about
by selenium is that transport of selenium and mercury
through the placenta membranes is inhibited (Parizek et
al., 1971).
So far only selenate or selenite compounds, and not
the naturally occurring selenium compounds in food, have
been studied in detail. However, Magos et al. (1984, 1987)
compared the distribution and form of mercury and protec-
tion against the nephrotoxic effects of mercury after
exposure to different forms or compounds of selenium. It
was concluded that dietary selenium is less efficient than
selenite as an antidote against mercurial nephrotoxicity.
Studies of selenium interaction with mercuric mercury
have mainly been carried out in rodents. Selenium metab-
olism in humans is different from that in most animals,
and selenium dependency in humans is comparatively less
than that in rodents. However, observations in workers
exposed to mercury vapour indicate that there is also a
selenium-mercury interaction in humans. Selenium and
mercury concentrations with a molar ratio of 1:1 have been
found in organs such as the brain, thyroid, and pituitary
(Kosta et al., 1975; Rossi et al., 1976). In renal
biopsies from two mercury-intoxicated patients, inclusion
bodies were seen in lysosomes of renal tubules, and it was
demonstrated that these inclusion bodies contained
selenium and mercury (Aoi et al., 1985). In 28 workers
exposed to mercury vapour, the selenium excretion in urine
was high compared to non-exposed workers (Alexander et
al., 1983). However, this was not confirmed by Suzuki et
al. (1986), who studied 57 workers exposed to mercury
vapour. They found a decrease in selenium excretion as
mercury excretion in the urine increased.
As discussed in section 6.2, ethanol inhibits the
enzyme catalase, which is the main enzyme responsible for
the oxidation of mercury in blood and tissues. Ethanol
consumption thus modifies the balance between oxidation
and reduction of mercury in tissues. As a consequence,
less mercury vapour is absorbed from the lungs, more
mercury exists unoxidized in the blood, and more mercury
is transported to the brain. It is unclear whether the
observed decrease of mercury in the brain is due to the
fact that less mercury is oxidized and retained in the
brain or that more retained mercury is lost from the brain
as a result of reduction following the intake of ethanol.
The mercury content of the brain following acute exposure
to mercury in acatalasaemic mice is greater than that of
mercury-exposed control mice (Ogata et al., 1987). This is
also the case with the fetal brain after exposure of preg-
nant female acatalasaemic mice (Ogata & Meguro, 1986).
8.7. Mechanisms of toxicity - mode of action
The neurotoxic effect seen after exposure to metallic
mercury vapour is attributable to the divalent mercury ion
formed through oxidation in the brain tissue. Interference
with enzyme function by binding to sulfhydryl groups is
one possible mechanism. Kark (1979) reviewed the available
evidence regarding the inhibitory effect of mercuric ions
on different enzyme systems ( in vitro and in vivo). Mer-
cury concentrations at which enzyme inhibition appears are
consistent with concentrations at which toxic effects on
the central nervous system are observed. The in vivo con-
ditions are, however, complicated. Ligands capable of
binding mercuric ions, e.g., sulfhydryl groups and seleno-
hydrol groups, are ubiquitous and associated with
proteins. These ligands may have a protective or scaveng-
ing effect, thereby preventing interference with important
receptors. Mercuric ions penetrate cell membranes to a
very limited degree. In contrast mercury vapour penetrates
more readily due to its lipophilicity. Miyamoto (1983)
demonstrated with frog nerve-muscle preparations that mer-
curic mercury penetrates the nerve cell membrane through
sodium and calcium channels, causing an irreversible
depolarization and an increase in transmitter release.
There is a subsequent irreversible block of transmitter
release. Transport through the cell membrane via the for-
mation of carrier complexes would also be a possibility,
although this has not been demonstrated. Mercury has been
found intracellularly in nerve cells after exposure to
mercury vapour (Cassano et al., 1966) and also after pro-
longed exposure to mercuric chloride in the rat (Moller-
Madsen & Danscher, 1986).
Mercuric ions react with DNA and RNA in vitro and may
change the tertiary structure of these molecules (Eichhorn
& Clark, 1963; Gruenwedel & Davidson, 1966). Inhibition
of protein synthesis has been observed in cell systems as
well as in cell-free systems at a mercury concentration
equivalent to 2 x 10-5 mol/litre (Nakada et al., 1980).
Similar mercury concentrations have been observed to
increase the pre-synaptic release of the transmitter sub-
stance acetylcholine (Kostial & Landeka, 1975; Manalis &
Cooper, 1975). An increase in the release of dopamine in
the rat brain after treatment with mercury (10-5 mol per
litre) at a level of 2 µg/g was observed by Bondy et al.
(1979).
Mercury may interfere with membrane structure in vitro
by hydrolysing specific lipids (Ganser & Kirschner,
1985), causing membrane lesions, and by reducing lipid
synthesis in nerve cells (Cloez et al., 1987). Further-
more, an irreversible interference with the post-synaptic
membrane has been observed (Manalis & Cooper, 1975; Juang,
1976).
Knowledge regarding the mechanism of mercury neurotox-
icity, following exposure to mercury vapour, is still
fragmentary. Little data has so far emerged concerning
morphological changes in the human or primate brain. Mer-
cury has been demonstrated in rats within the motor nuclei
of the rhombencephalon and the cerebral cortex, the
highest concentrations occurring in striated areas and
within the deep nuclei of the cerebellum. A proportion-
ately high level was seen in the anterior horn motor
neurones of the spinal cord. The localization was gener-
ally interneuronal, but was also seen in the cytoplasm of
glial and ependymal cells (Moller-Madsen & Danscher,
1986). In human cases of mercury poisoning, chromatolysis
in scattered neurones was observed in the occipital lobe
of the brain and there was a loss of Purkinje cells
(Takahata et al., 1970). Davis et al. (1974) demonstrated,
in two human cases, mercury in the cytoplasm of neurones
in the nuclei olivaris and dentatus, in Purkinje cells,
in anterior horn cells of the spinal cord, and in the
neurones of the substantia nigra. In both cases a decrease
in the number of neurones of the granular cell layer in
the cerebellum and, possibly, also of Purkinje cells was
seen.
The mechanism behind the nephrotoxic effect of inor-
ganic mercury is discussed in sections 8.2 and 9.3. Here
it can be summarized that two types of renal injury have
been observed. The first is a glomerular injury caused by
an auto-immune reaction induced by mercury and resulting
in antibody formation against the glomerular tissue, depo-
sition of immune complex, glomerular nephritis, protein-
uria, and nephrotic syndrome. Alternatively, immune com-
plexes containing other mercury-induced anti-bodies may be
deposited in the glomeruli. The second is a renal tubular
damage affecting the proximal tubules and developing in
parallel with the accumulation of mercury in the renal
tubular cells. This damage results in a loss of renal
tubular enzymes, such as gamma-glutamyl transferase, and
lysosome enzymes, such as beta-galactosidase, beta-glucur-
onidase and N-acetyl-beta-glucosaminidase (Foa et al.,
1976), and in decreased reabsorption leading to an
increased secretion of endogenous trace elements such as
zinc and copper (Chmielnicka et al., 1986). An early
effect is an inhibition of protein synthesis. A swel-
ling of the endoplasmic reticulum with disaggregation
of polyribosomes is observed in electron microscopy.
Eventually, renal tubular necrosis and renal failure
develop (Wessel, 1967; Gritzka & Trump, 1968; Barnes
et al., 1980; Pezerovic et al., 1981).
The immunotoxic effect of inorganic mercury is prob-
ably the least understood effect of exposure to inorganic
mercury. Mercuric mercury has been observed as a potent
stimulator of human T lymphocytes in vitro (Schöpf et al.,
1969; Nordlind & Henze, 1984; Nordlind, 1985). Mercury is
initially bound to lymphocyte membranes, but it has also
been demonstrated that there is an uptake of mercury by
the nuclei (Nordlind, 1985) at levels likely to occur in
blood following exposure to mercury vapour. It can be
speculated that this phenomenon may be related to the
rather generalized syndromes observed in children, such as
acrodynia or "Pink disease" (Warkany, 1966; Skerfving &
Vostal, 1972) and the rather generalized and unspecific
syndromes reported to be related to dental amalgam fill-
ings, but with an unproven relation to mercury exposure
(section 9.7).
9. EFFECTS ON HUMANS
WHO (1976) dealt primarily with effects of occu-
pational exposure to mercury vapour. Apart from accidental
exposure, there was little information on exposure to in-
organic mercury among the general population. Recent data
indicate that the release of mercury vapour from amalgam
fillings may dominate exposure to inorganic mercury among
the general population (Elinder et al., 1988; WHO, 1990).
Other sources are a fish-rich diet (biotransformation of
alkyl mercury present in some species of fish resulting in
the accumulation of inorganic mercury), environmental pol-
lution in the vicinity of industrial sources, toxic waste
sites, and accidental spillage. Mercury-containing phar-
maceuticals may also be significant sources of exposure
for some populations.
The present review will focus on possible chronic
effects of long-term, low-level occupational exposure and,
for the general population, of mercury released from
amalgam. However, a brief review of acute effects will be
given. In general, the available information is presented
according to the effects on organs or organ systems. Some-
times this is not possible, as is the case for the combi-
nation of a number of unspecific symptoms that have been
associated by some with exposure from dental amalgams.
Whenever dose-response relationships are discussed, it
should be borne in mind that data on exposure levels in
the past are scarce. Quality assurance data are with few
exceptions not reported. In most studies personal samplers
were not used but only static samplers, which can be a
source of considerable error in the estimated absorbed
dose, as demonstrated by Stopford et al. (1978) and Roels
et al. (1987).
Data related to response rates are sometimes difficult
to interpret. The studies are as a rule cross-sectional,
and so selection bias, which may lead to either an under-
estimation or an overestimation of risks, can occur. The
numbers of exposed subjects and controls are often small.
For quite a few of the parameters studied, the results may
be influenced by the investigator and "interviewer bias"
is thus possible.
9.1. Acute toxicity
Workers acutely exposed (4-8 h) to calculated elemen-
tal mercury levels of 1.1 to 44 mg/m3 due to an accident
exhibited chest pains, dyspnoea, cough, haemoptysis,
impairment of pulmonary function, and evidence of inter-
stitial pneumonitis (McFarland & Reigel, 1978). Acute mer-
cury poisoning with mercurial pneumonitis was reported
among four men after they were exposed to mercury vapour
while attempting home gold ore purification using a gold-
mercury amalgam and sulfuric acid (Levin et al., 1988).
Urine mercury levels were 169 to 520 µg/litre in two
cases. Probably there was also substantial exposure to
sulfur dioxide and sulfuric acid.
Troen et al. (1951) reported 18 cases of human poison-
ing following oral ingestion of single doses of mercuric
chloride, nine of which resulted in death. The lethal
doses ranged from 29 mg/kg body weight to at least
50 mg/kg. The most common autopsy findings in these cases
were gastrointestinal lesions (ranging from mild gastritis
to severe necrotizing ulceration of the mucosa) and renal
lesions that had resulted in renal failure.
9.2. Effects on the nervous system
Most information focuses on effects on the central
nervous system following occupational exposure. The
central nervous system is the critical organ for mercury
vapour exposure (WHO, 1976). Acute exposure has given rise
to psychotic reactions characterized by delirium, halluci-
nations, and suicidal tendency. Occupational exposure has
resulted in erethism, with irritability, excitability,
excessive shyness, and insomnia as the principal features
of a broad-ranging functional disturbance. With continuing
exposure, a fine tremor develops, initially involving the
hands and later spreading to the eyelids, lips, and
tongue, causing violent muscular spasms in the most severe
cases. The tremor is reflected in the handwriting which
has a characteristic appearance. In milder cases, erethism
and tremor regress slowly over a period of years following
removal from exposure. Decreased nerve conduction velocity
in mercury-exposed workers has been demonstrated (Bidstrup
et al., 1951). Long-term, low-level exposure has been
found to be associated with less pronounced symptoms of
erethism, characterized by fatigue, irritability, loss of
memory, vivid dreams, and depression (WHO, 1976).
9.2.1. Relations between mercury in the central nervous
system and effects/response
There is very little information on brain mercury
levels in cases of mercury poisoning, and nothing that
makes it possible to estimate a no-observed-effect level
or a dose-response curve. Furthermore, the available data
are uncertain as they are not accompanied by analytical
quality control information.
Brigatti (1949) reported mercury concentrations of
6-9 mg/kg in the brain of two workers exposed to mercury
vapour and with signs of mercury poisoning 2 years before
they died. In two similar cases, Takahata et al. (1970)
reported 5-34 mg mercury/kg in different parts of the
brain. In two fatal cases of poisoning, which followed
many years of exposure to high doses of calomel as a
laxative, mercury concentrations of about 4 mg/kg in the
frontal lobe cortex were reported, and in one of the cases
a concentration of 106 mg/kg was found in the inferior
olive (Davis et al., 1974; Wands et al., 1974). The symp-
toms were dementia, erethism, colitis, and renal failure.
A dose of two tablet laxatives, each containing 120 mg of
USP-grade mercurous chloride, had been taken daily for 25
years in one case and for 6 years in the other.
9.2.2. Relations between mercury in air, urine or blood and
effects/response
9.2.2.1 Occupational exposure
WHO (1976) found no evidence of the classical symptoms
of mercurialism, erethism, intentional tremor, or gingi-
vitis below a time-weighted occupational exposure to
mercury in air of 100 µg/m3. A report on cottage
industry mercury smelting in China (Wu et al., 1989) has
confirmed the classical symptoms at exposure levels of up
to 600 µg/m3. Symptoms such as loss of appetite and
psychological disturbance have also been found to occur at
mercury levels below 100 µg/m3 (WHO, 1976).
Zeglio (1958) observed that following cessation of
exposure, symptoms and signs of neurological impairment
regress slowly in the milder cases of poisoning affecting
the nervous system. However, in the more severe cases,
neurological impairment persists and may become exacer-
bated.
One of the studies on which WHO (1976) based its con-
clusions was carried out by Smith et al. (1970). This
study covered the prevalence of several medical findings
in 567 workers in a number of chloralkali factories in the
USA in relation to mercury vapour exposure (Fig. 6). The
authors concluded that the data showed no significant
signs or symptoms in persons exposed to mercury vapour at
or below a level of 0.1 mg/m3. Subjective symptoms
appeared to increase at lower exposure levels, but the
authors questioned this finding because of the confounding
effect of alcohol. In a follow-up of part of this study,
Bunn et al. (1986) did not find significant differences in
the frequency of objective or subjective findings related
to mercury exposure, which generally was said to range
from 50 to 100 µg/m3 (time-weighted average). The
report, however, did not give sufficient information about
several methodological questions, including quality assur-
ance aspects and possible confounding variables.
Later studies, covering industries where exposure has
been high, have pointed towards the importance of urine
mercury peaks in excess of 500 µg/litre for the develop-
ment of neurological signs and symptoms (Langolf et al.,
1978, 1981). Urine mercury peaks in excess of 100 µg per
litre have been associated with impaired performance in
mechanical and visual memory tasks and psychomotor ability
tests (Forzi et al., 1976).
There is also a report (Albers et al., 1988) that, as
long as 20 to 35 years after exposure, subjects who had
experienced urine mercury peak levels above 600 µg per
litre demonstrated significantly decreased strength,
decreased coordination, increased tremor, decreased sen-
sation, and increased prevalence of Babinski and snout
reflexes when compared with control subjects. Furthermore,
subjects with reported clinical polyneuropathy had sig-
nificantly higher peak levels of mercury in urine than the
subjects without those signs. The reported signs of the
presence of an upper motor neuron lesion will require
further investigation. Many measures demonstrating sig-
nificant differences between exposed and unexposed
subjects were age dependant, but a multiple regression
analysis showed that the association between neurological
signs and mercury exposure remained after allowing for age
(Albers et al., 1988).
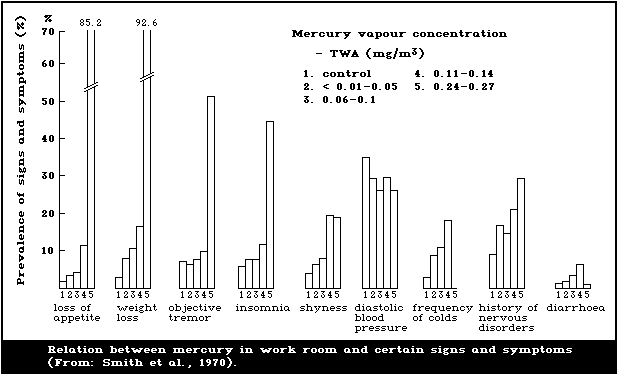 Several reports address studies where the investi-
gators have looked into possible effects at much lower
exposure levels. In a study of 142 exposed and 65 control
subjects, Miller et al. (1975) examined subclinical
effects related to exposure to inorganic mercury in the
chloralkali industry and in a factory for the manufacture
of magnetic materials. Mercury levels in urine varied from
normal to over 1000 µg/litre. Neurological examination
found evidence of eyelid fasciculation, hyperactive deep
tendon reflexes and dermatographia, but these findings did
not correlate with urinary mercury levels or length of
exposure. However, a power spectral analysis of forearm
tremor, by which it was possible to quantify tremor fre-
quency distribution and amplitude, showed a significant
increase in average tremor frequency with elevated urinary
mercury level. An effect was observed at urine concen-
trations above about 50 µg/litre.
Roels et al. (1982) studied psychomotor performance in
workers in a chloralkali plant and a factory for the manu-
facture of electric batteries. The results suggested that
preclinical psychomotor dysfunction related to the central
nervous system occurs when blood mercury levels rise to
values between 10 and 20 µg/litre and when mercury in
urine exceeds 50 µg/g creatinine. However, even in a sub-
group with urinary mercury levels below 50 µg/g creati-
nine, some parameters differed from those of a control
group.
In a further study, Roels et al. (1985) examined 131
male workers and 54 female workers exposed to metallic
mercury vapour in various Belgian factories. The controls
used were 114 non-exposed male workers and 48 female
workers. The subjects in the control and exposed groups
were closely matched with respect to age, weight, and
height. Also several other confounding factors were kept
under control. One criterion for the inclusion of exposed
workers was that they should have been uninterruptedly
exposed to mercury vapour for at least 1 year prior to the
study. A large number of questions were asked in order to
detect symptoms mainly related to nervous system disturb-
ances. A self-administered questionnaire, which was
completed the next day by the examiner, was the basis for
this information. A large number of CNS tests were used,
such as reaction time, flicker fusion, colour discrimi-
nation, short-term memory, and hand tremor. Mercury was
measured in urine and blood. Renal function was studied
using various tests, including total protein and beta-2-
microglobulin in blood and urine, and retinol-binding pro-
tein, albumin, and the lysosomal enzyme beta-galactosidase
in urine. Several symptoms mainly related to the central
nervous system (memory disturbances, depressive feelings,
fatigue, irritability) were more prevalent in the exposed
subjects than in the controls. The means and 95 percen-
tiles for urine mercury levels in non-exposed and exposed
subjects were, respectively, 52 and 147 µg/g creatinine
for males and 37 and 63 µg/g creatinine for females. The
symptoms were, however, not related to exposure par-
ameters. The authors therefore considered it possible
that the reporting of these symptoms was influenced by
knowledge of exposure to mercury vapour. There were no
significant disturbances in short-term memory, simple
reaction time, critical flicker fusion, and colour dis-
crimination ability that were related to the mercury
exposure. However, an effect on hand tremor was observed
in males. The prevalence was 5% in the non-exposed group
and 15% in the exposed group. Duration of exposure seemed
to be more important than exposure intensity, but an
increased prevalence of tremor was apparent in both the
groups with the lowest exposure (urine mercury level of
5-50 µg/g creatinine) and that with the shortest exposure
duration (1-4 years).
Among 26 workers exposed to mercury vapour in a chlor-
alkali plant or in the production of fluorescent tubes and
acetaldehyde, there was an increased incidence of reports
of hand tremor, compared with a control group (Fawer et
al., 1983). The exposure, based on personal air sampling,
was on average only 26 µg/m3. The mean urine concen-
tration was 20 µg/g creatinine (11.3 µmol/mol creati-
nine). Similar results were reported in a study by Verberk
et al. (1986), where 21 workers in a fluorescent lamp pro-
duction factory were examined. The excretion of mercury
in urine varied between 15 and 95 µg/g creatinine, the
average value being 35 µg/g creatinine. No control group
was examined, but with increasing exposure effects were
seen in several tremor parameters. The effects were
reported to occur irrespective of cigarette or alcohol
consumption or age.
Piikivi et al. (1984) reported decreased verbal intel-
ligence and memory in a group of 36 chloralkali workers
compared with a control group. Such effects were seen more
frequently in a subgroup where the urine mercury level was
above 56 µg/litre than in a subgroup where levels were
below this value. Piikivi & Hänninen (1989) made refined
analyses of the results of another study on 60 chloralkali
workers and matched controls. The exposed workers had an
average urine mercury concentration of 84.1 nmol/litre.
Neither the perceptual-motor, memory, nor learning abili-
ties of the mercury-exposed workers showed any disturb-
ances when compared to the controls. However, the exposed
workers reported statistically significantly more disturb-
ances of memory than the controls. According to multi-
variate analysis of variance, the memory disorders were
significantly associated with the form of workshift but
not with the level of exposure. In a further study
(Piikivi & Tolonen, 1989), EEG changes were found in a
group of 40 workers, compared to matched controls, after
several years of exposure to an average metallic mercury
vapour level of about 25 µg/m3 air, corresponding to a
urine mercury level of about 20 µg/litre. The EEG was
significantly slower and more attenuated in exposed
workers.
Results from the studies of Schiele et al. (1979),
Triebig et al. (1981), and Triebig & Schaller (1982) indi-
cate effects on cognitive functions and memory. However,
it is not possible to draw conclusions concerning dose-
response relationships from these studies. In a study by
Schuckmann (1979), 39 chloralkali workers with an average
urinary mercury concentration of about 100 µg/litre were
compared with a control group. There was no evidence of
changes in psychomotor activity. Smith et al. (1983)
studied effects on short-term memory in one group of 26
male chloralkali workers with an exposure corresponding
to urine mercury levels averaging 195 µg/litre and one
group of 60 male workers where the average urine mercury
level was 108 µg/litre. The severity of the effects was
found to be related to the intensity of mercury exposure.
There are some reports (Levine et al., 1982; Shapiro
et al., 1982; Singer et al., 1987; Zampollo et al., 1987)
that elemental mercury vapour causes peripheral neuropathy
at urinary levels of 50-100 µg/litre. Levine et al.
(1982) found a dose-response relationship between urine
mercury concentrations above 50 µg/litre and nerve con-
duction tests.
9.2.2.2 General population exposure
The exposure of the general population is typically
low, but occasionally may be raised to the level of occu-
pational exposure and can even result in adverse health
effects. Thus mishandling of liquid mercury, mercury
dispersed from jars, broken thermometers, fluorescent
lamps, and ingestion of mercury batteries have resulted in
severe intoxication and occasionally acute pneumonitis.
Children of mercury workers can also be exposed to mercury
vapour from contaminated work clothes. Hudson et al.
(1987) reported considerable mercury exposure among
children of mercury workers from a thermometer plant. The
median urine mercury level of 23 workers' children was
25 µg/litre compared with a value of 5 µg/litre among
39 controls. Three of the workers' children had urine mer-
cury levels above 50 µg/litre; one was above 100 µg per
litre. Mercury levels in workers' homes had a median
value of 0.24 µg/m3 compared with 0.05 µg/m3 in
non-workers' homes. No signs of mercury intoxication were
reported, based on a questionnaire to parents and on a
neurological examination that included assessment of
tremor by spectral power analysis. Urine protein was
measured only by dipstick. The reported air mercury levels
do not explain the high urinary concentrations. There must
have been exposure from sources that were not identified,
e.g., clothes. It is not known what measures were taken
to avoid contamination of sampling bottles.
Children who are exposed to mercury vapour from
interior latex paint may develop acrodynia. In 1989, a
4-year-old boy developed severe acrodynia 10 days after
the inside of his home was painted with 64 litres of
interior latex paint containing 945 mg mercury/litre. He
sequentially developed leg cramps, a generalized rash,
pruritis, sweating, tachycardia, an intermittent low-grade
fever, marked personality change, erythema and desqua-
mation of the hands, feet, and nose, weakness of the
pelvic and pectoral girdles, and lower extremity nerve
dysfunction. The level of mercury in a 24-h collection of
urine was 65 µg/litre.
9.3. Effects on the kidney
The kidney is the critical organ following the inges-
tion of inorganic bivalent mercury salts. Oliguria,
anuria, and death from renal failure resulting from acute
tubular necrosis has occurred not infrequently in the past
following the ingestion of mercuric chloride either acci-
dentally or with suicidal intent, and such cases have also
followed therapeutic administration of mercurials. At the
other extreme, organic mercurials have until recent years
been used extensively in medical practice as effective
diuretics in the management of congestive cardiac failure.
Occupational exposure to metallic mercury has for long
been associated with the development of proteinuria, both
in workers with other evidence of mercury poisoning and in
those without such evidence. An increased prevalence of
proteinuria in mercury workers, compared with a control
group, and a significant correlation between urinary mer-
cury excretion and protein excretion have been demon-
strated (Joselow & Goldwater, 1967). Less commonly, occu-
pational exposure has been followed by the nephrotic
syndrome (Kazantzis et al., 1962). Such cases have also
followed the therapeutic administration of mercurials,
although the role of mercury in some of these reported
cases, where other etiological factors may have been oper-
ative, is less clear. Two children developed the nephrotic
syndrome following a spillage of mercury in their bedroom
from a broken thermometer (Agner & Jans, 1978). The cur-
rent evidence suggests that the nephrotic syndrome follow-
ing absorption of mercury compounds results from an
immunotoxic response.
9.3.1. Immunological effects
WHO (1976) stated that effects of elemental mercury
vapour on the kidney had been reported only at doses
higher than those associated with the onset of CNS signs
and symptoms. Since then several new studies have been
carried out, and kidney effects have been seen at lower
exposure levels. Simultaneously, experimental studies on
animals have shown that inorganic mercury may induce
auto-immune glomerulonephritis in all species tested but
not in all strains, indicating a genetic predisposition.
Kazantzis (1978) and Filliastre et al. (1988) reviewed
the role of hypersensitivity and the immune response in
influencing susceptibility to metal toxicity, and gave
evidence of several case histories of clinical kidney
disease after exposure to mercury, occupationally as well
as among the general population. Of 60 adult African women
using skin-lightening creams containing inorganic mercury,
26 developed the nephrotic syndrome (Barr et al., 1972).
Kibukamusoke et al. (1974) reported one case of membranous
nephropathy, due to the use of skin-lightening cream,
where immunofluorescence showed finely granular IgG, IgM,
and C3 complement deposits. IgG and C3 complement deposits
were reported also by Lindqvist et al. (1974) in eight
cases with nephrotic syndrome. The authors also observed
similar kidney changes in two rabbits after application
(3 times per week for more than three months) of skin-
lightening cream to the skin area between the ears. The
rabbits developed proteinuria and died.
9.3.2. Relations between mercury in organs and effects/response
Only very limited information is available. In the
report by Davis et al. (1974) referred to in section
9.1.1, kidney mercury concentrations of 422 mg/kg and
25 mg/kg were measured in two fatal cases of poisoning.
However, no information was given on whether or not
adverse effects on the kidney were observed.
9.3.3. Relations between mercury in air, urine and/or blood
and effect/response
Foa et al. (1976) examined chloralkali industry
workers exposed to mercury vapour concentrations of 0.06
to 0.3 mg/m3. There were 15 cases of glomerular protein-
uria among 81 workers examined. Increased levels of
certain lysosomal enzymes were found in plasma, and this
effect was observed even in a group where the average
urine mercury level was only 35 µg/litre.
Stewart et al. (1977) examined 21 laboratory assist-
ants exposed not only to metallic mercury vapour but also
to mercuric mercury and formaldehyde and found increased
urinary excretion of protein. Air concentrations of mer-
cury were 10-50 µg/m3, and the median urine mercury
excretion rate was 53 µg/24 h (about 35 µg/litre urine).
Preventive measures were taken and in a follow-up study of
nine subjects one year later there was no evidence of
proteinuria.
Buchet et al. (1980) examined a group of 63 workers in
a chloralkali plant and found, compared with a control
group, increased plasma and urinary concentrations of the
enzyme beta-galactosidase, increased urinary excretion of
proteins with high relative molecular mass, and slightly
increased beta-2-microglobulin concentration in the plasma
without a concomitant increase in urinary concentration.
The urinary excretion of transferrin, albumin, and
beta-galactosidase was significantly correlated with the
urine concentration of mercury. The likelihood of finding
effects increased in workers with urine and/or blood mer-
cury concentrations of over 50 µg/g creatinine or
30 µg/litre blood. The data indicated an increased con-
centration of beta-galactosidase even in the group of
workers with an average urine mercury concentration of
about 20 µg/g creatinine. According to the authors the
results suggest that mercury vapour exposure may lead to a
slight glomerular dysfunction in some workers, and their
hypothesis is that the glomerular dysfunction is a result
of an auto-immune reaction.
The same research group (Roels et al., 1982) studied
the prevalence of proteinuria among 43 workers exposed to
metallic mercury vapour (median urine and blood mercury
levels of 71 µg/g creatinine and 21 µg/litre, respect-
ively) in two other factories (section 9.2.2). Increased
total proteinuria and albuminuria was slightly more preva-
lent in the mercury-exposed group than in the control
group.
No evidence of renal dysfunction (proteinuria, albumi-
nuria, retinol-binding proteins, aminoaciduria, creati-
nine, and beta-2-microglobulin in serum) was found among 62
exposed workers in a chloralkali plant and a zinc-mercury
amalgam factory, compared with a control group (Lauwerys
et al., 1983). The mean urine mercury concentration in the
exposed group was 56 µg/g creatinine. In eight exposed
workers, but in none of the controls, antibodies against
laminin, a non-collagen glycoprotein in the glomerular
basal membrane, were found. However, in a later study of
workers in another chloralkali plant and in a battery fac-
tory (Bernard et al., 1987), the prevalence of circulating
anti-laminin antibodies was not increased.
Stonard et al. (1983) examined a group of about 100
chloralkali industry workers with an average urine mercury
level of 67 µg/g creatinine. They found no evidence of
renal dysfunction and no increased excretion of proteins.
An increase in circulating immune complexes was found but
there were no anti-glomerular basement membrane antibodies
in the serum.
Roels et al. (1985) examined the renal function of 185
workers exposed to metallic mercury vapour (see also
section 9.2.2.1). Slight tubular effects were detected in
both male and female workers, in the form of an increased
urinary beta-galactosidase activity and an increased urinary
excretion of retinol-binding proteins. The effects were
dose related. Some increase in the prevalence of abnormal
values was seen even at mean urine mercury levels of about
30 µg/g creatinine. However, there was not, as was the
case for tremor, a dose-response relationship concerning
the length of the exposure period.
Rosenman et al. (1986) reported that urinary
N-acetyl-beta-glucosaminidase (NAG) enzyme levels increased
with increasing urine mercury levels over the range of
100-250 µg/litre. In a study of chloralkali industry
workers, there was a slight increase in the urine NAG con-
centration among exposed workers (average urine mercury
level of 50 µg/litre), compared with a control group
(Langworth, 1987).
Another way of studying kidney effects is to measure
the brush-border protein (BB-50) concentration in the
urine. This indicates the loss of organic tissue rather
than functional changes in the kidney cells. The urinary
BB-50 concentration was studied in 40 workers, with an
average urine mercury concentration of 46 µg/g creati-
nine and who were exposed for an average of 7 years (Mutti
et al., 1985), and 36 matched control workers. There was
no difference between exposed and non-exposed workers in
average urinary albumin or retinol-binding protein. How-
ever, when the 20 workers with urinary mercury above
50 µg/g creatinine were analysed separately, a shift of
the BB-50 distribution towards higher values was found by
a chi-square test (p = 0.07).
A study of 509 infants exposed to phenylmercury from
contaminated diapers (Gotelli et al., 1985) showed a clear
dose-response relationship between inorganic mercury in
urine and urinary excretion of gamma-glutamyl transpeptidase,
an enzyme in the brush borders of the renal tubular cells.
Since phenylmercury compounds are known to be rapidly
degraded to inorganic mercury in animals (Magos et al.,
1982), it is likely that the renal effect in the infants
was caused by inorganic mercury. Apart from the increased
enzyme excretion, the children with the highest exposure
also had increased 24-h urine volume. The enzyme excretion
increased at a urine mercury excretion of 4 µg/kg body
weight and the urine volume increased at 14 µg/kg body
weight.
9.4. Skin reactions
9.4.1. Contact dermatitis
Primary hypersensitivity to metallic mercury is con-
sidered rare (Burrows, 1986). However, Thiomersal (sodium
ethylmercurithiosalicilate) and ammoniated mercury have
been found to be common sensitizers in a survey on the
epidemiology of contact dermatitis (North American Contact
Dermatitis Group, 1973), Thiomersal being the third com-
monest sensitizer (after nickel and chromium) in the gen-
eral population. Both aryl- and alkylmercurial seed dress-
ings have also been shown to be potent skin sensitizers.
Mercury compounds give rise to a type IV cell-mediated
delayed hypersensitivity reaction (Coombs & Gell, 1975).
There have been a few cases of allergic dermatitis
among dental personnel (White & Brandt, 1976; Rudzki,
1979; Ancona et al., 1982). Patch testing of dental
students (White & Brandt, 1976) indicated that the preva-
lence rate of mercury hypersensitivity increased by class
from prefreshmen to seniors, successive values being 2.0%,
5.2%, 4.1%, 10.3%, and 10.8%. However, in a subsequent
study (Miller et al., 1987), similar results were not
found, but positive results from patch testing increased
in relation to the number of amalgam restorations in the
students. The overall percentage of positive reactions to
mercuric chloride was very high (32%), which may indicate
methodological problems. In this study, as in the study of
White & Brandt (1976), the patch testing was carried out
with 0.5 ml of a 0.1% aqueous solution of mercuric chloride.
Symptoms have occasionally been reported to relate to
amalgam fillings (Frykholm, 1957; Thomson & Russell, 1970;
Duxbury et al., 1982; SOS, 1987). In most cases the main
symptoms were facial dermatitis, sometimes with erythema-
tous and urticarial rashes. Symptoms from the mouth (oral
lichen planus) occasionally occurred. The symptoms started
a few hours after the insertion of amalgam. Nakayama et
al. (1983) reported 15 cases of generalized dermatitis
caused by mercury after exposure from broken thermometers
or dental treatment. In another very recent study
employing epicutaneous testing, positive mercury hypersen-
sitivity reactions were confined to subjects having pre-
existing amalgam restorations (Stenman & Bergman, 1989).
Finne et al. (1982) performed patch tests on 29
patients with amalgam fillings and oral lichen planus.
Contact allergy was found in 62% of the subjects, compared
with 3.2% in a control group. In four of the patients, all
the amalgam restorations were removed and replaced by gold
and composite materials. The lesions healed completely in
three of these patients after an observation period of one
year, and in the remaining case there was considerable
improvement.
When peripheral blood lymphocytes from non-atopic
subjects were cultured in the presence of pokeweed mitogen
and mercuric chloride, a significant enhancement of the
production of total IgE was observed, whereas the pro-
duction of IgM and IgA remained unaffected (Kimata et al.,
1983).
9.4.2. Pink disease and other skin manifestations
In the 1940s, "Pink disease" (acrodynia) was
reported in children below 5 years of age as a result of
the use of mercurous chloride in teething powder and oint-
ments. Affected children became irritable and generally
miserable and had difficulty in sleeping. Profuse
sweating, photophobia, and generalized rash followed. The
extremities became cold, painful, red, and swollen, and
the skin desquamated. Neither the occurrence of this
disease nor its severity was dose related. The urinary
excretion of mercury in affected children was elevated but
below the toxic level. After the withdrawal of teething
powder preparations by the main United Kingdom manufac-
turers in 1953, there was a dramatic decline in the occur-
rence of Pink disease. Calomel is not the only mercurial
that can cause Pink disease. Mercury dispersed from broken
fluorescent bulbs (Tunnessen et al., 1987), long-term
injection of gamma-globulin preserved with ethylmercurithio-
salicylate (Matheson et al., 1980), and the use of nappies
treated with phenylmercury (Gotelli et al., 1985) have
also been responsible for Pink disease. Exposure to mer-
cury vapour may be associated with the mucocutaneous lymph
node syndrome or "Kawasaki disease", which has many
similarities with Pink disease (Orlowski & Mercer, 1980).
Although the pathogenesis of Pink disease and Kawasaki
disease is unknown, there is good evidence that Kawasaki
disease is immunologically mediated, increased serum IgE
concentrations and eosinophilia having been reported
(Kusakawa & Heiner, 1976; Orlowski & Mercer, 1980; Adler
et al., 1982). In Kawasaki disease, urinary mercury
excretion is not always elevated, whereas it is in Pink
disease.
9.5. Carcinogenicity
Inorganic mercury is generally not considered to be
carcinogenic in humans (Kazantzis, 1981; Kazantzis &
Lilly, 1986). However, recent observations have shown an
excess risk of glioblastoma among Swedish dentists and
dental nurses (Ahlbom et al., 1986). Based on the Swedish
Cancer Environment Registry covering the years 1961-1979,
a standardized morbidity ratio of 2.1 was observed (with
95% confidence limits 1.3-3.4). The authors concluded that
the most probable origin is some occupational factor com-
mon to dentists and dental nurses, e.g., amalgam, chloro-
form, or radiography.
Cragle et al. (1984) published results of a mortality
study of men exposed to elemental mercury. It was a retro-
spective cohort study of 5663 workers selected from about
14 000 workers in the Y-12 plant in Oak Ridge, USA, orig-
inally working on the Manhattan Project but later in a
programme to produce large quantities of enriched lithium.
Elemental mercury was used in the lithium isotope separ-
ation process. Mercury urinalysis testing started in mid-
1953. Urine concentrations were not reported, but air
mercury levels in 50-80% of the samples taken during the
early 1950s were above 100 µg/m3. The workers studied
were divided into three groups: two exposed groups and one
non-exposed group. It is not possible to evaluate the
design as no valid exposure and selection data were pre-
sented. In all three groups, elevated SMRs (2.3, 1.2, 2.1)
for tumours of the central nervous system were found.
However, a statistically significant increase was reported
only for the group consisting of 3260 non-mercury workers.
9.6. Mutagenicity and related end-points
WHO (1976) did not report any studies showing that in-
organic mercury was genotoxic to humans. However, relevant
data have since been reported. Popescu et al. (1979) com-
pared four men exposed to elemental mercury vapour with a
control group and found an increased prevalence of chromo-
somal aberrations. Verschaeve et al. (1976) and Verschaeve
& Susanne (1979) showed an increase in aneuploidy after
exposure to very low concentrations of metallic mercury
vapour, but this could not be repeated in a later study
(Verschaeve et al., 1979). Similarly, Mabille et al.
(1984) did not find an increase of structural chromosomal
aberrations in peripheral blood lymphocytes of workers
exposed to metallic mercury vapour.
9.7. Dental amalgam and general health
During recent years there has been intense debate in
some countries (e.g., Sweden and USA) on the possible
health hazards of dental amalgams (Ziff, 1984; Penzer,
1986; SOS, 1987; Berglund, 1989). Those who claim that
mercury from amalgam may cause severe health hazards refer
to information on the release of mercury from amalgam and
subsequent uptake into the body due to inhalation and
swallowing of mercury. They also claim that a large number
of people suffer from a variety of complaints and that
their symptoms are caused by mercury. Those who deny a
causal relation between dental amalgams and health effects
point out that amalgam has been used for many years with
no proven health effects. Furthermore, the uptake of
mercury from amalgam is considerably less than has been
associated with effects after occupational exposure to
mercury (Fan, 1987).
There are many people with sometimes clearly incapaci-
tating complaints who believe that these are caused by
dental amalgam. Reports describing different types of
symptoms or other effects (Hansson, 1986; Johansson &
Lindh, 1987; Siblerud, 1988) do not allow any conclusions
to be reached concerning their cause. This was also the
opinion of a Swedish Task Group (SOS, 1987). The sympto-
matic picture is highly diverse and characterized by a
variety of different symptoms. Some studies reported that
patients improved after their amalgam fillings were re-
placed by another dental filling material. However, these
reports have not been controlled for potential placebo
effects.
Recently results from one epidemiological study have
been reported by Ahlqwist et al. (1988). The data collec-
tion was carried out during 1980-1981. The majority of
participants (85%) consisted of individuals who had
already in 1968-69 participated in a longitudinal descrip-
tive study of different diseases among women in the city
of Gothenburg, Sweden. The remainder were included to
expand the age strata and obtain a sample representative
of women of the same age in the general population.
Altogether 1024 women (aged 38-72 years) participated in
the study, which covered a dental examination with an
orthopantomogram and a medical examination including a
standardized self-administered questionnaire regarding
different symptoms or complaints. The dental and medical
examinations were made by different people and without
mentioning possible relations between amalgam and health
risks. No positive correlations were found between number
of amalgam fillings and number of symptoms or between
number of amalgam fillings and prevalence of specified
single symptoms or complaints. On the contrary, there were
several age-matched significant correlations in the
opposite direction. Some of these correlations (abdominal
pain, poor appetite) disappeared when adjustment was made
for number of teeth. Risk ratios (including 95% confidence
limits) for women with 20 fillings or more compared to
women with 0-4 fillings are given in Table 5. The authors
concluded that their results do not support a correlation
between number of surfaces with amalgam fillings and vari-
ous symptoms studied on the population level. They do not
exclude the possibility of a connection between amalgam
fillings and special symptoms and complaints on the indi-
vidual level, but, if such a connection exists, it has a
low prevalence among the general population.
Table 5. Risk ratio for a specific symptom or
complaint for 460 women with 20 or more fillings
compared to 193 women with 0-4 fillingsa
----------------------------------------------------
Risk ratio analysis
Symptom or complaint Risk 95% confidence
ratio limits for
risk ratio
----------------------------------------------------
Dizziness 0.70 0.46-1.07
Eye complaints 1.01 0.64-1.57
Hearing defects 0.66 0.41-1.07
Headache 1.22 0.83-1.80
General fatigue 0.79 0.55-1.15
Sleep disturbances 1.38 0.94-2.03
Nervous symptoms 0.80 0.52-1.25
Sweating 0.86 0.57-1.32
Breathlessness 0.65 0.41-1.03
Chest pain 0.62 0.39-0.99
Cough 0.71 0.47-1.08
Irritability 0.68 0.45-1.02
Over-exertion 0.60 0.38-0.96
Reduced mental
concentration capacity 0.74 0.47-1.18
Restlessness 0.70 0.45-1.09
Depressive symptoms 0.74 0.50-1.07
Readiness to crying 0.72 0.46-1.11
Reduced ability to relax 1.15 0.79-1.69
Abdominal pain 0.64 0.42-0.98
----------------------------------------------------
Table 5 (contd.)
----------------------------------------------------
Risk ratio analysis
Symptom or complaint Risk 95% confidence
ratio limits for
risk ratio
----------------------------------------------------
Indisposition 1.00 0.57-1.76
Diarrhoea 0.62 0.32-1.18
Constipation 0.82 0.50-1.37
Poor appetite 0.33 0.16-0.68
Loss of weight 0.27 0.10-0.70
Overweight 0.76 0.52-1.12
Sensitivity to cold 0.78 0.50-1.21
Micturation disturbances 0.66 0.30-1.43
Joint complaints 0.97 0.66-1.43
Back complaints 0.74 0.51-1.07
Leg complaints 0.74 0.51-1.09
----------------------------------------------------
a The analysis was confined to dentulous women. Age
was taken into consideration by means of the
Mantel-Haenszel procedure. Modified from Ahlqwist
et al. (1988).
Lavstedt & Sundberg (1989) investigated possible
associations between dental amalgam and a range of symp-
toms by re-examining certain dental, medical, and socio-
logical data originally collected from 1204 subjects in
1970 (i.e. prior to the present debate on dental amalgam).
Standardization was made for various confounding factors,
such as gender, social group, and smoking habits. There
was no statistically significant increase in the percent-
age of individuals with symptoms in groups with increased
numbers of amalgam fillings after controlling for con-
founding factors. The authors pointed out that the study
did not exclude a causal association on the individual
level. One strength of the study was that practically all
of the examinations were carried out by one single
investigator.
9.8. Reproduction, embryotoxicity, and teratogenicity
9.8.1. Occupational exposure
9.8.1.1 In males
McFarland & Reigel (1978) reported medical findings in
nine men exposed accidentally for less than 8 h when more
than 10 ml of mercury was instantly vapourized at a high
temperature. Air mercury concentrations were estimated to
be 44.3 mg/m3. Even if these theoretical estimates are
very uncertain, the exposure must have been extremely
high. Six of the cases developed symptoms of acute
poisoning. During a follow-up lasting several years they
also showed signs of chronic poisoning. A loss of libido,
which persisted for at least several years, was reported
in all six cases.
Lauwerys et al. (1985) compared the fertility of 103
male workers, exposed to elemental mercury vapour in a
zinc-mercury amalgam factory, a chloralkali plant, and in
plants manufacturing electrical equipment, with 101 well-
matched controls. The exposed group had an average blood
mercury level of 14.6 µg/litre (SD 11.6 µg/litre) and an
average urine mercury concentration of 52.4 µg/g creati-
nine (SD 46.7 µg/g). In the exposed group, 59 children
were born compared to 65.8 expected (as calculated from
data in the control group). However, the difference was
not statistically significant.
In a study carried out at a US Department of Energy
plant that used very large quantities of elemental mercury
from 1953 to 1963, reproductive outcomes were studied
among 247 male workers exposed to metallic mercury vapour
(Alcser et al., 1989). As controls, 255 plant workers
whose job did not require exposure to elemental mercury
were used. No associations were demonstrated between mer-
cury exposure and decreased fertility, increased rates of
major malformations among the offspring, or serious child-
hood disease. There was an association between exposure
and miscarriage, which disappeared however after control-
ling for the number of previous miscarriages before
exposure began. The 95% adjusted confidence limits were
0.97-1.18. The authors pointed out certain problems with
the study. The most serious methodological weakness in the
evaluation was the necessity for subject recall of events
that occurred 20 to 50 years previously. Another problem,
which was not considered, was possible exposure to other
toxic substances in the control group. The higher fre-
quency of miscarriages among the exposed group prior to
the exposure period could not be explained.
9.8.1.2 In females
There have been reports of increased menstrual dis-
turbances in women exposed industrially or in dentistry to
elemental mercury vapour. A study by De Rosis et al.
(1985) examined a group of 106 women exposed to low levels
of mercury (average values not exceeding 10 µg/m3) and
a control group of 241 unexposed women in another factory
with similar working conditions. The percentage of women
having normal menstrual cycles at the start of employment
was very similar in both groups of women. During their
period of employment more women in the exposed group
noticed changes in the menstrual cycle than women in the
control group. The age-standardized ratio of abnormal
cycle frequency in exposed women to that in the control
group was 1.4. The information was obtained by means of
interviews, but these were not carried out on a blind
basis. Therefore, according to the authors, the results
neither prove nor exclude the possibility that occu-
pational exposure to this concentration of mercury has a
negative effect on the female reproductive system.
There have been reports which suggest that inorganic
mercury compounds cause spontaneous abortion. Goncharuk
(1977) reported that during a 4-year period 17% of 168 ex-
posed workers in a mercury smelting plant had experienced
spontaneous abortion (average exposure, 80 µg mercury
per m3), compared with 5% among 178 controls. Toxaemia
during pregnancy was reported in 35% of the exposed and
2% of the unexposed workers. Gordon (1981) reported a
slightly elevated incidence of spontaneous abortions among
dentists. The results can not be interpreted with cer-
tainty, however, due to a non-response rate of almost 50%.
The study of De Rosis et al. (1985), referred to earlier
in this section, revealed no difference in the age-
standardized rate of spontaneous abortion between mercury-
exposed and unexposed female workers. Two other studies
of female dental staff also reported no increased abortion
rate when compared with age-standardized controls. In one
of the studies (Heidam, 1984), the upper 95% confidence
limit for odds ratio was about 2. In the other study
(Brodsky et al., 1985), a comparison was made between a
"low" exposure group and a "high" exposure group. The
low exposure group comprised dental assistants preparing
less than 40 amalgam restorations per week and the high
exposure group those preparing more than 40 fillings a
week. The assumed difference in exposure between the
groups was not validated by measurements.
Two studies, one from Poland and one from Sweden, both
dealing with spontaneous abortions and malformations, are
of particular interest. The Polish study (Sikorski et al.,
1987) revealed a high frequency of malformations among
dental staff. Of 117 pregnancies in the mercury-exposed
group, 28 pregnancies in 19 women led to reproductive
failure, such as spontaneous abortion (19 cases, 16.2%),
stillbirth (3 cases, 2.6%), congenital malformations (5
cases of spina bifida, 5.1%; one case of intra-atrial
defect). This contrasts, in non-exposed controls, with
seven cases of adverse pregnancy outcome (11.1%) in five
women out of a total of 63 pregnancies (30 women). There
were no malformations among the controls (R. Sikorski,
personal communication to the IPCS). The age distribution
of exposed and control women and the number of pregnancies
before exposure or effect were not reported, which makes
it difficult to interpret the data. For most countries the
average rate of spina bifida is 5-10 (or less) per 10 000
births (International Clearing-house for Birth Defects
Monitoring System, 1985) over a wide age span. Sikorski et
al. (1987) reported a correlation between mercury levels
in hair and reproductive failure in the exposed group. The
meaning of this correlation is difficult to interpret, as
hair is not a good indicator of exposure to metallic mer-
cury vapour, due to several factors, including the possi-
bility of external contamination (section 6.5.2). There is
reason to believe, however, that the exposure was higher
in the group with higher hair mercury levels (R. Sikorski,
personal communication to the IPCS). Only 13.6% of the
women studied used automatic amalgamators. The remaining
86.4% prepared the amalgam in an open mortar and almost
never in separate rooms.
The Swedish study was reported by Ericson & Källén
(1989), linking data from the Swedish National registers
for birth records, malformations, and occupation.
Altogether, 8157 children born to dentists (1360), dental
nurses (6340), and dental technicians (457) were compared
with the total number of births in Sweden during the
observation period (1976, 1982-1986). The analysis took
into consideration different confounding variables, such
as the age of the mother and number of children. The
study also examined the occurrence of stillbirths and
spontaneous and induced abortions treated in hospitals.
There was no tendency towards an elevated rate of malfor-
mation, abortion, or stillbirth. The study did not verify
the high risk of spina bifida described in the Polish
study (Sikorski et al., 1987). In spite of the large study
group, the upper 95% confidence limit for the risk ratio
of spina bifida was high (2.1), and the upper confidence
limit of the absolute risk was about 1 per 1000 births.
Therefore, the study is not a strong argument against an
effect. In addition, a sample of 3991 pregnancies from
the 1960s (among them there were 13 dentists, 65 dental
assistants, and 6 dental technicians) was studied but no
effects on spontaneous abortion rate or malformation was
seen. The authors point out, however, that the only mal-
formed infant observed had anencephaly and that both
parents worked as dental technicians.
Among dental nurses, there was a significant excess of
children with a birth weight of 2 to 2.5 kg. In total
there were 274 children weighing less than 2.5 kg (against
an expected number of 233), which gives a risk ratio for
low birth weight of 1.2 (with a 95% confidence interval of
1.0-1.3). No similar excess in low birth weight was seen
among dentists or dental technicians. Possible confounding
socioeconomic factors, e.g., smoking, were not studied,
but the authors suggested that these findings could be
explained by differences in socioeconomic status.
During recent years much interest has focused on
subclinical developmental changes in children exposed in
utero or in early childhood to methylmercury and lead. No
similar studies have been reported for inorganic mercury.
10. EVALUATION OF HUMAN HEALTH RISKS
Mercury exists in different forms, including elemental
mercury, inorganic mercury and organic mercury compounds.
They have some properties in common but differ in metab-
olism and toxicity. Biotransformation takes place in the
body, particularly the transformation of metallic mercury
vapour to mercuric compounds, which means that some of the
effects of inorganic mercury could also be expected after
exposure to metallic mercury vapour. There are, however,
no empirical data showing whether or not inorganic mercury
formed due to a biotransformation has similar toxicity and
metabolism to that of inorganic mercury accumulated in the
body as a result of exposure to inorganic mercury itself.
It would be prudent to consider, until more information is
available, that, with the exception of acute renal tubular
cell damage, the two forms of inorganic mercury have simi-
lar toxicity.
10.1. Exposure levels and routes
10.1.1. Mercury vapour
Human long-term exposure to mercury vapour is primar-
ily encountered in an occupational setting and in cases
where the metal has been handled inappropriately in the
home. Continual low-level exposure to mercury vapour
occurs in the mouth in the presence of dental amalgam
fillings. The amount of mercury vapour released intra-
orally depends on the number, surface area, and mechanical
loading of the amalgam restorations. Atmospheric levels of
mercury found in the workplace, e.g., chloralkali plants,
are usually below 50 µg/m3. Levels above 50 µg/m3 and
even exceeding 100 µg/m3 can be found in work environ-
ments where good industrial hygiene has not been practiced
or in home operations, in which the highest levels would
be expected. Values for air concentration (in µg/m3) are
approximately the same as those for urine mercury concen-
tration (expressed in µg/g creatinine). The use of
urinary mercury excretion makes it possible to compare
intake from the working atmosphere and release from dental
amalgam.
Occupational exposure to mercury vapour in dentistry
has been well established, reported exposure levels being
4-30 µg/m3, on average, with up to 150-170 µg/m3 in
some clinics. The values for mean intraoral mercury vapour
concentration derived from dental amalgam have been
reported to be in the range of 3 to 29 µg/m3.
Approximately 80% of inhaled mercury vapour is
absorbed from the lungs, while uptake of mercury vapour
via the skin is about 1% of uptake by inhalation. No data
are available on the possible oral mucosal absorption of
mercury vapour. Mercury vapour can cross the plancental
barrier, thus exposing the developing fetus.
10.1.2. Inorganic mercury compounds
The major source of high exposure to humans from mer-
curic compounds involves medicaments, both traditional and
alternative, and skin-lightening creams and soaps. There
is some evidence from occupational settings where chlorine
is used that a part of the mercury vapour can be trans-
formed in the atmosphere and absorbed as an aerosol of
mercuric mercury. Mercuric mercury is to a great extent
deposited in the placenta, where it causes damage that may
lead to adverse effects on the fetus.
Mercurous mercury, in the form of calomel, has for
long been used in therapeutics. The mercury content in the
brain of two daily users of calomel, ingesting 240 mg
mercurous chloride per day, was 4-106 mg/kg brain, indi-
cating that elemental mercury is formed from mercurous
chloride following ingestion.
10.2. Toxic effects
10.2.1. Mercury vapour
Over-exposure to mercury vapour gives rise to neuro-
logical effects with initially a fine high-frequency
intention tremor and neurobehavioural impairment. Periph-
eral nerve involvement has also been observed. Proteinuria
and lysosomal enzymes in the urine of exposed workers
indicate an effect on the kidney of chloralkali workers;
the presence of mercuric mercury may have contributed to
this effect. The nephrotic syndrome has been reported
among chloralkali workers. Pink disease, skin allergy, and
mucocutaneous lymph node syndrome (Kawasaki disease) in
children, have also been observed after exposure to mer-
cury vapour.
Hypersensitivity skin reactions have been described
after exposure to metallic mercury vapour from mercury
amalgam materials. No data supporting carcinogenic effects
of mercury vapour have been reported. There has been a
report of excess risk of glioblastoma observed in Swedish
dental personnel. At this stage, however, it has not been
possible to associate this excess risk with any specific
group of dental materials. The standard of published
epidemiological studies is such that it remains an open
question whether mercury vapour can adversely affect the
menstrual cycle or fetal development in the absence of the
well-known signs of mercury intoxication.
10.2.2. Inorganic mercury compounds
Information on pulmonary deposition and absorption of
mercuric mercury aerosols is lacking. However, it is
likely that significant absorption takes place directly
from the lung and probably from the gastrointestinal tract
after mucociliary clearance from the lung.
Most adverse effects of mercuric compounds in humans
have been reported after oral ingestion or skin absorp-
tion. However, only limited information is available as
far as dose-effect relationships are concerned. From
animal experiments it is possible to identify the critical
lowest effect levels that are likely to result in protein-
uria in humans after chronic exposure. Proteinuria in
humans is believed to be produced through the formation of
mercuric-mercury-induced autoimmune glomerulonephritis.
The production and deposition of IgG antibodies to glomer-
ular basement membrane can be considered the first step in
the formation of this glomerulonephritis. No data on
possible carcinogenic effects of mercuric mercury have
been reported.
10.3. Dose-response relationships
10.3.1. Mercury vapour
The risk assessment of exposure to mercury vapour is
hindered by the heterogeneity of published data, problems
with the estimation of exposure (e.g., lack of speciation
and methodological uncertainties), uncertainty concerning
the reliability of subjective symptoms, and the selection
of control groups for comparison with low exposure groups.
Nevertheless the data presented in section 9 allow a
broad characterization to be made.
a) When exposure is above 80 µg/m3, corresponding to
a urine mercury level of 100 µg/g creatinine (section
6.5.2), the probability of developing the classical
neurological signs of mercury intoxication (tremor,
erethism) and proteinuria is high.
b) Exposure in the range of 25 to 80 µg/m3, corre-
sponding to a level of 30 to 100 µg mercury/g creati-
nine, increases the incidence of certain less severe
toxic effects that do not lead to overt clinical
impairment. These subtle effects are defects in psy-
chomotor performance, objectively detectable tremor,
and evidence of impaired nerve conduction velocity,
which are present only in particularly sensitive indi-
viduals. The occurrence of several subjective symp-
toms, such as fatigue, irritability, and loss of
appetite, is also increased. In a few studies, tremor,
recorded electrophysiologically, has been observed at
low urine concentrations (down to 25-35 µg/g creati-
nine). Other studies did not show such an effect.
Although the incidence of some signs was increased in
this exposure range, most studies did not find a dose-
response relationship. Some of the exposed people
develop proteinuria (proteins of low relative molecu-
lar mass and microalbuminuria). The available studies
are generally of small size and low statistical power.
c) Appropriate epidemiological data covering exposure
levels corresponding to less than 30-50 µg mercury
per g creatinine are not available. Since a specific
no-observed-effect level cannot be established and if
larger populations are exposed to low concentrations
of mercury, it cannot be excluded that mild adverse
effects may occur in certain sensitive individuals.
Some studies have found miscarriages and abortions
after occupational exposure to mercury, but other studies
did not confirm these effects. The WHO Study Group in 1980
stated: "The exposure of women of child-bearing age to
mercury vapour should be as low as possible. The Group was
not in a position to recommend a specific value" (WHO,
1980). This statement is still prudent and will remain so
until new data become available.
10.3.2. Inorganic mercury compounds
The risk assessment of exposure to inorganic mercury
compounds is hindered by a lack of adequate human data
dealing with the relationship between dose and effects/
responses. For this reason, more human research is needed
in order to arrive at the goal of a human risk evaluation
of inorganic mercuric mercury compounds at low levels.
The intake of gram doses of mercuric chloride causes
severe to lethal renal tubular damage and necrosis of the
gastrointestinal mucous membrane. At lower dose levels,
less pronounced tubular damage occurs, reflected in amino-
aciduria, increased diuresis, and loss of renal enzymes in
the urine.
A special problem in the risk assessment of mercury is
the fact that mercury can give rise to allergic and immu-
notoxic reactions, which are partly genetically regulated.
There may well be a small fraction of the population that
is particularly sensitive, as has been observed in animal
studies. A consequence of an immunological etiology is
that it is not scientifically possible to set a level for
mercury, e.g., in blood or urine, below which mercury-
related symptoms will not occur in individual cases, since
dose-response studies for groups of immunologically sensi-
tive individuals are not yet available.
Based upon the evaluation in animals, the most sensi-
tive adverse effect for inorganic mercury risk assessment
is the formation of mercuric-mercury-induced auto-immune
glomerulonephritis, the first step being the production
and deposition of IgG antibodies to the glomerular base-
ment membrane. The Brown Norway rat is a good test species
for the study of mercuric-mercury-induced auto-immune
glomerulonephritis (although this effect has also been
observed in rabbits), and it is the best animal model for
the study of mercury-induced kidney damage at present. The
group of studies presented in Table 4 (section 8.2.2.6)
was selected for consideration of mercuric mercury risk
assessment. A no-observed-adverse-effect level (NOAEL)
could not be determined from these animal studies. The
lowest-observed-adverse-effect level (LOAEL) was found in
the subcutaneous exposure study by Druet et al. (1978). In
this study, subcutaneous doses of mercuric chloride (0.05
mg/kg body weight three times per week) were administered
for 12 weeks and resulted in antibodies being bound to the
glomerular basement membrane of the rat kidney.
Using this animal LOAEL (0.05 mg/kg), equivalent human
oral and inhalation LOAEL values for kidney effects can be
determined as follows:
Average daily subcutaneous dose:
(0.05 mg/kg) x 3 days x 0.739
= ----------------------------- = 0.0158 mg/kg per day
7 days
where: 0.05 mg/kg = dose of HgCl2 injected subcutaneously
into rats
3 days = number of days per week that doses were
administered
7 days = number of days per week
0.739 = fraction of HgCl2 that is Hg2+ ion.
Although the calculated values have been rounded, the un-
rounded values were always used in subsequent calculations.
Human oral exposure equivalent determination:
(0.0158 mg/kg per day) x 70 kg x 100%
= ------------------------------------- = 15.8 mg/day
7%
where: 0.0158 mg/kg per day = average daily subcutaneous
dose of Hg2+
70 kg = assumed body weight of an adult human
100% = assumed percentage of Hg2+ absorbed from
the subcutaneous route of exposure
7% = assumed percentage of Hg2+ absorbed from the
oral route of exposure
Human inhalation exposure equivalent determination:
a) For 24-h general population exposure:
(0.0158 mg/kg per day) x 70 kg x 100%
------------------------------------- = 0.069 mg/m3
(20 m3/day) x 80%
where: 0.0158 mg/kg per day = average daily subcutaneous
dose of Hg2+
70 kg = assumed body weight of an adult
20 m3/day = assumed volume of air inhaled during
a 24-h period
100% = assumed percentage of Hg2+ absorbed from
subcutaneous route of exposure
80% = assumed percentage of Hg0 absorbed from the
lung.
b) For 8-h work-day exposure:
(0.0158 mg/kg per day) x 70 kg x 100%
------------------------------------- = 0.139 mg/m3
(10 m3/day) x 80%
where: 0.0158 mg/kg per day = average daily subcutaneous
dose of Hg2+
70 kg = assumed body weight of an adult
10 m3 = assumed volume of air inhaled during a
work-day
100% = assumed percentage of Hg2+ absorbed from
subcutaneous route of exposure
80% = assumed percentage of Hg0 absorbed from the
lung.
These inhalation LOAEL values calculated for kidney
effects are well within the range of mercury vapour
exposures in humans where neurological and renal effects
have been observed.
11. RECOMMENDATIONS FOR FURTHER RESEARCH
Further research is required in the following areas:
1. Determination of the exposure to different chemical
forms of mercury at low levels of exposure, including
the development of microtechniques for speciation of
small quantities of mercury in biological materials
and of analytical quality assurance techniques.
2. The pharmacokinetics of mercury release from amalgam
restorations in relation to time, diet, technical and
physiological conditions, and the development of tests
for identifying specially sensitive individuals (e.g.,
local mucosa reactions, intra-oral electrochemical
measurements, immunotoxicity).
3. The use of mercury compounds in pharmaceuticals and
cosmetics.
4. The binding, biotransformation, and transport of dif-
ferent forms of mercury, both in animals and humans,
including interactions with selenium.
5. The transplacental transport of mercury and specific
distribution in fetal organs, fetotoxic effects, and
developmental effects with emphasis on neurobehav-
ioural effects.
6. Research on the neurobehavioural effects of mercury in
the occupationally exposed population (dentists, etc.).
7. The epidemiology of the role of mercury in inducing
glomerulonephritis in the general population.
8. The prevalence of immunological effects and hypersen-
sitivity in low-dose exposure to mercury with or with-
out subjective symptoms.
9. A case-control study of brain tumours, in particular
glioblastoma, and exposure to mercury.
Measures to decrease the exposure of the general popu-
lation to mercury-containing pharmaceuticals and cosmetics
should be promoted.
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
The human health risks of inorganic mercury compounds
were previously evaluated in Environmental Health Criteria
1: Mercury (WHO, 1976). More recent evaluations by the
International Programme on Chemical Safety (IPCS) have
dealt mainly with the health risks of methylmercury
exposure (WHO, 1990). A WHO review of the occupational
health risks of inorganic mercury (WHO, 1980) and an IPCS
review of the environmental aspects of mercury (WHO, 1989)
have been published. The recommended health-based occu-
pational exposure limit for metallic mercury vapour (WHO,
1980) is 25 µg mercury/m3 air (TWA, long-term exposure)
and 500 µg mercury/m3 air (peaks, short-term exposure).
The equivalent value for long-term exposure to inorganic
mercury compounds is 50 µg mercury/m3 air (TWA) (WHO,
1980). A maximum individual urine mercury concentration
of 50 µg/g creatinine has also been recommended (WHO,
1980).
Regulatory standards established by national bodies in
various countries and in the European Community are sum-
marized in the data profile of the International Register
of Potentially Toxic Chemicals (IRPTC, 1987).
REFERENCES
ABRAHAM, J.E., SVARE, C.W., & FRANK, C.W. (1984) The effect of
dental amalgam restorations on blood mercury levels. J. dent. Res.,
63(1): 71-73.
ADA (1985) Dental amalgam, Chicago, American Dental Association.
ADLER, R., BOXSTEIN, D., SCHAFF, P., & KELLY, D. (1982) Metallic
mercury vapour poisoning stimulating mucocutaneous lymph node
syndrome. J. Pediatr., 101(6): 967-968.
AGNER, E. & JANS, H. (1978) Mercury poisoning and nephrotic
syndrome in two young siblings. Lancet, II: 951.
AGOCS, M.M., ETZEL, R.A., PARRISH, R.G., PASCHAL, D.C.,
CAMPAGNA, P.R., COHEN, D.S., KILBOURNE, E.M., & HESSE, J.L. (in
press) Mercury exposure from interior latex paint. N. Engl. J. Med.
AHLBOM, A., NORELL, S., NYLANDER, M., & RODVALL, Y. (1986)
Dentists, dental nurses, and brain tumours. Br. med. J., 292: 662.
AHLQWIST, M., BENGTSSON, C., FURUNES, B., HOLLENDER, L., &
LAPIDUS, L. (1988) Number of amalgam tooth fillings in
relation to subjectively experienced symptoms in a study of
Swedish women. Community dent. oral Epidemiol., 16: 227-231.
AITIO, A. (1988) Biological monitoring. In: Clarkson, T.,
Friberg, L., Nordberg, G., & Sager, P., ed. Biological monitoring
of toxic metals, New York, London, Plenum Press, pp. 75-83.
AITIO, A., VALKONEN, S., KIVISTO, H., & YRJANHEIKKI, E. (1983)
Effect of occupational mercury exposure on plasma lysosomal
hydrolases. Int. Arch. occup. environ. Health, 53: 139-147.
ALBERS, J.W., KALLENBACH, L.R., FINE, L.J., LANGOLF, G.D.,
WOLFE, R.A., DONOFRIO, P.D., ALESSI, A.G., STOLP-SMITH, K.A.,
BROMBERG, M.B., & THE MERCURY WORKERS STUDY GROUP (1988)
Neurological abnormalities associated with remote occupational
elemental mercury exposure. Ann. Neurol., 24: 651-659.
ALCSER, K.H., BRIX, K.A., FINE, L.J., KALLENBACH, L.R., &
WOLFE, R.A. (1989) Occupational mercury exposure and male
reproductive health. Am. J. Ind. Med., 15(5): 517-529.
ALEXANDER, J., THOMASSEN, Y., & AASETH, J. (1983) Increased
urinary excretion of selenium among workers exposed to elemental
mercury vapor. J. appl. Toxicol., 3(3): 143-145.
ANCONA, A., RAMOS, M., SUAREZ, R., & MACOTELA, E. (1982)
Mercury sensitivity in a dentist. Contact dermatitis, 8: 218.
ANDERSEN, O., RONNE, M., & NORDBERG, G.F. (1983) Effects of
inorganic metal salts on chromosome length in human lymphocytes.
Hereditas, 98: 65-70.
ANDRES, P. (1984) IgA-IgG disease in the intestine of Brown-
Norway rats ingesting mercuric chloride. Clin. Immunol.
Immunopathol., 30: 488-494.
AOI, T., HIGUCHI, T., KIDOKORO, R., FUKUMURA, R., YAGI, A.,
OHGUCHI, S., SASA, A., HAYASHI, H., SAKAMOTO, N., & HANAICHI, T.
(1985) An association of mercury with selenium in inorganic mercury
intoxication. Hum. Toxicol., 4: 637-642.
ARONSSON, M.A., LIND, B., NYLANDER, M., & NORDBERG, M. (1989)
Dental amalgam and mercury. Biol. Metals, 2: 25-30.
ARVIDSON, B. (1987) Research note. Retrograde axonal transport of
mercury. Exper. Neurol., 98: 198-203.
ASHE, W., LARGENT, E., DUTRA, F., HUBBARD, D., & BLACKSTONE, M.
(1953) Behaviour of mercury in the animal organism following
inhalation. Ind. med. occup. Med., 22: 19-43.
ATSDR (1989) Toxicological profile for mercury. Atlanta, Agency
for Toxic Substances and Disease Registry (US Public Health
Service).
ATEN, J., BOSMAN, C.B., ROZING, J., STIJNEN, T., HOEDEMAEKER,
Ph.J., & WEENING, J.J. (1988) Mercuric chloride-induced
autoimmunity in the Brown Norway rat: cellular kinetics and major
histocompatibility complex antigen expression. Am. J. Pathol.,
133(1): 127-138.
BARIETY, J., DRUET, P., LALIBERTE, F., & SAPIN, C.
(1971) Glomerulonephritis with gamma- and beta1C-globulin
deposits induced in rats by mercuric chloride. Am. J. Pathol.,
65:293-302.
BARNES, J.L., MCDOWELL, E.M., MCNEIL, J.S., FLAMENBAUM, W., &
TRUMP, B.F. (1980) Studies on the pathophysiology of acute
renal failure. IV. Protective effect of dithiothreitol
following administration of mercuric chloride in the rat. Virchows
Arch. cell Pathol., 32: 201-232.
BARR, R.D., REES, P.H., CORDY, P.E., KUNGU, A., WOODGER, B.A., &
CAMERON, H.M. (1972) Nephrotic syndrome in adult Africans in
Nairobi. Br. med. J., 2: 131-134.
BARR, R.D., WOODGER, B.A., & REES, P.H. (1973) Levels of mercury
in urine correlated with the use of skin lightening creams. Am.
J. clin. Pathol., 59: 36-40.
BATTISTONE, G.C., HEFFERREN, J.J., MILLER, R.A., & CUTRIGHT, D.E.
(1976) Mercury: its relation to the dentist's health and
dental practice characteristics. J. Am. Dent. Assoc., 92: 1182-1188.
BAUER, J.G. & FIRST, H.A. (1982) The toxicity of mercury in
dental amalgam. California dent. J., 47-61.
BELLON, B., CAPRON, M., DRUET, E., VERROUST, P., VIAL, M.C.,
SAPIN, C., GIRARD, J.F., FOIDART, J.M., MAHIEU, P., & DRUET, P.
(1982) Mercuric chloride induced autoimmune disease in Brown-
Norway rats: Sequential search for anti-basement membrane
antibodies and circulating immune complexes. Eur. J. clin.
Invest., 12: 127-133.
BERGLUND, A., POHL, L., OLSSON, S., & BERGMAN, M. (1988)
Determination of the rate of release of intra-oral mercury vapor
from amalgam. J. dent. Res., 67(9): 1235-1242.
BERGLUND, F. (1989) [Amalgam poisoning, a medical
reality.] Läkartidningen, 86(1-2): 41-42 ( in Swedish).
BERLIN, M. & JOHANSSON, L.G. (1964) Mercury in mouse brain
after inhalation of mercury vapour and after intravenous
injection of mercury salts. Nature, London, 204: 85-86.
BERLIN, M., FAZACKERLEY, J., & NORDBERG, G. (1969) The uptake of
mercury in the brains of mammals exposed to mercury vapor and
mercuric salts. Arch. environ. Health, 18: 719-729.
BERLIN, M., CARLSON, J., & NORSETH, T. (1975) Dose-dependence of
methyl-mercury metabolism. A study of distribution:
biotransformation and excretion in the Squirrel Monkey. Arch.
environ. Health, 30: 307-313.
BERNARD, A.M., ROELS, H.R., FOIDART, J.M., & LAUWERYS, R.L. (1987)
Search for anti-laminin antibodies in the serum of workers
exposed to cadmium, mercury vapour or lead. Int. Arch. occup.
environ. Health, 59: 303-309.
BERNARD, S.R. & PURDUE, P. (1984) Metabolic models for
methyl and inorganic mercury. Health Phys., 46(3): 695-699.
BERNAUDIN, J.F., DRUET, E., DRUET, P., & MASSE, R. (1981)
Inhalation or ingestion of organic or inorganic mercurials
produces auto-immune disease in rats. Clin. Immunol. Immunopathol.,
20: 129-135.
BIDSTRUP, P.L., BONNEL, J.A., HARVEY, D.G., & LOCKET, S. (1951)
Chronic mercury poisoning in men repairing direct-current meters.
Lancet, 2: 856-861.
BONDY, S.C., ANDERSON, C.L., HARRINGTON, M.E., & PRASAD, K.N.
(1979) The effects of organic and inorganic lead and mercury on
neurotransmitter high-affinity transport and release mechanisms.
Environ. Res., 19: 102-111.
BOWMAN, C., MASON, D.W., PUSEY, C.D., & LOCKWOOD, C.M.
(1984) Autoregulation of autoantibody synthesis in mercuric
chloride nephritis in the Brown-Norway rat. I. A role for T
suppressor cells. Eur. J. Immunol., 14: 464-470.
BRADY, J.A., GEMMITI-NUNN, D., POLAN, A.K., MITCHELL, D., WEIL,
R., & VIANNA, N.J. (1980) The relationship of dental practice
characteristics to blood mercury levels. NY State dent. J., 46: 420-
424.
BRIGATTI, L. (1949) [Mercury levels in human organs with and
without mercury exposure.] Med. Lav., 40(10): 233-239 (in Italian).
BRODSKY, J.B., COHEN, E.N., WHITCHER, C., BROWN, B.W., Jr, &
WU, M.L. (1985) Occupational exposure to mercury in dentistry
and pregnancy outcome. J. Am. Dent. Assoc., 111: 779-780.
BRUNE, D. (1981) Corrosion of amalgams. Scand. J. dent. Res., 89:
506-514.
BRUNE, D. & EVJE, D.M. (1985) Man's mercury loading from a dental
amalgam. Sci. total Environ., 44: 51-63.
BUCHET, J.P., ROELS, H., BERNARD, A., Jr, & LAUWERYS, R. (1980)
Assessment of renal function of workers exposed to inorganic lead,
cadmium or mercury vapor. J. occup. Med., 22(11): 741-750.
BUCHWALD, H. (1972) Exposure of dental workers to mercury. Am.
Ind. Hyg. Assoc. J., 33: 492-502.
BUNN, W.B., MCGILL, C.M., BARBER, T.E., CROMER, J.W., Jr, &
GOLDWATER, L.J. (1986) Mercury exposure in chloralkali plants.
Am. Ind. Hyg. Assoc. J., 47(5): 249-254.
BURK, R.F., FOSTER, K.A., GREENFIELD, P.M., & KIKER, K.W. (1974)
Binding of simultaneously administered inorganic selenium and
mercury to a rat plasma protein (37894). Proc. Soc. Exp. Biol.
Med., 145: 782-785.
BURROWS, D. (1986) Hypersensitivity to mercury, nickel and
chromium in relation to dental materials. Int. dent. J., 36: 30-34.
CANTONI, O. & COSTA, M. (1983) Correlations of DNA strand breaks
and their repair with cell survival following acute exposure to
mercury (II) and X-rays. Mol. Pharmacol. 24: 84-89.
CANTONI, O., EVANS, R.M., & COSTA, M. (1982) Similarity in the
acute cytotoxic response of mammalian cells to mercury (II) and
X-rays: DNA damage and glutathione depletion. Biochem. Bioophys.
Res. Commun., 108(2): 614-619.
CANTONI, O., CHRISTIE, N.T., ROBISON, S.H., & COSTA, M.
(1984a) Characterization of DNA lesions produced by HgCl2 in cell
culture systems. Chem.-biol. Interact., 49: 209-224.
CANTONI, O., CHRISTIE, N.T., SWANN, A., DRATH, D.B., COSTA, M.
(1984b) Mechanism of HgCl2 cytotoxicity in cultured
mammalian cells. Mol. Pharmacol., 26: 360-368.
CASSANO, G.B., AMADUCCI, L., & VIOLA, P.L. (1966) Distribution of
mercury (H203) in the brain of chronically intoxicated mice
(autoradiographic study). Riv. Pat. nerv. ment., 87: 214-225.
CENTERS FOR DISEASE CONTROL (1990) Mercury exposure from
interior latex paint. CDC Mortal. Morb. Wkly Rep., 39(8): 125-126.
CHALOPING, J.M. & LOCKWOOD, C.M. (1984) Autoregulation of
autoantibody synthesis in mercuric chloride nephritis in the
Brown Norway rat. II. Presence of antigen-augmentable plaque-
forming cells in the spleen is associated with humoral
factors behaving as auto-anti-idiotypic antibodies. Eur. J.
Immunol., 14: 470-475.
CHANG, S.B., SIEW, C., & GRUNINGER, S.E. (1987) Examination of
blood levels of mercurials in practicing dentists using cold-
vapor atomic absorption spectrometry. J. anal. Toxicol., 11: 149-153.
CHEN, R.W., WHANGER, P.D., & FANG, S.C. (1974) Diversion of
mercury binding in rat tissues by selenium: A possible mechanism
of protection. Pharmacol. Res. Commun., 6(6): 571-579.
CHERIAN, M.G., HURSH, J.B., CLARKSON, T.W., & ALLEN, J. (1978)
Radioactive mercury distribution in biological fluids and
excretion in human subjects after inhalation of mercury vapor.
Arch. environ. Health, 33: 109-114.
CHMIELNICKA, J., BRZEZNICKA, E., & SNIADY, A. (1986) Kidney
concentrations and urinary excretion of mercury, zinc and
copper following the administration of mercuric chloride and
sodium selenite to rats. Arch. Toxicol., 59: 16-20.
CHOWDHURY, A.R., VACHHRAJANI, K.D., MAKHIJA, S., & KASHYAP, S.K.
(1986) Histomorphometric and biochemical changes in the
testicular tissues of rats treated with mercuric chloride.
Biomed. Biochim. Acta, 45(7): 949-956.
CHRISTIE, N.T., CANTONI, O., EVANS, R.M., MEYN, R.E., & COSTA, M.
(1984) Use of mammalian DNA repair-deficient mutants to assess
the effects of toxic metal compounds on DNA. Biochem. Pharmacol.,
33(10): 1661-1670.
CHRISTIE, N.T., CANTONI, O., SUGIYAMA, M., CATTEBENI, F., &
COSTA, M. (1986) Differences in the effects of Hg(II) on DNA
repair induced in Chinese hamster ovary cells by ultraviolet or
X-rays. Mol. Pharmacol., 29: 173-178.
CLARKSON, T.W. & KILPPER, R.W. (1978) The metabolism of inhaled
vapor in animals and man. In: Clarkson, T.W., ed. Heavy metals
as environmental hazards to man, Rochester, New York,
Environmental Health Sciences Center Program Project, pp. 449A,
449B.
CLARKSON, T.W., MAGOS, L., & GREENWOOD, M.R. (1972) The
transport of elemental mercury into fetal tissues. Biol. Neonate,
21: 239-244.
CLARKSON, T.W., FRIBERG, L., HURSH, J.B., & NYLANDER, M.
(l988a) The prediction of intake of mercury vapor from amalgams.
In: Clarkson, T.W., Friberg, L., Nordberg, G.F., & Sager, P.,
ed. Biological monitoring of metals, New York, London, Plenum
Press, pp. 247-264.
CLARKSON, T.W., HURSH, J.B., SAGER, P.R., & SYVERSEN, T.L.M.
(1988b) Mercury. In: Clarkson, T.W., Friberg, L., Nordberg, G.F., &
Sager, P., ed. Biological monitoring of metals, New York, London,
Plenum Press, pp. 199-246.
CLARKSON, T.W., FRIBERG. L., NORDBERG., G.F., & SAGER, P. ed.
(1988c) Biological monitoring of metals, New York, London, Plenum
Press, 686 pp.
CLOEZ, I., DUMONT, O., PICIOTTI, M., & BOURRE, J.M. (1987)
Alterations of lipid synthesis in the normal and dysmyelinating
trembler mouse sciatic nerve by heavy metals (Hg, Pb, Mn, Cu, Ni).
Toxicology, 46: 65-71.
COOLEY, R.L. & BARKMEIER, W.W. (1978) Mercury vapor emitted
during ultraspeed cutting of amalgam. J. Indiana Dent. State
Assoc., 57(2): 28-31.
COOMBS, R.R.A. & GELL, P.G.H. (1975) Classification of allergic
reactions responsible for clinical hypersensitivity and disease.
In: Gell, P.G.H., Coombs, R.R.A., & Lachmann, P.J. ed. Clinical
aspects of immunology, 3rd ed., Oxford, London, Blackwell
Scientific Publications, pp. 761-781.
COYLE, P. & HARTELY, T. (1981) Automated determination of mercury
in urine and blood by the Magos reagent and cold vapor
atomic absorption spectrometry. Anal. Chem., 53: 354-356.
CRAGLE, D.L., HOLLIS, D.R., QUALTERS, J.R., TANKERSLEY, W.G., &
FRY, S.A. (1984) A mortality study of men exposed to elemental
mercury. J. occup. Med., 26(11): 817-821.
CRC (1972) Handbook of chemistry and physics, 32nd ed.,
Cleveland, Ohio, CRC Press.
CROSS, J.D., DALE, I.M., GOOLVARD, L., LENIHAN, J.M.A., & SMITH, H.
(1978) Methyl mercury in blood of dentists. Lancet, II: 312-313.
DASTON, G.P., KAVLOCK, R.J., ROGERS, E.H., & CARVER, B. (1983)
Toxicity of mercuric chloride to the developing rat kidney. I.
Postnatal ontogeny of renal sensitivity. Toxicol. appl. Pharmacol.,
71: 24-41.
DAVIS, L.E., WANDS, J.R., WEISS, S.A., PRICE, D.L., & GIRLING, E.F.
(1974) Central nervous system intoxication from mercurous
chloride laxatives. Quantitative, histochemical and
ultrastructural studies. Arch. Neurol., 30: 428-431.
DEAN, R.B. & SUESS, M.J. (1985) The risk to health of chemicals in
sewage sludge applied to land. Waste Manage. and Res., 3: 251-278.
DE ROSIS, F., ANASTASIO, S.P., SELVAGGI, L., BELTRAME, A., &
MORIANI, G. (1985) Female reproductive health in two lamp
factories: Effects of exposure to inorganic mercury vapour and
stress factors. Br. J. ind. Med., 42: 488-494.
DIAMOND, G.L. (1988) Biological monitoring of urine for exposure
to toxic metals. In: Clarkson, T.W., Friberg, L., Nordberg, G.F.,
& Sager, P., ed. Biological monitoring of metals, New York,
London, Plenum Press, pp. 515-529.
DRUET, E., SAPIN, C., GUNTHER, E., FEINGOLD, N., & DRUET, P.
(1977) Mercuric chloride-induced anti-glomerular basement membrane
antibodies in the rat. Eur. J. Immunol., 7: 348-351.
DRUET, E., SAPIN, C., FOURNIE, G., MANDET, C., GUNTHER, E., &
DRUET, P. (1982) Genetic control of susceptibility to
mercury-induced immune nephritis in various strains of rat.
Clin. Immunol. Immunopathol., 25: 203-212.
DRUET, P., DRUET, E., POTDEVIN, F., & SAPIN, C. (1978)
Immune type glomerulonephritis induced by HgCl2 in the Brown
Norway rat. Ann. Immunol., 129C: 777-792.
DRUET, P., TEYCHENNE, P., MANDET, C., BASCOU, C., & DRUET, E.
(1981) Immune-type glomerulonephritis induced in the Brown-
Norway rat with mercury-containing pharmaceutical products.
Nephron, 28: 145-148.
DUMAREY, R., DAMS, R., & HOSTE, J. (1985) Comparison of the
collection and desorption efficiency of activated charcoal,
silver, and gold for the determination of vapor-phase
atmospheric mercury. Anal. Chem., 57(13): 2638-2643.
DUNN, J.D., CLARKSON, T.W., & MAGOS, L. (1978) Ethanol-
increased exhalation of mercury in mice. Br. J. ind. Med., 35: 241-244.
DUNN, J.D., CLARKSON, T.W., & MAGOS, L. (1981a) Ethanol reveals
novel mercury detoxification step in tissues. Science, 213: 1123-1125.
DUNN, J.D., CLARKSON, T.W., & MAGOS, L. (1981b) Interaction of
ethanol and inorganic mercury: Generation of mercury vapor in vivo.
J. Pharmacol. exp. Ther., 216(1): 19-23.
DUXBURY, A.J., EAD, R.D., MCMURROUGH, S., & WATTS, D.C. (1982)
Allergy to mercury in dental amalgam. Br. dent. J., 152: 47-48.
EDIMED (1989) [The pharmaceutical informer. Italian directory of
drugs and manufacturers], 50th ed., Milano, Organizzazione
Editoriale Medico Farmaceutica (in Italian).
EGGLESTON, D.W. & NYLANDER, M. (1987) Correlation of dental
amalgam with mercury in brain tissue. J. prosthet. Dent., 58(6):
704-707.
EHRENBERG, R.L., BLAIR SMITH, A., MCMANUS, K.P., HANNON, W.H.,
BRIGHTWELL, W.S., LOWRY, L.K., ANGER, W.K., VOGT, R.L., BRONDUM,
J., & HUDSON, P.J. (1986) Health hazard evaluation report,
Cincinnati, Ohio, National Institute for Occupational Safety and
Health, 65 pp. (HETA, 83-465-1674).
EICHHORN, G.L. & CLARK, P. (1963) The reaction of mercury
(II) with nucleosides. J. Am. Chem. Soc., 85: 4020-4024.
ELINDER, G.-G., GERHARDSSON, L., & OBERDOERSTER, G. (1988)
Biological monitoring of metals. In: Clarkson, T.W., Friberg, L.,
Nordberg, G.F., & Sager, P., ed. Biological monitoring of metals,
New York, London, Plenum Press, pp. 1-71.
ENWONWU, C.O. (1987) Potential health hazard of use of
mercury in dentistry: Critical review of the literature. Environ.
Res., 42: 251-274.
ERICSON, A. & KALLEN, B. (1988) Pregnancy outcome in women
working as dentists, dental assistants or dental technicians.
Arch. occup. environ. Health, 61(5), 329-333.
ETO, K., TOKUNAGA, H., ITAI, Y., TAKIZAWA, Y., & SUDA, H.
(1988) [An autopsy of fetal Minamata disease which takes a
long course.] Report presented at a Conference on the
Comprehensive Study on Minamata Disease, 27 February, 1988,
sponsored by the Environmental Agency, Japan (in Japanese).
EYBL, V., SYKORA, J., & MERTL, F. (1969) [Influence of sodium
selenite, sodium tellurite and sodium sulphite on the retention
and distribution of mercury in mice.] Arch. Toxikol., 25:
296-305 (in German with English summary).
FAN, P.L. (1987) Safety of amalgam. Cal. dent. Assoc. J.,
September: 34-36.
FARANT, J.P., BRISSETTE, D., MONCION, L., BIGRAS, L., &
CHARTRAND, A. (1981) Improved cold-vapor atomic absorption
technique for the microdetermination of total and inorganic
mercury in biological samples. J. anal. Toxicol., 5: 47-51.
FAWER, R.F., DE RIBAUPIERRE, Y., GUILLEMIN, M.P., BERODE, M., &
LOB, M. (1983) Measurement of hand tremor induced by
industrial exposure to metallic mercury. Br. J. ind. Med., 40: 204-
208.
FILLIASTRE, J.-P., DRUET, P., & MERY, J.-P. (1988)
Proteinuric nephropathies associated with drugs and substances of
abuse. In: Cameron, J.S. & Glassok, R.J. ed. The nephrotic
syndrome, New York, Basel, Marcel Dekker Inc., pp. 697-742.
FINNE, K., GORANSSON, K., & WINCKLER, L. (1982) Oral lichen
planus and contact allergy to mercury. Int. J. oral Surg., 11: 236-239.
FOA, V., CAIMI, L., AMANTE, L., ANTONINI, C., GATTINONI, A.,
TETTAMANTI, G., LOMBARDO, A., & GIULIANI, A. (1976) Patterns of
some lysosomal enzymes in the plasma and of proteins in urine
of workers exposed to inorganic mercury. Int. Arch. occup. environ.
Health, 37: 115-124.
FOOTE, R.S. (1972) Mercury vapor concentrations inside buildings.
Science, 177: 513-514.
FORZI, M., CASSITTO, M.G., BULGHERONI, C., & FOA, V. (1976)
Psychological measures in workers occupationally exposed to
mercury vapours. A validation study. In: Adverse effects of
environmental chemicals and psychotoxic drugs, Amsterdam,
Oxford, New York, Elsevier Science Publishers, Vol. 2, pp.
165-172.
FRIBERG, L. (1956) Studies on the accumulation, metabolism and
excretion of inorganic mercury (Hg203) after prolonged
subcutaneous administration to rats. Acta pharmacol. toxicol., 12:
411-427.
FRIBERG, L. (1988) Quality assurance. In: Clarkson, T.W.,
Friberg, L., Nordberg, G.F., & Sager, P., ed. Biological
monitoring of metals, New York, London, Plenum Press,
pp. 103-126.
FRIBERG, L. & NYLANDER, M. (1987) [The release and uptake of
metallic mercury vapour from amalgam.] In: Mercury/amalgam -
health risks. Report by an expert group. Stockholm, National
Board of Health and Welfare, Report series Socialstyrelsen
Redovisar 1987, 10, pp. 65-79 (in Swedish with English summary).
FRIBERG, L. & VOSTAL, J. ed. (1972) [The release and uptake of
metallic mercury vapour from amalgam.] In: [Mercury/amalgam -
Health risks], Stockholm, National Board of Health and Welfare,
pp. 65-79 (Report series Socialstyrelsen Redovisar 10) (in Swedish
with English summary).
FRIBERG, L., SKOG, E., & WAHLBERG, J.E. (1961) Resorption of
mercuric chloride and methyl mercury dicyandiamide in guinea-
pigs through normal skin and through skin pre-treated with
acetone, alkylarylsulphonate and soap. Acta dermatovenereol., 41:
40-52.
FRIBERG, L., KULLMAN, L., LIND, B., & NYLANDER, M. (1986) [Mercury
in the central nervous system in relation to amalgam fillings.]
Laekartidningen, 83: 519-522 (in Swedish).
FRYKHOLM, K.O. (1957) Mercury from dental amalgam. Its toxic and
allergic effects and some comments on occupational hygiene. Acta
odont. Scand., 22: 1-108.
FUKATSU, A., BRENTJENS, J.R., KILLEN, P.D., KLEINMAN, H.K.,
MARTIN, G.R., & ANDRES, G.A. (1987) Studies on the formation
of glomerular immune deposits in Brown Norway rats injected
with mercuric chloride. Clin. Immunol. Immunopathol., 45: 35-47.
FUKINO, H., HIRAI, M., MEI HSUEH, Y., MORIYASU, S., & YAMANE, Y.
(1986) Mechanism of protection by zinc against mercuric chloride
toxicity in rats: Effects of zinc and mercury on glutathionine
metabolism. J. Toxicol. environ. Health, 19: 75-89.
FUKUDA, K. (1971) Metallic mercury induced tremor in rabbits and
mercury content of the central nervous system. Br. J. ind. Med., 28:
308-311.
GALE, T.F. (1980) Cardiac and non-cardiac malformations
produced by mercury in hamsters. Bull. environ. Contam. Toxicol.,
25: 726-732.
GALE, T.F. (1981) The embryotoxic response produced by inorganic
mercury in different strains of hamsters. Environ. Res., 24: 152-161.
GALE, T.F. (1984) The amelioration of mercury-induced embryotoxic
effects by simultaneous treatment with zinc. Environ. Res., 35: 405-
411.
GALE, T.F. & FERM, V.H. (1971) Embryopathic effects of mercuric
salts. Life Sci., 10: 1341-1347.
GANSER, A.L. & KIRSCHNER, D.A. (1985) The interaction of
mercurials with myelin: Comparison of in vitro and in vivo
effects. Neurotoxicology, 6(1): 63-78.
GAY, D.D., COX, R.D., & REINHARDT, J.W. (1979) Chewing releases
mercury from fillings. Lancet, 8123: 985-986.
GELLER, S.A. (1976) Subacute and chronic tissue reaction to
metallic mercury: Two cases and a review of the literature. Mt
Sinai J. Med., 43: 534-541.
GLEICHMANN, E., PALS, S.T., ROLINK, A.G., RADASZKIEWICZ, T., &
GLEICHMANN, H. (1984) Graft-versus-host reactions: clues to the
etiopathology of a spectrum of immunological diseases. Immunol.
Today, 5(11): 324-332.
GONCHARUK, G.A. (1977) [Problems relating to occupational hygiene
in women in production of mercury.] Gig. Tr. prof. Zabol., 5: 17-20 (in
Russian).
GORDON, H. (1981) Pregnancy in female dentists - a mercury
hazard? In: Proceedings of the International Conference on
Mercury Hazards in Dental Practice, Glasgow, 2-4 September, (Paper
31).
GOTELLI, C. (1989) Methylmercury in a gold mining region. Proc.
Nat. Inst. Environm. Health Sci. (in print).
GOTELLI, C.A., ASTOLFI, E., COX, C., CERNICHIARI, E., &
CLARKSON, T.W. (1985) Early biochemical effects of an organic
mercury fungicide on infants: "dose makes the poison". Science,
227: 638-640.
GRITZKA, T.L. & TRUMP, B.F. (1968) Renal tubular lesions
caused by mercuric chloride. Am. J. Pathol., 52(6): 1225-1277.
GRONKA, P.A., BOBKOSKIE, R.L., TOMCHICK, G.J., BACH, F., &
RAKOW, A.B. (1970) Mercury vapor exposures in dental offices. J.
Am. Dent. Assoc., 81: 923-925.
GRUENWEDEL, D.W. & DAVIDSON, N. (1966) Complexing and denaturation
of DNA by methylmercuric hydroxide. J. mol. Biol., 21: 129-144.
HAHN, L.J., KLOIBER, R., VIMY, M.J., TAKAHASHI, Y., &
LORSHEIDER, F.L. (1989) Dental "silver" tooth fillings: a source
of mercury exposure revealed by whole-body image scan and
tissue analysis. FASEB J., 3(14): 2641-2646.
HALBACH, S. & CLARKSON, T.W. (1978) Enzymatic oxidation of mercury
vapors by erythrocytes. Biochem. Biophys. Acta, 523(2): 522-531.
HANDKE, J.L. & PRYOR, P. (1981) Health hazard evaluation
report, Cincinnati, Ohio, National Institute for Occupational
Safety and Health, 26 pp. (HHE 78-034-930).
HANSEN, J.C., KRISTENSEN, P., & AL-MASRI, S.N. (1981)
Mercury/selenium interaction. A comparative study on pigs. Nord.
vet. Med., 33: 57-64.
HANSEN, J.C., RESKE-NIELSEN, E., THORLACIUS-USSING, O., RUNGBY,
J., & DANSHER, G. (1989) Distribution of dietary mercury in a
dog. Quantitation and localization of total mercury in organs
and central nervous system. Sci. tot. Environ., 78: 23-43.
HANSSON, M. (1986) [Changes in health after removal of toxic
dental filling materials.] TF-BLADET, 1: 3-30 (in Swedish).
HARRIS, D., NICOLS, J.J., STARK, R., & HILL, K. (1978) The dental
working environment and the risk of mercury exposure. J. Am.
Dent. Assoc., 97: 811-815.
HATCH, W.R. & OTT, W.L. (1968) Determination of sub-microgram
quantities of mercury by atomic absorption spectrophotometry.
Anal. Chem., 40: 2085-2087.
HEIDAM, L.Z. (1984) Spontaneous abortions among dental assistants,
factory workers, painters, and gardening workers: A follow up
study. J. Epidemiol. community Health, 38: 149-155.
HEINTZE, U., EDWARDSSON, S., DERAND, T., & BIRKHED, D. (1983)
Methylation of mercury from dental amalgam and mercuric chloride
by oral streptococci in vitro . Scand. J. dent. Res., 9l: 150-152.
HIRSCH, F., COUDERC, J., SAPIN, C., FOURNIE, G., & DRUET, P.
(1982) Polyclonal effect of HgCl2 in the rat, its possible
role in an experimental autoimmune disease. Eur. J. Immunol., 12:
620-625.
HIRSHMAN, S.Z., FEINGOLD, M., & BOYLEN, G. (1963) Mercury in
house paint as a cause of acrodynia: effect of therapy
with N-acetyl-D,L-penicillamine. N. Engl. J. Med., 269: 889-893.
HOLT, D. & WEBB, M. (1986) The toxicity and teratogenicity of
mercuric mercury in the pregnant rat. Arch. Toxicol., 58: 243-248.
HUBERLANT, J.M., ROELS, H., BUCHET, J.P., BERNARD, A., &
LAUWERYS, R. (1983) Evaluation de l'exposition au mercure et
de ses répercussions éventuelles sur la santé du personnel
d'une trentaine de cabinets dentaires. Cah. Méd. Trav., 20(2):
109-127.
HUDSON, P.J., VOGT, R.L., BRONDUM, J., WITHERELL, L., MYERS,
G., & PASCHAL, D.C. (1987) Elemental mercury exposure among
children of thermometer plant workers. Pediatrics, 79(6): 935-938.
HULTMAN, P. & ENESTROM, S. (1987) The induction of immune complex
deposits in mice by peroral and parenteral administration of
mercuric chloride: Strain dependent susceptibility. Clin. exp.
Immunol., 67: 283-292.
HULTMAN, P. & ENESTROM, S. (1988) Mercury induced antinuclear
antibodies in mice: characterization and correlation with
renal immune complex deposits. Clin. exp. Immunol., 71: 269-274.
HURSH, J.B. (1985) Particle coefficients of mercury (203Hg) vapour
between air and biological fluids. J. appl. Toxicol., 5: 327-332.
HURSH, J.B., CLARKSON, T.W., CHERIAN, M.G., VOSTAL, J.V., &
MALLIE, R.V. (1976) Clearance of mercury (Hg-197, Hg-203) vapor
inhaled by human subjects. Arch. environ. Health, 31: 302-309.
HURSH, J.B., GREENWOOD, M.R., CLARKSON, T.W., ALLEN, J., &
DEMUTH, S. (1980) The effect of ethanol on the fate of mercury
vapor inhaled by man. J. Pharmacol. exp. Ther., 214: 520-527.
HURSH, J.B., SICHAK, S.P., & CLARKSON, T.W. (1988) In vitro
oxidation of mercury by the blood. Pharmacol. Toxicol., 63: 266-273.
HURSH, J.B., CLARKSON, T.W., MILES, E., & GOLDSMITH, L.A.
(1989) Percutaneous absorption of mercury vapour by man. Arch.
environ. Health, 44(2): 120-127.
IARC (1987) Overall evaluation of carcinogenicity: An updating
of IARC Monographs Volumes 1 to 42, Lyon, International Agency
for Research on Cancer, 440 pp. (IARC Monographs on the Evaluation
of Carcinogenic Risk to Humans, Suppl. 7).
ICRP (1980) Metabolic data for mercury. Ann. int. Comm. rad.
Prot., 30/Part 2, 4: 59-63.
IMURA, N. & NAGANUMA, A. (1978) Interaction of inorganic
mercury and selenite in rabbit blood after intravenous
administration. J. Pharm. Dyn., 1: 67-73.
INTERNATIONAL CLEARING-HOUSE FOR BIRTH DEFECTS MONITORING SYSTEM
(1985) Annual report. Sidney, National Perinatal Statistics Unit,
University of Sidney, 71 p. (ISSN 0743-5703).
ISHIHARA, N., URUSHIYAMA, K., & SUZUKI, T. (1977) Inorganic and
organic mercury in blood, urine and hair in low level mercury
vapour exposure. Int. Arch. occup. environ. Health, 40: 249-253.
JACOBS, M. & GOLDWATER, L.J. (1965) Absorption and excretion of
mercury in man: VIII. Mercury exposure from house paint - a
controlled study on humans. Arch. environ. Health, 11: 582-587.
JOHANSSON, E. & LINDH, U. (1987) Mercury in blood cells -
altered elemental profiles. Toxic events in human exposure.
Biol. Trace Elem. Res., 12: 309-321.
JOHNSON, S.L. & POND, W.G. (1974) Inorganic vs. organic Hg
toxicity in growing rats: Protection by dietary Se but not Zn.
Nutr. Rep. Int., 9(2): 135-147.
JOKSTAD, A. (1987) [Mercury exposure of dentists]. J. Norw. Dent.
Assoc., 97: 498-507 (in Norwegian).
JOSELOW, M.M. & GOLDWATER, L.J. (1967) Absorption and excretion of
mercury in man. XII. Relationship between urinary mercury and
proteinuria. Arch. environ. Health, 15: 155.
JOSELOW, M.M., GOLDWATER, L.J., ALVAREZ, A., & HERNDON, J.
(1968) Absorption and excretion of mercury in man. Arch. environ.
Health, 17: 39-43.
JUANG, M.S. (1976) An electrophysiological study of the
action of methylmercuric chloride and mercuric chloride on
the sciatic nerve-sartorius muscle preparation of the frog.
Toxicol. appl. Pharmacol., 37: 339-348.
JUGO, S. (1976) Retention and distribution of 203HgCl2 in
suckling and adult rats. Health Phys., 30: 240-241.
KANEMATSU, N., HARA, M., & KADA, T. (1980) Rec assay and
mutagenicity studies on metal compounds. Mutat. Res., 77: 109-116.
KARK, P. (1979) Clinical and neurochemical aspects of inorganic
mercury intoxication. In: Handbook of clinical neurology,
Amsterdam, Oxford, New York, Elsevier Science Publishers, Vol. 36,
pp. 147-197.
KAZANTZIS, G. (1978) The role of hypersensitivity and the immune
response in influencing susceptibility to metal toxicity.
Environ. Health Perspect., 25: 111-118.
KAZANTZIS, G. (1981) Role of cobalt, iron, lead, manganese,
mercury, platinum, selenium, and titanium in carcinogenesis.
Environ. Health Perspect., 40: 143-161.
KAZANTZIS, G. & LILLY, L.J. (1986) Mutagenic and carcinogenic
effects of metals. In: Friberg, L., Nordberg, G.F., & Vouk, V.,
ed. Handbook on the toxicology of metals, Amsterdam, Oxford,
New York, Elsevier Science Publishers, Vol. 1, pp. 319-390.
KAZANTZIS, G., SCHILLER, K.F.R., ASSCHER, A.W., & DREW, R.G.
(1962) Albuminuria as the nephrotic syndrome following exposure
to mercury and its compounds. Q. J. Med., 31(124): 403-418.
KELMAN, G.R. (1978) Notes and miscellanea: Urinary mercury
excretion in dental personnel. Br. J. ind. Med., 35: 262-265.
KHAYAT, A. & DENCKER, L. (1982) Fetal uptake and distribution of
metallic mercury vapor in the mouse: Influence of ethanol and
aminotriazole. Biol. Res. Pregn., 3(1): 38-46.
KHAYAT, A. & DENCKER, L. (1983a) Whole body and liver
distribution of inhaled mercury vapor in the mouse: Influence of
ethanol and aminotriazole pretreatment. J. appl. Toxicol., 3: 66-74.
KHAYAT, A. & DENCKER, L. (1983b) Interactions between selenium and
mercury in mice: Marked retention in the lung after
inhalation of metallic mercury. Chem. biol. Interact., 46: 283-298.
KHAYAT, A. & DENCKER, L. (1984) Organ and cellular distribution of
inhaled metallic mercury in the rat and marmoset monkey
(Callithrix jacchus): Influence of ethyl alcohol pretreatment.
Acta pharmacol. toxicol., 55: 145-152.
KIBUKAMUSOKE, J.W., DAVIES, D.R., & HUTT, M.S.R. (1974)
Membranous nephropathy due to skin-lightening cream. Br. med. J.,
2: 646-647.
KIMATA, H., SHINOMIYA, K., & MIKAWA, H. (1983) Selective
enhancement of human IgE production in vitro by synergy of
pokeweed mitogen and mercuric chloride. Clin. exp. Immunol., 53:
183-191.
KISHI, R., HASHIMOTO, K., SHIMIZU, S., & KOBAYASHI, M. (1978)
Behavioral changes and mercury concentrations in tissues of rats
exposed to mercury vapor. Toxicol. appl. Pharmacol., 46: 555-566.
KITAMURA, S., SUMINO, K., HAYAKAWA, K., & SHIBATA, T. (1976)
Mercury content in human tissues from Japan. In: Nordberg, G.F.,
ed. Effects and dose-response relationships of toxic metals,
Amsterdam, Oxford, New York, Elsevier Science Publishers,
pp. 290-298.
KNEIP, T.J. & FRIBERG, L. (1986) Sampling and analytical
methods. In: Friberg, L., Nordberg, G.F., & Vouk, V.B., ed.
Handbook on the toxicology of metals, 2nd ed., Amsterdam,
Oxford, New York, Elsevier Science Publishers, Vol. 1,
pp. 36-67.
KNOFLACH, P., ALBINI, B., & WEISER, M.M. (1986) Autoimmune disease
induced by oral administration of mercuric chloride in Brown-Norway
rats. Toxicol. Pathol., 14(2): 188-193.
KOMSTA-SZUMSKA, E. & CHMIELNICKA, J. (1977) Binding of
mercury and selenium in subcellular fractions of rat liver and
kidneys following separate and joint administration. Arch.
Toxicol., 38: 217-228.
KOSTA, L., BYRNE, A.R., & ZELENKO, V. (1975) Correlation between
selenium and mercury in man following exposure to inorganic
mercury. Nature, London, 254: 238-239.
KOSTIAL, K. & LANDEKA, M. (1975) The action of mercury ions on the
release of acetylcholine from presynaptic nerve endings.
Experientia, Basel, 31(7): 834-835.
KOSTIAL, K., KELLO, D., JUGO, S., RABAR, I., & MALJKOVIC, T.
(1978) Influence of age on metal metabolism and toxicity.
Environ. Health Perspect., 25: 81-86.
KOSTIAL, K., SIMONOVIC, I., RABAR, I., BLANUSA, M., & LANDEKA, M.
(1983) Age and intestinal retention of mercury and cadmium in
rats. Environ. Res., 31: 111-115.
KUSAKAWA, S. & HEINER, D.C. (1976) Elevated levels of
immunoglobin E in the acute febrile mucocutaneous lymph node
syndrome. Pediatr. Res., 10: 108-112.
LAMBETH, LONDON BOROUGH (1988) Mercury soaps and skin
lightening cream Directorate of environmental and consumer
services, London.
LAMPERTI, A.A. & PRINTZ, R.H. (1973) Effects of mercuric chloride
on the reproductive cycle of the female hamster. Biol. Reprod., 8:
378-387.
LAMPERTI, A.A. & PRINTZ, R.H. (1974) Localization, accumulation,
and toxic effects of mercuric chloride on the reproductive axis
of the female hamster. Biol. Reprod., 11: 180-186.
LAMPERTI, A. & NIEWENHUIS, R. (1976) The effects of mercury
on the structure and function of the hypothalamo-pituitary axis
in the hamster. Cell Tissue Res., 170: 315-324.
LANGAN, D.C., FAN, P.L., & HOOS, A.A. (1987) The use of
mercury in dentistry: A critical review of the recent
literature. J. Am. Dent. Assoc., 115: 867-880.
LANGOLF, G.D., CHAFFIN, D.B., HENDERSON, R., & WHITTLE, H.P. (1978)
Evaluation of workers exposed to elemental mercury using
quantitative tests of tremor and neuromuscular functions. Am. Ind.
Hyg. Assoc. J., 39: 976-984.
LANGOLF, G.D., SMITH, P.J., HENDERSON, R., & WHITTLE, H.P.
(1981) Measurements of neurological functions in the evaluations
of exposure to neurotoxic agents. Ann. occup. Hyg., 24(3): 293-296.
LANGWORTH, S. (1987) Renal function in workers exposed to
inorganic mercury. In: Occupational health in the chemical
industry: papers presented at the XXII ICOH congress, Sidney,
Australia, 27 September-2 October 1987. Copenhagen, World Health
Organization, p. 237.
LANGWORTH, S., ELINDER, C.-G., & AKESSON, A. (1988) Mercury
exposure from dental fillings. I Mercury concentrations in blood
and urine. Swed. dent. J., 12: 69-70.
LAUWERYS, R., BERNARD, A., ROELS, H., BUCHET, J.P., GENNART, J.P.,
MAHIEU, P., & FOIDART, J.M. (1983) Anti-laminin antibodies in
workers exposed to mercury vapour. Toxicol. Lett., 17: 113-116.
LAUWERYS, R., ROELS, H., GENET, P., TOUSSAINT, G., BOUCKAERT,
A., & DE COOMAN, S. (1985) Fertility of male workers exposed to
mercury vapor or to manganese dust: A questionnaire study.
Am. J. ind. Med., 7: 171-176.
LAUWERYS, R., BONNIER, C., EVRARD, P., GENNART, J., & BERNARD, A.
(1987) Prenatal and early postnatal intoxication by inorganic
mercury resulting from the maternal use of mercury-containing
soap. Hum. Toxicol., 6: 253-256.
LAVSTEDT, S. & SUNDBERG, H. (1989) [Medical diagnosis and disease
symptoms related to amalgam fillings.] Tandläkartidningen, 3: 81-88 (in
Swedish).
LEE, I.P. & DIXON, R.L. (1975) Effects of mercury on
spermatogenesis studied by velocity sedimentation cell separation
and serial mating. J. Pharmacol. exp. Ther., 194(1): 171-181.
LEE, S.A. (1984) Health hazard evaluation report, Cincinnati,
Ohio, National Institute for Occupational Safety and Health, 10
pp. (HETA 83-338-1399).
LEONARD, A., JACQUET, P., & LAUWERYS, R.R. (1983)
Mutagenicity and teratogenicity of mercury compounds. Mutat. Res.,
114: 1-18.
LEVIN, M., JACOBS, J., & POLOS, P.G. (1988) Acute mercury
poisoning and mercurial pneumonitis from gold ore purification.
Chest, 94: 554-556.
LEVINE, S.P., CAVENDER, G.D., LANGOLF, G.D., & ALBERS, J.W.
(1982) Elemental mercury exposure: peripheral neurotoxicity. Br.
J. ind. Med., 39: 136-139.
LIND, B,. FRIBERG, L., & NYLANDER, M. (1988) Preliminary
studies on methylmercury biotransformation and clearance in the
brain of primates: II. Demethylation of mercury in brain. J. Trace
Elem. exp. Med., 1: 49-56.
LINDBERG, S., STOKES, P., GOLDBERG, E., & WREN, C. (1987) Group
Report: Mercury. In: Hutchinson, T.W. & Meemz, K.M. ed. Lead,
mercury, cadmium and arsenic in the environment, New York,
Chichester, Brisbane, Toronto, John Wiley & Sons, pp. 17-34 (Scope
31).
LINDQVIST, K.J., MAKENE, W.J., SHABA, J.K., & NANTULYA, V.
(1974) Immunofluorescence and electron microscopic studies of
kidney biopsies from patients with nephrotic syndrome, possibly
induced by skin lightening creams containing mercury. East Afr.
med. J., 51: 168-169.
LINDQVIST, O, JERNELOV, A., JOHANNSON, K., & RODHE, R. (1984)
Mercury in the swedish environment: global and local sources.
Solna, National Environmental Protection Board, 105 pp. (Report
No. 1816).
LINDSTEDT, G., GOTTBERG, I., HOLMGREN, B., JONSSON, T., &
KARLSSON, G. (1979) Individual mercury exposure of chloralkali
workers and its relation to blood and urinary mercury levels.
Scand. J. Work Environ. Health, 5: 59-69.
LINKE, W.F. (1958) Inorganic and metal-organic compounds, New
York, Van Nostrand Reinhold Company Inc.
LUNDBORG, M., LIND, B., & CAMNER, P. (1984) Ability of rabbit
alveolar macrophages to dissolve metals. Exp. Lung Res., 7: 11-22.
MABILLE, V., ROELS, H., JACQUET, P., LEONARD, A., & LAUWERYS, R.
(1984) Cytogenetic examination of leucocytes of workers
exposed to mercury vapour. Int. Arch. occup. environ. Health, 53:
257-260.
MCCAMMON, C.S., Jr, EDWARDS, S.L., DELON HULL, R., & WOODFIN, W.J.
(1980) A comparison of four personal sampling methods for the
determination of mercury vapor. Am. Ind. hyg. Assoc. J., 41: 528-531.
MCFARLAND, R.B. & REIGEL, H. (1978) Chronic mercury poisoning
from a single brief exposure. J. occup. Med., 20(8): 532-534.
MACKERT, J.R., Jr, (1987) Factors affecting estimation of dental
amalgam mercury exposure from measurements of mercury vapor
levels in intra-oral and expired air. J. dent. Res., 66(12): 1775-1780.
MCNERNEY, J.J, BUSECK, P.R., & HANSON, R.C. (1972) Mercury
detection by means of thin gold films. Science, 178: 611-612.
MAGNUSSON, J. & RAMEL, C. (1986) Genetic variation in the
susceptibility to mercury and other metal compounds in Drosophila
melanogaster. Teratog. Carcinog. Mutagen., 6: 289-192.
MAGOS, L. (1971) Selective atomic-absorption determination of
inorganic mercury and methylmercury in undigested biological
samples. Analyst, 96: 847-853.
MAGOS, L. & CLARKSON, T.W. (1972) Atomic absorption
determination of total, inorganic and organic mercury in blood. J.
Assoc. Off. Anal. Chem., 5(5): 966-971.
MAGOS, L., WEBB, M., & BUTLER, W.H. (1974) The effect of
cadmium pretreatment on the nephrotoxic action and kidney uptake
of mercury in male and female rats. Br. J. exp. Pathol., 55: 589-594.
MAGOS, L., HALBACH, S., & CLARKSON, T.W. (1978) Role of catalase
in the oxidation of mercury vapor. Biochem. Pharmacol., 27: 1373-1377.
MAGOS, L., SPARROW, S., & SNOWDEN, R. (1982) The
comparative renotoxicology of phenylmercury and mercuric chloride.
Arch. Toxicol., 50: 133-139.
MAGOS, L., CLARKSON, T.W., & HUDSON, A.R. (1984) Differences
in the effects of selenite and biological selenium on the
chemical form and distribution of mercury after the simultaneous
administration of HgCl2 and selenium to rats. J. Pharmacol. exp.
Ther., 228(2): 478-483.
MAGOS, L., BROWN, A.W., SPARROW, S., BAILEY, E., SNOWDEN, R.T., &
SKIPP, W.R. (1985) The comparative toxicology of ethyl- and
methylmercury. Arch. Toxicol., 57: 260-267.
MAGOS, L., CLARKSON, T.W., SPARROW, S., & HUDSON, A.R. (1987)
Comparison of the protection given by selenite,
selenomethionine and biological selenium against the renotoxicity
of mercury. Arch. Toxicol., 60: 422-426.
MANALIS, R.S. & COOPER, G.P. (1975) Evoked transmitter release
increased by inorganic mercury at frog neuromuscular junction.
Nature, London, 257: 690-691.
MARAFANTE, E., LUNDBORG, M., VAHTER, M., & CAMNER, P. (1987)
Dissolution of two arsenic compounds by rabbit alveolar
macrophages in vitro . Fundam. appl. Toxicol., 8: 382-388.
MARZULLI, F.N. & BROWN, D.W.C. (1972) Potential systemic
hazards of topically applied mercurials. J. Soc. cosmet. Chem., 23:
875-886.
MATHESON, D.S., CLARKSON, T.W., & GELFAND, E.W. (1980) Mercury
toxicity (acrodynia) induced by long-term injection of
gammaglobulin. J. Pediatr., 97(1): 153-155.
MATTIUSSI, R., ARMELI, G., & BAREGGI, V. (1982) Statistical study
of the correlation between mercury exposure (TWA) and
urinary mercury concentrations in chloralkali workers. Am. J.
ind. Med., 3: 335-339.
MENGEL, H. & KARLOG, O. (1980) Studies on the interaction and
distribution of selenite, mercuric, methoxyethyl mercuric and
methyl mercuric chloride in rats. II. Analysis of the soluble
proteins and the precipitates of liver and kidney homogenates.
Acta pharmacol. toxicol., 46: 25-31.
MILLER, E.G., PERRY, W.L., & WAGNER, M.J. (1987) Prevalence of
mercury hypersensitivity in dental students. J. prosthet. Dent., 58:
235-237.
MILLER, J.M., CHAFFIN, D.B., & SMITH, R.G. (1975) Subclinical
psychomotor and neuromuscular changes in workers exposed to
inorganic mercury. Am. Ind. Hyg. Assoc. J., 36: 725-733.
MINAGAWA, K., HIRASAWA, F., HOZUMI, S., CHUANG, K., & TAKIZAWA, Y.
(1979) [Rapid determinations of total mercury and methylmercury
in biological samples.] Akita J. Med., 5: 263-271 (in Japanese
with English summary).
MIRTSCHEWA, J., NURNBERGER, W., HALLMANN, B., STILLER-WINKLER,
R., & GLEICHMANN, E. (1987) Genetically determined
susceptibility of mice to HgCl2-induced antinuclear antibodies
(ANA) and glomerulo-nephritis. Immunobiology, 175: 323-324.
MIYAMA, T. & SUZUKI, T. (1971) Release of mercury from organs and
breakage of carbon-mercury bond in tissues due to formalin
fixation. Jpn. J. ind. Health, 13(6): 56-57.
MIYAMOTO, M.D. (1983) Hg2+ causes neurotoxicity at an
intracellular site following entry through Na and Ca channels.
Brain Res., 267: 375-379.
MOLIN, M., BERGMAN, B., MARKLUND, S.L., SCHUTZ, A., & SKERFVING, S.
(1990) Mercury, selenium and glutathione peroxidase before and
after amalgam removal in man. Acta Odontol. Scand., 48(3): 189-202.
MOLLER-MADSEN, B. & DANSCHER, G. (1986) Localization of mercury in
CNS of the rat. Environ. Res., 41: 29-43.
MORIMOTO, K., IIJIMA, S., & KOIZUMI, A. (1982) Selenite
prevents the induction of sister-chromatid exchanges by
methylmercury and mercuric chloride in human whole-blood
cultures. Mutat. Res., 102: 183-192.
MORNER, S. & NILSSON, T. (1986) [Mercury discharge from
crematories in Gothenburg], City of Gothenburg, Environmental
and Health Protection Agency (Report 1986.1) (in Swedish).
MORROW, P.E., GIBB, F.R., & JOHNSON, L. (1964) Clearance of
insoluble dust from the lower respiratory tract. Health Phys., 10:
543-555.
MOTTET, K. & BURBACHER, T. M. (1988) Preliminary studies on
methylmercury biotransformation and clearance in the brain of
primates: I. Experimental design and general observations J. Trace
Elem. exp. Med., 1: 41-47.
MULLER, H.,VON, SCHUBERT, H., & VERBEEK, W. (1980) [Report
on the relationship between Hg concentration in urine and
blood and Hg concentration in workroom air.] Arbeitsmed.
Sozialmed. Präventivmed., 15: 64-67 (in German with English
summary).
MURAMATSU, Y. & PARR, R.M. (1985) Survey of currently available
reference materials for use in connection with the determination
of trace elements in biological and environmental materials.
Vienna, International Atomic Energy Agency (Report No.
IAEA/RL/128).
MUTTI, A., LUCERTINI, S., FORNARI, M., FRANCHINI, I., BERNARD, A.,
ROELS, H., & LAUWERYS, R. (1985) Urinary excretion of a brush
border antigen revealed by monoclonal antibodies in subjects
occupationally exposed to heavy metals. In: Proceedings of the
International Conference on Heavy Metals in the Environment,
Luxembourg, Commission of the European Communities, Vol. 1, pp.
565-567.
NAGANUMA, A. & IMURA, N. (1980) Bis(methylmercuric) selenide as a
reaction product from methylmercury and selenite in rabbit
blood. Res. Commun. chem. Pathol. Pharmacol., 27(1): 163-173.
NAKAAKI, K., FUKABORI, S., & TADA, O. (1975) [An experimental
study on inorganic mercury vapour exposure.] J. Sci. Labour,
51(12): 705-716 (in Japanese with English summary).
NAKAAKI, K., FUKABORI, S., & TADA, O. (1978) On the evaluation of
mercury exposure. J. sci. Labour, 54: 1-8.
NAKADA, S., NOMOTO, A., & IMURA, N. (1980) Effect of
methylmercury and inorganic mercury on protein synthesis in
mammalian cells. Ecotoxicol. environ. Saf., 4: 184-190.
NAKAYAMA, H., NIKI, F., SHONO, M., & HADA, S. (1983) Mercury
exanthem. Contact dermatitis, 9: 411-417.
NALEWAY, C., SAKAGUCHI, R., MITCHELL, E., MULLER, T., AYER,
W.A., & HEFFERREN, J.J. (1985) Urinary mercury levels in US
dentists, 1975-1983: Review of Health Assessment Program. J. Am.
Dent. Assoc., 111: 37-42.
NATIONAL ACADEMY OF SCIENCES (1978) An assessment of mercury
in the environment, Washington, DC, National Academy of
Sciences, National Research Council.
NEWTON, D. & FRY, F.A. (1978) The retention and
distribution of radioactive mercuric oxide following accidental
inhalation. Ann. occup. Hyg., 21: 21-32.
NILSEN, A., NYBERG, K., & CAMNER, P. (1988) Intraphagosomal pH in
alveolar macrophages after phagocytosis in vivo and in vitro of
fluorescein-labeled yeast particles. Exp. Lung Res., 13: 197-207.
NILSSON, B. & NILSSON, B. (1986a) Mercury in dental practice.
I. The working environment of dental personnel and their
exposure to mercury vapor. Swed. dent. J., 10: 1-14.
NILSSON, B. & NILSSON, B. (1986b) Mercury in dental practice. II.
Urinary mercury excretion in dental personnel. Swed. dent. J., 10:
221-232.
NISHIYAMA, S., TAGUCHI, T., & ONOSAKA, S. (1987) Induction of
zincthionein by estradiol and protective effects on inorganic
mercury-induced renal toxicity. Biochem. Pharmacol., 36(20): 3387-3391.
NIXON, G.S., WHITTLE, C.A., & WOODFIN, A. (1981) Mercury levels in
dental surgeries and dental personnel. Br. dent. J., 151: 149-154.
NORDLIND, K. (1985) Binding and uptake of mercuric chloride in
human lymphoid cells. Int. Arch. Allergy appl. Immunol., 77: 405-408.
NORDLIND, K. & HENZE, A. (1984) Stimulating effect of mercuric
chloride and nickel sulfate on DNA synthesis of thymocytes and
peripheral blood lymphocytes in children. Int. Arch. Allergy appl.
Immunol., 73: 162-165.
NORTH AMERICAN CONTACT DERMATITIS GROUP (1973) Epidemiology of
contact dermatitis in North America, 1972. Arch. Dermatol., 108: 537-
540.
NRIAGU, J.O. (1979) The biogeochemistry of mercury in the
environment, Amsterdam, Oxford, New York, Elsevier Science
Publishers.
NYGAARD, S.F. & HANSEN, J.C. (1978) Mercury-selenium
interaction at concentrations of selenium and of mercury vapours
as prevalent in nature. Bull. environ. Contam. Toxicol., 20: 20-23.
NYLANDER, M., FRIBERG, L., & LIND, B. (1987) Mercury concentrations
in the human brain and kidneys in relation to exposure from
dental amalgam fillings. Swed. dent. J., 11: 179-187.
NYLANDER, M., FRIBERG, L., LIND, B., & AAKERBERG, S. (1988)
Effects of breathing patterns on mercury release. In: Clarkson,
T.W., Friberg, L., Nordberg, G.F., & Sager, P.R., ed. Biological
monitoring of toxic metals, New York, London, Plenum Press, pp.
263-264.
NYLANDER, M., FRIBERG, L., EGGLESTON, D., & BJORKMAN, L. (1989)
Mercury accumulation in tissues from dental staff and controls
in relation to exposure. Swed. dent. J., 13: 235-243.
OBERLY, T.J., PIPER, C.E., & MCDONALD, D.S. (1982) Mutagenicity of
metals in the L5178Y mouse lymphoma assay. J. Toxicol. environ.
Health, 9: 367-376.
OGATA, M. & MEGURO, T. (1986) Foetal distribution of inhaled
mercury vapor in normal and acatalasaemic mice. Physiol. Chem.
Phys. med. NMR, 18: 165-170.
OGATA, M., KENMOTSU, K., HIROTA, N., MEGURO, T., & AIKOH, H.
(1987) Reduction of mercuric ion and exhalation of mercury in
acatalasemic and normal mice. Arch. environ. Health, 42(1): 26-30.
OKAMOTO, K., (1988) Biological reference materials from the
National Institute for Environmental Studies (Japan). Fresenius
Z. anal. Chem., 332: 524-527.
OLSSON, B. & BERGMAN, M. (1987) Letter to the Editor. J. dent.
Res., 66(7): 1288-1291.
OLSTAD, M.L., HOLLAND, R.I., WANDEL, N., & HENSTEN PETTERSEN, A.
(1987) Correlation between amalgam restorations and mercury
concentrations in urine. J. dent. Res., 66(6): 1179-1182.
ORLOWSKI, J.P. & MERCER, R.D. (1980) Urine mercury levels in
Kawasaki disease. Pediatrics, 66(4): 633-636.
OTT, K.H.R., LOH, F., KRONCKE, A., SCHALLER, K.-H., VALENTIN,
H., & WELTLE, D. (1984) [Mercury burden due to amalgam
fillings.] Dtsch. Zahnärztl. Z., 39: 199-205 (in German with
English summary).
PACYNA, J.M. (1987) Atmospheric emissions of arsenic, cadmium,
lead and mercury from high temperature processes in power
generation and industry. In: Hutchinson, T.C. & Meema, K.M. ed.
Lead, mercury, cadmium and arsenic in the environment, New York,
Chichester, Brisbane, Toronto, John Wiley & Sons, pp. 69-88 (Scope
31).
PAN, S.K., IMURA, N., YAMAMURA, Y., YOSHIDA, M., & SUZUKI, T.
(1980) Urinary methylmercury excretion in persons exposed to
elemental mercury vapor. Tohoku J. exp. Med., 130: 91-95.
PARIZEK, J. & OSTADALOVA, I. (1967) The protective effect of small
amounts of selenite in sublimate intoxication. Experientia, Basel,
23(2): 142-143.
PARIZEK, J., OSTADALOVA, I., KALOUSKOVA, J., BABICKY, A.,
PAVLIK, L., & BIBR, B. (1971) Effect of mercuric compounds on the
maternal transmission of selenium in the pregnant and lactating
rat. J. Reprod. Fertil., 25: 157-170.
PARR, R.M., MURAMATSU, Y., & CLEMENTS, S.A. (1987) Survey and
evaluation of available biological reference materials for trace
element analysis. Fresenius Z. anal. Chem., 326: 601-608.
PARR, R.M., SCHELENZ, R., & BALLESTRA, S. (1988) IAEA biological
reference materials. Fresenius Z. anal. Chem., 332: 518-523.
PATON, G.R. & ALLISON, A.C. (1972) Chromosome damage in
human cell cultures induced by metal salts. Mutat. Res., 16: 332-336.
PATTERSON, J.E., WEISSBERG, B.G., & DENNISON, P.J. (1985) Mercury
in human breath from dental amalgams. Bull. environ. Contam.
Toxicol., 34: 459-468.
PELLETIER, L., PASQUIER, R., HIRSH, F., SAPIN, C., & DRUET, P.
(1986) Autoreactive T cells in mercury-induced autoimmune
disease: in vitro demonstration. J. Immunol., 137(8): 2548-2554.
PELLETIER, L., PASQUIER, R., VIAL, M.C., MANDET, C., MOUTIER, R.,
SALOMON, J.C., & DRUET, P. (1987a) Mercury-induced autoimmune
glomerulonephritis: Requirement for T-cells. Nephrol. Dial.
Transplant., 1: 211-218.
PELLETIER, L., GALCERAN, M., PASQUIER, R., RONCO, P.,
VERROUST, P., BARIETY, J., & DRUET, P. (1987b) Down modulation of
Heymann's nephritis by mercuric chloride. Kidney Int., 32: 227-232.
PELLETIER, L., PASQUIER, R., ROSSERT, J., & DRUET, P. (1987c)
HgCl2 induces nonspecific immunosuppression in Lewis rats. Eur. J.
Immunol., 17: 49-54.
PELLETIER, L., PASQUIER, R., GUETTIER, C., VIAL, M.C., MANDET, C.,
NOCHY, D., & DRUET, P. (1988a) HgCl2 induces T and B cells to
proliferate and differentiate in BN rats. Clin. exp. Immunol., 71:
336-342.
PELLETIER, L., PASQUIER, R., ROSSERT, J., VIAL, M.C., MANDET, C., &
DRUET, P. (1988b) Autoreactive T-cells in mercury-induced
autoimmunity. Ability to induce the autoimmune disease. J.
Immunol., 140(3): 750-754.
PELLETIER, L., ROSSERT, J., PASQUIER, R., VILLARROYA, H.,
BELAIR, M.F., VIAL, M.C., ORIOL, R., & DRUET, P. (1988c) Effect of
HgCl2 on experimental allergic encephalomyelitis in Lewis rats.
HgCl2 induces down modulation of the disease. Eur. J. Immunol., 18:
243-247.
PENZER, V. (1986) Amalgam toxicity: grand deception. Int. J.
Orthod., 24: 21-24.
PETER, F. & STRUNC, G. (1984) Semiautomated analysis for mercury
in whole blood, urine and hair by on-stream generation of cold
vapor. Clin. Chem., 30(6): 893-895.
PEZEROVIC, Dz., NARANCSIK, P., & GAMULIN, S. (1981) Effects of
mercury bichloride on mouse kidney polyribosome structure and
function. Arch. Toxicol., 48: 167-172.
PIIKIVI, L. & HANNINEN, H. (1989) Subjective symptoms and
psychological performance of chlorin-alkali workers. Scand. J.
Work. Environ. Health, 15: 69-74.
PIIKIVI, L. & TOLONEN, U. (1989) EEG findings in chlor-alkali
workers subjected to low long term exposure to mercury vapour.
Brit. J. ind. Med., 46: 370-375.
PIIKIVI, L., HANNINEN, H., MARTELIN, T., & MANTERE, P.
(1984) Psychological performance and long-term exposure to mercury
vapors. Scand. J. Work environ. Health, 10: 35-41.
PIOTROWSKI, J.K., TROJANOWSKA, B., & MOGILNICKA, E.M. (1975)
Excretion kinetics and variability in urinary mercury in workers
exposed to mercury vapor. Internat. Arch. occup. environ. Health,
35: 245-256.
POLAK, L., BARNES, J.M., & TURK, J.L. (1968) The genetic
control of contact sensitization to inorganic metal
compounds in guinea-pigs. Immunology, 14: 707-711.
POPESCU, H.I., NEGRU, L., & LANCRANJAN, I. (1979) Chromosome
aberrations induced by occupational exposure to mercury. Arch.
environ. Health., 34(6): 461-463.
PRICE, J.H. & WISSEMAN, C.L. (1977) Hazard evaluation and
technical assistance report, Cincinnati, Ohio, National Institute
for Occupational Safety and Health, 17 pp. (TA 77-52).
PRITCHARD, J.G., McMULLIN, J.F., & SIKONDARI, A.H. (1982) The
prevalence of high levels of mercury in dentists' hair. Br. dent.
J., 153: 333-336.
PROUVOST-DANON, A., ABADIE, A., SAPIN, C., BAZIN, H., & DRUET, P.
(1981) Induction of IgE synthesis and potentiation of anti-
ovalbumin IgE antibody response by HgCl2 in the rat. J. Immunol.,
126(2): 699-702.
RAHOLA, T., HATTULA, T., KOROLAINEN, A., & MIETTINEN, J.K.
(1973) Elimination of free and protein-bound ionic mercury
(203Hg2+) in man. Ann. clin. Res., 5: 214-219.
RAMEL, C. (1972) Genetic effects. In: Friberg, L. & Vostal,
J., ed. Mercury in the environment, Cleveland, Ohio, CRC Press,
Chapter 9, pp. 169-181.
RASBERRY, S.D. (1987) Biological reference materials from the US
National Bureau of Standards - an update. Fresenius Z. anal. Chem.,
326: 609-612.
REINHARDT, J.W., KAI, CHIU CHAN, & SCHULEIN, T.M. (1983)
Mercury vaporization during amalgam removal. J. prosthet. Dent.,
50(1): 62-64.
REUTER, R., TRESSARS, G., VOHR, H.-W., GLEICHMANN, E., &
LUHRMANN, R. (1989) Mercuric chloride induces autoantibodies
to small nuclear ribonucleoprotein in susceptible mice. Proc.
Natl Acad. Sci. (USA), 86: 237-241.
RICE, D.C. (1989) Brain and tissue levels of mercury after
chronic methylmercury exposure in the monkey. J. Toxicol.
environ. Health, 27: 189-190.
RICHARDS, J.M. & WARREN, P.J. (1985) Mercury vapour released
during the removal of old amalgam restorations. Br. dent. J., 159:
231-232.
RIZZO, A.M. & FURST, A. (1972) Mercury teratogenesis in the
rat. Proc. West. Pharmacol. Soc., 15: 52-54.
ROBINSON, G.C.J., BALAZS, T., & EGOROV, I.K. (1986) Mercuric
chloride-, gold sodium thiomalate-, and D-penicillamine-
induced antinuclear antibodies in mice. Toxicol. appl.
Pharmacol., 86: 159-169.
ROBISON, S.H., CANTONI, O., & COSTA, M. (1984) Analysis of metal-
induced DNA lesions and DNA-repair replication in mammalian
cells. Mutat. Res., 131: 173-181.
ROELS, H., LAUWERYS, R., BUCHET, J.P., BERNARD, A.,
BARTHELS, A., OVERSTEYNS, M., & GAUSSIN, J. (1982) Comparison
of renal function and psychomotor performance in workers
exposed to elemental mercury. Int. Arch. occup. environ. Health,
50: 77-93.
ROELS, H., GENNART, J.-P., LAUWERYS, R., BUCHET, J.-P.,
MALCHAIRE, J., & BERNARD, A. (1985) Surveillance of workers
exposed to mercury vapour: Validation of a previously proposed
biological threshold limit value for mercury concentration in
urine. Am. J. ind. Med., 7: 45-71.
ROELS, H., ABDELADIM, S., CEULEMANS, E., & LAUWERYS, R.
(1987) Relationships between the concentrations of mercury in air
and in blood or urine in workers exposed to mercury vapour. Ann.
occup. Hyg., 31(2): 135-145.
ROMAN-FRANCO, A.A., TURIELLO, M., ALBINI, B., OSSI, E.,
MILGROM, F., & ANDRES, G.A. (1978) Anti-basement membrane
antibodies and antigen-antibody complexes in rabbits injected
with mercuric chloride. Clin. Immunol. Immunopathol., 9: 464-481.
ROSENMAN, K.D., VALCIUKAS, J.A., GLICKMAN, L., MEYERS, B.R.,
CINOTTI, A. (1986) Sensitive indicators of inorganic mercury
toxicity. Arch. environ. Health, 41(4): 208-215.
ROSSI, L.C., CLEMENTE, G.F., & SANTARONI, G. (1976) Mercury and
selenium distribution in a defined area and in its population.
Arch. environ. Health, 160-165.
ROWLAND, I.R., GRASSO, P., & DAVIES, M.J. (1975) The
methylation of mercuric chloride by human intestinal bacteria.
Experentia, Basel, 31(9): 1064-1065.
ROZALSKI, M. & WIERZBICKI, R. (1983) Effect of mercuric
chloride on cultured rat fibroblasts: Survival, protein
biosynthesis and binding of mercury to chromatin. Biochem.
Pharmacol., 32(13): 2124-2126.
RUDZKI, E. (1979) Occupational dermatitis among health service
workers. Derm. Beruf Umwelt, 27(4): 112.
SAPIN, C., DRUET, E., & DRUET, P. (1977) Induction of anti-
glomerular basement membrane antibodies in the Brown-Norway rat by
mercuric chloride. Clin. exp. Immunol., 28: 173-179.
SAPIN, C., HIRSH, F., DELAPORTE, J.P., BAZIN, H., & DRUET, P.
(1984) Polyclonal IgE increase after HgCl2 injections in BN and
LEW rats: a genetic analysis. Immunogenetics, 20: 227-236.
SAPOTA, A., PIOTROWSKI, J., & BARANSKI, B. (1974)
Levels of metallothionein in the fetuses and tissues of
pregnant rats exposed to mercury vapors. Med. Pr., 25: 129-136
(in Polish) (The original is not available. Quoted from US
Public Health Service, Agency for Toxic Substances and Disease
Registry, 1989).
SCHIELE, R. (1988) [Statement on uptake of mercury from
amalgam]. In: Institut der Deutschen Zahnärzte (IDZ), ed.
[Amalgam - for and against], Köln, Deutscher Arzte-Verlag, pp. 123-
133 (in German).
SCHIELE, R., SCHALLER, K.-H., & GROBE, T. (1979) [Studies of
persons occupationally exposed to mercury.] Arbeitsmed. Sozialmed.
Präventivmed., 10: 226-229 (in German).
SCHNEIDER, M. (1974) An environmental study of mercury
contamination in dental offices. J. Am. Dent. Assoc., 89: 1092-1098.
SCHROEDER, H. & MITCHENER, M. (1975) Life-term effects of
mercury, methylmercury, and nine other trace metals on mice. J.
Nutr., 105: 452-458.
SCHUCKMANN, F. (1979) Study of preclinical changes in workers
exposed to inorganic mercury in chloralkali plants. Int. Arch.
occup. environ. Health, 44: 193-200.
SCHOPF, E., SCHULZ, K.H., & ISENSEE, I. (1969)
[Investigations on lymphocyte transformation in mercury
sensitivity. Nonspecific transformation due to mercury
compounds.] Arch. klin. exp. Dermatol., 234: 420-433 (in German with
English summary).
SECO, J.M. (1987) Big mercury deposit found in Spain. Financ.
Times, Oct. 30.
SHAPIRO, I.M., CORNBLATH, D.R., SUMNER, A.J., UZZELL, B.,
SPITZ, L.K., SHIP, I.I., & BLOCH, P. (1982) Neurophysiological
and neuropsychological function in mercury-exposed dentists.
Lancet, I: 1147-1150.
SIBBET, D.J., MOYER, R., & MILLY, G. (1972) Emission of mercury
from latex paint. Proceedings of the Division of Water, Air,
and Waste Chemistry, Boston, American Chemical Society, pp. 20-26.
SIBLERUD, R.L. (1988) The relationship between dental amalgam and
health. Dissertation, Colorado State University (partial
fulfillment of a Ph.D. degree requirement in physiology).
SIKORSKI, R., JUSZKIEWICZ, T., PASZKOWSKI, T., & SZPRENGIER-
JUSZKIEWICZ, T. (1987) Women in dental surgeries: Reproductive
hazards in occupational exposure to metallic mercury. Int. Arch.
occup. environ. Health, 59: 551-557.
SINCLAIR, P.M., TURNER, P.R.C., & JOHNS, R.B. (1980) Mercury
levels in dental students and faculty measured by neutron
activation analysis. J. prosthet. Dent., 43: 581-585.
SINGER, R., VALCIUKAS, J.A., & ROSENMAN, K.D. (1987)
Peripheral neurotoxicity in workers exposed to inorganic mercury
compounds. Arch. environ. Health, 42: 181-184.
SKARE, I. (1972) Microdetermination of mercury in biological
samples. Part III. Automated determination of mercury in urine,
fish and blood samples. Analyst, 97: 148-155.
SKARE, I. & ENGQVIST, A. (1986) [Mercury in air. Part I.
Evaluation of solid adsorbents for exposure monitoring of
mercury vapour], Solna, National Institute of Occupational
Health, 47 pp. (Arbete och Hälsa 1986:38) (in Swedish with
English summary).
SKERFVING, S. (1988) Mercury in women exposed to methylmercury
through fish consumption, and in their newborn babies and
breast milk. Bull. environ. Contam. Toxicol., 41: 475-482.
SKERFVING, S. & BERLIN, M. (1985) [Nordic expert group for
limit value documentation 59. Inorganic mercury], Solna,
National Institute of Occupational Health, 80 pp. (Arbete och
Hälsa) (in Swedish with English summary).
SKERFVING, S. & VOSTAL, J. (1972) Symptoms and signs of
intoxication. In: Friberg, L. & Vostal, J., ed. Mercury in the
environment, Cleveland, Ohio, CRC Press, pp. 93-107.
SKOG, E. & WAHLBERG, J.E. (1964) A comparative investigation
of the percutaneous absorption of metal compounds in the guinea-
pig by means of the radioactive isotopes: 31Cr, 36Co, 63Zn,
110mCd, 203Hg. J. invest. Dermatol., 43: 187-192.
SKUBA, A.R.N. (1984) Survey for mercury vapour in Manitoba dental
offices (Summer 1983). Can. Dent. Assoc. J., (7): 517-522.
SLEMR, F., HEINZ, G.H., CAMARDESE, M.B., HILL, E.F., MOORE,
J.F., & MURRAY, H.C. (1981) Latitudinal distribution of mercury
over the Atlantic Ocean. J. geophys. Res., 86: 1159-1166.
SMITH, P.J., LANGOLF, G.D., & GOLDBERG, J. (1983) Effects of
occupational exposure to elemental mercury on short term memory.
Br. J. ind. Med., 40: 413-419.
SMITH, R.G., VORWALD, A.J., PATIL, L.S., & MOONEY, T.F., Jr,
(1970) Effects of exposure to mercury in the manufacture of
chlorine. Am. Ind. Hyg. Assoc. J., 31: 687-700.
SNAPP, K.R., BOYER, D.B., PETERSON, L.C., & SVARE, C.W.
(1989) The contribution of dental amalgam to mercury in blood.
J. dent. Res., 68: 780-785.
SOS (1987) [Mercury/amalgam - Health risks.] Stockholm, National
Board of Health and Welfare (Report by the LEK-Committee) (in
Swedish with English summary).
STEFFEK, A.J., CLAYTON, R., SIEW, C., & VERRUSIO, A.C. (1987)
Effects of elemental mercury vapor exposure on pregnant Sprague-
Dawley rats. J. dent. Res., 66: 239.
STENMAN, E. & BERGMAN, M. (1989) Hypersensitivity reactions to
dental materials in a referred group of patients. Scand. J. dent.
Res., 97: 76-83.
STEWART, W.K., GUIRGIS, H.A., SANDERSON, J., & TAYLOR, W. (1977)
Urinary mercury excretion and proteinuria in pathology
laboratory staff. Br. J. ind. Med., 34: 26-31.
STOCK, A. (1939) [Chronic mercury and amalgam intoxication.]
Zahnärztl. Rundsch., 10: 371-377, 403-700 (in German).
STONARD, M.D., CHATER, B.V., DUFFIELD, D.P., NEVITT, A.L.,
O'SULLIVAN, J.J., & STEEL, G.T. (1983) An evaluation of renal
function in workers occupationally exposed to mercury vapour.
Int. Arch. occup. environ. Health, 52: 177-189.
STOPFORD, W., BUNDY, S.D., GOLDWATER, L.J., & BIRRIKOFER, K.A.
(1978) Microenvironmental exposure to mercury vapor. Am. Ind. Hyg.
Assoc. J., 39: 379-384.
SUGATA, Y. & CLARKSON, T.W. (1979) Exhalation of mercury -
further evidence for an oxidation-reduction cycle in mammalian
tissues. Biochem. Pharmacol., 28: 3474-3476.
SUGITA, M. (1978) The biological half-time of heavy metals. The
existence of a third "slowest" component. Int. Arch. occup.
environ. Health, 41: 25-40.
SUMMERS, A.O., WIREMAN, J., VIMY, M.J., & LORSCHEIDER, F.L.
(1990) Increased mercury resistance in monkey gingival and
intestinal bacterial flora after placement of dental "silver"
fillings. Physiologist, 33(4): abstract 116.
SUTER, K. (1975) Studies on the dominant-lethal and fertility
effects of the heavy metal compounds methylmercuric hydroxide,
mercuric chloride, and cadmium chloride in male and female mice.
Mutat. Res., 30: 363-374.
SUZUKI, T., SHISHIDO, S., & ISHIHARA, N. (1976) Interaction of
inorganic to organic mercury in their metabolism in human body.
Int. Arch. occup. environ. Health, 38: 103-113.
SUZUKI, T., TAKEMOTO, T.I., SHISHIDO, S., & KANI, K. (1977)
Mercury in human amniotic fluid. Scand. J. Work Environ. Health, 9:
32-35.
SUZUKI, T., HIMENO, S., HONGO, T., & WATANABE, C. (1986)
Mercuryselenium interaction in workers exposed to elemental
mercury vapor. J. appl. Toxicol., 6: 149-153.
SVARE, C.W., PETERSON, L.C., REINHARDT, J.W., BOYER, D.B.,
FRANK, C.W., GAY, D.D., & COX, R.D. (1981) The effect of dental
amalgams on mercury levels in expired air. J. dent. Res., 60(9): 1668-
1671.
SWEDISH ENVIRONMENTAL PROTECTION BOARD (1986) Mercury:
occurrence and turnover of mercury in the environment, Stockholm,
National Environmental Protection Board, (Mercury Report No. 3).
TAKAHATA, N., HAYASHI, H., WATANABE, S., & ANSO, T. (1970)
Accumulation of mercury in the brains of two autopsy cases with
chronic inorganic mercury poisoning. Folia psychiatr. neurol. Jpn.,
24(1): 59-69.
TAKIZAWA, Y. (1986) Mercury content in recognized patients
and non-recognized patients exposed to methylmercury from Minamata
Bay in the last ten years. In: Tsubaki, T. & Takahashi, H.,
ed. Recent advances in Minamata disease studies, Tokyo, Kodansha
Ltd., pp. 24-39.
THOMSON, J. & RUSSELL, J.A. (1970) Dermatitis due to mercury
following amalgam dental restorations. Br. J. Dermatol., 82: 292-297.
TRIEBIG, G. & SCHALLER, K.-H. (1982) Neurotoxic effects in mercury-
exposed workers. Neurobehav. Toxicol. Teratol., 4: 717-720.
TRIEBIG, G., SCHALLER, K.-H., & VALENTIN, H. (1981)
[Investigations on neurotoxicity of chemical substances at the
workplace. I. Determination of motor and sensory nerve conduction
velocity in persons occupationally exposed to mercury.] Int.
Arch. occup. environ. Health, 48: 119-129 (in German with English
summary).
TROEN, P., KAUFMAN, S.A., & KATZ, K.H. (1951) Mercuric
bichloride poisoning. New Eng. J. Med., 244(13): 459-463.
TUNNESSEN, W.W., Jr, MCMAHON, K.J., & BASER, M. (1987) Acrodynia:
exposure to mercury from fluorescent light bulbs. Pediatrics, 79(5):
786-789.
UMEDA, M. & NISHIMURA, M. (1979) Inducibility of chromosomal
aberrations by metal compounds in cultured mammalian cells. Mutat.
Res., 67: 221-229.
UNEP/WHO (1984) Principles and procedures for quality
assurance in environmental pollution: Exposure monitoring,
Geneva, World Health Organization (EFP/HEAL/84.4).
US ENVIRONMENTAL PROTECTION AGENCY (1989) Integrated risk
information system (IRIS): Mercury (inorganic) file data 9/01/81,
Cincinnati, Ohio, US Environmental Protection Agency,
Environmental Criteria and Assessment Office.
VAHTER, M. (1982) Assessment of human exposure to lead and cadmium
through biological monitoring, Stockholm, National Swedish
Institute of Environmental Medicine and Department of
Environmental Hygiene, Karolinska Institute, 136 pp. (Prepared for
the United Nations Environment Programme and the World Health
Organization).
VELASQUEZ, E., HASSAN, A., BELMAR, R., DRUCKER, E., & MICHAELS, D.
(1980) Occupational mercury poisoning - Nicaragua. CDC Mortal.
Morb. Wkly Rep., 29: 393-395.
VERBERK, M.M., SALLE, H.J.A., & KEMPER, C.H. (1986) Tremor in
workers with low exposure to metallic mercury. Am. Ind. Hyg. Assoc.
J., 47: 559-562.
VERSCHAEVE, L. & SUSANNE, C. (1979) Genetic hazards of mercury
exposure in dental surgery. Mutat. Res., 64: 149.
VERSCHAEVE, L., KIRSCH-VOLDERS, M., SUSANNE, C.,
GROETENBRIEL, C., HAUSTERMANS, R., LECOMTE, A., & ROOSSELS, D.
(1976) Genetic damage induced by occupationally low mercury
exposure. Environ. Res., 12: 303-316.
VERSCHAEVE, L., TASSIGNON, J.-P., LEFEVRE, M., DE STOOP, P., &
SUSANNE, C. (1979) Cytogenetic investigation on leucocytes of
workers exposed to metallic mercury. Environ. Mutagen., 1: 259-268.
VERSCHAEVE, I., KIRSCH-VOLDERS, M., & SUSANNE, C. (1984) Mercury-
induced segregational errors of chromosomes in human lymphocytes
and in Indian muntjac cells. Toxicol. Let., 21: 247-253.
VERSCHAEVE, I., KIRSCH-VOLDERS, M., HENS, L., & SUSANNE, C.
(1985) Comparative in vitro cytogenetic studies in mercury-
exposed human lymphocytes. Mutat. Res., 157: 221-226.
VILLALUZ, M.G. (1988) Environmental impact assessment of small
scale gold mining operations in Davao del Norte. Quezon City,
University of the Philippines, College of Engineering (Thesis).
VIMY, M.J. & LORSCHEIDER, F.L. (1985a) Intra-oral air mercury
released from dental amalgam. J. dent. Res., 64(8): 1069-1071.
VIMY, M.J. & LORSCHEIDER, F.L. (1985b) Serial measurements of
intra-oral air mercury: Estimation of daily dose from dental
amalgam. J. dent. Res., 64(8): 1072-1085.
VIMY, M.J., LUFT, A.J., & LORSCHEIDER, F.L. (1986) Estimation of
mercury body burden from dental amalgam: Computer simulation
of a metabolic compartmental model. J. dent. Res., 65(12): 1415-1419.
VIMY, M.J., TAKAHASHI, Y., & LORSCHEIDER, F.L. (1990a)
Maternal-fetal distribution of mercury (Hg203) released from
dental amalgam tooth restorations in sheep. Am. J. Physiol., 258
(in press)
VIMY, M.J., BOYD, N.D., HOOPER, D.E., & LORSCHEIDER, F.L.
(1990b) Glomerular filtration impairment by mercury released
from dental "silver" fillings in sheep. Physiologist, 33(4): abstract
94.
WAHLBERG, J.E. (1965) "Disappearance measurements", a method
for studying percutaneous absorption of isotope-labeled
compounds emitting gamma-rays. Acta dermato-venereol., 45: 397-414.
WALLINGFORD, K.M. (1982) Health hazard evaluation report,
Cincinnati, Ohio, National Institute for Occupational Safety and
Health, 7 pp. (HETA 82-364-1243).
WANDS, J.R., WEISS, S.W., YARDLEY, J.H., & MADDREY, W.C. (1974)
Chronic inorganic mercury poisoning due to laxative abuse. A
clinical and ultrastructural study. Am. J. Med., 57: 92-101.
WARKANY, J. (1966) Acrodynia - postmorten of a disease. Am.
J. Dis. Child., 112: 147-156.
WATANABE, T., SHIMADA, T., & ENDO, A. (1982) Effects of mercury
compounds on ovulation and meiotic and mitotic chromosomes in
female golden hamster. Teratology, 25: 381-384.
WEENING, J.J., FLEUREN, G.J., & HOEDEMAEKER, Ph.J. (1978)
Demonstration of antinuclear antibodies in mercuric chloride-
induced glomerulopahty in the rat. Lab. Invest., 39(4): 405-411.
WEENING, J.J., HOEDEMAEKER, J., & BAKKER, W.W. (1981)
Immunoregulation and antinuclear antibodies in mercury-induced
glomerulopathy in the rat. Clin. exp. Immunol., 45: 64-71.
WESSEL, W. (1967) [Electron microscopic contribution to the
acute and chronic mercury bichloride and viomycin poisoning of
the kidney.] Verh. Dtsch. Ges. Pathol., 51: 313-316 (In German with
English summary).
WHITE, R.R. & BRANDT, R.L. (1976) Development of mercury
hypersensitivity among dental students. J. Am. Dent. Assoc., 92: 1204-
1207.
WHO (1976) Environmental Health Criteria 1: Mercury, Geneva, World
Health Organization, 131 pp.
WHO (1980) Recommended health-based limits in occupational
exposure to heavy metals. Report of a WHO Study Group,
Geneva, World Health Organization, 116 pp. (WHO Technical Report
Series, No. 647).
WHO (1986) Guidelines for integrated air, water, food and
biological exposure monitoring, Geneva, World Health
Organization, UNEP/WHO Global Environmental Monitoring System,
HEAL Project (document PEP/86.6).
WHO (1989) Environmental Health Criteria 86: Mercury -
Environmental aspects, Geneva, World Health Organization, 115 pp.
WHO (1990) Environmental Health Criteria 101: Methylmercury,
Geneva, World Health Organization, 144 pp.
WOLFF, M., OSBORNE, J.W., & HANSON, A.L. (1983) Mercury
toxicity and dental amalgam. Neurotoxicology, 4(3): 201-204.
WU, D.L. (1989) [Atmospheric pollution and mercury poisoning
caused by peasants mercury smelting.] Chin. J. prev. Med., 23:
83-86 (in Chinese with summary in English).
YOSHIDA, M. & YAMAMURA, Y. (1982) Elemental mercury in urine from
workers exposed to mercury vapour. Int. Arch. occup. environ.
Health, 51: 99-104.
YOSHIDA, M., YAMAMURA, Y., & SATOH, H. (1986) Distribution of
mercury in guinea-pig offspring after in utero exposure to mercury
vapor during late gestation. Arch. Toxicol., 58: 225-228.
YOSHIDA, M., AOYAMA, H., SATOH, H., & YAMAMURA, Y. (1987)
Binding of mercury to metallothionein-like protein in fetal
liver of the guinea pig following in utero exposure to mercury
vapor. Toxicol. Lett., 37: 1-6.
YOSHIDA, M., SATOH, H., KOJIMA, S., & YAMAMURA, Y. (1989)
Distribution of mercury in neonatal guinea-pigs after exposure to
mercury vapours. Bull. environ. Contam. Toxicol., 43(5): 697-704.
YOSHIKAWA, H. & OHTA, H. (1982) Interaction of metals and
metallothionein. In: Foulkes, E.C., ed. Biological roles of
metallothionein, Amsterdam, Oxford, New York, Elsevier Science
Publishers, pp. 11-23.
ZAMPOLLO, A., BARUFFINI, A., CIRLA, A.M., PISATI, G., & ZEDDA, S.
(1987) Subclinical inorganic mercury neuropathy:
Neurophysiological investigations in 17 occupationally exposed
subjects. Ital. J. neurol. Sci., 8: 249-254.
ZASUKHINA, G.D., VASILYEVA, I.M., SDIRKOVA, N.I., KRASOVSKY,
G.N., VASYUKOVICH, L.Ya., KENESARIEV, U.I., & BUTENKO, P.G.
(1983) Mutagenic effect of thallium and mercury salts on rodent
cells with different repair activities. Mutat. Res., 124: 163-173.
ZEGLIO, P. (1958) [Long-term effects of mercurialism]. Lav. Um.,
X: 420-422 (in Italian).
ZIFF, S. (1984) Silver dental fillings - The toxic time bomb.
Can the mercury in your dental fillings poison you? New York,
Aurora Press, 150 pp.
RESUME ET CONCLUSIONS
La présente monographie est essentiellement consacrée
aux risques pour la santé humaine imputables au mercure
minéral; elle passe en revue les résultats des recherches
qui ont été publiés depuis la parution des Critères
d'hygiène de l'environnement No 1: Mercure (WHO, 1977).
Depuis 1977, on dispose de nouvelles données concernant la
présence de mercure dans les amalgames dentaires et dans
les savons éclaircissants, qui constituent deux importants
sujets de préoccupation. Dans la présente monographie on
insiste principalement sur l'exposition résultant de ces
deux utilisations, mais on étudie également les données
cinétiques et toxicologiques fondamentales susceptibles
d'être utiles dans l'étude de l'ensemble des effets du
mercure minéral.
En ce qui concerne la santé humaine, les problèmes
liés au transport, à la bioaccumulation et la transfor-
mation du mercure minéral à l'échelon planétaire provien-
nent presque exclusivement de sa conversion en méthylmer-
cure et de l'exposition ultérieure au méthylmercure par
l'intermédiaire des fruits de mer ou autres denrées ali-
mentaires. Les aspects environnementaux et écologiques
généraux du mercure minéral sont récapitulés dans la
présente monographie. On pourra trouver un exposé plus
détaillé de ces questions dans les Critères d'hygiène de
l'environnement No 86: Mercure - aspects écologiques (WHO,
1989) ainsi que dans les Critères d'hygiène de l'environ-
nement No 101: Méthylmercure (WHO, 1990).
1. Identité
Le mercure existe aux trois degrés d'oxydation sui-
vants: Hg0 (mercure métallique); Hg2++ (mercure mercu-
reux) et Hg++ (mercure mercurique). Il peut former des
dérivés organométalliques dont quelques-uns sont utilisés
dans l'industrie et en agriculture.
2. Propriétés physiques et chimiques
Le mercure élémentaire a une très forte tension de
vapeur. Dans l'atmosphère saturée à 20 °C, sa concen-
tration est 200 fois plus élevée que celle qui est actuel-
lement admise sur les lieux de travail.
La solubilité dans l'eau augmente selon la séquence:
mercure élémentaire < chlorure mercureux < chlorure de
méthylmercure < chlorure mercurique. Le mercure élémen-
taire ainsi que les dérivés halogénés des composés alkyl-
mercuriels sont solubles dans les solvants apolaires.
Les vapeurs de mercure sont plus solubles dans le
plasma, le sang total et l'hémoglobine que dans l'eau
distillée où la solubilité est très faible. Les dérivés
organométalliques sont stables mais certains d'entre eux
sont facilement dégradés par les organismes vivants.
3. Méthodes d'analyse
Les méthodes les plus couramment utilisées pour le
dosage du mercure total et du mercure minéral sont
l'absorption atomique de la vapeur froide (CVAA) et
l'activation neutronique. On trouvera un exposé détaillé
des méthodes d'analyse dans les Critères d'hygiène de
l'environnement No 1: Mercure (WHO, 1977) ainsi que dans
le No 101: Méthylmercure (WHO, 1990).
Pour toutes ces méthodes d'analyse, une assurance
minutieuse de la qualité est indispensable.
3.1 Analyses, prélèvements et conservation des urines
Pour l'analyse de routine des divers milieux, on a
recours à la spectrophotométrie d'absorption atomique sans
flamme. Il faut être spécialement prudent lors du choix
des anticoagulants utilisés pour les prélèvements sanguins
afin d'éviter toute contamination par des dérivés mercu-
riels. Des précautions particulières sont également néces-
saires lors du prélèvement et de la conservation des
urines car la croissance bactérienne peut modifier la con-
centration des nombreuses formes de mercure susceptibles
d'être présentes dans les urines. Pour éviter l'altération
des échantillons d'urine, la meilleure méthode consiste à
les additionner d'acide chlorhydrique ou d'un bactéricide
puis de congeler l'échantillon. Il est recommandé de
procéder à une correction en concentration relativement à
la densité des urines ou à la teneur en créatinine.
3.2 Analyses et échantillonnage de l'air
Le dosage du mercure dans l'air peut s'effectuer soit
par des méthodes à lecture instantanée soit par des
méthodes qui comportent deux phases distinctes: échantil-
lonnage et analyse. Les méthodes à lecture instantanée
peuvent être utilisées pour le dosage des vapeurs de mer-
cure. Pour le dosage du mercure total, l'échantillonnage
s'effectue en milieu oxydant acide ou sur hopcalite.
Le dosage par absorption atomique de la vapeur froide
(CVAA) est la méthode la plus fréquemment utilisée.
4. Sources d'exposition humaine et environnementale
4.1 Etat naturel
Le mercure présent dans la nature provient principale-
ment du dégazage de la croûte terrestre, des éruptions
volcaniques et de l'évaporation des étendues d'eau.
Les émissions d'origine naturelle sont de l'ordre de
2700 à 6000 tonnes par an.
4.2 Sources d'origine humaine
On estime à 10 000 tonnes la quantité de mercure
extraite chaque année dans le monde. Cette activité
entraîne un certain nombre de pertes dans l'environnement
ainsi qu'une décharge directe dans l'atmosphère. Parmi les
autres sources importantes de pollution par le mercure, on
compte l'utilisation des combustibles fossiles, le gril-
lage des minerais métalliques sulfurés, le raffinage de
l'or, la production de ciment, l'incinération des déchets
et diverses opérations métallurgiques.
Une installation de production électrolytique de
chlore et de soude donne normalement lieu à des émissions
de mercure de l'ordre de 450 g par tonne de soude causti-
que produite.
La quantité totale libérée annuellement dans l'atmos-
phère de la planète par suite d'activités humaines atteint
quelque 3000 tonnes.
5. Usages
Le mercure est principalement utilisé comme cathode
dans l'électrolyse du chlorure de sodium. Etant donné que
les produits de cette électrolyse sont contaminés par du
mercure, leur emploi dans des opérations industrielles
ultérieures provoque la contamination d'autres produits.
Le mercure est également utilisé dans l'industrie élec-
trique, pour la fabrication d'instruments de mesure
utilisés dans les ménages ou dans l'industrie ainsi que
pour la fabrication d'instruments de laboratoire et
d'appareils médicaux. Certains médicaments contiennent du
mercure minéral. On utilise également une très grande
quantité de mercure pour l'extraction de l'or.
Les amalgames utilisés en art dentaire pour l'obtura-
tion des dents contiennent une grande quantité de mercure
mélangée (en proportion de 1:1) avec un alliage pulvéru-
lent à base d'argent, d'étain, de cuivre et de zinc.
L'amalgame au cuivre, utilisé essentiellement pour les
soins dentaires aux enfants, contient jusqu'à 70% de
mercure et jusqu'à 30% de cuivre. Il peut en résulter une
exposition du dentiste, de ses assistants et des patients
au mercure.
Certaines personnes de couleur utilisent des crèmes et
des savons à base de mercure pour s'éclaircir la peau.
Ces produits sont désormais interdits dans la Communauté
européenne, en Amérique du Nord et dans de nombreux pays
d'Afrique mais on fabrique encore des savons à base de
mercure dans plusieurs pays d'Europe. Les savons contien-
nent jusqu'à 3% d'iodure de mercure et les crèmes jusqu'à
10% de mercure ammoniacal.
6. Transport, distribution et transformation dans l'environnement
Le mercure émis dans l'atmosphère sous forme de vapeur
est transformé en dérivés solubles et il se dépose avec
les précipitations sur le sol et dans l'eau. La vapeur de
mercure peut subsister jusqu'à trois ans dans l'atmos-
phère, cette période étant réduite à quelques semaines
dans le cas des formes solubles.
La première étape du processus de bioaccumulation
aquatique consiste dans la transformation du mercure
minéral en méthylmercure. Cette transformation s'opère
soit par voie non enzymatique soit sous l'action de micro-
organismes. Le méthylmercure pénètre dans la chaîne ali-
mentaire des espèces prédatrices où il subit une bioampli-
fication.
7. Exposition humaine
C'est principalement par l'intermédiaire des aliments
et des amalgames dentaires que la population générale est
exposée au mercure. En fonction de l'importance de sa
concentration dans l'air et dans l'eau, la dose totale
ingérée quotidiennement peut s'en trouver notablement
augmentée. Le poisson constitue la source principale
d'exposition humaine au méthylmercure. Des études expéri-
mentales récentes ont montré que le mercure libéré dans la
cavité buccale à partir d'un amalgame l'est sous forme de
vapeur. La mastication augmente la vitesse de libération
de ces vapeurs. Un certain nombre d'études ont montré
qu'il y avait une corrélation entre le nombre d'obtura-
tions au moyen d'un amalgame ou de surfaces recouvertes
d'amalgame et la teneur en mercure des tissus, mesurée
après autopsie, ainsi que la teneur en mercure du sang,
des urines et du plasma. L'absorption de mercure calculée
à partir de la quantité d'amalgame et l'accumulation
effectivement observée présente d'importantes variations
individuelles. Il est donc difficile de procéder à des
estimations précises de la quantité de mercure provenant
des amalgames dentaires qui finit par se fixer dans
l'organisme. Des études expérimentales effectuées sur des
moutons ont permis d'étudier plus en détail la distri-
bution du mercure provenant des amalgames dentaires.
L'utilisation de savons et de crèmes pour s'éclaircir
la peau peut également donner lieu à une importante expo-
sition.
On a étudié l'exposition professionnelle au mercure
minéral dans les unités d'électrolyse du chlorure de
sodium, dans les mines de mercure, les fabriques de
thermomètres, les raffineries et les cabinets dentaires.
Pour tous ces types d'exposition professionnelle, on a
relevé d'importantes quantités de mercure, mais celles-ci
varient en fonction des ambiances de travail.
8. Cinétique et métabolisme
Les études sur l'homme et l'animal montrent qu'après
inhalation de vapeur de mercure, la proportion retenue par
l'organisme est d'environ 80% alors qu'elle est inférieure
à 1% lorsque le mercure métallique est ingéré sous forme
liquide, ce qui témoigne d'une faible absorption dans les
voies digestives. Après inhalation, les aérosols de
mercure minéral se déposent dans les voies respiratoires
et sont absorbés à une vitesse qui dépend de la taille des
particules. Il est probable que les composés minéraux du
mercure sont absorbés dans les voies digestives dans une
proportion qui est en moyenne inférieure à 10%, mais là
encore les variations individuelles sont considérables.
L'absorption est beaucoup plus élevée chez les ratons
nouveau-nés.
C'est principalement au niveau des reins que se dépose
le mercure après administration de vapeur de mercure
élémentaire ou de dérivés minéraux du mercure (cela repré-
sente 50 à 90% de la charge totale de l'organisme chez
l'animal). Après inhalation de mercure élémentaire on
observe que la quantité de mercure qui passe dans le
cerveau, chez des souris et des singes, est nettement plus
élevée qu'après injection intraveineuse équivalente sous
forme de sels mercuriques. Chez l'homme, le rapport
hématies/plasma est plus élevé (> 1) après administration
de mercure élémentaire qu'après administration d'un sel
mercurique et la quantité de mercure qui traverse la
barrière placentaire est plus importante. Seule une faible
fraction de la quantité de mercure administrée sous forme
de sels bivalents pénètre dans l'organisme du foetus de
rat.
Plusieurs types de transformation métabolique peuvent
se produire:
* oxydation du mercure métallique en mercure (II);
* réduction du mercure (II) en mercure métallique;
* méthylation du mercure minéral;
* conversion du méthylmercure en mercure minéral bivalent.
L'oxydation des vapeurs de mercure métallique en
mercure ionique bivalent (section 6.1.1) n'est pas suffi-
samment rapide pour empêcher le passage du mercure
élémentaire à travers la barrière hémo-méningée, à travers
le placenta ou d'autres tissus. Dans ces tissus, l'oxyda-
tion piège le mercure qui s'accumule dans le cerveau et
les tissus du foetus.
La réduction du mercure (II) en mercure (0) a été
observée tant chez l'animal (rats et souris) que chez
l'homme. Inversement, la décomposition d'organomercuriels
tels que le méthylmercure constitue une source de mercure
(II).
C'est principalement par la voie fécale et par la voie
urinaire que s'élimine chez l'homme le mercure minéral
encore qu'il puisse être exhalé en petites quantités sous
forme élémentaire. Il peut également se produire une
déplétion tissulaire par transfert des tissus maternels à
ceux du foetus.
La demi-vie biologique, qui pour la majeure partie du
mercure s'étend de quelques jours à quelques semaines,
peut être très longue - jusqu'à plusieurs années - pour la
fraction restante. Ces demi-vies très longues ont été
observées tant chez l'animal que chez l'homme. Il se
produit une interaction complexe entre le mercure et
certains autres éléments, notamment le sélénium. Il se
pourrait que la très longue demi-vie d'élimination d'une
fraction du mercure s'explique par la formation d'un
complexe avec le sélénium.
8.1 Valeurs de référence et valeurs normales
Les quelques données dont on dispose à propos de
mineurs décédés montrent que plusieurs années après
l'arrêt de l'exposition, la concentration du mercure dans
le cerveau était encore de plusieurs mg/kg, avec des
valeurs encore plus élevées dans certaines zones. Toute-
fois cette analyse n'ayant pas fait l'objet d'un contrôle
de qualité, les données demeurent incertaines. Chez un
petit nombre de dentistes examinés après leur mort et qui
ne présentaient pas de symptômes d'hydrargyrisme, on a
observé que les teneurs en mercure allaient de très
faibles concentrations jusqu'à des valeurs de quelques
centaines de µg/kg dans le cortex du lobe occipital et
d'environ 100 µg/kg à quelques mg/kg dans l'hypophyse.
L'examen post-mortem de sujets qui n'étaient pas pro-
fessionnellement exposés au mercure mais étaient porteurs
d'un certain nombre d'obturations au moyen d'amalgame, a
montré qu'un nombre modéré (environ 25) de surfaces recou-
vertes d'amalgame augmentent en moyenne la concentration
cérébrale du mercure d'à peu près 10 µg/kg. L'augmen-
tation correspondante au niveau des reins, déterminée au
moyen d'un nombre très limité d'analyses est probablement
de 300 à 400 µg/kg. Toutefois les variations individuel-
les sont considérables.
La concentration du mercure dans les urines et le sang
peut être utilisée comme indicateur de l'exposition, à
condition que celle-ci soit relativement constante,
qu'elle soit prolongée et déterminée sur un groupe de
sujets. Les données récentes sont plus fiables que celles
dont il est fait état dans les Critères d'hygiène de
l'environnement No 1: Mercure (WHO, 1977). Après exposi-
tion professionnelle à des quantités de mercure d'environ
40 µg/m3 d'air, on observe des concentrations urinaires
d'environ 50 µg/g de créatinine. Ce rapport (5:4) entre
les concentrations urinaires et les concentrations
atmosphériques est beaucoup plus faible que le rapport de
3:1 auquel étaient parvenus les experts de WHO (1977). La
différence peut s'expliquer en partie par des variations
dans les techniques d'échantillonnage utilisées pour
calculer l'exposition atmosphérique. Une exposition de
l'ordre de 40 µg/m3 d'air correspond à environ
15-20 µg de mercure par litre de sang. Toutefois, il peut
être difficile d'évaluer l'exposition à de faibles concen-
trations de mercure inorganique par analyse du sang
lorsqu'il y a exposition simultanée au méthylmercure. Pour
lever la difficulté, on peut procéder au dosage du mercure
dans le plasma ou doser simultanément le mercure minéral
et le méthylmercure. Le méthylmercure est beaucoup moins
gênant lorsqu'on procède à une analyse d'urine car il
n'est excrété dans les urines qu'en très faible proportion.
9. Effets chez l'homme
Une exposition aiguë au mercure par inhalation de
vapeurs peut occasionner des douleurs thoraciques, de la
dyspnée, de la toux, une hémoptysie et quelquefois provo-
quer une pneumonie interstitielle mortelle. L'ingestion
de dérivés mercuriques, en particulier de chlorure
mercurique, peut provoquer une gastro-entérite ulcérative
et une nécrose tubulaire aiguë suspectible d'entraîner la
mort par anurie si l'on ne dispose pas de moyens de
dialyse.
En cas d'exposition aux vapeurs de mercure, c'est le
système nerveux central qui constitue l'organe critique.
L'exposition subaiguë peut entraîner des réactions psycho-
tiques caractérisées par un délire, des hallucinations et
une tendance suicidaire. L'exposition professionnelle peut
conduire à des troubles fonctionnels très variés dont
l'éréthisme constitue la caractéristique essentielle.
Lorsque l'exposition se poursuit, on voit apparaître de
légers tremblements, initialement au niveau des mains.
Dans les cas bénins d'éréthisme, ces tremblements régres-
sent lentement en quelques années après cessation de
l'exposition. On a constaté chez des travailleurs exposés
au mercure une diminution de la vitesse de conduction
nerveuse. Des symptômes d'éréthisme moins prononcés ont
été observés à la suite d'une exposition prolongée à de
faibles concentrations.
On connaît très mal les concentrations de mercure dans
le cerveau dans les cas d'hydrargyrisme et on ne peut pas
évaluer la dose sans effet observable ni tracer une courbe
dose-réponse.
Lorsque le taux d'excrétion urinaire du mercure atte-
int 100 µg/g de créatinine, il existe une forte probabi-
lité pour qu'apparaissent les signes neurologiques classi-
ques de l'hydraargyrisme (tremblements, éréthisme) et l'on
note une forte protéinurie. Une exposition de 30 à 100 µg
de mercure/g de créatinine entraîne une incidence accrue
de certains effets toxiques de moindre gravité qui ne se
traduisent pas par une détérioration clinique manifeste.
Dans quelques études, on a observé des tremblements,
enregistrés par voie électrophysiologique, à des concen-
trations faibles dans l'urine (pouvant s'abaisser jusqu'à
25-35 µg/g de créatinine). En revanche, d'autres études
n'ont pas mis cet effet en évidence. Certaines des
personnes exposées font une protéinurie (protéines de
faibles masse moléculaire relative et micro-albuminurie).
On ne dispose pas de données épidémiologiques appropriées
pour les taux d'exposition qui correspondent à moins de
30-50 µg de mercure/g de créatinine.
L'exposition de la population générale est en principe
faible mais dans certains cas, elle peut atteindre les
valeurs constatées dans les ambiances de travail et même
conduire à des intoxications. C'est ainsi que des erreurs
de manipulation de mercure liquide ont pu conduire à de
graves intoxications.
Après ingestion de sels de mercure bivalent, c'est le
rein qui est l'organe critique. On sait depuis longtemps
que l'exposition professionnelle au mercure métallique
entraîne une protéinurie tant chez les travailleurs
présentant des signes d'hydrargyrisme que chez ceux qui
sont asymptomatiques. On observe moins fréquemment un
syndrome néphrotique, syndrome qui peut également se pro-
duire après utilisation de crèmes à base de mercure pour
s'éclaircir la peau et même après une exposition acciden-
telle. Il semblerait d'après les données actuelles que ce
syndrome néphrotique soit dû à une réaction immunotoxique.
Jusqu'à ces derniers temps, on n'avait signalé d'effets
néphrotoxiques de la vapeur de mercure qu'à des doses
supérieures à celles qui entraînent l'apparition de
symptômes centraux. Cependant des études nouvelles font
état d'effets rénaux à des concentrations plus faibles.
L'expérimentation animale montre que le mercure minéral
peut provoquer une glomérulonéphrite auto-immune. Cet
effet s'observe chez toutes les espèces à l'exception de
certaines souches, ce qui indique l'existence d'une pré-
disposition génétique. Une étiologie immunologique a pour
conséquence, en l'absence d'études dose-réponse sur des
groupes d'individus immunologiquement réceptifs,
l'impossibilité d'établir scientifiquement la dose limite
de mercure (par exemple dans le sang ou les urines) en
dessous de laquelle (dans les cas individuels) il n'y aura
pas de symptômes d'hydrargyrisme.
Les vapeurs de mercure et les dérivés mercuriels
peuvent provoquer des dermatites de contact. Des produits
pharmaceutiques à base de mercure ont provoqué des cas
d'acrodynie infantile et on tient l'exposition aux vapeurs
de mercure pour responsable de la maladie de "Kawasaki".
Certaines études, contrairement à d'autres, ont mis en
évidence des effets sur le cycle menstruel et sur le
développement foetal. Il ressort des études épidémiologi-
ques qui ont été publiées qu'il n'y a pour l'instant pas
de réponse à la question de savoir si, en l'absence des
signes bien connus de l'intoxication mercurielle, la
vapeur de mercure peut avoir des effets nocifs sur le
cycle menstruel ou le développement foetal.
Récemment, on a beaucoup débattu de la sécurité des
amalgames utilisés en art dentaire et certains ont avancé
que l'emploi d'amalgames à base de mercure comportait de
graves dangers pour la santé. Les rapports qui font état
de différents types de symptômes, de même que les résul-
tats des quelques études épidémiologiques qui ont été
effectuées, ne sont pas concluants.
RESUMEN Y CONCLUSIONES
La presente monografía se centra principalmente en el
riesgo que representa el mercurio inorgánico para la salud
humana y en ella se examinan los informes de investigación
aparecidos desde la publicación de Criterios de Salud
Ambiental 1: Mercurio (WHO, 1976) (versión española publi-
cada en 1978). Desde 1976, han ido apareciendo nuevos
datos de investigación sobre dos importantes cuestiones
de salud relacionadas con el mercurio inorgánico, a saber,
el mercurio presente en la amalgama de uso odontológico y
en los jabones destinados a aclarar la piel. La presente
monografía se centra en la exposición a esas dos fuentes,
pero se examinan los aspectos cinéticos y toxicológicos
elementales teniendo presentes todos los aspectos del
mercurio inorgánico.
Los efectos sobre la salud humana relacionados con el
transporte mundial, la bioacumulación y la transformación
del mercurio inorgánico se derivan casi exclusivamente de
la conversión de los compuestos de mercurio en metilmer-
curio y de la exposición al metilmercurio en los alimentos
de origen marino y otros alimentos. En la presente mono-
grafía se han resumido los aspectos ambientales y ecológi-
cos mundiales del mercurio inorgánico. Pueden encontrarse
descripciones más detalladas en Criterios de Salud Ambien-
tal 86: Mercurio - Aspectos Ambientales (WHO, 1989) y
Criterios de Salud Ambiental 101: Metilmercurio (WHO,
1990).
1. Identificación
El mercurio existe en tres estados: Hg0 (metálico);
Hg2++ (mercurioso); y Hg++ (mercúrico). Puede formar
compuestos organometálicos, algunos de los cuales tienen
usos industriales y agrícolas.
2. Propiedades físicas y químicas
El mercurio elemental tiene una presión de vapor
sumamente elevada. La atmósfera saturada a 20 °C tiene una
concentración más de 200 veces superior a la de la concen-
tración comúnmente aceptada para la exposición profesional.
La solubilidad en el agua aumenta en el orden sigui-
ente: mercurio elemental < cloruro mercurioso < cloruro de
metilmercurio < cloruro mercúrico. El mercurio elemental
y los haluros de compuestos alquilmercuriales son solubles
en disolventes no polares.
El vapor de mercurio es más soluble en plasma, sangre
entera y hemoglobina que en agua destilada, donde sólo se
disuelve ligeramente. Los compuestos organometálicos son
estables, aunque algunos son fácilmente descompuestos por
los organismos vivos.
3. Métodos analíticos
Los métodos analíticos más utilizados para cuantificar
los compuestos de mercurio total e inorgánico son la
absorción atómica sobre vapor frío (AAVF) y la activación
de neutrones. Puede encontrarse información detallada
sobre los métodos analíticos en Criterios de Salud
Ambiental 1: Mercurio (WHO, 1978) y en Criterios de Salud
Ambiental 101: Metilmercurio (WHO, 1990).
Todo análisis del mercurio requiere una rigurosa
garantía de calidad.
3.1 Análisis, muestreo y conservación de la orina
La espectrofotometría de absorción atómica sin llama
se utiliza en los análisis ordinarios para los diversos
medios. Debe tenerse especial cuidado al elegir el anti-
coagulante para el muestreo de sangre a fin de evitar la
contaminación por compuestos de mercurio. También debe
procederse con suma precaución en el muestreo y el
almacenamiento de la orina, puesto que el crecimiento
bacteriano es capaz de modificar la concentración de las
numerosas formas de mercurio que pueden estar presentes.
La adición de ácido clorhídrico o sustancias bactericidas
y la congelación son los mejores métodos para impedir la
alteración de las muestras de orina. Se recomienda corre-
gir la concentración por referencia a la densidad de la
orina o al contenido de creatinina.
3.2 Análisis y muestreo del aire
Los métodos analíticos del mercurio en el aire pueden
dividirse en métodos de lectura inmediata y métodos con
etapas separadas de muestreo y análisis. Los métodos de
lectura inmediata pueden utilizarse para cuantificar el
vapor de mercurio elemental. El muestreo en medios
acidoxidantes o con hopcalita se usan para cuantificar el
mercurio total.
La técnica (AAVF) es el método analítico más frecuente.
4. Fuentes de exposición humana y medioambiental
4.1 Fuentes naturales
Las principales fuentes naturales del mercurio son la
desgasificación de la corteza terrestre, las emisiones
volcánicas y la evaporación de las masas acuáticas
naturales.
Las emisiones naturales son del orden de 2700-6000
toneladas al año.
4.2 Fuentes debidas a la actividad humana
Se estima que la extracción minera del mercurio pro-
duce en todo el mundo alrededor de 10 000 toneladas/año.
Estas actividades originan ciertas pérdidas de mercurio y
descargas directas a la atmósfera. Otras fuentes import-
antes son la utilización de combustibles fósiles, la
fundición de metales con minerales de sulfuro, el refinado
del oro, la producción de cemento, la incineración de
desechos y las aplicaciones industriales de los metales.
La emisión normal específica de las industrias de
compuestos alcalinos del cloro es de aproximadamente 450 g
de mercurio por tonelada de sosa cáustica producida.
La cantidad y descarga mundial total de mercurio en la
atmósfera debida a las actividades humanas representa
hasta 3000 toneladas/año.
5. Usos
Uno de los principales usos del mercurio es como
cátodo en la electrólisis del cloruro sódico. Como los
compuestos químicos resultantes quedan contaminados con
mercurio, su utilización en otras actividades industriales
origina la contaminación de otros productos. El mercurio
se emplea en la industria eléctrica, en instrumentos de
control en el hogar y la industria, y en instrumental
médico y de laboratorio. Algunos agentes terapéuticos
contienen mercurio inorgánico. En la extracción de oro se
utilizan grandes cantidades de mercurio.
La amalgama odontológica de plata para la obturación
de dientes contiene grandes cantidades de mercurio,
mezclado (en la proporción 1:1) con polvo de aleación
(plata, estaño, cobre, zinc). La amalgama de cobre, que se
utiliza sobre todo en odontología pediátrica, contiene
hasta el 70% de mercurio y hasta el 30% de cobre. Estos
usos pueden causar la exposición del dentista, los
ayudantes y también de los pacientes.
Algunas personas de piel oscura utilizan cremas y
jabones que contienen mercurio para conseguir un tono de
piel más claro. La distribución de esos productos está
actualmente prohibida en la CEE, en América del Norte y en
muchos países africanos, pero en varios países europeos se
sigue fabricando jabón con mercurio. Estos jabones con-
tienen hasta un 3% de yoduro de mercurio y las cremas
contienen mercurio amoniacal (hasta el 10%).
6. Transporte, distribución y transformación en el medio ambiente
El vapor de mercurio emitido se convierte en formas
solubles que son depositadas por la lluvia en el suelo y
el agua. El tiempo de persistencia atmosférica para el
vapor de mercurio es de hasta tres años, mientras que el
de las formas solubles es de sólo algunas semanas.
El cambio de especiación del mercurio desde las formas
inorgánicas hasta las metiladas es la primera etapa del
proceso de bioacumulación acuática. Esto puede suceder
sin el concurso de enzimas o mediante la acción micro-
biana. El metilmercurio ingresa en la cadena alimentaria
de las especies predadoras en las que se produce biomagni-
ficación.
7. Exposición humana
La población general está principalmente expuesta al
mercurio por la dieta y la amalgama odontológica.
Atendiendo a las concentraciones en la atmósfera y en el
agua, pueden producirse contribuciones importantes a la
ingesta diaria de mercurio total. El pescado es una de
las fuentes principales de exposición humana al metilmer-
curio. En estudios experimentales recientes se ha demo-
strado que el mercurio se libera en forma de vapor desde
las restauraciones con amalgama en la boca. La tasa de
liberación de este vapor de mercurio aumenta, por ejemplo,
al masticar. Varios estudios han correlacionado el número
de obturaciones con amalgama odontológica o de superficies
de amalgama con el contenido de mercurio en tejidos
obtenidos en la autopsia humana, así como en muestras de
sangre, orina y plasma. Tanto la ingesta prevista de
mercurio a partir de la amalgama como la acumulación
observada de mercurio demuestran importantes variaciones
individuales. Así pues, resulta difícil cuantificar con
exactitud la liberación y la ingestión de mercurio por el
cuerpo humano a partir de las restauraciones odontológicas
con amalgama. Los estudios experimentales realizados en
ovejas han examinado con mayor detalle la distribución del
mercurio liberado de las restauraciones con amalgama.
El uso de jabón y cremas para aclarar la piel puede
ser origen de una importante exposición al mercurio.
La exposición profesional al mercurio inorgánico se ha
estudiado en plantas industriales de productos cloral-
calinos, minas de mercurio, fábricas de termómetros,
refinerías y consultorios odontológicos. Se han comunicado
elevados niveles de mercurio respecto de todas estas situ-
aciones de exposición profesional, si bien los niveles
varían en virtud de las condiciones del entorno laboral.
8. Cinética y metabolismo
Los resultados de los estudios realizados tanto en
personas como en animales indican que alrededor del 80%
del vapor de mercurio metálico inhalado es retenido por el
organismo, mientras que el mercurio metálico líquido se
absorbe mal en el tracto gastroinstestinal (menos del 1%).
Los aerosoles de mercurio inorgánico inhalados se depo-
sitan en el tracto respiratorio y son absorbidos a una
velocidad que depende del tamaño de las partículas. Los
compuestos de mercurio inorgánico probablemente son
absorbidos desde el tracto gastrointestinal humano hasta
un nivel inferior al 10%, en promedio, pero la variación
individual es considerable. La absorción es mucho más
elevada en la rata recién nacida.
El riñón es el depósito principal de mercurio tras la
administración de vapor de mercurio elemental o de compu-
estos de mercurio inorgánico (50-90% de la carga corporal
de los animales). De modo significativo, más mercurio es
transportado al cerebro del ratón y el mono tras la inha-
lación de mercurio elemental que tras la inyección intra-
venosa de dosis equivalentes de la forma mercúrica. El
cociente hematíes:plasma en el hombre es mayor (> 1) tras
la administración de mercurio elemental que tras la de
mercurio mercúrico, y la proporción de mercurio que atra-
viesa la barrera placentaria es mayor. Sólo una pequeña
fracción del mercurio bivalente administrado ingresa en el
feto de la rata.
Pueden producirse varias formas de transformación
metabólica:
* oxidación del mercurio metálico a mercurio bivalente;
* reducción del mercurio bivalente a mercurio metálico;
* metilación del mercurio inorgánico;
* conversión del metilmercurio en mercurio inorgánico
bivalente.
La oxidación de vapor de mercurio metálico a mercurio
iónico bivalente (sección 6.1.1) no es lo bastante rápida
como para impedir el paso de mercurio elemental a través
de la barrera hematoencefálica, la placenta y otros
tejidos. La oxidación en esos tejidos sirve como filtro
para retener el mercurio y lleva a su acumulación en el
cerebro y los tejidos fetales.
La reducción del mercurio bivalente a Hg0 se ha
demostrado tanto en animales (ratones y ratas) como en el
hombre. La descomposición de los compuestos organomer-
curiales, incluido el metilmercurio, es también una fuente
de mercurio mercúrico.
Las rutas fecal y urinaria son las principales vías de
eliminación del mercurio inorgánico en el hombre, si bien
se exhala una pequeña cantidad de mercurio elemental. Una
forma de eliminación de mercurio es la transferencia del
mercurio materno al feto.
La semivida biológica, que dura sólo unos cuantos días
o semanas para la mayor parte del mercurio absorbido, es
sumamente larga, probablemente de años, para una parte del
mercurio. Esas largas semividas se han observado en exper-
imentos realizados con animales así como en el hombre.
Existe una complicada interacción entre el mercurio y
algunos elementos, incluido el selenio. La formación de
un complejo con el selenio puede ser responsable de la
larga semivida que tiene una parte del mercurio.
8.1 Valores de referencia y normales
La limitada información de que se dispone sobre mine-
ros fallecidos muestra la existencia de concentraciones de
mercurio en el cerebro de varios mg/kg, años después de
finalizar la exposición, con valores aún más altos en
algunas partes del cerebro. No obstante, la falta de
control de la calidad en el análisis hace inciertos estos
datos. Entre un pequeño número de dentistas fallecidos,
sin síntomas conocidos de intoxicación por mercurio, los
niveles de éste variaron desde concentraciones muy bajas
hasta varios cientos de µg/kg en la corteza del lóbulo
occipital y desde unos 100 µg/kg hasta unos cuantos mg/kg
en la hipófisis.
De las autopsias realizadas en sujetos no expuestos
profesionalmente pero con un número variable de obtura-
ciones con amalgama, se desprende que un número moderado
(alrededor de 25) de superficies de amalgama pueden, en
promedio, aumentar la concentración de mercurio en el
cerebro en unos 10 µg/kg. El aumento correspondiente en
el riñón, basado en un número muy limitado de análisis, es
probablemente de 300-400 µg/kg. Sin embargo, la variación
individual es considerable.
Los niveles de mercurio en la orina y la sangre pueden
usarse como indicadores de la exposición, siempre que ésta
sea relativamente constante a largo plazo y se evalúe en
un grupo. Los datos de exposición recientes son más
fiables que los que se citan en Criterios de Salud Ambien-
tal 1: Mercurio (WHO, 1978). Se observan niveles en la
orina de unos 50 µg/g de creatinina tras la exposición
profesional a unos 40 µg de mercurio por m3 de aire.
Esta relación (5:4) entre orina y niveles atmosféricos es
mucho más baja que la de 3:1 estimada por la WHO (1976).
La diferencia puede deberse en parte a la distinta técnica
de muestreo para evaluar la exposición atmosférica. Una
exposición de 40 µg de mercurio/m3 de aire corresponderá
a unos 15-20 µg de mercurio/litro de sangre. Sin embargo,
la interferencia debida a la exposición al metilmercurio
puede hacer más difícil evaluar la exposición a bajas
concentraciones de mercurio inorgánico por medio de
análisis de sangre. Una forma de salvar esos problemas es
analizar el mercurio en el plasma o analizar tanto el
mercurio inorgánico como el metilmercurio. El problema de
la interferencia debida al metilmercurio es mucho menor
cuando se analiza la orina, puesto que el metilmercurio se
excreta con la orina en grado sumamente reducido.
9. Efectos en el hombre
La exposición aguda por inhalación de vapores de
mercurio puede verse seguida por dolores de pecho, disnea,
tos, hemoptisis, y a veces pneumonitis intersticial que
puede provocar la muerte. La ingestión de compuestos
mercúricos, en particular cloruro mercúrico, ha provocado
casos de gastroenteritis ulcerativa y necrosis tubular
aguda, con muerte por anuria en los casos en que no se
dispuso de diálisis.
El sistema nervioso central es el órgano crítico para
la exposición al vapor de mercurio. La exposición subaguda
ha dado origen a reacciones psicóticas caracterizadas por
delirio, alucinaciones y tendencias suicidas. La exposi-
ción profesional origina eretismo como principal carac-
terística de un trastorno funcional de amplio espectro.
Si prosigue la exposición, se presenta un temblor fino,
que al principio afecta a las manos. En los casos más
leves el eretismo y el temblor desaparecen poco a poco a
lo largo de varios años, una vez interrumpida la exposi-
ción. Se ha demostrado en trabajadores expuestos al
mercurio una menor velocidad de conducción nerviosa. La
exposición a bajos niveles durante periodos largos se ha
asociado a síntomas de eretismo menos pronunciados.
Se dispone de muy poca información sobre los niveles
cerebrales de mercurio en los casos de envenenamiento, y
no se sabe nada que permita estimar una concentración
carente de efectos observados o una curva dosis-respuesta.
Cuando el nivel de excreción urinaria de mercurio es
de 100 µg/g de creatinina, hay una probabilidad muy
alta de que aparezcan los signos neurológicos clásicos de
la intoxicación por mercurio (temblor, eretismo) y
proteinuria. Una exposición correspondiente a 30 hasta
100 µg de mercurio/g de creatinina aumenta la incidencia
de algunos efectos tóxicos menos graves que no provocan
trastornos clínicos manifiestos. En algunos estudios se
han observado temblores, electrofisiológicamente regis-
trados, a concentraciones urinarias reducidas (tan bajas
como 25-35 µg/g de creatinina). En otros estudios no se
observó ese efecto. Algunas de las personas expuestas
presentan proteinuria (proteínas de baja masa molecular
relativa y microalbuminuria). No se dispone de datos
epidemiológicos adecuados sobre los niveles de exposición
que corresponden a menos de 30-50 µg de mercurio/g de
creatinina.
Aunque la exposición de la población general es por lo
general reducida, en ocasiones puede elevarse hasta el
nivel de exposición profesional y puede incluso llegar a
ser tóxica. Así, la manipulación incorrecta de mercurio
líquido ha dado origen a casos graves de intoxicación.
El riñón es el órgano crítico tras la ingestión de
sales de mercurio bivalente inorgánico. La exposición
profesional a mercurio metálico se asocia desde hace
tiempo a la aparición de proteinuria, tanto en obreros con
otros síntomas de envenenamiento por mercurio como en
aquéllos sin esos síntomas. En otros casos menos frecu-
entes, la exposición profesional se ha visto seguida del
síndrome nefrótico, que también se ha producido tras el
uso de cremas para aclarar la piel con mercurio inorgá-
nico, e incluso tras la exposición accidental. Las pruebas
actuales sugieren que este síndrome nefrótico se debe a
una respuesta inmunotóxica. Hasta hace poco, los efectos
del vapor de mercurio elemental en el riñón se habían
comunicado sólo con respecto a dosis más elevadas que las
asociadas a la aparición de signos y síntomas del sistema
nervioso central. En los nuevos estudios, no obstante, se
han notificado efectos en el riñón con niveles inferiores
de exposición. Los estudios experimentales con animales
han demostrado que el mercurio puede inducir glomerulone-
fritis autoinmune en todas las especies ensayadas, pero no
en todas las estirpes, lo que indica una predisposición
genética. Una de las consecuencias de la etiología inmuno-
lógica es que, en ausencia de estudios de la dosis-
respuesta en grupos de individuos inmunológicamente sensi-
bles, resulta científicamente imposible establecer un
nivel de mercurio (por ejemplo, en la sangre o la orina)
por debajo del cual (en casos individuales) no se
producirán síntomas relacionados con el mercurio.
Tanto los vapores de mercurio metálico como los
compuestos de mercurio han dado origen a dermatitis de
contacto. Los productos farmacéuticos con mercurio han
sido responsables de la "enfermedad rosada" en los
niños, y la exposición al vapor de mercurio ha sido
responsable de la enfermedad de "Kawasaki". En algunos
estudios, pero no en todos, se han comunicado efectos en
el ciclo menstrual y/o en el desarrollo del feto. El nivel
de los estudios epidemiológicos publicados aún no permite
saber si los vapores de mercurio pueden afectar negativa-
mente al ciclo menstrual o al desarrollo del feto sin que
se observen los conocidos síntomas de la intoxicación por
mercurio.
Hace poco ha habido una intensa polémica sobre la
inocuidad de las amalgamas odontológicas y se ha afirmado
que el mercurio de la amalgama puede plantear graves peli-
gros para la salud. Los informes en los que se describen
distintos tipos de síntomas y signos y los resultados de
los escasos estudios epidemiológicos realizados no son
concluyentes.
Several reports address studies where the investi-
gators have looked into possible effects at much lower
exposure levels. In a study of 142 exposed and 65 control
subjects, Miller et al. (1975) examined subclinical
effects related to exposure to inorganic mercury in the
chloralkali industry and in a factory for the manufacture
of magnetic materials. Mercury levels in urine varied from
normal to over 1000 µg/litre. Neurological examination
found evidence of eyelid fasciculation, hyperactive deep
tendon reflexes and dermatographia, but these findings did
not correlate with urinary mercury levels or length of
exposure. However, a power spectral analysis of forearm
tremor, by which it was possible to quantify tremor fre-
quency distribution and amplitude, showed a significant
increase in average tremor frequency with elevated urinary
mercury level. An effect was observed at urine concen-
trations above about 50 µg/litre.
Roels et al. (1982) studied psychomotor performance in
workers in a chloralkali plant and a factory for the manu-
facture of electric batteries. The results suggested that
preclinical psychomotor dysfunction related to the central
nervous system occurs when blood mercury levels rise to
values between 10 and 20 µg/litre and when mercury in
urine exceeds 50 µg/g creatinine. However, even in a sub-
group with urinary mercury levels below 50 µg/g creati-
nine, some parameters differed from those of a control
group.
In a further study, Roels et al. (1985) examined 131
male workers and 54 female workers exposed to metallic
mercury vapour in various Belgian factories. The controls
used were 114 non-exposed male workers and 48 female
workers. The subjects in the control and exposed groups
were closely matched with respect to age, weight, and
height. Also several other confounding factors were kept
under control. One criterion for the inclusion of exposed
workers was that they should have been uninterruptedly
exposed to mercury vapour for at least 1 year prior to the
study. A large number of questions were asked in order to
detect symptoms mainly related to nervous system disturb-
ances. A self-administered questionnaire, which was
completed the next day by the examiner, was the basis for
this information. A large number of CNS tests were used,
such as reaction time, flicker fusion, colour discrimi-
nation, short-term memory, and hand tremor. Mercury was
measured in urine and blood. Renal function was studied
using various tests, including total protein and beta-2-
microglobulin in blood and urine, and retinol-binding pro-
tein, albumin, and the lysosomal enzyme beta-galactosidase
in urine. Several symptoms mainly related to the central
nervous system (memory disturbances, depressive feelings,
fatigue, irritability) were more prevalent in the exposed
subjects than in the controls. The means and 95 percen-
tiles for urine mercury levels in non-exposed and exposed
subjects were, respectively, 52 and 147 µg/g creatinine
for males and 37 and 63 µg/g creatinine for females. The
symptoms were, however, not related to exposure par-
ameters. The authors therefore considered it possible
that the reporting of these symptoms was influenced by
knowledge of exposure to mercury vapour. There were no
significant disturbances in short-term memory, simple
reaction time, critical flicker fusion, and colour dis-
crimination ability that were related to the mercury
exposure. However, an effect on hand tremor was observed
in males. The prevalence was 5% in the non-exposed group
and 15% in the exposed group. Duration of exposure seemed
to be more important than exposure intensity, but an
increased prevalence of tremor was apparent in both the
groups with the lowest exposure (urine mercury level of
5-50 µg/g creatinine) and that with the shortest exposure
duration (1-4 years).
Among 26 workers exposed to mercury vapour in a chlor-
alkali plant or in the production of fluorescent tubes and
acetaldehyde, there was an increased incidence of reports
of hand tremor, compared with a control group (Fawer et
al., 1983). The exposure, based on personal air sampling,
was on average only 26 µg/m3. The mean urine concen-
tration was 20 µg/g creatinine (11.3 µmol/mol creati-
nine). Similar results were reported in a study by Verberk
et al. (1986), where 21 workers in a fluorescent lamp pro-
duction factory were examined. The excretion of mercury
in urine varied between 15 and 95 µg/g creatinine, the
average value being 35 µg/g creatinine. No control group
was examined, but with increasing exposure effects were
seen in several tremor parameters. The effects were
reported to occur irrespective of cigarette or alcohol
consumption or age.
Piikivi et al. (1984) reported decreased verbal intel-
ligence and memory in a group of 36 chloralkali workers
compared with a control group. Such effects were seen more
frequently in a subgroup where the urine mercury level was
above 56 µg/litre than in a subgroup where levels were
below this value. Piikivi & Hänninen (1989) made refined
analyses of the results of another study on 60 chloralkali
workers and matched controls. The exposed workers had an
average urine mercury concentration of 84.1 nmol/litre.
Neither the perceptual-motor, memory, nor learning abili-
ties of the mercury-exposed workers showed any disturb-
ances when compared to the controls. However, the exposed
workers reported statistically significantly more disturb-
ances of memory than the controls. According to multi-
variate analysis of variance, the memory disorders were
significantly associated with the form of workshift but
not with the level of exposure. In a further study
(Piikivi & Tolonen, 1989), EEG changes were found in a
group of 40 workers, compared to matched controls, after
several years of exposure to an average metallic mercury
vapour level of about 25 µg/m3 air, corresponding to a
urine mercury level of about 20 µg/litre. The EEG was
significantly slower and more attenuated in exposed
workers.
Results from the studies of Schiele et al. (1979),
Triebig et al. (1981), and Triebig & Schaller (1982) indi-
cate effects on cognitive functions and memory. However,
it is not possible to draw conclusions concerning dose-
response relationships from these studies. In a study by
Schuckmann (1979), 39 chloralkali workers with an average
urinary mercury concentration of about 100 µg/litre were
compared with a control group. There was no evidence of
changes in psychomotor activity. Smith et al. (1983)
studied effects on short-term memory in one group of 26
male chloralkali workers with an exposure corresponding
to urine mercury levels averaging 195 µg/litre and one
group of 60 male workers where the average urine mercury
level was 108 µg/litre. The severity of the effects was
found to be related to the intensity of mercury exposure.
There are some reports (Levine et al., 1982; Shapiro
et al., 1982; Singer et al., 1987; Zampollo et al., 1987)
that elemental mercury vapour causes peripheral neuropathy
at urinary levels of 50-100 µg/litre. Levine et al.
(1982) found a dose-response relationship between urine
mercury concentrations above 50 µg/litre and nerve con-
duction tests.
9.2.2.2 General population exposure
The exposure of the general population is typically
low, but occasionally may be raised to the level of occu-
pational exposure and can even result in adverse health
effects. Thus mishandling of liquid mercury, mercury
dispersed from jars, broken thermometers, fluorescent
lamps, and ingestion of mercury batteries have resulted in
severe intoxication and occasionally acute pneumonitis.
Children of mercury workers can also be exposed to mercury
vapour from contaminated work clothes. Hudson et al.
(1987) reported considerable mercury exposure among
children of mercury workers from a thermometer plant. The
median urine mercury level of 23 workers' children was
25 µg/litre compared with a value of 5 µg/litre among
39 controls. Three of the workers' children had urine mer-
cury levels above 50 µg/litre; one was above 100 µg per
litre. Mercury levels in workers' homes had a median
value of 0.24 µg/m3 compared with 0.05 µg/m3 in
non-workers' homes. No signs of mercury intoxication were
reported, based on a questionnaire to parents and on a
neurological examination that included assessment of
tremor by spectral power analysis. Urine protein was
measured only by dipstick. The reported air mercury levels
do not explain the high urinary concentrations. There must
have been exposure from sources that were not identified,
e.g., clothes. It is not known what measures were taken
to avoid contamination of sampling bottles.
Children who are exposed to mercury vapour from
interior latex paint may develop acrodynia. In 1989, a
4-year-old boy developed severe acrodynia 10 days after
the inside of his home was painted with 64 litres of
interior latex paint containing 945 mg mercury/litre. He
sequentially developed leg cramps, a generalized rash,
pruritis, sweating, tachycardia, an intermittent low-grade
fever, marked personality change, erythema and desqua-
mation of the hands, feet, and nose, weakness of the
pelvic and pectoral girdles, and lower extremity nerve
dysfunction. The level of mercury in a 24-h collection of
urine was 65 µg/litre.
9.3. Effects on the kidney
The kidney is the critical organ following the inges-
tion of inorganic bivalent mercury salts. Oliguria,
anuria, and death from renal failure resulting from acute
tubular necrosis has occurred not infrequently in the past
following the ingestion of mercuric chloride either acci-
dentally or with suicidal intent, and such cases have also
followed therapeutic administration of mercurials. At the
other extreme, organic mercurials have until recent years
been used extensively in medical practice as effective
diuretics in the management of congestive cardiac failure.
Occupational exposure to metallic mercury has for long
been associated with the development of proteinuria, both
in workers with other evidence of mercury poisoning and in
those without such evidence. An increased prevalence of
proteinuria in mercury workers, compared with a control
group, and a significant correlation between urinary mer-
cury excretion and protein excretion have been demon-
strated (Joselow & Goldwater, 1967). Less commonly, occu-
pational exposure has been followed by the nephrotic
syndrome (Kazantzis et al., 1962). Such cases have also
followed the therapeutic administration of mercurials,
although the role of mercury in some of these reported
cases, where other etiological factors may have been oper-
ative, is less clear. Two children developed the nephrotic
syndrome following a spillage of mercury in their bedroom
from a broken thermometer (Agner & Jans, 1978). The cur-
rent evidence suggests that the nephrotic syndrome follow-
ing absorption of mercury compounds results from an
immunotoxic response.
9.3.1. Immunological effects
WHO (1976) stated that effects of elemental mercury
vapour on the kidney had been reported only at doses
higher than those associated with the onset of CNS signs
and symptoms. Since then several new studies have been
carried out, and kidney effects have been seen at lower
exposure levels. Simultaneously, experimental studies on
animals have shown that inorganic mercury may induce
auto-immune glomerulonephritis in all species tested but
not in all strains, indicating a genetic predisposition.
Kazantzis (1978) and Filliastre et al. (1988) reviewed
the role of hypersensitivity and the immune response in
influencing susceptibility to metal toxicity, and gave
evidence of several case histories of clinical kidney
disease after exposure to mercury, occupationally as well
as among the general population. Of 60 adult African women
using skin-lightening creams containing inorganic mercury,
26 developed the nephrotic syndrome (Barr et al., 1972).
Kibukamusoke et al. (1974) reported one case of membranous
nephropathy, due to the use of skin-lightening cream,
where immunofluorescence showed finely granular IgG, IgM,
and C3 complement deposits. IgG and C3 complement deposits
were reported also by Lindqvist et al. (1974) in eight
cases with nephrotic syndrome. The authors also observed
similar kidney changes in two rabbits after application
(3 times per week for more than three months) of skin-
lightening cream to the skin area between the ears. The
rabbits developed proteinuria and died.
9.3.2. Relations between mercury in organs and effects/response
Only very limited information is available. In the
report by Davis et al. (1974) referred to in section
9.1.1, kidney mercury concentrations of 422 mg/kg and
25 mg/kg were measured in two fatal cases of poisoning.
However, no information was given on whether or not
adverse effects on the kidney were observed.
9.3.3. Relations between mercury in air, urine and/or blood
and effect/response
Foa et al. (1976) examined chloralkali industry
workers exposed to mercury vapour concentrations of 0.06
to 0.3 mg/m3. There were 15 cases of glomerular protein-
uria among 81 workers examined. Increased levels of
certain lysosomal enzymes were found in plasma, and this
effect was observed even in a group where the average
urine mercury level was only 35 µg/litre.
Stewart et al. (1977) examined 21 laboratory assist-
ants exposed not only to metallic mercury vapour but also
to mercuric mercury and formaldehyde and found increased
urinary excretion of protein. Air concentrations of mer-
cury were 10-50 µg/m3, and the median urine mercury
excretion rate was 53 µg/24 h (about 35 µg/litre urine).
Preventive measures were taken and in a follow-up study of
nine subjects one year later there was no evidence of
proteinuria.
Buchet et al. (1980) examined a group of 63 workers in
a chloralkali plant and found, compared with a control
group, increased plasma and urinary concentrations of the
enzyme beta-galactosidase, increased urinary excretion of
proteins with high relative molecular mass, and slightly
increased beta-2-microglobulin concentration in the plasma
without a concomitant increase in urinary concentration.
The urinary excretion of transferrin, albumin, and
beta-galactosidase was significantly correlated with the
urine concentration of mercury. The likelihood of finding
effects increased in workers with urine and/or blood mer-
cury concentrations of over 50 µg/g creatinine or
30 µg/litre blood. The data indicated an increased con-
centration of beta-galactosidase even in the group of
workers with an average urine mercury concentration of
about 20 µg/g creatinine. According to the authors the
results suggest that mercury vapour exposure may lead to a
slight glomerular dysfunction in some workers, and their
hypothesis is that the glomerular dysfunction is a result
of an auto-immune reaction.
The same research group (Roels et al., 1982) studied
the prevalence of proteinuria among 43 workers exposed to
metallic mercury vapour (median urine and blood mercury
levels of 71 µg/g creatinine and 21 µg/litre, respect-
ively) in two other factories (section 9.2.2). Increased
total proteinuria and albuminuria was slightly more preva-
lent in the mercury-exposed group than in the control
group.
No evidence of renal dysfunction (proteinuria, albumi-
nuria, retinol-binding proteins, aminoaciduria, creati-
nine, and beta-2-microglobulin in serum) was found among 62
exposed workers in a chloralkali plant and a zinc-mercury
amalgam factory, compared with a control group (Lauwerys
et al., 1983). The mean urine mercury concentration in the
exposed group was 56 µg/g creatinine. In eight exposed
workers, but in none of the controls, antibodies against
laminin, a non-collagen glycoprotein in the glomerular
basal membrane, were found. However, in a later study of
workers in another chloralkali plant and in a battery fac-
tory (Bernard et al., 1987), the prevalence of circulating
anti-laminin antibodies was not increased.
Stonard et al. (1983) examined a group of about 100
chloralkali industry workers with an average urine mercury
level of 67 µg/g creatinine. They found no evidence of
renal dysfunction and no increased excretion of proteins.
An increase in circulating immune complexes was found but
there were no anti-glomerular basement membrane antibodies
in the serum.
Roels et al. (1985) examined the renal function of 185
workers exposed to metallic mercury vapour (see also
section 9.2.2.1). Slight tubular effects were detected in
both male and female workers, in the form of an increased
urinary beta-galactosidase activity and an increased urinary
excretion of retinol-binding proteins. The effects were
dose related. Some increase in the prevalence of abnormal
values was seen even at mean urine mercury levels of about
30 µg/g creatinine. However, there was not, as was the
case for tremor, a dose-response relationship concerning
the length of the exposure period.
Rosenman et al. (1986) reported that urinary
N-acetyl-beta-glucosaminidase (NAG) enzyme levels increased
with increasing urine mercury levels over the range of
100-250 µg/litre. In a study of chloralkali industry
workers, there was a slight increase in the urine NAG con-
centration among exposed workers (average urine mercury
level of 50 µg/litre), compared with a control group
(Langworth, 1987).
Another way of studying kidney effects is to measure
the brush-border protein (BB-50) concentration in the
urine. This indicates the loss of organic tissue rather
than functional changes in the kidney cells. The urinary
BB-50 concentration was studied in 40 workers, with an
average urine mercury concentration of 46 µg/g creati-
nine and who were exposed for an average of 7 years (Mutti
et al., 1985), and 36 matched control workers. There was
no difference between exposed and non-exposed workers in
average urinary albumin or retinol-binding protein. How-
ever, when the 20 workers with urinary mercury above
50 µg/g creatinine were analysed separately, a shift of
the BB-50 distribution towards higher values was found by
a chi-square test (p = 0.07).
A study of 509 infants exposed to phenylmercury from
contaminated diapers (Gotelli et al., 1985) showed a clear
dose-response relationship between inorganic mercury in
urine and urinary excretion of gamma-glutamyl transpeptidase,
an enzyme in the brush borders of the renal tubular cells.
Since phenylmercury compounds are known to be rapidly
degraded to inorganic mercury in animals (Magos et al.,
1982), it is likely that the renal effect in the infants
was caused by inorganic mercury. Apart from the increased
enzyme excretion, the children with the highest exposure
also had increased 24-h urine volume. The enzyme excretion
increased at a urine mercury excretion of 4 µg/kg body
weight and the urine volume increased at 14 µg/kg body
weight.
9.4. Skin reactions
9.4.1. Contact dermatitis
Primary hypersensitivity to metallic mercury is con-
sidered rare (Burrows, 1986). However, Thiomersal (sodium
ethylmercurithiosalicilate) and ammoniated mercury have
been found to be common sensitizers in a survey on the
epidemiology of contact dermatitis (North American Contact
Dermatitis Group, 1973), Thiomersal being the third com-
monest sensitizer (after nickel and chromium) in the gen-
eral population. Both aryl- and alkylmercurial seed dress-
ings have also been shown to be potent skin sensitizers.
Mercury compounds give rise to a type IV cell-mediated
delayed hypersensitivity reaction (Coombs & Gell, 1975).
There have been a few cases of allergic dermatitis
among dental personnel (White & Brandt, 1976; Rudzki,
1979; Ancona et al., 1982). Patch testing of dental
students (White & Brandt, 1976) indicated that the preva-
lence rate of mercury hypersensitivity increased by class
from prefreshmen to seniors, successive values being 2.0%,
5.2%, 4.1%, 10.3%, and 10.8%. However, in a subsequent
study (Miller et al., 1987), similar results were not
found, but positive results from patch testing increased
in relation to the number of amalgam restorations in the
students. The overall percentage of positive reactions to
mercuric chloride was very high (32%), which may indicate
methodological problems. In this study, as in the study of
White & Brandt (1976), the patch testing was carried out
with 0.5 ml of a 0.1% aqueous solution of mercuric chloride.
Symptoms have occasionally been reported to relate to
amalgam fillings (Frykholm, 1957; Thomson & Russell, 1970;
Duxbury et al., 1982; SOS, 1987). In most cases the main
symptoms were facial dermatitis, sometimes with erythema-
tous and urticarial rashes. Symptoms from the mouth (oral
lichen planus) occasionally occurred. The symptoms started
a few hours after the insertion of amalgam. Nakayama et
al. (1983) reported 15 cases of generalized dermatitis
caused by mercury after exposure from broken thermometers
or dental treatment. In another very recent study
employing epicutaneous testing, positive mercury hypersen-
sitivity reactions were confined to subjects having pre-
existing amalgam restorations (Stenman & Bergman, 1989).
Finne et al. (1982) performed patch tests on 29
patients with amalgam fillings and oral lichen planus.
Contact allergy was found in 62% of the subjects, compared
with 3.2% in a control group. In four of the patients, all
the amalgam restorations were removed and replaced by gold
and composite materials. The lesions healed completely in
three of these patients after an observation period of one
year, and in the remaining case there was considerable
improvement.
When peripheral blood lymphocytes from non-atopic
subjects were cultured in the presence of pokeweed mitogen
and mercuric chloride, a significant enhancement of the
production of total IgE was observed, whereas the pro-
duction of IgM and IgA remained unaffected (Kimata et al.,
1983).
9.4.2. Pink disease and other skin manifestations
In the 1940s, "Pink disease" (acrodynia) was
reported in children below 5 years of age as a result of
the use of mercurous chloride in teething powder and oint-
ments. Affected children became irritable and generally
miserable and had difficulty in sleeping. Profuse
sweating, photophobia, and generalized rash followed. The
extremities became cold, painful, red, and swollen, and
the skin desquamated. Neither the occurrence of this
disease nor its severity was dose related. The urinary
excretion of mercury in affected children was elevated but
below the toxic level. After the withdrawal of teething
powder preparations by the main United Kingdom manufac-
turers in 1953, there was a dramatic decline in the occur-
rence of Pink disease. Calomel is not the only mercurial
that can cause Pink disease. Mercury dispersed from broken
fluorescent bulbs (Tunnessen et al., 1987), long-term
injection of gamma-globulin preserved with ethylmercurithio-
salicylate (Matheson et al., 1980), and the use of nappies
treated with phenylmercury (Gotelli et al., 1985) have
also been responsible for Pink disease. Exposure to mer-
cury vapour may be associated with the mucocutaneous lymph
node syndrome or "Kawasaki disease", which has many
similarities with Pink disease (Orlowski & Mercer, 1980).
Although the pathogenesis of Pink disease and Kawasaki
disease is unknown, there is good evidence that Kawasaki
disease is immunologically mediated, increased serum IgE
concentrations and eosinophilia having been reported
(Kusakawa & Heiner, 1976; Orlowski & Mercer, 1980; Adler
et al., 1982). In Kawasaki disease, urinary mercury
excretion is not always elevated, whereas it is in Pink
disease.
9.5. Carcinogenicity
Inorganic mercury is generally not considered to be
carcinogenic in humans (Kazantzis, 1981; Kazantzis &
Lilly, 1986). However, recent observations have shown an
excess risk of glioblastoma among Swedish dentists and
dental nurses (Ahlbom et al., 1986). Based on the Swedish
Cancer Environment Registry covering the years 1961-1979,
a standardized morbidity ratio of 2.1 was observed (with
95% confidence limits 1.3-3.4). The authors concluded that
the most probable origin is some occupational factor com-
mon to dentists and dental nurses, e.g., amalgam, chloro-
form, or radiography.
Cragle et al. (1984) published results of a mortality
study of men exposed to elemental mercury. It was a retro-
spective cohort study of 5663 workers selected from about
14 000 workers in the Y-12 plant in Oak Ridge, USA, orig-
inally working on the Manhattan Project but later in a
programme to produce large quantities of enriched lithium.
Elemental mercury was used in the lithium isotope separ-
ation process. Mercury urinalysis testing started in mid-
1953. Urine concentrations were not reported, but air
mercury levels in 50-80% of the samples taken during the
early 1950s were above 100 µg/m3. The workers studied
were divided into three groups: two exposed groups and one
non-exposed group. It is not possible to evaluate the
design as no valid exposure and selection data were pre-
sented. In all three groups, elevated SMRs (2.3, 1.2, 2.1)
for tumours of the central nervous system were found.
However, a statistically significant increase was reported
only for the group consisting of 3260 non-mercury workers.
9.6. Mutagenicity and related end-points
WHO (1976) did not report any studies showing that in-
organic mercury was genotoxic to humans. However, relevant
data have since been reported. Popescu et al. (1979) com-
pared four men exposed to elemental mercury vapour with a
control group and found an increased prevalence of chromo-
somal aberrations. Verschaeve et al. (1976) and Verschaeve
& Susanne (1979) showed an increase in aneuploidy after
exposure to very low concentrations of metallic mercury
vapour, but this could not be repeated in a later study
(Verschaeve et al., 1979). Similarly, Mabille et al.
(1984) did not find an increase of structural chromosomal
aberrations in peripheral blood lymphocytes of workers
exposed to metallic mercury vapour.
9.7. Dental amalgam and general health
During recent years there has been intense debate in
some countries (e.g., Sweden and USA) on the possible
health hazards of dental amalgams (Ziff, 1984; Penzer,
1986; SOS, 1987; Berglund, 1989). Those who claim that
mercury from amalgam may cause severe health hazards refer
to information on the release of mercury from amalgam and
subsequent uptake into the body due to inhalation and
swallowing of mercury. They also claim that a large number
of people suffer from a variety of complaints and that
their symptoms are caused by mercury. Those who deny a
causal relation between dental amalgams and health effects
point out that amalgam has been used for many years with
no proven health effects. Furthermore, the uptake of
mercury from amalgam is considerably less than has been
associated with effects after occupational exposure to
mercury (Fan, 1987).
There are many people with sometimes clearly incapaci-
tating complaints who believe that these are caused by
dental amalgam. Reports describing different types of
symptoms or other effects (Hansson, 1986; Johansson &
Lindh, 1987; Siblerud, 1988) do not allow any conclusions
to be reached concerning their cause. This was also the
opinion of a Swedish Task Group (SOS, 1987). The sympto-
matic picture is highly diverse and characterized by a
variety of different symptoms. Some studies reported that
patients improved after their amalgam fillings were re-
placed by another dental filling material. However, these
reports have not been controlled for potential placebo
effects.
Recently results from one epidemiological study have
been reported by Ahlqwist et al. (1988). The data collec-
tion was carried out during 1980-1981. The majority of
participants (85%) consisted of individuals who had
already in 1968-69 participated in a longitudinal descrip-
tive study of different diseases among women in the city
of Gothenburg, Sweden. The remainder were included to
expand the age strata and obtain a sample representative
of women of the same age in the general population.
Altogether 1024 women (aged 38-72 years) participated in
the study, which covered a dental examination with an
orthopantomogram and a medical examination including a
standardized self-administered questionnaire regarding
different symptoms or complaints. The dental and medical
examinations were made by different people and without
mentioning possible relations between amalgam and health
risks. No positive correlations were found between number
of amalgam fillings and number of symptoms or between
number of amalgam fillings and prevalence of specified
single symptoms or complaints. On the contrary, there were
several age-matched significant correlations in the
opposite direction. Some of these correlations (abdominal
pain, poor appetite) disappeared when adjustment was made
for number of teeth. Risk ratios (including 95% confidence
limits) for women with 20 fillings or more compared to
women with 0-4 fillings are given in Table 5. The authors
concluded that their results do not support a correlation
between number of surfaces with amalgam fillings and vari-
ous symptoms studied on the population level. They do not
exclude the possibility of a connection between amalgam
fillings and special symptoms and complaints on the indi-
vidual level, but, if such a connection exists, it has a
low prevalence among the general population.
Table 5. Risk ratio for a specific symptom or
complaint for 460 women with 20 or more fillings
compared to 193 women with 0-4 fillingsa
----------------------------------------------------
Risk ratio analysis
Symptom or complaint Risk 95% confidence
ratio limits for
risk ratio
----------------------------------------------------
Dizziness 0.70 0.46-1.07
Eye complaints 1.01 0.64-1.57
Hearing defects 0.66 0.41-1.07
Headache 1.22 0.83-1.80
General fatigue 0.79 0.55-1.15
Sleep disturbances 1.38 0.94-2.03
Nervous symptoms 0.80 0.52-1.25
Sweating 0.86 0.57-1.32
Breathlessness 0.65 0.41-1.03
Chest pain 0.62 0.39-0.99
Cough 0.71 0.47-1.08
Irritability 0.68 0.45-1.02
Over-exertion 0.60 0.38-0.96
Reduced mental
concentration capacity 0.74 0.47-1.18
Restlessness 0.70 0.45-1.09
Depressive symptoms 0.74 0.50-1.07
Readiness to crying 0.72 0.46-1.11
Reduced ability to relax 1.15 0.79-1.69
Abdominal pain 0.64 0.42-0.98
----------------------------------------------------
Table 5 (contd.)
----------------------------------------------------
Risk ratio analysis
Symptom or complaint Risk 95% confidence
ratio limits for
risk ratio
----------------------------------------------------
Indisposition 1.00 0.57-1.76
Diarrhoea 0.62 0.32-1.18
Constipation 0.82 0.50-1.37
Poor appetite 0.33 0.16-0.68
Loss of weight 0.27 0.10-0.70
Overweight 0.76 0.52-1.12
Sensitivity to cold 0.78 0.50-1.21
Micturation disturbances 0.66 0.30-1.43
Joint complaints 0.97 0.66-1.43
Back complaints 0.74 0.51-1.07
Leg complaints 0.74 0.51-1.09
----------------------------------------------------
a The analysis was confined to dentulous women. Age
was taken into consideration by means of the
Mantel-Haenszel procedure. Modified from Ahlqwist
et al. (1988).
Lavstedt & Sundberg (1989) investigated possible
associations between dental amalgam and a range of symp-
toms by re-examining certain dental, medical, and socio-
logical data originally collected from 1204 subjects in
1970 (i.e. prior to the present debate on dental amalgam).
Standardization was made for various confounding factors,
such as gender, social group, and smoking habits. There
was no statistically significant increase in the percent-
age of individuals with symptoms in groups with increased
numbers of amalgam fillings after controlling for con-
founding factors. The authors pointed out that the study
did not exclude a causal association on the individual
level. One strength of the study was that practically all
of the examinations were carried out by one single
investigator.
9.8. Reproduction, embryotoxicity, and teratogenicity
9.8.1. Occupational exposure
9.8.1.1 In males
McFarland & Reigel (1978) reported medical findings in
nine men exposed accidentally for less than 8 h when more
than 10 ml of mercury was instantly vapourized at a high
temperature. Air mercury concentrations were estimated to
be 44.3 mg/m3. Even if these theoretical estimates are
very uncertain, the exposure must have been extremely
high. Six of the cases developed symptoms of acute
poisoning. During a follow-up lasting several years they
also showed signs of chronic poisoning. A loss of libido,
which persisted for at least several years, was reported
in all six cases.
Lauwerys et al. (1985) compared the fertility of 103
male workers, exposed to elemental mercury vapour in a
zinc-mercury amalgam factory, a chloralkali plant, and in
plants manufacturing electrical equipment, with 101 well-
matched controls. The exposed group had an average blood
mercury level of 14.6 µg/litre (SD 11.6 µg/litre) and an
average urine mercury concentration of 52.4 µg/g creati-
nine (SD 46.7 µg/g). In the exposed group, 59 children
were born compared to 65.8 expected (as calculated from
data in the control group). However, the difference was
not statistically significant.
In a study carried out at a US Department of Energy
plant that used very large quantities of elemental mercury
from 1953 to 1963, reproductive outcomes were studied
among 247 male workers exposed to metallic mercury vapour
(Alcser et al., 1989). As controls, 255 plant workers
whose job did not require exposure to elemental mercury
were used. No associations were demonstrated between mer-
cury exposure and decreased fertility, increased rates of
major malformations among the offspring, or serious child-
hood disease. There was an association between exposure
and miscarriage, which disappeared however after control-
ling for the number of previous miscarriages before
exposure began. The 95% adjusted confidence limits were
0.97-1.18. The authors pointed out certain problems with
the study. The most serious methodological weakness in the
evaluation was the necessity for subject recall of events
that occurred 20 to 50 years previously. Another problem,
which was not considered, was possible exposure to other
toxic substances in the control group. The higher fre-
quency of miscarriages among the exposed group prior to
the exposure period could not be explained.
9.8.1.2 In females
There have been reports of increased menstrual dis-
turbances in women exposed industrially or in dentistry to
elemental mercury vapour. A study by De Rosis et al.
(1985) examined a group of 106 women exposed to low levels
of mercury (average values not exceeding 10 µg/m3) and
a control group of 241 unexposed women in another factory
with similar working conditions. The percentage of women
having normal menstrual cycles at the start of employment
was very similar in both groups of women. During their
period of employment more women in the exposed group
noticed changes in the menstrual cycle than women in the
control group. The age-standardized ratio of abnormal
cycle frequency in exposed women to that in the control
group was 1.4. The information was obtained by means of
interviews, but these were not carried out on a blind
basis. Therefore, according to the authors, the results
neither prove nor exclude the possibility that occu-
pational exposure to this concentration of mercury has a
negative effect on the female reproductive system.
There have been reports which suggest that inorganic
mercury compounds cause spontaneous abortion. Goncharuk
(1977) reported that during a 4-year period 17% of 168 ex-
posed workers in a mercury smelting plant had experienced
spontaneous abortion (average exposure, 80 µg mercury
per m3), compared with 5% among 178 controls. Toxaemia
during pregnancy was reported in 35% of the exposed and
2% of the unexposed workers. Gordon (1981) reported a
slightly elevated incidence of spontaneous abortions among
dentists. The results can not be interpreted with cer-
tainty, however, due to a non-response rate of almost 50%.
The study of De Rosis et al. (1985), referred to earlier
in this section, revealed no difference in the age-
standardized rate of spontaneous abortion between mercury-
exposed and unexposed female workers. Two other studies
of female dental staff also reported no increased abortion
rate when compared with age-standardized controls. In one
of the studies (Heidam, 1984), the upper 95% confidence
limit for odds ratio was about 2. In the other study
(Brodsky et al., 1985), a comparison was made between a
"low" exposure group and a "high" exposure group. The
low exposure group comprised dental assistants preparing
less than 40 amalgam restorations per week and the high
exposure group those preparing more than 40 fillings a
week. The assumed difference in exposure between the
groups was not validated by measurements.
Two studies, one from Poland and one from Sweden, both
dealing with spontaneous abortions and malformations, are
of particular interest. The Polish study (Sikorski et al.,
1987) revealed a high frequency of malformations among
dental staff. Of 117 pregnancies in the mercury-exposed
group, 28 pregnancies in 19 women led to reproductive
failure, such as spontaneous abortion (19 cases, 16.2%),
stillbirth (3 cases, 2.6%), congenital malformations (5
cases of spina bifida, 5.1%; one case of intra-atrial
defect). This contrasts, in non-exposed controls, with
seven cases of adverse pregnancy outcome (11.1%) in five
women out of a total of 63 pregnancies (30 women). There
were no malformations among the controls (R. Sikorski,
personal communication to the IPCS). The age distribution
of exposed and control women and the number of pregnancies
before exposure or effect were not reported, which makes
it difficult to interpret the data. For most countries the
average rate of spina bifida is 5-10 (or less) per 10 000
births (International Clearing-house for Birth Defects
Monitoring System, 1985) over a wide age span. Sikorski et
al. (1987) reported a correlation between mercury levels
in hair and reproductive failure in the exposed group. The
meaning of this correlation is difficult to interpret, as
hair is not a good indicator of exposure to metallic mer-
cury vapour, due to several factors, including the possi-
bility of external contamination (section 6.5.2). There is
reason to believe, however, that the exposure was higher
in the group with higher hair mercury levels (R. Sikorski,
personal communication to the IPCS). Only 13.6% of the
women studied used automatic amalgamators. The remaining
86.4% prepared the amalgam in an open mortar and almost
never in separate rooms.
The Swedish study was reported by Ericson & Källén
(1989), linking data from the Swedish National registers
for birth records, malformations, and occupation.
Altogether, 8157 children born to dentists (1360), dental
nurses (6340), and dental technicians (457) were compared
with the total number of births in Sweden during the
observation period (1976, 1982-1986). The analysis took
into consideration different confounding variables, such
as the age of the mother and number of children. The
study also examined the occurrence of stillbirths and
spontaneous and induced abortions treated in hospitals.
There was no tendency towards an elevated rate of malfor-
mation, abortion, or stillbirth. The study did not verify
the high risk of spina bifida described in the Polish
study (Sikorski et al., 1987). In spite of the large study
group, the upper 95% confidence limit for the risk ratio
of spina bifida was high (2.1), and the upper confidence
limit of the absolute risk was about 1 per 1000 births.
Therefore, the study is not a strong argument against an
effect. In addition, a sample of 3991 pregnancies from
the 1960s (among them there were 13 dentists, 65 dental
assistants, and 6 dental technicians) was studied but no
effects on spontaneous abortion rate or malformation was
seen. The authors point out, however, that the only mal-
formed infant observed had anencephaly and that both
parents worked as dental technicians.
Among dental nurses, there was a significant excess of
children with a birth weight of 2 to 2.5 kg. In total
there were 274 children weighing less than 2.5 kg (against
an expected number of 233), which gives a risk ratio for
low birth weight of 1.2 (with a 95% confidence interval of
1.0-1.3). No similar excess in low birth weight was seen
among dentists or dental technicians. Possible confounding
socioeconomic factors, e.g., smoking, were not studied,
but the authors suggested that these findings could be
explained by differences in socioeconomic status.
During recent years much interest has focused on
subclinical developmental changes in children exposed in
utero or in early childhood to methylmercury and lead. No
similar studies have been reported for inorganic mercury.
10. EVALUATION OF HUMAN HEALTH RISKS
Mercury exists in different forms, including elemental
mercury, inorganic mercury and organic mercury compounds.
They have some properties in common but differ in metab-
olism and toxicity. Biotransformation takes place in the
body, particularly the transformation of metallic mercury
vapour to mercuric compounds, which means that some of the
effects of inorganic mercury could also be expected after
exposure to metallic mercury vapour. There are, however,
no empirical data showing whether or not inorganic mercury
formed due to a biotransformation has similar toxicity and
metabolism to that of inorganic mercury accumulated in the
body as a result of exposure to inorganic mercury itself.
It would be prudent to consider, until more information is
available, that, with the exception of acute renal tubular
cell damage, the two forms of inorganic mercury have simi-
lar toxicity.
10.1. Exposure levels and routes
10.1.1. Mercury vapour
Human long-term exposure to mercury vapour is primar-
ily encountered in an occupational setting and in cases
where the metal has been handled inappropriately in the
home. Continual low-level exposure to mercury vapour
occurs in the mouth in the presence of dental amalgam
fillings. The amount of mercury vapour released intra-
orally depends on the number, surface area, and mechanical
loading of the amalgam restorations. Atmospheric levels of
mercury found in the workplace, e.g., chloralkali plants,
are usually below 50 µg/m3. Levels above 50 µg/m3 and
even exceeding 100 µg/m3 can be found in work environ-
ments where good industrial hygiene has not been practiced
or in home operations, in which the highest levels would
be expected. Values for air concentration (in µg/m3) are
approximately the same as those for urine mercury concen-
tration (expressed in µg/g creatinine). The use of
urinary mercury excretion makes it possible to compare
intake from the working atmosphere and release from dental
amalgam.
Occupational exposure to mercury vapour in dentistry
has been well established, reported exposure levels being
4-30 µg/m3, on average, with up to 150-170 µg/m3 in
some clinics. The values for mean intraoral mercury vapour
concentration derived from dental amalgam have been
reported to be in the range of 3 to 29 µg/m3.
Approximately 80% of inhaled mercury vapour is
absorbed from the lungs, while uptake of mercury vapour
via the skin is about 1% of uptake by inhalation. No data
are available on the possible oral mucosal absorption of
mercury vapour. Mercury vapour can cross the plancental
barrier, thus exposing the developing fetus.
10.1.2. Inorganic mercury compounds
The major source of high exposure to humans from mer-
curic compounds involves medicaments, both traditional and
alternative, and skin-lightening creams and soaps. There
is some evidence from occupational settings where chlorine
is used that a part of the mercury vapour can be trans-
formed in the atmosphere and absorbed as an aerosol of
mercuric mercury. Mercuric mercury is to a great extent
deposited in the placenta, where it causes damage that may
lead to adverse effects on the fetus.
Mercurous mercury, in the form of calomel, has for
long been used in therapeutics. The mercury content in the
brain of two daily users of calomel, ingesting 240 mg
mercurous chloride per day, was 4-106 mg/kg brain, indi-
cating that elemental mercury is formed from mercurous
chloride following ingestion.
10.2. Toxic effects
10.2.1. Mercury vapour
Over-exposure to mercury vapour gives rise to neuro-
logical effects with initially a fine high-frequency
intention tremor and neurobehavioural impairment. Periph-
eral nerve involvement has also been observed. Proteinuria
and lysosomal enzymes in the urine of exposed workers
indicate an effect on the kidney of chloralkali workers;
the presence of mercuric mercury may have contributed to
this effect. The nephrotic syndrome has been reported
among chloralkali workers. Pink disease, skin allergy, and
mucocutaneous lymph node syndrome (Kawasaki disease) in
children, have also been observed after exposure to mer-
cury vapour.
Hypersensitivity skin reactions have been described
after exposure to metallic mercury vapour from mercury
amalgam materials. No data supporting carcinogenic effects
of mercury vapour have been reported. There has been a
report of excess risk of glioblastoma observed in Swedish
dental personnel. At this stage, however, it has not been
possible to associate this excess risk with any specific
group of dental materials. The standard of published
epidemiological studies is such that it remains an open
question whether mercury vapour can adversely affect the
menstrual cycle or fetal development in the absence of the
well-known signs of mercury intoxication.
10.2.2. Inorganic mercury compounds
Information on pulmonary deposition and absorption of
mercuric mercury aerosols is lacking. However, it is
likely that significant absorption takes place directly
from the lung and probably from the gastrointestinal tract
after mucociliary clearance from the lung.
Most adverse effects of mercuric compounds in humans
have been reported after oral ingestion or skin absorp-
tion. However, only limited information is available as
far as dose-effect relationships are concerned. From
animal experiments it is possible to identify the critical
lowest effect levels that are likely to result in protein-
uria in humans after chronic exposure. Proteinuria in
humans is believed to be produced through the formation of
mercuric-mercury-induced autoimmune glomerulonephritis.
The production and deposition of IgG antibodies to glomer-
ular basement membrane can be considered the first step in
the formation of this glomerulonephritis. No data on
possible carcinogenic effects of mercuric mercury have
been reported.
10.3. Dose-response relationships
10.3.1. Mercury vapour
The risk assessment of exposure to mercury vapour is
hindered by the heterogeneity of published data, problems
with the estimation of exposure (e.g., lack of speciation
and methodological uncertainties), uncertainty concerning
the reliability of subjective symptoms, and the selection
of control groups for comparison with low exposure groups.
Nevertheless the data presented in section 9 allow a
broad characterization to be made.
a) When exposure is above 80 µg/m3, corresponding to
a urine mercury level of 100 µg/g creatinine (section
6.5.2), the probability of developing the classical
neurological signs of mercury intoxication (tremor,
erethism) and proteinuria is high.
b) Exposure in the range of 25 to 80 µg/m3, corre-
sponding to a level of 30 to 100 µg mercury/g creati-
nine, increases the incidence of certain less severe
toxic effects that do not lead to overt clinical
impairment. These subtle effects are defects in psy-
chomotor performance, objectively detectable tremor,
and evidence of impaired nerve conduction velocity,
which are present only in particularly sensitive indi-
viduals. The occurrence of several subjective symp-
toms, such as fatigue, irritability, and loss of
appetite, is also increased. In a few studies, tremor,
recorded electrophysiologically, has been observed at
low urine concentrations (down to 25-35 µg/g creati-
nine). Other studies did not show such an effect.
Although the incidence of some signs was increased in
this exposure range, most studies did not find a dose-
response relationship. Some of the exposed people
develop proteinuria (proteins of low relative molecu-
lar mass and microalbuminuria). The available studies
are generally of small size and low statistical power.
c) Appropriate epidemiological data covering exposure
levels corresponding to less than 30-50 µg mercury
per g creatinine are not available. Since a specific
no-observed-effect level cannot be established and if
larger populations are exposed to low concentrations
of mercury, it cannot be excluded that mild adverse
effects may occur in certain sensitive individuals.
Some studies have found miscarriages and abortions
after occupational exposure to mercury, but other studies
did not confirm these effects. The WHO Study Group in 1980
stated: "The exposure of women of child-bearing age to
mercury vapour should be as low as possible. The Group was
not in a position to recommend a specific value" (WHO,
1980). This statement is still prudent and will remain so
until new data become available.
10.3.2. Inorganic mercury compounds
The risk assessment of exposure to inorganic mercury
compounds is hindered by a lack of adequate human data
dealing with the relationship between dose and effects/
responses. For this reason, more human research is needed
in order to arrive at the goal of a human risk evaluation
of inorganic mercuric mercury compounds at low levels.
The intake of gram doses of mercuric chloride causes
severe to lethal renal tubular damage and necrosis of the
gastrointestinal mucous membrane. At lower dose levels,
less pronounced tubular damage occurs, reflected in amino-
aciduria, increased diuresis, and loss of renal enzymes in
the urine.
A special problem in the risk assessment of mercury is
the fact that mercury can give rise to allergic and immu-
notoxic reactions, which are partly genetically regulated.
There may well be a small fraction of the population that
is particularly sensitive, as has been observed in animal
studies. A consequence of an immunological etiology is
that it is not scientifically possible to set a level for
mercury, e.g., in blood or urine, below which mercury-
related symptoms will not occur in individual cases, since
dose-response studies for groups of immunologically sensi-
tive individuals are not yet available.
Based upon the evaluation in animals, the most sensi-
tive adverse effect for inorganic mercury risk assessment
is the formation of mercuric-mercury-induced auto-immune
glomerulonephritis, the first step being the production
and deposition of IgG antibodies to the glomerular base-
ment membrane. The Brown Norway rat is a good test species
for the study of mercuric-mercury-induced auto-immune
glomerulonephritis (although this effect has also been
observed in rabbits), and it is the best animal model for
the study of mercury-induced kidney damage at present. The
group of studies presented in Table 4 (section 8.2.2.6)
was selected for consideration of mercuric mercury risk
assessment. A no-observed-adverse-effect level (NOAEL)
could not be determined from these animal studies. The
lowest-observed-adverse-effect level (LOAEL) was found in
the subcutaneous exposure study by Druet et al. (1978). In
this study, subcutaneous doses of mercuric chloride (0.05
mg/kg body weight three times per week) were administered
for 12 weeks and resulted in antibodies being bound to the
glomerular basement membrane of the rat kidney.
Using this animal LOAEL (0.05 mg/kg), equivalent human
oral and inhalation LOAEL values for kidney effects can be
determined as follows:
Average daily subcutaneous dose:
(0.05 mg/kg) x 3 days x 0.739
= ----------------------------- = 0.0158 mg/kg per day
7 days
where: 0.05 mg/kg = dose of HgCl2 injected subcutaneously
into rats
3 days = number of days per week that doses were
administered
7 days = number of days per week
0.739 = fraction of HgCl2 that is Hg2+ ion.
Although the calculated values have been rounded, the un-
rounded values were always used in subsequent calculations.
Human oral exposure equivalent determination:
(0.0158 mg/kg per day) x 70 kg x 100%
= ------------------------------------- = 15.8 mg/day
7%
where: 0.0158 mg/kg per day = average daily subcutaneous
dose of Hg2+
70 kg = assumed body weight of an adult human
100% = assumed percentage of Hg2+ absorbed from
the subcutaneous route of exposure
7% = assumed percentage of Hg2+ absorbed from the
oral route of exposure
Human inhalation exposure equivalent determination:
a) For 24-h general population exposure:
(0.0158 mg/kg per day) x 70 kg x 100%
------------------------------------- = 0.069 mg/m3
(20 m3/day) x 80%
where: 0.0158 mg/kg per day = average daily subcutaneous
dose of Hg2+
70 kg = assumed body weight of an adult
20 m3/day = assumed volume of air inhaled during
a 24-h period
100% = assumed percentage of Hg2+ absorbed from
subcutaneous route of exposure
80% = assumed percentage of Hg0 absorbed from the
lung.
b) For 8-h work-day exposure:
(0.0158 mg/kg per day) x 70 kg x 100%
------------------------------------- = 0.139 mg/m3
(10 m3/day) x 80%
where: 0.0158 mg/kg per day = average daily subcutaneous
dose of Hg2+
70 kg = assumed body weight of an adult
10 m3 = assumed volume of air inhaled during a
work-day
100% = assumed percentage of Hg2+ absorbed from
subcutaneous route of exposure
80% = assumed percentage of Hg0 absorbed from the
lung.
These inhalation LOAEL values calculated for kidney
effects are well within the range of mercury vapour
exposures in humans where neurological and renal effects
have been observed.
11. RECOMMENDATIONS FOR FURTHER RESEARCH
Further research is required in the following areas:
1. Determination of the exposure to different chemical
forms of mercury at low levels of exposure, including
the development of microtechniques for speciation of
small quantities of mercury in biological materials
and of analytical quality assurance techniques.
2. The pharmacokinetics of mercury release from amalgam
restorations in relation to time, diet, technical and
physiological conditions, and the development of tests
for identifying specially sensitive individuals (e.g.,
local mucosa reactions, intra-oral electrochemical
measurements, immunotoxicity).
3. The use of mercury compounds in pharmaceuticals and
cosmetics.
4. The binding, biotransformation, and transport of dif-
ferent forms of mercury, both in animals and humans,
including interactions with selenium.
5. The transplacental transport of mercury and specific
distribution in fetal organs, fetotoxic effects, and
developmental effects with emphasis on neurobehav-
ioural effects.
6. Research on the neurobehavioural effects of mercury in
the occupationally exposed population (dentists, etc.).
7. The epidemiology of the role of mercury in inducing
glomerulonephritis in the general population.
8. The prevalence of immunological effects and hypersen-
sitivity in low-dose exposure to mercury with or with-
out subjective symptoms.
9. A case-control study of brain tumours, in particular
glioblastoma, and exposure to mercury.
Measures to decrease the exposure of the general popu-
lation to mercury-containing pharmaceuticals and cosmetics
should be promoted.
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
The human health risks of inorganic mercury compounds
were previously evaluated in Environmental Health Criteria
1: Mercury (WHO, 1976). More recent evaluations by the
International Programme on Chemical Safety (IPCS) have
dealt mainly with the health risks of methylmercury
exposure (WHO, 1990). A WHO review of the occupational
health risks of inorganic mercury (WHO, 1980) and an IPCS
review of the environmental aspects of mercury (WHO, 1989)
have been published. The recommended health-based occu-
pational exposure limit for metallic mercury vapour (WHO,
1980) is 25 µg mercury/m3 air (TWA, long-term exposure)
and 500 µg mercury/m3 air (peaks, short-term exposure).
The equivalent value for long-term exposure to inorganic
mercury compounds is 50 µg mercury/m3 air (TWA) (WHO,
1980). A maximum individual urine mercury concentration
of 50 µg/g creatinine has also been recommended (WHO,
1980).
Regulatory standards established by national bodies in
various countries and in the European Community are sum-
marized in the data profile of the International Register
of Potentially Toxic Chemicals (IRPTC, 1987).
REFERENCES
ABRAHAM, J.E., SVARE, C.W., & FRANK, C.W. (1984) The effect of
dental amalgam restorations on blood mercury levels. J. dent. Res.,
63(1): 71-73.
ADA (1985) Dental amalgam, Chicago, American Dental Association.
ADLER, R., BOXSTEIN, D., SCHAFF, P., & KELLY, D. (1982) Metallic
mercury vapour poisoning stimulating mucocutaneous lymph node
syndrome. J. Pediatr., 101(6): 967-968.
AGNER, E. & JANS, H. (1978) Mercury poisoning and nephrotic
syndrome in two young siblings. Lancet, II: 951.
AGOCS, M.M., ETZEL, R.A., PARRISH, R.G., PASCHAL, D.C.,
CAMPAGNA, P.R., COHEN, D.S., KILBOURNE, E.M., & HESSE, J.L. (in
press) Mercury exposure from interior latex paint. N. Engl. J. Med.
AHLBOM, A., NORELL, S., NYLANDER, M., & RODVALL, Y. (1986)
Dentists, dental nurses, and brain tumours. Br. med. J., 292: 662.
AHLQWIST, M., BENGTSSON, C., FURUNES, B., HOLLENDER, L., &
LAPIDUS, L. (1988) Number of amalgam tooth fillings in
relation to subjectively experienced symptoms in a study of
Swedish women. Community dent. oral Epidemiol., 16: 227-231.
AITIO, A. (1988) Biological monitoring. In: Clarkson, T.,
Friberg, L., Nordberg, G., & Sager, P., ed. Biological monitoring
of toxic metals, New York, London, Plenum Press, pp. 75-83.
AITIO, A., VALKONEN, S., KIVISTO, H., & YRJANHEIKKI, E. (1983)
Effect of occupational mercury exposure on plasma lysosomal
hydrolases. Int. Arch. occup. environ. Health, 53: 139-147.
ALBERS, J.W., KALLENBACH, L.R., FINE, L.J., LANGOLF, G.D.,
WOLFE, R.A., DONOFRIO, P.D., ALESSI, A.G., STOLP-SMITH, K.A.,
BROMBERG, M.B., & THE MERCURY WORKERS STUDY GROUP (1988)
Neurological abnormalities associated with remote occupational
elemental mercury exposure. Ann. Neurol., 24: 651-659.
ALCSER, K.H., BRIX, K.A., FINE, L.J., KALLENBACH, L.R., &
WOLFE, R.A. (1989) Occupational mercury exposure and male
reproductive health. Am. J. Ind. Med., 15(5): 517-529.
ALEXANDER, J., THOMASSEN, Y., & AASETH, J. (1983) Increased
urinary excretion of selenium among workers exposed to elemental
mercury vapor. J. appl. Toxicol., 3(3): 143-145.
ANCONA, A., RAMOS, M., SUAREZ, R., & MACOTELA, E. (1982)
Mercury sensitivity in a dentist. Contact dermatitis, 8: 218.
ANDERSEN, O., RONNE, M., & NORDBERG, G.F. (1983) Effects of
inorganic metal salts on chromosome length in human lymphocytes.
Hereditas, 98: 65-70.
ANDRES, P. (1984) IgA-IgG disease in the intestine of Brown-
Norway rats ingesting mercuric chloride. Clin. Immunol.
Immunopathol., 30: 488-494.
AOI, T., HIGUCHI, T., KIDOKORO, R., FUKUMURA, R., YAGI, A.,
OHGUCHI, S., SASA, A., HAYASHI, H., SAKAMOTO, N., & HANAICHI, T.
(1985) An association of mercury with selenium in inorganic mercury
intoxication. Hum. Toxicol., 4: 637-642.
ARONSSON, M.A., LIND, B., NYLANDER, M., & NORDBERG, M. (1989)
Dental amalgam and mercury. Biol. Metals, 2: 25-30.
ARVIDSON, B. (1987) Research note. Retrograde axonal transport of
mercury. Exper. Neurol., 98: 198-203.
ASHE, W., LARGENT, E., DUTRA, F., HUBBARD, D., & BLACKSTONE, M.
(1953) Behaviour of mercury in the animal organism following
inhalation. Ind. med. occup. Med., 22: 19-43.
ATSDR (1989) Toxicological profile for mercury. Atlanta, Agency
for Toxic Substances and Disease Registry (US Public Health
Service).
ATEN, J., BOSMAN, C.B., ROZING, J., STIJNEN, T., HOEDEMAEKER,
Ph.J., & WEENING, J.J. (1988) Mercuric chloride-induced
autoimmunity in the Brown Norway rat: cellular kinetics and major
histocompatibility complex antigen expression. Am. J. Pathol.,
133(1): 127-138.
BARIETY, J., DRUET, P., LALIBERTE, F., & SAPIN, C.
(1971) Glomerulonephritis with gamma- and beta1C-globulin
deposits induced in rats by mercuric chloride. Am. J. Pathol.,
65:293-302.
BARNES, J.L., MCDOWELL, E.M., MCNEIL, J.S., FLAMENBAUM, W., &
TRUMP, B.F. (1980) Studies on the pathophysiology of acute
renal failure. IV. Protective effect of dithiothreitol
following administration of mercuric chloride in the rat. Virchows
Arch. cell Pathol., 32: 201-232.
BARR, R.D., REES, P.H., CORDY, P.E., KUNGU, A., WOODGER, B.A., &
CAMERON, H.M. (1972) Nephrotic syndrome in adult Africans in
Nairobi. Br. med. J., 2: 131-134.
BARR, R.D., WOODGER, B.A., & REES, P.H. (1973) Levels of mercury
in urine correlated with the use of skin lightening creams. Am.
J. clin. Pathol., 59: 36-40.
BATTISTONE, G.C., HEFFERREN, J.J., MILLER, R.A., & CUTRIGHT, D.E.
(1976) Mercury: its relation to the dentist's health and
dental practice characteristics. J. Am. Dent. Assoc., 92: 1182-1188.
BAUER, J.G. & FIRST, H.A. (1982) The toxicity of mercury in
dental amalgam. California dent. J., 47-61.
BELLON, B., CAPRON, M., DRUET, E., VERROUST, P., VIAL, M.C.,
SAPIN, C., GIRARD, J.F., FOIDART, J.M., MAHIEU, P., & DRUET, P.
(1982) Mercuric chloride induced autoimmune disease in Brown-
Norway rats: Sequential search for anti-basement membrane
antibodies and circulating immune complexes. Eur. J. clin.
Invest., 12: 127-133.
BERGLUND, A., POHL, L., OLSSON, S., & BERGMAN, M. (1988)
Determination of the rate of release of intra-oral mercury vapor
from amalgam. J. dent. Res., 67(9): 1235-1242.
BERGLUND, F. (1989) [Amalgam poisoning, a medical
reality.] Läkartidningen, 86(1-2): 41-42 ( in Swedish).
BERLIN, M. & JOHANSSON, L.G. (1964) Mercury in mouse brain
after inhalation of mercury vapour and after intravenous
injection of mercury salts. Nature, London, 204: 85-86.
BERLIN, M., FAZACKERLEY, J., & NORDBERG, G. (1969) The uptake of
mercury in the brains of mammals exposed to mercury vapor and
mercuric salts. Arch. environ. Health, 18: 719-729.
BERLIN, M., CARLSON, J., & NORSETH, T. (1975) Dose-dependence of
methyl-mercury metabolism. A study of distribution:
biotransformation and excretion in the Squirrel Monkey. Arch.
environ. Health, 30: 307-313.
BERNARD, A.M., ROELS, H.R., FOIDART, J.M., & LAUWERYS, R.L. (1987)
Search for anti-laminin antibodies in the serum of workers
exposed to cadmium, mercury vapour or lead. Int. Arch. occup.
environ. Health, 59: 303-309.
BERNARD, S.R. & PURDUE, P. (1984) Metabolic models for
methyl and inorganic mercury. Health Phys., 46(3): 695-699.
BERNAUDIN, J.F., DRUET, E., DRUET, P., & MASSE, R. (1981)
Inhalation or ingestion of organic or inorganic mercurials
produces auto-immune disease in rats. Clin. Immunol. Immunopathol.,
20: 129-135.
BIDSTRUP, P.L., BONNEL, J.A., HARVEY, D.G., & LOCKET, S. (1951)
Chronic mercury poisoning in men repairing direct-current meters.
Lancet, 2: 856-861.
BONDY, S.C., ANDERSON, C.L., HARRINGTON, M.E., & PRASAD, K.N.
(1979) The effects of organic and inorganic lead and mercury on
neurotransmitter high-affinity transport and release mechanisms.
Environ. Res., 19: 102-111.
BOWMAN, C., MASON, D.W., PUSEY, C.D., & LOCKWOOD, C.M.
(1984) Autoregulation of autoantibody synthesis in mercuric
chloride nephritis in the Brown-Norway rat. I. A role for T
suppressor cells. Eur. J. Immunol., 14: 464-470.
BRADY, J.A., GEMMITI-NUNN, D., POLAN, A.K., MITCHELL, D., WEIL,
R., & VIANNA, N.J. (1980) The relationship of dental practice
characteristics to blood mercury levels. NY State dent. J., 46: 420-
424.
BRIGATTI, L. (1949) [Mercury levels in human organs with and
without mercury exposure.] Med. Lav., 40(10): 233-239 (in Italian).
BRODSKY, J.B., COHEN, E.N., WHITCHER, C., BROWN, B.W., Jr, &
WU, M.L. (1985) Occupational exposure to mercury in dentistry
and pregnancy outcome. J. Am. Dent. Assoc., 111: 779-780.
BRUNE, D. (1981) Corrosion of amalgams. Scand. J. dent. Res., 89:
506-514.
BRUNE, D. & EVJE, D.M. (1985) Man's mercury loading from a dental
amalgam. Sci. total Environ., 44: 51-63.
BUCHET, J.P., ROELS, H., BERNARD, A., Jr, & LAUWERYS, R. (1980)
Assessment of renal function of workers exposed to inorganic lead,
cadmium or mercury vapor. J. occup. Med., 22(11): 741-750.
BUCHWALD, H. (1972) Exposure of dental workers to mercury. Am.
Ind. Hyg. Assoc. J., 33: 492-502.
BUNN, W.B., MCGILL, C.M., BARBER, T.E., CROMER, J.W., Jr, &
GOLDWATER, L.J. (1986) Mercury exposure in chloralkali plants.
Am. Ind. Hyg. Assoc. J., 47(5): 249-254.
BURK, R.F., FOSTER, K.A., GREENFIELD, P.M., & KIKER, K.W. (1974)
Binding of simultaneously administered inorganic selenium and
mercury to a rat plasma protein (37894). Proc. Soc. Exp. Biol.
Med., 145: 782-785.
BURROWS, D. (1986) Hypersensitivity to mercury, nickel and
chromium in relation to dental materials. Int. dent. J., 36: 30-34.
CANTONI, O. & COSTA, M. (1983) Correlations of DNA strand breaks
and their repair with cell survival following acute exposure to
mercury (II) and X-rays. Mol. Pharmacol. 24: 84-89.
CANTONI, O., EVANS, R.M., & COSTA, M. (1982) Similarity in the
acute cytotoxic response of mammalian cells to mercury (II) and
X-rays: DNA damage and glutathione depletion. Biochem. Bioophys.
Res. Commun., 108(2): 614-619.
CANTONI, O., CHRISTIE, N.T., ROBISON, S.H., & COSTA, M.
(1984a) Characterization of DNA lesions produced by HgCl2 in cell
culture systems. Chem.-biol. Interact., 49: 209-224.
CANTONI, O., CHRISTIE, N.T., SWANN, A., DRATH, D.B., COSTA, M.
(1984b) Mechanism of HgCl2 cytotoxicity in cultured
mammalian cells. Mol. Pharmacol., 26: 360-368.
CASSANO, G.B., AMADUCCI, L., & VIOLA, P.L. (1966) Distribution of
mercury (H203) in the brain of chronically intoxicated mice
(autoradiographic study). Riv. Pat. nerv. ment., 87: 214-225.
CENTERS FOR DISEASE CONTROL (1990) Mercury exposure from
interior latex paint. CDC Mortal. Morb. Wkly Rep., 39(8): 125-126.
CHALOPING, J.M. & LOCKWOOD, C.M. (1984) Autoregulation of
autoantibody synthesis in mercuric chloride nephritis in the
Brown Norway rat. II. Presence of antigen-augmentable plaque-
forming cells in the spleen is associated with humoral
factors behaving as auto-anti-idiotypic antibodies. Eur. J.
Immunol., 14: 470-475.
CHANG, S.B., SIEW, C., & GRUNINGER, S.E. (1987) Examination of
blood levels of mercurials in practicing dentists using cold-
vapor atomic absorption spectrometry. J. anal. Toxicol., 11: 149-153.
CHEN, R.W., WHANGER, P.D., & FANG, S.C. (1974) Diversion of
mercury binding in rat tissues by selenium: A possible mechanism
of protection. Pharmacol. Res. Commun., 6(6): 571-579.
CHERIAN, M.G., HURSH, J.B., CLARKSON, T.W., & ALLEN, J. (1978)
Radioactive mercury distribution in biological fluids and
excretion in human subjects after inhalation of mercury vapor.
Arch. environ. Health, 33: 109-114.
CHMIELNICKA, J., BRZEZNICKA, E., & SNIADY, A. (1986) Kidney
concentrations and urinary excretion of mercury, zinc and
copper following the administration of mercuric chloride and
sodium selenite to rats. Arch. Toxicol., 59: 16-20.
CHOWDHURY, A.R., VACHHRAJANI, K.D., MAKHIJA, S., & KASHYAP, S.K.
(1986) Histomorphometric and biochemical changes in the
testicular tissues of rats treated with mercuric chloride.
Biomed. Biochim. Acta, 45(7): 949-956.
CHRISTIE, N.T., CANTONI, O., EVANS, R.M., MEYN, R.E., & COSTA, M.
(1984) Use of mammalian DNA repair-deficient mutants to assess
the effects of toxic metal compounds on DNA. Biochem. Pharmacol.,
33(10): 1661-1670.
CHRISTIE, N.T., CANTONI, O., SUGIYAMA, M., CATTEBENI, F., &
COSTA, M. (1986) Differences in the effects of Hg(II) on DNA
repair induced in Chinese hamster ovary cells by ultraviolet or
X-rays. Mol. Pharmacol., 29: 173-178.
CLARKSON, T.W. & KILPPER, R.W. (1978) The metabolism of inhaled
vapor in animals and man. In: Clarkson, T.W., ed. Heavy metals
as environmental hazards to man, Rochester, New York,
Environmental Health Sciences Center Program Project, pp. 449A,
449B.
CLARKSON, T.W., MAGOS, L., & GREENWOOD, M.R. (1972) The
transport of elemental mercury into fetal tissues. Biol. Neonate,
21: 239-244.
CLARKSON, T.W., FRIBERG, L., HURSH, J.B., & NYLANDER, M.
(l988a) The prediction of intake of mercury vapor from amalgams.
In: Clarkson, T.W., Friberg, L., Nordberg, G.F., & Sager, P.,
ed. Biological monitoring of metals, New York, London, Plenum
Press, pp. 247-264.
CLARKSON, T.W., HURSH, J.B., SAGER, P.R., & SYVERSEN, T.L.M.
(1988b) Mercury. In: Clarkson, T.W., Friberg, L., Nordberg, G.F., &
Sager, P., ed. Biological monitoring of metals, New York, London,
Plenum Press, pp. 199-246.
CLARKSON, T.W., FRIBERG. L., NORDBERG., G.F., & SAGER, P. ed.
(1988c) Biological monitoring of metals, New York, London, Plenum
Press, 686 pp.
CLOEZ, I., DUMONT, O., PICIOTTI, M., & BOURRE, J.M. (1987)
Alterations of lipid synthesis in the normal and dysmyelinating
trembler mouse sciatic nerve by heavy metals (Hg, Pb, Mn, Cu, Ni).
Toxicology, 46: 65-71.
COOLEY, R.L. & BARKMEIER, W.W. (1978) Mercury vapor emitted
during ultraspeed cutting of amalgam. J. Indiana Dent. State
Assoc., 57(2): 28-31.
COOMBS, R.R.A. & GELL, P.G.H. (1975) Classification of allergic
reactions responsible for clinical hypersensitivity and disease.
In: Gell, P.G.H., Coombs, R.R.A., & Lachmann, P.J. ed. Clinical
aspects of immunology, 3rd ed., Oxford, London, Blackwell
Scientific Publications, pp. 761-781.
COYLE, P. & HARTELY, T. (1981) Automated determination of mercury
in urine and blood by the Magos reagent and cold vapor
atomic absorption spectrometry. Anal. Chem., 53: 354-356.
CRAGLE, D.L., HOLLIS, D.R., QUALTERS, J.R., TANKERSLEY, W.G., &
FRY, S.A. (1984) A mortality study of men exposed to elemental
mercury. J. occup. Med., 26(11): 817-821.
CRC (1972) Handbook of chemistry and physics, 32nd ed.,
Cleveland, Ohio, CRC Press.
CROSS, J.D., DALE, I.M., GOOLVARD, L., LENIHAN, J.M.A., & SMITH, H.
(1978) Methyl mercury in blood of dentists. Lancet, II: 312-313.
DASTON, G.P., KAVLOCK, R.J., ROGERS, E.H., & CARVER, B. (1983)
Toxicity of mercuric chloride to the developing rat kidney. I.
Postnatal ontogeny of renal sensitivity. Toxicol. appl. Pharmacol.,
71: 24-41.
DAVIS, L.E., WANDS, J.R., WEISS, S.A., PRICE, D.L., & GIRLING, E.F.
(1974) Central nervous system intoxication from mercurous
chloride laxatives. Quantitative, histochemical and
ultrastructural studies. Arch. Neurol., 30: 428-431.
DEAN, R.B. & SUESS, M.J. (1985) The risk to health of chemicals in
sewage sludge applied to land. Waste Manage. and Res., 3: 251-278.
DE ROSIS, F., ANASTASIO, S.P., SELVAGGI, L., BELTRAME, A., &
MORIANI, G. (1985) Female reproductive health in two lamp
factories: Effects of exposure to inorganic mercury vapour and
stress factors. Br. J. ind. Med., 42: 488-494.
DIAMOND, G.L. (1988) Biological monitoring of urine for exposure
to toxic metals. In: Clarkson, T.W., Friberg, L., Nordberg, G.F.,
& Sager, P., ed. Biological monitoring of metals, New York,
London, Plenum Press, pp. 515-529.
DRUET, E., SAPIN, C., GUNTHER, E., FEINGOLD, N., & DRUET, P.
(1977) Mercuric chloride-induced anti-glomerular basement membrane
antibodies in the rat. Eur. J. Immunol., 7: 348-351.
DRUET, E., SAPIN, C., FOURNIE, G., MANDET, C., GUNTHER, E., &
DRUET, P. (1982) Genetic control of susceptibility to
mercury-induced immune nephritis in various strains of rat.
Clin. Immunol. Immunopathol., 25: 203-212.
DRUET, P., DRUET, E., POTDEVIN, F., & SAPIN, C. (1978)
Immune type glomerulonephritis induced by HgCl2 in the Brown
Norway rat. Ann. Immunol., 129C: 777-792.
DRUET, P., TEYCHENNE, P., MANDET, C., BASCOU, C., & DRUET, E.
(1981) Immune-type glomerulonephritis induced in the Brown-
Norway rat with mercury-containing pharmaceutical products.
Nephron, 28: 145-148.
DUMAREY, R., DAMS, R., & HOSTE, J. (1985) Comparison of the
collection and desorption efficiency of activated charcoal,
silver, and gold for the determination of vapor-phase
atmospheric mercury. Anal. Chem., 57(13): 2638-2643.
DUNN, J.D., CLARKSON, T.W., & MAGOS, L. (1978) Ethanol-
increased exhalation of mercury in mice. Br. J. ind. Med., 35: 241-244.
DUNN, J.D., CLARKSON, T.W., & MAGOS, L. (1981a) Ethanol reveals
novel mercury detoxification step in tissues. Science, 213: 1123-1125.
DUNN, J.D., CLARKSON, T.W., & MAGOS, L. (1981b) Interaction of
ethanol and inorganic mercury: Generation of mercury vapor in vivo.
J. Pharmacol. exp. Ther., 216(1): 19-23.
DUXBURY, A.J., EAD, R.D., MCMURROUGH, S., & WATTS, D.C. (1982)
Allergy to mercury in dental amalgam. Br. dent. J., 152: 47-48.
EDIMED (1989) [The pharmaceutical informer. Italian directory of
drugs and manufacturers], 50th ed., Milano, Organizzazione
Editoriale Medico Farmaceutica (in Italian).
EGGLESTON, D.W. & NYLANDER, M. (1987) Correlation of dental
amalgam with mercury in brain tissue. J. prosthet. Dent., 58(6):
704-707.
EHRENBERG, R.L., BLAIR SMITH, A., MCMANUS, K.P., HANNON, W.H.,
BRIGHTWELL, W.S., LOWRY, L.K., ANGER, W.K., VOGT, R.L., BRONDUM,
J., & HUDSON, P.J. (1986) Health hazard evaluation report,
Cincinnati, Ohio, National Institute for Occupational Safety and
Health, 65 pp. (HETA, 83-465-1674).
EICHHORN, G.L. & CLARK, P. (1963) The reaction of mercury
(II) with nucleosides. J. Am. Chem. Soc., 85: 4020-4024.
ELINDER, G.-G., GERHARDSSON, L., & OBERDOERSTER, G. (1988)
Biological monitoring of metals. In: Clarkson, T.W., Friberg, L.,
Nordberg, G.F., & Sager, P., ed. Biological monitoring of metals,
New York, London, Plenum Press, pp. 1-71.
ENWONWU, C.O. (1987) Potential health hazard of use of
mercury in dentistry: Critical review of the literature. Environ.
Res., 42: 251-274.
ERICSON, A. & KALLEN, B. (1988) Pregnancy outcome in women
working as dentists, dental assistants or dental technicians.
Arch. occup. environ. Health, 61(5), 329-333.
ETO, K., TOKUNAGA, H., ITAI, Y., TAKIZAWA, Y., & SUDA, H.
(1988) [An autopsy of fetal Minamata disease which takes a
long course.] Report presented at a Conference on the
Comprehensive Study on Minamata Disease, 27 February, 1988,
sponsored by the Environmental Agency, Japan (in Japanese).
EYBL, V., SYKORA, J., & MERTL, F. (1969) [Influence of sodium
selenite, sodium tellurite and sodium sulphite on the retention
and distribution of mercury in mice.] Arch. Toxikol., 25:
296-305 (in German with English summary).
FAN, P.L. (1987) Safety of amalgam. Cal. dent. Assoc. J.,
September: 34-36.
FARANT, J.P., BRISSETTE, D., MONCION, L., BIGRAS, L., &
CHARTRAND, A. (1981) Improved cold-vapor atomic absorption
technique for the microdetermination of total and inorganic
mercury in biological samples. J. anal. Toxicol., 5: 47-51.
FAWER, R.F., DE RIBAUPIERRE, Y., GUILLEMIN, M.P., BERODE, M., &
LOB, M. (1983) Measurement of hand tremor induced by
industrial exposure to metallic mercury. Br. J. ind. Med., 40: 204-
208.
FILLIASTRE, J.-P., DRUET, P., & MERY, J.-P. (1988)
Proteinuric nephropathies associated with drugs and substances of
abuse. In: Cameron, J.S. & Glassok, R.J. ed. The nephrotic
syndrome, New York, Basel, Marcel Dekker Inc., pp. 697-742.
FINNE, K., GORANSSON, K., & WINCKLER, L. (1982) Oral lichen
planus and contact allergy to mercury. Int. J. oral Surg., 11: 236-239.
FOA, V., CAIMI, L., AMANTE, L., ANTONINI, C., GATTINONI, A.,
TETTAMANTI, G., LOMBARDO, A., & GIULIANI, A. (1976) Patterns of
some lysosomal enzymes in the plasma and of proteins in urine
of workers exposed to inorganic mercury. Int. Arch. occup. environ.
Health, 37: 115-124.
FOOTE, R.S. (1972) Mercury vapor concentrations inside buildings.
Science, 177: 513-514.
FORZI, M., CASSITTO, M.G., BULGHERONI, C., & FOA, V. (1976)
Psychological measures in workers occupationally exposed to
mercury vapours. A validation study. In: Adverse effects of
environmental chemicals and psychotoxic drugs, Amsterdam,
Oxford, New York, Elsevier Science Publishers, Vol. 2, pp.
165-172.
FRIBERG, L. (1956) Studies on the accumulation, metabolism and
excretion of inorganic mercury (Hg203) after prolonged
subcutaneous administration to rats. Acta pharmacol. toxicol., 12:
411-427.
FRIBERG, L. (1988) Quality assurance. In: Clarkson, T.W.,
Friberg, L., Nordberg, G.F., & Sager, P., ed. Biological
monitoring of metals, New York, London, Plenum Press,
pp. 103-126.
FRIBERG, L. & NYLANDER, M. (1987) [The release and uptake of
metallic mercury vapour from amalgam.] In: Mercury/amalgam -
health risks. Report by an expert group. Stockholm, National
Board of Health and Welfare, Report series Socialstyrelsen
Redovisar 1987, 10, pp. 65-79 (in Swedish with English summary).
FRIBERG, L. & VOSTAL, J. ed. (1972) [The release and uptake of
metallic mercury vapour from amalgam.] In: [Mercury/amalgam -
Health risks], Stockholm, National Board of Health and Welfare,
pp. 65-79 (Report series Socialstyrelsen Redovisar 10) (in Swedish
with English summary).
FRIBERG, L., SKOG, E., & WAHLBERG, J.E. (1961) Resorption of
mercuric chloride and methyl mercury dicyandiamide in guinea-
pigs through normal skin and through skin pre-treated with
acetone, alkylarylsulphonate and soap. Acta dermatovenereol., 41:
40-52.
FRIBERG, L., KULLMAN, L., LIND, B., & NYLANDER, M. (1986) [Mercury
in the central nervous system in relation to amalgam fillings.]
Laekartidningen, 83: 519-522 (in Swedish).
FRYKHOLM, K.O. (1957) Mercury from dental amalgam. Its toxic and
allergic effects and some comments on occupational hygiene. Acta
odont. Scand., 22: 1-108.
FUKATSU, A., BRENTJENS, J.R., KILLEN, P.D., KLEINMAN, H.K.,
MARTIN, G.R., & ANDRES, G.A. (1987) Studies on the formation
of glomerular immune deposits in Brown Norway rats injected
with mercuric chloride. Clin. Immunol. Immunopathol., 45: 35-47.
FUKINO, H., HIRAI, M., MEI HSUEH, Y., MORIYASU, S., & YAMANE, Y.
(1986) Mechanism of protection by zinc against mercuric chloride
toxicity in rats: Effects of zinc and mercury on glutathionine
metabolism. J. Toxicol. environ. Health, 19: 75-89.
FUKUDA, K. (1971) Metallic mercury induced tremor in rabbits and
mercury content of the central nervous system. Br. J. ind. Med., 28:
308-311.
GALE, T.F. (1980) Cardiac and non-cardiac malformations
produced by mercury in hamsters. Bull. environ. Contam. Toxicol.,
25: 726-732.
GALE, T.F. (1981) The embryotoxic response produced by inorganic
mercury in different strains of hamsters. Environ. Res., 24: 152-161.
GALE, T.F. (1984) The amelioration of mercury-induced embryotoxic
effects by simultaneous treatment with zinc. Environ. Res., 35: 405-
411.
GALE, T.F. & FERM, V.H. (1971) Embryopathic effects of mercuric
salts. Life Sci., 10: 1341-1347.
GANSER, A.L. & KIRSCHNER, D.A. (1985) The interaction of
mercurials with myelin: Comparison of in vitro and in vivo
effects. Neurotoxicology, 6(1): 63-78.
GAY, D.D., COX, R.D., & REINHARDT, J.W. (1979) Chewing releases
mercury from fillings. Lancet, 8123: 985-986.
GELLER, S.A. (1976) Subacute and chronic tissue reaction to
metallic mercury: Two cases and a review of the literature. Mt
Sinai J. Med., 43: 534-541.
GLEICHMANN, E., PALS, S.T., ROLINK, A.G., RADASZKIEWICZ, T., &
GLEICHMANN, H. (1984) Graft-versus-host reactions: clues to the
etiopathology of a spectrum of immunological diseases. Immunol.
Today, 5(11): 324-332.
GONCHARUK, G.A. (1977) [Problems relating to occupational hygiene
in women in production of mercury.] Gig. Tr. prof. Zabol., 5: 17-20 (in
Russian).
GORDON, H. (1981) Pregnancy in female dentists - a mercury
hazard? In: Proceedings of the International Conference on
Mercury Hazards in Dental Practice, Glasgow, 2-4 September, (Paper
31).
GOTELLI, C. (1989) Methylmercury in a gold mining region. Proc.
Nat. Inst. Environm. Health Sci. (in print).
GOTELLI, C.A., ASTOLFI, E., COX, C., CERNICHIARI, E., &
CLARKSON, T.W. (1985) Early biochemical effects of an organic
mercury fungicide on infants: "dose makes the poison". Science,
227: 638-640.
GRITZKA, T.L. & TRUMP, B.F. (1968) Renal tubular lesions
caused by mercuric chloride. Am. J. Pathol., 52(6): 1225-1277.
GRONKA, P.A., BOBKOSKIE, R.L., TOMCHICK, G.J., BACH, F., &
RAKOW, A.B. (1970) Mercury vapor exposures in dental offices. J.
Am. Dent. Assoc., 81: 923-925.
GRUENWEDEL, D.W. & DAVIDSON, N. (1966) Complexing and denaturation
of DNA by methylmercuric hydroxide. J. mol. Biol., 21: 129-144.
HAHN, L.J., KLOIBER, R., VIMY, M.J., TAKAHASHI, Y., &
LORSHEIDER, F.L. (1989) Dental "silver" tooth fillings: a source
of mercury exposure revealed by whole-body image scan and
tissue analysis. FASEB J., 3(14): 2641-2646.
HALBACH, S. & CLARKSON, T.W. (1978) Enzymatic oxidation of mercury
vapors by erythrocytes. Biochem. Biophys. Acta, 523(2): 522-531.
HANDKE, J.L. & PRYOR, P. (1981) Health hazard evaluation
report, Cincinnati, Ohio, National Institute for Occupational
Safety and Health, 26 pp. (HHE 78-034-930).
HANSEN, J.C., KRISTENSEN, P., & AL-MASRI, S.N. (1981)
Mercury/selenium interaction. A comparative study on pigs. Nord.
vet. Med., 33: 57-64.
HANSEN, J.C., RESKE-NIELSEN, E., THORLACIUS-USSING, O., RUNGBY,
J., & DANSHER, G. (1989) Distribution of dietary mercury in a
dog. Quantitation and localization of total mercury in organs
and central nervous system. Sci. tot. Environ., 78: 23-43.
HANSSON, M. (1986) [Changes in health after removal of toxic
dental filling materials.] TF-BLADET, 1: 3-30 (in Swedish).
HARRIS, D., NICOLS, J.J., STARK, R., & HILL, K. (1978) The dental
working environment and the risk of mercury exposure. J. Am.
Dent. Assoc., 97: 811-815.
HATCH, W.R. & OTT, W.L. (1968) Determination of sub-microgram
quantities of mercury by atomic absorption spectrophotometry.
Anal. Chem., 40: 2085-2087.
HEIDAM, L.Z. (1984) Spontaneous abortions among dental assistants,
factory workers, painters, and gardening workers: A follow up
study. J. Epidemiol. community Health, 38: 149-155.
HEINTZE, U., EDWARDSSON, S., DERAND, T., & BIRKHED, D. (1983)
Methylation of mercury from dental amalgam and mercuric chloride
by oral streptococci in vitro . Scand. J. dent. Res., 9l: 150-152.
HIRSCH, F., COUDERC, J., SAPIN, C., FOURNIE, G., & DRUET, P.
(1982) Polyclonal effect of HgCl2 in the rat, its possible
role in an experimental autoimmune disease. Eur. J. Immunol., 12:
620-625.
HIRSHMAN, S.Z., FEINGOLD, M., & BOYLEN, G. (1963) Mercury in
house paint as a cause of acrodynia: effect of therapy
with N-acetyl-D,L-penicillamine. N. Engl. J. Med., 269: 889-893.
HOLT, D. & WEBB, M. (1986) The toxicity and teratogenicity of
mercuric mercury in the pregnant rat. Arch. Toxicol., 58: 243-248.
HUBERLANT, J.M., ROELS, H., BUCHET, J.P., BERNARD, A., &
LAUWERYS, R. (1983) Evaluation de l'exposition au mercure et
de ses répercussions éventuelles sur la santé du personnel
d'une trentaine de cabinets dentaires. Cah. Méd. Trav., 20(2):
109-127.
HUDSON, P.J., VOGT, R.L., BRONDUM, J., WITHERELL, L., MYERS,
G., & PASCHAL, D.C. (1987) Elemental mercury exposure among
children of thermometer plant workers. Pediatrics, 79(6): 935-938.
HULTMAN, P. & ENESTROM, S. (1987) The induction of immune complex
deposits in mice by peroral and parenteral administration of
mercuric chloride: Strain dependent susceptibility. Clin. exp.
Immunol., 67: 283-292.
HULTMAN, P. & ENESTROM, S. (1988) Mercury induced antinuclear
antibodies in mice: characterization and correlation with
renal immune complex deposits. Clin. exp. Immunol., 71: 269-274.
HURSH, J.B. (1985) Particle coefficients of mercury (203Hg) vapour
between air and biological fluids. J. appl. Toxicol., 5: 327-332.
HURSH, J.B., CLARKSON, T.W., CHERIAN, M.G., VOSTAL, J.V., &
MALLIE, R.V. (1976) Clearance of mercury (Hg-197, Hg-203) vapor
inhaled by human subjects. Arch. environ. Health, 31: 302-309.
HURSH, J.B., GREENWOOD, M.R., CLARKSON, T.W., ALLEN, J., &
DEMUTH, S. (1980) The effect of ethanol on the fate of mercury
vapor inhaled by man. J. Pharmacol. exp. Ther., 214: 520-527.
HURSH, J.B., SICHAK, S.P., & CLARKSON, T.W. (1988) In vitro
oxidation of mercury by the blood. Pharmacol. Toxicol., 63: 266-273.
HURSH, J.B., CLARKSON, T.W., MILES, E., & GOLDSMITH, L.A.
(1989) Percutaneous absorption of mercury vapour by man. Arch.
environ. Health, 44(2): 120-127.
IARC (1987) Overall evaluation of carcinogenicity: An updating
of IARC Monographs Volumes 1 to 42, Lyon, International Agency
for Research on Cancer, 440 pp. (IARC Monographs on the Evaluation
of Carcinogenic Risk to Humans, Suppl. 7).
ICRP (1980) Metabolic data for mercury. Ann. int. Comm. rad.
Prot., 30/Part 2, 4: 59-63.
IMURA, N. & NAGANUMA, A. (1978) Interaction of inorganic
mercury and selenite in rabbit blood after intravenous
administration. J. Pharm. Dyn., 1: 67-73.
INTERNATIONAL CLEARING-HOUSE FOR BIRTH DEFECTS MONITORING SYSTEM
(1985) Annual report. Sidney, National Perinatal Statistics Unit,
University of Sidney, 71 p. (ISSN 0743-5703).
ISHIHARA, N., URUSHIYAMA, K., & SUZUKI, T. (1977) Inorganic and
organic mercury in blood, urine and hair in low level mercury
vapour exposure. Int. Arch. occup. environ. Health, 40: 249-253.
JACOBS, M. & GOLDWATER, L.J. (1965) Absorption and excretion of
mercury in man: VIII. Mercury exposure from house paint - a
controlled study on humans. Arch. environ. Health, 11: 582-587.
JOHANSSON, E. & LINDH, U. (1987) Mercury in blood cells -
altered elemental profiles. Toxic events in human exposure.
Biol. Trace Elem. Res., 12: 309-321.
JOHNSON, S.L. & POND, W.G. (1974) Inorganic vs. organic Hg
toxicity in growing rats: Protection by dietary Se but not Zn.
Nutr. Rep. Int., 9(2): 135-147.
JOKSTAD, A. (1987) [Mercury exposure of dentists]. J. Norw. Dent.
Assoc., 97: 498-507 (in Norwegian).
JOSELOW, M.M. & GOLDWATER, L.J. (1967) Absorption and excretion of
mercury in man. XII. Relationship between urinary mercury and
proteinuria. Arch. environ. Health, 15: 155.
JOSELOW, M.M., GOLDWATER, L.J., ALVAREZ, A., & HERNDON, J.
(1968) Absorption and excretion of mercury in man. Arch. environ.
Health, 17: 39-43.
JUANG, M.S. (1976) An electrophysiological study of the
action of methylmercuric chloride and mercuric chloride on
the sciatic nerve-sartorius muscle preparation of the frog.
Toxicol. appl. Pharmacol., 37: 339-348.
JUGO, S. (1976) Retention and distribution of 203HgCl2 in
suckling and adult rats. Health Phys., 30: 240-241.
KANEMATSU, N., HARA, M., & KADA, T. (1980) Rec assay and
mutagenicity studies on metal compounds. Mutat. Res., 77: 109-116.
KARK, P. (1979) Clinical and neurochemical aspects of inorganic
mercury intoxication. In: Handbook of clinical neurology,
Amsterdam, Oxford, New York, Elsevier Science Publishers, Vol. 36,
pp. 147-197.
KAZANTZIS, G. (1978) The role of hypersensitivity and the immune
response in influencing susceptibility to metal toxicity.
Environ. Health Perspect., 25: 111-118.
KAZANTZIS, G. (1981) Role of cobalt, iron, lead, manganese,
mercury, platinum, selenium, and titanium in carcinogenesis.
Environ. Health Perspect., 40: 143-161.
KAZANTZIS, G. & LILLY, L.J. (1986) Mutagenic and carcinogenic
effects of metals. In: Friberg, L., Nordberg, G.F., & Vouk, V.,
ed. Handbook on the toxicology of metals, Amsterdam, Oxford,
New York, Elsevier Science Publishers, Vol. 1, pp. 319-390.
KAZANTZIS, G., SCHILLER, K.F.R., ASSCHER, A.W., & DREW, R.G.
(1962) Albuminuria as the nephrotic syndrome following exposure
to mercury and its compounds. Q. J. Med., 31(124): 403-418.
KELMAN, G.R. (1978) Notes and miscellanea: Urinary mercury
excretion in dental personnel. Br. J. ind. Med., 35: 262-265.
KHAYAT, A. & DENCKER, L. (1982) Fetal uptake and distribution of
metallic mercury vapor in the mouse: Influence of ethanol and
aminotriazole. Biol. Res. Pregn., 3(1): 38-46.
KHAYAT, A. & DENCKER, L. (1983a) Whole body and liver
distribution of inhaled mercury vapor in the mouse: Influence of
ethanol and aminotriazole pretreatment. J. appl. Toxicol., 3: 66-74.
KHAYAT, A. & DENCKER, L. (1983b) Interactions between selenium and
mercury in mice: Marked retention in the lung after
inhalation of metallic mercury. Chem. biol. Interact., 46: 283-298.
KHAYAT, A. & DENCKER, L. (1984) Organ and cellular distribution of
inhaled metallic mercury in the rat and marmoset monkey
(Callithrix jacchus): Influence of ethyl alcohol pretreatment.
Acta pharmacol. toxicol., 55: 145-152.
KIBUKAMUSOKE, J.W., DAVIES, D.R., & HUTT, M.S.R. (1974)
Membranous nephropathy due to skin-lightening cream. Br. med. J.,
2: 646-647.
KIMATA, H., SHINOMIYA, K., & MIKAWA, H. (1983) Selective
enhancement of human IgE production in vitro by synergy of
pokeweed mitogen and mercuric chloride. Clin. exp. Immunol., 53:
183-191.
KISHI, R., HASHIMOTO, K., SHIMIZU, S., & KOBAYASHI, M. (1978)
Behavioral changes and mercury concentrations in tissues of rats
exposed to mercury vapor. Toxicol. appl. Pharmacol., 46: 555-566.
KITAMURA, S., SUMINO, K., HAYAKAWA, K., & SHIBATA, T. (1976)
Mercury content in human tissues from Japan. In: Nordberg, G.F.,
ed. Effects and dose-response relationships of toxic metals,
Amsterdam, Oxford, New York, Elsevier Science Publishers,
pp. 290-298.
KNEIP, T.J. & FRIBERG, L. (1986) Sampling and analytical
methods. In: Friberg, L., Nordberg, G.F., & Vouk, V.B., ed.
Handbook on the toxicology of metals, 2nd ed., Amsterdam,
Oxford, New York, Elsevier Science Publishers, Vol. 1,
pp. 36-67.
KNOFLACH, P., ALBINI, B., & WEISER, M.M. (1986) Autoimmune disease
induced by oral administration of mercuric chloride in Brown-Norway
rats. Toxicol. Pathol., 14(2): 188-193.
KOMSTA-SZUMSKA, E. & CHMIELNICKA, J. (1977) Binding of
mercury and selenium in subcellular fractions of rat liver and
kidneys following separate and joint administration. Arch.
Toxicol., 38: 217-228.
KOSTA, L., BYRNE, A.R., & ZELENKO, V. (1975) Correlation between
selenium and mercury in man following exposure to inorganic
mercury. Nature, London, 254: 238-239.
KOSTIAL, K. & LANDEKA, M. (1975) The action of mercury ions on the
release of acetylcholine from presynaptic nerve endings.
Experientia, Basel, 31(7): 834-835.
KOSTIAL, K., KELLO, D., JUGO, S., RABAR, I., & MALJKOVIC, T.
(1978) Influence of age on metal metabolism and toxicity.
Environ. Health Perspect., 25: 81-86.
KOSTIAL, K., SIMONOVIC, I., RABAR, I., BLANUSA, M., & LANDEKA, M.
(1983) Age and intestinal retention of mercury and cadmium in
rats. Environ. Res., 31: 111-115.
KUSAKAWA, S. & HEINER, D.C. (1976) Elevated levels of
immunoglobin E in the acute febrile mucocutaneous lymph node
syndrome. Pediatr. Res., 10: 108-112.
LAMBETH, LONDON BOROUGH (1988) Mercury soaps and skin
lightening cream Directorate of environmental and consumer
services, London.
LAMPERTI, A.A. & PRINTZ, R.H. (1973) Effects of mercuric chloride
on the reproductive cycle of the female hamster. Biol. Reprod., 8:
378-387.
LAMPERTI, A.A. & PRINTZ, R.H. (1974) Localization, accumulation,
and toxic effects of mercuric chloride on the reproductive axis
of the female hamster. Biol. Reprod., 11: 180-186.
LAMPERTI, A. & NIEWENHUIS, R. (1976) The effects of mercury
on the structure and function of the hypothalamo-pituitary axis
in the hamster. Cell Tissue Res., 170: 315-324.
LANGAN, D.C., FAN, P.L., & HOOS, A.A. (1987) The use of
mercury in dentistry: A critical review of the recent
literature. J. Am. Dent. Assoc., 115: 867-880.
LANGOLF, G.D., CHAFFIN, D.B., HENDERSON, R., & WHITTLE, H.P. (1978)
Evaluation of workers exposed to elemental mercury using
quantitative tests of tremor and neuromuscular functions. Am. Ind.
Hyg. Assoc. J., 39: 976-984.
LANGOLF, G.D., SMITH, P.J., HENDERSON, R., & WHITTLE, H.P.
(1981) Measurements of neurological functions in the evaluations
of exposure to neurotoxic agents. Ann. occup. Hyg., 24(3): 293-296.
LANGWORTH, S. (1987) Renal function in workers exposed to
inorganic mercury. In: Occupational health in the chemical
industry: papers presented at the XXII ICOH congress, Sidney,
Australia, 27 September-2 October 1987. Copenhagen, World Health
Organization, p. 237.
LANGWORTH, S., ELINDER, C.-G., & AKESSON, A. (1988) Mercury
exposure from dental fillings. I Mercury concentrations in blood
and urine. Swed. dent. J., 12: 69-70.
LAUWERYS, R., BERNARD, A., ROELS, H., BUCHET, J.P., GENNART, J.P.,
MAHIEU, P., & FOIDART, J.M. (1983) Anti-laminin antibodies in
workers exposed to mercury vapour. Toxicol. Lett., 17: 113-116.
LAUWERYS, R., ROELS, H., GENET, P., TOUSSAINT, G., BOUCKAERT,
A., & DE COOMAN, S. (1985) Fertility of male workers exposed to
mercury vapor or to manganese dust: A questionnaire study.
Am. J. ind. Med., 7: 171-176.
LAUWERYS, R., BONNIER, C., EVRARD, P., GENNART, J., & BERNARD, A.
(1987) Prenatal and early postnatal intoxication by inorganic
mercury resulting from the maternal use of mercury-containing
soap. Hum. Toxicol., 6: 253-256.
LAVSTEDT, S. & SUNDBERG, H. (1989) [Medical diagnosis and disease
symptoms related to amalgam fillings.] Tandläkartidningen, 3: 81-88 (in
Swedish).
LEE, I.P. & DIXON, R.L. (1975) Effects of mercury on
spermatogenesis studied by velocity sedimentation cell separation
and serial mating. J. Pharmacol. exp. Ther., 194(1): 171-181.
LEE, S.A. (1984) Health hazard evaluation report, Cincinnati,
Ohio, National Institute for Occupational Safety and Health, 10
pp. (HETA 83-338-1399).
LEONARD, A., JACQUET, P., & LAUWERYS, R.R. (1983)
Mutagenicity and teratogenicity of mercury compounds. Mutat. Res.,
114: 1-18.
LEVIN, M., JACOBS, J., & POLOS, P.G. (1988) Acute mercury
poisoning and mercurial pneumonitis from gold ore purification.
Chest, 94: 554-556.
LEVINE, S.P., CAVENDER, G.D., LANGOLF, G.D., & ALBERS, J.W.
(1982) Elemental mercury exposure: peripheral neurotoxicity. Br.
J. ind. Med., 39: 136-139.
LIND, B,. FRIBERG, L., & NYLANDER, M. (1988) Preliminary
studies on methylmercury biotransformation and clearance in the
brain of primates: II. Demethylation of mercury in brain. J. Trace
Elem. exp. Med., 1: 49-56.
LINDBERG, S., STOKES, P., GOLDBERG, E., & WREN, C. (1987) Group
Report: Mercury. In: Hutchinson, T.W. & Meemz, K.M. ed. Lead,
mercury, cadmium and arsenic in the environment, New York,
Chichester, Brisbane, Toronto, John Wiley & Sons, pp. 17-34 (Scope
31).
LINDQVIST, K.J., MAKENE, W.J., SHABA, J.K., & NANTULYA, V.
(1974) Immunofluorescence and electron microscopic studies of
kidney biopsies from patients with nephrotic syndrome, possibly
induced by skin lightening creams containing mercury. East Afr.
med. J., 51: 168-169.
LINDQVIST, O, JERNELOV, A., JOHANNSON, K., & RODHE, R. (1984)
Mercury in the swedish environment: global and local sources.
Solna, National Environmental Protection Board, 105 pp. (Report
No. 1816).
LINDSTEDT, G., GOTTBERG, I., HOLMGREN, B., JONSSON, T., &
KARLSSON, G. (1979) Individual mercury exposure of chloralkali
workers and its relation to blood and urinary mercury levels.
Scand. J. Work Environ. Health, 5: 59-69.
LINKE, W.F. (1958) Inorganic and metal-organic compounds, New
York, Van Nostrand Reinhold Company Inc.
LUNDBORG, M., LIND, B., & CAMNER, P. (1984) Ability of rabbit
alveolar macrophages to dissolve metals. Exp. Lung Res., 7: 11-22.
MABILLE, V., ROELS, H., JACQUET, P., LEONARD, A., & LAUWERYS, R.
(1984) Cytogenetic examination of leucocytes of workers
exposed to mercury vapour. Int. Arch. occup. environ. Health, 53:
257-260.
MCCAMMON, C.S., Jr, EDWARDS, S.L., DELON HULL, R., & WOODFIN, W.J.
(1980) A comparison of four personal sampling methods for the
determination of mercury vapor. Am. Ind. hyg. Assoc. J., 41: 528-531.
MCFARLAND, R.B. & REIGEL, H. (1978) Chronic mercury poisoning
from a single brief exposure. J. occup. Med., 20(8): 532-534.
MACKERT, J.R., Jr, (1987) Factors affecting estimation of dental
amalgam mercury exposure from measurements of mercury vapor
levels in intra-oral and expired air. J. dent. Res., 66(12): 1775-1780.
MCNERNEY, J.J, BUSECK, P.R., & HANSON, R.C. (1972) Mercury
detection by means of thin gold films. Science, 178: 611-612.
MAGNUSSON, J. & RAMEL, C. (1986) Genetic variation in the
susceptibility to mercury and other metal compounds in Drosophila
melanogaster. Teratog. Carcinog. Mutagen., 6: 289-192.
MAGOS, L. (1971) Selective atomic-absorption determination of
inorganic mercury and methylmercury in undigested biological
samples. Analyst, 96: 847-853.
MAGOS, L. & CLARKSON, T.W. (1972) Atomic absorption
determination of total, inorganic and organic mercury in blood. J.
Assoc. Off. Anal. Chem., 5(5): 966-971.
MAGOS, L., WEBB, M., & BUTLER, W.H. (1974) The effect of
cadmium pretreatment on the nephrotoxic action and kidney uptake
of mercury in male and female rats. Br. J. exp. Pathol., 55: 589-594.
MAGOS, L., HALBACH, S., & CLARKSON, T.W. (1978) Role of catalase
in the oxidation of mercury vapor. Biochem. Pharmacol., 27: 1373-1377.
MAGOS, L., SPARROW, S., & SNOWDEN, R. (1982) The
comparative renotoxicology of phenylmercury and mercuric chloride.
Arch. Toxicol., 50: 133-139.
MAGOS, L., CLARKSON, T.W., & HUDSON, A.R. (1984) Differences
in the effects of selenite and biological selenium on the
chemical form and distribution of mercury after the simultaneous
administration of HgCl2 and selenium to rats. J. Pharmacol. exp.
Ther., 228(2): 478-483.
MAGOS, L., BROWN, A.W., SPARROW, S., BAILEY, E., SNOWDEN, R.T., &
SKIPP, W.R. (1985) The comparative toxicology of ethyl- and
methylmercury. Arch. Toxicol., 57: 260-267.
MAGOS, L., CLARKSON, T.W., SPARROW, S., & HUDSON, A.R. (1987)
Comparison of the protection given by selenite,
selenomethionine and biological selenium against the renotoxicity
of mercury. Arch. Toxicol., 60: 422-426.
MANALIS, R.S. & COOPER, G.P. (1975) Evoked transmitter release
increased by inorganic mercury at frog neuromuscular junction.
Nature, London, 257: 690-691.
MARAFANTE, E., LUNDBORG, M., VAHTER, M., & CAMNER, P. (1987)
Dissolution of two arsenic compounds by rabbit alveolar
macrophages in vitro . Fundam. appl. Toxicol., 8: 382-388.
MARZULLI, F.N. & BROWN, D.W.C. (1972) Potential systemic
hazards of topically applied mercurials. J. Soc. cosmet. Chem., 23:
875-886.
MATHESON, D.S., CLARKSON, T.W., & GELFAND, E.W. (1980) Mercury
toxicity (acrodynia) induced by long-term injection of
gammaglobulin. J. Pediatr., 97(1): 153-155.
MATTIUSSI, R., ARMELI, G., & BAREGGI, V. (1982) Statistical study
of the correlation between mercury exposure (TWA) and
urinary mercury concentrations in chloralkali workers. Am. J.
ind. Med., 3: 335-339.
MENGEL, H. & KARLOG, O. (1980) Studies on the interaction and
distribution of selenite, mercuric, methoxyethyl mercuric and
methyl mercuric chloride in rats. II. Analysis of the soluble
proteins and the precipitates of liver and kidney homogenates.
Acta pharmacol. toxicol., 46: 25-31.
MILLER, E.G., PERRY, W.L., & WAGNER, M.J. (1987) Prevalence of
mercury hypersensitivity in dental students. J. prosthet. Dent., 58:
235-237.
MILLER, J.M., CHAFFIN, D.B., & SMITH, R.G. (1975) Subclinical
psychomotor and neuromuscular changes in workers exposed to
inorganic mercury. Am. Ind. Hyg. Assoc. J., 36: 725-733.
MINAGAWA, K., HIRASAWA, F., HOZUMI, S., CHUANG, K., & TAKIZAWA, Y.
(1979) [Rapid determinations of total mercury and methylmercury
in biological samples.] Akita J. Med., 5: 263-271 (in Japanese
with English summary).
MIRTSCHEWA, J., NURNBERGER, W., HALLMANN, B., STILLER-WINKLER,
R., & GLEICHMANN, E. (1987) Genetically determined
susceptibility of mice to HgCl2-induced antinuclear antibodies
(ANA) and glomerulo-nephritis. Immunobiology, 175: 323-324.
MIYAMA, T. & SUZUKI, T. (1971) Release of mercury from organs and
breakage of carbon-mercury bond in tissues due to formalin
fixation. Jpn. J. ind. Health, 13(6): 56-57.
MIYAMOTO, M.D. (1983) Hg2+ causes neurotoxicity at an
intracellular site following entry through Na and Ca channels.
Brain Res., 267: 375-379.
MOLIN, M., BERGMAN, B., MARKLUND, S.L., SCHUTZ, A., & SKERFVING, S.
(1990) Mercury, selenium and glutathione peroxidase before and
after amalgam removal in man. Acta Odontol. Scand., 48(3): 189-202.
MOLLER-MADSEN, B. & DANSCHER, G. (1986) Localization of mercury in
CNS of the rat. Environ. Res., 41: 29-43.
MORIMOTO, K., IIJIMA, S., & KOIZUMI, A. (1982) Selenite
prevents the induction of sister-chromatid exchanges by
methylmercury and mercuric chloride in human whole-blood
cultures. Mutat. Res., 102: 183-192.
MORNER, S. & NILSSON, T. (1986) [Mercury discharge from
crematories in Gothenburg], City of Gothenburg, Environmental
and Health Protection Agency (Report 1986.1) (in Swedish).
MORROW, P.E., GIBB, F.R., & JOHNSON, L. (1964) Clearance of
insoluble dust from the lower respiratory tract. Health Phys., 10:
543-555.
MOTTET, K. & BURBACHER, T. M. (1988) Preliminary studies on
methylmercury biotransformation and clearance in the brain of
primates: I. Experimental design and general observations J. Trace
Elem. exp. Med., 1: 41-47.
MULLER, H.,VON, SCHUBERT, H., & VERBEEK, W. (1980) [Report
on the relationship between Hg concentration in urine and
blood and Hg concentration in workroom air.] Arbeitsmed.
Sozialmed. Präventivmed., 15: 64-67 (in German with English
summary).
MURAMATSU, Y. & PARR, R.M. (1985) Survey of currently available
reference materials for use in connection with the determination
of trace elements in biological and environmental materials.
Vienna, International Atomic Energy Agency (Report No.
IAEA/RL/128).
MUTTI, A., LUCERTINI, S., FORNARI, M., FRANCHINI, I., BERNARD, A.,
ROELS, H., & LAUWERYS, R. (1985) Urinary excretion of a brush
border antigen revealed by monoclonal antibodies in subjects
occupationally exposed to heavy metals. In: Proceedings of the
International Conference on Heavy Metals in the Environment,
Luxembourg, Commission of the European Communities, Vol. 1, pp.
565-567.
NAGANUMA, A. & IMURA, N. (1980) Bis(methylmercuric) selenide as a
reaction product from methylmercury and selenite in rabbit
blood. Res. Commun. chem. Pathol. Pharmacol., 27(1): 163-173.
NAKAAKI, K., FUKABORI, S., & TADA, O. (1975) [An experimental
study on inorganic mercury vapour exposure.] J. Sci. Labour,
51(12): 705-716 (in Japanese with English summary).
NAKAAKI, K., FUKABORI, S., & TADA, O. (1978) On the evaluation of
mercury exposure. J. sci. Labour, 54: 1-8.
NAKADA, S., NOMOTO, A., & IMURA, N. (1980) Effect of
methylmercury and inorganic mercury on protein synthesis in
mammalian cells. Ecotoxicol. environ. Saf., 4: 184-190.
NAKAYAMA, H., NIKI, F., SHONO, M., & HADA, S. (1983) Mercury
exanthem. Contact dermatitis, 9: 411-417.
NALEWAY, C., SAKAGUCHI, R., MITCHELL, E., MULLER, T., AYER,
W.A., & HEFFERREN, J.J. (1985) Urinary mercury levels in US
dentists, 1975-1983: Review of Health Assessment Program. J. Am.
Dent. Assoc., 111: 37-42.
NATIONAL ACADEMY OF SCIENCES (1978) An assessment of mercury
in the environment, Washington, DC, National Academy of
Sciences, National Research Council.
NEWTON, D. & FRY, F.A. (1978) The retention and
distribution of radioactive mercuric oxide following accidental
inhalation. Ann. occup. Hyg., 21: 21-32.
NILSEN, A., NYBERG, K., & CAMNER, P. (1988) Intraphagosomal pH in
alveolar macrophages after phagocytosis in vivo and in vitro of
fluorescein-labeled yeast particles. Exp. Lung Res., 13: 197-207.
NILSSON, B. & NILSSON, B. (1986a) Mercury in dental practice.
I. The working environment of dental personnel and their
exposure to mercury vapor. Swed. dent. J., 10: 1-14.
NILSSON, B. & NILSSON, B. (1986b) Mercury in dental practice. II.
Urinary mercury excretion in dental personnel. Swed. dent. J., 10:
221-232.
NISHIYAMA, S., TAGUCHI, T., & ONOSAKA, S. (1987) Induction of
zincthionein by estradiol and protective effects on inorganic
mercury-induced renal toxicity. Biochem. Pharmacol., 36(20): 3387-3391.
NIXON, G.S., WHITTLE, C.A., & WOODFIN, A. (1981) Mercury levels in
dental surgeries and dental personnel. Br. dent. J., 151: 149-154.
NORDLIND, K. (1985) Binding and uptake of mercuric chloride in
human lymphoid cells. Int. Arch. Allergy appl. Immunol., 77: 405-408.
NORDLIND, K. & HENZE, A. (1984) Stimulating effect of mercuric
chloride and nickel sulfate on DNA synthesis of thymocytes and
peripheral blood lymphocytes in children. Int. Arch. Allergy appl.
Immunol., 73: 162-165.
NORTH AMERICAN CONTACT DERMATITIS GROUP (1973) Epidemiology of
contact dermatitis in North America, 1972. Arch. Dermatol., 108: 537-
540.
NRIAGU, J.O. (1979) The biogeochemistry of mercury in the
environment, Amsterdam, Oxford, New York, Elsevier Science
Publishers.
NYGAARD, S.F. & HANSEN, J.C. (1978) Mercury-selenium
interaction at concentrations of selenium and of mercury vapours
as prevalent in nature. Bull. environ. Contam. Toxicol., 20: 20-23.
NYLANDER, M., FRIBERG, L., & LIND, B. (1987) Mercury concentrations
in the human brain and kidneys in relation to exposure from
dental amalgam fillings. Swed. dent. J., 11: 179-187.
NYLANDER, M., FRIBERG, L., LIND, B., & AAKERBERG, S. (1988)
Effects of breathing patterns on mercury release. In: Clarkson,
T.W., Friberg, L., Nordberg, G.F., & Sager, P.R., ed. Biological
monitoring of toxic metals, New York, London, Plenum Press, pp.
263-264.
NYLANDER, M., FRIBERG, L., EGGLESTON, D., & BJORKMAN, L. (1989)
Mercury accumulation in tissues from dental staff and controls
in relation to exposure. Swed. dent. J., 13: 235-243.
OBERLY, T.J., PIPER, C.E., & MCDONALD, D.S. (1982) Mutagenicity of
metals in the L5178Y mouse lymphoma assay. J. Toxicol. environ.
Health, 9: 367-376.
OGATA, M. & MEGURO, T. (1986) Foetal distribution of inhaled
mercury vapor in normal and acatalasaemic mice. Physiol. Chem.
Phys. med. NMR, 18: 165-170.
OGATA, M., KENMOTSU, K., HIROTA, N., MEGURO, T., & AIKOH, H.
(1987) Reduction of mercuric ion and exhalation of mercury in
acatalasemic and normal mice. Arch. environ. Health, 42(1): 26-30.
OKAMOTO, K., (1988) Biological reference materials from the
National Institute for Environmental Studies (Japan). Fresenius
Z. anal. Chem., 332: 524-527.
OLSSON, B. & BERGMAN, M. (1987) Letter to the Editor. J. dent.
Res., 66(7): 1288-1291.
OLSTAD, M.L., HOLLAND, R.I., WANDEL, N., & HENSTEN PETTERSEN, A.
(1987) Correlation between amalgam restorations and mercury
concentrations in urine. J. dent. Res., 66(6): 1179-1182.
ORLOWSKI, J.P. & MERCER, R.D. (1980) Urine mercury levels in
Kawasaki disease. Pediatrics, 66(4): 633-636.
OTT, K.H.R., LOH, F., KRONCKE, A., SCHALLER, K.-H., VALENTIN,
H., & WELTLE, D. (1984) [Mercury burden due to amalgam
fillings.] Dtsch. Zahnärztl. Z., 39: 199-205 (in German with
English summary).
PACYNA, J.M. (1987) Atmospheric emissions of arsenic, cadmium,
lead and mercury from high temperature processes in power
generation and industry. In: Hutchinson, T.C. & Meema, K.M. ed.
Lead, mercury, cadmium and arsenic in the environment, New York,
Chichester, Brisbane, Toronto, John Wiley & Sons, pp. 69-88 (Scope
31).
PAN, S.K., IMURA, N., YAMAMURA, Y., YOSHIDA, M., & SUZUKI, T.
(1980) Urinary methylmercury excretion in persons exposed to
elemental mercury vapor. Tohoku J. exp. Med., 130: 91-95.
PARIZEK, J. & OSTADALOVA, I. (1967) The protective effect of small
amounts of selenite in sublimate intoxication. Experientia, Basel,
23(2): 142-143.
PARIZEK, J., OSTADALOVA, I., KALOUSKOVA, J., BABICKY, A.,
PAVLIK, L., & BIBR, B. (1971) Effect of mercuric compounds on the
maternal transmission of selenium in the pregnant and lactating
rat. J. Reprod. Fertil., 25: 157-170.
PARR, R.M., MURAMATSU, Y., & CLEMENTS, S.A. (1987) Survey and
evaluation of available biological reference materials for trace
element analysis. Fresenius Z. anal. Chem., 326: 601-608.
PARR, R.M., SCHELENZ, R., & BALLESTRA, S. (1988) IAEA biological
reference materials. Fresenius Z. anal. Chem., 332: 518-523.
PATON, G.R. & ALLISON, A.C. (1972) Chromosome damage in
human cell cultures induced by metal salts. Mutat. Res., 16: 332-336.
PATTERSON, J.E., WEISSBERG, B.G., & DENNISON, P.J. (1985) Mercury
in human breath from dental amalgams. Bull. environ. Contam.
Toxicol., 34: 459-468.
PELLETIER, L., PASQUIER, R., HIRSH, F., SAPIN, C., & DRUET, P.
(1986) Autoreactive T cells in mercury-induced autoimmune
disease: in vitro demonstration. J. Immunol., 137(8): 2548-2554.
PELLETIER, L., PASQUIER, R., VIAL, M.C., MANDET, C., MOUTIER, R.,
SALOMON, J.C., & DRUET, P. (1987a) Mercury-induced autoimmune
glomerulonephritis: Requirement for T-cells. Nephrol. Dial.
Transplant., 1: 211-218.
PELLETIER, L., GALCERAN, M., PASQUIER, R., RONCO, P.,
VERROUST, P., BARIETY, J., & DRUET, P. (1987b) Down modulation of
Heymann's nephritis by mercuric chloride. Kidney Int., 32: 227-232.
PELLETIER, L., PASQUIER, R., ROSSERT, J., & DRUET, P. (1987c)
HgCl2 induces nonspecific immunosuppression in Lewis rats. Eur. J.
Immunol., 17: 49-54.
PELLETIER, L., PASQUIER, R., GUETTIER, C., VIAL, M.C., MANDET, C.,
NOCHY, D., & DRUET, P. (1988a) HgCl2 induces T and B cells to
proliferate and differentiate in BN rats. Clin. exp. Immunol., 71:
336-342.
PELLETIER, L., PASQUIER, R., ROSSERT, J., VIAL, M.C., MANDET, C., &
DRUET, P. (1988b) Autoreactive T-cells in mercury-induced
autoimmunity. Ability to induce the autoimmune disease. J.
Immunol., 140(3): 750-754.
PELLETIER, L., ROSSERT, J., PASQUIER, R., VILLARROYA, H.,
BELAIR, M.F., VIAL, M.C., ORIOL, R., & DRUET, P. (1988c) Effect of
HgCl2 on experimental allergic encephalomyelitis in Lewis rats.
HgCl2 induces down modulation of the disease. Eur. J. Immunol., 18:
243-247.
PENZER, V. (1986) Amalgam toxicity: grand deception. Int. J.
Orthod., 24: 21-24.
PETER, F. & STRUNC, G. (1984) Semiautomated analysis for mercury
in whole blood, urine and hair by on-stream generation of cold
vapor. Clin. Chem., 30(6): 893-895.
PEZEROVIC, Dz., NARANCSIK, P., & GAMULIN, S. (1981) Effects of
mercury bichloride on mouse kidney polyribosome structure and
function. Arch. Toxicol., 48: 167-172.
PIIKIVI, L. & HANNINEN, H. (1989) Subjective symptoms and
psychological performance of chlorin-alkali workers. Scand. J.
Work. Environ. Health, 15: 69-74.
PIIKIVI, L. & TOLONEN, U. (1989) EEG findings in chlor-alkali
workers subjected to low long term exposure to mercury vapour.
Brit. J. ind. Med., 46: 370-375.
PIIKIVI, L., HANNINEN, H., MARTELIN, T., & MANTERE, P.
(1984) Psychological performance and long-term exposure to mercury
vapors. Scand. J. Work environ. Health, 10: 35-41.
PIOTROWSKI, J.K., TROJANOWSKA, B., & MOGILNICKA, E.M. (1975)
Excretion kinetics and variability in urinary mercury in workers
exposed to mercury vapor. Internat. Arch. occup. environ. Health,
35: 245-256.
POLAK, L., BARNES, J.M., & TURK, J.L. (1968) The genetic
control of contact sensitization to inorganic metal
compounds in guinea-pigs. Immunology, 14: 707-711.
POPESCU, H.I., NEGRU, L., & LANCRANJAN, I. (1979) Chromosome
aberrations induced by occupational exposure to mercury. Arch.
environ. Health., 34(6): 461-463.
PRICE, J.H. & WISSEMAN, C.L. (1977) Hazard evaluation and
technical assistance report, Cincinnati, Ohio, National Institute
for Occupational Safety and Health, 17 pp. (TA 77-52).
PRITCHARD, J.G., McMULLIN, J.F., & SIKONDARI, A.H. (1982) The
prevalence of high levels of mercury in dentists' hair. Br. dent.
J., 153: 333-336.
PROUVOST-DANON, A., ABADIE, A., SAPIN, C., BAZIN, H., & DRUET, P.
(1981) Induction of IgE synthesis and potentiation of anti-
ovalbumin IgE antibody response by HgCl2 in the rat. J. Immunol.,
126(2): 699-702.
RAHOLA, T., HATTULA, T., KOROLAINEN, A., & MIETTINEN, J.K.
(1973) Elimination of free and protein-bound ionic mercury
(203Hg2+) in man. Ann. clin. Res., 5: 214-219.
RAMEL, C. (1972) Genetic effects. In: Friberg, L. & Vostal,
J., ed. Mercury in the environment, Cleveland, Ohio, CRC Press,
Chapter 9, pp. 169-181.
RASBERRY, S.D. (1987) Biological reference materials from the US
National Bureau of Standards - an update. Fresenius Z. anal. Chem.,
326: 609-612.
REINHARDT, J.W., KAI, CHIU CHAN, & SCHULEIN, T.M. (1983)
Mercury vaporization during amalgam removal. J. prosthet. Dent.,
50(1): 62-64.
REUTER, R., TRESSARS, G., VOHR, H.-W., GLEICHMANN, E., &
LUHRMANN, R. (1989) Mercuric chloride induces autoantibodies
to small nuclear ribonucleoprotein in susceptible mice. Proc.
Natl Acad. Sci. (USA), 86: 237-241.
RICE, D.C. (1989) Brain and tissue levels of mercury after
chronic methylmercury exposure in the monkey. J. Toxicol.
environ. Health, 27: 189-190.
RICHARDS, J.M. & WARREN, P.J. (1985) Mercury vapour released
during the removal of old amalgam restorations. Br. dent. J., 159:
231-232.
RIZZO, A.M. & FURST, A. (1972) Mercury teratogenesis in the
rat. Proc. West. Pharmacol. Soc., 15: 52-54.
ROBINSON, G.C.J., BALAZS, T., & EGOROV, I.K. (1986) Mercuric
chloride-, gold sodium thiomalate-, and D-penicillamine-
induced antinuclear antibodies in mice. Toxicol. appl.
Pharmacol., 86: 159-169.
ROBISON, S.H., CANTONI, O., & COSTA, M. (1984) Analysis of metal-
induced DNA lesions and DNA-repair replication in mammalian
cells. Mutat. Res., 131: 173-181.
ROELS, H., LAUWERYS, R., BUCHET, J.P., BERNARD, A.,
BARTHELS, A., OVERSTEYNS, M., & GAUSSIN, J. (1982) Comparison
of renal function and psychomotor performance in workers
exposed to elemental mercury. Int. Arch. occup. environ. Health,
50: 77-93.
ROELS, H., GENNART, J.-P., LAUWERYS, R., BUCHET, J.-P.,
MALCHAIRE, J., & BERNARD, A. (1985) Surveillance of workers
exposed to mercury vapour: Validation of a previously proposed
biological threshold limit value for mercury concentration in
urine. Am. J. ind. Med., 7: 45-71.
ROELS, H., ABDELADIM, S., CEULEMANS, E., & LAUWERYS, R.
(1987) Relationships between the concentrations of mercury in air
and in blood or urine in workers exposed to mercury vapour. Ann.
occup. Hyg., 31(2): 135-145.
ROMAN-FRANCO, A.A., TURIELLO, M., ALBINI, B., OSSI, E.,
MILGROM, F., & ANDRES, G.A. (1978) Anti-basement membrane
antibodies and antigen-antibody complexes in rabbits injected
with mercuric chloride. Clin. Immunol. Immunopathol., 9: 464-481.
ROSENMAN, K.D., VALCIUKAS, J.A., GLICKMAN, L., MEYERS, B.R.,
CINOTTI, A. (1986) Sensitive indicators of inorganic mercury
toxicity. Arch. environ. Health, 41(4): 208-215.
ROSSI, L.C., CLEMENTE, G.F., & SANTARONI, G. (1976) Mercury and
selenium distribution in a defined area and in its population.
Arch. environ. Health, 160-165.
ROWLAND, I.R., GRASSO, P., & DAVIES, M.J. (1975) The
methylation of mercuric chloride by human intestinal bacteria.
Experentia, Basel, 31(9): 1064-1065.
ROZALSKI, M. & WIERZBICKI, R. (1983) Effect of mercuric
chloride on cultured rat fibroblasts: Survival, protein
biosynthesis and binding of mercury to chromatin. Biochem.
Pharmacol., 32(13): 2124-2126.
RUDZKI, E. (1979) Occupational dermatitis among health service
workers. Derm. Beruf Umwelt, 27(4): 112.
SAPIN, C., DRUET, E., & DRUET, P. (1977) Induction of anti-
glomerular basement membrane antibodies in the Brown-Norway rat by
mercuric chloride. Clin. exp. Immunol., 28: 173-179.
SAPIN, C., HIRSH, F., DELAPORTE, J.P., BAZIN, H., & DRUET, P.
(1984) Polyclonal IgE increase after HgCl2 injections in BN and
LEW rats: a genetic analysis. Immunogenetics, 20: 227-236.
SAPOTA, A., PIOTROWSKI, J., & BARANSKI, B. (1974)
Levels of metallothionein in the fetuses and tissues of
pregnant rats exposed to mercury vapors. Med. Pr., 25: 129-136
(in Polish) (The original is not available. Quoted from US
Public Health Service, Agency for Toxic Substances and Disease
Registry, 1989).
SCHIELE, R. (1988) [Statement on uptake of mercury from
amalgam]. In: Institut der Deutschen Zahnärzte (IDZ), ed.
[Amalgam - for and against], Köln, Deutscher Arzte-Verlag, pp. 123-
133 (in German).
SCHIELE, R., SCHALLER, K.-H., & GROBE, T. (1979) [Studies of
persons occupationally exposed to mercury.] Arbeitsmed. Sozialmed.
Präventivmed., 10: 226-229 (in German).
SCHNEIDER, M. (1974) An environmental study of mercury
contamination in dental offices. J. Am. Dent. Assoc., 89: 1092-1098.
SCHROEDER, H. & MITCHENER, M. (1975) Life-term effects of
mercury, methylmercury, and nine other trace metals on mice. J.
Nutr., 105: 452-458.
SCHUCKMANN, F. (1979) Study of preclinical changes in workers
exposed to inorganic mercury in chloralkali plants. Int. Arch.
occup. environ. Health, 44: 193-200.
SCHOPF, E., SCHULZ, K.H., & ISENSEE, I. (1969)
[Investigations on lymphocyte transformation in mercury
sensitivity. Nonspecific transformation due to mercury
compounds.] Arch. klin. exp. Dermatol., 234: 420-433 (in German with
English summary).
SECO, J.M. (1987) Big mercury deposit found in Spain. Financ.
Times, Oct. 30.
SHAPIRO, I.M., CORNBLATH, D.R., SUMNER, A.J., UZZELL, B.,
SPITZ, L.K., SHIP, I.I., & BLOCH, P. (1982) Neurophysiological
and neuropsychological function in mercury-exposed dentists.
Lancet, I: 1147-1150.
SIBBET, D.J., MOYER, R., & MILLY, G. (1972) Emission of mercury
from latex paint. Proceedings of the Division of Water, Air,
and Waste Chemistry, Boston, American Chemical Society, pp. 20-26.
SIBLERUD, R.L. (1988) The relationship between dental amalgam and
health. Dissertation, Colorado State University (partial
fulfillment of a Ph.D. degree requirement in physiology).
SIKORSKI, R., JUSZKIEWICZ, T., PASZKOWSKI, T., & SZPRENGIER-
JUSZKIEWICZ, T. (1987) Women in dental surgeries: Reproductive
hazards in occupational exposure to metallic mercury. Int. Arch.
occup. environ. Health, 59: 551-557.
SINCLAIR, P.M., TURNER, P.R.C., & JOHNS, R.B. (1980) Mercury
levels in dental students and faculty measured by neutron
activation analysis. J. prosthet. Dent., 43: 581-585.
SINGER, R., VALCIUKAS, J.A., & ROSENMAN, K.D. (1987)
Peripheral neurotoxicity in workers exposed to inorganic mercury
compounds. Arch. environ. Health, 42: 181-184.
SKARE, I. (1972) Microdetermination of mercury in biological
samples. Part III. Automated determination of mercury in urine,
fish and blood samples. Analyst, 97: 148-155.
SKARE, I. & ENGQVIST, A. (1986) [Mercury in air. Part I.
Evaluation of solid adsorbents for exposure monitoring of
mercury vapour], Solna, National Institute of Occupational
Health, 47 pp. (Arbete och Hälsa 1986:38) (in Swedish with
English summary).
SKERFVING, S. (1988) Mercury in women exposed to methylmercury
through fish consumption, and in their newborn babies and
breast milk. Bull. environ. Contam. Toxicol., 41: 475-482.
SKERFVING, S. & BERLIN, M. (1985) [Nordic expert group for
limit value documentation 59. Inorganic mercury], Solna,
National Institute of Occupational Health, 80 pp. (Arbete och
Hälsa) (in Swedish with English summary).
SKERFVING, S. & VOSTAL, J. (1972) Symptoms and signs of
intoxication. In: Friberg, L. & Vostal, J., ed. Mercury in the
environment, Cleveland, Ohio, CRC Press, pp. 93-107.
SKOG, E. & WAHLBERG, J.E. (1964) A comparative investigation
of the percutaneous absorption of metal compounds in the guinea-
pig by means of the radioactive isotopes: 31Cr, 36Co, 63Zn,
110mCd, 203Hg. J. invest. Dermatol., 43: 187-192.
SKUBA, A.R.N. (1984) Survey for mercury vapour in Manitoba dental
offices (Summer 1983). Can. Dent. Assoc. J., (7): 517-522.
SLEMR, F., HEINZ, G.H., CAMARDESE, M.B., HILL, E.F., MOORE,
J.F., & MURRAY, H.C. (1981) Latitudinal distribution of mercury
over the Atlantic Ocean. J. geophys. Res., 86: 1159-1166.
SMITH, P.J., LANGOLF, G.D., & GOLDBERG, J. (1983) Effects of
occupational exposure to elemental mercury on short term memory.
Br. J. ind. Med., 40: 413-419.
SMITH, R.G., VORWALD, A.J., PATIL, L.S., & MOONEY, T.F., Jr,
(1970) Effects of exposure to mercury in the manufacture of
chlorine. Am. Ind. Hyg. Assoc. J., 31: 687-700.
SNAPP, K.R., BOYER, D.B., PETERSON, L.C., & SVARE, C.W.
(1989) The contribution of dental amalgam to mercury in blood.
J. dent. Res., 68: 780-785.
SOS (1987) [Mercury/amalgam - Health risks.] Stockholm, National
Board of Health and Welfare (Report by the LEK-Committee) (in
Swedish with English summary).
STEFFEK, A.J., CLAYTON, R., SIEW, C., & VERRUSIO, A.C. (1987)
Effects of elemental mercury vapor exposure on pregnant Sprague-
Dawley rats. J. dent. Res., 66: 239.
STENMAN, E. & BERGMAN, M. (1989) Hypersensitivity reactions to
dental materials in a referred group of patients. Scand. J. dent.
Res., 97: 76-83.
STEWART, W.K., GUIRGIS, H.A., SANDERSON, J., & TAYLOR, W. (1977)
Urinary mercury excretion and proteinuria in pathology
laboratory staff. Br. J. ind. Med., 34: 26-31.
STOCK, A. (1939) [Chronic mercury and amalgam intoxication.]
Zahnärztl. Rundsch., 10: 371-377, 403-700 (in German).
STONARD, M.D., CHATER, B.V., DUFFIELD, D.P., NEVITT, A.L.,
O'SULLIVAN, J.J., & STEEL, G.T. (1983) An evaluation of renal
function in workers occupationally exposed to mercury vapour.
Int. Arch. occup. environ. Health, 52: 177-189.
STOPFORD, W., BUNDY, S.D., GOLDWATER, L.J., & BIRRIKOFER, K.A.
(1978) Microenvironmental exposure to mercury vapor. Am. Ind. Hyg.
Assoc. J., 39: 379-384.
SUGATA, Y. & CLARKSON, T.W. (1979) Exhalation of mercury -
further evidence for an oxidation-reduction cycle in mammalian
tissues. Biochem. Pharmacol., 28: 3474-3476.
SUGITA, M. (1978) The biological half-time of heavy metals. The
existence of a third "slowest" component. Int. Arch. occup.
environ. Health, 41: 25-40.
SUMMERS, A.O., WIREMAN, J., VIMY, M.J., & LORSCHEIDER, F.L.
(1990) Increased mercury resistance in monkey gingival and
intestinal bacterial flora after placement of dental "silver"
fillings. Physiologist, 33(4): abstract 116.
SUTER, K. (1975) Studies on the dominant-lethal and fertility
effects of the heavy metal compounds methylmercuric hydroxide,
mercuric chloride, and cadmium chloride in male and female mice.
Mutat. Res., 30: 363-374.
SUZUKI, T., SHISHIDO, S., & ISHIHARA, N. (1976) Interaction of
inorganic to organic mercury in their metabolism in human body.
Int. Arch. occup. environ. Health, 38: 103-113.
SUZUKI, T., TAKEMOTO, T.I., SHISHIDO, S., & KANI, K. (1977)
Mercury in human amniotic fluid. Scand. J. Work Environ. Health, 9:
32-35.
SUZUKI, T., HIMENO, S., HONGO, T., & WATANABE, C. (1986)
Mercuryselenium interaction in workers exposed to elemental
mercury vapor. J. appl. Toxicol., 6: 149-153.
SVARE, C.W., PETERSON, L.C., REINHARDT, J.W., BOYER, D.B.,
FRANK, C.W., GAY, D.D., & COX, R.D. (1981) The effect of dental
amalgams on mercury levels in expired air. J. dent. Res., 60(9): 1668-
1671.
SWEDISH ENVIRONMENTAL PROTECTION BOARD (1986) Mercury:
occurrence and turnover of mercury in the environment, Stockholm,
National Environmental Protection Board, (Mercury Report No. 3).
TAKAHATA, N., HAYASHI, H., WATANABE, S., & ANSO, T. (1970)
Accumulation of mercury in the brains of two autopsy cases with
chronic inorganic mercury poisoning. Folia psychiatr. neurol. Jpn.,
24(1): 59-69.
TAKIZAWA, Y. (1986) Mercury content in recognized patients
and non-recognized patients exposed to methylmercury from Minamata
Bay in the last ten years. In: Tsubaki, T. & Takahashi, H.,
ed. Recent advances in Minamata disease studies, Tokyo, Kodansha
Ltd., pp. 24-39.
THOMSON, J. & RUSSELL, J.A. (1970) Dermatitis due to mercury
following amalgam dental restorations. Br. J. Dermatol., 82: 292-297.
TRIEBIG, G. & SCHALLER, K.-H. (1982) Neurotoxic effects in mercury-
exposed workers. Neurobehav. Toxicol. Teratol., 4: 717-720.
TRIEBIG, G., SCHALLER, K.-H., & VALENTIN, H. (1981)
[Investigations on neurotoxicity of chemical substances at the
workplace. I. Determination of motor and sensory nerve conduction
velocity in persons occupationally exposed to mercury.] Int.
Arch. occup. environ. Health, 48: 119-129 (in German with English
summary).
TROEN, P., KAUFMAN, S.A., & KATZ, K.H. (1951) Mercuric
bichloride poisoning. New Eng. J. Med., 244(13): 459-463.
TUNNESSEN, W.W., Jr, MCMAHON, K.J., & BASER, M. (1987) Acrodynia:
exposure to mercury from fluorescent light bulbs. Pediatrics, 79(5):
786-789.
UMEDA, M. & NISHIMURA, M. (1979) Inducibility of chromosomal
aberrations by metal compounds in cultured mammalian cells. Mutat.
Res., 67: 221-229.
UNEP/WHO (1984) Principles and procedures for quality
assurance in environmental pollution: Exposure monitoring,
Geneva, World Health Organization (EFP/HEAL/84.4).
US ENVIRONMENTAL PROTECTION AGENCY (1989) Integrated risk
information system (IRIS): Mercury (inorganic) file data 9/01/81,
Cincinnati, Ohio, US Environmental Protection Agency,
Environmental Criteria and Assessment Office.
VAHTER, M. (1982) Assessment of human exposure to lead and cadmium
through biological monitoring, Stockholm, National Swedish
Institute of Environmental Medicine and Department of
Environmental Hygiene, Karolinska Institute, 136 pp. (Prepared for
the United Nations Environment Programme and the World Health
Organization).
VELASQUEZ, E., HASSAN, A., BELMAR, R., DRUCKER, E., & MICHAELS, D.
(1980) Occupational mercury poisoning - Nicaragua. CDC Mortal.
Morb. Wkly Rep., 29: 393-395.
VERBERK, M.M., SALLE, H.J.A., & KEMPER, C.H. (1986) Tremor in
workers with low exposure to metallic mercury. Am. Ind. Hyg. Assoc.
J., 47: 559-562.
VERSCHAEVE, L. & SUSANNE, C. (1979) Genetic hazards of mercury
exposure in dental surgery. Mutat. Res., 64: 149.
VERSCHAEVE, L., KIRSCH-VOLDERS, M., SUSANNE, C.,
GROETENBRIEL, C., HAUSTERMANS, R., LECOMTE, A., & ROOSSELS, D.
(1976) Genetic damage induced by occupationally low mercury
exposure. Environ. Res., 12: 303-316.
VERSCHAEVE, L., TASSIGNON, J.-P., LEFEVRE, M., DE STOOP, P., &
SUSANNE, C. (1979) Cytogenetic investigation on leucocytes of
workers exposed to metallic mercury. Environ. Mutagen., 1: 259-268.
VERSCHAEVE, I., KIRSCH-VOLDERS, M., & SUSANNE, C. (1984) Mercury-
induced segregational errors of chromosomes in human lymphocytes
and in Indian muntjac cells. Toxicol. Let., 21: 247-253.
VERSCHAEVE, I., KIRSCH-VOLDERS, M., HENS, L., & SUSANNE, C.
(1985) Comparative in vitro cytogenetic studies in mercury-
exposed human lymphocytes. Mutat. Res., 157: 221-226.
VILLALUZ, M.G. (1988) Environmental impact assessment of small
scale gold mining operations in Davao del Norte. Quezon City,
University of the Philippines, College of Engineering (Thesis).
VIMY, M.J. & LORSCHEIDER, F.L. (1985a) Intra-oral air mercury
released from dental amalgam. J. dent. Res., 64(8): 1069-1071.
VIMY, M.J. & LORSCHEIDER, F.L. (1985b) Serial measurements of
intra-oral air mercury: Estimation of daily dose from dental
amalgam. J. dent. Res., 64(8): 1072-1085.
VIMY, M.J., LUFT, A.J., & LORSCHEIDER, F.L. (1986) Estimation of
mercury body burden from dental amalgam: Computer simulation
of a metabolic compartmental model. J. dent. Res., 65(12): 1415-1419.
VIMY, M.J., TAKAHASHI, Y., & LORSCHEIDER, F.L. (1990a)
Maternal-fetal distribution of mercury (Hg203) released from
dental amalgam tooth restorations in sheep. Am. J. Physiol., 258
(in press)
VIMY, M.J., BOYD, N.D., HOOPER, D.E., & LORSCHEIDER, F.L.
(1990b) Glomerular filtration impairment by mercury released
from dental "silver" fillings in sheep. Physiologist, 33(4): abstract
94.
WAHLBERG, J.E. (1965) "Disappearance measurements", a method
for studying percutaneous absorption of isotope-labeled
compounds emitting gamma-rays. Acta dermato-venereol., 45: 397-414.
WALLINGFORD, K.M. (1982) Health hazard evaluation report,
Cincinnati, Ohio, National Institute for Occupational Safety and
Health, 7 pp. (HETA 82-364-1243).
WANDS, J.R., WEISS, S.W., YARDLEY, J.H., & MADDREY, W.C. (1974)
Chronic inorganic mercury poisoning due to laxative abuse. A
clinical and ultrastructural study. Am. J. Med., 57: 92-101.
WARKANY, J. (1966) Acrodynia - postmorten of a disease. Am.
J. Dis. Child., 112: 147-156.
WATANABE, T., SHIMADA, T., & ENDO, A. (1982) Effects of mercury
compounds on ovulation and meiotic and mitotic chromosomes in
female golden hamster. Teratology, 25: 381-384.
WEENING, J.J., FLEUREN, G.J., & HOEDEMAEKER, Ph.J. (1978)
Demonstration of antinuclear antibodies in mercuric chloride-
induced glomerulopahty in the rat. Lab. Invest., 39(4): 405-411.
WEENING, J.J., HOEDEMAEKER, J., & BAKKER, W.W. (1981)
Immunoregulation and antinuclear antibodies in mercury-induced
glomerulopathy in the rat. Clin. exp. Immunol., 45: 64-71.
WESSEL, W. (1967) [Electron microscopic contribution to the
acute and chronic mercury bichloride and viomycin poisoning of
the kidney.] Verh. Dtsch. Ges. Pathol., 51: 313-316 (In German with
English summary).
WHITE, R.R. & BRANDT, R.L. (1976) Development of mercury
hypersensitivity among dental students. J. Am. Dent. Assoc., 92: 1204-
1207.
WHO (1976) Environmental Health Criteria 1: Mercury, Geneva, World
Health Organization, 131 pp.
WHO (1980) Recommended health-based limits in occupational
exposure to heavy metals. Report of a WHO Study Group,
Geneva, World Health Organization, 116 pp. (WHO Technical Report
Series, No. 647).
WHO (1986) Guidelines for integrated air, water, food and
biological exposure monitoring, Geneva, World Health
Organization, UNEP/WHO Global Environmental Monitoring System,
HEAL Project (document PEP/86.6).
WHO (1989) Environmental Health Criteria 86: Mercury -
Environmental aspects, Geneva, World Health Organization, 115 pp.
WHO (1990) Environmental Health Criteria 101: Methylmercury,
Geneva, World Health Organization, 144 pp.
WOLFF, M., OSBORNE, J.W., & HANSON, A.L. (1983) Mercury
toxicity and dental amalgam. Neurotoxicology, 4(3): 201-204.
WU, D.L. (1989) [Atmospheric pollution and mercury poisoning
caused by peasants mercury smelting.] Chin. J. prev. Med., 23:
83-86 (in Chinese with summary in English).
YOSHIDA, M. & YAMAMURA, Y. (1982) Elemental mercury in urine from
workers exposed to mercury vapour. Int. Arch. occup. environ.
Health, 51: 99-104.
YOSHIDA, M., YAMAMURA, Y., & SATOH, H. (1986) Distribution of
mercury in guinea-pig offspring after in utero exposure to mercury
vapor during late gestation. Arch. Toxicol., 58: 225-228.
YOSHIDA, M., AOYAMA, H., SATOH, H., & YAMAMURA, Y. (1987)
Binding of mercury to metallothionein-like protein in fetal
liver of the guinea pig following in utero exposure to mercury
vapor. Toxicol. Lett., 37: 1-6.
YOSHIDA, M., SATOH, H., KOJIMA, S., & YAMAMURA, Y. (1989)
Distribution of mercury in neonatal guinea-pigs after exposure to
mercury vapours. Bull. environ. Contam. Toxicol., 43(5): 697-704.
YOSHIKAWA, H. & OHTA, H. (1982) Interaction of metals and
metallothionein. In: Foulkes, E.C., ed. Biological roles of
metallothionein, Amsterdam, Oxford, New York, Elsevier Science
Publishers, pp. 11-23.
ZAMPOLLO, A., BARUFFINI, A., CIRLA, A.M., PISATI, G., & ZEDDA, S.
(1987) Subclinical inorganic mercury neuropathy:
Neurophysiological investigations in 17 occupationally exposed
subjects. Ital. J. neurol. Sci., 8: 249-254.
ZASUKHINA, G.D., VASILYEVA, I.M., SDIRKOVA, N.I., KRASOVSKY,
G.N., VASYUKOVICH, L.Ya., KENESARIEV, U.I., & BUTENKO, P.G.
(1983) Mutagenic effect of thallium and mercury salts on rodent
cells with different repair activities. Mutat. Res., 124: 163-173.
ZEGLIO, P. (1958) [Long-term effects of mercurialism]. Lav. Um.,
X: 420-422 (in Italian).
ZIFF, S. (1984) Silver dental fillings - The toxic time bomb.
Can the mercury in your dental fillings poison you? New York,
Aurora Press, 150 pp.
RESUME ET CONCLUSIONS
La présente monographie est essentiellement consacrée
aux risques pour la santé humaine imputables au mercure
minéral; elle passe en revue les résultats des recherches
qui ont été publiés depuis la parution des Critères
d'hygiène de l'environnement No 1: Mercure (WHO, 1977).
Depuis 1977, on dispose de nouvelles données concernant la
présence de mercure dans les amalgames dentaires et dans
les savons éclaircissants, qui constituent deux importants
sujets de préoccupation. Dans la présente monographie on
insiste principalement sur l'exposition résultant de ces
deux utilisations, mais on étudie également les données
cinétiques et toxicologiques fondamentales susceptibles
d'être utiles dans l'étude de l'ensemble des effets du
mercure minéral.
En ce qui concerne la santé humaine, les problèmes
liés au transport, à la bioaccumulation et la transfor-
mation du mercure minéral à l'échelon planétaire provien-
nent presque exclusivement de sa conversion en méthylmer-
cure et de l'exposition ultérieure au méthylmercure par
l'intermédiaire des fruits de mer ou autres denrées ali-
mentaires. Les aspects environnementaux et écologiques
généraux du mercure minéral sont récapitulés dans la
présente monographie. On pourra trouver un exposé plus
détaillé de ces questions dans les Critères d'hygiène de
l'environnement No 86: Mercure - aspects écologiques (WHO,
1989) ainsi que dans les Critères d'hygiène de l'environ-
nement No 101: Méthylmercure (WHO, 1990).
1. Identité
Le mercure existe aux trois degrés d'oxydation sui-
vants: Hg0 (mercure métallique); Hg2++ (mercure mercu-
reux) et Hg++ (mercure mercurique). Il peut former des
dérivés organométalliques dont quelques-uns sont utilisés
dans l'industrie et en agriculture.
2. Propriétés physiques et chimiques
Le mercure élémentaire a une très forte tension de
vapeur. Dans l'atmosphère saturée à 20 °C, sa concen-
tration est 200 fois plus élevée que celle qui est actuel-
lement admise sur les lieux de travail.
La solubilité dans l'eau augmente selon la séquence:
mercure élémentaire < chlorure mercureux < chlorure de
méthylmercure < chlorure mercurique. Le mercure élémen-
taire ainsi que les dérivés halogénés des composés alkyl-
mercuriels sont solubles dans les solvants apolaires.
Les vapeurs de mercure sont plus solubles dans le
plasma, le sang total et l'hémoglobine que dans l'eau
distillée où la solubilité est très faible. Les dérivés
organométalliques sont stables mais certains d'entre eux
sont facilement dégradés par les organismes vivants.
3. Méthodes d'analyse
Les méthodes les plus couramment utilisées pour le
dosage du mercure total et du mercure minéral sont
l'absorption atomique de la vapeur froide (CVAA) et
l'activation neutronique. On trouvera un exposé détaillé
des méthodes d'analyse dans les Critères d'hygiène de
l'environnement No 1: Mercure (WHO, 1977) ainsi que dans
le No 101: Méthylmercure (WHO, 1990).
Pour toutes ces méthodes d'analyse, une assurance
minutieuse de la qualité est indispensable.
3.1 Analyses, prélèvements et conservation des urines
Pour l'analyse de routine des divers milieux, on a
recours à la spectrophotométrie d'absorption atomique sans
flamme. Il faut être spécialement prudent lors du choix
des anticoagulants utilisés pour les prélèvements sanguins
afin d'éviter toute contamination par des dérivés mercu-
riels. Des précautions particulières sont également néces-
saires lors du prélèvement et de la conservation des
urines car la croissance bactérienne peut modifier la con-
centration des nombreuses formes de mercure susceptibles
d'être présentes dans les urines. Pour éviter l'altération
des échantillons d'urine, la meilleure méthode consiste à
les additionner d'acide chlorhydrique ou d'un bactéricide
puis de congeler l'échantillon. Il est recommandé de
procéder à une correction en concentration relativement à
la densité des urines ou à la teneur en créatinine.
3.2 Analyses et échantillonnage de l'air
Le dosage du mercure dans l'air peut s'effectuer soit
par des méthodes à lecture instantanée soit par des
méthodes qui comportent deux phases distinctes: échantil-
lonnage et analyse. Les méthodes à lecture instantanée
peuvent être utilisées pour le dosage des vapeurs de mer-
cure. Pour le dosage du mercure total, l'échantillonnage
s'effectue en milieu oxydant acide ou sur hopcalite.
Le dosage par absorption atomique de la vapeur froide
(CVAA) est la méthode la plus fréquemment utilisée.
4. Sources d'exposition humaine et environnementale
4.1 Etat naturel
Le mercure présent dans la nature provient principale-
ment du dégazage de la croûte terrestre, des éruptions
volcaniques et de l'évaporation des étendues d'eau.
Les émissions d'origine naturelle sont de l'ordre de
2700 à 6000 tonnes par an.
4.2 Sources d'origine humaine
On estime à 10 000 tonnes la quantité de mercure
extraite chaque année dans le monde. Cette activité
entraîne un certain nombre de pertes dans l'environnement
ainsi qu'une décharge directe dans l'atmosphère. Parmi les
autres sources importantes de pollution par le mercure, on
compte l'utilisation des combustibles fossiles, le gril-
lage des minerais métalliques sulfurés, le raffinage de
l'or, la production de ciment, l'incinération des déchets
et diverses opérations métallurgiques.
Une installation de production électrolytique de
chlore et de soude donne normalement lieu à des émissions
de mercure de l'ordre de 450 g par tonne de soude causti-
que produite.
La quantité totale libérée annuellement dans l'atmos-
phère de la planète par suite d'activités humaines atteint
quelque 3000 tonnes.
5. Usages
Le mercure est principalement utilisé comme cathode
dans l'électrolyse du chlorure de sodium. Etant donné que
les produits de cette électrolyse sont contaminés par du
mercure, leur emploi dans des opérations industrielles
ultérieures provoque la contamination d'autres produits.
Le mercure est également utilisé dans l'industrie élec-
trique, pour la fabrication d'instruments de mesure
utilisés dans les ménages ou dans l'industrie ainsi que
pour la fabrication d'instruments de laboratoire et
d'appareils médicaux. Certains médicaments contiennent du
mercure minéral. On utilise également une très grande
quantité de mercure pour l'extraction de l'or.
Les amalgames utilisés en art dentaire pour l'obtura-
tion des dents contiennent une grande quantité de mercure
mélangée (en proportion de 1:1) avec un alliage pulvéru-
lent à base d'argent, d'étain, de cuivre et de zinc.
L'amalgame au cuivre, utilisé essentiellement pour les
soins dentaires aux enfants, contient jusqu'à 70% de
mercure et jusqu'à 30% de cuivre. Il peut en résulter une
exposition du dentiste, de ses assistants et des patients
au mercure.
Certaines personnes de couleur utilisent des crèmes et
des savons à base de mercure pour s'éclaircir la peau.
Ces produits sont désormais interdits dans la Communauté
européenne, en Amérique du Nord et dans de nombreux pays
d'Afrique mais on fabrique encore des savons à base de
mercure dans plusieurs pays d'Europe. Les savons contien-
nent jusqu'à 3% d'iodure de mercure et les crèmes jusqu'à
10% de mercure ammoniacal.
6. Transport, distribution et transformation dans l'environnement
Le mercure émis dans l'atmosphère sous forme de vapeur
est transformé en dérivés solubles et il se dépose avec
les précipitations sur le sol et dans l'eau. La vapeur de
mercure peut subsister jusqu'à trois ans dans l'atmos-
phère, cette période étant réduite à quelques semaines
dans le cas des formes solubles.
La première étape du processus de bioaccumulation
aquatique consiste dans la transformation du mercure
minéral en méthylmercure. Cette transformation s'opère
soit par voie non enzymatique soit sous l'action de micro-
organismes. Le méthylmercure pénètre dans la chaîne ali-
mentaire des espèces prédatrices où il subit une bioampli-
fication.
7. Exposition humaine
C'est principalement par l'intermédiaire des aliments
et des amalgames dentaires que la population générale est
exposée au mercure. En fonction de l'importance de sa
concentration dans l'air et dans l'eau, la dose totale
ingérée quotidiennement peut s'en trouver notablement
augmentée. Le poisson constitue la source principale
d'exposition humaine au méthylmercure. Des études expéri-
mentales récentes ont montré que le mercure libéré dans la
cavité buccale à partir d'un amalgame l'est sous forme de
vapeur. La mastication augmente la vitesse de libération
de ces vapeurs. Un certain nombre d'études ont montré
qu'il y avait une corrélation entre le nombre d'obtura-
tions au moyen d'un amalgame ou de surfaces recouvertes
d'amalgame et la teneur en mercure des tissus, mesurée
après autopsie, ainsi que la teneur en mercure du sang,
des urines et du plasma. L'absorption de mercure calculée
à partir de la quantité d'amalgame et l'accumulation
effectivement observée présente d'importantes variations
individuelles. Il est donc difficile de procéder à des
estimations précises de la quantité de mercure provenant
des amalgames dentaires qui finit par se fixer dans
l'organisme. Des études expérimentales effectuées sur des
moutons ont permis d'étudier plus en détail la distri-
bution du mercure provenant des amalgames dentaires.
L'utilisation de savons et de crèmes pour s'éclaircir
la peau peut également donner lieu à une importante expo-
sition.
On a étudié l'exposition professionnelle au mercure
minéral dans les unités d'électrolyse du chlorure de
sodium, dans les mines de mercure, les fabriques de
thermomètres, les raffineries et les cabinets dentaires.
Pour tous ces types d'exposition professionnelle, on a
relevé d'importantes quantités de mercure, mais celles-ci
varient en fonction des ambiances de travail.
8. Cinétique et métabolisme
Les études sur l'homme et l'animal montrent qu'après
inhalation de vapeur de mercure, la proportion retenue par
l'organisme est d'environ 80% alors qu'elle est inférieure
à 1% lorsque le mercure métallique est ingéré sous forme
liquide, ce qui témoigne d'une faible absorption dans les
voies digestives. Après inhalation, les aérosols de
mercure minéral se déposent dans les voies respiratoires
et sont absorbés à une vitesse qui dépend de la taille des
particules. Il est probable que les composés minéraux du
mercure sont absorbés dans les voies digestives dans une
proportion qui est en moyenne inférieure à 10%, mais là
encore les variations individuelles sont considérables.
L'absorption est beaucoup plus élevée chez les ratons
nouveau-nés.
C'est principalement au niveau des reins que se dépose
le mercure après administration de vapeur de mercure
élémentaire ou de dérivés minéraux du mercure (cela repré-
sente 50 à 90% de la charge totale de l'organisme chez
l'animal). Après inhalation de mercure élémentaire on
observe que la quantité de mercure qui passe dans le
cerveau, chez des souris et des singes, est nettement plus
élevée qu'après injection intraveineuse équivalente sous
forme de sels mercuriques. Chez l'homme, le rapport
hématies/plasma est plus élevé (> 1) après administration
de mercure élémentaire qu'après administration d'un sel
mercurique et la quantité de mercure qui traverse la
barrière placentaire est plus importante. Seule une faible
fraction de la quantité de mercure administrée sous forme
de sels bivalents pénètre dans l'organisme du foetus de
rat.
Plusieurs types de transformation métabolique peuvent
se produire:
* oxydation du mercure métallique en mercure (II);
* réduction du mercure (II) en mercure métallique;
* méthylation du mercure minéral;
* conversion du méthylmercure en mercure minéral bivalent.
L'oxydation des vapeurs de mercure métallique en
mercure ionique bivalent (section 6.1.1) n'est pas suffi-
samment rapide pour empêcher le passage du mercure
élémentaire à travers la barrière hémo-méningée, à travers
le placenta ou d'autres tissus. Dans ces tissus, l'oxyda-
tion piège le mercure qui s'accumule dans le cerveau et
les tissus du foetus.
La réduction du mercure (II) en mercure (0) a été
observée tant chez l'animal (rats et souris) que chez
l'homme. Inversement, la décomposition d'organomercuriels
tels que le méthylmercure constitue une source de mercure
(II).
C'est principalement par la voie fécale et par la voie
urinaire que s'élimine chez l'homme le mercure minéral
encore qu'il puisse être exhalé en petites quantités sous
forme élémentaire. Il peut également se produire une
déplétion tissulaire par transfert des tissus maternels à
ceux du foetus.
La demi-vie biologique, qui pour la majeure partie du
mercure s'étend de quelques jours à quelques semaines,
peut être très longue - jusqu'à plusieurs années - pour la
fraction restante. Ces demi-vies très longues ont été
observées tant chez l'animal que chez l'homme. Il se
produit une interaction complexe entre le mercure et
certains autres éléments, notamment le sélénium. Il se
pourrait que la très longue demi-vie d'élimination d'une
fraction du mercure s'explique par la formation d'un
complexe avec le sélénium.
8.1 Valeurs de référence et valeurs normales
Les quelques données dont on dispose à propos de
mineurs décédés montrent que plusieurs années après
l'arrêt de l'exposition, la concentration du mercure dans
le cerveau était encore de plusieurs mg/kg, avec des
valeurs encore plus élevées dans certaines zones. Toute-
fois cette analyse n'ayant pas fait l'objet d'un contrôle
de qualité, les données demeurent incertaines. Chez un
petit nombre de dentistes examinés après leur mort et qui
ne présentaient pas de symptômes d'hydrargyrisme, on a
observé que les teneurs en mercure allaient de très
faibles concentrations jusqu'à des valeurs de quelques
centaines de µg/kg dans le cortex du lobe occipital et
d'environ 100 µg/kg à quelques mg/kg dans l'hypophyse.
L'examen post-mortem de sujets qui n'étaient pas pro-
fessionnellement exposés au mercure mais étaient porteurs
d'un certain nombre d'obturations au moyen d'amalgame, a
montré qu'un nombre modéré (environ 25) de surfaces recou-
vertes d'amalgame augmentent en moyenne la concentration
cérébrale du mercure d'à peu près 10 µg/kg. L'augmen-
tation correspondante au niveau des reins, déterminée au
moyen d'un nombre très limité d'analyses est probablement
de 300 à 400 µg/kg. Toutefois les variations individuel-
les sont considérables.
La concentration du mercure dans les urines et le sang
peut être utilisée comme indicateur de l'exposition, à
condition que celle-ci soit relativement constante,
qu'elle soit prolongée et déterminée sur un groupe de
sujets. Les données récentes sont plus fiables que celles
dont il est fait état dans les Critères d'hygiène de
l'environnement No 1: Mercure (WHO, 1977). Après exposi-
tion professionnelle à des quantités de mercure d'environ
40 µg/m3 d'air, on observe des concentrations urinaires
d'environ 50 µg/g de créatinine. Ce rapport (5:4) entre
les concentrations urinaires et les concentrations
atmosphériques est beaucoup plus faible que le rapport de
3:1 auquel étaient parvenus les experts de WHO (1977). La
différence peut s'expliquer en partie par des variations
dans les techniques d'échantillonnage utilisées pour
calculer l'exposition atmosphérique. Une exposition de
l'ordre de 40 µg/m3 d'air correspond à environ
15-20 µg de mercure par litre de sang. Toutefois, il peut
être difficile d'évaluer l'exposition à de faibles concen-
trations de mercure inorganique par analyse du sang
lorsqu'il y a exposition simultanée au méthylmercure. Pour
lever la difficulté, on peut procéder au dosage du mercure
dans le plasma ou doser simultanément le mercure minéral
et le méthylmercure. Le méthylmercure est beaucoup moins
gênant lorsqu'on procède à une analyse d'urine car il
n'est excrété dans les urines qu'en très faible proportion.
9. Effets chez l'homme
Une exposition aiguë au mercure par inhalation de
vapeurs peut occasionner des douleurs thoraciques, de la
dyspnée, de la toux, une hémoptysie et quelquefois provo-
quer une pneumonie interstitielle mortelle. L'ingestion
de dérivés mercuriques, en particulier de chlorure
mercurique, peut provoquer une gastro-entérite ulcérative
et une nécrose tubulaire aiguë suspectible d'entraîner la
mort par anurie si l'on ne dispose pas de moyens de
dialyse.
En cas d'exposition aux vapeurs de mercure, c'est le
système nerveux central qui constitue l'organe critique.
L'exposition subaiguë peut entraîner des réactions psycho-
tiques caractérisées par un délire, des hallucinations et
une tendance suicidaire. L'exposition professionnelle peut
conduire à des troubles fonctionnels très variés dont
l'éréthisme constitue la caractéristique essentielle.
Lorsque l'exposition se poursuit, on voit apparaître de
légers tremblements, initialement au niveau des mains.
Dans les cas bénins d'éréthisme, ces tremblements régres-
sent lentement en quelques années après cessation de
l'exposition. On a constaté chez des travailleurs exposés
au mercure une diminution de la vitesse de conduction
nerveuse. Des symptômes d'éréthisme moins prononcés ont
été observés à la suite d'une exposition prolongée à de
faibles concentrations.
On connaît très mal les concentrations de mercure dans
le cerveau dans les cas d'hydrargyrisme et on ne peut pas
évaluer la dose sans effet observable ni tracer une courbe
dose-réponse.
Lorsque le taux d'excrétion urinaire du mercure atte-
int 100 µg/g de créatinine, il existe une forte probabi-
lité pour qu'apparaissent les signes neurologiques classi-
ques de l'hydraargyrisme (tremblements, éréthisme) et l'on
note une forte protéinurie. Une exposition de 30 à 100 µg
de mercure/g de créatinine entraîne une incidence accrue
de certains effets toxiques de moindre gravité qui ne se
traduisent pas par une détérioration clinique manifeste.
Dans quelques études, on a observé des tremblements,
enregistrés par voie électrophysiologique, à des concen-
trations faibles dans l'urine (pouvant s'abaisser jusqu'à
25-35 µg/g de créatinine). En revanche, d'autres études
n'ont pas mis cet effet en évidence. Certaines des
personnes exposées font une protéinurie (protéines de
faibles masse moléculaire relative et micro-albuminurie).
On ne dispose pas de données épidémiologiques appropriées
pour les taux d'exposition qui correspondent à moins de
30-50 µg de mercure/g de créatinine.
L'exposition de la population générale est en principe
faible mais dans certains cas, elle peut atteindre les
valeurs constatées dans les ambiances de travail et même
conduire à des intoxications. C'est ainsi que des erreurs
de manipulation de mercure liquide ont pu conduire à de
graves intoxications.
Après ingestion de sels de mercure bivalent, c'est le
rein qui est l'organe critique. On sait depuis longtemps
que l'exposition professionnelle au mercure métallique
entraîne une protéinurie tant chez les travailleurs
présentant des signes d'hydrargyrisme que chez ceux qui
sont asymptomatiques. On observe moins fréquemment un
syndrome néphrotique, syndrome qui peut également se pro-
duire après utilisation de crèmes à base de mercure pour
s'éclaircir la peau et même après une exposition acciden-
telle. Il semblerait d'après les données actuelles que ce
syndrome néphrotique soit dû à une réaction immunotoxique.
Jusqu'à ces derniers temps, on n'avait signalé d'effets
néphrotoxiques de la vapeur de mercure qu'à des doses
supérieures à celles qui entraînent l'apparition de
symptômes centraux. Cependant des études nouvelles font
état d'effets rénaux à des concentrations plus faibles.
L'expérimentation animale montre que le mercure minéral
peut provoquer une glomérulonéphrite auto-immune. Cet
effet s'observe chez toutes les espèces à l'exception de
certaines souches, ce qui indique l'existence d'une pré-
disposition génétique. Une étiologie immunologique a pour
conséquence, en l'absence d'études dose-réponse sur des
groupes d'individus immunologiquement réceptifs,
l'impossibilité d'établir scientifiquement la dose limite
de mercure (par exemple dans le sang ou les urines) en
dessous de laquelle (dans les cas individuels) il n'y aura
pas de symptômes d'hydrargyrisme.
Les vapeurs de mercure et les dérivés mercuriels
peuvent provoquer des dermatites de contact. Des produits
pharmaceutiques à base de mercure ont provoqué des cas
d'acrodynie infantile et on tient l'exposition aux vapeurs
de mercure pour responsable de la maladie de "Kawasaki".
Certaines études, contrairement à d'autres, ont mis en
évidence des effets sur le cycle menstruel et sur le
développement foetal. Il ressort des études épidémiologi-
ques qui ont été publiées qu'il n'y a pour l'instant pas
de réponse à la question de savoir si, en l'absence des
signes bien connus de l'intoxication mercurielle, la
vapeur de mercure peut avoir des effets nocifs sur le
cycle menstruel ou le développement foetal.
Récemment, on a beaucoup débattu de la sécurité des
amalgames utilisés en art dentaire et certains ont avancé
que l'emploi d'amalgames à base de mercure comportait de
graves dangers pour la santé. Les rapports qui font état
de différents types de symptômes, de même que les résul-
tats des quelques études épidémiologiques qui ont été
effectuées, ne sont pas concluants.
RESUMEN Y CONCLUSIONES
La presente monografía se centra principalmente en el
riesgo que representa el mercurio inorgánico para la salud
humana y en ella se examinan los informes de investigación
aparecidos desde la publicación de Criterios de Salud
Ambiental 1: Mercurio (WHO, 1976) (versión española publi-
cada en 1978). Desde 1976, han ido apareciendo nuevos
datos de investigación sobre dos importantes cuestiones
de salud relacionadas con el mercurio inorgánico, a saber,
el mercurio presente en la amalgama de uso odontológico y
en los jabones destinados a aclarar la piel. La presente
monografía se centra en la exposición a esas dos fuentes,
pero se examinan los aspectos cinéticos y toxicológicos
elementales teniendo presentes todos los aspectos del
mercurio inorgánico.
Los efectos sobre la salud humana relacionados con el
transporte mundial, la bioacumulación y la transformación
del mercurio inorgánico se derivan casi exclusivamente de
la conversión de los compuestos de mercurio en metilmer-
curio y de la exposición al metilmercurio en los alimentos
de origen marino y otros alimentos. En la presente mono-
grafía se han resumido los aspectos ambientales y ecológi-
cos mundiales del mercurio inorgánico. Pueden encontrarse
descripciones más detalladas en Criterios de Salud Ambien-
tal 86: Mercurio - Aspectos Ambientales (WHO, 1989) y
Criterios de Salud Ambiental 101: Metilmercurio (WHO,
1990).
1. Identificación
El mercurio existe en tres estados: Hg0 (metálico);
Hg2++ (mercurioso); y Hg++ (mercúrico). Puede formar
compuestos organometálicos, algunos de los cuales tienen
usos industriales y agrícolas.
2. Propiedades físicas y químicas
El mercurio elemental tiene una presión de vapor
sumamente elevada. La atmósfera saturada a 20 °C tiene una
concentración más de 200 veces superior a la de la concen-
tración comúnmente aceptada para la exposición profesional.
La solubilidad en el agua aumenta en el orden sigui-
ente: mercurio elemental < cloruro mercurioso < cloruro de
metilmercurio < cloruro mercúrico. El mercurio elemental
y los haluros de compuestos alquilmercuriales son solubles
en disolventes no polares.
El vapor de mercurio es más soluble en plasma, sangre
entera y hemoglobina que en agua destilada, donde sólo se
disuelve ligeramente. Los compuestos organometálicos son
estables, aunque algunos son fácilmente descompuestos por
los organismos vivos.
3. Métodos analíticos
Los métodos analíticos más utilizados para cuantificar
los compuestos de mercurio total e inorgánico son la
absorción atómica sobre vapor frío (AAVF) y la activación
de neutrones. Puede encontrarse información detallada
sobre los métodos analíticos en Criterios de Salud
Ambiental 1: Mercurio (WHO, 1978) y en Criterios de Salud
Ambiental 101: Metilmercurio (WHO, 1990).
Todo análisis del mercurio requiere una rigurosa
garantía de calidad.
3.1 Análisis, muestreo y conservación de la orina
La espectrofotometría de absorción atómica sin llama
se utiliza en los análisis ordinarios para los diversos
medios. Debe tenerse especial cuidado al elegir el anti-
coagulante para el muestreo de sangre a fin de evitar la
contaminación por compuestos de mercurio. También debe
procederse con suma precaución en el muestreo y el
almacenamiento de la orina, puesto que el crecimiento
bacteriano es capaz de modificar la concentración de las
numerosas formas de mercurio que pueden estar presentes.
La adición de ácido clorhídrico o sustancias bactericidas
y la congelación son los mejores métodos para impedir la
alteración de las muestras de orina. Se recomienda corre-
gir la concentración por referencia a la densidad de la
orina o al contenido de creatinina.
3.2 Análisis y muestreo del aire
Los métodos analíticos del mercurio en el aire pueden
dividirse en métodos de lectura inmediata y métodos con
etapas separadas de muestreo y análisis. Los métodos de
lectura inmediata pueden utilizarse para cuantificar el
vapor de mercurio elemental. El muestreo en medios
acidoxidantes o con hopcalita se usan para cuantificar el
mercurio total.
La técnica (AAVF) es el método analítico más frecuente.
4. Fuentes de exposición humana y medioambiental
4.1 Fuentes naturales
Las principales fuentes naturales del mercurio son la
desgasificación de la corteza terrestre, las emisiones
volcánicas y la evaporación de las masas acuáticas
naturales.
Las emisiones naturales son del orden de 2700-6000
toneladas al año.
4.2 Fuentes debidas a la actividad humana
Se estima que la extracción minera del mercurio pro-
duce en todo el mundo alrededor de 10 000 toneladas/año.
Estas actividades originan ciertas pérdidas de mercurio y
descargas directas a la atmósfera. Otras fuentes import-
antes son la utilización de combustibles fósiles, la
fundición de metales con minerales de sulfuro, el refinado
del oro, la producción de cemento, la incineración de
desechos y las aplicaciones industriales de los metales.
La emisión normal específica de las industrias de
compuestos alcalinos del cloro es de aproximadamente 450 g
de mercurio por tonelada de sosa cáustica producida.
La cantidad y descarga mundial total de mercurio en la
atmósfera debida a las actividades humanas representa
hasta 3000 toneladas/año.
5. Usos
Uno de los principales usos del mercurio es como
cátodo en la electrólisis del cloruro sódico. Como los
compuestos químicos resultantes quedan contaminados con
mercurio, su utilización en otras actividades industriales
origina la contaminación de otros productos. El mercurio
se emplea en la industria eléctrica, en instrumentos de
control en el hogar y la industria, y en instrumental
médico y de laboratorio. Algunos agentes terapéuticos
contienen mercurio inorgánico. En la extracción de oro se
utilizan grandes cantidades de mercurio.
La amalgama odontológica de plata para la obturación
de dientes contiene grandes cantidades de mercurio,
mezclado (en la proporción 1:1) con polvo de aleación
(plata, estaño, cobre, zinc). La amalgama de cobre, que se
utiliza sobre todo en odontología pediátrica, contiene
hasta el 70% de mercurio y hasta el 30% de cobre. Estos
usos pueden causar la exposición del dentista, los
ayudantes y también de los pacientes.
Algunas personas de piel oscura utilizan cremas y
jabones que contienen mercurio para conseguir un tono de
piel más claro. La distribución de esos productos está
actualmente prohibida en la CEE, en América del Norte y en
muchos países africanos, pero en varios países europeos se
sigue fabricando jabón con mercurio. Estos jabones con-
tienen hasta un 3% de yoduro de mercurio y las cremas
contienen mercurio amoniacal (hasta el 10%).
6. Transporte, distribución y transformación en el medio ambiente
El vapor de mercurio emitido se convierte en formas
solubles que son depositadas por la lluvia en el suelo y
el agua. El tiempo de persistencia atmosférica para el
vapor de mercurio es de hasta tres años, mientras que el
de las formas solubles es de sólo algunas semanas.
El cambio de especiación del mercurio desde las formas
inorgánicas hasta las metiladas es la primera etapa del
proceso de bioacumulación acuática. Esto puede suceder
sin el concurso de enzimas o mediante la acción micro-
biana. El metilmercurio ingresa en la cadena alimentaria
de las especies predadoras en las que se produce biomagni-
ficación.
7. Exposición humana
La población general está principalmente expuesta al
mercurio por la dieta y la amalgama odontológica.
Atendiendo a las concentraciones en la atmósfera y en el
agua, pueden producirse contribuciones importantes a la
ingesta diaria de mercurio total. El pescado es una de
las fuentes principales de exposición humana al metilmer-
curio. En estudios experimentales recientes se ha demo-
strado que el mercurio se libera en forma de vapor desde
las restauraciones con amalgama en la boca. La tasa de
liberación de este vapor de mercurio aumenta, por ejemplo,
al masticar. Varios estudios han correlacionado el número
de obturaciones con amalgama odontológica o de superficies
de amalgama con el contenido de mercurio en tejidos
obtenidos en la autopsia humana, así como en muestras de
sangre, orina y plasma. Tanto la ingesta prevista de
mercurio a partir de la amalgama como la acumulación
observada de mercurio demuestran importantes variaciones
individuales. Así pues, resulta difícil cuantificar con
exactitud la liberación y la ingestión de mercurio por el
cuerpo humano a partir de las restauraciones odontológicas
con amalgama. Los estudios experimentales realizados en
ovejas han examinado con mayor detalle la distribución del
mercurio liberado de las restauraciones con amalgama.
El uso de jabón y cremas para aclarar la piel puede
ser origen de una importante exposición al mercurio.
La exposición profesional al mercurio inorgánico se ha
estudiado en plantas industriales de productos cloral-
calinos, minas de mercurio, fábricas de termómetros,
refinerías y consultorios odontológicos. Se han comunicado
elevados niveles de mercurio respecto de todas estas situ-
aciones de exposición profesional, si bien los niveles
varían en virtud de las condiciones del entorno laboral.
8. Cinética y metabolismo
Los resultados de los estudios realizados tanto en
personas como en animales indican que alrededor del 80%
del vapor de mercurio metálico inhalado es retenido por el
organismo, mientras que el mercurio metálico líquido se
absorbe mal en el tracto gastroinstestinal (menos del 1%).
Los aerosoles de mercurio inorgánico inhalados se depo-
sitan en el tracto respiratorio y son absorbidos a una
velocidad que depende del tamaño de las partículas. Los
compuestos de mercurio inorgánico probablemente son
absorbidos desde el tracto gastrointestinal humano hasta
un nivel inferior al 10%, en promedio, pero la variación
individual es considerable. La absorción es mucho más
elevada en la rata recién nacida.
El riñón es el depósito principal de mercurio tras la
administración de vapor de mercurio elemental o de compu-
estos de mercurio inorgánico (50-90% de la carga corporal
de los animales). De modo significativo, más mercurio es
transportado al cerebro del ratón y el mono tras la inha-
lación de mercurio elemental que tras la inyección intra-
venosa de dosis equivalentes de la forma mercúrica. El
cociente hematíes:plasma en el hombre es mayor (> 1) tras
la administración de mercurio elemental que tras la de
mercurio mercúrico, y la proporción de mercurio que atra-
viesa la barrera placentaria es mayor. Sólo una pequeña
fracción del mercurio bivalente administrado ingresa en el
feto de la rata.
Pueden producirse varias formas de transformación
metabólica:
* oxidación del mercurio metálico a mercurio bivalente;
* reducción del mercurio bivalente a mercurio metálico;
* metilación del mercurio inorgánico;
* conversión del metilmercurio en mercurio inorgánico
bivalente.
La oxidación de vapor de mercurio metálico a mercurio
iónico bivalente (sección 6.1.1) no es lo bastante rápida
como para impedir el paso de mercurio elemental a través
de la barrera hematoencefálica, la placenta y otros
tejidos. La oxidación en esos tejidos sirve como filtro
para retener el mercurio y lleva a su acumulación en el
cerebro y los tejidos fetales.
La reducción del mercurio bivalente a Hg0 se ha
demostrado tanto en animales (ratones y ratas) como en el
hombre. La descomposición de los compuestos organomer-
curiales, incluido el metilmercurio, es también una fuente
de mercurio mercúrico.
Las rutas fecal y urinaria son las principales vías de
eliminación del mercurio inorgánico en el hombre, si bien
se exhala una pequeña cantidad de mercurio elemental. Una
forma de eliminación de mercurio es la transferencia del
mercurio materno al feto.
La semivida biológica, que dura sólo unos cuantos días
o semanas para la mayor parte del mercurio absorbido, es
sumamente larga, probablemente de años, para una parte del
mercurio. Esas largas semividas se han observado en exper-
imentos realizados con animales así como en el hombre.
Existe una complicada interacción entre el mercurio y
algunos elementos, incluido el selenio. La formación de
un complejo con el selenio puede ser responsable de la
larga semivida que tiene una parte del mercurio.
8.1 Valores de referencia y normales
La limitada información de que se dispone sobre mine-
ros fallecidos muestra la existencia de concentraciones de
mercurio en el cerebro de varios mg/kg, años después de
finalizar la exposición, con valores aún más altos en
algunas partes del cerebro. No obstante, la falta de
control de la calidad en el análisis hace inciertos estos
datos. Entre un pequeño número de dentistas fallecidos,
sin síntomas conocidos de intoxicación por mercurio, los
niveles de éste variaron desde concentraciones muy bajas
hasta varios cientos de µg/kg en la corteza del lóbulo
occipital y desde unos 100 µg/kg hasta unos cuantos mg/kg
en la hipófisis.
De las autopsias realizadas en sujetos no expuestos
profesionalmente pero con un número variable de obtura-
ciones con amalgama, se desprende que un número moderado
(alrededor de 25) de superficies de amalgama pueden, en
promedio, aumentar la concentración de mercurio en el
cerebro en unos 10 µg/kg. El aumento correspondiente en
el riñón, basado en un número muy limitado de análisis, es
probablemente de 300-400 µg/kg. Sin embargo, la variación
individual es considerable.
Los niveles de mercurio en la orina y la sangre pueden
usarse como indicadores de la exposición, siempre que ésta
sea relativamente constante a largo plazo y se evalúe en
un grupo. Los datos de exposición recientes son más
fiables que los que se citan en Criterios de Salud Ambien-
tal 1: Mercurio (WHO, 1978). Se observan niveles en la
orina de unos 50 µg/g de creatinina tras la exposición
profesional a unos 40 µg de mercurio por m3 de aire.
Esta relación (5:4) entre orina y niveles atmosféricos es
mucho más baja que la de 3:1 estimada por la WHO (1976).
La diferencia puede deberse en parte a la distinta técnica
de muestreo para evaluar la exposición atmosférica. Una
exposición de 40 µg de mercurio/m3 de aire corresponderá
a unos 15-20 µg de mercurio/litro de sangre. Sin embargo,
la interferencia debida a la exposición al metilmercurio
puede hacer más difícil evaluar la exposición a bajas
concentraciones de mercurio inorgánico por medio de
análisis de sangre. Una forma de salvar esos problemas es
analizar el mercurio en el plasma o analizar tanto el
mercurio inorgánico como el metilmercurio. El problema de
la interferencia debida al metilmercurio es mucho menor
cuando se analiza la orina, puesto que el metilmercurio se
excreta con la orina en grado sumamente reducido.
9. Efectos en el hombre
La exposición aguda por inhalación de vapores de
mercurio puede verse seguida por dolores de pecho, disnea,
tos, hemoptisis, y a veces pneumonitis intersticial que
puede provocar la muerte. La ingestión de compuestos
mercúricos, en particular cloruro mercúrico, ha provocado
casos de gastroenteritis ulcerativa y necrosis tubular
aguda, con muerte por anuria en los casos en que no se
dispuso de diálisis.
El sistema nervioso central es el órgano crítico para
la exposición al vapor de mercurio. La exposición subaguda
ha dado origen a reacciones psicóticas caracterizadas por
delirio, alucinaciones y tendencias suicidas. La exposi-
ción profesional origina eretismo como principal carac-
terística de un trastorno funcional de amplio espectro.
Si prosigue la exposición, se presenta un temblor fino,
que al principio afecta a las manos. En los casos más
leves el eretismo y el temblor desaparecen poco a poco a
lo largo de varios años, una vez interrumpida la exposi-
ción. Se ha demostrado en trabajadores expuestos al
mercurio una menor velocidad de conducción nerviosa. La
exposición a bajos niveles durante periodos largos se ha
asociado a síntomas de eretismo menos pronunciados.
Se dispone de muy poca información sobre los niveles
cerebrales de mercurio en los casos de envenenamiento, y
no se sabe nada que permita estimar una concentración
carente de efectos observados o una curva dosis-respuesta.
Cuando el nivel de excreción urinaria de mercurio es
de 100 µg/g de creatinina, hay una probabilidad muy
alta de que aparezcan los signos neurológicos clásicos de
la intoxicación por mercurio (temblor, eretismo) y
proteinuria. Una exposición correspondiente a 30 hasta
100 µg de mercurio/g de creatinina aumenta la incidencia
de algunos efectos tóxicos menos graves que no provocan
trastornos clínicos manifiestos. En algunos estudios se
han observado temblores, electrofisiológicamente regis-
trados, a concentraciones urinarias reducidas (tan bajas
como 25-35 µg/g de creatinina). En otros estudios no se
observó ese efecto. Algunas de las personas expuestas
presentan proteinuria (proteínas de baja masa molecular
relativa y microalbuminuria). No se dispone de datos
epidemiológicos adecuados sobre los niveles de exposición
que corresponden a menos de 30-50 µg de mercurio/g de
creatinina.
Aunque la exposición de la población general es por lo
general reducida, en ocasiones puede elevarse hasta el
nivel de exposición profesional y puede incluso llegar a
ser tóxica. Así, la manipulación incorrecta de mercurio
líquido ha dado origen a casos graves de intoxicación.
El riñón es el órgano crítico tras la ingestión de
sales de mercurio bivalente inorgánico. La exposición
profesional a mercurio metálico se asocia desde hace
tiempo a la aparición de proteinuria, tanto en obreros con
otros síntomas de envenenamiento por mercurio como en
aquéllos sin esos síntomas. En otros casos menos frecu-
entes, la exposición profesional se ha visto seguida del
síndrome nefrótico, que también se ha producido tras el
uso de cremas para aclarar la piel con mercurio inorgá-
nico, e incluso tras la exposición accidental. Las pruebas
actuales sugieren que este síndrome nefrótico se debe a
una respuesta inmunotóxica. Hasta hace poco, los efectos
del vapor de mercurio elemental en el riñón se habían
comunicado sólo con respecto a dosis más elevadas que las
asociadas a la aparición de signos y síntomas del sistema
nervioso central. En los nuevos estudios, no obstante, se
han notificado efectos en el riñón con niveles inferiores
de exposición. Los estudios experimentales con animales
han demostrado que el mercurio puede inducir glomerulone-
fritis autoinmune en todas las especies ensayadas, pero no
en todas las estirpes, lo que indica una predisposición
genética. Una de las consecuencias de la etiología inmuno-
lógica es que, en ausencia de estudios de la dosis-
respuesta en grupos de individuos inmunológicamente sensi-
bles, resulta científicamente imposible establecer un
nivel de mercurio (por ejemplo, en la sangre o la orina)
por debajo del cual (en casos individuales) no se
producirán síntomas relacionados con el mercurio.
Tanto los vapores de mercurio metálico como los
compuestos de mercurio han dado origen a dermatitis de
contacto. Los productos farmacéuticos con mercurio han
sido responsables de la "enfermedad rosada" en los
niños, y la exposición al vapor de mercurio ha sido
responsable de la enfermedad de "Kawasaki". En algunos
estudios, pero no en todos, se han comunicado efectos en
el ciclo menstrual y/o en el desarrollo del feto. El nivel
de los estudios epidemiológicos publicados aún no permite
saber si los vapores de mercurio pueden afectar negativa-
mente al ciclo menstrual o al desarrollo del feto sin que
se observen los conocidos síntomas de la intoxicación por
mercurio.
Hace poco ha habido una intensa polémica sobre la
inocuidad de las amalgamas odontológicas y se ha afirmado
que el mercurio de la amalgama puede plantear graves peli-
gros para la salud. Los informes en los que se describen
distintos tipos de síntomas y signos y los resultados de
los escasos estudios epidemiológicos realizados no son
concluyentes.