
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 3
Lead
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of either the World Health Organization or the United Nations
Environment Programme
Published under the joint sponsorship of
the United Nations Environment Programme
and the World Health Organization
World Health Organization
Geneva 1977
ISBN 92 4 154063 X
(c) World Health Organization 1977
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. For rights of reproduction or
translation of WHO publications, in part or in toto, application
should be made to the Office of Publications, World Health
Organization, Geneva, Switzerland. The World Health Organization
welcomes such applications.
The designations employed and the presentation of the material in
this publication do not imply the expression of any opinion whatsoever
on the part of the Secretariat of the World Health Organization
concerning the legal status of any country, territory, city or area or
of its authorities, or concerning the delimitation of its frontiers or
boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar nature
that are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR LEAD
1. SUMMARY AND RECOMMENDATIONS FOR FURTHER RESEARCH
1.1. Summary
1.1.1. Analytical problems
1.1.2. Sources and pathways of exposure
1.1.3. Metabolism
1.1.4. Experimental studies on the effects of lead
1.1.5. Clinical and epidemiological studies on the effects
of lead. Evaluation of health risk to man from
exposure to lead
1.2. Recommendations for further research
1.2.1. Analytical methods
1.2.2. Sources of lead intake
1.2.3. Epidemiological studies
1.2.4. Interactions of lead with other environmental
factors
1.2.5. Significance of biological effects
2. PROPERTIES AND ANALYTICAL METHODS
2.1. Physical and chemical properties of lead and its compounds
2.2. Analytical procedures
2.2.1. Sampting
2.2.2. Analytical methods for lead
2.2.3. Methods for the measurement of some biochemical
effects of lead
3. SOURCES OF LEAD IN THE ENVIRONMENT
3.1. Natural occurrence
3.1.1. Rocks
3.1.2. Soils
3.1.3. Water
3.1.4. Air
3.1.5. Plants
3.1.6. Environmental contamination from natural sources
3.2. Production of lead
3.2.1. Lead mining
3.2.2. Smelting and refining
3.2.3. Environmental pollution from production
3.3. Consumption and uses of lead and its compounds
3.3.1. Storage battery industry
3.3.2. Alkyllead fuel additives
3.3.3. Cable industry
3.3.4. Chemical industry
3.3.5. Miscellaneous
3.3.6. Environmental pollution from consumption and uses of
lead
3.4. Waste disposal
3.5. Miscellaneous sources of environmental pollution
4. ENVIRONMENTAL TRANSPORT AND DISTRIBUTION
5. ENVIRONMENTAL LEVELS AND EXPOSURES
5.1. Exposure of the general population
5.1.1. Air
5.1.2. Water
5.1.3. Food
5.1.4. Miscellaneous
5.2. Exposure of infants and young children
5.2.1. Soil, dust, and taint
5.2.2. Miscellaneous
5.3. Occupational exposures
5.3.1. Lead mining, smelting and refining
5.3.2. Electric storage battery manufacturing
5.3.3. Shipbreaking and welding
5.3.4. Printing
5.3.5. Alkyllead manufacture
5.3.6. Other industrial exposures
5.4. Blood lead concentrations of various populations
5.4.1. Adult populations
5.4.2. Children
6. METABOLISM OF LEAD
6.1. Absorption
6.1.1. Absorption by inharation
6.1.1.1 Human studies
6.1.1.2 The relationship of air lead to blood lead
in the general population
6.1.1.3 The relationship of air lead to blood lead
in occupational exposure
6.1.1.4 Animal studies
6.1.2. Absorption of lead from the gastrointestinal tract
6.1.2.1 Human studies
6.1.2.2 The relationship of oral intake of lead to
blood lead levels in man
6.1.2.3 Animal studies
6.2. Distribution and retention
6.2.1. Human studies
6.2.2. Studies in animals
6.3. Elimination of lead
6.3.1. Human studies
6.3.2. Animal studies
6.4. "The metabolism of alkyllead compounds
7. EXPERIMENTAL STUDIES ON THE EFFECTS OF LEAD
7.1. Animal studies
7.1.1. Haemopoietic system
7.1.2. Nervous system
7.1.2.1 Inorganic lead
7.1.2.2 Alkyllead compounds
7.1.3. Renal system
7.1.4. Gastrointestinal tract
7.1.5. Cardiovascular system
7.1.6. Respiratory system
7.1.7. Reproductive system
7.1.8. Endocrine organs
7.1.9. Carcinogenicity
7.1.9.1 Inorganic lead compounds
7.l.9.2 Alkyllead compounds
7.1.10. Mutagenicity
7.1.11. Teratogenicity
7.2. Acquisition of tolerance to lead
7.3. Factors influencing lead toxicity
7.3.1. Age and sex
7.3.2. Seasonal variations
7.3.3. Nutrition
7.3.4. Intercurrent disease, alcohol, and other metals
7.4. Human studies
8. EFFECTS OF LEAD ON MAN--EPIDEMIOLOGICAL AND CLINICAL STUDIES
8.1. Retrospective studies of lead-exposed populations
8.1.1. Epidemiology of lead poisoning in industry
8.1.2. Epidemiology of lead poisoning in the general adult
population
8.1.3. Epidemiology of lead poisoning in infants and young
children
8.2. Clinical and epidemiological studies of the effects of lead
on specific organs and systems
8.2.1. Haemopoietic system
8.2.1.1 delta-aminolevulinic acid dehydratase (ALAD)
8.2.1.2 Free erythrocyte porphyrins (FEP)
8.2.1.3 delta-aminolevulinic acid excretion in urine
(ALA-U)
8.2.1.4 Coproporphyrin excretion in urine (CP-U)
8.2.1.5 Effects of lead on cell morphology
8.2.1.6 Effects of lead on erythrocyte survival
8.2.1.7 Effects of lead on haem synthesis
8.2.1.8 Relationship between lead exposure and
anaemia
8.2.2. Nervous system
8.2.2.1 Central nervous system
8.2.2.2 Peripheral nervous system
8.2.3. Renal system
8.2.4. Gastrointestinal tract
8.2.5. Liver
8.2.6. Cardiovascular system
8.2.7. Reproduction
8.2.8. Endocrine organs
8.2.9. Carcinogenicity
8.2.10. Effects on chromosomes
8.2.11. Teratogenicity
8.3. Factors influencing lead toxicity
8.3.1. Acquisition of tolerance to lead
8.3.2. Age
8.3.3. Seasonal variation
8.3.4. Nutrition
8.3.5. Intercurrent disease, alcohol, and other metals
9. EVALUATION OF HEALTH RISKS TO MAN FROM EXPOSURE TO LEAD AND ITS
COMPOUNDS
9.1. Relative contributions of air, food, water and other
exposures to total intake
9.1.1. Adult members of general population groups
9.1.2. Infants and children
9.1.3. Occupationally exposed population groups
9.2. Evaluation of haematological effects
9.3. Dose-effect relationships
9.4. Dose-response relationships
9.5. Diagnosis of lead poisoning and indices of exposure and/or
effects for epidemiological studies
9.5.1. Concentration of lead in blood (Pb-B)
9.5.2. Aminolevulinic acid dehydratase (ALAD)
9.5.3. Aminolevulinic acid (ALA) and coproporphyrin (CP)
excretion in the urine
9.5.4. Lead excretion in the urine
9.5.5. Haematological changes (stippled cells, anaemia)
9.5.6. Lead in tissues (teeth and hair)
9.5.7. Some practical aspects
9.5.7.1 General population studies
9.5.7.2 Occupationally-exposed persons
9.5.7.3 Reliability of the sampling and analytical
methods
9.6. The problem of alkyllead compounds
REFERENCES
NOTE TO READERS OF THE CRITERIA DOCUMENTS
While every effort has been made to present information in the
criteria documents as accurately as possible without unduly delaying
their publication, mistakes might have occurred and are likely to
occur in the future. In the interest of all users of the environmental
health criteria documents, readers are kindly requested to communicate
any errors found to the Division of Environmental Health, World Health
Organization, 1211 Geneva 27, Switzerland, in order that they may be
included in corrigenda which will appear in subsequent volumes.
In addition, experts in any particular field dealt with in the
criteria documents are kindly requested to make available to the WHO
Secretariat any important published information that may have
inadvertently been omitted and which may change the evaluation of
health risks from exposure to the environmental agent under
examination, so that that information may be considered in the event
of updating and re-evaluating the conclusions contained in the
criteria documents.
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR LEAD
Geneva, 29 April-5 May 1975
Participants
Professor M Berlin, Department of Environmental Health, University of
Lund, Sweden
Professor A. David, Centre of Industrial Hygiene and Occupational
Diseases, Institute of Hygiene and Epidemiology, Prague,
Czechoslovakia (Vice-Chairman)
Dr. F. A. Fairweather, Division of Chemical Contamination of Food and
Environmental Pollution, Department of Health and Social Security,
London, England
Professor R. A. Goyer, Department of Pathology, University of Western
Ontario, London, Ontario, Canada (Chairman)
Dr. L. Graovac-Leposavic, Institute of Occupational and Radiological
Health, Belgrade, Yugoslavia
Dr. R. J. M. Horton, Environmental Protection Agency, Research
Triangle Park, NC, USA
Dr. C. H. Nordman, Institute of Occupational Health, Helsinki, Finland
(Rapporteur)
Dr. H. Sakabe, Department of Industrial Physiology, National Institute
of Industrial Hygiene, Kawasaki, Japan
Professor H. W. Schlipköter, Institute of Air Hygiene and Silicosis,
Düsseldorf, Federal Republic of Germany
Professor N. Ju. Tarasenko, First Moscow Medical Institute, Moscow,
USSR
Professor R. L. Zielhuis, Coronel Laboratory, Faculty of Medicine,
University of Amsterdam, Amsterdam, Netherlands
Representatives of other agencies
Dr. A. Berlin, Health Protection Directorate, Commission of the
European Communities, Centre Louvigny, Luxembourg
Professor R. Bourdon, International Union of Pure and Applied
Chemistry, Commission on Toxicology, Laboratoire de Biochimie-
Toxicologie, Centre Anti-Poison de l'Hôpital Fernand Widal, Paris,
France
Dr. D. Djordjevic, Occupational Safety and Health Branch,
International Labour Office, Geneva, Switzerland
Dr. R. Morf, International Union of Pure and Applied Chemistry,
Liaison Officer with WHO, 8311 Kyburg Zh, Switzerland
Secretariat
Professor Paul B. Hammond, Department of Environmental Health,
University of Cincinnati, The Kettering Laboratory, Cincinnati,
Ohio, USA (Temporary Adviser)
Dr. Y. Hasegawa, Medical Officer, Control of Environmental Pollution
and Hazards, Division of Environmental Health, World Health
Organization, Geneva, Switzerland
Dr. J. E. Korneev, Scientist, Control of Environmental Pollution and
Hazards, Division of Environmental Health, World Health
Organization, Geneva, Switzerland
Dr. V. Krichagin, Scientist, Promotion of Environmental Health, WHO
Regional Office for Europe, Copenhagen, Denmark
Dr. B. Marshall, Medical Officer, Occupational Health, Division of
Environmental Health, World Health Organization, Geneva.
Professor L. A. Timofievskaja, Institute of Occupational Health,
Moscow, USSR (Temporary Adviser)
Dr. V. B. Vouk, Chief, Control of Environmental Pollution and Hazards,
Division of Environmental Health, World Health Organization,
Geneva (Secretary)
List of abbreviations
ALA delta-aminolevulinic acid
ALA-U delta-aminolevulinic acid in urine
ALAD porphobilinogen synthase (EC 4.2.1.24), delta-aminolevulinate
dehydratase, delta-aminolevulinic acid dehydratase
ALAS delta-aminolevulinate synthase (EC 2.3.1.37), aminolevulinic
acid synthetase
CP coproporphyrins
CP-U coproporphyrin in urine
CPG coproporphyrinogen III
EDTA ethylenediaminetetraacetic acid
FEP free erythrocyte porphyrins
Hb haemoglobin
LD50 median lethal dose
PP protoporphyrin IX
PBG porphobilinogen
Pb-B lead in blood
Pb-U lead in urine
RBC red blood cells
SGOT aspartate aminotransferase (EC 2.6.1.1), serum glutamic
oxaloacetic transaminase
ENVIRONMENTAL HEALTH CRITERIA FOR LEAD
A WHO Task Group on Environmental Health Criteria for Lead met in
Geneva from 29 April to 5 May 1975. Dr B. H. Dieterich, Director,
Division of Environmental Health, opened the meeting on behalf of the
Director-General. The Task Group reviewed and revised the second draft
criteria document and made an evaluation of the health risks from
exposure to lead and its compounds.
The first and second drafts were prepared by Professor Paul B.
Hammond of the Department of Environmental Health, The Kettering
Laboratory, University of Cincinnati, Ohio, USA. The comments on which
the second draft was based were received from the national focal
points for the WHO Environmental Health Criteria Programme in
Bulgaria, Czechoslovakia, Federal Republic of Germany, Greece, Japan,
The Netherlands, New Zealand, Poland, Sweden, USA, and the USSR, and
from the United Nations Educational, Scientific and Cultural
Organization (UNESCO), Paris, from the United Nations Industrial
Development Organization (UNIDO), Vienna, from the Centro Panamericano
de Ingenieria Sanitaria y Ciencias del Ambiente (CEPIS) at Lima, Peru,
and from the Health Protection Directorate of the Commission of the
European Communities (CEC), Luxembourg. Comments were also received,
at the request of the Secretariat from: Professor R. Goyer and
Professor H. Warren, Canada; Professor J. Teisinger, Czechoslovakia;
Dr S. Hernberg, Finland; Dr K. Cramer and Dr B. Haeger-Aronsen,
Sweden; Dr D. Barltrop, Professor B. Clayton, Professor R. Lane, and
Professor P. J. Lawther, United Kingdom; Dr J. J. Chisholm, Professor
H. L. Margulis, and Dr G. Ter Haar, United States of America; and Dr
D. Djuric and Professor K. Kostial, Yugoslavia.
Valuable comments were received on the third draft, resulting from
the task group, from: Mr Joseph E. Faggan, Director of Petroleum
Chemicals Research, Ethyl Corporation, Ferndale, Michigan, USA, and
from Mr R. L. Stubbs, Director-General, Lead Development Association,
London and Chairman, Statistical Committee, International Lead and
Zinc Study Group.
The collaboration of these national institutions, international
organizations, WHO collaborating centres, and individual experts is
gratefully acknowledged. Without their assistance this document would
not have been completed. The Secretariat wishes to thank, in
particular, Professor Hammond for his continued help in all phases of
the preparation of the document, and Dr H. Nordman of the Institute of
Occupational Health, Helsinki, who assisted the Secretariat in the
final scientific editing of the document.
This document is based primarily on original publications listed
in the reference section. However, several recent publications broadly
reviewing health aspects of lead and its compounds have also been
used. These include publications by Kehoe (1961), NAS-NRC (1972), NRC-
Canada (1973), Goyer & Rhyne (1973), WHO Working Group (1973), Inter-
Department Working Group on Heavy Metals (1974), SCEP (1974),
Nordberg, ed. (1976). In addition, the document draws on comprehensive
and useful data from the proceedings of several symposia and meetings,
e.g. the "International Symposium on Environmental Aspects of Lead",
Amsterdam, 1972, arranged by the Commission of the European
Communities and the US Environmental Protection Agency; the
"International Symposium on Recent Advances in the Assessment of the
Health Effects of Environmental Pollution", Paris, 1974, jointly
organized by the Commission of the European Communities, US
Environmental Protection Agency, and the World Health Organization;
the University of Missouri's Annual Conferences on Trace Substances in
Environmental Health, Columbia, Missouri, 1967-1975; and the
"International Symposium on Environmental Lead Research", Dubrovnik,
1975, organized by the Institute for Medical Research and Occupational
Health, under the auspices of the Yugoslav Academy of Sciences and
Arts.
Details of the WHO Environmental Health Criteria Programme,
including some of the terms frequently used in the documents, may be
found in the introduction to the publication "Environmental Health
Criteria 1-Mercury", published by the World Health Organization,
Geneva, in 1976.
1. SUMMARY AND RECOMMENDATIONS FOR FURTHER RESEARCH
1.1 Summary
1.1.1 Analytical problems
The procurement of environmental and biological samples requires
careful consideration of the special problems relating to the
particular material to be analysed. In air sampling, it is most
important to ensure that the sampler is placed at the breathing zone
of the population group under study. For all sampling procedures and
particularly for blood, external contamination is a major problem.
The most successful analytical method in recent years has been
atomic absorption spectroscopy. It has proved to be versatile and
sufficiently sensitive for most purposes, but reliable results,
particularly for biological specimens such as blood, can be obtained
only after considerable experience has been acquired.
Determinations of haem intermediates and of porphobilinogen
synthase (EC 4.2.1.24) (ALAD)a,b activity in blood are important
methods for estimating the biological consequences of overexposure to
lead. There is a great need for standardization of both these methods
and of ways of expressing the results.
1.1.2 Sources and pathways of exposure
The major sources of lead in the environment that are of
significance for the health of man, arise from the industrial and
other technological uses of lead. The major dispersive non-recoverable
use of lead is in the manufacture and application of alkyllead fuel
additives. Because of current legislative actions with respect to the
maximum permissible concentration of lead in gasoline, the consumption
of lead for the production of alkyllead additives decreased from 1973
to 1975 and a further decline for the latter half of the 1970s may
occur as more cars equipped with catalysts which require lead-free
gasoline will come into use.
a "In the first instance, enzymes are named according to the 1972
recommendations of the Commission on Enzyme Nomenclature but
throughout the rest of the document the more familiar names or
abbreviations are used.
b Formerly known as delta-aminolevulinate dehydratase or
delta-aminolevulinic acid dehydratase.
From a mass balance point of view, the transport and distribution
of lead from stationary or mobile sources is mainly via air.
Although large amounts are probably also discharged into soil and
water, lead tends to localize near the points of such discharge. Lead
that is discharged into the air over areas of high traffic density
falls out mainly within the immediate metropolitan zone. The fraction
that remains airborne (about 20%, based on very limited data) is
widely dispersed. Residence time for these small particles is of the
order of days and is influenced by rainfall. In spite of widespread
dispersion, with consequent dilution, there is evidence of lead
accumulation at points extremely remote from human activity, e.g. in
glacial strata in Greenland.
The biota acquires lead both by surface deposition and by
secondary transfer from soil to plants and from plants to animals.
However, the impact of man-made lead pollution on the lead content of
plants and animals is not perceptible except in localized areas of
intense air pollution, e.g. around smelters and in the immediate
vicinity of roads with heavy traffic.
The concentration of lead in air varies from 2-4 µg/m3 in large
cities with dense automobile traffic to less than 0.2 µg/m3 in most
suburban areas and still less in rural areas. The concentration of
lead in drinking water is generally less than 10 µg/litre, but in some
areas where the water is soft (low in calcium and magnesium) and
where, at the same time, lead pipes and lead-lined water storage tanks
are used, the concentration may reach 2000-3000 µg/litre. At this
concentration (and even at concentrations of several hundred µg/litre)
a perceptible rise in the body burden of lead occurs, which is
reflected in elevated values of lead in the blood (Pb-B).
The contribution of food to man's exposure to lead is highly
variable. Some recent studies in the USA have estimated the daily oral
intake in food and beverages to be about 100 µg whereas earlier
studies and some recent European studies indicated the intake to be in
the range of 200-500 µg/day. However, a recent Swedish study reported
volumes of the order of 20 µg/day. No specific category of food has
been identified as being especially high in lead content other than
wine and foods that are stored in lead-soldered cans or lead-glazed
pottery. Processed milk contains considerably more lead than fresh
cow's milk which has a similar concentration to human milk. The
reported lead concentrations range from less than 5 µg/litre to
12 µ/litre. If this information is correct, milk could be a
significant source of lead for infants.
Various miscellaneous sources of lead have been identified as
being highly hazardous. These include lead-glazed ceramics used for
beverage storage, illicitly-distilled whisky, and discarded automobile
battery casings when used for fuel.
In certain countries, gross overexposure of some infants and young
children has been recorded. The major sources are lead-based paint in
old houses and in the soil surrounding these homes, and the soil
surrounding lead smelters. Lead in street dust due to atmospheric
fallout, and miscellaneous lead-containing objects chewed or eaten by
children are other possible sources of exposure, but their relative
importance is not clear.
The highest exposure occurs in workers who come into contact with
lead during mining, smelting, and various manufacturing processes
where lead is used. The major pathway of exposure is inhalation. The
concentration of air lead in the working environment of smelters and
storage battery factories often exceeds 1000 µg/m3. For other
industries, data are either not available or indicate a lower level of
exposure.
Extensive surveys have been made on blood concentrations in both
adults and young children. Such data are useful indicators of overall
exposure to lead.
1.1.3 Metabolism
A number of studies have been made which indicate that 35% of the
lead inhaled by man is deposited in the lungs. The relative importance
of the mucociliary escalator mechanism and of direct absorption from
pulmonary deposition is poorly understood and the contribution of
airborne lead to total daily intake cannot be estimated from metabolic
data. But when sustained Pb-B is used as a measure of lead absorption,
it can be assumed from human data that continuous exposure to 1 µg of
lead per m3 of air would contribute lead levels of about
1.0-2.0 µg/100 ml of blood.
About 10% of lead taken in from food and beverages is absorbed.
However, using data from several sources, the dietary contribution to
Pb-B can only be roughly estimated as 6-18 µg of lead per 100 ml of
blood per 100 µg of dietary lead intake.
From both animal and human studies, the general features of lead
distribution and excretion are fairly clearly defined. The body burden
of lead can be subdivided into a large, slow-turnover compartment and
a smaller more rapidly-exchanging compartment. Anatomically, the
larger compartment is mainly located in bones. The amount of lead in
this compartment increases throughout life. The smaller compartment
consists of the soft tissues and includes the blood. Lead levels in
soft tissues and in blood continue to increase up to early adulthood
and then change little. Elimination of lead from the body is mainly by
way of the urine (about 76%) and the gastrointestinal tract (about
16%). The other 8% is excreted by miscellaneous routes (sweat,
exfoliation of the skin, loss of hair) about which little is known.
Alkyllead compounds (tetraethyllead and tetramethyllead) are
dealkylated both to trialkyl derivatives and to inorganic lead.
Details of alkyllead metabolism have been learned from animal studies
and have not been defined in man.
1.1.4 Experimental studies on the effects of lead
The extensive animal studies that have been conducted concerning
the biological effects of lead indicate that, with rare exceptions,
the toxic phenomena that have been observed in man have also been
successfully reproduced in animals. Although animal studies have
provided a more profound understanding of the effects of lead than
could be learned from studies of man himself, they have not been of
much use in the elucidation of dose-effect and dose-response
relationships in man.
Major differences that have been noted are as follows: (1) benign
and malignant tumour induction has occurred in rats and mice exposed
to lead acetate and in rats exposed to lead subacetate and lead
phosphate but carcinogenic effects have not been seen in man; (2)
clear-cut reductions in fertility have been observed in experimental
animals but not in man, although data have been reported which suggest
that this might be so; (3) hyperactivity and other behaviourial
disturbances have been observed in rats, mice, and sheep without prior
encephalopathy. This is especially important because of current
suspicions that widespread, slight brain damage occurs in young
children with relatively low exposure not preceded by encephalopathy.
Evidence also exists for compensatory increases in ALAD in animals
with continuing exposure to lead whereas all human studies to date
have been negative in this respect.
1.1.5 Clinical and epidemiological studies on the effects of lead:
Evaluation of health risk to man from exposure to lead
Studies of the effects of lead on man may be divided into two
general types. The first type is the retrospective study of the causes
of mortality in lead-exposed populations in contrast with those in
matched control groups. Several studies showed that at high exposure
levels (Pb-B>80 µg/ 100 mla), a slightly higher number of deaths
occurred due to cerebrovascular disease and chronic nephritis. In one
study, where the mortality rate due to cancer was observed, no
statistically significant differences were found between the
industrially exposed workers and the control group.
a In this document, the concentrations of lead in blood are
expressed in µg/100 ml although in some original papers the values
are given in µg/100 g. For practical purposes, the difference of
about 5% can be neglected.
The second type of study concerns morbidity rates due to the
effects of lead on specific organs and systems. In some cases, it has
been possible to estimate the level of the exchangeable body burden
(expressed as Pb-B) at which a given intensity of effect (dose-
response relationship) has been observed in certain sections of a
selected group. For other effects it has only been possible to specify
the Pb-B level at which no effect was observed in reasonably large
groups of people (no-detected-effect level).
The haematopoietic system shows effects at lower Pb-B levels than
any other system. The effects are, in order of sensitivity: inhibition
of erythrocyte ALAD, elevation of erythrocyte protoporphyrin IX (FEP),
rise in urinary delta-aminolevulinic acid (ALA) and coproporphyrin
(CP) excretion, inhibition of erythrocyte sodium-potassium adenosine
triphosphatase (EC 3.6.1.3) (Na-K-ATP'ase), and fall in haemoglobin
level. A fall in haemoglobin level is clearly an indication of adverse
effects. The no-detected-effect level for this effect is a Pb-B
concentration equivalent to 50 µg/100 ml in adults and 40 µg/100 ml in
children.
The effects of inorganic lead on the central nervous system have
been under intensive investigation in recent years, particularly with
regard to subtle effects on behaviour, mainly in children, but also to
some extent in adults. Substantial doubts remain as to the validity of
some of the studies because the relationship between the exposure to
lead at the time the damage occurs and at the time the effects are
first observed is not known. Nevertheless, a no-detected-effect level
has been specified that is lower than for classical lead
encephalopathy. The no-detected-effect level is estimated to be at
Pb-B values of about 60-70 µg/100 ml for adults and of about
50-60 µg/100 ml for children.
The renal effects of lead are of two general types. The first is
tubular, characterized by the Fanconi triad of aminoaciduria,
hyperphosphaturia, and glycosuria. It occurs with relatively short-
term exposure and is reversible. The second type of renal effect is
characterized anatomically by sclerotic changes and interstitial
fibrosis. Functionally, filtration capacity is reduced. These changes
are of a progressive nature and may lead to renal failure. It is
probable that exposures leading to this type of nephropathy are rarely
encountered even in industry today. A no-detected-effect level cannot
be specified.
The problem of the toxic effects of alkyllead is almost entirely
restricted to workers who are occupationally exposed. There is very
little information concerning dose-effect and dose-response
relationships and even the frequency of occurrence of toxic effects
and their relation to specific work activities is not well documented.
1.2 Recommendations for Further Research
1.2.1 Analytical methods
One of the major needs is for the standardization of analytical
methods, particularly with regard to the haem intermediates, ALAD, and
erythrocyte Na-K-ATP'ase. At the present time, it is often impossible
to compare studies conducted in one laboratory with those of another.
This is particularly true for enzymatic methods that give different
results depending on pH, oxygen tension, and the presence or absence
of other factors, e.g. other metals that can influence the action of
lead. It is of equal importance that a standard mode of expressing
results be introduced in order to achieve valid interlaboratory
comparisons. Thus, measurements involving urine should be expressed
per unit of creatinine excreted per unit time; this would probably
take body mass into consideration.
In view of the highly variable results that have been obtained in
the interlaboratory comparisons conducted to date, more cooperative
efforts should be undertaken and maintained on a continuous basis. It
is recommended that all published data include interlaboratory
comparison results for the methods used. International standard
specimens of the commonly investigated biological media with reliably
determined concentrations of lead should be developed and made
available to investigators.
Finally, standardized methods of statistical treatment of
analytical data should be adopted and adhered to.
1.2.2 Sources of lead intake
It is apparent that the estimations of lead in the diet of man
vary greatly. Future studies should include specifications concerning
the characteristics of the individuals for whom lead consumption data
are being reported, including sex, age, weight, and physical activity.
Since the ultimate purpose of food studies is to evaluate the
contribution made to the total dose, it is important that future
reports also include the observed Pb-B levels and, preferably, other
indices, such as delta-aminolevulinic acid in urine (ALA-U), PP and
ALAD in erythrocytes. Food studies should also include estimates of
the lead concentration of various components of the total diet. Only
with such studies will it be possible to arrive at decisions regarding
the control of lead in foods.
More precise information is available concerning the contribution
of airborne lead to Pb-B and although this seems to be a minor
contributor to Pb-B for the general population compared with diet,
additional studies are needed both in occupational situations, and for
the general population. The studies should be of a relatively long-
term nature and should be done, as far as possible, with personal air
samplers maintained in operation continuously throughout the day
during the period of study.
There is a great need to study the sources of lead affecting
infants and young children including the contributions of food, milk
and other beverages, and air, and also miscellaneous sources, e.g.
paint, soil, and dust.
1.2.3 Epidemiological studies
Prospective studies are needed of the health effects of both
inorganic and organolead compounds, with particular reference to a
more thorough estimation of the nature of the lead exposure, Pb-B
levels, and measurable effects. It would seem particularly useful to
make further studies on occupational groups, beginning at the time of
their entry into the high lead environment.
1.2.4 Interactions of lead with other environmental factors
In both epidemiological studies and in experimental studies on
animals, not enough emphasis has been placed on the environmental
variables that can affect man's response to lead. The list of such
variables is long and is documented in this report. Particular
attention should be paid to the influence of other metals, air
pollutants, and the nutritional status of the subjects, since these
factors have been identified as interacting with lead either in regard
to its deposition in the body or in regard to its biological effects
in target organs.
1.2.5 Significance of biological effects
Numerous abnormalities have been identified, the toxic
significance of which is obscure, e.g. elevated free erythrocyte PP
and marginal erythrocyte ALAD inhibition. There is an urgent need to
study the significance of these findings in relation to human health.
2. PROPERTIES AND ANALYTICAL METHODS
2.1 Physical and Chemical Properties of Lead and its Compounds
Lead (atomic number, 82; atomic weight, 207.19; specific gravity,
11.34) is a bluish or silvery grey soft metal. The melting point is
327.5°C and the boiling point at atmospheric pressure 1740°C. It has
four naturally occurring isotopes (208, 206, 207, and 204 in order of
abundance), but the isotopic ratios for various mineral sources are
sometimes substantially different. This property has been used to
carry out non-radioactive-tracer environmental and metabolic studies.
Although lead has four electrons in its valence shell, only two
ionize readily. The usual oxidation state of lead in inorganic
compounds is therefore + 2 rather than + 4. The inorganic salts of
lead (II), lead sulfide, and the oxides of lead are generally poorly
soluble. Exceptions are the nitrate, the chlorate and, to a much
lesser degree, the chloride (Table 1). Some of the salts formed with
organic acids, e.g. lead oxalate, are also insoluble.
Under appropriate conditions of synthesis, stable compounds are
formed in which lead is directly bound to a carbon atom.
Tetraethyllead and tetramethyllead are well-known organolead
compounds. They are of great importance owing to their extensive use
as fuel additives. Both are colourless liquids. Their volatility is
lower than for most gasoline components. The boiling point of
tetramethyllead is 110°C and that of tetraethyllead is 200°C. By
contrast, the boiling point range for gasoline hydrocarbons is
20-200°C. Thus evaporation of gasoline tends to concentrate
tetraethyllead and tetramethyllead in the liquid residue.
Both tetramethyllead and tetraethyllead decompose at, or somewhat
below, the boiling point. Analysis of automobile exhaust gases shows
that the ratio of tetramethyllead to tetraethyllead increases as the
engine warms up, indicating that tetramethyllead is more thermostable
than tetraethyl-lead (Laveskog, 1971). These compounds are also
decomposed by ultraviolet light and trace chemicals in air such as
halogens, acids, or oxidizing agents (Snyder, 1967).
2.2 Analytical Procedures
2.2.1 Sampling
Particular attention should be paid to the cleanliness of the
instruments and the purity of chemicals to prevent the appearance of
artifacts due to the secondary contamination by lead, especially in
the sampling of foods and biological media.
Table 1. Some physical and chemical data on lead and selected lead compoundsa
Solubility
Name Synonym and Molecular Melting Boiling in cold Soluble in
formula weight point (°C) point (°C) water (g/litre)
lead Pb 207.19 327.502 1740 insoluble HNO3; hot concentrated H2SO4
acetate Pb(C2H3O2)2 325.28 280 -- 443 hot water; glycerine; alcohol
(slightly)
azide Pb(N3)2 291.23 explodes 350 0.23 acetic acid; hot water
(0.9 g/litre)
carbonate cerrusite PbCO3 267.20 315 (decomposes) 0.0011 acid; alkali; decomposes in
hot water
chlorate Pb(ClO3)2 374.09 230 (decomposes) very soluble alcohol
chloride cotunite PbCl2 278.10 501 950 9.9 NH4 salts; slightly in dilute
HCl and in NH3; hot water
(33.4 g/litre)
chromate crocoite, chrome 328.18 844 decomposes 0.000058 alcohol; alkali
yellow PbCrO4
nitrate Pb(NO3)2 331.20 470 (decomposes) 376.5 alcohol; alkali; NH3; hot water
(1270 g/litre)
ortophosphate Pb3(PO4)2 811.51 1014 0.00014 alkali; HNO3
oxalete PbC2O4 295.21 300 (decomposes) 0.0016 HNO3
oxide: di- plattnerite PbO2 239.19 290 (decomposes) insoluble dilute HCl; acetic acid (slightly)
mono- litharge PbO 223.19 888 0.017 HNO3; alkali; NH4Cl
red mioium Pb3O4 685.57 500 (decomposes) insoluble HCl; acetic acid
sesqui- Pb2O3 462.38 370 (decomposes) insoluble decomposes in acid and hot water
stearate Pb(C18H35O2)2 774.15 115.7 0.5 hot water (0.6 g/litre); ether
(0.05 g/litre)
sulfate anglesite PbSO4 303.25 1170 0.0425 NH4 salts; concentrated H2SO4
(slightly)
sulfide galena PbS 239.25 1114 0.00086 acid
Table 1. (Cont'd)
Solubility
Name Synonym and Molecular Melting Boiling in cold Soluble in
formula weight point (°C) point (°C) water (g/litre)
tetraethyllead Pb(C2H5)4 323.44 -136.80 200 insoluble benzene; petroleum; alcohol; ether
decomposes;
91
tetramethyllead Pb(CH3)4 267.3 -27.5 110 insoluble benzene; petroleum; alcohol; ether
a Adapted from Weast, R. C., ed. Handbook of Chemistry and Physics, 55th edition, Cleveland, Ohio, Chemical Rubber Company, 1974.
In air sampling, high-volume samplers are preferable for accuracy
(when it is necessary), but the low-volume technique is also useful
for obtaining extensive data. As in all sampling for suspended
particulate matter, the accuracy of volume meters should be checked
periodically. The size of the pores of filters for collecting lead-
containing particles should be small, possibly less than 0.2 µm for
glass-fibre filters (Lee & Goransen, 1972). Liquid scrubbers
containing iodine monochloride and solid scrubbers with activated
carbon, cristobalite, or iodine crystals have been used for sampling
organic lead compounds in air, in the range of about 1 µg/m3 or less
(Snyder, 1967; ASTM, 1970; Laveskog, 1971; Coleville & Hickman, 1973;
Purdue et al., 1973) up to 10 µg/m3 (Harrison et al, 1974).
Depending on the purpose of sampling, care should be taken to
select the appropriate site for sampling devices and to achieve the
best possible sampling conditions by:
-- estimating the required amount of particulates before deciding on
the sample volume and the sampling procedure;
-- placing the sampling devices in the appropriate position (e.g.
breathing air level, level of inlet tubes of house ventilators,
window level in the case of a traffic-laden town street, at a
reasonable distance from the highway in uninhabited zones, etc),
-- taking the samples at appropriate rates and volumes (e.g. daily
breathing volumes, daily ventilating capacities of installations)
and for a sufficient time to make possible the estimation of the
average concentration (e.g. during a work shift, or a 24-hour or
longer period for general population exposure);
-- taking into account the use of appropriate areas (cattle grazing,
recreational zones, children's playgrounds etc.)
In addition, whenever possible, a procedure should be used that
makes it possible to evaluate particle-size distribution and the
physico-chemical properties of the lead compounds involved, including
the shape of the particles and the state of their aggregation.
Stationary samplers can provide general indices of the exposure of
individuals within a certain area. For estimating exposure through
inhalation, personal samplers are highly desirable (Azar et al.,
1973).
Techniques for sampling water are less complex than for air. The
major question is whether or not the water should be filtered before
analysis since it is known that lead occurs in water both in the
particulate fraction and in solution. For most purposes at least, it
is reasonable to sample water without any fractionation of the
material collected.
However, in some cases it may be necessary to determine the
biological availability for absorption of the various forms of lead
that occur in water, and in soil. The latter is a dust source and may
be a food contamination source as well.
The preparation of soil and soil dust samples for lead analysis
usually involves drying (at 100°c), homogenization by grinding, and
sieving (Thornton & Webb, 1975; Bolter et al., 1975).
For the study of lead in foods, two general methods have been
used. These are the duplicate portions technique and the equivalent
composite technique (theoretical diet). These two general techniques
and others have been reviewed recently with reference to their
advantages and disadvantages (Pekkarinen, 1970). The duplicate
portions technique involves the collection for analysis of duplicates
of the meals actually consumed by the individual. When carried out
over a long enough period, the technique has the advantage of defining
variability in consumption. Kehoe (1961) used this method for the
daily determination of lead consumption over long periods.
Considerable variation in lead consumption was found in individuals
even when consumption was averaged for four- or eight-week collection
periods. The disadvantages of the method are the expense and the
exacting nature of the method of collecting samples; these factors
tend to limit the numbers of individuals included in such studies.
The equivalent composite technique consists of formulating the
ingredients of meals typical for subpopulations and analysing them.
The advantages are economy and ease of collection. This approach may
or may not include the cooking process. The disadvantage is
uncertainty as to how typical or representative the formulation is.
Even when the cooking process is included, there may be significant
differences in the manner of preparation for the study in comparison
with that carried out under actual home conditions.
The main problem in the sampling of body fluids and tissues for
lead analysis is potential secondary contamination with lead. Special
precautions must be taken to ensure that all blood-collecting and
blood-storage materials are as free from lead as possible. All glass
equipment involved in blood collection and storage should be made of
lead-free silicate glass, rinsed first in mineral acid, then with
copious amounts of glass-distilled or deionized water. Polypropylene
syringes have been recommended (NAS-NRC, 1972). Needles should be of
stainless steel with polypropylene hubs. Blood is often drawn directly
from the needle into vacuum tubes. It is wise to confirm periodically
the absence of significant amounts of lead in the anticoagulant used
in the blood container, although this has not been reported as a
problem.
New analytical techniques make it possible to determine lead
concentrations in microlitre quantities of blood. The trend towards
the procurement of micro-samples of blood by skin prick increases the
hazard of secondary contamination of the blood. Only one systematic
investigation on the significance of this problem has been reported.
Mitchell et al. (1974) describe a procedure whereby sample
contamination appears to be avoided. This is achieved by spraying
collodion over the cleansed skin before lancing. The correlation
between the concentration of lead in micro-samples and in macro-
samples obtained by venipuncture was fairly good (r = 0.92). The same
general precautions must be taken in the collection of urine samples
as in the collection of blood samples.
Ceramic surfaces are analysed to determine the quantity of lead
likely to be leached by different foods and beverages. In all cases
acetic acid solutions are used but the concentrations vary from 1 to
4%. The temperature of the tests ranges from 20 to 100°C and the
duration from 30 minutes to more than 24 hours (Laurs, 1976; Merwin,
1976).
2.2.2 Analytical methods for lead
The analytical methods currently in use for the estimation of lead
content are of two general types, destructive and non-destructive. In
the former, the sample is first oxidized to destroy all organic
matter. The ash is then usually dissolved in an aqueous medium, either
for further preparative steps or for direct instrumental analysis.
Non-destructive methods are of more recent origin and are still too
complicated for routine studies. They include X-ray fluorescence
analysis and fast neutron activation. In selecting methods,
consideration must be given to the cost of the equipment and the time
involved in performing the analyses.
The oldest and best known of the general methods currently in wide
use are those based on the formation of the red complex that lead
forms with dithizone (diphenylthiocarbazone). Numerous specific
procedures have been developed based on the spectrophotometric
determination of lead dithizonate. A typical example is the "US Public
Health Service" method commonly used for the determination of lead in
biological materials (NAS-NRC, 1972). The method has evolved over many
years. A study of its reliability was reported by Keenan et al.
(1963). An interlaboratory comparison was made of analyses of blood
and urine with and without the addition of lead. Ten laboratories
participated in the study. For blood, the concentration of lead
calculated in the principal laboratory was 20 µg/100 ml. The average
reported by the participating laboratories was 26 µg/100 ml with a
standard deviation of ± 0.82 µg/100 ml. For samples of blood to which
lead was added, the average result was right on the mark,
70 µg/100 ml ± 0.78. For "spiked" urine, determined by the primary
laboratory to contain 750 µg/litre, the average reported result was
679 ± 5.5 µg/litre.
Perhaps no method of instrumental analysis for lead has enjoyed
such a rapid acceptance in recent years as atomic absorption
spectroscopy. In conventional atomic absorption spectroscopy, the
source of heat is a flame into which the sample solution is aspirated.
More recently, various procedures have been developed whereby the
receptacle containing the sample is heated electrically. This type of
modified procedure is termed flameless atomic absorption spectroscopy.
The main advantage of this approach is that sample size is reduced
from the millilitre to the microlitre range with no commensurate loss
of sensitivity. Another advantage is that the heated receptacle can be
used for ashing the sample immediately prior to the spectrophotometric
analysis. Numerous reports have appeared describing various kinds of
flameless instrumentation and their application in the analysis of the
lead content of blood and other materials (Cernik, 1974; Delves, 1970;
Ediger & Coleman, 1973; Matousek & Stevens, 1971; Kubasik et al.,
1972; Hwang et al., 1971; Sansoni et al., 1973; Schramel, 1973;
Schramel, 1974). It has been reported that the analytical capabilities
of this method for determining lead in whole blood are comparable with
that of the conventional flame atomic absorption method (Kubasik et
al., 1972; Hicks et al., 1973).
Electroanalytical methods have also been found useful for lead
determinations. These include polarography and, more recently, anodic
stripping voltametry. The polarographic method was developed
specifically for lead by Teisinger (1935). The low sensitivity of the
method as applied to lead in blood and urine required working close to
the detection limits. This is obviously a disadvantage when
determining the normal levels of lead in blood and urine. Various
modifications of the original method have been used for the evaluation
of industrial exposures (Weber, 1947; Baker, 1950; Brezina & Zuman,
1958). This method found wide application until more effective masking
procedures were developed to increase the specificity of the dithizone
method. Anodic stripping voltametry is gaining in popularity for lead
analysis. Results have been compared using a dithizone method, an
atomic absorption method, and anodic stripping voltametry (Matson,
1971). Generally, there was good agreement between all three methods
in the estimation of the lead contents of blood and urine. In another
study, anodic stripping voltametry was compared with atomic absorption
spectroscopy and polarography for the analysis of lead in blood and
urine (Horiuchi et al., 1968). The authors concluded that there were
no significant differences between the results obtained by the various
methods. Anodic stripping voltametry has also been compared with
conventional and flameless atomic absorption spectroscopy and with
potentiometric determination using ion-specific electrodes to estimate
the lead content of water (Kempf & Sonnenborn, 1973).
Two non-destructive methods for lead analysis have been under
investigation in recent years. These are neutron activation and X-ray
fluorescence. The first of these is not likely to find wide
application for lead analysis in the near future because of the cost
and the need for access to a fast neutron source. Its advantage is
that the concentration of many elements can be determined
simultaneously.
X-ray fluorescence is also theoretically capable of detecting,
non-destructively, all elements in a substance. A major obstacle to
the wide application of this method is the profound matrix effect of
the substances being analysed. Another problem is the backscatter from
the exciting source. These design problems and approaches to their
solution have been discussed recently by Kneip & Laurer (1972). Lead
analysis by means of X-ray fluorescence with proton excitation has
been successfully used with biological samples (Möller et al., 1974).
It has also been used as the standard method for the determination of
lead on filters from air sampling equipment by the Warren Springs
Laboratory in the United Kingdom. In the USA, the most extensive
application of X-ray fluorescence for lead analysis has been for
estimating the concentration and amount of lead on the walls of
houses. For this purpose, several portable units have been designed
and are being used in surveys of dwellings for hazardous
concentrations of lead. Since the instruments in question scan
surfaces, instrument response is in terms of lead detected per unit
area and not per unit weight or volume of paint film. This creates
difficulties, since the thickness of the total paint film varies
depending on how many times a surface has been painted. Ordinances
should perhaps be revised to specify tolerances based on surface area.
The accuracy of these instruments is severely limited. These factors
have been studied using one of the commercially available instruments
(Spurgeon, 1973). In another report from the US National Bureau of
Standards (Rasberry, 1973), four commercial instruments were tested as
received from the manufacturer. It was found that all the instruments
had a detection limit below 1 mg/cm2, but that between 1 and
6.6 mg/cm2, errors as large as 30-50% occurred. It is difficult to
evaluate the adequacy of such instruments since it is not at all clear
where the cut-off is between hazardous and non-hazardous amounts of
lead per unit area of paint film. Thus, if the cut-off were known to
be at or above 1 mg/cm2, the instruments would clearly be useful.
The accuracy and precision of various methods for the lead
analysis of biological materials have been appraised in a number of
interlaboratory comparison programmes both at the national (Keppler et
al., 1970; Donovan et al., 1971) and international levels (Berlin et
al., 1973). In general, these published studies have indicated that
the accuracy of the measurements is unsatisfactory, with less than
half of the laboratories performing adequately. More recently, in a
programme involving sixty-six European laboratories, it was observed
that even when only the laboratories that measured lead in blood and
urine with a precision of greater than 10% were selected, the
interlaboratory variability still remained high. It is possible that
the performance could be improved by rapid distribution of the sample
and by improved sample preparation techniques, e.g. by subjecting
blood samples to ultrasonic irradiation prior to despatch to
participating laboratories.
The paper punch disc microtechnique (Cernik & Sayers, 1971;
Cernik, 1974) was used in a population survey of blood lead content
performed in Western Ireland (Grimes et al., 1975). Over 400 duplicate
samples were analysed double-blind by one laboratory. The assay showed
a satisfactory agreement with the results obtained by other
laboratories using various techniques.
Comparisons have also been reported of the agreement between
results obtained by the same investigator using different analytical
methods. Yeager et al. (1971) compared the results obtained using a
standard dithizone procedure and flame atomic absorption spectroscopy.
The results from common digests of the same material were compared.
The materials included blood, urine, tissue, faeces, food, and bone.
Since the two methods are based on entirely different analytical
principles, a straight line with a slope equal to 1 and an intercept
equal to 0, obtained when the results of atomic absorption
spectroscopy analyses were plotted against the results of the
dithizone method, suggested that the two methods were equally
accurate.
These studies show that blood sample preparation is important to
ensure sufficient homogeneity for microanalytical techniques.
2.2.3 Methods for the measurement of some biochemical effects of lead
The classic method for the urinary delta-aminolevulinic acid (ALA)
determination was developed by Mauzerall & Granick (1956). The major
procedural difficulty was separation from interfering substances. A
number of modifications and simplifications have been made by several
authors (Davis & Andelman, 1967; Grabecki et al., 1967; Williams &
Few, 1967; Sun et al., 1969; Tomokumi & Ogata, 1972).
The original Mauzerall & Granick method does not discriminate
between ALA and aminoacetone, a fact that these authors were careful
to point out. This is probably not very important when ALA excretion
is greatly increased due to lead exposure, but for marginal
elevations, it may be a serious problem. In healthy humans on a normal
diet, the urinary excretion of ALA and that of aminoacetone are nearly
equal (Marver et al., 1966). These authors and also Urata & Granick
(1963) separated ALA from aminoacetone by chromatography.
One interlaboratory comparison study of ALA methods has been
reported (Berlin et al., 1973). The methods used by the laboratories
were those of Mauzerall & Granick (1956), Davis & Andelman (1967) and
of Grabecki et al. (1967). The results using the Grabecki method were
significantly higher than those using the Mauzerall & Granick method.
Results with the Davis & Andelman method gave a mean value
intermediate between the other two. The coefficients of variation were
quite high: 33%, Grabecki; 28%, Mauzerall & Granick; and 49%, Davis &
Andelman. It should also be noted that in the case of the Grabecki
method, the colorimetric reaction was influenced by various
interfering substances in the individual urine samples. This source of
error was not considered in the interlaboratory comparison (Mappes,
1972).
Comparisons have also been reported between these different
techniques by Roels et al. (1974) who evaluated the critical factors
in the urine preparation which affected the different methods. The
ionic strength and pH of the urine can affect the results of some of
the methods.
In the methods used for the determination of ALAD activity, the
amount of porphobilinogen (PBG) formed per unit time by a standard
amount of enzyme source is measured. Limited data indicate that ALAD
in blood is stable for several hours, even at room temperature
(Hernberg et al., 1970); however, storage at lower temperatures
improves the stability. The major variables reported to influence the
activity of the enzyme are pH (Nikkanen et al., 1972), oxygen tension
(Gibson et al., 1955), the nature of the anticoagulant (Collier,
1971), and the presence or absence of activators (Bonsignore et al.,
1965; Collier, 1971; Granick et al., 1973; Hapke & Prigge, 1973).
Measurement of ALAD activity in erythrocytes is a relatively simple
procedure that can be conducted without sophisticated equipment. This
makes it attractive as a measure of the haematological effects of
exposure to lead. A number of investigators have shown it to be fairly
specific for lead.
In its simplest and most frequently used form, the method of
Bonsignore et al. (1965) requires the incubation of a mixture of
blood, ALA, and water under aerobic conditions at 38°C. However, many
investigators have modified the procedure and results from different
laboratories are not necessarily comparable. In a recent
interlaboratory comparison (Berlin et al., 1973), nine participants
used various modifications of the Bonsignore method. Thus, it was only
possible to compare the activity ratios between different blood
samples. For two blood samples this ratio showed a coefficient of
variation of only 13%.
Recently a "European standardized method" has been developed,
tested in a collaborative study, and agreed upon by nineteen
laboratories. The results of these tests compare very favourably with
blood lead determinations. The interlaboratory coefficient of
variation for ALAD was 10% (Berlin et al., 1974).
Porphyrins exhibit intense fluorescence when excited by light at
approximately 400 nm (Soret band). They may be quantitatively
determined either by measurement of light absorption in the Soret band
region or by the measurement of fluorescence (Sassa et al., 1973;
Chisolm, 1974).
A number of methods have been reported for the measurement of
protoporphyrin IX. Some of these methods discriminate between
different porphyrins, measuring specifically the concentration of
protoporphyrin IX in erythrocytes (Schwartz & Wikoff, 1952; Wranne,
1960; Schlegel et al., 1972; Granick et al., 1972; Sassa et al.,
1973). Other methods measure the total concentration of free
erythrocyte porphyrins including copro- and uro-porphyrins (Kammholtz
et al., 1972; Piomelli, 1973; Schiele et al., 1974b). It is, however,
scarcely necessary to make a distinction between the two kinds of
procedure as over 90% of the free erythrocyte porphyrins are made up
of protoporphyrin IX (Baloh, 1974). A particular advantage of the more
recently developed procedures for the measurement of FEP is that they
can be performed on microcapillary samples of blood (Kammholz, 1972;
Granick et al., 1972; Sassa et al., 1973: Piomelli, 1973; Schiele et
al., 1974a). The Piomelli procedure utilizes two successive
extractions into ethylacetate-acetic acid with subsequent transfer of
porphyrins into hydrochloric acid. The procedure of Granick et al.
(1972) is simpler. Ethylacetate-acetic acid and hydrochloric acid are
successively added to the sample of blood. In both procedures the
ethylacetate serves to remove and retain interfering impurities in the
blood. The data obtained by these two methods are not strictly
comparable.
All the methods described measure protoporphyrin in the free base
form. Lamola & Yamane (1974) have recently demonstrated that the
protoporphyrin IX associated with iron deficiency and lead
intoxication is present as a zinc chelate. This is not so in the case
of erythropoietic porphyria. On the basis of these observations they
developed a fluorimetric method for zinc chelate (Lamola et al.,
1975). The major advantage of this method is its simplicity and
rapidity. Microlitre samples are analysed fluorimetrically, after
dilution, without any extraction steps.
The measurement of coproporphyrins in urine is generally done by
extraction of the porphyrins into either ethylacetate-acetic acid
(Sano & Rimington, 1963) or diethyl ether (Askevold, 1951) followed by
transfer into hydrochloric acid. Absorbance is then measured at 401 nm
with the corrections recommended by Rimington & Sveinsson (1950). The
method is apparently specific, since uroporphyrins, the most likely
source of interference, are not extracted into the organic phase under
these conditions (Rimington & Sveinsson, 1950). An alternative method
has been reported whereby the fluorescence of the hydrochloric acid
extract is measured after adsorption on to magnesium hydroxide
(Djuric, 1964). Certain precautions are necessary if urine is to be
analysed for coproporphyrins. Coproporphyrins are unstable in acid
urine and, furthermore they are light-sensitive (Schwartz et al.,
1951). They may be stored safely in the dark at 4°C if the pH is
maintained between 6.5 and 8.5.
3. SOURCES OF LEAD IN THE ENVIRONMENT
3.1 Natural Occurrence
3.1.1 Rocks
Lead occurs naturally in the earth's crust in the concentration of
about 13 mg/kg. As with all elements, there are some areas with much
higher concentrations including the lead ore deposits scattered
throughout the world.
The most important sources of lead are igneous and metamorphic
rocks, with lead concentrations in the range of 10-20 mg/kg (Wedepohl,
1956, Vinogradov, 1956, 1962; Turekian & Wedepohl, 1961). The
concentration of lead in sedimentary rocks is of the same order of
magnitude. The lead content of carbonaceous shales from the United
States of America and Europe ranges from 10mg/kg to 70mg/kg (Wedepohl,
1971; Davidson & Lakin, 1962). The lead contents of shale and
sandstone are similar but that of phosphate rocks is higher, and may
exceed 100 mg/kg (Sheldon et al., 1953). Unconsolidated sediments in
bodies of freshwater and in shallow marine areas have a similar lead
content to shales. Deep marine sediments have quite a high lead
content by comparison, commonly containing 100-200mg/kg (Riley &
Skirrow, 1965).
The lead content of coal is relatively low. However, when
expressed on an ash-weight basis, the concentration is generally
higher than that of igneous, metamorphic, and sedimentary rocks, but
not more than ten-fold (Abernethy et al., 1969).
3.1.2 Soils
Surface soils are in direct contact with the contemporary
environment; thus, special care must be taken to distinguish between
soils that acquire lead only from natural sources and soils that are
polluted by man. Acidic soils generally have a lower lead content than
alkaline soils. The nature of the organic matter in soil also has a
considerable influence on its lead content. Some organic matter is
rich in chelating components, and it binds lead, either promoting its
movement out of the soil or fixing the metal, depending on the
solubility properties of the complex. Although all of these factors no
doubt play a role in determining the lead content of specific soils,
the concentrations usually encountered in areas, remote from human
activity, are similar to concentrations found in rocks, with an
average range of 5-25 mg/kg (Swaine, 1955). More recent data from
various parts of the world have confirmed this estimate.
3.1.3 Water
Analyses of groundwater have revealed lead concentrations varying
from 1 to 60 µg/litre (Kehoe et al., 1933, 1944; Bagchi et al., 1940).
Most data refer to water that has been filtered to remove particulate
matter. Colloidal lead is only partially removed by filtration and to
different degrees. Water that is pumped from the ground is usually not
filtered prior to analysis. The content of colloidal material is
probably insignificant in such samples owing to natural filtration
which removes colloidal particles fairly effectively.
There have been a large number of investigations concerning the
concentration of lead in natural surface waters. From the data
available, Livingstone (1963) estimated that the global mean lead
content in lakes and rivers is 1-10 µg/litre. Although this estimate
includes man-made pollution, it probably still represents a fair
approximation of natural conditions since water flowing through the
ecosystems has a considerable self-cleaning capacity.
The concentration of lead in sea water has been found to be lower
than in freshwaters. Tatsumoto & Patterson (1963) report
0.08-0.4 µg/litre in seawaters off the coast of California. In deep
waters the concentration was even lower. According to Chow (1968)
surface waters off Bermuda, which are free from continental
influences, have lead concentrations averaging 0.07µg/litre, while
central Atlantic waters contain an average of 0.05 µg/litre. Although
there seem to be somewhat higher lead concentrations in the surface
waters of the Pacific and the Mediterranean, compared with the central
Atlantic, the concentrations at depths below the 1000-m level are very
similar, i.e. around 0.03-0.04 µg/litre (Chow, 1968).
3.1.4 Air
The atmospheric concentration of lead measured at points most
remote from civilization is of the order of 0.0001-0.001 µg/m3
(Jernigan et al., 1971; Chow et al., 1969; Egorov et al, 1970;
Murozumi et al., 1969). The sampling sites in these studies were
mainly over remote areas of oceans and over Greenland. Patterson
(1965) estimated from geochemical data that the concentration of lead
in air of natural origin is about 0.0006 µg/m3. If that is a correct
estimate, even the air over uninhabited, remote, continental areas may
be contaminated by human activities. For example, Chow et al. (1972)
reported that the concentration of lead in the air over remote,
uninhabited mountains of southern California had a concentration of
0.008 µg/m3.
3.1.5 Plants
Lead occurs naturally in all plants, as well as in soil, air, and
water. Extremely variable concentrations of lead in plants have been
reported but nevertheless, certain generalizations have been made.
Warren & Delavault (1962) have concluded that the normal concentration
of lead in leaves and twigs of woody plants is 2.5 mg/kg on a dry
weight basis. For vegetables and cereals they estimated normal
concentrations to be 0.1-1.0 mg/kg dry weight. Mitchell (1963) found
that the usual concentration of lead in pasture grasses was 1.0 mg/kg
dry weight. These figures should be multiplied by a factor of 20 to
convert concentration on a dry weight basis to an ash weight basis.
3.1.6 Environmental contamination from natural sources
The contribution of natural sources of lead to lead concentrations
in the environment is small. As regards exposure of man, these sources
are negligible. Through various breakdown processes, rocks yield lead
which is transferred to the biosphere and the atmosphere and
ultimately back to the earth's crust in the form of sedimentary rocks.
Soluble lead has for thousands of years entered the oceans with river
discharges, and the amount has been estimated by Patterson (1965) at
some 17 000 tonnes per year. Sources contributing to airborne lead are
silicate dusts, volcanic halogen aerosols, forest fires, sea salts
aerosol, meteoric and meteoritic smoke, and lead derived from the
decay of radon. The last mentioned source generates the lead isotope
210Pb in trace amounts, the mean air residence time of which has
been calculated to be about four weeks; the radioactive half-life is
22 years (Hill, 1960).
3.2 Production of Lead
3.2.1 Lead mining
Lead is produced from ores and recycled lead products. Lead occurs
in a variety of minerals the most important of which are galena (PbS),
cerrusite (PbCO3) and anglesite (PbSO4). Galena is by far the most
important source of primary lead. It occurs mostly in deposits
associated with other minerals, particularly those containing zinc.
Mixed lead and zinc ores account for about 70% of total primary lead
supplies. Ores containing mainly lead account for about 20% and the
remaining 10% is obtained as a by-product from other deposits, mainly
zinc and copper-zinc deposits (Federal Institute for Minerals Research
and German Institute for Economic Research, 1972). The proportions of
various metals may differ in the ores of different countries. Silver
is the most important of the other metals frequently present in lead
deposits but copper may also be present in concentrations high enough
to be commercially important. Other minor constituents of lead ores
are gold, bismuth, antimony, arsenic, cadmium, tin, gallium, thallium,
indium, germanium, and tellurium. The lead content of ores is
comparatively low, i.e. 3-8%, but even ores with lower lead contents
may be commercially valuable.
The level of world mine production of lead concentrates from ores
has increased in recent years. According to the International Lead and
Zinc Study Group and the World Bureau of Metal Statistics, the world
mine production of lead (lead content) was about 3.6 million tonnes in
1975, as compared with about 2.6 million tonnes in 1965. These figures
include production estimates for socialist countries with a planned
economy made by the World Bureau of Metal Statistics. The most
important lead mining countries, producing over 100 000 tonnes each in
1975, were Australia (10% of the total world output), Bulgaria (3%),
Canada (9.6%), China (3.8%), Mexico (4.5%), Peru (5.5%), United States
of America (16%), USSR (14.5%), and Yugoslavia (3.5%). In addition,
some other countries had a production of over 2% of the world total,
e.g. Ireland, Japan, Democratic People's Republic of Korea, Morocco,
Poland, Spain, and Sweden. There are about 40 countries producing only
small amounts each, making together only some 12% of the world
production. One estimate of proven lead reserves of the world is 93
million tonnes of lead metal content. (Federal Institute for Minerals
Research and German Institute for Economic Research, 1972.)
3.2.2 Smelting and refining
Smelting and refining is classified as primary or secondary, the
former producing refined lead from concentrates (primary lead); the
latter recovering lead from scrap (secondary lead). The raw materials
for secondary lead are process (new) scrap arising during
manufacturing processes, and recycled (old) scrap which arises when
lead-containing manufactured goods are discarded. Old material makes
up the bulk of the scrap, the most important source being storage
batteries, which account for 70-80% of the total supply of scrap.
Secondary lead accounts for about half the consumption in the
United States of America and it has been estimated that about 35% of
the total world lead supply comes from secondary sources (Federal
Institute for Minerals Research and German Institute for Economic
Research, 1972). Table 2 gives the production of lead ore, the total
metal production, and the consumption of some industrialized
countries.
Table 2. Lead production and consumption in some industrialized countries (kilotonnes)a
Country Lead ore production Metal production Consumption
(metal content) (refined metal)
1973 1974 1975b 1973 1974 1975b 1973 1974 1975b
EUROPE 1134 1134 1069 2054 2115 1871 2118 2125 1831
Belgium - - - 98 95 103 52 64 54
Bulgaria 105 110 108 100 105 108 80 85 91
Denmark - - - 13 15 13 19 23 20
France 25 24 22 186 178 150 214 199 188
Germany, Federal
Republic of 40 35 37 300 319 260 290 260 210
Ireland 53 34 55 - - - 1 3 2
Italy 27 24 27 100 112 70 234 242 200
Netherlands - - - 25 26 20 38 41 38
Poland 70 70 72 68 70 66 87 90 40
Spain 64 65 58 120 102 85 121 116 90
Sweden 74 73 69 42 41 37 34 36 32
UK - - - 265 277 229 282 266 238
USSR 570 590 504 640 660 600 600 620 544
Yugoslavia 106 109 117 97 115 130 66 80 84
AFRICA 223 183 178 116 117 93 65 66 75
Morocco 90 86 - 1 1 - - - -
South Africa 63 55 53 64 64 49 27 31 39
AMERICA 1430 1412 1379 1666 1677 1565 1718 1706 1339
Canada 388 314 348 187 127 172 69 63 55
Mexico 168 169 163 177 204 179 88 83 74
Peru 199 201 185 83 80 72 10 9 10
USA 570 616 575 1100 1128 1008 1423 1374 1027
Table 2. (Cont'd)
Country Lead ore production Metal production Consumption
(metal content) (refined metal)
1973 1974 1975b 1973 1974 1975b 1973 1974 1975b
ASIA 273 284 291 413 423 395 457 412 386
Democratic Republic
of Korea 90 100 100 60 65 60 20 20 20
Japan 53 44 51 228 228 195 267 217 186
People's Republic
of China 130 140 140 125 130 140 170 175 180
OCEANIA 396 360 384 221 225 191 82 79 75
Australia 396 360 384 221 225 191 74 72 68
Other countries 55 53 48 102 108 88 189 203 233
TOTALS 3617 3569 3497 4642 4723 4260 4883 4882 4154
a Sources: International Lead, Zinc Study Group, and World Bureau of Metal Statistics.
b Estimated.
3.2.3 Environmental pollution from production
Mining, smelting, and refining, as well as the manufacture of
lead-containing compounds and goods, can give rise to lead emissions.
According to a study of the industrial sources of air pollution by
lead in the USA, Davis (1973) reported that 9% of the total of 18 000
tonnes generated from such sources was attributable to the production
of primary lead.
Smelters of lead ores are well known to create pollution problems
in local areas. Their influence on the surrounding air and soil
depends to a large extent on the height of the stack, the trapping
devices in the stacks, the topography, and other local features. The
emissions can cover a considerable area. The zone of air pollution for
one large smelter in the USA extended to approximately 5 km from the
smelter while soil contamination extended as far as 10 km (Landrigan
et al., 1975b). The larger area of the zone of soil pollution compared
to the zone of air pollution probably was due to the fact that current
emission control devices are more effective than earlier ones used to
be. The opposite situation was found around the Mezica mine and
smelter in Yugoslavia (Djuric et al., 1971; Kerin, 1972, 1973). In
this case, the zone of air pollution extended as far as 10 km from the
smelter stack. Soil was grossly contaminated (>200 mg/kg) as far away
as 7 km. There was also heavy pollution of water courses through
effluents.
Secondary smelters producing lead from scrap are comparatively
small, numerous, and frequently situated close to human settlements.
Several studies showed that pollution in the surroundings of such
smelters had been severe enough to produce an increase in the intake
of lead by people living nearby (section 5.1.1).
3.3 Consumption and Uses of Lead and its Compounds
Figures for the consumption of lead are available for most
industrialized countries. The estimated total world consumption of
lead in 1975 was about 4.1 million tonnes (Table 2). The use of lead
is greatly influenced by the growth of the automobile industry which
in 1974 took about 56% of total consumption. Table 3 is compiled from
statistics of lead consumption for the Federal Republic of Germany,
France, Italy, Japan, the United Kingdom, and the United States of
America. There has been a notable increase in the consumption for
batteries over the period 1969-1974.
3.3.1 Storage battery industry
The manufacture of electric storage batteries is responsible for
the largest consumption of lead (Table 3). This industry uses both
metallic lead in the form of a lead-antimony alloy, and lead oxides in
about equal proportions. The metallic lead is in the grids and lugs,
while the oxides, litharge (PbO), red lead (Pb304), and grey oxide
(PbO2), are used in the active material that is pasted on the
plates. The demand for lead batteries decreased in 1974 and 1975
concomitantly with the decline in total consumption (Table 2) as a
result of the economic recession in several of the major lead-
producing countries. However, the fall in the demand for batteries has
also been attributed to the longer life-time of batteries, (Stubbs,
1975) which in 1967 was considered to be about 29 months (US Bureau of
Mines, 1969) but according to Stubbs is, at present, close to 4 years.
The battery industry also constitutes the major source of lead for
secondary lead production. It has been estimated that up to 80% of the
lead in storage batteries is recovered at secondary smelters
(Ziegfeld, 1964).
Table 3. Percentage of total lead consumption by different
industries in six major industrial countries
Industry 1969a 1974b
Batteries 35.9 44
Alkyllead 12.0 12.0
Cable sheathing 10.9 9.2
Chemical pigments 10.9 12.0
Alloys 8.1 10.8
Semi-manufacturers 16.5 12.0
a Federal Institute for Minerals Research and German Institute
for Economic Research, 1972.
b Based on data provided by Stubbs, R. L., Lead Development
Association, London.
The lead battery is likely to retain its position as a convenient
source of electricity in the foreseeable future. The nickel-cadmium
battery does offer some advantages but is about three times more
expensive. Better battery design, improvements in the electrical
systems in cars and lower mileages because of higher gasoline costs
are factors that may retard the growth rate for lead consumption by
the battery industry. New applications for batteries may, on the other
hand, increase demand.
3.3.2 Alkyllead fuel additives
Alkyllead compounds have been in use as anti-knock additives in
gasoline for almost 50 years. Use of these compounds (almost
exclusively tetraethyllead and tetramethyllead) increased steadily up
to 1973 (Table 4). In 1973, the world consumption of refined lead for
the manufacture of lead additives was about 380 000 tonnes
(International Lead and Zinc Study Group, 1976). The moderate decrease
in consumption in 1974 was almost entirely attributable to a decrease
of 22 000 tonnes in the use of lead for gasoline additives in the USA.
A further decline in the consumption was estimated in the USA in 1975,
amounting to some 50 000 tonnes (Table 4); thus, the consumption in
1975 declined by 30% in comparison with the 1973 consumption (Stubbs,
1975). In the USA, the manufacture of alkylleads is, after batteries,
the largest lead consuming industry. By comparison, lead additives
make up only 6% of the European market for lead (International Lead
and Zinc Study Group, 1973). The decrease in the use of lead for fuel
additives is likely to continue in the latter half of the 1970s as
more cars fitted with catalysts requiring lead-free gasoline will come
into use, The regulations on the maximum permissible concentrations of
lead in gasoline will further affect the consumption of lead in fuels.
The US Environmental Protection Agency's reduction programme aiming at
0.13 g of lead per litre of gasoline by 1 January 1979 was ratified in
March 1976 by the US Court of Appeals. The maximum permissible level
in the Federal Republic of Germany has been 0.15 g of lead per litre
since 1 January 1976, and in Japan has been, 0.31 g of lead per litre
since July 1971. Some European countries introduced limits of 0.4 g of
lead per litre (e.g. Austria, Norway, Sweden, Switzerland) but most
European governments have deferred their decision because of the
economic implications of lowering the lead content (International Lead
and Zinc Study Group, 1976).
Table 4. Consumption of refined lead for the manufacture of
alkylleads (kilotonnes)a
Country 1972 1973 1974 1975b
USA 253 249 227 175
Europe: (total) (87) (89) (89) (91)
France 13 14 14 14
Germany, Federal Republic of 9 9 10 9
Italy 15 12 10 10
United Kingdom 50 54 56 58
Others n.a.b 40b 40b 35b
Total 340 378 357 301
a From: International Lead and Zinc Study Group, 1976
b Estimated data; n.a.=not available.
3.3.3 Cable industry
The relative importance of the cable industry as a lead consumer
has declined considerably (Table 3), mainly owing to the introduction
of plastic sheathing/insulation. However, the total amount of lead
used is still notable (Table 5). The use of lead in cable production
is comparatively greater in Europe and several developing countries
than in the United States of America. Alloys used for cable sheathing
contain small amounts of many other elements including cadmium,
tellurium, copper, antimony, and arsenic.
3.3.4 Chemical industry
Although a wide range of lead pigments are still produced they are
increasingly being substituted by other, less toxic, pigments. Red
lead (minium) is used extensively in the painting of structural steel
work and lead chromate is often used as a yellow pigment. The use of
lead for pigment manufacture in 1974 is given in Table 5.
Lead arsenate was, at one time, an important insecticide but is
now little used and current consumption figures are not available.
Table 5. Consumption of lead in cables and pigments in five
industrial countries in 1974 (kilotonnes)a
Country Cable Pigments
France 40 32
Germany, Federal Republic of 52 80
Italy 50 47
Japan 21 50
United Kingdom 44 35
Total 205 244
a Data from International Lead Zinc Study Group statistics.
The use of lead for the manufacture of alkyllead additives was
discussed in section 3.3.2. The petroleum industry also uses a small
amount of litharge dissolved in sodium hydroxide solution to remove
sulfur compounds in the refining of petroleum.
3.3.5 Miscellaneous
Industries producing semi-manufactured components account for an
important proportion of the total consumption. The surface of lead
oxidises readily and is then very resistant to corrosion. The building
and construction industries use lead sheet for roofing and other
flashings, wall cladding, and sound insulation. Lead also forms alloys
readily and is used in solder, bearing metals, brasses, type metal,
collapsible tubes, and for radiation shielding. The ammunition
industry is another major consumer of lead. There are many minor uses
of lead compounds but these account for only a very small proportion
of total lead consumption.
3.3.6 Environmental pollution from consumption and uses of lead
The combustion of alkyllead additives in motor fuels accounts for
the major part of all inorganic lead emissions. The consumption of
lead for the manufacture of alkylleads was estimated at 380 000 tonnes
in 1973 and 300 000 tonnes in 1975 (section 3.3.2). Of this amount,
over 70% is like to enter the environment immediately after
combustion, the rest being trapped in the crank case oil and in the
exhaust system of the vehicles (Davis, 1973; Huntzicker et al., 1975).
Moreover, part of the lead retained in the lubricating oil will enter
the environment through different pathways (section 3.4). The degree
of pollution from the combustion of alkyllead naturally differs from
country to country, depending on the car density. The importance of
alkyllead combustion is exceptionally high in the USA, where 20% of
the total lead consumed is for the manufacture of alkyllead compounds,
the corresponding values in 1969 being only 5% for France and 11% for
Italy and the United Kingdom. The estimated total world emissions from
this source were, according to the figures mentioned above, at least
266 000 tonnes in 1973 and 210 000 tonnes in 1975.
In the study by Davis (1973) on lead emissions into the air from
industrial sources in the USA, 11% (1900 tonnes) was attributed to the
processing of alkyllead additives. The manufacture of storage
batteries emitted smaller amounts (480 tonnes) and emissions were
still smaller in the production of lead oxide, lead pigments, type
metal, solder, etc. The amounts of effluent from these industries were
not studied. The dispersion of lead through the exhausts of workrooms
should also be considered. These emissions although not very large may
still contribute significantly to the pollution of the surrounding
areas. The possibility of contamination of the home environment
through working clothes should be borne in mind.
The magnitude of the pollution arising from the vast number of
lead containing items that are subjected to weathering or are
decomposed in the course of time is difficult to appraise. According
to one estimate, about 50% of paint is removed from surfaces protected
by lead pigments in a period of about seven years before re-painting
(Patterson, 1965). Heavy contamination of the dust and soil around
houses painted with lead paints has been consistently reported (Ter
Haar & Aranow, 1974).
Only an unknown, but probably small fraction of the lead used in
metallic form for the production of sheeting, cable, printing metal,
etc. is ever released into the environment. Contamination of domestic
water supplies, foods, and beverages resulting from the use of lead
pipes, PVC pipes, glazed ceramics, and from cans with lead containing
solders may under certain conditions be hazardous to man's health
(sections 5.1.2 and 5.1.4).
The lead content in tobacco has been attributed to lead residues
present in the soils of tobacco fields as a result of the former use
of lead arsenate as an insecticide (section 5.1.4).
3.4 Waste Disposal
A substantial part of lead wastes are remelted in secondary
smelters (see section 3.2.2).
Municipal incinerators have recently been investigated for lead
emissions. An unknown proportion of the non-recycled, lead containing,
consumer products, e.g. collapsible tubes, bottle caps, cable scrap,
battery casings, and products painted with lead pigments, are
incinerated. Depending on the type of furnace and on purification
devices, these emissions may be considerable (Davies, 1973; Mattsson &
Jaakkola, 1974).
Waste lubricating oil has been contaminated through the combustion
of lead alkyls. Over 50% of the oil is dumped or used as road oil. In
1970, the total amount of waste oil generated in the USA was about
2400 million litres. Waste crankcase oil contains about 1% lead. Thus,
the estimated amount of lead discharged into the environment from this
source in the USA was nearly twice the amount originating from, for
instance, the production of primary lead (Davis, 1973).
The extent of environmental pollution by lead arising from the
incineration of sewage and sludge is not known.
3.5 Miscellaneous Sources of Environmental Pollution
When studying all industrial sources emitting lead into air, Davis
(1973) reported that out of a total of 18 000 tonnes, copper smelting
accounted for 8% and the production of steel and iron another 8%.
Smaller amounts were generated in the production of primary zinc and
also in the production of cement.
Coal contains small amounts of lead with a wide range of
concentrations in different coals. Concentrations found by Abernethy
et al. (1969) in coal from various districts in the USA ranged from
0.6 to 33.1 mg/kg. According to Patterson (1965) about 5% of the ash
leaving boilers as stable fly-ash aerosols is made up of small
particles of a few micrometres. This silicate matter contains about
100 mg of lead per kg. Large quantities of coal are burnt to produce
steam in power stations, steel works, and in manufacturing industries.
Small amounts of lead are generated from burning oil, which also
has a very broad range of lead concentrations. The average
concentrations in oil appear to be below 0.5 mg/kg (Davis, 1973). The
possible future use of sewage sludge as fertilizer is discussed in
section 4.
4. ENVIRONMENTAL TRANSPORT AND DISTRIBUTION
From a mass-balance point of view, the transport and distribution
of lead from stationary or mobile sources into other environmental
media is mainly through the atmosphere. Large discharges may also
occur directly into natural waters and on to the land but, in such
cases, lead tends to localize near the points of discharge owing to
the very low solubility of the compounds that are formed upon contact
with soil and water. The mass transfer of lead from air to other media
is as yet poorly defined and the various mechanisms involved in the
removal of lead from air are not fully understood. Although some data
indicate that an important proportion of the lead may be removed
through sedimentation (Atkins, 1969) the most efficient clearing
mechanism is probably rain (Ter Haar et al., 1967). In a study of the
concentration of lead in rainfall at 32 stations in the United States
of America the average was 34 µg/litre (Lazrus et al., 1970). Most of
these data were collected in areas with a high population density.
Over rural areas of the USA the concentration was found to be
approximately 18 µg/litre (Ter Haar et al., 1967).
Lead is rapidly removed from water when it passes through soil and
bottom sediments. This is due to the high capacity of organic matters
to bind the lead firmly. Because of this clearing mechanism, lead
concentrations in both natural waters and water supplies are generally
low (section 5.1.2).
Table 6. Distribution of lead from motor vehicles in the Los
Angeles basina
Environmental area Fractional
fallout
Retained in car 0.25
Near fallout 0.40
Far fallout 0.08
Airborne 0.24
Unaccounted for 0.03
a Adapted from Huntzicker et al., 1975.
An attempt was made to account for the lead emitted by automobiles
in the Los Angeles Basin (Huntzicker et al., 1975) which is an area of
exceptionally dense motor traffic. Limited environmental monitoring
data tended to confirm the approximate correctness of the
calculations. The transport pattern was classified as "near fallout",
"far fallout" and "airborne". "Near fallout" was defined as the
deposition in the immediate vicinity of roadways. "Far fallout" was
defined as the fallout away from roadways, but within the basin, and
"airborne" designated small particles carried away from the basin and
ultimately deposited elsewhere. The data are shown in Table 6 and
indicate that most of the emission was deposited within the basin. The
fallout figures are calculated from the estimated behaviour of the
airborne particles based on particle size distribution. If these
results are approximately valid for other metropolitan areas, soil and
water pollution from automobile emission fallout is predominantly
limited to the immediate metropolitan area. The particles carried away
from the area by air transport are probably widely dispersed and
diluted since the atmospheric retention time of small particles is
probably fairly long. It has been estimated that the residence time of
airborne particles ranges from 6 days to 2 weeks in the lower
troposphere and from 2 to 4 weeks in the upper troposphere (SCEP,
1970). Residence time will vary with a number of factors such as wind
currents and rainfall. Yamamoto et al. (1968) demonstrated that
atmospheric turbidity varied inversely with rainfall, owing to the
washout effect of rain.
In spite of the great dilution of airborne lead that occurs during
transport from centres of human activity, there is evidence indicating
that a long-term global accumulation of lead has occurred. This long-
term accumulation has been studied in glacial ice and snow deposits.
Studies in Greenland showed that ice formed in about 1750 had lead
concentrations 25 times greater than ice estimated to have been formed
in about 800 B.C. From 1750, the concentration increased steadily to
about 1940. From 1940 to the present day, the rate of increase has
risen even more sharply. The most recent ice layers examined (about
1968) had a concentration 400 times greater than the natural
background. Similar studies in the region of the Antarctic have also
shown a rise, but it has not been so dramatic (Murozumi et al., 1969).
Jaworowski (1968) conducted studies of Polish glaciers similar to
those conducted in Greenland. He observed an approximate 16-fold
increase in the lead concentration over the past 100 years.
Chronological increases in the lead content of Swedish mosses have
also been reported from 1860 to 1968 (Ruhling & Tyler, 1968). These
increases, about 4-fold in the past hundred years, were thought to
reflect first the increase in coal combustion and later the
introduction of leaded gasoline.
The transfer of air lead to the biota may be direct or indirect.
For plants, the fallout contribution may be direct via the above
ground parts, or it may be indirect by way of the soil. The pattern
and degree of lead accumulation appears to be substantially influenced
by the state of growth. Mitchell & Reith (1966) found that the lead
content of certain plants increased 10-fold or more from the period of
active growth to the time when growth ceased in the late fall. Some
trees apparently have the capacity to accumulate high concentrations
of lead. Kennedy (1960) reported that the tips of larches, firs, and
white pines contained 100 mg of lead per kg dry weight, when grown in
the lead mining areas of Idaho where the soil lead concentration was
20 000 mg/kg. The total concentration of lead in soil does not
correlate well with the concentration in the plant but a correlation
does exist when adjustment is made for the degree to which the soil
lead can be brought into an aqueous solution of ammonium lactate and
acetic acid (Kerin et al., 1972).
Thus, there is no doubt that plants acquire lead from the soil and
air, but interspecies differences are prominent (Dedolph et al.,
1970). It does not seem likely, however, that lead deposited on the
leaves of plants transfers readily to other parts. Thus, Ter Haar
(1970) showed in greenhouse studies that atmospheric lead at
1.45 µg/m3 did not influence the lead content of tomatoes, beans,
carrots, potatoes, wheat, and cabbage heads, but did have an effect on
the lead content of lettuce and bean leaves.
Transfer of lead from plants to animals is not well-defined.
However, the concentration of lead in meat and eggs is quite similar,
on a wet weight basis, to the concentration found in vegetables and
grains (Schroeder et al., 1961). There is no evidence of biological
accumulation proceeding from plants to animals.
Much remains to be learned about the environmental transport and
distribution of lead. The potential pathways of lead from air to man
are indicated in Fig. 1. Special attention should be given to the
potential transfer of the fallout lead in cities that is washed into
the sewage systems. Sewage sludge is currently being considered for
use as fertilizer. Most cities have dual sewage system, i.e., storm
and sanitary sewers, and it was shown in the report of the US
Environmental Protection Agency Office of Research and Monitoring
(1972) that storm runoff is far from being clean and probably warrants
being treated in many instances. However, lead is not currently viewed
as a hazard in this case because sludges have a high phosphate content
which tends to minimize the bio-availability of the lead for plants
(Chaney, 1973).
Little information exists with regard to the biotransformation of
lead by microorganisms in the environment. However, Wong and his
collaborators (1975) have reported that microorganisms in lake
sediments can transform certain inorganic and organic lead compounds
into volatile tetramethyllead. The authors were not able to explain
completely the pathways of this transformation. A possible mechanism
for the conversion of trimethyllead acetate into tetramethyllead in
anaerobic systems was presented by Jarvie et al. (1975), who proposed
that this takes place through the formation of an intermediate sulfide
which decomposes into tetramethyllead. There is need for further
research along these lines.
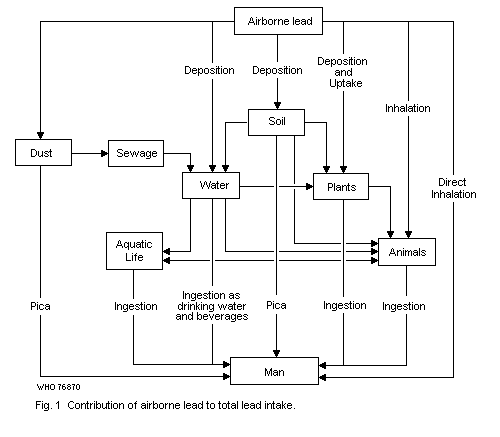 5. ENVIRONMENTAL LEVELS AND EXPOSURES
In the preceding chapter the general pattern of the environmental
transport and distribution of lead was described. This chapter is more
specifically concerned with the different circumstances under which
people are exposed to lead to a degree that may be hazardous to their
health.
5.1 Exposure of the General Population
The general population is exposed to lead by ingestion of food,
and water, and by inhalation. In addition, children are exposed by
eating non-food items, and those working in the lead industries suffer
exposure over and above their exposure as members of the general
population. These categories of exposure will be considered
separately.
5.1.1 Air
The highest concentrations of lead in ambient air are found in
dense population centres. The larger the city, the higher the ambient
air lead concentration. As one moves away from the centre of the city,
the concentration falls progressively. For urban stations, an average
concentration of 1.1 µg/m3 has been reported; for non-urban stations
(near the city) the average was 0.21 µg/m3; for stations somewhat
farther removed it was 0.10 µg/m3, and for remote areas,
0.02 µg/m3 (McMullen et al., 1970). Air over streets with heavy
traffic contained more lead than air over streets with light traffic,
and considerably more than the ambient air over rural areas.
There is a clear pattern in this picture, the non-urban sites
showing less than 0.5 µg/m3, while the urban sites have values
ranging from 1 to 5-10 µg/m3. The highest levels have been recorded
on highways during rush hours, 14-25 µg/m3 (WHO Expert Committee,
1969).
The results of continuous monitoring for 1971-72, in 27 European
cities, by 43 uniform sampling stations are summarized in Table 7
(Commission of European Communities, 1973).
The ambient air lead levels at 15 national sampling stations in
Japan in 1973 were 0.30 µg/m3 for the average 24-hour value,
2.72 µg/m3 for the maximum 24-hour value, and 0.01 µg/m3 for the
minimum (Environment Agency, Japan, 1975).
Table 7. Air lead concentrations in some cities of the European Community (1971-72)a
Location Continuous measurements Traffic-hour measurements
Non-urban monthly averages < 0.5 µg/m3 -
daily maxima < 1 µg/m3 -
Small cities
residential areas monthly averages < 1 µg/m3 -
daily maxima < 2 µg/m3
traffic areas - monthly averages < 3 µg/m3
individual measurements < 8 µg/m3
Metropolitan areas
residential areas monthly averages < 2 µg/m3 individual measurements < 4 µg/m3
daily averages
up to 8 µg/m3
traffic areas monthly averages monthly averages < 10 µg/m3
up to 6.5 µg/m3
daily values up to 10 µg/m3 single measurements
up to 20 µg/m3
a Data from the Commission of the European Communities (1973).
People who live in close proximity to dense automobile traffic are
exposed to appreciably higher concentrations than others. In Los
Angeles, California, where general ambient air levels are unusually
high, the monthly mean concentration near traffic was as high as
6.4 µg/m3 (US Department of Health, Education and Welfare, 1965).
This is in contrast to the general ambient air level of 2-4 µg/m3
reported for that city (Tepper & Levin, 1972). There is also a diurnal
pattern whereby the concentration rises and falls in approximate
proportion to the vehicular traffic activity (US Department of Health,
Education and Welfare, 1965; Lahmann, 1969; Heller & Kettner, 1969;
Chovin et al., 1973). Most studies report that a seasonal variation
also occurs (Tepper & Levin, 1972; Georgii & Jost, 1971).
Nearly all air lead measurements in communities have been made
outdoors. Only a small number of indoor concentration studies are
available (e.g. Fugas et al., 1973; Yocom et al., 1971; Daines et al.,
1972). Indoor levels vary from slightly lower than, to about 1/3 of,
comparable outdoor levels. Higher indoor levels are found only in lead
industry environments. In the absence of specific data, reference
should be made to the much more voluminous literature available on the
penetration of undifferentiated particulate pollution into buildings.
This was reviewed by Benson (1972). In general, very small particles
enter buildings readily, and exist there at levels similar to those
outside. Larger particles, near stationary sources and very close to
roadways, penetrate buildings less readily.
Studies of air lead concentrations over a number of years or even
a decade, at the same or similar locations, have produced quite
variable results. Occasionally air lead levels have declined as in
Cincinnati, Ohio (US Department of Health, Education and Welfare,
1965). This was attributed to greatly decreased coal consumption. The
US National Air Surveillance Network, which has most of its stations
in city centres, has shown little change in large cities and variable
behaviour in smaller cities (NAS-NRC, 1972). Tepper & Levin (1972) and
Chow & Earl (1970) have shown considerable increases in air lead
levels at a number of stations in large cities. In 1967, Ott et al.
(1970) developed a predictive model of increasing automotive pollution
based on carbon monoxide emission patterns. Since air lead comes
largely from vehicular sources, this report should be considered when
changes in air lead with time are evaluated.
The respiratory uptake of lead from air depends on total lead
concentration, particle size distribution, particle shape, chemical
composition, physicochemical properties, and respiratory volume
(section 6.1.1).
The particle size distribution of lead in ambient air has been
studied by a number of investigators. As regards pulmonary deposition
and absorption, the mass median equivalent diametera rather than the
microscopic particle size is considered appropriate. Robinson & Ludwig
(1967) reported a mass median equivalent diameter of 0.25 µm, with 25%
of the particles smaller than 0.16 µm and 25% larger than 0.43 µm.
These data were representative of a variety of areas in Los Angeles,
the San Francisco Bay area, Cincinnati, Chicago, and Philadelphia.
There was little variation from one city to another. Other studies
conducted in the United States of America gave similar results
(Mueller, 1970; Robinson et al., 1963). More recently Lee et al.
(1972) have reported mass median equivalent diameters of 0.42-0.69 µm
for six United States cities. Jost et al. (1973) reported that 50% of
particles had mass median equivalent diameters of less than 0.4 µm and
20% of more than 0.5 µm.
Not much is known about the chemical form in which general ambient
air lead occurs. Ter Haar & Bayard (1971) studied the composition of
airborne lead particulates with an electron microprobe analyser. They
studied particulates collected directly from the exhaust pipe of a car
and also from air at various distances from a busy highway. Their
results (Table 8) indicate that car exhaust lead is initially composed
of halides that are converted to oxides, sulfates, and carbonates with
aging.
Alkyllead vapours occur in ambient air because some of the
alkyllead in gasoline escapes combustion. Purdue et al. (1973) have
recently reported on the organic lead concentration in an underground
parking garage and in the general ambient air of six major cities in
the USA. In the parking garage, the total air lead level was
11.7 µg/m3, of which 16.7% was organic lead. In the six major
cities, the organic lead concentration was about 10% of total lead.
There is some uncertainty as to the accuracy of the organic lead data
since the concentrations found approached the detection limit of the
method. In another study in Los Angeles, using a different method for
trapping organic lead, approximately 2% of the lead was found to be
organic (Snyder, 1967). Differences found in the concentration of
organic lead relative to particulate lead can perhaps be explained in
part by differences in proximity to the emitting source. Laveskog
(1971) made repeated studies over several months on the presence of
organic lead in the air at a number of locations in Stockholm. The
a Mass median equivalent diameter = equivalent diameter above and
below which the weights of all larger and smaller particles are
equal.
levels were uniformly low (under 10% of the total lead) except for 2
brief periods. These occurred near a gasoline station, and were
attributed to the evaporation of spilled fuel. Colwill & Hickman
(1973) verified this concept in similar studies near gasoline handling
installations.
The air in the vicinity of lead smelters may be appreciably
polluted and thus can affect the general population. A detailed study
has recently been made of the environmental impact of a large ore
smelter near El Paso, Texas (Landrigan et al., 1975b). The annual mean
concentration in 1971 was approximately 80 µg/m3 in the immediate
vicinity of the smelter and fell off rapidly, attaining a near-
background level of 1 µg/m3 about 5 km away. Approximately 42% of
the particle mass had an aerodynamic diameter of less than 2 µm. In a
similar study conducted near a smelter and a mine in the Meza Valley,
Yugoslavia, the air lead concentration, 10 km away, ranged from 1.3 to
24.0 µg/m3 (Djuric et al., 1971). Five air sampling stations were
located at various distances within 10 km of the smelter stack. About
45% of the particles had a diameter equal to or less than 0.3 µm and
an additional 25% were in the range of 0.31-0.8 µm. Although not
specified, it is probable that these particle sizes were expressed as
aerodynamic diameters, since the authors refer to this range as being
of optimum size for absorption. The extensive pollution in some
directions was probably due to the topography. The Meza Valley is only
a few hundred metres wide and depending on wind direction, the lead
particles can be conveyed long distances.
Roberts et al. (1974) reported the lead levels in air, dust, soil,
and in the blood of children living near two secondary smelters in
totonto. Monthly mean air lead levels (1.0-5.3 µg/m3) were about
twice those encountered in other parts of the city, but were subject
to greater daily variation. Lead levels in dustfall of
200-1500 mg/m2/month, and soil levels of 16 000-40 000 mg/kg of soil
near the smelter declined to background levels within 300-400 m of the
stacks. From 13 to 30% of the children living within 300 m of the
stacks had blood lead levels of over 40 µg/100 ml.
Pollution of the surrounding country by a secondary lead smelter
has been reported to have affected the lead absorption of adults
living 1-4 km from the main emitter (Nordman et al., 1973). Air lead
concentrations were not reported. The dustfall lead concentrations
ranged from 10 mg/m2/month (4 km from the chimney) to
200 mg/m2/month (200 m from the chimney). There was a correlation
between blood lead concentration and degree oferythrocyte ALAD
inhibition on the one hand and the proximity of habitation to the
smelter on the other. A correlation between Pb-B values and monthly
dustfall lead was also demonstrated.
Table 8. Composition of airborne lead particles by electron microprobe analyser.a
Percentage of total
particles counted
Sample PbCl2 PbBr2 PbBrCl Pb(OH) Pb(OH) (PbO)2 (PbO)2
Cl Br PbCl2 PbBr2
Exhaust pipe
Zero time 10.4 5.5 32.0 7.7 2.2 5.2 1.1
18 h 8.3 0.5 12.0 7.2 0.1 5.6 0.1
Eight Mile Road
Near road 11.2 4.0 4.4 4.0 2.0 2.8 0.7
400 yards 10.5 0.7 0.6 8.8 1.1 5.6 0.3
Rural site 5.4 0.1 1.6 4.0 - 1.5 -
Percentage of total
particles counted
Sample (PbO)2 PbCO3 Pb3 PbOx (PbO)2 PbO PbSO4
PbBrCl (PO4)2 PbCO3 PbSO4
Exhaust pipe
Zero time 31.4 1.2 - 2.2 1.0 - 0.1
18 h 1.6 13.8 - 21.2 29.6 0.1 -
Eight Mile Road
Near road 2.0 15.6 0.2 12.0 37.9 1.0 2.2
400 yards 0.6 14.6 0.3 25.0 21.3 4.6 6.0
Rural site 1.0 30.2 - 20.5 27.5 5.0 3.2
a From: Ter Haar & Bayard (1971).
5.1.2 Water
Man's exposure to lead through water is generally low in
comparison with exposure through air and food (WHO Working Group,
1973). The lead concentration in the water supplies of most of the 100
largest American cities, as determined in 1962, ranged from a trace to
62 µg/litre (Durfor & Becker, 1964). Since 1962, continuous monitoring
of American water supplies has indicated that the US Public Health
Service prescribed limit of 50 µg/litre has not been exceeded (NAS-
NRC, 1972). In another study, only 41 out of 2595 samples of tap water
contained more than 50 µg/litre, and 25% contained no measurable
amount of lead (McCabe, 1970).
Under some circumstances, the concentration of lead in drinking
water can become extremely high. Gajdos & Gajdos-Török (1973)
described two cases of severe clinical lead poisoning attributable to
a municipal water supply that contained lead levels of 2.6 mg/litre.
In another case, in rural Scotland, four people developed clinical
lead poisoning and others showed biochemical evidence of grossly
elevated lead exposure (Goldberg, 1974). The concentration of lead in
the domestic water supply was 2-3 mg/litre. In this case, the reason
for the extreme contamination was that the water was stored in lead
tanks. In another study, conducted in Glasgow, Scotland, it was shown
that lead pipes in the plumbing of homes can also result in high
concentrations of lead in soft water (water low in calcium and
magnesium) (Beattie et al., 1972a). Homes with both lead-lined water
storage tanks and lead pipes had the highest concentration. The
plumbo-solvency of water standing in lead pipes is influenced
significantly by several factors. The solvency increases about four-
fold with increasing acidity over the pH range from 6 to 4. Increases
of a somewhat lesser degree were also noted with increasing alkalinity
over the range of pH from 8 to 10 (Moore, 1973). The same author also
pointed out the increasing plumbo-solvency of water with increasing
temperature and with decreasing calcium concentration. Quite recently
it was shown that lead concentrations in tap water were highly
dependent on the volume of water flushed through the system before
sampling. The concentrations were also considerably lower when a 95/5
(tin/lead) solder had been used in the copper piping instead of the
50/50 or 60/40 solders (Wong & Berrang, 1976).
When water was left standing overnight in plastic pipes, some
degree of leaching of lead into the water was observed (Heusgem &
DeGraeve, 1973). The source of lead in this case was probably lead
stearate which is used as a stabilizer in the manufacture of polyvinyl
plastics. The problem of plastic pipes has been discussed recently
(Schaller et al., 1968; Packham, 1971). Packham did not feel that
there was a hazard associated with the use of such material in
domestic water supply systems. But more study is needed, particularly
of situations in which water stands in the pipes for prolonged
periods.
Lead levels in surface and ground waters were recently reviewed by
a WHO Working Group (1973). Natural surface waters have been reported
to contain usually less than 0.1 mg/litre (Kopp & Kronen, 1965). In
unpolluted areas the concentrations are of the order of 1 µg/litre or
less (Zukovickaja et al., 1966). Some rivers in France were recently
analyzed by Servant (1973) who found that, in the Midi-Pyrenees
region, the mean concentration of dissolved lead varied from 6.7 to
10.4 µg/litre.
5.1.3 Food
The contribution of food to man's exposure to lead has been under
study for many years, beginning with the study of Kehoe et al. (1933)
who found lead in every item of food in both industrial and primitive
societies. The concentration of lead in various items of food is best
described as highly variable. In fact, there seems to be about as much
variation within specific items of food as between different
categories of foods. For example, Schroeder et al. (1961) found that
the range was 0-1.5 mg/kg for condiments, 0.2-2.5 mg/kg for fish and
seafood, 0-0.37 mg/kg for meat and eggs, 0-1.39 mg/kg for grains, and
0-1.3 mg/kg for vegetables.
Estimates of actual consumption of lead in food and beverages have
been made using two general approaches. Some investigators have used
the duplicate portions sampling method. Others have derived
theoretical intakes based on nutritional tables and known
concentrations of lead in the dietary components (composites
technique, see section 2.2.1). The results of studies using the two
methods are given in Table 9. In general, the composites technique
appears to yield somewhat higher mean values for adults than the
duplicate portion technique. There are considerable differences in the
daily intakes reported from different countries. Whether these
differences are real or due to factors associated with the methods
used remains to be assessed. Inadequacies in sampling and in the
analytical methods used may account for a considerable part of the
differences; few of the studies cited present any evidence of
interlaboratory quality control of the analytical assays. Most
estimates do not specify the age, sex, or level of physical activity
assumed in arriving at the estimates. These are very important
determinants of dietary intake. Thus, the calorie requirement for a
25-year-old male in the United States of America is approximately 2900
calories, whereas it is only 1900 calories for women, age 35-55
(Altman & Dittmer, 1968). Horiuchi et al. (1956) were quite aware of
the vast differences in food intake among different categories of
adults and between adults and children. They made an effort to take
these factors into account in developing their estimates of lead
intake from dietary sources.
Daily faecal lead excretion can also be used as a means of
estimating daily lead ingestion, since only approximately 10% of
dietary lead is absorbed (Kehoe, 1961). This approach, which was used
Table 9. Dietary lead intake.
Method Age Sex Activity Lead/day (µg) No. of References
subjects
range average
Duplicate adult male sedentary 120-350 218 9 Kehoe, 1961
portions adult male sedentary 74-216 113 17 Coulston et al., 1972b
21-30 years 4 male - 237-306 274 5 Thompson, 1971
1 female
adult - - 4.8-83.0 17.8 - Schütz et al., 1971
adult male medium- 119-360 231 35 Nordman, 1975
heavy
adult female medium 89-305 178 36 Nordman, 1975
3 months- - 40-210b 8 Alexander et al, 1973
8.5 years
Composites adult male heavy work - 455 - Horiuchi et al., 1956
technique adult male medium - 299 - Horiuchi el al., 1956
10 years male - 254 - Horiuchi et al., 1956
10 months male - 126 - Horiuchi et al., 1956
18 years male medium - 57-233a - Kolbye et al., 1974
adult male medium - 139 - NRC (Canada), 1973
adult male medium - 518 - Lehnert et al., 1969
adult male medium - 505 - Zurlo at al., 1970
a See page 52
b Ranging from 40 µg in a breast-fed infant to 210 µg in an 8´-year-old child, as calculated using ICRP
by Tepper & Levin (1972) (section 6.1.2.2), presents some uncertainty
regarding actual absorption and neglects contributions to faecal lead
from gastrointestinal secretion, which cannot be estimated.
A factor that is usually ignored is the occurrence of lead in
various foods at concentrations below the practical detection limits.
Thus, Kolbye et al. (1974) arrived at different estimates based on the
assumptions they made regarding whether or not lead was present in all
items eaten. There was uncertainty as to how to cope with the problem
of lead concentrations reported as "zero" or "trace" in certain
samples. When "zeros" and "traces" were accepted as meaning absolutely
no lead, the estimated daily lead intake was 57.4 µg for an 18-year-
old man. This seemed unduly low, particularly in the light of the fact
that the faecal lead excretion of normal American women is
90-150 µg/day (Tepper & Levin, 1972). Certain assumptions were
therefore made regarding the "zeros" and "traces". If it was assumed
only that "traces" really represented 0.09 mg/kg, the calculated
intake became 159 µg/day. When the additional assumption was made that
"zero" had a finite value of 0.05 mg/kg, the calculated daily intake
of an 18-year-old male became 233 µg. Another source of error in
establishing how much is consumed relates to food preparation. Lead
may be either added to the diet or removed in the course of
preparation. Both Horiuchi et al. (1956) and the report from the
British Ministry of Agriculture, Fisheries and Foods (1972) took
special pains to explain how this problem was handled.
Published reports on lead levels in wine (Truhaut et al., 1964;
Zurlo & Griffini, 1973) show that important variations occur from
sample to sample. Considering ordinary wines there does not seem to be
any significant difference between white, red, and rosé (Truhaut et
al., 1964). Average lead concentrations of 130-190 µg/litre could be
calculated (range 60-255 µg/litre), but even higher mean values
(299 µg/litre) have recently been reported (Boudene et al., 1975).
Wine therefore is likely to be a substantial source of lead for some
people, and may account for part of the differences in Pb-B levels
(section 5.4) and in daily dietary lead intake between various
countries.
The concentration of lead in milk is a matter of special concern
because milk is a major dietary constituent for infants. Human breast
milk has been reported to contain 12 µg/litre (Murthy & Rhea, 1971)
and <5 µg/litre (Lamm & Rosen, 1974). Cow's milk has been reported to
have a similar concentration, when taken directly from cows for
analysis; 9 µg/litre (Hammond & Aronson, 1964). The concentration of
lead in processed cow's milk is higher than in human milk or in milk
obtained directly from cows. The types of processing vary considerably
as does the degree of apparent lead contamination. Thus whole milk
concentrations are only moderately elevated. Mitchell & Aldous (1974)
reported an average of 40 µg/litre in whole bulk milk and Kehoe (1961)
reported 20-40 µg/litre for local USA market milk. By contrast,
evaporated milk and formulas have still higher concentrations.
Mitchell & Aldous (1974) reported an average of 202 µg/litre for
evaporated milk. Somewhat higher values were reported by Murthy & Rhea
(1971) (330-870 µg/litre) and somewhat lower by Lamm & Rosen (1974)
(110 ± 11 µg/litre).
A major contribution to the lead content of processed milk as well
as of other food products appears to be lead solder used in the seams
and caps of cans. It has been shown that foods preserved in such cans
frequently have much higher concentrations of lead than do the same
items packed in glass containers (Mitchell & Aldous, 1974).
Although plants do not take lead up from the soil readily, fruits
and vegetables grown in areas exposed to smelter emissions may be
appreciably contaminated. Kerin (1972) determined lead in the total
diet of peasants near a smelter and found that the daily ingestion of
lead with food was 670-2640 µg.
5.1.4 Miscellaneous
The intake of lead in food, air, and water is a major concern as
regards the general population because of the pervasive nature of
these exposures. Another frequent exposure source, smoking, probably
makes a small contribution to the lead burden (section 5.4.1 ).
However, surprisingly little information is available concerning the
concentration of lead in smoking tobacco. Cogbill & Hobbs (1957)
reported the concentration of lead in two separate brands of
cigarettes and in a composite sample of five brands. Concentrations
were 19, 80, and 39 mg/kg at 58%, relative humidity or 21, 84, and
41 µg per cigarette. The amount of lead transferred to mainstream
smoke was 1.0, 3.3, and 1.9 µg per cigarette which represented 4.8,
3.9, and 6% transfer. Arsenic/lead ratios found in the tobacco
indicated that the source of lead was probably lead arsenate. At one
time lead arsenate was used extensively as an insecticide in American
tobacco fields but other pesticides rapidly replaced it shortly after
World War II. Residues of lead arsenate have probably persisted in the
fields and could contaminate plants externally. More recently,
Szadkowski et al. (1969) reported 0.483 ± 0.267 µg of lead per
cigarette in the total smoke for eight brands of cigarette. This
represented 19%, of the total lead in the tobacco or 2.6 µg per
cigarette. No distinction was made between mainstream and side-stream
smokea. Untabulated data from a study by Menden et al. (1972)
indicated that only about 2% of lead in non-filter types of cigarettes
was transferred to the mainstream smoke. The average content of lead
in commercial cigarettes was given as 10.40-12.15 µg/cigarette
a "i.e. the smoke which drills off the burning end of cigarette
between puffs.
(Petering & Menden, private communication). Most of the lead was found
in the ash; the lead content of the sidestream of individual
cigarettes varied considerably with a maximum value of 16%. Assuming
an average lead content ranging from 2.5 to 12.2 (Lehnert et al.,
1967; Szadkowski et al., 1969; Rabinowitz, 1974; Petering & Menden,
private communication) and a 2% transfer to the mainstream smoke
(Menden et al., 1972) are fair estimates, and without taking into
account the possible contribution from the sidestream smoke, a crude
assessment of the direct inhalation intake of lead from smoking 20
cigarettes a day would be about 1-5 µg.
Certain other sources of exposure are important. These sources do
not affect any major segment of the population but collectively they
no doubt account for the majority of the cases of clinical lead
poisoning in the general population.
The presence of high concentrations of lead in illicitly distilled
whisky occurs commonly in the USA and causes poisoning in adults. The
condensers used in homemade stills are often discarded automobile
radiators. These contain substantial amounts of lead in the soldered
joints. The concentration of lead in the final product frequently
exceeds 10 mg/litre. The problem of lead poisoning from this source
exists predominantly in the southeastern parts of the USA where
illicit whisky production is most common.
Another source of poisoning is improperly glazed earthenware
vessels. Improper glazing results in the leaching of lead into the
vessel, particularly when the contents are acidic. Cases of poisoning,
both fatal and non-fatal, have been recorded from the use of
improperly glazed pottery. Klein et al. (1970) reported two cases (one
fatal) in which apple juice stored in the incriminated vessel for 3
days contained 1300 mg/litre. In another case the ceramic mug
responsible was used for drinking colaa, pH 2.7 (Harris & Elsea,
1967). After two hours of standing in the mug, the cola contained lead
levels of 6.8 mg/litre. It was estimated that this patient drank 3.2
mg of lead per night in this fashion for two years. Other cases have
been reported from Yugoslavia (Beritic & Stahuljak, 1961) and from the
United Kingdom (Whitehead & Prior, 1960). The problem involves the
storage of acid materials in the vessels. In a test of the leaching of
lead from commercial and handcrafted pottery, Klein et al. (1970)
found that 4% acetic acid allowed to stand at room temperature in the
vessels for 18 hours often acquired concentrations of lead in excess
of 100 mg/litre. In fact, in more than half of the cases, the
concentration of lead exceeded 7 mg/litre.
a A popular carbonated non-alcoholic beverage.
Another source of lead poisoning in the general population is the
use of discarded storage battery casings for fuel. There is some
uncertainty as to whether the cases of poisoning that have been
recorded (Williams et al., 1933; Gillet, 1955) were due to inhalation
of lead fumes or to hand-to-mouth transfer of fallout material. The
prevalence of children in the number of recorded cases supports the
argument for hand-to-mouth transfer.
Because of the wide variety of applications of lead, additional
potential hazards are still being identified. For example, the use of
lead wire core wicks in candles was only recently called to the
attention of the USA authorities (Bridbord, unpublished results,
1973).
5.2 Exposure of Infants and Young Children
5.2.1 Soil, dust, and paint
The young child of pre-school age is exposed to special hazards
from environmental sources of lead. This is because such children
frequently exhibit the habit of licking, chewing, or actually eating
foreign objects. Lead-based paints have long been considered the major
source of excessive lead intake in young children. Thus, Sachs (1974)
reported that 80% of patients seen because of evidence of excessive
lead absorption had a history of eating paint or plaster and in
another 10% X-ray examination revealed paint in the abdomen. The
author also was of the opinion that if X-ray examinations had been
repeated at each visit to the clinic, evidence of paint ingestion
would have appeared in all patients. A similar view was expressed by
Chisholm & Harrison (1956). In their series of 105 children whose
homes were investigated, 102 of the homes contained at least one
source of paint containing 5% lead or more. Of even greater
significance was the fact that the painted surfaces identified as
sources were flaking.
Other investigators have attempted to assess the importance of
paint as a source of excessive lead exposure. Griggs et al. (1964)
found a positive correlation between the presence of elevated urinary
lead or coproporphyrin and the presence of flaking paint in the homes.
Nonetheless, in many instances the homes of children with abnormal
urine had no flaking paint indoors. Unfortunately data were not given
as to the number of children with abnormal urine and no evidence of
flaking paint indoors or outdoors. Guinee (1973) reported that in an
extensive survey of the homes of children having blood lead
concentrations equal to or greater than 60 µg/100 ml, 75% of the homes
had at least one surface in which the paint contained more than 1%
lead. Furthermore, children with elevated blood lead concentrations
were more likely to live in homes where the painted surfaces were
cracked than children with low blood lead values.
All of these studies indicate that lead in painted surfaces in
houses is almost certainly the major source of lead for infants and
young children. Some other studies suggest that the issue is not that
clear-cut. Greenfield et al. (1973) reported that, in one study, 18
out of 19 rural children with elevated blood lead concentrations lived
in homes having at least one accessible painted surface containing 1%
or more of lead, whereas paint containing 1% or more of lead could be
found on accessible surfaces in only 60% of the homes of inner city
children with excessive lead exposure. The implication is that sources
of lead other than paint were often responsible for the exposure of
city children. Two equally rational interpretations are that an
insufficient number of surfaces were tested in the children's homes or
that children often spend time in several homes, some of which might
not have been tested for lead-based paint.
Studies of sources of lead all too often ignore the fact that
painted surfaces on the outside of houses are a potential source of
lead or, for that matter, that the soil surrounding the houses may
have accumulated substantial concentrations of lead from the
weathering of outer walls. With regard to the latter, Fairey & Gray
(1970) reported that the concentration of lead in the soil near homes
where paediatric lead poisoning had occurred was over 1000 mg/kg in 27
out of 30 cases. By contrast, only 30 out of 170 soil samples taken
from yards selected at random (and not associated with cases of lead
poisoning) had concentrations of lead in excess of 1000 mg/kg.
Bertinuson & Clark (1973) have reported extremely high soil lead
values close to residences in the older section of Cincinnati. In one
case, because the distance across the yard from the base of the house
to a road with heavy traffic was sufficient, it was possible to assess
the relative contributions of lead from car exhaust and lead from the
weathering of the house. The gradient ranged from 12 000 mg/kg
adjacent to the house, down to 400 mg/kg, about 10 m from the road.
This suggested that weathering of painted surfaces of the house could
have been the major source of soil lead in this instance. Although the
high concentrations of lead in the soil in the vicinity of houses may
be due to weathering of lead-based paint, it is possible that in many
cases it is also due to the accumulation of combusted alkyllead from
car exhaust. In this connexion, recently-reported data of Ter Haar &
Aranow (1974) are particularly informative. They surveyed the profile
of lead in soil, extending from the base of 36 urban residences out to
the street gutters. Eighteen of the residences were of brick
construction and 18 were of frame construction. In summary form, their
data were as shown in Table 10. The data reflect the likelihood of the
major contribution of weathered lead-based paint to soil lead. But
they also strongly suggest that vehicular sources make a significant
and sometimes very substantial contribution to soil lead near the
sidewalks.
Table 10. Lead in dirt in Detroit (mg/kg dry dirt)a
Location Painted frame houses Brick houses
Mean Range Mean Range
Within 0.6 m of house
front 2349 (126-17 590) 351 (78-1030)
back 1586 (162-4951) 501 (72-2350)
sides 2257 (140-7284) 426 (91-1160)
1846 (104-7000) 595 (40-2290)
3 m from house
front 447 (58-1530) 156 (39-316)
back 425 (149-1410) 200 (72-480)
Near sidewalk 627 (152-1958) 324 (86-1130)
curb 572 (320-1957) 612 (147-2420)
gutter 966 (415-1827) 1213 (304-3170)
a Adapted from Ter Haar & Aronow (1974)
Street dust has also been found to contain high concentrations of
lead. Using recent data from 77 midwestern cities in the USA, it was
calculated that the concentration of lead averaged 1636 mg/kg dust in
residential areas, 2413 mg/kg in commercial areas, and 1512mg/kg in
industrial areas (Hunt et al., 1971).
In order for soil or street dust to be a significant source of
lead for man, it is of course, necessary that it be ingested and/or
inhaled. Evidence regarding the likelihood that young children would
ingest soil or street dust is extremely fragmentary. However, in a
recent study of 58 children with increased lead burdens, it was found
that 37 had a history of eating dirt and sand, compared with 34 eating
plaster, 20 eating paint flakes, 15 chewing on furniture, 14 chewing
window sills, and 7 eating wallpaper (Pueschel et al., 1972). Further
inferential evidence as to the possible significance of soil and dust
as a source of lead is to be found in the recent Smeltertown episode
near El Paso, Texas, referred to earlier (Landrigan et al., 1975b).
This town is the site of a large smelter which processes lead ores,
among others. The young children in the town have high blood lead
concentrations. In a sample of 14 children of 1-5 years of age,
78.6-100% were found to have lead concentrations equal to or greater
than 40 µg/100 ml blood. The concentration of lead in the surface soil
of Smeltertown has a median value of 3700 mg/kg. One is tempted to
conclude that the blood lead levels of these young children increased
owing to ingestion of this soil. However, the picture is somewhat
confounded by the fact that older children also showed a high
incidence of elevated blood lead concentrations but to a lesser
degree; and older children are not generally considered to exhibit
pica. Smeltertown adults had normal blood lead concentrations. Intake
of lead by inhalation would probably have affected adults as well as
children. Thus, it is likely that lead intake by the children was by
direct oral intake. The painted surfaces in the residences were seldom
in a flaking condition and were not found to be more than two or three
layers thick, in contrast to the multiple layers usually found in city
slum areas where lead poisoning is prevalent. The information
available therefore suggests that the sources of lead were soil and
dust. Indeed, there was a highly significant correlation between the
concentration of lead in the blood of the children and the
concentration of lead in household dust.
The presence of high concentrations of lead in soil is not
necessarily hazardous. Thus, children living on soils containing lead
levels of up to 8000 mg/kg showed only minimal elevations in blood
lead concentration (Barltrop et al., 1974). This was found to be so
even among the children with pica for soil. Perhaps climatic
differences are important. Smeltertown in Texas is extremely dry and
dusty whereas the region studied by Barltrop and coworkers was in
England, where the soil is presumably not as accessible to children
owing to the relatively heavy cover of vegetation. The play behaviour
of children also determines to a certain extent their exposure to lead
(Einbrodt et al., 1974).
Since dust and dirt occur indoors as well as outdoors, some
attention has been directed recently to the significance of indoor
dust. Transfer of lead-bearing house dust to the hands of young
children has recently been demonstrated (Sayre et al., 1974). The
house dust of inner city old houses contained far more lead than the
dust of newer, suburban houses. Furthermore, the hands of the children
in inner city houses were heavily contaminated with lead, whereas the
hands of suburban children were not. It is not at all certain that the
source of lead in the house dust was fallout from car exhaust. New
housing in the inner city had very little lead in dust. The inference
is that the lead was probably from the painted surfaces, since the
paint in old houses has high concentrations of lead whereas the paint
in new houses in the same area generally has a low lead content. But
even the presence of lead-containing dust on children's hands provides
little information concerning hazard since the critical question is
how much is actually transferred from the hands to the digestive
tract.
5.2.2 Miscellaneous
Facial cosmetics have long been a source of lead poisoning in
Oriental countries. Kato (1932) discussed the problems encountered in
Japan. Face powders, pastes, and liquids were found to contain as much
as 67% lead. Exposure of children was considered to be by inhalation
of powders, or ingestion of powders and other formulations. More
recently, there have been several reports of infant poisoning from a
mascara-like cosmetic used by Indian and Pakistani women (Warley et
al., 1968; Alexander & Delves, 1972). This substance may contain as
much as 88% lead sulfide.
Another source of lead exposure for young children is coloured
newsprint (Hankin et al., 1973). It has been found that the coloured
inks used in magazine illustrations contain extremely high
concentrations of lead. Coloured pages were found to have lead
concentrations of 1140-3170 mg/kg.
Children and other family members may be exposed to lead
contamination at home by work clothing being worn at home or brought
home for cleaning, or by small pieces of metal which may be brought in
(InterDepartmental Working Group on Heavy Metals, 1974).
5.3 Occupational Exposures
It is among the workers who smelt, refine, and use lead in
manufacturing items of commerce that the highest and most prolonged
exposures are found. Lead poisoning among these people was common at
one time. Today, workers, management, and physicians are generally
aware of the danger of lead and know how to handle the problem; so,
the incidence and severity of poisoning have decreased substantially
in recent years. However, much still remains to be done to eliminate
lead poisoning completely as an occupational disease. The major hazard
today seems to be in small enterprises (Engel et al., 1971) and in
some large industries where adequate industrial hygiene programmes do
not exist or are difficult to implement, or where awareness of the
existence of hazardous circumstances may be lacking.
A recent WHO study of occupational health problems in the Andean
countries (El Batawi, unpublished results, 1974) showed that, in Chile
for instance, among 580 workers exposed to lead, 21.9% had an
increased level of ALA in the urine. In Colombia, 3370 workers exposed
to lead were examined, of whom 4.30% were considered to be suffering
from lead poisoning.
The major route of lead exposure in industry is by inhalation. The
generation of lead-bearing dusts and fumes is inevitable. The workers'
clothes may also be an important source of exposure. Even the lesser
problem of oral intake of lead is really a consequence of the
generation of airborne dusts which settle out from the air on to food,
water, or other objects that are transferred to the mouth in one
fashion or another. Thus, good housekeeping and, above all, good
ventilation have a strong impact on exposure. An industrial process
may be quite safe in one factory and quite hazardous in another solely
because of differences in ventilation engineering or because of
differences in housekeeping practices and worker education.
5.3.1 Lead mining, smelting, and refining
The lead mining hazards depend, to some extent, on the solubility
of the lead from the ores. The lead sulfide (PbS) in galena is
insoluble and absorption through the lung is slight. However, in the
stomach, some lead sulfide may be converted to slightly soluble lead
chloride which may then be absorbed in moderate quantities.
The process of lead smelting and refining probably has the
greatest potential for hazardous exposure of all the lead industries.
The most hazardous operations are those in which molten lead and lead
alloys are brought to high temperatures, resulting in the vaporization
of lead. This is because condensed lead vapour has, to a substantial
degree, a small (< 5 µm), respirable particle size range. Thus,
although the total air lead concentration may be greater in the
vicinity of ore proportioning bins than it is in the vicinity of a
blast furnace in a primary smelter, the amount of particle mass in the
respirable size range may be much greater near the latter.
As an example, we can consider the processes involved in the
preparation of lead bullion in typical primary lead ore smelters in
Salt Lake City, Utah. The various processes are essentially grinding
and smelting. The main operations are: (1) ore proportioning; (2)
nodulizing and sintering; (3) blast furnace; (4) drossing and
reverberation. Air lead concentrations have been determined using
personal monitors worn by workers at the various stations. These data
are summarized in Table 11. Similar data for primary lead smelters
elsewhere are not available. However, it is evident that lead exposure
in primary smelters may be extremely high. The hazard to the workers
in the example cited would be extremely serious were it not for the
fact that the use of respirators is mandatory in these particular
smelters.
Comparable data are not available for exposures in secondary
smelters. Secondary smelters are to be found in or near most large
cities. They depend on the local supply of lead scrap in the form of
discarded electric storage batteries, cable casings, pipes, and other
materials for their supply of lead. The nature of the operation is
similar to the one described for primary smelters, except that no ore-
processing is involved. Tola (1974) has recently reported on hazards
in secondary lead smelters in Finland. The work practices involved
were not described. Thus, it was not indicated whether or not these
Table 11. Air lead concentrations in three primary lead smelters (µg/m3)a
Smelter Year Locationb Meansc Mean Range
of (all values)
means
A 1972-75 (1) 610, 1930, 2860 1800 250-3670
(2) 970, 470, 450 630 250-1380
(3) 860, 950, 320 710 200-1700
(4) 1220, 350, 950 840 260-1640
B 1973-74 (1) 1310, 2330, 4720 2790 370-5160
(2) 2740, 3460, 770 2320 310-7570
(3) 860, 140, 530 510 120-1560
(4) 1270, 540, 5730, 4050 2900 60-7220
C 1973-74 (1) - - -
(2) 3850, 8740, 830 4470 < 10-31 200
(3) 1320, 230 780 90-1340
(4) 80 80
a Data provided by M. Varner, American Smelting and Refining Co.,
Salt Lake City, Utah, U.S.A.
b Locations: (1) Ore proportioning; (2) nodulizing and sintering;
(3) blast furnace; (4) drossing and reverberation.
c Determined with personal monitors on separate occasions.
Each sampling period was 5-7 hours.
workers wore respirators on the job. But whatever the work practices
may have been, they were not adequate. Out of 20 smelters and
founders, 16 had blood lead concentrations equal to or greater than
70 µg/100 ml.
Foundries in which molten lead is alloyed with other metals have
also been sources of high atmospheric exposure. In one such operation
the concentration of lead was 280-290 µg/m3 (Berg & Zenz, 1967).
5.3.2 Electric storage battery manufacturing
The electric storage battery industry has been studied fairly
carefully with reference to the nature and degree of lead exposure.
Within the manufacturing process, there are numerous specific
operations that are hazardous by virtue of the resultant high air lead
concentrations. Plate casting is a molten metal operation. The hazard
here is from spillage of dross, resulting in dusty floors. Mixing of
lead oxide paste runs parallel to grid casting. Here, as in subsequent
operations, the major hazard is from lead oxide dust, particularly
when loading the mixer with lead oxide powder. Ventilation is needed
during loading and frequent clean-up is necessary to prevent the
accumulation of dust. Pasting of the plates follows, either by hand or
by machine. In either case the hazard is from dust which accumulates
as the paste dries. The plates are then cured, oven dried and removed
for the forming process. Although the plates must be welded into
circuits, the temperature is not high enough to generate significant
concentrations of lead fumes. Once more, the main problem is lead
oxide dust, although the amount of handling involved generally does
not require ventilation. After another drying process, the plates are
stacked to make elements, either by hand or machine. In both cases the
process is dusty and ventilation is needed, but particularly with
machine stacking. The stacks are then burned to weld together the
positive and negative lugs. This is done in a ventilated burning box.
Final assembly and finishing are low-hazard operations that do not
require ventilation if conducted with care.
Reports have appeared concerning the air lead concentrations
associated with the various phases of battery manufacture. The data
summarized in Table 12, show that oxide mixing is probably the most
hazardous occupation, followed by machine pasting, assuming that the
same accumulative time is spent at each activity. This conclusion is
borne out by the result of a recent study. The blood lead
concentration was found to be most elevated and the erythrocyte ALAD
activity was most depressed among men engaged in oxide mixing and
pasting (Tola et al., 1971).
The data cited above for air lead concentrations in the lead
smelting and refining industry and in the electric storage battery
industry may not of course be wholly representative of these
industries. But they are sufficiently alarming to suggest that
respirators must be worn in most of these operations, as indeed they
were in the case of the smelters from which the data were gathered.
5.3.5 Shipbreaking and welding
Any process in which lead-containing metals are heated with
torches to high temperatures are potentially hazardous. This is due to
the formation of lead fumes with a high fraction of the airborne mass
existing in the respirable particle size range. As an example, steel
structures are coated with lead-based paint prior to final assembly.
Thus, Tabershaw et al. (1943) found the average air lead concentration
in the breathing zone of welders of structural steel to be
1200 µg/m3. Welding can also be a hazard on occasion, when the
coating is so-called zinc silicate, since zinc silicate can contain
Table 12. Air lead concentrations (µg/m3) in electric storage battery manufacturing
Operation Elkins, Tsuchiya & Williams, et al., Engels &
1950a Harashima, 1965a 1969b Kuhnen, 1973c,d
mean mean range mean S.E mean range
Oxide mixing 730 2000 250-13 000 - - 5400 180-21 600
Plate casting 260 500 200-620 50 3 - -
Pasting, hand 750 - - 150 29 710 100-2700
Pasting, machine - - - 220 25 1100 80-13 500
Forming - - - 130 13 220 30-2200
Stacking and
breaking 500 - - - - 880 110-1500
a Air sampling time not stated.
b Personal air samplers worn for full work shift for 2 weeks
c Air sampling time 40-60 minutes.
d Approximations derived by collation of various sub-categories from authors' data
substantial concentrations of lead. Welding of zinc silicate-coated
steel can give rise to breathing zone concentrations of lead far in
excess of 150 µg/m3, the current threshold limit value in the USA
(Pegues, 1960). Even the welding of galvanized steel creates
concentrations of 400-500 µg/m3. These high values were recorded
under conditions of poor ventilation. With good ventilation, welding
of zinc silicate-coated steel resulted in lead concentrations of
180 µg/m3 near the welder's nose and 70 µg/100 ml in his blood.
The recovery of scrap metal from the dismantling of ships requires
extensive cutting of steel plates with electric torches. These plates
are heavily coated with lead-based paint. Consequently, the evolution
of lead fumes and their inhalation by the shipbreakers commonly
results in lead intoxication. Air samples collected near the breathing
zone of shipbreakers show that lead concentrations of as much as
2700 µg/m3 are attained, even in the open (Rieke, 1969).
5.3.4 Printing
The hazard in a printing establishment is probably in direct
proportion to the dispersion of lead oxide dust, secondary to the
remelt operation. An early study was reported by Brandt & Reichenbach
(1943) in which melting pots were located in a variety of places where
used type was discarded. These pots were maintained at temperatures
ranging from 318°C to 477°C. The highest air lead concentration
recorded was 570 µg/m3, and the i highest average concentration for
any room was 200 µg/m3. Although working methods and industrial
hygienic conditions have probably changed considerably since this
report was published, a marginal degree of hazard still prevails.
Tsuchiya & Harashima (1965) reported a range of lead levels of
30-360 µg/m3 at breathing level in several printing shops in Japan.
Biological monitoring of workers in the printing industry has been
reported. It was found that four of those engaged in smelting had
blood lead concentrations greater than 50 µg/100 ml (Hernberg et al,
1969). There was only one blood lead value greater than 70 µg/100 ml
among the 28 workers studied.
5.3.5 Alkyllead manufacture
Tetraethyllead was first distributed as an additive to automobile
fuel in 1923. Tetramethyllead was introduced in 1960. Today, the
annual production of these two alkyllead compounds accounts for
approximately 12% of total lead consumption by industry (see 3.3).
Inevitably, workers engaged in the manufacture of these compounds are
exposed to both inorganic and alkyllead. Some exposure also occurs at
the petroleum refineries where tetraethyllead and tetramethyllead are
blended into gasoline.
The process of tetraethyllead manufacture consists of reacting a
sodium-lead alloy with ethyl chloride. The alloy is made by combining
molten lead with elemental sodium. The alloy is then transported to
the autoclaves in hoppers. After the autoclave has been charged, ethyl
chloride is added over several hours. The reaction takes place at
about 75°C for a further period of 30-60 minutes. Steam distillation
is then applied to remove residual ethyl chloride. The lead sludge is
recovered, purified by smelting and re-used. The process generally in
use for the manufacture of tetramethyllead is basically the same as
for tetraethyllead. The final step is blending with dyes and
scavengers. The product is shipped either in drums or tanker lorries.
Although there is a potential hazard from skin absorption of
tetraethyl and tetramethyllead, this is guarded against by the use of
protective clothing. In a recent study, a good correlation was found
between the organic air lead concentration in a plant and the rate of
lead excretion in the urine (Linch et al., 1970). The average
concentration of organic lead was 0.179 mg/m3 for the tetramethyl
lead operation and 0.120 mg/m3 for the tetraethyllead operation. The
somewhat higher level registered for tetramethyllead was probably
because the reaction between the organic reagent and the lead alloy
takes place at a somewhat higher temperature and pressure than that
employed in tetraethyllead production. Categories of hazard have been
established based on the frequency with which workers are removed from
exposure because of excessive urinary lead excretion (Table 13).
Table 13. Degree of hazard from lead exposure in the alkyllead
industrya
High Moderate Low
1. smelting furnaces 1. drumming plant 1. blending
2. charging autoclaves 2. steam distillation 2. pressure vessel
3. unloading and movement 3. alloying inspection
of lead pigs
4. lead recovery 4. autoclave area
5. maintenance
a Data provided by: M. R. Zavon, Medical Director, Ethyl
Corporation, Ferndale, Michigan, USA.
No exposure data are available for the blenders who mix
tetraethyllead and tetramethyllead with gasoline at the refineries,
but some exposure is likely to occur. Even at the filling stations
where gasoline is pumped into cars, the concentration of organic lead
in the vicinity of the pumps is appreciably greater than in the
ambient air. Organic lead concentrations of 0.2-1.5 µg/fm3 were
found in the vicinity of pumps (Colwill & Hickman, 1973; Harrison et
al., 1974), and the concentration of tetraalkyllead emitted from the
exhaust pipe of cars varied from 50 to 1000 µg/m3 when the engine
was idling (Laveskog, 1971).
5.3.6 Other industrial exposures
The diversity and extent of the industrial applications of lead
makes it impossible to consider all cases. Furthermore, in most
instances the actual exposure levels have not been assessed. Some
technological applications of lead are too recent to have provided
much industrial hygiene experience. For example, the use of lead
stearate as a stabilizer in the manufacture of poly(vinylchloride) is
emerging as a new hazard. In the 1971 Annual Report of the British
Chief Inspector of Factories, the number of reported cases of lead
poisoning in the plastics industry was second only to that in the lead
smelting industry (HM Chief Inspector of Factories, 1973). Other
individual cases have been reported in recent years (Scarlato et al.,
1969; Maljkovic, 1971). Lead stearate is milled and mixed with the
poly(vinylchloride) and the plasticizer, to the extent of about 1-3%.
It seems probable that the source of the problem is the dust that is
generated in the mixing process. It appears too, that lead exposure
occurs in the rubber tire industry (Sakurai et al., 1974), probably as
a result of using lead dithiocarbamate as an accelerator in rubber
manufacture.
Drawing from his own experiences and knowledge of the field,
Hernberg (1973) has provided a classification of hazard for common
industrial activities where lead is used (Table 14).
5.4 Blood Lead Concentrations of Various Populations
Under certain conditions, blood lead levels (Pb-B) are a useful
indicator of exposure and are therefore discussed in this section
dealing with environmental levels and exposures (see also section
6.1.1.2).
5.4.1 Adult populations
A great deal of data is available on the blood lead levels of
adult populations. By far the major proportion of these studies have
reported that Pb-B mean values for occupationally unexposed, rural,
and urban, populations range from 10 to 25 µg/100 ml (Hofreuter et
al., 1961; US Department of Health, Education and Welfare, 1965; Butt
et al., 1964; Holmquist, 1966; Lehnert et al., 1970; Horiuchi, 1970;
Tepper & Levin, 1972; McLaughlin et al., 1973; Tsuchiya et al., 1975).
Studies relating to populations from northern Italy have consistently
revealed somewhat higher mean values, ranging from 24 to 35 µg/100 ml
(Zurlo et al., 1970; Secchi et al., 1971; Secchi & Alessio, 1974).
Table 14. Relative hazard of lead poisoning in some occupations or operationsa
High hazard Moderate or slight hazard
Primary and secondary lead smelting Lead mining
Welding and cutting of lead-painted metal constructions Plumbing
Welding of galvanized or zinc silicate coated sheets Cable making
Shipbreaking Wire parenting
Nonferrous foundries Lead casting
Storage battery manufacture: pasting, assembling, Type founding in printing shops
welding of battery connectors Stereotype setting
Production of lead paints Assembling of cars
Spray painting Automobile repair
Mixing (by hand) of lead stabilizers into poly(vinyl Shot making
chloride)
Mixing (by hand) of crystal glass mass Welding (occasionally)
Sanding or scraping of lead paint Lead glass blowing
Burning of lead in enamelling workshops Pottery/glass making
Repair of automobile radiators
a From: Hernberg, 1973.
Similar Pb-B levels were also reported from rural and urban population
groups in France (Boudene et al., 1975). In contrast, relatively low
values (8.5 µg/100 ml) have been reported for 50 women from southern
Sweden (Haeger-Aronsen et al., 1971). These are consistent with
recently reported values for the Finnish female general population,
ranging from 7.9 (rural), to 9.7 (urban) µg/100 ml (Nordman, 1975).
As a rule, the Pb-B levels of urban populations, and of people
heavily exposed to automobile exhausts, have been found to be higher
than those of rural populations or of populations living in areas with
less traffic (Hofreuter et al., 1961; US Department of Health,
Education and Welfare, 1965; Lehnert et al., 1970; Tepper & Levin,
1972) (Table 15). In one recent study, Pb-B levels were determined
among adults before and after the opening of a motorway interchange
with a high traffic density. Pb-B levels were found to be considerably
higher among men and women living in the immediate vicinity of the
interchange after it was opened than before (Waldron, 1975). In the
evaluation of the results of this study, allowance must be made for
the facts that no control group was studied, the procedure of drawing
blood samples was changed after opening the interchange, the sampling
took place at different times of the year and no data were given
pertaining to the control of the analytical method used (atomic
absorption spectroscopy). Thus, the possibility of systematic errors
cannot be ruled out. On the other hand, Stopps (1969) found that the
Pb-B levels of people living in various places remote from
civilization had group means of 12-23 µg/100ml, values not
significantly different from group means reported for people living in
urban areas of highly industrialized countries. No information was
given in the report concerning procedures or quality control of the
analytical methods.
A distinct increase in the lead absorption has been recorded in
people living in the vicinity of lead smelters (Secchi et al., 1971;
Nordman et al., 1973; Martin et al., 1975; Graovac-Leposavic et al.,
1973).
Men have higher Pb-B levels than women (NAS-NRC, 1972). This
difference does not appear to be totally attributable to the higher
haematocrit values of men (Tepper & Levin, 1972; Nordman, 1975). At
least part of the difference is likely to be accounted for by the
higher food consumption of men.
No association has been established between Pb-B levels and age in
adults (NAS-NRC, 1972; Tepper & Levin, 1972; Nordman, 1975).
The influence of cigarette smoking is not fully evaluated; some
researchers have reported higher Pb-B levels for smokers than for non-
smokers (Hofreuter et al., 1961; US Department of Health, Education
and Welfare, 1965; Tepper & Levin, 1972), while others have been
unable to confirm such an association (Lehnert et al., 1967; Jones et
al., 1972; McLaughlin & Stopps, 1973; Nordman, 1975; Tsuchiya et al.,
1975).
Table 15. Summary of concentration of lead in blood of selected
groups of males, USAa
Mean No. of Identity of groups
(µg/100 ml) subjects
11 9 Suburban nonsmokers, Philadelphia
12 16 Residents of rural California county
13 10 Commuter nonsmokers, Philadelphia
15 14 Suburban smokers, Philadelphia
19 291 Aircraft employees, Los Angeles
19 88 City employees, Pasadena
21 33 Commuter smokers, Philadelphia
21 36 City Health Dept. employees, Cincinnati
21 155 Policemen, Los Angeles
22 11 Live and work downtown, nonsmokers, Philadelphia
23 140 Post Office employees, Cincinnati
24 30 Policemen, nonsmokers, Philadelphia
25 191 Firemen, Cincinnati
25 123 All policemen, Cincinnati
25 55 Live and work downtown, smokers, Philadelphia
26 83 Police, smokers, Philadelphia
27 86 Refinery handlers of gasoline, Cincinnati (1956)
28 130 Service station attendants, Cincinnati (1956)
30 40 Traffic police, Cincinnati
30 60 Tunnel employees, Boston
31 17 Traffic police, Cincinnati (1956)
31 14 Drivers of cars, Cincinnati
33 45 Drivers of cars, Cincinnati (1956)
34 48 Parking lot attendants, Cincinnati (1956)
38 152 Garage mechanics, Cincinnati (1956)
a From: US Department of Health, Education and Welfare, 1965.
5.4.2 Children
European studies of Pb-B levels in children indicate that, in
general, the values are similar to or possibly even lower than those
in adults. Pb-B levels of 200 children aged 4-13 years in rural
western Ireland have been reported to be below 13 µg/100ml with 45% of
the results below 10 µg/100ml (Grimes et al., 1975). A group of 363
children aged from 8 days to 8 years was surveyed in the
Nuremberg/Erlangen area. The children displayed a mean Pb-B level of
3.3 ± 2.6 p.g/100 ml in the first year of life; the Pb-B level
increased year by year and reached a mean of 11.5 ± 4.9 µg/100 ml at
the age of 6-8 years (Haas et al., 1972a). However, most of the
available data on Pb-B levels in children have been obtained as a
result of case-finding programmes conducted in the USA. In one study,
the average blood level of 230 children, aged 1-5 years, in two rural
counties was found to be 22.8 + 11.0 µg/100 ml (Cohen et al., 1973).
An upward correction was made for all haematocrit values below 40%;
more than half of the children lived in older houses (more than 25
years old) one-quarter of which had flaking paint or holes in the
plaster.
There has been great concern in the USA that a very large number
of inner city children have abnormally elevated Pb-B levels. The
concern is for children in the blood lead range of 40-80 µg/100 ml.
Thus, Blanksma et al. (1969) reported that in 1967, and 1968, 8%, and
3.8%, respectively, of Chicago slum children had Pb-B concentrations
in excess of 49 µg/100 ml. This study involved 68 744 children, the
majority of whom were between 1 and 6 years of age. The problem is not
limited to large cities. Fine et al. (1972) reported on a survey of 14
Illinois communities with populations ranging from 9641 (Robbins) to
126 963 (Peoria). Of a total of 6151 children, 18.6% had Pb-B levels
higher than 39 µg/100 ml and 3.1% had levels higher than 59 µg/100 ml.
Some of the communities were in the Chicago urban complex, but a
considerable number were not. There did not appear to be any great
difference in the percentage of children having an excessive
concentration of lead among the Chicago urban communities as compared
with the downstate and western Illinois communities. The findings are
certainly not unique to Illinois. In a recent survey, 34% of 343
children in an impoverished area of Boston had Pb-B levels in excess
of 39 µg/100ml and 12% were over 49 µg/100ml (Pueschel et al., 1972).
Similar data have been gathered recently in New York City and
elsewhere.
6. METABOLISM OF LEAD
6.1 Absorptiona
The absorption of lead from environmental sources is not solely
dependent on the amount of lead presented to the portals of entry per
unit time. It is also dependent on the physical and chemical state in
which the metal is presented and it is influenced by host factors such
as age and physiological status. The amount of food eaten and the
amount of air breathed, with the proportionate ingestion or inhalation
of lead, are functions of metabolic activity. Men engaged in heavy
work breathe more air and eat more food than sedentary individuals of
the same weight, and children eat almost as much food and breathe
almost as much air as middle-aged adults.
6.1.1 Absorption by inhalation
A large amount of information has accumulated regarding the
factors that determine the degree of deposition and retention of
inhaled aerosols in general (Task Group on Lung Dynamics, 1966). With
appropriate knowledge of the aerodynamic characteristics of lead
aerosols, it would be possible to make reasonable predictions from the
lung model developed by the ICRP Task Group on Lung Dynamics,
concerning the fractional deposition that would occur in the human
airways. It would also be possible to predict the pattern of regional
deposition in the airways. Unfortunately, the knowledge necessary for
making accurate predictions is not available, particularly in the case
of industrial exposure.
The ICRP lung model would predict that approximately 35% of the
lead inhaled in general ambient air would be deposited in the airways,
since the aerodynamic diameterb of the lead particles is
approximately 0.1-1.0 µm (see section 5.1.1). The lung model would
also predict that regional deposition would be predominantly in the
alveolar bed and in the deeper regions of the tracheobronchial system.
Furthermore, it would predict that fractional deposition of lead dusts
generated in an industrial environment would be greater than it would
be for lead in general ambient air, however, the deposition would be
a In this document, absorption and uptake are used synonymously.
b Aerodynamic diameter = diameter of a unit density sphere with the
same settling velocity as the particle in question (Task Group on
Lung Dynamics, 1966).
mainly in the nasopharynx rather than in the pulmonary bed or
tracheobronchial region, owing to the larger particle size. Industrial
lead fumes, such as those generated in the process of cutting metals
with electric torches, would be of small particle size and would
behave accordingly. But even the lead aerosols breathed by the general
population are not well enough characterized to predict deposition.
This is particularly true for the very small particles (<0.1 µm)
which are largely deposited by diffusion (Lawther, 1972).
The adequacy of the ICRP lung deposition model is open to
question, at least for small particles. The model predicts a total
airway deposition of 40-50% for 0.5-µm particles, whereas a study in
human volunteers indicated a deposition of only 6-16% depending on the
rate and depth of respiration (Muir & Davies, 1967).
Predictions concerning the characteristics of airway clearance of
lead aerosols using the ICRP lung model are even more difficult to
make than predictions regarding deposition. The lung model would
predict that the fate of lead deposited in the airways would vary
greatly depending on its solubility characteristics and on the
inherent toxicity of the particles to the clearance mechanism (lung
macrophages and cilia). The chemical forms of lead in air are both
numerous and variable, depending on the source and on residence time
in the air (see section 5.1.1). In many types of industrial exposure,
lead is probably mainly in the form of lead oxide.
6.1.1.1 Human studies
Actual studies on the fractional deposition of particles in the
respiratory tract of man have not been extensive, especially in the
case of lead. Kehoe (1961) studied the deposition of lead in human
volunteers with an air lead level of 150 µg/m3. The source of lead
was combusted tetraethyllead which produced lead (III) oxide
(Pb2O3) in the air. Subjects breathed air containing particles
with an average diameter of 0.05 µm viewed under the electron
microscope, with 90% ranging from 0.02-0.09 µm. A diameter of 0.05 µm
for lead (III) oxide as seen under the electron microscope represents
a mass median equivalent diameter of approximately 0.26 µm (NAS-NRC,
1972). Subjects also breathed air containing particles having an
average diameter of 0.9 µm (mass median equivalent diameter = 2.9 µm);
36% of the smaller particles, and 46% of the larger particles, were
deposited.
Nozaki (1966) also reported on lung deposition of inhaled lead in
man. Lead fumes were generated in a high-frequency induction furnace
and were inhaled at a concentration of 10 000 µg/m3. Particle size
was closely controlled according to the method of Homma (1966). The
results (see Table 16) were similar to those of Kehoe (1961) and were
reasonably consistent with the ICRP lung deposition model (Task Group
on Lung Dynamics, 1966).
These data suggest that an estimate of 30 ± 10% deposition is
reasonable for the usual general ambient air situation and that lead
oxide deposition characteristics will vary considerably, depending on
the particle size and on the depth and frequency of respiration.
However, one cannot predict the contribution of airborne lead to
the body burden of lead on the basis of deposition studies alone.
Regional deposition probably varies greatly from one exposure
situation to another, that is, the industrial setting versus the
ambient environment. Also, the nature of lung clearance is unknown and
is difficult to study. Nevertheless, it is possible to determine
short-term lung clearance by carrying out gamma ray lung scans
following inhalation of 212Pb. Such a study in man has been reported
(Hursh & Mercer, 1970) but its relevance to the rate of clearance of
the chemical and physical forms of lead usually inhaled by man is
highly questionable. Such radioactive lead studies involve the
adsorption of 212Pb atoms on carrier aerosol particles. The
desorption of lead atoms from aerosol nuclei under these artificial
circumstances may be quite significant, and the estimated rate may be
totally unlike the clearance rate for ambient air lead particles.
Kehoe (1961) has reported that when a subject breathed large-
particle aerosols of lead (III) oxide (approximately 2.9 µm mass
median equivalent diameter) for many weeks at 150 µg/m3 a very
substantial increase in faecal excretion occurred, probably reflecting
the fact that the particles were largely trapped in the nasopharynx
and swallowed. When the same subject inhaled air with a lead
concentration of 150 µg/m3, with the lead in small particles
(approximately 0.26 µm mass median equivalent diameter), only a small
rise in faecal lead excretion was observed.
During inhalation of particulate air pollutants, the lead dust
comes into contact with lung cells, which are primarily responsible
for phagocytosis. It must be remembered that alveolar macrophages are
damaged in vitro by inorganic lead compounds (Beck et al., 1973),
and that similar effects have been demonstrated in vivo in rats and
guinea-pigs (section 6.1.1.4). It seems possible, therefore, that the
lung defence mechanisms are, to some extent, impaired in an
environment with a high air lead concentration, and that the rate of
absorption of inhaled particles under such circumstances is affected.
In summary, studies of airway deposition and clearance of lead in
man have not, as yet, provided any clear indication of the daily
absorption to be expected under realistic conditions. They have only
emphasized the necessity to consider other kinds of data to obtain
this information.
Since the concentration of lead in the blood is thought to reflect
current and recent lead exposure, the degree of lead intake from air
should be reflected in this factor.
Table 16. Deposition of lead fumes in the airways of human subjectsa
10 respirations/min: 1350 cm3 tidal air 30 respirations/min: 450 cm3 tidal air
Particle diameterb % Deposition Particle diameterb % Deposition
(µm) (µm)
1.0 63.2 1.0 35.5
0.6 59.0 0.6 33.5
0.4 50.9 0.4 33.0
0.2 48.1 0.2 29.9
0.1 39.3 0.1 27.9
0.08 40.0 0.08 26.5
0.05 42.5 0.05 21.0
a Adapted from Nozaki, 1966.
b Mass median equivalent diameter.
6.1.1.2 The relationship of air lead to blood lead in the general
population
The risk to man from lead in air has become a matter of
considerable concern in recent years. Studies of lead deposition and
retention in the airways of man have not been very enlightening. A
more indirect but nonetheless useful approach to the problem starts
from the assumption that the concentration of lead in the blood is
proportional to the concurrent level of total uptake by way of the
several portals of entry. It follows that each environmental source
(mainly air, food and water) would contribute to the blood lead
concentration in direct proportion to its contribution to the total
daily lead uptake. Up to the present time, such a relationship has
never been rigorously demonstrated. Goldsmith & Hexter (1967)
developed a linear regression plot of log Pb-B versus log lead
concentration in air. The air lead samples were not necessarily taken
at the same time and place as the blood samples. Thus, the regression
line was calculated on the basis of rather imprecise information.
However, data from experimental human subjects breathing known high
concentrations of lead oxide were found to fit the regression line
rather well. A cogent criticism is the fact that the validity of the
air lead data as applied to the specific blood lead data is very
uncertain. The contribution of air lead to blood lead, as inferred
from the Goldsmith-Hexter curve, is about 1.3 µg of lead per 100 ml of
blood per 1 µg of lead per m3 of air. Other epidemiological studies
have been made of the relationship between air lead and blood lead.
Azar et al. (1973) monitored the inhaled air of 150 individuals using
personal air samplers continuously, 24 hours per day. The air lead
exposure ranged from 2 µg/m3 to 9 µg/m3. There was a significant
correlation between log air lead level and log blood lead level, when
data from all the cities involved were pooled. The contribution of air
lead to blood lead was found to be somewhat less (approximately 1.0 µg
of lead per 100 ml of blood per 1 p.g of lead per m3 of air over the
range of air lead concentrations studied), than was estimated from the
Goldsmith-Hexter curve.
Another recent epidemiological investigation which examined the
relationship between air lead and blood lead levels was the Seven
Cities Study (Tepper & Levin, 1972). No significant correlation was
found between air lead and blood lead levels over an air lead range of
0.17-3.39 µg/m3. A major deficiency was the fact that the air data
were obtained from fixed outdoor sampling stations in the 11 cities
involved.
Two studies have been reported recently in which the relationship
between blood lead and air lead levels was investigated in human
volunteers. In one study, 14 male volunteers were exposed to a lead
oxide aerosol for 23 hours per day at an average concentration of
10.9 µg/m3 for up to 17 weeks. Blood lead concentrations and other
parameters were measured before, during, and following the exposure
period. A plateau of blood lead concentration was attained during the
exposure, and a return to pre- or near pre-exposure levels was
observed during the post-exposure period. The air contribution to the
Pb-B levels was approximately 1.4 µg of lead per 100 ml blood per 1 µg
of lead per m3 of air (Coulston et al., 1972b). In another study,
male volunteers inhaled an air lead concentration of 3.2 µg/m3. The
blood lead level increased from 18 µg to 25 µg/100 ml, that is
approximately 2 µg of lead per 100 ml blood per 1 µg of lead per m3
(Coulston et al., 1972c). Rabinowitz (1974) reported a study of a
single volunteer using stable lead isotope tracers in which the sudden
removal of the normal lead in air by filtration resulted in a
reduction of the blood lead concentration from approximately
14.5 µg/100ml to approximately 11.3 µg/100 ml over a period of 40 days
(Fig. 2). The average air lead levels were estimated taking into
account measurements made indoors and outdoors, and the time spent in
both locations. Prior to the experiment (day 109), the average air
lead concentration was 1.6 µg/m3 and during the experiment (day
109-148) it was 0.2 µg/m3. In calculating the contribution of air
lead to blood lead at day 148, allowance should be made for the fact
that the concentration of lead of normal isotopic composition was
decreasing prior to the removal of lead from the air. If this is taken
into account the contribution of air lead to blood lead at day 148
would be about 1.7 µg of lead per 100 ml of blood per 1.4 µg of lead
per m3 of air, or 1.2 µg of lead per 100 ml of blood per 1 µg of
lead per m3 of air. It is unfortunate that it was not possible to
follow the blood lead concentrations for a longer period of time after
removal of lead from room air, since a new steady state had not been
fully achieved. The study is also of limited value for application to
the general population because only one individual was studied.
On the other hand, the Coulston study was deficient in that the
form of air lead breathed (lead (III) oxide) may be deposited in, and
cleared from, the airways in a significantly different manner from
lead, as it actually occurs in general ambient air.
In conclusion it seems, that there is probably a perceptible
effect of air lead on blood lead in the range of air lead
concentrations applicable to the general population. The data
available suggest that with blood lead levels in the range found in
the general population, air lead levels may contribute from 1.0 to
2.0 µg of lead per 100 ml of blood per 1 µg/m3 of air.
5. ENVIRONMENTAL LEVELS AND EXPOSURES
In the preceding chapter the general pattern of the environmental
transport and distribution of lead was described. This chapter is more
specifically concerned with the different circumstances under which
people are exposed to lead to a degree that may be hazardous to their
health.
5.1 Exposure of the General Population
The general population is exposed to lead by ingestion of food,
and water, and by inhalation. In addition, children are exposed by
eating non-food items, and those working in the lead industries suffer
exposure over and above their exposure as members of the general
population. These categories of exposure will be considered
separately.
5.1.1 Air
The highest concentrations of lead in ambient air are found in
dense population centres. The larger the city, the higher the ambient
air lead concentration. As one moves away from the centre of the city,
the concentration falls progressively. For urban stations, an average
concentration of 1.1 µg/m3 has been reported; for non-urban stations
(near the city) the average was 0.21 µg/m3; for stations somewhat
farther removed it was 0.10 µg/m3, and for remote areas,
0.02 µg/m3 (McMullen et al., 1970). Air over streets with heavy
traffic contained more lead than air over streets with light traffic,
and considerably more than the ambient air over rural areas.
There is a clear pattern in this picture, the non-urban sites
showing less than 0.5 µg/m3, while the urban sites have values
ranging from 1 to 5-10 µg/m3. The highest levels have been recorded
on highways during rush hours, 14-25 µg/m3 (WHO Expert Committee,
1969).
The results of continuous monitoring for 1971-72, in 27 European
cities, by 43 uniform sampling stations are summarized in Table 7
(Commission of European Communities, 1973).
The ambient air lead levels at 15 national sampling stations in
Japan in 1973 were 0.30 µg/m3 for the average 24-hour value,
2.72 µg/m3 for the maximum 24-hour value, and 0.01 µg/m3 for the
minimum (Environment Agency, Japan, 1975).
Table 7. Air lead concentrations in some cities of the European Community (1971-72)a
Location Continuous measurements Traffic-hour measurements
Non-urban monthly averages < 0.5 µg/m3 -
daily maxima < 1 µg/m3 -
Small cities
residential areas monthly averages < 1 µg/m3 -
daily maxima < 2 µg/m3
traffic areas - monthly averages < 3 µg/m3
individual measurements < 8 µg/m3
Metropolitan areas
residential areas monthly averages < 2 µg/m3 individual measurements < 4 µg/m3
daily averages
up to 8 µg/m3
traffic areas monthly averages monthly averages < 10 µg/m3
up to 6.5 µg/m3
daily values up to 10 µg/m3 single measurements
up to 20 µg/m3
a Data from the Commission of the European Communities (1973).
People who live in close proximity to dense automobile traffic are
exposed to appreciably higher concentrations than others. In Los
Angeles, California, where general ambient air levels are unusually
high, the monthly mean concentration near traffic was as high as
6.4 µg/m3 (US Department of Health, Education and Welfare, 1965).
This is in contrast to the general ambient air level of 2-4 µg/m3
reported for that city (Tepper & Levin, 1972). There is also a diurnal
pattern whereby the concentration rises and falls in approximate
proportion to the vehicular traffic activity (US Department of Health,
Education and Welfare, 1965; Lahmann, 1969; Heller & Kettner, 1969;
Chovin et al., 1973). Most studies report that a seasonal variation
also occurs (Tepper & Levin, 1972; Georgii & Jost, 1971).
Nearly all air lead measurements in communities have been made
outdoors. Only a small number of indoor concentration studies are
available (e.g. Fugas et al., 1973; Yocom et al., 1971; Daines et al.,
1972). Indoor levels vary from slightly lower than, to about 1/3 of,
comparable outdoor levels. Higher indoor levels are found only in lead
industry environments. In the absence of specific data, reference
should be made to the much more voluminous literature available on the
penetration of undifferentiated particulate pollution into buildings.
This was reviewed by Benson (1972). In general, very small particles
enter buildings readily, and exist there at levels similar to those
outside. Larger particles, near stationary sources and very close to
roadways, penetrate buildings less readily.
Studies of air lead concentrations over a number of years or even
a decade, at the same or similar locations, have produced quite
variable results. Occasionally air lead levels have declined as in
Cincinnati, Ohio (US Department of Health, Education and Welfare,
1965). This was attributed to greatly decreased coal consumption. The
US National Air Surveillance Network, which has most of its stations
in city centres, has shown little change in large cities and variable
behaviour in smaller cities (NAS-NRC, 1972). Tepper & Levin (1972) and
Chow & Earl (1970) have shown considerable increases in air lead
levels at a number of stations in large cities. In 1967, Ott et al.
(1970) developed a predictive model of increasing automotive pollution
based on carbon monoxide emission patterns. Since air lead comes
largely from vehicular sources, this report should be considered when
changes in air lead with time are evaluated.
The respiratory uptake of lead from air depends on total lead
concentration, particle size distribution, particle shape, chemical
composition, physicochemical properties, and respiratory volume
(section 6.1.1).
The particle size distribution of lead in ambient air has been
studied by a number of investigators. As regards pulmonary deposition
and absorption, the mass median equivalent diametera rather than the
microscopic particle size is considered appropriate. Robinson & Ludwig
(1967) reported a mass median equivalent diameter of 0.25 µm, with 25%
of the particles smaller than 0.16 µm and 25% larger than 0.43 µm.
These data were representative of a variety of areas in Los Angeles,
the San Francisco Bay area, Cincinnati, Chicago, and Philadelphia.
There was little variation from one city to another. Other studies
conducted in the United States of America gave similar results
(Mueller, 1970; Robinson et al., 1963). More recently Lee et al.
(1972) have reported mass median equivalent diameters of 0.42-0.69 µm
for six United States cities. Jost et al. (1973) reported that 50% of
particles had mass median equivalent diameters of less than 0.4 µm and
20% of more than 0.5 µm.
Not much is known about the chemical form in which general ambient
air lead occurs. Ter Haar & Bayard (1971) studied the composition of
airborne lead particulates with an electron microprobe analyser. They
studied particulates collected directly from the exhaust pipe of a car
and also from air at various distances from a busy highway. Their
results (Table 8) indicate that car exhaust lead is initially composed
of halides that are converted to oxides, sulfates, and carbonates with
aging.
Alkyllead vapours occur in ambient air because some of the
alkyllead in gasoline escapes combustion. Purdue et al. (1973) have
recently reported on the organic lead concentration in an underground
parking garage and in the general ambient air of six major cities in
the USA. In the parking garage, the total air lead level was
11.7 µg/m3, of which 16.7% was organic lead. In the six major
cities, the organic lead concentration was about 10% of total lead.
There is some uncertainty as to the accuracy of the organic lead data
since the concentrations found approached the detection limit of the
method. In another study in Los Angeles, using a different method for
trapping organic lead, approximately 2% of the lead was found to be
organic (Snyder, 1967). Differences found in the concentration of
organic lead relative to particulate lead can perhaps be explained in
part by differences in proximity to the emitting source. Laveskog
(1971) made repeated studies over several months on the presence of
organic lead in the air at a number of locations in Stockholm. The
a Mass median equivalent diameter = equivalent diameter above and
below which the weights of all larger and smaller particles are
equal.
levels were uniformly low (under 10% of the total lead) except for 2
brief periods. These occurred near a gasoline station, and were
attributed to the evaporation of spilled fuel. Colwill & Hickman
(1973) verified this concept in similar studies near gasoline handling
installations.
The air in the vicinity of lead smelters may be appreciably
polluted and thus can affect the general population. A detailed study
has recently been made of the environmental impact of a large ore
smelter near El Paso, Texas (Landrigan et al., 1975b). The annual mean
concentration in 1971 was approximately 80 µg/m3 in the immediate
vicinity of the smelter and fell off rapidly, attaining a near-
background level of 1 µg/m3 about 5 km away. Approximately 42% of
the particle mass had an aerodynamic diameter of less than 2 µm. In a
similar study conducted near a smelter and a mine in the Meza Valley,
Yugoslavia, the air lead concentration, 10 km away, ranged from 1.3 to
24.0 µg/m3 (Djuric et al., 1971). Five air sampling stations were
located at various distances within 10 km of the smelter stack. About
45% of the particles had a diameter equal to or less than 0.3 µm and
an additional 25% were in the range of 0.31-0.8 µm. Although not
specified, it is probable that these particle sizes were expressed as
aerodynamic diameters, since the authors refer to this range as being
of optimum size for absorption. The extensive pollution in some
directions was probably due to the topography. The Meza Valley is only
a few hundred metres wide and depending on wind direction, the lead
particles can be conveyed long distances.
Roberts et al. (1974) reported the lead levels in air, dust, soil,
and in the blood of children living near two secondary smelters in
totonto. Monthly mean air lead levels (1.0-5.3 µg/m3) were about
twice those encountered in other parts of the city, but were subject
to greater daily variation. Lead levels in dustfall of
200-1500 mg/m2/month, and soil levels of 16 000-40 000 mg/kg of soil
near the smelter declined to background levels within 300-400 m of the
stacks. From 13 to 30% of the children living within 300 m of the
stacks had blood lead levels of over 40 µg/100 ml.
Pollution of the surrounding country by a secondary lead smelter
has been reported to have affected the lead absorption of adults
living 1-4 km from the main emitter (Nordman et al., 1973). Air lead
concentrations were not reported. The dustfall lead concentrations
ranged from 10 mg/m2/month (4 km from the chimney) to
200 mg/m2/month (200 m from the chimney). There was a correlation
between blood lead concentration and degree oferythrocyte ALAD
inhibition on the one hand and the proximity of habitation to the
smelter on the other. A correlation between Pb-B values and monthly
dustfall lead was also demonstrated.
Table 8. Composition of airborne lead particles by electron microprobe analyser.a
Percentage of total
particles counted
Sample PbCl2 PbBr2 PbBrCl Pb(OH) Pb(OH) (PbO)2 (PbO)2
Cl Br PbCl2 PbBr2
Exhaust pipe
Zero time 10.4 5.5 32.0 7.7 2.2 5.2 1.1
18 h 8.3 0.5 12.0 7.2 0.1 5.6 0.1
Eight Mile Road
Near road 11.2 4.0 4.4 4.0 2.0 2.8 0.7
400 yards 10.5 0.7 0.6 8.8 1.1 5.6 0.3
Rural site 5.4 0.1 1.6 4.0 - 1.5 -
Percentage of total
particles counted
Sample (PbO)2 PbCO3 Pb3 PbOx (PbO)2 PbO PbSO4
PbBrCl (PO4)2 PbCO3 PbSO4
Exhaust pipe
Zero time 31.4 1.2 - 2.2 1.0 - 0.1
18 h 1.6 13.8 - 21.2 29.6 0.1 -
Eight Mile Road
Near road 2.0 15.6 0.2 12.0 37.9 1.0 2.2
400 yards 0.6 14.6 0.3 25.0 21.3 4.6 6.0
Rural site 1.0 30.2 - 20.5 27.5 5.0 3.2
a From: Ter Haar & Bayard (1971).
5.1.2 Water
Man's exposure to lead through water is generally low in
comparison with exposure through air and food (WHO Working Group,
1973). The lead concentration in the water supplies of most of the 100
largest American cities, as determined in 1962, ranged from a trace to
62 µg/litre (Durfor & Becker, 1964). Since 1962, continuous monitoring
of American water supplies has indicated that the US Public Health
Service prescribed limit of 50 µg/litre has not been exceeded (NAS-
NRC, 1972). In another study, only 41 out of 2595 samples of tap water
contained more than 50 µg/litre, and 25% contained no measurable
amount of lead (McCabe, 1970).
Under some circumstances, the concentration of lead in drinking
water can become extremely high. Gajdos & Gajdos-Török (1973)
described two cases of severe clinical lead poisoning attributable to
a municipal water supply that contained lead levels of 2.6 mg/litre.
In another case, in rural Scotland, four people developed clinical
lead poisoning and others showed biochemical evidence of grossly
elevated lead exposure (Goldberg, 1974). The concentration of lead in
the domestic water supply was 2-3 mg/litre. In this case, the reason
for the extreme contamination was that the water was stored in lead
tanks. In another study, conducted in Glasgow, Scotland, it was shown
that lead pipes in the plumbing of homes can also result in high
concentrations of lead in soft water (water low in calcium and
magnesium) (Beattie et al., 1972a). Homes with both lead-lined water
storage tanks and lead pipes had the highest concentration. The
plumbo-solvency of water standing in lead pipes is influenced
significantly by several factors. The solvency increases about four-
fold with increasing acidity over the pH range from 6 to 4. Increases
of a somewhat lesser degree were also noted with increasing alkalinity
over the range of pH from 8 to 10 (Moore, 1973). The same author also
pointed out the increasing plumbo-solvency of water with increasing
temperature and with decreasing calcium concentration. Quite recently
it was shown that lead concentrations in tap water were highly
dependent on the volume of water flushed through the system before
sampling. The concentrations were also considerably lower when a 95/5
(tin/lead) solder had been used in the copper piping instead of the
50/50 or 60/40 solders (Wong & Berrang, 1976).
When water was left standing overnight in plastic pipes, some
degree of leaching of lead into the water was observed (Heusgem &
DeGraeve, 1973). The source of lead in this case was probably lead
stearate which is used as a stabilizer in the manufacture of polyvinyl
plastics. The problem of plastic pipes has been discussed recently
(Schaller et al., 1968; Packham, 1971). Packham did not feel that
there was a hazard associated with the use of such material in
domestic water supply systems. But more study is needed, particularly
of situations in which water stands in the pipes for prolonged
periods.
Lead levels in surface and ground waters were recently reviewed by
a WHO Working Group (1973). Natural surface waters have been reported
to contain usually less than 0.1 mg/litre (Kopp & Kronen, 1965). In
unpolluted areas the concentrations are of the order of 1 µg/litre or
less (Zukovickaja et al., 1966). Some rivers in France were recently
analyzed by Servant (1973) who found that, in the Midi-Pyrenees
region, the mean concentration of dissolved lead varied from 6.7 to
10.4 µg/litre.
5.1.3 Food
The contribution of food to man's exposure to lead has been under
study for many years, beginning with the study of Kehoe et al. (1933)
who found lead in every item of food in both industrial and primitive
societies. The concentration of lead in various items of food is best
described as highly variable. In fact, there seems to be about as much
variation within specific items of food as between different
categories of foods. For example, Schroeder et al. (1961) found that
the range was 0-1.5 mg/kg for condiments, 0.2-2.5 mg/kg for fish and
seafood, 0-0.37 mg/kg for meat and eggs, 0-1.39 mg/kg for grains, and
0-1.3 mg/kg for vegetables.
Estimates of actual consumption of lead in food and beverages have
been made using two general approaches. Some investigators have used
the duplicate portions sampling method. Others have derived
theoretical intakes based on nutritional tables and known
concentrations of lead in the dietary components (composites
technique, see section 2.2.1). The results of studies using the two
methods are given in Table 9. In general, the composites technique
appears to yield somewhat higher mean values for adults than the
duplicate portion technique. There are considerable differences in the
daily intakes reported from different countries. Whether these
differences are real or due to factors associated with the methods
used remains to be assessed. Inadequacies in sampling and in the
analytical methods used may account for a considerable part of the
differences; few of the studies cited present any evidence of
interlaboratory quality control of the analytical assays. Most
estimates do not specify the age, sex, or level of physical activity
assumed in arriving at the estimates. These are very important
determinants of dietary intake. Thus, the calorie requirement for a
25-year-old male in the United States of America is approximately 2900
calories, whereas it is only 1900 calories for women, age 35-55
(Altman & Dittmer, 1968). Horiuchi et al. (1956) were quite aware of
the vast differences in food intake among different categories of
adults and between adults and children. They made an effort to take
these factors into account in developing their estimates of lead
intake from dietary sources.
Daily faecal lead excretion can also be used as a means of
estimating daily lead ingestion, since only approximately 10% of
dietary lead is absorbed (Kehoe, 1961). This approach, which was used
Table 9. Dietary lead intake.
Method Age Sex Activity Lead/day (µg) No. of References
subjects
range average
Duplicate adult male sedentary 120-350 218 9 Kehoe, 1961
portions adult male sedentary 74-216 113 17 Coulston et al., 1972b
21-30 years 4 male - 237-306 274 5 Thompson, 1971
1 female
adult - - 4.8-83.0 17.8 - Schütz et al., 1971
adult male medium- 119-360 231 35 Nordman, 1975
heavy
adult female medium 89-305 178 36 Nordman, 1975
3 months- - 40-210b 8 Alexander et al, 1973
8.5 years
Composites adult male heavy work - 455 - Horiuchi et al., 1956
technique adult male medium - 299 - Horiuchi el al., 1956
10 years male - 254 - Horiuchi et al., 1956
10 months male - 126 - Horiuchi et al., 1956
18 years male medium - 57-233a - Kolbye et al., 1974
adult male medium - 139 - NRC (Canada), 1973
adult male medium - 518 - Lehnert et al., 1969
adult male medium - 505 - Zurlo at al., 1970
a See page 52
b Ranging from 40 µg in a breast-fed infant to 210 µg in an 8´-year-old child, as calculated using ICRP
by Tepper & Levin (1972) (section 6.1.2.2), presents some uncertainty
regarding actual absorption and neglects contributions to faecal lead
from gastrointestinal secretion, which cannot be estimated.
A factor that is usually ignored is the occurrence of lead in
various foods at concentrations below the practical detection limits.
Thus, Kolbye et al. (1974) arrived at different estimates based on the
assumptions they made regarding whether or not lead was present in all
items eaten. There was uncertainty as to how to cope with the problem
of lead concentrations reported as "zero" or "trace" in certain
samples. When "zeros" and "traces" were accepted as meaning absolutely
no lead, the estimated daily lead intake was 57.4 µg for an 18-year-
old man. This seemed unduly low, particularly in the light of the fact
that the faecal lead excretion of normal American women is
90-150 µg/day (Tepper & Levin, 1972). Certain assumptions were
therefore made regarding the "zeros" and "traces". If it was assumed
only that "traces" really represented 0.09 mg/kg, the calculated
intake became 159 µg/day. When the additional assumption was made that
"zero" had a finite value of 0.05 mg/kg, the calculated daily intake
of an 18-year-old male became 233 µg. Another source of error in
establishing how much is consumed relates to food preparation. Lead
may be either added to the diet or removed in the course of
preparation. Both Horiuchi et al. (1956) and the report from the
British Ministry of Agriculture, Fisheries and Foods (1972) took
special pains to explain how this problem was handled.
Published reports on lead levels in wine (Truhaut et al., 1964;
Zurlo & Griffini, 1973) show that important variations occur from
sample to sample. Considering ordinary wines there does not seem to be
any significant difference between white, red, and rosé (Truhaut et
al., 1964). Average lead concentrations of 130-190 µg/litre could be
calculated (range 60-255 µg/litre), but even higher mean values
(299 µg/litre) have recently been reported (Boudene et al., 1975).
Wine therefore is likely to be a substantial source of lead for some
people, and may account for part of the differences in Pb-B levels
(section 5.4) and in daily dietary lead intake between various
countries.
The concentration of lead in milk is a matter of special concern
because milk is a major dietary constituent for infants. Human breast
milk has been reported to contain 12 µg/litre (Murthy & Rhea, 1971)
and <5 µg/litre (Lamm & Rosen, 1974). Cow's milk has been reported to
have a similar concentration, when taken directly from cows for
analysis; 9 µg/litre (Hammond & Aronson, 1964). The concentration of
lead in processed cow's milk is higher than in human milk or in milk
obtained directly from cows. The types of processing vary considerably
as does the degree of apparent lead contamination. Thus whole milk
concentrations are only moderately elevated. Mitchell & Aldous (1974)
reported an average of 40 µg/litre in whole bulk milk and Kehoe (1961)
reported 20-40 µg/litre for local USA market milk. By contrast,
evaporated milk and formulas have still higher concentrations.
Mitchell & Aldous (1974) reported an average of 202 µg/litre for
evaporated milk. Somewhat higher values were reported by Murthy & Rhea
(1971) (330-870 µg/litre) and somewhat lower by Lamm & Rosen (1974)
(110 ± 11 µg/litre).
A major contribution to the lead content of processed milk as well
as of other food products appears to be lead solder used in the seams
and caps of cans. It has been shown that foods preserved in such cans
frequently have much higher concentrations of lead than do the same
items packed in glass containers (Mitchell & Aldous, 1974).
Although plants do not take lead up from the soil readily, fruits
and vegetables grown in areas exposed to smelter emissions may be
appreciably contaminated. Kerin (1972) determined lead in the total
diet of peasants near a smelter and found that the daily ingestion of
lead with food was 670-2640 µg.
5.1.4 Miscellaneous
The intake of lead in food, air, and water is a major concern as
regards the general population because of the pervasive nature of
these exposures. Another frequent exposure source, smoking, probably
makes a small contribution to the lead burden (section 5.4.1 ).
However, surprisingly little information is available concerning the
concentration of lead in smoking tobacco. Cogbill & Hobbs (1957)
reported the concentration of lead in two separate brands of
cigarettes and in a composite sample of five brands. Concentrations
were 19, 80, and 39 mg/kg at 58%, relative humidity or 21, 84, and
41 µg per cigarette. The amount of lead transferred to mainstream
smoke was 1.0, 3.3, and 1.9 µg per cigarette which represented 4.8,
3.9, and 6% transfer. Arsenic/lead ratios found in the tobacco
indicated that the source of lead was probably lead arsenate. At one
time lead arsenate was used extensively as an insecticide in American
tobacco fields but other pesticides rapidly replaced it shortly after
World War II. Residues of lead arsenate have probably persisted in the
fields and could contaminate plants externally. More recently,
Szadkowski et al. (1969) reported 0.483 ± 0.267 µg of lead per
cigarette in the total smoke for eight brands of cigarette. This
represented 19%, of the total lead in the tobacco or 2.6 µg per
cigarette. No distinction was made between mainstream and side-stream
smokea. Untabulated data from a study by Menden et al. (1972)
indicated that only about 2% of lead in non-filter types of cigarettes
was transferred to the mainstream smoke. The average content of lead
in commercial cigarettes was given as 10.40-12.15 µg/cigarette
a "i.e. the smoke which drills off the burning end of cigarette
between puffs.
(Petering & Menden, private communication). Most of the lead was found
in the ash; the lead content of the sidestream of individual
cigarettes varied considerably with a maximum value of 16%. Assuming
an average lead content ranging from 2.5 to 12.2 (Lehnert et al.,
1967; Szadkowski et al., 1969; Rabinowitz, 1974; Petering & Menden,
private communication) and a 2% transfer to the mainstream smoke
(Menden et al., 1972) are fair estimates, and without taking into
account the possible contribution from the sidestream smoke, a crude
assessment of the direct inhalation intake of lead from smoking 20
cigarettes a day would be about 1-5 µg.
Certain other sources of exposure are important. These sources do
not affect any major segment of the population but collectively they
no doubt account for the majority of the cases of clinical lead
poisoning in the general population.
The presence of high concentrations of lead in illicitly distilled
whisky occurs commonly in the USA and causes poisoning in adults. The
condensers used in homemade stills are often discarded automobile
radiators. These contain substantial amounts of lead in the soldered
joints. The concentration of lead in the final product frequently
exceeds 10 mg/litre. The problem of lead poisoning from this source
exists predominantly in the southeastern parts of the USA where
illicit whisky production is most common.
Another source of poisoning is improperly glazed earthenware
vessels. Improper glazing results in the leaching of lead into the
vessel, particularly when the contents are acidic. Cases of poisoning,
both fatal and non-fatal, have been recorded from the use of
improperly glazed pottery. Klein et al. (1970) reported two cases (one
fatal) in which apple juice stored in the incriminated vessel for 3
days contained 1300 mg/litre. In another case the ceramic mug
responsible was used for drinking colaa, pH 2.7 (Harris & Elsea,
1967). After two hours of standing in the mug, the cola contained lead
levels of 6.8 mg/litre. It was estimated that this patient drank 3.2
mg of lead per night in this fashion for two years. Other cases have
been reported from Yugoslavia (Beritic & Stahuljak, 1961) and from the
United Kingdom (Whitehead & Prior, 1960). The problem involves the
storage of acid materials in the vessels. In a test of the leaching of
lead from commercial and handcrafted pottery, Klein et al. (1970)
found that 4% acetic acid allowed to stand at room temperature in the
vessels for 18 hours often acquired concentrations of lead in excess
of 100 mg/litre. In fact, in more than half of the cases, the
concentration of lead exceeded 7 mg/litre.
a A popular carbonated non-alcoholic beverage.
Another source of lead poisoning in the general population is the
use of discarded storage battery casings for fuel. There is some
uncertainty as to whether the cases of poisoning that have been
recorded (Williams et al., 1933; Gillet, 1955) were due to inhalation
of lead fumes or to hand-to-mouth transfer of fallout material. The
prevalence of children in the number of recorded cases supports the
argument for hand-to-mouth transfer.
Because of the wide variety of applications of lead, additional
potential hazards are still being identified. For example, the use of
lead wire core wicks in candles was only recently called to the
attention of the USA authorities (Bridbord, unpublished results,
1973).
5.2 Exposure of Infants and Young Children
5.2.1 Soil, dust, and paint
The young child of pre-school age is exposed to special hazards
from environmental sources of lead. This is because such children
frequently exhibit the habit of licking, chewing, or actually eating
foreign objects. Lead-based paints have long been considered the major
source of excessive lead intake in young children. Thus, Sachs (1974)
reported that 80% of patients seen because of evidence of excessive
lead absorption had a history of eating paint or plaster and in
another 10% X-ray examination revealed paint in the abdomen. The
author also was of the opinion that if X-ray examinations had been
repeated at each visit to the clinic, evidence of paint ingestion
would have appeared in all patients. A similar view was expressed by
Chisholm & Harrison (1956). In their series of 105 children whose
homes were investigated, 102 of the homes contained at least one
source of paint containing 5% lead or more. Of even greater
significance was the fact that the painted surfaces identified as
sources were flaking.
Other investigators have attempted to assess the importance of
paint as a source of excessive lead exposure. Griggs et al. (1964)
found a positive correlation between the presence of elevated urinary
lead or coproporphyrin and the presence of flaking paint in the homes.
Nonetheless, in many instances the homes of children with abnormal
urine had no flaking paint indoors. Unfortunately data were not given
as to the number of children with abnormal urine and no evidence of
flaking paint indoors or outdoors. Guinee (1973) reported that in an
extensive survey of the homes of children having blood lead
concentrations equal to or greater than 60 µg/100 ml, 75% of the homes
had at least one surface in which the paint contained more than 1%
lead. Furthermore, children with elevated blood lead concentrations
were more likely to live in homes where the painted surfaces were
cracked than children with low blood lead values.
All of these studies indicate that lead in painted surfaces in
houses is almost certainly the major source of lead for infants and
young children. Some other studies suggest that the issue is not that
clear-cut. Greenfield et al. (1973) reported that, in one study, 18
out of 19 rural children with elevated blood lead concentrations lived
in homes having at least one accessible painted surface containing 1%
or more of lead, whereas paint containing 1% or more of lead could be
found on accessible surfaces in only 60% of the homes of inner city
children with excessive lead exposure. The implication is that sources
of lead other than paint were often responsible for the exposure of
city children. Two equally rational interpretations are that an
insufficient number of surfaces were tested in the children's homes or
that children often spend time in several homes, some of which might
not have been tested for lead-based paint.
Studies of sources of lead all too often ignore the fact that
painted surfaces on the outside of houses are a potential source of
lead or, for that matter, that the soil surrounding the houses may
have accumulated substantial concentrations of lead from the
weathering of outer walls. With regard to the latter, Fairey & Gray
(1970) reported that the concentration of lead in the soil near homes
where paediatric lead poisoning had occurred was over 1000 mg/kg in 27
out of 30 cases. By contrast, only 30 out of 170 soil samples taken
from yards selected at random (and not associated with cases of lead
poisoning) had concentrations of lead in excess of 1000 mg/kg.
Bertinuson & Clark (1973) have reported extremely high soil lead
values close to residences in the older section of Cincinnati. In one
case, because the distance across the yard from the base of the house
to a road with heavy traffic was sufficient, it was possible to assess
the relative contributions of lead from car exhaust and lead from the
weathering of the house. The gradient ranged from 12 000 mg/kg
adjacent to the house, down to 400 mg/kg, about 10 m from the road.
This suggested that weathering of painted surfaces of the house could
have been the major source of soil lead in this instance. Although the
high concentrations of lead in the soil in the vicinity of houses may
be due to weathering of lead-based paint, it is possible that in many
cases it is also due to the accumulation of combusted alkyllead from
car exhaust. In this connexion, recently-reported data of Ter Haar &
Aranow (1974) are particularly informative. They surveyed the profile
of lead in soil, extending from the base of 36 urban residences out to
the street gutters. Eighteen of the residences were of brick
construction and 18 were of frame construction. In summary form, their
data were as shown in Table 10. The data reflect the likelihood of the
major contribution of weathered lead-based paint to soil lead. But
they also strongly suggest that vehicular sources make a significant
and sometimes very substantial contribution to soil lead near the
sidewalks.
Table 10. Lead in dirt in Detroit (mg/kg dry dirt)a
Location Painted frame houses Brick houses
Mean Range Mean Range
Within 0.6 m of house
front 2349 (126-17 590) 351 (78-1030)
back 1586 (162-4951) 501 (72-2350)
sides 2257 (140-7284) 426 (91-1160)
1846 (104-7000) 595 (40-2290)
3 m from house
front 447 (58-1530) 156 (39-316)
back 425 (149-1410) 200 (72-480)
Near sidewalk 627 (152-1958) 324 (86-1130)
curb 572 (320-1957) 612 (147-2420)
gutter 966 (415-1827) 1213 (304-3170)
a Adapted from Ter Haar & Aronow (1974)
Street dust has also been found to contain high concentrations of
lead. Using recent data from 77 midwestern cities in the USA, it was
calculated that the concentration of lead averaged 1636 mg/kg dust in
residential areas, 2413 mg/kg in commercial areas, and 1512mg/kg in
industrial areas (Hunt et al., 1971).
In order for soil or street dust to be a significant source of
lead for man, it is of course, necessary that it be ingested and/or
inhaled. Evidence regarding the likelihood that young children would
ingest soil or street dust is extremely fragmentary. However, in a
recent study of 58 children with increased lead burdens, it was found
that 37 had a history of eating dirt and sand, compared with 34 eating
plaster, 20 eating paint flakes, 15 chewing on furniture, 14 chewing
window sills, and 7 eating wallpaper (Pueschel et al., 1972). Further
inferential evidence as to the possible significance of soil and dust
as a source of lead is to be found in the recent Smeltertown episode
near El Paso, Texas, referred to earlier (Landrigan et al., 1975b).
This town is the site of a large smelter which processes lead ores,
among others. The young children in the town have high blood lead
concentrations. In a sample of 14 children of 1-5 years of age,
78.6-100% were found to have lead concentrations equal to or greater
than 40 µg/100 ml blood. The concentration of lead in the surface soil
of Smeltertown has a median value of 3700 mg/kg. One is tempted to
conclude that the blood lead levels of these young children increased
owing to ingestion of this soil. However, the picture is somewhat
confounded by the fact that older children also showed a high
incidence of elevated blood lead concentrations but to a lesser
degree; and older children are not generally considered to exhibit
pica. Smeltertown adults had normal blood lead concentrations. Intake
of lead by inhalation would probably have affected adults as well as
children. Thus, it is likely that lead intake by the children was by
direct oral intake. The painted surfaces in the residences were seldom
in a flaking condition and were not found to be more than two or three
layers thick, in contrast to the multiple layers usually found in city
slum areas where lead poisoning is prevalent. The information
available therefore suggests that the sources of lead were soil and
dust. Indeed, there was a highly significant correlation between the
concentration of lead in the blood of the children and the
concentration of lead in household dust.
The presence of high concentrations of lead in soil is not
necessarily hazardous. Thus, children living on soils containing lead
levels of up to 8000 mg/kg showed only minimal elevations in blood
lead concentration (Barltrop et al., 1974). This was found to be so
even among the children with pica for soil. Perhaps climatic
differences are important. Smeltertown in Texas is extremely dry and
dusty whereas the region studied by Barltrop and coworkers was in
England, where the soil is presumably not as accessible to children
owing to the relatively heavy cover of vegetation. The play behaviour
of children also determines to a certain extent their exposure to lead
(Einbrodt et al., 1974).
Since dust and dirt occur indoors as well as outdoors, some
attention has been directed recently to the significance of indoor
dust. Transfer of lead-bearing house dust to the hands of young
children has recently been demonstrated (Sayre et al., 1974). The
house dust of inner city old houses contained far more lead than the
dust of newer, suburban houses. Furthermore, the hands of the children
in inner city houses were heavily contaminated with lead, whereas the
hands of suburban children were not. It is not at all certain that the
source of lead in the house dust was fallout from car exhaust. New
housing in the inner city had very little lead in dust. The inference
is that the lead was probably from the painted surfaces, since the
paint in old houses has high concentrations of lead whereas the paint
in new houses in the same area generally has a low lead content. But
even the presence of lead-containing dust on children's hands provides
little information concerning hazard since the critical question is
how much is actually transferred from the hands to the digestive
tract.
5.2.2 Miscellaneous
Facial cosmetics have long been a source of lead poisoning in
Oriental countries. Kato (1932) discussed the problems encountered in
Japan. Face powders, pastes, and liquids were found to contain as much
as 67% lead. Exposure of children was considered to be by inhalation
of powders, or ingestion of powders and other formulations. More
recently, there have been several reports of infant poisoning from a
mascara-like cosmetic used by Indian and Pakistani women (Warley et
al., 1968; Alexander & Delves, 1972). This substance may contain as
much as 88% lead sulfide.
Another source of lead exposure for young children is coloured
newsprint (Hankin et al., 1973). It has been found that the coloured
inks used in magazine illustrations contain extremely high
concentrations of lead. Coloured pages were found to have lead
concentrations of 1140-3170 mg/kg.
Children and other family members may be exposed to lead
contamination at home by work clothing being worn at home or brought
home for cleaning, or by small pieces of metal which may be brought in
(InterDepartmental Working Group on Heavy Metals, 1974).
5.3 Occupational Exposures
It is among the workers who smelt, refine, and use lead in
manufacturing items of commerce that the highest and most prolonged
exposures are found. Lead poisoning among these people was common at
one time. Today, workers, management, and physicians are generally
aware of the danger of lead and know how to handle the problem; so,
the incidence and severity of poisoning have decreased substantially
in recent years. However, much still remains to be done to eliminate
lead poisoning completely as an occupational disease. The major hazard
today seems to be in small enterprises (Engel et al., 1971) and in
some large industries where adequate industrial hygiene programmes do
not exist or are difficult to implement, or where awareness of the
existence of hazardous circumstances may be lacking.
A recent WHO study of occupational health problems in the Andean
countries (El Batawi, unpublished results, 1974) showed that, in Chile
for instance, among 580 workers exposed to lead, 21.9% had an
increased level of ALA in the urine. In Colombia, 3370 workers exposed
to lead were examined, of whom 4.30% were considered to be suffering
from lead poisoning.
The major route of lead exposure in industry is by inhalation. The
generation of lead-bearing dusts and fumes is inevitable. The workers'
clothes may also be an important source of exposure. Even the lesser
problem of oral intake of lead is really a consequence of the
generation of airborne dusts which settle out from the air on to food,
water, or other objects that are transferred to the mouth in one
fashion or another. Thus, good housekeeping and, above all, good
ventilation have a strong impact on exposure. An industrial process
may be quite safe in one factory and quite hazardous in another solely
because of differences in ventilation engineering or because of
differences in housekeeping practices and worker education.
5.3.1 Lead mining, smelting, and refining
The lead mining hazards depend, to some extent, on the solubility
of the lead from the ores. The lead sulfide (PbS) in galena is
insoluble and absorption through the lung is slight. However, in the
stomach, some lead sulfide may be converted to slightly soluble lead
chloride which may then be absorbed in moderate quantities.
The process of lead smelting and refining probably has the
greatest potential for hazardous exposure of all the lead industries.
The most hazardous operations are those in which molten lead and lead
alloys are brought to high temperatures, resulting in the vaporization
of lead. This is because condensed lead vapour has, to a substantial
degree, a small (< 5 µm), respirable particle size range. Thus,
although the total air lead concentration may be greater in the
vicinity of ore proportioning bins than it is in the vicinity of a
blast furnace in a primary smelter, the amount of particle mass in the
respirable size range may be much greater near the latter.
As an example, we can consider the processes involved in the
preparation of lead bullion in typical primary lead ore smelters in
Salt Lake City, Utah. The various processes are essentially grinding
and smelting. The main operations are: (1) ore proportioning; (2)
nodulizing and sintering; (3) blast furnace; (4) drossing and
reverberation. Air lead concentrations have been determined using
personal monitors worn by workers at the various stations. These data
are summarized in Table 11. Similar data for primary lead smelters
elsewhere are not available. However, it is evident that lead exposure
in primary smelters may be extremely high. The hazard to the workers
in the example cited would be extremely serious were it not for the
fact that the use of respirators is mandatory in these particular
smelters.
Comparable data are not available for exposures in secondary
smelters. Secondary smelters are to be found in or near most large
cities. They depend on the local supply of lead scrap in the form of
discarded electric storage batteries, cable casings, pipes, and other
materials for their supply of lead. The nature of the operation is
similar to the one described for primary smelters, except that no ore-
processing is involved. Tola (1974) has recently reported on hazards
in secondary lead smelters in Finland. The work practices involved
were not described. Thus, it was not indicated whether or not these
Table 11. Air lead concentrations in three primary lead smelters (µg/m3)a
Smelter Year Locationb Meansc Mean Range
of (all values)
means
A 1972-75 (1) 610, 1930, 2860 1800 250-3670
(2) 970, 470, 450 630 250-1380
(3) 860, 950, 320 710 200-1700
(4) 1220, 350, 950 840 260-1640
B 1973-74 (1) 1310, 2330, 4720 2790 370-5160
(2) 2740, 3460, 770 2320 310-7570
(3) 860, 140, 530 510 120-1560
(4) 1270, 540, 5730, 4050 2900 60-7220
C 1973-74 (1) - - -
(2) 3850, 8740, 830 4470 < 10-31 200
(3) 1320, 230 780 90-1340
(4) 80 80
a Data provided by M. Varner, American Smelting and Refining Co.,
Salt Lake City, Utah, U.S.A.
b Locations: (1) Ore proportioning; (2) nodulizing and sintering;
(3) blast furnace; (4) drossing and reverberation.
c Determined with personal monitors on separate occasions.
Each sampling period was 5-7 hours.
workers wore respirators on the job. But whatever the work practices
may have been, they were not adequate. Out of 20 smelters and
founders, 16 had blood lead concentrations equal to or greater than
70 µg/100 ml.
Foundries in which molten lead is alloyed with other metals have
also been sources of high atmospheric exposure. In one such operation
the concentration of lead was 280-290 µg/m3 (Berg & Zenz, 1967).
5.3.2 Electric storage battery manufacturing
The electric storage battery industry has been studied fairly
carefully with reference to the nature and degree of lead exposure.
Within the manufacturing process, there are numerous specific
operations that are hazardous by virtue of the resultant high air lead
concentrations. Plate casting is a molten metal operation. The hazard
here is from spillage of dross, resulting in dusty floors. Mixing of
lead oxide paste runs parallel to grid casting. Here, as in subsequent
operations, the major hazard is from lead oxide dust, particularly
when loading the mixer with lead oxide powder. Ventilation is needed
during loading and frequent clean-up is necessary to prevent the
accumulation of dust. Pasting of the plates follows, either by hand or
by machine. In either case the hazard is from dust which accumulates
as the paste dries. The plates are then cured, oven dried and removed
for the forming process. Although the plates must be welded into
circuits, the temperature is not high enough to generate significant
concentrations of lead fumes. Once more, the main problem is lead
oxide dust, although the amount of handling involved generally does
not require ventilation. After another drying process, the plates are
stacked to make elements, either by hand or machine. In both cases the
process is dusty and ventilation is needed, but particularly with
machine stacking. The stacks are then burned to weld together the
positive and negative lugs. This is done in a ventilated burning box.
Final assembly and finishing are low-hazard operations that do not
require ventilation if conducted with care.
Reports have appeared concerning the air lead concentrations
associated with the various phases of battery manufacture. The data
summarized in Table 12, show that oxide mixing is probably the most
hazardous occupation, followed by machine pasting, assuming that the
same accumulative time is spent at each activity. This conclusion is
borne out by the result of a recent study. The blood lead
concentration was found to be most elevated and the erythrocyte ALAD
activity was most depressed among men engaged in oxide mixing and
pasting (Tola et al., 1971).
The data cited above for air lead concentrations in the lead
smelting and refining industry and in the electric storage battery
industry may not of course be wholly representative of these
industries. But they are sufficiently alarming to suggest that
respirators must be worn in most of these operations, as indeed they
were in the case of the smelters from which the data were gathered.
5.3.5 Shipbreaking and welding
Any process in which lead-containing metals are heated with
torches to high temperatures are potentially hazardous. This is due to
the formation of lead fumes with a high fraction of the airborne mass
existing in the respirable particle size range. As an example, steel
structures are coated with lead-based paint prior to final assembly.
Thus, Tabershaw et al. (1943) found the average air lead concentration
in the breathing zone of welders of structural steel to be
1200 µg/m3. Welding can also be a hazard on occasion, when the
coating is so-called zinc silicate, since zinc silicate can contain
Table 12. Air lead concentrations (µg/m3) in electric storage battery manufacturing
Operation Elkins, Tsuchiya & Williams, et al., Engels &
1950a Harashima, 1965a 1969b Kuhnen, 1973c,d
mean mean range mean S.E mean range
Oxide mixing 730 2000 250-13 000 - - 5400 180-21 600
Plate casting 260 500 200-620 50 3 - -
Pasting, hand 750 - - 150 29 710 100-2700
Pasting, machine - - - 220 25 1100 80-13 500
Forming - - - 130 13 220 30-2200
Stacking and
breaking 500 - - - - 880 110-1500
a Air sampling time not stated.
b Personal air samplers worn for full work shift for 2 weeks
c Air sampling time 40-60 minutes.
d Approximations derived by collation of various sub-categories from authors' data
substantial concentrations of lead. Welding of zinc silicate-coated
steel can give rise to breathing zone concentrations of lead far in
excess of 150 µg/m3, the current threshold limit value in the USA
(Pegues, 1960). Even the welding of galvanized steel creates
concentrations of 400-500 µg/m3. These high values were recorded
under conditions of poor ventilation. With good ventilation, welding
of zinc silicate-coated steel resulted in lead concentrations of
180 µg/m3 near the welder's nose and 70 µg/100 ml in his blood.
The recovery of scrap metal from the dismantling of ships requires
extensive cutting of steel plates with electric torches. These plates
are heavily coated with lead-based paint. Consequently, the evolution
of lead fumes and their inhalation by the shipbreakers commonly
results in lead intoxication. Air samples collected near the breathing
zone of shipbreakers show that lead concentrations of as much as
2700 µg/m3 are attained, even in the open (Rieke, 1969).
5.3.4 Printing
The hazard in a printing establishment is probably in direct
proportion to the dispersion of lead oxide dust, secondary to the
remelt operation. An early study was reported by Brandt & Reichenbach
(1943) in which melting pots were located in a variety of places where
used type was discarded. These pots were maintained at temperatures
ranging from 318°C to 477°C. The highest air lead concentration
recorded was 570 µg/m3, and the i highest average concentration for
any room was 200 µg/m3. Although working methods and industrial
hygienic conditions have probably changed considerably since this
report was published, a marginal degree of hazard still prevails.
Tsuchiya & Harashima (1965) reported a range of lead levels of
30-360 µg/m3 at breathing level in several printing shops in Japan.
Biological monitoring of workers in the printing industry has been
reported. It was found that four of those engaged in smelting had
blood lead concentrations greater than 50 µg/100 ml (Hernberg et al,
1969). There was only one blood lead value greater than 70 µg/100 ml
among the 28 workers studied.
5.3.5 Alkyllead manufacture
Tetraethyllead was first distributed as an additive to automobile
fuel in 1923. Tetramethyllead was introduced in 1960. Today, the
annual production of these two alkyllead compounds accounts for
approximately 12% of total lead consumption by industry (see 3.3).
Inevitably, workers engaged in the manufacture of these compounds are
exposed to both inorganic and alkyllead. Some exposure also occurs at
the petroleum refineries where tetraethyllead and tetramethyllead are
blended into gasoline.
The process of tetraethyllead manufacture consists of reacting a
sodium-lead alloy with ethyl chloride. The alloy is made by combining
molten lead with elemental sodium. The alloy is then transported to
the autoclaves in hoppers. After the autoclave has been charged, ethyl
chloride is added over several hours. The reaction takes place at
about 75°C for a further period of 30-60 minutes. Steam distillation
is then applied to remove residual ethyl chloride. The lead sludge is
recovered, purified by smelting and re-used. The process generally in
use for the manufacture of tetramethyllead is basically the same as
for tetraethyllead. The final step is blending with dyes and
scavengers. The product is shipped either in drums or tanker lorries.
Although there is a potential hazard from skin absorption of
tetraethyl and tetramethyllead, this is guarded against by the use of
protective clothing. In a recent study, a good correlation was found
between the organic air lead concentration in a plant and the rate of
lead excretion in the urine (Linch et al., 1970). The average
concentration of organic lead was 0.179 mg/m3 for the tetramethyl
lead operation and 0.120 mg/m3 for the tetraethyllead operation. The
somewhat higher level registered for tetramethyllead was probably
because the reaction between the organic reagent and the lead alloy
takes place at a somewhat higher temperature and pressure than that
employed in tetraethyllead production. Categories of hazard have been
established based on the frequency with which workers are removed from
exposure because of excessive urinary lead excretion (Table 13).
Table 13. Degree of hazard from lead exposure in the alkyllead
industrya
High Moderate Low
1. smelting furnaces 1. drumming plant 1. blending
2. charging autoclaves 2. steam distillation 2. pressure vessel
3. unloading and movement 3. alloying inspection
of lead pigs
4. lead recovery 4. autoclave area
5. maintenance
a Data provided by: M. R. Zavon, Medical Director, Ethyl
Corporation, Ferndale, Michigan, USA.
No exposure data are available for the blenders who mix
tetraethyllead and tetramethyllead with gasoline at the refineries,
but some exposure is likely to occur. Even at the filling stations
where gasoline is pumped into cars, the concentration of organic lead
in the vicinity of the pumps is appreciably greater than in the
ambient air. Organic lead concentrations of 0.2-1.5 µg/fm3 were
found in the vicinity of pumps (Colwill & Hickman, 1973; Harrison et
al., 1974), and the concentration of tetraalkyllead emitted from the
exhaust pipe of cars varied from 50 to 1000 µg/m3 when the engine
was idling (Laveskog, 1971).
5.3.6 Other industrial exposures
The diversity and extent of the industrial applications of lead
makes it impossible to consider all cases. Furthermore, in most
instances the actual exposure levels have not been assessed. Some
technological applications of lead are too recent to have provided
much industrial hygiene experience. For example, the use of lead
stearate as a stabilizer in the manufacture of poly(vinylchloride) is
emerging as a new hazard. In the 1971 Annual Report of the British
Chief Inspector of Factories, the number of reported cases of lead
poisoning in the plastics industry was second only to that in the lead
smelting industry (HM Chief Inspector of Factories, 1973). Other
individual cases have been reported in recent years (Scarlato et al.,
1969; Maljkovic, 1971). Lead stearate is milled and mixed with the
poly(vinylchloride) and the plasticizer, to the extent of about 1-3%.
It seems probable that the source of the problem is the dust that is
generated in the mixing process. It appears too, that lead exposure
occurs in the rubber tire industry (Sakurai et al., 1974), probably as
a result of using lead dithiocarbamate as an accelerator in rubber
manufacture.
Drawing from his own experiences and knowledge of the field,
Hernberg (1973) has provided a classification of hazard for common
industrial activities where lead is used (Table 14).
5.4 Blood Lead Concentrations of Various Populations
Under certain conditions, blood lead levels (Pb-B) are a useful
indicator of exposure and are therefore discussed in this section
dealing with environmental levels and exposures (see also section
6.1.1.2).
5.4.1 Adult populations
A great deal of data is available on the blood lead levels of
adult populations. By far the major proportion of these studies have
reported that Pb-B mean values for occupationally unexposed, rural,
and urban, populations range from 10 to 25 µg/100 ml (Hofreuter et
al., 1961; US Department of Health, Education and Welfare, 1965; Butt
et al., 1964; Holmquist, 1966; Lehnert et al., 1970; Horiuchi, 1970;
Tepper & Levin, 1972; McLaughlin et al., 1973; Tsuchiya et al., 1975).
Studies relating to populations from northern Italy have consistently
revealed somewhat higher mean values, ranging from 24 to 35 µg/100 ml
(Zurlo et al., 1970; Secchi et al., 1971; Secchi & Alessio, 1974).
Table 14. Relative hazard of lead poisoning in some occupations or operationsa
High hazard Moderate or slight hazard
Primary and secondary lead smelting Lead mining
Welding and cutting of lead-painted metal constructions Plumbing
Welding of galvanized or zinc silicate coated sheets Cable making
Shipbreaking Wire parenting
Nonferrous foundries Lead casting
Storage battery manufacture: pasting, assembling, Type founding in printing shops
welding of battery connectors Stereotype setting
Production of lead paints Assembling of cars
Spray painting Automobile repair
Mixing (by hand) of lead stabilizers into poly(vinyl Shot making
chloride)
Mixing (by hand) of crystal glass mass Welding (occasionally)
Sanding or scraping of lead paint Lead glass blowing
Burning of lead in enamelling workshops Pottery/glass making
Repair of automobile radiators
a From: Hernberg, 1973.
Similar Pb-B levels were also reported from rural and urban population
groups in France (Boudene et al., 1975). In contrast, relatively low
values (8.5 µg/100 ml) have been reported for 50 women from southern
Sweden (Haeger-Aronsen et al., 1971). These are consistent with
recently reported values for the Finnish female general population,
ranging from 7.9 (rural), to 9.7 (urban) µg/100 ml (Nordman, 1975).
As a rule, the Pb-B levels of urban populations, and of people
heavily exposed to automobile exhausts, have been found to be higher
than those of rural populations or of populations living in areas with
less traffic (Hofreuter et al., 1961; US Department of Health,
Education and Welfare, 1965; Lehnert et al., 1970; Tepper & Levin,
1972) (Table 15). In one recent study, Pb-B levels were determined
among adults before and after the opening of a motorway interchange
with a high traffic density. Pb-B levels were found to be considerably
higher among men and women living in the immediate vicinity of the
interchange after it was opened than before (Waldron, 1975). In the
evaluation of the results of this study, allowance must be made for
the facts that no control group was studied, the procedure of drawing
blood samples was changed after opening the interchange, the sampling
took place at different times of the year and no data were given
pertaining to the control of the analytical method used (atomic
absorption spectroscopy). Thus, the possibility of systematic errors
cannot be ruled out. On the other hand, Stopps (1969) found that the
Pb-B levels of people living in various places remote from
civilization had group means of 12-23 µg/100ml, values not
significantly different from group means reported for people living in
urban areas of highly industrialized countries. No information was
given in the report concerning procedures or quality control of the
analytical methods.
A distinct increase in the lead absorption has been recorded in
people living in the vicinity of lead smelters (Secchi et al., 1971;
Nordman et al., 1973; Martin et al., 1975; Graovac-Leposavic et al.,
1973).
Men have higher Pb-B levels than women (NAS-NRC, 1972). This
difference does not appear to be totally attributable to the higher
haematocrit values of men (Tepper & Levin, 1972; Nordman, 1975). At
least part of the difference is likely to be accounted for by the
higher food consumption of men.
No association has been established between Pb-B levels and age in
adults (NAS-NRC, 1972; Tepper & Levin, 1972; Nordman, 1975).
The influence of cigarette smoking is not fully evaluated; some
researchers have reported higher Pb-B levels for smokers than for non-
smokers (Hofreuter et al., 1961; US Department of Health, Education
and Welfare, 1965; Tepper & Levin, 1972), while others have been
unable to confirm such an association (Lehnert et al., 1967; Jones et
al., 1972; McLaughlin & Stopps, 1973; Nordman, 1975; Tsuchiya et al.,
1975).
Table 15. Summary of concentration of lead in blood of selected
groups of males, USAa
Mean No. of Identity of groups
(µg/100 ml) subjects
11 9 Suburban nonsmokers, Philadelphia
12 16 Residents of rural California county
13 10 Commuter nonsmokers, Philadelphia
15 14 Suburban smokers, Philadelphia
19 291 Aircraft employees, Los Angeles
19 88 City employees, Pasadena
21 33 Commuter smokers, Philadelphia
21 36 City Health Dept. employees, Cincinnati
21 155 Policemen, Los Angeles
22 11 Live and work downtown, nonsmokers, Philadelphia
23 140 Post Office employees, Cincinnati
24 30 Policemen, nonsmokers, Philadelphia
25 191 Firemen, Cincinnati
25 123 All policemen, Cincinnati
25 55 Live and work downtown, smokers, Philadelphia
26 83 Police, smokers, Philadelphia
27 86 Refinery handlers of gasoline, Cincinnati (1956)
28 130 Service station attendants, Cincinnati (1956)
30 40 Traffic police, Cincinnati
30 60 Tunnel employees, Boston
31 17 Traffic police, Cincinnati (1956)
31 14 Drivers of cars, Cincinnati
33 45 Drivers of cars, Cincinnati (1956)
34 48 Parking lot attendants, Cincinnati (1956)
38 152 Garage mechanics, Cincinnati (1956)
a From: US Department of Health, Education and Welfare, 1965.
5.4.2 Children
European studies of Pb-B levels in children indicate that, in
general, the values are similar to or possibly even lower than those
in adults. Pb-B levels of 200 children aged 4-13 years in rural
western Ireland have been reported to be below 13 µg/100ml with 45% of
the results below 10 µg/100ml (Grimes et al., 1975). A group of 363
children aged from 8 days to 8 years was surveyed in the
Nuremberg/Erlangen area. The children displayed a mean Pb-B level of
3.3 ± 2.6 p.g/100 ml in the first year of life; the Pb-B level
increased year by year and reached a mean of 11.5 ± 4.9 µg/100 ml at
the age of 6-8 years (Haas et al., 1972a). However, most of the
available data on Pb-B levels in children have been obtained as a
result of case-finding programmes conducted in the USA. In one study,
the average blood level of 230 children, aged 1-5 years, in two rural
counties was found to be 22.8 + 11.0 µg/100 ml (Cohen et al., 1973).
An upward correction was made for all haematocrit values below 40%;
more than half of the children lived in older houses (more than 25
years old) one-quarter of which had flaking paint or holes in the
plaster.
There has been great concern in the USA that a very large number
of inner city children have abnormally elevated Pb-B levels. The
concern is for children in the blood lead range of 40-80 µg/100 ml.
Thus, Blanksma et al. (1969) reported that in 1967, and 1968, 8%, and
3.8%, respectively, of Chicago slum children had Pb-B concentrations
in excess of 49 µg/100 ml. This study involved 68 744 children, the
majority of whom were between 1 and 6 years of age. The problem is not
limited to large cities. Fine et al. (1972) reported on a survey of 14
Illinois communities with populations ranging from 9641 (Robbins) to
126 963 (Peoria). Of a total of 6151 children, 18.6% had Pb-B levels
higher than 39 µg/100 ml and 3.1% had levels higher than 59 µg/100 ml.
Some of the communities were in the Chicago urban complex, but a
considerable number were not. There did not appear to be any great
difference in the percentage of children having an excessive
concentration of lead among the Chicago urban communities as compared
with the downstate and western Illinois communities. The findings are
certainly not unique to Illinois. In a recent survey, 34% of 343
children in an impoverished area of Boston had Pb-B levels in excess
of 39 µg/100ml and 12% were over 49 µg/100ml (Pueschel et al., 1972).
Similar data have been gathered recently in New York City and
elsewhere.
6. METABOLISM OF LEAD
6.1 Absorptiona
The absorption of lead from environmental sources is not solely
dependent on the amount of lead presented to the portals of entry per
unit time. It is also dependent on the physical and chemical state in
which the metal is presented and it is influenced by host factors such
as age and physiological status. The amount of food eaten and the
amount of air breathed, with the proportionate ingestion or inhalation
of lead, are functions of metabolic activity. Men engaged in heavy
work breathe more air and eat more food than sedentary individuals of
the same weight, and children eat almost as much food and breathe
almost as much air as middle-aged adults.
6.1.1 Absorption by inhalation
A large amount of information has accumulated regarding the
factors that determine the degree of deposition and retention of
inhaled aerosols in general (Task Group on Lung Dynamics, 1966). With
appropriate knowledge of the aerodynamic characteristics of lead
aerosols, it would be possible to make reasonable predictions from the
lung model developed by the ICRP Task Group on Lung Dynamics,
concerning the fractional deposition that would occur in the human
airways. It would also be possible to predict the pattern of regional
deposition in the airways. Unfortunately, the knowledge necessary for
making accurate predictions is not available, particularly in the case
of industrial exposure.
The ICRP lung model would predict that approximately 35% of the
lead inhaled in general ambient air would be deposited in the airways,
since the aerodynamic diameterb of the lead particles is
approximately 0.1-1.0 µm (see section 5.1.1). The lung model would
also predict that regional deposition would be predominantly in the
alveolar bed and in the deeper regions of the tracheobronchial system.
Furthermore, it would predict that fractional deposition of lead dusts
generated in an industrial environment would be greater than it would
be for lead in general ambient air, however, the deposition would be
a In this document, absorption and uptake are used synonymously.
b Aerodynamic diameter = diameter of a unit density sphere with the
same settling velocity as the particle in question (Task Group on
Lung Dynamics, 1966).
mainly in the nasopharynx rather than in the pulmonary bed or
tracheobronchial region, owing to the larger particle size. Industrial
lead fumes, such as those generated in the process of cutting metals
with electric torches, would be of small particle size and would
behave accordingly. But even the lead aerosols breathed by the general
population are not well enough characterized to predict deposition.
This is particularly true for the very small particles (<0.1 µm)
which are largely deposited by diffusion (Lawther, 1972).
The adequacy of the ICRP lung deposition model is open to
question, at least for small particles. The model predicts a total
airway deposition of 40-50% for 0.5-µm particles, whereas a study in
human volunteers indicated a deposition of only 6-16% depending on the
rate and depth of respiration (Muir & Davies, 1967).
Predictions concerning the characteristics of airway clearance of
lead aerosols using the ICRP lung model are even more difficult to
make than predictions regarding deposition. The lung model would
predict that the fate of lead deposited in the airways would vary
greatly depending on its solubility characteristics and on the
inherent toxicity of the particles to the clearance mechanism (lung
macrophages and cilia). The chemical forms of lead in air are both
numerous and variable, depending on the source and on residence time
in the air (see section 5.1.1). In many types of industrial exposure,
lead is probably mainly in the form of lead oxide.
6.1.1.1 Human studies
Actual studies on the fractional deposition of particles in the
respiratory tract of man have not been extensive, especially in the
case of lead. Kehoe (1961) studied the deposition of lead in human
volunteers with an air lead level of 150 µg/m3. The source of lead
was combusted tetraethyllead which produced lead (III) oxide
(Pb2O3) in the air. Subjects breathed air containing particles
with an average diameter of 0.05 µm viewed under the electron
microscope, with 90% ranging from 0.02-0.09 µm. A diameter of 0.05 µm
for lead (III) oxide as seen under the electron microscope represents
a mass median equivalent diameter of approximately 0.26 µm (NAS-NRC,
1972). Subjects also breathed air containing particles having an
average diameter of 0.9 µm (mass median equivalent diameter = 2.9 µm);
36% of the smaller particles, and 46% of the larger particles, were
deposited.
Nozaki (1966) also reported on lung deposition of inhaled lead in
man. Lead fumes were generated in a high-frequency induction furnace
and were inhaled at a concentration of 10 000 µg/m3. Particle size
was closely controlled according to the method of Homma (1966). The
results (see Table 16) were similar to those of Kehoe (1961) and were
reasonably consistent with the ICRP lung deposition model (Task Group
on Lung Dynamics, 1966).
These data suggest that an estimate of 30 ± 10% deposition is
reasonable for the usual general ambient air situation and that lead
oxide deposition characteristics will vary considerably, depending on
the particle size and on the depth and frequency of respiration.
However, one cannot predict the contribution of airborne lead to
the body burden of lead on the basis of deposition studies alone.
Regional deposition probably varies greatly from one exposure
situation to another, that is, the industrial setting versus the
ambient environment. Also, the nature of lung clearance is unknown and
is difficult to study. Nevertheless, it is possible to determine
short-term lung clearance by carrying out gamma ray lung scans
following inhalation of 212Pb. Such a study in man has been reported
(Hursh & Mercer, 1970) but its relevance to the rate of clearance of
the chemical and physical forms of lead usually inhaled by man is
highly questionable. Such radioactive lead studies involve the
adsorption of 212Pb atoms on carrier aerosol particles. The
desorption of lead atoms from aerosol nuclei under these artificial
circumstances may be quite significant, and the estimated rate may be
totally unlike the clearance rate for ambient air lead particles.
Kehoe (1961) has reported that when a subject breathed large-
particle aerosols of lead (III) oxide (approximately 2.9 µm mass
median equivalent diameter) for many weeks at 150 µg/m3 a very
substantial increase in faecal excretion occurred, probably reflecting
the fact that the particles were largely trapped in the nasopharynx
and swallowed. When the same subject inhaled air with a lead
concentration of 150 µg/m3, with the lead in small particles
(approximately 0.26 µm mass median equivalent diameter), only a small
rise in faecal lead excretion was observed.
During inhalation of particulate air pollutants, the lead dust
comes into contact with lung cells, which are primarily responsible
for phagocytosis. It must be remembered that alveolar macrophages are
damaged in vitro by inorganic lead compounds (Beck et al., 1973),
and that similar effects have been demonstrated in vivo in rats and
guinea-pigs (section 6.1.1.4). It seems possible, therefore, that the
lung defence mechanisms are, to some extent, impaired in an
environment with a high air lead concentration, and that the rate of
absorption of inhaled particles under such circumstances is affected.
In summary, studies of airway deposition and clearance of lead in
man have not, as yet, provided any clear indication of the daily
absorption to be expected under realistic conditions. They have only
emphasized the necessity to consider other kinds of data to obtain
this information.
Since the concentration of lead in the blood is thought to reflect
current and recent lead exposure, the degree of lead intake from air
should be reflected in this factor.
Table 16. Deposition of lead fumes in the airways of human subjectsa
10 respirations/min: 1350 cm3 tidal air 30 respirations/min: 450 cm3 tidal air
Particle diameterb % Deposition Particle diameterb % Deposition
(µm) (µm)
1.0 63.2 1.0 35.5
0.6 59.0 0.6 33.5
0.4 50.9 0.4 33.0
0.2 48.1 0.2 29.9
0.1 39.3 0.1 27.9
0.08 40.0 0.08 26.5
0.05 42.5 0.05 21.0
a Adapted from Nozaki, 1966.
b Mass median equivalent diameter.
6.1.1.2 The relationship of air lead to blood lead in the general
population
The risk to man from lead in air has become a matter of
considerable concern in recent years. Studies of lead deposition and
retention in the airways of man have not been very enlightening. A
more indirect but nonetheless useful approach to the problem starts
from the assumption that the concentration of lead in the blood is
proportional to the concurrent level of total uptake by way of the
several portals of entry. It follows that each environmental source
(mainly air, food and water) would contribute to the blood lead
concentration in direct proportion to its contribution to the total
daily lead uptake. Up to the present time, such a relationship has
never been rigorously demonstrated. Goldsmith & Hexter (1967)
developed a linear regression plot of log Pb-B versus log lead
concentration in air. The air lead samples were not necessarily taken
at the same time and place as the blood samples. Thus, the regression
line was calculated on the basis of rather imprecise information.
However, data from experimental human subjects breathing known high
concentrations of lead oxide were found to fit the regression line
rather well. A cogent criticism is the fact that the validity of the
air lead data as applied to the specific blood lead data is very
uncertain. The contribution of air lead to blood lead, as inferred
from the Goldsmith-Hexter curve, is about 1.3 µg of lead per 100 ml of
blood per 1 µg of lead per m3 of air. Other epidemiological studies
have been made of the relationship between air lead and blood lead.
Azar et al. (1973) monitored the inhaled air of 150 individuals using
personal air samplers continuously, 24 hours per day. The air lead
exposure ranged from 2 µg/m3 to 9 µg/m3. There was a significant
correlation between log air lead level and log blood lead level, when
data from all the cities involved were pooled. The contribution of air
lead to blood lead was found to be somewhat less (approximately 1.0 µg
of lead per 100 ml of blood per 1 p.g of lead per m3 of air over the
range of air lead concentrations studied), than was estimated from the
Goldsmith-Hexter curve.
Another recent epidemiological investigation which examined the
relationship between air lead and blood lead levels was the Seven
Cities Study (Tepper & Levin, 1972). No significant correlation was
found between air lead and blood lead levels over an air lead range of
0.17-3.39 µg/m3. A major deficiency was the fact that the air data
were obtained from fixed outdoor sampling stations in the 11 cities
involved.
Two studies have been reported recently in which the relationship
between blood lead and air lead levels was investigated in human
volunteers. In one study, 14 male volunteers were exposed to a lead
oxide aerosol for 23 hours per day at an average concentration of
10.9 µg/m3 for up to 17 weeks. Blood lead concentrations and other
parameters were measured before, during, and following the exposure
period. A plateau of blood lead concentration was attained during the
exposure, and a return to pre- or near pre-exposure levels was
observed during the post-exposure period. The air contribution to the
Pb-B levels was approximately 1.4 µg of lead per 100 ml blood per 1 µg
of lead per m3 of air (Coulston et al., 1972b). In another study,
male volunteers inhaled an air lead concentration of 3.2 µg/m3. The
blood lead level increased from 18 µg to 25 µg/100 ml, that is
approximately 2 µg of lead per 100 ml blood per 1 µg of lead per m3
(Coulston et al., 1972c). Rabinowitz (1974) reported a study of a
single volunteer using stable lead isotope tracers in which the sudden
removal of the normal lead in air by filtration resulted in a
reduction of the blood lead concentration from approximately
14.5 µg/100ml to approximately 11.3 µg/100 ml over a period of 40 days
(Fig. 2). The average air lead levels were estimated taking into
account measurements made indoors and outdoors, and the time spent in
both locations. Prior to the experiment (day 109), the average air
lead concentration was 1.6 µg/m3 and during the experiment (day
109-148) it was 0.2 µg/m3. In calculating the contribution of air
lead to blood lead at day 148, allowance should be made for the fact
that the concentration of lead of normal isotopic composition was
decreasing prior to the removal of lead from the air. If this is taken
into account the contribution of air lead to blood lead at day 148
would be about 1.7 µg of lead per 100 ml of blood per 1.4 µg of lead
per m3 of air, or 1.2 µg of lead per 100 ml of blood per 1 µg of
lead per m3 of air. It is unfortunate that it was not possible to
follow the blood lead concentrations for a longer period of time after
removal of lead from room air, since a new steady state had not been
fully achieved. The study is also of limited value for application to
the general population because only one individual was studied.
On the other hand, the Coulston study was deficient in that the
form of air lead breathed (lead (III) oxide) may be deposited in, and
cleared from, the airways in a significantly different manner from
lead, as it actually occurs in general ambient air.
In conclusion it seems, that there is probably a perceptible
effect of air lead on blood lead in the range of air lead
concentrations applicable to the general population. The data
available suggest that with blood lead levels in the range found in
the general population, air lead levels may contribute from 1.0 to
2.0 µg of lead per 100 ml of blood per 1 µg/m3 of air.
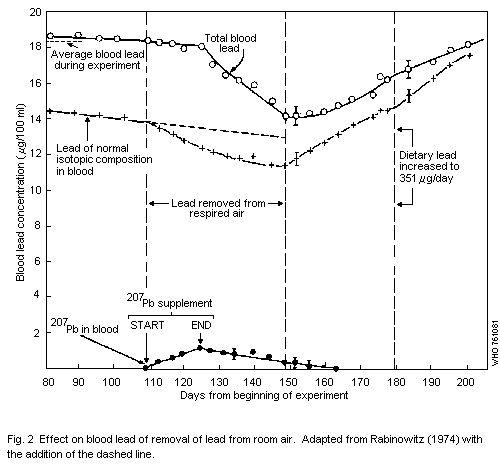 6.1.1.3 The relationship of air lead to blood lead in occupational
exposure
There is very little precise information concerning the
relationship between the concentration of lead in air (Pb-A) and Pb-B
levels in subjects who are occupationally exposed. The air sampling
technique used in the study of this relationship is of great
importance. Personal monitors should be used since in most industrial
situations the air lead concentrations to which individuals are
exposed may be highly variable, depending on the particular tasks
being performed and on the individual's work habits.
Only one study has been reported in which the subjects wore
personal monitors and in which the estimated individual Pb-A could be
related to Pb-B and some biochemical tests (Williams et al., 1969). In
this study, workers in various departments of an electric storage
battery factory wore personal samplers for the full work shift for two
weeks. There were considerable variations in the measured
concentrations of air lead both among departments and among individual
personal samples. Relevant data are presented in Table 17.
Using the data reported by Williams et al. (1969) an attempt was
made to estimate very crudely the potential contribution of Pb-A to
Pb-B in subjects who were occupationally exposed to lead. Several
arbitrary assumptions were made in this estimation:
Table 17. Means and standard errors of measured lead in air and
Pb-B levels in different departments of an electric
storage battery factorya
Department No. Pb in air (µg/m3) Pb-B (µg/100 ml)
mean S.E. mean S.E.
Machine pasting 6 218 25 74.2 4.7
Hand pasting 8 150 29 63.2 9.2
Forming 9 134 13 63.0 2.7
Casting 6 52 3 - -
Plastics dept. A 5 12 0.8 27.2 1.4
Plastics dept. B 5 9 0.8 29.1 1.6
a Adapted from Williams et al., 1969.
(1) that the weekly time-weighted average concentration of lead in air
(c) is a good measure of the effective inhalation exposure,
irrespective of the probable differences in breathing rates during
work hours. For a 40-hour working week c = 0.24 (Pb-A)o + 0.76
(Pb-A)a, subscripts o and a referring to the occupational
and ambient concentration of lead in air.
(2) that (Pb-A)a was 1 µg/m3 and that it had contributed
1.4 µg/100 ml to the measured Pb-B values (see 6.1.1.2), and that
for each further increase of Pb-A= 1 µg/m3, the increase in Pb-B
would be 1.4 µg/100 ml in the range of Pb-A values up to about
10 µg/m3.
(3) that the contributions of the occupational inhalation exposure,
non-occupational inhalation exposure, and exposures from other
sources (such as food) to the Pb-B levels are additive.
A further oversimplification was that the probable differences in
the chemical composition and physical characteristics of air-borne
lead in occupational and non-occupational environments were completely
neglected.
The contribution, (Pb-B)F, of non-inhalation exposures such as
food intake to the measured levels of lead in blood was assumed to be
the same for all workers and constant over the two week period of
observation. It was calculated from the data of Table 17 for the
workers in plastics departments A and B used as control groups, by
subtracting the estimated contribution of c to blood lead from the
measured Pb-B values, and taking the mean, i.e. (Pb-B)F=´[27.2-
(3.6 × 1.4)+ 29.1 - (2.9 × 1.4)] = 23.6 µg/100 ml. (Pb-B)o was then
obtained by subtracting 23.6 from measured Pb-B values for all other
departments.
The results are shown in Table 18.
Table 18. Estimation of blood lead levels potentially derived from
effective inhalation exposure c
Department Measured Pb-B (Pb-B)o c (Pb-B)o/c
µg/100 ml µg/100 ml µg/m3
Machine pasting 74.2 50.6 53.1 0.96
Hand pasting 63.2 39.6 36.8 1.1
Forming 63.0 39.4 32.9 1.2
Casting -- -- 13.2 --
Plastics A 27.2 3.6 3.6 1.0
Plastics B 29.1 5.5 2.9 1.5
From these calculations it would appear that an increase of
1 µg/m3 in the weekly time-weighted average concentration of lead in
air would correspond to an increase of approximately 1 µg/100 ml in
Pb-B.
A similar but somewhat lower figure for the air lead contribution
to Pb-B levels can be arrived at using data from a study, parts of
which are reported in two different publications (Prpic-Majic et al.,
1973; Fugas et al., 1973). From their data, they calculated that the
time-weighted average concentration of respirable lead particles for
52 workers in unspecified lead trades was 35 µg/m3. Their average
Pb-B level was 44.3 µg/100 ml, while the Pb-B level of a control
population living in an air environment of 0.2 µg/m3 was
22.4 µg/100 ml. Assuming the Pb-B levels due to non-air sources to be
the same for the two groups, i.e. 22.1 µg/100 ml (total (22.4) minus
the ambient air contribution to Pb-B (0.2 × 1.4 = 0.3)), the air
contribution to the Pb-B level for the industrially exposed group
would be 44.3-22.1 or 22.2µg/100ml. Since the air lead concentration
was 35 µg/m3, 1 µg of lead per m3 contributes 0.6 µg of lead per
100 ml of blood.
Another possible method of estimating the contribution of Pb-A to
Pb-B in the occupationally exposed subjects is to find first a
functional relationship that fits the Pb-B data from Table 17 and
c. A power function 1n y= 1n 18.9 + 0.34 1n c gives a good fit
in the range of c = 10 to c = 50 (correlation coefficient
r = 0.994), and enables the estimation of the increase in Pb-B per
unit increase in c. The results of these calculations are shown in
Table 19. Although still a gross oversimplification, this method seems
to give more realistic results because it reflects the fact that, at
the higher Pb-A level, Pb-B does not increase linearly with Pb-A, and
that therefore, the expected increase in Pb-B per unit increase in
c (dy/d c, Table 19) gets smaller and smaller as Pb-A levels grow.
Table 19. Power curve fita to the plot of Pb-B against the
time-weighted average concentration of lead in air (c)
Department c Measured Pb-B y dy /dc
µg/m3 µg/100 ml µg/100 ml
Machine pasting 53.1 74.2 73.2 0.47
Hand pasting 36.8 63.2 64.6 0.60
Forming 32.9 63.0 62.2 0.64
Casting 13.2 -- 45.6 1.09
-- (10) -- 41.3 1.41
Plastics A 3.6 27.2 29.3 --
Plastics B 2.9 29.1 27.2 --
a " y = 18.9 c 0.34 = Pb-B calculated.
6.1.1.4 Animal studies
Animal studies have been useful in the development of the ICRP
lung deposition and clearance models, but they have not contributed
much to resolution of the specific questions concerning the fate of
inhaled lead in man. However, observations made on the effects of
inhaled lead on lung macrophages are of special interest. A pronounced
reduction in the number of lung macrophages has been demonstrated in
rats and guinea-pigs owing to inhalation of lead (III) oxide at both
10 and 150 µg/m3 (Bingham et al., 1968). Maximum reduction occurred
within approximately one week. This phenomenon has also been reported
by others (Beck et al., 1973; Bruch et al., 1973, 1975). These
observations suggest that, with high air lead concentrations at least,
the lung clearance mechanism may not be functioning as effectively in
diverting lead deposited in the lower airways to the gastrointestinal
tract as the ICRP lung clearance model predicts. Thus, Pott &
Brockhaus (1971 ) reported that large doses of lead bromide solution
and of lead oxide suspension administered intratracheally to rats
(1.5 mg of lead oxide per dose on 8 successive days) were retained by
the body as completely as intravenous doses. However, at ´ of this
dose, retention was significantly less.
6.1.2 Absorption of lead from the gastrointestinal tract
6.1.2.1 Human studies
The uptake of lead from the gastrointestinal tract has been
studied fairly extensively, but as with the uptake of lead from air,
the evidence concerning a number of important points is somewhat
uncertain. Long-term balance studies conducted by Kehoe (1961) showed
that the daily excretion of lead into the urine was a little less than
10%, of the intake from food and beverages. He surmised that this
fraction represented the amount absorbed from the gastrointestinal
tract. In estimates made on this basis, the amount of urinary lead
that could have originated from the air is disregarded, as well as the
fact that some of the lead absorbed from the gastrointestinal tract is
re-excreted into the bowel.
Recent studies by Rabinowitz et al. (1974), using orally
administered 204Pb, indicate that the absorption of lead
incorporated into the diet is a little less than 10%, which is
consistent with Kehoe's conclusions based on a different experimental
approach.
Attention has been directed recently towards the absorption of
lead from the gastrointestinal tract in infants and young children. In
a study of eight normal children, from 3 months to 8.5 years of age,
Alexander et al. (1973) found a high degree of lead absorption (53%).
There did not appear to be any significant reduction in fractional
retention within the age range studied. This work is subject to
criticism because of the large scatter of values and because the
conclusions were based on 3-day balances, a period that is probably
insufficient for reaching any reliable conclusions.
6.1.2.2 The relationship of oral intake of lead to blood lead levels
in man
It would be of great interest to be able to relate oral intake of
lead to blood lead levels. It is obvious that, as the intake of lead
increases, blood lead levels will rise, but a quantitative expression
of this relationship at any particular level of lead intake has not
been determined. Table 20 compares daily oral lead intake (µg/day) and
Pb-B levels (µg/100 ml) found in adult populations without known
excessive exposure to lead, from several parts of the world.
Table 20. Comparison of daily oral lead intake with Pb-B levels
Study design Oral intake Pb-Ba Pb-B per Reference
(µg/day) (µg/100 ml) 100 µg
oral Pb
Duplicate portion 113 (men) 20.7 18.3 Coulston et al., 1972b
Faecal excretion 119b (women) 15.3 13.0 Tepper & Levin, 1972
Duplicate portion 230 (men) 12.3 5.4 Nordman, 1975
Duplicate portion 180 (women) 7.9 4.4 Nordman, 1975
Composites technique 505 (men) 34.6 6.8 Zurlo & Griffini, 1973c
a "Contributions of air to Pb-B levels are not reported in most of these studies and
could not be subtracted from total Pb-B levels.
b Calculated from daily faecal excretion of 108 µg of lead assuming gastrointestinal
absorption of 10%.
c Pb-B levels from Secchi et al. (1971).
From the data in Table 20 it is not possible to draw any reliable
conclusions regarding the contribution of foods and beverages to Pb-B
levels. The contribution is calculated to be greater in the two
American studies than in the European ones. One of these two American
studies (Tepper & Levin, 1972) was actually of faecal lead excretion,
not of dietary lead. But even if this study were discounted, there
remains a considerable discrepancy between the other American study
(Coulston et al., 1972b) and the European studies, which cannot be
explained.
Each of these studies involved a different number of subjects and
involved different analytical techniques. It is also probable that
there was also exposure from other environmental sources.
At levels of lead intake above 1000 µg per day, the rise in blood
lead level does not appear to increase linearly with dose, but, in
fact, may fit a logarithmic function.
From data published by Kehoe (1961) concerning balance studies on
human volunteers, a single individual with a total daily lead intake
of 600 µg had blood lead levels in the range of 30-35 µg/100 ml
registered over several months, which is consistent with the
relationships suggested in Table 20. However, individuals with larger
daily additions of lead did not have proportionately higher blood lead
levels. A single individual with oral lead intake of 3300 µg per day
had a blood lead level in the 50-60 µg/100 ml range, again followed up
for several months.
For children, the dietary contribution to blood lead is more
difficult to estimate than for adults. Because of the higher
absorption of lead, particularly in infants, the contribution of
dietary lead to blood lead levels may be higher than for adults.
6.1.2.3 Animal studies
The effect of age on gastrointestinal absorption of lead has been
studied in experimental animals. The absorption of lead from food has
been investigated in many animal studies. Values between 5 and 10% are
usual (Port & Brockhaus, 1971; Schlipkoter & Pott, 1973; Horiuchi,
1970).
Kostial et al. (1971) demonstrated that 5-7 day old rats absorb at
least 55% of single oral tracer doses of 203Pb. In an extension of
these studies, Forbes & Reina (1972) observed that the
gastrointestinal absorption of tracer doses of 212Pb, 85Sr and
59Fe was high prior to weaning and decreased rapidly thereafter. In
the case of lead, absorption which was 83% at 16 days, decreased
gradually to 74% on the day of weaning (22 days) and rapidly
thereafter to about 16% at 89 days. The addition of tracer doses of
metals to the diet is, however, an artificial situation. Results might
have been quite different had appreciable amounts of carrier lead been
included. Nevertheless, these observations are consistent with those
reported in young children.
Certain dietary factors have also been shown to influence the
gastrointestinal absorption of lead. Kello & Kostial (1973) have shown
that milk enhances lead absorption in 6-week-old-rats. Fasting
enhances lead absorption, at least as determined by Garber & Wei
(1974) in mice. Low dietary levels of calcium and of vitamin D enhance
lead absorption (Sobel et al., 1938b; Six & Goyer, 1970). It has also
been demonstrated that rats on an iron-deficient diet accumulate more
lead in their bodies than do rats on an iron-sufficient diet (Six &
Goyer, 1972). This seems particularly significant in the light of the
fact that young children in socially and economically deficient homes
have a high incidence of anaemia and excessively high blood lead
concentrations.
The absorption of lead ingested in the form of paint has received
attention because of the hazard of lead-based paint to young children.
Recent data from experiments on rats indicate that lead chromate and
lead naphthenate incorporated into dried paint films are substantially
available for absorption, although to a somewhat lesser degree than
lead naphthenate in oil or lead nitrate in aqueous solution (Gage &
Litchfield, 1968, 1969).
6.2 Distribution and Retention
As with all substances entering the body, a single dose of lead
distributes initially in accordance with the rate of delivery of blood
to the various organs and systems. Redistribution then occurs to
organs and systems in proportion to their respective affinities for
lead. Under conditions of continuous intake over long periods of time,
a near-steady state is achieved with respect to intercompartmental
distribution.
Perturbations in the pattern of distribution occur when large,
short-term peaks of lead intake are superimposed on this well-defined
pattern of long-term distribution.
6.2.1 Human studies
The kinetics of lead distribution and accumulation in man have not
been well defined in man directly. However, from autopsy data, the
general pattern of lead metabolism is clearly discernible. Above all,
it is clear that lead has a strong tendency to localize and accumulate
in bone. The accumulation of lead in the human body begins in fetal
life (Horiuchi et al., 1959; Barltrop, 1969). Lead is readily
transferred across the placenta and the concentration of lead in the
blood of newborn children is similar to that of their mothers,
indicating mother-fetus equilibration processes (Haas et al., 1972b;
Hower et al., 1975). The distribution of lead in fetal tissue is quite
similar to the distribution in adults (Barltrop, 1969).
The total lead content of the body may reach more than 200 mg in
men aged 60-70 years, but is lower for women. Barry & Mossman (1970)
calculated that in non-occupationally exposed adults, 94-95% of the
total body lead (body burden) was in the bones. A similar estimate was
made by Schroeder & Tipton (1968), by Horiuchi et al. (1959), and by
Horiguchi & Utsunomiya (1973). These recent reports serve to reaffirm
the long-recognized affinity of lead for bone. They also provide the
additional observation that the concentration of lead in bones
increases throughout most of life. This is in contrast to soft
tissues. Most soft tissues do not show a significant age-related
change in lead concentration after the second decade of life (Barry,
1975). This is also true of the concentration of lead in whole blood
(US Department of Health, Education and Welfare, 1965; Horiuchi &
Takada, 1954) and in blood serum (Butt et al., 1964). Thus, it appears
that the skeleton is a repository for lead that reflects the long-term
accumulative human exposure, whereas the body fluids and soft tissues
equilibrate reasonably fast and therefore reflect current and recent
exposure. Little is known as to whether the mobilization of lead lying
inactive in the bones can occur so rapidly that signs of poisoning
appear. There is need for more studies in this field.
The concentration of lead in the blood is of prime importance in
the evaluation of lead exposure. It is relied upon as an aid to the
diagnosis of poisoning and as an index of exposure to assess hazardous
conditions both in occupationally-exposed people and in the general
population. It has long been known that lead circulating in the blood
is mainly found in the erythrocytes (Cantarow & Trumper, 1944). The
concentration of lead in erythrocytes is about 16 times greater than
in plasma (Butt et al, 1964). The nature of the association of lead
with the erythrocyte is not clearly understood. Numerous studies have
been reported concerning the in vitro addition of lead to
erythrocytes suspended in plasma or saline solutions. But the validity
of such studies is open to serious question. Thus, Clarkson & Kench
(1958) found that lead added in vitro was readily removed by EDTA,
whereas residual lead present in the cells prior to the addition of
lead could not be removed. This suggests a difference in regard to:
(1) the degree of binding, (2) the site of binding in or on the cell,
or (3) the type of binding of the lead. Recent studies indicate that
lead is mainly bound to human erythrocyte protein, notably to
haemaglobin, rather than to stroma (Barltrop & Smith, 1971, 1972).
The rate of equilibration of lead in blood with sources of input
and with other body compartments has been studied in man by Rabinowitz
et al. (1973, 1974) using a stable lead isotope tracer (204Pb). The
data reported indicate that with a constant daily oral input of
204Pb, a virtually constant concentration of the tracer in the blood
is achieved after approximately 110 days. Upon withdrawal of the
tracer 204Pb from the diet, the 204Pb concentration in the blood
disappears with a half-time of approximately 19 days. The kinetics of
disappearance and accumulation suggest that first order rate processes
of exchange are involved with regard to this relatively mobile
compartment. Tola et al. (1973) also provided data which indicate that
the concentration of lead in the blood rises fairly rapidly to a new
steady state level when men are newly introduced into an occupational
lead environment. The time required for the blood lead concentration
to achieve a new plateau reflecting the new environment is about 60
days.
The body burden of lead increases from birth to old age (Schroeder
& Tipton, 1968; Barry & Mossman, 1970; Barry, 1975). When data for
various specific organs and systems are examined, it becomes evident
that there are two general pools of lead within the total organism.
The major one, in terms of total lead, consists of bone. This pool is
clearly highly accumulative. As a consequence, lead in bone
accumulates through most of the life span. Other organs and systems
are much less accumulative and, to different degrees, tend to
stabilize relatively early in adult life reflecting a greater turnover
rate of lead compared with that in bone.
There is good reason to make a distinction between total body
burden and exchangeable body burden since the organs and systems
comprising the exchangeable body burden are the ones having the
greater toxicological significance. It is also extremely important to
note that lead in whole blood is a part of the exchangeable fraction
of the body burden. Among adults in the general population there is no
age-related difference in regard either to the concentration of lead
in whole blood or in blood serum. Thus, in a general way, the Pb-B
level reflects the concentration of lead in soft tissues, and long-
term changes in Pb-B levels with changes in exposure levels are
probably accompanied by corresponding long-term changes in the rest of
the exchangeable pool.
Nuclear inclusion bodies containing lead have been found in man
subjected to lead exposure (Cramer et al., 1974; Galle & Morel-
Maroger, 1965; Richet et al., 1966) as well as in experimental animals
(see section 7.1.3). Although most frequently reported to occur in the
kidney, they have been found in other organs as well. There is a
suggestion from limited data that inclusion bodies are associated with
short-term lead exposure and not with long-term exposure (Cramer et
al., 1974).
The concentration of lead in deciduous teeth has received special
attention because they are readily available from young children and
because they provide a long-term record of lead exposure, much as is
the case with bone. Dentine in the area adjacent to the pulp is
particularly useful in this respect because it is laid down from the
time of eruption to the time the tooth is shed. It has been reported
that the concentration of lead in dentine is considerably lower in
suburban schoolchildren than it is in children in areas of high lead
exposure (Needleman & Shapiro, 1974).
There has been some interest in the possible use of hair lead as
an index of exposure. Unfortunately, there is no reliable information,
as yet, to indicate just how hair analyses should be interpreted in
relation to the frequency and degree of exposure.
6.2.2 Studies in animals
Animal studies have been particularly useful in defining more
precisely the nature of the kinetics of lead distribution and removal
from various tissues. Following administration of a single dose of
lead to rats, the concentration of lead in soft tissues is relatively
high and falls rapidly, mainly as a result of transfer into the bone
(Hammond, 1971). The distribution characteristics of lead were found
to be independent of the dose of lead over a wide range. The rate
constants for the elimination of lead from various tissues in rats
following a single dose of lead have been described by Castellino &
Aloj (1964). The rate of elimination was much slower from bone than
from other tissues. In studies on rats, Bolanowska et al. (1964) noted
that the rate of elimination of a single dose of lead from the body by
spontaneous excretion became slower with time, reflecting
progressively decreasing mobility of the residual body burden. This is
no doubt mainly due to the fact that as lead becomes progressively
more deeply buried in the bone matrix, its exchangeability with other
compartments and its availability for excretion decrease.
Rather striking age-related differences have been observed
concerning the distribution and retention of lead in rats (Momcilovic
& Kostial, 1974). The rate of elimination of a single tracer dose of
203Pb from the whole body, blood, and kidney was faster in adults
than in sucklings. In the case of the brain, there was actually a
slight increase in the 203Pb content of the brain of the sucklings
while the content was falling in other soft tissues. Numerous animal
studies have also demonstrated placental transfer of lead to the fetus
(see Carpenter, 1974, for relevant literature).
The intracellular distribution of lead has been studied in rat
tissue, mainly by cell fractionation techniques (Castellino & Aloj,
1969; Barltrop et al., 1971). Lead has an affinity for membranes of
the cell, particularly mitochondria. These organelles undergo
functional and ultrastructural changes in organs showing lead effects,
e.g. renal tubular cells (Goyer & Krall, 1969). Little lead is found
in lysosomes (Barltrop et al., 1971) in contrast with the
intracellular distribution of many other metals, e.g. mercury, copper,
iron.
There are few studies indicating the concentration of lead in
target organs that will produce effects. Formation of nuclear
inclusion bodies is observed in rats with renal lead concentrations of
about 10 mg/kg (wet weight) of kidney (Goyer et al., 1970a). Other
effects of lead were found to occur at higher levels of organ
concentration. Death in cattle is associated with lead levels of about
50 mg/kg of kidney cortex (wet weight) (Allcroft & Blaxter, 1950).
The concept of estimating the lowest level of metal accumulation
that results in adverse effects in a target organ has not been well-
explored in the case of lead. This is in contrast with cadmium where
estimates have been made of the minimum concentrations of cadmium in
the kidney cortex at which evidence of renal damage appears (Friberg
et al., 1974).
6.3 Elimination of Lead
The elimination of lead from the body is thought to be mainly by
way of the urine and the gastrointestinal tract. Little is known about
the miscellaneous routes of excretion such as sweat, exfoliation of
skin, and loss of hair.
6.3.1 Human studies
An approximation of the relative contributions of the various
routes to lead excretion in man has been given by Rabinowitz et al.
(1973). This study refers to only one non-occupationally exposed human
subject. Excretions via the kidneys and the gastrointestinal tract
were measured directly. Loss via other routes, e.g. hair, fingernails,
and sweat, was estimated from data on the efflux of 204Pb from the
blood compartment. Losses per day were as follows:
urine 38 µg (76%)
gastrointestinal secretions 8 µg (16%)
hair, nails, sweat, other 4 µg (8%)
The figure of 38 µg for daily urinary excretion is consistent
with the data of Teisinger & Srbova (1959). They reported an average
daily urinary lead excretion of 31 µg.
The mechanism of urinary lead excretion in man is not well
understood. However, the studies of Vostal (1966) provide strong
evidence that the process of renal clearance of lead is essentially
glomerular filtration. Extrapolation of a curve of glomerular
filtration rate plotted against lead excretion rate resulted in zero
lead excretion at zero filtration. The form of lead appearing in the
urine has not been defined. One study suggests that the form in which
lead appears in the urine depends on whether exposure to lead is
normal or elevated. Thus, in lead workers with high urinary lead
excretion, it has been found that only one-half to two-thirds of the
urine lead can be precipitated with co-precipitating agents such as
oxalate, phosphate, or carbonate. By contrast, virtually all the lead
in the urine of people with normal lead exposure can be co-
precipitated (Dinischiotu et al., 1960). This suggests that a stable
lead chelate species arises with elevated exposure. Nuclear inclusion
bodies or lead-protein complexes are found in the urine of children
with acute lead poisoning (Landing & Nakai, 1959).
The rate of biliary excretion of lead in man is not known.
The biological half-time of lead is extremely difficult to
estimate. The constantly decreasing availability of the major stores
of lead in osseous tissue makes it virtually impossible to describe
the rate of loss from the body in simple terms. It is at least clear
that, in man, clearance of one-half of a body burden of lead would
require a number of years.
6.3.2 Animal studies
Animal data on the routes of lead excretion suggest a
considerable species variation. In rats (Castellino et al., 1966) and
in sheep (Blaxter & Cowie, 1946) excretion by biliary and transmucosal
routes is greater than urinary excretion. On the other hand, the ratio
of urinary to gastrointestinal lead excretion in the baboon is 2:1
(Eisenbud & Wrenn, 1970). Vostal (1966) studied the mechanism of lead
excretion in dogs. In mild chronic intoxication, excretion was by
glomerular filtration, without evidence of any tubular secretion or
reabsorption. With more severe poisoning, there was evidence of renal
tubular reabsorption. Evidence was also presented for a tubular
secretory mechanism in the chicken.
6.4 The Metabolism of Alkyllead Compounds
The characteristic toxic effects of tetraethyllead and
tetramethyllead are not caused by the tetraalkyl compounds themselves,
but rather by the trialkyl derivatives formed by dealkylation in the
liver (Cremer, 1959; Cremer & Callaway, 1961). Tetraethyllead is
initially converted mainly to triethyllead and partly to inorganic
lead (Bolanowska, 1968). The triethyllead concentration in organs then
falls only slowly. Even after several days, there is no significant
reduction. The behaviour of tetramethyllead is quite similar to the
behaviour of tetraethyllead. Tetramethyllead is much less toxic
probably because it is dealkylated to the trialkyl toxic form much
more slowly than is the case with tetraethyllead (Cremer, 1965).
Since both these compounds have toxic and biochemical effects
unlike those of inorganic lead, it is not to be expected that the
biochemical tests used in assessing inorganic lead exposure would have
the same significance as in exposure to organic lead. Indeed, in
severe cases of tetraethyllead poisoning, urinary coproporphyrins and
ALA excretion are usually not elevated, and free erythrocyte
porphyrins are only moderately and inconstantly elevated (Gutniak et
al., 1964; Beattie et al., 1972b). These biochemical tests are
therefore of little use in short-term exposure situations. But, in
long-term exposure situations, it is possible that some of them may be
useful. Indeed, Robinson (1974) has shown that in workers industrially
exposed to tetraethyllead, the urinary excretion of ALA is increased,
but not to the same degree as in workers exposed to inorganic lead who
have equivalent levels of total urinary lead excretion (organic plus
inorganic). This suggests that some portion of total urinary lead is
reflecting alkyllead exposure. Bolanowska et al. (1967) demonstrated
that, in three fatal cases of tetraethyllead poisoning, the ratio of
inorganic lead to triethyllead ranged from 67:1 to 18:1 in the urine.
But this ratio did not reflect the ratio of inorganic to triethyllead
in tissues at all accurately. In tissues, including the brain, the
ratios were approximately 1:1.
7. EXPERIMENTAL STUDIES ON THE EFFECTS OF LEAD
The major part of published experimental work on animals
describes or aims to explain pathological or pathophysiological
changes caused by lead. It does not contribute much to the
understanding of the relationship between the dose administered, its
distribution in a period of time, and the biological effect. The doses
used in most animal experiments have, as a rule, been far above the
levels that can occur in environmental or occupational contact with
lead, with the exception of accidental ingestion of soluble lead
compounds.
7.1 Animal Studies
7.1.1 Haemopoietic system
Experimental studies on the effects of lead on blood and
haemopoiesis have been carried out essentially to study pathogenic
mechanisms. There are few studies dealing with the relationship
between the lead dose and blood changes.
There is a great deal of evidence showing that lead inhibits
several enzymes that participate in haem synthesis. Inhibition of
these enzymes is invoked to explain the rises in haem intermediates
that occur as a result of lead exposure. Thus, the rise in erythrocyte
protoporphyrin is readily explained on the basis of the well known
inhibitory effect of lead on the mitochondrial enzyme ferrochelatase
(EC 4.99.1.1) (haem synthetase). This action was first proposed by
Rimington (1938) as the probable explanation for the anaemia in lead
poisoning. Numerous studies have since confirmed that lead is indeed a
rather potent inhibitor of haem synthetase (Dresel & Falk, 1954;
Goldberg et al., 1956; Klein, 1962).
Although specific inhibition of the enzyme haem synthetase is
usually invoked to explain the accumulation of protoporphyrin, it is
also possible that the availability of iron for coupling with
protoporphyrin is inhibited by lead. It has been shown that lead
interferes with the transfer of iron from transferrin to human
reticulocytes (see section 8.2.1). Further support for the idea that
lead interferes with the availability of iron is to be found in
studies showing that lead causes accumulation of iron as "ferruginous
micelles" in developing erythrocytes (Bessis & Jensen, 1965).
Mitochondrial damage was evident in these studies, suggesting the
possibility that globin synthesis may be compromised, along with haem
synthesis.
The increased excretion of coproporphyrin III in urine is
suggestive of an inhibition by lead of the enzyme coproporphyrinogen
oxidase (EC 1.3.3.3), which converts coproporphyrinogen III to
protoporphyrin IX (PP) (Goldberg, 1972). There is no supportive
evidence showing a direct inhibitory effect on this enzyme. One would
imagine that inhibition of coproporphyrinogen oxidase (EC 1.3.3.3)
would result in decreased blood levels of protoporphyrin IX, however,
the opposite is true. Perhaps the concurrent rise in erythrocyte
protoporphyrin, ALA excretion, and excretion coproporphyrin III in
urine can be explained on the basis of delta-aminolevulinate synthase
(EC 2.3.1.37) (ALAS) stimulation. Stimulation of ALAS activity by lead
acetate in vivo has been demonstrated in the avian hepatocyte,
probably due to impairment of haem synthesis (Strand et al., 1972). By
contrast, Gajdos & Gajdos-Török (1969) found no change in the ALAS
activity of bone marrow or liver in experimental lead intoxication of
rabbits.
Animal studies have been reported concerning ALAD inhibition by
lead in tissues concurrently with inhibition in the circulating
erythrocytes. This has been shown in the blood, brain, and liver of
suckling rats (Miller et al., 1970). After 3040 days of exposure,
erythrocyte ALAD underwent an 80-90% reduction. The blood lead
concentration in these rats is not given but can be estimated from
data in the report. It is stated that a maximum of 3 ml of blood was
obtained from each rat. It also appears that the blood specimens each
contained about 4.5 µg of lead. Therefore the blood lead concentration
must have been at least 150 µg of lead per 100 ml of blood, and was
probably nearer to 200.
In other studies, in which long-term lead exposure of rats
resulted in about 50% inhibition of erythrocyte ALAD, there was no
inhibition of brain or liver ALAD (Coulston et al., 1972a): this may
be due to the fact that the exposure levels were lower in this study
than in the others cited.
The question of the significance of lead exposure in relation to
haemoglobin formation has been studied in dogs by Maxfield et al.
(1972). These authors were mainly concerned with the question of
whether the depression of ALAD activity in the peripheral blood was in
any way associated with depressed formation of haemoglobin. Dogs were
given lead over a period and the ALAD activity fell to a very low
level. But the ability of the dogs to regenerate haemoglobin after
removal of half of the circulating blood volume remained essentially
normal. Although this indicates that the inhibition of ALAD activity
in peripheral blood may not be significant, it should be pointed out
that the lead exposure was not sufficiently high to cause any
substantial rise in ALA excretion in the urine. ALA excretion was only
approximately two-three times the baseline level.
There is evidence that the synthesis of globin is affected by
lead in animals as well as in man. In an in vitro study, it was
shown that the incorporation of 14C-glycine into globin in duck
erythrocytes was reduced by 25% by a lead concentration of
5 × 10-4 M (Kassenaar et al., 1957). The reduction of 14C-glycine
incorporation into haem was considerably greater.
Relatively little is known about the effects of lead on the
formation or activity of other haem-containing compounds in the body.
There is some evidence, however, that lead can inhibit formation of
cytochrome P-450, a haemoprotein intimately involved in the drug-
metabolizing mixed function oxidase system of hepatic microsomes
(Alvares et al., 1972). Long-term lead administration has also been
shown to affect the activity of cytochrome c oxidase (EC 1.9.3.1)
(Makagev & Verbolovic, 1967; Verbolovic, 1965). The effects seen were
of a mixed nature, involving first stimulation then depression of
activity. A decrease was also observed in the myoglobin concentration
of some muscle groups. It is not clear from these studies whether the
effects were due to inhibition of haem synthesis or of protein
synthesis.
Administration of lead to rats (over a period of 6 months) in
doses of 2-4 g per rat, resulted in a change in cytochrome c oxidase
(EC 1.9.3.1) activity and in the amount of haemoglobin. The magnitude
of the change increased with larger doses (Verbolovic, 1965). Dogs
were given a solution of lead acetate over a 2-year period resulting
in a reduction in the activity of cytochrome c oxidase (EC 1.9.3.1)
that was in proportion to the dose of lead administered (Makagev &
Verbolovic, 1967).
By means of electron microscopy, Pernis et al. (1964) showed
grossly swollen mitochondria in the erythrocytes of lead-poisoned
guinea-pigs, diverse vacuolar formations, and aggregates of molecules
of ferritin. Electron microscopy of erythrocytes of rabbits receiving
an intravenous dose of 20 mg/kg of 2% lead acetate solution showed
vacuolization of the cytoplasm and swelling of the plasma membrane. An
intensive vacuolization in thrombocytes and a reduction in the
quantity of organelles, particularly those containing serotonin, also
took place. In addition, a swelling of mitochondria after the complete
disruption of cristae was noted (Hacirov, 1972). An experiment on rats
showed ultramicroscopic changes of mitochondria in the red bone marrow
cells in the early stages of poisoning.
7.1.2 Nervous system
7.1.2.1 Inorganic lead
In view of recent concern about subtle impairments of cerebral
function at sub-encephalopathic levels of lead exposure, there has
been a renewed interest in lead and its toxic effects. High doses of
lead will produce encephalopathy; this has been reported in cats (Aub
et al., 1926) and dogs (Staples, 1955).
The brains showed histopathological features similar to those
described in human encephalopathy. Others have since reproduced this
syndrome in the rat (Thomas et al., 1971; Michaelson, 1973; Clasen et
al., 1974) and in the mouse (Rosenblum et al., 1968; Silbergeld &
Goldberg, 1974). The effects may be explained on the basis of
retardation of brain development (Michaelson, 1973; Krigman et al.,
1974).
Paraplegia was reported in suckling rats by Pentschew & Garro
(1966). The disease was produced by transfer of lead from the mother's
milk until weaning, with subsequent post-weaning feeding of lead to
the young.
Behavioural abnormalities such as excessive self-grooming and
aggressiveness occur, even when the lead intake is reduced to a point
where paraplegia no longer occurs (Michaelson & Sauerhoff, 1974). It
was estimated that the minimum daily lead intake causing behavioural
effects (Michaelson & Sauerhoff, 1974) rose from 0.08 mg/kg body
weight at birth to 3 mg/kg at day 16 as a result of suckling. Post-
weaning, this minimum intake rose from 50 mg/kg at day 20 to 60 mg/kg
at day 28. From day 16 to day 20, intake was difficult to estimate
since the infant rats were eating and suckling to different degrees.
Other studies in rats (Snowdon, 1973) and sheep (Carson et al., 1974)
indicate that offspring of mothers exposed to lead during pregnancy
show learning defects. Older animals are refractory to this type of
effect (Brown et al., 1971).
Future behavioural studies should probably be extended to include
sub-human primates since it has been shown that the histological and
clinical features of lead encephalopathy can be produced in both
infant and adult baboons (Cohen et al., 1972; Hopkins & Dayan, 1974).
Studies of lead neuropathy in animals indicate that demyelination
and axonal degeneration are more consistent findings than neuronal
damage in the anterior horn cells or dorsal root ganglia of the spinal
cord (Lampert & Schochet, 1968; Schlaepfer, 1969; Fullerton, 1966).
This is consistent with findings in man. The slowing of nerve
conduction found in man has also been produced experimentally in the
guinea-pig (Fullerton, 1966).
It is known that lead interferes in some manner with synaptic
transmission in the peripheral nervous system and that the effects can
be reversed by calcium (Kostial & Vouk, 1957). But, in addition, an
increased frequency of miniature end-plate potentials has been
reported (Manalis & Cooper, 1973). Neuromuscular blockade has also
been demonstrated in the rat phrenic nerve-hemidiaphragm preparation
(Silbergeld et al., 1974). Again, as in the other studies, the effect
was antagonized by calcium. The significance of these findings with
regard to the central nervous system remains to be determined.
Studies at the biochemical level have been very limited. It has
been shown, using the "Pentschew model", that incorporation of
14C-glucose carbon into dicarboxylic amino-acids of the brain is
reduced (Patel et al., 1974a, 1974b). These results were interpreted
to indicate delayed brain maturation.
Recent work in dogs (Stowe et al., 1973) has mapped the variation
in lead concentrations in different parts of the brain of lead-
poisoned dogs. The studies show a relationship between areas of the
most marked histological change and high lead concentration. Male pups
from the same litter were fed a purified diet, low in calcium and
phosphorus, with and without 100 mg/kg of lead as lead acetate from
the age of 6-18 weeks. The concentration of lead in the various brain
segments is given in Table 21.
Table 21. Distribution of lead in the brains of control and
lead-intoxicated dogsa
Brain segment Lead concentration (mg/kg of wet tissue)
Control Lead intoxicated
Cerebellum 0.160 ± 0.052 0.587 ± 0.113
Medulla 0.155 ± 0.007 0.713 ± 0.112
Frontal white 0.053 ± 0.027 0.920 ± 0.156
Thalamus <0.10 1.023 ± 0.142
Occipital white 0.020 ± 0.012 1.030 ± 0.115
Caudate 0.120 ± 0.083 1.613 ± 0.345
Frontal grey 0.033 ± 0.015 1.767 ± 0.254
Occipital grey 0.080 ± 0.071 2.357 ± 0.181
a "Adapted from Stowe et al., 1973.
7.1.2.2 Alkyllead compounds
Unlike the case with inorganic lead, intoxication by
tetraethyllead in juvenile or adult rats caused a characteristic
encephalopathy, involving restlessness, ataxia, combativeness
progressing to convulsions, coma, and death (Davis et al., 1963). In
dogs there was extensive muscular tremor and twitching, which
progressed to convulsions, coma, and death.
Biochemical studies of the respiration of brain slices incubated
with inorganic lead compared with triethyllead (the active metabolite
of tetraethyllead) have substantiated the fundamental difference in
the action of alkyllead compounds on the brain (Cremer, 1959). The
toxic moieties in tetraethyllead and tetramethyllead poisoning are the
trialkyl metabolites and not the inorganic lead ion.
Essentially there is no qualitative difference between the toxic
effects of tetramethyllead and tetraethyllead. However, there is a
quantitative difference in that the inhalation LC50 for
tetramethyllead (8870 mg/m3) is about 10 times higher than that for
tetraethyllead (850 mg/m3) (Cremer & Callaway, 1961). The
intravenous LD50 for tetraethyllead is about 10 mg of lead per kg of
body weight in the rat (Cremer, 1959). This is in contrast to the
intravenous LD 50 for inorganic lead, which is approximately 70 mg/kg
in the rat (Fried et al., 1956). The precise manner in which the
trialkyllead ion acts to cause altered brain function is not clearly
known but the mechanism may involve inhibition of amine oxidase
(flavin-containing) (EC 1.4.3.4) (monoamine oxidase) (Galzigna et al.,
1964). In rabbits, administration of toxic doses of tetraethyllead
results in a loss of copper, iron, and zinc in certain areas of the
brain (Niklowitz & Yeager, 1973), suggesting that triethyllead may act
by displacing certain essential trace metals from metalloenzymes in
the brain.
Tetramethyllead injected into rats in overtly neurotoxic doses
did not depress ability to learn a simple task (Bullock et al., 1966).
7.1.3 Renal system
Animal studies have contributed to an understanding of the order
of appearance of the various manifestations of renal toxicity in lead
exposure. The spectrum and train of events as related to the exposure
time and to the dose of lead have recently been reviewed (Goyer &
Rhyne, 1973). In the earliest stage of renal response to lead
exposure, reversible tubular effects occur. These include the
appearance of intranuclear inclusion bodies, which is probably a
mechanism for sequestration of lead. These bodies have been isolated
and found to be composed of a lead-protein complex. The protein is
insoluble in physiological solutions and is rich in acidic amino
acids. It has not been characterized further (Moore & Goyer, 1974).
The intranuclear inclusion bodies appear to have a high and specific
affinity for lead compared with that for calcium, iron, zinc, copper
or cadmium and about 90% of the lead in the kidney is associated with
them (Goyer et al., 1970a; 1970b).
The appearance of these bodies is accompanied by amino aciduria,
glycosuria, and hyperphosphaturia. Morphological and functional
changes in tubular epithelial cells also occur at this stage,
including impaired respiratory and phosphorylative ability.
After further lead administration, more severe changes occur in
the renal tubular epithelium such as hyperplasia and cystic changes.
There is a progressive increase in interstitial fibrous tissue and
atrophy of tubular cells. These are irreversible changes that lead to
a third stage of renal failure, manifested by azotaemia and
hyperuricaemia. Sclerotic glomeruli appear, but the hypertension seen
in some cases of chronic lead nephropathy in man has not been
reproduced in experimental animals. The sequence described above in
animals is probably generally valid for man.
7.1.4 Gastrointestinal tract
The effects of lead on the gastrointestinal tract have been
studied in some detail in the guinea-pig (Mambeeva & Ahmiedova, 1967).
Spastic contractions occurred from the stomach to the jejunum.
Inhibitory effects were also noted, accounting for the frequent
constipation seen to accompany lead colic.
7.1.5 Cardiovascular system
Experimental animal data on the question of hypertension are
conflicting. Among rats given 70 mg of lead acetate per day orally,
only a few survived 40 days and all were hypertensive (Griffith &
Landauer, 1944). Hypertension has also been produced in the rabbit
(Beckmann, 1925). Others have not seen hypertension with lead exposure
in rats (Padilla et al., 1969) or dogs (Fours & Page, 1942). From all
the above animal studies it seems that hypertension can occur with
heavy lead exposure.
There are conflicting reports regarding whether lead can cause
atherosclerosis in experimental animals. Sroczynski et al. (1967)
observed increased serum lipoprotein, cholesterol, and cholesterol
deposits in the aortas of both rats and rabbits receiving large doses
of lead. On the other hand, Prerovska (1973) did not produce
atherosclerotic lesions in rabbits using similar doses of lead given
over an even longer period of time.
Cardiac myopathy has also been shown experimentally in lead-
intoxicated rabbits (Kosmider & Sroczynski, 1961). The mechanism for
this effect is not known.
Kuz'minskaja (1964) and Mironcik & Timofeeva (1974) observed that
rabbits receiving lead after a cholesterol load showed more intense
sclerotic changes in the aorta and myocardium than rabbits on a normal
diet without lead, or than rabbits given cholesterol alone.
Makasev & Krivdina (1972) observed a phased change in the
permeability of blood vessels (first phase-increased permeability;
second phase-decreased permeability) in rats, rabbits and dogs, that
received a solution of lead acetate. A phased change in the content of
catecholamines in the myocardium and in the blood vessels was observed
in subacute lead poisoning in dogs (Mambeeva & Kobkova, 1969). This
effect appears to be a link in the complex mechanism of the
cardiovascular pathology of lead poisoning.
7.1.6 Respiratory system
Alveolar macrophages from guinea-pigs are damaged in vitro by
inorganic lead compounds (3 µg/1 × 106 cells) thus releasing a
rapidly occurring lysis and a slowly developing, coarsely blistered
vacuolization. More than 90% of the cells are damaged within 20 hours
(Beck et al., 1973).
Similar effects seem to occur in the organism, since in rats that
had inhaled 10 µg lead/m3 for 3-12 months, the number of macrophages
that could be flushed from the lungs was reduced by 60% (Bingham et
al., 1968).
Electron microscope investigations of the lungs of rats that had
been exposed for 14 days to concentrations of 100-200 µg of lead
oxide/m3 revealed toxic effects in the alveolar macrophages and the
type I alveolar epithelial cells. The structures of the endoplasmatic
reticulum and mitochondria were changed (Bruch et al., 1973).
In the lungs, the alveolar macrophages have the capacity to
degrade noxious substances and are important for other defence
reactions. The ability of alveolar macrophages of guinea-pigs that had
inhaled concentrations of 70-170 µg of lead per m3 of air for four
days, to degrade benzopyrene was distinctly decreased, the benzopyrene
3-monooxygenase (1.14.14.2) activity being only about 10% of the
original value. The activity returned to normal after three days
without any lead exposure (Bruch et al., 1975). The elimination of
bacteria from the lungs was also reduced, when rats were exposed to
70 µg of lead per m3 of air (Schlipkoter et al., 1977).
7.1.7 Reproductive system
Animal studies support the contention that behavioural
deficiencies can occur in infants and newborn as a result of
intrauterine exposure to lead via their mothers (see section 7.1.2).
Others have shown a reduction in the numbers and size of offspring
(Dalldorf & Williams, 1945; Puhae et al., 1963). Data in rabbits (Cole
& Bachhuber, 1914), guinea-pigs (Weller, 1915), and rats (Stowe &
Goyer, 1971) indicate that paternally-transmitted effects can occur,
including reductions in litter size, weights of offspring, and in
survival rate. Several investigators have reported that oral
administration of lead to animals even at doses in the microgram per
kilogram range can cause changes in spermatogenesis (Egorova et al.,
1966; Golubovid et al., 1968), and an increase in testicular RNA and
DNA content (Golubovic & Gnevkovskaja, 1967; Golubovic et al., 1968).
7.1.8 Endocrine organs
The effects of lead on thyroid function that have been reported
in man have also been demonstrated experimentally in rats (Zel'tser,
1962; Sandstead, 1967).
7.1.9 Carcinogenicity
7.1.9.1 Inorganic lead compounds
The carcinogenic risk to man of lead salts and the relevant
studies in animals have recently been discussed in an IARC publication
(IARC, 1972).
The induction of benign and malignant renal neoplasms has been
observed in both Swiss mice and rats fed on diets containing 100 or
1000 mg of basic lead acetate (Pb(C2H302)2. 2Pb(OH)2) per kg
of diet (Van Esch & Kroes, 1969; Van Esch et al., 1962; Mao & Molner,
1967; Azar et al., 1973). Similar results were observed in rats fed
1000mg of lead acetate (Pb(C2H302)2. 3H20) per kg of diet
(Boyland et al., 1962). In addition to renal neoplasms, tumours of the
testes, the adrenal, thyroid, pituitary, and prostrate glands and of
the brain have been reported in rats fed lead acetate or basic lead
acetate, but the results await confirmation (Zawirska & Medras, 1968;
Oyasu et al., 1970). Rats given intraperitoneal or subcutaneous
injections of lead phosphate also developed renal tumours. Total doses
of 120-680 mg of lead were effective (Zollinger, 1953; Roe et al.,
1965). No kidney tumours were reported in hamsters fed 100 or 500 mg
of basic lead acetate per kg of diet for up to 2 years (van Esch &
Kroes, 1969).
In Syrian golden hamsters given a combination of lead oxide and
benzo[a]pyrene intratracheally once weekly for 10 weeks, lung adenomas
occurred in 11/26 animals within 60 weeks. One adenocarcinoma of the
lung was also observed. Such tumours did not occur in animals given
the same dose of lead oxide or benzo[a]pyrene alone (Kobayashi &
Okamoto, 1974).
7.1.9.2 Alkyllead compounds
Epstein & Mantel (1968) reported that subcutaneous injection of
0.6 mg of tetraethyllead (given as 4 equally divided doses) to Swiss
mice between birth and 21 days of age produced malignant lymphomas in
1/26 males and 5/41 females, compared with 1/39 and 0/48 controls. In
treated females, the tumours were observed between 36 and 51 weeks
after the first injection. The significance of this finding in female
mice is difficult to assess since this tumour occurs frequently and
with variable prevalence in untreated mice of this strain.
7.1.10 Mutagenicity
Chromosomes from leukocyte cultures from mice fed 1% lead acetate
in the diet showed an increased number of gap-break type aberrations
(Muro 8,: Goyer, 1969). These changes involved single chromatids,
suggesting that injury followed DNA replication.
7.1.11 Teratogenicity
There have not been any adequate animal studies to provide
evidence to support the suggestion that lead may have a teratogenic
effect.
7.2 Acquisition of Tolerance to Lead
Although human studies suggest that there is no acquired
tolerance in regard to haem-synthesis mechanisms, there may be for
other toxic effects. In this regard, it is interesting to note that
the blood lead level at which cattle develop severe encephalopathy
from eating paint is often less than 80 µg/100 ml (Hammond et al.,
1956). However, in cattle receiving 5-6 mg of lead per kg per day
orally, the concentration of lead in the blood exceeded 100 µg/100 ml
within 2-4 months and remained at about that level for as long as four
years with continuous administration, without any apparent harm to the
animals (Allcroft, 1951). In these studies, haemoglobin did not fall
until a terminal illness developed. Hapke (1974) found that in cattle
and sheep the sensitivity to acutely toxic amounts of lead was reduced
by a pretreatment with lead for 5 months. Goyer et al. (1972) have
suggested from their studies on rats that the intranuclear inclusion
bodies that develop during lead exposure serve as a protective
mechanism by binding lead in the kidney, making it less toxic. But in
the recent study of Cramer et al. (1974) (see section 7.1.3) it was
shown that renal intranuclear inclusion bodies are present only in
workers exposed to lead for a relatively short period of time. Thus,
if inclusion bodies serve some protective function, it is only during
a limited period of exposure. The formation of the cadmium-binding
protein, metallothionein, which appears to have a protective role in
cadmium exposure, is induced by a number of metals but not by lead
(Webb, 1972).
7.3 Factors Influencing Lead Toxicity
7.3.1 Age and sex
It has recently been reported that the intraperitoneal lethal
dose of lead in rats is significantly lower for adult male rats than
for adult female rats (Kostial et al., 1974). In the same study, it
was observed that the lethal dose in mg/kg body weight for 3-week-old
rats was about the same as for adult females.
7.3.2 Seasonal variations
The same seasonal pattern of high incidence of poisoning has been
reported in dogs belonging to urban families as has been reported in
children (Zook et al., 1969). It has also been shown experimentally in
rats and mice (Baetjer, 1959; Baetjer & Horiguchi, 1963) and in
rabbits (Blackman, 1937; Horiuchi et al., 1964) that susceptibility is
greater at high ambient temperatures than at normal temperatures.
7.3.3 Nutrition
Experimental studies have shown that nutritional factors may
influence the absorption of lead from the gastro-intestinal tract and
thus alter susceptibility to the toxic effects of lead (Goyer &
Mahaffey, 1972). Low phosphorous and calcium in the diet (Sobel et
al., 1938b; Six & Goyer, 1970), high vitamin D (Sobel et al., 1938a),
and low iron (Mahaffey, 1974) all enhance lead absorption. The amount
and the composition of dietary protein may also influence lead
toxicity. Low protein diets appear to increase the susceptibility to
lead intoxication as compared to high protein diets (Baernstein &
Grand, 1942; Goyer & Mahaffey, 1972).
The significance of these findings for the susceptibility of
people to lead poisoning has not been established. However, many
children, even in developed countries like the USA, have sub-optimal
dietary intakes of calcium, iron, and other nutrients (US Department
of Health, Education and Welfare, cited by Mahaffey, 1974). This may
have a bearing on the problem of increased lead absorption frequently
found in children in poor, urban areas.
7.3.4 Intercurrent disease, alcohol, and other metals
High lead exposure increases the susceptibility of mice to
Salmonella typhimurium infection (Hemphill et al., 1971). Lead
administration also increases the susceptibility of rats (Filkins &
Buchanan, 1973; Selye et al., 1966; Erve & Schumer, 1972), mice
(Clercq de & Merigan, 1969), and baboons (Hoffman et al., 1974) to
endotoxin shock, but such studies have been performed using extremely
large intravenous doses of lead simultaneously with the endotoxin.
Administration of ethanol (10% ad libitum in drinking water)
had no effect on the toxicity of lead to rats as measured by urinary
ALA excretion, renal weight, or lead concentration in the kidneys,
liver, or bones (Mahoffey, 1974).
Very little is known about metal interactions and how they might
affect the toxicity of lead, except at the nutritional level (see
section 7.3.3). Beyond that, a synergistic effect has been noted
between lead and cadmium with regard to experimental teratogenesis
(Ferm, 1969). It was also found that zinc, given in the diet with
lead, protected horses against the toxic effects of lead. Probably,
this effect was not due to inhibition of lead absorption. Zinc
supplementation actually caused an increase in the lead content of
liver and kidney, but a decrease in the lead content of brain and bone
(Willoughby et al., 1972). It might be inferred that zinc displaced
lead from lead-inhibited enzymes that are zinc-dependent, such as ALAD
(Cheh & Nellands, 1973). A dose-dependent effect of zinc, antagonistic
to the depression of ALAD by lead, has recently been shown in vivo
and in vitro as well as an in vitro antagonism of zinc on the
cytotoxic effect of lead on macrophages (Schlipköter et al. 1975;
Ruiter de et al. 1977).
7.4 Human Studies
Planned experimental studies on the effects of lead in man are
sparse. Kehoe (1961), in his famous experimental studies, in which
human volunteers were exposed to a known amount of lead over various
periods of time, confined himself to studying the lead balance only,
and did not report on the effects of lead.
Three subjects ingested 1 and 3 mg of lead daily, in the form of
lead (II) nitrate, for 33 weeks. The ALA-U, CP-U, and erythrocyte
protoporphyrin IX were measured regularly while Pb-B and Pb-U
measurements were performed at irregular intervals (Schlegel et al.,
1973). Exposure from food and ambient air was not controlled during
the experiment. A rise in FEP was obtained with both doses and a rise
in ALA-U and CP-U only with the 3 mg dose. Evaluation of the results
obtained in this study is difficult, partly because of the small
number of subjects studied and partly because the results were rather
erratic.
Coulston et al. (1972b; 1972c) conducted two exposure chamber
experiments on male volunteers (see section 6.1.1.2). The volunteers
were exposed to air lead concentrations with an average of 10.9 and
3.2 µg/m3 for up to 17 weeks. In the 10.9 µg/m3 exposure study, 24
volunteers participated, 6 of whom served as controls. In order to
control dietary lead exposure, total diet for one full day was
collected at intervals of eight days; the results indicated an average
lead intake of about 110 µg/day only. The variables measured were the
Pb-B, ALAD, ALA-U, and CP-U. Blood lead levels increased in all of the
exposed men and appeared to stabilize after about 2 weeks of exposure.
The mean Pb-B level at that time was about double the pre-exposure
mean, i.e., an increase from 19 to 37 µg/100 ml. A concomitant
increase of the urinary excretion of lead was reported; the faecal
excretion remained unchanged however. The rise in blood lead levels
was followed by a decrease in ALAD activity, which after 5 weeks of
exposure was about 50% of the pre-exposure level. No change in ALA-U
and CP-U was reported. Five months after the termination of the
exposure, all but one of the participants had Pb-B values similar to
those before exposure. The ALAD activity returned to normal almost
immediately after cessation of exposure. No changes in the haemoglobin
level were noted during the experiment. In the 3.2 µg/m3 experiment
a rise in the Pb-B level from 20 to 26 µg/100 ml was obtained,
followed by a slight decrease in ALAD activity, which after five weeks
of exposure was about 85% of the pre-exposure level. Other changes
were not reported.
In a recent experimental study, a greater susceptibility to
inorganic lead was demonstrated in females (Stuik, 1974; Stuik &
Zielhuis, 1975). The volunteers were healthy male and female students
aged 18-26 years. Groups of 5 males and 5 females received 20 µg of
lead per kg per day orally for 21 days. Lead was administered as lead
acetate in glycerol.
The control blood lead levels remained fairly constant at
approximately 17 µg/100 ml during the experiment. The exposed male
subjects showed an increase from 20.6 µg/100 ml to 40.0 µg/100 ml at
the end of the second week of exposure (40.9 µg/100 ml in the third
week). The blood lead in females rose from 12.7 µg/100 ml to
30.4 µg/100 ml, the highest level being reached in the first part of
the third week.
The protoporphyrin IX content of the erythrocytes showed no
change in either the control or the exposed male group. However, in
the female group, it showed a rise beginning in the third week and
rising to 48.0 µg/100 ml erythrocytes. The findings were confirmed in
a second experiment.
It is suggested that the increase of the erythrocyte
protoporphyrin IX was a result of interference in the use of iron in
the formation of haemoglobin. The synergism of lead exposure and iron
deficiency might be suggested as being responsible for the increased
response of FEP in females but this will have to be tested further in
experimental and epidemiological work.
8. EFFECTS OF LEAD ON MAN-EPIDEMIOLOGICAL AND CLINICAL STUDIES
Two types of study characterizing the effects of lead on man have
been reported:
-- retrospective studies of the causes of mortality and morbidity in
lead-exposed populations compared with unexposed populations, and
-- studies of the effects of lead on specific organs and systems.
The findings from these two types of study will be considered
separately. In both cases, the main objective will be to establish, as
far as possible, the dose of, or exposure to lead which is associated
with specified effects, and the frequency of such effects.
From the toxicological point of view, "the dose should be defined
as the amount or concentration of a given chemical at the site of
effect, i.e. where its presence leads to a given effect" (Nordberg
ed., 1976). The application of this definition is difficult because
the dose as defined above can rarely be measured directly and has to
be estimated in various ways. In experiments, it is estimated from the
amount injected or ingested or from dermal and other topical
applications (using appropriate absorption factors and body
distribution factors). In inhalation experiments it is estimated from
the concentration as measured in air, the time of exposure, and the
relevant deposition, retention, and absorption factors (if available).
The same considerations apply for dose estimation from occupational
exposure where, in addition to inhalation, the possible dermal
exposure, ingestion during work-time, and exposure which workers are
subject to as members of the general population, should be taken into
account. The dose for the general population is estimated from
inhalation of air, ingestion of food, water, and other beverages, and
various other contacts, including drugs and consumer products,
smoking, and in children, ingestion of soil, settled dust, and paint
chips. A more direct way of estimating the dose is from measurements
in body tissues and fluids such as blood, urine, faeces, sweat, or
hair. Other organs, tissues, cells, and subcellular elements can be
used for this purpose in animal experiments or in autopsy or biopsy
material.
Although the biological effects of lead on man have been
characterized in some detail, the precise doses of lead responsible
for the effects are rarely, if ever, known. With all its acknowledged
shortcomings, the Pb-B level is the vital link between exposure and an
effect. In section 6, an effort was made to define, as far as
possible, the relationship between the lead in air and in the diet and
Pb-B levels. The main objective of this section is to establish the
relationship between Pb-B levels and biological effects. Only in this
way is it possible to estimate the possible biological consequences of
specific levels of lead in environmental media.
Some biological effects of lead bear a close relationship to
concurrent Pb-B levels, others do not. Thus, the degree of ALAD
inhibition in peripheral blood rises and falls more or less
concurrently with the Pb-B level, while some renal effects of lead are
the consequence of an exposure to lead that may have occurred at a
point remote in time and which is not reflected in the Pb-B level at
the time the effect is first manifested clinically. The fidelity with
which the Pb-B level reflects lead concentrations in target organs is
subject to serious problems of analytical error as described in
section 3.
Beyond these considerations, there is the additional problem of
variation in the inherent susceptibility of individuals, and the
influence of co-existent variables that may modify this
susceptibility, such as nutritional status, age, and presence or
absence of diseases such as alcoholism. For all the above reasons, the
Pb-B level cannot be used as a reliable indication of dose or exposure
in dealing with individual patients. They should be used only in
assessing population group exposures at which effects may occur in a
certain proportion of individuals.
Other tests for assessing dose have been proposed, e.g. lead
excretion in response to chelating agents. Regardless of potential
merits and special applications, most information relating health
effects to dose has been obtained using Pb-B levels as an estimate or
index of dose.
8.1 Retrospective Studies of Lead-exposed Populations
8.1.1 Epidemiology of lead poisoning in industry
In many countries there has been a considerable improvement over
the past forty years with respect to hygienic conditions in the lead-
using industries. The exposure of workers to lead was considerably
higher before 1930 than after. In the United Kingdom, the number of
reported cases of poisoning fell dramatically in the decade 1920-30
(Lane, 1964). Against this background, it is useful to consider the
studies of Dingwall-Fordyce & Lane (1963). They found a higher than
expected incidence of death due to cerebrovascular disease among men
with past high lead exposure. The men studied retired from work
between 1926 and 1960. All those studied had at least 25 years of
service. Men in the heavy exposure category had an average urine lead
concentration of 100-250 µg/litrea over the last 20 years of
a 100 µg/litre corresponds to a Pb-B level of approximately
60 µg/100 ml and 250 µg/litre corresponds to a Pb-B level of
approximately 120 µg/100 ml (Williams et al., 1969).
employment. Men in the moderate exposure group had urine lead
concentrations in the normal range. The third group had no exposure.
As can be seen from Table 22, in the heavy exposure group deaths from
cerebrovascular diseases (cerebral haemorrhage, thrombosis, and
arteriosclerosis) were much higher than normal.
The data also suggest that in this group the excessive death rate
was most pronounced among men who retired prior to 1951 when exposure
conditions were probably considerably worse than they were later. In
the same study, it was found that the death rate from malignant
neoplasms was not above the expected rate in any exposure grade.
Unfortunately, the incidence of death due to chronic nephritis was not
reported. A very similar survey was reported by Malcolm (1971) in
which the subjects studied had, with few exceptions, been exposed to
lead at moderate levels (average Pb-B level-65 µg/100 ml). There was
no statistically significant excess mortality in any of the following
disease categories: heart disease, chest disease, cerebrovascular
accidents, cancer, renal disease, and "miscellaneous".
A recent American study is in general agreement with the
conclusions of the British investigators concerning longevity and
causes of death in the lead industries as they have operated over the
last 25-30 years (Tabershaw & Cooper, 1974; Cooper & Gaffrey, 1975).
The subjects were 1356 workers employed in the lead battery and
smelter industries from 1946 to 1970. Both blood lead levels and
urinary lead excretion were quite high. For example, 78.7% of 47
smelter workers had Pb-B levels of 80 µg/100 ml or more, from 1946 to
1961. The figure was still 13.5% after 1965 (489 total workers
sampled). The percentage of battery workers with Pb-B levels above
this was somewhat lower. But for all the various categories of
duration of employment and type of work, 81.5-95.7% of the Pb-B levels
were equal to or greater than 40 µg/100 ml. About 50% of the workers
were employed for more than 10 years. The total mortality in this
group was approximately the same as in the general population. The
authors concluded that there was no evidence that work associated with
lead increased the risk of death due to the major categories of
cardiovascular and renal diseases. However, when chronic renal disease
(chronic nephritis or other renal sclerosis) was segregated as a
separate cause of death, there did appear to be a significant excess
number of deaths. Thus, among smelter workers, the ratio of observed
deaths to expected deaths was 7:2.8 and among the battery workers the
ratio was 14:8.6. A similar association was found for a category of
death classified as "other hypertensive disease": 7:1.9 among smelter
workers and 13:6.3 among battery workers. For the two disease
categories this adds up to 21 excess deaths out of 1267 for whom cause
of death was listed. The authors emphasize that many of the workers in
the study group were probably exposed to air lead concentrations
considerably in excess of 0.15 mg/m3.
Table 22. Deaths from cerebrovascular disease in retired and employed workers from a lead industrya
Status Year of Grade of exposure
death
None Medium Heavy
Expected Observed Expected Observed Expected Observed
incidence incidence incidence incidence incidence incidence
Retired 1926-50 0.7 0 0.2 3 0.8 5
1951-61 7.2 6 3.2 3 8.5 19
1926-61 7.9 6 3.4 6 9.3 24b
Employed 1946-61 3.2 3 3.1 3 5.6 9
a Adapted from Dingwall-Fordyce & Lane, 1963.
b P <0.001.
Although most epidemiological studies on occupational exposure
have been carried out on industrial populations, one extensive study
on orchard workers in the Wenatchee area of the state of Washington,
has been reported (Neal et al., 1941). This study was somewhat
complicated by the fact that exposure was to lead arsenate. In view of
the known toxicity of arsenic, studies were included on the combined
toxicities of lead and arsenic in animals. No synergism was found in
these animal studies (Fairhall & Miller, 1941). The blood lead
concentrations of the orchard workers and their families are
summarized in Table 23.
This study may have been crude in comparison to some more recent
ones, but it had the rather unique merit of examining health effects
not only in men, but also in women and children. Furthermore, the
exposure levels, as reflected in the urine and blood data of Table 23,
were only slightly higher than the approximate upper limit for people
living in highly polluted cities today. The study was concerned with
weight, blood pressure, diseases of the cardiovascular system, skin
disorders, eye irritation, chronic nervous diseases, blood dyscrasias,
kidney diseases, neoplastic diseases, and fertility. There was no
evidence, based on data available at the time, that the health profile
of these people was any different from that of the general population.
In 1968, a follow-up study was undertaken of the people who had
participated in the original study (Nelson et al., 1973). Over 97% of
the original participants were successfully traced. There had been 452
deaths among the 1231 original participants. A life table method of
analysis of the standard mortality ratio was used. The overall
mortality was less than the average for the state of Washington. The
standard mortality ratios of exposed groups were not consistent with
the exposure gradient. The mortality pattern for increasing duration
of exposure was not consistent either.
8.1.2 Epidemiology of lead poisoning in the general adult population
Adequate studies of the relationship between lead exposure and
health status in the general adult population have not been carried
out. The limitations that apply to the epidemiological studies of
occupational groups are magnified when applied to the general
population. The range of exposure levels is smaller between sub-groups
of the general adult population and their socioeconomic,
physiological, and health profiles are probably more diverse.
8.1.3 Epidemiology of lead poisoning in infants and young children
There has been only one study reported of general mortality and
disease-specific morbidity rate in children exposed to lead. The
Wenatchee study referred to in section 8.1.1 included 146 children
under the age of 15. As with the adults in this study, no abnormal
pattern of disease incidence was noted. These children had moderately
high lead exposure (see Table 23).
Table 23. Urine and blood lead content of persons in the Wenatchee study according to
severity of exposurea
Group Urine lead content Blood lead content
No. Average, S.D. No. Average, S.D.
analyses µg/litre µg/litre analyses µg/100 ml µg/100 ml
Low exposure
men 146 35 21 148 26 11
women 123 28 19 124 26 10
Intermediate exposure
men 102 43 30 108 30 11
women 25 27 15 27 22 10
High exposure
men 386 88 60 329 44 16
women 61 46 25 58 34 13
Children under
15 years
boys 81 53 39 17 37 15
girls 65 54 40 14 36 10
a "From Neal et. al., (1941).
8.2 Clinical and Epidemiological Studies of the Effects of Lead on
Specific Organs and Systems
In the following discussion of the effects of lead on various
organs and systems, consideration will be given to dose-effect and
dose-response relationships. The word "dose" as used here will refer
to Pb-B levels, as described in the introductory remarks of this
chapter.
The diversity of the effects of lead on haemoglobin formation and
the complexity of the process itself make it difficult to determine
which inhibitory effect is most sensitive and what is their relative
importance at different levels of exposure (or dose).
Dose-effect refers to the relationship between dose and the
intensity of a specified effect in an individual, e.g. Pb-B level
versus percentage inhibition of blood ALAD.
Dose-response refers to the relationship between the dose and the
proportion of a population showing a defined effect, specified as to
the level of intensity, e.g. the proportion of a population showing
more than 50% inhibition of blood ALAD at a Pb-B of 20 µg/100 ml.
Some effects of lead are not graded, for example, the effects on
the kidney and the central nervous system are usually reported in all-
or-none terms, i.e. a certain proportion of individuals in a
population are reported to have shown the effect at a given range of
Pb-B concentrations. With many effects of lead it is difficult to
specify a dose-response or a dose-effect relationship because the
available data are inadequate.
8.2.1 Haemopoietic system
The evidence for disturbances in haem synthesis is clearly shown
in man by the appearance of abnormal concentrations of haem precursors
in blood and urine. The levels of lead exposure at which these various
manifestations of disturbed haem synthesis first appear have been
studied extensively in man. The sequence of reactions affected by
lead, and the consequences thereof, are shown in Fig. 5.
Lead interferes with the biosynthesis of haem at several
enzymatic steps, with the use of iron, and with globin synthesis in
erythrocytes. Inhibition of ALAD and haem synthetase is well
documented, and accumulation of the substrates of these enzymes (ALA
and PP) is characteristic of human lead poisoning. Inhibition of ALAS
is based on experimental evidence only. Whether there is enzymatic
inhibition or whether other factors affect the conversion of
coproporphyrinogen III (CPG) to protoporphyrin IX (PP) is not clear;
nevertheless, increased urinary excretion of coproporphyrin III is
6.1.1.3 The relationship of air lead to blood lead in occupational
exposure
There is very little precise information concerning the
relationship between the concentration of lead in air (Pb-A) and Pb-B
levels in subjects who are occupationally exposed. The air sampling
technique used in the study of this relationship is of great
importance. Personal monitors should be used since in most industrial
situations the air lead concentrations to which individuals are
exposed may be highly variable, depending on the particular tasks
being performed and on the individual's work habits.
Only one study has been reported in which the subjects wore
personal monitors and in which the estimated individual Pb-A could be
related to Pb-B and some biochemical tests (Williams et al., 1969). In
this study, workers in various departments of an electric storage
battery factory wore personal samplers for the full work shift for two
weeks. There were considerable variations in the measured
concentrations of air lead both among departments and among individual
personal samples. Relevant data are presented in Table 17.
Using the data reported by Williams et al. (1969) an attempt was
made to estimate very crudely the potential contribution of Pb-A to
Pb-B in subjects who were occupationally exposed to lead. Several
arbitrary assumptions were made in this estimation:
Table 17. Means and standard errors of measured lead in air and
Pb-B levels in different departments of an electric
storage battery factorya
Department No. Pb in air (µg/m3) Pb-B (µg/100 ml)
mean S.E. mean S.E.
Machine pasting 6 218 25 74.2 4.7
Hand pasting 8 150 29 63.2 9.2
Forming 9 134 13 63.0 2.7
Casting 6 52 3 - -
Plastics dept. A 5 12 0.8 27.2 1.4
Plastics dept. B 5 9 0.8 29.1 1.6
a Adapted from Williams et al., 1969.
(1) that the weekly time-weighted average concentration of lead in air
(c) is a good measure of the effective inhalation exposure,
irrespective of the probable differences in breathing rates during
work hours. For a 40-hour working week c = 0.24 (Pb-A)o + 0.76
(Pb-A)a, subscripts o and a referring to the occupational
and ambient concentration of lead in air.
(2) that (Pb-A)a was 1 µg/m3 and that it had contributed
1.4 µg/100 ml to the measured Pb-B values (see 6.1.1.2), and that
for each further increase of Pb-A= 1 µg/m3, the increase in Pb-B
would be 1.4 µg/100 ml in the range of Pb-A values up to about
10 µg/m3.
(3) that the contributions of the occupational inhalation exposure,
non-occupational inhalation exposure, and exposures from other
sources (such as food) to the Pb-B levels are additive.
A further oversimplification was that the probable differences in
the chemical composition and physical characteristics of air-borne
lead in occupational and non-occupational environments were completely
neglected.
The contribution, (Pb-B)F, of non-inhalation exposures such as
food intake to the measured levels of lead in blood was assumed to be
the same for all workers and constant over the two week period of
observation. It was calculated from the data of Table 17 for the
workers in plastics departments A and B used as control groups, by
subtracting the estimated contribution of c to blood lead from the
measured Pb-B values, and taking the mean, i.e. (Pb-B)F=´[27.2-
(3.6 × 1.4)+ 29.1 - (2.9 × 1.4)] = 23.6 µg/100 ml. (Pb-B)o was then
obtained by subtracting 23.6 from measured Pb-B values for all other
departments.
The results are shown in Table 18.
Table 18. Estimation of blood lead levels potentially derived from
effective inhalation exposure c
Department Measured Pb-B (Pb-B)o c (Pb-B)o/c
µg/100 ml µg/100 ml µg/m3
Machine pasting 74.2 50.6 53.1 0.96
Hand pasting 63.2 39.6 36.8 1.1
Forming 63.0 39.4 32.9 1.2
Casting -- -- 13.2 --
Plastics A 27.2 3.6 3.6 1.0
Plastics B 29.1 5.5 2.9 1.5
From these calculations it would appear that an increase of
1 µg/m3 in the weekly time-weighted average concentration of lead in
air would correspond to an increase of approximately 1 µg/100 ml in
Pb-B.
A similar but somewhat lower figure for the air lead contribution
to Pb-B levels can be arrived at using data from a study, parts of
which are reported in two different publications (Prpic-Majic et al.,
1973; Fugas et al., 1973). From their data, they calculated that the
time-weighted average concentration of respirable lead particles for
52 workers in unspecified lead trades was 35 µg/m3. Their average
Pb-B level was 44.3 µg/100 ml, while the Pb-B level of a control
population living in an air environment of 0.2 µg/m3 was
22.4 µg/100 ml. Assuming the Pb-B levels due to non-air sources to be
the same for the two groups, i.e. 22.1 µg/100 ml (total (22.4) minus
the ambient air contribution to Pb-B (0.2 × 1.4 = 0.3)), the air
contribution to the Pb-B level for the industrially exposed group
would be 44.3-22.1 or 22.2µg/100ml. Since the air lead concentration
was 35 µg/m3, 1 µg of lead per m3 contributes 0.6 µg of lead per
100 ml of blood.
Another possible method of estimating the contribution of Pb-A to
Pb-B in the occupationally exposed subjects is to find first a
functional relationship that fits the Pb-B data from Table 17 and
c. A power function 1n y= 1n 18.9 + 0.34 1n c gives a good fit
in the range of c = 10 to c = 50 (correlation coefficient
r = 0.994), and enables the estimation of the increase in Pb-B per
unit increase in c. The results of these calculations are shown in
Table 19. Although still a gross oversimplification, this method seems
to give more realistic results because it reflects the fact that, at
the higher Pb-A level, Pb-B does not increase linearly with Pb-A, and
that therefore, the expected increase in Pb-B per unit increase in
c (dy/d c, Table 19) gets smaller and smaller as Pb-A levels grow.
Table 19. Power curve fita to the plot of Pb-B against the
time-weighted average concentration of lead in air (c)
Department c Measured Pb-B y dy /dc
µg/m3 µg/100 ml µg/100 ml
Machine pasting 53.1 74.2 73.2 0.47
Hand pasting 36.8 63.2 64.6 0.60
Forming 32.9 63.0 62.2 0.64
Casting 13.2 -- 45.6 1.09
-- (10) -- 41.3 1.41
Plastics A 3.6 27.2 29.3 --
Plastics B 2.9 29.1 27.2 --
a " y = 18.9 c 0.34 = Pb-B calculated.
6.1.1.4 Animal studies
Animal studies have been useful in the development of the ICRP
lung deposition and clearance models, but they have not contributed
much to resolution of the specific questions concerning the fate of
inhaled lead in man. However, observations made on the effects of
inhaled lead on lung macrophages are of special interest. A pronounced
reduction in the number of lung macrophages has been demonstrated in
rats and guinea-pigs owing to inhalation of lead (III) oxide at both
10 and 150 µg/m3 (Bingham et al., 1968). Maximum reduction occurred
within approximately one week. This phenomenon has also been reported
by others (Beck et al., 1973; Bruch et al., 1973, 1975). These
observations suggest that, with high air lead concentrations at least,
the lung clearance mechanism may not be functioning as effectively in
diverting lead deposited in the lower airways to the gastrointestinal
tract as the ICRP lung clearance model predicts. Thus, Pott &
Brockhaus (1971 ) reported that large doses of lead bromide solution
and of lead oxide suspension administered intratracheally to rats
(1.5 mg of lead oxide per dose on 8 successive days) were retained by
the body as completely as intravenous doses. However, at ´ of this
dose, retention was significantly less.
6.1.2 Absorption of lead from the gastrointestinal tract
6.1.2.1 Human studies
The uptake of lead from the gastrointestinal tract has been
studied fairly extensively, but as with the uptake of lead from air,
the evidence concerning a number of important points is somewhat
uncertain. Long-term balance studies conducted by Kehoe (1961) showed
that the daily excretion of lead into the urine was a little less than
10%, of the intake from food and beverages. He surmised that this
fraction represented the amount absorbed from the gastrointestinal
tract. In estimates made on this basis, the amount of urinary lead
that could have originated from the air is disregarded, as well as the
fact that some of the lead absorbed from the gastrointestinal tract is
re-excreted into the bowel.
Recent studies by Rabinowitz et al. (1974), using orally
administered 204Pb, indicate that the absorption of lead
incorporated into the diet is a little less than 10%, which is
consistent with Kehoe's conclusions based on a different experimental
approach.
Attention has been directed recently towards the absorption of
lead from the gastrointestinal tract in infants and young children. In
a study of eight normal children, from 3 months to 8.5 years of age,
Alexander et al. (1973) found a high degree of lead absorption (53%).
There did not appear to be any significant reduction in fractional
retention within the age range studied. This work is subject to
criticism because of the large scatter of values and because the
conclusions were based on 3-day balances, a period that is probably
insufficient for reaching any reliable conclusions.
6.1.2.2 The relationship of oral intake of lead to blood lead levels
in man
It would be of great interest to be able to relate oral intake of
lead to blood lead levels. It is obvious that, as the intake of lead
increases, blood lead levels will rise, but a quantitative expression
of this relationship at any particular level of lead intake has not
been determined. Table 20 compares daily oral lead intake (µg/day) and
Pb-B levels (µg/100 ml) found in adult populations without known
excessive exposure to lead, from several parts of the world.
Table 20. Comparison of daily oral lead intake with Pb-B levels
Study design Oral intake Pb-Ba Pb-B per Reference
(µg/day) (µg/100 ml) 100 µg
oral Pb
Duplicate portion 113 (men) 20.7 18.3 Coulston et al., 1972b
Faecal excretion 119b (women) 15.3 13.0 Tepper & Levin, 1972
Duplicate portion 230 (men) 12.3 5.4 Nordman, 1975
Duplicate portion 180 (women) 7.9 4.4 Nordman, 1975
Composites technique 505 (men) 34.6 6.8 Zurlo & Griffini, 1973c
a "Contributions of air to Pb-B levels are not reported in most of these studies and
could not be subtracted from total Pb-B levels.
b Calculated from daily faecal excretion of 108 µg of lead assuming gastrointestinal
absorption of 10%.
c Pb-B levels from Secchi et al. (1971).
From the data in Table 20 it is not possible to draw any reliable
conclusions regarding the contribution of foods and beverages to Pb-B
levels. The contribution is calculated to be greater in the two
American studies than in the European ones. One of these two American
studies (Tepper & Levin, 1972) was actually of faecal lead excretion,
not of dietary lead. But even if this study were discounted, there
remains a considerable discrepancy between the other American study
(Coulston et al., 1972b) and the European studies, which cannot be
explained.
Each of these studies involved a different number of subjects and
involved different analytical techniques. It is also probable that
there was also exposure from other environmental sources.
At levels of lead intake above 1000 µg per day, the rise in blood
lead level does not appear to increase linearly with dose, but, in
fact, may fit a logarithmic function.
From data published by Kehoe (1961) concerning balance studies on
human volunteers, a single individual with a total daily lead intake
of 600 µg had blood lead levels in the range of 30-35 µg/100 ml
registered over several months, which is consistent with the
relationships suggested in Table 20. However, individuals with larger
daily additions of lead did not have proportionately higher blood lead
levels. A single individual with oral lead intake of 3300 µg per day
had a blood lead level in the 50-60 µg/100 ml range, again followed up
for several months.
For children, the dietary contribution to blood lead is more
difficult to estimate than for adults. Because of the higher
absorption of lead, particularly in infants, the contribution of
dietary lead to blood lead levels may be higher than for adults.
6.1.2.3 Animal studies
The effect of age on gastrointestinal absorption of lead has been
studied in experimental animals. The absorption of lead from food has
been investigated in many animal studies. Values between 5 and 10% are
usual (Port & Brockhaus, 1971; Schlipkoter & Pott, 1973; Horiuchi,
1970).
Kostial et al. (1971) demonstrated that 5-7 day old rats absorb at
least 55% of single oral tracer doses of 203Pb. In an extension of
these studies, Forbes & Reina (1972) observed that the
gastrointestinal absorption of tracer doses of 212Pb, 85Sr and
59Fe was high prior to weaning and decreased rapidly thereafter. In
the case of lead, absorption which was 83% at 16 days, decreased
gradually to 74% on the day of weaning (22 days) and rapidly
thereafter to about 16% at 89 days. The addition of tracer doses of
metals to the diet is, however, an artificial situation. Results might
have been quite different had appreciable amounts of carrier lead been
included. Nevertheless, these observations are consistent with those
reported in young children.
Certain dietary factors have also been shown to influence the
gastrointestinal absorption of lead. Kello & Kostial (1973) have shown
that milk enhances lead absorption in 6-week-old-rats. Fasting
enhances lead absorption, at least as determined by Garber & Wei
(1974) in mice. Low dietary levels of calcium and of vitamin D enhance
lead absorption (Sobel et al., 1938b; Six & Goyer, 1970). It has also
been demonstrated that rats on an iron-deficient diet accumulate more
lead in their bodies than do rats on an iron-sufficient diet (Six &
Goyer, 1972). This seems particularly significant in the light of the
fact that young children in socially and economically deficient homes
have a high incidence of anaemia and excessively high blood lead
concentrations.
The absorption of lead ingested in the form of paint has received
attention because of the hazard of lead-based paint to young children.
Recent data from experiments on rats indicate that lead chromate and
lead naphthenate incorporated into dried paint films are substantially
available for absorption, although to a somewhat lesser degree than
lead naphthenate in oil or lead nitrate in aqueous solution (Gage &
Litchfield, 1968, 1969).
6.2 Distribution and Retention
As with all substances entering the body, a single dose of lead
distributes initially in accordance with the rate of delivery of blood
to the various organs and systems. Redistribution then occurs to
organs and systems in proportion to their respective affinities for
lead. Under conditions of continuous intake over long periods of time,
a near-steady state is achieved with respect to intercompartmental
distribution.
Perturbations in the pattern of distribution occur when large,
short-term peaks of lead intake are superimposed on this well-defined
pattern of long-term distribution.
6.2.1 Human studies
The kinetics of lead distribution and accumulation in man have not
been well defined in man directly. However, from autopsy data, the
general pattern of lead metabolism is clearly discernible. Above all,
it is clear that lead has a strong tendency to localize and accumulate
in bone. The accumulation of lead in the human body begins in fetal
life (Horiuchi et al., 1959; Barltrop, 1969). Lead is readily
transferred across the placenta and the concentration of lead in the
blood of newborn children is similar to that of their mothers,
indicating mother-fetus equilibration processes (Haas et al., 1972b;
Hower et al., 1975). The distribution of lead in fetal tissue is quite
similar to the distribution in adults (Barltrop, 1969).
The total lead content of the body may reach more than 200 mg in
men aged 60-70 years, but is lower for women. Barry & Mossman (1970)
calculated that in non-occupationally exposed adults, 94-95% of the
total body lead (body burden) was in the bones. A similar estimate was
made by Schroeder & Tipton (1968), by Horiuchi et al. (1959), and by
Horiguchi & Utsunomiya (1973). These recent reports serve to reaffirm
the long-recognized affinity of lead for bone. They also provide the
additional observation that the concentration of lead in bones
increases throughout most of life. This is in contrast to soft
tissues. Most soft tissues do not show a significant age-related
change in lead concentration after the second decade of life (Barry,
1975). This is also true of the concentration of lead in whole blood
(US Department of Health, Education and Welfare, 1965; Horiuchi &
Takada, 1954) and in blood serum (Butt et al., 1964). Thus, it appears
that the skeleton is a repository for lead that reflects the long-term
accumulative human exposure, whereas the body fluids and soft tissues
equilibrate reasonably fast and therefore reflect current and recent
exposure. Little is known as to whether the mobilization of lead lying
inactive in the bones can occur so rapidly that signs of poisoning
appear. There is need for more studies in this field.
The concentration of lead in the blood is of prime importance in
the evaluation of lead exposure. It is relied upon as an aid to the
diagnosis of poisoning and as an index of exposure to assess hazardous
conditions both in occupationally-exposed people and in the general
population. It has long been known that lead circulating in the blood
is mainly found in the erythrocytes (Cantarow & Trumper, 1944). The
concentration of lead in erythrocytes is about 16 times greater than
in plasma (Butt et al, 1964). The nature of the association of lead
with the erythrocyte is not clearly understood. Numerous studies have
been reported concerning the in vitro addition of lead to
erythrocytes suspended in plasma or saline solutions. But the validity
of such studies is open to serious question. Thus, Clarkson & Kench
(1958) found that lead added in vitro was readily removed by EDTA,
whereas residual lead present in the cells prior to the addition of
lead could not be removed. This suggests a difference in regard to:
(1) the degree of binding, (2) the site of binding in or on the cell,
or (3) the type of binding of the lead. Recent studies indicate that
lead is mainly bound to human erythrocyte protein, notably to
haemaglobin, rather than to stroma (Barltrop & Smith, 1971, 1972).
The rate of equilibration of lead in blood with sources of input
and with other body compartments has been studied in man by Rabinowitz
et al. (1973, 1974) using a stable lead isotope tracer (204Pb). The
data reported indicate that with a constant daily oral input of
204Pb, a virtually constant concentration of the tracer in the blood
is achieved after approximately 110 days. Upon withdrawal of the
tracer 204Pb from the diet, the 204Pb concentration in the blood
disappears with a half-time of approximately 19 days. The kinetics of
disappearance and accumulation suggest that first order rate processes
of exchange are involved with regard to this relatively mobile
compartment. Tola et al. (1973) also provided data which indicate that
the concentration of lead in the blood rises fairly rapidly to a new
steady state level when men are newly introduced into an occupational
lead environment. The time required for the blood lead concentration
to achieve a new plateau reflecting the new environment is about 60
days.
The body burden of lead increases from birth to old age (Schroeder
& Tipton, 1968; Barry & Mossman, 1970; Barry, 1975). When data for
various specific organs and systems are examined, it becomes evident
that there are two general pools of lead within the total organism.
The major one, in terms of total lead, consists of bone. This pool is
clearly highly accumulative. As a consequence, lead in bone
accumulates through most of the life span. Other organs and systems
are much less accumulative and, to different degrees, tend to
stabilize relatively early in adult life reflecting a greater turnover
rate of lead compared with that in bone.
There is good reason to make a distinction between total body
burden and exchangeable body burden since the organs and systems
comprising the exchangeable body burden are the ones having the
greater toxicological significance. It is also extremely important to
note that lead in whole blood is a part of the exchangeable fraction
of the body burden. Among adults in the general population there is no
age-related difference in regard either to the concentration of lead
in whole blood or in blood serum. Thus, in a general way, the Pb-B
level reflects the concentration of lead in soft tissues, and long-
term changes in Pb-B levels with changes in exposure levels are
probably accompanied by corresponding long-term changes in the rest of
the exchangeable pool.
Nuclear inclusion bodies containing lead have been found in man
subjected to lead exposure (Cramer et al., 1974; Galle & Morel-
Maroger, 1965; Richet et al., 1966) as well as in experimental animals
(see section 7.1.3). Although most frequently reported to occur in the
kidney, they have been found in other organs as well. There is a
suggestion from limited data that inclusion bodies are associated with
short-term lead exposure and not with long-term exposure (Cramer et
al., 1974).
The concentration of lead in deciduous teeth has received special
attention because they are readily available from young children and
because they provide a long-term record of lead exposure, much as is
the case with bone. Dentine in the area adjacent to the pulp is
particularly useful in this respect because it is laid down from the
time of eruption to the time the tooth is shed. It has been reported
that the concentration of lead in dentine is considerably lower in
suburban schoolchildren than it is in children in areas of high lead
exposure (Needleman & Shapiro, 1974).
There has been some interest in the possible use of hair lead as
an index of exposure. Unfortunately, there is no reliable information,
as yet, to indicate just how hair analyses should be interpreted in
relation to the frequency and degree of exposure.
6.2.2 Studies in animals
Animal studies have been particularly useful in defining more
precisely the nature of the kinetics of lead distribution and removal
from various tissues. Following administration of a single dose of
lead to rats, the concentration of lead in soft tissues is relatively
high and falls rapidly, mainly as a result of transfer into the bone
(Hammond, 1971). The distribution characteristics of lead were found
to be independent of the dose of lead over a wide range. The rate
constants for the elimination of lead from various tissues in rats
following a single dose of lead have been described by Castellino &
Aloj (1964). The rate of elimination was much slower from bone than
from other tissues. In studies on rats, Bolanowska et al. (1964) noted
that the rate of elimination of a single dose of lead from the body by
spontaneous excretion became slower with time, reflecting
progressively decreasing mobility of the residual body burden. This is
no doubt mainly due to the fact that as lead becomes progressively
more deeply buried in the bone matrix, its exchangeability with other
compartments and its availability for excretion decrease.
Rather striking age-related differences have been observed
concerning the distribution and retention of lead in rats (Momcilovic
& Kostial, 1974). The rate of elimination of a single tracer dose of
203Pb from the whole body, blood, and kidney was faster in adults
than in sucklings. In the case of the brain, there was actually a
slight increase in the 203Pb content of the brain of the sucklings
while the content was falling in other soft tissues. Numerous animal
studies have also demonstrated placental transfer of lead to the fetus
(see Carpenter, 1974, for relevant literature).
The intracellular distribution of lead has been studied in rat
tissue, mainly by cell fractionation techniques (Castellino & Aloj,
1969; Barltrop et al., 1971). Lead has an affinity for membranes of
the cell, particularly mitochondria. These organelles undergo
functional and ultrastructural changes in organs showing lead effects,
e.g. renal tubular cells (Goyer & Krall, 1969). Little lead is found
in lysosomes (Barltrop et al., 1971) in contrast with the
intracellular distribution of many other metals, e.g. mercury, copper,
iron.
There are few studies indicating the concentration of lead in
target organs that will produce effects. Formation of nuclear
inclusion bodies is observed in rats with renal lead concentrations of
about 10 mg/kg (wet weight) of kidney (Goyer et al., 1970a). Other
effects of lead were found to occur at higher levels of organ
concentration. Death in cattle is associated with lead levels of about
50 mg/kg of kidney cortex (wet weight) (Allcroft & Blaxter, 1950).
The concept of estimating the lowest level of metal accumulation
that results in adverse effects in a target organ has not been well-
explored in the case of lead. This is in contrast with cadmium where
estimates have been made of the minimum concentrations of cadmium in
the kidney cortex at which evidence of renal damage appears (Friberg
et al., 1974).
6.3 Elimination of Lead
The elimination of lead from the body is thought to be mainly by
way of the urine and the gastrointestinal tract. Little is known about
the miscellaneous routes of excretion such as sweat, exfoliation of
skin, and loss of hair.
6.3.1 Human studies
An approximation of the relative contributions of the various
routes to lead excretion in man has been given by Rabinowitz et al.
(1973). This study refers to only one non-occupationally exposed human
subject. Excretions via the kidneys and the gastrointestinal tract
were measured directly. Loss via other routes, e.g. hair, fingernails,
and sweat, was estimated from data on the efflux of 204Pb from the
blood compartment. Losses per day were as follows:
urine 38 µg (76%)
gastrointestinal secretions 8 µg (16%)
hair, nails, sweat, other 4 µg (8%)
The figure of 38 µg for daily urinary excretion is consistent
with the data of Teisinger & Srbova (1959). They reported an average
daily urinary lead excretion of 31 µg.
The mechanism of urinary lead excretion in man is not well
understood. However, the studies of Vostal (1966) provide strong
evidence that the process of renal clearance of lead is essentially
glomerular filtration. Extrapolation of a curve of glomerular
filtration rate plotted against lead excretion rate resulted in zero
lead excretion at zero filtration. The form of lead appearing in the
urine has not been defined. One study suggests that the form in which
lead appears in the urine depends on whether exposure to lead is
normal or elevated. Thus, in lead workers with high urinary lead
excretion, it has been found that only one-half to two-thirds of the
urine lead can be precipitated with co-precipitating agents such as
oxalate, phosphate, or carbonate. By contrast, virtually all the lead
in the urine of people with normal lead exposure can be co-
precipitated (Dinischiotu et al., 1960). This suggests that a stable
lead chelate species arises with elevated exposure. Nuclear inclusion
bodies or lead-protein complexes are found in the urine of children
with acute lead poisoning (Landing & Nakai, 1959).
The rate of biliary excretion of lead in man is not known.
The biological half-time of lead is extremely difficult to
estimate. The constantly decreasing availability of the major stores
of lead in osseous tissue makes it virtually impossible to describe
the rate of loss from the body in simple terms. It is at least clear
that, in man, clearance of one-half of a body burden of lead would
require a number of years.
6.3.2 Animal studies
Animal data on the routes of lead excretion suggest a
considerable species variation. In rats (Castellino et al., 1966) and
in sheep (Blaxter & Cowie, 1946) excretion by biliary and transmucosal
routes is greater than urinary excretion. On the other hand, the ratio
of urinary to gastrointestinal lead excretion in the baboon is 2:1
(Eisenbud & Wrenn, 1970). Vostal (1966) studied the mechanism of lead
excretion in dogs. In mild chronic intoxication, excretion was by
glomerular filtration, without evidence of any tubular secretion or
reabsorption. With more severe poisoning, there was evidence of renal
tubular reabsorption. Evidence was also presented for a tubular
secretory mechanism in the chicken.
6.4 The Metabolism of Alkyllead Compounds
The characteristic toxic effects of tetraethyllead and
tetramethyllead are not caused by the tetraalkyl compounds themselves,
but rather by the trialkyl derivatives formed by dealkylation in the
liver (Cremer, 1959; Cremer & Callaway, 1961). Tetraethyllead is
initially converted mainly to triethyllead and partly to inorganic
lead (Bolanowska, 1968). The triethyllead concentration in organs then
falls only slowly. Even after several days, there is no significant
reduction. The behaviour of tetramethyllead is quite similar to the
behaviour of tetraethyllead. Tetramethyllead is much less toxic
probably because it is dealkylated to the trialkyl toxic form much
more slowly than is the case with tetraethyllead (Cremer, 1965).
Since both these compounds have toxic and biochemical effects
unlike those of inorganic lead, it is not to be expected that the
biochemical tests used in assessing inorganic lead exposure would have
the same significance as in exposure to organic lead. Indeed, in
severe cases of tetraethyllead poisoning, urinary coproporphyrins and
ALA excretion are usually not elevated, and free erythrocyte
porphyrins are only moderately and inconstantly elevated (Gutniak et
al., 1964; Beattie et al., 1972b). These biochemical tests are
therefore of little use in short-term exposure situations. But, in
long-term exposure situations, it is possible that some of them may be
useful. Indeed, Robinson (1974) has shown that in workers industrially
exposed to tetraethyllead, the urinary excretion of ALA is increased,
but not to the same degree as in workers exposed to inorganic lead who
have equivalent levels of total urinary lead excretion (organic plus
inorganic). This suggests that some portion of total urinary lead is
reflecting alkyllead exposure. Bolanowska et al. (1967) demonstrated
that, in three fatal cases of tetraethyllead poisoning, the ratio of
inorganic lead to triethyllead ranged from 67:1 to 18:1 in the urine.
But this ratio did not reflect the ratio of inorganic to triethyllead
in tissues at all accurately. In tissues, including the brain, the
ratios were approximately 1:1.
7. EXPERIMENTAL STUDIES ON THE EFFECTS OF LEAD
The major part of published experimental work on animals
describes or aims to explain pathological or pathophysiological
changes caused by lead. It does not contribute much to the
understanding of the relationship between the dose administered, its
distribution in a period of time, and the biological effect. The doses
used in most animal experiments have, as a rule, been far above the
levels that can occur in environmental or occupational contact with
lead, with the exception of accidental ingestion of soluble lead
compounds.
7.1 Animal Studies
7.1.1 Haemopoietic system
Experimental studies on the effects of lead on blood and
haemopoiesis have been carried out essentially to study pathogenic
mechanisms. There are few studies dealing with the relationship
between the lead dose and blood changes.
There is a great deal of evidence showing that lead inhibits
several enzymes that participate in haem synthesis. Inhibition of
these enzymes is invoked to explain the rises in haem intermediates
that occur as a result of lead exposure. Thus, the rise in erythrocyte
protoporphyrin is readily explained on the basis of the well known
inhibitory effect of lead on the mitochondrial enzyme ferrochelatase
(EC 4.99.1.1) (haem synthetase). This action was first proposed by
Rimington (1938) as the probable explanation for the anaemia in lead
poisoning. Numerous studies have since confirmed that lead is indeed a
rather potent inhibitor of haem synthetase (Dresel & Falk, 1954;
Goldberg et al., 1956; Klein, 1962).
Although specific inhibition of the enzyme haem synthetase is
usually invoked to explain the accumulation of protoporphyrin, it is
also possible that the availability of iron for coupling with
protoporphyrin is inhibited by lead. It has been shown that lead
interferes with the transfer of iron from transferrin to human
reticulocytes (see section 8.2.1). Further support for the idea that
lead interferes with the availability of iron is to be found in
studies showing that lead causes accumulation of iron as "ferruginous
micelles" in developing erythrocytes (Bessis & Jensen, 1965).
Mitochondrial damage was evident in these studies, suggesting the
possibility that globin synthesis may be compromised, along with haem
synthesis.
The increased excretion of coproporphyrin III in urine is
suggestive of an inhibition by lead of the enzyme coproporphyrinogen
oxidase (EC 1.3.3.3), which converts coproporphyrinogen III to
protoporphyrin IX (PP) (Goldberg, 1972). There is no supportive
evidence showing a direct inhibitory effect on this enzyme. One would
imagine that inhibition of coproporphyrinogen oxidase (EC 1.3.3.3)
would result in decreased blood levels of protoporphyrin IX, however,
the opposite is true. Perhaps the concurrent rise in erythrocyte
protoporphyrin, ALA excretion, and excretion coproporphyrin III in
urine can be explained on the basis of delta-aminolevulinate synthase
(EC 2.3.1.37) (ALAS) stimulation. Stimulation of ALAS activity by lead
acetate in vivo has been demonstrated in the avian hepatocyte,
probably due to impairment of haem synthesis (Strand et al., 1972). By
contrast, Gajdos & Gajdos-Török (1969) found no change in the ALAS
activity of bone marrow or liver in experimental lead intoxication of
rabbits.
Animal studies have been reported concerning ALAD inhibition by
lead in tissues concurrently with inhibition in the circulating
erythrocytes. This has been shown in the blood, brain, and liver of
suckling rats (Miller et al., 1970). After 3040 days of exposure,
erythrocyte ALAD underwent an 80-90% reduction. The blood lead
concentration in these rats is not given but can be estimated from
data in the report. It is stated that a maximum of 3 ml of blood was
obtained from each rat. It also appears that the blood specimens each
contained about 4.5 µg of lead. Therefore the blood lead concentration
must have been at least 150 µg of lead per 100 ml of blood, and was
probably nearer to 200.
In other studies, in which long-term lead exposure of rats
resulted in about 50% inhibition of erythrocyte ALAD, there was no
inhibition of brain or liver ALAD (Coulston et al., 1972a): this may
be due to the fact that the exposure levels were lower in this study
than in the others cited.
The question of the significance of lead exposure in relation to
haemoglobin formation has been studied in dogs by Maxfield et al.
(1972). These authors were mainly concerned with the question of
whether the depression of ALAD activity in the peripheral blood was in
any way associated with depressed formation of haemoglobin. Dogs were
given lead over a period and the ALAD activity fell to a very low
level. But the ability of the dogs to regenerate haemoglobin after
removal of half of the circulating blood volume remained essentially
normal. Although this indicates that the inhibition of ALAD activity
in peripheral blood may not be significant, it should be pointed out
that the lead exposure was not sufficiently high to cause any
substantial rise in ALA excretion in the urine. ALA excretion was only
approximately two-three times the baseline level.
There is evidence that the synthesis of globin is affected by
lead in animals as well as in man. In an in vitro study, it was
shown that the incorporation of 14C-glycine into globin in duck
erythrocytes was reduced by 25% by a lead concentration of
5 × 10-4 M (Kassenaar et al., 1957). The reduction of 14C-glycine
incorporation into haem was considerably greater.
Relatively little is known about the effects of lead on the
formation or activity of other haem-containing compounds in the body.
There is some evidence, however, that lead can inhibit formation of
cytochrome P-450, a haemoprotein intimately involved in the drug-
metabolizing mixed function oxidase system of hepatic microsomes
(Alvares et al., 1972). Long-term lead administration has also been
shown to affect the activity of cytochrome c oxidase (EC 1.9.3.1)
(Makagev & Verbolovic, 1967; Verbolovic, 1965). The effects seen were
of a mixed nature, involving first stimulation then depression of
activity. A decrease was also observed in the myoglobin concentration
of some muscle groups. It is not clear from these studies whether the
effects were due to inhibition of haem synthesis or of protein
synthesis.
Administration of lead to rats (over a period of 6 months) in
doses of 2-4 g per rat, resulted in a change in cytochrome c oxidase
(EC 1.9.3.1) activity and in the amount of haemoglobin. The magnitude
of the change increased with larger doses (Verbolovic, 1965). Dogs
were given a solution of lead acetate over a 2-year period resulting
in a reduction in the activity of cytochrome c oxidase (EC 1.9.3.1)
that was in proportion to the dose of lead administered (Makagev &
Verbolovic, 1967).
By means of electron microscopy, Pernis et al. (1964) showed
grossly swollen mitochondria in the erythrocytes of lead-poisoned
guinea-pigs, diverse vacuolar formations, and aggregates of molecules
of ferritin. Electron microscopy of erythrocytes of rabbits receiving
an intravenous dose of 20 mg/kg of 2% lead acetate solution showed
vacuolization of the cytoplasm and swelling of the plasma membrane. An
intensive vacuolization in thrombocytes and a reduction in the
quantity of organelles, particularly those containing serotonin, also
took place. In addition, a swelling of mitochondria after the complete
disruption of cristae was noted (Hacirov, 1972). An experiment on rats
showed ultramicroscopic changes of mitochondria in the red bone marrow
cells in the early stages of poisoning.
7.1.2 Nervous system
7.1.2.1 Inorganic lead
In view of recent concern about subtle impairments of cerebral
function at sub-encephalopathic levels of lead exposure, there has
been a renewed interest in lead and its toxic effects. High doses of
lead will produce encephalopathy; this has been reported in cats (Aub
et al., 1926) and dogs (Staples, 1955).
The brains showed histopathological features similar to those
described in human encephalopathy. Others have since reproduced this
syndrome in the rat (Thomas et al., 1971; Michaelson, 1973; Clasen et
al., 1974) and in the mouse (Rosenblum et al., 1968; Silbergeld &
Goldberg, 1974). The effects may be explained on the basis of
retardation of brain development (Michaelson, 1973; Krigman et al.,
1974).
Paraplegia was reported in suckling rats by Pentschew & Garro
(1966). The disease was produced by transfer of lead from the mother's
milk until weaning, with subsequent post-weaning feeding of lead to
the young.
Behavioural abnormalities such as excessive self-grooming and
aggressiveness occur, even when the lead intake is reduced to a point
where paraplegia no longer occurs (Michaelson & Sauerhoff, 1974). It
was estimated that the minimum daily lead intake causing behavioural
effects (Michaelson & Sauerhoff, 1974) rose from 0.08 mg/kg body
weight at birth to 3 mg/kg at day 16 as a result of suckling. Post-
weaning, this minimum intake rose from 50 mg/kg at day 20 to 60 mg/kg
at day 28. From day 16 to day 20, intake was difficult to estimate
since the infant rats were eating and suckling to different degrees.
Other studies in rats (Snowdon, 1973) and sheep (Carson et al., 1974)
indicate that offspring of mothers exposed to lead during pregnancy
show learning defects. Older animals are refractory to this type of
effect (Brown et al., 1971).
Future behavioural studies should probably be extended to include
sub-human primates since it has been shown that the histological and
clinical features of lead encephalopathy can be produced in both
infant and adult baboons (Cohen et al., 1972; Hopkins & Dayan, 1974).
Studies of lead neuropathy in animals indicate that demyelination
and axonal degeneration are more consistent findings than neuronal
damage in the anterior horn cells or dorsal root ganglia of the spinal
cord (Lampert & Schochet, 1968; Schlaepfer, 1969; Fullerton, 1966).
This is consistent with findings in man. The slowing of nerve
conduction found in man has also been produced experimentally in the
guinea-pig (Fullerton, 1966).
It is known that lead interferes in some manner with synaptic
transmission in the peripheral nervous system and that the effects can
be reversed by calcium (Kostial & Vouk, 1957). But, in addition, an
increased frequency of miniature end-plate potentials has been
reported (Manalis & Cooper, 1973). Neuromuscular blockade has also
been demonstrated in the rat phrenic nerve-hemidiaphragm preparation
(Silbergeld et al., 1974). Again, as in the other studies, the effect
was antagonized by calcium. The significance of these findings with
regard to the central nervous system remains to be determined.
Studies at the biochemical level have been very limited. It has
been shown, using the "Pentschew model", that incorporation of
14C-glucose carbon into dicarboxylic amino-acids of the brain is
reduced (Patel et al., 1974a, 1974b). These results were interpreted
to indicate delayed brain maturation.
Recent work in dogs (Stowe et al., 1973) has mapped the variation
in lead concentrations in different parts of the brain of lead-
poisoned dogs. The studies show a relationship between areas of the
most marked histological change and high lead concentration. Male pups
from the same litter were fed a purified diet, low in calcium and
phosphorus, with and without 100 mg/kg of lead as lead acetate from
the age of 6-18 weeks. The concentration of lead in the various brain
segments is given in Table 21.
Table 21. Distribution of lead in the brains of control and
lead-intoxicated dogsa
Brain segment Lead concentration (mg/kg of wet tissue)
Control Lead intoxicated
Cerebellum 0.160 ± 0.052 0.587 ± 0.113
Medulla 0.155 ± 0.007 0.713 ± 0.112
Frontal white 0.053 ± 0.027 0.920 ± 0.156
Thalamus <0.10 1.023 ± 0.142
Occipital white 0.020 ± 0.012 1.030 ± 0.115
Caudate 0.120 ± 0.083 1.613 ± 0.345
Frontal grey 0.033 ± 0.015 1.767 ± 0.254
Occipital grey 0.080 ± 0.071 2.357 ± 0.181
a "Adapted from Stowe et al., 1973.
7.1.2.2 Alkyllead compounds
Unlike the case with inorganic lead, intoxication by
tetraethyllead in juvenile or adult rats caused a characteristic
encephalopathy, involving restlessness, ataxia, combativeness
progressing to convulsions, coma, and death (Davis et al., 1963). In
dogs there was extensive muscular tremor and twitching, which
progressed to convulsions, coma, and death.
Biochemical studies of the respiration of brain slices incubated
with inorganic lead compared with triethyllead (the active metabolite
of tetraethyllead) have substantiated the fundamental difference in
the action of alkyllead compounds on the brain (Cremer, 1959). The
toxic moieties in tetraethyllead and tetramethyllead poisoning are the
trialkyl metabolites and not the inorganic lead ion.
Essentially there is no qualitative difference between the toxic
effects of tetramethyllead and tetraethyllead. However, there is a
quantitative difference in that the inhalation LC50 for
tetramethyllead (8870 mg/m3) is about 10 times higher than that for
tetraethyllead (850 mg/m3) (Cremer & Callaway, 1961). The
intravenous LD50 for tetraethyllead is about 10 mg of lead per kg of
body weight in the rat (Cremer, 1959). This is in contrast to the
intravenous LD 50 for inorganic lead, which is approximately 70 mg/kg
in the rat (Fried et al., 1956). The precise manner in which the
trialkyllead ion acts to cause altered brain function is not clearly
known but the mechanism may involve inhibition of amine oxidase
(flavin-containing) (EC 1.4.3.4) (monoamine oxidase) (Galzigna et al.,
1964). In rabbits, administration of toxic doses of tetraethyllead
results in a loss of copper, iron, and zinc in certain areas of the
brain (Niklowitz & Yeager, 1973), suggesting that triethyllead may act
by displacing certain essential trace metals from metalloenzymes in
the brain.
Tetramethyllead injected into rats in overtly neurotoxic doses
did not depress ability to learn a simple task (Bullock et al., 1966).
7.1.3 Renal system
Animal studies have contributed to an understanding of the order
of appearance of the various manifestations of renal toxicity in lead
exposure. The spectrum and train of events as related to the exposure
time and to the dose of lead have recently been reviewed (Goyer &
Rhyne, 1973). In the earliest stage of renal response to lead
exposure, reversible tubular effects occur. These include the
appearance of intranuclear inclusion bodies, which is probably a
mechanism for sequestration of lead. These bodies have been isolated
and found to be composed of a lead-protein complex. The protein is
insoluble in physiological solutions and is rich in acidic amino
acids. It has not been characterized further (Moore & Goyer, 1974).
The intranuclear inclusion bodies appear to have a high and specific
affinity for lead compared with that for calcium, iron, zinc, copper
or cadmium and about 90% of the lead in the kidney is associated with
them (Goyer et al., 1970a; 1970b).
The appearance of these bodies is accompanied by amino aciduria,
glycosuria, and hyperphosphaturia. Morphological and functional
changes in tubular epithelial cells also occur at this stage,
including impaired respiratory and phosphorylative ability.
After further lead administration, more severe changes occur in
the renal tubular epithelium such as hyperplasia and cystic changes.
There is a progressive increase in interstitial fibrous tissue and
atrophy of tubular cells. These are irreversible changes that lead to
a third stage of renal failure, manifested by azotaemia and
hyperuricaemia. Sclerotic glomeruli appear, but the hypertension seen
in some cases of chronic lead nephropathy in man has not been
reproduced in experimental animals. The sequence described above in
animals is probably generally valid for man.
7.1.4 Gastrointestinal tract
The effects of lead on the gastrointestinal tract have been
studied in some detail in the guinea-pig (Mambeeva & Ahmiedova, 1967).
Spastic contractions occurred from the stomach to the jejunum.
Inhibitory effects were also noted, accounting for the frequent
constipation seen to accompany lead colic.
7.1.5 Cardiovascular system
Experimental animal data on the question of hypertension are
conflicting. Among rats given 70 mg of lead acetate per day orally,
only a few survived 40 days and all were hypertensive (Griffith &
Landauer, 1944). Hypertension has also been produced in the rabbit
(Beckmann, 1925). Others have not seen hypertension with lead exposure
in rats (Padilla et al., 1969) or dogs (Fours & Page, 1942). From all
the above animal studies it seems that hypertension can occur with
heavy lead exposure.
There are conflicting reports regarding whether lead can cause
atherosclerosis in experimental animals. Sroczynski et al. (1967)
observed increased serum lipoprotein, cholesterol, and cholesterol
deposits in the aortas of both rats and rabbits receiving large doses
of lead. On the other hand, Prerovska (1973) did not produce
atherosclerotic lesions in rabbits using similar doses of lead given
over an even longer period of time.
Cardiac myopathy has also been shown experimentally in lead-
intoxicated rabbits (Kosmider & Sroczynski, 1961). The mechanism for
this effect is not known.
Kuz'minskaja (1964) and Mironcik & Timofeeva (1974) observed that
rabbits receiving lead after a cholesterol load showed more intense
sclerotic changes in the aorta and myocardium than rabbits on a normal
diet without lead, or than rabbits given cholesterol alone.
Makasev & Krivdina (1972) observed a phased change in the
permeability of blood vessels (first phase-increased permeability;
second phase-decreased permeability) in rats, rabbits and dogs, that
received a solution of lead acetate. A phased change in the content of
catecholamines in the myocardium and in the blood vessels was observed
in subacute lead poisoning in dogs (Mambeeva & Kobkova, 1969). This
effect appears to be a link in the complex mechanism of the
cardiovascular pathology of lead poisoning.
7.1.6 Respiratory system
Alveolar macrophages from guinea-pigs are damaged in vitro by
inorganic lead compounds (3 µg/1 × 106 cells) thus releasing a
rapidly occurring lysis and a slowly developing, coarsely blistered
vacuolization. More than 90% of the cells are damaged within 20 hours
(Beck et al., 1973).
Similar effects seem to occur in the organism, since in rats that
had inhaled 10 µg lead/m3 for 3-12 months, the number of macrophages
that could be flushed from the lungs was reduced by 60% (Bingham et
al., 1968).
Electron microscope investigations of the lungs of rats that had
been exposed for 14 days to concentrations of 100-200 µg of lead
oxide/m3 revealed toxic effects in the alveolar macrophages and the
type I alveolar epithelial cells. The structures of the endoplasmatic
reticulum and mitochondria were changed (Bruch et al., 1973).
In the lungs, the alveolar macrophages have the capacity to
degrade noxious substances and are important for other defence
reactions. The ability of alveolar macrophages of guinea-pigs that had
inhaled concentrations of 70-170 µg of lead per m3 of air for four
days, to degrade benzopyrene was distinctly decreased, the benzopyrene
3-monooxygenase (1.14.14.2) activity being only about 10% of the
original value. The activity returned to normal after three days
without any lead exposure (Bruch et al., 1975). The elimination of
bacteria from the lungs was also reduced, when rats were exposed to
70 µg of lead per m3 of air (Schlipkoter et al., 1977).
7.1.7 Reproductive system
Animal studies support the contention that behavioural
deficiencies can occur in infants and newborn as a result of
intrauterine exposure to lead via their mothers (see section 7.1.2).
Others have shown a reduction in the numbers and size of offspring
(Dalldorf & Williams, 1945; Puhae et al., 1963). Data in rabbits (Cole
& Bachhuber, 1914), guinea-pigs (Weller, 1915), and rats (Stowe &
Goyer, 1971) indicate that paternally-transmitted effects can occur,
including reductions in litter size, weights of offspring, and in
survival rate. Several investigators have reported that oral
administration of lead to animals even at doses in the microgram per
kilogram range can cause changes in spermatogenesis (Egorova et al.,
1966; Golubovid et al., 1968), and an increase in testicular RNA and
DNA content (Golubovic & Gnevkovskaja, 1967; Golubovic et al., 1968).
7.1.8 Endocrine organs
The effects of lead on thyroid function that have been reported
in man have also been demonstrated experimentally in rats (Zel'tser,
1962; Sandstead, 1967).
7.1.9 Carcinogenicity
7.1.9.1 Inorganic lead compounds
The carcinogenic risk to man of lead salts and the relevant
studies in animals have recently been discussed in an IARC publication
(IARC, 1972).
The induction of benign and malignant renal neoplasms has been
observed in both Swiss mice and rats fed on diets containing 100 or
1000 mg of basic lead acetate (Pb(C2H302)2. 2Pb(OH)2) per kg
of diet (Van Esch & Kroes, 1969; Van Esch et al., 1962; Mao & Molner,
1967; Azar et al., 1973). Similar results were observed in rats fed
1000mg of lead acetate (Pb(C2H302)2. 3H20) per kg of diet
(Boyland et al., 1962). In addition to renal neoplasms, tumours of the
testes, the adrenal, thyroid, pituitary, and prostrate glands and of
the brain have been reported in rats fed lead acetate or basic lead
acetate, but the results await confirmation (Zawirska & Medras, 1968;
Oyasu et al., 1970). Rats given intraperitoneal or subcutaneous
injections of lead phosphate also developed renal tumours. Total doses
of 120-680 mg of lead were effective (Zollinger, 1953; Roe et al.,
1965). No kidney tumours were reported in hamsters fed 100 or 500 mg
of basic lead acetate per kg of diet for up to 2 years (van Esch &
Kroes, 1969).
In Syrian golden hamsters given a combination of lead oxide and
benzo[a]pyrene intratracheally once weekly for 10 weeks, lung adenomas
occurred in 11/26 animals within 60 weeks. One adenocarcinoma of the
lung was also observed. Such tumours did not occur in animals given
the same dose of lead oxide or benzo[a]pyrene alone (Kobayashi &
Okamoto, 1974).
7.1.9.2 Alkyllead compounds
Epstein & Mantel (1968) reported that subcutaneous injection of
0.6 mg of tetraethyllead (given as 4 equally divided doses) to Swiss
mice between birth and 21 days of age produced malignant lymphomas in
1/26 males and 5/41 females, compared with 1/39 and 0/48 controls. In
treated females, the tumours were observed between 36 and 51 weeks
after the first injection. The significance of this finding in female
mice is difficult to assess since this tumour occurs frequently and
with variable prevalence in untreated mice of this strain.
7.1.10 Mutagenicity
Chromosomes from leukocyte cultures from mice fed 1% lead acetate
in the diet showed an increased number of gap-break type aberrations
(Muro 8,: Goyer, 1969). These changes involved single chromatids,
suggesting that injury followed DNA replication.
7.1.11 Teratogenicity
There have not been any adequate animal studies to provide
evidence to support the suggestion that lead may have a teratogenic
effect.
7.2 Acquisition of Tolerance to Lead
Although human studies suggest that there is no acquired
tolerance in regard to haem-synthesis mechanisms, there may be for
other toxic effects. In this regard, it is interesting to note that
the blood lead level at which cattle develop severe encephalopathy
from eating paint is often less than 80 µg/100 ml (Hammond et al.,
1956). However, in cattle receiving 5-6 mg of lead per kg per day
orally, the concentration of lead in the blood exceeded 100 µg/100 ml
within 2-4 months and remained at about that level for as long as four
years with continuous administration, without any apparent harm to the
animals (Allcroft, 1951). In these studies, haemoglobin did not fall
until a terminal illness developed. Hapke (1974) found that in cattle
and sheep the sensitivity to acutely toxic amounts of lead was reduced
by a pretreatment with lead for 5 months. Goyer et al. (1972) have
suggested from their studies on rats that the intranuclear inclusion
bodies that develop during lead exposure serve as a protective
mechanism by binding lead in the kidney, making it less toxic. But in
the recent study of Cramer et al. (1974) (see section 7.1.3) it was
shown that renal intranuclear inclusion bodies are present only in
workers exposed to lead for a relatively short period of time. Thus,
if inclusion bodies serve some protective function, it is only during
a limited period of exposure. The formation of the cadmium-binding
protein, metallothionein, which appears to have a protective role in
cadmium exposure, is induced by a number of metals but not by lead
(Webb, 1972).
7.3 Factors Influencing Lead Toxicity
7.3.1 Age and sex
It has recently been reported that the intraperitoneal lethal
dose of lead in rats is significantly lower for adult male rats than
for adult female rats (Kostial et al., 1974). In the same study, it
was observed that the lethal dose in mg/kg body weight for 3-week-old
rats was about the same as for adult females.
7.3.2 Seasonal variations
The same seasonal pattern of high incidence of poisoning has been
reported in dogs belonging to urban families as has been reported in
children (Zook et al., 1969). It has also been shown experimentally in
rats and mice (Baetjer, 1959; Baetjer & Horiguchi, 1963) and in
rabbits (Blackman, 1937; Horiuchi et al., 1964) that susceptibility is
greater at high ambient temperatures than at normal temperatures.
7.3.3 Nutrition
Experimental studies have shown that nutritional factors may
influence the absorption of lead from the gastro-intestinal tract and
thus alter susceptibility to the toxic effects of lead (Goyer &
Mahaffey, 1972). Low phosphorous and calcium in the diet (Sobel et
al., 1938b; Six & Goyer, 1970), high vitamin D (Sobel et al., 1938a),
and low iron (Mahaffey, 1974) all enhance lead absorption. The amount
and the composition of dietary protein may also influence lead
toxicity. Low protein diets appear to increase the susceptibility to
lead intoxication as compared to high protein diets (Baernstein &
Grand, 1942; Goyer & Mahaffey, 1972).
The significance of these findings for the susceptibility of
people to lead poisoning has not been established. However, many
children, even in developed countries like the USA, have sub-optimal
dietary intakes of calcium, iron, and other nutrients (US Department
of Health, Education and Welfare, cited by Mahaffey, 1974). This may
have a bearing on the problem of increased lead absorption frequently
found in children in poor, urban areas.
7.3.4 Intercurrent disease, alcohol, and other metals
High lead exposure increases the susceptibility of mice to
Salmonella typhimurium infection (Hemphill et al., 1971). Lead
administration also increases the susceptibility of rats (Filkins &
Buchanan, 1973; Selye et al., 1966; Erve & Schumer, 1972), mice
(Clercq de & Merigan, 1969), and baboons (Hoffman et al., 1974) to
endotoxin shock, but such studies have been performed using extremely
large intravenous doses of lead simultaneously with the endotoxin.
Administration of ethanol (10% ad libitum in drinking water)
had no effect on the toxicity of lead to rats as measured by urinary
ALA excretion, renal weight, or lead concentration in the kidneys,
liver, or bones (Mahoffey, 1974).
Very little is known about metal interactions and how they might
affect the toxicity of lead, except at the nutritional level (see
section 7.3.3). Beyond that, a synergistic effect has been noted
between lead and cadmium with regard to experimental teratogenesis
(Ferm, 1969). It was also found that zinc, given in the diet with
lead, protected horses against the toxic effects of lead. Probably,
this effect was not due to inhibition of lead absorption. Zinc
supplementation actually caused an increase in the lead content of
liver and kidney, but a decrease in the lead content of brain and bone
(Willoughby et al., 1972). It might be inferred that zinc displaced
lead from lead-inhibited enzymes that are zinc-dependent, such as ALAD
(Cheh & Nellands, 1973). A dose-dependent effect of zinc, antagonistic
to the depression of ALAD by lead, has recently been shown in vivo
and in vitro as well as an in vitro antagonism of zinc on the
cytotoxic effect of lead on macrophages (Schlipköter et al. 1975;
Ruiter de et al. 1977).
7.4 Human Studies
Planned experimental studies on the effects of lead in man are
sparse. Kehoe (1961), in his famous experimental studies, in which
human volunteers were exposed to a known amount of lead over various
periods of time, confined himself to studying the lead balance only,
and did not report on the effects of lead.
Three subjects ingested 1 and 3 mg of lead daily, in the form of
lead (II) nitrate, for 33 weeks. The ALA-U, CP-U, and erythrocyte
protoporphyrin IX were measured regularly while Pb-B and Pb-U
measurements were performed at irregular intervals (Schlegel et al.,
1973). Exposure from food and ambient air was not controlled during
the experiment. A rise in FEP was obtained with both doses and a rise
in ALA-U and CP-U only with the 3 mg dose. Evaluation of the results
obtained in this study is difficult, partly because of the small
number of subjects studied and partly because the results were rather
erratic.
Coulston et al. (1972b; 1972c) conducted two exposure chamber
experiments on male volunteers (see section 6.1.1.2). The volunteers
were exposed to air lead concentrations with an average of 10.9 and
3.2 µg/m3 for up to 17 weeks. In the 10.9 µg/m3 exposure study, 24
volunteers participated, 6 of whom served as controls. In order to
control dietary lead exposure, total diet for one full day was
collected at intervals of eight days; the results indicated an average
lead intake of about 110 µg/day only. The variables measured were the
Pb-B, ALAD, ALA-U, and CP-U. Blood lead levels increased in all of the
exposed men and appeared to stabilize after about 2 weeks of exposure.
The mean Pb-B level at that time was about double the pre-exposure
mean, i.e., an increase from 19 to 37 µg/100 ml. A concomitant
increase of the urinary excretion of lead was reported; the faecal
excretion remained unchanged however. The rise in blood lead levels
was followed by a decrease in ALAD activity, which after 5 weeks of
exposure was about 50% of the pre-exposure level. No change in ALA-U
and CP-U was reported. Five months after the termination of the
exposure, all but one of the participants had Pb-B values similar to
those before exposure. The ALAD activity returned to normal almost
immediately after cessation of exposure. No changes in the haemoglobin
level were noted during the experiment. In the 3.2 µg/m3 experiment
a rise in the Pb-B level from 20 to 26 µg/100 ml was obtained,
followed by a slight decrease in ALAD activity, which after five weeks
of exposure was about 85% of the pre-exposure level. Other changes
were not reported.
In a recent experimental study, a greater susceptibility to
inorganic lead was demonstrated in females (Stuik, 1974; Stuik &
Zielhuis, 1975). The volunteers were healthy male and female students
aged 18-26 years. Groups of 5 males and 5 females received 20 µg of
lead per kg per day orally for 21 days. Lead was administered as lead
acetate in glycerol.
The control blood lead levels remained fairly constant at
approximately 17 µg/100 ml during the experiment. The exposed male
subjects showed an increase from 20.6 µg/100 ml to 40.0 µg/100 ml at
the end of the second week of exposure (40.9 µg/100 ml in the third
week). The blood lead in females rose from 12.7 µg/100 ml to
30.4 µg/100 ml, the highest level being reached in the first part of
the third week.
The protoporphyrin IX content of the erythrocytes showed no
change in either the control or the exposed male group. However, in
the female group, it showed a rise beginning in the third week and
rising to 48.0 µg/100 ml erythrocytes. The findings were confirmed in
a second experiment.
It is suggested that the increase of the erythrocyte
protoporphyrin IX was a result of interference in the use of iron in
the formation of haemoglobin. The synergism of lead exposure and iron
deficiency might be suggested as being responsible for the increased
response of FEP in females but this will have to be tested further in
experimental and epidemiological work.
8. EFFECTS OF LEAD ON MAN-EPIDEMIOLOGICAL AND CLINICAL STUDIES
Two types of study characterizing the effects of lead on man have
been reported:
-- retrospective studies of the causes of mortality and morbidity in
lead-exposed populations compared with unexposed populations, and
-- studies of the effects of lead on specific organs and systems.
The findings from these two types of study will be considered
separately. In both cases, the main objective will be to establish, as
far as possible, the dose of, or exposure to lead which is associated
with specified effects, and the frequency of such effects.
From the toxicological point of view, "the dose should be defined
as the amount or concentration of a given chemical at the site of
effect, i.e. where its presence leads to a given effect" (Nordberg
ed., 1976). The application of this definition is difficult because
the dose as defined above can rarely be measured directly and has to
be estimated in various ways. In experiments, it is estimated from the
amount injected or ingested or from dermal and other topical
applications (using appropriate absorption factors and body
distribution factors). In inhalation experiments it is estimated from
the concentration as measured in air, the time of exposure, and the
relevant deposition, retention, and absorption factors (if available).
The same considerations apply for dose estimation from occupational
exposure where, in addition to inhalation, the possible dermal
exposure, ingestion during work-time, and exposure which workers are
subject to as members of the general population, should be taken into
account. The dose for the general population is estimated from
inhalation of air, ingestion of food, water, and other beverages, and
various other contacts, including drugs and consumer products,
smoking, and in children, ingestion of soil, settled dust, and paint
chips. A more direct way of estimating the dose is from measurements
in body tissues and fluids such as blood, urine, faeces, sweat, or
hair. Other organs, tissues, cells, and subcellular elements can be
used for this purpose in animal experiments or in autopsy or biopsy
material.
Although the biological effects of lead on man have been
characterized in some detail, the precise doses of lead responsible
for the effects are rarely, if ever, known. With all its acknowledged
shortcomings, the Pb-B level is the vital link between exposure and an
effect. In section 6, an effort was made to define, as far as
possible, the relationship between the lead in air and in the diet and
Pb-B levels. The main objective of this section is to establish the
relationship between Pb-B levels and biological effects. Only in this
way is it possible to estimate the possible biological consequences of
specific levels of lead in environmental media.
Some biological effects of lead bear a close relationship to
concurrent Pb-B levels, others do not. Thus, the degree of ALAD
inhibition in peripheral blood rises and falls more or less
concurrently with the Pb-B level, while some renal effects of lead are
the consequence of an exposure to lead that may have occurred at a
point remote in time and which is not reflected in the Pb-B level at
the time the effect is first manifested clinically. The fidelity with
which the Pb-B level reflects lead concentrations in target organs is
subject to serious problems of analytical error as described in
section 3.
Beyond these considerations, there is the additional problem of
variation in the inherent susceptibility of individuals, and the
influence of co-existent variables that may modify this
susceptibility, such as nutritional status, age, and presence or
absence of diseases such as alcoholism. For all the above reasons, the
Pb-B level cannot be used as a reliable indication of dose or exposure
in dealing with individual patients. They should be used only in
assessing population group exposures at which effects may occur in a
certain proportion of individuals.
Other tests for assessing dose have been proposed, e.g. lead
excretion in response to chelating agents. Regardless of potential
merits and special applications, most information relating health
effects to dose has been obtained using Pb-B levels as an estimate or
index of dose.
8.1 Retrospective Studies of Lead-exposed Populations
8.1.1 Epidemiology of lead poisoning in industry
In many countries there has been a considerable improvement over
the past forty years with respect to hygienic conditions in the lead-
using industries. The exposure of workers to lead was considerably
higher before 1930 than after. In the United Kingdom, the number of
reported cases of poisoning fell dramatically in the decade 1920-30
(Lane, 1964). Against this background, it is useful to consider the
studies of Dingwall-Fordyce & Lane (1963). They found a higher than
expected incidence of death due to cerebrovascular disease among men
with past high lead exposure. The men studied retired from work
between 1926 and 1960. All those studied had at least 25 years of
service. Men in the heavy exposure category had an average urine lead
concentration of 100-250 µg/litrea over the last 20 years of
a 100 µg/litre corresponds to a Pb-B level of approximately
60 µg/100 ml and 250 µg/litre corresponds to a Pb-B level of
approximately 120 µg/100 ml (Williams et al., 1969).
employment. Men in the moderate exposure group had urine lead
concentrations in the normal range. The third group had no exposure.
As can be seen from Table 22, in the heavy exposure group deaths from
cerebrovascular diseases (cerebral haemorrhage, thrombosis, and
arteriosclerosis) were much higher than normal.
The data also suggest that in this group the excessive death rate
was most pronounced among men who retired prior to 1951 when exposure
conditions were probably considerably worse than they were later. In
the same study, it was found that the death rate from malignant
neoplasms was not above the expected rate in any exposure grade.
Unfortunately, the incidence of death due to chronic nephritis was not
reported. A very similar survey was reported by Malcolm (1971) in
which the subjects studied had, with few exceptions, been exposed to
lead at moderate levels (average Pb-B level-65 µg/100 ml). There was
no statistically significant excess mortality in any of the following
disease categories: heart disease, chest disease, cerebrovascular
accidents, cancer, renal disease, and "miscellaneous".
A recent American study is in general agreement with the
conclusions of the British investigators concerning longevity and
causes of death in the lead industries as they have operated over the
last 25-30 years (Tabershaw & Cooper, 1974; Cooper & Gaffrey, 1975).
The subjects were 1356 workers employed in the lead battery and
smelter industries from 1946 to 1970. Both blood lead levels and
urinary lead excretion were quite high. For example, 78.7% of 47
smelter workers had Pb-B levels of 80 µg/100 ml or more, from 1946 to
1961. The figure was still 13.5% after 1965 (489 total workers
sampled). The percentage of battery workers with Pb-B levels above
this was somewhat lower. But for all the various categories of
duration of employment and type of work, 81.5-95.7% of the Pb-B levels
were equal to or greater than 40 µg/100 ml. About 50% of the workers
were employed for more than 10 years. The total mortality in this
group was approximately the same as in the general population. The
authors concluded that there was no evidence that work associated with
lead increased the risk of death due to the major categories of
cardiovascular and renal diseases. However, when chronic renal disease
(chronic nephritis or other renal sclerosis) was segregated as a
separate cause of death, there did appear to be a significant excess
number of deaths. Thus, among smelter workers, the ratio of observed
deaths to expected deaths was 7:2.8 and among the battery workers the
ratio was 14:8.6. A similar association was found for a category of
death classified as "other hypertensive disease": 7:1.9 among smelter
workers and 13:6.3 among battery workers. For the two disease
categories this adds up to 21 excess deaths out of 1267 for whom cause
of death was listed. The authors emphasize that many of the workers in
the study group were probably exposed to air lead concentrations
considerably in excess of 0.15 mg/m3.
Table 22. Deaths from cerebrovascular disease in retired and employed workers from a lead industrya
Status Year of Grade of exposure
death
None Medium Heavy
Expected Observed Expected Observed Expected Observed
incidence incidence incidence incidence incidence incidence
Retired 1926-50 0.7 0 0.2 3 0.8 5
1951-61 7.2 6 3.2 3 8.5 19
1926-61 7.9 6 3.4 6 9.3 24b
Employed 1946-61 3.2 3 3.1 3 5.6 9
a Adapted from Dingwall-Fordyce & Lane, 1963.
b P <0.001.
Although most epidemiological studies on occupational exposure
have been carried out on industrial populations, one extensive study
on orchard workers in the Wenatchee area of the state of Washington,
has been reported (Neal et al., 1941). This study was somewhat
complicated by the fact that exposure was to lead arsenate. In view of
the known toxicity of arsenic, studies were included on the combined
toxicities of lead and arsenic in animals. No synergism was found in
these animal studies (Fairhall & Miller, 1941). The blood lead
concentrations of the orchard workers and their families are
summarized in Table 23.
This study may have been crude in comparison to some more recent
ones, but it had the rather unique merit of examining health effects
not only in men, but also in women and children. Furthermore, the
exposure levels, as reflected in the urine and blood data of Table 23,
were only slightly higher than the approximate upper limit for people
living in highly polluted cities today. The study was concerned with
weight, blood pressure, diseases of the cardiovascular system, skin
disorders, eye irritation, chronic nervous diseases, blood dyscrasias,
kidney diseases, neoplastic diseases, and fertility. There was no
evidence, based on data available at the time, that the health profile
of these people was any different from that of the general population.
In 1968, a follow-up study was undertaken of the people who had
participated in the original study (Nelson et al., 1973). Over 97% of
the original participants were successfully traced. There had been 452
deaths among the 1231 original participants. A life table method of
analysis of the standard mortality ratio was used. The overall
mortality was less than the average for the state of Washington. The
standard mortality ratios of exposed groups were not consistent with
the exposure gradient. The mortality pattern for increasing duration
of exposure was not consistent either.
8.1.2 Epidemiology of lead poisoning in the general adult population
Adequate studies of the relationship between lead exposure and
health status in the general adult population have not been carried
out. The limitations that apply to the epidemiological studies of
occupational groups are magnified when applied to the general
population. The range of exposure levels is smaller between sub-groups
of the general adult population and their socioeconomic,
physiological, and health profiles are probably more diverse.
8.1.3 Epidemiology of lead poisoning in infants and young children
There has been only one study reported of general mortality and
disease-specific morbidity rate in children exposed to lead. The
Wenatchee study referred to in section 8.1.1 included 146 children
under the age of 15. As with the adults in this study, no abnormal
pattern of disease incidence was noted. These children had moderately
high lead exposure (see Table 23).
Table 23. Urine and blood lead content of persons in the Wenatchee study according to
severity of exposurea
Group Urine lead content Blood lead content
No. Average, S.D. No. Average, S.D.
analyses µg/litre µg/litre analyses µg/100 ml µg/100 ml
Low exposure
men 146 35 21 148 26 11
women 123 28 19 124 26 10
Intermediate exposure
men 102 43 30 108 30 11
women 25 27 15 27 22 10
High exposure
men 386 88 60 329 44 16
women 61 46 25 58 34 13
Children under
15 years
boys 81 53 39 17 37 15
girls 65 54 40 14 36 10
a "From Neal et. al., (1941).
8.2 Clinical and Epidemiological Studies of the Effects of Lead on
Specific Organs and Systems
In the following discussion of the effects of lead on various
organs and systems, consideration will be given to dose-effect and
dose-response relationships. The word "dose" as used here will refer
to Pb-B levels, as described in the introductory remarks of this
chapter.
The diversity of the effects of lead on haemoglobin formation and
the complexity of the process itself make it difficult to determine
which inhibitory effect is most sensitive and what is their relative
importance at different levels of exposure (or dose).
Dose-effect refers to the relationship between dose and the
intensity of a specified effect in an individual, e.g. Pb-B level
versus percentage inhibition of blood ALAD.
Dose-response refers to the relationship between the dose and the
proportion of a population showing a defined effect, specified as to
the level of intensity, e.g. the proportion of a population showing
more than 50% inhibition of blood ALAD at a Pb-B of 20 µg/100 ml.
Some effects of lead are not graded, for example, the effects on
the kidney and the central nervous system are usually reported in all-
or-none terms, i.e. a certain proportion of individuals in a
population are reported to have shown the effect at a given range of
Pb-B concentrations. With many effects of lead it is difficult to
specify a dose-response or a dose-effect relationship because the
available data are inadequate.
8.2.1 Haemopoietic system
The evidence for disturbances in haem synthesis is clearly shown
in man by the appearance of abnormal concentrations of haem precursors
in blood and urine. The levels of lead exposure at which these various
manifestations of disturbed haem synthesis first appear have been
studied extensively in man. The sequence of reactions affected by
lead, and the consequences thereof, are shown in Fig. 5.
Lead interferes with the biosynthesis of haem at several
enzymatic steps, with the use of iron, and with globin synthesis in
erythrocytes. Inhibition of ALAD and haem synthetase is well
documented, and accumulation of the substrates of these enzymes (ALA
and PP) is characteristic of human lead poisoning. Inhibition of ALAS
is based on experimental evidence only. Whether there is enzymatic
inhibition or whether other factors affect the conversion of
coproporphyrinogen III (CPG) to protoporphyrin IX (PP) is not clear;
nevertheless, increased urinary excretion of coproporphyrin III is
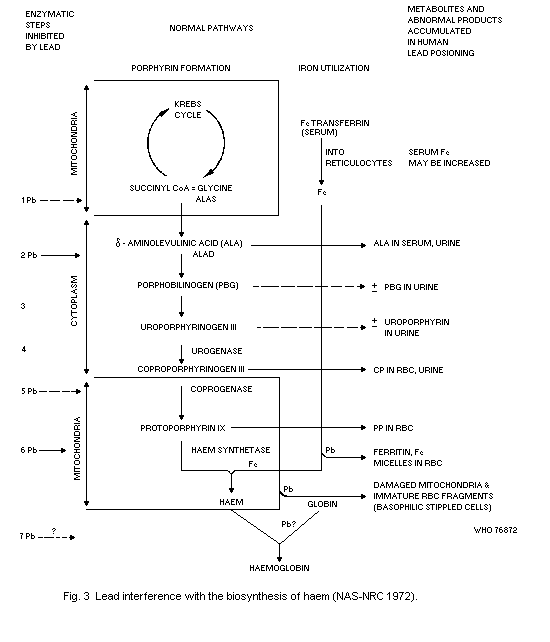 prominent in human lead poisoning. Minor increases in porphobilinogen
(PBG) and uroporphyrins in urine are occasionally reported in severe
lead poisoning. Although the in vivo mechanisms are not clear,
nonhaem iron (ferritin and iron micelles) accumulates in red blood
cells with damaged mitochondria and other fragments not found in
normal mature erythrocytes. Serum iron may be increased in persons
with lead poisoning, but without iron-deficiency states. Globin
synthesis in red blood cells is apparently impaired, although the
mechanisms responsible for reduced globin synthesis remain unknown.
The evidence available suggests that mild anaemia with a small
reduction in blood haemoglobin may occur at, or slightly above, dose
levels that are associated with minimal increases in urinary excretion
of ALA, (Tola et al., 1973).
Increased urinary excretion of ALA is accompanied by an elevation
of the concentration in plasma in adults (Cramer et al., 1974) and in
children (Chisolm, 1968a). This could indicate either an increased
rate of ALA formation or a decrease in the rate of use of ALA. In view
of the well-known inhibition of the enzyme ALAD, most authorities
favour the view that elevated plasma levels reflect decreased use of
ALA. The alternative possibility is that ALA formation is increased,
presumably by increased formation or activity of the enzyme ALA-
synthetase (ALAS). This may in fact be a significant factor. Berk et
al. (1970) studied the rate of haem labelling in one case of lead
poisoning with anaemia. They observed an increase in the rate of
14C-glycine incorporation into the "early labelled peak" of
stercobilin, and into haemin, indicating an increased rate of haem
synthesis in response to an anaemia due to increased erythrocyte
destruction. Coproporphyrin (CP) and ALA excretion were both elevated.
This indicates that haem biosynthesis may be increased in lead
poisoning in spite of increased excretion of haem precursors.
It is also possible that the rate-limiting step in the
pathogenesis of lead-induced anaemia may involve globin synthesis
rather than haem synthesis. White & Harvey (1972) reported that the
incorporation of 3H-leucine into alpha- and ß-chain globins of
reticulocytes was differentially affected in a pair of 3-year-old
twins with clinical lead poisoning accompanied by anaemia. The
radioactivity associated with the different globin chains shifted
systematically as the blood haemoglobin values of the children
returned towards normal.
The major effects of lead on haemopoiesis that are readily
measured in man, are on the rate of excretion of ALA or CP in the
urine, on the concentration of PP in the blood, and on the degree of
inhibition of ALAD in the blood. None have been evaluated in relation
to the fidelity with which they reflect the actual amount of lead
absorbed per unit time, but they have been evaluated extensively with
reference to their correlation with the concentration of lead in the
blood. The literature since 1955, concerned with these
interrelationships, has been reviewed recently by Zeilhuis (1971).
8.2.1.1 delta-aminolevulinic acid dehydratase (ALAD)
The effect of lead that most closely correlates with the
concentration of lead in the blood is the inhibition of erythrocyte
ALAD activity. Within the range of lead exposure encountered in the
general population, the higher the concentration of lead in the blood,
the lower the activity of the enzyme. Above this range, enzyme
inhibition is almost complete and changes little with increasing dose.
The relationship between Pb-B levels and ALAD activity was first
reported by Makao et al. (1968) in a group of twelve men industrially
exposed to lead. Later, Hernberg et al. (1970) reported on a much
larger population of adults having a wide range of lead exposures.
Granick et al. (1973) suggest the interesting possibility of
correcting for individual variations in total ALAD by calculating the
ratio of activity with, versus activity without, enzyme reactivation
using dithiothreitol as a reactivator. This calculation presumably
expresses the inhibitory activity of lead for the particular sample.
The normalization procedure improved the correlation between ALAD and
blood lead. They found that the average no-effect Pb-B level for
inhibitory effects in children, using this correction procedure, was
about 15 µg/100 ml. Tola (1973) reached a similar conclusion from his
study of 1370 workers. His observations suggested that the average
threshold was at a Pb-B level of 10-20 µg/100 ml. However, a recent
study on the Finnish general population puts the existence of a no-
effect level into some doubt. In their study, Nordman & Hernberg
(1975) obtained a statistically significant correlation between ALAD
activity and Pb-B values not exceeding 10 µg/100 ml (Pb-B mean value
8.4 µg/100 ml).
Based on data concerning male workers and children, Zielhuis
(1975) calculated a dose-response relationship for over 40% and over
70% inhibition of ALAD (see Table 24).
8.2.1.2 Free erythrocyte porphyrins (FEP)
The most recently identified biochemical correlate of blood lead
concentration is the erythrocyte protoporphyrin concentration. Some of
the analytical methods in use (see section 2.2.3) measure the
protoporphyrin IX concentration in erythrocytes, while others measure
the free erythrocyte porphyrins, more than 90% of which, however,
consists of protoporphyrin IX (Baloh, 1974). A correlation between FEP
and Pb-B levels has been reported for industrial workers (Haeger-
Aronsen, 1971). The dose-effect relationship is linear if log FEP is
plotted against Pb-B. Two reports have appeared showing this
relationship (Piomelli, 1973; Sassa et al., 1973). In both cases, the
subjects were young children with a wide range of blood lead values.
For the data reported by Sassa et al. (1973) the correlation of the
logarithm of the protoporphyrin IX values and the blood lead
concentrations was fairly good ( r = 0.72). When only the data for
children having had a constant blood lead level for three months or
Table 24. Percentage of adults and children with more than 40% and
70% inhibition of the mean ALAD activity found in control
subjects with Pb-B < 14 µg/100 mla
Pb-B level adults children
(µg/100 ml)
No. > 40% > 70% No. > 40% > 70%
14 -- -- -- 9 11 0
15-24 30 13 3 37 73 8
25-34 26 62 12 24 88 13
35-44 32 97 22 10 90 50
45-54 53 100 68 -- -- --
55-64 37 100 92 -- -- --
65-74 43 100 95 -- -- --
221 80
a "From Zielhuis (1975).
longer were used, the correlation was much better ( r = 0.91). The
point was made by the authors that the elevation of erythrocyte
protoporphyrin IX reflected an inhibitory effect of lead on haem
synthesis that occurs in erythroid cells in the bone marrow, whereas
the absorption of lead by blood elements takes place both in
circulating cells and in erythroid cells.
In recent years, it has become evident that the increase of FEP
occurs at lower Pb-B levels than the increase in ALA in the urine
(Stuik, 1974; Roels et al., 1975). In addition, the same authors
observed that women were more sensitive than men with regard to the
effect of lead on erythrocyte protoporphyrin IX. In women the effect
was evident at a lower Pb-B level than in men, and the rate of
increase in erythrocyte protoporphyrin IX with increasing Pb-B was
greater than in men. From the results of a recent preliminary survey,
children appear to display an FEP response to lead resembling that of
women (Roels et al., 1975). Based on these limited data, for 109 men,
49 women, and for 219 children, Zielhuis (1975) calculated the dose-
response relationship (see Tables 25, 26, and 27).
Table 25. Percentage of adult female subjects with
FEP levels that exceeded those found in
control subjects with Pb-B < 20 µg/100ml.
Pb-B level No. % with FEP level
(µg/100 ml) higher than normal
11-20 28 4
21-30 9 33
31-40 8 90
41-50 )
51-60 ) 4 100
61-70 )
49
a From: Zielhuis, 1975.
Table 26. Percentage of adult male subjects with
FEP levels that exceeded those found
in control subjects with Pb-B <20 pg/100 ml.
Pb-B level No. % with FEP level
(pg/100 ml) higher than normal
11-20 26 0
21 30 43 7
31-40 32 19
41-50 4
51-60 2 100
61 70 2
109
a "From: Zielhuis, 1 975.
Table 27. Percentage of children with FEP
levels that exceeded those found in
control subjects with Pb-B < 20 µg/100 ml.
Pb-B level No. % with FEP level
(µg/100 ml) higher than normal
20 87 5
21-30 72 21
31-40 24 29
41-50 14 )
51-60 12 ) 64
61-70 10 )
219
a From: Zielhuis, 1975.
8.2.1.3 delta-aminolevulinic acid excretion in urine (ALA-U)
The rate of ALA excretion in urine has long been used as a
measure of a biological effect of lead. The most recent studies of
this relationship in industrially exposed subjects indicate that the
logarithm of the ALA concentration in urine increases linearly with
Pb-B levels from 40 µg/100 ml (Selander & Cramer, 1970; Haeger-
Aronsen, 1971; Soliman et al., 1973). Chisolm (1973) reported a good
correlation in children of log ALA excreted in urine per 24 hours
per m2 of body surface and Pb-B levels over a wide range of blood
lead values. In occupational exposure, the excretion of ALA in urine,
at a given Pb-B level was higher in women than in men (Roels et al.,
1975).
Using diagrams published by Haeger-Aronsen (1971) and by Selander
& Cramer (1970) for 207 adult males, Zielhuis (1975) calculated the
dose-response relationships for levels of ALA excretion greater than
5 mg/litre and greater than 10 mg/litre (see Table 28). Some of the
dose-response relationships shown in Tables 24-28 are illustrated in
Fig. 4.
8.2.1.4 Coproporphyrin excretion in urine (CP-U)
Although there is some uncertainty, ALA-U is probably somewhat
more sensitive to the effects of lead exposure than CP-U (Haeger-
Aronsen, 1960; Djuric et al., 1966). ALA-U is also more lead-specific
than CP-U. Data are insufficient for estimating dose-response
relationships.
prominent in human lead poisoning. Minor increases in porphobilinogen
(PBG) and uroporphyrins in urine are occasionally reported in severe
lead poisoning. Although the in vivo mechanisms are not clear,
nonhaem iron (ferritin and iron micelles) accumulates in red blood
cells with damaged mitochondria and other fragments not found in
normal mature erythrocytes. Serum iron may be increased in persons
with lead poisoning, but without iron-deficiency states. Globin
synthesis in red blood cells is apparently impaired, although the
mechanisms responsible for reduced globin synthesis remain unknown.
The evidence available suggests that mild anaemia with a small
reduction in blood haemoglobin may occur at, or slightly above, dose
levels that are associated with minimal increases in urinary excretion
of ALA, (Tola et al., 1973).
Increased urinary excretion of ALA is accompanied by an elevation
of the concentration in plasma in adults (Cramer et al., 1974) and in
children (Chisolm, 1968a). This could indicate either an increased
rate of ALA formation or a decrease in the rate of use of ALA. In view
of the well-known inhibition of the enzyme ALAD, most authorities
favour the view that elevated plasma levels reflect decreased use of
ALA. The alternative possibility is that ALA formation is increased,
presumably by increased formation or activity of the enzyme ALA-
synthetase (ALAS). This may in fact be a significant factor. Berk et
al. (1970) studied the rate of haem labelling in one case of lead
poisoning with anaemia. They observed an increase in the rate of
14C-glycine incorporation into the "early labelled peak" of
stercobilin, and into haemin, indicating an increased rate of haem
synthesis in response to an anaemia due to increased erythrocyte
destruction. Coproporphyrin (CP) and ALA excretion were both elevated.
This indicates that haem biosynthesis may be increased in lead
poisoning in spite of increased excretion of haem precursors.
It is also possible that the rate-limiting step in the
pathogenesis of lead-induced anaemia may involve globin synthesis
rather than haem synthesis. White & Harvey (1972) reported that the
incorporation of 3H-leucine into alpha- and ß-chain globins of
reticulocytes was differentially affected in a pair of 3-year-old
twins with clinical lead poisoning accompanied by anaemia. The
radioactivity associated with the different globin chains shifted
systematically as the blood haemoglobin values of the children
returned towards normal.
The major effects of lead on haemopoiesis that are readily
measured in man, are on the rate of excretion of ALA or CP in the
urine, on the concentration of PP in the blood, and on the degree of
inhibition of ALAD in the blood. None have been evaluated in relation
to the fidelity with which they reflect the actual amount of lead
absorbed per unit time, but they have been evaluated extensively with
reference to their correlation with the concentration of lead in the
blood. The literature since 1955, concerned with these
interrelationships, has been reviewed recently by Zeilhuis (1971).
8.2.1.1 delta-aminolevulinic acid dehydratase (ALAD)
The effect of lead that most closely correlates with the
concentration of lead in the blood is the inhibition of erythrocyte
ALAD activity. Within the range of lead exposure encountered in the
general population, the higher the concentration of lead in the blood,
the lower the activity of the enzyme. Above this range, enzyme
inhibition is almost complete and changes little with increasing dose.
The relationship between Pb-B levels and ALAD activity was first
reported by Makao et al. (1968) in a group of twelve men industrially
exposed to lead. Later, Hernberg et al. (1970) reported on a much
larger population of adults having a wide range of lead exposures.
Granick et al. (1973) suggest the interesting possibility of
correcting for individual variations in total ALAD by calculating the
ratio of activity with, versus activity without, enzyme reactivation
using dithiothreitol as a reactivator. This calculation presumably
expresses the inhibitory activity of lead for the particular sample.
The normalization procedure improved the correlation between ALAD and
blood lead. They found that the average no-effect Pb-B level for
inhibitory effects in children, using this correction procedure, was
about 15 µg/100 ml. Tola (1973) reached a similar conclusion from his
study of 1370 workers. His observations suggested that the average
threshold was at a Pb-B level of 10-20 µg/100 ml. However, a recent
study on the Finnish general population puts the existence of a no-
effect level into some doubt. In their study, Nordman & Hernberg
(1975) obtained a statistically significant correlation between ALAD
activity and Pb-B values not exceeding 10 µg/100 ml (Pb-B mean value
8.4 µg/100 ml).
Based on data concerning male workers and children, Zielhuis
(1975) calculated a dose-response relationship for over 40% and over
70% inhibition of ALAD (see Table 24).
8.2.1.2 Free erythrocyte porphyrins (FEP)
The most recently identified biochemical correlate of blood lead
concentration is the erythrocyte protoporphyrin concentration. Some of
the analytical methods in use (see section 2.2.3) measure the
protoporphyrin IX concentration in erythrocytes, while others measure
the free erythrocyte porphyrins, more than 90% of which, however,
consists of protoporphyrin IX (Baloh, 1974). A correlation between FEP
and Pb-B levels has been reported for industrial workers (Haeger-
Aronsen, 1971). The dose-effect relationship is linear if log FEP is
plotted against Pb-B. Two reports have appeared showing this
relationship (Piomelli, 1973; Sassa et al., 1973). In both cases, the
subjects were young children with a wide range of blood lead values.
For the data reported by Sassa et al. (1973) the correlation of the
logarithm of the protoporphyrin IX values and the blood lead
concentrations was fairly good ( r = 0.72). When only the data for
children having had a constant blood lead level for three months or
Table 24. Percentage of adults and children with more than 40% and
70% inhibition of the mean ALAD activity found in control
subjects with Pb-B < 14 µg/100 mla
Pb-B level adults children
(µg/100 ml)
No. > 40% > 70% No. > 40% > 70%
14 -- -- -- 9 11 0
15-24 30 13 3 37 73 8
25-34 26 62 12 24 88 13
35-44 32 97 22 10 90 50
45-54 53 100 68 -- -- --
55-64 37 100 92 -- -- --
65-74 43 100 95 -- -- --
221 80
a "From Zielhuis (1975).
longer were used, the correlation was much better ( r = 0.91). The
point was made by the authors that the elevation of erythrocyte
protoporphyrin IX reflected an inhibitory effect of lead on haem
synthesis that occurs in erythroid cells in the bone marrow, whereas
the absorption of lead by blood elements takes place both in
circulating cells and in erythroid cells.
In recent years, it has become evident that the increase of FEP
occurs at lower Pb-B levels than the increase in ALA in the urine
(Stuik, 1974; Roels et al., 1975). In addition, the same authors
observed that women were more sensitive than men with regard to the
effect of lead on erythrocyte protoporphyrin IX. In women the effect
was evident at a lower Pb-B level than in men, and the rate of
increase in erythrocyte protoporphyrin IX with increasing Pb-B was
greater than in men. From the results of a recent preliminary survey,
children appear to display an FEP response to lead resembling that of
women (Roels et al., 1975). Based on these limited data, for 109 men,
49 women, and for 219 children, Zielhuis (1975) calculated the dose-
response relationship (see Tables 25, 26, and 27).
Table 25. Percentage of adult female subjects with
FEP levels that exceeded those found in
control subjects with Pb-B < 20 µg/100ml.
Pb-B level No. % with FEP level
(µg/100 ml) higher than normal
11-20 28 4
21-30 9 33
31-40 8 90
41-50 )
51-60 ) 4 100
61-70 )
49
a From: Zielhuis, 1975.
Table 26. Percentage of adult male subjects with
FEP levels that exceeded those found
in control subjects with Pb-B <20 pg/100 ml.
Pb-B level No. % with FEP level
(pg/100 ml) higher than normal
11-20 26 0
21 30 43 7
31-40 32 19
41-50 4
51-60 2 100
61 70 2
109
a "From: Zielhuis, 1 975.
Table 27. Percentage of children with FEP
levels that exceeded those found in
control subjects with Pb-B < 20 µg/100 ml.
Pb-B level No. % with FEP level
(µg/100 ml) higher than normal
20 87 5
21-30 72 21
31-40 24 29
41-50 14 )
51-60 12 ) 64
61-70 10 )
219
a From: Zielhuis, 1975.
8.2.1.3 delta-aminolevulinic acid excretion in urine (ALA-U)
The rate of ALA excretion in urine has long been used as a
measure of a biological effect of lead. The most recent studies of
this relationship in industrially exposed subjects indicate that the
logarithm of the ALA concentration in urine increases linearly with
Pb-B levels from 40 µg/100 ml (Selander & Cramer, 1970; Haeger-
Aronsen, 1971; Soliman et al., 1973). Chisolm (1973) reported a good
correlation in children of log ALA excreted in urine per 24 hours
per m2 of body surface and Pb-B levels over a wide range of blood
lead values. In occupational exposure, the excretion of ALA in urine,
at a given Pb-B level was higher in women than in men (Roels et al.,
1975).
Using diagrams published by Haeger-Aronsen (1971) and by Selander
& Cramer (1970) for 207 adult males, Zielhuis (1975) calculated the
dose-response relationships for levels of ALA excretion greater than
5 mg/litre and greater than 10 mg/litre (see Table 28). Some of the
dose-response relationships shown in Tables 24-28 are illustrated in
Fig. 4.
8.2.1.4 Coproporphyrin excretion in urine (CP-U)
Although there is some uncertainty, ALA-U is probably somewhat
more sensitive to the effects of lead exposure than CP-U (Haeger-
Aronsen, 1960; Djuric et al., 1966). ALA-U is also more lead-specific
than CP-U. Data are insufficient for estimating dose-response
relationships.
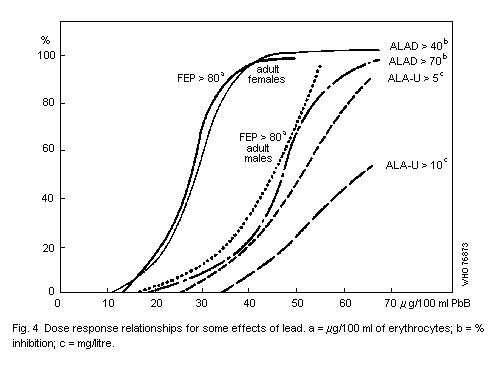 Table 28. Percentage of male adults with ALA-U
levels > 5 mg/litre and >10 mg/litre
according to Pb-B level
Pb-B level No. ALA-U level (mg/litre)
(pg/100 ml)
>5 >10
11-20 17 0 0
21-30 27 0 0
31-40 36 14 3
41-50 55 33 11
51-60 38 74 37
61-70 34 88 50
207
a From: Zielhuis, 1975.
8.2.1.5 Effects of lead on cell morphology
Punctate basophilia occurs in lead poisoning, but a quantitative
relationship between the number of stippled cells and Pb-B levels is
not to be expected (Zielhuis, 1971). Too many variables are involved
in the preparation of smears. The same is probably true of
reticulocyte counts.
8.2.1.6 Effects of lead on erythrocyte survival
Increased rate of erythrocyte breakdown (decreased erythrocyte
life) is often, but not consistently, seen in cases of anaemia due to
lead poisoning. When erythrocytes are exposed to lead in vitro, they
exhibit increased osmotic resistance and increased mechanical
fragility (Waldron, 1966). They also show inhibition of Na-K-ATPase
with increased loss of intracellular potassium (Hasan & Hernberg,
1966; Secchi et al., 1973). These effects have been cited to explain
the fact that in many instances the anaemia in lead poisoning is
accompanied by a shortening of the erythrocyte life span. It is
presumed that one or more of these effects is responsible for the
sensitivity of erythrocytes to spontaneous haemolysis. Erythrocyte
survival time was reduced on the average by 20% in 17 occupationally-
exposed workers, only 3 of whom showed clinical signs of poisoning
(Hernberg, 1967). The author postulated that shortened cell life was
due to the loss of membrane integrity secondary to Na-K-ATPase
inhibition. Anaemia does not necessarily accompany a shortened red
cell life span, and the correlation between blood haemoglobin and life
span was not good in this particular study. The kinetics of
disappearance of labelled cells indicated a shortening of life span by
increased random destruction of cells of all ages. Leikin & Eng (1963)
determined erythrocyte survival in 7 cases of lead poisoning in
children. In 3 cases the erythrocyte survival time was shortened. All
patients were mildly to moderately anaemic. It would seem from these
and other studies that the anaemia in lead poisoning cannot be
explained solely on the basis of reduced erythrocyte survival time.
8.2.1.7 Effects of lead on haem synthesis
The two general points of attack that have been identified are on
haem synthesis and on globin synthesis. Of the two, the effects on
haem synthesis are better understood. It is generally recognized, too,
that manifestations of disturbed haem synthesis often occur in the
absence of frank anaemia. These disturbances may also be significant
for the numerous other haem-dependent enzymatic reactions essential
for normal body functions. Thus, cytochromes, cytochrome c oxidase
(EC 1.9.3.1), and hydroperoxidases are all part of electron transfer
systems requiring haem.
Little is known about the effects of lead on the formation or
activity of other haem-containing compounds. It has been reported that
treatment with EDTA reversed the prolonged antipyrine half-life seen
in two cases of clinical lead poisoning (Alvares et al., 1975). The
authors suggested that in these cases, lead may have significantly
inhibited the synthesis of cytochrome P-450.
8.2.1.7 Relationship between lead exposure and anaemia
It is well known that anaemia is a characteristic early toxic
effect of lead in man. The Pb-B threshold level for this effect is
still not certain. Williams (1966) reported that anaemia did not occur
in industrial workers with Pb-B levels below 110 µg/100 ml. Cooper et
al. (1973) reported that the average haemoglobin level (Hb) was not
decreased at Pb-B levels of up to 100 µg/100 ml and Sakurai et al.
(1974) did not observe any decrease of Hb or erythrocyte
concentrations in workers at Pb-B levels of up to 50 µg/100 ml. On the
other hand, Tola et al. (1973) reported a slight effect of lead on Hb
at an average Pb-B level of about 50 µg/100 ml. This conclusion was
drawn from analysis of the sequential change in Hb among workers newly
introduced into an "industrial lead environment". This approach to the
analysis of the effect of lead on Hb is certainly more sensitive for
detecting an interaction between Pb-B levels and Hb than is a single
Hb determination in a population of lead-exposed persons. Allowance
must, however, be made for the possibility that sequential change in
Hb may be due to seasonal effects independent of lead exposure
(Coulthard, 1958).
Children appear to be more sensitive to lead anaemia than adults.
Thus, Betts et al. (1973) found a significant negative correlation
between Hb and Pb-B levels; a decrease in Hb was evident in 36% of
children with Pb-B levels from 37 to 60 µg/100 ml, compared with only
14% in children with Pb-B levels less than 37 µg/100 ml. Pueschel et
al. (1972) observed a curvilinear decrease in Hb between Pb-B levels
of 40 and 130 µg/100 ml in children between 1 and 6 years old. On the
other hand, McNeil & Ptaznik (1975) found no anaemia in children with
Pb-B levels considerably higher than 40 µg/100 ml. Nutritional
differences may explain the discrepancy. But this does not invalidate
the proposition that for some groups of children a reduction in Hb may
occur at a Pb-B level of approximately 40 µg/100 ml.
8.2.2 Nervous system
8.2.2.1 Central nervous system
Inorganic lead compounds. The effects of lead on the nervous system
vary with the duration and intensity of exposure. Distinction must
also be made between the effects on the central nervous system and the
effects on peripheral nerves. Further questions have been raised
concerning the inherent differences in the sensitivity of the nervous
system of adults and the nervous system of infants and young children.
There is no doubt that lead effects on the brain are much more
commonly associated with childhood lead poisoning than with poisoning
as it is seen in adults. But it is also possible that these
differences are related to the intensity of exposure at the time the
cases are identified rather than to any difference in inherent
sensitivity.
With chronic lead exposure, striking effects may occur referred
to as lead encephalopathy. There are numerous detailed descriptions of
adult lead encephalopathy (Crutcher, 1963; Whitfield et al., 1972;
Teisinger & Styblova, 1961; Aub et al., 1926; Cantarow & Trumpet,
1944). The major features are dullness, restlessness, irritability,
headaches, muscular tremor, hallucinations, and loss of memory and
ability to concentrate. These signs and symptoms may progress to
delirium, mania, convulsions, paralysis, and coma. The signs and
symptoms of encephalopathy in infants and young children are quite
similar to those reported to occur in adults.
The brain lesions in fatal cases of lead poisoning are cerebral
oedema and changes in cerebral blood vessels. The normal convolutions
of the cerebral hemispheres are often obliterated. Capillary
endothelial cells are usually swollen (Pentschew, 1965). Extravasation
of red blood cells and perivascular haemorrhage occur rather commonly
and patchy neuronal loss, serous exudate, glial proliferation, and
occasional areas of demyelinization are all characteristic of lead
poisoning (Blackman, 1937; Okazaki et al., 1963; Whitfield et al.,
1972). But not all deaths due to lead encephalopathy are accompanied
by histological lesions of the central nervous system (Pentschew,
1965).
Neurological sequelae can occur in severe or repeated episodes of
lead encephalopathy. The sequelae are no different qualitatively from
those that occur following traumatic or infectious cerebral injury.
The occurrence of permanent sequelae seems to be much more common
among young children than among adults. Approximately one-fourth of
the children who survived an attack of acute lead encephalopathy
sustained permanent sequelae (Byers, 1959; Chisolm & Harrison, 1956;
Smith, 1964). At least this was true prior to the introduction of
current therapeutic practices such as those described by Chisolm
(1968a). The incidence of sequelae appears to have been substantially
reduced in recent years, but central nervous system sequelae may still
occur if therapy is initiated only after the onset of encephalopathy
(Chisolm, 1973). The most severe sequelae are cortical atrophy,
hydrocephalus, convulsive seizures, and idiocy. More commonly, the
sequelae are of a more subtle nature. Learning ability may be impaired
due to motor incoordination, lack of sensory perception, or inability
to concentrate. Such subtle disturbances have also been claimed to
occur in children with high lead exposure, but in the absence of a
history of encephalopathy (Byers & Lord, 1943; Cohen & Ahrens, 1959).
The major concern today is that young children with elevated lead
exposure, as reflected in Pb-B levels of 40-80 µg/100 ml, may be
experiencing subtle neurological damage without ever exhibiting
classical signs of lead encephalopathy. Studies have been reported of
the neurological status of children with Pb-B values in this range. In
view of the possible long-term effects of lead on the brain,
association between Pb-B and neurological status at the time of
evaluation may give a false impression concerning the level of lead
exposure when the damage was initiated. Exposure levels at the time of
examination may be lower than at the time toxic effects occurred.
Thus, the Pb-B level-effect association may underestimate the dose
responsible for the effect.
Burdé de la et al. (1972) and Peuschel et al. (1972) observed
dysfunction of the central nervous system (irritability, clumsiness,
fine motor dysfunction, impaired concept formation, etc.) in 70 and 58
children, respectively, whose Pb-B levels were always, in all cases,
above 40 µg/100 ml. Albert et al. (1974) studied the psychological
profiles and educational performances of children, 5-15 years of age,
who had histories of lead exposure early in childhood. Those who had
been treated for lead poisoning, with or without encephalopathy,
exhibited a higher incidence of diagnosed mental disorders and of poor
school performance than those who had no such history, even when their
history showed elevated lead exposure early in childhood.
Kotok (1972) established that development deficiencies (using the
Denver Development Screening test, which, according to the author is a
somewhat insensitive measure of development) in a group of
asymptomatic children with elevated lead levels (58-137 µg/100 ml)
were identical to those of a control group similar in age, sex, ethnic
group, environment, neonatal condition, and presence of pica, but
whose Pb-B levels were lower (20-55 µg/100 ml). The deficiencies could
be correlated with inadequacies in the children's environment. Klein
et al. (1974) pointed out that in many studies, pica is not used as a
controlled variable. In his view, pica may be part of a behavioural
deficiency syndrome. In such a case the child would have the
behavioural deficiency regardless of whether or not he ingested lead-
containing objects. Indeed, there is evidence that among mentally
subnormal children whose mental deficiency is unrelated to excessive
lead absorption there is a high incidence of both pica and of
moderately elevated Pb-B levels (Bicknell et al., 1968). In this
study, 67% of the children, whose subnormal state antedated pica, had
Pb-B levels from 39 to 88 µg/100 ml, with a mean of 48 µg/100 ml. By
contrast, among the subnormal group without pica all but one had a
Pb-B level of less than 36 µg/100 ml. The study did not exclude the
possibility that an excessive lead exposure could have aggravated the
pre-existent subnormal state.
Recently McNeil & Ptasnik (1975) published an initial evaluation
of the long-term effects of elevated Pb-B levels in asymptomatic
children, living in El Paso, USA. In 138 out of 206 children aged from
21 months to 18 years (median 9 years), who volunteered (possibility
of selection) to participate, the authors could not find any evidence
of non-specific complaints, hyperactivity, or of abnormal psychometric
testing values, if compared with a matched control group. There
existed a significant difference in one personality test; however this
was explained by geographic isolation and other factors and not by
lead exposure. The average Pb-B levels were, respectively,
50 µg/100 ml (range 14-93) and 16 µg/100 ml (range 10-28).
More recently another psychological evaluation of the El Paso
subjects was published by Landrigan et al. (1975a). Forty-six
children, aged from 3 to 15 years, with Pb-B levels of 40-60 µg/100 ml
were compared with 78 ethnically and socioeconomically similar
controls with Pb-B levels below 40 µg/100 ml. The "Wechsler
Intelligence Scale" showed that the age adjusted I.Q. was
significantly lower in the first group. In addition, the lead exposed
group also showed a significant slowing in the finger-wrist tapping
test. The full-scale I.Q., verbal I.Q., and the behavioural and
hyperactivity ratings did not differ. In this study, unfortunately,
there were differences in age and sex between the study and control
group which might account for the positive findings. It seems
therefore that we have two studies of this situation that come to
different conclusions regarding the possible effects of lead on
neurological and psychological functions.
Another approach has been to identify children with neurological
or behavioural disorders of obscure etiology and to determine whether
they show evidence of current or past elevated lead exposure (David et
al., 1972; Moncrieff et al., 1964; Gibson et al., 1967).
The work of David et al. (1972) is of particular interest because
the neurological abnormality described was one that was reproduced
experimentally in animals (see section 7.1.2). These workers reported
occurrence of hyperactivity among children who had essentially normal
blood lead concentrations, but who excreted abnormally large amounts
of lead when treated with penicillamine. The children had no history
of earlier lead encephalopathy. This study has been criticized because
of statistical inadequacies (Bullpitt, 1972).
Lansdown et al. (1974) examined a population of schoolchildren in
London (less than 17 years of age); there was no relationship between
Pb-B levels and intelligence (Wechsler test), reading (Butt test), and
behaviour (e.g. hyperactivity as rated by the teachers). The authors
suggested that social factors were more important than exposure to
lead in determining mental development. The design of the study has
also been criticized. Neither Landsown's nor David's study are
conclusive.
Morgan & Repko (1974) reported preliminary results of an
extensive study of behavioural functions in 190 lead-exposed workers
(Pb-B = 60.48 ± 16.96 µg/100 ml). In 68% of the subjects the Pb-B
level was less than 80 µg/100 ml. The majority of the subjects were
exposed for between 5 and 20 years. The authors examined 36 non-
independent measures of general performance. In addition, 44 measures
of sensory, psychomotor, and psychological functions were obtained.
Preliminary analysis suggested that Pb-B levels correlated with
several reaction-time measures and ALAD correlated with measures from
strength-endurance-recovery tasks. Both Pb-B levels and ALAD
correlated with eye-hand co-ordination. This study, therefore,
suggested that below a Pb-B level of 80 µg/100 ml some behavioural
changes did occur in adult workers. In addition, variability of
performance increased with increasing Pb-B levels. Only during periods
of high-demand performance did a worker's capacity decrease due to
lead exposure. The authors themselves stressed that this preliminary
analysis still has to be confirmed by further work.
Alkyllead compounds. The encephalopathy of alkyllead intoxication is
somewhat different from that due to inorganic lead exposure. In
documented adult cases of poisoning the most frequent findings suggest
a psychiatric problem. Hallucinations, tremor, delirium, insomnia,
delusions, headaches, and violent mood swings are the most commonly
reported symptoms (Boyd et al., 1957; Machie, 1935). The course of the
intoxication runs from 1 to 10 weeks. Although alkyllead compounds are
notorious for their high lethality, recovery is fairly complete among
survivors (Akatsuka, 1973). Convulsions and coma apparently occur only
in the most severe cases. There is insufficient information to
establish dose-effect and dose-response relationships.
8.2.2.2 Peripheral nervous system
Inorganic lead has toxic effects on the peripheral nervous
system. The older lead literature cites the frequent occurrence of
lead palsy in occupational exposure to lead. The manifestations are
mainly weakness of the extensor muscles, particularly those used most
heavily. While motor function is mainly affected, hyperaesthesia,
analgesia, and anaesthesia of affected areas have also been reported.
Catton et al. (1970) found evidence of reduced nerve conduction
velocity in about one-third of a group of 19 occupationally-exposed
men of whom only one showed any other overt signs of lead toxicity.
The most prominent finding of Seppalainen & Hernberg (1972) in
lead workers (Pb-B levels 80-120 µg/100 ml) without any clinical
neurological signs was reduced motor conduction velocity of the slower
fibres of the ulnar nerves; electromyographic changes included a
diminished number of motor units on maximum contraction and
fibrillations. Similar although less pronounced effects were reported
by Seppäläinen et al. (1975) in 26 workers whose Pb-B levels had never
exceeded 70 µg/100 ml (exposure time 13 months-17 years). Furthermore,
in lead workers with Pb-B levels of 2-273 µg/100 ml, Araki & Honma
(1976) reported statistically significant negative correlations
between nerve conduction velocity and Pb-B, ALAD, and lead
mobilization test values, respectively. More recently, Seppäläinen et
al. (unpublished resultsa) reported a dose-response relationship
between abnormally low conduction velocities, defined as values 2
standard deviations below the mean of an unexposed reference group,
and the highest Pb-B recorded during employment (2-20 years). The
results indicate that nerve conduction impairment is induced in some
workers at Pb-B's exceeding 50 µg/100 ml.
8.2.3 Renal system
The effects of lead on the kidney have been studied extensively.
Two general types of effect have been described. The first is rather
clear-cut renal tubular damage characterized by generalized
aminoaciduria, hypophosphataemia with relative hyperphosphaturia, and
a Reported at the Second International Workshop Permissible Levels
for Occupational Exposure to Inorganic Lead, 21-23 September 1976.
University of Amsterdam, The Netherlands. To be published shortly
in Int. Arch. Occup. Health.
glycosuria, which has been studied in some detail in children with
clinical lead poisoning (Chisolm, 1962). The condition is
characterized by decreased tubular reabsorption of glucose and
alpha-amino acids and therefore reflects proximal tubular damage.
Aminoaciduria was seen more consistently in Chisolm's studies than the
other two manifestations of tubular damage. Thus, the amino acid
transport system is probably more sensitive to the toxic actions of
lead than the transport systems for glucose and phosphate. Limited
data indicate that aminoaciduria is terminated by chelation (Chisolm,
1968b).
In a group of children with slight lead-related neurological
signs, generalized aminoaciduria was found in 8/43 children with Pb-B
levels of 40-120 µg/100 ml (Pueschel et al., 1972). A similar renal
tubular syndrome has been reported to occur in industrially exposed
adults (Clarkson & Kench, 1956; Goyer et al., 1972). In neither of
these studies were Pb-B levels reported. However, Clarkson & Kench
observed signs of lead poisoning (colic and punctate basophilia) in
conjunction with aminoaciduria.
In a group of 7 carefully studied lead-exposed workers,
aminoaciduria was not present. Inulin clearance and renal blood flow
were also normal at the time of examination. For these cases, the
average Pb-B level was 100 µg/100 ml and the minimum was 71 µg/100 ml.
These workers had been exposed for up to 20 years (Cramer et al.,
1974). All had markedly elevated urinary ALA excretion. Interestingly,
some of these workers with prolonged exposure had diffuse interstitial
and peritubular fibrosis as determined by renal biopsy. These
pathological findings are associated with quite a different kind of
renal effect which is seen with prolonged lead exposure. It is
commonly referred to as chronic lead nephropathy. Chronic nephropathy
is characterized by slow development of contracted kidneys with
arteriosclerotic changes, interstitial fibrosis, glomerular atrophy,
and hyaline degeneration of the vessels. This progressive disease
sometimes ends in renal failure. There is evidence that it occurs in
industrially exposed workers, in long-term drinkers of lead-
contaminated whisky, and among middle-aged people who had developed
clinical lead poisoning much earlier in life. Currently, it is only
rarely encountered in occupational exposure.
This renal syndrome can develop and progress to renal failure
long after abnormal lead exposure has terminated. As early as 1897, it
was noted that deaths from chronic nephritis were much more frequent
among people under 30 years of age in Queensland than in other
sections of Australia. The first serious attempt to document a
suspected relationship to earlier childhood lead poisoning was
reported by Nye (1929). Further evidence of a causal relationship
between chronic nephropathy and childhood lead exposure was provided
later (Henderson, 1958). It was shown that people dying of chronic
nephropathy in Queensland usually had a high concentration of lead in
their bones (Henderson & Inglis, 1957). Emmerson (1963) later
demonstrated abnormally elevated lead excretion in response to EDTA
among surviving middle-aged cases of chronic nephropathy. Tepper
(1963), however, was unable to find evidence of chronic nephropathy
among young American adults with a history of childhood lead
poisoning. The Americans had probably been exposed for a much shorter
period of time than the Australians. Other unknown factors may also
have played a role.
The Australian cases involved childhood exposure with an apparent
latency of 10-30 years for the development of renal insufficiency. But
there is evidence that the same effect can result from continuous,
prolonged high lead exposure among adults (Lilis et al., 1968; Richet
et al., 1966; Danilovic, 1958; Morgan et al., 1966; Albahary et al.,
1965; Albahary, 1964). In these cases, lead exposure was higher than
is commonly encountered in industry today.
In a series of 102 cases of lead poisoning studied by Lilis et
al. (1968), 18 cases of clinically verified chronic nephropathy were
found. For the whole series, the mean Pb-B level was approximately
80 µg/100 ml with a range of 42-141 µg/100 ml. Nephropathy was more
common among patients who had been exposed to lead for more than 10
years than among those who had been exposed for less than 10 years.
In the Danilovic (1958) study 7/23 cases had Pb-B levels of about
100-200 µg/100 ml. In the studies of Albahary et al. (1965) Pb-B
levels were not reported. But exposure levels must have been quite
high since the mean ALA excretion was about 37 mg/24 h for 29 workers.
It seems likely, from all available evidence, that a prolonged
high-level lead exposure is necessary, even in childhood, to produce
this progressive chronic nephropathy.
One interesting feature of this syndrome of chronic renal
insufficiency is the frequent association with gout (Emmerson, 1963;
Morgan et al., 1966). Although uric acid excretion is largely
dependent upon tubular secretion, it is not at all certain that
tubular secretion is inhibited. As a matter of fact, a study by
Emmerson et al. (1971) of 13 cases of renal insufficiency due to lead
nephropathy failed to reveal any alteration in uric acid secretion.
The authors suggested an increased tubular reabsorption to account for
the observed decreased clearance of uric acid.
In summary, proximal tubular effects can occur in children and
adults with subtle signs of lead poisoning.
Prolonged exposure to lead leading to a Pb-B level of more than
70 µg/100 ml may give rise to chronic irreversible nephropathy.
However, little is known about dose-effect relationships or about
time-effect relationships for lead-induced chronic interstitial
nephritis.
8.2.4 Gastrointestinal tract
As a symptom of lead poisoning, colic is a fairly consistent
early warning of potentially more serious effects likely to occur with
prolonged periods of exposure. It is most commonly encountered in
industrial exposure. But it is probably also common in lead-poisoned
infants and young children. The occurrence of colic at relatively low
exposure levels in industry is well-known. Although it has been
reported that 13/64 industrially exposed men with presumably lead-
related colic and constipation had blood lead levels from somewhat
less than 40 µg to 80 µg/100 ml (Beritic, 1971), it was also reported
that in every case the diagnosis of lead colic was confirmed by the
findings of high CP-U, excessive basophilic stippling,
reticulocytosis, and various degrees of anaemia. This is consistent
with the general observation that lead colic seems to be accompanied
by other signs of poisoning. There are not enough data available to
establish a dose-response relationship for this lead effect.
8.2.5 Liver
There is no definite evidence for the effects of lead on the
liver. Dodic et al. (1971) reported signs of impaired liver function
in 11 out of 91 patients hospitalized for lead poisoning. Liver damage
was more frequent in cases of severe lead poisoning in 7 out of 18
patients. However, the authors did not provide any information on Pb-B
levels or on indices of disturbed porphyrin metabolism which would
enable the assessment of the stage of lead poisoning. In a laboratory
study of 301 workers in lead smelting and refining, Cooper et al.
(1973) found 11.5% increased aspartate aminotransferase (EC 2.6.1.1),
(SGOT)a values (above 50 U/litreb) in subjects with a Pb-B level
below 70 µg/100 ml, 20% in those with a Pb-B level of about
70 µg/100 ml, and 50% in workers with a Pb-B level above
100 µg/100 ml. The correlation between Pb-B levels and SGOT values was
statistically significant. However, in the absence of information on
the possible influence of diet, infections, or personal habits, the
authors did not draw any definite conclusions concerning the etiology
of these changes.
a Formerly known as serum glutamic oxaloacetic transaminase.
b = 50 × 1.67 × 10-5 mol/(m3.s)
8.2.6 Cardiovascular system
Increased capillary permeability occurs in acute lead
encephalopathy (section 8.2.2.1). Under conditions of long-term lead
exposure at high levels, arteriosclerotic changes have been
demonstrated in the kidney (section 8.2.3). Dingwell-Fordyce & Lane
(1963) reported a marked increase in the cerebrovascular mortality
rate as compared with the expected rate among heavily exposed lead
workers (section 8.1.1). This observation applied to men exposed to
lead during the first quarter of this century, when working conditions
were quite bad. There was no similar increase in the mortality rate
for men employed more recently. Hypertension is an important element
in the etiology of cerebrovascular deaths. Cramer & Dahlberg (1966)
studied the incidence of hypertension in a population of 364
industrially-exposed men, 273 of whom had a long-term exposure to
lead. They subdivided these workers into "lead affected" and "non-
lead-affected" groups, on the basis of the urinary coproporphyrin
test. There was no statistically significant difference between the
groups. Nor was the incidence higher than expected for non-exposed men
in Sweden. This is contrary to the earlier findings of Vigdortchik
(1935) and to the observations of Monaenkova & Glotova (1969). The
disparity may have been due to differences in lead exposure. Other
reports on the question do not show hypertension to be unduly
prevalent among workers exposed to lead (Dressen et al., 1941; Lane,
1949). It is not clear whether vascular effects of lead in man are the
result of an action on blood vessels directly, or whether the effects
are secondary to renal effects.
There is a good evidence that signs of clinical lead poisoning
sometimes include evidence of a toxic action on the heart. Cases have
been described in adults and in children, always with clinical signs
of poisoning. There is of course the possibility that the coexistence
of lead poisoning and myocarditis is coincidental. But in many cases
the electrocardiographic abnormalities disappeared with chelation
therapy, suggesting that lead may have been the original etiological
factor (Myerson & Eisenhauer, 1963; Silver & Rodriguez-Torres, 1968;
Freeman, 1965). In a review of 5 fatal cases of lead poisoning in
young children, heart failure was concluded to be the proximate cause
of death in 2 cases (Kline, 1960). Kosmider & Petelenz (1962) examined
38 adults over 46 years of age with chronic lead poisoning. They found
that 66% had electrocardiographic changes, which was four times the
expected rate for that age group. Orlova (1954) also reported
electrocardiographic abnormalities in cases of lead poisoning.
Dimitrova (1972) reported cardiac abnormalities in workers with
undefined degrees of lead intoxication. There was a correlation of
urinary excretion of lead with duration of systolic contraction and
with isometric tension. Lead mobilization by EDTA accentuated these
effects on the heart. No dose-effect relationships are apparent from
the limited data available.
8.2.7 Reproduction
There is no epidemiological evidence of an effect of lead on the
fertility of women or on in utero fetal development, but there are
numerous reports in the older literature of stillbirths and
miscarriages among women working in the lead trade (Cantarow &
Trumper, 1944; Oliver, 1914). These reports probably contributed to
the promulgation of legislation forbidding the employment of women in
the lead trades in many countries. Panova (1972) reported that women
working in lead industries had a higher incidence, compared with a
control group, of ovulatory dysfunction -- mainly an ovulatory cycles
and cycles with luteal abnormality. A relationship was reported
between ALA-U and the incidence of anovulatory cycles. The effect was
seen at 8-10 mg ALA/litre of urine.
There are not any reliable data to indicate that infertility in
women results from exposure of the male partner to lead.
Some of the early reports on lead poisoning (Oliver, 1914)
suggested that reproductive failures such as sterility and
miscarriages occurred even among the non-working wives of
industrially-exposed men. The reproductive capability of 150
occupationally exposed men was recently studied by Lancranjan et al.
(1975). The results indicated that both lead poisoning and moderately
increased lead absorption decreased the fertility of men. An increased
frequency of asthenospermia, hypospermia, and teratospermia was found.
No interference with the hypothalamopituitary axis was demonstrated;
thus, hypofertility was thought to be due to the toxic effect of lead
on the gonads.
8.2.8 Endocrine organs
Impairment of thyroid function and of adrenal function has been
reported in cases of lead poisoning (Monaenkova, 1957; Sandstead et
al, 1969; Sandstead et al., 1970; Pines, 1965).
There is some evidence suggesting that lead may cause a
derangement of tryptophan metabolism. This is based on the observation
that urinary excretion of 5-hydroxyindoleacetic acid was increased in
227 children living near a lead smelter (Ghelberg, 1966).
Unfortunately, the 5-hydroxyindoleacetic acid determinations were not
quantitative. Furthermore, blood lead values or other indices of
exposure were not determined. Urbanowicz et al. (1969) noted a rise in
5-hydroxyindoleacetic acid excretion in workers heavily exposed to
lead (ALA-U--33.7 mg/litre of urine). The rise preceded the rise in
ALA-U and CP-U. Dugandzic et al. (1973) also noted a rise in
5-hydroxyindoleacetic acid excretion in moderately exposed workers
(ALA-U--28.2 ± 22.6 mg/litre of urine). More recently Schiele et al.
(1974a), using another analytical method, reported that they were
unable to find any significant elevation in 5-hydroxyindoleacetic acid
excretion in workers with relatively high blood lead levels
(88.5 ± 16.1 µg/100 ml).
8.2.9 Carcinogenicity
Dingwall-Fordyce & Lane (1963) did not find any evidence of an
increased incidence of malignant diseases in their follow-up study of
267 workers (section 8.1.1).
In a more recent study of the causes of mortality among lead
smelter and lead battery workers, it was concluded that while the
incidence of malignant neoplasms was somewhat greater than expected,
the difference was not statistically significant (Tabershaw & Cooper,
1974; Cooper & Gaffey, 1975). This seems to support the conclusion of
a IARC Working Group that there is no evidence to suggest that
exposure to lead salts causes cancer of any site in man (IARC, 1972).
8.2.10 Effects on chromosomes
The literature is controversial as regards chromosomal
abnormalities induced by exposure to lead. On the one hand,
chromosomal aberrations have been reported to result from lead
exposure corresponding to mean Pb-B values of 38-75 µg/100 ml in
various groups studied (Forni & Secchi, 1973; Schwanitz et al., 1970).
Moreover, Deknudt et al. (1973) reported chromosomal aberrations in a
group of 14 male workers with signs of lead poisoning. The authors
concluded that, although the workers were exposed to zinc and cadmium
as well as lead, the lead ought to be considered responsible for the
aberrations. On the other hand, Schwanitz et al. (1975) were not able
to corroborate their own findings among occupationally exposed workers
and O'Riordan & Evans (1974) did not find any significant increase in
chromosomal aberrations in shipbreakers with Pb-B values ranging from
40 to over 120 µg/100 ml. Schmid et al. (1972) did not find any
evidence of lead-induced chromosome aberrations in a study on human
peripheral lymphocytes in vivo and in vitro; furthermore,
Bauchinger et al. (1972) did not find any abnormalities in the
chromosomes of policemen with elevated Pb-B levels.
In a recent report, Bauchinger et al. (1976) found that
chromosomal aberrations were significantly increased in a group of 24
male workers occupied in zinc electrolysis and exposed to zinc, lead,
and cadmium. The workers had clearly elevated Pb-B and blood cadmium
levels in comparison with a control group. The authors pointed out the
similarity between this group and the group studied by Deknudt et al.
(1973) as regards combined exposure. However, referring to studies
indicating mutagenicity of cadmium (Oehlkers, 1953; Shiraishi et al.,
1972; Shiraishi, 1975), Bauchinger and his colleagues were inclined to
consider cadmium as being mainly responsible for the aberrations. They
also emphasized the possibility of a synergistic effect of several
metals on the chromosomes. Thus, the question as to whether
chromosomal abnormalities occur as a result of lead exposure in man
remains open. Furthermore, the human health significance of
chromosomal abnormalities seen in lymphocyte cultures, as observed in
some of these studies, is not yet known.
8.2.11 Teratogenicity
There is practically no information in the literature to suggest
that lead is teratogenic for man (Wilson, 1973). Only one case has
been reported of neuromuscular abnormalities and failure to grow in a
child attributed to lead poisoning as a result of the consumption by
the pregnant mother of illicit whisky (Palmisano et al., 1969).
8.3 Factors influencing Lead Toxicity
8.3.1 Acquisition of tolerance to lead
Experience in industry does not suggest that, with continuous
lead exposure, the human body becomes less reactive to lead. There
have been two studies in which the biochemical parameters of lead
exposure were followed for a long period after the initiation of
industrial lead exposure. Tola et al. (1973) found that erythrocyte
ALAD fell to a stable level in about 21 days, as the concentration of
lead in the blood increased correspondingly. Then both blood lead and
blood ALAD remained essentially stable for the next three months.
There was no return toward normal values to suggest development of
tolerance. Urbanowicz (1971) followed ALA-U and CP-U levels in 60
workers for 24 months after they first became industrially exposed.
There was a build-up of both biochemical effects for several months.
But the levels then stabilized for the remainder of the two-year
period. These studies suggest that the toxicologically-active fraction
of the body burden during steady, long-term exposure remains
essentially unchanged.
8.3.2 Age
Young children absorb lead more readily than older people. It
also seems that children are more susceptible than adults in the sense
that toxic effects occur at lower blood lead concentrations. The
susceptibility of old people in comparison with younger adults has not
been studied.
8.3.3 Seasonal variations
It has long been recognized that the incidence of severe lead
intoxication in children is highest during the summer months (Baetjer,
1959; NAS-NRC, 1972). The observation that urinary excretion of lead
increases in late summer may have some bearing (Kehoe, 1961).
8.3.4 Nutrition
There are few reports of studies that point to nutritional
variables as having a distinct effect on lead toxicity in man (NAS-
NRC, 1972; Goyer & Rhyne, 1974). Iron deficiency and lead exposure
both affect porphyrin metabolism at the point where protoporphyrin IX
is converted to haem. An additive effect results.
8.3.5 Intercurrent disease, alcohol, and other metals
Little is known about the effects of intercurrent diseases on the
toxicity of lead or about the effect of lead on the susceptibility of
people to other diseases. People with haemoglobin and erythrocyte
anomalies, such as sickle cell anaemia and thalassaemia, would
probably be more sensitive to the effects of lead exposure, as would
perhaps people with renal damage. It is also possible that an
interaction may exist between lead exposure and infectious disease
processes, although reliable human data are not available to prove the
point.
The effect of ethanol on lead toxicity is of some interest
because the encephalopathy of illicitly-distilled whisky drinkers
could conceivably involve an interaction of lead and the alcohol
consumed. Furthermore, it has been suggested that heavy drinkers among
industrially-exposed men may be more prone to lead toxicity than non-
drinkers (Cramer, 1966; Candani & Farina, 1972).
9. EVALUATION OF HEALTH RISKS TO MAN FROM EXPOSURE TO LEAD AND
ITS COMPOUNDS
The evaluation of health risks to man from exposure to lead and
its compounds involves the following considerations:
(1) the significance of different environmental sources of lead and
of pathways of exposure;
(2) the probability of occurrence of biological effects at different
levels and rates of lead intake;
(3) the significance for human health of the various known biological
effects of lead;
(4) the validity and limitations of various indicators of lead
exposure and of resultant effects.
These considerations have been used in arriving at the
conclusions which are summarized in this chapter.
9.1 Relative Contributions of Air, Food, Water, and Other Exposures
to Total Intake
9.1.1 Adult members of general population groups
For the general population, the major contribution of lead to the
total daily intake is from food, but water and air may provide
significant contributions under certain conditions. Separate
consideration must be given to occupationally-exposed persons in whom
both the total lead intake and the relative contributions of dietary
and airborne lead are quite different.
The inhalation of airborne lead contributes comparatively little
to the Pb-B level in the general population. This follows from the
fact that the lead concentration in ambient air seldom exceeds
3 µg/m3 when averaged over months and from the conclusions reached
in section 6 that the contribution of airborne lead to Pb-B levels is
probably within the range of 1.0 to about 2.0 µg/100 ml for every
1 µg/m3 of air. Although deposition and retention of different forms
of lead in air may vary, estimates of Pb-B levels from the
concentrations of lead in air are similar for the ambient air and for
the air in the work environment.
Even if we assume a concentration of 1 µg of lead per cubic metre
of air contributes as much as 2.0 µg/100 ml of blood, and that the
ambient air concentration of lead is as high as 4.5 µg/m3, the total
contribution of airborne lead would not exceed 9.0 µg/100 ml. This is
still less than two-thirds of the value estimated by a WHO Expert
Committee (1973). The discrepancy arises from the different approaches
used in making the estimate. The WHO Expert Committee's estimate was
based on lung deposition figures for lead obtained using the ICRP
model (Task Group on Lung Dynamics, 1966). However, the ICRP lung
model probably overestimates deposition for particles smaller than
0.5 µm (aerodynamic diameter) (Mercer, 1975), and the assumption that
all the lead that is deposited is absorbed is probably also incorrect.
Dietary intake of lead varies with eating habits and the lead
content of water sources. The majority of estimates from various
countries suggest that the daily oral lead intake from food by adults
ranges from approximately 100 µg to more than 500 µg; most studies
show lead intake from dietary sources to be 200-300 µg/day. Relating
blood lead levels to known daily oral lead intake suggests that each
100 µg of oral lead intake contributes about 6-18 µg of lead/100 ml of
blood. This source of lead therefore accounts for a very large
fraction of the blood lead levels found in the general adult
population with Pb-B values below 25 µg/100 ml.
The quantity of lead intake directly related to the lead content
of drinking water is difficult to estimate. Assuming a lead
concentration in drinking water of 50 µg/litre (which is the upper
limit generally found in the absence of lead pipes or other lead
contributing factors) and a daily intake of one litre of water, 50 pg
of total dietary lead could be attributed to water. This may be
regarded as an upper limit but it must also be pointed out that lead
in water ingested independently of food may be more readily absorbed
and may provide a relatively greater contribution to the blood lead
level than lead in food.
In assessing the relative contributions of air and diet to Pb-B
levels, attention is called to the possibility that air may be a
significant source of dietary lead through fallout. However, there are
no data to confirm this assumption.
Improperly glazed pottery and illicit whisky have been cited as
potential sources of excessive lead exposure for members of the
general population.
Smoking one packet of 20 cigarettes would result in the direct
inhalation of about 1-5 µg of lead but this only indicates the order
of magnitude.
9.1.2 Infants and children
Infants and preschool children are a high-risk group with regard
to lead intake and absorption. Relative contributions from food,
water, and air are difficult to estimate because of the different diet
(e.g. milk) and more active metabolic rate of young children. Also,
intestinal absorption of lead by young children and, in particular, by
infants may be greater than by adults. Tolerable intake of lead for
preschool children should be less than the 3 mg/week recommended
provisionally for adults by a WHO Expert Committee on Food Additives
(1972).
A special hazard for young children is the ingestion of non-food
items, particularly lead-containing paint from surfaces in homes and
lead-contaminated dust and soil.
9.1.3 Occupationally exposed population groups
Because of the variability of occupational exposure, no general
conclusions are possible but precautions against excessive exposure
must be exercised in view of the possibility of extremely high
occupational lead exposures, as cited in section 5.
9.2 Evaluation of Haematological Effects
Based on information presented in section 8, the following
conclusions have been reached concerning the significance of different
effects on haematopoiesis.
Inhibition of ALAD activity in erythrocytes. The health significance
of decreased ALAD activity is still open to discussion. Although
inhibition of ALAD in erythrocytes is to a certain extent paralleled
by a decrease in other organs, e.g. liver and brain, no effect on
health of this decrease has ever been established. Inhibition of ALAD
is generally regarded as a good indicator of lead absorption but not
of health impairment.
Increased excretion of ALA and CP in urine, and increase of FEP are
indicators of impaired haematopoiesis. Although at moderate levels of
increase, no evidence has been brought forward to show that the vital
functions of haematopoiesis are impaired, resulting, for example, in a
reduced life-span of erythrocytes or anaemia, any increase should be
regarded with suspicion and particularly so when it is more than twice
the level found in non-exposed population groups. Because free
erythrocyte protoporphyrins are also increased in the case of iron
deficiency, this test may provide a better indication of impaired
haematopoiesis in exposed iron deficient population groups (especially
children) than the excretion of ALA and CP. Moreover, females and
children appear to have an earlier and steeper increase of FEP than
males for the same levels of Pb-B.
Effects on erythrocyte membrane, as evidenced by shortened life-span
and a decrease of Na-K-ATPase clearly can result in adverse health
effects since anaemia may occur. Anaemia, expressed by decreased
haemoglobin level, may be regarded as a consequence of disturbed haem
and globin synthesis and of the decreased life-span of erythrocytes
and has clear adverse health consequences.
9.3 Dose-Effect Relationships
At present Pb-B levels are the best available indicator of the
dose. It should, however, be recognized that Pb-B does not reflect the
type of exposure. The dose-effect relationships based on Pb-B levels
should generally be used for long-term exposure.
As stated in section 8 (page 99) a dose-effect relationship
refers in this report to the relationship between the dose as
estimated by Pb-B levels and the intensity of a specified effect in
individual subjects. For most effects, not enough data are available
to present adequate dose-effect curves; however, for some effects,
some points on the dose-effect curve can be tentatively estimated; for
other effects, the data available only permit a statement referring to
the Pb-B level below which such an effect has not been reported. This
level is referred to as the no-detected-effect level. The degree of
confidence that can be placed on such estimates will vary depending on
the sample size and the number of studies reporting no effect.
ALAD activity in erythrocytes. There is a negative linear
relationship between the logarithm of ALAD and Pb-B levels. Increase
in Pb-B levels is paralleled by a decrease in ALAD levels in the Pb-B
range up to about 60 µg/100 ml. For higher Pb-B values, the ALAD
activity levels off at a very low level of enzyme activity. The no-
detected-effect level for Pb-B is probably about 10 µg/100 ml but may
be even lower.
ALA and CP in urine; PP in erythrocytes. There is a positive linear
relationship between the logarithm of ALA (CP, FEP) and Pb-B levels;
the no-detected-effect level for ALA and CP is about 40 µg/100 ml; for
FEP the no-detected-effect level in females is about 20-30 µg/100 ml,
in males it is about 25-35 µg/100 ml; and in iron-deficient children
in particular, it may be about 20-25 µg/100 ml.
Effects on the erythrocyte membrane start to occur at higher Pb-B
levels, probably higher than 50-60 µg/100 ml; a study by Secchi et al.
(1974), on Na-K-ATPase, however, reports a lower no-detected-effect
level of between 30 and 40 µg/100 ml.
Anaemia. Some authors maintain that the no-effect level in workers
is above a Pb-B level of 100µg/100ml; others, however, report a slight
decrease in the haemoglobin level, at a mean level of Pb-B of about
50 µg/100 ml. In some population groups and particularly in iron-
deficient children, the no-detected-effect level is at an approximate
Pb-B level of 40 µg/100 ml.
Nervous system effects. The data on effects of lead compounds on the
nervous system lead to the following tentative conclusions in regard
to prolonged exposures:
(1) From Pb-B levels of approximately 40 µg/100 ml, the probability
of the occurrence of subclinical peripheral electrophysiological
changes increases.
(2) From approximately 50 µg/100 ml in children, the probability of
noticeable brain dysfunction increases; in adults the level is
probably somewhat higher (60-70 µg/100 ml).
(3) From approximately 60 µg/100 ml in children the probability of
acute or chronic encephalopathy increases; in adults this level
is higher, probably above 80 µg/100 ml.
(4) The potential effects of lead on the nervous system constitute
one of the main concerns, particularly in children. More
carefully considered prospective studies should be carried out
taking into account various interacting variables such as
nutrition, socioeconomic status, and parental care in order to
establish better founded dose-effect and dose-response
relationships.
(5) No dose-effect or dose-response relationships can be established
for alkyllead exposure on the basis of currently available
information.
The present no-detected-effect level for sub-clinical neuropathy
appears to be a Pb-B value of 40-50 µg/100 ml. For minimal brain
dysfunction it is probably 50-60 µg/100 ml in children and
60-70 µg/100 ml in adults, and for acute or chronic encephalopathy,
60-70 µg/100 ml in children, and over 80 µg/100 ml in adults. The
establishment of relationships between Pb-B levels and effect is
especially difficult in children because the effect may be detected
months or years after the critical exposure occurred.
Renal function. Apparently prolonged exposure to Pb-B levels greater
than 70 µg/100 ml is necessary to produce nephropathy; a no-detected-
effect level cannot be given. The problem is non-correspondence in
time between the determination of Pb-B level and the detection of
effect.
Aminoaciduria, reflecting impaired amino acid transport through
the renal tubules may occur in children and adults with increased lead
absorption. The present data do not allow a no-detected-effect level
to be estimated, but indicate that this effect is unlikely to be found
in association with Pb-B levels below some 90-100 µg/100 ml (Chisolm,
1968b, Cramer et al., 1974).
Changes in blood constituents such as calcium, phosphorus, glucose,
cholesterol, total proteins, serum albumins, alkaline phosphatase
(EC 3.1.3.1), lactate acid dehydrogenase, and urea nitrogen, could not
be found in male workers with a median Pb-B level of 63 µg/100 ml; 37%
showed an effect with a Pb-B level greater than 70 µg/100 ml (Cooper
et al., 1973). There was an indication of increased bilirubin at a
Pb-B level of about 70 µg/100 ml. An increased pyruvate level after
glucose administration was reported in 50% of children with Pb-B
levels of 40-60 µg/100 ml (Moncrieff et al., 1964).
The general pattern of morbidity and mortality in workers does not
appear to be affected if the Pb-B level never exceeds 70 µg/100 ml.
In assessing reported dose-effect relationships and no-detected-
effect levels, one should take into account the fact that the
available data are limited. Even from a theoretical viewpoint, the
establishment of a definite no-effect level is not possible, because
one can hardly ever expect to cover the whole range of susceptibility
in human populations. Nevertheless, the available data suggest that
the no-detected-effect levels given above are on the conservative
side.
Table 29 summarizes the no-detected-effect levels discussed. For
some of these effects, it is possible to elaborate dose-response
relationships. These cases are considered in section 9.4.
Table 29. No-detected effect levels in terms of Pb-B µg of lead per 100 ml of blood)
No detected Effect Population
effect level
< 10 Erythrocyte ALAD inhibition adults, children
20-25 FEP children
20-30 FEP adult, female
25-35 FEP adult, male
30-40 Erythrocyte ATPase inhibition general
40 ALA excretion in urine adults, children
40 CP excretion in urine adults
40 Anaemia children
40-50 Peripheral neuropathy adults
50 Anaemia adults
50-60 Minimal brain dysfunction children
60-70 Minimal brain dysfunction adults
60-70 Encephalopathy children
> 80 Encephalopathy adults
9.4 Dose-response Relationships
A dose-response relationship considers the observed relative
frequency of occurrence of a specified effecta in a group of
subjects at a given dose level. As in the case of dose-effect
relationships, the data available to evaluate a dose-response
relationship are either limited or non-existent. The available
information on dose-response relationships has been presented in
section 8. In this section, attention is paid to the 5% response
levels, i.e. that level of Pb-B at which not more than 5% of the group
considered is expected to show the specified intensity of a specified
effect. The 5% level has had to be stipulated, because not enough data
are available to state the Pb-B levels for 0.5%, 1%, etc. Further
investigations have to be carried out to enlarge the amount of data
available. For further discussion see Zielhuis (1975). His review
suggests the 5% response levels recorded in Table 30, which are in
accordance with the data discussed in section 8. These response levels
are also in agreement with those suggested by Hernberg (1975).
Table 30. Pb-B levels at which no more than 5% of the population will show
the indicated intensity of effect
Biochemical effect Intensity of effect Population Pb-B
(µg/100 ml)
ALAD inhibition perceptible inhibition adult, children 10
> 40% inhibition adults 15-20
> 70% inhibition adults 30
> 70% inhibition children 25-30
ALA-U perceptible increase adults, children 40
> 10 mg/litre adults, children 50
FEP perceptible increase adult males 30
adult females 25
children 20
a From: Zielhuis, 1975.
a Graded effects may be specified in terms of their intensity.
9.5 Diagnosis of lead Poisoning and Indices of Exposure and/or
Effects for Epidemiological Studies
For epidemiological studies and for the detection of the early
effects of lead in occupational exposure of individuals, the following
tests have been used:
(1) Lead levels in blood.
(2) Excretion of lead in urine spontaneously or after administration
of chelating agents.
(3) Lead levels in tissues (teeth, bones, hair, etc.).
(4) Activity of ALAD in blood.
(5) Indices of disturbed porphyrin metabolism: ALA and/or CP in
urine, Protoporphyrin IX in erythrocytes.
(6) Haematological indices such as basophilic stippling and
haemoglobin levels.
(7) Early (sub-clinical) symptoms and signs of other damage (e.g. to
the nervous system or the kidneys).
(8) Clinical evidence of poisoning.
The criteria used by individual investigators correspond to the
premises and purposes of their studies, for example, Pb-B for
evaluating lead levels in the general population, and clinical signs
of poisoning to assess morbidity caused by occupational exposure.
The following considerations should be kept in mind when using
and interpreting the results.
9.5.1 Concentration of lead in blood (Pb-B)
Pb-B reflects the current state of the dynamic equilibrium
between the amounts of lead entering the organism, transported in the
blood, and deposited in the tissues (including the bones). To date,
insufficient information has been collected about the quantitative
aspects of these processes, but from the data available, it may be
stated that:
(a) After a single inhalation of a soluble lead compound, the
concentration of lead in the body will change in the same way as after
an intravenous injection, i.e. there will be a rapid increase in Pb-B
levels followed by a slower decrease; initially there will be a rapid
elimination in the urine and a slow deposition in the tissues with
subsequent redistribution according to the metabolism of lead in the
various organs and systems.
(b) During long-term exposure at a constant rate, an equilibrium
between the amount of lead absorbed, deposited, and excreted develops
over a long period (weeks to months, according to the daily doses
received), which can be considered as a steady state.
(c) There are only limited data as to how quickly this
equilibrium (and Pb-B) changes when irregular variations in the dose
of lead received (e.g. air lead concentrations) occur.
A long-term steady state probably exists normally in non-
occupationally exposed general adult populations, at least in the Pb-B
level. However, no direct evidence for this assumption is available.
In occupationally exposed persons, a steady state cannot be
assumed because of the well known and marked variations of air lead
concentrations in the working environment and of Pb-B levels in
occupationally exposed individuals from one time to another and among
the individuals in the same work place. Occasional exposure to a high
lead concentration in the air could raise the Pb-B level for some time
without contributing significantly to the body burden and to the
biological effects.
If Pb-B is to be used as an indicator of the degree of
environmental lead exposure the above-mentioned facts must be taken
into account, as well as the analytical method used and the
limitations (accuracy, precision, sensitivity, limits of detection).
9.5.2 Aminolevulinic acid dehydratase (ALAD)
For ALAD the same conditions can apply as for Pb-B. The behaviour
of ALAD activity will follow closely the level of Pb-B up to
50-60 µg/100 ml.
9.5.3 Aminolevulinic acid (ALA) and coproporphyrin (CP) excretion
in the urine
ALA and CP in urine are not so dependent on the current state of
lead exposure and absorption as the Pb-B, although their excretion
diminishes relatively quickly when exposure ceases; they reflect more
the average short-term level of lead exposure and have proved useful
in this way. ALA and CP estimates have found broad recognition as
indices of lead absorption and as indicators of early effects they
reflect individual susceptibility to lead.
9.5.4 Lead excretion in the urine
An elevated rate of spontaneous lead excretion in the urine is
indicative of high lead absorbed, but a normal rate of excretion does
not serve as a reliable means of excluding the possibility of
excessive absorption. Lead excretion in urine is dependent on the Pb-B
level but is also influenced by other-mostly unknown-factors, so that
no direct conclusions about exposure and the extent of absorption can
be derived from lead levels in urine (even in a 24-hour sample).
The excretion of lead provoked by chelating agents such as
calcium disodium ethylenediamintetraacetate is thought to reflect the
biologically active portion of the body burden. It is probably a more
sensitive index of over exposure and excess absorption than the Pb-B
level since clearly elevated values have been reported in cases of
only marginally elevated Pb-B levels.
9.5.5 Haematological changes (stippled cells, anaemia)
These are not sensitive indices of over-exposure or excess
absorption. They are not very useful for the early detection of
possible health impairment.
9.5.6 Lead in tissues (teeth and hair)
These have been used as indicators of integrated long-term
exposure and have the advantage that samples are easy to procure. As
yet, the amount of information concerning the interpretation of the
values obtained is inadequate for their evaluation as indices of
exposure or dose.
9.5.7 Some practical aspects
9.5.7.1 General population studies
The Pb-B level is the epidemiological index of choice, assuming
that a reasonable approximation of a steady state exposure exists.
ALAD activity estimates are equally useful for such studies or as
epidemiological indices of lead absorption. The decision to use Pb-B
or ALAD depends on the laboratory facilities available. Signs of lead
effects other than ALAD inhibition are not to be expected at Pb-B
levels below 20 µg/100 ml. Lead in deciduous teeth and hair is
potentially useful as an indicator of integrated exposure in infants
but needs more study.
9.5.7.2 Occupationally-exposed persons
For screening the exposure of groups of workers, any method can
be used that has the required sensitivity and specificity. Economic
and time factors will determine the choice of test. When using the
Pb-B level, the conditions of sampling must be well defined, taking
into account the factors influencing the variations of the Pb-B
concentrations. ALA and CP estimations in urine are widely used since
they are simple, avoid the possibility of external contamination, and
may provide a better picture of the integral exposure. ALAD activity
is only useful at Pb-B levels below about 60 µg/100 ml. For early
detection of the signs of lead effects in individuals, ALA-U or CP-U
tests are the best established screening methods. When abnormal values
are found, further tests (including clinical and laboratory
investigations) will have to be applied to evaluate the kind of
disturbance and the degree of health risk (WHO Study Group, 1975).
9.5.7.3 Reliability of sampling and analytical methods
The evaluation of the pollution of the environment by lead and of
the health effects on man which might result, depends on the
reliability of sampling procedures and analytical methods used.
The methods of sampling for different environmental media, and
the possible exposure pathways of man have been discussed in section
3. The great spatial and temporal variability of these environmental
media and their diversity make the accurate assessment of total
exposure a difficult task. Unless elaborate schemes are set up and
extreme precautions are taken, the total exposure of a population
group cannot be evaluated with an error of less than about 50%, taking
into account tile analytical uncertainties.
In the determination of the dose received or the effects on
haematopoiesis observed, the sampling problem is relatively minor but
the accuracy and precision of analytical techniques play an important
role. An evaluation with up to 20% relative precision is seldom
achieved under normal operational conditions.
9.6 The Problem of Alkyllead Compounds
The principal risk of alkyllead compounds is in occupational
exposure, either by inhalation or by absorption through the skin.
Acute toxicity results in an encephalopathy that differs greatly from
the effects of inorganic lead on the central nervous system. Some
components of the toxic effects are probably due to the alkyl compound
as a whole rather than its lead component. Workmen at greatest risk
are those involved in mixing fuel additives, although other workmen
engaged in related occupations such as the cleaning of storage tanks
where inhalation is possible, are also at high risk. Over-exposure of
the general population to alkyllead compounds has not been documented.
REFERENCES
ABERNETHY, R. F., PETERSON, M. J., & GIBSON, F. H. (1969)
Spectrochemical analyses of coal ash for trace elements. U.S.
Bur. Mines, Rep. Inv. 7281.
AKATSUKA, K. (1973) Tetraalkyl lead poisoning. Jpn. d. ind. Health,
15: 3-66. ALBAHARY, C. (1964) Les troubles porphyriques dans le
saturnisme. Arch. Mal. prof., 25: 495-507.
ALBAHARY, C., RICHET, G., GUILLAUME, J., & MOREL-MAROGER, L. (1965) Le
rein dans le saturnisme professionnel. Arch. Mal. prof.,
26: 5-19.
ALBERT, R. E., SHORE, R. E., SAYERS, A. J., STREHLOW, C., KNEIP, T.
Y., PASTERNACK, B. S., FRIEDHOEF, A. J., COVAN, F., & CIMINO, J.
A. (1974) Follow-up of children overexposed to lead. Environ.
Health. Perspect., Exp. Issue No. 7, 33-41.
ALEXANDER, F. W., & DELVES, H. T. (1972) Death from acute lead
poisoning. Arch. Dis. Child., 47: 446-448.
ALEXANDER, F. W., DELVES, H. T., & CLAYTON, B. E. (1973) The uptake
and excretion by children of lead and other contaminants. In:
Proceedings of the International Symposium; Environmental Health
Aspects of Lead, Amsterdam, 2-6 October 1972. Luxembourg,
Commission of the European Communities, pp. 319-330.
ALLCROFT, R. (1951) Lead poisoning in cattle and sheep. Vet. Rec.,
63: 583-590.
ALLCROFT, R., & BLAXTER, R. L. (1950) Lead as a nutritional hazard to
farm livestock V. The toxicity of lead to cattle and sheep and an
evaluation of the lead hazard under farm conditions. J. Comp.
Pathol., 60(3): 209-218.
ALTMAN, P. U., & DETTMER, D. S., ed. (1968) Nutritional standards in
man. In: Metabolism. Bethesda, MD, Fed. Am. Soc. Exp. Biol.,
pp. 95-96.
ALVARES, A. P., KAPELNER, S., SASSA, S., & Kappas, A. (1975) Drug
metabolism in normal children, lead-poisoned children and normal
adults. Clin. Pharm. Ther., 17(2): 179-183.
ALVARES, A. P., LEIGH, S., CONN, J., & KAPPAS, A. (1972) Lead and
methyl mercury: effects of acute exposure of cytochrome P-450 and
the mixed function oxidase system in the liver. J. exp. Med.,
135 (6): 1406-1409.
ARAKI, S, & HONMA, T. (1976) Relationships between lead absorption and
peripheral nerve conduction velocities in lead workers. Scand.
J. Work environ. Health, 4: 225-231.
ASKEVOLD, R. (1951) Routine analysis of porphyrines in urine.
J. clin. Lab. Invest., 3: 318-319.
ASTM (1970) Book of standards. Tentative method of test for lead in
the atmosphere. Philadelphia, American Society for Testing and
Materials, vol. 23, pp. 830-836.
ATKINS, P. R. (1969) Lead in suburban environment. J. air Pollut.
Contr. Ass., 19: 591-594.
AUB, J. C., FAIRHALL, L. T., MINOT, A. S., & REZNIKOFF, P. (1926) Lead
poisoning. Baltimore, Williams and Wilkins Co., pp. 206 (Medicine
Monographs Vol. VII).
AZAR, A., SNEE, R., & HABIBI, K. (1973) Relationship of community
levels of air lead and indices of lead absorption. In:
Proceedings of the International Symposium; Environmental Health
Aspects of Lead, Amsterdam, 2-6 October 1972. Luxembourg,
Commission of the European Communities, pp. 581-594.
BAERNSTEIN, H. D, & GRAND, J. A. (1942) The relation of protein intake
to lead poisoning in rats. J. Pharmacol. exp. Ther., 74: 18-24.
BAETJER, A. M. (1959) Effects of season and temperature on childhood
plumbism. Ind. Med. Surg., 28: 137-144.
BAETJER, A. M., & HORIGUCHI, S. (1963) Effects of environmental
temperature and dehydration on lead poisoning in laboratory
animals. Amsterdam, Excerpta Medica, pp. 795-797 (International
Congress Series, No. 62).
BAETJER, A.M., Joardar, S. N. D., & MCQUARY, W. A. (1960) Effect of
environmental temperature and humidity on lead poisoning in
animals. Arch. environ. Health, 1: 463-477.
BAGCHI, R. B., GANGULY, H. D., & SIRDAR, J. M. (1940) Lead in food.
Ind. J. med. Res., 28: 441-445.
BAKER, R. W. R. (1950) Polarographic determination of lead in urine.
Biochem. J., 46: 606-612
BALOH, R. W. (1974) Laboratory diagnosis of increased lead absorption.
Arch. environ. Health, 28: 198-208.
BARLTROP, D. (1969) Transfer of lead to the human foetus. In:
Barltrop, D. & Burland, W. L., ed., Mineral metabolism in
pediatrics. Philadelphia, Davis Co., pp. 135-151.
BARLTROP, D., & SMITH, A. (1971) Interaction of lead with
erythrocytes. Experientia (Basel) 27: 92-95.
BARLTROP, D., & SMITU, A. (1972) Lead binding to haemoglobin.
Experientia (Basel), 28: 76-77.
BARLTROP, D., BARRET, A. J., & DINGLE, J. T. (1971 ) Subcellular
distribution of lead in the rat. J. Lab. clin. Med.
77: 705-712.
BARLTROP, D., STREHLOW, C. D., THORNTON, J., & WEBB, J. S. (1974)
Significance of high soil lead concentrations for childhood lead
problem. Environ. Health Perspect., Exp. Issue No. 7, 75-83.
BARRY, P. S. I. (1975) A comparison of concentrations of lead in human
tissues. Brit. J. ind. Med. 32: 119-139.
BARRY, P.S., & MOSSMAN, D. B. (1970) Lead concentrations in human
tissues. Brit. J. ind. Med. 27: 339-351.
BAUCHINGER, M., SCHMID, E, EINBRODT, H. J., & DRESP, J. (1976)
Chromosome aberrations in lymphocytes after occupational exposure
to lead and cadmium. Mutat. Res., 40: 57-62.
BAUCHINGER, M., SCHMID, E., & SCHMIDT, D. (1972) Chromosomenanalyse
bei Verkehrspolizisten mit erhöhter Bleilast. Mutat. Res.,
16: 407-412.
BEATTIE, A.D., MOORE, M. R., DEVENAY, W. I., MILLER, A. R., &
GOLDBERG, A. (1972a) Environmental lead pollution in an urban
soft-water area. Brit. med. J., 2: 491-493.
BEATTIE, A.D., MOORE, M. R., & GOLDBERG, A. (1972b) Tetraethyllead
poisoning. Lancet, 2: 12-15.
BECK, E.G., MANOJLOVIC, N., & FISHER, A. B. (1973) Die Zytotoxizität
von Blei. In: Proceedings of the International Symposium;
Environmental Health Aspects of lead, Amsterdam, 2-6 October,
1972. Luxembourg, Commission of the European Communities,
pp. 451-461.
BECKMAN, K. (1925) Uber die Beziehungen zwischen Blutdruck,
Kapillardruck und Nierenveranderungen im Tiereexperiment.
Arch. klin. Med., 149, 177-188.
BENSON, F. (1972) Indoor-outdoor air pollution relationships. US EPA
Publ. No. AP-112.
BERG, B., & ZENZ, C. (1967) Environmental and clinical control of lead
exposure in a non-ferrous foundry. Am. ind. Hyg. Assoc. J.,
28: 175-178.
BERITIC, T. (1971) Lead concentration found in human blood in
association with lead colic. Arch. environ. Health,
23: 289-291.
BERITIC, T., & STAHULJAK, D. (1961) Lead poisoning from lead-glazed
pottery. Lancet, 1: 669.
BERK, P. D., TSCHUNDY, D. P., SHEPLEY, L. A., WAGGONER, J. G., &
BERLIN, N. I. (1970) Hematologic and biochemical studies in a
case of lead poisoning. Am. J. Med., 48: 137-144.
BERLIN, A., DEL CASTILHO, P., & SMEETS, J. (1973) European
intercomparison programmes. In: Proceedings of the
International Symposium; Environmental Health Aspects of Lead,
Amsterdam, 2-6 October 1972. Luxembourg, Commission of the
European Community, pp. 1033-1046.
BERLIN, A., SCHALLER, K. H., & SMEETS, J. (1975) Standardization of
ALAD activity determinations at the European level;
Intercalibration and applications. In: Proceedings of CEC-EPA-
WHO International Symposium; Recent Advances in the Assessment
of the Health Effects of Environmental Pollution, Paris, 24-28
June 1974. Luxembourg, Commission of the European Communities,
pp. 1087-1101.
BERTINUSON, J. R., & CLARK, C. S. (1973) The contribution to lead
content of soils from urban housing. Interface, 2: 6.
BESSIS, M. C., & JENSEN, W. N. (1965) Sideroblastic anaemia,
mitochondria and erythroblastic iron. Brit. J. Haematol.,
11: 49-51.
BETTS, P. R., ASTLEY, R., & RAINE, D. N. (1973) Lead intoxication in
children in Birmingham. Brit. reed. J., 1: 402-406.
BICKNELL, J., CLAYTON, B. E., & DELVES, H. T. (1968) Lead in mentally
retarded children. J. Mental Def. Res., 12: 282-293.
BINGHAM, E., PFITZER, E. A., BARKLEY, W., & RADFORD, E. P. (1968)
Alveolar macrophages: reduced number in rats after prolonged
inhalation of lead sesquioxide. Science, 162: 1297-1299.
BLACKMAN, S.S. (1937) The lesions of lead encephalitis in children.
Bull. Johns Hopkins Hosp., 61(1): 1-61.
BLANKSMA, L. A., SACHS, H. K., MURRAY, E. F., & O'CONNEL, M. J. (1969)
Incidence of high blood lead levels in Chicago children.
Pediatrics, 44: 661-665.
BLAXTER, K. L., & COWIC, A. T. (1946) Excretion of lead in the bile.
Nature (Lond.), 157: 588.
BOLANOWSKA, W. (1968) Distribution and excretion of triethyllead in
rats. Brit. J. ind. Med., 25: 203-208.
BOLANOWSKA, W., PIOTROWSKI, J., & CARCZYNSKI, H. (1967) Triethyllead
in the biological materia in cases of acute tetraethyllead
poisoning. Archiv J. Toxikol. 22: 278-282.
BOLANOWSKA, W., PIOTROWSKI, J., & TROJANOWSKA, B. (1964) The kinetics
of distribution and excretion of lead (PB210) in rats. In:
Proceedings of the 14th International Congress of Occupational
Health, Madrid, 16-21 Sept., pp. 420-422.
BOLTER, E., BUTZ, T., & ARSENEAU, J. F. (1975) Mobilization of heavy
metals by organic acids in the soils of a lead mining and
smelting district. In: Proceedings of the IXth Annual
Conference on Trace Substances in Environmental Health,
Columbia, MO, 10-12 June 1975, pp. 107-112.
BONSIGNORE, D., CALISANO, P., & CARTASEGNA, C. (1965) Un semplice
metodo per la determinazione della delta-amino-levulinico-
deidratasi nel sangue. Med. Lav., 56: 199-205.
BOUDENE, C., ARSAC, F., & MEININGER, J. (1975) Etude des taux de
plomb dans fair et dans la population en France. In:
International Symposium on Environmental Lead Research,
Dubrovnik, 14-15 May 1975. Arch. industr. Hyg. Toxicol.,
26: supplement, pp. 179-189.
BOYD, P. R., WALKER, G., & HENDERSON, J. N. (1957) The treatment of
tetraethyl lead poisoning. Lancet, 1: 181-185
BOYLAND, E., DUKES, C. E., GROVE, P. L., & MITCHLEY, B.C. V. (1962)
The induction of renal tumours by feeding lead acetate to rats.
Brit. J. Cancer, 16: 283-288.
BRANDT, A.D., & REICHENBACH, G. S. (1943) Lead exposures at the
government printing office. J. ind. Hyg. Toxicol., 25
(10): 445-450.
BREZINA, M., & ZUMAN, P. (1958) Polarography in medicine,
biochemistry and pharmacy. New York, Interscience Publishers
Inc.
BRITISH MINISTRY OF AGRICULTURE, FISHERIES AND FOOD (1972) Survey of
lead in food. Working Party on the Monitoring of Foodstuffs for
Heavy Metals: Second Report. London, Her Majesty's Stationery
Office, pp. 31.
BROWN S., DRAGAN, N., & VOGEL, W. (1971) Effects of lead acetate on
learning and memory in rats. Arch. environ. Health,
22: 370-372.
BRUCH, J., BROCKHAUS, A., & DEHNEN, W. (1973) Elektronmikroskopische
Beobachtungen an Rattenlungen nach Exposition reit
partikelförmigem Blei. In: Proceedings of the International
Symposium; Environmental Health Aspects of Lead, Amsterdam, 2-6
October 1972. Luxembourg, Commission of the European
Communities, pp. 221-229.
BRUCH, J., BROCKHAUS, A., & DEHNEN, W. (1975). Local effects of
inhaled lead compounds on the lung. In: Proceedings of CEC-
EPA-WHO International Symposium; Recent Advances in the
Assessment of the Health Effects of Environmental Pollution,
Paris, 24-28 June 1974. Luxembourg, Commission of the European
Communities, pp. 781-793.
BULLOCK, J. D., WEY, R. J., ZAIA, J. A., ZAREMBOK, I., & SCHROEDER, H.
A. (1966) Effect of tetraethyllead on learning and memory in
rats. Arch. environ. Health, 13: 21-22.
BULPITT, G. J. (1972) Lead and hyperactivity. Lancet, II: 1144.
BURDÉ, B. DE LA, & CHOATE, M. S.(1972) Does asymptomatic lead exposure
in children have latent sequelae? J. Pediar., 81(6): 1088-1091.
BUTT, E. U., NUSBAUM, R. E., GILMOUR, T. C., DIDIO, S. L., & Sister
MARIANO (1964) Trace metal levels in human serum and blood.
Arch. environ. Health, 8: 52-57.
BYERS, R. K. (1959) Lead poisoning. Pediatrics, 23: 585-603.
BYERS, R. K., & LORD, E. E. (1943) Late effects of lead poisoning on
mental development. Am. J. Dis. Child., 66: 471-494.
CANTAROW, A., & TRUMPER, M. (1944) Lead poisoning. Baltimore,
Williams and Wilkins Co., p. 8.
CANDANI, A., & FARINA, G. (1972) Influence of alcoholic beverages
consumption on lead-induced changes of haemebiosynthesis.
Med. Lay., 63, 22-28.
CARPENTER, S. (1974) Placental permeability of lead. Environ. Health,
Perspect. Exp. Issue, No. 7, 129-133.
CARSON, T. L., VAN GELDER, G. A., KARAS, G. G., & BUCK, W. B. (1974)
Development of behavioural tests for the assessment of neurologic
effects of lead in sheep. Environ. Health Perspect., Exp. Issue
No. 7, 233-239.
CASTELLINO, N., & ALOJ, S. (1964) Kinetics of the distribution and
excretion of lead in the rat. Brit. J. ind. Med., 21: 308-314.
CASTELLINO, N., & ALOJ, S. (1969) Intracellular distribution of lead
in the liver and kidney of the rat. Brit. J. ind. Med.,
26: 139-143.
CASTELLINO, N., LIAMANNA, P., & CRIECO, B. (1966) Biliary excretion of
lead in the rat. Brit. J. ind. Med., 23: 237-239.
CATTON, M. J., HARRISON, M. J. G., FULLERTON, P.M., & KAZANTZIS, O.
(1970) Subclinical neuropathy in lead workers. Brit. med. J.,
2: 80-82.
CERNIK, A. A. (1974) Determination of blood lead using a 4.0 mm paper
punched disc carbon sampling cup technique. Brit. J. ind. Med.,
31: 239-245.
CERNIK, A. A., & SAYERS, M. H. P. (1971) Determination of lead in
capillary blood using a paper punched disc atomic absorption
technique application to the supervision of lead workers.
Brit. J. ind. Med., 28(4): 392-398.
CHANEY, R. L. (1973) Crop and food chain effects of toxic elements in
sludges and effluents. Recycling municipal sludges and effluents
on land July 9-13, 1973 Illinois, pp. 129-141.
CHEH, A., & NEILANDS, J. B. (1973) Zinc, an essential metal for beef
liver 6-aminolevulinate dehydratase. Biochem. biophys. Res.
Comm., 55(4): 1060-1063.
CHISOLM, J. J. (1962) Aminoaciduria as a manifestation of renal
tubular injury in lead intoxication and a comparison with
patterns of aminoaciduria seen in other diseases. J. Pediatr.,
60: 1-17.
CHISOLM, J. J. (1968a) Lead poisoning (plumbism). In: H. L. Barnett,
ed., Pediatrics (14th Ed.) New York, Appelton-Century-Crofts,
pp. 313-319.
CHISOLM, J. J. (1968b) The use of chelating agents in the treatment of
acute and chronic lead intoxication in childhood. J. Pediatr.,
73: 1-38.
CHISOLM, J. J. (1973) Management of increased lead absorption and lead
poisoning in children. New Engl. J. Med., 289: 1016-1018.
CHISOLM, J. J. (1974) Chelation therapy in children with subclinical
plumbism. Pediatrics, 53: 441-443.
CHISOLM, J. J., & HARRISON, H. E. (1956) The exposure of children to
lead. Pediatrics, 18: 943-958.
CHOVIN, P., DUFFAUD, J., MERKEZ, F., FAYART, M., & TRUFFERT, L. (1973)
Resultats d'une annee d'étude de la teneur en plomb particulaire
et l'atmosphère Symposium: Environmental Health Aspects of Lead,
Amsterdam 2-6 October 1972. Luxembourg, Commission of the
European Communities, pp. 1003-1015.
CHOW, T. J. (1968) Isotope analysis of seawater by mass spectrometry.
J. Water Pollut. Control Fed., 40: 399-411.
CHOW, T. J., & EARL, J. L. (1970) Lead aerosols in the atmosphere:
Increasing concentrations. Science, 169: 577-580.
CHOW, T. J., & BENNET, C. F. (1969) Lead aerosols in marine
atmosphere. Environ. Sci. Technol., 3: 737-740.
CHOW, T. J., EARL, J. L., & SNYDER, C. B. (1972) Lead aerosol
baseline: Concentration at White Mountain and Liaguna Mountain.
Science, 178: 401-402.
CLARKSON, T. W., & KENCH, J. E. (1956) Urinary excretion of amino
acids by men absorbing heavy metals. Biochem. J., 62: 361-372.
CLARKSON, T. W., & KENCH, J. E. (1958) Uptake of lead by human
erythrocytes in vitro. Biochem. J., 69: 432-439.
CLASEN, R. A., HARTMAN, J. F., STARR, A. J., COOGAN, PH. S., PANDOLFI,
S., LAING, J., BECER, R., & HASS, G. U. (1974) Electron
microscopic and chemical studies of the vascular changes and
edema of lead encephalopathy. Am. J. Pathol., 74(2): 215-233.
CLERCQ, E. DE, & MERIGAN, T. C. (1969) An active interferon inducer
obtained from Haemophilus influenzae type B. J. Immun.,
103: 899-906.
COGBILL, E. C., & HOBBS, M. E. (1957) Transfer of metallic
constituents of cigarettes to the main-stream smoke. Tob. Sci.,
1: 68-73.
COHEN, G. J., & AHRENS, W. E. (1959) Chronic lead poisoning.
J. Pediatr., 54(2): 271-284.
COHEN, G. J., BOWERS, G. N., & LEPOW, M. (1973) Epidemiology of lead
poisoning. J. Am. Med. Ass., 266: 1430-1433.
COHEN, N., KNEIP, T. J., GOLDSTEIN, D. H., & MUCHMORE, E. A. S. (1972)
The juvenile baboon as a model for studies of lead poisoning in
children. J. Med. Primatol., 1: 142-155.
COLE, L. J., & BACHHUBER, L. J. (1914) The effect of lead on the germ
cells of the male rabbit and fowl as indicated by their progeny.
Proc. Soc. Exp. Biol. Med., 12: 24-29.
COLLIER, H. B. (1971) A study of the determination of delta-
aminolevulinate hydrolyase (delta-aminolevulinate dehydratase)
activity in hemolysates of human erythrocytes. Clin. Biochem.,
4: 222-232.
COLWILL, D. M., & HICKMAN, A. J. (1973) The concentration of volatile
and particulate lead compounds in the atmosphere: measurements
at four road sites. Transport and Road Research Laboratory
Report LR 545 Crowthorne, Berkshire, pp. 5.
COMMISSION OF THE EUROPEAN COMMUNITIES (1973) Air lead concentrations
in the European Community. Yearly Report: April 1971-March 1972.
Luxembourg.
COOPER, W. C., & GAFFEY, W. R. (1975) Mortality of lead workers.
J. occup. Med., 17: 100-107.
COOPER, W. C., TABERSHAW, I. R., & NELSON, K. W. (1973) Laboratory
studies of workers in lead smelting and refining. In:
Proceedings of the International symposium: Environmental Health
Aspects of lead, Amsterdam, 2-6 October 1972, Luxembourg,
Commission of the European Communities, pp. 517-529.
COULSTON, F., GOLDBERG, L., GRIFFIN, T. B., & RUSSELL, J. C. (1972a)
The effects of continuous exposure to airborne lead. 1. Exposure
of rats and monkeys to particulate lead at a level of
21.5 µg/m3. Final Report to the US Environmental Protection
Agency.
COULSTON, F., GOLDBERG, L., GRIFFIN, T. B., & RUSSELL, J. C. (1972b)
The effects of continuous exposure to airborne lead. 2. Exposure
of man to particulate lead at a level of 10.9 µg/m3. Final
Report to the US Environmental Protection Agency.
COULSTON, F., GOLDBERG, L., GRIFFIN, T. B., & RUSSELL, J. C. (1972c)
The effects of continuous exposure to airborne lead. 4. Exposure
of man to particulate lead at a level of 3.2 µg/m3. Final
Report to the US Environmental Protection Agency.
COULTHARD, A. J. (1958) Animal cycle of blood haemoglobin levels.
Clin. Chim. Acta, 2: 226-233.
CRAMER, K. (1966) Predisposing factors for lead poisoning. Acta. Med.
scand., (Suppl. 445): 179: 56-59.
CRAMER, K., & DAHLBERG, L. (1966) Incidence of hypertension among lead
workers. Brit. J. ind. Med., 23: 101-104.
CRAMER, K., GOYER, R. A., JAGENBURG, R., & WILSON, M. H. (1974) Renal
ultrastructure renal function, and parameters of lead toxicity in
workers with different period of lead exposure. Brit. J. ind.
Med., 31: 113-127.
CREMER, J. E. (1959) Biochemical studies on the toxicity of tetraethyl
lead and other organo-lead compounds. Brit. J. ind. Med.,
16: 191-199.
CREMER, J. E. (1965) Toxicology and biochemistry of alkyl lead
compounds. Occup. Health Rev., 17: 14-19.
CREMER, J. E, & CALLAWAY, S. (1961) Further studies on the toxicity of
some tetra- and trialkyl lead compounds. Brit. J. ind. Med.,
18: 277-282.
CRUTCHER, J. C. (1963) Clinical manifestation and therapy of acute
lead intoxication due to the ingestion of illicitly distilled
alcohol. Ann. intern. Med., 59: 707-715.
DAINES, R. H., SMITH, D. W., FELICIANO, A., & TROUT, J. R. (1972) Air
levels of lead inside and outside of homes. Ind. Med. Surg.,
41: 26-28.
DALLDORF, G., & WILLIAMS, R. R. (1945) Impairment of reproduction in
rats by ingestion of lead. Science, 102: 668.
DANILOVIC, V. (1958) Chronic nephritis due to ingestion of lead-
contaminated flour. Brit. Ted. J., 1: 27-28.
DAVID, O. J., CLARK, J., & VOELLER, K. (1972) Lead and hyperactivity.
Lancet, 2: 900-903.
DAVIDSON, D. F., & LAKIN, H. W. (1962) Metal content of some black
shales of the western United States. U.S. Geological Survey
Professional Paper 450C, p. C74.
DAVIS, J. R., & ANDELMAN, S. L. (1967) Urinary delta aminolevulinic
acid levels in lead poisoning. A modified method for the rapid
determination of urinary delta-aminolevulinic acid using
disposable ion-exchange chromatography columns. Arch. environ.
Health, 15: 53-59.
DAVIS, R. K., HORTON, A. W., LARSON, E. E., & STEMMER, K. L. (1963)
Inhalation of tetramethyllead and tetraethyllead. Arch. environ.
Health, 6: 473-479.
DAVIS, W. E. (1973) Emission study of industrial sources of lead air
pollutants 1970. US EPA, Document APTD-1543, pp. 1-123.
DEDOLPH, R. G., TER HAAER, R., HOLTZMAN, R., & LUCAS, H. J. (1970)
Sources of lead in perennial ryegrass and radishes. Environ.
Sci. Technol, 4: 217-223.
DEKNUDT, O., LEONARD, A., & IVANOV, B. (1973) Chromosome aberrations
observed in male workers occupationally exposed to lead.
Environ. Physiol. Biochem, 3: 132-138.
DELVES, H. T. (1970) A micro sampling method for the rapid
determination of lead in blood by atomic absorption spectrometry.
Analyst, 95: 431.
DIMITROVA, M. (1972) Modifications of the contractile function of the
myocard in chronic lead poisoning. Arch. Mal. prof. Med. Trav.,
33: 383-387.
DINGWALL-FORDYCE, J., & LANE, R. E. (1963) A follow-up study of lead
workers. Brit. J. ind. Med., 20: 313-315.
DINISCHIOUTU, O. T., NESTORESCU, B., RADIELESCU, J. C., JONESCU, C.,
PREDA, N., & HUTZA, G. (1960) Studies on the chemical forms of
urinary lead. Brit. J. ind. Med, 17: 141-145.
DJURIC, D. (1964) Fluorimetric determination of porphyrius. Arch.
environ. Health, 9: 742-744.
DJURIC, D., KERIN, Z., GRAOVAC-LEPOSAVIC, L., NOVAK, L., & KOP, M.
(1971) Environmental contamination by lead from a mine and
smelter; A preliminary Report. Arch. environ. Health,
23: 275-279.
DJURIC, D., NOVAK, L., MILIC, S., & KALIC-FILIPOVlC, D. (1966) Delta-
ALA as an early sign of lead exposure. Med. Lav., 57: 161-166.
DODIC, S., VIDAKOVIC, A., PERISIC, V., & STEFANOVIC, S. (1971) Stanje
jetre u pojedinih profesionalnih intoksikacija. In: III
Jugoslovanski Kongres Medicine Dela, Ljubljana, 1971.
Ljubljana, 20-24 September, 1971, Jzdange "Lek", pp. 285-293.
DONOVAN, D. T., VOUGHT, V. M., & RAKOW, A. B. (1971) Laboratories
which conduct lead analysis on biologic specimens. Arch.
environ. Health, 23: 111-113.
DRESEL, E. J. B., & FALK, J. E. (1954) Studies on the biosynthesis of
blood pigments. Haem synthesis in hemolysed erythrocytes of
chicken blood. Biochem. J., 56: 156-163.
DRESSEN, W. C., EDWARDS, T. J., REINHART, W. H., PAGE, R. T., WEBSTER,
S. H., ARMSTRONG, D. W., & SAYERS, R. R. (1941) The control of
the lead hazard in the storage battery industry. Publ. Health
Bull. (Wash.), No. 262.
DUGANDZIC, M., STANKOVIC, B., MILOVANOVIC, LY., & KORICANAC, Z. (1973)
Urinary excretion of 5-hydroxindolacetic acid in lead exposed
persons. Archiv. Hig. Rada., 24: 37-43.
DURFOR, C., & BECKER, E. (1964) Selected data on public supplies of
the 100 largest cities in the United States, 1962. J. Am. Water
Works Assoc., 56: 237-246.
EDIGER, R. D., & COLEMAN, R. L. (1973) An evaluation of anodic
stripping voltamentry and non-flame atomic absorption as routine
analytical tools. In: Proceedings of the 6th Annual Conference
on Trace Substances in the Environment, Columbia, MO., 13-15
June 1972, pp. 279-283.
EGOROV, V. V., ZHIGALOVSKAJA, T. N., & MALAKHOV, S. G. (1970)
Microelement content of surface air above the continent and the
ocean. J. geophys. Res., 75: 18, 3650-3656.
EGOROVA, G. M., IVANOV, N. G., & SANOCSKIJ, J. V. (1966) Specificity
of the effect of lead on spermatogenesis. Toksikol., nov. prom.
him., vescestv., 8: 33-41.
EINBRODT, H. J., ROSMANITH, J., & SCHROEDER, A. (1974) Beim Spielen,
Schulkind Vergiftet, Umwelt, 3: 726.
EISENBUD, M., & WRENN, M. E. (1970) Radioactivity studies annual
report NYO-3086-10 VI., 1970. Springfield, VA, National
Technical Information Service.
ELKINS, H. B. (1959) The chemistry of industrial toxicology. New
York, J. Wiley & Sons (2nd edition), pp. 51-52.
EMMERSON, B. T. (1963) Chronic lead nephropathy: The diagnostic use of
calcium EDTA and the association with gout. Austr. Ann. Med.,
12: 310-324.
EMMERSON, B. T., MIROSH, W., & DOUGLAS, J. B. (1971) The relative
contributions of tubular reabsorption and secretion to urate
excretion in lead nephropathy. Aust. N.Z.J. Med., 1: 353-362.
ENGELS, L. H. VON, & KÜHNEN, G. (1973) Staubschutz in der
Akkumulatoren-Industrie Bonn, Staubforschungsinstitut des
Hauptverbandes der gewarblichen Berufsgenossenschaften, STF-
Report No. 2, pp. 27.
ENGEL, R. E., HAMMER, D. J., HORTON, R. J. M., LANE, N. M., & PLUMLEE,
L. A. (1971) Environmental lead and public health. Research
Triangle, Park, NC Environmental Protection Agency. Air Pollution
Control Office Publication No. AP 90, pp. 1-34.
ENVIRONMENT AGENCY, JAPAN (1975) Results of air pollution survey.
Tokyo, pp. 148-153.
EPSTEIN, S.S., & MANTEL, N. (1968) Carcinogenicity of tetraethyllead.
Experientia Basel, 24: 580-581.
ERVE, P. R., & SCHUMER, W. (1972) Endotoxin sensitivity of
adrenalectomized rats treated with lead acetate. Res. J.
Reticuloend. Soc., 11(38): 427.
FAIREY, F. S., & GREY, J. W. (1970 Soil lead and pediatric lead
poisoning in Charleston, S.C. J. S. Carolina med. Assoc.,
66: 79-82.
FAIRHALL, L. T., & MILLER, J. W. (1941) A study of the relative
toxicity of the molecular components of lead arsenate. Public
Health Rep. (Wash.), 56: 1610-1625.
FEDERAL INSTITUTE FOR MINERALS RESEARCH AND GERMAN INSTITUTE FOR
ECONOMIC RESEARCH (1972). Supply and demand for lead. (Translated
into English by Lead Development Association, 34 Berkeley Square,
London W1X 6AJ, April, 1972), pp. 1-47.
FERM, V. H. (1969) The synteratogenic effect of lead and cadmium.
Experientia Basel 25: 56-57.
FILKINS, J.P., & BUCHANEN, B. J. (1973) Effects of lead acetate on
sensitivity to shock, intravascular carbon and endotoxin
clearances, and hepatic endotoxin detoxification. Proc. Soc.
Exp. Biol. Med., 142: 471-475.
FINE, P. R., THOMAS, C. W., SUHS, R. H., COHNBERG, R. E., & FLASHNER,
B. A. (1972) Pediatric blood lead levels: A study in 14 Illinois
cities of intermediate population. J. Am. Med. Assoc., 221
(13): 1475-1479.
FORBES, G. B., & REINA, J. C. (1972) Effect of age on gastrointestinal
absorption (Fe, Sr, Pb) in the rat. J. Nutr, 102: 647-652).
FORNI, A., & SECCHI, C. C. (1973) Chromosome changes in preclinical
and clinical lead poisoning and correlation with biochemical
findings. In: Proceedings of the International Symposium,
Environmental Aspects of lead, Amsterdam, 2-6 October 1972.
Luxembourg, Commission of the European Communities, pp. 473-483.
FOUTS, P. J., & PAGE, J. H. (1942) The effect of chronic lead
poisoning on arterial blood pressure in dogs. Am. Heart J.,
24: 329-331.
FREEMAN, R. (1965) Reversible myocarditis due to chronic lead
poisoning in childhood. Arch. Dis. Child., 40: 389-393.
FRIBERG, L., PISCATOR, M., NORDBERG, G., & KJELLSTROM, T. (1974)
Cadmium in the environment. 2nd ed. Ohio, Chemical Rubber
Company.
FRIED, J. F., ROSENTHAL, N. W., & SCHUBERT, J. (1956) Induced
accumulation of titrate in therapy of experimental lead
poisoning. Proc. Soc. Exp. Biol. Med., 92: 331-333.
FUGAS, M., WILDER, B., PAUKOVIC, R., HRSAK, J., & STEINER-SKREB, D.
(1973) Concentration levels and particle size distribution of
lead in the air of an urban and an industrial area as a basis
for the calculation of population exposure. In: Proceedings of
the International Symposium; Environmental Health Aspects of
Lead, Amsterdam, 2-6 October 1972. Luxembourg, Commission of
the European Communities, pp. 961-968.
FULLERTON, P.M. (1966) Chronic peripheral neuropathy produced by lead
poisoning in guinea pigs. J. Neuropathol. exp. Neurol.,
25: 214-236.
GAGE, J. C., & LITCHFIELD, M. H. (1968) The migration of lead from
polymers in the rat gastro-intestinal tract. Food Cosmet.
Toxic., 6: 329-338.
GAGE, J. C., & LITCHFIELD, M. H. (1969) The migration of lead from
paint films in the rat gastro-intestinal tract. J. Oil Col.
Chem. Assoc., 52: 236-243.
GAJDOS, A., & GAJDOS-TÖRÖK, M. (1969 L'activite de l'acide delta-
aminolevulinique synthetase hépatique et médullaire au cours du
saturnisme experimental du lapin. C.R. Soc. Biol., 163: 60-63.
GAJDOS, A., & GAJDOS-TÖRÖK, M. (1973) Erreur du diagnostique
differentiel entre l'intoxication par le plomb et la porphyrie
intermittente aiguë. In: Proceedings of the International
Symposium; Environmental Health Aspects of Lead, Amsterdam, 2-6
October, 1972. Luxembourg, Commission of the European
Communities, pp. 501-505.
GALLE, P., & MOREL-MAROGEN, L. (1965) Les lesions renales du
saturnisme humain et experimental. Nephron, 2: 273-286.
GALZIGNA, L., BRUGNONE, F., & CORSI, G. C. (1964) Excretion of
5-hydroxyindole acetic acid in experimental tetraethyllead
poisoning. Med. Lavoro., 55: 102-106.
GARBER, B. T., & WEI, E. (1974) Influence of dietary factors on the
gastrointestinal absorption of lead. Toxic. appl. Pharm.,
27: 685-691.
GEORGII, H. W., & JOST, D. (1971) On the lead-concentration in an
urban aerosol. Atmosph. Environ., 5: 725-727.
GHELBERG, N. W., GORGAN, J., & CHECIN, J. (1966) 5-hydroxyindoleacetic
acid excretion in a population chronically exposed to low lead
concentration in the atmosphere. Igiena (Buc) 15: 87-92.
GIBSON, K. G., NEUBERGER, A., & SCOTT, J. J. (1955) The purification
and properties of delta-aminolaevulic acid dehydrase. Biochem.
J., 61: 618-629.
GIBSON, S. L. M., LAM, C. N., MCCRAE, W. M., & GOLDBERG, A. (1967)
Blood lead levels in normal and mentally retarded children.
Arch. Dis. Child., 42: 573-578.
GILLET, J. A. (1955) An outbreak of lead poisoning in the Canklow
district of Rotterdam. Lancet, 1: 1118-1121.
GOLDBERG, A. (1972) Lead poisoning and haem biosynthesis. Brit. J.
Haematol., 23: 521-523.
GOLDBERG, A. (1974) Drinking water as a source of lead pollution.
Environ. Health Perspect., Exp. Issue No. 7, pp. 103-107.
GOLDBERG, A., ASHENBRUCKER, H., CARTWRIGHT, G. E., & WINTROBE, M. M.
(1956) Studies on the biosynthesis of heme in vitro by arian
erythrocytes. Blood, 11: 821-833.
GOLDBERG, E. D. (1976) The health of the oceans. Chapter 5. Heavy
metals: lead. Paris, The Unesco Press, pp. 109-111.
GOLDSMITH, J. R., & HEXTER, A. C. (1967) Respiratory exposure to lead
epidemiological experimental dose-response relationships.
Science, 158: 132-134.
GOLUBOVIC, E. Y., & GEVKOVSKAJA, T. V. (1967) Effects of ethyleneimine
and lead on several sides of nucleic acids changes in the
spermary of rats. Toksikol. nov. prom. him. vescestv, 9: 86-91.
GOLUBOVIC, E. Y., AVHIMENKO, M. M., & CIRKOVA, E. M. (1968)
Biochemical and morphological changes in testicles of rats under
the effect of low doses of lead. Toksikol. nov. prom. him.
vescestv, 10: 64-72 (in Russian).
GOYER, R. A., & KRALL, K. (1969) Ultrastructural transformation in
mitochondria isolated from kidneys of normal and lead-intoxicated
rats. J. cell. Biol., 41: 393-400.
GOYER, R. A., & MAHAFFEY, K. R. (1972) Susceptibility to lead
toxicity. Environ. Health Perspect., No. 2: 73-80.
GOYER, R. A., & RHYNE, B.C. (1973) Pathological effects of lead.
Intern. Rev. exp. Pathol., 12: 1-77.
GOYER, R. A., LEONARD, D. L., MORRE, J. F., RHYNE, B., & KRIGMAN, M.
R. (1970a) Lead dosage and the role of the intranuclear inclusion
body. Arch. environ. Health, 20: 705-711.
GOYER, R. A., MAY, P., CATES, M. M., & KRIGMAN, M. R. (1970b) Lead and
protein content of isolated inclusion bodies from kidneys of lead
poisoned rats. Lab. Invest., 22: 245-251.
GOYER, R. A., TSUCHIYA, K., LEONARD, D. L., & KAHYO, H. (1972)
Aminoaciduria in Japanese workers in the lead and cadmium
industries. Am. J. clin. Pathol., 57: 635-642.
GRABECKI, J., HADUCH, T., & URBANOWICZ, H. (1967) Die einfachen
Bestimmungsmethoden der delta-Amino-lävulinsäure in Harn. Int.
Arch. Gewerbpath. Gewerbhyg., 23: 226-240.
GRANICK, J. L., SASSA, S., GRANICK, S., LEVERE, R. D., & KAPPAS, A.
(1973) Studies in lead poisoning II Correlation between the ratio
of activated to inactivated aminolevulinic acid dehydratase of
whole blood and the blood lead level. Biochem. Med.,
8: 149-159.
GRANICK, S., SASSA, S., GRANICK, J. L., LEVERE, R. D., & KAPPAS, A.
(1972) Assays for porphyrins. Proc. Nat. Acad. Sci. USA,
69: 2382-2385.
GRAOVAC-LEPOSAVIC, L., DJURIC, D., VALJAREVIC, V., SENICAR, L., MILIC,
S., & DELIC, V. (1973) Environmental lead contamination of Meza
Valley-Study on lead exposure of populations. In: Proceedings
of the International Symposium: Environmental Health Aspects of
Lead, Amsterdam, 2-6 October 1972. Luxembourg, Commission of
the European Communities, pp. 685-703.
GREENFIELD, S. M., BRIDBORD, K., BARTH, D., & ENGEL, R. (1973) The
changing perspectives regarding lead as an environmental
pollutant. In: Proceedings of the International Symposium:
Environmental Health Aspects of lead, Amsterdam, 2-6 October
1972. Luxembourg, Commission of the European Communities,
pp. 19-27.
GRIFFITH, J. Q., & LANDAUER, M. A. (1944) The effect of chronic lead
poisoning on arterial blood pressure in rats. Am. Heart J.,
28: 295-297.
GRIGGS, R. C., SUNSHINE, J., NEWILL, V. NEWTON, B. W., BUCHANAN, S., &
RASCH, C. A. (1964) Environmental factors in childhood lead
poisoning. J. Am. Med. Assoc., 187: 703-707.
GRIMES, H., SAYERS, M. H. P., CERNIK, A. A., BERLIN, a., RECHT, P., &
SMEETS, J. (1975) Note on the lead exposure of children:
determinations carried out on behalf of the Commission in
Western Ireland. Luxembourg, Commission of the European
Communities (Report V/F/1491/75), pp. 7.
GUINEE, V. F. (1973) Epidemiologic studies of lead exposure in New
York city. In: Proceedings of the International Symposium;
Environmental Health Aspects of Lead, Amsterdam, 2-6 October
1972. Luxembourg, Commission of the European Communities,
pp. 763-770.
GUTNIAK, O., KOZIOLOWA, H., & KOWALSKI, E. (1964) Free protoporphyrin
content of erythrocytes in chronic tetraethyl lead poisoning.
Lancet, 1: 1137-1138.
HAAS, T., MACHE, K., SCHALLER, K.-H., MACHE, W., & VALENTIN, H.
(1972a) Investigations into ecological lead levels in
children.(Untersuchungen über die ökologische Bleibelastung im
Kindesalter). Zbl. Bakt. Hyg. I. Abt. Orig., B 156: 353-360.
HAAS, T., WIEK, A. O., SCHALLER, K. H., MACHE, K., & VALENTIN, R.
(1972b) Die usuelle Bleibelastung bei Neugeborenen und ihren
Muttern. Zbl. Bakt. Hyg., I. Abt. Orig., B 155, 341-349.
HACIROV, D. G. (1972) Data on electronmicroscopic investigations of
erythrocytes and thrombocytes under saturnism. Naucn. Tr. Sev.-
Oset. Med. Inst., 36(4): 59-63.
HAEGER-ARONSEN, B. (1960) Studies on urinary excretion of delta-
aminolaevulic acid and other haem precursors in lead workers and
lead-intoxicated rabbits. Scand. J. clin. Lab. Invest., 12
(Suppl. 47) 1.
HAEGER-ARONSEN, B. (1971) An assessment of the laboratory tests used
to monitor the exposure of lead workers. Brit. J. ind. Med.,
28: 52-58.
HAEGER-ARONSEN, B., ABDULLA, M., & FRISTEDT, B. (1971) Effect of lead
on delta-aminolevulinic acid dehydrase activity in red blood
cells. Arch. environ. Health, 23: 440-445.
HAMMOND, P. B. (1971) The effects of chelating agents on the tissue
distribution and excretion of lead. Toxicol. Pharmacol.,
18: 296-310.
HAMMOND, P. B., & ARONSON, A. L. (1964) Lead poisoning in cattle and
horses in the vicinity of a smelter. Ann. N.Y. Acad. Sci.,
III: 595-611.
HAMMOND, P. B., WRIGHT, H. N., & ROEPKE, M. H. (1956) A method for the
detection of lead in bovine blood and liver. University of
Minnesota. Agric. Exp. Stat. Techn. Bull., 221: 3-14.
HANKIN, L., HEICHEL, G. H., & BOTSFORD, R. A. (1973) Lead poisoning
from coloured printing inks. Clin. Pediatr., 12: 654-655.
HAPKE, H. J. (1974) Effects and damage by lead, cadmium and zinc on
useful animals. Staub Reinhaltung der Luft, 34(1): 8-11.
HAPKE, H. J., & PRIGGE, E. (1973) Interactions of lead and glutathione
with delta-aminolevulinic acid dehydratase. Arch. Toxicol.,
31: 153-161.
HARRIS, R. W., & ELSEA, W. R. (1967) Ceramic glaze as a source of lead
poisoning. J. Am. Med. Assoc., 202: 544-549.
HARRISON, R. M., PERRY, R., & SLATER, D. H. (1974) An adsorption
technique for the determination of organic lead in street air.
Atmos. Environ., 8: 1187-1194.
HASAN, J., & HERNBERG, S. (1966) Interactions of inorganic lead with
human blood cells. Work environ. Health, 2: 26-44.
HELLER, A., & KETTNER, H. (1969) Probenahme und Bestimmune kleinster
Bleimengen in der Luft. Wasser-Boden- und Lufthygiene, No. 29,
3-50.
HEMPHILL, F. E., KAEBERLE, M. L., & BUCK, W. B. (1971) Lead
suppression of mouse resistance to salmonella typhimurium.
Science, 172: 1031-1032.
HENDERSON, D. A. (1958) The etiology of chronic nephritis in
Queensland. Med. d. Austr, 1: 377-386.
HENDERSON, D. A., & INGLIS, J. A. (1957) The lead content of bone in
chronic Bright's disease. Austr. Ann. Med, 6: 145-154.
HERNBERG, S. (1967) Lifespan, potassium fluxes and membrane ATPases of
erythrocytes from subjects exposed to inorganic lead. Work
environ. Health, 3, suppl. 1, pp. 1-74.
HERNBERG, S. (1973) Prevention of occupational poisoning from
inorganic lead. Work environ. Health, 10: 53-61.
HERNBERG, S. (1975) Lead In: Zenz, C. (ed.) Occupational Medicine
Yearbook Pt 4: The Chemical Occupational Environment, Chicago,
pp. 715-769.
HERNBERG, S., LILIUS, H., MELLIN, G., & NIKKANEN, J. (1969) Lead
exposure of workers in printing shops. Work environ. Health,
6(2): 5-8.
HERNBERG, S., NIKKANEN, J., MELLIN, G., & LILIUS, H. (1970) delta-
aminolevulinic acid dehydrase as a measure of lead exposure.
Arch. environ. Health, 21: 140-145.
HEUSGEM, C., & DEGRAEVE, J. (1973) Importance de l'apport alimentaire
en plomb dans l'est de la Belgique. In: Proceedings of the
International Symposium; Environmental Health Aspects of Lead,
Amsterdam, 2-6 October 1972. Luxembourg, Commission of the
European Communities, pp. 85-91.
HICKS, J. M., GUTIERREZ, A. N., & WORTHY, B. E. (1973) Evaluation of
the Delves micro-system for blood lead analysis. Clin. Chem.,
19: 322-325.
HILL, C. R. (1960) Lead-210 and polonium-210 in grass. Nature
(Lond.), 187: 211-212.
H M CHIEF INSPECTOR OF FACTORIES (1972) H.M. Great Britain Department
of Employment. Annual Report. London, Her Majesty's Stationery
Office.
HOFFMAN, E. O., DI LUZIO, N.R., HOLPER, K;, BRETTSCHNEIDER, L., &
COOVER, J. (1974). Ultrastructural changes in the liver of
baboons following lead and endotoxin administration. Lab.
Invest., 30: 311-319.
HOFREUTER, D. H., CATCOTT, E. J., KEENAN, R. G., & XINTARAS, A. B.
(1961) The public health significance of atmospheric lead. Arch.
environ. Health, 3: 568-574.
HOLMQVIST, J. (1966) Normal lead values in blood. 15th International
Congress of Occupational Health. Vienna 19-24 Sept., A-III-1,
pp. 179-183.
HOMMA, K. (1966) Experimental study of preparing metal fumes. Ind.
Health, 4: 129-137.
HOPKINS, A. P., & DAYAN, A. D. (1974) The pathology of experimental
lead encephalopathy in the baboon (Papio amibis). Brit. J. ind.
Med., 31: 128-133.
HORIGUCHI, S., & UTSUNOMIYA, T. (1973) An estimate of the body burden
of lead in the healthy Japanese population. An attempt to assume
absorption and excretion of lead in the healthy Japanese
population, Part 2. Osaka City, med. J., 19: 1-5.
HORIUCHI, K., (1970) Lead in the environment and its effect on man in
Japan. Osaka City, med. J., 16: 1.
HORIUCHI, K., & TAKADA, J. (1954) Studies on the industrial lead
poisoning. I. Absorption, transportation, deposition and
excretion of lead, 1. Normal limits of lead in the blood, urine
and feces among healthy Japanese urban inhabitants. Osaka City,
reed. J., 1: 117-125.
HORIUCHI, K., HORIGUCHI, T., KASAHARA, A., MORIOKA, T., UTSUNOMYA, T.,
& SHINAGAWA, K. (1964) Influences of temperature on manifestation
of symptoms in industrial poisoning. 1. Influences of high
temperature in lead poisoning. Jpn. J. ind. Health, 6: 170-171.
HORIUCHI, K., HORIGUCHI, S., SHINAGAWA, K., TAKADA, F., & TERAMOTO, K.
(1968) A polarographic method for the determination of a small
amount of lead in biological materials. Osaka City, med. J,
14: 113-118.
HORIUCHI, K., HORIGUCHI, S., & SUEKANE, M. (1959) Studies on the
industrial lead poisoning. I. Absorption, transportation,
deposition and excretion of lead. 6. The lead contents in organ
tissues of the normal Japanese. Osaka City, reed. J., 5: 41-70.
HORIUCHI, K., YAMAMOTO, T., & TAMORI, E. (1956) Studies on the
industrial lead poisoning. I. Absorption, transportation,
deposition and excretion of lead. 2. A study on the lead content
in daily food in Japan. Osaka City, med. J., 3: 84-113.
HOWER, J., PRINZ, B., GONO, E., & REUSMANN, G. (1975) Untersuchungen
zum Zusammenhangzwischen dem Blutbleispiegel bei Neugeboren en
und der Bleiimmissionsbelastung der Mütter am Wohnort. In:
Proceedings of CEC-EPA-WHO International Symposium; Recent
Advances in the Assessment of the Health Effect of Environmental
Pollution, Paris, 24-28 June 1974. Luxembourg, Commission of
the European Communities, pp. 591-603.
HUNT, W. F., PINKERTON, C., MCNULTY, O., & CREASON, J. (1971) A study
in trace element pollution in 77 midwestern cities. In: ed. D.
D. Hemphill Proceedings of the University of Missouri's 4th
Annual Conference on Trace Substances in Environmental Health,
Columbia, Missouri, 23-25 June 1970. University of Missouri,
Columbia, 1971, pp. 56-68.
HUNTZICKER, J. J., FRIEDLANDER, S. K., & DAVIDSON, C. J. (1975)
Material balance for automobile-emitted lead in Los Angeles
basin. Environ. Sci. Technol., 9: 448-457.
HURSH, J. B., & MERCER, T. T. (1970) Measurement of Pb212 loss rate
from human lungs. J. appl. Physiol., 28: 268.
HWANG, J. Y., ULLUCCI, P. A., SMITH, S. B., & MALENFANT, A. L. (1971)
Microdetermination of lead in blood by flameless atomic
absorption spectrometry. Anal. Chem., 43: 1319-1321.
IARC (1972) Monographs. Evaluation of carcinogenic risk of chemicals
to man. Vol. 1, Lyon, International Agency for Research on
Cancer, pp. 184.
INTER-DEPARTMENT WORKING GROUP ON HEAVY METALS (1974) Lead in the
environment and its significance to man. Report for the
Department of the Environment Central Unit on Environmental
Pollution. London, Her Majesty's Stationery Office, pp. 1-47,
Pollution Paper No. 2.
INTERNATIONAL LEAD AND ZINC STUDY GROUP (1973) Lead in gasoline: A
review of the current situation, International Lead and Zinc
Study Group, United Nations, New York, pp. 1-37. First addendum
1974; Second addendum 1975; Third addendum 1976.
IUPAC-IUB COMMISSION ON BIOCHEMICAL NOMENCLATURE (1973) Enzyme
nomenclature. Recommendations (1972) of the Commission on
Biochemical Nomenclature on the nomenclature and classification
of enzymes together with their units and the symbols of enzyme
kinetics. Amsterdam, Elsevier.
JARVIE, A. W. P., MARKALL, R. N., & POTTER, H. R. (1975) Chemical
alkylation of lead. Nature (Lond.), 255: 217-218.
JAWOROSKI, Z. (1968) Stable lead in fossil ice and bones. Nature
(Lond.), 217: 152-153.
JERNIGAN, E. L., RAY, B. J., & DUCE, R. A. (1971) Lead and bromine in
atmospheric particulate matter on Oahu, Hawaii. Atmosph.
Environ., 5: 881-886.
JONES, R. D., COMMINS, B. T., & CERNIK, A. A. (1972) Blood lead in
carboxyhemoglobin levels in London taxi drivers. Lancet,
2: 302-303.
JOST, D., MULLER, J., & JENDRICKE, U. (1973) Lead in atmospheric
aerosol. In: Proceedings of the International Symposium;
Environmental Health Aspects of Lead, Amsterdam, 2-6 October
1972. Luxembourg, Commission of the European Communities,
pp. 941-948.
KAMMHOLZ, L. P, THATCHER, L. G., BLODGETT, F. M., & GOOD, T. A. (1972)
Rapid protoporphyrin quantitation for detection of lead
poisoning. Pediatrics, 50: 625-631.
KASSENAAR, A., MORELL, H., & LONDON, I. U. (1957) The incorporation of
glycine into globin and the synthesis of heme in vitro in duck
erythrocytes. J. Biol. Chem., 229: 423-435.
KATO, K. (1932) Lead meningitis in infants. Am. J. Dis. Child.,
44: 569-591.
KEENAN, R. C., BYERS, D. H., SALTZMAN, B. E., & HYSLOP, F. L. (1963)
The "USPHS" method for determining lead in air and in biological
materials. Am. Ind. Hyg. Assoc., 24: 481-491.
KEHOE, R. A. (1961) The metabolism of lead in health and disease. The
Harben Lectures, 1960. J. Roy. Inst. Publ. Health Hyg.,
24: 81-96, 101-120, 129-143, 177-203.
KEHOE, R. A., CHOLAK, J., & LARGENT, E. J. (1944) The concentrations
of certain trace metals in drinking water. J. Am. Water Works.
Assoc., 36: 637-644.
KEHOE, R. A., THAMAN, F., & CHOLAK, J. (1933) Lead absorption and
excretion in relation to the diagnosis of lead poisoning.
J. ind. Hyg., 15: 320.
KELLO, D., & KOSTIAL, K. (1973) The effect of milk diet on lead
metabolism in rats. Environ. Res., 6: 355-360.
KEMPF, TH., & SONNEBORN, M. (1973) Vergleich von Methoden zur
Bestimmung von Spurenmetallen in Wasserkreislauf. Z. anal.
Chem., 267: 267-270.
KENNEDY, V. C. (1960) Geochemical studies in the Coeur d'Alene
district Shoshone County, Idaho. U.S. geol Survey Bull.,
1098-A.
KEPPLER, J. F., MAXFIELD, M. E., MOAS, W. D., TIETJEN, C., & LINCH, A.
L. (1970) Interlaboratory evaluation of the reliability of blood
lead analyses. Am. Ind. Hyg. Assoc. J., 31: 412-429.
KERIN, Z. (1972) Tägliche Bleiaufnahme Kit der Bauernkost aus dem
Emissionsgebiet einer Bleihutte. Protectio vitae, 71: 22-23.
KERIN, Z. (1973) Lead in new-fallen snow near a lead smelter. Arch.
environ. Health, 26: 256-260.
KERIN, Z., KERIN, D., & DJURIC, D. (1972) Lead contamination of
environment in Meza Valley. Lead content of the soil. Int. Arch.
Arbeitsmed., 29: 129-138.
KLEIN, U. C., SAYRE, J. W., & KOTOK, D. (1974) Am. J. Dis. Childh.,
127: 805-807. porphyrin in duck erythrocytes. Am. J. Physiol.,
203: 971-974.
KLEIN, M., NANER, R., HARPUR, E., & CORBIN, R. (1970) Earthenware
containers as a source of fatal lead poisoning. Case study and
public-health considerations. New Eng. J. Med., 283: 669-672.
KLEIN, U. C., SAYRE, J. W., & KOTOK, D. (1974). Am. J. Dis. Child.,
127: 805-807.
KLINE, T. S. (1960) Myocardial changes in lead poisoning. A.M.A.J.
Dis. Child., 99: 48-54.
KNEIP, T. J., & LAUREN, G. R. (1972) Isotope excited X-ray
fluorescence. Anal. Chem., 44: 57A.
KOBAYASHI, N., & OKAMOTO, T. (1974) Effects of lead oxide on the
induction of lung tumors in Syrian hamsters. J. Nat. Cancer
Inst., 52(5): 1605-1610.
KOLBYE, A. C., MAHAFFEY, R., FIORINO, A., CORNELIUSSEN, P. C., &
JELINEK, C. F. (1974) Food exposures to lead. Environ. Health
Perspect., Exp. Issue No. 7, pp. 65-75.
KOPP, J. F., & KRONER, P. T. (1970) Trace metals in water of the
United States: a 5-year summary of trace metals in rivers and
lakes of the US. (Oct. 1962-Sept. 1967) Cincinnati, Ohio, US
Department of the Interior.
KOSMIDER, S., & PETELNZ, T. (1962) Zmiany elektrokardiograficzne u
starszych osob z przewleklym zawodowym zatruciem olowiem. Pol.
Arch. Med. Wewn, 32: 437-442.
KOSMIDER, S., & SROCZYNSKI, J. (1961) Zmiany elektrokardiograficzne w
przewleklym doswiadczalney olowiej u krolikow
(Electrocardiographic changes in chronic experimental plumbism in
rabbits). Postepy Hig. i Med. Dosw., 15: 353-357.
KOSTIAL, K., & VOUK, V. B. (1957) Lead ions and synaptic transmission
in the superior cervical ganglion of the cat. Brit. J.
Pharmacol., Chemother., 12: 219-222.
KOSTIAL, K., MALJKOVIC, T., & JUGO, S. (1974) Lead acetate toxicity in
rats in relation to age and sex. Arch. Toxicol., 31: 265-269.
KOSTIAL, K., SIMONOVIC, I., & PISONIC, U. (1971) Lead absorption from
the intestine in newborn rats. Nature (Lond.), 233: 564.
KOTOK, D. (1972) Development of children with elevated blood lead
levels; A controlled study. J. Pediatr., 80: 57-61.
KRIGMAN, U. R., DRUSE, U. J., TAYLOR, T. D., WILSON, U. H., NEWELL, L.
R., & HOGAN, E. L. (1974) Lead encephalopathy in the developing
rat: Effect upon myelination. J. Neuropath. exp. Neurol.,
33(1): 58-74.
KUBASIK, N. P., VALOSIN, U. T., & MURRAY, U. H. (1972) Carbon rod
atomizer applied to measurement of lead in whole blood by atomic
absorption spectrophotometry. Clin. Chem., 18: 410-412.
KUZ'MINSKAJA, T. N. (1964) Experimental atherosclerosis against a
background of Pb-intoxication. Arch Patol., 26(9): 21-24.
LAHMANN, E. (1969) Untersuchungen über Luftverunreinigungen dutch den
Kraftverkehr. Wasser-boden-und Lufthyg., No. 28, 3-80.
LAMM, S. H., & ROSEN, J. F. (1974) Lead contamination in milk fed to
infants' 1972-1973. Pediatrics, 53: 137-141.
LAMOLA, A. A., & YAMANE, T. (1974) Zinc protoporphyrin in the
erythocytes of patients with lead intoxication and iron
deficiency anaemia. Science, 186: 936-938.
LAMOLA, A. A., JOSELOW, M., & YAMANE, T. (1975) Zinc protoporphyrin
(ZPP): A simple sensitive, fluorometric screening test for lead
poisoning. Clin. Chem., 21: 93-97.
LAMPTER, P. W., & SCHOCHET, S.S. (1968) Demylination and remyelination
in lead neuropathy. Electron microscopic studies. J. Neuropath.
exp. Neurol., 25: 527-545.
LANCRANJAN, I., POPESCU, H. I., GAVANESCU, O., KLEPSCH, I., &
SERBANESCU, M. (1975). Reproductive ability of workmen
occupationally exposed to lead. Arch. environ. Health,
30: 396-401.
LANE, R. E. (1949) The care of the lead worker. Brit. J. ind. Med.,
6: 125-143.
LANE, R. E. (1964) Health control in inorganic lead industries. A
follow-up of exposed workers. Arch. environ. Health, 8: 55.
LANDING, B. H., & NAKAI, H. (1959) Histochemical properties of renal
lead inclusions and their demonstration in urinary sediment. Am.
J. clin. Path., 31: 499-503.
LANDRIGAN, P. J., BALCH, R. W., BARTHEL, W. F., WHITWORTH, R. H.,
STAEHLING, N. W., & ROSENBLUM, B. F. (1975a) Neurophysiological
dysfunction in children with chronic low-level lead absorption.
Lancet, 1: 708-712.
LANDRIGAN, P. J., GEHLBACH, S. H., ROSENBLUM, B. F., SHOULTS, J. M.,
CANDELARIA, R. M. BARTHEL, W. F., LIDDLE, J. A., SMREK, A. L.,
STAEHLING, N. W., & SANDERS, J. F. (1975b) Epidemic lead
absorption near an ore smelter; The role of particulate lead.
New Engl. J. Med., 292: 123-129.
LANSDOWN, R. G., CLAYTON, B. E., GRAHAM, P. J., SHEPHERD, J., DELVES,
H. T., & TURNER, W. C. (1974) Blood-lead levels, behaviour and
intelligence: a population study. Lancet, 1: 538-541.
LAURS, A. J. (1976) Review of European test methods for measuring
lead and cadmium release. In: Proceedings of the International
Conference on Ceramic Foodware Safety, Geneva, 1974. New York,
Lead Industries Association Inc., pp. 27-36.
LAVESKOG, A. (1971) A method for determination of tetramethyl lead
and tetraethyl lead in air. Proceedings of the Second
International Clean Air Congress, H. H. England and W. T.
Beery, eds. New York, Acad. Press, pp. 549-557.
LAWTHER, P. J., COMMINS, B. T., ELLISON, J. MEK., & BILES, B. (1972)
Airborne lead and its uptake by inhalation. In: Hepple, P. ed.,
Lead in the environment. Essex, UK, Applied Science Publishers
Ltd., pp. 8-28.
LAZRUS, A. L., LORANGE, E., & LODGE, J.P. (1970) Lead and other
metalions in US precipitation. Environ, Sci. Technol.,
4: 55-58.
LEAD DEVELOPMENT ASSOCIATION (1976) Lead In Gasoline Bulletin No. 12,
Lead Development Association, 34 Berkeley Square, London W1X 6AJ,
30 March 1976.
LEE, R. E., & GORANSON, S. (1972) National air surveillance cascade
impactor network. I. Size distribution measurements of suspended
particulate matter in air. Environ. Sci. Technol.,
6: 1019-1024.
LEE, R. E., GORANSON, S.S., ENRIONE, R. E., & MORGAN, G. B. (1972) The
NASH cascade impactor network, II Size distribution of trace-
metal components. Env. Sci. Technol., 6: 1025-1030.
LEHNERT, G., MASTALL, H. SZADKOWSKI, D., & SCHALLER, K. H. (1970)
Berufliche Bleibelastung durch Autoabgase in Grosstadtstrassen.
Dtsch. Med. Wschr., 95: 1097-1099.
LEHNERT, G., SCHALLER, K. H., KÜHNER, A., & SZADKOWSKI, D. (1967)
Auswirkungen des Zigarettenrauchens aus dem Blutbleisspiegel.
Int. Arch. Geweberpath. Geweberhyg., 23: 358-363.
LEHNERT, G., SCHALLER, K. H., & SZADKOWSKI, D. (1969) Eine
zuverlässige Schnellmethode zur Bleibestimmung in kleinen
Blutmengen. Z. Klin. Chem. Klin. Biochem., 7: 310.
LEIKIN, S., & ENG, G. (1963) Erythrokmetic studies of the anemia of
lead poisoning. Pediatrics, 31: 996-1002.
LILIS, R., GAVRILESCU, N., NESTORESCU, B., DURIMTIU, C., & ROVENTA, A.
(1968) Nephropathy in chronic lead poisoning. Brit. J. ind.
Med., 25: 196-202.
LINCH, A. L., WIEST, E.G., & CARTER, U. D. (1970) Evaluation of
tetraalkyl lead exposure by personnel monitor surveys. Am. Ind.
Hyg. Assoc. Y., 31: 170-179.
LIVINGSTONE, D. A. (1963) Data of geochemistry. Sixth Edition. Chapter
of chemical composition of rivers and lakes. US Geol. Survey
Prof. Paper 440.
MACHLE, W. E. (1935) Tetraethyl lead intoxication and poisoning by
related compounds of lead. J. Am. Med. Assoc., 105: 578-585.
MAHOFFEY, K. (1974) Nutritional factors and susceptibility to lead
toxicity. Environ. Health Perspect., Exp. Issue No. 7,
pp. 107-113.
MAKASEV, K. K., & KRIVDINA, L. V. (1972) Status of the interstitial
tissues of vascular walls and their penetration under the lead
poisoning. Tr. Naucn-issl. In-ta Kraev. Pat. Kaz. SSR,
23: 11-13.
MAKASEV, K. K., & VERBOLOVIC, V. P. (1967) Succinic dehydrogenase and
cytochrome oxidase in the duodenum during lead poisoning. Izv.
Akad. Nauk Kazahst. SSR Set. Biol., 5(2): 59-64.
MALCOLM, D. (1971) Prevention of long-term sequelae following the
absorption of lead. Arch. environ. Health, 23: 292-298.
MALJKOVIC, J. (1971) A case of occupational poisoning with lead
carbonate and stearate. Sigurnost u pogonu., 13: 123-124.
MAMBEEVA, A. A., & AHMEDOVA, A. S. (1967) Changes of reactivity of the
small intestine in healthy and lead-intoxicated homeotherms to
histamine under the influence of solutions of lead acetate.
Biull. eksper. Biol., 32: 34-37.
MAMBEEVA, A. A., & KOBKOVA, I. D. (1969) The concentration of
catecholamines in tissues of the cardiovascular system in
experimental lead intoxication. Izv. Akad, Nauk Kazahst. SSR.
Ser. Biol., No. 1: 77-82.
MANALIS, R. S., & COOPER, G. I. (1973) Presynaptic and postsynaptic
effects of lead at the frog neuromuscular junction. Nature
(Lond.), 243: 354-355.
MAO, P., & MOLNAR, J. J. (1967) The fine structure and histochemistry
of lead-induced renal tumours in rats. Am. J. Pathol.,
50: 571-603.
MAPPES, R. (1972) Eine Fehlerquelle und ihre Kompensation bei der
delta-Amino-lävulinsäurebestimmung im Harn nach Grabecki. Int.
Arch. Arbeitsmed., 30: 81-86.
MARTIN, A. E., FAIRWEATHER, F. A., BUXTON, R. S. J., & ROOTS, L, M.
(1975) Recent epidemiological studies of environmental lead of
industrial origin. In: Proceedings of the CEP-EPA-WHO
International Symposium; Recent Advances in the Assessment of
the Health Effects of Environmental Pollution, Paris. 24-28 June
1974. Luxembourg, Commission of the European Communities.
pp. 1113-1120.
MARVER, H. S., TSCHUDY, D. P., PERLROTH, M. G., COLLINS, A., & HUNTER,
G. JR. (1966) The determination of aminoketones in biological
fluids. Anal. Biochem., 14: 53-60.
MATOUSEK, J. F., & STEVENS, B. J. (1971) Biological applications of
the carbon rod atomizer in atomic absorption spectroscopy. I.
Preliminary studies on magnesium, iron, copper, lead and zinc in
blood in plasma. Clin. Chem., 17: 363-368.
MATSON, W. R. (1971) Rapid sub-nanogram simultaneous analysis. In:
Proceedings of the University of Missouri's 4th Annual
Conference on Trace Substances in Environmental Health,
Columbia, Missouri, 23-25 June 1970. Columbia, ed. D. D.
Hemphill, University of Missouri, pp. 396-406.
MATTSSON, R., & JAAKKOLA, T. (1974) Lead in the Helsinki air.
Ympäristö ja Terveys, 5: 721.
MAUZERALL, D., & GRANICK, S. (1956) The occurrence and determination
of delta- aminolevulinic acid and porphobilinogen in urine.
J. Biol. Chem., 219: 435-446.
MAXFIELD, M. E., STOPPS, G. J., BARNES, J.P., SNEE, R. D., & AZAR, A.
(1972) Effect of lead on blood regeneration following acute
hemorrhage in dogs. Am. Ind. Hyg. Assoc. J., 33: 326-337.
MCCABE, L. J. (1970) Corrosion by soft water. Amer. Water Works
Seminar on Corrosion by Soft Water. Wash. DC, pp. 9.
MCLAUGHLIN, M., & STOPPS, G. J.(1973) Smoking and Lead. Arch. environ
Health, 26: 131-136.
MCLAUGHLIN, M., LINCH, A. L., & SNEE, R. D. (1973) Longitudinal
studies of lead levels in US population. Arch. environ. Health,
27: 305-312.
MCMULLEN, T. B, FAORO, R. B., & MORGAN, G. B. (1970) Profile of
pollutant fractions in non-urban suspended particulate matter.
J. Air Pollut. Control Assoc., 20: 369-372.
MCNEIL, J. L., & PTASNIK, J. A. (1975) Evaluation of long-term
effects of elevated blood lead concentrations in asymptomatic
children. In: Proceedings of CEC-EPA-WHO International
Symposium; Recent Advances in the Assessment of the Health
Effects of Environmental Pollution, Paris, 24-28 June 1974.
Luxembourg, Commission of the European Communities, pp. 571-579.
MENDEN, R. E., ELIA, V. J., MICHAEL, L. W., & PETERING, H. C. (1972)
Distribution of Cd and Ni of tobacco during cigarette smoking.
Env. Sci. Technol., 6: 830-832.
MERCER, T. T. (1975) The deposition model of the Task Group on Lung
Dynamics: A comparison with recent experimental data. Health
Phys., 29: 673-680.
MERWIN, B. W. (1976) Review of U.S. standards on test methods for
lead and cadmium release. In: Proceedings of the International
Conference on Ceramic Foodware Safety, Geneva, 1974. New York,
Lead Industries Association Inc., pp. 36-40.
MICHAELSON, J. A. (1973) Effect of inorganic lead on levels of RNA,
DNA and protein on developing neonatal rats. Toxicol. appl.
Pharmacol., 26: 539-548.
MICHAELSON, J. A., & SAUERHOFF, M. W. (1974) An improved model of lead
induced brain dysfunction in the suckling rat. Toxicol. appl.
Pharmacol., 28: 88-96.
MILLAR, J. A., BATTISTINI, V., GUMMING, R. L. C., CARSWELL, F., &
GOLDBERG, A. (1970) Lead and delta-aminolevulinic acid
dehydratase levels in mentally retarded children and in lead-
poisoned suckling rats. Lancet, 2: 695-698.
MIRONCIK, L. M., & TIMOFEEVA, N. D. (1974) Lead effects on the
experimental athero-sclerosis process and several respiratory
ferments of the myocard. Probl. Gig. i organiz zdravoochr. v
Uzbekistane, 2: 77-78.
MITCHELL, D. G., & ALDOUS, K. M. (1974) Lead content of foodstuffs.
Environ. Health Perspect., Exp. Issue No. 7, pp. 59-65.
MITCHELL, D. G., ALDOUS, K. M., & RYAN, F. J. (1974) Mass screening
for lead poisoning, capillary blood sampling and automated Delves
cup atomic absorption analysis. N.Y. State, J. Med., 74: 1599.
MITCHELL, R. L. (1963) Soil aspects of trace element problems in
plants and animals. J. Roy. Agric. Soc., 124: 75-86.
MITCHELL, R. L., & REITH, J. W. S. (1966) The lead content of pasture
herbage. J. Sci. Food Agric., 17: 437-440.
MÖLLER, B., AKSELLSSON, R., BERLIN, M., FISCHBEIN, A., HAMMARSTRÖM, L.
(1974) Sammanfatting av föredrag rid Läkaresfällskapets
Riksstämma 27-30 Nov., 1974 (Omgivningshygien No. 7).
MOMCILOVIC, B., & KOSTIAL, K. (1974) Kinetics of lead retention and
distribution in suckling and adult rats. Environ. Res.,
8: 214-220.
MONAENKOVA, A.M. (1957) Functional state of the thyroid gland in
chronic intoxication from several industrial poisons (Pb, Hg).
Gig. Trud. Prof. Zabol., 2: 44-48.
MONAENKOVA, A.M., & GLOTOVA, K. V. (1969) Cardiovascular changes in
some chronic intoxications with industrial poisons (lead, carbon
disulphide, benzene). Gig. Trud. Prof. Zabol, 2: 32-35.
MONCRIEFF, A. A., KOUMIDES, O. P., CLAYTON, B. E., PATRICK, A.D.,
RENWICK, A. G. C., & ROBERTS, G. E. (1964) Lead poisoning in
children. Arch. Dis. Child., 39: 1-13.
MOORE, J. F., & GOYER, R. A. (1974) Lead-induced inclusion bodies;
Composition and probable role in lead metabolism. Environ.
Health Perspect., Exp. Issue No. 7, pp. 121-129.
MOORE, M. R. (1973) Plumbosolvency of waters. Nature Lond.,
243: 222-223.
MORGAN, B. B., & REPKO, J. D. (1974) Evaluation of behavioural
functions in workers exposed to lead. In: C. Xintaras et al.,
eds, Behavioural toxicology, early detection of occupational
hazards. Washington, U.S. Department of Health, Education and
Welfare.
MORGAN, J. M., HARTLEY, M. W., & MILLER, R. E. (1966) Nephropathy in
chronic lead poisoning. Arch. intern. Med., 118: 17-29.
MUELLER, P. K. (1970) Discussion (characterization of particulate lead
in vehicle exhaust experimental techniques). Environ. Sci.
Technol., 4: 248-251.
MUIR, D.C. F, & DAVIES, C. N. (1967) The deposition of 0.5 µ diameter
aerosols in the lungs of man. Ann. occup. Hyg., 10: 161-174.
MURO, L. A., & GOYER, R. A. (1969) Chromosome damage in experimental
lead poisoning. Arch. Pathol., 87: 660-663.
MUROZUMI, M., CHOW, T. J., PATTERSON, C. C. (1969) Chemical
concentrations of pollutant lead aerosols, terrestrial dusts and
sea salts in Greenland and Antarctic snow strata. Geochem.
Cosmochim. Acta, 33: 1247-1294.
MURTHY, G. M., & RHEA, U.S. (1971) Cadmium, copper, iron, lead,
manganese and zinc in evaporated milk, infant products and human
milk. J. Dairy Sci., 54: 1001-1005.
MYERSON, R. M., & EISENHAUER, J. H. (1963) Atrioventricular conduction
defects in lead poisoning. Am. d. Cardiol., 11: 409-412.
NAKAO, K., WADA, O, & YANO, Y. (1968) Delta-aminolevulinic acid
dehydratase activity in erythrocytes for the evaluation of lead
poisoning. Clin. Chim. Acta., 19: 319-325.
NAS-NRC (1972) Airborne lead in perspective. Washington, DC, Nat.
Aced. Sci.
NEAL, P. A., DRESSEN, W. C., EDWARDS, T. J., REINHART, W. H., WEBSTER,
S. H., CASTBERG, H. T., & FAIRHALL, L. T. (1941) A study of the
effect of lead arsenate exposure on orchardists and consumers of
sprayed fruit. US publ. Health Bull., No. 267.
NEEDLEMAN, H. L., & SHAPIRO, J. M. (1974) Dentine lead levels in
asymptomatic Philadelphia school children: Subclinical exposure
in high and low risk groups. Environ. Health, Perspect, Exp.
Issue. No. 7, pp. 27-33.
NELSON, W. C., LYKINS, M. H., MACKEY, J., NEWILL, V. A., FINKLEA, J.
F., & HAMMER, D. J. (1973) Mortality among orchard workers
exposed to lead arsenate spray: A cohort study. J. chron. Dis.,
26: 105-118.
NIKKANEN, J., HERNBERG, S., & TOLA, S. (1972) Modifications of the
delta-aminolevulinic acid dehydratase test and their significance
for assessing different intensities of lead exposure. Work-
environ.-Health, 9: 46-52.
NIKLOWITZ, W. J., & YEAGER, D. W. (1973) Interference of Pb with
essential brain tissue Cu, Fe and Zn as main determinant in
experimental tetraethyllead encephalopathy. Life Sci.,
13: 897-905.
NORDBERG, G. F., ed. (1976) Effects and dose-response relationships of
toxic metals. Proceedings from an international meeting
organized by the subcommittee on the Toxicology of Metals of the
Permanent Commission and International Association on
Occupational Health, Tokyo, 18-23 November 1974. Amsterdam,
Elsevier, 1976, p. 15.
NORDMAN, C. H. (1975) Environment lead exposure in Finland. A study
on selected population groups. Doctoral Thesis, University of
Helsinki, pp. 1-118.
NORDMAN, C. H., & HERNBERG, S. (1975) Blood lead levels and
erythrocyte delta-aminolevulinic acid dehydratase activity of
selected population groups in Helsinki. Scand. J. Work-Environ.-
Health, 1: 219-232.
NORDMAN, C. H., HERNBERG, S., NIKKANEN, J., & RYHANEN, A. (1973) Blood
lead levels and erythrocyte-delta-aminolevulinic acid dehydratase
activity in people living around a secondary lead smelter. Work-
Environ.-Health, 10: 19-25.
NOZAKI, K. (1966) Method for studies on inhaled particles in human
respiratory system and retention of lead fume. Ind. Health
(Jpn), 4: 118-128.
NRC, CANADA (1973) Lead in the Canadian Environment. Associate
Committee on Scientific Criteria for Environmental Quality.
NYE, L. J. J. (1929) An investigation of the extraordinary incidence
of chronic nephritis in young people in Queensland. Med. J.
Aust., 2: 145-159.
OEHLKERS, F. (1953). Chromosome breaks influenced by chemicals.
Heredity, 6 (suppl.) 95-105.
OKAZAKI, H., ARONSON, S. M., DIMAIO, D. J., & OLVERA, J. E. (1963)
Acute lead encephalopathy of childhood. Trans. Am. neurol.
Assoc., 88: 248-250.
OLIVER, T. (1914) Lead Poisoning. London, H. K. Lewis.
ORLOVA, A. A. (1954) Changes in cardiac activity of patients with lead
and mercury intoxication. Trud. Akad. Med. Nauk. SSSR.,
31: 102-112.
O'RIORDAN, M. L., & EVANS, H. J. (1974) Absence of significant
chromosome damage in males occupationally exposed to lead.
Nature (Lond.), 247: 50-53.
OTT, W., CLARKE, J. F., & OZOLINS, G. (1970) Calculating future
carbon monoxide emissions and concentrations from urban traffic
data. Durham, DHEW Publ. no. AP-41.
OYASU, R., BATTIFORA, H. A., CLASEN, R. A., MCDONALD, J. H., & HASS,
G. M. (1970) Induction of cerebral gliomas in rats with dietary
lead subacutate and 2-acetylamino-fluorene. Cancer Res.,
30: 1248-1261.
PACKHAM, R. F. (1971) The leaching of toxic stabilizers for
unplasticized PVC water pipe: P. 1 A critical study of laboratory
test procedures, P.2. A survey of lead levels in PVC distribution
systems. Water Treat. Exam., 20: 108-124; 144-166.
PADILLA, F., SHAPIRO, A. P., & JENSEN, W. N. (1969) Effect of chronic
lead intoxication on blood pressure in the rat. Am. J. reed.
Sci., 258: 359-365.
PALMISANO, P. A., SNEED, R. C., & CASSADY, O. (1969) Untaxed whisky
and fetal lead exposure. J. Pediatr., 75: 869.
PANOVA, Z. (1972) Early changes in the ovarian function of women in
occupational contact with inorganic lead. Works United Res.
Inst. Hyg. ind. Saf. (Sofia), 23: 161-166.
PATEL, A. J., MICHAELSON, J. A., CREMER, J. E., & BALAZS, R.
(1974a)The metabolism of (C14) glucose by the brains of
suckling rats intoxicated with inorganic lead. J. Neurochem.,
22: 581-590.
PATEL, A. J., MICHAELSON, J. A., CREMER, J. E., & BALAZS, R. (1974b)
Changes within metabolic compartments in the brains of young rats
ingesting lead. J. Neurochem., 22: 591-598.
PATTERSON, C. C. (1965) Contaminated and natural lead environments of
man. Arch environ. Health, 11: 344-363.
PEGUES, W. L. (1960) Lead fume from welding on galvanized and zinc-
silicate coated steels. Ind. Hyg. J., 21: 252-255.
PEKKARINEN, M. (1970) Methodology in the collection of food
consumption data. World Rev. Nut. Diet., 12: 145-170.
PENTSCHEW, A. (1965) Morphology and morphogenesis of lead
encephalopathy. Acta Neuropathol., 5: 133-160.
PENTSCHEW, A., & GARRO, F. (1966) Lead encephalo-myelopathy of the
suckling rat and its implications on the porphyrinopathic nervous
diseases. Acta Neuropathol., 6: 266-278.
PERNIS, B., DE PETRIS, S., BEARD, R. R., & KARLSHAD, G. (1964) The
ultrastructure of red cells in experimental lead-poisoning.
Med. Lav., 55: 81-101.
PINES, A. G. (1965) Indexes of general reactivity in saturnine. Vrac.
Delo, 3: 93-96.
PIOMELLI, S. (1973) A micromethod for free erythrocyte porphyrin: the
FEP test. J. Lab. clin. Med., 81: 932-940.
POTT, F., & BROCKHAUS, A. (1971) Vergleich der enteralen und
pulmonalen Resorptionsquote von Bleiverbindungen. Zentralbl.
Bakt. Hyg. J. Orig. B., 155: 1-17.
PREROVSKA, I. (1973) Einfluss von Blei auf biochemische Veränderungen
im Serum und Veränderungen in der Aderwand im Hinblick auf
Atherosklerose. In: Proceedings of the International
Symposium. Environmental Health Aspects of Lead, Amsterdam, 2-6
October 1972. Luxembourg, Commission of the European
Communities, pp. 551-561.
PRPIC-MAJIC, D., MUELLER, P. K., LEW, V. C., & TWISS, S. (1973)
delta-aminolevulinic acid dehydratase stability in human blood.
Am. ind. Hyg. Assoc. J., 315-319.
PUESCHEL, S. M., KOPITO, L., & SCHWACHMAN, H. (1972) A screening and
follow up study of children with an increased lead burden.
J. Am. Med. Assoc., 333: 462-466.
PUHAC, J., HRGOVIC, N, STANKOVIC, M., & POPOVIC, S. (1963) Laboratory
investigations of the possibility of application of lead nitrates
compounds as a raticide means by decreasing reproductive
capability of rats. Acta Vet. (Beograd), 13: 3-9.
PURDUE, L. J., ENRIONE, R. E., THOMPSON, R. J., & BONFIELD, B. A.
(1973) Determination of organic and total lead in the atmosphere
by atomic absorption spectrometry. Anal. Chem, 45: 527-530.
RABINOWITZ, M. B. (1974) Lead contamination of the biosphere by human
activity. A stable isotope study. Ph.D. Thesis, University of
California, Los Angeles.
RABINOWITZ, M. B., WETHERILL, G. W., & KOPPLE, J. D. (1973) Lead
metabolism in the normal human: stable isotope studies. Science,
182: 725-727.
RABINOWITZ, M.B., WETHERILL, G. W., & KOPPLE, J. D. (1974) Studies of
human lead metabolism by use of stable isotope tracers. Environ.
Health. Perspect, Exp. Issue No. 7, pp. 145-155.
RASBERRY, S. D. (1973) Investigation of portable X-ray Fluorescence
analyzers for determining lead on painted surfaces. Appl.
Spectrosc., 27(2): 102-108.
RICHET, G., ALBAHARY, C., MOREL-MAROGER, L., GUILLAUME, P., & GALLE,
P. (1966) Les altérations rénales dans 23 cas de saturnisme
professionnel. Bull. Mere. Soc. Med. Hop. Paris, 117: 441-466.
RIEKE, F. E. (1969) Lead intoxication in shipbuilding and
shipscrapping 1941-1968. Arch. environ. Health, 19: 521-539.
RILEY, J. P., & SKIRROW, C. (1965) Chemical oceanography. New York,
Academic Press, vol. 2.
RIMINGTON, C. (1938) An enzymic theory of haemopoiesis. C.R. Lab
Carlsberg. Ser. Chim., 22: 454-464.
RIMINGTON, C., & SVEINSSON, S. L. (1950) The spectrophotometric
determination of uroporphyrin. Scand. J. clin. Lab. Invest.,
2: 209-216.
ROBERTS, T. M., HUTCHINSON, T. C., PACIGA, J., CHATTOPADHYAY, A.,
JERVIS, R. E., VAN-LOON, J., & PARKINSON, D. R. (1974) Lead
contamination around secondary smelters: Estimation of dispersal
and accumulation by humans. Science, 186: 1120-1123.
ROBINSON, E., & LUDWIG, F. L. (1967) Particle size distribution of
urban lead aerosols. J. Air Pollut. Control Assoc.,
17: 664-669.
ROBINSON, E., LUDWIG, F. L., DEVries, J. E, & HOPKINS, T. E. (1963)
Variations of atmospheric lead concentrations and type with
particle size. Final Report PA-4211. Cal. Stanford Res. Inst.
Menlo Park, pp. 1-80.
ROBINSON, T. R. (1974) Delta-aminolevulinic acid and lead in urine of
lead antiknock workers. Arch. Environ. Health, 28: 133-139.
ROE, F. Y., BOYLAND, C. E., DUKES, C. E., & MITCHLEY, B. V. C. (1965)
Failure of testosterone or xanthopterin to influence the
induction of renal neoplasms by lead in rats. Brit. J. Cancer,
19: 860-866.
ROELS, H. A., BUCHET, J.P., & LAUWERYS, R. R. (1974) Inhibition of
human erythrocyte delta-aminolevulinate dehydratase by lead.
Int. Arch. Arbeitsmed., 3: 277-281.
ROELS, H. A., LAUWERYS, R. R., BUCHET, J.P., & VRELUST, M. (1975)
Response of free erythrocyte porphyrin and urinary delta-
aminolevulinic acid in men and women moderately exposed to lead.
Int. Arch. Arbeitsmed., 34: 97-108.
ROSENBLUM, W. J., & JOHNSON, M. G. (1968) Neuropathologic changes
produced in suckling mice by adding lead to the maternal diet.
Arch. Pathol., 85: 640-648.
RÜHLING, A., & TYLER, G. (1968) An ecological approach to the lead
problem. Bot. Notiser, 121: 321-342.
RUITER, N. DE, SEEMAYER, N., & MANOJLOVIC, N. (1977) Einfluss yon
Zink-Ionen auf die toxische Wirkung von Bleichlorid (PbC12)
untersucht an Mäusemakrophagen in vitro. Zentraibl. Bakt. Hyg.,
I. Abt. Orig. B., 164: 90-98.
SACHS, H. K. (1974) Effect of a screening program on changing patterns
of lead poisoning. Environ. Health Perspect., Exp. Issue No. 7,
pp. 41-47.
SAKURAI, H., SUGITA, M., & TSUCHIYA, K. (1974) Biological response and
subjective symptoms in low level lead exposure. Arch. environ.
Health, 29: 157-163.
SANDSTEAD, H. H. (1967) Effect of chronic lead intoxication on in
vivo I-131 uptake by the rat thyroid. Proc. Soc. Exp. Biol.
Med. 124: 18-20.
SANDSTEAD, H. H., ORTH, D. N., ABE, K., & STIEL, J. (1970) Lead
intoxication: Effect on pituitary and adrenal function in man.
Clin. Res., 18: 76 (abstract).
SANDSTEAD, H. H., STANT, E.G., & BRILL, H. B. (1969) Lead intoxication
and the thyroid. Arch. int. Med., 123: 632-635.
SANO, S., & RIMINGTON, C. (1963) Excretion of various porphyrins and
their corresponding porphyrinogens by rabbits after intravenous
injection. Biochem. J., 86: 203-212.
SANSONI, B., KRACKE, W., DIETL, F., & FISHER, J. (1973)
Mikrospurenbestimmung von Blei in verschiedenartigen
Umweltproben durch fiammenlose Atomabsorption nach externer
Nassveraschung reit H202/Fe2. In: Proceedings of the
International Symposium; Environmental Health Aspects of lead,
Amsterdam, 2-6 October 1972. Luxembourg, Commission of the
European Communities, pp. 1107-1116.
SASSA, S., GRANICK, J. L., GRANICK, S., KAPPAS, A., & LEVERE, R. D.
(1973) Studies in lead poisoning. I. Microanalysis of erythrocyte
protoporphyrin levels by spectrofluorometry in the detection of
chronic lead intoxication in the subclinical range. Biochem.
Med., 8: 135-148.
SAYRE, J. W., CHARNEY, E., VOSTAL, J., & PLESS, B. (1974) House and
land dust as a potential source in childhood lead exposure. Am.
J. Dis. Child., 127: 167-170.
SCARLATO, G., SMIRNE, S., & POLONI, A. E. (1969) L'encefalopatia
saturnina acuta dell'adulto. Acta Neurol., 24: 578-580.
SCEP (1970) Study of critical environmental problems. Man's impact on
the global environment. Cambridge, Mass. London, England. Mit
Press.
SCHALLER, K. H., LINDNER, K., & LEHNERT, G. (1968)
Atomabsorptionsspektrometrische Schwermetallbestimmung im
Trinkwasser. Arch. Hyg. Bakteriol, 152: 298-301.
SCHIELE, R., SCHALLER, K. H., & VALENTIN, H. (1974a) The influence of
lead upon the tryptophan metabolism. Kiln. Wschr., 52: 401-404.
SCHIELE, R., SCHALLER, K. H., & WAGNER, H. M. (1974b) Die Bestimmung
der freien Erythrozytenporphyrine als schneller Suchtest einer
erhöhten Bleiexposition und seine Validität im vergleich zum
Blutbleispiegel und zur Delta-Aminolävulinsäure-Dehydratase-
Aktivität. SchrReihe Ven. Wasser-Baden. Lufthyg. Berlin,
41: 231-240.
SCHLAEPFER, W. W. (1969) Experimental lead neuropathy: A disease of
the supporting cells in the peripheral system. J. Neuropathol.
exp. Neurology, 28: 401-418.
SCHLEGEL, H., KUFNER, G., & LEINIBERGER, H. (1972) Die Praxis der
Verhütung yon Bleibeschädigungen in der metallverarbeitenden
Industrie. In: Kommission für Umweltge-faren des
Bundesgesundheitsamtes, Arbeitsgruppe Blei und Umwelt, Berlin,
pp. 67-69.
SCHLEGEL, H., KUFNER, G., & LEINBERGER, H. (1973) Das Verhalten
verschiedener Parameter der Hämsynthesestörung am Menschen bei
experimenteller Aufnahme anorganischer Bleiverbindungen. In:
Proceedings of the International Symposium; Environmental Health
Aspects of Lead, Amsterdam, 2-6 October 1972. Luxembourg,
Commission of the European Communities, pp. 569-579.
SCHLIPKÖTER, H. W., & POTT, F. (1973) Die pulmonale Resorption yon
Bleistaub. In: Proceedings of the International Symposium;
Environmental Health Aspects of Lead, Amsterdam, 2-6 October
1972. Luxembourg, Commission of the European Communities,
pp. 403-412.
SCHLIPKÖTER, H. W., GHELERTER, L., & OST, B. (1975) Untersuchungen zur
Kombination-swirkung von Zink und Blei. Zentralbl. Bakt. Hyg. L
Abt. Orig. B., 160: 130-138.
SCHLIPKÖTER, H. W., IDEL, H., STILLER-WINKLER, R., & KRAUSE, G. H. M.
(1977) Die Beeinflussung der Infektionsresistenz durch inhalierte
Noxen. Zentralbl. Bakt. Hyg., I. Abt. Orig. B. (in press).
SCHMID, E., BAUCHINGER, M., PIETRUCK, S., & HALL, O. (1972) Die
cytogenetische Wirkung yon Blei in menschlichen peripheren
Lymphocyten in vitro und in vivo. Mutat. Res. 16: 401-406.
SCHRAMEL, P. (1973) Determination of eight metals in the international
biological standard by flameless atomic-absorption spectrometry.
Analytica Chimica Acta., 67: 69-77.
SCHRAMEL, P. (1974) The application of peak integration in flameless
atomic-absorption spectrometry. Analytica Chimica Acta.,
72: 414-418.
SCHROEDER, H. A., & TIPTON, I. H. (1968) The human body burden of
lead. Arch. environ. 17: 965-978.
SCHROEDER, H. A., & BALASSA, J. J. (1961) Abnormal trace metals in
man: lead. J. chron. Dis., 14: 408-425.
SCHUTZ, A., DENCKER, I., & NORDEN, A. (1971) Bly i kosten-undersokning
med dubbelportionsteknik i Dalby. Svenska Lakeresallskapet,
Medicinska Rikstamman (sammanfattningar), p. 424.
SCHWARTZ, S., & WIKOFF, H. (1952) The relation of erythrocyte
coproporphyrin and protoporphyrin to erythropoiesis. J. Biol.
Chem., 194: 563-573.
SCHWARTZ, S., ZIEVE, L, & WATSON, C. J. (1951) An improved method for
the determination of urinary coproporphyrin and an evaluation of
factors influencing the analysis. J. Lab. Clin. Med.,
37: 843-859.
SCHWANITZ, O., GEBHART, E., ROTT, H. D., SCHALLER, K. H., ESSING, H.
G., LAUER, O., & PRESTELE, H. (1975) Chromosome investigations in
subjects with occupational lead exposure. Dtsch. Med. Wschr,
100: 1007-1011.
SCHWANITZ, G., LEHNERT, G., & GEBHART, E. (1970) Chromosomenschäden
bei beruflicher Bleibelastung. Dtsch. Med. Wschr.,
95: 1636-1641.
SECCHI, G. C., & ALESSIO, L. (1974) Laboratory results of some
biological measures in workers exposed to lead. Arch. environ.
Health, 29: 351-355.
SECCHI, G. C., ALESSIO, L., & CAMBIOGGHI, G. (1971) Ricerche
sull'attivita ALA-deidrasica eritrocitaria di soggetti non
esposti a contatto professionale con plombo ed abitanti in zone
rutall ed urbane. Med. Lav., 62: 435-450.
SECCHI, G. C., ALESSIO, L., & CAMBIOGGHI, G. (1973) Na/K-ATPase
activity of erythrocyte membranes. Arch. environ. Health,
28: 131-132.
SECCHI, G. C., ERBA, L., & CAMBIOGGHI, G. (1974) Delta-aminolevulinic
acid dehydratase activity of erythrocytes and liver tissue in
man. Arch. environ. Health, 28: 130-133.
SELANDER, S., & CRAMER, K. (1970) Interrelationships between lead in
blood, lead in urine and ALA in urine during lead work. Brit. J.
ind. Med., 27: 28-39.
SELYE, H., TUCHWEBER, B., & BERTOK, L. (1966) Effect of lead acetate
on the susceptibility of rats to bacterial endotoxins.
J. Bacteriol., 91: 884-890.
SEPPÄLÄINEN, H. M., & HERNBERG, S. (1972) Sensitive technique for
detecting subclinical lead neuropathy. Brit. J. ind. Med.,
29: 443-449.
SEPPÄLÄINEN, A.M., TOLA, S., HERNBERG, S., & KOCK, B. (1975)
Subclinical neuropathy at "safe" levels of lead exposure. Arch.
environ. Health, 30: 180-183.
SERVANT, J. (1973) Transport du plomb dans l'environnement. In:
Proceedings of the International Symposium; Environmental Health
Aspects of Lead, Amsterdam, 2-6 October 1972. Luxembourg,
Commission of the European Communities, pp. 155-165.
SHELDON, R. P., WARNER, M. A., THOMPSON, M. E., & PEIRCE, H. W. (1953)
Stratigraphic sections of the phosphoria formation in Idaho,
1949, Part I. U.S. geol. Survey eric., 304: pt. I, p. 1.
SHIRAISHI, Y. (1975) Cytogenetic studies of 12 patients with Itai-Itai
disease. Humangentik, 27: 31-44.
SHIRAISHI, Y., KURAHASHI, H., & YOSIDA, T. H. (1972) Chromosomal
aberrations in cultured human leucocytes induced by cadmium
sulfide. Proc. Jpn. Acad., 48: 133-137.
SILBERGELD, E. K., & GOLDBERG, A.M. (1974) Lead-induced behavioral
dysfunction: an animal model of hyperactivity. Exp. Neurol.,
42: 146-157.
SILBERGELD, E. K., FALES, J. T., & GOLDBERG, A.M. (1974) Evidence for
a functional effect of lead on neuromuscular function. Nature
(Lond.), 247: 49-50.
SILVER, W., & RODRIQUEZ-TORRES, R. (1968) Electrocardiographic studies
in children with lead poisoning. Pediatrics, 41: 1124-1127.
SIX, K. M., & GOYER, R. A. (1970) Experimental enhancement of lead
toxicity by low dietary calcium. J. Lab. Clin. Med.
76: 933-942.
SIX, K. M., & GOYER, R. A. (1972) The influence of iron deficiency on
tissue content and toxicity of ingested lead in the rat. J. Lab.
Clin. Med., 79: 128-136.
SMITH, H. D. (1964). Pediatric lead poisoning. Arch. environ. Health,
8: 256-261.
SNOWDON, C. T. (1973) Learning deficits in lead-injected rats.
Pharmacol. Biochem. Behavior, 1: 599-603.
SNYDER, L. J. (1967) Determination of trace amounts of organic lead in
air. Anal. Chem., 39: 591-595.
SOBEL, A. E., GAWRON, O., & KRAMER, B. (1938a) Influence of vitamin D
in experimental lead poisoning. Proc. Soc. Exp. Biol. Med.,
38: 433-435.
SOBEL, A. E., WEXLER, I. B., PETROVSKY, D. D., & KRAMER, B. (1938b)
Influence of dietary calcium and phosphorus upon action of
vitamin D in experimental lead poisoning. Proc. Soc. exp. Biol.
Med., 38: 435-437.
SOLIMAN, M., EL-SADIK, Y., & EL-WASSEF, A. (1973) Evaluation of some
parameters of lead exposure and possible correlation between
them. In: Proceedings of the International Symposium;
Environmental Health Aspects of lead, Amsterdam, 2-4 October
1972. Luxembourg, Commission of the European Communities,
pp. 531-543.
SOLOMINA, V. F. (1961) Effect of lead acetate and silica on the
development of experimental skin cancer. Isvest. Akad. Nauk
Kazakh. SSR. Set. Med. i Fiziol., 2(16): 55-67.
SPURGEON, J. O. (1973) Response characteristics of a portable X-ray
fluorescence lead detector; detection of lead in paint. Report to
the Department of Housing and Urban Development by the National
Bureau of Standards, N13 SIR, pp. 73-231.
SROCZYNSKI, J., ZAJUSZ, K., KOSSMANN, S., & WEGIEL, A. (1967) Effect
of experimental lead poisoning on the development of
arteriosclerosis. Pol. Arch. Med. Wewn., 38 (5): 641-646.
STAPLES, E. L. J. (1955) Experimental lead-poisoning in dogs. N.Z.
vet. J., 3: 39-46.
STOPPS, O. J. (1969) Discussion on epidemiological bases for possible
air quality criteria for lead. Air Pollut. Control Assoc.,
19: 719-721.
STOWE, H. D., & GOYER, R. A. (1971) The reproductive ability and
progeny of F, lead-toxic rats. Fertil. Steril., 22: 755-760.
STOWE, H. D., GOYER, R. A., & KRIGMAN, M. R. (1973) Experimental oral
lead toxicity in young dogs. Arch. Pathol., 95: 106-116.
STRAND, L. J., MANNING, J., & MARVER, H. S. (1972) The induction of
delta-aminolevulinic acid synthetase in cultured liver cells.
J. biol. Chem., 247: 2820-2824.
STUBBS, R. L. (1975) Lead and zinc in 1975, Mining Annual Review-1976,
pp. 45-49.
STUIK, E. J. (1974) Biological response of male and female volunteers
to inorganic lead. Int. Arch. Arbeitsmed., 33: 89-97.
STUIK, E. J., & ZIELHUIS, R. L. (1975) Increased susceptibility of
females to inorganic lead. In: Proceedings of CEC-EPA-WHO
International Symposium; Recent Advances in the Assessment of
the Health Effects of Environmental Pollution, Paris, 24-28 June
1974. Luxembourg, Commission of the European Communities,
pp. 537-545.
SUN, M. W., STEIN, E., & GRUEN, F. W. (1969) A single column method
for the determination of urinary delta-aminolevulinic acid.
Clin. Chem., 15: 183-189.
SWAINE, D. J. (1955) The trace-element content of soils.
Commonwealth Bur. Soil. Sci. Technol. Comm. No. 48.
SZADKOWSKI, D., SCHULTZE, H., SCHALLER, K. H., & LEHNERT, G. (1969)
Zur ökologischen Bedeutung des Schwermetallgehaltes von
Zigaretten. Arch. Hyg. Bakt., 153: 1-8.
TABERSHAW, J. R, & COOPER, W. C. (1974) Health study of lead workers.
A report prepared for the International Lead and Zinc Research
Organization.
TABERSHAW, J. R., RUOTOLO, B. P. W., & GLAESON, R. P. (1943) Plumbism
resulting from oxyacetylene cutting of painted structural steel.
J. ind. Hyg. Toxicol., 25: 189-191.
TASK GROUP ON LUNG DYNAMICS (1966) Deposition and retention models for
internal dosimetry of the human respiratory tract. Health Phys.,
12: 173-207.
TATSUMOTO, M., & PATTERSON, C. C., (1963) The concentration of common
lead in sea water. Earth Sci. Meteorics, 74-89.
TEISINGER, J. (1935) Biochemical reactions of lead in blood. Biochem.
Zeitschrift, 277: 3-4, 178-185.
TEISINGER, J., & SRBOVA, J. (1959) The value of mobilization of lead
by calcium ethylene-diamine-tetra-acetate in the diagnosis of
lead poisoning. Brit. J. ind. Med., 16: 148-152.
TEISINGER, J., & STYBLOVA, V. (1961) Neurological picture of chronic
lead poisoning. Acta Universitatis Carolinae-Med. Supp. 14,
199-206.
TEPPER, L. B. (1963) Renal function subsequent to childhood plumbism.
Arch. environ. Health, 7: 76-85.
TEPPER, L. B., & LEVIN, L. S. (1972) A survey of air and population
lead levels in selected American communities. Final report to the
US EPA.
TER HAAR, G. L. (1970) Air as a source of lead in edible crops.
Environ. Sci. Technol., 4: 226-229.
TER HAAR, G. L., & ARONOW, R. (1974) New information on lead in dirt
and dust as related to the childhood lead problem. Environ.
Health Perspect. Experim. Issue No. 7, 83-89.
HAAR, G. L., & BAYARD, M. A. (1971) Composition of airborne lead
particles. Nature (Lond.), 232: 553-554.
TER HAAR, G. L., HOLTZMAN, R. B., & LUCAS, H. F. (1967) Lead and lead-
210 in rainwater. Nature (Lond.), 216: 353-355.
THOMAS, J. A., DALLENBACH, F. D., & THOMAS, M. (1971) Considerations
on the development of experimental lead encephalopathy. Virchows
Arch. Abt. d. Path. Anat., 352: 61-74.
THOMPSON, J. A. (1971) Balance between intake and output of lead in
normal individuals. Brit. J. ind. Med., 28: 189-194.
THORNTON, I. & WEBB, J. S. (1975) Trace elements in soils and surface
waters contaminated by plast metaliferous mining in parts of
England. In: Proceedings of the IXth Annual Conference on
Trace Substances in Environmental Health, Columbia, MO, 10-12
June 1975, pp. 77-88.
TOLA, S. (1973) Effect of blood lead concentration, age, sex and
exposure time on the erythrocyte delta-aminolevulinic acid
dehydratase activity. Work environ. Health, 10: 26-35.
TOLA, S. (1974) Occupational lead exposure in Finland. III: Lead scrap
smelteries and scrap metal shops. Work Environ. Health,
11: 114-118.
TOLA, S., HERNBERG, S., AsP, S., & NIKKANEN, J. (1973) Parameters
indicative of absorption and biological effect in new lead
exposure: A prospective study. Brit. J. ind. Med., 30: 134-141.
TOLA, S., HERNBERG, S., NIKKANEN, J., & VALKONEN, S. (1971)
Occupational lead exposure in Finland: 1. Electric storage
battery manufacturing and repair. Work environ. Health,
3: 81-85.
TOMOKUNI, K., & OGATA, M. (1972) Simple method for determination of
urinary delta-aminolevulinic acid as an index of lead exposure.
Clin. Chem., 18: 1534-1536.
TRUHAUT, R., BOUDERIC, C., & ALBAHARY, C. (1964) Röle possible de la
consommation exagérée de vin dans l'etiologie du saturnisme.
Bull. WHO, 31: 127-129.
TSUCHIYA, K., & HARASHIMA, S. (1965) Lead exposure and the derivation
of maximum allowable concentrations and threshold limit values.
Brit. J. ind. Med., 22: 181-186.
TSUCHIYA, K., SUGITA, M., SEKI, Y., KOBAYASHI, Y., HORI, M., & BIN
PARK, C. (1975) Study of lead concentrations in atmosphere and
population in Japan. In: Coulston et al., ed., Environmental
quality and safety. Supplement Vol. II, Lead. Stuttgart, Georg
Thieme and NY, Academic Press, pp. 95-146.
TUREKIAN, N. K., & WEDEPOHL, K. H. (1961) Distribution of the elements
in some major units of the earth's crust. Geol. Soc. Am. Bull,
72: 175-191.
UNITED NATIONS, STATISTICAL OFFICE (1975) Statistical Yearbook 1974,
Twenty-sixth issue, United Nations Department of Economic and
Social Affairs, Statistical Office, New York, p. 190.
URATA, G., & GRANICK, S. (1963) Biosynthesis of alpha-aminoketones and
the metabolism of aminoacetone. J. Biol. Chem., 238: 811-820.
URBANOWICZ, H. (1971) Occupational exposure to inorganic compounds of
lead. Arch. environ. Health, 23: 284-288.
URBANOWICZ, H., GRABECKI, J., & KOZIELSKA, J. (1969) The urinary
excretion of 5-hydroxyindoleacetic acid in industrial lead
exposure. Med. Lav., 60: 582-586.
US BUREAU OF MINES (1969) Minerals Yearbook 1968, vol. I-II; Metals,
minerals, and fuels. Washington, pp. 631-660.
US DHEW (1965) Survey of lead in the atmosphere of three urban
communities. USPHS Publ. No. 999 AP 12.
US ENVIRONMENTAL PROTECTION AGENCY OFFICE OF RESEARCH AND MONITORING
(1972) Water pollution aspects of street surface contaminants.
Washington DC (EPA-R2-72-081).
VAN ESCH, G. J., & KROES, R. (1969) The induction of renal tumours by
feeding basic lead acetate to mice and hamsters. Brit. J.
Cancer, 23: 765-771.
VAN ESCH, G. J., VAN GENDEVEN, H., & VINK, H. H. (1962) The induction
of renal tumours by feeding of basic lead acetate to rats. Brit.
J. Cancer, 16: 289-297.
VERBOLOVIC, V. P. (1965) Peculiarities of cytochrome oxidase activity
and of the localization of myoglobin during lead poisoning.
T. Kazahsk. Inst. Kraevoi Pat. Akad. bled. Nauk SSSR,
14: 74-80.
VIGDORTCHIK, N. A. (1935) Lead intoxication in the etiology of
hypertonia. J. ind. Hyg., 17(1): 1-6.
VINOGRADOV, A. P. (1956) Regularity of distribution of chemical
elements in the earth's crust. Geohimija, p. 6 (English
translation in Geochemistry, 1956, pp. 1-43).
VINOGRADOV, A. P. (1962) Average contents of chemical elements in the
principal types of igneous rocks of the earth's crust.
Geohimija, p. 555 (English translation in Geochemistry, 1962,
pp. 641-664).
VOSTAL, J. (1966) Study of the renal excretory mechanisms of heavy
metals. 15th International Congress Occupational Health,
Vienna, 19-24 September, v. 3., pp. 61-64.
WALDRON, H. A. (1966) The anemia of lead poisoning. A review. Brit.
J. ind. Med., 23: 82-100.
WALDRON, H. A. (1975) Lead levels in blood of residents near the
M6-A38(M) interchange, Birmingham. Nature (Lond.),
253: 345-346.
WARLEY, M. A., BLACKLEDGE, P., & O'GORMAN, P. (1968) Lead poisoning
from eye cosmetics. Brit. med. J., 1: 117.
WARREN, H. V., & DELAVAULT, R. E. (1962) Lead in some food crops and
trees. J. Sci. Food Agric., 13: 96-98.
WEAST, R. C., ed. (1974) Handbook of Chemistry and Physics, 55th
ed., Cleveland, CRC Press, pp. B-100-B-101.
WEBB, M. (1972) Binding of cadmium ions by rat liver and kidney.
Biochem. Pharmacol., 21: 2751-2765.
WEBER, H. J. (1947) Some experiences with polarographic methods in
controlling a lead hazard in brass foundries. J. ind. Hyg.
Toxicol., 28: 158-167.
WEDEPOHL, K. H. (1956) Untersuchungen zur Geochemie des Bleis.
Geochim. Acta, 10: 69-148.
WEDEPOHL, K. H. (1971) Zinc and lead in common sedimentary rocks.
Econ. Geol., 66: 240-242.
WELLER, C. V. (1915) The blastopthoric effect of chronic lead
poisoning. J. med. Res., 3: 271-293.
WHITE, J. M., & HARVEY, D. R. (1972) Defective synthesis of alpha- and
ß-globin chains in lead poisoning. Nature (Lond.), 236: 71-73.
WHITEHEAD, T. P., & PRIOR, A. P. (1960) Lead poisoning from homemade
wine. Lancet, 2: 1343-1344.
WHITFIELD, C. L., CHIEN, L. T., & WHITEHEAD, J. D. (1972) Lead
encephalopathy in adults. Am. d. Med., 52: 289-298.
WHO EXPERT COMMITTEE (1969) Urban air pollution with particular
reference to motor vehicles. WHO Technical Report Series, No.
410, p. 19.
WHO EXPERT COMMITTEE ON FOOD ADDITIVES (1972) Lead. Sixteenth Report,
pp. 16-20.
WHO EXPERT COMMITTEE (1973) Trace elements in human nutrition.
WHO Technical Report Series No. 532, p. 47.
WHO STUDY GROUP (1975) Early detection of health impairment in
occupational exposure to health hazards. WHO Technical Report
Series No. 571, pp. 55-61.
WHO WORKING GROUP (1973) Technical document on lead. In: The hazards
to health of persistent substances in water. Annexes to the
Report of a WHO Working Group, Copenhagen, WHO Regional Office
for Europe, pp. 61-110 (Document EURO 3109 w(1)).
WILLIAMS, H., SCHULZE, W. H., ROTHCHILD, H. B., BROWN, A. S. & SMITH,
F. R. (1933) Lead poisoning from the burning of battery casings.
J. Am. Med. Assoc., 100: 1485-1489.
WILLIAMS, U. K. (1966) Blood lead and haemoglobin in lead absorption.
Brit. J. ind. Med., 23: 105-111.
WILLIAMS, M. K, & FEW, J. D. (1967) A simplified procedure for the
determination of urinary delta-aminoloevulinic acid. Brit.
J. ind., Med., 24: 294-296.
WILLIAMS, M. K., KING, E., & WALFORD, J. (1969) An investigation of
lead absorption in an electric accumulator factory with the use
of personal samplers. Brit. J. ind. Med., 26: 202-216.
WILLOUGHBY, R. A., MACDONALD, E., MCSHERRY, B. J., & BROWN, G. (1972)
The interaction of toxic amounts of lead and zinc fed to young
growing horses. Vet. Rec., 91: 382-383.
WILSON, J. G. (1973) Environment and birth defects. New York,
Academic Press, p. 78.
WONG, C. S., & BERRANG, P. (1976) Contamination of tap water by lead
pipe and solder. Bull. environm. Contain. Toxicol.,
15: 530-534.
WONG, P. T. S., CHATS, Y. K., & LUXON, P. L. (1975) Methylation of
lead in the environment. Nature (Lond.), 253: 263-264.
WRANNE, L., (1960) Free erythrocytes copro- and protoporphyrin: A
methodological and clinical study. Acta. Paediatrica., 49
(suppl. 124).
YAMAMOTO, Y., TANAKA, M., & ARAO, K. (1968) Hemispherical distribution
of turbidity coefficient as estimated from direct solar radiation
measurements. J. Meteorol. Soc. Jpn, 46: 287-300.
YEAGER, D. W., CHOLAK, J., & HENDERSON, E. W. (197 l) Determination of
lead in biological and related material by atomic absorption
spectrophotometry. Environ. Sci. Technol., 5: 1020-1022.
YOCOM, J. E., CLINK, W. L., & COTE, W. A. (1971) Indoor/outdoor air
quality relationships. J. Air Pollut. Control Assoc.,
21: 251-259.
ZAWIRSKA, B., & MEDRAS, K. (1968) Tumoren und Störungen des
Porphyrinstoffwechsels bei Ratten Tit chronischer experimenteller
Bleiintoxikation. Zentralbl. Allg. Path., 111(1): 1-12.
ZEL'TSER, M. E. (1962) The functional state of thyroid gland in lead
poisoning. Tr. Just. kraevoi Patol. Akad. Nauk Kaz. SSR,
10: 116-120.
ZIEGFIELD, R. L. (1964) Importance and uses of lead. Arch. environ.
Health, 8: 202-212.
ZIELHUIS, R. L. (1971) Interrelationship of biochemical responses to
the absorption of inorganic lead. Arch. environ. Health,
23: 299-311.
ZIELHUIS, R. L. (1974) Dose response relationship for inorganic lead.
Report to the Director, Health Protection EEC.
ZIELHUIS, R. L. (1975) Dose response relationships for inorganic lead.
I. Biochemical and haematological responses. II. Subjective and
functional responses. Chronic sequelae. No-response levels.
Int. Arch. occup. Health, 35: 1-18, 19-35.
ZOLLINGER, H. U. (1953) Durch chronische Bleivergiftung erzeugter
Nierenadenome und Carcinome bei Ratten und ihre Beziehungen zu
den entsprechenden Neubildungen des Menschen. Virchows Arch.
Bd., 323: 694-710.
ZOOK, B. C., CARPENTER, J. L., & LEEDS, E. B. (1969) Lead poisoning in
dogs. J. Am. vet. Med. Assoc., 155: 1329-1342.
ZUKOVICKAJA, A. L., ZAMUATKINA A. A., & LUKASEV, K. I. (1966) Trace
elements in the water of the upper Dnepr river. Dokl. Akad. Nauk
BSSR, 10: 891-893.
ZURLO, N., & GRIFFINI, A. M. (1973) Le plomb dans les aliments et
dans les boissons consommes à Milan. In: Proceedings of the
International Symposium; Environmental Health Aspects of Lead,
Amsterdam, 2-6 October 1972. Luxembourg, Commission of the
European Communities, pp. 93-99.
ZURLO, N., GRIFFINI, H. M., & VIGLIANI, E. C. (1970) The content of
lead in blood and urine of adults, living in Milan, not
occupationally exposed to lead. Am. ind. Hyg. Assoc. J.
31: 92-95.
Table 28. Percentage of male adults with ALA-U
levels > 5 mg/litre and >10 mg/litre
according to Pb-B level
Pb-B level No. ALA-U level (mg/litre)
(pg/100 ml)
>5 >10
11-20 17 0 0
21-30 27 0 0
31-40 36 14 3
41-50 55 33 11
51-60 38 74 37
61-70 34 88 50
207
a From: Zielhuis, 1975.
8.2.1.5 Effects of lead on cell morphology
Punctate basophilia occurs in lead poisoning, but a quantitative
relationship between the number of stippled cells and Pb-B levels is
not to be expected (Zielhuis, 1971). Too many variables are involved
in the preparation of smears. The same is probably true of
reticulocyte counts.
8.2.1.6 Effects of lead on erythrocyte survival
Increased rate of erythrocyte breakdown (decreased erythrocyte
life) is often, but not consistently, seen in cases of anaemia due to
lead poisoning. When erythrocytes are exposed to lead in vitro, they
exhibit increased osmotic resistance and increased mechanical
fragility (Waldron, 1966). They also show inhibition of Na-K-ATPase
with increased loss of intracellular potassium (Hasan & Hernberg,
1966; Secchi et al., 1973). These effects have been cited to explain
the fact that in many instances the anaemia in lead poisoning is
accompanied by a shortening of the erythrocyte life span. It is
presumed that one or more of these effects is responsible for the
sensitivity of erythrocytes to spontaneous haemolysis. Erythrocyte
survival time was reduced on the average by 20% in 17 occupationally-
exposed workers, only 3 of whom showed clinical signs of poisoning
(Hernberg, 1967). The author postulated that shortened cell life was
due to the loss of membrane integrity secondary to Na-K-ATPase
inhibition. Anaemia does not necessarily accompany a shortened red
cell life span, and the correlation between blood haemoglobin and life
span was not good in this particular study. The kinetics of
disappearance of labelled cells indicated a shortening of life span by
increased random destruction of cells of all ages. Leikin & Eng (1963)
determined erythrocyte survival in 7 cases of lead poisoning in
children. In 3 cases the erythrocyte survival time was shortened. All
patients were mildly to moderately anaemic. It would seem from these
and other studies that the anaemia in lead poisoning cannot be
explained solely on the basis of reduced erythrocyte survival time.
8.2.1.7 Effects of lead on haem synthesis
The two general points of attack that have been identified are on
haem synthesis and on globin synthesis. Of the two, the effects on
haem synthesis are better understood. It is generally recognized, too,
that manifestations of disturbed haem synthesis often occur in the
absence of frank anaemia. These disturbances may also be significant
for the numerous other haem-dependent enzymatic reactions essential
for normal body functions. Thus, cytochromes, cytochrome c oxidase
(EC 1.9.3.1), and hydroperoxidases are all part of electron transfer
systems requiring haem.
Little is known about the effects of lead on the formation or
activity of other haem-containing compounds. It has been reported that
treatment with EDTA reversed the prolonged antipyrine half-life seen
in two cases of clinical lead poisoning (Alvares et al., 1975). The
authors suggested that in these cases, lead may have significantly
inhibited the synthesis of cytochrome P-450.
8.2.1.7 Relationship between lead exposure and anaemia
It is well known that anaemia is a characteristic early toxic
effect of lead in man. The Pb-B threshold level for this effect is
still not certain. Williams (1966) reported that anaemia did not occur
in industrial workers with Pb-B levels below 110 µg/100 ml. Cooper et
al. (1973) reported that the average haemoglobin level (Hb) was not
decreased at Pb-B levels of up to 100 µg/100 ml and Sakurai et al.
(1974) did not observe any decrease of Hb or erythrocyte
concentrations in workers at Pb-B levels of up to 50 µg/100 ml. On the
other hand, Tola et al. (1973) reported a slight effect of lead on Hb
at an average Pb-B level of about 50 µg/100 ml. This conclusion was
drawn from analysis of the sequential change in Hb among workers newly
introduced into an "industrial lead environment". This approach to the
analysis of the effect of lead on Hb is certainly more sensitive for
detecting an interaction between Pb-B levels and Hb than is a single
Hb determination in a population of lead-exposed persons. Allowance
must, however, be made for the possibility that sequential change in
Hb may be due to seasonal effects independent of lead exposure
(Coulthard, 1958).
Children appear to be more sensitive to lead anaemia than adults.
Thus, Betts et al. (1973) found a significant negative correlation
between Hb and Pb-B levels; a decrease in Hb was evident in 36% of
children with Pb-B levels from 37 to 60 µg/100 ml, compared with only
14% in children with Pb-B levels less than 37 µg/100 ml. Pueschel et
al. (1972) observed a curvilinear decrease in Hb between Pb-B levels
of 40 and 130 µg/100 ml in children between 1 and 6 years old. On the
other hand, McNeil & Ptaznik (1975) found no anaemia in children with
Pb-B levels considerably higher than 40 µg/100 ml. Nutritional
differences may explain the discrepancy. But this does not invalidate
the proposition that for some groups of children a reduction in Hb may
occur at a Pb-B level of approximately 40 µg/100 ml.
8.2.2 Nervous system
8.2.2.1 Central nervous system
Inorganic lead compounds. The effects of lead on the nervous system
vary with the duration and intensity of exposure. Distinction must
also be made between the effects on the central nervous system and the
effects on peripheral nerves. Further questions have been raised
concerning the inherent differences in the sensitivity of the nervous
system of adults and the nervous system of infants and young children.
There is no doubt that lead effects on the brain are much more
commonly associated with childhood lead poisoning than with poisoning
as it is seen in adults. But it is also possible that these
differences are related to the intensity of exposure at the time the
cases are identified rather than to any difference in inherent
sensitivity.
With chronic lead exposure, striking effects may occur referred
to as lead encephalopathy. There are numerous detailed descriptions of
adult lead encephalopathy (Crutcher, 1963; Whitfield et al., 1972;
Teisinger & Styblova, 1961; Aub et al., 1926; Cantarow & Trumpet,
1944). The major features are dullness, restlessness, irritability,
headaches, muscular tremor, hallucinations, and loss of memory and
ability to concentrate. These signs and symptoms may progress to
delirium, mania, convulsions, paralysis, and coma. The signs and
symptoms of encephalopathy in infants and young children are quite
similar to those reported to occur in adults.
The brain lesions in fatal cases of lead poisoning are cerebral
oedema and changes in cerebral blood vessels. The normal convolutions
of the cerebral hemispheres are often obliterated. Capillary
endothelial cells are usually swollen (Pentschew, 1965). Extravasation
of red blood cells and perivascular haemorrhage occur rather commonly
and patchy neuronal loss, serous exudate, glial proliferation, and
occasional areas of demyelinization are all characteristic of lead
poisoning (Blackman, 1937; Okazaki et al., 1963; Whitfield et al.,
1972). But not all deaths due to lead encephalopathy are accompanied
by histological lesions of the central nervous system (Pentschew,
1965).
Neurological sequelae can occur in severe or repeated episodes of
lead encephalopathy. The sequelae are no different qualitatively from
those that occur following traumatic or infectious cerebral injury.
The occurrence of permanent sequelae seems to be much more common
among young children than among adults. Approximately one-fourth of
the children who survived an attack of acute lead encephalopathy
sustained permanent sequelae (Byers, 1959; Chisolm & Harrison, 1956;
Smith, 1964). At least this was true prior to the introduction of
current therapeutic practices such as those described by Chisolm
(1968a). The incidence of sequelae appears to have been substantially
reduced in recent years, but central nervous system sequelae may still
occur if therapy is initiated only after the onset of encephalopathy
(Chisolm, 1973). The most severe sequelae are cortical atrophy,
hydrocephalus, convulsive seizures, and idiocy. More commonly, the
sequelae are of a more subtle nature. Learning ability may be impaired
due to motor incoordination, lack of sensory perception, or inability
to concentrate. Such subtle disturbances have also been claimed to
occur in children with high lead exposure, but in the absence of a
history of encephalopathy (Byers & Lord, 1943; Cohen & Ahrens, 1959).
The major concern today is that young children with elevated lead
exposure, as reflected in Pb-B levels of 40-80 µg/100 ml, may be
experiencing subtle neurological damage without ever exhibiting
classical signs of lead encephalopathy. Studies have been reported of
the neurological status of children with Pb-B values in this range. In
view of the possible long-term effects of lead on the brain,
association between Pb-B and neurological status at the time of
evaluation may give a false impression concerning the level of lead
exposure when the damage was initiated. Exposure levels at the time of
examination may be lower than at the time toxic effects occurred.
Thus, the Pb-B level-effect association may underestimate the dose
responsible for the effect.
Burdé de la et al. (1972) and Peuschel et al. (1972) observed
dysfunction of the central nervous system (irritability, clumsiness,
fine motor dysfunction, impaired concept formation, etc.) in 70 and 58
children, respectively, whose Pb-B levels were always, in all cases,
above 40 µg/100 ml. Albert et al. (1974) studied the psychological
profiles and educational performances of children, 5-15 years of age,
who had histories of lead exposure early in childhood. Those who had
been treated for lead poisoning, with or without encephalopathy,
exhibited a higher incidence of diagnosed mental disorders and of poor
school performance than those who had no such history, even when their
history showed elevated lead exposure early in childhood.
Kotok (1972) established that development deficiencies (using the
Denver Development Screening test, which, according to the author is a
somewhat insensitive measure of development) in a group of
asymptomatic children with elevated lead levels (58-137 µg/100 ml)
were identical to those of a control group similar in age, sex, ethnic
group, environment, neonatal condition, and presence of pica, but
whose Pb-B levels were lower (20-55 µg/100 ml). The deficiencies could
be correlated with inadequacies in the children's environment. Klein
et al. (1974) pointed out that in many studies, pica is not used as a
controlled variable. In his view, pica may be part of a behavioural
deficiency syndrome. In such a case the child would have the
behavioural deficiency regardless of whether or not he ingested lead-
containing objects. Indeed, there is evidence that among mentally
subnormal children whose mental deficiency is unrelated to excessive
lead absorption there is a high incidence of both pica and of
moderately elevated Pb-B levels (Bicknell et al., 1968). In this
study, 67% of the children, whose subnormal state antedated pica, had
Pb-B levels from 39 to 88 µg/100 ml, with a mean of 48 µg/100 ml. By
contrast, among the subnormal group without pica all but one had a
Pb-B level of less than 36 µg/100 ml. The study did not exclude the
possibility that an excessive lead exposure could have aggravated the
pre-existent subnormal state.
Recently McNeil & Ptasnik (1975) published an initial evaluation
of the long-term effects of elevated Pb-B levels in asymptomatic
children, living in El Paso, USA. In 138 out of 206 children aged from
21 months to 18 years (median 9 years), who volunteered (possibility
of selection) to participate, the authors could not find any evidence
of non-specific complaints, hyperactivity, or of abnormal psychometric
testing values, if compared with a matched control group. There
existed a significant difference in one personality test; however this
was explained by geographic isolation and other factors and not by
lead exposure. The average Pb-B levels were, respectively,
50 µg/100 ml (range 14-93) and 16 µg/100 ml (range 10-28).
More recently another psychological evaluation of the El Paso
subjects was published by Landrigan et al. (1975a). Forty-six
children, aged from 3 to 15 years, with Pb-B levels of 40-60 µg/100 ml
were compared with 78 ethnically and socioeconomically similar
controls with Pb-B levels below 40 µg/100 ml. The "Wechsler
Intelligence Scale" showed that the age adjusted I.Q. was
significantly lower in the first group. In addition, the lead exposed
group also showed a significant slowing in the finger-wrist tapping
test. The full-scale I.Q., verbal I.Q., and the behavioural and
hyperactivity ratings did not differ. In this study, unfortunately,
there were differences in age and sex between the study and control
group which might account for the positive findings. It seems
therefore that we have two studies of this situation that come to
different conclusions regarding the possible effects of lead on
neurological and psychological functions.
Another approach has been to identify children with neurological
or behavioural disorders of obscure etiology and to determine whether
they show evidence of current or past elevated lead exposure (David et
al., 1972; Moncrieff et al., 1964; Gibson et al., 1967).
The work of David et al. (1972) is of particular interest because
the neurological abnormality described was one that was reproduced
experimentally in animals (see section 7.1.2). These workers reported
occurrence of hyperactivity among children who had essentially normal
blood lead concentrations, but who excreted abnormally large amounts
of lead when treated with penicillamine. The children had no history
of earlier lead encephalopathy. This study has been criticized because
of statistical inadequacies (Bullpitt, 1972).
Lansdown et al. (1974) examined a population of schoolchildren in
London (less than 17 years of age); there was no relationship between
Pb-B levels and intelligence (Wechsler test), reading (Butt test), and
behaviour (e.g. hyperactivity as rated by the teachers). The authors
suggested that social factors were more important than exposure to
lead in determining mental development. The design of the study has
also been criticized. Neither Landsown's nor David's study are
conclusive.
Morgan & Repko (1974) reported preliminary results of an
extensive study of behavioural functions in 190 lead-exposed workers
(Pb-B = 60.48 ± 16.96 µg/100 ml). In 68% of the subjects the Pb-B
level was less than 80 µg/100 ml. The majority of the subjects were
exposed for between 5 and 20 years. The authors examined 36 non-
independent measures of general performance. In addition, 44 measures
of sensory, psychomotor, and psychological functions were obtained.
Preliminary analysis suggested that Pb-B levels correlated with
several reaction-time measures and ALAD correlated with measures from
strength-endurance-recovery tasks. Both Pb-B levels and ALAD
correlated with eye-hand co-ordination. This study, therefore,
suggested that below a Pb-B level of 80 µg/100 ml some behavioural
changes did occur in adult workers. In addition, variability of
performance increased with increasing Pb-B levels. Only during periods
of high-demand performance did a worker's capacity decrease due to
lead exposure. The authors themselves stressed that this preliminary
analysis still has to be confirmed by further work.
Alkyllead compounds. The encephalopathy of alkyllead intoxication is
somewhat different from that due to inorganic lead exposure. In
documented adult cases of poisoning the most frequent findings suggest
a psychiatric problem. Hallucinations, tremor, delirium, insomnia,
delusions, headaches, and violent mood swings are the most commonly
reported symptoms (Boyd et al., 1957; Machie, 1935). The course of the
intoxication runs from 1 to 10 weeks. Although alkyllead compounds are
notorious for their high lethality, recovery is fairly complete among
survivors (Akatsuka, 1973). Convulsions and coma apparently occur only
in the most severe cases. There is insufficient information to
establish dose-effect and dose-response relationships.
8.2.2.2 Peripheral nervous system
Inorganic lead has toxic effects on the peripheral nervous
system. The older lead literature cites the frequent occurrence of
lead palsy in occupational exposure to lead. The manifestations are
mainly weakness of the extensor muscles, particularly those used most
heavily. While motor function is mainly affected, hyperaesthesia,
analgesia, and anaesthesia of affected areas have also been reported.
Catton et al. (1970) found evidence of reduced nerve conduction
velocity in about one-third of a group of 19 occupationally-exposed
men of whom only one showed any other overt signs of lead toxicity.
The most prominent finding of Seppalainen & Hernberg (1972) in
lead workers (Pb-B levels 80-120 µg/100 ml) without any clinical
neurological signs was reduced motor conduction velocity of the slower
fibres of the ulnar nerves; electromyographic changes included a
diminished number of motor units on maximum contraction and
fibrillations. Similar although less pronounced effects were reported
by Seppäläinen et al. (1975) in 26 workers whose Pb-B levels had never
exceeded 70 µg/100 ml (exposure time 13 months-17 years). Furthermore,
in lead workers with Pb-B levels of 2-273 µg/100 ml, Araki & Honma
(1976) reported statistically significant negative correlations
between nerve conduction velocity and Pb-B, ALAD, and lead
mobilization test values, respectively. More recently, Seppäläinen et
al. (unpublished resultsa) reported a dose-response relationship
between abnormally low conduction velocities, defined as values 2
standard deviations below the mean of an unexposed reference group,
and the highest Pb-B recorded during employment (2-20 years). The
results indicate that nerve conduction impairment is induced in some
workers at Pb-B's exceeding 50 µg/100 ml.
8.2.3 Renal system
The effects of lead on the kidney have been studied extensively.
Two general types of effect have been described. The first is rather
clear-cut renal tubular damage characterized by generalized
aminoaciduria, hypophosphataemia with relative hyperphosphaturia, and
a Reported at the Second International Workshop Permissible Levels
for Occupational Exposure to Inorganic Lead, 21-23 September 1976.
University of Amsterdam, The Netherlands. To be published shortly
in Int. Arch. Occup. Health.
glycosuria, which has been studied in some detail in children with
clinical lead poisoning (Chisolm, 1962). The condition is
characterized by decreased tubular reabsorption of glucose and
alpha-amino acids and therefore reflects proximal tubular damage.
Aminoaciduria was seen more consistently in Chisolm's studies than the
other two manifestations of tubular damage. Thus, the amino acid
transport system is probably more sensitive to the toxic actions of
lead than the transport systems for glucose and phosphate. Limited
data indicate that aminoaciduria is terminated by chelation (Chisolm,
1968b).
In a group of children with slight lead-related neurological
signs, generalized aminoaciduria was found in 8/43 children with Pb-B
levels of 40-120 µg/100 ml (Pueschel et al., 1972). A similar renal
tubular syndrome has been reported to occur in industrially exposed
adults (Clarkson & Kench, 1956; Goyer et al., 1972). In neither of
these studies were Pb-B levels reported. However, Clarkson & Kench
observed signs of lead poisoning (colic and punctate basophilia) in
conjunction with aminoaciduria.
In a group of 7 carefully studied lead-exposed workers,
aminoaciduria was not present. Inulin clearance and renal blood flow
were also normal at the time of examination. For these cases, the
average Pb-B level was 100 µg/100 ml and the minimum was 71 µg/100 ml.
These workers had been exposed for up to 20 years (Cramer et al.,
1974). All had markedly elevated urinary ALA excretion. Interestingly,
some of these workers with prolonged exposure had diffuse interstitial
and peritubular fibrosis as determined by renal biopsy. These
pathological findings are associated with quite a different kind of
renal effect which is seen with prolonged lead exposure. It is
commonly referred to as chronic lead nephropathy. Chronic nephropathy
is characterized by slow development of contracted kidneys with
arteriosclerotic changes, interstitial fibrosis, glomerular atrophy,
and hyaline degeneration of the vessels. This progressive disease
sometimes ends in renal failure. There is evidence that it occurs in
industrially exposed workers, in long-term drinkers of lead-
contaminated whisky, and among middle-aged people who had developed
clinical lead poisoning much earlier in life. Currently, it is only
rarely encountered in occupational exposure.
This renal syndrome can develop and progress to renal failure
long after abnormal lead exposure has terminated. As early as 1897, it
was noted that deaths from chronic nephritis were much more frequent
among people under 30 years of age in Queensland than in other
sections of Australia. The first serious attempt to document a
suspected relationship to earlier childhood lead poisoning was
reported by Nye (1929). Further evidence of a causal relationship
between chronic nephropathy and childhood lead exposure was provided
later (Henderson, 1958). It was shown that people dying of chronic
nephropathy in Queensland usually had a high concentration of lead in
their bones (Henderson & Inglis, 1957). Emmerson (1963) later
demonstrated abnormally elevated lead excretion in response to EDTA
among surviving middle-aged cases of chronic nephropathy. Tepper
(1963), however, was unable to find evidence of chronic nephropathy
among young American adults with a history of childhood lead
poisoning. The Americans had probably been exposed for a much shorter
period of time than the Australians. Other unknown factors may also
have played a role.
The Australian cases involved childhood exposure with an apparent
latency of 10-30 years for the development of renal insufficiency. But
there is evidence that the same effect can result from continuous,
prolonged high lead exposure among adults (Lilis et al., 1968; Richet
et al., 1966; Danilovic, 1958; Morgan et al., 1966; Albahary et al.,
1965; Albahary, 1964). In these cases, lead exposure was higher than
is commonly encountered in industry today.
In a series of 102 cases of lead poisoning studied by Lilis et
al. (1968), 18 cases of clinically verified chronic nephropathy were
found. For the whole series, the mean Pb-B level was approximately
80 µg/100 ml with a range of 42-141 µg/100 ml. Nephropathy was more
common among patients who had been exposed to lead for more than 10
years than among those who had been exposed for less than 10 years.
In the Danilovic (1958) study 7/23 cases had Pb-B levels of about
100-200 µg/100 ml. In the studies of Albahary et al. (1965) Pb-B
levels were not reported. But exposure levels must have been quite
high since the mean ALA excretion was about 37 mg/24 h for 29 workers.
It seems likely, from all available evidence, that a prolonged
high-level lead exposure is necessary, even in childhood, to produce
this progressive chronic nephropathy.
One interesting feature of this syndrome of chronic renal
insufficiency is the frequent association with gout (Emmerson, 1963;
Morgan et al., 1966). Although uric acid excretion is largely
dependent upon tubular secretion, it is not at all certain that
tubular secretion is inhibited. As a matter of fact, a study by
Emmerson et al. (1971) of 13 cases of renal insufficiency due to lead
nephropathy failed to reveal any alteration in uric acid secretion.
The authors suggested an increased tubular reabsorption to account for
the observed decreased clearance of uric acid.
In summary, proximal tubular effects can occur in children and
adults with subtle signs of lead poisoning.
Prolonged exposure to lead leading to a Pb-B level of more than
70 µg/100 ml may give rise to chronic irreversible nephropathy.
However, little is known about dose-effect relationships or about
time-effect relationships for lead-induced chronic interstitial
nephritis.
8.2.4 Gastrointestinal tract
As a symptom of lead poisoning, colic is a fairly consistent
early warning of potentially more serious effects likely to occur with
prolonged periods of exposure. It is most commonly encountered in
industrial exposure. But it is probably also common in lead-poisoned
infants and young children. The occurrence of colic at relatively low
exposure levels in industry is well-known. Although it has been
reported that 13/64 industrially exposed men with presumably lead-
related colic and constipation had blood lead levels from somewhat
less than 40 µg to 80 µg/100 ml (Beritic, 1971), it was also reported
that in every case the diagnosis of lead colic was confirmed by the
findings of high CP-U, excessive basophilic stippling,
reticulocytosis, and various degrees of anaemia. This is consistent
with the general observation that lead colic seems to be accompanied
by other signs of poisoning. There are not enough data available to
establish a dose-response relationship for this lead effect.
8.2.5 Liver
There is no definite evidence for the effects of lead on the
liver. Dodic et al. (1971) reported signs of impaired liver function
in 11 out of 91 patients hospitalized for lead poisoning. Liver damage
was more frequent in cases of severe lead poisoning in 7 out of 18
patients. However, the authors did not provide any information on Pb-B
levels or on indices of disturbed porphyrin metabolism which would
enable the assessment of the stage of lead poisoning. In a laboratory
study of 301 workers in lead smelting and refining, Cooper et al.
(1973) found 11.5% increased aspartate aminotransferase (EC 2.6.1.1),
(SGOT)a values (above 50 U/litreb) in subjects with a Pb-B level
below 70 µg/100 ml, 20% in those with a Pb-B level of about
70 µg/100 ml, and 50% in workers with a Pb-B level above
100 µg/100 ml. The correlation between Pb-B levels and SGOT values was
statistically significant. However, in the absence of information on
the possible influence of diet, infections, or personal habits, the
authors did not draw any definite conclusions concerning the etiology
of these changes.
a Formerly known as serum glutamic oxaloacetic transaminase.
b = 50 × 1.67 × 10-5 mol/(m3.s)
8.2.6 Cardiovascular system
Increased capillary permeability occurs in acute lead
encephalopathy (section 8.2.2.1). Under conditions of long-term lead
exposure at high levels, arteriosclerotic changes have been
demonstrated in the kidney (section 8.2.3). Dingwell-Fordyce & Lane
(1963) reported a marked increase in the cerebrovascular mortality
rate as compared with the expected rate among heavily exposed lead
workers (section 8.1.1). This observation applied to men exposed to
lead during the first quarter of this century, when working conditions
were quite bad. There was no similar increase in the mortality rate
for men employed more recently. Hypertension is an important element
in the etiology of cerebrovascular deaths. Cramer & Dahlberg (1966)
studied the incidence of hypertension in a population of 364
industrially-exposed men, 273 of whom had a long-term exposure to
lead. They subdivided these workers into "lead affected" and "non-
lead-affected" groups, on the basis of the urinary coproporphyrin
test. There was no statistically significant difference between the
groups. Nor was the incidence higher than expected for non-exposed men
in Sweden. This is contrary to the earlier findings of Vigdortchik
(1935) and to the observations of Monaenkova & Glotova (1969). The
disparity may have been due to differences in lead exposure. Other
reports on the question do not show hypertension to be unduly
prevalent among workers exposed to lead (Dressen et al., 1941; Lane,
1949). It is not clear whether vascular effects of lead in man are the
result of an action on blood vessels directly, or whether the effects
are secondary to renal effects.
There is a good evidence that signs of clinical lead poisoning
sometimes include evidence of a toxic action on the heart. Cases have
been described in adults and in children, always with clinical signs
of poisoning. There is of course the possibility that the coexistence
of lead poisoning and myocarditis is coincidental. But in many cases
the electrocardiographic abnormalities disappeared with chelation
therapy, suggesting that lead may have been the original etiological
factor (Myerson & Eisenhauer, 1963; Silver & Rodriguez-Torres, 1968;
Freeman, 1965). In a review of 5 fatal cases of lead poisoning in
young children, heart failure was concluded to be the proximate cause
of death in 2 cases (Kline, 1960). Kosmider & Petelenz (1962) examined
38 adults over 46 years of age with chronic lead poisoning. They found
that 66% had electrocardiographic changes, which was four times the
expected rate for that age group. Orlova (1954) also reported
electrocardiographic abnormalities in cases of lead poisoning.
Dimitrova (1972) reported cardiac abnormalities in workers with
undefined degrees of lead intoxication. There was a correlation of
urinary excretion of lead with duration of systolic contraction and
with isometric tension. Lead mobilization by EDTA accentuated these
effects on the heart. No dose-effect relationships are apparent from
the limited data available.
8.2.7 Reproduction
There is no epidemiological evidence of an effect of lead on the
fertility of women or on in utero fetal development, but there are
numerous reports in the older literature of stillbirths and
miscarriages among women working in the lead trade (Cantarow &
Trumper, 1944; Oliver, 1914). These reports probably contributed to
the promulgation of legislation forbidding the employment of women in
the lead trades in many countries. Panova (1972) reported that women
working in lead industries had a higher incidence, compared with a
control group, of ovulatory dysfunction -- mainly an ovulatory cycles
and cycles with luteal abnormality. A relationship was reported
between ALA-U and the incidence of anovulatory cycles. The effect was
seen at 8-10 mg ALA/litre of urine.
There are not any reliable data to indicate that infertility in
women results from exposure of the male partner to lead.
Some of the early reports on lead poisoning (Oliver, 1914)
suggested that reproductive failures such as sterility and
miscarriages occurred even among the non-working wives of
industrially-exposed men. The reproductive capability of 150
occupationally exposed men was recently studied by Lancranjan et al.
(1975). The results indicated that both lead poisoning and moderately
increased lead absorption decreased the fertility of men. An increased
frequency of asthenospermia, hypospermia, and teratospermia was found.
No interference with the hypothalamopituitary axis was demonstrated;
thus, hypofertility was thought to be due to the toxic effect of lead
on the gonads.
8.2.8 Endocrine organs
Impairment of thyroid function and of adrenal function has been
reported in cases of lead poisoning (Monaenkova, 1957; Sandstead et
al, 1969; Sandstead et al., 1970; Pines, 1965).
There is some evidence suggesting that lead may cause a
derangement of tryptophan metabolism. This is based on the observation
that urinary excretion of 5-hydroxyindoleacetic acid was increased in
227 children living near a lead smelter (Ghelberg, 1966).
Unfortunately, the 5-hydroxyindoleacetic acid determinations were not
quantitative. Furthermore, blood lead values or other indices of
exposure were not determined. Urbanowicz et al. (1969) noted a rise in
5-hydroxyindoleacetic acid excretion in workers heavily exposed to
lead (ALA-U--33.7 mg/litre of urine). The rise preceded the rise in
ALA-U and CP-U. Dugandzic et al. (1973) also noted a rise in
5-hydroxyindoleacetic acid excretion in moderately exposed workers
(ALA-U--28.2 ± 22.6 mg/litre of urine). More recently Schiele et al.
(1974a), using another analytical method, reported that they were
unable to find any significant elevation in 5-hydroxyindoleacetic acid
excretion in workers with relatively high blood lead levels
(88.5 ± 16.1 µg/100 ml).
8.2.9 Carcinogenicity
Dingwall-Fordyce & Lane (1963) did not find any evidence of an
increased incidence of malignant diseases in their follow-up study of
267 workers (section 8.1.1).
In a more recent study of the causes of mortality among lead
smelter and lead battery workers, it was concluded that while the
incidence of malignant neoplasms was somewhat greater than expected,
the difference was not statistically significant (Tabershaw & Cooper,
1974; Cooper & Gaffey, 1975). This seems to support the conclusion of
a IARC Working Group that there is no evidence to suggest that
exposure to lead salts causes cancer of any site in man (IARC, 1972).
8.2.10 Effects on chromosomes
The literature is controversial as regards chromosomal
abnormalities induced by exposure to lead. On the one hand,
chromosomal aberrations have been reported to result from lead
exposure corresponding to mean Pb-B values of 38-75 µg/100 ml in
various groups studied (Forni & Secchi, 1973; Schwanitz et al., 1970).
Moreover, Deknudt et al. (1973) reported chromosomal aberrations in a
group of 14 male workers with signs of lead poisoning. The authors
concluded that, although the workers were exposed to zinc and cadmium
as well as lead, the lead ought to be considered responsible for the
aberrations. On the other hand, Schwanitz et al. (1975) were not able
to corroborate their own findings among occupationally exposed workers
and O'Riordan & Evans (1974) did not find any significant increase in
chromosomal aberrations in shipbreakers with Pb-B values ranging from
40 to over 120 µg/100 ml. Schmid et al. (1972) did not find any
evidence of lead-induced chromosome aberrations in a study on human
peripheral lymphocytes in vivo and in vitro; furthermore,
Bauchinger et al. (1972) did not find any abnormalities in the
chromosomes of policemen with elevated Pb-B levels.
In a recent report, Bauchinger et al. (1976) found that
chromosomal aberrations were significantly increased in a group of 24
male workers occupied in zinc electrolysis and exposed to zinc, lead,
and cadmium. The workers had clearly elevated Pb-B and blood cadmium
levels in comparison with a control group. The authors pointed out the
similarity between this group and the group studied by Deknudt et al.
(1973) as regards combined exposure. However, referring to studies
indicating mutagenicity of cadmium (Oehlkers, 1953; Shiraishi et al.,
1972; Shiraishi, 1975), Bauchinger and his colleagues were inclined to
consider cadmium as being mainly responsible for the aberrations. They
also emphasized the possibility of a synergistic effect of several
metals on the chromosomes. Thus, the question as to whether
chromosomal abnormalities occur as a result of lead exposure in man
remains open. Furthermore, the human health significance of
chromosomal abnormalities seen in lymphocyte cultures, as observed in
some of these studies, is not yet known.
8.2.11 Teratogenicity
There is practically no information in the literature to suggest
that lead is teratogenic for man (Wilson, 1973). Only one case has
been reported of neuromuscular abnormalities and failure to grow in a
child attributed to lead poisoning as a result of the consumption by
the pregnant mother of illicit whisky (Palmisano et al., 1969).
8.3 Factors influencing Lead Toxicity
8.3.1 Acquisition of tolerance to lead
Experience in industry does not suggest that, with continuous
lead exposure, the human body becomes less reactive to lead. There
have been two studies in which the biochemical parameters of lead
exposure were followed for a long period after the initiation of
industrial lead exposure. Tola et al. (1973) found that erythrocyte
ALAD fell to a stable level in about 21 days, as the concentration of
lead in the blood increased correspondingly. Then both blood lead and
blood ALAD remained essentially stable for the next three months.
There was no return toward normal values to suggest development of
tolerance. Urbanowicz (1971) followed ALA-U and CP-U levels in 60
workers for 24 months after they first became industrially exposed.
There was a build-up of both biochemical effects for several months.
But the levels then stabilized for the remainder of the two-year
period. These studies suggest that the toxicologically-active fraction
of the body burden during steady, long-term exposure remains
essentially unchanged.
8.3.2 Age
Young children absorb lead more readily than older people. It
also seems that children are more susceptible than adults in the sense
that toxic effects occur at lower blood lead concentrations. The
susceptibility of old people in comparison with younger adults has not
been studied.
8.3.3 Seasonal variations
It has long been recognized that the incidence of severe lead
intoxication in children is highest during the summer months (Baetjer,
1959; NAS-NRC, 1972). The observation that urinary excretion of lead
increases in late summer may have some bearing (Kehoe, 1961).
8.3.4 Nutrition
There are few reports of studies that point to nutritional
variables as having a distinct effect on lead toxicity in man (NAS-
NRC, 1972; Goyer & Rhyne, 1974). Iron deficiency and lead exposure
both affect porphyrin metabolism at the point where protoporphyrin IX
is converted to haem. An additive effect results.
8.3.5 Intercurrent disease, alcohol, and other metals
Little is known about the effects of intercurrent diseases on the
toxicity of lead or about the effect of lead on the susceptibility of
people to other diseases. People with haemoglobin and erythrocyte
anomalies, such as sickle cell anaemia and thalassaemia, would
probably be more sensitive to the effects of lead exposure, as would
perhaps people with renal damage. It is also possible that an
interaction may exist between lead exposure and infectious disease
processes, although reliable human data are not available to prove the
point.
The effect of ethanol on lead toxicity is of some interest
because the encephalopathy of illicitly-distilled whisky drinkers
could conceivably involve an interaction of lead and the alcohol
consumed. Furthermore, it has been suggested that heavy drinkers among
industrially-exposed men may be more prone to lead toxicity than non-
drinkers (Cramer, 1966; Candani & Farina, 1972).
9. EVALUATION OF HEALTH RISKS TO MAN FROM EXPOSURE TO LEAD AND
ITS COMPOUNDS
The evaluation of health risks to man from exposure to lead and
its compounds involves the following considerations:
(1) the significance of different environmental sources of lead and
of pathways of exposure;
(2) the probability of occurrence of biological effects at different
levels and rates of lead intake;
(3) the significance for human health of the various known biological
effects of lead;
(4) the validity and limitations of various indicators of lead
exposure and of resultant effects.
These considerations have been used in arriving at the
conclusions which are summarized in this chapter.
9.1 Relative Contributions of Air, Food, Water, and Other Exposures
to Total Intake
9.1.1 Adult members of general population groups
For the general population, the major contribution of lead to the
total daily intake is from food, but water and air may provide
significant contributions under certain conditions. Separate
consideration must be given to occupationally-exposed persons in whom
both the total lead intake and the relative contributions of dietary
and airborne lead are quite different.
The inhalation of airborne lead contributes comparatively little
to the Pb-B level in the general population. This follows from the
fact that the lead concentration in ambient air seldom exceeds
3 µg/m3 when averaged over months and from the conclusions reached
in section 6 that the contribution of airborne lead to Pb-B levels is
probably within the range of 1.0 to about 2.0 µg/100 ml for every
1 µg/m3 of air. Although deposition and retention of different forms
of lead in air may vary, estimates of Pb-B levels from the
concentrations of lead in air are similar for the ambient air and for
the air in the work environment.
Even if we assume a concentration of 1 µg of lead per cubic metre
of air contributes as much as 2.0 µg/100 ml of blood, and that the
ambient air concentration of lead is as high as 4.5 µg/m3, the total
contribution of airborne lead would not exceed 9.0 µg/100 ml. This is
still less than two-thirds of the value estimated by a WHO Expert
Committee (1973). The discrepancy arises from the different approaches
used in making the estimate. The WHO Expert Committee's estimate was
based on lung deposition figures for lead obtained using the ICRP
model (Task Group on Lung Dynamics, 1966). However, the ICRP lung
model probably overestimates deposition for particles smaller than
0.5 µm (aerodynamic diameter) (Mercer, 1975), and the assumption that
all the lead that is deposited is absorbed is probably also incorrect.
Dietary intake of lead varies with eating habits and the lead
content of water sources. The majority of estimates from various
countries suggest that the daily oral lead intake from food by adults
ranges from approximately 100 µg to more than 500 µg; most studies
show lead intake from dietary sources to be 200-300 µg/day. Relating
blood lead levels to known daily oral lead intake suggests that each
100 µg of oral lead intake contributes about 6-18 µg of lead/100 ml of
blood. This source of lead therefore accounts for a very large
fraction of the blood lead levels found in the general adult
population with Pb-B values below 25 µg/100 ml.
The quantity of lead intake directly related to the lead content
of drinking water is difficult to estimate. Assuming a lead
concentration in drinking water of 50 µg/litre (which is the upper
limit generally found in the absence of lead pipes or other lead
contributing factors) and a daily intake of one litre of water, 50 pg
of total dietary lead could be attributed to water. This may be
regarded as an upper limit but it must also be pointed out that lead
in water ingested independently of food may be more readily absorbed
and may provide a relatively greater contribution to the blood lead
level than lead in food.
In assessing the relative contributions of air and diet to Pb-B
levels, attention is called to the possibility that air may be a
significant source of dietary lead through fallout. However, there are
no data to confirm this assumption.
Improperly glazed pottery and illicit whisky have been cited as
potential sources of excessive lead exposure for members of the
general population.
Smoking one packet of 20 cigarettes would result in the direct
inhalation of about 1-5 µg of lead but this only indicates the order
of magnitude.
9.1.2 Infants and children
Infants and preschool children are a high-risk group with regard
to lead intake and absorption. Relative contributions from food,
water, and air are difficult to estimate because of the different diet
(e.g. milk) and more active metabolic rate of young children. Also,
intestinal absorption of lead by young children and, in particular, by
infants may be greater than by adults. Tolerable intake of lead for
preschool children should be less than the 3 mg/week recommended
provisionally for adults by a WHO Expert Committee on Food Additives
(1972).
A special hazard for young children is the ingestion of non-food
items, particularly lead-containing paint from surfaces in homes and
lead-contaminated dust and soil.
9.1.3 Occupationally exposed population groups
Because of the variability of occupational exposure, no general
conclusions are possible but precautions against excessive exposure
must be exercised in view of the possibility of extremely high
occupational lead exposures, as cited in section 5.
9.2 Evaluation of Haematological Effects
Based on information presented in section 8, the following
conclusions have been reached concerning the significance of different
effects on haematopoiesis.
Inhibition of ALAD activity in erythrocytes. The health significance
of decreased ALAD activity is still open to discussion. Although
inhibition of ALAD in erythrocytes is to a certain extent paralleled
by a decrease in other organs, e.g. liver and brain, no effect on
health of this decrease has ever been established. Inhibition of ALAD
is generally regarded as a good indicator of lead absorption but not
of health impairment.
Increased excretion of ALA and CP in urine, and increase of FEP are
indicators of impaired haematopoiesis. Although at moderate levels of
increase, no evidence has been brought forward to show that the vital
functions of haematopoiesis are impaired, resulting, for example, in a
reduced life-span of erythrocytes or anaemia, any increase should be
regarded with suspicion and particularly so when it is more than twice
the level found in non-exposed population groups. Because free
erythrocyte protoporphyrins are also increased in the case of iron
deficiency, this test may provide a better indication of impaired
haematopoiesis in exposed iron deficient population groups (especially
children) than the excretion of ALA and CP. Moreover, females and
children appear to have an earlier and steeper increase of FEP than
males for the same levels of Pb-B.
Effects on erythrocyte membrane, as evidenced by shortened life-span
and a decrease of Na-K-ATPase clearly can result in adverse health
effects since anaemia may occur. Anaemia, expressed by decreased
haemoglobin level, may be regarded as a consequence of disturbed haem
and globin synthesis and of the decreased life-span of erythrocytes
and has clear adverse health consequences.
9.3 Dose-Effect Relationships
At present Pb-B levels are the best available indicator of the
dose. It should, however, be recognized that Pb-B does not reflect the
type of exposure. The dose-effect relationships based on Pb-B levels
should generally be used for long-term exposure.
As stated in section 8 (page 99) a dose-effect relationship
refers in this report to the relationship between the dose as
estimated by Pb-B levels and the intensity of a specified effect in
individual subjects. For most effects, not enough data are available
to present adequate dose-effect curves; however, for some effects,
some points on the dose-effect curve can be tentatively estimated; for
other effects, the data available only permit a statement referring to
the Pb-B level below which such an effect has not been reported. This
level is referred to as the no-detected-effect level. The degree of
confidence that can be placed on such estimates will vary depending on
the sample size and the number of studies reporting no effect.
ALAD activity in erythrocytes. There is a negative linear
relationship between the logarithm of ALAD and Pb-B levels. Increase
in Pb-B levels is paralleled by a decrease in ALAD levels in the Pb-B
range up to about 60 µg/100 ml. For higher Pb-B values, the ALAD
activity levels off at a very low level of enzyme activity. The no-
detected-effect level for Pb-B is probably about 10 µg/100 ml but may
be even lower.
ALA and CP in urine; PP in erythrocytes. There is a positive linear
relationship between the logarithm of ALA (CP, FEP) and Pb-B levels;
the no-detected-effect level for ALA and CP is about 40 µg/100 ml; for
FEP the no-detected-effect level in females is about 20-30 µg/100 ml,
in males it is about 25-35 µg/100 ml; and in iron-deficient children
in particular, it may be about 20-25 µg/100 ml.
Effects on the erythrocyte membrane start to occur at higher Pb-B
levels, probably higher than 50-60 µg/100 ml; a study by Secchi et al.
(1974), on Na-K-ATPase, however, reports a lower no-detected-effect
level of between 30 and 40 µg/100 ml.
Anaemia. Some authors maintain that the no-effect level in workers
is above a Pb-B level of 100µg/100ml; others, however, report a slight
decrease in the haemoglobin level, at a mean level of Pb-B of about
50 µg/100 ml. In some population groups and particularly in iron-
deficient children, the no-detected-effect level is at an approximate
Pb-B level of 40 µg/100 ml.
Nervous system effects. The data on effects of lead compounds on the
nervous system lead to the following tentative conclusions in regard
to prolonged exposures:
(1) From Pb-B levels of approximately 40 µg/100 ml, the probability
of the occurrence of subclinical peripheral electrophysiological
changes increases.
(2) From approximately 50 µg/100 ml in children, the probability of
noticeable brain dysfunction increases; in adults the level is
probably somewhat higher (60-70 µg/100 ml).
(3) From approximately 60 µg/100 ml in children the probability of
acute or chronic encephalopathy increases; in adults this level
is higher, probably above 80 µg/100 ml.
(4) The potential effects of lead on the nervous system constitute
one of the main concerns, particularly in children. More
carefully considered prospective studies should be carried out
taking into account various interacting variables such as
nutrition, socioeconomic status, and parental care in order to
establish better founded dose-effect and dose-response
relationships.
(5) No dose-effect or dose-response relationships can be established
for alkyllead exposure on the basis of currently available
information.
The present no-detected-effect level for sub-clinical neuropathy
appears to be a Pb-B value of 40-50 µg/100 ml. For minimal brain
dysfunction it is probably 50-60 µg/100 ml in children and
60-70 µg/100 ml in adults, and for acute or chronic encephalopathy,
60-70 µg/100 ml in children, and over 80 µg/100 ml in adults. The
establishment of relationships between Pb-B levels and effect is
especially difficult in children because the effect may be detected
months or years after the critical exposure occurred.
Renal function. Apparently prolonged exposure to Pb-B levels greater
than 70 µg/100 ml is necessary to produce nephropathy; a no-detected-
effect level cannot be given. The problem is non-correspondence in
time between the determination of Pb-B level and the detection of
effect.
Aminoaciduria, reflecting impaired amino acid transport through
the renal tubules may occur in children and adults with increased lead
absorption. The present data do not allow a no-detected-effect level
to be estimated, but indicate that this effect is unlikely to be found
in association with Pb-B levels below some 90-100 µg/100 ml (Chisolm,
1968b, Cramer et al., 1974).
Changes in blood constituents such as calcium, phosphorus, glucose,
cholesterol, total proteins, serum albumins, alkaline phosphatase
(EC 3.1.3.1), lactate acid dehydrogenase, and urea nitrogen, could not
be found in male workers with a median Pb-B level of 63 µg/100 ml; 37%
showed an effect with a Pb-B level greater than 70 µg/100 ml (Cooper
et al., 1973). There was an indication of increased bilirubin at a
Pb-B level of about 70 µg/100 ml. An increased pyruvate level after
glucose administration was reported in 50% of children with Pb-B
levels of 40-60 µg/100 ml (Moncrieff et al., 1964).
The general pattern of morbidity and mortality in workers does not
appear to be affected if the Pb-B level never exceeds 70 µg/100 ml.
In assessing reported dose-effect relationships and no-detected-
effect levels, one should take into account the fact that the
available data are limited. Even from a theoretical viewpoint, the
establishment of a definite no-effect level is not possible, because
one can hardly ever expect to cover the whole range of susceptibility
in human populations. Nevertheless, the available data suggest that
the no-detected-effect levels given above are on the conservative
side.
Table 29 summarizes the no-detected-effect levels discussed. For
some of these effects, it is possible to elaborate dose-response
relationships. These cases are considered in section 9.4.
Table 29. No-detected effect levels in terms of Pb-B µg of lead per 100 ml of blood)
No detected Effect Population
effect level
< 10 Erythrocyte ALAD inhibition adults, children
20-25 FEP children
20-30 FEP adult, female
25-35 FEP adult, male
30-40 Erythrocyte ATPase inhibition general
40 ALA excretion in urine adults, children
40 CP excretion in urine adults
40 Anaemia children
40-50 Peripheral neuropathy adults
50 Anaemia adults
50-60 Minimal brain dysfunction children
60-70 Minimal brain dysfunction adults
60-70 Encephalopathy children
> 80 Encephalopathy adults
9.4 Dose-response Relationships
A dose-response relationship considers the observed relative
frequency of occurrence of a specified effecta in a group of
subjects at a given dose level. As in the case of dose-effect
relationships, the data available to evaluate a dose-response
relationship are either limited or non-existent. The available
information on dose-response relationships has been presented in
section 8. In this section, attention is paid to the 5% response
levels, i.e. that level of Pb-B at which not more than 5% of the group
considered is expected to show the specified intensity of a specified
effect. The 5% level has had to be stipulated, because not enough data
are available to state the Pb-B levels for 0.5%, 1%, etc. Further
investigations have to be carried out to enlarge the amount of data
available. For further discussion see Zielhuis (1975). His review
suggests the 5% response levels recorded in Table 30, which are in
accordance with the data discussed in section 8. These response levels
are also in agreement with those suggested by Hernberg (1975).
Table 30. Pb-B levels at which no more than 5% of the population will show
the indicated intensity of effect
Biochemical effect Intensity of effect Population Pb-B
(µg/100 ml)
ALAD inhibition perceptible inhibition adult, children 10
> 40% inhibition adults 15-20
> 70% inhibition adults 30
> 70% inhibition children 25-30
ALA-U perceptible increase adults, children 40
> 10 mg/litre adults, children 50
FEP perceptible increase adult males 30
adult females 25
children 20
a From: Zielhuis, 1975.
a Graded effects may be specified in terms of their intensity.
9.5 Diagnosis of lead Poisoning and Indices of Exposure and/or
Effects for Epidemiological Studies
For epidemiological studies and for the detection of the early
effects of lead in occupational exposure of individuals, the following
tests have been used:
(1) Lead levels in blood.
(2) Excretion of lead in urine spontaneously or after administration
of chelating agents.
(3) Lead levels in tissues (teeth, bones, hair, etc.).
(4) Activity of ALAD in blood.
(5) Indices of disturbed porphyrin metabolism: ALA and/or CP in
urine, Protoporphyrin IX in erythrocytes.
(6) Haematological indices such as basophilic stippling and
haemoglobin levels.
(7) Early (sub-clinical) symptoms and signs of other damage (e.g. to
the nervous system or the kidneys).
(8) Clinical evidence of poisoning.
The criteria used by individual investigators correspond to the
premises and purposes of their studies, for example, Pb-B for
evaluating lead levels in the general population, and clinical signs
of poisoning to assess morbidity caused by occupational exposure.
The following considerations should be kept in mind when using
and interpreting the results.
9.5.1 Concentration of lead in blood (Pb-B)
Pb-B reflects the current state of the dynamic equilibrium
between the amounts of lead entering the organism, transported in the
blood, and deposited in the tissues (including the bones). To date,
insufficient information has been collected about the quantitative
aspects of these processes, but from the data available, it may be
stated that:
(a) After a single inhalation of a soluble lead compound, the
concentration of lead in the body will change in the same way as after
an intravenous injection, i.e. there will be a rapid increase in Pb-B
levels followed by a slower decrease; initially there will be a rapid
elimination in the urine and a slow deposition in the tissues with
subsequent redistribution according to the metabolism of lead in the
various organs and systems.
(b) During long-term exposure at a constant rate, an equilibrium
between the amount of lead absorbed, deposited, and excreted develops
over a long period (weeks to months, according to the daily doses
received), which can be considered as a steady state.
(c) There are only limited data as to how quickly this
equilibrium (and Pb-B) changes when irregular variations in the dose
of lead received (e.g. air lead concentrations) occur.
A long-term steady state probably exists normally in non-
occupationally exposed general adult populations, at least in the Pb-B
level. However, no direct evidence for this assumption is available.
In occupationally exposed persons, a steady state cannot be
assumed because of the well known and marked variations of air lead
concentrations in the working environment and of Pb-B levels in
occupationally exposed individuals from one time to another and among
the individuals in the same work place. Occasional exposure to a high
lead concentration in the air could raise the Pb-B level for some time
without contributing significantly to the body burden and to the
biological effects.
If Pb-B is to be used as an indicator of the degree of
environmental lead exposure the above-mentioned facts must be taken
into account, as well as the analytical method used and the
limitations (accuracy, precision, sensitivity, limits of detection).
9.5.2 Aminolevulinic acid dehydratase (ALAD)
For ALAD the same conditions can apply as for Pb-B. The behaviour
of ALAD activity will follow closely the level of Pb-B up to
50-60 µg/100 ml.
9.5.3 Aminolevulinic acid (ALA) and coproporphyrin (CP) excretion
in the urine
ALA and CP in urine are not so dependent on the current state of
lead exposure and absorption as the Pb-B, although their excretion
diminishes relatively quickly when exposure ceases; they reflect more
the average short-term level of lead exposure and have proved useful
in this way. ALA and CP estimates have found broad recognition as
indices of lead absorption and as indicators of early effects they
reflect individual susceptibility to lead.
9.5.4 Lead excretion in the urine
An elevated rate of spontaneous lead excretion in the urine is
indicative of high lead absorbed, but a normal rate of excretion does
not serve as a reliable means of excluding the possibility of
excessive absorption. Lead excretion in urine is dependent on the Pb-B
level but is also influenced by other-mostly unknown-factors, so that
no direct conclusions about exposure and the extent of absorption can
be derived from lead levels in urine (even in a 24-hour sample).
The excretion of lead provoked by chelating agents such as
calcium disodium ethylenediamintetraacetate is thought to reflect the
biologically active portion of the body burden. It is probably a more
sensitive index of over exposure and excess absorption than the Pb-B
level since clearly elevated values have been reported in cases of
only marginally elevated Pb-B levels.
9.5.5 Haematological changes (stippled cells, anaemia)
These are not sensitive indices of over-exposure or excess
absorption. They are not very useful for the early detection of
possible health impairment.
9.5.6 Lead in tissues (teeth and hair)
These have been used as indicators of integrated long-term
exposure and have the advantage that samples are easy to procure. As
yet, the amount of information concerning the interpretation of the
values obtained is inadequate for their evaluation as indices of
exposure or dose.
9.5.7 Some practical aspects
9.5.7.1 General population studies
The Pb-B level is the epidemiological index of choice, assuming
that a reasonable approximation of a steady state exposure exists.
ALAD activity estimates are equally useful for such studies or as
epidemiological indices of lead absorption. The decision to use Pb-B
or ALAD depends on the laboratory facilities available. Signs of lead
effects other than ALAD inhibition are not to be expected at Pb-B
levels below 20 µg/100 ml. Lead in deciduous teeth and hair is
potentially useful as an indicator of integrated exposure in infants
but needs more study.
9.5.7.2 Occupationally-exposed persons
For screening the exposure of groups of workers, any method can
be used that has the required sensitivity and specificity. Economic
and time factors will determine the choice of test. When using the
Pb-B level, the conditions of sampling must be well defined, taking
into account the factors influencing the variations of the Pb-B
concentrations. ALA and CP estimations in urine are widely used since
they are simple, avoid the possibility of external contamination, and
may provide a better picture of the integral exposure. ALAD activity
is only useful at Pb-B levels below about 60 µg/100 ml. For early
detection of the signs of lead effects in individuals, ALA-U or CP-U
tests are the best established screening methods. When abnormal values
are found, further tests (including clinical and laboratory
investigations) will have to be applied to evaluate the kind of
disturbance and the degree of health risk (WHO Study Group, 1975).
9.5.7.3 Reliability of sampling and analytical methods
The evaluation of the pollution of the environment by lead and of
the health effects on man which might result, depends on the
reliability of sampling procedures and analytical methods used.
The methods of sampling for different environmental media, and
the possible exposure pathways of man have been discussed in section
3. The great spatial and temporal variability of these environmental
media and their diversity make the accurate assessment of total
exposure a difficult task. Unless elaborate schemes are set up and
extreme precautions are taken, the total exposure of a population
group cannot be evaluated with an error of less than about 50%, taking
into account tile analytical uncertainties.
In the determination of the dose received or the effects on
haematopoiesis observed, the sampling problem is relatively minor but
the accuracy and precision of analytical techniques play an important
role. An evaluation with up to 20% relative precision is seldom
achieved under normal operational conditions.
9.6 The Problem of Alkyllead Compounds
The principal risk of alkyllead compounds is in occupational
exposure, either by inhalation or by absorption through the skin.
Acute toxicity results in an encephalopathy that differs greatly from
the effects of inorganic lead on the central nervous system. Some
components of the toxic effects are probably due to the alkyl compound
as a whole rather than its lead component. Workmen at greatest risk
are those involved in mixing fuel additives, although other workmen
engaged in related occupations such as the cleaning of storage tanks
where inhalation is possible, are also at high risk. Over-exposure of
the general population to alkyllead compounds has not been documented.
REFERENCES
ABERNETHY, R. F., PETERSON, M. J., & GIBSON, F. H. (1969)
Spectrochemical analyses of coal ash for trace elements. U.S.
Bur. Mines, Rep. Inv. 7281.
AKATSUKA, K. (1973) Tetraalkyl lead poisoning. Jpn. d. ind. Health,
15: 3-66. ALBAHARY, C. (1964) Les troubles porphyriques dans le
saturnisme. Arch. Mal. prof., 25: 495-507.
ALBAHARY, C., RICHET, G., GUILLAUME, J., & MOREL-MAROGER, L. (1965) Le
rein dans le saturnisme professionnel. Arch. Mal. prof.,
26: 5-19.
ALBERT, R. E., SHORE, R. E., SAYERS, A. J., STREHLOW, C., KNEIP, T.
Y., PASTERNACK, B. S., FRIEDHOEF, A. J., COVAN, F., & CIMINO, J.
A. (1974) Follow-up of children overexposed to lead. Environ.
Health. Perspect., Exp. Issue No. 7, 33-41.
ALEXANDER, F. W., & DELVES, H. T. (1972) Death from acute lead
poisoning. Arch. Dis. Child., 47: 446-448.
ALEXANDER, F. W., DELVES, H. T., & CLAYTON, B. E. (1973) The uptake
and excretion by children of lead and other contaminants. In:
Proceedings of the International Symposium; Environmental Health
Aspects of Lead, Amsterdam, 2-6 October 1972. Luxembourg,
Commission of the European Communities, pp. 319-330.
ALLCROFT, R. (1951) Lead poisoning in cattle and sheep. Vet. Rec.,
63: 583-590.
ALLCROFT, R., & BLAXTER, R. L. (1950) Lead as a nutritional hazard to
farm livestock V. The toxicity of lead to cattle and sheep and an
evaluation of the lead hazard under farm conditions. J. Comp.
Pathol., 60(3): 209-218.
ALTMAN, P. U., & DETTMER, D. S., ed. (1968) Nutritional standards in
man. In: Metabolism. Bethesda, MD, Fed. Am. Soc. Exp. Biol.,
pp. 95-96.
ALVARES, A. P., KAPELNER, S., SASSA, S., & Kappas, A. (1975) Drug
metabolism in normal children, lead-poisoned children and normal
adults. Clin. Pharm. Ther., 17(2): 179-183.
ALVARES, A. P., LEIGH, S., CONN, J., & KAPPAS, A. (1972) Lead and
methyl mercury: effects of acute exposure of cytochrome P-450 and
the mixed function oxidase system in the liver. J. exp. Med.,
135 (6): 1406-1409.
ARAKI, S, & HONMA, T. (1976) Relationships between lead absorption and
peripheral nerve conduction velocities in lead workers. Scand.
J. Work environ. Health, 4: 225-231.
ASKEVOLD, R. (1951) Routine analysis of porphyrines in urine.
J. clin. Lab. Invest., 3: 318-319.
ASTM (1970) Book of standards. Tentative method of test for lead in
the atmosphere. Philadelphia, American Society for Testing and
Materials, vol. 23, pp. 830-836.
ATKINS, P. R. (1969) Lead in suburban environment. J. air Pollut.
Contr. Ass., 19: 591-594.
AUB, J. C., FAIRHALL, L. T., MINOT, A. S., & REZNIKOFF, P. (1926) Lead
poisoning. Baltimore, Williams and Wilkins Co., pp. 206 (Medicine
Monographs Vol. VII).
AZAR, A., SNEE, R., & HABIBI, K. (1973) Relationship of community
levels of air lead and indices of lead absorption. In:
Proceedings of the International Symposium; Environmental Health
Aspects of Lead, Amsterdam, 2-6 October 1972. Luxembourg,
Commission of the European Communities, pp. 581-594.
BAERNSTEIN, H. D, & GRAND, J. A. (1942) The relation of protein intake
to lead poisoning in rats. J. Pharmacol. exp. Ther., 74: 18-24.
BAETJER, A. M. (1959) Effects of season and temperature on childhood
plumbism. Ind. Med. Surg., 28: 137-144.
BAETJER, A. M., & HORIGUCHI, S. (1963) Effects of environmental
temperature and dehydration on lead poisoning in laboratory
animals. Amsterdam, Excerpta Medica, pp. 795-797 (International
Congress Series, No. 62).
BAETJER, A.M., Joardar, S. N. D., & MCQUARY, W. A. (1960) Effect of
environmental temperature and humidity on lead poisoning in
animals. Arch. environ. Health, 1: 463-477.
BAGCHI, R. B., GANGULY, H. D., & SIRDAR, J. M. (1940) Lead in food.
Ind. J. med. Res., 28: 441-445.
BAKER, R. W. R. (1950) Polarographic determination of lead in urine.
Biochem. J., 46: 606-612
BALOH, R. W. (1974) Laboratory diagnosis of increased lead absorption.
Arch. environ. Health, 28: 198-208.
BARLTROP, D. (1969) Transfer of lead to the human foetus. In:
Barltrop, D. & Burland, W. L., ed., Mineral metabolism in
pediatrics. Philadelphia, Davis Co., pp. 135-151.
BARLTROP, D., & SMITH, A. (1971) Interaction of lead with
erythrocytes. Experientia (Basel) 27: 92-95.
BARLTROP, D., & SMITU, A. (1972) Lead binding to haemoglobin.
Experientia (Basel), 28: 76-77.
BARLTROP, D., BARRET, A. J., & DINGLE, J. T. (1971 ) Subcellular
distribution of lead in the rat. J. Lab. clin. Med.
77: 705-712.
BARLTROP, D., STREHLOW, C. D., THORNTON, J., & WEBB, J. S. (1974)
Significance of high soil lead concentrations for childhood lead
problem. Environ. Health Perspect., Exp. Issue No. 7, 75-83.
BARRY, P. S. I. (1975) A comparison of concentrations of lead in human
tissues. Brit. J. ind. Med. 32: 119-139.
BARRY, P.S., & MOSSMAN, D. B. (1970) Lead concentrations in human
tissues. Brit. J. ind. Med. 27: 339-351.
BAUCHINGER, M., SCHMID, E, EINBRODT, H. J., & DRESP, J. (1976)
Chromosome aberrations in lymphocytes after occupational exposure
to lead and cadmium. Mutat. Res., 40: 57-62.
BAUCHINGER, M., SCHMID, E., & SCHMIDT, D. (1972) Chromosomenanalyse
bei Verkehrspolizisten mit erhöhter Bleilast. Mutat. Res.,
16: 407-412.
BEATTIE, A.D., MOORE, M. R., DEVENAY, W. I., MILLER, A. R., &
GOLDBERG, A. (1972a) Environmental lead pollution in an urban
soft-water area. Brit. med. J., 2: 491-493.
BEATTIE, A.D., MOORE, M. R., & GOLDBERG, A. (1972b) Tetraethyllead
poisoning. Lancet, 2: 12-15.
BECK, E.G., MANOJLOVIC, N., & FISHER, A. B. (1973) Die Zytotoxizität
von Blei. In: Proceedings of the International Symposium;
Environmental Health Aspects of lead, Amsterdam, 2-6 October,
1972. Luxembourg, Commission of the European Communities,
pp. 451-461.
BECKMAN, K. (1925) Uber die Beziehungen zwischen Blutdruck,
Kapillardruck und Nierenveranderungen im Tiereexperiment.
Arch. klin. Med., 149, 177-188.
BENSON, F. (1972) Indoor-outdoor air pollution relationships. US EPA
Publ. No. AP-112.
BERG, B., & ZENZ, C. (1967) Environmental and clinical control of lead
exposure in a non-ferrous foundry. Am. ind. Hyg. Assoc. J.,
28: 175-178.
BERITIC, T. (1971) Lead concentration found in human blood in
association with lead colic. Arch. environ. Health,
23: 289-291.
BERITIC, T., & STAHULJAK, D. (1961) Lead poisoning from lead-glazed
pottery. Lancet, 1: 669.
BERK, P. D., TSCHUNDY, D. P., SHEPLEY, L. A., WAGGONER, J. G., &
BERLIN, N. I. (1970) Hematologic and biochemical studies in a
case of lead poisoning. Am. J. Med., 48: 137-144.
BERLIN, A., DEL CASTILHO, P., & SMEETS, J. (1973) European
intercomparison programmes. In: Proceedings of the
International Symposium; Environmental Health Aspects of Lead,
Amsterdam, 2-6 October 1972. Luxembourg, Commission of the
European Community, pp. 1033-1046.
BERLIN, A., SCHALLER, K. H., & SMEETS, J. (1975) Standardization of
ALAD activity determinations at the European level;
Intercalibration and applications. In: Proceedings of CEC-EPA-
WHO International Symposium; Recent Advances in the Assessment
of the Health Effects of Environmental Pollution, Paris, 24-28
June 1974. Luxembourg, Commission of the European Communities,
pp. 1087-1101.
BERTINUSON, J. R., & CLARK, C. S. (1973) The contribution to lead
content of soils from urban housing. Interface, 2: 6.
BESSIS, M. C., & JENSEN, W. N. (1965) Sideroblastic anaemia,
mitochondria and erythroblastic iron. Brit. J. Haematol.,
11: 49-51.
BETTS, P. R., ASTLEY, R., & RAINE, D. N. (1973) Lead intoxication in
children in Birmingham. Brit. reed. J., 1: 402-406.
BICKNELL, J., CLAYTON, B. E., & DELVES, H. T. (1968) Lead in mentally
retarded children. J. Mental Def. Res., 12: 282-293.
BINGHAM, E., PFITZER, E. A., BARKLEY, W., & RADFORD, E. P. (1968)
Alveolar macrophages: reduced number in rats after prolonged
inhalation of lead sesquioxide. Science, 162: 1297-1299.
BLACKMAN, S.S. (1937) The lesions of lead encephalitis in children.
Bull. Johns Hopkins Hosp., 61(1): 1-61.
BLANKSMA, L. A., SACHS, H. K., MURRAY, E. F., & O'CONNEL, M. J. (1969)
Incidence of high blood lead levels in Chicago children.
Pediatrics, 44: 661-665.
BLAXTER, K. L., & COWIC, A. T. (1946) Excretion of lead in the bile.
Nature (Lond.), 157: 588.
BOLANOWSKA, W. (1968) Distribution and excretion of triethyllead in
rats. Brit. J. ind. Med., 25: 203-208.
BOLANOWSKA, W., PIOTROWSKI, J., & CARCZYNSKI, H. (1967) Triethyllead
in the biological materia in cases of acute tetraethyllead
poisoning. Archiv J. Toxikol. 22: 278-282.
BOLANOWSKA, W., PIOTROWSKI, J., & TROJANOWSKA, B. (1964) The kinetics
of distribution and excretion of lead (PB210) in rats. In:
Proceedings of the 14th International Congress of Occupational
Health, Madrid, 16-21 Sept., pp. 420-422.
BOLTER, E., BUTZ, T., & ARSENEAU, J. F. (1975) Mobilization of heavy
metals by organic acids in the soils of a lead mining and
smelting district. In: Proceedings of the IXth Annual
Conference on Trace Substances in Environmental Health,
Columbia, MO, 10-12 June 1975, pp. 107-112.
BONSIGNORE, D., CALISANO, P., & CARTASEGNA, C. (1965) Un semplice
metodo per la determinazione della delta-amino-levulinico-
deidratasi nel sangue. Med. Lav., 56: 199-205.
BOUDENE, C., ARSAC, F., & MEININGER, J. (1975) Etude des taux de
plomb dans fair et dans la population en France. In:
International Symposium on Environmental Lead Research,
Dubrovnik, 14-15 May 1975. Arch. industr. Hyg. Toxicol.,
26: supplement, pp. 179-189.
BOYD, P. R., WALKER, G., & HENDERSON, J. N. (1957) The treatment of
tetraethyl lead poisoning. Lancet, 1: 181-185
BOYLAND, E., DUKES, C. E., GROVE, P. L., & MITCHLEY, B.C. V. (1962)
The induction of renal tumours by feeding lead acetate to rats.
Brit. J. Cancer, 16: 283-288.
BRANDT, A.D., & REICHENBACH, G. S. (1943) Lead exposures at the
government printing office. J. ind. Hyg. Toxicol., 25
(10): 445-450.
BREZINA, M., & ZUMAN, P. (1958) Polarography in medicine,
biochemistry and pharmacy. New York, Interscience Publishers
Inc.
BRITISH MINISTRY OF AGRICULTURE, FISHERIES AND FOOD (1972) Survey of
lead in food. Working Party on the Monitoring of Foodstuffs for
Heavy Metals: Second Report. London, Her Majesty's Stationery
Office, pp. 31.
BROWN S., DRAGAN, N., & VOGEL, W. (1971) Effects of lead acetate on
learning and memory in rats. Arch. environ. Health,
22: 370-372.
BRUCH, J., BROCKHAUS, A., & DEHNEN, W. (1973) Elektronmikroskopische
Beobachtungen an Rattenlungen nach Exposition reit
partikelförmigem Blei. In: Proceedings of the International
Symposium; Environmental Health Aspects of Lead, Amsterdam, 2-6
October 1972. Luxembourg, Commission of the European
Communities, pp. 221-229.
BRUCH, J., BROCKHAUS, A., & DEHNEN, W. (1975). Local effects of
inhaled lead compounds on the lung. In: Proceedings of CEC-
EPA-WHO International Symposium; Recent Advances in the
Assessment of the Health Effects of Environmental Pollution,
Paris, 24-28 June 1974. Luxembourg, Commission of the European
Communities, pp. 781-793.
BULLOCK, J. D., WEY, R. J., ZAIA, J. A., ZAREMBOK, I., & SCHROEDER, H.
A. (1966) Effect of tetraethyllead on learning and memory in
rats. Arch. environ. Health, 13: 21-22.
BULPITT, G. J. (1972) Lead and hyperactivity. Lancet, II: 1144.
BURDÉ, B. DE LA, & CHOATE, M. S.(1972) Does asymptomatic lead exposure
in children have latent sequelae? J. Pediar., 81(6): 1088-1091.
BUTT, E. U., NUSBAUM, R. E., GILMOUR, T. C., DIDIO, S. L., & Sister
MARIANO (1964) Trace metal levels in human serum and blood.
Arch. environ. Health, 8: 52-57.
BYERS, R. K. (1959) Lead poisoning. Pediatrics, 23: 585-603.
BYERS, R. K., & LORD, E. E. (1943) Late effects of lead poisoning on
mental development. Am. J. Dis. Child., 66: 471-494.
CANTAROW, A., & TRUMPER, M. (1944) Lead poisoning. Baltimore,
Williams and Wilkins Co., p. 8.
CANDANI, A., & FARINA, G. (1972) Influence of alcoholic beverages
consumption on lead-induced changes of haemebiosynthesis.
Med. Lay., 63, 22-28.
CARPENTER, S. (1974) Placental permeability of lead. Environ. Health,
Perspect. Exp. Issue, No. 7, 129-133.
CARSON, T. L., VAN GELDER, G. A., KARAS, G. G., & BUCK, W. B. (1974)
Development of behavioural tests for the assessment of neurologic
effects of lead in sheep. Environ. Health Perspect., Exp. Issue
No. 7, 233-239.
CASTELLINO, N., & ALOJ, S. (1964) Kinetics of the distribution and
excretion of lead in the rat. Brit. J. ind. Med., 21: 308-314.
CASTELLINO, N., & ALOJ, S. (1969) Intracellular distribution of lead
in the liver and kidney of the rat. Brit. J. ind. Med.,
26: 139-143.
CASTELLINO, N., LIAMANNA, P., & CRIECO, B. (1966) Biliary excretion of
lead in the rat. Brit. J. ind. Med., 23: 237-239.
CATTON, M. J., HARRISON, M. J. G., FULLERTON, P.M., & KAZANTZIS, O.
(1970) Subclinical neuropathy in lead workers. Brit. med. J.,
2: 80-82.
CERNIK, A. A. (1974) Determination of blood lead using a 4.0 mm paper
punched disc carbon sampling cup technique. Brit. J. ind. Med.,
31: 239-245.
CERNIK, A. A., & SAYERS, M. H. P. (1971) Determination of lead in
capillary blood using a paper punched disc atomic absorption
technique application to the supervision of lead workers.
Brit. J. ind. Med., 28(4): 392-398.
CHANEY, R. L. (1973) Crop and food chain effects of toxic elements in
sludges and effluents. Recycling municipal sludges and effluents
on land July 9-13, 1973 Illinois, pp. 129-141.
CHEH, A., & NEILANDS, J. B. (1973) Zinc, an essential metal for beef
liver 6-aminolevulinate dehydratase. Biochem. biophys. Res.
Comm., 55(4): 1060-1063.
CHISOLM, J. J. (1962) Aminoaciduria as a manifestation of renal
tubular injury in lead intoxication and a comparison with
patterns of aminoaciduria seen in other diseases. J. Pediatr.,
60: 1-17.
CHISOLM, J. J. (1968a) Lead poisoning (plumbism). In: H. L. Barnett,
ed., Pediatrics (14th Ed.) New York, Appelton-Century-Crofts,
pp. 313-319.
CHISOLM, J. J. (1968b) The use of chelating agents in the treatment of
acute and chronic lead intoxication in childhood. J. Pediatr.,
73: 1-38.
CHISOLM, J. J. (1973) Management of increased lead absorption and lead
poisoning in children. New Engl. J. Med., 289: 1016-1018.
CHISOLM, J. J. (1974) Chelation therapy in children with subclinical
plumbism. Pediatrics, 53: 441-443.
CHISOLM, J. J., & HARRISON, H. E. (1956) The exposure of children to
lead. Pediatrics, 18: 943-958.
CHOVIN, P., DUFFAUD, J., MERKEZ, F., FAYART, M., & TRUFFERT, L. (1973)
Resultats d'une annee d'étude de la teneur en plomb particulaire
et l'atmosphère Symposium: Environmental Health Aspects of Lead,
Amsterdam 2-6 October 1972. Luxembourg, Commission of the
European Communities, pp. 1003-1015.
CHOW, T. J. (1968) Isotope analysis of seawater by mass spectrometry.
J. Water Pollut. Control Fed., 40: 399-411.
CHOW, T. J., & EARL, J. L. (1970) Lead aerosols in the atmosphere:
Increasing concentrations. Science, 169: 577-580.
CHOW, T. J., & BENNET, C. F. (1969) Lead aerosols in marine
atmosphere. Environ. Sci. Technol., 3: 737-740.
CHOW, T. J., EARL, J. L., & SNYDER, C. B. (1972) Lead aerosol
baseline: Concentration at White Mountain and Liaguna Mountain.
Science, 178: 401-402.
CLARKSON, T. W., & KENCH, J. E. (1956) Urinary excretion of amino
acids by men absorbing heavy metals. Biochem. J., 62: 361-372.
CLARKSON, T. W., & KENCH, J. E. (1958) Uptake of lead by human
erythrocytes in vitro. Biochem. J., 69: 432-439.
CLASEN, R. A., HARTMAN, J. F., STARR, A. J., COOGAN, PH. S., PANDOLFI,
S., LAING, J., BECER, R., & HASS, G. U. (1974) Electron
microscopic and chemical studies of the vascular changes and
edema of lead encephalopathy. Am. J. Pathol., 74(2): 215-233.
CLERCQ, E. DE, & MERIGAN, T. C. (1969) An active interferon inducer
obtained from Haemophilus influenzae type B. J. Immun.,
103: 899-906.
COGBILL, E. C., & HOBBS, M. E. (1957) Transfer of metallic
constituents of cigarettes to the main-stream smoke. Tob. Sci.,
1: 68-73.
COHEN, G. J., & AHRENS, W. E. (1959) Chronic lead poisoning.
J. Pediatr., 54(2): 271-284.
COHEN, G. J., BOWERS, G. N., & LEPOW, M. (1973) Epidemiology of lead
poisoning. J. Am. Med. Ass., 266: 1430-1433.
COHEN, N., KNEIP, T. J., GOLDSTEIN, D. H., & MUCHMORE, E. A. S. (1972)
The juvenile baboon as a model for studies of lead poisoning in
children. J. Med. Primatol., 1: 142-155.
COLE, L. J., & BACHHUBER, L. J. (1914) The effect of lead on the germ
cells of the male rabbit and fowl as indicated by their progeny.
Proc. Soc. Exp. Biol. Med., 12: 24-29.
COLLIER, H. B. (1971) A study of the determination of delta-
aminolevulinate hydrolyase (delta-aminolevulinate dehydratase)
activity in hemolysates of human erythrocytes. Clin. Biochem.,
4: 222-232.
COLWILL, D. M., & HICKMAN, A. J. (1973) The concentration of volatile
and particulate lead compounds in the atmosphere: measurements
at four road sites. Transport and Road Research Laboratory
Report LR 545 Crowthorne, Berkshire, pp. 5.
COMMISSION OF THE EUROPEAN COMMUNITIES (1973) Air lead concentrations
in the European Community. Yearly Report: April 1971-March 1972.
Luxembourg.
COOPER, W. C., & GAFFEY, W. R. (1975) Mortality of lead workers.
J. occup. Med., 17: 100-107.
COOPER, W. C., TABERSHAW, I. R., & NELSON, K. W. (1973) Laboratory
studies of workers in lead smelting and refining. In:
Proceedings of the International symposium: Environmental Health
Aspects of lead, Amsterdam, 2-6 October 1972, Luxembourg,
Commission of the European Communities, pp. 517-529.
COULSTON, F., GOLDBERG, L., GRIFFIN, T. B., & RUSSELL, J. C. (1972a)
The effects of continuous exposure to airborne lead. 1. Exposure
of rats and monkeys to particulate lead at a level of
21.5 µg/m3. Final Report to the US Environmental Protection
Agency.
COULSTON, F., GOLDBERG, L., GRIFFIN, T. B., & RUSSELL, J. C. (1972b)
The effects of continuous exposure to airborne lead. 2. Exposure
of man to particulate lead at a level of 10.9 µg/m3. Final
Report to the US Environmental Protection Agency.
COULSTON, F., GOLDBERG, L., GRIFFIN, T. B., & RUSSELL, J. C. (1972c)
The effects of continuous exposure to airborne lead. 4. Exposure
of man to particulate lead at a level of 3.2 µg/m3. Final
Report to the US Environmental Protection Agency.
COULTHARD, A. J. (1958) Animal cycle of blood haemoglobin levels.
Clin. Chim. Acta, 2: 226-233.
CRAMER, K. (1966) Predisposing factors for lead poisoning. Acta. Med.
scand., (Suppl. 445): 179: 56-59.
CRAMER, K., & DAHLBERG, L. (1966) Incidence of hypertension among lead
workers. Brit. J. ind. Med., 23: 101-104.
CRAMER, K., GOYER, R. A., JAGENBURG, R., & WILSON, M. H. (1974) Renal
ultrastructure renal function, and parameters of lead toxicity in
workers with different period of lead exposure. Brit. J. ind.
Med., 31: 113-127.
CREMER, J. E. (1959) Biochemical studies on the toxicity of tetraethyl
lead and other organo-lead compounds. Brit. J. ind. Med.,
16: 191-199.
CREMER, J. E. (1965) Toxicology and biochemistry of alkyl lead
compounds. Occup. Health Rev., 17: 14-19.
CREMER, J. E, & CALLAWAY, S. (1961) Further studies on the toxicity of
some tetra- and trialkyl lead compounds. Brit. J. ind. Med.,
18: 277-282.
CRUTCHER, J. C. (1963) Clinical manifestation and therapy of acute
lead intoxication due to the ingestion of illicitly distilled
alcohol. Ann. intern. Med., 59: 707-715.
DAINES, R. H., SMITH, D. W., FELICIANO, A., & TROUT, J. R. (1972) Air
levels of lead inside and outside of homes. Ind. Med. Surg.,
41: 26-28.
DALLDORF, G., & WILLIAMS, R. R. (1945) Impairment of reproduction in
rats by ingestion of lead. Science, 102: 668.
DANILOVIC, V. (1958) Chronic nephritis due to ingestion of lead-
contaminated flour. Brit. Ted. J., 1: 27-28.
DAVID, O. J., CLARK, J., & VOELLER, K. (1972) Lead and hyperactivity.
Lancet, 2: 900-903.
DAVIDSON, D. F., & LAKIN, H. W. (1962) Metal content of some black
shales of the western United States. U.S. Geological Survey
Professional Paper 450C, p. C74.
DAVIS, J. R., & ANDELMAN, S. L. (1967) Urinary delta aminolevulinic
acid levels in lead poisoning. A modified method for the rapid
determination of urinary delta-aminolevulinic acid using
disposable ion-exchange chromatography columns. Arch. environ.
Health, 15: 53-59.
DAVIS, R. K., HORTON, A. W., LARSON, E. E., & STEMMER, K. L. (1963)
Inhalation of tetramethyllead and tetraethyllead. Arch. environ.
Health, 6: 473-479.
DAVIS, W. E. (1973) Emission study of industrial sources of lead air
pollutants 1970. US EPA, Document APTD-1543, pp. 1-123.
DEDOLPH, R. G., TER HAAER, R., HOLTZMAN, R., & LUCAS, H. J. (1970)
Sources of lead in perennial ryegrass and radishes. Environ.
Sci. Technol, 4: 217-223.
DEKNUDT, O., LEONARD, A., & IVANOV, B. (1973) Chromosome aberrations
observed in male workers occupationally exposed to lead.
Environ. Physiol. Biochem, 3: 132-138.
DELVES, H. T. (1970) A micro sampling method for the rapid
determination of lead in blood by atomic absorption spectrometry.
Analyst, 95: 431.
DIMITROVA, M. (1972) Modifications of the contractile function of the
myocard in chronic lead poisoning. Arch. Mal. prof. Med. Trav.,
33: 383-387.
DINGWALL-FORDYCE, J., & LANE, R. E. (1963) A follow-up study of lead
workers. Brit. J. ind. Med., 20: 313-315.
DINISCHIOUTU, O. T., NESTORESCU, B., RADIELESCU, J. C., JONESCU, C.,
PREDA, N., & HUTZA, G. (1960) Studies on the chemical forms of
urinary lead. Brit. J. ind. Med, 17: 141-145.
DJURIC, D. (1964) Fluorimetric determination of porphyrius. Arch.
environ. Health, 9: 742-744.
DJURIC, D., KERIN, Z., GRAOVAC-LEPOSAVIC, L., NOVAK, L., & KOP, M.
(1971) Environmental contamination by lead from a mine and
smelter; A preliminary Report. Arch. environ. Health,
23: 275-279.
DJURIC, D., NOVAK, L., MILIC, S., & KALIC-FILIPOVlC, D. (1966) Delta-
ALA as an early sign of lead exposure. Med. Lav., 57: 161-166.
DODIC, S., VIDAKOVIC, A., PERISIC, V., & STEFANOVIC, S. (1971) Stanje
jetre u pojedinih profesionalnih intoksikacija. In: III
Jugoslovanski Kongres Medicine Dela, Ljubljana, 1971.
Ljubljana, 20-24 September, 1971, Jzdange "Lek", pp. 285-293.
DONOVAN, D. T., VOUGHT, V. M., & RAKOW, A. B. (1971) Laboratories
which conduct lead analysis on biologic specimens. Arch.
environ. Health, 23: 111-113.
DRESEL, E. J. B., & FALK, J. E. (1954) Studies on the biosynthesis of
blood pigments. Haem synthesis in hemolysed erythrocytes of
chicken blood. Biochem. J., 56: 156-163.
DRESSEN, W. C., EDWARDS, T. J., REINHART, W. H., PAGE, R. T., WEBSTER,
S. H., ARMSTRONG, D. W., & SAYERS, R. R. (1941) The control of
the lead hazard in the storage battery industry. Publ. Health
Bull. (Wash.), No. 262.
DUGANDZIC, M., STANKOVIC, B., MILOVANOVIC, LY., & KORICANAC, Z. (1973)
Urinary excretion of 5-hydroxindolacetic acid in lead exposed
persons. Archiv. Hig. Rada., 24: 37-43.
DURFOR, C., & BECKER, E. (1964) Selected data on public supplies of
the 100 largest cities in the United States, 1962. J. Am. Water
Works Assoc., 56: 237-246.
EDIGER, R. D., & COLEMAN, R. L. (1973) An evaluation of anodic
stripping voltamentry and non-flame atomic absorption as routine
analytical tools. In: Proceedings of the 6th Annual Conference
on Trace Substances in the Environment, Columbia, MO., 13-15
June 1972, pp. 279-283.
EGOROV, V. V., ZHIGALOVSKAJA, T. N., & MALAKHOV, S. G. (1970)
Microelement content of surface air above the continent and the
ocean. J. geophys. Res., 75: 18, 3650-3656.
EGOROVA, G. M., IVANOV, N. G., & SANOCSKIJ, J. V. (1966) Specificity
of the effect of lead on spermatogenesis. Toksikol., nov. prom.
him., vescestv., 8: 33-41.
EINBRODT, H. J., ROSMANITH, J., & SCHROEDER, A. (1974) Beim Spielen,
Schulkind Vergiftet, Umwelt, 3: 726.
EISENBUD, M., & WRENN, M. E. (1970) Radioactivity studies annual
report NYO-3086-10 VI., 1970. Springfield, VA, National
Technical Information Service.
ELKINS, H. B. (1959) The chemistry of industrial toxicology. New
York, J. Wiley & Sons (2nd edition), pp. 51-52.
EMMERSON, B. T. (1963) Chronic lead nephropathy: The diagnostic use of
calcium EDTA and the association with gout. Austr. Ann. Med.,
12: 310-324.
EMMERSON, B. T., MIROSH, W., & DOUGLAS, J. B. (1971) The relative
contributions of tubular reabsorption and secretion to urate
excretion in lead nephropathy. Aust. N.Z.J. Med., 1: 353-362.
ENGELS, L. H. VON, & KÜHNEN, G. (1973) Staubschutz in der
Akkumulatoren-Industrie Bonn, Staubforschungsinstitut des
Hauptverbandes der gewarblichen Berufsgenossenschaften, STF-
Report No. 2, pp. 27.
ENGEL, R. E., HAMMER, D. J., HORTON, R. J. M., LANE, N. M., & PLUMLEE,
L. A. (1971) Environmental lead and public health. Research
Triangle, Park, NC Environmental Protection Agency. Air Pollution
Control Office Publication No. AP 90, pp. 1-34.
ENVIRONMENT AGENCY, JAPAN (1975) Results of air pollution survey.
Tokyo, pp. 148-153.
EPSTEIN, S.S., & MANTEL, N. (1968) Carcinogenicity of tetraethyllead.
Experientia Basel, 24: 580-581.
ERVE, P. R., & SCHUMER, W. (1972) Endotoxin sensitivity of
adrenalectomized rats treated with lead acetate. Res. J.
Reticuloend. Soc., 11(38): 427.
FAIREY, F. S., & GREY, J. W. (1970 Soil lead and pediatric lead
poisoning in Charleston, S.C. J. S. Carolina med. Assoc.,
66: 79-82.
FAIRHALL, L. T., & MILLER, J. W. (1941) A study of the relative
toxicity of the molecular components of lead arsenate. Public
Health Rep. (Wash.), 56: 1610-1625.
FEDERAL INSTITUTE FOR MINERALS RESEARCH AND GERMAN INSTITUTE FOR
ECONOMIC RESEARCH (1972). Supply and demand for lead. (Translated
into English by Lead Development Association, 34 Berkeley Square,
London W1X 6AJ, April, 1972), pp. 1-47.
FERM, V. H. (1969) The synteratogenic effect of lead and cadmium.
Experientia Basel 25: 56-57.
FILKINS, J.P., & BUCHANEN, B. J. (1973) Effects of lead acetate on
sensitivity to shock, intravascular carbon and endotoxin
clearances, and hepatic endotoxin detoxification. Proc. Soc.
Exp. Biol. Med., 142: 471-475.
FINE, P. R., THOMAS, C. W., SUHS, R. H., COHNBERG, R. E., & FLASHNER,
B. A. (1972) Pediatric blood lead levels: A study in 14 Illinois
cities of intermediate population. J. Am. Med. Assoc., 221
(13): 1475-1479.
FORBES, G. B., & REINA, J. C. (1972) Effect of age on gastrointestinal
absorption (Fe, Sr, Pb) in the rat. J. Nutr, 102: 647-652).
FORNI, A., & SECCHI, C. C. (1973) Chromosome changes in preclinical
and clinical lead poisoning and correlation with biochemical
findings. In: Proceedings of the International Symposium,
Environmental Aspects of lead, Amsterdam, 2-6 October 1972.
Luxembourg, Commission of the European Communities, pp. 473-483.
FOUTS, P. J., & PAGE, J. H. (1942) The effect of chronic lead
poisoning on arterial blood pressure in dogs. Am. Heart J.,
24: 329-331.
FREEMAN, R. (1965) Reversible myocarditis due to chronic lead
poisoning in childhood. Arch. Dis. Child., 40: 389-393.
FRIBERG, L., PISCATOR, M., NORDBERG, G., & KJELLSTROM, T. (1974)
Cadmium in the environment. 2nd ed. Ohio, Chemical Rubber
Company.
FRIED, J. F., ROSENTHAL, N. W., & SCHUBERT, J. (1956) Induced
accumulation of titrate in therapy of experimental lead
poisoning. Proc. Soc. Exp. Biol. Med., 92: 331-333.
FUGAS, M., WILDER, B., PAUKOVIC, R., HRSAK, J., & STEINER-SKREB, D.
(1973) Concentration levels and particle size distribution of
lead in the air of an urban and an industrial area as a basis
for the calculation of population exposure. In: Proceedings of
the International Symposium; Environmental Health Aspects of
Lead, Amsterdam, 2-6 October 1972. Luxembourg, Commission of
the European Communities, pp. 961-968.
FULLERTON, P.M. (1966) Chronic peripheral neuropathy produced by lead
poisoning in guinea pigs. J. Neuropathol. exp. Neurol.,
25: 214-236.
GAGE, J. C., & LITCHFIELD, M. H. (1968) The migration of lead from
polymers in the rat gastro-intestinal tract. Food Cosmet.
Toxic., 6: 329-338.
GAGE, J. C., & LITCHFIELD, M. H. (1969) The migration of lead from
paint films in the rat gastro-intestinal tract. J. Oil Col.
Chem. Assoc., 52: 236-243.
GAJDOS, A., & GAJDOS-TÖRÖK, M. (1969 L'activite de l'acide delta-
aminolevulinique synthetase hépatique et médullaire au cours du
saturnisme experimental du lapin. C.R. Soc. Biol., 163: 60-63.
GAJDOS, A., & GAJDOS-TÖRÖK, M. (1973) Erreur du diagnostique
differentiel entre l'intoxication par le plomb et la porphyrie
intermittente aiguë. In: Proceedings of the International
Symposium; Environmental Health Aspects of Lead, Amsterdam, 2-6
October, 1972. Luxembourg, Commission of the European
Communities, pp. 501-505.
GALLE, P., & MOREL-MAROGEN, L. (1965) Les lesions renales du
saturnisme humain et experimental. Nephron, 2: 273-286.
GALZIGNA, L., BRUGNONE, F., & CORSI, G. C. (1964) Excretion of
5-hydroxyindole acetic acid in experimental tetraethyllead
poisoning. Med. Lavoro., 55: 102-106.
GARBER, B. T., & WEI, E. (1974) Influence of dietary factors on the
gastrointestinal absorption of lead. Toxic. appl. Pharm.,
27: 685-691.
GEORGII, H. W., & JOST, D. (1971) On the lead-concentration in an
urban aerosol. Atmosph. Environ., 5: 725-727.
GHELBERG, N. W., GORGAN, J., & CHECIN, J. (1966) 5-hydroxyindoleacetic
acid excretion in a population chronically exposed to low lead
concentration in the atmosphere. Igiena (Buc) 15: 87-92.
GIBSON, K. G., NEUBERGER, A., & SCOTT, J. J. (1955) The purification
and properties of delta-aminolaevulic acid dehydrase. Biochem.
J., 61: 618-629.
GIBSON, S. L. M., LAM, C. N., MCCRAE, W. M., & GOLDBERG, A. (1967)
Blood lead levels in normal and mentally retarded children.
Arch. Dis. Child., 42: 573-578.
GILLET, J. A. (1955) An outbreak of lead poisoning in the Canklow
district of Rotterdam. Lancet, 1: 1118-1121.
GOLDBERG, A. (1972) Lead poisoning and haem biosynthesis. Brit. J.
Haematol., 23: 521-523.
GOLDBERG, A. (1974) Drinking water as a source of lead pollution.
Environ. Health Perspect., Exp. Issue No. 7, pp. 103-107.
GOLDBERG, A., ASHENBRUCKER, H., CARTWRIGHT, G. E., & WINTROBE, M. M.
(1956) Studies on the biosynthesis of heme in vitro by arian
erythrocytes. Blood, 11: 821-833.
GOLDBERG, E. D. (1976) The health of the oceans. Chapter 5. Heavy
metals: lead. Paris, The Unesco Press, pp. 109-111.
GOLDSMITH, J. R., & HEXTER, A. C. (1967) Respiratory exposure to lead
epidemiological experimental dose-response relationships.
Science, 158: 132-134.
GOLUBOVIC, E. Y., & GEVKOVSKAJA, T. V. (1967) Effects of ethyleneimine
and lead on several sides of nucleic acids changes in the
spermary of rats. Toksikol. nov. prom. him. vescestv, 9: 86-91.
GOLUBOVIC, E. Y., AVHIMENKO, M. M., & CIRKOVA, E. M. (1968)
Biochemical and morphological changes in testicles of rats under
the effect of low doses of lead. Toksikol. nov. prom. him.
vescestv, 10: 64-72 (in Russian).
GOYER, R. A., & KRALL, K. (1969) Ultrastructural transformation in
mitochondria isolated from kidneys of normal and lead-intoxicated
rats. J. cell. Biol., 41: 393-400.
GOYER, R. A., & MAHAFFEY, K. R. (1972) Susceptibility to lead
toxicity. Environ. Health Perspect., No. 2: 73-80.
GOYER, R. A., & RHYNE, B.C. (1973) Pathological effects of lead.
Intern. Rev. exp. Pathol., 12: 1-77.
GOYER, R. A., LEONARD, D. L., MORRE, J. F., RHYNE, B., & KRIGMAN, M.
R. (1970a) Lead dosage and the role of the intranuclear inclusion
body. Arch. environ. Health, 20: 705-711.
GOYER, R. A., MAY, P., CATES, M. M., & KRIGMAN, M. R. (1970b) Lead and
protein content of isolated inclusion bodies from kidneys of lead
poisoned rats. Lab. Invest., 22: 245-251.
GOYER, R. A., TSUCHIYA, K., LEONARD, D. L., & KAHYO, H. (1972)
Aminoaciduria in Japanese workers in the lead and cadmium
industries. Am. J. clin. Pathol., 57: 635-642.
GRABECKI, J., HADUCH, T., & URBANOWICZ, H. (1967) Die einfachen
Bestimmungsmethoden der delta-Amino-lävulinsäure in Harn. Int.
Arch. Gewerbpath. Gewerbhyg., 23: 226-240.
GRANICK, J. L., SASSA, S., GRANICK, S., LEVERE, R. D., & KAPPAS, A.
(1973) Studies in lead poisoning II Correlation between the ratio
of activated to inactivated aminolevulinic acid dehydratase of
whole blood and the blood lead level. Biochem. Med.,
8: 149-159.
GRANICK, S., SASSA, S., GRANICK, J. L., LEVERE, R. D., & KAPPAS, A.
(1972) Assays for porphyrins. Proc. Nat. Acad. Sci. USA,
69: 2382-2385.
GRAOVAC-LEPOSAVIC, L., DJURIC, D., VALJAREVIC, V., SENICAR, L., MILIC,
S., & DELIC, V. (1973) Environmental lead contamination of Meza
Valley-Study on lead exposure of populations. In: Proceedings
of the International Symposium: Environmental Health Aspects of
Lead, Amsterdam, 2-6 October 1972. Luxembourg, Commission of
the European Communities, pp. 685-703.
GREENFIELD, S. M., BRIDBORD, K., BARTH, D., & ENGEL, R. (1973) The
changing perspectives regarding lead as an environmental
pollutant. In: Proceedings of the International Symposium:
Environmental Health Aspects of lead, Amsterdam, 2-6 October
1972. Luxembourg, Commission of the European Communities,
pp. 19-27.
GRIFFITH, J. Q., & LANDAUER, M. A. (1944) The effect of chronic lead
poisoning on arterial blood pressure in rats. Am. Heart J.,
28: 295-297.
GRIGGS, R. C., SUNSHINE, J., NEWILL, V. NEWTON, B. W., BUCHANAN, S., &
RASCH, C. A. (1964) Environmental factors in childhood lead
poisoning. J. Am. Med. Assoc., 187: 703-707.
GRIMES, H., SAYERS, M. H. P., CERNIK, A. A., BERLIN, a., RECHT, P., &
SMEETS, J. (1975) Note on the lead exposure of children:
determinations carried out on behalf of the Commission in
Western Ireland. Luxembourg, Commission of the European
Communities (Report V/F/1491/75), pp. 7.
GUINEE, V. F. (1973) Epidemiologic studies of lead exposure in New
York city. In: Proceedings of the International Symposium;
Environmental Health Aspects of Lead, Amsterdam, 2-6 October
1972. Luxembourg, Commission of the European Communities,
pp. 763-770.
GUTNIAK, O., KOZIOLOWA, H., & KOWALSKI, E. (1964) Free protoporphyrin
content of erythrocytes in chronic tetraethyl lead poisoning.
Lancet, 1: 1137-1138.
HAAS, T., MACHE, K., SCHALLER, K.-H., MACHE, W., & VALENTIN, H.
(1972a) Investigations into ecological lead levels in
children.(Untersuchungen über die ökologische Bleibelastung im
Kindesalter). Zbl. Bakt. Hyg. I. Abt. Orig., B 156: 353-360.
HAAS, T., WIEK, A. O., SCHALLER, K. H., MACHE, K., & VALENTIN, R.
(1972b) Die usuelle Bleibelastung bei Neugeborenen und ihren
Muttern. Zbl. Bakt. Hyg., I. Abt. Orig., B 155, 341-349.
HACIROV, D. G. (1972) Data on electronmicroscopic investigations of
erythrocytes and thrombocytes under saturnism. Naucn. Tr. Sev.-
Oset. Med. Inst., 36(4): 59-63.
HAEGER-ARONSEN, B. (1960) Studies on urinary excretion of delta-
aminolaevulic acid and other haem precursors in lead workers and
lead-intoxicated rabbits. Scand. J. clin. Lab. Invest., 12
(Suppl. 47) 1.
HAEGER-ARONSEN, B. (1971) An assessment of the laboratory tests used
to monitor the exposure of lead workers. Brit. J. ind. Med.,
28: 52-58.
HAEGER-ARONSEN, B., ABDULLA, M., & FRISTEDT, B. (1971) Effect of lead
on delta-aminolevulinic acid dehydrase activity in red blood
cells. Arch. environ. Health, 23: 440-445.
HAMMOND, P. B. (1971) The effects of chelating agents on the tissue
distribution and excretion of lead. Toxicol. Pharmacol.,
18: 296-310.
HAMMOND, P. B., & ARONSON, A. L. (1964) Lead poisoning in cattle and
horses in the vicinity of a smelter. Ann. N.Y. Acad. Sci.,
III: 595-611.
HAMMOND, P. B., WRIGHT, H. N., & ROEPKE, M. H. (1956) A method for the
detection of lead in bovine blood and liver. University of
Minnesota. Agric. Exp. Stat. Techn. Bull., 221: 3-14.
HANKIN, L., HEICHEL, G. H., & BOTSFORD, R. A. (1973) Lead poisoning
from coloured printing inks. Clin. Pediatr., 12: 654-655.
HAPKE, H. J. (1974) Effects and damage by lead, cadmium and zinc on
useful animals. Staub Reinhaltung der Luft, 34(1): 8-11.
HAPKE, H. J., & PRIGGE, E. (1973) Interactions of lead and glutathione
with delta-aminolevulinic acid dehydratase. Arch. Toxicol.,
31: 153-161.
HARRIS, R. W., & ELSEA, W. R. (1967) Ceramic glaze as a source of lead
poisoning. J. Am. Med. Assoc., 202: 544-549.
HARRISON, R. M., PERRY, R., & SLATER, D. H. (1974) An adsorption
technique for the determination of organic lead in street air.
Atmos. Environ., 8: 1187-1194.
HASAN, J., & HERNBERG, S. (1966) Interactions of inorganic lead with
human blood cells. Work environ. Health, 2: 26-44.
HELLER, A., & KETTNER, H. (1969) Probenahme und Bestimmune kleinster
Bleimengen in der Luft. Wasser-Boden- und Lufthygiene, No. 29,
3-50.
HEMPHILL, F. E., KAEBERLE, M. L., & BUCK, W. B. (1971) Lead
suppression of mouse resistance to salmonella typhimurium.
Science, 172: 1031-1032.
HENDERSON, D. A. (1958) The etiology of chronic nephritis in
Queensland. Med. d. Austr, 1: 377-386.
HENDERSON, D. A., & INGLIS, J. A. (1957) The lead content of bone in
chronic Bright's disease. Austr. Ann. Med, 6: 145-154.
HERNBERG, S. (1967) Lifespan, potassium fluxes and membrane ATPases of
erythrocytes from subjects exposed to inorganic lead. Work
environ. Health, 3, suppl. 1, pp. 1-74.
HERNBERG, S. (1973) Prevention of occupational poisoning from
inorganic lead. Work environ. Health, 10: 53-61.
HERNBERG, S. (1975) Lead In: Zenz, C. (ed.) Occupational Medicine
Yearbook Pt 4: The Chemical Occupational Environment, Chicago,
pp. 715-769.
HERNBERG, S., LILIUS, H., MELLIN, G., & NIKKANEN, J. (1969) Lead
exposure of workers in printing shops. Work environ. Health,
6(2): 5-8.
HERNBERG, S., NIKKANEN, J., MELLIN, G., & LILIUS, H. (1970) delta-
aminolevulinic acid dehydrase as a measure of lead exposure.
Arch. environ. Health, 21: 140-145.
HEUSGEM, C., & DEGRAEVE, J. (1973) Importance de l'apport alimentaire
en plomb dans l'est de la Belgique. In: Proceedings of the
International Symposium; Environmental Health Aspects of Lead,
Amsterdam, 2-6 October 1972. Luxembourg, Commission of the
European Communities, pp. 85-91.
HICKS, J. M., GUTIERREZ, A. N., & WORTHY, B. E. (1973) Evaluation of
the Delves micro-system for blood lead analysis. Clin. Chem.,
19: 322-325.
HILL, C. R. (1960) Lead-210 and polonium-210 in grass. Nature
(Lond.), 187: 211-212.
H M CHIEF INSPECTOR OF FACTORIES (1972) H.M. Great Britain Department
of Employment. Annual Report. London, Her Majesty's Stationery
Office.
HOFFMAN, E. O., DI LUZIO, N.R., HOLPER, K;, BRETTSCHNEIDER, L., &
COOVER, J. (1974). Ultrastructural changes in the liver of
baboons following lead and endotoxin administration. Lab.
Invest., 30: 311-319.
HOFREUTER, D. H., CATCOTT, E. J., KEENAN, R. G., & XINTARAS, A. B.
(1961) The public health significance of atmospheric lead. Arch.
environ. Health, 3: 568-574.
HOLMQVIST, J. (1966) Normal lead values in blood. 15th International
Congress of Occupational Health. Vienna 19-24 Sept., A-III-1,
pp. 179-183.
HOMMA, K. (1966) Experimental study of preparing metal fumes. Ind.
Health, 4: 129-137.
HOPKINS, A. P., & DAYAN, A. D. (1974) The pathology of experimental
lead encephalopathy in the baboon (Papio amibis). Brit. J. ind.
Med., 31: 128-133.
HORIGUCHI, S., & UTSUNOMIYA, T. (1973) An estimate of the body burden
of lead in the healthy Japanese population. An attempt to assume
absorption and excretion of lead in the healthy Japanese
population, Part 2. Osaka City, med. J., 19: 1-5.
HORIUCHI, K., (1970) Lead in the environment and its effect on man in
Japan. Osaka City, med. J., 16: 1.
HORIUCHI, K., & TAKADA, J. (1954) Studies on the industrial lead
poisoning. I. Absorption, transportation, deposition and
excretion of lead, 1. Normal limits of lead in the blood, urine
and feces among healthy Japanese urban inhabitants. Osaka City,
reed. J., 1: 117-125.
HORIUCHI, K., HORIGUCHI, T., KASAHARA, A., MORIOKA, T., UTSUNOMYA, T.,
& SHINAGAWA, K. (1964) Influences of temperature on manifestation
of symptoms in industrial poisoning. 1. Influences of high
temperature in lead poisoning. Jpn. J. ind. Health, 6: 170-171.
HORIUCHI, K., HORIGUCHI, S., SHINAGAWA, K., TAKADA, F., & TERAMOTO, K.
(1968) A polarographic method for the determination of a small
amount of lead in biological materials. Osaka City, med. J,
14: 113-118.
HORIUCHI, K., HORIGUCHI, S., & SUEKANE, M. (1959) Studies on the
industrial lead poisoning. I. Absorption, transportation,
deposition and excretion of lead. 6. The lead contents in organ
tissues of the normal Japanese. Osaka City, reed. J., 5: 41-70.
HORIUCHI, K., YAMAMOTO, T., & TAMORI, E. (1956) Studies on the
industrial lead poisoning. I. Absorption, transportation,
deposition and excretion of lead. 2. A study on the lead content
in daily food in Japan. Osaka City, med. J., 3: 84-113.
HOWER, J., PRINZ, B., GONO, E., & REUSMANN, G. (1975) Untersuchungen
zum Zusammenhangzwischen dem Blutbleispiegel bei Neugeboren en
und der Bleiimmissionsbelastung der Mütter am Wohnort. In:
Proceedings of CEC-EPA-WHO International Symposium; Recent
Advances in the Assessment of the Health Effect of Environmental
Pollution, Paris, 24-28 June 1974. Luxembourg, Commission of
the European Communities, pp. 591-603.
HUNT, W. F., PINKERTON, C., MCNULTY, O., & CREASON, J. (1971) A study
in trace element pollution in 77 midwestern cities. In: ed. D.
D. Hemphill Proceedings of the University of Missouri's 4th
Annual Conference on Trace Substances in Environmental Health,
Columbia, Missouri, 23-25 June 1970. University of Missouri,
Columbia, 1971, pp. 56-68.
HUNTZICKER, J. J., FRIEDLANDER, S. K., & DAVIDSON, C. J. (1975)
Material balance for automobile-emitted lead in Los Angeles
basin. Environ. Sci. Technol., 9: 448-457.
HURSH, J. B., & MERCER, T. T. (1970) Measurement of Pb212 loss rate
from human lungs. J. appl. Physiol., 28: 268.
HWANG, J. Y., ULLUCCI, P. A., SMITH, S. B., & MALENFANT, A. L. (1971)
Microdetermination of lead in blood by flameless atomic
absorption spectrometry. Anal. Chem., 43: 1319-1321.
IARC (1972) Monographs. Evaluation of carcinogenic risk of chemicals
to man. Vol. 1, Lyon, International Agency for Research on
Cancer, pp. 184.
INTER-DEPARTMENT WORKING GROUP ON HEAVY METALS (1974) Lead in the
environment and its significance to man. Report for the
Department of the Environment Central Unit on Environmental
Pollution. London, Her Majesty's Stationery Office, pp. 1-47,
Pollution Paper No. 2.
INTERNATIONAL LEAD AND ZINC STUDY GROUP (1973) Lead in gasoline: A
review of the current situation, International Lead and Zinc
Study Group, United Nations, New York, pp. 1-37. First addendum
1974; Second addendum 1975; Third addendum 1976.
IUPAC-IUB COMMISSION ON BIOCHEMICAL NOMENCLATURE (1973) Enzyme
nomenclature. Recommendations (1972) of the Commission on
Biochemical Nomenclature on the nomenclature and classification
of enzymes together with their units and the symbols of enzyme
kinetics. Amsterdam, Elsevier.
JARVIE, A. W. P., MARKALL, R. N., & POTTER, H. R. (1975) Chemical
alkylation of lead. Nature (Lond.), 255: 217-218.
JAWOROSKI, Z. (1968) Stable lead in fossil ice and bones. Nature
(Lond.), 217: 152-153.
JERNIGAN, E. L., RAY, B. J., & DUCE, R. A. (1971) Lead and bromine in
atmospheric particulate matter on Oahu, Hawaii. Atmosph.
Environ., 5: 881-886.
JONES, R. D., COMMINS, B. T., & CERNIK, A. A. (1972) Blood lead in
carboxyhemoglobin levels in London taxi drivers. Lancet,
2: 302-303.
JOST, D., MULLER, J., & JENDRICKE, U. (1973) Lead in atmospheric
aerosol. In: Proceedings of the International Symposium;
Environmental Health Aspects of Lead, Amsterdam, 2-6 October
1972. Luxembourg, Commission of the European Communities,
pp. 941-948.
KAMMHOLZ, L. P, THATCHER, L. G., BLODGETT, F. M., & GOOD, T. A. (1972)
Rapid protoporphyrin quantitation for detection of lead
poisoning. Pediatrics, 50: 625-631.
KASSENAAR, A., MORELL, H., & LONDON, I. U. (1957) The incorporation of
glycine into globin and the synthesis of heme in vitro in duck
erythrocytes. J. Biol. Chem., 229: 423-435.
KATO, K. (1932) Lead meningitis in infants. Am. J. Dis. Child.,
44: 569-591.
KEENAN, R. C., BYERS, D. H., SALTZMAN, B. E., & HYSLOP, F. L. (1963)
The "USPHS" method for determining lead in air and in biological
materials. Am. Ind. Hyg. Assoc., 24: 481-491.
KEHOE, R. A. (1961) The metabolism of lead in health and disease. The
Harben Lectures, 1960. J. Roy. Inst. Publ. Health Hyg.,
24: 81-96, 101-120, 129-143, 177-203.
KEHOE, R. A., CHOLAK, J., & LARGENT, E. J. (1944) The concentrations
of certain trace metals in drinking water. J. Am. Water Works.
Assoc., 36: 637-644.
KEHOE, R. A., THAMAN, F., & CHOLAK, J. (1933) Lead absorption and
excretion in relation to the diagnosis of lead poisoning.
J. ind. Hyg., 15: 320.
KELLO, D., & KOSTIAL, K. (1973) The effect of milk diet on lead
metabolism in rats. Environ. Res., 6: 355-360.
KEMPF, TH., & SONNEBORN, M. (1973) Vergleich von Methoden zur
Bestimmung von Spurenmetallen in Wasserkreislauf. Z. anal.
Chem., 267: 267-270.
KENNEDY, V. C. (1960) Geochemical studies in the Coeur d'Alene
district Shoshone County, Idaho. U.S. geol Survey Bull.,
1098-A.
KEPPLER, J. F., MAXFIELD, M. E., MOAS, W. D., TIETJEN, C., & LINCH, A.
L. (1970) Interlaboratory evaluation of the reliability of blood
lead analyses. Am. Ind. Hyg. Assoc. J., 31: 412-429.
KERIN, Z. (1972) Tägliche Bleiaufnahme Kit der Bauernkost aus dem
Emissionsgebiet einer Bleihutte. Protectio vitae, 71: 22-23.
KERIN, Z. (1973) Lead in new-fallen snow near a lead smelter. Arch.
environ. Health, 26: 256-260.
KERIN, Z., KERIN, D., & DJURIC, D. (1972) Lead contamination of
environment in Meza Valley. Lead content of the soil. Int. Arch.
Arbeitsmed., 29: 129-138.
KLEIN, U. C., SAYRE, J. W., & KOTOK, D. (1974) Am. J. Dis. Childh.,
127: 805-807. porphyrin in duck erythrocytes. Am. J. Physiol.,
203: 971-974.
KLEIN, M., NANER, R., HARPUR, E., & CORBIN, R. (1970) Earthenware
containers as a source of fatal lead poisoning. Case study and
public-health considerations. New Eng. J. Med., 283: 669-672.
KLEIN, U. C., SAYRE, J. W., & KOTOK, D. (1974). Am. J. Dis. Child.,
127: 805-807.
KLINE, T. S. (1960) Myocardial changes in lead poisoning. A.M.A.J.
Dis. Child., 99: 48-54.
KNEIP, T. J., & LAUREN, G. R. (1972) Isotope excited X-ray
fluorescence. Anal. Chem., 44: 57A.
KOBAYASHI, N., & OKAMOTO, T. (1974) Effects of lead oxide on the
induction of lung tumors in Syrian hamsters. J. Nat. Cancer
Inst., 52(5): 1605-1610.
KOLBYE, A. C., MAHAFFEY, R., FIORINO, A., CORNELIUSSEN, P. C., &
JELINEK, C. F. (1974) Food exposures to lead. Environ. Health
Perspect., Exp. Issue No. 7, pp. 65-75.
KOPP, J. F., & KRONER, P. T. (1970) Trace metals in water of the
United States: a 5-year summary of trace metals in rivers and
lakes of the US. (Oct. 1962-Sept. 1967) Cincinnati, Ohio, US
Department of the Interior.
KOSMIDER, S., & PETELNZ, T. (1962) Zmiany elektrokardiograficzne u
starszych osob z przewleklym zawodowym zatruciem olowiem. Pol.
Arch. Med. Wewn, 32: 437-442.
KOSMIDER, S., & SROCZYNSKI, J. (1961) Zmiany elektrokardiograficzne w
przewleklym doswiadczalney olowiej u krolikow
(Electrocardiographic changes in chronic experimental plumbism in
rabbits). Postepy Hig. i Med. Dosw., 15: 353-357.
KOSTIAL, K., & VOUK, V. B. (1957) Lead ions and synaptic transmission
in the superior cervical ganglion of the cat. Brit. J.
Pharmacol., Chemother., 12: 219-222.
KOSTIAL, K., MALJKOVIC, T., & JUGO, S. (1974) Lead acetate toxicity in
rats in relation to age and sex. Arch. Toxicol., 31: 265-269.
KOSTIAL, K., SIMONOVIC, I., & PISONIC, U. (1971) Lead absorption from
the intestine in newborn rats. Nature (Lond.), 233: 564.
KOTOK, D. (1972) Development of children with elevated blood lead
levels; A controlled study. J. Pediatr., 80: 57-61.
KRIGMAN, U. R., DRUSE, U. J., TAYLOR, T. D., WILSON, U. H., NEWELL, L.
R., & HOGAN, E. L. (1974) Lead encephalopathy in the developing
rat: Effect upon myelination. J. Neuropath. exp. Neurol.,
33(1): 58-74.
KUBASIK, N. P., VALOSIN, U. T., & MURRAY, U. H. (1972) Carbon rod
atomizer applied to measurement of lead in whole blood by atomic
absorption spectrophotometry. Clin. Chem., 18: 410-412.
KUZ'MINSKAJA, T. N. (1964) Experimental atherosclerosis against a
background of Pb-intoxication. Arch Patol., 26(9): 21-24.
LAHMANN, E. (1969) Untersuchungen über Luftverunreinigungen dutch den
Kraftverkehr. Wasser-boden-und Lufthyg., No. 28, 3-80.
LAMM, S. H., & ROSEN, J. F. (1974) Lead contamination in milk fed to
infants' 1972-1973. Pediatrics, 53: 137-141.
LAMOLA, A. A., & YAMANE, T. (1974) Zinc protoporphyrin in the
erythocytes of patients with lead intoxication and iron
deficiency anaemia. Science, 186: 936-938.
LAMOLA, A. A., JOSELOW, M., & YAMANE, T. (1975) Zinc protoporphyrin
(ZPP): A simple sensitive, fluorometric screening test for lead
poisoning. Clin. Chem., 21: 93-97.
LAMPTER, P. W., & SCHOCHET, S.S. (1968) Demylination and remyelination
in lead neuropathy. Electron microscopic studies. J. Neuropath.
exp. Neurol., 25: 527-545.
LANCRANJAN, I., POPESCU, H. I., GAVANESCU, O., KLEPSCH, I., &
SERBANESCU, M. (1975). Reproductive ability of workmen
occupationally exposed to lead. Arch. environ. Health,
30: 396-401.
LANE, R. E. (1949) The care of the lead worker. Brit. J. ind. Med.,
6: 125-143.
LANE, R. E. (1964) Health control in inorganic lead industries. A
follow-up of exposed workers. Arch. environ. Health, 8: 55.
LANDING, B. H., & NAKAI, H. (1959) Histochemical properties of renal
lead inclusions and their demonstration in urinary sediment. Am.
J. clin. Path., 31: 499-503.
LANDRIGAN, P. J., BALCH, R. W., BARTHEL, W. F., WHITWORTH, R. H.,
STAEHLING, N. W., & ROSENBLUM, B. F. (1975a) Neurophysiological
dysfunction in children with chronic low-level lead absorption.
Lancet, 1: 708-712.
LANDRIGAN, P. J., GEHLBACH, S. H., ROSENBLUM, B. F., SHOULTS, J. M.,
CANDELARIA, R. M. BARTHEL, W. F., LIDDLE, J. A., SMREK, A. L.,
STAEHLING, N. W., & SANDERS, J. F. (1975b) Epidemic lead
absorption near an ore smelter; The role of particulate lead.
New Engl. J. Med., 292: 123-129.
LANSDOWN, R. G., CLAYTON, B. E., GRAHAM, P. J., SHEPHERD, J., DELVES,
H. T., & TURNER, W. C. (1974) Blood-lead levels, behaviour and
intelligence: a population study. Lancet, 1: 538-541.
LAURS, A. J. (1976) Review of European test methods for measuring
lead and cadmium release. In: Proceedings of the International
Conference on Ceramic Foodware Safety, Geneva, 1974. New York,
Lead Industries Association Inc., pp. 27-36.
LAVESKOG, A. (1971) A method for determination of tetramethyl lead
and tetraethyl lead in air. Proceedings of the Second
International Clean Air Congress, H. H. England and W. T.
Beery, eds. New York, Acad. Press, pp. 549-557.
LAWTHER, P. J., COMMINS, B. T., ELLISON, J. MEK., & BILES, B. (1972)
Airborne lead and its uptake by inhalation. In: Hepple, P. ed.,
Lead in the environment. Essex, UK, Applied Science Publishers
Ltd., pp. 8-28.
LAZRUS, A. L., LORANGE, E., & LODGE, J.P. (1970) Lead and other
metalions in US precipitation. Environ, Sci. Technol.,
4: 55-58.
LEAD DEVELOPMENT ASSOCIATION (1976) Lead In Gasoline Bulletin No. 12,
Lead Development Association, 34 Berkeley Square, London W1X 6AJ,
30 March 1976.
LEE, R. E., & GORANSON, S. (1972) National air surveillance cascade
impactor network. I. Size distribution measurements of suspended
particulate matter in air. Environ. Sci. Technol.,
6: 1019-1024.
LEE, R. E., GORANSON, S.S., ENRIONE, R. E., & MORGAN, G. B. (1972) The
NASH cascade impactor network, II Size distribution of trace-
metal components. Env. Sci. Technol., 6: 1025-1030.
LEHNERT, G., MASTALL, H. SZADKOWSKI, D., & SCHALLER, K. H. (1970)
Berufliche Bleibelastung durch Autoabgase in Grosstadtstrassen.
Dtsch. Med. Wschr., 95: 1097-1099.
LEHNERT, G., SCHALLER, K. H., KÜHNER, A., & SZADKOWSKI, D. (1967)
Auswirkungen des Zigarettenrauchens aus dem Blutbleisspiegel.
Int. Arch. Geweberpath. Geweberhyg., 23: 358-363.
LEHNERT, G., SCHALLER, K. H., & SZADKOWSKI, D. (1969) Eine
zuverlässige Schnellmethode zur Bleibestimmung in kleinen
Blutmengen. Z. Klin. Chem. Klin. Biochem., 7: 310.
LEIKIN, S., & ENG, G. (1963) Erythrokmetic studies of the anemia of
lead poisoning. Pediatrics, 31: 996-1002.
LILIS, R., GAVRILESCU, N., NESTORESCU, B., DURIMTIU, C., & ROVENTA, A.
(1968) Nephropathy in chronic lead poisoning. Brit. J. ind.
Med., 25: 196-202.
LINCH, A. L., WIEST, E.G., & CARTER, U. D. (1970) Evaluation of
tetraalkyl lead exposure by personnel monitor surveys. Am. Ind.
Hyg. Assoc. Y., 31: 170-179.
LIVINGSTONE, D. A. (1963) Data of geochemistry. Sixth Edition. Chapter
of chemical composition of rivers and lakes. US Geol. Survey
Prof. Paper 440.
MACHLE, W. E. (1935) Tetraethyl lead intoxication and poisoning by
related compounds of lead. J. Am. Med. Assoc., 105: 578-585.
MAHOFFEY, K. (1974) Nutritional factors and susceptibility to lead
toxicity. Environ. Health Perspect., Exp. Issue No. 7,
pp. 107-113.
MAKASEV, K. K., & KRIVDINA, L. V. (1972) Status of the interstitial
tissues of vascular walls and their penetration under the lead
poisoning. Tr. Naucn-issl. In-ta Kraev. Pat. Kaz. SSR,
23: 11-13.
MAKASEV, K. K., & VERBOLOVIC, V. P. (1967) Succinic dehydrogenase and
cytochrome oxidase in the duodenum during lead poisoning. Izv.
Akad. Nauk Kazahst. SSR Set. Biol., 5(2): 59-64.
MALCOLM, D. (1971) Prevention of long-term sequelae following the
absorption of lead. Arch. environ. Health, 23: 292-298.
MALJKOVIC, J. (1971) A case of occupational poisoning with lead
carbonate and stearate. Sigurnost u pogonu., 13: 123-124.
MAMBEEVA, A. A., & AHMEDOVA, A. S. (1967) Changes of reactivity of the
small intestine in healthy and lead-intoxicated homeotherms to
histamine under the influence of solutions of lead acetate.
Biull. eksper. Biol., 32: 34-37.
MAMBEEVA, A. A., & KOBKOVA, I. D. (1969) The concentration of
catecholamines in tissues of the cardiovascular system in
experimental lead intoxication. Izv. Akad, Nauk Kazahst. SSR.
Ser. Biol., No. 1: 77-82.
MANALIS, R. S., & COOPER, G. I. (1973) Presynaptic and postsynaptic
effects of lead at the frog neuromuscular junction. Nature
(Lond.), 243: 354-355.
MAO, P., & MOLNAR, J. J. (1967) The fine structure and histochemistry
of lead-induced renal tumours in rats. Am. J. Pathol.,
50: 571-603.
MAPPES, R. (1972) Eine Fehlerquelle und ihre Kompensation bei der
delta-Amino-lävulinsäurebestimmung im Harn nach Grabecki. Int.
Arch. Arbeitsmed., 30: 81-86.
MARTIN, A. E., FAIRWEATHER, F. A., BUXTON, R. S. J., & ROOTS, L, M.
(1975) Recent epidemiological studies of environmental lead of
industrial origin. In: Proceedings of the CEP-EPA-WHO
International Symposium; Recent Advances in the Assessment of
the Health Effects of Environmental Pollution, Paris. 24-28 June
1974. Luxembourg, Commission of the European Communities.
pp. 1113-1120.
MARVER, H. S., TSCHUDY, D. P., PERLROTH, M. G., COLLINS, A., & HUNTER,
G. JR. (1966) The determination of aminoketones in biological
fluids. Anal. Biochem., 14: 53-60.
MATOUSEK, J. F., & STEVENS, B. J. (1971) Biological applications of
the carbon rod atomizer in atomic absorption spectroscopy. I.
Preliminary studies on magnesium, iron, copper, lead and zinc in
blood in plasma. Clin. Chem., 17: 363-368.
MATSON, W. R. (1971) Rapid sub-nanogram simultaneous analysis. In:
Proceedings of the University of Missouri's 4th Annual
Conference on Trace Substances in Environmental Health,
Columbia, Missouri, 23-25 June 1970. Columbia, ed. D. D.
Hemphill, University of Missouri, pp. 396-406.
MATTSSON, R., & JAAKKOLA, T. (1974) Lead in the Helsinki air.
Ympäristö ja Terveys, 5: 721.
MAUZERALL, D., & GRANICK, S. (1956) The occurrence and determination
of delta- aminolevulinic acid and porphobilinogen in urine.
J. Biol. Chem., 219: 435-446.
MAXFIELD, M. E., STOPPS, G. J., BARNES, J.P., SNEE, R. D., & AZAR, A.
(1972) Effect of lead on blood regeneration following acute
hemorrhage in dogs. Am. Ind. Hyg. Assoc. J., 33: 326-337.
MCCABE, L. J. (1970) Corrosion by soft water. Amer. Water Works
Seminar on Corrosion by Soft Water. Wash. DC, pp. 9.
MCLAUGHLIN, M., & STOPPS, G. J.(1973) Smoking and Lead. Arch. environ
Health, 26: 131-136.
MCLAUGHLIN, M., LINCH, A. L., & SNEE, R. D. (1973) Longitudinal
studies of lead levels in US population. Arch. environ. Health,
27: 305-312.
MCMULLEN, T. B, FAORO, R. B., & MORGAN, G. B. (1970) Profile of
pollutant fractions in non-urban suspended particulate matter.
J. Air Pollut. Control Assoc., 20: 369-372.
MCNEIL, J. L., & PTASNIK, J. A. (1975) Evaluation of long-term
effects of elevated blood lead concentrations in asymptomatic
children. In: Proceedings of CEC-EPA-WHO International
Symposium; Recent Advances in the Assessment of the Health
Effects of Environmental Pollution, Paris, 24-28 June 1974.
Luxembourg, Commission of the European Communities, pp. 571-579.
MENDEN, R. E., ELIA, V. J., MICHAEL, L. W., & PETERING, H. C. (1972)
Distribution of Cd and Ni of tobacco during cigarette smoking.
Env. Sci. Technol., 6: 830-832.
MERCER, T. T. (1975) The deposition model of the Task Group on Lung
Dynamics: A comparison with recent experimental data. Health
Phys., 29: 673-680.
MERWIN, B. W. (1976) Review of U.S. standards on test methods for
lead and cadmium release. In: Proceedings of the International
Conference on Ceramic Foodware Safety, Geneva, 1974. New York,
Lead Industries Association Inc., pp. 36-40.
MICHAELSON, J. A. (1973) Effect of inorganic lead on levels of RNA,
DNA and protein on developing neonatal rats. Toxicol. appl.
Pharmacol., 26: 539-548.
MICHAELSON, J. A., & SAUERHOFF, M. W. (1974) An improved model of lead
induced brain dysfunction in the suckling rat. Toxicol. appl.
Pharmacol., 28: 88-96.
MILLAR, J. A., BATTISTINI, V., GUMMING, R. L. C., CARSWELL, F., &
GOLDBERG, A. (1970) Lead and delta-aminolevulinic acid
dehydratase levels in mentally retarded children and in lead-
poisoned suckling rats. Lancet, 2: 695-698.
MIRONCIK, L. M., & TIMOFEEVA, N. D. (1974) Lead effects on the
experimental athero-sclerosis process and several respiratory
ferments of the myocard. Probl. Gig. i organiz zdravoochr. v
Uzbekistane, 2: 77-78.
MITCHELL, D. G., & ALDOUS, K. M. (1974) Lead content of foodstuffs.
Environ. Health Perspect., Exp. Issue No. 7, pp. 59-65.
MITCHELL, D. G., ALDOUS, K. M., & RYAN, F. J. (1974) Mass screening
for lead poisoning, capillary blood sampling and automated Delves
cup atomic absorption analysis. N.Y. State, J. Med., 74: 1599.
MITCHELL, R. L. (1963) Soil aspects of trace element problems in
plants and animals. J. Roy. Agric. Soc., 124: 75-86.
MITCHELL, R. L., & REITH, J. W. S. (1966) The lead content of pasture
herbage. J. Sci. Food Agric., 17: 437-440.
MÖLLER, B., AKSELLSSON, R., BERLIN, M., FISCHBEIN, A., HAMMARSTRÖM, L.
(1974) Sammanfatting av föredrag rid Läkaresfällskapets
Riksstämma 27-30 Nov., 1974 (Omgivningshygien No. 7).
MOMCILOVIC, B., & KOSTIAL, K. (1974) Kinetics of lead retention and
distribution in suckling and adult rats. Environ. Res.,
8: 214-220.
MONAENKOVA, A.M. (1957) Functional state of the thyroid gland in
chronic intoxication from several industrial poisons (Pb, Hg).
Gig. Trud. Prof. Zabol., 2: 44-48.
MONAENKOVA, A.M., & GLOTOVA, K. V. (1969) Cardiovascular changes in
some chronic intoxications with industrial poisons (lead, carbon
disulphide, benzene). Gig. Trud. Prof. Zabol, 2: 32-35.
MONCRIEFF, A. A., KOUMIDES, O. P., CLAYTON, B. E., PATRICK, A.D.,
RENWICK, A. G. C., & ROBERTS, G. E. (1964) Lead poisoning in
children. Arch. Dis. Child., 39: 1-13.
MOORE, J. F., & GOYER, R. A. (1974) Lead-induced inclusion bodies;
Composition and probable role in lead metabolism. Environ.
Health Perspect., Exp. Issue No. 7, pp. 121-129.
MOORE, M. R. (1973) Plumbosolvency of waters. Nature Lond.,
243: 222-223.
MORGAN, B. B., & REPKO, J. D. (1974) Evaluation of behavioural
functions in workers exposed to lead. In: C. Xintaras et al.,
eds, Behavioural toxicology, early detection of occupational
hazards. Washington, U.S. Department of Health, Education and
Welfare.
MORGAN, J. M., HARTLEY, M. W., & MILLER, R. E. (1966) Nephropathy in
chronic lead poisoning. Arch. intern. Med., 118: 17-29.
MUELLER, P. K. (1970) Discussion (characterization of particulate lead
in vehicle exhaust experimental techniques). Environ. Sci.
Technol., 4: 248-251.
MUIR, D.C. F, & DAVIES, C. N. (1967) The deposition of 0.5 µ diameter
aerosols in the lungs of man. Ann. occup. Hyg., 10: 161-174.
MURO, L. A., & GOYER, R. A. (1969) Chromosome damage in experimental
lead poisoning. Arch. Pathol., 87: 660-663.
MUROZUMI, M., CHOW, T. J., PATTERSON, C. C. (1969) Chemical
concentrations of pollutant lead aerosols, terrestrial dusts and
sea salts in Greenland and Antarctic snow strata. Geochem.
Cosmochim. Acta, 33: 1247-1294.
MURTHY, G. M., & RHEA, U.S. (1971) Cadmium, copper, iron, lead,
manganese and zinc in evaporated milk, infant products and human
milk. J. Dairy Sci., 54: 1001-1005.
MYERSON, R. M., & EISENHAUER, J. H. (1963) Atrioventricular conduction
defects in lead poisoning. Am. d. Cardiol., 11: 409-412.
NAKAO, K., WADA, O, & YANO, Y. (1968) Delta-aminolevulinic acid
dehydratase activity in erythrocytes for the evaluation of lead
poisoning. Clin. Chim. Acta., 19: 319-325.
NAS-NRC (1972) Airborne lead in perspective. Washington, DC, Nat.
Aced. Sci.
NEAL, P. A., DRESSEN, W. C., EDWARDS, T. J., REINHART, W. H., WEBSTER,
S. H., CASTBERG, H. T., & FAIRHALL, L. T. (1941) A study of the
effect of lead arsenate exposure on orchardists and consumers of
sprayed fruit. US publ. Health Bull., No. 267.
NEEDLEMAN, H. L., & SHAPIRO, J. M. (1974) Dentine lead levels in
asymptomatic Philadelphia school children: Subclinical exposure
in high and low risk groups. Environ. Health, Perspect, Exp.
Issue. No. 7, pp. 27-33.
NELSON, W. C., LYKINS, M. H., MACKEY, J., NEWILL, V. A., FINKLEA, J.
F., & HAMMER, D. J. (1973) Mortality among orchard workers
exposed to lead arsenate spray: A cohort study. J. chron. Dis.,
26: 105-118.
NIKKANEN, J., HERNBERG, S., & TOLA, S. (1972) Modifications of the
delta-aminolevulinic acid dehydratase test and their significance
for assessing different intensities of lead exposure. Work-
environ.-Health, 9: 46-52.
NIKLOWITZ, W. J., & YEAGER, D. W. (1973) Interference of Pb with
essential brain tissue Cu, Fe and Zn as main determinant in
experimental tetraethyllead encephalopathy. Life Sci.,
13: 897-905.
NORDBERG, G. F., ed. (1976) Effects and dose-response relationships of
toxic metals. Proceedings from an international meeting
organized by the subcommittee on the Toxicology of Metals of the
Permanent Commission and International Association on
Occupational Health, Tokyo, 18-23 November 1974. Amsterdam,
Elsevier, 1976, p. 15.
NORDMAN, C. H. (1975) Environment lead exposure in Finland. A study
on selected population groups. Doctoral Thesis, University of
Helsinki, pp. 1-118.
NORDMAN, C. H., & HERNBERG, S. (1975) Blood lead levels and
erythrocyte delta-aminolevulinic acid dehydratase activity of
selected population groups in Helsinki. Scand. J. Work-Environ.-
Health, 1: 219-232.
NORDMAN, C. H., HERNBERG, S., NIKKANEN, J., & RYHANEN, A. (1973) Blood
lead levels and erythrocyte-delta-aminolevulinic acid dehydratase
activity in people living around a secondary lead smelter. Work-
Environ.-Health, 10: 19-25.
NOZAKI, K. (1966) Method for studies on inhaled particles in human
respiratory system and retention of lead fume. Ind. Health
(Jpn), 4: 118-128.
NRC, CANADA (1973) Lead in the Canadian Environment. Associate
Committee on Scientific Criteria for Environmental Quality.
NYE, L. J. J. (1929) An investigation of the extraordinary incidence
of chronic nephritis in young people in Queensland. Med. J.
Aust., 2: 145-159.
OEHLKERS, F. (1953). Chromosome breaks influenced by chemicals.
Heredity, 6 (suppl.) 95-105.
OKAZAKI, H., ARONSON, S. M., DIMAIO, D. J., & OLVERA, J. E. (1963)
Acute lead encephalopathy of childhood. Trans. Am. neurol.
Assoc., 88: 248-250.
OLIVER, T. (1914) Lead Poisoning. London, H. K. Lewis.
ORLOVA, A. A. (1954) Changes in cardiac activity of patients with lead
and mercury intoxication. Trud. Akad. Med. Nauk. SSSR.,
31: 102-112.
O'RIORDAN, M. L., & EVANS, H. J. (1974) Absence of significant
chromosome damage in males occupationally exposed to lead.
Nature (Lond.), 247: 50-53.
OTT, W., CLARKE, J. F., & OZOLINS, G. (1970) Calculating future
carbon monoxide emissions and concentrations from urban traffic
data. Durham, DHEW Publ. no. AP-41.
OYASU, R., BATTIFORA, H. A., CLASEN, R. A., MCDONALD, J. H., & HASS,
G. M. (1970) Induction of cerebral gliomas in rats with dietary
lead subacutate and 2-acetylamino-fluorene. Cancer Res.,
30: 1248-1261.
PACKHAM, R. F. (1971) The leaching of toxic stabilizers for
unplasticized PVC water pipe: P. 1 A critical study of laboratory
test procedures, P.2. A survey of lead levels in PVC distribution
systems. Water Treat. Exam., 20: 108-124; 144-166.
PADILLA, F., SHAPIRO, A. P., & JENSEN, W. N. (1969) Effect of chronic
lead intoxication on blood pressure in the rat. Am. J. reed.
Sci., 258: 359-365.
PALMISANO, P. A., SNEED, R. C., & CASSADY, O. (1969) Untaxed whisky
and fetal lead exposure. J. Pediatr., 75: 869.
PANOVA, Z. (1972) Early changes in the ovarian function of women in
occupational contact with inorganic lead. Works United Res.
Inst. Hyg. ind. Saf. (Sofia), 23: 161-166.
PATEL, A. J., MICHAELSON, J. A., CREMER, J. E., & BALAZS, R.
(1974a)The metabolism of (C14) glucose by the brains of
suckling rats intoxicated with inorganic lead. J. Neurochem.,
22: 581-590.
PATEL, A. J., MICHAELSON, J. A., CREMER, J. E., & BALAZS, R. (1974b)
Changes within metabolic compartments in the brains of young rats
ingesting lead. J. Neurochem., 22: 591-598.
PATTERSON, C. C. (1965) Contaminated and natural lead environments of
man. Arch environ. Health, 11: 344-363.
PEGUES, W. L. (1960) Lead fume from welding on galvanized and zinc-
silicate coated steels. Ind. Hyg. J., 21: 252-255.
PEKKARINEN, M. (1970) Methodology in the collection of food
consumption data. World Rev. Nut. Diet., 12: 145-170.
PENTSCHEW, A. (1965) Morphology and morphogenesis of lead
encephalopathy. Acta Neuropathol., 5: 133-160.
PENTSCHEW, A., & GARRO, F. (1966) Lead encephalo-myelopathy of the
suckling rat and its implications on the porphyrinopathic nervous
diseases. Acta Neuropathol., 6: 266-278.
PERNIS, B., DE PETRIS, S., BEARD, R. R., & KARLSHAD, G. (1964) The
ultrastructure of red cells in experimental lead-poisoning.
Med. Lav., 55: 81-101.
PINES, A. G. (1965) Indexes of general reactivity in saturnine. Vrac.
Delo, 3: 93-96.
PIOMELLI, S. (1973) A micromethod for free erythrocyte porphyrin: the
FEP test. J. Lab. clin. Med., 81: 932-940.
POTT, F., & BROCKHAUS, A. (1971) Vergleich der enteralen und
pulmonalen Resorptionsquote von Bleiverbindungen. Zentralbl.
Bakt. Hyg. J. Orig. B., 155: 1-17.
PREROVSKA, I. (1973) Einfluss von Blei auf biochemische Veränderungen
im Serum und Veränderungen in der Aderwand im Hinblick auf
Atherosklerose. In: Proceedings of the International
Symposium. Environmental Health Aspects of Lead, Amsterdam, 2-6
October 1972. Luxembourg, Commission of the European
Communities, pp. 551-561.
PRPIC-MAJIC, D., MUELLER, P. K., LEW, V. C., & TWISS, S. (1973)
delta-aminolevulinic acid dehydratase stability in human blood.
Am. ind. Hyg. Assoc. J., 315-319.
PUESCHEL, S. M., KOPITO, L., & SCHWACHMAN, H. (1972) A screening and
follow up study of children with an increased lead burden.
J. Am. Med. Assoc., 333: 462-466.
PUHAC, J., HRGOVIC, N, STANKOVIC, M., & POPOVIC, S. (1963) Laboratory
investigations of the possibility of application of lead nitrates
compounds as a raticide means by decreasing reproductive
capability of rats. Acta Vet. (Beograd), 13: 3-9.
PURDUE, L. J., ENRIONE, R. E., THOMPSON, R. J., & BONFIELD, B. A.
(1973) Determination of organic and total lead in the atmosphere
by atomic absorption spectrometry. Anal. Chem, 45: 527-530.
RABINOWITZ, M. B. (1974) Lead contamination of the biosphere by human
activity. A stable isotope study. Ph.D. Thesis, University of
California, Los Angeles.
RABINOWITZ, M. B., WETHERILL, G. W., & KOPPLE, J. D. (1973) Lead
metabolism in the normal human: stable isotope studies. Science,
182: 725-727.
RABINOWITZ, M.B., WETHERILL, G. W., & KOPPLE, J. D. (1974) Studies of
human lead metabolism by use of stable isotope tracers. Environ.
Health. Perspect, Exp. Issue No. 7, pp. 145-155.
RASBERRY, S. D. (1973) Investigation of portable X-ray Fluorescence
analyzers for determining lead on painted surfaces. Appl.
Spectrosc., 27(2): 102-108.
RICHET, G., ALBAHARY, C., MOREL-MAROGER, L., GUILLAUME, P., & GALLE,
P. (1966) Les altérations rénales dans 23 cas de saturnisme
professionnel. Bull. Mere. Soc. Med. Hop. Paris, 117: 441-466.
RIEKE, F. E. (1969) Lead intoxication in shipbuilding and
shipscrapping 1941-1968. Arch. environ. Health, 19: 521-539.
RILEY, J. P., & SKIRROW, C. (1965) Chemical oceanography. New York,
Academic Press, vol. 2.
RIMINGTON, C. (1938) An enzymic theory of haemopoiesis. C.R. Lab
Carlsberg. Ser. Chim., 22: 454-464.
RIMINGTON, C., & SVEINSSON, S. L. (1950) The spectrophotometric
determination of uroporphyrin. Scand. J. clin. Lab. Invest.,
2: 209-216.
ROBERTS, T. M., HUTCHINSON, T. C., PACIGA, J., CHATTOPADHYAY, A.,
JERVIS, R. E., VAN-LOON, J., & PARKINSON, D. R. (1974) Lead
contamination around secondary smelters: Estimation of dispersal
and accumulation by humans. Science, 186: 1120-1123.
ROBINSON, E., & LUDWIG, F. L. (1967) Particle size distribution of
urban lead aerosols. J. Air Pollut. Control Assoc.,
17: 664-669.
ROBINSON, E., LUDWIG, F. L., DEVries, J. E, & HOPKINS, T. E. (1963)
Variations of atmospheric lead concentrations and type with
particle size. Final Report PA-4211. Cal. Stanford Res. Inst.
Menlo Park, pp. 1-80.
ROBINSON, T. R. (1974) Delta-aminolevulinic acid and lead in urine of
lead antiknock workers. Arch. Environ. Health, 28: 133-139.
ROE, F. Y., BOYLAND, C. E., DUKES, C. E., & MITCHLEY, B. V. C. (1965)
Failure of testosterone or xanthopterin to influence the
induction of renal neoplasms by lead in rats. Brit. J. Cancer,
19: 860-866.
ROELS, H. A., BUCHET, J.P., & LAUWERYS, R. R. (1974) Inhibition of
human erythrocyte delta-aminolevulinate dehydratase by lead.
Int. Arch. Arbeitsmed., 3: 277-281.
ROELS, H. A., LAUWERYS, R. R., BUCHET, J.P., & VRELUST, M. (1975)
Response of free erythrocyte porphyrin and urinary delta-
aminolevulinic acid in men and women moderately exposed to lead.
Int. Arch. Arbeitsmed., 34: 97-108.
ROSENBLUM, W. J., & JOHNSON, M. G. (1968) Neuropathologic changes
produced in suckling mice by adding lead to the maternal diet.
Arch. Pathol., 85: 640-648.
RÜHLING, A., & TYLER, G. (1968) An ecological approach to the lead
problem. Bot. Notiser, 121: 321-342.
RUITER, N. DE, SEEMAYER, N., & MANOJLOVIC, N. (1977) Einfluss yon
Zink-Ionen auf die toxische Wirkung von Bleichlorid (PbC12)
untersucht an Mäusemakrophagen in vitro. Zentraibl. Bakt. Hyg.,
I. Abt. Orig. B., 164: 90-98.
SACHS, H. K. (1974) Effect of a screening program on changing patterns
of lead poisoning. Environ. Health Perspect., Exp. Issue No. 7,
pp. 41-47.
SAKURAI, H., SUGITA, M., & TSUCHIYA, K. (1974) Biological response and
subjective symptoms in low level lead exposure. Arch. environ.
Health, 29: 157-163.
SANDSTEAD, H. H. (1967) Effect of chronic lead intoxication on in
vivo I-131 uptake by the rat thyroid. Proc. Soc. Exp. Biol.
Med. 124: 18-20.
SANDSTEAD, H. H., ORTH, D. N., ABE, K., & STIEL, J. (1970) Lead
intoxication: Effect on pituitary and adrenal function in man.
Clin. Res., 18: 76 (abstract).
SANDSTEAD, H. H., STANT, E.G., & BRILL, H. B. (1969) Lead intoxication
and the thyroid. Arch. int. Med., 123: 632-635.
SANO, S., & RIMINGTON, C. (1963) Excretion of various porphyrins and
their corresponding porphyrinogens by rabbits after intravenous
injection. Biochem. J., 86: 203-212.
SANSONI, B., KRACKE, W., DIETL, F., & FISHER, J. (1973)
Mikrospurenbestimmung von Blei in verschiedenartigen
Umweltproben durch fiammenlose Atomabsorption nach externer
Nassveraschung reit H202/Fe2. In: Proceedings of the
International Symposium; Environmental Health Aspects of lead,
Amsterdam, 2-6 October 1972. Luxembourg, Commission of the
European Communities, pp. 1107-1116.
SASSA, S., GRANICK, J. L., GRANICK, S., KAPPAS, A., & LEVERE, R. D.
(1973) Studies in lead poisoning. I. Microanalysis of erythrocyte
protoporphyrin levels by spectrofluorometry in the detection of
chronic lead intoxication in the subclinical range. Biochem.
Med., 8: 135-148.
SAYRE, J. W., CHARNEY, E., VOSTAL, J., & PLESS, B. (1974) House and
land dust as a potential source in childhood lead exposure. Am.
J. Dis. Child., 127: 167-170.
SCARLATO, G., SMIRNE, S., & POLONI, A. E. (1969) L'encefalopatia
saturnina acuta dell'adulto. Acta Neurol., 24: 578-580.
SCEP (1970) Study of critical environmental problems. Man's impact on
the global environment. Cambridge, Mass. London, England. Mit
Press.
SCHALLER, K. H., LINDNER, K., & LEHNERT, G. (1968)
Atomabsorptionsspektrometrische Schwermetallbestimmung im
Trinkwasser. Arch. Hyg. Bakteriol, 152: 298-301.
SCHIELE, R., SCHALLER, K. H., & VALENTIN, H. (1974a) The influence of
lead upon the tryptophan metabolism. Kiln. Wschr., 52: 401-404.
SCHIELE, R., SCHALLER, K. H., & WAGNER, H. M. (1974b) Die Bestimmung
der freien Erythrozytenporphyrine als schneller Suchtest einer
erhöhten Bleiexposition und seine Validität im vergleich zum
Blutbleispiegel und zur Delta-Aminolävulinsäure-Dehydratase-
Aktivität. SchrReihe Ven. Wasser-Baden. Lufthyg. Berlin,
41: 231-240.
SCHLAEPFER, W. W. (1969) Experimental lead neuropathy: A disease of
the supporting cells in the peripheral system. J. Neuropathol.
exp. Neurology, 28: 401-418.
SCHLEGEL, H., KUFNER, G., & LEINIBERGER, H. (1972) Die Praxis der
Verhütung yon Bleibeschädigungen in der metallverarbeitenden
Industrie. In: Kommission für Umweltge-faren des
Bundesgesundheitsamtes, Arbeitsgruppe Blei und Umwelt, Berlin,
pp. 67-69.
SCHLEGEL, H., KUFNER, G., & LEINBERGER, H. (1973) Das Verhalten
verschiedener Parameter der Hämsynthesestörung am Menschen bei
experimenteller Aufnahme anorganischer Bleiverbindungen. In:
Proceedings of the International Symposium; Environmental Health
Aspects of Lead, Amsterdam, 2-6 October 1972. Luxembourg,
Commission of the European Communities, pp. 569-579.
SCHLIPKÖTER, H. W., & POTT, F. (1973) Die pulmonale Resorption yon
Bleistaub. In: Proceedings of the International Symposium;
Environmental Health Aspects of Lead, Amsterdam, 2-6 October
1972. Luxembourg, Commission of the European Communities,
pp. 403-412.
SCHLIPKÖTER, H. W., GHELERTER, L., & OST, B. (1975) Untersuchungen zur
Kombination-swirkung von Zink und Blei. Zentralbl. Bakt. Hyg. L
Abt. Orig. B., 160: 130-138.
SCHLIPKÖTER, H. W., IDEL, H., STILLER-WINKLER, R., & KRAUSE, G. H. M.
(1977) Die Beeinflussung der Infektionsresistenz durch inhalierte
Noxen. Zentralbl. Bakt. Hyg., I. Abt. Orig. B. (in press).
SCHMID, E., BAUCHINGER, M., PIETRUCK, S., & HALL, O. (1972) Die
cytogenetische Wirkung yon Blei in menschlichen peripheren
Lymphocyten in vitro und in vivo. Mutat. Res. 16: 401-406.
SCHRAMEL, P. (1973) Determination of eight metals in the international
biological standard by flameless atomic-absorption spectrometry.
Analytica Chimica Acta., 67: 69-77.
SCHRAMEL, P. (1974) The application of peak integration in flameless
atomic-absorption spectrometry. Analytica Chimica Acta.,
72: 414-418.
SCHROEDER, H. A., & TIPTON, I. H. (1968) The human body burden of
lead. Arch. environ. 17: 965-978.
SCHROEDER, H. A., & BALASSA, J. J. (1961) Abnormal trace metals in
man: lead. J. chron. Dis., 14: 408-425.
SCHUTZ, A., DENCKER, I., & NORDEN, A. (1971) Bly i kosten-undersokning
med dubbelportionsteknik i Dalby. Svenska Lakeresallskapet,
Medicinska Rikstamman (sammanfattningar), p. 424.
SCHWARTZ, S., & WIKOFF, H. (1952) The relation of erythrocyte
coproporphyrin and protoporphyrin to erythropoiesis. J. Biol.
Chem., 194: 563-573.
SCHWARTZ, S., ZIEVE, L, & WATSON, C. J. (1951) An improved method for
the determination of urinary coproporphyrin and an evaluation of
factors influencing the analysis. J. Lab. Clin. Med.,
37: 843-859.
SCHWANITZ, O., GEBHART, E., ROTT, H. D., SCHALLER, K. H., ESSING, H.
G., LAUER, O., & PRESTELE, H. (1975) Chromosome investigations in
subjects with occupational lead exposure. Dtsch. Med. Wschr,
100: 1007-1011.
SCHWANITZ, G., LEHNERT, G., & GEBHART, E. (1970) Chromosomenschäden
bei beruflicher Bleibelastung. Dtsch. Med. Wschr.,
95: 1636-1641.
SECCHI, G. C., & ALESSIO, L. (1974) Laboratory results of some
biological measures in workers exposed to lead. Arch. environ.
Health, 29: 351-355.
SECCHI, G. C., ALESSIO, L., & CAMBIOGGHI, G. (1971) Ricerche
sull'attivita ALA-deidrasica eritrocitaria di soggetti non
esposti a contatto professionale con plombo ed abitanti in zone
rutall ed urbane. Med. Lav., 62: 435-450.
SECCHI, G. C., ALESSIO, L., & CAMBIOGGHI, G. (1973) Na/K-ATPase
activity of erythrocyte membranes. Arch. environ. Health,
28: 131-132.
SECCHI, G. C., ERBA, L., & CAMBIOGGHI, G. (1974) Delta-aminolevulinic
acid dehydratase activity of erythrocytes and liver tissue in
man. Arch. environ. Health, 28: 130-133.
SELANDER, S., & CRAMER, K. (1970) Interrelationships between lead in
blood, lead in urine and ALA in urine during lead work. Brit. J.
ind. Med., 27: 28-39.
SELYE, H., TUCHWEBER, B., & BERTOK, L. (1966) Effect of lead acetate
on the susceptibility of rats to bacterial endotoxins.
J. Bacteriol., 91: 884-890.
SEPPÄLÄINEN, H. M., & HERNBERG, S. (1972) Sensitive technique for
detecting subclinical lead neuropathy. Brit. J. ind. Med.,
29: 443-449.
SEPPÄLÄINEN, A.M., TOLA, S., HERNBERG, S., & KOCK, B. (1975)
Subclinical neuropathy at "safe" levels of lead exposure. Arch.
environ. Health, 30: 180-183.
SERVANT, J. (1973) Transport du plomb dans l'environnement. In:
Proceedings of the International Symposium; Environmental Health
Aspects of Lead, Amsterdam, 2-6 October 1972. Luxembourg,
Commission of the European Communities, pp. 155-165.
SHELDON, R. P., WARNER, M. A., THOMPSON, M. E., & PEIRCE, H. W. (1953)
Stratigraphic sections of the phosphoria formation in Idaho,
1949, Part I. U.S. geol. Survey eric., 304: pt. I, p. 1.
SHIRAISHI, Y. (1975) Cytogenetic studies of 12 patients with Itai-Itai
disease. Humangentik, 27: 31-44.
SHIRAISHI, Y., KURAHASHI, H., & YOSIDA, T. H. (1972) Chromosomal
aberrations in cultured human leucocytes induced by cadmium
sulfide. Proc. Jpn. Acad., 48: 133-137.
SILBERGELD, E. K., & GOLDBERG, A.M. (1974) Lead-induced behavioral
dysfunction: an animal model of hyperactivity. Exp. Neurol.,
42: 146-157.
SILBERGELD, E. K., FALES, J. T., & GOLDBERG, A.M. (1974) Evidence for
a functional effect of lead on neuromuscular function. Nature
(Lond.), 247: 49-50.
SILVER, W., & RODRIQUEZ-TORRES, R. (1968) Electrocardiographic studies
in children with lead poisoning. Pediatrics, 41: 1124-1127.
SIX, K. M., & GOYER, R. A. (1970) Experimental enhancement of lead
toxicity by low dietary calcium. J. Lab. Clin. Med.
76: 933-942.
SIX, K. M., & GOYER, R. A. (1972) The influence of iron deficiency on
tissue content and toxicity of ingested lead in the rat. J. Lab.
Clin. Med., 79: 128-136.
SMITH, H. D. (1964). Pediatric lead poisoning. Arch. environ. Health,
8: 256-261.
SNOWDON, C. T. (1973) Learning deficits in lead-injected rats.
Pharmacol. Biochem. Behavior, 1: 599-603.
SNYDER, L. J. (1967) Determination of trace amounts of organic lead in
air. Anal. Chem., 39: 591-595.
SOBEL, A. E., GAWRON, O., & KRAMER, B. (1938a) Influence of vitamin D
in experimental lead poisoning. Proc. Soc. Exp. Biol. Med.,
38: 433-435.
SOBEL, A. E., WEXLER, I. B., PETROVSKY, D. D., & KRAMER, B. (1938b)
Influence of dietary calcium and phosphorus upon action of
vitamin D in experimental lead poisoning. Proc. Soc. exp. Biol.
Med., 38: 435-437.
SOLIMAN, M., EL-SADIK, Y., & EL-WASSEF, A. (1973) Evaluation of some
parameters of lead exposure and possible correlation between
them. In: Proceedings of the International Symposium;
Environmental Health Aspects of lead, Amsterdam, 2-4 October
1972. Luxembourg, Commission of the European Communities,
pp. 531-543.
SOLOMINA, V. F. (1961) Effect of lead acetate and silica on the
development of experimental skin cancer. Isvest. Akad. Nauk
Kazakh. SSR. Set. Med. i Fiziol., 2(16): 55-67.
SPURGEON, J. O. (1973) Response characteristics of a portable X-ray
fluorescence lead detector; detection of lead in paint. Report to
the Department of Housing and Urban Development by the National
Bureau of Standards, N13 SIR, pp. 73-231.
SROCZYNSKI, J., ZAJUSZ, K., KOSSMANN, S., & WEGIEL, A. (1967) Effect
of experimental lead poisoning on the development of
arteriosclerosis. Pol. Arch. Med. Wewn., 38 (5): 641-646.
STAPLES, E. L. J. (1955) Experimental lead-poisoning in dogs. N.Z.
vet. J., 3: 39-46.
STOPPS, O. J. (1969) Discussion on epidemiological bases for possible
air quality criteria for lead. Air Pollut. Control Assoc.,
19: 719-721.
STOWE, H. D., & GOYER, R. A. (1971) The reproductive ability and
progeny of F, lead-toxic rats. Fertil. Steril., 22: 755-760.
STOWE, H. D., GOYER, R. A., & KRIGMAN, M. R. (1973) Experimental oral
lead toxicity in young dogs. Arch. Pathol., 95: 106-116.
STRAND, L. J., MANNING, J., & MARVER, H. S. (1972) The induction of
delta-aminolevulinic acid synthetase in cultured liver cells.
J. biol. Chem., 247: 2820-2824.
STUBBS, R. L. (1975) Lead and zinc in 1975, Mining Annual Review-1976,
pp. 45-49.
STUIK, E. J. (1974) Biological response of male and female volunteers
to inorganic lead. Int. Arch. Arbeitsmed., 33: 89-97.
STUIK, E. J., & ZIELHUIS, R. L. (1975) Increased susceptibility of
females to inorganic lead. In: Proceedings of CEC-EPA-WHO
International Symposium; Recent Advances in the Assessment of
the Health Effects of Environmental Pollution, Paris, 24-28 June
1974. Luxembourg, Commission of the European Communities,
pp. 537-545.
SUN, M. W., STEIN, E., & GRUEN, F. W. (1969) A single column method
for the determination of urinary delta-aminolevulinic acid.
Clin. Chem., 15: 183-189.
SWAINE, D. J. (1955) The trace-element content of soils.
Commonwealth Bur. Soil. Sci. Technol. Comm. No. 48.
SZADKOWSKI, D., SCHULTZE, H., SCHALLER, K. H., & LEHNERT, G. (1969)
Zur ökologischen Bedeutung des Schwermetallgehaltes von
Zigaretten. Arch. Hyg. Bakt., 153: 1-8.
TABERSHAW, J. R, & COOPER, W. C. (1974) Health study of lead workers.
A report prepared for the International Lead and Zinc Research
Organization.
TABERSHAW, J. R., RUOTOLO, B. P. W., & GLAESON, R. P. (1943) Plumbism
resulting from oxyacetylene cutting of painted structural steel.
J. ind. Hyg. Toxicol., 25: 189-191.
TASK GROUP ON LUNG DYNAMICS (1966) Deposition and retention models for
internal dosimetry of the human respiratory tract. Health Phys.,
12: 173-207.
TATSUMOTO, M., & PATTERSON, C. C., (1963) The concentration of common
lead in sea water. Earth Sci. Meteorics, 74-89.
TEISINGER, J. (1935) Biochemical reactions of lead in blood. Biochem.
Zeitschrift, 277: 3-4, 178-185.
TEISINGER, J., & SRBOVA, J. (1959) The value of mobilization of lead
by calcium ethylene-diamine-tetra-acetate in the diagnosis of
lead poisoning. Brit. J. ind. Med., 16: 148-152.
TEISINGER, J., & STYBLOVA, V. (1961) Neurological picture of chronic
lead poisoning. Acta Universitatis Carolinae-Med. Supp. 14,
199-206.
TEPPER, L. B. (1963) Renal function subsequent to childhood plumbism.
Arch. environ. Health, 7: 76-85.
TEPPER, L. B., & LEVIN, L. S. (1972) A survey of air and population
lead levels in selected American communities. Final report to the
US EPA.
TER HAAR, G. L. (1970) Air as a source of lead in edible crops.
Environ. Sci. Technol., 4: 226-229.
TER HAAR, G. L., & ARONOW, R. (1974) New information on lead in dirt
and dust as related to the childhood lead problem. Environ.
Health Perspect. Experim. Issue No. 7, 83-89.
HAAR, G. L., & BAYARD, M. A. (1971) Composition of airborne lead
particles. Nature (Lond.), 232: 553-554.
TER HAAR, G. L., HOLTZMAN, R. B., & LUCAS, H. F. (1967) Lead and lead-
210 in rainwater. Nature (Lond.), 216: 353-355.
THOMAS, J. A., DALLENBACH, F. D., & THOMAS, M. (1971) Considerations
on the development of experimental lead encephalopathy. Virchows
Arch. Abt. d. Path. Anat., 352: 61-74.
THOMPSON, J. A. (1971) Balance between intake and output of lead in
normal individuals. Brit. J. ind. Med., 28: 189-194.
THORNTON, I. & WEBB, J. S. (1975) Trace elements in soils and surface
waters contaminated by plast metaliferous mining in parts of
England. In: Proceedings of the IXth Annual Conference on
Trace Substances in Environmental Health, Columbia, MO, 10-12
June 1975, pp. 77-88.
TOLA, S. (1973) Effect of blood lead concentration, age, sex and
exposure time on the erythrocyte delta-aminolevulinic acid
dehydratase activity. Work environ. Health, 10: 26-35.
TOLA, S. (1974) Occupational lead exposure in Finland. III: Lead scrap
smelteries and scrap metal shops. Work Environ. Health,
11: 114-118.
TOLA, S., HERNBERG, S., AsP, S., & NIKKANEN, J. (1973) Parameters
indicative of absorption and biological effect in new lead
exposure: A prospective study. Brit. J. ind. Med., 30: 134-141.
TOLA, S., HERNBERG, S., NIKKANEN, J., & VALKONEN, S. (1971)
Occupational lead exposure in Finland: 1. Electric storage
battery manufacturing and repair. Work environ. Health,
3: 81-85.
TOMOKUNI, K., & OGATA, M. (1972) Simple method for determination of
urinary delta-aminolevulinic acid as an index of lead exposure.
Clin. Chem., 18: 1534-1536.
TRUHAUT, R., BOUDERIC, C., & ALBAHARY, C. (1964) Röle possible de la
consommation exagérée de vin dans l'etiologie du saturnisme.
Bull. WHO, 31: 127-129.
TSUCHIYA, K., & HARASHIMA, S. (1965) Lead exposure and the derivation
of maximum allowable concentrations and threshold limit values.
Brit. J. ind. Med., 22: 181-186.
TSUCHIYA, K., SUGITA, M., SEKI, Y., KOBAYASHI, Y., HORI, M., & BIN
PARK, C. (1975) Study of lead concentrations in atmosphere and
population in Japan. In: Coulston et al., ed., Environmental
quality and safety. Supplement Vol. II, Lead. Stuttgart, Georg
Thieme and NY, Academic Press, pp. 95-146.
TUREKIAN, N. K., & WEDEPOHL, K. H. (1961) Distribution of the elements
in some major units of the earth's crust. Geol. Soc. Am. Bull,
72: 175-191.
UNITED NATIONS, STATISTICAL OFFICE (1975) Statistical Yearbook 1974,
Twenty-sixth issue, United Nations Department of Economic and
Social Affairs, Statistical Office, New York, p. 190.
URATA, G., & GRANICK, S. (1963) Biosynthesis of alpha-aminoketones and
the metabolism of aminoacetone. J. Biol. Chem., 238: 811-820.
URBANOWICZ, H. (1971) Occupational exposure to inorganic compounds of
lead. Arch. environ. Health, 23: 284-288.
URBANOWICZ, H., GRABECKI, J., & KOZIELSKA, J. (1969) The urinary
excretion of 5-hydroxyindoleacetic acid in industrial lead
exposure. Med. Lav., 60: 582-586.
US BUREAU OF MINES (1969) Minerals Yearbook 1968, vol. I-II; Metals,
minerals, and fuels. Washington, pp. 631-660.
US DHEW (1965) Survey of lead in the atmosphere of three urban
communities. USPHS Publ. No. 999 AP 12.
US ENVIRONMENTAL PROTECTION AGENCY OFFICE OF RESEARCH AND MONITORING
(1972) Water pollution aspects of street surface contaminants.
Washington DC (EPA-R2-72-081).
VAN ESCH, G. J., & KROES, R. (1969) The induction of renal tumours by
feeding basic lead acetate to mice and hamsters. Brit. J.
Cancer, 23: 765-771.
VAN ESCH, G. J., VAN GENDEVEN, H., & VINK, H. H. (1962) The induction
of renal tumours by feeding of basic lead acetate to rats. Brit.
J. Cancer, 16: 289-297.
VERBOLOVIC, V. P. (1965) Peculiarities of cytochrome oxidase activity
and of the localization of myoglobin during lead poisoning.
T. Kazahsk. Inst. Kraevoi Pat. Akad. bled. Nauk SSSR,
14: 74-80.
VIGDORTCHIK, N. A. (1935) Lead intoxication in the etiology of
hypertonia. J. ind. Hyg., 17(1): 1-6.
VINOGRADOV, A. P. (1956) Regularity of distribution of chemical
elements in the earth's crust. Geohimija, p. 6 (English
translation in Geochemistry, 1956, pp. 1-43).
VINOGRADOV, A. P. (1962) Average contents of chemical elements in the
principal types of igneous rocks of the earth's crust.
Geohimija, p. 555 (English translation in Geochemistry, 1962,
pp. 641-664).
VOSTAL, J. (1966) Study of the renal excretory mechanisms of heavy
metals. 15th International Congress Occupational Health,
Vienna, 19-24 September, v. 3., pp. 61-64.
WALDRON, H. A. (1966) The anemia of lead poisoning. A review. Brit.
J. ind. Med., 23: 82-100.
WALDRON, H. A. (1975) Lead levels in blood of residents near the
M6-A38(M) interchange, Birmingham. Nature (Lond.),
253: 345-346.
WARLEY, M. A., BLACKLEDGE, P., & O'GORMAN, P. (1968) Lead poisoning
from eye cosmetics. Brit. med. J., 1: 117.
WARREN, H. V., & DELAVAULT, R. E. (1962) Lead in some food crops and
trees. J. Sci. Food Agric., 13: 96-98.
WEAST, R. C., ed. (1974) Handbook of Chemistry and Physics, 55th
ed., Cleveland, CRC Press, pp. B-100-B-101.
WEBB, M. (1972) Binding of cadmium ions by rat liver and kidney.
Biochem. Pharmacol., 21: 2751-2765.
WEBER, H. J. (1947) Some experiences with polarographic methods in
controlling a lead hazard in brass foundries. J. ind. Hyg.
Toxicol., 28: 158-167.
WEDEPOHL, K. H. (1956) Untersuchungen zur Geochemie des Bleis.
Geochim. Acta, 10: 69-148.
WEDEPOHL, K. H. (1971) Zinc and lead in common sedimentary rocks.
Econ. Geol., 66: 240-242.
WELLER, C. V. (1915) The blastopthoric effect of chronic lead
poisoning. J. med. Res., 3: 271-293.
WHITE, J. M., & HARVEY, D. R. (1972) Defective synthesis of alpha- and
ß-globin chains in lead poisoning. Nature (Lond.), 236: 71-73.
WHITEHEAD, T. P., & PRIOR, A. P. (1960) Lead poisoning from homemade
wine. Lancet, 2: 1343-1344.
WHITFIELD, C. L., CHIEN, L. T., & WHITEHEAD, J. D. (1972) Lead
encephalopathy in adults. Am. d. Med., 52: 289-298.
WHO EXPERT COMMITTEE (1969) Urban air pollution with particular
reference to motor vehicles. WHO Technical Report Series, No.
410, p. 19.
WHO EXPERT COMMITTEE ON FOOD ADDITIVES (1972) Lead. Sixteenth Report,
pp. 16-20.
WHO EXPERT COMMITTEE (1973) Trace elements in human nutrition.
WHO Technical Report Series No. 532, p. 47.
WHO STUDY GROUP (1975) Early detection of health impairment in
occupational exposure to health hazards. WHO Technical Report
Series No. 571, pp. 55-61.
WHO WORKING GROUP (1973) Technical document on lead. In: The hazards
to health of persistent substances in water. Annexes to the
Report of a WHO Working Group, Copenhagen, WHO Regional Office
for Europe, pp. 61-110 (Document EURO 3109 w(1)).
WILLIAMS, H., SCHULZE, W. H., ROTHCHILD, H. B., BROWN, A. S. & SMITH,
F. R. (1933) Lead poisoning from the burning of battery casings.
J. Am. Med. Assoc., 100: 1485-1489.
WILLIAMS, U. K. (1966) Blood lead and haemoglobin in lead absorption.
Brit. J. ind. Med., 23: 105-111.
WILLIAMS, M. K, & FEW, J. D. (1967) A simplified procedure for the
determination of urinary delta-aminoloevulinic acid. Brit.
J. ind., Med., 24: 294-296.
WILLIAMS, M. K., KING, E., & WALFORD, J. (1969) An investigation of
lead absorption in an electric accumulator factory with the use
of personal samplers. Brit. J. ind. Med., 26: 202-216.
WILLOUGHBY, R. A., MACDONALD, E., MCSHERRY, B. J., & BROWN, G. (1972)
The interaction of toxic amounts of lead and zinc fed to young
growing horses. Vet. Rec., 91: 382-383.
WILSON, J. G. (1973) Environment and birth defects. New York,
Academic Press, p. 78.
WONG, C. S., & BERRANG, P. (1976) Contamination of tap water by lead
pipe and solder. Bull. environm. Contain. Toxicol.,
15: 530-534.
WONG, P. T. S., CHATS, Y. K., & LUXON, P. L. (1975) Methylation of
lead in the environment. Nature (Lond.), 253: 263-264.
WRANNE, L., (1960) Free erythrocytes copro- and protoporphyrin: A
methodological and clinical study. Acta. Paediatrica., 49
(suppl. 124).
YAMAMOTO, Y., TANAKA, M., & ARAO, K. (1968) Hemispherical distribution
of turbidity coefficient as estimated from direct solar radiation
measurements. J. Meteorol. Soc. Jpn, 46: 287-300.
YEAGER, D. W., CHOLAK, J., & HENDERSON, E. W. (197 l) Determination of
lead in biological and related material by atomic absorption
spectrophotometry. Environ. Sci. Technol., 5: 1020-1022.
YOCOM, J. E., CLINK, W. L., & COTE, W. A. (1971) Indoor/outdoor air
quality relationships. J. Air Pollut. Control Assoc.,
21: 251-259.
ZAWIRSKA, B., & MEDRAS, K. (1968) Tumoren und Störungen des
Porphyrinstoffwechsels bei Ratten Tit chronischer experimenteller
Bleiintoxikation. Zentralbl. Allg. Path., 111(1): 1-12.
ZEL'TSER, M. E. (1962) The functional state of thyroid gland in lead
poisoning. Tr. Just. kraevoi Patol. Akad. Nauk Kaz. SSR,
10: 116-120.
ZIEGFIELD, R. L. (1964) Importance and uses of lead. Arch. environ.
Health, 8: 202-212.
ZIELHUIS, R. L. (1971) Interrelationship of biochemical responses to
the absorption of inorganic lead. Arch. environ. Health,
23: 299-311.
ZIELHUIS, R. L. (1974) Dose response relationship for inorganic lead.
Report to the Director, Health Protection EEC.
ZIELHUIS, R. L. (1975) Dose response relationships for inorganic lead.
I. Biochemical and haematological responses. II. Subjective and
functional responses. Chronic sequelae. No-response levels.
Int. Arch. occup. Health, 35: 1-18, 19-35.
ZOLLINGER, H. U. (1953) Durch chronische Bleivergiftung erzeugter
Nierenadenome und Carcinome bei Ratten und ihre Beziehungen zu
den entsprechenden Neubildungen des Menschen. Virchows Arch.
Bd., 323: 694-710.
ZOOK, B. C., CARPENTER, J. L., & LEEDS, E. B. (1969) Lead poisoning in
dogs. J. Am. vet. Med. Assoc., 155: 1329-1342.
ZUKOVICKAJA, A. L., ZAMUATKINA A. A., & LUKASEV, K. I. (1966) Trace
elements in the water of the upper Dnepr river. Dokl. Akad. Nauk
BSSR, 10: 891-893.
ZURLO, N., & GRIFFINI, A. M. (1973) Le plomb dans les aliments et
dans les boissons consommes à Milan. In: Proceedings of the
International Symposium; Environmental Health Aspects of Lead,
Amsterdam, 2-6 October 1972. Luxembourg, Commission of the
European Communities, pp. 93-99.
ZURLO, N., GRIFFINI, H. M., & VIGLIANI, E. C. (1970) The content of
lead in blood and urine of adults, living in Milan, not
occupationally exposed to lead. Am. ind. Hyg. Assoc. J.
31: 92-95.