
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 108
NICKEL
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
First draft prepared by Dr. R.F. Hertel,
Dr. T. Maass and Ms V.R. Muller,
Fraunhofer Institute of Toxicology and Aerosol Research, Germany
World Health Orgnization
Geneva, 1991
The International Programme on Chemical Safety (IPCS) is a
joint venture of the United Nations Environment Programme, the
International Labour Organisation, and the World Health
Organization. The main objective of the IPCS is to carry out and
disseminate evaluations of the effects of chemicals on human health
and the quality of the environment. Supporting activities include
the development of epidemiological, experimental laboratory, and
risk-assessment methods that could produce internationally
comparable results, and the development of manpower in the field of
toxicology. Other activities carried out by the IPCS include the
development of know-how for coping with chemical accidents,
coordination of laboratory testing and epidemiological studies, and
promotion of research on the mechanisms of the biological action of
chemicals.
WHO Library Cataloguing in Publication Data
Nickel.
(Environmental health criteria ; 108)
1.Nickel-adverse effects 2.Nickel-toxicity 3.Environmental exposure
I.Series
ISBN 92 4 157108 X (NLM Classification: QV 290)
ISSN 0250-863X
The World Health Organization welcomes requests for permission
to reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made
to the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1991
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material
in this publication do not imply the expression of any opinion
whatsoever on the part of the Secretariat of the World Health
Organization concerning the legal status of any country, territory,
city or area or of its authorities, or concerning the delimitation
of its frontiers or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar
nature that are not mentioned. Errors and omissions excepted, the
names of proprietary products are distinguished by initial capital
letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR NICKEL
1. SUMMARY AND CONCLUSIONS
1.1. Identity, physical and chemical properties, and
analytical methods
1.2. Sources of human and environmental exposure
1.3. Environmental transport, distribution, and transformation
1.4. Environmental levels and human exposure
1.5. Kinetics and metabolism in human beings and animals
1.6. Effects on organisms in the environment
1.7. Effects on experimental animals and in vitro test systems
1.8. Effects on human beings
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1. Identity, physical and chemical properties of nickel and
nickel compounds
2.1.1. Nickel carbonate hydroxide
2.1.2. Nickel carbonyl
2.1.3. Nickel chloride and nickel chloride hexahydrate
2.1.4. Nickel hydroxide
2.1.5. Nickel nitrate
2.1.6. Nickel oxide
2.1.7. Nickel sulfate
2.1.8. Nickel sulfide
2.1.9. Nickel subsulfide
2.2. Analytical methods
2.2.1. Determination of trace amounts
2.2.2. Sample collection
2.2.3. Sample pretreatment
2.2.4. Analytical methods
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.1.1. Rocks
3.1.2. Soils
3.1.3. Water
3.1.4. Fossil fuels
3.1.5. Air
3.2. Man-made sources
3.2.1. Production, use, and disposal
3.2.1.1 Primary production
3.2.1.2 Intermediate products and end-use
3.2.1.3 World production levels and trends
3.2.1.4 Emissions from the primary nickel
industry
3.2.1.5 Emissions from the intermediate nickel
industry
3.2.1.6 Emissions from the combustion of fossil
fuels
3.2.1.7 Emissions from sewage sludge and waste
incineration
3.2.1.8 Miscellaneous emission sources
3.2.1.9 Waste disposal
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1. Transport and distribution between media
4.1.1. Air
4.1.2. Water
4.1.3. Rocks and soil
4.1.4. Vegetation and wildlife
4.2. Uptake and bioaccumulation
4.2.1. Terrestrial organisms
4.2.2. Aquatic organisms
4.3. Biomagnification
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Air
5.1.2. Drinking-water
5.1.3. Food
5.1.4. Terrestrial and aquatic organisms
5.2. General population exposure
5.2.1. Oral
5.2.2. Inhalation
5.2.3. Dermal
5.3. Iatrogenic exposure
5.4. Occupational exposure
6. KINETICS AND METABOLISM
6.1. Absorption
6.1.1. Absorption via the respiratory tract
6.1.1.1 Particulate nickel
6.1.1.2 Nickel carbonyl
6.1.2. Absorption via the gastrointestinal tract
6.1.2.1 Experimental animals
6.1.2.2 Human beings
6.1.2.3 Factors influencing gastrointestinal
absorption
6.1.3. Absorption through the skin
6.1.3.1 Experimental animals
6.1.3.2 Human beings
6.1.4. Other routes of absorption
6.1.4.1 Experimental animals
6.1.4.2 Human beings
6.1.5. Transplacental transfer
6.1.5.1 Experimental animals
6.1.5.2 Human beings
6.1.6. Nickel carbonyl
6.2. Distribution, retention, and elimination
6.2.1. Transport
6.2.2. Tissue distribution
6.2.2.1 Experimental animals
6.2.2.2 Kinetics of metabolism
6.2.2.3 Nickel carbonyl
6.2.2.4 Nickel levels in human beings
6.2.2.5 Pathological states influencing nickel
levels
6.3. Elimination and excretion
6.3.1. Experimental animals
6.3.2. Human beings
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
7.1. Microorganisms
7.2. Aquatic algae and plants
7.3. Aquatic invertebrates
7.4. Fish
7.5. Terrestrial organisms
7.5.1. Plants
7.5.2. Animals
7.5.3. Essentiality of nickel for bacteria and plants
7.6. Population and ecosystem effects
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO AND OTHER TEST
SYSTEMS
8.1. Animals
8.1.1. Essentiality
8.1.1.1 Nickel deficiency symptoms
8.1.2. Acute exposures
8.1.2.1 Nickel carbonyl
8.1.2.2 Other nickel compounds
8.1.2.3 Possible mechanisms of acute nickel
toxicity
8.1.3. Short- and long-term exposures
8.1.3.1 Effects on the respiratory tract
8.1.4. Relationship of nickel toxicity and mixed metal
exposure
8.1.5. Endocrine effects
8.1.6. Cardiovascular effects
8.1.7. Effects on the immune system
8.1.8. Skin and eye irritation and contact hypersensitivity
8.1.8.1 Skin and eye irritation
8.1.8.2 Contact hypersensitivity
8.1.9. Reproduction, embryotoxicity, and teratogenicity
8.1.9.1 Effects on the male reproductive system
8.1.9.2 Effects on the female reproductive system
8.1.10. Embryotoxicity and teratogenicity
8.2. Mutagenicity and related end-points
8.2.1. Mutagenesis in bacteria and mammalian cells
8.2.2. Chromosomal aberration and sister chromatid
exchange (SCE)
8.2.3. Mammalian cell transformation
8.3. Other test systems
8.4. Carcinogenicity
8.4.1. Inhalation
8.4.2. Oral
8.4.3. Other routes
8.4.4. Interactions with known carcinogens
8.4.5. Possible mechanisms of nickel carcinogenesis
8.4.6. Factors influencing nickel carcinogenesis
9. EFFECTS ON HUMAN BEINGS
9.1. Systemic effects
9.1.1. Acute toxicity - poisoning incidents
9.1.1.1 Nickel carbonyl
9.1.1.2 Other nickel compounds
9.1.2. Short- and long-term exposure
9.1.2.1 Respiratory effects
9.1.2.2 Renal effects
9.1.2.3 Cardiovascular effects
9.1.2.4 Other effects
9.2. Skin and eye irritation and contact hypersensitivity
9.2.1. Skin and eye irritancy
9.2.2. Contact hypersensitivity
9.3. Reproduction, embryotoxicity and teratogenicity
9.4. Genetic effects in exposed workers
9.5. Carcinogenicity
9.5.1. Epidemiological studies
9.5.1.1 Nickel refining industry
9.5.1.2 Nickel alloy manufacturing
9.5.1.3 Nickel plating industry
9.5.1.4 Welding
9.5.1.5 Nickel powder
9.5.1.6 Nickel-cadmium battery manufacturing
9.5.1.7 Case-control studies
9.5.2. Carcinogenicity of metal alloys in orthopaedic
prostheses
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1. Exposure
10.2. Human health effects
10.3. Environmental effects
11. RECOMMENDATIONS
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
REFERENCES
RESUME ET CONCLUSIONS
RESUMEN Y CONCLUSIONES
WHO TASK GROUP ON NICKEL
Members
Professor D.A. Calamari, Institute of Agricultural Entomology,
University of Milan, Milan, Italy
Dr R.F. Hertel, Fraunhofer Institute of Toxicology and Aerosol
Research (ITA), Hanover, Germany (Rapporteur)
Professor S.M. Hopfer, University of Connecticut School of
Medicine, Farmington, Connecticut, USA
Professor B.A. Katsnelson, Occupational Health Research
Institute, Sverdlovsk, USSR
Professor Yasushi Kodama, Department of Environmental Health,
School of Medicine, University of Occupational and Environmental
Health, Kitakyushu City, Japan
Professor V. Yu. Kogan, Occupational Health Research Institute,
Erevan, USSR (Vice-Chairman)
Ms V.R. Müller, Fraunhofer Institute of Toxicology and Aerosol
Research (ITA), Hanover, Germany
Dr G.D. Nielsen, Department of Environmental Medicine, Odense
University, Odense, Denmark
Professor T. Norseth, National Institute of Occupational Health,
Oslo, Norway (Chairman)
Dr J. Pastuszka, Institute of Environmental Protection, Katowice,
Poland
Professor J. Peto, Section of Epidemiology, Institute of Cancer
Research, Belmont, Surrey, United Kingdom
Dr E.A. Soyombo, Environmental and Occupational Health Division,
Federal Ministry of Health, Lagos, Nigeria
Dr S.H.H. Swierenga, Genetic Toxicology Section, Bureau of Drug
Research, Health Protection Branch, Health and Welfare Canada,
Tunney's Pasture, Ottawa, Ontario, Canada
Dr A.P. Tossavainen, Institute of Occupational Health, Helsinki,
Finland
Representatives of nongovernmental organizations
Professor N. Izmerov, Institute of Industrial Hygiene and
Occupational Diseases, Moscow, USSR, representing the International
Commission on Occupational Health (ICOH)
Observers
Professor A. Horie, Department of Environmental Health, School of
Medicine, University of Occupational and Environmental Health,
Kitakyushu City, Japan
Dr J. Ishmael, Central Toxicology Laboratory, ICI plc,
Macclesfield, Cheshire, United Kingdom
Professor M.I. Mikheev, Institute for Advanced Medical Studies,
Leningrad, USSR
Dr L.G. Morgan, INCO Europe Limited, Swansea, United Kingdom
Dr M. Richold, Unilever Research, Colworth Laboratory, Bedford,
United Kingdom
Professor A.V. Roscin, Central Institute for Advanced Medical
Studies, Moscow, USSR
Secretariat
Dr A. Aitio, International Agency for Research on Cancer, Lyon,
France
Dr E. Smith, International Programme on Chemical Safety, Division
of Environmental Health, World Health Organization, Geneva,
Switzerland
NOTE TO READERS OF THE CRITERIA DOCUMENTS
Every effort has been made to present information in the
criteria documents as accurately as possible without unduly
delaying their publication. In the interest of all users of the
environmental health criteria documents, readers are kindly
requested to communicate any errors that may have occurred to the
Manager of the International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland, in order that they may be
included in corrigenda, which will appear in subsequent volumes.
* * *
A detailed data profile and a legal file can be obtained from
the International Register of Potentially Toxic Chemicals, Palais
des Nations, 1211 Geneva 10, Switzerland (Telephone no.
7988400/7985850).
ENVIRONMENTAL HEALTH CRITERIA FOR NICKEL
A WHO Task Group on Environmental Health Criteria for Nickel
met at the Leningradskaya Hotel, Moscow, USSR, from 17 to 21 April
1989, under the auspices of the USSR State Committee for
Environmental Protection, Centre for International Projects. Dr
S.N. Morozov welcomed the participants on behalf of the host
institution and Dr E. Smith opened the meeting on behalf of the
three cooperating organizations of the IPCS (ILO/UNEP/WHO). The
Task Group reviewed and revised the draft criteria document and
made an evaluation of the health risks of exposure to nickel.
The first draft of this document was prepared by Dr R.F.
Hertel, Dr J. Maass, and Ms V. Müller, Fraunhofer Institute of
Toxicology and Aerosol Research, Hanover, Germany. This draft was
reviewed in the light of international comments by a Working Group
comprising Dr V. Bencko, Prague, Czechoslovakia, Dr M. Piscator,
Stockholm, Sweden, and Dr F.W. Sunderman, Farmington, Connecticut,
USA, with the assistance of Dr R.F. Hertel, Ms V. Müller and Dr G.
Rosner. The revised draft resulting from this Working Group was
submitted for the Task Group review. Dr E. Smith, IPCS Central
Unit, was responsible for the overall scientific content of the
document and for the organization of the meetings, and Mrs M.O.
Head of Oxford, England, was responsible for the editing.
The efforts of all who helped in the preparation and
finalization of the document are gratefully acknowledged.
* * *
Financial support for the Task Group was provided by the United
Nations Environment Programme, through the USSR Commission for
UNEP. Partial financial support for the publication of this
criteria document was kindly provided by the United States
Department of Health and Human Services, through a contract from
the National Institute of Environmental Health Sciences, Research
Triangle Park, North Carolina, USA, a WHO Collaborating Centre for
Environmental Health Effects.
1. SUMMARY AND CONCLUSIONS
1.1. Identity, physical and chemical properties, and analytical
methods
Nickel is a metallic element belonging to group VIIIb of the
periodic table. It is resistant to alkalis, but generally
dissolves in dilute oxidizing acids. Nickel carbonate, nickel
sulfide, and nickel oxide are insoluble in water, whereas nickel
chloride, nickel sulfate, and nickel nitrate are water soluble.
Nickel carbonyl is a volatile colourless liquid that decomposes at
temperatures above 50 °C. The prevalent ionic form is nickel (II).
In biological systems, dissolved nickel may form complex components
with various ligands and bind to organic material.
The most commonly used methods for the analysis of biological
and environmental materials are atomic absorption spectroscopy and
voltammetry. In order to obtain reliable results, especially in
the ultratrace range, specific procedures have to be followed to
minimize the risk of contamination during sample collection,
storage, processing, and analysis. Depending on sample
pretreatment, extraction and enrichment procedures, detection
limits of 1-100 ng/litre can be achieved in biological materials
and water.
1.2. Sources of human and environmental exposure
Nickel is a ubiquitous trace metal and occurs in soil, water,
air, and in the biosphere. The average content in the earth's
crust is about 0.008%. Farm soils contain between 3 and 1000 mg
nickel/kg. Levels in natural waters have been found to range from
2 to 10 µg/litre (fresh water) and from 0.2 to 0.7 µg/litre
(marine). Atmospheric nickel concentrations in remote areas range
from <0.1 to 3 ng/m3.
Nickel ore deposits are accumulations of nickel sulfide
minerals (mostly pentlandite) and laterites. Nickel is extracted
from the mined ore by pyro- and hydro-metallurgical refining
processes. Most of the nickel is used for the production of
stainless steel and other nickel alloys with high corrosion and
temperature resistance. Nickel alloys and nickel platings are used
in vehicles, processing machinery, armaments, tools, electrical
equipment, household appliances, and coinage. Nickel compounds are
also used as catalysts, pigments, and in batteries. Global mining
production of nickel was approximately 67 million kg in 1985. The
primary sources of nickel emissions into the ambient air are the
combustion of coal and oil for heat or power generation, the
incineration of waste and sewage sludge, nickel mining and primary
production, steel manufacture, electroplating, and miscellaneous
sources, such as cement manufacturing. In polluted air, the
predominant nickel compounds appear to be nickel sulfate, oxides,
and sulfides, and to a lesser extent, metallic nickel.
Nickel from various industrial processes and other sources
finally reaches waste water. Residues from waste-water treatment
are disposed of by deep well injection, ocean dumping, land
treatment, and incineration. Effluents from waste-water treatment
plants have been reported to contain up to 0.2 mg nickel/litre.
1.3 Environmental transport, distribution, and transformation
Nickel, which is emitted into the environment from both natural
and man-made sources, is circulated throughout all environmental
compartments by means of chemical and physical processes, and is
biologically transported by living organisms.
Atmospheric nickel is considered to exist mainly in the form of
particulate aerosols containing different concentrations of nickel,
depending on the source. The highest nickel concentrations in
ambient air are usually found in the smallest particles. Nickel
carbonyl is unstable in air and decomposes to form nickel oxide.
The transport and distribution of nickel particles to, or
between, different environmental compartments is strongly
influenced by particle size and meteorological conditions.
Particle size distribution is primarily a function of the emitting
sources. In general, particles from man-made sources are smaller
than natural dust particles.
Nickel is introduced into the hydrosphere by removal from the
atmosphere, by surface run-off, by discharge of industrial and
municipal waste, and also following natural erosion of soils and
rocks. In rivers, nickel is mainly transported in the form of a
precipitated coating on particles and in association with organic
matter; in lakes, it is transported in the ionic form, also mainly
in association with organic matter. Nickel may also be absorbed on
to clay particles and via uptake by biota. Absorption processes
may be reversed leading to release of nickel from the sediment.
Part of the nickel is transported via rivers and streams to the
ocean. Riverine suspended particulate input is estimated to be 135
x 107 kg/year.
Depending on the soil type, nickel may exhibit a high mobility
within the soil profile finally reaching ground water and, thus,
rivers and lakes. Acid rain has a pronounced tendency to mobilize
nickel from the soil. Terrestrial plants take up nickel from soil
primarily via the roots. The amount of nickel uptake from soil
depends on various geochemical and physical parameters including
the type of soil, the soil pH and humidity, the organic matter
content of the soil, and the concentration of extractable nickel.
The best known example of nickel accumulation is the increased
nickel levels, in excess of 1 mg/kg dry weight, found in a number
of plant species ("hyperaccumulators") growing on relatively
infertile serpentine soils. Nickel levels above 50 mg/kg dry
weight are toxic for most plants. Accumulation and toxic effects
have been observed in vegetables grown on sewage sludge-treated
soils and in vegetation close to nickel-emitting sources. High
concentration factors have been found in aquatic plants.
Laboratory studies showed that nickel had little capacity for
accumulation in all the fish studied. In uncontaminated waters,
the range of concentrations reported in whole fish (on a wet-weight
basis) ranged from 0.02 to 2 mg/kg. These values could be up to 10
times higher in fish from contaminated waters. In wildlife, nickel
is found in many organs and tissues, due to dietary uptake by
herbivorous animals and their carnivorous predators. However,
there is no evidence for the biomagnification of nickel in the food
chain.
1.4 Environmental levels and human exposure
Nickel levels in terrestrial and aquatic organisms can vary
over several orders of magnitude. Typical atmospheric nickel
levels for human exposure range from about 5 to 35 ng/m3 at rural
and urban sites, leading to a nickel uptake via inhalation of 0.1-
0.7 µg/day. Drinking-water generally contains less than 10 µg
nickel/litre, but occasionally nickel may be released from the
plumbing fittings, resulting in concentrations of up to 500 µg
nickel/litre.
Nickel concentrations in food are usually below 0.5 mg/kg fresh
weight. Cocoa, soybeans, some dried legumes, various nuts, and
oatmeal contain high concentrations of nickel. Daily intake of
nickel from food will vary widely, because of different dietary
habits, and can range from 100 to 800 µg/day; the mean dietary
nickel intake in most countries is 100-300 µg/day. Release of
nickel from kitchen utensils may contribute significantly to oral
intake. Pulmonary intake of 2-23 µg nickel/day can result from
smoking 40 cigarettes a day.
Dermal exposure in the general environment is important for the
induction and maintenance of contact hypersensitivity caused by
daily skin contact with nickel-plated objects or nickel-containing
alloys (e.g., jewellery, coins, clips).
Iatrogenic exposure to nickel results from implants and
prostheses made from nickel-containing alloys, from intravenous or
dialysis fluids, and from radiographic contrast media. An
estimated average intravenous nickel uptake from dialysis fluids is
100 µg per treatment.
In the working environment, airborne nickel concentrations can
vary from a few µg/m3 to, occasionally, a few mg/m3, depending on
the process involved and the nickel content of the material being
handled.
Throughout the world, millions of workers are exposed to
nickel-containing dusts and fumes during welding, plating and
grinding, mining, nickel refining, and in steel plants, foundries,
and other metal industries.
Dermal exposure to nickel may occur in a wide range of jobs,
either by direct exposure to dissolved nickel, e.g., in refining,
electroplating, and electroforming industries or by handling
nickel-containing tools. Wet cleaning work may involve exposure to
nickel, because of the amounts of nickel that become dissolved in
the washing water.
1.5 Kinetics and metabolism in human beings and animals
Nickel can be absorbed in human beings and animals via
inhalation or ingestion, or percutaneously. Respiratory absorption
with secondary gastrointestinal absorption of nickel (insoluble and
soluble) is the major route of entry during occupational exposure.
A significant quantity of inhaled material is swallowed following
mucociliary clearance from the respiratory tract. Poor personal
hygiene and work practices can contribute to gastrointestinal
exposure. Percutaneous absorption is negligible, quantitatively,
but is important in the pathogenesis of contact hypersensitivity.
Absorption is related to the solubility of the compound, following
the general relationships nickel carbonyl > soluble nickel
compounds > insoluble nickel compounds. Nickel carbonyl is the
most rapidly and completely absorbed nickel compound in both
animals and human beings. Studies in which nickel was administered
via inhalation are limited. Studies on hamsters and rats with
insoluble nickel oxide showed poor absorption, with retention of
much of the material in the lung after several weeks. In contrast,
absorption of soluble nickel chloride or amorphous nickel sulfide
was rapid. Nickel is transported in the blood, principally bound
to albumin.
Gastrointestinal absorption of nickel is variable and depends
on the composition of the diet. In a recent study on human
volunteers, absorption of nickel was 27% from water compared with
less than 1% from food. All body secretions are potential routes
of excretion including urine, bile, sweat, tears, milk, and
mucociliary fluid. Non-absorbed nickel is eliminated in the
faeces. Transplacental transfer has been demonstrated in rodents.
Following parenteral administration of nickel salts, the highest
nickel accumulation occurs in the kidney, endocrine glands, lung,
and liver: high concentrations are also observed in the brain
following administration of nickel carbonyl. Data on nickel
excretion suggest a two-compartment model. Nickel concentrations
in the serum and urine of healthy non-occupationally exposed adults
are 0.2 µg/litre (range: 0.05-1.1 µg/litre) and l.5 µg/g creatinine
(range: 0.5-4.0 mg/g creatinine), respectively. Increased
concentrations of nickel are seen in both of these fluids following
occupational exposure. The body burden of nickel in a non-exposed,
70-kg adult is 0.5 µg.
1.6 Effects on organisms in the environment
In microorganisms, growth was generally inhibited at nickel
concentrations in the medium of 1-5 mg/litre in the case of
actinomycetes, yeast, and marine and non-marine eubacteria and at
levels of 5-1000 mg/litre in filamentous fungi. In algae, no growth
was observed at approximately 0.05-5 mg nickel/litre. Abiotic
factors, such as the pH, hardness, temperature, and salinity of the
medium and the presence of organic and inorganic particles,
influence the toxicity of nickel.
Nickel toxicity in aquatic invertebrates varies considerably
according to species and abiotic factors. A 96-h LC50 of 0.5 mg
nickel/litre has been found for Daphnia spp., while, in molluscs,
96-h LC50 values were around 0.2 mg/litre in two freshwater snail
species and 1100 mg/litre in a bivalve.
In fish, 96-h LC50 values generally fall within the range of
4-20 mg nickel/litre, but can be higher in some species. Long-term
studies on fish, and fish development, in soft water demonstrated
some effects on rainbow trout at levels as low as 0.05 mg
nickel/litre. In terrestrial plants, nickel levels above 50 mg/kg
dry weight are usually toxic. Copper was found to act
toxicologically in a synergistic way, whereas calcium reduced the
toxicity of nickel. Data on the effects of nickel on terrestrial
animals are limited.
Earthworms seem to be relatively insensitive to nickel, if the
medium is rich in microorganisms and organic matter, thus, making
the nickel less available to the earthworms. Nickel has not been
considered as a broad scale global contaminant; however, ecological
changes, such as decreases in the number and diversity of species,
have been observed near nickel-emitting sources. Microecosystem
studies have shown that addition of nickel to soil disturbs the
nitrogen cycle.
1.7 Effects on experimental animals and in vitro test systems
Nickel is essential for the catalytic activity of some plant
and bacterial enzymes. Slow weight gain, anaemia, and decreased
viability of offspring have been described in some animal species
after dietary deprivation of nickel.
The most acutely toxic nickel compound is nickel carbonyl, the
lung being the target organ; pulmonary oedema may occur within 4 h
following exposure. The acute toxicity of other nickel species is
low.
Though contact allergy to nickel is very common in human
beings, experimental sensitization in animals is only successful
under special conditions. Long-term inhalation exposure to
metallic nickel, nickel oxide, or nickel subsulfide caused mucosal
damage and inflammatory reaction in the respiratory tract in rats,
mice, and guinea-pigs. Epithelial hyperplasia was observed in rats
after inhalation exposure to aerosols of nickel chloride or nickel
oxide.
High-level, long-term exposure to nickel oxide led to gradually
progressive pneumoconiosis in rats. Inflammatory reaction,
sometimes accompanied by slight fibrosis, was observed in rabbits
after high-level exposure to nickel-graphite dust. Pulmonary
fibrosis was seen in rats exposed to nickel subsulfide.
Nickel salts, administered parenterally, induced a rapid
transitory hyperglycaemia in rats, rabbits, and chickens. These
changes may be associated with effects on alpha and beta cells in
the islets of Langerhans. Nickel also decreased the release of
prolactin. Nickel chloride, given orally or by inhalation, has
been reported to decrease iodine uptake by the thyroid.
Nickel salts, given intravenously, decreased blood flow in the
coronary arteries in the dog; high concentrations of nickel
decreased the contractility of dog myocardium in vitro.
Nickel chloride affects the T-cell system and suppresses the
activity of natural killer cells. Parenteral administration of
nickel chloride and nickel subsulfide have been reported to cause
intrauterine mortality and decreased weight gain in rats and mice.
Inhalation exposure to nickel carbonyl caused fetal death and
decreased weight gain, and was teratogenic in rats and hamsters.
Information on maternal toxicity was not given in any of these
studies. Nickel carbonyl has been reported to cause dominant
lethal mutations in rats.
Several inorganic nickel compounds were tested for mutagenicity
in various test systems. Nickel compounds were generally inactive
in bacterial mutagenesis assays, except where fluctuation tests
were used. Mutations were observed in several cultured mammalian
cell types. Nickel compounds inhibited DNA synthesis in a wide
variety of organisms. In addition, nickel compounds induced
chromosomal aberrations and sister chromatid exchange (SCE) in both
mammalian and human cultured cells. Chromosomal aberrations, but
not sister chromatid exchange (except in one study on electrolysis
workers), were observed in human beings, occupationally exposed to
either insoluble or soluble nickel compounds. Nickel induced cell
transformation in vitro.
In an inhalation study, nickel subsulfide induced benign and
malignant pulmonary tumours in rats. A few pulmonary tumours were
seen in rats in a series of inhalation studies with nickel
carbonyl. There was no significant increase in lung tumours in
rats in an adequate inhalation study with metallic nickel.
Inhalation exposure to black nickel oxide did not induce lung
tumours in Syrian golden hamsters (a species resistant to lung
carcinogenesis). Adequate carcinogenicity studies on inhalation
exposure to other nickel compounds were not available. However,
nickel subsulfide, metallic nickel powder, and an unspecified
nickel oxide induced benign and malignant lung tumours in rats
after repeated intratracheal instillations.
Nickel carbonyl, nickelocene, and a large number of slightly
soluble or insoluble nickel compounds, including nickel subsulfide,
carbonate, chromate, hydroxide, sulfides, selenides, arsenides,
telluride, antimonide, various unidentified oxide preparations, two
nickel-copper oxides, metallic nickel, and various nickel alloys,
induced local mesenchymal tumours in a variety of experimental
animals after intramuscular, subcutaneous, intraperitoneal,
intrapleural, intraocular, intraosseous, intrarenal, intra-articular,
intratesticular or intra-adipose administration. No local
carcinogenic response was seen in single-dose studies with some
nickel alloys, colloidal nickel hydroxide, or with two specimens of
nickel oxide, especially prepared for carcinogenicity testing by
calcining at 735 °C or 1045 °C.
Nickel sulfate and nickel acetate, but not nickel chloride,
induced tumours of the peritoneal cavity in rats after repeated
intraperitoneal administration.
Metallic nickel and a very large number of nickel compounds
have been tested for carcinogenicity by parenteral routes of
administration; with few exceptions, they caused local tumours.
Only nickel subsulfide has been shown convincingly to cause
cancer after inhalation exposure. However, the number of adequate
inhalation studies is very small.
In studies using repeated intratracheal instillation, nickel
powder, nickel oxide, and nickel subsulfide caused pulmonary
tumours.
When nickel sulfate and nickel chloride, which had not induced
local tumours in intramuscular studies, were tested using repeated
intraperitoneal administration, they elicited a carcinogenic
response.
1.8 Effects on human beings
In terms of human health, nickel carbonyl is the most acutely
toxic nickel compound. The effects of acute nickel carbonyl
poisoning include frontal headache, vertigo, nausea, vomiting,
insomnia, and irritability, followed by pulmonary symptoms similar
to those of a viral pneumonia. Pathological pulmonary lesions
include haemorrhage, oedema, and cellular derangement. The liver,
kidneys, adrenal glands, spleen, and brain are also affected.
Cases of nickel poisoning have also been reported in patients
dialysed with nickel-contaminated dialysate and in electroplaters
who accidentally ingested water contaminated with nickel sulfate
and nickel chloride.
Chronic effects such as rhinitis, sinusitis, nasal septal
perforations, and asthma have been reported in nickel refinery and
nickel plating workers. Some authors reported pulmonary changes
with fibrosis in workers inhaling nickel dust. In addition, nasal
dysplasia has been reported in nickel refinery workers. Nickel
contact hypersensitivity has been documented extensively in both
the general population and in a number of occupations in which
workers were exposed to soluble nickel compounds. In several
countries, it has been reported that 10% of the female population
and 1% of the male population are sensitive to nickel. Of these,
40-50% have vesicular hand eczema, which, in some cases, can be
very severe and lead to loss of working ability. Oral nickel
intake may aggravate vesicular hand eczema and, possibly, also
eczema arising on other parts of the body where there has not been
any skin contact with nickel.
Prostheses, or other surgical implants, made from nickel-
containing alloys have been reported to cause nickel sensitization
or to aggravate existing dermatitis.
Nephrotoxic effects, such as renal oedema with hyperaemia and
parenchymatous degeneration, have been reported in cases of
accidental industrial exposure to nickel carbonyl. Transient
nephrotoxic effects have been recorded after accidental ingestion
of nickel salts.
Very high risks of lung and nasal cancer have been reported in
nickel refinery workers employed in the high-temperature roasting
of sulfide ores, involving substantial exposure to nickel
subsulfide, oxide, and, perhaps, sulfate. Similar risks have been
reported in processes involving exposure to soluble nickel
(electrolysis, copper sulfate extraction, hydrometallurgy), often
combined with some nickel oxide exposure, but with low nickel
subsulfide exposure. The risk to miners and other refinery workers
has been reported to be much lower. Cancer rates have generally
been close to normal in stainless steel welding and nickel-using
industries, with the exception of those involving exposure to
chromium, particularly electroplating. However, nickel/cadmium
battery workers exposed to high levels of both nickel and cadmium
may have suffered a slightly increased risk of lung cancer.
Excesses of various cancers other than lung and nasal cancers,
such as renal, gastric, or prostatic cancer, have occasionally been
reported in nickel workers, but none has been found consistently.
The epidemiological data can be used to address two important
questions: (i) whether specific nickel compounds have been shown to
be carcinogenic; and (ii) whether low-exposure cohorts provide
upper limits of risk at specified exposure levels.
(a) Soluble nickel
There was evidence of a cancer hazard in workers exposed to
soluble nickel concentrations of the order of 1-2 mg/m3, both in
electrolysis and in the preparation of soluble salts. These
workers were also exposed to other nickel compounds, but often at
lower levels than in other high-risk processes. In the absence of
historical exposure measurements it is impossible to draw
unequivocal conclusions, but the evidence that soluble nickel is
carcinogenic is certainly strong. Refinery dust sometimes contains
a substantial proportion of nickel sulfate in addition to nickel
subsulfide. This raises the possibility that the very high cancer
risk observed in workers employed in the high-temperature oxidation
of nickel subsulfide may be partly due to soluble nickel.
(b) Nickel subsulfide
In refinery areas where cancer risks were high, exposure to
nickel subsulfide almost always occurred together with exposure to
the oxide and, perhaps, sulfate (see above). Thus, it is difficult
to demonstrate from epidemiological data alone, that nickel
subsulfide is carcinogenic, though this seems likely.
(c) Nickel oxide
Nickel oxide was present in almost all circumstances in which
cancer risks were elevated, together with one or more other forms
of nickel (nickel subsulfide, soluble nickel, metallic nickel). As
for nickel subsulfide, it is difficult to either demonstrate or
disprove its suspected carcinogenicity on the basis of
epidemiological data alone.
(d) Metallic nickel
No increased cancer risk has been demonstrated in workers
exposed exclusively to metallic nickel. The combined data on
nickel alloy workers and gaseous diffusion workers, all of whom
were exposed to average concentrations of the order of 0.5 mg
nickel/m3, show no excess risk, though the total number of lung
cancers in these cohorts was too small to exclude a small increase
in risk at this level.
(e) Conclusion
Although some, and perhaps all, forms of nickel may be
carcinogenic, there is little or no detectable risk in most sectors
of the nickel industry at current exposure levels; this includes
some processes that were associated, in the past, with very high
lung and nasal cancer risks. Long-term exposure to soluble nickel
at concentrations of the order of 1 mg/m3 may cause a marked
increase in the relative risk of lung cancer, but the relative risk
among workers exposed to average metallic nickel levels of about
0.5 mg/m3 is approximately 1. The cancer risk at a given exposure
level may be higher for soluble nickel compounds than for metallic
nickel and, possibly, than for other forms as well. The absence of
any marked lung cancer risk among nickel platers is not surprising,
as the average exposures to soluble nickel are very much lower than
those in electrolytic refining or nickel salt processing.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1. Identity, and physical and chemical properties of nickel and
nickel compounds
Nickel is a silvery white metal belonging to Group VIIIb of the
periodic table. Nickel is slightly more resistant to oxidation than
iron and cobalt, with a standard potential of -0.236 V relative to
the hydrogen electrode (Stoeppler, 1980). Several hundreds of
nickel compounds have been identified and characterized. Nickel has
a specific density of 8.90 g/cm3, a melting point of 1555 °C, and
a boiling point of 2837 °C (Table 1). It is insoluble in water,
soluble in dilute nitric acid and aqua regia, and slightly soluble
in hydrochloric and sulfuric acid. Nickel usually has an oxidation
state of two, but also occurs as relatively stable tri- and
tetravalent ions (Stoeppler, 1980). Several binary nickel compounds
are commercially and environmentally significant. A brief
description of the chemistry of some of these compounds is given
below. Physical and chemical properties of nickel and its compounds
are summarized in Table 1.
Nickel forms complexes (chelates) that are insoluble in water,
but soluble in organic solvents. These compounds are often very
stable and play an important role in trace analysis. For example,
nickel dimethylglyoxime is the compound that makes possible the
separation of nickel from cobalt, which is similar in its chemical
and analytical behaviour (Stoeppler, 1980). Divided nickel (Raney
nickel) absorbs up to seventeen times its volume of hydrogen and
can act as an catalyst (Lewis & Ott, 1970).
2.1.1. Nickel carbonate hydroxide
Nickel carbonate hydroxide (2NiCO3 x 3Ni(OH)2 x 4H2O) is
insoluble in water, but soluble in ammonia and in dilute acids.
The composition of basic nickel carbonate can vary. The most
common forms range from 2NiCO3 x 3Ni(OH)2 x XH2O to NiCO3 x Ni(OH)3
x XH2O. The tetrahydrate occurs in nature as zaratite. It is used
in nickel plating, as a catalyst for the hardening of fats, and in
colours and glazes for ceramics (Windholz et al., 1983). High
purity nickel carbonate is used in electronic components (IARC, 1976).
2.1.2. Nickel carbonyl
Nickel carbonyl (Ni(CO)4) is a colourless volatile liquid and
is formed when nickel powder is treated with carbon monoxide at
about 50 °C. It is used for the production of pure nickel by
thermal deposition at atmospheric pressure and at 200-250 °C
(Stoeppler, 1980). The carbonyl is insoluble in water, but soluble
in most organic solvents (Windholz et al., 1983).
Table 1. Physical properties of nickel and nickel compoundsa
_____________________________________________________________________________________________________
Name Chemical Relative Appearance Density Melting Boiling Solubility
formula molecular (g/cm3) point point (water; other
mass (°C) (°C) solvents)
_____________________________________________________________________________________________________
Nickel Ni 58.70 lustrous, white, 8.90 1555 2837 insoluble
face-centered cubic 1455b
crystals
Nickel Ni(CH3CO2)2 176.80 green crystalline 1.744 -c - soluble;
acetate mass or powder soluble
in alcohol
Nickel Ni3(AsO4)2 453.97 yellow-green powder 4.982 - - insoluble;
arsenate soluble
in acids
Nickel NiBr2 218.53 yellow-green, - loses - soluble;
bromide deliquescent crystals H2O - soluble
at 200 in alcohol
Nickel 2NiCO3 118.70 light-green crystals - decomposes - insoluble;
carbonate soluble
in acids
Nickel Ni(CO)4 170.73 colourless, volatile 1.318 -19.3 43 insoluble;
carbonyl liquid (17 °C) soluble in
organic
solvents
Nickel NiCl2 129.61 yellow, deliquescent 3.55d 987d soluble
chloride crystals
Nickel NiCl2 x 237.70d green, monoclinic, - - - soluble;
chloride 6H2O deliquescent crystals soluble
hexahydrate in alcohol
_____________________________________________________________________________________________________
Table 1 (contd.)
_____________________________________________________________________________________________________
Name Chemical Relative Appearance Density Melting Boiling Solubility
formula molecular (g/cm3) point point (water; other
mass (°C) (°C) solvents)
_____________________________________________________________________________________________________
Nickel NiF2 96.69 yellow-green, 4.72 - - slightly
fluoride tetragonal crystals soluble
Nickel Ni(OH)2 92.72 green powder - decomposes - insoluble;
hydroxide above 200 soluble in
acids and
ammonia
Nickel 2NiCO3 x 587.67b green powder - - - insoluble;
hydroxy- 3Ni(OH)2 x soluble in
carbonate 4H2O acids
tetrahydrate
Nickel Ni(NO3)2 182.72 green, deliquescent 2.05 56.7 137 soluble;
nitrate crystals soluble
in alcohol
Nickel NiO 74.69 green or black 6.67d 1990d - insoluble;
oxide powder soluble in
acid
Table 1 (contd.)
_____________________________________________________________________________________________________
Name Chemical Relative Appearance Density Melting Boiling Solubility
formula molecular (g/cm3) point point (water; other
mass (°C) (°C) solvents)
_____________________________________________________________________________________________________
Nickel Ni3(PO4)3 366.07 light-green powder - - - insoluble;
phosphate soluble in
acid
Nickel NiSO4 154.77 alpha blue-green, - 53.3(a-b) - soluble
sulfate tetragonal crystals loses
beta green, water soluble
monoclinic crystals at 280
beta-Nickel NiS 90.77d trigonal crystalse 5.3d 797d - insoluble
sulfide
Nickel Ni3S2 240.26b pale, yellowish 5.82b 790b - insoluble;
subsulfide bronze metallicb soluble in
nitric acidb
_____________________________________________________________________________________________________
a From: Windholz et al. (1983).
b From: Weast (1981).
c Data not available.
d From: Blankenstein & Starck (1979).
e From: Neumüller (1985).
2.1.3. Nickel chloride and nickel chloride hexahydrate
Nickel chloride (NiCl2) and nickel chloride hexahydrate
(NiCl2 x 6H2O) are both soluble in water. The anhydrate salt is used
as an absorbent for ammonia in gas masks and in nickel plating
(Windholz et al., 1983).
2.1.4. Nickel hydroxide
Nickel hydroxide (Ni(OH)2) is insoluble in water but soluble in
acids (Windholz et al., 1983). When dissolved in ammonia it forms
complexes. It is used as electrode material for secondary cells
(Blankenstein, 1979).
2.1.5. Nickel nitrate
Nickel nitrate (Ni(NO3)2) dissolves easily in water and
alcohol. It is used in nickel plating and nickel-cadmium batteries
(Neumüller, 1985).
2.1.6. Nickel oxide
Nickel oxide (NiO) includes several nickel-oxygen compounds,
which differ in stoichiometry, and chemical and physical properties
(see Table 32 in section 8.4.3). The different nickel oxides, and
also the nickel-copper oxides present in the nickel refining
industry, have different biological properties (Sunderman et al.,
1987a).
Nickel oxide is insoluble in water. The solubility in acids
and other properties depend on the method of preparation. Nickel
oxide is an important raw material for smelting and alloy-producing
processes. It is also used as a catalyst and in glass colours
(Blankenstein & Starck, 1979). Nickel oxide exists in two forms.
Black nickel oxide is chemically reactive and forms simple salts in
the presence of acids. Green nickel oxide is an inert and
refractory material. It is used primarily in metallurgical
operations.
2.1.7. Nickel sulfate
Nickel sulfate (NiSO4), which exists as a hexahydrate in the
alpha-form, changes into the beta-form at 53.3 °C (Windholz et al.,
1983). It is produced by dissolving nickel oxide or hydroxide in
sulfuric acid (Neumüller, 1985). It is the main component of the
electrolyte solution in electrolytic refining and is a raw material
for the production of catalysts. It is also used in fabrication of
jewellery.
2.1.8. Nickel sulfide
Nickel sulfide (NiS) occurs naturally as millerite. It is
insoluble in water and is of importance in catalyst production and
in the hydrogenation of sulfur compounds in the oil industry
(Blankenstein, 1979).
2.1.9. Nickel subsulfide
Nickel subsulfide (Ni3S2) exists at high-temperatures in a
bronze-yellow form (beta-Ni3S2). At lower temperatures, it
transforms to the green beta-form, which is stable at normal
temperature, and may be formed electrolytically. The grey mineral
heazlewoodite is the same modification, but has been named alpha-
nickel subsulfide. Nickel subsulfide may be formed during the
production of nickel from sulfide ores.
2.2. Analytical methods
A variety of methods has been used to determine nickel
concentrations in different media. Methods are summarized in
Table 2.
2.2.1. Determination of trace amounts
The use of very sensitive instrumental methods has shown that
detection limits are not so much set by the capabilities of the
instrument as by contamination from different sources. Sources of
contamination include laboratory air, laboratory equipment and
construction material, reagents and the analyst. In order to
obtain reliable results, especially when determining trace (mg/kg)
and ultratrace (µg/kg) amounts, specific procedures concerning
contamination control during sample collection, storage, processing,
and analysis must be adhered to.
Besides contamination control during sample processing, the
establishment of the level of accuracy of the analytical procedure
is of great importance. Thus, analysis of certified reference
materials is recommended. Recovery experiments to check the
analytical procedure include the spiking of samples with known
amounts of nickel.
2.2.2. Sample collection
Great care must be taken to minimize the risk of contamination
during sample collection by the use of suitable procedures (Nieboer
& Jusys, 1983; Boyer & Howitz, 1986). Persons who handle samples
should wear talc-free gloves to avoid nickel contamination from
sweat. When collecting liquid samples, e.g., sea water, fresh
water, or urine, acid-washed polyethylene containers should be
used. As stainless steel is a source of nickel contamination,
Teflon(R), intravenous catheters are recommended for blood
collection. Tissues should be dissected with plastic forceps and
obsidian scalpels (Sunderman et al., 1985).
Collection of airborne particulate nickel involves pumping a
known volume of air through a membrane filter, which usually
consists of cellulose, PVC, or glass fibre (Mackenzie Peers, 1986).
Equipment of the air sampling system with a cyclone, cascade, or
cascade impactor allows sampling of respirable particulate nickel
(Roy, 1985).
Table 2. Analytical methods for nickel determinationa
_________________________________________________________________________________________________________
Medium Sample treatment Analytical method Detection Comment Reference
limit
_________________________________________________________________________________________________________
Biological materials
Serum, urine sampling in analysis of extract 0.4 µg/ suitable for monitoring Mikac-Devic
urine PE-bottles; blood by EAAS, with litre occupational exposure; et al. (1977)
collection with graphite atomizer, interference from high
PE-catheter and using a deuterium iron contents possible
PE-syringe; wet background corrector
digestion;
extraction as
furildioxime into
MIBK
Liver, wet digestion; analysis of extract by ns removal of iron and copper Dornemann
kidney evaporation to EAAS, with graphite as N-nitrosophenylhydr- & Kleist
(animal) dryness; atomizer oxylamine-chelates (1980)
dissolution;
extraction as
hexamethylenedi-
thiocarbamate-
chelate into
diisopropylketone
and xylene
Food filtration; wet analysis of extract by 1 ng/ rapid and inexpensive Pilhar et
digestion DPV, with HMDE litre method; higher sensitivity al. (1981)
extraction as than AAS
DMG-chelate
Tissues, wet digestion; analysis of extract ns good agreement between Szathmary
body evaporation to and of digested sample results from EAAS & Daldrup
fluids dryness; by EAAS, with graphite determination and results (1982)
extraction as atomizer; analysis of from GC-determination
DDTC-chelate into extract by GC with FID
MIBK
_________________________________________________________________________________________________________
Table 2 (contd.)
_________________________________________________________________________________________________________
Medium Sample treatment Analytical method Detection Comment Reference
limit
_________________________________________________________________________________________________________
Whole wet digestion; analysis of extract by 0.1 ng/ suitable for routine Ostapczuk
blood, evaporation to DPV, with HMDE litre determination in a variety et al.
urine, dryness and of biological materials (1983)
saliva, dissolution;
liver, extraction as
nails DMG-chelate
Hair washing in analysis by AAS ns more convenient for Bencko et
redistilled sampling and storage than al. (1986)
acetone, then in other biological materials
deionized water
and again in
redistilled acetone;
repeat twice
Serum, blood collection analysis by EAAS, with 0.05 µg/ suitable for routine Sunderman
whole with PE-cannula Zeeman background litre determination et al.
blood and PP-syringe; corrector (1984a)
wet digestion
Body wet digestion; analysis by SWV at ns more sensitive method Ostapczuk et
fluids, extraction as DMG- HMDE compared with DPV al. (1985a)
tissues chelate
Tissue sampling with analysis of sample by 10 ng/g minimal nickel Sunderman
plastic forceps EAAS, with Zeeman dry weight contamination et al.
and obsidian background corrector (1985)
scalpel; wet
digestion
Urine sampling in PE- analysis of extract ns suitable for monitoring Long-zhu
bottles; wet by EAAS, with graphite occupational exposure & Zhe-ming
digestion using atomizer (1985)
nitric acid,
perchloric acid,
and ascorbic acid;
extraction as APDC-
chelate into MIBK
_________________________________________________________________________________________________________
Table 2 (contd.)
_________________________________________________________________________________________________________
Medium Sample treatment Analytical method Detection Comment Reference
limit
_________________________________________________________________________________________________________
Urine filtration; automated 0.1 ng/ convenient for Bond et al.
acidification; determination by 10 µlitre determination of (1986)
complexation with electrochemical sample multimetals (normal
HPDC within determination, to occupational
automated following separation exposure levels)
analytical system by HPLC (reversed after direct injection
phase) of sample
Urine sampling in PE- analysis by EAAS, 0.45 µg/ direct analysis of Sunderman
bottles; with Zeeman background litre sample et al.
acidification; corrector (1986a)
centrifugation
Plasma dilution of sample analysis by EAAS, with 0.09 µg/ no sample pretreatment Andersen et
with nitric acid Zeeman background litre necessary al. (1986)
and Triton X-100 corrector
Lung freeze drying analysis by EAAS, with lower suitable method for the Baumgardt et
tissue following graphite atomizer ng/g routine determination al (1986)
collection; wet amounts of trace elements
digestion;
evaporation to
dryness; dilution
Blood, enzymatic analysis by EAAS, with 0.1 µg/ lower risk of Christensen
serum, digestion of blood Zeeman background litre contamination with & Pedersen
sweat, and serum; corrector pretreatment method used (1986)
urine ultrasonic
treatment of urine
and sweat
_________________________________________________________________________________________________________
Table 2 (contd.)
_________________________________________________________________________________________________________
Medium Sample treatment Analytical method Detection Comment Reference
limit
_________________________________________________________________________________________________________
Food
Food- wet digestion; analysis of extract 0.048- interference by high Evans et al.
stuffs dilution; by FAAS 0.061 copper content (1978)
extraction as APDC- mg/kg
DDDC-chelate into
n-methylpentane-
2-one
Fish freeze-drying; analysis of dissolved ns Ikebe &
shell- low temperature sample by FAAS Tanaka (1979)
fish ashing
Food- wet digestion; analysis of extract ns accurate and inexpensive Valenta et
stuffs evaporation to by DPASV, with HMDE method al. (1981)
dryness;
dissolution;
extraction as
DMG-chelate
Dried dry ashing; analysis of diluted 5 ng/ sensitive and accurate Meyer &
milk extraction as extract by DPV, with sample method, less interference Neeb (1985)
powder DMG-chelate; HMDE compared with FID-GC
dilution
Dried dry ashing; analysis of solution 100 ng/ interference by higher Meyer &
milk extraction as Na- by GC, with FID µlitre iron content in presence Neeb (1985)
powder FDEDTC-chelate in sample of low copper content is
chloroform; possible
evaporation and
dissolution in
chloroform
_________________________________________________________________________________________________________
Table 2 (contd.)
_________________________________________________________________________________________________________
Medium Sample treatment Analytical method Detection Comment Reference
limit
_________________________________________________________________________________________________________
Citrus wet digestion; analysis of extract µg/g simultaneous determination Ichinoki &
leaves, extraction as HMDE by HPLC of nickel, molybdenum, Yamazaki
rice chelates into zinc, and copper (1985)
flour chloroform
(both
standard
reference
materials)
Water
Sea extraction with analysis of extract ns probably only ionic form Danielsson
water APDC and DDTC into by EAAS with graphite of total nickel is et al. (1978)
Freon TF and back- atomizer using a measured; stability of
extraction into deuterium background extract is good
nitric acid corrector
Sea PE bottles or analysis of extract concentration factor 200 Bruland &
water Teflon-coated PVC by EAAS with graphite Franks (1979)
ball-valve furnace
samplers;
a) double extraction 10 ng/
with APDC and DDTC litre
into chloroform; (instrumental
back extraction detection
into nitric acid; limit)
evaporation of
back-extract and
redissolution into
nitric acid
_________________________________________________________________________________________________________
Table 2 (contd.)
_________________________________________________________________________________________________________
Medium Sample treatment Analytical method Detection Comment Reference
limit
_________________________________________________________________________________________________________
Sea b) concentration on 15 ng/ inefficient concentration Bruland &
water Chelex-100 resin litre factor by Chelex-100 Franks (1979)
(contd.) (instru-
mental
detection
limit)
Sea PVC samplers; analysis of ns probably only ionic form Frache et al.
water storage in PE extract by FAAS of total nickel is measured (1980)
containers;
filtration;
extraction with
ADPC into MIBK
Sea adsorption on analysis of resin 0.077 µg/ rapid and inexpensive Yoshimura et
water PAN-resin phase by ion-exchange litre method al. (1980)
calorimetry
Fresh 0.34 µg/
water litre
Fresh buffered analysis of extract 2 µg/ suitable for routine water Flora &
water, extraction as by DPP, with HMDE litre analysis Nieboer
drink- DMG-chelate (1980)
ing
water
Sea UV-irradiation; analysis of extract 1 µg/ rapid and inexpensive Pilhar et
water, extraction as by DPV, with HMDE litre method; high sensitivity al. (1981)
fresh DMG-chelate
water,
waste
water
_________________________________________________________________________________________________________
Table 2 (contd.)
_________________________________________________________________________________________________________
Medium Sample treatment Analytical method Detection Comment Reference
limit
_________________________________________________________________________________________________________
Fresh UV-irradiation; analysis of ns enrichment factor Wilson &
water enrichment by electrolyte by FAAS decreases at higher DiNunzio
(river) Donnan dialysis calcium concentration (1981)
Aqueous extraction as analysis of extract 1-2 µg/ in case of high copper and Gemmer-Colos
solution heptoxime-chelate; by DPP with HMDE litre iron concentrations, et al. (1981)
evaporation and extraction with NH4OH is
redissolution in necessary to prevent
toluene/methanol/ interference
LiCl
Sea preconcentration analysis of diluted 0.05 mg/ concentration factor 200 Watanabe et
water by complexation extract by ICP-AES litre al. (1981)
with 8-hydroxy-
quinoline;
adsorption on
C18-bonded silica
gel; evaporation of
eluate to dryness,
dissolution in
nitric acid
Sea preconcentration analysis by EAAS with ns concentration factor 50 Sturgeon et
water by complexation graphite furnace al. (1982)
with 8-hydroxy-
quinoline;
adsorption on
C18-bonded silica
gel; evaporation of
eluate to dryness,
dissolution in
nitric acid
_________________________________________________________________________________________________________
Table 2 (contd.)
_________________________________________________________________________________________________________
Medium Sample treatment Analytical method Detection Comment Reference
limit
_________________________________________________________________________________________________________
Indus- complexation with separation of chelate 0.5 ng suitable for automated Bond &
trial APDC and DDTC with by HPCC (reversed (electro- monitoring of nickel and Wallace
plant automated phase) followed by chemical) copper (1983)
solu- analytical system electrochemical and 0.1 ng
tions spectrophotometric (spectro-
detection within photo-
automated system metric)
Sea preconcentration analysis of eluate 1 µg/ 50-fold preconcentration McLaren et
water by adsorption on by ICP-MS litre al. (1985)
immobilized
8-hydroxyquinoline
Sea coprecipitation analysis of dissolved 60 ng/ 200-fold preconcentration, Akagi et al.
water with gallium precipitate by ICP-AES litre appropriate for multi- (1985b)
element analysis
Fresh filtration; analysis by DPCSV, 0.4 µg/ elimination of Weidenauer
water oxidative UV- with HMDE litre interference caused by & Lieser
river photolysis dissolved organic matter (1985)
by UV-photolysis
Drink- evaporation onto analysis by PIXE 1.2 µg/ suitable for multi-element Ali et al.
ing cellulose matrix, litre analysis (1985)
water grinding and
pelletizing of
residue
Waste dilution; ion-chromatographic ns Tanaka (1985)
water, separation of analysis, with anion
plating metal ions as separator
solution EDTA-complexes
_________________________________________________________________________________________________________
Table 2 (contd.)
_________________________________________________________________________________________________________
Medium Sample treatment Analytical method Detection Comment Reference
limit
_________________________________________________________________________________________________________
Sea extraction with analysis of diluted 17 µg/ inexpensive method; Carvajal &
water DDTC into extracts by GC, with litre to appropriate for multimetal Zienius
(synth.) chloroform FID by GC with ECD 0.2 µg/ analyses (1986)
litre
(depending
on type of
column)
Rain filtration; analysis by DPSV, 0.24 mg/ rapid, inexpensive and Vos et al.
water acidification; with HMDE litre sensitive method for (1986)
extraction as DMG- multielement analysis
chelate
Soil
Rock wet digestion with analysis of extract 5-200 mg/ appropriate for iron, Sanzolone
material HF and HNO3; by FAAS kg molybdenum, and calcium- et al. (1979)
(stand- extraction as rich geological materials
ard DDTC-chelate into
refer- MIBK
ence
material)
River wet digestion; analysis of diluted 0.1 mg/ elimination of Abo-Rady
sedi- filtration and sample by FAAS using kg interference of matrix (1979a)
ments, dilution a deuterium background effects by use of
rock corrector deuterium background
material, detector
plants
Soil wet digestion; analysis of 4 mg/kg concentration factor 5; Schmidt &
extraction as re-extracts by EAAS re- reduction of interference Dietl (1981)
APDC-chelate into with zirconium coated extract by re-extraction
MIBK; re- graphite atomizer
extraction with
nitric acid
_________________________________________________________________________________________________________
Table 2 (contd.)
_________________________________________________________________________________________________________
Medium Sample treatment Analytical method Detection Comment Reference
limit
_________________________________________________________________________________________________________
Soil acid digestion analysis by ICP-AES 0.010- suitable for multielement Church
0.015 mg/ analysis (1981)
(depending
on spectral
path)
Soil wet digestion; analysis by ASWV 0.08 µg/ more sensitive and rapid Ostapczuk
extraction as ml analyte method for determination et al.
DMG-chelate solution of heavy metals than DPV (1985b)
Air
Air adsorption on analysis by FAAS 1 µg/ suitable for determining US NIOSH
cellulose ester sample occupational exposure (1977b)
membrane filter;
wet digestion
Air adsorption on analysis by ICP-AES 1 µg/ suitable for simultaneous Mackenzie
cellulose ester sample multielement analysis Peers (1986)
membrane filter;
wet digestion;
evaporation to
dryness; dilution
Air adsorption in analysis by 1 µg/m3 nickel carbonyl is Stedman
alcoholic iodine colorimetry measured, interference by (1986a)
solution; gaseous nickel compounds
extraction as
furildioxime
chelate into
chloroform
Air direct sampling analysis by 0.2 µg/m3 allows continuous Stedman
into chemilumin- chemiluminescence measuring of nickel (1986b)
escence detector; carbonyl
mixing of sampling
with carbon
monoxide
_________________________________________________________________________________________________________
Table 2 (contd.)
_________________________________________________________________________________________________________
Medium Sample treatment Analytical method Detection Comment Reference
limit
_________________________________________________________________________________________________________
Various materials
Steel extraction as analysis by DPP, 1 µg/kg Copper can be determined Weinzierl
DMG-chelate; with HMDE simultaneously Umland (1982)
complexation of
Fe3+ and Mn2+ with
triethanolamine
solution
City wet digestion; analysis by ICP-AES 25 µg/ multistep digestion Taylor et al.
waste evaporation to litre procedure necessary (1985)
incin- near dryness; analyte because of difficult
erator dilution solution matrix
ash filtration
(standard
reference
material)
_________________________________________________________________________________________________________
a Abbreviations:
APDC ammonium pyrolidinedithiocarbamate GC gas-chromatography
ASWV adsorption square wave voltammetry HMDE hanging mercury drop electrode
DDDC diethylammonium diethyldithiocarbamate HPLC high-performance liquid chromatography
DDTC diethyldithiocarbamate ICP-AES inductively coupled plasma atomic
DMG dimethylgyoxime emission spectroscopy
DPASV differential pulse aniodic stripping ICP-MS inductively coupled plasma mass
voltammetry spectroscopy
DPCSV differential pulse cathodic stripping MIBK methyl isobutyl ketone
voltammetry NaFDEDTC natrium (ditrifluorethylene)dithio-
DPP differential pulse polarography carbamate
DPV differential pulse voltammetry ns not specified
EAAS electrothermal atomic absorption PAN [1-(2-pyridylazo)-2-naphthol]
spectrometry PE polyethylene
ECD electron-capture detector PIXE particle-induced X-ray emission
FAAS flame atomic absorption spectroscopy PP polypropylene
FID flame ionization detector PVC polyvinyl chloride
TPP tetraphenylporphine
Volatile nickel compounds, such as nickel carbonyl, can be
absorbed in an alcoholic iodine solution through which the air
being sampled is passed (NIOSH, 1977a; Stedman, 1986a).
2.2.3. Sample pretreatment
Prior to the determination of nickel in biological and
environmental materials, the organic constituents must be oxidized
or removed to avoid interference during analysis. The most common
methods include wet digestion, i.e., oxidation of organic matter by
reagents, such as nitric acid, sulfuric acid, perchloric acid, or
hydrogen peroxide, or combinations of these compounds, and dry
ashing, which ensures oxidation of organic matter by the action of
oxygen and high temperatures. Puchyr & Shapiro (1986) developed an
extraction method for food samples that involved low-temperature
HCl/HNO3-leaching followed by filtration. This method proved to be
very efficient and less hazardous and less time-consuming than
common wet or dry digestion techniques. Organic substances,
dissolved in natural waters, and certain liquid foods are
successfully decomposed by oxidative ultraviolet (UV) photolysis
(Pilhar et al., 1981; Weidenauer & Lieser, 1985).
As nickel concentrations are often low in relation to
analytical detection limits, preconcentration steps are introduced,
which may also separate nickel from substances interfering with
analysis. Techniques very frequently employed include chelate
extraction with dithiocarbamates, dimethylglyoxime, furildioxime,
or 8-hydroxyquinoline into organic non-polar solvents. Tanaka
(1985) used EDTA as a complexing agent prior to determination of
nickel in waste water and plating solution: Gemmer-Colos et al.
(1981) reported complete extraction of nickel-heptoxime from an
aqueous nickel solution at low pH values. Interfering cobalt and
iron ions were eliminated by treatment of the extract with ammonia.
Another preconcentration technique, prior to analysis of nickel in
fresh and sea water, is the use of chelating ion-exchanged resins,
e.g., Chelex 100(R), (Bruland et al., 1979) or 1-(2-pyridylazo)-2-
naphthol (PAN) (Yoshimura et al., 1980). Brajter & Slonawska
(1986) considered Chelex-P(R), a dibasic phosphate ester of
cellulose, as very efficient for the preconcentration of nickel in
water samples. A less time-consuming method for the
preconcentration of nickel in sea water was developed by Watanabe
et al. (1981), Sturgeon et al. (1982), and McLaren et al. (1985).
It involved complexation of the trace metals by 8-hydroxyquinoline
followed by adsorption on C18 chemically bonded silica gel. Wan et
al. (1985) achieved a greater enrichment factor, smaller sample
volume, and removal of interfering humic substances when
preconcentrating nickel and other trace metals in natural waters on
XAD-7 regions (cross-linked polymer of methylmethacrylate) in a
two-step procedure at two different pH values. A very efficient
preconcentration method was developed by Burba & Willmer (1985) in
which trace metals in natural waters were enriched on metal
hydroxide coated cellulose, using iron hydroxide and indium
hydroxide. The use of gallium hydroxide as a coprecipitation agent
for multi-element determination in sea water, and zirconium
hydroxide as a coprecipitation agent for multi-element
determination in sea and fresh water has been described (Akagi et
al., 1985a,b). Zirconium caused spectral interferences in the
inductively coupled plasma atomic emission spectrometry, whereas
coprecipitation with gallium proved to be more efficient with lower
limits of detection in subsequent analysis.
2.2.4. Analytical methods
The two most commonly used analytical methods for nickel are
atomic absorption spectroscopy and voltammetry.
In biological samples, such as tissues and body fluids, nickel
concentrations are routinely determined by electrothermal atomic
absorption spectroscopy (EAAS). Acid digestion is required before
analysis of biological samples, which is commonly followed by an
enrichment step. The IUPAC Subcommittee on the Environmental and
Occupational Toxicology of Nickel (Sunderman, 1980) developed a
reference method for the determination of nickel in serum or urine
by EAAS, after acid digestion and the subsequent extraction of
nickel with ammonium pyrrolidine dithiocarbamate (APDC) into methyl
isobutyl ketone (MIBK).
The introduction of a Zeeman-compensated system improved
background compensation and permitted a more rapid and direct
determination of nickel levels with considerably lower detection
limits, which was suitable for routine use. Sunderman et al.
(1984a, 1985) applied EAAS with Zeeman background correction for the
direct determination of nickel in acid-digested serum (detection
limit, 0.05 µg/litre), in whole blood, and in acid-digested tissue
homogenates (detection limit, 10 ng/g dry weight). The suitability
of this method for the direct determination of nickel in acidified
urine with a detection limit of 0.5 µg/litre has been demonstrated
(Sunderman et al., 1986a). Andersen et al. (1986a) presented an
even more direct method, which only required dilution of the human
plasma prior to quantification by Zeeman-corrected EAAS. The limit
of detection was 0.09 µg/litre. Recent progress in voltammetry has
made this method the most sensitive. Ostapczuk et al. (1983) used
a new voltammetric method for the determination of nickel in a
variety of biological materials following acid digestion of the
sample. The method was based on the application of differential
pulse voltammetry (DPV) after prior interfacial accumulation by an
adsorption layer of nickel-dimethylglyoxime chelate at the hanging
mercury drop electrode (HMDE). The measurement of nickel
concentrations as low as 1 ng/litre was possible using this method,
which was also suitable for analysing food samples (Meyer & Neeb,
1985). Though it requires time-consuming sample digestion
procedures, voltammetry is more sensitive, more rapid, and less
costly than EAAS (Ostapczuk et al., 1983). An isotope dilution gas
chromatography-mass spectrometric method for the detection of
nickel in biological materials at the ng/litre level was recently
introduced by Aggarwal et al. (1988). The method depends on the
preparation of a thermally stable and volatile chelate (chelating
agents: sodium diethyldithiocarbonate or lithium
bis(trifluoroethyl) dithiocarbamate) followed by on-column
injection into a gas chromatographic column and electron
ionization of the eluted chelate in the mass spectrometer.
Analysis for nickel in natural water is frequently performed by
EAAS following preconcentration. Large concentration factors
(200:1) provide detection limits as low as 10 ng/litre in sea-water
analysis (Bruland et al., 1979). Inductively-coupled plasma atomic
emission spectroscopy (ICP-AES) is gaining importance in
simultaneous multi-element determination. Provided that there is
sufficient enrichment, nickel concentrations as low as 60 ng/litre
can be determined in natural waters (Akagi et al., 1985a).
Pilhar et al. (1981) presented DPV-HMDE with prior chelate
adsorption at the electrode as a simple, rapid, and inexpensive
procedure for determining nickel levels in natural waters and waste
water, with a detection limit of 1 ng/litre. This method is also
suitable for determining the nickel contents of various kinds of
food (Valenta et al., 1981; Meyer & Neeb, 1985). Particle-induced
X-ray emission makes possible the detection of various trace metals
in water at the ng/litre level (Ali et al., 1985).
Atomic absorption spectroscopy is the most widely used method
of analysis for nickel in soil. The sample must undergo acid
digestion and may be submitted to enrichment procedures. Detection
limits are in the mg/kg range (Abo-Rady, 1979a; Sanzolone et al.,
1979; Schmidt & Dietl, 1981) . Voltammetry, which has been
successfully used for the determination of nickel in a variety of
biological samples, has also been applied in the analysis of acid-
digested soil samples, using square wave voltammetry as the more
efficient method (Ostapczuk et al., 1985b).
Determination of nickel in air samples has been performed using
different methods (NIOSH, 1977b). However, flame atomic absorption
spectroscopy (FAAS) is the most commonly used analytical technique
for measuring the nickel concentration in air samples. Following
an acid digestion procedure, 1 µg of nickel in 1 ml sample can be
detected by this method. Interference by a 100-fold excess of
iron, manganese, chromium, copper, cobalt or zinc can be minimized
by proper burner elevation and the use of an oxidizing flame.
A technique suitable for the simultaneous determination of
several metals in air has been reported (Mackenzie Peers, 1986).
Following acid digestion of the absorbing cellulose ester membrane
filter the extracted sample was analysed by ICP-AES with a
detection limit of 1 µg/sample.
Volatile nickel carbonyl in air can be determined by
colorimetry, as a coloured furildioxime-chelate (Stedman, 1986a),
or directly, by photometric detection of chemiluminescence
(Stedman, 1986b). Detection limits are 1 µg/m3 and 0.2 µg/m3,
respectively.
Electron microscopy and X-ray microanalysis can be used for the
determination of nickel in single dust particles, such as welding
fumes and grinding dusts.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
Nickel is a ubiquitous element and has been detected in
different media in all parts of the biosphere. It is the fifth
most abundant element by weight after iron, oxygen, magnesium, and
silicon, and the 24th most abundant element in the earth's crust.
However, the average concentration of nickel in the earth's crust
is only about 0.008% (Mason, 1952). Meteorites have been found to
contain 5-50% nickel. Nickel-enriched nodules have been discovered
on the ocean floor (NAS, 1975).
3.1.1. Rocks
Most nickel occurs in the ferromagnesium minerals of igneous
and metamorphic rocks, e.g., olivine [(MgFe)2 x SiO4]. Normal nickel
concentrations in igneous rocks range from 2 to 60 mg/kg (acidic
rocks), 50-200 mg/kg (basic rocks), and 10-2000 mg/kg (ultramafic
rocks) (Boyle, 1981). Among the major sedimentary rocks, shale and
carbonate rocks contain an average of 50 mg nickel/kg; sandstone
contains only 1 mg nickel/kg (NAS, 1975).
The most commercially important nickel ore deposits are
accumulations of nickel sulfide minerals in ultramafic igneous
rocks. Such deposits are found in Australia, Canada, and the USSR.
The ores are composed almost entirely of pentlandite [(Fe,Ni)9S8],
chalcopyrite (CuFeS2), and pyrrhotite (FexSx+1), and usually
contain 1-4% nickel (Duke, 1980). Other nickel ore deposits are
formed by the weathering of ultramafic ferromagnesium silicate
rocks in humid tropical areas. The residual soil (laterite)
developing during the weathering process may contain up to 10 times
the amount of nickel in the original rock (Duke, 1980). The
nickeliferous lateritic weathering profile is characterized by two
deposits, an upper oxide zone and a silicate zone, both varying in
proportion. The oxide zone is composed of iron oxides containing
nickel in solid solution. In the silicate zone, also called the
garnierite zone, nickel is found in the mineral serpentine
Mg3Si2O5(OH)5 substituting for magnesium. The nickel content of
lateritic ores is approximately 1-3% (Duke, 1980). Important
deposits are located in Brazil, Cuba, Dominican Republic,
Guatemala, Indonesia, New Caledonia, and the Philippines.
3.1.2. Soils
In glacial areas, nickel-containing components may have been
dispersed over wide areas; thus, the nickel contents of the soil
can differ considerably from the nickel content of the underlying
bedrock.
In unweathered glacial sediments, nickel occurs in the same
mineral phases as those in which it is found in the rocks, i.e.,
sulfides and silicates. Weathering of rocks and soils leads to
nickel release from nickeliferous minerals. The nickel released is
largely retained in the weathered material in association with clay
particles and therefore not considered to be very mobile in the
superficial environment (Duke, 1980).
Nickel can exist in soils in several forms (Hutchinson et al.,
1981) including:
(a) inorganic crystalline minerals or precipitates (e.g., in the
lattice of aluminium silicates);
(b) complexed or adsorbed on organic cation surfaces (e.g., organic
matter) or on inorganic cation exchange surfaces (e.g., clay
minerals); and
(c) water-soluble, free-ion, or chelated metal complexes in soil
solution.
In a soil-water system, nickel may form complexes with
inorganic ligands (Cl-, OH-, SO42-, or NH3) (Richter & Theiss,
1980) and organic ligands (e.g., humic or fulvic acids) (Nriagu,
1980). Agricultural soils of the world contain between 3 and 1000
mg nickel/kg (NAS, 1975). In forest floor samples collected from
78 sites in 9 states in the northeastern USA, nickel was present at
concentrations in the range of 8.5-15 mg/kg (Friedland et al., 1986).
3.1.3. Water
Nickel occurs in aquatic systems as soluble salts adsorbed on
clay particles or organic matter (detritus, algae, bacteria), or
associated with organic particles, such as humic and fulvic acids
and proteins.
Nickel may enter surface waters from three natural sources
(Boyle, 1981), i.e., as particulate matter in rainwater, through
the dissolution of primary bedrock minerals, and from secondary
soil phases.
The fate of nickel in freshwater and sea water is affected by
several factors including pH, pE, ionic strength, type and
concentration of organic and inorganic ligands, and the presence of
solid surfaces for adsorption (Snodgras, 1980).
In natural waters, at a pH range of 5-9, the divalent ion Ni2+
(Ni(H2O)62+) is the dominant form. In this pH range, nickel may
also be adsorbed on iron and manganese oxides, or form complexes
with inorganic ligands (OH-, SO42-, Cl- or NH3) (Richter & Theiss,
1980).
If sulfate concentrations are sufficiently high, nickel sulfate
may be the predominant soluble form; under anaerobic conditions,
sulfide is the major factor controlling the solubility of nickel
(Richter & Theiss, 1980). Nickel concentrations of 0.228-0.693
µg/litre, determined for a vertical open-ocean water profile, were
considered to reflect the actual nickel concentration in this
medium (Bruland et al., 1979). Concentrations of nickel in
freshwater systems are generally less than 2-10 µg/litre (Stokes,
1981). For nickel levels in drinking-water see section 5.1.2.
3.1.4. Fossil fuels
Nickel occurs in both coal and crude oil in minor quantities,
originating from vegetation and from percolating waters containing
nickel leached from rocks. The average value in some Canadian
coals was found to be 15 mg nickel/kg (Hawley, 1955). The nickel
contents in some Western Canadian crude oils, analysed by Hodgson
(1954), were in the range of 0.09-76.6 mg/kg.
3.1.5. Air
Atmospheric nickel is considered to exist mainly in the form of
aerosols with different nickel concentrations in particles
depending on the type of source (Schmidt & Andren, 1980). Major
natural sources include the aerosols constantly produced by the
oceanic surface, windblown soil dusts, and volcanic ash. Nickel is
released from plants during growth, at different levels, depending
on soil composition. Forest fires produce nickel-containing smoke
particles. A part of atmospheric nickel originates from meteoric
dusts.
Atmospheric nickel concentrations for remote areas that are
considered to be relatively free from man-made nickel emissions are
in the range of <0.1-1 ng/m3 (marine) and 1-3 ng/m3 (continental)
(Schmidt & Andren, 1980). The wide variation in ambient nickel
concentrations reflects the influence of nickel emissions from
distant sources being transported by means of meteorological
processes.
Nickel from natural sources, excluding volcanic dust and forest
fires, is probably in the form of the oxide (Barrie, 1981).
3.2. Man-made sources
3.2.1. Production, use, and disposal
3.2.1.1 Primary production
The methods for the extraction and refining of nickel minerals
depend on the mineralogical and geological characteristics of the
ore. To date, nickel has mainly been extracted from sulfide and
laterite ores.
Nickel sulfide ores are mostly mined underground using
drilling, blasting, and other techniques. Milling procedures
include liberation, flotation, and magnetic separation. Liberation
of the sulfides from the gangue includes grinding of the rock
material. Then the sulfides are concentrated by flotation
processes. Flotation involves streaming air bubbles through an
aqueous slurry of the ore particles in a flotation cell. The
particles that are not wetted by the liquid adhere to the air
bubbles, rise to the surface of the slurry, and can be removed.
The addition of different chemicals to the flotation medium allows
the selective flotation of nickel- and copper-rich fractions.
Most of the pyrrhotite (both lump ore and ground ore) can be
separated magnetically, because of its magnetic properties.
Laterite nickel deposits are mined from surface pits using
earth-moving equipment.
Both sulfide ore concentrates and laterite ores are subjected
to pyro- and hydrometallurgical processes. The pyrometallurgical
processing basically involves three operations, i.e., roasting,
smelting, and converting.
During roasting, the concentrate is oxidized by hot air. Most
of the iron is oxidized, while nickel, copper, and cobalt remain
combined with sulfur. Part of the sulfur is removed as gaseous
sulfur dioxide.
The roasted product is smelted in a furnace together with a
siliceous flux to obtain two immiscible phases, an iron-rich
silicate slag and a nickel-rich sulfide matte, which also contains
iron, copper, and cobalt.
The matte is treated in a "converter" where more sulfur is
driven off and the remaining iron is oxidized and removed as slag.
The matte is allowed to cool and treated in different ways. It may
be, for example, cast into anodes for electrolytic refining or
cooled slowly to facilitate crystallization to nickel sulfide,
copper sulfide and a nickel-copper alloy containing the desired
metals. These three phases can then be separated by flotation and
magnetic separation. The species of nickel likely to be present
during roasting, smelting, and converting include the ore, nickel
subsulfides, nickel copper sulfides, nickel oxides, nickel-copper
oxides, arsenides, and anhydrous nickel sulfate. The extraction of
nickel from laterite ores is similar to the extraction of nickel
from sulfide ores with the exception that sulfur (commonly gypsum)
has to be added. The molten matte is charged into a converter
where the iron is oxidized and the sulfur combines with nickel to
form Ni3S2.
Smelting to ferronickel is essentially the same as matte
smelting, except that no sulfur is added. It is often applied to
laterite ores. The resulting iron-nickel alloy contains 20-50%
nickel (Duke, 1980).
Most of the nickel matte obtained from sulfide or laterite ore
smelting undergoes further refining techniques, such as electro-,
vapo-, or hydrometallurgical refining, but a part of the matte is
roasted to marketable nickel oxide sinter.
Hydrometallurgical refining can be applied both to laterite ore
and sulfide ore or sulfide ore concentrates. Soluble nickel amines
are formed during pressure leaching of the sulfide ore concentrate
with strong ammoniacal solution at a moderately elevated
temperature. The saturated solution is boiled to drive off ammonia
and precipitate copper as sulfide. Sulfur is oxidized. Nickel and
cobalt are recovered as pure metal powders by reduction with
hydrogen under pressure.
Laterite ores must first be reduced. The reduced ore is
leached with an ammonia-ammonium carbonate solution. Nickel
dissolves as nickel amine. The saturated solution is heated by
steam, ammonia is driven off, and nickel is precipitated as a basic
carbonate.
Pure nickel (99.9%) can be produced by electrolytic refining.
Generally, an impure metal anode (produced by reducing nickel
oxide) and a cathode starting sheet are placed in an acidic
electrolytic solution. When a current flows, nickel and other
metals are dissolved from the anode. The electrolyte is then
removed, purified and returned to the cathode compartment, where
nickel is deposited on the cathode.
During vapometallurgical refining, impure metal obtained by the
reduction of nickel oxide is subjected to the action of carbon
monoxide forming volatile nickel carbonyl [Ni(CO)4] (carbonyl or
Mond process). This reaction is reversed by heat and the nickel
carbonyl decomposes to pure nickel metal and carbon monoxide. The
carbonyl process produces the purest nickel (99.97% or more).
The smelting and refining processes yield various marketable
forms of nickel of different purities (Table 3).
Table 3. Commercial forms of primary nickela
------------------------------------------------------------------------------------------------
Type Composition (%)
Nickel Carbon Copper Iron Sulfur Cobalt Oxygen Silicon Chromium
------------------------------------------------------------------------------------------------
Pure unwrought nickel
Cathode >99.9 0.01 0.005 0.002 0.001 - - - -
Pellets >99.97 <0.1 0.001 0.0015 0.0003 5 x 10-5 - - -
Powder 99.74 <0.1 - <0.1 <0.01 - <0.15 - -
Briquettes 99.9 0.01 0.001 0.002 0.0035 0.03 - - -
Rondelles 99.25 0.022 0.046 0.087 0.004 0.37 0.042 - -
Ferronickelb 20-50c 1.5-1.8 - Rest <0.3 -c - 1.8-4 1.2-1.8
Nickel oxide 76.0 - 0.75 0.3 0.006 1.0 Rest - -
------------------------------------------------------------------------------------------------
a Modified from: Corrick (1977).
b Ranges used to denote variable grades produced.
c Cobalt included with nickel (1-2%).
3.2.1.2 Intermediate products and end-use
Most of the nickel produced is used in the production of alloys
(Table 4). In the production of nickel steel alloys, steel scrap,
limestone, iron oxide ore, and nickel are charged into a furnace
(open-hearth furnace, electric arc furnace and cupola) where the
steel and iron alloys are melted. After final adjustment of the
carbon and alloy contents, the steel is cast into moulds. Non-
ferrous melting is commonly performed in a reverberatory furnace.
Table 4. Consumption of nickel by
intermediate product and end-use industry in
1985 in the USA
---------------------------------------------
Index Consumptiona
(% of total)
---------------------------------------------
Intermediate product
Stainless and alloy steels 42
Nonferrous alloys 36
Electroplating 18
End-use industry
Transportation 23
Chemical industry 15
Electrical equipment 12
Construction 10
Fabricating metal products 9
Petroleum 8
Household appliances 8
Machinery 8
Other 7
---------------------------------------------
a Data from: US Bureau of Mines (1986).
The forming and shaping of ingots, after the casting of the
alloy, is performed by hot-working, grinding, and welding. Hot-
working includes the reduction of the cross section, e.g., by
forging or rolling. The resulting product may be cut and then
extruded to the desired form. Grinding is necessary to condition
the metal surface for further processing, e.g., welding. Welding
techniques, such as electric-arc, electric-spot oxyacetylene-torch,
or furnace-brazing, are used to fabricate assembled shapes. In
special cases, forming of parts may also be performed by sintering,
e.g., by sintering nickel powder from the Mond process.
The addition of nickel to steel and cast iron yields an alloy
with increased strength and toughness and resistance to corrosion.
Stainless steel is used in the chemical and food-processing
industries. Because of their ferromagnetic properties, iron-nickel
alloys are important materials for the electrical industry.
Various medical devices, such as prostheses or orthopaedic
implants, are made from stainless steel.
Nickel-copper alloys exhibit the highest mechanical strength
and resistance to corrosion and are used in the chemical and
machine industries (pipes, nozzles, machine parts for the food and
textile industries). Their high resistance to corrosion makes
these alloys a valuable material for the shipbuilding industry.
The nickel-copper alloy containing 77-63% nickel is known as Monel
metal.
Nickel alloys containing chromium, molybdenum, aluminium,
cobalt, titanium, or combinations of these elements are of special
industrial importance, because of their high-temperature
resistance.
Nickel-chromium alloys are used for jet engine components, in
nuclear reactors, and for turbine blades. Superalloy is an
extremely high-temperature resistant alloy containing 10-20%
cobalt. It is used in turbine blades and other engine components
of jets, ships, and racing vehicles, where extreme mechanical and
high-temperature resistance is required.
In plating, nickel gives a hard, tarnish resistant surface that
can be polished, which makes the finished product suitable for
consumer items, such as automobile components, household furniture,
and plumbing fixtures. Normally nickel-plated consumer items are
covered with a thin layer of chromium plating.
Other important uses of nickel are in nickel-cadmium batteries,
electronic equipment, and computers. Nickel compounds are used as
catalysts in the manufacture of organic chemicals, petroleum
refining, and edible oil hardening. They are also constituents of
pigments and colours for ceramics and glassware, and of marine
anti-fouling paint. In the glass industry, nickel is used in
moulds for bottles. Nickel compounds are also used as a coating
for pressure sensitive papers. In the United Kingdom, "silver"
coinage (5 p, 10 p, and 20 p) is based on cupro-nickel alloys
containing approximately 20% nickel. Coinage from other countries
contains higher levels, e.g., Canadian 10 cents (99.8% nickel), and
French 1 and 2 francs (99.8-99.9% nickel).
The production of secondary nickel in the form of scrap
recovery is a major source of nickel. Recycled scrap is generally
melted and refined and subsequently used for the production of
steels and alloys, similar in composition to those in which it
entered the recycling process. Thus, scrap recycling processes are
analogous with those used in primary production.
3.2.1.3 World production levels and trends
The development of global mine production during this decade is
shown in Table 5.
Table 5. Global mine production of nickel, by countrya,b (short tonnes of nickel)
--------------------------------------------------------------------------------------------------------
Country or territory 1980 1981 1982 1983c 1984d 1985d
--------------------------------------------------------------------------------------------------------
Albania (content of ore)d 6 100 6 200 6 400 6 400 6 600 6 600
Australia (content of concentrate) 81 927 81 963e 96 510 84 465 82 900 81 000
Botswana (content of matte) 17 022 18 200 19 573 20 079 19 300 19 000
Brazil (content of ore) 2 504e 2 573e 5 306 11 840 12 100 12 000
Burma (content of speiss) 15 22 22d 22d 22 -
Canadaf 203 709 176 642 97 824 134 300 192 000 195 000
Chinad 12 000 12 000 13 200e 14 300e 15 400 16 000
Colombia (content of ferroalloys) - - 1 100 15 000 15 400 10 000
Cuba (content of oxide, sinter, sulfide) 40 338 42 489 39 790 41 500d,e 35 050 40 000
Dominican Republic 18 019 20 601 5 838 23 369 26 698g 27 000
Finland (content of concentrate) 7 199 7 566 6 852 5 418 5 500 6 000
German Democratic Republicd 3 000 3 000 2 800 2 400 2 300
Greece (recoverable content of ore)h 16 796 17 200 5 500d,e 18 500d,e 18 400 16 000
Guatemala 7 650 - - - - -
Indonesia (content of ore)h 58 738 53 848 50 578 54 430 68 900 70 000
Morocco (content of nickel ore and 148 144e 140 - -
cobalt ore)
New Caledonia (recoverable content or ore) 95 451 86 079 66 250 43 542 45 200 44 000
Norway (content of concentrate)d 2 200e 7 700e 3 900e 4 000e 3 900e
Philippines 51 934 32 239 22 183 17 522 18 300 25 000
Poland (content of ore)d 2 300 2 300 2 300 2 300 2 300 -
South Africa, Republic of 28 239 29 100 24 250d 22 600d 27 600 27 000
USSR (content of ore)d 170 000 174 000 182 000 187 000 192 000 197 000
USA (content of ore shipped) 14 653 12 099 3 203 - 14 540g 6 900
Yugoslavia (content of ore)d 2 200 4 400 4 400e 3 300e 4 400 3 000
Zimbabwe (content of concentrate) 16 617 14 350 14 671 11 186 11 080 11 000
--------------------------------------------------------------------------------------------------------
Total 858 850e 804 715e 674 590 723 473 819 890 821 000
--------------------------------------------------------------------------------------------------------
a From: US Bureau of Mines (1985; 1986).
b As far as possible, this table represents recoverable mine production of nickel. Where data relate
to some more highly processed form of nickel, the figure given has been used in place of an
unreported actual mine output, to provide some indication of the magnitude of mine output. See notes
in parentheses and footnotes.
c Preliminary.
d Estimated.
e Revised.
f Refined nickel and nickel content.
g Reported figure.
h Includes a small amount of cobalt not reported and not recovered separately.
The nickel market weakened considerably from 1981 to 1983,
because of a reduction in demand arising from a recession in the
economy. In 1984, production and demand increased again. From a
1983 base, the US Bureau of Mines (1986) estimated that there would
be an increase in the average annual demand of about 2.5%, up
to 1990.
The identified world deposits with an average nickel content of
approximately 1% or more, contain 143 million tonnes of nickel (US
Bureau of Mines, 1986). In addition, there are extensive deep-sea
resources of nickel in manganese nodules, particularly in the
Pacific Ocean (US Bureau of Mines, 1986).
At present, there are only a few actual and potential
substitutes for nickel, e.g., aluminium, coated steel, titanium,
and plastic for industrial purposes, and platinum, cobalt, and
copper for catalytic uses. However, the use of these substitutes
results in increased costs and a lower quality end-product (US
Bureau of Mines, 1986).
3.2.1.4 Emissions from the primary nickel industry
Data on the loss of nickel into the environment during
production are limited. The smelting and roasting stages of ore
refining and alloy production may be considered as the more
important sources of nickel emission, because these processes
generate flue dust, i.e., fine particulate matter that is swept
from roasters and reverberatory furnaces by air and combustion
gases that pass through these units.
During an environmental study initiated by the Ontario Ministry
of Environment, trace metal emission rates from two nickel smelters
were calculated on the basis of the results of chimney stack
emission tests (Chan & Lusis, 1986).
The annual emissions of nickel during the study period are
given in Table 6. Annual emissions from a 381-m stack of one
smelter that emits particulates and gases from pyrometallurgical
smelting processes are listed in Table 7. This was considered the
most significant emission source.
Data on the chemical forms of nickel released into the
atmosphere from production processes are practically non-existent.
In most cases, statements are based on assumptions.
Species of nickel emitted into the air from mining garnierite
and processing it to produce ferronickel at a facility in the USA,
were assumed to be in the form of silicates, as in the ore, but
were expected to be minimal (Radian Corporation, 1984). Depending
on the temperature reached during drying and calcining, some nickel
on the surface of ore fragments may become oxidized and emitted as
iron-nickel oxide (Radian Corporation, 1984). Emissions during
roasting and smelting would probably be in the form of nickel oxide
combined with iron oxide as a ferrite (Radian Corporation, 1984;
Warner, 1984).
Table 6. Yearly emission (in tonnes)
of nickel (Sudbury Basin, Canada) for
the period 1973-81a,b
______________________________________
Source Variation Nickel
______________________________________
INCO 381-m stack Maximum 342
Average 228
Minimum 53
INCO 194-m stack Maximum 226
Average
Minimum
INCO Smelter Maximum 40
(low level) Average 31
Minimum 15
Falconbridge Maximum
93-m stack Average 9.6
Minimum
______________________________________
a From: Chan & Lusis (1986).
b Basis: 365 x 24 h/day production.
Table 7. Average measured emissions
of nickel from a 381-m stack (Canada)
(in kg/h)a
______________________________________
Year Emission
______________________________________
1973 48
1974 55
1975 15
1976 22
1977 33
1978 20
1979 12
1980 44
Average 31
______________________________________
a From: Chan & Lusis (1986).
When producing nickel from the sulfide ore, the process of
roasting the concentrated ore may lead to the formation of small
amounts of nickel sulfate and the emission of fine particles that
are sulfated as they are carried through the flues (Warner, 1984).
According to the investigations of Radian Corporation (1984),
emissions from matte refining processes at a US nickel refinery are
expected to be predominantly in the form of subsulfide, as the
processed matte is sulfide, and metallic nickel. A refinery dust
sample from a Canadian nickel refinery was calculated to contain
20% nickel sulfate, 57% nickel sulfide, and 6.3% nickel oxide
(Gilman & Ruckerbauer, 1962). Warner (1984) reported a nickel
content of 5-10% (10% of which was water-soluble) in flue dusts
from a Canadian smelter; most of these dusts are captured and
recycled.
3.2.1.5 Emissions from the intermediate nickel industry
Fumes from stainless steel melting processes were found to
contain 5% of total nickel in a water-soluble form. Chemically, it
occurs in fumes from stainless steel manufacturing mainly as the
metallic alloyed element in the iron matrix or in small amounts as
nickel oxide (Koponen et al., 1981). Nickel emissions into the
atmosphere can occur potentially from electroplating, and from
grinding, polishing, and cutting operations performed on the
finished product and scrap metal. However, in the case of
electroplating, they are considered to be very low or non-existent,
or are retained in the workplace area (Radian Corporation, 1984).
Grinding, polishing, and cutting operations could release
metallic nickel into the working environment with possible emission
to the outside atmosphere as a result of work-area ventilation
(Radian Corporation, 1984).
3.2.1.6 Emissions from the combustion of fossil fuels
The major source of airborne nickel is the combustion of fossil
fuels containing trace amounts of nickel (section 3.1.4).
Combustion sources include facilities burning coal and oil for
power generation or space heating.
Krishnan & Hellwig (1982) estimated emissions of trace metals
in the USA from various coal and oil combustion sources (Table 8)
and showed that nickel was a substantial trace pollutant. Nickel
was the only trace metal emitted at a significant rate from
domestic oil-fired boilers. The combustion of oil is a much more
significant source of nickel emissions than the combustion of coal
and is estimated to contribute 76-98% of the total nickel emissions
from coal and oil combustion in the USA (Krishnan & Hellwig, 1982).
A quantitative assessment of source contributions to inhalable
particulate matter in metropolitan Boston revealed a high
correlation between inhalable nickel particles (aerodynamic
diameter, 2.5-15 µm) and residual oil combustion (Thurston &
Spengler, 1985).
Cass & McRae (1983) evaluated routine air monitoring data from
sites in the South Coast Air Basin of California, in order to
relate sources to particular trace elements determined in the
samples. Eighty-one percent of fine nickel emissions (aerodynamic
diameter <10 µm) were calculated to arise from fuel oil fly ash.
However, a similar study by Kowalczyk et al. (1982) failed to
assign nickel particulate to any specific type of source.
Table 8. Emissions of trace metals in the USA from coal and oil combustion (metric tonnes
per year)a
-----------------------------------------------------------------------------------------------
Trace Utility boilersb Industrial boilersc Commercial boilersc Residential boilersb
(>264 GJ/h input) (>26 GJ/h input) (>26 GJ/h input) (>422 MJ/h input)
Coal Oil Coal Oil Coal Oil Coal Oil
-----------------------------------------------------------------------------------------------
Arsenic 149.1 144.7 214.8 54.7 99.3 84.6 60.3 3.2
Beryllium 19.2 7.0 6.2 2.2 3.7 0.1 0.8 4.1
Cadmium 7.7 219.7 4.9 83.9 2.7 128.4 1.6 23.1
Chromium 561.3 87.5 33.3 33.1 7.9 76.8 7.8 2.3
Lead 360.0 61.1 113.3 23.6 58.2 36.5 36.8 19.9
Manganese 407.2 33.7 31.5 7.5 22.8 11.6 163.9 1.2
Mercury 86.9 3.1 4.6 1.0 1.3 1.5 1.0 2.5
Nickel 281.1 877.3 34.0 363.1 14.1 818.0 7.8 216.4
Selenium 120.9 30.9 44.4 11.7 17.4 18.1 14.5 21.2
Vanadium 390.0 4637.0 29.4 1505.0 12.0 3293.0 7.8 6.1
-----------------------------------------------------------------------------------------------
a From: Krishnan & Hellwig (1982).
b 1978.
c 1977.
Fly ash emitted from combustion sources has been analysed, in
order to gain information on the chemical species of nickel present
in air. Henry & Knapp (1980) analysed fly ash samples from the
stacks of oil-fired and coal-fired power plants. In fly ash
samples from oil-fired plants, 60-100% of the nickel components
were water soluble, whereas, with one exception, samples from coal-
fired plants contained 20-80% water-soluble material. As the
sulfate ion was the only major ion detected in the water-soluble
phase, it was concluded that nickel sulfate is the predominant form
of nickel in emissions from oil-fired and coal-fired power plants.
This conclusion was confirmed by Fourier transform infrared
analysis (Gendreau et al., 1980).
Analysis of filter-collected fly ash from five oil-fired units
revealed the presence of metals, including nickel, as sulfates in
the soluble phase (Dietz & Wieser, 1983). As the sulfate amount
measured by ion chromatography was, on average, 17% less than the
sulfate amount expected from stoichiometric considerations, Dietz &
Wieser (1983) suggested that some of the soluble nickel might have
been present as partially soluble oxide or very finely dispersed
particles of metal oxide.
Major components in the insoluble phase of fly ash samples from
oil-fired utility boilers were determined by X-ray diffraction to
be oxides of iron, aluminium, calcium, and silicon, and possibly
nickel oxide (Henry & Knapp, 1980).
Hulett et al. (1980) studied the 100-200 µm fractions of fly
ash specimens from 4 coal-fired power plants. They separated the
ash magnetically into 3 insoluble fractions, i.e., glass, mullite-
quartz, and magnetic spinel. Chemical determination showed that
90% of the nickel was present in the magnetic spinel phase. The
nickel was assumed to be in the form of a substituted spinel,
Fe3-xNixO4.
The results of studies by Hansen & Fisher (1980) and Hansen et
al. (1984) indicated that most of the nickel, present in coal
combustion fly ash particles, was soluble and associated primarily
with sulfate.
Thus, nickel emissions into the atmosphere from coal and oil
combustion are considered to be composed predominantly of nickel
sulfate, with smaller amounts of nickel oxide and nickel combined
with other metals in complex oxides.
Another potentially important source of nickel in the
environment is the combustion of diesel oil, which can contain 2 mg
nickel/litre (2 ppm) (Fishbein, 1981). The vapour phase of diesel
engine exhaust may also contain nickel carbonyl. In urban air near
a busy intersection, Filkova & Jäger (1986), using EAAS, measured
nickel carbonyl concentrations in the range of 0-14.1 ng/m3.
3.2.1.7 Emissions from sewage sludge and waste incineration
Estimates made by Schmidt & Andren (1980) (section 4.1.1)
indicated that, after fuel combustion, and nickel mining and
refining, waste and sewage sludge incineration is the next major
source of nickel emissions.
Evaluation of emission data from sewage sludge incinerators
indicated that less than 1% of the nickel contained in the sludge
feed was emitted as a fume, while the major part was emitted as fly
ash (Gerstle & Albrinck, 1982). Dewling et al. (1980) noted that
80% of the nickel in the feed sludge of a fluidized bed, wastewater
sludge incinerator in north-west Bergen was retained in the ash.
Emission rates may vary widely, depending on combustion
temperature, sludge composition, pollution control devices, and
type of incinerator (Gerstle & Albrinck, 1982; Samela et al.,
1986). Nickel species present in emissions from sewage sludge and
waste incineration were analysed by Henry and co-workers (1982).
The water-soluble phase of sewage sludge incinerator emissions
contained mainly sulfate ions, indicating that the water-soluble
nickel existed in the sulfate form. The soluble phase of refuse
incinerator emissions also contained chloride ions, suggesting that
nickel can be present in this phase as the chloride or sulfate.
The insoluble phases of emissions from the two sources were similar
and it is highly probable that the nickel may exist as complex
oxides and iron spinels.
3.2.1.8 Miscellaneous emission sources
Nickel can be emitted during cement manufacturing and asbestos
mining and milling, because nickel is a natural component of the
minerals used in these operations.
During cement manufacturing, nickel is emitted, either as a
component of the clays, limestones, and shales, used as raw
materials, or as an oxide formed in the high temperature process
kilns. Swedish cement was found to contain 5-59 mg nickel/kg
(Wahlberg et al., 1977).
Crude chrysotile asbestos fibres from different mines in Canada
(Quebec) contained 63-389 mg nickel/kg. In the host rock, the
nickel content was 265-3075 mg/kg. Milled fibres are enriched by a
factor of 4 (Barbeau et al., 1985). Nickel emitted into the air is
expected to be in the form of silicates.
3.2.1.9 Waste disposal
Nickel from various industrial processes and other sources
reaches waste water.
Klein et al. (1974) examined the major sources of nickel
flowing into the New York City municipal wastewater collection
system. The electroplating industry was found to be the dominant
source of nickel (62%) in wastewater treatment plants. The total
daily amount of nickel discharged into the sewers by electroplating
firms was estimated to be 508 kg. The effluent of an
electroplating factory in India contained 578.12 mg nickel/litre
(Ajmal & Khan, 1985). Residential sources contribute 25% of the
nickel in waste water, 3% comes from other industrial sources, and
10% from run-off. The total amount of nickel reaching the harbour
of New York City, estimated to be 978 kg/day, originated as 43%
from the treatment plant effluents, 30% from run-off, 20% from
untreated waste water and 7% from sludge. Nickel concentrations in
the influents of 12 wastewater treatment plants ranged from 0.05
to 0.31 mg/litre.
In the raw sewage of 25 full-scale municipal sewage-treatment
plants, the nickel concentration varied between undetectable and
0.69 mg/litre (Sung et al., 1986).
Conventional treatment of mixed waste water consists of
hydroxide precipitation of the metals at an alkaline pH, followed
by removal of the resulting solids by sedimentation and, sometimes,
filtration. Chen et al. (1974) reported a removal efficiency of
the secondary treatment process (sludge activation and
sedimentation) of 25-57%. The final effluent contained 0.14-0.177
mg nickel/litre. Sung et al. (1986) measured nickel levels in the
influents and effluents of 25 sewage-treatment plants in the USA.
In treatment plants with 50% minimum removal efficiency, 50% or
more of the nickel was removed in the primary effluent at 5% of the
plants; 50% or more was removed in the secondary effluent in 11% of
the plants, and 50% or more was removed in the discharge of 10% of
the plants.
Finally, residues from wastewater treatment are disposed of by
deep-well injection, ocean dumping, land treatment, landfill, or
incineration. Deep-well disposal is limited to residual liquids
containing low levels of suspended solids and is most applicable to
scrubber water blow-down. Ocean dumping is a source of
contamination for coastal areas. Land treatment, such as sewage
sludge treatment of agricultural soils, is a potential source of
soil, and subsequent food plant, contamination.
Leachates from landfills may contaminate ground water and may
contain 1.85-8.2 mg nickel/litre (Hrudey, 1985). Incineration of
sewage sludge gives rise to considerable air emissions.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1. Transport and distribution between media
Nickel is introduced into the environment from both natural and
man-made sources (section 3). It is circulated throughout all
environmental compartments (atmosphere, pedosphere, hydrosphere,
and biosphere) by means of chemical and physical processes, such as
wet and dry deposition, and, to a much lesser extent, by means of
the biological transport mechanisms of living organisms.
4.1.1. Air
Nickel is emitted into the atmosphere from various natural
sources, as indicated in Table 9. As only limited data are
available concerning the relative quantities emitted, estimates
have been made. Estimated emission values vary depending on the
impact that is attributed to individual sources. Barrie (1981)
considered sea spray to be a major contributor of atmospheric
nickel. Generally, soil and volcanoes appear to be major sources
and may contribute 40-50% of airborne nickel from natural sources.
Table 9. Global emission of nickel from
natural sources to the atmosphere.
Emission rate (106 kg/year)
------------------------------------------------
Source Nriagu Schmidt & Barrie
(1980) Andren (1980) (1981)
------------------------------------------------
Soil dust 20 4.8 7.5-37.5
Volcanoes 3.8 2.5 10-60
Vegetation 1.6 0.82 1.5-20
Forest fires -a 0.19 0.3-15
Meteoric dust - 0.18 -
Sea salt - - 27
Sea aerosol - 0.009 -
Total 26b 8.5 46-160
________________________________________________
a No data available.
b Total includes 0.6 for "others".
Estimated man-made inputs into the atmosphere exceed the
natural inputs (Tables 9 and 10). It has been estimated that the
total amount of nickel that has been dispersed into the world
ecosystems through the atmosphere is 1.0 x 109 kg (Nriagu, 1979).
As indicated in Table 10, combustion of oil and incineration of
waste contribute more than 70% of the nickel from man-made sources,
followed by nickel mining and refining with 17%.
Table 10. Global emission of nickel from
man-made sources to the atmospherea
------------------------------------------
Source Emission rate
(106 kg/year)
------------------------------------------
Residual oil combustion 17
Fuel oil combustion 9.7
Nickel mining and 7.2
refining
Municipal incinerators 5.1
Steel production 1.2
Gasoline and diesel 0.9
fuel combustion
Nickel alloy production 0.7
Coal burning 0.66
Cast iron production 0.3
Sewage sludge 0.048
incineration
Copper-Nickel alloy 0.04
production
Total 42.85
------------------------------------------
a Adapted from: Schmidt & Andren (1980).
The transport and distribution of nickel particulates to, or
between, different environmental compartments is strongly
influenced by particle size and meteorological conditions.
Particle size is primarily a function of the emitting source.
Generally, particles from man-made sources are finer than dust
particles of natural origin, e.g., soil (Beijer & Jernelöv, 1986).
As the highest nickel concentrations are found in the smallest
particles collected from ambient air (Lee & von Lehmden, 1973;
Natusch et al., 1974), these particles are of special environmental
and toxicological significance. There is evidence that fine
particulate matter, which has a longer residence time in the
atmosphere, is carried a long distance, whereas larger particles
are deposited near the emission source (Beijer & Jernelöv, 1986).
Schmidt & Andren (1980) estimated an atmospheric residence time for
nickel particulates of 5.4-7.9 days.
There are no data on the chemical forms of nickel from natural
sources in the atmosphere. When considering the composition of the
source, a part of airborne nickel may exist as pentlandite
((FeNi)9S8) and garnierite (a silicate mixture) (Schmidt & Andren,
1980). The chemical composition of nickel compounds released from
man-made sources differs from that of compounds from natural
sources because of the different processes involved. Flue dust
analysis revealed a predominance of oxides and sulfates (section
3). Alterations in the chemical forms, following distribution in
the atmosphere, have not been investigated.
The fate of nickel carbonyl, the only gaseous nickel compound
of environmental importance, may be deduced from its chemical
properties. Nickel carbonyl is unstable in air and decomposes to
form nickel carbonate. At 25 °C, the lifetime of ng/m3
concentrations of nickel carbonyl is about one minute, increasing
by one minute for each mg/m3 of carbon monoxide present (Stedman &
Hikade, 1980).
4.1.2. Water
Nickel is introduced into the hydrosphere by removal from the
atmosphere (wet and dry deposition of naturally and
anthropogenically released nickel), by surface run-off, by
discharge of industrial and municipal waste, and following the
natural erosion of soils and rocks.
In surface or ground waters not polluted by human beings, the
nickel content often reflects the weathering process of the parent
soil or rock. However, data are insufficient to separate natural
geochemical effects from man-made influences. Keller & Pitblado
(1986) demonstrated a relationship between elevated nickel levels
in Sudbury area lakes and nickel-emitting sources.
In rivers, nickel is transported mainly as a precipitated
coating on particles and in association with organic matter; in
lakes, the ionic form and the association with organic matter are
predominant (Snodgras, 1980).
Nickel may be deposited in the sediment by such processes as
precipitation, complexation, adsorption on clay particles, and via
uptake by biota. Because of microbial activity or changes in
physical and chemical parameters, including pH, ionic strength, and
particle concentration, sorption processes may be reversed (Di Toro
et al., 1986) leading to release of nickel from the sediment.
Part of the nickel is transported via rivers and streams to the
ocean. Riverine suspended particulate input is estimated to be 135
x 107 kg/year (Nriagu, 1980). Industrial and municipal waste and
atmospheric fallout contribute 0.38 x 107 kg/year and 2.5 x 107
kg/year, respectively (Nriagu, 1980). Possible routes of removal of
nickel particulate are scavenging by ferromanganese phases on the
ocean floor or algae. The average residence time of nickel in the
deep ocean is calculated to be 2.3 x 104 years (Nriagu, 1980).
4.1.3. Rocks and soil
Nickel occurs naturally in several types of rocks (section 3.1)
and it may enter the surface environment by the chemical and
mechanical degradation of the rock to soil. Nickel is fractionated
within the different components of the soil profile, depending on
the type of soil and soil chemical conditions. In residual soils
nickel is preferentially adsorbed on alkali and alkaline earth
cations in the clay minerals (Boyle, 1981).
Depending on soil type, nickel may exhibit a high mobility
within the soil profile as demonstrated by Heinrichs & Mayer (1980)
for a brown forest soil in the Solling ecosystem, a relatively
unpolluted area. The soil profile in the study area revealed an
accumulation of nickel in the top organic layer, and a
concentration increasing with depth in the subsequent mineral
layer, indicating a high mobility of nickel. Similar results were
obtained for peat muck soil profiles (Sapek & Sapek, 1980). In the
Solling forest ecosystem study, comparison of flux data and
ecosystem output (seepage) and input (precipitation) showed that
nickel was balanced within the ecosystem and that the forest
ecosystem was neither a source nor a sink in the geochemical cycle
of nickel. However, this balance may be disturbed in case of heavy
pollution.
Water solubility, and thus bioavailability to plants, is
affected by soil pH; decreases in pH generally mobilize nickel.
Most nickel compounds are relatively soluble at pH values <6.5,
whereas nickel exists predominantly as insoluble nickel hydroxides
at pH values >6.7. Therefore, acid rain has a pronounced tendency
to mobilize nickel from soil and increase nickel concentrations in
ground water, leading eventually to increased uptake and potential
toxicity for microorganisms, plants, and animals (Sunderman &
Oskarsson, 1988). Other factors are the numbers of organic and
inorganic cation exchange sites, the adsorption strength, and the
relative numbers of other cations competing with nickel for cation
exchange sites (Hutchinson et al., 1981). Depending on the
geochemical characteristics of soils, nickel may become distributed
among the different soil compartments finally reaching ground water
and, thus, rivers and lakes. Transport of nickel to river systems
and oceans is also mediated by erosion and run-off processes.
The main man-made sources of nickel contamination in soils are
emissions from nickel smelting and refining and disposal of
contaminated sewage sludge. Atmospheric input of nickel into the
soil and inputs by waste disposal and through the application of
fertilizers are estimated to be 5.5 x 104 kg/year and 1.4 x 104
kg/year, respectively (Nriagu, 1980). On the basis of the nickel
pool in soils and the denudation of nickel from continents, the
residence time of nickel in soils is calculated to be approximately
3500 years (Nriagu, 1980).
4.1.4. Vegetation and wildlife
Terrestrial plants take up nickel from soil primarily via the
roots. The nickel concentrations in most natural vegetation range
from 0.05 to 5 mg/kg dry weight (NAS, 1975). The amount of nickel
uptake from the soil depends on various geochemical and physical
parameters including the type of soil, soil pH, humidity, the
organic matter content of the soil, and the concentration of
extractable nickel.
Although nickel concentrations exceeding 50 mg/kg (on a dry
weight basis) are usually toxic for plants (NAS, 1975), nickel-
tolerant species growing on serpentine soils can accumulate nickel
levels that are orders of magnitude higher.
Aquatic plants are also known to take up and accumulate nickel
(Jenkins, 1980a). As algae are at the lower end of many
foodchains, this fact needs special consideration. The highest
nickel levels found in aquatic algae and spermatophytes in
contaminated areas were 150.9 mg/kg dry weight or 690 mg/kg wet
weight, respectively, exceeding normal levels by more than 10 times
(Jenkins, 1980a).
Investigations of nickel distribution and cycling in the
Solling ecosystem indicated a steady state, i.e., a balance between
atmospheric input and binding in the soil and phytomass (Heinrichs
& Mayer, 1980). This steady state may be disturbed by human
impact, e.g., increase of atmospheric nickel input or acidity of
rainfall. Nickel is released into the atmosphere from plant
exudates and forest fires. It migrates into soil and surface
waters following the decay and mineralization of plants.
Since nickel occurs in virtually all plants at different
levels, it may be taken up by herbivorous terrestrial and aquatic
animals and their predators. Increased nickel levels in vegetation
can give rise to increased nickel contents in grazing animals and
their predators. For example, nickel levels were found to be
higher in the organs of wild ruminants than in those of domestic
animals, because of the higher nickel content in their grazing
areas (Groppel et al., 1980).
4.2. Uptake and bioaccumulation
4.2.1. Terrestrial organisms
The best known phenomenon of nickel accumulation in plants is
the considerably increased nickel levels found in certain species
growing on infertile serpentine soils. Approximately 70 nickel
hyperaccumulators, i.e., species with nickel concentrations
exceeding 1000 mg/kg, are known, most of them belonging to the
genus Allyssum. Investigations of nickel-accumulating
Flacourtiaceae in New Caledonia revealed nickel levels in the range
of 1000-50 000 mg/kg dry weight (Jaffre et al., 1979). Yang et al.
(1985) found a significant inverse correlation between the contents
of nickel and the nutrients manganese, boron, and sodium in plant
material, thus indicating the role of high nickel concentrations in
serpentine substrates as controlling factors in nutrient uptake.
Nickel hyperaccumulators are of scientific interest, because of the
possible use of vegetation, regrowing on mine dumps, for nickel
exploitation (Brooks, 1980).
The major source of nickel accumulation in terrestrial plants
is the increased occurrence of nickel in soils. High levels of
nickel in soils result from nickel-emitting industrial sources
(Rutherford & Bray, 1979; Polemio et al., 1982; Alloway & Morgan,
1986; Gignac & Beckett, 1986) and sewage sludge treatment (Berrow &
Burridge, 1981; Chang et al., 1984). Nickel was found to be more
available to plants from soils treated with sewage sludge than from
inorganically polluted soils (Alloway & Morgan, 1986). The total
nickel content in sludge samples collected from different municipal
sewage treatment plants in the USA was in the range of 21-1990
mg/kg dry weight (Sung et al., 1986). Coker & Matthews (1983)
reported a nickel content of 10-2000 mg/kg dry weight in sewage
sludge applied to land in the United Kingdom in 1977. The
persistence of nickel in acetic acid-extractable form in soil was
found to be 10 years (Berrow & Burridge, 1981). Nickel in polluted
soils was found to be highly concentrated in the upper organic soil
horizon (Chang et al., 1982; Brown et al., 1983a; Chang et al.,
1984), probably because of the high cation exchange capacity of the
surface organic layer (Hutchinson et al., 1981).
In a research project initiated by the Federal Environmental
Protection Agency of Germany, vegetables and plant crops were grown
on soils that were polluted with nickel (average 558 mg/kg soil)
through sewage sludge application (Grössman, 1988). Levels in
green vegetables, different types of cabbage, and onions ranged
from 10.8 to 65 mg nickel/kg dry weight. In beans and peas, nickel
levels ranged from 42 to 65.1 mg/kg and 16.5 to 23.4 mg/kg dry
weight, respectively. In root vegetables, nickel was accumulated
to a lesser extent; concentrations of 7.95-26.9 mg nickel/kg dry
weight were measured. Increasing nickel concentrations in the soil
resulted in increasing nickel accumulation in the plants. This
also held true for maize used as fodder for animals. However, in
maize, nickel levels were generally lower ranging from 2.32 to 4.27
mg/kg dry weight in kernels, 6.69 to 10.7 mg/kg in leaves, and 4.33
to 5.53 mg/kg in stems. The nickel concentration in the soil was
745 mg/kg.
Reddy & Dunn (1984) grew soya beans on sewage sludge-treated
soil in glass houses and found increasing nickel levels in plant
tissues with increasing rates of sludge application. Concentrations
were greater in leaves than in stems and ranged from 2.1 to 8.5
mg/kg dry weight in leaves and from 1.2 to 6.2 mg/kg dry weight in
stems, following application of 0-8.4 kg nickel/ha.
Keefer et al. (1986) found accumulation of nickel in both the
edible and inedible parts of vegetables grown on soils treated with
different types of sewage sludge. Nickel loading of the soil was
in the range of 2-2540 kg/ha. Nickel concentrations detected in
cabbage heads were 2.04-22.1 mg/kg dry weight (control, 3.78
mg/kg). Radish roots and tops contained 1.28-12.3 mg nickel/kg
(control, 1.64 mg/kg) and 3.12-18.3 mg/kg (control, 3.16 mg/kg),
respectively. In green bean leaves and pods, nickel levels were
4.68-14.0 mg/kg (control, 4.00 mg/kg) and 5.0-11.0 mg/kg (control
5.04 mg/kg). The water-soluble nickel concentration in sewage
sludges was related to nickel uptake in the species tested. Soil
characteristics, such as texture, drainage status, and sorptive
capacity, play a dominant role in nickel availability to plants.
When vegetables were grown in greenhouse pots on sewage sludge-
treated soils of a calcareous loam, a clay, and a sandy loam type,
the highest nickel accumulation occurred in cabbage grown on clay
and lettuce grown on sandy loam (Alloway & Morgan, 1986). Gignac &
Beckett (1986) found a negative correlation between the nickel
content of peat and the percentage organic content.
Increased acidity of soils resulting, e.g., from SO2- emission,
enhanced nickel solubility and uptake by plants (Hutchinson &
Whitby, 1977; Brown et al., 1983b; Sanders et al., 1986). Liming
of soil can reduce nickel uptake by plants (Machelett & Podlesak,
1980; Francis et al., 1985).
Nickel accumulation in plants growing in the vicinity of a
nickel smelter was investigated by Hutchinson & Whitby (1977). The
nickel contents of foliage from Comptonia peregrina, Deschampsia
flexuosa, Acer rubrum, and Betula papyrifera, growing at a distance
of 1.6 km from the Coniston nickel smelter near Sudbury, Ontario,
were 113 mg/kg, 902 mg/kg, 109 mg/kg, and 148 mg/kg dry weight,
respectively. The corresponding nickel concentration in the upper
soil surface was 2.679 mg/kg dry weight. The nickel contents of
the soil and the plant leaves declined with increasing distance
from the smelter. Similar observations were reported by Gignac &
Beckett (1986) for vascular plant species, sphagnum species, and
bryophytes growing near Sudbury.
Determination of nickel concentrations in plant species growing
on a copper mine spoil heap demonstrated relative bioconcentration
values (mg nickel per kg in plants/mg nickel per kg in EDTA soil
fraction) of 2.7 (leaves) and 1.4 (branches) in Thlaspi montanum,
and 2.0 (leaves) and 0.9 (branches) in Phlox austromontana (Hobbs &
Streit, 1986).
Animals grazing on nickel-contaminated vegetation accumulated
nickel in various organs. Wild ruminants grazing near nickel-
emitting industrial sources accumulated nickel in the ribs and
kidneys at levels of from 1.13-1.50 mg/kg dry weight and 0.47-0.86
mg/kg dry weight, respectively. The nickel contents of their
winter grazing were determined to be in the range of 3.12-14.49
mg/kg dry weight (Groppel et al., 1980). Highly elevated nickel
levels (27 times the control value) were detected in primary flight
feathers of mallard and black duck in the Sudbury district (20-140
km from a nickel smelter) (Ranta et al., 1978).
4.2.2. Aquatic organisms
In general, aquatic organisms resorb metals over their entire
surface. They also incorporate metals from their food. In rooted
aquatic plants, metals can be absorbed not only by the roots but
also by submerged stems and leaves (Mortimer, 1985). Most groups
of aquatic organisms include some species capable of accumulating
nickel (Jenkins, 1980a). The highest levels have been found in
aquatic organisms near sources of pollution, especially nickel
smelters.
Clark et al. (1981) studied the accumulation and depuration of
nickel by the duckweed Lemna perpusilla. Plants collected from a
fly-ash pond were allowed to depurate in dechlorinated tap water at
20 °C for 14 days. Accumulation was then examined over a 10-day
period and depuration over the following 8 days. During the 14-day
depuration period, nickel concentrations fell from 160 mg/kg dry
weight to less than 40 mg/kg dry weight and, in clean water,
remained at this level. When exposed to 0.1 mg nickel/litre in the
water, duckweed accumulated nickel to levels of about 800 mg/kg dry
weight at the end of a 10-day exposure period. The peak
bioaccumulation concentration of nickel occurred 2 days after the
depuration period began, with most nickel elimination occurring in
the 2 succeeding days. After 8 days, the nickel level was down to
the original value of <160 mg/kg. Because the concentration of
nickel in the water of the fly-ash pond was also about 0.1
mg/litre, greater accumulation occurred in the laboratory than in
the field.
Cowgill (1976) found that Euglena gracilis accumulated nickel
to a concentration of 1.8 mg/kg dry weight when exposed to 8.9 x
104 mg nickel/litre in spring water. A biological concentration
factor of about 2000 was calculated.
In a study by Hutchinson & Czyrska (1975), Lemna minor was
collected from 23 ponds and lakes in Southern Ontario in which the
mean content of nickel in the water was 0.027 mg/litre. The plants
contained 5.4-35.1 mg nickel/kg dry weight, equivalent to
concentration factors of 200-1300. The authors also cultured Lemna
minor in a growth medium containing 0.01-1.00 mg nickel/litre at a
temperature of 24±52 °C and a pH of 6.8, for 3 weeks. Nickel
accumulation ranged from 4000 (0.01 mg nickel/litre in the growth
medium) to 6134 (0.5 mg nickel/litre in the growth medium). There
was a correlation between levels of nickel in the plant and levels
in water. Nickel accumulation was greater in the presence of
copper.
Elodea densa, cultivated at 21-25 °C in a flowing water system
with a constant nickel concentration in the medium of 0.01
mg/litre, showed an accumulation factor of 200 after 12 days
(Mortimer, 1985).
The highest concentration factor reported, approximately 20 000,
was found in an aquatic ecosystem study, conducted by Hutchinson et
al. (1975), in periphyton algae sampled from a section of the
metal-contaminated Wanapitei river in the Sudbury area. Analysis
of aquatic macrophytes, which were collected from metal-
contaminated rivers in this area, indicated a species specificity
for uptake and a significant correlation between total nickel
content in the sediment and water and in rooted macrophytes.
Watras et al. (1985) studied the accumulation of nickel in two
levels of a simple aquatic food chain using Scenedesmus obliquus
and Daphnia magna. The algae accumulated nickel to concentrations
30-300 times the ambient concentration. In Daphnia, concentration
factors were only 2-12. There was little difference in
accumulation from incubation in 63Ni-labelled medium without algae
or from incubation in labelled medium with labelled algae. The
data indicated that direct uptake from the medium rather than
uptake from ingested algae was the primary accumulation mechanism.
These results confirmed earlier studies by Hall (1982), who
described nickel accumulation in Daphnia magna as the sum of five
processes occurring in the various body components, namely,
adsorption to, and desorption from, body and tissue surfaces,
absorption, retention or storage, and excretion.
In the course of the aquatic ecosystem study performed by
Hutchinson et al. (1975), nickel levels were determined in aquatic
animals. Accumulation factors in animals were lower than in
aquatic vegetation and were found to be 643 in zooplankton, 929 in
crayfish, and 262 in clams. In fish species caught in the
Wanapitei river (42 mg nickel/litre), nickel levels in muscle
tissue were lower than those in the liver, kidneys, and gills. The
predatory yellow pickerel exhibited the highest nickel levels with
51.6 mg/kg wet weight in kidney tissue, giving a concentration
value of 229.
Calamari et al. (1982) reported nickel levels of 2.9 mg/kg wet
weight in liver, 4.0 mg/kg in kidneys, and 0.8 mg/kg in muscle in
Salmo gairdneri, after 180 days exposure to 1 mg nickel/litre in
the water. Nickel levels at the start of the study were 1.5, 1.5,
and 0.5 mg/kg, in liver, kidneys, and muscle, respectively. The
authors also found, by means of a toxicokinetic model, that
theoretical asymptotic values for liver, kidney, and muscle should
be reached in 397, 313, and 460 days, respectively, yielding
bioconcentration values of 3.1, 4.2, and 1.0, respectively.
Laboratory studies showed that nickel had little capacity for
accumulation in all the fish species studied. However, it was also
demonstrated that this relatively low concentration of nickel in
tissues could cause biochemical damage. The range of
concentrations reported in whole fish in uncontaminated waters, on
a wet-weight basis, is 0.2-2 mg/kg. This value could be increased
by a factor of ten in contaminated areas (Calamari et al., 1984).
White et al. (1986) investigated nickel levels in coots (Fulica
americana) resting and feeding by a pond that was used for the
disposal of fly ash from a nearby coal-fired power plant. Though
the nickel concentration in the pond sediment was much higher than
the concentration in the water (which was below detection limit,
except at one collection period), accumulation of nickel in coot
livers was not observed in 2 years of plant operation.
4.3. Biomagnification
Accumulation factors in different trophic levels of aquatic
food chains suggest that biomagnification of nickel along the food
chain, at least in aquatic ecosystems, does not occur.
Hutchinson et al. (1975), in their investigations on nickel
compartmentation in an aquatic ecosystem, found large concentration
factors in the vegetation and decreasing factors in the higher
trophic levels. In a small food chain consisting of an alga
(Scenedesmus obliquus) and a zooplankton species (Daphnia magna),
there was no biomagnification (Watras et al., 1985). Because
nickel in aquatic ecosystems decreases in concentration with
increasing levels of the food chain, biomagnification does not
occur.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Air
Owing to the large number of sources releasing nickel into the
atmosphere and their uneven distribution over the globe, ambient
nickel concentrations may vary over several orders of magnitude.
Urban and rural areas usually exhibit air nickel levels ranging
from 5 to 35 ng/m3 (Bennett, 1984). Higher values were recorded in
heavily industrialized areas and larger cities. Nickel
concentrations, monitored continuously over one year in 4 American
cities, were found to be in the range of 18-42 ng/m3 (Saltzman et
al., 1985). In the vicinity of a nickel smelter in the Sudbury
area, Ontario, levels of 124 ng/m3 were measured (Chan & Lusis,
1986). Atmospheric concentrations at Spitsbergen, measured during
two months, ranged between approximately 1 and 2 ng nickel/m3
(Pacyna et al., 1985). In the Canadian Arctic, the annual mean
concentration was 0.38 ng/m3. The winter mean was 0.62 ng/m3,
indicating a seasonal cycle (Hoff & Barrie, 1986). Assuming a
ventilation rate of 20 m3 and air concentrations of 5-35 ng/m3, the
amount of nickel entering the human respiratory tract is in the
range of 0.1-0.7 µg/day.
The distribution of nickel among suspended particulates in the
air will determine the fraction that is inhalable. Data on the
size distribution of nickel particulates are limited. Lee & von
Lehmden (1973) summarized data on the size of nickel particulates
in urban air and found mass median diameters of 0.83-1.67 µm, 28-
55% of the particles being <1 µm. A more recent summary of size
distribution of trace elements in different areas yielded a mass
median diameter for nickel particulates of 0.98 µm (Milford &
Davidson, 1985). Particles of less than about 1 µm are deposited
predominantly in the alveolar regions of the lung (Stern et al.,
1984). Nickel was found to be most concentrated in the smallest
particles emitted from coal-fired power plants (Natusch et al.,
1974). Particles of a mass median diameter of 0.65-1.1 µm
contained 1600 mg nickel/kg while 4.7-11 µm particles contained
about 400 mg nickel/kg.
Another important route of nickel exposure is tobacco smoking.
Cigarette tobacco may contain approximately 1.3-4.0 mg nickel/kg.
About 0.04-0.58 µg nickel is released with the main stream smoke
(gas phase and particle phase) of one cigarette (Szadowski et al.,
1969a; Menden et al., 1972; Gutenmann et al., 1982). Smoking 40
cigarettes per day may result in the inhalation of 2-23 µg nickel
per day. The formation of nickel carbonyl in the main stream smoke
is suspected (Sunderman & Sunderman, 1961a; Stahley, 1973), but it
could not be detected using Fourier-transform infrared spectrometry
(detection limit 0.1 µg/litre) (Alexander et al., 1983).
5.1.2. Drinking-water
On the basis of determinations of nickel concentrations in 969
water supplies in the USA during 1965-70, the average concentration
of nickel in water samples taken at the consumer's tap was 4.8
µg/litre (NAS, 1975).
In Italy, nickel levels in drinking-water were mostly below 10
µg/litre (Clemente et al., 1980). Schumann (1980) measured levels
of 6.8-10.9 µg/litre in the German Democratic Republic. Leaching
processes from water taps and fixtures contribute to nickel levels
already present in drinking-water. Between 18 and 900 mg of nickel
were leached from 10 used water taps, which had been filled, in an
inverted position, with 15 ml deionized water, and left overnight
for 16 h (Strain et al., 1980). In Denmark, levels of up to 490
µg/litre were observed, when water was left standing overnight in
nickel-containing plumbing fittings (Andersen et al., 1983).
In areas where nickel is mined, as much as 200 µg nickel/litre
has been recorded in drinking-water (McNeely et al., 1972).
Assuming a daily intake of 1.5 litres water and a level of 5-10
µg nickel/litre, the mean daily intake of nickel from water for
adults would be between 7.5 and 15 µg.
5.1.3. Food
Nickel is ingested by human beings through the consumption of
plants and animals that contain nickel. Nickel levels were
determined, using EAAS, in foods in the Netherlands by Ellen et al.
(1978). They were found to be less than 0.5 mg/kg fresh weight in
most products, except for cacao products and nuts, which contained
nickel levels of up to 9.8 and 5.1 mg/kg, respectively. Smart &
Sherlock (1987) reported that nickel levels, determined using FAAS,
in English meat, fruit, and vegetables were of the order of > 0.2
mg/kg fresh weight. Aquatic organisms, e.g., molluscs and fish,
may contain relatively large amounts of nickel, if the nickel
concentration in the water is high (Table 11).
Table 11. Nickel concentrations in aquatic organisms
------------------------------------------------------------------------------------------
Species Tissue Locality Concentrationa Reference
or (mg/kg)
organ
------------------------------------------------------------------------------------------
Plants
Lemna minor whole Southern Ontario; ponds 5.4-35.1 D Hutchinson &
(duckweed) and lakes Czyrska (1975)
Eichhornia crassipes whole Yamuna river, India; 4.4-83.0 D Ajmal et al.
near big cities (1985)
------------------------------------------------------------------------------------------
Table 11 (contd.)
------------------------------------------------------------------------------------------
Species Tissue Locality Concentrationa Reference
or (mg/kg)
organ
------------------------------------------------------------------------------------------
Fontiralis antipyretrica Augraben river, Italy; 5.5-10.6 D Dallinger &
near motorway Kautzky (1985)
Leiferer Graben river, 17.7-29.2 D
Italy; metal industry
area
Ranunculus fluitans Augraben river, Italy; 69 D Dallinger &
near motorway Kautzky (1985)
Animals
Penaeus semisulcatusb body AD-Damman, Saudi Arabia; 0.54 W Sadiq et al.
(shrimp) tissue sewage outfall area (1982)
Paphia undulatab \ Gulf of Thailand 1.30-2.00 W Phillips &
(clam) | Muttarasin
| whole (1985)
| soft
Anadara granosab | parts 0.65-2.31 W
(cockle) |
/
Perna viridisb \ Gulf of Thailand 0.26-4.74 W Phillips &
(green mussel) | Muttarasin
| whole (1985)
| soft
Crassostrea | parts 0.60-2.52 W
commercialisb |
(rock oyster) /
Crassostrea virginicab body Estuary of Mississippi; 2.1 D Byrne &
(oyster) tissue pollutants from rivers, Deleon (1986)
Rangia cuneatab bayous, municipal and
(clam) agricultural run-off
------------------------------------------------------------------------------------------
Table 11 (contd.)
------------------------------------------------------------------------------------------
Species Tissue Locality Concentrationa Reference
or (mg/kg)
organ
------------------------------------------------------------------------------------------
Elliptio complanata shell Great Lakes, Ontario 6.56-7.6 D Dermott &
(bivalve) tissue 6.79-10.7 D Lum (1986)
Mytilus edulisb tissue Eastern Scheldt, 0.24-1.0 W Vos et al.
(mussel) Netherlands, 1977-1980 (1986)
Crangon crangonb 0.13-0.70 W
(shrimp)
Salmo truttab muscle Leine river, Germany 0.220-0.220 W Abo-Rady
(brook trout) liver municipal discharge area 0.327-0.469 W (1979b)
body 0.359-0.477 W
tissue
Leuciscus cephalusa muscle River Danube, Federal 0.5-1.2 W Wachs (1982)
(chub) Republic of Germany
Vimba vimba (vimba bream)
Abramis brama (bream)
Esox lucius (pike)
Psetta maximaa (turbot) muscle Southern Baltic, Poland 0.24 W Falandysz (1985)
Pleuronectes platessab 0.19 W
(plaice)
Platichthys flesusb 0.14 W
(flounder)
Heteropnuestes fossilisb muscle Yamura river, India; 1.9-32.7 D Ajmal et al.
near big cities (1985)
Basilichthys bonariensisb whole Lago Poopo, Bolivia; tin 1.37 D Beveridge et
(pejerry) carcass mining and refining area al. (1985)
------------------------------------------------------------------------------------------
Table 11 (contd.)
------------------------------------------------------------------------------------------
Species Tissue Locality Concentration Reference
or (mg/kg)
organ
------------------------------------------------------------------------------------------
Orestias luteusb 2.85 D
(killifish)
Salmo gairdnerib gills Augraben river, Italy; 14.5 D Dallinger &
(rainbow trout) near motorway Kautzky (1985)
liver 5.8 D
kidney 7.9 D
muscle 5.9 D
gonads 3.6 D
gills Leifer Graben river, 21.8 D
Italy; metal industry
area
liver 8.4 D
kidney 11.5 D
muscle 7.8 D
gonads 9.9 D
Salvelinus namaycushb kidney Lakes near Sudbury, 2.0-5.1 D Bradley &
(lake trout) Ontario Morris (1986)
Perca flavescensb muscle 2.8;3.4 D
(yellow perch) liver 2.2;2.9 D
white suckerb liver 16.4 D
Gadus morhua muscle Southern Baltic Sea 0.081 W Falandysz
(cod) (1986a)
Clupea harengus muscle Southern Baltic Sea 0.10 W Falandysz
(herring) (1986b)
Solea solea edible Coast of Netherlands, 0.01-0.05 W Vos et al.
(sole) parts 1977-80 (1986)
------------------------------------------------------------------------------------------
Table 11 (contd.)
------------------------------------------------------------------------------------------
Species Tissue Locality Concentrationa Reference
or (mg/kg)
organ
------------------------------------------------------------------------------------------
Gadus morhua 0.03-0.22 W
(cod)
Clupea harengus 0.02-0.12 W
(herring)
Anguilla anguilla 0.02-0.31 W
(eel)
Stenella coeruleoalba cart- Kii peninsula, Japan 0.029-0.009 D Honda et al.
(stripped dolphin) ilage (1984)
skull 0.025-0.083 D
verte- 0.024-0.057 D
brae
ribs 0.036-0.44 D
------------------------------------------------------------------------------------------
a D = dry weight.
W = wet weight.
b Intended for human consumption.
Nickel levels were determined in 234 foods from a 1984 US FDA
Total Diet Study, using ICP-AES (Pennington & Jones, 1987); 91% of
the foods contained nickel levels of less than 0.4 mg/kg and 66.2%
contained levels of less than 0.1 mg/kg. Only seven foods had
values exceeding 1 mg/kg. Foods highest in nickel included nuts,
legumes, items containing chocolate, canned foods, and grain
products.
Food processing and storage methods apparently add to the
nickel levels already present in foodstuffs through: leaching from
nickel-containing alloys in food-processing equipment made from
stainless steel; the milling of flour; and the catalytic
hydrogenation of fats and oils using nickel catalysts (NAS, 1975).
Grandjean (1984) estimated that leaching from cooking ware, kitchen
utensils, and water piping could occasionally add 1 mg to the daily
intake of nickel, i.e., much more than the intake resulting from
nickel in food and beverages.
Schroeder et al. (1962) calculated the average oral intake of
nickel by American adults to be about 300-600 µg/day, but more
recent estimates are lower. Myron et al. (1978) determined the
nickel content of 9 typical diets in the USA and calculated an
average intake of 165 µg/day. Clemente et al. (1980) evaluated the
available data and derived a mean intake value of 200-300 µg/day
for the normal adult in the Western countries. However, because of
the wide variation in the nickel contents of single food items, the
daily intake may vary considerably (100-800 µg/day) as a function
of dietary habits. Levels of 250-270 µg/day, determined by Smart &
Sherlock (1987) for United Kingdom diets, are within this range.
This includes an estimated 100 µg/day for nickel migrating from
metal cookware. An actual measured value using glass cookware for
food preparation was only 140-150 µg/day. The maximum daily nickel
intake from an average Danish diet was calculated to be 900 µg/day
(Nielsen & Flyvholm, 1984).
5.1.4. Terrestrial and aquatic organisms
Levels in terrestrial and aquatic organisms may vary over
several orders of magnitude, according to the species and
environmental factors (Jenkins, 1980a,b). Tables 11 and 12,
include data on nickel levels in tissue samples of different
organisms, including plants and animals relevant to human
nutrition.
5.2. General population exposure
The general population is exposed to nickel species through the
oral, inhalation, and dermal routes.
5.2.1. Oral
Nickel is ingested through the consumption of foodstuffs and
beverages that contain nickel and through the ingestion of inhaled
material returned to the pharynx by mucociliary clearance. Nickel
levels in European and USA foodstuffs generally ranged from less
than 0.1 mg/kg to 0.5 mg/kg, though a few foodstuffs, e.g., nuts
and legumes, contained levels of more than 1 mg/kg (section 5.1.3).
The results of dietary studies in the United Kingdom and the USA
showed average intakes from food of 200-300 µg nickel/day. In the
United Kingdom, it has been estimated that 100 µg nickel/day is
contributed by nickel-containing cooking utensils and that the
level in food ranges from 100 to 200 µg/day. Drinking-water may
contain nickel derived from its source, from environmental
contamination, and from nickel-containing plumbing fittings.
Concentrations in uncontaminated water may range from 5 to 11
µg/litre (section 5.1.2). Assuming a nickel level of 5-10 µg/litre
and a daily intake of 1.5 litres water, a mean daily intake of
nickel from water would be 7.5-15 µg.
5.2.2. Inhalation
Measured atmospheric nickel concentrations have been in the
ranges of 18-42 ng/m3 in industrial areas and 5-35 ng/m3 in non-
industrial urban and rural areas. Taking a concentration range of
5-35 ng/m3 and a daily ventilation rate of 20 m3, 0.1-0.7 µg
nickel/day will enter the respiratory tract; absorption will then
be a function of the nickel species and the physical state.
Cigarette smoking also contributes to the inhalation of nickel; for
example, smoking 40 cigarettes daily may result in the inhalation
of 2-23 µg nickel (section 5.1.1).
Table 12. Nickel concentration in terrestrial organisms
--------------------------------------------------------------------------------------------------
Species Tissue or Locality Concentrationa Reference
organ (mg/kg)
--------------------------------------------------------------------------------------------------
Plants
Aster tripoliumb shoots Salt marsh in Netherlands; 0.3-4.9 D Beeftink &
Halimione portulacoides discharge from urban and 0.1-3.3 D Nieuwenhuize
Limonium vulgare industrial areas 0.8-5.4 D (1982)
Plantago maritima 0.9-4.5 D
Puccinellia maritima 0.5-5.0 D
Salicornia europaeab 0.4-4.2 D
Spartina anglica 0.2-1.8 D
Suaeda maritima 0.3-4.6 D
Triglochin martim 0.5-6.2 D
Peanuts kernels <0.14-14 W Wolnik et
Soybeans beans (without 0.35-29 W al. (1983)
pod)
Sweet corn kernels <0.026-0.35 W
Field corn kernels <0.2-1.1 W Wolnik et
Spinach leaves <0.02-0.3 W al. (1985)
Onions peeled bulb <0.02-0.16 W
Tomatoes fruit <0.002- W
0.255
Rice kernels <0.2-1.2 W
Carrots root <0.02-0.46 W
Thlaspi montanum leaves Grand Canyon, Arizona; 54 D Hobbs &
shoots copper mine spoil 27 D Streit (1986)
Phlox austromontana leaves 39 D
shoots <17 D
Juniperus osteosperma leaves 8 D
shoots 17 D
--------------------------------------------------------------------------------------------------
Table 12 (contd.)
--------------------------------------------------------------------------------------------------
Species Tissue or Locality Concentrationa Reference
organ (mg/kg)
--------------------------------------------------------------------------------------------------
Animals
Lagopus lagopus plumage remote areas in Ontario 0.91 D Parker (1985)
(willow ptarmigan)
Bonasa umbellus 0.74 D
(ruffed grouse)
Canachites canadensis 1.09 D
(spruce grouse)
Egretta alba muscle Central Korea 0.03-0.04 W Honda et
(great white egret) bone 0.03-0.19 W al. (1985)
feather 0.14-1.59 W
Sterna kirundo liver Providence, Rhode Island; ND-1.0 D Custer et
(common tern) former electroplating al. (1986)
industry centre
Fulica americana liver Texas; fly ash pond 0.06-0.24 Wc White et
(coot) control site 0.07-0.22 W al. (1986)
Microtus pensylvannicus liver Sudbury, Ontario; ND-9.9 Dd Cloutier et
kidney nickel and copper mine ND-18.4 D al. (1986)
muscle tailings control site 5.9 D
liver ND-8.7 D
kidney ND-25.3 D
muscle 5.5 D
--------------------------------------------------------------------------------------------------
a D = dry weight.
W = wet weight.
ND = not detected.
b Intended for human consumption.
c No statistically significant difference between sites.
d Nickel levels in most samples were below detection limit; site effects were not observed.
5.2.3. Dermal
This route of exposure is of particular significance for nickel
contact hypersensitivity. Because of the ubiquity of nickel-
containing commodities in an industrial society, dermal exposure to
nickel is almost continuous in the general population. This is
reflected by the constantly increasing number of patients with
nickel contact dermatitis (Dooms-Goossens et al., 1980). Sources
of environmental exposure include jewellery, coinage, clothing,
fasteners, tools, cooking utensils, and stainless steel kitchen
utensils (NAS, 1975). Even jewellery made of white gold containing
2-15% of nickel may cause eczema (Fischer, 1984).
A number of common sources of nickel dermatitis are listed in
Table 13. The amount of nickel released from these items depends
on the corrosion resistance of the object and the presence of sweat
and other fluids in the environment; because of its chloride
content and relative low pH, sweat can dissolve nickel. The
solution of nickel in synthetic sweat was examined for 15 different
nickel alloys and surfaces; a high release of nickel ions was
documented for nickel-plated items (both electrolytically and
chemically plated), nickel-iron alloy (65% nickel), German silver
(10-20% nickel), Monel 400(R) alloy (66% nickel), Nicrobraze LM(R)
alloy (83% nickel), and coin alloy (25% nickel). These nickel
objects were also tested for their ability to provoke nickel
allergy in sensitized persons. Nine of the materials tested caused
a medium to severe degree of allergic skin reactions, and the
amount of nickel dissolved was related to the degree of the
allergic reaction (Kato & Samitz, 1975).
Table 13. Non-occupational exposure to nickela
------------------------------------------------------
Source causing dermatitis Location
------------------------------------------------------
Earrings Earlobes
Garter clasps, metal chairs Thighs
Thimbles, crochet and knitting Fingers
needles, scissors, nickel coins,
cover of fountain pen
Handles of car doors, baby Palms
carriage, umbrella, refrigerator,
and handbags
Clasp of necklace, zipper Neck
Watch band, bracelet Wrists
Wire support of brassiere cup Breast
Handbags Antecubital area
Bobby pins Side of face
Spectacle frames Back of ears
Nickel coin (patient had rubbing Side of nose
habit)
Eyelash curler Eyelids
Zipper Axilla
Safety pin Pubic area
Eyelets of shoe Dorsum of foot
------------------------------------------------------
Table 13 (contd.)
------------------------------------------------------
Source causing dermatitis Location
------------------------------------------------------
Metal arch support Plantar aspect
of foot
Bobby pins held in mouth, metal Lips
lipstick cases
Hair gripsb Scalp
Jeans buttonsc Belly
------------------------------------------------------
a Adapted from: Fisher & Shapiro (1956).
b From: Cronin (1980).
c From: Fisher (1985).
Fisher's dimethyglyoxime spot test can be used, in most cases,
to identify nickel alloys and nickel-plated surfaces releasing
nickel ions. Important exceptions are Inconnel 600(R), which has a
low nickel release to synthetic sweat, but was positive when tested
on nickel sensitive patients, and Nicrobraze(R), which has a high
nickel release and was positive when tested on patients (Menné et
al. 1987b).
Stainless steel, which contains about 10% nickel, is very
resistant because of the protective effect of chromium oxides on
the surface. It did not release noticeable amounts of nickel ions
into synthetic sweat and did not produce skin reactions in nickel-
sensitive patients (Kato & Samitz, 1975; Menné et al., 1987b).
However, nickel can be released from stainless steel in a very
corrosive environment or by prolonged treatment in a sweat
solution.
5.3. Iatrogenic exposure
Iatrogenic exposures to nickel arise from: (a) nickel-
containing implants (joint replacements, intraosseous pins, cardiac
valve replacements, cardiac pacemaker wires, and dental
prostheses); (b) intravenous fluids and medications that are
contaminated with nickel; and (c) haemodialysis with nickel
contamination of the dialysate fluid (Sunderman, 1986; Hopfer et
al., 1989). Marek & Treharne (1982) investigated nickel release
from surgical implant alloys (14 and 36% nickel) into Ringer's
solution. Nickel release from the alloy shavings declined after a
few days and reached a constant value of approximately 0.3 ng/cm2
per day. Thus, a prosthesis with a surface area of 200 cm2 would
release about 60 ng nickel per day or 22 µg/year. The significance
of nickel release from implants has been questioned (Fisher, 1977;
Burrows et al., 1981 ). Hypernickelaemia (serum-nickel >1.1
µg/litre) occurred in only one out of 13 patients with stainless
steel hip prostheses. This patient suffered from mild renal
insufficiency, suggesting an impaired ability to excrete nickel
absorbed from the prosthesis (Linden et al., 1985; Sunderman,
1986).
Brune (1986) compiled data on the amounts of different metals
released from dental alloys in natural or artificial saliva in in
vitro and in vivo tests. The quantities released were calculated
on the basis of a standard man with a specified number or area of
dental restorations. Nickel was released in vitro from a metal
alloy with a high nickel content (ca 80%), at the same level as
from food and drink. Nickel release from cobalt-based alloys (0.1-
0.2% nickel) was less than 2 µg per day.
Contamination of dialysate fluids may produce parenteral
exposure to nickel. Hopfer et al. (1985) demonstrated that serum-
nickel concentrations in haemodialysis patients may, on average, be
11-22 times the mean concentration in serum from healthy subjects.
During normal dialysis, the average intravenous nickel uptake has
been estimated to be 100 µg per treatment (Sunderman, 1983a).
Hypernickelaemia has also been observed in patients following
intravenous administration of meglumine diatrizoate (Renografin-
76(R), a radiographic contrast medium) for coronary arteriography.
Nickel analysis revealed levels of 144±44 µg nickel/litre contrast
medium. Approximately half an hour after arteriography, the
incremental serum-nickel concentration in patients averaged
1.81±0.39 µg/litre. The authors recommended that nickel
concentrations in radiographic contrast media should not exceed 10
µg/litre (Leach & Sunderman, 1987).
5.4. Occupational exposure
Nickel concentrations may be significantly higher in the
working environment than normal atmospheric air levels. US NIOSH
(1977b) estimated that about 250 000 individuals in the USA were
occupationally exposed to inorganic nickel. A list of occupations,
identified as involving exposure to nickel, is presented in Table
14 (US NIOSH, 1977b).
Table 14. Occupations with potential exposure to nickela
-------------------------------------------------------------
Battery makers, storage Nickel-alloy makers
Cashiers Nickel miners
Catalyst workers Nickel refiners
Cemented-carbide makers Nickel smelters
Ceramic makers Oil hydrogenators
Disinfectant makers Organic-chemical synthesizers
Dyers Paint makers
Electroplaters Penpoint makers
Enamellers Petroleum-refinery workers
Gas-mask makers Spark-plug makers
Glass makersb Stainless-steel makers
Ink makers Textile dyers
Jewellers Vacuum-tube makers
Magnet makers Varnish makers
Metallizers Welders
Mond-process workers
-------------------------------------------------------------
a Adapted from: US NIOSH (1977b).
b From: Raithel et al. (1981).
Representative exposure data are difficult to obtain, and most
published values of occupational exposure were gained during
biological monitoring studies. US NIOSH (1977b) compiled data on
the concentrations of nickel in air, in smelting and refining
operations, the alloy industry, welding operations, and other
processes; the concentrations ranged from a few µg/m3 to several
mg/m3. Recent exposure data from the primary and secondary nickel
industries are summarized in Table 15. Levels may vary
considerably, according to the individual operations, or areas of a
manufacturing process. For example, in alloy production an average
concentration of airborne nickel during pickling and handling was
determined to be 0.008 mg/m3, whereas, during grinding,the average
airborne nickel level was 0.298 mg/m3 (Warner, 1984). Generally,
the concentration of nickel in the material being handled and the
operation being performed affect the concentration of nickel in
air. Levels of airborne nickel are expected to be higher in dusty
operations involving fine, dry particulates, e.g., metal powder or
salts. Welding operations can create airborne nickel levels of
0.004-0.24 mg/m3. A higher exposure, especially to metallic
nickel, exists in the user industries. Improvements in operational
techniques and ventilation reduce nickel concentrations in the air
of work-places (Boysen et al., 1982; Coenen et al., 1986).
In the occupational environment, nickel dust may also enter the
body through oral exposure, because of poor personal hygiene or
inadequate work practices. This route of exposure was reported in
a battery factory where high faecal levels of nickel were related
to dusty working conditions (Adamsson et al., 1980).
The dermal route of exposure is of significance to workers
sensitized to nickel. Occupational dermal exposure to nickel may
occur in battery makers, nickel-catalyst makers, ceramics makers,
duplicating machine workers, dyers, electronics workers,
electroplaters, ink-makers, jewellers, spark-plug makers, and
rubber workers (NAS, 1975). In some occupations, the skin may be
directly exposed to dissolved nickel, e.g., in the electroplating
and electroforming industry (Wall & Calnan, 1980). Nickel contact
dermatitis was observed in hairdressers and was attributed to the
handling of nickel-bearing tools and contact with liquids
throughout the day (Wahlberg, 1975). Nickel contact dermatitis has
been reported in hospital cleaning personnel (Gawkrodger, 1986a).
The level of nickel in the water they used for washing surfaces
increased during the cleaning process by transfer from cleaned
areas on wash cloths. In water from used cloths, a mean level of
90 µg nickel/litre was found (Clemmensen et al., 1981).
Table 15. Occupational exposure to nickel
---------------------------------------------------------------------------------------------
Process/Operations Average nickel Number Chemical formb Reference
concentration in of
air (mg/m3)a samplesb
---------------------------------------------------------------------------------------------
Nickel mining and refining
Mining 0.025 A ns mineral form Warner (1984)
Concentrating 0.03-0.13 P ns mineral form Warner (1984)
Roasting/smelting <0.1 A ns nickel subsulfide Boysen et
nickel oxide al. (1982)
Roasting/smelting 0.048; 0.075 A ns mineral form, Warner (1984)
nickel subsulfide,
nickel oxide
combined with iron
oxide, nickel
sulfate
Converting 0.033; 0.037 A ns nickel subsulfide Warner (1984)
Smelting to ferronickel 0.0022-0.274 P >79 nickel oxide Warner (1984)
0.0047-0.029 A combined with iron
oxide,
0.005-0.193 P nickel subsulfide
Hydrometallurgical refining 0.029-0.336 A,P 577 nickel subsulfide, Warner (1984)
nickel oxide,
metallic nickel,
nickel sulfate,
nickel chloride
Electrolytic refining <0.1 A ns ns Boysen et
al. (1982)
0.34; 0.19 A ns as under Hydro- Warner (1984)
metallurgical
refining
---------------------------------------------------------------------------------------------
Table 15 (contd.)
---------------------------------------------------------------------------------------------
Process/Operations Average nickel Number Chemical formb Reference
concentration in of
air (mg/m3)a samplesb
---------------------------------------------------------------------------------------------
Use of primary nickel products
Stainless steel welding 0.004 A 35 ns Wilson et
0.313 P 7 ns al. (1981)
0.07-0.24c P 182 ns Coenen et
0.02-0.22c A 280 ns al. (1986)
Stainless steel production 0.014-0.134 P 40d nickel oxide, Warner (1984)
metallic nickel
High nickel alloy 0.008-0.298 P 1530 nickel oxide, Warner (1984)
production metallic nickel
Foundry operations <0.3 P ns nickel oxide, Bernacki et
(melting, casting, grinding) nickel alloys al. (1978b)
0.013-0.310 P 217 nickel oxide, Warner (1984)
nickel alloys
Electroplating 0.03-0.16 P 25 nickel sulfate Tola et al.
(1979)
0.005-0.016 P 15 ns Bernacki et
al. (1980)
<0.003-<0.011 A,P 48 various nickel Warner (1984)
salts
Metal sintering
Furnace maintenance 0.001-0.168 P 20 ns Lichty & Zey
Metal powder mixing 0.006-1.28 P 6 ns (1985)
---------------------------------------------------------------------------------------------
Table 15 (contd.)
---------------------------------------------------------------------------------------------
Process/Operations Average nickel Number Chemical formb Reference
concentration in of
air (mg/m3)a samplesb
---------------------------------------------------------------------------------------------
Nickel-cadmium battery 0.012-0.033 P 213 ns Adamsson et
manufacturing al. (1980)
0.02-1.91 P 36 ns Warner (1984)
Glass production 0.03-3.8 A ns ns Raithel et
0.07-2.622 P al. (1981)
---------------------------------------------------------------------------------------------
a A = area samples; P = personal samples.
b ns = not specified.
c 90% value.
d Number of companies reporting exposure.
6. KINETICS AND METABOLISM
Health hazards associated with exposure to nickel in the
occupational environment have resulted primarily from inhalation.
For this reason, deposition, retention, and clearance of nickel
from the human respiratory tract are of special importance.
However, in addition to this main exposure through inhaled air
(ambient and at the work-place), human beings are also exposed to
nickel in drinking-water and food, and through skin contact, which
is of special concern in view of resulting adverse effects, namely,
nickel contact dermatitis.
6.1. Absorption
Human exposure to nickel originates from a variety of sources
and is highly variable. Nickel and its inorganic compounds can be
absorbed via the gastrointestinal tract as well as the respiratory
passages. Under certain circumstances, the skin is a qualitatively
important route by which nickel enters the body. However,
percutaneous absorption is less important for the systemic effects
of nickel than for the allergenic responses to it. Placental
transfer is of importance because of the effects on the fetus.
Knowledge of this route of absorption makes it possible to estimate
the contribution to the body burden at birth. The diverse routes
of parenteral administration of nickel compounds are mainly of
interest in toxicity studies on animals and are particularly useful
in assessing the kinetics of nickel transport, distribution, and
excretion.
The relative amounts of nickel absorbed by an organism are
determined, not only by the quantities inhaled, ingested, or
administered, but also by the physical and chemical characteristics
of the nickel compound. Solubility is an important factor in all
routes of absorption. Soluble salts of nickel dissociate readily
in the aqueous environment of biological membranes, thus
facilitating their transport as metal ions. Conversely, insoluble
nickel compounds are relatively poorly absorbed.
Kuehn & Sunderman (1982) incubated 17 nickel compounds in
water, rat serum, and renal cytosol for 72 h at 37 °C.
Concentrations of dissolved nickel were determined by
electrothermal atomic absorption and dissolution half-times were
calculated. Eleven of the nickel compounds (Ni, beta-NiS,
amorphous NiS, alpha-Ni3S2, NiSe, Ni3Se2, NiTe, NiAs, Ni11As8,
Ni5As2, and NiFeS4) dissolved more rapidly in serum or cytosol than
in water. Dissolution of 4 of the compounds (NiO, NiSb, NiFe
alloy, and NiTiO3) was not detectable in any of the media (half-
time, >11 years). One compound (NiAsS) had approximately equal
dissolution half-times in the 3 media. Because of precipitation,
the half-time value for NiS2 could not be determined. These
findings were in close agreement with the elimination half-time (24
days) obtained from elimination of 63Ni in the urine and faeces of
rats, after intramuscular injection of alpha-63Ni3S2. The authors
suggested that the in vitro dissolution half-times of nickel
compounds might be used to predict their in vivo elimination half-
times, since the dissolution process is rate limiting for the
metabolism and elimination of the compounds.
It is possible that other factors, such as host, nutritional
and physiological status, or stage of development, also play a
role, but these have not been studied.
Several studies on the dissolution kinetics of nickel
subsulfide have been performed. Autoradiographic observations
(Kasprzak, 1974) showed that extracellular particles of 63Ni
subsulfide or Ni335S2 could persist at the site of injection for
many months, without detectable alterations, and could eventually
become surrounded by neoplastic tissue. Intracellular localization
of 63Ni or 35S was not detected within muscle or tumour cells.
This finding was confirmed quantitatively by Sunderman et al.
(1976b), who measured the elimination of 63Ni following 63Ni
subsulfide administration to rats, and by Oskarsson (1979), who
carried out whole-body autoradiography in mice. After 20 weeks, as
much as 19% of the 63Ni dose was found at the site of injection,
while the retention of nickel in organs distant from the injection
site was less than 0.1% (Sunderman et al., 1976b). Addition of
manganese to the administered nickel subsulfide, which
significantly decreased the carcinogenicity of the latter, did not
affect the gross elimination of 63Ni. The role of manganese was to
effect the subcellular partition of soluble 63Ni derived from 63Ni
subsulfide. Ultrafiltered homogenates of muscle tissue injected
with 63Ni derived from 63Ni subsulfide + Mn contained less nickel
than those injected with 63Ni subsulfide alone (Sunderman et al.,
1976b). Nickel dissolution kinetics, similar to those in vivo,
were obtained when leaching of 63Ni was measured during two weeks
of in vitro incubation of 63Ni subsulfide with rat serum or aqueous
triethylenetetramine, but not with water (Kasprzak & Sunderman,
1977). This finding suggested that the interaction of nickel
subsulfide with body fluids could be of the same nature under both
in vivo and in vitro conditions. Evaluation of the kinetic curves
and the X-ray diffractometry of the sediments following incubation
of nickel subsulfide in the three media (Kasprzak & Sunderman,
1977) revealed that solubilization of nickel required the presence
of oxygen and involved two reactions:
(a) 2 alpha-Ni3S2 + O2 + 2H2O <-> 4 beta-NiS + 2Ni(OH)2
(b) beta-NiS + 202 <-> Ni2+ + SO2-
When complexing agents, i.e., proteins, amino-acids, etc., are
present, Ni2+ and Ni(OH)2 form water-soluble Ni(II)-complexes and
undergo fast mobilization and elimination. The remaining beta-Ni
monosulfide constitutes a surface coating on the nickel subsulfide
particles and requires more oxygen for the further dissolution of
nickel.
6.1.1. Absorption via the respiratory tract
Respiratory absorption of nickel is normally the principal
route for its entry into the human body, under conditions of
occupational exposure. It usually involves the inhalation of one
of the following substances: dust of relatively insoluble nickel
compounds, aerosols derived from nickel solutions (soluble nickel),
and gaseous forms containing nickel (usually nickel carbonyl). The
inhalation route is also of importance in exposure from the general
environment, including tobacco smoke. It has been reported that
cigarette smoke may contain nickel carbonyl (section 5.1.1).
The relative amounts of inhaled nickel absorbed from various
compartments of the pulmonary tract are a function of both the
chemical and physical forms.
6.1.1.1 Particulate nickel
The Task Group on Lung Dynamics (1966) considered that the
respiratory absorption of nickel compounds in particulate form was
influenced by three processes in the lung, namely deposition,
mucociliary clearance, and alveolar clearance. This Group
developed deposition and clearance models for man for inhaled
particulate matter of whatever chemical origin, as a function of
particulate size, chemical category, and compartmentalization
within the respiratory tract. Nickel oxide and nickel halides are
classified as compounds having moderate retention in the lungs and
a clearance time of weeks. Although this model approach has its
limitations, it can be of some value in assessing deposition and
clearance rates for nickel compounds of known particle size and
chemical composition.
Removal of material deposited in the lung depends on its
solubility characteristics and is slow for metallic nickel or
nickel oxide dust, faster for soluble nickel salts, and most rapid
for the volatile and lipid-soluble nickel carbonyl.
Nickel has a tendency to accumulate in lung tissue and in the
regional lymph nodes. Thus, only part of the nickel retained will
be transferred to the blood, depending on the solubility of the
nickel compound.
Absorption from the pulmonary tract of nickel in particulate
matter is considerably less than that of nickel carbonyl. Smaller
particles penetrate deeper in the respiratory tract than larger
particles and the relative absorption is greater. Soluble nickel
compounds are absorbed quickly, making them less available for
mucociliary clearance. A solubility model may be the most accurate
means of evaluating the rate of absorption of the dust retained in
the alveoli (Mercer, 1967; Morrow, 1970).
(a) Experimental animals
There are few animal studies dealing with respiratory
absorption, and data on the pulmonary uptake of nickel in
particulate form are limited.
Wehner & Craig (1972) exposed Syrian golden hamsters to nickel
oxide (nickel oxide not specified) particles with a mass mean
aerodynamic diameter (MMAD) of 1.0-2.5 µm, and observed that
inhalation for 2 days (7 h/day) at a concentration of 10-190 mg/m3
air resulted in a deposition of 20% of the inhaled amount. On the
10th day after exposure, more than 75% of the nickel oxide was
still present in the lungs, and, even after 45 days, approximately
50% of the total amount inhaled still remained. As no significant
quantities of nickel oxide were found in the liver and kidney at
any time after exposure, absorption seemed to be negligible during
this period. In ancillary studies, Wehner et al. (1975) exposed
hamsters for 61 days to nickel oxide aerosol (nickel oxide not
specified) for 7 h/day and cigarette smoke (nose-only exposure of
approximately 10 min duration, twice before, and once after, the
daily 7-h dust exposure). It was found that the inhalation of
cigarette smoke did not change either the deposition or the
clearance pattern of the nickel oxide.
In a later study, Wehner et al. (1979) exposed Syrian golden
hamsters to a highly respirable aerosol (MMAD = 2.8 µm) of nickel-
enriched fly ash (NEFA; nickel content 9%), at concentrations of
220 mg/m3 (one 6-h exposure) and 190 mg/m3 (for 60 days, 6 h/day).
In the acute exposure, approximately 95% (6.8 µg nickel = 75 µg
NEFA) of the total deposited amount (7 µg nickel = 78 µg NEFA) was
found in the deep lung, 1 month after exposure, indicating a very
slow clearance. The findings also show, that nickel in the NEFA is
retained and does not leach appreciably from the NEFA into tissue
fluids. This assumption is supported by the observation that the
average nickel content remained practically the same from 7 days
after exposure (7 µg) to 30 days after exposure (6.8 µg), which
would not be the case if the nickel were to leach from NEFA. In
the 2-month study, the deposition was 5.7 mg NEFA or 510 mg nickel
on the third day after exposure.
In another study, Wehner et al. (1981) exposed Syrian golden
hamsters to a high (70 mg/m3) and a low (17 mg/m3) respirable NEFA
concentration, for up to 20 months. The NEFA contained
approximately 6% nickel. Exposure resulted in heavy deposits of
NEFA in the lungs of 731±507 µg and 91±65 µg nickel/lung in the 70
mg/m3 and 17 mg/m3 exposure groups, respectively).
The short- and long-term NEFA studies showed that the time from
the end of exposure to sacrifice (64 h-7 days) was not long enough
to allow for mucociliary clearance of the nickel deposited in the
ciliated part of the respiratory tract. In addition, clearance
mechanisms may be disturbed by the repeated 6-h exposures to high
aerosol concentrations, making them decreasingly efficient and
resulting in the retention of larger quantities of material.
However, trace elements are not homogeneously distributed, even
in particles of similar size, and the mean content of a given trace
element in fly ash is determined by relatively few particles with a
very high content of that element. This means that cells in the
respiratory tract or the lung would not come into contact with fly
ash particles containing 0.03% nickel, but instead with particles
containing many times that quantity. It should be noted that
respirable NEFA is emitted from coal-fired power plants (Natusch et
al., 1974).
Leslie et al. (1976) exposed mice for 4 h to nickel-containing
welding fume aerosols. Particle size and nickel content were
determined. The nickel content was highest (8.4 µg/m3) with
particles 0.5-1.0 µm in diameter. It was reported that no
clearance of lung-deposited nickel had occurred by 24 h, nor was
there any elevation in blood-nickel levels, indicating that there
had been no absorption into the bloodstream. When rats were
exposed through inhalation to nickel oxide aerosol (0.4-70 mg/m3)
for 6-7 h, 5 days/week, for a maximum of 3 months, the fraction
deposited in the lung significantly decreased with increasing mass
median diameter and slightly decreased with increasing exposure
concentration (Kodama et al., 1985).
The data obtained by Wehner et al., showing poor absorption and
retention of the greatest portion of inhaled nickel oxide in the
lungs, are supported by the study of Rittmann et al. (1981), who
exposed Wistar rats via inhalation to nickel oxide, produced by the
pyrolysis of nickel acetate at 500-600 °C (50 µg/m3 for 15 weeks).
They found a half-life for the clearance of nickel from the deep
tract of 36 days. The half-life for clearance from the
tracheobronchiolar compartment was less than one day.
Valentine & Fisher (1984) administered 63Ni3S2 intratracheally
to mice (11.7 µg 63Ni3S2/animal). During the initial phase of
clearance, 38% of the instilled dose was cleared, with a
biological half-time of 1.2 days in the final phase. Four hours
after instillation, the total lung burden was 85% of the
administered dose. Thirty-five days after exposure, 10% of the
administered dose was still retained by the lung.
In a study by Graham et al. (1978), mice were exposed, through
inhalation, to nickel chloride (particle diameter = 3 µm, 644 µg
nickel/m3) for 2 h. Clearance of 70% ( 5.77 mg nickel/kg dry
weight) of the deposited fraction (8.06±0.506 mg nickel/kg dry
weight) was found in the lung on the fourth day after exposure. In
rats that had received 1 mg nickel, administered intratracheally as
a single dose of 63Ni chloride, most of the administered dose was
found in the kidney (53%) and the lung (30%), the rest being
distributed among the adrenals, liver, pancreas, spleen, heart, and
testes (Clary, 1975). As clearance by 3 days was faster in the
kidney, the lungs became the organ with the highest 63Ni level (64%
of the total amount deposited; kidney: 19%). Lung clearance within
6 h was 27%, which means that 70% of the material originally
deposited had been absorbed.
Appreciable amounts of radioactive nickel, administered to male
rats intratracheally as the chloride (1.27 µg nickel), were
absorbed (Carvalho & Ziemer, 1982). Twenty-one days after
exposure, the only measurable activity was in the lungs and
kidneys. For example, 1 day after exposure, 29% of the initial
burden was retained in the lungs, decreasing to 0.1% on day 21.
Following intratracheal instillation of nickel carbonate in
mice (0.05 mg/animal), most of the nickel was eliminated after 12
days (Furst & Al-Mahrouq, 1981).
Medinsky et al (1987) administered nickel sulfate solution
intratracheally to rats at doses of 1 µg, 11.2 µg or 105.7 µg
nickel/rat. After 4 h, 49%, 21%, or 8%, respectively, of the
instilled dose/g tissue was found in the lungs.
An important factor for retention in the lung is the solubility
of the nickel compounds. Insoluble forms, such as nickel oxide and
metallic nickel, seem to be retained in the lung for a longer time,
whereas the more soluble nickel salts are absorbed. They are also
solubilized in the fluids and mucus cleared from the lung by the
mucociliary mechanisms into the alimentary tract.
(b) Human beings
Particulate nickel can be taken up from ambient air and from
cigarette smoke. Respiratory absorption of nickel in particulate
form is the major route of entry, under conditions of occupational
exposure.
The amount of nickel absorbed from the air is expected to vary
according to ambient atmospheric levels. Schroeder (1970)
calculated that 75% of respiratory nickel intake is retained in the
body and 25% is expired, depending on the particle size
distribution. About 50% of the inhaled nickel would be deposited
on the bronchial mucosa (and swept upward by mucociliary transport
to be swallowed), and 25% in the pulmonary parenchyma.
6.1.1.2 Nickel carbonyl
In the toxicology of nickel, a special position is occupied by
nickel carbonyl, a volatile, liquid compound. After nickel
carbonyl inhalation, removal of nickel deposited in the lung is the
most rapid, compared with the clearance of all other compounds,
indicating an extensive absorption and clearance. Since the
alveolar cells are covered by a phospolipid layer, the lipid
solubility of nickel carbonyl vapours is of importance for their
penetration of the alveolar membrane. This explains why nickel
carbonyl is the only one of the nickel compounds to cause acute
symptoms of poisoning, when inhaled.
(a) Experimental animals
Because of its industrial importance, nickel carbonyl
absorption through inhalation has been studied extensively in
experimental animal species including the dog, cat, rabbit (Armit,
1908; Tedeschi & Sunderman, 1957; Sunderman et al., 1961; Mikheyev,
1971), rat (Barnes & Denz, 1951; Sunderman et al., 1957, 1961;
Ghiringhelli & Agamennone, 1957; Sunderman & Selin, 1968; Sunderman
et al., 1968) and mouse (Oskarsson & Tjälve, 1979a). Animals
received single doses ranging from 200 to 3050 mg nickel/m3 air for
periods ranging from 5 to 240 min. Sunderman et al. (1957)
administered concentrations of 30 or 60 mg nickel/m3 air for 30-min
periods, 3 times/weeks, for 3 or 52 weeks. In all the studies,
nickel was found in the respiratory tissues, brain, liver, kidneys,
urinary bladder, adrenals, renal cortex, heart, diaphragm, and
blood, from where it was rapidly mobilized after exposure (within 2
days). Sunderman & Selin (1968) reported that, 24 h after
inhalation of 63Ni carbonyl, the partition of the body burden of
63Ni was: viscera 50%, muscle and gut 30%; bone and connective
tissue 16%, and nervous tissue 4%. During 2-4 h following exposure
of rats or rabbits to nickel carbonyl, the lung was the major
excretory organ for the compound (Sunderman & Selin , 1968;
Mikheyev, 1971). Elimination of nickel has been reported to be
mainly via the urine: 62% (after 3 days) in rabbit (Mikheyev,
1971); 75% in dog, cat, rabbit (Armit, 1908); 90% in rat and dog
(Tedeschi & Sunderman, 1957). Sunderman & Selin (1968) indicated
that 26% of the inhaled amount was excreted via the urine in 4
days. Since the same amount or more might have been exhaled during
the same period, the authors speculated that at least 50% of the
inhaled dose could have been absorbed.
(b) Human beings
The extensive absorption of nickel carbonyl by human beings
after respiratory exposure has been demonstrated by the
measurements of enhanced nickel concentrations in organs of workers
who died from nickel carbonyl poisoning (Brandes, 1934; Bayer,
1939; Sunderman & Kincaid, 1954; Ludewigs & Thiess, 1970;
Sunderman, 1971; National Academy of Sciences, 1975) and of
increased levels in the blood, serum, plasma, and urine in nickel-
refining workers (Kincaid et al., 1956; Sorinson et al., 1958;
Hagedorn-Götz et al., 1977). In general, the highest tissue
concentrations after inhalation of nickel carbonyl have been found
in the lungs; lower concentrations have been measured in the
kidneys, liver, and brain. However, there are no firm data on the
dose levels of nickel carbonyl that are toxic for human beings.
Experience suggests that, not only is there considerable
interpersonal variation, but also that a certain degree of
resistance can develop. Exposure to concentrations estimated to
have been of the order of 0.5 mg NiCO4/m3 for half an hour have
caused severe illness (Morgan, 1989, personal communication).
6.1.2. Absorption via the gastrointestinal tract
Absorption of nickel from the gastrointestinal tract occurs
after ingestion of food, beverages, or drinking-water. In the
occupational environment, an appreciable amount of nickel dust may
be swallowed via the mucociliary clearance mechanisms; insufficient
personal hygiene, poor work practices, and inadequate work-place
conditions may also increase this uptake. Gastrointestinal intake
of nickel leaching from nickel-containing dental alloys is of
limited importance.
The rate of nickel absorption from the gastrointestinal tract
is dependent on its chemical form. While soluble nickel compounds
(e.g., NiSO4) are better absorbed than relatively insoluble ones,
the contribution of the poorly soluble compounds to the total
nickel absorption may be more significant, since they are more
soluble in the acidic gastric fluids.
6.1.2.1 Experimental animals
Nickel is poorly absorbed from ordinary diets and is eliminated
mainly in the faeces. This has been shown in a nickel balance
study on dogs (Tedeschi & Sunderman, 1957), in which the nickel
intake in the food was equal to the output in the urine and faeces.
The results also indicated that an average of 90% (1.01±0.44 mg) of
the amount of nickel ingested (1.12±0.16 mg) was eliminated in the
faeces. In rats, even at very high intakes of nickel (approx. 14.5
mg) from different sources, over periods of 4, 8, 12, and 16 days,
nickel was absorbed poorly (0.15 mg, i.e., 1%) and was eliminated
mainly in the faeces (13.9 mg = 96%). Levels of retained nickel
averaged 0.5 mg (range: 0.29-0.8 mg), i.e., 3.5% (Phatak &
Patwardhan, 1950, 1952).
Schroeder et al. (1969, 1974) did not find any measurable
absorption of nickel in mice that were given 5 mg nickel/litre in
the drinking-water, throughout their lives.
It was reported by Elakhovskaya (1972) that nickel, given
orally to rats as the chloride in the drinking-water (0.005, 0.5,
or 5 mg/litre), was eliminated mainly in the faeces. Ho & Furst
(1973) reported that intubation in rats of 63Ni (as the chloride)
in 0.1 NHCl led to 3-6% absorption of the labelled nickel,
regardless of the administered dose (1.8 µg/animal, or 4, 16, and
64 mg nickel/kg body weight). From these two studies
(Elakhovskaya, 1972; Ho & Furst, 1973), it can be concluded that
very little nickel in water or beverages is bioavailable. Phatak &
Patwardhan (1950) showed that large doses are required to overcome
the intestinal absorption-limiting mechanism.
The mechanism of nickel absorption in the perfused rat jejunum
was studied by Foulkes & McMullen (1986). In step 1, the
absorption process (uptake from lumen of perfused jejunum)
proceeded at a rate linearly dependent on the concentration up to
about 20 µmol nickel/litre perfusate. At higher levels, it
approached apparent saturation. In step 2 of nickel absorption
(movement of nickel from mucosa into the body), nickel was not
appreciably retained in the mucosa.
6.1.2.2 Human beings
The intake of nickel via the gastrointestinal tract in human
beings can be high, compared with that of other trace elements.
Although the daily dietary intake may range up to 900 µg nickel,
average values have been estimated to be around 200 µg (section
5.1.3).
Nodiya (1972) performed nickel balance studies on 10 male
volunteers, aged 17 years, who ingested a mean of 289±23 mg
nickel/day (range 251-309 mg) and found that faecal elimination of
nickel averaged around 89% (258±23 mg/day).
In human volunteers who ingested nickel sulfate in the
drinking-water or food, at doses of between 12 and 50 µg/kg body
weight (one treatment), the amount of nickel absorbed averaged
27±17% of the dose ingested in water compared with 0.7±0.4% of the
same dose ingested in food (Sunderman et al., 1989a).
6.1.2.3 Factors influencing gastrointestinal absorption
In assessing toxicity from ingested nickel, it is important to
keep in mind possible factors that might change the absorption rate.
(a) Bioavailability
The experimental data obtained by Ho & Furst (1973) and
Elakhovskaya (1972) did not indicate any change in absorption
efficiency in rats, whether the nickel was taken up from liquids or
drinking-water (section 6.1.2.1). However, Cronin et al. (1980)
reported that ingestion of a soluble nickel compound during fasting
resulted in high urinary elimination rates of 4-20% of the dose.
Fifteen female volunteers, who received single oral doses of 2.5,
1.25, or 0.6 mg nickel (as the sulfate, in gelatine-lactose
capsules, together with 100 ml water), excreted 95-206 µg, 62-253
µg, and 48-89 µg nickel, respectively, (normal value 9 µg) in the
urine. Nickel naturally occurring in food items also increases
urinary nickel elimination; a diet containing 850 µg nickel
resulted in an increased nickel elimination corresponding to about
1% of the amount in the diet (Nielsen et al., 1987a).
Solomons et al. (1982) estimated the bioavailability of nickel
in human subjects by the serial determination of the changes in
plasma-nickel concentrations following a standard dose of 22.4 mg
of nickel sulfate hexahydrate (5 mg nickel), given in each of two
standard meals, as well as in the drinking-water and 5 beverages
(cow's milk, coffee, tea, orange juice, and Coca Cola(R)). The
plasma-nickel concentration was stable in the fasting state and
after an unlabelled test meal, but was elevated after the standard
dose of nickel in water. It did not rise above fasting levels in
the two labelled standard meals. When 5 mg of nickel was added to
each of the 5 beverages, the rise in the plasma concentration was
significantly suppressed with all but Coca Cola(R). These results
indicate that nickel absorption may be suppressed by binding or
chelating substances, competitive inhibitors, or redox reagents; on
the other hand absorption is often enhanced by substances that
increase pH, solubility, or oxidation, or by chelating agents that
are actively absorbed. Such compounds, which were constituents of
the meals and beverages studied by Solomons et al. (1982), include:
ascorbic acid, citric acid, pectins (from orange juice), which
affect trace mineral absorption; tannins (in tea and coffee), which
inhibit absorption of iron and zinc; ascorbic acid, which
suppresses nickel absorption; and complexing agents, such as
NaFeEDTA and EDTA, which depress plasma-nickel levels.
Sunderman et al. (1989a) studied the kinetics of nickel
absorption, distribution, and elimination in healthy human
volunteers who ingested NiSO4 in the drinking-water or added to
food (section 6.1.2.2). Nickel levels were determined, using
electrothermal atomic absorption spectrophotometry, in samples of
serum, urine, and faeces collected 2 days before, and 4 days after,
a specified NiSO4 dose (12 µg Ni/kg, N = 4; 18 µg Ni/kg, N = 4, or
50 µg Ni/kg, N = 1). Absorbed nickel averaged 27±17% (mean±SD) of
the dose ingested in water versus 0.7±0.4% of the same dose
ingested in food (40-fold difference). The results of this study
confirmed that dietary constituents profoundly reduce the
bioavailability of the Ni2+ for gastrointestinal absorption.
Approximately one-quarter of the nickel ingested in drinking-water
after an overnight fast was absorbed from the human intestine and
excreted in urine, compared to only 1% of nickel ingested in food.
The kinetic parameters provided by this study reduce the
uncertainty of toxicological risk assessments of human exposures to
nickel in the drinking-water and food.
Nickel absorption and distribution can be influenced by other
factors. An example is disulfiram, which is used for alcohol
aversion therapy, and which is immediately metabolized into two
molecules of DDC (diethyldithiocarbamate). Following oral exposure
of mice to 57Ni (3 µg/kg), the residual body burdens of nickel
after 22 h and 48 h were increased several fold in groups receiving
clinically effective doses of DDC, either orally or intraperitoneally,
compared with controls. The organ distribution was considerably
changed compared with the control values: at 48 h, the amount
deposited in the brain was at least 100 times greater than the
control value; deposition in the kidneys, liver, and lungs was also
increased (Nielsen et al., 1987b).
(b) Nickel/iron interaction
Becker et al. (1980) suggested that iron might affect nickel
absorption. Using isolated intestinal segments of rats in an in
vitro test system, they found that nickel ions had their own
transport system located in the proximal part of the small
intestine, thus making it likely that iron nutrition could affect
nickel absorption. Forth & Rummel (1971) found that the transfer
of nickel from the mucosal to the serosal side was elevated in
iron-deficient intestinal segments.
6.1.3. Absorption through the skin
Percutaneous absorption is of negligible significance,
quantitatively, but is clinically important in the pathogenesis of
contact dermatitis. Because of the ubiquity of nickel-containing
objects in industrialized society, nickel-sensitive patients
frequently face considerable problems in the work-place in a wide
range of jobs, and also because of contact with nickel-containing
material in household and everyday items.
6.1.3.1 Experimental animals
The majority of studies on the dermal uptake of nickel do not
permit the calculation of absorption data. Norgaard (1957), using
guinea-pigs and rabbits (two of each species), applied 10 µl of a
5% solution of 57Ni (as the sulfate heptahydrate) to a shaved area
of 5 x 5 cm on the animal's back and measured the radioactivity in
the organs and body fluids, 24 h after the application. The
relative distribution levels in the urine, blood, kidney, and
liver, measured as impulses/min using a Geiger-Müller counter, are
shown in Table 16. The findings demonstrate that nickel absorption
took place through the skin of the two animal species examined.
Table 16. Radioactivity (impulses/min) in organs, blood,
and urine of rabbits and guinea-pigs, 24 h after
application of 57Ni to the skina
--------------------------------------------------------------
Organ or Rabbit 1 Rabbit 2 Guinea-pig 1 Guinea-pig 2
body fluid
--------------------------------------------------------------
Urine, 5 ml 72 15
Blood, 10 ml 25 9 4 4
Kidney 26 22 2.6 2.6
Liver 5 4 2 1.4
--------------------------------------------------------------
a From: Norgaard (1957)
Mathur et al. (1977) found that nickel (as the sulfate) was
absorbed systemically in male albino rats. Nickel sulfate in
saline solution was applied daily, for 15 or 30 days, at doses
equivalent to 40, 60, or 100 mg nickel/kg body weight. There were
no clinical symptoms of toxicity in any of the animals and no gross
changes were noted at autopsy. However, in rats sacrificed at 15
days, the livers of those that had received 60 or 100 mg nickel/kg
body weight showed microscopic changes consisting of swollen
hepatocytes and feathery degeneration; the testes were normal. In
rats sacrificed at 30 days, the liver changes were more marked,
with focal necrosis in those that had received 60 or 100 mg
nickel/kg body weight; the testes showed tubular damage and
degeneration.
In a study on dermal absorption, Lloyd (1980) applied 63Ni
chloride (40 µCi) to the shaven flanks of guinea-pigs and reported
that a small amount of the applied dose passed through the skin and
appeared in the plasma. After 4, 12, and 24 h exposure, 0.005,
0.07, and 0.05%, respectively, of the total 63Ni dose were found in
the plasma, and 0.009, 0.21, and 0.51%, respectively, of the
absorbed nickel, were measured in urine. In excised skin, levels
of 1.94, 7.30, and 5.33%, respectively, were found. Using micro-
autoradiography of 63Ni chloride exposed skin (2 µCi; periods of
1/2-48 h, shaved flanks of guinea-pigs), Lloyd (1980) also found
that the radioactive nickel accumulated within 1 h in the highly
keratinized areas, the stratum corneum, and hair shafts, showing a
route of entry via the hair follicles and sweat glands. Increased
radioactivity was also measured in the serum and urine. Wells
(1956) had also reported that nickel ions can penetrate via the
sweat ducts and hair follicle ostia and that they have a special
affinity for keratin. Based on histochemical evidence, it was
suggested that nickel is bound by the carboxyl groups of keratin
(Samitz & Katz, 1976).
The greatest accumulation of nickel was found in the Malpighian
layer, the sweat glands, and the walls of the blood vessels. Wells
(1956) reported that nickel sulfate did not penetrate the skin,
because the stratum corneum was a barrier to its penetration.
Samitz & Pomerantz (1958) found that the extent of penetration
(nickel sulfate, 0.5% in aqueous solution) was enhanced by sweat
and sodium lauryl sulfate (1% aqueous solution) in animals (species
not indicated). However, there was no evidence of the actual
absorption of nickel sulfate.
6.1.3.2 Human beings
Nickel/epidermal interactions were studied in vitro in
diffusion cells by Samitz & Katz (1976), who found that the
diffusion of 63Ni (as the sulfate, specific activity 1 µCi/ml; 0.1,
0.01, or 0.001 mol/litre in physiological salt solution) through
the human epidermis was only slight after 17, 24, and 90 h,
respectively. Diffusion did not take place within the first 5 h.
Sweat or surfactants (0.2% in physiological saline solution)
slightly enhanced the diffusion of nickel (0.002 mol nickel
sulfate/litre, containing 0.2 8 µCi 63Ni/ml).
Spruit et al. (1965) using human cadaver skin reported that
nickel ions (from nickel chloride) penetrate, and are bound by, the
dermis. He suggested that nickel bound by the dermis can serve as
a reservoir for the subsequent release of nickel ions. In a study
(using 57Ni as an indicator) on normal and nickel-hypersensitive
persons, it was shown that when 10 µlitre of a 5, 2.5, 1.25, or
0.68% solution of nickel sulfate was applied to the skin, about two
thirds of the nickel was absorbed in 24 h (as measured with a
Geiger-Müller counter) (Norgaard, 1955). The absolute amount
absorbed was highest during the first few hours following
application. Absorption was the same in normal and in
hypersensitive patients. In hypersensitive patients, the
eczematous reaction appeared at the time when only about 10% of the
quantity of nickel absorbed was left on the skin. However, the
findings have not been verified by examining nickel levels in the
skin, other organs, and in body fluids. Kolpakov (1963), using
skin from persons who had died suddenly from accidental injury,
found that the Malphigian layer of the epidermis, the dermis, and
the hypodermis were readily permeable to nickel sulfate.
Permeation of nickel from nickel chloride and nickel sulfate
solution through the human skin was determined in vitro in
diffusion cells (Fullerton et al., 1986). The permeation process
was slow with a lag time of around 50 h. Without occlusion, the
amount of nickel permeation was negligible. Permeation of nickel
ions was faster from a nickel chloride solution than from a nickel
sulfate solution. After about 200 h, 13-43% of the nickel from a
nickel chloride solution and about 4.7% from a nickel sulfate
solution were present in the skin matrix.
6.1.4. Other routes of absorption
Parenterally injected nickel is only of practical interest in
toxicity studies, where it is particularly useful in assessing the
kinetics of nickel transport, distribution, and elimination.
6.1.4.1 Experimental animals
Bergman et al. (1980a) implanted specimens of non-precious
dental casting alloys containing 70-75% nickel (by weight)
subcutaneously in the neck region of mice. After 5 months of
exposure, most of the nickel, released from the implants through
electrochemical corrosion, had accumulated in the soft tissue
capsule at a concentration of 123 mg/kg (wet weight), whereas only
0.31 mg/kg (wet weight) appeared in the kidney; other tissues
contained amounts that were more than 10 times less.
Samitz & Katz (1975) found that nickel was leached from
stainless steel spheres (nickel content not indicated) implanted in
incisions in both hind legs of 3 rabbits. Taking biopsies, from
both legs, at the site of implant and at distances of 1, 2, 3, 4,
and 5 cm around it, nickel was found only in the tissue near the
implant (1 cm distance); levels ranged from 34.3 to 39.8 mg/kg (wet
weight) after 3 weeks, and from 46.2 to 53.3 mg/kg (wet weight)
after 6 weeks. Whether nickel was translocated to organs and the
blood was not examined.
6.1.4.2 Human beings
In human beings, absorption may occur from a variety of
implanted nickel-containing medications, metallic devices, and
prostheses, which release nickel by leaching (section 5.3).
However, leaching from implanted metals is difficult to assess in
human beings, because of the few and conflicting experimental data.
Leaching of nickel from nickel-coated containers may contaminate
intravenous fluids (section 5.3).
6.1.5. Transplacental transfer
Transplacental transfer provides an initial body burden that
will be augmented by later environmental exposures. For this
reason, and, in view of the possible adverse effects associated
with the exposure of pregnant women to nickel during early
pregnancy, transplacental transfer is important. Placental
transfer is influenced by gestational age and the availability of
nickel in the maternal blood. Species differences in placental
structure and implantation, which may possibly influence nickel
transfer, must also be considered.
6.1.5.1 Experimental animals
Several reports indicate that transplacental transfer of nickel
occurs in animals. In a study by Phatak & Padwardhan (1950), the
newborn offspring of rats, fed nickel in various chemical forms
(nickel carbonate, nickel catalyst, nickel soapa) at dietary
concentrations of 250-1000 mg/kg, showed whole-body levels of 22-30
mg/kg and 12-17 mg/kg body weight, when dams received 1000 mg
nickel/kg diet, and 500 mg/kg, respectively.
__________________________________________________________________
a Nickel soap was prepared by neutralizing mixed fatty acids
(obtained by saponification of refined groundnut oil) with
nickel carbonate.
Lu et al. (1981) injected pregnant mice (ICR strain, 12-14
weeks old) intraperitoneally, on day 16 of gestation, with a single
0.1 ml dose of nickel chloride solution (equivalent to 4.6 mg
nickel/kg). The kinetics of nickel chloride in the fetal tissues
showed a different pattern from that in maternal tissues. The
concentration of nickel in the maternal blood and the placenta were
found to be at a maximum (19.8 and 3.9 mg/kg, respectively) 2 h
after injection. The maximum concentration in fetal tissues (1.1
mg/kg) was reached 8 h after injection, and only a slight and
gradual decrease in concentration was observed up to 24 h. The
concentration began to decrease rapidly between 24 and 48 h. The
biological half-life was calculated to be 8.9 h in the rapid phase
and 33 h in the slow phase. In this study, the mean concentration
of nickel in the placenta was less than those in the maternal
kidneys and blood, but higher than those in the maternal liver and
spleen after 24-48 h.
In a study by Lu et al. (1979), 10 pregnant ICR mice given an
intraperitoneal injection of nickel chloride solution, equivalent
to 4.6 mg nickel/kg body weight, on day 8 of gestation, were
sacrificed after 4 h and the embryos removed. The concentration of
nickel retained in embryonic tissues was 800 times higher in the
exposed animals than in the controls.
When pregnant mice were given a single intraperitoneal
injection of 63Ni chloride (50 µCi; 0.14 mg/kg body weight) on day
18 of gestation, passage of 63Ni from mother to fetus was rapid and
concentrations in fetal tissues were generally higher than those in
the dam (Jacobsen et al., 1978).
Olsen & Jonsen (1979a) investigated nickel uptake and retention
after intraperitoneal injection of 0.5 ml 63Ni chloride in a 16-day
pregnant mouse. They observed that placental transfer of nickel
occurred throughout gestation (in the visceral yolk sac during
early gestation 10-11 days, and in the visceral yolk sac and
chorioallantoic placenta during late gestation). A significant
uptake of nickel was seen on days 5-6 of gestation. Fetal
accumulation of nickel took place up to day 16 of gestation.
Nickel was distributed throughout the tissues in the early embryo;
distribution became more differentiated with increasing gestation
and became similar to that in the dam.
Sunderman et al. (1978a) administered 63Ni intramuscularly to
groups of pregnant Fischer 344 rats on days 8 or 18 of gestation
and determined maternal and fetal tissue concentrations by
autoradiography. In the fetuses of dams injected on day 8 of
gestation, the mean 63Ni concentration in the embryos and
membranes, after 24 h, was equivalent to the 63Ni concentrations in
the maternal lungs, adrenals, and ovaries. In dams injected on day
18 of gestation, there was localization of 63Ni in the placentas
and 63Ni was present in the yolk sacs and fetuses, after 24 h. The
fetal organ with the highest concentration of 63Ni was the urinary
bladder, suggesting that there was renal elimination of the 63Ni
that had entered the fetuses on day 18 of gestation.
Nadeenko et al. (1979) administered nickel to rats in the
drinking-water for 7 months before, and during, pregnancy. The
nickel contents increased in the placentas, but not in the fetuses.
Dostal et al. (1989) studied the effects of nickel on lactating
rats, their suckling pups, and the transfer of nickel via the milk.
Dose-dependent increases were observed in the concentrations of
nickel in the milk and plasma, 4 h after a single subcutaneous
injection of nickel chloride at 10, 50, or 100 µmol/kg body weight
to lactating dams giving a milk/plasma nickel ratio of 0.02. Peak
plasma nickel concentrations in the dams occurred 4 h after the
injection, while the peak concentration in the milk was observed at
12 h and remained elevated at 24 h. Daily subcutaneous injections
of dams with 50-100 µmol/kg body weight for 4 days increased the
milk/plasma nickel ratio to 0.10. Significant alterations in milk
composition included increased solids and lipids (42% and 110%,
respectively) and decreased milk protein and lactose (29% and 62%,
respectively). In multiple dose studies where 50 or 100 µmol
nickel chloride/kg body weight were given subcutaneously, once
daily, on days 12-15 of lactation, the plasma-nickel concentrations
in suckling pups, sacrificed 4-6 h after the third daily injection
of 50 or 100 µmol/kg body weight to the dams, were 24 and 48
µg/litre, respectively. Liver weights were decreased in the pups
whose dams received 100 µmol/kg body weight, but no changes in
hepatic lipid peroxidation or thymus weight were reported.
6.1.5.2 Human beings
Nickel has been shown to cross the human placenta; it has been
found in both the fetal tissue (Schroeder et al., 1962) and the
umbilical cord serum, where the average concentration from 12
newborn babies was 3±1.2 µg/litre (range 1.7-4.9 µg/litre) and was
identical with that in the mother's serum, immediately after
delivery (McNeely et al., 1971b). Measurable concentrations have
been found in various fetal tissues. Stack et al. (1976) found a
mean nickel concentration of 23 mg/kg (SD = 7.2) in developing
teeth from 26 cases of stillbirth and neonatal death, while enamel
and dentine of developing teeth from 4 fetuses showed levels
ranging from 11 to 19 mg/kg for dentine and 12 to 20 mg/kg for
enamel. In tissues such as liver, kidney, brain, heart, lung,
skeletal muscle, and bone, nickel was found in mean concentrations
similar to those in adults ranging from 0.24 to 0.69 mg/kg dry
matter (SD ranging from 0.16 to 0.6) (Casey & Robinson, 1978). The
passage of nickel across the human placental barrier is of
relevance because of the presence of female workers in industry.
Appreciable amounts of nickel have been found in breast milk
(Stovbun et al., 1962; Medvedeva, 1965).
6.1.6. Nickel carbonyl
Nickel carbonyl absorption and toxicity is primarily of concern
in occupational inhalation exposure (section 6.1.1.2). Other
routes of absorption are not of practical significance. Absorption
by the gastrointestinal route could be of importance in case of
accidental intake, but no data are available. Because of its
lipid-soluble properties, nickel carbonyl may be absorbed dermally,
but this has not been demonstrated. Parenteral absorption is only
of experimental significance in studying nickel metabolism.
6.2. Distribution, retention, and elimination
The distribution of nickel in the body and its mode of
elimination are relevant in view of the occupational and non-
occupational exposures to nickel resulting from its wide industrial
applications. Studies on the distribution of nickel in the tissues
of animals are useful for the understanding of the interaction
between nickel and biological materials and, consequently, of its
toxic and carcinogenic effects.
Nickel is concentrated in the kidneys, liver, and lungs; it is
excreted primarily in the urine. Nickel can also be found in the
urine of non-occupationally exposed persons. Since the
bioavailability of nickel and the rate of elimination depend very
much on the nature of the nickel compound, urinary excretion may
not always be an appropriate measure of exposure.
6.2.1. Transport
A few nickel-binding serum proteins have been identified in in
vivo and in vitro studies using labelled nickel chloride. Nickel
has been found in three major fractions of human and rabbit serum:
(a) macroglobulin-bound nickel;
(b) albumin-bound nickel; and
(c) nickel bound to ultrafiltrable ligands (e.g., amino acids)
(Nomoto et al., 1971; Hendel & Sunderman, 1972; van Soestbergen
& Sunderman, 1972; Callan & Sunderman, 1973).
Albumin is the principal transport protein for nickel in human,
bovine, rabbit, and rat sera (van Soestbergen & Sunderman, 1972;
Callan & Sunderman, 1973).
A metalloprotein, designated nickeloplasmin, has been isolated
from the sera of rabbits and man (Nomoto et al., 1971; Decsy &
Sunderman, 1974). It is a macroglobulin with an estimated relative
molecular mass of 7 x 105 and contains approximately 0.8 g atomic
nickel/mole. Disc gel- and immunoelectrophoresis have shown that
purified nickeloplasmin is an alpha-2-macroglobulin in rabbit serum
and a 9.5 S alpha-glycoprotein in human serum. These results have
been confirmed, using more refined and sensitive techniques (Nomoto
& Sunderman, 1988).
Ultrafiltrable, nickel-binding ligands play an important role
in extracellular transport and in the elimination of nickel in
urine (Hendel & Sunderman, 1972; van Soestbergen & Sunderman, 1972;
Asato et al., 1975). The low-relative-molecular-mass, nickel-
binding constituent of human serum has been identified as the amino
acid, L-histidine (Lucassen & Sarkar, 1979; Glennon & Sarker, 1982).
In an in vitro system, L-histidine was found to have a
greater affinity for nickel than serum-albumin. Nickel-binding to
human albumin became evident only when no more L-histidine was
available. In vivo, the concentration of albumin was much higher
than the concentration of L-histidine and most of the nickel was
associated with albumin. The equilibrium between L-histidine-
nickel and serum-albumin-nickel may be biologically significant.
The L-histidine nickel complex, which has a much smaller molecular
size than the albumin-nickel complex, may mediate the transport
through a biological membrane by virtue of the equilibrium between
these two molecular species of nickel. The equilibrium in favour
of the L-histidine-nickel complex may be an explanation for the
rapid urinary excretion of nickel observed by Onkelinx et al.
(1973) and Sarkar (1980). The exchange and transfer of nickel
between L-histidine and albumin appear to be mediated by a ternary
complex in the form of albumin-nickel-L-histidine. The amount of
nickel in each compartment varies from species to species and this
may be due, in part, to species variation in the affinity of
albumin for nickel (Hendel & Sunderman, 1972; Callan & Sunderman,
1973).
6.2.2. Tissue distribution
Parameters, such as the nickel compound administered, the dose,
the number of administrations, the length of time between exposure
and sacrifice, the strain of animal, and the route of absorption,
may strongly influence the organ distribution pattern. Thus, the
uptake of nickel by organs has differed in the reports of various
investigators.
6.2.2.1 Experimental animals
Levels of nickel in the tissues of experimental animals have
been determined following exposure to various nickel compounds
under different experimental conditions (Table 17). The studies
(in most cases with radioactive nickel, 63Ni) indicate that nickel
is widely distributed and rapidly eliminated. Administration of
divalent nickel salts intravenously, intraperitoneally, or
subcutaneously, as single or repeated injections, led to the
highest accumulation in the kidney, endocrine glands, lung, and
liver. Concentrations in nerve tissue were low; this is consistent
with the observed low neurotoxic potential of divalent nickel salts.
There was also slight uptake by bone, consistent with the rapid and
extensive elimination of nickel from the organism. Low concentrations
are retained in soft and mineral tissue.
A large amount of nickel was found in the guinea-pig kidney and
pituitary gland, after daily subcutaneous injections of 1 mg nickel
chloride/kg body weight for 5 days (Clary, 1975). In a study by
Parker & Sunderman (1974), substantial concentrations of 63Ni in
the pituitary gland of the rabbit were found following single or
repeated intravenous injections of 63Ni chloride. The 63Ni level
in the pituitary gland was second only to that in the kidneys. The
findng that nickel is particularly localized in the pituitary gland
may have physiological significance. LaBella et al. (1973a) suggested
that nickel may exert a direct, specific inhibitory action on
prolactin-secreting cells in the anterior pituitary gland.
Table 17. Relative tissue distribution of nickel in experimental animalsa
-------------------------------------------------------------------------------------------------
Species (Number) Dosage and stpb Relative distributionc Reference
-------------------------------------------------------------------------------------------------
Rat (4) 617 µg/kg (single iv kidney > lung > adrenal > ovary > Smith &
injection) 2 h heart > gastrointestinal tract > skin > Hackley
eye > pancreas > spleen = liver > muscle (1968)
teeth > bone > brain = fat
Rabbit (3) 240 µg/kg (single iv kidney > pituitary > serum > whole Parker &
injection) 2 h blood > skin > lung > heart > Sunderman
testis > pancreas > adrenal > (1974)
duodenum > bone > spleen > liver >
muscle > spinal cord > cerebellum >
medulla oblongata = hypothalamus
Rabbit (4) 4.5 µg/kg (34-38 daily kidney > pituitary > spleen > lung > Parker &
consecutive injections) skin > testis > serum = pancreas = Sunderman
24 h adrenal > sclearae > duodenum = liver > (1974)
whole blood > heart > bone > iris >
muscle > cornea = cerebellum =
hypothalamus > medulla oblongata >
spinal cord > retina > lens >
vitreous humor
Rat (4-11) 3.3 or 6.5 mg/kg kidney > adrenal > lung > heart > Chausmer
(single iv injection) pancreas > small intestine > eye > (1976)
2 h thymus > muscle > epididymis > ovary >
liver > spleen > bone > brain >
incisor > fat
Mouse (12) 38.3 µg or 76.6 µg/kg kidney > lung > sternal cartilage Oskarsson &
(single iv injection) liver > pancreas Tjälve
1 h (1979a)
Mouse (3) 0.5 mg/kg (single iv kidney > urinary
bladder (urine) > Bergman et
injection) 2 h lung > skin > cartilage > eye (retina) > al. (1980a)
hair follicle > blood > oral epithelium >
gastric epithelium > tooth enamel > tooth
dentine > salivary glands > liver >
gastric mucosa > pancreas > spleen >
brown fat > bone > muscle
-------------------------------------------------------------------------------------------------
Table 17 (contd.)
-------------------------------------------------------------------------------------------------
Species (Number) Dosage and stpb Relative distributionc Reference
-------------------------------------------------------------------------------------------------
Mouse (8) 6.2 mg/kg (one ip kidney > lung > plasma > liver > Wase et al.
injection) 2 h erythrocytes > spleen > bladder > (1954)
heart > brain > carcass (muscle, bone,
and fat)
Rat (18) 3 or 6 mg/kg (7 or 14 7 or 14 x 3 mg/kg: heart > spleen > Mathur et
injections as NiSO4 x kidney > testis > bone > liver al. (1978)
6H2O) 48 h following
last injection 7 x 6 mg/kg: heart > spleen > kidney >
testis > liver > bone
14 x 6 mg/kg: heart > spleen > kidney >
bone > testis > liver
Rat 82 µg/kg (ip) 6 h kidney > spleen > lung > heart > liver > Sarkar
muscle (1980)
Rat (5) 6 mg/kg (single ip kidney > serum > heart > liver Tandon
injection) 24 h (1982)
Mouse 50 mCi (single ip 6 days after injection: lung > kidney > Herlant-
injection) skin > small intestine > spleen > Peers et
liver > uterus > brain > stomach > al. (1982)
heart > whole blood > muscle > serum
Mouse 100 µCi (7 successive 24 h after injection: lung > kidney > Herlant-
ip injections at 24 h uterus > skin > stomach > spleen > Peers et
intervals) small intestine > liver > brain > al. (1982)
heart > serum > muscle > whole blood
Mouse (5) 1 or 24 (3 times/week) 1 injection: kidney > lung > pancreas > Kasprzak &
ip injections of 42.5 spleen > liver > brain > heart > blood Poirier
µmol 63Ni acetate/kg (1983)
48 h 24 injections: pancreas > lung > heart >
kidney > spleen > liver > brain > blood
-------------------------------------------------------------------------------------------------
Table 17 (contd.)
-------------------------------------------------------------------------------------------------
Species (Number) Dosage and stpb Relative distributionc Reference
-------------------------------------------------------------------------------------------------
Mouse (10) specimen of non- soft tissue capsule around implants > Bergman et
precious dental casting kidney > lung > spleen > liver > al. (1980b)
alloy implanted pancreas > heart > blood
subcutaneously in the
neck region (10 x 5 x
1 mm) containing 71%
nickel, exposure time
5 months
Guinea-pig (6) 1 mg/kg (subcutaneously kidney > pituitary > lung liver > Clary
for 5 days) 6 h spleen > heart > adrenal > testis > (1975)
pancreas > medulla oblongata =
cerebrum = cerebellum
Rat (5) 7.5 µg/kg (one lung > kidney > blood > skin > bone = Carvalho &
intratracheal spleen = testes = liver > heart Ziemer
injection ) 1 h (1982)
Rat (30) 1 mg/animal (one kidney > lung > adrenal > liver > Clary
intratracheal pancreas = spleen = heart > testes (1975)
injection) 6 h
Hamster (51) 53.2 % 11.1 mg nickel lung > liver > kidney Wehner et
oxide/m3 (inhalation) al. (1975)
life-span exposure
Rat (12) 25, 50, or 100 mg 100 mg: bones > heart > kidney > blood > Phatak &
NiCO3/100 g diet spleen > intestine > testes > skin > Padwardhan
week 9 liver (1950)
50 mg: bones > testes > spleen > intestine
heart > liver > kidney > blood > skin
25 mg: bones > intestine > testes >
kidney > heart > spleen > blood > skin >
liver
-------------------------------------------------------------------------------------------------
Table 17 (contd.)
-------------------------------------------------------------------------------------------------
Species (Number) Dosage and stpb Relative distributionc Reference
-------------------------------------------------------------------------------------------------
Rat (72) 100 nmol NiCl2 lung > mediastinal lymph nodes > English
(single intratracheal kidney > ovaries > blood > femur > et al.
injection) 0.5 h heart = adrenals > skin = pancreas > (1981)
duodenum > pituitary > liver > spleen
100 nmol NiO lung > mediastinal lymph nodes >
(single intratracheal kidney > heart > femur > duodenum >
injection) 0.5 h kidney > pancreas > ovaries > spleen >
blood > adrenals > skin > pituitary >
liver
Calf (12) 62.5, 250, 1000 mg 1000 mg/kg: serum > kidney > vitreous O'Dell et
NiCO3/kg dietary humor > lung > testis > bile > tongue > al. (1971)
supplementation for pancreas > rib > spleen > brain >
8 weeks liver > heart
250 mg/kg: lung > serum > kidney
62.5 mg/kg: lung > kidney > liver >
testis
Rat (24) 100, 500, 1000 mg/kg liver > heart > kidney > testis Whanger
diet as nickel acetate (1973)
for 6 weeks
Rat (64) 5.4 mg/kg in food and spleen > heart > kidney > lung Schroeder
and water for life liver et al.
(1974)
Lamb (12) 65 µg/kg or 5 mg 65 mg/kg: kidney > lung > spleen > Spears
nickel/kg in diet for heart > liver > brain > testis et al.
97 days; on day 94: (1978)
single oral dose of 5 mg/g: kidney > spleen > lung >
40 µCi 63Ni/kg liver > testis > heart > brain
-------------------------------------------------------------------------------------------------
Table 17 (contd.)
-------------------------------------------------------------------------------------------------
Species (Number) Dosage and stpb Relative distributionc Reference
-------------------------------------------------------------------------------------------------
Rat (45) 1, 11.2, or 105.7 µg lung > trachea > larynx > kidney > Medinsky
sulfate/rat (single urinary bladder > adrenal glands > et al.
intratracheal injection) blood > large intestine > thyroid (1987)
4, 24, or 96 h
Rat (15) 2.5 µg/animal orally trachea > nasopharynx > skull bone > Huang et
for 30 days as nickel oesophagus > spleen > kidneys > al. (1986)
sulfate lungs > heart
-------------------------------------------------------------------------------------------------
a Partially adapted from: NAS (1975).
b Nickel given as 63Ni chloride unless other compound indicated; stp = sacrifice time
post exposure.
c Distribution in decreasing nickel concentration.
Studies by Herlant-Peers et al. (1982) and Kasprzak & Poirier
(1983) showed that the number of administrations and the time
interval between nickel injection and sacrifice of the animals
influenced the distribution pattern of nickel chloride injected
intraperitoneally (Table 17, Fig. 1).
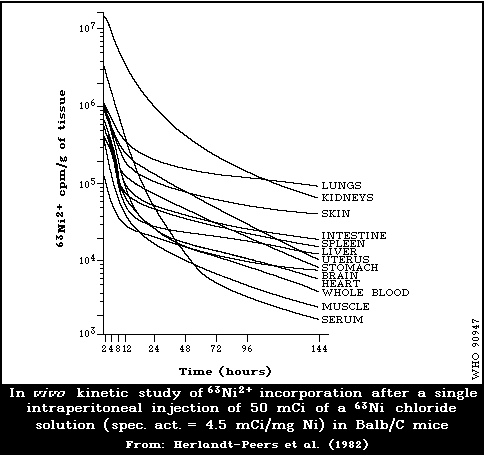 The most striking findings of the study by Kasprzak et al.
(1983) were the high accumulation of nickel in the pancreas and the
decreasing nickel accumulation in the kidney and heart, following
multiple intraperitoneal injections of nickel acetate. The first
finding would link nickel with zinc and insulin metabolism and
relate the commonly observed nickel-induced elevation of serum
glucose to this interaction (Clary, 1975; Sunderman et al., 1976a).
The second finding suggests the development of some detoxifying
mechanisms in the kidney and heart during prolonged exposure to
nickel. Huang et al. (1986) administered 2.5 µg nickel
sulfate/animal to rats, orally, for 30 days. The nickel contents
in the trachea, nasopharynx, oesophagus, lungs, skull, bone, heart,
spleen, and kidneys of rats fed with nickel were significantly
higher than those in the control animals.
After exposing rats to nickel at a concentration of 5 mg/litre
in the drinking-water for their lifetime, Schroeder et al. (1974)
did not find any measurable accumulation of nickel in tissues.
When rats were fed nickel carbonate, nickel soaps, or metallic
nickel catalyst, tissue accumulation was significant only in the
case of the carbonate (Phatak & Padwardhan, 1950). O'Dell et al.
(1971) fed calves supplemental dietary nickel at levels of 62.5,
250, or 1000 mg/kg and found pronounced increases in nickel levels
in the pancreas, testes, and bone at the highest dietary level.
While comparison of data for monogastric and ruminant animals may
not be valid, these data indicate that the skeleton is the main
storage depot for nickel, even though the nickel concentration in
bone differs greatly between the studies by Phatak & Padwardhan
(1950) and O'Dell et al. (1971). The data agree on the limited
capacity of the liver to store nickel (which is in contrast to most
of the trace elements). A major difference between the data for
rats and calves is in the nickel level in the heart muscle. Nickel
concentrations in this tissue were somewhat elevated in rats (250
mg/kg), but not in calves.
Similar studies on weanling rats (Whanger, 1973) and lambs
(Spears et al., 1978) given soluble nickel salts (acetate or 63Ni
chloride) in the diet at various levels up to 1000 mg nickel/kg
showed the highest accumulation in the kidney. As the nickel dose
increased, the nickel contents of the tissues (kidney, liver,
heart, lung,and testes) also increased. In rats, treated
intratracheally, the distribution was virtually analogous (except
for the pituitary gland), though, as expected, the lung (rather
than the kidney) showed the highest accumulation (Clary, 1975;
Carvalho & Ziemer, 1982).
6.2.2.2 Kinetics of metabolism
Following inhalation of high concentrations of nickel oxide
(10-190 mg/m3) by hamsters (7 h daily, repeated exposures for up to
3 months), 20% of the inhaled amount of nickel oxide was still
present in the lungs 3-4 days after exposure. Complete clearance
of this oxide was estimated to take weeks to months; 75% of the
nickel oxide was still present in the lungs 10 days after exposure
and 40% was still present 100 days after exposure (Wehner & Craig,
1972; Wehner et al., 1975). The lungs retained more than 99% of
the nickel oxide deposited there. The liver and kidney retained
small amounts of 0.21 and 0.04%, respectively. Kodama et al (1985)
exposed rats to nickel oxide by inhalation at concentrations of
0.4-70 mg/m3 for 6-7 h/day, 5 days/week, for a maximum of 3 months.
Deposition of nickel oxide in the lungs ranged from 2.3±0.9 to
23.4±1%. The deposition fraction significantly decreased with
increase in the mass median diameter of the particles, and slightly
decreased with increasing exposure concentration. The clearance
rate was estimated to be approximately 100 µg/year.
In contrast to the prolonged retention of nickel oxide in the
lung after inhalation, the more soluble nickel chloride was rapidly
cleared after a single intratracheal injection (1 mg/kg body
weight) in rats (Clary, 1975). Six h after exposure, the kidneys
showed the greatest amount of nickel followed, in order, by the
lung and adrenals, with decreasing amounts in the pancreas, spleen,
heart, and testes. Other tissues, such as whole brain, thymus,
eyes, and femur showed only trace amounts. By 3 days, 90% of the
injected nickel had been excreted, mainly in the urine (75%).
Carvalho & Ziemer (1982) studied the deposition, clearance, and
distribution of 63Ni after intratracheal instillation in rats of
very low dose levels (1.27 µg 63Ni per animal); the highest
concentrations of 63Ni were retained in the lungs and kidneys.
These were the only organs containing measurable amounts of 63Ni,
21 days after exposure. Urinary excretion was the main route of
elimination (78.5% of the initial deposition within 3 days). On
day 21, almost all the 63Ni (96.5% of the initial body burden) had
been excreted in the urine. The lungs retained 29% of their
initial deposition (35 min after exposure), decreasing to less than
1% on day 21. Medinsky et al. (1987) gave 1, 11.2, or 105.7 µg
nickel sulfate/animal, by intratracheal instillation, to Fischer
344 rats. Urinary excretion accounted for 50% of the dose, at
doses of 1 and 11.2 µg/rat, and 80% at a dose of 105.7 µg/rat. The
half-time for urinary excretion of nickel increased from 4.6 h at
the highest dose to 23 h at the lowest dose. Faecal elimination of
the initial dose was 30% (1 and 11.2 µg doses) or 13% (105.7 µg
dose). Over 50% of the nickel remaining in the body at the end of
96 h was in the lungs. The half-time for lung clearance of nickel
sulfate ranged from 21 h (highest dose) to 36 h (lowest dose). The
results of this study indicate that the differences in the lung
clearance of soluble nickel compounds reported by Carvalho & Ziemer
(1982) and Clary (1975) can be explained by differences in
instilled doses.
Tanaka et al. (1985, 1988) estimated the biological half-time
of nickel monosulfide (amorphous) aerosol and of green nickel oxide
in rats exposed through inhalation. The biological half-time of NiS
(A) in the rat lung was 20-h, while that of the oxide was 21 months
(particle size 4.0 µm) or 11.5 months (particle size 1.2 µm).
In several studies, 63Ni-labelled nickel salts have been used
to study the distribution and elimination of nickel after
parenteral injection (Wase et al., 1954; Smith & Hackley, 1968;
Parker & Sunderman, 1974; Clary, 1975; Mathur et al., 1978;
Oskarsson & Tjälve, 1979b; Bergman et al., 1980b; Tandon, 1982).
Using this route of administration, most nickel was excreted in the
urine, causing a high labelling of the kidneys, which may be
related to the role of the kidneys in nickel clearance. However,
there have been different observations on its localization in other
organs. Smith & Hackley (1968) found a good correlation between
the blood volume of each tissue and the amount of nickel retained
by that tissue. Mathur et al. (1978) investigated the effects of
dose and duration of exposure on the relative distribution and
found that, while a single exposure to nickel may not have a
lasting effect on the body tissues, regular exposures could have a
cumulative effect, particularly on the kidneys and the heart. An
autoradiographic distribution study by Bergman et al. (1980b) on
the albino mouse showed that, between 30 min and 24 h, there were
high concentrations of 63Ni in the urogenital, circulatory, and
respiratory organs. Accumulation was also found in cartilage,
lacrimal glands, and the skin. After one day, the distribution
pattern changed, so that the highest concentrations were in the
lungs, kidneys, central nervous system, skin, and the epithelia of
the oral cavity and the oesophageal part of the stomach, with the
long residence time of 3 weeks; accumulation was highest in the
lung, central nervous system, kidneys, hard tissues (teeth,
cartilage, bone), and the skin. A distribution study by Bergman et
al. (1980a), who implanted specimens of non-precious dental casting
alloys subcutaneously in the neck region of mice, did not yield any
information about the dynamic pattern of the release of nickel, the
uptake in various tissues and organs, or its elimination.
Metabolic data from nickel-balance studies carried out by
Phatak & Padwardhan (1950) who fed rats, nickel carbonate, nickel
soaps, or nickel catalyst (250, 500, or 1000 mg/kg in the diet for
2 months) demonstrated that appreciable quantities of nickel from
all the nickel-containing diets were retained. Retention from the
nickel carbonate diet was greater than that from the other two
nickel preparations. This can be attributed to the ready
solubility of the compound in the stomach and the easier absorption
from the intestine. The proportion of the ingested nickel found in
the faeces was lowest in the carbonate group. The amount of nickel
excreted in the urine was only slightly higher in this group than
in the other two.
O'Dell et al. (1971) fed calves a basal diet supplemented with
nickel (as the carbonate) at levels similar to those used by Phatak
& Padwardhan (1950) and for the same period of time. The results
of this study showed that the absorption and tissue retention of
dietary nickel can be increased and that the increase is related to
the rate of nickel intake as well as to the total nickel intake.
(a) Kinetic modelling
The whole-body kinetics of nickel chloride (or other soluble
metal salts) can be studied by injecting animals with suitable
radiotracers, following the metal concentration in plasma as a
function of time after injection and measuring the amounts
eliminated in urinary and faecal collections. In general, data
obtained in this fashion can be analysed mathematically. This
allows the formulation of compartment models describing the
metabolism of nickel in terms of distribution volumes, clearances
by elimination and clearances by exchange. Nickel metabolism is
characterized by a typical distribution and elimination pattern
(Fig. 2, Table 18). Onkelinx et al. (1973) injected 63NiCl2
intravenously in male and female Wistar rats (17 µg/animal) and New
Zealand albino rabbits (816 µg/animal) and measured the radioactive
label in the urine and faeces, 3 days after injection, and, in the
blood, at intervals ranging from 1 h to 7 or 9 days. In the rats,
during the first day, 68% 63Ni was excreted in the urine and, after
3 days, 78%. In the rabbit (2 animals), 9% of the administered
dose was excreted in the urine, 5 h after injection, and 78% during
the first day (Fig. 3). Faecal elimination of 63Ni in the rat was
15% of the administered dose during the first 3 days following
injection; faecal elimination was not determined in the rabbits.
Sunderman et al. (1976a) administered 2173 µg 63Ni chloride/animal
to Fischer rats by intraperitoneal injection. Blood samples were
taken at intervals between 10 min and 24 h after injection. Urine
and faeces were collected at intervals between 6 h and 5 days after
injection. Both studies showed that absorption and elimination
fitted a 2-compartment modela in both species, comprising a rapid
clearance phase from plasma or serum during the first 2 days and a
much slower phase between the third and seventh days.
Chausmer (1976) determined tissue exchangeable pools (in rats
injected intravenously with 63Ni chloride) at a number of intervals
following injection, and found (after performing a compartmental
analysis of tissue exchangeable pools by computer evaluation of the
percentage retained radioactive nickel) a rapid intracellular
compartment having a half-life of several h in most tissues. The
slower compartment had a half-life of several days. The kidney
(followed by the lung, liver, and spleen) was found to have the
largest rapidly exchangeable pool, 16 h after injection, with a
two-compartment distribution, whereas bone had the best fit with a
single compartment.
The most striking findings of the study by Kasprzak et al.
(1983) were the high accumulation of nickel in the pancreas and the
decreasing nickel accumulation in the kidney and heart, following
multiple intraperitoneal injections of nickel acetate. The first
finding would link nickel with zinc and insulin metabolism and
relate the commonly observed nickel-induced elevation of serum
glucose to this interaction (Clary, 1975; Sunderman et al., 1976a).
The second finding suggests the development of some detoxifying
mechanisms in the kidney and heart during prolonged exposure to
nickel. Huang et al. (1986) administered 2.5 µg nickel
sulfate/animal to rats, orally, for 30 days. The nickel contents
in the trachea, nasopharynx, oesophagus, lungs, skull, bone, heart,
spleen, and kidneys of rats fed with nickel were significantly
higher than those in the control animals.
After exposing rats to nickel at a concentration of 5 mg/litre
in the drinking-water for their lifetime, Schroeder et al. (1974)
did not find any measurable accumulation of nickel in tissues.
When rats were fed nickel carbonate, nickel soaps, or metallic
nickel catalyst, tissue accumulation was significant only in the
case of the carbonate (Phatak & Padwardhan, 1950). O'Dell et al.
(1971) fed calves supplemental dietary nickel at levels of 62.5,
250, or 1000 mg/kg and found pronounced increases in nickel levels
in the pancreas, testes, and bone at the highest dietary level.
While comparison of data for monogastric and ruminant animals may
not be valid, these data indicate that the skeleton is the main
storage depot for nickel, even though the nickel concentration in
bone differs greatly between the studies by Phatak & Padwardhan
(1950) and O'Dell et al. (1971). The data agree on the limited
capacity of the liver to store nickel (which is in contrast to most
of the trace elements). A major difference between the data for
rats and calves is in the nickel level in the heart muscle. Nickel
concentrations in this tissue were somewhat elevated in rats (250
mg/kg), but not in calves.
Similar studies on weanling rats (Whanger, 1973) and lambs
(Spears et al., 1978) given soluble nickel salts (acetate or 63Ni
chloride) in the diet at various levels up to 1000 mg nickel/kg
showed the highest accumulation in the kidney. As the nickel dose
increased, the nickel contents of the tissues (kidney, liver,
heart, lung,and testes) also increased. In rats, treated
intratracheally, the distribution was virtually analogous (except
for the pituitary gland), though, as expected, the lung (rather
than the kidney) showed the highest accumulation (Clary, 1975;
Carvalho & Ziemer, 1982).
6.2.2.2 Kinetics of metabolism
Following inhalation of high concentrations of nickel oxide
(10-190 mg/m3) by hamsters (7 h daily, repeated exposures for up to
3 months), 20% of the inhaled amount of nickel oxide was still
present in the lungs 3-4 days after exposure. Complete clearance
of this oxide was estimated to take weeks to months; 75% of the
nickel oxide was still present in the lungs 10 days after exposure
and 40% was still present 100 days after exposure (Wehner & Craig,
1972; Wehner et al., 1975). The lungs retained more than 99% of
the nickel oxide deposited there. The liver and kidney retained
small amounts of 0.21 and 0.04%, respectively. Kodama et al (1985)
exposed rats to nickel oxide by inhalation at concentrations of
0.4-70 mg/m3 for 6-7 h/day, 5 days/week, for a maximum of 3 months.
Deposition of nickel oxide in the lungs ranged from 2.3±0.9 to
23.4±1%. The deposition fraction significantly decreased with
increase in the mass median diameter of the particles, and slightly
decreased with increasing exposure concentration. The clearance
rate was estimated to be approximately 100 µg/year.
In contrast to the prolonged retention of nickel oxide in the
lung after inhalation, the more soluble nickel chloride was rapidly
cleared after a single intratracheal injection (1 mg/kg body
weight) in rats (Clary, 1975). Six h after exposure, the kidneys
showed the greatest amount of nickel followed, in order, by the
lung and adrenals, with decreasing amounts in the pancreas, spleen,
heart, and testes. Other tissues, such as whole brain, thymus,
eyes, and femur showed only trace amounts. By 3 days, 90% of the
injected nickel had been excreted, mainly in the urine (75%).
Carvalho & Ziemer (1982) studied the deposition, clearance, and
distribution of 63Ni after intratracheal instillation in rats of
very low dose levels (1.27 µg 63Ni per animal); the highest
concentrations of 63Ni were retained in the lungs and kidneys.
These were the only organs containing measurable amounts of 63Ni,
21 days after exposure. Urinary excretion was the main route of
elimination (78.5% of the initial deposition within 3 days). On
day 21, almost all the 63Ni (96.5% of the initial body burden) had
been excreted in the urine. The lungs retained 29% of their
initial deposition (35 min after exposure), decreasing to less than
1% on day 21. Medinsky et al. (1987) gave 1, 11.2, or 105.7 µg
nickel sulfate/animal, by intratracheal instillation, to Fischer
344 rats. Urinary excretion accounted for 50% of the dose, at
doses of 1 and 11.2 µg/rat, and 80% at a dose of 105.7 µg/rat. The
half-time for urinary excretion of nickel increased from 4.6 h at
the highest dose to 23 h at the lowest dose. Faecal elimination of
the initial dose was 30% (1 and 11.2 µg doses) or 13% (105.7 µg
dose). Over 50% of the nickel remaining in the body at the end of
96 h was in the lungs. The half-time for lung clearance of nickel
sulfate ranged from 21 h (highest dose) to 36 h (lowest dose). The
results of this study indicate that the differences in the lung
clearance of soluble nickel compounds reported by Carvalho & Ziemer
(1982) and Clary (1975) can be explained by differences in
instilled doses.
Tanaka et al. (1985, 1988) estimated the biological half-time
of nickel monosulfide (amorphous) aerosol and of green nickel oxide
in rats exposed through inhalation. The biological half-time of NiS
(A) in the rat lung was 20-h, while that of the oxide was 21 months
(particle size 4.0 µm) or 11.5 months (particle size 1.2 µm).
In several studies, 63Ni-labelled nickel salts have been used
to study the distribution and elimination of nickel after
parenteral injection (Wase et al., 1954; Smith & Hackley, 1968;
Parker & Sunderman, 1974; Clary, 1975; Mathur et al., 1978;
Oskarsson & Tjälve, 1979b; Bergman et al., 1980b; Tandon, 1982).
Using this route of administration, most nickel was excreted in the
urine, causing a high labelling of the kidneys, which may be
related to the role of the kidneys in nickel clearance. However,
there have been different observations on its localization in other
organs. Smith & Hackley (1968) found a good correlation between
the blood volume of each tissue and the amount of nickel retained
by that tissue. Mathur et al. (1978) investigated the effects of
dose and duration of exposure on the relative distribution and
found that, while a single exposure to nickel may not have a
lasting effect on the body tissues, regular exposures could have a
cumulative effect, particularly on the kidneys and the heart. An
autoradiographic distribution study by Bergman et al. (1980b) on
the albino mouse showed that, between 30 min and 24 h, there were
high concentrations of 63Ni in the urogenital, circulatory, and
respiratory organs. Accumulation was also found in cartilage,
lacrimal glands, and the skin. After one day, the distribution
pattern changed, so that the highest concentrations were in the
lungs, kidneys, central nervous system, skin, and the epithelia of
the oral cavity and the oesophageal part of the stomach, with the
long residence time of 3 weeks; accumulation was highest in the
lung, central nervous system, kidneys, hard tissues (teeth,
cartilage, bone), and the skin. A distribution study by Bergman et
al. (1980a), who implanted specimens of non-precious dental casting
alloys subcutaneously in the neck region of mice, did not yield any
information about the dynamic pattern of the release of nickel, the
uptake in various tissues and organs, or its elimination.
Metabolic data from nickel-balance studies carried out by
Phatak & Padwardhan (1950) who fed rats, nickel carbonate, nickel
soaps, or nickel catalyst (250, 500, or 1000 mg/kg in the diet for
2 months) demonstrated that appreciable quantities of nickel from
all the nickel-containing diets were retained. Retention from the
nickel carbonate diet was greater than that from the other two
nickel preparations. This can be attributed to the ready
solubility of the compound in the stomach and the easier absorption
from the intestine. The proportion of the ingested nickel found in
the faeces was lowest in the carbonate group. The amount of nickel
excreted in the urine was only slightly higher in this group than
in the other two.
O'Dell et al. (1971) fed calves a basal diet supplemented with
nickel (as the carbonate) at levels similar to those used by Phatak
& Padwardhan (1950) and for the same period of time. The results
of this study showed that the absorption and tissue retention of
dietary nickel can be increased and that the increase is related to
the rate of nickel intake as well as to the total nickel intake.
(a) Kinetic modelling
The whole-body kinetics of nickel chloride (or other soluble
metal salts) can be studied by injecting animals with suitable
radiotracers, following the metal concentration in plasma as a
function of time after injection and measuring the amounts
eliminated in urinary and faecal collections. In general, data
obtained in this fashion can be analysed mathematically. This
allows the formulation of compartment models describing the
metabolism of nickel in terms of distribution volumes, clearances
by elimination and clearances by exchange. Nickel metabolism is
characterized by a typical distribution and elimination pattern
(Fig. 2, Table 18). Onkelinx et al. (1973) injected 63NiCl2
intravenously in male and female Wistar rats (17 µg/animal) and New
Zealand albino rabbits (816 µg/animal) and measured the radioactive
label in the urine and faeces, 3 days after injection, and, in the
blood, at intervals ranging from 1 h to 7 or 9 days. In the rats,
during the first day, 68% 63Ni was excreted in the urine and, after
3 days, 78%. In the rabbit (2 animals), 9% of the administered
dose was excreted in the urine, 5 h after injection, and 78% during
the first day (Fig. 3). Faecal elimination of 63Ni in the rat was
15% of the administered dose during the first 3 days following
injection; faecal elimination was not determined in the rabbits.
Sunderman et al. (1976a) administered 2173 µg 63Ni chloride/animal
to Fischer rats by intraperitoneal injection. Blood samples were
taken at intervals between 10 min and 24 h after injection. Urine
and faeces were collected at intervals between 6 h and 5 days after
injection. Both studies showed that absorption and elimination
fitted a 2-compartment modela in both species, comprising a rapid
clearance phase from plasma or serum during the first 2 days and a
much slower phase between the third and seventh days.
Chausmer (1976) determined tissue exchangeable pools (in rats
injected intravenously with 63Ni chloride) at a number of intervals
following injection, and found (after performing a compartmental
analysis of tissue exchangeable pools by computer evaluation of the
percentage retained radioactive nickel) a rapid intracellular
compartment having a half-life of several h in most tissues. The
slower compartment had a half-life of several days. The kidney
(followed by the lung, liver, and spleen) was found to have the
largest rapidly exchangeable pool, 16 h after injection, with a
two-compartment distribution, whereas bone had the best fit with a
single compartment.
 ________________________________________________________________
a Compartment I: central compartment including the plasma and from
which elimination takes place.
Compartment II: hypothetical volume that is connected to
compartment I by reversible exchange.
Table 18. Parameters of the two-compartment model of Ni(II)
metabolisma
-------------------------------------------------------------------
Parameters Symbol Units Wistar Fischer Rabbitb
ratb ratc
-------------------------------------------------------------------
Compartment I V1 ml 75.1 59.2 697
Compartment II V2 ml 8.3 31.6 265
Exchange I-II fe ml/h 0.12 0.92 2.31
Total excretory ft ml/h 8.14 7.73 61.2
clearance
Urinary clearance fu ml/h 6.39 6.70 54.1
Faecal clearance fd ml/h 1.28 0.75 -d
Clearance into skin fs ml/h 0.47 0.28 -d
Average body weight wt g 208 165 3400
Injected dose µg 17 (iv) 2173 (ip) 816 (iv)
(µg Ni/animal)
-------------------------------------------------------------------
a From: Onkelinx & Sunderman (1980).
b From: Onkelinx et al. (1973).
c From: Sunderman et al. (1976a).
d Measurements of faecal 63Ni(II) were not performed in rabbits;
hence fd and fs could not be calculated.
________________________________________________________________
a Compartment I: central compartment including the plasma and from
which elimination takes place.
Compartment II: hypothetical volume that is connected to
compartment I by reversible exchange.
Table 18. Parameters of the two-compartment model of Ni(II)
metabolisma
-------------------------------------------------------------------
Parameters Symbol Units Wistar Fischer Rabbitb
ratb ratc
-------------------------------------------------------------------
Compartment I V1 ml 75.1 59.2 697
Compartment II V2 ml 8.3 31.6 265
Exchange I-II fe ml/h 0.12 0.92 2.31
Total excretory ft ml/h 8.14 7.73 61.2
clearance
Urinary clearance fu ml/h 6.39 6.70 54.1
Faecal clearance fd ml/h 1.28 0.75 -d
Clearance into skin fs ml/h 0.47 0.28 -d
Average body weight wt g 208 165 3400
Injected dose µg 17 (iv) 2173 (ip) 816 (iv)
(µg Ni/animal)
-------------------------------------------------------------------
a From: Onkelinx & Sunderman (1980).
b From: Onkelinx et al. (1973).
c From: Sunderman et al. (1976a).
d Measurements of faecal 63Ni(II) were not performed in rabbits;
hence fd and fs could not be calculated.
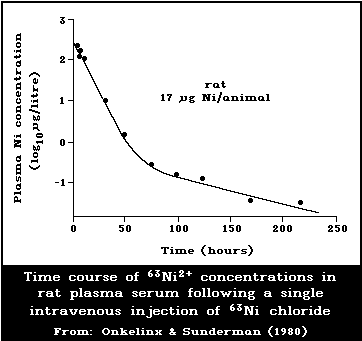
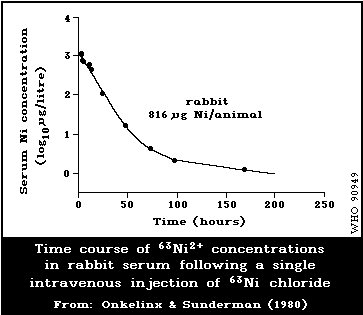 The distribution and elimination of nickel, given to animals as
63Ni chloride, has been studied extensively. Most of the
introduced nickel is rapidly excreted in the urine (65-87% in 24
h), the rest undergoing much slower elimination (76-90% in 5 days)
(Sunderman & Selin, 1968; Sunderman et al., 1976a).
The 2-compartment model described by Onkelinx & Sunderman
(1980) also provides a satisfactory fit for experimental data from
human volunteers who ingested nickel sulfate in the drinking-water
or food at doses of 12, 18, or 50 µg nickel/kg body weight
(Sunderman et al., 1989a). Faecal elimination of nickel during 4
days following treatment averaged 76±19% of the dose ingested in
water versus 102±20% of the dose ingested in food. The elimination
half-time for absorbed nickel averaged 28±9 h (range 17-48 h).
Renal clearance was determined to be 8.3±2.0 ml/min per 1.73 m2 in
human beings who had ingested nickel sulfate in water, and 5.8±4.3
ml/min per 1.73 m2 in those who had received nickel sulfate in
food. The difference was not statistically significant.
6.2.2.3 Nickel carbonyl
There are few studies on the fate of nickel carbonyl in
experimental animals. After the administration of nickel carbonyl,
deposition occurs in the lung and in tissues, such as the brain,
liver, and adrenals, and part of the administered dose of nickel is
recovered in the urine (Armit, 1908; Barnes & Denz, 1951; Sunderman
& Selin, 1968). It was assumed earlier that nickel carbonyl was
rapidly dissociated in the lung and that the nickel was then
transported to other tissues. However, the results of several
studies (Sunderman & Selin, 1968; Sunderman et al., 1968; Kasprzak
& Sunderman, 1969) have indicated that unchanged nickel carbonyl is
present in the blood several hours after administration and can
pass across the pulmonary alveoli in either direction without
decomposition. It was suggested by Kasprzak & Sunderman (1969)
that the nickel carbonyl that was not exhaled underwent a slow
intracellular decomposition to NiO and CO. The released NiO was
then oxidized to Ni2+, which might become bound to nucleic acids or
proteins, or to albumin in the plasma and, ultimately, would be
excreted in the urine; the released CO would become bound to
haemoglobin and finally exhaled.
Oskarsson & Tjälve (1979a) studied the distribution of
intravenously administered 63Ni and 14C-labelled nickel carbonyl
(63Ni(CO4) and Ni(14CO4)) in mice by whole-body autoradiography and
liquid scintillation counting. Radioactivity in the animals given
14C-labelled carbonyl was mainly confined to the blood, indicating
the formation of 14CO-haemoglobin. This confirms the findings of
Kasprzak & Sunderman (1969). After the administration of 63Ni-
labelled carbonyl, the highest level of 63Ni was found in the lung,
followed by the brain, spinal cord, heart, diaphragm, brown fat,
adrenals, and corpora lutea. Additional studies showed that nickel
was present in the lung, brain, heart, and blood as the cation.
6.2.2.4 Nickel levels in human beings
In human beings, wide variations have been reported in body
nickel levels. This makes it difficult to appraise and compare the
results obtained by various investigators. In addition to
variations in the geographical origin of data and individual
dietary and smoking habits, major differences can be attributed to
the analytical methods employed. Only limited comparisons can be
made using variants of spectrography, atomic absorption
spectrometry, photometric methods, and special analytical
techniques. Furthermore, no uniform reference samples have been
used. Nickel values from tissue analyses have been related to ash
or dry weight as well as to wet weight. The normal ranges of
nickel concentrations in body fluids or tissues (serum, blood,
lung, kidney) are not significantly influenced by age, sex, or
pregnancy (McNeely et al., 1971; Turhan et al., 1983; Zober et al.,
1984).
(a) Body fluids, hair, and nasal mucosa
The levels of nickel in biological fluids, hair, and some other
materials increase remarkably in persons with increased
occupational or environmental exposure and decline rapidly when
exposure is reduced or stopped (Tables 19, 20, 21, and 22). Thus,
measurements of nickel, particularly in the urine, serum, or hair,
may serve as indices of exposure.
Data for normal nickel values in urine, blood, plasma, and
serum, published in the last three decades, vary widely. Lower
levels have been obtained by later investigators, because of the
use of more sensitive analytical methods. Reference values in
specimens from healthy, non-exposed persons are listed in Table 19.
Because of doubts about the reliability of older studies, only
recent data have been included.
Table 19. Normal nickel concentrations in specimens from healthy non-exposed
adults
--------------------------------------------------------------------------------
Specimen No. of Nickel concentrations Units References
subjects mean ± SD range
(m/f)
--------------------------------------------------------------------------------
Whole blood 30 (15,15) 0.34 ± 0.28 < 0.05-1.05 µg/litre Linden et al.
(1985)
Serum 10 (6, 4) 0.32 ± 0.17 0.1-0.6 µg/litre Sunderman et
al. (1989a)
Serum 43 (22, 21) 0.2 ± 0.2 < 0.05-1.0 µg/litre Hopfer et al.
(1989)
Serum 30 (15, 15) 0.28 ± 0.24 < 0.05-1.08 µg/litre Linden et al.
(1985)
Lymphocytes 10 (4, 6) 0.72 ± 0.75 < 0.05-1.10 µg/1010 Wills et
cells al. (1985)
Urine (spot 34 (18, 16) 2.0 ± 1.5 0.5-6.1 µg/litre Sunderman
collection) 2.8 ± 1.9 0.5-8.8 µg/litre et al.
(1.024 (1986a)
sp.gr.)a
Urine (24-h 50 (24, 26) 2.2 ± 1.2 0.7-5.2 µg/litre Sunderman
collection) 2.6 ± 1.4 0.5-6.4 µg/day (1977)
Faeces (3-day 10 (6, 4) 14.2 ± 2.7 10.8-18.7 mg/kg Horak &
collection) (dry Sunderman
weight) (1973)
258 ± 126 80-540 µg/day
Faeces 10 (6, 4) 1.5 ± 0.5 1.0-2.2 mg/kg Sunderman
(wet et al.
weight) (1989a)
Sweat 14 (6, 8) 51 ± 38 8-158 µg/litre Christensen
et al.
(1979)
Bile 5(b) 2.3 ± 0.8 15-33 µg/litre Rezuke et
al. (1987)
Saliva 38 (32, 6) 1.9 ± 1.0 0.8-4.5 µg/litre Catalanatto
et al.
(1977)
Hair 102 (b) 0.29 0.0-13.0 mg/kg Bencko et
(net al. (1986)
weight)
--------------------------------------------------------------------------------
Table 19 (contd.)
--------------------------------------------------------------------------------
Specimen No. of Nickel concentrations Units References
subjects mean ± SD range
(m/f)
--------------------------------------------------------------------------------
Hair 22 (15, 7) 0.22 ± 0.08 0.13-0.51 mg/kg Nechay &
(dry Sunderman
weight) (1973)
Hair 905 (437, -c 0.26-2.70 mg/kg Tagaki et
468) (wet al. (1986)
weight)
Nasal mucosa 57 (57m) 0.13 ± 0.20 <0.53d mg/kg Torjussen &
(wet Andersen
weight) (1979)
--------------------------------------------------------------------------------
a Urine nickel concentrations, factored to specific gravity = 1.024.
b Sex not indicated.
c Range of means of samples from five countries.
d Upper 95th percentile of nickel concentrations in nasal biopsies from
non-exposed subjects.
A large number of workers exposed to various nickel compounds
have been found to have elevated levels of nickel in the urine.
These include those working in: nickel refineries (Morgan, 1960;
Kemka, 1971; Norseth, 1975; Hogetveit & Barton, 1976, 1977;
Bernacki et al., 1978a; Hogetveit et al., 1978; Morgan & Rouge,
1979; Torjussen & Andersen, 1979; Hogetveit et al., 1980; Boysen et
al., 1982), the welding of nickel alloy steels (Norseth, 1975;
Bernacki et al., 1978a; Grandjean et al., 1980; Polednak, 1981;
Kalliomäki et al., 1981), nickel electroplating plants (Tandon et
al., 1977; Tola et al., 1979; Bernacki et al., 1980; Tossavainen et
al., 1980), nickel battery factories (Bernacki et al., 1978a;
Adamsson et al., 1980), different occupations in shipyards
(Grandjean et al., 1980), the pigment industry (Tandon et al.,
1977), the glass industry (Raithel et al., 1981), nickel carbonyl
processing in nickel refining (Kincaid et al., 1956; Sorinson et
al., 1958; Morgan, 1960; Nomoto & Sunderman, 1970; Hagedorn-Götz et
al., 1977), aircraft mechanics and metal spraying (Bernacki et al.,
1978a). The results of the investigations of Bernacki et al.
(1978a), who analysed urine samples from nickel-exposed workers in
10 occupational groups, are listed in Table 20.
Serum or plasma nickel levels have been determined in workers
in the following occupations: nickel refining (Hogetveit & Barton,
1976, 1977; Hogetveit et al., 1978, 1980; Torjussen & Andersen,
1979; Boysen et al., 1982), welding (Grandjean et al., 1980),
electroplating (Tola et al., 1979; Tossavainen et al., 1980),
battery manufacture (Adamsson et al., 1980) and shipyards
(Grandjean et al., 1980), and the Mond process in nickel refining
(Sorinson et al., 1958; Nomoto & Sunderman, 1970).
Table 20. Nickel concentrations in urine of workers in various occupational groupsa
-------------------------------------------------------------------------------------
Occupation No. Description Concentration
Atmospheric Urineb Creatinineb
nickel (µg/m3) (µg/litre) (µg/litre)
-------------------------------------------------------------------------------------
External 9 abrasive wheel 1.6±3.0 5.4±2.4 3.5±1.6
grinders grinding of exteriors (0.02-9.5) (2.1-8.8) (1.7-6.1)
of articles made of
nickel alloys
Arc welders 10 DC arc welding of 6.0±14.3 6.3±4.1c 5.6±6.2
aircraft made of (0.2-46) (1.6-14) (1.1-17)
nickel alloys
Bench mechanics 8 assembling, fitting, 52±94 12.2±13.6c 7.2±6.8c
and finishing parts (0.01-252) (1.4-41) (0.7-20)
made of nickel alloys
Nickel battery 6 fabricating nickel- Not 11.7±7.75d 10.2±6.4d
workers cadmium or nickel- measured (3.4-25) (7.2-23)
zinc electrical
storage batteries
Metal sprayers 5 flame-spraying 2.4±2.6 17.2±9.8d 16.0±21.9
nickel-containing (0.04-2.1) (1.4-26) (1.4-54)
powders in phase on
to aircraft parts
Electroplaters 11 intermittent exposure 0.8±0.9 10.5±8.1d 5.9±5.0c
to nickel in combined (0.04-2.1) (1.3-30) (1.0-20)
electro-deposition
operations involving
silver, cadmium,
chromium plating, as
well as nickel
Nickel platers 21 full-time work in Not 27.5±21.2e 19.0±14.7e
nickel plating measured (3.6-65) (2.4-47)
operations
Nickel refinery 15 workers in a nickel 489±560 222±226e 124±109e
refinery using (20-2200) (8.6-8.3) (6.1-287)
electrolytic processes
-----------------------------------------------------------------------------------
a From: Bernacki et al. (1978a).
b Mean SD with range in parentheses.
c P < 0.05 versus controls, calculated by t-test.
d P < 0.01 versus controls.
e P < 0.001 versus controls.
The highest nickel concentrations were found in the body fluids
of nickel refinery workers. Concentrations in workers in
electroplating shops, battery factories, and aircraft engineering
works were lower (Table 10). After occupational exposure to nickel
in electroplating processes, biological half-times ranging from 13
to 39 h for nickel in urine and from 20 to 34 h for nickel in
plasma have been reported (Tossavainen et al., 1980).
Serum specimens of 22 residents of Sudbury, Ontario, who had
been environmentally exposed to nickel (including air and tap
water), contained nickel concentrations ranging from 0.2 to 1.3
µg/litre (mean 0.6±0.3 µg/litre). These were significantly higher
than the serum levels of residents without environmental exposure
(Hopfer et al., 1989).
Nickel determinations in blood and urine, are widely used and
accepted methods for monitoring nickel exposure. Although more
data are available for urine, no clear-cut choice can be made
between the use of blood or urine.
Grandjean et al. (1980, 1988) reported that analyses of nickel
concentrations in both urine and plasma samples should be obtained
to assess worker exposure. There were significantly higher ratios
of plasma/urine nickel levels in painters and lower ratios in
welders, compared with other workers in the shipyard. These
differences probably reflected the different toxicokinetic
characteristics of the nickel compounds to which the workers were
exposed.
The correlation between exposure levels and nickel
concentrations in body fluids is poor in most studies (Table 21).
The closest positive relationships of nickel concentrations in body
fluids with ambient air levels were found by Norseth (1975) and
Rahkonen et al. (1983) in welders, and by Tola et al. (1979) and
Bernacki et al. (1980) in electroplaters.
Table 21. Studies on the correlation between nickel concentrations
in the air and in biological fluids in occupational exposure to
nickel compoundsa
-------------------------------------------------------------------
Exposure Biological Correlation Reference
matrix coefficient
(r)
-------------------------------------------------------------------
welding urine 0.85 Norseth (1975)
roasting-smelting urine none
welding urine none Bernacki et al.
bench mechanics urine none (1978a)
electroplating urine none
metal spraying urine none
refinery urine none
roasting-smelting plasma -0.11 Hogetveit et al.
urine 0.14 (1978)
electrolysis plasma 0.21
urine 0.31
refinery (other) plasma 0.67
urine 0.47
-------------------------------------------------------------------
Table 21 (contd.)
-------------------------------------------------------------------
Exposure Biological Correlation Reference
matrix coefficient
(r)
-------------------------------------------------------------------
refinery (nickel urine 0.49; 0.55 Morgan & Rouge
salts) (1979)
refinery:
Mond process urine 0.01
calciner urine 0.22
powder plant urine -0.05
electroplating plasma 0.83 Tola et al.
urine 0.82b (1979)
urine 0.96c
electroplating urine 0.70d Bernacki et al.
(1980)
battery urine significante Adamsson et al.
manufacturing (1980)
welding blood 0.56 Rahkonen et al.
urine 0.95 (1983)
-------------------------------------------------------------------
a From: Aitio (1984).
b Afternoon.
c Next morning.
d After shift.
e P < 0.01.
There seem to be at least three reasons for the inconsistencies
in the correlation between exposure levels and biological
measurements of nickel:
(a) exposure is not to a single chemical species, but to a variety
of nickel compounds of very different solubility, absorption,
transportation, and elimination rates. The same workers may be
simultaneously exposed to both insoluble and readily soluble
compounds, having half-lives ranging from days (Tola et al.,
1979) to years (Torjussen & Andersen, 1979). The influence of
the nickel species in ambient air on the concentration in body
fluids has been shown in a study by Bernacki et al. (1978a),
who found widely varying ratios of air/urine levels in
different occupational groups (Table 22);
(b) differences in personal working habits and hygiene;
(c) failure to standardize sampling methods.
Table 22. Nickel concentrations in serum, urine, nasal mucosa, and personal air samples
from workers at the Falconbridge Nickel Refinery in Kristiansand, Norwaya
-----------------------------------------------------------------------------------------
Category of No. of Plasma nickel Urine nickel Nasal mucosal Air nickel
subjects/work subjects (µg/litre) (µg/litre) nickel (mg/m3)
mean±SD mean±SD (µg/100g) mean±SD
(wet weight)
-----------------------------------------------------------------------------------------
First studyb
Roasting/smelting 24 7.2±2.8 65±58 0.86±1.20
Electrolysis 90 11.9±8.0 129±106 0.23±0.40
Other process 13 6.4±1.9 45±27 0.42±0.49
Second studyc
Controls 57 1.9±1.4 4.9±4.2 13±20
Roasting/smelting 97 5.2±2.7 34±35 467±595
Electrolysis 144 8.1±6.0 73±85 178±235
Other process 77 4.3±2.2 22±18 211±301
-----------------------------------------------------------------------------------------
a From: Sunderman et al. (1986b).
b From: Hogetveit et al. (1978).
c From: Torjussen & Andersen (1979).
The nickel contents of hair and nasal mucosa have been
determined in occupationally-exposed persons. Theoretically, the
nasal mucosa is one of the target tissues for nickel
carcinogenicity. However, practical problems of sampling and
standardization preclude the routine use of these measurements.
Torjussen & Andersen (1979) analysed biopsy specimens of nasal
mucosa from 318 nickel workers, 15 retired nickel workers, and 57
unexposed controls. The results showed that nickel exposure led to
significantly raised nickel concentrations in the nasal mucosa in
both active and retired nickel workers (2.74±4.12 mg/kg wet weight,
and 1.14±1.78 mg/kg wet weight, respectively, versus 0.13±0.2 mg/kg
wet weight in the controls). The average nickel concentration in
the nasal mucosa was highest in workers exposed to the highest
atmospheric nickel concentration, inhaled as nickel subsulfide and
nickel oxide dust. Workers exposed to aerosols of soluble nickel
components, such as the chloride and sulfate, at a lower
atmospheric nickel concentration, had the highest mean nickel
concentrations in the plasma and urine and the lowest in the nasal
mucosa. The mucosal concentration was significantly correlated
with the duration of nickel exposure.
For hair, normal values ranged between 0.13 mg/kg (dry weight)
and 2.7 mg/kg (wet weight). In hair samples from 45
occupationally-exposed adults, Bencko et al. (1986) found nickel
concentrations ranging from 1.6 to 3.5 mg/kg (mean 2.39 mg/kg) in
welders and from 42.7 to 2140 mg/kg (mean 222.5 mg/kg) in nickel
smelter workers. Control values ranged from 0 to 13 mg/kg (mean
0.29 mg/kg). In an accidental case of exposure to nickel carbonyl,
nickel concentrations in hair samples from 5 workers ranged from 4
to 48.1 mg/kg (Hagedorn-Götz et al., 1977).
Although hair has been studied as a rapid, non-invasive
measurement of exposure/absorption relationships, conflicting
values have been obtained. Furthermore, the use of hair as an
internal exposure index is controversial because of various
factors, such as the external contamination of the hair surface,
different sampling methods, and non-standardized cleaning methods.
(b) Tissues
There are few data on human tissue concentrations of nickel.
Spectrographic analyses indicate that the retained nickel is widely
distributed in very low concentrations in the body. Information on
normal nickel levels in organs and tissues is presented in Tables
23 and 24.
Table 23. Reference values for nickel concentrations in human autopsy tissuesa
--------------------------------------------------------------------------------------
Nickel concentrations
Tissue No. of Wet weight (µg/kg) Dry weight (µg/kg) Reference
subjects Mean±SD Range Mean±SD Range
--------------------------------------------------------------------------------------
Lung 4 16±8 (8-24) 86±56 (33-146) Sunderman et al. (1971)
9 132±99 (50-290) Chen et al. (1977)
41 7±10 (<1-70) Zober et al. (1984)
15 180±105 (43-361) Seemann et al. (1985)
9 18±12 (7-46) 173±94 (71-371) Rezuke et al. (1987)
15 44±56 (16-242) Raithel (1987)
Kidney 6 125±54 (50-120) Chen et al. (1977)
36 14±27 (<1-165) Zober et al. (1984)
18 34±22 (<5-84) Seemann et al. (1985)
10 9±6 (3-25) 62±43 (19-171) Rezuke et al. (1987)
Liver 4 9±3 (5-13) 32±12 (21-48) Sunderman et al. (1971)
23 18±21 (<5-86) Seemann et al. (1985)
10 10±7 (8-21) 50±31 (11-102) Rezuke et al. (1987)
Heart 4 6±2 (4-8) 23±6 (16-30) Sunderman et al. (1971)
9 8±5 (1-14) 54±40 (10-110) Rezuke et al. (1987)
Spleen 22 23±20 (<5-85) Seemann et al. (1985)
10 7±5 (1-15) 37±31 (9-95) Rezuke et al. (1987)
--------------------------------------------------------------------------------------
a Adapted from: Rezuke et al. (1987).
Table 24. Normal nickel concentration in Japanese human
tissues (mg/kg wet weight)a
---------------------------------------------------------------
Tissue Sex/ Median Average Range Mean±SD
number
---------------------------------------------------------------
Rib M/6 0.19
0.230 0.13-0.35 0.23±0.07
F/6 0.27
Lung M/15 0.21
0.160 0.04-0.44 0.16±0.09
F/15 <0.10
Small M/5 0.11
intestine 0.120 0.05-0.29 0.13±0.07
F/5 0.15
Large M/5 0.14
intestine 0.111 0.04-0.30 0.14±0.10
F/5 0.15
Trachea M/3 0.09
0.098 0.06-0.11 0.09±0.02
F/1 0.11
Kidney M/14 0.10
0.081 0.01-0.30 0.10±0.07
F/14 0.10
Skin M/4 0.09
0.072 0.02-0.22 0.10±0.08
F/2 0.14
Muscle M/5 0.11
0.070 0.02-0.27 0.10±0.08
F/5 0.09
Liver M/14 0.10
0.068 0.03-0.22 0.08±0.05
F/13 0.05
Cerebrum M/2 0.06
0.025 0.02-0.11 0.05±0.11
F/1 0.03
Cerebellum M/1
NMb <0.03 NIc NM
F/1
Heart NM NCd NC NM
Pancreas M/6
NM <0.10 NM NM
F/2
---------------------------------------------------------------
Table 24 (contd.)
---------------------------------------------------------------
Tissue Sex/ Median Average Range Mean±SD
number
---------------------------------------------------------------
Spleen M/1 <0.30
NM NM NM
Adrenal M/1 <0.10
glands NM NM NM
Testis M/1 0.05
Ovary NM - NM NM
Fat F/3 NM <0.01 NI NM
---------------------------------------------------------------
a From: Sumino et al. (1975).
b NM = not measured.
c NI = not indicated.
d NC = not calculated because there were less than 5 samples
available or there was no mean.
Generally, there is no significant influence of sex or age on
human organ levels of nickel (McNeely et al., 1971a; Turhan et al.,
1983; Zober et al., 1984). The ribs, liver, and kidneys in babies
up to the age of 3 months, were found to accumulate significantly
more nickel than those in persons between 1 and 90 years of age
(Schneider et al., 1980). Few data exist on the organ levels of
nickel in occupationally exposed persons. In lung autopsy samples
from 4 deceased persons living in the vicinity of a nickel-
processing industry in the German Democratic Republic, the mean
nickel concentration was 2135±1867 mg/kg dry weight (Schneider et
al., 1980).
(c) Body burden
One assessment of nickel metabolism in human beings indicated
that the body burden of nickel in normal adults averaged 0.5 mg (7
µg/kg for a 70-kg adult person) (Bennett, 1984). However, in a
study on 30 Japanese subjects, Sumino et al. (1975) calculated a
total body burden of about 5.7 mg (for a body weight of 55 kg), and
Schroeder et al. (1962) indicated a body burden of 10 mg nickel for
an adult person. Bennett (1984) concluded that the oral intake of
nickel averaged 170 µg/day, of which approximately 5% would be
absorbed (8.5 µg/day). Inhalation of nickel averaged 0.4 µg/day
for urban dwellers, of which 35% was retained (0.07-0.14 µg/day);
this involves the assumption that 70% of the nickel absorbed into
the blood is promptly excreted by the kidneys and that the
remaining 30% is deposited in the tissues, with a mean retention
time of 200 days (Bennett, 1984).
6.2.2.5 Pathological states influencing nickel levels
Nickel metabolism is known to be altered in several common
diseases, as well as in some physiological states. Alonzo & Pell
(1963) observed increased nickel concentrations in the serum of 19
out of 20 patients with acute myocardial infarction, sampled within
24 h of admission to the hospital. Sunderman et al. (1970, 1971)
reported increased nickel concentrations in the serum of 25 out of
35 patients with acute myocardial infarction, sampled 12-36 h after
onset of symptoms. The frequent occurrence of hypernickelaemia
after acute myocardial infarction has been confirmed by studies in
the Federal Republic of Germany (Völlkopf et al., 1981), Pakistan
(Khan et al., 1984), the United Kingdom (Howard, 1980), the USA
(Leach et al., 1985), and the USSR (Nozdryukhina, 1978). McNeely
et al. (1971a) showed that hypernickelaemia is not specific for
myocardial infarction, because nickel concentrations in serum are
also increased in patients with cerebral stroke and thermal burns,
as well as in patients with myocardial ischaemia without
infarction. Hypernickelaemia has been observed in patients with
unstable angina pectoris, without infarction, and in patients
suffering from coronary atherosclerosis, who developed cardiac
ischaemia during treadmill exercise (Leach et al., 1985).
Volini et al. (1968) observed increased nickel concentrations
in the liver in both the early and advanced stages of hepatic
cirrhosis. In a patient suffering from aspartylglycosaminurea, a
10-fold increased concentration of nickel in the hepatic tissue was
reported by Palo & Savolainen (1973).
Significantly decreased serum-nickel levels have been measured
in steel-mill workers exposed to extreme heat (Szadkowski et al.,
1969b).
In an investigation by Rubanyi et al. (1982a), serum-nickel
levels in postpartum mothers were found to be reduced by 60%.
However, a significant 20-fold elevation in the concentration of
nickel was observed immediately after delivery of the infant, but
before delivery of the placenta. Nomoto et al. (1983) did not
confirm the occurrence of hypernickelaemia. Post-operative
hypernickelaemia and nickeluresis were observed in patients
following total knee and hip arthroplasty with porous coated nickel
alloy prostheses (Sunderman et al., 1989b).
Nickel concentrations in the serum, whole blood, and urine from
61 patients with chronic alcoholism were elevated 17-, 15-, and 39-
fold, respectively, after 4 months to 4 years of disulfiram
treatment. Disulfiram (tetraethyl-thiuram-disulfide), a nickel-
chelating agent, is used in alcoholism therapy (Hopfer et al., 1987).
6.3. Elimination and excretion
The elimination routes for nickel in human beings and animals
depend, in part, on the chemical form of the compound and the mode
of intake. In general, relatively low gastrointestinal absorption
explains the elimination of dietary nickel in the faeces. In human
beings and animals, urinary excretion is usually the major
clearance route for absorbed nickel. Other routes of elimination
are of minor importance. All body secretions appear to have the
ability to excrete nickel; it has been found in saliva, sweat,
tears, and milk. Biliary excretion is minimal in animals, but may
be significant in human beings. Hair is an excretory tissue for
nickel.
6.3.1. Experimental animals
In experimental animals, urinary excretion is the main
clearance route for nickel compounds, introduced parenterally.
Only a small portion of an injected dose is excreted via the
gastrointestinal tract. Wase et al. (1954) studied the
distribution and elimination of 63Ni in mice using a high dose (102
µg 63Ni/animal), administered intraperitoneally, and found faecal
and urinary elimination in the ratio of 30:70%. The primary route
of elimination of supplemental dietary nickel (carbonate) fed to
calves was faecal (Tedeschi & Sunderman, 1957; O'Dell et al.,
1971).
Biliary excretion of nickel was minimal following subcutaneous
injection of 0.1 mg 63Ni, as nickel chloride, in rats (Marzouk &
Sunderman, 1985).
In rats or rabbits, after inhalation of nickel carbonyl,
Sunderman & Selin (1968) and Mikheyev (1971) found the lungs were
the major excretory organ besides excretion via the urine, 2-4 h
after exposure. Other studies indicated that up to 90% nickel was
excreted in the urine (Armit, 1908; Tedeschi & Sunderman, 1957;
Sunderman & Selin, 1968; Mikheyev, 1971) and 38% was exhaled via
the lungs as nickel carbonyl (Sunderman & Selin, 1968).
After intravenous injection of 14C-nickel carbonyl in rats,
Kasprzak & Sunderman (1969) found that 30% of the 14C was excreted
in the expired air as 14C-nickel carbonyl and 50% as 14C-carbon
monoxide.
Nickel is excreted in the urine, not as the free metal, but
bound to a protein that is similar to, or a fragment of, the
soluble low relative molecular mass glycoprotein associated with
nickel in renal tissue (Verma et al., 1980; Abdulwajid & Sarkar,
1983).
6.3.2. Human beings
As human beings take up most nickel via ingestion, it is
eliminated unabsorbed, mainly in the faeces (Drinker et al., 1924;
Tedeschi & Sunderman, 1957; Sunderman et al., 1963; Nodiya, 1972).
Horak & Sunderman (1973) found that the faecal elimination of
nickel in 10 healthy adults averaged 258 µg/day (SD 126) or 14.2
mg/kg, (dry weight) (SD 2.7), thus, the normal faecal elimination
of nickel was approximately 100 times greater than the normal
urinary excretion (2.6 µg/day (SD 1.4) or 2.2 µg/litre (SD 1.2)).
The urinary nickel levels of persons occupationally exposed to
appreciable nickel concentrations, via inhalation at the work-
place, are raised significantly. Positive correlations have been
reported between air and urinary nickel concentrations in workers
in the nickel industry (section 6.2.2.4). Urinary nickel
concentrations of normal and exposed persons are given in Tables 19
and 20. The large amounts of nickel also found in the faeces, in
some cases, indicated that, either retrograde loss from the lung
into the oesophagus, or considerable oral exposure via contaminated
surfaces, had occurred.
Nickel concentrations in samples of human bile (section
6.2.2.4) suggest that biliary excretion of nickel may be
quantitatively significant in human beings (Rezuke et al., 1987).
Sweat may constitute an excretory route of significance under
conditions of physical exertion. Hohnadel et al. (1973)
demonstrated that, in sauna bathers, the mean concentrations of
nickel in the sweat from healthy men and women were significantly
higher than the mean concentrations in the urine (men: 52 µg/litre,
SD=36; women: 131 µg/litre, SD=65). Under conditions of profuse
sweating, appreciable losses of nickel occurred. This may account
for the diminished concentrations of serum nickel that were
reported by Szadkowski et al. (1969b) in blast-furnace workers who
were exposed to extreme heat over a long period.
The role of nickel deposition in human hair as an excretory
mechanism has been studied (section 6.2.2.4).
Measurements of salivary nickel were performed by Catalanatto
et al. (1977) on specimens of parotid saliva from 38 healthy
adults. The concentrations of nickel in saliva averaged 1.9 ± 1.0
µg/litre (range 0.8-4.5 µg/litre). There was no significant
correlation between the concentrations of salivary nickel and
protein. No significant differences were observed between the mean
concentrations of nickel in saliva samples from men and women.
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
7.1. Microorganisms
Nickel is considered essential for certain metabolic processes
in bacteria. Bartha & Ordal (1965) demonstrated a nickel
requirement in the "Knallgas" bacterium Alcalignes entrophus. A
nickel requirement was reported by van Baalen & O'Donnell (1978)
for the blue green algae Oscillatoria sp.
Fungi and microorganisms demonstrate a fairly wide variety of
sensitivity to nickel, but are generally more tolerant than the
higher organisms. The toxic effects of nickel on microorganisms,
including eubacteria (non-marine and marine), actinomycetes,
yeasts, and filamentous fungi, were studied by Babich & Stotzky
(1982a,b; 1983). Filamentous fungi varied considerably in their
response to nickel, growth of Achyla sp. being inhibited at 5 mg
nickel/litre whereas Aspergillus niger and Gliocladium sp. were
only affected at concentrations as high as 1000 mg nickel/litre.
With actinomycetes and eubacteria, there was less variability in
toxicity. Concentrations of nickel inhibiting growth ranged from 5
to 30 mg nickel/litre, with the exception of Caulobacter leidyi,
which exhibited some growth at 100 mg nickel/litre. Growth
inhibition in yeasts occurred at 1-40 mg nickel/litre. In all
microorganisms, toxicity increased as the pH decreased.
Babich & Stotzky (1983) investigated the influence of various
factors on the toxicity of nickel for eubacteria, an actinomycete,
and yeasts. Reductions in cell number occurred at 5 or 10 mg/litre
and viable cells were eliminated at 10-50 mg/litre, though some
species were unaffected after 24 h at 100 mg/litre. Reductions in
pH from 6.8 to 5.3 enhanced the toxicity of 75 mg nickel/litre in
some species, but not in others. The toxicity of nickel (100
mg/litre) for marine microbes was reduced by increasing the
salinity and decreasing the temperature. Addition of a simulated
sediment (a mixture of organic and inorganic particles from soil)
reduced toxic effects after exposure to 100 mg nickel/litre. In
freshwater microbes, addition of a clay mineral (50 mg/litre)
provided protection against the toxicity of 10 mg nickel/litre.
This effect was probably because of the adsorption of nickel on the
particulates. Increasing the hardness of the water, by adding
calcium carbonate at 200 or 400 mg/litre, reduced the toxicity of
10 mg nickel/litre. Long-term studies indicated that microbial
survival was greater in marine than in fresh water. Bringmann et
al. (1980) reported that, in fairly hard water (approximately 150
mg CaCO3/litre) and a pH of 6.9, a nickel concentration of 0.82
mg/litre reduced the numbers of the saprozoic flagellate Chilomonas
paramecium.
Thus, fungi and microorganisms have a wide range of
sensitivities to nickel, but are usually more tolerant than higher
organisms.
7.2. Aquatic algae and plants
Nickel at concentrations of 0.05-0.1 mg/litre inhibited the
growth of algae, though some species may be more tolerant (Spencer,
1980). Upitis et al. (1980) reported growth inhibition in blue-
green algae at concentrations of 1-5 mg nickel/litre. The
chlorophyll content was found to be significantly reduced leading
to discoloration of the cells. Concentrations of 10-30 mg
nickel/litre were lethal for Chlorella sp. The same authors
investigated the influence of various environmental factors on the
toxicity of nickel for Chlorella. Nickel inhibition could be
overcome by the addition of ethylenediaminetetramine (EDTA) (40
mg/litre) and also by the addition of zinc (10 mg/litre) to a
medium containing 5 mg nickel/litre. A synergistic effect of
copper and nickel was demonstrated by Hutchinson (1973) for
Chlorella vulgaris, Scenedesmus acuminata, Haematococcus capensis,
and Chlamydomonas eugametos.
A nickel concentration of 0.1 mg/litre at 20 °C inhibited the
growth of 4 species of green algae, Pediastrum tetras,
Ankistrodesmus falcatus, Scenedesmus quadricauda, and S. dimorpha.
However, a concentration of 0.6 mg/litre did not affect the blue-
green alga Anabaena cylindrica, though it reduced the rate of
growth of Anabaena flos-aquae (Spencer & Greene, 1981). Stokes
(1975) studied Scenedesmus acutiformis var. alternans, from a lake
in a nickel-mining and smelting area. The lake water contained
about 2.5 mg nickel/litre. At a nickel concentration of 1.9
mg/litre, the Scenedesmus grew at 53% of the rate of controls grown
in clean water and, at 3.0 mg/litre, growth was still 18% of the
control rate.
A nickel concentration of 0.125 mg/litre inhibited the growth
of Anabaena inequalis, but a concentration of 10 mg/litre was
required to inhibit photosynthesis, and 20 mg/litre, to inhibit
nitrogenase activity (Stratton & Corke, 1979).
Chiaudani & Vighi (1978) exposed Selenastrum capricornutum to
nickel in a standard medium with, and without, EDTA. At 24 °C and
a pH of 6.9-6.3 the 7-day EC50 (inhibition of growth to 50% of
control values) was 0.9925 mg nickel/litre. When 0.3 mg EDTA/litre
was added to medium, the 7-day EC50 of nickel was increased to
0.013 mg/litre. In a further study, the addition of 0.04 mg
nickel/litre to the water samples did not inhibit growth as much as
was predicted from the laboratory studies.
When the diatom Navicula pelliculosa was exposed to 0.1 mg
nickel/litre (of which all but 0.2% was said to be Ni2+) for 14
days, growth was retarded (50% of control value) (Fezy et al.,
1979).
Hutchinson & Czyrska (1975) exposed Lemna minor (Valdiviana),
for 3 weeks, to nickel concentrations ranging from 0.01 to 1.0
mg/litre in an artificial medium at pH 6.8, and a temperature of
24±2 °C, with 16 h of light per 24 h. They found that a
concentration of 0.05 mg/litre stimulated growth, and that
concentrations greater than 0.1 mg/litre inhibited growth. At 1 mg
nickel/litre, growth was prevented. Nickel uptake and toxicity
were enhanced by the presence of copper.
The same authors examined Lemna minor from 23 sites, where the
mean concentration of nickel in the water was 0.027 mg/litre. The
plants contained from 5.4 to 35.1 mg nickel/kg (dry weight),
equivalent to bioaccumulation factors (BCFs) of 200 and 1300.
Lemna, grown on a culture medium containing 0.01-1 mg nickel/litre
at pH 6.8 and a temperature of 24±2 °C for 3 weeks, accumulated
nickel concentrations ranging from 40 mg/kg dry weight, at 0.01
mg/litre, to 3067 mg/kg, at 0.5 mg/litre.
Clark et al. (1981) studied the accumulation and depuration of
nickel by Lemna perpusilla. Plants collected from a fly ash basin
(nickel concentration, 0.1 mg/litre) were allowed to depurate in
dechlorinated tap water at 20 °C for a 14-day depuration period.
Concentrations of nickel fell from about 160 mg/kg dry weight to
about half of this value. In the accumulation studies, Lemna
accumulated nickel readily, particularly at the lowest ambient
concentration of 0.1 mg/litre, and levels reached 500-600 mg/kg in
10 days. After a return to depuration conditions, the nickel
concentration in the plants fell to 160 mg/kg, in 8 days.
Euglena gracilis, exposed to 8.9 x 10-4 mg nickel/litre in
spring-water, accumulated the metal to a concentration of 1.8
mg/kg, a BCF of about 2000 (Cowgill, 1976).
Ipomea aquatica took up 200 mg nickel/kg dry plant in 48 h,
mostly in the roots, from water containing 5 mg nickel/litre (BCF =
40) (Low & Lee, 1981).
It is noted that, under laboratory conditions, the growth of a
macrophyte (Lemna) was inhibited at a concentration of 0.1
mg/litre, but the growth of algae was inhibited at concentrations
as low as 0.04 mg/litre. However, in natural waters, a nickel
concentration of 0.04 mg/litre had a less inhibiting effect.
7.3. Aquatic invertebrates
Timourian & Watchmaker (1972) investigated the uptake of nickel
chloride and its effects on the development of sea urchin embryos.
After fertilization, sea urchin eggs exhibited increased rates of
nickel uptake that appeared to be a result of an active transport
mechanism. When exposed to 59-590 mg nickel/litre, gastrulation of
embryos was prevented. Embryos grown in 0.59-5.9 mg nickel/litre
were able to gastrulate, but failed to develop dorsoventral symmetry.
Acute and long-term toxicity studies performed by Powlesland &
George (1986) revealed a different sensitivity to nickel in
different developmental stages of Chironomus riparis larvae. First
instar larvae were found to be significantly more sensitive to
nickel than second instars with 48-h LC50 values of 79.5 mg/litre
and 169 mg/litre, respectively. Longer term toxicity tests (30
days), in which larvae were allowed to develop from eggs until just
prior to pupation, indicated that nickel concentrations up to 25
mg/litre appeared to have little effect on the percentage hatch.
However, the growth of larvae was significantly reduced at 2.5
mg/litre. A threshold concentration for the effect of nickel on
growth was estimated to be 1.1 mg nickel/litre.
Bryant et al. (1985) investigated the effects of temperature
and salinity on the toxicity of nickel in two estuarine
invertebrates. In the amphipod Corophium volutator, and the
bivalve Macoma baltia, 96-h LC50 values varied from 5 to 54 mg
nickel/litre and from 95 to 1100 mg nickel/litre, respectively. A
decrease in salinity from 35 to 5 mg/litre resulted in greater
toxicity in both species. Toxicity also increased in Corophium
volutator with an increase in temperature from 5 to 15 °C.
Mathis & Cummings (1973) measured nickel levels in sediments,
water, and biota in a river. The water contained the lowest
concentrations of nickel (<0.01 mg/litre) and the sediments, the
highest (3-124 mg/kg). Two species of tubificid worms (Limnodrilus
hoffmeisteri and Tubifex tubifex) contained 4-18 mg nickel/kg wet
weight. Three species of clam were examined: in order of
increasing nickel content (mg/kg on a wet-weight basis) they were
Quadrula quadrula (0.4-1.6), Amblema plicata (0.4-2.3) and
Fusconaia flava (0.7-3.0). Neither worms nor clams were starved
before being examined, and nickel may also have been present in
their gut contents. Brkovic-Popovic & Popovic (1977) studied the
effects of nickel and other heavy metals on the survival of Tubifex
tubifex in water of different pH and hardness. At a hardness of
0.1 mg CaCO3/litre, the 48-h LC50 was 0.082 mg nickel/litre.
Increases in hardness to 34.2 mg CaCO3/litre and 261 mg CaCO3/litre
increased the 48-h LC50 to 8.7 mg nickel/litre and 61.4 mg
nickel/litre, respectively, thus decreasing the toxic effects.
A 64-h LC50 was determined for Daphnia magna of 0.32 mg
nickel/litre, at a temperature of 25 °C and a hardness of around
100 mg CaCO3/litre (Anderson, 1950). Baudouin & Scoppa (1974),
using Daphnia hyalina, estimated a 48-h LC50 value of 1.9 mg
nickel/litre at a temperature of 10 °C, pH 6.2, and hardness of 58
mg CaCO3/litre.
Exposure of Daphnia magna to nickel sulfate at concentrations
ranging from 5 to 10 µg nickel/litre for 3 generations resulted in
extermination (Lazareva, 1985).
Hall (1982) exposed Daphnia magna to 0.25 mg nickel/litre,
including 63Ni, at pH 6.9, and a temperature of 18-21 °C in water
with a hardness of 60 mg CaCO3/litre. The uptake of nickel was
initially rapid (about 12 mg in 80 h). Depuration also occurred,
and 25-33% of the nickel was lost from the animal in the exuviae,
shed on moulting. Gut tissue did not accumulate nickel until after
the first 5 h of exposure, suggesting that the oral route was not
important for nickel.
Daphnia, exposed for 3 weeks to 0.125 mg nickel/litre in water,
at a temperature of 18±1 °C, hardness of 42.3-45.3 mg CaCO3/litre,
and pH 7.74, had 43% lower weights than control Daphnia, 9% less
proteins, and the glutamic oxalacetic transaminase activity was
reduced by 26%. A 16% impairment of reproduction occurred at 0.03
mg nickel/litre with a 50% impairment at 0.095 mg nickel/litre
(Biesinger & Christensen, 1972).
Cowgill (1976) reared Daphnia magna and Daphnia pulex for 3
months on Euglena gracilis, which had been cultured in spring water
containing 8.9 x 10-4 mg nickel/litre. The algal cells contained
1.8 mg nickel/kg, the Daphnia magna, 3.6 mg nickel/kg, and the D.
pulex, 4.2 mg nickel/kg, giving BCF values of 2020 and 4050.
The acute effects of nickel on the freshwater snails Juga
plicifera and Physa gyrina were studied by Nebeker et al. (1986).
The 96-h LC50 values were 0.237 mg nickel/litre and 0.239 mg/litre,
respectively. A no-observed-effect-level of 0.124 mg nickel/litre
was determined for Juga plicifera. Data published for other
species of snails did not indicate a pronounced effect of hardness
on nickel toxicity (Nebeker et al., 1986). The eggs and adults of
the snail Amnicola were exposed to nickel in water at a temperature
of 17 °C, pH 7.6, and a hardness of 50 mg/litre with 6.2 mg
dissolved oxygen/litre. The 24-h LC50s were 26.9 mg/litre for eggs
and 21.1 mg/litre for adults, whereas at 96 h, the LC50s were 11.4
and 14.3 mg/litre, respectively (Rehwoldt et al., 1973).
Nickel influenced the rate of filtration in the marine bivalve
Villorita cyprinoides (Abraham et al., 1986). Rates of filtration
decreased exponentially with increasing nickel levels. The EC50,
i.e., the concentration that reduced the rate of filtration by 50%,
was 0.003 mg nickel/litre. A 96-h LC50 was 0.061 mg nickel/litre.
It is concluded that the nickel concentrations causing mortality in
acutely exposed invertebrates were generally similar to those for
fish, but that Daphnia sp. appeared more sensitive, with LC50
values of less than 2 mg nickel/litre.
7.4. Fish
Sensitivity to nickel varies considerably among fish species.
However, 96-h median lethal concentrations generally fall within
the ranges of 4-14 and 24-44 mg nickel/litre for tests conducted in
soft, and hard water, respectively. For example, in water of
hardness 100, 125, and 174 mg CaCO3/litre, the LC50s for rainbow
trout, exposed from fertilization to 4 days after hatching, were
0.05, 0.06, and 0.09 mg/litre, respectively (Birge & Black, 1980).
Pickering & Henderson (1966) compared the toxicity of nickel
chloride in waters of 2 levels of hardness (total hardness 20 or
300 mg CaCO3/litre). The 96-h LC50 was 4.9 mg/litre for fathead
minnow (Pimephales promelas) and 5.3 mg/litre for bluegill sunfish
(Lepomis macrochirus) in soft water, and 43.5 mg/litre and 39.6
mg/litre, respectively, in hard water. Rainbow trout (Salmo
gairdneri) showed a 4-fold increase in sensitivity between hard and
soft water, the 48-h LC50 changing from about 80 to about 20
mg/litre (Brown, 1968).
In rainbow trout, a 48-h LC50 for nickel sulfate of 263
mg/litre was determined in a static test (Osterreichisches
Forschungs-Zentrum Seibersdorf, 1983). The water had a hardness of
402 mg CaCO3/litre, a pH of 7.6, and a temperature of about 15 °C.
The first signs of toxicity were observed at a concentration of 85
mg nickel sulfate/litre.
Using the same test method and corresponding test conditions,
Butz (1984) demonstrated that a decrease in water hardness from 270
to 49 mg CaCO3/litre resulted in a 2.6-fold increase in toxicity.
Rehwoldt et al. (1971, 1972) studied the effects of temperature
on the toxicity of nickel for 6 warm-water species of fish: banded
killifish (Fundulus diaphanus), striped bass (Roccus saxatilis),
pumpkin seed (Lepomis gibbosus), white perch (Roccus americanus),
American eel (Anguilla rostrata) and carp (Cyprinus carpio). There
was a wide range of sensitivity among these species, with 96-h LC50
values at 17 °C ranging from 6.2 to 46.2 mg nickel/litre. However,
each species showed very little variation in sensitivity at
temperatures of 17 and 28 °C. At 28 °C and a hardness of 55 mg
CaCO3/litre, Roccus saxatilis and Lepomis gibbosus were the most
sensitive, having 96-h LC50s of 6.3 and 8.0 mg nickel/litre,
respectively, while Fundulus diaphanus was the most tolerant (46.1
mg nickel/litre). Roccus americanus, Anguilla rostrata, and
Cyprinus carpio were intermediate in their response, but relatively
sensitive, the 96-h LC50s being 13.7, 13.9, and 10.4 mg
nickel/litre, respectively (Rehwoldt et al., 1972).
In static tests in softer water (20 mg CaCO3/litre), Pimephales
promelas, Lepomis macrochirus, Carassius auratus, and Lebistes
reticulatus showed similar levels of sensitivity in terms of 96-h
LC50s with LC50 values of 4.9, 5.3, 9.8, and 4.5 mg nickel/litre,
respectively (Pickering & Henderson, 1966).
In a flow-through test, rainbow trout (Salmo gairdneri) were
less sensitive than other fish species with a 48-h LC50 of 20 mg
nickel/litre, in soft water (Brown, 1968). In field studies, Hale
(1977) reported a 96-h LC50 for rainbow trout of 35.5 mg
nickel/litre in continuous-flow tests in water with a hardness of
82-132 mg CaCO3/litre. Arillo et al. (1982) found that rainbow
trout (Salmo gairdneri), exposed to nickel, showed a reduction in
glucidic stores. This effect is consistent with direct metal
interaction on both membranes and enzyme thiolic groups of the
pancreatic cells. Other effects, similar to those found with other
metals, such as damage to the secondary lamellae of gills (Hughes
et al., 1979) and sialic acid depletion in the gills (Arillo et
al., 1982) have been described.
A 96-h LC50 of 118.3 mg nickel/litre was determined for the
marine grey mullet (Chelon labrosus) (Taylor et al., 1985a).
The toxicity of nickel(II) chloride was studied in 2 estuarine
fish species (US EPA, 1987). In tidewater silverside (Menidia
peninsulae) larvae, a 96-h LC50 was 38 mg/litre. For adult spot-
fish (Leiostomus xanthums), the 96-h LC50 was 70 mg/litre.
In short-term tests in soft water, the most sensitive species
of freshwater fish were killed by exposure to concentrations of
about 4-20 mg nickel/litre. Higher LC50 values of nickel have been
found for different species of fish in harder waters, ranging from
about 30 to 80 mg nickel/litre. From the limited data available,
it appears that hardness has the greatest effect on toxicity, while
other determinants have not been proved to have any significant
effects. In acute tests, there are interspecies differences in
sensitivity, but these are within a single order of magnitude.
Shaw & Brown (1971) did not observe any effects on rainbow
trout eggs fertilized in water containing 1 mg nickel/litre and
then maintained in clean water. In a life-cycle study on fathead
minnow, in water with a hardness of 210 mg CaCO3/litre, pH 7.8, and
an average temperature of 18 °C, Pickering (1974) found that nickel
concentrations of 0.38 mg/litre and less (0.18 and 0.08 mg/litre)
did not have any adverse effects on survival, growth, and
reproduction. However, a concentration of 0.73 mg nickel/litre had
a statistically significant effect on the number of eggs produced
per spawning and on the hatchability of these eggs, though it did
not affect the survival and growth of the first generation of fish.
In carp (Cyprinus carpio) eggs and larvae, the 72-h LC50s were
6.1 and 8.4 mg/litre, respectively, while the 257-h LC50 for larvae
was 0.75 mg nickel/litre in water with a hardness of 128 mg
CaCO3/litre, pH of 7.4, and a temperature of 25 °C. A
concentration of 3 mg nickel/litre caused an increased incidence of
abnormal larvae (23% compared with 8.6% in the controls) and 32.7%
of embryos failed to hatch, compared with 5.9% in the controls
(Blaylock & Frank, 1979).
Birge et al. (1978) exposed rainbow trout and largemouth bass
(Micropterus salmoides) from fertilization to 4 days after
hatching, to 11 trace metals found in coal, at a temperature of 12-
13 °C, equivalent to a period of exposure of 28 days for rainbow
trout and 8 days for bass. The LC50 values for these periods were
0.05 mg nickel/litre for rainbow trout and 2.06 mg nickel/litre for
bass. The water used in the tests had a hardness of about 100 mg
CaCO3/litre and a pH of 7.2-7.8. For rainbow trout and goldfish,
teratic larvae were observed at exposure levels that did not
significantly affect egg hatchability. In water from a natural
source with a pH of 7.8 and 174 mg CaCO3/litre hardness, an LC50
for rainbow trout was 0.09 mg nickel/litre whereas in dechlorinated
tap water (pH 7.6, hardness 125 mg CaCO3/litre) the LC50 was 0.06
mg nickel/litre.
Using water with a hardness of about 100 mg CaCO3/litre and pH
7.2-7.8, Birge & Black (1980) found that the LC50 from
fertilization to 4 days after hatching was 0.71 mg nickel/litre for
channel catfish (Ictalurus punctatus) and 2.78 mg nickel/litre for
goldfish (Carassius auratus).
Calamari et al. (1982) found that, during the long-term
exposure of fish to 1 mg nickel/litre, continuous uptake of nickel
occurred for 180 days. The concentrations found were: liver, about
2.9 mg nickel/kg wet weight, kidneys, 4.0 mg/kg, and muscle, 0.8
mg/kg, while, at the beginning of the study, the levels had been
1.5, 1.5, and 9.5 mg nickel/kg, respectively. Toxicokinetic
modelling indicated that theoretical asymptotic values for the
liver, kidney, and muscle should be reached in 397, 313, and 460
days, respectively, at which times the calculated bioconcentration
factors (BCF tissue concentration/environmental concentration) were
3.1, 4.2, and 1.9, respectively. Release was slower in clean water
and the proportions of nickel remaining after 90 days in the liver,
kidney, and muscle were 25, 41, and 31%, respectively. These data
suggest that nickel had little capacity for accumulation in the
tissues examined. However, even these relatively low
concentrations are toxic (Arillo et al., 1982).
7.5. Terrestrial organisms
7.5.1. Plants
Nickel is ubiquitous in plant tissues. There is evidence that
nickel is a required nutrient in a number of plant species. The
urease enzyme of jack bean (Canavalia ensiformis) has been shown to
be a nickel metalloenzyme (Dixon et al., 1975).
Although nickel levels above 50 mg/kg in plants are usually
toxic, a number of plant species may tolerate higher levels
(section 4.2.1).
In general, the effects of long-term, low-level exposure to
nickel are only manifested in growth decrements with no visible
signs. Nickel toxicity in plants is characterized by chlorosis and
necrosis of the leaves, stunting of the roots, deformation of
various plant organs, and wilting (Brooks, 1980; Prokipcak &
Ormrod, 1986).
Apart from the solubility of nickel ions or nickel complexes,
other factors can affect nickel toxicity. Of special interest is
the presence of other heavy metals, which can act synergistically,
and the ameliorating effects of calcium.
A synergistic effect of nickel and copper on the growth of bush
beans was demonstrated by Wallace & Berry (1983). When barley was
grown on loam soil with an elevated level of each of the 6 trace
elements, lithium (13 mg/kg soil, dry weight), zinc (200 mg/kg),
copper (200 mg/kg), nickel (100 mg/kg), and cadmium (100 mg/kg)
there was no reduction in yield when they were applied singly.
However, when all 6 were applied together, at the concentrations
applied singly, there was a 40% reduction in yield, probably
because of depressed phosphorus levels (Wallace et al., 1980a).
Investigations by Prokipcak & Ormrod (1986) of the growth
responses of tomato and soy bean to combinations of nickel, copper,
and ozone, indicated that the nature of the joint action of these
chemicals is very complex and depends on species, the
concentrations of the metals and ozone, and, to a lesser extent,
the duration of exposure. In the first study, nickel was added to
the nutrient solution of tomato and soy bean plants at 1.5, 7.5, or
37.5 mg/litre for 6 days, beginning on day 14 after seeding. The
plants were then exposed to 0.15 or 0.30 µlitre atmospheric
ozone/litre. Growth variables were markedly reduced by nickel, but
ozone response depended on the nickel level. In the second study,
0.3 or 1.5 mg nickel/litre was provided from the 5th or 14th day
onwards. There was little effect of duration of nickel treatment
on growth. Increasing nickel levels and increasing ozone levels
decreased growth, but there was no interaction. In the third
study, treatments with 1.5 or 3.0 mg nickel/litre were combined
with 3.0 or 6.0 mg copper/litre, prior to treatment with 0.25 ml
atmospheric ozone/litre. There were complex interactive effects of
all 3 compounds on tomato plant growth, but not on soy bean plant
growth.
Calcium can reduce nickel toxicity. For example, when soy
beans were grown in nutrient solution containing 1.2 mg
nickel/litre, leaf yield depression was 74% at 4 mg calcium/litre,
but only 45% at 400 mg calcium/litre (Wallace et al., 1980b).
7.5.2. Animals
Few data are available on the effects of nickel on terrestrial
animals. Most data are derived from laboratory animals and
indicate that nickel is an essential element in some species.
As land application of wastes is a common method of
fertilization, studies were performed to evaluate the impact of
heavy metals on the soil ecosystem, using the earthworm Eisenia
foetida as a test organism. Following a 14-day exposure to nickel
nitrate in artificial soil, the LC50 was calculated to be 757 mg/kg
(Neuhauser et al., 1985). Hartenstein et al. (1981) determined
the level at which added concentrations of heavy metals would cause
an activated sludge to induce toxic effects on Eisenia foetida.
Nickel was found to inhibit growth and to induce death at
concentrations of 1200-12000 mg/kg dry weight. These
concentrations seemed very high and the authors concluded that
nickel might have been accumulated by the large population of
microorganisms in the rich organic matrix, part of which might not
be ingestible or digestible by earthworms. This would enable
earthworms to grow in the presence of high nickel concentrations.
7.5.3. Essentiality of nickel for bacteria and plants
Evidence for specific biochemical functions of nickel has come
from studies of microbial systems. Nickel is involved, in some
way, in the "Knallgas" reaction, which is mediated by a number of
bacteria of different genera (Tabillion et al., 1980; Friedrich et
al., 1981; Albracht et al., 1982). The reduction of carbon dioxide
to acetate, carried out by acetogenic bacteria, is dependent on
nickel, which is needed to activate the enzyme carbon monoxide
dehydrogenase (Diekert & Thauer, 1980; Drake, 1982). Diekert &
Ritter (1982) demonstrated that Acetobacterium woodii growth on
fructose was stimulated by, but not dependent on, nickel, unlike
CO2 reduction. A number of studies have established that nickel is
the core metal in the tetrapyrrole ("Factor F430"), found in
methanogenic bacteria, and is essential for the growth of these
organisms.
In plants, Dixon et al. (1975) showed that nickel is essential
at the active site of urease in jack beans (Canavalia ensiformis),
for its enzymatic activity.
7.6. Population and ecosystem effects
Few data are available that identify nickel as a specific cause
for effects at the population level, because nickel is generally
associated with other, often more toxic, trace metals or pollutants
that could be involved in the effects.
Gradual ecological changes have been observed near sources
emitting nickel and other trace metals, resulting in a decrease in
the number and diversity of species (Hutchinson & Whitby, 1977;
Gignac & Beckett, 1986). Yan et al. (1985) investigated 39 lakes
in Ontario and found that the tracheophyte richness of acidic lakes
decreased with increasing nickel and copper levels.
In acidic copper-, and nickel-contaminated lakes near Sudbury,
Ontario, species richness and community biomasses were reduced in
Crustacean zooplankton communities (Yan & Strus, 1980).
DeCantazaro & Hutchinson (1985a,b) demonstrated that the
addition of nickel to microecosystems and incubated soil samples
from boreal jack pine forests could disrupt nitrogen cycling.
Nickel additions of 100-500 mg/kg soil were shown to stimulate
nitrification and nitrogen mineralization, resulting in loss of
nitrogen by leaching. The authors concluded that loss of nitrogen,
which is probably the nutrient most limiting to growth in a boreal
forest ecosystem, could have serious ecological consequences for
forests in the vicinity of nickel smelters.
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO AND OTHER TEST
SYSTEMS
Various studies have indicated that nickel is an essential
element in a number of experimental animal species and that it may
also have a physiological role in human beings. However, nickel
deficiency has not been demonstrated in human beings, and the
possible nickel requirement is probably very low. While the
elucidation of nickel essentiality is in progress, it has not
reached the stage where it can be quantified in relation to nickel
deficiency.
8.1. Animals
8.1.1. Essentiality
Earlier studies in trace-element nutritional research did not
demonstrate any consistent effects of nickel deficiency (Schroeder,
1968; Smith, 1969; Nielsen & Säuberlich, 1970; Wellenreiter et al.,
1970; Nielsen & Higgs, 1971; Schroeder et al., 1974), in part,
because of the technical difficulties of controlling nickel intake
due to its ubiquity. Since 1975, diets and environments have been
devised for adequately controlled studies on nickel metabolism and
nutrition, and the effects of deprivation have been described for
17 animal species, including: chicken, cow, goat, mini-pig, pig,
rat, and sheep.
Nickel is a component of several enzyme systems (certainly
urease and some hydrogenases) and it seems essential for the well-
being of several animal species (Spears et al., 1978).
8.1.1.1 Nickel deficiency symptoms
(a) Growth
In goats (Anke et al., 1977, 1978, 1980, 1986), pigs (Anke et
al., 1977, 1986; Spears, 1984; Spears et al., 1984), and rats
(Nielsen et al., 1975b; Schnegg & Kirchgessner, 1975a, 1980), a
nickel-deficient diet resulted in significantly decreased growth.
The growth depression depended on the nickel level and the duration
of administration and only became evident after intrauterine nickel
deficiency, i.e., in the second or later generations. In addition,
species-specific differences seemed to exist (Anke et al., 1977).
(b) Reproduction and mortality
In goats, mini-pigs, and rats, reproduction was decreased only
insignificantly by intrauterine nickel deficiency (Anke et al.,
1977; Schnegg & Kirchgessner, 1975a, 1980). Conception and
abortion rates as well as the number of offspring were not
influenced by nickel deficiency, but kidding in nickel-deficient
goats and farrowing in nickel-deficient sows occurred later (Anke
et al., 1974). Furthermore, at the end of the lactation period,
significantly fewer offspring of nickel-deficient goats were still
alive compared with control animals. Schnegg & Kirchgessner (1980)
did not find any increase in mortality in intrauterinely nickel-
deficient rats, whereas Nielsen et al. (1975b) found it to a
remarkable extent. Smaller litter size has been observed in both
rats and pigs (Anke et al., 1974; Nielsen et al., 1975b; Schnegg &
Kirchgessner, 1975a).
(c) Histological parameters
Nielsen & Sauberlich (1970) described a nickel-deficiency
syndrome in chickens, characterized by changes in the pigmentation
of the shank skin, thicker bones, swollen joints, and a light-
coloured liver. However, these findings are not consistent with
those observed by other authors (Sunderman et al., 1972; Nielsen,
1974). Sunderman et al. (1972) and Nielsen & Ollerich (1974)
observed ultrastructural lesions in the hepatocytes of chickens.
In mini-pigs fed a nickel-deficient diet, Anke (1974) observed
parakeratosis-like damage to the epithelium. Skin eruptions were
also seen in nickel-deficient goats; the hair of the animals was
brittle, there were fissures of the mouth and legs (Anke et al.,
1976, 1980b). Offspring of nickel-deficient rats had an anaemic
appearance (Schnegg & Kirchgessner, 1975a, 1980a; Nielsen et al.,
1979a,b).
(d) Rumen activity
Nickel seems to be essential for ruminants, because urease
activity in the rumen depends on nickel. Spears & Hatfield (1977)
demonstrated disturbances in metabolic parameters in lambs
maintained on a low-nickel diet, including reduced oxygen
consumption in liver homogenate preparations, increased activity of
alanine transaminase, decreased levels of serum proteins, and
enhanced urinary nitrogen excretion. In a follow-up study, Spears
et al. (1978) found that these animals had significantly lower
microbial urease activity. It was possible to increase urease
activity in the rumen contents by means of nickel supplementation.
(e) Disturbance of iron metabolism
Schnegg & Kirchgessner (1975b; 1976a,b) showed that nickel
deficiency in rats led to a reduced iron content in organs, reduced
haemoglobin and haematocrit values, and anaemia. Iron
supplementation did not cure this anaemia (Nielsen et al., 1979a;
Nielsen & Shuler, 1981), indicating a markedly impaired iron
absorption. Nickel-deficient goats eliminated 33% more iron via
the faeces than control animals (Anke et al., 1980). Spears et
al., (1984) found that additional nickel could improve the iron
status of neonatal pigs. The mechanism through which nickel might
enhance iron absorption is still unclear. While nickel might act
enzymatically to convert ferric to ferrous iron (a form more
soluble for absorption), it might also promote the absorption of
iron by enhancing its complexing with a molecule that can be
absorbed (Nielsen, 1984).
(f) Nickel/calcium interaction
Anke (1974) found that nickel-deficient mini-pigs excreted more
calcium renally than corresponding control animals. The skeletons
of nickel-deficient animals contained less calcium than those of
animals on a nickel-rich diet. Kirchgessner & Schnegg (1980a)
confirmed this effect of nickel deficiency in 30-day-old rats and
showed that more magnesium, instead of calcium, was incorporated
into bones.
(g) Nickel/zinc interaction
Analysis showed that different organs and body fluids of
nickel-deficient goats and pigs suffering from parakeratosis-like
changes of the skin and hair, were not only poor in calcium, but
also in zinc. There were single cases of dwarfism in goats (Anke,
1974; Anke et al., 1980, 1981). In rats, nickel deficiency also
resulted in a significantly decreased zinc concentration in organs,
demonstrated by the reduced size of the organs (Nielsen & Shuler,
1979; Kirchgessner & Schnegg, 1980a).
(h) Enzyme activities
The effects of nickel deficiency on enzyme activity have been
studied in rats by Schnegg & Kirchgessner (1975b, 1977a,b,c,d) and
Kirchgessner & Schnegg (1979, 1980b). They found that, as a rule,
the activity of a number of dehydrogenases and transaminases
decreased by 40-75% (malate dehydrogenase (MDH), isocitrate
dehydrogenase (ICDH), lactic dehydrogenase (LDH), glucose-6-
phosphate dehydrogenase (G6PDH), glutamate dehydrogenase (GLDH),
glutamic oxalate transaminase (GOT), glutamic pyruvic transaminase
(GPT)) with LDH and G6PDH being influenced by secondary iron
deficiency. Kirchgessner & Schnegg (1979, 1980b) also measured a
significant 50% reduction in the activity of alpha-amylases in the
liver and pancreas. The results of other studies suggest that
nickel may serve as a co-factor for the activation of calcineurin,
a calmodulin-dependent phosphoprotein phosphatase (King et al.,
1985).
(i) Substrate and metabolite concentrations
Nickel deficiency mainly affects carbohydrate metabolism, and
this has been demonstrated in nickel-deficient rats by Schnegg &
Kirchgessner (1977c). The glucose and glycogen contents of the
liver in nickel-deficient rats was reduced by 90% and the
triglycerides decreased by 40% compared with those in control
animals. Similar values were found in the serum of the rats, and
also in that of goats. Anke et al. (1980b) reported a reduced
triglyceride level in the serum of ruminants, but the cholesterol
level was unchanged. The significantly increased alpha-lipoprotein
and reduced beta-lipoprotein concentrations were probably connected
with a normal cholesterol level, and a disturbance of triglyceride
metabolism, because the alpha-fraction was rich in cholesterol and
the beta-fraction, rich in triglyceride.
The influence of nickel deficiency on the plasma cholesterol
concentration and on the fat content of the liver has been studied
(Nielsen, 1971; Sunderman et al., 1972; Nielsen et al., 1974,
1975a,b; Schnegg & Kirchgessner, 1977c). However, the results were
inconsistent, because the cholesterol level was not affected in
nickel-deficient animals. Nielsen (1971) found a decrease in the
fat content of the liver in chickens, but Anke et al. (1977) did
not find this in mini-pigs.
8.1.2. Acute exposures
8.1.2.1 Nickel carbonyl
Acute lethal concentrations of nickel carbonyl for laboratory
animals are shown in Table 25. The lethal doses range from an LC50
of 0.1 mg nickel carbonyl/litre air for a 20-min inhalation
exposure of the rat, to an LC50 of 2.5 mg/litre air for the dog
following inhalation exposure of 30 min. The LD50s via other
routes range from 13 to 65 mg/kg, the intraperitoneal route being
the most toxic.
Animals acutely exposed to nickel carbonyl vapour show
pulmonary effects and lesions similar to those observed in human
cases of industrial poisoning. The lung is the primary target organ
for nickel carbonyl in animals, and pulmonary effects are rapid at
high exposure levels, oedema occurring within 1 h of exposure.
Subsequently, proliferation and hyperplasia of the bronchial
epithelium and alveolar lining cells develop. Several days after
exposure, severe intra-alveolar oedema with focal haemorrhage and
alveolar cell degeneration occur. In animals that survive the
acute effects, regression of cytological changes with fibroblastic
proliferation within the alveolar interstitium occurs.
Pathological lesions in other organs after acute exposure of
animals to nickel carbonyl are less severe than those in the lung.
However, focal haemorrhage, congestion, oedema, hydropic
degeneration, mild inflammation, and vacuolization have been
reported in the brain, liver, kidney, adrenals, spleen, and
pancreas. In hepatic parenchymal cells, dilatation of rough
endoplasmic reticulum is the most prominent and consistent
ultrastructural abnormality. Nucleolar alterations also develop in
hepatocytes, 2-24 h after exposure to nickel carbonyl.
Pathological lesions of tubules and glomeruli have been seen in
rats exposed to nickel carbonyl (Kincaid et al., 1953; Sunderman et
al., 1961; Hackett & Sunderman, 1967).
8.1.2.2 Other nickel compounds
LD50 data for some other nickel compounds are listed in Table
26.
Table 25. Acute toxicity studies on nickel carbonyl in experimental animalsa
------------------------------------------------------------------------------------------------------------------------------
Species Route Lethal dose Observations in surviving Observation References
animals period after
exposure
------------------------------------------------------------------------------------------------------------------------------
Rabbit inhalation LC80 = 1.4 mg/litre 50 min Lungs: intra-alveolar 1-5 days Armit (1908)
Cat LC80 = 3.0 mg/litre 75 min haemorrhage, oedema, and (rabbit)
Dog LC80 = 2.7 mg/litre 75 min exudate and alveolar cell
degeneration
Adrenals: haemorrhages
Brain: perivascular
leukocytosis and neuronal
degeneration
Rat inhalation LC80 = 0.9 mg/litre 30 min Lungs: at 2-12 h, capillary 2 h-several Barnes & Denz
congestion and interstitial months (1951)
oedema, at 1-3 days, massive
intra-alveolar oedema, at
4-10 days, pulmonary
consolidation and interstitial
fibrosis
Mouse inhalation LC50 = 0.067 mg/litre 30 min Lungs: at 1 h, pulmonary 0.2 h-6 days Kincaid et al.
Rat LC50 = 0.24 mg/litre 30 min congestion and oedema, at (rat) (1953)
Cat LC50 = 0.19 mg/litre 30 min 12 h-6 days, interstitial
pneumonities with focal
atelectasis and necrosis, and
peribronchial congestion;
Liver, spleen, kidneys,
pancreas: parenchymal cellular
degeneration with focal
necrosis
Mouse inhalation LC100 = 0.2 mg/litre 120 min Sanotskii (1955)
Mouse LC100 = 0.01 mg/litre 120 min
Rat inhalation LC100 = 0.3 mg/litre 20 min Ghirinhgelli (1957)
Rat LC50 = 0.1 mg/litre 20 min
------------------------------------------------------------------------------------------------------------------------------
Table 25 (contd.)
------------------------------------------------------------------------------------------------------------------------------
Species Route Lethal dose Observations in surviving Observation References
animals period after
exposure
------------------------------------------------------------------------------------------------------------------------------
Mouse inhalation LC80 = 0.048 mg/litre 30 min West & Sunderman
Rat LC65 = 0.50 mg/litre 30 min (1958)
Dog inhalation LC90 = 2.5 mg/litre 30 min Sunderman et al.
(1961)
Rat inhalation Lungs: at 1-2 days intra- 1-6 days Sunderman et al.
Dog alveolar oedema and swelling 1-7 days (1961)
of alveolar lining cells, at
3-5 days inflammation,
atelectasis, and interstitial
fibroblastic proliferation;
Kidneys and adrenals: hyperaemia
and haemorrhage
Rat inhalation LC30 = 0.51 mg/litre 30 min Sunderman (1964)
Rat intravenous LD50 = 22 mg/kg Lungs: at 1-4 h, perivascular 1 h-21 days Hackett &
Rat subcutaneous LD50 = 21 mg/kg oedema, at 2-5 days severe Sunderman (1967)
Rat intraperi- LD50 = 13 mg/kg pneumonitis with intra-alveolar
toneal oedema, haemorrhage, subpleural
consolidation, hypertrophy and
hyperplasia of alveolar lining
cells, and focal adenomatous
proliferation, at 8 days,
interstitial fibroblastic
proliferation;
Liver, kidneys, adrenals:
congestion, vacuolization,
and oedema
------------------------------------------------------------------------------------------------------------------------------
Table 25 (contd.)
------------------------------------------------------------------------------------------------------------------------------
Species Route Lethal dose Observations in surviving Observation References
animals period after
exposure
------------------------------------------------------------------------------------------------------------------------------
Rat intravenous 65 mg/kg Lung: ultrastructural 0.5 h-8 days Hackett &
alterations, including Sunderman (1968)
oedema of endothelial cells
at 6 h and massive hypertrophy
of membranous and granular
pneumocytes at 2-6 days;
Liver: ultrastructural
alterations of hepatocytes
including nucleolar distortions
at 2-24 h, dilatation of rough
endoplasmic reticulum at 1-4
days, and cytoplasmic inclusion
bodies at 4-6 days
Mouse inhalation LC100 = 0.1 mg/litre 120 min Sanina (1968)
------------------------------------------------------------------------------------------------------------------------------
a From: NAS (1975)
Table 26. Acute toxicity of nickel compounds in experimental animals
_______________________________________________________________________________________
Species (sex) Substance Route of LD50 (mg/kg) and References
administration confidence limits
_______________________________________________________________________________________
Rat (male) nickel acetate oral 360 (410-316) Haro et al.
(female) 350 (403-304) (1968)
Mouse (male) 410 (500-336)
(female) 420 (515-336)
Rat intraperitoneal 23 (28-19) Haro et al.
(1968)
Mouse 32 (37-28)
Rat (female) nickel chloride intraperitoneal 29 Horak et al.
(1976)
(pregnant intramuscular 71 Sunderman et
female) 98 al. (1978a)
intraperitoneal 38 (34-41) Mas et al.
(1985)
Rat (male) nickelocene oral 490 (510-471) Haro et al.
(female) 500 (525-474) (1968)
Rat intraperitoneal 50 (59-42)
Mouse oral 600 (660-545)
intraperitoneal 86 (102-72)
_______________________________________________________________________________________
Diarrhoea, respiratory distress, and lethargy were noted in
rats and mice dying 2-3 hours after receiving nickel acetate or
nickelocene by the oral or intraperitoneal route (Haro et al.,
1968).
Benson et al. (1986) investigated the effects of single
intratracheal doses of nickel subsulfide (3.2, 32, or 320 µg/kg
body weight), nickel oxide (3, 30, or 300 µg/kg body weight),
nickel sulfate (10.5, 105.2, or 1052 µg/kg body weight), and nickel
chloride (9.5, 95.2, or 952 µg/kg body weight) on rats; 24 h after
dosing, no effects were observed. However, at 7 days, multifocal
alveolitis with some type II hyperplasia was observed in animals
treated with nickel chloride, nickel sulfate, or nickel subsulfide.
Lung lavage fluid contained increased numbers of neutrophils and
macrophages in the medium- and high-dose groups of nickel chloride
and nickel sulfate and in the highest nickel subsulfide dose group.
While increased levels of enzymes, total protein, and sialic acid
occurred in rats exposed to nickel chloride, nickel sulfate, or
nickel subsulfide, no such changes were seen in rats receiving
nickel oxide.
Effects on kidney function in rabbits were studied by Foulkes &
Blanck (1984), who reported a reduction in the maximum tubular
transport rate for aspartase following ip injection of 20 µmol
nickel chloride/kg body weight. Intraperitoneal injection of 3 or
6 mg nickel/kg body weight in male Wistar rats induced a decrease
in Bowman's space, dilated tubules, loss of brush border, flattened
epithelia, and some regenerative activity (Sanford et al., 1988).
Intraperitoneal administration of NiCl2 to mice and rats caused
a rapid decrease of body temperature (Gordon, 1989; Gordon & Stead,
1986; Gordon et al., 1989). The nickel chloride treatment resulted
in hypothermia that lasted for more than 1 h, with a reduction in
colonic temperature of 3-4 °C at 20 °C ambient temperature. The
Ni2+-induced hypothermia was accentuated at lower ambient
temperature (10 °C) and ameliorated at higher ambient temperature
(30 °C).
Hopfer & Sunderman (1988) monitored core body temperature and
physical activity by radiotelemetry from a thermistor probe
implanted in the peritoneal cavity. After injection of nickel
chloride (250 µmol/kg body weight), core body temperature
diminished to a minimum at 1.5 h and returned to baseline at 4 h;
core body temperature at 1.5 h after dosing averaged 3.0 ± 0.5 °C
below the simultaneous value in control rats. During the 8-80 h
following dosing, the mean body temperature of NiCl2-treated rats
did not differ from that of the controls, but the amplitude of the
diurnal cycle of body temperature was dampened and the acrophase of
the temperature cycle was delayed from 10.32 pm to 03.00 am. These
parameters returned towards the control values during the 80-152 h
period following dosing.
Acute thymic involution occurred in male Fischer 344 rats
following a single subcutaneous injection of nickel chloride (500
µmol/kg body weight) (Knight et al., 1987). In nickel-treated
rats, the mean thymic weight, which was significantly decreased at
24 h, continued to diminish at 48 h and reached 24% of the control
value at 72 h. The concentration of lipoperoxides in the thymus
increased 2-fold at 48 h and 7-fold at 72 h. Histological
examination showed marked degenerative changes. Gitlitz et al.
(1975) noted aminoaciduria and proteinuria in rats given single
injections of nickel chloride (2 mg/kg body weight), the response
being dose-dependent. Proteinuria was seen initially with
aminoaciduria at higher doses. These effects, transitory in
duration, were associated with morphological changes in the
glomeruli.
8.1.2.3 Possible mechanisms of acute nickel toxicity
The mechanisms of nickel toxicity are not well understood, but
studies by Knight et al. (1987), Sunderman et al. (1987), and
Sunderman (1987) suggest that acute Ni2+ toxicity in rats is
associated with lipid peroxidation in target organs. The chemical
reactions whereby Ni2+ induces lipid peroxidation in vivo have not
yet been explained, however, Sunderman (1987) proposed the
following four possible mechanisms:
i. An indirect mechanism owing to Ni2+ displacement of iron and
copper from intracellular binding sites;
ii. An indirect mechanism, by which Ni2+ inhibits cellular
defences against peroxidative damage, mediated by catalase,
superoxide dismutase, glutathione peroxidase, aldehyde
dehydrogenase, or other enzymes that protect against free-radical
injury or that metabolize products of lipid peroxidation;
iii. Generation of oxygen-free radicals by the redox couple:
Ni2+/Ni3+
Ni2+ + H2 --> Ni3+ + OH- + OH- (Fenton reaction)
Ni3+ + O-2 --> Ni2+ + O2
H2O2 + O-2 --> OH- + OH- + O2 (Haber-Weiss reaction)
iv. Ni2+ may accelerate the degradation of lipid hydroperoxides to
form lipid-oxygen radicals, propagating autocatalytic peroxidation
of polyenoic fatty acids.
Experiments performed by Inoue & Kawanishi (1989) using
electron spin resonance (ESR) (spin traps 5,5-dimethylpyroline- N-
oxide and alpha-(4-pyridyl 1-oxide)- N-tert-butylnitrone) indicate
that hydroxyl radical adducts are produced in vitro by the
decomposition of hydrogen peroxide in the presence of the nickel
(II) oligopeptide Gly Gly His. These investigators suggested that
Gly Gly His plus hydrogen peroxide produce superoxide in addition
to the hydroxyl radical. The experimental findings support the
conclusion that the nickel-dependent formation of an activated
oxygen species is a primary molecular event in acute nickel
toxicity and carcinogenicity.
8.1.3. Short- and long-term exposures
8.1.3.1 Effects on the respiratory tract
(a) In vivo studies
Data on the chronic respiratory effects of nickel carbonyl are
summarized in section 8.1.2.1.
Long-term inhalation studies have been performed on guinea-
pigs, rats, and mice (Hueper, 1958), rats (Bingham et al., 1972),
and hamsters (Wehner et al., 1975, 1981). Exposure extended over
more than one and a half years in all studies. The compounds
tested were metallic nickel powder, nickel subsulfide, nickel
oxide, and nickel-enriched fly ash. Hueper (1958) exposed animals
to metallic nickel dust at 15 mg/m3, and noted nasal sinus
inflammation and ulcers in rats, and signs of lung irritation in
guinea-pigs and rats. A common finding was an accumulation of
adenomatoid cell formations. In mice, there were signs of lung
irritation, but not to the same extent as in guinea-pigs and rats.
Bingham et al. (1972) used aerosols of soluble nickel chloride
at 109 µg/m3 and nickel oxide at 120 µg/m3 and observed hyperplasia
of bronchiolar and bronchial epithelium with peribronchial
lymphocyte infiltrates.
Ottolenghi et al. (1974) found a number of lung changes, such
as abscesses as well as metaplastic changes, when rats were exposed
for 78 weeks to nickel subsulfide by inhalation. Wehner et al.
(1975) exposed rats to a concentration of 53 mg nickel oxide/m3
(type of oxide not specified) for the life span and found that
particulate material accumulated on the alveolar septa. Emphysema
was observed early in the exposure period. With longer exposure,
the cellular response increased and pneumoconiosis developed
gradually. However, there was no reduction in the life span.
Wehner et al. (1981) exposed hamsters through inhalation to nickel-
enriched fly ash or fly ash at concentrations of 17 or 70 µg/m3 for
up to 20 months. Lung weights and volumes were significantly
increased in the 70 µg/m3 fly ash exposure group. The severity of
anthracosis, interstitial reaction, and bronchiolization was dose-
dependent.
Friberg (1950) exposed rabbits for 6 months to nickel-graphite
dust (nickel hydrate, nickel content approx. 50%) at a
concentration of 100 mg/m3 (3 h/day, 5 days/week) and found
emphysematous and inflammatory changes in the nasal mucous
membranes and the trachea, bronchitis, and sometimes slight
fibrosis in the lung.
Port et al. (1975) reported that intratracheal injection of a
suspension of nickel oxide (5 mg, particle size <5 µm, type of
nickel oxide not specified) into Syrian hamsters, treated 48 h
previously with influenza A/PR/8 virus, resulted in significantly
increased mortality compared with controls. Surviving animals at
this and lower doses showed mild to severe acute interstitial
infiltration by polymorphonuclear cells and macrophages, several
weeks later. Additional pathological changes included bronchial
epithelial hyperplasia, focal proliferative pleuritis, and
adenomatosis.
Short-term inhalation studies (12 days) with nickel sulfate
(3.5-60 mg/m3) and nickel subsulfide (0.6-10 mg/m3) were performed
on rats and mice (Benson et al., 1987, 1988). Nickel sulfate
caused lesions in the lung, nose, bronchial, and mediastinal lymph
nodes in the surviving animals at 3.5 mg/m3. In the nickel
subsulfide-exposed animals, similar changes occurred in the
respiratory tract with extensive lesions in the lung, including
necrotizing pneumonia. Emphysema developed in rats exposed to 5 or
10 mg/m3 and fibrosis was seen in mice exposed to 5 mg nickel
subsulfide/m3. Degeneration of the respiratory epithelium and
atrophy of the olfactory epithelium occurred in all nickel
subsulfide dose groups except in mice exposed to 0.6 mg/m3.
Clinical signs included laboured respiration, emaciation,
dehydration, and reduced body weight gain in rats and mice exposed
to nickel sulfate or nickel subsulfide. A 12-day inhalation
exposure of rats and mice to nickel oxide (1.2-30 mg/m3) also
caused lung lesions in the higher dose groups. Lung lesions
included hyperplasia of alveolar macrophages, focal suppurative
inflammation, focal interstitial cellular infiltrate and particles
in alveoli and alveolar macrophages. In mice, lung lesions were
less severe (Dunnick et al., 1988). On the basis of these 12-day
studies, relative toxicity ranking was NiSO4 x 6H2O >Ni3S2 >NiO
(Dunnick et al., 1988).
The effects of nickel on the cellular respiratory system
defence mechanisms were studied by exposing guinea-pigs, rats, or
rabbits to various concentrations (0.05-2 mg/m3, for 1-8 months) of
nickel dust, nickel oxide, or nickel chloride (Waters et al., 1975;
Graham et al., 1975a; Aranyi et al., 1979; Johansson et al., 1980,
1981, 1983a,b; Castranova et al., 1980; Casarett-Bruce et al.,
1981; Lundborg & Camner, 1982, 1984; Wiernik et al., 1983; Murthy
et al., 1983; Takenaka et al., 1985; Glaser et al., 1986). It was
concluded that the overall effects had some features in common with
the pulmonary alveolar proteinosis described in human beings.
There was no difference in the effect pattern between exposure to
insoluble and soluble nickel compounds. Effects, such as changes in
the morphology and function of alveolar macrophages and type II
alveolar epithelial cells, and in the composition of lung lavage,
were found, with the severity of effects depending on the
concentration and duration of exposure.
The phagocytic activity of alveolar macrophages was increased
in rabbits following 4 weeks inhalation of metallic nickel dust
(0.5-2.0 mg/m3; Camner et al., 1978; Yarstrand et al., 1978) and in
rats following 1-4 months inhalation of nickel oxide (produced by
pyrolysis of nickel acetate at 550 °C) (Spiegelberg et al., 1984).
No change in the phagocytic activity of alveolar macrophages was
observed in rats following exposure to 0.13 mg metallic nickel
dust/m3 for 4-8 months (Johansson et al., 1983a), and in rabbits
following exposure to nickel chloride aerosols (0.3 mg/m3) for 1
month (Wiernik et al., 1983).
Alveolar macrophages varied in size and, ultrastructurally, had
an active cell surface with numerous slender microvilli and long
protrusions. Laminated structures were regularly found in the
alveolar macrophages as well as in the lung fluid (Camner et al.,
1978; Johansson et al., 1980, 1983a; Wiernik et al., 1983).
Spiegelberg et al. (1984) found an increase in size and number of
polynucleated cells in the lungs of exposed rats. Morphometric
studies on the lungs of exposed rabbits showed an increase (up to
3-fold) in the volume density of type II cells. Cells contained
many lamellar bodies and vesicles; the endoplasmic reticulum had a
slightly dilated appearance (Johansson & Camner, 1980; Johansson et
al., 1981, 1983b). There were changes in the pulmonary lipid
content and composition of the lung fluid of rabbits inhaling
metallic nickel dust at levels ranging from 0.13 to 1.7 mg/m3
(Casarett-Bruce et al., 1981; Curstedt et al., 1983, 1984). There
was a twofold increase in the concentration of total phospholipids,
mainly due to an elevated level of phosphatidylcholines, especially
disaturated species, and phosphatidylinositols. Lundborg & Camner
(1982, 1984) exposed rabbits to nickel dust (0.1 mg/m3, for 4 and 6
months) and nickel chloride (0.2-0.6 mg/m3 for 4-6 weeks) and found
markedly reduced lysosomal enzyme activity compared to control
animals. When rats were exposed to aerosols of nickel oxide (type
of oxide not specified) at 120 µg/m3 or nickel chloride at 109
µg/m3, Murthy et al. (1983) found that various hydrolytic enzymes
were reduced in alveolar macrophages, but, in contrast, the enzyme
activities were significantly increased in lung lavage.
Parenteral administration of nickel chloride can also cause
toxic effects in alveolar macrophages, as demonstrated by Sunderman
et al. (1989c) in rats that had received single subcutaneous
injections of 8-65 mg (62-500 µmol) nickel chloride/kg. Alveolar
macrophages showed morphological and biochemical signs of
activation, functional impairment, and lipid peroxidation. In
alveolar macrophages from 63NiCl2-treated rats, 63Ni was primarily
located in the cytoplasm bound to high- and low-relative molecular-
mass constituents.
At the ciliated epithelium level, nickel significantly
depressed normal ciliary activity in a hamster tracheal ring assay
following in vivo exposure to 100 µg nickel/m3 as nickel chloride
for 2 h (Adalis et al., 1978; Olsen & Jonsen, 1979b).
Fisher et al. (1984) evaluated the effects of particle size and
dose regimen on the toxicity of intratracheally instilled nickel
subsulfide in mice, and showed the importance of physical form in
the evaluation of pulmonary toxicity. The median lethal dose for a
single exposure to larger particles (MMAD 13.3 µm) was 12 times
that of fine particles (MMAD 1.8 µm), i.e., 50 versus 4 mg/kg.
Repeated exposures (once/week for 4 weeks) resulted in a 2-fold
greater median lethal dose of coarse particles compared with fine
particles, i.e., 2 versus 1 mg/kg. Thus, repeated exposure to fine
particles resulted in a cumulative lethality equivalent to the
single exposure, while coarse particle lethality was enhanced with
repeated exposure. Alveolar macrophages from mice exposed to fine
nickel subsulfide showed depressed cellular function, 14 days after
a single administration.
Reichrtova et al. (1986a,b) performed 2 inhalation studies on
rats to compare the effects of different types of exposure on
alveolar macrophages. In both studies, the nickel content of the
aerosol was only 0.36% (NiO), thus, the effects are unlikely to be
nickel-specific responses. Increases in the alveolar macrophage
count and lysosomal enzyme activities (acid phosphatase and beta-
glucuronidase) were found in Wistar rats, exposed to an aerosol of
solid waste from a nickel refinery dump, under chamber conditions,
for 6 months (aerosol concentration 0.1 g/m3, 4 h/day, 5
days/week). However, the activity of alveolar macrophage plasma
membrane enzymes decreased. Cells contained in lung lavage had a
pleomorphic appearance. When rabbits were environmentally exposed
at a biomonitoring station, situated in the direction of the
prevailing wind from a nickel refinery dump, for 6 months, with an
average dust fallout of 5.5 g/m2 in 30 days, the number of alveolar
macrophages was significantly increased (Reichrtova et al., 1986b).
A significant increase in lysosomal enzyme activity also occurred,
but these changes were not as pronounced as the effects obtained in
the chamber studies on rats. In contrast to the increased alveolar
macrophage activity, a significant reduction occurred in antibody-
mediated rosette formation by alveolar macrophages in the
environmentally exposed rabbits.
(b) In vitro cytotoxicity studies
Alveolar macrophages exposed to nickel (1.1 mmol/litre) in
vitro showed depressed phagocytic activity (Graham et al., 1975a;
Aranyi et al., 1979). Waters et al. (1975) correlated changes in
cell viability with changes in the morphology and the specific
activity of a lysosomal enzyme (acid phosphatase). At 4.0
mmol/litre, cell viability was reduced to approximately 50% of that
of controls. Castranova et al. (1980) demonstrated that nickel
affected oxygen metabolism in the rat alveolar macrophages; at
rest, and during phagocytosis, oxygen consumption and glucose
metabolism were depressed.
The in vitro cytotoxicity of nickel chloride on a human
pulmonary epithelium cell line (A549) was reported by Dubreuil et
al. (1984). Nickel chloride in amounts ranging from 0.1 to 1.0
mmol/litre produced decreases in cell growth rate and in the levels
of cell adenosine triphosphate (ATP), and loss of viability, at the
highest concentration. No changes were seen in transmission
electron microscopy preparations.
In vitro toxicity studies on bovine alveolar macrophages
indicated that nickel subsulfide was 10 times more toxic than
solubilized nickel subsulfide or soluble nickel chloride (Fisher et
al., 1984).
Nickel subsulfide, nickel sulfate, nickel chloride and nickel
oxide were tested for their relative toxicity in beagle dog and rat
alveolar macrophages in vitro. Toxicity ranking was Ni3S2 >NiCl2
ca NiSO4 >NiO (Benson et al., 1986).
8.1.4. Relationship of nickel toxicity and mixed metal exposure
As both nickel production and some end uses of nickel involve
mixed exposure of workers to nickel and copper, nickel and
chromium, nickel, chromium, and manganese, and so on, the problem
of the combined toxic effects of these metals is of great practical
importance. However, the relevant information is rather scarce.
Johansson et al. (1988) showed that the cytotoxic effects of a
trivalent chromium salt on alveolar macrophages of the rabbit
impaired their dealing with pulmonary surfactant, the latter being
hyperproduced as a result of nickel's action on the type II
alveolar epithelial cells. However, analysis of these experimental
data showed that, as far as the cytotoxicity of these metals for
alveolar macrophages was concerned, their joint effect was
antagonistic.
Davydova et al. (1981) demonstrated that subadditivity or even
clear antagonism was the main type of combined acute toxicity for
rats of nickel and chromium, nickel and manganese, and even a
triple exposure of rats to nickel and cobalt. On the contrary, the
long-term exposure of rats to nickel and cobalt in the drinking-
water demonstrated the additive long-term toxicity of these two
metals (Nadeenko et al., 1988).
8.1.5. Endocrine effects
Bertrand & Macheboeuf (1926a,b) reported that parenteral
administration of nickel chloride or nickel sulfate to rabbits or
dogs antagonized the hyperglycaemic action of insulin. Later
investigators observed that after parenteral (iv or ip) injection
to rabbits, rats, or chickens, or oral administration to rabbits,
there was a rapid increase in plasma glucose concentrations, which
returned to normal within 4 h (Kadota & Kurita, 1955; Gordynia,
1969; Clary & Vignati, 1973; Freeman & Langslow, 1973; Horak &
Sunderman, 1975a,b). In the pancreatic islets of Langerhans,
Kadota & Kurita (1955) noted marked damage of alpha-cells, and, to
a lesser degree, degranulation and vacuolization of beta-cells.
Ashraf & Sybers (1974) noted lysis of pancreatic exocrine cells in
rats fed nickel acetate (0.1%). In adrenalectomized or
hypophysectomized rats, the hyperglycaemic effect was greatly
depressed, but was not completely prevented. Concurrent
administration of insulin antagonized the hyperglycaemic effect
(Horak & Sunderman, 1975a,b). LaBella et al. (1973a,b) showed
that nickel also affects the hypothalamus of animals. Nickel salts
specifically inhibited the release of prolactin in vivo (in the
rat) and in vitro from bovine pituitary. Subcutaneous injection of
10 or 20 mg nickel chloride/kg in rats initially produced a drop in
serum prolactin over the short term, but resulted in a sustained
elevation of the hormone after 1 day, which lasted up to 4 days.
The elevation was due to reduced levels of the prolactin-inhibiting
factor (Clemons & Garcia, 1981). Carlson (1984) demonstrated that
nickel antagonized the stimulation of both prolactin and growth
hormone by barium; thus, the basis of antagonism may be the
competitive inhibition of calcium uptake. Dormer et al. (1973)
showed that the nickel ion is a potent inhibitor of secretion in
vitro in the parotid gland, (amylase), the islets of Langerhans
(insulin), and the pituitary gland (growth hormone). Inhibition of
growth hormone secretion at nickel concentrations comparable to
those observed by LaBella et al. (1973b) to enhance release, may
reflect differences in tissue preparation prior to assay. Dormer
et al. (1973) suggested that nickel may block exocytosis by
interfering with either secretory granule migration or membrane
fusion and microvilli formation.
The effects of nickel on thyroid function were reported by
Lestrovoi et al. (1974). Nickel chloride, given orally (0.5-5.0
mg/kg per day, for 2-4 weeks) or by inhalation (0.05-0.5 mg/m3) to
rats, significantly decreased iodine uptake by the thyroid, the
effect being more pronounced with inhaled nickel.
8.1.6. Cardiovascular effects
The serum nickel level increased in patients with acute
myocardial infarction, stroke, and burns (D'Alonzo & Pell, 1963;
McNeely et al., 1971; Leach et al., 1985).
Rubanyi & Kovach (1980) found that micromolar concentrations of
nickel chloride (0.1-1 µmol/litre) increased cardiac contractility
in the isolated rat heart. At higher doses, there was depressed
myocardial contractile performance. Ultrastructural damage was
found (Rubanyi et al., 1980).
Nickel chloride at a concentration of 1 µmol/litre in the
perfusate produced tonic contraction in the isolated canine
coronary artery (Rubanyi et al., 1982b).
Nickel is released from the ischaemic dog myocardium (Rubanyi
et al., 1981); exogenous nickel, in doses comparable to the amount
released endogenously from the heart, induced coronary
vasoconstriction in rat and dog hearts. In further studies, the
possible involvement of Na/K ATPase inhibition (Rubanyi et al.,
1982c) and/or stimulation of adrenergic receptors in the coronary
vessels (Rubanyi et al., 1982d) were discussed as mechanisms of
nickel-induced coronary vasoconstriction. Rubanyi & Inovay (1982)
studied the effects of nickel ions on spontaneous, electrically-,
and norepinephrine-stimulated isometric contractions in the
isolated portal vein of the rat. They found that low
concentrations of nickel (1-10 µmol/litre) inhibited spontaneous
isometric force development and decreased basal tone, but
significantly increased the frequency of contractions. Inhibition
of the effect of selective stimulation of adrenergic nerves was
significantly more pronounced than the depression of contractions
evoked by exogenous norepinephrine.
In the in situ heart (open-chest anaesthetized dogs), a
decrease in coronary vascular flow with a low intravenous dose of
nickel (0.02 mg nickel chloride/kg body weight) was reported,
while, at higher dose levels (0.2, 2, or 20 mg nickel chloride/kg
body weight), further reduction in coronary blood flow, depression
of heart rate, and a decrease in left ventricular contractility
were observed. Coronary vasoconstriction may be regarded as a
local action of nickel on coronary vessels (Ligeti et al., 1980).
In another in situ study on dogs (intravenous bolus injection
of 0.02 mg nickel/kg, or intracoronary infusion of 0.04 mg nickel
chloride/min per kg body weight), Rubanyi et al. (1984) found that
vasoconstriction was induced when coronary arteries were dilated by
low-flow ischaemia, arterial hypoxaemia, and adenosine infusion.
Nickel inhibited post-occlusion reactive hyperaemia and
vasorelaxation in response to arterial hypoxaemia or intracoronary
infusion of adenosine. It was postulated that vasoactivity might
be related to the existence of positive feedback loops triggered by
alterations in the level of nickel.
Endogenous nickel release from damaged tissues and its
implications for ischaemic heart disease have been examined with
respect to the pathology of acute carbon monoxide poisoning and
acute burn injury. Significant endogenous nickel ion accumulation
was noted in the heart muscle of rats and rabbits, when the CO-Hb
level was above 30% (Balogh et al., 1983).
8.1.7. Effects on the immune system
The effects of nickel on alveolar macrophages have been
described in section 8.1.3.1.
Koller (1980) noted that nickel exposure of animals could
reduce host resistance to both viral and bacterial infections, and
suppress the phagocytic capacity of macrophages.
Other cellular and humoral immune responses following nickel
treatment were studied by Smialowicz et al. (1984, 1985). Single
or multiple intramuscular injections of nickel chloride in mice
caused a significant reduction in a variety of T-lymphocytes and
natural killer cell-mediated immune functions. Suppression of the
lymphoproliferative responses to the T-cell mitogens,
phytohaemagglutinin and concanavalin A, and a reduction in the
number of theta-positive T-lymphocytes were observed in the spleens
of nickel chloride-injected mice (18.3 and 36.6 mg/kg body weight).
Reductions in the primary antibody response to T-lymphocyte-
dependent antigen sheep red blood cells, but not T-lymphocyte-
independent antigen polyvinyl-pyrrolidone, were observed following
a single injection of 18.3 mg nickel chloride/kg (Smialowicz et
al., 1984).
Smialowicz et al. (1985) demonstrated that suppression of
natural killer cell activity could be detected by in vitro and in
vivo assays and that reduction of natural killer cell activity was
not associated with either a significant reduction in spleen
cellularity or the production of suppressor cells. A further
demonstration of the effect of nickel chloride on natural killer
cell activity was the enhancement of the development of lung tumour
colonies in mice injected with B16-F10 melanoma cells, following a
single injection of 18.3 mg nickel chloride/kg. Unlike nickel
chloride, manganese chloride was found to enhance natural killer
cell activity, when injected into mice (Smialowicz, 1985). No
alteration in natural killer cell activity was observed in mice
injected with magnesium, calcium, or zinc (Smialowicz et al.,
1987). Manganese chloride was considered to have an antagonistic
effect on nickel chloride-suppression of natural killer cell
activity.
The effects of nickel compounds on natural killer cells are of
particular interest, because of the suspected function of these
cells in nonspecific defence against certain types of infections
and tumours.
The studies performed by Smialowicz et al. (1984, 1985, 1986,
1987) confirmed the findings of other investigators on the immuno-
suppressive effects of nickel salts on circulatory antibody titres
to T1 phage in rats (Figoni & Treagan, 1975), on antibody response
to sheep erythrocytes (Graham et al., 1975b), on interferon
production in vitro (Treagan & Furst, 1970) and in vivo in mice
(Gainer, 1977), and on susceptibility to induced pulmonary
infection in mice following inhalation of nickel chloride (Adkins
et al., 1979).
In cynomolgus monkeys that had been previously immunized and
repeatedly challenged with sheep red blood cells, instillation of
10.6 mg nickel subsulfide/kg lung in one immunized and one control
lobe of each animal increased target cell killing by conjugate-
forming natural killer cells and decreased macrophage phagocytic
activity (Haley et al., 1987).
The results of in vitro studies have shown that nickel can
replace magnesium, which is essential for the proper functioning of
the complement system, in both the classical and the alternative
pathway. Nickel has been shown to result in a more efficient
formation of the C3b,Bb enzyme, which theoretically may lead to
disturbance of a well balanced complement cascade. The
significance of this finding in vivo is not known (Fishelson et
al., 1983).
8.1.8. Skin and eye irritation and contact hypersensitivity
8.1.8.1 Skin and eye irritation
Repeated skin application of 40-100 mg nickel/kg (as nickel
sulfate), daily for 30 days, produced skin atrophy, acanthosis, and
hyperkeratinization in rats (Mathur et al., 1977). No data are
available on the eye irritancy of soluble nickel salts or on the
skin and eye irritancy of insoluble nickel compounds.
8.1.8.2 Contact hypersensitivity
Experimental sensitization to nickel in guinea-pigs has been
reported (Walthard, 1926; Stewart & Cromia, 1934; Vinson & Choman,
1960; Jansen et al., 1964; Gross et al., 1968; Magnusson & Kligman,
1970). Nilzén & Wikström (1955) reported a method for sensitizing
laboratory animals to nickel by repeated topical applications of
aqueous nickel sulfate solutions containing sodium lauryl sulfate.
However, Samitz & Pomerantz (1958) were unable to demonstrate
sensitization with this technique and attributed the effect to
local irritation, rather than true allergenic reaction. Samitz et
al., (1975) were unable to induce sensitization in guinea-pigs
using nickel compounds from the complexation of the nickel ion with
amino acids or guinea-pig skin extracts. Furthermore,
sensitization of experimental animals was not found by Hunziker
(1960) or Bühler (1965).
The sensitivity of guinea-pigs was increased by repeated intra-
dermal injections and skin painting with nickel sulfate solutions
during the sensitization phase. The responses were significantly
greater than in control animals (Wahlberg, 1976).
Turk & Parker (1977) reported sensitization to nickel
manifested as allergic-type granuloma formation. This required the
use of Freund's complete adjuvant followed by weekly intradermal
injections of 25µg of the salt after 2 weeks. Delayed
hypersensitivity reactions developed in 2 out of 5 animals at 5
weeks when a split-adjuvant method was used. Suppression of the
delayed hypersensitivity occurred when intratracheal intubation of
nickel sulfate was also performed on these animals (Parker & Turk,
1978). Möller (1984) reported that mice could easily be sensitized
to a potent antigen, such as picryl chloride, but response to
nickel could only be achieved by repeated epicutaneous application
of a strong (20%) nickel salt solution for 3 weeks.
The study of the allergic properties of nickel in experimental
animals is a problem, because of difficulties involved in
reproducing the phenomenon of allergodermatosis and because of lack
of a uniform approach to reproducing the experimental contact
allergic dermatitis model. Duyeva (1983) recommended a single
intracutaneous administration of 100-200 µg nickel chloride in the
ear of the guinea-pig. Sensitization developed as early as days 4-
10, reaching a peak on day 20.
8.1.9. Reproduction, embryotoxicity, and teratogenicity
8.1.9.1 Effects on the male reproductive system
Data on the effects of nickel on the male reproductive system
are limited. Hoey (1966) examined the effects of nickel sulfate on
the testis and epididymis of Fischer rats and reported histological
changes in the testis and adnexae. Following a single
intracutaneous injection of 0.04 µmol nickel sulfate/kg body weight,
histological examination, 18 h after exposure, revealed shrinkage
of the tubules and complete degeneration of the spermatozoa.
Infertility was observed in rats after 120 days of daily ingestion
of 25 mg nickel sulfate/kg (Waltschewa et al., 1972).
Mathur et al. (1977) studied the dermal exposure of male rats
to nickel sulfate at daily levels of 40, 60, or 100 mg nickel/kg
body weight for 15 and 30 days. Tubular damage and spermatozoal
degeneration were observed in the testis following exposure to 60
mg nickel/kg for 30 days. These changes were more severe with
exposure at 100 mg/kg for 30 days. There were no effects on the
testis following exposure to 40 mg/kg for 30 days or at any dose
level after 15 days exposure.
In vitro embryo cultures were used to study the effects of
nickel nitrate on male germ cells (Jacquet & Mayence, 1982).
BALB/C mice were injected intraperitoneally with 40 or 56 mg nickel
nitrate/kg body weight and were then allowed to mate with
superovulated females, at weekly intervals, for 5 weeks following
treatment. The embryos were isolated and those at the 2-cell stage
were cultured for 3 days. These embryos were then classified
according to their ability to develop to the blastocyst stage. A
dose of 40 mg/kg body weight did not affect the fertilizing
capacity of the spermatozoa or the ability of the fertilized egg to
cleave, but a dose of 56 mg nickel nitrate/kg body weight yielded a
significant proportion of uncleaved unfertilized eggs. Cleaved
eggs from this treatment group were able to develop into
blastocysts, suggesting that nickel nitrate treatment reduced the
fertilizing capacity of the spermatozoa, presumably because of a
toxic effect of nickel on spermatogenesis. Deknudt & Leonard
(1982) conducted a dominant lethal mutation test for nickel
chloride and nickel sulfate in BALB/C mice. Both compounds
produced a reduction in implantations in the matings performed 2,
3, or 4 weeks after treatment, but the post-implantation loss was
not increased. It was considered that this was attributable to a
toxic effect of nickel treatment on male germ cells.
8.1.9.2 Effects on the female reproductive system
Insertion of nickel wire into one uterine horn of rats on day 3
of pregnancy produced a decrease in the number of implants and an
increase in the number of resorption sites, compared with the
untreated contralateral horn (Chang et al., 1970).
Nickel has been reported to localize within the pituitary and
the hypothalamus of rats and to inhibit prolactin secretion (La Bella
et al., 1973a,b). It may, therefore, modify interactions between
the hypothalamus and the pituitary, needed to maintain pregnancy.
No effects on fertility were observed in 3 generations, when
breeding rats were exposed to 5 mg nickel/litre as a soluble salt
(not specified) in the drinking-water (Schroeder & Mitchener, 1971).
8.1.10. Embryotoxicity and teratogenicity
Nickel has been shown to cross the placental barrier and enter
the fetus (section 6.1.5).
Sunderman et al. (1978a) studied the embryo- and fetotoxicity
of nickel chloride and nickel subsulfide in Fischer 344 rats.
Single intramuscular injection of nickel chloride (12 or 16 mg
nickel/kg body weight) on day 8 of gestation significantly reduced
the mean number of live pups per dam and resulted in reduced body
weight in fetuses on day 20 of gestation and in pups, 4-8 weeks
after birth. When nickel chloride was administered in repeated
intramuscular doses of 1.5 or 2.0 mg/kg body weight on days 6-10 of
gestation, the higher dose caused significant intrauterine
mortality, but did not cause any reduction in the mean body weight
of live pups. Intramuscular injection of nickel subsulfide (80 mg
nickel/kg body weight) reduced the mean number of live pups per
dam. Exposure to nickel chloride or nickel subsulfide did not
produce any skeletal or visceral anomalies.
Lu et al. (1979) observed a dose-related increase in fetal
deaths and a higher incidence of malformations in pregnant CD-1
mice following intraperitoneal injection of single doses of nickel
chloride (1.2, 2.3, 3.5, 4.6, 5.7, or 6.9 mg nickel/kg) between
days 7 and 11 of gestation. Fetal death and some general
malformation (not described in detail) were reported in hamsters
after intravenous injection of nickel acetate at 30 mg/kg body
weight on day 8 of pregnancy (Ferm, 1972). Nadeenko et al. (1979)
demonstrated a dose-dependent embryotoxic effect of 27Ni given to
female rats for 7 months (before and during pregnancy) in
concentrations ranging from 5 x 10-1 to 5 x 10-4 mg/kg body weight.
The effects of nickel chloride on early embryogenesis were
studied by Storeng & Jonsen (1980) in vitro. Mouse embryos at the
2-, 4-, and 8-cell stages were cultured in media containing nickel
chloride hexahydrate at a concentration of 10-1000 µmol/litre. A
concentration of nickel chloride hexahydrate of 10 µmol/litre
adversely affected the development of 2-cell stage embryos whereas
a concentration of 300 µmol/litre was needed to affect 8-cell stage
embryos. No effect was observed at 100 µmol/litre. In a
subsequent study (Storeng & Jonsen, 1981), a single intraperitoneal
injection of nickel chloride hexahydrate (20 mg/kg) was given to
mice on days 1-6 of gestation. On day 19, implantation frequency
in females treated with nickel chloride hexahydrate on day 1 was
significantly lower compared with the controls. Litter size was
significantly reduced in females treated on days 1, 3, and 5 of
gestation. The incidence of abnormalities, such as haematomas and
exencephaly, in the fetuses of treated females was higher
(statistical significance not reported) than in the controls.
Sunderman et al. (1983) used intrarenal injection of nickel
subsulfide to assess the effects on the progeny of rats.
Administration of 30 mg nickel subsulfide/kg by intrarenal
injection, 1 week prior to breeding, produced intense
erythrocytosis in the dams, but not in the pups. These findings
indicate that the release of maternal erythropoietin by the
maternal kidneys, caused by nickel subsulfide, did not stimulate
erythropoiesis in the pups. Nickel subsulfide was associated with
a significant decrease in mean body weights of pups, 2 and 4 weeks
postpartum.
In a series of studies, Sunderman et al. (1978b,c, 1979a,
1980, 1983) demonstrated that nickel carbonyl, administered by
inhalation or injection before, or a few days after, implantation
produced various types of malformations in hamsters and rats.
Intravenous injection of nickel carbonyl (11 mg nickel/kg body
weight) into Fischer rats on day 7 of gestation caused fetal
mortality, reduced body weight in live pups, and a 16% incidence of
fetal malformations, including anophthalmia, microphthalmia, cystic
lungs, and hydronephrosis (Sunderman et al., 1983). There was no
maternal toxicity. Similar effects were observed in rats following
inhalation of nickel carbonyl at a concentration of 0.16 g/m3 on
day 7 or 8 of gestation and a concentration of 0.3 g/m3 on day 7 of
gestation (Sunderman et al., 1979a).
In a dominant lethal mutation test on male rats, administration
of nickel carbonyl, by the inhalation route (50 mg nickel/m3 for 15
min), 2-6 weeks prior to breeding, did not affect fertilization
rates or reproductive yield; administration of nickel carbonyl by
intravenous injection during the same period (22 mg nickel/kg)
caused reduced numbers of live pups in litters sired during the
fifth week (Sunderman et al., 1983). In hamsters, inhalation
exposure to 60 mg nickel carbonyl/m3, for 15 min on day 4 or 5 of
gestation, led to decreased fetal viability and increased numbers
of fetuses with malformations including cystic lungs, exencephaly,
and haemorrhages in serous cavities (Sunderman et al., 1980).
Gilani & Marano (1980) injected nickel chloride into chicken
eggs (0.02 and 0.7 mg per egg) on days 0, 1, 2, or 3 of incubation.
Examination on day 8 revealed a number of malformations, such as
exencephaly, everted viscera, short and twisted neck or limbs,
microphthalmia, haemorrhage, and reduced body size. The incidence
of malformations was highest in embryos treated on day 2.
When rats were exposed long-term to nickel chloride or nickel
sulfate in the diet or drinking-water, an increased frequency of
runts and greater prenatal and neonatal mortality were observed
(Schroeder & Mitchener, 1971; Ambrose et al., 1976; Nadeenko et
al., 1979). Berman & Rehnberg (1983) observed spontaneous
abortions, loss of fetal mass in survivors, and loss of maternal
mass in mice.
8.2. Mutagenicity and related end-points
Genotoxicity data on nickel and nickel compounds are summarized
in Table 27.
The data collectively show that nickel compounds are generally
inactive in bacterial assays, but active in systems using
eukaryotic organisms, and that positive responses were observed
regardless of the nickel compounds used; particularly when these
were compared at equitoxic concentrations (Hansen & Stern, 1983;
Swierenga & McLean, 1985). The ability of nickel compounds to
inhibit DNA synthesis and excision repair should also be noted
(Table 27).
Table 27. Summary of the genotoxic effects of nickel compounds
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
PROKARYOTIC SYSTEMS
Prophage Prophage induction Ni(CH3CO3)2 160-640 µmol/litre + (weak) Rossman et al.
(1984)
T4 Bacteriophage Bacteriophage-forward NiSO4 300 mg/litre - Corbett et al.
mutation (1970)
Salmonella
TA1535 Salmonella typhimurium NiCl2 0.01-0.1 g/litre + LaVelle &
reverse mutation Witmer (1981)
(fluctuation test)
TA1535 S. typhimurium - Haworth et al.
reverse mutation (1983)
TA1525 S. typhimurium NiCl2 - Arlauskas et
reverse mutation NiSO4 al. (1985)
TA1537 S. typhimurium - Haworth et al.
reverse mutation (1983)
TA1537 S. typhimurium NiCl2 - Arlauskas et
Reverse mutation NiSO4 al. (1985)
TA1535 S. typhimurium Ni salts 10-1000 µg/plate + (weak) Saichenko &
reverse mutation Sharapora
(1987)
TA98 S. typhimurium - Haworth et al.
reverse mutation (1983)
TA98 S. typhimurium NiCl2 - Arlauskas et
reverse mutation NiSO4 al. (1985)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
TA100 S. typhimurium - Haworth et al.
reverse mutation (1983)
TA100 S. typhimurium NiCl2 - Arlauskas et
reverse mutation NiSO4 al. (1985)
Several strains S. typhimurium Ni(CH3CO2)2 - DeFlora et al.
reverse mutation NiCl2 - (1984)
Ni(NO3)2 -
Frameshift Co-mutagenesis with NiCl2 + Ogawa et al.
tester strains 9-aminoacridine (1987)
Co-mutagenesis with Ni(II) + Dubins &
alkylating agents (see Lavelle (1986)
pair substitution)
Escherichia coli
WP2 E. coli, reverse NiCl2 0, 5, 10, 25 - Green et al.
mutation mg/litre (1976)
WP2 E. coli (fluctuation) NiCl2 50 mmol/litre - Nishioka (1975)
WP2 uvra E. coli (fluctuation) NiCl2 50 mmol/litre -
CM571 E. coli (fluctuation) NiCl2 50 mmol/litre -
WP67 Differential toxicity NiCl2 0, 200, 500, + dose Tweats et al.
assay 1000 mg/litre response (1981)
CM871 Differential toxicity NiCl2 0, 200, 500, + dose Tweats et al.
assay 1000 mg/litre response (1981)
WP2 (repair Differential toxicity NiCl2 0, 200, 500, + dose
deficient) assay 1000 mg/litre response
Differential toxicity NiCl2 0, 200, 500, + dose
assay 1000 mg/litre response
WP2 E. coli, reverse NiCl2 - Arlauskas et
mutation NiSO4 - al. (1985)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Bacillus subtilis
H17 (rec+) B. subtilis, NiCl2 50 mmol/litre - Nishioka (1975)
M45 (rec-) rec. strains
differential toxicity
H17 (rec+) B. subtilis, NiCl2 5-50 mmol/litre - Kanematsu et
M45 (rec-) rec. strains NiO 5-50 mmol/litre - al. (1980)
differential toxicity Ni2O3 5-50 mmol/litre -
Corynebacterium
SP887 Reverse mutation NiCl2 0.03-10 mg/litre + dose Pikalet &
fluctuation test 8 doses response Necasek (1983)
at > 5.0
mg/litre
Salmonella
G46 (his) Host-mediated assay NiCl2 50 mg/kg - Buselmaier et
in mouse (NMRI strain) al. (1972)
Serrati marcescens
a21 (leu-) Host-mediated assay NiCl2 50 mg/kg - Buselmaier et
in mouse (NMRI strain) al. (1972)
Paramecium
Paramecium species Ni3S2 + at 0.5 Smith-Sonneborn
mutation µg/ml et al. (1983)
NiS particles + at 0.5
(1.8 µm) µg/ml
Paramecium species NiS particles + at 0.57 Smith-Sonneborn
chromosome aberration (1.8 µm) µg/ml et al. (1983)
Yeast (Saccharomyces)
D7 (diploid S. cerevisiae, gene NiCl2 3 or 10 mmol/litre + Fukunaga et al.
strain) conversion 24 h (1982)
D7 (diploid S. cerevisiae, gene NiSO4 5, 10, 20, 40 ? Singh (1984)
strain) conversion mmol/litre
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Yeast 19 haploid S. cervisiae, growth NiCl2 26 mmol/litre + Egilsson et al.
strains inhibition (1979)
Vicia faba
Chromosome aberrations NiO + Glaess (1956a)
Mitotic effects NiO + Glaess (1956b)
Vicia faba Mitotic effects NiCl2 + Komczynski et
Ni(NO3)2 + al. (1963)
NiSO4 +
Pisum Chromosome aberrations Ni(NO3)2 + Van Rosen (1954)
Allium cepa Chromosome aberrations NiO + Levan (1945)
INSECT SYSTEM
Drosophila
D. melanogaster Sex-linked recessive NiSO4 200, 300, 400 + Rodriguez-
white males lethal mutations mg/litre in 5% Arnaiz & Ramos
BASC females sucrose i.p. (1986)
D. melanogaster Sex chromosome NiSO4 200, 300, 400 + (weak)
XC2Y B/SC8Y loss assay mg/litre in 5%
males sucrose i.p.
Y2Wa females
D. melanogaster Somatic eye colour Ni(NO3)2 0.14 mmol/litre - Rasmuson (1985)
eggs from C(1)DX test system NiCl2 0.21 mmol/litre -
y,w,f females X
SC Z W+f males
D. melanogaster mutation Ni(NO3)2 3.4-6.9 mmol/litre +? Vogel (1984)
NiCl2 4.2 mmol/litre -
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
MAMMALIAN CELLS: DNA DAMAGE
Rat
Sprague-Dawley DNA strand breaks, NiCO3 5-40 mg/kg + Ciccarelli et
in vivo X links, alkaline al. (1981)
elution, in vivo Ciccarelli &
exposure Wetterhahn
(1985)
Primary DNA strand breaks, (+) Sina et al.
hepatocyte X links (1983)
Hamster
CHO DNA strand breaks NiCl2 100-500 µmol/litre + Robison &
alkaline sucrose Ni3S2 10 mg/litre + Costa (1982)
gradients (cryst.)
NiS (cryst.) 1-10 mg/litre +
NiS 10 mg/litre -
(amorphous)
CHO DNA strand breaks + Costa et al.
X links (1982)
CHO DNA strand breaks Ni3S2 10 mg/litre, 24 h + Patierno &
X links NiCl2 0.25-1.0 mmol/litre + Costa (1985)
Human
Normal human DNA strand breaks NiSO4 10-2000 mg/litre - Fornace (1982)
fibroblasts alkaline elution
XP cells
Peripheral DNA strand breaks NiCl2 0.05 mmol/litre ? McLean et al.
lymphocytes FADU technique (1982)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
MAMMALIAN CELLS: DNA BINDING
Hamster
CHO Binding to DNA in 63NiS 10 mg/litre + Harnett et al.
vitro (radioactive 63NiCl2 10 mg/litre + (10x (1982)
precursor) less)
CHO Binding to DNA in vivo 63NiCl2 + to Hui & Sunderman
liver & (1980)
63Ni(CO)4 + kidney
DNA
CHO Binding to RNA, 63NiS 10 mg/litre + Harnett et al.
protein 63NiCl2 10 mg/litre + (10x (1982)
less)
L132 pulmonary Binding, X-ray Ni3S2 Ni bound Chirali et
cells microprobe analysis to al. (1982)
phosphate
groups of
DNA, RNA
of cell
membranes
Human
HeLa Binding, colorimetric Ni Kovacs &
assay (dimethyl localized Darvas (1982)
glyoxime) in
centrioles
Mouse
FM3A cells Inhibition of DNA Ni(CH3CO2)2 0.6, 0.8, 1.0 + Umeda &
(mouse mammary synthesis mmol/litre Nishimura
carcinoma) (1979)
Nishimura &
Umeda (1979)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
CBA strain Inhibition of DNA NiSO4 15-30% LD50 - kidney Amlacher &
synthesis in vivo epithelium Rudolf (1981)
+ hepatic
epithelium
Rat
Fischer rat Inhibition of DNA NiCl2 + Basrur &
embryo cells synthesis; 3H TdR Gilman (1967)
uptake
Rat liver Inhibition of DNA Ni3S2 5, 10, 10, mg/litre + equal Swierenga &
epithelial cell synthesis; 3H TdR NiCl2 up to 150 mg/litre + potency McLean (1985)
line T51B uptake at
equitoxic
doses
Hamster
CHO S-phase inhibition, NiCl2 40-120 µmol/litre + Costa et al.
flow cytometry Ni metal 10-20 mg/litre + (1980b, 1982)
Ni3S2 1-10 mg/litre +
(cryst.)
Ni3Se2 1-5 mg/litre +
(cryst.)
NiO 5 mg/litre +
Human
HeLa Inhibition of DNA NiCl2 - Painter &
synthesis; 3H TdR uptake Howard (1982)
Bronchial Inhibition of DNA NiSO4 + Lechner et al.
epithelial cells synthesis; 3H TdR uptake (1984)
MAMMALIAN CELLS: DNA REPAIR
Rat
Fischer rat UDS NiCl2 Inhibition Swierenga et
primary of MMS- al. (1987)
hepatocytes induced
UDS
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Hamster
CHO Repair induction NiCl2 100-1000 µmol/litre + Robison &
SHE Cesium chloride Ni3S2 10 mg/litre + Costa (1982)
gradients (cryst.)
NiS (cryst.) 1-10 mg/litre +
NiS 5-10 mg/litre -
(amorphous)
MAMMALIAN CELLS: GENE MUTATION
Mouse
L5178Y mouse Thymidine kinase locus NiCl2 0.16-0.53 mmol/ + dose Amacher &
lymphoma cells litre (5 concs.) response Paillet
(toxic) (1980)
L5178Y mouse Thymidine kinase locus NiSO4 300-800 mg/litre + (weak) McGregor et
lymphoma cells at toxic al. (1988)
levels
Rat
T51B rat liver HPRT locus Ni3S2 5-20 mg/litre + Swierenga &
epithelial cells McLean (1985)
NRK, normal rat Frameshift and base NiCl2 20-40 µg/litre + Biggart &
kidney cells, pair substitution Murphy (1988)
with integrated
viral gene
6m2 murine Gene expression NiCl2 20-160 µmol/litre + dose
sarcoma virus- dependent
infected cells
Hamster
V79 HPRT locus NiCl2 0.4, 0.8 mmol/litre - Miyaki et al.
(1979)
V79 HPRT locus NiCl2 + Hartwig &
Beyersmann
(1989)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
V79, HGPRT with HPRT locus Ni(II) + Christie &
integrated E.coli Tummolo (1988)
gpt gene
CHO HPRT locus NiCl2 ? Hsie et al.
(1979)
CHO HPRT locus Ni3S2 + (weak) Costa et al.
NiS + (weak) (1980)
SHE Ouabain resistance NiSO4 0.019 mmol/litre - Rivedal &
+ as Sanner (1980)
comutagen
with BaP
MAMMALIAN CELLS: SISTER CHROMATID EXCHANGE (SCE)
Mouse \
FM3A cells mouse SCE NiCl2 6 x 10-4-10-3 mol/ + | Nishimura &
mammary carcinoma litre | Umeda (1979)
|
FM3A cells mouse Ni(CH3CO2)2 6 x 10-4-10-3 mol/ + | recovery
mammary carcinoma litre | period
NiK2(CN)4 1-1.6 x 10-3 mol/ + | included
litre |
NiS 4-8 x 10-4 mol/ + |
litre /
NiSO4 2.3 x 10-6 to +
2.3 x 10-3 mol/litre
P338D1 mouse SCE NiSO4 10-4 + Andersen (1983)
macrophage line
Hamster
Don cells SCE NiSO4 0.19 mmol/litre + at LC50 Ohno et al.
NiCl2 0.13 mmol/litre + at LC50 (1982)
CHO SCE NiSO4 0.95, 2.85 µmol/ + at 0.75 Deng & Qu
litre mg/litre (1981)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
CHO SCE NiS (cryst.) 5-20 mg/litre + Sen & Costa
NiCl2 1-1000 µmol/litre + (1985)
SHE SCE NiSO4 3.8, 9.5, 19 µmol/ + dose Larramendy
litre response et al. (1981)
Human
Peripheral blood SCE NiCl2 0.0-1-1.0 mmol/ + dose Newman et al.
lymphocytes, litre response (1982)
in vitro at 0.1
mmol/litre
Peripheral blood SCE NiSO4 0.0023-2.3 mmol/ + dose Wulf (1980)
lymphocytes, litre response
in vitro 0.02, 0.2
mmol/litre
Peripheral blood SCE NiSO4 0.95, 2.85 µmol/ + dose Deng & Qu
lymphocytes, litre response (1981)
in vitro
Peripheral blood SCE NiSO4 9.5, 19.0 µmol/ + dose Larramendy et
lymphocytes, litre response al. (1981)
in vitro
Peripheral blood SCE Ni3S2 10-4 mol/litre + (weak) Andersen (1983)
lymphocytes,
in vitro
Peripheral blood SCE Ni3S2 0.001-100 mg/litre ? Saxholm et al.
lymphocytes (1981)
Peripheral blood SCE nickel 2-20 mg/litre + (weak) Djachenko
lymphocytes (1989)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
MAMMALIAN CELLS: CHROMOSOME ABERRATIONS (CA)
Mouse
In vivo CA, in Ni-induced NiS (cryst.) + aneup- Christie et al.
rhabdomyosarcomas loidy, (1988)
marker
chromosome
BalbC in vivo Micronucleus Ni(NO3)2 56 mg/kg - Deknudt &
NiCl2 25 mg/kg - Leonard (1982)
FM3A cells CA Ni(CH3CO2)2 0.2, 0.3, 0.6, + gaps Umeda &
mammary carcinoma 1 mmol/litre; 24, Nishimura
48 h recovery (1979)
NiCl2 0.2, 0.3, 0.6, ? gaps
1 mmol/litre; 24,
48 h recovery
NiS 0.2, 0.3, 0.6, 1 + gaps,
mmol/litre; 24, breaks,
48 h recovery exchanges
FM3A cells CA Ni(CH3CO2)2 0.6, 0.8, 1.0 mmol/ + gaps, Nishimura &
mammary carcinoma litre; 6, 24, 48 h breaks, Umeda (1979)
recovery exchanges
NiCl2 0.6, 0.8, 1.0 mmol/ + gaps,
litre; 6, 24, 48 h breaks,
recovery exchanges
NiS 0.6, 0.8, 1.0 mmol/ + gaps,
litre; 6, 24, 48 h breaks,
recovery exchanges
FM3A cells CA Ni(CH3CO2)2 10-4 mol/litre + gaps Morita et al.
mammary carcinoma (2-3 days) breaks, (1985)
NiCl2 exchanges
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Rat
In vivo CA, rat bone marrow NiSO4 3, 6 mg/kg for - Mathur et al.
CA, spermatogonial cells 7 and 14 days (1978)
Hamster
Chinese hamster CA NiCl2 1/5, 1/10, 1/25 + dose Chorvatocvicova
in vivo LD50 response (1983)
1/10, 1/5
Chinese hamster CA NiCl2 1-1000 µmol/litre dose Sen & Costa
in vitro 2 h + 24 h recovery response (1985)
gaps,
breaks,
exchanges
NiS (cryst.) 5-20 mg/litre + at 20 mg/
litre, gaps,
breaks,
exchanges
Syrian hamster CA NiSO4 0.019 mmol/litre + gaps, Larramendy et
in vitro 24 hr breaks, al. (1981)
exchanges,
minutes,
dicentrics
Human
Peripheral blood CA NiSO4 0.19 mmol/litre + breaks, Larramendy et
lymphocytes rings, al. (1981)
in vitro minutes
Peripheral blood CA Ni powder - Paton & Allison
lymphocytes NiO (1972)
in vitro
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Peripheral blood CA Ni(CH3CO2)2 10-100 mg/litre - Voroshilin et
lymphocytes al. (1977)
in vitro
Peripheral blood CA NiCl2 2-20 mg/litre + Djachenko
lymphocytes (1989)
in vitro
MAMMALIAN CELLS: CELL TRANSFORMATION
Mouse
C3H10T Transformed foci Ni3S2 0.001, 0.1 mg/litre + Saxholm et al.
(1981)
Mouse embryo Inhibition of NiS (cryst.) 1-20 mg/litre + Sonnenfeld et
fibroblasts interferon production NiS (amorph.) 1-20 mg/litre - al. (1983)
Rat
HRT cells Initiation & promotion NiSO4 40 µmol/litre + Eker & Sanner
(hereditary renal test (1983)
tumour)
Rat embryo cells Transformed foci NiSO4 0.19, 0.38 mmol/ + Traul et al.
infected with litre (1981)
Rauscher leukaemia
virus
NRK (normal rat Transformed foci NiSO4 38-152 µmol/litre + max at Wilson &
kidney) cells, 10 mg/ Khoobyarian
infected with litre (1982)
Maloney murine
sarcoma virus
T51B rat liver Calcium independence, alphaNi3S2 2.5 mg/litre, + Swierenga et
epithelial cells growth control and long-term al. (1989)
morphology, differen- exposure
tiation induction
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Rat tracheal Transformed foci alphaNi3S2 5 mg/litre, 24 h + Feren & Reith
primary epithelial (1988)
cells
Hamster
SHE Transformed foci NiSO4 10, 19, 38 µmol/ + dose DiPaulo &
litre response Costa (1979)
Ni3S2 1.0, 2.5, 5.0 mg/ + dose
litre response
NiS up to 20 mg/litre -
(amorphous)
SHE Transformed foci NiSO4 3.8, 19, 76 µmol/ + at 19, Rivedal &
litre 76 µmol/ Sanner (1980)
litre co-
carcinogen
with BaP
SHE Transformed foci NiSO4 19 µmol/litre + at 19, Rivedal &
+ BAP 76 µmol/ Sanner (1981)
litre co-
carcinogen
with BaP
SHE Transformed foci NiSO4 19 µmol/litre + with Rivedal et al.
cigarette (1980)
smoke
extract
SHE Transformed foci Ni3S2 0.1, 1.0 mg/litre + at Costa et al.
(crystalline) subtoxic (1979)
doses,
confirmed
in nude
mice
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Hamster
SHE Transformed foci NiS 0.1, 1.0 mg/litre Costa et al.
(amorphous) (1979)
\
SHE Transformed foci Ni 5, 10, 20 mg/litre + | Costa et al.
NiO, Ni2O3 + | (1981a)b
NiS (cryst.) + | crystalline
NiS (amorph.) + | compounds
Ni3S2 (cryst.) + | more
Ni3S2 (cryst.) + | potent
/
SHE Transformed foci Inco Black 5, 10, 20 mg/litre + Sunderman et
NiO (jet al. (1987a)
black, 5 µm
particle size)
\
BHK-21 (baby Anchorage independence alphaNi3S2 5-20 mg/litre + | Hansen & Stern
hamster kidney) Ni2O3 5-20 mg/litre + | (1983)
NiO 37.5-150 mg/litre + | anchorage
Ni(CH3CO2)2 100-400 mg/litre + | independence
MIG nickel 100-400 mg/litre + | at equitoxic
welding fume / concentrations
Human
Bronchial Growth control and NiSO4 5-20 mg/litre + Lechner et al.
epithelial cells morphology (1984)
\
Foreskin Anchorage independence Ni(CH3)CO2)2 10 µmol/litre + | Biedermann &
fibroblasts NiO 50 µmol/litre + | Landolph (1986)
NiSO4 10 µmol/litre + | anchorage
Ni3S2 10 µmol/litre + | independence at
/ LC50 concentrations
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
MAMMALIAN CELLS: OTHER TESTS
Balb/c mouse Dominant lethal NiCl2 25 mg/kg - pre- DeKnudt &
in vivo Ni(NO3)2 56 mg/kg implant- Leonard (1982)
ation loss
observed
Balb/c mouse Dominant lethal Ni(NO3)2 8.1, 11.3 mg/kg - pre- Jacquet &
in vivo implant- Mayence (1982)
ation loss
observed
Rat Dominant lethal NiCl2 0.005-50 mg/kg - pre- Saichenko et
for 24 weeks implant- al. (1985)
ation loss
observed
Rat embryo cells Spindle disturbance NiCl2 1 mg Ni/litre + Swierenga &
in vitro culture medium Basrur (1968)
Human lymphocytes Spindle disturbance NiSO4 10-3 mol/litre + Andersen (1985)
NIH3T3 cells Inhibition of NiSO4 0.5-20 mmol/litre + dose Miki et al.
intercellular response (1987)
communication
(radioisotope transfer)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Human Induction of free Ni3S2 + more Evans et al.
polymorphonuclear radicals potent (1988)
leukocytes (chemiluminescent NiO +
technique)
---------------------------------------------------------------------------------------------------------
a No figures means no concentration data given.
b Transformation experiments with this system have been repeated numerous times for mechanistic studies.
Findings include:
- crystalline but not amorphous nickel compounds are actively phagocytosed in vitro (Costa &
Mollenhauer, (1980b);
- reduction of amorphous compounds with LiA1H4 enchances phagocytosis (Costa & Mollenhauer, 1980b);
- phagocytosis is Ca-dependent (Heck & Costa, 1982);
- phagocytosed particles are contained in cytoplasmic vesicles where they slowly dissolve, releasing
nickel ions (Evans et al., 1982);
- nickel compounds selectively damage heterochromatin (Sen & Costa, 1985);
- NiS (cryst.) and NiCl2 preferentially transform male SHE cells (Costa et al., 1981a);
- observed damage includes deletion of long arm of X chromosome (Conway & Costa, 1989).
8.2.1. Mutagenesis in bacteria and mammalian cells
Nickel compounds were generally inactive in bacterial mutation
assays. In some cases, where standard procedures were modified by,
for example, using fluctuation assays (LaVelle & Witmer 1981;
Pikalek & Necasek, 1983) or co-mutagenesis tests with other
carcinogenic agents (Dubins & LaVelle, 1986; Ogawa et al., 1987),
positive results were obtained.
The mutagenic effects of nickel chloride at the hypoxanthine-
guanine phosphoriboxyl transferase (HGPRT) locus were studied in
cultured V79 Chinese hamster cells and Chinese hamster ovary (CHO)
cells (Hsie et al., 1979). Hsie et al. (1979) claimed positive
results without reporting the detailed data. Miyaki et al. (1979)
obtained a negative response. However, Hartwig & Beyersmann (1989)
obtained a positive response with the same cell line, but the
response occurred only in a serum-free medium.
Amacher & Paillet (1980) studied nickel chloride at the
thymidine kinase (TK) locus in L51784Y mouse lymphoma cells and
found a dose-dependent increase in the number of mutants of up to 4
times that in the controls. The possibility that this response was
due to chromosome damage, also detected in this assay, cannot be
excluded.
Swierenga & McLean (1985) used epithelial cells from rat liver
(T51B) to study the genotoxic effects of nickel chloride and also
of nickel subsulfide, either as an aqueous suspension of washed
particles or as an aqueous solution. All produced increased
numbers of mutations at the hypoxanthine-guanine phosphoriboxyl
transferase (HGPRT) locus, over a range of concentrations.
In all the gene mutation studies using mammalian cells, any
response following exposure to nickel compounds was associated with
considerable cell toxicity.
8.2.2. Chromosomal aberration and sister chromatid exchange (SCE)
Nishimura & Umeda (1979) compared the effects of nickel
chloride, nickel acetate, potassium cyanonickelate and nickel
sulfide on the induction of chromosomal aberrations in cultured
FM3A mouse mammary carcinoma cells. The 4 compounds elicited
similar inhibitory effects on the synthesis of protein, RNA, and
DNA. The chromosomal aberrations were manifested as breaks,
exchanges, and fragmentations. Treatment of Chinese hamster ovary
(CHO) cells with crystalline nickel sulfide and nickel chloride
induced chromosomal aberrations including gaps, breaks, and
exchanges. In both cases, the heterochromatic centromeric regions
of the chromosomes were preferred; nickel sulfide also caused
selective fragmentation at the heterochromatic long arms of the
X-chromosome.
Chromosomal damage and increased SCE were induced in Syrian
hamster embryo (SHE) cells and human lymphocyte cultures treated
with nickel sulfate (Larramendy et al., 1981).
Wulf (1980) showed that nickel sulfate and nickel subsulfide
could produce SCE in human lymphocytes in vitro. A significant
increase in SCE in human lymphocytes exposed to nickel subsulfide
was reported by Saxholm et al. (1981). Newman et al. (1982)
observed increased SCE in human lymphocytes exposed to nickel
chloride. Ohno et al. (1982) investigated the induction of SCE by
nickel sulfate and nickel chloride in Don Chinese hamster cells and
found significantly increased frequencies of SCE.
8.2.3. Mammalian cell transformation
These assays may not necessarily have a "genotoxic" endpoint as
they also predict the carcinogenic potential of compounds that act
by non-genotoxic mechanisms.
DiPaolo & Casto (1979) studied the capacity of nickel sulfate,
nickel subsulfide, and amorphous nickel monosulfide to induce
morphological transformation in Syrian hamster embryo (SHE) cells
in vitro. Amorphous nickel monosulfide did not produce
transformation at plate concentrations as high as 20 µg/ml medium.
Nickel sulfate, tested at plate concentrations of 2.5, 5, and 10
µg/ml medium, caused transformation in a dose-dependent manner.
Nickel subsulfide produced a higher percentage of transformations,
in a dose-dependent manner, than nickel sulfate, when tested at
plate concentrations of 1.0, 2.5, and 5 µg/ml medium. In a study by
Costa et al. (1979), nickel subsulfide caused morphological
transformation, in a dose-dependent manner, of Syrian hamster fetal
cells, at plate concentrations of 0.1, 1.0, 5.0, and 10.0 µg/ml
medium, but, under the same experimental conditions, nickel
monosulfide, at plate concentrations of 0.1, 1.0, and 5.0 µg/ml
medium, did not.
Costa et al. (1981) demonstrated that the incidence of
morphological transformations of SHE and CHO cells, exposed to
various nickel compounds, was directly correlated with
phagocytosis. Subcutaneous implantation of transformed SHE cells
led to the development of fibrosarcomas in nude mice (Costa et al.,
1979). Nickel sulfate caused cell transformation in cultured Syrian
hamster embryo cells (Pienta et al., 1977).
Sunderman et al. (1987a) compared nickel oxides with different
physical and chemical properties in the SHE cell transformation
assay, and in vivo responses (Table 28). Three out of the 4
compounds that were active in the transformation assay were also
positive in vivo when injected intramuscularly.
Table 28. Comparison of activity of some nickel oxides with different physiochemical
characteristics in in vitro transformation assays and in vivo carcinogenesis
---------------------------------------------------------------------------------------
Compound Concentration Resulta No. of tumours
observed (20 mg
Ni/rat im)b
---------------------------------------------------------------------------------------
Nickel oxides Calcination
temperature
INCO Black (jet black) 650 °C 5, 10, 20 mg/litre + 6/15
Grey black 735 °C Particle size 5 µm + 0/15
Green 1045 °C - 0/15
Nickel copper oxides Ni-Cu ratio
Maroon 2.5:1 5, 10, 20 mg/litre + 13/15
Red-brown 5.2:1 Particle size 5 µm + 15/15
alphaNiS (positive + 15/15
control)
---------------------------------------------------------------------------------------
a Sunderman et al. (1987a).
b Sunderman et al. (1988a).
Hansen & Stern (1984) compared the abilities of welding fume,
nickel powder, nickel acetate, black nickel oxide, black nickel
oxide catalyst (a commercial catalyst for organic reactions, which
is a mixture of nickel(II) and nickel(IV) oxides (NiO1.4 x 3H2O),
and nickel subsulfide, in the in vitro transformation of Syrian
baby hamster kidney BHK-21 cells. Although a wide range of
transformation potency was found, the compounds produced the same
number of transformed colonies at the same degree of toxicity (50%
survival). The authors concluded that this indicated that if
toxicity is a direct measure of net available nickel, then nickel
or the nickel ion is the ultimate transforming agent. In a
subsequent BHK-21 mammalian cell assay, Stern et al. (1985)
determined the 50% toxicity of the soluble and insoluble fraction
of nickel welding fume and found that only the insoluble fraction
showed a transformation potential.
Synergistic effects of nickel compounds and benzo (a)pyrene on
morphological transformation of SHE cells have been reported.
Costa & Mollenhauer (1980c) found that pretreatment of the cells
with benzo (a)pyrene enhanced the cellular uptake of nickel
subsulfide. Treatment with nickel sulfate and benzo (a)pyrene
resulted in a transformation frequency of 10.7% compared with 0.5%
and 0.6%, respectively, for the individual substances (Rivedal &
Sanner, 1980).
8.3. Other test systems
Smith-Sonneborn et al. (1986) used the ciliated protozoan
Paramecium to quantify the effects of pure nickel powder, iron-
nickel powder, and nickel subsulfide. Genotoxicity was indicated
by significant increases in the fraction of non-viable offspring
(presumed index of lethal mutation) found after autogamy in parents
from the nickel-treated groups compared with the controls. Only
nickel subsulfide consistently induced a significant increase in
offspring lethality.
Rodriguez-Arnaiz & Ramos (1986) studied the mutagenic potential
of nickel sulfate in the Drosophila sex-linked recessive lethal
(SLRL) assay in vivo. Nickel sulfate induced SLRL in a dose-
dependent way, whereas sex chromosome loss was only detectable in
significant numbers at the highest concentration.
8.4. Carcinogenicity
8.4.1. Inhalation
Studies on inhalation exposure are summarized in Table 29.
Hueper (1958) and Hueper & Payne (1962) studied the effects of
exposure to airborne concentrations of elemental nickel. Hueper
(1958) exposed 20 female C57BL mice, 50 male and 50 female Wistar
rats, 60 female Bethesda black rats, and 32 male and 10 female
guinea-pigs to an atmosphere containing 99% pure nickel powder
(particle size 4 µm or less) at 15 mg/m3, for an exposure period of
6 h/day, 4-5 days/week, for up to 21 months. There were no control
groups. There were no lung tumours, but 2 lymphosarcomas were seen
in the mice. However, most animals died before 15 months. Fifteen
out of 50 rats of both strains that were studied histologically
showed adenomatoid formations in the lung, which were classified as
benign neoplasms. At death, most of the guinea-pigs exhibited
adenomatoid proliferations. One animal had an intra-alveolar
carcinoma. The results of a later study (Hueper & Payne, 1962) on
rats and hamsters did not reveal lung tumours following inhalation
exposure to 99% pure nickel together with 56-98 mg sulfur
dioxide/m3 (20-35 ppm) and powdered limestone.
In studies by Kim et al. (1969) a group of 77 male Wistar rats
was exposed to metallic nickel dust, equivalent to 3.1 mg/m3 for 6
h a day, 5 days/week, over 21 months, followed by exposure to air
alone; 98% of the dust particles were less than 2 µm in diameter.
Sub-groups were exposed to the dust for various periods followed by
periods of recovery. Two exposed rats developed lung tumours of a
carcinoid pattern and a similar tumour was found in one rat in the
unexposed control group.
Table 29. Experimental animal studies on the carcinogenicity studies of nickel compounds administered by inhalation or
tracheal instillation
---------------------------------------------------------------------------------------------------------------------------
Nickel compound Animal Dosage schedule Lung tumours detected Comments References
(group size)
---------------------------------------------------------------------------------------------------------------------------
Inhalation studies
Nickel powder C57BL female 15 mg/m3, 6 h/day, no lung tumours, all animals died Hueper (1958)
mice (20) 4-5 days/week for 2 lymphosarcomas by week 60, no
60 weeks control group
Nickel powder Wistar rats 15 mg/m3, 6 h/day, numerous multicentric histology on 50 Heuper (1958)
(108); Bethesda 4-5 days/week, for adenomatoid animals only, no
black rats (60) 60 weeks proliferations in 15 no control group
animals
Nickel powder Guinea-pigs 15 mg/m3, 6 h/day, adenomatous alveolar all animals died Heuper (1958)
(42) 4-5 days/week, for lesions in almost all by 21 months
60 weeks animals
Nickel powder + Bethesda black level not specified no lung tumours Heuper & Payne
powdered rats (120) (1962)
limestone + Hamsters (100)
sulfur dioxide
Nickel powder Wistar rats 3.1 mg/m3, 6 h/day, carcinoid lung tumour Kim et al.
(77) 5 days/week for 21 in 2 exposed and in 1 (1969)
months (?) control rat
Ni(CO4) Wistar rats 30 mg/m3, 30 min/ 1 lung carcinoma no lung tumours Sunderman et al.
(64) day, 3 days/week, in a control group (1959)
for 52 weeks of 41 animals
Ni(CO4) Wistar rats 60 mg/m3, 30 min/ 1 lung carcinoma 9 animals survived Sunderman et al.
(32) day, 3 days/week, for 2 years (1959)
for 52 weeks
Ni(CO4) Wistar rats single exposure to 2 lung carcinomas Sunderman et al.
(80) 250 mg/m3 (30 min?) (1959)
---------------------------------------------------------------------------------------------------------------------------
Table 29 (contd.)
---------------------------------------------------------------------------------------------------------------------------
Nickel compound Animal Dosage schedule Lung tumours detected Comments References
(group size)
---------------------------------------------------------------------------------------------------------------------------
Ni(CO4) Wistar rats 30 mg/m3, 30 min/ 1 lung adenocarcinoma Sunderman &
(64) day, 3 days/week, Donelly (1965)
for lifetime
Ni(CO4) Wistar rats single exposure to 1 lung adenocarcinoma 214/285 animals Sunderman &
(285) 600 mg/m3 for 30 min died within 3 weeks Donelly (1965)
Nickel F344 rats 0.97 mg/m3, 6 h/day, 14 malignant and 15 survival: 5% in Ottolenghi et
subsulfide (226) 5 days/week, for 78 benign lung tumours in nickel-exposed al. (1974)
weeks, followed by the exposed, 1 rats; 31% in
observation for 30 malignant and 1 benign controls
weeks in the control animals
P <0.01
Black nickel Syrian golden 53.2 mg/m3, 7 h/day, no significant increase massive Wehner et al.
oxide hamsters (102) 5 days/week, for in incidence of pneumoconiosis in (1975)
2 years respiratory tumours exposed animals;
part of animals also
exposed to tobacco
smoke
Green nickel Wistar rats 0.6 mg/m3, 6 h/day, 1 adenocarcinoma and 5 control animals Horie et al.
oxide (6) 5 days/week, for 4 1 adenomatous pulmonary (1985)
weeks, followed by lesion
observation for 80
weeks
Green nickel Wistar rats 8 mg/m3, 6 h/day, 1 adenomatous lesion Horie et al.
oxide (8) 5 days/week, for 4 in lungs (1985)
weeks, followed by
observation for 80
weeks
Nickel-enriched Syrian golden fly ash containing no lung tumours 20 of the animals Wehner et al.
coal fly ash (Ni hamster (102) 6% Ni, 70 mg/m3, removed from (1981)
acetate added to 6 h/day, 5 days/ exposure for other
coal before week, for 20 months studies
burning)
---------------------------------------------------------------------------------------------------------------------------
Table 29 (contd.)
---------------------------------------------------------------------------------------------------------------------------
Nickel compound Animal Dosage schedule Lung tumours detected Comments References
(group size)
---------------------------------------------------------------------------------------------------------------------------
Nickel-enriched Syrian golden fly ash containing no lung tumours Wehner et al.
coal fly ash (Ni hamsters 6% Ni, 17 mg/m3, (1981)
acetate added to 6 h/day, 5 days/week,
coal before for 20 months
burning)
fly ash containing no lung tumours Wehner et al.
0.3% Ni, 70 mg/m3, (1981)
6 h/day, 5 days/week,
for 20 months
Nickel oxide Wister rats 60 µg/m3 continuous no lung tumours Alveolar Glaser et al.
(60) exposure for 18 proteinosis in (1986)
months nickel-exposed
animals
200 µg/m3 continuous no lung tumours
exposure, doe, 18
months
Intratracheal instillation
Nickel powder Wistar rats 0.3 mg Ni x 20, at 1 adenoma, 1 no lung tumours in Pott et al.
(39) weekly intervals adenocarcinoma, 8 in 40 control (1987)
squamous cell animals
carcinomas
Wistar rats 0.9 mg Ni x 10, at 3 adenocarcinomas, Pott et al.
(32) weekly intervals 4 squamous cell (1987)
carcinomas, 1 mixed
tumour
Nickel oxide Wistar rats 5 mg x 10, at 4 adenocarcinomas, Pott et al.
(not specified) (37) weekly intervals 4 squamous cell (1987)
carcinomas, 2 mixed
tumours
Wistar rats 15 mg x 10, at 12 squamous cell
(38) weekly intervals carcinomas
---------------------------------------------------------------------------------------------------------------------------
Table 29 (contd.)
---------------------------------------------------------------------------------------------------------------------------
Nickel compound Animal Dosage schedule Lung tumours detected Comments References
(group size)
---------------------------------------------------------------------------------------------------------------------------
Black nickel Hamsters 4 mg x 30, at no lung tumours 3 hamsters in both Farrell & Davis
oxide (50) weekly intervals groups survived for (1974)
12 months
Black nickel Rats (26) single 1 squamous cell the nickel oxide Saknyn & Blokhin
oxide administration carcinoma of the dust contained (1978)
20-40 mg lung 64.7% NiO, 0.13%
NiS, 0.18% Ni; no
pulmonary tumours
in 47 controls
Nickel B6C3F1 mice 0.024, 0.056, 0.156, no neoplastic or Fisher et al.
subsulfide (20) 0.412, or 1.1 mg/kg, non-neoplastic (1986)
four doses at weekly lesions
intervals, follow-up
of 27 months
Ni3S2 Rats (47) 0.063 mg x 15, at 2 adenocarcinomas, no pulmonary Pott et al.
weekly intervals 5 squamous cell tumours in control (1987)
carcinomas rats
Ni3S2 Rats (45) 0.125 mg x 15, at 3 adenocarcinomas, 6 no pulmonary Pott et al.
weekly intervals squamous cell tumours in control (1987)
carcinomas, 4 mixed rats
tumours
Ni3S2 Rats (40) 0.25 mg x 15, at 7 adenocarcinomas, no pulmonary Pott et al.
weekly intervals 4 squamous cell tumours in control (1987)
carcinomas, 1 mixed rats
tumours
Powder Hamsters, 0.8 mg powder, 1 lung tumour (nickel no pulmonary Muhle et al.
Ni3S2 (30-40 per sex) 0.1 mg alphaNi3S2, powder group) tumours in positive (1988)
Pentlandite 3 mg pentlandite, 1 lung tumour control group
Cr-Ni(55) Alloy 3 or 9 mg Cr-Ni(55) (pentlandite group)
Cr(55) Alloy alloy, 9 mg Cr(55)
alloy; 12 times at
14-day intervals
---------------------------------------------------------------------------------------------------------------------------
Ottolenghi et al. (1974) found a significantly higher incidence
of pulmonary hyperplastic and neoplastic lesions in 226 Fischer 344
rats of both sexes exposed to nickel subsulfide (0.97 mg nickel/m3;
70% of particles smaller than 1 µm) for 6 h/day, 5 days/week, over
78 weeks. The overall incidence of lung tumours (adenoma and
adenocarcinomas) in treated animals was 14% compared with 1% in the
controls. The nickel sulfide-exposed rats also had a higher
incidence of respiratory tract inflammation.
The pathogenic effect of inhaled black nickel oxide was
investigated in hamsters by Wehner et al. (1975). Random-bred ENG:
ELA Syrian golden hamsters (102 males) were exposed to respirable
aerosols of nickel oxide (count median diameter 0.3 µm) at a
concentration of 53.2 mg/m3 for 7 h/day, on 5 days/week, for up to
2 years. Half of the animals were also exposed (nose-only) to
cigarette smoke for 10 min, 3 times a day. Histopathological
examination revealed increasing cellular response, proliferative
and inflammatory, in animals dying late in the study.
Histopathologically, there were no marked differences between the
nickel-oxide-plus cigarette smoke and the nickel-oxide-only exposed
groups. It was concluded that there was neither a significant
carcinogenic effect of nickel oxide nor a co-carcinogenic effect of
cigarette smoke. However, though not statistically significant, 3
malignant musculoskeletal tumours were found among nickel oxide-
exposed hamsters (Wehner et al., 1975).
Wehner et al. (1981) exposed 4 groups, each of 102 male Syrian
golden hamsters (outbred LAK:LVG), through inhalation to nickel-
enriched fly ash (NEFA) for 6 h/day, 5 days/week, for up to 20
months. The first group was exposed to 70 mg NEFA/m3 aerosol (4 mg
nickel/m3), the second group to 17 mg NEFA/m3 aerosol (1 mg
nickel/m3), the third group was exposed to 70 mg fly ash (FA)/m3
aerosol containing 0.21 mg nickel/m3, and the fourth group was
exposed to filtered air (control group). Five animals from each
group were killed after 4, 8, 12, and 16 months of exposure.
Additional groups of 5 animals were withdrawn from exposure at the
same time intervals and maintained for observation up to the
twentieth month of the study, when all the animals were killed.
Dust deposition, interstitial reaction, and bronchiolization in the
lungs were higher in the high-NEFA and FA groups than in the low-
NEFA group, indicating that dust quantity rather than actual nickel
content may be the major factor in determining tissue response.
While 2 malignant pulmonary tumours were found in 2 hamsters of the
high-NEFA group, no statistically significant carcinogenesis was
observed.
Horie et al. (1985) studied the carcinogenic effects of
inhalation of 8.0 or 0.6 mg green nickel oxide/m3 on male Wistar
rats (5-8 rats/dose group). The exposure time was 6 h/day, 5
days/week, for 1 month. Animals were killed 20 months after
exposure. There was one adenocarcinoma in a low-dose animal. In a
study by Glaser et al. (1986), male Wistar rats were exposed
continuously for 18 months to nickel oxide aerosols (60 or 200 µg
nickel/m3). The nickel oxide aerosols were generated by
atomization of aqueous nickel acetate solutions and subsequent
pyrolysis. No lung tumours were observed.
Sunderman et al. (1959) and Sunderman & Donelly (1965) reported
carcinogenesis in rats following inhalation exposure to nickel
carbonyl. In the first study (Sunderman et al., 1959), groups of
64 and 32 male Wistar rats were exposed to 30 and 60 mg nickel
carbonyl/m3, respectively, for 30 min, 3 days/week, for one year.
A further group was exposed once to 250 mg nickel carbonyl/m3.
Four out of the 9 animals that survived 2 years developed neoplasms
of the lung; 2 of these animals were in the single-exposure group,
the other 2 rats were in the repeated-exposure groups. No
pulmonary tumours were seen in 41 control animals, the death rate
of which was similar to that in the animals exposed to nickel
carbonyl. Sunderman & Donelly (1965), using 285 male Wistar rats,
observed pulmonary adenocarcinoma with metastases in one of the 35
rats that survived 2 or more years after a single 30-min inhalation
of 600 mg nickel carbonyl/m3. In a further group of 64 male rats,
exposed to repeated inhalation of nickel carbonyl (30 mg/m3 for 30
min, 3 days/week, until death), one pulmonary adenocarcinoma with
metastases developed among 8 rats that survived 2 or more years.
The control animals did not show any tumours. Because of the
rarity of spontaneous pulmonary malignancies in Wistar rats, it was
suggested that the tumours observed in the two studies were due to
inhalation of nickel carbonyl. Survivability of treated and
control animals was poor in both studies. Statistical analysis
could not be performed because of small sample size.
Kasprzak et al. (1973) did not find any lung tumours in 13 rats
following intratracheal instillation of 5 mg nickel subsulfide.
Thirty weekly intratracheal injections of 4 mg black nickel oxide
did not produce lung tumours in 50 hamsters, but only 3 animals
survived (Farrell & Davis, 1974). In a study by Saknyn & Blokhin
(1978) 26 rats were exposed to 20-40 mg nickel oxide by
intratracheal instillation; 1 lung carcinoma was found. In a
series of studies, Pott et al. (1987) examined the potential
carcinogenic effect of a number of dusts, including nickel oxide,
nickel powder, and nickel subsulfide, at various concentrations, in
rats. All 3 nickel compounds produced lung tumours, including
adenocarcinomas and squamous cell carcinomas, nickel subsulfide
exhibiting the strongest effect in relation to dose. Lung tumour
incidence was 14.9% (7 out of 47 animals), 28.9% (12 out of 45
animals), and 30% (12 out of 40 animals) following 15 weekly
intratracheal injections each containing 0.063 mg, 0.125 mg, and
0.25 mg nickel, respectively. Nickel powder was given
intratracheally in 20 weekly doses of 0.3 mg nickel; an additional
group was given 10 weekly doses of 0.9 mg nickel. Lung tumours
occurred in 10 out of 39 animals (25.6%) and in 8 out of 32 animals
(25.0%), respectively. Ten weekly instillations of nickel oxide,
each corresponding to a nickel content of 5 or 15 mg, produced
27.0% (10/37 animals) or 31.6% (12/38 animals) lung tumours,
respectively. No lung tumours occurred in the controls (40
animals). In a study with nickel subsulfide using the
intratracheal route, no increase in tumour incidence was observed
compared with the controls. Doses ranged from 0.024 to 1.1 mg/kg,
administered once a week for 4 weeks (Fisher et al., 1984).
Muhle et al. (1988) administered nickel powder (cumulative dose
9.6 mg), nickel subsulfide (cumulative dose 1.2 mg), pentlandite
(cumulative dose 36 mg), chromium-nickel-steel (cumulative dose 36
or 108 mg) and chromium-containing stainless steel (cumulative dose
108 mg) to Syrian golden hamsters (30-40 animals per sex and dose
group) by intratracheal instillation, 12 times every 14 days. One
lung tumour was found after treatment with nickel powder and one
tumour after treatment with pentlandite.
8.4.2. Oral
Schroeder et al. (1964, 1974) and Schroeder & Mitchener (1975)
studied the carcinogenic potential for mice and rats of nickel
salts in the drinking-water. Levels of 5 mg nickel/litre in the
drinking-water did not produce a significantly higher incidence of
tumours compared with controls.
Ambrose et al. (1976) added nickel sulfate hexahydrate fines
(22.3% nickel) to the diet of Wistar derived rats for 2 years, at
concentrations of 0, 100, 1000, or 2500 mg nickel/kg. There was no
carcinogenic response.
8.4.3. Other routes
The results of carcinogenesis studies of nickel compounds in
rodents are summarized in Table 30.
Table 30. Experimental carcinogenesis studies of various nickel compounds
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Ni3S2 Fischer im Form of implant Gilman & Herchen
rats - implants 500 mg 14/17 is important (1963)
- diffusion chambers
10 mg 14/17
- powder 10 mg 19/20
- chips 500 mg 5/7
Ni3S2 Rat 1 or 3 mg Ni3S2/ 1 mg: 9/60 tumour No tumours in Yarita &
gelatin pellet (6 carcinomas, 3 control Nettesheim
implanted 4 weeks sarcomas) transplanted (1978)
post-grafting in 3 mg: 45/60 tumours tracheas
tracheas grafted (1 carcinoma, 44
under dorsal skin of sarcomas)
isogenic recipients
Ni3S2 Rats Intratesticular, 16/19 (fibrosarcomas, No tumours in Damjanov et al.
0.6-10 mg rhabdomyosarcomas, controls (1978)
malignant
histiocytomas)
Ni3S2 Fischer intrarenal Renal cancers in No tumours in Sunderman et al.
rats 18/24 rats at 10 mg controls (1979b)
dosage and 5/18 rats
at 5 mg dosage
Ni3S2 NMRI mice im, sc Local sarcomas in 19 Oskarsson et al.
of 32 mice at 10 mg (1979)
dosage
Ni3S2 Pregnant im, on day 6 Local sarcomas in Sunderman et al.
rats all dams; no excess (1981)
tumours in progeny
---------------------------------------------------------------------------------------------------------
Table 30 (contd.)
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Ni3S2 Rabbits im Leiomyosarcomas with Hildebrand &
myosin light-chains Tetaert (1981);
typical of fetal Hildebrand &
smooth muscle, Biserte (1979a,b)
rhabdomyosarcomas
Ni3S2 Fischer intraocular Retinoblastomas, Albert et al.
rats gliomas, and (1980, 1982)
melanomas
Ni3S2 Fischer & im Rhabdomyosarcomas, Yamashiro et al.
hooded leiomyosarcomas, (1980, 1983)
rats fibrosarcomas and
lymphosarcomas
Ni3S2 Fischer im Ni3S2 carcinogenesis Sunderman &
rats inhibited by McCully (1983)
simultaneous injection
of manganese dust
Ni3S2, Wistar im Sarcomas induced by Kasprzak et al.
Ni(OH)2, rats Ni3S2 and crystalline (1983)
NiSO4 Ni(OH)2, but not by
NiSO4 or amorphous
Ni(OH)2
Various Fischer im Sarcomas induced by Sunderman
nickel rats nickel compounds (1984)
compounds
Ni3S2, NiO Wistar & intrapleural Rhabdomyosarcomas, Skaug et al.
Ni3S2 Fischer implantation malignant tumours of (1985)
rats pluripotential origin Lumb et al.
(1985)
Ni3S2 Fischer im Rhabdomyosarcomas, Kasprzak &
rats fibrosarcomas Poirier (1985)
---------------------------------------------------------------------------------------------------------
Table 30 (contd.)
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Ni3S2 F344 rats sc, im, if, ia Malignant soft No synovial Shibata et al.
tissue tumours in sarcomas (1985a)
18/19 (sc), 19/20
(im), 9/20 (if),
16/19 (ia)
Ni3S2 Male im (single dose) Mg carbonate A possible Kasprzak et al.
Fischer inhibited Ni3S2 involvement of (1987)
rats carcinogenicity NK and phagocytic
10 groups from 100 to 55% cells in this
20 each inhibition
Ni3S2 Fischer im Local sarcomas Simultaneous Kasprzak et al.
rats admin. of ZnO (1988)
or Zn acetate
delayed the onset
of sarcomas
Ni3S2 F344 NCT im Local sarcomas Kasprzak &
100% Rodriguez (1988)
Ni3S2 + Rat im Local sarcomas Interaction role Kasprzak &
iron: of inflammation, Rodriguez
Fe-/Ni = 0.5 60% active oxygen (1988)
Fe-/Ni = 1.0 40% species, enzyme
Fe-/Ni = 2.0 10% system
Local sarcomas
Fe3+ = 0.5 95% Kasprzak &
Fe3+ = 1.0 45% Rodriguez
Fe3+ = 2.0 35% (1988)
---------------------------------------------------------------------------------------------------------
Table 30 (contd.)
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Ni3S2 + 100% Iron effect is Kasprzak &
remote purely local Rodriguez
Fe- or (1988)
Fe3+/Ni = 2
Ni3S2 in vitro Deactivation of Kasprzak &
cell free catalase and Rodriguez
glutathione (1988)
peroxidase
Ni3S2 Japanese intraocular Malignant Mitsumasa
common melanoma-like (1987)
newts tumour in 7/8
(Cynops) at 400-1100
pyrrhogaster µg/newt
NiO Wistar im 26 local tumours No controls used Gilman
rats 20 mg in 32 rats (80%) (1962)
NiO Swiss im 35% mice with
mice 5 mg tumours
NiO C3H mice im 23% mice with
5 mg tumours
NiO NIH black im implants 4 sarcomas in 35 Payne
rats 7 mg rats after 18 months (1964)
Nickel Rats Instillation in Rhabdomyosarcomas Negative study Eilertsen
oxides and pleural cavity in 8/220 (4/20 in et al.
nickel- positive control) (1988)
copper
oxides
---------------------------------------------------------------------------------------------------------
Table 30 (contd.)
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Nickel Hooded im, 52/66 rats with Gilman &
refinery Wistar sarcomas Ruckerbauer
flue dust rats 20 or 30 mg, one or 8/20 rats with (1962)
thighs sarcomas
Mice im, 23/40 mice with
10 mg each thigh sarcomas
Feinstein Rats ip 6/39 rats with injection Saknyn &
dust 90-150 mg dust/rat site sarcomas Blokhin (1978)
Nickel Hooded im, 28.3 mg in 10/10 rats with Heath &
powder rats 0.4 ml fowl serum local rhabdomyosarcomas Daniel (1964)
Nickel Osborne- Intrafemoral, 4/17 rats with Hueper (1952)
powder Mendel 21 mg tumours, 1 squamous
rats cell carcinoma,
3 osteosarcomas
Wistar Intrafemoral 28% of treated rats No tumours in
rats implant, 50 mg with tumours of control rats
injected thighs
Dutch Intrafemoral 1/6 rabbits with
rabbits implant, 54 mg/kg fibrosarcomas
C57BL iv, No tumours
mice weekly for 2 weeks
Rabbits iv, No tumours
6 times of a 1% Ni
suspension in 2.5%
gelatin at a rate
of 0.5 ml/kg
---------------------------------------------------------------------------------------------------------
Table 30 (contd.)
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Wistar iv, 7/25 rats with
rats 6 times of 0.5% Ni tumours
suspension in saline
at 0.5 ml/kg
Nickel Fischer im, 5 monthly 38/50 rats with Furst &
powder rats injections of 5 mg fibrosarcomas Schlauder
Ni in 0.2 ml (1971)
trioctanoin
Hamsters im, 5 monthly 2/50 hamsters with Furst &
injections of 5 mg fibrosarcomas Schlauder
Ni in 0.2 ml (1971)
trioctanoin
Nickel Fischer im, 3.6 mg/rat 0/10 rats with local Sunderman &
powder rats tumours Maenza (1976)
14.4 mg/rat 2/10 rats with local
tumours
Nickel Fischer Intrathoracic, Mesothelioma, Only 10 animals Furst et al.
powder 344 rats 5 monthly doses of pleural cavity in (1973)
5 mg/animal 2 out of 10 rats
Nickel Fischer ip, 16 injections 30-50% Furst &
powder 344 rats twice monthly, 5 mg/ intraperitoneal Cassetta
animal tumours (1973)
Nickel Fischer Intrathoracic 40% mesothelioma Furst &
powder 344 rats 5 monthly doses, of Cassetta
5 mg/animal (1973)
---------------------------------------------------------------------------------------------------------
Table 30 (contd.)
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Ni(CO)4 Sprague- iv, 6 x 50 µl/kg 19/21 rats with Lau et al.
Dawley (9 mg Ni/kg) malignant tumours (1972)
rats at various sites,
2/47 rats with
pulmonary lymphomas
< 0.05
NiCO3 NIH black Muscle implant, 4/35 implant site Payne
rats 7 mg sarcomas (1964)
Nickelocene Fischer im injections, 18/50 rats or 21/50 No tumours Furst &
rats 12 x 12 mg or rats with in controls Schlauder
12 x 25 mg fibrosarcomas (1971)
nickelocene
Hamsters im injections, 4/29 hamsters with No tumours at
1 x 25 mg or fibrosarcomas at 8 x 5 mg
8 x 5 mg 1 x 25 mg
nickelocene
Ni(CH3CO2)2 Mice, ip, 24 injections, Lung adenomas and Stoner
Strain A 3/week at 72, 180, adenocarcinomas et al.
360 mg/kg (significant for (1976)
360 mg/kg group)
Ni(CH3CO2)2 Mice, ip Average 1.5 lung Poirier
Strain A adenomas per mouse et al. (1984)
Ni(CH3CO2)2 F344 rats sc, 150 mg/kg Injection site Teraki &
sarcomas Uchiumi (1988)
---------------------------------------------------------------------------------------------------------
Table 30 (contd.)
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Nickel Wistar ip 46/48, 48/47 and Pott et al.
powder rats 27/42 rats with (1987)
NiO, Ni3S2 sarcomas,
mesotheliomas, or
carcinomas
Nickel Wistar ip High incidence of Pott et al.
powder rats sarcomas and (1987)
NiO, Ni3S2 mesotheliomas in
NiCl2, Ni3S2-, nickel
Ni(CH3CO2)2 powder-, nickel
NiSO4, alloy-, and NiO-
NiCO3 nickel groups
alloy
---------------------------------------------------------------------------------------------------------
a ia = intra-articular
if = intra-retroperitoneal fat
im = intramuscular
ip = intraperitoneal
iv = intravenous
sc = subcutaneous.
Nickel subsulfide is the nickel compound that has been most
studied. All routes of administration, except submaxillary gland
injection into Fischer rats and local application to the cheek
pouches of Syrian hamsters (Sunderman et al., 1978d), produced
tumours. Sunderman (1983b) and Gilman & Yamashiro (1985) reviewed
experimental data on factors influencing the carcinogenetic
activity of nickel subsulfide and other nickel compounds, including
species and strain, susceptibility of organs, route of
administration, physical form of nickel compound, and dose-response
characteristics. Dose-effect relationships could be demonstrated
for nickel subsulfide in rats following intramuscular and
intrarenal injections and in hamsters following intramuscular and
intratesticular injections. Intraocular and intramuscular routes
of administration yielded the highest tumour incidence. Local
tumours after intraocular injection included malignant melanomas,
retinoblastomas, gliomas, a phakocarcinoma, and unclassified
tumours. Following intramuscular injection, local tumours were
mostly rhabdomyosarcomas, fibrosarcomas, and undifferentiated
sarcomas. Rats are more susceptible to the induction of sarcomas
by nickel subsulfide than mice, hamsters, or rabbits, though
Sunderman (1983b) considered that absolute species susceptibility
could not be defined, because of differences in experimental
conditions. For strain differences in rats, Gilman & Yamashiro
(1985) ranked susceptibility to tumour induction by nickel
subsulfide as: Hooded > Wistar > Fischer > Sprague-Dawley.
Yarita & Nettesheim (1978) implanted nickel subsulfide pellets into
heterotopic tracheas grafted in Fischer 344 rats and reported
tracheal tumours including sarcomas (2 fibrosarcomas, 1
leiomyosarcoma) and carcinomas (5 squamous cell carcinoma, 1
undifferentiated carcinoma). Malignant testicular neoplasms
developed in Fischer 344 rats after intratesticular injection of
10 mg nickel subsulfide (Damjanov et al., 1978). Gilman & Herchen
(1963) found corresponding numbers of rhabdomyosarcomas in rats
after intramuscular implantation of Ni2S3 in implants of different
forms, injection of powder, or exposure in a diffusion chamber.
Shibata et al. (1989) injected nickel subsulfide (single injection,
0.5 mg) subcutaneously (sc), intramuscularly (im), in
retroperitoneal fat, and intra-articularly (ia), in male Fischer
rats, and observed the animals for 48 weeks. Malignant soft-tissue
tumours developed in 18/19 (sc) 19/28 (im) 9/20 (retroperitoneally),
and 16/19 (ia) animals. No synovial sarcomas were detected.
Magnesium carbonate was observed to decrease sarcoma incidence due
to Ni3S2 from 100 to 55% in a dose-related manner, when both
compounds were injected into the thigh muscles of male Fischer 344
rats (Kasprzak et al., 1987). Kasprzak et al. (1988) found that,
40 weeks after injection, zinc, either as zinc oxide or zinc
acetate, delayed the onset, but not the final incidence of local
sarcomas when injected intramuscularly with Ni3S2. All of the rats
given Ni3S2 developed tumours compared with only 60% of those that
received Ni3S2 plus zinc. Kasprzak & Rodriguez (1988) found that
iron counteracted the ability of nickel subsulfide to induce cancer
and that it inhibited catalase and glutathione peroxidase, leading
to a build up of hydrogen peroxide (H2O2), which may be related to
carcinogenesis.
Iron can decompose H2O2 so reversing the action of nickel
subsulfide. Mitsumasa (1987) administered Ni3S2 to lentectomized
Japanese common newts (Cynops pyrrhogaster). A single injection of
40-100 µg/newt produced malignant melanoma-like tumours in the
injected eyes of 7 or 8 newts.
Nickel oxide produced local tumours (mainly rhabdomyosarcomas,
fibrosarcomas, and undifferentiated sarcomas) following intramuscular
injections in rats and mice (Gilman, 1962; Sunderman, 1984) and a
few tumours (not specified) following implantation in rats (Payne,
1964). It was also carcinogenic when administered by intrapleural
injection in Wistar rats (Skaug et al., 1985). Eilertsen et al.
(1988) tested the carcinogenicity in the rat of a variety of nickel
oxides given by the intrapleural route but obtained negative
results.
When Gilman & Ruckerbauer (1962) injected nickel refinery flue
dust (20% nickel sulfate, 57% nickel subsulfide, 6.3% nickel oxide)
into the thigh muscles of rats and mice, they found a high
induction rate of sarcomas. Saknyn & Blokhin (1978) injected
"feinstein" dust (an intermediate product of nickel refining
containing nickel sulfide, nickel oxide, and metallic nickel)
intraperitoneally at a dose of 90-150 mg/rat and found sarcomas at
the injection sites in 6 out of 39 rats.
Intramuscular injection of nickel powder produced sarcomas in
rats at the injection sites (Heath & Daniel, 1964; Furst &
Schlauder, 1971; Sunderman & Maenza, 1976; Sunderman, 1984). In
hamsters, intramuscular injection of nickel powder produced
sarcomas at the site of injection, but the incidence was low (2 out
of 50 animals) (Furst & Schlauder, 1971). Intravenous injection of
nickel powder produced local sarcomas in rats, but not in mice and
rabbits (Hueper, 1955). No controls were used. Hueper (1952,
1955) injected nickel powder into the femurs of rats and rabbits
and observed sarcomas at the site of injection. Intrathoracic
injections of nickel powder produced mesotheliomas in Fischer 344
rats (Furst et al., 1973; Furst & Cassetta, 1973). Following
intraperitoneal injection of nickel powder, tumours were seen in
Fischer 344 rats (Furst & Cassetta, 1973).
Besides its carcinogenic potential in inhalation studies,
administration of nickel carbonyl to rats by repeated intravenous
injection was also associated with an increased tumour rate (Lau et
al., 1972). Malignant tumours included pulmonary lymphomas,
undifferentiated sarcomas in the lung, pleura, liver, pancreas,
uterus, and abdominal wall, fibrosarcomas in the neck, pinna, and
orbit, carcinomas of liver, breast, and kidney, and one
haemangioendothelioma.
Local sarcomas were induced in rats with repeated intramuscular
injections of crystalline nickel hydroxide, but not with colloidal
nickel hydroxideor nickel sulfate, under similar experimental
conditions (Kasprzak et al., 1983).
Implantation of 7 mg nickel carbonate hydroxide/rat produced
local sarcomas in 4 out of 35 rats (Payne, 1964).
Nickelocene, an organic nickel compound used as a laboratory
reagent, induced fibrosarcomas in rats and hamsters, when injected
intramuscularly (Furst & Schlauder, 1971).
It was reported by Stoner et al. (1976) that 24 intraperitoneal
injections of nickel acetate to strain A mice (total dose 72-360
mg/kg body weight) induced lung tumours at the highest dose.
Poirier et al. (1984) observed an increased incidence of lung
adenomas in Strain A mice following intraperitoneal administration
of nickel acetate, though these authors also observed an increase
in tumour incidence in animals treated with calcium acetate or lead
acetate. Teraki & Uchiumi (1988) reported that nickel acetate
causes injection site tumours in rats.
Single intraperitoneal doses of nickel powder, nickel
subsulfide, or an unspecified nickel oxide induced a high frequency
(46 out of 48 animals, 27 out of 42 animals, and 46 out of 47
animals, respectively) of abdominal cavity tumours in Wistar rats
(Pott et al., 1987). Pott et al. (1988) investigated nickel
carcinogenicity by intraperitoneal injection in rats and produced
a variety of responses (see Table 30). They concluded that the
model is good for screening nickel compounds and alloys for
carcinogenicity.
In a study by Sunderman (1984), 18 nickel compounds were
investigated under standardized experimental conditions to relate
their physical, chemical, and biological properties to their
carcinogenic activity. Marked differences were observed in the
incidence of sarcomas in male Fischer rats within 2 years following
single intramuscular injections of equivalent doses (14 mg
nickel/rat). The results of these bioassays are summarized in
Table 31. They provide an adequate basis for ranking the relative
carcinogenicity of different nickel compounds in animals. Sarcoma
incidences in these bioassays were significantly correlated with
the mass-fractions of nickel in the respective compounds. They
were not significantly related to the dissolution half-times of the
nickel compounds in rat serum or renal cytosol (Kuehn & Sunderman,
1982), or to the susceptibilities of the compounds to phagocytosis
by rat peritoneal macrophages in vitro (Kuehn et al., 1982). On
the other hand, there was a strong correlation between the sarcoma
incidences and the capacity of nickel compounds to induce
erythrocytosis after intrarenal administration to rats, suggesting
that nickel-stimulated production of erythropoietin and nickel
carcinogenesis may be related in some way (Sunderman & Hopfer,
1983).
Table 31. Summary of survival data and sarcoma incidences in carcinogenesis tests of 18 nickel compoundsa
-----------------------------------------------------------------------------------------------------------------------------
Category Test substance Survivors at Rats with local Median Median Rats with metastases/
2 years/total sarcomas/total tumour survival rats with sarcomas
number of rats number of rats latency period (% survival)
(% survival) (% survival) (weeks) (weeks)
-----------------------------------------------------------------------------------------------------------------------------
Controls Glycerol vehicle 25/40 (63%) 0/40 (0%) - >100 -
Penicillin vehicle 24/44 (55%) 0/44 (0%) - >100 -
All controls 49/84 (58%) 0/84 (0%) - >100 -
Class A Nickel subsulfide (alphaNi3S2) 0/9d (0%) 9/9d (100%) 30 39c 5/9 (56%)
Nickel monosulfide (BNiS) 0/14d (0%) 14/14d (100%) 40 48c 10/14 (71%)
Nickel ferrosulfide (Ni4FeS4) 0/15d (0%) 15/15d (100%) 16 32c 10/15 (67%)
Class B Nickel oxide (NiO) 0/15d (0%) 14/15d (93%) 49 58c 4/14 (29%)
Nickel subselenide (Ni3Se2) 0/23d (0%) 21/23d (91%) 28 38c 18/21 (86%)
Nickel sulfarsenide (NiAsS) 0/16d (0%) 14/16d (88%) 40 57c 10/14 (71%)
Nickel disulfide (NiS2) 0/14d (0%) 12/14d (86%) 36 47c 6/12 (50%)
Nickel subarsenide (Ni5As2) 0/20d (0%) 17/20d (85%) 22 44c 9/17 (53%)
Class C Nickel dust 4/20c (20%) 13/20d (65%) 34 42c 6/13 (40%)
Nickel antimonide (NiSb) 9/29c (31%) 17/29 (59%) 20 66c 10/17 (59%)
Nickel telluride (NiTe) 12/26 (46%) 14/26d (54%) 17 80c 8/14 (57%)
Nickel monoselenide (NiSe) 7/16 (44%) 8/16d (50%) 56 72c 3/8 (38%)
Nickel subarsenide (Ni11As8) 5/16b (31%) 8/16d (50%) 33 88c 6/8 (75%)
Class D Amorphous nickel monosulfide 5/25c (20%) 3/25c (12%) 41 71c 3/3 (100%)
(NiS)
Nickel chromate (NiCrO4) 10/16 (63%) 1/16 (6%) 72 >100 1/1 (100%)
Class E Nickel monoarsenide (NiAs) 13/20 (65%) 0/20 (0%) - >100 -
Nickel titanate (NiTiO3) 11/20 (55%) 0/20 (0%) - >100 -
Feronickel alloy (NiFe1,6) 11/16 (75%) 0/20 (0%) - >100 -
----------------------------------------------------------------------------------------------------------------------------
a From Sunderman (1984d).
b P < 0.05 versus corresponding vehicle controls.
c P < 0.01 versus corresponding vehicle controls.
d P < 0.001 versus corresponding vehicle controls.
Six nickel oxides and 4 nickel-copper oxides were categorized
according to the criteria shown in Table 32 (Sunderman et al.,
1987a). These compounds were assayed as follows: (a) in vitro
dissolution in water and body fluids; (b) in vitro phagocytosis
tests in Chinese hamster ovary and C3H-10T1/2 cells; (c)
morphological transformation and cytotoxicity tests in cultured
Syrian hamster embryos (SHE) cells; (d) erythropoiesis stimulation
assay by intrarenal administration to Fischer 344 rats; and (e)
scoring the renal histopathological responses in rats killed 3
months after injection. The extent of agreement among the
physical, chemical, and biological attributes of the test compounds
was tested by Kendalls' non-parametric test for concordance of
rankings. On the basis of a highly significant concordance of
ranked results in the assays ( P <0.001), 6 biological attributes of
the compounds were identified: (i) dissolution half-times in rat
serum and renal cytosol; (ii) phagocytosis by C3H-10T1/2 cells;
(iii) morphological transformation of SHE cells; (iv)
erythropoiesis stimulation in rats; (v) induction of tubular
hyperplasia in rat kidneys; and (vi) induction of arteriosclerosis
in rat kidneys. A strong rank correlation between results of the
cell transformation and erythropoiesis stimulation was observed.
The presence of high surface area and demonstrable Ni(III) were
the two physicochemical characteristics associated with the
greatest biological effects of the nickel oxides.
Compounds A, B, and F (nickel oxides) and compounds H and I
(nickel-copper oxides) were selected for carcinogenesis testing in
male Fischer 344 rats (20 mg, intramuscular injection, right thigh)
(Sunderman et al., 1988a). Injection site sarcomas (524 months)
developed in 6 out of 15 rats that received compound A (INCO black
NiO), versus 0 out of 15 rats that received compound B (NiO
calcined at 735 °C), and 0 out of 15 rats that received compound F
(NiO calcined at 1045 °C). Sarcomas developed in 13 out of 15 rats
that received compound H (NiO/CuO, 2.5/1 ratio) and 15 out of 15
rats that received compound I (NiO/CuO, 5.2/1 ratio). Injection
site sarcomas developed in 0 out of 15 vehicle control rats and 15
out of 15 positive controls that received intramuscular injection
of Ni3S2.
8.4.4. Interactions with known carcinogens
Several studies have been performed to investigate the possible
interactions of different nickel species with known carcinogens,
either in classical two-stage carcinogenesis studies, or using
simultaneous administration.
In a two-stage carcinogenesis assay, orally administered nickel
chloride enhanced the renal carcinogenicity of N-ethyl- N-hydroxyethyl
nitrosamine in rats, but not the hepatocarcinogenicity of N-
nitrosodiethylamine, the gastric carcinogenicity of N-methyl-
N-nitro- N-nitrosoguanidine, the pancreatic carcinogenicity of N-
nitrosobis(2-oxopropyl)amine, or the skin carcinogenicity of 7,12-
dimethylbenzanthracene (Hayashi et al., 1985).
Table 32. Physical and chemical characteristics of some test compounds: 6 Ni oxides and 4 Ni-Cu oxidesa
-------------------------------------------------------------------------------------------------------------------------
Test Calcin- Surface Elemental composition (% by weight) Constitutents and Relative
compound ation area speciation (% by weight) height of
and colour temp. (m2/g) XRD peaks
(°C) Ni Cu Ob S Fe Co Ni(III) SO4 Ni3S2 CuO NiOc for Ni
oxidesd
-------------------------------------------------------------------------------------------------------------------------
A (jet black) <650 41.5 77.7 <.001 22.3 <0.02 <0.01 <0.01 0.81 <0.06 NDe ND SSf 0.31
B (grey-black) 735 3.0 78.1 <.001 21.9 <0.02 <0.01 <0.01 0.05 <0.06 ND ND SS 0.74
C (grey) 800 1.4 78.2 <.001 21.8 <0.02 <0.01 <0.01 0.03 <0.06 ND ND SS 0.87
D (grey) 850 0.7 78.2 <.001 21.8 <0.02 <0.01 <0.01 <0.03 <0.06 ND ND SS 0.88
E (dark green) 918 0.2 78.4 <.001 21.6 <0.02 <0.01 <0.01 <0.03 <0.06 ND ND SS 0.90
F (green) 1045 <0.2 78.6 <.001 21.4 <0.02 <0.01 <0.01 <0.03 <0.06 ND ND SS 1.00
G (dark maroon) 850 <0.2 43.8 27.9 23.4 0.69 2.98 1.17 g 1.86 0.3 14 77
H (maroon) 850 <0.2 51.7 20.6 23.6 1.02 2.02 1.09 g 2.67 0.5 3 89
I (red-brown) 850 <0.2 62.6 12.6 22.3 0.30 1.20 1.04 g 0.06 1.0 ND 95
J (brown) 850 <0.2 68.8 6.9 22.1 0.42 0.75 0.97 g 0.06 1.5 ND 96
-------------------------------------------------------------------------------------------------------------------------
a From: Sunderman et al. (1987a)
b Estimated by subtracting the percentages for other elemental constituents from 100%.
c NiO (bunsenite) can accommodate up to 25% CuO (tenorite) as a solid solution within its crystal lattice at 850C.
d Heights of XRD peaks of NiO (bunsenite) at 1.48, 2.09, and 2.41 angstrom, relative to peak heights obtained with
compound F.
e ND = not detected by XRD.
f SS = sole species detected by XRD.
g Presence of sulfur in compounds G-J precluded assays for Ni(III).
Single intratracheal injections of nickel subsulfide did not
increase the carcinogenicity of benzo (a)pyrene, also instilled
intratracheally, in rats (Kasprzak et al., 1973).
Metallic nickel powder seemed to enhance the lung
carcinogenicity of 20-methylcholanthrene (MC) when both were
administered intratracheally to rats. The tumour incidences in
different groups, 12 weeks after the instillation were: (i) 5 mg
MC: 2/7; (ii) 5 mg MC + 10 mg Ni: 3/5; (iii) 1 mg MC: 1/8; (iv) 1
mg MC + 10 mg Ni 2/7; and (v) 10 mg Ni only: 0/7 (Mukubo, 1978).
Nickel oxide, administered intratracheally, did not enhance the
carcinogenic response to diethylnitrosamine in the lungs or nasal
cavity of hamsters (Farrell & Davis, 1974).
In studies on combined intramuscular injection of nickel
subsulfide and benzo (a)pyrene, latency times to local tumour
development were shorter, and the proportion of malignant tumours
higher, than when either carcinogen was injected alone (Maenza et
al., 1971).
In a two-stage carcinogenicity study, Ou et al. (1981) gave a
single injection of dinitrosopiperazine (9 mg) to rats, followed by
administration of nickel sulfate, either in the drinking-water (3.7
mg/day), or topically in the nasopharynx as a gelatin solution
(0.02 ml of 0.5% NiSO4). Two out of 12 rats in both groups
receiving both nickel and the nitrosamine developed nasopharyngeal
tumours (1 squamous cell carcinoma, 1 fibrosarcoma, 1 papilloma, 1
"early carcinoma"), while none was seen in the 24 animals treated
with the nitrosamine alone, or in the two groups of 12 animals
treated with nickel sulfate. The same authors (abstract only)
reported an extension of the study, using topical nickel sulfate as
a promoter. Five out of 22 rats given the combined treatment
developed carcinomas of the nasopharynx, nasal cavity, or hard
palate, while these tumours were not found in the other groups (Liu
et al., 1983).
Ou et al. (1983) reported a study in which N-nitrosopiperazine
was given to female rats on the 18th day of pregnancy, and the pups
were given nickel sulfate orally. Five out of 21 pups with
combined treatment, and 1 out of 4 with nitrosamine treatment only
developed nasopharyngeal carcinoma, and two other pups developed
other tumours.
8.4.5. Possible mechanisms of nickel carcinogenesis
Both in vivo and in vitro studies have shown varying
carcinogenic responses, depending on the bioavailability of certain
nickel compounds. Several factors influencing bioavailability have
been suggested, such as the physical form of the compound
(crystalline or amorphous), solubility in water or serum, and its
ability to be phagocytosed by target cells (Costa et al., 1982) or
pass through calcium channels (Sunderman, 1989a). In vitro studies
have shown that various nickel compounds had similar transforming
potencies at equitoxic concentrations (Hansen & Stern, 1984).
The transformation potency of a number of particulate nickel
compounds has been correlated with phagocytosis (Costa et al.,
1981b). The incidence of sarcomas and the capacity of nickel
compounds to produce erythrocytosis are also related (Sunderman &
Hopfer, 1983; Sunderman et al., 1987a). Erythrocytosis can be
induced in rats by intrarenal injection of nickel subsulfide,
probably due to increased production of the stimulating hormone
erythropoietin (Jasmin & Ripolle, 1976; Morse et al., 1977; Hopfer
et al., 1978). Intrarenal injection of nickel subsulfide caused
renal cancers (Sunderman et al., 1984b). Induction of
erythrocytosis and carcinogenesis in rats by nickel subsulfide were
both inhibited by the administration of manganese dust (Sunderman
et al., 1976b). In vitro studies have demonstrated that nickel
compounds cause (a) DNA damage in a wide variety of organisms, (b)
mutagenicity, possibly related to cytotoxicity, in mammalian cells,
and (c) clastogenicity and morphological transformation in
mammalian cells including human cells. From in vitro and in vivo
studies, it is known that nickel interacts with DNA and proteins,
causing lesions, such as DNA-protein (possibly heterochromatin)
cross-links and DNA single strand breaks (Ciccarelli et al., 1981;
Ciccarelli & Wetterhahn, 1982; Robison et al., 1982; Patierno et
al., 1985), resulting in DNA damage and other cytotoxic effects
that can lead to altered gene expression in surviving cells. For
example, Sen & Costa (1986) showed that nickel preferentially
induced decondensation (presumably by DNA-protein cross-linking)
and fragmentation of the long arm of the X-chromosome in CHO cells.
Other consequences of cross-linking, such as the appearance of
irreversible, but heritable, lesions of intermediate filaments (a
family of cytoskeletal proteins linking the nucleus with the cell
membrane), related to disturbance of normal growth control
processes, have been observed early after nickel intoxication in
vitro (Swierenga & McLean, 1985; Swierenga et al., 1987, 1989).
Nickel-induced DNA damage/alterations that have been reported
include: conformational transitions of the DNA from the normal
right-handed B-helix to the left-handed Z helix (van de Sande et
al., 1982; Boutayre et al., 1984), infidelity of DNA synthesis, as
shown with cell-free systems using synthetic templates (Sirover &
Loeb, 1977; Zakour et al, 1981), and inhibition of DNA synthesis in
a wide variety of organisms extending to inhibition of DNA excision
repair (Table 27). The latter may be an important mechanism in the
ability of nickel to enhance the carcinogenic activity of other
compounds (Maenza et al., 1971; Kurokawa et al., 1985). Other in
vitro evidence indicates that nickel disrupts intercellular
communication, a property of tumour promoters (Miki et al., 1987).
Studies by Nieboer et al. (1986) indicated that, in the
presence of certain peptides, the Ni3+/Ni2+ redox couple can
participate in dioxygen radical reactions in vitro. As chemical
promoters of cancer appear to have the capacity to stimulate
phagocytic cells to produce dioxygen radicals, which may damage
DNA, proteins and lipids, and as metal ions catalyze processes
involving molecular oxygen, this observed effect of nickel may have
implications for nickel carcinogenesis.
Sunderman & Barber (1988) have suggested a mechanism for the
interaction of nickel with DNA. This model proposes that the Zn2+
binding sites of DNA binding proteins known as "finger loop
domains", which have been identified on some proto-oncogens, are
likely molecular targets for metal toxicity. As Ni2+ has a similar
ionic radius to Zn2+, substitution is possible, interfering with
regulation of gene expression. The replacement of Zn2+ in finger
loops might affect the conformation and stability of DNA-binding
structures interfering with expression and induce site-specific
free radical reactions, causing cleavage of DNA, formation of DNA-
protein cross links, and disturbances of mitosis.
8.4.6. Factors influencing nickel carcinogenesis
Maenza et al. (1971) reported a synergistic action between
nickel subsulfide and benzo (a)pyrene. After intramuscular
injection, the latency period for sarcoma induction was
significantly reduced. Kasprzak et al. (1973) found an increased
incidence of premalignant changes in the lungs of rats receiving
intratracheal injections with a mixture of benzo (a)pyrene and
nickel subsulfide compared with groups receiving one compound at a
time.
Manganese in metallic form has been shown to antagonize the
tumourigenic effect of nickel subsulfide by intramuscular injection
(Sunderman et al., 1976b) and intrarenal injection (Sunderman et
al., 1979b). Kasprzak & Poirier (1985) found that simultaneous
intramuscular injection of magnesium carbonate inhibited the
development of muscle tumours induced by nickel subsulfide.
Calcium carbonate, under the same experimental conditions, was
ineffective. Poirier et al. (1984) reported that both magnesium
acetate and calcium acetate inhibited lung adenoma formation in
Strain A mice treated with intraperitoneal nickel acetate.
Manganese, magnesium, calcium, copper, and zinc were shown by
Kasprzak & Poirier (1985) and Kasprzak et al. (1986) to inhibit
binding of nickel to DNA. They also found that amino acids were
very effective inhibitors of nickel binding to DNA, but their
eventual role in moderating nickel carcinogenicity has not been
investigated.
When rats with muscular implantations of nickel subsulfide were
injected intraperitoneally with sodium diethyldithiocarbamate,
tumour incidence was significantly reduced compared with that in
rats that had received the nickel subsulfide implant alone
(Sunderman et al., 1984b).
9. EFFECTS ON HUMAN BEINGS
9.1. Systemic effects
9.1.1. Acute toxicity - poisoning incidents
9.1.1.1 Nickel carbonyl
In terms of human health, the most acutely toxic nickel
compound is nickel carbonyl. The clinical manifestations of nickel
carbonyl poisoning have been described in detail by Sunderman
(1970) and Vuopala et al. (1970). A summary of 179 cases of nickel
carbonyl poisoning in China since 1961 has been published by Shi
(1986).
The acute toxic effects occur in 2 stages, immediate and
delayed. The immediate symptomatology includes frontal headache,
vertigo, nausea, vomiting, insomnia, and irritability, followed by
an asymptomatic interval before the onset of delayed pulmonary
symptoms. These include constrictive chest pains, dry coughing,
dyspnoea, cyanosis, tachycardia, occasional gastrointestinal
symptoms, sweating, visual disturbances, and weakness. The
symptomatology resembles that of a viral pneumonia. In men who
died, pulmonary haemorrhage and oedema or pneumonitis were observed
accompanied by derangement of alveolar cells, degeneration of the
bronchial epithelium, and the appearance of a fibrinous intra-
alveolar exudate. The pathology of the pulmonary lesions was
similar to that observed in animal studies. Other affected organs
included the liver, kidneys, adrenal glands, and spleen, where
parenchymal degeneration was observed. Cerebral oedema and
punctate cerebral haemorrhages were noted in men dying after
inhalation of nickel carbonyl. Shi (1986) reported leukocytosis in
25 out of 179 cases. Urinary nickel concentrations were determined
in 27 cases and ranged from 0.003 to 0.66 mg/litre. Air
concentrations were measured and were reported to have exceeded 50
mg nickel carbonyl /m3 with exposure periods ranging from 30 min to
more than 2 h. Recovery time was 7-40 days, depending on the
severity of exposure. In some cases, symptoms persisted for 3-6
months. In lethal cases, death occurred between the third and
thirteenth days following exposure.
9.1.1.2 Other nickel compounds
Information on poisoning with other nickel compounds is
limited. Daldrup et al. (1983) reported a case of fatal nickel
poisoning. A 2 1/2-year-old girl ingested about 15 g of nickel
sulfate crystals. On admission to hospital, she was somnolent with
wide and unresponsive pupils, a high pulse rate, and pulmonary
rhonchi. Cardiac arrest occurred after 4 h. Autopsy findings
revealed increased nickel levels (7.5 mg/kg in blood, 50 mg/litre
in urine, 25 mg/kg in liver tissue). Nickel poisoning was reported
in a group of 23 dialysed patients, when leaching from a nickel-
plated stainless steel water heater tank contaminated the
dialysate. Symptoms occurred during, and after, dialysis, at
plasma-nickel concentrations of approximately 3 mg/litre. Symptoms
included nausea (37 out of 37), vomiting (31 out of 37), weakness
(29 out of 37), headache (22 out of 37), and palpitation (2 out of
37). Recovery occurred spontaneously, generally 3-13 h after
cessation of dialysis (Webster et al., 1980).
Thirty-two workers in an electroplating plant accidentally
drank water contaminated with nickel sulfate and nickel chloride
(1.63 g nickel/litre). Twenty workers rapidly developed symptoms
(e.g., nausea, vomiting, abdominal discomfort, diarrhoea,
giddiness, lassitude, headache, cough, shortness of breath) that
typically lasted a few hours, but persisted for 1-2 days in 7
cases. The nickel doses in workers with symptoms were estimated to
range from 0.5 to 2.5 g. In 15 exposed workers, who were
investigated on day 1 after exposure, serum-nickel concentrations
ranged from 12.8 to 1340 µg/litre (average 286 µg/litre) with
urinary nickel concentrations ranging from 0.23 to 37.1 mg/litre
(average 5.8 mg/litre) compared with control values for nickel-
plating workers of 2.0-6.5 µg/litre (average 4.0 µg/litre) for
serum nickel and 22-70 µg/litre (average 50 µg/litre) for urinary
nickel. Laboratory tests showed transiently elevated levels of
blood reticulocytes (7 workers), urine-albumin (3 workers), and
serum-bilirubin (2 workers) (Sunderman et al., 1988b).
9.1.2. Short- and long-term exposure
9.1.2.1 Respiratory effects
A chemical engineer, who had been exposed for a long period to
low levels of nickel carbonyl, developed asthma and Löffler's
syndrome. In addition to pulmonary infiltrations and eosinophilia,
which are markers in Löffler's syndrome, the patient had an
eczematous dermatitis of the hands (Sunderman & Sunderman, 1961b).
There have been systematic investigations of the health state
of the respiratory tract of workers in the nickel industry.
Tatarskaya (1960) examined 486 workers from nickel-refining plants,
exposed mainly to nickel sulfate during electrorefining operations.
Exposure levels were not given. Rhinitis was observed in 10-16%,
chronic rhinitis in 5.3%, nasal septal erosions in 13%,
perforations in 6.1%, and ulceration in 1.4% of the workers. The
frequencies of hypo-osmia and anosmia were 30.6 and 32.9%,
respectively. Kucharin (1970) studied 302 workers who had been
exposed to nickel sulfate, accompanied by sulfuric acid vapour, for
at least 10 years, at concentrations of 0.02-4.5 mg/m3. Clinical
and radiological examination revealed chronic sinusitis in 83% of
the workers. Among these workers, severe injuries were apparent,
such as septal erosion and perforation in 41.4 and 5.6%, respectively.
Hypo- or anosmia was noted in 46% of the workers with chronic
sinusitis. In a nickel refinery where hydrometallurgical methods
were used, 37 out of 151 workers (24.5%) exhibited pathological
changes of the nasopharynx. Septal erosions were noted in 14
workers (9.3%). Exposure concentrations of soluble nickel salts,
during the years 1966 and 1970, ranged from 0.035 to 1.65 mg/m3
(Sushenko & Rafikova, 1972).
Other studies on the nasopharyngeal effects of inhalation of
nickel salt aerosols, conducted in order to detect precancerous
lesions, are described in the section 9.5.
Data are available on the development of pulmonary changes with
fibrosis in workers inhaling nickel dust or fumes. Zislin et al.
(1969) studied respiratory function in 13 workers, who had been
exposed to nickel dust for periods of 12.9-21.7 years. Exposure
levels were not given. There was decreased pulmonary residual
capacity, increased respiratory frequency, and radiography showed a
diffuse fibrosis, considered to be nickel pneumoconiosis.
Industrial dusts, collected in the workrooms of nickel
refineries, induced the development of pneumoconiosis of an
interstitial type, together with bronchitis, in experimental
animals (Saknyn et al., 1978; Grebnev & Yelnichnykh, 1983). The
fibrogenicity of these dusts did not depend on their silica
content, and the dust that was almost pure nickel suboxide (Ni2O3)
was fibrogenic, when instilled intratracheally. Pneumoconiosis of
an interstitial and micronodular type had been detected in a number
of workers in the same industry (Zislin et al., 1969).
Peto et al. (1984) reported mortality experience in Welsh
nickel refinery workers. Men first employed before 1925, who had
worked for 5 or more years in the process areas where lung and
nasal cancer risks were highest, also suffered a significantly
elevated mortality from non-malignant respiratory disease (20
deaths, 11.1 expected); less heavily exposed workers suffered no
such excess (43 deaths, 51.2 expected).
Jones & Warner (1972) studied respiratory symptoms, chest X-
rays, and lung ventilation capacities in 12 steelworkers, who had
been employed as de-seamers of steel ingots for periods of up to 16
years. Total fume concentration at the workplace ranged from 1.3
to 294.1 mg/m3, either from iron oxide, chromium oxide, and nickel
oxide in proportions of 6:1:1 (stainless steel fume) or 98.8% iron
oxide (special steel fume). Airborne nickel oxide concentrations
of 0.15-34 mg/m3 were calculated from these values. Two of the
workers had measurable loss of pulmonary function. In 5 men,
definite signs of pneumoconiosis were detected radiographically.
Effects on the respiratory system have also been observed in
welders. Between 1955 and 1979, Zober (1981a,b) found a total of
47 cases of histologically verified fibrotic changes in the lungs
of electric arc welders. Though the findings were very
heterogeneous, indicating a diversity of causes, welding fumes were
found to be a concomitant cause of fibrotic changes in 20 cases.
Zober (1982) studied 40 welders (22, non-smoking and 18, smoking),
who were using metal inert gas and electric arc welding techniques
on chromium- and nickel-containing materials. Concentrations of
airborne nickel did not exceed 0.5 mg/m3, except in one working
process, where there was a concentration of 1.2 mg nickel/m3.
Respiratory-tract effects, including acute bronchitis and
bronchitic pulmonary rhonchi, were increased, mainly in welders who
smoked tobacco.
Asthmatic lung disease in nickel-plating workers exposed to
soluble nickel has been reported (Tolot et al., 1956; McConnell et
al., 1973; Block & Yeung, 1982; Malo et al., 1982; Dolovich et al.,
1984). Cirla et al. (1985) examined 12 workers with respiratory
problems in a nickel-plating industry. In 6 workers, allergic
asthma could be provoked by the inhalation of nickel sulfate
aerosols. Novey et al. (1983) reported that a metal-plating
(chromium and nickel) worker developed acute asthma in response to
inhalational challenge with nickel sulfate and chromium sulfate.
Radioimmuno-assays incorporating the challenge materials revealed
specific IgE antibodies to the provocative agents.
Five stainless steel welders, suffering from respiratory
distress, developed asthmatic symptoms during provocation tests
with stainless steel fumes (Keskinen et al., 1980).
Inhalation testing using 0.01-0.001% nickel chloride solution
has been carried out to detect hypersensitivity to nickel and its
compounds in patients occupationally exposed to nickel and
suffering from atopic bronchial asthma. In inhalation testing, it
is necessary to take pneumotachometric readings before, and after,
the test, at various times over a period of 24 h (Izmerov, 1983).
9.1.2.2 Renal effects
Sanford et al. (1988) assessed different parameters for kidney
dysfunction in urine samples from electrolytic nickel refinery
workers (renal clearance, protein, specific gravity, and
qualitative "dipstick" tests). In male workers who accidentally
ingested drinking-water contaminated with nickel sulfate and
chloride, Sunderman et al. (1988b) found elevated urine-albumin
concentrations (68, 40, and 27 mg/g creatinine), which returned to
normal on the fifth day after exposure. The findings suggested a
mild transient nephrotoxicity (section 9.1.1).
9.1.2.3 Cardiovascular effects
Patients with acute myocardial infarction and unstable angina
pectoris develop high serum-nickel levels, which may be followed by
coronary vasoconstriction (Leach et al., 1985). The mechanism and
source of nickel release are not yet known, but Sunderman (1983a)
recommended that the amount of nickel in intravenous solutions
should not exceed 5 µg/kg per day. Peto et al. (1984) observed
elevated cerebrovascular mortality (16 deaths, 8.5 expected) among
nickel refinery workers who had worked for 5 years in the process
area, where lung and nasal cancer rates were highest. Mortality
from all other cardiovascular causes was similar to local
population rates.
9.1.2.4 Other effects
In a study by Shi et al. (1986), nickel carbonyl refinery
workers were compared with a control group. Exposure ranged from
0.007 to 0.52 mg Ni(CO4)/m3. A decrease in the MAO (serum
monoamine oxidase) activity, and EEG abnormalities were observed in
the most severely exposed. A number of non-specific symptoms in
persons exposed for long periods to low concentrations
(unspecified) were reported, but no details were given.
Immune reactions elicited in the sera of individuals exposed to
nickel and cobalt were assessed according to changes in the
concentrations of the serum immunoglobulins, IgG, IgA, and IgM, and
the serum proteins, alpha2 macroglobulin (A2M), transferin (TRF),
alpha1-antitrypsin (A1AT), ceruloplasmin (CPL), and lysozyme
(LYS) (Bencko et al., 1983). Examinations were carried out on
workers occupationally exposed to nickel (38 individuals) or cobalt
(35 individuals) and in groups of non-occupationally exposed
children living in areas with different levels of air pollution
from a nearby source of nickel and cobalt emissions.
Non-exposed controls were represented by a group of 42 male
adults, matched by age, and by a group of 48 children from a non-
polluted area. Significantly increased average values were
obtained for IgG, IgA, and IgM, in the group of workers exposed to
nickel, for IgA, in workers exposed to cobalt, and for A1AT, A2M,
CPL and LYS, in both groups of occupationally-exposed adults
( P >0.001 and P >0.005, respectively). Among non-occupationally
exposed children, the most highly exposed group had significantly
elevated average values for A2M and A1AT, which were higher than
those recorded in the groups of "less exposed" and control children
( P >0.02 and P >0.05, respectively) (Bencko et al., 1983).
9.2. Skin and eye irritation and contact hypersensitivity
9.2.1. Skin and eye irritancy
Primary skin irritation reactions to nickel salts in solution
were observed when human volunteers were patch tested. When
aqueous solutions of nickel chloride were applied to the back, the
threshold concentrations for irritancy were 1% with occlusion and
10% without occlusion (Vandenberg & Epstein, 1963).
A 5% aqueous solution of nickel sulfate was also irritant in
some individuals, when applied to the back, under occlusion (Kalimo
& Lammintausta, 1984). On the unoccluded human forearm, the
threshold concentrations of aqueous nickel sulfate producing
irritation were 20% on unbroken skin and 0.13% on scarified skin,
the nickel solutions being applied once daily for 3 days (Frosch &
Kligman, 1976).
9.2.2. Contact hypersensitivity
Nickel and its water-soluble salts are potent skin sensitizers.
Nickel ions pass the skin barrier and bind to carrier proteins to
form the allergen. In the pathogenesis of allergic contact
dermatitis (a type IV reaction), two steps can be recognized.
First, sensitization, after which, subsequent exposure of fully
sensitized skin, even to minute quantities of nickel or its water-
soluble salts, will elicit or provoke an eczematous response which
begins within 12 h, is fully developed at 48-72 h, and then
subsides. After the elicitation phase, the sensitivity will
remain, usually for years, and frequently for a lifetime.
Sensitization is only known to occur after dermal exposure to
nickel, whereas provocation can take place after exposure of the
skin and after ingestion of nickel. The clinical manifestations of
nickel-associated allergic contact dermatitis comprise both primary
and secondary eruptions. The primary eruptions usually appear at
the site of sensitization, and the secondary eruptions will
frequently be more or less generalized, often symmetrical,
maculopapular vesicules of the pompholyx type eczema, which affects
the flexures of the elbows, the sides of the neck, the axillae, the
eyelids, and the genital area. A vesicular hand eczema can also
develop. Nickel contact dermatitis has been documented in the
general population and in nickel workers, especially in the nickel-
plating industry (NAS, 1975). Nickel contact hypersensitivity was
originally considered to be a problem in occupational medicine, but
more recent epidemiological and clinical data indicate that it
could also be a problem in the general population, because of the
increasing number of nickel-containing commodities in everyday use.
The prevalence of nickel sensitivity in the general population can
be evaluated by patch-testing with 5% nickel sulfate in petrolatum
or by an interview technique. Peltonen (1979) and Prystowsky et
al. (1979) studied volunteers from different subpopulations in
Finland and the USA. The prevalence of nickel sensitivity was
about 10% for women and about 1% for men. In women, Prystowsky et
al. (1979) found a high correlation of sensitivity to nickel with
the practice of ear piercing. Peltonen (1979) reported that hand
eczema is very common in patients with nickel sensitivity.
Christensen & Möller (1975a) reported that pompholyx was the
predominant type of hand eczema. Nickel allergy was investigated
in a stratified sample of the Danish female population using an
interview technique (Menné et al., 1982). Of those responding,
14.5% reported a history of nickel allergy. The prevalence rate
was highest in the younger age groups and declined after the age of
50 years. Although the use of the interview technique had certain
limitations, the data were in good agreement with data obtained
through other test methods (e.g., patch testing).
Edman & Möller (1982) reported a study on a patient population
of 8933, who had been patch-tested over a 12-year period. Nickel
sensitization increased during this period in both male and female
patients, females producing a higher rate of positive reactions
than males.
Oral provocation of hand eczema in nickel-allergic patients
with vesicular hand eczema has been studied (Nielsen, in press).
Oral provocation, using nickel sulfate in lactose capsules, was
performed by Christensen & Möller (1975), Kaaber et al. (1978),
Cronin et al. (1980), Jordan & King (1979), Burrows et al. (1981),
and Gawkrodger et al. (1986). The last three studies were double-
blind, weeks with provocation and weeks with placebo following each
other randomly. Evaluation of the eczema was performed in the 3
days following exposure and it was assumed that no delayed response
occurred. The doses ranged from 0.5 to 5.6 mg nickel; lower doses
were used in the double-blind studies. An exacerbation of the
eczema was observed with doses of 2.5 mg nickel or more. The
response to nickel sulfate in lactose differed from nickel
contained in the diet. A provocation study was performed with a
diet naturally containing about 500 µg nickel. Two conclusions were
drawn: (i) a diet naturally high in nickel content (chocolate cake,
soya bean stew, and oatmeal) resulted in an exacerbation of
vesicular hand eczema, when ingesting the diet daily for 4 days,
(ii) exacerbation of hand eczema was observed on day 7 after
finishing the 4-day provocation period. This delayed reaction may
be the reason why reactions to nickel were not reported in previous
double-blind studies in which weeks with nickel exposure and weeks
with placebo randomly followed each other.
The response to oral intake of nickel salts may not be uniform
and hyposensitization may be possible. In 2 controlled studies,
doses of 5.0 mg nickel sulfate given once a week, for 6 weeks,
significantly lowered the degree of contact allergy, as measured by
patch test reactions before, and after, nickel administration
(Sjövall et al., 1987). Further adaptation to oral doses of 2.24
mg nickel sulfate was observed after the volunteer subjects were
given gradually increasing daily doses over a 3-month period
(Santucci et al., 1988). These reported responses to the oral
intake of inorganic nickel do not necessarily contradict one
another, but the mechanisms are not known. However, they indicate
that hand eczema can benefit from a diet low in nickel.
Allergic reaction as well as urticarial and eczematous
dermatitis in nickel-sensitive persons can be produced by implanted
prostheses and other surgical devices made from nickel-containing
alloys (NAS, 1975; Fisher, 1977). Deutman et al. (1977) found
nickel sensitivity in 11 out of 85 patients following hip
prosthesis implantation. Three of the patients had had previous
implants. Oakley et al. (1987) described 4 patients with a history
of metal intolerance, who developed dermatitis following surgical
wound closure with disposable skin clips (nickel content about
10%). Two positive reactions were reported by Waterman & Schrik
(1985) in 85 patch-tested patients, who had undergone hip
arthroplasty.
Metal alloys, used in dental surgery, contain various amounts
of nickel. Data on the relationship between nickel allergy and
dental alloys are limited. In a retrospective study, Moffa et al.
(1983) found that the incidence of nickel allergy in patients with
intraoral exposure to nickel alloys was not significantly higher
than that in patients with no intraoral exposure.
Treatment of nickel-sensitive patients who were suffering from
hand eczema of the pompholyx type, with the nickel-chelating agent
diethyldithiocarbamate was successful in 10 out of 11 patients
(Christensen & Kristensen, 1982). However, hepatotoxicity was
observed in some patients, and this form of treatment is no longer
used.
9.3. Reproduction, embryotoxicity, and teratogenicity
No data are available on the reproductive and developmental
effects of nickel in human beings.
9.4. Genetic effects in exposed workers
Elevated levels of chromosome aberrations, mainly gaps, were
reported by Waksvik & Boysen (1982) in 2 groups of nickel workers,
but SCE was not observed; one group was involved in nickel
crushing/roasting/smelting processes and exposed mainly to nickel
oxide and nickel sulfide, and the other was involved in
electrolysis and exposed mainly to nickel chloride and nickel
sulfate. In a further study on 9 ex-nickel workers, who had been
exposed for an average duration of 25 years, but retired for at
least 8 years, Waksvik et al. (1984) showed the persistence of a
low level of gaps, as well as chromatid breaks, when plasma nickel
levels were still 2 µg/litre. Deng et al. (1983, 1988) showed
elevated levels of both SCEs and chromosome aberrations (gaps,
breaks, fragments) in electroplating workers. However, these
effects were not observed in a study by Decheng et al. (1987) on
workers exposed to nickel carbonyl. Low exposure and good
workplace conditions were cited by the latter authorities.
9.5. Carcinogenicity
9.5.1. Epidemiological studies
9.5.1.1 Nickel refining industry
(a) Clydach nickel refinery, Clydach (Wales)
The existence of an increased risk of nasal cancer in workers
from the Clydach nickel refinery was recognized in 1928. Processes
at Clydach involved: the crushing and grinding of nickel copper
matte, received from Canada, which consisted of the phases Ni3S2,
CuS, and metallic nickel and copper, in a 3:4:1 ratio (Boldt &
Queneau, 1967); the calcining of the crushed matte to produce
nickel and copper oxides (the copper oxide is dissolved in the
metal oxide and described as (Ni,Cu)O (Boldt & Queneau, 1967)); the
extraction of the copper by sulfuric acid; reduction to nickel
oxide; and refining with nickel carbonyl. The first
epidemiological study on Clydach plant workers was carried out by
Hill in 1939 (quoted by Morgan, 1958). Hill found that the
observed to expected ratios (O/E) of deaths were 16:1 for lung
cancer and 22:1 for nasal cancer in the period 1929-38. Morgan
(1958) and Doll (1958) extended the observation period from 1902 to
1957 and identified 131 deaths from lung cancer and 61 deaths from
nasal cancer in 9340 workers. Excess mortality occurred only in
workers first employed before 1924. The highest risk of lung
cancer appeared to have occurred in the calcination and copper
sulfate departments. Deaths from nasal cancer occurred most
frequently in the calcination department. There were no nasal
cancers among workers entering employment after 1924. The decline
in the numbers of deaths from nasal and lung cancer was attributed
to improvements in work practices and refinery processes in the
early 1920s. These improvements included the wearing of gauze
masks, improvements in ventilation, and the use of arsenic-free
sulfuric acid in refining. Follow-up studies were carried out by
Doll et al. (1970) on a cohort of 819 workers, who had been
employed for at least 5 years at Clydach between 1902 and 1944, and
who were still employed in 1934. Thirty-nine cases of nasal cancer
and 113 cases of lung cancer were identified. The reported O/E
ratio for lung cancer was 10.5 for workers starting between 1910
and 1915, 1.8 for workers employed between 1925 and 1929, and 1.1
for those employed between 1930 and 1944. In this study, no nasal
cancer was reported in workers starting employment after 1925.
Doll et al. (1977) extended the follow-up period to 1971 and
confirmed that, since 1925, the risk of respiratory cancer had
decreased sharply; they attributed this to the improvements in work
practices and refinery processes.
Cuckle et al. (1980) studied the causes of death in a small
cohort of 297 men who were mainly exposed to soluble nickel salts
between 1937 and 1960. They did not find any nasal sinus cancers.
Thirteen cases of lung cancer were found, a significant excess
above regional rates (expected number 7.54), but not significant
when compared with the rates for England and Wales. The small size
of the group limited this study.
Estimated concentration data for different nickel species in
different areas of Clydach nickel refinery are shown in Table 33.
Table 33. Estimated concentration (mg/m3)
before 1930, of major forms of nickel in
high-risk areas of the Clydach nickel refinery,
South Walesa
--------------------------------------------------
Metal Oxide Ni3S2 Soluble
--------------------------------------------------
Calcining 5.3 18.8 6.8 0.8
Furnaces 5.6 6.4 2.6 0.4
Copper plant - 13.1 0.4 1.1
Hydrometallurgy 0.5 0.9 0.1 1.4
--------------------------------------------------
a From: Peto (1988).
Peto et al. (1984) extended the follow-up to 1981. The cohort
included 968 men. One hundred and fifty-nine lung cancer deaths
and 56 nasal cancer deaths were identified. The duration of
employment in 4 areas showed a statistically significant
association with lung or nasal cancer or both. These were the
calcining furnace area, the calcining/crushing area, the copper
sulfate area, and the furnace area. Peto et al. (1984) also
related age, time of exposure, and exposure levels, to lung and
nasal sinus cancer incidence in pre-1925 workers. Exposure level
was defined on the basis of duration of work in high-risk areas.
Both lung and nasal cancer showed increasing risk with increasing
duration of work in high-exposure areas. Nasal cancer showed a
strong positive relationship with age at first exposure, whereas
lung cancer did not. The latency period was 20-50 years for nasal
cancer and somewhat shorter for lung cancer. The risk of nasal
cancer was highest for workers with first exposure between 1910 and
1915 and declined thereafter, whereas the risk for lung cancer was
highest for workers between 1920 and 1925.
(b) Falconbridge refinery, Kristiansand (Norway)
Loken (1950) reported 3 cases of lung cancer in workers from
the Falconbridge refinery. This plant began operations in 1910,
processing matte from Ontario via roasting, smelting, and
electrolysis. Although workers in the roasting/smelting
departments were, and are, exposed to sulfides, oxides, and nickel-
copper oxides in dust and fumes, the processes have changed in
recent years and exposure levels have been reduced dramatically.
The electrolytic workers are exposed to aerosols containing, inter
alia, nickel sulfate and nickel chloride (Hogetveit & Barton,
1976). These workers are, as cited by Hogetveit & Barton (1976),
"exposed largely to soluble nickel aerosols". That the soluble
form predominates in the exposure of electrolysis workers is also
supported by the results in Table 34. This table shows that, even
when the air values are lower in the electrolysis department than
in roasting/smelting department, the plasma and urine values are
higher.
Table 34. Summary of biological tests and personal sampler measurementsa
---------------------------------------------------------------------------
Departments Number of Mean Mean Mean
employees plasma ± SD urine ± SD concentration
(µg Ni/litre) (µg Ni/m3) in air ± SD
(µg Ni/m3)
---------------------------------------------------------------------------
No. 01-19 24 72 ± 2.8 65.0 ± 57.6 0.86 ± 1.20
(Roasting-
smelting)
No. 22-44 90 11.9 ± 8.0 129.2 ± 105.6 0.23 ± 0.42
(Electrolytic)
Other process 13 6.4 ± 1.9 44.6 ± 26.7 0.42 ± 0.49
departments
---------------------------------------------------------------------------
CORRELATION BETWEEN RESULTS
Departments Number of Plasma vs. air Plasma vs. air Plasma vs. urine
employees Corr. factor R Corr. factor R Corr. factor R
-----------------------------------------------------------------------------
No. 01-19 24 -0.55 -0.11 -0.71 +0.14 3.72 +0.76
No.22-44 90 1.98 0.21 2.99 0.31 7.30 0.77
Other 13 2.41 0.67 1.70 0.47 2.27 0.63
-----------------------------------------------------------------------------
a Modified from Hogetveit et al. (1978).
The first epidemiological study was carried out on a cohort of
1916 workers who were employed for at least 3 years before 1961
(Pedersen et al., 1973). Exposure was defined according to
department or work in which there had been the longest employment.
The authors used 4 work categories: roasting and smelting,
electrolysis, other processes, other work groups. All occupational
groups were associated with an excess risk of respiratory cancer.
Risk was greatest in the roasting and smelting department and the
electrolysis department. Workers in the electrolysis department
showed the highest risk of lung cancer (O/E=26/3.6) and an excess
risk of nasal cavity cancer (O/E=6/0.2). The risk of nasal cancer
was greatest (O/E=5/0.1) in the roasting and smelting department,
but there was also an increased risk of lung cancer in this
department (O/E=12/2.5).
Magnus et al. (1982) updated the study of Pedersen et al.
(1973) and confirmed the results. They also analysed data on
smoking habits and found evidence for an additive effect of smoking
and nickel exposure.
The cohort was further followed up by Andersen (1988). During
the follow-up period 1953-87, 163 new cases of respiratory cancer
were observed against 42 expected. There was a decreasing trend in
ratio between observed/expected from 6.9 in 1926-35 to 2.4 among
those first employed in 1956-65. No risk for any other cancer was
recorded, in fact, the observed number is lower than the expected
number in all periods (207/248 all periods combined). No cases of
nasal cancer have been observed in workers employed after 1955, but
the expected number is only 0.20.
Following technological improvements in the plant, Andersen
(1988) formed a new cohort of 1237 employees with a total
employment of one year or more, the first entry in 1968 or later,
and exposure estimated according to nickel species and exposure
concentration. For entry in the period 1968-72, 7 new cases of
lung cancer were found, versus 1 expected. Actual measurements
reported in 1979 (Torjussen & Andersen, 1979) were 0.1-0.5 mg/m3
for electrolysis workers and 0.1-1.0 mg/m3 for the roasting/
smelting category. Estimated concentrations for the period 1968-77
were about 0.5-2.0 mg/m3 and about 2.0-5.0 mg/m3, respectively. In
the electrolysis department, the exposure was mainly to soluble
nickel, even if there might have been up to 60% NiS and NiO present
in some working areas. No exposure to soluble compounds was
considered to take place in the roasting/smelting department, but
the possible presence of NiSO4, as an intermediate, was not
appreciated.
The risk of lung cancer for a 1953-84 cohort of 3250 workers
was also estimated according to department and to cumulative
exposure to different nickel species (nickel subsulfide, nickel
oxide, soluble nickel). Although an increased risk was found for
both electrolysis and roasting, smelting, and calcining, the
highest risk and steepest dose-response relationship (based on
exposure time) was found for electrolysis, and the highest risk was
found for the highest cumulative exposure to soluble nickel. It
must be remembered, however, that most workers were exposed to more
than one form of nickel.
Torjussen et al. (1979a,b) examined nasal biopsy specimens from
318 active and 15 retired workers with at least 8 years of
employment. Epithelial dysplasia was found in 14 (14%) of the 97
roasting/smelting workers, in 16 (11%) of the 144 electrolysis
workers, in 8 (10%) of the 77 non-process workers and in 7 (47%) of
the retired workers. One out of 57 controls, a carpenter, had
dysplasia. Two co-workers from the roasting/smelting department
had nasal carcinoma. Based on the methods employed by Torjussen et
al. (1979a,b), histopathological reinvestigations of nasal biopsy
specimens from workers, previously included in the Torjussen study,
were carried out by Boysen et al. (1980, 1982, 1984) and similar
results were found. The histopathological changes in the nasal
epithelium, including dysplasia, persisted in active workers,
despite reduction of airborne nickel concentrations in the working
environment. The frequency of dysplasia was also still elevated in
retired workers. The real significance of nasal epithelial
dysplasia is not known, but it could be considered as a
precancerous lesion because of its prevalence in groups with
increased risk of nasal cancer. The role of nasal biopsies in
identifying and monitoring occupational nickel exposure is unclear.
(c) International Nickel Company (INCO), Ontario
Sulfide nickel ore is mined and refined at several locations
using different processes. The Ontario Health Department studied a
cohort of 2355 refinery workers followed from 1930 to 1957
(Sutherland, 1959, reviewed by Mastromatteo, 1967). There were 7
deaths from sinus cancer (0.19 expected) and 19 from lung cancer
(8.45 expected). Nearly all excess deaths from sinus cancer and
lung cancer occurred in workers exposed to furnace dusts in
calcining and sintering. Mastromatteo (1967) extended Sutherland's
study to the period between 1930 and 1960. The risk of nasal
cancer was still elevated (16 sinus cancer deaths observed, 0.166
expected) as well as the risk of lung cancer (37 lung cancer deaths
observed, 12.71 expected). According to Mastromatteo (1967), the
investigations of Sutherland led to major process changes, among
them the termination of the sinter plant operations, in 1962. The
Sutherland study was updated by Rigaut(1986 ) and the US EPA,
(1986a) to cover the period between 1930 and 1970. By this time,
sinus and lung cancer deaths amounted to 24 (SMR=5106) and 76 (SMR=
1861), respectively.
On the basis of the unpublished work by Sutherland (1959),
Chovil et al. (1981) identified a cohort of 495 men, who had been
employed at the Copper Cliff sinter plant for a period between 1948
and 1962, and were alive in 1963. However, only 75% of the cohort
were successfully followed through 1977 and 1978. Six cases of
sinus cancer were identified (no expected numbers given), and 54
cases of lung cancer of which 37 were fatal, compared with 4.25
expected deaths. Despite some methodological weaknesses, an excess
risk of lung and sinus cancer is suggested.
A large cohort study was carried out by Roberts et al. (1984).
The cohort consisted of 54724 INCO workers employed in Port
Colborne refinery and in the extraction and refining of copper and
nickel in two Sudbury sinter plants (Coniston smelter and Copper
Cliff plant). The cohort was followed for mortality between 1950
and 1976. In Sudbury workers not exposed to sintering operations,
the SMRs for nasal and lung cancer were 144 (O/E=3/2.08) for nasal
cancer and 108 (O/E=222/204.98), respectively, compared with SMRs of
2174 (O/E=2/0.09) for nasal cancer and 463 (O/E= 42/9.08) for lung
cancer in workers exposed to sintering operations. In workers from
the Port Colborne leaching, calcining, and sintering departments,
SMRs for nasal and lung cancer were 9412 (O/E=16/0.17) and 298
(O/E=49/16.44), respectively, compared with SMRs of 78
(O/E=13/16.65) for lung cancer in men who had never worked in this
department. A single case of nasal cancer was observed compared to
0.16 expected, but, on further investigation, it was found that
this man had had 20 years exposure to roasting/calcining operations
elsewhere. Nasal and lung cancer mortality was found to increase
with increasing duration of exposure. Lung cancer was already seen
in excess 5-10 years after first exposure in Sudbury, but, in Port
Colborne, it did not become apparent until 20 years after the first
exposure. Nasal cancer began to appear after 15 years. Apparently
the nasal cancer risk was higher at Port Colborne than at Sudbury,
while lung cancer was more frequent at Sudbury.
Airborne nickel levels were much higher in the sintering
department than in other areas of these refineries. Warner (1985)
reported that a 40-h sample, taken on the operating floor of the
Copper Cliff Sinter Plant in 1960, contained 46.4 mg total dust/m3.
The major nickel compounds in the dust were Ni3S2, NiO, and NiSO4.
Samples taken between 1948 and 1952 from dusty air escaping from
roof monitors contained over 100 mg total dust/m3, but it is not
known if these were representative of exposures in working areas.
Other processes are believed to have been very much less dusty.
Mean levels in most areas were generally well below 1 mg nickel/m3
in later years, though substantially higher levels occasionally
occurred, particularly in matte grinding and in the handling of
metallic nickel powders (Warner, 1984).
The composition of a sample of dust collected in 1959 from the
calcining and sintering rooms of the Port Colborne, Ontario,
refinery is shown in Table 35 (Gilman & Yamashiro, 1985). The
major constituents were nickel subsulfide (57%), nickel sulfate
(20%), nickel oxide (6%), and copper oxide (3%).
Table 35. Composition of dust collected in
1959 in the calcining and sintering of the
Port Colborne (INCO) nickel refinery Ontarioa
---------------------------------------------
Compound Percentage
---------------------------------------------
CuO 3.4
NiSO4 x 6H2O 20.0
Ni3S2 57.0
NiO 6.3
CoO 1.0
Fe2O3 1.8
SiO2 1.2
Miscellaneous 2.0
Moisture 7.3
---------------------------------------------
a From: Gilman & Yamashiro (1985).
Kaldor et al. (1986) analysed the same data using a case-
control approach. In addition to the four areas identified by Peto
et al. (1984), employment in nickel sulfate production was
associated with increased lung and nasal cancer risk. Lung and
nasal cancer mortality and exposure levels for various nickel
compounds for men employed in major production areas were reported
by Peto (1988). There were 5 lung cancer deaths (1.5 expected) and
4 nasal cancer deaths among men, employed for 5 or more years in
hydrometallurgy (nickel sulfate production), who had spent less
than a year in other high-risk areas (copper sulfate, calcining,
furnaces). The principal exposure in hydrometallurgy was to
soluble nickel (about 1.4 mg/m3) with lower levels of nickel oxide
(0.9 mg/m3), nickel subsulfide (0.1 mg/m3), and metallic nickel
(0.5 mg/m3). These estimates were provided by the technical staff
of the refinery, but no measurements were made to verify them.
(d) Falconbridge, Ltd., Falconbridge (Ontario)
A mortality study was carried out on a cohort of 11 594
workers, employed for at least 6 months by the Falconbridge nickel
mines, Sudbury, Ontario, between 1950 and 1976 (Shannon et al.,
1984). In this plant, sintering techniques included a low
temperature step. Nasal cancer was not observed. A slight excess
of lung cancer (SMR=123, O/E=46/37.5) in both mines and service
occupations was not statistically significant. However, laryngeal
cancer deaths were significantly increased in miners (SMR=261,
O/E=5/1.92). A slight, though not significant, increase in deaths
from prostate cancer was noted in smelter workers.
(e) Sherritt Gordon Mines Ltd., Fort Saskatchewan, Alberta
Sherritt Gordon Mines, Ltd., established hydrometallurgical
nickel refining in 1954. Nickel exposures were mainly in the form
of oxide ore concentrate, nickel metal, and some soluble compounds.
Egedahl & Rice (1984) conducted a study of cancer incidence and
mortality with a cohort of 720 workers employed in
hydrometallurgical refining and did not find any excess of
respiratory cancer. Two cases of renal cancer occurred in workers
with 11 and 16 years exposure to nickel concentrate, soluble nickel
substances, and nickel metal, prior to developing their illness.
Both were cigarette smokers.
(f) Nickel refinery, Huntington (West Virginia)
Copper-nickel matte was refined in Huntington during 1922-47
and a mortality study was performed on 1855 men, employed before
1947 who had worked for at least one year at the plant (Enterline &
Marsh, 1982). Concentrations of airborne nickel in the area where
nickel was crushed, ground, and handled, were estimated to range
from 20 to 350 mg/m3 and from 5 to 15 mg/m3 in the calcining area.
The SMR for nasal cancer was 2443.5 with 2 observed and 0.08
expected cases. Two nasal cancers were observed among nickel-
exposed non-refinery workers. There was no significant increase in
deaths from lung cancer among refinery workers (SMR=118.5 with 8
observed and 6.75 expected cases) and it was suggested that the
nickel exposures at the plant may have been considerably lower than
in other plants. However, for refinery and non-refinery workers
combined, and for all respiratory cancer cases combined, there was
a dose-relationship with accumulated nickel exposure.
(g) Oxide ore refinery, New Caledonia
Nickel oxide ore has been mined on the South Pacific Island of
New Caledonia since 1866 and smelted there since 1885. Lessard et
al. (1978) identified 92 cases of lung cancer that had occurred in
this area between 1970 and 1974. The observed rate was 3-7 times
higher than rates in other Central or South Pacific areas, but
similar to those seen in industrialized countries. A significant
increase in lung cancer rates was observed with decreasing distance
from the refinery. The authors found that, in New Caledonia, there
was a 3-fold risk of lung cancer among workers compared with non-
workers, independent of age and tobacco smoking, and considered
that these results supported the etiological role of nickel in lung
cancer. Goldberg et al. (1985a,b) carried out a retrospective
cohort study and a case-control study and reported no excess of
respiratory cancer in workers of the Society Le Nickel (SLN)
compared to the general population of New Caledonia; however, these
studies have weaknesses in statistical power and definition of the
control population. Goldberg et al. (1988) concluded that, in a
10-year period, there was no excess of either lung or sinonasal
cancer in the nickel workers, when compared with the island
population on an age-matched basis. Both studies were considered
to have methodological problems, but the Goldberg et al. (1988)
study had more comprehensive health data.
(h) Other nickel refineries
In the Soviet Union, Saknyn & Shabynina (1970, 1973) reported
an increase in lung and gastric cancer in workers in a nickel
refinery plant in the Urals. The refinery was engaged in the
preparation of nickel oxide ore, roasting, and smelting. They
stated that the lung cancer death rate from 1955 to 1967 exceeded
that of the nearby urban population by 180% and that the death rate
from sarcomas and stomach cancer was also increased. Excess lung
cancer was seen in workers in roasting/reducing and smelting, but
also in electrolysis, where exposure was principally to nickel
sulfate and nickel chloride, and, in the cobalt shop, where
exposure was to various soluble salts of nickel and cobalt.
Tatarskaya (1965) reported 2 cases of nasal cancer among
workers engaged in the electrolytic refining of nickel.
In workers in a Czech plant, where nickel oxide ore had been
refined since 1963, 8 lung tumours were diagnosed between 1971 and
1980 (Olejar et al., 1982). All workers had smoked more than 25
cigarettes per day. These results must be treated with caution as
the latency period from 1963 was very short.
Rockstroh (1959) reported 45 cases of lung cancer in workers in
a nickel refinery in Aue in the German Democratic Republic between
1928 and 1956. Exposure was very complex: nickel, arsenic, cobalt,
copper, bismuth, and benzo (a)pyrene, and workers were often
involved in different processes, during their employment. One case
of lung cancer was found among office personnel, but the size of
the comparison group was not indicated.
9.5.1.2 Nickel alloy manufacturing
In a nickel alloy manufacturing plant in Hereford, England, the
alloys are manufactured from materials consisting of metallic
nickel and other metals (iron, copper, cobalt, chromium,
molybdenum). Workers are exposed to metallic nickel and oxidic
nickel. The average airborne nickel concentrations ranged from
0.84 mg/m3 in the melting, fettling, and pickling area to 0.04
mg/m3 in the process, stock handling, distribution, and warehouse.
Cox et al. (1981) studied a cohort of 1925 men with at least 5
years of employment between 1953 and 1978. He did not find any
cases of nasal sinus cancer. The lung cancer death rate was not
increased compared with the numbers expected from national and
local mortality rates. However, the study period was too short to
exclude the risk of respiratory cancer completely.
Redmond (1984) reported a cohort mortality study on 28 261
workers employed at 12 high nickel alloy production plants between
1956 and 1960, including a follow-up to 1977. The predominant
nickel species, to which the workers were exposed, appeared to have
been nickel dust and nickel oxide. Average levels of airborne
nickel ranged from 0.064 mg/m3 in the cold working area to 1.5
mg/m3 in the powder metallurgy area (Redmond et al., 1983, internal
report). Overall, no statistically significant increased risk was
observed for cancers of the lung, nasal sinuses, larynx, or kidney.
Another study dealt with the standardized proportionate
mortality ratios of 851 foundrymen from 26 nickel/chromium alloy
foundries compared with 141 unexposed foundrymen during the period
1968-79 (Cornell & Landis, 1984). There were no cases of nasal
cancer. The percentages of respiratory cancer in the exposed group
were not significantly different from the percentages of
respiratory cancer in the unexposed group. When compared to
national mortality data, the observed numbers of lung cancer and
all cancer deaths in exposed men were not significantly different
from the expected values.
Cornell (1984) performed a proportional mortality analysis on
4487 workers in 12 US plants engaged in the production of stainless
steel and low nickel alloys. All deaths were included for, at
least, a 5-year period, up to 1977. No cases of nasal cancer were
observed and there was no excess of lung cancer. However, details
of the study, such as the definition of exposure and latency
period, were not provided.
9.5.1.3 Nickel plating industry
Silverstein et al. (1981) conducted a case-control study on
deaths among workers at a Scandinavian die-casting and
electroplating plant. The major operations in the plant were zinc
alloy die-casting, buffing, polishing of zinc and steel parts, and
electroplating with copper, nickel, and chrome. There were 238
deaths among workers with at least 10 years of employment. The
proportionate mortality ratio (PMR) (38 deaths, PMR=209) for lung
cancer was significantly increased. However, because of
heterogeneous exposure, the increased risk of lung cancer
mortality could not be exclusively associated with nickel exposure.
The authors subsequently initiated a case-control study on the
28 white male and 10 white female lung cancer deaths. Cases and
controls were compared for length of employment in individual
departments. Odds ratios were estimated for work in 14 different
departments. For males in three departments, the odds ratio
increased with duration of work in the departments. This trend was
most significant for the department where die-casting and plating
were the major activities.
A study was carried out by Burges (1980) on a group of 508
nickel platers in an engineering firm in England. Nickel plating
was done in a separate shop from chrome plating. The cohort was
divided into groups: nickel bath workers with less than one year of
exposure, more than one year of exposure, and other exposed
workers. In the short-exposure group (less than one year), no
excess mortality was found, whereas, in the longer-exposure group
(more than one year), a significant excess of deaths was found for
stomach cancer (O/E = 4/0.6, SMR = 623) and for non-malignant
respiratory disease (O/E = 8/2.8, SMR = 286). Only one lung cancer
death was observed in the longer-exposure group compared with 1.7
expected cases. The results of the study suggest a higher incidence
of gastric cancer in nickel platers. Sorahan et al. (1987) reported
a study on mortality in nickel and chromium platers in the United
Kingdom. While an excess of certain cancers was associated with
chromium bath workers, it was stated that nickel exposure was not a
confounding factor.
9.5.1.4 Welding
Stern (1983) and Langard & Stern (1984) concluded that the
excess risk of lung cancer in welders is approximately 30% greater
than that in the non-welding population. It was suggested that the
greater risk of lung cancer found among stainless steel welders, in
some studies, could be related to high concentrations of nickel and
hexavalent chromium in stainless steel welding fumes.
The role of nickel compounds as a cause of respiratory cancer
is difficult to assess, because chromates and other carcinogens may
occur in the work-place air during the welding of stainless steel
and other nickel-containing alloys (IARC, 1990).
9.5.1.5 Nickel powder
In the Oak Ridge Gaseous Diffusion Plant, Tennessee, metallic
nickel is used in the production of barrier material for the
isotopic enrichment of uranium by gaseous diffusion. The median
nickel concentration in air between 1948 and 1963 ranged between
0.1 and 1.0 mg/m3 (Godbold & Tompkins, 1979). In a cohort of 814
workers, Godbold & Tompkins (1979) did not find any increased risk
of respiratory cancer during the period 1948-72. In a follow-up
study by Cragle et al. (1984) covering the years from 1972 to 1977,
the lung cancer rate did not increase. The SMR for men employed
for more than 5 years was 91, based on 8 deaths.
9.5.1.6 Nickel-cadmium battery manufacturing
Sorahan & Waterhouse (1983) examined 3025 nickel-cadmium
battery workers for a possible excess of cancer mortality.
Overall, the SMRs showed a statistically significant excess of
respiratory cancer deaths, but it was not possible to attribute the
excess to nickel or to cadmium, since exposure to both occurred at
high levels. In studies by Kjellstrom et al. (1979) and Andersen
et al. (1983), the mortality was examined in men working at a
nickel-cadmium battery-manufacturing plant with mixed exposure to
both metals and nickel hydroxide. Increased respiratory, renal,
and prostate cancer mortality were noted, though there was no
increase in overall cancer mortality. There were two cases of
nasopharyngeal cancer. One started employment before 1948, the
other between 1948 and 1961, when exposure to nickel powder was
very high, exceeding 100 mg nickel/m3 in the work area. The low
risk ratio for lung cancer rendered the studies inconclusive for
the effects of nickel compounds.
9.5.1.7 Case-control studies
The case-referent design has been used to associate the
incidence of certain cancer types with occupations in nickel-
producing or nickel-using industries.
In a British study (Acheson et al., 1981), an excess risk for
nasal cancer was found in furnace and foundry workers, partly,
because of several cases among Clydach refinery workers.
Bernacki et al. (1978b) carried out a small case-control study
at an aircraft-engine factory where high nickel alloys were used.
Nickel exposure was not found to be associated with lung cancer
cases. Roush et al. (1980), using data from the Connecticut Tumor
Registry and from City Directories, determined the occupational
exposures of individuals reported to have cancer of the nasal
sinuses in the years 1935-75. The study did not reveal any
increase in nasal cancer deaths in occupations classified as having
nickel exposure. A case-control study of 204 laryngeal cancer
patients and 204 neighbourhood controls did not show any relation
to occupational exposure to nickel, while tobacco, alcohol, and
asbestos were major risk factors (Burch et al., 1981). However,
Olsen & Sabroe (1984) found a statistically significant excess risk
of laryngeal cancer related to potential nickel exposure from
alloys, battery chemicals, and chemicals used in the production of
plastics. A case-referent study covering the entire Montreal
population showed a statistically significant odds ratio of 3.1 for
lung cancer and nickel exposure and some indication of a dose-
response relationship (Gerin et al., 1984), though individuals
exposed to nickel were always exposed to chromium as well.
9.5.2. Carcinogenicity of metal alloys in orthopaedic prostheses
It was concluded by Sunderman (1989b) that case reports of
sarcomas at sites of metal implants in human beings and domestic
animals have implicated carcinogenesis as a potential, albeit rare,
complication of metal orthopaedic prostheses (Table 36). Since
physicians and veterinarians are not obliged to report adverse
reactions to prostheses, the actual incidences of sarcomas in
association with metal implants cannot be reliably estimated, and
the possibility that the apparent associations are coincidental
cannot be excluded. Carcinogenesis bioassays in rodents have shown
that pure nickel or cobalt powders induce sarcomas at injection
sites, and transformation assays in mammalian cells have yielded
positive results with nickel powder, suggesting that surgical
implantation of alloys containing these metals may pose
carcinogenic risks.
Table 36. Patients with tumours at sites of metal implantsa
----------------------------------------------------------------------------------------------------
Patient age Time lapse Implant type Tumour type Reference
[years] to tumour
implant (years)
(site)
----------------------------------------------------------------------------------------------------
12 (humerus) 30 Steel plate (FeCrNi) and 4 Ewing's sarcoma McDougall (1956)
screws (FeCr), much corrosion
37 (tibia) 3 Osteosynthesis plate and 2 Osteochondrosarcoma Delgado (1958)
screws, alloy unknown
53 (femur) 3 Steel plate and nail (6 Giant cell tumour Castleman &
months) then Moore McNeely (1956)
prosthesis and trochanteric
wires
58 (tibia) 26 Steel (Type 316) plate and Haemangiosarcoma Dube & Fisher
screws (Types 304 and 316) (1972)
56 (hip) 2.5 Mckee metal-to-metal Fibrosarcoma Arden & Bywaters
arthroplasty, alloy not (1978)
stated
4 (hip) 7 Sherman (CoCrMo) plate and Ewing's sarcoma Tayton (1980)
6 screws (implanted 1 year)
31 (tibia) 17 Sherman (CoCrMo) plate and Histiocytic McDonald (1981)
screws lymphoma
75 (hip) 2 Charnley-Muller arthroplasty Malignant fibrous Bago-Granell et
and trochanteric wires, alloy histiocytoma al. (1984)
not stated
63 (hip) 2.4 McKee-Farrar arthroplasty Malignant fibrous Swann (1984)
(CoCrMo) histiocytoma
----------------------------------------------------------------------------------------------------
Table 36 (contd.)
----------------------------------------------------------------------------------------------------
Patient age Time lapse Implant type Tumour type Reference
[years] to tumour
implant (years)
(site)
----------------------------------------------------------------------------------------------------
75 (hip) 5 Ring arthroplasty (CoCrMo) Osteogenic sarcoma Penman & Ring
(1984)
76 (knee) 4.5 CoCrMo arthroplasty Epithelioid sarcoma Weber (1986)
14 (hip) 29 Sherman screw (CoCrMo) Malignant fibrous Hughes et al.
histiocytoma (1987)
40 (hip) 13 2 Screws (unspecified alloy) Undifferentiated Ryu et al.
for 12 years; then hip sarcoma (1987)
prosthesis (CoCrMo stem and
Al2O3 head)
----------------------------------------------------------------------------------------------------
a Modified from: Sanderman (1989b).
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1. Exposure
Nickel is an ubiquitous element and has been detected in
different media in all parts of the biosphere.
Nickel is introduced into the environment from both natural and
man-made sources and is circulated throughout all environmental
compartments by means of chemical and physical processes, as well
as through the biological transport mechanisms of living organisms.
Acid rain may leach nickel as well as other metals from plants
and soil.
Atmospheric nickel is considered to exist mainly in the form of
particulate aerosols.
Nickel is introduced into the hydrosphere by removal from the
atmosphere, by surface run-off, by discharge of industrial and
municipal wastes, and also following the natural erosion of soils
and rocks.
A major source of nickel in the environment is the combustion
of fossil fuels, particularly coal.
Uncontrolled emissions and waste disposal may have adverse
effects on the environment.
Both the chemical and physical forms of nickel and its salts
strongly influence bioavailability and toxicity.
Nickel from soil and water is absorbed and metabolized by
plants and microorganisms and these small quantities of nickel are
widely present in all foods and water.
Some foods, such as pulses and cocoa products, contain
relatively high amounts of nickel, but these quantities have not
been correlated with adverse health effects.
10.2. Human health effects
Nickel is normally present in human tissues and, under
conditions of high exposure, these levels may increase
significantly.
The general population is exposed to nickel via the diet and
objects containing nickel, especially jewellery and coins.
Occupational exposure to nickel is important.
Inhalation is an important route of exposure to nickel and its
salts in relation to health risks. The gastrointestinal route is
of lesser importance.
Nickel absorption from the gastrointestinal tract is poor,
though nickel in the drinking-water is absorbed to a greater extent
in an empty stomach. This may be a risk for sensitized persons.
Smoking tobacco may contribute to nickel intake, but there is
no agreement on the chemical nature of nickel in tobacco smoke and
its health significance.
Target organs are the respiratory system, especially the nasal
cavities and sinuses, and the immune system.
The percutaneous absorption of nickel is minor, but it is
important in sensitization.
Nickel and its salts are potent skin sensitizers and possible
respiratory sensitizers in human beings. Nickel dermatitis is a
common result of nickel exposure, especially in women.
Primary skin and eye irritation reactions to high
concentrations of soluble nickel salts have also been reported.
Acute toxicity is a minor risk from nickel or its compounds,
with the exception of nickel carbonyl.
There is no convincing evidence that nickel salts produce point
mutations in bacterial systems. However, some nickel salts are
clastogenic in vitro, producing chromosome aberrations,
(transformation) and sister chromatid exchanges in mammalian cells.
There is a lack of evidence of a carcinogenic risk from oral
exposure to nickel, but the possibility that it acts as a promoter
has been raised.
There is evidence of a carcinogenic risk through the inhalation
of nickel metal dusts and some nickel compounds.
Very high concentrations of nickel are required to produce
teratogenic or genotoxic effects.
The consequences of the effects of nickel on the immune system
are not clear.
10.3. Environmental effects
Nickel is accumulated by plants. Growth retardation has been
reported in some species, at high nickel concentrations.
There is no evidence that nickel may undergo biotransformation,
though it does undergo complexation.
Nickel has been shown to be essential for the nutrition of many
microorganisms, a variety of plants, and for some vertebrates.
11. RECOMMENDATIONS
1. The nomenclature for nickel compounds should be further
standardized.
2. Analytical methods should be developed and standardized to
facilitate speciation of nickel compounds in environmental
media, biological materials, the workplace, and atmospheric
emissions.
3. Studies are recommended on the kinetics and mass transfer of
nickel, in order to elucidate the biogeochemical cycle on a
global scale and its potential for long-range transport.
4. The use of nickel in consumer products that may release nickel
when in contact with skin should be regulated. The
specification and testing requirements should be standardized.
5. Studies should be performed on the absorption and cellular
uptake, transport, and metabolism of well characterized nickel
species, following different routes and types of administration.
6. Nickel compounds, to which human beings are exposed, should be
tested for carcinogenicity in adequate, long-term, inhalation
studies on animals.
7. Further large-scale cohort studies with well characterized
exposures, including biological monitoring, are needed in all
sectors of industry, to establish more accurate upper limits of
cancer risk.
8. Priority should be given to improving industrial hygiene in
occupations where exposure to high levels of soluble nickel
compounds may occur.
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
The carcinogenic risk of nickel and inorganic nickel compounds
has been evaluated by the International Agency for Research on
Cancer (IARC). An International Working Group evaluated
epidemiological and experimental carcinogenicity data on nickel and
certain nickel compounds encountered in the nickel refining
industry (IARC, 1990). The evaluations are summarized below:
-----------------------------------------------------------------------
Degree of evidence Overall
for carcinogenicitya evaluation
Human Animal
-----------------------------------------------------------------------
Nickel and nickel compounds
Nickel compounds 1
Nickel salts L
Nickel sulfate S
Combination of nickel oxides
and sulfides encountered in
the nickel refining industry S
Nickel monoxides S
Nickel trioxides I
Nickel sulfide, amorphous I
Nickel sulfides, crystalline S
Nickel antimonide L
Nickel arsenides L
Nickel carbonyl L
Nickel hydroxides S
Nickelocene L
Nickel selenides L
Nickel telluride L
Nickel titanate I
Metallic nickel I S 2B
Nickel alloys I L
-----------------------------------------------------------------------
a S : Sufficient evidence of carcinogenicity.
L : Limited evidence of carcinogenicity.
I : Inadequate evidence of carcinogenicity.
1 : The agent is carcinogenic for human beings.
2B : The agent is possibly carcinogenic for human beings.
In the Guidelines for drinking-water quality (WHO, 1984), no
guideline value was set for nickel in drinking-water, as the
toxicological data available at the time indicated that a guideline
value was not necessary.
In the Air quality guidelines for Europe (WHO, 1988), no safe
level was recommended for nickel, because of its carcinogenic
properties. At an air concentration of nickel dust of 1 µg/m3, a
conservative estimate of the lifetime risk is 4 x 10-4.
REFERENCES
ABDULWAJID, A.W. & SARKAR, B. (1983) Nickel-sequestering renal
glycoprotein. Proc. Natl Acad. Sci. (USA), 80(14): 4509-4512.
ABO-RADY, M.D. (1979a) Determination of heavy metals in two
biological and two geological standard materials by atomic
absorption spectroscopy. Fresenius Z. anal. Chem., 286: 380-382.
ABO-RADY, M.D. (1979b) The levels of heavy metals (Cd, Cu, Hg, Ni,
Pb, Zn) in brook trout from the river Leine in the area of
Göttingen (West Germany). Z. Lebensm. Unters. Forsch., 168(4): 259-263.
ABRAHAM, T.J., SALIH, K.Y. & CHACKO, J. (1986) Effects of heavy
metals on the filtration rate of bivalve Villorita cyprinoides
(Hanley) var. cochinensis. Indian J. mar. Sci., 15(3): 195-196.
ACHESON, E.D., COWDELL, R.H. & RANG, E.H. (1981) Nasal cancer in
England and Wales: an occupational survey. Medicine, 38(3): 218-224.
ADALIS, D., GARDNER, D.E. & MILLER, F.J. (1978) Cytotoxic effects
of nickel on ciliated epithelium. Am. Rev. respir. Dis. 118(2):
347-354.
ADAMSSON, E., LIND, B., NILSSON, B. & PISCATOR, M. (1980) Urinary
and fecal elimination of nickel in relation to air-borne nickel in
a battery factory., In: Brown, S.S. & Sunderman, F.W. Jr, ed., Nickel
toxicology: Proceedings of the 2nd International Conference on
Nickel Toxicology, Swansea, 3-5 September,1980, London, New York,
Academic Press, pp. 103-106.
ADKINS, B., RICHARDS, J.H. & GARDNER, D.E. (1979) Enhancement of
experimental respiratory infection following nickel inhalation.
Environ. Res., 20(1): 33-42.
AGGARWAL, S.K., KINTER, M., WILLS, M.R., SAVORY, J. & HEROLD, D.A.,
(1988) Isotope dilution gas chromatography - mass spectrometric
method for the determination of nickel in biological materials. In:
Fourth International Conference on Nickel Metabolism and
Toxicology: Abstracts, Espoo, Finland, 5-9 September, 1988,
Helsinki, Institute of Occupational Health (Abstract II.1).
AITIO, A. (1984) Biological monitoring of occupational exposure to
nickel. In: Nickel in the human environment. Proceedings of a
Joint Symposium, Lyon, 8-11 March, 1983, Lyon, International Agency
for Research on Cancer, pp. 497-505 (IARC Scientific Publications
No. 53).
AJMAL, M. & KHAN, A.U. (1985) Effect of electroplating factory
effluent on germination and growth of hyacinth bean and mustard.
Environ. Res., 38(2): 248-255.
AJMAL, M., KHAN, M.A. & NOMANI, A.A. (1985) Distribution of heavy
metals in plants and fish of the Yamuna river (India). Environ.
monit. Assess., 5(4): 361-367.
AKAGI, T., FUWA, K. & HARAGUCHI, H. (1985a) Simultaneous multi-
element determination of trace metals in seawater by inductively
coupled plasma atomic emission spectrometry after coprecipitation
with gallium. Anal. Chim. Acta, 177: 139-155.
AKAGI, T., NOJIMI, Y., MATSUI, M. & HARAGUCHI, H. (1985b)
Zirconium coprecipitation for simultaneous multi-element
determination of trace metals in seawater by inductively coupled
plasma atomic emission spectrometry. Appl. Spectroscop. 39(4):
662-667.
ALBERT, D.M., GONDER, J.R., PAPALE, J., CRAFT, J.L., DOHLMAN, H.G.,
REID, M.C. & SUNDERMAN, F.W. Jr (1980) Induction of ocular
neoplasms in Fischer rats by intraocular injection of nickel
subsulfide. In: Brown, S.S. & Sunderman, F.W. Jr, ed. Nickel
toxicology. Proceedings of the 2nd International Conference on
Nickel Toxicology, Swansea, 3-5 September, London, New York,
Academic Press, pp. 55-58.
ALBERT, D.M., GONDER, J.R., PAPALE, J., CRAFT, J.L., DOHLMAN, H.G.,
REID, M.C. & SUNDERMAN, F.W. Jr (1982) Induction of ocular
neoplasms in Fischer rats intraocular injection of nickel
subsulfide. Invest. Opthalmol. vis. Sci., 22(6): 768-782.
ALBRACHT, S.P.J., GRAF, E.G. & THAUER, R.K. (1982) The ERP
properties of nickel in hydrogenase from Methanobacterium
thermoautotrophicum. FEBS Lett., 140: 311-313.
ALEXANDER, A.J., GOGGIN, P.L. & COOKE, M. (1983) A Fourier-
transform infrared spectrometic study of the pyrosynthesis of
nickel tetracarbonyl and iron trentacarbonyl by combustion of
tobacco. Analyt. chim. Acta, 151: 1-12.
ALI, M., BISWAS, S.K., AKHTER, S. & KHAN, A.H. (1985) Multi-
element analysis of water residue: a PIXE measurement. Fresenius
Z. anal. Chem., 322(8): 755-760.
ALLOWAY, B.J. & MORGAN, H. (1986) The behaviour and availability of
cadmium, nickel, and lead in polluted soils. In: Assink, J.W. &
van den Brink, W.J., ed. Contaminated soil. Dordrecht, Boston,
Lancaster, Martinus Nifhoff Publishers, pp. 101-113.
AMACHER, D. & PAILLET, S. (1980) Induction of trifluorothymidine-
resistant mutants by metal ions in L5178Y/TK+/- cells. Mutat. Res.,
78: 279-288.
AMBROSE, A.M., LARSON P.S., BORZELLECA, J.R. & HENNIGA, G.R. Jr
(1976) Long-term toxicologic assessment of nickel in rats and dogs.
J. food Sci. Technol., 13: 181-187.
AMLACHER, E. & RUDOLPH, CH. (1981) The thymidine incorporation
inhibiting screening system (TSS) to test carcinogenic substances
(a nuclear DNA synthesis suppressive short-term test). Arch.
Geschwulstforsch., 51(7): 605-610.
ANDERSEN, A. (1988) Recent follow-up of respiratory cancer in a
Norwegian nickel refinery. In: Fourth International Conference on
Nickel Metabolism and Toxicology: Abstracts, Espoo, Finland, 5-9
September, 1988, Helsinki, Institute of Occupational Health, p. 49.
ANDERSEN, J.R., GAMMELGAARD, B. & REIMERT, S. (1986a) Direct
determination of nickel in human plasma by Zeeman-corrected atomic
absorption spectrometry. Analyst, 111(6): 721-722.
ANDERSEN, K.E., NIELSEN, G.D., FLYVHOLM, M.A., FREGERT, S. &
GRUVBERGE, B. (1983) Nickel in tap water. Contact Dermatit., 9(2):
140-143.
ANDERSEN, O. (1983) Effects of coal combustion products and metal
compounds on sister chromatid exchange (SCE) in a macrophage-like
cell line. Environ. Health Perspect., 47: 239-253.
ANDERSEN, O. (1985) Evaluation of the spindle-inhibiting effect of
Ni++ by quantitation of chromosomal super-contraction. Res. Commun.
chem. Pathol. Pharmacol., 50: 379-386.
ANDERSON, B.G. (1950) The apparent thresholds of toxicity to
Daphnia magna for chlorides of various metals when added to Lake
Erie water. Serv. works J., 16: 96-113.
ANDERSSON, K., ELINDER, C.G., HOGSTEDT, C., KJELLSTROM, T. &
SPANG, G. (1984) Mortality among cadmium and nickel-exposed workers
in a Swedish battery factory. Toxicol. environ. Chem., 9(1): 53-62.
ANKE, M. (1974) [The significance of micronutrients for animal
performance.] Akad. Landwirtschaftswiss. DDR Tagungsber., 132: 197-218
(in German).
ANKE, M., GRUN, M., DITTRICH, G., GROPPEL, B. & HENNIG, A. (1974)
Low nickel rations for growth and reproduction in pigs. In:
Hoekstra, W.G. ed. Trace element metabolism in animals,
Baltimore, Maryland, University Park Press, Vol. 2, pp. 715-718.
ANKE, M., GRUN, M. & PARTSCHEFELD, M. (1976) [Nickel, a new
essential trace element.] Arch. Tierernähr., 26: 740-741 (in German).
ANKE, M., HENNIG, A., GRUN, M., PARTSCHEFELD, M., GROPPEL, B. &
LUDKE, H. (1977) [Nickel, an essential trace element. 1st
communication: The supply of nickel as affecting the live weight
gains, food consumption and body composition of growing pigs and
goats.] Arch. Tierernähr., 27: 25-38 (in German).
ANKE, M., PARTSCHEFELD, M., GRUN, M. & GROPPEL, B. (1978) [Nickel
- an essential trace element. 3rd communication: The influence of
nickel content on the reproductive performance of female animals.]
Arch. Tierernähr., 28: 83-90 (in German).
ANKE, M., KRONEMANN, H., GROPPEL, B., HENNIG, A., MEISSNER, D. &
SCHNEIDER, H.-J. (1980) The influence of nickel deficiency on
growth, reproduction, longevity and different biochemical
parameters of goats. In: Anke, M. Schneider, H.-J. & Brûckner,
Chr., ed. [3. Trace Element Symposium: Nickel, Jena, 7-11 July,
1980,] Jena, Friedr.-Schiller Univ. pp. 3-10.
ANKE, M., GRUN, M., HOFFMANN, G., GROPPEL, B., GRUHN, K. & FAUST,
H. (1981) Zinc metabolism in ruminants suffering from nickel
deficiency. Mengen-Spurenelemente, 1: 189-196.
ANKE, M., GROPPEL, B., KRONEMANN, H. & GRUN, M. (1984) Nickel - an
essential element. In: Nickel in the human environment, Proceedings
of a Joint Symposium, Lyon, France, 8-11 March, 1983, Lyon,
International Agency for Research on Cancer, pp. 339-365 (IARC
Scientific Publications No. 53).
ARANYI, C., MILLER, F.J., ANDRES, S., EHRLICH, R., FENTERS, J.,
GARDNER, F.E. & WATERS, M.D. (1979) Cytotoxicity to alveolar
macrophages of trace metals adsorbed to fly fish. Environ. Res.,
20: 14-23.
ARDEN, G.P. & BYWATERS, E.G.L. (1978) Tissue reaction. In: Arden,
G.B. & Ansel, B.M., ed. Surgical management of juvenile chronic
polyarthritis. London, New York, Academic Press, pp. 263-275.
ARILLO, A., MARGIOCCO, C., MELODIA, F. & MENSI, P. (1982)
Biochemical effects of long-term exposure to Cr, Cd, Ni on rainbow
trout ( Salmo gairdneri Rich): influence of sex and season.
Chemosphere, 11:47-57.
ARLAUSKAS, A., BAKER, R., BONIN, A.M., TANDON, R.K., CRISP, P.T. &
ELLIS, J. (1985) Mutagenicity of metal ions in bacteria. Environ.
Res., 36(2): 379-388.
ARMIT, H.W. (1908) The toxicology of nickel carbonyl, Part II. J.
Hyg., 8: 565-600.
ASATO, N., SOESTBERGEN, M.V. & SUNDERMAN F.W. Jr (1975) Binding of
63Ni(II) to ultrafiltrable constituents of rabbit serum in vivo
and in vitro. Clin. Chem., 21(4): 521-527.
ASHRAF, M. & SYBERS, H.D. (1974) Lysis of pancreatic exocrine cells
and other lesions in rats fed nickel acetate. Am. J. Pathol., 74: 199.
BABICH, H. & STOTZKY, G. (1982a) Nickel toxicity to microbes:
effect of pH and implications for acid rain. Environ. Res., 29:
335-350.
BABICH, H. & STOTZKY, G. (1982b) Nickel toxicity to fungi:
Influence of some environmental factors. Ecotoxicol. environ. Saf.,
6: 577-589.
BABICH, H. & STOTZKY, G. (1983) Temperature, pH, salinity,
hardness, and particulates mediate nickel toxicity to eubacteria,
an actinomycete, and yeasts in lake, simulated estuarine, and sea
waters. Aquat. Toxicol., 3: 195-208.
BAGO-GRANELL, J., AGUIRRE-CANYADELL, M., NARDI, J. & TALLADA, N.
(1984) Malignant fibrous histiocytoma of bone at the site of a
total hip arthroplasty. J. bone joint Surg. B., 66: 38-40.
BALOGH, I., SOMOGYI, E., SOTONYI, P., POGATSA, G., RUBANYI, G. &
BELLUS, E. (1983) Electron-cytochemical detection of endogenous
nickel in the myocardium in acute carbon monoxide poisoning.
Applicability of a new cytochemical technique in forensic medicine.
Z. Rechtsmed., 90(1): 7-14.
BARBEAU, C., DUPUIS, M. & ROY, J.C. (1985) Metallic elements in
crude and milled chrysotile asbestos from Quebec. Environ. Res.,
38(2): 275-282.
BARNES, J.M. & DENZ (1951) The effects of 2,3-dimercapto-propanol
(BAL) on experimental nickel carbonyl poisoning. Br. J. ind. Med.,
8: 117-126.
BARRIE, L.A. (1981) Atmospheric nickel in Canada. In: Effects of
nickel in the Canadian environment, Ottawa, National Research
Council of Canada, pp. 55-76 (Publication No. 18568).
BASRUR, P.K. & GILMAN, J.P.W. (1967) Morphologic and synthetic
response of normal and tumor muscle cultures to nickel sulfide.
Cancer Res., 27: 1168-1177.
BAUDOUIN, M., F. & SCOPPA, P. (1974) Acute toxicity of various
metals to freshwater zooplankton. Bull. environ. Contam. Toxicol.,
121(6):745-751.
BAUMGARDT, B., JACKWERTH, E., OTTO, H. & TOLG, G. (1986)
Contribution to the trace analytical investigation of human lung
tissue. Fresenius Z. anal. Chem., 323(5): 481-486.
BAYER, O. (1939) [Toxicology, pathology and clinical aspects of
nickel carbonyl poisoning.] Arch. Gewerbepathol. Gewerbehyg., 9: 592-
606 (in German).
BECKER, P., DORSTELMANN, K., FROMBERGER, W. & FORTH, W. (1980) [On
the absorption of cobalt(II) and nickel(II) ions by isolated
intestinal segments in vitro of rats.] In: Anke, M., Schneider,
H.-J. & Brückner, Chr., ed. [3. Trace Element Symposium: Nickel,
Jena, German Democratic Republic, 7-11 July, 1980,] Jena,
Friedrich-Schiller University, pp. 79-85 (in German).
BEEFTINK, W.G. & NIEUWENHUIZE, J. (1982) Heavy-metal accumulation
in salt marshes from the Western and Eastern Scheldt. Sci. total
Environ., 25: 199-223.
BEIJER, K. & JERNELOV, A. (1986) Sources, transport and
transformation of metals in the environment. In: Friberg, L.,
Nordberg, G.F. & Vouk, V.B., ed. Handbook on the toxicology of
metals, Amsterdam, New York, Oxford, Elsevier Science Publishers,
Vol. I, pp. 68-84.
BENCKO, V., WAGNER, V., WAGNEROVA, M. & REICHRTOVA, E. (1983)
Immuno-biochemical findings in groups of individuals occupationally
and non-occupationally exposed to immissions containing nickel and
cobalt. J. Hyg. Epidemiol. Microbiol. Immunol. 27: 387-394
BENCKO, V., GEIST, T., ARBETOVA, D., DHARMADIKARI, D.M. &
SVANDOVA, E. (1986) Biological monitoring of environmental
pollution and human exposure to some trace elements. J. Hyg.
Epidemiol. Microbiol. Immunol., 30: 1-10.
BENNETT, B.G. (1984) Environmental nickel pathways to man. In:
Nickel in the human environment, Proceedings of a Joint Symposium,
Lyon, 8-11 March, 1983, Lyon, International Agency for Research on
Cancer, pp. 487-495 (IARC Scientific Publications No. 53).
BENSON, J.M., HENDERSON, R.F. & MACLELLAN, R.O. (1986) Comparative
toxicity of four nickel compounds to canine and rodent alocolar
macrophages in vitro. J. Toxicol. environ. Health, 19: 105-110.
BENSON, J.M., CARPENTER, R.L., HAHN, F.F., HALEY, P.J., HANSON,
R.L., HOBBS, C.H., PICKRELL, J.A. & DUNNICK, J.K. (1987)
Comparative inhalation toxicity of nickel subsulfide to F344/N rats
and B6C3F1 mice exposed for twelve days. Fundam. appl. Toxicol.,
9: 251-265.
BENSON, J.M., BURT, D.G., CARPENTER, R.L., EIDSON, A.F., HAHN,
F.F., HALEY, P.J., HANSON, R.L., HOBBS, C.H., PICKRELL, J.A. &
DUNNICK, J.K. (1988) Comparative inhalation toxicity of nickel
subsulfide to F344/N rats and B6C3F1 mice exposed for twelve days.
Fundam. appl. Toxicol., 10: 164-178.
BERGMAN, B., BERGMAN, M., MAGNUSSON, B., SOREMARK, R. & TODA, Y.,
(1980a) The distribution of nickel in mice. An autoradiographic
study. J. oral Rehabil., 7(4): 319-324.
BERGMAN, M., BERGMAN, B. & SOREMARK, R. (1980b) Tissue
accumulation of nickel released due to electrochemical corrosion of
non-precious dental casting alloys. J. oral Rehabil., 7(4): 325-330.
BERMAN, E. & REHNBERG, B. (1983) Fetotoxic effects of nickel in
drinking water in mice. Washington, DC, US Environmental
Protection Agency, 11 pp (EPA-600/1- 83-007).
BERNACKI, E.J., PARSONS, G.E., ROY, B.R., MIKAC-DEVIC, M., KENNEDY,
C.D. & SUNDERMAN, F.W. Jr (1978a) Urine nickel concentrations in
nickel-exposed workers. Ann. clin. lab. Sci., 8(3): 184-189.
BERNACKI, E.J., PARSONS, G.E. & SUNDERMAN, F.W. Jr (1978b)
Investigation of exposure to nickel and lung cancer mortality: case
control study at aircraft engine factory. Ann. clin. lab. Sci.,
8(3): 190-194.
BERNACKI, E.J., ZYGOWICZ, E. & SUNDERMAN, F.W. Jr (1980)
Fluctuations of nickel concentrations in urine of electroplating
workers. Ann. clin. lab. Sci., 10(1): 33-39.
BERROW, M.C. & BURRIDGE, J.C. (1981) Persistence of metals in
available form in sewage sludge-treated soils under field
conditions. In: Proceedings of International Conference on Heavy
Metals in the Environment, Amsterdam, Sept. 1981, Edinburgh, CEP
Consultants Ltd. pp. 202-205.
BERTRAND, G. & MACHEBOEUF, M. (1926a) Chimie biologique. Influence
du nickel et du cobalt sur l'action exercée par l'insuline chez le
lapin. C. R. Séances Acad. Sci., 182: 1504-1507.
BERTRAND, G. & MACHEBOEUF, M. (1926b) Chimie biologique. Influence
du nickel et du cobalt sur l'action exercée par l'insuline chez le
chien. C. R. Séances Acad. Sci., 183: 5-9.
BEVERIDGE, M.C., STAFFORD, E. & COUTTS, R. (1985) Metal
concentrations in the commercially exploited fishes of an endorheic
saline lake in the tin-silver province of Bolivia. Aquacult. Fish.
Manage., 16(1): 41-53.
BIEDERMANN, K. & LANDOLPH, J.R. (1986) Induction of anchorage
independence in human diploid foreskin fibroblasts by metal salts.
Carcinogenesis, 542: 137.
BIESINGER, K.E. & CHRISTENSEN, G.M. (1972) Effects of various
metals on survival, growth, reproduction, and metabolism of Daphnia
magna. J. Fish Res. Board. Can., 29(12): 1691-1700.
BIGGART, N.W. & MURPHY, E.C. Jr (1988) Analysis of metal-induced
mutations altering the expression or structure of a retroviral gene
in a mammalian cell line. Mutat. Res., 198: 115-129.
BINGHAM, E.W., BARKLEY, W., ZERWAS, M., STEMMER, K. & TAYLOR, P.,
(1972) Responses of alveolar macrophages to metals. I. Inhalation
of lead and nickel. Arch. environ. Health, 25: 406-414.
BIRGE, W.J. (1978) Embryo-larval bioassays on inorganic coal
elements and in situ Biomonitoring of coal-waste effluents. In:
Surface mining and fish/wildlife needs in the eastern USA.
Proceedings of a Symposium, December 1978. Washington, DC, US Fish
and Wildlife Service, pp. 97-108 (9098.910 FWS/ OBS-78/81).
BIRGE, W.J. & BLACK, J.A. (1980) Aquatic toxicology of nickel. In:
Nriagu, J.O., ed. Nickel in the environment, New York, Chichester,
Brisbane, Toronto. John Wiley and Sons, pp. 349-406.
BLAYLOCK, B.G. & FRANK, M.L. (1979) A comparison of the toxicity of
nickel to the developing eggs and larvae of carp ( Cyprinus carpio).
Bull. environ. Contam. Toxicol., 21:604-611.
BLANKENSTEIN, K. & STARCK, H.C. (1979) [Nickel compounds.] In:
Bartholomé, E., Biekert, E., Hellmann, H., Ley, H., Weigert, W. &
Weise, E., ed. [Ullmanns encyclopaedia of technical chemistry,]
Weinheim, New York, Verlag Chemie, pp. 293-302 (in German).
BLOCK, G.T. & YEUNG, M. (1982) Asthma induced by nickel. J. Am.
Med. Assoc., 247(11): 1600-1602.
BOLDT, J.R. & QUENEAU, P. (1967) The winning of nickel, its
geology, mining, and extractive metallurgy, London, Methuen & Co.
Ltd., 465 pp.
BOND, A.M. & WALLACE, G.G. (1983) Automated determination of nickel
and copper by liquid chromatography with electrochemical and
spectrophotometric detection. Anal. Chem., 55: 718-723.
BOND, A.M., KNIGHT, R.W., REUST, J.J.B., TUCKER, D.J. & WALLACE,
G.G. (1986) Determination of metals in urine by direct injection of
sample, high-performance liquid chromatography and electrochemical
or spectrophotometric detection. Anal. chim. Acta, 182: 47-59.
BOUTAYRE, P., PIZZORNI, L., LIQUIER, J., TABOURY, J. &
TAILLANDIER, E. (1984) Z-form induction in DNA by carcinogenic
nickel compounds: an optical spectroscopy study. In: Nickel in the
human environment. Proceedings of a Joint Symposium, Lyon, 8-11
March 1983, Lyon, International Agency for Research on Cancer, pp.
227-234 (IARC Scientific Publications No. 53).
BOYER, K.W. & HOWITZ, W. (1986) Special considerations in trace
element analysis of food and biological materials. In:
Environmental carcinogens - selected methods of analysis, Vol. 8:
Some metals: As, Be, Cd, Cr, Ni, Pb, Se, Zn, Lyon, International
Agency for Research on Cancer, pp. 191-220 (IARC Scientific
Publications No. 71).
BOYLE, R.W. (1981) Geochemistry of nickel. In: Effects of nickel in
the Canadian environment, Ottawa, National Research Council of
Canada, pp. 31-44 (Publication No. NRCC 18568).
BOYSEN, M., WAKSVIK, H., SOLBERG, L.A., REITH, A. & HOGETVEIT,
A.C. (1980) Histopathological follow-up studies and chromosome
analysis in nickel workers. In: Brown, S.S. & Sunderman, F.W. Jr,
ed., Nickel Toxicology. Proceedings of the 2nd International
Conference on Nickel Toxicology, Swansea, 3-5 September 1980,
London, New York, Academic Press, pp. 35-38.
BOYSEN, M., SOLBERG, L.A., ANDERSEN, I., HOGETVEIT, A.C. &
TORJUSSEN, W. (1982) Nasal histology and nickel concentration in
plasma and urine after improvements in the work environment at a
nickel refinery in Norway. Scand. J. Work Environ. Health, 8(4):
283-290.
BOYSEN, M., SOLBERG, L.A., TORJUSSEN, W., POPPE, S. & HOGETVEIT,
A.C. (1984) Histological changes, rhinoscopical findings and nickel
concentration in plasma and urine in retired nickel workers. Acta
otolaryngol., 97(1-2): 105-115.
BRADLEY, R.W. & MORRIS, J.R. (1986) Heavy metals in fish from a
series of metal-contaminated lakes near Sudbury, Ontario. Water Air
Soil Pollut., 27(3-4): 341-354.
BRAJTER, K. & SLONAWSKA, K. (1986) The efficiency of Cellex-P for
the preconcentration of lead and other trace metals from waters.
Anal. Chim. Acta, 185: 271-277.
BRANDES, W.W. (1934) Nickel carbonyl poisoning. J. Am. Med. Assoc.,
102: 1204-1206.
BRINGMANN, G., KUHN, R. & WINTER, A. (1980) [Determination of the
biological effect of water pollutants on protozoa. 3. Saprozoic
flagellates.] Z. Wasser Abwasser Forsch., 13(5):170-173 (in German
with English summary).
BRKOVIC-POPOVIC, L. & POPOVIC, M. (1977) Effects of heavy metals on
survival and respiration rate of tubificid worms: Part 1. Effects
on survival. Environ. Pollut., 13: 65-72.
BROOKS, R.R. (1980) Accumulation of nickel by terrestrial plants.
In: Nriagu, J.O., ed. Nickel in the environment, New York,
Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 407-429.
BROWN, K.W., THOMAS, J.C. & SLOWEY, J.F. (1983a) The movement of
metals applied to soils in sewage effluent. Water air soil Pollut.,
19(1): 43-54.
BROWN, K.W., THOMAS, J.C. & SLOWEY, J.F. (1983b) Metal
accumulation by Bermuda grass grown on four diverse soils amended
with secondarily treated sewage effluent. Water air soil Pollut.,
20(4): 431-446.
BROWN, V.M. (1968) The calculation of the acute toxicity of
mixtures of poisons to rainbow trout. Water Res., 2: 723-733.
BRULAND, K.W., FRANKS, R.P., KNAUER, G.A. & MARTIN, J.H. (1979)
Sampling and analytical methods for the determination of copper,
cadmium, zinc, and nickel at the nanogram per liter level in
seawater. Anal. chim. Acta, 105: 233-245.
BRUNE, D. (1986) Metal release from dental biomaterials.
Biomaterials, 7(3): 163-175.
BRYANT, V., NEWBERY, D.M., MCCLUSKY, D.S. & CAMPBELL, R. (1985)
Effect of temperature and salinity on the toxicity of nickel and
zinc to two estuarine invertebrates ( Corophium volutator, Macoma
balthica). Mar. Ecol. Prog. Ser., 24(1-2): 139-153.
BUHLER, E.V. (1965) Delayed contact hypersensitivity in the guinea-
pig. Arch. Dermatol., 91: 171-175.
BURBA, P. & WILLMER, P.G. (1985) Analytical multielement
preconcentration on metal hydroxide-coated cellulose. Fresenius Z.
anal. Chem. 321(2): 109-118.
BURCH, J.D., HOWE, G.R., MILLER, A.B. & SEMENCIW, R. (1981)
Tobacco, alcohol, asbestos, and nickel in the etiolgy of cancer of
the larynx: a case-control study. J. Natl Cancer Inst., 67(6): 1219-
1224.
BURGES, D. (1980) Mortality study of nickel platers. In: Brown,
S.S. & Sunderman, F.J. Jr, ed., Nickel toxicology. Proceedings of
the 2nd International Conference on Nickel Toxicology, Swansea, 3-5
September, 1980, London, New York, Academic Press, pp. 15-18.
BURROWS, D., CRESWELL, S. & MERRETT, J.D. (1981) Nickel, hands and
hip prostheses. Br. J. Dermatol., 105(4): 437-443.
BUSELMAIER, W., ROHRBORN, G. & PROPPING, P. (1972) [Mutagenicity
investigations with pesticides in the host-mediated assay and the
dominant lethal test in mice.] Biol. Zbl., 91: 311-325 (in German).
BUTZ, I. (1984) [Effect of dilution water on the acute toxicity of
nickel sulfate to rainbow trout.] Wasser Abwasser, 28: 41-56
(in German).
BYRNE, C.J. & DELEON, I.R. (1986) Trace metal residues in biota and
sediments from Lake Pontchartrain, Lousiana, USA. Bull. environ.
Contam. Toxicol., 37(1): 151-158.
CALAMARI, D., GAGGINO, G.F. & PACCHETTI, G. (1982) Toxokinetics of
low levels of Cd, Cr, Ni and their mixture in long-term treatment
of Salmo gairdneri Rich. Chemosphere, 11(1): 59-70.
CALAMARI, D., LLOYD, R., SOLBE, J.F. & SOLBE, L.G. (1984) Water
quality criteria for European freshwater fish: Report on nickel and
freshwater fish. Rome, Food and Agriculture Organization of the
United Nations 20 pp (European Inland Fisheries Advisory Commission
(EIFAC) Technical Paper 45).
CALLAN, W.M. & SUNDERMAN, F.W. Jr (1973) Species variations in
binding of 63NiCl2 by serum albumin. Res. Commun. chem. Pathol.
Pharmacol., 5: 459-472.
CALNAN, C.D. (1956) Nickel dermatitis. Br. J. Dermatol., 68: 229-236.
CAMNER, P., JOHANSSON, A. & LUNDBERG, M. (1978) Alveolar
macrophages in rabbits exposed to nickel dust. Environ. Res., 16:
226-235.
CARLSON, H.E. (1984) Inhibition of prolactin and growth hormone
secretion by nickel. Life Sci., 35(17): 1747-1754.
CARVAJAL, N.J. & ZIENIUS, R.H. (1986) Gas chromatographic analysis
of trace metals isolated from aqueous solutions as
diethyldithiocarbamates. J. Chromatogr., 355: 107-116.
CARVALHO, S.M. & ZIEMER, P.L. (1982) Distribution and clearance of
63Ni administered as 63NiCl2 in the rat: intratracheal study.
Arch. environ. Contam. Toxicol., 11: 245-248.
CASARETT-BRUCE, M., CAMNER, P. & CURSTEDT, T. (1981) Changes in
pulmonary lipid composition of rabbits exposed to nickel dust.
Environ. Res., 26(2): 353-362.
CASEY, C.E. & ROBINSON, M.F. (1978) Copper, manganese, zinc,
nickel, cadmium and lead in human foetal tissues. Brit. J. Nutr.,
39: 639-646.
CASS, G.R. & MCRAE, G.J. (1983) Source-receptor reconciliation of
routine air monitoring data for trace metals: An emission
inventory-assisted approach. Environ. Sci. Technol., 17(3): 129-139.
CASTLEMAN, B. & MCNEELY, B.U. (1965) Case records of the
Massachusetts General Hospital, Case 38-1965. New Engl. J. Med.,
273: 494-504.
CASTRANOVA, V. BOWMAN, L., REASOR, M.J. & MILES, P.R. (1980)
Effects of heavy metal ions on selected oxidative metabolic
processes in rat alveolar macrophages. Toxicol. appl. Pharmacol.,
53(1): 14-23.
CATALANATTO, F.A., SUNDERMAN, F.W. Jr, & MCINTOSH, T.R. (1977)
Nickel concentrations in human parotid saliva. Ann. clin. lab.
Sci., 7(2): 146-151.
CHAN, W.H. & LUSIS, M.A. (1986) Smelting operations and trace
metals in air and precipitation in the Sudbury Basin. Adv.
environ. Sci. Technol., 17: 113-143.
CHANG, A.C., PAGE, A.L. & BINGHAM, F.T. (1982) Heavy metal
absorption by winter wheat following termination of cropland sludge
applications. J. environ. Qual., 11(4): 705-708.
CHANG, A.C., WARNECKE, J.E., LUND, L.J. & PAGE, A.L. (1984)
Accumulation of heavy metals in sewage sludge-related soils. J.
environ. Qual., 13(1): 87-91.
CHANG, C.C., TATUM, H.J. & KINCL, F.A. (1970) The effect of
intrauterine copper and other metals on implantation in rats and
hamsters. Fertil. Steril., 21: 274-278.
CHAUSMER, A.B. (1976) Measurement of exchangeable nickel in the
rat. Nutr. Rep. Int., 14: 323-326.
CHEN, J.R., FRANCISCO, R.B. & MILLER, T.E. (1977) Légionnaires'
disease: nickel levels. Science, 196: 906-908.
CHEN, K.Y., YOUNG, C.S., JAN, T.K. & ROHATGI, N. (1974) Trace
metals in wastewater effluents. J. Water Pollut. Fed., 46(12): 2663-2675.
CHIAUDANI, G. & VIGHI, M. (1978) The use of Selenastrum
capricornutum batch cultures in toxicity studies. Mitt. Int. Ver.
Limnol., 21: 316- 329.
CHORVATOVICOVA, D. (1983) The effect of nickel chloride on the
level of chromosome aberrations in Chinese hamster Cricetulus
griseus. Biologica, 38: 1107-1112.
CHOVIL, A., SUTHERLAND, R.B. & HALLIDAY, M. (1981) Respiratory
cancer in a cohort of nickel sinter plant workers. Br. J. ind.
Med., 38(4): 327-333.
CHRISTENSEN, J.M. & PEDERSEN, L.M. (1986) Enzymatic digestion of
whole blood for improved determination of cadmium, nickel and
chromium by electrothermal atomic absorption spectrophotometry:
measurements in patients with rheumatoid arthritis and in normal
humans. Acta pharmacol. toxicol., 59(7): 399-402.
CHRISTENSEN, O.B & KRISTENSEN, M. (1982) Treatment with disulfiram
in chronic nickel hand dermatitis. Contact Dermatit., 8(1): 59-63.
CHRISTENSEN, O.B. & MOELLER, H. (1975a) Nickel allergy and hand
eczema. Contact Dermatit., 1(3): 129-135.
CHRISTENSEN, O.B. & MOELLER, H. (1975b) External and internal
exposure to the antigen in the hand eczema of nickel allergy.
Contact dermatit., 1(3): 136-141.
CHRISTENSEN, O.B., MOELLER, H., ANDRASKO, K. & LAGESSON, V.,
(1979) Nickel concentration of blood, urine and sweat after oral
administration. Contact Dermatit., 5: 312-316.
CHRISTIE, N.T. & TUMMOLO, D. (1988) The role of Ni(II) in
mutation. In: Fourth International Conference on Nickel Metabolism
and Toxicology: Abstracts, Espoo, 5-9 September 1988, Helsinki,
Institute of Occupational Health, p. 33.
CHRISTIE, N.T., TUMMOLO, D.M., BIGGART, N.W. & MURPHY, E.C. Jr
(1988) Chromosomal changes in cell lines from mouse tumors induced
by nickel sulfide and methylcholanthre. Cell Biol. Toxicol., 4: 427.
CHURCH, S.E. (1981) Multi-element analysis of fifty-four
geochemical reference samples using inductively coupled plasma
atomic-emission spectrometry. Geostand. Newsl., V: 133-160.
CICCARELLI, R.B. & WETTERHAHN, K.E. (1982) Nickel distribution and
DNA lesions induced in rat tissues by the carcinogen nickel
carbonate. Cancer Res., 42: 3544-3549.
CICCARELLI, R.B. & WETTERHAHN, K.E. (1985) In vitro interaction of
63nickel(II) with chromatin and DNA from rat kidney and liver
nuclei. Chem.-biol. Interact., 52(3): 347-360.
CICCARELLI, R.B., HAMPTON, T.H. & JENNETTE, K.W. (1981) Nickel
carbonate induces DNA-protein crosslinks and DNA strand breaks in
rat kidney. Cancer Lett., 12(4): 349-354.
CIRLA, A.M., BERNABEO, F., OTTOBONI, F. & RATTI, R. (1985) Nickel-
induced occupational asthma - immunological and clinical aspects.
In: Brown, S.S. & Sunderman, F.W. Jr, ed. Progress in Nickel
toxicology. Proceedings of the 3rd International Congress on Nickel
Metabolism and Toxicology, Paris, 4-7 September, 1984, Oxford,
Blackwell Scientific Publications, pp. 165-168.
CLARK, J.R., VAN HASSEL, J.H., NICHOLSEN, R.B., CHERRY, D.S. &
CAIRNS, J. (1981) Accumulation and depuration of metals by duckweed
(Lemna perpusilla). Ecotoxicol. environ. Safety, 5: 87-96.
CLARY, J.J. (1975) Nickel chloride-induced metabolic changes in the
rat and guinea-pig. Toxicol. appl. Pharmacol., 31: 55-65.
CLARY, J.J. & VIGNATI, I. (1973) Nickel chloride-induced changes in
metabolism in the rat. Toxicol. appl. Pharmacol., 25: 467-468.
CLEMENTE, G.F., CIGNA ROSSI, L. & SANTARONI, G.P. (1980) Nickel in
foods and dietary intake of nickel. In: Nriagu, J.O., ed. Nickel in
the environment, New York, Chichester, Brisbane, Toronto, John
Wiley and Sons, pp. 493-498.
CLEMMENSEN, O.J., MENNE, T., KAABER, K. & SOLGAARD, P. (1981)
Exposure of nickel and the relevance of nickel sensitivity among
hospital cleaners. Contact Dermatit., 7(1): 14-18.
CLEMONS, G.K. & GARCIA, J.F. (1981) Neuroendocrine effects of acute
nickel chloride administration in rats. Toxicol. appl. Pharmacol.,
61: 343-348.
CLOUTIER, N.R., CLULOW, F.V., LIM, T.P. & DAVE, N.K. (1986) Metal
(copper, iron, cobalt, zinc, lead) and radium-226 levels in tissues
of meadow voles Microtus pennsylvanicus living on nickel and
uranium mine tailings in Ontario, Canada: Site, sex age and season
effects with calculation of average skeletal radiation dose.
Environ. Pollut., A41(4): 295-314.
COENEN, W., GROTHE, I. & KUHNEN, G. (1986) Exposure to welding
fumes in the workplace with regard to nickel and chromates. Int.
Congr. Ser. Excerpta Med., 676: 149-152.
COKER, E.G. & MATTHEWS, P.J. (1983) Metals in sewage sludge and
their potential effects in agriculture. Water Sci. Technol., 15:
209-217.
CONWAY, K. & COSTA, M. (1989) The involvement of hereto-chromatic
damage in nickel-induced transformation. In: Costa, M. &
Rossmann, T.G., ed., Proceedings of the First International Meeting
on the Molecular Mechanisms of Metal Toxicity and Carcinogenicity,
Urbino, Italy, September 1988. Clifton, New Jersey, Humana Press
Inc., pp.437-444 (Biological Trace Element Research, Vol. 21).
CORBETT, T.H., HEIDELBERGER, C. & DOVE, W.F. (1970) Determination
of the mutagenic activity to bacteriophage T4 of carcinogenic and
noncarcinogenic compounds. Mol. Pharmacol., 6: 667-679.
CORNELL, R.G. (1984) Mortality patterns among stainless-steel
workers. In: Nickel in the human environment, Proceedings of a
Joint Symposium, Lyon, 8-11 March, 1983, Lyon, International Agency
for Research on Cancer, pp. 65-71 (IARC Scientific Publications No.
53).
CORNELL, R.G. & LANDIS, J.R. (1984) Mortality patterns among
nickel/chromium alloy foundry workers. In: Nickel in the human
environment, Proceedings of a Joint Symposium, Lyon, 8-11 March,
1983, Lyon, International Agency for Research on Cancer, pp. 87-93
(IARC Scientific Publications No. 53).
CORRICK, J.D. (1977) Mineral commodity profiles: Nickel,
Washington, DC, US Department of the Interior, Bureau of Mines, 19
pp. (MCP-4).
COSTA, M. & MOLLENHAUER, H.H. (1980a) Carcinogenic activity of
particulate nickel compounds is proportional to their cellular
uptake. Science, 209(4455): 515-517.
COSTA, M. & MOLLENHAUER, H.H. (1980b) Phagocytosis of nickel
subsulfide particles during the early stage of neoplastic
transformation in tissue culture. Cancer Res., 40: 2688-2694.
COSTA, M., NYE, J.S., SUNDERMAN, F.W. Jr, ALLPASS, P.R. & GONDOS,
B. (1979) Induction of sarcomas in nude mice by implantation of
Syrian hamster fetal cells exposed in vitro to nickel subsulfide.
Cancer Res., 39: 3591-3597.
COSTA, M., JONES, M.K. & LINDBERG, O. (1980) Metal carcinogenesis
in tissue culture. In: Martell, A.E., ed. Inorganic chemistry in
biology and medicine. Washington, DC, pp. 45-73 (ACS Symp. Series
140).
COSTA, M., ABBRACCHIO, M.P. & SIMMONS-HANSEN, J. (1981a) Factors
influencing the phagocytosis, neoplastic transformation, and
cytoxicity of particulate nickel compounds in tissue culture
systems. Toxicol. appl. Pharmacol., 60: 313-313.
COSTA, M., SIMMONS-HANSEN, J., BEDROSSIAN, C.W., BONURA, J. &
CAPRIOLI, R.M. (1981b) Phagocytosis, cellular distribution, and
carcinogenic activity of particulate nickel compounds in tissue
culture. Cancer Res., 41(7): 2868-2876.
COSTA, M., HECK, J.D. & ROBINSON, S.H. (1982) Selective
phagocytosis of crystalline nickel sulfide particles and DNA
strandbreaks as a mechanism for the induction of cellular
transformation. Cancer Res., 42: 2757-2763.
COWGILL, U.M. (1976) The chemical composition of two species of
Daphnia, their algal food and their environment. Sci. total
Environ., 6: 79-102.
COX, J.E., DOLL, R., SCOTT, W.A. & SMITH, S. (1981) Mortality of
nickel workers: experience of men working with metallic nickel. Br.
J. ind. Med., 38(3): 235- 239.
CRAGLE, D.L., HOLLIS, D.R., NEWPORT, T.H. & SHY, C.M. (1984) A
retrospective cohort mortality study among workers occupationally
exposed to metallic nickel powder at the Oak-Ridge Tennessee USA
gaseous diffusion plant. In: Nickel in the human environment,
Proceedings of a Joint Symposium, Lyon, 8-11 March, 1983, Lyon,
International Agency for Research on Cancer, pp. 57-64 (IARC
Scientific Publications No. 53).
CRONIN, E. (1980) Contact dermatitis. Edinburgh, London, New York,
Churchill Livingstone, pp. 279-390.
CRONIN, E., DI MICHIEL, A.D. & BROWN, S.S. (1980) Oral challenge
in nickel-sensitive women with hand eczema. In: Brown, S.S. &
Sunderman, F.W. Jr, ed. Nickel toxicology. Proceedings of the 2nd
International Conference on Nickel Toxicology, Swansea, 3-5
September, 1980, London, New York, Academic Press, pp. 149-152.
CROSSMANN, G. (1988) [The cycle in the system soil-plant-animal on
locations with extremely high soil contaminations by cadmium and
nickel caused by sewage-sludge.] Berlin, Federal Agency of
Environmental Protection, 95 pp (Research report No. 106 07 043)
(in German).
CUCKLE, H., DOLL, R. & MORGAN, L.G. (1980) Mortality study of men
working with soluble nickel compounds. In: Brown, S.S. & Sunderman,
F.W. Jr, ed. Nickel toxicology. Proceedings of the 2nd
International Conference on Nickel Toxicology, Swansea, 3-5
September, 1980, London, New York, Academic Press, pp. 11-14.
CURSTEDT, T., HAGMAN, M., ROBERTSON, B. & CAMNER, P. (1983) Rabbit
lungs after long-term exposure to low nickel dust concentration.
Environ. Res., 30: 89-94.
CURSTEDT, T., CASARETT-BRUCE, M. & CANNER, P. (1984) Changes in
glycerophosphatides and their ether analogs in lung lavage of
rabbits exposed to nickel dust. Exp. mol. Pathol., 41: 26-34
CUSTER, T.W., FRANSON, J.C., MOORE, J.F. & MYERS, J.E. (1986)
Reproductive success and heavy metal contamination in Rhode Island
common terns. Environ. Pollut., A41(1): 33-52.
DALDRUP, T., HAARHOFF, K. & SZATHMARY, S.C. (1983) [Fatal nickel
sulfate poisoning.] Beitr. gerichtl. Med., 41: 141-144 (in German).
DALLINGER, R. & KAUTZKY, H. (1985) The importance of contaminated
food for the uptake of heavy metals by rainbow trout ( Salmo
gairdneri): A field study. Oecologia, 67(1): 82-89.
D'ALONZO, C.A. & PELL, S. (1963) A study of trace metals in
myocardial infarction. Arch. environ. Health, 6: 381-385.
DAMJANOV, I., SUNDERMAN, F.W. Jr, MITCHELL, J.M. & ALLPASS, P.R.
(1978) The induction of testicular sarcomas in Fischer rats by
intratesticular injection of nickel subsulfide. Cancer Res., 38:
268-276.
DANIELSSON, L.-G., MAGNUSSON, B. & WESTERLUND, S. (1978) An
improved metal extraction procedure for the determination of trace
metals in seawater by atomic absorption spectrometry with
electrothermal atomization. Anal. Chim. Acta, 98: 47-57.
DAVYDOVA, V.I., NEIZVESTNOVA, E.M., BLOKHIN, V.A. & SERGOVA, N.V.
(1981) Toxicological evaluation of manganese, chromium and nickel.
Gig. i Sanit., 7: 20-22.
DECATANZARO, J.B. & HUTCHINSON, T.C. (1985a) Effects of nickel
addition on nitrogen mineralization, nitrification, and nitrogen
leaching in some boreal forest soils. Water air soil Pollut., 24(2):
153-164.
DECATANZARO, J.B. & HUTCHINSON, T.C. (1985b) Leaching and
distribution of nitrogen and nickel in nickel-perturbed jack pine
forest microcosms. Water air soil Pollut., 26(3): 281-292.
DECHENG, C., MING, J., LING, H., SHAN, W., ZIQING, X. & XINSHUI,
Z. (1987) Cytogenic analysis in workers occupationally exposed to
nickel carbonyl. Mutat. Res., 188: 149-152.
DECSY, M.I. & SUNDERMAN, F.W. Jr. (1974) Binding of 63Ni to rabbit
serum alpha-macroglobulin in vivo and in vitro. Bioinorg. Chem.
3(2): 95-105.
DEFLORA, S., ZANACCI, P., CAMOIRANO, A., BENICELLI, C. & BADOLATI,
G.S. (1984) Genotoxic activity and potency of 135 compounds in the
Ames reversion test and in a bacterial DNA-repair test. Mutat.
Res., 133: 161-198.
DEKNUDT, G. & LEONARD, A. (1982) Mutagenicity tests with nickel
salts in the male mouse. Toxicology, 25(4): 289-292.
DELGADO, E.A. (1958) Sarcoma following a surgically treated
fractured tibia. Clin. Orthop., 12: 315-318.
DENG, C. & QU, B. (1981) The cytogenetic effects of nickel
sulphate. Acta genet. sin., 8: 212-215.
DENG, C., QU, B., HUANG, J., ZHUO, Z.L., XIAN, H., YAO, M., CHEN,
M.Y., LI, Z.X., SHENG, S.Y. & YEI, Z.F. (1983) [Cytogenetic
effects of electroplating workers.] Acta scientiae circumstantiae,
3: 267-271 (in Chinese with English abstract).
DENG, C., LEE, H.H., XIAN, H., YAO, M., HUANG, J. & QU, B. (1988)
Chromosomal aberrations and sister chromatid exchanges of
peripheral blood lymphocytes in Chinese electroplating workers. J.
trace Elem. exp. Med., 1: 57-62.
DERMOTT, R.M. & LUM, K.R. (1986) Metal concentrations in the annual
shell layers of the bivalve Elliptio complanata. Environ. Pollut.
B12(2): 131-144.
DEUTMAN, R., MULDER, T.J., BRIAN, R. & NATER, J.P. (1977) Metal
sensitivity before and after total hip arthroplasty. J. bone joint
Surg., 59(7): 862-865.
DEWLING, R., MANGANELLI, R. & BAER, G. (1980) Fate and behaviour
of selected heavy metals in incinerated sludge. J. Water Pollut.
Control Fed., 52: 2552-2557.
DIEKERT, G. & RITTER, M. (1982) Nickel requirement of
Acetobacterium woodii. J. Bacteriol. 151: 1043-1045.
DIEKERT, G. & THAUER, R.K. (1980) The effect of nickel on carbon
monoxide dehydrogenase formation in Clostridium thermoaceticum and
Clostridium formicoaceticum. FEMS microbiol. Lett., 7: 187-189.
DIETZ, R.N. & WIESER, R.F. (1983) Sulfate formation in oil-fired
power plant plumes. Vol. 1: Parameters affecting primary sulfate
emissions and a model for predicting emissions and plume opacity,
Upton, NY, Brookhaven National Laboratories (Report No. EA-3231).
DIPAOLO, J.A. & CASTO, B.C. (1979) Quantitative studies of in
vitro morphological transformation of Syrian golden hamster
cells by inorganic metal salts. Cancer Res., 39: 1008-1013.
DITORO, D.M., MAHONY, J.D., KRICHGRABER, P.R., O'BYRNE, A.L. &
PASQUALE, L.R. (1986) Effect of nonreversibility, particle
concentration and ionic strength on heavy metal sorption. Environ.
Sci. Technol., 20(1): 55-61.
DIXON, N.E., GAZZOLA, C., BEAKELEY, R.L. & ZERNER, B. (1975) Jack
bean urease (EC 3.5.1.5). A metalloenzyme. A simple biological role
for nickel? J. Am. Chem. Soc., 97: 4130-4133.
DJACHENKO, O.Z (in press) Effects of nickel and manganese on the
chromosome aberrations and sister chromatid exchanges in human
lymphocytes in vitro. In: Domnin, S.G. & Shcherbakov, S.V. ed.
Problems of labour hygiene in steel and coloured metals industry,
Moscow, Erisman Institute of Hygiene.
DOLL, R. (1958) Cancer of the lung and nose in nickel workers. Br.
J. ind. Med., 15: 217-223.
DOLL, R., MORGAN, L.G. & SPEIZER, F.E. (1970) Cancers of the lung
and nasal sinuses in nickel workers. Br. J. Cancer, 24(4): 623-632.
DOLL, R., MATTHEWS, J.D. & MORGAN, L.G. (1977) Cancers of the lung
and nasal sinuses in nickel workers: A reassessment of the period
of risk. Br. J. ind. Med.,34(2): 102-105.
DOLOVICH, J., EVANS, S.L. & NIEBOER, E. (1984) Occupational asthma
from nickel sensitivity: I. Humam serum albumin in the antigenic
determinant. Br. J. ind. Med., 41(1): 51-55.
DOOMS-GOOSSENS, A., CEUTERICK, A., VANMAELE, N. & DEGREEF, H.
(1980) Follow-up study of patients with contact dermatitis caused
by chromates, nickel and cobalt. Dermatologica, 160(4): 249-260.
DORMER, R.L., KERBEY, A.L., MCPHERSON, M., MANLEY, S., ASHCROFT,
S.J.H., SCHOFIELD, J.G. & RANDLE, P.J. (1973) The effect of nickel
on secretory systems. Studies on the release of amylase, insulin
and growth hormone. Biochem. J., 140: 135-142.
DORNEMANN, A. & KLEIST, H. (1980) Nickel in liver and kidney
samples. In: Brown, S.S. & Sunderman, F.W. Jr. ed. Nickel
toxicology, Proceedings of the 2nd International Conference on
Nickel Toxicology, Swansea, 3-5 September, 1980, London, New York,
Academic Press, pp. 175-178.
DOSTAL, L.A., HOPFER, S.M., LIN, S.M. & SUNDERMAN, F.W. Jr (1989)
Effects of nickel chloride on lactating rats and their suckling
poups, and the transfer of nickel through rat milk. Toxicol. appl.
Pharmacol.,101: 220-231.
DRAKE, H.L. (1982) Occurrence of nickel in carbon monoxide
dehydrogenase from Clostridium thermoaceticum. J. Bacteriol. 149:
561-566.
DRINKER, K.R., FIARHALL, J.T., RAY, G.B. & DRINKER, C.K. (1924)
The hygienic significance of nickel. J. ind. Hyg., 6: 307-360.
DUBE, V.E. & FISHER, D.E. (1972) Hemangioendothelioma of the leg
following metallic fixation of the tibia. Cancer, 30: 1260-1266.
DUBINS, J.S. & LAVELLE, J.M. (1986) Nickel(II), genotoxicity:
potentiation of mutagenesis of simple alkylating agents. Mutat.
Res., 162(2): 187-199.
DUBREUIL, A., BOULEY, G., DURET, S., MESTRE, J.C. & BOUDENE, C.
(1984) In vitro cytotoxicity of nickel chloride on a human
pulmonary epithelial cell line. Arch. Toxicol., Suppl. 7: 391-393.
DUKE, J.M. (1980) Nickel in rocks and ores. In: Nriagu, J.O. ed.
Nickel in the environment, New York, Chichester, Brisbane, Toronto,
John Wiley and Sons, pp. 27-50.
DUNNICK, J.K., BENSON, J.M., HOBB, C.H., HAHN, F.F., CHENG, Y.S. &
EIDSON, A.F. (1988) Comparative toxicity of nickel oxide, nickel
sulfate hexahydrate, and nickel subsulfide after 12 days of
inhalation exposure to F344/N rats and B6C3F1 mice. Toxicology, 50:
145-156.
DUYEVA, L.A. (1983) [Allergenicity of metals.] In: [Metals.
Hygienic aspects of environmental evaluation and improvement].
Moscow, Meditsina, pp. 28-41 (in Russian).
EDMAN, B. & MOELLER, H. (1982) Trends and forecasts for standard
allergens in a 12-year patch test material. Contact Dermatit., 8(2):
95-104.
EGEDAHL, R. & RICE, E. (1984) Cancer incidence at a
hydrometallurgical nickel refinery. In: Nickel in the human
environment, Proceedings of a Joint Symposium, Lyon, 8-11 March,
1983, Lyon, International Agency for Research on Cancer, pp. 47-56
(IARC Scientific Publications No. 53).
EGILSSON, V., EVANS, I.H. & WILKIE, D. (1979) Toxic and mutagenic
effects of carcinogens on the mitochondria of Saccharomyces
cerevisiae. Molec. gen. Genet., 174: 39-46.
EILERTSEN, E., SKAUG, V. & NORSETH, T. (1988) Tumor induction in
rats after intrapleural instillation of various nickel oxides. In:
Fourth International Conference on Nickel Metabolism and
Toxicology, Abstracts, Espoo, Finland, 5-9 September 1988.
Helsinki, Institute of Occupational Health, p. 43.
EKER, P. & SANNER, T. (1983) Assay for initiators and promoters of
carcinogenesis based on attachment-independent survival of cells in
aggregates. Cancer Res., 43: 320-323.
ELAKHOVSKAYA, N.P. (1972) [Metabolism of nickel entering the body
with drinking water.] Gig.i Sanit., 37(6): 20-22 (in Russian).
ELLEN, G., VAN DEN BOSCH-TIBBESMA, G. & DOUMA, F.F. (1978) Nickel
content in various Dutch foodstuffs. Z. Lebensm. Unters. Forsch.
166(3): 145-147.
ENGLISH, J.C., PARKER, R.D., SHARMA, R.P. & OBERG, S.G. (1981)
Toxicokinetics of nickel in rats after intratracheal administration
of a soluble and insoluble form. Am. Ind. Hyg. Assoc. J., 42(7):
486-492.
ENTERLINE, P.E. & MARSH, G.M. (1982) Mortality among workers in a
nickel refinery and alloy manufacturing plant in West Virginia. J.
Natl Cancer Inst., 68: 925-933.
EVANS, R.M., DAVIES, P.J.A. & COSTA, M. (1982) Video time-lapse
microscopy of phagocytosis and intracellular fate of crystalline
nickel sulfide particles in cultured mammalian cells. Cancer Res.
42: 2829-2735.
EVANS, R.M., YANO, E., MORGAN, L.G. & URANO, N. (1988)
Chemiluminescent detection of free oxygen radical generation by
stimulated PMN in vitro: effects of nickel compounds. In: Fourth
International Conference on Nickel Metabolism and Toxicology,
Abstracts, Espoo, 5-9 September l988, Helsinki, Institute of
Occupational Health, p. 37.
EVANS, W.H., READ, J.I. & LUCAS, B.E. (1978) Evaluation of a
method for the determination of total cadmium, lead and nickel in
foodstuffs using measurement by flame atomic-absorption
spectrophotometry. Analyst, 103(1227): 580-594.
FALANDYSZ, J. (1985) Trace metals in flatfish from the southern
Baltic, 1983. Z. Lebensm. Unters. Forsch., 181(2): 117-120.
FALANDYSZ, J. (1986a) Trace metals in cod from the southern Baltic,
1983. Z. Lebensm. Unters. Forsch., 182(3): 228-231.
FALANDYSZ, J. (1986b) Trace metals in herring from the southern
Baltic, 1983. Z. Lebensm. Unters. Forsch., 182(1): 36-39.
FARRELL, R.L. & DAVIS, G.W. (1974) The effects of particulates on
respiratory carcinogenesis by diethylnitrosamine. In: Karbe, E. &
Park, I.F. ed. Experimental lung cancer, carcinogenesis, and
bioassays, Berlin, Heidelberg, New York, Springer Verlag, pp. 219-
233.
FEREN, K. & REITH, A. (1988). Phagocytosis and transformation
testing of Ni3S2 using primary epithelial respiratory cells.
Fourth International Conference on Nickel Metabolism and
Toxicology, Abstracts, Espoo, Finland, 5-9 September 1988,
Helsinki, Institute of Occupational Health, p.64.
FERM, V.H. (1972) The teratogenic effects of metals on mammalian
embryos. Adv. Teratol., 5: 51-75.
FEZY, J.S., SPENCER, D.F. & GREENE, R.W. (1979) The effect of
nickel on the growth of the freshwater diatom Navicula pelliculosa.
Environ. Pollut., 20:131-137.
FIGONI, R. & TREAGAN, L. (1975) Inhibition effect of nickel and
chromium upon antibody response of rats immunization with T-1
phage. Res. Commun. chem. Pathol. Pharmacol., 11: 335-338.
FILKOVA, L. & JAGER, J. (1986) Nonoccupational exposure to nickel
tetracarbonyl. Cesk. Hyg., 31(5): 255-259.
FISHBEIN, L. (1981) Sources, transport and alterations of metal
compounds: An overview. I. Arsenic, beryllium, cadmium, chromium,
and nickel. Environ. Health Perspect., 40: 43-64.
FISCHER, T., FREGERT, S., GREENBERGER, B. & RYSTEDT, I. (1984)
Contact sensitivity to nickel in white gold. Contact Dermatit., 10:
213-24.
FISHELSON, Z. PANBURN, M.K. & MULLER-EBERHARD, H.J. (1983) C3
convertase of the alternative complement pathway. Demonstration
of an active, stable C3b,Bb(Ni) complex. J. biol. Chem., 258(12): 7411-
7415.
FISHER, A.A. (1977) Allergic dermatitis presumably due to metallic
foreign bodies containing nickel or cobalt. Cutis, 19(3): 285-286.
FISHER, A.A. (1985) Nickel dermatitis in men. Cutis, 35(5): 424-426.
FISHER, A.A. & SHAPIRO, A. (1956) Allergic eczematous contact
dermatitis due to metallic nickel. J. Am. Med. Assoc., 161: 717-721.
FISHER, G.L., CHRISP, C.E., MCNEILL, K.L., MCNEILL, D.A., DEMOCKO,
C. & FINCH, G.L. (1984) Mechanistic evaluations of the pulmonary
toxicology of nickel subsulfide. Adv. mod. environ. Toxicol., 6: 49-
60.
FISHER, G.L., CRISP, C.E. & MCNEILL, D.A. (1986) Lifetime effects
of intertracheally instilled nickel subsulfide on B6C3F1 mice.
Environ. Res, 40: 313-320.
FISHER, N.S. (1986) On the reactivity of metals for marine
phytoplankton. Limnol. Oceanogr., 31(2): 443-449.
FLORA, C.J. & NIEBOER, E. (1980) Determination of nickel by
differential pulse polarography at a dropping mercury electrode.
Anal. Chem., 52: 1013-1020.
FORNACE, A.J. Jr (1982) Detection of DNA single-strand breaks
produced during the repair of damage by DNA-protein cross-linking
agents. Cancer Res., 42: 145-149.
FORTH, W. & RUMMEL, W. (1971) Absorption of iron and chemically
related metals in vitro and in vivo: specificity of the iron-
binding system in mucosa of the jejunum. In: Shoryna, S.C. &
Waldron-Edward, D. ed. Intestinal absorption of metal ions, trace
elements and radionuclides, New York, Pergamon Press, pp. 173-191.
FOULKES, E.C. & BLANCK, S. (1984) The selective action of nickel
on tubule function in rabbit kidneys. Toxicology, 33: 245-249.
FOULKES, E.C. & McMULLEN, D.M. (1986) On the mechanism of nickel
absorption in the rat jejunum. Toxicology, 38: 35-42.
FRACHE, R., BAFFI, F., DADONE, A., SCARPONI, G. & DAGNINO, I.
(1980) The determination of heavy metals in the Ligurian sea. II.
The geographical and vertical distribution of Cd, Cu, and Ni. Deep-
Sea Res., 27A: 1079-1082.
FRANCIS, C.W., DAVIS, E.C. & GOYERT, J.C. (1985) Plant uptake of
trace elements from coal gasification ashes. J. environ. Qual.
14(4): 561-569.
FREEMAN, B.M. & LANGSLOW, D.R. (1973) Response of plasma glucose,
free fatty acids and glucagon to cobalt and nickel chlorides by
Gallus domesticus. Comp. Biochem. Physiol., 46A: 427-436.
FRIBERG, L. (1950) Health hazards in the manufacture of alcaline
accumulation. Acta med. Scand., 138(240): 124.
FRIEDLAND, A.J., JOHNSON, A.H. & SICCAMA, T.G. (1986) Zinc, Cu, Ni
and Cd in the forest floor in the Northeastern United States.
Water air soil Pollut., 29: 233-243
FRIEDRICH, B., HEINE, E., FINK, A. & FRIEDRICH, C.G. (1981) Nickel
requirement for active hydrogenase formation in Alcaligenes
eutrophus. J. Bacteriol., 145: 1144-1149.
FROSCH, P.J. & KLIGMAN, A.M. (1976) The chamber-scarification test
for irritancy. Contact Dermatit., 2: 314-324.
FUKUNAGA, M., KURACHI, Y. & MIZYGUCHI, Y. (1982) Action of some
metal ions on yeast chromosomes. Chem. pharm. Bull. (Tokyo), 30:
3017-3019.
FULLERTON, A., ANDERSEN, J.R., HOELGAARD, A. & MENNE, T. (1986)
Permeation of nickel salts through human skin in vitro. Contact
Dermatit., 15: 183-177.
FURST, A. & AL-MAHROUQ, H. (1981) Excretion of nickel following
intratracheal administration of the carbonate. Proc. West.
Pharmacol. Soc., 24: 119-121.
FURST, A. & CASSETTA, D. (1973) Carcinogenicity of nickel by
different routes. Proc. Am. Assoc. Cancer Res., 121: 31.
FURST, A. & SCHLAUDER, M.C. (1971) The hamster as a model for
metal carcinogenesis. Proc. West. Pharmacol. Soc., 14: 68-71.
FURST, A., CASSETTA, D.M. & SASMORE, D.P. (1973) Rapid induction
of pleural mesotheliomas in the rat. Proc. West. Pharmacol. Soc.
16: 150-153.
GAINER, J.H. (1977) Effects of heavy metals and/or of deficiency of
zinc on mortality rates in mice infected with encephalomyocarditis
virus. Am. J. vet. Res., 38: 869-872.
GAWKRODGER, D.J., LLOYD, M. & HUNTER, J. (1986a) Occupational skin
disease in hospital cleaning and kitchen workers. Contact
Dermatit., 15(3): 132-135.
GAWKRODGER, D.J., COOK, S.W., FELL, G.S. & HUNTER, J.A.A. (1986b)
Nickel dermatitis: the reaction to oral nickel challenge. Br. J.
Dermatol., 115: 33-38.
GEMMER-COLOS, U., TUSS, H., SAUR, D. & NEEB, R. (1981)
[Polarographic determination of Nickel in the ppb-range after
extracting nickel as dioxime.] Fresenius Z. anal. Chem., 307: 347-351
(in German).
GENDREAU, R.M., JAKOBSEN, R.J. & HENRY, W.M. (1980) Fourier
transform infrared spectroscopy for inorganic compound speciation.
Environ. Sci. Technol., 14: 990-995.
GERIN, M., SIEMIATYCKI, J., RICHARDSON, L., PELLERIN, J., LAKHANI,
R. & DEWAR, R. (1984) Nickel and cancer associations from a
multicancer occupation exposure case-referent study - preliminary
findings. In: Nickel in the Human Environment, Proceedings of a
Joint Symposium, Lyon, 8-11 March, 1983, Lyon, International Agency
for Research on Cancer, pp. 105-115 (IARC Scientific Publications
No. 53).
GERSTLE, R.W. & ALBRINCK, D.N. (1982) Atmospheric emissions of
metals from sewage sludge incineration. J. Air Pollut. Control
Assoc., 32(11): 1119-1123.
GHIRINGHELLI, L. (1957) [Therapeutic utility of 2-3
dimercaptopropanol and thiotic acid in rats poisoned with nickel
carbonyl.] Attiv. Soc. Lomb. Med. Biol., 12: 24-26 (in Italian).
GHIRINGHELLI, J. & AGAMENNONE, M. (1957) [The metabolism of nickel
in animals experimentally poisoned with nickel carbonyl.] Med.
Lav., 48: 187-194 (in Italian).
GIGNAC, L.D. & BECKETT, P.J. (1986) The effect of smelting
operations on peatlands near Sudbury, Ontario, Canada. Can. J.
Bot., 64(6): 1138-1147.
GILANI, S.H. & MARANO, M. (1980) Congenital abnormalities in
nickel poisoning in chick embryos. Arch. environ. Contam.
Toxicol., 9(1): 17-22.
GILMAN, J.P.W. (1962) Metal carcinogenesis. II. A study of the
carcinogenic activity of cobalt, copper, iron and nickel compounds.
Cancer Res., 22: 158- 162.
GILMAN, J.P.W. & HERCHEN, H. (1963). The effect of physical form
of implant on nickel sulphide tumourigenesis in the rat. Acta
Union int. Cancer, 19: 615-619.
GILMAN, J.P.W. & RUCKERBAUER, G.M. (1962) Metal carcinogenesis. I.
Observations on the carcinogenicity of a refinery dust, cobalt
oxide, and colloidal thorium dioxide. Cancer Res., 22: 152-157.
GILMAN, J.P.W. & YAMASHIRO, S. (1985) Muscle tumorigenesis by
nickel compounds. In: Brown, S.S. & Sunderman, F.W. Jr, ed.
Progress in Nickel toxicology. Proceedings of the 3rd International
Conference on Nickel Metabolism and Toxicology, Paris, 4-7
September, 1984, Oxford, Blackwell Scientific Publications,
pp. 9-22.
GITLITZ, P.H., SUNDERMAN, F.W. & GOLDBLATT, P.J. (1975)
Aminoaciduria and proteinuria in rats after single intraperitoneal
injection of Ni(II). Toxicol. appl. Pharmacol., 34: 430-440.
GLAESS, E. (1956a) [Distribution of fragmentations and achromatic
areas on chromosomes of Vicia faba following treatment with heavy
metal salts]. Chromosome, 8: 260-284 (in German).
GLAESS, E. (1956b) [Investigations on the effects of heavy metal
salts on root top mitosis of Vicia faba.] Ztschr. Botanik, 44:
1-58 (in German).
GLASER, U., HOCHRAINER, D., OLDIGES, H. & TAKENAKA, S. (1986)
Long-term inhalation studies with NiO and As2O3 aerosols in Wistar
rats. In: Stern, R.M., Berlin, A., Fletcher, A.C. & Jarvisalo,
J. ed. Health hazards and biological effects of welding fumes and
gases. Proceedings of The International Conference on Health
Hazards and Biological Effects of Welding Fumes and Gases,
Copenhagen, 18-21 February, 1985, Amsterdam, New York, Oxford,
Elsevier Science Publishers, pp. 325-328.
GLENNON, J.D. & SARKAR, B. (1982) Nickel(II) transport in human
blood serum. Studies of nickel(II) binding to human albumin and to
native-sequence peptide, and ternary-complex formation with L-
histidine. Biochem. J., 203(1): 15-23.
GODBOLD, J.H. Jr & TOMPKINS, E.A. (1979) A long-term mortality
study of workers occupationally exposed to metallic nickel at the
Oak Ridge Gaseous Diffusion Plant. J. occup. Med., 21(12): 799-806.
GOLDBERG, M., FUHRER, R., BRODEUR, J.-M., GOLDBERG, P., SEGNAN, N.,
LECLERC, A., CHASTANG, J.-F., BOURBONNAIS, R. & FRANCOIS, D.
(1985a) Cancer des voies respiratoires parmi les travailleurs d'une
entreprise d'extraction et du raffinage du nickel en Nouvelle
Calédonie: Une étude cas-témoins au sein d' une cohorte. In: Brown,
S.S. & Sunderman, J.W. Jr, ed. Progress in Nickel toxicology,
Proceedings of the 3rd International Conference on Nickel
Metabolism and Toxicology, Paris, 4-7 September, 1984, Oxford,
Blackwell Scientific Publications, pp. 215-218.
GOLDBERG, P., BLANC, M., FUHRER, R., SEGNAN, N., BRODEUR, J.-M.,
LECLERC, A., FRANCOIS, D., DAB, W., GODARD, C. & GOLDBERG, M.
(1985b) Incidence des cancers respiratoires chez les travailleurs
d'une entreprise d'extraction et de raffinage du nickel et dans
la population générale en Nouvelle Calédonie. In: Brown, S.S. &
Sunderman, F.W. Jr, ed. Progress in Nickel toxicology. Proceedings
of the 3rd International Conference on Nickel Metabolism and
Toxicology, Paris, 4-7 September, 1984, Oxford, Blackwell
Scientific Publications, pp. 211-214.
GOLDBERG, M., GOLDBERG, P., LECLERC, A., CHASTANG, J.F., MARNE,
M.J., GUEZIEC, J., LAVIGNE, F., DUBOURDIEU, D. & HUERRE, M. (1988)
A 7-year survey of respiratory cancers among nickel workers in New
Caledonia (1978-1984). In: Fourth International Conference on
Nickel Metabolism and Toxicology, Abstracts, Espoo, Finland, 5-9
September 1988, Helsinki, Institute of Occupational Health, p. 42.
GORDON, C.J. (1989) Effect of nickel chloride on body temperature
and behavioral thermoregulation in the rat. Neurotoxicol. Teratol.,
11: 317-320.
GORDON, C.J. & STEAD, A.G. (1986) Effect of nickel and cadmium
chloride on autonomic and behavioral thermoregulation in mice.
Neurotoxicology, 7: 97-106.
GORDON, C.J., FOGELSON, L. & STEAD, A.G. (1989) Temperature
regulation following nickel intoxication in the mouse: effect of
ambient temperature. Comp. Biochem. Physiol., 92: 73-76.
GORDYNIA, R.I. (1969) [Effect of a ration containing a nickel salt
additive on carbohydrate metabolism in experimental animals.] Vopr.
Racion. Pitan., 5: 167-170 (in Russian).
GRAHAM, J.A., GARDNER, D.E., WATERS, M.D. & COFFIN, D.L. (1975a)
Effect of trace metals on phagocytosis by alveolar macrophages.
Infect. Immun., 11: 1278-1283.
GRAHAM, J.A., GARDNER, D.E., MILLER, F.J., DANIELS, M.J. & COFFIN,
D.L. (1975b) Effect of nickel chloride on primary antibody
production in the spleen. Environ. Health Perspect., 12: 109-113.
GRAHAM, J.A., MILLER, F.J., DANIELS, M.J., PAYNE, E.A. & GARDNER,
D.E. (1978) Influence of cadmium, nickel, and chromium on primary
immunity in mice. Environ. Res., 16: 77-87.
GRANDJEAN, P. (1984) Human exposure to nickel. In: Nickel in the
human environment. Proceedings of a Joint Symposium, Lyon, 8-11
March, 1983, Lyon, International Agency for Research on Cancer, pp.
469-485 (IARC Scientific Publications No. 53).
GRANDJEAN, P., SELIKOFF, I.J., SHEN, S.K., SUNDERMAN, F.W. Jr
(1980) Nickel concentrations in plasma and urine of shipyard
workers. Am J. ind. Med., 1(2): 181-189.
GRANDJEAN, P., ANDERSEN, O. & NIELSEN, G.D. (1988) Nickel. In:
Alession, L. Berlin, A. Boris, M. & Roi, R. ed. Biological
indicators for the assessment of human exposure to industrial
chemicals. ISPRA (Varese), IPSRA Establishment, Joint Research
Centre, Commission of the European Communities, pp. 56-80.
GREBNEV, V.L. & YELNICHNYKH, L.N. (1983) Experimental study of the
action of nickel-containing aerosols in preparatory workshops of
nickel production on parenchymour organs. Non-specific action of
harmful occupation factors on workers' organism, Sverdlovsk,
Medical College, pp. 90-95.
GREEN, M.H.L., MURIEL, W.J. & BRIDGES, B.A. (1976). Use of a
simplified fluctuation test to detect low levels of mutagens.
Mutat. Res., 38: 33-42.
GROPPEL, B., ANKE, M., RIEDEL, E. & GRUN, M. (1980) [The nickel
supply and status of wild ruminants in the GDR.] In: Anke, M.
Schneider, H.-J. & Brûckner, Chr. ed. [3. Trace Element
Symposium: Nickel, Jena, German Democratic Republic, 7-11 July,
1980,] Jena, Friedrich-Schiller University, pp. 269-276 (in German).
GROSS, P.R., KATZ, S.A. & SAMITZ, M.H. (1968) Sensitization of
guinea-pigs to chromium salts. J. invest. Dermatol., 50: 424-427.
GUTENMANN, W.H., BACHE, C.A., LISK, D.J., HOFFMANN, D., ADAMS,
J.D. & ELFVING, D.C. (1982) Cadmium and nickel in smoke of
cigarettes prepared from tobacco cultured on municipal sludge-
amended soil. J. Toxicol. environ. Health, 10(3): 423-431.
HACKETT, R.L. & SUNDERMAN, F.W. Jr (1967) Acute pathological
reactions to administration of nickel carbonyl. Arch. environ.
Health, 14(4): 604-613.
HACKETT, R.L. & SUNDERMAN, F.W. Jr (1968) Pulmonary alveolar
reaction to nickel carbonyl: Ultrastructural and histochemical
studies. Arch. environ. Health, 16(3): 349-362.
HACKETT, R.L. & SUNDERMAN, F.W. Jr (1969) Nickel carbonyl. Effects
upon the ultrastructure of hepatic parenchymal cells. Arch.
environ. Health, 19: 337- 343.
HAGEDORN-GOTZ, H., KUPPERS, G. & STOPPLER, M. (1977) On nickel
contents in urine and hair in a case of exposure to nickel
carbonyl. Arch. Toxicol., 38(4): 275-285.
HALE, J.G. (1977) Toxicity of metal mining wastes. Bull. environ.
Contam. Toxicol., 17:66-73.
HALEY, P.Y., BICE, D.E., MUGGENBURG, B.A., HAHN, F.F. & BENJAMIN,
S.A. (1987) Immunopathologic effects of nickel subsulfide on the
primate pulmonary system. Toxicol. appl. Pharmacol., 88: 1-12
HALL, T.M. (1982) Free ionic nickel accumulation and localization
in the freshwater zooplankton Daphnia magna. Limnol. Oceanogr.
27(4): 718-727.
HANSEN, K. & STERN, R.M. (1983) In vitro toxicity and
transformation potency of nickel compounds. Environ. Health
Perspect., 51: 223-226.
HANSEN, K. & STERN, R.M. (1984) Toxicity and transformation potency
of nickel compounds in BHK cells in vitro. In: Nickel in the human
environment. Proceedings of a Joint Symposium, Lyon, 8-11 March,
1983, Lyon, International Agency for Research on Cancer, pp. 193-
200 (IARC Scientific Publications No. 53).
HANSEN, L.D. & FISHER, G.L. (1980) Elemental distribution in coal
fly ash particles. Environ. Sci. Technol., 14(9): 1111-1117.
HANSEN, L.D., SILBERMAN, D., FISHER, G.L. & EATOUGH, D.J. (1984)
Chemical speciation of elements in stack-collected, respirable-size
coal fly ash. Environ. Sci. Technol., 18: 181-186.
HARO, R.T., FURST, A. & FALK, H.L. (1968) Studies on the acute
toxicity of nickelocene. Proc. West. Pharmacol. Soc., 11: 39-42.
HARNETT, P.B., ROBISON, S.H., SWARTZENDRUBER, D.E. & COSTA, M.
(1982) Comparison of protein, RNA, and DNA binding and cell-cycle-
specific growth inhibitory effects of nickel compounds in cultured
cells. Toxicol. appl. Pharmacol., 64: 20-30.
HARTENSTEIN, R., NEUHAUSER, E.F. & NARAHARA, A. (1981) Effects of
heavy metal and other elemental additives to activated sludge on
growth of Eisenia foetida. J. environ. Qual., 10(3): 372-377.
HARTWIG, A. & BEYERSMANN, D. (1989) Enhancement of UV-induced
mutagenesis and sister-chromatid exchanges by nickel ions in V79
cells: evidence for inhibition of DNA repair. Mutat. Res., 217: 65-73.
HAWLEY, J.E. (1955) Spectographic study of some Nova Scotia coals.
Can. min. metall. Bull., 48: 712-726.
HAWORTH, S., LAWLOR, T., MORTELMANS, K., SPECK, W. & ZEIGER, E.
(1983) Salmonella mutagenicity test results in 250 chemicals.
Environ. mol. Mutagen., 5: 3-142.
HAYASHI, YL, TAKAHASHI, M., MAEKAWA, A., KUROKAWA, Y. & KOLUBO, T.
(1985) Screening of environmental pollutants for promotion effects
on carcinogenesis. Annu. Rep. Minist. Health Welfare Jpn 1982-
1984, 20: 1-10.
HEATH, J.C. & DANIEL, M.R. (1964) The production of malignant
tumors by nickel in the rat. Br. J. Cancer, 18: 261-264.
HECK, J.D. & COSTA, M. (1982) Extracellular requirements for the
endocytosis of carcinogenic crystalline nickel sulfide particles by
facultative phagocytes. Toxicol. Lett., 12: 243-250.
HEINRICHS, H. & R. MAYER, R. (1980) Distribution and cycling of
nickel in forest ecosystems. In: Nriagu, J.O. ed. Nickel in the
environment, New York, Chichester, Brisbane, Toronto, John Wiley
and Sons, pp. 431-455.
HENDEL, R.C. & SUNDERMAN, F.W. Jr (1972) Species variations in the
properties of ultrafiltrable and protein-bound serum nickel. Res.
Commun. chem. Pathol. Pharmacol., 4(1): 141-146.
HENRY, W.M. & KNAPP, K.T. (1980) Compound forms of fossil fuel fly
ash emissions. Environ. Sci. Technol., 14(4): 450-456.
HENRY, W.M., BARBOUR, R.L., JAKOBSEN, R.J. & SCHUMAKER, P.M.
(1982) Inorganic compound identification of fly ash emissions from
municipal incinerators. Final Report, Washington, DC, US
Environmental Protection Agency, 32 pp (Report prepared by Battelle
Columbus Laboratories, Ohio, under the EPA Contract 68-02-2296)
(EPA-600/3-82-095. PB83-146175).
HERLANT-PEERS, M.C., HILDEBRAND, H.F. & BISERTE, G. (1982) 63Ni(II)
incorporation into lung and liver cytosol of Balb/C mouse. An in
vitro and in vivo study. Zentralbl. Bakteriol. Mikrobiol. Hyg. (B),
176(4): 368-382.
HILDEBRAND, H.F. & BISERTE, G. (1979a) Cylindrical laminated bodies
in nickel subsulphide-induced rhabdomyosarcoma in rabbits. Eur. J.
cell Biol., 19: 276-280.
HILDEBRAND, H.F. & BISERTE, G. (1979b) Nickel sub-sulphide-induced
leiomyosarcoma in rabbit white skeletal muscle. A light
microscopical and ultrastructural study. Cancer, 43: 1358-1374.
HILDEBRAND, H.F. & TETAERT, D. (1981) Ni3S2-induced
leiomyosarcomas in rabbit skeletal muscle: analysis of the tumoral
myosin and its significance in the retrodifferentiation concept.
Oncodev. Biol. Med., 2: 101-108.
HO, W. & FURST, A. (1973) Nickel excretion by rats following a
single treatment. Proc. West. Pharmacol. Soc., 16: 245-248.
HOBBS, R.J. & STREIT, B. (1986) Heavy metal concentrations in
plants growing on a copper mine spoil heap in the Grand Canyon,
Arizona. Am. Mid. Nat., 115(2): 277-281.
HODGSON, G.W. (1954) Vanadium, nickel, and iron trace metals in
crude oil of Western Canada. Bull. Am. Assoc. Pet. Geol., 38(12):
2537-2554.
HOEY, M.J. (1966) The effects of metallic salts on the histology
and functioning of the rat testis. J. reprod. Fertil., 12: 461-471.
HOFF, R.M. & BARRIE, L.A. (1986) Air chemistry observations in the
Canadian arctic. Water Sci. Tech., 18: 97-107.
HOGETVEIT, A.C. & BARTON, R.T. (1976) Preventive health program
for nickel workers. J. occup. Med., 18(12): 805-808.
HOGETVEIT, A.C. & BARTON, R.T. (1977) Monitoring nickel exposure in
refinery workers. In: Brown, S.S. ed. Clinical chemistry and
chemical toxicology of metals, Amsterdam, Oxford, New York,
Elsevier Science Publishers, Vol. 1, pp. 265-268.
HOGETVEIT, A.C., BARTON, R.T. & KOSTOL, L.C. (1978) Plasma nickel
as a primary index of exposure in nickel refining. Ann. occup.
Hyg., 21: 113-120.
HOGETVEIT, A.C., BARTON, R.T. & ANDERSEN, I. (1980) Variations of
nickel in plasma and urine during the work period. J. occup. Med.
22: 597-600.
HOHNADEL, D.C., SUNDERMAN, F.W. Jr, NECHAY, M.W. & MCNEELY, M.D.
(1973) Atomic absorption spectrometry of nickel, copper, zinc and
lead in sweat collected from healthy subjects during sauna bathing.
Clin. Chem., 19: 1288-1292.
HONDA, K., FUJISE, Y., TATSUKAWA, R. & ITANO, K. (1984)
Composition of chemical components in bone of striped dolphin,
Stenella coeruleoalba: Distribution characteristics of heavy metals
in various bones. Agric. biol. Chem., 48(3): 677-680.
HONDA, K., MIN BYUNG, Y. & TATSUKAWA, R. (1985) Heavy metal
distribution in organs and tissues of the eastern great white egret
Egretta alba modesta. Bull. environ. Contam. Toxicol., 35(6): 781-790.
HOPFER, S.M. & SUNDERMAN, F.W. Jr (1988) Hypothermia and deranged
circadian rhythm of core body temperature in nickel chloride-
treated rats. Res. Commun. chem. Pathol. Pharmacol., 62: 495-505.
HOPFER, S.M., SUNDERMAN, F.W. Jr, FREDRICKSON, R.N. & MORSE, F.E.
(1978) Nickel induced erythrocytosis: efficacies of nickel
compounds and susceptabilities of rat strains. Ann. clin. lab.
Sci., 8: 396-402.
HOPFER, S.M., LINDEN, J.V., CRISOSTOMO, C., CATALANATTO, F.A.,
GALEN, M. & SUNDERMAN, F.W. Jr (1985) Hypernickelemia in
hemodialysis patients. In: Brown, S.S. & Sunderman, F.W. Jr, ed.
Progress in Nickel toxicology. Proceedings of the 3rd International
Conference on Nickel Metabolism and Toxicology, Paris, 4-7
September, 1984, Oxford, Blackwell Scientific Publications, pp.
133-136.
HOPFER, S.M., LINDEN, J.V., REZUKE, W.N., O'BRIEN, J.E., SMITH, L.
WATTERS, F. & SUNDERMAN, F.W. Jr (1987) Increased nickel
concentrations in body fluids of patients with chronic alcoholism
during disulfiram therapy. Res. Commun. chem. Pathol. Pharmacol.
55: 101-109.
HOPFER, S.M., FAY, W.P. & SUNDERMAN, F.W. Jr (1989) Serum nickel
concentration in hemodialysis patients with environmental nickel
exposure. Ann. clin. lab. Sci.
HORAK, E. & SUNDERMANN, F.W. Jr (1973) Fecal nickel excretion by
healthy adults. Clin. Chem., 19(4): 429-430.
HORAK, E. & SUNDERMAN, F.W. Jr (1975a) Effects of Ni(II), other
divalent metal ions, and glucagon upon plasma glucose
concentrations in normal, adrenalectomized and hypophysectomized
rats. Toxicol. appl. Pharmacol., 32: 316-329.
HORAK, E. & SUNDERMAN, F.W. Jr (1975b) Effects of Ni(II) upon
plasma glucagon and glucose in rats. Toxicol. appl. Pharmacol., 33:
388-391.
HORIE, A., HARATAKE, J., TANAKA, I., KODAMA, Y. & TSUCHIYA, K.
(1985) Electron microscopical findings, with special reference to
cancer in rats caused by inhalation of nickel oxide. Biol. trace
Res., 7: 223-239.
HOWARD, J.M. (1980) Serum nickel in myocardial infarction. Clin.
Chem., 26(10): 1515.
HRUDEY, S.E. (1985) Residues from hazardous waste treatment.
Effluent Water Treat. J., 25(1): 7-12.
HSI, A.W., O'NEILL, J. & SAN SEBASTIAN, J.R. (1979a) Quantitative
mammalian cell genetic toxicology: study of the cytotoxicity and
mutagenicity of 70 individual environmental agents related to
energy technologies and 3 subfractions of a crude synthetic oil in
the CHO/HGPRT system. Environ. Sci. Res., 15: 291-315.
HSIE, A.W., JOHNSON, N.P., COUCH, D.B., SAN SEBASTIAN, I., O'NEILL,
I.P., HOESCHELE, I.D., RAHN, R.O. & FORBES, N.L. (1979b)
Quantitative mammalian cell mutagenesis and a preliminary study of
the mutagenic potential of metallic compounds. In: Kharasch, N.
ed. Trace metals in health and disease, New York, Raven Press, pp.
55-69.
HUANG, J., LU, Y., LIU, G., ZHENG, G., MEI, C., LI, Z., SHENG, S.
& YE, Z. (1986) The distribution of trace elements in rats. Acta
zool. sin., 32: 35- 39.
HUEPER, W.C. (1952) Experimental studies in metal cancerogenesis.
I. Nickel cancer in rats. Texas Rep. Biol. Med., 10: 167-186.
HUEPER, W.C. (1955) Experimental studies in metal cancerigenesis.
IV. Cancer produced by parenterally introduced metallic nickel. J.
Natl Cancer Inst., 16: 55-67.
HUEPER, W.C. (1958) Experimental studies in metal cancerigenesis.
IX. Pulmonary lesions in guinea-pigs and rats exposed to prolonged
inhalation of powdered metallic nickel. Arch. Pathol., 65: 600-607.
HUEPER, W.C. & PAYNE, W.W. (1962) Experimental studies in metal
carcinogenesis. Arch. environ. Health, 5: 445-462.
HUGHES, G.M., PERRY, S.F. & BROWN, V.M. (1979) A morphometric
study of effects of nickel, chromium and cadmium on the secondary
lamellae of rainbow trout gills. Water Res., 13:665-679.
HUGHES, A.W., SHERLOCK, D.A., HAMBLEN, D.L. & REID, R. (1987)
Sarcoma at the site of a single hip screw. J. bone joint Surg.
B69: 470-472.
HUI, G. & SUNDERMAN, F.W. Jr (1980) Effects of nickel compounds on
incorporation of the (3H) thymidine into DNA in rat liver and
kidney. Carcinogenesis, 1: 297-304.
HULETT, L.D., WEINBURGER, A.J., NORTHCUTT, K.J. & FERGUSON, M.
(1980) Chemical species in fly ash from coal-burning power plants.
Science, 210: 1356-1358.
HUNZIKER, N. (1960) De l'eczéma experimental. Quelques expériences
concernant la sensibilisation du cobaye au nickel. Dermatologica,
121: 307-312.
HUTCHINSON, T.C. (1973) Comparative studies of the toxicity of
heavy metals to phytoplankton and their synergistic interactions.
Water Pollut. Res. Can., 8: 68-90.
HUTCHINSON, T.C. & CZYRSKA, H. (1975) Heavy metal toxicity and
synergism in floating aquatic weeds. Verh. Int. Ver. Limnol., 19:
2102-2111.
HUTCHINSON, T.C. & WHITBY, L.M. (1977) The effects of acid
rainfall and heavy metal particulates on a boreal forest ecosystem
near the Sudbury smelting region of Canada. Water air soil Pollut.
7: 421-438.
HUTCHINSON, T.C., FEDORENKO, A., FITCHKO, J., KUJA, A., VANLOON,
J. & LICHWA, J. (1975) Movement and compartmentation of nickel and
copper in an aquatic ecosystem. In: Nriagu, J.O. ed.
Environmental biogeochemistry, Ann Arbor, Michigan, Ann Arbor
Science Publishers Inc. Vol. 2, pp. 565-585.
HUTCHINSON, T.C., FREEDMAN, B. & WHITBY, L. (1981) Nickel in
Canadian soils and vegetation. In: Effects of nickel in the
Canadian environment, Ottawa, National Research Council of Canada,
pp. 119-157 (Publication No. NRCC 18568).
IARC (1976) Cadmium, nickel, some epoxides, miscellaneous
industrial chemicals and general considerations on volatile
anaesthetics. Lyon International Agency for Research on Cancer,
pp. 75-112 (IARC Monographs on the Evaluation of Carcinogenic Risk
of Chemicals to Man, Vol. 11).
IARC (1989) Mortality and cancer incidence follow-up of an
historical cohort of European welders. Lyon, International Agency
for Research on Cancer (Internal Technical Report No. 89/003).
IARC (1990) Nickel and nickel compounds. In: Chromium, nickel and
welding. Lyon, International Agency for Research on Cancer, pp.
257-445 (IARC Monographs on the Evaluation of Carcinogenic Risks to
Humans, Vol. 49).
ICHINOKI, S. & YAMAZAKI, M. (1985) Simultaneous determination of
nickel, lead, zinc, and copper in citrus leaves and rice flour by
liquid chromatography with hexamethylene dithiocarbamate
extraction. Anal. Chem., 57(12): 2219-2222.
IKEBE, K. & TANAKA, R. (1979) Determination of vanadium and nickel
in marine samples by flameless and flame atomic absorption
spectrophotometry. Bull. environ. Contam. Toxicol., 21(4-5): 526-532.
INOUE, S. & KAWANISHI, S. (1989) ESR evidence for superoxide,
hydroxyl radicals and singlet oxygen produced from hydrogen
peroxide and nickel(ii) complex of glycylglycyl-L-histidine.
Biochem. Biophys. Res. Commun., 159(2): 445-451.
IZMEROV, N.F. (1983) [Occupational diseases: guidelines.] Moscow,
Meditsina, pp. 319-329 (in Russian).
JACOBSEN, N., ALFHEIM, I. & JONSEN, J. (1978) Nickel and strontium
distribution in some mouse tissues. Passage through placenta and
mammary glands. Res. Commun. chem. Pathol. Pharmacol., 20(3): 571-584.
JACQUET, P. & MAYENCE, A. (1982) Application of the in vitro
embryo culture to the study of the mutagenic effets of nickel in
male germ cells. Toxicol. Lett., 11(1-2): 193-197.
JAFFRE, T., KERSTEN, W., BROOKS, R.R. & REEVES, R.D. (1979) Nickel
uptake by Flacourtiaceae of New Caledonia. Proc. R. Soc. Lond.
(Biol), 205(1160): 385-394.
JANSEN, L.H., BERRENS, L. & VAN DELDEN, J. (1964) Contact
sensitivity to simple chemicals: The role of intermediates in the
process of sensitization. Naturwissenschaften, 51: 387-388.
JARSTRAND, C., LUNDBORG, M., WIRNIK, A. & CAMNER, P. (1978)
Alveolar macrophage function in nickel dust exposed rabbits.
Toxicology, 11(4): 353-359.
JASMIN, G. & RIOPELLE, J.L. (1976) Renal carcinomas and
erythrocytosis in rats following intrarenal injection of nickel
subsulfide. Lab. Invest., 35: 71-78.
JENKINS, D.W. (1980a) Nickel accumulation in aquatic biota. In:
Nriagu, J.O. ed. Nickel in the environment, New York, Chichester,
Brisbane, Toronto, John Wiley and Sons, pp. 283-337.
JENKINS, D.W. (1980b) Nickel accumulation in terrestrial wildlife.
In: Nriagu, J.O. ed. Nickel in the environment, New York,
Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 457-462.
JOHANSSON, A. & CAMNER, P. (1980) Effects of nickel dust on rabbit
alveolar epithelium. Environ. Res., 22: 510-516.
JOHANSSON, A., CAMNER, P., JARSTRAND, C. & WIERNIK, A. (1980)
Morphology and function of alveolar macrophages after longterm
nickel exposure. Environ. Res., 23: 170-180.
JOHANSSON, A., CAMNER, P. & ROBERTSON, B. (1981) Effects of
longterm nickel dust exposure on rabbit alveolar epithelium.
Environ. Res., 25: 391-403.
JOHANSSON, A., CAMNER, P., JARSTRAND, C. & WIERNIK, A. (1983a)
Rabbit lungs after long-term exposure to low nickel dust
concentration. II. Effects on morphology and function. Environ.
Res., 30: 142-151.
JOHANSSON, A., CURSTEDT, T., ROBERTSON, B. & CAMNER, P. (1983b)
Rabbit lung after inhalation of soluble nickel. II. Effects on lung
tissue and phospholipids. Environ. Res., 31: 399-412.
JOHANSSON, A., WIERNIK, A., LUNDBORG, M., JARSTRAND, C. & CAMNER,
P. (1988) Alveolar macrophages in rabbits after combined exposure
to nickel and trivalent chromium. Environ. Res., 46: 120-132.
JONES, J.G. & WARNER, C.G. (1972) Chronic exposure to iron oxide,
chromium oxide, and nickel oxide fumes in metal dressers in a
steelworks. Br. J. ind. Med., 29: 168-177.
JORDAN, W.P. & KING, S.E. (1979) Nickel feeding in nickel-sensitive
patients with hand eczema. J. Am. Acad. Dermatol., 1(6): 506-508.
KAABER, K., VEIEN, N.K. & TJELL, J.C. (1978) Low nickel diet in
the treatment of patients with chronic nickel dermatitis. Br. J.
Dermatol., 98: 197-201.
KADOTA, I. & KURITA, M. (1955) Hyperglycemia and islet cell damage
caused by nickelous chloride. Metabolism, 4: 337-342.
KALDOR, J., PETO, J., EASTON, D., DOLL, R., HERMON, C. & MORGAN,
L. (1986) Models for respiratory cancer in nickel refinery workers.
J. Natl Cancer Inst., 77: 841-848.
KALIMO, K. & LAMMINTAUSTA, K. (1984) 24 and 48 h allergen exposure
in patch testing. Contact Dermatit., 10: 25-29.
KALLIOMAKI, P.L., RAHKONEN, E., VAARANEN, V., KALLIOMAKI, K. &
AITTONIEMI, K. (1981) Lung-retained contaminants, urinary chromium
and nickel among stainless steel welders. Int. Arch. occup.
environ. Health, 49(1): 67-75.
KANEMATSU, N., HARA, M. & KADA, T. (1980) REC assay and
mutagenicity studies on metal compounds. Mutat. Res., 77: 109-116.
KASPRZAK, K.S. (1974) An autoradiographic study of nickel
carcinogenesis in rats following injection of 63Ni3S2 and Ni335S2.
Res. Commun. chem. Pathol. Pharmacol., 8(1): 141-150.
KASPRZAK, K.S. & POIRIER, L.A. (1983) Effects of calcium, magnesium
and sodium acetates on tissue distribution of nickel (II) in Strain
A mice. In: Brown, S.S. & Savory, J. ed. Chemical toxicology and
clinical chemistry of metals. Proceedings of the 2nd International
Conference, Montreal, 19-22 July, 1983, Oxford, Blackwell
Scientific Publications, pp. 374-376.
KASPRZAK, K.S. & POIRIER, L.A. (1985) Effects of calcium and
magnesium salts on nickel subsulfide carcinogenesis in Fisher rats.
In: Brown, S.S. & Sunderman, F. W. Jr, ed. Progress in nickel
toxicology. Proceedings of the 3rd International Conference on
Nickel Metabolism and Toxicology, Paris, 4-7 September, 1984,
Oxford, Blackwell Scientific Publications, pp. 29-32.
KASPRZAK, K.S. & SUNDERMAN, F.W. Jr (1969) The metabolism of nickel
carbonyl-14C. Toxicol. appl. Pharmacol., 15(2): 295-303.
KASPRZAK, K.S. & SUNDERMAN, F.W. Jr (1977) Mechanisms of
dissolution of nickel subsulfide in rat serum. Res. Commun. chem.
Pathol. Pharmacol., 16(1): 95-108.
KASPRZAK, K.S., MARCHOW, L. & BREBOROWICZ, J. (1973) Pathological
reactions in rat lungs following intratracheal injection of nickel
subsulfide and 3,4-benzpyrene. Res. Commun. chem. Pathol.
Pharmacol., 6(1): 237-245.
KASPRZAK, K.S., GABRYEL, P. & JARCZEWSKA, K. (1983)
Carcinogenicity of nickel(II)hydroxides and nickel(II) sulfate in
Wistar rats and its relation to the in vitro dissolution rates.
Carcinogenesis, 4(3): 275-279.
KASPRZAK, K.S., WAALKES, M.P. & POIRIER, L.A. (1986) Antagonism by
essential divalent metals and amino acides of nickel(II)-DNA
binding in vitro. Toxicol. appl. Pharmacol., 82: 336-343.
KASPRZAK, K.S. & RODRIGUEZ, R.E. (1988) Nickel-iron interactions in
carcinogenesis: dose effect and involvement of active oxygen and
related enzymes. In: Fourth International Conference on Nickel
Metabolism and Toxicology, Abstracts, Espoo, Finland,5-9 September
1988, Helsinki, Institute of Occupational Health, p. 59.
KASPRZAK, K.S., KOVATCH, R.M. & POIRIER, L.A. (1988) Inhibitory
effect of zinc on nickel subsulfide carcinogenesis in Fischer rats.
Toxicology, 52: 153-262.
KASPRZAK, K.S., WARD, J.M., POIRIER, L.A., REICHARDT, D.A., DENN,
A.C. III, & REYNOLDS, C.W. (1987) Nickel-magnesium interactions in
carcinogenesis: dose effects and involvement of natural killer
cells. Carcinogenesis, 8(7): 1005-1011.
KATZ, S.A. & SAMITZ, M.H. (1975) Leaching of nickel from stainless
steel consumer commodities. Acta dermatol., 55: 113-115.
KEEFER, R.F., SINGH, R.N. & HORVATH, D.J. (1986) Chemical
composition of vegetables grown on an agricultural soil amended
with sewage sludges. Environ. Qual., 15(2): 146-152.
KELLER, W. & PITBLADO, J.R. (1986) Water quality changes in Sudbury
area lakes: A comparison of synoptic surveys in 1974-1976 and 1981-
1983. Water air soil Pollut., 29(3): 285-296.
KEMKA, R. (1971) [Determination of the nickel and cobalt contents
in urine and the atmosphere.] Prac. Lek., 23: 80-85 (in Czech).
KESKINEN, H., KALLIOMAKI, P.L. & ALANKO, K. (1980) Occupational
asthma due to stainless steel welding fume. Clin. Allergy, 10:
151-159.
KHAN, S.N., RAHMAN, M.A. & SAMAD, A. (1984) Trace elements in
serum from Pakistani patients with acute and chronic ischemic heart
disease and hypertension. Clin. Chem., 30: 644-648.
KIM, M.K., FISHER, A.M. & MACKAY, R.J. (1969) Pulmonary effects of
metallic dusts - nickel and iron. Toronto, University of Toronto,
School of Hygiene, Department of Physiological Hygiene, 24 pp.
KINCAID, J.F., STRONG, J.S. & SUNDERMAN, F.W. (1953) Nickel
poisoning. I. Experimental study of the effects of acute and
subacute exposure to nickel carbonyl. Arch. ind. Hyg. occup. Med.
8: 48-60.
KINCAID, J.F., STANLEY, E.L., BECKWORTH, C.H. & SUNDERMAN, F.W.
(1956) Nickel poisoning. III. Procedures for detection, prevention,
and treatment of nickel carbonyl exposure including a method for
the determination of nickel in biologic materials. Am. J. clin.
Pathol., 26: 107-119.
KING, M.M., LYNN, K.K. & HUANG, C.Y. (1985) Activation of the
calmodulin-dependant phosphoprotein phosphatase by nickel ions In:
Brown, S.S. & Sunderman, F.W. Jr, ed. Progress in nickel
toxicology. Proceedings of the 3rd International Conference on
Nickel Metabolism and Toxicology, Paris, 4-7 September, 1984,
Oxford, Blackwell Scientific Publications, pp. 117-122.
KIRCHGESSNER, M. & SCHNEGG, A. (1979) [Activity of proteases
leucinaryl-amidase and a-amylase in pancreas at nickel deficiency.]
Nutr. Metab., 23: 62-64 (in German).
KIRCHGESSNER, M. & SCHNEGG, A. (1980a) Biochemical and
physiological effects of nickel deficiency. In: Nriagu, J.O. ed.
Nickel in the environment, New York, Chichester, Brisbane, Toronto,
John Wiley and Sons, pp. 635-652.
KIRCHGESSNER, M. & SCHNEGG, A. (1980b) [Hydrocarbon metabolism in
nickel deficiency.] In: Anke, M., Schneider, H.-J. & Brûckner,
Chr. ed. [3. Trace Element Symposium: Nickel, Jena, German
Democratic Republic, 7-11 July, 1980,] Jena, Friedrich-Schiller
University, pp. 23-26 (in German).
KJELLSTROM, T., FRIBERG, A. & RAHNSTER, B. (1979) Mortality and
cancer morbidity among cadmium exposed workers. Environ. Health
Perspect., 28: 199- 204.
KLEIN, L.A., LANG, M., NASH, N. & KIRSCHNER, S.L. (1974) Sources
of metals in New York City wastewater. J. Water Pollut. Control
Fed., 46(12): 2653-2662.
KNIGHT, Y.A., REZUKE, W.N., WONG, S.H-Y., HOPFER, S.M., ZAHARIA, O. &
SUNDERMAN, F.W. Jr (1987) Acute thymic involution and increased
lipoperoxides in thymus of nickel chloride-treated rats. Res. Commun.
chem. Pathol. Pharmacol., 55: 291-302.
KODAMA, Y., TANAKA, I., MATSUNO, K., ISHIMATSU, S., HORIE, A. &
TSUCHIYA, K. (1985) Pulmonary deposition and clearance of inhaled
nickel oxide aerosol. In: Brown, S.S. & Sunderman, F.W. Jr, ed.
Progress in nickel toxicology, Proceedings of the 3rd International
Conference on Nickel Metabolism and Toxicology, Paris, 4-7 September
1984, Oxford, Blackwell Scientific Publications, pp. 81-84.
KOLLER, L.D. (1980) Immunotoxicology of heavy metals. Int. J.
Immunopharmacol., 2: 269-279.
KOLPAKOV, F.I. (1963) [Permeability of skin to nickel compounds.]
Arkh. Patol., 25(6): 38-45 (in Russian).
KOMCZYNSKI, L., NOWAK, H. & REJNIAK, L. (1963) Effect of cobalt,
nickel and iron on mitosis in the roots of the broad bean ( Vicia
faba). Nature (Lond.), 198: 1016-1017.
KOPONEN, M., GUSTAFSSON, T., KALLIOMAKI, P.L. & PYY, L. (1981)
Chromium and nickel aerosols in stainless steel manufacturing,
grinding and welding. Am. Ind. Hyg. Assoc. J., 42(8): 596-601.
KOVACS, P. & DARVAS, Z. (1982) Studies on the Ni content of the
centriole. Acta histochem., 71: 169-173.
KOWALCZYK, G.S., GORDON, G.E. & RHEINGROVER, S.W. (1982)
Identification of atmospheric particulate sources in Washington,
DC, using chemical element balances. Env. Sci. Technol., 16(2):
79-89.
KRISHNAN, E.R. & HELLWIG, G.V. (1982) Trace emissions from coal and
oil combustion. Environ. Prog., 1(4): 290-295.
KUCHARIN, G.M. (1970) [Occupational disorders of the nose and nasal
sinuses in workers in an electrolytic nickel refining plant.] Gig.
Tr. prof. Zabol., 14: 38-40 (in Russian).
KUEHN, K. & SUNDERMAN, F.W. Jr (1982) Dissolution half-times of
nickel compounds in water, rat serum, and renal cytosol. J. inorg.
Biochem., 17(1): 29-39.
KUEHN, K., FRASER, O.B. & SUNDERMAN, F.W. Jr (1982) Phagocytosis
of particulate nickel compounds by rat peritoneal macrophage in
vitro. Carcinogenesis, 3(3): 321-326.
KUROKAWA, Y., MATSUSHIMA, M., IMAZAWA, T., TAKAMURA, N., TAKAHASHI,
M. & HAYASHI, Y. (1985) Promoting effect of metal compounds on rat
renal tumorigenesis. J. Am. Coll. Toxicol., 4: 321-330.
LABELLA, F., DULAR, R., LEMON, P., VIVIAN, S. & QUEEN, G. (1973a)
Prolactin secretion is specifically inhibited by nickel. Nature
(Lond.), 245: 331-332.
LABELLA, F., DULAR, R., VIVIAN, S. & QUEEN, G. (1973b) Pituitary
hormone releasing or inhibiting activity of metal ions present in
hypothalamic extracts. Biochem. Biophys. Res. Commun., 52: 786-791.
LANGARD, S. & STERN, R.M. (1984) Nickel in welding fumes - A cancer
hazard to welders? A review of epidemiological studies on cancer in
welders. In: Nickel in the human environment, Proceedings of a
Joint Symposium, Lyon, 8-11 March, 1983, Lyon, International Agency
for Research on Cancer, pp. 95-103 (IARC Scientific Publications
No. 53).
LARRAMENDY, M.L., POPESCU, N.C. & DIPAOLO, J.A. (1981) Induction
by inorganic metal salts of sister chromatid exchanges and
chromosome aberrations in human and Syrian hamster cell strains.
Environ. Mutagen., 3: 597-606.
LAU, T.J., HACKETT, R.L. & SUNDERMAN, F.W. Jr (1972) The
carcinogenicity of intravenous nickel carbonyl in rats. Cancer
Res., 32: 2253-2258.
LAVELLE, J.M. & WITMER, C.M. (1981) Mutagenicity of NiCl2 and the
analysis of mutagenicity of metal ions in a bacterial fluctuation
test. Environ. Mutagen., 3: 320-321.
LAZAREVA, L.P. (1985) Changes in biological characteristics of
Daphnia magna from chronic action of copper and nickel at low
concentrations. Gidrobiol. Zh., 21: 59-62.
LEACH, C.A. Jr & SUNDERMAN, F.W. Jr (1987) Hypernickelemia
following coronary arteriography, caused by nickel in the
radiographic contrast medium. Ann. clin. lab. Sci., 17(3):
137-143.
LEACH, C.A. Jr, LINDEN, J.V., HOPFER, S.M., CRISOSTOMO, M.C. &
SUNDERMAN, F.W. Jr (1985) Nickel concentrations in serum of
patients with acute myocardial infarction or unstable angina
pectoris. Clin. Chem., 31(4): 556-560.
LECHNER, J.F., TOKIWA, T., MCCLENDON, I.A. & HAUGEN, A. (1984)
Effects of nickel sulfate on growth and differentiation of normal
human bronchial epithelial cells. Carcinogenesis, 5: 1697-1703.
LEE, R.E. & VON LEHMDEN, D.J. (1973) Trace metal pollution in the
environment. J. Air. Pollut. Control Assoc., 23: 853-857.
LESLIE, A.C.D., WINCHESTER, J.W., LEYSIEFFER, F.W. & AHLBERG, M.S.
(1976) Prediction of health effects of pollution aerosols. Trace
Subst. environ. Health, 10: 497-504.
LESSARD, R., REED, D., MAHEUX, B. & LAMBERT, J. (1978) Lung cancer
in New Caledonia, a nickel smelting island. J. occup. Med., 20(12):
815-817.
LESTROVOI, A.P., ITSKOVA, A.I. & ELISEEV, I.N. (1974) [Effect of
nickel on the iodine fixation of the thyroid gland when
administered perorally and by inhalation.] Gig. i Sanit., 10: 105-106
(in Russian).
LEVAN, A. (1945) Cytological reactions induced by inorganic salt
solutions. Nature (Lond.), 3973: 751-751.
LEWIS, C.L. & OTT, W.L. (1970) Analytical chemistry of nickel,
Oxford, New York, Pergamon Press, 232 pp.
LICHTY, P.D. & ZEY, J.N. (1985) Health Hazard Evaluation Report:
General Motors Corporation, Dayton, Ohio. Cincinnati, Ohio,
National Institute for Occupational Safety and Health, 37 pp (HETA
84-060-1645).
LIGETI, L., RUBANYI, G., KOLLER, A. & KOVACH, A.G.B. (1980)
[Effect of nickel ions on hemodynamics, cardiac performance and
coronary blood flow in anaesthetized dogs.] In: Anke, M.,
Schneider, H.-J. & Brückner, Chr. ed. [3. Trace Element Symposium
Nickel, Jena, German Democratic Republic, 7-11 July, 1980], Jena,
Friedrich-Schiller University, pp. 117-122 (in German).
LINDEN, J.V., HOPFER, S.M., GOSSLING, H.R. & SUNDERMAN, F.W. Jr
(1985) Blood nickel concentrations in patients with stainless-steel
hip prostheses. Ann. clin. lab. Sci., 15(6): 459-464.
LIU, Y. ET AL. (1983) [The role of nickel sulfate in inducing
nasoharyngeal carcinoma (NPC) in rats]. Guangzhou, Cancer Institute
of Zhongshan Medical College, WHO Collaborating Centre for Research
on Cancer, pp. 48-49 (Cancer Research Reports, Vol. 4)(in Chinese
with English abstract).
LLOYD, G.K. (1980) Dermal absorption and conjugation of nickel in
relation to the induction of allergic contact dermatitis -
preliminary results. In: Brown, S.S. & Sunderman, F.W. ed. Nickel
toxicology, Proceedings of the 2nd International Conference on
Nickel Toxicology, Swansea, 3-5 September, 1980, London, New York,
Academic Press, pp. 145-148.
LOKEN, A.C. (1950) [Carcinoma of the lung in nickel workers.]
Tidskr. Nor. Laegeforen., 70: 376-378 (in Norwegian).
LONG-ZHU, J. & ZHE-MING, N. (1985) Determination of nickel in urine
and other biological samples by graphite furnace atomic absorption
spectrometry. Fresenius Z. anal. Chem., 321: 72-76.
LOW, K.S. & LEE, C.K. (1981) Copper, zinc, nickel and chromium
uptake by "Kangkong Air" ( Ipomea acquatica Forsk). Pertanika,
4(1):16-20.
LU, C.C., MATSUMOTO, N. & IIJIMA, S. (1979) Teratogenic effects of
nickel chloride on embryonic mice and its transfer to embryonic
mice. Teratology, 19(2): 137-142.
LU, C.C., MATSUMOTO, N. & IIJIMA, S. (1981) Placental transfer and
body distribution of nickel chloride in pregnant mice. Toxicol.
appl. Pharmacol., 59(3): 409-413.
LUCASSEN, M. & SARKAR, B. (1979) Nickel(II)-binding constituents of
human blood serum. J. Toxicol. environ. Health, 5(5): 897-905.
LUDEWIGS, H.-J. & THIESS, A.M. (1970) [Knowledge in occupational
medicine of nickel carbonyl poisoning.] Zentralbl. Arbeitsmed.
Arbeitsschutz, 20: 329-339 (in German).
LUMB, G.D., SUNDERMAN, F.W. Jr, & SCHNEIDER, H.P. (1985) Nickel-
induced malignant tumors. Ann. clin. lab. Sci., 15(5): 374-380.
LUNDBORG, M. & CAMNER, P. (1982) Decreased level of lysozyme in
rabbit lung lavage fluid after inhalation of low nickel
concentrations. Toxicology, 22(4): 353-358.
LUNDBORG, M. & CAMNER, P. (1984) Lysozyme levels in rabbit lung
after inhalation of nickel, cadmium, cobalt, and copper chlorides.
Environ. Res., 34: 335-342.
MACHELETT, B. & PODLESAK, W. (1980) [Nickel uptake by oat and
mustard plants as a function of lime status of the soil.] In: Anke,
M. Schneider, H.J. & Brûckner, Chr. Jr ed. [3. Trace Element
Symposium Nickel, Jena, German Democratic Republic, 7-11 July 1980,]
Jena, Friedrich-Schiller University, pp. 207- 213 (in German).
MACKENZIE PEERS, A. (1986) Determination of arsenic, chromium,
nickel, cadmium, lead, beryllium and selenium in air and airborne
particulates by inductively coupled argon atomic emission
spectroscopy. NIOSH 7300 adapted by Mackenzie Peers, A. In:
Environmental carcinogens - selected methods of analysis. Vol. 8:
Some metals: As, Be, Cd, Cr, Ni, Pb, Se, Zn, Lyon, International
Agency for Research on Cancer, pp. 225-230 (IARC Scientific
Publications No. 71).
MCCONNELL, L.H., FINK, J.N., SCHLUETER, D.P. & SCHMIDT, M.G. Jr
(1973) Asthma caused by nickel sensitivity. Ann. intern. Med.
78(6): 888-890.
MCDONALD, I. (1981) Malignant lymphoma associated with internal
fixation of a fractured tibia. Cancer, 48: 1009-1011.
MCDOUGALL, A. (1956) Malignant tumor at site of bone plating. J.
bone joint Surg. B,. 38: 709-713.
MCGREGOR, D.B., BROWN, A., CATTANACH, P., EDWARDS, I., McBRIDE, D.
RIACH, C. & CASPARY, W.J. (1988) Responses of the L5178Y tK+/tK-
mouse lymphoma cell forward mutation assay: III. 72 coded
chemicals. Environ. molec. Mutagen., 12: 85-154.
MCLAREN, J.W., MYKYTIUK, A.P., WILLIE, S.N. & BERMAN, S.S. (1985)
Determination of trace metals in seawater by inductively coupled
plasma mass spectrometry with preconcentration on silica-
immobilized 8-hydroxyquinoline. Anal. Chem., 57: 2907-2911.
MCLEAN, J.R., MCWILLIAMS, R.S., KAPLAN, J.G. & BIRNBOIM, H.C.
(1982) Rapid detection of DNA strand breaks in human peripheral
blood cells and animal organs following treatment with physical and
chemical agents. In: Bora, K.C., Douglas, G.R. & Nestmann, E.R.
ed. Chemical mutagenesis, human population monitoring and genetic
risk assessment. Amsterdam, Oxford, New York, Elsevier Science
Publishers, pp. 137-141 (Progress in Mutation Research, Vol. 3).
McNEELY, M.D., SUNDERMAN, F.W. Jr, NECHAY, M.W. & LEVINE, H.
(1971a) Abnormal concentrations of nickel in serum in cases of
myocardial infarction, stroke, burns, hepatic cirrhosis, and
uremia. Clin. Chem., 17(11): 1123-1128.
MCNEELY, M.D., NECHAY, M.W. & SUNDERMAN, F.W. Jr (1971b)
Measurement of nickel in serum and urine as indices of
environmental exposure to nickel. Clin. Chem., 18: 992-995.
MAENZA, R.M., PRADHAN, A.M. & SUNDERMAN, F.W. Jr (1971) Rapid
induction of sarcomas in rats by combination of nickel sulfide and
3,4-benzypyrene. Cancer Res., 31: 2067-2071.
MAGNUS, K., ANDERSEN, A. & HOGETVEIT, A.C. (1982) Cancer of
respiratory organs among workers at a nickel refinery in Norway.
Int. J. Cancer, 30(6): 681-685.
MAGNUSSON, B. & KLIGMAN, A.M. (1970) Allergic contact dermatitis in
the guinea pig. Springfield, Illinois, Charles C. Thomas, 141 pp.
MALO, J.L., CARTIER, A., DOEPNER, M., NIEBOER, E., EVANS, S. &
DOLOVICH, J. (1982) Occupational asthma caused by nickel sulfate.
J. Allergy clin. Immunol., 69: 55-59.
MAREK, M. & TREHARNE, R.W. (1982) An in vitro study of the release
of nickel from two surgical implant alloys. Clin. Orthop., 167: 291-
295.
MARZOUK, A. & SUNDERMAN, F.W. Jr (1985) Biliary excretion of
nickel. Toxicol. Lett., 27: 65-71.
MAS, A., HOLT, D. & WEBB, M.C. (1985) The acute toxicology and
teratogenicity of nickel in pregnant rats. Toxicology, 35: 47-57.
MASON, B. (1952) Principles of geochemistry, New York, John Wiley &
Sons, London, Chapman & Hall, Limited, 276 pp.
MASTROMATTEO, E. (1967) Yant memorial lecture: Nickel: A review of
its occupational health aspects. J. occup. Med., 9(3): 127-136.
MASTROMATTEO, E. (1986) Nickel. Am Ind. Hyg. Assoc. J.
47(10): 589-601.
MATHIS, B.J. & CUMMINGS, T.F. (1973) Selected metals in sediments,
water, and biota in the Illionois River. J. Water Pollut. Control
Fed., 45(7):1573- 1583.
MATHUR, A.K., DATTA, K.K. TANDON, S.K. & DIKSHITH, T.S. (1977)
Effect of nickel sulphate on male rats. Bull. environ. Contam.
Toxicol., 17: 241-247.
MATHUR, A.K., DIKSHITH, T.S., LAL, M.M. & TANDON, S.K. (1978)
Distribution of nickel and cytogenetic changes in poisoned rats.
Toxicology, 10(2): 105-113.
MEDINSKY, M.A., BENSON, J.M. & HOBBS, C.H. (1987) Lung clearance
and disposition of the 63Ni in F344/N rats after intratracheal
instillation of nickel sulfate solutions. Environ. Res., 43: 168-178.
MEDVEDEVA, V.J. (1965) [Nickel content in blood of a new born baby
and in venous and retroplacental blood and milk of the mother.]
Vest. Akad. Navuk. Belarusk. SSR. Ser. biyal Navuk, 2: 114-115 (in
Russian).
MENDEN, E.E., ELIA, V.J., MICHAEL, L.W. & PETERING, H.G. (1972)
Distribution of cadmium and nickel of tobacco during cigarette
smoking. Environ. Sci. Technol., 6: 830-832.
MENNE, T., BORGAN, O. & GREEN, A. (1982) Nickel allergy and hand
dermatitis in a stratified sample of the Danish female population:
an epidemiological study including a statistic appendix. Acta
dermatovenereol., 62(1): 35-41.
MENNE, T., ANDERSEN, K.E., KAABER, K., OSMUNDSEN, P.E., ANDERSEN,
J.R., YOING, F. & VALEUR, G. (1987) Evaluation of the
dimethylglyoxime stick test for the detection of nickel. Derm.
Beruf Umwelt, 35: 128-130.
MERCER, T.T. (1967) On the role of particle size in the dissolution
of lung burdens. Health Phys., 13: 1211-1221.
MERTZ, W. (1981) The essential trace elements. Science, 213:
1332-1338.
MEYER, A. & NEEB, R. (1985) Determination of cobalt and nickel in
some biological matrices - comparison of chelate gas-chromatography
and adsorption voltammetry. Fresenius Z. anal. Chem., 321(3):
235-241.
MIKAC-DEVIC, D. SUNDERMAN, F.W. Jr, & NOMOTO, S. (1977)
Furildioxime method for nickel analysis in serum and urine by
electrothermal atomic absorption spectrometry. Clin. Chem., 23(6):
948-956.
MIKHEYEV, M.J. (1971) [Distribution and elimination of nickel
carbonyl.] Gig. Tr. prof. Zabol., 15: 35-38 (in Russian).
MIKI, H., KASPRZAK, K.S., KENNEY, S. & HEINE, U.K. (1987)
Inhibition of intercellular communication by nickel(II):
antagonistic effect of magnesium. Carcinogenesis, 8(11):
1757-1760.
MILFORD, J.B. & DAVIDSON, C.I. (1985) The sizes of particulate
trace elements in the atmosphere - a review. J. air Pollut., 35(12):
1249-1260.
MITSUMASA, O. (1987) Induction of ocular tumor by nickel subsulfide
in the Japanese common newt, Cynops pyrrhogaster. Cancer Res., 47:
5213-5217.
MIYAKI, M., AKAMATSU, N., ONO, T. & KOYAMA, H. (1979) Mutagenicity
of metal cations in cultured cells from Chinese hamsters. Mutat.
Res., 68: 259-263.
MOELLER, H. (1984) Attempts to induce contact allergy to nickel in
the mouse. Contact Dermatit., 10: 65-68.
MOFFA, I.P., ELLISON, J.E. & HAMILTON, J.C. (1983) Incidence of
nickel sensitivity in dental patients. J. dent. Res., 62: 199.
MORGAN, J.G.A. (1958) Some observations on the incidence of
respiratory cancer in nickel workers. Br. J. ind. Med., 15: 224-234.
MORGAN, J.G.A. (1960) A simplified method for the estimation of
nickel in urine. Br. J. ind. Med., 17: 209-212.
MORGAN, L.G. & ROUGE, P.J. (1979) A study into the correlation
between atmospheric and biological monitoring of nickel in nickel
refinery workers. Ann. occup. Hyg., 22(3): 311-317.
MORITA, H., NODA, K. & UMEDA (1985) Mutagenicities of nickel and
cobalt compounds in a mammalian cell line. Mutat. Res., 147: 265-266.
MORROW, P.E. (1970) Models for the study of particle retention and
elimination in the lung. Inhalation carcinogenesis, Oak Ridge,
Tennessee, US Atomic Energy Commission, Division of Technical
Information, pp. 103-116 (AEC Symposium Series, No. 18).
MORSE, E.E., LEE, T.Y., REISS, R.F. & SUNDERMAN, F.W. Jr (1977)
Dose-response and time-response study of erythrocytosis in rats
after intrarenal injection of nickel subsulfide. Ann. clin. lab.
Sci., 7: 17-24.
MORTIMER, D.C. (1985) Freshwater aquatic macrophytes as heavy metal
monitors the Ottawa river experience Canada. Environ. monit.
Assess., 5(3): 311-324.
MUHLE, H., BELLMANN, B. & TAKENAKA, S. (1988) Chronic effects of
intratracheally instilled nickel containing particles in hamsters.
In: Fourth International Conference on Nickel Metabolism and
Toxicology: Abstracts, Espoo, Finland, 5-9 September 1988,
Helsinki, Institute of Occupational Health, p. 41.
MUKUBO, K. (1978) Studies on experimental lung tumor by the
chemical carcinogens and inorganic substances. III.
Histopathological studies on lung tumor induced by pertracheal
vinyl tube infusion of 20-methylcholanthrene combined with chromium
and nickel powder. J. Nara Med. Ass., 29: 321-340.
MURTHY, R.C., BARKLEY, W., HOLLINGSWORTH, C. & BINGHAM, E. (1983)
Enzymatic changes in alveolar macrophages of rats exposed to lead
and nickel by inhalation. J. Am. Coll. Toxicol., 2: 193-199.
MYRON, D.R., ZIMMERMAN, T.J., SHULER, T.R., KLEVAY L.M., LEE, D.E.
& NIELSEN, F.H. (1978) Intake of nickel and vanadium by humans. A
survey of selected diets. Am. J. clin. Nutr., 31(3): 527-531.
NADEENKO, V.G., LENCHENKO, V.G., ARKHIPENKO, T.A., SAICHENKO, S.P.
& PETROVA, N.N. (1979) [Embryotoxic effect of nickel entering the
body via drinking water.] Gig. i Sanit., 6: 86-88 (in Russian).
NADEENKO, V.G., LECHENKO, V.G. & SAICHENKO, S.P. (2988) [Combined
action of nickel and cobalt administered with drinking water.] In:
Domnin, S.G. & Tartakovskaya, L.Ya. ed. [Combined effects of
physical and chemical factors of industrial environment], Moscow,
Erisman Institute of Hygiene, pp. 84-90 (in Russian).
NAS (1975) Nickel. Washington, DC, National Academy of Sciences,
277 pp.
NATUSCH, D.F.S., WALLACE, J.R. & EVANS, C.A. (1974) Toxic trace
elements: preferential concentration in respirable particles.
Science, 183: 202-204.
NEBEKER, A.V., STINCHFIELD, A., SAVONEN, C. & CHAPMAN, G.A. (1986)
Effects of copper, nickel and zinc on three species of Oregon
freshwater snails. Environ. Toxicol. Chem., 5(9): 807-811.
NECHAY, M.W. & SUNDERMAN, F.W. Jr (1973) Measurements of nickel in
hair by atomic absorption spectrometry. Ann. clin. lab. Sci., 3(1):
30-35.
NEUHAUSER, E.F., LOEHR, R.C. & MALECKI, M.R. (1985) Contact and
artificial soil tests using earthworms to evaluate the impact of
wastes in soil. In: Petros, J.K., Lacy, W.J. & Conway, R.A. ed.
Hazardous and industrial solid waste testing: Fourth Symposium,
Philadelphia, American Society for Testing and Materials, pp. 192-
203 (ASTM STP 886).
NEUMULLER, O.A. (1985) Römpp's chemical encyclopaedia, 8th ed.
Stuttgart, Franck'sche Verlagsbuchhandlung, Vol. 4, 3230 pp.
NEWMAN, S.M., SUMMIT, R.L. & NUNEZ, L. J. (1982) Incidence of
nickel-induced sister chromatid exchange. Mutat. Res., 101: 67-75.
NIEBOER, E. & JUSYS, A.A. (1983) Contamination control in routine
ultratrace analysis of toxic metals. In: Brown, S.S. & Savory, J.
ed. Chemical toxicology and clinical chemistry of metals,
Proceedings of the 2nd International Conference, Montreal, 19-22
July, 1983, London, New York, Blackwell Scientific Publications,
pp. 3-16.
NIEBOER, E., MAXWELL, R.J., ROSSETTO, F.E., STAFFORD, A.R. &
STETSKO, P.J. (1986) Concepts in nickel carcinogenesis. In: Xavier,
A.V. ed. Frontiers of bioinorganic chemistry. 2nd ed. Weinheim,
VCH Verlag, pp. 142-151.
NIELSEN, F.H. (1971) Studies on the essentiality of nickel. In:
Mertz, W. & Cornatzer, W.E. ed. New trace elements in nutrition,
New York, Basel, Marcel Dekker, pp. 215-253.
NIELSEN, F.H. (1974) Essentiality and function of nickel. In:
Hoekstra, W.G., Suttie, J.W., Ganther, H.E. & Mertz, W. ed. Trace
element metabolism in animals. Proceedings of the 2nd International
Symposium on Trace Element Metabolism in Animals, Madison,
Wisconsin, 18-22 June, 1973, Baltimore, Maryland, University Park
Press, pp. 381-395.
NIELSEN, F.H. (1984) Ultratrace elements in nutrition. Annu. Rev.
Nutr., 4: 21-41.
NIELSEN, F.H. & HIGGS, D.J. (1971) Further studies involving a
nickel deficiency in chicks. Proc. trace Subst. environ. Health, 4:
241-246.
NIELSEN, F.H. & OLLERICH, D.A. (1974) Nickel: a new essential trace
element. Fed. Proc., 33: 1767-1772.
NIELSEN, F.H. & SAUBERLICH, H.E. (1970) Evidence of a possible
requirement for nickel by the chick. Proc. Soc. Exp. Biol. Med.
134(3): 845-849.
NIELSEN, F.H. & SHULER, T.R. (1979) Effect of dietary nickel and
iron on the trace element content of rat liver. Biol. trace elem.
Res., 1: 337-346.
NIELSEN, F.H. & SHULER, T.R. (1981) Effect of form of iron on
nickel deprivation in the rat. Liver content of copper, manganese
and zinc. Biol. trace elem. Res., 3: 245-256.
NIELSEN, F.H., OLLERICH, D.A., FOSMIRE, G.J. & SANDSTEAD, H.H.
(1974) Nickel deficiency in chicks and rats: effects on liver
morphology, function and polysomal integrity. Adv. exp. Med. Biol.
48: 389-403.
NIELSEN, F.H., MYRON, D.R., GIVAND, S.H. & OLLERICH, D. A. (1975a)
Nickel deficiency and nickel-rhodium interaction in chicks. J.
Nutr., 105: 1607-1619.
NIELSEN, F.H., MYRON, O.R., GIVAND, S.H., ZIMMRMANN, T.J. &
OLLERICH, D.A. (1975b) Nickel deficiency in rats. J. Nutr., 105: 1620-
1630.
NIELSEN, F.H., SHULER, T.R., ZIMMERMANN, T.J., COLLINGS, M.E. &
UTHUS, E.O. (1979a) Interaction between nickel and iron in the rat.
Biol. trace elem. Res., 1: 325-335.
NIELSEN, F.H., ZIMMERMAN, T.J., COLLINGS, M.E. & MYRON, D.R.
(1979b) Nickel deprivation in rats: Nickel-iron interactions. J.
Nutr., 109(9): 1623-1632.
NIELSEN, G.D. (in press) Oral challenge of nickel-allergic patients
with hand eczema. In: Nieboer, E. & Aitio, A. ed. Nickel and human
health: Current perspectives. New York, Chichester, Brisbane,
Toronto, John Wiley & Sons (Advances in environmental science and
technology Series).
NIELSEN, G.D. & FLYVHOLM, M. (1984) Risks of high nickel intake
with diet. In: Nickel in the human environment. Proceedings of a
Joint Symposium, Lyon, 8-11 March 1983, Lyon, International Agency
for Research on Cancer, pp. 333-338 (IARC Scientific Publications
No. 53).
NIELSEN, G.D., JORGENSEN, P.J., KEIDING, K. & GRANDJEAN, P.
(1987a) Urinary nickel excretion before and after loading with
naturally occurring nickel. In: Trace elements in human health and
disease. Second Nordic Symposium, Odense, Denmark, 17-21 August
1987: Symposium Abstracts, Odense, Odense University (Abstract C3).
NIELSEN, G.D., ENDERSEN, O. & GRANDJEAN, P. (1987b) Effect of
diethyldithiocarbamate on toxicokinetics of 57Ni in mice. In: Trace
elements in human health and disease. Extended abstracts from the
second Nordic Symposium, Odense, 17-21 August 1987, Copenhagen,
World Health Organization, Regional Office for Europe, pp. 78-81
(Environmental Health Series No. 20).
NILZEN, A. & WIKSTROM, K. (1955) The influence of lauryl sulphate
on the sensitization of guinea-pigs to chrome and nickel. Acta
dermatovenereol., 35: 292-299.
NISHIMURA, M. & UMEDA, M. (1979) Induction of chromosomal
aberrations in cultured mammalian cells by nickel compounds.
Mutat. Res., 68: 337-349.
NISHIOKA, H. (1975) Mutagenic activities of metal compounds in
bacteria. Mutat. Res., 31: 185-189.
NODIYA, P.J. (1972) [Cobalt and nickel balance in students of an
occupational technical school.] Gig. i Sanit., 37: 108-109 (in Russian).
NOMOTO, S. & SUNDERMAN, F.W. (1970) Atomic absorption spectrometry
of nickel in serum, urine, and other biological materials. Clin.
Chem., 16: 477-485.
NOMOTO, S. & SUNDERMAN, F.W. Jr (1988) Presence of nickel in alpha-
2-macroglobulin isolated from human serum by high performance
liguid chromatography. Ann. clin. lab. Sci., 18: 78-84.
NOMOTO, S., HIRABAYASHI, T. & FUKUDA, T. (1983) Serum nickel
concentrations in women during pregnancy, parturition, and post-
partum. In: Brown, S.S. & Savory, J. ed. Chemical toxicology and
clinical chemistry of metals, Proceedings of the 2nd International
Conference, Montreal, 19-22 July, 1983, London, New York, Blackwell
Scientific Publications, pp. 351-352.
NOMOTO, S., MCNEELY, M.D. & SUNDERMAN, F.W. (1971) Isolation of a
nickel a2-macroglobulin from rabbit serum. Biochemistry, 10(9):
1647-1651.
NORGAARD, O. (1955) Investigations with radioactive Ni57 into the
resorption of nickel through the skin in normal and in nickel-
hypersensitive persons. Acta dermtovenereol., 35: 111-117.
NORGAARD, O. (1957) Investigations with radioactive nickel, cobalt
and sodium on the resorption through the skin in rabbits, guinea-
pigs and man. Acta dermatovenereol., 37: 440-445.
NORSETH, T. (1975) Urinary excretion of nickel as an index of
nickel exposure in welders and nickel refinery workers. Int. Congr.
Occup. Health, 18: 327.
NOVEY, H.S., HABIB, M. & WELLS, I.D. (1983) Asthma and IgE
antibodies induced by chromium and nickel salts. J. Allergy clin.
Immunol., 72: 407-412
NOZDRYUKHINA, L.R. (1978) Use of blood trace elements for diagnosis
of heart and liver disease. In: Kirchgessner, M. ed. Trace element
metabolism in man and animals. Proceedings of the 3rd International
Symposium, Freising, Federal Republic of Germany, July 1977,
Freising-Weihenstephan, Technical University Munich, pp. 336-339.
NRIAGU, J.O. (1979) Global inventory of natural and anthropogenic
emissions of trace metals to the atmosphere. Nature (Lond.),
279(5712): 409-411.
NRIAGU, J.O. (1980) Global cycle and properties of nickel. In:
Nriagu, J.O. ed. Nickel in the environment, New York, Chichester,
Brisbane, Toronto, John Wiley and Sons, pp. 1-26.
OAKLEY, A.M.M., IVE, F.A. & CARR, M.M.C. (1987) Skin clips are
contraindicated when there is nickel allergy. J. R. Soc. Med., 80:
290-291.
O'DELL, G.D., MILLER, W.J., MOORE, S.L., KING, W.A., ELLERS, J.C.
& JURECEK, H. (1971) Effect of dietary nickel level on excretion
and nickel content of tissues in male calves. J. anim. Sci., 32(4):
769-773.
OGAWA, H.I., TSURUTA, S., NIYITANI, Y., MINO, H., SAKATA, K. &
KATO, Y. (1987) Mutagenesis of metal salts in combination with 9-
amonoacridine in Salmonella typhimurium. Jpn. J. Genet., 62: 159.
OHNO, H., HANAOKA, F. & YAMADA, M.A. (1982) Inducibility of
sister-chromatid exchanges by heavy-metal ions. Mutat. Res., 104:
141-145.
OLEJAR, S., OLEJAROVA, E. & VRABEL, K. (1982) [Lung tumors in
workers in a nickel refinery.] Prac. Lek., 34(8): 80-282 (in Czech).
OLSEN, I. & JONSEN, J. (1979a) Whole-body autoradiography of 63Ni
in mice throughout gestation. Toxicology, 12(2): 165-172.
OLSEN, I. & JONSEN, J. (1979b) Effect of cadmium acetate, copper
sulfate and nickel chloride on organ cultures of mouse trachea.
Acta pharmacol. toxicol., 44: 120-127.
OLSEN, J. & SABROE, S. (1984) Occupational causes of laryngeal
cancer. J. Epidemiol. community Health, 38(2): 117-121.
ONKELINX, C. & SUNDERMAN, F.W. (1980) Modeling of nickel
metabolism. In: Nriagu, J.O. ed. Nickel in the environment, New
York, Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 525-
545.
ONKELINX, C., BECKER, J. & SUNDERMAN, F.W. (1973) Compartmental
analysis of the metabolism of 63Ni(II) in rats and rabbits. Res.
Commun. chem. Pathol. Pharmacol., 6(2): 663-676.
OSKARSSON, A. & TJALVE, H. (1979a) The distribution and metabolism
of nickel carbonyl in mice. Br. J. ind. Med., 36(4): 326-335.
OSKARSSON, A. & TJALVE, H. (1979b) An autoradiographic study on the
distribution of 63NiCl2 in mice. Ann. clin. lab. Sci., 9(1):
47-59.
OSKARSSON, A., ANDERSSON, Y. & TJALVE, H. (1979) Fate of nickel
subsulfide during carcinogenesis studied by autoradiography and X-
ray powder diffraction. Cancer Res., 39(10): 4175-4182.
OSTAPCZUK, P., VALENTA, P., STOEPPLER, M. & NURNBERG, H.W. (1983)
Voltammetric determination of nickel and cobalt in body fluids and
other biological materials. In: Brown, S.S. & Savory, J. ed.
Chemical toxicology and clinical chemistry of metals, Proceedings
of the 2nd International Conference, Montreal, 19-22 July, 1983;
London, New York, Blackwell Scientific Publications, pp. 61-64.
OSTAPCZUK, P., FRONING, M., STOEPPLER, M. & NUERNBERG, H.W.
(1985a) Square wave voltammetry: A new approach for the sensitive
determination of nickel and cobalt in human samples. In: Brown,
S.S. & Sunderman, F.W. Jr ed. Progress in Nickel toxicology.
Proceedings of the 3rd International Conference on Nickel
Metabolism and Toxicology, Paris, 4-7 September, 1984, Oxford,
Blackwell Scientific Publications, pp. 129-132.
OSTAPCZUK, P., FRONING, M., STOEPPLER, M. & NURNBERG, H.W. (1985b)
[Determination of heavy metals in environmental samples and biotic
material by means of square wave voltammetry.] Fresenius Z. anal.
Chem., 320: 645 pp. (in German).
OSTERREICHISCHES FORSCHUNGSZENTRUM SEIBERSDORF GMBH (1983)
[Assessment of the acute toxicity of nickel sulfate to rainbow
trout.] Vienna, Austrian Research Centre, 21 pp. (Report No. 4213,
BL-424/83) (in German).
OTTOLENGHI, A.D., HASEMAN, J.K., PAYNE, W.W., FALK, H.L. &
MACFARLAND, H.N. (1974) Inhalation studies of nickel sulfide in
pulmonary carcinogenesis of rats. J. Natl Cancer Inst., 54(5):
1165-1172.
OU, B., LIU, Y., HUANG, X. & FENG, G. (1981) [The promoting action
of nickel in the induction of nasopharyngeal carcinoma in rats.]
Guangzhou, Cancer Institute of Zhongshan Medical College, WHO
Collaborating Centre for Research on Cancer, pp. 3-8 (Cancer
Research Reports, Vol. 2) (in Chinese with English abstract).
OU, B., LIU, Y. & SHENG, G. (1983) [Tumor induction in next
generation of dinitrosopiperazine-treated pregnant rats.]
Guangzhou, Cancer Institute of Zgongshan Medical College, WHO
Collaborating Centre for Research on Cancer pp. 44-45, (Cancer
Research Reports, Vol. 4) (in Chinese with English abstract).
PACYNA, J.M., OTTAR, B., TOMZA, U. & MAENHAUT, W. (1985) Long-
range transport of trace elements to Ny Alesund, Spitsbergen.
Atmos. Environ. 19(16): 857-865.
PAINTER, R.B. & HOWARD, R. (1982) The HeLa DNA-synthesis inhibition
test as a rapid screen for mutagenic carcinogens. Mutat. Res., 92:
427-437.
PALO, J. & SAVOLAINEN, H. (1973) Biochemical diagnosis of aspartyl-
glycosaminuria. Ann. clin. Res., 5: 156-162.
PARKER, D. & TURK, J.L. (1978) Delay in the development of the
allergic response to metals following intratracheal instillation.
Int. Arch. Allergy appl. Immunol., 57: 289-293.
PARKER, G.H. (1985) Copper, nickel, and iron in plumage of three
upland gamebird species from non-contaminated environments. Bull.
environ. Contam. Toxicol., 35(6): 776-780.
PARKER, K. & SUNDERMAN, F.W. Jr (1974) Distribution of 63Ni in
rabbit tissues following intravenous injection of 63NiCl2. Res.
Commun. chem. Pathol. Pharmacol., 7: 755-762.
PATIERNO, S.R. & COSTA, M. (1985) DNA-protein cross-links induced
by nickel compounds in intact cultured mammalian cells Chem.-biol.
Interact., 55: 75-91.
PATIERNO, S.R., SUGIYAMA, M., BASILIKON, J.P. & COSTA, M. (1985)
Preferential DNA-protein cross-linking by NiCl2 in magnesium-
insoluble regions of fractionated Chinese hamster ovary cell
chromatin. Cancer Res., 45(11): 5787-55794.
PATON, G.R. & ALLISON, A.C. (1972) Chromosome damage in human cell
cultures induced by metal salts. Mutat. Res., 16: 332-336.
PAYNE, W.W. (1964) Carcinogenicity of nickel compounds in
experimental animals. Proc. Am. Assoc. Cancer Res., 5: 50
(Abstract 197).
PEDERSEN, E., HOGETVEIT, A.C. & ANDERSEN, A. (1973) Cancer of
respiratory organs among workers at a nickel refinery in Norway.
Int. J. Cancer, 12(1): 32-41.
PELTONEN, L. (1979) Nickel sensitivity in the general population.
Contact Dermatit., 5(1): 27-32.
PENMAN, H.G. & RING, P.A. (1984) Osteosarcoma in association with
total hip replacement. J. bone joint Surg., B66: 632-634.
PENNINGTON, J.A.T. & JONES, J.W. (1987) Molybdenum, nickel, cobalt,
vanadium and strontium in total diets. J. Am. Diet. Assoc., 87(12):
1644-1650.
PETO, J. (1988) The international nickel speciation research
project. In: Fourth International Conference on Nickel Metabolism
and Toxicology: Abstracts, Espoo, Finland, 5-9 September 1988,
Helsinki, Institute of Occupational Health, p. 50.
PETO, J., CUCKLE, H., DOLL, R., HERMON, C. & MORGAN, L.G. (1984)
Respiratory cancer mortality of Welsh nickel refinery workers. In:
Nickel in the human environment. Proceedings of a Joint Symposium,
Lyon, 8-11 March, 1983, Lyon, International Agency for Research on
Cancer, pp. 37-46 (IARC Scientific Publications No. 53).
PHATAK, S.S. & PATWARDHAN, V.N. (1950) Toxicity of nickel. J. Sci.
ind. Res., 9B: 70-76.
PHATAK, S.S. & PATWARDHAN, V.N. (1952) Toxicity of nickel.
Accumulation of nickel in rats fed on nickel-containing diets and
its elimination. J. Sci. ind. Res., 11B: 173-176.
PHILLIPS, D.J.H. & MUTTARASIN, K. (1985) Trace metals in bivalve
molluscs from Thailand. Mar. environ. Res., 15(3): 215-234.
PICKERING, Q.H. (1974) Chronic toxicity of nickel to the fathead
minnow. J. Water Pollut. Control Fed., 46: 760-765.
PICKERING, Q.H. & HENDERSON, C. (1966) The acute toxicity of some
heavy metals to different species of warm water fishes. Air Water
Pollut. Int. J., 10: 453-463.
PIENTA, R.J., POILEY, J.A. & LEBHERZ, W.B. (1977) Morphological
transformation of early passage golden hamster embryo cells derived
from cryopreserved primary cultures as a reliable in vitro bioassay
for identifying diverse carcinogens. Int. J. Cancer, 19: 642-655.
PIHLAR, B., VALENTA, P. & NURNBERG, H.W. (1981) New high-performance
analytical procedure for the voltammetric determination of nickel
in routine analysis of waters, biological materials and food.
Fresenius Z. anal. Chem., 307: 337-346.
PIKALEK, P. & NECASEK, J. (1983) The mutagenic activity of nickel
in Corynebacterium sp. Folia microbiol., 28: 17-21.
POIRIER, L.A., THEISS, J.C., ARNOLD, L.J. & SHIMKIN, M.B. (1984)
Inhibition by magnesium and calcium acetates of lead subacetate-
and nickel acetate-induced lung tumors in strain A mice. Cancer
Res., 44: 1520-1522.
POLEDNAK, A.P. (1981) Mortality among welders, including a group
exposed to nickel oxides. Arch. environ. Health, 36(5): 235-242.
POLEMIO, M., SENESI, N. & BUFO, S.A. (1982) Soil contamination by
metals; a survey in industrial and rural areas of Southern Italy.
Sci. total Environ., 25: 71-80.
PORT, C.D., FENTERS, J.D., EHRLICH, R., COFFIN, D.L. & GARDNER, D.
(1975) Interaction of nickel oxide and influenza infection in the
hamster. Environ. Health Perspect., 10: 268.
POTT, F., ZIEM, U., REIFFER, F.-J., HUTH, F., ERNST, H. & MOHR, U.
(1987) Carcinogenicity studies on fibres, metal compounds, and some
other dusts in rats. Exp. Pathol., 32: 129-152.
POTT, F., RIPPE, M., ROLLER, M., ROSENBRUCH, M. & HUTH, F. (1988)
Carcinogenicity studies on nickel compounds and nickel alloys after
intraperitoneal injection in rats. In: Fourth International
Conference on Nickel Metabolism and Toxicology: Abstracts, Espoo,
Finland, 5-9 September 1988, Helsinki, Institute of Occupational
Health, p. 42.
POWLESLAND, C. & GEORGE, I.C. (1986) Acute and chronic toxicity of
nickel to larvae of Chironomus riparis (Meigen). Environ. Pollut.
42(1): 47-64.
PROKIPCAK, B. & ORMROD, D.P. (1986) Visible injury and growth
responses of tomato and soybean to combinations of nickel, copper
and ozone. Water air soil Pollut., 27(3-4): 329-340.
PRYSTOWSKY, S.D., ALLEN, A.M., SMITH, R.W., NONOMURA, J.H., ODOM,
R.B. & AKERS, W.A. (1979) Allergic contact hypersensivity to
nickel, neomycin, ethylenediamine, and benzocaine. Relationships
between age, sex, history of exposure, and reactivity to standard
patch tests and use tests in a general population. Arch. Dermatol.
115(8): 959-965.
PUCHYR, R. & SHAPIRO, R. (1986) Determination of trace elements in
food by HCl-HNO3 leaching and flame atomic absorption spectroscopy.
J. Assoc. Off. Anal. Chem., 69(5): 868-870.
RADIAN CORPORATION (1984) Locating and estimating emissions from
sources of nickel. Research Triangle Park, North Carolina, US
Environmental Protection Agency, Office of Air Quality Planning and
Standards, 188 pp (EPA-450/4-84-007F).
RAHKONEN, E., JUNTTILA, M.-L., KALLIOMAKI, P.-L., OLKINUORA, M.,
KOPONEN, M. & KALLIOMAKI, K. (1983) Evaluation of biological
monitoring among stainless steel welders. Int. Arch. occup.
environ. Health, 52: 243-255.
RAITHEL, H.J. (1987) [Research report nickel. Investigations on
occupational nickel exposure and strain-situations of 837 persons.]
Sankt Augustin, Federation of Industrial Cooperative Societies, 199
pp (in German).
RAITHEL, H.J., MAYER, P., SCHALLER, K.H., MOHRMANN, W., WELTLE, D.
& VALENTIN, H. (1981) [Exposure to nickel in glass industry
workers. I. analysis and quantification of external and internal
nickel load.] Zentralbl. Arbeitsmed. Arbeitsschutz prophyl.
Ergonomie, 31(8): 332-339 (in German).
RANTA, W.B., TOMASSINI, F.D. & NIEBOER, E. (1978) Elevation of
copper and nickel levels in primaries from black and mallard ducks
collected in the Sudbury district, Ontario. Can. J. Zool., 56(4):
581-586.
RASMUSON, A. (1985) Mutagenic effects of some water-soluble metal
compounds in a somatic eye-color test system in Drosophila
melanogaster. Mutat. Res., 157: 157-162.
REDDY, M.R. & DUNN, S.J. (1984) Accumulation of heavy metals by
soybean from sludge-amended soil. Env. Pollution, B7: 281-296.
REDMOND, C.K. (1984) Site-specific cancer mortality among workers
involved in the production of high nickel alloys. In: Nickel in the
human environment. Proceedings of a Joint Symposium, Lyon, 8-11
March, 1983, Lyon, International Agency for Research on Cancer, pp.
73-86 (IARC Scientific Publications No. 53).
REDMOND, C.K., LEGASSE, A.A. & BASS, G. (1983) Cancer mortality in
workers in the high nickel alloy industries. Pittsburgh,
Pennsylvania, University of Pittsburgh, Graduate School of Public
Health, Department of Biostatistics, 145 pp.
REHWOLDT, R., BIDA, G. & NERRIE, B. (1971) Acute toxicity of
copper, nickel and zinc ions to some Hudson river fish species.
Bull. environ. Contam. Toxicol., 6: 445-448.
REHWOLDT, R., MENAPACE, L.W., NERRIE, B. & ALESSANDRELLO, D. (1972)
The effect of increased temperature upon the acute toxicity of some
heavy metal ions. Bull. environ. Contam. Toxicol., 8: 91-96.
REHWOLDT, R., LASKO, L., SHAW, C. & WIRHOWSKI, E. (1973) The acute
toxicity of some heavy metal ions toward benthic organisms. Bull.
environ. Contam. Toxicol., 10:291-294.
REICHRTOVA, E., KOVACIKOVA, Z., TAKAC, L. & ORAVEC, C. (1986a) The
effect of metal particles from a nickel refinery dump on alveolar
macrophages. Part 1. Chamber exposure of Wistar rats. Environ.
Pollut., A40: 87-94.
REICHRTOVA, E., TAKAC, L. & KOVACIKOVA, Z. (1986b) The effect of
metal particles from a nickel refinery dump on alveolar
macrophages. Part 2. Environmental exposure of rabbits. Environ.
Pollut., A40: 101-107.
REZUKE, W.N., KNIGHT, J.A. & SUNDERMAN, F.W. Jr (1987) Reference
values for nickel concentrations in human tissue and bile. Ann. J.
ind. Med., 11: 419-426.
RICHTER, O.R. & THEIS, T.L. (1980) Nickel speciation in a
soil/water system. In: Nriagu, J.O. ed. Nickel in the
environment, New York, Chichester, Brisbane, Toronto, John Wiley
and Sons, pp. 189-202.
RIGAUT, J.-P. (1983) Preliminary report on nickel health criteria,
Luxemburg, Commission of the European Communities, 1009 pp.
(CCE/LUX/V/E/24/83)
RITTMANN, D., NOCHRAINER, D., OBERDORSTER, G., KORDEL, W.,
SPIEGELBERG, T., KONIG, H., FRITSCHE, V., KUHNEN, H. & FINGERHUT,
R. (1981) [Inhalation experiment on rats and pathophysiological
effect of nickel. Environmental research plan of the Federal
Ministry of the Interior.] Berlin, Federal Ministry of the Interior
(Research Report No. 107 06 00702) (in German).
RIVEDAL, E. & SANNER, T. (1980) Synergistic effect on morphological
transformation of hamster embryo cells by nickel sulphate and
benz( a)pyrene. Cancer Lett., 8(3): 203-208.
RIVEDAL, E. & SANNER, T. (1981) Metal salts as promoters of in
vitro morphological transformation of hamster embryo cells
initiated by benzo( a)pyrene. Cancer Res., 41: 2950-2953.
RIVEDAL, E., HEMSTAD, J. & SANNER, T. (1980) Synergistic effects
of cigarette smoke extracts, benz( a)pyrene and nickel sulphate on
morphological transformation of hamster embryo cells. In:
Holmstedt, B., Lauwerys, R., Mercier, M. & Roberfroid, M., ed.
Mechanisms of toxicity and hazard evaluation. Proceedings of the
2nd International Congress on Toxicology, Brussels, Belgium, 6-11
July 1980. Amsterdam, New York, Oxford, Elsevier/North Holland
Biomedical Press, pp. 259-263 (Developments in Toxicology and
Environmental Science, Vol. 8).
ROBERTS, R.S., JULIAN, J.A., MUIR, D.C. & SHANNON, H.S. (1984)
Cancer mortality associated with the high temperature oxidation of
nickel subsulfide. In: Nickel in the human environment, Proceedings
of a Joint Symposium, Lyon, 8-11 March, 1983, Lyon, International
Agency for Research on Cancer, pp. 23-35 (IARC Scientific
Publications No. 53).
ROBISON, S.H. & COSTA, M. (1982) The induction of DNA strand
breakage of nickel compounds in cultured Chinese hamster ovary
cells. Cancer Lett., 15: 35-40.
ROBISON, S.H., CANTONI, O. & COSTA, M. (1982) Strand breakage and
decreased molecular weight of DNA induced by specific metal
compounds. Carcinogenesis, 3: 657-662.
ROCKSTROH, H. (1959) [On the etiology of bronchial cancer in
arsenic processing nickel smelting plants.] Arch.
Geschwulstforsch., 14: 151-162 (in German).
RODRIGUEZ-ARNAIZ, R. & RAMOS, P. (1986) Mutagenicity of nickel
sulphate in Drosophila melanogaster. Mutat. Res., 170(3): 115-117.
ROSSMAN, T.G., MOLINA, M. & MEYER, L.W. (1984) The genetic
toxicology of metal compounds: I. Induction of prophage in E.
coli WP2s. Environ. Mutagen., 6: 59-69.
ROUSH, C.C., MEIGS, I.W., KELLY, J.A., FLANNERY, I.T. & BURDO, H.
(1980) Sinonasal cancer and occupation: a case-control study. Am.
J. Epidemiol., 111: 183-193.
ROY, B.R. (1985) Nepera 82-06 sampling method for inorganic nickel.
In: Brown, S.S. & Sunderman, F.W. Jr, ed. Progress in Nickel
toxicology. Proceedings of the 3rd International Conference on
Nickel Metabolism and Toxicology, Paris, 4-7 September, 1984,
Oxford, Blackwell Scientific Publications, pp. 191-194.
RUBANYI, G. & INOVAY, J. (1982) Effect of nickel ions on
spontaneous electrically and norepinephrine stimulated isometric
contractions in the isolated portal vein of the rat. Acta physiol.
Acad. Sci. Hung., 59: 181-186.
RUBANYI, G. & KOVACH, A.G.B. (1980) Cardiovascular actions of
nickel ions. Acta physiol. Acad. Sci. Hung., 55: 345-353.
RUBANYI, G., BALOGH, I., KOVACH, A.G.B., SOMOGYI, E. & SOTONYI, P.
(1980) Effect of nickel ions on ultrastructure of isolated perfused
rat heart. J. mol. cell Cardiol., 12: 609-618.
RUBANYI, G., LIGETI, L. & KOLLER, A. (1981) Nickel is released
from the ischemic myocardium and contracts coronary vessels by a
Ca-dependent mechanism. J. mol. cell Cardiol., 13(11): 1023-1026.
RUBANYI, G., BIRTALAN, I., GERGELY, A. & KOVACH, A.G. (1982a)
Serum nickel concentration in women during pregnancy, during
parturition, and post partum. Am. J. Obstet. Gynecol., 143: 167-169.
RUBANYI, G., KALABAY, L., PATAKI, T. & HAJDU, K. (1982b) Nickel
induces vasoconstriction in the isolated canine coronary artery by
a tonic Ca2+-activation mechanism. Acta physiol. Acad. Sci. Hung.
59: 155-159.
RUBANYI, G., BAKOS, M., HAJDU, K. & PATAKI, T. (1982c) Dependence
of nickel induced coronary vasoconstriction on the activity of the
electrogenic Na+, K+-pump. Acta physiol. Acad. Sci. Hung., 59:
169-174.
RUBANYI, G., HAJDU, K., PATAKI, T. & BAKOS, M. (1982d) The role of
adrenergic receptors in Ni2+-induced coronary vasoconstriction.
Acta Physiol. Acad. Sci. Hung., 59: 161-167.
RUBANYI, G., LIGETI, L., KOLLER, A. & KOVACH, A.G. (1984) Possible
role of nickel ions in the pathogenesis of ischemic coronary
vasoconstriction in the dog heart. J. mol. cell Cardiol., 16: 533-546.
RUTHERFORD, G.K. & BRAY, C.R. (1979) Extent and distribution of
soil heavy metal contamination near a nickel smelter at Coniston,
Ontario. J. environ. Qual., 8(2): 219-222.
RYU, R.K.N., BOVILL, E.G. Jr, SKINNER, H.B. & MURREY, W.R. (1987)
Soft tissue sarcoma associated with aluminum oxide ceramic total
hip arthroplasty. Clin. Orthop. relat. Res., 216: 207-212.
SADIQ, M., ZAIDI, T.H., UL-HODA, A. & MIAN, A.A. (1982) Heavy
metal concentrations in shrimp, crab, and sediment obtained from
Ad-Dammam sewage outfall area. Bull. environ. Contam. Toxicol.
29(3): 313-320.
SAICHENKO, S.P. (1985) Experimental evaluation of genetic danger of
metals administered with drinking water. In: Domnin, S.C. & Kogan,
F.V. ed. Problems of occupational pathology and toxicology in
mining and metallurgy. Moscow, Erisman Institute of Hygiene, pp.
75-80.
SAICHENKO, S.P. & SHARAPOVA, N.Z (1987) About the mutagenicity of
some metals for Salmonella typhimurium. In: Domnin, S.C. &
Shcherbakov S.V. ed. Problems of hygiene and occupational
pathology in colored metals and steel industry. Moscow, Erisman
Institute of Hygiene, pp. 97-101.
SAKNYN, A.V. & BLOKHIN, V.A. (1978) [Development of malignant
tumors in rats under the influence of nickel-containing aerosols.]
Vopr. Onkol., 24(4): 44-48 (in Russian).
SAKNYN, A.V. & SHABYNINA, N.K. (1970) [Some statistical material on
carcinogenic danger in the production of nickel from oxidized
ores.] Gig. Tr. prof. Zabol., 14(11): 10-13 (in Russian).
SAKNYN, A.V. & SHABYNINA, N.K. (1973) [Epidemiology of malignant
neoplasms in nickel plants.] Gig. Tr. prof. Zabol., 9: 25-28
(in Russian).
SAKNYN, A.V., SIPATOV, G.YA., YELNICHUYKH, L.N. & STARKOV, P.S.
(1978) [Hygienic characterization of dusts in primary nickel
production.] In: Domnin, S.G. ed. Occupational disease of dust
aetiology. Erisman Institute of Hygiene, Moscow, Vol. 4, pp. 18-23.
SALTZMANN, B.E., CHOLAK, J., SCHAFER, L.J., YEAGER, D.W., MEINERS,
B.O., SVETLIK, J. & ZWILLENBERG, M.L. (1985) Concentrations of six
metals in the air of eight cities. Environ. Sci. Technol., 19(4):
328-333.
SAMELA, D., TSOUMPAS, G.M. & WALSHANS, G.K. (1986) Environmental
aspects of the combustion of sewage sludge in a utility boiler.
Environ. Prog., 5(2): 110-115.
SAMITZ, M.H. & KATZ, S.A. (1975) Nickel dermatitis hazards from
prostheses. In vivo and in vitro solubilization studies. Br. J.
Dermatol., 92: 287.
SAMITZ, M.H. & KATZ, S.A. (1976) Nickel-epidermal interactions:
diffusion and binding. Environ. Res., 11(1): 34-39.
SAMITZ, M.H. & POMERANTZ, H. (1958) Studies of the effects on the
skin of nickel and chromium salts. Arch. ind. Health, 18: 473-479.
SAMITZ, M.H., KATZ, S.A., SCHEINER, D.M. & LEWIS, J.E. (1975)
Attempts to induce sensitization in guinea-pigs with nickel
complexes. Acta dermtovenereol., 55: 475-480.
SANDE, J.H. VAN DE, MACINTOSH, L.P. & JOVIN, T.M. (1982) Mn2+ and
other transition metals at low concentration induce the right-to-
left helical transformation of poly (d(G-C)). EMBO J., 1: 777-782.
SANDERS, J.R., MCGRATH, S.P. & ADAMS, T.M. (1986) Zink, copper and
nickel concentrations in ryegrass grown on sewage sludge-
contaminated soils of different pH. J. Sci. Food Agric., 37:
961-968.
SANFORD, W.E., NIEBOER, E., BACH, P., STACE, B., GREGG, N. &
DOBROTA, M. (1988) The renal clearance and toxicity of nickel. In:
Fourth International Conference on Nickel Metabolism and
Toxicology, Abstracts, Espoo, Finland, 5-9 September 1988,
Helsinki, Institute of Occupational Health, p.17.
SANINA, J.P. (1968) [Toxicology of nickel carbonyl.] Toxikol. Nov.
Prom. Khim. Veshchestv., 10: 144-149 (in Russian).
SANOTSKII, J.V. (1955) [Action mechanism of nickel carbonyl.]
Farmakol. Toksikol., 18(2): 48-50 (in Russian).
SANTUCCI, B., CRISTAUDO, A., CANNISTRACI, C. & PICARDO, M. (1988)
Nickel sensitivity: effects of prolonged oral intake of the
element. Contact Dermatit., 19: 202-295.
SANZOLONE, R.F., CHAO, T.T. & CRENSHAW, G.L. (1979) Atomic
absorption spectrometric determination of cobalt, nickel, and
copper in geological materials with matrix marking and chelation-
extraction. Anal. chim. Acta, 105: 247-253.
SAPEK, B. & SAPEK, A. (1980) [Nickel content in the profiles of
organic soils.] In: Anke, M., Schneider, H.-J. & Brückner, Chr.
ed. [3. Trace Element Symposium: Nickel, Jena, German Democratic
Republic, 7-11 July, 1980,] Jena, Friedrich-Schiller University, pp.
185-191 (in German).
SARKAR, B. (1980) Nickel in blood and kidney. In: Brown, S.S. &
Sunderman, F.W. Jr, ed. Nickel toxicology, Proceedings of the 2nd
International Conference on Nickel Toxicology, Swansea, 3-5
September, 1980, London, New York, Academic Press, pp. 81-84.
SAXHOLM, H.J.K., REITH, A. & BROEGGER, A. (1981) Oncogenic
transformation and cell lysis in C3H/10T 1/2 cells and increased
sister chromatid exchange in human lymphocytes by nickel
subsulfide. Cancer Res., 41(10): 4136-4139.
SCHMIDT, J.A. & ANDREN, A.W. (1980) The atmospheric chemistry of
nickel. In: Nriagu, J.O. ed. Nickel in the environment, New York,
Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 93-135.
SCHMIDT, W. & DIETL, F. (1981) [Determination of nickel and cobalt
in digested soils by means of flameless atomic absorption with
zirconium coated graphite tubes.] Fresenius Z. anal. Chem., 308:
129-132 (in German with English summary).
SCHNEGG, A. & KIRCHGESSNER, M. (1975a) [The essentiality of nickel
for the growth of animals.] Z. Tierphysiol. Tierernähr.
Futtermittelkd., 36: 63-74 (in German).
SCHNEGG, A. & KIRCHGESSNER, M. (1975b) [Changes in the hemoglobin
content, erythrocyte content and hematocrit in nickel deficiency.]
Nutr. Metabol., 19: 268-278 (in German).
SCHNEGG, A. & KIRCHGESSNER, M. (1976a) [Interaction of nickel with
iron, copper and zinc.]. Arch. Tierernähr., 26(8): 543-549
(in German with English summary).
SCHNEGG, A. & KIRCHGESSNER, M. (1976b) [Absorption and metabolic
efficiency of iron during nickel deficiency.] Int. Z. Vitam.
Forsch., 46: 96-99 (in German with English summary).
SCHNEGG, A. & KIRCHGESSNER, M. (1977a) [Changes in enzyme
activities in liver and kidney with nickel and iron deficiency,
respectively.] Z. Tierphysiol. Tierernährg. Futtermittelkd., 38:
200-205 (in German with English summary).
SCHNEGG, A. & KIRCHGESSNER, M. (1977b) [Alkaline and acid
phosphatase activity in liver and serum during nickel versus Fe-
deficiency.] Int. Z. Vitam. Forsch., 47: 274-276 (in German with
English summary).
SCHNEGG, A. & KIRCHGESSNER, M. (1977c) [Changes of various
substrate concentrations in the serum and liver in Ni- or Fe-
deficiency.] Z. Tierphysiol. Tierernährg. Futtermittelkd., 39(5-6):
247-251 (in German with English summary).
SCHNEGG, A. & KIRCHGESSNER, M. (1977d) [Differential diagnosis of
Fe and Ni deficiency by determination of some enzyme activities.]
Zbl. Vet. Med., 24 A: 242-247 (in German).
SCHNEGG, A. & KIRCHGESSNER, M. (1980) [On the essentiallity of
nickel for animal growth.] In: Anke, M. Schneider, H.-J. &
Brückner, Chr. ed. [3. Trace Element Symposium: Nickel, Jena,
German Democratic Republic, 7-11 July, 1980,] Jena, Friedrich-
Schiller University, pp. 11-16 (in German).
SCHNEIDER, H.-J., ANKE, M. & KLINGER, G. (1980) [The nickel-status
of human beings.] In: Anke, M. Schneider, H.-J. & Brückner, Chr.
ed. [3. Trace Element Symposium: Nickel, Jena, German Democratic
Republic, 7-11 July, 1980,] Jena, Friedrich-Schiller-University, pp.
277-283 (in German).
SCHROEDER, H.A. (1968) Serum cholesterol levels in rats fed
thirteen trace elements. J. Nutr., 94: 475-480.
SCHROEDER, H.A. (1970) A sensible look at air pollution by metals.
Arch. environ. Health, 21: 798-806.
SCHROEDER, H.A. & MITCHENER, M. (1971) Toxic effects of trace
elements on the reproduction of mice and rats. Arch. environ.
Health, 23: 102-106.
SCHROEDER, H.A. & MITCHENER, M. (1975) Life time effects of
mercury, methyl mercury and nine other trace metals on mice. J.
Nutr., 105: 452-458.
SCHROEDER, H.A., BALASSA, J.J. & TIPTON, J.H. (1962) Abnormal
trace metals in man - nickel. J. chron. Dis., 15: 51-65.
SCHROEDER, H.A., BALASSA, J.J. & VINTON, W.H. Jr (1964) Chromium,
lead, cadmium, nickel and titanium in mice: effect on mortality,
tumors and tissue levels. J. Nutr., 83: 239-250.
SCHROEDER, H.A., MITCHENER, M. & NASON, A.P. (1974) Life-term
effects of nickel in rats: survival, tumors, interactions with
trace elements and tissue levels. J. Nutr., 104: 239-243.
SCHUMANN, H. (1980) [Nickel contamination of surface waters and
drinking water and some influencing factors.] In: Anke, M.,
Schneider, H.-J. & Brückner, Chr. ed. [3. Trace Element Symposium
Nickel, Jena, German Democratic Republic, 7-11 July, 1980.] Jena,
Friedrich-Schiller University, pp. 155-161 (in German).
SEEMANN, J., WITTIG, P., KOLLMEIER, H. & ROTHE, G. (1985)
Analytical measurements of Cd, Pb, Zn, Cr and Ni in human tissues.
Lab. Med., 9: 294-299.
SEN, P. & COSTA, M. (1985) Induction of chromosomal damage in
Chinese hamster ovary cells by soluble and particulate nickel
compounds: preferential fragmentation of the heterochromatic long
arm of the X-chromosome by carcinogenic crystalline NiS particles.
Cancer Res., 45(5): 2320-2325.
SEN, P. & COSTA, M. (1986) Pathway of nickel uptake influences its
interaction with heterochromatic DNA. Toxicol. appl. Pharmacol.
84(2): 278-285.
SHANNON, H.S., JULIAN, J.A., MUIR, D.C.F., ROBERTS, R.S. &
CECUTTI, A.C. (1984) A mortality study of Falconbridge workers. In:
Nickel in the human environment, Proceedings of a Joint Symposium,
Lyon, 8-11 March, 1983, Lyon, International Agency for Research on
Cancer, pp. 117-124 (IARC Scientific Publications No. 53).
SHAW, T.L. & BROWN, V.M. (1971) Heavy metals and the fertilization
of rainbow trout eggs. Nature (Lond.), 230: 251.
SHI, Z. (1986) Acute nickel carbonyl poisoning: a report of 179
cases. Br. J. ind. Med., 43: 422-424.
SHI, Z., LATA, A. & HAN, Y. (1986) A study of serum monoamine
oxidase (MAO) activity and EEG in nickel carbonyl workers. Br. J.
ind. Med., 43: 425-426.
SHIBATA, M., IZUMI, K., SANO, N., AKAGI, A. & OTSUKA, H. (1989)
Induction of soft tissue tumours in F344 rats by subcutaneous,
intramuscular, intra-articular, and retroperitoneal injection of
nickel sulphide (Ni3S2). J. Pathol., 157: 263-274.
SILVERSTEIN, M., MIRER, F., KOTELCHUCK, D., SILVERSTEIN, B. &
BENNETT, M. (1981) Mortality among workers in a diecasting and
electroplating plant. Scand. J. Work Environ. Health, 7: 156-165.
SINA. J.F., BEAN, C.L., DYSART, G.R., TAYLOR, V.I. & BRADLEY,
M.O. (1983) Evaluation of the alkaline elution/rat hepatocyte assay
as a predictor of carcinogenic/mutagenic potential. Mutat. Res.
113: 357-391.
SINGH, I. (1984) Induction of gene conversion and reverse mutation
by manganese sulphate and nickel sulphate in Saccharomyces
cerevisiae. Mutat. Res., 137: 47-49.
SIROVER, M.A. & LOEB, L.A. (1977) On the fidelity of DNA
replication. Effect of metal activators during synthesis with avian
myeloblastosis virus DNA polymerase. J. biol. Chem., 252: 3605-3610.
SJOVALL, P., CHRISTENSEN, O.B. & MOLLER, H. (1987) Oral
hyposensitization in nickel allergy. J. Am. Acad. Dermatol., 17: 774-
778.
SKAUG, V., GYLSETH, B., REISS, A.P. & NORSETH, T. (1985) Tumor
induction in rats after intrapleural injection of nickel subsulfide
and nickel oxide. In: Brown, S.S. & Sunderman, F.W. Jr, ed. Nickel
toxicology, Proceedings of the 2nd International Conference on
Nickel Toxicology, Swansea, 3-5 September, 1980, London, New York,
Academic Press, pp. 37-40.
SMART, G.A. & SHERLOCK, J.C. (1987) Nickel in foods and the diet.
Food Addit. Contam., 4(1): 61-71.
SMIALOWICZ, R.J. (1985) The effect of nickel and manganese on
natural killer cell activity. In: Brown, S.S. & Sunderman, F.W. Jr,
ed. Progress in Nickel toxicology. Proceedings of the 3rd
International Conference on Nickel Metabolism and Toxicology,
Paris, France, 4-7 September, 1984, Oxford, Blackwell Scientific
Publications, pp. 161-164.
SMIALOWICZ, R.J., ROGERS, R.R., RIDDLE, M.M. & STOTT, G.A. (1984)
Immunologic effects of nickel: 1. Suppression of cellular and human
immunity. Environ. Res., 33: 413-427.
SMIALOWICZ, R.J., RODGERS, R.R., RIDDLE, M.M., GARNER, R.J., ROWE,
D.G. & LUEBKE, R.W. (1985) Immunologic effects of nickel. II.
Suppression of natural killer cell activity. Environ. Res., 36: 56-66.
SMIALOWICZ, R.J., ROGERS, R.R., RIDDLE, M.M., ROWE, D.G. & LUEBKE,
R.W. (1986) Immunological studies in mice following in utero
exposure to NiCl2. Toxicology, 38: 293-303.
SMIALOWICZ, R.J., ROGERS, R.R., RIDDLE, M.M., LUEBKE, R.W.,
FOGELSON, L.D. & ROWE, D.G. (1987) Effects of manganese, calcium,
magnesium, and zinc on nickel-induced suppression of murine natural
killer cell activity. J. Toxicol. environ. Health, 20(1-2): 67-80.
SMITH, J.C. (1969) A controlled environment system for trace
element deficiency studies. Trace Subst. environ. Health, 2: 223-242.
SMITHE, J.C. & HACKLEY, B. (1968) Distribution and excretion of
nickel-63 administered intravenously to rats. J. Nutr., 95: 541-546.
SMITH-SONNEBORN, J., PALIZZI, R.A., McCANN, E.A. & FISHER, G.L.
(1983) Bioassay of genotoxic effects of environmental particles in
a feeding ciliate. Environ. Res., 32: 474-479.
SMITH-SONNEBORN, J., LEIBOVITZ, B., DONATHAN, R. & FISHER, G.L.
(1986) Bioassay of environmental nickel dusts in a particle feeding
ciliate. Environ. Mutagen., 8(4): 621-626.
SNODGRAS, W. (1980) Distribution and behaviour of nickel in the
aquatic environment. In: Nriagu, J.O. ed. Nickel in the
environment, New York, Chichester, Brisbane, Toronto, John Wiley
and Sons, pp. 203-274.
SOLOMONS, N.W., VITERI, F., SHULER, T.R. & NIELSEN, F.H. (1982)
Bioavailability of nickel in man: effects of foods and chemically-
defined dietary constituents on the absorption of inorganic nickel.
J. Nutr., 112(1): 39-50.
SONNENFELD, G., STREIPS, U.N. & COSTA, M. (1983) Differential
effects of amorphous and crystalline nickel sulfide on murine
alpha/beta interferon production. Environ. Res., 32: 474-479.
SORAHAN, T. & WATERHOUSE, J.A.W. (1983) Mortality study of nickel-
cadmium battery workers by the method of regression models in life
tables. Br. J. ind. Med., 40: 293-300.
SORAHAN, T., BURGES, D.C.L. & WATERHOUSE, J.A.H. (1987) A
mortality study of nickel/chromium platers. Br. J. ind. Med., 44:
250-258.
SORINSON, S.N., KORNILOVA, A.P. & ARTEMEVA, A.M. (1958)
[Concentrations of nickel in blood and urine of workers in the
nickel carbonyl industry.] Gig. i Sanit., 23(9): 69-72 (in Russian).
SPEARS, J.W. (1984) Nickel as a "newer trace element" in the
nutrition of domestic animals. J. anim. Sci., 59: 823-835.
SPEARS, J.W. & HATFIELD, E.E. (1977) Role of nickel in animal
nutrition. Feedstuffs, 49: 24-28.
SPEARS, J.W., HATFIELD, E.E., FORBES, R.M. & KOENIG, S.E. (1978)
Studies on the role of nickel in the ruminant. J. Nutr., 108:
313-320.
SPEARS, J.W., JONES, E.E., SAMSELL, L.J. & ARMSTRONG, W.D. (1984)
Effect of dietary nickel on growth, urease activity, blood
parameters and tissue mineral concentrations in the neonatal pig.
J. Nutr., 114: 845-854.
SPENCER, D.F. (1980) Nickel and aquatic algae. In: Nriagu, J.O.
ed. Nickel in the environment, New York, Chichester, Brisbane,
Toronto, John Wiley and Sons, pp. 339-347.
SPENCER, D.F. & GREENE, R.W. (1981) Effects of nickel on seven
species of freshwater algae. Environ. Pollut., A25: 241-247.
SPIEGELBERG, T., KORDEL, W. & HOCHRAINER, D. (1984) Effects of NiO
inhalation on alveolar macrophages and the humoral immune systems
of rats. Ecotoxicol. environ. Safety, 8: 516-525.
SPRUIT, D., MALI, J.W.H. & DE GROOT, N. (1965) The interaction of
nickel ions with human cadaverous dermis. J. Invest. Dermatol., 44:
103-106.
STACK, M.V., BURKITT, A.J. & NICKLESS, G. (1976) Trace metals in
teeth at birth (1957-1963 and 1972-1973). Bull. environ. Contam.
Toxicol., 16: 764-766.
STAHLY, E.E. (1973) Some considerations of metal carbonyls in
tobacco smoke. Chem. Ind., 12: 620-623.
STEDMAN, D.H. (1986a) Method 5 - determination of nickel carbonyl
in air colorimetry. In: O'Neill, I.K. Schuller, P. & Fishbein,
L. ed. Environmental carcinogens - selected methods of analysis.
Vol. 8: Some metals: As,Be, Cd, Cr, Ni, Pb, Se, Zn. Lyon,
International Agency for Research on Cancer, pp. 261-267 (IARC
Scientific Publications No. 71).
STEDMAN, D.H. (1986b) Method 6 - determination of nickel carbonyl
in air by chemiluminescence. In: O'Neill, J.K. Schuller, P. &
Fishbein, L. ed. Environmental carcinogens - selected methods of
analysis. Vol. 8: Some metals: As, Be, Cd, Cr, Ni, Pb, Se, Zn.
Lyon, International Agency for Research on Cancer, pp. 269-273
(IARC Scientific Publications No. 71).
STEDMAN, D.HJ. & HIKADE, D.A. (1980) The rate of decay of traces of
nickel carbonyl in air. In: Brown, S.S. & Sunderman, F.W. Jr, ed.
Nickel toxicology, Proceedings of the 2nd International Conference
on Nickel Toxicology, Swansea, 3-5 September, 1980, London, New
York, Academic Press, pp. 183-186.
STERN, A.C., BOUBEL, R.W., TURNER, D.B. & FOX, D.L. (1984)
Fundamentals of air pollution, 2nd ed. Orlando, San Diego, San
Francisco, Academic Press, pp. 103-113.
STERN, R.M. (1983) Assessment of risk of lung cancer for welders.
Arch. env. Health, 38(3): 148-156.
STERN, R.M., HANSEN, K. & THOMSEN, E. (1985) An in vitro
genotoxicity assay to complement in vivo inhalation studies and for
occupational monitoring of metallic aerosols and welding fumes. In:
Brown, S.S. & Sunderman, F.W. Jr, ed. Progress in nickel
toxicology. Proceedings of the 3rd International Conference on
Nickel Metabolism and Toxicology, Paris, 4-7 September, 1984,
Oxford, Blackwell Scientific Publications, pp. 77-80.
STEWART, S.G. & CROMIA, F.E. (1934) Experimental nickel dermatitis.
J. Allergy, 5: 575-582.
STOEPPLER, M. (1980) Analysis of nickel in biological materials and
natural waters. In: Nriagu, J.O. ed. Nickel in the environment, New
York, Chichester, Brisbane, Toronto, John Wiley & Sons, pp. 661-821.
STOKES, P. (1975) Uptake and accumulation of copper and nickel by
metal-tolerant strains of Scenedesmus. Int. Ver. Limnol., 19:
128-137.
STOKES, P.M. (1981) Nickel in aquatic systems. In: Effects of
nickel in the Canadian environment, Ottawa, National Research
Council of Canada, pp. 77-117 (Publication No. NRCC-18568).
STONER, G.O., SHIMKIN, M.B., TROXELL, M.C., THOMPSON, T.L. &
TERRY, L.S. (1976) Test for carcinogenicity of metallic compounds
by the pulmonary tumor response in strain of mice. Cancer Res., 36:
1744-1747.
STORENG, R. & JONSEN, J. (1980) Effect of nickel chloride and
cadmium acetate on the development of preimplantation mouse embryos
in vitro. Toxicology, 17: 183-187.
STORENG, R. & JONSEN, J. (1981) Nickel toxicity in early
embryogenesis in mice. Toxicology, 20(1): 45-51.
STOVBUN, A.T., YATSYUK, M.D., POMARENKO, U.I. & YAKOVLEVA, L.S.
(1962) [Data on the trace element composition of human milk and
various modifications of cow's milk.] Nauk. Zap. Ivano.-Frankivskii
Derzk. Med. Inst., 5: 38-40 (in Russian).
STRAIN, W.H., VARNES, A.W., DAVIS, B.R. & KARK, E.C. (1980)
[Nickel in drinking and household water.] In: Anke, M., Schneider,
H.-J. & Brûckner, Chr. ed. [3. Trace Element Symposium: Nickel,
Jena, German Democratic Republic, 7- 11 July, 1980,] Jena,
Friedrich-Schiller University, pp. 149-154 (in German).
STRATTON, G.W. & CORKE, C.T. (1979) The effect of nickel on the
growth, photosynthesis and nitrogenase activity of Anabaena
inequalis. Can. J. Microbiol., 25: 1094-1099.
STURGEON, R.E., BERMAN, S.S. & WILLIE, S.N. (1982) Concentration
of trace metals from sea-water by complexation with 8-
hydroxyquinoline and adsorption in C18-bonded silica gel. Talanta,
29: 167-171.
SUMINO, K., HAYAKAWA, K., SHIBATA, T. & KITAMURA, S. (1975) Heavy
metals in normal Japanese tissues. Arch. environ. Health, 30: 487-494.
SUNDERMAN, F.W. (1964) Nickel and copper mobilization by sodium
diethyldithiocarbamate. J. New Drugs, 4: 154-161.
SUNDERMAN, F.W. (1970) Nickel poisoning. In: Sunderman, F.W. &
Sunderman, F.W. Jr, ed. Laboratory diagnosis of diseases caused by
toxic agents, St. Louis, Missouri, Warren H. Green Inc. pp. 387-
396.
SUNDERMAN, F.W. (1971) The treatment of acute nickel carbonyl
poisoning with sodium diethyldithiocarbamate. Ann. clin. Res., 3(3):
182-185.
SUNDERMAN, F.W. Jr (1977) The metabolism and toxicology of nickel.
In: Brown, S.S. & Savory, J. ed. Chemical toxicology and clinical
chemistry of metals. Proceedings of the 2nd International
Conference, Montreal, 19-22 July, 1983, London, New York, Blackwell
Scientific Publications, pp. 231-259.
SUNDERMAN, F.W. Jr (1980) Analytical biochemistry of nickel.
International Union of Pure and Applied Chemistry, Subcommittee on
environmental and Occupational Toxicology of Nickel. Pure appl.
Chem., 52: 527-544.
SUNDERMAN, F.W. Jr (1983a) Potential toxicity from nickel
contamination of intravenous fluids. Ann. clin. lab. Sci., 13(1): 1-4.
SUNDERMAN, F.W. Jr (1983b) Organs and species specificity in nickel
subsulfide carcinogenesis. In: Langenbach, R., Nesnow, S. & Rice,
J.M. ed. Organ and species specificity in chemical carcinogenesis,
New York, Plenum Press, pp. 107-127 (Basic life sciences. Vol. 24).
SUNDERMAN, F.W. Jr (1984) Carcinogenicity of nickel compounds in
animals. In: Nickel in the human environment. Proceedings of a
Joint Symposium, Lyon, 8-11 March, 1983, Lyon, International Agency
for Research on Cancer, pp. 127-142 (IARC Scientific Publications
No. 53).
SUNDERMAN, F.W. Jr (1986) Iatrogenic exposures to toxic metals. 9th
Annual meeting of the National Academy of Clinical Biochemistry on
Metal Metabolism and Disease, Atlanta, TA, USA, July 9-20, 1985.
Clin. Physiol. Biochem., 4(1): 112.
SUNDERMAN, F.W. Jr (1987) Lipid peroxidation as a mechanism of
acute nickel toxicity. Toxicol. environ. Chem., 15: 59-69.
SUNDERMAN, F.W. Jr (1989a) Mechanisms of nickel carcinogensis.
Scand. J. Work Environ. Health, 15: 1-12.
SUNDERMAN, F.W. Jr (1989b) Carcinogencity of metal alloys in
orthopaedic prostheses: clinical and experimental studies. Fundam.
appl. Toxicol., 13: 1-12.
SUNDERMAN, F.W. Jr & BARBER, A. M. (1988) Finger loops, ongogens,
and metals. Ann. clin. lab. Sci., 18: 267-288.
SUNDERMAN, F.W. Jr & DONELLY, A.J. (1965) Studies of nickel
carcinogenesis metastasizing pulmonary tumors in rats induced by
the inhalation of nickel carbonyl. Am. J. Pathol., 46: 1027-1041.
SUNDERMAN, F.W. Jr & HOPFER, S.M. (1983) Correlation between the
carcinogenic activities of nickel compounds and their potencies for
stimulating erythropoiesis in rats. In: Saker, B. ed. Biological
aspects of metals and metal-related diseases, New York, Raven
Press, pp. 171-181.
SUNDERMAN, F.W. & KINCAID, J.F. (1954) Nickel poisoning II.
Studies on patients suffering from acute exposure to vapors of
nickel carbonyl. J. Am. Med. Assoc., 155(10): 889-894.
SUNDERMAN, F.W. Jr & MCCULLY, K.S. (1983) Effects of manganese
compounds on carcinogenicity of nickel subsulfide in rats.
Carcinogenesis, 4(4): 461-465.
SUNDERMAN, F.W. Jr & MAENZA, R.M. (1976) Comparisons of
carcinogenicities of nickel compounds in rats. Res. Commun. chem.
Pathol. Pharmacol., 14: 319-330.
SUNDERMAN, F.W. Jr & OSKARSSON, A. (1988) Nickel. In: Merian, ed.
Metals and their compounds in the environment, Weinheim, VCH
Verlagsgesellschaft, pp. 1-19.
SUNDERMAN, F.W. Jr & SELIN, C.E. (1968) The metabolism of nickel-63
carbonyl. Toxicol. appl. Pharmacol., 12: 207-217.
SUNDERMAN, F.W. & SUNDERMAN, F.W. Jr (1961a) Nickel poisoning. XI.
Implication of nickel as a pulmonary carcinogen in tobacco smoke.
Am. J. clin. Pathol., 35: 203-209.
SUNDERMAN, F.W. & SUNDERMAN, F.W. Jr (1961b) Löffler's syndrome
associated with nickel sensitivity. Arch. int. Med., 107: 405-408.
SUNDERMAN, F.W. Jr, KINCAID, J.F., DONNELLY, A.J. & WEST, B.
(1957) Nickel poisoning. IV. Chronic exposure of rats to nickel
carbonyl: a report after one year of observation. Arch. ind.
Health, 16: 480-485.
SUNDERMAN, F.W., DONELLY, A.J., WEST, B. & J.F. KINCAID (1959)
Nickel poisoning. IX. Carcinogenesis in rats exposed to nickel
carbonyl. Arch. ind. Health, 20: 36-41.
SUNDERMAN, F.W., RANGE, C.L., SUNDERMAN, F.W. Jr, DONELLY, A.J. &
LUCYSZYN, G.W. (1961) Nickel poisoning. XII. Metabolic and
pathologic changes in acute pneumonitis from nickel carbonyl. Am.
J. clin. Pathol., 36: 477-491.
SUNDERMAN, F.W. Jr, WHITE, J.C. & SUNDERMAN, F.W. (1963) Metabolic
balance studies in hepatolenticular degeneration treated with
diethyldithiocarbamate. Am. J. Med., 34: 875-888.
SUNDERMAN, F.W. Jr, ROSZEL, N.O. & CLARK, R.J. (1968) Gas
chromatography of nickel carbonyl in blood and breath. Arch.
environ. Health, 16(6): 836-843.
SUNDERMAN, F.W. Jr, NOMOTO, S., PRADHAN, A.M., LEVINE, H.,
BERNSTEIN, S.H. & HIRSCH, R. (1970) Increased concentrations of
serum nickel after acute myocardial infarction. J. Med., 283
(17): 896-899.
SUNDERMAN, F.W. Jr, NOMOTO, S. & NECHAY, M. (1971) Nickel
metabolism in myocardial infarction. II. Measurements of nickel in
human tissues. Trace Subst. environ. Health, 4: 352-356.
SUNDERMAN, F.W. Jr, NOMOTO, S., MORANG, R., NECHAY, M.W., BURKE,
C.N. & NIELSEN, S.W. (1972) Nickel deprivation in chicks. J.
Nutr., 102: 259-268.
SUNDERMAN, F.W. Jr, KASPRZAK, K., HORAK, E., GITLITZ, P. &
ONKELINX, C. (1976a) Effects of triethylenetetramin upon the
metabolism and toxicity of 63NiCl2 in rats. Toxicol. appl.
Pharmacol., 38: 177-188.
SUNDERMAN, F.W. Jr, KASPRZAK, K.S., LAU, T.J., MINGHETTI, P.P.,
MAECIZA, R.M., BECKER, N., ONKELINX, C. & GOLDBLATT, P.J. (1976b)
Effects of manganese on carcinogenicity and metabolism of nickel
subsulfide. Cancer Res., 36: 1790-1800.
SUNDERMAN, F.W. Jr, SHEN, S.K., MITCHELL, J.M., ALLPASS, P.R. &
DAMJANOV, I. (1978a) Embryotoxicity and fetal toxicity of nickel in
rats. Toxicol. appl. Pharmacol., 43(2): 381-390.
SUNDERMAN, F.W. Jr, ALLPASS, P. & MITCHELL, J. (1978b) Recent
progress in nickel carcinogenesis. Ann. Ist. Super. Sanita, 22(2):
669-679.
SUNDERMAN, F.W. Jr, ALLPASS, P. & MITCHELL, J. (1978c) Ophthalmic
malformations in rats following prenatal exposure to inhalation of
nickel carbonyl. Ann. clin. lab. Sci., 8: 499-500.
SUNDERMAN, F.W. Jr, MITCHELL, J., ALLPASS, P. & BASELT, R. (1978d)
Embryotoxicity and teratogenicity of nickel carbonyl in rats.
Toxicol. appl. Pharmacol., 45: 345.
SUNDERMAN, F.W. Jr, MAENZA, R.M., ALPASS, P.R., MITCHELL, J.M.,
DAMJANOV, I. & GOLDBLATT, P.J. (1978e) Carcinogenicity of nickel
subsulfide in Fischer rats and Syrian hamsters after administration
by various routes. Adv. exp. Med. Biol., 91: 57-67.
SUNDERMAN, F.W. Jr, ALLPASS, P.R., MITCHELL, J.M., BASELT, R.C. &
ALBERT, D.M. (1979a) Eye malformations in rats: induction by
prenatal exposure to nickel carbonyl. Science, 203: 550-553.
SUNDERMAN, F.W. Jr, MAENZA, R.M., HOPFER, S.M., MITCHELL, J.M.,
ALLPASS, P.R. & DAMJANOV, I. (1979b) Induction of renal cancers in
rats by intrarenal injection of nickel subsulfide. J. environ.
Pathol. Toxicol., 2(6): 1511-1527.
SUNDERMAN, F.W. Jr, SHEN, S.K., REID, M.C. & ALLPASS, P.R. (1980)
Teratogenicity and embryotoxicity of nickel carbonyl in Syrian
hamsters. Teratog. Carcinog. Mutagen., 1(2): 223-233.
SUNDERMAN, F.W. Jr, MCCULLY, K.S. & RINEHIMER, L.A. (1981)
Negative test for transplacental carcinogenicity of nickel
subsulfide in Fischer rats. Res. Commun. chem. Pathol. Pharmacol.
31(3): 545-554.
SUNDERMAN, F.W. Jr, REID, M.C., SHEN, S.K. & KEVORKIAN, C.B.
(1983) Embryotoxicity and teratogenicity of nickel compounds. In:
Clarkson, T.W., Nordberg, G.F. & Sager, P.R. ed. Reproductive and
developmental toxicity of metals. New York, London, Plenum Press,
pp. 399-416.
SUNDERMAN, F.W. Jr, CRISOSTOMO, M.C., REID, M.C., HOPFER, S.M. &
NOMOTO, S. (1984a) Rapid analysis of nickel in serum and whole
blood by electrothermal atomic absorption spectrophotometry. Ann.
clin. lab. Sci., 14(3): 232-241.
SUNDERMAN, F.W. Jr, MACCULLY, K.S. & HOPFER, S.M. (1984b)
Association between erythrocytosis and renal cancers in rats
following intrarenal injection of nickel compounds. Carcinogenesis,
5: 1511-1517.
SUNDERMAN, F.W. Jr, MARZOUK, A., CRISOSTOMO, M.C. & WEATHERBY,
D.R. (1985) Electrothermal atomic absorption spectrophotometry of
nickel in tissue homogenates. Ann. clin. lab. Sci., 15(4): 299-307.
SUNDERMAN, F.W. Jr, HOPFER, S.M., CRISOSTOMO, M.C. & STOEPPLER, M.
(1986a) Rapid analysis of nickel in urine by electrothermal atomic
absorption spectrophotometry. Ann. clin. lab. Sci., 16(3): 219-230.
SUNDERMAN, F.W. Jr, AITIO, A., MORGAN, L.G. & NORSETH, T. (1986b)
Biological monitoring of nickel. Toxicol. ind. Health, 2(1): 17-78.
SUNDERMAN, F.W. Jr, HOPFER, S.M., KNIGHT, J.A., McCEILLY, K.S.,
CECUTTI, A.G., THORNHILL, A.G., CONWAY, K., MILLER, C., PATIERNO,
S.R. & COSTA, M. (1987a) Physiochemical characteristics and
biological effects of nickel oxides. Carcinogenesis, 8: 305-313.
SUNDERMAN, F.W. Jr, AN, L.L., ZAHARIA, O., WONG, S.H.-Y. & HOPFER,
S.M. (1987b) Acute depletion of pulmonary lavage cells and enhanced
lipid peroxidation in alveolar macrophages following parenteral
administration of nickel chloride to rats. In: Rand, N.Y. & Raper,
C. ed. Pharmacology, Amsterdam, New York, Oxford, Elsevier Science
Publishers, pp. 817-820.
SUNDERMAN, F.W. Jr, HOPFER, S.M. & KNIGHT, J.A. (1988a) Biological
reactivity of nickel oxides and related compounds. In: Fourth
International Conference on Nickel Metabolism and Toxicology:
Abstracts, Espoo, Finland, 5-9 September 1988, Helsinki, Institute
of Occupational Health, p. 15.
SUNDERMAN, F.W. Jr, DINGLE, B., HOPFER, S.M. & SWIFT, T. (1988b)
Acute nickel toxicity in electroplating workers who accidentally
ingested a solution of nickel sulfate and nickel chloride. Ann. J.
ind. Med., 14: 257-266.
SUNDERMAN, F.W. Jr, HOPFER, S.M., SWEENEY, K.C., MARCUS, A.H.,
MOST, B.M. & CREASON, J. (1989a) Nickel absorption and kinetics in
human volunteers. Proc. Soc. exp. Biol. Med., 191: 5-11.
SUNDERMAN, F.W. Jr, HOPFER, S.M., SWIFT, T., REZUKE, W.N., ZIEBKA,
L., HIGHAM, P., EDWARDS, B., FOLCIK, M. & GOSSLING, H.R. (1989b)
Cobalt, chrominium and nickel concentrations in body fluids of
patients with porous-coated knee or hip prostheses. J. Orthop.
Res., 7(3): 307-315.
SUNDERMAN, F.W. Jr, HOPFER, F.W. Jr, LIN, S.-M., PLOWMAN, M.C.,
STOJANOVIC, T., WONG, S.H.-Y., ZAHARIA, O. & ZIEBKA, L. (1989c)
Toxicity to alveolar macrophages in rats following parenteral
injection of nickel chloride. Toxicol. appl. Pharmacol., 100:
107-118.
SUNG, J.F.C., NEVISSI, A.E. & DEWALLE, F.B. (1986) Concentration
and removal efficiency of major and trace elements in municipal
wastewater. J. environ. Sci. Health, A21(5): 435-448.
SUSHENKO, O.V. & RAFIKOVA, K.E. (1972) [Questions of work hygiene
in hydrometallurgy of copper, nickel and cobalt in a sulfide ore.]
Gig. Tr. prof. Zabol., 16: 42-45 (in Russian).
SUTHERLAND, R.B. (1959) Respiratory cancer mortality in workers
employed in an Ontario nickel refinery covering the period 1939-
1957, Ottawa, Ontario Department of Health, Division of Industrial
Hygiene (Unpublished report, November, 1959).
SWANN, M. (1984) Malignant soft-tissue tumour at the site of a
total hip replacement. J. bone joint Surg., B66: 629-631.
SWIERENGA, S.H. & BASRUR, P.K. (1968) Effect of nickel on cultured
rat embryo muscle cells. Lab. Invest., 19: 663-674.
SWIERENGA, S.H. & MCLEAN, J.R. (1985) Further insights into
mechanisms of nickel-induced DNA damage studies with cultured rat
liver cells. In: Brown, S.S. & Sunderman, F.W. Jr, ed. Progress
in Nickel toxicology. Proceedings of the 3rd International
Conference on Nickel Metabolism and Toxicology, Paris, 4-7
September, 1984, Oxford, Blackwell Scientific Publications, pp.
101-104.
SWIERENGA, S.H.H., GILMAN, J.P.W. & MCCLEAN, J.R. (1987) Cancer
risk from inorganics. Cancer Metastasis Rev., 6: 113-154.
SWIERENGA, S.H.H., MARCEAU, N., KATSUMA, Y., FRENCH, S.W., MULLER,
R. & LEE, F. (1989) Altered cytokeratin expression and
differentiation induction during neoplastic transformation of
cultured rat liver cells by nickel subsulfide. Cell Biol. Toxicol.
5(3): 271-286.
SZADKOWSKI, D., SCHULTZE, H., SCHALLER, K.H. & LEHNERT, G. (1969a)
[On the significance of heavy metal contents in cigarettes.] Arch.
Hyg., 153: 1-8 (in German).
SZADKOWSKI, D., KOHLER, G. & LEHNERT, G. (1969b) [Electrolyte-
changes in serum and electrical-mechanical heart action in
industrial heat stress.] Aertzl. Forsch. 23(8): 271-284 (in German).
SZATHMARY, S.C. & DALDRUP, T. (1982) [Determination of nickel by
GC, GC-MS and AAS in biological materials in a case of lettral
poisoning.] Fresenius Z. anal. Chem., 313: 48 (in German).
TABILLION, R., WEBER, F. & KALTWASSER, H. (1980) Nickel
requirement for chemolithotropic growth in hydrogen oxidizing
bacteria. Arch. Microbiol., 124: 131-136.
TAGAKI, Y., MATSUDA, S., IMAI, S., OHMORI, Y., MASUDA, T., VINSON,
J.A., MEHRA, M.C., PURI, B.K. & KANIEWSKI, A. (1986) Trace
elements in human air. Bull. environ. Contam. Toxicol., 36:
793-899.
TAKENAKA, S., HOCHRAINER, D. & OLDIGES, H. (1985) Alveolar
proteinosis induced in rats by long-term inhalation of nickel
oxide. In: Brown, S.S. & Sunderman, F.W. Jr, ed. Progress in
nickel toxicology. Proceedings of the 3rd International Conference
on Nickel Metabolism and Toxicology, Paris, 4-7 September, 1984,
Oxford, Blackwell Scientific Publications, pp. 89-92.
TANAKA, I. (1985) Ion-chromatographic determination of divalent
metal ions (Co, Ni, Cu, Zn, Cd) using EDTA as complexing agent.
Fresenius Z. anal. Chem., 320(2): 125-127.
TANAKA, I., ISHIMATSU, S., MATSUNO, K., KODAMA, Y. & TSYCHIYA, K.
(1985) Biological half time of deposited nickel oxide aerosol in
rat lung by inhalation. Biol. Trace Elem. Res., 8: 203-210.
TANAKA, I., ISHIMATSU, S., HARATAKE, J., HORIE, A. & KODAMA, Y.
(1988) Biological half-time in rats exposed to nickel monosulfide
(amorphous) aerosol by inhalation. Biol. Trace Elem. Res., 17:
237-246.
TANDON, S.K. (1982) Disposition of nickel-63 in the rat. Toxicol.
Lett., 10(1): 71-73.
TANDON, S.K., MATHUR, A.K. & GAUR, J.S. (1977) Urinary excretion
of chromium and nickel among electroplaters and pigment industry
workers. Int. Arch. occup. environ. Health, 40(1): 71-76.
TASK GROUP ON LUNG DYNAMICS (1966) Deposition and retention models
for internal dosimetry of the human respiratory tract. Health
Phys., 12: 173- 207.
TATARSKAYA, A.A. (1960) [Occupational disease of upper respiratory
tract in persons employed in electrolytic nickel refining
departments.] Gig. Tr. prof. Zabol., 6: 35-38 (in Russian).
TATARSKAYA, A.A. (1965) [On the problem of occupational cancer of
the upper respiratory tract in the nickel-refining industry.] Gig.
Tr. prof. Zabol., 9(2): 22-25 (in Russian).
TAYLOR, D., MADDOCK, B.G. & MANCE, G. (1985a) The acute toxicity
of nine gray list metals (arsenic, boron, chromium, copper, lead,
nickel, tin, vanadium, and zinc) to two marine fish species: dab
( Limanda limanda) and grey mullet (Chelon labrosus). Aquat.
Toxicol., 7(3): 135-144.
TAYLOR, P., DAMS, R. & HOSTE, R. (1985b) The determination of
cadmium, chromium, copper, nickel and zinc in city waste
incinerator ash using inductively coupled plasma-atomic emission
spectrometry. J. anal. Let., 18(A19): 2361-2368.
TAYTON, K.J.J. (1980) Ewing's sarcoma at the site of a metal plate.
Cancer, 45: 413-415.
TEDESCHI, R.E. & SUNDERMAN, F.W. (1957) Nickel poisoning. V. The
metabolism of nickel under normal conditions and after exposure to
nickel carbonyl. Arch. ind. Health, 16: 486-488.
TERAKI, Y. & UCHIUMI, A. (1988) Experimental studies on metal
carcinogenesis: a comparative assessment of nickel and lead. In:
Fourth International Conference on Nickel Metabolism and
Toxicology, Abstracts, Espoo, Finland, 5-9 September, 1988,
Helsinki, Institute of Occupational Health, p. 5.
THURSTON, G.D. & SPENGLER, J.D. (1985) A quantitative assessment
of source contributions to inhalable particulate matter pollution
in metropolitan Boston. Atmos. Environ., 19(1): 9-25.
TIMOURIAN, H. & WATCHMAKER, G. (1972) Nickel uptake by sea urchin
embryos and their subsequent development. J. exp. Zool., 182:
379-388.
TOLA, S., KILPIOE, J. & VIRTAMO, M. (1979) Urinary and plasma
concentrations of nickel as indicators of exposure to nickel in an
electroplating shop. J. occup. Med., 21(3): 184-188.
TOLOT, F., BROUDEUR, P. & NEULAT, G. (1956) Troubles pulmonaires
asthmatiformes chez des ouvriers exposés à l'inhalation de chrome,
nickel et aniline. Arch. Mal. prof. Méd. Trav. Séc. soc., 18:
291-293.
TORJUSSEN, W. & ANDERSEN, I. (1979) Nickel concentrations in nasal
mucosa, plasma, and urine in active and retired nickel workers.
Ann. clin. lab. Sci., 9(4): 289-298.
TORJUSSEN, W., SOLBERG, L.A. & HOGETVEIT, A.C. (1979a)
Histopathologic changes of nasal mucosa in nickel workers: a pilot
study. Cancer, 44(3): 963- 974.
TORJUSSEN, W., SOLBERG, L.A. & HOGETVEIT, A.C. (1979b)
Histopathological changes of the nasal mucosa in active and retired
nickel workers. Br. J. Cancer, 40(4): 568-580.
TOSSAVAINEN, A., NURMINEN, M., MUTANEN, P. & TOLA, S. (1980)
Application of mathematical modelling for assessing the biological
half-times of chromium and nickel in field studies. Br. J. ind.
Med., 37(3): 285-291.
TRAUL, K.A., HINK, R.J., WOLFF, J.S. & WLODYMR, K. (1981) Chemical
carcinogenesis in vitro: an improved method for chemical
transformation in Rauscher leukemia virus-infected rat embryo
cells. J. appl. Toxicol., 1(1): 32-37.
TREAGAN, L. & FURST, A. (1970) Inhibition of interferon synthesis
in mammalian cell cultures after nickel treatment. Res. Commun.
chem. Pathol. Pharmacol., 1: 395-401.
TURHAN, U., HAIN, C., WOLLBURG, C. & SZADKOWSKY, D. (1983)
[Chromium and nickel content in human lung. Medical aspects.] In:
[Proceedings of the 1983 Annual Congress of the German Association
for Occupational Medecine, Göttingen], Stuttgart, Gentner, pp. 477-
480 (in German).
TURK, J.L. & PARKER, D. (1977) Sensitization with Cr, Ni, and Zn
salts and allergic type granuloma formation in the guinea-pig. J.
invest. Dermatol., 68: 341-345.
TWEATS, D.J., GREEN, M.H.L. & MURIEL, W.J.A. (1981) A differential
killing assay for mutagens and carcinogens based on an improved
repair-deficient strain of E. coli. Carcinogenesis, 2: 189-194.
UMEDA, M. & NISHIMURA, M. (1979) Inducibility of chromosomal
aberrations by metal compounds in cultured mammalian cells. Mutat.
Res., 67: 221-229.
UPITIS, V.V., NOLLENDORF, A.F. & PAKALNE, D.S. (1980) [Relevance
of nickel for culturing microalgae.] In: Anke, M., Schneider, H.-
J. & Brückner, Chr. ed. [3. Trace Element Symposium Nickel,
Jena, German Democratic Republic, 7-11 July, 1980,] Jena, Friedrich-
Schiller University, pp. 171-183 (in German).
US BUREAU OF MINES (1985) Nickel. In: Minerals yearbook 1984,
Washington, DC, Department of the Interior, Vol. I, pp. 665-684.
US BUREAU OF MINES (1986) Nickel. In: Mineral commodity summaries
1986. An up-to-date summary of 87 nonfuel mineral commodities,
Wahington, DC, Department of the Interior, pp. 108-109.
US EPA (1986) Health assessment document for nickel and nickel
compounds, Washington DC, US Environmental Protection Agency, 438
pp (EPA/600/8-83/012 FF).
US EPA (1987) Acute toxicity handbook of chemicals to estuarine
organisms, Gulf Breeze, Florida, US Environmental Protection
Agency, 274 pp (EPA/600/8-87/017).
US NIOSH (1977a) Special occupational hazard review and control
recommendations for nickel carbonyl. Cincinatti, Ohio, US National
Institute for Occupational Safety and Health, 45 pp (DHEW (NIOSH)
Publication No. 77-184).
US NIOSH (1977b) Criteria for a recommended standard occupational
exposure to inorganic nickel. Cincinatti, Ohio, US National
Institute for Occupational Safety and Health, 282 pp (prepared by
Stanford Research Institute, Menlo Park) (DHEW (NIOSH) Publication
No. 77-164).
VAN SOESTBERGEN, M. & SUNDERMAN, F.W. (1972) 63Ni complexes in
rabbit serum and urine after injection of 63NiCl2. Clin. Chem.
18(12): 1478-1484.
VALENTA, P. OSTAPCZUK, P.H. PIKLAR, B. & NURNBERG, H.W. (1981)
New applications of voltammetry in the determination of toxic trace
metals in food. Proceedings of the 3rd CEC/WHO International
Conference on Heavy Metals in the Environment, Amsterdam, 1981.
Edinburgh, CEP Consultants Ltd, pp. 619-621.
VALENTINE, R. & FISHER, G.L. (1984) Pulmonary clearance of
intratracheally administered 63Ni3S2 in strain A/J mice. Environ.
Res., 34: 328-334.
VAN BAALEN, C. & O'DONNELL, R. (1978) Isolation of a nickel-
dependent blue-green algae. J. gen. Microbiol., 105: 351-353.
VANDENBERG, J. & EPSTEIN,W. (1963) Experimental skin contact
sensitisation in man. J. invest. Dermatol., 41: 413-416.
VAN ROSEN, G. (1954) Breaking of chromosomes by the action of
elements of the periodical system and by some other principles.
Hereditas, 40: 258-263.
VANDENBERG,J. & EPSTEIN, W. (1963) Experimental skin contact
sensitisation in man. J. invest. Dermatol., 41: 413-416.
VERMA, H.S., BEHARI, J.R. & TANDON, S.K. (1980) Pattern of urinary
63Ni excretion in rats. Toxicol. Lett., 5(3-4): 223-226.
VINSON, L.J. & CHOMAN, B.R. (1960) Percutaneous absorption and
surface active agents. J. Soc. Cosmet. Chem., 11: 127-137.
VOGEL, E. (1984) The relation between mutational pattern and
concentration by chemical mutagens in Drosophila. In: Screening
tests in chemical carcinogenesis. Proceedings of a workshop
organized by IARC, and the Commission of the European Communities,
Brussels, 9-12 June, 1975, Lyon, International Agency for Research
on Cancer, pp. 117-137 (IARC Scientific Publications No. 12).
VOLLKOPF, U., GROBENSKI, Z. & WELZ, B. (1981) Determination of
nickel in serum using graphite furnace atomic absorption. At.
Spectrosci., 2: 68-70.
VOLINI, F. DE LA, HUERGA, J. & KENT, G. (1968) Trace metal studies
in liver disease using atomic absorption spectrometry. In:
Sunderman, F.W. & Sunderman, F.W. Jr, ed. Laboratory diagnosis of
liver diseases, St. Louis, Missouri, Warren H. Green, pp. 199-206.
VOROSHILIN, B.I., PLOTKO, E.G., NIKIPHOROVA, V.YA. & FINK, T.V.
(1977) Cytogenetic effects of some inorganic chemicals on human
leukocytes in cultures. In: Problems of genetics and selection in
Urals (information data). Sverdlorsk. The Urals Scientific Center
of the USSR Academy of Sciences, pp. 46-47.
VOS, L., KOMY, Z., REGGERS, G., ROEKENS, E. & VAN GRIEKEN, R.
(1986) Determination of trace metals in rain water by differential-
pulse stripping voltammetry. Anal. chim. Acta, 184: 271-280.
VUOPALA, U., HUHTI, E., TAKKUNEN, J. & HUIKKO, M. (1970) Nickel
carbonyl poisoning. Report of 25 cases. Ann. clin. Res., 2: 214-222.
WACHS, B. (1982) Concentration of heavy metals in fishes from the
river Danube. Z. Wasser Abwasser Forsch., 15(2): 43-84.
WAHLBERG, J.E. (1975) Nickel allergy and atopy in hairdressers.
Contact Dermatit., 1(3): 161-165.
WAHLBERG, J.E. (1976) Sensitization and testing of guinea-pigs with
nickel sulfate. Dermatology, 152: 321-330.
WAHLBERG, J.E., LINDSTEDT, G. & EINARSSON, O. (1977) Chromium,
cobalt and nickel in Swedish cement, detergents, mould and cutting
oils. Berufsdermatosen, 25(6): 220-228.
WAKSVIK, H. & BOYSEN, M. (1982) Cytogenetic analyses of
lymphocytes from workers in a nickel refinery. Mutat. Res., 103:
185-190.
WAKSVIK, H., BOYSEN, M. & HOGETVEIT, A. Chr. (1984) Increased
incidence of chromosomal aberrations in peripheral lymphocytes of
retired nickel workers. Carcinogenesis, 5(11): 1525-1527.
WALL, L.M. & CALNAN, C.D. (1980) Occupational nickel dermatitis in
the electroforming industry. Contact Dermatit., 6(6): 414-420.
WALLACE, A., ROMNEY, E.M., KINNEAR, J. & ALEXANDER, G.U. (1980a)
Single and multiple trace metal excess effects on three different
plant species. J. Plant Nutr., 2: 11-23.
WALLACE, A., ROMNEY, E.M., MULLER, R.T. & ALEXANDER, G.U. (1980b)
Calcium-trace metal interactions in soybean plants. J. Plant Nutr.
2: 79-86.
WALTHARD, B. (1926) [Experimental nickel idiosyncrasy in laboratory
animals.] Schweiz. Med. Wochenschr., 7: 603-604 (in German).
WALTSCHEWA, W., SLATEWA, M. & MICHAILOW, I. (1972) [Testicular
changes due to long-term administration of nickel sulphate in
rats.] Exp. Pathol., 6(3): 116-120 (in German).
WAN, C.C., CHIANG, S. & CORSINI, A. (1985) Two-column method for
preconcentration of trace metals in natural waters on acrylate
resin. Anal. Chem., 57: 719-723.
WARNER, J.S. (1984) Occupational exposure to airborne nickel in
producing and using primary nickel products. In: Nickel in the
human environment, Proceedings of a Joint Symposium, Lyon, 8-11
March, 1983, Lyon, International Agency for Research on Cancer, pp.
419-437 (IARC Scientific Publications No. 53).
WARNER, J.S. (1985) Estimating past exposures to airborne nickel
compounds in the Copper Cliff Sinter Plant. In: Brown, S.S. &
Sunderman, F.W. Jr, ed. Progress in nickel toxicology. Proceedings
of the 3rd International Conference on Nickel Metabolism and
Toxicology, Paris, 4-7 September, 1984, Oxford, Blackwell
Scientific Publications, pp. 203-206.
WASE, A.W., GOSS, D.M. & BOYD, M.J. (1954) The metabolism of
nickel. I. Spatial and temporal distribution of Ni63 in the mouse.
Arch. Biochem. Biophys., 51: 1-4.
WATANABE, H., GOTO, K., TAGUCHI, S., MCLAREN, J.W., BERMAN, S.S. &
RUSSELL, O.S. (1981) Preconcentration of trace elements in sea-
water by complexation with 8-hydroxyquinoline and adsorption on
C18-bonded silica gel. Anal. Chem., 53: 738-739.
WATERMAN, A.H. & SCHRIK, J.J. (1985) Allergy in hip arthroplasty.
Contact Dermatit., 13(5): 294-301.
WATERS, M.D., GARDNER, D.E. & COFFIN, D.L. (1975) Toxicity of
metallic chlorides and oxides for rabbit alveolar macrophages in
vitro. Environ. health Perspect., 10: 267-268.
WATRAS, C.J., MACFARLANE, J. & MOREL, F.M.M. (1985) Nickel
accumulation by Scenedesmus and Daphnia: Food-chain transport and
geochemical implications. Can. J. Fish. aquat. Sci., 42(4): 724-730.
WEAST, R.C. (1981) CRC Handbook of chemistry and physics, 62nd ed.
Boca Raton, Florida, CRC Press, pp. B123-B124.
WEBER, P.C. (1986) Epithelioid sarcoma in association with total
knee replacement. J. bone joint Surg., B68: 824-826.
WEBSTER, J.D., PARKER, T.F., ALFREY, A.C., SMYTHE, W.R., KUBO, H.,
NEAL, G. & HULL, A.R. (1980) Acute nickel intoxication by
dialysis. Ann. intern. Med., 92: 631-633.
WEHNER, A.P. & CRAIG, D.K. (1972) Toxicology of inhaled NiO and
CoO in Syrian golden hamsters. Am. Ind. Hyg. Assoc. J., 33: 146-155.
WEHNER, A.P., BUSCH, R.H., OLSON, R.J. & CRAIG, D.K. (1975)
Chronic inhalation of nickel oxide and cigarette smoke by hamsters.
Am. Ind. Hyg. Assoc. J., 36(11): 801-810.
WEHNER, A.P., MOSS, O.R., MILLIMAN, E.M., DAGLE, G.E. & SCHIRMER,
R.E. (1979) Acute and subchronic inhalation exposures of hamsters
to nickel-enriched fly ash. Environ. Res., 19(2): 355-370.
WEHNER, A.P., DAGLE, G.E. & MILLIMAN, E.M. (1981) Chronic
inhalation exposure of hamsters to nickel-enriched fly ash.
Environ. Res., 26(1): 195- 216.
WEIDENAUER, M. & LIESER, K.H. (1985) [Determination of trace
elements in river water by means of voltammetry.] Fresenius Z.
anal. Chem., 320: 550-555 (in German).
WEINZIERL, J. & UMLAND, F. (1982) [Determination of Co (and Ni) in
the ppb-range by differential pulse polarography.] Fresenius Z.
anal. Chem., 312: 608-610 (in German).
WELLENREITER, R.H., ULLREY, D.E. & MILLER, E.R. (1970) Nutritional
studies with nickel. In: Mills, C.F. ed. Trace element metabolism
in animals. Proceedings of 1st WAAP/IBP International Symposium,
Aberdeen, Scotland, July 1969, Edinburgh, London, Livingstone, pp.
52-58.
WELLS, G.C. (1956) Effects of nickel on the skin. Br. J. Dermatol.
68: 237-242.
WEST, B. & SUNDERMAN, F.W. (1958) Nickel poisoning. VII. The
therapeutic effectiveness of alkyl dithiocarbamates in experimental
animals exposed to nickel carbonyl. Am. J. med. Sci., 236: 15-25.
WHANGER, P.D. (1973) Effects of dietary nickel on enzyme activities
and mineral contents in rats. Toxicol. appl. Pharmacol., 25: 323-331.
WHITE, D.H., KING, K.A., MITCHELL, L.A. & MULHERN, B.M. (1986)
Trace elements in sediments, water, and american coots ( Fulica
americana) at a coal-fired power plant in Texas. Bull environ.
Contam. Toxicol., 36(3): 376-383.
WHO (1984) Guidelines for drinking-water quality. Vol. 1 -
Recommendations, Geneva, World Health Organization, p. 57.
WHO (1988) Air quality guidelines for Europe, Copenhagen, World
Health Organization, pp. 285-296 (WHO Regional Publications,
European Series No 23).
WIERNIK, A., JOHANSSON, A., JARSTRAND, C. & CAMNER, P. (1983)
Rabbit lung after inhalation of soluble nickel. I. Effects on
alveolar macrophages. Environ. Res., 30: 129-141.
WILLS, M.R., BROWN, C.S., BERTHOLF, R.L. & SAVORY, J. (1985) Serum
and lymphocyte aluminium and nickel in chronic renal failure. Clin.
chim. Acta, 145: 193-196.
WILSON, J.D., STENZEL, M.R., LOMBARDOZZI, K.L. & NICHOLS, C.L.C.
(1981) Monitoring personal exposure to stainless steel welding
fumes in confined spaces at a petrochemical plant. Am. Ind. Hyg.
Assoc. J., 42: 431-436.
WILSON, L.W. & DINUNZIO, J.E. (1981) Enrichment of nickel and
cobalt in natural hard water by Donnan Dialysis. Anal. Chem., 53:
692-695.
WILSON, W.W. & KHOOBYARIAN, N. (1982) Potential identification of
chemical carcinogens in a viral transformation system. Chem. biol.
Interact., 38: 253- 259.
WINDHOLZ, M., BUDAVARI, S., BLUMETTI, R.F. & OTTERBEIN, E.S.
(1983) The Merck Index, 10th ed. Rahway, New Jersey, Merck & Co.
Inc.
WOLNIK, K.A., FRICKE, F.L., CAPAR, S.C., BRAUDE, G.L., MEYER, M.W.,
SATZGER, R.D. & KUENNEN, R.W. (1983) Elements in major raw
agricultural crops in the United States. 2. Other elements in
lettuce, peanuts, potatoes, soybeans, sweet corn and wheat. J.
Agric. food Chem., 31: 1244-1249.
WOLNIK, K.A., FRICKE, F.L., CAPAR, S.G., MEYER, M.W., SATZGER,
R.D., BONNIN, E. & GASTON, C.M. (1985) Elements in major raw
agricultural crops in the United States. 3. Cadmium, lead, and
eleven other elements in carrots, field corn, onions, rice, spinach
and tomatoes. J. Agric. food. Chem., 33: 807-811.
WULF, H.C. (1980) Sister chromatid exchanges in human lymphocytes
exposed to nickel and lead. Danish med. Bull., 27: 40-42.
YAMASHIRO, S., GILMAN, J.P.W., HULLAND, T.J. & ABANDOWITZ, H.M.
(1980) Nickel sulphide-induced rhabdomyosarcomata in rats. Acta
pathol. Jpn., 30(1): 9-22.
YAMASHIRO, S., BASRUR, P.K., GILMAN, J.P., HULLAND, T.J. &
FUJIMOTO, Y. (1983) Ultrastructural study of Ni3S2-induced tumors
in rats. Acta pathol. Jpn., 33(1): 45-58.
YAN, N.D. & STRUS, R. (1980) Crustacean zooplankton communities of
acidic, metal-contaminated lakes near Sudbury, Ontario. Can. J.
Fish. aquat. Sci., 37(12): 2282-2294.
YAN, N.D., MILLER, G.E., WILE, I. & HITCHIN, G.G. (1985) Richness
of aquatic macrophyte floras of soft water lakes of differing pH
and trace metal content in Ontario, Canada. Aquat. Bot., 23(1): 27-40.
YANG, X.H., BROOKS, R.R., JAFFRE, T. & LEE, J. (1985) Elemental
levels and relationships in the flacourtiaceae of New Caledonia and
their significance for the evaluation of the serpentine problem.
Plant Soil, 87(2): 281-292.
YARITA, T. & NETTESHEIM, P. (1978) Carcinogenicity of nickel
subsulfide for respiratory tract mucosa. Cancer Res., 38: 3140-3145.
YOSHIMURA, K., THOSHIMITSU, Y. & OHASKI, S. (1980) Ion-exchanger
colorimetry - VI micro-determination of nickel in natural water.
Talanta, 27: 693-697.
ZAKOUR, R.A., TKESHELASHVILI, L.K., SHERMAN, C.W., KOPLITZ, L.M. &
LOEB, L.A. (1981) Metal-induced infidelity of DNA synthesis. J.
cancer Res. clin. Oncol., 99: 187-196.
ZISLIN, D.M., GANYUSKINA, S.M., DUBINIA, E.S. & TYUSHNYAKOVA, N.V.
(1969) [Residual air in a complex evaluation of the respiratory
system function in initial and suspected pneumonconiosis.] Gig.
Tr. prof. Zabol., 13: 26-29 (in Russian).
ZOBER, A. (1981a) [Symptoms and findings at the bronchopulmonary
system of electric arc welders. II. communication: pulmonary
fibrosis.] Zentralbl. Bakteriol. Hyg. I. Abt. Orig. B, 173: 120-148
(in German).
ZOBER, A. (1981b) [Symptoms and findings at the bronchopulmonary
system of electric arc welders. I. communication: epidemiology.]
Zentralbl. Bakt. Hyg. I. Abt. Orig. B, 173: 92-119 (in German).
ZOBER, A. (1982) [Possible dangers to the respiratory tract from
welding fumes.] Schweissen Schneiden, 34: 77-81 (in German).
ZOBER, A., KICK, K., SCHALLER, K.H., SCHELLMANN, B. & VALENTIN, H.
(1984) [Nickel and chromium content of selected human organs and
body fluids.] Zentralbl. Bakteriol. Hyg. I. Abt. Orig. B , 179: 80-95
(in German).
RESUME ET CONCLUSIONS
1. Identité, propriétés physiques et chimiques et méthodes
d'analyse
Le nickel est un élément métallique qui appartient au groupe
VIIIb de la classification périodique. Il résiste aux alcalis mais
se dissout en général dans les acides oxydants dilués. Le
carbonate, le sulfure et l'oxyde sont insolubles dans l'eau, alors
que le chlorure, le sulfate et le nitrate sont solubles. Le nickel
carbonyle est un liquide volatil incolore qui se décompose au-
dessus de 50 °C. Sous forme ionique, le nickel existe
essentiellement à l'état de nickel(II). Dans les systèmes
biologiques, le nickel une fois dissous peut former des complexes
avec divers ligands et se fixer aux matières organiques.
L'analyse des échantillons biologiques et environnementaux se
fait le plus fréquemment par spectroscopie d'absorption atomique et
voltammétrie. Pour obtenir des résultats fiables, en particulier
dans le domaine des ultratraces (ultramicrotraces), il faut suivre
un mode opératoire précis pour réduire au minimum le risque de
contamination lors des prélèvements, de la conservation, du
traitement, et de l'analyse des échantillons. Selon les
traitements préalables subis par l'échantillon et les méthodes
d'extraction ou d'enrichissement utilisées, on peut atteindre des
limites de détection de 1 à 100 ng/litre dans les produits
biologiques et l'eau.
2. Sources d'exposition humaine et environnementale
Le nickel est présent à l'état de traces un peu partout dans le
sol, l'eau, l'air et la biosphère. La teneur moyenne de la croûte
terrestre en nickel est d'environ 0,008%. Les terrains agricoles
en contiennent entre 3 et 1000 mg/kg. Dans les eaux naturelles,
les concentrations varient de 2 à 10 µg/litre (eau douce) et de 0,2
à 0,7 µg/litre (eau de mer). Dans l'atmosphère, les concentrations
en nickel varient de 0,1 à 3 ng/m3, au-dessus des zones écartées.
Les gisements de nickel consistent en accumulations de minéraux
sulfurés (essentiellement de la pentlandite) et en latérites. Le
nickel est extrait de ces minerais par affinage pyro- et hydro-
métallurgique. Le nickel est principalement utilisé pour la
production d'aciers inoxydables qui sont très résistants à la
corrosion et aux températures élevées. On l'utilise sous forme
d'alliages et de dépôts galvaniques pour la confection de pièces
d'automobiles, de machines-outils, d'armements, d'outils, de
matériel électrique, d'appareils ménagers, et de pièces de monnaie.
Les dérivés du nickel sont également utilisés comme catalyseurs,
comme pigments, et dans les batteries d'accumulateurs. En 1985, la
production minière mondiale de nickel a été d'environ 67 millions
de kg. Les principales émissions de nickel dans l'air ambiant
proviennent de la combustion du charbon et du mazout pour le
chauffage et la production d'énergie,de l'incinération des déchets
et des boues de stations d'épuration, de la production minière et
de la métallurgie du nickel, de la fabrication de l'acier, du
nickelage, et de diverses autres sources, comme les cimenteries.
Dans les polluants atmosphériques, le nickel est présent
principalement sous forme de sulfates, d'oxydes, de sulfures, et,
dans une moindre mesure, de nickel métallique.
Le nickel rejeté lors de diverses opérations industrielles ou
autres finit par aboutir dans les eaux usées. Après traitement de
ces dernières, les résidus sont éliminés par injection dans des
puits profonds, décharge en mer, enfouissement dans le sol et
incinération. Les effluents rejetés par les usines de traitement
des eaux usées contiendraient jusqu'à 0,2 mg de nickel par litre.
3. Transport, distribution, et transformation dans l'environnement
Le nickel qui pénètre dans l'environnement peut être d'origine
naturelle ou artificielle et il circule à travers tous les
compartiments du milieu, soit sous l'effet de processus
physicochimiques, soit par transport biologique au sein
d'organismes vivants.
On estime que le nickel atmosphérique est essentiellement
présent sous forme d'aérosols particulaires dont la teneur en
nickel varie en fonction de leur origine. C'est en général les
particules les plus petites qui présentent les teneurs les plus
élevées en nickel. Le nickel carbonyle est instable à l'air et il
se décompose pour former de l'oxyde de nickel.
Le transport et la distribution des particules de nickel en
direction ou à l'intérieur des divers compartiments du milieu
dépend beaucoup de la granulométrie des particules et des
conditions météorologiques. Cette granulométrie dépend à son tour
principalement de la source émettrice. En général, les particules
d'origine artificielle sont de dimension plus réduite que celles
qui proviennent de la poussière naturelle.
Le nickel peut pénétrer dans l'hydrosphère soit à partir de
l'atmosphère, soit par lessivage superficiel, soit encore par
décharge de déchets industriels ou municipaux ou érosion naturelle
des sols et des roches. Dans les cours d'eau, le nickel est
principalement transporté par des particules enrobées de matières
précipitées ou en association avec des matières organiques; dans
les lacs, le transport est assuré sous forme ionique,
essentiellement en association avec des matières organiques. Le
nickel peut également s'adsorber sur des particules d'argile ou
être fixé par des organismes vivants. A l'inverse, il peut se
désorber et s'échapper ainsi des sédiments où il était fixé. Une
partie du nickel ainsi transporté par les cours d'eau aboutit à
l'océan. On estime que l'apport sous forme de particules en
suspension dans les cours d'eau est d'environ 135 x 107 kg/an.
Selon le type de sol, le nickel peut présenter une forte
mobilité et parvenir jusqu'aux eaux souterraines, puis aux lacs et
aux rivières. C'est principalement par leurs racines que les
plantes terrestres fixent le nickel présent dans le sol. La
quantité ainsi fixée dépend de divers paramètres géochimiques et
physiques, notamment le type de sol, son pH, sa teneur en eau et en
matières organiques, ainsi que la concentration en nickel extractible.
L'exemple le mieux connu d'accumulation de nickel est fourni par un
certain nombre d'espèces végétales "hyperaccumulatrices", qui
concentrent le nickel à raison de plus de 1mg/kg de poids sec, et
qui poussent sur des sols serpentinifères relativement stériles. A
partir de 50 mg/kg de poids sec, le nickel est toxique pour la
plupart des végétaux. On a observé une accumulation de nickel et
des effets toxiques chez les végétaux cultivés sur sols traités par
des boues de stations d'épuration ainsi que dans la végétation
située à proximité de sources d'émission. Certaines plantes
aquatiques concentrent fortement le nickel. Des études de
laboratoire portant sur des poissons ont montré que le nickel
n'avait guère tendance à s'accumuler, quelques que soient les
espèces étudiées. Dans des eaux non polluées, les concentrations
relevées dans le poisson entier (et rapportées au poids de tissus
frais), variaient de 0,02 à 2 mg/kg. Ces valeurs pourraient être
jusqu'à 10 fois supérieures dans les poissons qui vivent en eau
polluée. Toutefois rien n'indique que le nickel subisse une
bioamplification tout au long de la chaîne alimentaire.
Les animaux sauvages, par exemple, les herbivores et leurs
prédateurs carnivores, absorbent du nickel par voie alimentaire,
qui se retrouve ensuite dans de nombreux organes et tissus.
4. Concentrations dans l'environnement et exposition humaine
Dans les organismes terrestres et aquatiques, les
concentrations de nickel peuvent varier de plusieurs ordres de
grandeur. L'homme est exposé à des concentrations atmosphériques
dont les valeurs caractéristiques vont de 5 à 35 ng/m3 en zones
rurales et urbaines, d'où une absorption par inhalation de 0,1 à
0,7 µg de nickel par jour. L'eau de boisson en contient
généralement moins de 10 µg/litre mais il arrive que du nickel soit
libéré dans l'eau d'adduction par les éléments de la plomberie et
atteigne des concentrations de 500 µg/litre.
Dans les produits alimentaires, les concentrations sont
généralement inférieures à 0,5 mg/kg en poids de substance fraîche.
Le cacao, les graines de soja, certains légumes secs, diverses
noix, et la farine d'avoine en contiennent de fortes
concentrations. L'ingestion journalière de nickel par la voie
alimentaire est très variable du fait de la diversité des habitudes
alimentaires: les quantités ingérées peuvent varier de 100 à 800
µg/jour; en moyenne l'apport alimentaire dans la plupart des pays
est de 100 à 300 µg/jour. La libération de nickel par les
ustensiles de cuisine peut augmenter sensiblement les quantités
ingérées. Chez les fumeurs, les quantités absorbées par voie
pulmonaire peuvent aller de 2 à 23 µg/jour pour une consommation
quotidienne de 40 cigarettes.
L'exposition cutanée dans l'environnement général joue un rôle
important dans l'apparition et l'entretien d'une hypersensibilité
de contact qui peut être due à un contact quotidien avec des objets
nickelés ou en alliage de nickel (par exemple des bijoux, des
pièces de monnaie, des agrafes).
Les implants et prothèses en alliage de nickel, les liquides
pour injection intraveineuse ou dialyse, et les produits de
contraste utilisés en radiographie peuvent entraîner une exposition
iatrogène au nickel. On estime qu'un malade sous dialyse reçoit à
chaque traitement environ 100 µg de nickel en moyenne par voie
intra-veineuse.
Dans les ambiances de travail, les concentrations de nickel en
suspension dans l'air peuvent varier de quelques µg jusqu'à,
parfois, plusieurs mg/m3, selon le processus industriel et la
teneur en nickel des produits manipulés.
Partout dans le monde, des millions de travailleurs sont
exposés à des poussières et des fumées contenant du nickel lors de
travaux tels que le soudage, le nickelage, le broyage, l'extraction
minière, l' affinage du nickel ainsi que dans les aciéries,
fonderies, et autres industries métallurgiques.Une exposition
cutanée au nickel peut intervenir à l'occasion des activités
professionnelles les plus diverses, qu'il s'agisse d'une exposition
directe à du nickel dissous, par exemple, lors de l'affinage, du
nickelage ou de l'électroformage ou lors de la manipulation
d'outils contenant du nickel. Le nettoyage par voie humide peut
comporter une exposition au nickel qui s'est dissous dans les eaux
de lavage.
5. Cinétique et métabolisme chez l'homme et l'animal
Chez l'homme et l'animal le nickel peut être absorbé par
inhalation, ingestion, ou par voie percutanée. En cas d'exposition
professionnelle, la principale voie de pénétration dans l'organisme
est l'absorption respiratoire avec résorption gastro-intestinale
secondaire (formes solubles et insolubles). Une proportion
importante des matières inhalées est avalée après avoir été
éliminée des voies respiratoires sous l'action de l'ascenseur
mucociliaire. Une hygiène personnelle insuffisante et de mauvaises
habitudes de travail peuvent accroître l'exposition par voie
digestive. L'absorption percutanée est négligeable
quantitativement mais joue un rôle important dans la pathogénèse de
l'hypersensibilité de contact. La résorption dépend de la
solubilité du composé et elle décroît en général dans l'ordre
suivant: nickel carbonyle > composés solubles > composés
insolubles. De tous les dérivés du nickel, c'est le nickel
carbonyle qui est le plus rapidement et le plus complètement
résorbé dans l'organisme de l'homme et des animaux. Les études
comportant sur l'administration de nickel par voie respiratoire
sont limitées. Des études au cours desquelles on a administré à
des hamsters et à des rats de l'oxyde de nickel insoluble ont
montré que celui-ci était faiblement résorbé, la plus grande partie
du produit étant retenue dans les poumons après plusieurs semaines.
En revanche, la résorption du chlorure de nickel soluble et du
sulfure de nickel amorphe ont été rapides. Dans le sang, le nickel
est principalement transporté par l'albumine.
Le nickel est plus ou moins résorbé au niveau gastrointestinal
en fonction de la composition de l'alimentation. Lors d'une étude
récente sur des volontaires humains, on a constaté que le nickel
était résorbé à hauteur de 27% à partir de l'eau, contre moins de
1% à partir de la nourriture. L'excrétion peut s'effectuer en
principe dans toutes les sécrétions et notamment l'urine, la bile,
la sueur, les larmes, le lait et le liquide muco-ciliaire. Le
nickel qui n'est pas résorbé est éliminé dans les matières fécales.
On a mis en évidence l'existence d'un transfert transplacentaire
chez des rongeurs. Après administration parentérale de sels de
nickel, on a constaté que c'est au niveau des reins, des glandes
endocrines, des poumons et du foie que l'accumulation de nickel
était la plus importante: des concentrations élevées ont été
également observées dans l'encéphale après administration de nickel
carbonyle. Les données relatives à l'excrétion correspondent à un
modèle bicompartimental. Chez des adultes en bonne santé exposés
en dehors de leur activité professionnelle, on a observé des
concentrations sériques et urinaires de 0,2 µg/litre (limites:
0,05-1,1 µg/litre) et de 1,5 µg/g de créatinine (limites: 0,5-4,0
µg/g de créatinine), respectivement. Après exposition
professionnelle, on a constaté une augmentation des concentrations
de nickel dans ces deux liquides. Pour un adulte non exposé de 70
kg, la charge de l'organisme en nickel est de 0,5 µg.
6. Effets sur les êtres vivants dans leur milieu naturel
Chez les microorganismes, on a constaté une inhibition de la
croissance lorsque la concentration en nickel dans le milieu était
de 1 à 5 mg/litre (cas des actinomycètes, des levures et des
eubactéries marines ou non marines) ou de 5 à 1000 mg/litre (cas des
champignons filamenteux). Dans le cas des algues, on n'a pas
observé de croissance à des concentrations d'environ 0,05-5 mg de
nickel par litre. La toxicité du nickel dépend également de
facteurs abiotiques tels que le pH, la dureté, la température et la
salinité du milieu ainsi que de la présence de particules
organiques ou minérales.
Chez les invertébrés aquatiques, la toxicité du nickel varie
dans de très importantes proportions selon l'espèce et les facteurs
abiotiques. Chez la daphnie, on a observé une CL50 à 96 h. de 0,5
mg/litre alors que chez trois mollusques, deux gastéropodes d'eau
douce et un bivalve, ce paramètre était respectivement égal à 0,2
mg/litre et 1100 mg/litre.
Chez les poissons, les valeurs de la CL50 à 96 h. sont
généralement comprises entre 4 et 20 mg/litre mais elles peuvent
être plus élevées chez certaines espèces. Des études à long terme
sur les poissons et leur développement effectuées dans des eaux
douces ont montré qu'à des concentrations ne dépassant pas
0,05 mg/litre, le nickel exerçait certains effets, en particulier
sur la truite arc-en-ciel. Pour les plantes terrestres, des
concentrations de nickel dépassant 50 mg/kg de poids à sec sont
généralement toxiques. On a observé que le cuivre potentialisait
la toxicité du nickel alors que le calcium la réduisait. On ne
dispose que de données limitées au sujet des effets du nickel sur
les animaux terrestres.
Les lombrics paraissent être relativement insensibles au
nickel, du moins si le milieu abonde en microorganismes et matières
organiques qui captent une partie du nickel aux dépens des vers.
On n'a pas envisagé la possibilité que le nickel constitue un
polluant majeur à l'échelon planétaire encore que des modifications
écologiques, par exemple une diminution du nombre et de la
diversité des espèces ait été observée à proximité de sources de
nickel. En outre, des études portant sur les microécosystèmes ont
montré qu'un apport de nickel dans le sol perturbait le cycle de
l'azote.
7. Effets sur les animaux d'experience et sur les systemes
d'épreuve in vitro
Le nickel joue un rôle essentiel dans l'activité catalytique de
certaines enzymes végétales et bactériennes. Chez certaines
espèces animales on a observé, après administration d'un régime
carencé en nickel, un ralentissement du gain de poids, de l'anémie
et une moindre viabilité de la progéniture.
Le dérivé du nickel qui présente la plus forte toxicité aiguë
est le nickel carbonyle dont le poumon constitue l'organe cible.
Ce composé peut provoquer un oedème pulmonaire en l'espace de 4
heures. Les autres dérivés du nickel présentent une faible
toxicité aiguë.
Si les allergies de contact sont très fréquentes chez l'homme,
on ne parvient à produire une sensibilisation expérimentale chez
l'animal que dans certaines conditions particulières. Chez le rat,
la souris et le cobaye on a provoqué des lésions des muqueuses et
des réactions inflammatoires des voies respiratoires en faisant
inhaler pendant une longue période à ces animaux du nickel
métallique, de l'oxyde et du sous-sulfure de nickel. On a observé
une hyperplasie de l'épithélium chez des rats après inhalation
d'aérosols de chlorure et d'oxyde de nickel.
Chez le rat, une exposition intense et prolongée à de l'oxyde
de nickel a provoqué une pneumoconiose progressive. Des réactions
inflammatoires quelquefois accompagnées d'une légère fibrose ont
été observées chez des lapins après exposition intense à la
poussière de nickel et de graphite. L'exposition de rats à du
sous-sulfure de nickel a provoqué une fibrose pulmonaire.
Des sels de nickel, administrés par voie parentérale à des
rats, des lapins et des poulets ont provoqué une hyperglycémie
transitoire rapide. Ce phénomène pourrait être lié aux effets qui
ont été observés sur les cellules alpha et béta des ilots de
Langerhans. Le nickel réduit également la libération de
prolactine. L'administration par voie orale ou par inhalation de
chlorure de nickel a réduit la fixation d'iode par la thyroïde.
Administrés par voie intraveineuse à des chiens, des sels de
nickel ont entraîné une réduction du débit sanguin des artères
coronaires; à forte concentration, le nickel diminue la
contractilité du myocarde du chien in vitro.
Le chlorure de nickel perturbe le système des cellules T et
inhibe l'activité des cellules tueuses naturelles (cellules NK).
On a observé que l'administration parentérale de chlorure et de
sous-sulfure de nickel entraînait la mort intrautérine des foetus
et réduisait le gain de poids chez le rat et la souris. Chez des
rats et des hamsters, l'inhalation de nickel carbonyle a provoqué
la mort des foetus et une diminution de leur poids, avec en outre
des effets tératogènes. Les études en question ne donnaient aucune
information sur la toxicité des composés du nickel pour les
femelles gestantes. On a fait état de mutations létales dominantes
obtenues chez des rats par exposition au nickel carbonyle.
Un certain nombre de dérivés minéraux du nickel ont fait
l'objet d'études de mutagénicité dans divers systèmes d'épreuve.
En général les dérivés du nickel se révèlent inactifs chez les
bactéries sauf dans le cas des tests de fluctuation. On a observé
des mutations chez plusieurs types de cellules mamaliennes en
culture. Les dérivés du nickel inhibent la synthèse de l'ADN chez
un grand nombre d'organismes. En outre, ces dérivés produisent des
aberrations chromosomiques et des échanges entre chromatides soeurs
tant dans les cellules mamaliennes que dans les cellules humaines
en culture. Chez des personnes exposées, de par leur profession, à
des composés insolubles ou solubles du nickel on a effectivement
observé des aberrations chromosomiques, mais pas d'échange entre
chromatides soeurs (sauf dans le cas d'une étude portant sur des
ouvriers travaillant dans des ateliers d'électrolyse). In vitro,
le nickel provoque la transformation des cellules.
Lors d'une étude d'inhalation, on a observé que du sous-sulfure
de nickel provoquait l'apparition de tumeurs pulmonaires bénignes
et malignes chez le rat. Quelques tumeurs pulmonaires ont
également été observées chez des rats soumis à une série d'études
où on leur faisait inhaler du nickel carbonyle. On n'a pas noté
d'augmentation significative des tumeurs pulmonaires chez des rats
soumis à l'inhalation de nickel métallique dans des conditions
d'étude satisfaisantes. L'inhalation d'oxyde noir n'a pas provoqué
non plus l'apparition de tumeurs pulmonaires chez des hamsters
dorés (espèce qui résiste à la cancérogénèse au niveau pulmonaire).
On ne dispose pas de bonnes études de cancérogénicité basées sur
l'exposition par inhalation à d'autres composés du nickel.
Toutefois on a constaté qu'après l'instillation intratrachéenne
réitérée de sous-sulfure de nickel, de poudre de nickel métallique
et d'un oxyde non précisé, des tumeurs pulmonaires bénignes se
formaient chez le rat.
Le nickel carbonyle, le nickelocène, ainsi qu'un grand nombre
de composés légèrement solubles ou insolubles comme le sous-sulfure
de nickel, le carbonate, le chromate, l'hydroxyde et divers
sulfures, des seléniures, des arséniures, des téllurures, des
antimoniures, différents oxydes non spécifiés, deux oxydes de
nickel et de cuivre, du nickel métallique et divers alliages, ont
provoqué l'apparition de tumeurs mésenchymateuses locales chez
divers animaux d'ex-périence après administration intramusculaire,
sous-cutanée, intrapéritonéale, intrapleurale, intraoculaire,
intraosseuse, intrarénale, intra-articulaire, intratesticulaire et
intra-adipeuse. On n'a pas observé d'effet cancérogène local lors
d'une étude portant sur des doses uniques de certains alliages de
nickel, d'hydroxyde colloïdal de nickel et de deux échantillons
d'oxyde, préparés spécialement en vue d'une étude de
cancérogénicité par calcination à 735 et 1045 °C.
On a provoqué l'apparition de tumeurs de la cavité péritonéale
chez des rats par administration intrapéritonéale réitérée de
sulfate et d'acétate de nickel; toutefois aucun effet n'a été
obtenu avec le chlorure de nickel.
On a étudié la cancérogénicité du nickel métallique et d'un
grand nombre de dérivés du nickel par administration parentérale; à
de rares exceptions près, toutes ces substances ont provoqué
l'apparition de tumeurs locales.
C'est seulement dans le cas du sous-sulfure de nickel qu'on a
des preuves convaincantes d'un pouvoir cancérogène après exposition
par voie respiratoire. Toutefois le nombre d'études de ce type qui
sont vraiment satisfaisantes reste très limité.
Lors d'études au cours desquelles on a procédé à des
instillations intratrachéennes répétées de poudre de nickel,
d'oxyde et de sous-sulfure de nickel, on a observé l'apparition de
tumeurs pulmonaires.
Trois sels solubles de nature différente qui n'avaient pas
produit de tumeurs locales lors d'études antérieures ont été
réétudiés en procédant cette fois à une administration
intrapéritonéale réitérée: deux de ces sels ont alors manifesté des
effets cancérogènes.
8. Effets sur l'homme
C'est le nickel carbonyle qui présente la toxicité aiguë la
plus forte pour l'homme. Elle se traduit par des céphalées
frontales, des vertiges, de la nausée, des vomissements, de
l'insomnie et de l'irritabilité, puis par des symptômes pulmonaires
évoquant une pneumonie d'origine virale. Parmi les lésions
pulmonaires patho-logiques qui ont été observées on peut citer des
hémorragies, de l'oedème et une désorganisation cellulaire. Le
foie, les reins, les surrénales, la rate et le cerveau sont
également affectés. On a aussi fait état d'intoxications par le
nickel chez des malades sous dialyse après contamination du liquide
de dialyse par des dérivés du nickel ainsi que chez des nickeleurs
qui avaient bu accidentellement de l'eau contaminée par du sulfate
et du chlorure de nickel.
On a signalé chez des nickeleurs et chez des ouvriers
d'ateliers d'affinage du nickel des effets chroniques tels que
rhinite, sinusite, perforation de la cloison nasale et asthme.
Certains auteurs font état d'anomalies pulmonaires, notamment une
fibrose, chez des ouvriers amenés à inhaler de la poussière de
nickel. En outre, on a signalé des cas de dysplasie nasale chez des
ouvriers travaillant dans des ateliers d'affinage du nickel.
L'hypersensibilité de contact est très largement documentée tant
dans la population générale que chez un certain nombre d'ouvriers
exposés de par leur profession à des dérivés solubles du nickel.
Dans plusieurs pays on indique que 10% de la population féminine et
1% de la population masculine présentent une sensibilité au nickel.
Parmi ces personnes, 40 à 50% présentent un eczéma vésiculeux des
mains qui peut être très grave dans certains cas et entraîner une
incapacité. L'ingestion de nickel peut aggraver l'eczéma siégeant
au niveau des mains ou d'autres régions du corps protégées de tout
contact avec du nickel.
On a également signalé que les prothèses et autres implants
chirurgicaux en alliage de nickel pouvaient produire une
sensibilisation à ce métal ou aggraver une dermatite préexistante.
Des effets néphrotoxiques, comme un oedème rénal avec hyperémie
et dégénérescence du parenchyme, ont été observés dans des cas
d'exposition industrielle accidentelle à du nickel carbonyle. Des
effets néphrotoxiques passagers ont été enregistrés après ingestion
accidentelle de sels de nickel.
On a fait état d'un risque très élevé de cancers pulmonaire et
nasal chez des ouvriers travaillant dans des ateliers d'affinage du
nickel où l'on effectue le grillage à mort des minerais sulfurés,
opération qui entraîne une exposition importante au sous-sulfure, à
l'oxyde et éventuellement au sulfate de nickel. L'exposition à des
dérivés solubles du nickel (électrolyse, extraction du sulfate de
cuivre, hydrométallurgie), souvent associée à une exposition à
l'oxyde de nickel mais sans exposition trop importante au sous-
sulfure, comporte des risques analogues. Pour les mineurs et les
ouvriers des ateliers d'affinage, le risque est beaucoup plus
faible. Chez les soudeurs d'acier inoxydable ou chez ceux qui
travaillent dans des industries employant du nickel, les taux de
cancer sont généralement proches de la normale, sauf dans le cas où
il y a également exposition au chrome, en particulier dans des
ateliers de galvanoplastie. Il semblerait cependant que les
ouvriers employés à la fabrication des accumulateurs au
nickel/cadmium, qui sont exposés à de fortes concentrations de
nickel et de cadmium, soient exposés à un risque légèrement plus
important de cancer du poumon.
On a signalé occasionnellement un excès de divers cancers
autres que ceux qui siègent au niveau des poumons ou du nez, par
exemple des cancers rénaux, gastriques ou prostatiques, chez des
travailleurs de l'industrie du nickel, mais cette surmorbidité n'a
pas été systématiquement constatée. On peut s'appuyer sur les
données épidémiologiques pour répondre à deux questions
importantes: i) certains dérivés du nickel sont-ils effectivement
cancérogènes, et ii) l'étude de cohortes faiblement exposées
permet-elle d'établir la limite supérieure du risque pour des
niveaux d'exposition donnés?
a) Dérivés solubles
On a mis en evidence un certain risque de cancer chez des
ouvriers exposés à des concentrations en sels solubles de l'ordre
de 1 à 2 mg/m3, qu'il s'agisse d'ouvriers d'ateliers d'électrolyse
ou travaillant à la préparation de sels solubles. Ces ouvriers
étaient également exposés à d'autres dérivés du nickel mais souvent
à des concentrations plus faibles que dans les processus à haut
risque. En l'absence d'une évaluation rétrospective de
l'exposition, il est impossible de tirer des conclusions
définitives de ces observations mais il existe certainement une
forte présomption d'une cancérogénicité des dérivés solubles du
nickel. Les poussières des ateliers d'affinage contiennent
quelques fois une proportion non négligeable de sulfate de nickel à
côté du sous-sulfure. Il se pourrait donc que le risque de cancer
très important qui a été observé chez les ouvriers qui travaillent
au grillage à mort des minerais sulfurés soit dû pour une part aux
dérivés solubles du nickel.
b) Sous-sulfure de nickel (sulfure nickeleux)
Dans les ateliers d'affinage où le risque de cancer est élevé,
on a constaté que l'exposition au sous-sulfure de nickel était
toujours accompagnée d'une exposition à l'oxyde et peut-être même
au sulfate (voir plus haut). Il est donc difficile à partir des
seules données épidémiologiques d'affirmer que le sous-sulfure est
cancérogène, encore que ce soit probable.
c) Oxyde de nickel
Dans tous les cas où l'on a observé un risque élevé de cancer,
il y avait présence d'oxyde de nickel accompagné d'une ou de
plusieurs autres formes (sous-sulfure, sels solubles, nickel
métallique). Comme dans le cas du sous-sulfure, il est difficile
d'affirmer ou d'écarter l'existence d'un pouvoir cancérogène sur la
base des seules données épidémiologiques.
d) Nickel métallique
On n'a pas constaté d'accroissement du risque de cancer chez
des ouvriers exposés uniquement à du nickel métallique. L'ensemble
des données relatives aux ouvriers qui travaillaient sur des
alliages du nickel ou dans des ateliers de diffusion gazeuse et qui
tous étaient exposés à des concentrations moyennes de l'ordre de
0,5 mg/m3, ne font pas ressortir d'élévation du risque, encore que
le nombre total de cancers du poumon parmi ces cohortes ait été
trop faible pour qu'on puisse exclure un léger accroissement du
risque à cette concentration.
e) Conclusion
Bien que quelques-uns, voire tous les dérivés du nickel
puissent être cancérogènes, il n'existe que peu ou pas de risque
décelable dans la plupart des secteurs de l'industrie du nickel aux
niveaux d'exposition actuels; cette observation vaut aussi pour
certains procédés dont on estimait jusqu'ici qu'ils comportaient un
risque très élevé de cancers pulmonaire et nasal. Une exposition
prolongée à des dérivés solubles du nickel à des concentrations de
l'ordre de 1 mg/m3 peut entraîner une augmentation sensible du
risque relatif de cancer pulmonaire mais ce risque est à peu près
égal à l'unité chez les ouvriers exposés à des concentrations en
nickel métallique de l'ordre de 0,5 mg/m3 en moyenne. Pour un
niveau d'exposition donné, le risque de cancer est plus important
dans le cas des composés solubles que dans le cas du nickel
métallique et peut-être des autres dérivés du nickel. L'absence
d'un risque notable de cancer pulmonaire chez les nickeleurs n'est
pas sur-prenante étant donné qu'en moyenne l'exposition aux dérivés
solubles est beaucoup plus faible dans les ateliers de
galvanoplastie que dans les ateliers d'affinage électrolytique ou
de traitement des sels de nickel.
RESUMEN Y CONCLUSIONES
1. Identidad, propiedades fisicas y químicas y métodos analíticos
El níquel es un elemento metálico perteneciente al grupo VIIIb
de la tabla periódica. Es resistente a los álcalis, pero en general
se disuelve bien en ácidos oxidantes diluidos. El carbonato de
níquel, el sulfuro de níquel y el óxido de níquel no son solubles
en agua, mientras que sí lo son el cloruro de níquel, el sulfato de
níquel y el nitrato de níquel. El carbonilo de níquel es un líquido
incoloro y volátil que se descompone por encima de los 50 °C. La
forma iónica predominante es la de níquel (II). En los sistemas
biológicos, el níquel disuelto puede formar componentes complejos
con diversos ligandos y unirse a materiales orgánicos.
Los métodos que más se utilizan en el análisis de materiales de
origen biológico y medio ambiental son la espectroscopía de
absorción atómica y la voltamperimetría. A fin de obtener
resultados fidedignos, sobre todo en el orden de las ultratrazas,
hay que adoptar procedimientos específicos para reducir al mínimo
el riesgo de contaminación durante la recogida, el almacenamiento,
el tratamiento y el análisis de muestras. En los materiales de
origen biológico y en el agua se puede llegar límites de detección
de 1-100 ng/litro, atendiendo a los procedimientos de tratamiento
previo, extracción y enriquecimiento de la muestra.
2. Fuentes de exposición humana y ambiental
El níquel es un metal traza muy ampliamente distribuido, que se
encuentra en el suelo, el agua, el aire y la biosfera. En la
corteza terrestre hay un porcentaje medio del 0,008%. Los terrenos
cultivables contienen entre 3 y 1000 mg de níquel/kg. En el agua
natural los niveles oscilan entre 2 y 10 µg/litro (agua dulce) y de
0,2 a 0,7 µg/litro (agua de mar). Las concentraciones atmosféricas
de níquel en zonas remotas son de 0,1 a 3 ng/m3.
Los yacimientos metalíferos de níquel están formados por
acumulaciones de minerales de sulfuro de níquel (principalmente
pentlandita) y lateritas. El níquel se extrae de la mena mediante
procesos de afinado piro e hidrometalúrgico. La mayor parte del
níquel obtenido se destina a la producción de acero inoxidable y a
otras aleaciones muy resistentes a la corrosión y a la temperatura.
Las aleaciones de níquel y el niquelado se utilizan en vehículos,
maquinaria industrial, armamento, herramientas, equipo eléctrico,
utensilios domésticos y la acuñación de monedas. También se
utilizan compuestos de níquel como catalizadores, pigmentos y en
las baterías. En 1985, la producción minera mundial de níquel fue
de unos 67 millones de kg. Las principales fuentes de emisión de
níquel a la atmósfera son la combustión de carbón y petróleo para
la obtención de calor o energía, la incineración de desechos y
fangos cloacales, la extracción minera y la producción primaria de
níquel, la fabricación de acero, la galvanoplastia y otras fuentes,
como la producción de cemento. En el aire contaminado pre-dominan
el sulfato, los óxidos y los sulfuros de níquel, y en menor
cantidad el níquel metálico.
El níquel procedente de los distintos procesos industriales y
de otras fuentes termina llegando a las aguas residuales. Los
restos que se obtienen del tratamiento de esas aguas se eliminan
mediante inyección en pozos profundos, vertido en los océanos,
tratamiento en tierra e incineración. Los efluentes de las
instalaciones de tratamiento de aguas residuales contienen, según
los datos disponibles, hasta 0,2 mg de níquel/litro.
3. Transporte, distribución y transformación en el medio ambiente
El níquel se introduce en el medio ambiente a partir de fuentes
tanto naturales como de origen humano y circula por todos sus
compartimentos mediante procesos físicos y químicos, así como por
el transporte biológico que efectúan los organismos vivos.
Se considera que en la atmosféra el níquel está principalmente
en forma de aerosoles de partículas con distintas concentraciones
del metal, en función de su procedencia. Las concentraciones más
altas de níquel en el aire suelen aparecer en las partículas más
pequeñas. El carbonilo de níquel es inestable en el aire y se
descompone formando óxido de níquel.
El transporte y la distribución de las partículas de níquel a
los diferentes compartimentos del medio ambiente o entre ellos
depende en gran medida del tamaño de las partículas y de las
condiciones meteorológicas. La distribución por tamaños de las
partículas depende sobre todo de las fuentes de emisión. En
general, las partículas de fuentes artificiales son más pequeñas
que las que se encuentran en el polvo natural.
El níquel ingresa en la hidrosfera por eliminación desde la
atmósfera, por la escorrentía superficial, por la descarga de
residuos industriales y municipales, y también tras la erosión
natural de los suelos y de las rocas. En los ríos, este elemento se
transporta principalmente en forma de revestimiento precipitado
sobre partículas y unido a materia orgánica; en los lagos, se
transporta en forma iónica y de manera predominante unido a materia
orgánica. La partículas de arcilla también lo pueden absorber, y a
la biota pasa por ingestión o absorción. El proceso de absorción se
puede invertir, lo que se traduce en una liberación del níquel del
sedimento. Los ríos y arroyos transportan una parte hasta el mar.
El aporte fluvial de partículas suspendidas se estima en 135 x 107
kg/año.
En función del tipo de suelo, el níquel puede presentar una
gran movilidad dentro del perfil edáfico hasta alcanzar las aguas
subterráneas y, con ello, los ríos y lagos. La lluvia ácida tiene
una marcada tendencia a movilizar el níquel del suelo. Las plantas
terrestres lo absorben principalmente por las raíces. La cantidad
absorbida depende de diversos parámetros geoquímicos y físicos,
entre los que figuran el tipo de suelo, su pH y humedad, su
contenido en materia orgánica y la concentración de níquel
extraíble. El caso más conocido de acumulación de níquel es el de
algunas especies de plantas ("hiperacumuladoras"), que crecen en
suelos serpentinosos relativamente poco fértiles y en las que se
han encontrade niveles superiores a 1 mg/kg de peso seco. Los
niveles de níquel superiores a 50 mg/kg de peso seco son tóxicos
para la mayoria de las plantas. Se han observado casos de
acumulación y efectos tóxicos en hortalizas cultivadas en suelos
tratados con fangos residuales y en la vegetación próxima a las
fuentes de emisión del metal. En plantas acuáticas se han detectado
factores de concentración elevados. Los estudios de laboratorio han
puesto de manifiesto que la capacidad de acumulación de níquel en
los peces examinados es escasa. En aguas no contaminadas, las
concentraciones halladas en los peces enteros (sobre la base del
peso fresco) oscilaron entre 0,02 y 2 mg/kg. Estos valores pueden
ser hasta diez veces superiores en los peces procedentes de aguas
contaminadas. Sin embargo, no hay pruebas de que se produzca
bioamplificación del níquel en la cadena de los alimentos.
El níquel se encuentra en muchos órganos y tejidos de animales
silvestres, debido a su ingestión con los alimentos por los
animales herbívoros y sus predadores carnívoros.
4. Niveles medioambientales y exposición humana
Los niveles de níquel en los organismos terrestres y acuáticos
pueden diferir en varios ordenes de magnitud. La exposición humana
a los valores habituales en la atmósfera, que oscilan entre
alrededor de 5 y 35 ng/m3 en zonas rurales y urbanas, hace que la
inhalación de este elemento sea de 0,1-0,7 µg/día. Aunque el agua
potable contiene menos de 10 µg/litro, en ocasiones los empalmes de
las tuberías pueden liberarlo, dando lugar a concentraciones de
hasta 500 µg/litro.
Su concentración en los alimentos suele estar por debajo de 0,5
mg/kg de peso fresco. El cacao, la soja, algunas legumbres secas,
diversos frutos secos y la harina de avena contienen elevadas
concentraciónes de níquel. Su ingestión diaria con los alimentos
varía ampliamente en función de los diferentes hábitos
alimentarios, y puede oscilar entre 100 y 800 µg/día; la ingestión
media de níquel en la dieta es, en la mayoría de los países, de
100-300 µg/día. Los utensilios de cocina liberan níquel, lo que
contribuye de manera considerable a su ingestión. Fumando 40
cigarrillos diarios pueden absorberse a través de los pulmones 2-23
µg/día.
La exposición cutánea en el medio ambiente general es
importante, porque induce y mantiene la hipersensibilidad por
contacto que produce en la piel el uso diario de objetos niquelados
o con aleaciones de este metal (por ejemplo, joyería, monedas,
"clips").
Puede haber exposición iatrogénica al níquel debida a las
implantaciones y las prótesis hechas con aleaciones que lo
contienen, a fluidos intravenosos o de diálisis y a los medios de
contraste radiográfico. Se estima que la absorción media de níquel
por vía intravenosa a partir de los fluidos de diálisis es de 100
µg por tratamiento.
En el ámbito laboral, la concentración en el aire puede variar
desde varios g/m3 a, en ocasiones, unos mg/m3, en función del
proceso de que se trate y del contenido en níquel del material que
se maneja.
Hay millones de trabajadores de todo el mundo expuestos a polvo
y humo que contienen níquel durante las operaciones de soldadura,
niquelado y pulimentado, la extracción minera, el afinado del
níquel, y en las acerías, fundiciones y otras industrias
metalúrgicas.
Hay una gran variedad de empleos en los que puede haber
exposición cutánea al níquel, sea por contacto directo con el metal
disuelto, como por ejemplo en las industrias de afinado,
galvanoplastia y electroconformación, o bien por el manejo de
herramientas que lo contienen. En los trabajos de limpieza con agua
puede haber una exposición a este metal, a causa de las cantidades
que lleva disueltas el agua de lavado.
5. Cinetica y metabolismo en el hombre y en los animales
El hombre y los animales pueden absorber níquel por inhalación,
por ingestión o por vía cutánea. La absorción respiratoria, con
asimilación gastrointestinal secundaria de níquel (soluble e
insoluble), es la principal vía de penetración durante la
exposición en el trabajo. Una cantidad importante del material
inhalado se traga con el fluido mucociliar procedente del tracto
respiratorio. La mala higiene personal y las prácticas laborales
deficientes pueden contribuir a la exposición gastrointestinal. La
absorción percutánea es cuantitativamente insignificante, pero es
importante en la patogenia de la hipersensibilidad por contacto. La
absorción está relacionada con la solubilidad del compuesto, y
sigue la relación general carbonilo de níquel > compuestos de
níquel solubles > compuestos de níquel insolubles. El carbonilo de
níquel es el compuesto de este metal que absorben de forma más
rápida y completa el hombre y los animales. Hay pocos estudios en
los que se haya administrado níquel por inhalación. En experimentos
con hámsters y ratas tratados con óxido de níquel insoluble se
observó una escasa absorción, con retención pulmonar de gran parte
del material después de varias semanas. En cambio, la absorción de
cloruro de níquel soluble o de sulfuro de níquel amorfo fue rápida.
El níquel es transportado en la sangre, principalmente ligado a la
albúmina.
Su absorción gastrointestinal es variable y depende de la
composición de la dieta. En un estudio reciente con un grupo de
voluntarios se encontró que la absorción de níquel a partir del
agua era del 27%, mientras que a partir de los alimentos era
inferior al 1%. Todas las secreciones orgánicas, como la orina, la
bilis, el sudor, las lágrimas, la leche y el fluido mucociliar, son
posibles vías de eliminación. El níquel que no se absorbe se
elimina con las heces. En roedores se ha demostrado que atraviesa
la placenta. Tras la administración parenteral de sales de níquel,
los niveles más altos de acumulación se producen en el riñon, las
glándulas endocrinas y el hígado; si lo que se administra es
carbonilo de níquel, también se observa una alta concentración en
el cerebro. Los datos sobre excreción del níquel sugieren un modelo
bicompartimental. Su concentración en el suero y la orina de
adultos sanos no expuestos en el trabajo es de 0,2 µg/litro
(oscilación: 0,05-1,1 µg/litro) y 1,5 µg/g de creatinina
(oscilación: 0,5-4,0 µg/g de creatinina), respectivamente. Se ha
observado que después de la exposición profesional, la
concentración aumenta en ambos fluidos. La carga corporal de níquel
en un adulto de 70 kg de peso no expuesto es de 0,5 µg.
6. Efectos en los seres vivos del medio ambiente
En los microorganismos, con concentraciones de 1-5 mg/litro de
níquel en el medio se inhibía, en general, el crecimiento en
actinomicetos, levaduras y eubacterias marinas y no marinas, y con
niveles de 5-1000 mg/litro, en hongos filamentosos. En el caso de
las algas, con una concentración aproximada de 0,05-5 mg de
níquel/litro no se observó crecimiento. Ciertos factores abióticos,
como el pH, la dureza, la temperatura y la salinidad del medio, así
como la presencia de partículas orgánicas e inorgánicas, influyen
en la toxicidad del níquel.
La toxicidad del níquel para los invertebrados acuáticos varía
considerablemente en función de las especies y de los factores
abióticos. En el género Daphnia se ha encontrado una CL50 a las 96
horas de 0,5 mg de níquel/litro, mientras que en moluscos los
valores de la CL50 a las 96 horas fueron de unos 0,2 mg/litro en
dos especies de caracol de agua dulce y de 1100 mg/litro en un
bivalvo.
En los peces, los valores de la CL50 a las 96 horas suelen
oscilar entre 4 y 20 mg de níquel/litro, pero pueden ser más altos
en algunas especies. Los estudios a largo plazo realizados sobre
los peces y su crecimiento en aguas blandas pusieron de manifiesto
que niveles tan bajos como 0,05 mg de níquel/litro tenían algunos
efectos en la trucha arco iris. En plantas terrestres se ha
encontrado que los niveles superiores a 50 mg/kg de peso seco
suelen ser tóxicos. Se ha observado que desde el punto de vista
toxicologica el cobre tiene un efecto sinérgico con el níquel,
mientras que el calcio reduce su toxicidad. Hay pocos datos acerca
de los efectos del níquel en animales terrestres.
La lombriz de tierra parece ser relativamente insensible a este
metal si el medio es rico en microorganismos y materia orgánica,
que dejan menos níquel disponible para aquella. Aunque el níquel no
se ha considerado un contaminante mundial en gran escala, en las
cercanías de fuentes emisoras se han observado cambios ecológicos,
como la disminución del número y la diversidad de especies. En
estudios de microecosistemas se ha puesto de manifiesto que la
adición de este elemento al suelo altera el ciclo del nitrógeno.
7. Efectos en los animales de experimentación y en sistemas de
prueba in vitro
El níquel es indispensable para la actividad catalítica de
algunas enzimas vegetales y bacterianas. En algunas especies
animales se ha señalado que una dieta carente de níquel provoca
lentitud en la ganacia de peso, anemia y una disminución de la
viabilidad de la descendencia.
El compuesto de toxicidad más aguda es el carbonilo de níquel,
cuyo órgano destinatario es el pulmón; dentro de las cuatro horas
siguientes a la exposición puede aparecer edema pulmonar. La
toxicidad aguda de los demás compuestos del níquel es baja.
Aunque la alergia de contacto causada por este metal es muy
frecuente en el hombre, en animales sólo se consigue inducir
sensibilización experimental en determinadas condiciones. La
exposición a un largo período de inhalación de níquel metálico,
óxido de níquel o subsulfuro de níquel causó lesiones en las
mucosas y una reacción inflamatoria del tracto respiratorio en
ratas, ratones y cobayos. En ratas expuestas a la inhalación de
aerosoles de cloruro de níquel u óxido de níquel se observó
hiperplasia epitelial.
La exposición a altos niveles de óxido de níquel durante un
período prolongado produjo una neumoconiosis progresiva en ratas.
En conejos expuestos a un nivel elevado de polvo de níquel-grafito
se observó una reacción inflamatoria, a veces acompañada de una
ligera fibrosis. Las ratas expuestas a subsulfuro de níquel
presentaron fibrosis pulmonar.
La administración de sales de níquel a ratas, conejos y pollos
por vía parenteral indujo una hiperglucemia rápida transitoria.
Estos cambios pueden estar relacionados con los efectos en las
células alfa y beta de los islotes de Langerhans. El níquel también
disminuyo la liberación de prolactina. Se ha informado de que la
administración por vía oral o respiratoria de cloruro de níquel
reduce la absorción de yodo por el tiroides.
La administración intravenosa de sales de níquel a perros hizo
disminuir el flujo sanguíneo en las arterias coronarias; la
concentración elevada de este metal redujo la contractibilidad del
miocardio de perro in vitro.
El cloruro de níquel afecta al sistema de células T y suprime
la actividad de las células destructoras naturales. Se ha observado
que la administración parenteral de cloruro de níquel y de
subsulfuro de níquel a ratas y ratones causa mortalidad
intrauterina y una disminución de la ganancia de peso. En ratas y
hámsters expuestos a inhalaciones de carbonilo de níquel se produjo
la muerte de fetos y una disminución de la ganancia de peso, y tuvo
también un efecto teratógenico. En ninguno de esos estudios se
informa acerca de la toxicidad materna. Se ha señalado que el
carbonilo de níquel ocasiona mutaciones letales dominantes en la
rata.
Se han hecho pruebas con distintos sistemas para determinar la
mutagenicidad de varios compuestos inorgánicos del níquel. Estos,
en general, se mostraron inactivos en los ensayos de mutagénesis
efectuados con bacterias, excepto en los casos en que se utilizaron
pruebas de fluctuación. Se observaron mutaciones en varios tipos de
células de mamíferos cultivadas. Los compuestos del níquel
inhibieron la síntesis de ADN en una gran variedad de organismos.
Además, produjeron aberraciones cromosómicas e intercambio de
cromátidas hermanas en cultivos de células tanto de mamíferos como
humanas. En personas profesionalmente expuestas a compuestos de
níquel insolubles o solubles se observaron aberraciones
cromosómicas, pero no intercambio de cromátidas hermanas (excepto
en un estudio sobre personas que trabajan en electrólisis). El
níquel indujo transformación celular in vitro.
En un estudio de inhalación en ratas, el subsulfuro de níquel
provocó la aparición de tumores pulmonares benignos y malignos. En
una serie de estudios de inhalación de carbonilo de níquel en la
rata, se observaron algunos tumores pulmonares. En un estudio de
inhalación de níquel metálico efectuado en ratas no se apreció un
aumento importante de tales tumores. La inhalación de óxido de
níquel negro no indujo la aparición de tumores pulmonares en
hámsters dorados sirios (especie resistente a la carcinogénesis
pulmonar). No se conocen estudios adecuados sobre la carcinogénesis
por inhalación de otros compuestos del níquel. Sin embargo, la
instilación intratraqueal repetida de subsulfuro de níquel, de
polvo de níquel metálico y de un óxido de níquel no especificado
produjo en ratas tumores pulmonares benignos y malignos.
El carbonilo de níquel, el niqueloceno y gran número de
compuestos del níquel ligeramente solubles o insolubles, entre
ellos el subsulfuro, el carbonato, el cromato, el hidróxido, los
sulfuros, los seleniuros, los arseniuros, el telururo, el
antimoniuro, diversas preparaciones de óxidos no identificadas, dos
óxidos de níquel y cobre, el níquel metálico y diversas aleaciones
de níquel provocaron tumores mesenquimáticos locales en distintos
animales de experimentación tras la administración por vía
intramuscular, subcutánea, intraperitoneal, intrapleural,
intraocular, intraósea, intrarrenal, intraarticular,
intratesticular o intraadiposa. No se observó respuesta
carcinogénica local en los estudios de dosis única con algunas
aleaciones de níquel, hidróxido de níquel coloidal o dos muestras
de óxido de níquel, especialmente preparados para pruebas de
carcinogénesis mediante calcinación a 735 °C o bien a 1045 °C.
La administración repetida de sulfato y acetato de níquel a
ratas por vía intraperitoneal indujo tumores en la cavidad
peritoneal, pero no la de cloruro de níquel.
Se han realizado pruebas de carcinogénesis mediante la
administración por vía parenteral de níquel metálico y de una gran
variedad de compuestos; salvo algunas excepciones, se produjeron
tumores locales.
Sólo en el caso del subsulfuro de níquel se ha demostrado de
manera convincente que la inhalación produce cáncer. Sin embargo,
hay muy pocos estudios adecuados sobre la exposición por vía
respiratoria.
En los estudios de aplicación de instilaciones intratraqueales
repetidas de polvo de níquel, óxido de níquel y subsulfuro de
níquel aparecieron tumores pulmonares.
Cuando se realizaron pruebas con tres sales solubles de níquel
distintas que en estudios anteriores no habían provocado la
aparición de tumores locales, utilizando la administración
intraperitoneal repetida, dos de las sales indujeron una respuesta
carcinogénica.
8. Efectos en la especie humana
En relación con la salud del ser humano, el carbonilo de níquel
es el compuesto del metal con toxicidad más aguda. Entre los
efectos de la intoxicación aguda que causa están dolor de cabeza
frontal, vértigo, náuseas, vómitos, insomnio e irritabilidad,
seguidos de trastornos pulmonares análogos a los producidos por una
neumonía vírica. Entre las lesiones patológicas de los pulmones
figuran hemorragias, edema y desorganización celular. También se
ven afectados el hígado, los riñones, las cápsulas suprarrenales,
el bazo y el cerebro. Ha habido asimismo casos de intoxicación en
pacientes sometidos a diálisis con contaminación de níquel y en
galvanizadores que accidentalmente han ingerido agua contaminada
por sulfato y cloruro de níquel.
Se han notificado casos de efectos crónicos, como rinitis,
sinusitis, perforación del tabique nasal y asma, entre trabajadores
del afinado del níquel y del niquelado. Algunos autores han
descrito alteraciones pulmonares con fibrosis en personas que en el
trabajo respiraban polvo de níquel. Además, se ha informado de
casos de displasia nasal en trabajadores del afinado del níquel.
Hay abundante documentación acerca de la hipersensibilidad por
contacto, tanto en la población en general como en algunos
trabajadores profesionalmente expuestos a compuestos solubles de
níquel. En varios países se ha comunicado que el 10% de la
población femenina y el 1% de la masculina son sensibles al níquel.
De ellos, el 40-50% tienen eccema vesicular en las manos; en
algunos casos puede ser muy grave y causa de incapacidad laboral.
La ingestión de níquel puede agravar la eccema vesicular de las
manos y, posiblemente, inducir también su aparición en otras partes
del cuerpo que no han tenido contacto cutáneo con este metal.
Se sabe que las prótesis y otras implantaciones quirúrgicas de
aleaciones con níquel causan sensibilización a este elemento o
agravan la dermatitis existente.
Se han descrito efectos nefrotóxicos, como edema renal con
hiperemia y degeneración parenquimatosa, en casos de exposición
industrial accidental a carbonilo de níquel. También se han
registrado efectos nefrotóxicos pasajeros por ingestión accidental
de sales de níquel.
Se ha informado de un elevadísimo riesgo de cáncer del pulmón y
la nariz en los trabajadores del afinado del níquel que se ocupan
de tostar a alta temperatura los minerales de sulfuro, lo que
supone una gran exposición al subsulfuro, el óxido y quizás el
sulfato de níquel. Se han señalado riesgos análogos en los procesos
que entrañan exposición al níquel soluble (electrólisis, extracción
de sulfato de cobre, hidrometalurgia), a menudo combinados con
alguna exposición al óxido de níquel pero muy poca al subsulfuro.
El riesgo de los mineros y otros trabajadores del afinado parece
ser mucho menor. El índice de cáncer entre los soldadores de acero
inoxidable y los trabajadores de industrias que usan níquel ha
sido, en general, casi normal, salvo cuando hay exposición al
cromo, particularmente en la galvanoplastia. Sin embargo, los
trabajadores que hacen baterías de níquel/cadmio y están expuestos
a altos niveles de ambos metales corren un riesgo ligeramente mayor
de contraer cáncer de pulmón.
Ocasionalmente se ha notificado en trabajadores del níquel una
frecuencia excesiva de otros tipos de cáncer distintos del de
pulmón y el nasal, como por ejemplo renal, gástrico o de próstata,
pero ninguno se ha encontrado de manera constante.
Se pueden utilizar los datos epidemiológicos para plantear dos
importantes cuestiones: i) si se ha demostrado que ciertos
compuestos de níquel que son carcinógenos; y ii) si las cohortes
sometidas a bajas exposiciónes presentan límites superiores de
riesgo a determinados niveles de exposición.
a) Níquel soluble
Se obtuiseron pruebas de que los trabajadores expuestos a
concentraciones de níquel soluble del orden de 1-2 mg/m3, tanto en
el sector de la electrólisis como en el de la preparación de sales
solubles, corrían riesgo de cáncer. Esos trabajadores estaban
expuestos también a otros compuestos de níquel, pero con frecuencia
a niveles más bajos que en otros procesos de alto riesgo. En
ausencia de mediciones anteriores de la exposición, es imposible
llegar a conclusiones definitivas, pero las pruebas de que el
níquel soluble es carcinógeno son ciertamente sólidas. El polvo del
afinado contiene a veces una proporción importante de sulfato de
níquel, además de subsulfuro. Esto plantea la posibilidad de que el
altísimo riesgo de cáncer observado en los trabajadores que se
ocupan de la oxidación de subsulfuro de níquel a alta temperatura
pueda deberse en parte al níquel soluble.
b) Subsulfuro de níquel
En las zonas de afinado donde el riesgo de contraer cáncer era
alto, la exposición al subsulfuro de níquel casi siempre iba
acompañada de la exposición al óxido y, quizás, al sulfato (véase
más arriba). Así pues, basándose solamente en los datos
epidemiológicos, es dificil demonstrar que el subsulfuro de níquele
sea carcinógeno, aunque parece probable.
c) Oxido de níquel
El óxido de níquel estaba presente en casi todas las
circunstancias en que el riesgo de cáncer era elevado, acompañado
de una o más formas del elemento (subsulfuro de níquel, níquel
soluble, níquel metálico). Como en el caso del subsulfuro de
níquel, es difícil afirmar o negar su carácter carcinogénico
basándose exclusivamente en los datos epidemiológicos.
d) Níquel metálico
No se ha demostrado que los trabajadores expuestos
exclusivamente al níquel metálico corran mayor riesgo de cáncer.
Los datos combinados relativos a trabajadores que se ocupan de
aleaciones de níquel y los dedicados a difusión gaseosa, todos
ellos expuestos a concentraciones medias del orden de 0,5 mg de
níquel/m3, no muestran un riesgo excesivo, aunque el número total
de casos de cáncer de pulmón en estas cohortes era demasiado
pequeño para excluir un pequeño aumento del riesgo a este nivel.
e) Conclusión
Aunque algunas, y quizás todas, las formas del níquel pueden
ser carcinógenas, con los niveles normales de exposición en muchos
sectores de la industria de este metal hay un riesgo pequeño o no
detectable; entre ellos se encuentran algunos procesos que en el
pasado estaban asociados con un riesgo muy alto de cáncer pulmonar
y nasal. La exposición prolongada a concentraciones del orden de 1
mg/m3 de níquel soluble puede ocasionar un importante aumento del
riesgo relativo de cáncer de pulmón, pero este riesgo entre los
trabajadores expuestos a un nivel medio de níquel metálico de
alrededor de 0,5 mg/m3 es aproximadamente 1. El riesgo de cáncer
con un determinado nivel de exposición puede ser más alto para los
compuestos de níquel solubles que para el níquel metálico, y
posiblemente, que para otras formas. No es extraña la ausencia de
cualquier riesgo pronunciado de cáncer pulmonar entre los
niqueladores, puesto que el promedio de exposición al níquel
soluble es mucho más bajo que el que se produce en el afinado
electrolítico o en el tratamiento de sales de níquel.
The distribution and elimination of nickel, given to animals as
63Ni chloride, has been studied extensively. Most of the
introduced nickel is rapidly excreted in the urine (65-87% in 24
h), the rest undergoing much slower elimination (76-90% in 5 days)
(Sunderman & Selin, 1968; Sunderman et al., 1976a).
The 2-compartment model described by Onkelinx & Sunderman
(1980) also provides a satisfactory fit for experimental data from
human volunteers who ingested nickel sulfate in the drinking-water
or food at doses of 12, 18, or 50 µg nickel/kg body weight
(Sunderman et al., 1989a). Faecal elimination of nickel during 4
days following treatment averaged 76±19% of the dose ingested in
water versus 102±20% of the dose ingested in food. The elimination
half-time for absorbed nickel averaged 28±9 h (range 17-48 h).
Renal clearance was determined to be 8.3±2.0 ml/min per 1.73 m2 in
human beings who had ingested nickel sulfate in water, and 5.8±4.3
ml/min per 1.73 m2 in those who had received nickel sulfate in
food. The difference was not statistically significant.
6.2.2.3 Nickel carbonyl
There are few studies on the fate of nickel carbonyl in
experimental animals. After the administration of nickel carbonyl,
deposition occurs in the lung and in tissues, such as the brain,
liver, and adrenals, and part of the administered dose of nickel is
recovered in the urine (Armit, 1908; Barnes & Denz, 1951; Sunderman
& Selin, 1968). It was assumed earlier that nickel carbonyl was
rapidly dissociated in the lung and that the nickel was then
transported to other tissues. However, the results of several
studies (Sunderman & Selin, 1968; Sunderman et al., 1968; Kasprzak
& Sunderman, 1969) have indicated that unchanged nickel carbonyl is
present in the blood several hours after administration and can
pass across the pulmonary alveoli in either direction without
decomposition. It was suggested by Kasprzak & Sunderman (1969)
that the nickel carbonyl that was not exhaled underwent a slow
intracellular decomposition to NiO and CO. The released NiO was
then oxidized to Ni2+, which might become bound to nucleic acids or
proteins, or to albumin in the plasma and, ultimately, would be
excreted in the urine; the released CO would become bound to
haemoglobin and finally exhaled.
Oskarsson & Tjälve (1979a) studied the distribution of
intravenously administered 63Ni and 14C-labelled nickel carbonyl
(63Ni(CO4) and Ni(14CO4)) in mice by whole-body autoradiography and
liquid scintillation counting. Radioactivity in the animals given
14C-labelled carbonyl was mainly confined to the blood, indicating
the formation of 14CO-haemoglobin. This confirms the findings of
Kasprzak & Sunderman (1969). After the administration of 63Ni-
labelled carbonyl, the highest level of 63Ni was found in the lung,
followed by the brain, spinal cord, heart, diaphragm, brown fat,
adrenals, and corpora lutea. Additional studies showed that nickel
was present in the lung, brain, heart, and blood as the cation.
6.2.2.4 Nickel levels in human beings
In human beings, wide variations have been reported in body
nickel levels. This makes it difficult to appraise and compare the
results obtained by various investigators. In addition to
variations in the geographical origin of data and individual
dietary and smoking habits, major differences can be attributed to
the analytical methods employed. Only limited comparisons can be
made using variants of spectrography, atomic absorption
spectrometry, photometric methods, and special analytical
techniques. Furthermore, no uniform reference samples have been
used. Nickel values from tissue analyses have been related to ash
or dry weight as well as to wet weight. The normal ranges of
nickel concentrations in body fluids or tissues (serum, blood,
lung, kidney) are not significantly influenced by age, sex, or
pregnancy (McNeely et al., 1971; Turhan et al., 1983; Zober et al.,
1984).
(a) Body fluids, hair, and nasal mucosa
The levels of nickel in biological fluids, hair, and some other
materials increase remarkably in persons with increased
occupational or environmental exposure and decline rapidly when
exposure is reduced or stopped (Tables 19, 20, 21, and 22). Thus,
measurements of nickel, particularly in the urine, serum, or hair,
may serve as indices of exposure.
Data for normal nickel values in urine, blood, plasma, and
serum, published in the last three decades, vary widely. Lower
levels have been obtained by later investigators, because of the
use of more sensitive analytical methods. Reference values in
specimens from healthy, non-exposed persons are listed in Table 19.
Because of doubts about the reliability of older studies, only
recent data have been included.
Table 19. Normal nickel concentrations in specimens from healthy non-exposed
adults
--------------------------------------------------------------------------------
Specimen No. of Nickel concentrations Units References
subjects mean ± SD range
(m/f)
--------------------------------------------------------------------------------
Whole blood 30 (15,15) 0.34 ± 0.28 < 0.05-1.05 µg/litre Linden et al.
(1985)
Serum 10 (6, 4) 0.32 ± 0.17 0.1-0.6 µg/litre Sunderman et
al. (1989a)
Serum 43 (22, 21) 0.2 ± 0.2 < 0.05-1.0 µg/litre Hopfer et al.
(1989)
Serum 30 (15, 15) 0.28 ± 0.24 < 0.05-1.08 µg/litre Linden et al.
(1985)
Lymphocytes 10 (4, 6) 0.72 ± 0.75 < 0.05-1.10 µg/1010 Wills et
cells al. (1985)
Urine (spot 34 (18, 16) 2.0 ± 1.5 0.5-6.1 µg/litre Sunderman
collection) 2.8 ± 1.9 0.5-8.8 µg/litre et al.
(1.024 (1986a)
sp.gr.)a
Urine (24-h 50 (24, 26) 2.2 ± 1.2 0.7-5.2 µg/litre Sunderman
collection) 2.6 ± 1.4 0.5-6.4 µg/day (1977)
Faeces (3-day 10 (6, 4) 14.2 ± 2.7 10.8-18.7 mg/kg Horak &
collection) (dry Sunderman
weight) (1973)
258 ± 126 80-540 µg/day
Faeces 10 (6, 4) 1.5 ± 0.5 1.0-2.2 mg/kg Sunderman
(wet et al.
weight) (1989a)
Sweat 14 (6, 8) 51 ± 38 8-158 µg/litre Christensen
et al.
(1979)
Bile 5(b) 2.3 ± 0.8 15-33 µg/litre Rezuke et
al. (1987)
Saliva 38 (32, 6) 1.9 ± 1.0 0.8-4.5 µg/litre Catalanatto
et al.
(1977)
Hair 102 (b) 0.29 0.0-13.0 mg/kg Bencko et
(net al. (1986)
weight)
--------------------------------------------------------------------------------
Table 19 (contd.)
--------------------------------------------------------------------------------
Specimen No. of Nickel concentrations Units References
subjects mean ± SD range
(m/f)
--------------------------------------------------------------------------------
Hair 22 (15, 7) 0.22 ± 0.08 0.13-0.51 mg/kg Nechay &
(dry Sunderman
weight) (1973)
Hair 905 (437, -c 0.26-2.70 mg/kg Tagaki et
468) (wet al. (1986)
weight)
Nasal mucosa 57 (57m) 0.13 ± 0.20 <0.53d mg/kg Torjussen &
(wet Andersen
weight) (1979)
--------------------------------------------------------------------------------
a Urine nickel concentrations, factored to specific gravity = 1.024.
b Sex not indicated.
c Range of means of samples from five countries.
d Upper 95th percentile of nickel concentrations in nasal biopsies from
non-exposed subjects.
A large number of workers exposed to various nickel compounds
have been found to have elevated levels of nickel in the urine.
These include those working in: nickel refineries (Morgan, 1960;
Kemka, 1971; Norseth, 1975; Hogetveit & Barton, 1976, 1977;
Bernacki et al., 1978a; Hogetveit et al., 1978; Morgan & Rouge,
1979; Torjussen & Andersen, 1979; Hogetveit et al., 1980; Boysen et
al., 1982), the welding of nickel alloy steels (Norseth, 1975;
Bernacki et al., 1978a; Grandjean et al., 1980; Polednak, 1981;
Kalliomäki et al., 1981), nickel electroplating plants (Tandon et
al., 1977; Tola et al., 1979; Bernacki et al., 1980; Tossavainen et
al., 1980), nickel battery factories (Bernacki et al., 1978a;
Adamsson et al., 1980), different occupations in shipyards
(Grandjean et al., 1980), the pigment industry (Tandon et al.,
1977), the glass industry (Raithel et al., 1981), nickel carbonyl
processing in nickel refining (Kincaid et al., 1956; Sorinson et
al., 1958; Morgan, 1960; Nomoto & Sunderman, 1970; Hagedorn-Götz et
al., 1977), aircraft mechanics and metal spraying (Bernacki et al.,
1978a). The results of the investigations of Bernacki et al.
(1978a), who analysed urine samples from nickel-exposed workers in
10 occupational groups, are listed in Table 20.
Serum or plasma nickel levels have been determined in workers
in the following occupations: nickel refining (Hogetveit & Barton,
1976, 1977; Hogetveit et al., 1978, 1980; Torjussen & Andersen,
1979; Boysen et al., 1982), welding (Grandjean et al., 1980),
electroplating (Tola et al., 1979; Tossavainen et al., 1980),
battery manufacture (Adamsson et al., 1980) and shipyards
(Grandjean et al., 1980), and the Mond process in nickel refining
(Sorinson et al., 1958; Nomoto & Sunderman, 1970).
Table 20. Nickel concentrations in urine of workers in various occupational groupsa
-------------------------------------------------------------------------------------
Occupation No. Description Concentration
Atmospheric Urineb Creatinineb
nickel (µg/m3) (µg/litre) (µg/litre)
-------------------------------------------------------------------------------------
External 9 abrasive wheel 1.6±3.0 5.4±2.4 3.5±1.6
grinders grinding of exteriors (0.02-9.5) (2.1-8.8) (1.7-6.1)
of articles made of
nickel alloys
Arc welders 10 DC arc welding of 6.0±14.3 6.3±4.1c 5.6±6.2
aircraft made of (0.2-46) (1.6-14) (1.1-17)
nickel alloys
Bench mechanics 8 assembling, fitting, 52±94 12.2±13.6c 7.2±6.8c
and finishing parts (0.01-252) (1.4-41) (0.7-20)
made of nickel alloys
Nickel battery 6 fabricating nickel- Not 11.7±7.75d 10.2±6.4d
workers cadmium or nickel- measured (3.4-25) (7.2-23)
zinc electrical
storage batteries
Metal sprayers 5 flame-spraying 2.4±2.6 17.2±9.8d 16.0±21.9
nickel-containing (0.04-2.1) (1.4-26) (1.4-54)
powders in phase on
to aircraft parts
Electroplaters 11 intermittent exposure 0.8±0.9 10.5±8.1d 5.9±5.0c
to nickel in combined (0.04-2.1) (1.3-30) (1.0-20)
electro-deposition
operations involving
silver, cadmium,
chromium plating, as
well as nickel
Nickel platers 21 full-time work in Not 27.5±21.2e 19.0±14.7e
nickel plating measured (3.6-65) (2.4-47)
operations
Nickel refinery 15 workers in a nickel 489±560 222±226e 124±109e
refinery using (20-2200) (8.6-8.3) (6.1-287)
electrolytic processes
-----------------------------------------------------------------------------------
a From: Bernacki et al. (1978a).
b Mean SD with range in parentheses.
c P < 0.05 versus controls, calculated by t-test.
d P < 0.01 versus controls.
e P < 0.001 versus controls.
The highest nickel concentrations were found in the body fluids
of nickel refinery workers. Concentrations in workers in
electroplating shops, battery factories, and aircraft engineering
works were lower (Table 10). After occupational exposure to nickel
in electroplating processes, biological half-times ranging from 13
to 39 h for nickel in urine and from 20 to 34 h for nickel in
plasma have been reported (Tossavainen et al., 1980).
Serum specimens of 22 residents of Sudbury, Ontario, who had
been environmentally exposed to nickel (including air and tap
water), contained nickel concentrations ranging from 0.2 to 1.3
µg/litre (mean 0.6±0.3 µg/litre). These were significantly higher
than the serum levels of residents without environmental exposure
(Hopfer et al., 1989).
Nickel determinations in blood and urine, are widely used and
accepted methods for monitoring nickel exposure. Although more
data are available for urine, no clear-cut choice can be made
between the use of blood or urine.
Grandjean et al. (1980, 1988) reported that analyses of nickel
concentrations in both urine and plasma samples should be obtained
to assess worker exposure. There were significantly higher ratios
of plasma/urine nickel levels in painters and lower ratios in
welders, compared with other workers in the shipyard. These
differences probably reflected the different toxicokinetic
characteristics of the nickel compounds to which the workers were
exposed.
The correlation between exposure levels and nickel
concentrations in body fluids is poor in most studies (Table 21).
The closest positive relationships of nickel concentrations in body
fluids with ambient air levels were found by Norseth (1975) and
Rahkonen et al. (1983) in welders, and by Tola et al. (1979) and
Bernacki et al. (1980) in electroplaters.
Table 21. Studies on the correlation between nickel concentrations
in the air and in biological fluids in occupational exposure to
nickel compoundsa
-------------------------------------------------------------------
Exposure Biological Correlation Reference
matrix coefficient
(r)
-------------------------------------------------------------------
welding urine 0.85 Norseth (1975)
roasting-smelting urine none
welding urine none Bernacki et al.
bench mechanics urine none (1978a)
electroplating urine none
metal spraying urine none
refinery urine none
roasting-smelting plasma -0.11 Hogetveit et al.
urine 0.14 (1978)
electrolysis plasma 0.21
urine 0.31
refinery (other) plasma 0.67
urine 0.47
-------------------------------------------------------------------
Table 21 (contd.)
-------------------------------------------------------------------
Exposure Biological Correlation Reference
matrix coefficient
(r)
-------------------------------------------------------------------
refinery (nickel urine 0.49; 0.55 Morgan & Rouge
salts) (1979)
refinery:
Mond process urine 0.01
calciner urine 0.22
powder plant urine -0.05
electroplating plasma 0.83 Tola et al.
urine 0.82b (1979)
urine 0.96c
electroplating urine 0.70d Bernacki et al.
(1980)
battery urine significante Adamsson et al.
manufacturing (1980)
welding blood 0.56 Rahkonen et al.
urine 0.95 (1983)
-------------------------------------------------------------------
a From: Aitio (1984).
b Afternoon.
c Next morning.
d After shift.
e P < 0.01.
There seem to be at least three reasons for the inconsistencies
in the correlation between exposure levels and biological
measurements of nickel:
(a) exposure is not to a single chemical species, but to a variety
of nickel compounds of very different solubility, absorption,
transportation, and elimination rates. The same workers may be
simultaneously exposed to both insoluble and readily soluble
compounds, having half-lives ranging from days (Tola et al.,
1979) to years (Torjussen & Andersen, 1979). The influence of
the nickel species in ambient air on the concentration in body
fluids has been shown in a study by Bernacki et al. (1978a),
who found widely varying ratios of air/urine levels in
different occupational groups (Table 22);
(b) differences in personal working habits and hygiene;
(c) failure to standardize sampling methods.
Table 22. Nickel concentrations in serum, urine, nasal mucosa, and personal air samples
from workers at the Falconbridge Nickel Refinery in Kristiansand, Norwaya
-----------------------------------------------------------------------------------------
Category of No. of Plasma nickel Urine nickel Nasal mucosal Air nickel
subjects/work subjects (µg/litre) (µg/litre) nickel (mg/m3)
mean±SD mean±SD (µg/100g) mean±SD
(wet weight)
-----------------------------------------------------------------------------------------
First studyb
Roasting/smelting 24 7.2±2.8 65±58 0.86±1.20
Electrolysis 90 11.9±8.0 129±106 0.23±0.40
Other process 13 6.4±1.9 45±27 0.42±0.49
Second studyc
Controls 57 1.9±1.4 4.9±4.2 13±20
Roasting/smelting 97 5.2±2.7 34±35 467±595
Electrolysis 144 8.1±6.0 73±85 178±235
Other process 77 4.3±2.2 22±18 211±301
-----------------------------------------------------------------------------------------
a From: Sunderman et al. (1986b).
b From: Hogetveit et al. (1978).
c From: Torjussen & Andersen (1979).
The nickel contents of hair and nasal mucosa have been
determined in occupationally-exposed persons. Theoretically, the
nasal mucosa is one of the target tissues for nickel
carcinogenicity. However, practical problems of sampling and
standardization preclude the routine use of these measurements.
Torjussen & Andersen (1979) analysed biopsy specimens of nasal
mucosa from 318 nickel workers, 15 retired nickel workers, and 57
unexposed controls. The results showed that nickel exposure led to
significantly raised nickel concentrations in the nasal mucosa in
both active and retired nickel workers (2.74±4.12 mg/kg wet weight,
and 1.14±1.78 mg/kg wet weight, respectively, versus 0.13±0.2 mg/kg
wet weight in the controls). The average nickel concentration in
the nasal mucosa was highest in workers exposed to the highest
atmospheric nickel concentration, inhaled as nickel subsulfide and
nickel oxide dust. Workers exposed to aerosols of soluble nickel
components, such as the chloride and sulfate, at a lower
atmospheric nickel concentration, had the highest mean nickel
concentrations in the plasma and urine and the lowest in the nasal
mucosa. The mucosal concentration was significantly correlated
with the duration of nickel exposure.
For hair, normal values ranged between 0.13 mg/kg (dry weight)
and 2.7 mg/kg (wet weight). In hair samples from 45
occupationally-exposed adults, Bencko et al. (1986) found nickel
concentrations ranging from 1.6 to 3.5 mg/kg (mean 2.39 mg/kg) in
welders and from 42.7 to 2140 mg/kg (mean 222.5 mg/kg) in nickel
smelter workers. Control values ranged from 0 to 13 mg/kg (mean
0.29 mg/kg). In an accidental case of exposure to nickel carbonyl,
nickel concentrations in hair samples from 5 workers ranged from 4
to 48.1 mg/kg (Hagedorn-Götz et al., 1977).
Although hair has been studied as a rapid, non-invasive
measurement of exposure/absorption relationships, conflicting
values have been obtained. Furthermore, the use of hair as an
internal exposure index is controversial because of various
factors, such as the external contamination of the hair surface,
different sampling methods, and non-standardized cleaning methods.
(b) Tissues
There are few data on human tissue concentrations of nickel.
Spectrographic analyses indicate that the retained nickel is widely
distributed in very low concentrations in the body. Information on
normal nickel levels in organs and tissues is presented in Tables
23 and 24.
Table 23. Reference values for nickel concentrations in human autopsy tissuesa
--------------------------------------------------------------------------------------
Nickel concentrations
Tissue No. of Wet weight (µg/kg) Dry weight (µg/kg) Reference
subjects Mean±SD Range Mean±SD Range
--------------------------------------------------------------------------------------
Lung 4 16±8 (8-24) 86±56 (33-146) Sunderman et al. (1971)
9 132±99 (50-290) Chen et al. (1977)
41 7±10 (<1-70) Zober et al. (1984)
15 180±105 (43-361) Seemann et al. (1985)
9 18±12 (7-46) 173±94 (71-371) Rezuke et al. (1987)
15 44±56 (16-242) Raithel (1987)
Kidney 6 125±54 (50-120) Chen et al. (1977)
36 14±27 (<1-165) Zober et al. (1984)
18 34±22 (<5-84) Seemann et al. (1985)
10 9±6 (3-25) 62±43 (19-171) Rezuke et al. (1987)
Liver 4 9±3 (5-13) 32±12 (21-48) Sunderman et al. (1971)
23 18±21 (<5-86) Seemann et al. (1985)
10 10±7 (8-21) 50±31 (11-102) Rezuke et al. (1987)
Heart 4 6±2 (4-8) 23±6 (16-30) Sunderman et al. (1971)
9 8±5 (1-14) 54±40 (10-110) Rezuke et al. (1987)
Spleen 22 23±20 (<5-85) Seemann et al. (1985)
10 7±5 (1-15) 37±31 (9-95) Rezuke et al. (1987)
--------------------------------------------------------------------------------------
a Adapted from: Rezuke et al. (1987).
Table 24. Normal nickel concentration in Japanese human
tissues (mg/kg wet weight)a
---------------------------------------------------------------
Tissue Sex/ Median Average Range Mean±SD
number
---------------------------------------------------------------
Rib M/6 0.19
0.230 0.13-0.35 0.23±0.07
F/6 0.27
Lung M/15 0.21
0.160 0.04-0.44 0.16±0.09
F/15 <0.10
Small M/5 0.11
intestine 0.120 0.05-0.29 0.13±0.07
F/5 0.15
Large M/5 0.14
intestine 0.111 0.04-0.30 0.14±0.10
F/5 0.15
Trachea M/3 0.09
0.098 0.06-0.11 0.09±0.02
F/1 0.11
Kidney M/14 0.10
0.081 0.01-0.30 0.10±0.07
F/14 0.10
Skin M/4 0.09
0.072 0.02-0.22 0.10±0.08
F/2 0.14
Muscle M/5 0.11
0.070 0.02-0.27 0.10±0.08
F/5 0.09
Liver M/14 0.10
0.068 0.03-0.22 0.08±0.05
F/13 0.05
Cerebrum M/2 0.06
0.025 0.02-0.11 0.05±0.11
F/1 0.03
Cerebellum M/1
NMb <0.03 NIc NM
F/1
Heart NM NCd NC NM
Pancreas M/6
NM <0.10 NM NM
F/2
---------------------------------------------------------------
Table 24 (contd.)
---------------------------------------------------------------
Tissue Sex/ Median Average Range Mean±SD
number
---------------------------------------------------------------
Spleen M/1 <0.30
NM NM NM
Adrenal M/1 <0.10
glands NM NM NM
Testis M/1 0.05
Ovary NM - NM NM
Fat F/3 NM <0.01 NI NM
---------------------------------------------------------------
a From: Sumino et al. (1975).
b NM = not measured.
c NI = not indicated.
d NC = not calculated because there were less than 5 samples
available or there was no mean.
Generally, there is no significant influence of sex or age on
human organ levels of nickel (McNeely et al., 1971a; Turhan et al.,
1983; Zober et al., 1984). The ribs, liver, and kidneys in babies
up to the age of 3 months, were found to accumulate significantly
more nickel than those in persons between 1 and 90 years of age
(Schneider et al., 1980). Few data exist on the organ levels of
nickel in occupationally exposed persons. In lung autopsy samples
from 4 deceased persons living in the vicinity of a nickel-
processing industry in the German Democratic Republic, the mean
nickel concentration was 2135±1867 mg/kg dry weight (Schneider et
al., 1980).
(c) Body burden
One assessment of nickel metabolism in human beings indicated
that the body burden of nickel in normal adults averaged 0.5 mg (7
µg/kg for a 70-kg adult person) (Bennett, 1984). However, in a
study on 30 Japanese subjects, Sumino et al. (1975) calculated a
total body burden of about 5.7 mg (for a body weight of 55 kg), and
Schroeder et al. (1962) indicated a body burden of 10 mg nickel for
an adult person. Bennett (1984) concluded that the oral intake of
nickel averaged 170 µg/day, of which approximately 5% would be
absorbed (8.5 µg/day). Inhalation of nickel averaged 0.4 µg/day
for urban dwellers, of which 35% was retained (0.07-0.14 µg/day);
this involves the assumption that 70% of the nickel absorbed into
the blood is promptly excreted by the kidneys and that the
remaining 30% is deposited in the tissues, with a mean retention
time of 200 days (Bennett, 1984).
6.2.2.5 Pathological states influencing nickel levels
Nickel metabolism is known to be altered in several common
diseases, as well as in some physiological states. Alonzo & Pell
(1963) observed increased nickel concentrations in the serum of 19
out of 20 patients with acute myocardial infarction, sampled within
24 h of admission to the hospital. Sunderman et al. (1970, 1971)
reported increased nickel concentrations in the serum of 25 out of
35 patients with acute myocardial infarction, sampled 12-36 h after
onset of symptoms. The frequent occurrence of hypernickelaemia
after acute myocardial infarction has been confirmed by studies in
the Federal Republic of Germany (Völlkopf et al., 1981), Pakistan
(Khan et al., 1984), the United Kingdom (Howard, 1980), the USA
(Leach et al., 1985), and the USSR (Nozdryukhina, 1978). McNeely
et al. (1971a) showed that hypernickelaemia is not specific for
myocardial infarction, because nickel concentrations in serum are
also increased in patients with cerebral stroke and thermal burns,
as well as in patients with myocardial ischaemia without
infarction. Hypernickelaemia has been observed in patients with
unstable angina pectoris, without infarction, and in patients
suffering from coronary atherosclerosis, who developed cardiac
ischaemia during treadmill exercise (Leach et al., 1985).
Volini et al. (1968) observed increased nickel concentrations
in the liver in both the early and advanced stages of hepatic
cirrhosis. In a patient suffering from aspartylglycosaminurea, a
10-fold increased concentration of nickel in the hepatic tissue was
reported by Palo & Savolainen (1973).
Significantly decreased serum-nickel levels have been measured
in steel-mill workers exposed to extreme heat (Szadkowski et al.,
1969b).
In an investigation by Rubanyi et al. (1982a), serum-nickel
levels in postpartum mothers were found to be reduced by 60%.
However, a significant 20-fold elevation in the concentration of
nickel was observed immediately after delivery of the infant, but
before delivery of the placenta. Nomoto et al. (1983) did not
confirm the occurrence of hypernickelaemia. Post-operative
hypernickelaemia and nickeluresis were observed in patients
following total knee and hip arthroplasty with porous coated nickel
alloy prostheses (Sunderman et al., 1989b).
Nickel concentrations in the serum, whole blood, and urine from
61 patients with chronic alcoholism were elevated 17-, 15-, and 39-
fold, respectively, after 4 months to 4 years of disulfiram
treatment. Disulfiram (tetraethyl-thiuram-disulfide), a nickel-
chelating agent, is used in alcoholism therapy (Hopfer et al., 1987).
6.3. Elimination and excretion
The elimination routes for nickel in human beings and animals
depend, in part, on the chemical form of the compound and the mode
of intake. In general, relatively low gastrointestinal absorption
explains the elimination of dietary nickel in the faeces. In human
beings and animals, urinary excretion is usually the major
clearance route for absorbed nickel. Other routes of elimination
are of minor importance. All body secretions appear to have the
ability to excrete nickel; it has been found in saliva, sweat,
tears, and milk. Biliary excretion is minimal in animals, but may
be significant in human beings. Hair is an excretory tissue for
nickel.
6.3.1. Experimental animals
In experimental animals, urinary excretion is the main
clearance route for nickel compounds, introduced parenterally.
Only a small portion of an injected dose is excreted via the
gastrointestinal tract. Wase et al. (1954) studied the
distribution and elimination of 63Ni in mice using a high dose (102
µg 63Ni/animal), administered intraperitoneally, and found faecal
and urinary elimination in the ratio of 30:70%. The primary route
of elimination of supplemental dietary nickel (carbonate) fed to
calves was faecal (Tedeschi & Sunderman, 1957; O'Dell et al.,
1971).
Biliary excretion of nickel was minimal following subcutaneous
injection of 0.1 mg 63Ni, as nickel chloride, in rats (Marzouk &
Sunderman, 1985).
In rats or rabbits, after inhalation of nickel carbonyl,
Sunderman & Selin (1968) and Mikheyev (1971) found the lungs were
the major excretory organ besides excretion via the urine, 2-4 h
after exposure. Other studies indicated that up to 90% nickel was
excreted in the urine (Armit, 1908; Tedeschi & Sunderman, 1957;
Sunderman & Selin, 1968; Mikheyev, 1971) and 38% was exhaled via
the lungs as nickel carbonyl (Sunderman & Selin, 1968).
After intravenous injection of 14C-nickel carbonyl in rats,
Kasprzak & Sunderman (1969) found that 30% of the 14C was excreted
in the expired air as 14C-nickel carbonyl and 50% as 14C-carbon
monoxide.
Nickel is excreted in the urine, not as the free metal, but
bound to a protein that is similar to, or a fragment of, the
soluble low relative molecular mass glycoprotein associated with
nickel in renal tissue (Verma et al., 1980; Abdulwajid & Sarkar,
1983).
6.3.2. Human beings
As human beings take up most nickel via ingestion, it is
eliminated unabsorbed, mainly in the faeces (Drinker et al., 1924;
Tedeschi & Sunderman, 1957; Sunderman et al., 1963; Nodiya, 1972).
Horak & Sunderman (1973) found that the faecal elimination of
nickel in 10 healthy adults averaged 258 µg/day (SD 126) or 14.2
mg/kg, (dry weight) (SD 2.7), thus, the normal faecal elimination
of nickel was approximately 100 times greater than the normal
urinary excretion (2.6 µg/day (SD 1.4) or 2.2 µg/litre (SD 1.2)).
The urinary nickel levels of persons occupationally exposed to
appreciable nickel concentrations, via inhalation at the work-
place, are raised significantly. Positive correlations have been
reported between air and urinary nickel concentrations in workers
in the nickel industry (section 6.2.2.4). Urinary nickel
concentrations of normal and exposed persons are given in Tables 19
and 20. The large amounts of nickel also found in the faeces, in
some cases, indicated that, either retrograde loss from the lung
into the oesophagus, or considerable oral exposure via contaminated
surfaces, had occurred.
Nickel concentrations in samples of human bile (section
6.2.2.4) suggest that biliary excretion of nickel may be
quantitatively significant in human beings (Rezuke et al., 1987).
Sweat may constitute an excretory route of significance under
conditions of physical exertion. Hohnadel et al. (1973)
demonstrated that, in sauna bathers, the mean concentrations of
nickel in the sweat from healthy men and women were significantly
higher than the mean concentrations in the urine (men: 52 µg/litre,
SD=36; women: 131 µg/litre, SD=65). Under conditions of profuse
sweating, appreciable losses of nickel occurred. This may account
for the diminished concentrations of serum nickel that were
reported by Szadkowski et al. (1969b) in blast-furnace workers who
were exposed to extreme heat over a long period.
The role of nickel deposition in human hair as an excretory
mechanism has been studied (section 6.2.2.4).
Measurements of salivary nickel were performed by Catalanatto
et al. (1977) on specimens of parotid saliva from 38 healthy
adults. The concentrations of nickel in saliva averaged 1.9 ± 1.0
µg/litre (range 0.8-4.5 µg/litre). There was no significant
correlation between the concentrations of salivary nickel and
protein. No significant differences were observed between the mean
concentrations of nickel in saliva samples from men and women.
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
7.1. Microorganisms
Nickel is considered essential for certain metabolic processes
in bacteria. Bartha & Ordal (1965) demonstrated a nickel
requirement in the "Knallgas" bacterium Alcalignes entrophus. A
nickel requirement was reported by van Baalen & O'Donnell (1978)
for the blue green algae Oscillatoria sp.
Fungi and microorganisms demonstrate a fairly wide variety of
sensitivity to nickel, but are generally more tolerant than the
higher organisms. The toxic effects of nickel on microorganisms,
including eubacteria (non-marine and marine), actinomycetes,
yeasts, and filamentous fungi, were studied by Babich & Stotzky
(1982a,b; 1983). Filamentous fungi varied considerably in their
response to nickel, growth of Achyla sp. being inhibited at 5 mg
nickel/litre whereas Aspergillus niger and Gliocladium sp. were
only affected at concentrations as high as 1000 mg nickel/litre.
With actinomycetes and eubacteria, there was less variability in
toxicity. Concentrations of nickel inhibiting growth ranged from 5
to 30 mg nickel/litre, with the exception of Caulobacter leidyi,
which exhibited some growth at 100 mg nickel/litre. Growth
inhibition in yeasts occurred at 1-40 mg nickel/litre. In all
microorganisms, toxicity increased as the pH decreased.
Babich & Stotzky (1983) investigated the influence of various
factors on the toxicity of nickel for eubacteria, an actinomycete,
and yeasts. Reductions in cell number occurred at 5 or 10 mg/litre
and viable cells were eliminated at 10-50 mg/litre, though some
species were unaffected after 24 h at 100 mg/litre. Reductions in
pH from 6.8 to 5.3 enhanced the toxicity of 75 mg nickel/litre in
some species, but not in others. The toxicity of nickel (100
mg/litre) for marine microbes was reduced by increasing the
salinity and decreasing the temperature. Addition of a simulated
sediment (a mixture of organic and inorganic particles from soil)
reduced toxic effects after exposure to 100 mg nickel/litre. In
freshwater microbes, addition of a clay mineral (50 mg/litre)
provided protection against the toxicity of 10 mg nickel/litre.
This effect was probably because of the adsorption of nickel on the
particulates. Increasing the hardness of the water, by adding
calcium carbonate at 200 or 400 mg/litre, reduced the toxicity of
10 mg nickel/litre. Long-term studies indicated that microbial
survival was greater in marine than in fresh water. Bringmann et
al. (1980) reported that, in fairly hard water (approximately 150
mg CaCO3/litre) and a pH of 6.9, a nickel concentration of 0.82
mg/litre reduced the numbers of the saprozoic flagellate Chilomonas
paramecium.
Thus, fungi and microorganisms have a wide range of
sensitivities to nickel, but are usually more tolerant than higher
organisms.
7.2. Aquatic algae and plants
Nickel at concentrations of 0.05-0.1 mg/litre inhibited the
growth of algae, though some species may be more tolerant (Spencer,
1980). Upitis et al. (1980) reported growth inhibition in blue-
green algae at concentrations of 1-5 mg nickel/litre. The
chlorophyll content was found to be significantly reduced leading
to discoloration of the cells. Concentrations of 10-30 mg
nickel/litre were lethal for Chlorella sp. The same authors
investigated the influence of various environmental factors on the
toxicity of nickel for Chlorella. Nickel inhibition could be
overcome by the addition of ethylenediaminetetramine (EDTA) (40
mg/litre) and also by the addition of zinc (10 mg/litre) to a
medium containing 5 mg nickel/litre. A synergistic effect of
copper and nickel was demonstrated by Hutchinson (1973) for
Chlorella vulgaris, Scenedesmus acuminata, Haematococcus capensis,
and Chlamydomonas eugametos.
A nickel concentration of 0.1 mg/litre at 20 °C inhibited the
growth of 4 species of green algae, Pediastrum tetras,
Ankistrodesmus falcatus, Scenedesmus quadricauda, and S. dimorpha.
However, a concentration of 0.6 mg/litre did not affect the blue-
green alga Anabaena cylindrica, though it reduced the rate of
growth of Anabaena flos-aquae (Spencer & Greene, 1981). Stokes
(1975) studied Scenedesmus acutiformis var. alternans, from a lake
in a nickel-mining and smelting area. The lake water contained
about 2.5 mg nickel/litre. At a nickel concentration of 1.9
mg/litre, the Scenedesmus grew at 53% of the rate of controls grown
in clean water and, at 3.0 mg/litre, growth was still 18% of the
control rate.
A nickel concentration of 0.125 mg/litre inhibited the growth
of Anabaena inequalis, but a concentration of 10 mg/litre was
required to inhibit photosynthesis, and 20 mg/litre, to inhibit
nitrogenase activity (Stratton & Corke, 1979).
Chiaudani & Vighi (1978) exposed Selenastrum capricornutum to
nickel in a standard medium with, and without, EDTA. At 24 °C and
a pH of 6.9-6.3 the 7-day EC50 (inhibition of growth to 50% of
control values) was 0.9925 mg nickel/litre. When 0.3 mg EDTA/litre
was added to medium, the 7-day EC50 of nickel was increased to
0.013 mg/litre. In a further study, the addition of 0.04 mg
nickel/litre to the water samples did not inhibit growth as much as
was predicted from the laboratory studies.
When the diatom Navicula pelliculosa was exposed to 0.1 mg
nickel/litre (of which all but 0.2% was said to be Ni2+) for 14
days, growth was retarded (50% of control value) (Fezy et al.,
1979).
Hutchinson & Czyrska (1975) exposed Lemna minor (Valdiviana),
for 3 weeks, to nickel concentrations ranging from 0.01 to 1.0
mg/litre in an artificial medium at pH 6.8, and a temperature of
24±2 °C, with 16 h of light per 24 h. They found that a
concentration of 0.05 mg/litre stimulated growth, and that
concentrations greater than 0.1 mg/litre inhibited growth. At 1 mg
nickel/litre, growth was prevented. Nickel uptake and toxicity
were enhanced by the presence of copper.
The same authors examined Lemna minor from 23 sites, where the
mean concentration of nickel in the water was 0.027 mg/litre. The
plants contained from 5.4 to 35.1 mg nickel/kg (dry weight),
equivalent to bioaccumulation factors (BCFs) of 200 and 1300.
Lemna, grown on a culture medium containing 0.01-1 mg nickel/litre
at pH 6.8 and a temperature of 24±2 °C for 3 weeks, accumulated
nickel concentrations ranging from 40 mg/kg dry weight, at 0.01
mg/litre, to 3067 mg/kg, at 0.5 mg/litre.
Clark et al. (1981) studied the accumulation and depuration of
nickel by Lemna perpusilla. Plants collected from a fly ash basin
(nickel concentration, 0.1 mg/litre) were allowed to depurate in
dechlorinated tap water at 20 °C for a 14-day depuration period.
Concentrations of nickel fell from about 160 mg/kg dry weight to
about half of this value. In the accumulation studies, Lemna
accumulated nickel readily, particularly at the lowest ambient
concentration of 0.1 mg/litre, and levels reached 500-600 mg/kg in
10 days. After a return to depuration conditions, the nickel
concentration in the plants fell to 160 mg/kg, in 8 days.
Euglena gracilis, exposed to 8.9 x 10-4 mg nickel/litre in
spring-water, accumulated the metal to a concentration of 1.8
mg/kg, a BCF of about 2000 (Cowgill, 1976).
Ipomea aquatica took up 200 mg nickel/kg dry plant in 48 h,
mostly in the roots, from water containing 5 mg nickel/litre (BCF =
40) (Low & Lee, 1981).
It is noted that, under laboratory conditions, the growth of a
macrophyte (Lemna) was inhibited at a concentration of 0.1
mg/litre, but the growth of algae was inhibited at concentrations
as low as 0.04 mg/litre. However, in natural waters, a nickel
concentration of 0.04 mg/litre had a less inhibiting effect.
7.3. Aquatic invertebrates
Timourian & Watchmaker (1972) investigated the uptake of nickel
chloride and its effects on the development of sea urchin embryos.
After fertilization, sea urchin eggs exhibited increased rates of
nickel uptake that appeared to be a result of an active transport
mechanism. When exposed to 59-590 mg nickel/litre, gastrulation of
embryos was prevented. Embryos grown in 0.59-5.9 mg nickel/litre
were able to gastrulate, but failed to develop dorsoventral symmetry.
Acute and long-term toxicity studies performed by Powlesland &
George (1986) revealed a different sensitivity to nickel in
different developmental stages of Chironomus riparis larvae. First
instar larvae were found to be significantly more sensitive to
nickel than second instars with 48-h LC50 values of 79.5 mg/litre
and 169 mg/litre, respectively. Longer term toxicity tests (30
days), in which larvae were allowed to develop from eggs until just
prior to pupation, indicated that nickel concentrations up to 25
mg/litre appeared to have little effect on the percentage hatch.
However, the growth of larvae was significantly reduced at 2.5
mg/litre. A threshold concentration for the effect of nickel on
growth was estimated to be 1.1 mg nickel/litre.
Bryant et al. (1985) investigated the effects of temperature
and salinity on the toxicity of nickel in two estuarine
invertebrates. In the amphipod Corophium volutator, and the
bivalve Macoma baltia, 96-h LC50 values varied from 5 to 54 mg
nickel/litre and from 95 to 1100 mg nickel/litre, respectively. A
decrease in salinity from 35 to 5 mg/litre resulted in greater
toxicity in both species. Toxicity also increased in Corophium
volutator with an increase in temperature from 5 to 15 °C.
Mathis & Cummings (1973) measured nickel levels in sediments,
water, and biota in a river. The water contained the lowest
concentrations of nickel (<0.01 mg/litre) and the sediments, the
highest (3-124 mg/kg). Two species of tubificid worms (Limnodrilus
hoffmeisteri and Tubifex tubifex) contained 4-18 mg nickel/kg wet
weight. Three species of clam were examined: in order of
increasing nickel content (mg/kg on a wet-weight basis) they were
Quadrula quadrula (0.4-1.6), Amblema plicata (0.4-2.3) and
Fusconaia flava (0.7-3.0). Neither worms nor clams were starved
before being examined, and nickel may also have been present in
their gut contents. Brkovic-Popovic & Popovic (1977) studied the
effects of nickel and other heavy metals on the survival of Tubifex
tubifex in water of different pH and hardness. At a hardness of
0.1 mg CaCO3/litre, the 48-h LC50 was 0.082 mg nickel/litre.
Increases in hardness to 34.2 mg CaCO3/litre and 261 mg CaCO3/litre
increased the 48-h LC50 to 8.7 mg nickel/litre and 61.4 mg
nickel/litre, respectively, thus decreasing the toxic effects.
A 64-h LC50 was determined for Daphnia magna of 0.32 mg
nickel/litre, at a temperature of 25 °C and a hardness of around
100 mg CaCO3/litre (Anderson, 1950). Baudouin & Scoppa (1974),
using Daphnia hyalina, estimated a 48-h LC50 value of 1.9 mg
nickel/litre at a temperature of 10 °C, pH 6.2, and hardness of 58
mg CaCO3/litre.
Exposure of Daphnia magna to nickel sulfate at concentrations
ranging from 5 to 10 µg nickel/litre for 3 generations resulted in
extermination (Lazareva, 1985).
Hall (1982) exposed Daphnia magna to 0.25 mg nickel/litre,
including 63Ni, at pH 6.9, and a temperature of 18-21 °C in water
with a hardness of 60 mg CaCO3/litre. The uptake of nickel was
initially rapid (about 12 mg in 80 h). Depuration also occurred,
and 25-33% of the nickel was lost from the animal in the exuviae,
shed on moulting. Gut tissue did not accumulate nickel until after
the first 5 h of exposure, suggesting that the oral route was not
important for nickel.
Daphnia, exposed for 3 weeks to 0.125 mg nickel/litre in water,
at a temperature of 18±1 °C, hardness of 42.3-45.3 mg CaCO3/litre,
and pH 7.74, had 43% lower weights than control Daphnia, 9% less
proteins, and the glutamic oxalacetic transaminase activity was
reduced by 26%. A 16% impairment of reproduction occurred at 0.03
mg nickel/litre with a 50% impairment at 0.095 mg nickel/litre
(Biesinger & Christensen, 1972).
Cowgill (1976) reared Daphnia magna and Daphnia pulex for 3
months on Euglena gracilis, which had been cultured in spring water
containing 8.9 x 10-4 mg nickel/litre. The algal cells contained
1.8 mg nickel/kg, the Daphnia magna, 3.6 mg nickel/kg, and the D.
pulex, 4.2 mg nickel/kg, giving BCF values of 2020 and 4050.
The acute effects of nickel on the freshwater snails Juga
plicifera and Physa gyrina were studied by Nebeker et al. (1986).
The 96-h LC50 values were 0.237 mg nickel/litre and 0.239 mg/litre,
respectively. A no-observed-effect-level of 0.124 mg nickel/litre
was determined for Juga plicifera. Data published for other
species of snails did not indicate a pronounced effect of hardness
on nickel toxicity (Nebeker et al., 1986). The eggs and adults of
the snail Amnicola were exposed to nickel in water at a temperature
of 17 °C, pH 7.6, and a hardness of 50 mg/litre with 6.2 mg
dissolved oxygen/litre. The 24-h LC50s were 26.9 mg/litre for eggs
and 21.1 mg/litre for adults, whereas at 96 h, the LC50s were 11.4
and 14.3 mg/litre, respectively (Rehwoldt et al., 1973).
Nickel influenced the rate of filtration in the marine bivalve
Villorita cyprinoides (Abraham et al., 1986). Rates of filtration
decreased exponentially with increasing nickel levels. The EC50,
i.e., the concentration that reduced the rate of filtration by 50%,
was 0.003 mg nickel/litre. A 96-h LC50 was 0.061 mg nickel/litre.
It is concluded that the nickel concentrations causing mortality in
acutely exposed invertebrates were generally similar to those for
fish, but that Daphnia sp. appeared more sensitive, with LC50
values of less than 2 mg nickel/litre.
7.4. Fish
Sensitivity to nickel varies considerably among fish species.
However, 96-h median lethal concentrations generally fall within
the ranges of 4-14 and 24-44 mg nickel/litre for tests conducted in
soft, and hard water, respectively. For example, in water of
hardness 100, 125, and 174 mg CaCO3/litre, the LC50s for rainbow
trout, exposed from fertilization to 4 days after hatching, were
0.05, 0.06, and 0.09 mg/litre, respectively (Birge & Black, 1980).
Pickering & Henderson (1966) compared the toxicity of nickel
chloride in waters of 2 levels of hardness (total hardness 20 or
300 mg CaCO3/litre). The 96-h LC50 was 4.9 mg/litre for fathead
minnow (Pimephales promelas) and 5.3 mg/litre for bluegill sunfish
(Lepomis macrochirus) in soft water, and 43.5 mg/litre and 39.6
mg/litre, respectively, in hard water. Rainbow trout (Salmo
gairdneri) showed a 4-fold increase in sensitivity between hard and
soft water, the 48-h LC50 changing from about 80 to about 20
mg/litre (Brown, 1968).
In rainbow trout, a 48-h LC50 for nickel sulfate of 263
mg/litre was determined in a static test (Osterreichisches
Forschungs-Zentrum Seibersdorf, 1983). The water had a hardness of
402 mg CaCO3/litre, a pH of 7.6, and a temperature of about 15 °C.
The first signs of toxicity were observed at a concentration of 85
mg nickel sulfate/litre.
Using the same test method and corresponding test conditions,
Butz (1984) demonstrated that a decrease in water hardness from 270
to 49 mg CaCO3/litre resulted in a 2.6-fold increase in toxicity.
Rehwoldt et al. (1971, 1972) studied the effects of temperature
on the toxicity of nickel for 6 warm-water species of fish: banded
killifish (Fundulus diaphanus), striped bass (Roccus saxatilis),
pumpkin seed (Lepomis gibbosus), white perch (Roccus americanus),
American eel (Anguilla rostrata) and carp (Cyprinus carpio). There
was a wide range of sensitivity among these species, with 96-h LC50
values at 17 °C ranging from 6.2 to 46.2 mg nickel/litre. However,
each species showed very little variation in sensitivity at
temperatures of 17 and 28 °C. At 28 °C and a hardness of 55 mg
CaCO3/litre, Roccus saxatilis and Lepomis gibbosus were the most
sensitive, having 96-h LC50s of 6.3 and 8.0 mg nickel/litre,
respectively, while Fundulus diaphanus was the most tolerant (46.1
mg nickel/litre). Roccus americanus, Anguilla rostrata, and
Cyprinus carpio were intermediate in their response, but relatively
sensitive, the 96-h LC50s being 13.7, 13.9, and 10.4 mg
nickel/litre, respectively (Rehwoldt et al., 1972).
In static tests in softer water (20 mg CaCO3/litre), Pimephales
promelas, Lepomis macrochirus, Carassius auratus, and Lebistes
reticulatus showed similar levels of sensitivity in terms of 96-h
LC50s with LC50 values of 4.9, 5.3, 9.8, and 4.5 mg nickel/litre,
respectively (Pickering & Henderson, 1966).
In a flow-through test, rainbow trout (Salmo gairdneri) were
less sensitive than other fish species with a 48-h LC50 of 20 mg
nickel/litre, in soft water (Brown, 1968). In field studies, Hale
(1977) reported a 96-h LC50 for rainbow trout of 35.5 mg
nickel/litre in continuous-flow tests in water with a hardness of
82-132 mg CaCO3/litre. Arillo et al. (1982) found that rainbow
trout (Salmo gairdneri), exposed to nickel, showed a reduction in
glucidic stores. This effect is consistent with direct metal
interaction on both membranes and enzyme thiolic groups of the
pancreatic cells. Other effects, similar to those found with other
metals, such as damage to the secondary lamellae of gills (Hughes
et al., 1979) and sialic acid depletion in the gills (Arillo et
al., 1982) have been described.
A 96-h LC50 of 118.3 mg nickel/litre was determined for the
marine grey mullet (Chelon labrosus) (Taylor et al., 1985a).
The toxicity of nickel(II) chloride was studied in 2 estuarine
fish species (US EPA, 1987). In tidewater silverside (Menidia
peninsulae) larvae, a 96-h LC50 was 38 mg/litre. For adult spot-
fish (Leiostomus xanthums), the 96-h LC50 was 70 mg/litre.
In short-term tests in soft water, the most sensitive species
of freshwater fish were killed by exposure to concentrations of
about 4-20 mg nickel/litre. Higher LC50 values of nickel have been
found for different species of fish in harder waters, ranging from
about 30 to 80 mg nickel/litre. From the limited data available,
it appears that hardness has the greatest effect on toxicity, while
other determinants have not been proved to have any significant
effects. In acute tests, there are interspecies differences in
sensitivity, but these are within a single order of magnitude.
Shaw & Brown (1971) did not observe any effects on rainbow
trout eggs fertilized in water containing 1 mg nickel/litre and
then maintained in clean water. In a life-cycle study on fathead
minnow, in water with a hardness of 210 mg CaCO3/litre, pH 7.8, and
an average temperature of 18 °C, Pickering (1974) found that nickel
concentrations of 0.38 mg/litre and less (0.18 and 0.08 mg/litre)
did not have any adverse effects on survival, growth, and
reproduction. However, a concentration of 0.73 mg nickel/litre had
a statistically significant effect on the number of eggs produced
per spawning and on the hatchability of these eggs, though it did
not affect the survival and growth of the first generation of fish.
In carp (Cyprinus carpio) eggs and larvae, the 72-h LC50s were
6.1 and 8.4 mg/litre, respectively, while the 257-h LC50 for larvae
was 0.75 mg nickel/litre in water with a hardness of 128 mg
CaCO3/litre, pH of 7.4, and a temperature of 25 °C. A
concentration of 3 mg nickel/litre caused an increased incidence of
abnormal larvae (23% compared with 8.6% in the controls) and 32.7%
of embryos failed to hatch, compared with 5.9% in the controls
(Blaylock & Frank, 1979).
Birge et al. (1978) exposed rainbow trout and largemouth bass
(Micropterus salmoides) from fertilization to 4 days after
hatching, to 11 trace metals found in coal, at a temperature of 12-
13 °C, equivalent to a period of exposure of 28 days for rainbow
trout and 8 days for bass. The LC50 values for these periods were
0.05 mg nickel/litre for rainbow trout and 2.06 mg nickel/litre for
bass. The water used in the tests had a hardness of about 100 mg
CaCO3/litre and a pH of 7.2-7.8. For rainbow trout and goldfish,
teratic larvae were observed at exposure levels that did not
significantly affect egg hatchability. In water from a natural
source with a pH of 7.8 and 174 mg CaCO3/litre hardness, an LC50
for rainbow trout was 0.09 mg nickel/litre whereas in dechlorinated
tap water (pH 7.6, hardness 125 mg CaCO3/litre) the LC50 was 0.06
mg nickel/litre.
Using water with a hardness of about 100 mg CaCO3/litre and pH
7.2-7.8, Birge & Black (1980) found that the LC50 from
fertilization to 4 days after hatching was 0.71 mg nickel/litre for
channel catfish (Ictalurus punctatus) and 2.78 mg nickel/litre for
goldfish (Carassius auratus).
Calamari et al. (1982) found that, during the long-term
exposure of fish to 1 mg nickel/litre, continuous uptake of nickel
occurred for 180 days. The concentrations found were: liver, about
2.9 mg nickel/kg wet weight, kidneys, 4.0 mg/kg, and muscle, 0.8
mg/kg, while, at the beginning of the study, the levels had been
1.5, 1.5, and 9.5 mg nickel/kg, respectively. Toxicokinetic
modelling indicated that theoretical asymptotic values for the
liver, kidney, and muscle should be reached in 397, 313, and 460
days, respectively, at which times the calculated bioconcentration
factors (BCF tissue concentration/environmental concentration) were
3.1, 4.2, and 1.9, respectively. Release was slower in clean water
and the proportions of nickel remaining after 90 days in the liver,
kidney, and muscle were 25, 41, and 31%, respectively. These data
suggest that nickel had little capacity for accumulation in the
tissues examined. However, even these relatively low
concentrations are toxic (Arillo et al., 1982).
7.5. Terrestrial organisms
7.5.1. Plants
Nickel is ubiquitous in plant tissues. There is evidence that
nickel is a required nutrient in a number of plant species. The
urease enzyme of jack bean (Canavalia ensiformis) has been shown to
be a nickel metalloenzyme (Dixon et al., 1975).
Although nickel levels above 50 mg/kg in plants are usually
toxic, a number of plant species may tolerate higher levels
(section 4.2.1).
In general, the effects of long-term, low-level exposure to
nickel are only manifested in growth decrements with no visible
signs. Nickel toxicity in plants is characterized by chlorosis and
necrosis of the leaves, stunting of the roots, deformation of
various plant organs, and wilting (Brooks, 1980; Prokipcak &
Ormrod, 1986).
Apart from the solubility of nickel ions or nickel complexes,
other factors can affect nickel toxicity. Of special interest is
the presence of other heavy metals, which can act synergistically,
and the ameliorating effects of calcium.
A synergistic effect of nickel and copper on the growth of bush
beans was demonstrated by Wallace & Berry (1983). When barley was
grown on loam soil with an elevated level of each of the 6 trace
elements, lithium (13 mg/kg soil, dry weight), zinc (200 mg/kg),
copper (200 mg/kg), nickel (100 mg/kg), and cadmium (100 mg/kg)
there was no reduction in yield when they were applied singly.
However, when all 6 were applied together, at the concentrations
applied singly, there was a 40% reduction in yield, probably
because of depressed phosphorus levels (Wallace et al., 1980a).
Investigations by Prokipcak & Ormrod (1986) of the growth
responses of tomato and soy bean to combinations of nickel, copper,
and ozone, indicated that the nature of the joint action of these
chemicals is very complex and depends on species, the
concentrations of the metals and ozone, and, to a lesser extent,
the duration of exposure. In the first study, nickel was added to
the nutrient solution of tomato and soy bean plants at 1.5, 7.5, or
37.5 mg/litre for 6 days, beginning on day 14 after seeding. The
plants were then exposed to 0.15 or 0.30 µlitre atmospheric
ozone/litre. Growth variables were markedly reduced by nickel, but
ozone response depended on the nickel level. In the second study,
0.3 or 1.5 mg nickel/litre was provided from the 5th or 14th day
onwards. There was little effect of duration of nickel treatment
on growth. Increasing nickel levels and increasing ozone levels
decreased growth, but there was no interaction. In the third
study, treatments with 1.5 or 3.0 mg nickel/litre were combined
with 3.0 or 6.0 mg copper/litre, prior to treatment with 0.25 ml
atmospheric ozone/litre. There were complex interactive effects of
all 3 compounds on tomato plant growth, but not on soy bean plant
growth.
Calcium can reduce nickel toxicity. For example, when soy
beans were grown in nutrient solution containing 1.2 mg
nickel/litre, leaf yield depression was 74% at 4 mg calcium/litre,
but only 45% at 400 mg calcium/litre (Wallace et al., 1980b).
7.5.2. Animals
Few data are available on the effects of nickel on terrestrial
animals. Most data are derived from laboratory animals and
indicate that nickel is an essential element in some species.
As land application of wastes is a common method of
fertilization, studies were performed to evaluate the impact of
heavy metals on the soil ecosystem, using the earthworm Eisenia
foetida as a test organism. Following a 14-day exposure to nickel
nitrate in artificial soil, the LC50 was calculated to be 757 mg/kg
(Neuhauser et al., 1985). Hartenstein et al. (1981) determined
the level at which added concentrations of heavy metals would cause
an activated sludge to induce toxic effects on Eisenia foetida.
Nickel was found to inhibit growth and to induce death at
concentrations of 1200-12000 mg/kg dry weight. These
concentrations seemed very high and the authors concluded that
nickel might have been accumulated by the large population of
microorganisms in the rich organic matrix, part of which might not
be ingestible or digestible by earthworms. This would enable
earthworms to grow in the presence of high nickel concentrations.
7.5.3. Essentiality of nickel for bacteria and plants
Evidence for specific biochemical functions of nickel has come
from studies of microbial systems. Nickel is involved, in some
way, in the "Knallgas" reaction, which is mediated by a number of
bacteria of different genera (Tabillion et al., 1980; Friedrich et
al., 1981; Albracht et al., 1982). The reduction of carbon dioxide
to acetate, carried out by acetogenic bacteria, is dependent on
nickel, which is needed to activate the enzyme carbon monoxide
dehydrogenase (Diekert & Thauer, 1980; Drake, 1982). Diekert &
Ritter (1982) demonstrated that Acetobacterium woodii growth on
fructose was stimulated by, but not dependent on, nickel, unlike
CO2 reduction. A number of studies have established that nickel is
the core metal in the tetrapyrrole ("Factor F430"), found in
methanogenic bacteria, and is essential for the growth of these
organisms.
In plants, Dixon et al. (1975) showed that nickel is essential
at the active site of urease in jack beans (Canavalia ensiformis),
for its enzymatic activity.
7.6. Population and ecosystem effects
Few data are available that identify nickel as a specific cause
for effects at the population level, because nickel is generally
associated with other, often more toxic, trace metals or pollutants
that could be involved in the effects.
Gradual ecological changes have been observed near sources
emitting nickel and other trace metals, resulting in a decrease in
the number and diversity of species (Hutchinson & Whitby, 1977;
Gignac & Beckett, 1986). Yan et al. (1985) investigated 39 lakes
in Ontario and found that the tracheophyte richness of acidic lakes
decreased with increasing nickel and copper levels.
In acidic copper-, and nickel-contaminated lakes near Sudbury,
Ontario, species richness and community biomasses were reduced in
Crustacean zooplankton communities (Yan & Strus, 1980).
DeCantazaro & Hutchinson (1985a,b) demonstrated that the
addition of nickel to microecosystems and incubated soil samples
from boreal jack pine forests could disrupt nitrogen cycling.
Nickel additions of 100-500 mg/kg soil were shown to stimulate
nitrification and nitrogen mineralization, resulting in loss of
nitrogen by leaching. The authors concluded that loss of nitrogen,
which is probably the nutrient most limiting to growth in a boreal
forest ecosystem, could have serious ecological consequences for
forests in the vicinity of nickel smelters.
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO AND OTHER TEST
SYSTEMS
Various studies have indicated that nickel is an essential
element in a number of experimental animal species and that it may
also have a physiological role in human beings. However, nickel
deficiency has not been demonstrated in human beings, and the
possible nickel requirement is probably very low. While the
elucidation of nickel essentiality is in progress, it has not
reached the stage where it can be quantified in relation to nickel
deficiency.
8.1. Animals
8.1.1. Essentiality
Earlier studies in trace-element nutritional research did not
demonstrate any consistent effects of nickel deficiency (Schroeder,
1968; Smith, 1969; Nielsen & Säuberlich, 1970; Wellenreiter et al.,
1970; Nielsen & Higgs, 1971; Schroeder et al., 1974), in part,
because of the technical difficulties of controlling nickel intake
due to its ubiquity. Since 1975, diets and environments have been
devised for adequately controlled studies on nickel metabolism and
nutrition, and the effects of deprivation have been described for
17 animal species, including: chicken, cow, goat, mini-pig, pig,
rat, and sheep.
Nickel is a component of several enzyme systems (certainly
urease and some hydrogenases) and it seems essential for the well-
being of several animal species (Spears et al., 1978).
8.1.1.1 Nickel deficiency symptoms
(a) Growth
In goats (Anke et al., 1977, 1978, 1980, 1986), pigs (Anke et
al., 1977, 1986; Spears, 1984; Spears et al., 1984), and rats
(Nielsen et al., 1975b; Schnegg & Kirchgessner, 1975a, 1980), a
nickel-deficient diet resulted in significantly decreased growth.
The growth depression depended on the nickel level and the duration
of administration and only became evident after intrauterine nickel
deficiency, i.e., in the second or later generations. In addition,
species-specific differences seemed to exist (Anke et al., 1977).
(b) Reproduction and mortality
In goats, mini-pigs, and rats, reproduction was decreased only
insignificantly by intrauterine nickel deficiency (Anke et al.,
1977; Schnegg & Kirchgessner, 1975a, 1980). Conception and
abortion rates as well as the number of offspring were not
influenced by nickel deficiency, but kidding in nickel-deficient
goats and farrowing in nickel-deficient sows occurred later (Anke
et al., 1974). Furthermore, at the end of the lactation period,
significantly fewer offspring of nickel-deficient goats were still
alive compared with control animals. Schnegg & Kirchgessner (1980)
did not find any increase in mortality in intrauterinely nickel-
deficient rats, whereas Nielsen et al. (1975b) found it to a
remarkable extent. Smaller litter size has been observed in both
rats and pigs (Anke et al., 1974; Nielsen et al., 1975b; Schnegg &
Kirchgessner, 1975a).
(c) Histological parameters
Nielsen & Sauberlich (1970) described a nickel-deficiency
syndrome in chickens, characterized by changes in the pigmentation
of the shank skin, thicker bones, swollen joints, and a light-
coloured liver. However, these findings are not consistent with
those observed by other authors (Sunderman et al., 1972; Nielsen,
1974). Sunderman et al. (1972) and Nielsen & Ollerich (1974)
observed ultrastructural lesions in the hepatocytes of chickens.
In mini-pigs fed a nickel-deficient diet, Anke (1974) observed
parakeratosis-like damage to the epithelium. Skin eruptions were
also seen in nickel-deficient goats; the hair of the animals was
brittle, there were fissures of the mouth and legs (Anke et al.,
1976, 1980b). Offspring of nickel-deficient rats had an anaemic
appearance (Schnegg & Kirchgessner, 1975a, 1980a; Nielsen et al.,
1979a,b).
(d) Rumen activity
Nickel seems to be essential for ruminants, because urease
activity in the rumen depends on nickel. Spears & Hatfield (1977)
demonstrated disturbances in metabolic parameters in lambs
maintained on a low-nickel diet, including reduced oxygen
consumption in liver homogenate preparations, increased activity of
alanine transaminase, decreased levels of serum proteins, and
enhanced urinary nitrogen excretion. In a follow-up study, Spears
et al. (1978) found that these animals had significantly lower
microbial urease activity. It was possible to increase urease
activity in the rumen contents by means of nickel supplementation.
(e) Disturbance of iron metabolism
Schnegg & Kirchgessner (1975b; 1976a,b) showed that nickel
deficiency in rats led to a reduced iron content in organs, reduced
haemoglobin and haematocrit values, and anaemia. Iron
supplementation did not cure this anaemia (Nielsen et al., 1979a;
Nielsen & Shuler, 1981), indicating a markedly impaired iron
absorption. Nickel-deficient goats eliminated 33% more iron via
the faeces than control animals (Anke et al., 1980). Spears et
al., (1984) found that additional nickel could improve the iron
status of neonatal pigs. The mechanism through which nickel might
enhance iron absorption is still unclear. While nickel might act
enzymatically to convert ferric to ferrous iron (a form more
soluble for absorption), it might also promote the absorption of
iron by enhancing its complexing with a molecule that can be
absorbed (Nielsen, 1984).
(f) Nickel/calcium interaction
Anke (1974) found that nickel-deficient mini-pigs excreted more
calcium renally than corresponding control animals. The skeletons
of nickel-deficient animals contained less calcium than those of
animals on a nickel-rich diet. Kirchgessner & Schnegg (1980a)
confirmed this effect of nickel deficiency in 30-day-old rats and
showed that more magnesium, instead of calcium, was incorporated
into bones.
(g) Nickel/zinc interaction
Analysis showed that different organs and body fluids of
nickel-deficient goats and pigs suffering from parakeratosis-like
changes of the skin and hair, were not only poor in calcium, but
also in zinc. There were single cases of dwarfism in goats (Anke,
1974; Anke et al., 1980, 1981). In rats, nickel deficiency also
resulted in a significantly decreased zinc concentration in organs,
demonstrated by the reduced size of the organs (Nielsen & Shuler,
1979; Kirchgessner & Schnegg, 1980a).
(h) Enzyme activities
The effects of nickel deficiency on enzyme activity have been
studied in rats by Schnegg & Kirchgessner (1975b, 1977a,b,c,d) and
Kirchgessner & Schnegg (1979, 1980b). They found that, as a rule,
the activity of a number of dehydrogenases and transaminases
decreased by 40-75% (malate dehydrogenase (MDH), isocitrate
dehydrogenase (ICDH), lactic dehydrogenase (LDH), glucose-6-
phosphate dehydrogenase (G6PDH), glutamate dehydrogenase (GLDH),
glutamic oxalate transaminase (GOT), glutamic pyruvic transaminase
(GPT)) with LDH and G6PDH being influenced by secondary iron
deficiency. Kirchgessner & Schnegg (1979, 1980b) also measured a
significant 50% reduction in the activity of alpha-amylases in the
liver and pancreas. The results of other studies suggest that
nickel may serve as a co-factor for the activation of calcineurin,
a calmodulin-dependent phosphoprotein phosphatase (King et al.,
1985).
(i) Substrate and metabolite concentrations
Nickel deficiency mainly affects carbohydrate metabolism, and
this has been demonstrated in nickel-deficient rats by Schnegg &
Kirchgessner (1977c). The glucose and glycogen contents of the
liver in nickel-deficient rats was reduced by 90% and the
triglycerides decreased by 40% compared with those in control
animals. Similar values were found in the serum of the rats, and
also in that of goats. Anke et al. (1980b) reported a reduced
triglyceride level in the serum of ruminants, but the cholesterol
level was unchanged. The significantly increased alpha-lipoprotein
and reduced beta-lipoprotein concentrations were probably connected
with a normal cholesterol level, and a disturbance of triglyceride
metabolism, because the alpha-fraction was rich in cholesterol and
the beta-fraction, rich in triglyceride.
The influence of nickel deficiency on the plasma cholesterol
concentration and on the fat content of the liver has been studied
(Nielsen, 1971; Sunderman et al., 1972; Nielsen et al., 1974,
1975a,b; Schnegg & Kirchgessner, 1977c). However, the results were
inconsistent, because the cholesterol level was not affected in
nickel-deficient animals. Nielsen (1971) found a decrease in the
fat content of the liver in chickens, but Anke et al. (1977) did
not find this in mini-pigs.
8.1.2. Acute exposures
8.1.2.1 Nickel carbonyl
Acute lethal concentrations of nickel carbonyl for laboratory
animals are shown in Table 25. The lethal doses range from an LC50
of 0.1 mg nickel carbonyl/litre air for a 20-min inhalation
exposure of the rat, to an LC50 of 2.5 mg/litre air for the dog
following inhalation exposure of 30 min. The LD50s via other
routes range from 13 to 65 mg/kg, the intraperitoneal route being
the most toxic.
Animals acutely exposed to nickel carbonyl vapour show
pulmonary effects and lesions similar to those observed in human
cases of industrial poisoning. The lung is the primary target organ
for nickel carbonyl in animals, and pulmonary effects are rapid at
high exposure levels, oedema occurring within 1 h of exposure.
Subsequently, proliferation and hyperplasia of the bronchial
epithelium and alveolar lining cells develop. Several days after
exposure, severe intra-alveolar oedema with focal haemorrhage and
alveolar cell degeneration occur. In animals that survive the
acute effects, regression of cytological changes with fibroblastic
proliferation within the alveolar interstitium occurs.
Pathological lesions in other organs after acute exposure of
animals to nickel carbonyl are less severe than those in the lung.
However, focal haemorrhage, congestion, oedema, hydropic
degeneration, mild inflammation, and vacuolization have been
reported in the brain, liver, kidney, adrenals, spleen, and
pancreas. In hepatic parenchymal cells, dilatation of rough
endoplasmic reticulum is the most prominent and consistent
ultrastructural abnormality. Nucleolar alterations also develop in
hepatocytes, 2-24 h after exposure to nickel carbonyl.
Pathological lesions of tubules and glomeruli have been seen in
rats exposed to nickel carbonyl (Kincaid et al., 1953; Sunderman et
al., 1961; Hackett & Sunderman, 1967).
8.1.2.2 Other nickel compounds
LD50 data for some other nickel compounds are listed in Table
26.
Table 25. Acute toxicity studies on nickel carbonyl in experimental animalsa
------------------------------------------------------------------------------------------------------------------------------
Species Route Lethal dose Observations in surviving Observation References
animals period after
exposure
------------------------------------------------------------------------------------------------------------------------------
Rabbit inhalation LC80 = 1.4 mg/litre 50 min Lungs: intra-alveolar 1-5 days Armit (1908)
Cat LC80 = 3.0 mg/litre 75 min haemorrhage, oedema, and (rabbit)
Dog LC80 = 2.7 mg/litre 75 min exudate and alveolar cell
degeneration
Adrenals: haemorrhages
Brain: perivascular
leukocytosis and neuronal
degeneration
Rat inhalation LC80 = 0.9 mg/litre 30 min Lungs: at 2-12 h, capillary 2 h-several Barnes & Denz
congestion and interstitial months (1951)
oedema, at 1-3 days, massive
intra-alveolar oedema, at
4-10 days, pulmonary
consolidation and interstitial
fibrosis
Mouse inhalation LC50 = 0.067 mg/litre 30 min Lungs: at 1 h, pulmonary 0.2 h-6 days Kincaid et al.
Rat LC50 = 0.24 mg/litre 30 min congestion and oedema, at (rat) (1953)
Cat LC50 = 0.19 mg/litre 30 min 12 h-6 days, interstitial
pneumonities with focal
atelectasis and necrosis, and
peribronchial congestion;
Liver, spleen, kidneys,
pancreas: parenchymal cellular
degeneration with focal
necrosis
Mouse inhalation LC100 = 0.2 mg/litre 120 min Sanotskii (1955)
Mouse LC100 = 0.01 mg/litre 120 min
Rat inhalation LC100 = 0.3 mg/litre 20 min Ghirinhgelli (1957)
Rat LC50 = 0.1 mg/litre 20 min
------------------------------------------------------------------------------------------------------------------------------
Table 25 (contd.)
------------------------------------------------------------------------------------------------------------------------------
Species Route Lethal dose Observations in surviving Observation References
animals period after
exposure
------------------------------------------------------------------------------------------------------------------------------
Mouse inhalation LC80 = 0.048 mg/litre 30 min West & Sunderman
Rat LC65 = 0.50 mg/litre 30 min (1958)
Dog inhalation LC90 = 2.5 mg/litre 30 min Sunderman et al.
(1961)
Rat inhalation Lungs: at 1-2 days intra- 1-6 days Sunderman et al.
Dog alveolar oedema and swelling 1-7 days (1961)
of alveolar lining cells, at
3-5 days inflammation,
atelectasis, and interstitial
fibroblastic proliferation;
Kidneys and adrenals: hyperaemia
and haemorrhage
Rat inhalation LC30 = 0.51 mg/litre 30 min Sunderman (1964)
Rat intravenous LD50 = 22 mg/kg Lungs: at 1-4 h, perivascular 1 h-21 days Hackett &
Rat subcutaneous LD50 = 21 mg/kg oedema, at 2-5 days severe Sunderman (1967)
Rat intraperi- LD50 = 13 mg/kg pneumonitis with intra-alveolar
toneal oedema, haemorrhage, subpleural
consolidation, hypertrophy and
hyperplasia of alveolar lining
cells, and focal adenomatous
proliferation, at 8 days,
interstitial fibroblastic
proliferation;
Liver, kidneys, adrenals:
congestion, vacuolization,
and oedema
------------------------------------------------------------------------------------------------------------------------------
Table 25 (contd.)
------------------------------------------------------------------------------------------------------------------------------
Species Route Lethal dose Observations in surviving Observation References
animals period after
exposure
------------------------------------------------------------------------------------------------------------------------------
Rat intravenous 65 mg/kg Lung: ultrastructural 0.5 h-8 days Hackett &
alterations, including Sunderman (1968)
oedema of endothelial cells
at 6 h and massive hypertrophy
of membranous and granular
pneumocytes at 2-6 days;
Liver: ultrastructural
alterations of hepatocytes
including nucleolar distortions
at 2-24 h, dilatation of rough
endoplasmic reticulum at 1-4
days, and cytoplasmic inclusion
bodies at 4-6 days
Mouse inhalation LC100 = 0.1 mg/litre 120 min Sanina (1968)
------------------------------------------------------------------------------------------------------------------------------
a From: NAS (1975)
Table 26. Acute toxicity of nickel compounds in experimental animals
_______________________________________________________________________________________
Species (sex) Substance Route of LD50 (mg/kg) and References
administration confidence limits
_______________________________________________________________________________________
Rat (male) nickel acetate oral 360 (410-316) Haro et al.
(female) 350 (403-304) (1968)
Mouse (male) 410 (500-336)
(female) 420 (515-336)
Rat intraperitoneal 23 (28-19) Haro et al.
(1968)
Mouse 32 (37-28)
Rat (female) nickel chloride intraperitoneal 29 Horak et al.
(1976)
(pregnant intramuscular 71 Sunderman et
female) 98 al. (1978a)
intraperitoneal 38 (34-41) Mas et al.
(1985)
Rat (male) nickelocene oral 490 (510-471) Haro et al.
(female) 500 (525-474) (1968)
Rat intraperitoneal 50 (59-42)
Mouse oral 600 (660-545)
intraperitoneal 86 (102-72)
_______________________________________________________________________________________
Diarrhoea, respiratory distress, and lethargy were noted in
rats and mice dying 2-3 hours after receiving nickel acetate or
nickelocene by the oral or intraperitoneal route (Haro et al.,
1968).
Benson et al. (1986) investigated the effects of single
intratracheal doses of nickel subsulfide (3.2, 32, or 320 µg/kg
body weight), nickel oxide (3, 30, or 300 µg/kg body weight),
nickel sulfate (10.5, 105.2, or 1052 µg/kg body weight), and nickel
chloride (9.5, 95.2, or 952 µg/kg body weight) on rats; 24 h after
dosing, no effects were observed. However, at 7 days, multifocal
alveolitis with some type II hyperplasia was observed in animals
treated with nickel chloride, nickel sulfate, or nickel subsulfide.
Lung lavage fluid contained increased numbers of neutrophils and
macrophages in the medium- and high-dose groups of nickel chloride
and nickel sulfate and in the highest nickel subsulfide dose group.
While increased levels of enzymes, total protein, and sialic acid
occurred in rats exposed to nickel chloride, nickel sulfate, or
nickel subsulfide, no such changes were seen in rats receiving
nickel oxide.
Effects on kidney function in rabbits were studied by Foulkes &
Blanck (1984), who reported a reduction in the maximum tubular
transport rate for aspartase following ip injection of 20 µmol
nickel chloride/kg body weight. Intraperitoneal injection of 3 or
6 mg nickel/kg body weight in male Wistar rats induced a decrease
in Bowman's space, dilated tubules, loss of brush border, flattened
epithelia, and some regenerative activity (Sanford et al., 1988).
Intraperitoneal administration of NiCl2 to mice and rats caused
a rapid decrease of body temperature (Gordon, 1989; Gordon & Stead,
1986; Gordon et al., 1989). The nickel chloride treatment resulted
in hypothermia that lasted for more than 1 h, with a reduction in
colonic temperature of 3-4 °C at 20 °C ambient temperature. The
Ni2+-induced hypothermia was accentuated at lower ambient
temperature (10 °C) and ameliorated at higher ambient temperature
(30 °C).
Hopfer & Sunderman (1988) monitored core body temperature and
physical activity by radiotelemetry from a thermistor probe
implanted in the peritoneal cavity. After injection of nickel
chloride (250 µmol/kg body weight), core body temperature
diminished to a minimum at 1.5 h and returned to baseline at 4 h;
core body temperature at 1.5 h after dosing averaged 3.0 ± 0.5 °C
below the simultaneous value in control rats. During the 8-80 h
following dosing, the mean body temperature of NiCl2-treated rats
did not differ from that of the controls, but the amplitude of the
diurnal cycle of body temperature was dampened and the acrophase of
the temperature cycle was delayed from 10.32 pm to 03.00 am. These
parameters returned towards the control values during the 80-152 h
period following dosing.
Acute thymic involution occurred in male Fischer 344 rats
following a single subcutaneous injection of nickel chloride (500
µmol/kg body weight) (Knight et al., 1987). In nickel-treated
rats, the mean thymic weight, which was significantly decreased at
24 h, continued to diminish at 48 h and reached 24% of the control
value at 72 h. The concentration of lipoperoxides in the thymus
increased 2-fold at 48 h and 7-fold at 72 h. Histological
examination showed marked degenerative changes. Gitlitz et al.
(1975) noted aminoaciduria and proteinuria in rats given single
injections of nickel chloride (2 mg/kg body weight), the response
being dose-dependent. Proteinuria was seen initially with
aminoaciduria at higher doses. These effects, transitory in
duration, were associated with morphological changes in the
glomeruli.
8.1.2.3 Possible mechanisms of acute nickel toxicity
The mechanisms of nickel toxicity are not well understood, but
studies by Knight et al. (1987), Sunderman et al. (1987), and
Sunderman (1987) suggest that acute Ni2+ toxicity in rats is
associated with lipid peroxidation in target organs. The chemical
reactions whereby Ni2+ induces lipid peroxidation in vivo have not
yet been explained, however, Sunderman (1987) proposed the
following four possible mechanisms:
i. An indirect mechanism owing to Ni2+ displacement of iron and
copper from intracellular binding sites;
ii. An indirect mechanism, by which Ni2+ inhibits cellular
defences against peroxidative damage, mediated by catalase,
superoxide dismutase, glutathione peroxidase, aldehyde
dehydrogenase, or other enzymes that protect against free-radical
injury or that metabolize products of lipid peroxidation;
iii. Generation of oxygen-free radicals by the redox couple:
Ni2+/Ni3+
Ni2+ + H2 --> Ni3+ + OH- + OH- (Fenton reaction)
Ni3+ + O-2 --> Ni2+ + O2
H2O2 + O-2 --> OH- + OH- + O2 (Haber-Weiss reaction)
iv. Ni2+ may accelerate the degradation of lipid hydroperoxides to
form lipid-oxygen radicals, propagating autocatalytic peroxidation
of polyenoic fatty acids.
Experiments performed by Inoue & Kawanishi (1989) using
electron spin resonance (ESR) (spin traps 5,5-dimethylpyroline- N-
oxide and alpha-(4-pyridyl 1-oxide)- N-tert-butylnitrone) indicate
that hydroxyl radical adducts are produced in vitro by the
decomposition of hydrogen peroxide in the presence of the nickel
(II) oligopeptide Gly Gly His. These investigators suggested that
Gly Gly His plus hydrogen peroxide produce superoxide in addition
to the hydroxyl radical. The experimental findings support the
conclusion that the nickel-dependent formation of an activated
oxygen species is a primary molecular event in acute nickel
toxicity and carcinogenicity.
8.1.3. Short- and long-term exposures
8.1.3.1 Effects on the respiratory tract
(a) In vivo studies
Data on the chronic respiratory effects of nickel carbonyl are
summarized in section 8.1.2.1.
Long-term inhalation studies have been performed on guinea-
pigs, rats, and mice (Hueper, 1958), rats (Bingham et al., 1972),
and hamsters (Wehner et al., 1975, 1981). Exposure extended over
more than one and a half years in all studies. The compounds
tested were metallic nickel powder, nickel subsulfide, nickel
oxide, and nickel-enriched fly ash. Hueper (1958) exposed animals
to metallic nickel dust at 15 mg/m3, and noted nasal sinus
inflammation and ulcers in rats, and signs of lung irritation in
guinea-pigs and rats. A common finding was an accumulation of
adenomatoid cell formations. In mice, there were signs of lung
irritation, but not to the same extent as in guinea-pigs and rats.
Bingham et al. (1972) used aerosols of soluble nickel chloride
at 109 µg/m3 and nickel oxide at 120 µg/m3 and observed hyperplasia
of bronchiolar and bronchial epithelium with peribronchial
lymphocyte infiltrates.
Ottolenghi et al. (1974) found a number of lung changes, such
as abscesses as well as metaplastic changes, when rats were exposed
for 78 weeks to nickel subsulfide by inhalation. Wehner et al.
(1975) exposed rats to a concentration of 53 mg nickel oxide/m3
(type of oxide not specified) for the life span and found that
particulate material accumulated on the alveolar septa. Emphysema
was observed early in the exposure period. With longer exposure,
the cellular response increased and pneumoconiosis developed
gradually. However, there was no reduction in the life span.
Wehner et al. (1981) exposed hamsters through inhalation to nickel-
enriched fly ash or fly ash at concentrations of 17 or 70 µg/m3 for
up to 20 months. Lung weights and volumes were significantly
increased in the 70 µg/m3 fly ash exposure group. The severity of
anthracosis, interstitial reaction, and bronchiolization was dose-
dependent.
Friberg (1950) exposed rabbits for 6 months to nickel-graphite
dust (nickel hydrate, nickel content approx. 50%) at a
concentration of 100 mg/m3 (3 h/day, 5 days/week) and found
emphysematous and inflammatory changes in the nasal mucous
membranes and the trachea, bronchitis, and sometimes slight
fibrosis in the lung.
Port et al. (1975) reported that intratracheal injection of a
suspension of nickel oxide (5 mg, particle size <5 µm, type of
nickel oxide not specified) into Syrian hamsters, treated 48 h
previously with influenza A/PR/8 virus, resulted in significantly
increased mortality compared with controls. Surviving animals at
this and lower doses showed mild to severe acute interstitial
infiltration by polymorphonuclear cells and macrophages, several
weeks later. Additional pathological changes included bronchial
epithelial hyperplasia, focal proliferative pleuritis, and
adenomatosis.
Short-term inhalation studies (12 days) with nickel sulfate
(3.5-60 mg/m3) and nickel subsulfide (0.6-10 mg/m3) were performed
on rats and mice (Benson et al., 1987, 1988). Nickel sulfate
caused lesions in the lung, nose, bronchial, and mediastinal lymph
nodes in the surviving animals at 3.5 mg/m3. In the nickel
subsulfide-exposed animals, similar changes occurred in the
respiratory tract with extensive lesions in the lung, including
necrotizing pneumonia. Emphysema developed in rats exposed to 5 or
10 mg/m3 and fibrosis was seen in mice exposed to 5 mg nickel
subsulfide/m3. Degeneration of the respiratory epithelium and
atrophy of the olfactory epithelium occurred in all nickel
subsulfide dose groups except in mice exposed to 0.6 mg/m3.
Clinical signs included laboured respiration, emaciation,
dehydration, and reduced body weight gain in rats and mice exposed
to nickel sulfate or nickel subsulfide. A 12-day inhalation
exposure of rats and mice to nickel oxide (1.2-30 mg/m3) also
caused lung lesions in the higher dose groups. Lung lesions
included hyperplasia of alveolar macrophages, focal suppurative
inflammation, focal interstitial cellular infiltrate and particles
in alveoli and alveolar macrophages. In mice, lung lesions were
less severe (Dunnick et al., 1988). On the basis of these 12-day
studies, relative toxicity ranking was NiSO4 x 6H2O >Ni3S2 >NiO
(Dunnick et al., 1988).
The effects of nickel on the cellular respiratory system
defence mechanisms were studied by exposing guinea-pigs, rats, or
rabbits to various concentrations (0.05-2 mg/m3, for 1-8 months) of
nickel dust, nickel oxide, or nickel chloride (Waters et al., 1975;
Graham et al., 1975a; Aranyi et al., 1979; Johansson et al., 1980,
1981, 1983a,b; Castranova et al., 1980; Casarett-Bruce et al.,
1981; Lundborg & Camner, 1982, 1984; Wiernik et al., 1983; Murthy
et al., 1983; Takenaka et al., 1985; Glaser et al., 1986). It was
concluded that the overall effects had some features in common with
the pulmonary alveolar proteinosis described in human beings.
There was no difference in the effect pattern between exposure to
insoluble and soluble nickel compounds. Effects, such as changes in
the morphology and function of alveolar macrophages and type II
alveolar epithelial cells, and in the composition of lung lavage,
were found, with the severity of effects depending on the
concentration and duration of exposure.
The phagocytic activity of alveolar macrophages was increased
in rabbits following 4 weeks inhalation of metallic nickel dust
(0.5-2.0 mg/m3; Camner et al., 1978; Yarstrand et al., 1978) and in
rats following 1-4 months inhalation of nickel oxide (produced by
pyrolysis of nickel acetate at 550 °C) (Spiegelberg et al., 1984).
No change in the phagocytic activity of alveolar macrophages was
observed in rats following exposure to 0.13 mg metallic nickel
dust/m3 for 4-8 months (Johansson et al., 1983a), and in rabbits
following exposure to nickel chloride aerosols (0.3 mg/m3) for 1
month (Wiernik et al., 1983).
Alveolar macrophages varied in size and, ultrastructurally, had
an active cell surface with numerous slender microvilli and long
protrusions. Laminated structures were regularly found in the
alveolar macrophages as well as in the lung fluid (Camner et al.,
1978; Johansson et al., 1980, 1983a; Wiernik et al., 1983).
Spiegelberg et al. (1984) found an increase in size and number of
polynucleated cells in the lungs of exposed rats. Morphometric
studies on the lungs of exposed rabbits showed an increase (up to
3-fold) in the volume density of type II cells. Cells contained
many lamellar bodies and vesicles; the endoplasmic reticulum had a
slightly dilated appearance (Johansson & Camner, 1980; Johansson et
al., 1981, 1983b). There were changes in the pulmonary lipid
content and composition of the lung fluid of rabbits inhaling
metallic nickel dust at levels ranging from 0.13 to 1.7 mg/m3
(Casarett-Bruce et al., 1981; Curstedt et al., 1983, 1984). There
was a twofold increase in the concentration of total phospholipids,
mainly due to an elevated level of phosphatidylcholines, especially
disaturated species, and phosphatidylinositols. Lundborg & Camner
(1982, 1984) exposed rabbits to nickel dust (0.1 mg/m3, for 4 and 6
months) and nickel chloride (0.2-0.6 mg/m3 for 4-6 weeks) and found
markedly reduced lysosomal enzyme activity compared to control
animals. When rats were exposed to aerosols of nickel oxide (type
of oxide not specified) at 120 µg/m3 or nickel chloride at 109
µg/m3, Murthy et al. (1983) found that various hydrolytic enzymes
were reduced in alveolar macrophages, but, in contrast, the enzyme
activities were significantly increased in lung lavage.
Parenteral administration of nickel chloride can also cause
toxic effects in alveolar macrophages, as demonstrated by Sunderman
et al. (1989c) in rats that had received single subcutaneous
injections of 8-65 mg (62-500 µmol) nickel chloride/kg. Alveolar
macrophages showed morphological and biochemical signs of
activation, functional impairment, and lipid peroxidation. In
alveolar macrophages from 63NiCl2-treated rats, 63Ni was primarily
located in the cytoplasm bound to high- and low-relative molecular-
mass constituents.
At the ciliated epithelium level, nickel significantly
depressed normal ciliary activity in a hamster tracheal ring assay
following in vivo exposure to 100 µg nickel/m3 as nickel chloride
for 2 h (Adalis et al., 1978; Olsen & Jonsen, 1979b).
Fisher et al. (1984) evaluated the effects of particle size and
dose regimen on the toxicity of intratracheally instilled nickel
subsulfide in mice, and showed the importance of physical form in
the evaluation of pulmonary toxicity. The median lethal dose for a
single exposure to larger particles (MMAD 13.3 µm) was 12 times
that of fine particles (MMAD 1.8 µm), i.e., 50 versus 4 mg/kg.
Repeated exposures (once/week for 4 weeks) resulted in a 2-fold
greater median lethal dose of coarse particles compared with fine
particles, i.e., 2 versus 1 mg/kg. Thus, repeated exposure to fine
particles resulted in a cumulative lethality equivalent to the
single exposure, while coarse particle lethality was enhanced with
repeated exposure. Alveolar macrophages from mice exposed to fine
nickel subsulfide showed depressed cellular function, 14 days after
a single administration.
Reichrtova et al. (1986a,b) performed 2 inhalation studies on
rats to compare the effects of different types of exposure on
alveolar macrophages. In both studies, the nickel content of the
aerosol was only 0.36% (NiO), thus, the effects are unlikely to be
nickel-specific responses. Increases in the alveolar macrophage
count and lysosomal enzyme activities (acid phosphatase and beta-
glucuronidase) were found in Wistar rats, exposed to an aerosol of
solid waste from a nickel refinery dump, under chamber conditions,
for 6 months (aerosol concentration 0.1 g/m3, 4 h/day, 5
days/week). However, the activity of alveolar macrophage plasma
membrane enzymes decreased. Cells contained in lung lavage had a
pleomorphic appearance. When rabbits were environmentally exposed
at a biomonitoring station, situated in the direction of the
prevailing wind from a nickel refinery dump, for 6 months, with an
average dust fallout of 5.5 g/m2 in 30 days, the number of alveolar
macrophages was significantly increased (Reichrtova et al., 1986b).
A significant increase in lysosomal enzyme activity also occurred,
but these changes were not as pronounced as the effects obtained in
the chamber studies on rats. In contrast to the increased alveolar
macrophage activity, a significant reduction occurred in antibody-
mediated rosette formation by alveolar macrophages in the
environmentally exposed rabbits.
(b) In vitro cytotoxicity studies
Alveolar macrophages exposed to nickel (1.1 mmol/litre) in
vitro showed depressed phagocytic activity (Graham et al., 1975a;
Aranyi et al., 1979). Waters et al. (1975) correlated changes in
cell viability with changes in the morphology and the specific
activity of a lysosomal enzyme (acid phosphatase). At 4.0
mmol/litre, cell viability was reduced to approximately 50% of that
of controls. Castranova et al. (1980) demonstrated that nickel
affected oxygen metabolism in the rat alveolar macrophages; at
rest, and during phagocytosis, oxygen consumption and glucose
metabolism were depressed.
The in vitro cytotoxicity of nickel chloride on a human
pulmonary epithelium cell line (A549) was reported by Dubreuil et
al. (1984). Nickel chloride in amounts ranging from 0.1 to 1.0
mmol/litre produced decreases in cell growth rate and in the levels
of cell adenosine triphosphate (ATP), and loss of viability, at the
highest concentration. No changes were seen in transmission
electron microscopy preparations.
In vitro toxicity studies on bovine alveolar macrophages
indicated that nickel subsulfide was 10 times more toxic than
solubilized nickel subsulfide or soluble nickel chloride (Fisher et
al., 1984).
Nickel subsulfide, nickel sulfate, nickel chloride and nickel
oxide were tested for their relative toxicity in beagle dog and rat
alveolar macrophages in vitro. Toxicity ranking was Ni3S2 >NiCl2
ca NiSO4 >NiO (Benson et al., 1986).
8.1.4. Relationship of nickel toxicity and mixed metal exposure
As both nickel production and some end uses of nickel involve
mixed exposure of workers to nickel and copper, nickel and
chromium, nickel, chromium, and manganese, and so on, the problem
of the combined toxic effects of these metals is of great practical
importance. However, the relevant information is rather scarce.
Johansson et al. (1988) showed that the cytotoxic effects of a
trivalent chromium salt on alveolar macrophages of the rabbit
impaired their dealing with pulmonary surfactant, the latter being
hyperproduced as a result of nickel's action on the type II
alveolar epithelial cells. However, analysis of these experimental
data showed that, as far as the cytotoxicity of these metals for
alveolar macrophages was concerned, their joint effect was
antagonistic.
Davydova et al. (1981) demonstrated that subadditivity or even
clear antagonism was the main type of combined acute toxicity for
rats of nickel and chromium, nickel and manganese, and even a
triple exposure of rats to nickel and cobalt. On the contrary, the
long-term exposure of rats to nickel and cobalt in the drinking-
water demonstrated the additive long-term toxicity of these two
metals (Nadeenko et al., 1988).
8.1.5. Endocrine effects
Bertrand & Macheboeuf (1926a,b) reported that parenteral
administration of nickel chloride or nickel sulfate to rabbits or
dogs antagonized the hyperglycaemic action of insulin. Later
investigators observed that after parenteral (iv or ip) injection
to rabbits, rats, or chickens, or oral administration to rabbits,
there was a rapid increase in plasma glucose concentrations, which
returned to normal within 4 h (Kadota & Kurita, 1955; Gordynia,
1969; Clary & Vignati, 1973; Freeman & Langslow, 1973; Horak &
Sunderman, 1975a,b). In the pancreatic islets of Langerhans,
Kadota & Kurita (1955) noted marked damage of alpha-cells, and, to
a lesser degree, degranulation and vacuolization of beta-cells.
Ashraf & Sybers (1974) noted lysis of pancreatic exocrine cells in
rats fed nickel acetate (0.1%). In adrenalectomized or
hypophysectomized rats, the hyperglycaemic effect was greatly
depressed, but was not completely prevented. Concurrent
administration of insulin antagonized the hyperglycaemic effect
(Horak & Sunderman, 1975a,b). LaBella et al. (1973a,b) showed
that nickel also affects the hypothalamus of animals. Nickel salts
specifically inhibited the release of prolactin in vivo (in the
rat) and in vitro from bovine pituitary. Subcutaneous injection of
10 or 20 mg nickel chloride/kg in rats initially produced a drop in
serum prolactin over the short term, but resulted in a sustained
elevation of the hormone after 1 day, which lasted up to 4 days.
The elevation was due to reduced levels of the prolactin-inhibiting
factor (Clemons & Garcia, 1981). Carlson (1984) demonstrated that
nickel antagonized the stimulation of both prolactin and growth
hormone by barium; thus, the basis of antagonism may be the
competitive inhibition of calcium uptake. Dormer et al. (1973)
showed that the nickel ion is a potent inhibitor of secretion in
vitro in the parotid gland, (amylase), the islets of Langerhans
(insulin), and the pituitary gland (growth hormone). Inhibition of
growth hormone secretion at nickel concentrations comparable to
those observed by LaBella et al. (1973b) to enhance release, may
reflect differences in tissue preparation prior to assay. Dormer
et al. (1973) suggested that nickel may block exocytosis by
interfering with either secretory granule migration or membrane
fusion and microvilli formation.
The effects of nickel on thyroid function were reported by
Lestrovoi et al. (1974). Nickel chloride, given orally (0.5-5.0
mg/kg per day, for 2-4 weeks) or by inhalation (0.05-0.5 mg/m3) to
rats, significantly decreased iodine uptake by the thyroid, the
effect being more pronounced with inhaled nickel.
8.1.6. Cardiovascular effects
The serum nickel level increased in patients with acute
myocardial infarction, stroke, and burns (D'Alonzo & Pell, 1963;
McNeely et al., 1971; Leach et al., 1985).
Rubanyi & Kovach (1980) found that micromolar concentrations of
nickel chloride (0.1-1 µmol/litre) increased cardiac contractility
in the isolated rat heart. At higher doses, there was depressed
myocardial contractile performance. Ultrastructural damage was
found (Rubanyi et al., 1980).
Nickel chloride at a concentration of 1 µmol/litre in the
perfusate produced tonic contraction in the isolated canine
coronary artery (Rubanyi et al., 1982b).
Nickel is released from the ischaemic dog myocardium (Rubanyi
et al., 1981); exogenous nickel, in doses comparable to the amount
released endogenously from the heart, induced coronary
vasoconstriction in rat and dog hearts. In further studies, the
possible involvement of Na/K ATPase inhibition (Rubanyi et al.,
1982c) and/or stimulation of adrenergic receptors in the coronary
vessels (Rubanyi et al., 1982d) were discussed as mechanisms of
nickel-induced coronary vasoconstriction. Rubanyi & Inovay (1982)
studied the effects of nickel ions on spontaneous, electrically-,
and norepinephrine-stimulated isometric contractions in the
isolated portal vein of the rat. They found that low
concentrations of nickel (1-10 µmol/litre) inhibited spontaneous
isometric force development and decreased basal tone, but
significantly increased the frequency of contractions. Inhibition
of the effect of selective stimulation of adrenergic nerves was
significantly more pronounced than the depression of contractions
evoked by exogenous norepinephrine.
In the in situ heart (open-chest anaesthetized dogs), a
decrease in coronary vascular flow with a low intravenous dose of
nickel (0.02 mg nickel chloride/kg body weight) was reported,
while, at higher dose levels (0.2, 2, or 20 mg nickel chloride/kg
body weight), further reduction in coronary blood flow, depression
of heart rate, and a decrease in left ventricular contractility
were observed. Coronary vasoconstriction may be regarded as a
local action of nickel on coronary vessels (Ligeti et al., 1980).
In another in situ study on dogs (intravenous bolus injection
of 0.02 mg nickel/kg, or intracoronary infusion of 0.04 mg nickel
chloride/min per kg body weight), Rubanyi et al. (1984) found that
vasoconstriction was induced when coronary arteries were dilated by
low-flow ischaemia, arterial hypoxaemia, and adenosine infusion.
Nickel inhibited post-occlusion reactive hyperaemia and
vasorelaxation in response to arterial hypoxaemia or intracoronary
infusion of adenosine. It was postulated that vasoactivity might
be related to the existence of positive feedback loops triggered by
alterations in the level of nickel.
Endogenous nickel release from damaged tissues and its
implications for ischaemic heart disease have been examined with
respect to the pathology of acute carbon monoxide poisoning and
acute burn injury. Significant endogenous nickel ion accumulation
was noted in the heart muscle of rats and rabbits, when the CO-Hb
level was above 30% (Balogh et al., 1983).
8.1.7. Effects on the immune system
The effects of nickel on alveolar macrophages have been
described in section 8.1.3.1.
Koller (1980) noted that nickel exposure of animals could
reduce host resistance to both viral and bacterial infections, and
suppress the phagocytic capacity of macrophages.
Other cellular and humoral immune responses following nickel
treatment were studied by Smialowicz et al. (1984, 1985). Single
or multiple intramuscular injections of nickel chloride in mice
caused a significant reduction in a variety of T-lymphocytes and
natural killer cell-mediated immune functions. Suppression of the
lymphoproliferative responses to the T-cell mitogens,
phytohaemagglutinin and concanavalin A, and a reduction in the
number of theta-positive T-lymphocytes were observed in the spleens
of nickel chloride-injected mice (18.3 and 36.6 mg/kg body weight).
Reductions in the primary antibody response to T-lymphocyte-
dependent antigen sheep red blood cells, but not T-lymphocyte-
independent antigen polyvinyl-pyrrolidone, were observed following
a single injection of 18.3 mg nickel chloride/kg (Smialowicz et
al., 1984).
Smialowicz et al. (1985) demonstrated that suppression of
natural killer cell activity could be detected by in vitro and in
vivo assays and that reduction of natural killer cell activity was
not associated with either a significant reduction in spleen
cellularity or the production of suppressor cells. A further
demonstration of the effect of nickel chloride on natural killer
cell activity was the enhancement of the development of lung tumour
colonies in mice injected with B16-F10 melanoma cells, following a
single injection of 18.3 mg nickel chloride/kg. Unlike nickel
chloride, manganese chloride was found to enhance natural killer
cell activity, when injected into mice (Smialowicz, 1985). No
alteration in natural killer cell activity was observed in mice
injected with magnesium, calcium, or zinc (Smialowicz et al.,
1987). Manganese chloride was considered to have an antagonistic
effect on nickel chloride-suppression of natural killer cell
activity.
The effects of nickel compounds on natural killer cells are of
particular interest, because of the suspected function of these
cells in nonspecific defence against certain types of infections
and tumours.
The studies performed by Smialowicz et al. (1984, 1985, 1986,
1987) confirmed the findings of other investigators on the immuno-
suppressive effects of nickel salts on circulatory antibody titres
to T1 phage in rats (Figoni & Treagan, 1975), on antibody response
to sheep erythrocytes (Graham et al., 1975b), on interferon
production in vitro (Treagan & Furst, 1970) and in vivo in mice
(Gainer, 1977), and on susceptibility to induced pulmonary
infection in mice following inhalation of nickel chloride (Adkins
et al., 1979).
In cynomolgus monkeys that had been previously immunized and
repeatedly challenged with sheep red blood cells, instillation of
10.6 mg nickel subsulfide/kg lung in one immunized and one control
lobe of each animal increased target cell killing by conjugate-
forming natural killer cells and decreased macrophage phagocytic
activity (Haley et al., 1987).
The results of in vitro studies have shown that nickel can
replace magnesium, which is essential for the proper functioning of
the complement system, in both the classical and the alternative
pathway. Nickel has been shown to result in a more efficient
formation of the C3b,Bb enzyme, which theoretically may lead to
disturbance of a well balanced complement cascade. The
significance of this finding in vivo is not known (Fishelson et
al., 1983).
8.1.8. Skin and eye irritation and contact hypersensitivity
8.1.8.1 Skin and eye irritation
Repeated skin application of 40-100 mg nickel/kg (as nickel
sulfate), daily for 30 days, produced skin atrophy, acanthosis, and
hyperkeratinization in rats (Mathur et al., 1977). No data are
available on the eye irritancy of soluble nickel salts or on the
skin and eye irritancy of insoluble nickel compounds.
8.1.8.2 Contact hypersensitivity
Experimental sensitization to nickel in guinea-pigs has been
reported (Walthard, 1926; Stewart & Cromia, 1934; Vinson & Choman,
1960; Jansen et al., 1964; Gross et al., 1968; Magnusson & Kligman,
1970). Nilzén & Wikström (1955) reported a method for sensitizing
laboratory animals to nickel by repeated topical applications of
aqueous nickel sulfate solutions containing sodium lauryl sulfate.
However, Samitz & Pomerantz (1958) were unable to demonstrate
sensitization with this technique and attributed the effect to
local irritation, rather than true allergenic reaction. Samitz et
al., (1975) were unable to induce sensitization in guinea-pigs
using nickel compounds from the complexation of the nickel ion with
amino acids or guinea-pig skin extracts. Furthermore,
sensitization of experimental animals was not found by Hunziker
(1960) or Bühler (1965).
The sensitivity of guinea-pigs was increased by repeated intra-
dermal injections and skin painting with nickel sulfate solutions
during the sensitization phase. The responses were significantly
greater than in control animals (Wahlberg, 1976).
Turk & Parker (1977) reported sensitization to nickel
manifested as allergic-type granuloma formation. This required the
use of Freund's complete adjuvant followed by weekly intradermal
injections of 25µg of the salt after 2 weeks. Delayed
hypersensitivity reactions developed in 2 out of 5 animals at 5
weeks when a split-adjuvant method was used. Suppression of the
delayed hypersensitivity occurred when intratracheal intubation of
nickel sulfate was also performed on these animals (Parker & Turk,
1978). Möller (1984) reported that mice could easily be sensitized
to a potent antigen, such as picryl chloride, but response to
nickel could only be achieved by repeated epicutaneous application
of a strong (20%) nickel salt solution for 3 weeks.
The study of the allergic properties of nickel in experimental
animals is a problem, because of difficulties involved in
reproducing the phenomenon of allergodermatosis and because of lack
of a uniform approach to reproducing the experimental contact
allergic dermatitis model. Duyeva (1983) recommended a single
intracutaneous administration of 100-200 µg nickel chloride in the
ear of the guinea-pig. Sensitization developed as early as days 4-
10, reaching a peak on day 20.
8.1.9. Reproduction, embryotoxicity, and teratogenicity
8.1.9.1 Effects on the male reproductive system
Data on the effects of nickel on the male reproductive system
are limited. Hoey (1966) examined the effects of nickel sulfate on
the testis and epididymis of Fischer rats and reported histological
changes in the testis and adnexae. Following a single
intracutaneous injection of 0.04 µmol nickel sulfate/kg body weight,
histological examination, 18 h after exposure, revealed shrinkage
of the tubules and complete degeneration of the spermatozoa.
Infertility was observed in rats after 120 days of daily ingestion
of 25 mg nickel sulfate/kg (Waltschewa et al., 1972).
Mathur et al. (1977) studied the dermal exposure of male rats
to nickel sulfate at daily levels of 40, 60, or 100 mg nickel/kg
body weight for 15 and 30 days. Tubular damage and spermatozoal
degeneration were observed in the testis following exposure to 60
mg nickel/kg for 30 days. These changes were more severe with
exposure at 100 mg/kg for 30 days. There were no effects on the
testis following exposure to 40 mg/kg for 30 days or at any dose
level after 15 days exposure.
In vitro embryo cultures were used to study the effects of
nickel nitrate on male germ cells (Jacquet & Mayence, 1982).
BALB/C mice were injected intraperitoneally with 40 or 56 mg nickel
nitrate/kg body weight and were then allowed to mate with
superovulated females, at weekly intervals, for 5 weeks following
treatment. The embryos were isolated and those at the 2-cell stage
were cultured for 3 days. These embryos were then classified
according to their ability to develop to the blastocyst stage. A
dose of 40 mg/kg body weight did not affect the fertilizing
capacity of the spermatozoa or the ability of the fertilized egg to
cleave, but a dose of 56 mg nickel nitrate/kg body weight yielded a
significant proportion of uncleaved unfertilized eggs. Cleaved
eggs from this treatment group were able to develop into
blastocysts, suggesting that nickel nitrate treatment reduced the
fertilizing capacity of the spermatozoa, presumably because of a
toxic effect of nickel on spermatogenesis. Deknudt & Leonard
(1982) conducted a dominant lethal mutation test for nickel
chloride and nickel sulfate in BALB/C mice. Both compounds
produced a reduction in implantations in the matings performed 2,
3, or 4 weeks after treatment, but the post-implantation loss was
not increased. It was considered that this was attributable to a
toxic effect of nickel treatment on male germ cells.
8.1.9.2 Effects on the female reproductive system
Insertion of nickel wire into one uterine horn of rats on day 3
of pregnancy produced a decrease in the number of implants and an
increase in the number of resorption sites, compared with the
untreated contralateral horn (Chang et al., 1970).
Nickel has been reported to localize within the pituitary and
the hypothalamus of rats and to inhibit prolactin secretion (La Bella
et al., 1973a,b). It may, therefore, modify interactions between
the hypothalamus and the pituitary, needed to maintain pregnancy.
No effects on fertility were observed in 3 generations, when
breeding rats were exposed to 5 mg nickel/litre as a soluble salt
(not specified) in the drinking-water (Schroeder & Mitchener, 1971).
8.1.10. Embryotoxicity and teratogenicity
Nickel has been shown to cross the placental barrier and enter
the fetus (section 6.1.5).
Sunderman et al. (1978a) studied the embryo- and fetotoxicity
of nickel chloride and nickel subsulfide in Fischer 344 rats.
Single intramuscular injection of nickel chloride (12 or 16 mg
nickel/kg body weight) on day 8 of gestation significantly reduced
the mean number of live pups per dam and resulted in reduced body
weight in fetuses on day 20 of gestation and in pups, 4-8 weeks
after birth. When nickel chloride was administered in repeated
intramuscular doses of 1.5 or 2.0 mg/kg body weight on days 6-10 of
gestation, the higher dose caused significant intrauterine
mortality, but did not cause any reduction in the mean body weight
of live pups. Intramuscular injection of nickel subsulfide (80 mg
nickel/kg body weight) reduced the mean number of live pups per
dam. Exposure to nickel chloride or nickel subsulfide did not
produce any skeletal or visceral anomalies.
Lu et al. (1979) observed a dose-related increase in fetal
deaths and a higher incidence of malformations in pregnant CD-1
mice following intraperitoneal injection of single doses of nickel
chloride (1.2, 2.3, 3.5, 4.6, 5.7, or 6.9 mg nickel/kg) between
days 7 and 11 of gestation. Fetal death and some general
malformation (not described in detail) were reported in hamsters
after intravenous injection of nickel acetate at 30 mg/kg body
weight on day 8 of pregnancy (Ferm, 1972). Nadeenko et al. (1979)
demonstrated a dose-dependent embryotoxic effect of 27Ni given to
female rats for 7 months (before and during pregnancy) in
concentrations ranging from 5 x 10-1 to 5 x 10-4 mg/kg body weight.
The effects of nickel chloride on early embryogenesis were
studied by Storeng & Jonsen (1980) in vitro. Mouse embryos at the
2-, 4-, and 8-cell stages were cultured in media containing nickel
chloride hexahydrate at a concentration of 10-1000 µmol/litre. A
concentration of nickel chloride hexahydrate of 10 µmol/litre
adversely affected the development of 2-cell stage embryos whereas
a concentration of 300 µmol/litre was needed to affect 8-cell stage
embryos. No effect was observed at 100 µmol/litre. In a
subsequent study (Storeng & Jonsen, 1981), a single intraperitoneal
injection of nickel chloride hexahydrate (20 mg/kg) was given to
mice on days 1-6 of gestation. On day 19, implantation frequency
in females treated with nickel chloride hexahydrate on day 1 was
significantly lower compared with the controls. Litter size was
significantly reduced in females treated on days 1, 3, and 5 of
gestation. The incidence of abnormalities, such as haematomas and
exencephaly, in the fetuses of treated females was higher
(statistical significance not reported) than in the controls.
Sunderman et al. (1983) used intrarenal injection of nickel
subsulfide to assess the effects on the progeny of rats.
Administration of 30 mg nickel subsulfide/kg by intrarenal
injection, 1 week prior to breeding, produced intense
erythrocytosis in the dams, but not in the pups. These findings
indicate that the release of maternal erythropoietin by the
maternal kidneys, caused by nickel subsulfide, did not stimulate
erythropoiesis in the pups. Nickel subsulfide was associated with
a significant decrease in mean body weights of pups, 2 and 4 weeks
postpartum.
In a series of studies, Sunderman et al. (1978b,c, 1979a,
1980, 1983) demonstrated that nickel carbonyl, administered by
inhalation or injection before, or a few days after, implantation
produced various types of malformations in hamsters and rats.
Intravenous injection of nickel carbonyl (11 mg nickel/kg body
weight) into Fischer rats on day 7 of gestation caused fetal
mortality, reduced body weight in live pups, and a 16% incidence of
fetal malformations, including anophthalmia, microphthalmia, cystic
lungs, and hydronephrosis (Sunderman et al., 1983). There was no
maternal toxicity. Similar effects were observed in rats following
inhalation of nickel carbonyl at a concentration of 0.16 g/m3 on
day 7 or 8 of gestation and a concentration of 0.3 g/m3 on day 7 of
gestation (Sunderman et al., 1979a).
In a dominant lethal mutation test on male rats, administration
of nickel carbonyl, by the inhalation route (50 mg nickel/m3 for 15
min), 2-6 weeks prior to breeding, did not affect fertilization
rates or reproductive yield; administration of nickel carbonyl by
intravenous injection during the same period (22 mg nickel/kg)
caused reduced numbers of live pups in litters sired during the
fifth week (Sunderman et al., 1983). In hamsters, inhalation
exposure to 60 mg nickel carbonyl/m3, for 15 min on day 4 or 5 of
gestation, led to decreased fetal viability and increased numbers
of fetuses with malformations including cystic lungs, exencephaly,
and haemorrhages in serous cavities (Sunderman et al., 1980).
Gilani & Marano (1980) injected nickel chloride into chicken
eggs (0.02 and 0.7 mg per egg) on days 0, 1, 2, or 3 of incubation.
Examination on day 8 revealed a number of malformations, such as
exencephaly, everted viscera, short and twisted neck or limbs,
microphthalmia, haemorrhage, and reduced body size. The incidence
of malformations was highest in embryos treated on day 2.
When rats were exposed long-term to nickel chloride or nickel
sulfate in the diet or drinking-water, an increased frequency of
runts and greater prenatal and neonatal mortality were observed
(Schroeder & Mitchener, 1971; Ambrose et al., 1976; Nadeenko et
al., 1979). Berman & Rehnberg (1983) observed spontaneous
abortions, loss of fetal mass in survivors, and loss of maternal
mass in mice.
8.2. Mutagenicity and related end-points
Genotoxicity data on nickel and nickel compounds are summarized
in Table 27.
The data collectively show that nickel compounds are generally
inactive in bacterial assays, but active in systems using
eukaryotic organisms, and that positive responses were observed
regardless of the nickel compounds used; particularly when these
were compared at equitoxic concentrations (Hansen & Stern, 1983;
Swierenga & McLean, 1985). The ability of nickel compounds to
inhibit DNA synthesis and excision repair should also be noted
(Table 27).
Table 27. Summary of the genotoxic effects of nickel compounds
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
PROKARYOTIC SYSTEMS
Prophage Prophage induction Ni(CH3CO3)2 160-640 µmol/litre + (weak) Rossman et al.
(1984)
T4 Bacteriophage Bacteriophage-forward NiSO4 300 mg/litre - Corbett et al.
mutation (1970)
Salmonella
TA1535 Salmonella typhimurium NiCl2 0.01-0.1 g/litre + LaVelle &
reverse mutation Witmer (1981)
(fluctuation test)
TA1535 S. typhimurium - Haworth et al.
reverse mutation (1983)
TA1525 S. typhimurium NiCl2 - Arlauskas et
reverse mutation NiSO4 al. (1985)
TA1537 S. typhimurium - Haworth et al.
reverse mutation (1983)
TA1537 S. typhimurium NiCl2 - Arlauskas et
Reverse mutation NiSO4 al. (1985)
TA1535 S. typhimurium Ni salts 10-1000 µg/plate + (weak) Saichenko &
reverse mutation Sharapora
(1987)
TA98 S. typhimurium - Haworth et al.
reverse mutation (1983)
TA98 S. typhimurium NiCl2 - Arlauskas et
reverse mutation NiSO4 al. (1985)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
TA100 S. typhimurium - Haworth et al.
reverse mutation (1983)
TA100 S. typhimurium NiCl2 - Arlauskas et
reverse mutation NiSO4 al. (1985)
Several strains S. typhimurium Ni(CH3CO2)2 - DeFlora et al.
reverse mutation NiCl2 - (1984)
Ni(NO3)2 -
Frameshift Co-mutagenesis with NiCl2 + Ogawa et al.
tester strains 9-aminoacridine (1987)
Co-mutagenesis with Ni(II) + Dubins &
alkylating agents (see Lavelle (1986)
pair substitution)
Escherichia coli
WP2 E. coli, reverse NiCl2 0, 5, 10, 25 - Green et al.
mutation mg/litre (1976)
WP2 E. coli (fluctuation) NiCl2 50 mmol/litre - Nishioka (1975)
WP2 uvra E. coli (fluctuation) NiCl2 50 mmol/litre -
CM571 E. coli (fluctuation) NiCl2 50 mmol/litre -
WP67 Differential toxicity NiCl2 0, 200, 500, + dose Tweats et al.
assay 1000 mg/litre response (1981)
CM871 Differential toxicity NiCl2 0, 200, 500, + dose Tweats et al.
assay 1000 mg/litre response (1981)
WP2 (repair Differential toxicity NiCl2 0, 200, 500, + dose
deficient) assay 1000 mg/litre response
Differential toxicity NiCl2 0, 200, 500, + dose
assay 1000 mg/litre response
WP2 E. coli, reverse NiCl2 - Arlauskas et
mutation NiSO4 - al. (1985)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Bacillus subtilis
H17 (rec+) B. subtilis, NiCl2 50 mmol/litre - Nishioka (1975)
M45 (rec-) rec. strains
differential toxicity
H17 (rec+) B. subtilis, NiCl2 5-50 mmol/litre - Kanematsu et
M45 (rec-) rec. strains NiO 5-50 mmol/litre - al. (1980)
differential toxicity Ni2O3 5-50 mmol/litre -
Corynebacterium
SP887 Reverse mutation NiCl2 0.03-10 mg/litre + dose Pikalet &
fluctuation test 8 doses response Necasek (1983)
at > 5.0
mg/litre
Salmonella
G46 (his) Host-mediated assay NiCl2 50 mg/kg - Buselmaier et
in mouse (NMRI strain) al. (1972)
Serrati marcescens
a21 (leu-) Host-mediated assay NiCl2 50 mg/kg - Buselmaier et
in mouse (NMRI strain) al. (1972)
Paramecium
Paramecium species Ni3S2 + at 0.5 Smith-Sonneborn
mutation µg/ml et al. (1983)
NiS particles + at 0.5
(1.8 µm) µg/ml
Paramecium species NiS particles + at 0.57 Smith-Sonneborn
chromosome aberration (1.8 µm) µg/ml et al. (1983)
Yeast (Saccharomyces)
D7 (diploid S. cerevisiae, gene NiCl2 3 or 10 mmol/litre + Fukunaga et al.
strain) conversion 24 h (1982)
D7 (diploid S. cerevisiae, gene NiSO4 5, 10, 20, 40 ? Singh (1984)
strain) conversion mmol/litre
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Yeast 19 haploid S. cervisiae, growth NiCl2 26 mmol/litre + Egilsson et al.
strains inhibition (1979)
Vicia faba
Chromosome aberrations NiO + Glaess (1956a)
Mitotic effects NiO + Glaess (1956b)
Vicia faba Mitotic effects NiCl2 + Komczynski et
Ni(NO3)2 + al. (1963)
NiSO4 +
Pisum Chromosome aberrations Ni(NO3)2 + Van Rosen (1954)
Allium cepa Chromosome aberrations NiO + Levan (1945)
INSECT SYSTEM
Drosophila
D. melanogaster Sex-linked recessive NiSO4 200, 300, 400 + Rodriguez-
white males lethal mutations mg/litre in 5% Arnaiz & Ramos
BASC females sucrose i.p. (1986)
D. melanogaster Sex chromosome NiSO4 200, 300, 400 + (weak)
XC2Y B/SC8Y loss assay mg/litre in 5%
males sucrose i.p.
Y2Wa females
D. melanogaster Somatic eye colour Ni(NO3)2 0.14 mmol/litre - Rasmuson (1985)
eggs from C(1)DX test system NiCl2 0.21 mmol/litre -
y,w,f females X
SC Z W+f males
D. melanogaster mutation Ni(NO3)2 3.4-6.9 mmol/litre +? Vogel (1984)
NiCl2 4.2 mmol/litre -
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
MAMMALIAN CELLS: DNA DAMAGE
Rat
Sprague-Dawley DNA strand breaks, NiCO3 5-40 mg/kg + Ciccarelli et
in vivo X links, alkaline al. (1981)
elution, in vivo Ciccarelli &
exposure Wetterhahn
(1985)
Primary DNA strand breaks, (+) Sina et al.
hepatocyte X links (1983)
Hamster
CHO DNA strand breaks NiCl2 100-500 µmol/litre + Robison &
alkaline sucrose Ni3S2 10 mg/litre + Costa (1982)
gradients (cryst.)
NiS (cryst.) 1-10 mg/litre +
NiS 10 mg/litre -
(amorphous)
CHO DNA strand breaks + Costa et al.
X links (1982)
CHO DNA strand breaks Ni3S2 10 mg/litre, 24 h + Patierno &
X links NiCl2 0.25-1.0 mmol/litre + Costa (1985)
Human
Normal human DNA strand breaks NiSO4 10-2000 mg/litre - Fornace (1982)
fibroblasts alkaline elution
XP cells
Peripheral DNA strand breaks NiCl2 0.05 mmol/litre ? McLean et al.
lymphocytes FADU technique (1982)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
MAMMALIAN CELLS: DNA BINDING
Hamster
CHO Binding to DNA in 63NiS 10 mg/litre + Harnett et al.
vitro (radioactive 63NiCl2 10 mg/litre + (10x (1982)
precursor) less)
CHO Binding to DNA in vivo 63NiCl2 + to Hui & Sunderman
liver & (1980)
63Ni(CO)4 + kidney
DNA
CHO Binding to RNA, 63NiS 10 mg/litre + Harnett et al.
protein 63NiCl2 10 mg/litre + (10x (1982)
less)
L132 pulmonary Binding, X-ray Ni3S2 Ni bound Chirali et
cells microprobe analysis to al. (1982)
phosphate
groups of
DNA, RNA
of cell
membranes
Human
HeLa Binding, colorimetric Ni Kovacs &
assay (dimethyl localized Darvas (1982)
glyoxime) in
centrioles
Mouse
FM3A cells Inhibition of DNA Ni(CH3CO2)2 0.6, 0.8, 1.0 + Umeda &
(mouse mammary synthesis mmol/litre Nishimura
carcinoma) (1979)
Nishimura &
Umeda (1979)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
CBA strain Inhibition of DNA NiSO4 15-30% LD50 - kidney Amlacher &
synthesis in vivo epithelium Rudolf (1981)
+ hepatic
epithelium
Rat
Fischer rat Inhibition of DNA NiCl2 + Basrur &
embryo cells synthesis; 3H TdR Gilman (1967)
uptake
Rat liver Inhibition of DNA Ni3S2 5, 10, 10, mg/litre + equal Swierenga &
epithelial cell synthesis; 3H TdR NiCl2 up to 150 mg/litre + potency McLean (1985)
line T51B uptake at
equitoxic
doses
Hamster
CHO S-phase inhibition, NiCl2 40-120 µmol/litre + Costa et al.
flow cytometry Ni metal 10-20 mg/litre + (1980b, 1982)
Ni3S2 1-10 mg/litre +
(cryst.)
Ni3Se2 1-5 mg/litre +
(cryst.)
NiO 5 mg/litre +
Human
HeLa Inhibition of DNA NiCl2 - Painter &
synthesis; 3H TdR uptake Howard (1982)
Bronchial Inhibition of DNA NiSO4 + Lechner et al.
epithelial cells synthesis; 3H TdR uptake (1984)
MAMMALIAN CELLS: DNA REPAIR
Rat
Fischer rat UDS NiCl2 Inhibition Swierenga et
primary of MMS- al. (1987)
hepatocytes induced
UDS
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Hamster
CHO Repair induction NiCl2 100-1000 µmol/litre + Robison &
SHE Cesium chloride Ni3S2 10 mg/litre + Costa (1982)
gradients (cryst.)
NiS (cryst.) 1-10 mg/litre +
NiS 5-10 mg/litre -
(amorphous)
MAMMALIAN CELLS: GENE MUTATION
Mouse
L5178Y mouse Thymidine kinase locus NiCl2 0.16-0.53 mmol/ + dose Amacher &
lymphoma cells litre (5 concs.) response Paillet
(toxic) (1980)
L5178Y mouse Thymidine kinase locus NiSO4 300-800 mg/litre + (weak) McGregor et
lymphoma cells at toxic al. (1988)
levels
Rat
T51B rat liver HPRT locus Ni3S2 5-20 mg/litre + Swierenga &
epithelial cells McLean (1985)
NRK, normal rat Frameshift and base NiCl2 20-40 µg/litre + Biggart &
kidney cells, pair substitution Murphy (1988)
with integrated
viral gene
6m2 murine Gene expression NiCl2 20-160 µmol/litre + dose
sarcoma virus- dependent
infected cells
Hamster
V79 HPRT locus NiCl2 0.4, 0.8 mmol/litre - Miyaki et al.
(1979)
V79 HPRT locus NiCl2 + Hartwig &
Beyersmann
(1989)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
V79, HGPRT with HPRT locus Ni(II) + Christie &
integrated E.coli Tummolo (1988)
gpt gene
CHO HPRT locus NiCl2 ? Hsie et al.
(1979)
CHO HPRT locus Ni3S2 + (weak) Costa et al.
NiS + (weak) (1980)
SHE Ouabain resistance NiSO4 0.019 mmol/litre - Rivedal &
+ as Sanner (1980)
comutagen
with BaP
MAMMALIAN CELLS: SISTER CHROMATID EXCHANGE (SCE)
Mouse \
FM3A cells mouse SCE NiCl2 6 x 10-4-10-3 mol/ + | Nishimura &
mammary carcinoma litre | Umeda (1979)
|
FM3A cells mouse Ni(CH3CO2)2 6 x 10-4-10-3 mol/ + | recovery
mammary carcinoma litre | period
NiK2(CN)4 1-1.6 x 10-3 mol/ + | included
litre |
NiS 4-8 x 10-4 mol/ + |
litre /
NiSO4 2.3 x 10-6 to +
2.3 x 10-3 mol/litre
P338D1 mouse SCE NiSO4 10-4 + Andersen (1983)
macrophage line
Hamster
Don cells SCE NiSO4 0.19 mmol/litre + at LC50 Ohno et al.
NiCl2 0.13 mmol/litre + at LC50 (1982)
CHO SCE NiSO4 0.95, 2.85 µmol/ + at 0.75 Deng & Qu
litre mg/litre (1981)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
CHO SCE NiS (cryst.) 5-20 mg/litre + Sen & Costa
NiCl2 1-1000 µmol/litre + (1985)
SHE SCE NiSO4 3.8, 9.5, 19 µmol/ + dose Larramendy
litre response et al. (1981)
Human
Peripheral blood SCE NiCl2 0.0-1-1.0 mmol/ + dose Newman et al.
lymphocytes, litre response (1982)
in vitro at 0.1
mmol/litre
Peripheral blood SCE NiSO4 0.0023-2.3 mmol/ + dose Wulf (1980)
lymphocytes, litre response
in vitro 0.02, 0.2
mmol/litre
Peripheral blood SCE NiSO4 0.95, 2.85 µmol/ + dose Deng & Qu
lymphocytes, litre response (1981)
in vitro
Peripheral blood SCE NiSO4 9.5, 19.0 µmol/ + dose Larramendy et
lymphocytes, litre response al. (1981)
in vitro
Peripheral blood SCE Ni3S2 10-4 mol/litre + (weak) Andersen (1983)
lymphocytes,
in vitro
Peripheral blood SCE Ni3S2 0.001-100 mg/litre ? Saxholm et al.
lymphocytes (1981)
Peripheral blood SCE nickel 2-20 mg/litre + (weak) Djachenko
lymphocytes (1989)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
MAMMALIAN CELLS: CHROMOSOME ABERRATIONS (CA)
Mouse
In vivo CA, in Ni-induced NiS (cryst.) + aneup- Christie et al.
rhabdomyosarcomas loidy, (1988)
marker
chromosome
BalbC in vivo Micronucleus Ni(NO3)2 56 mg/kg - Deknudt &
NiCl2 25 mg/kg - Leonard (1982)
FM3A cells CA Ni(CH3CO2)2 0.2, 0.3, 0.6, + gaps Umeda &
mammary carcinoma 1 mmol/litre; 24, Nishimura
48 h recovery (1979)
NiCl2 0.2, 0.3, 0.6, ? gaps
1 mmol/litre; 24,
48 h recovery
NiS 0.2, 0.3, 0.6, 1 + gaps,
mmol/litre; 24, breaks,
48 h recovery exchanges
FM3A cells CA Ni(CH3CO2)2 0.6, 0.8, 1.0 mmol/ + gaps, Nishimura &
mammary carcinoma litre; 6, 24, 48 h breaks, Umeda (1979)
recovery exchanges
NiCl2 0.6, 0.8, 1.0 mmol/ + gaps,
litre; 6, 24, 48 h breaks,
recovery exchanges
NiS 0.6, 0.8, 1.0 mmol/ + gaps,
litre; 6, 24, 48 h breaks,
recovery exchanges
FM3A cells CA Ni(CH3CO2)2 10-4 mol/litre + gaps Morita et al.
mammary carcinoma (2-3 days) breaks, (1985)
NiCl2 exchanges
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Rat
In vivo CA, rat bone marrow NiSO4 3, 6 mg/kg for - Mathur et al.
CA, spermatogonial cells 7 and 14 days (1978)
Hamster
Chinese hamster CA NiCl2 1/5, 1/10, 1/25 + dose Chorvatocvicova
in vivo LD50 response (1983)
1/10, 1/5
Chinese hamster CA NiCl2 1-1000 µmol/litre dose Sen & Costa
in vitro 2 h + 24 h recovery response (1985)
gaps,
breaks,
exchanges
NiS (cryst.) 5-20 mg/litre + at 20 mg/
litre, gaps,
breaks,
exchanges
Syrian hamster CA NiSO4 0.019 mmol/litre + gaps, Larramendy et
in vitro 24 hr breaks, al. (1981)
exchanges,
minutes,
dicentrics
Human
Peripheral blood CA NiSO4 0.19 mmol/litre + breaks, Larramendy et
lymphocytes rings, al. (1981)
in vitro minutes
Peripheral blood CA Ni powder - Paton & Allison
lymphocytes NiO (1972)
in vitro
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Peripheral blood CA Ni(CH3CO2)2 10-100 mg/litre - Voroshilin et
lymphocytes al. (1977)
in vitro
Peripheral blood CA NiCl2 2-20 mg/litre + Djachenko
lymphocytes (1989)
in vitro
MAMMALIAN CELLS: CELL TRANSFORMATION
Mouse
C3H10T Transformed foci Ni3S2 0.001, 0.1 mg/litre + Saxholm et al.
(1981)
Mouse embryo Inhibition of NiS (cryst.) 1-20 mg/litre + Sonnenfeld et
fibroblasts interferon production NiS (amorph.) 1-20 mg/litre - al. (1983)
Rat
HRT cells Initiation & promotion NiSO4 40 µmol/litre + Eker & Sanner
(hereditary renal test (1983)
tumour)
Rat embryo cells Transformed foci NiSO4 0.19, 0.38 mmol/ + Traul et al.
infected with litre (1981)
Rauscher leukaemia
virus
NRK (normal rat Transformed foci NiSO4 38-152 µmol/litre + max at Wilson &
kidney) cells, 10 mg/ Khoobyarian
infected with litre (1982)
Maloney murine
sarcoma virus
T51B rat liver Calcium independence, alphaNi3S2 2.5 mg/litre, + Swierenga et
epithelial cells growth control and long-term al. (1989)
morphology, differen- exposure
tiation induction
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Rat tracheal Transformed foci alphaNi3S2 5 mg/litre, 24 h + Feren & Reith
primary epithelial (1988)
cells
Hamster
SHE Transformed foci NiSO4 10, 19, 38 µmol/ + dose DiPaulo &
litre response Costa (1979)
Ni3S2 1.0, 2.5, 5.0 mg/ + dose
litre response
NiS up to 20 mg/litre -
(amorphous)
SHE Transformed foci NiSO4 3.8, 19, 76 µmol/ + at 19, Rivedal &
litre 76 µmol/ Sanner (1980)
litre co-
carcinogen
with BaP
SHE Transformed foci NiSO4 19 µmol/litre + at 19, Rivedal &
+ BAP 76 µmol/ Sanner (1981)
litre co-
carcinogen
with BaP
SHE Transformed foci NiSO4 19 µmol/litre + with Rivedal et al.
cigarette (1980)
smoke
extract
SHE Transformed foci Ni3S2 0.1, 1.0 mg/litre + at Costa et al.
(crystalline) subtoxic (1979)
doses,
confirmed
in nude
mice
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Hamster
SHE Transformed foci NiS 0.1, 1.0 mg/litre Costa et al.
(amorphous) (1979)
\
SHE Transformed foci Ni 5, 10, 20 mg/litre + | Costa et al.
NiO, Ni2O3 + | (1981a)b
NiS (cryst.) + | crystalline
NiS (amorph.) + | compounds
Ni3S2 (cryst.) + | more
Ni3S2 (cryst.) + | potent
/
SHE Transformed foci Inco Black 5, 10, 20 mg/litre + Sunderman et
NiO (jet al. (1987a)
black, 5 µm
particle size)
\
BHK-21 (baby Anchorage independence alphaNi3S2 5-20 mg/litre + | Hansen & Stern
hamster kidney) Ni2O3 5-20 mg/litre + | (1983)
NiO 37.5-150 mg/litre + | anchorage
Ni(CH3CO2)2 100-400 mg/litre + | independence
MIG nickel 100-400 mg/litre + | at equitoxic
welding fume / concentrations
Human
Bronchial Growth control and NiSO4 5-20 mg/litre + Lechner et al.
epithelial cells morphology (1984)
\
Foreskin Anchorage independence Ni(CH3)CO2)2 10 µmol/litre + | Biedermann &
fibroblasts NiO 50 µmol/litre + | Landolph (1986)
NiSO4 10 µmol/litre + | anchorage
Ni3S2 10 µmol/litre + | independence at
/ LC50 concentrations
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
MAMMALIAN CELLS: OTHER TESTS
Balb/c mouse Dominant lethal NiCl2 25 mg/kg - pre- DeKnudt &
in vivo Ni(NO3)2 56 mg/kg implant- Leonard (1982)
ation loss
observed
Balb/c mouse Dominant lethal Ni(NO3)2 8.1, 11.3 mg/kg - pre- Jacquet &
in vivo implant- Mayence (1982)
ation loss
observed
Rat Dominant lethal NiCl2 0.005-50 mg/kg - pre- Saichenko et
for 24 weeks implant- al. (1985)
ation loss
observed
Rat embryo cells Spindle disturbance NiCl2 1 mg Ni/litre + Swierenga &
in vitro culture medium Basrur (1968)
Human lymphocytes Spindle disturbance NiSO4 10-3 mol/litre + Andersen (1985)
NIH3T3 cells Inhibition of NiSO4 0.5-20 mmol/litre + dose Miki et al.
intercellular response (1987)
communication
(radioisotope transfer)
---------------------------------------------------------------------------------------------------------
Table 27 (contd.)
---------------------------------------------------------------------------------------------------------
Species/Strain Test system Compound Concentrationa Result Reference
---------------------------------------------------------------------------------------------------------
Human Induction of free Ni3S2 + more Evans et al.
polymorphonuclear radicals potent (1988)
leukocytes (chemiluminescent NiO +
technique)
---------------------------------------------------------------------------------------------------------
a No figures means no concentration data given.
b Transformation experiments with this system have been repeated numerous times for mechanistic studies.
Findings include:
- crystalline but not amorphous nickel compounds are actively phagocytosed in vitro (Costa &
Mollenhauer, (1980b);
- reduction of amorphous compounds with LiA1H4 enchances phagocytosis (Costa & Mollenhauer, 1980b);
- phagocytosis is Ca-dependent (Heck & Costa, 1982);
- phagocytosed particles are contained in cytoplasmic vesicles where they slowly dissolve, releasing
nickel ions (Evans et al., 1982);
- nickel compounds selectively damage heterochromatin (Sen & Costa, 1985);
- NiS (cryst.) and NiCl2 preferentially transform male SHE cells (Costa et al., 1981a);
- observed damage includes deletion of long arm of X chromosome (Conway & Costa, 1989).
8.2.1. Mutagenesis in bacteria and mammalian cells
Nickel compounds were generally inactive in bacterial mutation
assays. In some cases, where standard procedures were modified by,
for example, using fluctuation assays (LaVelle & Witmer 1981;
Pikalek & Necasek, 1983) or co-mutagenesis tests with other
carcinogenic agents (Dubins & LaVelle, 1986; Ogawa et al., 1987),
positive results were obtained.
The mutagenic effects of nickel chloride at the hypoxanthine-
guanine phosphoriboxyl transferase (HGPRT) locus were studied in
cultured V79 Chinese hamster cells and Chinese hamster ovary (CHO)
cells (Hsie et al., 1979). Hsie et al. (1979) claimed positive
results without reporting the detailed data. Miyaki et al. (1979)
obtained a negative response. However, Hartwig & Beyersmann (1989)
obtained a positive response with the same cell line, but the
response occurred only in a serum-free medium.
Amacher & Paillet (1980) studied nickel chloride at the
thymidine kinase (TK) locus in L51784Y mouse lymphoma cells and
found a dose-dependent increase in the number of mutants of up to 4
times that in the controls. The possibility that this response was
due to chromosome damage, also detected in this assay, cannot be
excluded.
Swierenga & McLean (1985) used epithelial cells from rat liver
(T51B) to study the genotoxic effects of nickel chloride and also
of nickel subsulfide, either as an aqueous suspension of washed
particles or as an aqueous solution. All produced increased
numbers of mutations at the hypoxanthine-guanine phosphoriboxyl
transferase (HGPRT) locus, over a range of concentrations.
In all the gene mutation studies using mammalian cells, any
response following exposure to nickel compounds was associated with
considerable cell toxicity.
8.2.2. Chromosomal aberration and sister chromatid exchange (SCE)
Nishimura & Umeda (1979) compared the effects of nickel
chloride, nickel acetate, potassium cyanonickelate and nickel
sulfide on the induction of chromosomal aberrations in cultured
FM3A mouse mammary carcinoma cells. The 4 compounds elicited
similar inhibitory effects on the synthesis of protein, RNA, and
DNA. The chromosomal aberrations were manifested as breaks,
exchanges, and fragmentations. Treatment of Chinese hamster ovary
(CHO) cells with crystalline nickel sulfide and nickel chloride
induced chromosomal aberrations including gaps, breaks, and
exchanges. In both cases, the heterochromatic centromeric regions
of the chromosomes were preferred; nickel sulfide also caused
selective fragmentation at the heterochromatic long arms of the
X-chromosome.
Chromosomal damage and increased SCE were induced in Syrian
hamster embryo (SHE) cells and human lymphocyte cultures treated
with nickel sulfate (Larramendy et al., 1981).
Wulf (1980) showed that nickel sulfate and nickel subsulfide
could produce SCE in human lymphocytes in vitro. A significant
increase in SCE in human lymphocytes exposed to nickel subsulfide
was reported by Saxholm et al. (1981). Newman et al. (1982)
observed increased SCE in human lymphocytes exposed to nickel
chloride. Ohno et al. (1982) investigated the induction of SCE by
nickel sulfate and nickel chloride in Don Chinese hamster cells and
found significantly increased frequencies of SCE.
8.2.3. Mammalian cell transformation
These assays may not necessarily have a "genotoxic" endpoint as
they also predict the carcinogenic potential of compounds that act
by non-genotoxic mechanisms.
DiPaolo & Casto (1979) studied the capacity of nickel sulfate,
nickel subsulfide, and amorphous nickel monosulfide to induce
morphological transformation in Syrian hamster embryo (SHE) cells
in vitro. Amorphous nickel monosulfide did not produce
transformation at plate concentrations as high as 20 µg/ml medium.
Nickel sulfate, tested at plate concentrations of 2.5, 5, and 10
µg/ml medium, caused transformation in a dose-dependent manner.
Nickel subsulfide produced a higher percentage of transformations,
in a dose-dependent manner, than nickel sulfate, when tested at
plate concentrations of 1.0, 2.5, and 5 µg/ml medium. In a study by
Costa et al. (1979), nickel subsulfide caused morphological
transformation, in a dose-dependent manner, of Syrian hamster fetal
cells, at plate concentrations of 0.1, 1.0, 5.0, and 10.0 µg/ml
medium, but, under the same experimental conditions, nickel
monosulfide, at plate concentrations of 0.1, 1.0, and 5.0 µg/ml
medium, did not.
Costa et al. (1981) demonstrated that the incidence of
morphological transformations of SHE and CHO cells, exposed to
various nickel compounds, was directly correlated with
phagocytosis. Subcutaneous implantation of transformed SHE cells
led to the development of fibrosarcomas in nude mice (Costa et al.,
1979). Nickel sulfate caused cell transformation in cultured Syrian
hamster embryo cells (Pienta et al., 1977).
Sunderman et al. (1987a) compared nickel oxides with different
physical and chemical properties in the SHE cell transformation
assay, and in vivo responses (Table 28). Three out of the 4
compounds that were active in the transformation assay were also
positive in vivo when injected intramuscularly.
Table 28. Comparison of activity of some nickel oxides with different physiochemical
characteristics in in vitro transformation assays and in vivo carcinogenesis
---------------------------------------------------------------------------------------
Compound Concentration Resulta No. of tumours
observed (20 mg
Ni/rat im)b
---------------------------------------------------------------------------------------
Nickel oxides Calcination
temperature
INCO Black (jet black) 650 °C 5, 10, 20 mg/litre + 6/15
Grey black 735 °C Particle size 5 µm + 0/15
Green 1045 °C - 0/15
Nickel copper oxides Ni-Cu ratio
Maroon 2.5:1 5, 10, 20 mg/litre + 13/15
Red-brown 5.2:1 Particle size 5 µm + 15/15
alphaNiS (positive + 15/15
control)
---------------------------------------------------------------------------------------
a Sunderman et al. (1987a).
b Sunderman et al. (1988a).
Hansen & Stern (1984) compared the abilities of welding fume,
nickel powder, nickel acetate, black nickel oxide, black nickel
oxide catalyst (a commercial catalyst for organic reactions, which
is a mixture of nickel(II) and nickel(IV) oxides (NiO1.4 x 3H2O),
and nickel subsulfide, in the in vitro transformation of Syrian
baby hamster kidney BHK-21 cells. Although a wide range of
transformation potency was found, the compounds produced the same
number of transformed colonies at the same degree of toxicity (50%
survival). The authors concluded that this indicated that if
toxicity is a direct measure of net available nickel, then nickel
or the nickel ion is the ultimate transforming agent. In a
subsequent BHK-21 mammalian cell assay, Stern et al. (1985)
determined the 50% toxicity of the soluble and insoluble fraction
of nickel welding fume and found that only the insoluble fraction
showed a transformation potential.
Synergistic effects of nickel compounds and benzo (a)pyrene on
morphological transformation of SHE cells have been reported.
Costa & Mollenhauer (1980c) found that pretreatment of the cells
with benzo (a)pyrene enhanced the cellular uptake of nickel
subsulfide. Treatment with nickel sulfate and benzo (a)pyrene
resulted in a transformation frequency of 10.7% compared with 0.5%
and 0.6%, respectively, for the individual substances (Rivedal &
Sanner, 1980).
8.3. Other test systems
Smith-Sonneborn et al. (1986) used the ciliated protozoan
Paramecium to quantify the effects of pure nickel powder, iron-
nickel powder, and nickel subsulfide. Genotoxicity was indicated
by significant increases in the fraction of non-viable offspring
(presumed index of lethal mutation) found after autogamy in parents
from the nickel-treated groups compared with the controls. Only
nickel subsulfide consistently induced a significant increase in
offspring lethality.
Rodriguez-Arnaiz & Ramos (1986) studied the mutagenic potential
of nickel sulfate in the Drosophila sex-linked recessive lethal
(SLRL) assay in vivo. Nickel sulfate induced SLRL in a dose-
dependent way, whereas sex chromosome loss was only detectable in
significant numbers at the highest concentration.
8.4. Carcinogenicity
8.4.1. Inhalation
Studies on inhalation exposure are summarized in Table 29.
Hueper (1958) and Hueper & Payne (1962) studied the effects of
exposure to airborne concentrations of elemental nickel. Hueper
(1958) exposed 20 female C57BL mice, 50 male and 50 female Wistar
rats, 60 female Bethesda black rats, and 32 male and 10 female
guinea-pigs to an atmosphere containing 99% pure nickel powder
(particle size 4 µm or less) at 15 mg/m3, for an exposure period of
6 h/day, 4-5 days/week, for up to 21 months. There were no control
groups. There were no lung tumours, but 2 lymphosarcomas were seen
in the mice. However, most animals died before 15 months. Fifteen
out of 50 rats of both strains that were studied histologically
showed adenomatoid formations in the lung, which were classified as
benign neoplasms. At death, most of the guinea-pigs exhibited
adenomatoid proliferations. One animal had an intra-alveolar
carcinoma. The results of a later study (Hueper & Payne, 1962) on
rats and hamsters did not reveal lung tumours following inhalation
exposure to 99% pure nickel together with 56-98 mg sulfur
dioxide/m3 (20-35 ppm) and powdered limestone.
In studies by Kim et al. (1969) a group of 77 male Wistar rats
was exposed to metallic nickel dust, equivalent to 3.1 mg/m3 for 6
h a day, 5 days/week, over 21 months, followed by exposure to air
alone; 98% of the dust particles were less than 2 µm in diameter.
Sub-groups were exposed to the dust for various periods followed by
periods of recovery. Two exposed rats developed lung tumours of a
carcinoid pattern and a similar tumour was found in one rat in the
unexposed control group.
Table 29. Experimental animal studies on the carcinogenicity studies of nickel compounds administered by inhalation or
tracheal instillation
---------------------------------------------------------------------------------------------------------------------------
Nickel compound Animal Dosage schedule Lung tumours detected Comments References
(group size)
---------------------------------------------------------------------------------------------------------------------------
Inhalation studies
Nickel powder C57BL female 15 mg/m3, 6 h/day, no lung tumours, all animals died Hueper (1958)
mice (20) 4-5 days/week for 2 lymphosarcomas by week 60, no
60 weeks control group
Nickel powder Wistar rats 15 mg/m3, 6 h/day, numerous multicentric histology on 50 Heuper (1958)
(108); Bethesda 4-5 days/week, for adenomatoid animals only, no
black rats (60) 60 weeks proliferations in 15 no control group
animals
Nickel powder Guinea-pigs 15 mg/m3, 6 h/day, adenomatous alveolar all animals died Heuper (1958)
(42) 4-5 days/week, for lesions in almost all by 21 months
60 weeks animals
Nickel powder + Bethesda black level not specified no lung tumours Heuper & Payne
powdered rats (120) (1962)
limestone + Hamsters (100)
sulfur dioxide
Nickel powder Wistar rats 3.1 mg/m3, 6 h/day, carcinoid lung tumour Kim et al.
(77) 5 days/week for 21 in 2 exposed and in 1 (1969)
months (?) control rat
Ni(CO4) Wistar rats 30 mg/m3, 30 min/ 1 lung carcinoma no lung tumours Sunderman et al.
(64) day, 3 days/week, in a control group (1959)
for 52 weeks of 41 animals
Ni(CO4) Wistar rats 60 mg/m3, 30 min/ 1 lung carcinoma 9 animals survived Sunderman et al.
(32) day, 3 days/week, for 2 years (1959)
for 52 weeks
Ni(CO4) Wistar rats single exposure to 2 lung carcinomas Sunderman et al.
(80) 250 mg/m3 (30 min?) (1959)
---------------------------------------------------------------------------------------------------------------------------
Table 29 (contd.)
---------------------------------------------------------------------------------------------------------------------------
Nickel compound Animal Dosage schedule Lung tumours detected Comments References
(group size)
---------------------------------------------------------------------------------------------------------------------------
Ni(CO4) Wistar rats 30 mg/m3, 30 min/ 1 lung adenocarcinoma Sunderman &
(64) day, 3 days/week, Donelly (1965)
for lifetime
Ni(CO4) Wistar rats single exposure to 1 lung adenocarcinoma 214/285 animals Sunderman &
(285) 600 mg/m3 for 30 min died within 3 weeks Donelly (1965)
Nickel F344 rats 0.97 mg/m3, 6 h/day, 14 malignant and 15 survival: 5% in Ottolenghi et
subsulfide (226) 5 days/week, for 78 benign lung tumours in nickel-exposed al. (1974)
weeks, followed by the exposed, 1 rats; 31% in
observation for 30 malignant and 1 benign controls
weeks in the control animals
P <0.01
Black nickel Syrian golden 53.2 mg/m3, 7 h/day, no significant increase massive Wehner et al.
oxide hamsters (102) 5 days/week, for in incidence of pneumoconiosis in (1975)
2 years respiratory tumours exposed animals;
part of animals also
exposed to tobacco
smoke
Green nickel Wistar rats 0.6 mg/m3, 6 h/day, 1 adenocarcinoma and 5 control animals Horie et al.
oxide (6) 5 days/week, for 4 1 adenomatous pulmonary (1985)
weeks, followed by lesion
observation for 80
weeks
Green nickel Wistar rats 8 mg/m3, 6 h/day, 1 adenomatous lesion Horie et al.
oxide (8) 5 days/week, for 4 in lungs (1985)
weeks, followed by
observation for 80
weeks
Nickel-enriched Syrian golden fly ash containing no lung tumours 20 of the animals Wehner et al.
coal fly ash (Ni hamster (102) 6% Ni, 70 mg/m3, removed from (1981)
acetate added to 6 h/day, 5 days/ exposure for other
coal before week, for 20 months studies
burning)
---------------------------------------------------------------------------------------------------------------------------
Table 29 (contd.)
---------------------------------------------------------------------------------------------------------------------------
Nickel compound Animal Dosage schedule Lung tumours detected Comments References
(group size)
---------------------------------------------------------------------------------------------------------------------------
Nickel-enriched Syrian golden fly ash containing no lung tumours Wehner et al.
coal fly ash (Ni hamsters 6% Ni, 17 mg/m3, (1981)
acetate added to 6 h/day, 5 days/week,
coal before for 20 months
burning)
fly ash containing no lung tumours Wehner et al.
0.3% Ni, 70 mg/m3, (1981)
6 h/day, 5 days/week,
for 20 months
Nickel oxide Wister rats 60 µg/m3 continuous no lung tumours Alveolar Glaser et al.
(60) exposure for 18 proteinosis in (1986)
months nickel-exposed
animals
200 µg/m3 continuous no lung tumours
exposure, doe, 18
months
Intratracheal instillation
Nickel powder Wistar rats 0.3 mg Ni x 20, at 1 adenoma, 1 no lung tumours in Pott et al.
(39) weekly intervals adenocarcinoma, 8 in 40 control (1987)
squamous cell animals
carcinomas
Wistar rats 0.9 mg Ni x 10, at 3 adenocarcinomas, Pott et al.
(32) weekly intervals 4 squamous cell (1987)
carcinomas, 1 mixed
tumour
Nickel oxide Wistar rats 5 mg x 10, at 4 adenocarcinomas, Pott et al.
(not specified) (37) weekly intervals 4 squamous cell (1987)
carcinomas, 2 mixed
tumours
Wistar rats 15 mg x 10, at 12 squamous cell
(38) weekly intervals carcinomas
---------------------------------------------------------------------------------------------------------------------------
Table 29 (contd.)
---------------------------------------------------------------------------------------------------------------------------
Nickel compound Animal Dosage schedule Lung tumours detected Comments References
(group size)
---------------------------------------------------------------------------------------------------------------------------
Black nickel Hamsters 4 mg x 30, at no lung tumours 3 hamsters in both Farrell & Davis
oxide (50) weekly intervals groups survived for (1974)
12 months
Black nickel Rats (26) single 1 squamous cell the nickel oxide Saknyn & Blokhin
oxide administration carcinoma of the dust contained (1978)
20-40 mg lung 64.7% NiO, 0.13%
NiS, 0.18% Ni; no
pulmonary tumours
in 47 controls
Nickel B6C3F1 mice 0.024, 0.056, 0.156, no neoplastic or Fisher et al.
subsulfide (20) 0.412, or 1.1 mg/kg, non-neoplastic (1986)
four doses at weekly lesions
intervals, follow-up
of 27 months
Ni3S2 Rats (47) 0.063 mg x 15, at 2 adenocarcinomas, no pulmonary Pott et al.
weekly intervals 5 squamous cell tumours in control (1987)
carcinomas rats
Ni3S2 Rats (45) 0.125 mg x 15, at 3 adenocarcinomas, 6 no pulmonary Pott et al.
weekly intervals squamous cell tumours in control (1987)
carcinomas, 4 mixed rats
tumours
Ni3S2 Rats (40) 0.25 mg x 15, at 7 adenocarcinomas, no pulmonary Pott et al.
weekly intervals 4 squamous cell tumours in control (1987)
carcinomas, 1 mixed rats
tumours
Powder Hamsters, 0.8 mg powder, 1 lung tumour (nickel no pulmonary Muhle et al.
Ni3S2 (30-40 per sex) 0.1 mg alphaNi3S2, powder group) tumours in positive (1988)
Pentlandite 3 mg pentlandite, 1 lung tumour control group
Cr-Ni(55) Alloy 3 or 9 mg Cr-Ni(55) (pentlandite group)
Cr(55) Alloy alloy, 9 mg Cr(55)
alloy; 12 times at
14-day intervals
---------------------------------------------------------------------------------------------------------------------------
Ottolenghi et al. (1974) found a significantly higher incidence
of pulmonary hyperplastic and neoplastic lesions in 226 Fischer 344
rats of both sexes exposed to nickel subsulfide (0.97 mg nickel/m3;
70% of particles smaller than 1 µm) for 6 h/day, 5 days/week, over
78 weeks. The overall incidence of lung tumours (adenoma and
adenocarcinomas) in treated animals was 14% compared with 1% in the
controls. The nickel sulfide-exposed rats also had a higher
incidence of respiratory tract inflammation.
The pathogenic effect of inhaled black nickel oxide was
investigated in hamsters by Wehner et al. (1975). Random-bred ENG:
ELA Syrian golden hamsters (102 males) were exposed to respirable
aerosols of nickel oxide (count median diameter 0.3 µm) at a
concentration of 53.2 mg/m3 for 7 h/day, on 5 days/week, for up to
2 years. Half of the animals were also exposed (nose-only) to
cigarette smoke for 10 min, 3 times a day. Histopathological
examination revealed increasing cellular response, proliferative
and inflammatory, in animals dying late in the study.
Histopathologically, there were no marked differences between the
nickel-oxide-plus cigarette smoke and the nickel-oxide-only exposed
groups. It was concluded that there was neither a significant
carcinogenic effect of nickel oxide nor a co-carcinogenic effect of
cigarette smoke. However, though not statistically significant, 3
malignant musculoskeletal tumours were found among nickel oxide-
exposed hamsters (Wehner et al., 1975).
Wehner et al. (1981) exposed 4 groups, each of 102 male Syrian
golden hamsters (outbred LAK:LVG), through inhalation to nickel-
enriched fly ash (NEFA) for 6 h/day, 5 days/week, for up to 20
months. The first group was exposed to 70 mg NEFA/m3 aerosol (4 mg
nickel/m3), the second group to 17 mg NEFA/m3 aerosol (1 mg
nickel/m3), the third group was exposed to 70 mg fly ash (FA)/m3
aerosol containing 0.21 mg nickel/m3, and the fourth group was
exposed to filtered air (control group). Five animals from each
group were killed after 4, 8, 12, and 16 months of exposure.
Additional groups of 5 animals were withdrawn from exposure at the
same time intervals and maintained for observation up to the
twentieth month of the study, when all the animals were killed.
Dust deposition, interstitial reaction, and bronchiolization in the
lungs were higher in the high-NEFA and FA groups than in the low-
NEFA group, indicating that dust quantity rather than actual nickel
content may be the major factor in determining tissue response.
While 2 malignant pulmonary tumours were found in 2 hamsters of the
high-NEFA group, no statistically significant carcinogenesis was
observed.
Horie et al. (1985) studied the carcinogenic effects of
inhalation of 8.0 or 0.6 mg green nickel oxide/m3 on male Wistar
rats (5-8 rats/dose group). The exposure time was 6 h/day, 5
days/week, for 1 month. Animals were killed 20 months after
exposure. There was one adenocarcinoma in a low-dose animal. In a
study by Glaser et al. (1986), male Wistar rats were exposed
continuously for 18 months to nickel oxide aerosols (60 or 200 µg
nickel/m3). The nickel oxide aerosols were generated by
atomization of aqueous nickel acetate solutions and subsequent
pyrolysis. No lung tumours were observed.
Sunderman et al. (1959) and Sunderman & Donelly (1965) reported
carcinogenesis in rats following inhalation exposure to nickel
carbonyl. In the first study (Sunderman et al., 1959), groups of
64 and 32 male Wistar rats were exposed to 30 and 60 mg nickel
carbonyl/m3, respectively, for 30 min, 3 days/week, for one year.
A further group was exposed once to 250 mg nickel carbonyl/m3.
Four out of the 9 animals that survived 2 years developed neoplasms
of the lung; 2 of these animals were in the single-exposure group,
the other 2 rats were in the repeated-exposure groups. No
pulmonary tumours were seen in 41 control animals, the death rate
of which was similar to that in the animals exposed to nickel
carbonyl. Sunderman & Donelly (1965), using 285 male Wistar rats,
observed pulmonary adenocarcinoma with metastases in one of the 35
rats that survived 2 or more years after a single 30-min inhalation
of 600 mg nickel carbonyl/m3. In a further group of 64 male rats,
exposed to repeated inhalation of nickel carbonyl (30 mg/m3 for 30
min, 3 days/week, until death), one pulmonary adenocarcinoma with
metastases developed among 8 rats that survived 2 or more years.
The control animals did not show any tumours. Because of the
rarity of spontaneous pulmonary malignancies in Wistar rats, it was
suggested that the tumours observed in the two studies were due to
inhalation of nickel carbonyl. Survivability of treated and
control animals was poor in both studies. Statistical analysis
could not be performed because of small sample size.
Kasprzak et al. (1973) did not find any lung tumours in 13 rats
following intratracheal instillation of 5 mg nickel subsulfide.
Thirty weekly intratracheal injections of 4 mg black nickel oxide
did not produce lung tumours in 50 hamsters, but only 3 animals
survived (Farrell & Davis, 1974). In a study by Saknyn & Blokhin
(1978) 26 rats were exposed to 20-40 mg nickel oxide by
intratracheal instillation; 1 lung carcinoma was found. In a
series of studies, Pott et al. (1987) examined the potential
carcinogenic effect of a number of dusts, including nickel oxide,
nickel powder, and nickel subsulfide, at various concentrations, in
rats. All 3 nickel compounds produced lung tumours, including
adenocarcinomas and squamous cell carcinomas, nickel subsulfide
exhibiting the strongest effect in relation to dose. Lung tumour
incidence was 14.9% (7 out of 47 animals), 28.9% (12 out of 45
animals), and 30% (12 out of 40 animals) following 15 weekly
intratracheal injections each containing 0.063 mg, 0.125 mg, and
0.25 mg nickel, respectively. Nickel powder was given
intratracheally in 20 weekly doses of 0.3 mg nickel; an additional
group was given 10 weekly doses of 0.9 mg nickel. Lung tumours
occurred in 10 out of 39 animals (25.6%) and in 8 out of 32 animals
(25.0%), respectively. Ten weekly instillations of nickel oxide,
each corresponding to a nickel content of 5 or 15 mg, produced
27.0% (10/37 animals) or 31.6% (12/38 animals) lung tumours,
respectively. No lung tumours occurred in the controls (40
animals). In a study with nickel subsulfide using the
intratracheal route, no increase in tumour incidence was observed
compared with the controls. Doses ranged from 0.024 to 1.1 mg/kg,
administered once a week for 4 weeks (Fisher et al., 1984).
Muhle et al. (1988) administered nickel powder (cumulative dose
9.6 mg), nickel subsulfide (cumulative dose 1.2 mg), pentlandite
(cumulative dose 36 mg), chromium-nickel-steel (cumulative dose 36
or 108 mg) and chromium-containing stainless steel (cumulative dose
108 mg) to Syrian golden hamsters (30-40 animals per sex and dose
group) by intratracheal instillation, 12 times every 14 days. One
lung tumour was found after treatment with nickel powder and one
tumour after treatment with pentlandite.
8.4.2. Oral
Schroeder et al. (1964, 1974) and Schroeder & Mitchener (1975)
studied the carcinogenic potential for mice and rats of nickel
salts in the drinking-water. Levels of 5 mg nickel/litre in the
drinking-water did not produce a significantly higher incidence of
tumours compared with controls.
Ambrose et al. (1976) added nickel sulfate hexahydrate fines
(22.3% nickel) to the diet of Wistar derived rats for 2 years, at
concentrations of 0, 100, 1000, or 2500 mg nickel/kg. There was no
carcinogenic response.
8.4.3. Other routes
The results of carcinogenesis studies of nickel compounds in
rodents are summarized in Table 30.
Table 30. Experimental carcinogenesis studies of various nickel compounds
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Ni3S2 Fischer im Form of implant Gilman & Herchen
rats - implants 500 mg 14/17 is important (1963)
- diffusion chambers
10 mg 14/17
- powder 10 mg 19/20
- chips 500 mg 5/7
Ni3S2 Rat 1 or 3 mg Ni3S2/ 1 mg: 9/60 tumour No tumours in Yarita &
gelatin pellet (6 carcinomas, 3 control Nettesheim
implanted 4 weeks sarcomas) transplanted (1978)
post-grafting in 3 mg: 45/60 tumours tracheas
tracheas grafted (1 carcinoma, 44
under dorsal skin of sarcomas)
isogenic recipients
Ni3S2 Rats Intratesticular, 16/19 (fibrosarcomas, No tumours in Damjanov et al.
0.6-10 mg rhabdomyosarcomas, controls (1978)
malignant
histiocytomas)
Ni3S2 Fischer intrarenal Renal cancers in No tumours in Sunderman et al.
rats 18/24 rats at 10 mg controls (1979b)
dosage and 5/18 rats
at 5 mg dosage
Ni3S2 NMRI mice im, sc Local sarcomas in 19 Oskarsson et al.
of 32 mice at 10 mg (1979)
dosage
Ni3S2 Pregnant im, on day 6 Local sarcomas in Sunderman et al.
rats all dams; no excess (1981)
tumours in progeny
---------------------------------------------------------------------------------------------------------
Table 30 (contd.)
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Ni3S2 Rabbits im Leiomyosarcomas with Hildebrand &
myosin light-chains Tetaert (1981);
typical of fetal Hildebrand &
smooth muscle, Biserte (1979a,b)
rhabdomyosarcomas
Ni3S2 Fischer intraocular Retinoblastomas, Albert et al.
rats gliomas, and (1980, 1982)
melanomas
Ni3S2 Fischer & im Rhabdomyosarcomas, Yamashiro et al.
hooded leiomyosarcomas, (1980, 1983)
rats fibrosarcomas and
lymphosarcomas
Ni3S2 Fischer im Ni3S2 carcinogenesis Sunderman &
rats inhibited by McCully (1983)
simultaneous injection
of manganese dust
Ni3S2, Wistar im Sarcomas induced by Kasprzak et al.
Ni(OH)2, rats Ni3S2 and crystalline (1983)
NiSO4 Ni(OH)2, but not by
NiSO4 or amorphous
Ni(OH)2
Various Fischer im Sarcomas induced by Sunderman
nickel rats nickel compounds (1984)
compounds
Ni3S2, NiO Wistar & intrapleural Rhabdomyosarcomas, Skaug et al.
Ni3S2 Fischer implantation malignant tumours of (1985)
rats pluripotential origin Lumb et al.
(1985)
Ni3S2 Fischer im Rhabdomyosarcomas, Kasprzak &
rats fibrosarcomas Poirier (1985)
---------------------------------------------------------------------------------------------------------
Table 30 (contd.)
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Ni3S2 F344 rats sc, im, if, ia Malignant soft No synovial Shibata et al.
tissue tumours in sarcomas (1985a)
18/19 (sc), 19/20
(im), 9/20 (if),
16/19 (ia)
Ni3S2 Male im (single dose) Mg carbonate A possible Kasprzak et al.
Fischer inhibited Ni3S2 involvement of (1987)
rats carcinogenicity NK and phagocytic
10 groups from 100 to 55% cells in this
20 each inhibition
Ni3S2 Fischer im Local sarcomas Simultaneous Kasprzak et al.
rats admin. of ZnO (1988)
or Zn acetate
delayed the onset
of sarcomas
Ni3S2 F344 NCT im Local sarcomas Kasprzak &
100% Rodriguez (1988)
Ni3S2 + Rat im Local sarcomas Interaction role Kasprzak &
iron: of inflammation, Rodriguez
Fe-/Ni = 0.5 60% active oxygen (1988)
Fe-/Ni = 1.0 40% species, enzyme
Fe-/Ni = 2.0 10% system
Local sarcomas
Fe3+ = 0.5 95% Kasprzak &
Fe3+ = 1.0 45% Rodriguez
Fe3+ = 2.0 35% (1988)
---------------------------------------------------------------------------------------------------------
Table 30 (contd.)
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Ni3S2 + 100% Iron effect is Kasprzak &
remote purely local Rodriguez
Fe- or (1988)
Fe3+/Ni = 2
Ni3S2 in vitro Deactivation of Kasprzak &
cell free catalase and Rodriguez
glutathione (1988)
peroxidase
Ni3S2 Japanese intraocular Malignant Mitsumasa
common melanoma-like (1987)
newts tumour in 7/8
(Cynops) at 400-1100
pyrrhogaster µg/newt
NiO Wistar im 26 local tumours No controls used Gilman
rats 20 mg in 32 rats (80%) (1962)
NiO Swiss im 35% mice with
mice 5 mg tumours
NiO C3H mice im 23% mice with
5 mg tumours
NiO NIH black im implants 4 sarcomas in 35 Payne
rats 7 mg rats after 18 months (1964)
Nickel Rats Instillation in Rhabdomyosarcomas Negative study Eilertsen
oxides and pleural cavity in 8/220 (4/20 in et al.
nickel- positive control) (1988)
copper
oxides
---------------------------------------------------------------------------------------------------------
Table 30 (contd.)
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Nickel Hooded im, 52/66 rats with Gilman &
refinery Wistar sarcomas Ruckerbauer
flue dust rats 20 or 30 mg, one or 8/20 rats with (1962)
thighs sarcomas
Mice im, 23/40 mice with
10 mg each thigh sarcomas
Feinstein Rats ip 6/39 rats with injection Saknyn &
dust 90-150 mg dust/rat site sarcomas Blokhin (1978)
Nickel Hooded im, 28.3 mg in 10/10 rats with Heath &
powder rats 0.4 ml fowl serum local rhabdomyosarcomas Daniel (1964)
Nickel Osborne- Intrafemoral, 4/17 rats with Hueper (1952)
powder Mendel 21 mg tumours, 1 squamous
rats cell carcinoma,
3 osteosarcomas
Wistar Intrafemoral 28% of treated rats No tumours in
rats implant, 50 mg with tumours of control rats
injected thighs
Dutch Intrafemoral 1/6 rabbits with
rabbits implant, 54 mg/kg fibrosarcomas
C57BL iv, No tumours
mice weekly for 2 weeks
Rabbits iv, No tumours
6 times of a 1% Ni
suspension in 2.5%
gelatin at a rate
of 0.5 ml/kg
---------------------------------------------------------------------------------------------------------
Table 30 (contd.)
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Wistar iv, 7/25 rats with
rats 6 times of 0.5% Ni tumours
suspension in saline
at 0.5 ml/kg
Nickel Fischer im, 5 monthly 38/50 rats with Furst &
powder rats injections of 5 mg fibrosarcomas Schlauder
Ni in 0.2 ml (1971)
trioctanoin
Hamsters im, 5 monthly 2/50 hamsters with Furst &
injections of 5 mg fibrosarcomas Schlauder
Ni in 0.2 ml (1971)
trioctanoin
Nickel Fischer im, 3.6 mg/rat 0/10 rats with local Sunderman &
powder rats tumours Maenza (1976)
14.4 mg/rat 2/10 rats with local
tumours
Nickel Fischer Intrathoracic, Mesothelioma, Only 10 animals Furst et al.
powder 344 rats 5 monthly doses of pleural cavity in (1973)
5 mg/animal 2 out of 10 rats
Nickel Fischer ip, 16 injections 30-50% Furst &
powder 344 rats twice monthly, 5 mg/ intraperitoneal Cassetta
animal tumours (1973)
Nickel Fischer Intrathoracic 40% mesothelioma Furst &
powder 344 rats 5 monthly doses, of Cassetta
5 mg/animal (1973)
---------------------------------------------------------------------------------------------------------
Table 30 (contd.)
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Ni(CO)4 Sprague- iv, 6 x 50 µl/kg 19/21 rats with Lau et al.
Dawley (9 mg Ni/kg) malignant tumours (1972)
rats at various sites,
2/47 rats with
pulmonary lymphomas
< 0.05
NiCO3 NIH black Muscle implant, 4/35 implant site Payne
rats 7 mg sarcomas (1964)
Nickelocene Fischer im injections, 18/50 rats or 21/50 No tumours Furst &
rats 12 x 12 mg or rats with in controls Schlauder
12 x 25 mg fibrosarcomas (1971)
nickelocene
Hamsters im injections, 4/29 hamsters with No tumours at
1 x 25 mg or fibrosarcomas at 8 x 5 mg
8 x 5 mg 1 x 25 mg
nickelocene
Ni(CH3CO2)2 Mice, ip, 24 injections, Lung adenomas and Stoner
Strain A 3/week at 72, 180, adenocarcinomas et al.
360 mg/kg (significant for (1976)
360 mg/kg group)
Ni(CH3CO2)2 Mice, ip Average 1.5 lung Poirier
Strain A adenomas per mouse et al. (1984)
Ni(CH3CO2)2 F344 rats sc, 150 mg/kg Injection site Teraki &
sarcomas Uchiumi (1988)
---------------------------------------------------------------------------------------------------------
Table 30 (contd.)
---------------------------------------------------------------------------------------------------------
Nickel Animals Routea Tumours Comments Reference
compound
---------------------------------------------------------------------------------------------------------
Nickel Wistar ip 46/48, 48/47 and Pott et al.
powder rats 27/42 rats with (1987)
NiO, Ni3S2 sarcomas,
mesotheliomas, or
carcinomas
Nickel Wistar ip High incidence of Pott et al.
powder rats sarcomas and (1987)
NiO, Ni3S2 mesotheliomas in
NiCl2, Ni3S2-, nickel
Ni(CH3CO2)2 powder-, nickel
NiSO4, alloy-, and NiO-
NiCO3 nickel groups
alloy
---------------------------------------------------------------------------------------------------------
a ia = intra-articular
if = intra-retroperitoneal fat
im = intramuscular
ip = intraperitoneal
iv = intravenous
sc = subcutaneous.
Nickel subsulfide is the nickel compound that has been most
studied. All routes of administration, except submaxillary gland
injection into Fischer rats and local application to the cheek
pouches of Syrian hamsters (Sunderman et al., 1978d), produced
tumours. Sunderman (1983b) and Gilman & Yamashiro (1985) reviewed
experimental data on factors influencing the carcinogenetic
activity of nickel subsulfide and other nickel compounds, including
species and strain, susceptibility of organs, route of
administration, physical form of nickel compound, and dose-response
characteristics. Dose-effect relationships could be demonstrated
for nickel subsulfide in rats following intramuscular and
intrarenal injections and in hamsters following intramuscular and
intratesticular injections. Intraocular and intramuscular routes
of administration yielded the highest tumour incidence. Local
tumours after intraocular injection included malignant melanomas,
retinoblastomas, gliomas, a phakocarcinoma, and unclassified
tumours. Following intramuscular injection, local tumours were
mostly rhabdomyosarcomas, fibrosarcomas, and undifferentiated
sarcomas. Rats are more susceptible to the induction of sarcomas
by nickel subsulfide than mice, hamsters, or rabbits, though
Sunderman (1983b) considered that absolute species susceptibility
could not be defined, because of differences in experimental
conditions. For strain differences in rats, Gilman & Yamashiro
(1985) ranked susceptibility to tumour induction by nickel
subsulfide as: Hooded > Wistar > Fischer > Sprague-Dawley.
Yarita & Nettesheim (1978) implanted nickel subsulfide pellets into
heterotopic tracheas grafted in Fischer 344 rats and reported
tracheal tumours including sarcomas (2 fibrosarcomas, 1
leiomyosarcoma) and carcinomas (5 squamous cell carcinoma, 1
undifferentiated carcinoma). Malignant testicular neoplasms
developed in Fischer 344 rats after intratesticular injection of
10 mg nickel subsulfide (Damjanov et al., 1978). Gilman & Herchen
(1963) found corresponding numbers of rhabdomyosarcomas in rats
after intramuscular implantation of Ni2S3 in implants of different
forms, injection of powder, or exposure in a diffusion chamber.
Shibata et al. (1989) injected nickel subsulfide (single injection,
0.5 mg) subcutaneously (sc), intramuscularly (im), in
retroperitoneal fat, and intra-articularly (ia), in male Fischer
rats, and observed the animals for 48 weeks. Malignant soft-tissue
tumours developed in 18/19 (sc) 19/28 (im) 9/20 (retroperitoneally),
and 16/19 (ia) animals. No synovial sarcomas were detected.
Magnesium carbonate was observed to decrease sarcoma incidence due
to Ni3S2 from 100 to 55% in a dose-related manner, when both
compounds were injected into the thigh muscles of male Fischer 344
rats (Kasprzak et al., 1987). Kasprzak et al. (1988) found that,
40 weeks after injection, zinc, either as zinc oxide or zinc
acetate, delayed the onset, but not the final incidence of local
sarcomas when injected intramuscularly with Ni3S2. All of the rats
given Ni3S2 developed tumours compared with only 60% of those that
received Ni3S2 plus zinc. Kasprzak & Rodriguez (1988) found that
iron counteracted the ability of nickel subsulfide to induce cancer
and that it inhibited catalase and glutathione peroxidase, leading
to a build up of hydrogen peroxide (H2O2), which may be related to
carcinogenesis.
Iron can decompose H2O2 so reversing the action of nickel
subsulfide. Mitsumasa (1987) administered Ni3S2 to lentectomized
Japanese common newts (Cynops pyrrhogaster). A single injection of
40-100 µg/newt produced malignant melanoma-like tumours in the
injected eyes of 7 or 8 newts.
Nickel oxide produced local tumours (mainly rhabdomyosarcomas,
fibrosarcomas, and undifferentiated sarcomas) following intramuscular
injections in rats and mice (Gilman, 1962; Sunderman, 1984) and a
few tumours (not specified) following implantation in rats (Payne,
1964). It was also carcinogenic when administered by intrapleural
injection in Wistar rats (Skaug et al., 1985). Eilertsen et al.
(1988) tested the carcinogenicity in the rat of a variety of nickel
oxides given by the intrapleural route but obtained negative
results.
When Gilman & Ruckerbauer (1962) injected nickel refinery flue
dust (20% nickel sulfate, 57% nickel subsulfide, 6.3% nickel oxide)
into the thigh muscles of rats and mice, they found a high
induction rate of sarcomas. Saknyn & Blokhin (1978) injected
"feinstein" dust (an intermediate product of nickel refining
containing nickel sulfide, nickel oxide, and metallic nickel)
intraperitoneally at a dose of 90-150 mg/rat and found sarcomas at
the injection sites in 6 out of 39 rats.
Intramuscular injection of nickel powder produced sarcomas in
rats at the injection sites (Heath & Daniel, 1964; Furst &
Schlauder, 1971; Sunderman & Maenza, 1976; Sunderman, 1984). In
hamsters, intramuscular injection of nickel powder produced
sarcomas at the site of injection, but the incidence was low (2 out
of 50 animals) (Furst & Schlauder, 1971). Intravenous injection of
nickel powder produced local sarcomas in rats, but not in mice and
rabbits (Hueper, 1955). No controls were used. Hueper (1952,
1955) injected nickel powder into the femurs of rats and rabbits
and observed sarcomas at the site of injection. Intrathoracic
injections of nickel powder produced mesotheliomas in Fischer 344
rats (Furst et al., 1973; Furst & Cassetta, 1973). Following
intraperitoneal injection of nickel powder, tumours were seen in
Fischer 344 rats (Furst & Cassetta, 1973).
Besides its carcinogenic potential in inhalation studies,
administration of nickel carbonyl to rats by repeated intravenous
injection was also associated with an increased tumour rate (Lau et
al., 1972). Malignant tumours included pulmonary lymphomas,
undifferentiated sarcomas in the lung, pleura, liver, pancreas,
uterus, and abdominal wall, fibrosarcomas in the neck, pinna, and
orbit, carcinomas of liver, breast, and kidney, and one
haemangioendothelioma.
Local sarcomas were induced in rats with repeated intramuscular
injections of crystalline nickel hydroxide, but not with colloidal
nickel hydroxideor nickel sulfate, under similar experimental
conditions (Kasprzak et al., 1983).
Implantation of 7 mg nickel carbonate hydroxide/rat produced
local sarcomas in 4 out of 35 rats (Payne, 1964).
Nickelocene, an organic nickel compound used as a laboratory
reagent, induced fibrosarcomas in rats and hamsters, when injected
intramuscularly (Furst & Schlauder, 1971).
It was reported by Stoner et al. (1976) that 24 intraperitoneal
injections of nickel acetate to strain A mice (total dose 72-360
mg/kg body weight) induced lung tumours at the highest dose.
Poirier et al. (1984) observed an increased incidence of lung
adenomas in Strain A mice following intraperitoneal administration
of nickel acetate, though these authors also observed an increase
in tumour incidence in animals treated with calcium acetate or lead
acetate. Teraki & Uchiumi (1988) reported that nickel acetate
causes injection site tumours in rats.
Single intraperitoneal doses of nickel powder, nickel
subsulfide, or an unspecified nickel oxide induced a high frequency
(46 out of 48 animals, 27 out of 42 animals, and 46 out of 47
animals, respectively) of abdominal cavity tumours in Wistar rats
(Pott et al., 1987). Pott et al. (1988) investigated nickel
carcinogenicity by intraperitoneal injection in rats and produced
a variety of responses (see Table 30). They concluded that the
model is good for screening nickel compounds and alloys for
carcinogenicity.
In a study by Sunderman (1984), 18 nickel compounds were
investigated under standardized experimental conditions to relate
their physical, chemical, and biological properties to their
carcinogenic activity. Marked differences were observed in the
incidence of sarcomas in male Fischer rats within 2 years following
single intramuscular injections of equivalent doses (14 mg
nickel/rat). The results of these bioassays are summarized in
Table 31. They provide an adequate basis for ranking the relative
carcinogenicity of different nickel compounds in animals. Sarcoma
incidences in these bioassays were significantly correlated with
the mass-fractions of nickel in the respective compounds. They
were not significantly related to the dissolution half-times of the
nickel compounds in rat serum or renal cytosol (Kuehn & Sunderman,
1982), or to the susceptibilities of the compounds to phagocytosis
by rat peritoneal macrophages in vitro (Kuehn et al., 1982). On
the other hand, there was a strong correlation between the sarcoma
incidences and the capacity of nickel compounds to induce
erythrocytosis after intrarenal administration to rats, suggesting
that nickel-stimulated production of erythropoietin and nickel
carcinogenesis may be related in some way (Sunderman & Hopfer,
1983).
Table 31. Summary of survival data and sarcoma incidences in carcinogenesis tests of 18 nickel compoundsa
-----------------------------------------------------------------------------------------------------------------------------
Category Test substance Survivors at Rats with local Median Median Rats with metastases/
2 years/total sarcomas/total tumour survival rats with sarcomas
number of rats number of rats latency period (% survival)
(% survival) (% survival) (weeks) (weeks)
-----------------------------------------------------------------------------------------------------------------------------
Controls Glycerol vehicle 25/40 (63%) 0/40 (0%) - >100 -
Penicillin vehicle 24/44 (55%) 0/44 (0%) - >100 -
All controls 49/84 (58%) 0/84 (0%) - >100 -
Class A Nickel subsulfide (alphaNi3S2) 0/9d (0%) 9/9d (100%) 30 39c 5/9 (56%)
Nickel monosulfide (BNiS) 0/14d (0%) 14/14d (100%) 40 48c 10/14 (71%)
Nickel ferrosulfide (Ni4FeS4) 0/15d (0%) 15/15d (100%) 16 32c 10/15 (67%)
Class B Nickel oxide (NiO) 0/15d (0%) 14/15d (93%) 49 58c 4/14 (29%)
Nickel subselenide (Ni3Se2) 0/23d (0%) 21/23d (91%) 28 38c 18/21 (86%)
Nickel sulfarsenide (NiAsS) 0/16d (0%) 14/16d (88%) 40 57c 10/14 (71%)
Nickel disulfide (NiS2) 0/14d (0%) 12/14d (86%) 36 47c 6/12 (50%)
Nickel subarsenide (Ni5As2) 0/20d (0%) 17/20d (85%) 22 44c 9/17 (53%)
Class C Nickel dust 4/20c (20%) 13/20d (65%) 34 42c 6/13 (40%)
Nickel antimonide (NiSb) 9/29c (31%) 17/29 (59%) 20 66c 10/17 (59%)
Nickel telluride (NiTe) 12/26 (46%) 14/26d (54%) 17 80c 8/14 (57%)
Nickel monoselenide (NiSe) 7/16 (44%) 8/16d (50%) 56 72c 3/8 (38%)
Nickel subarsenide (Ni11As8) 5/16b (31%) 8/16d (50%) 33 88c 6/8 (75%)
Class D Amorphous nickel monosulfide 5/25c (20%) 3/25c (12%) 41 71c 3/3 (100%)
(NiS)
Nickel chromate (NiCrO4) 10/16 (63%) 1/16 (6%) 72 >100 1/1 (100%)
Class E Nickel monoarsenide (NiAs) 13/20 (65%) 0/20 (0%) - >100 -
Nickel titanate (NiTiO3) 11/20 (55%) 0/20 (0%) - >100 -
Feronickel alloy (NiFe1,6) 11/16 (75%) 0/20 (0%) - >100 -
----------------------------------------------------------------------------------------------------------------------------
a From Sunderman (1984d).
b P < 0.05 versus corresponding vehicle controls.
c P < 0.01 versus corresponding vehicle controls.
d P < 0.001 versus corresponding vehicle controls.
Six nickel oxides and 4 nickel-copper oxides were categorized
according to the criteria shown in Table 32 (Sunderman et al.,
1987a). These compounds were assayed as follows: (a) in vitro
dissolution in water and body fluids; (b) in vitro phagocytosis
tests in Chinese hamster ovary and C3H-10T1/2 cells; (c)
morphological transformation and cytotoxicity tests in cultured
Syrian hamster embryos (SHE) cells; (d) erythropoiesis stimulation
assay by intrarenal administration to Fischer 344 rats; and (e)
scoring the renal histopathological responses in rats killed 3
months after injection. The extent of agreement among the
physical, chemical, and biological attributes of the test compounds
was tested by Kendalls' non-parametric test for concordance of
rankings. On the basis of a highly significant concordance of
ranked results in the assays ( P <0.001), 6 biological attributes of
the compounds were identified: (i) dissolution half-times in rat
serum and renal cytosol; (ii) phagocytosis by C3H-10T1/2 cells;
(iii) morphological transformation of SHE cells; (iv)
erythropoiesis stimulation in rats; (v) induction of tubular
hyperplasia in rat kidneys; and (vi) induction of arteriosclerosis
in rat kidneys. A strong rank correlation between results of the
cell transformation and erythropoiesis stimulation was observed.
The presence of high surface area and demonstrable Ni(III) were
the two physicochemical characteristics associated with the
greatest biological effects of the nickel oxides.
Compounds A, B, and F (nickel oxides) and compounds H and I
(nickel-copper oxides) were selected for carcinogenesis testing in
male Fischer 344 rats (20 mg, intramuscular injection, right thigh)
(Sunderman et al., 1988a). Injection site sarcomas (524 months)
developed in 6 out of 15 rats that received compound A (INCO black
NiO), versus 0 out of 15 rats that received compound B (NiO
calcined at 735 °C), and 0 out of 15 rats that received compound F
(NiO calcined at 1045 °C). Sarcomas developed in 13 out of 15 rats
that received compound H (NiO/CuO, 2.5/1 ratio) and 15 out of 15
rats that received compound I (NiO/CuO, 5.2/1 ratio). Injection
site sarcomas developed in 0 out of 15 vehicle control rats and 15
out of 15 positive controls that received intramuscular injection
of Ni3S2.
8.4.4. Interactions with known carcinogens
Several studies have been performed to investigate the possible
interactions of different nickel species with known carcinogens,
either in classical two-stage carcinogenesis studies, or using
simultaneous administration.
In a two-stage carcinogenesis assay, orally administered nickel
chloride enhanced the renal carcinogenicity of N-ethyl- N-hydroxyethyl
nitrosamine in rats, but not the hepatocarcinogenicity of N-
nitrosodiethylamine, the gastric carcinogenicity of N-methyl-
N-nitro- N-nitrosoguanidine, the pancreatic carcinogenicity of N-
nitrosobis(2-oxopropyl)amine, or the skin carcinogenicity of 7,12-
dimethylbenzanthracene (Hayashi et al., 1985).
Table 32. Physical and chemical characteristics of some test compounds: 6 Ni oxides and 4 Ni-Cu oxidesa
-------------------------------------------------------------------------------------------------------------------------
Test Calcin- Surface Elemental composition (% by weight) Constitutents and Relative
compound ation area speciation (% by weight) height of
and colour temp. (m2/g) XRD peaks
(°C) Ni Cu Ob S Fe Co Ni(III) SO4 Ni3S2 CuO NiOc for Ni
oxidesd
-------------------------------------------------------------------------------------------------------------------------
A (jet black) <650 41.5 77.7 <.001 22.3 <0.02 <0.01 <0.01 0.81 <0.06 NDe ND SSf 0.31
B (grey-black) 735 3.0 78.1 <.001 21.9 <0.02 <0.01 <0.01 0.05 <0.06 ND ND SS 0.74
C (grey) 800 1.4 78.2 <.001 21.8 <0.02 <0.01 <0.01 0.03 <0.06 ND ND SS 0.87
D (grey) 850 0.7 78.2 <.001 21.8 <0.02 <0.01 <0.01 <0.03 <0.06 ND ND SS 0.88
E (dark green) 918 0.2 78.4 <.001 21.6 <0.02 <0.01 <0.01 <0.03 <0.06 ND ND SS 0.90
F (green) 1045 <0.2 78.6 <.001 21.4 <0.02 <0.01 <0.01 <0.03 <0.06 ND ND SS 1.00
G (dark maroon) 850 <0.2 43.8 27.9 23.4 0.69 2.98 1.17 g 1.86 0.3 14 77
H (maroon) 850 <0.2 51.7 20.6 23.6 1.02 2.02 1.09 g 2.67 0.5 3 89
I (red-brown) 850 <0.2 62.6 12.6 22.3 0.30 1.20 1.04 g 0.06 1.0 ND 95
J (brown) 850 <0.2 68.8 6.9 22.1 0.42 0.75 0.97 g 0.06 1.5 ND 96
-------------------------------------------------------------------------------------------------------------------------
a From: Sunderman et al. (1987a)
b Estimated by subtracting the percentages for other elemental constituents from 100%.
c NiO (bunsenite) can accommodate up to 25% CuO (tenorite) as a solid solution within its crystal lattice at 850C.
d Heights of XRD peaks of NiO (bunsenite) at 1.48, 2.09, and 2.41 angstrom, relative to peak heights obtained with
compound F.
e ND = not detected by XRD.
f SS = sole species detected by XRD.
g Presence of sulfur in compounds G-J precluded assays for Ni(III).
Single intratracheal injections of nickel subsulfide did not
increase the carcinogenicity of benzo (a)pyrene, also instilled
intratracheally, in rats (Kasprzak et al., 1973).
Metallic nickel powder seemed to enhance the lung
carcinogenicity of 20-methylcholanthrene (MC) when both were
administered intratracheally to rats. The tumour incidences in
different groups, 12 weeks after the instillation were: (i) 5 mg
MC: 2/7; (ii) 5 mg MC + 10 mg Ni: 3/5; (iii) 1 mg MC: 1/8; (iv) 1
mg MC + 10 mg Ni 2/7; and (v) 10 mg Ni only: 0/7 (Mukubo, 1978).
Nickel oxide, administered intratracheally, did not enhance the
carcinogenic response to diethylnitrosamine in the lungs or nasal
cavity of hamsters (Farrell & Davis, 1974).
In studies on combined intramuscular injection of nickel
subsulfide and benzo (a)pyrene, latency times to local tumour
development were shorter, and the proportion of malignant tumours
higher, than when either carcinogen was injected alone (Maenza et
al., 1971).
In a two-stage carcinogenicity study, Ou et al. (1981) gave a
single injection of dinitrosopiperazine (9 mg) to rats, followed by
administration of nickel sulfate, either in the drinking-water (3.7
mg/day), or topically in the nasopharynx as a gelatin solution
(0.02 ml of 0.5% NiSO4). Two out of 12 rats in both groups
receiving both nickel and the nitrosamine developed nasopharyngeal
tumours (1 squamous cell carcinoma, 1 fibrosarcoma, 1 papilloma, 1
"early carcinoma"), while none was seen in the 24 animals treated
with the nitrosamine alone, or in the two groups of 12 animals
treated with nickel sulfate. The same authors (abstract only)
reported an extension of the study, using topical nickel sulfate as
a promoter. Five out of 22 rats given the combined treatment
developed carcinomas of the nasopharynx, nasal cavity, or hard
palate, while these tumours were not found in the other groups (Liu
et al., 1983).
Ou et al. (1983) reported a study in which N-nitrosopiperazine
was given to female rats on the 18th day of pregnancy, and the pups
were given nickel sulfate orally. Five out of 21 pups with
combined treatment, and 1 out of 4 with nitrosamine treatment only
developed nasopharyngeal carcinoma, and two other pups developed
other tumours.
8.4.5. Possible mechanisms of nickel carcinogenesis
Both in vivo and in vitro studies have shown varying
carcinogenic responses, depending on the bioavailability of certain
nickel compounds. Several factors influencing bioavailability have
been suggested, such as the physical form of the compound
(crystalline or amorphous), solubility in water or serum, and its
ability to be phagocytosed by target cells (Costa et al., 1982) or
pass through calcium channels (Sunderman, 1989a). In vitro studies
have shown that various nickel compounds had similar transforming
potencies at equitoxic concentrations (Hansen & Stern, 1984).
The transformation potency of a number of particulate nickel
compounds has been correlated with phagocytosis (Costa et al.,
1981b). The incidence of sarcomas and the capacity of nickel
compounds to produce erythrocytosis are also related (Sunderman &
Hopfer, 1983; Sunderman et al., 1987a). Erythrocytosis can be
induced in rats by intrarenal injection of nickel subsulfide,
probably due to increased production of the stimulating hormone
erythropoietin (Jasmin & Ripolle, 1976; Morse et al., 1977; Hopfer
et al., 1978). Intrarenal injection of nickel subsulfide caused
renal cancers (Sunderman et al., 1984b). Induction of
erythrocytosis and carcinogenesis in rats by nickel subsulfide were
both inhibited by the administration of manganese dust (Sunderman
et al., 1976b). In vitro studies have demonstrated that nickel
compounds cause (a) DNA damage in a wide variety of organisms, (b)
mutagenicity, possibly related to cytotoxicity, in mammalian cells,
and (c) clastogenicity and morphological transformation in
mammalian cells including human cells. From in vitro and in vivo
studies, it is known that nickel interacts with DNA and proteins,
causing lesions, such as DNA-protein (possibly heterochromatin)
cross-links and DNA single strand breaks (Ciccarelli et al., 1981;
Ciccarelli & Wetterhahn, 1982; Robison et al., 1982; Patierno et
al., 1985), resulting in DNA damage and other cytotoxic effects
that can lead to altered gene expression in surviving cells. For
example, Sen & Costa (1986) showed that nickel preferentially
induced decondensation (presumably by DNA-protein cross-linking)
and fragmentation of the long arm of the X-chromosome in CHO cells.
Other consequences of cross-linking, such as the appearance of
irreversible, but heritable, lesions of intermediate filaments (a
family of cytoskeletal proteins linking the nucleus with the cell
membrane), related to disturbance of normal growth control
processes, have been observed early after nickel intoxication in
vitro (Swierenga & McLean, 1985; Swierenga et al., 1987, 1989).
Nickel-induced DNA damage/alterations that have been reported
include: conformational transitions of the DNA from the normal
right-handed B-helix to the left-handed Z helix (van de Sande et
al., 1982; Boutayre et al., 1984), infidelity of DNA synthesis, as
shown with cell-free systems using synthetic templates (Sirover &
Loeb, 1977; Zakour et al, 1981), and inhibition of DNA synthesis in
a wide variety of organisms extending to inhibition of DNA excision
repair (Table 27). The latter may be an important mechanism in the
ability of nickel to enhance the carcinogenic activity of other
compounds (Maenza et al., 1971; Kurokawa et al., 1985). Other in
vitro evidence indicates that nickel disrupts intercellular
communication, a property of tumour promoters (Miki et al., 1987).
Studies by Nieboer et al. (1986) indicated that, in the
presence of certain peptides, the Ni3+/Ni2+ redox couple can
participate in dioxygen radical reactions in vitro. As chemical
promoters of cancer appear to have the capacity to stimulate
phagocytic cells to produce dioxygen radicals, which may damage
DNA, proteins and lipids, and as metal ions catalyze processes
involving molecular oxygen, this observed effect of nickel may have
implications for nickel carcinogenesis.
Sunderman & Barber (1988) have suggested a mechanism for the
interaction of nickel with DNA. This model proposes that the Zn2+
binding sites of DNA binding proteins known as "finger loop
domains", which have been identified on some proto-oncogens, are
likely molecular targets for metal toxicity. As Ni2+ has a similar
ionic radius to Zn2+, substitution is possible, interfering with
regulation of gene expression. The replacement of Zn2+ in finger
loops might affect the conformation and stability of DNA-binding
structures interfering with expression and induce site-specific
free radical reactions, causing cleavage of DNA, formation of DNA-
protein cross links, and disturbances of mitosis.
8.4.6. Factors influencing nickel carcinogenesis
Maenza et al. (1971) reported a synergistic action between
nickel subsulfide and benzo (a)pyrene. After intramuscular
injection, the latency period for sarcoma induction was
significantly reduced. Kasprzak et al. (1973) found an increased
incidence of premalignant changes in the lungs of rats receiving
intratracheal injections with a mixture of benzo (a)pyrene and
nickel subsulfide compared with groups receiving one compound at a
time.
Manganese in metallic form has been shown to antagonize the
tumourigenic effect of nickel subsulfide by intramuscular injection
(Sunderman et al., 1976b) and intrarenal injection (Sunderman et
al., 1979b). Kasprzak & Poirier (1985) found that simultaneous
intramuscular injection of magnesium carbonate inhibited the
development of muscle tumours induced by nickel subsulfide.
Calcium carbonate, under the same experimental conditions, was
ineffective. Poirier et al. (1984) reported that both magnesium
acetate and calcium acetate inhibited lung adenoma formation in
Strain A mice treated with intraperitoneal nickel acetate.
Manganese, magnesium, calcium, copper, and zinc were shown by
Kasprzak & Poirier (1985) and Kasprzak et al. (1986) to inhibit
binding of nickel to DNA. They also found that amino acids were
very effective inhibitors of nickel binding to DNA, but their
eventual role in moderating nickel carcinogenicity has not been
investigated.
When rats with muscular implantations of nickel subsulfide were
injected intraperitoneally with sodium diethyldithiocarbamate,
tumour incidence was significantly reduced compared with that in
rats that had received the nickel subsulfide implant alone
(Sunderman et al., 1984b).
9. EFFECTS ON HUMAN BEINGS
9.1. Systemic effects
9.1.1. Acute toxicity - poisoning incidents
9.1.1.1 Nickel carbonyl
In terms of human health, the most acutely toxic nickel
compound is nickel carbonyl. The clinical manifestations of nickel
carbonyl poisoning have been described in detail by Sunderman
(1970) and Vuopala et al. (1970). A summary of 179 cases of nickel
carbonyl poisoning in China since 1961 has been published by Shi
(1986).
The acute toxic effects occur in 2 stages, immediate and
delayed. The immediate symptomatology includes frontal headache,
vertigo, nausea, vomiting, insomnia, and irritability, followed by
an asymptomatic interval before the onset of delayed pulmonary
symptoms. These include constrictive chest pains, dry coughing,
dyspnoea, cyanosis, tachycardia, occasional gastrointestinal
symptoms, sweating, visual disturbances, and weakness. The
symptomatology resembles that of a viral pneumonia. In men who
died, pulmonary haemorrhage and oedema or pneumonitis were observed
accompanied by derangement of alveolar cells, degeneration of the
bronchial epithelium, and the appearance of a fibrinous intra-
alveolar exudate. The pathology of the pulmonary lesions was
similar to that observed in animal studies. Other affected organs
included the liver, kidneys, adrenal glands, and spleen, where
parenchymal degeneration was observed. Cerebral oedema and
punctate cerebral haemorrhages were noted in men dying after
inhalation of nickel carbonyl. Shi (1986) reported leukocytosis in
25 out of 179 cases. Urinary nickel concentrations were determined
in 27 cases and ranged from 0.003 to 0.66 mg/litre. Air
concentrations were measured and were reported to have exceeded 50
mg nickel carbonyl /m3 with exposure periods ranging from 30 min to
more than 2 h. Recovery time was 7-40 days, depending on the
severity of exposure. In some cases, symptoms persisted for 3-6
months. In lethal cases, death occurred between the third and
thirteenth days following exposure.
9.1.1.2 Other nickel compounds
Information on poisoning with other nickel compounds is
limited. Daldrup et al. (1983) reported a case of fatal nickel
poisoning. A 2 1/2-year-old girl ingested about 15 g of nickel
sulfate crystals. On admission to hospital, she was somnolent with
wide and unresponsive pupils, a high pulse rate, and pulmonary
rhonchi. Cardiac arrest occurred after 4 h. Autopsy findings
revealed increased nickel levels (7.5 mg/kg in blood, 50 mg/litre
in urine, 25 mg/kg in liver tissue). Nickel poisoning was reported
in a group of 23 dialysed patients, when leaching from a nickel-
plated stainless steel water heater tank contaminated the
dialysate. Symptoms occurred during, and after, dialysis, at
plasma-nickel concentrations of approximately 3 mg/litre. Symptoms
included nausea (37 out of 37), vomiting (31 out of 37), weakness
(29 out of 37), headache (22 out of 37), and palpitation (2 out of
37). Recovery occurred spontaneously, generally 3-13 h after
cessation of dialysis (Webster et al., 1980).
Thirty-two workers in an electroplating plant accidentally
drank water contaminated with nickel sulfate and nickel chloride
(1.63 g nickel/litre). Twenty workers rapidly developed symptoms
(e.g., nausea, vomiting, abdominal discomfort, diarrhoea,
giddiness, lassitude, headache, cough, shortness of breath) that
typically lasted a few hours, but persisted for 1-2 days in 7
cases. The nickel doses in workers with symptoms were estimated to
range from 0.5 to 2.5 g. In 15 exposed workers, who were
investigated on day 1 after exposure, serum-nickel concentrations
ranged from 12.8 to 1340 µg/litre (average 286 µg/litre) with
urinary nickel concentrations ranging from 0.23 to 37.1 mg/litre
(average 5.8 mg/litre) compared with control values for nickel-
plating workers of 2.0-6.5 µg/litre (average 4.0 µg/litre) for
serum nickel and 22-70 µg/litre (average 50 µg/litre) for urinary
nickel. Laboratory tests showed transiently elevated levels of
blood reticulocytes (7 workers), urine-albumin (3 workers), and
serum-bilirubin (2 workers) (Sunderman et al., 1988b).
9.1.2. Short- and long-term exposure
9.1.2.1 Respiratory effects
A chemical engineer, who had been exposed for a long period to
low levels of nickel carbonyl, developed asthma and Löffler's
syndrome. In addition to pulmonary infiltrations and eosinophilia,
which are markers in Löffler's syndrome, the patient had an
eczematous dermatitis of the hands (Sunderman & Sunderman, 1961b).
There have been systematic investigations of the health state
of the respiratory tract of workers in the nickel industry.
Tatarskaya (1960) examined 486 workers from nickel-refining plants,
exposed mainly to nickel sulfate during electrorefining operations.
Exposure levels were not given. Rhinitis was observed in 10-16%,
chronic rhinitis in 5.3%, nasal septal erosions in 13%,
perforations in 6.1%, and ulceration in 1.4% of the workers. The
frequencies of hypo-osmia and anosmia were 30.6 and 32.9%,
respectively. Kucharin (1970) studied 302 workers who had been
exposed to nickel sulfate, accompanied by sulfuric acid vapour, for
at least 10 years, at concentrations of 0.02-4.5 mg/m3. Clinical
and radiological examination revealed chronic sinusitis in 83% of
the workers. Among these workers, severe injuries were apparent,
such as septal erosion and perforation in 41.4 and 5.6%, respectively.
Hypo- or anosmia was noted in 46% of the workers with chronic
sinusitis. In a nickel refinery where hydrometallurgical methods
were used, 37 out of 151 workers (24.5%) exhibited pathological
changes of the nasopharynx. Septal erosions were noted in 14
workers (9.3%). Exposure concentrations of soluble nickel salts,
during the years 1966 and 1970, ranged from 0.035 to 1.65 mg/m3
(Sushenko & Rafikova, 1972).
Other studies on the nasopharyngeal effects of inhalation of
nickel salt aerosols, conducted in order to detect precancerous
lesions, are described in the section 9.5.
Data are available on the development of pulmonary changes with
fibrosis in workers inhaling nickel dust or fumes. Zislin et al.
(1969) studied respiratory function in 13 workers, who had been
exposed to nickel dust for periods of 12.9-21.7 years. Exposure
levels were not given. There was decreased pulmonary residual
capacity, increased respiratory frequency, and radiography showed a
diffuse fibrosis, considered to be nickel pneumoconiosis.
Industrial dusts, collected in the workrooms of nickel
refineries, induced the development of pneumoconiosis of an
interstitial type, together with bronchitis, in experimental
animals (Saknyn et al., 1978; Grebnev & Yelnichnykh, 1983). The
fibrogenicity of these dusts did not depend on their silica
content, and the dust that was almost pure nickel suboxide (Ni2O3)
was fibrogenic, when instilled intratracheally. Pneumoconiosis of
an interstitial and micronodular type had been detected in a number
of workers in the same industry (Zislin et al., 1969).
Peto et al. (1984) reported mortality experience in Welsh
nickel refinery workers. Men first employed before 1925, who had
worked for 5 or more years in the process areas where lung and
nasal cancer risks were highest, also suffered a significantly
elevated mortality from non-malignant respiratory disease (20
deaths, 11.1 expected); less heavily exposed workers suffered no
such excess (43 deaths, 51.2 expected).
Jones & Warner (1972) studied respiratory symptoms, chest X-
rays, and lung ventilation capacities in 12 steelworkers, who had
been employed as de-seamers of steel ingots for periods of up to 16
years. Total fume concentration at the workplace ranged from 1.3
to 294.1 mg/m3, either from iron oxide, chromium oxide, and nickel
oxide in proportions of 6:1:1 (stainless steel fume) or 98.8% iron
oxide (special steel fume). Airborne nickel oxide concentrations
of 0.15-34 mg/m3 were calculated from these values. Two of the
workers had measurable loss of pulmonary function. In 5 men,
definite signs of pneumoconiosis were detected radiographically.
Effects on the respiratory system have also been observed in
welders. Between 1955 and 1979, Zober (1981a,b) found a total of
47 cases of histologically verified fibrotic changes in the lungs
of electric arc welders. Though the findings were very
heterogeneous, indicating a diversity of causes, welding fumes were
found to be a concomitant cause of fibrotic changes in 20 cases.
Zober (1982) studied 40 welders (22, non-smoking and 18, smoking),
who were using metal inert gas and electric arc welding techniques
on chromium- and nickel-containing materials. Concentrations of
airborne nickel did not exceed 0.5 mg/m3, except in one working
process, where there was a concentration of 1.2 mg nickel/m3.
Respiratory-tract effects, including acute bronchitis and
bronchitic pulmonary rhonchi, were increased, mainly in welders who
smoked tobacco.
Asthmatic lung disease in nickel-plating workers exposed to
soluble nickel has been reported (Tolot et al., 1956; McConnell et
al., 1973; Block & Yeung, 1982; Malo et al., 1982; Dolovich et al.,
1984). Cirla et al. (1985) examined 12 workers with respiratory
problems in a nickel-plating industry. In 6 workers, allergic
asthma could be provoked by the inhalation of nickel sulfate
aerosols. Novey et al. (1983) reported that a metal-plating
(chromium and nickel) worker developed acute asthma in response to
inhalational challenge with nickel sulfate and chromium sulfate.
Radioimmuno-assays incorporating the challenge materials revealed
specific IgE antibodies to the provocative agents.
Five stainless steel welders, suffering from respiratory
distress, developed asthmatic symptoms during provocation tests
with stainless steel fumes (Keskinen et al., 1980).
Inhalation testing using 0.01-0.001% nickel chloride solution
has been carried out to detect hypersensitivity to nickel and its
compounds in patients occupationally exposed to nickel and
suffering from atopic bronchial asthma. In inhalation testing, it
is necessary to take pneumotachometric readings before, and after,
the test, at various times over a period of 24 h (Izmerov, 1983).
9.1.2.2 Renal effects
Sanford et al. (1988) assessed different parameters for kidney
dysfunction in urine samples from electrolytic nickel refinery
workers (renal clearance, protein, specific gravity, and
qualitative "dipstick" tests). In male workers who accidentally
ingested drinking-water contaminated with nickel sulfate and
chloride, Sunderman et al. (1988b) found elevated urine-albumin
concentrations (68, 40, and 27 mg/g creatinine), which returned to
normal on the fifth day after exposure. The findings suggested a
mild transient nephrotoxicity (section 9.1.1).
9.1.2.3 Cardiovascular effects
Patients with acute myocardial infarction and unstable angina
pectoris develop high serum-nickel levels, which may be followed by
coronary vasoconstriction (Leach et al., 1985). The mechanism and
source of nickel release are not yet known, but Sunderman (1983a)
recommended that the amount of nickel in intravenous solutions
should not exceed 5 µg/kg per day. Peto et al. (1984) observed
elevated cerebrovascular mortality (16 deaths, 8.5 expected) among
nickel refinery workers who had worked for 5 years in the process
area, where lung and nasal cancer rates were highest. Mortality
from all other cardiovascular causes was similar to local
population rates.
9.1.2.4 Other effects
In a study by Shi et al. (1986), nickel carbonyl refinery
workers were compared with a control group. Exposure ranged from
0.007 to 0.52 mg Ni(CO4)/m3. A decrease in the MAO (serum
monoamine oxidase) activity, and EEG abnormalities were observed in
the most severely exposed. A number of non-specific symptoms in
persons exposed for long periods to low concentrations
(unspecified) were reported, but no details were given.
Immune reactions elicited in the sera of individuals exposed to
nickel and cobalt were assessed according to changes in the
concentrations of the serum immunoglobulins, IgG, IgA, and IgM, and
the serum proteins, alpha2 macroglobulin (A2M), transferin (TRF),
alpha1-antitrypsin (A1AT), ceruloplasmin (CPL), and lysozyme
(LYS) (Bencko et al., 1983). Examinations were carried out on
workers occupationally exposed to nickel (38 individuals) or cobalt
(35 individuals) and in groups of non-occupationally exposed
children living in areas with different levels of air pollution
from a nearby source of nickel and cobalt emissions.
Non-exposed controls were represented by a group of 42 male
adults, matched by age, and by a group of 48 children from a non-
polluted area. Significantly increased average values were
obtained for IgG, IgA, and IgM, in the group of workers exposed to
nickel, for IgA, in workers exposed to cobalt, and for A1AT, A2M,
CPL and LYS, in both groups of occupationally-exposed adults
( P >0.001 and P >0.005, respectively). Among non-occupationally
exposed children, the most highly exposed group had significantly
elevated average values for A2M and A1AT, which were higher than
those recorded in the groups of "less exposed" and control children
( P >0.02 and P >0.05, respectively) (Bencko et al., 1983).
9.2. Skin and eye irritation and contact hypersensitivity
9.2.1. Skin and eye irritancy
Primary skin irritation reactions to nickel salts in solution
were observed when human volunteers were patch tested. When
aqueous solutions of nickel chloride were applied to the back, the
threshold concentrations for irritancy were 1% with occlusion and
10% without occlusion (Vandenberg & Epstein, 1963).
A 5% aqueous solution of nickel sulfate was also irritant in
some individuals, when applied to the back, under occlusion (Kalimo
& Lammintausta, 1984). On the unoccluded human forearm, the
threshold concentrations of aqueous nickel sulfate producing
irritation were 20% on unbroken skin and 0.13% on scarified skin,
the nickel solutions being applied once daily for 3 days (Frosch &
Kligman, 1976).
9.2.2. Contact hypersensitivity
Nickel and its water-soluble salts are potent skin sensitizers.
Nickel ions pass the skin barrier and bind to carrier proteins to
form the allergen. In the pathogenesis of allergic contact
dermatitis (a type IV reaction), two steps can be recognized.
First, sensitization, after which, subsequent exposure of fully
sensitized skin, even to minute quantities of nickel or its water-
soluble salts, will elicit or provoke an eczematous response which
begins within 12 h, is fully developed at 48-72 h, and then
subsides. After the elicitation phase, the sensitivity will
remain, usually for years, and frequently for a lifetime.
Sensitization is only known to occur after dermal exposure to
nickel, whereas provocation can take place after exposure of the
skin and after ingestion of nickel. The clinical manifestations of
nickel-associated allergic contact dermatitis comprise both primary
and secondary eruptions. The primary eruptions usually appear at
the site of sensitization, and the secondary eruptions will
frequently be more or less generalized, often symmetrical,
maculopapular vesicules of the pompholyx type eczema, which affects
the flexures of the elbows, the sides of the neck, the axillae, the
eyelids, and the genital area. A vesicular hand eczema can also
develop. Nickel contact dermatitis has been documented in the
general population and in nickel workers, especially in the nickel-
plating industry (NAS, 1975). Nickel contact hypersensitivity was
originally considered to be a problem in occupational medicine, but
more recent epidemiological and clinical data indicate that it
could also be a problem in the general population, because of the
increasing number of nickel-containing commodities in everyday use.
The prevalence of nickel sensitivity in the general population can
be evaluated by patch-testing with 5% nickel sulfate in petrolatum
or by an interview technique. Peltonen (1979) and Prystowsky et
al. (1979) studied volunteers from different subpopulations in
Finland and the USA. The prevalence of nickel sensitivity was
about 10% for women and about 1% for men. In women, Prystowsky et
al. (1979) found a high correlation of sensitivity to nickel with
the practice of ear piercing. Peltonen (1979) reported that hand
eczema is very common in patients with nickel sensitivity.
Christensen & Möller (1975a) reported that pompholyx was the
predominant type of hand eczema. Nickel allergy was investigated
in a stratified sample of the Danish female population using an
interview technique (Menné et al., 1982). Of those responding,
14.5% reported a history of nickel allergy. The prevalence rate
was highest in the younger age groups and declined after the age of
50 years. Although the use of the interview technique had certain
limitations, the data were in good agreement with data obtained
through other test methods (e.g., patch testing).
Edman & Möller (1982) reported a study on a patient population
of 8933, who had been patch-tested over a 12-year period. Nickel
sensitization increased during this period in both male and female
patients, females producing a higher rate of positive reactions
than males.
Oral provocation of hand eczema in nickel-allergic patients
with vesicular hand eczema has been studied (Nielsen, in press).
Oral provocation, using nickel sulfate in lactose capsules, was
performed by Christensen & Möller (1975), Kaaber et al. (1978),
Cronin et al. (1980), Jordan & King (1979), Burrows et al. (1981),
and Gawkrodger et al. (1986). The last three studies were double-
blind, weeks with provocation and weeks with placebo following each
other randomly. Evaluation of the eczema was performed in the 3
days following exposure and it was assumed that no delayed response
occurred. The doses ranged from 0.5 to 5.6 mg nickel; lower doses
were used in the double-blind studies. An exacerbation of the
eczema was observed with doses of 2.5 mg nickel or more. The
response to nickel sulfate in lactose differed from nickel
contained in the diet. A provocation study was performed with a
diet naturally containing about 500 µg nickel. Two conclusions were
drawn: (i) a diet naturally high in nickel content (chocolate cake,
soya bean stew, and oatmeal) resulted in an exacerbation of
vesicular hand eczema, when ingesting the diet daily for 4 days,
(ii) exacerbation of hand eczema was observed on day 7 after
finishing the 4-day provocation period. This delayed reaction may
be the reason why reactions to nickel were not reported in previous
double-blind studies in which weeks with nickel exposure and weeks
with placebo randomly followed each other.
The response to oral intake of nickel salts may not be uniform
and hyposensitization may be possible. In 2 controlled studies,
doses of 5.0 mg nickel sulfate given once a week, for 6 weeks,
significantly lowered the degree of contact allergy, as measured by
patch test reactions before, and after, nickel administration
(Sjövall et al., 1987). Further adaptation to oral doses of 2.24
mg nickel sulfate was observed after the volunteer subjects were
given gradually increasing daily doses over a 3-month period
(Santucci et al., 1988). These reported responses to the oral
intake of inorganic nickel do not necessarily contradict one
another, but the mechanisms are not known. However, they indicate
that hand eczema can benefit from a diet low in nickel.
Allergic reaction as well as urticarial and eczematous
dermatitis in nickel-sensitive persons can be produced by implanted
prostheses and other surgical devices made from nickel-containing
alloys (NAS, 1975; Fisher, 1977). Deutman et al. (1977) found
nickel sensitivity in 11 out of 85 patients following hip
prosthesis implantation. Three of the patients had had previous
implants. Oakley et al. (1987) described 4 patients with a history
of metal intolerance, who developed dermatitis following surgical
wound closure with disposable skin clips (nickel content about
10%). Two positive reactions were reported by Waterman & Schrik
(1985) in 85 patch-tested patients, who had undergone hip
arthroplasty.
Metal alloys, used in dental surgery, contain various amounts
of nickel. Data on the relationship between nickel allergy and
dental alloys are limited. In a retrospective study, Moffa et al.
(1983) found that the incidence of nickel allergy in patients with
intraoral exposure to nickel alloys was not significantly higher
than that in patients with no intraoral exposure.
Treatment of nickel-sensitive patients who were suffering from
hand eczema of the pompholyx type, with the nickel-chelating agent
diethyldithiocarbamate was successful in 10 out of 11 patients
(Christensen & Kristensen, 1982). However, hepatotoxicity was
observed in some patients, and this form of treatment is no longer
used.
9.3. Reproduction, embryotoxicity, and teratogenicity
No data are available on the reproductive and developmental
effects of nickel in human beings.
9.4. Genetic effects in exposed workers
Elevated levels of chromosome aberrations, mainly gaps, were
reported by Waksvik & Boysen (1982) in 2 groups of nickel workers,
but SCE was not observed; one group was involved in nickel
crushing/roasting/smelting processes and exposed mainly to nickel
oxide and nickel sulfide, and the other was involved in
electrolysis and exposed mainly to nickel chloride and nickel
sulfate. In a further study on 9 ex-nickel workers, who had been
exposed for an average duration of 25 years, but retired for at
least 8 years, Waksvik et al. (1984) showed the persistence of a
low level of gaps, as well as chromatid breaks, when plasma nickel
levels were still 2 µg/litre. Deng et al. (1983, 1988) showed
elevated levels of both SCEs and chromosome aberrations (gaps,
breaks, fragments) in electroplating workers. However, these
effects were not observed in a study by Decheng et al. (1987) on
workers exposed to nickel carbonyl. Low exposure and good
workplace conditions were cited by the latter authorities.
9.5. Carcinogenicity
9.5.1. Epidemiological studies
9.5.1.1 Nickel refining industry
(a) Clydach nickel refinery, Clydach (Wales)
The existence of an increased risk of nasal cancer in workers
from the Clydach nickel refinery was recognized in 1928. Processes
at Clydach involved: the crushing and grinding of nickel copper
matte, received from Canada, which consisted of the phases Ni3S2,
CuS, and metallic nickel and copper, in a 3:4:1 ratio (Boldt &
Queneau, 1967); the calcining of the crushed matte to produce
nickel and copper oxides (the copper oxide is dissolved in the
metal oxide and described as (Ni,Cu)O (Boldt & Queneau, 1967)); the
extraction of the copper by sulfuric acid; reduction to nickel
oxide; and refining with nickel carbonyl. The first
epidemiological study on Clydach plant workers was carried out by
Hill in 1939 (quoted by Morgan, 1958). Hill found that the
observed to expected ratios (O/E) of deaths were 16:1 for lung
cancer and 22:1 for nasal cancer in the period 1929-38. Morgan
(1958) and Doll (1958) extended the observation period from 1902 to
1957 and identified 131 deaths from lung cancer and 61 deaths from
nasal cancer in 9340 workers. Excess mortality occurred only in
workers first employed before 1924. The highest risk of lung
cancer appeared to have occurred in the calcination and copper
sulfate departments. Deaths from nasal cancer occurred most
frequently in the calcination department. There were no nasal
cancers among workers entering employment after 1924. The decline
in the numbers of deaths from nasal and lung cancer was attributed
to improvements in work practices and refinery processes in the
early 1920s. These improvements included the wearing of gauze
masks, improvements in ventilation, and the use of arsenic-free
sulfuric acid in refining. Follow-up studies were carried out by
Doll et al. (1970) on a cohort of 819 workers, who had been
employed for at least 5 years at Clydach between 1902 and 1944, and
who were still employed in 1934. Thirty-nine cases of nasal cancer
and 113 cases of lung cancer were identified. The reported O/E
ratio for lung cancer was 10.5 for workers starting between 1910
and 1915, 1.8 for workers employed between 1925 and 1929, and 1.1
for those employed between 1930 and 1944. In this study, no nasal
cancer was reported in workers starting employment after 1925.
Doll et al. (1977) extended the follow-up period to 1971 and
confirmed that, since 1925, the risk of respiratory cancer had
decreased sharply; they attributed this to the improvements in work
practices and refinery processes.
Cuckle et al. (1980) studied the causes of death in a small
cohort of 297 men who were mainly exposed to soluble nickel salts
between 1937 and 1960. They did not find any nasal sinus cancers.
Thirteen cases of lung cancer were found, a significant excess
above regional rates (expected number 7.54), but not significant
when compared with the rates for England and Wales. The small size
of the group limited this study.
Estimated concentration data for different nickel species in
different areas of Clydach nickel refinery are shown in Table 33.
Table 33. Estimated concentration (mg/m3)
before 1930, of major forms of nickel in
high-risk areas of the Clydach nickel refinery,
South Walesa
--------------------------------------------------
Metal Oxide Ni3S2 Soluble
--------------------------------------------------
Calcining 5.3 18.8 6.8 0.8
Furnaces 5.6 6.4 2.6 0.4
Copper plant - 13.1 0.4 1.1
Hydrometallurgy 0.5 0.9 0.1 1.4
--------------------------------------------------
a From: Peto (1988).
Peto et al. (1984) extended the follow-up to 1981. The cohort
included 968 men. One hundred and fifty-nine lung cancer deaths
and 56 nasal cancer deaths were identified. The duration of
employment in 4 areas showed a statistically significant
association with lung or nasal cancer or both. These were the
calcining furnace area, the calcining/crushing area, the copper
sulfate area, and the furnace area. Peto et al. (1984) also
related age, time of exposure, and exposure levels, to lung and
nasal sinus cancer incidence in pre-1925 workers. Exposure level
was defined on the basis of duration of work in high-risk areas.
Both lung and nasal cancer showed increasing risk with increasing
duration of work in high-exposure areas. Nasal cancer showed a
strong positive relationship with age at first exposure, whereas
lung cancer did not. The latency period was 20-50 years for nasal
cancer and somewhat shorter for lung cancer. The risk of nasal
cancer was highest for workers with first exposure between 1910 and
1915 and declined thereafter, whereas the risk for lung cancer was
highest for workers between 1920 and 1925.
(b) Falconbridge refinery, Kristiansand (Norway)
Loken (1950) reported 3 cases of lung cancer in workers from
the Falconbridge refinery. This plant began operations in 1910,
processing matte from Ontario via roasting, smelting, and
electrolysis. Although workers in the roasting/smelting
departments were, and are, exposed to sulfides, oxides, and nickel-
copper oxides in dust and fumes, the processes have changed in
recent years and exposure levels have been reduced dramatically.
The electrolytic workers are exposed to aerosols containing, inter
alia, nickel sulfate and nickel chloride (Hogetveit & Barton,
1976). These workers are, as cited by Hogetveit & Barton (1976),
"exposed largely to soluble nickel aerosols". That the soluble
form predominates in the exposure of electrolysis workers is also
supported by the results in Table 34. This table shows that, even
when the air values are lower in the electrolysis department than
in roasting/smelting department, the plasma and urine values are
higher.
Table 34. Summary of biological tests and personal sampler measurementsa
---------------------------------------------------------------------------
Departments Number of Mean Mean Mean
employees plasma ± SD urine ± SD concentration
(µg Ni/litre) (µg Ni/m3) in air ± SD
(µg Ni/m3)
---------------------------------------------------------------------------
No. 01-19 24 72 ± 2.8 65.0 ± 57.6 0.86 ± 1.20
(Roasting-
smelting)
No. 22-44 90 11.9 ± 8.0 129.2 ± 105.6 0.23 ± 0.42
(Electrolytic)
Other process 13 6.4 ± 1.9 44.6 ± 26.7 0.42 ± 0.49
departments
---------------------------------------------------------------------------
CORRELATION BETWEEN RESULTS
Departments Number of Plasma vs. air Plasma vs. air Plasma vs. urine
employees Corr. factor R Corr. factor R Corr. factor R
-----------------------------------------------------------------------------
No. 01-19 24 -0.55 -0.11 -0.71 +0.14 3.72 +0.76
No.22-44 90 1.98 0.21 2.99 0.31 7.30 0.77
Other 13 2.41 0.67 1.70 0.47 2.27 0.63
-----------------------------------------------------------------------------
a Modified from Hogetveit et al. (1978).
The first epidemiological study was carried out on a cohort of
1916 workers who were employed for at least 3 years before 1961
(Pedersen et al., 1973). Exposure was defined according to
department or work in which there had been the longest employment.
The authors used 4 work categories: roasting and smelting,
electrolysis, other processes, other work groups. All occupational
groups were associated with an excess risk of respiratory cancer.
Risk was greatest in the roasting and smelting department and the
electrolysis department. Workers in the electrolysis department
showed the highest risk of lung cancer (O/E=26/3.6) and an excess
risk of nasal cavity cancer (O/E=6/0.2). The risk of nasal cancer
was greatest (O/E=5/0.1) in the roasting and smelting department,
but there was also an increased risk of lung cancer in this
department (O/E=12/2.5).
Magnus et al. (1982) updated the study of Pedersen et al.
(1973) and confirmed the results. They also analysed data on
smoking habits and found evidence for an additive effect of smoking
and nickel exposure.
The cohort was further followed up by Andersen (1988). During
the follow-up period 1953-87, 163 new cases of respiratory cancer
were observed against 42 expected. There was a decreasing trend in
ratio between observed/expected from 6.9 in 1926-35 to 2.4 among
those first employed in 1956-65. No risk for any other cancer was
recorded, in fact, the observed number is lower than the expected
number in all periods (207/248 all periods combined). No cases of
nasal cancer have been observed in workers employed after 1955, but
the expected number is only 0.20.
Following technological improvements in the plant, Andersen
(1988) formed a new cohort of 1237 employees with a total
employment of one year or more, the first entry in 1968 or later,
and exposure estimated according to nickel species and exposure
concentration. For entry in the period 1968-72, 7 new cases of
lung cancer were found, versus 1 expected. Actual measurements
reported in 1979 (Torjussen & Andersen, 1979) were 0.1-0.5 mg/m3
for electrolysis workers and 0.1-1.0 mg/m3 for the roasting/
smelting category. Estimated concentrations for the period 1968-77
were about 0.5-2.0 mg/m3 and about 2.0-5.0 mg/m3, respectively. In
the electrolysis department, the exposure was mainly to soluble
nickel, even if there might have been up to 60% NiS and NiO present
in some working areas. No exposure to soluble compounds was
considered to take place in the roasting/smelting department, but
the possible presence of NiSO4, as an intermediate, was not
appreciated.
The risk of lung cancer for a 1953-84 cohort of 3250 workers
was also estimated according to department and to cumulative
exposure to different nickel species (nickel subsulfide, nickel
oxide, soluble nickel). Although an increased risk was found for
both electrolysis and roasting, smelting, and calcining, the
highest risk and steepest dose-response relationship (based on
exposure time) was found for electrolysis, and the highest risk was
found for the highest cumulative exposure to soluble nickel. It
must be remembered, however, that most workers were exposed to more
than one form of nickel.
Torjussen et al. (1979a,b) examined nasal biopsy specimens from
318 active and 15 retired workers with at least 8 years of
employment. Epithelial dysplasia was found in 14 (14%) of the 97
roasting/smelting workers, in 16 (11%) of the 144 electrolysis
workers, in 8 (10%) of the 77 non-process workers and in 7 (47%) of
the retired workers. One out of 57 controls, a carpenter, had
dysplasia. Two co-workers from the roasting/smelting department
had nasal carcinoma. Based on the methods employed by Torjussen et
al. (1979a,b), histopathological reinvestigations of nasal biopsy
specimens from workers, previously included in the Torjussen study,
were carried out by Boysen et al. (1980, 1982, 1984) and similar
results were found. The histopathological changes in the nasal
epithelium, including dysplasia, persisted in active workers,
despite reduction of airborne nickel concentrations in the working
environment. The frequency of dysplasia was also still elevated in
retired workers. The real significance of nasal epithelial
dysplasia is not known, but it could be considered as a
precancerous lesion because of its prevalence in groups with
increased risk of nasal cancer. The role of nasal biopsies in
identifying and monitoring occupational nickel exposure is unclear.
(c) International Nickel Company (INCO), Ontario
Sulfide nickel ore is mined and refined at several locations
using different processes. The Ontario Health Department studied a
cohort of 2355 refinery workers followed from 1930 to 1957
(Sutherland, 1959, reviewed by Mastromatteo, 1967). There were 7
deaths from sinus cancer (0.19 expected) and 19 from lung cancer
(8.45 expected). Nearly all excess deaths from sinus cancer and
lung cancer occurred in workers exposed to furnace dusts in
calcining and sintering. Mastromatteo (1967) extended Sutherland's
study to the period between 1930 and 1960. The risk of nasal
cancer was still elevated (16 sinus cancer deaths observed, 0.166
expected) as well as the risk of lung cancer (37 lung cancer deaths
observed, 12.71 expected). According to Mastromatteo (1967), the
investigations of Sutherland led to major process changes, among
them the termination of the sinter plant operations, in 1962. The
Sutherland study was updated by Rigaut(1986 ) and the US EPA,
(1986a) to cover the period between 1930 and 1970. By this time,
sinus and lung cancer deaths amounted to 24 (SMR=5106) and 76 (SMR=
1861), respectively.
On the basis of the unpublished work by Sutherland (1959),
Chovil et al. (1981) identified a cohort of 495 men, who had been
employed at the Copper Cliff sinter plant for a period between 1948
and 1962, and were alive in 1963. However, only 75% of the cohort
were successfully followed through 1977 and 1978. Six cases of
sinus cancer were identified (no expected numbers given), and 54
cases of lung cancer of which 37 were fatal, compared with 4.25
expected deaths. Despite some methodological weaknesses, an excess
risk of lung and sinus cancer is suggested.
A large cohort study was carried out by Roberts et al. (1984).
The cohort consisted of 54724 INCO workers employed in Port
Colborne refinery and in the extraction and refining of copper and
nickel in two Sudbury sinter plants (Coniston smelter and Copper
Cliff plant). The cohort was followed for mortality between 1950
and 1976. In Sudbury workers not exposed to sintering operations,
the SMRs for nasal and lung cancer were 144 (O/E=3/2.08) for nasal
cancer and 108 (O/E=222/204.98), respectively, compared with SMRs of
2174 (O/E=2/0.09) for nasal cancer and 463 (O/E= 42/9.08) for lung
cancer in workers exposed to sintering operations. In workers from
the Port Colborne leaching, calcining, and sintering departments,
SMRs for nasal and lung cancer were 9412 (O/E=16/0.17) and 298
(O/E=49/16.44), respectively, compared with SMRs of 78
(O/E=13/16.65) for lung cancer in men who had never worked in this
department. A single case of nasal cancer was observed compared to
0.16 expected, but, on further investigation, it was found that
this man had had 20 years exposure to roasting/calcining operations
elsewhere. Nasal and lung cancer mortality was found to increase
with increasing duration of exposure. Lung cancer was already seen
in excess 5-10 years after first exposure in Sudbury, but, in Port
Colborne, it did not become apparent until 20 years after the first
exposure. Nasal cancer began to appear after 15 years. Apparently
the nasal cancer risk was higher at Port Colborne than at Sudbury,
while lung cancer was more frequent at Sudbury.
Airborne nickel levels were much higher in the sintering
department than in other areas of these refineries. Warner (1985)
reported that a 40-h sample, taken on the operating floor of the
Copper Cliff Sinter Plant in 1960, contained 46.4 mg total dust/m3.
The major nickel compounds in the dust were Ni3S2, NiO, and NiSO4.
Samples taken between 1948 and 1952 from dusty air escaping from
roof monitors contained over 100 mg total dust/m3, but it is not
known if these were representative of exposures in working areas.
Other processes are believed to have been very much less dusty.
Mean levels in most areas were generally well below 1 mg nickel/m3
in later years, though substantially higher levels occasionally
occurred, particularly in matte grinding and in the handling of
metallic nickel powders (Warner, 1984).
The composition of a sample of dust collected in 1959 from the
calcining and sintering rooms of the Port Colborne, Ontario,
refinery is shown in Table 35 (Gilman & Yamashiro, 1985). The
major constituents were nickel subsulfide (57%), nickel sulfate
(20%), nickel oxide (6%), and copper oxide (3%).
Table 35. Composition of dust collected in
1959 in the calcining and sintering of the
Port Colborne (INCO) nickel refinery Ontarioa
---------------------------------------------
Compound Percentage
---------------------------------------------
CuO 3.4
NiSO4 x 6H2O 20.0
Ni3S2 57.0
NiO 6.3
CoO 1.0
Fe2O3 1.8
SiO2 1.2
Miscellaneous 2.0
Moisture 7.3
---------------------------------------------
a From: Gilman & Yamashiro (1985).
Kaldor et al. (1986) analysed the same data using a case-
control approach. In addition to the four areas identified by Peto
et al. (1984), employment in nickel sulfate production was
associated with increased lung and nasal cancer risk. Lung and
nasal cancer mortality and exposure levels for various nickel
compounds for men employed in major production areas were reported
by Peto (1988). There were 5 lung cancer deaths (1.5 expected) and
4 nasal cancer deaths among men, employed for 5 or more years in
hydrometallurgy (nickel sulfate production), who had spent less
than a year in other high-risk areas (copper sulfate, calcining,
furnaces). The principal exposure in hydrometallurgy was to
soluble nickel (about 1.4 mg/m3) with lower levels of nickel oxide
(0.9 mg/m3), nickel subsulfide (0.1 mg/m3), and metallic nickel
(0.5 mg/m3). These estimates were provided by the technical staff
of the refinery, but no measurements were made to verify them.
(d) Falconbridge, Ltd., Falconbridge (Ontario)
A mortality study was carried out on a cohort of 11 594
workers, employed for at least 6 months by the Falconbridge nickel
mines, Sudbury, Ontario, between 1950 and 1976 (Shannon et al.,
1984). In this plant, sintering techniques included a low
temperature step. Nasal cancer was not observed. A slight excess
of lung cancer (SMR=123, O/E=46/37.5) in both mines and service
occupations was not statistically significant. However, laryngeal
cancer deaths were significantly increased in miners (SMR=261,
O/E=5/1.92). A slight, though not significant, increase in deaths
from prostate cancer was noted in smelter workers.
(e) Sherritt Gordon Mines Ltd., Fort Saskatchewan, Alberta
Sherritt Gordon Mines, Ltd., established hydrometallurgical
nickel refining in 1954. Nickel exposures were mainly in the form
of oxide ore concentrate, nickel metal, and some soluble compounds.
Egedahl & Rice (1984) conducted a study of cancer incidence and
mortality with a cohort of 720 workers employed in
hydrometallurgical refining and did not find any excess of
respiratory cancer. Two cases of renal cancer occurred in workers
with 11 and 16 years exposure to nickel concentrate, soluble nickel
substances, and nickel metal, prior to developing their illness.
Both were cigarette smokers.
(f) Nickel refinery, Huntington (West Virginia)
Copper-nickel matte was refined in Huntington during 1922-47
and a mortality study was performed on 1855 men, employed before
1947 who had worked for at least one year at the plant (Enterline &
Marsh, 1982). Concentrations of airborne nickel in the area where
nickel was crushed, ground, and handled, were estimated to range
from 20 to 350 mg/m3 and from 5 to 15 mg/m3 in the calcining area.
The SMR for nasal cancer was 2443.5 with 2 observed and 0.08
expected cases. Two nasal cancers were observed among nickel-
exposed non-refinery workers. There was no significant increase in
deaths from lung cancer among refinery workers (SMR=118.5 with 8
observed and 6.75 expected cases) and it was suggested that the
nickel exposures at the plant may have been considerably lower than
in other plants. However, for refinery and non-refinery workers
combined, and for all respiratory cancer cases combined, there was
a dose-relationship with accumulated nickel exposure.
(g) Oxide ore refinery, New Caledonia
Nickel oxide ore has been mined on the South Pacific Island of
New Caledonia since 1866 and smelted there since 1885. Lessard et
al. (1978) identified 92 cases of lung cancer that had occurred in
this area between 1970 and 1974. The observed rate was 3-7 times
higher than rates in other Central or South Pacific areas, but
similar to those seen in industrialized countries. A significant
increase in lung cancer rates was observed with decreasing distance
from the refinery. The authors found that, in New Caledonia, there
was a 3-fold risk of lung cancer among workers compared with non-
workers, independent of age and tobacco smoking, and considered
that these results supported the etiological role of nickel in lung
cancer. Goldberg et al. (1985a,b) carried out a retrospective
cohort study and a case-control study and reported no excess of
respiratory cancer in workers of the Society Le Nickel (SLN)
compared to the general population of New Caledonia; however, these
studies have weaknesses in statistical power and definition of the
control population. Goldberg et al. (1988) concluded that, in a
10-year period, there was no excess of either lung or sinonasal
cancer in the nickel workers, when compared with the island
population on an age-matched basis. Both studies were considered
to have methodological problems, but the Goldberg et al. (1988)
study had more comprehensive health data.
(h) Other nickel refineries
In the Soviet Union, Saknyn & Shabynina (1970, 1973) reported
an increase in lung and gastric cancer in workers in a nickel
refinery plant in the Urals. The refinery was engaged in the
preparation of nickel oxide ore, roasting, and smelting. They
stated that the lung cancer death rate from 1955 to 1967 exceeded
that of the nearby urban population by 180% and that the death rate
from sarcomas and stomach cancer was also increased. Excess lung
cancer was seen in workers in roasting/reducing and smelting, but
also in electrolysis, where exposure was principally to nickel
sulfate and nickel chloride, and, in the cobalt shop, where
exposure was to various soluble salts of nickel and cobalt.
Tatarskaya (1965) reported 2 cases of nasal cancer among
workers engaged in the electrolytic refining of nickel.
In workers in a Czech plant, where nickel oxide ore had been
refined since 1963, 8 lung tumours were diagnosed between 1971 and
1980 (Olejar et al., 1982). All workers had smoked more than 25
cigarettes per day. These results must be treated with caution as
the latency period from 1963 was very short.
Rockstroh (1959) reported 45 cases of lung cancer in workers in
a nickel refinery in Aue in the German Democratic Republic between
1928 and 1956. Exposure was very complex: nickel, arsenic, cobalt,
copper, bismuth, and benzo (a)pyrene, and workers were often
involved in different processes, during their employment. One case
of lung cancer was found among office personnel, but the size of
the comparison group was not indicated.
9.5.1.2 Nickel alloy manufacturing
In a nickel alloy manufacturing plant in Hereford, England, the
alloys are manufactured from materials consisting of metallic
nickel and other metals (iron, copper, cobalt, chromium,
molybdenum). Workers are exposed to metallic nickel and oxidic
nickel. The average airborne nickel concentrations ranged from
0.84 mg/m3 in the melting, fettling, and pickling area to 0.04
mg/m3 in the process, stock handling, distribution, and warehouse.
Cox et al. (1981) studied a cohort of 1925 men with at least 5
years of employment between 1953 and 1978. He did not find any
cases of nasal sinus cancer. The lung cancer death rate was not
increased compared with the numbers expected from national and
local mortality rates. However, the study period was too short to
exclude the risk of respiratory cancer completely.
Redmond (1984) reported a cohort mortality study on 28 261
workers employed at 12 high nickel alloy production plants between
1956 and 1960, including a follow-up to 1977. The predominant
nickel species, to which the workers were exposed, appeared to have
been nickel dust and nickel oxide. Average levels of airborne
nickel ranged from 0.064 mg/m3 in the cold working area to 1.5
mg/m3 in the powder metallurgy area (Redmond et al., 1983, internal
report). Overall, no statistically significant increased risk was
observed for cancers of the lung, nasal sinuses, larynx, or kidney.
Another study dealt with the standardized proportionate
mortality ratios of 851 foundrymen from 26 nickel/chromium alloy
foundries compared with 141 unexposed foundrymen during the period
1968-79 (Cornell & Landis, 1984). There were no cases of nasal
cancer. The percentages of respiratory cancer in the exposed group
were not significantly different from the percentages of
respiratory cancer in the unexposed group. When compared to
national mortality data, the observed numbers of lung cancer and
all cancer deaths in exposed men were not significantly different
from the expected values.
Cornell (1984) performed a proportional mortality analysis on
4487 workers in 12 US plants engaged in the production of stainless
steel and low nickel alloys. All deaths were included for, at
least, a 5-year period, up to 1977. No cases of nasal cancer were
observed and there was no excess of lung cancer. However, details
of the study, such as the definition of exposure and latency
period, were not provided.
9.5.1.3 Nickel plating industry
Silverstein et al. (1981) conducted a case-control study on
deaths among workers at a Scandinavian die-casting and
electroplating plant. The major operations in the plant were zinc
alloy die-casting, buffing, polishing of zinc and steel parts, and
electroplating with copper, nickel, and chrome. There were 238
deaths among workers with at least 10 years of employment. The
proportionate mortality ratio (PMR) (38 deaths, PMR=209) for lung
cancer was significantly increased. However, because of
heterogeneous exposure, the increased risk of lung cancer
mortality could not be exclusively associated with nickel exposure.
The authors subsequently initiated a case-control study on the
28 white male and 10 white female lung cancer deaths. Cases and
controls were compared for length of employment in individual
departments. Odds ratios were estimated for work in 14 different
departments. For males in three departments, the odds ratio
increased with duration of work in the departments. This trend was
most significant for the department where die-casting and plating
were the major activities.
A study was carried out by Burges (1980) on a group of 508
nickel platers in an engineering firm in England. Nickel plating
was done in a separate shop from chrome plating. The cohort was
divided into groups: nickel bath workers with less than one year of
exposure, more than one year of exposure, and other exposed
workers. In the short-exposure group (less than one year), no
excess mortality was found, whereas, in the longer-exposure group
(more than one year), a significant excess of deaths was found for
stomach cancer (O/E = 4/0.6, SMR = 623) and for non-malignant
respiratory disease (O/E = 8/2.8, SMR = 286). Only one lung cancer
death was observed in the longer-exposure group compared with 1.7
expected cases. The results of the study suggest a higher incidence
of gastric cancer in nickel platers. Sorahan et al. (1987) reported
a study on mortality in nickel and chromium platers in the United
Kingdom. While an excess of certain cancers was associated with
chromium bath workers, it was stated that nickel exposure was not a
confounding factor.
9.5.1.4 Welding
Stern (1983) and Langard & Stern (1984) concluded that the
excess risk of lung cancer in welders is approximately 30% greater
than that in the non-welding population. It was suggested that the
greater risk of lung cancer found among stainless steel welders, in
some studies, could be related to high concentrations of nickel and
hexavalent chromium in stainless steel welding fumes.
The role of nickel compounds as a cause of respiratory cancer
is difficult to assess, because chromates and other carcinogens may
occur in the work-place air during the welding of stainless steel
and other nickel-containing alloys (IARC, 1990).
9.5.1.5 Nickel powder
In the Oak Ridge Gaseous Diffusion Plant, Tennessee, metallic
nickel is used in the production of barrier material for the
isotopic enrichment of uranium by gaseous diffusion. The median
nickel concentration in air between 1948 and 1963 ranged between
0.1 and 1.0 mg/m3 (Godbold & Tompkins, 1979). In a cohort of 814
workers, Godbold & Tompkins (1979) did not find any increased risk
of respiratory cancer during the period 1948-72. In a follow-up
study by Cragle et al. (1984) covering the years from 1972 to 1977,
the lung cancer rate did not increase. The SMR for men employed
for more than 5 years was 91, based on 8 deaths.
9.5.1.6 Nickel-cadmium battery manufacturing
Sorahan & Waterhouse (1983) examined 3025 nickel-cadmium
battery workers for a possible excess of cancer mortality.
Overall, the SMRs showed a statistically significant excess of
respiratory cancer deaths, but it was not possible to attribute the
excess to nickel or to cadmium, since exposure to both occurred at
high levels. In studies by Kjellstrom et al. (1979) and Andersen
et al. (1983), the mortality was examined in men working at a
nickel-cadmium battery-manufacturing plant with mixed exposure to
both metals and nickel hydroxide. Increased respiratory, renal,
and prostate cancer mortality were noted, though there was no
increase in overall cancer mortality. There were two cases of
nasopharyngeal cancer. One started employment before 1948, the
other between 1948 and 1961, when exposure to nickel powder was
very high, exceeding 100 mg nickel/m3 in the work area. The low
risk ratio for lung cancer rendered the studies inconclusive for
the effects of nickel compounds.
9.5.1.7 Case-control studies
The case-referent design has been used to associate the
incidence of certain cancer types with occupations in nickel-
producing or nickel-using industries.
In a British study (Acheson et al., 1981), an excess risk for
nasal cancer was found in furnace and foundry workers, partly,
because of several cases among Clydach refinery workers.
Bernacki et al. (1978b) carried out a small case-control study
at an aircraft-engine factory where high nickel alloys were used.
Nickel exposure was not found to be associated with lung cancer
cases. Roush et al. (1980), using data from the Connecticut Tumor
Registry and from City Directories, determined the occupational
exposures of individuals reported to have cancer of the nasal
sinuses in the years 1935-75. The study did not reveal any
increase in nasal cancer deaths in occupations classified as having
nickel exposure. A case-control study of 204 laryngeal cancer
patients and 204 neighbourhood controls did not show any relation
to occupational exposure to nickel, while tobacco, alcohol, and
asbestos were major risk factors (Burch et al., 1981). However,
Olsen & Sabroe (1984) found a statistically significant excess risk
of laryngeal cancer related to potential nickel exposure from
alloys, battery chemicals, and chemicals used in the production of
plastics. A case-referent study covering the entire Montreal
population showed a statistically significant odds ratio of 3.1 for
lung cancer and nickel exposure and some indication of a dose-
response relationship (Gerin et al., 1984), though individuals
exposed to nickel were always exposed to chromium as well.
9.5.2. Carcinogenicity of metal alloys in orthopaedic prostheses
It was concluded by Sunderman (1989b) that case reports of
sarcomas at sites of metal implants in human beings and domestic
animals have implicated carcinogenesis as a potential, albeit rare,
complication of metal orthopaedic prostheses (Table 36). Since
physicians and veterinarians are not obliged to report adverse
reactions to prostheses, the actual incidences of sarcomas in
association with metal implants cannot be reliably estimated, and
the possibility that the apparent associations are coincidental
cannot be excluded. Carcinogenesis bioassays in rodents have shown
that pure nickel or cobalt powders induce sarcomas at injection
sites, and transformation assays in mammalian cells have yielded
positive results with nickel powder, suggesting that surgical
implantation of alloys containing these metals may pose
carcinogenic risks.
Table 36. Patients with tumours at sites of metal implantsa
----------------------------------------------------------------------------------------------------
Patient age Time lapse Implant type Tumour type Reference
[years] to tumour
implant (years)
(site)
----------------------------------------------------------------------------------------------------
12 (humerus) 30 Steel plate (FeCrNi) and 4 Ewing's sarcoma McDougall (1956)
screws (FeCr), much corrosion
37 (tibia) 3 Osteosynthesis plate and 2 Osteochondrosarcoma Delgado (1958)
screws, alloy unknown
53 (femur) 3 Steel plate and nail (6 Giant cell tumour Castleman &
months) then Moore McNeely (1956)
prosthesis and trochanteric
wires
58 (tibia) 26 Steel (Type 316) plate and Haemangiosarcoma Dube & Fisher
screws (Types 304 and 316) (1972)
56 (hip) 2.5 Mckee metal-to-metal Fibrosarcoma Arden & Bywaters
arthroplasty, alloy not (1978)
stated
4 (hip) 7 Sherman (CoCrMo) plate and Ewing's sarcoma Tayton (1980)
6 screws (implanted 1 year)
31 (tibia) 17 Sherman (CoCrMo) plate and Histiocytic McDonald (1981)
screws lymphoma
75 (hip) 2 Charnley-Muller arthroplasty Malignant fibrous Bago-Granell et
and trochanteric wires, alloy histiocytoma al. (1984)
not stated
63 (hip) 2.4 McKee-Farrar arthroplasty Malignant fibrous Swann (1984)
(CoCrMo) histiocytoma
----------------------------------------------------------------------------------------------------
Table 36 (contd.)
----------------------------------------------------------------------------------------------------
Patient age Time lapse Implant type Tumour type Reference
[years] to tumour
implant (years)
(site)
----------------------------------------------------------------------------------------------------
75 (hip) 5 Ring arthroplasty (CoCrMo) Osteogenic sarcoma Penman & Ring
(1984)
76 (knee) 4.5 CoCrMo arthroplasty Epithelioid sarcoma Weber (1986)
14 (hip) 29 Sherman screw (CoCrMo) Malignant fibrous Hughes et al.
histiocytoma (1987)
40 (hip) 13 2 Screws (unspecified alloy) Undifferentiated Ryu et al.
for 12 years; then hip sarcoma (1987)
prosthesis (CoCrMo stem and
Al2O3 head)
----------------------------------------------------------------------------------------------------
a Modified from: Sanderman (1989b).
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1. Exposure
Nickel is an ubiquitous element and has been detected in
different media in all parts of the biosphere.
Nickel is introduced into the environment from both natural and
man-made sources and is circulated throughout all environmental
compartments by means of chemical and physical processes, as well
as through the biological transport mechanisms of living organisms.
Acid rain may leach nickel as well as other metals from plants
and soil.
Atmospheric nickel is considered to exist mainly in the form of
particulate aerosols.
Nickel is introduced into the hydrosphere by removal from the
atmosphere, by surface run-off, by discharge of industrial and
municipal wastes, and also following the natural erosion of soils
and rocks.
A major source of nickel in the environment is the combustion
of fossil fuels, particularly coal.
Uncontrolled emissions and waste disposal may have adverse
effects on the environment.
Both the chemical and physical forms of nickel and its salts
strongly influence bioavailability and toxicity.
Nickel from soil and water is absorbed and metabolized by
plants and microorganisms and these small quantities of nickel are
widely present in all foods and water.
Some foods, such as pulses and cocoa products, contain
relatively high amounts of nickel, but these quantities have not
been correlated with adverse health effects.
10.2. Human health effects
Nickel is normally present in human tissues and, under
conditions of high exposure, these levels may increase
significantly.
The general population is exposed to nickel via the diet and
objects containing nickel, especially jewellery and coins.
Occupational exposure to nickel is important.
Inhalation is an important route of exposure to nickel and its
salts in relation to health risks. The gastrointestinal route is
of lesser importance.
Nickel absorption from the gastrointestinal tract is poor,
though nickel in the drinking-water is absorbed to a greater extent
in an empty stomach. This may be a risk for sensitized persons.
Smoking tobacco may contribute to nickel intake, but there is
no agreement on the chemical nature of nickel in tobacco smoke and
its health significance.
Target organs are the respiratory system, especially the nasal
cavities and sinuses, and the immune system.
The percutaneous absorption of nickel is minor, but it is
important in sensitization.
Nickel and its salts are potent skin sensitizers and possible
respiratory sensitizers in human beings. Nickel dermatitis is a
common result of nickel exposure, especially in women.
Primary skin and eye irritation reactions to high
concentrations of soluble nickel salts have also been reported.
Acute toxicity is a minor risk from nickel or its compounds,
with the exception of nickel carbonyl.
There is no convincing evidence that nickel salts produce point
mutations in bacterial systems. However, some nickel salts are
clastogenic in vitro, producing chromosome aberrations,
(transformation) and sister chromatid exchanges in mammalian cells.
There is a lack of evidence of a carcinogenic risk from oral
exposure to nickel, but the possibility that it acts as a promoter
has been raised.
There is evidence of a carcinogenic risk through the inhalation
of nickel metal dusts and some nickel compounds.
Very high concentrations of nickel are required to produce
teratogenic or genotoxic effects.
The consequences of the effects of nickel on the immune system
are not clear.
10.3. Environmental effects
Nickel is accumulated by plants. Growth retardation has been
reported in some species, at high nickel concentrations.
There is no evidence that nickel may undergo biotransformation,
though it does undergo complexation.
Nickel has been shown to be essential for the nutrition of many
microorganisms, a variety of plants, and for some vertebrates.
11. RECOMMENDATIONS
1. The nomenclature for nickel compounds should be further
standardized.
2. Analytical methods should be developed and standardized to
facilitate speciation of nickel compounds in environmental
media, biological materials, the workplace, and atmospheric
emissions.
3. Studies are recommended on the kinetics and mass transfer of
nickel, in order to elucidate the biogeochemical cycle on a
global scale and its potential for long-range transport.
4. The use of nickel in consumer products that may release nickel
when in contact with skin should be regulated. The
specification and testing requirements should be standardized.
5. Studies should be performed on the absorption and cellular
uptake, transport, and metabolism of well characterized nickel
species, following different routes and types of administration.
6. Nickel compounds, to which human beings are exposed, should be
tested for carcinogenicity in adequate, long-term, inhalation
studies on animals.
7. Further large-scale cohort studies with well characterized
exposures, including biological monitoring, are needed in all
sectors of industry, to establish more accurate upper limits of
cancer risk.
8. Priority should be given to improving industrial hygiene in
occupations where exposure to high levels of soluble nickel
compounds may occur.
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
The carcinogenic risk of nickel and inorganic nickel compounds
has been evaluated by the International Agency for Research on
Cancer (IARC). An International Working Group evaluated
epidemiological and experimental carcinogenicity data on nickel and
certain nickel compounds encountered in the nickel refining
industry (IARC, 1990). The evaluations are summarized below:
-----------------------------------------------------------------------
Degree of evidence Overall
for carcinogenicitya evaluation
Human Animal
-----------------------------------------------------------------------
Nickel and nickel compounds
Nickel compounds 1
Nickel salts L
Nickel sulfate S
Combination of nickel oxides
and sulfides encountered in
the nickel refining industry S
Nickel monoxides S
Nickel trioxides I
Nickel sulfide, amorphous I
Nickel sulfides, crystalline S
Nickel antimonide L
Nickel arsenides L
Nickel carbonyl L
Nickel hydroxides S
Nickelocene L
Nickel selenides L
Nickel telluride L
Nickel titanate I
Metallic nickel I S 2B
Nickel alloys I L
-----------------------------------------------------------------------
a S : Sufficient evidence of carcinogenicity.
L : Limited evidence of carcinogenicity.
I : Inadequate evidence of carcinogenicity.
1 : The agent is carcinogenic for human beings.
2B : The agent is possibly carcinogenic for human beings.
In the Guidelines for drinking-water quality (WHO, 1984), no
guideline value was set for nickel in drinking-water, as the
toxicological data available at the time indicated that a guideline
value was not necessary.
In the Air quality guidelines for Europe (WHO, 1988), no safe
level was recommended for nickel, because of its carcinogenic
properties. At an air concentration of nickel dust of 1 µg/m3, a
conservative estimate of the lifetime risk is 4 x 10-4.
REFERENCES
ABDULWAJID, A.W. & SARKAR, B. (1983) Nickel-sequestering renal
glycoprotein. Proc. Natl Acad. Sci. (USA), 80(14): 4509-4512.
ABO-RADY, M.D. (1979a) Determination of heavy metals in two
biological and two geological standard materials by atomic
absorption spectroscopy. Fresenius Z. anal. Chem., 286: 380-382.
ABO-RADY, M.D. (1979b) The levels of heavy metals (Cd, Cu, Hg, Ni,
Pb, Zn) in brook trout from the river Leine in the area of
Göttingen (West Germany). Z. Lebensm. Unters. Forsch., 168(4): 259-263.
ABRAHAM, T.J., SALIH, K.Y. & CHACKO, J. (1986) Effects of heavy
metals on the filtration rate of bivalve Villorita cyprinoides
(Hanley) var. cochinensis. Indian J. mar. Sci., 15(3): 195-196.
ACHESON, E.D., COWDELL, R.H. & RANG, E.H. (1981) Nasal cancer in
England and Wales: an occupational survey. Medicine, 38(3): 218-224.
ADALIS, D., GARDNER, D.E. & MILLER, F.J. (1978) Cytotoxic effects
of nickel on ciliated epithelium. Am. Rev. respir. Dis. 118(2):
347-354.
ADAMSSON, E., LIND, B., NILSSON, B. & PISCATOR, M. (1980) Urinary
and fecal elimination of nickel in relation to air-borne nickel in
a battery factory., In: Brown, S.S. & Sunderman, F.W. Jr, ed., Nickel
toxicology: Proceedings of the 2nd International Conference on
Nickel Toxicology, Swansea, 3-5 September,1980, London, New York,
Academic Press, pp. 103-106.
ADKINS, B., RICHARDS, J.H. & GARDNER, D.E. (1979) Enhancement of
experimental respiratory infection following nickel inhalation.
Environ. Res., 20(1): 33-42.
AGGARWAL, S.K., KINTER, M., WILLS, M.R., SAVORY, J. & HEROLD, D.A.,
(1988) Isotope dilution gas chromatography - mass spectrometric
method for the determination of nickel in biological materials. In:
Fourth International Conference on Nickel Metabolism and
Toxicology: Abstracts, Espoo, Finland, 5-9 September, 1988,
Helsinki, Institute of Occupational Health (Abstract II.1).
AITIO, A. (1984) Biological monitoring of occupational exposure to
nickel. In: Nickel in the human environment. Proceedings of a
Joint Symposium, Lyon, 8-11 March, 1983, Lyon, International Agency
for Research on Cancer, pp. 497-505 (IARC Scientific Publications
No. 53).
AJMAL, M. & KHAN, A.U. (1985) Effect of electroplating factory
effluent on germination and growth of hyacinth bean and mustard.
Environ. Res., 38(2): 248-255.
AJMAL, M., KHAN, M.A. & NOMANI, A.A. (1985) Distribution of heavy
metals in plants and fish of the Yamuna river (India). Environ.
monit. Assess., 5(4): 361-367.
AKAGI, T., FUWA, K. & HARAGUCHI, H. (1985a) Simultaneous multi-
element determination of trace metals in seawater by inductively
coupled plasma atomic emission spectrometry after coprecipitation
with gallium. Anal. Chim. Acta, 177: 139-155.
AKAGI, T., NOJIMI, Y., MATSUI, M. & HARAGUCHI, H. (1985b)
Zirconium coprecipitation for simultaneous multi-element
determination of trace metals in seawater by inductively coupled
plasma atomic emission spectrometry. Appl. Spectroscop. 39(4):
662-667.
ALBERT, D.M., GONDER, J.R., PAPALE, J., CRAFT, J.L., DOHLMAN, H.G.,
REID, M.C. & SUNDERMAN, F.W. Jr (1980) Induction of ocular
neoplasms in Fischer rats by intraocular injection of nickel
subsulfide. In: Brown, S.S. & Sunderman, F.W. Jr, ed. Nickel
toxicology. Proceedings of the 2nd International Conference on
Nickel Toxicology, Swansea, 3-5 September, London, New York,
Academic Press, pp. 55-58.
ALBERT, D.M., GONDER, J.R., PAPALE, J., CRAFT, J.L., DOHLMAN, H.G.,
REID, M.C. & SUNDERMAN, F.W. Jr (1982) Induction of ocular
neoplasms in Fischer rats intraocular injection of nickel
subsulfide. Invest. Opthalmol. vis. Sci., 22(6): 768-782.
ALBRACHT, S.P.J., GRAF, E.G. & THAUER, R.K. (1982) The ERP
properties of nickel in hydrogenase from Methanobacterium
thermoautotrophicum. FEBS Lett., 140: 311-313.
ALEXANDER, A.J., GOGGIN, P.L. & COOKE, M. (1983) A Fourier-
transform infrared spectrometic study of the pyrosynthesis of
nickel tetracarbonyl and iron trentacarbonyl by combustion of
tobacco. Analyt. chim. Acta, 151: 1-12.
ALI, M., BISWAS, S.K., AKHTER, S. & KHAN, A.H. (1985) Multi-
element analysis of water residue: a PIXE measurement. Fresenius
Z. anal. Chem., 322(8): 755-760.
ALLOWAY, B.J. & MORGAN, H. (1986) The behaviour and availability of
cadmium, nickel, and lead in polluted soils. In: Assink, J.W. &
van den Brink, W.J., ed. Contaminated soil. Dordrecht, Boston,
Lancaster, Martinus Nifhoff Publishers, pp. 101-113.
AMACHER, D. & PAILLET, S. (1980) Induction of trifluorothymidine-
resistant mutants by metal ions in L5178Y/TK+/- cells. Mutat. Res.,
78: 279-288.
AMBROSE, A.M., LARSON P.S., BORZELLECA, J.R. & HENNIGA, G.R. Jr
(1976) Long-term toxicologic assessment of nickel in rats and dogs.
J. food Sci. Technol., 13: 181-187.
AMLACHER, E. & RUDOLPH, CH. (1981) The thymidine incorporation
inhibiting screening system (TSS) to test carcinogenic substances
(a nuclear DNA synthesis suppressive short-term test). Arch.
Geschwulstforsch., 51(7): 605-610.
ANDERSEN, A. (1988) Recent follow-up of respiratory cancer in a
Norwegian nickel refinery. In: Fourth International Conference on
Nickel Metabolism and Toxicology: Abstracts, Espoo, Finland, 5-9
September, 1988, Helsinki, Institute of Occupational Health, p. 49.
ANDERSEN, J.R., GAMMELGAARD, B. & REIMERT, S. (1986a) Direct
determination of nickel in human plasma by Zeeman-corrected atomic
absorption spectrometry. Analyst, 111(6): 721-722.
ANDERSEN, K.E., NIELSEN, G.D., FLYVHOLM, M.A., FREGERT, S. &
GRUVBERGE, B. (1983) Nickel in tap water. Contact Dermatit., 9(2):
140-143.
ANDERSEN, O. (1983) Effects of coal combustion products and metal
compounds on sister chromatid exchange (SCE) in a macrophage-like
cell line. Environ. Health Perspect., 47: 239-253.
ANDERSEN, O. (1985) Evaluation of the spindle-inhibiting effect of
Ni++ by quantitation of chromosomal super-contraction. Res. Commun.
chem. Pathol. Pharmacol., 50: 379-386.
ANDERSON, B.G. (1950) The apparent thresholds of toxicity to
Daphnia magna for chlorides of various metals when added to Lake
Erie water. Serv. works J., 16: 96-113.
ANDERSSON, K., ELINDER, C.G., HOGSTEDT, C., KJELLSTROM, T. &
SPANG, G. (1984) Mortality among cadmium and nickel-exposed workers
in a Swedish battery factory. Toxicol. environ. Chem., 9(1): 53-62.
ANKE, M. (1974) [The significance of micronutrients for animal
performance.] Akad. Landwirtschaftswiss. DDR Tagungsber., 132: 197-218
(in German).
ANKE, M., GRUN, M., DITTRICH, G., GROPPEL, B. & HENNIG, A. (1974)
Low nickel rations for growth and reproduction in pigs. In:
Hoekstra, W.G. ed. Trace element metabolism in animals,
Baltimore, Maryland, University Park Press, Vol. 2, pp. 715-718.
ANKE, M., GRUN, M. & PARTSCHEFELD, M. (1976) [Nickel, a new
essential trace element.] Arch. Tierernähr., 26: 740-741 (in German).
ANKE, M., HENNIG, A., GRUN, M., PARTSCHEFELD, M., GROPPEL, B. &
LUDKE, H. (1977) [Nickel, an essential trace element. 1st
communication: The supply of nickel as affecting the live weight
gains, food consumption and body composition of growing pigs and
goats.] Arch. Tierernähr., 27: 25-38 (in German).
ANKE, M., PARTSCHEFELD, M., GRUN, M. & GROPPEL, B. (1978) [Nickel
- an essential trace element. 3rd communication: The influence of
nickel content on the reproductive performance of female animals.]
Arch. Tierernähr., 28: 83-90 (in German).
ANKE, M., KRONEMANN, H., GROPPEL, B., HENNIG, A., MEISSNER, D. &
SCHNEIDER, H.-J. (1980) The influence of nickel deficiency on
growth, reproduction, longevity and different biochemical
parameters of goats. In: Anke, M. Schneider, H.-J. & Brûckner,
Chr., ed. [3. Trace Element Symposium: Nickel, Jena, 7-11 July,
1980,] Jena, Friedr.-Schiller Univ. pp. 3-10.
ANKE, M., GRUN, M., HOFFMANN, G., GROPPEL, B., GRUHN, K. & FAUST,
H. (1981) Zinc metabolism in ruminants suffering from nickel
deficiency. Mengen-Spurenelemente, 1: 189-196.
ANKE, M., GROPPEL, B., KRONEMANN, H. & GRUN, M. (1984) Nickel - an
essential element. In: Nickel in the human environment, Proceedings
of a Joint Symposium, Lyon, France, 8-11 March, 1983, Lyon,
International Agency for Research on Cancer, pp. 339-365 (IARC
Scientific Publications No. 53).
ARANYI, C., MILLER, F.J., ANDRES, S., EHRLICH, R., FENTERS, J.,
GARDNER, F.E. & WATERS, M.D. (1979) Cytotoxicity to alveolar
macrophages of trace metals adsorbed to fly fish. Environ. Res.,
20: 14-23.
ARDEN, G.P. & BYWATERS, E.G.L. (1978) Tissue reaction. In: Arden,
G.B. & Ansel, B.M., ed. Surgical management of juvenile chronic
polyarthritis. London, New York, Academic Press, pp. 263-275.
ARILLO, A., MARGIOCCO, C., MELODIA, F. & MENSI, P. (1982)
Biochemical effects of long-term exposure to Cr, Cd, Ni on rainbow
trout ( Salmo gairdneri Rich): influence of sex and season.
Chemosphere, 11:47-57.
ARLAUSKAS, A., BAKER, R., BONIN, A.M., TANDON, R.K., CRISP, P.T. &
ELLIS, J. (1985) Mutagenicity of metal ions in bacteria. Environ.
Res., 36(2): 379-388.
ARMIT, H.W. (1908) The toxicology of nickel carbonyl, Part II. J.
Hyg., 8: 565-600.
ASATO, N., SOESTBERGEN, M.V. & SUNDERMAN F.W. Jr (1975) Binding of
63Ni(II) to ultrafiltrable constituents of rabbit serum in vivo
and in vitro. Clin. Chem., 21(4): 521-527.
ASHRAF, M. & SYBERS, H.D. (1974) Lysis of pancreatic exocrine cells
and other lesions in rats fed nickel acetate. Am. J. Pathol., 74: 199.
BABICH, H. & STOTZKY, G. (1982a) Nickel toxicity to microbes:
effect of pH and implications for acid rain. Environ. Res., 29:
335-350.
BABICH, H. & STOTZKY, G. (1982b) Nickel toxicity to fungi:
Influence of some environmental factors. Ecotoxicol. environ. Saf.,
6: 577-589.
BABICH, H. & STOTZKY, G. (1983) Temperature, pH, salinity,
hardness, and particulates mediate nickel toxicity to eubacteria,
an actinomycete, and yeasts in lake, simulated estuarine, and sea
waters. Aquat. Toxicol., 3: 195-208.
BAGO-GRANELL, J., AGUIRRE-CANYADELL, M., NARDI, J. & TALLADA, N.
(1984) Malignant fibrous histiocytoma of bone at the site of a
total hip arthroplasty. J. bone joint Surg. B., 66: 38-40.
BALOGH, I., SOMOGYI, E., SOTONYI, P., POGATSA, G., RUBANYI, G. &
BELLUS, E. (1983) Electron-cytochemical detection of endogenous
nickel in the myocardium in acute carbon monoxide poisoning.
Applicability of a new cytochemical technique in forensic medicine.
Z. Rechtsmed., 90(1): 7-14.
BARBEAU, C., DUPUIS, M. & ROY, J.C. (1985) Metallic elements in
crude and milled chrysotile asbestos from Quebec. Environ. Res.,
38(2): 275-282.
BARNES, J.M. & DENZ (1951) The effects of 2,3-dimercapto-propanol
(BAL) on experimental nickel carbonyl poisoning. Br. J. ind. Med.,
8: 117-126.
BARRIE, L.A. (1981) Atmospheric nickel in Canada. In: Effects of
nickel in the Canadian environment, Ottawa, National Research
Council of Canada, pp. 55-76 (Publication No. 18568).
BASRUR, P.K. & GILMAN, J.P.W. (1967) Morphologic and synthetic
response of normal and tumor muscle cultures to nickel sulfide.
Cancer Res., 27: 1168-1177.
BAUDOUIN, M., F. & SCOPPA, P. (1974) Acute toxicity of various
metals to freshwater zooplankton. Bull. environ. Contam. Toxicol.,
121(6):745-751.
BAUMGARDT, B., JACKWERTH, E., OTTO, H. & TOLG, G. (1986)
Contribution to the trace analytical investigation of human lung
tissue. Fresenius Z. anal. Chem., 323(5): 481-486.
BAYER, O. (1939) [Toxicology, pathology and clinical aspects of
nickel carbonyl poisoning.] Arch. Gewerbepathol. Gewerbehyg., 9: 592-
606 (in German).
BECKER, P., DORSTELMANN, K., FROMBERGER, W. & FORTH, W. (1980) [On
the absorption of cobalt(II) and nickel(II) ions by isolated
intestinal segments in vitro of rats.] In: Anke, M., Schneider,
H.-J. & Brückner, Chr., ed. [3. Trace Element Symposium: Nickel,
Jena, German Democratic Republic, 7-11 July, 1980,] Jena,
Friedrich-Schiller University, pp. 79-85 (in German).
BEEFTINK, W.G. & NIEUWENHUIZE, J. (1982) Heavy-metal accumulation
in salt marshes from the Western and Eastern Scheldt. Sci. total
Environ., 25: 199-223.
BEIJER, K. & JERNELOV, A. (1986) Sources, transport and
transformation of metals in the environment. In: Friberg, L.,
Nordberg, G.F. & Vouk, V.B., ed. Handbook on the toxicology of
metals, Amsterdam, New York, Oxford, Elsevier Science Publishers,
Vol. I, pp. 68-84.
BENCKO, V., WAGNER, V., WAGNEROVA, M. & REICHRTOVA, E. (1983)
Immuno-biochemical findings in groups of individuals occupationally
and non-occupationally exposed to immissions containing nickel and
cobalt. J. Hyg. Epidemiol. Microbiol. Immunol. 27: 387-394
BENCKO, V., GEIST, T., ARBETOVA, D., DHARMADIKARI, D.M. &
SVANDOVA, E. (1986) Biological monitoring of environmental
pollution and human exposure to some trace elements. J. Hyg.
Epidemiol. Microbiol. Immunol., 30: 1-10.
BENNETT, B.G. (1984) Environmental nickel pathways to man. In:
Nickel in the human environment, Proceedings of a Joint Symposium,
Lyon, 8-11 March, 1983, Lyon, International Agency for Research on
Cancer, pp. 487-495 (IARC Scientific Publications No. 53).
BENSON, J.M., HENDERSON, R.F. & MACLELLAN, R.O. (1986) Comparative
toxicity of four nickel compounds to canine and rodent alocolar
macrophages in vitro. J. Toxicol. environ. Health, 19: 105-110.
BENSON, J.M., CARPENTER, R.L., HAHN, F.F., HALEY, P.J., HANSON,
R.L., HOBBS, C.H., PICKRELL, J.A. & DUNNICK, J.K. (1987)
Comparative inhalation toxicity of nickel subsulfide to F344/N rats
and B6C3F1 mice exposed for twelve days. Fundam. appl. Toxicol.,
9: 251-265.
BENSON, J.M., BURT, D.G., CARPENTER, R.L., EIDSON, A.F., HAHN,
F.F., HALEY, P.J., HANSON, R.L., HOBBS, C.H., PICKRELL, J.A. &
DUNNICK, J.K. (1988) Comparative inhalation toxicity of nickel
subsulfide to F344/N rats and B6C3F1 mice exposed for twelve days.
Fundam. appl. Toxicol., 10: 164-178.
BERGMAN, B., BERGMAN, M., MAGNUSSON, B., SOREMARK, R. & TODA, Y.,
(1980a) The distribution of nickel in mice. An autoradiographic
study. J. oral Rehabil., 7(4): 319-324.
BERGMAN, M., BERGMAN, B. & SOREMARK, R. (1980b) Tissue
accumulation of nickel released due to electrochemical corrosion of
non-precious dental casting alloys. J. oral Rehabil., 7(4): 325-330.
BERMAN, E. & REHNBERG, B. (1983) Fetotoxic effects of nickel in
drinking water in mice. Washington, DC, US Environmental
Protection Agency, 11 pp (EPA-600/1- 83-007).
BERNACKI, E.J., PARSONS, G.E., ROY, B.R., MIKAC-DEVIC, M., KENNEDY,
C.D. & SUNDERMAN, F.W. Jr (1978a) Urine nickel concentrations in
nickel-exposed workers. Ann. clin. lab. Sci., 8(3): 184-189.
BERNACKI, E.J., PARSONS, G.E. & SUNDERMAN, F.W. Jr (1978b)
Investigation of exposure to nickel and lung cancer mortality: case
control study at aircraft engine factory. Ann. clin. lab. Sci.,
8(3): 190-194.
BERNACKI, E.J., ZYGOWICZ, E. & SUNDERMAN, F.W. Jr (1980)
Fluctuations of nickel concentrations in urine of electroplating
workers. Ann. clin. lab. Sci., 10(1): 33-39.
BERROW, M.C. & BURRIDGE, J.C. (1981) Persistence of metals in
available form in sewage sludge-treated soils under field
conditions. In: Proceedings of International Conference on Heavy
Metals in the Environment, Amsterdam, Sept. 1981, Edinburgh, CEP
Consultants Ltd. pp. 202-205.
BERTRAND, G. & MACHEBOEUF, M. (1926a) Chimie biologique. Influence
du nickel et du cobalt sur l'action exercée par l'insuline chez le
lapin. C. R. Séances Acad. Sci., 182: 1504-1507.
BERTRAND, G. & MACHEBOEUF, M. (1926b) Chimie biologique. Influence
du nickel et du cobalt sur l'action exercée par l'insuline chez le
chien. C. R. Séances Acad. Sci., 183: 5-9.
BEVERIDGE, M.C., STAFFORD, E. & COUTTS, R. (1985) Metal
concentrations in the commercially exploited fishes of an endorheic
saline lake in the tin-silver province of Bolivia. Aquacult. Fish.
Manage., 16(1): 41-53.
BIEDERMANN, K. & LANDOLPH, J.R. (1986) Induction of anchorage
independence in human diploid foreskin fibroblasts by metal salts.
Carcinogenesis, 542: 137.
BIESINGER, K.E. & CHRISTENSEN, G.M. (1972) Effects of various
metals on survival, growth, reproduction, and metabolism of Daphnia
magna. J. Fish Res. Board. Can., 29(12): 1691-1700.
BIGGART, N.W. & MURPHY, E.C. Jr (1988) Analysis of metal-induced
mutations altering the expression or structure of a retroviral gene
in a mammalian cell line. Mutat. Res., 198: 115-129.
BINGHAM, E.W., BARKLEY, W., ZERWAS, M., STEMMER, K. & TAYLOR, P.,
(1972) Responses of alveolar macrophages to metals. I. Inhalation
of lead and nickel. Arch. environ. Health, 25: 406-414.
BIRGE, W.J. (1978) Embryo-larval bioassays on inorganic coal
elements and in situ Biomonitoring of coal-waste effluents. In:
Surface mining and fish/wildlife needs in the eastern USA.
Proceedings of a Symposium, December 1978. Washington, DC, US Fish
and Wildlife Service, pp. 97-108 (9098.910 FWS/ OBS-78/81).
BIRGE, W.J. & BLACK, J.A. (1980) Aquatic toxicology of nickel. In:
Nriagu, J.O., ed. Nickel in the environment, New York, Chichester,
Brisbane, Toronto. John Wiley and Sons, pp. 349-406.
BLAYLOCK, B.G. & FRANK, M.L. (1979) A comparison of the toxicity of
nickel to the developing eggs and larvae of carp ( Cyprinus carpio).
Bull. environ. Contam. Toxicol., 21:604-611.
BLANKENSTEIN, K. & STARCK, H.C. (1979) [Nickel compounds.] In:
Bartholomé, E., Biekert, E., Hellmann, H., Ley, H., Weigert, W. &
Weise, E., ed. [Ullmanns encyclopaedia of technical chemistry,]
Weinheim, New York, Verlag Chemie, pp. 293-302 (in German).
BLOCK, G.T. & YEUNG, M. (1982) Asthma induced by nickel. J. Am.
Med. Assoc., 247(11): 1600-1602.
BOLDT, J.R. & QUENEAU, P. (1967) The winning of nickel, its
geology, mining, and extractive metallurgy, London, Methuen & Co.
Ltd., 465 pp.
BOND, A.M. & WALLACE, G.G. (1983) Automated determination of nickel
and copper by liquid chromatography with electrochemical and
spectrophotometric detection. Anal. Chem., 55: 718-723.
BOND, A.M., KNIGHT, R.W., REUST, J.J.B., TUCKER, D.J. & WALLACE,
G.G. (1986) Determination of metals in urine by direct injection of
sample, high-performance liquid chromatography and electrochemical
or spectrophotometric detection. Anal. chim. Acta, 182: 47-59.
BOUTAYRE, P., PIZZORNI, L., LIQUIER, J., TABOURY, J. &
TAILLANDIER, E. (1984) Z-form induction in DNA by carcinogenic
nickel compounds: an optical spectroscopy study. In: Nickel in the
human environment. Proceedings of a Joint Symposium, Lyon, 8-11
March 1983, Lyon, International Agency for Research on Cancer, pp.
227-234 (IARC Scientific Publications No. 53).
BOYER, K.W. & HOWITZ, W. (1986) Special considerations in trace
element analysis of food and biological materials. In:
Environmental carcinogens - selected methods of analysis, Vol. 8:
Some metals: As, Be, Cd, Cr, Ni, Pb, Se, Zn, Lyon, International
Agency for Research on Cancer, pp. 191-220 (IARC Scientific
Publications No. 71).
BOYLE, R.W. (1981) Geochemistry of nickel. In: Effects of nickel in
the Canadian environment, Ottawa, National Research Council of
Canada, pp. 31-44 (Publication No. NRCC 18568).
BOYSEN, M., WAKSVIK, H., SOLBERG, L.A., REITH, A. & HOGETVEIT,
A.C. (1980) Histopathological follow-up studies and chromosome
analysis in nickel workers. In: Brown, S.S. & Sunderman, F.W. Jr,
ed., Nickel Toxicology. Proceedings of the 2nd International
Conference on Nickel Toxicology, Swansea, 3-5 September 1980,
London, New York, Academic Press, pp. 35-38.
BOYSEN, M., SOLBERG, L.A., ANDERSEN, I., HOGETVEIT, A.C. &
TORJUSSEN, W. (1982) Nasal histology and nickel concentration in
plasma and urine after improvements in the work environment at a
nickel refinery in Norway. Scand. J. Work Environ. Health, 8(4):
283-290.
BOYSEN, M., SOLBERG, L.A., TORJUSSEN, W., POPPE, S. & HOGETVEIT,
A.C. (1984) Histological changes, rhinoscopical findings and nickel
concentration in plasma and urine in retired nickel workers. Acta
otolaryngol., 97(1-2): 105-115.
BRADLEY, R.W. & MORRIS, J.R. (1986) Heavy metals in fish from a
series of metal-contaminated lakes near Sudbury, Ontario. Water Air
Soil Pollut., 27(3-4): 341-354.
BRAJTER, K. & SLONAWSKA, K. (1986) The efficiency of Cellex-P for
the preconcentration of lead and other trace metals from waters.
Anal. Chim. Acta, 185: 271-277.
BRANDES, W.W. (1934) Nickel carbonyl poisoning. J. Am. Med. Assoc.,
102: 1204-1206.
BRINGMANN, G., KUHN, R. & WINTER, A. (1980) [Determination of the
biological effect of water pollutants on protozoa. 3. Saprozoic
flagellates.] Z. Wasser Abwasser Forsch., 13(5):170-173 (in German
with English summary).
BRKOVIC-POPOVIC, L. & POPOVIC, M. (1977) Effects of heavy metals on
survival and respiration rate of tubificid worms: Part 1. Effects
on survival. Environ. Pollut., 13: 65-72.
BROOKS, R.R. (1980) Accumulation of nickel by terrestrial plants.
In: Nriagu, J.O., ed. Nickel in the environment, New York,
Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 407-429.
BROWN, K.W., THOMAS, J.C. & SLOWEY, J.F. (1983a) The movement of
metals applied to soils in sewage effluent. Water air soil Pollut.,
19(1): 43-54.
BROWN, K.W., THOMAS, J.C. & SLOWEY, J.F. (1983b) Metal
accumulation by Bermuda grass grown on four diverse soils amended
with secondarily treated sewage effluent. Water air soil Pollut.,
20(4): 431-446.
BROWN, V.M. (1968) The calculation of the acute toxicity of
mixtures of poisons to rainbow trout. Water Res., 2: 723-733.
BRULAND, K.W., FRANKS, R.P., KNAUER, G.A. & MARTIN, J.H. (1979)
Sampling and analytical methods for the determination of copper,
cadmium, zinc, and nickel at the nanogram per liter level in
seawater. Anal. chim. Acta, 105: 233-245.
BRUNE, D. (1986) Metal release from dental biomaterials.
Biomaterials, 7(3): 163-175.
BRYANT, V., NEWBERY, D.M., MCCLUSKY, D.S. & CAMPBELL, R. (1985)
Effect of temperature and salinity on the toxicity of nickel and
zinc to two estuarine invertebrates ( Corophium volutator, Macoma
balthica). Mar. Ecol. Prog. Ser., 24(1-2): 139-153.
BUHLER, E.V. (1965) Delayed contact hypersensitivity in the guinea-
pig. Arch. Dermatol., 91: 171-175.
BURBA, P. & WILLMER, P.G. (1985) Analytical multielement
preconcentration on metal hydroxide-coated cellulose. Fresenius Z.
anal. Chem. 321(2): 109-118.
BURCH, J.D., HOWE, G.R., MILLER, A.B. & SEMENCIW, R. (1981)
Tobacco, alcohol, asbestos, and nickel in the etiolgy of cancer of
the larynx: a case-control study. J. Natl Cancer Inst., 67(6): 1219-
1224.
BURGES, D. (1980) Mortality study of nickel platers. In: Brown,
S.S. & Sunderman, F.J. Jr, ed., Nickel toxicology. Proceedings of
the 2nd International Conference on Nickel Toxicology, Swansea, 3-5
September, 1980, London, New York, Academic Press, pp. 15-18.
BURROWS, D., CRESWELL, S. & MERRETT, J.D. (1981) Nickel, hands and
hip prostheses. Br. J. Dermatol., 105(4): 437-443.
BUSELMAIER, W., ROHRBORN, G. & PROPPING, P. (1972) [Mutagenicity
investigations with pesticides in the host-mediated assay and the
dominant lethal test in mice.] Biol. Zbl., 91: 311-325 (in German).
BUTZ, I. (1984) [Effect of dilution water on the acute toxicity of
nickel sulfate to rainbow trout.] Wasser Abwasser, 28: 41-56
(in German).
BYRNE, C.J. & DELEON, I.R. (1986) Trace metal residues in biota and
sediments from Lake Pontchartrain, Lousiana, USA. Bull. environ.
Contam. Toxicol., 37(1): 151-158.
CALAMARI, D., GAGGINO, G.F. & PACCHETTI, G. (1982) Toxokinetics of
low levels of Cd, Cr, Ni and their mixture in long-term treatment
of Salmo gairdneri Rich. Chemosphere, 11(1): 59-70.
CALAMARI, D., LLOYD, R., SOLBE, J.F. & SOLBE, L.G. (1984) Water
quality criteria for European freshwater fish: Report on nickel and
freshwater fish. Rome, Food and Agriculture Organization of the
United Nations 20 pp (European Inland Fisheries Advisory Commission
(EIFAC) Technical Paper 45).
CALLAN, W.M. & SUNDERMAN, F.W. Jr (1973) Species variations in
binding of 63NiCl2 by serum albumin. Res. Commun. chem. Pathol.
Pharmacol., 5: 459-472.
CALNAN, C.D. (1956) Nickel dermatitis. Br. J. Dermatol., 68: 229-236.
CAMNER, P., JOHANSSON, A. & LUNDBERG, M. (1978) Alveolar
macrophages in rabbits exposed to nickel dust. Environ. Res., 16:
226-235.
CARLSON, H.E. (1984) Inhibition of prolactin and growth hormone
secretion by nickel. Life Sci., 35(17): 1747-1754.
CARVAJAL, N.J. & ZIENIUS, R.H. (1986) Gas chromatographic analysis
of trace metals isolated from aqueous solutions as
diethyldithiocarbamates. J. Chromatogr., 355: 107-116.
CARVALHO, S.M. & ZIEMER, P.L. (1982) Distribution and clearance of
63Ni administered as 63NiCl2 in the rat: intratracheal study.
Arch. environ. Contam. Toxicol., 11: 245-248.
CASARETT-BRUCE, M., CAMNER, P. & CURSTEDT, T. (1981) Changes in
pulmonary lipid composition of rabbits exposed to nickel dust.
Environ. Res., 26(2): 353-362.
CASEY, C.E. & ROBINSON, M.F. (1978) Copper, manganese, zinc,
nickel, cadmium and lead in human foetal tissues. Brit. J. Nutr.,
39: 639-646.
CASS, G.R. & MCRAE, G.J. (1983) Source-receptor reconciliation of
routine air monitoring data for trace metals: An emission
inventory-assisted approach. Environ. Sci. Technol., 17(3): 129-139.
CASTLEMAN, B. & MCNEELY, B.U. (1965) Case records of the
Massachusetts General Hospital, Case 38-1965. New Engl. J. Med.,
273: 494-504.
CASTRANOVA, V. BOWMAN, L., REASOR, M.J. & MILES, P.R. (1980)
Effects of heavy metal ions on selected oxidative metabolic
processes in rat alveolar macrophages. Toxicol. appl. Pharmacol.,
53(1): 14-23.
CATALANATTO, F.A., SUNDERMAN, F.W. Jr, & MCINTOSH, T.R. (1977)
Nickel concentrations in human parotid saliva. Ann. clin. lab.
Sci., 7(2): 146-151.
CHAN, W.H. & LUSIS, M.A. (1986) Smelting operations and trace
metals in air and precipitation in the Sudbury Basin. Adv.
environ. Sci. Technol., 17: 113-143.
CHANG, A.C., PAGE, A.L. & BINGHAM, F.T. (1982) Heavy metal
absorption by winter wheat following termination of cropland sludge
applications. J. environ. Qual., 11(4): 705-708.
CHANG, A.C., WARNECKE, J.E., LUND, L.J. & PAGE, A.L. (1984)
Accumulation of heavy metals in sewage sludge-related soils. J.
environ. Qual., 13(1): 87-91.
CHANG, C.C., TATUM, H.J. & KINCL, F.A. (1970) The effect of
intrauterine copper and other metals on implantation in rats and
hamsters. Fertil. Steril., 21: 274-278.
CHAUSMER, A.B. (1976) Measurement of exchangeable nickel in the
rat. Nutr. Rep. Int., 14: 323-326.
CHEN, J.R., FRANCISCO, R.B. & MILLER, T.E. (1977) Légionnaires'
disease: nickel levels. Science, 196: 906-908.
CHEN, K.Y., YOUNG, C.S., JAN, T.K. & ROHATGI, N. (1974) Trace
metals in wastewater effluents. J. Water Pollut. Fed., 46(12): 2663-2675.
CHIAUDANI, G. & VIGHI, M. (1978) The use of Selenastrum
capricornutum batch cultures in toxicity studies. Mitt. Int. Ver.
Limnol., 21: 316- 329.
CHORVATOVICOVA, D. (1983) The effect of nickel chloride on the
level of chromosome aberrations in Chinese hamster Cricetulus
griseus. Biologica, 38: 1107-1112.
CHOVIL, A., SUTHERLAND, R.B. & HALLIDAY, M. (1981) Respiratory
cancer in a cohort of nickel sinter plant workers. Br. J. ind.
Med., 38(4): 327-333.
CHRISTENSEN, J.M. & PEDERSEN, L.M. (1986) Enzymatic digestion of
whole blood for improved determination of cadmium, nickel and
chromium by electrothermal atomic absorption spectrophotometry:
measurements in patients with rheumatoid arthritis and in normal
humans. Acta pharmacol. toxicol., 59(7): 399-402.
CHRISTENSEN, O.B & KRISTENSEN, M. (1982) Treatment with disulfiram
in chronic nickel hand dermatitis. Contact Dermatit., 8(1): 59-63.
CHRISTENSEN, O.B. & MOELLER, H. (1975a) Nickel allergy and hand
eczema. Contact Dermatit., 1(3): 129-135.
CHRISTENSEN, O.B. & MOELLER, H. (1975b) External and internal
exposure to the antigen in the hand eczema of nickel allergy.
Contact dermatit., 1(3): 136-141.
CHRISTENSEN, O.B., MOELLER, H., ANDRASKO, K. & LAGESSON, V.,
(1979) Nickel concentration of blood, urine and sweat after oral
administration. Contact Dermatit., 5: 312-316.
CHRISTIE, N.T. & TUMMOLO, D. (1988) The role of Ni(II) in
mutation. In: Fourth International Conference on Nickel Metabolism
and Toxicology: Abstracts, Espoo, 5-9 September 1988, Helsinki,
Institute of Occupational Health, p. 33.
CHRISTIE, N.T., TUMMOLO, D.M., BIGGART, N.W. & MURPHY, E.C. Jr
(1988) Chromosomal changes in cell lines from mouse tumors induced
by nickel sulfide and methylcholanthre. Cell Biol. Toxicol., 4: 427.
CHURCH, S.E. (1981) Multi-element analysis of fifty-four
geochemical reference samples using inductively coupled plasma
atomic-emission spectrometry. Geostand. Newsl., V: 133-160.
CICCARELLI, R.B. & WETTERHAHN, K.E. (1982) Nickel distribution and
DNA lesions induced in rat tissues by the carcinogen nickel
carbonate. Cancer Res., 42: 3544-3549.
CICCARELLI, R.B. & WETTERHAHN, K.E. (1985) In vitro interaction of
63nickel(II) with chromatin and DNA from rat kidney and liver
nuclei. Chem.-biol. Interact., 52(3): 347-360.
CICCARELLI, R.B., HAMPTON, T.H. & JENNETTE, K.W. (1981) Nickel
carbonate induces DNA-protein crosslinks and DNA strand breaks in
rat kidney. Cancer Lett., 12(4): 349-354.
CIRLA, A.M., BERNABEO, F., OTTOBONI, F. & RATTI, R. (1985) Nickel-
induced occupational asthma - immunological and clinical aspects.
In: Brown, S.S. & Sunderman, F.W. Jr, ed. Progress in Nickel
toxicology. Proceedings of the 3rd International Congress on Nickel
Metabolism and Toxicology, Paris, 4-7 September, 1984, Oxford,
Blackwell Scientific Publications, pp. 165-168.
CLARK, J.R., VAN HASSEL, J.H., NICHOLSEN, R.B., CHERRY, D.S. &
CAIRNS, J. (1981) Accumulation and depuration of metals by duckweed
(Lemna perpusilla). Ecotoxicol. environ. Safety, 5: 87-96.
CLARY, J.J. (1975) Nickel chloride-induced metabolic changes in the
rat and guinea-pig. Toxicol. appl. Pharmacol., 31: 55-65.
CLARY, J.J. & VIGNATI, I. (1973) Nickel chloride-induced changes in
metabolism in the rat. Toxicol. appl. Pharmacol., 25: 467-468.
CLEMENTE, G.F., CIGNA ROSSI, L. & SANTARONI, G.P. (1980) Nickel in
foods and dietary intake of nickel. In: Nriagu, J.O., ed. Nickel in
the environment, New York, Chichester, Brisbane, Toronto, John
Wiley and Sons, pp. 493-498.
CLEMMENSEN, O.J., MENNE, T., KAABER, K. & SOLGAARD, P. (1981)
Exposure of nickel and the relevance of nickel sensitivity among
hospital cleaners. Contact Dermatit., 7(1): 14-18.
CLEMONS, G.K. & GARCIA, J.F. (1981) Neuroendocrine effects of acute
nickel chloride administration in rats. Toxicol. appl. Pharmacol.,
61: 343-348.
CLOUTIER, N.R., CLULOW, F.V., LIM, T.P. & DAVE, N.K. (1986) Metal
(copper, iron, cobalt, zinc, lead) and radium-226 levels in tissues
of meadow voles Microtus pennsylvanicus living on nickel and
uranium mine tailings in Ontario, Canada: Site, sex age and season
effects with calculation of average skeletal radiation dose.
Environ. Pollut., A41(4): 295-314.
COENEN, W., GROTHE, I. & KUHNEN, G. (1986) Exposure to welding
fumes in the workplace with regard to nickel and chromates. Int.
Congr. Ser. Excerpta Med., 676: 149-152.
COKER, E.G. & MATTHEWS, P.J. (1983) Metals in sewage sludge and
their potential effects in agriculture. Water Sci. Technol., 15:
209-217.
CONWAY, K. & COSTA, M. (1989) The involvement of hereto-chromatic
damage in nickel-induced transformation. In: Costa, M. &
Rossmann, T.G., ed., Proceedings of the First International Meeting
on the Molecular Mechanisms of Metal Toxicity and Carcinogenicity,
Urbino, Italy, September 1988. Clifton, New Jersey, Humana Press
Inc., pp.437-444 (Biological Trace Element Research, Vol. 21).
CORBETT, T.H., HEIDELBERGER, C. & DOVE, W.F. (1970) Determination
of the mutagenic activity to bacteriophage T4 of carcinogenic and
noncarcinogenic compounds. Mol. Pharmacol., 6: 667-679.
CORNELL, R.G. (1984) Mortality patterns among stainless-steel
workers. In: Nickel in the human environment, Proceedings of a
Joint Symposium, Lyon, 8-11 March, 1983, Lyon, International Agency
for Research on Cancer, pp. 65-71 (IARC Scientific Publications No.
53).
CORNELL, R.G. & LANDIS, J.R. (1984) Mortality patterns among
nickel/chromium alloy foundry workers. In: Nickel in the human
environment, Proceedings of a Joint Symposium, Lyon, 8-11 March,
1983, Lyon, International Agency for Research on Cancer, pp. 87-93
(IARC Scientific Publications No. 53).
CORRICK, J.D. (1977) Mineral commodity profiles: Nickel,
Washington, DC, US Department of the Interior, Bureau of Mines, 19
pp. (MCP-4).
COSTA, M. & MOLLENHAUER, H.H. (1980a) Carcinogenic activity of
particulate nickel compounds is proportional to their cellular
uptake. Science, 209(4455): 515-517.
COSTA, M. & MOLLENHAUER, H.H. (1980b) Phagocytosis of nickel
subsulfide particles during the early stage of neoplastic
transformation in tissue culture. Cancer Res., 40: 2688-2694.
COSTA, M., NYE, J.S., SUNDERMAN, F.W. Jr, ALLPASS, P.R. & GONDOS,
B. (1979) Induction of sarcomas in nude mice by implantation of
Syrian hamster fetal cells exposed in vitro to nickel subsulfide.
Cancer Res., 39: 3591-3597.
COSTA, M., JONES, M.K. & LINDBERG, O. (1980) Metal carcinogenesis
in tissue culture. In: Martell, A.E., ed. Inorganic chemistry in
biology and medicine. Washington, DC, pp. 45-73 (ACS Symp. Series
140).
COSTA, M., ABBRACCHIO, M.P. & SIMMONS-HANSEN, J. (1981a) Factors
influencing the phagocytosis, neoplastic transformation, and
cytoxicity of particulate nickel compounds in tissue culture
systems. Toxicol. appl. Pharmacol., 60: 313-313.
COSTA, M., SIMMONS-HANSEN, J., BEDROSSIAN, C.W., BONURA, J. &
CAPRIOLI, R.M. (1981b) Phagocytosis, cellular distribution, and
carcinogenic activity of particulate nickel compounds in tissue
culture. Cancer Res., 41(7): 2868-2876.
COSTA, M., HECK, J.D. & ROBINSON, S.H. (1982) Selective
phagocytosis of crystalline nickel sulfide particles and DNA
strandbreaks as a mechanism for the induction of cellular
transformation. Cancer Res., 42: 2757-2763.
COWGILL, U.M. (1976) The chemical composition of two species of
Daphnia, their algal food and their environment. Sci. total
Environ., 6: 79-102.
COX, J.E., DOLL, R., SCOTT, W.A. & SMITH, S. (1981) Mortality of
nickel workers: experience of men working with metallic nickel. Br.
J. ind. Med., 38(3): 235- 239.
CRAGLE, D.L., HOLLIS, D.R., NEWPORT, T.H. & SHY, C.M. (1984) A
retrospective cohort mortality study among workers occupationally
exposed to metallic nickel powder at the Oak-Ridge Tennessee USA
gaseous diffusion plant. In: Nickel in the human environment,
Proceedings of a Joint Symposium, Lyon, 8-11 March, 1983, Lyon,
International Agency for Research on Cancer, pp. 57-64 (IARC
Scientific Publications No. 53).
CRONIN, E. (1980) Contact dermatitis. Edinburgh, London, New York,
Churchill Livingstone, pp. 279-390.
CRONIN, E., DI MICHIEL, A.D. & BROWN, S.S. (1980) Oral challenge
in nickel-sensitive women with hand eczema. In: Brown, S.S. &
Sunderman, F.W. Jr, ed. Nickel toxicology. Proceedings of the 2nd
International Conference on Nickel Toxicology, Swansea, 3-5
September, 1980, London, New York, Academic Press, pp. 149-152.
CROSSMANN, G. (1988) [The cycle in the system soil-plant-animal on
locations with extremely high soil contaminations by cadmium and
nickel caused by sewage-sludge.] Berlin, Federal Agency of
Environmental Protection, 95 pp (Research report No. 106 07 043)
(in German).
CUCKLE, H., DOLL, R. & MORGAN, L.G. (1980) Mortality study of men
working with soluble nickel compounds. In: Brown, S.S. & Sunderman,
F.W. Jr, ed. Nickel toxicology. Proceedings of the 2nd
International Conference on Nickel Toxicology, Swansea, 3-5
September, 1980, London, New York, Academic Press, pp. 11-14.
CURSTEDT, T., HAGMAN, M., ROBERTSON, B. & CAMNER, P. (1983) Rabbit
lungs after long-term exposure to low nickel dust concentration.
Environ. Res., 30: 89-94.
CURSTEDT, T., CASARETT-BRUCE, M. & CANNER, P. (1984) Changes in
glycerophosphatides and their ether analogs in lung lavage of
rabbits exposed to nickel dust. Exp. mol. Pathol., 41: 26-34
CUSTER, T.W., FRANSON, J.C., MOORE, J.F. & MYERS, J.E. (1986)
Reproductive success and heavy metal contamination in Rhode Island
common terns. Environ. Pollut., A41(1): 33-52.
DALDRUP, T., HAARHOFF, K. & SZATHMARY, S.C. (1983) [Fatal nickel
sulfate poisoning.] Beitr. gerichtl. Med., 41: 141-144 (in German).
DALLINGER, R. & KAUTZKY, H. (1985) The importance of contaminated
food for the uptake of heavy metals by rainbow trout ( Salmo
gairdneri): A field study. Oecologia, 67(1): 82-89.
D'ALONZO, C.A. & PELL, S. (1963) A study of trace metals in
myocardial infarction. Arch. environ. Health, 6: 381-385.
DAMJANOV, I., SUNDERMAN, F.W. Jr, MITCHELL, J.M. & ALLPASS, P.R.
(1978) The induction of testicular sarcomas in Fischer rats by
intratesticular injection of nickel subsulfide. Cancer Res., 38:
268-276.
DANIELSSON, L.-G., MAGNUSSON, B. & WESTERLUND, S. (1978) An
improved metal extraction procedure for the determination of trace
metals in seawater by atomic absorption spectrometry with
electrothermal atomization. Anal. Chim. Acta, 98: 47-57.
DAVYDOVA, V.I., NEIZVESTNOVA, E.M., BLOKHIN, V.A. & SERGOVA, N.V.
(1981) Toxicological evaluation of manganese, chromium and nickel.
Gig. i Sanit., 7: 20-22.
DECATANZARO, J.B. & HUTCHINSON, T.C. (1985a) Effects of nickel
addition on nitrogen mineralization, nitrification, and nitrogen
leaching in some boreal forest soils. Water air soil Pollut., 24(2):
153-164.
DECATANZARO, J.B. & HUTCHINSON, T.C. (1985b) Leaching and
distribution of nitrogen and nickel in nickel-perturbed jack pine
forest microcosms. Water air soil Pollut., 26(3): 281-292.
DECHENG, C., MING, J., LING, H., SHAN, W., ZIQING, X. & XINSHUI,
Z. (1987) Cytogenic analysis in workers occupationally exposed to
nickel carbonyl. Mutat. Res., 188: 149-152.
DECSY, M.I. & SUNDERMAN, F.W. Jr. (1974) Binding of 63Ni to rabbit
serum alpha-macroglobulin in vivo and in vitro. Bioinorg. Chem.
3(2): 95-105.
DEFLORA, S., ZANACCI, P., CAMOIRANO, A., BENICELLI, C. & BADOLATI,
G.S. (1984) Genotoxic activity and potency of 135 compounds in the
Ames reversion test and in a bacterial DNA-repair test. Mutat.
Res., 133: 161-198.
DEKNUDT, G. & LEONARD, A. (1982) Mutagenicity tests with nickel
salts in the male mouse. Toxicology, 25(4): 289-292.
DELGADO, E.A. (1958) Sarcoma following a surgically treated
fractured tibia. Clin. Orthop., 12: 315-318.
DENG, C. & QU, B. (1981) The cytogenetic effects of nickel
sulphate. Acta genet. sin., 8: 212-215.
DENG, C., QU, B., HUANG, J., ZHUO, Z.L., XIAN, H., YAO, M., CHEN,
M.Y., LI, Z.X., SHENG, S.Y. & YEI, Z.F. (1983) [Cytogenetic
effects of electroplating workers.] Acta scientiae circumstantiae,
3: 267-271 (in Chinese with English abstract).
DENG, C., LEE, H.H., XIAN, H., YAO, M., HUANG, J. & QU, B. (1988)
Chromosomal aberrations and sister chromatid exchanges of
peripheral blood lymphocytes in Chinese electroplating workers. J.
trace Elem. exp. Med., 1: 57-62.
DERMOTT, R.M. & LUM, K.R. (1986) Metal concentrations in the annual
shell layers of the bivalve Elliptio complanata. Environ. Pollut.
B12(2): 131-144.
DEUTMAN, R., MULDER, T.J., BRIAN, R. & NATER, J.P. (1977) Metal
sensitivity before and after total hip arthroplasty. J. bone joint
Surg., 59(7): 862-865.
DEWLING, R., MANGANELLI, R. & BAER, G. (1980) Fate and behaviour
of selected heavy metals in incinerated sludge. J. Water Pollut.
Control Fed., 52: 2552-2557.
DIEKERT, G. & RITTER, M. (1982) Nickel requirement of
Acetobacterium woodii. J. Bacteriol. 151: 1043-1045.
DIEKERT, G. & THAUER, R.K. (1980) The effect of nickel on carbon
monoxide dehydrogenase formation in Clostridium thermoaceticum and
Clostridium formicoaceticum. FEMS microbiol. Lett., 7: 187-189.
DIETZ, R.N. & WIESER, R.F. (1983) Sulfate formation in oil-fired
power plant plumes. Vol. 1: Parameters affecting primary sulfate
emissions and a model for predicting emissions and plume opacity,
Upton, NY, Brookhaven National Laboratories (Report No. EA-3231).
DIPAOLO, J.A. & CASTO, B.C. (1979) Quantitative studies of in
vitro morphological transformation of Syrian golden hamster
cells by inorganic metal salts. Cancer Res., 39: 1008-1013.
DITORO, D.M., MAHONY, J.D., KRICHGRABER, P.R., O'BYRNE, A.L. &
PASQUALE, L.R. (1986) Effect of nonreversibility, particle
concentration and ionic strength on heavy metal sorption. Environ.
Sci. Technol., 20(1): 55-61.
DIXON, N.E., GAZZOLA, C., BEAKELEY, R.L. & ZERNER, B. (1975) Jack
bean urease (EC 3.5.1.5). A metalloenzyme. A simple biological role
for nickel? J. Am. Chem. Soc., 97: 4130-4133.
DJACHENKO, O.Z (in press) Effects of nickel and manganese on the
chromosome aberrations and sister chromatid exchanges in human
lymphocytes in vitro. In: Domnin, S.G. & Shcherbakov, S.V. ed.
Problems of labour hygiene in steel and coloured metals industry,
Moscow, Erisman Institute of Hygiene.
DOLL, R. (1958) Cancer of the lung and nose in nickel workers. Br.
J. ind. Med., 15: 217-223.
DOLL, R., MORGAN, L.G. & SPEIZER, F.E. (1970) Cancers of the lung
and nasal sinuses in nickel workers. Br. J. Cancer, 24(4): 623-632.
DOLL, R., MATTHEWS, J.D. & MORGAN, L.G. (1977) Cancers of the lung
and nasal sinuses in nickel workers: A reassessment of the period
of risk. Br. J. ind. Med.,34(2): 102-105.
DOLOVICH, J., EVANS, S.L. & NIEBOER, E. (1984) Occupational asthma
from nickel sensitivity: I. Humam serum albumin in the antigenic
determinant. Br. J. ind. Med., 41(1): 51-55.
DOOMS-GOOSSENS, A., CEUTERICK, A., VANMAELE, N. & DEGREEF, H.
(1980) Follow-up study of patients with contact dermatitis caused
by chromates, nickel and cobalt. Dermatologica, 160(4): 249-260.
DORMER, R.L., KERBEY, A.L., MCPHERSON, M., MANLEY, S., ASHCROFT,
S.J.H., SCHOFIELD, J.G. & RANDLE, P.J. (1973) The effect of nickel
on secretory systems. Studies on the release of amylase, insulin
and growth hormone. Biochem. J., 140: 135-142.
DORNEMANN, A. & KLEIST, H. (1980) Nickel in liver and kidney
samples. In: Brown, S.S. & Sunderman, F.W. Jr. ed. Nickel
toxicology, Proceedings of the 2nd International Conference on
Nickel Toxicology, Swansea, 3-5 September, 1980, London, New York,
Academic Press, pp. 175-178.
DOSTAL, L.A., HOPFER, S.M., LIN, S.M. & SUNDERMAN, F.W. Jr (1989)
Effects of nickel chloride on lactating rats and their suckling
poups, and the transfer of nickel through rat milk. Toxicol. appl.
Pharmacol.,101: 220-231.
DRAKE, H.L. (1982) Occurrence of nickel in carbon monoxide
dehydrogenase from Clostridium thermoaceticum. J. Bacteriol. 149:
561-566.
DRINKER, K.R., FIARHALL, J.T., RAY, G.B. & DRINKER, C.K. (1924)
The hygienic significance of nickel. J. ind. Hyg., 6: 307-360.
DUBE, V.E. & FISHER, D.E. (1972) Hemangioendothelioma of the leg
following metallic fixation of the tibia. Cancer, 30: 1260-1266.
DUBINS, J.S. & LAVELLE, J.M. (1986) Nickel(II), genotoxicity:
potentiation of mutagenesis of simple alkylating agents. Mutat.
Res., 162(2): 187-199.
DUBREUIL, A., BOULEY, G., DURET, S., MESTRE, J.C. & BOUDENE, C.
(1984) In vitro cytotoxicity of nickel chloride on a human
pulmonary epithelial cell line. Arch. Toxicol., Suppl. 7: 391-393.
DUKE, J.M. (1980) Nickel in rocks and ores. In: Nriagu, J.O. ed.
Nickel in the environment, New York, Chichester, Brisbane, Toronto,
John Wiley and Sons, pp. 27-50.
DUNNICK, J.K., BENSON, J.M., HOBB, C.H., HAHN, F.F., CHENG, Y.S. &
EIDSON, A.F. (1988) Comparative toxicity of nickel oxide, nickel
sulfate hexahydrate, and nickel subsulfide after 12 days of
inhalation exposure to F344/N rats and B6C3F1 mice. Toxicology, 50:
145-156.
DUYEVA, L.A. (1983) [Allergenicity of metals.] In: [Metals.
Hygienic aspects of environmental evaluation and improvement].
Moscow, Meditsina, pp. 28-41 (in Russian).
EDMAN, B. & MOELLER, H. (1982) Trends and forecasts for standard
allergens in a 12-year patch test material. Contact Dermatit., 8(2):
95-104.
EGEDAHL, R. & RICE, E. (1984) Cancer incidence at a
hydrometallurgical nickel refinery. In: Nickel in the human
environment, Proceedings of a Joint Symposium, Lyon, 8-11 March,
1983, Lyon, International Agency for Research on Cancer, pp. 47-56
(IARC Scientific Publications No. 53).
EGILSSON, V., EVANS, I.H. & WILKIE, D. (1979) Toxic and mutagenic
effects of carcinogens on the mitochondria of Saccharomyces
cerevisiae. Molec. gen. Genet., 174: 39-46.
EILERTSEN, E., SKAUG, V. & NORSETH, T. (1988) Tumor induction in
rats after intrapleural instillation of various nickel oxides. In:
Fourth International Conference on Nickel Metabolism and
Toxicology, Abstracts, Espoo, Finland, 5-9 September 1988.
Helsinki, Institute of Occupational Health, p. 43.
EKER, P. & SANNER, T. (1983) Assay for initiators and promoters of
carcinogenesis based on attachment-independent survival of cells in
aggregates. Cancer Res., 43: 320-323.
ELAKHOVSKAYA, N.P. (1972) [Metabolism of nickel entering the body
with drinking water.] Gig.i Sanit., 37(6): 20-22 (in Russian).
ELLEN, G., VAN DEN BOSCH-TIBBESMA, G. & DOUMA, F.F. (1978) Nickel
content in various Dutch foodstuffs. Z. Lebensm. Unters. Forsch.
166(3): 145-147.
ENGLISH, J.C., PARKER, R.D., SHARMA, R.P. & OBERG, S.G. (1981)
Toxicokinetics of nickel in rats after intratracheal administration
of a soluble and insoluble form. Am. Ind. Hyg. Assoc. J., 42(7):
486-492.
ENTERLINE, P.E. & MARSH, G.M. (1982) Mortality among workers in a
nickel refinery and alloy manufacturing plant in West Virginia. J.
Natl Cancer Inst., 68: 925-933.
EVANS, R.M., DAVIES, P.J.A. & COSTA, M. (1982) Video time-lapse
microscopy of phagocytosis and intracellular fate of crystalline
nickel sulfide particles in cultured mammalian cells. Cancer Res.
42: 2829-2735.
EVANS, R.M., YANO, E., MORGAN, L.G. & URANO, N. (1988)
Chemiluminescent detection of free oxygen radical generation by
stimulated PMN in vitro: effects of nickel compounds. In: Fourth
International Conference on Nickel Metabolism and Toxicology,
Abstracts, Espoo, 5-9 September l988, Helsinki, Institute of
Occupational Health, p. 37.
EVANS, W.H., READ, J.I. & LUCAS, B.E. (1978) Evaluation of a
method for the determination of total cadmium, lead and nickel in
foodstuffs using measurement by flame atomic-absorption
spectrophotometry. Analyst, 103(1227): 580-594.
FALANDYSZ, J. (1985) Trace metals in flatfish from the southern
Baltic, 1983. Z. Lebensm. Unters. Forsch., 181(2): 117-120.
FALANDYSZ, J. (1986a) Trace metals in cod from the southern Baltic,
1983. Z. Lebensm. Unters. Forsch., 182(3): 228-231.
FALANDYSZ, J. (1986b) Trace metals in herring from the southern
Baltic, 1983. Z. Lebensm. Unters. Forsch., 182(1): 36-39.
FARRELL, R.L. & DAVIS, G.W. (1974) The effects of particulates on
respiratory carcinogenesis by diethylnitrosamine. In: Karbe, E. &
Park, I.F. ed. Experimental lung cancer, carcinogenesis, and
bioassays, Berlin, Heidelberg, New York, Springer Verlag, pp. 219-
233.
FEREN, K. & REITH, A. (1988). Phagocytosis and transformation
testing of Ni3S2 using primary epithelial respiratory cells.
Fourth International Conference on Nickel Metabolism and
Toxicology, Abstracts, Espoo, Finland, 5-9 September 1988,
Helsinki, Institute of Occupational Health, p.64.
FERM, V.H. (1972) The teratogenic effects of metals on mammalian
embryos. Adv. Teratol., 5: 51-75.
FEZY, J.S., SPENCER, D.F. & GREENE, R.W. (1979) The effect of
nickel on the growth of the freshwater diatom Navicula pelliculosa.
Environ. Pollut., 20:131-137.
FIGONI, R. & TREAGAN, L. (1975) Inhibition effect of nickel and
chromium upon antibody response of rats immunization with T-1
phage. Res. Commun. chem. Pathol. Pharmacol., 11: 335-338.
FILKOVA, L. & JAGER, J. (1986) Nonoccupational exposure to nickel
tetracarbonyl. Cesk. Hyg., 31(5): 255-259.
FISHBEIN, L. (1981) Sources, transport and alterations of metal
compounds: An overview. I. Arsenic, beryllium, cadmium, chromium,
and nickel. Environ. Health Perspect., 40: 43-64.
FISCHER, T., FREGERT, S., GREENBERGER, B. & RYSTEDT, I. (1984)
Contact sensitivity to nickel in white gold. Contact Dermatit., 10:
213-24.
FISHELSON, Z. PANBURN, M.K. & MULLER-EBERHARD, H.J. (1983) C3
convertase of the alternative complement pathway. Demonstration
of an active, stable C3b,Bb(Ni) complex. J. biol. Chem., 258(12): 7411-
7415.
FISHER, A.A. (1977) Allergic dermatitis presumably due to metallic
foreign bodies containing nickel or cobalt. Cutis, 19(3): 285-286.
FISHER, A.A. (1985) Nickel dermatitis in men. Cutis, 35(5): 424-426.
FISHER, A.A. & SHAPIRO, A. (1956) Allergic eczematous contact
dermatitis due to metallic nickel. J. Am. Med. Assoc., 161: 717-721.
FISHER, G.L., CHRISP, C.E., MCNEILL, K.L., MCNEILL, D.A., DEMOCKO,
C. & FINCH, G.L. (1984) Mechanistic evaluations of the pulmonary
toxicology of nickel subsulfide. Adv. mod. environ. Toxicol., 6: 49-
60.
FISHER, G.L., CRISP, C.E. & MCNEILL, D.A. (1986) Lifetime effects
of intertracheally instilled nickel subsulfide on B6C3F1 mice.
Environ. Res, 40: 313-320.
FISHER, N.S. (1986) On the reactivity of metals for marine
phytoplankton. Limnol. Oceanogr., 31(2): 443-449.
FLORA, C.J. & NIEBOER, E. (1980) Determination of nickel by
differential pulse polarography at a dropping mercury electrode.
Anal. Chem., 52: 1013-1020.
FORNACE, A.J. Jr (1982) Detection of DNA single-strand breaks
produced during the repair of damage by DNA-protein cross-linking
agents. Cancer Res., 42: 145-149.
FORTH, W. & RUMMEL, W. (1971) Absorption of iron and chemically
related metals in vitro and in vivo: specificity of the iron-
binding system in mucosa of the jejunum. In: Shoryna, S.C. &
Waldron-Edward, D. ed. Intestinal absorption of metal ions, trace
elements and radionuclides, New York, Pergamon Press, pp. 173-191.
FOULKES, E.C. & BLANCK, S. (1984) The selective action of nickel
on tubule function in rabbit kidneys. Toxicology, 33: 245-249.
FOULKES, E.C. & McMULLEN, D.M. (1986) On the mechanism of nickel
absorption in the rat jejunum. Toxicology, 38: 35-42.
FRACHE, R., BAFFI, F., DADONE, A., SCARPONI, G. & DAGNINO, I.
(1980) The determination of heavy metals in the Ligurian sea. II.
The geographical and vertical distribution of Cd, Cu, and Ni. Deep-
Sea Res., 27A: 1079-1082.
FRANCIS, C.W., DAVIS, E.C. & GOYERT, J.C. (1985) Plant uptake of
trace elements from coal gasification ashes. J. environ. Qual.
14(4): 561-569.
FREEMAN, B.M. & LANGSLOW, D.R. (1973) Response of plasma glucose,
free fatty acids and glucagon to cobalt and nickel chlorides by
Gallus domesticus. Comp. Biochem. Physiol., 46A: 427-436.
FRIBERG, L. (1950) Health hazards in the manufacture of alcaline
accumulation. Acta med. Scand., 138(240): 124.
FRIEDLAND, A.J., JOHNSON, A.H. & SICCAMA, T.G. (1986) Zinc, Cu, Ni
and Cd in the forest floor in the Northeastern United States.
Water air soil Pollut., 29: 233-243
FRIEDRICH, B., HEINE, E., FINK, A. & FRIEDRICH, C.G. (1981) Nickel
requirement for active hydrogenase formation in Alcaligenes
eutrophus. J. Bacteriol., 145: 1144-1149.
FROSCH, P.J. & KLIGMAN, A.M. (1976) The chamber-scarification test
for irritancy. Contact Dermatit., 2: 314-324.
FUKUNAGA, M., KURACHI, Y. & MIZYGUCHI, Y. (1982) Action of some
metal ions on yeast chromosomes. Chem. pharm. Bull. (Tokyo), 30:
3017-3019.
FULLERTON, A., ANDERSEN, J.R., HOELGAARD, A. & MENNE, T. (1986)
Permeation of nickel salts through human skin in vitro. Contact
Dermatit., 15: 183-177.
FURST, A. & AL-MAHROUQ, H. (1981) Excretion of nickel following
intratracheal administration of the carbonate. Proc. West.
Pharmacol. Soc., 24: 119-121.
FURST, A. & CASSETTA, D. (1973) Carcinogenicity of nickel by
different routes. Proc. Am. Assoc. Cancer Res., 121: 31.
FURST, A. & SCHLAUDER, M.C. (1971) The hamster as a model for
metal carcinogenesis. Proc. West. Pharmacol. Soc., 14: 68-71.
FURST, A., CASSETTA, D.M. & SASMORE, D.P. (1973) Rapid induction
of pleural mesotheliomas in the rat. Proc. West. Pharmacol. Soc.
16: 150-153.
GAINER, J.H. (1977) Effects of heavy metals and/or of deficiency of
zinc on mortality rates in mice infected with encephalomyocarditis
virus. Am. J. vet. Res., 38: 869-872.
GAWKRODGER, D.J., LLOYD, M. & HUNTER, J. (1986a) Occupational skin
disease in hospital cleaning and kitchen workers. Contact
Dermatit., 15(3): 132-135.
GAWKRODGER, D.J., COOK, S.W., FELL, G.S. & HUNTER, J.A.A. (1986b)
Nickel dermatitis: the reaction to oral nickel challenge. Br. J.
Dermatol., 115: 33-38.
GEMMER-COLOS, U., TUSS, H., SAUR, D. & NEEB, R. (1981)
[Polarographic determination of Nickel in the ppb-range after
extracting nickel as dioxime.] Fresenius Z. anal. Chem., 307: 347-351
(in German).
GENDREAU, R.M., JAKOBSEN, R.J. & HENRY, W.M. (1980) Fourier
transform infrared spectroscopy for inorganic compound speciation.
Environ. Sci. Technol., 14: 990-995.
GERIN, M., SIEMIATYCKI, J., RICHARDSON, L., PELLERIN, J., LAKHANI,
R. & DEWAR, R. (1984) Nickel and cancer associations from a
multicancer occupation exposure case-referent study - preliminary
findings. In: Nickel in the Human Environment, Proceedings of a
Joint Symposium, Lyon, 8-11 March, 1983, Lyon, International Agency
for Research on Cancer, pp. 105-115 (IARC Scientific Publications
No. 53).
GERSTLE, R.W. & ALBRINCK, D.N. (1982) Atmospheric emissions of
metals from sewage sludge incineration. J. Air Pollut. Control
Assoc., 32(11): 1119-1123.
GHIRINGHELLI, L. (1957) [Therapeutic utility of 2-3
dimercaptopropanol and thiotic acid in rats poisoned with nickel
carbonyl.] Attiv. Soc. Lomb. Med. Biol., 12: 24-26 (in Italian).
GHIRINGHELLI, J. & AGAMENNONE, M. (1957) [The metabolism of nickel
in animals experimentally poisoned with nickel carbonyl.] Med.
Lav., 48: 187-194 (in Italian).
GIGNAC, L.D. & BECKETT, P.J. (1986) The effect of smelting
operations on peatlands near Sudbury, Ontario, Canada. Can. J.
Bot., 64(6): 1138-1147.
GILANI, S.H. & MARANO, M. (1980) Congenital abnormalities in
nickel poisoning in chick embryos. Arch. environ. Contam.
Toxicol., 9(1): 17-22.
GILMAN, J.P.W. (1962) Metal carcinogenesis. II. A study of the
carcinogenic activity of cobalt, copper, iron and nickel compounds.
Cancer Res., 22: 158- 162.
GILMAN, J.P.W. & HERCHEN, H. (1963). The effect of physical form
of implant on nickel sulphide tumourigenesis in the rat. Acta
Union int. Cancer, 19: 615-619.
GILMAN, J.P.W. & RUCKERBAUER, G.M. (1962) Metal carcinogenesis. I.
Observations on the carcinogenicity of a refinery dust, cobalt
oxide, and colloidal thorium dioxide. Cancer Res., 22: 152-157.
GILMAN, J.P.W. & YAMASHIRO, S. (1985) Muscle tumorigenesis by
nickel compounds. In: Brown, S.S. & Sunderman, F.W. Jr, ed.
Progress in Nickel toxicology. Proceedings of the 3rd International
Conference on Nickel Metabolism and Toxicology, Paris, 4-7
September, 1984, Oxford, Blackwell Scientific Publications,
pp. 9-22.
GITLITZ, P.H., SUNDERMAN, F.W. & GOLDBLATT, P.J. (1975)
Aminoaciduria and proteinuria in rats after single intraperitoneal
injection of Ni(II). Toxicol. appl. Pharmacol., 34: 430-440.
GLAESS, E. (1956a) [Distribution of fragmentations and achromatic
areas on chromosomes of Vicia faba following treatment with heavy
metal salts]. Chromosome, 8: 260-284 (in German).
GLAESS, E. (1956b) [Investigations on the effects of heavy metal
salts on root top mitosis of Vicia faba.] Ztschr. Botanik, 44:
1-58 (in German).
GLASER, U., HOCHRAINER, D., OLDIGES, H. & TAKENAKA, S. (1986)
Long-term inhalation studies with NiO and As2O3 aerosols in Wistar
rats. In: Stern, R.M., Berlin, A., Fletcher, A.C. & Jarvisalo,
J. ed. Health hazards and biological effects of welding fumes and
gases. Proceedings of The International Conference on Health
Hazards and Biological Effects of Welding Fumes and Gases,
Copenhagen, 18-21 February, 1985, Amsterdam, New York, Oxford,
Elsevier Science Publishers, pp. 325-328.
GLENNON, J.D. & SARKAR, B. (1982) Nickel(II) transport in human
blood serum. Studies of nickel(II) binding to human albumin and to
native-sequence peptide, and ternary-complex formation with L-
histidine. Biochem. J., 203(1): 15-23.
GODBOLD, J.H. Jr & TOMPKINS, E.A. (1979) A long-term mortality
study of workers occupationally exposed to metallic nickel at the
Oak Ridge Gaseous Diffusion Plant. J. occup. Med., 21(12): 799-806.
GOLDBERG, M., FUHRER, R., BRODEUR, J.-M., GOLDBERG, P., SEGNAN, N.,
LECLERC, A., CHASTANG, J.-F., BOURBONNAIS, R. & FRANCOIS, D.
(1985a) Cancer des voies respiratoires parmi les travailleurs d'une
entreprise d'extraction et du raffinage du nickel en Nouvelle
Calédonie: Une étude cas-témoins au sein d' une cohorte. In: Brown,
S.S. & Sunderman, J.W. Jr, ed. Progress in Nickel toxicology,
Proceedings of the 3rd International Conference on Nickel
Metabolism and Toxicology, Paris, 4-7 September, 1984, Oxford,
Blackwell Scientific Publications, pp. 215-218.
GOLDBERG, P., BLANC, M., FUHRER, R., SEGNAN, N., BRODEUR, J.-M.,
LECLERC, A., FRANCOIS, D., DAB, W., GODARD, C. & GOLDBERG, M.
(1985b) Incidence des cancers respiratoires chez les travailleurs
d'une entreprise d'extraction et de raffinage du nickel et dans
la population générale en Nouvelle Calédonie. In: Brown, S.S. &
Sunderman, F.W. Jr, ed. Progress in Nickel toxicology. Proceedings
of the 3rd International Conference on Nickel Metabolism and
Toxicology, Paris, 4-7 September, 1984, Oxford, Blackwell
Scientific Publications, pp. 211-214.
GOLDBERG, M., GOLDBERG, P., LECLERC, A., CHASTANG, J.F., MARNE,
M.J., GUEZIEC, J., LAVIGNE, F., DUBOURDIEU, D. & HUERRE, M. (1988)
A 7-year survey of respiratory cancers among nickel workers in New
Caledonia (1978-1984). In: Fourth International Conference on
Nickel Metabolism and Toxicology, Abstracts, Espoo, Finland, 5-9
September 1988, Helsinki, Institute of Occupational Health, p. 42.
GORDON, C.J. (1989) Effect of nickel chloride on body temperature
and behavioral thermoregulation in the rat. Neurotoxicol. Teratol.,
11: 317-320.
GORDON, C.J. & STEAD, A.G. (1986) Effect of nickel and cadmium
chloride on autonomic and behavioral thermoregulation in mice.
Neurotoxicology, 7: 97-106.
GORDON, C.J., FOGELSON, L. & STEAD, A.G. (1989) Temperature
regulation following nickel intoxication in the mouse: effect of
ambient temperature. Comp. Biochem. Physiol., 92: 73-76.
GORDYNIA, R.I. (1969) [Effect of a ration containing a nickel salt
additive on carbohydrate metabolism in experimental animals.] Vopr.
Racion. Pitan., 5: 167-170 (in Russian).
GRAHAM, J.A., GARDNER, D.E., WATERS, M.D. & COFFIN, D.L. (1975a)
Effect of trace metals on phagocytosis by alveolar macrophages.
Infect. Immun., 11: 1278-1283.
GRAHAM, J.A., GARDNER, D.E., MILLER, F.J., DANIELS, M.J. & COFFIN,
D.L. (1975b) Effect of nickel chloride on primary antibody
production in the spleen. Environ. Health Perspect., 12: 109-113.
GRAHAM, J.A., MILLER, F.J., DANIELS, M.J., PAYNE, E.A. & GARDNER,
D.E. (1978) Influence of cadmium, nickel, and chromium on primary
immunity in mice. Environ. Res., 16: 77-87.
GRANDJEAN, P. (1984) Human exposure to nickel. In: Nickel in the
human environment. Proceedings of a Joint Symposium, Lyon, 8-11
March, 1983, Lyon, International Agency for Research on Cancer, pp.
469-485 (IARC Scientific Publications No. 53).
GRANDJEAN, P., SELIKOFF, I.J., SHEN, S.K., SUNDERMAN, F.W. Jr
(1980) Nickel concentrations in plasma and urine of shipyard
workers. Am J. ind. Med., 1(2): 181-189.
GRANDJEAN, P., ANDERSEN, O. & NIELSEN, G.D. (1988) Nickel. In:
Alession, L. Berlin, A. Boris, M. & Roi, R. ed. Biological
indicators for the assessment of human exposure to industrial
chemicals. ISPRA (Varese), IPSRA Establishment, Joint Research
Centre, Commission of the European Communities, pp. 56-80.
GREBNEV, V.L. & YELNICHNYKH, L.N. (1983) Experimental study of the
action of nickel-containing aerosols in preparatory workshops of
nickel production on parenchymour organs. Non-specific action of
harmful occupation factors on workers' organism, Sverdlovsk,
Medical College, pp. 90-95.
GREEN, M.H.L., MURIEL, W.J. & BRIDGES, B.A. (1976). Use of a
simplified fluctuation test to detect low levels of mutagens.
Mutat. Res., 38: 33-42.
GROPPEL, B., ANKE, M., RIEDEL, E. & GRUN, M. (1980) [The nickel
supply and status of wild ruminants in the GDR.] In: Anke, M.
Schneider, H.-J. & Brûckner, Chr. ed. [3. Trace Element
Symposium: Nickel, Jena, German Democratic Republic, 7-11 July,
1980,] Jena, Friedrich-Schiller University, pp. 269-276 (in German).
GROSS, P.R., KATZ, S.A. & SAMITZ, M.H. (1968) Sensitization of
guinea-pigs to chromium salts. J. invest. Dermatol., 50: 424-427.
GUTENMANN, W.H., BACHE, C.A., LISK, D.J., HOFFMANN, D., ADAMS,
J.D. & ELFVING, D.C. (1982) Cadmium and nickel in smoke of
cigarettes prepared from tobacco cultured on municipal sludge-
amended soil. J. Toxicol. environ. Health, 10(3): 423-431.
HACKETT, R.L. & SUNDERMAN, F.W. Jr (1967) Acute pathological
reactions to administration of nickel carbonyl. Arch. environ.
Health, 14(4): 604-613.
HACKETT, R.L. & SUNDERMAN, F.W. Jr (1968) Pulmonary alveolar
reaction to nickel carbonyl: Ultrastructural and histochemical
studies. Arch. environ. Health, 16(3): 349-362.
HACKETT, R.L. & SUNDERMAN, F.W. Jr (1969) Nickel carbonyl. Effects
upon the ultrastructure of hepatic parenchymal cells. Arch.
environ. Health, 19: 337- 343.
HAGEDORN-GOTZ, H., KUPPERS, G. & STOPPLER, M. (1977) On nickel
contents in urine and hair in a case of exposure to nickel
carbonyl. Arch. Toxicol., 38(4): 275-285.
HALE, J.G. (1977) Toxicity of metal mining wastes. Bull. environ.
Contam. Toxicol., 17:66-73.
HALEY, P.Y., BICE, D.E., MUGGENBURG, B.A., HAHN, F.F. & BENJAMIN,
S.A. (1987) Immunopathologic effects of nickel subsulfide on the
primate pulmonary system. Toxicol. appl. Pharmacol., 88: 1-12
HALL, T.M. (1982) Free ionic nickel accumulation and localization
in the freshwater zooplankton Daphnia magna. Limnol. Oceanogr.
27(4): 718-727.
HANSEN, K. & STERN, R.M. (1983) In vitro toxicity and
transformation potency of nickel compounds. Environ. Health
Perspect., 51: 223-226.
HANSEN, K. & STERN, R.M. (1984) Toxicity and transformation potency
of nickel compounds in BHK cells in vitro. In: Nickel in the human
environment. Proceedings of a Joint Symposium, Lyon, 8-11 March,
1983, Lyon, International Agency for Research on Cancer, pp. 193-
200 (IARC Scientific Publications No. 53).
HANSEN, L.D. & FISHER, G.L. (1980) Elemental distribution in coal
fly ash particles. Environ. Sci. Technol., 14(9): 1111-1117.
HANSEN, L.D., SILBERMAN, D., FISHER, G.L. & EATOUGH, D.J. (1984)
Chemical speciation of elements in stack-collected, respirable-size
coal fly ash. Environ. Sci. Technol., 18: 181-186.
HARO, R.T., FURST, A. & FALK, H.L. (1968) Studies on the acute
toxicity of nickelocene. Proc. West. Pharmacol. Soc., 11: 39-42.
HARNETT, P.B., ROBISON, S.H., SWARTZENDRUBER, D.E. & COSTA, M.
(1982) Comparison of protein, RNA, and DNA binding and cell-cycle-
specific growth inhibitory effects of nickel compounds in cultured
cells. Toxicol. appl. Pharmacol., 64: 20-30.
HARTENSTEIN, R., NEUHAUSER, E.F. & NARAHARA, A. (1981) Effects of
heavy metal and other elemental additives to activated sludge on
growth of Eisenia foetida. J. environ. Qual., 10(3): 372-377.
HARTWIG, A. & BEYERSMANN, D. (1989) Enhancement of UV-induced
mutagenesis and sister-chromatid exchanges by nickel ions in V79
cells: evidence for inhibition of DNA repair. Mutat. Res., 217: 65-73.
HAWLEY, J.E. (1955) Spectographic study of some Nova Scotia coals.
Can. min. metall. Bull., 48: 712-726.
HAWORTH, S., LAWLOR, T., MORTELMANS, K., SPECK, W. & ZEIGER, E.
(1983) Salmonella mutagenicity test results in 250 chemicals.
Environ. mol. Mutagen., 5: 3-142.
HAYASHI, YL, TAKAHASHI, M., MAEKAWA, A., KUROKAWA, Y. & KOLUBO, T.
(1985) Screening of environmental pollutants for promotion effects
on carcinogenesis. Annu. Rep. Minist. Health Welfare Jpn 1982-
1984, 20: 1-10.
HEATH, J.C. & DANIEL, M.R. (1964) The production of malignant
tumors by nickel in the rat. Br. J. Cancer, 18: 261-264.
HECK, J.D. & COSTA, M. (1982) Extracellular requirements for the
endocytosis of carcinogenic crystalline nickel sulfide particles by
facultative phagocytes. Toxicol. Lett., 12: 243-250.
HEINRICHS, H. & R. MAYER, R. (1980) Distribution and cycling of
nickel in forest ecosystems. In: Nriagu, J.O. ed. Nickel in the
environment, New York, Chichester, Brisbane, Toronto, John Wiley
and Sons, pp. 431-455.
HENDEL, R.C. & SUNDERMAN, F.W. Jr (1972) Species variations in the
properties of ultrafiltrable and protein-bound serum nickel. Res.
Commun. chem. Pathol. Pharmacol., 4(1): 141-146.
HENRY, W.M. & KNAPP, K.T. (1980) Compound forms of fossil fuel fly
ash emissions. Environ. Sci. Technol., 14(4): 450-456.
HENRY, W.M., BARBOUR, R.L., JAKOBSEN, R.J. & SCHUMAKER, P.M.
(1982) Inorganic compound identification of fly ash emissions from
municipal incinerators. Final Report, Washington, DC, US
Environmental Protection Agency, 32 pp (Report prepared by Battelle
Columbus Laboratories, Ohio, under the EPA Contract 68-02-2296)
(EPA-600/3-82-095. PB83-146175).
HERLANT-PEERS, M.C., HILDEBRAND, H.F. & BISERTE, G. (1982) 63Ni(II)
incorporation into lung and liver cytosol of Balb/C mouse. An in
vitro and in vivo study. Zentralbl. Bakteriol. Mikrobiol. Hyg. (B),
176(4): 368-382.
HILDEBRAND, H.F. & BISERTE, G. (1979a) Cylindrical laminated bodies
in nickel subsulphide-induced rhabdomyosarcoma in rabbits. Eur. J.
cell Biol., 19: 276-280.
HILDEBRAND, H.F. & BISERTE, G. (1979b) Nickel sub-sulphide-induced
leiomyosarcoma in rabbit white skeletal muscle. A light
microscopical and ultrastructural study. Cancer, 43: 1358-1374.
HILDEBRAND, H.F. & TETAERT, D. (1981) Ni3S2-induced
leiomyosarcomas in rabbit skeletal muscle: analysis of the tumoral
myosin and its significance in the retrodifferentiation concept.
Oncodev. Biol. Med., 2: 101-108.
HO, W. & FURST, A. (1973) Nickel excretion by rats following a
single treatment. Proc. West. Pharmacol. Soc., 16: 245-248.
HOBBS, R.J. & STREIT, B. (1986) Heavy metal concentrations in
plants growing on a copper mine spoil heap in the Grand Canyon,
Arizona. Am. Mid. Nat., 115(2): 277-281.
HODGSON, G.W. (1954) Vanadium, nickel, and iron trace metals in
crude oil of Western Canada. Bull. Am. Assoc. Pet. Geol., 38(12):
2537-2554.
HOEY, M.J. (1966) The effects of metallic salts on the histology
and functioning of the rat testis. J. reprod. Fertil., 12: 461-471.
HOFF, R.M. & BARRIE, L.A. (1986) Air chemistry observations in the
Canadian arctic. Water Sci. Tech., 18: 97-107.
HOGETVEIT, A.C. & BARTON, R.T. (1976) Preventive health program
for nickel workers. J. occup. Med., 18(12): 805-808.
HOGETVEIT, A.C. & BARTON, R.T. (1977) Monitoring nickel exposure in
refinery workers. In: Brown, S.S. ed. Clinical chemistry and
chemical toxicology of metals, Amsterdam, Oxford, New York,
Elsevier Science Publishers, Vol. 1, pp. 265-268.
HOGETVEIT, A.C., BARTON, R.T. & KOSTOL, L.C. (1978) Plasma nickel
as a primary index of exposure in nickel refining. Ann. occup.
Hyg., 21: 113-120.
HOGETVEIT, A.C., BARTON, R.T. & ANDERSEN, I. (1980) Variations of
nickel in plasma and urine during the work period. J. occup. Med.
22: 597-600.
HOHNADEL, D.C., SUNDERMAN, F.W. Jr, NECHAY, M.W. & MCNEELY, M.D.
(1973) Atomic absorption spectrometry of nickel, copper, zinc and
lead in sweat collected from healthy subjects during sauna bathing.
Clin. Chem., 19: 1288-1292.
HONDA, K., FUJISE, Y., TATSUKAWA, R. & ITANO, K. (1984)
Composition of chemical components in bone of striped dolphin,
Stenella coeruleoalba: Distribution characteristics of heavy metals
in various bones. Agric. biol. Chem., 48(3): 677-680.
HONDA, K., MIN BYUNG, Y. & TATSUKAWA, R. (1985) Heavy metal
distribution in organs and tissues of the eastern great white egret
Egretta alba modesta. Bull. environ. Contam. Toxicol., 35(6): 781-790.
HOPFER, S.M. & SUNDERMAN, F.W. Jr (1988) Hypothermia and deranged
circadian rhythm of core body temperature in nickel chloride-
treated rats. Res. Commun. chem. Pathol. Pharmacol., 62: 495-505.
HOPFER, S.M., SUNDERMAN, F.W. Jr, FREDRICKSON, R.N. & MORSE, F.E.
(1978) Nickel induced erythrocytosis: efficacies of nickel
compounds and susceptabilities of rat strains. Ann. clin. lab.
Sci., 8: 396-402.
HOPFER, S.M., LINDEN, J.V., CRISOSTOMO, C., CATALANATTO, F.A.,
GALEN, M. & SUNDERMAN, F.W. Jr (1985) Hypernickelemia in
hemodialysis patients. In: Brown, S.S. & Sunderman, F.W. Jr, ed.
Progress in Nickel toxicology. Proceedings of the 3rd International
Conference on Nickel Metabolism and Toxicology, Paris, 4-7
September, 1984, Oxford, Blackwell Scientific Publications, pp.
133-136.
HOPFER, S.M., LINDEN, J.V., REZUKE, W.N., O'BRIEN, J.E., SMITH, L.
WATTERS, F. & SUNDERMAN, F.W. Jr (1987) Increased nickel
concentrations in body fluids of patients with chronic alcoholism
during disulfiram therapy. Res. Commun. chem. Pathol. Pharmacol.
55: 101-109.
HOPFER, S.M., FAY, W.P. & SUNDERMAN, F.W. Jr (1989) Serum nickel
concentration in hemodialysis patients with environmental nickel
exposure. Ann. clin. lab. Sci.
HORAK, E. & SUNDERMANN, F.W. Jr (1973) Fecal nickel excretion by
healthy adults. Clin. Chem., 19(4): 429-430.
HORAK, E. & SUNDERMAN, F.W. Jr (1975a) Effects of Ni(II), other
divalent metal ions, and glucagon upon plasma glucose
concentrations in normal, adrenalectomized and hypophysectomized
rats. Toxicol. appl. Pharmacol., 32: 316-329.
HORAK, E. & SUNDERMAN, F.W. Jr (1975b) Effects of Ni(II) upon
plasma glucagon and glucose in rats. Toxicol. appl. Pharmacol., 33:
388-391.
HORIE, A., HARATAKE, J., TANAKA, I., KODAMA, Y. & TSUCHIYA, K.
(1985) Electron microscopical findings, with special reference to
cancer in rats caused by inhalation of nickel oxide. Biol. trace
Res., 7: 223-239.
HOWARD, J.M. (1980) Serum nickel in myocardial infarction. Clin.
Chem., 26(10): 1515.
HRUDEY, S.E. (1985) Residues from hazardous waste treatment.
Effluent Water Treat. J., 25(1): 7-12.
HSI, A.W., O'NEILL, J. & SAN SEBASTIAN, J.R. (1979a) Quantitative
mammalian cell genetic toxicology: study of the cytotoxicity and
mutagenicity of 70 individual environmental agents related to
energy technologies and 3 subfractions of a crude synthetic oil in
the CHO/HGPRT system. Environ. Sci. Res., 15: 291-315.
HSIE, A.W., JOHNSON, N.P., COUCH, D.B., SAN SEBASTIAN, I., O'NEILL,
I.P., HOESCHELE, I.D., RAHN, R.O. & FORBES, N.L. (1979b)
Quantitative mammalian cell mutagenesis and a preliminary study of
the mutagenic potential of metallic compounds. In: Kharasch, N.
ed. Trace metals in health and disease, New York, Raven Press, pp.
55-69.
HUANG, J., LU, Y., LIU, G., ZHENG, G., MEI, C., LI, Z., SHENG, S.
& YE, Z. (1986) The distribution of trace elements in rats. Acta
zool. sin., 32: 35- 39.
HUEPER, W.C. (1952) Experimental studies in metal cancerogenesis.
I. Nickel cancer in rats. Texas Rep. Biol. Med., 10: 167-186.
HUEPER, W.C. (1955) Experimental studies in metal cancerigenesis.
IV. Cancer produced by parenterally introduced metallic nickel. J.
Natl Cancer Inst., 16: 55-67.
HUEPER, W.C. (1958) Experimental studies in metal cancerigenesis.
IX. Pulmonary lesions in guinea-pigs and rats exposed to prolonged
inhalation of powdered metallic nickel. Arch. Pathol., 65: 600-607.
HUEPER, W.C. & PAYNE, W.W. (1962) Experimental studies in metal
carcinogenesis. Arch. environ. Health, 5: 445-462.
HUGHES, G.M., PERRY, S.F. & BROWN, V.M. (1979) A morphometric
study of effects of nickel, chromium and cadmium on the secondary
lamellae of rainbow trout gills. Water Res., 13:665-679.
HUGHES, A.W., SHERLOCK, D.A., HAMBLEN, D.L. & REID, R. (1987)
Sarcoma at the site of a single hip screw. J. bone joint Surg.
B69: 470-472.
HUI, G. & SUNDERMAN, F.W. Jr (1980) Effects of nickel compounds on
incorporation of the (3H) thymidine into DNA in rat liver and
kidney. Carcinogenesis, 1: 297-304.
HULETT, L.D., WEINBURGER, A.J., NORTHCUTT, K.J. & FERGUSON, M.
(1980) Chemical species in fly ash from coal-burning power plants.
Science, 210: 1356-1358.
HUNZIKER, N. (1960) De l'eczéma experimental. Quelques expériences
concernant la sensibilisation du cobaye au nickel. Dermatologica,
121: 307-312.
HUTCHINSON, T.C. (1973) Comparative studies of the toxicity of
heavy metals to phytoplankton and their synergistic interactions.
Water Pollut. Res. Can., 8: 68-90.
HUTCHINSON, T.C. & CZYRSKA, H. (1975) Heavy metal toxicity and
synergism in floating aquatic weeds. Verh. Int. Ver. Limnol., 19:
2102-2111.
HUTCHINSON, T.C. & WHITBY, L.M. (1977) The effects of acid
rainfall and heavy metal particulates on a boreal forest ecosystem
near the Sudbury smelting region of Canada. Water air soil Pollut.
7: 421-438.
HUTCHINSON, T.C., FEDORENKO, A., FITCHKO, J., KUJA, A., VANLOON,
J. & LICHWA, J. (1975) Movement and compartmentation of nickel and
copper in an aquatic ecosystem. In: Nriagu, J.O. ed.
Environmental biogeochemistry, Ann Arbor, Michigan, Ann Arbor
Science Publishers Inc. Vol. 2, pp. 565-585.
HUTCHINSON, T.C., FREEDMAN, B. & WHITBY, L. (1981) Nickel in
Canadian soils and vegetation. In: Effects of nickel in the
Canadian environment, Ottawa, National Research Council of Canada,
pp. 119-157 (Publication No. NRCC 18568).
IARC (1976) Cadmium, nickel, some epoxides, miscellaneous
industrial chemicals and general considerations on volatile
anaesthetics. Lyon International Agency for Research on Cancer,
pp. 75-112 (IARC Monographs on the Evaluation of Carcinogenic Risk
of Chemicals to Man, Vol. 11).
IARC (1989) Mortality and cancer incidence follow-up of an
historical cohort of European welders. Lyon, International Agency
for Research on Cancer (Internal Technical Report No. 89/003).
IARC (1990) Nickel and nickel compounds. In: Chromium, nickel and
welding. Lyon, International Agency for Research on Cancer, pp.
257-445 (IARC Monographs on the Evaluation of Carcinogenic Risks to
Humans, Vol. 49).
ICHINOKI, S. & YAMAZAKI, M. (1985) Simultaneous determination of
nickel, lead, zinc, and copper in citrus leaves and rice flour by
liquid chromatography with hexamethylene dithiocarbamate
extraction. Anal. Chem., 57(12): 2219-2222.
IKEBE, K. & TANAKA, R. (1979) Determination of vanadium and nickel
in marine samples by flameless and flame atomic absorption
spectrophotometry. Bull. environ. Contam. Toxicol., 21(4-5): 526-532.
INOUE, S. & KAWANISHI, S. (1989) ESR evidence for superoxide,
hydroxyl radicals and singlet oxygen produced from hydrogen
peroxide and nickel(ii) complex of glycylglycyl-L-histidine.
Biochem. Biophys. Res. Commun., 159(2): 445-451.
IZMEROV, N.F. (1983) [Occupational diseases: guidelines.] Moscow,
Meditsina, pp. 319-329 (in Russian).
JACOBSEN, N., ALFHEIM, I. & JONSEN, J. (1978) Nickel and strontium
distribution in some mouse tissues. Passage through placenta and
mammary glands. Res. Commun. chem. Pathol. Pharmacol., 20(3): 571-584.
JACQUET, P. & MAYENCE, A. (1982) Application of the in vitro
embryo culture to the study of the mutagenic effets of nickel in
male germ cells. Toxicol. Lett., 11(1-2): 193-197.
JAFFRE, T., KERSTEN, W., BROOKS, R.R. & REEVES, R.D. (1979) Nickel
uptake by Flacourtiaceae of New Caledonia. Proc. R. Soc. Lond.
(Biol), 205(1160): 385-394.
JANSEN, L.H., BERRENS, L. & VAN DELDEN, J. (1964) Contact
sensitivity to simple chemicals: The role of intermediates in the
process of sensitization. Naturwissenschaften, 51: 387-388.
JARSTRAND, C., LUNDBORG, M., WIRNIK, A. & CAMNER, P. (1978)
Alveolar macrophage function in nickel dust exposed rabbits.
Toxicology, 11(4): 353-359.
JASMIN, G. & RIOPELLE, J.L. (1976) Renal carcinomas and
erythrocytosis in rats following intrarenal injection of nickel
subsulfide. Lab. Invest., 35: 71-78.
JENKINS, D.W. (1980a) Nickel accumulation in aquatic biota. In:
Nriagu, J.O. ed. Nickel in the environment, New York, Chichester,
Brisbane, Toronto, John Wiley and Sons, pp. 283-337.
JENKINS, D.W. (1980b) Nickel accumulation in terrestrial wildlife.
In: Nriagu, J.O. ed. Nickel in the environment, New York,
Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 457-462.
JOHANSSON, A. & CAMNER, P. (1980) Effects of nickel dust on rabbit
alveolar epithelium. Environ. Res., 22: 510-516.
JOHANSSON, A., CAMNER, P., JARSTRAND, C. & WIERNIK, A. (1980)
Morphology and function of alveolar macrophages after longterm
nickel exposure. Environ. Res., 23: 170-180.
JOHANSSON, A., CAMNER, P. & ROBERTSON, B. (1981) Effects of
longterm nickel dust exposure on rabbit alveolar epithelium.
Environ. Res., 25: 391-403.
JOHANSSON, A., CAMNER, P., JARSTRAND, C. & WIERNIK, A. (1983a)
Rabbit lungs after long-term exposure to low nickel dust
concentration. II. Effects on morphology and function. Environ.
Res., 30: 142-151.
JOHANSSON, A., CURSTEDT, T., ROBERTSON, B. & CAMNER, P. (1983b)
Rabbit lung after inhalation of soluble nickel. II. Effects on lung
tissue and phospholipids. Environ. Res., 31: 399-412.
JOHANSSON, A., WIERNIK, A., LUNDBORG, M., JARSTRAND, C. & CAMNER,
P. (1988) Alveolar macrophages in rabbits after combined exposure
to nickel and trivalent chromium. Environ. Res., 46: 120-132.
JONES, J.G. & WARNER, C.G. (1972) Chronic exposure to iron oxide,
chromium oxide, and nickel oxide fumes in metal dressers in a
steelworks. Br. J. ind. Med., 29: 168-177.
JORDAN, W.P. & KING, S.E. (1979) Nickel feeding in nickel-sensitive
patients with hand eczema. J. Am. Acad. Dermatol., 1(6): 506-508.
KAABER, K., VEIEN, N.K. & TJELL, J.C. (1978) Low nickel diet in
the treatment of patients with chronic nickel dermatitis. Br. J.
Dermatol., 98: 197-201.
KADOTA, I. & KURITA, M. (1955) Hyperglycemia and islet cell damage
caused by nickelous chloride. Metabolism, 4: 337-342.
KALDOR, J., PETO, J., EASTON, D., DOLL, R., HERMON, C. & MORGAN,
L. (1986) Models for respiratory cancer in nickel refinery workers.
J. Natl Cancer Inst., 77: 841-848.
KALIMO, K. & LAMMINTAUSTA, K. (1984) 24 and 48 h allergen exposure
in patch testing. Contact Dermatit., 10: 25-29.
KALLIOMAKI, P.L., RAHKONEN, E., VAARANEN, V., KALLIOMAKI, K. &
AITTONIEMI, K. (1981) Lung-retained contaminants, urinary chromium
and nickel among stainless steel welders. Int. Arch. occup.
environ. Health, 49(1): 67-75.
KANEMATSU, N., HARA, M. & KADA, T. (1980) REC assay and
mutagenicity studies on metal compounds. Mutat. Res., 77: 109-116.
KASPRZAK, K.S. (1974) An autoradiographic study of nickel
carcinogenesis in rats following injection of 63Ni3S2 and Ni335S2.
Res. Commun. chem. Pathol. Pharmacol., 8(1): 141-150.
KASPRZAK, K.S. & POIRIER, L.A. (1983) Effects of calcium, magnesium
and sodium acetates on tissue distribution of nickel (II) in Strain
A mice. In: Brown, S.S. & Savory, J. ed. Chemical toxicology and
clinical chemistry of metals. Proceedings of the 2nd International
Conference, Montreal, 19-22 July, 1983, Oxford, Blackwell
Scientific Publications, pp. 374-376.
KASPRZAK, K.S. & POIRIER, L.A. (1985) Effects of calcium and
magnesium salts on nickel subsulfide carcinogenesis in Fisher rats.
In: Brown, S.S. & Sunderman, F. W. Jr, ed. Progress in nickel
toxicology. Proceedings of the 3rd International Conference on
Nickel Metabolism and Toxicology, Paris, 4-7 September, 1984,
Oxford, Blackwell Scientific Publications, pp. 29-32.
KASPRZAK, K.S. & SUNDERMAN, F.W. Jr (1969) The metabolism of nickel
carbonyl-14C. Toxicol. appl. Pharmacol., 15(2): 295-303.
KASPRZAK, K.S. & SUNDERMAN, F.W. Jr (1977) Mechanisms of
dissolution of nickel subsulfide in rat serum. Res. Commun. chem.
Pathol. Pharmacol., 16(1): 95-108.
KASPRZAK, K.S., MARCHOW, L. & BREBOROWICZ, J. (1973) Pathological
reactions in rat lungs following intratracheal injection of nickel
subsulfide and 3,4-benzpyrene. Res. Commun. chem. Pathol.
Pharmacol., 6(1): 237-245.
KASPRZAK, K.S., GABRYEL, P. & JARCZEWSKA, K. (1983)
Carcinogenicity of nickel(II)hydroxides and nickel(II) sulfate in
Wistar rats and its relation to the in vitro dissolution rates.
Carcinogenesis, 4(3): 275-279.
KASPRZAK, K.S., WAALKES, M.P. & POIRIER, L.A. (1986) Antagonism by
essential divalent metals and amino acides of nickel(II)-DNA
binding in vitro. Toxicol. appl. Pharmacol., 82: 336-343.
KASPRZAK, K.S. & RODRIGUEZ, R.E. (1988) Nickel-iron interactions in
carcinogenesis: dose effect and involvement of active oxygen and
related enzymes. In: Fourth International Conference on Nickel
Metabolism and Toxicology, Abstracts, Espoo, Finland,5-9 September
1988, Helsinki, Institute of Occupational Health, p. 59.
KASPRZAK, K.S., KOVATCH, R.M. & POIRIER, L.A. (1988) Inhibitory
effect of zinc on nickel subsulfide carcinogenesis in Fischer rats.
Toxicology, 52: 153-262.
KASPRZAK, K.S., WARD, J.M., POIRIER, L.A., REICHARDT, D.A., DENN,
A.C. III, & REYNOLDS, C.W. (1987) Nickel-magnesium interactions in
carcinogenesis: dose effects and involvement of natural killer
cells. Carcinogenesis, 8(7): 1005-1011.
KATZ, S.A. & SAMITZ, M.H. (1975) Leaching of nickel from stainless
steel consumer commodities. Acta dermatol., 55: 113-115.
KEEFER, R.F., SINGH, R.N. & HORVATH, D.J. (1986) Chemical
composition of vegetables grown on an agricultural soil amended
with sewage sludges. Environ. Qual., 15(2): 146-152.
KELLER, W. & PITBLADO, J.R. (1986) Water quality changes in Sudbury
area lakes: A comparison of synoptic surveys in 1974-1976 and 1981-
1983. Water air soil Pollut., 29(3): 285-296.
KEMKA, R. (1971) [Determination of the nickel and cobalt contents
in urine and the atmosphere.] Prac. Lek., 23: 80-85 (in Czech).
KESKINEN, H., KALLIOMAKI, P.L. & ALANKO, K. (1980) Occupational
asthma due to stainless steel welding fume. Clin. Allergy, 10:
151-159.
KHAN, S.N., RAHMAN, M.A. & SAMAD, A. (1984) Trace elements in
serum from Pakistani patients with acute and chronic ischemic heart
disease and hypertension. Clin. Chem., 30: 644-648.
KIM, M.K., FISHER, A.M. & MACKAY, R.J. (1969) Pulmonary effects of
metallic dusts - nickel and iron. Toronto, University of Toronto,
School of Hygiene, Department of Physiological Hygiene, 24 pp.
KINCAID, J.F., STRONG, J.S. & SUNDERMAN, F.W. (1953) Nickel
poisoning. I. Experimental study of the effects of acute and
subacute exposure to nickel carbonyl. Arch. ind. Hyg. occup. Med.
8: 48-60.
KINCAID, J.F., STANLEY, E.L., BECKWORTH, C.H. & SUNDERMAN, F.W.
(1956) Nickel poisoning. III. Procedures for detection, prevention,
and treatment of nickel carbonyl exposure including a method for
the determination of nickel in biologic materials. Am. J. clin.
Pathol., 26: 107-119.
KING, M.M., LYNN, K.K. & HUANG, C.Y. (1985) Activation of the
calmodulin-dependant phosphoprotein phosphatase by nickel ions In:
Brown, S.S. & Sunderman, F.W. Jr, ed. Progress in nickel
toxicology. Proceedings of the 3rd International Conference on
Nickel Metabolism and Toxicology, Paris, 4-7 September, 1984,
Oxford, Blackwell Scientific Publications, pp. 117-122.
KIRCHGESSNER, M. & SCHNEGG, A. (1979) [Activity of proteases
leucinaryl-amidase and a-amylase in pancreas at nickel deficiency.]
Nutr. Metab., 23: 62-64 (in German).
KIRCHGESSNER, M. & SCHNEGG, A. (1980a) Biochemical and
physiological effects of nickel deficiency. In: Nriagu, J.O. ed.
Nickel in the environment, New York, Chichester, Brisbane, Toronto,
John Wiley and Sons, pp. 635-652.
KIRCHGESSNER, M. & SCHNEGG, A. (1980b) [Hydrocarbon metabolism in
nickel deficiency.] In: Anke, M., Schneider, H.-J. & Brûckner,
Chr. ed. [3. Trace Element Symposium: Nickel, Jena, German
Democratic Republic, 7-11 July, 1980,] Jena, Friedrich-Schiller
University, pp. 23-26 (in German).
KJELLSTROM, T., FRIBERG, A. & RAHNSTER, B. (1979) Mortality and
cancer morbidity among cadmium exposed workers. Environ. Health
Perspect., 28: 199- 204.
KLEIN, L.A., LANG, M., NASH, N. & KIRSCHNER, S.L. (1974) Sources
of metals in New York City wastewater. J. Water Pollut. Control
Fed., 46(12): 2653-2662.
KNIGHT, Y.A., REZUKE, W.N., WONG, S.H-Y., HOPFER, S.M., ZAHARIA, O. &
SUNDERMAN, F.W. Jr (1987) Acute thymic involution and increased
lipoperoxides in thymus of nickel chloride-treated rats. Res. Commun.
chem. Pathol. Pharmacol., 55: 291-302.
KODAMA, Y., TANAKA, I., MATSUNO, K., ISHIMATSU, S., HORIE, A. &
TSUCHIYA, K. (1985) Pulmonary deposition and clearance of inhaled
nickel oxide aerosol. In: Brown, S.S. & Sunderman, F.W. Jr, ed.
Progress in nickel toxicology, Proceedings of the 3rd International
Conference on Nickel Metabolism and Toxicology, Paris, 4-7 September
1984, Oxford, Blackwell Scientific Publications, pp. 81-84.
KOLLER, L.D. (1980) Immunotoxicology of heavy metals. Int. J.
Immunopharmacol., 2: 269-279.
KOLPAKOV, F.I. (1963) [Permeability of skin to nickel compounds.]
Arkh. Patol., 25(6): 38-45 (in Russian).
KOMCZYNSKI, L., NOWAK, H. & REJNIAK, L. (1963) Effect of cobalt,
nickel and iron on mitosis in the roots of the broad bean ( Vicia
faba). Nature (Lond.), 198: 1016-1017.
KOPONEN, M., GUSTAFSSON, T., KALLIOMAKI, P.L. & PYY, L. (1981)
Chromium and nickel aerosols in stainless steel manufacturing,
grinding and welding. Am. Ind. Hyg. Assoc. J., 42(8): 596-601.
KOVACS, P. & DARVAS, Z. (1982) Studies on the Ni content of the
centriole. Acta histochem., 71: 169-173.
KOWALCZYK, G.S., GORDON, G.E. & RHEINGROVER, S.W. (1982)
Identification of atmospheric particulate sources in Washington,
DC, using chemical element balances. Env. Sci. Technol., 16(2):
79-89.
KRISHNAN, E.R. & HELLWIG, G.V. (1982) Trace emissions from coal and
oil combustion. Environ. Prog., 1(4): 290-295.
KUCHARIN, G.M. (1970) [Occupational disorders of the nose and nasal
sinuses in workers in an electrolytic nickel refining plant.] Gig.
Tr. prof. Zabol., 14: 38-40 (in Russian).
KUEHN, K. & SUNDERMAN, F.W. Jr (1982) Dissolution half-times of
nickel compounds in water, rat serum, and renal cytosol. J. inorg.
Biochem., 17(1): 29-39.
KUEHN, K., FRASER, O.B. & SUNDERMAN, F.W. Jr (1982) Phagocytosis
of particulate nickel compounds by rat peritoneal macrophage in
vitro. Carcinogenesis, 3(3): 321-326.
KUROKAWA, Y., MATSUSHIMA, M., IMAZAWA, T., TAKAMURA, N., TAKAHASHI,
M. & HAYASHI, Y. (1985) Promoting effect of metal compounds on rat
renal tumorigenesis. J. Am. Coll. Toxicol., 4: 321-330.
LABELLA, F., DULAR, R., LEMON, P., VIVIAN, S. & QUEEN, G. (1973a)
Prolactin secretion is specifically inhibited by nickel. Nature
(Lond.), 245: 331-332.
LABELLA, F., DULAR, R., VIVIAN, S. & QUEEN, G. (1973b) Pituitary
hormone releasing or inhibiting activity of metal ions present in
hypothalamic extracts. Biochem. Biophys. Res. Commun., 52: 786-791.
LANGARD, S. & STERN, R.M. (1984) Nickel in welding fumes - A cancer
hazard to welders? A review of epidemiological studies on cancer in
welders. In: Nickel in the human environment, Proceedings of a
Joint Symposium, Lyon, 8-11 March, 1983, Lyon, International Agency
for Research on Cancer, pp. 95-103 (IARC Scientific Publications
No. 53).
LARRAMENDY, M.L., POPESCU, N.C. & DIPAOLO, J.A. (1981) Induction
by inorganic metal salts of sister chromatid exchanges and
chromosome aberrations in human and Syrian hamster cell strains.
Environ. Mutagen., 3: 597-606.
LAU, T.J., HACKETT, R.L. & SUNDERMAN, F.W. Jr (1972) The
carcinogenicity of intravenous nickel carbonyl in rats. Cancer
Res., 32: 2253-2258.
LAVELLE, J.M. & WITMER, C.M. (1981) Mutagenicity of NiCl2 and the
analysis of mutagenicity of metal ions in a bacterial fluctuation
test. Environ. Mutagen., 3: 320-321.
LAZAREVA, L.P. (1985) Changes in biological characteristics of
Daphnia magna from chronic action of copper and nickel at low
concentrations. Gidrobiol. Zh., 21: 59-62.
LEACH, C.A. Jr & SUNDERMAN, F.W. Jr (1987) Hypernickelemia
following coronary arteriography, caused by nickel in the
radiographic contrast medium. Ann. clin. lab. Sci., 17(3):
137-143.
LEACH, C.A. Jr, LINDEN, J.V., HOPFER, S.M., CRISOSTOMO, M.C. &
SUNDERMAN, F.W. Jr (1985) Nickel concentrations in serum of
patients with acute myocardial infarction or unstable angina
pectoris. Clin. Chem., 31(4): 556-560.
LECHNER, J.F., TOKIWA, T., MCCLENDON, I.A. & HAUGEN, A. (1984)
Effects of nickel sulfate on growth and differentiation of normal
human bronchial epithelial cells. Carcinogenesis, 5: 1697-1703.
LEE, R.E. & VON LEHMDEN, D.J. (1973) Trace metal pollution in the
environment. J. Air. Pollut. Control Assoc., 23: 853-857.
LESLIE, A.C.D., WINCHESTER, J.W., LEYSIEFFER, F.W. & AHLBERG, M.S.
(1976) Prediction of health effects of pollution aerosols. Trace
Subst. environ. Health, 10: 497-504.
LESSARD, R., REED, D., MAHEUX, B. & LAMBERT, J. (1978) Lung cancer
in New Caledonia, a nickel smelting island. J. occup. Med., 20(12):
815-817.
LESTROVOI, A.P., ITSKOVA, A.I. & ELISEEV, I.N. (1974) [Effect of
nickel on the iodine fixation of the thyroid gland when
administered perorally and by inhalation.] Gig. i Sanit., 10: 105-106
(in Russian).
LEVAN, A. (1945) Cytological reactions induced by inorganic salt
solutions. Nature (Lond.), 3973: 751-751.
LEWIS, C.L. & OTT, W.L. (1970) Analytical chemistry of nickel,
Oxford, New York, Pergamon Press, 232 pp.
LICHTY, P.D. & ZEY, J.N. (1985) Health Hazard Evaluation Report:
General Motors Corporation, Dayton, Ohio. Cincinnati, Ohio,
National Institute for Occupational Safety and Health, 37 pp (HETA
84-060-1645).
LIGETI, L., RUBANYI, G., KOLLER, A. & KOVACH, A.G.B. (1980)
[Effect of nickel ions on hemodynamics, cardiac performance and
coronary blood flow in anaesthetized dogs.] In: Anke, M.,
Schneider, H.-J. & Brückner, Chr. ed. [3. Trace Element Symposium
Nickel, Jena, German Democratic Republic, 7-11 July, 1980], Jena,
Friedrich-Schiller University, pp. 117-122 (in German).
LINDEN, J.V., HOPFER, S.M., GOSSLING, H.R. & SUNDERMAN, F.W. Jr
(1985) Blood nickel concentrations in patients with stainless-steel
hip prostheses. Ann. clin. lab. Sci., 15(6): 459-464.
LIU, Y. ET AL. (1983) [The role of nickel sulfate in inducing
nasoharyngeal carcinoma (NPC) in rats]. Guangzhou, Cancer Institute
of Zhongshan Medical College, WHO Collaborating Centre for Research
on Cancer, pp. 48-49 (Cancer Research Reports, Vol. 4)(in Chinese
with English abstract).
LLOYD, G.K. (1980) Dermal absorption and conjugation of nickel in
relation to the induction of allergic contact dermatitis -
preliminary results. In: Brown, S.S. & Sunderman, F.W. ed. Nickel
toxicology, Proceedings of the 2nd International Conference on
Nickel Toxicology, Swansea, 3-5 September, 1980, London, New York,
Academic Press, pp. 145-148.
LOKEN, A.C. (1950) [Carcinoma of the lung in nickel workers.]
Tidskr. Nor. Laegeforen., 70: 376-378 (in Norwegian).
LONG-ZHU, J. & ZHE-MING, N. (1985) Determination of nickel in urine
and other biological samples by graphite furnace atomic absorption
spectrometry. Fresenius Z. anal. Chem., 321: 72-76.
LOW, K.S. & LEE, C.K. (1981) Copper, zinc, nickel and chromium
uptake by "Kangkong Air" ( Ipomea acquatica Forsk). Pertanika,
4(1):16-20.
LU, C.C., MATSUMOTO, N. & IIJIMA, S. (1979) Teratogenic effects of
nickel chloride on embryonic mice and its transfer to embryonic
mice. Teratology, 19(2): 137-142.
LU, C.C., MATSUMOTO, N. & IIJIMA, S. (1981) Placental transfer and
body distribution of nickel chloride in pregnant mice. Toxicol.
appl. Pharmacol., 59(3): 409-413.
LUCASSEN, M. & SARKAR, B. (1979) Nickel(II)-binding constituents of
human blood serum. J. Toxicol. environ. Health, 5(5): 897-905.
LUDEWIGS, H.-J. & THIESS, A.M. (1970) [Knowledge in occupational
medicine of nickel carbonyl poisoning.] Zentralbl. Arbeitsmed.
Arbeitsschutz, 20: 329-339 (in German).
LUMB, G.D., SUNDERMAN, F.W. Jr, & SCHNEIDER, H.P. (1985) Nickel-
induced malignant tumors. Ann. clin. lab. Sci., 15(5): 374-380.
LUNDBORG, M. & CAMNER, P. (1982) Decreased level of lysozyme in
rabbit lung lavage fluid after inhalation of low nickel
concentrations. Toxicology, 22(4): 353-358.
LUNDBORG, M. & CAMNER, P. (1984) Lysozyme levels in rabbit lung
after inhalation of nickel, cadmium, cobalt, and copper chlorides.
Environ. Res., 34: 335-342.
MACHELETT, B. & PODLESAK, W. (1980) [Nickel uptake by oat and
mustard plants as a function of lime status of the soil.] In: Anke,
M. Schneider, H.J. & Brûckner, Chr. Jr ed. [3. Trace Element
Symposium Nickel, Jena, German Democratic Republic, 7-11 July 1980,]
Jena, Friedrich-Schiller University, pp. 207- 213 (in German).
MACKENZIE PEERS, A. (1986) Determination of arsenic, chromium,
nickel, cadmium, lead, beryllium and selenium in air and airborne
particulates by inductively coupled argon atomic emission
spectroscopy. NIOSH 7300 adapted by Mackenzie Peers, A. In:
Environmental carcinogens - selected methods of analysis. Vol. 8:
Some metals: As, Be, Cd, Cr, Ni, Pb, Se, Zn, Lyon, International
Agency for Research on Cancer, pp. 225-230 (IARC Scientific
Publications No. 71).
MCCONNELL, L.H., FINK, J.N., SCHLUETER, D.P. & SCHMIDT, M.G. Jr
(1973) Asthma caused by nickel sensitivity. Ann. intern. Med.
78(6): 888-890.
MCDONALD, I. (1981) Malignant lymphoma associated with internal
fixation of a fractured tibia. Cancer, 48: 1009-1011.
MCDOUGALL, A. (1956) Malignant tumor at site of bone plating. J.
bone joint Surg. B,. 38: 709-713.
MCGREGOR, D.B., BROWN, A., CATTANACH, P., EDWARDS, I., McBRIDE, D.
RIACH, C. & CASPARY, W.J. (1988) Responses of the L5178Y tK+/tK-
mouse lymphoma cell forward mutation assay: III. 72 coded
chemicals. Environ. molec. Mutagen., 12: 85-154.
MCLAREN, J.W., MYKYTIUK, A.P., WILLIE, S.N. & BERMAN, S.S. (1985)
Determination of trace metals in seawater by inductively coupled
plasma mass spectrometry with preconcentration on silica-
immobilized 8-hydroxyquinoline. Anal. Chem., 57: 2907-2911.
MCLEAN, J.R., MCWILLIAMS, R.S., KAPLAN, J.G. & BIRNBOIM, H.C.
(1982) Rapid detection of DNA strand breaks in human peripheral
blood cells and animal organs following treatment with physical and
chemical agents. In: Bora, K.C., Douglas, G.R. & Nestmann, E.R.
ed. Chemical mutagenesis, human population monitoring and genetic
risk assessment. Amsterdam, Oxford, New York, Elsevier Science
Publishers, pp. 137-141 (Progress in Mutation Research, Vol. 3).
McNEELY, M.D., SUNDERMAN, F.W. Jr, NECHAY, M.W. & LEVINE, H.
(1971a) Abnormal concentrations of nickel in serum in cases of
myocardial infarction, stroke, burns, hepatic cirrhosis, and
uremia. Clin. Chem., 17(11): 1123-1128.
MCNEELY, M.D., NECHAY, M.W. & SUNDERMAN, F.W. Jr (1971b)
Measurement of nickel in serum and urine as indices of
environmental exposure to nickel. Clin. Chem., 18: 992-995.
MAENZA, R.M., PRADHAN, A.M. & SUNDERMAN, F.W. Jr (1971) Rapid
induction of sarcomas in rats by combination of nickel sulfide and
3,4-benzypyrene. Cancer Res., 31: 2067-2071.
MAGNUS, K., ANDERSEN, A. & HOGETVEIT, A.C. (1982) Cancer of
respiratory organs among workers at a nickel refinery in Norway.
Int. J. Cancer, 30(6): 681-685.
MAGNUSSON, B. & KLIGMAN, A.M. (1970) Allergic contact dermatitis in
the guinea pig. Springfield, Illinois, Charles C. Thomas, 141 pp.
MALO, J.L., CARTIER, A., DOEPNER, M., NIEBOER, E., EVANS, S. &
DOLOVICH, J. (1982) Occupational asthma caused by nickel sulfate.
J. Allergy clin. Immunol., 69: 55-59.
MAREK, M. & TREHARNE, R.W. (1982) An in vitro study of the release
of nickel from two surgical implant alloys. Clin. Orthop., 167: 291-
295.
MARZOUK, A. & SUNDERMAN, F.W. Jr (1985) Biliary excretion of
nickel. Toxicol. Lett., 27: 65-71.
MAS, A., HOLT, D. & WEBB, M.C. (1985) The acute toxicology and
teratogenicity of nickel in pregnant rats. Toxicology, 35: 47-57.
MASON, B. (1952) Principles of geochemistry, New York, John Wiley &
Sons, London, Chapman & Hall, Limited, 276 pp.
MASTROMATTEO, E. (1967) Yant memorial lecture: Nickel: A review of
its occupational health aspects. J. occup. Med., 9(3): 127-136.
MASTROMATTEO, E. (1986) Nickel. Am Ind. Hyg. Assoc. J.
47(10): 589-601.
MATHIS, B.J. & CUMMINGS, T.F. (1973) Selected metals in sediments,
water, and biota in the Illionois River. J. Water Pollut. Control
Fed., 45(7):1573- 1583.
MATHUR, A.K., DATTA, K.K. TANDON, S.K. & DIKSHITH, T.S. (1977)
Effect of nickel sulphate on male rats. Bull. environ. Contam.
Toxicol., 17: 241-247.
MATHUR, A.K., DIKSHITH, T.S., LAL, M.M. & TANDON, S.K. (1978)
Distribution of nickel and cytogenetic changes in poisoned rats.
Toxicology, 10(2): 105-113.
MEDINSKY, M.A., BENSON, J.M. & HOBBS, C.H. (1987) Lung clearance
and disposition of the 63Ni in F344/N rats after intratracheal
instillation of nickel sulfate solutions. Environ. Res., 43: 168-178.
MEDVEDEVA, V.J. (1965) [Nickel content in blood of a new born baby
and in venous and retroplacental blood and milk of the mother.]
Vest. Akad. Navuk. Belarusk. SSR. Ser. biyal Navuk, 2: 114-115 (in
Russian).
MENDEN, E.E., ELIA, V.J., MICHAEL, L.W. & PETERING, H.G. (1972)
Distribution of cadmium and nickel of tobacco during cigarette
smoking. Environ. Sci. Technol., 6: 830-832.
MENNE, T., BORGAN, O. & GREEN, A. (1982) Nickel allergy and hand
dermatitis in a stratified sample of the Danish female population:
an epidemiological study including a statistic appendix. Acta
dermatovenereol., 62(1): 35-41.
MENNE, T., ANDERSEN, K.E., KAABER, K., OSMUNDSEN, P.E., ANDERSEN,
J.R., YOING, F. & VALEUR, G. (1987) Evaluation of the
dimethylglyoxime stick test for the detection of nickel. Derm.
Beruf Umwelt, 35: 128-130.
MERCER, T.T. (1967) On the role of particle size in the dissolution
of lung burdens. Health Phys., 13: 1211-1221.
MERTZ, W. (1981) The essential trace elements. Science, 213:
1332-1338.
MEYER, A. & NEEB, R. (1985) Determination of cobalt and nickel in
some biological matrices - comparison of chelate gas-chromatography
and adsorption voltammetry. Fresenius Z. anal. Chem., 321(3):
235-241.
MIKAC-DEVIC, D. SUNDERMAN, F.W. Jr, & NOMOTO, S. (1977)
Furildioxime method for nickel analysis in serum and urine by
electrothermal atomic absorption spectrometry. Clin. Chem., 23(6):
948-956.
MIKHEYEV, M.J. (1971) [Distribution and elimination of nickel
carbonyl.] Gig. Tr. prof. Zabol., 15: 35-38 (in Russian).
MIKI, H., KASPRZAK, K.S., KENNEY, S. & HEINE, U.K. (1987)
Inhibition of intercellular communication by nickel(II):
antagonistic effect of magnesium. Carcinogenesis, 8(11):
1757-1760.
MILFORD, J.B. & DAVIDSON, C.I. (1985) The sizes of particulate
trace elements in the atmosphere - a review. J. air Pollut., 35(12):
1249-1260.
MITSUMASA, O. (1987) Induction of ocular tumor by nickel subsulfide
in the Japanese common newt, Cynops pyrrhogaster. Cancer Res., 47:
5213-5217.
MIYAKI, M., AKAMATSU, N., ONO, T. & KOYAMA, H. (1979) Mutagenicity
of metal cations in cultured cells from Chinese hamsters. Mutat.
Res., 68: 259-263.
MOELLER, H. (1984) Attempts to induce contact allergy to nickel in
the mouse. Contact Dermatit., 10: 65-68.
MOFFA, I.P., ELLISON, J.E. & HAMILTON, J.C. (1983) Incidence of
nickel sensitivity in dental patients. J. dent. Res., 62: 199.
MORGAN, J.G.A. (1958) Some observations on the incidence of
respiratory cancer in nickel workers. Br. J. ind. Med., 15: 224-234.
MORGAN, J.G.A. (1960) A simplified method for the estimation of
nickel in urine. Br. J. ind. Med., 17: 209-212.
MORGAN, L.G. & ROUGE, P.J. (1979) A study into the correlation
between atmospheric and biological monitoring of nickel in nickel
refinery workers. Ann. occup. Hyg., 22(3): 311-317.
MORITA, H., NODA, K. & UMEDA (1985) Mutagenicities of nickel and
cobalt compounds in a mammalian cell line. Mutat. Res., 147: 265-266.
MORROW, P.E. (1970) Models for the study of particle retention and
elimination in the lung. Inhalation carcinogenesis, Oak Ridge,
Tennessee, US Atomic Energy Commission, Division of Technical
Information, pp. 103-116 (AEC Symposium Series, No. 18).
MORSE, E.E., LEE, T.Y., REISS, R.F. & SUNDERMAN, F.W. Jr (1977)
Dose-response and time-response study of erythrocytosis in rats
after intrarenal injection of nickel subsulfide. Ann. clin. lab.
Sci., 7: 17-24.
MORTIMER, D.C. (1985) Freshwater aquatic macrophytes as heavy metal
monitors the Ottawa river experience Canada. Environ. monit.
Assess., 5(3): 311-324.
MUHLE, H., BELLMANN, B. & TAKENAKA, S. (1988) Chronic effects of
intratracheally instilled nickel containing particles in hamsters.
In: Fourth International Conference on Nickel Metabolism and
Toxicology: Abstracts, Espoo, Finland, 5-9 September 1988,
Helsinki, Institute of Occupational Health, p. 41.
MUKUBO, K. (1978) Studies on experimental lung tumor by the
chemical carcinogens and inorganic substances. III.
Histopathological studies on lung tumor induced by pertracheal
vinyl tube infusion of 20-methylcholanthrene combined with chromium
and nickel powder. J. Nara Med. Ass., 29: 321-340.
MURTHY, R.C., BARKLEY, W., HOLLINGSWORTH, C. & BINGHAM, E. (1983)
Enzymatic changes in alveolar macrophages of rats exposed to lead
and nickel by inhalation. J. Am. Coll. Toxicol., 2: 193-199.
MYRON, D.R., ZIMMERMAN, T.J., SHULER, T.R., KLEVAY L.M., LEE, D.E.
& NIELSEN, F.H. (1978) Intake of nickel and vanadium by humans. A
survey of selected diets. Am. J. clin. Nutr., 31(3): 527-531.
NADEENKO, V.G., LENCHENKO, V.G., ARKHIPENKO, T.A., SAICHENKO, S.P.
& PETROVA, N.N. (1979) [Embryotoxic effect of nickel entering the
body via drinking water.] Gig. i Sanit., 6: 86-88 (in Russian).
NADEENKO, V.G., LECHENKO, V.G. & SAICHENKO, S.P. (2988) [Combined
action of nickel and cobalt administered with drinking water.] In:
Domnin, S.G. & Tartakovskaya, L.Ya. ed. [Combined effects of
physical and chemical factors of industrial environment], Moscow,
Erisman Institute of Hygiene, pp. 84-90 (in Russian).
NAS (1975) Nickel. Washington, DC, National Academy of Sciences,
277 pp.
NATUSCH, D.F.S., WALLACE, J.R. & EVANS, C.A. (1974) Toxic trace
elements: preferential concentration in respirable particles.
Science, 183: 202-204.
NEBEKER, A.V., STINCHFIELD, A., SAVONEN, C. & CHAPMAN, G.A. (1986)
Effects of copper, nickel and zinc on three species of Oregon
freshwater snails. Environ. Toxicol. Chem., 5(9): 807-811.
NECHAY, M.W. & SUNDERMAN, F.W. Jr (1973) Measurements of nickel in
hair by atomic absorption spectrometry. Ann. clin. lab. Sci., 3(1):
30-35.
NEUHAUSER, E.F., LOEHR, R.C. & MALECKI, M.R. (1985) Contact and
artificial soil tests using earthworms to evaluate the impact of
wastes in soil. In: Petros, J.K., Lacy, W.J. & Conway, R.A. ed.
Hazardous and industrial solid waste testing: Fourth Symposium,
Philadelphia, American Society for Testing and Materials, pp. 192-
203 (ASTM STP 886).
NEUMULLER, O.A. (1985) Römpp's chemical encyclopaedia, 8th ed.
Stuttgart, Franck'sche Verlagsbuchhandlung, Vol. 4, 3230 pp.
NEWMAN, S.M., SUMMIT, R.L. & NUNEZ, L. J. (1982) Incidence of
nickel-induced sister chromatid exchange. Mutat. Res., 101: 67-75.
NIEBOER, E. & JUSYS, A.A. (1983) Contamination control in routine
ultratrace analysis of toxic metals. In: Brown, S.S. & Savory, J.
ed. Chemical toxicology and clinical chemistry of metals,
Proceedings of the 2nd International Conference, Montreal, 19-22
July, 1983, London, New York, Blackwell Scientific Publications,
pp. 3-16.
NIEBOER, E., MAXWELL, R.J., ROSSETTO, F.E., STAFFORD, A.R. &
STETSKO, P.J. (1986) Concepts in nickel carcinogenesis. In: Xavier,
A.V. ed. Frontiers of bioinorganic chemistry. 2nd ed. Weinheim,
VCH Verlag, pp. 142-151.
NIELSEN, F.H. (1971) Studies on the essentiality of nickel. In:
Mertz, W. & Cornatzer, W.E. ed. New trace elements in nutrition,
New York, Basel, Marcel Dekker, pp. 215-253.
NIELSEN, F.H. (1974) Essentiality and function of nickel. In:
Hoekstra, W.G., Suttie, J.W., Ganther, H.E. & Mertz, W. ed. Trace
element metabolism in animals. Proceedings of the 2nd International
Symposium on Trace Element Metabolism in Animals, Madison,
Wisconsin, 18-22 June, 1973, Baltimore, Maryland, University Park
Press, pp. 381-395.
NIELSEN, F.H. (1984) Ultratrace elements in nutrition. Annu. Rev.
Nutr., 4: 21-41.
NIELSEN, F.H. & HIGGS, D.J. (1971) Further studies involving a
nickel deficiency in chicks. Proc. trace Subst. environ. Health, 4:
241-246.
NIELSEN, F.H. & OLLERICH, D.A. (1974) Nickel: a new essential trace
element. Fed. Proc., 33: 1767-1772.
NIELSEN, F.H. & SAUBERLICH, H.E. (1970) Evidence of a possible
requirement for nickel by the chick. Proc. Soc. Exp. Biol. Med.
134(3): 845-849.
NIELSEN, F.H. & SHULER, T.R. (1979) Effect of dietary nickel and
iron on the trace element content of rat liver. Biol. trace elem.
Res., 1: 337-346.
NIELSEN, F.H. & SHULER, T.R. (1981) Effect of form of iron on
nickel deprivation in the rat. Liver content of copper, manganese
and zinc. Biol. trace elem. Res., 3: 245-256.
NIELSEN, F.H., OLLERICH, D.A., FOSMIRE, G.J. & SANDSTEAD, H.H.
(1974) Nickel deficiency in chicks and rats: effects on liver
morphology, function and polysomal integrity. Adv. exp. Med. Biol.
48: 389-403.
NIELSEN, F.H., MYRON, D.R., GIVAND, S.H. & OLLERICH, D. A. (1975a)
Nickel deficiency and nickel-rhodium interaction in chicks. J.
Nutr., 105: 1607-1619.
NIELSEN, F.H., MYRON, O.R., GIVAND, S.H., ZIMMRMANN, T.J. &
OLLERICH, D.A. (1975b) Nickel deficiency in rats. J. Nutr., 105: 1620-
1630.
NIELSEN, F.H., SHULER, T.R., ZIMMERMANN, T.J., COLLINGS, M.E. &
UTHUS, E.O. (1979a) Interaction between nickel and iron in the rat.
Biol. trace elem. Res., 1: 325-335.
NIELSEN, F.H., ZIMMERMAN, T.J., COLLINGS, M.E. & MYRON, D.R.
(1979b) Nickel deprivation in rats: Nickel-iron interactions. J.
Nutr., 109(9): 1623-1632.
NIELSEN, G.D. (in press) Oral challenge of nickel-allergic patients
with hand eczema. In: Nieboer, E. & Aitio, A. ed. Nickel and human
health: Current perspectives. New York, Chichester, Brisbane,
Toronto, John Wiley & Sons (Advances in environmental science and
technology Series).
NIELSEN, G.D. & FLYVHOLM, M. (1984) Risks of high nickel intake
with diet. In: Nickel in the human environment. Proceedings of a
Joint Symposium, Lyon, 8-11 March 1983, Lyon, International Agency
for Research on Cancer, pp. 333-338 (IARC Scientific Publications
No. 53).
NIELSEN, G.D., JORGENSEN, P.J., KEIDING, K. & GRANDJEAN, P.
(1987a) Urinary nickel excretion before and after loading with
naturally occurring nickel. In: Trace elements in human health and
disease. Second Nordic Symposium, Odense, Denmark, 17-21 August
1987: Symposium Abstracts, Odense, Odense University (Abstract C3).
NIELSEN, G.D., ENDERSEN, O. & GRANDJEAN, P. (1987b) Effect of
diethyldithiocarbamate on toxicokinetics of 57Ni in mice. In: Trace
elements in human health and disease. Extended abstracts from the
second Nordic Symposium, Odense, 17-21 August 1987, Copenhagen,
World Health Organization, Regional Office for Europe, pp. 78-81
(Environmental Health Series No. 20).
NILZEN, A. & WIKSTROM, K. (1955) The influence of lauryl sulphate
on the sensitization of guinea-pigs to chrome and nickel. Acta
dermatovenereol., 35: 292-299.
NISHIMURA, M. & UMEDA, M. (1979) Induction of chromosomal
aberrations in cultured mammalian cells by nickel compounds.
Mutat. Res., 68: 337-349.
NISHIOKA, H. (1975) Mutagenic activities of metal compounds in
bacteria. Mutat. Res., 31: 185-189.
NODIYA, P.J. (1972) [Cobalt and nickel balance in students of an
occupational technical school.] Gig. i Sanit., 37: 108-109 (in Russian).
NOMOTO, S. & SUNDERMAN, F.W. (1970) Atomic absorption spectrometry
of nickel in serum, urine, and other biological materials. Clin.
Chem., 16: 477-485.
NOMOTO, S. & SUNDERMAN, F.W. Jr (1988) Presence of nickel in alpha-
2-macroglobulin isolated from human serum by high performance
liguid chromatography. Ann. clin. lab. Sci., 18: 78-84.
NOMOTO, S., HIRABAYASHI, T. & FUKUDA, T. (1983) Serum nickel
concentrations in women during pregnancy, parturition, and post-
partum. In: Brown, S.S. & Savory, J. ed. Chemical toxicology and
clinical chemistry of metals, Proceedings of the 2nd International
Conference, Montreal, 19-22 July, 1983, London, New York, Blackwell
Scientific Publications, pp. 351-352.
NOMOTO, S., MCNEELY, M.D. & SUNDERMAN, F.W. (1971) Isolation of a
nickel a2-macroglobulin from rabbit serum. Biochemistry, 10(9):
1647-1651.
NORGAARD, O. (1955) Investigations with radioactive Ni57 into the
resorption of nickel through the skin in normal and in nickel-
hypersensitive persons. Acta dermtovenereol., 35: 111-117.
NORGAARD, O. (1957) Investigations with radioactive nickel, cobalt
and sodium on the resorption through the skin in rabbits, guinea-
pigs and man. Acta dermatovenereol., 37: 440-445.
NORSETH, T. (1975) Urinary excretion of nickel as an index of
nickel exposure in welders and nickel refinery workers. Int. Congr.
Occup. Health, 18: 327.
NOVEY, H.S., HABIB, M. & WELLS, I.D. (1983) Asthma and IgE
antibodies induced by chromium and nickel salts. J. Allergy clin.
Immunol., 72: 407-412
NOZDRYUKHINA, L.R. (1978) Use of blood trace elements for diagnosis
of heart and liver disease. In: Kirchgessner, M. ed. Trace element
metabolism in man and animals. Proceedings of the 3rd International
Symposium, Freising, Federal Republic of Germany, July 1977,
Freising-Weihenstephan, Technical University Munich, pp. 336-339.
NRIAGU, J.O. (1979) Global inventory of natural and anthropogenic
emissions of trace metals to the atmosphere. Nature (Lond.),
279(5712): 409-411.
NRIAGU, J.O. (1980) Global cycle and properties of nickel. In:
Nriagu, J.O. ed. Nickel in the environment, New York, Chichester,
Brisbane, Toronto, John Wiley and Sons, pp. 1-26.
OAKLEY, A.M.M., IVE, F.A. & CARR, M.M.C. (1987) Skin clips are
contraindicated when there is nickel allergy. J. R. Soc. Med., 80:
290-291.
O'DELL, G.D., MILLER, W.J., MOORE, S.L., KING, W.A., ELLERS, J.C.
& JURECEK, H. (1971) Effect of dietary nickel level on excretion
and nickel content of tissues in male calves. J. anim. Sci., 32(4):
769-773.
OGAWA, H.I., TSURUTA, S., NIYITANI, Y., MINO, H., SAKATA, K. &
KATO, Y. (1987) Mutagenesis of metal salts in combination with 9-
amonoacridine in Salmonella typhimurium. Jpn. J. Genet., 62: 159.
OHNO, H., HANAOKA, F. & YAMADA, M.A. (1982) Inducibility of
sister-chromatid exchanges by heavy-metal ions. Mutat. Res., 104:
141-145.
OLEJAR, S., OLEJAROVA, E. & VRABEL, K. (1982) [Lung tumors in
workers in a nickel refinery.] Prac. Lek., 34(8): 80-282 (in Czech).
OLSEN, I. & JONSEN, J. (1979a) Whole-body autoradiography of 63Ni
in mice throughout gestation. Toxicology, 12(2): 165-172.
OLSEN, I. & JONSEN, J. (1979b) Effect of cadmium acetate, copper
sulfate and nickel chloride on organ cultures of mouse trachea.
Acta pharmacol. toxicol., 44: 120-127.
OLSEN, J. & SABROE, S. (1984) Occupational causes of laryngeal
cancer. J. Epidemiol. community Health, 38(2): 117-121.
ONKELINX, C. & SUNDERMAN, F.W. (1980) Modeling of nickel
metabolism. In: Nriagu, J.O. ed. Nickel in the environment, New
York, Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 525-
545.
ONKELINX, C., BECKER, J. & SUNDERMAN, F.W. (1973) Compartmental
analysis of the metabolism of 63Ni(II) in rats and rabbits. Res.
Commun. chem. Pathol. Pharmacol., 6(2): 663-676.
OSKARSSON, A. & TJALVE, H. (1979a) The distribution and metabolism
of nickel carbonyl in mice. Br. J. ind. Med., 36(4): 326-335.
OSKARSSON, A. & TJALVE, H. (1979b) An autoradiographic study on the
distribution of 63NiCl2 in mice. Ann. clin. lab. Sci., 9(1):
47-59.
OSKARSSON, A., ANDERSSON, Y. & TJALVE, H. (1979) Fate of nickel
subsulfide during carcinogenesis studied by autoradiography and X-
ray powder diffraction. Cancer Res., 39(10): 4175-4182.
OSTAPCZUK, P., VALENTA, P., STOEPPLER, M. & NURNBERG, H.W. (1983)
Voltammetric determination of nickel and cobalt in body fluids and
other biological materials. In: Brown, S.S. & Savory, J. ed.
Chemical toxicology and clinical chemistry of metals, Proceedings
of the 2nd International Conference, Montreal, 19-22 July, 1983;
London, New York, Blackwell Scientific Publications, pp. 61-64.
OSTAPCZUK, P., FRONING, M., STOEPPLER, M. & NUERNBERG, H.W.
(1985a) Square wave voltammetry: A new approach for the sensitive
determination of nickel and cobalt in human samples. In: Brown,
S.S. & Sunderman, F.W. Jr ed. Progress in Nickel toxicology.
Proceedings of the 3rd International Conference on Nickel
Metabolism and Toxicology, Paris, 4-7 September, 1984, Oxford,
Blackwell Scientific Publications, pp. 129-132.
OSTAPCZUK, P., FRONING, M., STOEPPLER, M. & NURNBERG, H.W. (1985b)
[Determination of heavy metals in environmental samples and biotic
material by means of square wave voltammetry.] Fresenius Z. anal.
Chem., 320: 645 pp. (in German).
OSTERREICHISCHES FORSCHUNGSZENTRUM SEIBERSDORF GMBH (1983)
[Assessment of the acute toxicity of nickel sulfate to rainbow
trout.] Vienna, Austrian Research Centre, 21 pp. (Report No. 4213,
BL-424/83) (in German).
OTTOLENGHI, A.D., HASEMAN, J.K., PAYNE, W.W., FALK, H.L. &
MACFARLAND, H.N. (1974) Inhalation studies of nickel sulfide in
pulmonary carcinogenesis of rats. J. Natl Cancer Inst., 54(5):
1165-1172.
OU, B., LIU, Y., HUANG, X. & FENG, G. (1981) [The promoting action
of nickel in the induction of nasopharyngeal carcinoma in rats.]
Guangzhou, Cancer Institute of Zhongshan Medical College, WHO
Collaborating Centre for Research on Cancer, pp. 3-8 (Cancer
Research Reports, Vol. 2) (in Chinese with English abstract).
OU, B., LIU, Y. & SHENG, G. (1983) [Tumor induction in next
generation of dinitrosopiperazine-treated pregnant rats.]
Guangzhou, Cancer Institute of Zgongshan Medical College, WHO
Collaborating Centre for Research on Cancer pp. 44-45, (Cancer
Research Reports, Vol. 4) (in Chinese with English abstract).
PACYNA, J.M., OTTAR, B., TOMZA, U. & MAENHAUT, W. (1985) Long-
range transport of trace elements to Ny Alesund, Spitsbergen.
Atmos. Environ. 19(16): 857-865.
PAINTER, R.B. & HOWARD, R. (1982) The HeLa DNA-synthesis inhibition
test as a rapid screen for mutagenic carcinogens. Mutat. Res., 92:
427-437.
PALO, J. & SAVOLAINEN, H. (1973) Biochemical diagnosis of aspartyl-
glycosaminuria. Ann. clin. Res., 5: 156-162.
PARKER, D. & TURK, J.L. (1978) Delay in the development of the
allergic response to metals following intratracheal instillation.
Int. Arch. Allergy appl. Immunol., 57: 289-293.
PARKER, G.H. (1985) Copper, nickel, and iron in plumage of three
upland gamebird species from non-contaminated environments. Bull.
environ. Contam. Toxicol., 35(6): 776-780.
PARKER, K. & SUNDERMAN, F.W. Jr (1974) Distribution of 63Ni in
rabbit tissues following intravenous injection of 63NiCl2. Res.
Commun. chem. Pathol. Pharmacol., 7: 755-762.
PATIERNO, S.R. & COSTA, M. (1985) DNA-protein cross-links induced
by nickel compounds in intact cultured mammalian cells Chem.-biol.
Interact., 55: 75-91.
PATIERNO, S.R., SUGIYAMA, M., BASILIKON, J.P. & COSTA, M. (1985)
Preferential DNA-protein cross-linking by NiCl2 in magnesium-
insoluble regions of fractionated Chinese hamster ovary cell
chromatin. Cancer Res., 45(11): 5787-55794.
PATON, G.R. & ALLISON, A.C. (1972) Chromosome damage in human cell
cultures induced by metal salts. Mutat. Res., 16: 332-336.
PAYNE, W.W. (1964) Carcinogenicity of nickel compounds in
experimental animals. Proc. Am. Assoc. Cancer Res., 5: 50
(Abstract 197).
PEDERSEN, E., HOGETVEIT, A.C. & ANDERSEN, A. (1973) Cancer of
respiratory organs among workers at a nickel refinery in Norway.
Int. J. Cancer, 12(1): 32-41.
PELTONEN, L. (1979) Nickel sensitivity in the general population.
Contact Dermatit., 5(1): 27-32.
PENMAN, H.G. & RING, P.A. (1984) Osteosarcoma in association with
total hip replacement. J. bone joint Surg., B66: 632-634.
PENNINGTON, J.A.T. & JONES, J.W. (1987) Molybdenum, nickel, cobalt,
vanadium and strontium in total diets. J. Am. Diet. Assoc., 87(12):
1644-1650.
PETO, J. (1988) The international nickel speciation research
project. In: Fourth International Conference on Nickel Metabolism
and Toxicology: Abstracts, Espoo, Finland, 5-9 September 1988,
Helsinki, Institute of Occupational Health, p. 50.
PETO, J., CUCKLE, H., DOLL, R., HERMON, C. & MORGAN, L.G. (1984)
Respiratory cancer mortality of Welsh nickel refinery workers. In:
Nickel in the human environment. Proceedings of a Joint Symposium,
Lyon, 8-11 March, 1983, Lyon, International Agency for Research on
Cancer, pp. 37-46 (IARC Scientific Publications No. 53).
PHATAK, S.S. & PATWARDHAN, V.N. (1950) Toxicity of nickel. J. Sci.
ind. Res., 9B: 70-76.
PHATAK, S.S. & PATWARDHAN, V.N. (1952) Toxicity of nickel.
Accumulation of nickel in rats fed on nickel-containing diets and
its elimination. J. Sci. ind. Res., 11B: 173-176.
PHILLIPS, D.J.H. & MUTTARASIN, K. (1985) Trace metals in bivalve
molluscs from Thailand. Mar. environ. Res., 15(3): 215-234.
PICKERING, Q.H. (1974) Chronic toxicity of nickel to the fathead
minnow. J. Water Pollut. Control Fed., 46: 760-765.
PICKERING, Q.H. & HENDERSON, C. (1966) The acute toxicity of some
heavy metals to different species of warm water fishes. Air Water
Pollut. Int. J., 10: 453-463.
PIENTA, R.J., POILEY, J.A. & LEBHERZ, W.B. (1977) Morphological
transformation of early passage golden hamster embryo cells derived
from cryopreserved primary cultures as a reliable in vitro bioassay
for identifying diverse carcinogens. Int. J. Cancer, 19: 642-655.
PIHLAR, B., VALENTA, P. & NURNBERG, H.W. (1981) New high-performance
analytical procedure for the voltammetric determination of nickel
in routine analysis of waters, biological materials and food.
Fresenius Z. anal. Chem., 307: 337-346.
PIKALEK, P. & NECASEK, J. (1983) The mutagenic activity of nickel
in Corynebacterium sp. Folia microbiol., 28: 17-21.
POIRIER, L.A., THEISS, J.C., ARNOLD, L.J. & SHIMKIN, M.B. (1984)
Inhibition by magnesium and calcium acetates of lead subacetate-
and nickel acetate-induced lung tumors in strain A mice. Cancer
Res., 44: 1520-1522.
POLEDNAK, A.P. (1981) Mortality among welders, including a group
exposed to nickel oxides. Arch. environ. Health, 36(5): 235-242.
POLEMIO, M., SENESI, N. & BUFO, S.A. (1982) Soil contamination by
metals; a survey in industrial and rural areas of Southern Italy.
Sci. total Environ., 25: 71-80.
PORT, C.D., FENTERS, J.D., EHRLICH, R., COFFIN, D.L. & GARDNER, D.
(1975) Interaction of nickel oxide and influenza infection in the
hamster. Environ. Health Perspect., 10: 268.
POTT, F., ZIEM, U., REIFFER, F.-J., HUTH, F., ERNST, H. & MOHR, U.
(1987) Carcinogenicity studies on fibres, metal compounds, and some
other dusts in rats. Exp. Pathol., 32: 129-152.
POTT, F., RIPPE, M., ROLLER, M., ROSENBRUCH, M. & HUTH, F. (1988)
Carcinogenicity studies on nickel compounds and nickel alloys after
intraperitoneal injection in rats. In: Fourth International
Conference on Nickel Metabolism and Toxicology: Abstracts, Espoo,
Finland, 5-9 September 1988, Helsinki, Institute of Occupational
Health, p. 42.
POWLESLAND, C. & GEORGE, I.C. (1986) Acute and chronic toxicity of
nickel to larvae of Chironomus riparis (Meigen). Environ. Pollut.
42(1): 47-64.
PROKIPCAK, B. & ORMROD, D.P. (1986) Visible injury and growth
responses of tomato and soybean to combinations of nickel, copper
and ozone. Water air soil Pollut., 27(3-4): 329-340.
PRYSTOWSKY, S.D., ALLEN, A.M., SMITH, R.W., NONOMURA, J.H., ODOM,
R.B. & AKERS, W.A. (1979) Allergic contact hypersensivity to
nickel, neomycin, ethylenediamine, and benzocaine. Relationships
between age, sex, history of exposure, and reactivity to standard
patch tests and use tests in a general population. Arch. Dermatol.
115(8): 959-965.
PUCHYR, R. & SHAPIRO, R. (1986) Determination of trace elements in
food by HCl-HNO3 leaching and flame atomic absorption spectroscopy.
J. Assoc. Off. Anal. Chem., 69(5): 868-870.
RADIAN CORPORATION (1984) Locating and estimating emissions from
sources of nickel. Research Triangle Park, North Carolina, US
Environmental Protection Agency, Office of Air Quality Planning and
Standards, 188 pp (EPA-450/4-84-007F).
RAHKONEN, E., JUNTTILA, M.-L., KALLIOMAKI, P.-L., OLKINUORA, M.,
KOPONEN, M. & KALLIOMAKI, K. (1983) Evaluation of biological
monitoring among stainless steel welders. Int. Arch. occup.
environ. Health, 52: 243-255.
RAITHEL, H.J. (1987) [Research report nickel. Investigations on
occupational nickel exposure and strain-situations of 837 persons.]
Sankt Augustin, Federation of Industrial Cooperative Societies, 199
pp (in German).
RAITHEL, H.J., MAYER, P., SCHALLER, K.H., MOHRMANN, W., WELTLE, D.
& VALENTIN, H. (1981) [Exposure to nickel in glass industry
workers. I. analysis and quantification of external and internal
nickel load.] Zentralbl. Arbeitsmed. Arbeitsschutz prophyl.
Ergonomie, 31(8): 332-339 (in German).
RANTA, W.B., TOMASSINI, F.D. & NIEBOER, E. (1978) Elevation of
copper and nickel levels in primaries from black and mallard ducks
collected in the Sudbury district, Ontario. Can. J. Zool., 56(4):
581-586.
RASMUSON, A. (1985) Mutagenic effects of some water-soluble metal
compounds in a somatic eye-color test system in Drosophila
melanogaster. Mutat. Res., 157: 157-162.
REDDY, M.R. & DUNN, S.J. (1984) Accumulation of heavy metals by
soybean from sludge-amended soil. Env. Pollution, B7: 281-296.
REDMOND, C.K. (1984) Site-specific cancer mortality among workers
involved in the production of high nickel alloys. In: Nickel in the
human environment. Proceedings of a Joint Symposium, Lyon, 8-11
March, 1983, Lyon, International Agency for Research on Cancer, pp.
73-86 (IARC Scientific Publications No. 53).
REDMOND, C.K., LEGASSE, A.A. & BASS, G. (1983) Cancer mortality in
workers in the high nickel alloy industries. Pittsburgh,
Pennsylvania, University of Pittsburgh, Graduate School of Public
Health, Department of Biostatistics, 145 pp.
REHWOLDT, R., BIDA, G. & NERRIE, B. (1971) Acute toxicity of
copper, nickel and zinc ions to some Hudson river fish species.
Bull. environ. Contam. Toxicol., 6: 445-448.
REHWOLDT, R., MENAPACE, L.W., NERRIE, B. & ALESSANDRELLO, D. (1972)
The effect of increased temperature upon the acute toxicity of some
heavy metal ions. Bull. environ. Contam. Toxicol., 8: 91-96.
REHWOLDT, R., LASKO, L., SHAW, C. & WIRHOWSKI, E. (1973) The acute
toxicity of some heavy metal ions toward benthic organisms. Bull.
environ. Contam. Toxicol., 10:291-294.
REICHRTOVA, E., KOVACIKOVA, Z., TAKAC, L. & ORAVEC, C. (1986a) The
effect of metal particles from a nickel refinery dump on alveolar
macrophages. Part 1. Chamber exposure of Wistar rats. Environ.
Pollut., A40: 87-94.
REICHRTOVA, E., TAKAC, L. & KOVACIKOVA, Z. (1986b) The effect of
metal particles from a nickel refinery dump on alveolar
macrophages. Part 2. Environmental exposure of rabbits. Environ.
Pollut., A40: 101-107.
REZUKE, W.N., KNIGHT, J.A. & SUNDERMAN, F.W. Jr (1987) Reference
values for nickel concentrations in human tissue and bile. Ann. J.
ind. Med., 11: 419-426.
RICHTER, O.R. & THEIS, T.L. (1980) Nickel speciation in a
soil/water system. In: Nriagu, J.O. ed. Nickel in the
environment, New York, Chichester, Brisbane, Toronto, John Wiley
and Sons, pp. 189-202.
RIGAUT, J.-P. (1983) Preliminary report on nickel health criteria,
Luxemburg, Commission of the European Communities, 1009 pp.
(CCE/LUX/V/E/24/83)
RITTMANN, D., NOCHRAINER, D., OBERDORSTER, G., KORDEL, W.,
SPIEGELBERG, T., KONIG, H., FRITSCHE, V., KUHNEN, H. & FINGERHUT,
R. (1981) [Inhalation experiment on rats and pathophysiological
effect of nickel. Environmental research plan of the Federal
Ministry of the Interior.] Berlin, Federal Ministry of the Interior
(Research Report No. 107 06 00702) (in German).
RIVEDAL, E. & SANNER, T. (1980) Synergistic effect on morphological
transformation of hamster embryo cells by nickel sulphate and
benz( a)pyrene. Cancer Lett., 8(3): 203-208.
RIVEDAL, E. & SANNER, T. (1981) Metal salts as promoters of in
vitro morphological transformation of hamster embryo cells
initiated by benzo( a)pyrene. Cancer Res., 41: 2950-2953.
RIVEDAL, E., HEMSTAD, J. & SANNER, T. (1980) Synergistic effects
of cigarette smoke extracts, benz( a)pyrene and nickel sulphate on
morphological transformation of hamster embryo cells. In:
Holmstedt, B., Lauwerys, R., Mercier, M. & Roberfroid, M., ed.
Mechanisms of toxicity and hazard evaluation. Proceedings of the
2nd International Congress on Toxicology, Brussels, Belgium, 6-11
July 1980. Amsterdam, New York, Oxford, Elsevier/North Holland
Biomedical Press, pp. 259-263 (Developments in Toxicology and
Environmental Science, Vol. 8).
ROBERTS, R.S., JULIAN, J.A., MUIR, D.C. & SHANNON, H.S. (1984)
Cancer mortality associated with the high temperature oxidation of
nickel subsulfide. In: Nickel in the human environment, Proceedings
of a Joint Symposium, Lyon, 8-11 March, 1983, Lyon, International
Agency for Research on Cancer, pp. 23-35 (IARC Scientific
Publications No. 53).
ROBISON, S.H. & COSTA, M. (1982) The induction of DNA strand
breakage of nickel compounds in cultured Chinese hamster ovary
cells. Cancer Lett., 15: 35-40.
ROBISON, S.H., CANTONI, O. & COSTA, M. (1982) Strand breakage and
decreased molecular weight of DNA induced by specific metal
compounds. Carcinogenesis, 3: 657-662.
ROCKSTROH, H. (1959) [On the etiology of bronchial cancer in
arsenic processing nickel smelting plants.] Arch.
Geschwulstforsch., 14: 151-162 (in German).
RODRIGUEZ-ARNAIZ, R. & RAMOS, P. (1986) Mutagenicity of nickel
sulphate in Drosophila melanogaster. Mutat. Res., 170(3): 115-117.
ROSSMAN, T.G., MOLINA, M. & MEYER, L.W. (1984) The genetic
toxicology of metal compounds: I. Induction of prophage in E.
coli WP2s. Environ. Mutagen., 6: 59-69.
ROUSH, C.C., MEIGS, I.W., KELLY, J.A., FLANNERY, I.T. & BURDO, H.
(1980) Sinonasal cancer and occupation: a case-control study. Am.
J. Epidemiol., 111: 183-193.
ROY, B.R. (1985) Nepera 82-06 sampling method for inorganic nickel.
In: Brown, S.S. & Sunderman, F.W. Jr, ed. Progress in Nickel
toxicology. Proceedings of the 3rd International Conference on
Nickel Metabolism and Toxicology, Paris, 4-7 September, 1984,
Oxford, Blackwell Scientific Publications, pp. 191-194.
RUBANYI, G. & INOVAY, J. (1982) Effect of nickel ions on
spontaneous electrically and norepinephrine stimulated isometric
contractions in the isolated portal vein of the rat. Acta physiol.
Acad. Sci. Hung., 59: 181-186.
RUBANYI, G. & KOVACH, A.G.B. (1980) Cardiovascular actions of
nickel ions. Acta physiol. Acad. Sci. Hung., 55: 345-353.
RUBANYI, G., BALOGH, I., KOVACH, A.G.B., SOMOGYI, E. & SOTONYI, P.
(1980) Effect of nickel ions on ultrastructure of isolated perfused
rat heart. J. mol. cell Cardiol., 12: 609-618.
RUBANYI, G., LIGETI, L. & KOLLER, A. (1981) Nickel is released
from the ischemic myocardium and contracts coronary vessels by a
Ca-dependent mechanism. J. mol. cell Cardiol., 13(11): 1023-1026.
RUBANYI, G., BIRTALAN, I., GERGELY, A. & KOVACH, A.G. (1982a)
Serum nickel concentration in women during pregnancy, during
parturition, and post partum. Am. J. Obstet. Gynecol., 143: 167-169.
RUBANYI, G., KALABAY, L., PATAKI, T. & HAJDU, K. (1982b) Nickel
induces vasoconstriction in the isolated canine coronary artery by
a tonic Ca2+-activation mechanism. Acta physiol. Acad. Sci. Hung.
59: 155-159.
RUBANYI, G., BAKOS, M., HAJDU, K. & PATAKI, T. (1982c) Dependence
of nickel induced coronary vasoconstriction on the activity of the
electrogenic Na+, K+-pump. Acta physiol. Acad. Sci. Hung., 59:
169-174.
RUBANYI, G., HAJDU, K., PATAKI, T. & BAKOS, M. (1982d) The role of
adrenergic receptors in Ni2+-induced coronary vasoconstriction.
Acta Physiol. Acad. Sci. Hung., 59: 161-167.
RUBANYI, G., LIGETI, L., KOLLER, A. & KOVACH, A.G. (1984) Possible
role of nickel ions in the pathogenesis of ischemic coronary
vasoconstriction in the dog heart. J. mol. cell Cardiol., 16: 533-546.
RUTHERFORD, G.K. & BRAY, C.R. (1979) Extent and distribution of
soil heavy metal contamination near a nickel smelter at Coniston,
Ontario. J. environ. Qual., 8(2): 219-222.
RYU, R.K.N., BOVILL, E.G. Jr, SKINNER, H.B. & MURREY, W.R. (1987)
Soft tissue sarcoma associated with aluminum oxide ceramic total
hip arthroplasty. Clin. Orthop. relat. Res., 216: 207-212.
SADIQ, M., ZAIDI, T.H., UL-HODA, A. & MIAN, A.A. (1982) Heavy
metal concentrations in shrimp, crab, and sediment obtained from
Ad-Dammam sewage outfall area. Bull. environ. Contam. Toxicol.
29(3): 313-320.
SAICHENKO, S.P. (1985) Experimental evaluation of genetic danger of
metals administered with drinking water. In: Domnin, S.C. & Kogan,
F.V. ed. Problems of occupational pathology and toxicology in
mining and metallurgy. Moscow, Erisman Institute of Hygiene, pp.
75-80.
SAICHENKO, S.P. & SHARAPOVA, N.Z (1987) About the mutagenicity of
some metals for Salmonella typhimurium. In: Domnin, S.C. &
Shcherbakov S.V. ed. Problems of hygiene and occupational
pathology in colored metals and steel industry. Moscow, Erisman
Institute of Hygiene, pp. 97-101.
SAKNYN, A.V. & BLOKHIN, V.A. (1978) [Development of malignant
tumors in rats under the influence of nickel-containing aerosols.]
Vopr. Onkol., 24(4): 44-48 (in Russian).
SAKNYN, A.V. & SHABYNINA, N.K. (1970) [Some statistical material on
carcinogenic danger in the production of nickel from oxidized
ores.] Gig. Tr. prof. Zabol., 14(11): 10-13 (in Russian).
SAKNYN, A.V. & SHABYNINA, N.K. (1973) [Epidemiology of malignant
neoplasms in nickel plants.] Gig. Tr. prof. Zabol., 9: 25-28
(in Russian).
SAKNYN, A.V., SIPATOV, G.YA., YELNICHUYKH, L.N. & STARKOV, P.S.
(1978) [Hygienic characterization of dusts in primary nickel
production.] In: Domnin, S.G. ed. Occupational disease of dust
aetiology. Erisman Institute of Hygiene, Moscow, Vol. 4, pp. 18-23.
SALTZMANN, B.E., CHOLAK, J., SCHAFER, L.J., YEAGER, D.W., MEINERS,
B.O., SVETLIK, J. & ZWILLENBERG, M.L. (1985) Concentrations of six
metals in the air of eight cities. Environ. Sci. Technol., 19(4):
328-333.
SAMELA, D., TSOUMPAS, G.M. & WALSHANS, G.K. (1986) Environmental
aspects of the combustion of sewage sludge in a utility boiler.
Environ. Prog., 5(2): 110-115.
SAMITZ, M.H. & KATZ, S.A. (1975) Nickel dermatitis hazards from
prostheses. In vivo and in vitro solubilization studies. Br. J.
Dermatol., 92: 287.
SAMITZ, M.H. & KATZ, S.A. (1976) Nickel-epidermal interactions:
diffusion and binding. Environ. Res., 11(1): 34-39.
SAMITZ, M.H. & POMERANTZ, H. (1958) Studies of the effects on the
skin of nickel and chromium salts. Arch. ind. Health, 18: 473-479.
SAMITZ, M.H., KATZ, S.A., SCHEINER, D.M. & LEWIS, J.E. (1975)
Attempts to induce sensitization in guinea-pigs with nickel
complexes. Acta dermtovenereol., 55: 475-480.
SANDE, J.H. VAN DE, MACINTOSH, L.P. & JOVIN, T.M. (1982) Mn2+ and
other transition metals at low concentration induce the right-to-
left helical transformation of poly (d(G-C)). EMBO J., 1: 777-782.
SANDERS, J.R., MCGRATH, S.P. & ADAMS, T.M. (1986) Zink, copper and
nickel concentrations in ryegrass grown on sewage sludge-
contaminated soils of different pH. J. Sci. Food Agric., 37:
961-968.
SANFORD, W.E., NIEBOER, E., BACH, P., STACE, B., GREGG, N. &
DOBROTA, M. (1988) The renal clearance and toxicity of nickel. In:
Fourth International Conference on Nickel Metabolism and
Toxicology, Abstracts, Espoo, Finland, 5-9 September 1988,
Helsinki, Institute of Occupational Health, p.17.
SANINA, J.P. (1968) [Toxicology of nickel carbonyl.] Toxikol. Nov.
Prom. Khim. Veshchestv., 10: 144-149 (in Russian).
SANOTSKII, J.V. (1955) [Action mechanism of nickel carbonyl.]
Farmakol. Toksikol., 18(2): 48-50 (in Russian).
SANTUCCI, B., CRISTAUDO, A., CANNISTRACI, C. & PICARDO, M. (1988)
Nickel sensitivity: effects of prolonged oral intake of the
element. Contact Dermatit., 19: 202-295.
SANZOLONE, R.F., CHAO, T.T. & CRENSHAW, G.L. (1979) Atomic
absorption spectrometric determination of cobalt, nickel, and
copper in geological materials with matrix marking and chelation-
extraction. Anal. chim. Acta, 105: 247-253.
SAPEK, B. & SAPEK, A. (1980) [Nickel content in the profiles of
organic soils.] In: Anke, M., Schneider, H.-J. & Brückner, Chr.
ed. [3. Trace Element Symposium: Nickel, Jena, German Democratic
Republic, 7-11 July, 1980,] Jena, Friedrich-Schiller University, pp.
185-191 (in German).
SARKAR, B. (1980) Nickel in blood and kidney. In: Brown, S.S. &
Sunderman, F.W. Jr, ed. Nickel toxicology, Proceedings of the 2nd
International Conference on Nickel Toxicology, Swansea, 3-5
September, 1980, London, New York, Academic Press, pp. 81-84.
SAXHOLM, H.J.K., REITH, A. & BROEGGER, A. (1981) Oncogenic
transformation and cell lysis in C3H/10T 1/2 cells and increased
sister chromatid exchange in human lymphocytes by nickel
subsulfide. Cancer Res., 41(10): 4136-4139.
SCHMIDT, J.A. & ANDREN, A.W. (1980) The atmospheric chemistry of
nickel. In: Nriagu, J.O. ed. Nickel in the environment, New York,
Chichester, Brisbane, Toronto, John Wiley and Sons, pp. 93-135.
SCHMIDT, W. & DIETL, F. (1981) [Determination of nickel and cobalt
in digested soils by means of flameless atomic absorption with
zirconium coated graphite tubes.] Fresenius Z. anal. Chem., 308:
129-132 (in German with English summary).
SCHNEGG, A. & KIRCHGESSNER, M. (1975a) [The essentiality of nickel
for the growth of animals.] Z. Tierphysiol. Tierernähr.
Futtermittelkd., 36: 63-74 (in German).
SCHNEGG, A. & KIRCHGESSNER, M. (1975b) [Changes in the hemoglobin
content, erythrocyte content and hematocrit in nickel deficiency.]
Nutr. Metabol., 19: 268-278 (in German).
SCHNEGG, A. & KIRCHGESSNER, M. (1976a) [Interaction of nickel with
iron, copper and zinc.]. Arch. Tierernähr., 26(8): 543-549
(in German with English summary).
SCHNEGG, A. & KIRCHGESSNER, M. (1976b) [Absorption and metabolic
efficiency of iron during nickel deficiency.] Int. Z. Vitam.
Forsch., 46: 96-99 (in German with English summary).
SCHNEGG, A. & KIRCHGESSNER, M. (1977a) [Changes in enzyme
activities in liver and kidney with nickel and iron deficiency,
respectively.] Z. Tierphysiol. Tierernährg. Futtermittelkd., 38:
200-205 (in German with English summary).
SCHNEGG, A. & KIRCHGESSNER, M. (1977b) [Alkaline and acid
phosphatase activity in liver and serum during nickel versus Fe-
deficiency.] Int. Z. Vitam. Forsch., 47: 274-276 (in German with
English summary).
SCHNEGG, A. & KIRCHGESSNER, M. (1977c) [Changes of various
substrate concentrations in the serum and liver in Ni- or Fe-
deficiency.] Z. Tierphysiol. Tierernährg. Futtermittelkd., 39(5-6):
247-251 (in German with English summary).
SCHNEGG, A. & KIRCHGESSNER, M. (1977d) [Differential diagnosis of
Fe and Ni deficiency by determination of some enzyme activities.]
Zbl. Vet. Med., 24 A: 242-247 (in German).
SCHNEGG, A. & KIRCHGESSNER, M. (1980) [On the essentiallity of
nickel for animal growth.] In: Anke, M. Schneider, H.-J. &
Brückner, Chr. ed. [3. Trace Element Symposium: Nickel, Jena,
German Democratic Republic, 7-11 July, 1980,] Jena, Friedrich-
Schiller University, pp. 11-16 (in German).
SCHNEIDER, H.-J., ANKE, M. & KLINGER, G. (1980) [The nickel-status
of human beings.] In: Anke, M. Schneider, H.-J. & Brückner, Chr.
ed. [3. Trace Element Symposium: Nickel, Jena, German Democratic
Republic, 7-11 July, 1980,] Jena, Friedrich-Schiller-University, pp.
277-283 (in German).
SCHROEDER, H.A. (1968) Serum cholesterol levels in rats fed
thirteen trace elements. J. Nutr., 94: 475-480.
SCHROEDER, H.A. (1970) A sensible look at air pollution by metals.
Arch. environ. Health, 21: 798-806.
SCHROEDER, H.A. & MITCHENER, M. (1971) Toxic effects of trace
elements on the reproduction of mice and rats. Arch. environ.
Health, 23: 102-106.
SCHROEDER, H.A. & MITCHENER, M. (1975) Life time effects of
mercury, methyl mercury and nine other trace metals on mice. J.
Nutr., 105: 452-458.
SCHROEDER, H.A., BALASSA, J.J. & TIPTON, J.H. (1962) Abnormal
trace metals in man - nickel. J. chron. Dis., 15: 51-65.
SCHROEDER, H.A., BALASSA, J.J. & VINTON, W.H. Jr (1964) Chromium,
lead, cadmium, nickel and titanium in mice: effect on mortality,
tumors and tissue levels. J. Nutr., 83: 239-250.
SCHROEDER, H.A., MITCHENER, M. & NASON, A.P. (1974) Life-term
effects of nickel in rats: survival, tumors, interactions with
trace elements and tissue levels. J. Nutr., 104: 239-243.
SCHUMANN, H. (1980) [Nickel contamination of surface waters and
drinking water and some influencing factors.] In: Anke, M.,
Schneider, H.-J. & Brückner, Chr. ed. [3. Trace Element Symposium
Nickel, Jena, German Democratic Republic, 7-11 July, 1980.] Jena,
Friedrich-Schiller University, pp. 155-161 (in German).
SEEMANN, J., WITTIG, P., KOLLMEIER, H. & ROTHE, G. (1985)
Analytical measurements of Cd, Pb, Zn, Cr and Ni in human tissues.
Lab. Med., 9: 294-299.
SEN, P. & COSTA, M. (1985) Induction of chromosomal damage in
Chinese hamster ovary cells by soluble and particulate nickel
compounds: preferential fragmentation of the heterochromatic long
arm of the X-chromosome by carcinogenic crystalline NiS particles.
Cancer Res., 45(5): 2320-2325.
SEN, P. & COSTA, M. (1986) Pathway of nickel uptake influences its
interaction with heterochromatic DNA. Toxicol. appl. Pharmacol.
84(2): 278-285.
SHANNON, H.S., JULIAN, J.A., MUIR, D.C.F., ROBERTS, R.S. &
CECUTTI, A.C. (1984) A mortality study of Falconbridge workers. In:
Nickel in the human environment, Proceedings of a Joint Symposium,
Lyon, 8-11 March, 1983, Lyon, International Agency for Research on
Cancer, pp. 117-124 (IARC Scientific Publications No. 53).
SHAW, T.L. & BROWN, V.M. (1971) Heavy metals and the fertilization
of rainbow trout eggs. Nature (Lond.), 230: 251.
SHI, Z. (1986) Acute nickel carbonyl poisoning: a report of 179
cases. Br. J. ind. Med., 43: 422-424.
SHI, Z., LATA, A. & HAN, Y. (1986) A study of serum monoamine
oxidase (MAO) activity and EEG in nickel carbonyl workers. Br. J.
ind. Med., 43: 425-426.
SHIBATA, M., IZUMI, K., SANO, N., AKAGI, A. & OTSUKA, H. (1989)
Induction of soft tissue tumours in F344 rats by subcutaneous,
intramuscular, intra-articular, and retroperitoneal injection of
nickel sulphide (Ni3S2). J. Pathol., 157: 263-274.
SILVERSTEIN, M., MIRER, F., KOTELCHUCK, D., SILVERSTEIN, B. &
BENNETT, M. (1981) Mortality among workers in a diecasting and
electroplating plant. Scand. J. Work Environ. Health, 7: 156-165.
SINA. J.F., BEAN, C.L., DYSART, G.R., TAYLOR, V.I. & BRADLEY,
M.O. (1983) Evaluation of the alkaline elution/rat hepatocyte assay
as a predictor of carcinogenic/mutagenic potential. Mutat. Res.
113: 357-391.
SINGH, I. (1984) Induction of gene conversion and reverse mutation
by manganese sulphate and nickel sulphate in Saccharomyces
cerevisiae. Mutat. Res., 137: 47-49.
SIROVER, M.A. & LOEB, L.A. (1977) On the fidelity of DNA
replication. Effect of metal activators during synthesis with avian
myeloblastosis virus DNA polymerase. J. biol. Chem., 252: 3605-3610.
SJOVALL, P., CHRISTENSEN, O.B. & MOLLER, H. (1987) Oral
hyposensitization in nickel allergy. J. Am. Acad. Dermatol., 17: 774-
778.
SKAUG, V., GYLSETH, B., REISS, A.P. & NORSETH, T. (1985) Tumor
induction in rats after intrapleural injection of nickel subsulfide
and nickel oxide. In: Brown, S.S. & Sunderman, F.W. Jr, ed. Nickel
toxicology, Proceedings of the 2nd International Conference on
Nickel Toxicology, Swansea, 3-5 September, 1980, London, New York,
Academic Press, pp. 37-40.
SMART, G.A. & SHERLOCK, J.C. (1987) Nickel in foods and the diet.
Food Addit. Contam., 4(1): 61-71.
SMIALOWICZ, R.J. (1985) The effect of nickel and manganese on
natural killer cell activity. In: Brown, S.S. & Sunderman, F.W. Jr,
ed. Progress in Nickel toxicology. Proceedings of the 3rd
International Conference on Nickel Metabolism and Toxicology,
Paris, France, 4-7 September, 1984, Oxford, Blackwell Scientific
Publications, pp. 161-164.
SMIALOWICZ, R.J., ROGERS, R.R., RIDDLE, M.M. & STOTT, G.A. (1984)
Immunologic effects of nickel: 1. Suppression of cellular and human
immunity. Environ. Res., 33: 413-427.
SMIALOWICZ, R.J., RODGERS, R.R., RIDDLE, M.M., GARNER, R.J., ROWE,
D.G. & LUEBKE, R.W. (1985) Immunologic effects of nickel. II.
Suppression of natural killer cell activity. Environ. Res., 36: 56-66.
SMIALOWICZ, R.J., ROGERS, R.R., RIDDLE, M.M., ROWE, D.G. & LUEBKE,
R.W. (1986) Immunological studies in mice following in utero
exposure to NiCl2. Toxicology, 38: 293-303.
SMIALOWICZ, R.J., ROGERS, R.R., RIDDLE, M.M., LUEBKE, R.W.,
FOGELSON, L.D. & ROWE, D.G. (1987) Effects of manganese, calcium,
magnesium, and zinc on nickel-induced suppression of murine natural
killer cell activity. J. Toxicol. environ. Health, 20(1-2): 67-80.
SMITH, J.C. (1969) A controlled environment system for trace
element deficiency studies. Trace Subst. environ. Health, 2: 223-242.
SMITHE, J.C. & HACKLEY, B. (1968) Distribution and excretion of
nickel-63 administered intravenously to rats. J. Nutr., 95: 541-546.
SMITH-SONNEBORN, J., PALIZZI, R.A., McCANN, E.A. & FISHER, G.L.
(1983) Bioassay of genotoxic effects of environmental particles in
a feeding ciliate. Environ. Res., 32: 474-479.
SMITH-SONNEBORN, J., LEIBOVITZ, B., DONATHAN, R. & FISHER, G.L.
(1986) Bioassay of environmental nickel dusts in a particle feeding
ciliate. Environ. Mutagen., 8(4): 621-626.
SNODGRAS, W. (1980) Distribution and behaviour of nickel in the
aquatic environment. In: Nriagu, J.O. ed. Nickel in the
environment, New York, Chichester, Brisbane, Toronto, John Wiley
and Sons, pp. 203-274.
SOLOMONS, N.W., VITERI, F., SHULER, T.R. & NIELSEN, F.H. (1982)
Bioavailability of nickel in man: effects of foods and chemically-
defined dietary constituents on the absorption of inorganic nickel.
J. Nutr., 112(1): 39-50.
SONNENFELD, G., STREIPS, U.N. & COSTA, M. (1983) Differential
effects of amorphous and crystalline nickel sulfide on murine
alpha/beta interferon production. Environ. Res., 32: 474-479.
SORAHAN, T. & WATERHOUSE, J.A.W. (1983) Mortality study of nickel-
cadmium battery workers by the method of regression models in life
tables. Br. J. ind. Med., 40: 293-300.
SORAHAN, T., BURGES, D.C.L. & WATERHOUSE, J.A.H. (1987) A
mortality study of nickel/chromium platers. Br. J. ind. Med., 44:
250-258.
SORINSON, S.N., KORNILOVA, A.P. & ARTEMEVA, A.M. (1958)
[Concentrations of nickel in blood and urine of workers in the
nickel carbonyl industry.] Gig. i Sanit., 23(9): 69-72 (in Russian).
SPEARS, J.W. (1984) Nickel as a "newer trace element" in the
nutrition of domestic animals. J. anim. Sci., 59: 823-835.
SPEARS, J.W. & HATFIELD, E.E. (1977) Role of nickel in animal
nutrition. Feedstuffs, 49: 24-28.
SPEARS, J.W., HATFIELD, E.E., FORBES, R.M. & KOENIG, S.E. (1978)
Studies on the role of nickel in the ruminant. J. Nutr., 108:
313-320.
SPEARS, J.W., JONES, E.E., SAMSELL, L.J. & ARMSTRONG, W.D. (1984)
Effect of dietary nickel on growth, urease activity, blood
parameters and tissue mineral concentrations in the neonatal pig.
J. Nutr., 114: 845-854.
SPENCER, D.F. (1980) Nickel and aquatic algae. In: Nriagu, J.O.
ed. Nickel in the environment, New York, Chichester, Brisbane,
Toronto, John Wiley and Sons, pp. 339-347.
SPENCER, D.F. & GREENE, R.W. (1981) Effects of nickel on seven
species of freshwater algae. Environ. Pollut., A25: 241-247.
SPIEGELBERG, T., KORDEL, W. & HOCHRAINER, D. (1984) Effects of NiO
inhalation on alveolar macrophages and the humoral immune systems
of rats. Ecotoxicol. environ. Safety, 8: 516-525.
SPRUIT, D., MALI, J.W.H. & DE GROOT, N. (1965) The interaction of
nickel ions with human cadaverous dermis. J. Invest. Dermatol., 44:
103-106.
STACK, M.V., BURKITT, A.J. & NICKLESS, G. (1976) Trace metals in
teeth at birth (1957-1963 and 1972-1973). Bull. environ. Contam.
Toxicol., 16: 764-766.
STAHLY, E.E. (1973) Some considerations of metal carbonyls in
tobacco smoke. Chem. Ind., 12: 620-623.
STEDMAN, D.H. (1986a) Method 5 - determination of nickel carbonyl
in air colorimetry. In: O'Neill, I.K. Schuller, P. & Fishbein,
L. ed. Environmental carcinogens - selected methods of analysis.
Vol. 8: Some metals: As,Be, Cd, Cr, Ni, Pb, Se, Zn. Lyon,
International Agency for Research on Cancer, pp. 261-267 (IARC
Scientific Publications No. 71).
STEDMAN, D.H. (1986b) Method 6 - determination of nickel carbonyl
in air by chemiluminescence. In: O'Neill, J.K. Schuller, P. &
Fishbein, L. ed. Environmental carcinogens - selected methods of
analysis. Vol. 8: Some metals: As, Be, Cd, Cr, Ni, Pb, Se, Zn.
Lyon, International Agency for Research on Cancer, pp. 269-273
(IARC Scientific Publications No. 71).
STEDMAN, D.HJ. & HIKADE, D.A. (1980) The rate of decay of traces of
nickel carbonyl in air. In: Brown, S.S. & Sunderman, F.W. Jr, ed.
Nickel toxicology, Proceedings of the 2nd International Conference
on Nickel Toxicology, Swansea, 3-5 September, 1980, London, New
York, Academic Press, pp. 183-186.
STERN, A.C., BOUBEL, R.W., TURNER, D.B. & FOX, D.L. (1984)
Fundamentals of air pollution, 2nd ed. Orlando, San Diego, San
Francisco, Academic Press, pp. 103-113.
STERN, R.M. (1983) Assessment of risk of lung cancer for welders.
Arch. env. Health, 38(3): 148-156.
STERN, R.M., HANSEN, K. & THOMSEN, E. (1985) An in vitro
genotoxicity assay to complement in vivo inhalation studies and for
occupational monitoring of metallic aerosols and welding fumes. In:
Brown, S.S. & Sunderman, F.W. Jr, ed. Progress in nickel
toxicology. Proceedings of the 3rd International Conference on
Nickel Metabolism and Toxicology, Paris, 4-7 September, 1984,
Oxford, Blackwell Scientific Publications, pp. 77-80.
STEWART, S.G. & CROMIA, F.E. (1934) Experimental nickel dermatitis.
J. Allergy, 5: 575-582.
STOEPPLER, M. (1980) Analysis of nickel in biological materials and
natural waters. In: Nriagu, J.O. ed. Nickel in the environment, New
York, Chichester, Brisbane, Toronto, John Wiley & Sons, pp. 661-821.
STOKES, P. (1975) Uptake and accumulation of copper and nickel by
metal-tolerant strains of Scenedesmus. Int. Ver. Limnol., 19:
128-137.
STOKES, P.M. (1981) Nickel in aquatic systems. In: Effects of
nickel in the Canadian environment, Ottawa, National Research
Council of Canada, pp. 77-117 (Publication No. NRCC-18568).
STONER, G.O., SHIMKIN, M.B., TROXELL, M.C., THOMPSON, T.L. &
TERRY, L.S. (1976) Test for carcinogenicity of metallic compounds
by the pulmonary tumor response in strain of mice. Cancer Res., 36:
1744-1747.
STORENG, R. & JONSEN, J. (1980) Effect of nickel chloride and
cadmium acetate on the development of preimplantation mouse embryos
in vitro. Toxicology, 17: 183-187.
STORENG, R. & JONSEN, J. (1981) Nickel toxicity in early
embryogenesis in mice. Toxicology, 20(1): 45-51.
STOVBUN, A.T., YATSYUK, M.D., POMARENKO, U.I. & YAKOVLEVA, L.S.
(1962) [Data on the trace element composition of human milk and
various modifications of cow's milk.] Nauk. Zap. Ivano.-Frankivskii
Derzk. Med. Inst., 5: 38-40 (in Russian).
STRAIN, W.H., VARNES, A.W., DAVIS, B.R. & KARK, E.C. (1980)
[Nickel in drinking and household water.] In: Anke, M., Schneider,
H.-J. & Brûckner, Chr. ed. [3. Trace Element Symposium: Nickel,
Jena, German Democratic Republic, 7- 11 July, 1980,] Jena,
Friedrich-Schiller University, pp. 149-154 (in German).
STRATTON, G.W. & CORKE, C.T. (1979) The effect of nickel on the
growth, photosynthesis and nitrogenase activity of Anabaena
inequalis. Can. J. Microbiol., 25: 1094-1099.
STURGEON, R.E., BERMAN, S.S. & WILLIE, S.N. (1982) Concentration
of trace metals from sea-water by complexation with 8-
hydroxyquinoline and adsorption in C18-bonded silica gel. Talanta,
29: 167-171.
SUMINO, K., HAYAKAWA, K., SHIBATA, T. & KITAMURA, S. (1975) Heavy
metals in normal Japanese tissues. Arch. environ. Health, 30: 487-494.
SUNDERMAN, F.W. (1964) Nickel and copper mobilization by sodium
diethyldithiocarbamate. J. New Drugs, 4: 154-161.
SUNDERMAN, F.W. (1970) Nickel poisoning. In: Sunderman, F.W. &
Sunderman, F.W. Jr, ed. Laboratory diagnosis of diseases caused by
toxic agents, St. Louis, Missouri, Warren H. Green Inc. pp. 387-
396.
SUNDERMAN, F.W. (1971) The treatment of acute nickel carbonyl
poisoning with sodium diethyldithiocarbamate. Ann. clin. Res., 3(3):
182-185.
SUNDERMAN, F.W. Jr (1977) The metabolism and toxicology of nickel.
In: Brown, S.S. & Savory, J. ed. Chemical toxicology and clinical
chemistry of metals. Proceedings of the 2nd International
Conference, Montreal, 19-22 July, 1983, London, New York, Blackwell
Scientific Publications, pp. 231-259.
SUNDERMAN, F.W. Jr (1980) Analytical biochemistry of nickel.
International Union of Pure and Applied Chemistry, Subcommittee on
environmental and Occupational Toxicology of Nickel. Pure appl.
Chem., 52: 527-544.
SUNDERMAN, F.W. Jr (1983a) Potential toxicity from nickel
contamination of intravenous fluids. Ann. clin. lab. Sci., 13(1): 1-4.
SUNDERMAN, F.W. Jr (1983b) Organs and species specificity in nickel
subsulfide carcinogenesis. In: Langenbach, R., Nesnow, S. & Rice,
J.M. ed. Organ and species specificity in chemical carcinogenesis,
New York, Plenum Press, pp. 107-127 (Basic life sciences. Vol. 24).
SUNDERMAN, F.W. Jr (1984) Carcinogenicity of nickel compounds in
animals. In: Nickel in the human environment. Proceedings of a
Joint Symposium, Lyon, 8-11 March, 1983, Lyon, International Agency
for Research on Cancer, pp. 127-142 (IARC Scientific Publications
No. 53).
SUNDERMAN, F.W. Jr (1986) Iatrogenic exposures to toxic metals. 9th
Annual meeting of the National Academy of Clinical Biochemistry on
Metal Metabolism and Disease, Atlanta, TA, USA, July 9-20, 1985.
Clin. Physiol. Biochem., 4(1): 112.
SUNDERMAN, F.W. Jr (1987) Lipid peroxidation as a mechanism of
acute nickel toxicity. Toxicol. environ. Chem., 15: 59-69.
SUNDERMAN, F.W. Jr (1989a) Mechanisms of nickel carcinogensis.
Scand. J. Work Environ. Health, 15: 1-12.
SUNDERMAN, F.W. Jr (1989b) Carcinogencity of metal alloys in
orthopaedic prostheses: clinical and experimental studies. Fundam.
appl. Toxicol., 13: 1-12.
SUNDERMAN, F.W. Jr & BARBER, A. M. (1988) Finger loops, ongogens,
and metals. Ann. clin. lab. Sci., 18: 267-288.
SUNDERMAN, F.W. Jr & DONELLY, A.J. (1965) Studies of nickel
carcinogenesis metastasizing pulmonary tumors in rats induced by
the inhalation of nickel carbonyl. Am. J. Pathol., 46: 1027-1041.
SUNDERMAN, F.W. Jr & HOPFER, S.M. (1983) Correlation between the
carcinogenic activities of nickel compounds and their potencies for
stimulating erythropoiesis in rats. In: Saker, B. ed. Biological
aspects of metals and metal-related diseases, New York, Raven
Press, pp. 171-181.
SUNDERMAN, F.W. & KINCAID, J.F. (1954) Nickel poisoning II.
Studies on patients suffering from acute exposure to vapors of
nickel carbonyl. J. Am. Med. Assoc., 155(10): 889-894.
SUNDERMAN, F.W. Jr & MCCULLY, K.S. (1983) Effects of manganese
compounds on carcinogenicity of nickel subsulfide in rats.
Carcinogenesis, 4(4): 461-465.
SUNDERMAN, F.W. Jr & MAENZA, R.M. (1976) Comparisons of
carcinogenicities of nickel compounds in rats. Res. Commun. chem.
Pathol. Pharmacol., 14: 319-330.
SUNDERMAN, F.W. Jr & OSKARSSON, A. (1988) Nickel. In: Merian, ed.
Metals and their compounds in the environment, Weinheim, VCH
Verlagsgesellschaft, pp. 1-19.
SUNDERMAN, F.W. Jr & SELIN, C.E. (1968) The metabolism of nickel-63
carbonyl. Toxicol. appl. Pharmacol., 12: 207-217.
SUNDERMAN, F.W. & SUNDERMAN, F.W. Jr (1961a) Nickel poisoning. XI.
Implication of nickel as a pulmonary carcinogen in tobacco smoke.
Am. J. clin. Pathol., 35: 203-209.
SUNDERMAN, F.W. & SUNDERMAN, F.W. Jr (1961b) Löffler's syndrome
associated with nickel sensitivity. Arch. int. Med., 107: 405-408.
SUNDERMAN, F.W. Jr, KINCAID, J.F., DONNELLY, A.J. & WEST, B.
(1957) Nickel poisoning. IV. Chronic exposure of rats to nickel
carbonyl: a report after one year of observation. Arch. ind.
Health, 16: 480-485.
SUNDERMAN, F.W., DONELLY, A.J., WEST, B. & J.F. KINCAID (1959)
Nickel poisoning. IX. Carcinogenesis in rats exposed to nickel
carbonyl. Arch. ind. Health, 20: 36-41.
SUNDERMAN, F.W., RANGE, C.L., SUNDERMAN, F.W. Jr, DONELLY, A.J. &
LUCYSZYN, G.W. (1961) Nickel poisoning. XII. Metabolic and
pathologic changes in acute pneumonitis from nickel carbonyl. Am.
J. clin. Pathol., 36: 477-491.
SUNDERMAN, F.W. Jr, WHITE, J.C. & SUNDERMAN, F.W. (1963) Metabolic
balance studies in hepatolenticular degeneration treated with
diethyldithiocarbamate. Am. J. Med., 34: 875-888.
SUNDERMAN, F.W. Jr, ROSZEL, N.O. & CLARK, R.J. (1968) Gas
chromatography of nickel carbonyl in blood and breath. Arch.
environ. Health, 16(6): 836-843.
SUNDERMAN, F.W. Jr, NOMOTO, S., PRADHAN, A.M., LEVINE, H.,
BERNSTEIN, S.H. & HIRSCH, R. (1970) Increased concentrations of
serum nickel after acute myocardial infarction. J. Med., 283
(17): 896-899.
SUNDERMAN, F.W. Jr, NOMOTO, S. & NECHAY, M. (1971) Nickel
metabolism in myocardial infarction. II. Measurements of nickel in
human tissues. Trace Subst. environ. Health, 4: 352-356.
SUNDERMAN, F.W. Jr, NOMOTO, S., MORANG, R., NECHAY, M.W., BURKE,
C.N. & NIELSEN, S.W. (1972) Nickel deprivation in chicks. J.
Nutr., 102: 259-268.
SUNDERMAN, F.W. Jr, KASPRZAK, K., HORAK, E., GITLITZ, P. &
ONKELINX, C. (1976a) Effects of triethylenetetramin upon the
metabolism and toxicity of 63NiCl2 in rats. Toxicol. appl.
Pharmacol., 38: 177-188.
SUNDERMAN, F.W. Jr, KASPRZAK, K.S., LAU, T.J., MINGHETTI, P.P.,
MAECIZA, R.M., BECKER, N., ONKELINX, C. & GOLDBLATT, P.J. (1976b)
Effects of manganese on carcinogenicity and metabolism of nickel
subsulfide. Cancer Res., 36: 1790-1800.
SUNDERMAN, F.W. Jr, SHEN, S.K., MITCHELL, J.M., ALLPASS, P.R. &
DAMJANOV, I. (1978a) Embryotoxicity and fetal toxicity of nickel in
rats. Toxicol. appl. Pharmacol., 43(2): 381-390.
SUNDERMAN, F.W. Jr, ALLPASS, P. & MITCHELL, J. (1978b) Recent
progress in nickel carcinogenesis. Ann. Ist. Super. Sanita, 22(2):
669-679.
SUNDERMAN, F.W. Jr, ALLPASS, P. & MITCHELL, J. (1978c) Ophthalmic
malformations in rats following prenatal exposure to inhalation of
nickel carbonyl. Ann. clin. lab. Sci., 8: 499-500.
SUNDERMAN, F.W. Jr, MITCHELL, J., ALLPASS, P. & BASELT, R. (1978d)
Embryotoxicity and teratogenicity of nickel carbonyl in rats.
Toxicol. appl. Pharmacol., 45: 345.
SUNDERMAN, F.W. Jr, MAENZA, R.M., ALPASS, P.R., MITCHELL, J.M.,
DAMJANOV, I. & GOLDBLATT, P.J. (1978e) Carcinogenicity of nickel
subsulfide in Fischer rats and Syrian hamsters after administration
by various routes. Adv. exp. Med. Biol., 91: 57-67.
SUNDERMAN, F.W. Jr, ALLPASS, P.R., MITCHELL, J.M., BASELT, R.C. &
ALBERT, D.M. (1979a) Eye malformations in rats: induction by
prenatal exposure to nickel carbonyl. Science, 203: 550-553.
SUNDERMAN, F.W. Jr, MAENZA, R.M., HOPFER, S.M., MITCHELL, J.M.,
ALLPASS, P.R. & DAMJANOV, I. (1979b) Induction of renal cancers in
rats by intrarenal injection of nickel subsulfide. J. environ.
Pathol. Toxicol., 2(6): 1511-1527.
SUNDERMAN, F.W. Jr, SHEN, S.K., REID, M.C. & ALLPASS, P.R. (1980)
Teratogenicity and embryotoxicity of nickel carbonyl in Syrian
hamsters. Teratog. Carcinog. Mutagen., 1(2): 223-233.
SUNDERMAN, F.W. Jr, MCCULLY, K.S. & RINEHIMER, L.A. (1981)
Negative test for transplacental carcinogenicity of nickel
subsulfide in Fischer rats. Res. Commun. chem. Pathol. Pharmacol.
31(3): 545-554.
SUNDERMAN, F.W. Jr, REID, M.C., SHEN, S.K. & KEVORKIAN, C.B.
(1983) Embryotoxicity and teratogenicity of nickel compounds. In:
Clarkson, T.W., Nordberg, G.F. & Sager, P.R. ed. Reproductive and
developmental toxicity of metals. New York, London, Plenum Press,
pp. 399-416.
SUNDERMAN, F.W. Jr, CRISOSTOMO, M.C., REID, M.C., HOPFER, S.M. &
NOMOTO, S. (1984a) Rapid analysis of nickel in serum and whole
blood by electrothermal atomic absorption spectrophotometry. Ann.
clin. lab. Sci., 14(3): 232-241.
SUNDERMAN, F.W. Jr, MACCULLY, K.S. & HOPFER, S.M. (1984b)
Association between erythrocytosis and renal cancers in rats
following intrarenal injection of nickel compounds. Carcinogenesis,
5: 1511-1517.
SUNDERMAN, F.W. Jr, MARZOUK, A., CRISOSTOMO, M.C. & WEATHERBY,
D.R. (1985) Electrothermal atomic absorption spectrophotometry of
nickel in tissue homogenates. Ann. clin. lab. Sci., 15(4): 299-307.
SUNDERMAN, F.W. Jr, HOPFER, S.M., CRISOSTOMO, M.C. & STOEPPLER, M.
(1986a) Rapid analysis of nickel in urine by electrothermal atomic
absorption spectrophotometry. Ann. clin. lab. Sci., 16(3): 219-230.
SUNDERMAN, F.W. Jr, AITIO, A., MORGAN, L.G. & NORSETH, T. (1986b)
Biological monitoring of nickel. Toxicol. ind. Health, 2(1): 17-78.
SUNDERMAN, F.W. Jr, HOPFER, S.M., KNIGHT, J.A., McCEILLY, K.S.,
CECUTTI, A.G., THORNHILL, A.G., CONWAY, K., MILLER, C., PATIERNO,
S.R. & COSTA, M. (1987a) Physiochemical characteristics and
biological effects of nickel oxides. Carcinogenesis, 8: 305-313.
SUNDERMAN, F.W. Jr, AN, L.L., ZAHARIA, O., WONG, S.H.-Y. & HOPFER,
S.M. (1987b) Acute depletion of pulmonary lavage cells and enhanced
lipid peroxidation in alveolar macrophages following parenteral
administration of nickel chloride to rats. In: Rand, N.Y. & Raper,
C. ed. Pharmacology, Amsterdam, New York, Oxford, Elsevier Science
Publishers, pp. 817-820.
SUNDERMAN, F.W. Jr, HOPFER, S.M. & KNIGHT, J.A. (1988a) Biological
reactivity of nickel oxides and related compounds. In: Fourth
International Conference on Nickel Metabolism and Toxicology:
Abstracts, Espoo, Finland, 5-9 September 1988, Helsinki, Institute
of Occupational Health, p. 15.
SUNDERMAN, F.W. Jr, DINGLE, B., HOPFER, S.M. & SWIFT, T. (1988b)
Acute nickel toxicity in electroplating workers who accidentally
ingested a solution of nickel sulfate and nickel chloride. Ann. J.
ind. Med., 14: 257-266.
SUNDERMAN, F.W. Jr, HOPFER, S.M., SWEENEY, K.C., MARCUS, A.H.,
MOST, B.M. & CREASON, J. (1989a) Nickel absorption and kinetics in
human volunteers. Proc. Soc. exp. Biol. Med., 191: 5-11.
SUNDERMAN, F.W. Jr, HOPFER, S.M., SWIFT, T., REZUKE, W.N., ZIEBKA,
L., HIGHAM, P., EDWARDS, B., FOLCIK, M. & GOSSLING, H.R. (1989b)
Cobalt, chrominium and nickel concentrations in body fluids of
patients with porous-coated knee or hip prostheses. J. Orthop.
Res., 7(3): 307-315.
SUNDERMAN, F.W. Jr, HOPFER, F.W. Jr, LIN, S.-M., PLOWMAN, M.C.,
STOJANOVIC, T., WONG, S.H.-Y., ZAHARIA, O. & ZIEBKA, L. (1989c)
Toxicity to alveolar macrophages in rats following parenteral
injection of nickel chloride. Toxicol. appl. Pharmacol., 100:
107-118.
SUNG, J.F.C., NEVISSI, A.E. & DEWALLE, F.B. (1986) Concentration
and removal efficiency of major and trace elements in municipal
wastewater. J. environ. Sci. Health, A21(5): 435-448.
SUSHENKO, O.V. & RAFIKOVA, K.E. (1972) [Questions of work hygiene
in hydrometallurgy of copper, nickel and cobalt in a sulfide ore.]
Gig. Tr. prof. Zabol., 16: 42-45 (in Russian).
SUTHERLAND, R.B. (1959) Respiratory cancer mortality in workers
employed in an Ontario nickel refinery covering the period 1939-
1957, Ottawa, Ontario Department of Health, Division of Industrial
Hygiene (Unpublished report, November, 1959).
SWANN, M. (1984) Malignant soft-tissue tumour at the site of a
total hip replacement. J. bone joint Surg., B66: 629-631.
SWIERENGA, S.H. & BASRUR, P.K. (1968) Effect of nickel on cultured
rat embryo muscle cells. Lab. Invest., 19: 663-674.
SWIERENGA, S.H. & MCLEAN, J.R. (1985) Further insights into
mechanisms of nickel-induced DNA damage studies with cultured rat
liver cells. In: Brown, S.S. & Sunderman, F.W. Jr, ed. Progress
in Nickel toxicology. Proceedings of the 3rd International
Conference on Nickel Metabolism and Toxicology, Paris, 4-7
September, 1984, Oxford, Blackwell Scientific Publications, pp.
101-104.
SWIERENGA, S.H.H., GILMAN, J.P.W. & MCCLEAN, J.R. (1987) Cancer
risk from inorganics. Cancer Metastasis Rev., 6: 113-154.
SWIERENGA, S.H.H., MARCEAU, N., KATSUMA, Y., FRENCH, S.W., MULLER,
R. & LEE, F. (1989) Altered cytokeratin expression and
differentiation induction during neoplastic transformation of
cultured rat liver cells by nickel subsulfide. Cell Biol. Toxicol.
5(3): 271-286.
SZADKOWSKI, D., SCHULTZE, H., SCHALLER, K.H. & LEHNERT, G. (1969a)
[On the significance of heavy metal contents in cigarettes.] Arch.
Hyg., 153: 1-8 (in German).
SZADKOWSKI, D., KOHLER, G. & LEHNERT, G. (1969b) [Electrolyte-
changes in serum and electrical-mechanical heart action in
industrial heat stress.] Aertzl. Forsch. 23(8): 271-284 (in German).
SZATHMARY, S.C. & DALDRUP, T. (1982) [Determination of nickel by
GC, GC-MS and AAS in biological materials in a case of lettral
poisoning.] Fresenius Z. anal. Chem., 313: 48 (in German).
TABILLION, R., WEBER, F. & KALTWASSER, H. (1980) Nickel
requirement for chemolithotropic growth in hydrogen oxidizing
bacteria. Arch. Microbiol., 124: 131-136.
TAGAKI, Y., MATSUDA, S., IMAI, S., OHMORI, Y., MASUDA, T., VINSON,
J.A., MEHRA, M.C., PURI, B.K. & KANIEWSKI, A. (1986) Trace
elements in human air. Bull. environ. Contam. Toxicol., 36:
793-899.
TAKENAKA, S., HOCHRAINER, D. & OLDIGES, H. (1985) Alveolar
proteinosis induced in rats by long-term inhalation of nickel
oxide. In: Brown, S.S. & Sunderman, F.W. Jr, ed. Progress in
nickel toxicology. Proceedings of the 3rd International Conference
on Nickel Metabolism and Toxicology, Paris, 4-7 September, 1984,
Oxford, Blackwell Scientific Publications, pp. 89-92.
TANAKA, I. (1985) Ion-chromatographic determination of divalent
metal ions (Co, Ni, Cu, Zn, Cd) using EDTA as complexing agent.
Fresenius Z. anal. Chem., 320(2): 125-127.
TANAKA, I., ISHIMATSU, S., MATSUNO, K., KODAMA, Y. & TSYCHIYA, K.
(1985) Biological half time of deposited nickel oxide aerosol in
rat lung by inhalation. Biol. Trace Elem. Res., 8: 203-210.
TANAKA, I., ISHIMATSU, S., HARATAKE, J., HORIE, A. & KODAMA, Y.
(1988) Biological half-time in rats exposed to nickel monosulfide
(amorphous) aerosol by inhalation. Biol. Trace Elem. Res., 17:
237-246.
TANDON, S.K. (1982) Disposition of nickel-63 in the rat. Toxicol.
Lett., 10(1): 71-73.
TANDON, S.K., MATHUR, A.K. & GAUR, J.S. (1977) Urinary excretion
of chromium and nickel among electroplaters and pigment industry
workers. Int. Arch. occup. environ. Health, 40(1): 71-76.
TASK GROUP ON LUNG DYNAMICS (1966) Deposition and retention models
for internal dosimetry of the human respiratory tract. Health
Phys., 12: 173- 207.
TATARSKAYA, A.A. (1960) [Occupational disease of upper respiratory
tract in persons employed in electrolytic nickel refining
departments.] Gig. Tr. prof. Zabol., 6: 35-38 (in Russian).
TATARSKAYA, A.A. (1965) [On the problem of occupational cancer of
the upper respiratory tract in the nickel-refining industry.] Gig.
Tr. prof. Zabol., 9(2): 22-25 (in Russian).
TAYLOR, D., MADDOCK, B.G. & MANCE, G. (1985a) The acute toxicity
of nine gray list metals (arsenic, boron, chromium, copper, lead,
nickel, tin, vanadium, and zinc) to two marine fish species: dab
( Limanda limanda) and grey mullet (Chelon labrosus). Aquat.
Toxicol., 7(3): 135-144.
TAYLOR, P., DAMS, R. & HOSTE, R. (1985b) The determination of
cadmium, chromium, copper, nickel and zinc in city waste
incinerator ash using inductively coupled plasma-atomic emission
spectrometry. J. anal. Let., 18(A19): 2361-2368.
TAYTON, K.J.J. (1980) Ewing's sarcoma at the site of a metal plate.
Cancer, 45: 413-415.
TEDESCHI, R.E. & SUNDERMAN, F.W. (1957) Nickel poisoning. V. The
metabolism of nickel under normal conditions and after exposure to
nickel carbonyl. Arch. ind. Health, 16: 486-488.
TERAKI, Y. & UCHIUMI, A. (1988) Experimental studies on metal
carcinogenesis: a comparative assessment of nickel and lead. In:
Fourth International Conference on Nickel Metabolism and
Toxicology, Abstracts, Espoo, Finland, 5-9 September, 1988,
Helsinki, Institute of Occupational Health, p. 5.
THURSTON, G.D. & SPENGLER, J.D. (1985) A quantitative assessment
of source contributions to inhalable particulate matter pollution
in metropolitan Boston. Atmos. Environ., 19(1): 9-25.
TIMOURIAN, H. & WATCHMAKER, G. (1972) Nickel uptake by sea urchin
embryos and their subsequent development. J. exp. Zool., 182:
379-388.
TOLA, S., KILPIOE, J. & VIRTAMO, M. (1979) Urinary and plasma
concentrations of nickel as indicators of exposure to nickel in an
electroplating shop. J. occup. Med., 21(3): 184-188.
TOLOT, F., BROUDEUR, P. & NEULAT, G. (1956) Troubles pulmonaires
asthmatiformes chez des ouvriers exposés à l'inhalation de chrome,
nickel et aniline. Arch. Mal. prof. Méd. Trav. Séc. soc., 18:
291-293.
TORJUSSEN, W. & ANDERSEN, I. (1979) Nickel concentrations in nasal
mucosa, plasma, and urine in active and retired nickel workers.
Ann. clin. lab. Sci., 9(4): 289-298.
TORJUSSEN, W., SOLBERG, L.A. & HOGETVEIT, A.C. (1979a)
Histopathologic changes of nasal mucosa in nickel workers: a pilot
study. Cancer, 44(3): 963- 974.
TORJUSSEN, W., SOLBERG, L.A. & HOGETVEIT, A.C. (1979b)
Histopathological changes of the nasal mucosa in active and retired
nickel workers. Br. J. Cancer, 40(4): 568-580.
TOSSAVAINEN, A., NURMINEN, M., MUTANEN, P. & TOLA, S. (1980)
Application of mathematical modelling for assessing the biological
half-times of chromium and nickel in field studies. Br. J. ind.
Med., 37(3): 285-291.
TRAUL, K.A., HINK, R.J., WOLFF, J.S. & WLODYMR, K. (1981) Chemical
carcinogenesis in vitro: an improved method for chemical
transformation in Rauscher leukemia virus-infected rat embryo
cells. J. appl. Toxicol., 1(1): 32-37.
TREAGAN, L. & FURST, A. (1970) Inhibition of interferon synthesis
in mammalian cell cultures after nickel treatment. Res. Commun.
chem. Pathol. Pharmacol., 1: 395-401.
TURHAN, U., HAIN, C., WOLLBURG, C. & SZADKOWSKY, D. (1983)
[Chromium and nickel content in human lung. Medical aspects.] In:
[Proceedings of the 1983 Annual Congress of the German Association
for Occupational Medecine, Göttingen], Stuttgart, Gentner, pp. 477-
480 (in German).
TURK, J.L. & PARKER, D. (1977) Sensitization with Cr, Ni, and Zn
salts and allergic type granuloma formation in the guinea-pig. J.
invest. Dermatol., 68: 341-345.
TWEATS, D.J., GREEN, M.H.L. & MURIEL, W.J.A. (1981) A differential
killing assay for mutagens and carcinogens based on an improved
repair-deficient strain of E. coli. Carcinogenesis, 2: 189-194.
UMEDA, M. & NISHIMURA, M. (1979) Inducibility of chromosomal
aberrations by metal compounds in cultured mammalian cells. Mutat.
Res., 67: 221-229.
UPITIS, V.V., NOLLENDORF, A.F. & PAKALNE, D.S. (1980) [Relevance
of nickel for culturing microalgae.] In: Anke, M., Schneider, H.-
J. & Brückner, Chr. ed. [3. Trace Element Symposium Nickel,
Jena, German Democratic Republic, 7-11 July, 1980,] Jena, Friedrich-
Schiller University, pp. 171-183 (in German).
US BUREAU OF MINES (1985) Nickel. In: Minerals yearbook 1984,
Washington, DC, Department of the Interior, Vol. I, pp. 665-684.
US BUREAU OF MINES (1986) Nickel. In: Mineral commodity summaries
1986. An up-to-date summary of 87 nonfuel mineral commodities,
Wahington, DC, Department of the Interior, pp. 108-109.
US EPA (1986) Health assessment document for nickel and nickel
compounds, Washington DC, US Environmental Protection Agency, 438
pp (EPA/600/8-83/012 FF).
US EPA (1987) Acute toxicity handbook of chemicals to estuarine
organisms, Gulf Breeze, Florida, US Environmental Protection
Agency, 274 pp (EPA/600/8-87/017).
US NIOSH (1977a) Special occupational hazard review and control
recommendations for nickel carbonyl. Cincinatti, Ohio, US National
Institute for Occupational Safety and Health, 45 pp (DHEW (NIOSH)
Publication No. 77-184).
US NIOSH (1977b) Criteria for a recommended standard occupational
exposure to inorganic nickel. Cincinatti, Ohio, US National
Institute for Occupational Safety and Health, 282 pp (prepared by
Stanford Research Institute, Menlo Park) (DHEW (NIOSH) Publication
No. 77-164).
VAN SOESTBERGEN, M. & SUNDERMAN, F.W. (1972) 63Ni complexes in
rabbit serum and urine after injection of 63NiCl2. Clin. Chem.
18(12): 1478-1484.
VALENTA, P. OSTAPCZUK, P.H. PIKLAR, B. & NURNBERG, H.W. (1981)
New applications of voltammetry in the determination of toxic trace
metals in food. Proceedings of the 3rd CEC/WHO International
Conference on Heavy Metals in the Environment, Amsterdam, 1981.
Edinburgh, CEP Consultants Ltd, pp. 619-621.
VALENTINE, R. & FISHER, G.L. (1984) Pulmonary clearance of
intratracheally administered 63Ni3S2 in strain A/J mice. Environ.
Res., 34: 328-334.
VAN BAALEN, C. & O'DONNELL, R. (1978) Isolation of a nickel-
dependent blue-green algae. J. gen. Microbiol., 105: 351-353.
VANDENBERG, J. & EPSTEIN,W. (1963) Experimental skin contact
sensitisation in man. J. invest. Dermatol., 41: 413-416.
VAN ROSEN, G. (1954) Breaking of chromosomes by the action of
elements of the periodical system and by some other principles.
Hereditas, 40: 258-263.
VANDENBERG,J. & EPSTEIN, W. (1963) Experimental skin contact
sensitisation in man. J. invest. Dermatol., 41: 413-416.
VERMA, H.S., BEHARI, J.R. & TANDON, S.K. (1980) Pattern of urinary
63Ni excretion in rats. Toxicol. Lett., 5(3-4): 223-226.
VINSON, L.J. & CHOMAN, B.R. (1960) Percutaneous absorption and
surface active agents. J. Soc. Cosmet. Chem., 11: 127-137.
VOGEL, E. (1984) The relation between mutational pattern and
concentration by chemical mutagens in Drosophila. In: Screening
tests in chemical carcinogenesis. Proceedings of a workshop
organized by IARC, and the Commission of the European Communities,
Brussels, 9-12 June, 1975, Lyon, International Agency for Research
on Cancer, pp. 117-137 (IARC Scientific Publications No. 12).
VOLLKOPF, U., GROBENSKI, Z. & WELZ, B. (1981) Determination of
nickel in serum using graphite furnace atomic absorption. At.
Spectrosci., 2: 68-70.
VOLINI, F. DE LA, HUERGA, J. & KENT, G. (1968) Trace metal studies
in liver disease using atomic absorption spectrometry. In:
Sunderman, F.W. & Sunderman, F.W. Jr, ed. Laboratory diagnosis of
liver diseases, St. Louis, Missouri, Warren H. Green, pp. 199-206.
VOROSHILIN, B.I., PLOTKO, E.G., NIKIPHOROVA, V.YA. & FINK, T.V.
(1977) Cytogenetic effects of some inorganic chemicals on human
leukocytes in cultures. In: Problems of genetics and selection in
Urals (information data). Sverdlorsk. The Urals Scientific Center
of the USSR Academy of Sciences, pp. 46-47.
VOS, L., KOMY, Z., REGGERS, G., ROEKENS, E. & VAN GRIEKEN, R.
(1986) Determination of trace metals in rain water by differential-
pulse stripping voltammetry. Anal. chim. Acta, 184: 271-280.
VUOPALA, U., HUHTI, E., TAKKUNEN, J. & HUIKKO, M. (1970) Nickel
carbonyl poisoning. Report of 25 cases. Ann. clin. Res., 2: 214-222.
WACHS, B. (1982) Concentration of heavy metals in fishes from the
river Danube. Z. Wasser Abwasser Forsch., 15(2): 43-84.
WAHLBERG, J.E. (1975) Nickel allergy and atopy in hairdressers.
Contact Dermatit., 1(3): 161-165.
WAHLBERG, J.E. (1976) Sensitization and testing of guinea-pigs with
nickel sulfate. Dermatology, 152: 321-330.
WAHLBERG, J.E., LINDSTEDT, G. & EINARSSON, O. (1977) Chromium,
cobalt and nickel in Swedish cement, detergents, mould and cutting
oils. Berufsdermatosen, 25(6): 220-228.
WAKSVIK, H. & BOYSEN, M. (1982) Cytogenetic analyses of
lymphocytes from workers in a nickel refinery. Mutat. Res., 103:
185-190.
WAKSVIK, H., BOYSEN, M. & HOGETVEIT, A. Chr. (1984) Increased
incidence of chromosomal aberrations in peripheral lymphocytes of
retired nickel workers. Carcinogenesis, 5(11): 1525-1527.
WALL, L.M. & CALNAN, C.D. (1980) Occupational nickel dermatitis in
the electroforming industry. Contact Dermatit., 6(6): 414-420.
WALLACE, A., ROMNEY, E.M., KINNEAR, J. & ALEXANDER, G.U. (1980a)
Single and multiple trace metal excess effects on three different
plant species. J. Plant Nutr., 2: 11-23.
WALLACE, A., ROMNEY, E.M., MULLER, R.T. & ALEXANDER, G.U. (1980b)
Calcium-trace metal interactions in soybean plants. J. Plant Nutr.
2: 79-86.
WALTHARD, B. (1926) [Experimental nickel idiosyncrasy in laboratory
animals.] Schweiz. Med. Wochenschr., 7: 603-604 (in German).
WALTSCHEWA, W., SLATEWA, M. & MICHAILOW, I. (1972) [Testicular
changes due to long-term administration of nickel sulphate in
rats.] Exp. Pathol., 6(3): 116-120 (in German).
WAN, C.C., CHIANG, S. & CORSINI, A. (1985) Two-column method for
preconcentration of trace metals in natural waters on acrylate
resin. Anal. Chem., 57: 719-723.
WARNER, J.S. (1984) Occupational exposure to airborne nickel in
producing and using primary nickel products. In: Nickel in the
human environment, Proceedings of a Joint Symposium, Lyon, 8-11
March, 1983, Lyon, International Agency for Research on Cancer, pp.
419-437 (IARC Scientific Publications No. 53).
WARNER, J.S. (1985) Estimating past exposures to airborne nickel
compounds in the Copper Cliff Sinter Plant. In: Brown, S.S. &
Sunderman, F.W. Jr, ed. Progress in nickel toxicology. Proceedings
of the 3rd International Conference on Nickel Metabolism and
Toxicology, Paris, 4-7 September, 1984, Oxford, Blackwell
Scientific Publications, pp. 203-206.
WASE, A.W., GOSS, D.M. & BOYD, M.J. (1954) The metabolism of
nickel. I. Spatial and temporal distribution of Ni63 in the mouse.
Arch. Biochem. Biophys., 51: 1-4.
WATANABE, H., GOTO, K., TAGUCHI, S., MCLAREN, J.W., BERMAN, S.S. &
RUSSELL, O.S. (1981) Preconcentration of trace elements in sea-
water by complexation with 8-hydroxyquinoline and adsorption on
C18-bonded silica gel. Anal. Chem., 53: 738-739.
WATERMAN, A.H. & SCHRIK, J.J. (1985) Allergy in hip arthroplasty.
Contact Dermatit., 13(5): 294-301.
WATERS, M.D., GARDNER, D.E. & COFFIN, D.L. (1975) Toxicity of
metallic chlorides and oxides for rabbit alveolar macrophages in
vitro. Environ. health Perspect., 10: 267-268.
WATRAS, C.J., MACFARLANE, J. & MOREL, F.M.M. (1985) Nickel
accumulation by Scenedesmus and Daphnia: Food-chain transport and
geochemical implications. Can. J. Fish. aquat. Sci., 42(4): 724-730.
WEAST, R.C. (1981) CRC Handbook of chemistry and physics, 62nd ed.
Boca Raton, Florida, CRC Press, pp. B123-B124.
WEBER, P.C. (1986) Epithelioid sarcoma in association with total
knee replacement. J. bone joint Surg., B68: 824-826.
WEBSTER, J.D., PARKER, T.F., ALFREY, A.C., SMYTHE, W.R., KUBO, H.,
NEAL, G. & HULL, A.R. (1980) Acute nickel intoxication by
dialysis. Ann. intern. Med., 92: 631-633.
WEHNER, A.P. & CRAIG, D.K. (1972) Toxicology of inhaled NiO and
CoO in Syrian golden hamsters. Am. Ind. Hyg. Assoc. J., 33: 146-155.
WEHNER, A.P., BUSCH, R.H., OLSON, R.J. & CRAIG, D.K. (1975)
Chronic inhalation of nickel oxide and cigarette smoke by hamsters.
Am. Ind. Hyg. Assoc. J., 36(11): 801-810.
WEHNER, A.P., MOSS, O.R., MILLIMAN, E.M., DAGLE, G.E. & SCHIRMER,
R.E. (1979) Acute and subchronic inhalation exposures of hamsters
to nickel-enriched fly ash. Environ. Res., 19(2): 355-370.
WEHNER, A.P., DAGLE, G.E. & MILLIMAN, E.M. (1981) Chronic
inhalation exposure of hamsters to nickel-enriched fly ash.
Environ. Res., 26(1): 195- 216.
WEIDENAUER, M. & LIESER, K.H. (1985) [Determination of trace
elements in river water by means of voltammetry.] Fresenius Z.
anal. Chem., 320: 550-555 (in German).
WEINZIERL, J. & UMLAND, F. (1982) [Determination of Co (and Ni) in
the ppb-range by differential pulse polarography.] Fresenius Z.
anal. Chem., 312: 608-610 (in German).
WELLENREITER, R.H., ULLREY, D.E. & MILLER, E.R. (1970) Nutritional
studies with nickel. In: Mills, C.F. ed. Trace element metabolism
in animals. Proceedings of 1st WAAP/IBP International Symposium,
Aberdeen, Scotland, July 1969, Edinburgh, London, Livingstone, pp.
52-58.
WELLS, G.C. (1956) Effects of nickel on the skin. Br. J. Dermatol.
68: 237-242.
WEST, B. & SUNDERMAN, F.W. (1958) Nickel poisoning. VII. The
therapeutic effectiveness of alkyl dithiocarbamates in experimental
animals exposed to nickel carbonyl. Am. J. med. Sci., 236: 15-25.
WHANGER, P.D. (1973) Effects of dietary nickel on enzyme activities
and mineral contents in rats. Toxicol. appl. Pharmacol., 25: 323-331.
WHITE, D.H., KING, K.A., MITCHELL, L.A. & MULHERN, B.M. (1986)
Trace elements in sediments, water, and american coots ( Fulica
americana) at a coal-fired power plant in Texas. Bull environ.
Contam. Toxicol., 36(3): 376-383.
WHO (1984) Guidelines for drinking-water quality. Vol. 1 -
Recommendations, Geneva, World Health Organization, p. 57.
WHO (1988) Air quality guidelines for Europe, Copenhagen, World
Health Organization, pp. 285-296 (WHO Regional Publications,
European Series No 23).
WIERNIK, A., JOHANSSON, A., JARSTRAND, C. & CAMNER, P. (1983)
Rabbit lung after inhalation of soluble nickel. I. Effects on
alveolar macrophages. Environ. Res., 30: 129-141.
WILLS, M.R., BROWN, C.S., BERTHOLF, R.L. & SAVORY, J. (1985) Serum
and lymphocyte aluminium and nickel in chronic renal failure. Clin.
chim. Acta, 145: 193-196.
WILSON, J.D., STENZEL, M.R., LOMBARDOZZI, K.L. & NICHOLS, C.L.C.
(1981) Monitoring personal exposure to stainless steel welding
fumes in confined spaces at a petrochemical plant. Am. Ind. Hyg.
Assoc. J., 42: 431-436.
WILSON, L.W. & DINUNZIO, J.E. (1981) Enrichment of nickel and
cobalt in natural hard water by Donnan Dialysis. Anal. Chem., 53:
692-695.
WILSON, W.W. & KHOOBYARIAN, N. (1982) Potential identification of
chemical carcinogens in a viral transformation system. Chem. biol.
Interact., 38: 253- 259.
WINDHOLZ, M., BUDAVARI, S., BLUMETTI, R.F. & OTTERBEIN, E.S.
(1983) The Merck Index, 10th ed. Rahway, New Jersey, Merck & Co.
Inc.
WOLNIK, K.A., FRICKE, F.L., CAPAR, S.C., BRAUDE, G.L., MEYER, M.W.,
SATZGER, R.D. & KUENNEN, R.W. (1983) Elements in major raw
agricultural crops in the United States. 2. Other elements in
lettuce, peanuts, potatoes, soybeans, sweet corn and wheat. J.
Agric. food Chem., 31: 1244-1249.
WOLNIK, K.A., FRICKE, F.L., CAPAR, S.G., MEYER, M.W., SATZGER,
R.D., BONNIN, E. & GASTON, C.M. (1985) Elements in major raw
agricultural crops in the United States. 3. Cadmium, lead, and
eleven other elements in carrots, field corn, onions, rice, spinach
and tomatoes. J. Agric. food. Chem., 33: 807-811.
WULF, H.C. (1980) Sister chromatid exchanges in human lymphocytes
exposed to nickel and lead. Danish med. Bull., 27: 40-42.
YAMASHIRO, S., GILMAN, J.P.W., HULLAND, T.J. & ABANDOWITZ, H.M.
(1980) Nickel sulphide-induced rhabdomyosarcomata in rats. Acta
pathol. Jpn., 30(1): 9-22.
YAMASHIRO, S., BASRUR, P.K., GILMAN, J.P., HULLAND, T.J. &
FUJIMOTO, Y. (1983) Ultrastructural study of Ni3S2-induced tumors
in rats. Acta pathol. Jpn., 33(1): 45-58.
YAN, N.D. & STRUS, R. (1980) Crustacean zooplankton communities of
acidic, metal-contaminated lakes near Sudbury, Ontario. Can. J.
Fish. aquat. Sci., 37(12): 2282-2294.
YAN, N.D., MILLER, G.E., WILE, I. & HITCHIN, G.G. (1985) Richness
of aquatic macrophyte floras of soft water lakes of differing pH
and trace metal content in Ontario, Canada. Aquat. Bot., 23(1): 27-40.
YANG, X.H., BROOKS, R.R., JAFFRE, T. & LEE, J. (1985) Elemental
levels and relationships in the flacourtiaceae of New Caledonia and
their significance for the evaluation of the serpentine problem.
Plant Soil, 87(2): 281-292.
YARITA, T. & NETTESHEIM, P. (1978) Carcinogenicity of nickel
subsulfide for respiratory tract mucosa. Cancer Res., 38: 3140-3145.
YOSHIMURA, K., THOSHIMITSU, Y. & OHASKI, S. (1980) Ion-exchanger
colorimetry - VI micro-determination of nickel in natural water.
Talanta, 27: 693-697.
ZAKOUR, R.A., TKESHELASHVILI, L.K., SHERMAN, C.W., KOPLITZ, L.M. &
LOEB, L.A. (1981) Metal-induced infidelity of DNA synthesis. J.
cancer Res. clin. Oncol., 99: 187-196.
ZISLIN, D.M., GANYUSKINA, S.M., DUBINIA, E.S. & TYUSHNYAKOVA, N.V.
(1969) [Residual air in a complex evaluation of the respiratory
system function in initial and suspected pneumonconiosis.] Gig.
Tr. prof. Zabol., 13: 26-29 (in Russian).
ZOBER, A. (1981a) [Symptoms and findings at the bronchopulmonary
system of electric arc welders. II. communication: pulmonary
fibrosis.] Zentralbl. Bakteriol. Hyg. I. Abt. Orig. B, 173: 120-148
(in German).
ZOBER, A. (1981b) [Symptoms and findings at the bronchopulmonary
system of electric arc welders. I. communication: epidemiology.]
Zentralbl. Bakt. Hyg. I. Abt. Orig. B, 173: 92-119 (in German).
ZOBER, A. (1982) [Possible dangers to the respiratory tract from
welding fumes.] Schweissen Schneiden, 34: 77-81 (in German).
ZOBER, A., KICK, K., SCHALLER, K.H., SCHELLMANN, B. & VALENTIN, H.
(1984) [Nickel and chromium content of selected human organs and
body fluids.] Zentralbl. Bakteriol. Hyg. I. Abt. Orig. B , 179: 80-95
(in German).
RESUME ET CONCLUSIONS
1. Identité, propriétés physiques et chimiques et méthodes
d'analyse
Le nickel est un élément métallique qui appartient au groupe
VIIIb de la classification périodique. Il résiste aux alcalis mais
se dissout en général dans les acides oxydants dilués. Le
carbonate, le sulfure et l'oxyde sont insolubles dans l'eau, alors
que le chlorure, le sulfate et le nitrate sont solubles. Le nickel
carbonyle est un liquide volatil incolore qui se décompose au-
dessus de 50 °C. Sous forme ionique, le nickel existe
essentiellement à l'état de nickel(II). Dans les systèmes
biologiques, le nickel une fois dissous peut former des complexes
avec divers ligands et se fixer aux matières organiques.
L'analyse des échantillons biologiques et environnementaux se
fait le plus fréquemment par spectroscopie d'absorption atomique et
voltammétrie. Pour obtenir des résultats fiables, en particulier
dans le domaine des ultratraces (ultramicrotraces), il faut suivre
un mode opératoire précis pour réduire au minimum le risque de
contamination lors des prélèvements, de la conservation, du
traitement, et de l'analyse des échantillons. Selon les
traitements préalables subis par l'échantillon et les méthodes
d'extraction ou d'enrichissement utilisées, on peut atteindre des
limites de détection de 1 à 100 ng/litre dans les produits
biologiques et l'eau.
2. Sources d'exposition humaine et environnementale
Le nickel est présent à l'état de traces un peu partout dans le
sol, l'eau, l'air et la biosphère. La teneur moyenne de la croûte
terrestre en nickel est d'environ 0,008%. Les terrains agricoles
en contiennent entre 3 et 1000 mg/kg. Dans les eaux naturelles,
les concentrations varient de 2 à 10 µg/litre (eau douce) et de 0,2
à 0,7 µg/litre (eau de mer). Dans l'atmosphère, les concentrations
en nickel varient de 0,1 à 3 ng/m3, au-dessus des zones écartées.
Les gisements de nickel consistent en accumulations de minéraux
sulfurés (essentiellement de la pentlandite) et en latérites. Le
nickel est extrait de ces minerais par affinage pyro- et hydro-
métallurgique. Le nickel est principalement utilisé pour la
production d'aciers inoxydables qui sont très résistants à la
corrosion et aux températures élevées. On l'utilise sous forme
d'alliages et de dépôts galvaniques pour la confection de pièces
d'automobiles, de machines-outils, d'armements, d'outils, de
matériel électrique, d'appareils ménagers, et de pièces de monnaie.
Les dérivés du nickel sont également utilisés comme catalyseurs,
comme pigments, et dans les batteries d'accumulateurs. En 1985, la
production minière mondiale de nickel a été d'environ 67 millions
de kg. Les principales émissions de nickel dans l'air ambiant
proviennent de la combustion du charbon et du mazout pour le
chauffage et la production d'énergie,de l'incinération des déchets
et des boues de stations d'épuration, de la production minière et
de la métallurgie du nickel, de la fabrication de l'acier, du
nickelage, et de diverses autres sources, comme les cimenteries.
Dans les polluants atmosphériques, le nickel est présent
principalement sous forme de sulfates, d'oxydes, de sulfures, et,
dans une moindre mesure, de nickel métallique.
Le nickel rejeté lors de diverses opérations industrielles ou
autres finit par aboutir dans les eaux usées. Après traitement de
ces dernières, les résidus sont éliminés par injection dans des
puits profonds, décharge en mer, enfouissement dans le sol et
incinération. Les effluents rejetés par les usines de traitement
des eaux usées contiendraient jusqu'à 0,2 mg de nickel par litre.
3. Transport, distribution, et transformation dans l'environnement
Le nickel qui pénètre dans l'environnement peut être d'origine
naturelle ou artificielle et il circule à travers tous les
compartiments du milieu, soit sous l'effet de processus
physicochimiques, soit par transport biologique au sein
d'organismes vivants.
On estime que le nickel atmosphérique est essentiellement
présent sous forme d'aérosols particulaires dont la teneur en
nickel varie en fonction de leur origine. C'est en général les
particules les plus petites qui présentent les teneurs les plus
élevées en nickel. Le nickel carbonyle est instable à l'air et il
se décompose pour former de l'oxyde de nickel.
Le transport et la distribution des particules de nickel en
direction ou à l'intérieur des divers compartiments du milieu
dépend beaucoup de la granulométrie des particules et des
conditions météorologiques. Cette granulométrie dépend à son tour
principalement de la source émettrice. En général, les particules
d'origine artificielle sont de dimension plus réduite que celles
qui proviennent de la poussière naturelle.
Le nickel peut pénétrer dans l'hydrosphère soit à partir de
l'atmosphère, soit par lessivage superficiel, soit encore par
décharge de déchets industriels ou municipaux ou érosion naturelle
des sols et des roches. Dans les cours d'eau, le nickel est
principalement transporté par des particules enrobées de matières
précipitées ou en association avec des matières organiques; dans
les lacs, le transport est assuré sous forme ionique,
essentiellement en association avec des matières organiques. Le
nickel peut également s'adsorber sur des particules d'argile ou
être fixé par des organismes vivants. A l'inverse, il peut se
désorber et s'échapper ainsi des sédiments où il était fixé. Une
partie du nickel ainsi transporté par les cours d'eau aboutit à
l'océan. On estime que l'apport sous forme de particules en
suspension dans les cours d'eau est d'environ 135 x 107 kg/an.
Selon le type de sol, le nickel peut présenter une forte
mobilité et parvenir jusqu'aux eaux souterraines, puis aux lacs et
aux rivières. C'est principalement par leurs racines que les
plantes terrestres fixent le nickel présent dans le sol. La
quantité ainsi fixée dépend de divers paramètres géochimiques et
physiques, notamment le type de sol, son pH, sa teneur en eau et en
matières organiques, ainsi que la concentration en nickel extractible.
L'exemple le mieux connu d'accumulation de nickel est fourni par un
certain nombre d'espèces végétales "hyperaccumulatrices", qui
concentrent le nickel à raison de plus de 1mg/kg de poids sec, et
qui poussent sur des sols serpentinifères relativement stériles. A
partir de 50 mg/kg de poids sec, le nickel est toxique pour la
plupart des végétaux. On a observé une accumulation de nickel et
des effets toxiques chez les végétaux cultivés sur sols traités par
des boues de stations d'épuration ainsi que dans la végétation
située à proximité de sources d'émission. Certaines plantes
aquatiques concentrent fortement le nickel. Des études de
laboratoire portant sur des poissons ont montré que le nickel
n'avait guère tendance à s'accumuler, quelques que soient les
espèces étudiées. Dans des eaux non polluées, les concentrations
relevées dans le poisson entier (et rapportées au poids de tissus
frais), variaient de 0,02 à 2 mg/kg. Ces valeurs pourraient être
jusqu'à 10 fois supérieures dans les poissons qui vivent en eau
polluée. Toutefois rien n'indique que le nickel subisse une
bioamplification tout au long de la chaîne alimentaire.
Les animaux sauvages, par exemple, les herbivores et leurs
prédateurs carnivores, absorbent du nickel par voie alimentaire,
qui se retrouve ensuite dans de nombreux organes et tissus.
4. Concentrations dans l'environnement et exposition humaine
Dans les organismes terrestres et aquatiques, les
concentrations de nickel peuvent varier de plusieurs ordres de
grandeur. L'homme est exposé à des concentrations atmosphériques
dont les valeurs caractéristiques vont de 5 à 35 ng/m3 en zones
rurales et urbaines, d'où une absorption par inhalation de 0,1 à
0,7 µg de nickel par jour. L'eau de boisson en contient
généralement moins de 10 µg/litre mais il arrive que du nickel soit
libéré dans l'eau d'adduction par les éléments de la plomberie et
atteigne des concentrations de 500 µg/litre.
Dans les produits alimentaires, les concentrations sont
généralement inférieures à 0,5 mg/kg en poids de substance fraîche.
Le cacao, les graines de soja, certains légumes secs, diverses
noix, et la farine d'avoine en contiennent de fortes
concentrations. L'ingestion journalière de nickel par la voie
alimentaire est très variable du fait de la diversité des habitudes
alimentaires: les quantités ingérées peuvent varier de 100 à 800
µg/jour; en moyenne l'apport alimentaire dans la plupart des pays
est de 100 à 300 µg/jour. La libération de nickel par les
ustensiles de cuisine peut augmenter sensiblement les quantités
ingérées. Chez les fumeurs, les quantités absorbées par voie
pulmonaire peuvent aller de 2 à 23 µg/jour pour une consommation
quotidienne de 40 cigarettes.
L'exposition cutanée dans l'environnement général joue un rôle
important dans l'apparition et l'entretien d'une hypersensibilité
de contact qui peut être due à un contact quotidien avec des objets
nickelés ou en alliage de nickel (par exemple des bijoux, des
pièces de monnaie, des agrafes).
Les implants et prothèses en alliage de nickel, les liquides
pour injection intraveineuse ou dialyse, et les produits de
contraste utilisés en radiographie peuvent entraîner une exposition
iatrogène au nickel. On estime qu'un malade sous dialyse reçoit à
chaque traitement environ 100 µg de nickel en moyenne par voie
intra-veineuse.
Dans les ambiances de travail, les concentrations de nickel en
suspension dans l'air peuvent varier de quelques µg jusqu'à,
parfois, plusieurs mg/m3, selon le processus industriel et la
teneur en nickel des produits manipulés.
Partout dans le monde, des millions de travailleurs sont
exposés à des poussières et des fumées contenant du nickel lors de
travaux tels que le soudage, le nickelage, le broyage, l'extraction
minière, l' affinage du nickel ainsi que dans les aciéries,
fonderies, et autres industries métallurgiques.Une exposition
cutanée au nickel peut intervenir à l'occasion des activités
professionnelles les plus diverses, qu'il s'agisse d'une exposition
directe à du nickel dissous, par exemple, lors de l'affinage, du
nickelage ou de l'électroformage ou lors de la manipulation
d'outils contenant du nickel. Le nettoyage par voie humide peut
comporter une exposition au nickel qui s'est dissous dans les eaux
de lavage.
5. Cinétique et métabolisme chez l'homme et l'animal
Chez l'homme et l'animal le nickel peut être absorbé par
inhalation, ingestion, ou par voie percutanée. En cas d'exposition
professionnelle, la principale voie de pénétration dans l'organisme
est l'absorption respiratoire avec résorption gastro-intestinale
secondaire (formes solubles et insolubles). Une proportion
importante des matières inhalées est avalée après avoir été
éliminée des voies respiratoires sous l'action de l'ascenseur
mucociliaire. Une hygiène personnelle insuffisante et de mauvaises
habitudes de travail peuvent accroître l'exposition par voie
digestive. L'absorption percutanée est négligeable
quantitativement mais joue un rôle important dans la pathogénèse de
l'hypersensibilité de contact. La résorption dépend de la
solubilité du composé et elle décroît en général dans l'ordre
suivant: nickel carbonyle > composés solubles > composés
insolubles. De tous les dérivés du nickel, c'est le nickel
carbonyle qui est le plus rapidement et le plus complètement
résorbé dans l'organisme de l'homme et des animaux. Les études
comportant sur l'administration de nickel par voie respiratoire
sont limitées. Des études au cours desquelles on a administré à
des hamsters et à des rats de l'oxyde de nickel insoluble ont
montré que celui-ci était faiblement résorbé, la plus grande partie
du produit étant retenue dans les poumons après plusieurs semaines.
En revanche, la résorption du chlorure de nickel soluble et du
sulfure de nickel amorphe ont été rapides. Dans le sang, le nickel
est principalement transporté par l'albumine.
Le nickel est plus ou moins résorbé au niveau gastrointestinal
en fonction de la composition de l'alimentation. Lors d'une étude
récente sur des volontaires humains, on a constaté que le nickel
était résorbé à hauteur de 27% à partir de l'eau, contre moins de
1% à partir de la nourriture. L'excrétion peut s'effectuer en
principe dans toutes les sécrétions et notamment l'urine, la bile,
la sueur, les larmes, le lait et le liquide muco-ciliaire. Le
nickel qui n'est pas résorbé est éliminé dans les matières fécales.
On a mis en évidence l'existence d'un transfert transplacentaire
chez des rongeurs. Après administration parentérale de sels de
nickel, on a constaté que c'est au niveau des reins, des glandes
endocrines, des poumons et du foie que l'accumulation de nickel
était la plus importante: des concentrations élevées ont été
également observées dans l'encéphale après administration de nickel
carbonyle. Les données relatives à l'excrétion correspondent à un
modèle bicompartimental. Chez des adultes en bonne santé exposés
en dehors de leur activité professionnelle, on a observé des
concentrations sériques et urinaires de 0,2 µg/litre (limites:
0,05-1,1 µg/litre) et de 1,5 µg/g de créatinine (limites: 0,5-4,0
µg/g de créatinine), respectivement. Après exposition
professionnelle, on a constaté une augmentation des concentrations
de nickel dans ces deux liquides. Pour un adulte non exposé de 70
kg, la charge de l'organisme en nickel est de 0,5 µg.
6. Effets sur les êtres vivants dans leur milieu naturel
Chez les microorganismes, on a constaté une inhibition de la
croissance lorsque la concentration en nickel dans le milieu était
de 1 à 5 mg/litre (cas des actinomycètes, des levures et des
eubactéries marines ou non marines) ou de 5 à 1000 mg/litre (cas des
champignons filamenteux). Dans le cas des algues, on n'a pas
observé de croissance à des concentrations d'environ 0,05-5 mg de
nickel par litre. La toxicité du nickel dépend également de
facteurs abiotiques tels que le pH, la dureté, la température et la
salinité du milieu ainsi que de la présence de particules
organiques ou minérales.
Chez les invertébrés aquatiques, la toxicité du nickel varie
dans de très importantes proportions selon l'espèce et les facteurs
abiotiques. Chez la daphnie, on a observé une CL50 à 96 h. de 0,5
mg/litre alors que chez trois mollusques, deux gastéropodes d'eau
douce et un bivalve, ce paramètre était respectivement égal à 0,2
mg/litre et 1100 mg/litre.
Chez les poissons, les valeurs de la CL50 à 96 h. sont
généralement comprises entre 4 et 20 mg/litre mais elles peuvent
être plus élevées chez certaines espèces. Des études à long terme
sur les poissons et leur développement effectuées dans des eaux
douces ont montré qu'à des concentrations ne dépassant pas
0,05 mg/litre, le nickel exerçait certains effets, en particulier
sur la truite arc-en-ciel. Pour les plantes terrestres, des
concentrations de nickel dépassant 50 mg/kg de poids à sec sont
généralement toxiques. On a observé que le cuivre potentialisait
la toxicité du nickel alors que le calcium la réduisait. On ne
dispose que de données limitées au sujet des effets du nickel sur
les animaux terrestres.
Les lombrics paraissent être relativement insensibles au
nickel, du moins si le milieu abonde en microorganismes et matières
organiques qui captent une partie du nickel aux dépens des vers.
On n'a pas envisagé la possibilité que le nickel constitue un
polluant majeur à l'échelon planétaire encore que des modifications
écologiques, par exemple une diminution du nombre et de la
diversité des espèces ait été observée à proximité de sources de
nickel. En outre, des études portant sur les microécosystèmes ont
montré qu'un apport de nickel dans le sol perturbait le cycle de
l'azote.
7. Effets sur les animaux d'experience et sur les systemes
d'épreuve in vitro
Le nickel joue un rôle essentiel dans l'activité catalytique de
certaines enzymes végétales et bactériennes. Chez certaines
espèces animales on a observé, après administration d'un régime
carencé en nickel, un ralentissement du gain de poids, de l'anémie
et une moindre viabilité de la progéniture.
Le dérivé du nickel qui présente la plus forte toxicité aiguë
est le nickel carbonyle dont le poumon constitue l'organe cible.
Ce composé peut provoquer un oedème pulmonaire en l'espace de 4
heures. Les autres dérivés du nickel présentent une faible
toxicité aiguë.
Si les allergies de contact sont très fréquentes chez l'homme,
on ne parvient à produire une sensibilisation expérimentale chez
l'animal que dans certaines conditions particulières. Chez le rat,
la souris et le cobaye on a provoqué des lésions des muqueuses et
des réactions inflammatoires des voies respiratoires en faisant
inhaler pendant une longue période à ces animaux du nickel
métallique, de l'oxyde et du sous-sulfure de nickel. On a observé
une hyperplasie de l'épithélium chez des rats après inhalation
d'aérosols de chlorure et d'oxyde de nickel.
Chez le rat, une exposition intense et prolongée à de l'oxyde
de nickel a provoqué une pneumoconiose progressive. Des réactions
inflammatoires quelquefois accompagnées d'une légère fibrose ont
été observées chez des lapins après exposition intense à la
poussière de nickel et de graphite. L'exposition de rats à du
sous-sulfure de nickel a provoqué une fibrose pulmonaire.
Des sels de nickel, administrés par voie parentérale à des
rats, des lapins et des poulets ont provoqué une hyperglycémie
transitoire rapide. Ce phénomène pourrait être lié aux effets qui
ont été observés sur les cellules alpha et béta des ilots de
Langerhans. Le nickel réduit également la libération de
prolactine. L'administration par voie orale ou par inhalation de
chlorure de nickel a réduit la fixation d'iode par la thyroïde.
Administrés par voie intraveineuse à des chiens, des sels de
nickel ont entraîné une réduction du débit sanguin des artères
coronaires; à forte concentration, le nickel diminue la
contractilité du myocarde du chien in vitro.
Le chlorure de nickel perturbe le système des cellules T et
inhibe l'activité des cellules tueuses naturelles (cellules NK).
On a observé que l'administration parentérale de chlorure et de
sous-sulfure de nickel entraînait la mort intrautérine des foetus
et réduisait le gain de poids chez le rat et la souris. Chez des
rats et des hamsters, l'inhalation de nickel carbonyle a provoqué
la mort des foetus et une diminution de leur poids, avec en outre
des effets tératogènes. Les études en question ne donnaient aucune
information sur la toxicité des composés du nickel pour les
femelles gestantes. On a fait état de mutations létales dominantes
obtenues chez des rats par exposition au nickel carbonyle.
Un certain nombre de dérivés minéraux du nickel ont fait
l'objet d'études de mutagénicité dans divers systèmes d'épreuve.
En général les dérivés du nickel se révèlent inactifs chez les
bactéries sauf dans le cas des tests de fluctuation. On a observé
des mutations chez plusieurs types de cellules mamaliennes en
culture. Les dérivés du nickel inhibent la synthèse de l'ADN chez
un grand nombre d'organismes. En outre, ces dérivés produisent des
aberrations chromosomiques et des échanges entre chromatides soeurs
tant dans les cellules mamaliennes que dans les cellules humaines
en culture. Chez des personnes exposées, de par leur profession, à
des composés insolubles ou solubles du nickel on a effectivement
observé des aberrations chromosomiques, mais pas d'échange entre
chromatides soeurs (sauf dans le cas d'une étude portant sur des
ouvriers travaillant dans des ateliers d'électrolyse). In vitro,
le nickel provoque la transformation des cellules.
Lors d'une étude d'inhalation, on a observé que du sous-sulfure
de nickel provoquait l'apparition de tumeurs pulmonaires bénignes
et malignes chez le rat. Quelques tumeurs pulmonaires ont
également été observées chez des rats soumis à une série d'études
où on leur faisait inhaler du nickel carbonyle. On n'a pas noté
d'augmentation significative des tumeurs pulmonaires chez des rats
soumis à l'inhalation de nickel métallique dans des conditions
d'étude satisfaisantes. L'inhalation d'oxyde noir n'a pas provoqué
non plus l'apparition de tumeurs pulmonaires chez des hamsters
dorés (espèce qui résiste à la cancérogénèse au niveau pulmonaire).
On ne dispose pas de bonnes études de cancérogénicité basées sur
l'exposition par inhalation à d'autres composés du nickel.
Toutefois on a constaté qu'après l'instillation intratrachéenne
réitérée de sous-sulfure de nickel, de poudre de nickel métallique
et d'un oxyde non précisé, des tumeurs pulmonaires bénignes se
formaient chez le rat.
Le nickel carbonyle, le nickelocène, ainsi qu'un grand nombre
de composés légèrement solubles ou insolubles comme le sous-sulfure
de nickel, le carbonate, le chromate, l'hydroxyde et divers
sulfures, des seléniures, des arséniures, des téllurures, des
antimoniures, différents oxydes non spécifiés, deux oxydes de
nickel et de cuivre, du nickel métallique et divers alliages, ont
provoqué l'apparition de tumeurs mésenchymateuses locales chez
divers animaux d'ex-périence après administration intramusculaire,
sous-cutanée, intrapéritonéale, intrapleurale, intraoculaire,
intraosseuse, intrarénale, intra-articulaire, intratesticulaire et
intra-adipeuse. On n'a pas observé d'effet cancérogène local lors
d'une étude portant sur des doses uniques de certains alliages de
nickel, d'hydroxyde colloïdal de nickel et de deux échantillons
d'oxyde, préparés spécialement en vue d'une étude de
cancérogénicité par calcination à 735 et 1045 °C.
On a provoqué l'apparition de tumeurs de la cavité péritonéale
chez des rats par administration intrapéritonéale réitérée de
sulfate et d'acétate de nickel; toutefois aucun effet n'a été
obtenu avec le chlorure de nickel.
On a étudié la cancérogénicité du nickel métallique et d'un
grand nombre de dérivés du nickel par administration parentérale; à
de rares exceptions près, toutes ces substances ont provoqué
l'apparition de tumeurs locales.
C'est seulement dans le cas du sous-sulfure de nickel qu'on a
des preuves convaincantes d'un pouvoir cancérogène après exposition
par voie respiratoire. Toutefois le nombre d'études de ce type qui
sont vraiment satisfaisantes reste très limité.
Lors d'études au cours desquelles on a procédé à des
instillations intratrachéennes répétées de poudre de nickel,
d'oxyde et de sous-sulfure de nickel, on a observé l'apparition de
tumeurs pulmonaires.
Trois sels solubles de nature différente qui n'avaient pas
produit de tumeurs locales lors d'études antérieures ont été
réétudiés en procédant cette fois à une administration
intrapéritonéale réitérée: deux de ces sels ont alors manifesté des
effets cancérogènes.
8. Effets sur l'homme
C'est le nickel carbonyle qui présente la toxicité aiguë la
plus forte pour l'homme. Elle se traduit par des céphalées
frontales, des vertiges, de la nausée, des vomissements, de
l'insomnie et de l'irritabilité, puis par des symptômes pulmonaires
évoquant une pneumonie d'origine virale. Parmi les lésions
pulmonaires patho-logiques qui ont été observées on peut citer des
hémorragies, de l'oedème et une désorganisation cellulaire. Le
foie, les reins, les surrénales, la rate et le cerveau sont
également affectés. On a aussi fait état d'intoxications par le
nickel chez des malades sous dialyse après contamination du liquide
de dialyse par des dérivés du nickel ainsi que chez des nickeleurs
qui avaient bu accidentellement de l'eau contaminée par du sulfate
et du chlorure de nickel.
On a signalé chez des nickeleurs et chez des ouvriers
d'ateliers d'affinage du nickel des effets chroniques tels que
rhinite, sinusite, perforation de la cloison nasale et asthme.
Certains auteurs font état d'anomalies pulmonaires, notamment une
fibrose, chez des ouvriers amenés à inhaler de la poussière de
nickel. En outre, on a signalé des cas de dysplasie nasale chez des
ouvriers travaillant dans des ateliers d'affinage du nickel.
L'hypersensibilité de contact est très largement documentée tant
dans la population générale que chez un certain nombre d'ouvriers
exposés de par leur profession à des dérivés solubles du nickel.
Dans plusieurs pays on indique que 10% de la population féminine et
1% de la population masculine présentent une sensibilité au nickel.
Parmi ces personnes, 40 à 50% présentent un eczéma vésiculeux des
mains qui peut être très grave dans certains cas et entraîner une
incapacité. L'ingestion de nickel peut aggraver l'eczéma siégeant
au niveau des mains ou d'autres régions du corps protégées de tout
contact avec du nickel.
On a également signalé que les prothèses et autres implants
chirurgicaux en alliage de nickel pouvaient produire une
sensibilisation à ce métal ou aggraver une dermatite préexistante.
Des effets néphrotoxiques, comme un oedème rénal avec hyperémie
et dégénérescence du parenchyme, ont été observés dans des cas
d'exposition industrielle accidentelle à du nickel carbonyle. Des
effets néphrotoxiques passagers ont été enregistrés après ingestion
accidentelle de sels de nickel.
On a fait état d'un risque très élevé de cancers pulmonaire et
nasal chez des ouvriers travaillant dans des ateliers d'affinage du
nickel où l'on effectue le grillage à mort des minerais sulfurés,
opération qui entraîne une exposition importante au sous-sulfure, à
l'oxyde et éventuellement au sulfate de nickel. L'exposition à des
dérivés solubles du nickel (électrolyse, extraction du sulfate de
cuivre, hydrométallurgie), souvent associée à une exposition à
l'oxyde de nickel mais sans exposition trop importante au sous-
sulfure, comporte des risques analogues. Pour les mineurs et les
ouvriers des ateliers d'affinage, le risque est beaucoup plus
faible. Chez les soudeurs d'acier inoxydable ou chez ceux qui
travaillent dans des industries employant du nickel, les taux de
cancer sont généralement proches de la normale, sauf dans le cas où
il y a également exposition au chrome, en particulier dans des
ateliers de galvanoplastie. Il semblerait cependant que les
ouvriers employés à la fabrication des accumulateurs au
nickel/cadmium, qui sont exposés à de fortes concentrations de
nickel et de cadmium, soient exposés à un risque légèrement plus
important de cancer du poumon.
On a signalé occasionnellement un excès de divers cancers
autres que ceux qui siègent au niveau des poumons ou du nez, par
exemple des cancers rénaux, gastriques ou prostatiques, chez des
travailleurs de l'industrie du nickel, mais cette surmorbidité n'a
pas été systématiquement constatée. On peut s'appuyer sur les
données épidémiologiques pour répondre à deux questions
importantes: i) certains dérivés du nickel sont-ils effectivement
cancérogènes, et ii) l'étude de cohortes faiblement exposées
permet-elle d'établir la limite supérieure du risque pour des
niveaux d'exposition donnés?
a) Dérivés solubles
On a mis en evidence un certain risque de cancer chez des
ouvriers exposés à des concentrations en sels solubles de l'ordre
de 1 à 2 mg/m3, qu'il s'agisse d'ouvriers d'ateliers d'électrolyse
ou travaillant à la préparation de sels solubles. Ces ouvriers
étaient également exposés à d'autres dérivés du nickel mais souvent
à des concentrations plus faibles que dans les processus à haut
risque. En l'absence d'une évaluation rétrospective de
l'exposition, il est impossible de tirer des conclusions
définitives de ces observations mais il existe certainement une
forte présomption d'une cancérogénicité des dérivés solubles du
nickel. Les poussières des ateliers d'affinage contiennent
quelques fois une proportion non négligeable de sulfate de nickel à
côté du sous-sulfure. Il se pourrait donc que le risque de cancer
très important qui a été observé chez les ouvriers qui travaillent
au grillage à mort des minerais sulfurés soit dû pour une part aux
dérivés solubles du nickel.
b) Sous-sulfure de nickel (sulfure nickeleux)
Dans les ateliers d'affinage où le risque de cancer est élevé,
on a constaté que l'exposition au sous-sulfure de nickel était
toujours accompagnée d'une exposition à l'oxyde et peut-être même
au sulfate (voir plus haut). Il est donc difficile à partir des
seules données épidémiologiques d'affirmer que le sous-sulfure est
cancérogène, encore que ce soit probable.
c) Oxyde de nickel
Dans tous les cas où l'on a observé un risque élevé de cancer,
il y avait présence d'oxyde de nickel accompagné d'une ou de
plusieurs autres formes (sous-sulfure, sels solubles, nickel
métallique). Comme dans le cas du sous-sulfure, il est difficile
d'affirmer ou d'écarter l'existence d'un pouvoir cancérogène sur la
base des seules données épidémiologiques.
d) Nickel métallique
On n'a pas constaté d'accroissement du risque de cancer chez
des ouvriers exposés uniquement à du nickel métallique. L'ensemble
des données relatives aux ouvriers qui travaillaient sur des
alliages du nickel ou dans des ateliers de diffusion gazeuse et qui
tous étaient exposés à des concentrations moyennes de l'ordre de
0,5 mg/m3, ne font pas ressortir d'élévation du risque, encore que
le nombre total de cancers du poumon parmi ces cohortes ait été
trop faible pour qu'on puisse exclure un léger accroissement du
risque à cette concentration.
e) Conclusion
Bien que quelques-uns, voire tous les dérivés du nickel
puissent être cancérogènes, il n'existe que peu ou pas de risque
décelable dans la plupart des secteurs de l'industrie du nickel aux
niveaux d'exposition actuels; cette observation vaut aussi pour
certains procédés dont on estimait jusqu'ici qu'ils comportaient un
risque très élevé de cancers pulmonaire et nasal. Une exposition
prolongée à des dérivés solubles du nickel à des concentrations de
l'ordre de 1 mg/m3 peut entraîner une augmentation sensible du
risque relatif de cancer pulmonaire mais ce risque est à peu près
égal à l'unité chez les ouvriers exposés à des concentrations en
nickel métallique de l'ordre de 0,5 mg/m3 en moyenne. Pour un
niveau d'exposition donné, le risque de cancer est plus important
dans le cas des composés solubles que dans le cas du nickel
métallique et peut-être des autres dérivés du nickel. L'absence
d'un risque notable de cancer pulmonaire chez les nickeleurs n'est
pas sur-prenante étant donné qu'en moyenne l'exposition aux dérivés
solubles est beaucoup plus faible dans les ateliers de
galvanoplastie que dans les ateliers d'affinage électrolytique ou
de traitement des sels de nickel.
RESUMEN Y CONCLUSIONES
1. Identidad, propiedades fisicas y químicas y métodos analíticos
El níquel es un elemento metálico perteneciente al grupo VIIIb
de la tabla periódica. Es resistente a los álcalis, pero en general
se disuelve bien en ácidos oxidantes diluidos. El carbonato de
níquel, el sulfuro de níquel y el óxido de níquel no son solubles
en agua, mientras que sí lo son el cloruro de níquel, el sulfato de
níquel y el nitrato de níquel. El carbonilo de níquel es un líquido
incoloro y volátil que se descompone por encima de los 50 °C. La
forma iónica predominante es la de níquel (II). En los sistemas
biológicos, el níquel disuelto puede formar componentes complejos
con diversos ligandos y unirse a materiales orgánicos.
Los métodos que más se utilizan en el análisis de materiales de
origen biológico y medio ambiental son la espectroscopía de
absorción atómica y la voltamperimetría. A fin de obtener
resultados fidedignos, sobre todo en el orden de las ultratrazas,
hay que adoptar procedimientos específicos para reducir al mínimo
el riesgo de contaminación durante la recogida, el almacenamiento,
el tratamiento y el análisis de muestras. En los materiales de
origen biológico y en el agua se puede llegar límites de detección
de 1-100 ng/litro, atendiendo a los procedimientos de tratamiento
previo, extracción y enriquecimiento de la muestra.
2. Fuentes de exposición humana y ambiental
El níquel es un metal traza muy ampliamente distribuido, que se
encuentra en el suelo, el agua, el aire y la biosfera. En la
corteza terrestre hay un porcentaje medio del 0,008%. Los terrenos
cultivables contienen entre 3 y 1000 mg de níquel/kg. En el agua
natural los niveles oscilan entre 2 y 10 µg/litro (agua dulce) y de
0,2 a 0,7 µg/litro (agua de mar). Las concentraciones atmosféricas
de níquel en zonas remotas son de 0,1 a 3 ng/m3.
Los yacimientos metalíferos de níquel están formados por
acumulaciones de minerales de sulfuro de níquel (principalmente
pentlandita) y lateritas. El níquel se extrae de la mena mediante
procesos de afinado piro e hidrometalúrgico. La mayor parte del
níquel obtenido se destina a la producción de acero inoxidable y a
otras aleaciones muy resistentes a la corrosión y a la temperatura.
Las aleaciones de níquel y el niquelado se utilizan en vehículos,
maquinaria industrial, armamento, herramientas, equipo eléctrico,
utensilios domésticos y la acuñación de monedas. También se
utilizan compuestos de níquel como catalizadores, pigmentos y en
las baterías. En 1985, la producción minera mundial de níquel fue
de unos 67 millones de kg. Las principales fuentes de emisión de
níquel a la atmósfera son la combustión de carbón y petróleo para
la obtención de calor o energía, la incineración de desechos y
fangos cloacales, la extracción minera y la producción primaria de
níquel, la fabricación de acero, la galvanoplastia y otras fuentes,
como la producción de cemento. En el aire contaminado pre-dominan
el sulfato, los óxidos y los sulfuros de níquel, y en menor
cantidad el níquel metálico.
El níquel procedente de los distintos procesos industriales y
de otras fuentes termina llegando a las aguas residuales. Los
restos que se obtienen del tratamiento de esas aguas se eliminan
mediante inyección en pozos profundos, vertido en los océanos,
tratamiento en tierra e incineración. Los efluentes de las
instalaciones de tratamiento de aguas residuales contienen, según
los datos disponibles, hasta 0,2 mg de níquel/litro.
3. Transporte, distribución y transformación en el medio ambiente
El níquel se introduce en el medio ambiente a partir de fuentes
tanto naturales como de origen humano y circula por todos sus
compartimentos mediante procesos físicos y químicos, así como por
el transporte biológico que efectúan los organismos vivos.
Se considera que en la atmosféra el níquel está principalmente
en forma de aerosoles de partículas con distintas concentraciones
del metal, en función de su procedencia. Las concentraciones más
altas de níquel en el aire suelen aparecer en las partículas más
pequeñas. El carbonilo de níquel es inestable en el aire y se
descompone formando óxido de níquel.
El transporte y la distribución de las partículas de níquel a
los diferentes compartimentos del medio ambiente o entre ellos
depende en gran medida del tamaño de las partículas y de las
condiciones meteorológicas. La distribución por tamaños de las
partículas depende sobre todo de las fuentes de emisión. En
general, las partículas de fuentes artificiales son más pequeñas
que las que se encuentran en el polvo natural.
El níquel ingresa en la hidrosfera por eliminación desde la
atmósfera, por la escorrentía superficial, por la descarga de
residuos industriales y municipales, y también tras la erosión
natural de los suelos y de las rocas. En los ríos, este elemento se
transporta principalmente en forma de revestimiento precipitado
sobre partículas y unido a materia orgánica; en los lagos, se
transporta en forma iónica y de manera predominante unido a materia
orgánica. La partículas de arcilla también lo pueden absorber, y a
la biota pasa por ingestión o absorción. El proceso de absorción se
puede invertir, lo que se traduce en una liberación del níquel del
sedimento. Los ríos y arroyos transportan una parte hasta el mar.
El aporte fluvial de partículas suspendidas se estima en 135 x 107
kg/año.
En función del tipo de suelo, el níquel puede presentar una
gran movilidad dentro del perfil edáfico hasta alcanzar las aguas
subterráneas y, con ello, los ríos y lagos. La lluvia ácida tiene
una marcada tendencia a movilizar el níquel del suelo. Las plantas
terrestres lo absorben principalmente por las raíces. La cantidad
absorbida depende de diversos parámetros geoquímicos y físicos,
entre los que figuran el tipo de suelo, su pH y humedad, su
contenido en materia orgánica y la concentración de níquel
extraíble. El caso más conocido de acumulación de níquel es el de
algunas especies de plantas ("hiperacumuladoras"), que crecen en
suelos serpentinosos relativamente poco fértiles y en las que se
han encontrade niveles superiores a 1 mg/kg de peso seco. Los
niveles de níquel superiores a 50 mg/kg de peso seco son tóxicos
para la mayoria de las plantas. Se han observado casos de
acumulación y efectos tóxicos en hortalizas cultivadas en suelos
tratados con fangos residuales y en la vegetación próxima a las
fuentes de emisión del metal. En plantas acuáticas se han detectado
factores de concentración elevados. Los estudios de laboratorio han
puesto de manifiesto que la capacidad de acumulación de níquel en
los peces examinados es escasa. En aguas no contaminadas, las
concentraciones halladas en los peces enteros (sobre la base del
peso fresco) oscilaron entre 0,02 y 2 mg/kg. Estos valores pueden
ser hasta diez veces superiores en los peces procedentes de aguas
contaminadas. Sin embargo, no hay pruebas de que se produzca
bioamplificación del níquel en la cadena de los alimentos.
El níquel se encuentra en muchos órganos y tejidos de animales
silvestres, debido a su ingestión con los alimentos por los
animales herbívoros y sus predadores carnívoros.
4. Niveles medioambientales y exposición humana
Los niveles de níquel en los organismos terrestres y acuáticos
pueden diferir en varios ordenes de magnitud. La exposición humana
a los valores habituales en la atmósfera, que oscilan entre
alrededor de 5 y 35 ng/m3 en zonas rurales y urbanas, hace que la
inhalación de este elemento sea de 0,1-0,7 µg/día. Aunque el agua
potable contiene menos de 10 µg/litro, en ocasiones los empalmes de
las tuberías pueden liberarlo, dando lugar a concentraciones de
hasta 500 µg/litro.
Su concentración en los alimentos suele estar por debajo de 0,5
mg/kg de peso fresco. El cacao, la soja, algunas legumbres secas,
diversos frutos secos y la harina de avena contienen elevadas
concentraciónes de níquel. Su ingestión diaria con los alimentos
varía ampliamente en función de los diferentes hábitos
alimentarios, y puede oscilar entre 100 y 800 µg/día; la ingestión
media de níquel en la dieta es, en la mayoría de los países, de
100-300 µg/día. Los utensilios de cocina liberan níquel, lo que
contribuye de manera considerable a su ingestión. Fumando 40
cigarrillos diarios pueden absorberse a través de los pulmones 2-23
µg/día.
La exposición cutánea en el medio ambiente general es
importante, porque induce y mantiene la hipersensibilidad por
contacto que produce en la piel el uso diario de objetos niquelados
o con aleaciones de este metal (por ejemplo, joyería, monedas,
"clips").
Puede haber exposición iatrogénica al níquel debida a las
implantaciones y las prótesis hechas con aleaciones que lo
contienen, a fluidos intravenosos o de diálisis y a los medios de
contraste radiográfico. Se estima que la absorción media de níquel
por vía intravenosa a partir de los fluidos de diálisis es de 100
µg por tratamiento.
En el ámbito laboral, la concentración en el aire puede variar
desde varios g/m3 a, en ocasiones, unos mg/m3, en función del
proceso de que se trate y del contenido en níquel del material que
se maneja.
Hay millones de trabajadores de todo el mundo expuestos a polvo
y humo que contienen níquel durante las operaciones de soldadura,
niquelado y pulimentado, la extracción minera, el afinado del
níquel, y en las acerías, fundiciones y otras industrias
metalúrgicas.
Hay una gran variedad de empleos en los que puede haber
exposición cutánea al níquel, sea por contacto directo con el metal
disuelto, como por ejemplo en las industrias de afinado,
galvanoplastia y electroconformación, o bien por el manejo de
herramientas que lo contienen. En los trabajos de limpieza con agua
puede haber una exposición a este metal, a causa de las cantidades
que lleva disueltas el agua de lavado.
5. Cinetica y metabolismo en el hombre y en los animales
El hombre y los animales pueden absorber níquel por inhalación,
por ingestión o por vía cutánea. La absorción respiratoria, con
asimilación gastrointestinal secundaria de níquel (soluble e
insoluble), es la principal vía de penetración durante la
exposición en el trabajo. Una cantidad importante del material
inhalado se traga con el fluido mucociliar procedente del tracto
respiratorio. La mala higiene personal y las prácticas laborales
deficientes pueden contribuir a la exposición gastrointestinal. La
absorción percutánea es cuantitativamente insignificante, pero es
importante en la patogenia de la hipersensibilidad por contacto. La
absorción está relacionada con la solubilidad del compuesto, y
sigue la relación general carbonilo de níquel > compuestos de
níquel solubles > compuestos de níquel insolubles. El carbonilo de
níquel es el compuesto de este metal que absorben de forma más
rápida y completa el hombre y los animales. Hay pocos estudios en
los que se haya administrado níquel por inhalación. En experimentos
con hámsters y ratas tratados con óxido de níquel insoluble se
observó una escasa absorción, con retención pulmonar de gran parte
del material después de varias semanas. En cambio, la absorción de
cloruro de níquel soluble o de sulfuro de níquel amorfo fue rápida.
El níquel es transportado en la sangre, principalmente ligado a la
albúmina.
Su absorción gastrointestinal es variable y depende de la
composición de la dieta. En un estudio reciente con un grupo de
voluntarios se encontró que la absorción de níquel a partir del
agua era del 27%, mientras que a partir de los alimentos era
inferior al 1%. Todas las secreciones orgánicas, como la orina, la
bilis, el sudor, las lágrimas, la leche y el fluido mucociliar, son
posibles vías de eliminación. El níquel que no se absorbe se
elimina con las heces. En roedores se ha demostrado que atraviesa
la placenta. Tras la administración parenteral de sales de níquel,
los niveles más altos de acumulación se producen en el riñon, las
glándulas endocrinas y el hígado; si lo que se administra es
carbonilo de níquel, también se observa una alta concentración en
el cerebro. Los datos sobre excreción del níquel sugieren un modelo
bicompartimental. Su concentración en el suero y la orina de
adultos sanos no expuestos en el trabajo es de 0,2 µg/litro
(oscilación: 0,05-1,1 µg/litro) y 1,5 µg/g de creatinina
(oscilación: 0,5-4,0 µg/g de creatinina), respectivamente. Se ha
observado que después de la exposición profesional, la
concentración aumenta en ambos fluidos. La carga corporal de níquel
en un adulto de 70 kg de peso no expuesto es de 0,5 µg.
6. Efectos en los seres vivos del medio ambiente
En los microorganismos, con concentraciones de 1-5 mg/litro de
níquel en el medio se inhibía, en general, el crecimiento en
actinomicetos, levaduras y eubacterias marinas y no marinas, y con
niveles de 5-1000 mg/litro, en hongos filamentosos. En el caso de
las algas, con una concentración aproximada de 0,05-5 mg de
níquel/litro no se observó crecimiento. Ciertos factores abióticos,
como el pH, la dureza, la temperatura y la salinidad del medio, así
como la presencia de partículas orgánicas e inorgánicas, influyen
en la toxicidad del níquel.
La toxicidad del níquel para los invertebrados acuáticos varía
considerablemente en función de las especies y de los factores
abióticos. En el género Daphnia se ha encontrado una CL50 a las 96
horas de 0,5 mg de níquel/litro, mientras que en moluscos los
valores de la CL50 a las 96 horas fueron de unos 0,2 mg/litro en
dos especies de caracol de agua dulce y de 1100 mg/litro en un
bivalvo.
En los peces, los valores de la CL50 a las 96 horas suelen
oscilar entre 4 y 20 mg de níquel/litro, pero pueden ser más altos
en algunas especies. Los estudios a largo plazo realizados sobre
los peces y su crecimiento en aguas blandas pusieron de manifiesto
que niveles tan bajos como 0,05 mg de níquel/litro tenían algunos
efectos en la trucha arco iris. En plantas terrestres se ha
encontrado que los niveles superiores a 50 mg/kg de peso seco
suelen ser tóxicos. Se ha observado que desde el punto de vista
toxicologica el cobre tiene un efecto sinérgico con el níquel,
mientras que el calcio reduce su toxicidad. Hay pocos datos acerca
de los efectos del níquel en animales terrestres.
La lombriz de tierra parece ser relativamente insensible a este
metal si el medio es rico en microorganismos y materia orgánica,
que dejan menos níquel disponible para aquella. Aunque el níquel no
se ha considerado un contaminante mundial en gran escala, en las
cercanías de fuentes emisoras se han observado cambios ecológicos,
como la disminución del número y la diversidad de especies. En
estudios de microecosistemas se ha puesto de manifiesto que la
adición de este elemento al suelo altera el ciclo del nitrógeno.
7. Efectos en los animales de experimentación y en sistemas de
prueba in vitro
El níquel es indispensable para la actividad catalítica de
algunas enzimas vegetales y bacterianas. En algunas especies
animales se ha señalado que una dieta carente de níquel provoca
lentitud en la ganacia de peso, anemia y una disminución de la
viabilidad de la descendencia.
El compuesto de toxicidad más aguda es el carbonilo de níquel,
cuyo órgano destinatario es el pulmón; dentro de las cuatro horas
siguientes a la exposición puede aparecer edema pulmonar. La
toxicidad aguda de los demás compuestos del níquel es baja.
Aunque la alergia de contacto causada por este metal es muy
frecuente en el hombre, en animales sólo se consigue inducir
sensibilización experimental en determinadas condiciones. La
exposición a un largo período de inhalación de níquel metálico,
óxido de níquel o subsulfuro de níquel causó lesiones en las
mucosas y una reacción inflamatoria del tracto respiratorio en
ratas, ratones y cobayos. En ratas expuestas a la inhalación de
aerosoles de cloruro de níquel u óxido de níquel se observó
hiperplasia epitelial.
La exposición a altos niveles de óxido de níquel durante un
período prolongado produjo una neumoconiosis progresiva en ratas.
En conejos expuestos a un nivel elevado de polvo de níquel-grafito
se observó una reacción inflamatoria, a veces acompañada de una
ligera fibrosis. Las ratas expuestas a subsulfuro de níquel
presentaron fibrosis pulmonar.
La administración de sales de níquel a ratas, conejos y pollos
por vía parenteral indujo una hiperglucemia rápida transitoria.
Estos cambios pueden estar relacionados con los efectos en las
células alfa y beta de los islotes de Langerhans. El níquel también
disminuyo la liberación de prolactina. Se ha informado de que la
administración por vía oral o respiratoria de cloruro de níquel
reduce la absorción de yodo por el tiroides.
La administración intravenosa de sales de níquel a perros hizo
disminuir el flujo sanguíneo en las arterias coronarias; la
concentración elevada de este metal redujo la contractibilidad del
miocardio de perro in vitro.
El cloruro de níquel afecta al sistema de células T y suprime
la actividad de las células destructoras naturales. Se ha observado
que la administración parenteral de cloruro de níquel y de
subsulfuro de níquel a ratas y ratones causa mortalidad
intrauterina y una disminución de la ganancia de peso. En ratas y
hámsters expuestos a inhalaciones de carbonilo de níquel se produjo
la muerte de fetos y una disminución de la ganancia de peso, y tuvo
también un efecto teratógenico. En ninguno de esos estudios se
informa acerca de la toxicidad materna. Se ha señalado que el
carbonilo de níquel ocasiona mutaciones letales dominantes en la
rata.
Se han hecho pruebas con distintos sistemas para determinar la
mutagenicidad de varios compuestos inorgánicos del níquel. Estos,
en general, se mostraron inactivos en los ensayos de mutagénesis
efectuados con bacterias, excepto en los casos en que se utilizaron
pruebas de fluctuación. Se observaron mutaciones en varios tipos de
células de mamíferos cultivadas. Los compuestos del níquel
inhibieron la síntesis de ADN en una gran variedad de organismos.
Además, produjeron aberraciones cromosómicas e intercambio de
cromátidas hermanas en cultivos de células tanto de mamíferos como
humanas. En personas profesionalmente expuestas a compuestos de
níquel insolubles o solubles se observaron aberraciones
cromosómicas, pero no intercambio de cromátidas hermanas (excepto
en un estudio sobre personas que trabajan en electrólisis). El
níquel indujo transformación celular in vitro.
En un estudio de inhalación en ratas, el subsulfuro de níquel
provocó la aparición de tumores pulmonares benignos y malignos. En
una serie de estudios de inhalación de carbonilo de níquel en la
rata, se observaron algunos tumores pulmonares. En un estudio de
inhalación de níquel metálico efectuado en ratas no se apreció un
aumento importante de tales tumores. La inhalación de óxido de
níquel negro no indujo la aparición de tumores pulmonares en
hámsters dorados sirios (especie resistente a la carcinogénesis
pulmonar). No se conocen estudios adecuados sobre la carcinogénesis
por inhalación de otros compuestos del níquel. Sin embargo, la
instilación intratraqueal repetida de subsulfuro de níquel, de
polvo de níquel metálico y de un óxido de níquel no especificado
produjo en ratas tumores pulmonares benignos y malignos.
El carbonilo de níquel, el niqueloceno y gran número de
compuestos del níquel ligeramente solubles o insolubles, entre
ellos el subsulfuro, el carbonato, el cromato, el hidróxido, los
sulfuros, los seleniuros, los arseniuros, el telururo, el
antimoniuro, diversas preparaciones de óxidos no identificadas, dos
óxidos de níquel y cobre, el níquel metálico y diversas aleaciones
de níquel provocaron tumores mesenquimáticos locales en distintos
animales de experimentación tras la administración por vía
intramuscular, subcutánea, intraperitoneal, intrapleural,
intraocular, intraósea, intrarrenal, intraarticular,
intratesticular o intraadiposa. No se observó respuesta
carcinogénica local en los estudios de dosis única con algunas
aleaciones de níquel, hidróxido de níquel coloidal o dos muestras
de óxido de níquel, especialmente preparados para pruebas de
carcinogénesis mediante calcinación a 735 °C o bien a 1045 °C.
La administración repetida de sulfato y acetato de níquel a
ratas por vía intraperitoneal indujo tumores en la cavidad
peritoneal, pero no la de cloruro de níquel.
Se han realizado pruebas de carcinogénesis mediante la
administración por vía parenteral de níquel metálico y de una gran
variedad de compuestos; salvo algunas excepciones, se produjeron
tumores locales.
Sólo en el caso del subsulfuro de níquel se ha demostrado de
manera convincente que la inhalación produce cáncer. Sin embargo,
hay muy pocos estudios adecuados sobre la exposición por vía
respiratoria.
En los estudios de aplicación de instilaciones intratraqueales
repetidas de polvo de níquel, óxido de níquel y subsulfuro de
níquel aparecieron tumores pulmonares.
Cuando se realizaron pruebas con tres sales solubles de níquel
distintas que en estudios anteriores no habían provocado la
aparición de tumores locales, utilizando la administración
intraperitoneal repetida, dos de las sales indujeron una respuesta
carcinogénica.
8. Efectos en la especie humana
En relación con la salud del ser humano, el carbonilo de níquel
es el compuesto del metal con toxicidad más aguda. Entre los
efectos de la intoxicación aguda que causa están dolor de cabeza
frontal, vértigo, náuseas, vómitos, insomnio e irritabilidad,
seguidos de trastornos pulmonares análogos a los producidos por una
neumonía vírica. Entre las lesiones patológicas de los pulmones
figuran hemorragias, edema y desorganización celular. También se
ven afectados el hígado, los riñones, las cápsulas suprarrenales,
el bazo y el cerebro. Ha habido asimismo casos de intoxicación en
pacientes sometidos a diálisis con contaminación de níquel y en
galvanizadores que accidentalmente han ingerido agua contaminada
por sulfato y cloruro de níquel.
Se han notificado casos de efectos crónicos, como rinitis,
sinusitis, perforación del tabique nasal y asma, entre trabajadores
del afinado del níquel y del niquelado. Algunos autores han
descrito alteraciones pulmonares con fibrosis en personas que en el
trabajo respiraban polvo de níquel. Además, se ha informado de
casos de displasia nasal en trabajadores del afinado del níquel.
Hay abundante documentación acerca de la hipersensibilidad por
contacto, tanto en la población en general como en algunos
trabajadores profesionalmente expuestos a compuestos solubles de
níquel. En varios países se ha comunicado que el 10% de la
población femenina y el 1% de la masculina son sensibles al níquel.
De ellos, el 40-50% tienen eccema vesicular en las manos; en
algunos casos puede ser muy grave y causa de incapacidad laboral.
La ingestión de níquel puede agravar la eccema vesicular de las
manos y, posiblemente, inducir también su aparición en otras partes
del cuerpo que no han tenido contacto cutáneo con este metal.
Se sabe que las prótesis y otras implantaciones quirúrgicas de
aleaciones con níquel causan sensibilización a este elemento o
agravan la dermatitis existente.
Se han descrito efectos nefrotóxicos, como edema renal con
hiperemia y degeneración parenquimatosa, en casos de exposición
industrial accidental a carbonilo de níquel. También se han
registrado efectos nefrotóxicos pasajeros por ingestión accidental
de sales de níquel.
Se ha informado de un elevadísimo riesgo de cáncer del pulmón y
la nariz en los trabajadores del afinado del níquel que se ocupan
de tostar a alta temperatura los minerales de sulfuro, lo que
supone una gran exposición al subsulfuro, el óxido y quizás el
sulfato de níquel. Se han señalado riesgos análogos en los procesos
que entrañan exposición al níquel soluble (electrólisis, extracción
de sulfato de cobre, hidrometalurgia), a menudo combinados con
alguna exposición al óxido de níquel pero muy poca al subsulfuro.
El riesgo de los mineros y otros trabajadores del afinado parece
ser mucho menor. El índice de cáncer entre los soldadores de acero
inoxidable y los trabajadores de industrias que usan níquel ha
sido, en general, casi normal, salvo cuando hay exposición al
cromo, particularmente en la galvanoplastia. Sin embargo, los
trabajadores que hacen baterías de níquel/cadmio y están expuestos
a altos niveles de ambos metales corren un riesgo ligeramente mayor
de contraer cáncer de pulmón.
Ocasionalmente se ha notificado en trabajadores del níquel una
frecuencia excesiva de otros tipos de cáncer distintos del de
pulmón y el nasal, como por ejemplo renal, gástrico o de próstata,
pero ninguno se ha encontrado de manera constante.
Se pueden utilizar los datos epidemiológicos para plantear dos
importantes cuestiones: i) si se ha demostrado que ciertos
compuestos de níquel que son carcinógenos; y ii) si las cohortes
sometidas a bajas exposiciónes presentan límites superiores de
riesgo a determinados niveles de exposición.
a) Níquel soluble
Se obtuiseron pruebas de que los trabajadores expuestos a
concentraciones de níquel soluble del orden de 1-2 mg/m3, tanto en
el sector de la electrólisis como en el de la preparación de sales
solubles, corrían riesgo de cáncer. Esos trabajadores estaban
expuestos también a otros compuestos de níquel, pero con frecuencia
a niveles más bajos que en otros procesos de alto riesgo. En
ausencia de mediciones anteriores de la exposición, es imposible
llegar a conclusiones definitivas, pero las pruebas de que el
níquel soluble es carcinógeno son ciertamente sólidas. El polvo del
afinado contiene a veces una proporción importante de sulfato de
níquel, además de subsulfuro. Esto plantea la posibilidad de que el
altísimo riesgo de cáncer observado en los trabajadores que se
ocupan de la oxidación de subsulfuro de níquel a alta temperatura
pueda deberse en parte al níquel soluble.
b) Subsulfuro de níquel
En las zonas de afinado donde el riesgo de contraer cáncer era
alto, la exposición al subsulfuro de níquel casi siempre iba
acompañada de la exposición al óxido y, quizás, al sulfato (véase
más arriba). Así pues, basándose solamente en los datos
epidemiológicos, es dificil demonstrar que el subsulfuro de níquele
sea carcinógeno, aunque parece probable.
c) Oxido de níquel
El óxido de níquel estaba presente en casi todas las
circunstancias en que el riesgo de cáncer era elevado, acompañado
de una o más formas del elemento (subsulfuro de níquel, níquel
soluble, níquel metálico). Como en el caso del subsulfuro de
níquel, es difícil afirmar o negar su carácter carcinogénico
basándose exclusivamente en los datos epidemiológicos.
d) Níquel metálico
No se ha demostrado que los trabajadores expuestos
exclusivamente al níquel metálico corran mayor riesgo de cáncer.
Los datos combinados relativos a trabajadores que se ocupan de
aleaciones de níquel y los dedicados a difusión gaseosa, todos
ellos expuestos a concentraciones medias del orden de 0,5 mg de
níquel/m3, no muestran un riesgo excesivo, aunque el número total
de casos de cáncer de pulmón en estas cohortes era demasiado
pequeño para excluir un pequeño aumento del riesgo a este nivel.
e) Conclusión
Aunque algunas, y quizás todas, las formas del níquel pueden
ser carcinógenas, con los niveles normales de exposición en muchos
sectores de la industria de este metal hay un riesgo pequeño o no
detectable; entre ellos se encuentran algunos procesos que en el
pasado estaban asociados con un riesgo muy alto de cáncer pulmonar
y nasal. La exposición prolongada a concentraciones del orden de 1
mg/m3 de níquel soluble puede ocasionar un importante aumento del
riesgo relativo de cáncer de pulmón, pero este riesgo entre los
trabajadores expuestos a un nivel medio de níquel metálico de
alrededor de 0,5 mg/m3 es aproximadamente 1. El riesgo de cáncer
con un determinado nivel de exposición puede ser más alto para los
compuestos de níquel solubles que para el níquel metálico, y
posiblemente, que para otras formas. No es extraña la ausencia de
cualquier riesgo pronunciado de cáncer pulmonar entre los
niqueladores, puesto que el promedio de exposición al níquel
soluble es mucho más bajo que el que se produce en el afinado
electrolítico o en el tratamiento de sales de níquel.