
IPCS/CEC EVALUATION OF ANTIDOTES SERIES
VOLUME 2
ANTIDOTES FOR POISONING BY CYANIDE
IPCS/CEC Evaluation of Antidotes Series
IPCS International Programme on Chemical Safety
CEC Commission of the European Communities
Volume 1 Naloxone, flumazenil and dantrolene as antidotes
Volume 2 Antidotes for poisoning by cyanide
This important new series will provide definitive and authoritative
guidance on the use of antidotes to treat poisoning. The
International Programme on Chemical Safety (IPCS) and the Commission
of the European Communities (CEC) (ILO/UNEP/WHO) have jointly
undertaken a major programme to evaluate antidotes used clinically
in the treatment of poisoning. The aim of this programme has been
to identify and evaluate for the first time in a scientific and
rigorous way the efficacy and use of a wide range of antidotes.
This series will therefore summarise and assess, on an antidote by
antidote basis, their clinical use, mode of action and efficacy. The
aim has been to provide an authoritative consensus statement which
will greatly assist in the selection and administration of an
appropriate antidote. This scientific assessment is complemented by
detailed clinical information on routes of administration,
contraindications, precautions and so on. The series will therefore
collate a wealth of useful information which will be of immense
practical use to clinical toxicologists and all those involved in the
treatment and management of poisoining.
Scientific Editors
T.J. MEREDITH
Department of Health, London, United Kingdom
D. JACOBSEN
Ulleval University Hospital, Oslo, Norway
J.A. HAINES
International Programme on Chemical Safety,
World Health Organization, Geneva, Switzerland
J-C. BERGER
Health and Safety Directorate,
Commission of the European Communities, Luxembourg
Guest Editor
A.N.P. van HEIJST
Formerly of the Dutch National Poison Control Centre,
Utrecht, The Netherlands
EUR 14280 EN
Published by Cambridge University Press on behalf of the World Health
Organization and of the Commission of the European Communities
CAMBRIDGE UNIVERSITY PRESS
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar
nature that are not mentioned.
Neither the Commission of the European Communities nor any person
acting on behalf of the Commission is responsible for the use which
might be made of the information contained in this report.
(c) World Health Organization, Geneva, 1993 and
ECSC-EEC-EAEC, Brussels-Luxembourg, 1993
First published 1993
Publication No. EUR 14280 EN of the Commission of the European
Communities, Dissemination of Scientific and Technical Knowledge
Unit, Directorate-General Information Technologies and Industries,
and Telecommunications, Luxembourg
ISBN 0 521 45458 1 hardback
CONTENTS
PREFACE
ABBREVIATIONS
1. OVERVIEW
1.1. Historical review
1.2. Potential sources of cyanide
1.2.1. Industrial sources
1.2.2. Non-industrial sources
1.2.3. Natural sources
1.2.4. Iatrogenic sources
1.3. Toxicity of cyanide in man
1.3.1. Acute poisoning
1.3.2. Chronic poisoning
1.4. Mechanism of toxicity
1.5. Clinical features
1.6. Laboratory findings
1.6.1. Lactic acidosis
1.6.2. Hyperglycaemia
1.6.3. Cyanide concentration in blood and plasma
1.7. Biological detoxification of cyanide
1.7.1. Thiocyanate toxicity
1.8. Protective measures for occupational exposure
1.9. Treatment
1.9.1. Supportive treatment
1.9.2. Antidotal treatment
1.9.2.1 Oxygen
1.9.2.2 Sodium thiosulfate
1.9.2.3 Amyl nitrite
1.9.2.4 Sodium nitrite
1.9.2.5 4-Dimethylaminophenol
1.9.2.6 Hydroxocobalamin
1.9.2.7 Dicobalt edetate
1.9.2.8 Antidotes to methaemoglobin-forming
agents
1.10. Summary of treatment recommendations
1.10.1. First aid and treatment measures at the site of
the incident
1.10.2. Hospital treatment
1.10.2.1 Severe poisoning
1.10.2.2 Moderately severe poisoning
1.10.2.3 Mild Poisoning
1.11. Summary of analytical aspects
1.12. Proposed areas for research
1.13. New developments in cyanide antidotes
1.13.1. Nonspecific agents
1.13.2. Sodium pyruvate
1.13.3. Ifenprodil
1.13.4. Rhodanese
1.13.5. Alpha-ketoglutaric acid
1.13.6. Stroma-free methaemoglobin solution
1.14. References
2. OXYGEN
2.1. Introduction
2.2. Name and chemical formula of antidote
2.3. Physico-chemical properties of molecular oxygen
2.4. Synthesis
2.5. Analytical methods
2.5.1. Quality control procedures
2.5.1.1 Tests
2.5.1.2 Assay for oxygen
2.5.2. Methods for identification
2.5.3. Methods for analysis of the antidote in
biological samples
2.5.3.1 In the gas phase
2.5.3.2 In solution
2.5.4. The saturation of haemoglobin by oxygen
2.6. Storage conditions
2.7. General properties
2.8. Animal studies
2.8.1. Pharmacokinetics
2.8.2. Pharmacodynamics
2.8.3. Toxicology
2.8.3.1 Mechanism of injury
2.9. Volunteer studies of pulmonary oxygen toxicity
2.10. Clinical studies of oxygen toxicity
2.10.1. Eyes
2.10.2. Central nervous system
2.11. Clinical studies - case reports
2.11.1. Patients treated alone with supportive therapy
and who survived
2.11.2. Hyperbaric oxygen therapy in cyanide poisoning
2.11.3. Cyanide poisoning due to smoke inhalation
2.12. Summary of evaluation
2.13. Model information sheet
2.13.1. Uses
2.13.2. Dosage and route
2.13.3. Precautions/contraindications
2.13.4. Adverse effects
2.13.5. Use in pregnancy and lactation
2.13.6. Storage
2.14. References
3. SODIUM THIOSULFATE
3.1. Introduction
3.1.1. Indications
3.1.2. Rationale for the choice of the antidote
3.1.3. Risk groups
3.2. Name and chemical formula of antidote
3.3. Physico-chemical properties
3.3.1. Melting point, boiling point
3.3.2. Solubility in vehicle for administration
3.3.3. Optical properties
3.3.4. Acidity
3.3.5. pKa
3.3.6. Stability
3.3.7. Refractive index, specific gravity
3.3.8. Loss of weight on drying
3.3.9. Excipients
3.3.10. Incompatibility
3.3.11. Other information
3.4. Synthesis
3.5. Analytical methods
3.5.1. Quality control procedures for sodium thiosulfate
3.5.2. Methods for identifying sodium thiosulfate
3.5.3. Assay
3.5.4. Methods for analysis of sodium thiosulfate in
biological samples
3.6. Shelf-life
3.7. General properties
3.7.1. Mechanism of antidotal activity
3.7.2. Other biochemical/pharmacological profiles
3.8. Animal studies
3.8.1. Pharmacokinetics
3.8.2. Pharmacodynamics
3.8.3. Toxicology
3.9. Volunteer studies
3.10. Clinical studies
3.11. Clinical studies - case reports
3.12. Summary of evaluation
3.12.1. Indications
3.12.2. Route of administration
3.12.3. Dose
3.12.4. Other consequential or supportive therapy
3.13. Model information sheet
3.13.1. Uses
3.13.2. Dosage and route of administration
3.13.3. Precautions and contraindications
3.13.4. Adverse effects
3.13.5. Use in pregnancy/lactation
3.13.6. Storage
3.14. References
4. HYDROXOCOBALAMIN
4.1. Introduction
4.2. Name and chemical formula of antidote
4.3. Physico-chemical properties
4.3.1. Characteristics
4.3.2. Melting-point
4.3.3. Solubility in vehicles for administration
4.3.4. Optical properties
4.3.5. Acidity
4.3.6. Stability in light
4.3.7. Thermal stability
4.3.8. Interference with other compounds
4.4. Synthesis
4.5. Analytical methods
4.5.1. Identification of hydroxocobalamin
4.5.1.1 UV spectroscopy
4.5.1.2 Colorimetric method
4.5.2. Quality controls
4.5.3. Raw materials
4.5.4. Finished galenic form
4.5.5. Measurement
4.5.5.1 In raw materials and in finished form
4.5.5.2 In biological samples
4.6. Shelf-life
4.7. General properties
4.8. Animal studies
4.8.1. Pharmacokinetics
4.8.2. Pharmacodynamics in the presence of the toxin
4.8.3. Toxicology
4.8.3.1 Acute toxicity
4.8.3.2 Sub-acute and chronic toxicity
4.9. Volunteer studies
4.10. Clinical studies
4.11. Clinical studies - case reports
4.12. Summary of evaluation
4.12.1. Indications
4.12.2. Advised route and dosage
4.12.3. Practical advice
4.12.4. Side effects
4.13. Model information sheet
4.13.1. Uses
4.13.2. Dosage and route
4.13.3. Precautions/contraindications
4.13.4. Adverse effects
4.13.5. Use in pregnancy and lactation
4.13.6. Storage
4.14. References
5. DICOBALT EDETATE
5.1. Introduction
5.2. Name and chemical formula
5.3. Physico-chemical properties
5.4. Synthesis
5.4.1. Source of materials
5.4.1.1 Cobalt carbonate
5.4.1.2 Ethylenediaminetetraacetic acid
5.4.1.3 Glucose
5.5. Analytical methods
5.5.1. Free cobalt
5.5.2. Dicobalt edetate
5.5.3. Analysis in biological fluids
5.6. Stability and shelf-life
5.7. General properties
5.8. Animal studies
5.8.1. Pharmacokinetics
5.8.2. Pharmacodynamics
5.8.2.1 Efficacy in animals
5.8.2.2 Comparison of dicobalt edetate with
other compounds
5.8.2.3 Interactions with other drugs
5.8.3. Toxicology
5.8.3.1 In vitro studies
5.8.3.2 Acute toxicity studies
5.8.3.3 Repeated dose toxicity
5.8.3.4 Circulatory effects in dogs
5.8.3.5 Other toxicity studies
5.9. Volunteer studies
5.10. Clinical trials
5.11. Clinical studies - case reports
5.11.1. Successful use
5.11.2. Use in pregnant women and children
5.11.3. Adverse effects
5.11.4. Use in combination with other antidotes
5.12. Summary of evaluation
5.12.1. Indications
5.12.2. Administration
5.12.3. Other consequential or supportive therapy
5.12.4. Contraindications
5.12.5. Comparison with other antidotes
5.13. Model information sheet
5.13.1. Uses
5.13.2. Dosage and route
5.13.3. Precautions/contraindications
5.13.4. Adverse effects
5.13.5. Use in pregnancy and lactation
5.13.6. Storage
5.14. References
6. AMYL NITRITE
6.1. Introduction
6.2. Name and chemical formula
6.3. Physico-chemical properties
6.4. Synthesis
6.5. Analytical methods
6.5.1. Identification
6.5.2. Purity
6.5.2.1 Acidity
6.5.2.2 Non-volatile residue
6.5.2.3 Assay for total nitrites
6.6. Shelf-life
6.7. General properties
6.8. Animal studies
6.8.1. Pharmacokinetics
6.8.2. Pharmacodynamics
6.8.3. Toxicology
6.9. Volunteer studies
6.10. Clinical studies
6.11. Clinical studies - case reports
6.12. Summary of evaluation
6.12.1. Indications
6.12.2. Advised routes and dose
6.12.3. Other consequential or supportive therapy
6.13. Model information sheet
6.13.1. Uses
6.13.2. Dosage and route
6.13.3. Precautions/contraindications
6.13.4. Storage
6.14. References
7. SODIUM NITRITE
7.1. Introduction
7.2. Name and chemical formula
7.3. Physico-chemical properties
7.4. Synthesis
7.5. Analytical methods
7.5.1. Quality control
7.5.1.1 Solid sodium nitrite
7.5.1.2 Sodium nitrite injection
7.5.1.3 Preparation of volumetric solutions
7.5.2. Identification
7.5.2.1 Sodium
7.5.2.2 Nitrite
7.5.3. Impurities
7.5.3.1 Preparation of sodium nitrite to test
7.5.3.2 Preparation of special reagents
7.5.3.3 Preparation of standard
7.5.3.4 Preparation of test
7.5.3.5 Preparation of monitor
7.5.3.6 Preparation of hydrogen sulfide test
solution
7.5.3.7 Test procedure
7.6. Shelf-life
7.7. General properties
7.7.1. Mode of action
7.7.2. Other relevant properties
7.8. Animal studies
7.8.1. Pharmacokinetics
7.8.2. Pharmacodynamics
7.8.3. Toxicology
7.9. Volunteer studies
7.9.1. Pharmacokinetics
7.9.2. Sodium nitrite poisoning
7.10. Clinical studies
7.11. Clinical studies - case reports
7.12. Summary of evaluation
7.12.1. Indications
7.12.2. Contraindications
7.12.3. Advised route and dosage
7.12.4. Other consequential or supportive therapy
7.13. Model information sheet
7.13.1. Uses
7.13.2. Dosage and route
7.13.3. Precautions/contraindications
7.13.4. Adverse effects
7.13.5. Use in pregnancy and lactation
7.13.6. Storage
7.14. References
8. 4-DIMETHYLAMINOPHENOL
8.1. Introduction
8.2. Name and chemical formula
8.3. Physico-chemical properties
8.4. Synthesis
8.5. Analytical methods
8.5.1. Identity
8.5.2. Quantification
8.5.3. Purity
8.5.4. Methods for analysis of 4-DMAP in biological
samples
8.6. Shelf-life
8.7. General properties
8.8. Animal studies
8.8.1. In vitro studies
8.8.1.1 Metabolism of 4-DMAP in the liver
8.8.1.2 Red cell metabolism of 4-DMAP
8.8.1.3 Toxic effects of 4-DMAP on
erythrocytes
8.8.1.4 Toxic effects of 4-DMAP on isolated
rat kidney tubules
8.8.1.5 Oxygen saturation and methaemoglobin
formation
8.8.2. Pharmacokinetics
8.8.3. Pharmacodynamics
8.8.4. Toxicology
8.8.4.1 Nephrotoxicity
8.8.4.2 Mutagenicity
8.9. Volunteer studies
8.9.1. Metabolism of 4-DMAP in the liver
8.9.2. Metabolism of 4-DMAP in erythrocytes
8.9.3. Adverse effects
8.10. Clinical studies
8.11. Clinical studies - case reports
8.12. Summary of evaluation
8.12.1. Indications
8.12.2. Recommended routes and dosage
8.12.3. Other consequential or supportive therapy
8.12.4. Areas of use where there is insufficient
information to make recommendations
8.13. Model information sheet
8.13.1. Uses
8.13.2. Dosage and route
8.13.3. Precautions/contraindications
8.13.4. Adverse effects
8.13.5. Use in pregnancy and lactation
8.13.6. Storage
8.14. References
9. METHYLENE BLUE AND TOLUIDINE BLUE
9.1. Methylene blue
9.1.1. Introduction
9.1.2. Name and chemical formula of antidote
9.1.3. Physico-chemical properties
9.1.4. Synthesis
9.1.5. Analytical methods
9.1.6. Shelf-life
9.1.7. General properties
9.1.8. Animal studies
9.1.9. Volunteer studies
9.1.10. Clinical studies
9.1.11. Clinical studies - case reports
9.1.12. Summary of evaluation
9.1.12.1 Indications
9.1.12.2 Advised route and dosage
9.1.12.3 Precautions and contraindications
9.1.12.4 Adverse effects
9.1.12.5 Other consequential or supportive
theory
9.1.13. Model information sheet
9.1.13.1 Uses
9.1.13.2 Dosage and route of administration
9.1.13.3 Precautions and contraindications
9.1.13.4 Adverse effects
9.1.13.5 Use in pregnancy/lactation
9.1.13.6 Storage
9.1.14. References
9.2. Toluidine blue
9.2.1. Introduction
9.2.2. Name and chemical formula of antidote
9.2.3. Physico-chemical properties
9.2.4. Synthesis
9.2.5. Analysis
9.2.5.1 Analysis of methaemoglobin
9.2.6. Stability
9.2.7. General properties
9.2.8. Animal studies
9.2.8.1 Pharmacokinetics
9.2.8.2 Pharmacodynamics
9.2.8.3 Toxicology
9.2.9. Volunteer studies
9.2.10. Clinical studies
9.2.11. Clinical studies - case reports
9.2.12. Summary of evaluations
9.2.13. Model information sheet
9.2.13.1 Indications
9.2.13.2 Side effects
9.2.13.3 Advised route and dose
9.2.13.4 Use in pregnancy and children
9.2.13.5 Storage
9.2.14. References
10. ANALYTICAL METHODS FOR CYANIDE ALONE AND IN
COMBINATION WITH CYANIDE ANTIDOTES IN BLOOD
10.1. Qualitative methods
10.1.1. Detection in blood with a detector tube
10.1.1.1 Principle
10.1.1.2 Materials
10.1.1.3 Procedure
10.1.1.4 Specificity
10.1.2. Spot test
10.1.2.1 Principle
10.1.2.2 Equipment
10.1.2.3 Chemicals
10.1.2.4 Reagents
10.1.2.5 Specimen collection
10.1.2.6 Procedure
10.1.2.7 Specificity
10.2. Quantitative methods
10.2.1. Gas chromatographic head space technique
10.2.1.1 Principle
10.2.1.2 Equipment
10.2.1.3 Chemicals
10.2.1.4 Solutions
10.2.1.5 Calibration standards
10.2.1.6 Specimen collection and sample
preparation
10.2.1.7 Operational parameters for gas
chromatography
10.2.1.8 Analytical determination
10.2.1.9 Calibration
10.2.1.10 Calculation of the analytical result
10.2.1.11 Reliability of the method
10.2.1.12 Detection limit
10.2.1.13 Specificity
10.2.2. Microdiffusion technique
10.2.2.1 Principle
10.2.2.2 Equipment
10.2.2.3 Chemicals
10.2.2.4 Solvents and reagents
10.2.2.5 Calibration standards
10.2.2.6 Specimen
10.2.2.7 Procedure
10.2.2.8 Reliability of the method
10.2.2.9 Detection limit
10.2.2.10 Specificity
10.3. References
WORKING GROUP ON ANTIDOTES TO POISONING BY CYANIDE
Members
Professor C. Bismuth, Hôpital Fernand Widal, Clinique Toxicologique,
Paris, France
Professor M. von Clarmann, Poisons Centre, Toxicology Department, 11
Med. Klinikrechts der Isar der Tecknischer Universität, Munich,
Germany
Dr A. van Dijk, Apotheek, Academisch Ziekenhuis, Utrecht, The
Netherlands
Professor M. Geldmacher von Mallinckrodt, Institut für
Rechtsmedizia, Erlangen, Germany
Dr A. Hall, Rocky Mountain Poison and Drug Center, Denver, Colorado,
USA
Professor A.N.P. van Heijst, Bosch en Duin, The Netherlands
Dr T.C. Marrs, Department of Health, London, United Kingdom
Dr T.J. Meredith, Department of Health, London, United Kingdom
(Rapporteur)
Dr A.C.G.M. Parren, Te Heerlen, The Netherlands
Dr H. Persson, Poison Information Centre, Karolinska Sjukhuset,
Stockholm, Sweden
Dr U. Taitelman, Rambam Medical Center, Haifa, Israel
Observers
Dr J. Aubrun, Rhone-Poulenc, Courbevoie, France
Dr A. Heath, Poisons Therapy Group, Department of Anaesthesia and
Intensive Care, Sahlgren's Hospital, Gothenburg, Sweden
Dr J. Henry, Poisons Unit, New Cross Hospital, London, United
Kingdom
Dr J.A. Vale, West Midlands Poisons Unit, Dudley Road Hospital,
Birmingham, United Kingdom
Secretariat
Dr J.-C. Berger, Health and Safety Directorate, Commission of the
European Communities, Luxembourg
Dr J.A. Haines, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland (Chairman)
Dr M. ten Ham, Pharmaceuticals Programme, World Health Organization,
Geneva, Switzerland
Dr M.-Th. van der Venne, Health and Safety Directorate, Commission
of the European Communities, Luxembourg
PREFACE
At a joint meeting of the World Federation of Associations of
Clinical Toxicology and Poison Control Centres, the International
Programme on Chemical Safety (IPCS), and the Commission of the
European Communities (CEC), held at the headquarters of the World
Health Organization in October 1985, the evaluation of antidotes
used in the treatment of poisonings was identified as a priority
area for international collaboration. During 1986, the IPCS and CEC
undertook the preparatory phase of a joint project on this subject.
For the purpose of the project an antidote was defined as a
therapeutic substance used to counteract the toxic action(s) of a
specified xenobiotic. Antidotes, as well as other agents used to
prevent the absorption of poisons, to enhance their elimination and
to treat their effects on body functions, were listed and
preliminarily classified according to the urgency of treatment and
efficacy in practice. With respect to efficacy in practice, they
were classified as: (1) those generally accepted as useful; (2)
those widely used and considered promising but not yet universally
accepted as useful and requiring further research concerning their
efficacy and/or their indications for use; and (3) those of
questionable usefulness. Additionally, certain antidotes or agents
used for specific purposes were considered to correspond to the WHO
criteria for essential drugs (see Criteria for the Selection of
Essential Drugs, WHO Technical Report Series 722, Geneva, 1985).
A methodology for the principles of evaluating antidotes and
agents used in the treatment of poisonings and a proforma for
preparing monographs on antidotes for specific toxins were drafted.
These were included in volume 1 of this series.
Monographs are being prepared, using the proforma, for those
antidotes and agents provisionally classified in category 1 as
regards efficacy in practice. For those classified in categories 2
and 3, where there are insufficient data or controversy regarding
efficacy in practice, it was agreed that further study was
necessary. Accordingly, several were selected for initial review
and evaluation, among which were antidotes used in the treatment of
poisoning by cyanide.
The review and evaluation of antidotes used in the treatment of
poisoning by cyanide was initiated at a joint meeting of the
European Association of Poison Control Centres and Clinical
Toxicologists (EAPCCT; formerly known as the European Association of
Poison Control Centres), the IPCS, and the CEC, organized by the
National Poison Information Centre of the Netherlands National
Institute of Public Health and Environmental Hygiene and held at the
University Hospital AZU, Utrecht, The Netherlands, 13-15 May 1987.
In preparation for this meeting, documents were drafted, using the
proforma, on oxygen by Dr U. Taitelman, sodium thiosulfate by Dr H.
Persson, hydroxocobalamin by Professor C. Bismuth, dicobalt edetate
by Dr T.C. Marrs, sodium nitrite by Dr A. Hall, and
4-dimethylaminophenol by Professor M. von Clarmann. Also in
preparation for the meeting, documents were drafted by Professor M.
Geldmacher von Mallinckrodt on the analytical assessment of cyanide
poisoning, by Dr A. van Dijk on the pharmaceutical aspects of
cyanide antidotes, and by Professor A.N.P. van Heijst (formerly
Director, Dutch National Poison Control Centre, Utrecht, the
Netherlands) on the clinical aspects of cyanide antidotes. The
documents presented by each author were discussed at the meeting and
participants gave their own experience and views. Experience of
industrial aspects of cyanide poisoning was presented by Dr A.C.G.M.
Parren.
The main meeting was followed by that of an IPCS/CEC working
group, consisting of the authors of documents, the meeting
rapporteur and a number of observers, at which a review was made of
the comments on the documents and of the additional material
presented at the main meeting. Based on the available material, an
evaluation was made of the different approaches to treatment of
cyanide poisoning depending on the type of cyanide exposure
(hydrogen cyanide, either alone or with carbon monoxide, cyanide
salts or cyanogenic glycosides), the state of intoxication and
number of patients, the location of the patient with respect to
treatment facilities, and special situations (e.g., inherited
metabolic and haemoglobin abnormalities). The group concentrated on
acute poisoning by cyanide, considering that there were insufficient
data for evaluating approaches to treatment of chronic cyanide
toxicity. Nevertheless, it was considered that a review of chronic
poisoning by cyanide, particularly in relation to cyanide ingestion
from food, was needed. It was agreed that traditional means of
treatment of cyanide poisoning would have to be revised, and that
any evaluation of approaches to treatment must also include
antidotes for methaemoglobin-forming agents. Concerning the
analytical aspects, it was noted that there was particular
difficulty in measuring the concentration of cyanide in blood if an
antidote had already been administered, a problem that is being
studied by a group of experts established under the auspices of the
German Research Association Commission on Clinical Analytical
Toxicology. A number of new cyanide antidotes in various stages of
research and development were discussed. An editorial group
consisting of Professor A.N.P. van Heijst (chairman of the meeting),
Dr T.J. Meredith (rapporteur), Dr J.A. Haines (IPCS, chairman of the
working group) and Dr J.-C. Berger (CEC) was established in order to
prepare a consolidated monograph on cyanide antidotes.
Draft documents were revised by their authors. Those on
methylene blue and toluidine blue were prepared by Dr Christina
Alonzo (CIAT, Montevideo, Uruguay) and Dr T.C. Marrs, respectively.
Subsequently Dr J.A. Vick (Food and Drug Administration, USA), who
was invited to the meeting but was unable to attend, prepared a
draft document on experience with the use of amyl nitrite in
treating cyanide poisoning in animals. Professor C. Bismuth and
Dr A. Hall drafted material on new antidotes under development for
clinical trials, and Dr A.C.G.M. Parren drafted material on
protective measures.
The editorial group met twice in Utrecht on 22-23 October 1987
and 20-22 July 1988. Material was checked and rearranged,
additional material was prepared for a number of the chapters and
the overview chapter was drafted. The efforts of all who helped in
the preparation and finalization of this monograph are gratefully
acknowledged.
ABBREVIATIONS
ATA atmosphere absolute
BE base excess
CNS central nervous system
CT computer tomography
4-DMAP 4-dimethylaminophenol
EDTA ethylenediaminetetraacetic acid
G6PD glucose-6-phosphate dehydrogenase
Hb haemoglobin
HMPS hexose monophosphate shunt
INN international non-proprietary name
LDLo lowest published lethal dose
MLD minimal lethal dose
NADH reduced nicotinamide adenine dinucleotide
NADPH reduced nicotinamide adenine dinucleotide phosphate
OHB12 hydroxocobalamin
LD50 Lethal Dose 50
USP United States Pharmacopoeia
B12 Vitamin B12
HbO2 Oxyhaemoglobin
AV atrioventricular
SNP sodium nitroprusside
VS volumetric solution
1. Overview
1.1 Historical Review
The recognition of cyanide as a poison in bitter almonds,
cherry laurel leaves, and cassava goes back to antiquity. An
inscription on an Egyptian papyrus in the Louvre Museum, Paris,
refers to the "penalty of the peach," and Dioscorides in the first
century A.D. was aware of the poisonous properties of bitter almonds
(Sykes, 1981).
The first description of cyanide poisoning was by Wepfer in
1679 and dealt with the effects of the administration of extract of
bitter almonds (Sykes, 1981). Two fatal cases of poisoning in
Ireland caused by drinking cherry laurel water, used as a flavouring
agent in cooking and to dilute brandy, led to the experiments of
Madden (1731). He showed that cherry laurel water contains a
poison; given orally, into the rectum, or by injection, it rapidly
killed dogs. It was not until 1786 that isolation of pure hydrogen
cyanide (HCN) from the dye Prussian blue was achieved by Scheele
(1786). The mechanism of toxicity of cyanide was explored by
Fontana (1795). Cyanide was obtained from bitter almonds by
Schrader (1802). The introduction of cyanide as a medicament to
treat coughs and lung diseases was suggested by Magendie (1817).
Indeed, it was not until 1948 that cherry laurel water was removed
from the British Pharmacopoeia! Attempts to antagonize the toxic
effects of cyanide were reported by Blake (1839 and 1840).
Hoppe-Seyler (1876) reported that cyanide inhibits tissue oxidation
reactions.
Antagonism between amyl nitrite and prussic acid was mentioned
by Pedigo (1888), and, as early as 1894, cobalt compounds were
advocated by Antal (1894) as cyanide antagonists. Sodium nitrite
was used as an antidote in experimental cyanide poisoning by
Mladoveanu & Gheorghiu (1929).
A biochemical mechanism for cyanide antagonism was described by
Chen et al. (1933, 1934). They suggested using a combination of
amyl nitrite, sodium nitrite and sodium thiosulfate, the latter
compound serving as a sulfur donor for rhodanese (thiosulfate sulfur
transferase). Rhodanese accelerates cyanide detoxification by
forming the metabolite thiocyanate. This represented the
development of one of the first antidotes based on scientific
toxicological reasoning. This combination of antidotes has stood
the test of time, and still represents one of the most efficacious
antidotal combinations for the treatment of cyanide intoxication.
Interest in cobalt compounds was renewed by Mushett et al.
(1952), who demonstrated in 1952 that hydroxocobalamin (vitamin
B12a) combined with cyanide to form cyanocobalamin (vitamin
B12).
Paulet (1960) subsequently reported that cobalt EDTA was more
effective as a cyanide antidote than the classic nitrite-thiosulfate
combination.
1.2 Potential Sources of Cyanide
1.2.1 Industrial sources
Hydrogen cyanide is used in the fumigation of ships, large
buildings, flour mills, private dwellings, freight cars, and
aeroplanes that have been infested by rodents or insects. It is
bound to a carrier, commonly diatomaceous earth, and blended with an
odorous or irritating product as a warning marker.
Cyanide salts are utilized in metal cleaning, hardening,
ore-extracting processes, and electroplating.
Halogenated cyanides (chloro-, bromo- and iodocyanide) in
contact with water produce the non-toxic cyanic acid. As a result
of contact with strong acids, hydrogen cyanide is liberated.
Nitriles are cyano-derivatives of organic compounds. Acetonitrile
is used as a solvent and is less toxic (LD50 = 120 mg/kg) than
hydrogen cyanide (LD50= 0.5 mg/kg), but often contains toxic
admixtures due to metabolism to inorganic cyanide. While aliphatic nitriles
metabolise to inorganic cyanide, the aromatic nitrile bond is stable
in vivo. Acrylonitrile is the raw material used for the
manufacture of plastics and synthetic fibres. Contact with skin
causes bullae formation. Pyrolysis generates hydrogen cyanide.
Acrylonitrile and propionitrile are less toxic (LD50 = 35 mg/kg)
than butyronitrile (LD50 = 10 mg/kg). Trichloroacetonitrile
(LD50 = 200 mg/kg) is used as an insecticide. The aromatic
nitriles, bromoxynil (LD50= 190 mg/kg) and ioxynil (LD50= 110
mg/kg), are used as herbicides.
Cyanamide, cyanoacetic acid, ferricyanide and ferrocyanide do
not release cyanide. They are therefore less toxic (LD50=
1000-2000 mg/kg) than the cyanogenic compounds above, though they
may cause toxicity by other means, e.g. cyanide in combination with
alcohol.
1.2.2 Non-industrial sources
Fires and automobile pollution-control devices with
malfunctioning catalytic converters (Voorhoeve et al., 1975)
generate cyanide. Natural substances, such as wool, silk, horse
hair, and tobacco, as well as modern synthetic materials, such as
polyurethane and polyacrylonitriles, release cyanide during
combustion (Levine et al., 1978; Birky et al., 1979; Anderson &
Harland, 1982; Clark et al., 1983; Alarie, 1985; Lowry et al., 1985)
(Table 1).
Table 1. Hydrogen cyanide generated by pyrolysis
µg HCN per
Material g material
paper 1100
cotton 130
wool 6300
nylon 780
polyurethane foam 1200
From: Montgomery et al. (1975)
1.2.3 Natural sources
Cyanide is found in foodstuffs such as cabbage, spinach, and
almonds, and as amygdalin in apple pips, peach, plum, cherry, and
almond kernels. In the kernels themselves, amygdalin seems to be
completely harmless as long as it is relatively dry. However, the
seeds contain an enzyme that is capable of catalysing the following
hydrolytic reaction when the seeds are crushed and moistened:
C20H27NO11 + 2H2O --> 2C6H12O6 + C6H5CHO + HCN
amygdalin glucose benzaldehyde hydrogen
cyanide
The reaction is slow in acid but rapid in alkaline solution.
Natural oil of bitter almonds contains 4% HCN. American white
lima beans contain 10 mg cyanide/100 g bean. The dried root of
cassava (tapioca) may contain 245 mg cyanide/100 g root. The
cyanide content in 100 g of cultivated apricot seeds has been found
to be about 9 mg and that in wild apricot seeds more than 200 mg.
1.2.4 Iatrogenic sources
Cyanide is also formed during nitroprusside therapy, especially
when it is prolonged, because tachyphylaxis sometimes requires the
use of higher doses than the recommended maximum of 10 µg/kg per min
(Smith & Kruszyna, 1974; MacRae & Owen, 1974; Piper, 1975; Atkins,
1977; Anon, 1978). Cyanide metabolises to thiocyanate. Thiocyanates
were used some years ago as
antihypertensive agents and they saw wide use because they were very
effective. However, a variety of subacute toxic effects, including
anorexia, fatigue, and gastrointestinal tract and CNS disturbances,
led to their disfavour.
Laetrile, amygdalin derived from apricot kernels, has been used
as an anticancer agent, but it is now obsolete because a therapeutic
effect could not be demonstrated in either retrospective or
prospective studies. Laetrile has caused fatal cyanide poisoning
(Sadoff et al., 1978).
1.3 Toxicity of Cyanide in Man
1.3.1 Acute poisoning
It is generally accepted that inhalation of approximately 50 ml
(at 1.85 mmol/l) of hydrogen cyanide gas is fatal within minutes.
Poisoning from hydrogen cyanide is more frequently
accidental than suicidal. Thus accidental cyanide poisoning may
occur in fumigators and chemists who use hydrogen cyanide during the
course of their work (Chen et al., 1944). In fires, a combination
of HCN and carbon monoxide (CO) toxicity, as a result of inhalation of
combustion products, may cause fatalities.
Suicidal ingestion of cyanide salts most commonly occurs in
personnel with occupational access to cyanide. The ingestion of as
little as 250 mg of an inorganic cyanide salt may be fatal (Peters
et al., 1982). However, death may be delayed for several hours
following the ingestion of cyanide on a full stomach; a first-pass
effect in the liver may also delay the onset of toxicity
(Naughton, 1974).
1.3.2 Chronic poisoning
Chronic low-dose neurotoxicity have been suggested by
epidemiological studies of populations ingesting naturally occurring
plant glycosides (Blanc et al, 1985). These glycosides are present
in a wide variety of plant species, most notably the cassava plant,
a major tropical foodstuff (Conn, 1973; Cook & Coursey, 1981;
Ministry of Health, Mozambique, 1984). Cassava has been associated
with tropical ataxic neuropathy (Cook & Coursey, 1981). Epidemic
spastic paraparesis has been associated with a combination of a high
cyanide and a low sulfur intake from diets dominated by
insufficiently processed cassava and lacking protein supplementary
food (Rosling, 1989). A neurotoxicological role for cyanide has
also been suggested in tobacco-associated amblyopia (Grant, 1980)
and in amygdalin-associated peripheral neuropathy (Kalyanaraman et
al., 1983). Long-term cyanide intoxication has been shown to be
associated both with thyroid gland enlargement and dysfunction in
case reports and in cohort studies of individuals exposed
occupationally (Blanc et al., 1985), through dietary intake (Cook &
Coursey, 1981), and experimentally (El Ghawabi et al., 1975).
1.4 Mechanism of Toxicity
Cyanide has a special affinity for the ferric ions that occur
in cytochrome oxidase, the terminal oxidative respiratory enzyme in
mitochondria. This enzyme is an essential catalyst for tissue
utilization of oxygen. When cytochrome oxidase is inhibited by
cyanide, histotoxic anoxia occurs as aerobic metabolism becomes
inhibited. In massive cyanide poisoning, the mechanism of toxicity
is more complex. It is possible that autonomic shock from the
release of biogenic amines may play a role by causing cardiac
failure (Burrows & Way, 1976). Cyanide could cause both pulmonary
arteriolar and/or coronary arterial vasoconstriction, which would
result, either directly or indirectly, in pump failure and a
decrease in cardiac output. This theory is supported by the sharp
increase in central venous pressure that was observed by Vick &
Froelich (1985) at a time when the arterial blood pressure fell
after the intravenous administration of sodium cyanide to dogs. The
observation that phenoxybenzamine, an alpha-adrenergic blocking
drug, partially prevented these early changes (Vick & Froelich,
1985) supports the concept of an early shock-like state not related
to inhibition of the cytochrome oxidase system. Inhalation of amyl
nitrite, a potent arteriolar vasodilating agent, resulted in the
survival of dogs in these experimental circumstances. This could
have been due to reversal of early cyanide-induced vasoconstriction
with restoration of normal cardiac function (Vick & Froelich, 1985).
1.5 Clinical Features
The smell of bitter almonds in expired air is an important sign
in cyanide poisoning. However, many people are unable to perceive
the odour of hydrocyanic acid (Kalmus & Hubbard, 1960). The
incidence of "non-smellers" is reported to be 18% among males and 5%
among females (Kirk & Stenhouse, 1953; Fukumoto et al., 1957).
Immediately after swallowing cyanide, very early symptoms, such
as irritation of the tongue and mucous membranes, may be
experienced. A blood-stained aspirate may be observed if gastric
lavage is performed. Early symptoms and signs that occur after
inhalation of HCN or the ingestion of cyanide salts include anxiety,
headache, vertigo, confusion, and hyperpnoea, followed by dyspnoea,
cyanosis, hypotension, bradycardia, and sinus or AV nodal
arrythmias.
In the secondary stage of poisoning, impaired consciousness,
coma and convulsions occur and the skin becomes cold, clammy, and
moist. The pulse becomes weaker and more rapid. Opisthotonos and
trismus may be observed. Late signs of cyanide toxicity include
hypotension, complex arrythmias, cardiovascular collapse, pulmonary
oedema, and death.
It should be emphasized that the bright-red coloration of the
skin or absence of cyanosis mentioned in textbooks (Gosselin et al.,
1984; Goldfrank et al., 1984) is seldom described in case reports of
cyanide poisonings. Theoretically this sign could be explained by
the high concentration of oxyhaemoglobin in the venous return, but,
especially in massive poisoning, cardiovascular collapse will
prevent this from occurring. Sometimes, cyanosis can be observed
initially, while later the patient may become bright pink (Hilmann
et al., 1974).
The pathogenesis of pulmonary oedema could be due to several
different mechanisms: (1) an intracellular metabolic process that
could injure the alveolar and capillary epithelium directly,
producing a capillary leak syndrome; (2) neurogenic pulmonary oedema
or, (3) most likely, a direct effect on the myocardium leading to
left ventricular failure and increased pulmonary venous pressure.
The brain is obviously the key organ involved in cyanide
poisoning and it has been shown that cyanide significantly increases
brain lactate and decreases brain ATP concentrations (Olsen & Klein,
1947).
1.6 Laboratory Findings
1.6.1 Lactic acidosis
Since oxidative phosphorylation is blocked, the rate of
glycolysis is markedly increased, which in turn leads to lactic
acidosis. The degree of lactic acidosis can be correlated with the
severity of cyanide poisoning (Trapp, 1970; Naughton, 1974).
1.6.2 Hyperglycaemia
A reversible toxic effect occurs on the pancreatic beta-cells,
which may occasionally give rise to an erroneous diagnosis of
hyperglycaemic diabetic coma.
1.6.3 Cyanide concentration in blood and plasma
Before intravenous treatment with antidotes is commenced, it is
necessary to collect a heparinized (not fluoride) blood sample for
determination of cyanide concentration. Results from samples
collected after treatment are totally unreliable. A quantitative
test employing a detector tube (see chapter 10) can be used if the
diagnosis is in doubt. The blood can also be used for a
quantitative test (see chapter 10), so that the severity of
poisoning can be evaluated. Therapeutic measures after antidotal
treatment should be based on the clinical condition of the patient
rather than on blood cyanide concentrations (Berlin, 1971; Vogel et
al., 1981; Peters et al., 1982). Since blood concentrations of up
to 0.005-0.04 mg/l have been recorded in healthy non-smokers, and
0.01-0.09 mg/l in smokers, only concentrations above these values
were previously considered to be toxic (Vogel et al., 1981; Peters
et al., 1982). Lundquist et al., (1985) reported even lower
concentration: non-smokers 3.4 µg/l (whole blood), 0.5 µg/l
(plasma), 6.0 µg/l (erythrocytes); smokers 8.6 µg/l (whole blood),
0.8 µg/l (plasma), 17.7 µg/l (erythrocytes).
Fatal cyanide poisoning has been reported with whole blood
concentrations of >3 mg/l and severe poisoning with 2 mg/l (Graham
et al., 1977). However, when cyanide enters the bloodstream, up to
98% quickly enters the red blood cells where it becomes tightly
bound. A plasma-to-blood ratio as high as 1:10 has been reported
and, as a consequence, the whole blood cyanide concentration may not
accurately reflect tissue concentrations of cyanide. Plasma levels
of cyanide may be of greater significance because severe toxicity
occurs in the presence of only modest concentrations (Vesey et al.,
1976). However, a serious drawback to the use of plasma cyanide
determinations in the assessment of poisoning is the pronounced
instability of cyanide in plasma (Lundquist et al., 1985).
1.7 Biological Detoxification of Cyanide
The major pathway of endogenous detoxification is conversion,
by means of thiosulfate, to thiocyanate. Minor routes of elimination
are excretion of hydrogen cyanide through the lungs and binding
to cysteine or hydroxocobalamin.
.Metabolic Detoxification of Cyanide;V02ANnew.BMP
The detoxification of cyanide occurs slowly at the rate of
0.017 mg/kg per min (McNamara, 1976). A sulfurtransferase
enzyme is needed to catalyse the transfer of a sulfur atom
from the donor thiosulfate to cyanide. The classical theory
indicating that mitochondrial thiosulfate sulfurtransferase
is the most important enzyme in this reaction is now in
doubt because thiosulfate penetrates lipid membranes slowly
and would, therefore, not be readily available as a
source of sulfur in cyanide poisoning. The modern concept assumes a
greater role for the serum albumin-sulfane complex, which is the
primary cyanide detoxification buffer operating in normal metabolism
(Sylvester et al., 1983). A further enzyme, beta-mercaptopyruvate
sulfurtransferase, also converts cyanide to thiocyanate (Vesey et
al., 1974). This enzyme is found in the erythrocytes, but in human
cells its activity is low.
1.7.1 Thiocyanate toxicity
The detoxification product of cyanide, thiocyanate, is excreted
in the urine. Thiocyanate concentrations are normally between
1-4 mg/l in the plasma of non-smokers and 3-12 mg/l in smokers. The
plasma half-life of thiocyanate in patients with normal renal
function is 4 h (Blaschle & Melmon, 1980), but in those with renal
insufficiency it is markedly prolonged and these patients are
therefore at increased risk of toxicity (Schulz et al., 1978).
Thiocyanate levels exceeding 100 mg/l are thought to be associated
with toxicity. Thiocyanate toxicity is characterized by weakness,
muscle spasm, nausea, disorientation, psychosis, hyper-reflexia, and
stupor (Smith, 1973; Michenfelder & Tinker, 1977). Lethal poisoning
at concentrations greater than 180 mg/l has been reported (Healy,
1931; Garvin, 1939; Russel & Stahl, 1942; Kessler & Hines, 1948;
Domalski et al., 1953). Haemodialysis is recommended as an
effective means of removing thiocyanate (Marbury et al., 1982).
Dialysance values of 82.8 ml/min ( in vivo) and 102.3 ml/min
( in vitro) have been recorded (Pahl & Vaziri, 1982). Little is
known about the protein-binding characteristics of thiocyanate, and
haemoperfusion may be more effective than haemodialysis.
1.8 Protective Measures for Occupational Exposure
Accidental exposure to cyanide, as either hydrogen cyanide or
cyanide salts, will occur primarily in the occupational context, and
appropriate preventive and protective measures need to be taken
wherever cyanides are manufactured or used. Many industrial accidents
occur as a result of mixing cyanide salts and acids, and care
should be taken when both are present on industrial premises.
As hydrogen cyanide may be generated during combustion of organic
substances, fire fighters may also be exposed occupationally.
The public may be affected in the case of a major industrial
emergency, or of a transport accident, involving the release of
cyanides. It is essential for local authorities in areas where
cyanides are used to have contingency plans that will enable them to
respond effectively. Adequate hospital facilities for treatment of
casualties must be available.
Proper maintenance of plant, good operating practice, and
industrial hygiene are essential for the prevention of cyanide
poisoning. Areas in the workplace where cyanides are used and
containers for storage and transport of cyanide should be clearly
marked. Work schedules should ensure that there are at least two
people in zones where cyanide could be released accidentally. There
should be showers and first-aid kits in these areas. Personnel
without proper training should not be allowed in the plant. Normal
industrial and laboratory hygiene measures for personnel handling
toxic materials, such as dirty and clean locker facilities and
showers, should be provided. Eating, drinking, and smoking should
not be allowed in the work area where cyanides are used but in
places specially reserved for these purposes.
Each employee working at a plant or laboratory that handles
cyanides, should receive instruction on the dangers of cyanides and
be trained in appropriate first-aid measures, as should
emergency-service personnel. They should be aware of the hazards
and informed about the possible routes of exposure (inhalation, skin
absorption, ingestion). Training should involve recognition of the
symptoms and signs of cyanide poisoning and how to achieve safe
removal of victims from the source of intoxication. Personnel
should also be able to guide a rescue or fire-fighting team to a
trapped intoxicated person. Rescue personnel should be able to put
on protective clothing quickly in an emergency. There should be
regular instruction sessions covering procedures for handling
cyanides and for rescue in case of accidents, as well as random
alarm exercises. First-aid training should include the essential
measures to be taken before medical help arrives, which may need to
be undertaken at the same time as removal of contaminated clothing
and decontamination of exposed skin and eyes. It should be realized
that further uptake of cyanide into the blood may occur after
showering because of continued skin absorption.
Each plant handling cyanide should have its own medical staff
trained in the emergency treatment of cyanide poisonings. The
atmospheric concentrations of hydrogen cyanide should be monitored
in plants where the gas is used or may be generated. Warning
devices are available for this purpose and should be installed. In
certain circumstances in which cyanide is used, it is possible to
add a warning gas, e.g., cyanogen chloride and chloropicrin have
been added to hydrogen cyanide used as a fumigant (Cousineau & Legg,
1935; Polson & Tattersall, 1969).
Filter respirators should be carried at all times by employees
working in zones where hydrogen cyanide may be released. At high
hydrogen cyanide concentrations, absorption occurs through the skin
and impermeable butyl rubber protective clothing is required.
Oxygen breathing apparatus may be needed.
In the case of an accident involving hydrogen cyanide there
should be both an acoustic and a visual alarm for the plant, which
may be activated by workers in zones where the gas is used. Each
worker should be aware of the emergency procedures to be followed
and the protective clothing and equipment to be used. If a large
number of victims is involved or if there is a danger to the public,
local authorities need to be warned, so that contingency plans are
put into effect and hospitals alerted.
For accidents at plants in remote areas where a qualified
physician is not readily available and there are no hospital
intensive care facilities, attending paramedical personnel should
have the authority and training to perform the special resuscitation
measures involved in treating cyanide poisonings, including rapid
endotracheal intubation and techniques for obtaining intravenous
access.
1.9 Treatment
1.9.1 Supportive treatment
Although effective antidotes are available, general supportive
measures should not be ignored and may be life-saving.
According to Jacobs (1984), who reported his personal
experience of 104 industrial poisoning cases, the use of specific
antidotes was indicated only in cases of severe intoxication with
deep coma, wide non-reactive pupils, and respiratory insufficiency
in combination with circulatory insufficiency. In patients with
moderately severe poisoning, who had suffered only a brief period of
unconsciousness, convulsions, vomiting, and cyanosis, therapy
consisted of intensive care and intravenous sodium thiosulfate. In
cases of mild intoxication with dizziness, nausea, and drowsiness,
rest and oxygen alone were used.
Peden et al. (1986) described nine patients poisoned by
hydrogen cyanide released by a leak from a valve. Three of them
were briefly unconscious but recovered rapidly after being moved
from the area where they had been working. The arterial whole-blood
cyanide concentrations on admission were 3.5, 3.1 and 2.8 mg/l,
respectively. The cyanide concentrations in the other cases ranged
between 2.6 and 0.93 mg/l. All recovered with supportive therapy
alone.
Between 1970 and 1984, three other men were treated similarly;
two were transiently unconscious, and in these cases the cyanide
concentrations 30 min after exposure were 7.7 and 4.7 mg/l. The
concentration in the other patient was 1.6 mg/l. All three patients
recovered without the use of cyanide antidotes. Small numbers of
comatose patients with potentially lethal blood concentrations on
admission, and who recovered without cyanide antidotes, have been
reported by Graham et al. (1977), Edwards & Thomas (1978), and Vogel
et al. (1981).
Even if a patient is unconscious, an antidote does not
necessarily have to be administered immediately unless vital signs
deteriorate.
A patient exposed to hydrogen cyanide who reaches hospital
fully conscious is only likely to require observation and
reassurance.
1.9.2 Antidotal treatment
1.9.2.1 Oxygen
It is difficult to understand how oxygen has a favourable
effect in cyanide poisoning, because inhibition of cytochrome
oxidase is non-competitive. However, oxygen has always been
regarded as an important first-aid measure in cyanide poisoning, and
there is now experimental evidence that oxygen has specific
antidotal activity. Oxygen accelerates the reactivation of
cytochrome oxidase and protects against cytochrome oxidase
inhibition by cyanide (Takano et al., 1980). Nevertheless, there
are other possible modes of action and those that are clinically
important have yet to be determined.
Hyperbaric oxygen is recommended for smoke inhalation victims
suffering from combined carbon monoxide and cyanide poisoning, since
these two agents are synergistically toxic. The use of hyperbaric
oxygen in pure cyanide poisoning remains controversial.
1.9.2.2 Sodium thiosulfate
The major route of cyanide detoxification in the body is
conversion to thiocyanate by rhodanese, although other
sulfurtransferases, such as beta-mercaptopyruvate sulfurtransferase,
may also be involved. This reaction requires a source of sulfane
sulfur, but endogenous supplies of this substance are limited.
Cyanide poisoning is an intramitochondrial process and an
intravenous supply of sulfur will only penetrate mitochondria
slowly. While sodium thiosulfate may be sufficient alone in mild to
moderately severe cases, it should be administered with other
antidotes in cases of severe poisoning. It is also the antidote of
choice when the diagnosis of cyanide intoxication is not certain,
for example in cases of smoke inhalation. Sodium thiosulfate is
assumed to be intrinsically nontoxic but the detoxification product
formed from cyanide, thiocyanate, may cause toxicity in patients
with renal insufficiency (see section 1.7).
1.9.2.3 Amyl nitrite
The administration of amyl nitrite by inhalation has been used
for many years as a simple first-aid measure that generates
methaemoglobin and which can be employed by lay personnel. Its use
was abandoned because the methaemoglobin concentration obtained with
amyl nitrite is no more than 7% and it is thought that at least 15%
is required to bind a potentially lethal dose of cyanide. However,
recent studies suggest that methaemoglobin formation plays only a
small role in the therapeutic effect of amyl nitrite, and
vasodilatation may be the most important mechanism of antidotal
action. Artificial respiration with amyl nitrite ampoules broken
into an Ambu bag proved to be life-saving in dogs severely poisoned
with cyanide. Amyl nitrite may therefore be reintroduced as a
first-aid measure.
1.9.2.4 Sodium nitrite
Nitrites generate methaemoglobin, which combines with cyanide
to form the nontoxic substance cyanmethaemoglobin. Methaemoglobin
does not have a higher affinity for cyanide than does cytochrome
oxidase, but there is a much larger potential source of
methaemoglobin than there is of cytochrome oxidase. The efficacy of
methaemoglobin is therefore primarily the result of mass action. A
drawback of methaemoglobin generation is the resultant impairment of
oxygen transport to cells and, ideally, the total amount of free
haemoglobin should be monitored to ensure aerobic metabolism of the
cells. Methaemoglobin can be measured very quickly, but this in
itself will not provide an accurate guide to the amount of
haemoglobin available for oxygen transport because the
cyanmethaemoglobin concentration is not taken into account.
Individuals deficient in glucose-6-phosphate dehydrogenase (G6PD)
are at great risk from sodium nitrite therapy because of the
likelihood of severe haemolysis, but the risk from amyl nitrite is
likely to be less because only low plasma concentrations are
achieved. Excess methaemoglobinaemia may be corrected with either
methylene or toluidine blue (see Chapter 9) or, preferably, where
feasible, by exchange transfusion.
1.9.2.5 4-Dimethylaminophenol (4-DMAP)
4-DMAP generates a methaemoglobin concentration of 30-50%
within a few minutes (Weger, 1968) and, theoretically, it should
therefore be valuable as a first-aid measure. However, the problems
associated with methaemoglobin formation, as described above for
nitrites, apply to 4-DMAP to an even greater extent. Furthermore,
it has very poor dose-response curve reproducibility. Haemolysis as
a result of 4-DMAP therapy has been observed in overdose as well as
following a correct therapeutic dose. Treatment with 4-DMAP is
contraindicated in patients with G6PD deficiency. Excess
methaemoglobinaemia may be corrected with either methylene or
toluidine blue (see section 1.9.2.8).
1.9.2.6 Hydroxocobalamin (vitamin Bl2a)
This antidote binds cyanide strongly to form cyanocobalamin
(vitamin B12) and, compared to nitrite and 4-DMAP therapy, it has
the great advantage of not interfering with tissue oxygenation. The
disadvantage of hydroxocobalamin as a cyanide antidote is the large
dose required for it to be effective. Detoxification of 1 mmol
cyanide (corresponding to 65 mg KCN) needs 1406 mg hydroxocobalamin.
In most countries it is only commercially available in formulations
of 1-2 mg per ampoule. In some countries, e.g., France, a
formulation is available that contains 4 g hydroxocobalamin powder
that has to be reconstituted with 80 ml of a 10% sodium thiosulfate
solution prior to use and administered intravenously in a minimum of
220 ml of 5% dextrose. Recorded side effects are anaphylactoid
reactions and acne. Some authors have reported a reduced antidotal
effect as a result of mixing hydroxocobalamin and sodium thiosulfate
in the same solution (Evans, 1964; Friedberg & Shukla, 1975).
Histological changes in the liver, myocardium, and kidney apparently
induced by hydroxocobalamin have been reported in animal
experiments (Hoebel et al., 1980), but their relevance to man has
not yet been established. Transient pink discoloration of mucous
membranes and urine is an unimportant and nontoxic side-effect.
1.9.2.7 Dicobalt edetate
This agent has been shown to be effective in the treatment of
cyanide poisoning in man, and in the United Kingdom it is the
current treatment of choice provided that cyanide toxicity is
definitely present. This is a strict criterion, because as a result
of the manufacturing process some free cobalt ions are always
present in solutions of dicobalt edetate. Cobalt ions are toxic and
the use of dicobalt edetate, in the absence of cyanide, will lead to
serious cobalt toxicity. There is evidence from animal experiments
that glucose protects against cobalt toxicity and it is recommended
that this be given at the same time as dicobalt edetate. Serious
adverse effects recorded include vomiting, urticaria, anaphylactic
shock, hypotension, and ventricular arrhythmias (Hilmann et al.,
1974; Naughton, 1974).
1.9.2.8 Antidotes to methaemoglobin-forming agents
Accurate determination of methaemoglobin and free haemoglobin
concentrations in the presence of cyanide is difficult.
Nevertheless, excess methaemoglobinaemia does undoubtedly occur on
occasions following the use of nitrites and 4-DMAP. Excess
methaemoglobin concentrations may be reduced by methylene or
toluidine blue. However, regeneration of haemoglobin will release
cyanide back into the circulation, leading to a recurrence of
toxicity.
1.10 Summary of Treatment Recommendations
The management of cyanide poisoning is determined by (i) the
nature of exposure, i.e. hydrogen cyanide (with or without carbon
monoxide), cyanide salts, aliphatic nitriles, cyanogenic glycosides;
(ii) the severity of poisoning; (iii) the number of patients
involved; (iv) the proximity of hospital facilities; (v) the
presence of risk factors, e.g., G6PD deficiency. Urgent specific
antidotal therapy is not indicated unless the patient is in a deep
coma, with dilated non-reactive pupils and deteriorating
cardio-respiratory function. A patient exposed to hydrogen cyanide
confwho reaches hospital fully conscious requires observation and
reassurance only.
In order to assess the severity of cyanide poisoning, it is
necessary to take a blood sample before the administration of
antidotes. Analytical results are otherwise unreliable.
1.10.1 First aid and treatment measures at the site of the incident
The doses given are for adults. Model Information Sheets
should be consulted for the pediatric dose and for the use of
antidotes in special-risk groups, e.g., G6PD-deficient patients.
The following should be undertaken:
(a) Trained personnel (wearing appropriate protective clothing
and breathing apparatus if hydrogen cyanide or liquid
cyanide preparations are involved) should
* terminate further exposure
* commence artificial ventilation with 100% oxygena
* administer 0.2-0.4 ml amyl nitrite via Ambu bag
(b) A physician (if immediately present on the scene) should
* terminate further exposure
* artificial ventilation with 100% oxygena
* administer 0.2-0.4 ml amyl nitrite via Ambu bag
In cases of unequivocal moderate to severe poisoning, the above
procedure should be followed by
50 ml of 25% sodium thiosulfate solution
(12.5 g) i.v. for 10 minutes
and either 20 ml of 1.5% dicobalt edetate solution
(300 mg) i.v. for 1 minute
or 10 ml of 40% hydroxocobalamin solution (4 g)
i.v. for 20 minutes
or 10 ml of 3% sodium nitrite solution (300 mg)
i.v. for 5-20 minutes
or 5 ml of 5% 4-DMAP solution (250 mg or
3-4 mg/kg) i.v. for 1 minute
a Oxygen should be administered using a mask and a bag with a
"non-return" valve to prevent inspiration of exhaled gases.
1.10.2 Hospital treatmenta
The doses given are for adults. Model Information Sheets
should be consulted for the pediatric doses and for the use of
antidotes in special-risk groups, e.g., G6PD-deficient patients.
1.10.2.1 Severe poisoning
Patients in deep coma with dilated non-reactive pupils and
deteriorating cardio-respiratory function (blood cyanide
concentrations 3 to 4 mg/l) should be given
* artificial ventilation with 100% oxygenb
* cardio-respiratory support
This should be followed by
50 ml of 25% sodium thiosulfate solution
(12.5 g) i.v. over 10 min
and either 20 ml of 1.5% dicobalt edetate solution
(300 mg) i.v. over 1 min
or 10 ml of 40% hydroxocobalamin solution (4 g)
i.v. over 20 min
or 10 ml of 3% sodium nitrite solution (300 mg)
i.v. over 5-20 min
or 5 ml of 5% 4-DMAP solution (250 mg or
3-4 mg/kg) i.v. over 1 min
a Hospital physicians must establish whether specific antidotal
therapy was administered at the time of the incident before
further doses are administered, especially in the case of
methaemoglobin-forming agents.
b Oxygen should be administered using a mask and a bag with a
"non-return" valve to prevent inspiration of exhaled gases.
1.10.2.2 Moderately severe poisoning
Patients who have suffered a short-lived period of
unconsciousness, convulsions, vomiting, and/or cyanosis (blood
cyanide concentrations 2-3 mg/l) should be given
* 100% oxygen, but for no longer than 12-24 h
* 50 ml of 25% sodium thiosulfate solution (12.5 g)
i.v. over 10 min
* observation in an intensive-care area
1.10.2.3 Mild poisoning
Patients with nausea, dizziness, drowsiness only (blood cyanide
concentrations < 2 mg/l) should be given
* oxygen
* reassurance
* bed rest
It should be noted that severely poisoned patients may
occasionally fail to respond to the initial dose of a specific
antidote. Whilst repeat doses of hydroxocobalamin and/or sodium
thiosulfate are unlikely to be associated with toxicity, expert
advice should be sought before a repeat dose of any other specific
antidote is administered. Intensive supportive therapy is of
paramount importance in these circumstances.
1.11 Summary of Analytical Aspects
There are many reliable methods for the detection and
qualitative determination of cyanide in biological material in cases
of suspected intoxication (see Chapter 10). They can be used as
"bedside methods" as well as for qualitative determination in cases
of acute poisoning but only before antidotes are administered.
Interference results from the presence of thiosulfate,
methaemoglobin, thiocyanates, and chelating agents during the course
of whole-blood cyanide analysis. For this reason, it may be more
appropriate to measure plasma rather than whole-blood cyanide
concentrations. However, the pronounced instability of cyanide in
plasma is a serious drawback (Lundquist et al., 1985).
Quantitative analysis of cyanide in blood or serum before the
administration of antidotes is a useful means of evaluating the
severity of poisoning. Evaluation of the efficacy of different
antidotes will not be possible before accurate methods of analysis
free from interference are developed.
When methaemoglobin-generating agents (nitrites or 4-DMAP) are
administered as antidotes in cyanide poisoning, it is necessary to
maintain an adequate concentration of free haemoglobin in order to
guarantee sufficient oxygen transport to allow aerobic tissue
metabolism.
Special instruments for rapid analysis of methaemoglobin in
hospitals do not provide information about the amount of haemoglobin
available for oxygen transport, because the multicomponent analysis
is invalidated by the presence of a haemoglobin derivate
(cyanmethaemoglobin). Since there is no satisfactory means of
quantifying cyanmethaemoglobin under these circumstances, therapy
with methaemoglobin-generating agents cannot be monitored at present
by laboratory methods.
1.12 Proposed Areas for Research
There are two areas of research where further work is needed as
a matter of urgency:
(a) Analytical techniques currently available for the
measurement of methaemoglobin do not permit accurate
estimation of the amount of free haemoglobin available for
oxygen transport, because cyanmethaemoglobin cannot be
quantified. A rapid and accurate technique for measuring
methaemoglobin and cyanmethaemoglobin concentrations in
conjunction is therefore needed to monitor the use of
methaemoglobin-generating cyanide antidotes.
(b) Reliable quantitative analytical methods for cyanide in
whole blood in the presence of one or more antidotes are
needed.
(c) Determination of cyanide concentration in plasma or serum
may be the best reflection of the tissue concentration of
cyanide, since cyanide trapped in erythrocytes will not
affect tissue utilization of oxygen. However, cyanide has
been shown to be very unstable in these body fluids. A
method to prevent this phenomenon is urgently needed.
(d) The intravenous injection of DMAP generates high
concentrations of methaemoglobin within minutes. However,
the absorption kinetics of DMAP administered
intramuscularly are not known with certainty, particularly
in patients who are shocked with poor muscle perfusion.
The efficacy of intramuscular DMAP as a first-aid measure
in cases of severe cyanide poisoning needs further
evaluation.
(e) Hydroxocobalamin has recently been reported to cause
histological changes in the liver, kidney, and myocardium
of animals. The relevance of these findings to man is not
known and further investigation is required.
(f) Enzyme systems other than cytochrome oxidase may be
inhibited. This may be the cause for the symptomatology
in acute severe cyanide poisoning.
1.13 New Developments in Cyanide Antidotes
Currently available cyanide antidotes have potentially
undesirable adverse effects, and none has been successful in all
cases of acute, severe cyanide poisoning. Various agents for the
treatment of cyanide poisoning are at the experimental stage of
development. However, these antidotes are not currently recommended
for administration in cases of human poisoning.
1.13.1 Nonspecific agents
Based on animal studies, certain nonspecific agents, such as
naloxone in huge doses (equivalent to 700 mg in a 70 kg human adult
compared with a usual therapeutic dose of 0.4 to 10 mg) (Leung et
al., 1986) or alpha-adrenergic blocking agents such as
chlorpromazine (which has no beneficial effect when administered
alone but variably enhances sodium nitrite and/or sodium thiosulfate
efficacy) (Kong et al., 1983; Petterson & Cohen, 1985), have been
suggested as adjunctive therapy. However, at the moment, there is
no accepted place for the use of these agents in human poisoning.
1.13.2 Sodium pyruvate
This agent re-establishes cellular respiration in tumour
tissues inactivated by cyanide and has some efficacy in experimental
animal poisoning. It may promote cyanide detoxification through
combination of the cyanide anion with a carbonyl radical, producing
cyanohydrin (Pronczuk de Garbino & Bismuth, 1981). Sodium pyruvate
acts rapidly and is well distributed to tissues, but clinical trials
in human cyanide poisoning have not been undertaken.
1.13.3 Ifenprodil
Ifenprodil is a 2-piperidine allonal derivative, which, in
experimental poisoning, affords some protection including decreased
respiratory distress, improved blood pressure, normalization of
cardiac rhythm, and lessened electroencephalographic abnormalities.
The mechanism of action is thought to be a direct stimulation of
mitochondrial respiratory function. At present, ifenprodil is in
the investigational stage and no human clinical trials have been
proposed (Pronczuk de Garbino & Bismuth, 1981).
1.13.4 Rhodanese
Rhodanese (thiosulfate-cyanide sulfurtransferase) is the
naturally-occurring cyanide-detoxifying enzyme (see section 1.7).
Although the availability of sulfane sulfur is the rate-limiting
factor, studies in dogs have indicated that there is enough
rhodanese present in the normal liver and muscle tissue to detoxify
about 500 grams of cyanide. When derived from hepatic tissue, the
enzyme is unstable and requires an optimal pH for cyanide
detoxification. Bacterial enzyme, derived from cultures of
Thiobacillus denitrificans, is more stable and has been studied
in experimental animals. It is efficacious in experimental cyanide
poisoning, but no human clinical applications have yet been proposed
(Pronczuk de Garbino & Bismuth, 1981).
1.13.5 Alpha-ketoglutaric acid
The cyanide ion can react with carbonyl groups to form
cyanohydrins, and this could represent an important detoxification
reaction. In rodents poisoned with cyanide and pretreated with
various antidotes, alpha-ketoglutaric acid was more effective than
either sodium nitrite or sodium thiosulfate, and nearly as effective
as sodium nitrite and sodium thiosulfate in combination (Moore et
al., 1986). The combination of alpha-ketoglutaric acid with sodium
nitrite plus sodium thiosulfate increased the cyanide LD50 from a
mean of 6.7 mg/kg in control animals to 119.4 mg/kg, whereas the
sodium nitrite/thiosulfate combination alone increased the mean
LD50 to only 35.0 mg/kg (alpha-ketoglutaric acid alone increased
the mean LD50 to 33.3 mg/kg). No methaemoglobin induction was
observed with alpha-ketoglutaric acid administration. Some tremors
were noted when this agent was administered alone. Tremors did not
occur when sodium thiosulfate was added to the treatment regimen,
and the LD50 value was increased to 101.3 mg/kg with this
combination (very close to the 19.4 mg/kg LD50 observed with
addition of both sodium nitrite and sodium thiosulfate to
alpha-ketoglutaric acid). While these studies demonstrated only
protective activity with prophylactic alpha-ketoglutaric acid
administration, they raise the possibility of another potentially
efficacious and safe antidote combination with sodium thiosulfate
(Moore et al., 1986). Alpha-ketoglutaric acid, especially in
combination with sodium thiosulfate, deserves further study.
1.13.6 Stroma-free methaemoglobin solution
Stroma-free methaemoglobin solution is prepared from outdated
red blood cells by the removal of all cellular membranes (stroma)
and induction of methaemoglobinaemia equivalent to 90% of the total
haemoglobin with potassium ferricyanide. Any excess potassium
ferricyanide is then removed by dialysis against saline. The
resultant preparation does not contain the antigenic components that
have been previously reported to cause renal failure and
coagulopathies. No rats given stroma-free methaemoglobin solution
alone died or had any adverse reactions. Concentrated stroma-free
methaemoglobin solution (200-300 g/l) was an effective experimental
antidote when administered 30 seconds after doses of cyanide up to 6
times the LD90. More dilute solutions were effective 90% of the
time following cyanide administration up to 4 times the LD90.
None of the animals in these studies were given any supportive
therapy. In animals administered an amount of stroma-free
methaemoglobin solution thought to be equivalent to the conversion
of 1.5% of the endogenous haemoglobin, a 90% survival rate was noted
when a cyanide LD100 was administered. Spectroscopic examination
of urine revealed cyanmethaemoglobin excretion (Ten Eyck et al.,
1984, 1985, 1986).
These studies suggest that methaemoglobin prepared exogenously
may be an effective cyanide antidote. Exogenously administered
methaemoglobin would not be expected to interfere with oxygen
transport and, unlike methaemoglobin-generating agents (Moore et
al., 1987), could even be used in smoke-inhalation victims with
elevated carboxyhaemoglobin levels. Since no adverse effects have
been noted, this agent may be a safe alternative to currently
available cyanide antidotes. However, no human studies have been
undertaken and extensive animal toxicology experiments have not yet
been reported.
1.14 References
Alarie Y (1985) The toxicity of smoke from polymeric materials
during thermal decomposition. Am Rev Pharmacol Toxicol, 25: 325-347.
Anderson RA & Harland WA (1982) Fire deaths in the Glasgow area.
III. The role of hydrogen cyanide. Med Sci Law, 22: 35-37.
Anon (1978) Controlled intravascular sodium nitroprusside treatment.
Br Med J, 6140: 784-785.
Antal J (1894) [Experimental studies on the treatment of cyanide
poisoning.] Ung Arch Med, 3:117-128 (in German).
Atkins D (1977) Cyanide toxicity following nitroprusside-induced
hypotension. Can Anaesth Soc J, 24: 651-660.
Berlin AM (1971) The treatment of cyanide poisoning in children.
Pediatrics, 46: 793.
Birky MM, Halpin BM, Caplan YH, Fisher RS, McAllister JM, & Dixon AM
(1979) Fire fatality study. Fire Mater, 3: 211-217.
Blake G (1839) Observations on the physiological effects of various
agents introduced into the circulation, as indicated by the
hemodynamometer. Edinburgh Med Surg J, 53: 35-49.
Blake G (1840) Observations and experiments in the mode in which
various poisonous agents act on the animal body. Edin Med Surg J 53:
35-49.
Blanc P, Hogan M, Malin K, Hryhorczuk D, Hessl S, & Bernard B (1985)
Cyanide intoxication among silver reclaiming workers. J Am Med
Assoc, 253: 367-371.
Blaschle TE & Melmon KL (1980) Antihypertensive agents and the drug
therapy of hypertension. In: Goodman LS & Gilma A ed. The
pharmacological basis of therapeutics. New York, MacMillan
Publishing Co., vol 6, pp 807-808.
Burrows GE & Way JL (1976) Antagonism of cyanide toxicity by
phenoxybenzamine. Fed Proc, 35: 533.
Chen KK, Rose CL, & Clowes GHA (1933) Methylene blue, nitrites and
sodium thiosulfate against cyanide poisoning. Proc Soc Exp Biol Med,
31: 250-252.
Chen KK, Rose CL, & Clowes GHA (1934) Comparative values of several
antidotes in cyanide poisoning. Am J Med Sci, 188: 767.
Chen KK, Rose CL, & Clowes GHA (1944) The modem treatment of cyanide
poisoning. J Indiana Med Assoc, 37: 344-350.
Clark CJ, Campbell D, & Reid WH (1983) Blood carboxyhaemoglobin and
cyanide levels in fire survivors. Lancet, 1: 1332-1335.
Conn EE (1973) Cyanogenic glucosides (Committee on Food Protection,
Food and Nutrition Board, National Research Council). In: Toxicants
occurring naturally in foods, 2nd ed. Washington, DC, National
Academy of Sciences, pp 299-308.
Cook RD & Coursey DB (1981) Cassava: a major cyanide-containing food
crop. In: Vennesland B, Conn E, Knowles CJ, Westley J, & Wissing F
ed. Cyanide in biology. New York, London, Academic Press, pp 11-28.
Cousineau A & Legg FG (1935) Hydrocyanic acid gas and other toxic
gases in commercial fumigation. Am J Public Health, 25: 277-294.
Domalski CA, Kolb LC, & Hines EA (1953) Deleterious reactions
secondary to thiocyanate therapy of hypertension. Proc Mayo Clin,
28: 272-280.
Edwards AC & Thomas IC (1978) Cyanide poisoning. Lancet, 1: 92.
El Ghawabi SH, Gaafar MA, El Saharti AA, Ahmed SH, Malash KK, & Fares
R (1975) Chronic cyanide exposure, a clinical, radio isotope and
laboratory study. Br J Ind Med, 32: 215-219.
Evans CL (1964) Cobalt compounds as antidotes for hydrocyanic acid.
Br J Pharmacol Chemother, 23: 455-475.
Fontana F (1795) Treatise on the venom of viper; on the American
poisons; and on the Cherry Laurel and some other vegetable poisons.
J Skinner (trans), London.
Friedberg KD & Shukla UR (1975) The efficiency of aquocobalamin as
an antidote in cyanide poisoning when given alone or combined with
sodium thiosulfate. Arch Toxicol, 33: 103-113.
Fukumoto Y, Nakajimo H, Uetake M, Matusyama A, & Yoshida T (1957) A
study on the sense of smell with respect to potassium cyanide solution
and its hereditary transmission. Jpn J Hum Genet, 2: 7-16.
Garvin CF (1939) The fatal toxic manifestations of the thiocyanates.
J Am Med Assoc, 112: 1125-1127.
Goldfrank L, Bresnitz EA, Weisman RS, & Lewin NA (1984) The inhaled
agents and other disorders of oxygen transport. In: Hanson W Jr ed.
Toxic emergencies. New York, Churchill Livingstone, pp 204-211.
Gosselin RE, Smith RP, & Hodge HC (1984) Cyanide. In: Clinical
toxicology of commercial products. Baltimore, London, Williams &
Wilkins Co., pp 123-130.
Graham DL, Laman D, Theodore J, & Robin ED (1977) Acute cyanide
poisoning complicated by lactic acidosis and pulmonary edema. Arch
Intern Med, 137: 1051.
Grant WM (1980) The peripheral visual system as a target. In: Spencer
PS & Schaumberg HH ed. Experimental and clinical neurotoxicology.
Baltimore, London, Williams & Wilkins Co., pp 77-91.
Healy JC (1931) Therapeutics and toxicology of sulfocyanates. New Engl
J Med, 205: 5481-5583.
Hilmann B, Bardham, KD, & Bain JTB (1974) The use of dicobalt edetate
(Kelocyanor) in cyanide poisoning. Postgrad Med J, 50: 171-174.
Hoebel M, Engeser P, Nemeth L, & Pill J (1980) The antidote effect of
thiosulfate and hydroxocobalamine in formation of nitroprusside
intoxication of rabbits. Arch Toxicol, 46: 207-13.
Hoppe-Seyler F (1876) [On fermentation processes and their
relationship to the life of organisms.] PfQgers Arch gesamte Physiol
Menschen Tieren, 12: 1-17 (in German).
Jacobs K (1984) [Report on experience with the administration of
4-DMAP in severe prussic acid poisoning. Consequences for medical
practice.] Zentralbl Arbeitsmed, 34: 274-277 (in German).
Kalmus H & Hubbard DJ (1960) The chemical senses in health and
disease. Springfield, Illinois, Charles C. Thomas.
Kalyanaraman UP, Kalyanaraman K, & Cullinan SA (1983) Neuropathy of
cyanide intoxication due to "laetrile" (amygdalin). Cancer, 51:
2126-2133.
Kessler DL & Hines LE (1948) Hazards of thiocyanate therapy in
hypertension. J Am Med Assoc, 138: 549-551.
Kirk RI & Stenhouse NS (1953) Ability to smell solutions of
potassium cyanide. Nature (Lond), 171: 698-699.
Kong A, Shen A, Burrows G, Sylvester D, Ison GE, & Way JL (1983)
Effect of chlorpromazine on cyanide intoxication. Toxicol Appl
Pharmacol, 71: 407-413.
Leung P, Sylvester DM, Chiou F, Way LI, Way EL, & Way JL (1986)
Stereospecific effect of naloxone hydrochloride on cyanide
intoxication. Toxicol Appl Pharmacol, 83: 525-530.
Levine MS, Radford MPH, & Radford EP (1978) Occupational exposures
to cyanide in Baltimore fire fighters. J Occup Med, 20: 53-56.
Lowry WT, Juarez L, Petty CS, & Roberts B (1985) Studies of toxic
gas production during actual structural fires in the Dallas area. J
Forensic Sci, 30: 59-72.
Lundquist P, Rosling H, & Sorbo B (1985) Determination of cyanide in
whole blood, erythrocytes and plasma. Clin Ther, 31: 591-595.
McNamara BP (1976) Estimation of the toxicity of hydrocyanid acid
vapors in man. (Edgewood Arsenal Technical Report No. EB-TR-76023)
(Army Department).
MacRae WR & Owen M (1974) Severe metabolic acidosis following
hypotension induced with sodium nitroprusside. Br J Anaesth, 46:
795-797.
Madden T (1731) Poisoning by laurel leaves. Philos Trans R Soc Lond
Biol Sci, 37: 84-99.
Magendie F (1817) Sur l'emploi de l'acide prussique dans le
traitement de plusieurs maladies de poitrine, et particulièrement
dans la phtisie pulmonaire. Ann Chim Phys, 6: 347-360.
Marbury TC, Sheppard JE, Gibbons K, & Lee CC (1982) Combined antidotal
and hemodialysis treatments for nitroprusside-induced cyanide
toxicity. J Toxicol Clin Toxicol, 19: 475-482.
Michenfelder JD & Tinker JH (1977) Cyanide toxicity and thiosulfate
protection during chronic administration of sodium nitroprusside in
the dogs: correlation with a human case. Anesthesiology, 47:
441-448.
Ministry of Health, Mozambique (1984) Mantakassa: an epidemic of
plastic paraparesis associated with chronic cyanid intoxication in a
cassava staple area of Mozambique. I.
Epidemiology and clinical and laboratory findings in patients. Bull
World Health Organ, 62: 477-488.
Mladoveanu C & Gheorghiu P (1929) Le nitrite de soude comme antidote
de l'empoisonnement experimental par le cyanure de potassium. C R
Soc Biol (Paris), 102: 164-166.
Montgomery R, Reinhart CF, & Terril JB (1975) Comments on fire
toxicity. Comb Toxicol, 2: 179-212.
Moore SJ, Norris JC, Ing KH, & Hume AS (1986) The efficacy of
alpha-ketoglutaric acid in the antagonism of cyanide intoxication.
Toxicol Appl Pharmacol, 81: 265-273.
Moore SJ, Norris JC, Walsh DA, & Hume AS (1987) The antidotal use of
methemoglobin-forming cyanide antagonists in concurrent carbon
monoxide/cyanide intoxication. J Pharmacol Exp Ther, 242: 70-73.
Mushett CW, Kelly KL, Boxer GE, & Rickards JC (1952) Antidotal
efficacy of vitamin B12a (hydroxocobalamin) in experimental
cyanide poisoning. Proc Soc Exp Biol Med, 81: 234-237.
Naughton M (1974) Acute cyanide poisoning. Anesth Intensive Care, 4:
351-356.
Olsen NS & Klein RJ (1947) Effect of cyanide on the concentration of
lactate and phosphates in brain. J Biol Chem, 167: 739.
Pahl MV & Vaziri ND (1982) In vivo and in vitro hemodialysis studies
of thiocyanate. J Toxicol Clin Toxicol, 19: 965-974.
Paulet G (1960) Les chelatés de cobalt dans le traitement de
l'intoxication cyanhydrique. Pathol Biol, 8: 255-266.
Peden NR, Taha A, McSofiey PD, Bryden GT, Murdoch IB, & Anderson JM
(1986) Industrial exposure to hydrogen cyanide: implications for
treatment. Br Med J, 293: 538.
Pedigo LG (1888) Antagonism between amyl nitrite and prussic acid.
Trans Med Soc Virginia, 19: 124-131.
Peters CG, Mundy JVB, & Rayner PR (1982) Acute cyanide poisoning.
Anaesthesia, 37: 582-586.
Petterson JC & Cohen SD (1985) Antagonism of cyanide poisoning by
chloro-promazine and sodium thiosulfate. Toxicol Appl Pharmacol, 81:
265-273.
Piper J (1975) Use and toxicity of nitroprusside. New Engl J Med,
292: 1081-1082.
Polson CJ & Tattersall RN (1969) Clinical toxicology. London,
Pitman, pp 128-159.
Pronczuk de Garbino J & Bismuth C (1981) Propositions thérapeutiques
actuelles en cas d'intoxication par les cyanures. Toxicol Eur Res,
III(2): 69-76.
Rosling H (1989) Cassava associated neurotoxicity in Africa. In:
Proceedings of the 5th International Congress of Toxicology. Volans
GN, Sims J, Sullivan FM, & Turner P ed. Brighton, Taylor & Francis, pp
605-614.
Russel WO & Stahl WC (1942) Fatal poisoning from potassium
thiocyanate treatment of hypertension. J Am Med Assoc, 119:
1177-1181.
Sadoff L, Fuchs J, & Hollander J (1978) Rapid death associated with
laetrile ingestion. J Am Med Assoc, 239: 1532.
Scheele CW (1786) Chemical essays. Beddves T (trans) Reprint 1966,
London, Dawson.
Schrader JCC (1802) Des expériences de M. Schrader sur l'acide
prussique continu dans les plantes. J Phys Chim Hist Nat Arts, 56:
224-226.
Schulz V, Dohring W, & Rathsac (1978) [Thiocyanate poisoning in
antihypertensive therapy with sodium nitroprusside.] Klin
Wochenschr, 56: 355-361 (in German).
Smith RP (1973) Cyanate and thiocyanate: Acute toxicity. Proc Soc
Exp Biol Med, 142: 1041-1044.
Smith RP & Kruszyna H (1974) Nitroprusside produces cyanide
poisoning via a reaction with hemoglobin. J Chem Exp Ther, 191:
557-563.
Sykes AH (1981) Early studies on the toxicology of cyanide. In:
Vennesland B, Conn EE, Knowles CJ, Westley J, & Wissing F ed.
Cyanide in biology. New York, London, Academic Press, pp 1-9.
Sylvester DM, Hayton WL, Morgan RL, & Way JL (1983) Effects of
thiosulfate on cyanide pharmacokinetics in dogs. Toxicol Appl
Pharmacol, 69: 265.
Takano T, Miyzaki Y, Nashimoto I, & Kobayashi K (1980) Effect of
hyperbaric oxygen on cyanide intoxication: in situ, changes in
intracellular oxidation reduction. Undersea Biomed Res, 7: 191-197.
Ten Eyck RP, Schaerdel AD, Lynett JE, Marks DH, Patrissi GA,
Ottinger WE, & Stansell MJ (1984) Stroma-free methemoglobin solution
as an antidote for cyanide poisoning: a preliminary study. Clin
Toxicol, 21: 343-358.
Ten Eyck RP, Schaerdel AD, & Ottinger WE (1985) Stroma-free
methemoglobin solution: an effective antidote for acute cyanide
poisoning. Am J Emergency Med, 3: 519-523.
Ten Eyck RP, Schaerdel AD, & Ottinger WE (1986) Comparison of
nitrite treatment and stroma-free methemoglobin solution as
antidotes for cyanide poisoning in a rat model. Clin Toxicol, 23:
477-487.
Trapp WG (1970) Massive cyanide poisoning with recovery. Can Med
Assoc J, 102: 3517.
Vesey CJ, Cole PV, Linnell JC, & Wilson J (1974) Some metabolic
effects of sodium nitroprusside in man. Br Med J, 2: 140-142.
Vesey CJ, Cole PV, & Simpson PJ (1976) Cyanide and thiocyanate
concentration following sodium nitroprusside infusion in man, Br J
Anaesth, 48: 651-659.
Vick JA & Froelich HL (1985) Studies on cyanide poisoning. Arch Int
Pharmacodyn, 273: 314-322.
Vogel SN, Sultan TR, & Ten Eyck RP (1981) Cyanide poisoning. Clin
Toxicol, 18: 367-383.
Voorhoeve RJH, Patel CKW, Trimble LE, & Kerl RJ (1975) Automobile
pollution control devices with malfunctional catalytic converters.
Science, 190: 149-151.
Weger N (1968) [Aminophenols as antidotes to prussic acid.] Arch
Toxikol, 24: 49-50 (in German).
2. OXYGEN
2.1 Introduction
Oxygen consumption is indispensable for human life. All organs
of the body will undergo successive dysfunction, permanent damage,
and then death if oxygen consumption falls below that necessary for
metabolic needs. A decrease in oxygen consumption may be due to
inadequate oxygen delivery to the cells or to an inability of the
cells to utilize oxygen.
The ambient atmospheric pressure at sea level is defined as one
atmosphere absolute (1 ATA), equivalent to an air pressure of
1.03 kg per sq. cm., i.e., 101 kPa or 760 mm of mercury. Molecular
oxygen constitutes about 21% of atmospheric air. Hence the partial
pressure of oxygen at sea level is 0.21 ATA.
Hyperoxia is the artificial elevation of the partial pressure
of oxygen in the body and can be produced by increasing the partial
pressure of oxygen from 0.21 to 1 ATA (normobaric oxygen therapy) or
above 1 ATA (hyperbaric oxygenation). Normobaric oxygenation is
commonly used in many conditions associated with decreased delivery
of oxygen to tissues, such as cardiac and pulmonary failure and
cardiopulmonary resuscitation.
Both normobaric and hyperbaric oxygen therapy have an antidotal
effect on carbon monoxide (CO), hydrogen sulfide (H2S), and
cyanide (CN) poisoning. However, hyperoxia may induce oxygen
toxicity related to the length of exposure and the partial pressure
of oxygen employed. The use of oxygen therapy is therefore limited
by oxygen toxicity.
2.2 Name and Chemical Formula of Antidote
Molecular oxygen is a diatomic molecule and is properly named
dioxygen (formula O2, relative molecular mass 16). More than
99.9% of atmospheric oxygen consists of molecules containing the
16O isotope. Trace quantities of 17O and 18O exist in
atmospheric air.
2.3 Physico-chemical Properties of Molecular Oxygen
Critical temperature 154.575 K
Critical density 0.4361 g/cm3
Critical pressure 50.14 ATA
Boiling point 90.188 K
Melting point 54.361 K
Solubility
in water, 25 °C 2.8%
in ethanol 22.6%
in plasma approx 3.0%
in whole blood 20.0%
2.4 Synthesis
Oxygen is obtained on a large scale by the liquefaction of air.
Modern manufacturing processes produce the gas at a concentration
much higher than 99.0% (v/v) of oxygen, this being the lowest
permissible concentration in medical oxygen allowed in the European
Pharmacopoeia. Contamination with carbon monoxide or carbon dioxide
is therefore very slight.
2.5 Analytical Methods
2.5.1 Quality control procedures
2.5.1.1 Tests
For the tests, deliver the sample to be examined at a rate of
4 l/h.
Carbon monoxide Carry out the test using 7.5 l of the
substance to be examined and 7.5 l of argon R for the blank. The
difference between the volumes of sodium thiosulfate (2 mmol/l) used
in the titrations should not be more than 0.4 ml (5 ppm v/v).
For the following three tests, pass the sample to be examined
through the appropriate reagent contained in a hermetically closed
flat-bottomed glass cylinder (with dimensions such that 50 ml of
liquid occupies a height of 12 cm to 14 cm) fitted with: (a) a
delivery tube terminated by a capillary 1 mm in internal diameter
and reaching to within 2 mm of the bottom of the cylinder; (b) an
outlet tube. Prepare the reference solutions in identical
cylinders.
Acidity or alkalinity
Test solution Pass 2.0 litres of the sample to be examined
through a mixture of 0.1 ml of hydrochloric acid (0.01 mol/l) and
50 ml of CO2-free water R.
Reference solution (a) 50 ml of CO2-free water R.
Reference solution (b) To 50 ml of CO2-free water R add
0.2 ml of hydrochloric acid (0.01 mol/l).
To each solution add 0.1 ml of a 0.02% m/v solution of methyl
red R in alcohol (70% v/v). The intensity of the colour of the test
solution should be between those of reference solutions (a) and (b).
Carbon dioxide Pass 1.0 litre through 50 ml of barium
hydroxide solution R (the solution to be used must be clear). Any
turbidity in the solution after passage of the gas should not be
more intense than that in a reference solution prepared by adding
1 ml of a 0.11% m/v solution of sodium hydrogen carbonate R in
CO2-free water R to 50 ml of barium hydroxide solution R
(300 ppm v/v).
Oxidizing substances Place in each of two cylinders 50 ml
of freshly prepared potassium iodide and starch solution R, and add
0.2 ml of glacial acetic acid R. Protect the cylinders from light.
Pass 5.0 l of the substance to be examined into one of the
cylinders. The test solution should remain colourless when compared
with the blank.
2.5.1.2 Assay for oxygen
Use a gas burette (see Fig. 1) of 25 ml capacity in the form of
a chamber having at its upper end a tube graduated in 0.2% divisions
between 95 and 100, and isolated at each end by a tap with a conical
barrel. The lower tap is joined to a tube with an olive-shaped
nozzle and is used to introduce the gas into the apparatus. A
cylindrical funnel above the upper tap is used to introduce the
absorbent solution. Wash the burette with water and dry. Open the
two taps. Connect the nozzle to the source of the sample to be
examined and set the flow rate to 1 l/min. Flush the burette by
passing the substance to be examined through it for 1 min. Close
the upper tap of the burette and immediately afterwards the lower
tap. Rapidly disconnect the burette from the source of the sample
to be examined and give a half turn to the upper tap to eliminate
any excess pressure in the burette. Keeping the burette vertical,
fill the funnel with a limited amount of freshly prepared mixture of
21 ml of a 56% m/v solution of potassium hydroxide R and 130 ml of a
20% m/v solution of sodium dithionite R. Open the upper tap slowly.
The solution absorbs the oxygen and enters the burette. Allow to
stand for 10 min without shaking. Read the level of the liquid
meniscus on the graduated part of the burette. This figure
represents the percentage v/v of oxygen.
2.5.2 Methods for identification
(a) Place a glowing splint of wood in the substance to be
examined. The splint bursts into flame.
(b) Shake with alkaline pyrogallol solution R. The sample to
be examined is absorbed and the solution becomes dark
brown.
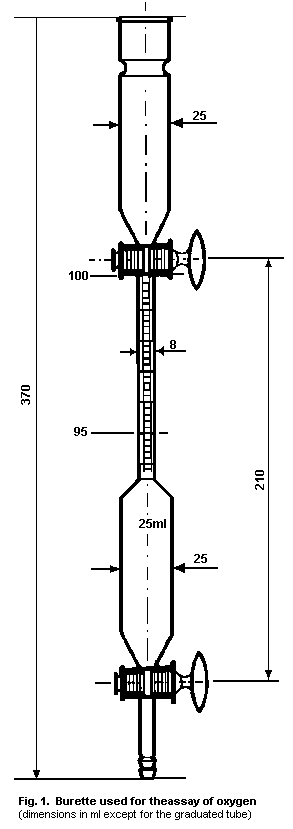 2.5.3 Methods for analysis of the antidote in biological samples
2.5.3.1 In the gas phase
(a) Chemical volumetric methods based on absorption of oxygen
by chemical reagents (such as alkaline pyrogallol) are
still in use. The skill of the operator is a major factor
in the accuracy of the results.
(b) Methods based on physical properties of oxygen:
* paramagnetic susceptibility;
* thermomagnetic (magnetic wind);
* differential pressure (Quincke);
* magnetic auto-balance (Faraday);
* electron capture;
* ultraviolet absorption;
* mass spectrometry.
2.5.3.2 In solution
Polarographic measurement is a sensitive method for measuring
the partial pressure of oxygen in solution. The Clark electrode is
commonly used for biological samples. This has a cathode made of a
platinum wire, while the anode is Ag/AgCl in phosphate buffer with
added KCl.
2.5.4 The saturation of haemoglobin by oxygen
The degree of saturation of haemoglobin by oxygen (HbO2 %)
can be measured by photometric procedures. This method is
particularly useful for the non-invasive monitoring of patients
(oximeters, pulse oximeters).
2.6 Storage Conditions
Oxygen should be stored under pressure in a suitable metal
container of a type permitted by the safety regulations of the
national authority. Valves and taps should not be lubricated with
oil or grease.
2.5.3 Methods for analysis of the antidote in biological samples
2.5.3.1 In the gas phase
(a) Chemical volumetric methods based on absorption of oxygen
by chemical reagents (such as alkaline pyrogallol) are
still in use. The skill of the operator is a major factor
in the accuracy of the results.
(b) Methods based on physical properties of oxygen:
* paramagnetic susceptibility;
* thermomagnetic (magnetic wind);
* differential pressure (Quincke);
* magnetic auto-balance (Faraday);
* electron capture;
* ultraviolet absorption;
* mass spectrometry.
2.5.3.2 In solution
Polarographic measurement is a sensitive method for measuring
the partial pressure of oxygen in solution. The Clark electrode is
commonly used for biological samples. This has a cathode made of a
platinum wire, while the anode is Ag/AgCl in phosphate buffer with
added KCl.
2.5.4 The saturation of haemoglobin by oxygen
The degree of saturation of haemoglobin by oxygen (HbO2 %)
can be measured by photometric procedures. This method is
particularly useful for the non-invasive monitoring of patients
(oximeters, pulse oximeters).
2.6 Storage Conditions
Oxygen should be stored under pressure in a suitable metal
container of a type permitted by the safety regulations of the
national authority. Valves and taps should not be lubricated with
oil or grease.
 The containers for medical oxygen are coded with a white colour
at the top according to ISO-32-19 (Fig 2) and carry the indication
"Medical Oxygen" carved in the iron material of the container. To
prevent connection with other medical gases, the containers are
provided with an international standardized "Pin Index Safety
System" ISO-407. This system is the same for medical and for
industrial oxygen so that in emergencies industrial oxygen can also
be used.
Regular control of gas pressure is necessary to prevent
shortage of oxygen in emergency situations.
Although the duration of storage will not alter the quality, it
is recommended that the gas should not be stored for more than six
months.
2.7 General Properties
In most circumstances, supplemental oxygen is indicated if
tissue hypoxia is imminent. In the case of cyanide poisoning,
however, cytochrome oxidase activity is inhibited and tissue
utilization of oxygen prevented. Even so, animal experiments
(Takano et al., 1980) suggest that inhibition of cytochrome oxidase
activity by cyanide is prevented, and recovery accelerated, in the
presence of oxygen.
2.8 Animal Studies
2.8.1 Pharmacokinetics
The bioavailability of oxygen depends on the following factors:
(a) the thickness of the alveolar membrane; as a result of
oedema fluid in the interstitial spaces of the membrane
oxygen cannot readily diffuse into the blood;
(b) haemoglobin configuration:
(i) oxygen transport by the blood to the tissues can be
disturbed by methaemoglobin-forming antidotes, e.g.,
sodium nitrite or 4-dimethylaminophenol, often used
in the treatment of cyanide poisoning;
(ii) oxygen transport is also hampered in carbon monoxide
poisoning, which occurs in combination with cyanide
poisoning following smoke inhalation.
(c) Oxygen utilization by tissues is prevented by the
inhibition of cytochrome oxidase activity in cyanide
poisoning.
2.8.2 Pharmacodynamics
Isom & Way (1982) studied the reduction of cytochrome oxidase
prepared from brains and livers of mice poisoned with potassium
cyanide. They examined several groups of animals and compared
exposure to air and to oxygen at 0.95 ATA, both with and without
sodium nitrite and sodium thiosulfate treatment. Cytochrome oxidase
was inhibited by 75% within 2 min of the injection of 5 KCN mg/kg.
The inhibition of cytochrome oxidase was similar whether the animals
inhaled air or 0.95 ATA O2, but the reactivation phase of
cytochrome oxidase was faster in oxygen-breathing animals. They
also found that, at a lower dose (4 KCN mg/kg), enzyme activity was
increased from 22% to 66% by increasing the partial pressure of
oxygen from 0.11 to 0.95 ATA O2. Oxygen shifted the dose-response
curve of brain cytochrome oxidase inhibition by KCN to the right:
the dose of KCN producing 50% inhibition was 24 mg/kg in
air-breathing animals and 55 mg/kg in animals breathing oxygen. The
authors also found that liver rhodanese activity was 15 times higher
than brain rhodanese activity and that treatment with sodium nitrite
and sodium thiosulfate protected liver, but not brain, cytochrome
oxidase at high doses of KCN.
Way et al. (1972) found that oxygen alone antagonized cyanide
toxicity. However, they found no significant benefit in increasing
the partial pressure of oxygen from 1 to 4 ATA O2. The authors
demonstrated that oxygen potentiates the effect of thiosulfate and
that the best results were obtained using hyperbaric oxygen together
with nitrite and thiosulfate.
Ivanov (1959) reported that hyperbaric oxygen re-established
normal EEG activity in animals poisoned with cyanide.
Takano et al. (1980) measured the reduction of pyridine
nucleotide in vivo as an indicator of cytochrome oxidase
inhibition. They found a protective effect of oxygen at 1 and 2 ATA
O2 when the doses of KCN injected into rabbits were within the
range that produced inhibition of cytochrome oxidase in animals
breathing air. At much higher doses, the protective effect of O2
at 1 or 2 ATA became progressively weaker.
Paulet (1955) described the protective effect of oxygen on the
chemoreceptors and on the clinical manifestations in dogs poisoned
by cyanide.
Cope & Tenn (1961) showed that infusion of KCN solution (2 g/l)
to anaesthetized dogs breathing air produced atrioventricular block.
Oxygen breathing reversed this phenomenon and re-established normal
sinus rhythm.
2.8.3 Toxicology
Oxygen toxicity was discovered in 1878 by Paul Bert, who
observed convulsions in animals treated with air at 15-20 ATA. He
then noted that when pure oxygen was used instead of air, one-fifth
of the pressure sufficed to produce the convulsions. He concluded
that oxygen toxicity is directly correlated to the product of
partial pressure of oxygen and administration period.
Pulmonary oxygen toxicity was first described in 1897 by
Lorrain Smith, who noted that mice exposed to oxygen at pressures
above 1 ATA died from respiratory failure and that there was great
variability both within and between animal species in the
susceptibility to oxygen damage.
The initial stage of acute pulmonary oxygen toxicity is termed
the exudative phase and is characterized by a perivascular and
interstitial inflammatory response that includes damage to the
capillary endothelium with exudation of oedema
fluid,polymorphonuclear leucocytes and, eventually, macrophages.
The oedema is associated with widening of the interalveolar septa
and a thickened air-blood tissue barrier. Alveolar haemorrhage
develops and there is fibrin deposition in the alveoli (hyaline
membranes). Loss of alveolar lining cells (type I lining cells) can
also occur during this phase. As the condition progresses, it
reaches a stage referred to as the proliferative phase,
characterized by resolution of the inflammatory exudate and
increased cellularity of the interstitium due to proliferation of
macrophages and fibroblasts. There is also proliferation of the
cuboidal type II alveolar lining cells that serve to
re-epithelialize the denuded alveolar basement membrane. By the
time a typical laboratory animal, such as the rat, is near death, a
substantial number of pulmonary capillaries have been destroyed and
embolization of some arterioles will have occurred. Depending on
the species, however, the destruction of type I alveolar lining
cells and the proliferation of the type II cells may not be
extensive.
The bronchial tree is damaged by oxygen toxicity: a
necrotizing bronchiolitis has been described in neonatal mice by
Ludwin et al. (1974). The bronchiolitis consists of degeneration
and necrosis of bronchiolar mucosal epithelium involving both
ciliated as well as non-ciliated (Clara) cells. Early change,
occurring within 72 h in neonatal mice, consist of loss of cilia,
cellular swelling associated with mitochondrial swelling, oedema of
the bronchiolar wall, and leucocytic infiltration of the bronchiolar
connective tissue. These changes are followed by necrosis of the
epithelial cells with subsequent sloughing. Necrosis of the mucosal
cells is maximal at 6 to 7 days. Cold-blooded animals are
relatively resistant to oxygen toxicity.
Guinea-pigs, dogs and rats have a median time to death (LT50)
in the range of 65-80 h while breathing 1 ATA oxygen. Monkeys have
an LT50 of about 100 h, while chicks survived for 28 days at
1 ATA. In general, there is a great interspecies variability in
susceptibility to hyperoxia.
2.8.3.1 Mechanism of injury
Free radicals (superoxide anion O-2 and hydroxyl radical
OH) are produced by reduction of oxygen and these and hydrogen
peroxide (H2O2) are responsible for most of the toxic effects of
oxygen. The free radicals react with cellular components and cause
oxidative damage by oxygen radicals of the unsaturated fatty acids,
which are abundant in the membranes of cells, and the sulfhydryl
groups present in many enzymes and cellular proteins. The cells are
protected against oxygen radicals by enzymes which convert the
radicals to less toxic molecules and to molecular oxygen. Haber &
Weiss (1934) described the production of hydroxyl free radicals from
the superoxide anion and hydrogen peroxide.
Hyperoxia also causes arteriolar vasoconstriction, this being
unrelated to the free radical mechanism.
2.9 Volunteer Studies of Pulmonary Oxygen Toxicity
Clark & Lambertsen (1971) studied normal humans exposed to
2 ATA O2. Symptoms began within 3-8 h in the form of mild
tracheal irritation and increased in intensity throughout the
exposure time. After 8-10 h, symptoms were characterized by
uncontrollable coughing, dyspnoea at rest, and a constant
tracheobronchial burning sensation. Decreased vital capacity was
the most constant and sensitive sign of pulmonary oxygen toxicity.
Decreased carbon monoxide diffusion capacity and increased
alveolar-arterial O2 difference were also observed.
Singer et al. (1970) compared two groups of patients treated
with 0.4 ATA O2 or 1 ATA O2 for up to 24 h and found no
immediate or delayed dysfunction. He concluded that humans can
tolerate 1 ATA O2 for up to 24 h without detectable pulmonary
dysfunction. Comroe et al. (1945) showed that inhalation of oxygen
at 0.5-1.0 ATA O2 for more than 24 h decreased vital capacity.
Clark & Lambertsen (1971) established oxygen pressure versus time
tolerance curves. It appears that no dysfunction is observed if up
to 0.5 ATA O2 is inhaled for many days and that the use of 1 ATA
O2 is safe for up to 24 h.
2.10 Clinical Studies of Oxygen Toxicity
All human cells are affected by oxygen toxicity, but the three
organs most susceptible to acute oxygen toxicity are the eyes in the
newborn, and the lung and central nervous system at all ages.
2.10.1 Eyes
The eyes of the newborn and, especially, those of the premature
newborn, are very sensitive to oxygen toxicity, which produces
retrolental fibroplasia.
Patz (1968a,b) described retrolental fibroplasia which occurred
in premature babies exposed to 0.30-0.45 ATA O2 for 10 days.
Initially, in the presence of high concentrations of oxygen, there
is vasoconstriction of arteries, arterioles and venules with
necrosis of the immature retinal vessels (referred to as the
vaso-obliterative phase). Following oxygen-induced
vaso-obliteration, there is a vaso-proliferation of new capillaries
in the inner layer of the retina. The proliferation of capillaries
extends into the vitreous behind the lens and eventually results in
retinal detachment, which represents the final phase.
2.10.2 Central nervous system (CNS)
Central nervous system oxygen toxicity is characterized by
grand-mal seizures. The manifestation of acute CNS toxicity in
humans consists not only of grand-mal seizures but also of focal
motor seizures, constriction of the visual fields, deafness,
hyperacuity, changes of mood, and visual and auditory
hallucinations. CNS oxygen toxicity in humans is only observed
during hyperbaric oxygen exposure.
Grand-mal convulsions have been described in humans exposed to
pressures above 2.8 ATA O2 for periods exceeding 2 h. Central
nervous system toxicity is rare if the O2 pressure is less than
2.5 ATA and if oxygen and air are used intermittently, e.g., 10 min
of air and 30 min of oxygen, as long as the total hyperbaric
exposure time does not exceed 2 h.
2.11 Clinical Studies - Case Reports
In most cases reported in the literature the evidence for
cyanide poisoning is lacking because blood concentrations of cyanide
are not reported. Moreover, the majority of cases of cyanide
poisoning are treated with oxygen in combination with other
antidotal therapy. The individual role of oxygen as a cyanide
antidote in man is therefore difficult to assess. The following
cases have been reported.
2.11.1 Patients treated alone with supportive therapy and who
survived
* An analytical chemist (48 years old) was found unconscious
with unrecordable blood pressure and cardiac arrest. The
blood cyanide level was 5.8 mg/l (Edwards & Thomas, 1978).
* A three-year-old child was treated with laetrile enemas.
On admission the child was cyanotic, lethargic, and
unresponsive. The blood cyanide level was 2.14 mg/l
(Ortega & Creek, 1978).
* A biochemist, aged 31 years, was found in deep coma with a
metabolic acidosis and a blood cyanide concentration of
2.3 mg/l (Vogel et al., 1981).
* Nine people were poisoned by hydrogen cyanide as a result
of a leak from a valve. Six of them lost consciousness
briefly. Blood cyanide concentrations were 2.8 to
7.7 mg/l (Peden et al., 1986).
2.11.2 Hyperbaric oxygen therapy in cyanide poisoning
Trapp (1970) reported acute cyanide poisoning successfully
treated with hyperbaric oxygen, while Litovitz et al. (1983)
reported the unsuccessful use of hyperbaric oxygen.
Trapp & Lepawsky (1983) reported five cases of acute cyanide
poisoning successfully treated by hyperbaric oxygen in the Vancouver
General Hospital, Canada.
2.11.3 Cyanide poisoning due to smoke inhalation
Four out of five smoke inhalation victims with mean blood
cyanide levels of 1.62 mg/l (62 µmol/l) are reported to have
survived after receiving hyperbaric oxygen therapy combined with the
administration of sodium nitrite and sodium thiosulfate (Hart et
al., 1985).
2.12 Summary of Evaluation
1. In acute cases of poisoning by inhalation of hydrogen
cyanide, either alone or in combination with carbon monoxide,
termination of exposure and evacuation of the patient from
contaminated areas is indicated most urgently and should be
performed by the rescuers using appropriate protective equipment.
Measures to support respiration, circulation, and cardiac function
should be commenced, if necessary. Patients should receive pure
humidified oxygen.
Following smoke inhalation, patients frequently develop acute
pulmonary failure due to the irritant effect of the smoke. They may
benefit from the use of continuous positive airway pressure while
breathing spontaneously or positive end-expiratory pressure if
ventilated artificially.
Other antidotes for cyanide poisoning, especially sodium
thiosulfate, should be administered intravenously as soon as
possible. Blood samples, which must be taken before the
administration of antidotes, should be sent for toxicological
analysis.
2. In acute cyanide poisoning by ingestion, decontamination of
the stomach should be performed as soon as possible after or during
first-aid therapy.
3. The final decision about the use of hyperbaric oxygen
therapy depends on the availability of chambers and the ability to
treat.
4. Oxygen has always been regarded as an important first-aid
measure even though, it is difficult to understand, on a theoretical
basis, how oxygen has a favourable effect in cyanide poisoning
because inhibition of cytochrome oxidase is non-competitive. There
is now experimental evidence that oxygen has specific antidotal
activity. Oxygen accelerates the reactivation of cytochrome oxidase
and protects against cytochrome oxidase inhibition by cyanide.
Nevertheless, there are other possible modes of action and those
which are clinically important have yet to be determined.
5. Hyperbaric oxygen is recommended for smoke inhalation
victims suffering from combined carbon monoxide and cyanide
poisoning, since these two agents are synergistically toxic. The
role of hyperbaric oxygen in pure cyanide poisoning remains
controversial.
2.13 Model Information Sheet
2.13.1 Uses
Oxygen is indicated for the treatment of patients poisoned by
cyanide, either alone or in combination with carbon monoxide
following smoke inhalation.
2.13.2 Dosage and route
In cases of severe poisoning (respiratory insufficiency and/or
deteriorating vital signs), artificial ventilation with 100% oxygen
is indicated but for no longer than 12-24 h, after which the
inspired oxygen concentration should be reduced. Similarly in
moderately severe cases, 100% oxygen is indicated but again for no
longer than 12-24 h. For mild cases 40% oxygen is indicated.
2.13.3 Precautions/contraindications
In order to prevent rebreathing of hydrogen cyanide from the
expired air of a patient, it is advisable to include a non-return
valve when a bag and mask is used.
2.13.4 Adverse effects
The alveolar membranes of the lung are damaged by 100% oxygen
if this is administered for more than 24 h.
2.13.5 Use in pregnancy and lactation
No contraindication.
2.13.6 Storage
Store under pressure in a suitable metal container of a type
permitted by the safety regulations of the national authority.
Valves and taps should not be lubricated with oil or grease.
The containers for medical oxygen are coded with a white colour
at the top according to ISO-32-19 (Fig. 2) and carry the indication
"Medical Oxygen" in the iron material of the container. To prevent
connection with other medical gases the containers are provided with
an international standardized "Pin Index Safety System" ISO-407.
This system is the same for medical and for industrial oxygen so
that in emergencies industrial oxygen can also be used.
Regular control of gas pressure is necessary to prevent
shortage of oxygen in emergency situations.
Although the duration of storage will not alter the quality it
is recommended that the gas should not be stored for more than six
months.
2.14 References
Clark JM & Lambertsen CJ (1971) Rate of development of pulmonary
O2 toxicity in man during O2 at 2.0 ATM. ABS. J Appl Physiol,
30: 730-752.
Clark CJ, Campbell D, & Reid WH (1981) Blood carboxyhaemoglobin and
cyanide levels in fire survivors. Lancet, 1: 1332-1335.
Comroe JH, Dripps RD, Dumke PR, & Deming M (1945) Oxygen toxicity:
The effect of inhalation of high concentrations of oxygen for
twenty-four hours on normal men at sea level and at a simulated
altitude of 18,000 feet. J Am Med Assoc, 128: 710-717.
Cope C & Tenn M (1961) The importance of oxygen in the treatment of
cyanide poisoning. J Am Med Assoc, 175: 109-112.
Edwards AC & Thomas ID (1978) Cyanide poisoning. Lancet, 1: 92-93.
Haber F & Weiss JJ (1934) The catalytic decomposition of hydrogen
peroxide by iron salts. Proc R Soc Lond, A147: 332-351.
Hart GB, Strauss MB, Lennon PA, & Whitcroft DD (1985) Treatment of
smoke inhalation by hyperbaric oxygen. J Emergency Med, 3: 211-215.
Isom GE & Way JL (1982) Effects of oxygen on the antagonism of
cyanide intoxication - cytochrome oxidase, in vitro. Toxicol Appl
Pharmacol, 74: 57-62.
Ivanov JP (1959) The effect of elevated oxygen pressure on animals
poisoned with potassium cyanide. Pharmacol Toxicol (USSR), 22:
476-479.
Litovitz TL, Larkin RF, & Myers RAM (1983) Cyanide poisoning treated
with hyperbaric oxygen. Am J Emergency Med, 1: 94-101.
Ludwin SK, Northway WH Jr, & Bensch KG (1974) Oxygen toxicity in the
newborn. Necrotizing bronchiolitis in mice exposed to 100% oxygen.
Lab Invest, 31: 421-435.
Ortega MA & Creek JE (1978) Acute cyanide poisoning following
administration of laetrile enemas. J Pediatr, 93: 1059.
Patz A (1968a) New role of ophthalmologist in prevention of
retrolental fibroplasia. Arch Ophthalmol (Chicago), 78: 565-568.
Patz A (1968b) Comment on oxygen therapy. Arch Ophthalmol (Chicago),
79: 507.
Paulet G (1955) Valeur et méchanisme d'action d'intoxication
cyanhydrique. Arch Int Physiol, 63: 340.
Peden NR, Taha A, McSorley PD, Bryden GT, Murdoch IB, & Anderson JM
(1986) Industrial exposure to hydrogen cyanide: Implications for
treatment. Br Med J, 293: 538.
Singer MM, Wright F, Stanley LK, Roc BB, & Hamilton WK (1970) Oxygen
toxicity in man. A prospective study in patients after open heart
surgery. New Engl J Med, 283: 1473.
Takano T, Miyzaki Y, Nashimoto I, & Kobayashi K (1980) Effect of
hyperbaric oxygen on cyanide intoxication: in situ changes in
intracellular oxidation reduction. Undersea Biomed Res, 7: 191-197.
Trapp WG (1970) Massive cyanide poisoning with recovery: A
Boxing-Day story. Can Med Assoc J, 102: 517.
Trapp WG & Lepawsky M (1983) 100% survival in five life-threatening
acute cyanide poisoning victims treated by a therapeutic spectrum
including hyperbaric oxygen. Proceedings of the 1st European
Conference on Hyperbaric Medicine Amsterdam, The Netherlands p 45.
Vogel SN, Sultan TR, & Ten Eyck R (1981) Cyanide poisoning. Clin
Toxicol, 18: 367-383.
Way JL, End E, Sheehy MH, De Miranda P, & Feitknecht UF (1972)
Effect of oxygen on cyanide intoxication. IV. Hyperbaric oxygen.
Toxicol Appl Pharmacol, 22: 415-421.
3. SODIUM THIOSULFATE
3.1 Introduction
3.1.1 Indications
The use of sodium thiosulfate as an antidote has been recorded
in the literature on poisoning due to cyanide, mustard gas, nitrogen
mustard, bromate, chlorate, bromine, iodine, cisplatin, and certain
drugs (Dactinomycin, Mechlorethamine, Mitomycin) when extravasated.
There are also some references to its effect in iodate and
hypochlorite toxicity. The principal role of sodium thiosulfate,
though, lies in the treatment of cyanide poisoning.
3.1.2 Rationale for the choice of the antidote
The effect of sodium thiosulfate as an antidote in cyanide
poisoning is well documented, and was first demonstrated by Lang
(1895). Some authors believe it to be a relatively slow-acting
antidote, though others have demonstrated that it acts more rapidly
than was previously thought, enabling the conversion of cyanide to
thiocyanate (Krapez et al., 1981). Thiosulfate helps to detoxify
cyanide in the presence of the enzyme rhodanese. However, rhodanese
is an intramitochondrial enzyme and thiosulfate has limited ability
to penetrate cell and mitochondrial membranes. Thus, distribution
of thiosulfate is almost exclusively extracellular (Cardozo &
Edelman, 1952), while its antidotal action has been thought to take
place intracellularly. This view is now being re-examined in the
light of recent experimental evidence (see section 3.8).
3.1.3 Risk groups
No special-risk groups can be identified as regards the use of
sodium thiosulfate. However, it should be noted that there is an
apparently reduced ability to convert cyanide to thiocyanate in some
diseases, e.g., toxic amblyopias (in particular tobacco amblyopia)
and Leber's hereditary optic atrophy (Wilson, 1965; Darby & Wilson,
1967). Abnormally low rhodanese activity in the liver has been
described in two patients with Leber's hereditary optic atrophy
(Grant, 1986).
3.2 Name and Chemical Formula of Antidote
International non-proprietary name: Natrii thiosulfas; Sodium
thiosulfate (thiosulphate); Thiosulfate de sodium; Natrium
thiosulfuricum; Natriumthiosulfat (Hager, 1977).
CAS number: 10102-17-7 for sodium thiosulfate, pentahydrate
(NIOSH, 1986); 7772-98-7 for sodium thiosulfate, anhydrous, (NIOSH,
1986).
IUPAC name: Sodium thiosulfate, pentahydrate
Manufacturer: Readily available in many countries.
Commercial names: Commercially available as sodium thiosulfate
or equivalent in many countries.
Formula: Na2S2O3.5H2O (Martindale, 1989)
Relative molecular mass: 248.2 (Martindale, 1989)
Specification of chemical salt used: sodium thiosulfate
contains not less that 99.0% and not more than the equivalent of
101.0% of Na2S2O3.5H2O (European Pharmacopoeia, 1980);
transparent, colourless crystals (European Pharmacopoeia, 1980);
colourless, odourless, (or almost odourless) monoclinic prismatic
crystals, or a coarse crystalline powder with a saline taste
(Martindale, 1989).
3.3 Physico-chemical Properties
3.3.1 Melting point, boiling point
Sodium thiosulfate dissolves in its own water of
crystallisation at about 49 °C (European Pharmacopoeia, 1980;
Martindale, 1989;). It loses all its water at 100 °C and decomposes
at higher temperatures (Windholz, 1983). Above 200-300 °C, it
decomposes to sulfate and pentasulfide (Kirk-Othmer, 1969; Hager,
1977). When it is heated to the point of decomposition, fumes of
sulfur oxides are emitted (Sax, 1984; PoisIndex, 1987).
3.3.2 Solubility in vehicle for administration
Highly soluble in water (2 parts sodium thiosulfate in 1 part
water) (Martindale, 1982; Windholz, 1983).
3.3.3 Optical properties
Inactive with respect to polarized light; absorbs ultraviolet
light at wavelengths less than 230 nm (information from the National
Corporation of Swedish Pharmacies).
3.3.4 Acidity
A 10% solution in water has a pH of 6.0-8.4 ( European
Pharmacopoeia, 1980; Martindale, 1989). The pH of injectable
thiosulfate (0.15 g/ml) is 8.2-8.8 (information from the National
Corporation of Swedish Pharmacies).
3.3.5 pKa
pKa1: 1.46-1.74 (an approximate value) (IUPAC, 1969).
3.3.6 Stability
Efflorescent in warm (>30 °C) dry air. Slightly hygroscopic
(delinquesent) in moist air (Windholz, 1983). Store in an airtight
container (Martindale, 1989).
Aqueous solutions have limited stability due to a tendency to
decompose slowly by the following reactions:
Na2S2O3 --> Na2SO3 + S (neutral or acidic
solutions)
Na2S2O3 + H2O --> Na2SO4+H2S (alkaline
solutions)
The first reaction is accelerated by acids and the second by
air or oxygen. Aqueous solutions of sodium thiosulfate decompose
more rapidly on heating. Storage with limited access to air and
light in a cool environment increases stability (Kirk-Othmer, 1969;
Martindale, 1989; Windholz, 1983).
Injectable thiosulfate stored in ampoules for three years shows
no significant change in composition.
3.3.7 Refractive index, specific gravity
No value for refractive index (injectable thiosulfate
[0.15 g/ml]) is available. The specific gravity for injectable
thiosulfate (0.15 g/ml) is 1.076 (information from the National
Corporation of Swedish Pharmacies).
3.3.8 Loss of weight on drying
The substance loses 35.5-37.0% of weight when it is dried at
105 °C (Pharmacopoea Nordica, 1964; Hager, 1977).
3.3.9 Excipients
For injectable thiosulfate (0.15 g/ml): sodium phosphate
dodecahydrate (Na2HPO4.12H2O) 1.21% (information from The
National Corporation of Swedish Pharmacies; see also section 3.6).
3.3.10 Incompatibility
Incompatibility with iodine, acids, lead, mercury and silver
salts (Windholz, 1983) and with salts of heavy metals, oxidizing
agents, and acids has been indicated. If sodium thiosulfate is
triturated with chlorates, nitrates, or permanganates, an explosion
may occur (Martindale, 1989).
3.3.11 Other information
Solutions are sterilized by autoclaving. A 2.98% solution is
iso-osmotic with serum (Martindale, 1989).
Table 2. Physical properties of sodium thiosulfate pentahydrate
Property Value
refractive index, nd20 1.4886
density, d425 1.750
heat of solution in water at 25 °C (kcal/mol) - 11.3
heat of formation (kcal/mol) -621.9
heat of fusion (kcal/mol) 11.85
specific heat at 21 °C (cal/g) 0.346
cryoscopic constant 42.6
dissociation pressure (mm Hg)
at 20 °C 6.0
at 35 °C 18.0
vapour pressure of saturated solution (mmHg)
at 33 °C 10.0
at 57 °C 42.0
at 90 °C 233.0
20
density of aqueous solution, d20, (Na2S2O3, by weight)
10% 1.0847
20% 1.1760
30% 1.2762
40% 1.3851
From: Kirk-Othmer (1969)
3.4 Synthesis
Sodium thiosulfate can be produced commercially by a number of
methods, of which the four principal processes may be classified as
follows (Kirk-Othmer, 1969).
(a) Air oxidation of sulfides, hydrosulfides, and polysulfides
of alkali and alkaline earth metals.
(b) Recovery from waste liquors resulting from the production
of sulfur black and other sulfur dyes.
(c) Reaction of sulfur dioxide and sulfides.
(d) Reaction of sulfur and sulfites.
Possible contaminants include chlorides, sulfates, sulfites,
and heavy metals.
3.5 Analytical Methods
3.5.1 Quality control procedures for sodium thiosulfate
(European Pharmacopoeia, 1980)
Tests for chlorides, sulfates, sulfites, sulfides, and heavy
metals. The pH of a 10% solution is 6.0-8.4; clear and colourless
(European Pharmacopoeia, 1980).
3.5.2 Methods for identifying sodium thiosulfate
(European Pharmacopoeia, 1980).
(a) Decolorization of iodinated potassium iodide solution R.
(b) In combination with silver nitrate solution R2, a white
precipitate forms, which rapidly turns yellowish and then
black.
(c) In combination with hydrochloric acid, a precipitate of
sulfur is formed and a gas is evolved which gives a blue
colour to starch iodate paper R.
(d) Reaction on sodium.
3.5.3 Assay
Dissolve 0.500g in 20ml of water and titrate with 0.1 N iodine,
using 1 ml of starch solution R, added towards the end of the
titration, as indicator.
1 ml of 0.1 N iodine is equivalent to 24.82 mg of Na2S2O3.5H2O
(European Pharmacopoeia, 1980).
3.5.4 Methods for analysis of sodium thiosulfate in
biological
samples
Gast et al. (1952) and Dixon (1962) described a method for the
measurement of thiosulfate, based on reduction by iodine, but this
is a nonspecific means of estimation.
Sörbo & Ohman (1978) described a method for the determination
of thiosulfate in urine. After the removal of interfering
compounds, including endogenous thiocyanate by ion exchange,
thiosulfate is converted to thiocyanate in the presence of cyanide
and cupric ions. The thiocyanate formed is concentrated by ion
exchange, eluted with an acid solution of ferric ions, and the
ferric thiocyanate complex is determined colorimetrically.
A method for determining biological thiols at the picomole
level, based upon the conversion of thiols to fluorescent
derivatives by reaction with monobromobimane and the separation of
the derivatives by reverse-phase high-performance liquid
chromatography, was described by Newton et al. (1981). A
modification of this method was used by Shea et al. (1984) for the
determination of thiosulfate levels in plasma and urine.
The methylene blue method (Ivankovich et al., 1983) is
considered specific for thiosulfate and sensitive to 1 µg/ml. The
analyses are undertaken directly on heparinized plasma or urine.
Potassium iodide, potassium bromide, and monobasic potassium
phosphate are added to the sample. Then potassium borohydride (in
sodium hydroxide) and acetone are added with stirring, followed by
ferric sulfate (with water) and N,N-dimethyl- p-phenylenediamine
sulfate in sulfuric acid. A blue colour develops and absorbance is
measured at 665 nm.
3.6 Shelf-life
According to studies at the National Corporation of Swedish
Pharmacies, which has followed the stability of injectable
thiosulfate (0.15 g/ml) (Sweden) for three years, no significant
changes occurred during the period of observation.
A solution without, or with only small amounts of, sodium
phosphate dodecahydrate is unstable. The National Corporation of
Swedish Pharmacies has found the composition described in
section 3.3.9 to be the most suitable. For information concerning
storage, see section 3.13.6.
3.7 General Properties
3.7.1 Mechanism of antidotal activity
The major route of detoxification of cyanide in the body is
conversion to thiocyanate. This reaction requires a source of
sulfane sulfur (divalent sulfur bonded to another sulfur) and is
catalysed by sulfur transferases. It has been suggested that there
is a physiological pool of cyanide-reactive sulfane sulfur bound to
albumin that might act as a buffer against endogenous cyanide
production (Westley et al., 1983; Way et al., 1984). Thiosulfate is
present in the body in only small quantities, derived mainly from
cystine and other mercapto compounds. The physiological reserves
available for detoxifying cyanide are therefore limited (Schulz et
al., 1979b).
Rhodanese
Na2S2O3 + CN- --> SCN- + Na2SO3.
The sulfurtransferases catalyse reactions where sulfane sulfur
is involved. Rhodanese is the sulfurtransferase which has been
studied most extensively. Rhodanese (thiosulfate: cyanide
sulfurtransferase; EC 2.8.1.1.) catalyses the transfer of sulfane
sulfur directly to cyanide. It is distributed throughout the body,
the highest concentrations being found in the liver, and is located
mainly in the matrix of mitochondria (Westley et al., 1983).
The existence of a thiocyanate oxidase that could oxidize
thiocyanate back to cyanide (Goldstein & Rieders, 1953) has been
questioned. However, this is now attributed to artifactual
formation of HCN during assay (Vesey, 1979).
Sodium thiosulfate contains the necessary divalent sulfur donor
bound to another sulfur and it is the main sulfur donor for
rhodanese in the conversion of cyanide to thiocyanate. Whereas
rhodanese is available in excess in the body, the lack of a suitable
sulfur donor is the rate-limiting factor for this route of
detoxification in cyanide poisoning. This is the rationale for the
administration of sodium thiosulfate in cyanide poisoning so that
the endogenous detoxification capacity of the body is enhanced.
3.7.2 Other biochemical/pharmacological profiles
As described above, sodium thiosulfate may be used for
indications other than cyanide poisoning, utilizing other properties
of this substance. These are not dealt with in this document.
3.8 Animal Studies
3.8.1 Pharmacokinetics
When high doses of thiosulfate are injected into mammals, the
greater part is eliminated unchanged by renal excretion but a
certain amount is oxidized to sulfate. This latter fraction
increases as the dose of thiosulfate decreases. The oxidation of
thiosulfate to sulfate occurs in the liver by a two-step enzymatic
pathway. Studies by Gilman et al. (1946) demonstrated that
intravenously injected thiosulfate is rapidly distributed in the
extracellular fluid space and that its renal excretion occurs by
glomerular filtration. Further animal experiments have shown that
tubular transport may also take place (Sörbo, 1972).
Thiosulfate is both secreted and reabsorbed in man and dog,
according to Bucht (1949) and Foulks et al. (1952). Clearance of
thiosulfate is low, but at high levels clearance equals the
glomerular filtration rate. This means that at high plasma levels
of thiosulfate, secretion Tm (transfer maximum) is similar to
reabsorption Tm, whereas at low plasma levels both filtered and
secreted thiosulfate are reabsorbed and thus there is a diminished
clearance value for thiosulfate.
The volume of distribution, as determined in seven dogs
weighing 8.5-14.4 kg was, on average, 3 1 (Cardozo & Edelman, 1952).
Kinetic studies have shown that there is a cationic site on
rhodanese for the anionic sulfur donor (Westley et al., 1983). Most
of an injected dose of thiosulfate is excreted unchanged.
Thiosulfate is thought to permeate slowly through cell membranes
(Himwich & Saunders, 1948; Sörbo, 1962).
According to Crompton et al. (1974), thiosulfate can utilize
the dicarboxylate carrier to enter the mitochondria, as shown in
experiments with rat liver mitochondria. This system is specific
for divalent anions.
It has been shown by Szczepkowski et al. (1961) that when using
labelled thiosulfate the two atoms of sulfur have different fates
during the course of metabolism in animals. In rats, the inner
sulfur atom is eliminated very quickly in the form of sulfate while
the outer atom is transformed into sulfate much more slowly,
probably going through a number of intermediate stages.
When an experimental animal is injected with thiosulfate
containing 35S in its sulfane position exclusively, the whole of
this can be found labelled in the plasma as quickly as a sample can
be obtained (Schneider & Westley, 1969).
Experiments on dogs (Michenfelder & Tinker, 1977; Schulz et
al., 1979b) have shown that the capacity of the endogenous reserves
of thiosulfate to detoxify cyanide is exceeded if sodium
nitroprusside is administered as a continuous infusion at a rate of
more than 0.5 mg/kg per h. When the experimental animals received
doses greater than 0.5 mg/kg per h, their blood cyanide
concentrations rose continuously. Experimental animals receiving
the same dosage under otherwise identical conditions, but with the
additional infusion of thiosulfate at six times (w/w) the sodium
nitroprusside dosage, showed no abnormal signs. The urinary volume
in the thiosulfate-treated dogs, over a 48 h period, was
approximately twice that of the untreated animals, presumably due to
the increased rate of formation of thiocyanate and the resultant
osmotic diuresis.
Similar results were obtained in experiments on rabbits (Höbel
et al., 1978).
3.8.2 Pharmacodynamics
After the induction of acute sodium nitroprusside (SNP)
intoxication in rabbits, bolus injections of thiosulfate and
hydroxocobalamin (B12a), at SNP/antidote molar ratios of 1:5, were
equally effective in reducing the early signs and severity of the
metabolic acidosis (Pill et al., 1980). During the subsequent
observation period, the base excess with B12a as an antidote was
found to be lower than with thiosulfate. When the two antidotes
were given in parallel with a highly toxic dose of SNP, sodium
thiosulfate proved to be superior to B12a. The authors suggested
that for clinical purposes, SNP should always be administered in
combination with thiosulfate (1:5).
One molecule of sodium nitroprusside contains five cyanide
ions. Thiosulfate should therefore be given in a molar ratio of at
least 5:1, which corresponds to a dose of four parts by weight of
sodium thiosulfate to one of SNP. Schulz et al. (1979b) suggested
that since thiosulfate is rapidly metabolized and eliminated from
the body, it is better to administer it in excess by continuous
infusion.
In studies by Ivankovich et al. (1980), dogs were given KCN in
a constant infusion (0.1 mg/kg per min). One group of animals
(n = 5) was given an infusion of sodium thiosulfate (12 mg/kg per h)
intravenously 10 min prior to and during KCN infusion; another
group (n = 5) was given an intravenous bolus injection of
thiosulfate (150 mg/kg). The infusion increased the amount of
cyanide needed to cause death and the bolus injection increased the
protection from lethality even further. It was shown that
thiosulfate alone is capable of providing complete protection
against both cyanide and cyanide-forming compounds when administered
simultaneously with these compounds as a continuous infusion.
Furthermore, thiosulfate treatment resulted in no noticeable adverse
haemodynamic or respiratory effects when given as either a bolus or
a constant infusion. When high plasma concentrations of thiosulfate
are available, the detoxification mechanism is rapid enough to
provide adequate protection. Since thiosulfate is rapidly
eliminated by the kidneys, this high plasma level of thiosulfate is
best maintained by constant infusion. The only deleterious effect
of such a constant infusion may be a lowering of plasma volume,
since thiosulfate acts as an osmotic diuretic at this dosage;
however, this is rarely important clinically. The authors stated
that true detoxification of cyanide was achieved with thiosulfate
alone and that thiosulfate appears to be the agent of choice,
resulting in the lowest cyanide concentrations in tissues and blood
and the fewest side effects.
Dogs given thiosulfate (75 mg/kg) intravenously 5 min before
the start of an infusion of SNP (1.5 mg/kg) had significantly lower
plasma and red cell cyanide concentrations, and significantly higher
plasma thiocyanate concentrations, than controls (Krapez et al.,
1981). These changes were associated with only minimal disturbance
of tissue oxygenation. The authors suggested that a bolus dose of
sodium thiosulfate (75 mg/kg) is an effective antidote with
negligible toxicity. This study demonstrates that thiosulfate acts
more rapidly than had been thought previously. The maintenance of
low blood cyanide concentrations, coupled with the rapid increase in
plasma thiocyanate concentrations and unimpaired tissue oxygenation
in those animals that received thiosulfate, strongly suggests that
cyanide was converted to thiocyanate as quickly as it was released
from the nitroprusside. In this study, no synergism was found
between sodium thiosulfate and hydroxocobalamin. Investigations by
Evans (1964) suggested a negative interaction between thiosulfate
and hydroxocobalamin, but others (Hall & Rumack, 1987) believe there
is antidotal synergy.
In a study by Vesey et al. (1985), a bolus dose of sodium
thiosulfate (150 mg/kg, i.e., 12 times the stoichiometric amount
theoretically required to "neutralize" the cyanide from the SNP
dose) was given to dogs at the end of a 60-min infusion of SNP
(3 mg/kg, i.e., near lethal dose). Compared with the controls,
there was an impressive reduction in the mean half-lives of plasma
cyanide (25.1 min as opposed to 74.1min) and red blood cell cyanide
concentrations (22.4 min as opposed to 203.6 min). Cyanide toxicity
may be delayed after SNP administration due to continued breakdown
and release of HCN. The authors suggested that red blood cells act
as a site for cyanide detoxication. Thiosulfate enhances the rate
of HCN metabolism and also limits its peripheral distribution in
dogs (Sylvester et al., 1981).
Chen et al. (1934) showed that sodium thiosulfate detoxified
sodium cyanide at up to 3 times the minimal lethal dose (MLD).
Sodium nitrite did so at up to 4 times the MLD and a combination of
the two at up to 20 times the MLD.
Differing doses of thiosulfate were given intraperitoneally to
mice at different times after the injection of sublethal or lethal
doses of cyanide (Schubert & Brill, 1968). When thiosulfate was
administered to mice 5 min after cyanide, the time for recovery from
cyanide toxicity was shortened considerably. Rats given thiosulfate
10 min after cyanide (when inhibition of liver cytochrome oxidase
was maximal) invariably recovered 5 to 10 min later instead of the
30 to 40 min normally required without treatment.
A pharmacokinetic analysis of cyanide distribution and
metabolism with and without intravenous sodium thiosulfate was
conducted in pretreated mongrel dogs (Sylvester et al., 1983). The
mechanism of thiosulfate protection appeared to be extremely rapid
formation of thiocyanate in the central compartment, which therefore
limited the amount of cyanide distributed to sites of toxicity.
Thiosulfate increased the rate of conversion of cyanide to
thiocyanate over 30-fold.
3.8.3 Toxicology
According to NIOSH (1986), the intravenous LD50 in the mouse
is 2350 mg/kg, whereas the intravenous lowest published lethal dose
(LDLo) in the dog is 3000 mg/kg (Dennis & Fletcher, 1966). When
dogs were given 3000 mg/kg of sodium thiosulfate pentahydrate
intravenously (Dennis & Fletcher, 1966), the following effects
developed rapidly: metabolic acidosis, hypoxaemia, hypernatraemia,
and changes in the ECG and in arterial and venous pressures. In
these experiments, an immediate and rapid rise in the serum sodium
concentration would be expected, since the sodium content in sodium
thiosulfate pentahydrate is about 24 mEq/3000 mg. Moreover, the
dogs that survived the injection showed a marked diuresis, which
would be expected from the large osmotic dose administered. The
authors suggested that sodium thiosulfate pentahydrate (1500 mg/kg)
given intravenously at a constant rate over a 30-min period is
tolerated well.
During chronic sodium nitroprusside (SNP) administration, the
simultaneous infusion of thiosulfate may present problems because of
enhanced plasma thiocyanate accumulation and the danger of
hypovolaemia (Michenfelder & Tinker, 1977). Vesey et al. (1985)
suggest that it would be sufficient to give a bolus dose of sodium
thiosulfate only if the SNP dose/dose-rate is excessive.
There appears to be no information concerning the
teratogenicity or mutagenicity of sodium thiosulfate.
3.9 Volunteer Studies
Ivankovich et al. (1983) studied the available thiosulfate pool
and the pharmacokinetics of administered thiosulfate in healthy
volunteer subjects. Plasma thiosulfate concentrations, sampled from
the volunteers were 11.3 (± 1.1) mg/l (n = 26) and urine thiosulfate
concentrations were 2.8 (± 0.2) mg/l (n = 24). Bile contained 137.2
(± 29.5) mg/l thiosulfate (n = 6, sampled during cholecystectomy).
Thiosulfate (150 mg/kg) was injected intravenously into 5 normal
male volunteers. Plasma thiosulfate concentrations after 5 min were
1012 (± 88.5) mg/l. The half-life of the distribution phase was
23 min and that of the elimination phase 182 min. The calculated
VD was 151 ml/kg body weight. Urine concentration, clearance, and
rate of thiosulfate excretion increased markedly after injection.
Total excretion was 42.6 (± 3.5)% of the injected dose at 180 min,
although urinary excretion did not increase much during the
elimination phase; at 18 h after injection it was 47.4 (± 2.4)%.
Bile thiosulfate excretion did not change after thiosulfate
injection and bile excretion of thiosulfate accounted for less than
0.1% of total thiosulfate excretion. This study demonstrated that
the plasma concentration of thiosulfate in normal males is about
10 mg/l, and that excretion amounts to approximately 3 mg/day,
compared with findings of Sörbo & Ohman (1978) who discovered a
renal excretion value of 31.7 (± 12.8) mmol/24 h (7.9 (± 3.2) mg/l
per 24h). The normal endogenous production of thiosulfate can
therefore be considered to be small and the ability to produce
increased amounts in response to poisoning is likely to be limited.
The VD is 150 ml/kg and a 70 kg man would therefore have a total
extracellular thiosulfate content of 125 mg. Similar human VD
values were found by Cardozo & Edelman (1952). "Therapeutic" doses
of thiosulfate (150 mg/kg according to these authors) would elevate
plasma concentrations about 100 times. Such high concentrations may
be necessary to increase the intracellular concentration and enable
rhodanese to detoxify cyanide at the mitochondrial membrane if
indeed this is the site of action of thiosulfate (see above).
Schulz (1984) and Schulz et al. (1982) found a serum half-life
for thiosulfate of about 15 min during SNP therapy. According to
Gladtke (1966), the elimination half-life of sodium thiosulfate is
about 40 min (children aged 4 months to 14 years).
Absorption of sodium thiosulfate after oral administration is
poor (Martindale 1989). The oral toxicity is low and single doses
of 5-18 g have only a laxative action. Nausea and vomiting have
also been reported (Sörbo, 1972; Poisindex, 1987). It has also been
shown that thiosulfate injected intravenously is rapidly distributed
in the extracellular fluid space. Its renal excretion occurs by
glomerular filtration and secretion (Bucht, 1949; Foulks et al.,
1952; Sörbo, 1972).
3.10 Clinical Studies
The use of sodium thiosulfate as a single antidote in cyanide
poisoning has been evaluated in only a few studies.
Schulz et al. (1982) studied cyanide toxicity resulting from
sodium nitroprusside (SNP) in therapeutic use with and without
sodium thiosulfate. Cyanide was analysed using the method of Asmus
and Garschagen (see chapter 10), and concentrations were expressed
as nmol cyanide/ml erythrocyte. Thiocyanate concentrations were
also measured; following the addition of ferric chloride,
thiocyanate reacted with elemental chlorine to form cyanogen
chloride. The remainder of the measurement was carried out as for
cyanide. Concentrations were expressed as nmol thiocyanate/ml
plasma throughout. Thiosulfate was measured in plasma using the
method of Gast et al. (1952) and Dixon (1962). Seventy patients
(aged 17-78 years) were studied.
In 51 patients, SNP was given for short periods, while in 19
patients, SNP was given over longer periods of up to 2 weeks,
usually in combination with sodium thiosulfate infusion. In seven
of these 19 patients, 1 g sodium thiosulfate was given intravenously
as a bolus injection during SNP treatment in order to study the
kinetics. The drugs were infused quickly in a ratio of 50 mg SNP to
500 mg sodium thiosulfate. The threshold dose for the release of
free cyanide into the bloodstream in this study was 2 µg SNP/kg per
min. It was calculated that 5 µg/kg per min for 10 h, 10 µg/kg per
min for 4 hr or 20 µg/kg per min for 1.5 h would cause potentially
toxic levels of cyanide. Sodium thiosulfate by infusion stopped
accumulation of cyanide and the elevated cyanide levels declined,
whereas thiocyanate levels increased. The simultaneous infusion of
thiosulfate and SNP prevented accumulation of cyanide. According to
Saunders & Himwich (1950), the optimum in vitro molar
cyanide/thiosulfate ratio for the rhodanese reaction is 1:3. As
none of the patients in this study showed clinical signs of
toxicity, an assessment of treatment efficacy was made on the basis
of analytical data.
Shea et al. (1984) found that sodium thiosulfate (12 g/m2)
could be given safely to humans over 6 h provided that cardiac and
renal functions were normal. Similarly, 2 g/m2 per h for 12 h has
been given without side-effects (Howell et al., 1982). The
elimination half-life determined by Shea et al. (1984) using a
one-compartment kinetic model was approximately 80 min.
Schulz et al. (1979a) studied the kinetics of thiocyanate in
healthy subjects and in patients with renal failure. In healthy
subjects, the elimination half-life of thiocyanate (oral
administration) was 1 to 5 days (mean 3 days). The average
half-life for patients with renal failure was 9 days, with
elimination constants increasing in proportion to the creatinine
clearance. These findings have special relevance to the use of
sodium nitroprusside in the treatment of patients with renal
insufficiency.
3.11 Clinical Studies - Case Reports
Numerous cases have been reported where sodium thiosulfate has
been used in conjunction with other antidotes in the treatment of
cyanide poisoning. The use of sodium thiosulfate alone, though, has
been reported only rarely.
A baby, weighing 4.4 kg, developed hypertension in the neonatal
period and was treated with SNP by infusion (2-5 µg/kg per min).
After 30 h the blood pressure fell and the child became acidotic.
The erythrocyte cyanide concentration was 400 nmol/ml
(life-threatening intoxication). Infusion of sodium thiosulfate in
a dose of 100 mg/kg promptly caused the cyanide level to fall to
one-tenth of its maximum value. It was concluded that the mixed
infusion of SNP and thiosulfate is always advisable, especially in
small children where the thiosulfate pool is small (Schulz & Roth,
1982).
A man aged 42 years was treated with SNP and sodium thiosulfate
in combination and observations were made to calculate appropriate
doses. The patient developed toxic cyanide levels (erythrocyte
cyanide concentration, 3.6 µg/ml) during long-term infusion of SNP,
and was treated successfully with sodium thiosulfate (the
erythrocyte cyanide concentration level dropped to 0.5 µg/ml within
7 h). It was suggested that thiosulfate should be given at a dose
at least four times that of SNP but, as some excess of thiosulfate
is desirable, it was suggested that 300-400 mg sodium thiosulfate be
given together with 60 mg SNP by continuous infusion (Schulz et al.,
1979b).
A boy aged 14 years was given SNP during surgery until the
desired clinical effect was achieved. After 5 h the patient
developed circulatory shock but responded promptly to the
administration of sodium thiosulfate (150 mg/kg) given over 15 min
(Perschau et al., 1977).
A man aged 30 years ingested an unknown amount of a
cyanide-containing insecticide in a glass of beer. He was found
unconscious, apnoeic, and cyanotic 30 min later. Resuscitative
measures were commenced and he was given sodium thiosulfate
intravenously. He regained consciousness but remained mute for 12
days and developed choreiform movements. On examination 16 years
later everything was normal with the exception of mildly dysarthric
speech and some minor motor disturbances. A computer tomography
(CT) scan showed bilateral symmetrical infarction of the globus
pallidus and infarction of the left cerebellar hemisphere (Finelli,
1981).
During a fire in an apartment the mother jumped out through the
window with a baby while twin brothers, aged 2.5 years, tried to get
out through the front door. They were subsequently found
unconscious just inside the door and remained so on admission to
hospital. Both children were severely acidotic with pH values of
6.77 and 6.9, respectively. Initial treatment consisted of 100%
oxygen, controlled ventilation, and the intravenous administration
of sodium thiosulfate (approximately 400 mg/kg body weight). A few
hours later hyperbaric oxygen was also employed. Carbon monoxide
concentrations were low, being 5.7% and 1.4%, and cyanide
concentrations in blood were 1.15 mg/l and 1.1 mg/l. One twin
remained more severely ill than the other, but both children were
discharged after 3 weeks without sequelae.
A man aged 28 years ingested an unknown amount of a cyanide
solution in an attempt at suicide. On admission to hospital,
shortly after ingestion, he was talkative and anxious with an
intense smell of bitter almonds. He was hyperventilating slightly
and had a mild metabolic acidosis (BE -6). He was given 15 g of
sodium thiosulfate by slow intravenous infusion. The patient did
not become unconscious but instead became calm and mentally clear.
The acidosis reverted without further treatment. The blood cyanide
concentration was 39.7 µmol/l (9.9 mg/l) and that of thiocyanate,
150 µmol/l (0.88 mg/l).
A man aged 32 years ingested an unknown amount of potassium
cyanide solution in a suicidal attempt. The patient became
unconscious after 10 min and was brought to hospital within 40 min
after ingestion. He was by then deeply unconscious, slightly
cyanotic, and unresponsive to pain. The patient was treated
immediately with oxygen, 8 g sodium thiosulfate intravenously, and
gastric lavage. There was striking improvement after 30 min and
Kelocyanor was then given, after which the patient became completely
conscious. The cyanide concentration in blood was 3.7 mg/l.
A 54-year-old man ingested 3 g potassium cyanide and arrived in
hospital unconscious and with respiratory arrest. The patient was
resuscitated and 12 g of sodium thiosulfate was given intravenously.
There was a moderately severe acidosis (pH 7.31, BE -11) and another
3 g of sodium thiosulfate was therefore given. After 30 min the
patient breathed spontaneously, after 90 min he moved his
extremities and eyes, and after 6 h he was fully alert. No sequelae
were observed. The cyanide concentration in blood was 0.26 mg/l.
3.12 Summary of Evaluation
3.12.1 Indications
Sodium thiosulfate is indicated in poisoning from cyanide,
chlorate, bromate, bromine, iodine, cisplatin, mustard gas, and
nitrogen mustard. However, this monograph deals only with the use
of sodium thiosulfate as an antidote in cyanide poisoning
3.12.2 Route of administration
In cyanide poisoning, sodium thiosulfate should be given
intravenously (absorption is poor after oral administration) as a
bolus injection or by infusion over at least 10 min. When used to
prevent cyanide poisoning during sodium nitroprusside therapy, it
may be given either simultaneously by continuous infusion or,
alternatively, as a slow bolus injection.
3.12.3 Dose
The recommended initial dose for adults in established cyanide
poisoning is 8 to 12.5 g (Chen et al., 1944; Chen & Rose, 1952), or
0.2 g/kg body weight (Sörbo, 1972). This dose is based on
individual cases where doses of this size have proven effective.
Experimental data and theoretical considerations support these
recommendations, though true validation is lacking.
For children relatively higher doses are generally recommended.
For children with normal haemoglobin concentrations, a dose of
approximately 410 mg/kg body wt has been suggested (Berlin, 1970)
and many handbooks suggest doses in the range 300-500 mg/kg body
weight. It should be noted that in those sources which make these
recommendations, sodium thiosulfate is used in combination with
other antidotes, especially sodium nitrite.
The risk of cyanide poisoning in patients undergoing treatment
with sodium nitroprusside is well documented. Sodium thiosulfate
has been found to be ideal in this situation, and it has been
recommended that the w/w ratio for SNP and sodium thiosulfate should
be at least 1:4 (Schulz et al., 1979b) and preferably, to obtain an
excess of thiosulfate, 1:5-6. The antidote may be given either by
continuous infusion, simultaneously with SNP (Schulz et al., 1982),
or by bolus injection.
3.12.4 Other consequential or supportive therapy
The capacity of sodium thiosulfate to enhance the
detoxification of cyanide in the body has been established in
animals and man. As an antidote in cyanide poisoning, sodium
thiosulfate alone, together with oxygen and necessary supportive
therapy, is probably sufficient in mild to moderately severe cases.
It is also valuable in doubtful cases of poisoning, where it may
have both therapeutic and diagnostic value. In severe poisoning,
sodium thiosulfate should be given together with other antidotes,
with which it acts synergistically.
3.13 Model Information Sheet
3.13.1 Uses
Sodium thiosulfate is indicated for use in cyanide poisoning.
3.13.2 Dosage and route of administration (cyanide poisoning)
The initial dose in adults is (8 to)12.5 g of sodium
thiosulfate given as an intravenous bolus injection/infusion over
10 (to 15) min. Alternatively, the total initial dose can be
calculated as 150-200 mg/kg body weight. Additional doses may be
indicated according to the clinical course.
The initial dose in children is 400 (300-500) mg/kg body
weight given intravenously as indicated above.
To prevent cyanide intoxication during SNP therapy, sodium
thiosulfate should be given either by simultaneous infusion of a
dose 5-6 times exceeding (w/w) the SNP dose or, alternatively, a
bolus injection may be employed.
3.13.3 Precautions and contraindications
There are no specific contraindications. The toxicity of
sodium thiosulfate is low and toxic effects should not be expected
unless doses far exceed those recommended. In patients with renal
insufficiency, dialysis can be considered for the more rapid
elimination of thiocyanate (during long-term treatment).
3.13.4 Adverse effects
Adverse effects are mild and of minor importance compared to
the risks associated with cyanide poisoning. Rapid injection of a
hyperosmolar sodium thiosulfate solution has caused nausea and
vomiting (Ivankovich et al., 1983). Hypotension has been reported,
due probably to the formation of thiocyanate, which is known to have
hypotensive properties (Done, 1961). Other side effects attributed
to excess thiocyanate production are nausea, headache, and
disorientation. When thiosulfate was injected into dogs (Vesey et
al., 1985) no side effects were seen other than transient
hypotension. Diuretic effects and osmotic disturbances are possible
side effects (Martindale, 1989).
3.13.5 Use in pregnancy/lactation
These aspects are seldom discussed in the literature and are of
little relevance in this context. In a life-saving situation, the
dosage recommended above should not be modified in the case of
pregnancy or lactation.
3.13.6 Storage
Injectable thiosulfate should be stored in ampoules. Storage
over three years does not cause any significant change in
composition. The solid substance may be stored in an airtight
container for five years without change.
3.14 References
Berlin CM (1970) The treatment of cyanide poisoning in children.
Pediatrics, 46: 793-796.
Bucht H (1949) On the tubular secretion of thiosulfate and
creatinine under the influence of caronamide. Scand J Clin Lab
Invest, 1: 270-276.
Cardozo RH & Edelman IS (1952) The volume of distribution of sodium
thiosulfate as a measure of the extracellular fluid space. J Clin
Invest, 31: 280-290.
Chen KK & Rose CL (1952) Nitrite and thiosulfate therapy in cyanide
poisoning. J Am Med Assoc, 149: 113-119.
Chen KK, Rose CL, & Clowes GHA (1934) Comparative values of several
antidotes in cyanide poisoning. Am J Med Sci, 188: 767-781.
Chen KK, Rose CL, & Clowes GHA (1944) The modern treatment of
cyanide poisoning. J Indiana Med Assoc, 37: 344-350.
Crompton M, Palmieri F, Capano M, & Quagliariello E (1974) The
transport of thiosulphate in rat liver mitochondria. FEBS Lett, 46:
247-250.
Darby PW & Wilson J (1967) Cyanide, smoking, and tobacco amblyopia.
Observations on the cyanide content of tobacco smoke. Br J
Ophthalmol, 51: 336-338.
Dennis DL & Fletcher WS (1966) Toxicity of sodium thiosulfate
(NCS-45624), a nitrogen mustard antagonist, in the dog. Cancer
Chemother Rep, 50: 255-257.
Dixon K (1962) Spectrophotometric determination of sodium
thiosulfate in body fluids by use of an iodine-amylose complex. Clin
Chim Acta, 7: 453-458.
Done AK (1961) Cyanide antidotes. In clinical pharmacology of
systemic antidotes. Clin Pharmacol Ther, 2: 765-768.
European Pharmacopoeia (1980) 2nd ed. Sainte-Ruffine, France,
Maisonneuve S.A.
Evans CL (1964) Cobalt compounds as antidotes for hydrocyanic acid.
Br J Pharmacol, 23: 455-475.
Finelli PF (1981) Changes in the basal ganglia following cyanide
poisoning. J Comput assisted Tomography, 5: 755-756.
Foulks J, Brazeau P, Koelle ES, & Gilman A (1952) Renal secretion of
thiosulfate in the dog. Am J Physiol, 168: 77-85.
Gast JH, Arai K, & Aldrich FL (1952) Quantitative studies on urinary
thiosulfate excretion by healthy human subjects. J Biol Chem, 196:
875-884.
Gilman A, Philips S, & Koelle ES (1946) The renal clearance of
thiosulfate with observations on its volume of distribution. Am J
Physiol, 146: 348-357.
Gladtke E (1966) [The thiosulfate space in the child.] Supplements
to the Archiv für Kinderheilkunde. Stuttgart, Ferdinand Enke Verlag
(in German).
Goldstein F & Rieders F (1953) Conversion of thiocyanate to cyanide
by an erythrocytic enzyme. Am J Physiol, 173: 287-290.
Grant WM (1986) Toxicology of the eye, 3rd ed. Springfield,
Illinois, Charles C. Thomas.
Hager HHJ (1977) [Hager's manual of pharmaceutical practice.]
Berlin, Heidelberg, New York, Springer-Verlag.
Hall AH & Rumack BH (1987) Hydroxocobalamin/sodium thiosulfate as a
cyanide antidote. J Emergency Med, 5: 115-121.
Himwich WA & Saunders JP (1948) Enzymatic conversion of cyanide to
thiocyanate. Am J. Physiol, 153: 348-354.
Höbel M, Kreye VAW, & Pill J (1978) Effect of sodium nitroprusside
alone and in combination with sodium thiosulfate on the acid-base
balance, and on thiocyanate and iron plasma levels in the rabbit.
Klin Wochenschr, 56(suppl 1): 147-152.
Howell SB, Pfeifle CE, Wung WE, Ohlsen RA, Lucas WE, Yon JL, & Green
M (1982) Intraperitoneal cisplatin with systemic thiosulfate
protection. Ann Intern Med, 97: 845-851.
IUPAC (1969) Dissociation constants of inorganic acids and bases in
aqueous solution. Oxford, United Kingdom, International Union of
Pure and Applied Chemistry.
Ivankovich AD, Braverman B, Kanuru RP, Heyman HJ, & Paulissian R
(1980) Cyanide antidotes and methods of their administration in
dogs: a comparative study. Anaesthesiology, 52: 210-216.
Ivankovich AD, Braverman B, Stephens TS, Shulman M, & Heyman HJ
(1983) Sodium thiosulfate disposition in humans: Relation to sodium
nitroprusside toxicity. Anesthesiology, 58: 11-17.
Kirk-Othmer (1969) Encyclopedia of chemical technology, 2nd ed. New
York, John Wiley & Sons, vol 20.
Krapez JR, Vesey CJ, Adams L, & Cole PV (1981) Effects of cyanide
antidotes used with sodium nitroprusside infusions: Sodium
thiosulfate and hydroxocobalamin given prophylactically to dogs. Br
J Anaesth, 53: 793-804.
Lang S (1895) [Prussic acid detoxification.] Arch Exp Pathol
Pharmakol, 36: 75-99 (in German).
Martindale (1989) In: Reynolds JEF ed. The extra pharmacopoeia, 29th
ed. London, The Pharmaceutical Press, p 855.
Michenfelder JD & Tinker JH (1977) Cyanide toxicity and thiosulfate
protection during chronic administration of sodium nitroprusside in
the dog: Correlation with a human case. Anesthesiology, 47: 441-448.
Newton GL, Dorian R, & Fahey RC (1981) Analysis of biological
thiols: Derivatization with monobromobimane and separation by
reverse-phase high performance liquid chromatography. Anal Biochem,
114: 383-387.
NIOSH (1986) Register of toxic effects of chemical substances.
Washington, DC, National Institute for Occupational Safety and
Health.
Perschau RA, Modell JH, Bright RW, & Shirley PD (1977) Suspected
sodium nitroprusside-induced cyanide intoxication. Anesth Analg,
56(4): 533-537.
Pharmacopoea Nordica (1964) Editio Svecica Stockholm,
Apotekarsocietetens förlag.
Pill H, Engeser P, Hobel M, & Kreye VAW (1980) Sodium nitroprusside:
Comparison of the antidotal effect of hydroxocobalamin and sodium
thiosulfate in rabbits. Dev Toxicol Environ Sci, 8: 423-426.
PoisIndex (1987) Microfiche data base, 53rd ed. Denver, Colorado,
Micromedex Inc.
Saunders JP & Himwich WA (1950) Properties of the transulfurase
responsible for conversion of cyanide to thiocyanate. Am J Physiol,
163: 404-409.
Sax NI (1984) Dangerous properties of industrial materials, 6th ed.
New York, Van Nostrand Reinhold Co.
Schneider JF & Westley J (1969) Metabolic interrelations of sulphur
in proteins, thiosulphate and cystein. J Biol Chem, 244: 5735-5744.
Schubert J & Brill WA (1968) Antagonism of experimental cyanide
toxicity in relation to the in vivo activity of cytochrome
oxidase. J Pharmacol Exp Ther, 162: 352-359.
Schulz V (1984) Clinical pharmacokinetics of nitroprusside, cyanide,
thiosulphate and thiocyanate. Clin Pharmacokinet, 9: 239-251.
Schulz V & Roth B (1982) Detoxification of cyanide in a new-born
child. Klin Wochenschr, 60: 527-528.
Schulz V, Bonn R, & Kindler J (1979a) Kinetics of elimination of
thiocyanate in 7 healthy subjects and in 8 subjects with renal
failure. Klin Wochenschr, 57: 243-247.
Schulz V, Bonn R, Kammerer H, Kriegel R, & Ecker N (1979b)
Counteraction of cyanide poisoning by thiosulphate when
administering sodium nitroprusside as a hypotensive treatment. Klin
Wochenschr, 57: 905-907.
Schulz V, Gross R, Pasch T, Busse J, & Loeschcke G (1982) Cyanide
toxicity of sodium nitroprusside in therapeutic use with and without
sodium thiosulphate. Klin Wochenschr, 60: 1393-1400.
Shea M, Koziol JA, & Howell SB (1984) Kinetics of sodium
thiosulfate, a cisplatin neutralizer. Clin Pharmacol Ther, 35:
419-425.
Sörbo B (1962) Enzymatic conversion of cyanide to thiocyanate. In:
Brodie BB & Erdos EG ed. Proceedings of the First International
Pharmacological Meeting, Stockholm, 1961. Volume VI: Metabolic
factors controlling duration of drug action. New York, Pergamon
Press, pp 121-128.
Sörbo B (1972) The pharmacology and toxicology of inorganic sulfur
compounds. In: Senning A ed. Sulfur in organic and inorganic
chemistry. New York, Marcel Dekker, vol 2, pp 156-158.
Sörbo B & Ohman S (1978) Determination of thiosulphate in urine.
Scand J Clin Lab Invest, 38: 521-527.
Sylvester DM, Sander C, Hayton WL, & Way JL (1981) Alteration of the
pharmaco-dynamics of sodium cyanide by sodium thiosulphate. Proc
West Pharmacol Soc, 24: 135.
Sylvester DM, Hayton WL, Morgan RL, & Way JL (1983) Effects of
thiosulfate on cyanide pharmacokinetics in dogs. Toxicol Appl
Pharmacol, 69: 265-271.
Szczepkowski TW, Skarzynski B, & Weber M (1961) The metabolic state
of thiosulphate. Nature (Lond), 189: 1007-1008.
United States Dispensatory (1973) 27th ed. Philadelphia, Toronto,
J.B. Lippincott Co.
Vesey CJ (1979) Letter to the editor. Clin Toxicol, 14: 307-309.
Vesey CJ, Krapez JR, Varley JG, & Cole PV (1985) The antidotal
action of thiosulfate following acute nitroprusside infusion in
dogs. Anesthesiology, 62: 415-421.
Way JL, Tamulinas CB, Leung P, Ray L, Nizamani S, Sylvester D, Way
JL, & Chiou F (1984) Pharmacologic and toxicologic basis of cyanide
antagonism. Proc West Pharmacol Soc, 27: 149-153.
Westley J, Adler H, Westley L, & Nishida C (1983) The
sulfurtransferases. Fundam Appl Toxicol, 3: 377-382.
Wilson J (1965) Leber's hereditary optic atrophy: a possible defect
of cyanide metabolism. Clin Sci, 29: 505-515.
Windholz M ed. (1983) The Merck index: An encyclopedia of chemicals,
drugs, and biologicals, 10th ed. Rahway, New Jersey, Merck and Co.,
Inc.
4. Hydroxocobalamin
4.1 Introduction
Hydroxocobalamin is one of a number of antidotes which may be
used in the treatment of cyanide poisoning. It acts as a chelating
agent for cyanide. Hydroxocobalamin is commonly used in conjunction
with sodium thiosulfate (Na2S2O3) (Trousse anticyanure,
Laboratoire Anphar-Rolland). Another (unregistered) product is made
by the Pharmacie Centrale des Hôpitaux de Paris. It consists of 5 g
hydroxycobalamin in 100 ml water.
4.2 Name and Chemical Formula of Antidote
Hydroxocobalamin (OHB12) is a natural form of vitamin B12
found in humans (vitamin B12a). This chemical is described in the
British Pharmacopoeia (1980), the Italian Pharmacopoeia (1985), the
Pharmacopoée Française (1988), and the United States Pharmacopeia
(1985).
CAS number: 13 422-51-0
Formula: (dimethyl-5,6-benzimadazolyl) hydroxocobamide
C62H89CoN13O15P
The containers for medical oxygen are coded with a white colour
at the top according to ISO-32-19 (Fig 2) and carry the indication
"Medical Oxygen" carved in the iron material of the container. To
prevent connection with other medical gases, the containers are
provided with an international standardized "Pin Index Safety
System" ISO-407. This system is the same for medical and for
industrial oxygen so that in emergencies industrial oxygen can also
be used.
Regular control of gas pressure is necessary to prevent
shortage of oxygen in emergency situations.
Although the duration of storage will not alter the quality, it
is recommended that the gas should not be stored for more than six
months.
2.7 General Properties
In most circumstances, supplemental oxygen is indicated if
tissue hypoxia is imminent. In the case of cyanide poisoning,
however, cytochrome oxidase activity is inhibited and tissue
utilization of oxygen prevented. Even so, animal experiments
(Takano et al., 1980) suggest that inhibition of cytochrome oxidase
activity by cyanide is prevented, and recovery accelerated, in the
presence of oxygen.
2.8 Animal Studies
2.8.1 Pharmacokinetics
The bioavailability of oxygen depends on the following factors:
(a) the thickness of the alveolar membrane; as a result of
oedema fluid in the interstitial spaces of the membrane
oxygen cannot readily diffuse into the blood;
(b) haemoglobin configuration:
(i) oxygen transport by the blood to the tissues can be
disturbed by methaemoglobin-forming antidotes, e.g.,
sodium nitrite or 4-dimethylaminophenol, often used
in the treatment of cyanide poisoning;
(ii) oxygen transport is also hampered in carbon monoxide
poisoning, which occurs in combination with cyanide
poisoning following smoke inhalation.
(c) Oxygen utilization by tissues is prevented by the
inhibition of cytochrome oxidase activity in cyanide
poisoning.
2.8.2 Pharmacodynamics
Isom & Way (1982) studied the reduction of cytochrome oxidase
prepared from brains and livers of mice poisoned with potassium
cyanide. They examined several groups of animals and compared
exposure to air and to oxygen at 0.95 ATA, both with and without
sodium nitrite and sodium thiosulfate treatment. Cytochrome oxidase
was inhibited by 75% within 2 min of the injection of 5 KCN mg/kg.
The inhibition of cytochrome oxidase was similar whether the animals
inhaled air or 0.95 ATA O2, but the reactivation phase of
cytochrome oxidase was faster in oxygen-breathing animals. They
also found that, at a lower dose (4 KCN mg/kg), enzyme activity was
increased from 22% to 66% by increasing the partial pressure of
oxygen from 0.11 to 0.95 ATA O2. Oxygen shifted the dose-response
curve of brain cytochrome oxidase inhibition by KCN to the right:
the dose of KCN producing 50% inhibition was 24 mg/kg in
air-breathing animals and 55 mg/kg in animals breathing oxygen. The
authors also found that liver rhodanese activity was 15 times higher
than brain rhodanese activity and that treatment with sodium nitrite
and sodium thiosulfate protected liver, but not brain, cytochrome
oxidase at high doses of KCN.
Way et al. (1972) found that oxygen alone antagonized cyanide
toxicity. However, they found no significant benefit in increasing
the partial pressure of oxygen from 1 to 4 ATA O2. The authors
demonstrated that oxygen potentiates the effect of thiosulfate and
that the best results were obtained using hyperbaric oxygen together
with nitrite and thiosulfate.
Ivanov (1959) reported that hyperbaric oxygen re-established
normal EEG activity in animals poisoned with cyanide.
Takano et al. (1980) measured the reduction of pyridine
nucleotide in vivo as an indicator of cytochrome oxidase
inhibition. They found a protective effect of oxygen at 1 and 2 ATA
O2 when the doses of KCN injected into rabbits were within the
range that produced inhibition of cytochrome oxidase in animals
breathing air. At much higher doses, the protective effect of O2
at 1 or 2 ATA became progressively weaker.
Paulet (1955) described the protective effect of oxygen on the
chemoreceptors and on the clinical manifestations in dogs poisoned
by cyanide.
Cope & Tenn (1961) showed that infusion of KCN solution (2 g/l)
to anaesthetized dogs breathing air produced atrioventricular block.
Oxygen breathing reversed this phenomenon and re-established normal
sinus rhythm.
2.8.3 Toxicology
Oxygen toxicity was discovered in 1878 by Paul Bert, who
observed convulsions in animals treated with air at 15-20 ATA. He
then noted that when pure oxygen was used instead of air, one-fifth
of the pressure sufficed to produce the convulsions. He concluded
that oxygen toxicity is directly correlated to the product of
partial pressure of oxygen and administration period.
Pulmonary oxygen toxicity was first described in 1897 by
Lorrain Smith, who noted that mice exposed to oxygen at pressures
above 1 ATA died from respiratory failure and that there was great
variability both within and between animal species in the
susceptibility to oxygen damage.
The initial stage of acute pulmonary oxygen toxicity is termed
the exudative phase and is characterized by a perivascular and
interstitial inflammatory response that includes damage to the
capillary endothelium with exudation of oedema
fluid,polymorphonuclear leucocytes and, eventually, macrophages.
The oedema is associated with widening of the interalveolar septa
and a thickened air-blood tissue barrier. Alveolar haemorrhage
develops and there is fibrin deposition in the alveoli (hyaline
membranes). Loss of alveolar lining cells (type I lining cells) can
also occur during this phase. As the condition progresses, it
reaches a stage referred to as the proliferative phase,
characterized by resolution of the inflammatory exudate and
increased cellularity of the interstitium due to proliferation of
macrophages and fibroblasts. There is also proliferation of the
cuboidal type II alveolar lining cells that serve to
re-epithelialize the denuded alveolar basement membrane. By the
time a typical laboratory animal, such as the rat, is near death, a
substantial number of pulmonary capillaries have been destroyed and
embolization of some arterioles will have occurred. Depending on
the species, however, the destruction of type I alveolar lining
cells and the proliferation of the type II cells may not be
extensive.
The bronchial tree is damaged by oxygen toxicity: a
necrotizing bronchiolitis has been described in neonatal mice by
Ludwin et al. (1974). The bronchiolitis consists of degeneration
and necrosis of bronchiolar mucosal epithelium involving both
ciliated as well as non-ciliated (Clara) cells. Early change,
occurring within 72 h in neonatal mice, consist of loss of cilia,
cellular swelling associated with mitochondrial swelling, oedema of
the bronchiolar wall, and leucocytic infiltration of the bronchiolar
connective tissue. These changes are followed by necrosis of the
epithelial cells with subsequent sloughing. Necrosis of the mucosal
cells is maximal at 6 to 7 days. Cold-blooded animals are
relatively resistant to oxygen toxicity.
Guinea-pigs, dogs and rats have a median time to death (LT50)
in the range of 65-80 h while breathing 1 ATA oxygen. Monkeys have
an LT50 of about 100 h, while chicks survived for 28 days at
1 ATA. In general, there is a great interspecies variability in
susceptibility to hyperoxia.
2.8.3.1 Mechanism of injury
Free radicals (superoxide anion O-2 and hydroxyl radical
OH) are produced by reduction of oxygen and these and hydrogen
peroxide (H2O2) are responsible for most of the toxic effects of
oxygen. The free radicals react with cellular components and cause
oxidative damage by oxygen radicals of the unsaturated fatty acids,
which are abundant in the membranes of cells, and the sulfhydryl
groups present in many enzymes and cellular proteins. The cells are
protected against oxygen radicals by enzymes which convert the
radicals to less toxic molecules and to molecular oxygen. Haber &
Weiss (1934) described the production of hydroxyl free radicals from
the superoxide anion and hydrogen peroxide.
Hyperoxia also causes arteriolar vasoconstriction, this being
unrelated to the free radical mechanism.
2.9 Volunteer Studies of Pulmonary Oxygen Toxicity
Clark & Lambertsen (1971) studied normal humans exposed to
2 ATA O2. Symptoms began within 3-8 h in the form of mild
tracheal irritation and increased in intensity throughout the
exposure time. After 8-10 h, symptoms were characterized by
uncontrollable coughing, dyspnoea at rest, and a constant
tracheobronchial burning sensation. Decreased vital capacity was
the most constant and sensitive sign of pulmonary oxygen toxicity.
Decreased carbon monoxide diffusion capacity and increased
alveolar-arterial O2 difference were also observed.
Singer et al. (1970) compared two groups of patients treated
with 0.4 ATA O2 or 1 ATA O2 for up to 24 h and found no
immediate or delayed dysfunction. He concluded that humans can
tolerate 1 ATA O2 for up to 24 h without detectable pulmonary
dysfunction. Comroe et al. (1945) showed that inhalation of oxygen
at 0.5-1.0 ATA O2 for more than 24 h decreased vital capacity.
Clark & Lambertsen (1971) established oxygen pressure versus time
tolerance curves. It appears that no dysfunction is observed if up
to 0.5 ATA O2 is inhaled for many days and that the use of 1 ATA
O2 is safe for up to 24 h.
2.10 Clinical Studies of Oxygen Toxicity
All human cells are affected by oxygen toxicity, but the three
organs most susceptible to acute oxygen toxicity are the eyes in the
newborn, and the lung and central nervous system at all ages.
2.10.1 Eyes
The eyes of the newborn and, especially, those of the premature
newborn, are very sensitive to oxygen toxicity, which produces
retrolental fibroplasia.
Patz (1968a,b) described retrolental fibroplasia which occurred
in premature babies exposed to 0.30-0.45 ATA O2 for 10 days.
Initially, in the presence of high concentrations of oxygen, there
is vasoconstriction of arteries, arterioles and venules with
necrosis of the immature retinal vessels (referred to as the
vaso-obliterative phase). Following oxygen-induced
vaso-obliteration, there is a vaso-proliferation of new capillaries
in the inner layer of the retina. The proliferation of capillaries
extends into the vitreous behind the lens and eventually results in
retinal detachment, which represents the final phase.
2.10.2 Central nervous system (CNS)
Central nervous system oxygen toxicity is characterized by
grand-mal seizures. The manifestation of acute CNS toxicity in
humans consists not only of grand-mal seizures but also of focal
motor seizures, constriction of the visual fields, deafness,
hyperacuity, changes of mood, and visual and auditory
hallucinations. CNS oxygen toxicity in humans is only observed
during hyperbaric oxygen exposure.
Grand-mal convulsions have been described in humans exposed to
pressures above 2.8 ATA O2 for periods exceeding 2 h. Central
nervous system toxicity is rare if the O2 pressure is less than
2.5 ATA and if oxygen and air are used intermittently, e.g., 10 min
of air and 30 min of oxygen, as long as the total hyperbaric
exposure time does not exceed 2 h.
2.11 Clinical Studies - Case Reports
In most cases reported in the literature the evidence for
cyanide poisoning is lacking because blood concentrations of cyanide
are not reported. Moreover, the majority of cases of cyanide
poisoning are treated with oxygen in combination with other
antidotal therapy. The individual role of oxygen as a cyanide
antidote in man is therefore difficult to assess. The following
cases have been reported.
2.11.1 Patients treated alone with supportive therapy and who
survived
* An analytical chemist (48 years old) was found unconscious
with unrecordable blood pressure and cardiac arrest. The
blood cyanide level was 5.8 mg/l (Edwards & Thomas, 1978).
* A three-year-old child was treated with laetrile enemas.
On admission the child was cyanotic, lethargic, and
unresponsive. The blood cyanide level was 2.14 mg/l
(Ortega & Creek, 1978).
* A biochemist, aged 31 years, was found in deep coma with a
metabolic acidosis and a blood cyanide concentration of
2.3 mg/l (Vogel et al., 1981).
* Nine people were poisoned by hydrogen cyanide as a result
of a leak from a valve. Six of them lost consciousness
briefly. Blood cyanide concentrations were 2.8 to
7.7 mg/l (Peden et al., 1986).
2.11.2 Hyperbaric oxygen therapy in cyanide poisoning
Trapp (1970) reported acute cyanide poisoning successfully
treated with hyperbaric oxygen, while Litovitz et al. (1983)
reported the unsuccessful use of hyperbaric oxygen.
Trapp & Lepawsky (1983) reported five cases of acute cyanide
poisoning successfully treated by hyperbaric oxygen in the Vancouver
General Hospital, Canada.
2.11.3 Cyanide poisoning due to smoke inhalation
Four out of five smoke inhalation victims with mean blood
cyanide levels of 1.62 mg/l (62 µmol/l) are reported to have
survived after receiving hyperbaric oxygen therapy combined with the
administration of sodium nitrite and sodium thiosulfate (Hart et
al., 1985).
2.12 Summary of Evaluation
1. In acute cases of poisoning by inhalation of hydrogen
cyanide, either alone or in combination with carbon monoxide,
termination of exposure and evacuation of the patient from
contaminated areas is indicated most urgently and should be
performed by the rescuers using appropriate protective equipment.
Measures to support respiration, circulation, and cardiac function
should be commenced, if necessary. Patients should receive pure
humidified oxygen.
Following smoke inhalation, patients frequently develop acute
pulmonary failure due to the irritant effect of the smoke. They may
benefit from the use of continuous positive airway pressure while
breathing spontaneously or positive end-expiratory pressure if
ventilated artificially.
Other antidotes for cyanide poisoning, especially sodium
thiosulfate, should be administered intravenously as soon as
possible. Blood samples, which must be taken before the
administration of antidotes, should be sent for toxicological
analysis.
2. In acute cyanide poisoning by ingestion, decontamination of
the stomach should be performed as soon as possible after or during
first-aid therapy.
3. The final decision about the use of hyperbaric oxygen
therapy depends on the availability of chambers and the ability to
treat.
4. Oxygen has always been regarded as an important first-aid
measure even though, it is difficult to understand, on a theoretical
basis, how oxygen has a favourable effect in cyanide poisoning
because inhibition of cytochrome oxidase is non-competitive. There
is now experimental evidence that oxygen has specific antidotal
activity. Oxygen accelerates the reactivation of cytochrome oxidase
and protects against cytochrome oxidase inhibition by cyanide.
Nevertheless, there are other possible modes of action and those
which are clinically important have yet to be determined.
5. Hyperbaric oxygen is recommended for smoke inhalation
victims suffering from combined carbon monoxide and cyanide
poisoning, since these two agents are synergistically toxic. The
role of hyperbaric oxygen in pure cyanide poisoning remains
controversial.
2.13 Model Information Sheet
2.13.1 Uses
Oxygen is indicated for the treatment of patients poisoned by
cyanide, either alone or in combination with carbon monoxide
following smoke inhalation.
2.13.2 Dosage and route
In cases of severe poisoning (respiratory insufficiency and/or
deteriorating vital signs), artificial ventilation with 100% oxygen
is indicated but for no longer than 12-24 h, after which the
inspired oxygen concentration should be reduced. Similarly in
moderately severe cases, 100% oxygen is indicated but again for no
longer than 12-24 h. For mild cases 40% oxygen is indicated.
2.13.3 Precautions/contraindications
In order to prevent rebreathing of hydrogen cyanide from the
expired air of a patient, it is advisable to include a non-return
valve when a bag and mask is used.
2.13.4 Adverse effects
The alveolar membranes of the lung are damaged by 100% oxygen
if this is administered for more than 24 h.
2.13.5 Use in pregnancy and lactation
No contraindication.
2.13.6 Storage
Store under pressure in a suitable metal container of a type
permitted by the safety regulations of the national authority.
Valves and taps should not be lubricated with oil or grease.
The containers for medical oxygen are coded with a white colour
at the top according to ISO-32-19 (Fig. 2) and carry the indication
"Medical Oxygen" in the iron material of the container. To prevent
connection with other medical gases the containers are provided with
an international standardized "Pin Index Safety System" ISO-407.
This system is the same for medical and for industrial oxygen so
that in emergencies industrial oxygen can also be used.
Regular control of gas pressure is necessary to prevent
shortage of oxygen in emergency situations.
Although the duration of storage will not alter the quality it
is recommended that the gas should not be stored for more than six
months.
2.14 References
Clark JM & Lambertsen CJ (1971) Rate of development of pulmonary
O2 toxicity in man during O2 at 2.0 ATM. ABS. J Appl Physiol,
30: 730-752.
Clark CJ, Campbell D, & Reid WH (1981) Blood carboxyhaemoglobin and
cyanide levels in fire survivors. Lancet, 1: 1332-1335.
Comroe JH, Dripps RD, Dumke PR, & Deming M (1945) Oxygen toxicity:
The effect of inhalation of high concentrations of oxygen for
twenty-four hours on normal men at sea level and at a simulated
altitude of 18,000 feet. J Am Med Assoc, 128: 710-717.
Cope C & Tenn M (1961) The importance of oxygen in the treatment of
cyanide poisoning. J Am Med Assoc, 175: 109-112.
Edwards AC & Thomas ID (1978) Cyanide poisoning. Lancet, 1: 92-93.
Haber F & Weiss JJ (1934) The catalytic decomposition of hydrogen
peroxide by iron salts. Proc R Soc Lond, A147: 332-351.
Hart GB, Strauss MB, Lennon PA, & Whitcroft DD (1985) Treatment of
smoke inhalation by hyperbaric oxygen. J Emergency Med, 3: 211-215.
Isom GE & Way JL (1982) Effects of oxygen on the antagonism of
cyanide intoxication - cytochrome oxidase, in vitro. Toxicol Appl
Pharmacol, 74: 57-62.
Ivanov JP (1959) The effect of elevated oxygen pressure on animals
poisoned with potassium cyanide. Pharmacol Toxicol (USSR), 22:
476-479.
Litovitz TL, Larkin RF, & Myers RAM (1983) Cyanide poisoning treated
with hyperbaric oxygen. Am J Emergency Med, 1: 94-101.
Ludwin SK, Northway WH Jr, & Bensch KG (1974) Oxygen toxicity in the
newborn. Necrotizing bronchiolitis in mice exposed to 100% oxygen.
Lab Invest, 31: 421-435.
Ortega MA & Creek JE (1978) Acute cyanide poisoning following
administration of laetrile enemas. J Pediatr, 93: 1059.
Patz A (1968a) New role of ophthalmologist in prevention of
retrolental fibroplasia. Arch Ophthalmol (Chicago), 78: 565-568.
Patz A (1968b) Comment on oxygen therapy. Arch Ophthalmol (Chicago),
79: 507.
Paulet G (1955) Valeur et méchanisme d'action d'intoxication
cyanhydrique. Arch Int Physiol, 63: 340.
Peden NR, Taha A, McSorley PD, Bryden GT, Murdoch IB, & Anderson JM
(1986) Industrial exposure to hydrogen cyanide: Implications for
treatment. Br Med J, 293: 538.
Singer MM, Wright F, Stanley LK, Roc BB, & Hamilton WK (1970) Oxygen
toxicity in man. A prospective study in patients after open heart
surgery. New Engl J Med, 283: 1473.
Takano T, Miyzaki Y, Nashimoto I, & Kobayashi K (1980) Effect of
hyperbaric oxygen on cyanide intoxication: in situ changes in
intracellular oxidation reduction. Undersea Biomed Res, 7: 191-197.
Trapp WG (1970) Massive cyanide poisoning with recovery: A
Boxing-Day story. Can Med Assoc J, 102: 517.
Trapp WG & Lepawsky M (1983) 100% survival in five life-threatening
acute cyanide poisoning victims treated by a therapeutic spectrum
including hyperbaric oxygen. Proceedings of the 1st European
Conference on Hyperbaric Medicine Amsterdam, The Netherlands p 45.
Vogel SN, Sultan TR, & Ten Eyck R (1981) Cyanide poisoning. Clin
Toxicol, 18: 367-383.
Way JL, End E, Sheehy MH, De Miranda P, & Feitknecht UF (1972)
Effect of oxygen on cyanide intoxication. IV. Hyperbaric oxygen.
Toxicol Appl Pharmacol, 22: 415-421.
3. SODIUM THIOSULFATE
3.1 Introduction
3.1.1 Indications
The use of sodium thiosulfate as an antidote has been recorded
in the literature on poisoning due to cyanide, mustard gas, nitrogen
mustard, bromate, chlorate, bromine, iodine, cisplatin, and certain
drugs (Dactinomycin, Mechlorethamine, Mitomycin) when extravasated.
There are also some references to its effect in iodate and
hypochlorite toxicity. The principal role of sodium thiosulfate,
though, lies in the treatment of cyanide poisoning.
3.1.2 Rationale for the choice of the antidote
The effect of sodium thiosulfate as an antidote in cyanide
poisoning is well documented, and was first demonstrated by Lang
(1895). Some authors believe it to be a relatively slow-acting
antidote, though others have demonstrated that it acts more rapidly
than was previously thought, enabling the conversion of cyanide to
thiocyanate (Krapez et al., 1981). Thiosulfate helps to detoxify
cyanide in the presence of the enzyme rhodanese. However, rhodanese
is an intramitochondrial enzyme and thiosulfate has limited ability
to penetrate cell and mitochondrial membranes. Thus, distribution
of thiosulfate is almost exclusively extracellular (Cardozo &
Edelman, 1952), while its antidotal action has been thought to take
place intracellularly. This view is now being re-examined in the
light of recent experimental evidence (see section 3.8).
3.1.3 Risk groups
No special-risk groups can be identified as regards the use of
sodium thiosulfate. However, it should be noted that there is an
apparently reduced ability to convert cyanide to thiocyanate in some
diseases, e.g., toxic amblyopias (in particular tobacco amblyopia)
and Leber's hereditary optic atrophy (Wilson, 1965; Darby & Wilson,
1967). Abnormally low rhodanese activity in the liver has been
described in two patients with Leber's hereditary optic atrophy
(Grant, 1986).
3.2 Name and Chemical Formula of Antidote
International non-proprietary name: Natrii thiosulfas; Sodium
thiosulfate (thiosulphate); Thiosulfate de sodium; Natrium
thiosulfuricum; Natriumthiosulfat (Hager, 1977).
CAS number: 10102-17-7 for sodium thiosulfate, pentahydrate
(NIOSH, 1986); 7772-98-7 for sodium thiosulfate, anhydrous, (NIOSH,
1986).
IUPAC name: Sodium thiosulfate, pentahydrate
Manufacturer: Readily available in many countries.
Commercial names: Commercially available as sodium thiosulfate
or equivalent in many countries.
Formula: Na2S2O3.5H2O (Martindale, 1989)
Relative molecular mass: 248.2 (Martindale, 1989)
Specification of chemical salt used: sodium thiosulfate
contains not less that 99.0% and not more than the equivalent of
101.0% of Na2S2O3.5H2O (European Pharmacopoeia, 1980);
transparent, colourless crystals (European Pharmacopoeia, 1980);
colourless, odourless, (or almost odourless) monoclinic prismatic
crystals, or a coarse crystalline powder with a saline taste
(Martindale, 1989).
3.3 Physico-chemical Properties
3.3.1 Melting point, boiling point
Sodium thiosulfate dissolves in its own water of
crystallisation at about 49 °C (European Pharmacopoeia, 1980;
Martindale, 1989;). It loses all its water at 100 °C and decomposes
at higher temperatures (Windholz, 1983). Above 200-300 °C, it
decomposes to sulfate and pentasulfide (Kirk-Othmer, 1969; Hager,
1977). When it is heated to the point of decomposition, fumes of
sulfur oxides are emitted (Sax, 1984; PoisIndex, 1987).
3.3.2 Solubility in vehicle for administration
Highly soluble in water (2 parts sodium thiosulfate in 1 part
water) (Martindale, 1982; Windholz, 1983).
3.3.3 Optical properties
Inactive with respect to polarized light; absorbs ultraviolet
light at wavelengths less than 230 nm (information from the National
Corporation of Swedish Pharmacies).
3.3.4 Acidity
A 10% solution in water has a pH of 6.0-8.4 ( European
Pharmacopoeia, 1980; Martindale, 1989). The pH of injectable
thiosulfate (0.15 g/ml) is 8.2-8.8 (information from the National
Corporation of Swedish Pharmacies).
3.3.5 pKa
pKa1: 1.46-1.74 (an approximate value) (IUPAC, 1969).
3.3.6 Stability
Efflorescent in warm (>30 °C) dry air. Slightly hygroscopic
(delinquesent) in moist air (Windholz, 1983). Store in an airtight
container (Martindale, 1989).
Aqueous solutions have limited stability due to a tendency to
decompose slowly by the following reactions:
Na2S2O3 --> Na2SO3 + S (neutral or acidic
solutions)
Na2S2O3 + H2O --> Na2SO4+H2S (alkaline
solutions)
The first reaction is accelerated by acids and the second by
air or oxygen. Aqueous solutions of sodium thiosulfate decompose
more rapidly on heating. Storage with limited access to air and
light in a cool environment increases stability (Kirk-Othmer, 1969;
Martindale, 1989; Windholz, 1983).
Injectable thiosulfate stored in ampoules for three years shows
no significant change in composition.
3.3.7 Refractive index, specific gravity
No value for refractive index (injectable thiosulfate
[0.15 g/ml]) is available. The specific gravity for injectable
thiosulfate (0.15 g/ml) is 1.076 (information from the National
Corporation of Swedish Pharmacies).
3.3.8 Loss of weight on drying
The substance loses 35.5-37.0% of weight when it is dried at
105 °C (Pharmacopoea Nordica, 1964; Hager, 1977).
3.3.9 Excipients
For injectable thiosulfate (0.15 g/ml): sodium phosphate
dodecahydrate (Na2HPO4.12H2O) 1.21% (information from The
National Corporation of Swedish Pharmacies; see also section 3.6).
3.3.10 Incompatibility
Incompatibility with iodine, acids, lead, mercury and silver
salts (Windholz, 1983) and with salts of heavy metals, oxidizing
agents, and acids has been indicated. If sodium thiosulfate is
triturated with chlorates, nitrates, or permanganates, an explosion
may occur (Martindale, 1989).
3.3.11 Other information
Solutions are sterilized by autoclaving. A 2.98% solution is
iso-osmotic with serum (Martindale, 1989).
Table 2. Physical properties of sodium thiosulfate pentahydrate
Property Value
refractive index, nd20 1.4886
density, d425 1.750
heat of solution in water at 25 °C (kcal/mol) - 11.3
heat of formation (kcal/mol) -621.9
heat of fusion (kcal/mol) 11.85
specific heat at 21 °C (cal/g) 0.346
cryoscopic constant 42.6
dissociation pressure (mm Hg)
at 20 °C 6.0
at 35 °C 18.0
vapour pressure of saturated solution (mmHg)
at 33 °C 10.0
at 57 °C 42.0
at 90 °C 233.0
20
density of aqueous solution, d20, (Na2S2O3, by weight)
10% 1.0847
20% 1.1760
30% 1.2762
40% 1.3851
From: Kirk-Othmer (1969)
3.4 Synthesis
Sodium thiosulfate can be produced commercially by a number of
methods, of which the four principal processes may be classified as
follows (Kirk-Othmer, 1969).
(a) Air oxidation of sulfides, hydrosulfides, and polysulfides
of alkali and alkaline earth metals.
(b) Recovery from waste liquors resulting from the production
of sulfur black and other sulfur dyes.
(c) Reaction of sulfur dioxide and sulfides.
(d) Reaction of sulfur and sulfites.
Possible contaminants include chlorides, sulfates, sulfites,
and heavy metals.
3.5 Analytical Methods
3.5.1 Quality control procedures for sodium thiosulfate
(European Pharmacopoeia, 1980)
Tests for chlorides, sulfates, sulfites, sulfides, and heavy
metals. The pH of a 10% solution is 6.0-8.4; clear and colourless
(European Pharmacopoeia, 1980).
3.5.2 Methods for identifying sodium thiosulfate
(European Pharmacopoeia, 1980).
(a) Decolorization of iodinated potassium iodide solution R.
(b) In combination with silver nitrate solution R2, a white
precipitate forms, which rapidly turns yellowish and then
black.
(c) In combination with hydrochloric acid, a precipitate of
sulfur is formed and a gas is evolved which gives a blue
colour to starch iodate paper R.
(d) Reaction on sodium.
3.5.3 Assay
Dissolve 0.500g in 20ml of water and titrate with 0.1 N iodine,
using 1 ml of starch solution R, added towards the end of the
titration, as indicator.
1 ml of 0.1 N iodine is equivalent to 24.82 mg of Na2S2O3.5H2O
(European Pharmacopoeia, 1980).
3.5.4 Methods for analysis of sodium thiosulfate in
biological
samples
Gast et al. (1952) and Dixon (1962) described a method for the
measurement of thiosulfate, based on reduction by iodine, but this
is a nonspecific means of estimation.
Sörbo & Ohman (1978) described a method for the determination
of thiosulfate in urine. After the removal of interfering
compounds, including endogenous thiocyanate by ion exchange,
thiosulfate is converted to thiocyanate in the presence of cyanide
and cupric ions. The thiocyanate formed is concentrated by ion
exchange, eluted with an acid solution of ferric ions, and the
ferric thiocyanate complex is determined colorimetrically.
A method for determining biological thiols at the picomole
level, based upon the conversion of thiols to fluorescent
derivatives by reaction with monobromobimane and the separation of
the derivatives by reverse-phase high-performance liquid
chromatography, was described by Newton et al. (1981). A
modification of this method was used by Shea et al. (1984) for the
determination of thiosulfate levels in plasma and urine.
The methylene blue method (Ivankovich et al., 1983) is
considered specific for thiosulfate and sensitive to 1 µg/ml. The
analyses are undertaken directly on heparinized plasma or urine.
Potassium iodide, potassium bromide, and monobasic potassium
phosphate are added to the sample. Then potassium borohydride (in
sodium hydroxide) and acetone are added with stirring, followed by
ferric sulfate (with water) and N,N-dimethyl- p-phenylenediamine
sulfate in sulfuric acid. A blue colour develops and absorbance is
measured at 665 nm.
3.6 Shelf-life
According to studies at the National Corporation of Swedish
Pharmacies, which has followed the stability of injectable
thiosulfate (0.15 g/ml) (Sweden) for three years, no significant
changes occurred during the period of observation.
A solution without, or with only small amounts of, sodium
phosphate dodecahydrate is unstable. The National Corporation of
Swedish Pharmacies has found the composition described in
section 3.3.9 to be the most suitable. For information concerning
storage, see section 3.13.6.
3.7 General Properties
3.7.1 Mechanism of antidotal activity
The major route of detoxification of cyanide in the body is
conversion to thiocyanate. This reaction requires a source of
sulfane sulfur (divalent sulfur bonded to another sulfur) and is
catalysed by sulfur transferases. It has been suggested that there
is a physiological pool of cyanide-reactive sulfane sulfur bound to
albumin that might act as a buffer against endogenous cyanide
production (Westley et al., 1983; Way et al., 1984). Thiosulfate is
present in the body in only small quantities, derived mainly from
cystine and other mercapto compounds. The physiological reserves
available for detoxifying cyanide are therefore limited (Schulz et
al., 1979b).
Rhodanese
Na2S2O3 + CN- --> SCN- + Na2SO3.
The sulfurtransferases catalyse reactions where sulfane sulfur
is involved. Rhodanese is the sulfurtransferase which has been
studied most extensively. Rhodanese (thiosulfate: cyanide
sulfurtransferase; EC 2.8.1.1.) catalyses the transfer of sulfane
sulfur directly to cyanide. It is distributed throughout the body,
the highest concentrations being found in the liver, and is located
mainly in the matrix of mitochondria (Westley et al., 1983).
The existence of a thiocyanate oxidase that could oxidize
thiocyanate back to cyanide (Goldstein & Rieders, 1953) has been
questioned. However, this is now attributed to artifactual
formation of HCN during assay (Vesey, 1979).
Sodium thiosulfate contains the necessary divalent sulfur donor
bound to another sulfur and it is the main sulfur donor for
rhodanese in the conversion of cyanide to thiocyanate. Whereas
rhodanese is available in excess in the body, the lack of a suitable
sulfur donor is the rate-limiting factor for this route of
detoxification in cyanide poisoning. This is the rationale for the
administration of sodium thiosulfate in cyanide poisoning so that
the endogenous detoxification capacity of the body is enhanced.
3.7.2 Other biochemical/pharmacological profiles
As described above, sodium thiosulfate may be used for
indications other than cyanide poisoning, utilizing other properties
of this substance. These are not dealt with in this document.
3.8 Animal Studies
3.8.1 Pharmacokinetics
When high doses of thiosulfate are injected into mammals, the
greater part is eliminated unchanged by renal excretion but a
certain amount is oxidized to sulfate. This latter fraction
increases as the dose of thiosulfate decreases. The oxidation of
thiosulfate to sulfate occurs in the liver by a two-step enzymatic
pathway. Studies by Gilman et al. (1946) demonstrated that
intravenously injected thiosulfate is rapidly distributed in the
extracellular fluid space and that its renal excretion occurs by
glomerular filtration. Further animal experiments have shown that
tubular transport may also take place (Sörbo, 1972).
Thiosulfate is both secreted and reabsorbed in man and dog,
according to Bucht (1949) and Foulks et al. (1952). Clearance of
thiosulfate is low, but at high levels clearance equals the
glomerular filtration rate. This means that at high plasma levels
of thiosulfate, secretion Tm (transfer maximum) is similar to
reabsorption Tm, whereas at low plasma levels both filtered and
secreted thiosulfate are reabsorbed and thus there is a diminished
clearance value for thiosulfate.
The volume of distribution, as determined in seven dogs
weighing 8.5-14.4 kg was, on average, 3 1 (Cardozo & Edelman, 1952).
Kinetic studies have shown that there is a cationic site on
rhodanese for the anionic sulfur donor (Westley et al., 1983). Most
of an injected dose of thiosulfate is excreted unchanged.
Thiosulfate is thought to permeate slowly through cell membranes
(Himwich & Saunders, 1948; Sörbo, 1962).
According to Crompton et al. (1974), thiosulfate can utilize
the dicarboxylate carrier to enter the mitochondria, as shown in
experiments with rat liver mitochondria. This system is specific
for divalent anions.
It has been shown by Szczepkowski et al. (1961) that when using
labelled thiosulfate the two atoms of sulfur have different fates
during the course of metabolism in animals. In rats, the inner
sulfur atom is eliminated very quickly in the form of sulfate while
the outer atom is transformed into sulfate much more slowly,
probably going through a number of intermediate stages.
When an experimental animal is injected with thiosulfate
containing 35S in its sulfane position exclusively, the whole of
this can be found labelled in the plasma as quickly as a sample can
be obtained (Schneider & Westley, 1969).
Experiments on dogs (Michenfelder & Tinker, 1977; Schulz et
al., 1979b) have shown that the capacity of the endogenous reserves
of thiosulfate to detoxify cyanide is exceeded if sodium
nitroprusside is administered as a continuous infusion at a rate of
more than 0.5 mg/kg per h. When the experimental animals received
doses greater than 0.5 mg/kg per h, their blood cyanide
concentrations rose continuously. Experimental animals receiving
the same dosage under otherwise identical conditions, but with the
additional infusion of thiosulfate at six times (w/w) the sodium
nitroprusside dosage, showed no abnormal signs. The urinary volume
in the thiosulfate-treated dogs, over a 48 h period, was
approximately twice that of the untreated animals, presumably due to
the increased rate of formation of thiocyanate and the resultant
osmotic diuresis.
Similar results were obtained in experiments on rabbits (Höbel
et al., 1978).
3.8.2 Pharmacodynamics
After the induction of acute sodium nitroprusside (SNP)
intoxication in rabbits, bolus injections of thiosulfate and
hydroxocobalamin (B12a), at SNP/antidote molar ratios of 1:5, were
equally effective in reducing the early signs and severity of the
metabolic acidosis (Pill et al., 1980). During the subsequent
observation period, the base excess with B12a as an antidote was
found to be lower than with thiosulfate. When the two antidotes
were given in parallel with a highly toxic dose of SNP, sodium
thiosulfate proved to be superior to B12a. The authors suggested
that for clinical purposes, SNP should always be administered in
combination with thiosulfate (1:5).
One molecule of sodium nitroprusside contains five cyanide
ions. Thiosulfate should therefore be given in a molar ratio of at
least 5:1, which corresponds to a dose of four parts by weight of
sodium thiosulfate to one of SNP. Schulz et al. (1979b) suggested
that since thiosulfate is rapidly metabolized and eliminated from
the body, it is better to administer it in excess by continuous
infusion.
In studies by Ivankovich et al. (1980), dogs were given KCN in
a constant infusion (0.1 mg/kg per min). One group of animals
(n = 5) was given an infusion of sodium thiosulfate (12 mg/kg per h)
intravenously 10 min prior to and during KCN infusion; another
group (n = 5) was given an intravenous bolus injection of
thiosulfate (150 mg/kg). The infusion increased the amount of
cyanide needed to cause death and the bolus injection increased the
protection from lethality even further. It was shown that
thiosulfate alone is capable of providing complete protection
against both cyanide and cyanide-forming compounds when administered
simultaneously with these compounds as a continuous infusion.
Furthermore, thiosulfate treatment resulted in no noticeable adverse
haemodynamic or respiratory effects when given as either a bolus or
a constant infusion. When high plasma concentrations of thiosulfate
are available, the detoxification mechanism is rapid enough to
provide adequate protection. Since thiosulfate is rapidly
eliminated by the kidneys, this high plasma level of thiosulfate is
best maintained by constant infusion. The only deleterious effect
of such a constant infusion may be a lowering of plasma volume,
since thiosulfate acts as an osmotic diuretic at this dosage;
however, this is rarely important clinically. The authors stated
that true detoxification of cyanide was achieved with thiosulfate
alone and that thiosulfate appears to be the agent of choice,
resulting in the lowest cyanide concentrations in tissues and blood
and the fewest side effects.
Dogs given thiosulfate (75 mg/kg) intravenously 5 min before
the start of an infusion of SNP (1.5 mg/kg) had significantly lower
plasma and red cell cyanide concentrations, and significantly higher
plasma thiocyanate concentrations, than controls (Krapez et al.,
1981). These changes were associated with only minimal disturbance
of tissue oxygenation. The authors suggested that a bolus dose of
sodium thiosulfate (75 mg/kg) is an effective antidote with
negligible toxicity. This study demonstrates that thiosulfate acts
more rapidly than had been thought previously. The maintenance of
low blood cyanide concentrations, coupled with the rapid increase in
plasma thiocyanate concentrations and unimpaired tissue oxygenation
in those animals that received thiosulfate, strongly suggests that
cyanide was converted to thiocyanate as quickly as it was released
from the nitroprusside. In this study, no synergism was found
between sodium thiosulfate and hydroxocobalamin. Investigations by
Evans (1964) suggested a negative interaction between thiosulfate
and hydroxocobalamin, but others (Hall & Rumack, 1987) believe there
is antidotal synergy.
In a study by Vesey et al. (1985), a bolus dose of sodium
thiosulfate (150 mg/kg, i.e., 12 times the stoichiometric amount
theoretically required to "neutralize" the cyanide from the SNP
dose) was given to dogs at the end of a 60-min infusion of SNP
(3 mg/kg, i.e., near lethal dose). Compared with the controls,
there was an impressive reduction in the mean half-lives of plasma
cyanide (25.1 min as opposed to 74.1min) and red blood cell cyanide
concentrations (22.4 min as opposed to 203.6 min). Cyanide toxicity
may be delayed after SNP administration due to continued breakdown
and release of HCN. The authors suggested that red blood cells act
as a site for cyanide detoxication. Thiosulfate enhances the rate
of HCN metabolism and also limits its peripheral distribution in
dogs (Sylvester et al., 1981).
Chen et al. (1934) showed that sodium thiosulfate detoxified
sodium cyanide at up to 3 times the minimal lethal dose (MLD).
Sodium nitrite did so at up to 4 times the MLD and a combination of
the two at up to 20 times the MLD.
Differing doses of thiosulfate were given intraperitoneally to
mice at different times after the injection of sublethal or lethal
doses of cyanide (Schubert & Brill, 1968). When thiosulfate was
administered to mice 5 min after cyanide, the time for recovery from
cyanide toxicity was shortened considerably. Rats given thiosulfate
10 min after cyanide (when inhibition of liver cytochrome oxidase
was maximal) invariably recovered 5 to 10 min later instead of the
30 to 40 min normally required without treatment.
A pharmacokinetic analysis of cyanide distribution and
metabolism with and without intravenous sodium thiosulfate was
conducted in pretreated mongrel dogs (Sylvester et al., 1983). The
mechanism of thiosulfate protection appeared to be extremely rapid
formation of thiocyanate in the central compartment, which therefore
limited the amount of cyanide distributed to sites of toxicity.
Thiosulfate increased the rate of conversion of cyanide to
thiocyanate over 30-fold.
3.8.3 Toxicology
According to NIOSH (1986), the intravenous LD50 in the mouse
is 2350 mg/kg, whereas the intravenous lowest published lethal dose
(LDLo) in the dog is 3000 mg/kg (Dennis & Fletcher, 1966). When
dogs were given 3000 mg/kg of sodium thiosulfate pentahydrate
intravenously (Dennis & Fletcher, 1966), the following effects
developed rapidly: metabolic acidosis, hypoxaemia, hypernatraemia,
and changes in the ECG and in arterial and venous pressures. In
these experiments, an immediate and rapid rise in the serum sodium
concentration would be expected, since the sodium content in sodium
thiosulfate pentahydrate is about 24 mEq/3000 mg. Moreover, the
dogs that survived the injection showed a marked diuresis, which
would be expected from the large osmotic dose administered. The
authors suggested that sodium thiosulfate pentahydrate (1500 mg/kg)
given intravenously at a constant rate over a 30-min period is
tolerated well.
During chronic sodium nitroprusside (SNP) administration, the
simultaneous infusion of thiosulfate may present problems because of
enhanced plasma thiocyanate accumulation and the danger of
hypovolaemia (Michenfelder & Tinker, 1977). Vesey et al. (1985)
suggest that it would be sufficient to give a bolus dose of sodium
thiosulfate only if the SNP dose/dose-rate is excessive.
There appears to be no information concerning the
teratogenicity or mutagenicity of sodium thiosulfate.
3.9 Volunteer Studies
Ivankovich et al. (1983) studied the available thiosulfate pool
and the pharmacokinetics of administered thiosulfate in healthy
volunteer subjects. Plasma thiosulfate concentrations, sampled from
the volunteers were 11.3 (± 1.1) mg/l (n = 26) and urine thiosulfate
concentrations were 2.8 (± 0.2) mg/l (n = 24). Bile contained 137.2
(± 29.5) mg/l thiosulfate (n = 6, sampled during cholecystectomy).
Thiosulfate (150 mg/kg) was injected intravenously into 5 normal
male volunteers. Plasma thiosulfate concentrations after 5 min were
1012 (± 88.5) mg/l. The half-life of the distribution phase was
23 min and that of the elimination phase 182 min. The calculated
VD was 151 ml/kg body weight. Urine concentration, clearance, and
rate of thiosulfate excretion increased markedly after injection.
Total excretion was 42.6 (± 3.5)% of the injected dose at 180 min,
although urinary excretion did not increase much during the
elimination phase; at 18 h after injection it was 47.4 (± 2.4)%.
Bile thiosulfate excretion did not change after thiosulfate
injection and bile excretion of thiosulfate accounted for less than
0.1% of total thiosulfate excretion. This study demonstrated that
the plasma concentration of thiosulfate in normal males is about
10 mg/l, and that excretion amounts to approximately 3 mg/day,
compared with findings of Sörbo & Ohman (1978) who discovered a
renal excretion value of 31.7 (± 12.8) mmol/24 h (7.9 (± 3.2) mg/l
per 24h). The normal endogenous production of thiosulfate can
therefore be considered to be small and the ability to produce
increased amounts in response to poisoning is likely to be limited.
The VD is 150 ml/kg and a 70 kg man would therefore have a total
extracellular thiosulfate content of 125 mg. Similar human VD
values were found by Cardozo & Edelman (1952). "Therapeutic" doses
of thiosulfate (150 mg/kg according to these authors) would elevate
plasma concentrations about 100 times. Such high concentrations may
be necessary to increase the intracellular concentration and enable
rhodanese to detoxify cyanide at the mitochondrial membrane if
indeed this is the site of action of thiosulfate (see above).
Schulz (1984) and Schulz et al. (1982) found a serum half-life
for thiosulfate of about 15 min during SNP therapy. According to
Gladtke (1966), the elimination half-life of sodium thiosulfate is
about 40 min (children aged 4 months to 14 years).
Absorption of sodium thiosulfate after oral administration is
poor (Martindale 1989). The oral toxicity is low and single doses
of 5-18 g have only a laxative action. Nausea and vomiting have
also been reported (Sörbo, 1972; Poisindex, 1987). It has also been
shown that thiosulfate injected intravenously is rapidly distributed
in the extracellular fluid space. Its renal excretion occurs by
glomerular filtration and secretion (Bucht, 1949; Foulks et al.,
1952; Sörbo, 1972).
3.10 Clinical Studies
The use of sodium thiosulfate as a single antidote in cyanide
poisoning has been evaluated in only a few studies.
Schulz et al. (1982) studied cyanide toxicity resulting from
sodium nitroprusside (SNP) in therapeutic use with and without
sodium thiosulfate. Cyanide was analysed using the method of Asmus
and Garschagen (see chapter 10), and concentrations were expressed
as nmol cyanide/ml erythrocyte. Thiocyanate concentrations were
also measured; following the addition of ferric chloride,
thiocyanate reacted with elemental chlorine to form cyanogen
chloride. The remainder of the measurement was carried out as for
cyanide. Concentrations were expressed as nmol thiocyanate/ml
plasma throughout. Thiosulfate was measured in plasma using the
method of Gast et al. (1952) and Dixon (1962). Seventy patients
(aged 17-78 years) were studied.
In 51 patients, SNP was given for short periods, while in 19
patients, SNP was given over longer periods of up to 2 weeks,
usually in combination with sodium thiosulfate infusion. In seven
of these 19 patients, 1 g sodium thiosulfate was given intravenously
as a bolus injection during SNP treatment in order to study the
kinetics. The drugs were infused quickly in a ratio of 50 mg SNP to
500 mg sodium thiosulfate. The threshold dose for the release of
free cyanide into the bloodstream in this study was 2 µg SNP/kg per
min. It was calculated that 5 µg/kg per min for 10 h, 10 µg/kg per
min for 4 hr or 20 µg/kg per min for 1.5 h would cause potentially
toxic levels of cyanide. Sodium thiosulfate by infusion stopped
accumulation of cyanide and the elevated cyanide levels declined,
whereas thiocyanate levels increased. The simultaneous infusion of
thiosulfate and SNP prevented accumulation of cyanide. According to
Saunders & Himwich (1950), the optimum in vitro molar
cyanide/thiosulfate ratio for the rhodanese reaction is 1:3. As
none of the patients in this study showed clinical signs of
toxicity, an assessment of treatment efficacy was made on the basis
of analytical data.
Shea et al. (1984) found that sodium thiosulfate (12 g/m2)
could be given safely to humans over 6 h provided that cardiac and
renal functions were normal. Similarly, 2 g/m2 per h for 12 h has
been given without side-effects (Howell et al., 1982). The
elimination half-life determined by Shea et al. (1984) using a
one-compartment kinetic model was approximately 80 min.
Schulz et al. (1979a) studied the kinetics of thiocyanate in
healthy subjects and in patients with renal failure. In healthy
subjects, the elimination half-life of thiocyanate (oral
administration) was 1 to 5 days (mean 3 days). The average
half-life for patients with renal failure was 9 days, with
elimination constants increasing in proportion to the creatinine
clearance. These findings have special relevance to the use of
sodium nitroprusside in the treatment of patients with renal
insufficiency.
3.11 Clinical Studies - Case Reports
Numerous cases have been reported where sodium thiosulfate has
been used in conjunction with other antidotes in the treatment of
cyanide poisoning. The use of sodium thiosulfate alone, though, has
been reported only rarely.
A baby, weighing 4.4 kg, developed hypertension in the neonatal
period and was treated with SNP by infusion (2-5 µg/kg per min).
After 30 h the blood pressure fell and the child became acidotic.
The erythrocyte cyanide concentration was 400 nmol/ml
(life-threatening intoxication). Infusion of sodium thiosulfate in
a dose of 100 mg/kg promptly caused the cyanide level to fall to
one-tenth of its maximum value. It was concluded that the mixed
infusion of SNP and thiosulfate is always advisable, especially in
small children where the thiosulfate pool is small (Schulz & Roth,
1982).
A man aged 42 years was treated with SNP and sodium thiosulfate
in combination and observations were made to calculate appropriate
doses. The patient developed toxic cyanide levels (erythrocyte
cyanide concentration, 3.6 µg/ml) during long-term infusion of SNP,
and was treated successfully with sodium thiosulfate (the
erythrocyte cyanide concentration level dropped to 0.5 µg/ml within
7 h). It was suggested that thiosulfate should be given at a dose
at least four times that of SNP but, as some excess of thiosulfate
is desirable, it was suggested that 300-400 mg sodium thiosulfate be
given together with 60 mg SNP by continuous infusion (Schulz et al.,
1979b).
A boy aged 14 years was given SNP during surgery until the
desired clinical effect was achieved. After 5 h the patient
developed circulatory shock but responded promptly to the
administration of sodium thiosulfate (150 mg/kg) given over 15 min
(Perschau et al., 1977).
A man aged 30 years ingested an unknown amount of a
cyanide-containing insecticide in a glass of beer. He was found
unconscious, apnoeic, and cyanotic 30 min later. Resuscitative
measures were commenced and he was given sodium thiosulfate
intravenously. He regained consciousness but remained mute for 12
days and developed choreiform movements. On examination 16 years
later everything was normal with the exception of mildly dysarthric
speech and some minor motor disturbances. A computer tomography
(CT) scan showed bilateral symmetrical infarction of the globus
pallidus and infarction of the left cerebellar hemisphere (Finelli,
1981).
During a fire in an apartment the mother jumped out through the
window with a baby while twin brothers, aged 2.5 years, tried to get
out through the front door. They were subsequently found
unconscious just inside the door and remained so on admission to
hospital. Both children were severely acidotic with pH values of
6.77 and 6.9, respectively. Initial treatment consisted of 100%
oxygen, controlled ventilation, and the intravenous administration
of sodium thiosulfate (approximately 400 mg/kg body weight). A few
hours later hyperbaric oxygen was also employed. Carbon monoxide
concentrations were low, being 5.7% and 1.4%, and cyanide
concentrations in blood were 1.15 mg/l and 1.1 mg/l. One twin
remained more severely ill than the other, but both children were
discharged after 3 weeks without sequelae.
A man aged 28 years ingested an unknown amount of a cyanide
solution in an attempt at suicide. On admission to hospital,
shortly after ingestion, he was talkative and anxious with an
intense smell of bitter almonds. He was hyperventilating slightly
and had a mild metabolic acidosis (BE -6). He was given 15 g of
sodium thiosulfate by slow intravenous infusion. The patient did
not become unconscious but instead became calm and mentally clear.
The acidosis reverted without further treatment. The blood cyanide
concentration was 39.7 µmol/l (9.9 mg/l) and that of thiocyanate,
150 µmol/l (0.88 mg/l).
A man aged 32 years ingested an unknown amount of potassium
cyanide solution in a suicidal attempt. The patient became
unconscious after 10 min and was brought to hospital within 40 min
after ingestion. He was by then deeply unconscious, slightly
cyanotic, and unresponsive to pain. The patient was treated
immediately with oxygen, 8 g sodium thiosulfate intravenously, and
gastric lavage. There was striking improvement after 30 min and
Kelocyanor was then given, after which the patient became completely
conscious. The cyanide concentration in blood was 3.7 mg/l.
A 54-year-old man ingested 3 g potassium cyanide and arrived in
hospital unconscious and with respiratory arrest. The patient was
resuscitated and 12 g of sodium thiosulfate was given intravenously.
There was a moderately severe acidosis (pH 7.31, BE -11) and another
3 g of sodium thiosulfate was therefore given. After 30 min the
patient breathed spontaneously, after 90 min he moved his
extremities and eyes, and after 6 h he was fully alert. No sequelae
were observed. The cyanide concentration in blood was 0.26 mg/l.
3.12 Summary of Evaluation
3.12.1 Indications
Sodium thiosulfate is indicated in poisoning from cyanide,
chlorate, bromate, bromine, iodine, cisplatin, mustard gas, and
nitrogen mustard. However, this monograph deals only with the use
of sodium thiosulfate as an antidote in cyanide poisoning
3.12.2 Route of administration
In cyanide poisoning, sodium thiosulfate should be given
intravenously (absorption is poor after oral administration) as a
bolus injection or by infusion over at least 10 min. When used to
prevent cyanide poisoning during sodium nitroprusside therapy, it
may be given either simultaneously by continuous infusion or,
alternatively, as a slow bolus injection.
3.12.3 Dose
The recommended initial dose for adults in established cyanide
poisoning is 8 to 12.5 g (Chen et al., 1944; Chen & Rose, 1952), or
0.2 g/kg body weight (Sörbo, 1972). This dose is based on
individual cases where doses of this size have proven effective.
Experimental data and theoretical considerations support these
recommendations, though true validation is lacking.
For children relatively higher doses are generally recommended.
For children with normal haemoglobin concentrations, a dose of
approximately 410 mg/kg body wt has been suggested (Berlin, 1970)
and many handbooks suggest doses in the range 300-500 mg/kg body
weight. It should be noted that in those sources which make these
recommendations, sodium thiosulfate is used in combination with
other antidotes, especially sodium nitrite.
The risk of cyanide poisoning in patients undergoing treatment
with sodium nitroprusside is well documented. Sodium thiosulfate
has been found to be ideal in this situation, and it has been
recommended that the w/w ratio for SNP and sodium thiosulfate should
be at least 1:4 (Schulz et al., 1979b) and preferably, to obtain an
excess of thiosulfate, 1:5-6. The antidote may be given either by
continuous infusion, simultaneously with SNP (Schulz et al., 1982),
or by bolus injection.
3.12.4 Other consequential or supportive therapy
The capacity of sodium thiosulfate to enhance the
detoxification of cyanide in the body has been established in
animals and man. As an antidote in cyanide poisoning, sodium
thiosulfate alone, together with oxygen and necessary supportive
therapy, is probably sufficient in mild to moderately severe cases.
It is also valuable in doubtful cases of poisoning, where it may
have both therapeutic and diagnostic value. In severe poisoning,
sodium thiosulfate should be given together with other antidotes,
with which it acts synergistically.
3.13 Model Information Sheet
3.13.1 Uses
Sodium thiosulfate is indicated for use in cyanide poisoning.
3.13.2 Dosage and route of administration (cyanide poisoning)
The initial dose in adults is (8 to)12.5 g of sodium
thiosulfate given as an intravenous bolus injection/infusion over
10 (to 15) min. Alternatively, the total initial dose can be
calculated as 150-200 mg/kg body weight. Additional doses may be
indicated according to the clinical course.
The initial dose in children is 400 (300-500) mg/kg body
weight given intravenously as indicated above.
To prevent cyanide intoxication during SNP therapy, sodium
thiosulfate should be given either by simultaneous infusion of a
dose 5-6 times exceeding (w/w) the SNP dose or, alternatively, a
bolus injection may be employed.
3.13.3 Precautions and contraindications
There are no specific contraindications. The toxicity of
sodium thiosulfate is low and toxic effects should not be expected
unless doses far exceed those recommended. In patients with renal
insufficiency, dialysis can be considered for the more rapid
elimination of thiocyanate (during long-term treatment).
3.13.4 Adverse effects
Adverse effects are mild and of minor importance compared to
the risks associated with cyanide poisoning. Rapid injection of a
hyperosmolar sodium thiosulfate solution has caused nausea and
vomiting (Ivankovich et al., 1983). Hypotension has been reported,
due probably to the formation of thiocyanate, which is known to have
hypotensive properties (Done, 1961). Other side effects attributed
to excess thiocyanate production are nausea, headache, and
disorientation. When thiosulfate was injected into dogs (Vesey et
al., 1985) no side effects were seen other than transient
hypotension. Diuretic effects and osmotic disturbances are possible
side effects (Martindale, 1989).
3.13.5 Use in pregnancy/lactation
These aspects are seldom discussed in the literature and are of
little relevance in this context. In a life-saving situation, the
dosage recommended above should not be modified in the case of
pregnancy or lactation.
3.13.6 Storage
Injectable thiosulfate should be stored in ampoules. Storage
over three years does not cause any significant change in
composition. The solid substance may be stored in an airtight
container for five years without change.
3.14 References
Berlin CM (1970) The treatment of cyanide poisoning in children.
Pediatrics, 46: 793-796.
Bucht H (1949) On the tubular secretion of thiosulfate and
creatinine under the influence of caronamide. Scand J Clin Lab
Invest, 1: 270-276.
Cardozo RH & Edelman IS (1952) The volume of distribution of sodium
thiosulfate as a measure of the extracellular fluid space. J Clin
Invest, 31: 280-290.
Chen KK & Rose CL (1952) Nitrite and thiosulfate therapy in cyanide
poisoning. J Am Med Assoc, 149: 113-119.
Chen KK, Rose CL, & Clowes GHA (1934) Comparative values of several
antidotes in cyanide poisoning. Am J Med Sci, 188: 767-781.
Chen KK, Rose CL, & Clowes GHA (1944) The modern treatment of
cyanide poisoning. J Indiana Med Assoc, 37: 344-350.
Crompton M, Palmieri F, Capano M, & Quagliariello E (1974) The
transport of thiosulphate in rat liver mitochondria. FEBS Lett, 46:
247-250.
Darby PW & Wilson J (1967) Cyanide, smoking, and tobacco amblyopia.
Observations on the cyanide content of tobacco smoke. Br J
Ophthalmol, 51: 336-338.
Dennis DL & Fletcher WS (1966) Toxicity of sodium thiosulfate
(NCS-45624), a nitrogen mustard antagonist, in the dog. Cancer
Chemother Rep, 50: 255-257.
Dixon K (1962) Spectrophotometric determination of sodium
thiosulfate in body fluids by use of an iodine-amylose complex. Clin
Chim Acta, 7: 453-458.
Done AK (1961) Cyanide antidotes. In clinical pharmacology of
systemic antidotes. Clin Pharmacol Ther, 2: 765-768.
European Pharmacopoeia (1980) 2nd ed. Sainte-Ruffine, France,
Maisonneuve S.A.
Evans CL (1964) Cobalt compounds as antidotes for hydrocyanic acid.
Br J Pharmacol, 23: 455-475.
Finelli PF (1981) Changes in the basal ganglia following cyanide
poisoning. J Comput assisted Tomography, 5: 755-756.
Foulks J, Brazeau P, Koelle ES, & Gilman A (1952) Renal secretion of
thiosulfate in the dog. Am J Physiol, 168: 77-85.
Gast JH, Arai K, & Aldrich FL (1952) Quantitative studies on urinary
thiosulfate excretion by healthy human subjects. J Biol Chem, 196:
875-884.
Gilman A, Philips S, & Koelle ES (1946) The renal clearance of
thiosulfate with observations on its volume of distribution. Am J
Physiol, 146: 348-357.
Gladtke E (1966) [The thiosulfate space in the child.] Supplements
to the Archiv für Kinderheilkunde. Stuttgart, Ferdinand Enke Verlag
(in German).
Goldstein F & Rieders F (1953) Conversion of thiocyanate to cyanide
by an erythrocytic enzyme. Am J Physiol, 173: 287-290.
Grant WM (1986) Toxicology of the eye, 3rd ed. Springfield,
Illinois, Charles C. Thomas.
Hager HHJ (1977) [Hager's manual of pharmaceutical practice.]
Berlin, Heidelberg, New York, Springer-Verlag.
Hall AH & Rumack BH (1987) Hydroxocobalamin/sodium thiosulfate as a
cyanide antidote. J Emergency Med, 5: 115-121.
Himwich WA & Saunders JP (1948) Enzymatic conversion of cyanide to
thiocyanate. Am J. Physiol, 153: 348-354.
Höbel M, Kreye VAW, & Pill J (1978) Effect of sodium nitroprusside
alone and in combination with sodium thiosulfate on the acid-base
balance, and on thiocyanate and iron plasma levels in the rabbit.
Klin Wochenschr, 56(suppl 1): 147-152.
Howell SB, Pfeifle CE, Wung WE, Ohlsen RA, Lucas WE, Yon JL, & Green
M (1982) Intraperitoneal cisplatin with systemic thiosulfate
protection. Ann Intern Med, 97: 845-851.
IUPAC (1969) Dissociation constants of inorganic acids and bases in
aqueous solution. Oxford, United Kingdom, International Union of
Pure and Applied Chemistry.
Ivankovich AD, Braverman B, Kanuru RP, Heyman HJ, & Paulissian R
(1980) Cyanide antidotes and methods of their administration in
dogs: a comparative study. Anaesthesiology, 52: 210-216.
Ivankovich AD, Braverman B, Stephens TS, Shulman M, & Heyman HJ
(1983) Sodium thiosulfate disposition in humans: Relation to sodium
nitroprusside toxicity. Anesthesiology, 58: 11-17.
Kirk-Othmer (1969) Encyclopedia of chemical technology, 2nd ed. New
York, John Wiley & Sons, vol 20.
Krapez JR, Vesey CJ, Adams L, & Cole PV (1981) Effects of cyanide
antidotes used with sodium nitroprusside infusions: Sodium
thiosulfate and hydroxocobalamin given prophylactically to dogs. Br
J Anaesth, 53: 793-804.
Lang S (1895) [Prussic acid detoxification.] Arch Exp Pathol
Pharmakol, 36: 75-99 (in German).
Martindale (1989) In: Reynolds JEF ed. The extra pharmacopoeia, 29th
ed. London, The Pharmaceutical Press, p 855.
Michenfelder JD & Tinker JH (1977) Cyanide toxicity and thiosulfate
protection during chronic administration of sodium nitroprusside in
the dog: Correlation with a human case. Anesthesiology, 47: 441-448.
Newton GL, Dorian R, & Fahey RC (1981) Analysis of biological
thiols: Derivatization with monobromobimane and separation by
reverse-phase high performance liquid chromatography. Anal Biochem,
114: 383-387.
NIOSH (1986) Register of toxic effects of chemical substances.
Washington, DC, National Institute for Occupational Safety and
Health.
Perschau RA, Modell JH, Bright RW, & Shirley PD (1977) Suspected
sodium nitroprusside-induced cyanide intoxication. Anesth Analg,
56(4): 533-537.
Pharmacopoea Nordica (1964) Editio Svecica Stockholm,
Apotekarsocietetens förlag.
Pill H, Engeser P, Hobel M, & Kreye VAW (1980) Sodium nitroprusside:
Comparison of the antidotal effect of hydroxocobalamin and sodium
thiosulfate in rabbits. Dev Toxicol Environ Sci, 8: 423-426.
PoisIndex (1987) Microfiche data base, 53rd ed. Denver, Colorado,
Micromedex Inc.
Saunders JP & Himwich WA (1950) Properties of the transulfurase
responsible for conversion of cyanide to thiocyanate. Am J Physiol,
163: 404-409.
Sax NI (1984) Dangerous properties of industrial materials, 6th ed.
New York, Van Nostrand Reinhold Co.
Schneider JF & Westley J (1969) Metabolic interrelations of sulphur
in proteins, thiosulphate and cystein. J Biol Chem, 244: 5735-5744.
Schubert J & Brill WA (1968) Antagonism of experimental cyanide
toxicity in relation to the in vivo activity of cytochrome
oxidase. J Pharmacol Exp Ther, 162: 352-359.
Schulz V (1984) Clinical pharmacokinetics of nitroprusside, cyanide,
thiosulphate and thiocyanate. Clin Pharmacokinet, 9: 239-251.
Schulz V & Roth B (1982) Detoxification of cyanide in a new-born
child. Klin Wochenschr, 60: 527-528.
Schulz V, Bonn R, & Kindler J (1979a) Kinetics of elimination of
thiocyanate in 7 healthy subjects and in 8 subjects with renal
failure. Klin Wochenschr, 57: 243-247.
Schulz V, Bonn R, Kammerer H, Kriegel R, & Ecker N (1979b)
Counteraction of cyanide poisoning by thiosulphate when
administering sodium nitroprusside as a hypotensive treatment. Klin
Wochenschr, 57: 905-907.
Schulz V, Gross R, Pasch T, Busse J, & Loeschcke G (1982) Cyanide
toxicity of sodium nitroprusside in therapeutic use with and without
sodium thiosulphate. Klin Wochenschr, 60: 1393-1400.
Shea M, Koziol JA, & Howell SB (1984) Kinetics of sodium
thiosulfate, a cisplatin neutralizer. Clin Pharmacol Ther, 35:
419-425.
Sörbo B (1962) Enzymatic conversion of cyanide to thiocyanate. In:
Brodie BB & Erdos EG ed. Proceedings of the First International
Pharmacological Meeting, Stockholm, 1961. Volume VI: Metabolic
factors controlling duration of drug action. New York, Pergamon
Press, pp 121-128.
Sörbo B (1972) The pharmacology and toxicology of inorganic sulfur
compounds. In: Senning A ed. Sulfur in organic and inorganic
chemistry. New York, Marcel Dekker, vol 2, pp 156-158.
Sörbo B & Ohman S (1978) Determination of thiosulphate in urine.
Scand J Clin Lab Invest, 38: 521-527.
Sylvester DM, Sander C, Hayton WL, & Way JL (1981) Alteration of the
pharmaco-dynamics of sodium cyanide by sodium thiosulphate. Proc
West Pharmacol Soc, 24: 135.
Sylvester DM, Hayton WL, Morgan RL, & Way JL (1983) Effects of
thiosulfate on cyanide pharmacokinetics in dogs. Toxicol Appl
Pharmacol, 69: 265-271.
Szczepkowski TW, Skarzynski B, & Weber M (1961) The metabolic state
of thiosulphate. Nature (Lond), 189: 1007-1008.
United States Dispensatory (1973) 27th ed. Philadelphia, Toronto,
J.B. Lippincott Co.
Vesey CJ (1979) Letter to the editor. Clin Toxicol, 14: 307-309.
Vesey CJ, Krapez JR, Varley JG, & Cole PV (1985) The antidotal
action of thiosulfate following acute nitroprusside infusion in
dogs. Anesthesiology, 62: 415-421.
Way JL, Tamulinas CB, Leung P, Ray L, Nizamani S, Sylvester D, Way
JL, & Chiou F (1984) Pharmacologic and toxicologic basis of cyanide
antagonism. Proc West Pharmacol Soc, 27: 149-153.
Westley J, Adler H, Westley L, & Nishida C (1983) The
sulfurtransferases. Fundam Appl Toxicol, 3: 377-382.
Wilson J (1965) Leber's hereditary optic atrophy: a possible defect
of cyanide metabolism. Clin Sci, 29: 505-515.
Windholz M ed. (1983) The Merck index: An encyclopedia of chemicals,
drugs, and biologicals, 10th ed. Rahway, New Jersey, Merck and Co.,
Inc.
4. Hydroxocobalamin
4.1 Introduction
Hydroxocobalamin is one of a number of antidotes which may be
used in the treatment of cyanide poisoning. It acts as a chelating
agent for cyanide. Hydroxocobalamin is commonly used in conjunction
with sodium thiosulfate (Na2S2O3) (Trousse anticyanure,
Laboratoire Anphar-Rolland). Another (unregistered) product is made
by the Pharmacie Centrale des Hôpitaux de Paris. It consists of 5 g
hydroxycobalamin in 100 ml water.
4.2 Name and Chemical Formula of Antidote
Hydroxocobalamin (OHB12) is a natural form of vitamin B12
found in humans (vitamin B12a). This chemical is described in the
British Pharmacopoeia (1980), the Italian Pharmacopoeia (1985), the
Pharmacopoée Française (1988), and the United States Pharmacopeia
(1985).
CAS number: 13 422-51-0
Formula: (dimethyl-5,6-benzimadazolyl) hydroxocobamide
C62H89CoN13O15P
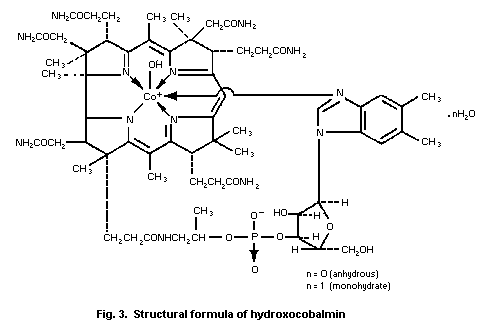 Structure: OHB12 contains a hydroxyl group attached to a
cobalt atom.
Relative molecular mass: 1346.47.
Contains not less than 95.0% and not more than 102.0% of
C62H89CoN13O15P, calculated on a dried weight basis (United States
Pharmacopeia, 1985)
Commercial names: The Anphar-Rolland Cyanide Antidote Kit
(containing OHB12 and Na2S2O3). Other proprietary formulations
currently available contain only 1 mg/ml. Another (unregistered)
product is produced by the Pharmacie Centrale des Hôpitaux de Paris.
It consists of 5 g hydroxycobalamin in 100 ml water.
4.3 Physico-chemical Properties
4.3.1 Characteristics
Dark red crystals or a red powder
Hygroscopic
Odourless.
4.3.2 Melting-point
Becomes brown at 200 °C
Not melted at 300 °C.
4.3.3 Solubility in vehicles for administration
Solubility in water: 1 part OHB12 in 50 parts water.
(Martindale, 1989).
4.3.4 Optical properties
Not relevant.
4.3.5 Acidity
The pH of a 0.2% solution in water is greater than 7.5
(Pharmacopée Française, 1988). The pH of a 2% solution in water is
8.0 - 10.0 (USP°XXI).
4.3.6 Stability in light
Must be protected from light.
4.3.7 Thermal stability
Must be stored at +4 °C (Anphar-Rolland).
Decreased activity has been observed with increasing
temperature.
Hydroxocobalamin solutions are sterilized by filtration.
4.3.8 Interference with other compounds
Complexes with basic substances. Incompatible with reducing
agents such as ascorbic acid, saccharose, sorbitol, and other B
vitamins (Mizoule, 1966). It has been suggested by Evans (1964)
that hydroxocobalamin interacts with sodium thiosulfate to form a
new compound, a thiosulfato-cobalamin, which can no longer (or less
firmly) fix the cyanide ion.
4.4 Synthesis
Routes of synthesis (confidential)
Manufacturing processes (confidential)
OHB12 (4 g) is lyophilized as a freeze-dried form and mixed
before use with one vial containing 8 g sodium thiosulfate. The
resulting solution is administered in 220 ml of 5% dextrose solution
by intravenous infusion over 20 min.
4.5 Analytical Methods
4.5.1 Identification of hydroxocobalamin
4.5.1.1 UV spectroscopy
The visible absorption spectrum of the solution, prepared as
for pH-dependent cobalamins, is maximal at 426 (±2)nm, 5l6 (±2)nm,
and 550 (±2)nm (United States Pharmacopeia, 1985).
OHB12 absorbs light at three other wavelengths: 274 nm,
351 nm, and 523 nm (British Pharmacopoeia, 1980; Pharmacopèe
Française, 1988).
4.5.1.2 Colorimetric method
Fuse a mixture of 1 mg of OHB12 and 50 mg of potassium
pyrosulfate in a porcelain crucible. Cool, break up the mass with a
glass rod, add 3 ml of water, and boil until dissolved. Add 1 drop
of phenolphthalein TS and sodium hydroxide (2 mol/l) drop by drop
until a pink colour appears. Add 0.5 g of sodium acetate, 0.5 ml of
acetic acid (1 mol/l), and 0.5 ml of nitroso R salt solution (1 in
100): a red or orange-red colour should appear immediately. Add
0.5 ml of hydrochloric acid and boil for l min: the red or
orange-red colour should persist (United States Pharmacopeia, 1985).
4.5.2 Quality controls
Information not available.
4.5.3 Raw materials
Foreign pigments, acidic impurities, acetate, chloride, and
sulfates should be lower than the following limits:
* Other cobalamins should constitute not more than 3.0% when
determined by chromatography (diethylaminoethylcellulose
column then carboxymethylcellulose column; and measurement
of absorption at 361 nm) (British Pharmacopoeia, 1980).
* Coloured impurities < 5.0% with descending paper
chromatography and measurement of absorption at 361 nm.
* Acidic impurities < 3.0% with a
diethylaminoethylcellulose column and measurement of
absorption at 351 and 361 nm.
* pH-dependent cobalamins between 95.0% and 102.0%,
calculated on a dry weight basis using a
spectrophotometric method (550 nm) with two solutions of
OHB12. One of these solutions, in a pH 4.0 buffer,
serves as the blank, the other solution should be in a pH
9.3 buffer (United States Pharmacopeia, 1985).
* Cyanocobalamin < 5.0%, calculated on a dry weight basis
and determined with a cobalamin radiotracer assay using a
cyanocobalamin tracer reagent.
4.5.4 Finished galenic form
* OHB12 is in freeze-dried form
* After reconstitution with thiosulfate:
- pH 10-12
- the liquid is a clear, deep-red solution.
4.5.5 Measurement
4.5.5.1 In raw materials and in finished form
* Spectrophotometric measurement at 351 nm (British
Pharmacopoeia, 1980; Pharmacopée Française, 1988)
* Radioimmunoassay with labelled cyanocobalamin (United
States Pharmacopeia, 1985).
4.5.5.2 In biological samples
Differential spectroscopy at two wavelengths, 351 nm (maximum
absorbance for OHB12) and 361 nm (maximum absorbance for
cyanocobalamin), is used to measure OHB12 and cyanocobalamin
levels in plasma and in urine (Baud et al., 1987).
4.6 Shelf-life
Keep in the dark, in a refrigerator at +4 °C, for no more than
three years.
4.7 General Properties
Kaczka et al. (1950) have demonstrated in in vitro studies
that the reaction of OHB12 with cyanide results in the
displacement of a hydroxyl group by a cyano group to form
cyanocobalamin, which is then excreted in the urine. Thus one
molecule of OHB12 binds one molecule of cyanide. Hydroxocobalamin
has been shown to be an effective antidote for experimental cyanide
poisoning in mice (Mushett et al., 1952), guinea-pigs (Posner et
al., 1976a), baboons (Posner et al., 1976b), and dogs (Rose et al.,
1965).
The antidotal effect of OHB12 is enhanced by the use of
thiosulfate in the same solution. Thiosulfate provides sulfur
radicals, which complex with cyanide to form thiocyanate using the
endogenous cyanide detoxification mechanism rhodanese (thiosulfate
sulfurtransferase). However, the action of thiosulfate may be too
slow to prevent death in serious cases of cyanide poisoning when
administered alone. The combination of OHB12 and thiosulfate was
shown to be more effective by Mizoule (1966), who studied it
extensively in experimental animals; other authors have reported
similar results (Motin et al., 1970; Luher et al., 1971; Yacoub et
al., 1974; Jouglard et al., 1974; Friedberg & Shukla, 1975; Racle et
al., 1976; Bismuth et al., 1984).
4.8 Animal Studies
4.8.1 Pharmacokinetics
No data are available.
4.8.2 Pharmacodynamics in the presence of the toxin
The antidotal action of OHB12 has been studied in animals,
firstly in the case of medium-term intoxication (Haguenoer et al.,
1975), and secondly in the prophylactic treatment of cyanide
intoxication (Posner et al., 1976a).
In the study by Haguenoer et. al. (1975), five groups of Wistar
male rats (mean weight, 330g) were used as follows:
* group 1 received injections of sodium chloride (0.9%) in
water (control group)
* group 2 received 1 ml of 50 mg/ml acetonitrile
* group 3 received 1 ml of 50 mg/ml acetonitrile plus 1 ml
of 5 mg/ml OHB12 (6 h later)
* group 4 received 1 ml of 1.12 mg/ml potassium cyanide
(KCN)
* group 5 received 1 ml of 1.12 mg/ml KCN plus 1 ml of
5 mg/ml OHB12 (6 h later).
In all, 28 intraperitoneal injections were given over a 6-week
period. Rats treated with OHB12 (groups 3 and 5) excreted less
free CN- in the urine than the others (groups 2 and 4); the
excretion of combined CN- was higher for groups 3 and 5. Urinary
excretion was higher in the case of KCN than acetonitrile. However,
a significant proportion of acetonitrile and cyanhydric acid (formed
from acetonitrile and KCN) was excreted by the lungs, and a large
amount of thiocyanate was excreted in the faeces.
In the second study (Posner et al., 1976a), nine groups of
healthy baboons (weighing 20 to 30 kg) were anaesthetized and
received nitroprusside by infusion, either alone (four animals) or
in combination with OHB12 (22.5 mg B12 per 1 mg nitroprusside)
(five baboons).
The infusion was continued for up to 2 h or until 500 mg of
nitroprusside had been administered. Cyanide concentrations
increased and the animals developed a severe metabolic acidosis.
OHB12 infused simultaneously with nitroprusside significantly
reduced the increase in cyanide concentrations and eliminated the
development of metabolic acidosis.
In a third study (Höbel et al., 1980), sodium nitroprusside
(SNP) and OHB12 were infused over 4 h in conscious rabbits in
molar ratios of 1:4, 1:5, or 1:8. Control animals received OHB12
only. Sodium thiosulfate was infused with SNP at molar ratios of
1:4, 1:5, and 1:10. SNP provoked a severe acidosis which was not
corrected by the lowest dose of OHB12: three deaths occurred among
ten animals. When the 1:5 ratio was administered, the acidosis was
less marked, but three out of seven animals succumbed. The highest
dose (1:8) prevented acidosis, but not death, in three out of eight
animals. All doses of OHB12 caused histological changes in the
liver, myocardium, and kidney. By contrast, sodium thiosulfate was
completely effective as an antidote in this study and did not give
rise to histological changes.
4.8.3 Toxicology
OHB12 has weak intrinsic toxicity and may be administered
safely in large doses (Mizoule, 1966).
4.8.3.1 Acute toxicity
The acute toxicity of OHB12 has been tested in mice and
rabbits. A solution containing 0.4-0.75 g or l g/kg was
administered to Swiss strain mice (0.20 ml per 20 mg body weight)
without any toxic effects either immediately or a few days later.
Doses of OHB12 (50 and 100 mg/kg body weight) were injected
intravenously into five rabbits (weighing 2-3 kg) without causing
side effects either immediately or 3 days later (Mizoule, 1966).
4.8.3.2 Sub-acute and chronic toxicity
The sub-acute and chronic toxicity of hydroxocobalamin has been
tested in rats and rabbits. Twelve young male Wistar rats (weight
43-53 g) received three subcutaneous injections weekly for a period
of 82 days. Six of these rats were treated with 50 mg OHB12
injections, while six received injections of sodium chloride (0.9%)
in water. No significant differences were reported in the behaviour
of the rats or in the haematological results. Rabbits received one
injection of OHB12 (50 mg/kg) every other day for a period of 10
days. No toxicity was reported (Mizoule, 1966).
4.9 Volunteer Studies
Data not available.
4.10 Clinical Studies
After intravenous infusion, OHB12 is bound sparingly to a
specific globulin transport plasma protein, transcobalamin II
(Herbert & Sullivan, 1964). Lesser amounts are bound to the storage
protein transcobalamin I (a glycoprotein) and to transcobalamin III
(an intermediate glycoprotein). A small amount may be free or very
loosely bound to protein. In the fasting state, the majority of
circulating vitamin B12 is bound to transcobalamin, and it is
stored principally in the liver and bone marrow.
OHB12 has a very short half-life (5 min), being rapidly
metabolized and excreted (Vesey & Cole, 1981; Cottrell et al.,
1982). Up to 50% of an administered dose of OHB12 is excreted
unchanged in the urine, which becomes red-orange in colour following
a dose of 4 g OHB12 (Goodman & Gilman, 1975).
4.11 Clinical Studies - Case Reports
Only a few studies in man have included measurement of free and
total cyanide concentrations. These include 12 cases of acute
cyanide poisoning (Baud et al., 1986; Danel et al., 1986a);
several cases of sodium nitroprusside poisoning in which OHB12 was
used prophylatically (Cottrell et al., 1982), and one case of
combined cyanide and carbon monoxide poisoning (Baud et al., 1987).
Baud et al. (1986) reported the case of a 55-year-old worker,
exposed to propionitrile by inhalation and dermal exposure, who was
treated with a combination of OHB12 and thiosulfate. The patient
rapidly lost consciousness: 25 min after exposure he was comatose
and unresponsive with a pulse rate of 90 beats/min and a blood
pressure of 17.3/9.3 kPa (130/70 mmHg). Laboratory investigations
revealed no abnormalities except for a base deficit of 14-16 mmol/l
on arterial blood gas analysis. The serum lactate concentration was
10 mmol/l. The patient received 4 g OHB12 and 8 g thiosulfate
(Trousse anticyanure, Laboratoire Anphar Rolland) intravenously in
just under 30 min. During infusion of the antidote, the patient
regained consciousness and became oriented. Complete reversion of
CNS depression and normalization of vital signs were observed during
the next hour.
Table 3. Blood cyanide and thiocyanate concentrations
Time after Cyanide Thiocyanate
intoxication
2 h 5.71 mg/l undetectable
2.5 h 0.93 mg/l 21.1 mg/l
(at the end of the
antidote infusion)
7 h 1.00 mg/l baseline level
a Personal communication by V. Danel, L. Barret, and J.L. Debru
Regional Hospital Centre and University, Grenoble, France
concerning four hydrogen cyanide intoxications treated with
OHB12 and sodium hyposulfite.
Baud et al. (1989) studied the toxicokinetics of OHB12 and
cyanocobalamin (CNB12),which were measured simultaneously in cases
of cyanide intoxication treated with OHB12. Plasma cobalamin
levels were measured using a differential spectrophotometric assay.
Whole blood cyanide levels were measured, using a
microdiffusion/colormetric assay, in samples taken before
administration of OHB12 by infusion (5 g over 30 min).
Source of cyanide Bromo Sodium Mercurous Smoke inhalation
cyanide cyanide cyanide
Route of absorption lung orally orally lung lung lung lung
Initial blood cyanide 13 260 217 22 123 129 208
(µmol/l)
Peak OHB12 (µmol/l) 325 15 83 514 74 131 0
Peak CNB12 (µmol/l) 39 275 226 0 215 221 259
After completion of the OHB12 infusion, low levels of OHB12
were observed concurrently with high levels of CNB12 in patients
with high whole blood cyanide levels, but not in those with low
cyanide concentrations. These findings suggest that OHB12 acts
rapidly as a cyanide antidote.
Cottrell et al. (1982) studied two groups of patients in whom
cyanide toxicity was induced by nitroprusside. Group I received
nitroprusside alone, while group II received nitroprusside and
OHB12. Red cell and plasma cyanide concentrations were,
respectively, 83.44% (±23.12) and 3.51 (±1.01) mg per 100 ml after
administration of nitroprusside alone and 33.18% (±17.29) and 2.18%
(±0.65) mg per 100 ml after administration of nitroprusside with
OHB12. OHB12 infusion resulted in lower blood cyanide
concentrations and base deficit than in the untreated group. The
dose of nitroprusside used in each group did not differ
significantly.
Following inhalation of fumes from a firm making plastics, a
man became poisoned by both cyanide and carbon monoxide (Baud et
al., 1989). On admission, the whole-blood cyanide concentration was
3.6 mg/l. He immediately received 4 g OHB12 and 8 g sodium
thiosulfate by intravenous infusion. A second injection had to be
administered 3 h after admission because of clinical relapse, again
with a satisfactory response.
4.12 Summary of Evaluation
4.12.1 Indications
* In the treatment of pernicious anaemia and vitamin B12
deficiency states by intramuscular administration.
* In cyanide poisoning as an antidote:
a) as therapeutic treatment after exposure to
acetonitrile, propionitrile, or potassium or sodium
cyanides by oral, inhalational, or dermal routes;
b) as prophylactic treatment during sodium nitroprusside
infusion.
4.12.2 Advised route and dosage
OHB12 is usually given intravenously in a 5% solution of
dextrose in water. The normal adult dose is 4 g OHB12, but the
dose may be increased in massive poisoning.
4.12.3 Practical advice
* To maintain normal blood pH and isotonicity, the OHB12
must be injected in more than 220 ml of 5% dextrose.
* OHB12 must not be administered with reducing or basic
substances (see section 4.3.8).
* When OHB12 is administered as a prolonged infusion,
aluminium foil should be used to protect it from light.
4.12.4 Side effects
* The most common side effect is an orange-red discoloration
of the skin, mucuous membranes, and urine that lasts about
12 h but is not consequential.
* Allergic reactions to OHB12 have been reported (Dally &
Gaultier, 1976).
4.13 Model Information Sheet
4.13.1 Uses
Hydroxocobalamin is used for the treatment of pernicious
anaemia and vitamin B12 deficiency states. In the treatment of
cyanide intoxication, hydroxocobalamin is indicated in two
circumstances. Firstly, it is used for the treatment of exposure to
hydrogen cyanide, inorganic cyanide salts, and acetonitrile and
propionitrile. Secondly, hydroxocobalamin is used prophylactically
to prevent cyanide intoxication as a result of sodium nitroprusside
given by infusion.
4.13.2 Dosage and route
Hydroxocobalamin is given intravenously in a 5% dextrose
solution. The usual adult dose is 4 g, which may be increased in
cases of massive cyanide poisoning.
4.13.3 Precautions/contraindications
Hydroxocobalamin should be diluted in more than 220 ml of 5%
dextrose solution. It must not be administered with reducing
substances or basic agents. In the case of prolonged infusion, the
solution must be protected from light. The possibility of an
anaphylactoid reaction to hydroxocobalamin should be borne in mind.
4.13.4 Adverse effects
The most common side effect is an orange-red discoloration of
the skin, mucous membranes, and urine that lasts for approximately
12 h. Allergic reactions have been reported. Histological
abnormalities in the liver, myocardium, and kidney of rabbits with
nitroprusside intoxication treated with hydroxocobalamin have been
described (Höbel et al., 1980).
4.13.5 Use in pregnancy and lactation
These aspects are seldom discussed in the literature, and are
of little relevance in this context. In a life-saving situation,
the dosage recommended above should not be modified during pregnancy
or lactation.
4.13.6 Storage
Hydroxocobalamin must be kept in the dark, in a refrigerator at
4 °C, for not more than 3 years.
4.14 References
Baud FJ, Bismuth C, Astier A, Djeghour H, & Aubriot D (1986)
Toxicocinétique des cyanures et des thyocyanates lors d'un accident
professionel au propionitrile. Arch Mal Prof Méd Trav Sécur Soc,
47(2): 85-86.
Baud FJ, Astier A, Toffis V, & Barriot P (1987) Plasma kinetics of
hydroxocobalamin and cyanocobalamin in a treated cyanide poisoning.
Paris, Hôpital Fernand Widal (Unpublished data).
Baud FJ, Toffis V, Certain B, Muzynski J, Astier A, & Bismuth C
(1989) Hydroxocobalamine antidote des cyanures. Vérification
cinétique de sa transformation en cyanocobalamine dans l'organisme
intoxiqué. Thérapie, 44: 469.
Bismuth C, Cantineau JP, Pontal P, Baud F, Garnier R, Poulos L, &
Bolo A (1984) Priorité de l'oxygénation dans l'intoxication
cyanhydrique: A propos de 25 cas. J Toxicol Méd, 4(2): 107-121.
British Pharmacopoeia (1980) London, Her Majesty's Stationery
Office.
Cottrell JE, Casthely P, Brodie JD, Patel K, Klein A, & Turndorf H
(1982) Prevention of nitroprusside-induced toxicity with
hydroxocobalamin. New Engl J. Med, 298: 809-811.
Dally S & Gaultier M (1976) Choc anaphylactique dû à
l'hydrocobalamine. Nouv Presse Méd, 30: 1917.
Evans CL (1964) Cobalt compounds as antidotes for hydrocyanic acid.
Br J Pharmacol, 23: 455-475.
Friedberg KD & Shukla UR (1975) The efficiency of aquocoblamine as
an antidote in cyanide poisoning when given alone or combined with
sodium thiosulfate. Arch Toxicol, 33: 103-113.
Goodman LS & Gilman A ed. (1975) The pharmacological basis of
therapeutics, 5th ed. New York, MacMillan Publishing Co., Inc., p
1334.
Haguenoer JM, Dequidt J, & Jacquemont MC (1975) Intoxications
expérimentales par l'acétonitrile. 4ème note: Influence de
l'hydroxocobalamine sur l'intoxication à moyen terme. J Eur Toxicol,
2: 113-121.
Herbert V & Sullivan LW (1964) Activity of coenzyme B12 in man.
Ann NY Acad Sci, 112: 855-870.
Höbel M, Engeser P, Nemeth L, & Pill J (1980) The antidote effect of
thiosulphate and hydroxocobalamin in formation of nitroprusside
intoxication of rabbits. Arch Toxicol, 46: 207-213.
Italian Pharmacopoeia (1985) 9th ed. Rome, Libreria della Stato.
Jouglard J, Nava G, Botta A, Michel-Manuel C, Poyen B, & Mattei JL
(1974) A propos d'une intoxication aiguë par le cyanure de potassium
traitée par l'hydroxocobalamine. Marseille Méd, 12: 617-624.
Kaczka EA, Wolf DE, Kuehl FA Jr, & Folkers K (1950) Vitamin B12:
reactions of cyano-cobalamin and related compounds. Science, 112:
354-355.
Luher F, Dusoleil P, & De Montgros J (1971) Action de
l'hydroxocobalamine à dose massive dans l'intoxication aiguë au
cyanure: A propos d'un cas. Arch Mal Prof, 32: 683-686.
Martindale (1989) In: Reynolds JEF ed. The extra pharmacopoeia, 29th
ed. London, The Pharmaceutical Press, pp 1260-1261.
Mizoule J (1966) Etude de l'action antidote de l'hydroxocobalamine à
l'égard de l'intoxication cyanhydrique. Paris, Université de Paris
(Thèse de Doctorat en Pharmacie).
Motin J, Bouletreau P, & Rouzioux JM (1970) Intoxication
cyanhydrique grave traitée avec succès par l'hydroxocobalamine. J
Méd Strasbourg, 1: 717-722.
Mushett CW, Kelly KL, Boxer EG, & Rickards JC (1952) Antidotal
efficacy of vitamin B12a (hydroxocobalamin) in experimental
cyanide poisoning. Proc Soc Exp Biol Med, 81: 234-237.
Pharmacopée Française (1988) 10th ed. Paris, La Commission nationale
de Pharmacopée.
Posner MA, Tobey RE, & McElroy H (1976a) Hydroxocobalamin therapy of
cyanide intoxication in guinea-pigs. Anaesthesiology, 44(2):
157-160.
Posner MA, Rodkey FL, & Tobey RE (1976b) Nitroprusside induced
cyanide poisoning: Antidotal effect of hydroxocobalamin.
Anaesthesiology, 44(4): 330-335.
Racle JP, Chausset R, & Dissait F (1976) L'intoxication cyanhydrique
aiguë. Aspects biologiques, toxicologiques, cliniques et
thérapeutiques. Rev Méd Clermont-Ferrand, 5: 371-382.
Rose CL, Worth RM, & Chen KK (1965) Hydroxocobalamin and acute
cyanide poisoning in dogs. Life Sci, 4: 1785-1789.
United States Pharmacopeia (1985) 21st ed. Rockville, Maryland,
United States Pharmacopeial Convention, Inc.
Vesey GJ & Cole PV (1981) Cyanide antagonist (letter). Can Anaesth
Soc J, 28: 290.
Yacoub M, Faure J, Morena H, Vincent M, & Faure H. (1974)
L'intoxication cyanhydrique aiguë: Données actuelles sur le
métabolisme du cyanure et le traitement par l'hydroxocobalamine.
J Eur Toxicol, 7: 22-29.
5. DICOBALT EDETATE (KELOCYANOR)
5.1 Introduction
The ability of cyanide to form complexes with cobalt is an
example of the propensity of cyanide to form stable complexes with
many transition metals (Sharpe, 1976). It is a property of cyanide
that has been known since the last century, but there is still room
for disagreement about the precise nature of the complex formed in
vivo when cobalt is used to treat cyanide poisoning. In vitro,
a complex of one cobalt and five cyanide atoms is formed on addition
of potassium cyanide to cobalt (II) salts (Cotton & Wilkinson,
1966).
Structure: OHB12 contains a hydroxyl group attached to a
cobalt atom.
Relative molecular mass: 1346.47.
Contains not less than 95.0% and not more than 102.0% of
C62H89CoN13O15P, calculated on a dried weight basis (United States
Pharmacopeia, 1985)
Commercial names: The Anphar-Rolland Cyanide Antidote Kit
(containing OHB12 and Na2S2O3). Other proprietary formulations
currently available contain only 1 mg/ml. Another (unregistered)
product is produced by the Pharmacie Centrale des Hôpitaux de Paris.
It consists of 5 g hydroxycobalamin in 100 ml water.
4.3 Physico-chemical Properties
4.3.1 Characteristics
Dark red crystals or a red powder
Hygroscopic
Odourless.
4.3.2 Melting-point
Becomes brown at 200 °C
Not melted at 300 °C.
4.3.3 Solubility in vehicles for administration
Solubility in water: 1 part OHB12 in 50 parts water.
(Martindale, 1989).
4.3.4 Optical properties
Not relevant.
4.3.5 Acidity
The pH of a 0.2% solution in water is greater than 7.5
(Pharmacopée Française, 1988). The pH of a 2% solution in water is
8.0 - 10.0 (USP°XXI).
4.3.6 Stability in light
Must be protected from light.
4.3.7 Thermal stability
Must be stored at +4 °C (Anphar-Rolland).
Decreased activity has been observed with increasing
temperature.
Hydroxocobalamin solutions are sterilized by filtration.
4.3.8 Interference with other compounds
Complexes with basic substances. Incompatible with reducing
agents such as ascorbic acid, saccharose, sorbitol, and other B
vitamins (Mizoule, 1966). It has been suggested by Evans (1964)
that hydroxocobalamin interacts with sodium thiosulfate to form a
new compound, a thiosulfato-cobalamin, which can no longer (or less
firmly) fix the cyanide ion.
4.4 Synthesis
Routes of synthesis (confidential)
Manufacturing processes (confidential)
OHB12 (4 g) is lyophilized as a freeze-dried form and mixed
before use with one vial containing 8 g sodium thiosulfate. The
resulting solution is administered in 220 ml of 5% dextrose solution
by intravenous infusion over 20 min.
4.5 Analytical Methods
4.5.1 Identification of hydroxocobalamin
4.5.1.1 UV spectroscopy
The visible absorption spectrum of the solution, prepared as
for pH-dependent cobalamins, is maximal at 426 (±2)nm, 5l6 (±2)nm,
and 550 (±2)nm (United States Pharmacopeia, 1985).
OHB12 absorbs light at three other wavelengths: 274 nm,
351 nm, and 523 nm (British Pharmacopoeia, 1980; Pharmacopèe
Française, 1988).
4.5.1.2 Colorimetric method
Fuse a mixture of 1 mg of OHB12 and 50 mg of potassium
pyrosulfate in a porcelain crucible. Cool, break up the mass with a
glass rod, add 3 ml of water, and boil until dissolved. Add 1 drop
of phenolphthalein TS and sodium hydroxide (2 mol/l) drop by drop
until a pink colour appears. Add 0.5 g of sodium acetate, 0.5 ml of
acetic acid (1 mol/l), and 0.5 ml of nitroso R salt solution (1 in
100): a red or orange-red colour should appear immediately. Add
0.5 ml of hydrochloric acid and boil for l min: the red or
orange-red colour should persist (United States Pharmacopeia, 1985).
4.5.2 Quality controls
Information not available.
4.5.3 Raw materials
Foreign pigments, acidic impurities, acetate, chloride, and
sulfates should be lower than the following limits:
* Other cobalamins should constitute not more than 3.0% when
determined by chromatography (diethylaminoethylcellulose
column then carboxymethylcellulose column; and measurement
of absorption at 361 nm) (British Pharmacopoeia, 1980).
* Coloured impurities < 5.0% with descending paper
chromatography and measurement of absorption at 361 nm.
* Acidic impurities < 3.0% with a
diethylaminoethylcellulose column and measurement of
absorption at 351 and 361 nm.
* pH-dependent cobalamins between 95.0% and 102.0%,
calculated on a dry weight basis using a
spectrophotometric method (550 nm) with two solutions of
OHB12. One of these solutions, in a pH 4.0 buffer,
serves as the blank, the other solution should be in a pH
9.3 buffer (United States Pharmacopeia, 1985).
* Cyanocobalamin < 5.0%, calculated on a dry weight basis
and determined with a cobalamin radiotracer assay using a
cyanocobalamin tracer reagent.
4.5.4 Finished galenic form
* OHB12 is in freeze-dried form
* After reconstitution with thiosulfate:
- pH 10-12
- the liquid is a clear, deep-red solution.
4.5.5 Measurement
4.5.5.1 In raw materials and in finished form
* Spectrophotometric measurement at 351 nm (British
Pharmacopoeia, 1980; Pharmacopée Française, 1988)
* Radioimmunoassay with labelled cyanocobalamin (United
States Pharmacopeia, 1985).
4.5.5.2 In biological samples
Differential spectroscopy at two wavelengths, 351 nm (maximum
absorbance for OHB12) and 361 nm (maximum absorbance for
cyanocobalamin), is used to measure OHB12 and cyanocobalamin
levels in plasma and in urine (Baud et al., 1987).
4.6 Shelf-life
Keep in the dark, in a refrigerator at +4 °C, for no more than
three years.
4.7 General Properties
Kaczka et al. (1950) have demonstrated in in vitro studies
that the reaction of OHB12 with cyanide results in the
displacement of a hydroxyl group by a cyano group to form
cyanocobalamin, which is then excreted in the urine. Thus one
molecule of OHB12 binds one molecule of cyanide. Hydroxocobalamin
has been shown to be an effective antidote for experimental cyanide
poisoning in mice (Mushett et al., 1952), guinea-pigs (Posner et
al., 1976a), baboons (Posner et al., 1976b), and dogs (Rose et al.,
1965).
The antidotal effect of OHB12 is enhanced by the use of
thiosulfate in the same solution. Thiosulfate provides sulfur
radicals, which complex with cyanide to form thiocyanate using the
endogenous cyanide detoxification mechanism rhodanese (thiosulfate
sulfurtransferase). However, the action of thiosulfate may be too
slow to prevent death in serious cases of cyanide poisoning when
administered alone. The combination of OHB12 and thiosulfate was
shown to be more effective by Mizoule (1966), who studied it
extensively in experimental animals; other authors have reported
similar results (Motin et al., 1970; Luher et al., 1971; Yacoub et
al., 1974; Jouglard et al., 1974; Friedberg & Shukla, 1975; Racle et
al., 1976; Bismuth et al., 1984).
4.8 Animal Studies
4.8.1 Pharmacokinetics
No data are available.
4.8.2 Pharmacodynamics in the presence of the toxin
The antidotal action of OHB12 has been studied in animals,
firstly in the case of medium-term intoxication (Haguenoer et al.,
1975), and secondly in the prophylactic treatment of cyanide
intoxication (Posner et al., 1976a).
In the study by Haguenoer et. al. (1975), five groups of Wistar
male rats (mean weight, 330g) were used as follows:
* group 1 received injections of sodium chloride (0.9%) in
water (control group)
* group 2 received 1 ml of 50 mg/ml acetonitrile
* group 3 received 1 ml of 50 mg/ml acetonitrile plus 1 ml
of 5 mg/ml OHB12 (6 h later)
* group 4 received 1 ml of 1.12 mg/ml potassium cyanide
(KCN)
* group 5 received 1 ml of 1.12 mg/ml KCN plus 1 ml of
5 mg/ml OHB12 (6 h later).
In all, 28 intraperitoneal injections were given over a 6-week
period. Rats treated with OHB12 (groups 3 and 5) excreted less
free CN- in the urine than the others (groups 2 and 4); the
excretion of combined CN- was higher for groups 3 and 5. Urinary
excretion was higher in the case of KCN than acetonitrile. However,
a significant proportion of acetonitrile and cyanhydric acid (formed
from acetonitrile and KCN) was excreted by the lungs, and a large
amount of thiocyanate was excreted in the faeces.
In the second study (Posner et al., 1976a), nine groups of
healthy baboons (weighing 20 to 30 kg) were anaesthetized and
received nitroprusside by infusion, either alone (four animals) or
in combination with OHB12 (22.5 mg B12 per 1 mg nitroprusside)
(five baboons).
The infusion was continued for up to 2 h or until 500 mg of
nitroprusside had been administered. Cyanide concentrations
increased and the animals developed a severe metabolic acidosis.
OHB12 infused simultaneously with nitroprusside significantly
reduced the increase in cyanide concentrations and eliminated the
development of metabolic acidosis.
In a third study (Höbel et al., 1980), sodium nitroprusside
(SNP) and OHB12 were infused over 4 h in conscious rabbits in
molar ratios of 1:4, 1:5, or 1:8. Control animals received OHB12
only. Sodium thiosulfate was infused with SNP at molar ratios of
1:4, 1:5, and 1:10. SNP provoked a severe acidosis which was not
corrected by the lowest dose of OHB12: three deaths occurred among
ten animals. When the 1:5 ratio was administered, the acidosis was
less marked, but three out of seven animals succumbed. The highest
dose (1:8) prevented acidosis, but not death, in three out of eight
animals. All doses of OHB12 caused histological changes in the
liver, myocardium, and kidney. By contrast, sodium thiosulfate was
completely effective as an antidote in this study and did not give
rise to histological changes.
4.8.3 Toxicology
OHB12 has weak intrinsic toxicity and may be administered
safely in large doses (Mizoule, 1966).
4.8.3.1 Acute toxicity
The acute toxicity of OHB12 has been tested in mice and
rabbits. A solution containing 0.4-0.75 g or l g/kg was
administered to Swiss strain mice (0.20 ml per 20 mg body weight)
without any toxic effects either immediately or a few days later.
Doses of OHB12 (50 and 100 mg/kg body weight) were injected
intravenously into five rabbits (weighing 2-3 kg) without causing
side effects either immediately or 3 days later (Mizoule, 1966).
4.8.3.2 Sub-acute and chronic toxicity
The sub-acute and chronic toxicity of hydroxocobalamin has been
tested in rats and rabbits. Twelve young male Wistar rats (weight
43-53 g) received three subcutaneous injections weekly for a period
of 82 days. Six of these rats were treated with 50 mg OHB12
injections, while six received injections of sodium chloride (0.9%)
in water. No significant differences were reported in the behaviour
of the rats or in the haematological results. Rabbits received one
injection of OHB12 (50 mg/kg) every other day for a period of 10
days. No toxicity was reported (Mizoule, 1966).
4.9 Volunteer Studies
Data not available.
4.10 Clinical Studies
After intravenous infusion, OHB12 is bound sparingly to a
specific globulin transport plasma protein, transcobalamin II
(Herbert & Sullivan, 1964). Lesser amounts are bound to the storage
protein transcobalamin I (a glycoprotein) and to transcobalamin III
(an intermediate glycoprotein). A small amount may be free or very
loosely bound to protein. In the fasting state, the majority of
circulating vitamin B12 is bound to transcobalamin, and it is
stored principally in the liver and bone marrow.
OHB12 has a very short half-life (5 min), being rapidly
metabolized and excreted (Vesey & Cole, 1981; Cottrell et al.,
1982). Up to 50% of an administered dose of OHB12 is excreted
unchanged in the urine, which becomes red-orange in colour following
a dose of 4 g OHB12 (Goodman & Gilman, 1975).
4.11 Clinical Studies - Case Reports
Only a few studies in man have included measurement of free and
total cyanide concentrations. These include 12 cases of acute
cyanide poisoning (Baud et al., 1986; Danel et al., 1986a);
several cases of sodium nitroprusside poisoning in which OHB12 was
used prophylatically (Cottrell et al., 1982), and one case of
combined cyanide and carbon monoxide poisoning (Baud et al., 1987).
Baud et al. (1986) reported the case of a 55-year-old worker,
exposed to propionitrile by inhalation and dermal exposure, who was
treated with a combination of OHB12 and thiosulfate. The patient
rapidly lost consciousness: 25 min after exposure he was comatose
and unresponsive with a pulse rate of 90 beats/min and a blood
pressure of 17.3/9.3 kPa (130/70 mmHg). Laboratory investigations
revealed no abnormalities except for a base deficit of 14-16 mmol/l
on arterial blood gas analysis. The serum lactate concentration was
10 mmol/l. The patient received 4 g OHB12 and 8 g thiosulfate
(Trousse anticyanure, Laboratoire Anphar Rolland) intravenously in
just under 30 min. During infusion of the antidote, the patient
regained consciousness and became oriented. Complete reversion of
CNS depression and normalization of vital signs were observed during
the next hour.
Table 3. Blood cyanide and thiocyanate concentrations
Time after Cyanide Thiocyanate
intoxication
2 h 5.71 mg/l undetectable
2.5 h 0.93 mg/l 21.1 mg/l
(at the end of the
antidote infusion)
7 h 1.00 mg/l baseline level
a Personal communication by V. Danel, L. Barret, and J.L. Debru
Regional Hospital Centre and University, Grenoble, France
concerning four hydrogen cyanide intoxications treated with
OHB12 and sodium hyposulfite.
Baud et al. (1989) studied the toxicokinetics of OHB12 and
cyanocobalamin (CNB12),which were measured simultaneously in cases
of cyanide intoxication treated with OHB12. Plasma cobalamin
levels were measured using a differential spectrophotometric assay.
Whole blood cyanide levels were measured, using a
microdiffusion/colormetric assay, in samples taken before
administration of OHB12 by infusion (5 g over 30 min).
Source of cyanide Bromo Sodium Mercurous Smoke inhalation
cyanide cyanide cyanide
Route of absorption lung orally orally lung lung lung lung
Initial blood cyanide 13 260 217 22 123 129 208
(µmol/l)
Peak OHB12 (µmol/l) 325 15 83 514 74 131 0
Peak CNB12 (µmol/l) 39 275 226 0 215 221 259
After completion of the OHB12 infusion, low levels of OHB12
were observed concurrently with high levels of CNB12 in patients
with high whole blood cyanide levels, but not in those with low
cyanide concentrations. These findings suggest that OHB12 acts
rapidly as a cyanide antidote.
Cottrell et al. (1982) studied two groups of patients in whom
cyanide toxicity was induced by nitroprusside. Group I received
nitroprusside alone, while group II received nitroprusside and
OHB12. Red cell and plasma cyanide concentrations were,
respectively, 83.44% (±23.12) and 3.51 (±1.01) mg per 100 ml after
administration of nitroprusside alone and 33.18% (±17.29) and 2.18%
(±0.65) mg per 100 ml after administration of nitroprusside with
OHB12. OHB12 infusion resulted in lower blood cyanide
concentrations and base deficit than in the untreated group. The
dose of nitroprusside used in each group did not differ
significantly.
Following inhalation of fumes from a firm making plastics, a
man became poisoned by both cyanide and carbon monoxide (Baud et
al., 1989). On admission, the whole-blood cyanide concentration was
3.6 mg/l. He immediately received 4 g OHB12 and 8 g sodium
thiosulfate by intravenous infusion. A second injection had to be
administered 3 h after admission because of clinical relapse, again
with a satisfactory response.
4.12 Summary of Evaluation
4.12.1 Indications
* In the treatment of pernicious anaemia and vitamin B12
deficiency states by intramuscular administration.
* In cyanide poisoning as an antidote:
a) as therapeutic treatment after exposure to
acetonitrile, propionitrile, or potassium or sodium
cyanides by oral, inhalational, or dermal routes;
b) as prophylactic treatment during sodium nitroprusside
infusion.
4.12.2 Advised route and dosage
OHB12 is usually given intravenously in a 5% solution of
dextrose in water. The normal adult dose is 4 g OHB12, but the
dose may be increased in massive poisoning.
4.12.3 Practical advice
* To maintain normal blood pH and isotonicity, the OHB12
must be injected in more than 220 ml of 5% dextrose.
* OHB12 must not be administered with reducing or basic
substances (see section 4.3.8).
* When OHB12 is administered as a prolonged infusion,
aluminium foil should be used to protect it from light.
4.12.4 Side effects
* The most common side effect is an orange-red discoloration
of the skin, mucuous membranes, and urine that lasts about
12 h but is not consequential.
* Allergic reactions to OHB12 have been reported (Dally &
Gaultier, 1976).
4.13 Model Information Sheet
4.13.1 Uses
Hydroxocobalamin is used for the treatment of pernicious
anaemia and vitamin B12 deficiency states. In the treatment of
cyanide intoxication, hydroxocobalamin is indicated in two
circumstances. Firstly, it is used for the treatment of exposure to
hydrogen cyanide, inorganic cyanide salts, and acetonitrile and
propionitrile. Secondly, hydroxocobalamin is used prophylactically
to prevent cyanide intoxication as a result of sodium nitroprusside
given by infusion.
4.13.2 Dosage and route
Hydroxocobalamin is given intravenously in a 5% dextrose
solution. The usual adult dose is 4 g, which may be increased in
cases of massive cyanide poisoning.
4.13.3 Precautions/contraindications
Hydroxocobalamin should be diluted in more than 220 ml of 5%
dextrose solution. It must not be administered with reducing
substances or basic agents. In the case of prolonged infusion, the
solution must be protected from light. The possibility of an
anaphylactoid reaction to hydroxocobalamin should be borne in mind.
4.13.4 Adverse effects
The most common side effect is an orange-red discoloration of
the skin, mucous membranes, and urine that lasts for approximately
12 h. Allergic reactions have been reported. Histological
abnormalities in the liver, myocardium, and kidney of rabbits with
nitroprusside intoxication treated with hydroxocobalamin have been
described (Höbel et al., 1980).
4.13.5 Use in pregnancy and lactation
These aspects are seldom discussed in the literature, and are
of little relevance in this context. In a life-saving situation,
the dosage recommended above should not be modified during pregnancy
or lactation.
4.13.6 Storage
Hydroxocobalamin must be kept in the dark, in a refrigerator at
4 °C, for not more than 3 years.
4.14 References
Baud FJ, Bismuth C, Astier A, Djeghour H, & Aubriot D (1986)
Toxicocinétique des cyanures et des thyocyanates lors d'un accident
professionel au propionitrile. Arch Mal Prof Méd Trav Sécur Soc,
47(2): 85-86.
Baud FJ, Astier A, Toffis V, & Barriot P (1987) Plasma kinetics of
hydroxocobalamin and cyanocobalamin in a treated cyanide poisoning.
Paris, Hôpital Fernand Widal (Unpublished data).
Baud FJ, Toffis V, Certain B, Muzynski J, Astier A, & Bismuth C
(1989) Hydroxocobalamine antidote des cyanures. Vérification
cinétique de sa transformation en cyanocobalamine dans l'organisme
intoxiqué. Thérapie, 44: 469.
Bismuth C, Cantineau JP, Pontal P, Baud F, Garnier R, Poulos L, &
Bolo A (1984) Priorité de l'oxygénation dans l'intoxication
cyanhydrique: A propos de 25 cas. J Toxicol Méd, 4(2): 107-121.
British Pharmacopoeia (1980) London, Her Majesty's Stationery
Office.
Cottrell JE, Casthely P, Brodie JD, Patel K, Klein A, & Turndorf H
(1982) Prevention of nitroprusside-induced toxicity with
hydroxocobalamin. New Engl J. Med, 298: 809-811.
Dally S & Gaultier M (1976) Choc anaphylactique dû à
l'hydrocobalamine. Nouv Presse Méd, 30: 1917.
Evans CL (1964) Cobalt compounds as antidotes for hydrocyanic acid.
Br J Pharmacol, 23: 455-475.
Friedberg KD & Shukla UR (1975) The efficiency of aquocoblamine as
an antidote in cyanide poisoning when given alone or combined with
sodium thiosulfate. Arch Toxicol, 33: 103-113.
Goodman LS & Gilman A ed. (1975) The pharmacological basis of
therapeutics, 5th ed. New York, MacMillan Publishing Co., Inc., p
1334.
Haguenoer JM, Dequidt J, & Jacquemont MC (1975) Intoxications
expérimentales par l'acétonitrile. 4ème note: Influence de
l'hydroxocobalamine sur l'intoxication à moyen terme. J Eur Toxicol,
2: 113-121.
Herbert V & Sullivan LW (1964) Activity of coenzyme B12 in man.
Ann NY Acad Sci, 112: 855-870.
Höbel M, Engeser P, Nemeth L, & Pill J (1980) The antidote effect of
thiosulphate and hydroxocobalamin in formation of nitroprusside
intoxication of rabbits. Arch Toxicol, 46: 207-213.
Italian Pharmacopoeia (1985) 9th ed. Rome, Libreria della Stato.
Jouglard J, Nava G, Botta A, Michel-Manuel C, Poyen B, & Mattei JL
(1974) A propos d'une intoxication aiguë par le cyanure de potassium
traitée par l'hydroxocobalamine. Marseille Méd, 12: 617-624.
Kaczka EA, Wolf DE, Kuehl FA Jr, & Folkers K (1950) Vitamin B12:
reactions of cyano-cobalamin and related compounds. Science, 112:
354-355.
Luher F, Dusoleil P, & De Montgros J (1971) Action de
l'hydroxocobalamine à dose massive dans l'intoxication aiguë au
cyanure: A propos d'un cas. Arch Mal Prof, 32: 683-686.
Martindale (1989) In: Reynolds JEF ed. The extra pharmacopoeia, 29th
ed. London, The Pharmaceutical Press, pp 1260-1261.
Mizoule J (1966) Etude de l'action antidote de l'hydroxocobalamine à
l'égard de l'intoxication cyanhydrique. Paris, Université de Paris
(Thèse de Doctorat en Pharmacie).
Motin J, Bouletreau P, & Rouzioux JM (1970) Intoxication
cyanhydrique grave traitée avec succès par l'hydroxocobalamine. J
Méd Strasbourg, 1: 717-722.
Mushett CW, Kelly KL, Boxer EG, & Rickards JC (1952) Antidotal
efficacy of vitamin B12a (hydroxocobalamin) in experimental
cyanide poisoning. Proc Soc Exp Biol Med, 81: 234-237.
Pharmacopée Française (1988) 10th ed. Paris, La Commission nationale
de Pharmacopée.
Posner MA, Tobey RE, & McElroy H (1976a) Hydroxocobalamin therapy of
cyanide intoxication in guinea-pigs. Anaesthesiology, 44(2):
157-160.
Posner MA, Rodkey FL, & Tobey RE (1976b) Nitroprusside induced
cyanide poisoning: Antidotal effect of hydroxocobalamin.
Anaesthesiology, 44(4): 330-335.
Racle JP, Chausset R, & Dissait F (1976) L'intoxication cyanhydrique
aiguë. Aspects biologiques, toxicologiques, cliniques et
thérapeutiques. Rev Méd Clermont-Ferrand, 5: 371-382.
Rose CL, Worth RM, & Chen KK (1965) Hydroxocobalamin and acute
cyanide poisoning in dogs. Life Sci, 4: 1785-1789.
United States Pharmacopeia (1985) 21st ed. Rockville, Maryland,
United States Pharmacopeial Convention, Inc.
Vesey GJ & Cole PV (1981) Cyanide antagonist (letter). Can Anaesth
Soc J, 28: 290.
Yacoub M, Faure J, Morena H, Vincent M, & Faure H. (1974)
L'intoxication cyanhydrique aiguë: Données actuelles sur le
métabolisme du cyanure et le traitement par l'hydroxocobalamine.
J Eur Toxicol, 7: 22-29.
5. DICOBALT EDETATE (KELOCYANOR)
5.1 Introduction
The ability of cyanide to form complexes with cobalt is an
example of the propensity of cyanide to form stable complexes with
many transition metals (Sharpe, 1976). It is a property of cyanide
that has been known since the last century, but there is still room
for disagreement about the precise nature of the complex formed in
vivo when cobalt is used to treat cyanide poisoning. In vitro,
a complex of one cobalt and five cyanide atoms is formed on addition
of potassium cyanide to cobalt (II) salts (Cotton & Wilkinson,
1966).
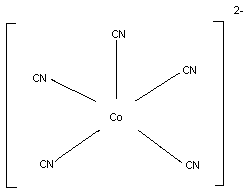 However, in the studies carried out in vivo by Evans (1964),
it was suggested that inorganic cobalt salts could bind six moles of
cyanide for each mole of cobalt. Evans further proposed that there
was a two stage reaction, six moles of cyanide reacting with one
mole of cobalt, forming first the cobaltocyanide ion, and then the
cobalticyanide ion.
Co2+ + 6 CN- --> [Co (CN)6] 4-
|
|--> e-
|
v
[Co (CN)6] 3-
The cobalticyanides are not very toxic, much less so than the
same amount of free cyanide, and have an LD50 in the region of
1 g/kg.
Although it was known that cobalt and cyanide formed stable
complexes, it was generally thought that inorganic cobalt salts were
too toxic for use in humans as cyanide antidotes. In particular,
those most investigated, the cobalt (II) salts, were known to be
toxic to the heart, liver, and kidney (Speijers et al., 1982). For
a long time, during which the preferred treatment for cyanide
poisoning was sodium nitrite and sodium thiosulfate, interest in the
anticyanide activity of cobalt compounds dwindled. A rekindling of
interest in cobalt compounds, especially organic complexes, was
brought about by the discovery by Mushett et al. (1952) that the
complex organocobalt vitamin, hydroxocobalamin, possessed
anticyanide activity in mice. Experimental work, notably by Paulet
(1957, 1958), led to the introduction of dicobalt edetate
(Kelocyanor).
5.2 Name and Chemical Formula
Kelocyanor is the proprietary name for a solution containing
dicobalt edetate and glucose. Dicobalt edetate has not been
crystallized. The structural formula was believed to be that of the
monocobalt salt of a monocobalt edetate anion (Fig. 4), but there is
reason to think that this does not fully represent the structure of
the active ingredient of Kelocyanor (see below). The CAS number is
36499-65-7, the INN is dicobalt edetate, and the IUPAC name is
dicobalt ethylenediamine- NNN'N'-tetraacetate. Other names are
cobalt edetate, cobalt tetracemate and cobalt EDTA.
However, in the studies carried out in vivo by Evans (1964),
it was suggested that inorganic cobalt salts could bind six moles of
cyanide for each mole of cobalt. Evans further proposed that there
was a two stage reaction, six moles of cyanide reacting with one
mole of cobalt, forming first the cobaltocyanide ion, and then the
cobalticyanide ion.
Co2+ + 6 CN- --> [Co (CN)6] 4-
|
|--> e-
|
v
[Co (CN)6] 3-
The cobalticyanides are not very toxic, much less so than the
same amount of free cyanide, and have an LD50 in the region of
1 g/kg.
Although it was known that cobalt and cyanide formed stable
complexes, it was generally thought that inorganic cobalt salts were
too toxic for use in humans as cyanide antidotes. In particular,
those most investigated, the cobalt (II) salts, were known to be
toxic to the heart, liver, and kidney (Speijers et al., 1982). For
a long time, during which the preferred treatment for cyanide
poisoning was sodium nitrite and sodium thiosulfate, interest in the
anticyanide activity of cobalt compounds dwindled. A rekindling of
interest in cobalt compounds, especially organic complexes, was
brought about by the discovery by Mushett et al. (1952) that the
complex organocobalt vitamin, hydroxocobalamin, possessed
anticyanide activity in mice. Experimental work, notably by Paulet
(1957, 1958), led to the introduction of dicobalt edetate
(Kelocyanor).
5.2 Name and Chemical Formula
Kelocyanor is the proprietary name for a solution containing
dicobalt edetate and glucose. Dicobalt edetate has not been
crystallized. The structural formula was believed to be that of the
monocobalt salt of a monocobalt edetate anion (Fig. 4), but there is
reason to think that this does not fully represent the structure of
the active ingredient of Kelocyanor (see below). The CAS number is
36499-65-7, the INN is dicobalt edetate, and the IUPAC name is
dicobalt ethylenediamine- NNN'N'-tetraacetate. Other names are
cobalt edetate, cobalt tetracemate and cobalt EDTA.
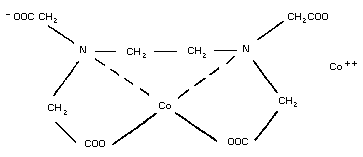 For reasons discussed in the text, this may only be an approximation
to the true structure of the material in Kelocyanor.
5.3 Physico-chemical Properties
The usual preparation of dicobalt edetate is Kelocyanor.a
Injectable Kelocyanor is a clear violet solution, produced in clear
glass 20-ml ampoules, of pH 4.0-7.5. It contains 0.196-0.240
g/100 ml free cobalt and 1.35-1.65 g/100 ml dicobalt edetate, as
well as 4 g glucose per ampoule. The refractive index of the
solution in the ampoule is 1.3638.
The optical rotation of Kelocyanor is + 9.25°: the optical
rotation of a dextrose solution of the same dextrose concentration
as Kelocyanor is +11°. Therefore the net optical rotation
attributable to the cobalt edetate is -1.75°. This would give a
specific rotation for dicobalt edetate of -116.7°. The relative
molecular mass of dicobalt edetate is 408. No information is
available on the specific gravity.
5.4 Synthesis
Dicobalt edetate is made by adding cobalt carbonate to sterile
water and then adding ethylenediaminetetraacetic acid. After the
addition of glucose, the resultant solution is cleared with
activated charcoal. Sterility and pyrogen tests are performed to
the requirements of the European Pharmacopoeia (1980). The volume
of solution in the ampoules is checked using method B (British
Pharmacopoeia, 1980). The material is not checked for the absence
of activated charcoal, but this would be expected to be removed
during filtration through a membrane with a pore diameter of
0.22 µm.
5.4.1 Source of materials
5.4.1.1 Cobalt carbonate
The current supplier to both the French and British drug
companies is Prolabo, BP No 200, 75526 Paris, Cedex 11, France. The
specification is as follows:
appearance: fine wine-coloured powder
identification: cobalt carbonate confirmed
loss on drying: about 14%
assay: about 50% cobalt
a L'Arguenon, Société d'Etudes et de Recherches Biologiques,
53 Rue Villiers de l'Isle Adam, 75020 Paris, France, UK
suppliers, Lipha Pharmaceuticals Ltd, Harrier House, Yiewsley,
Middlesex UB7 7QG.
5.4.1.2 Ethylenediaminetetraacetic acid (EDTA)
The current supplier is BDH Ltd, Poole, Dorset, BH12 4NN,
England. The material complies with the US National Formulary (see
USNF, 1985).
5.4.1.3 Glucose
This complies with the European Pharmacopoeia.
5.5 Analytical Methods
5.5.1 Free cobalt
In duplicate, pipette 5.0 ml of the solution in the ampoule
into a 250- ml conical flask. Add 100 mg of murexide compound
mixture and 100 ml water. Stir using a magnetic stirrer. Add
dilute ammonia drop by drop until the solution just turns yellow
(usually 1 drop). Titrate with disodium edetate (0.02 mol/l) to a
violet end-point. During the titration, the pH of the solution
falls drastically and it turns red. It is necessary to add 1 drop
of dilute ammonia to restore the yellow colour. Check the end point
of the titration with 1 drop of ammonia.
To prepare the murexide mixture, grind 10 mg murexide and
990 mg sodium chloride in a pestle and mortar. The dilute ammonia
solution consists of 5.5 ml concentrated ammonia diluted to 100 ml
with water.
The following equation is used to calculate the concentration
of free cobalt (g/100 ml):
T x 1.179 x F x 20
1000
where T = titre (ml)
F = factor for disodium edetate at 0.02 mol/l
1 ml disodium edetate (0.02 mol/l) equiv. 1.179 mg cobalt
5.5.2 Dicobalt edetate
In duplicate, pipette 5.0 ml of the solution in the ampoules
into a 250-ml conical flask. Add 0.65 g potassium cyanide, stir,
and allow to stand for 5 min. Add 85 ml of water, 10 ml lead
acetate solution, and 1 ml glacial acetic acid. Stir for 15 min,
using a magnetic stirrer. Add 100 mg xylenol orange compound
mixture and titrate slowly with disodium edetate (0.02 mol/l) to a
clear yellow end-point (T1). Carry out a blank titration,
omitting the sample solution (T2).
Xylenol orange compound mixture is prepared by grinding 10 mg
xylenol orange with 990 mg potassium nitrate in a pestle and mortar.
The lead acetate solution consists of 1.5 g lead acetate dissolved
in water and make up to 100 ml. The following equation is used to
calculate the concentration of dicobalt edetate (g/100 ml):
(T2-T1) x F x 4.06
25
where T1 = sample titre
T2 = blank titre
F = factor for disodium edetate (0.02 mol/l)
5.5.3 Analysis in biological fluids
Since dicobalt edetate has not been fully characterized, it is
difficult to say whether analysis would be meaningful. Methods are
available for the analysis of cobalt in biological fluids by
spectrophotometry or atomic absorption flame photometry (Thiers et
al., 1955; Mulford, 1966; Christian, 1969; Lewis et al., 1985).
Dicobalt edetate interferes with the Feldstein & Klendshoj
(1954) method of analysis of biological fluids for cyanide
(Ballantyne & Marrs, 1987). See chapter 10 for further details.
5.6 Stability and Shelf-life
The shelf-life is 3 years at 25 °C, and the material should be
stored in the dark. The drug has been stored for up to 3 years at
room temperature 20-25, 30 and 37 °C (Lipha Pharmaceuticals,
1987)a, the batches having been tested at regular intervals
(12 months maximum) for compliance with the finished product
specification with respect to appearance, extractable volume, pH,
and free cobalt and dicobalt edetate content. The only deviation
noted was that the colour of the solution became lighter at 30 °C
and above. All the other variables remained within the
specification. If stored in the light, the drug bleaches in about
one month. The nature of the chemical change that brings about the
loss of colour is not known.
a Personal communication from Lipha Pharmaceuticals Ltd, West
Drayton, Middlesex, United Kingdom.
5.7 General Properties
Cyanide blocks intracellular respiration by binding to
cytochrome oxidase. Cobalt forms a stable complex with cyanide, the
effect being direct and peripheral (Mercker & Bastian, 1959).
Although some cobalt compounds are reputed to inhibit methaemoglobin
reductase (Hagler & Coppes, 1982), it is unlikely that
methaemoglobinaemia contributes meaningfully to the anticyanide
action of dicobalt edetate.
5.8 Animal Studies
5.8.1 Pharmacokinetics
Formal pharmacokinetic studies have not been carried out on
dicobalt edetate. Some of the effects observed with cobalt
compounds, including dicobalt edetate, are probably centrally
mediated (Bartelheimer, 1962a). Thus, it is likely that after
Kelocyanor injection, cobalt or dicobalt edetate crosses the
blood-brain barrier. In the mouse, the cyanide-cobalt complex is
excreted in the urine (Frankenberg & Sörbo, 1975).
5.8.2 Pharmacodynamics
5.8.2.1 Efficacy in animals
The introduction of dicobalt edetate followed from the work of
Paulet (1957, 1958, 1960a,b, 1961, 1965) and Paulet et al. (1960),
who studied the antidotal effects and toxicity of various cobalt
compounds. The aim of studies on cobalt antidotes at this time was
to find a compound that retained the ability of cobalt to bind
cyanide, yet lacked its toxic effects. The compounds investigated
by Paulet (1960a,b) included certain cobalt (II) salts, namely the
chloride, glutamate, and gluconate. Paulet (1960a) also studied the
cobalt chelates, dicobalt edetate, cobalt histidine, and disodium
monocobalt edetate and found that the salts, as well as all except
the last named of the chelates, possessed appreciable anticyanide
activity. Dicobalt edetate and cobalt histidine were the most
promising, and in one study dicobalt edetate seemed preferable
(Paulet, 1960a, 1961). This study was undertaken in anaesthetized
dogs infused with sodium cyanide (0.1 mg/kg per min): both dicobalt
edetate and cobalt histidine were capable of resuscitating the dogs
in secondary apnoea, but the dose of cobalt histidine required was
considerably higher than that of dicobalt edetate. In the same
reports, the effective doses of the two compounds were compared with
their respective intraperitoneal LD50 values in mice, the result
favouring dicobalt edetate. However, if the dog results had been
tabulated against the rat intravenous LD50 values (Tauberger &
Klimmer, 1963), cobalt histidine might have been preferred, this
antidote having a much higher intravenous LD50 than dicobalt
edetate.
Evans (1964) investigated the stoichiometry of the reaction
between dicobalt edetate or other cobalt compounds on the one hand,
and cyanide on the other. He studied their antidotal effects using
various doses of both toxicant and treatment in both mice and
rabbits, designing his experiments in such a way that it was
possible to calculate the cyanide/cobalt molar ratios above which
the antidote would no longer be efficacious. He found that, while
most cobalt (II) salts would be effective against six moles of
cyanide/mole cobalt, dicobalt edetate only possessed efficacy at
molar ratios of up to two. Whilst on a mass basis, dicobalt
edetate is about half as toxic as cobalt (II) salts, on a molar
basis it is not conspicuously less toxic than cobalt acetate. It
had been thought that dicobalt edetate was the monocobalt (II) salt
of a monocobalt edetate anion, one cobalt being completely
unavailable for cyanide binding by virtue of its complexing with
EDTA, the other being fully available. In fact the study of Evans
(1964) suggested that one cobalt is completely unavailable whilst,
of the coordination sites of the second, only two are available to
bind cyanide. In that case, the structure given in Fig 4 does not
fully describe the structure of dicobalt edetate. It seems likely,
however, that the second cobalt atom is required for anticyanide
activity, since disodium monocobalt edetate, which is a compound of
very low toxicity (Eybl et al., 1959), is almost ineffective as a
cyanide antidote (Paulet, 1958). It may be that the same property,
ionizability, is responsible for cyanide binding and for cobalt
toxicity. In that case, complexing cobalt would decrease the
therapeutic effect in parallel with the toxicity. That this is not
the case is suggested by, firstly, the lack of toxicity of
hydroxocobalamin, which is nevertheless a successful cyanide
antidote and, secondly, the many years of successful use of
dicobalt edetate.
5.8.2.2 Comparison of dicobalt edetate with other compounds
A number of research groups have compared cobalt edetate with
other cyanide antidotes, from the point of view both of antidotal
efficacy (Table 4) and also of toxicity. For example, using dogs
and rabbits, Paulet (1960a, 1961) compared the cobalt chelate with
sodium nitrite and found the former superior. He also carried out a
comparison with both elements of the sodium nitrite/thiosulfate
treatment, again demonstrating that the cobalt compound was more
effective. Terzic & Milosevic (1963) compared sodium nitrite and
dicobalt edetate. They studied the effective doses of the two
compounds, as well as their LD50 values, and concluded that cobalt
edetate was superior. Unfortunately their study was conducted in
mice: these animals especially, but also other small laboratory
animals, are extremely insensitive to methaemoglobin-producing
chemicals by virtue of the very high activity of NADH-linked
methaemoglobin reductase in murine erythrocytes (Calabrese, 1983).
This difference renders extrapolation of these results to man almost
impossible. Two research groups have attempted to compare dicobalt
edetate with 4-DMAP in dogs. Klimmek et al. (1979a) found better
survival with 4-DMAP when the antidotes were given 4 min after the
poisoning but similar antidotal efficacy when they were given 1 min
afterwards. Marrs et al. (1985) found that dicobalt edetate
successfully treated up to about three times the LD50 of cyanide,
when given at the moment of apnoea, whilst 4-DMAP ensured survival
at about six time the LD50 under the same conditions.
5.8.2.3 Interactions with other drugs
The only interactions that have been studied in animals are
those with other cyanide antidotes. The effect, in experimental
animals, is generally additive. This is despite the theoretical
risk that cobalt toxicity might be exacerbated by other antidotes
that prevent cyanide from binding to cobalt. Antidotal combinations
including dicobalt edetate have been studied by Evans (1964) and
Frankenberg & Sörbo (1975).
Table 4. Studies of the efficacy of dicobalt edetate
compared with other cyanide antidotes
Comparison of dicobalt Species Author
edetate with
Sodium nitrite Dog Paulet (1960 a,b)
Sodium nitrite/thiosulfate Dog Paulet (1960 a,b)
Sodium nitrite Mouse Terzic & Milosevic (1963)
4-DMAP Dog Klimmek et al. (1979a)
4-DMAP Dog Marrs et al. (1985)
Thiosulfate/rhodanese Rabbit Atkinson et al. (1974)
Other cobalt antidotes Mouse/Rabbit Evans (1964)
5.8.3 Toxicology
Extensive toxicology was not carried out on dicobalt edetate
before its introduction. Published information is largely confined
to acute toxicity testing, but it is believed that the
organ-specific toxicity is similar to that of cobalt salts and that
it is ameliorated by chelation (Paulet, 1960b).
5.8.3.1 In vitro studies
Dicobalt edetate gives negative results in the Ames test
(Morris, 1987)
5.8.3.2 Acute toxicity studies
The acute toxicity of dicobalt edetate has been studied
exclusively in small laboratory animals (Table 5). An interesting
feature is that the acute toxicity is decreased by the presence of
glucose or of cyanide. Thus, Paulet (1960a,b, 1961) observed that,
in the mouse, the intravenous LD50 of dicobalt edetate (50 mg/kg)
was increased threefold by giving it in hypertonic glucose. The
LD50 was increased to 82 mg/kg or 103 mg/kg, respectively, by the
simultaneous administration of 2.5 or 5 mg NaCN/kg.
5.8.3.3 Repeated dose toxicity
Bartelheimer (1962b) gave intraperitoneal injections of cobalt
edetate daily for 14 days at doses of 30, 40, or 50% of the LD50
to rats, surviving animals being observed for 4 weeks. One of six
and two of six of the high and middle groups, respectively, died,
whilst none of the low-dose group did so. The majority of the rats
in the two higher groups suffered diarrhoea and weight loss,
although some recovery was seen in the latter half of the exposure
period. Autopsy and histological examination of the dead animals
showed necrosis of the intestinal mucous membrane. No pathological
changes were seen in the liver, kidney, or heart. Late effects were
not observed in the survivors.
Table 5. The acute toxicity of dicobalt edetate
Species Route LD50 (mg/kg) Reference
Mouse iv 50 Paulet (1961)
Mouse iv 71 Evans (1964)
Mouse ip 213 Paulet (1961)
Mouse ip 225 Terzic & Milosevic (1963)
Rat iv 43 Tauberger & Klimmer (1963)
Rat ip 100 Bartelheimer (1962b)
5.8.3.4 Circulatory effects in dogs
Klimmek et al. (1979b), during their studies comparing dicobalt
edetate with 4-dimethylaminophenol (4-DMAP), found that the former
caused circulatory depression and hyperventilation. This was
accompanied by metabolic acidosis.
5.8.3.5 Other toxicity studies
No information is available on reproductive toxicology or
carcinogenicity. No mammalian cell or in vivo mutagenicity test has
been published.
5.9 Volunteer Studies
No human volunteer studies have been carried out.
5.10 Clinical Trials
Clinical trials have not been undertaken.
5.11 Clinical Studies - Case Reports
5.11.1 Successful use
There are a number of case reports of the successful use of
dicobalt edetate against different types of cyanide poisoning,
including inhalation of HCN (e.g. Bain & Knowles, 1967; Nagler et
al., 1978) and sodium cyanide (Hillman et al., 1974), and potassium
cyanide ingestion (Hoang The Dan et al., 1981; Klaui et al., 1984).
The use of dicobalt edetate in poisoning with fused sodium cyanide
has also been reported (Bourrelier & Paulet, 1971). The case
reported by Bain & Knowles involved a patient who was semicomatose
but who became fully conscious immediately after the first of two
doses of 10 ml of Kelocyanor: the blood cyanide concentration just
before the antidote injection was found to be 5.1 mg/l. In the
report by Nagler et al. (1978), three workers were poisoned as a
result of the accidental addition of sodium cyanide to an acid bath.
In each case the patients had definite evidence of cyanide
poisoning, yet recovered. A blood cyanide level of 5.25 mg/l was
found in the patient reported by Hoang The Dan et al. (1981), yet
the subject recovered without sequelae.
5.11.2 Use in pregnant women and children
No data exist on the use of dicobalt edetate in pregnant women
or children.
5.11.3 Adverse effects
A number of adverse effects have been reported, generally
following the inappropriate administration of dicobalt edetate. It
has been proposed that their occurrence can be minimized by using
strict clinical criteria for giving the antidote (Bryson, 1978,1987;
Tyrer, 1981; Aw & Bishop, 1981; Peden et al., 1986). Nevertheless,
it has been suggested that adverse reactions to dicobalt edetate may
not be confined to instances of inappropriate administration (Dodds
& McKnight, 1985). Typical case reports of adverse reactions
include that of Froneman (1975), who reported a massive urticarial
reaction affecting the face, eyelids, and lips; it was questioned if
the subject had, in fact, consumed any cyanide. Four adverse
reactions, where the diagnosis of cyanide poisoning was
questionable, were reported by Tyrer (1981). Three of the patients
had a variety of symptoms and signs, including convulsions, oedema
of the face and neck, urticaria, chest pains, dyspnoea, and
hypotension. Hypotension was also reported by Daunderer et al.
(1974) (see section 5.11.4). A patient described by McKiernan
(1980) had a normal blood cyanide level, in spite of severe cyanide
burns. He reacted to Kelocyanor by developing facial and laryngeal
oedema. A case reported by Yacoub et al. (1974) was complicated by
the use of other antidotes; this patient developed urticaria.
5.11.4 Use in combination with other antidotes
Reference has been made above (section 5.8.3) to animal studies
that suggest that the use of dicobalt edetate in combination with
other antidotes could be beneficial. Thus, Jeretin (1963) used it
with sodium nitrite and thiosulfate, and Daunderer et al. (1974)
successfully resuscitated with 4-DMAP and Kelocyanor a patient who
had ingested about 10 g potassium cyanide.
5.12 Summary of Evaluation
5.12.1 Indications
The only indication for the use of dicobalt edetate is acute
cyanide poisoning. It should not be used except where there is
definite indication of poisoning. Reactions are likely to occur if
the drug is given inappropriately.
5.12.2 Administration
It is recommended that one ampoule should be given over one
min. A second or third may be given in the case of inadequate
response. Because glucose reduces the toxicity of dicobalt edetate
(Paulet et al., 1960; Paulet, 1961), the recommendation is that the
injections should be immediately followed by 50 ml dextrose
(500 g/l).
5.12.3 Other consequential or supportive therapy
See section 1.10
5.12.4 Contraindications
Dicobalt edetate should not be used in suspected cyanide
poisoning unaccompanied by signs of poisoning such as impairment or
loss of consciousness.
5.12.5 Comparison with other antidotes
Dicobalt edetate probably crosses the blood-brain barrier,
while methaemoglobin quite clearly does not. Moreover, both DMAP
and nitrite require follow-up with a sulfur donor; with dicobalt
edetate the cyanide is excreted with cobalt in the urine. In these
two respects dicobalt edetate seems to offer some advantages. It is
in clinical use that reservations have arisen, almost entirely
because of adverse reactions. Whereas occupational health
practitioners seem to be particularly impressed with dicobalt
edetate (Davison, 1969; Bryson, 1987), it is in hospital practice
that problems are likely to occur. The probable reason for this is
that, at cyanide-using facilities, not only is the antidote to hand,
but also occupational health personnel are familiar with the
presentation of cyanide poisoning and the hazards of inappropriate
use of antidotes. A particular problem of this form of poisoning is
that insubstantial or suspected cyanide poisoning may produce panic,
which is mistaken for incipient intoxication. It is then that the
antidote is given (Anon, 1977). Provided it is given only when the
patient is unconscious many of the problems can be avoided. While
in the past it was suggested that in casualty departments Kelocyanor
should be given to all patients with symptoms (Anon, 1977), this
policy is no longer advocated. Were the use of dicobalt edetate to
be confined to cases of unequivocal poisoning with cyanide, it seems
likely that adverse reactions would be far less frequent.
5.13 Model Information Sheet
5.13.1 Uses
Dicobalt edetate is a specific antidote for use in acute
cyanide poisoning.
5.13.2 Dosage and route
It should be given in the form of ampoules of Kelocyanor, which
also contain glucose. One ampoule (300 mg dicobalt edetate) should
be given intravenously over 1 min. If the response is inadequate, a
second ampoule may be given. The use of Kelocyanor should be
followed immediately by 50 ml Dextrose IV infusion (500 g/l).
5.13.3 Precautions/contraindications
Kelocyanor should not be administered in the absence of
cyanide. Fully conscious patients, especially those exposed to HCN
by inhalation, are unlikely to require Kelocyanor.
5.13.4 Adverse effects
These include facial, laryngeal, and neck oedema, chest pain,
vomiting, and rashes.
5.13.5 Use in pregnancy and lactation
There is no experience of use in this situation. In
life-saving situations the recommended dosage should not be
modified.
5.13.6 Storage
This antidote should be stored at a temperature below 25 °C.
The shelf-life is 3 years.
5.14 References
Anon (1977) Which antidote for cyanide? Lancet, 2: 1167.
Atkinson A, Rutter DA, & Sargeant K (1974) Enzyme antidote for
experimental cyanide poisoning. Lancet, 2: 1446.
Aw TC & Bishop CM (1981) Cyanide poisoning. J Soc Occup Med, 31:
173-175.
Bain JTB & Knowles EL (1967) Successful treatment of cyanide
poisoning. Br Med J, 2: 763.
Ballantyne B & Marrs TC (1987) Postmortem features and criteria for
the diagnosis of acute cyanide poisoning. In: Ballantyne B & Marrs
TC ed. Clinical and experimental toxicology of cyanides. Bristol,
United Kingdom, John Wright, pp 217-247.
Bartelheimer EW (1962a) [Analysis of acute cobalt poisoning in
experimental animals.] Naunyn-Schmiedebergs Arch Exp Pathol
Pharmakol, 243: 237-253 (in German).
Bartelheimer EW (1962b) [Differences between the toxic and
cyanide-antagonistic efficacy of cobalt chelate compounds
(Co-histidine and CO2 - EDTA.] Naunyn-Schmiedebergs Arch Exp
Pathol Pharmakol, 243: 254-268 (in German).
British Pharmacopoeia (1980) London, Her Majesty's Stationery
Office, vol. 2, p 579.
Bourrelier J & Paulet G (1971) Intoxication cyanhydrique consécutive
à des brûlures graves par cyanure de sodium fondu. Presse Méd, 79:
1013-1014.
Bryson D (1978) Cyanide poisoning. Lancet, 1: 92.
Bryson D (1987) Occupational cyanide poisoning. In: Ballantyne B &
Marrs TC ed. The clinical and experimental toxicology of cyanides.
Bristol, United Kingdom, John Wright, pp 348-358.
Calabrese EJ (1983) Principles of animal extrapolation. New York,
John Wiley & Sons.
Christian GD (1969) Medicine, trace elements, and atomic absorption
spectroscopy. Anal Chem, 41: 24A.
Cotton FA & Wilkinson G (1966) Advanced inorganic chemistry, 2nd ed.
New York, London, Interscience Publishers, p 869.
Daunderer M, Theml H, & Weger N (1974) [Treatment of prussic acid
poisoning with 4-dimethylaminophenol (4-DMAP).] Med Klin, 69:
1626-1631 (in German).
Davison V (1969) Cyanide poisoning. Kelocyanor - a new treatment.
Occup Health, 21: 306-308.
Dodds C & McKnight C (1985) Cyanide toxicity after immersion and the
hazards of dicobalt edetate. Br Med J, 291: 785-786.
European Pharmacopoeia (1980) 2nd ed. Sainte Ruffine, France,
Maisonneuve SA.
Evans CL (1964) Cobalt compounds as antidotes for hydrocyanic acid.
Br J Pharmacol, 23: 455-475.
Eybl V, Sykora J, & Kocher Z (1959) [The influence of EDTA on the
effects of cobalt in vivo.] Naunyn-Schmiedebergs Arch Exp Pathol
Pharmakol, 236: 199-201 (in German).
Feldstein MA & Klendshoj WJ (1954) The determination of cyanide in
biologic fluids by microdiffusion analysis. J Lab Chem Med, 44:
166-170.
Frankenberg L & Sörbo B (1975) Effect of cyanide antidotes on the
metabolic conversion of cyanide to thiocyanate. Arch Toxicol, 33:
81-89.
Froneman JPC (1975) A first experience in the use of Kelocyanor.
Proc Mine Off Assoc (Johannesburg), 54: 43.
Hagler L & Coppes RI (1982) Inhibition of methemoglobin and
metmyoglobin reduction by cobalt. Biochem Pharmacol, 31: 1779-1782.
Hillman B, Bardhan KD, & Bain JTB (1974) The use of dicobalt edetate
(Kelocyanor) in cyanide poisoning. Postgrad Med J, 50: 171-174.
Hoang The Dan P, Bretsztajn A, Pourriat JL, Lapandry C, & Cupa M
(1981) Un cas d'intoxication aiguë au cyanure. Anesth Anal Réanim,
38: 161-162.
Jeretin S (1963) [A case of successful recovery of a patient
suffering from potassium cyanide poisoning.] Zdrav Vestn, 32: 3-4
(in Slovene).
Klaui H, Russi E, & Bauman PC (1984) [Cyanide poisoning.] Schweiz
Med Wochenschr, 114: 983-989 (in German).
Klimmek R, Fladerer H, & Weger N (1979a) Circulation, respiration
and blood homeostasis in cyanide-poisoned dogs after treatment with
4-dimethylaminophenol or cobalt compounds. Arch Toxicol, 43:
121-133.
Klimmek R, Fladerer H, Szinicz L, Weger N, & Kiese M (1979b) Effects
of 4-dimethylaminophenol and CO2 EDTA on circulation, respiration,
and blood homeostasis in dogs. Arch Toxicol, 42: 75-84.
Lewis SA, O'Haver TC, & Harnley JM (1985) Determination of metals at
the microgram-per-liter level in blood serum by simultaneous
multielement atomic absorption spectrometry with graphite furnace
atomization. Anal Chem, 57: 2-5.
McKiernan MJC (1980) Emergency treatment of cyanide poisoning.
Lancet, 2: 1980.
Marrs TC, Swanston DW, & Bright JE (1985) 4-Dimethylaminophenol and
dicobalt edetate (Kelocyanor) in the treatment of experimental
cyanide poisoning. Hum Toxicol, 4: 591-600.
Mercker H & Bastian G (1959) [Cobalt compounds for prussic acid
detoxification.] Naunyn-Schmiedebergs Arch Exp Pathol Pharmakol,
236: 449-458 (in German).
Morris B (1987) Postmortem features and criteria for the diagnosis
of acute lethal cyanide poisoning In: Ballantyne B & Marrs TC ed.
Clinical and experimental toxicology of cyanides. Bristol, United
Kingdom, John Wright, pp 217-247.
Mulford CE (1966) Solvent extraction techniques for atomic
absorption spectroscopy. At Absorpt Newsl, 5: 88-90.
Mushett CW, Kelly KL, Boxer GE, & Rickards JC (1952) Antidotal
efficacy of vitamin B12a (hydroxo-cobalamin) in experimental
cyanide poisoning. Proc Soc Exp Biol Med (NY), 81: 234, 237.
Nagler J, Provoost RA, & Parizel G (1978) Hydrogen cyanide
poisoning: treatment with cobalt EDTA. J Occup Med, 20: 414-416.
Paulet G (1957) Valeur des sels organiques du cobalt dans le
traitement de l'intoxication cyanhydrique. C R Soc Biol (Paris),
151: 1932-1935.
Paulet G (1958) Intoxication cyanhydrique et chélates de cobalt. J
Physiol (Paris), 50: 438-442.
Paulet G (1960a) L'intoxication cyanhydrique et son traitement.
Paris, Masson SA.
Paulet G (1960b) Les chélates de cobalt dans le traitement de
l'intoxication cyanhydrique. Pathol Biol, 8: 256-266.
Paulet G (1961) Nouvelles perspectives dans le traitement de
l'intoxication cyanhydrique. Arch Mal Prof, 22: 102-127.
Paulet G (1965) Au sujet du traitement de l'intoxication
cyanhydrique par les chélates de cobalt. Urgence, 11: 611-613.
Paulet G, Chary R, Bocquet P, & Fouilhoux M (1960) Valeur comparée
du nitrite de sodium et des chélates de cobalt dans le traitement de
l'intoxication cyanhydrique chez l'animal (chien-lapin)
non-anesthésié. Arch Int Pharmacodyn, 127: 104-117.
Peden NR, Taha A, McSorley PD, Bryden GT, Murdoch IB, & Anderson JM
(1986) Industrial exposure to cyanide: implications for treatment.
Br Med J, 293: 538.
Sharpe AT (1976) The chemistry of cyano complexes of the transition
metals. New York, London, Academic Press.
Speijers GJA, Krajnc EI, Berkvens JM, & Van Logten MJ (1982) Acute
oral toxicity of inorganic cobalt compounds in rats. Food Chem
Toxicol, 20: 311-314.
Tauberger G & Klimmer OR (1963) [Animal experimental studies of some
cobalt compounds after intravenous injection.] Arch Int Pharmacodyn,
143: 219-239 (in German).
Terzic M & Milosevic M (1963) Action protectrice de
l'éthylène-diamine-tétra-acétate-dicobaltique dans l'intoxication
cyanée. Thérapie, 18: 55-61.
Thiers RE, Williams JF, & Yoe JH (1955) Separation and determination
of millimicrogram amounts of cobalt. Anal Chem, 27: 1725-1731.
Tyrer FH (1981) Treatment of cyanide poisoning. J Soc Occup Med, 31:
65-66.
United States National Formulary (1985) 16th ed. Rockville,
Maryland, US Pharmacopeial Convention Inc., p 1559.
Yacoub M, Faure J, Morena H, Vincent M, & Gaure H (1974)
L'intoxication cyanhydrique aiguë. Données actuelles sur le
métabolisme du cyanure et le traitement par hydroxocobalamine. J Eur
Toxicol, 7: 22-29.
6. AMYL NITRITE
6.1 Introduction
Amyl nitrite by inhalation has been used for many years as a
simple first-aid measure for cyanide poisoning. It generates
methaemoglobin and may be administered by lay personnel.
Methaemoglobin combines with cyanide to form non-toxic
cyanmethaemoglobin. However, amyl nitrite is not a powerful
methaemoglobin-forming agent in humans and therefore it has
generally been used in combination with intravenous sodium nitrite.
Of 44 patients poisoned with cyanide, and of which 43 recovered
[Chen et al., 1944 (15 patients); Wolfsie, 1951 (12 patients);
Miller & Toops, 1951 (1 patient); and Chen & Rose, 1952 (16
patients)], only 7 patients were treated with amyl nitrite alone; 2
were treated with amyl nitrite in combination with sodium
thiosulfate.
Recently there has been a resurgence of interest in amyl
nitrite. Artificial respiration with amyl nitrite broken into an
Ambu bag was reported by Vick & Froelich (1985) as being life-saving
in dogs severely poisoned with cyanide before significant formation
of methaemoglobin occurred (Vick & Froelich, 1985). The vasogenic
effects of amyl nitrite may therefore play a role in its antidotal
effect in cyanide poisoning.
6.2 Name and Chemical Formula
Chemical name: Amyl nitrite, Amylis nitris, Nitris amylicus
Molecular formula: C5H11NO2
Structural formula:
Amyl nitrite is a mixture of nitrite esters of
2-methylbutan-l-ol and 3-methylbutan-l-ol.
ONO ONO
| |
CH2 CH2
nitrate ester of | | nitrate ester of
2-methylbutan-l-ol HCCH3 CH2 3-methylbutan-l-ol
| |
CH2 HCCH3
| |
CH3 CH3
It contains not less than 85% and not more than 103% of
C5H11NO2 (Martindale, 1989; United States Pharmacopeia, 1985).
Relative molecular mass: 117.15
Appearance: yellow, transparent fluid
CAS numbers: 8017-89-8 and 110-46-3
IUPAC name: -
6.3 Physio-chemical Properties
Boiling point: 96 °C
Solubility: practically insoluble in water;
miscible with alcohol, chloroform,
ether, and light petroleum
Boiling point: no data available
Acidity: no data available
pK: no data available
Stability in light: protect from light
Thermal stability: it is volatile even at low
temperatures and is liable to
decompose with the evolution of
nitrogen, particularly if it has
become acidic. Flash point 10 °C
(closed-cup test)
Specific gravity: 0.8970-0.880
Refractive index: 1.386-1.389 (Nederlandse
Pharmacopee, 1958)
Explosive properties: amyl nitrite forms an explosive
mixture with air and oxygen. It is
very inflammable and must not be
used where it may be ignited.
6.4 Synthesis
Amyl nitrite is generally manufactured by the action of sodium
nitrate and sulfuric acid on the appropriate alcohols.
6.5 Analytical Methods
6.5.1 Identification
a) To a mixture of 2 drops of amyl nitrite and 2 drops of
water, add 2 ml of sulfuric acid and dilute with water.
The odour of amyl valerate should become apparent.
b) To a few drops of amyl nitrite, add a mixture of 1 ml of
hydrochloric acid (3 mol/l). A greenish-brown colour
should be produced (United States Pharmacopeia, 1985).
6.5.2 Purity
6.5.2.1 Acidity
To 0.30 ml in a glass-stoppered cylinder, add a mixture of
0.60 ml of sodium hydroxide (0.1 mol/l), 10 ml of water, and 1 drop
of phenolphthalein TS, and invert the cylinder three times. The red
tint of the water layer should remain perceptible (United States
Pharmacopeia, 1985).
6.5.2.2 Non-volatile residue
Allow 10 ml to evaporate at room temperature in a weighed
evaporating dish within a well-ventilated hood, and dry the residue
at 105 °C for 1 h. The weight of the residue should not exceed 2 mg
(0.02%) (United States Pharmacopeia, 1985).
6.5.2.3 Assay for total nitrites
Inject an aliquot of amyl nitrite of suitable volume, but not
more than 2 ml, into a suitable gas chromatograph equipped with a
thermal conductivity detector. Under typical conditions, the
instrument should contain a column (2 m x 3 mm) packed with a methyl
polysiloxane oil, 25% by weight on suitable calcine diatomite. The
column should be maintained at 80 °C, the injection port and
detector block maintained at 10 °C above the temperature of the
column, and helium used as a carrier gas at a flow rate of about
60 ml per min. The percentage of total nitrites is calculated from
the area under the curve; a total of not less than 97% should be
found.
6.6 Shelf-life
The following precautions should be taken.
* Protect from light.
* Insist that the suppliers only provide ampoules that
have at least 18 month shelf-life left of the nominal
24-month total life.
* Take reasonable precautions to store at the lowest
possible temperature compatible with immediate access
in case of emergency; refrigerated storage is not
necessary.
* Inspect monthly for any broken tubes (Beasley et
al., 1978).
As indicated in section 6.3, amyl nitrite forms an explosive
mixture with air and oxygen. It is highly inflammable and must not
be used where it may be ignited. Storage for a prolonged period or
at an excessive temperature presents the following hazards:
* breakage of the capsule by the build up of pressure
internally and loss of amyl nitrite before use;
* explosive distribution of glass splinters when the
ampoule is broken for use;
* loss of amyl nitrite by chemical decomposition.
6.7 General Properties
The principal pharmacological action of amyl nitrite is to
cause the relaxation of vascular smooth muscle. Peripheral venous
resistance is decreased as a result of a selective action on venous
capacitance vessels with resultant venous pooling of blood and
decreased venous return to the heart. The vasodilating effect of
amyl nitrite on arteriolar resistance is less than that on the
venous side. As a result of this combined action, both venous
filling pressure (preload) and, to a lesser extent, arterial
impedance (afterload) are reduced (AHFS, 1988).
It has been postulated that cyanide causes pulmonary and/or
coronary arteriolar vasoconstriction, which results directly or
indirectly in pump failure and an observed decrease in cardiac
output (Sarnoff, 1980a). This theory is supported by the reported
observations of Vick & Froelich (1985) that a sharp increase in
central venous pressure occurs at a time when the arterial blood
pressure is falling. Following the administration of amyl nitrite
by inhalation, central venous pressure decreased rapidly while the
arterial blood pressure increased. The apparent life-saving effect
of this drug in dogs could have been due to a reversal of early
cyanide-induced vasoconstriction and the restoration of normal
cardiac function. Subsequently, amyl nitrite causes the formation of
methaemoglobin, which may have an additional antidotal action.
a Personal communication by S.J. Sarnoff, Survival Technology
Inc., Bethesda, USA.
6.8 Animal Studies
6.8.1 Pharmacokinetics
Amyl nitrite is readily absorbed into the circulation from
mucous membranes, the rate of absorption being greatest from the
lungs. It is rapidly inactivated by hydrolysis to isoamyl alcohol
and nitrite. Amyl nitrite is rapidly hydrolysed in the
gastrointestinal tract and is therefore pharmacologically inactive
when administered orally (Martindale, 1989).
6.8.2 Pharmacodynamics
Amyl nitrite has proved to be effective in experimental animals
poisoned by cyanide. Indeed, antagonism between amyl nitrite and
hydrocyanic acid was mentioned as long ago as l888 by Pedigo.
Chen et al. (1933) demonstrated that nine to ten inhalations of
amyl nitrite were effective in dogs against 4 times the lethal dose
of cyanide (12-24 mg/kg) (sodium nitrite was also given
subcutaneously).
Amyl nitrite had a significant therapeutic effect on dogs
exposed to cyanogen chloride (CNCl) at Ct values (the product of the
concentration of the gas in mg/m3, and the length of exposure in
min) ranging from 3300 to 6300 mg.min.m-3. However, no such
effect was observed in the groups subjected to higher exposures, at
Ct values ranging from 6900 to 11800 mg.min.m-3 (Jandorf &
Bodansky, 1946).
Amyl nitrite treatment of mice exposed to CNCl resulted in a
statistically significant increase in the number of survivors
(Jandorf & Bodansky, 1946).
Amyl nitrite administered by inhalation increased the median
lethal subcutaneous dose of sodium cyanide in dogs from 5.36 (+0.28)
to 24.5 (+1.2) mg/kg (Chen & Rose, 1952).
Part of the antidotal effect may result from methaemoglobin
formation. Any attempt to correlate the therapeutic effect of amyl
nitrite with the degree of methaemoglobinaemia encounters
difficulties. After exposure to HCN or CNCl, the blood of treated
animals contains, in addition to haemoglobin and oxyhaemoglobin,
cyanmethaemoglobin and methaemoglobin. To date, it has not been
possible to quantify the amount of cyanmethaemoglobin. However,
experiments have been undertaken to estimate the degree of
methaemoglobinaemia produced by inhalation of amyl nitrite in
animals not previously exposed to cyanide.
When ten mice were placed in a chamber containing approximately
12 mg amyl nitrite per l, the methaemoglobin levels 10 seconds,
30 seconds, and 1, 2, and 4 min after the beginning of the exposure,
were 1.0, 1.8, 5.6, 14, and 30%, respectively (Jandorf & Bodansky,
1946).
A cone holding one crushed ampoule containing 0.3 ml amyl
nitrite was fitted over the muzzle of each of four dogs
anaesthetized with pentobarbital, and kept in place for 3 min.
Under these conditions, 16, 18, 20, and 32% methaemoglobinaemia were
found in the four dogs at the end of amyl nitrite inhalation
(Jandorf & Bodansky, 1946).
Bastian & Mercker (1959) recorded a methaemoglobin level of 60%
in mice placed in an atmosphere of amyl nitrite (0.112% v/v) for
15 min. Thereafter the degree of methaemoglobinaemia remained
constant and it appeared that an equilibrium existed between the
formation and reduction of methaemoglobin, the level depending on
the concentration of amyl nitrite in the inspired air.
In cats, inhalation of amyl nitrite (0.06 and 0.12% v/v)
resulted in methaemoglobin levels of 30% and 70%, respectively. At
the higher concentration of amyl nitrite, a fall in blood pressure
from 10.7 to 2.7 kPa (80 to 20 mmHg) was observed (Bastian &
Mercker, 1959).
Part of the antidotal effect of amyl nitrite may be vasogenic
in origin, rather than being due to methaemoglobin formation (Way et
al., 1984). Amyl nitrite given after cyanide administration was
reported by Vick & Froelich (1985) to reverse both cardiovascular
changes and respiratory paralysis in 24 of 30 dogs. These changes
occurred before significant methaemoglobin formation and suggest
that early death caused by cyanide could be due in part to
cardiovascular and respiratory failure.
6.8.3 Toxicology
Syncope or shock induced by large doses of amyl nitrite are the
result of a pooling of blood in dilated capacitance vessels. Reflex
tachycardia normally occurs, but a vaso-vagal reflex may induce a
transient bradycardia immediately before collapse (Wilkins et al.,
1937).
The degree of methaemoglobinaemia caused by inhalation of amyl
nitrite is insufficient to affect oxygen transport (see section
6.8.3).
Amyl nitrite also causes a substantial increase in
cerebrospinal fluid pressure (Norcross, 1938), and a fall in pO2
in brain tissue has been recorded using an oxygen electrode
(Rosemann et al., 1946).
6.9 Volunteer Studies
Inhalation of therapeutic doses of amyl nitrite in man did not
result in methaemoglobin formation (Mathes & Gross, 1939).
When six volunteers inhaled 0.1 ml amyl nitrite 10 times for
20 seconds once per min, the median concentration of methaemoglobin
was 3.45%, with a maximum of 6% and a minimum of 1.4%. During and
shortly after inhalation, a small fall in blood pressure was
observed, followed by immediate recovery. The pulse rate rose from
40 to 120 beats per min. Repeated use of the same square of gauze
without application of a new ampoule of amyl nitrite did not have
any effect (Bastian & Mercker, 1959).
6.10 Clinical Studies
No controlled human clinical trials have been undertaken.
6.11 Clinical Studies - Case Reports
No case reports are available.
6.12 Summary of Evaluation
6.12.1 Indications
Amyl nitrite is used as a first-aid measure in cases of cyanide
poisoning where the patients are in deep coma, with dilated
non-reactive pupils and deteriorating cardio-respiratory function.
6.12.2 Advised routes and dose
In adults, administer 0.2-0.4 ml amyl nitrite, via an Ambu bag,
prior to artificial ventilation. In children, administer a maximum
of 0.1 ml, via an Ambu bag, prior to artificial ventilation.
6.12.3 Other consequential or supportive therapy
First-aid therapy with amyl nitrite must be followed by
additional antidotal treatment (see chapter 1). However, supportive
therapy is the most important aspect of the treatment of cyanide
poisoning. Special attention should be paid to the circulation.
The vasodilating action of amyl nitrite may lead to hypotension,
which should be treated immediately with plasma expanders.
6.13 Model Information Sheet
6.13.1 Uses
Amyl nitrite is used as a first-aid measure in cases of cyanide
poisoning where patients are in deep coma, with dilated non-reactive
pupils and deteriorating cardio-respiratory function.
6.13.2 Dosage and route
In adults, 0.2-0.4 ml amyl nitrite should be administered via
an Ambu bag prior to artificial respiration.
In children, a maximum of 0.l ml should be administered via an
Ambu bag prior to artificial respiration.
6.13.3 Precautions/contraindications
Special attention should be paid to the circulation. The
vasodilating action of amyl nitrite may lead to hypotension, which
should be treated immediately with plasma expanders.
6.13.4 Storage
* Protect from light.
* Insist that the suppliers provide only ampoules that
have at least 18 months shelf-life left of the
nominal 24-months total life.
* Take reasonable precautions to store at the lowest
possible temperature compatible with immediate access
in case of emergency; refrigerated storage is not
necessary.
* Inspect monthly for any broken tubes.
Amyl nitrite forms an explosive mixture with air and oxygen.
It is highly inflammable and must not be used where it may be
ignited.
6.14 References
American Hospital Formulary Service (AHFS) (1988) In: McEnvoy KG,
Litvak K, Welsh OH, Campbell JF, Ziegler K, Douglas PM, Shannon-Lass
EP, Thomson K, McQuerrie GM, Schmadel LK, Fredrickson MK, Mendham
NA, & Morisseau AL ed. Drug Information, Bethesda, American Society
of Hospital Pharmacists.
Bastian G & Mercker H (1959) [The efficacy of amyl nitrite
inhalation in the treatment of cyanide poisoning.]
Naunyn-Schmiedebergs Arch Exp Pathol Pharmakol, 237: 285-295 (in
German).
Beasley RWR, Blown RJ, Lunau FW, & Taylor BM (1978) Amyl nitrite and
all that. J Soc Occup Med, 28: 142-143.
Chen KK & Rose CL (1952) Nitrite and thiosulfate therapy in cyanide
poisoning. J Am Med Assoc, 149: 113-119.
Chen KK, Rose CL, & Clowes GHA (1933) Amyl nitrite and cyanide
poisoning. J Am Med Assoc, 100: 1920-1922.
Chen KK, Rose CL, & Clowes GHA (1944) The modern treatment of
cyanide poisoning. J Ind Hyg Toxicol, 37: 344-350.
Jandorf BJ & Bodansky O (1946) Therapeutic and prophylactic effect
of methemoglobinemia in inhalation poisoning by hydrogen cyanide and
cyanogen chloride. J Ind Hyg Toxicol, 28: 124-132.
Martindale (1989) In: Reynolds JEF ed. The extra pharmacopoeia, 29th
ed. London, The Pharmaceutical Press, pp 1492-1493.
Mathes K & Gross F (1939) [The determination of methaemoglobin and
cyanmethaemoglobin in circulating blood.] Naunyn-Schmiedebergs Arch
Exp Pathol Pharmakol, 191: 701 (in German).
Miller MH & Toops TC (1951) Acute cyanide poisoning. Recovery with
sodium thiosulfate therapy. J Indiana Med Assoc, 44: 1164.
Nederlandse Pharmacopee (1958) 6th ed. The Hague, Staatsdrukkerij.
Norcross NC (1938) Intracerebral blood flow. Arch Neurol Psychiatry
(Chicago), 40: 291.
Pedigo LG (1888) Antagonism between amyl nitrite and Prussic acid.
Trans Med Soc Virginia, 19: 124-131.
Rosemann E, Goodman CW, & McCulloch WS (1946) Rapid changes in
cerebral oxygen tension induced by altering the oxygenation and
circulation of the blood. J Neurophysiol, 9: 33.
United States Pharmacopeia (1985) 21st ed. Rockville, Maryland,
United States Pharmacopeial Convention, Inc.
Vick JA & Froelich HL (1985) Studies on cyanide poisoning. Arch Int
Pharmacodyn, 273: 314-322.
Way JL, Sylvester D, Morgan RL, Isom GE, & Burrows CB (1984) Recent
perspectives on the toxidynamic basis of cyanide antagonism. Fundam
Appl Toxicol, 4: S231-S239.
Wilkins RW, Haynes FW, & Weiss S (1937) Role of venous system in
circulatory collapse induced by sodium nitrite. J Clin Invest, 16:
85-91.
Wolfsie JH (1951) Treatment of cyanide poisoning in industry. Am Med
Assoc Arch Ind Hyg, 4: 417-425.
7. SODIUM NITRITE
7.1 Introduction
Methaemoglobin has a great affinity for cyanide in vitro (Hug
& Marenzi, 1933a). Sodium nitrite is an inducer of methaemoglobin
(Hug, 1933; Hug & Marenzi, 1933b) and has been in clinical use for
over 50 years as an antidote for acute cyanide poisoning, most often
in combination with amyl nitrite and sodium thiosulfate (Viana et
al., 1934; Chen & Rose, 1952; Hall & Rumack, 1986).
Potential side-effects of sodium nitrite infusion are
hypotension and excessive methaemoglobin formation (Viana et al.,
1934; Berlin, 1970; Feihl et al., 1982; Hall & Rumack, 1986).
Patients who are already hypotensive are at special risk, and those
with certain types of congenital methaemoglobinaemia are
theoretically more at risk from excessive methaemoglobin induction.
Children administered large doses of sodium nitrite have developed
excessive methaemoglobinaemia (Berlin, 1970). Victims of smoke
inhalation may have both elevated carboxyhaemoglobin levels and
cyanide poisoning (Hart et al., 1985), and may develop further
hypoxia from methaemoglobin induction (Becker, 1985; Jones et al.,
1987).
7.2 Name and Chemical Formula
Sodium nitrite, otherwise known as diazotizing salts, dusitan
sodny (Czech), erinitrit, filmerine, natrium nitrit (German),
nitrite de sodium (French), nitrous acid sodium salt, sodii nitris,
natrii nitris, natrium nitrosum, sodium nitrite (DOT), NCI-C02084,
NIOSH No. RA 1225000, CAS 7632-00-0 (Sax, 1984; NIOSH, 1983;
Windholz et al., 1983; Martindale, 1982), is supplied in the USA by
Eli Lilly & Co., Indianapolis, Indiana, as a component of the Lilly
Cyanide Antidote Package [R] (Product Information, 1984). It is
sold in Australia by Orapharm under the proprietary name of OAR
(Martindale, 1989). The relative molecular mass of sodium nitrite
is 69.00, and the chemical formula is NaNO2 (Sax, 1984).
7.3 Physico-chemical Properties
Sodium nitrite has a melting point of 271 °C (Windholz et al.,
1983). It is soluble in either 1.5 parts of cold water or 0.6 parts
of boiling water (Windholz et al., 1983). Solutions may be made in
water (1:1.5) or alcohol 1:160 (Martindale, 1989). Sodium nitrite
is slightly soluble in ether (ITI, 1985). Acids will decompose
sodium nitrite and cause the evolution of brownish N2O3 fumes
(Windholz et al., 1983). Aqueous solutions are alkaline to litmus,
with a pH between 7 and 9 (Windholz et al., 1983; United States
Pharmacopeia, 1985). The specific gravity is 2.17 (ITI, 1985). A
10% solution of sodium nitrite has a density of 1.065 (Izmerov,
1982). Excipients are apparently not added.
On drying over silica gel for 4 h, solid sodium nitrite loses
no more than 0.25% of its weight (United States Pharmacopeia, 1985).
Injectable sodium nitrite solution is made by diluting solid
sodium nitrite with "Water for Injection" (United States
Pharmacopeia, 1985). The final solution is sterile and contains not
less than 95% and not more than 105% of the labelled amount of
sodium nitrite (United States Pharmacopeia, 1985). Sodium nitrite
is supplied in the Lilly Cyanide Antidote Package [R] in two 10 ml
vials of a 3% solution (30 mg/ml; 300 mg per vial) (Product
Information, 1984).
Sodium nitrite is stable in light, but slowly oxidizes in air
to nitrate (Windholz et al., 1983). It is incompatible with
acetanilide, antipyrine, caffeine, citrate, chlorates,
hypophosphites, iodides, mercury salts, morphine, oxidizing agents,
permanganate, phenazone, sulfites, tannic acid, and vegetable
astringent decoctions, infusions, or tinctures (Martindale, 1989;
Windholz et al., 1983).
7.4 Synthesis
Commercial sodium nitrite is derived from sodium hydroxide and
nitrogen oxides by one of three common methods (Izmerov, 1982).
One method uses a low concentration of nitrogen oxides from gas
formed during dilute nitric acid production, which is absorbed by a
sodium hydroxide solution.
A second method (the "hot method") uses sodium hydroxide
absorption of high concentrations of nitrogen oxides at atmospheric
or higher pressure.
A third method (or "two-staged method") combines features of
the first two.
Pharmaceutical sodium nitrite is produced by reduction of
sodium nitrate, previously heated until fused, with sulfur dioxide,
lead, or a sulfite (Harvey, 1980). Alternatively, it can be derived
from sodium nitrate by nitric oxide absorption (the nitric oxide
being obtained from ammonia by catalytic oxidation in a sodium
carbonate solution) (Harvey, 1980). The sodium nitrite mixture
obtained is lixiviated with water and filtered, then partially
evaporated and allowed to crystallize (Surgenor, 1970).
Injectable sodium nitrite solution is prepared by dissolving
solid sodium nitrite in "Water for Injection" to a final
concentration of 3% (30 mg/ml) (Product Information, 1984; United
States Pharmacopeia, 1985). It is then sterilized by autoclaving or
membrane filtration (Martindale, 1989). The final delivery form
meets both United States and European Pharmacopoeia requirements for
pyrogens and sterility.
The United States Pharmacopeia specifies that no more than
0.002% of heavy metals may be present (United States Pharmacopeia,
1985). "Top grade" sodium nitrite from some processes has a sodium
nitrite concentration of at least 99%, with at most 1.4% moisture
content and impurities of not more than 0.8% sodium nitrate, 0.3%
calcinated residue, and 0.17% sodium chloride (Izmerov, 1982).
7.5 Analytical Methods
7.5.1 Quality control
7.5.1.1 Solid sodium nitrite (United States Pharmacopeia, 1985)
Sodium nitrite (1g) is dissolved in water, making up to 100 ml.
Then 10 ml of this solution is pipetted into a mixture containing
50.0 ml of 0.1 N potassium permanganate volumetric solution (VS)
(0.1 mol/l), 100 ml of water, and 5 ml of sulfuric acid, and the
pipette tip is immersed beneath the surface of this mixture during
transfer. The liquid is warmed to 40 °C and allowed to stand for
5 min. After the addition of 25.0 ml oxalic acid VS (0.05 mol/l),
the resultant mixture is heated to 80 °C and titrated with potassium
permanganate (0.1 mol/l), equivalent to 3.45 mg sodium nitrite.
7.5.1.2 Sodium nitrite injection (United States Pharmacopeia, 1985)
The method described in 7.5.1.1 is followed except that, in
place of the 10 ml sodium nitrite solution, an accurately measured
volume of sodium nitrite injection containing about 150 mg sodium
nitrite is used.
7.5.1.3 Preparation of volumetric solutions (VS) (United States
Pharmacopeia, 1985)
(a) Potassium permanganate VS (0.1 mol/l)
KMnO4; relative molecular mass, 158.03; 3.161 grams in
1000 ml
About 3.3 g of potassium permanganate is dissolved in
1000 ml of water and boiled for about 15 min in a suitable
flask. The flask is then stoppered and allowed to stand
for at least 2 days. It is then filtered through a
fine-porosity, sintered glass crucible, which may be lined
with a pledget of glass wool. The solution is then
standardized by weighing accurately 200 mg of sodium
oxalate, previously dried to constant weight at 110 °C.
This is dissolved in 250 ml of water. Sulfuric acid
(7 ml) is added and the mixture is heated to about 70 °C.
The potassium permanganate solution is slowly added from a
burette with constant stirring until a pale-pink colour
persisting for about 15 seconds is produced. At the
termination of titration, the temperature should be no
lower than 60 °C. The molarity is calculated, with each
6.700 mg of sodium oxalate equivalent to 1 ml of potassium
permanganate (0.1 mol/l). The solution should be stored
in glass-stoppered amber-coloured bottles, and contact
with organic materials such as rubber, which will reduce
the potassium permanganate, should be avoided.
(b) Oxalic Acid VS (0.05 mol/l)
H2C2O4 . 2H2O; relative molecular mass 126.07;
6.303 g in 1000 ml
6.45 g of oxalic acid is dissolved in water and made up to
1000 ml. The solution is standardized by titration
against potassium permanganate VS (1 mol/l) as described
above. The solution should be stored protected from light
in glass-stoppered bottles.
7.5.2 Identification (United States Pharmacopeia, 1985)
A solution of sodium nitrite responds to tests for sodium and
for nitrite.
7.5.2.1 Sodium
After conversion to chloride or nitrate, solutions of sodium
compounds with five times their volume of cobalt-uranyl acetate test
solution (TS) yield a precipitate that is golden yellow in colour
and forms after several min of agitation. Cobalt-uranyl acetate
test solution is prepared by dissolving 40 g of uranyl acetate in a
mixture of 30 g of glacial acetic acid and enough water to make
500 ml. A solution is also prepared of 200 g of cobaltous acetate
in a mixture of 30 g of glacial acetic acid and enough water to make
500 ml. Both solutions are prepared with warming, mixed while still
warm, and cooled to 20 °C. The temperature is maintained at 20 °C
for about 2 h, allowing the separation of excess salts, and then the
resultant solution is filtered through a dry filter. Sodium
compounds also impart an intense yellow coloration to a non-luminous
flame.
7.5.2.2 Nitrite
Treatment of nitrite-containing solutions with diluted mineral
acids or acetic acid (6 mol/l) produces brownish-red fumes, and the
solution will give a blue coloration on starch-iodide paper.
7.5.3 Impurities (United States Pharmacopeia, 1985)
7.5.3.1 Preparation of sodium nitrite to test
Sodium nitrite (1 g) is dissolved in 6 ml hydrochloric acid
(3 mol/l) and then evaporated to dryness on a steam bath. The
residue is reduced to a coarse powder, which is then heated on the
steam bath until the hydrochloric acid odour is no longer
perceptible. This residue is then dissolved in 23 ml of water, and
2 ml of acetic acid (1 mol/l) is added.
The test is for metallic impurities that are coloured by the
sulfide ion under specified test conditions, in terms of the
percentage of lead (by weight) in the test substance. Limits for
sodium nitrite are 0.002%.
7.5.3.2 Preparation of special reagents
Lead nitrate stock solution is prepared by dissolving 159.8 mg
lead nitrate in 100 ml of water and adding 1 ml of nitric acid.
This solution is then diluted to 1000 ml with water and stored in
glass containers free of lead soluble salts. Standard lead solution
is prepared by diluting 10.0 ml of the lead nitrate stock solution
with water to a volume of 100.0 ml, so that each 1 ml of standard
lead solution contains the equivalent of 10 µg of lead. A
comparison solution prepared with 100 µl of standard lead solution
per g of sodium nitrate contains the equivalent of 1 part of lead
per million parts of sodium nitrate.
7.5.3.3 Preparation of standard
Pipette 2 ml of standard lead solution (containing 20 mg of
lead) into a 50 ml colour comparison tube. Dilute with water to
25 ml and adjust pH to between 3.0 and 4.0 with acetic acid
(1 mol/l) or ammonium hydroxide (6 mol/l) using narrow-range pH
paper as an indicator. Dilute to 40 ml with water and mix.
7.5.3.4 Preparation of test
Pipette 25 ml of the prepared sodium nitrite test material (see
section 7.5.3.1) into a 50 ml colour comparison tube. Using
narrow-range pH indicator paper, adjust pH to between 3.0 and 4.0
with either acetic acid (1 mol/l) or ammonium hydroxide (6 mol/l).
Then dilute to 40 ml with water and mix.
7.5.3.5 Preparation of monitor
Place 25 ml of solution prepared as in section 7.5.3.4 above
into a 50 ml colour comparison tube and add 2.0 ml of the standard
lead solution. Using short-range pH indicator paper, adjust pH to
between 3.0 and 4.0 with either (1 mol/l) acetic acid or ammonium
hydroxide (6 mol/l). Then dilute to 40 ml with water and mix.
7.5.3.6 Preparation of hydrogen sulfide test solution (TS)
Pass hydrogen sulfide gas into cold water making a saturated
solution that should possess a strong odour of hydrogen sulfide.
Store in a cold, dark area in small, dark amber-coloured bottles
filled nearly to the top.
7.5.3.7 Test procedure
Add 10 ml of freshly prepared hydrogen sulfide TS solution to
each colour comparison tube. Allow to stand for 5 min, then view
downwards over a white surface. The colour of the solution
described in section 7.5.3.4 should not be darker than that of the
solution described in section 7.5.3.3. The colour intensity of the
solution described in section 7.5.3.5 should be equal to or greater
than that of the solution described in section 7.5.3.3.
7.6 Shelf-life
The shelf-life of sodium nitrite solution for injection is
5 years, which might be somewhat reduced in very hot climates though
stability testing at elevated temperatures has not been undertaken
(Personal Communication, Eli Lilly & Co., March 1987). The supplier
recommends that sodium nitrite be stored at controlled room
temperatures of 15 - 30 °C (Product Information, 1984). Experiments
with stored ampoules showed that sodium nitrite is stable in water
for more than 21 years (Chen & Rose, 1956).
7.7 General Properties
7.7.1 Mode of action
Sodium nitrite has seldom been utilized alone for the treatment
of cyanide poisoning. Rather, it is used in combination with
intravenous sodium thiosulfate and, often, amyl nitrite by
inhalation. These combinations have been shown to be more
efficacious in experimental poisoning than any of the compounds
alone, demonstrating true antidotal synergy (Hug, 1933; Chen et al.,
1933a; Chen et al., 1934; Chen & Rose, 1952; Way et al., 1984).
Sodium nitrite induces methaemoglobin in vitro, which binds
cyanide (Hug & Marenzi, 1933a). In animal studies with dogs and
rabbits, sodium nitrite induced methaemoglobinaemia, with levels
averaging 50% following a dose of 40 mg/kg (Hug & Marenzi, 1933a;
Hug & Marenzi, 1933b). Sodium nitrite alone has been shown to be
efficacious in dogs with experimental cyanide poisoning (Chen et
al., 1933a; Chen et al., 1934; Chen & Rose, 1952). However, dog
blood has been shown to be more sensitive to methaemoglobin
induction by sodium nitrite in vitro than human blood (Paulet &
Menoret, 1954). Methaemoglobin can be an efficacious cyanide
antidote, as shown by results in experimental poisoning in rats
treated with an exogenously prepared stroma-free methaemoglobin
solution (Ten Eyck et al., 1986). Human red blood cells, with
methaemoglobin induced in vitro, were injected intraperitoneally
to cyanide-poisoned mice and had antidotal efficacy (Kruszyna et
al., 1982). However, animals pretreated with methylene blue did not
develop significant methaemoglobin levels and had unchanged sodium
nitrite antidotal efficacy in experimental cyanide poisoning (Holmes
& Way, 1982). Other mechanisms, including vasodilatation with
changes in local capillary blood flow, have been postulated to
account for part or all of the antidotal action of sodium nitrite
(Way et al., 1984; Cohen & Guzzardi, 1984).
Alternative mechanisms of action are suggested by the fact that
sodium nitrite relaxes maximally contracted vascular smooth muscle
in vitro (Kruszyna et al., 1985). The contracted vascular smooth
muscle relaxation was not reversed in this model by the addition of
cyanide (Kruszyna et al., 1985). In mice, methaemoglobin levels
induced by sodium nitrite were more efficacious in protecting
against cyanide poisoning than were comparable methaemoglobin levels
induced by either hydroxylamine or 4-dimethylaminophenol (Kruszyna
et al., 1982). Equivalent amounts of methaemoglobin induced
in vitro by these three agents did not bind significantly
different amounts of cyanide (Kruszyna et al., 1982), suggesting
that sodium nitrite may have some other mechanism of action. The
methaemoglobinaemia induced in these mice by sodium nitrite peaked
later and at a lower level than the methaemoglobinaemia induced by
hydroxylamine or 4-dimethylaminophenol, but was longer lasting
(Kruszyna et al., 1982). This may explain differences in efficacy
noted in this animal study, as cyanide initially bound to
methaemoglobin induced by the other agents may have been rapidly
released to exacerbate the poisoning (Kruszyna et al., 1982).
The oxidation of haemoglobin to methaemoglobin by sodium
nitrite is favoured when the oxygen partial pressure is high and
haemoglobin is in the oxyhaemoglobin state (Tomoda & Yoneyama,
1979). If methaemoglobin induction accounts for all, or part, of
the antidotal action of sodium nitrite, this might explain the
potentiation noted with concomitant oxygen administration (Mansouri,
1985), although other investigators have not reached this conclusion
(Way et al., 1966).
The mechanism of action of sodium nitrite is not fully
understood at present. Methaemoglobin induced by sodium nitrite and
measurable with a co-oximeter, however, does not bind cyanide at the
time of measurement (cyanmethaemoglobin cannot be measured by these
instruments). Patients have survived seriously symptomatic acute
cyanide poisoning, despite developing measurable methaemoglobin
levels of 10% or less (Hall et al., 1987; DiNapoli et al., 1989).
Administering futher sodium nitrite to a patient who has already had
a satisfactory clinical improvement in order to produce a
"therapeutic methaemoglobin level" of 25%, as has somethimes been
recommended (Jones et al., 1987), is both unnecessary and
potentially dangerous (DiNapoli et al., 1989).
7.7.2 Other relevant properties
Sodium nitrite is a vasodilating agent and has been used in the
past for the treatment of angina (Martindale, 1989). Rapid
intravenous administration may cause hypotension (Viana et al.,
1934; Hall & Rumack, 1986). This effect can be avoided by beginning
the infusion slowly and increasing the infusion rate cautiously with
careful blood pressure monitoring.
7.8 Animal Studies
7.8.1 Pharmacokinetics
Sodium nitrite blood levels have not been measured during
antidote therapy and most pharmacokinetic parameters have not been
determined. Most studies have focused on the levels of
methaemoglobin induced by sodium nitrite.
In one study into fetal methaemoglobin formation, when pregnant
rats were administered sodium nitrite orally or by intraperitoneal
injection, peak maternal sodium nitrite blood levels were usually
produced within the first 20 to 30 min, while peak methaemoglobin
levels were observed a few minutes later (Gruener et al., 1973).
Peak sodium nitrite blood concentrations ranged from 3.9 mg/l
(56.52 µmol/l), with an administered dose of 2.5 mg/kg, to 32.5 mg/l
(471.01 µmol/l) with an administered dose of 30 mg/kg, and
corresponded to peak maternal methaemoglobin levels of 3.4%
(2.5 mg/kg dose) to 60.2% (30 mg/kg dose) (Gruener et al., 1973).
Sodium nitrite produced an equimolar conversion of haemoglobin
to methaemoglobin in vitro in canine blood (Hug & Marenzi, 1933a).
However, only about 74% of an administered intravenous dose was
involved in methaemoglobin formation in dogs in vivo (Hug &
Marenzi, 1933b). Dogs administered 40 mg/kg of sodium nitrite
intravenously developed approximately 50% methaemoglobinaemia (Hug &
Marenzi, 1933b). In these experiments using sodium nitrite alone,
all of the administered dose of potassium cyanide was recovered in
the urine as thiocyanate over a 4-day period (Hug & Marenzi, 1933b).
In a dog with acute cyanide poisoning, given sodium nitrite
(22.5 mg/kg) immediately and an additional 11.25 mg/kg at 29 and
54 min after the poisoning, peak methaemoglobin levels of 65% at
30 min and 60% at 4 h were observed (Chen & Rose, 1952). The
methaemoglobin concentration decreased rapidly after each peak,
reaching 20% about 30 min after the first peak, and 4-5 h after the
second peak (Chen & Rose, 1952). Methaemoglobin disappeared from
the circulation 11 h after the first injection of sodium nitrite
(Chen & Rose, 1952).
In mice administered sodium nitrite 75 (mg/kg)
intraperitoneally, methaemoglobin levels peaked at 30-35% about
20 min after the injection (Kruszyna et al., 1982). The levels
remained in this range until 40 min after sodium nitrite
administration and then decreased slowly, reaching 25%
methaemoglobinaemia at 1 h and 10% at 2 h (Kruszyna et al., 1982).
These findings contrast with studies undertaken in mice with other
methaemoglobin-inducing agents (4-dimethylaminophenol and
hydroxylamine), which produced higher peak methaemoglobin levels
more quickly (Kruszyna et al., 1982). Following the peak level, the
degree of methaemoglobinemia then decreased faster than that induced
by sodium nitrite (Kruszyna et al., 1982).
In vitro studies have indicated that canine blood is more
susceptible to sodium nitrite-induced methaemoglobin conversion than
human blood (Paulet & Menoret, 1954). Rodent blood is more
resistant to methaemoglobin formation than human blood (Kruszyna et
al., 1982).
7.8.2 Pharmacodynamics
Sodium nitrite, at a minimum dose of 5 mg/kg, was shown to be
an effective antidote for experimental cyanide poisoning in dogs,
while 200 mg/kg was efficacious in rabbits (Hug, 1932; Hug, 1933).
In other experiments, sodium nitrite alone was shown to be an
effective cyanide antidote in dogs (Chen et al., 1934), though the
combination of sodium nitrite with a sulfur donor allowed dogs to
tolerate doses of cyanide greater than those antagonized by the two
agents separately (Chen et al., 1933a; Chen et al., 1933b; Hug,
1933; Chen et al., 1934). This antidotal synergy has been noted in
numerous studies since the 1930s (e.g., Way et al., 1972). Sodium
nitrite has been shown to have protective effects against rat brain
cytochrome oxidase inhibition in vivo (Piantadosi et al., 1983).
Two studies have reported data that are inconsistent with the
classical antidotal mechanism of action, namely methaemoglobin
induction. When comparable levels of methaemoglobinaemia were
induced in mice by 4-dimethylaminophenol, hydroxylamine, or sodium
nitrite, cyanide was antagonized better by the administration of
sodium nitrite (Kruszyna et al., 1982). Following pre-treatment
with methylene blue prior to cyanide poisoning, mice had unchanged
sodium nitrite antidotal efficacy, even though they did not develop
significant methaemoglobinaemia (Holmes & Way, 1982).
Sodium nitrite causes relaxation of vascular smooth muscle
in vitro (Kruszyna et al., 1985). Sodium nitrite alone did not
have any effect on either the respiratory or urinary excretion of
14C-labelled sodium cyanide in mice (Burrows et al., 1982). Other
postulated antidotal mechanisms include vasodilatation with improved
capillary blood flow (Way et al., 1984; Cohen & Guzzardi, 1984).
7.8.3 Toxicology
A rather large body of sub-acute and chronic toxicity data
exists for sodium nitrite (Musil, 1966; Izmerov, 1982). Effects
including increased methaemoglobin levels, neuromuscular
excitability, thyroid dysfunction, changes in conditioned reflexes,
elevated serum protein and albumin concentrations, possible effects
on hepatic drug metabolism systems, prolonged prothrombin time, and
decreased erythrocyte counts have been noted in chronic and subacute
animal exposures (Musil, 1966; Izmerov, 1982; Gosselin et al.,
1984). In antidote administration, most subjects would receive only
one dose, or at most a few doses over a short period of time, and
these chronic and subacute animal studies would not seem to be
particularly relevant.
Pre-treatment of rats with sodium nitrite potentiates carbon
tetrachloride hepatotoxicity (Suarez & Bhonsle, 1978). A
synergistic effect on acute hepatic necrosis in mice has been
produced by oral administration of a combination of sodium nitrite
and various secondary amines (Asahina et al., 1971). A second
abnormal haemoglobin derivative, nitrosohaemoglobin, has been
produced in rats following intraperitoneal sodium nitrite injection
(Rein et al., 1968).
Sodium nitrite is listed as a non-carcinogen, but as giving a
positive result in the Ames test, by the International Agency for
Research on Cancer (IARC) (Kuroki et al., 1980). It can react with
secondary amines either in vitro or in vivo to produce
potentially carcinogenic nitrosamines (Izmerov, 1982). In vitro
studies have shown induction of volatile mutagens in the faeces of
normal human volunteers following treatment with sodium nitrite
(MacDonald & Rao, 1982). Administration of sodium nitrite and
morpholine together to rats produced hepatic and forestomach tumours
(Mirvish et al., 1983). However, a cohort study of workers
chronically exposed to a combination of sodium nitrite and various
amines in cutting fluids did not show an increased cancer morbidity
(Jarvholm et al., 1986). Epidemiological data suggest a
relationship between the consumption of large quantities of
nitrite-nitrate-treated foodstuffs, a diet poor in ascorbic acid,
and the development of gastric, oral cavity, and oesophageal cancers
(Weisburger et al., 1983).
Sodium nitrite administered orally or intraperitoneally to
pregnant rats produced peak fetal sodium nitrite blood levels
ranging from trace amounts, with a 2.5 mg/kg maternal dose and a
corresponding maternal blood level of 3.9 mg/l (56-52 µmol/l), to
9.4 mg/l (136.23 µmol/l) with a 30 mg/kg maternal dose and a
corresponding maternal blood level of 32.5 mg/l (471.01 µmol/l)
(Gruener et al., 1973). Fetal methaemoglobin levels ranged from
1.2% (with a maternal sodium nitrite dose of 2.5 mg/kg and a
corresponding maternal peak methaemoglobin level of 3.4%) to 27.2%
(with a maternal sodium nitrite dose of 30 mg/kg and a corresponding
maternal peak methaemoglobin level of 60.2%) (Gruener et al., 1973).
Fetal effects other than methaemoglobin induction were not reported.
Sodium nitrite was shown not to be teratogenic in several
animal species when used as a cardiovascular agent or as a fungicide
in combination with iodine (Schardein, 1985). In combination with
ethylurea, sodium nitrite forms the potent teratogen ethylnitrosurea
(Schardein, 1985). The median effective dose (ED50) for mortality
in a chicken embryo study was 22 µmol/egg (Korhonen et al., 1983).
A maximum of 27% of the embryos were noted to be malformed (Korhonen
et al., 1983). A combination of sodium nitrite (50 mg/kg) and
methyl urea (30 mg/kg), administered orally to rats on the 9th day
of pregnancy, killed over 40% of the fetuses and produced
malformations in about 15% of the surviving fetuses (Izmerov, 1982).
Administration of ascorbic acid blocked both the embryotoxic and
teratogenic effects (Izmerov, 1982). Sodium nitrite doses of
100 mg/kg were teratogenic in rats (Izmerov, 1982). Cyanosis was
observed in 25% of newborn rats at birth following maternal
administration of sodium nitrite (100 mg/kg) and in 6% of newborn
rats with a maternal dose of 5 mg/kg (Izmerov, 1982).
As sodium nitrite is only likely to be administered once, or at
most a few times, as an antidote, the main concern would appear to
be induction of fetal methaemoglobinaemia if administered to a
pregnant patient. The risk to the fetus from maternal cyanide
poisoning would seem to override the risk from possible fetal
methaemoglobin induction. At clinically relevant doses (300 mg, or
about 4.3 mg/kg for a 70-kg subject) (Product Information, 1984),
data from a rat study suggested that the peak fetal methaemoglobin
level would be in the neighbourhood of 2.7% (Gruener et al., 1973).
This amount of fetal methaemoglobinaemia is not likely to be
clinically significant. However, based on in vitro comparisons of
methaemoglobin reductase activity, the human fetus may have a weaker
defense mechanism against the development of significant
methaemoglobinaemia than has the rat fetus (Gruener et al., 1973).
Some representative animal toxicity values for sodium nitrite
are: LD50 rat (oral), 85 mg/kg; LDLo hamster (oral), 3 mg/kg;
LD50 mouse (oral), 175 mg/kg; LD50 rat (intravenous), 65 mg/kg;
LDLo dog (oral), 330 mg/kg; LDLo dog (intravenous), 15 mg/kg; LDLo
rabbit (intravenous), 80 mg/kg (Sax, 1984; ITI, 1985). A
comparative human intravenous antidotal dose is 300 mg (or about
4.3 mg/kg for a 70-kg subject) (Product Information, 1984).
7.9 Volunteer Studies
7.9.1 Pharmacokinetics
Sodium nitrite is rapidly absorbed following oral
administration (Martindale, 1989). About 60% of the absorbed
nitrite ion is metabolized in the body, partially to ammonia, while
the rest of the absorbed dose is excreted unchanged in the urine
(Martindale, 1982). The nitrite ion disappears from the circulatory
system quickly, but little is known about its fate (Baselt, 1982).
An intravenous injection of 400 mg in humans produced a peak
methaemoglobin concentration of 10.1%, while 600 mg produced a peak
methaemoglobin level of 17.5% (Chen & Rose, 1952). A later study
in normal volunteers showed that a methaemoglobin level of 6% was
produced by the intravenous injection of 1 mg sodium nitrite per kg
(about 300 mg, 10 ml of a 3% solution of sodium nitrite) (Weger,
1968). In this same study, injecting 12 mg/kg (about 900 mg, 30 ml
of a 3 % solution of sodium nitrite) resulted in a 30%
methaemoglobinaemia, but also in clinical shock (Weger, 1968).
7.9.2 Sodium nitrite poisoning
Most human toxicity data relates to accidental or suicidal
ingestion of sodium nitrite from industrial, laboratory, or food
additive sources. Fatal poisoning has occurred when sodium nitrite
has been substituted for table salt and used as seasoning
(MacQuiston, 1936; Padberg & Martin, 1939) or when food contaminated
with motor vehicle cooling fluid was consumed (Ten Brink et al.,
1982). A case of suicide from the ingestion of sodium nitrite has
been reported, with a post-mortem methaemoglobin level of 90%
(Standefer et al., 1979). Ingestion of sausages cured with sodium
nitrite produced symptomatic poisoning with syncope, hypotension,
and methaemoglobinaemia and a decreased arterial oxygen saturation
(Bakshi et al., 1967). Two patients who mistakenly ingested sodium
nitrite instead of table salt developed methaemoglobin levels of 34%
and 54%, but recovered after being administered methylene blue and
supplemental oxygen (Aquanno et al., 1981).
The range of toxicity of sodium nitrite is difficult to
determine, as the ingested dose is seldom known. A patient who
ingested 14.5 g complained of transient darkening of both visual
fields (Grant, 1986). A 17-month-old child died with a
methaemoglobin level greater than 90% after being given 450 mg
(32 mg/kg) intravenously in the mistaken impression that acute
cyanide poisoning was present (Berlin, 1970). A two-month-old
infant developed severe poisoning but survived after ingesting
130 mg sodium nitrite (Gosselin et al., 1984). The mean lethal oral
dose in adults is probably in the neighbourhood of 1 g if no
treatment is given (Gosselin et al., 1984), though survival has
followed the ingestion of 1 g (Baselt, 1982).
Treatment of acquired methaemoglobinaemia from sodium nitrite
poisoning in circumstances similar to those described above may
involve only supplemental oxygenation and observation if the patient
is asymptomatic and the methaemoglobin level is 20-30% or less (Hall
et al., 1986a). With higher methaemoglobin levels or in symptomatic
patients, intravenous infusion of methylene blue at a usual dose of
0.1-0.2 ml/kg of a 1% solution (1-2 mg/kg) may be necessary (Hall et
al., 1986b). Toluidine blue may be used when methylene blue is not
available. Exchange transfusion may be required if severely
poisoned patients are not responsive to the above measures (Hall et
al., 1986a). Experimental animal studies have suggested that
hyperbaric oxygen is both efficacious (Goldstein & Doull, 1971) and
not efficacious (Sheehy & Way, 1974) in reducing nitrite-induced
methaemoglobinaemia. Hyperbaric oxygen can be used to maintain
tissue oxygenation while exchange transfusion is prepared (Hall et
al., 1986a).
If the therapeutic administration of sodium nitrite in cyanide
poisoning produces excessive methaemoglobinaemia (Viana et al.,
1934; Berlin, 1970; Lasch & El Shawa, 1981; Feihl et al., 1982),
there is some controversy about appropriate treatment. The
administration of methylene blue has been recommended (Product
Information, 1984), and toluidine blue has also been used. However,
exchange transfusion rather than methylene or toluidine blue
administration has been suggested because conversion of
cyanmethaemoglobin back to normal haemoglobin could theoretically
release bound cyanide and worsen the poisoning (Rumack, 1987). This
controversy is unresolved at present.
7.10 Clinical Studies
No controlled human clinical trials have been performed to
compare the efficacy of various cyanide antidotes in acute human
poisoning.
7.11 Clinical Studies - Case Reports
In the absence of controlled clinical trials, anecdotal reports
of human poisoning treated with sodium nitrite are all that can be
evaluated. To compound the problem, only a single case of acute
cyanide poisoning treated solely with sodium nitrite has been
reported (Mota, 1933). All other reported patients were given more
than one antidote. Many patients have received a combination of
sodium nitrite and sodium thiosulfate, or amyl nitrite and sodium
nitrite plus sodium thiosulfate (Chen & Rose, 1952; Chen & Rose,
1956). In other cases, sodium nitrite has been administered in
various combinations with sodium thiosulfate, methylene blue,
dicobalt-EDTA (Kelocyanor [R]), and hydroxocobalamin (Motin et al.,
1970; Lutier et al., 1971; Yacoub et al., 1974). The clinical
efficacy of sodium nitrite used alone in acute cyanide poisoning is
therefore impossible to separate from its use in an antidotal
combination, and, judging from various animal studies showing
antidotal synergism between sodium nitrite and sodium thiosulfate
(Chen et al., 1934; Chen & Rose, 1952; Way et al., 1972), it should
probably not be used alone.
The first reported cases of human acute cyanide poisoning to be
treated with a combination of sodium nitrite and sodium thiosulfate
were described by Viana et al. (1934). Their first patient ingested
about 5 g of potassium cyanide and became comatose with a weak pulse
and respiratory distress, but recovered after being given 1500 mg of
sodium nitrite and 18 g of sodium thiosulfate. Their second patient
ingested about 2 g of potassium cyanide and developed coma,
respiratory distress, and convulsions, but recovered after being
given 750 mg of sodium nitrite and 12 g of sodium thiosulfate. No
cyanide assays were undertaken. Hypotension and cyanosis developed
in both cases (Viana et al., 1934).
The largest case series of acute cyanide poisoning treated with
the amyl nitrite/sodium nitrite/sodium thiosulfate antidote
combination was assembled by Chen & Rose (1952, 1956) comprising a
total of 49 patients. One patient who was moribund before being
administered these antidotes did not recover (Chen & Rose, 1952).
Only historical evidence of cyanide poisoning was available in these
cases; no cyanide assays were reported (Chen & Rose, 1952; Chen &
Rose, 1956).
Survival following acute cyanide poisoning from potassium and
sodium cyanide (De Busk & Seidl, 1969; Stewart, 1974; Feihl et al.,
1982; Peters et al., 1982; Wood, 1982; Litovitz et al., 1983; Wesson
et al., 1985; Hall et al., 1987), cyanogenic plants (Sayre &
Kaymakcalan, 1964; Rubino, 1978; Lasch & El Shawa, 1981; Shragg et
al., 1983), and laetrile (Moss et al., 1981; Beamer et al., 1983;
Hall et al., 1986b) has been reported. However, not all patients
with severe cyanide poisoning given the sodium nitrite/sodium
thiosulfate antidote combination have survived (Chen & Rose, 1952;
Braico et al., 1979; Litovitz et al., 1983). Patients with severe
acute cyanide poisoning (including coma, convulsions, and metabolic
acidosis) and blood cyanide levels, measured at various times after
exposure, ranging from 0.26 to 40 mg/l (10 to 1539 µmol/l) have
survived following the administration of sodium nitrite and sodium
thiosulfate in combination with supportive measures (Rubino, 1978;
Moss et al., 1981; Peters et al., 1982; Feihl et al, 1982; Litovitz
et al., 1983; Shragg et al., 1983; Wesson, et al., 1985; Hall et
al., 1987; Hall et al., 1986b). Patients poisoned with cyanide have
survived with supportive treatment only (Graham et al., 1977; Vogel
et al., 1981; Brivet et al., 1983), but the highest reported blood
cyanide level in a patient who survived without receiving specific
antidote treatment was 3.8 mg/l (147 µmol/l) (Edwards & Thomas,
1978). This suggests that patients with more severe cyanide
poisoning may have a better chance of recovery with both supportive
measures and specific antidotes than with supportive measures alone
(Hall & Rumack, 1986).
Administration of the sodium nitrite/sodium thiosulfate
antidote combination in five smoke inhalation victims with carbon
monoxide and cyanide poisoning and mean blood cyanide levels of
1.62 mg/l (62 µmol/l) has been reported (Hart et al., 1985). These
five patients also received hyperbaric oxygen therapy. There were
four survivors and one fatality in this series.
7.12 Summary of Evaluation
7.12.1 Indications
Sodium nitrite is indicated for use in acute cyanide poisoning.
It should not be used except where there is definite indication of
severe poisoning, such as loss of consciousness and deteriorating
vital functions. It is usually administered with sodium thiosulfate
and its administration may be preceded by the use of amyl nitrite.
7.12.2 Contraindications
Sodium nitrite should not be administered to asymptomatic
patients following exposure to cyanide. It should not be
administered to patients with smoke inhalation and combined carbon
monoxide and cyanide poisoning unless hyperbaric oxygen therapy is
available and such therapy has already been initiated.
G6DP-deficient individuals are theoretically at great risk from
nitrite therapy because of the likelihood of severe haemolysis,
although no such cases have been reported.
7.12.3 Advised route and dosage
Sodium nitrite is administered intravenously in an initial
adult dose of 300 mg (10 ml of a 3% solution) (Chen & Rose, 1952;
Hall & Rumack, 1986). While many authors recommend that this dose
be infused over a 5-min period, hypotension from the vasodilating
properties of sodium nitrite may occur. Hypotension may be avoided
by diluting sodium nitrite with normal saline and infusing the
medication over a 20-min period with frequent blood pressure
monitoring (Hall, 1986). The rate of infusion may be increased if
no hypotension occurs (Hall, 1986). Methaemoglobin levels should be
monitored to avoid excessive methaemoglobin induction (Berlin, 1970)
(see chapter 10). The paediatric dose for an average child,
frequently quoted in the literature and based on in vitro
experiments with canine blood, is 0.33 ml of a 3% solution per kg
body weight (about 10 mg/kg), administered intravenously with the
precautions noted above (Hall & Rumack, 1986). A pediatric dose of
approximately 0.2 ml/kg of a 3% solution has also been recommended
(Product Information, 1984). If there is reason to suspect anaemia,
tables devised to calculate a reduced dose, taking into account the
relatively lesser amount of haemoglobin present, should be consulted
(Berlin, 1970).
The adult dose may be lethal for a child. With a haemoglobin
level of 12 g/100 ml, only 10 mg/kg per body weight can be
administered immediately and 5 mg/kg repeated within 30 min if
necessary (Berlin, 1970). However, the calculation of Berlin was
based on a therapeutic level of methaemoglobinaemia of 15 mg/kg.
This may be too high because Weger (1968) observed severe
circulatory collapse in volunteers after 12 mg/kg was given
intravenously. In the same study, the administration of 4 mg/kg
(10 ml of a 3% solution NaNO2 solution, i.e., 300 mg) induced only
6% methaemoglobinaemia. As the adult dose of about 4 mg/kg has been
shown to be effective clinically, it may be safer to begin treatment
in children with sodium nitrite at 4 mg/kg (about 0.13 ml of a 3%
solution per kg body weight) and to administer additional sodium
nitrite only if excessive methaemoglobinaemia is not present and a
satisfactory clinical response has not occurred.
In both adults and children, another sodium nitrite dose of 50%
the initial amount administered may be repeated 30 min after the
first dose if there is inadequate clinical improvement (Hall &
Rumack, 1986).
7.12.4 Other consequential or supportive therapy
See section 1.10.
7.13 Model Information Sheet
7.13.1 Uses
Sodium nitrite is indicated for the treatment of acute cyanide
poisoning.
7.13.2 Dosage and route
The adult dose is 300 mg (10 ml of a 3% solution) infused
intravenously at the fastest rate possible without causing
hypotension (usually over 5 to 20 min). The initial paediatric dose
is 0.13-0.33 ml of a 3% solution per kg body weight (about
4-10 mg/kg). It is advisable to begin with lower doses in children
and increase to the desired effect. If anaemia is suspected,
standard tables should be consulted to calculate a reduced
paediatric sodium nitrite dose.
In both adults and children, another dose of one-half the
initial amount administered may be repeated 30 min after the first
dose if there is inadequate clinical improvement.
7.13.3 Precautions/contraindications
Excessive methaemoglobinaemia may occur, especially when doses
larger than those recommended are administered to children.
Hypotension may occur following rapid administration of sodium
nitrite, owing to its vasodilating properties. Blood pressure
should be monitored carefully during sodium nitrite administration,
and the infusion rate slowed if hypotension occurs.
Patients with smoke inhalation and combined carbon monoxide and
cyanide poisoning with elevated carboxyhaemoglobin levels should not
be given sodium nitrite unless treatment in a hyperbaric oxygen
chamber is available and such treatment has been initiated.
G6PD-deficient individuals are theoretically at great risk from
sodium nitrite therapy because of the likelihood of severe
haemolysis, although no such cases have been reported.
7.13.4 Adverse effects
Excessive methaemoglobinaemia may occur, especially with doses
exceeding those recommended. Hypotension may occur with rapid
intravenous infusion.
7.13.5 Use in pregnancy and lactation
There are no reported cases of the use of sodium nitrite during
pregnancy or lactation. Animal experiments indicate that some
sodium nitrite crosses the placenta and that fetal
methaemoglobinaemia may be induced. The risk to the fetus from
severe maternal cyanide poisoning would seem to be greater than the
risk of fetal methaemoglobin induction. No animal studies have
addressed the question of sodium nitrite excretion in breast milk or
its possible effects on the nursing infant.
7.13.6 Storage
Sodium nitrite should be stored at a controlled temperature of
15 to 30 °C (59 to 86 °F). The shelf-life is 5 years.
7.14 References
Aquanno JJ, Chan KM, & Dietzler DN (1981) Accidental poisoning of
two laboratory technologists with sodium nitrite. Clin Chem, 27:
1145-1146.
Asahina S, Friedman MA, Arnold E, Millar GN, Mishkin M, Bishop Y, &
Epstein SS (1971) Acute synergistic toxicity and hepatic necrosis
following oral administration of sodium nitrite and secondary amines
to mice. Cancer Res, 31: 1201-1205.
Bakshi SP, Fahey JL, & Pierce LE (1967) Sausage cyanosis - Acquired
methemoglobinemic nitrite poisoning. New Engl J Med, 277: 1072.
Baselt RC (1982) Cyanide and nitrite. In: Disposition of toxic drugs
and chemicals in man, 2nd ed. Davis, California, Biomedical
Publications, pp 209-214, 555-557.
Beamer WC, Shealy RM, & Prough DS (1983) Acute cyanide poisoning
from laetrile ingestion. Ann Emergency Med, 12: 449-451.
Becker CM (1985) The role of cyanide in fires. Vet Hum Toxicol, 27:
487-490.
Berlin AM (1970) The treatment of cyanide poisoning in children.
Pediatrics, 46: 793-796.
Braico KT, Humbert JR, Terplan KL, & Lehotay JM (1979) Laetrile
intoxication: Report of a fatal case. New Engl J Med, 300: 238-240.
Brivet F, Delfraissy JF, Duche M, Bertrand P, & Dormont J (1983)
Acute cyanide poisoning: Recovery with non-specific supportive
therapy. Intensive Care Med, 9: 33-35.
Burrows GE, Liu DHW, Isom GE, & Way JL (1982) Effect of antagonists
on the physiologic disposition of sodium cyanide. J Toxicol Environ
Health, 10: 181-189.
Chen KK & Rose CL (1952) Nitrite and thiosulfate therapy in cyanide
poisoning. J Am Med Assoc, 149: 113-119.
Chen KK & Rose CL (1956) Treatment of acute cyanide poisoning. J Am
Med Assoc, 162: 1154-1155.
Chen KK, Rose CL, & Clowes GHA (1933a) Methylene blue, nitrites, and
sodium thiosulphate against cyanide poisoning. Proc Soc Exp Biol
Med, 31 : 250-251.
Chen KK, Rose CL, & Clowes GHA (1933b) Potentiation of antidotal
action of sodium tetrathionate and sodium nitrite in cyanide
poisoning. Proc Soc Exp Biol Med, 31: 252-253.
Chen KK, Rose CL, & Clowes GHA (1934) Comparative values of several
antidotes in cyanide poisoning. Am J Med Sci, 188: 767-781.
Cohen MA & Guzzardi LJ (1984) A letter on the treatment of cyanide
poisoning. Vet Hum Toxicol, 26: 503-504.
De Busk RF & Seidl LG (1969) Attempted suicide by cyanide: A report
of two cases. California Med, 110: 394-396.
DiNapoli J, Hall AH, Orake R, & Rumack BH (1989) Cyanide and arsenic
poisoning by intravenous injection. Ann Emergency Med, 18: 308-311.
Edwards AC & Thomas ID (1978) Cyanide poisoning. Lancet, 1: 92-93.
Feihl F, Domenighetti G, & Perret Cl (1982) Intoxication massive au
cyanure avec évolution favorable. Schweiz Med Wochenschr, 112:
1280-1282.
Goldstein GM & Doull J (1971) Treatment of nitrite-induced
methemoglobinemia with hyperbaric oxygen. Proc Soc Exp Biol Med,
138: 137-139.
Gosselin RE, Smith RP, & Hodge HC ed. (1984) Nitrite. In: Clinical
toxicology of commercial products, 5th ed. Baltimore, Maryland,
Williams and Wilkins, pp 314-319.
Graham DL, Lamand D, Theodore J, & Robin ED (1977) Acute cyanide
poisoning complicated by lactic acidosis and pulmonary edema. Arch
Intern Med, 137: 1051-1055.
Grant WM (1986) Sodium nitrite. In: Toxicology of the eye, 3rd ed.
Springfield, Illinois, Charles C. Thomas, p 840.
Gruener N, Shuval HI, Behroozi K, Cohen S, & Shechter H (1973)
Methemoglobinemia induced by transplacental passage of nitrites in
rats. Bull Environ Contam Toxicol, 9: 44-48.
Hall AH (1986) Cyanide poisoning: Dealing with unexpected menace.
Emergency Med, 18: 919-205.
Hall AH & Rumack BH (1986) Clinical toxicology of cyanide. Ann
Emergency Med, 15: 1067-1074.
Hall AH, Kulig KW, & Rumack BH (1986a) Drug- and chemical-induced
methaemoglobinaemia: Clinical features and management. Med Toxicol,
1: 253-260.
Hall AH, Linden CH, Kulig KW, & Rumack BH (1986b) Cyanide poisoning
from laetrile ingestion: Role of nitrite therapy. Pediatrics, 78(2):
269-272.
Hall AH, Doutre W, Ludden T, Kulig K, & Rumack BH (1987)
Nitrite/thiosulfate treated acute cyanide poisoning: Estimated
kinetics after antidote. Clin Toxicol, 25: 121-133.
Hart GB, Strauss MB, Lennon PA, & Whitcraft DD (1985) Treatment of
smoke inhalation by hyperbaric oxygen. J Emergency Med, 3: 211-215.
Harvey SC (1980) Sodium nitrite. In: Osol A ed. Remington's
pharmaceutical science, 16th ed. Easton, Pennsylvania, Mack
Publishing Co., p 782.
Holmes RK & Way JL (1982) Mechanism of cyanide antagonism by sodium
nitrite. Pharmacologist, 24: 182 (Abstract).
Hug E (1932) L'intoxication par l'acide cyanhydrique. Action
antidote du bleu de méthylène, du nitrite de sodium et du sulfure de
sodium. C R Soc Biol (Paris), 111: 88-90.
Hug E (1933) Action combinée du nitrite de sodium dans le traitement
de l'intoxication cyanhydrique chez le lapin. C R Soc Biol (Paris),
114: 87-89.
Hug E & Marenzi AD (1933a) La fixation de l'acide cyanhydrique par
les hématies qui contiennent de la méthémoglobine. C R Soc Biol
(Paris), 114: 84-86.
Hug E & Marenzi AD (1933b) Mécanisme de l'action antidote du sodium
vis-à-vis de l'intoxication par l'acide cyanhydrique. C R Soc Biol
(Paris), 114: 86-87.
ITI (1985) Sodium nitrite. In: Toxic and hazardous industrial
chemicals safety manual. Tokyo, The International Technical
Information Institute, p 485.
Izmerov NF ed. (1982) IRPTC Scientific Reviews of Soviet Literature
on Toxicity and Hazards of Chemicals No. 22: Nitrites. Moscow,
Centre of International Projects, GKNT, 32 pp.
Jarvholm B, Lavenius B, & Sallsten G (1986) Cancer morbidity in
workers exposed to cutting fluids containing nitrites and amines. Br
J. Ind Med, 43: 563-565.
Jones J, McMullen MJ, & Dougherty J (1987) Toxic Smoke Inhalation:
cyanide poisoning in fire victims. Am J Emerg Med, 5: 318-321.
Korhonen A, Hemminki K, & Vainio H (1983) Toxicity of rubber
chemicals towards three-day chicken embryos. Scand J Work Environ
Health, 9: 115-119.
Kruszyna R, Kruszyna H, & Smith RP (1982) Comparison of
hydroxylamine, 4-dimethylaminophenol and nitrite protection against
cyanide poisoning in mice. Arch Toxicol, 49: 191-202.
Kruszyna H, Kruszyna R, & Smith RP (1985) Cyanide and sulfide
interact with nitrogenous compounds to influence the relaxation of
various smooth muscles. Proc Soc Exp Biol Med, 179: 44-49.
Kuroki T, Abbondandolo A, Drevon C, Huberman E, & Laval F (1980)
Mutagenesis assays with mammalian cells. In: Long-term and
short-term screening assays for carcinogens: A critical appraisal.
Lyons, International Agency for Research on Cancer, pp 107-133 (IARC
Monographs on the Evaluation of the Carcinogenic Risk of Chemicals
to Humans, Supplement 2).
Lasch EE & El Shawa R (1981) Multiple cases of cyanide poisoning by
apricot kernels in children from Gaza. Pediatrics, 68(1): 5-7.
Litovitz TL, Larkin RF, & Myers RAM (1983) Cyanide poisoning treated
with hyperbaric oxygen. Am J Emergency Med, 1: 94-101.
Lutier F, Dusoleil P, & De Montgros J (1971) Action de
l'hydroxocobalamine à dose massive dans l'intoxication aiguë au
cyanure. (A propos d'un cas). Arch Mal Prof, 32: 683-686.
MacDonald IA & Rao BG (1982) Nitrite-induced volatile fecal
mutagens: Some preliminary studies. Cancer, 49: 1405-1408.
MacQuiston TAC (1936) Fatal poisoning by sodium nitrite. Lancet, 2:
1153-1154.
Mansouri A (1985) Methemoglobinemia. Am J Med Sci, 298: 200-209.
Martindale (1989) In: Reynolds JEF ed. The extra pharmacopoeia, 29th
ed. London, The Pharmaceutical Press, p 854.
Mirvish SS, Salmasi S, Cohen SM, Patil K, & Mahboubi E (1983) Liver
and forestomach tumours and other forestomach lesions in rats
treated with morpholine and sodium nitrite, with and without sodium
ascorbate. J Natl Cancer Inst, 71: 81-85.
Moss M, Khalil N, & Gray J (1981) Deliberate self-poisoning with
laetrile. Can Med Assoc J, 125: 1126-1128.
Mota MM (1933) [On a case of intoxication by potassium cyanide
treated with sodium nitrite with survival.] Rev Med Rosario, 23:
670-674 (in Spanish).
Motin J, Bouletreau P, & Rouzioux JM (1970) Intoxication
cyanhydrique grave traitée avec succès par l'hydroxocobalamine. J
Med Strasbourg, 1: 717-722.
Musil J (1966) [The effects of chronic sodium nitrite intoxication
in rats.] Acta Biol Med Ger, 16: 388-394 (in German).
Padberg LR & Martin T (1939) Three fatal cases of sodium nitrite
poisoning. J Am Med Assoc, 113: 1733.
Paulet G & Menoret P (1954) Etude comparée de la sensibilité du sang
de l'homme (frais ou conservé) et du sang de chien à divers
méthémoglobinisants. C R Soc Biol (Paris), 148: 1014-1017.
Peters CG, Mundy JVB, & Rayner PR (1982) Acute cyanide poisoning:
The treatment of a suicide attempt. Anaesthesia, 37: 582-586.
Piantadosi CA, Sylvia AK, & Jobsis FF (1983) Cyanide-induced
cytochrome a,a3 oxidation-reduction responses in rat brain in vivo.
J. Clin Invest, 72: 1224-1233.
Product Information (1984) Product information for the cyanide
antidote package[R]. Indianapolis, Indiana, Eli Lilly & Co.
Rein H, Ristau O, Kupfer E, & Jung F (1968) [Nitrosohaemoglobin from
poisoning with sodium nitrite.] Folia Haematol, 90: 176-181 (in
German).
Rubino JM (1978) Cyanide toxicity: Report of a case. Connecticut
Poison Inf Bull, 3: 1-6.
Rumack BH ed. (1987) Cyanide poisoning management. In:
Poisindex[R]: Poison information system. Denver, Colorado,
Micromedex 52: 1-F4.
Sax NI (1984) Sodium nitrite. In: Dangerous properties of industrial
materials, 6th ed. New York, Van Nostrand Reinhold Co., pp
2442-2443.
Sayre JW & Kaymakcalan S (1964) Cyanide poisoning from apricot seeds
among children in central Turkey. New Engl J Med, 270: 1113-1115.
Schardein JL (1985) Chemically induced birth defects. New York,
Marcel Dekker, pp 74, 759, 810-824.
Sheehy MH & Way JL (1974) Nitrite intoxication: Protection with
methylene blue and oxygen. Toxicol Appl Pharmacol, 30: 221-226.
Shragg TA, Albertson TE, & Fischer CJ (1983) Cyanide poisoning after
bitter almond ingestion. West J. Med, 136: 65-69.
Standefer JC, Jones AM, Street E, & Inserra R (1979) Death
associated with nitrite ingestion: Report of a case. J Forensic Sci,
24: 678-771.
Stewart R (1974) Cyanide poisoning. Clin Toxicol, 7: 561-564.
Suarez KA & Bhonsle P (1978) Enhanced hepatotoxicity of carbon
tetrachloride following sodium nitrite pre-treatment. Arch Int
Pharmacodyn, 234: 329-334.
Surgenor DM (1970) Sodium nitrite USP. In: Osol E ed. Remington's
pharmaceutical sciences, 14th ed. Easton, Pennsylvania, Mack
Publishing Co., pp 846-847.
Ten Brink WAG, Wiezer JHA, Luijpen AFMG, Van Heijst ANP, Pikaar SA,
& Seldenrijk R (1982) Nitrate poisoning caused by food contaminated
with cooling fluid. J Toxicol Clin Toxicol, 19: 139-147.
Ten Eyck RP, Schaerdel AD, & Ottinger WE (1986) Comparison of
nitrite treatment and stroma-free methemoglobin solution as
antidotes for cyanide poisoning in a rat model. Clin Toxicol, 23:
477-487.
Tomoda A & Yoneyama Y (1979) On the relationship of hemoglobin
oxidation with the conformation of hemoglobin. Experientia (Basel),
35: 15-16.
United States Pharmacopeia (1985) Sodium nitrite and sodium nitrite
injection. In: United States Pharmacopoeia, 21st ed. Rockville,
Maryland, United States Pharmacopeial Convention, Inc., pp 974,
1186-1187, 1189-1190, 1206.
US NIOSH (1983) Registry of toxic effects of chemical substances
1981-1982. Atlanta, Georgia, National Institute of Occupational
Safety and Health, p 857.
Viana C, Cagnoli H, & Cedan J (1934) Action du nitrite de sodium
dans l'intoxication au cyanure. C R Soc Biol (Paris), 115:
1649-1651.
Vogel SN, Sultan TR, & Ten Eyck RP (1981) Cyanide poisoning. Clin
Toxicol, 18: 367-383.
Way JL, Gibbon SL, & Sheehy MH (1966) Effect of oxygen on cyanide
intoxication. I. Prophylactic protection. J Pharmacol Exp Ther, 153:
381-385.
Way JL, End E, Sheehy MH, De Miranda P, Feitknecht UF, Bachand R,
Gibbon SC, & Burrows GE (1972) Effects of oxygen on cyanide
intoxication. IV. Hyperbaric oxygen. Toxicol Appl Pharmacol, 22:
415-421.
Way JL, Sylvester D, Morgan RL, Isom GE, Burrows GE, Tamulinas CB, &
Way JL (1984) Recent perspectives on the toxicodynamic basis of
cyanid antagonism. Fundam Appl Toxicol, 4: S231-S239.
Weger N (1968) [Aminophenol as a cyanide antidote.] Arch Toxikol,
24: 49-50 (in German).
Weisburger JH, Horn CL, & Barnes WS (1983) Possible genotoxic
carcinogens in foods in relation to cancer causation. Semin Oncol,
10: 330-341.
Wesson DE, Foley R, Sabatini S, Wharton J, Kapusnik J, & Kurtzman NA
(1985) Treatment of acute cyanide intoxication with hemodialysis. Am
J Nephrol, 5: 121-126.
Windholz M, Budavari S, & Blumetti RF, ed. (1983) Sodium nitrite.
In: The Merck index, 10th ed. Rahway, New Jersey, Merck & Co., Inc.,
p 8480.
Wood GC (1982) Acute cyanide intoxication: Diagnosis and management.
Clin Toxicol Consultant, 4: 140-149.
Yacoub M, Faure J, Morena H, Vincent M, & Gaure H (1974)
L'intoxication cyanhydrique aiguë. Données actuelles sur le
métabolisme du cyanure et le traitement par hydroxocobalamine. J Eur
Toxicol, 7: 22-29.
8. 4-DIMETHYLAMINOPHENOL (4-DMAP)
8.1 Introduction
Cyanide has a special affinity for ferric ions, which are found
in cytochrome oxidase, the terminal oxidative respiratory enzyme in
mitochondria. Blood contains a substantial quantity of ferrous ions
within haemoglobin and it is possible to convert these readily to
ferric ions, for example, by the use of nitrites as suggested by
Chen et al. (1933). A disadvantage of the use of nitrites is the
fact that the concentration of methaemoglobin rises only slowly
after intravenous administration. In six experiments on volunteers,
using the dose of sodium nitrite suggested by Chen (4 mg/kg), a
methaemoglobin level of 6% resulted after 40 min (Kiese & Weger,
1969). This was considered to be too low a concentration of
methaemoglobin after too long a time. Experiments with several
aminophenols showed that the use of 4-dimethylaminophenol (4-DMAP)
resulted in rapid and controlled formation of methaemoglobin. An
intravenous dose of 3.25 mg/kg resulted in a methaemoglobin
concentration of 30% in 10 min, a methaemoglobin level of 15% being
reached within one min.
8.2 Name and Chemical Formula
Chemical name: Dimethyl( para)aminophenol
hydrochloride
Formula:
For reasons discussed in the text, this may only be an approximation
to the true structure of the material in Kelocyanor.
5.3 Physico-chemical Properties
The usual preparation of dicobalt edetate is Kelocyanor.a
Injectable Kelocyanor is a clear violet solution, produced in clear
glass 20-ml ampoules, of pH 4.0-7.5. It contains 0.196-0.240
g/100 ml free cobalt and 1.35-1.65 g/100 ml dicobalt edetate, as
well as 4 g glucose per ampoule. The refractive index of the
solution in the ampoule is 1.3638.
The optical rotation of Kelocyanor is + 9.25°: the optical
rotation of a dextrose solution of the same dextrose concentration
as Kelocyanor is +11°. Therefore the net optical rotation
attributable to the cobalt edetate is -1.75°. This would give a
specific rotation for dicobalt edetate of -116.7°. The relative
molecular mass of dicobalt edetate is 408. No information is
available on the specific gravity.
5.4 Synthesis
Dicobalt edetate is made by adding cobalt carbonate to sterile
water and then adding ethylenediaminetetraacetic acid. After the
addition of glucose, the resultant solution is cleared with
activated charcoal. Sterility and pyrogen tests are performed to
the requirements of the European Pharmacopoeia (1980). The volume
of solution in the ampoules is checked using method B (British
Pharmacopoeia, 1980). The material is not checked for the absence
of activated charcoal, but this would be expected to be removed
during filtration through a membrane with a pore diameter of
0.22 µm.
5.4.1 Source of materials
5.4.1.1 Cobalt carbonate
The current supplier to both the French and British drug
companies is Prolabo, BP No 200, 75526 Paris, Cedex 11, France. The
specification is as follows:
appearance: fine wine-coloured powder
identification: cobalt carbonate confirmed
loss on drying: about 14%
assay: about 50% cobalt
a L'Arguenon, Société d'Etudes et de Recherches Biologiques,
53 Rue Villiers de l'Isle Adam, 75020 Paris, France, UK
suppliers, Lipha Pharmaceuticals Ltd, Harrier House, Yiewsley,
Middlesex UB7 7QG.
5.4.1.2 Ethylenediaminetetraacetic acid (EDTA)
The current supplier is BDH Ltd, Poole, Dorset, BH12 4NN,
England. The material complies with the US National Formulary (see
USNF, 1985).
5.4.1.3 Glucose
This complies with the European Pharmacopoeia.
5.5 Analytical Methods
5.5.1 Free cobalt
In duplicate, pipette 5.0 ml of the solution in the ampoule
into a 250- ml conical flask. Add 100 mg of murexide compound
mixture and 100 ml water. Stir using a magnetic stirrer. Add
dilute ammonia drop by drop until the solution just turns yellow
(usually 1 drop). Titrate with disodium edetate (0.02 mol/l) to a
violet end-point. During the titration, the pH of the solution
falls drastically and it turns red. It is necessary to add 1 drop
of dilute ammonia to restore the yellow colour. Check the end point
of the titration with 1 drop of ammonia.
To prepare the murexide mixture, grind 10 mg murexide and
990 mg sodium chloride in a pestle and mortar. The dilute ammonia
solution consists of 5.5 ml concentrated ammonia diluted to 100 ml
with water.
The following equation is used to calculate the concentration
of free cobalt (g/100 ml):
T x 1.179 x F x 20
1000
where T = titre (ml)
F = factor for disodium edetate at 0.02 mol/l
1 ml disodium edetate (0.02 mol/l) equiv. 1.179 mg cobalt
5.5.2 Dicobalt edetate
In duplicate, pipette 5.0 ml of the solution in the ampoules
into a 250-ml conical flask. Add 0.65 g potassium cyanide, stir,
and allow to stand for 5 min. Add 85 ml of water, 10 ml lead
acetate solution, and 1 ml glacial acetic acid. Stir for 15 min,
using a magnetic stirrer. Add 100 mg xylenol orange compound
mixture and titrate slowly with disodium edetate (0.02 mol/l) to a
clear yellow end-point (T1). Carry out a blank titration,
omitting the sample solution (T2).
Xylenol orange compound mixture is prepared by grinding 10 mg
xylenol orange with 990 mg potassium nitrate in a pestle and mortar.
The lead acetate solution consists of 1.5 g lead acetate dissolved
in water and make up to 100 ml. The following equation is used to
calculate the concentration of dicobalt edetate (g/100 ml):
(T2-T1) x F x 4.06
25
where T1 = sample titre
T2 = blank titre
F = factor for disodium edetate (0.02 mol/l)
5.5.3 Analysis in biological fluids
Since dicobalt edetate has not been fully characterized, it is
difficult to say whether analysis would be meaningful. Methods are
available for the analysis of cobalt in biological fluids by
spectrophotometry or atomic absorption flame photometry (Thiers et
al., 1955; Mulford, 1966; Christian, 1969; Lewis et al., 1985).
Dicobalt edetate interferes with the Feldstein & Klendshoj
(1954) method of analysis of biological fluids for cyanide
(Ballantyne & Marrs, 1987). See chapter 10 for further details.
5.6 Stability and Shelf-life
The shelf-life is 3 years at 25 °C, and the material should be
stored in the dark. The drug has been stored for up to 3 years at
room temperature 20-25, 30 and 37 °C (Lipha Pharmaceuticals,
1987)a, the batches having been tested at regular intervals
(12 months maximum) for compliance with the finished product
specification with respect to appearance, extractable volume, pH,
and free cobalt and dicobalt edetate content. The only deviation
noted was that the colour of the solution became lighter at 30 °C
and above. All the other variables remained within the
specification. If stored in the light, the drug bleaches in about
one month. The nature of the chemical change that brings about the
loss of colour is not known.
a Personal communication from Lipha Pharmaceuticals Ltd, West
Drayton, Middlesex, United Kingdom.
5.7 General Properties
Cyanide blocks intracellular respiration by binding to
cytochrome oxidase. Cobalt forms a stable complex with cyanide, the
effect being direct and peripheral (Mercker & Bastian, 1959).
Although some cobalt compounds are reputed to inhibit methaemoglobin
reductase (Hagler & Coppes, 1982), it is unlikely that
methaemoglobinaemia contributes meaningfully to the anticyanide
action of dicobalt edetate.
5.8 Animal Studies
5.8.1 Pharmacokinetics
Formal pharmacokinetic studies have not been carried out on
dicobalt edetate. Some of the effects observed with cobalt
compounds, including dicobalt edetate, are probably centrally
mediated (Bartelheimer, 1962a). Thus, it is likely that after
Kelocyanor injection, cobalt or dicobalt edetate crosses the
blood-brain barrier. In the mouse, the cyanide-cobalt complex is
excreted in the urine (Frankenberg & Sörbo, 1975).
5.8.2 Pharmacodynamics
5.8.2.1 Efficacy in animals
The introduction of dicobalt edetate followed from the work of
Paulet (1957, 1958, 1960a,b, 1961, 1965) and Paulet et al. (1960),
who studied the antidotal effects and toxicity of various cobalt
compounds. The aim of studies on cobalt antidotes at this time was
to find a compound that retained the ability of cobalt to bind
cyanide, yet lacked its toxic effects. The compounds investigated
by Paulet (1960a,b) included certain cobalt (II) salts, namely the
chloride, glutamate, and gluconate. Paulet (1960a) also studied the
cobalt chelates, dicobalt edetate, cobalt histidine, and disodium
monocobalt edetate and found that the salts, as well as all except
the last named of the chelates, possessed appreciable anticyanide
activity. Dicobalt edetate and cobalt histidine were the most
promising, and in one study dicobalt edetate seemed preferable
(Paulet, 1960a, 1961). This study was undertaken in anaesthetized
dogs infused with sodium cyanide (0.1 mg/kg per min): both dicobalt
edetate and cobalt histidine were capable of resuscitating the dogs
in secondary apnoea, but the dose of cobalt histidine required was
considerably higher than that of dicobalt edetate. In the same
reports, the effective doses of the two compounds were compared with
their respective intraperitoneal LD50 values in mice, the result
favouring dicobalt edetate. However, if the dog results had been
tabulated against the rat intravenous LD50 values (Tauberger &
Klimmer, 1963), cobalt histidine might have been preferred, this
antidote having a much higher intravenous LD50 than dicobalt
edetate.
Evans (1964) investigated the stoichiometry of the reaction
between dicobalt edetate or other cobalt compounds on the one hand,
and cyanide on the other. He studied their antidotal effects using
various doses of both toxicant and treatment in both mice and
rabbits, designing his experiments in such a way that it was
possible to calculate the cyanide/cobalt molar ratios above which
the antidote would no longer be efficacious. He found that, while
most cobalt (II) salts would be effective against six moles of
cyanide/mole cobalt, dicobalt edetate only possessed efficacy at
molar ratios of up to two. Whilst on a mass basis, dicobalt
edetate is about half as toxic as cobalt (II) salts, on a molar
basis it is not conspicuously less toxic than cobalt acetate. It
had been thought that dicobalt edetate was the monocobalt (II) salt
of a monocobalt edetate anion, one cobalt being completely
unavailable for cyanide binding by virtue of its complexing with
EDTA, the other being fully available. In fact the study of Evans
(1964) suggested that one cobalt is completely unavailable whilst,
of the coordination sites of the second, only two are available to
bind cyanide. In that case, the structure given in Fig 4 does not
fully describe the structure of dicobalt edetate. It seems likely,
however, that the second cobalt atom is required for anticyanide
activity, since disodium monocobalt edetate, which is a compound of
very low toxicity (Eybl et al., 1959), is almost ineffective as a
cyanide antidote (Paulet, 1958). It may be that the same property,
ionizability, is responsible for cyanide binding and for cobalt
toxicity. In that case, complexing cobalt would decrease the
therapeutic effect in parallel with the toxicity. That this is not
the case is suggested by, firstly, the lack of toxicity of
hydroxocobalamin, which is nevertheless a successful cyanide
antidote and, secondly, the many years of successful use of
dicobalt edetate.
5.8.2.2 Comparison of dicobalt edetate with other compounds
A number of research groups have compared cobalt edetate with
other cyanide antidotes, from the point of view both of antidotal
efficacy (Table 4) and also of toxicity. For example, using dogs
and rabbits, Paulet (1960a, 1961) compared the cobalt chelate with
sodium nitrite and found the former superior. He also carried out a
comparison with both elements of the sodium nitrite/thiosulfate
treatment, again demonstrating that the cobalt compound was more
effective. Terzic & Milosevic (1963) compared sodium nitrite and
dicobalt edetate. They studied the effective doses of the two
compounds, as well as their LD50 values, and concluded that cobalt
edetate was superior. Unfortunately their study was conducted in
mice: these animals especially, but also other small laboratory
animals, are extremely insensitive to methaemoglobin-producing
chemicals by virtue of the very high activity of NADH-linked
methaemoglobin reductase in murine erythrocytes (Calabrese, 1983).
This difference renders extrapolation of these results to man almost
impossible. Two research groups have attempted to compare dicobalt
edetate with 4-DMAP in dogs. Klimmek et al. (1979a) found better
survival with 4-DMAP when the antidotes were given 4 min after the
poisoning but similar antidotal efficacy when they were given 1 min
afterwards. Marrs et al. (1985) found that dicobalt edetate
successfully treated up to about three times the LD50 of cyanide,
when given at the moment of apnoea, whilst 4-DMAP ensured survival
at about six time the LD50 under the same conditions.
5.8.2.3 Interactions with other drugs
The only interactions that have been studied in animals are
those with other cyanide antidotes. The effect, in experimental
animals, is generally additive. This is despite the theoretical
risk that cobalt toxicity might be exacerbated by other antidotes
that prevent cyanide from binding to cobalt. Antidotal combinations
including dicobalt edetate have been studied by Evans (1964) and
Frankenberg & Sörbo (1975).
Table 4. Studies of the efficacy of dicobalt edetate
compared with other cyanide antidotes
Comparison of dicobalt Species Author
edetate with
Sodium nitrite Dog Paulet (1960 a,b)
Sodium nitrite/thiosulfate Dog Paulet (1960 a,b)
Sodium nitrite Mouse Terzic & Milosevic (1963)
4-DMAP Dog Klimmek et al. (1979a)
4-DMAP Dog Marrs et al. (1985)
Thiosulfate/rhodanese Rabbit Atkinson et al. (1974)
Other cobalt antidotes Mouse/Rabbit Evans (1964)
5.8.3 Toxicology
Extensive toxicology was not carried out on dicobalt edetate
before its introduction. Published information is largely confined
to acute toxicity testing, but it is believed that the
organ-specific toxicity is similar to that of cobalt salts and that
it is ameliorated by chelation (Paulet, 1960b).
5.8.3.1 In vitro studies
Dicobalt edetate gives negative results in the Ames test
(Morris, 1987)
5.8.3.2 Acute toxicity studies
The acute toxicity of dicobalt edetate has been studied
exclusively in small laboratory animals (Table 5). An interesting
feature is that the acute toxicity is decreased by the presence of
glucose or of cyanide. Thus, Paulet (1960a,b, 1961) observed that,
in the mouse, the intravenous LD50 of dicobalt edetate (50 mg/kg)
was increased threefold by giving it in hypertonic glucose. The
LD50 was increased to 82 mg/kg or 103 mg/kg, respectively, by the
simultaneous administration of 2.5 or 5 mg NaCN/kg.
5.8.3.3 Repeated dose toxicity
Bartelheimer (1962b) gave intraperitoneal injections of cobalt
edetate daily for 14 days at doses of 30, 40, or 50% of the LD50
to rats, surviving animals being observed for 4 weeks. One of six
and two of six of the high and middle groups, respectively, died,
whilst none of the low-dose group did so. The majority of the rats
in the two higher groups suffered diarrhoea and weight loss,
although some recovery was seen in the latter half of the exposure
period. Autopsy and histological examination of the dead animals
showed necrosis of the intestinal mucous membrane. No pathological
changes were seen in the liver, kidney, or heart. Late effects were
not observed in the survivors.
Table 5. The acute toxicity of dicobalt edetate
Species Route LD50 (mg/kg) Reference
Mouse iv 50 Paulet (1961)
Mouse iv 71 Evans (1964)
Mouse ip 213 Paulet (1961)
Mouse ip 225 Terzic & Milosevic (1963)
Rat iv 43 Tauberger & Klimmer (1963)
Rat ip 100 Bartelheimer (1962b)
5.8.3.4 Circulatory effects in dogs
Klimmek et al. (1979b), during their studies comparing dicobalt
edetate with 4-dimethylaminophenol (4-DMAP), found that the former
caused circulatory depression and hyperventilation. This was
accompanied by metabolic acidosis.
5.8.3.5 Other toxicity studies
No information is available on reproductive toxicology or
carcinogenicity. No mammalian cell or in vivo mutagenicity test has
been published.
5.9 Volunteer Studies
No human volunteer studies have been carried out.
5.10 Clinical Trials
Clinical trials have not been undertaken.
5.11 Clinical Studies - Case Reports
5.11.1 Successful use
There are a number of case reports of the successful use of
dicobalt edetate against different types of cyanide poisoning,
including inhalation of HCN (e.g. Bain & Knowles, 1967; Nagler et
al., 1978) and sodium cyanide (Hillman et al., 1974), and potassium
cyanide ingestion (Hoang The Dan et al., 1981; Klaui et al., 1984).
The use of dicobalt edetate in poisoning with fused sodium cyanide
has also been reported (Bourrelier & Paulet, 1971). The case
reported by Bain & Knowles involved a patient who was semicomatose
but who became fully conscious immediately after the first of two
doses of 10 ml of Kelocyanor: the blood cyanide concentration just
before the antidote injection was found to be 5.1 mg/l. In the
report by Nagler et al. (1978), three workers were poisoned as a
result of the accidental addition of sodium cyanide to an acid bath.
In each case the patients had definite evidence of cyanide
poisoning, yet recovered. A blood cyanide level of 5.25 mg/l was
found in the patient reported by Hoang The Dan et al. (1981), yet
the subject recovered without sequelae.
5.11.2 Use in pregnant women and children
No data exist on the use of dicobalt edetate in pregnant women
or children.
5.11.3 Adverse effects
A number of adverse effects have been reported, generally
following the inappropriate administration of dicobalt edetate. It
has been proposed that their occurrence can be minimized by using
strict clinical criteria for giving the antidote (Bryson, 1978,1987;
Tyrer, 1981; Aw & Bishop, 1981; Peden et al., 1986). Nevertheless,
it has been suggested that adverse reactions to dicobalt edetate may
not be confined to instances of inappropriate administration (Dodds
& McKnight, 1985). Typical case reports of adverse reactions
include that of Froneman (1975), who reported a massive urticarial
reaction affecting the face, eyelids, and lips; it was questioned if
the subject had, in fact, consumed any cyanide. Four adverse
reactions, where the diagnosis of cyanide poisoning was
questionable, were reported by Tyrer (1981). Three of the patients
had a variety of symptoms and signs, including convulsions, oedema
of the face and neck, urticaria, chest pains, dyspnoea, and
hypotension. Hypotension was also reported by Daunderer et al.
(1974) (see section 5.11.4). A patient described by McKiernan
(1980) had a normal blood cyanide level, in spite of severe cyanide
burns. He reacted to Kelocyanor by developing facial and laryngeal
oedema. A case reported by Yacoub et al. (1974) was complicated by
the use of other antidotes; this patient developed urticaria.
5.11.4 Use in combination with other antidotes
Reference has been made above (section 5.8.3) to animal studies
that suggest that the use of dicobalt edetate in combination with
other antidotes could be beneficial. Thus, Jeretin (1963) used it
with sodium nitrite and thiosulfate, and Daunderer et al. (1974)
successfully resuscitated with 4-DMAP and Kelocyanor a patient who
had ingested about 10 g potassium cyanide.
5.12 Summary of Evaluation
5.12.1 Indications
The only indication for the use of dicobalt edetate is acute
cyanide poisoning. It should not be used except where there is
definite indication of poisoning. Reactions are likely to occur if
the drug is given inappropriately.
5.12.2 Administration
It is recommended that one ampoule should be given over one
min. A second or third may be given in the case of inadequate
response. Because glucose reduces the toxicity of dicobalt edetate
(Paulet et al., 1960; Paulet, 1961), the recommendation is that the
injections should be immediately followed by 50 ml dextrose
(500 g/l).
5.12.3 Other consequential or supportive therapy
See section 1.10
5.12.4 Contraindications
Dicobalt edetate should not be used in suspected cyanide
poisoning unaccompanied by signs of poisoning such as impairment or
loss of consciousness.
5.12.5 Comparison with other antidotes
Dicobalt edetate probably crosses the blood-brain barrier,
while methaemoglobin quite clearly does not. Moreover, both DMAP
and nitrite require follow-up with a sulfur donor; with dicobalt
edetate the cyanide is excreted with cobalt in the urine. In these
two respects dicobalt edetate seems to offer some advantages. It is
in clinical use that reservations have arisen, almost entirely
because of adverse reactions. Whereas occupational health
practitioners seem to be particularly impressed with dicobalt
edetate (Davison, 1969; Bryson, 1987), it is in hospital practice
that problems are likely to occur. The probable reason for this is
that, at cyanide-using facilities, not only is the antidote to hand,
but also occupational health personnel are familiar with the
presentation of cyanide poisoning and the hazards of inappropriate
use of antidotes. A particular problem of this form of poisoning is
that insubstantial or suspected cyanide poisoning may produce panic,
which is mistaken for incipient intoxication. It is then that the
antidote is given (Anon, 1977). Provided it is given only when the
patient is unconscious many of the problems can be avoided. While
in the past it was suggested that in casualty departments Kelocyanor
should be given to all patients with symptoms (Anon, 1977), this
policy is no longer advocated. Were the use of dicobalt edetate to
be confined to cases of unequivocal poisoning with cyanide, it seems
likely that adverse reactions would be far less frequent.
5.13 Model Information Sheet
5.13.1 Uses
Dicobalt edetate is a specific antidote for use in acute
cyanide poisoning.
5.13.2 Dosage and route
It should be given in the form of ampoules of Kelocyanor, which
also contain glucose. One ampoule (300 mg dicobalt edetate) should
be given intravenously over 1 min. If the response is inadequate, a
second ampoule may be given. The use of Kelocyanor should be
followed immediately by 50 ml Dextrose IV infusion (500 g/l).
5.13.3 Precautions/contraindications
Kelocyanor should not be administered in the absence of
cyanide. Fully conscious patients, especially those exposed to HCN
by inhalation, are unlikely to require Kelocyanor.
5.13.4 Adverse effects
These include facial, laryngeal, and neck oedema, chest pain,
vomiting, and rashes.
5.13.5 Use in pregnancy and lactation
There is no experience of use in this situation. In
life-saving situations the recommended dosage should not be
modified.
5.13.6 Storage
This antidote should be stored at a temperature below 25 °C.
The shelf-life is 3 years.
5.14 References
Anon (1977) Which antidote for cyanide? Lancet, 2: 1167.
Atkinson A, Rutter DA, & Sargeant K (1974) Enzyme antidote for
experimental cyanide poisoning. Lancet, 2: 1446.
Aw TC & Bishop CM (1981) Cyanide poisoning. J Soc Occup Med, 31:
173-175.
Bain JTB & Knowles EL (1967) Successful treatment of cyanide
poisoning. Br Med J, 2: 763.
Ballantyne B & Marrs TC (1987) Postmortem features and criteria for
the diagnosis of acute cyanide poisoning. In: Ballantyne B & Marrs
TC ed. Clinical and experimental toxicology of cyanides. Bristol,
United Kingdom, John Wright, pp 217-247.
Bartelheimer EW (1962a) [Analysis of acute cobalt poisoning in
experimental animals.] Naunyn-Schmiedebergs Arch Exp Pathol
Pharmakol, 243: 237-253 (in German).
Bartelheimer EW (1962b) [Differences between the toxic and
cyanide-antagonistic efficacy of cobalt chelate compounds
(Co-histidine and CO2 - EDTA.] Naunyn-Schmiedebergs Arch Exp
Pathol Pharmakol, 243: 254-268 (in German).
British Pharmacopoeia (1980) London, Her Majesty's Stationery
Office, vol. 2, p 579.
Bourrelier J & Paulet G (1971) Intoxication cyanhydrique consécutive
à des brûlures graves par cyanure de sodium fondu. Presse Méd, 79:
1013-1014.
Bryson D (1978) Cyanide poisoning. Lancet, 1: 92.
Bryson D (1987) Occupational cyanide poisoning. In: Ballantyne B &
Marrs TC ed. The clinical and experimental toxicology of cyanides.
Bristol, United Kingdom, John Wright, pp 348-358.
Calabrese EJ (1983) Principles of animal extrapolation. New York,
John Wiley & Sons.
Christian GD (1969) Medicine, trace elements, and atomic absorption
spectroscopy. Anal Chem, 41: 24A.
Cotton FA & Wilkinson G (1966) Advanced inorganic chemistry, 2nd ed.
New York, London, Interscience Publishers, p 869.
Daunderer M, Theml H, & Weger N (1974) [Treatment of prussic acid
poisoning with 4-dimethylaminophenol (4-DMAP).] Med Klin, 69:
1626-1631 (in German).
Davison V (1969) Cyanide poisoning. Kelocyanor - a new treatment.
Occup Health, 21: 306-308.
Dodds C & McKnight C (1985) Cyanide toxicity after immersion and the
hazards of dicobalt edetate. Br Med J, 291: 785-786.
European Pharmacopoeia (1980) 2nd ed. Sainte Ruffine, France,
Maisonneuve SA.
Evans CL (1964) Cobalt compounds as antidotes for hydrocyanic acid.
Br J Pharmacol, 23: 455-475.
Eybl V, Sykora J, & Kocher Z (1959) [The influence of EDTA on the
effects of cobalt in vivo.] Naunyn-Schmiedebergs Arch Exp Pathol
Pharmakol, 236: 199-201 (in German).
Feldstein MA & Klendshoj WJ (1954) The determination of cyanide in
biologic fluids by microdiffusion analysis. J Lab Chem Med, 44:
166-170.
Frankenberg L & Sörbo B (1975) Effect of cyanide antidotes on the
metabolic conversion of cyanide to thiocyanate. Arch Toxicol, 33:
81-89.
Froneman JPC (1975) A first experience in the use of Kelocyanor.
Proc Mine Off Assoc (Johannesburg), 54: 43.
Hagler L & Coppes RI (1982) Inhibition of methemoglobin and
metmyoglobin reduction by cobalt. Biochem Pharmacol, 31: 1779-1782.
Hillman B, Bardhan KD, & Bain JTB (1974) The use of dicobalt edetate
(Kelocyanor) in cyanide poisoning. Postgrad Med J, 50: 171-174.
Hoang The Dan P, Bretsztajn A, Pourriat JL, Lapandry C, & Cupa M
(1981) Un cas d'intoxication aiguë au cyanure. Anesth Anal Réanim,
38: 161-162.
Jeretin S (1963) [A case of successful recovery of a patient
suffering from potassium cyanide poisoning.] Zdrav Vestn, 32: 3-4
(in Slovene).
Klaui H, Russi E, & Bauman PC (1984) [Cyanide poisoning.] Schweiz
Med Wochenschr, 114: 983-989 (in German).
Klimmek R, Fladerer H, & Weger N (1979a) Circulation, respiration
and blood homeostasis in cyanide-poisoned dogs after treatment with
4-dimethylaminophenol or cobalt compounds. Arch Toxicol, 43:
121-133.
Klimmek R, Fladerer H, Szinicz L, Weger N, & Kiese M (1979b) Effects
of 4-dimethylaminophenol and CO2 EDTA on circulation, respiration,
and blood homeostasis in dogs. Arch Toxicol, 42: 75-84.
Lewis SA, O'Haver TC, & Harnley JM (1985) Determination of metals at
the microgram-per-liter level in blood serum by simultaneous
multielement atomic absorption spectrometry with graphite furnace
atomization. Anal Chem, 57: 2-5.
McKiernan MJC (1980) Emergency treatment of cyanide poisoning.
Lancet, 2: 1980.
Marrs TC, Swanston DW, & Bright JE (1985) 4-Dimethylaminophenol and
dicobalt edetate (Kelocyanor) in the treatment of experimental
cyanide poisoning. Hum Toxicol, 4: 591-600.
Mercker H & Bastian G (1959) [Cobalt compounds for prussic acid
detoxification.] Naunyn-Schmiedebergs Arch Exp Pathol Pharmakol,
236: 449-458 (in German).
Morris B (1987) Postmortem features and criteria for the diagnosis
of acute lethal cyanide poisoning In: Ballantyne B & Marrs TC ed.
Clinical and experimental toxicology of cyanides. Bristol, United
Kingdom, John Wright, pp 217-247.
Mulford CE (1966) Solvent extraction techniques for atomic
absorption spectroscopy. At Absorpt Newsl, 5: 88-90.
Mushett CW, Kelly KL, Boxer GE, & Rickards JC (1952) Antidotal
efficacy of vitamin B12a (hydroxo-cobalamin) in experimental
cyanide poisoning. Proc Soc Exp Biol Med (NY), 81: 234, 237.
Nagler J, Provoost RA, & Parizel G (1978) Hydrogen cyanide
poisoning: treatment with cobalt EDTA. J Occup Med, 20: 414-416.
Paulet G (1957) Valeur des sels organiques du cobalt dans le
traitement de l'intoxication cyanhydrique. C R Soc Biol (Paris),
151: 1932-1935.
Paulet G (1958) Intoxication cyanhydrique et chélates de cobalt. J
Physiol (Paris), 50: 438-442.
Paulet G (1960a) L'intoxication cyanhydrique et son traitement.
Paris, Masson SA.
Paulet G (1960b) Les chélates de cobalt dans le traitement de
l'intoxication cyanhydrique. Pathol Biol, 8: 256-266.
Paulet G (1961) Nouvelles perspectives dans le traitement de
l'intoxication cyanhydrique. Arch Mal Prof, 22: 102-127.
Paulet G (1965) Au sujet du traitement de l'intoxication
cyanhydrique par les chélates de cobalt. Urgence, 11: 611-613.
Paulet G, Chary R, Bocquet P, & Fouilhoux M (1960) Valeur comparée
du nitrite de sodium et des chélates de cobalt dans le traitement de
l'intoxication cyanhydrique chez l'animal (chien-lapin)
non-anesthésié. Arch Int Pharmacodyn, 127: 104-117.
Peden NR, Taha A, McSorley PD, Bryden GT, Murdoch IB, & Anderson JM
(1986) Industrial exposure to cyanide: implications for treatment.
Br Med J, 293: 538.
Sharpe AT (1976) The chemistry of cyano complexes of the transition
metals. New York, London, Academic Press.
Speijers GJA, Krajnc EI, Berkvens JM, & Van Logten MJ (1982) Acute
oral toxicity of inorganic cobalt compounds in rats. Food Chem
Toxicol, 20: 311-314.
Tauberger G & Klimmer OR (1963) [Animal experimental studies of some
cobalt compounds after intravenous injection.] Arch Int Pharmacodyn,
143: 219-239 (in German).
Terzic M & Milosevic M (1963) Action protectrice de
l'éthylène-diamine-tétra-acétate-dicobaltique dans l'intoxication
cyanée. Thérapie, 18: 55-61.
Thiers RE, Williams JF, & Yoe JH (1955) Separation and determination
of millimicrogram amounts of cobalt. Anal Chem, 27: 1725-1731.
Tyrer FH (1981) Treatment of cyanide poisoning. J Soc Occup Med, 31:
65-66.
United States National Formulary (1985) 16th ed. Rockville,
Maryland, US Pharmacopeial Convention Inc., p 1559.
Yacoub M, Faure J, Morena H, Vincent M, & Gaure H (1974)
L'intoxication cyanhydrique aiguë. Données actuelles sur le
métabolisme du cyanure et le traitement par hydroxocobalamine. J Eur
Toxicol, 7: 22-29.
6. AMYL NITRITE
6.1 Introduction
Amyl nitrite by inhalation has been used for many years as a
simple first-aid measure for cyanide poisoning. It generates
methaemoglobin and may be administered by lay personnel.
Methaemoglobin combines with cyanide to form non-toxic
cyanmethaemoglobin. However, amyl nitrite is not a powerful
methaemoglobin-forming agent in humans and therefore it has
generally been used in combination with intravenous sodium nitrite.
Of 44 patients poisoned with cyanide, and of which 43 recovered
[Chen et al., 1944 (15 patients); Wolfsie, 1951 (12 patients);
Miller & Toops, 1951 (1 patient); and Chen & Rose, 1952 (16
patients)], only 7 patients were treated with amyl nitrite alone; 2
were treated with amyl nitrite in combination with sodium
thiosulfate.
Recently there has been a resurgence of interest in amyl
nitrite. Artificial respiration with amyl nitrite broken into an
Ambu bag was reported by Vick & Froelich (1985) as being life-saving
in dogs severely poisoned with cyanide before significant formation
of methaemoglobin occurred (Vick & Froelich, 1985). The vasogenic
effects of amyl nitrite may therefore play a role in its antidotal
effect in cyanide poisoning.
6.2 Name and Chemical Formula
Chemical name: Amyl nitrite, Amylis nitris, Nitris amylicus
Molecular formula: C5H11NO2
Structural formula:
Amyl nitrite is a mixture of nitrite esters of
2-methylbutan-l-ol and 3-methylbutan-l-ol.
ONO ONO
| |
CH2 CH2
nitrate ester of | | nitrate ester of
2-methylbutan-l-ol HCCH3 CH2 3-methylbutan-l-ol
| |
CH2 HCCH3
| |
CH3 CH3
It contains not less than 85% and not more than 103% of
C5H11NO2 (Martindale, 1989; United States Pharmacopeia, 1985).
Relative molecular mass: 117.15
Appearance: yellow, transparent fluid
CAS numbers: 8017-89-8 and 110-46-3
IUPAC name: -
6.3 Physio-chemical Properties
Boiling point: 96 °C
Solubility: practically insoluble in water;
miscible with alcohol, chloroform,
ether, and light petroleum
Boiling point: no data available
Acidity: no data available
pK: no data available
Stability in light: protect from light
Thermal stability: it is volatile even at low
temperatures and is liable to
decompose with the evolution of
nitrogen, particularly if it has
become acidic. Flash point 10 °C
(closed-cup test)
Specific gravity: 0.8970-0.880
Refractive index: 1.386-1.389 (Nederlandse
Pharmacopee, 1958)
Explosive properties: amyl nitrite forms an explosive
mixture with air and oxygen. It is
very inflammable and must not be
used where it may be ignited.
6.4 Synthesis
Amyl nitrite is generally manufactured by the action of sodium
nitrate and sulfuric acid on the appropriate alcohols.
6.5 Analytical Methods
6.5.1 Identification
a) To a mixture of 2 drops of amyl nitrite and 2 drops of
water, add 2 ml of sulfuric acid and dilute with water.
The odour of amyl valerate should become apparent.
b) To a few drops of amyl nitrite, add a mixture of 1 ml of
hydrochloric acid (3 mol/l). A greenish-brown colour
should be produced (United States Pharmacopeia, 1985).
6.5.2 Purity
6.5.2.1 Acidity
To 0.30 ml in a glass-stoppered cylinder, add a mixture of
0.60 ml of sodium hydroxide (0.1 mol/l), 10 ml of water, and 1 drop
of phenolphthalein TS, and invert the cylinder three times. The red
tint of the water layer should remain perceptible (United States
Pharmacopeia, 1985).
6.5.2.2 Non-volatile residue
Allow 10 ml to evaporate at room temperature in a weighed
evaporating dish within a well-ventilated hood, and dry the residue
at 105 °C for 1 h. The weight of the residue should not exceed 2 mg
(0.02%) (United States Pharmacopeia, 1985).
6.5.2.3 Assay for total nitrites
Inject an aliquot of amyl nitrite of suitable volume, but not
more than 2 ml, into a suitable gas chromatograph equipped with a
thermal conductivity detector. Under typical conditions, the
instrument should contain a column (2 m x 3 mm) packed with a methyl
polysiloxane oil, 25% by weight on suitable calcine diatomite. The
column should be maintained at 80 °C, the injection port and
detector block maintained at 10 °C above the temperature of the
column, and helium used as a carrier gas at a flow rate of about
60 ml per min. The percentage of total nitrites is calculated from
the area under the curve; a total of not less than 97% should be
found.
6.6 Shelf-life
The following precautions should be taken.
* Protect from light.
* Insist that the suppliers only provide ampoules that
have at least 18 month shelf-life left of the nominal
24-month total life.
* Take reasonable precautions to store at the lowest
possible temperature compatible with immediate access
in case of emergency; refrigerated storage is not
necessary.
* Inspect monthly for any broken tubes (Beasley et
al., 1978).
As indicated in section 6.3, amyl nitrite forms an explosive
mixture with air and oxygen. It is highly inflammable and must not
be used where it may be ignited. Storage for a prolonged period or
at an excessive temperature presents the following hazards:
* breakage of the capsule by the build up of pressure
internally and loss of amyl nitrite before use;
* explosive distribution of glass splinters when the
ampoule is broken for use;
* loss of amyl nitrite by chemical decomposition.
6.7 General Properties
The principal pharmacological action of amyl nitrite is to
cause the relaxation of vascular smooth muscle. Peripheral venous
resistance is decreased as a result of a selective action on venous
capacitance vessels with resultant venous pooling of blood and
decreased venous return to the heart. The vasodilating effect of
amyl nitrite on arteriolar resistance is less than that on the
venous side. As a result of this combined action, both venous
filling pressure (preload) and, to a lesser extent, arterial
impedance (afterload) are reduced (AHFS, 1988).
It has been postulated that cyanide causes pulmonary and/or
coronary arteriolar vasoconstriction, which results directly or
indirectly in pump failure and an observed decrease in cardiac
output (Sarnoff, 1980a). This theory is supported by the reported
observations of Vick & Froelich (1985) that a sharp increase in
central venous pressure occurs at a time when the arterial blood
pressure is falling. Following the administration of amyl nitrite
by inhalation, central venous pressure decreased rapidly while the
arterial blood pressure increased. The apparent life-saving effect
of this drug in dogs could have been due to a reversal of early
cyanide-induced vasoconstriction and the restoration of normal
cardiac function. Subsequently, amyl nitrite causes the formation of
methaemoglobin, which may have an additional antidotal action.
a Personal communication by S.J. Sarnoff, Survival Technology
Inc., Bethesda, USA.
6.8 Animal Studies
6.8.1 Pharmacokinetics
Amyl nitrite is readily absorbed into the circulation from
mucous membranes, the rate of absorption being greatest from the
lungs. It is rapidly inactivated by hydrolysis to isoamyl alcohol
and nitrite. Amyl nitrite is rapidly hydrolysed in the
gastrointestinal tract and is therefore pharmacologically inactive
when administered orally (Martindale, 1989).
6.8.2 Pharmacodynamics
Amyl nitrite has proved to be effective in experimental animals
poisoned by cyanide. Indeed, antagonism between amyl nitrite and
hydrocyanic acid was mentioned as long ago as l888 by Pedigo.
Chen et al. (1933) demonstrated that nine to ten inhalations of
amyl nitrite were effective in dogs against 4 times the lethal dose
of cyanide (12-24 mg/kg) (sodium nitrite was also given
subcutaneously).
Amyl nitrite had a significant therapeutic effect on dogs
exposed to cyanogen chloride (CNCl) at Ct values (the product of the
concentration of the gas in mg/m3, and the length of exposure in
min) ranging from 3300 to 6300 mg.min.m-3. However, no such
effect was observed in the groups subjected to higher exposures, at
Ct values ranging from 6900 to 11800 mg.min.m-3 (Jandorf &
Bodansky, 1946).
Amyl nitrite treatment of mice exposed to CNCl resulted in a
statistically significant increase in the number of survivors
(Jandorf & Bodansky, 1946).
Amyl nitrite administered by inhalation increased the median
lethal subcutaneous dose of sodium cyanide in dogs from 5.36 (+0.28)
to 24.5 (+1.2) mg/kg (Chen & Rose, 1952).
Part of the antidotal effect may result from methaemoglobin
formation. Any attempt to correlate the therapeutic effect of amyl
nitrite with the degree of methaemoglobinaemia encounters
difficulties. After exposure to HCN or CNCl, the blood of treated
animals contains, in addition to haemoglobin and oxyhaemoglobin,
cyanmethaemoglobin and methaemoglobin. To date, it has not been
possible to quantify the amount of cyanmethaemoglobin. However,
experiments have been undertaken to estimate the degree of
methaemoglobinaemia produced by inhalation of amyl nitrite in
animals not previously exposed to cyanide.
When ten mice were placed in a chamber containing approximately
12 mg amyl nitrite per l, the methaemoglobin levels 10 seconds,
30 seconds, and 1, 2, and 4 min after the beginning of the exposure,
were 1.0, 1.8, 5.6, 14, and 30%, respectively (Jandorf & Bodansky,
1946).
A cone holding one crushed ampoule containing 0.3 ml amyl
nitrite was fitted over the muzzle of each of four dogs
anaesthetized with pentobarbital, and kept in place for 3 min.
Under these conditions, 16, 18, 20, and 32% methaemoglobinaemia were
found in the four dogs at the end of amyl nitrite inhalation
(Jandorf & Bodansky, 1946).
Bastian & Mercker (1959) recorded a methaemoglobin level of 60%
in mice placed in an atmosphere of amyl nitrite (0.112% v/v) for
15 min. Thereafter the degree of methaemoglobinaemia remained
constant and it appeared that an equilibrium existed between the
formation and reduction of methaemoglobin, the level depending on
the concentration of amyl nitrite in the inspired air.
In cats, inhalation of amyl nitrite (0.06 and 0.12% v/v)
resulted in methaemoglobin levels of 30% and 70%, respectively. At
the higher concentration of amyl nitrite, a fall in blood pressure
from 10.7 to 2.7 kPa (80 to 20 mmHg) was observed (Bastian &
Mercker, 1959).
Part of the antidotal effect of amyl nitrite may be vasogenic
in origin, rather than being due to methaemoglobin formation (Way et
al., 1984). Amyl nitrite given after cyanide administration was
reported by Vick & Froelich (1985) to reverse both cardiovascular
changes and respiratory paralysis in 24 of 30 dogs. These changes
occurred before significant methaemoglobin formation and suggest
that early death caused by cyanide could be due in part to
cardiovascular and respiratory failure.
6.8.3 Toxicology
Syncope or shock induced by large doses of amyl nitrite are the
result of a pooling of blood in dilated capacitance vessels. Reflex
tachycardia normally occurs, but a vaso-vagal reflex may induce a
transient bradycardia immediately before collapse (Wilkins et al.,
1937).
The degree of methaemoglobinaemia caused by inhalation of amyl
nitrite is insufficient to affect oxygen transport (see section
6.8.3).
Amyl nitrite also causes a substantial increase in
cerebrospinal fluid pressure (Norcross, 1938), and a fall in pO2
in brain tissue has been recorded using an oxygen electrode
(Rosemann et al., 1946).
6.9 Volunteer Studies
Inhalation of therapeutic doses of amyl nitrite in man did not
result in methaemoglobin formation (Mathes & Gross, 1939).
When six volunteers inhaled 0.1 ml amyl nitrite 10 times for
20 seconds once per min, the median concentration of methaemoglobin
was 3.45%, with a maximum of 6% and a minimum of 1.4%. During and
shortly after inhalation, a small fall in blood pressure was
observed, followed by immediate recovery. The pulse rate rose from
40 to 120 beats per min. Repeated use of the same square of gauze
without application of a new ampoule of amyl nitrite did not have
any effect (Bastian & Mercker, 1959).
6.10 Clinical Studies
No controlled human clinical trials have been undertaken.
6.11 Clinical Studies - Case Reports
No case reports are available.
6.12 Summary of Evaluation
6.12.1 Indications
Amyl nitrite is used as a first-aid measure in cases of cyanide
poisoning where the patients are in deep coma, with dilated
non-reactive pupils and deteriorating cardio-respiratory function.
6.12.2 Advised routes and dose
In adults, administer 0.2-0.4 ml amyl nitrite, via an Ambu bag,
prior to artificial ventilation. In children, administer a maximum
of 0.1 ml, via an Ambu bag, prior to artificial ventilation.
6.12.3 Other consequential or supportive therapy
First-aid therapy with amyl nitrite must be followed by
additional antidotal treatment (see chapter 1). However, supportive
therapy is the most important aspect of the treatment of cyanide
poisoning. Special attention should be paid to the circulation.
The vasodilating action of amyl nitrite may lead to hypotension,
which should be treated immediately with plasma expanders.
6.13 Model Information Sheet
6.13.1 Uses
Amyl nitrite is used as a first-aid measure in cases of cyanide
poisoning where patients are in deep coma, with dilated non-reactive
pupils and deteriorating cardio-respiratory function.
6.13.2 Dosage and route
In adults, 0.2-0.4 ml amyl nitrite should be administered via
an Ambu bag prior to artificial respiration.
In children, a maximum of 0.l ml should be administered via an
Ambu bag prior to artificial respiration.
6.13.3 Precautions/contraindications
Special attention should be paid to the circulation. The
vasodilating action of amyl nitrite may lead to hypotension, which
should be treated immediately with plasma expanders.
6.13.4 Storage
* Protect from light.
* Insist that the suppliers provide only ampoules that
have at least 18 months shelf-life left of the
nominal 24-months total life.
* Take reasonable precautions to store at the lowest
possible temperature compatible with immediate access
in case of emergency; refrigerated storage is not
necessary.
* Inspect monthly for any broken tubes.
Amyl nitrite forms an explosive mixture with air and oxygen.
It is highly inflammable and must not be used where it may be
ignited.
6.14 References
American Hospital Formulary Service (AHFS) (1988) In: McEnvoy KG,
Litvak K, Welsh OH, Campbell JF, Ziegler K, Douglas PM, Shannon-Lass
EP, Thomson K, McQuerrie GM, Schmadel LK, Fredrickson MK, Mendham
NA, & Morisseau AL ed. Drug Information, Bethesda, American Society
of Hospital Pharmacists.
Bastian G & Mercker H (1959) [The efficacy of amyl nitrite
inhalation in the treatment of cyanide poisoning.]
Naunyn-Schmiedebergs Arch Exp Pathol Pharmakol, 237: 285-295 (in
German).
Beasley RWR, Blown RJ, Lunau FW, & Taylor BM (1978) Amyl nitrite and
all that. J Soc Occup Med, 28: 142-143.
Chen KK & Rose CL (1952) Nitrite and thiosulfate therapy in cyanide
poisoning. J Am Med Assoc, 149: 113-119.
Chen KK, Rose CL, & Clowes GHA (1933) Amyl nitrite and cyanide
poisoning. J Am Med Assoc, 100: 1920-1922.
Chen KK, Rose CL, & Clowes GHA (1944) The modern treatment of
cyanide poisoning. J Ind Hyg Toxicol, 37: 344-350.
Jandorf BJ & Bodansky O (1946) Therapeutic and prophylactic effect
of methemoglobinemia in inhalation poisoning by hydrogen cyanide and
cyanogen chloride. J Ind Hyg Toxicol, 28: 124-132.
Martindale (1989) In: Reynolds JEF ed. The extra pharmacopoeia, 29th
ed. London, The Pharmaceutical Press, pp 1492-1493.
Mathes K & Gross F (1939) [The determination of methaemoglobin and
cyanmethaemoglobin in circulating blood.] Naunyn-Schmiedebergs Arch
Exp Pathol Pharmakol, 191: 701 (in German).
Miller MH & Toops TC (1951) Acute cyanide poisoning. Recovery with
sodium thiosulfate therapy. J Indiana Med Assoc, 44: 1164.
Nederlandse Pharmacopee (1958) 6th ed. The Hague, Staatsdrukkerij.
Norcross NC (1938) Intracerebral blood flow. Arch Neurol Psychiatry
(Chicago), 40: 291.
Pedigo LG (1888) Antagonism between amyl nitrite and Prussic acid.
Trans Med Soc Virginia, 19: 124-131.
Rosemann E, Goodman CW, & McCulloch WS (1946) Rapid changes in
cerebral oxygen tension induced by altering the oxygenation and
circulation of the blood. J Neurophysiol, 9: 33.
United States Pharmacopeia (1985) 21st ed. Rockville, Maryland,
United States Pharmacopeial Convention, Inc.
Vick JA & Froelich HL (1985) Studies on cyanide poisoning. Arch Int
Pharmacodyn, 273: 314-322.
Way JL, Sylvester D, Morgan RL, Isom GE, & Burrows CB (1984) Recent
perspectives on the toxidynamic basis of cyanide antagonism. Fundam
Appl Toxicol, 4: S231-S239.
Wilkins RW, Haynes FW, & Weiss S (1937) Role of venous system in
circulatory collapse induced by sodium nitrite. J Clin Invest, 16:
85-91.
Wolfsie JH (1951) Treatment of cyanide poisoning in industry. Am Med
Assoc Arch Ind Hyg, 4: 417-425.
7. SODIUM NITRITE
7.1 Introduction
Methaemoglobin has a great affinity for cyanide in vitro (Hug
& Marenzi, 1933a). Sodium nitrite is an inducer of methaemoglobin
(Hug, 1933; Hug & Marenzi, 1933b) and has been in clinical use for
over 50 years as an antidote for acute cyanide poisoning, most often
in combination with amyl nitrite and sodium thiosulfate (Viana et
al., 1934; Chen & Rose, 1952; Hall & Rumack, 1986).
Potential side-effects of sodium nitrite infusion are
hypotension and excessive methaemoglobin formation (Viana et al.,
1934; Berlin, 1970; Feihl et al., 1982; Hall & Rumack, 1986).
Patients who are already hypotensive are at special risk, and those
with certain types of congenital methaemoglobinaemia are
theoretically more at risk from excessive methaemoglobin induction.
Children administered large doses of sodium nitrite have developed
excessive methaemoglobinaemia (Berlin, 1970). Victims of smoke
inhalation may have both elevated carboxyhaemoglobin levels and
cyanide poisoning (Hart et al., 1985), and may develop further
hypoxia from methaemoglobin induction (Becker, 1985; Jones et al.,
1987).
7.2 Name and Chemical Formula
Sodium nitrite, otherwise known as diazotizing salts, dusitan
sodny (Czech), erinitrit, filmerine, natrium nitrit (German),
nitrite de sodium (French), nitrous acid sodium salt, sodii nitris,
natrii nitris, natrium nitrosum, sodium nitrite (DOT), NCI-C02084,
NIOSH No. RA 1225000, CAS 7632-00-0 (Sax, 1984; NIOSH, 1983;
Windholz et al., 1983; Martindale, 1982), is supplied in the USA by
Eli Lilly & Co., Indianapolis, Indiana, as a component of the Lilly
Cyanide Antidote Package [R] (Product Information, 1984). It is
sold in Australia by Orapharm under the proprietary name of OAR
(Martindale, 1989). The relative molecular mass of sodium nitrite
is 69.00, and the chemical formula is NaNO2 (Sax, 1984).
7.3 Physico-chemical Properties
Sodium nitrite has a melting point of 271 °C (Windholz et al.,
1983). It is soluble in either 1.5 parts of cold water or 0.6 parts
of boiling water (Windholz et al., 1983). Solutions may be made in
water (1:1.5) or alcohol 1:160 (Martindale, 1989). Sodium nitrite
is slightly soluble in ether (ITI, 1985). Acids will decompose
sodium nitrite and cause the evolution of brownish N2O3 fumes
(Windholz et al., 1983). Aqueous solutions are alkaline to litmus,
with a pH between 7 and 9 (Windholz et al., 1983; United States
Pharmacopeia, 1985). The specific gravity is 2.17 (ITI, 1985). A
10% solution of sodium nitrite has a density of 1.065 (Izmerov,
1982). Excipients are apparently not added.
On drying over silica gel for 4 h, solid sodium nitrite loses
no more than 0.25% of its weight (United States Pharmacopeia, 1985).
Injectable sodium nitrite solution is made by diluting solid
sodium nitrite with "Water for Injection" (United States
Pharmacopeia, 1985). The final solution is sterile and contains not
less than 95% and not more than 105% of the labelled amount of
sodium nitrite (United States Pharmacopeia, 1985). Sodium nitrite
is supplied in the Lilly Cyanide Antidote Package [R] in two 10 ml
vials of a 3% solution (30 mg/ml; 300 mg per vial) (Product
Information, 1984).
Sodium nitrite is stable in light, but slowly oxidizes in air
to nitrate (Windholz et al., 1983). It is incompatible with
acetanilide, antipyrine, caffeine, citrate, chlorates,
hypophosphites, iodides, mercury salts, morphine, oxidizing agents,
permanganate, phenazone, sulfites, tannic acid, and vegetable
astringent decoctions, infusions, or tinctures (Martindale, 1989;
Windholz et al., 1983).
7.4 Synthesis
Commercial sodium nitrite is derived from sodium hydroxide and
nitrogen oxides by one of three common methods (Izmerov, 1982).
One method uses a low concentration of nitrogen oxides from gas
formed during dilute nitric acid production, which is absorbed by a
sodium hydroxide solution.
A second method (the "hot method") uses sodium hydroxide
absorption of high concentrations of nitrogen oxides at atmospheric
or higher pressure.
A third method (or "two-staged method") combines features of
the first two.
Pharmaceutical sodium nitrite is produced by reduction of
sodium nitrate, previously heated until fused, with sulfur dioxide,
lead, or a sulfite (Harvey, 1980). Alternatively, it can be derived
from sodium nitrate by nitric oxide absorption (the nitric oxide
being obtained from ammonia by catalytic oxidation in a sodium
carbonate solution) (Harvey, 1980). The sodium nitrite mixture
obtained is lixiviated with water and filtered, then partially
evaporated and allowed to crystallize (Surgenor, 1970).
Injectable sodium nitrite solution is prepared by dissolving
solid sodium nitrite in "Water for Injection" to a final
concentration of 3% (30 mg/ml) (Product Information, 1984; United
States Pharmacopeia, 1985). It is then sterilized by autoclaving or
membrane filtration (Martindale, 1989). The final delivery form
meets both United States and European Pharmacopoeia requirements for
pyrogens and sterility.
The United States Pharmacopeia specifies that no more than
0.002% of heavy metals may be present (United States Pharmacopeia,
1985). "Top grade" sodium nitrite from some processes has a sodium
nitrite concentration of at least 99%, with at most 1.4% moisture
content and impurities of not more than 0.8% sodium nitrate, 0.3%
calcinated residue, and 0.17% sodium chloride (Izmerov, 1982).
7.5 Analytical Methods
7.5.1 Quality control
7.5.1.1 Solid sodium nitrite (United States Pharmacopeia, 1985)
Sodium nitrite (1g) is dissolved in water, making up to 100 ml.
Then 10 ml of this solution is pipetted into a mixture containing
50.0 ml of 0.1 N potassium permanganate volumetric solution (VS)
(0.1 mol/l), 100 ml of water, and 5 ml of sulfuric acid, and the
pipette tip is immersed beneath the surface of this mixture during
transfer. The liquid is warmed to 40 °C and allowed to stand for
5 min. After the addition of 25.0 ml oxalic acid VS (0.05 mol/l),
the resultant mixture is heated to 80 °C and titrated with potassium
permanganate (0.1 mol/l), equivalent to 3.45 mg sodium nitrite.
7.5.1.2 Sodium nitrite injection (United States Pharmacopeia, 1985)
The method described in 7.5.1.1 is followed except that, in
place of the 10 ml sodium nitrite solution, an accurately measured
volume of sodium nitrite injection containing about 150 mg sodium
nitrite is used.
7.5.1.3 Preparation of volumetric solutions (VS) (United States
Pharmacopeia, 1985)
(a) Potassium permanganate VS (0.1 mol/l)
KMnO4; relative molecular mass, 158.03; 3.161 grams in
1000 ml
About 3.3 g of potassium permanganate is dissolved in
1000 ml of water and boiled for about 15 min in a suitable
flask. The flask is then stoppered and allowed to stand
for at least 2 days. It is then filtered through a
fine-porosity, sintered glass crucible, which may be lined
with a pledget of glass wool. The solution is then
standardized by weighing accurately 200 mg of sodium
oxalate, previously dried to constant weight at 110 °C.
This is dissolved in 250 ml of water. Sulfuric acid
(7 ml) is added and the mixture is heated to about 70 °C.
The potassium permanganate solution is slowly added from a
burette with constant stirring until a pale-pink colour
persisting for about 15 seconds is produced. At the
termination of titration, the temperature should be no
lower than 60 °C. The molarity is calculated, with each
6.700 mg of sodium oxalate equivalent to 1 ml of potassium
permanganate (0.1 mol/l). The solution should be stored
in glass-stoppered amber-coloured bottles, and contact
with organic materials such as rubber, which will reduce
the potassium permanganate, should be avoided.
(b) Oxalic Acid VS (0.05 mol/l)
H2C2O4 . 2H2O; relative molecular mass 126.07;
6.303 g in 1000 ml
6.45 g of oxalic acid is dissolved in water and made up to
1000 ml. The solution is standardized by titration
against potassium permanganate VS (1 mol/l) as described
above. The solution should be stored protected from light
in glass-stoppered bottles.
7.5.2 Identification (United States Pharmacopeia, 1985)
A solution of sodium nitrite responds to tests for sodium and
for nitrite.
7.5.2.1 Sodium
After conversion to chloride or nitrate, solutions of sodium
compounds with five times their volume of cobalt-uranyl acetate test
solution (TS) yield a precipitate that is golden yellow in colour
and forms after several min of agitation. Cobalt-uranyl acetate
test solution is prepared by dissolving 40 g of uranyl acetate in a
mixture of 30 g of glacial acetic acid and enough water to make
500 ml. A solution is also prepared of 200 g of cobaltous acetate
in a mixture of 30 g of glacial acetic acid and enough water to make
500 ml. Both solutions are prepared with warming, mixed while still
warm, and cooled to 20 °C. The temperature is maintained at 20 °C
for about 2 h, allowing the separation of excess salts, and then the
resultant solution is filtered through a dry filter. Sodium
compounds also impart an intense yellow coloration to a non-luminous
flame.
7.5.2.2 Nitrite
Treatment of nitrite-containing solutions with diluted mineral
acids or acetic acid (6 mol/l) produces brownish-red fumes, and the
solution will give a blue coloration on starch-iodide paper.
7.5.3 Impurities (United States Pharmacopeia, 1985)
7.5.3.1 Preparation of sodium nitrite to test
Sodium nitrite (1 g) is dissolved in 6 ml hydrochloric acid
(3 mol/l) and then evaporated to dryness on a steam bath. The
residue is reduced to a coarse powder, which is then heated on the
steam bath until the hydrochloric acid odour is no longer
perceptible. This residue is then dissolved in 23 ml of water, and
2 ml of acetic acid (1 mol/l) is added.
The test is for metallic impurities that are coloured by the
sulfide ion under specified test conditions, in terms of the
percentage of lead (by weight) in the test substance. Limits for
sodium nitrite are 0.002%.
7.5.3.2 Preparation of special reagents
Lead nitrate stock solution is prepared by dissolving 159.8 mg
lead nitrate in 100 ml of water and adding 1 ml of nitric acid.
This solution is then diluted to 1000 ml with water and stored in
glass containers free of lead soluble salts. Standard lead solution
is prepared by diluting 10.0 ml of the lead nitrate stock solution
with water to a volume of 100.0 ml, so that each 1 ml of standard
lead solution contains the equivalent of 10 µg of lead. A
comparison solution prepared with 100 µl of standard lead solution
per g of sodium nitrate contains the equivalent of 1 part of lead
per million parts of sodium nitrate.
7.5.3.3 Preparation of standard
Pipette 2 ml of standard lead solution (containing 20 mg of
lead) into a 50 ml colour comparison tube. Dilute with water to
25 ml and adjust pH to between 3.0 and 4.0 with acetic acid
(1 mol/l) or ammonium hydroxide (6 mol/l) using narrow-range pH
paper as an indicator. Dilute to 40 ml with water and mix.
7.5.3.4 Preparation of test
Pipette 25 ml of the prepared sodium nitrite test material (see
section 7.5.3.1) into a 50 ml colour comparison tube. Using
narrow-range pH indicator paper, adjust pH to between 3.0 and 4.0
with either acetic acid (1 mol/l) or ammonium hydroxide (6 mol/l).
Then dilute to 40 ml with water and mix.
7.5.3.5 Preparation of monitor
Place 25 ml of solution prepared as in section 7.5.3.4 above
into a 50 ml colour comparison tube and add 2.0 ml of the standard
lead solution. Using short-range pH indicator paper, adjust pH to
between 3.0 and 4.0 with either (1 mol/l) acetic acid or ammonium
hydroxide (6 mol/l). Then dilute to 40 ml with water and mix.
7.5.3.6 Preparation of hydrogen sulfide test solution (TS)
Pass hydrogen sulfide gas into cold water making a saturated
solution that should possess a strong odour of hydrogen sulfide.
Store in a cold, dark area in small, dark amber-coloured bottles
filled nearly to the top.
7.5.3.7 Test procedure
Add 10 ml of freshly prepared hydrogen sulfide TS solution to
each colour comparison tube. Allow to stand for 5 min, then view
downwards over a white surface. The colour of the solution
described in section 7.5.3.4 should not be darker than that of the
solution described in section 7.5.3.3. The colour intensity of the
solution described in section 7.5.3.5 should be equal to or greater
than that of the solution described in section 7.5.3.3.
7.6 Shelf-life
The shelf-life of sodium nitrite solution for injection is
5 years, which might be somewhat reduced in very hot climates though
stability testing at elevated temperatures has not been undertaken
(Personal Communication, Eli Lilly & Co., March 1987). The supplier
recommends that sodium nitrite be stored at controlled room
temperatures of 15 - 30 °C (Product Information, 1984). Experiments
with stored ampoules showed that sodium nitrite is stable in water
for more than 21 years (Chen & Rose, 1956).
7.7 General Properties
7.7.1 Mode of action
Sodium nitrite has seldom been utilized alone for the treatment
of cyanide poisoning. Rather, it is used in combination with
intravenous sodium thiosulfate and, often, amyl nitrite by
inhalation. These combinations have been shown to be more
efficacious in experimental poisoning than any of the compounds
alone, demonstrating true antidotal synergy (Hug, 1933; Chen et al.,
1933a; Chen et al., 1934; Chen & Rose, 1952; Way et al., 1984).
Sodium nitrite induces methaemoglobin in vitro, which binds
cyanide (Hug & Marenzi, 1933a). In animal studies with dogs and
rabbits, sodium nitrite induced methaemoglobinaemia, with levels
averaging 50% following a dose of 40 mg/kg (Hug & Marenzi, 1933a;
Hug & Marenzi, 1933b). Sodium nitrite alone has been shown to be
efficacious in dogs with experimental cyanide poisoning (Chen et
al., 1933a; Chen et al., 1934; Chen & Rose, 1952). However, dog
blood has been shown to be more sensitive to methaemoglobin
induction by sodium nitrite in vitro than human blood (Paulet &
Menoret, 1954). Methaemoglobin can be an efficacious cyanide
antidote, as shown by results in experimental poisoning in rats
treated with an exogenously prepared stroma-free methaemoglobin
solution (Ten Eyck et al., 1986). Human red blood cells, with
methaemoglobin induced in vitro, were injected intraperitoneally
to cyanide-poisoned mice and had antidotal efficacy (Kruszyna et
al., 1982). However, animals pretreated with methylene blue did not
develop significant methaemoglobin levels and had unchanged sodium
nitrite antidotal efficacy in experimental cyanide poisoning (Holmes
& Way, 1982). Other mechanisms, including vasodilatation with
changes in local capillary blood flow, have been postulated to
account for part or all of the antidotal action of sodium nitrite
(Way et al., 1984; Cohen & Guzzardi, 1984).
Alternative mechanisms of action are suggested by the fact that
sodium nitrite relaxes maximally contracted vascular smooth muscle
in vitro (Kruszyna et al., 1985). The contracted vascular smooth
muscle relaxation was not reversed in this model by the addition of
cyanide (Kruszyna et al., 1985). In mice, methaemoglobin levels
induced by sodium nitrite were more efficacious in protecting
against cyanide poisoning than were comparable methaemoglobin levels
induced by either hydroxylamine or 4-dimethylaminophenol (Kruszyna
et al., 1982). Equivalent amounts of methaemoglobin induced
in vitro by these three agents did not bind significantly
different amounts of cyanide (Kruszyna et al., 1982), suggesting
that sodium nitrite may have some other mechanism of action. The
methaemoglobinaemia induced in these mice by sodium nitrite peaked
later and at a lower level than the methaemoglobinaemia induced by
hydroxylamine or 4-dimethylaminophenol, but was longer lasting
(Kruszyna et al., 1982). This may explain differences in efficacy
noted in this animal study, as cyanide initially bound to
methaemoglobin induced by the other agents may have been rapidly
released to exacerbate the poisoning (Kruszyna et al., 1982).
The oxidation of haemoglobin to methaemoglobin by sodium
nitrite is favoured when the oxygen partial pressure is high and
haemoglobin is in the oxyhaemoglobin state (Tomoda & Yoneyama,
1979). If methaemoglobin induction accounts for all, or part, of
the antidotal action of sodium nitrite, this might explain the
potentiation noted with concomitant oxygen administration (Mansouri,
1985), although other investigators have not reached this conclusion
(Way et al., 1966).
The mechanism of action of sodium nitrite is not fully
understood at present. Methaemoglobin induced by sodium nitrite and
measurable with a co-oximeter, however, does not bind cyanide at the
time of measurement (cyanmethaemoglobin cannot be measured by these
instruments). Patients have survived seriously symptomatic acute
cyanide poisoning, despite developing measurable methaemoglobin
levels of 10% or less (Hall et al., 1987; DiNapoli et al., 1989).
Administering futher sodium nitrite to a patient who has already had
a satisfactory clinical improvement in order to produce a
"therapeutic methaemoglobin level" of 25%, as has somethimes been
recommended (Jones et al., 1987), is both unnecessary and
potentially dangerous (DiNapoli et al., 1989).
7.7.2 Other relevant properties
Sodium nitrite is a vasodilating agent and has been used in the
past for the treatment of angina (Martindale, 1989). Rapid
intravenous administration may cause hypotension (Viana et al.,
1934; Hall & Rumack, 1986). This effect can be avoided by beginning
the infusion slowly and increasing the infusion rate cautiously with
careful blood pressure monitoring.
7.8 Animal Studies
7.8.1 Pharmacokinetics
Sodium nitrite blood levels have not been measured during
antidote therapy and most pharmacokinetic parameters have not been
determined. Most studies have focused on the levels of
methaemoglobin induced by sodium nitrite.
In one study into fetal methaemoglobin formation, when pregnant
rats were administered sodium nitrite orally or by intraperitoneal
injection, peak maternal sodium nitrite blood levels were usually
produced within the first 20 to 30 min, while peak methaemoglobin
levels were observed a few minutes later (Gruener et al., 1973).
Peak sodium nitrite blood concentrations ranged from 3.9 mg/l
(56.52 µmol/l), with an administered dose of 2.5 mg/kg, to 32.5 mg/l
(471.01 µmol/l) with an administered dose of 30 mg/kg, and
corresponded to peak maternal methaemoglobin levels of 3.4%
(2.5 mg/kg dose) to 60.2% (30 mg/kg dose) (Gruener et al., 1973).
Sodium nitrite produced an equimolar conversion of haemoglobin
to methaemoglobin in vitro in canine blood (Hug & Marenzi, 1933a).
However, only about 74% of an administered intravenous dose was
involved in methaemoglobin formation in dogs in vivo (Hug &
Marenzi, 1933b). Dogs administered 40 mg/kg of sodium nitrite
intravenously developed approximately 50% methaemoglobinaemia (Hug &
Marenzi, 1933b). In these experiments using sodium nitrite alone,
all of the administered dose of potassium cyanide was recovered in
the urine as thiocyanate over a 4-day period (Hug & Marenzi, 1933b).
In a dog with acute cyanide poisoning, given sodium nitrite
(22.5 mg/kg) immediately and an additional 11.25 mg/kg at 29 and
54 min after the poisoning, peak methaemoglobin levels of 65% at
30 min and 60% at 4 h were observed (Chen & Rose, 1952). The
methaemoglobin concentration decreased rapidly after each peak,
reaching 20% about 30 min after the first peak, and 4-5 h after the
second peak (Chen & Rose, 1952). Methaemoglobin disappeared from
the circulation 11 h after the first injection of sodium nitrite
(Chen & Rose, 1952).
In mice administered sodium nitrite 75 (mg/kg)
intraperitoneally, methaemoglobin levels peaked at 30-35% about
20 min after the injection (Kruszyna et al., 1982). The levels
remained in this range until 40 min after sodium nitrite
administration and then decreased slowly, reaching 25%
methaemoglobinaemia at 1 h and 10% at 2 h (Kruszyna et al., 1982).
These findings contrast with studies undertaken in mice with other
methaemoglobin-inducing agents (4-dimethylaminophenol and
hydroxylamine), which produced higher peak methaemoglobin levels
more quickly (Kruszyna et al., 1982). Following the peak level, the
degree of methaemoglobinemia then decreased faster than that induced
by sodium nitrite (Kruszyna et al., 1982).
In vitro studies have indicated that canine blood is more
susceptible to sodium nitrite-induced methaemoglobin conversion than
human blood (Paulet & Menoret, 1954). Rodent blood is more
resistant to methaemoglobin formation than human blood (Kruszyna et
al., 1982).
7.8.2 Pharmacodynamics
Sodium nitrite, at a minimum dose of 5 mg/kg, was shown to be
an effective antidote for experimental cyanide poisoning in dogs,
while 200 mg/kg was efficacious in rabbits (Hug, 1932; Hug, 1933).
In other experiments, sodium nitrite alone was shown to be an
effective cyanide antidote in dogs (Chen et al., 1934), though the
combination of sodium nitrite with a sulfur donor allowed dogs to
tolerate doses of cyanide greater than those antagonized by the two
agents separately (Chen et al., 1933a; Chen et al., 1933b; Hug,
1933; Chen et al., 1934). This antidotal synergy has been noted in
numerous studies since the 1930s (e.g., Way et al., 1972). Sodium
nitrite has been shown to have protective effects against rat brain
cytochrome oxidase inhibition in vivo (Piantadosi et al., 1983).
Two studies have reported data that are inconsistent with the
classical antidotal mechanism of action, namely methaemoglobin
induction. When comparable levels of methaemoglobinaemia were
induced in mice by 4-dimethylaminophenol, hydroxylamine, or sodium
nitrite, cyanide was antagonized better by the administration of
sodium nitrite (Kruszyna et al., 1982). Following pre-treatment
with methylene blue prior to cyanide poisoning, mice had unchanged
sodium nitrite antidotal efficacy, even though they did not develop
significant methaemoglobinaemia (Holmes & Way, 1982).
Sodium nitrite causes relaxation of vascular smooth muscle
in vitro (Kruszyna et al., 1985). Sodium nitrite alone did not
have any effect on either the respiratory or urinary excretion of
14C-labelled sodium cyanide in mice (Burrows et al., 1982). Other
postulated antidotal mechanisms include vasodilatation with improved
capillary blood flow (Way et al., 1984; Cohen & Guzzardi, 1984).
7.8.3 Toxicology
A rather large body of sub-acute and chronic toxicity data
exists for sodium nitrite (Musil, 1966; Izmerov, 1982). Effects
including increased methaemoglobin levels, neuromuscular
excitability, thyroid dysfunction, changes in conditioned reflexes,
elevated serum protein and albumin concentrations, possible effects
on hepatic drug metabolism systems, prolonged prothrombin time, and
decreased erythrocyte counts have been noted in chronic and subacute
animal exposures (Musil, 1966; Izmerov, 1982; Gosselin et al.,
1984). In antidote administration, most subjects would receive only
one dose, or at most a few doses over a short period of time, and
these chronic and subacute animal studies would not seem to be
particularly relevant.
Pre-treatment of rats with sodium nitrite potentiates carbon
tetrachloride hepatotoxicity (Suarez & Bhonsle, 1978). A
synergistic effect on acute hepatic necrosis in mice has been
produced by oral administration of a combination of sodium nitrite
and various secondary amines (Asahina et al., 1971). A second
abnormal haemoglobin derivative, nitrosohaemoglobin, has been
produced in rats following intraperitoneal sodium nitrite injection
(Rein et al., 1968).
Sodium nitrite is listed as a non-carcinogen, but as giving a
positive result in the Ames test, by the International Agency for
Research on Cancer (IARC) (Kuroki et al., 1980). It can react with
secondary amines either in vitro or in vivo to produce
potentially carcinogenic nitrosamines (Izmerov, 1982). In vitro
studies have shown induction of volatile mutagens in the faeces of
normal human volunteers following treatment with sodium nitrite
(MacDonald & Rao, 1982). Administration of sodium nitrite and
morpholine together to rats produced hepatic and forestomach tumours
(Mirvish et al., 1983). However, a cohort study of workers
chronically exposed to a combination of sodium nitrite and various
amines in cutting fluids did not show an increased cancer morbidity
(Jarvholm et al., 1986). Epidemiological data suggest a
relationship between the consumption of large quantities of
nitrite-nitrate-treated foodstuffs, a diet poor in ascorbic acid,
and the development of gastric, oral cavity, and oesophageal cancers
(Weisburger et al., 1983).
Sodium nitrite administered orally or intraperitoneally to
pregnant rats produced peak fetal sodium nitrite blood levels
ranging from trace amounts, with a 2.5 mg/kg maternal dose and a
corresponding maternal blood level of 3.9 mg/l (56-52 µmol/l), to
9.4 mg/l (136.23 µmol/l) with a 30 mg/kg maternal dose and a
corresponding maternal blood level of 32.5 mg/l (471.01 µmol/l)
(Gruener et al., 1973). Fetal methaemoglobin levels ranged from
1.2% (with a maternal sodium nitrite dose of 2.5 mg/kg and a
corresponding maternal peak methaemoglobin level of 3.4%) to 27.2%
(with a maternal sodium nitrite dose of 30 mg/kg and a corresponding
maternal peak methaemoglobin level of 60.2%) (Gruener et al., 1973).
Fetal effects other than methaemoglobin induction were not reported.
Sodium nitrite was shown not to be teratogenic in several
animal species when used as a cardiovascular agent or as a fungicide
in combination with iodine (Schardein, 1985). In combination with
ethylurea, sodium nitrite forms the potent teratogen ethylnitrosurea
(Schardein, 1985). The median effective dose (ED50) for mortality
in a chicken embryo study was 22 µmol/egg (Korhonen et al., 1983).
A maximum of 27% of the embryos were noted to be malformed (Korhonen
et al., 1983). A combination of sodium nitrite (50 mg/kg) and
methyl urea (30 mg/kg), administered orally to rats on the 9th day
of pregnancy, killed over 40% of the fetuses and produced
malformations in about 15% of the surviving fetuses (Izmerov, 1982).
Administration of ascorbic acid blocked both the embryotoxic and
teratogenic effects (Izmerov, 1982). Sodium nitrite doses of
100 mg/kg were teratogenic in rats (Izmerov, 1982). Cyanosis was
observed in 25% of newborn rats at birth following maternal
administration of sodium nitrite (100 mg/kg) and in 6% of newborn
rats with a maternal dose of 5 mg/kg (Izmerov, 1982).
As sodium nitrite is only likely to be administered once, or at
most a few times, as an antidote, the main concern would appear to
be induction of fetal methaemoglobinaemia if administered to a
pregnant patient. The risk to the fetus from maternal cyanide
poisoning would seem to override the risk from possible fetal
methaemoglobin induction. At clinically relevant doses (300 mg, or
about 4.3 mg/kg for a 70-kg subject) (Product Information, 1984),
data from a rat study suggested that the peak fetal methaemoglobin
level would be in the neighbourhood of 2.7% (Gruener et al., 1973).
This amount of fetal methaemoglobinaemia is not likely to be
clinically significant. However, based on in vitro comparisons of
methaemoglobin reductase activity, the human fetus may have a weaker
defense mechanism against the development of significant
methaemoglobinaemia than has the rat fetus (Gruener et al., 1973).
Some representative animal toxicity values for sodium nitrite
are: LD50 rat (oral), 85 mg/kg; LDLo hamster (oral), 3 mg/kg;
LD50 mouse (oral), 175 mg/kg; LD50 rat (intravenous), 65 mg/kg;
LDLo dog (oral), 330 mg/kg; LDLo dog (intravenous), 15 mg/kg; LDLo
rabbit (intravenous), 80 mg/kg (Sax, 1984; ITI, 1985). A
comparative human intravenous antidotal dose is 300 mg (or about
4.3 mg/kg for a 70-kg subject) (Product Information, 1984).
7.9 Volunteer Studies
7.9.1 Pharmacokinetics
Sodium nitrite is rapidly absorbed following oral
administration (Martindale, 1989). About 60% of the absorbed
nitrite ion is metabolized in the body, partially to ammonia, while
the rest of the absorbed dose is excreted unchanged in the urine
(Martindale, 1982). The nitrite ion disappears from the circulatory
system quickly, but little is known about its fate (Baselt, 1982).
An intravenous injection of 400 mg in humans produced a peak
methaemoglobin concentration of 10.1%, while 600 mg produced a peak
methaemoglobin level of 17.5% (Chen & Rose, 1952). A later study
in normal volunteers showed that a methaemoglobin level of 6% was
produced by the intravenous injection of 1 mg sodium nitrite per kg
(about 300 mg, 10 ml of a 3% solution of sodium nitrite) (Weger,
1968). In this same study, injecting 12 mg/kg (about 900 mg, 30 ml
of a 3 % solution of sodium nitrite) resulted in a 30%
methaemoglobinaemia, but also in clinical shock (Weger, 1968).
7.9.2 Sodium nitrite poisoning
Most human toxicity data relates to accidental or suicidal
ingestion of sodium nitrite from industrial, laboratory, or food
additive sources. Fatal poisoning has occurred when sodium nitrite
has been substituted for table salt and used as seasoning
(MacQuiston, 1936; Padberg & Martin, 1939) or when food contaminated
with motor vehicle cooling fluid was consumed (Ten Brink et al.,
1982). A case of suicide from the ingestion of sodium nitrite has
been reported, with a post-mortem methaemoglobin level of 90%
(Standefer et al., 1979). Ingestion of sausages cured with sodium
nitrite produced symptomatic poisoning with syncope, hypotension,
and methaemoglobinaemia and a decreased arterial oxygen saturation
(Bakshi et al., 1967). Two patients who mistakenly ingested sodium
nitrite instead of table salt developed methaemoglobin levels of 34%
and 54%, but recovered after being administered methylene blue and
supplemental oxygen (Aquanno et al., 1981).
The range of toxicity of sodium nitrite is difficult to
determine, as the ingested dose is seldom known. A patient who
ingested 14.5 g complained of transient darkening of both visual
fields (Grant, 1986). A 17-month-old child died with a
methaemoglobin level greater than 90% after being given 450 mg
(32 mg/kg) intravenously in the mistaken impression that acute
cyanide poisoning was present (Berlin, 1970). A two-month-old
infant developed severe poisoning but survived after ingesting
130 mg sodium nitrite (Gosselin et al., 1984). The mean lethal oral
dose in adults is probably in the neighbourhood of 1 g if no
treatment is given (Gosselin et al., 1984), though survival has
followed the ingestion of 1 g (Baselt, 1982).
Treatment of acquired methaemoglobinaemia from sodium nitrite
poisoning in circumstances similar to those described above may
involve only supplemental oxygenation and observation if the patient
is asymptomatic and the methaemoglobin level is 20-30% or less (Hall
et al., 1986a). With higher methaemoglobin levels or in symptomatic
patients, intravenous infusion of methylene blue at a usual dose of
0.1-0.2 ml/kg of a 1% solution (1-2 mg/kg) may be necessary (Hall et
al., 1986b). Toluidine blue may be used when methylene blue is not
available. Exchange transfusion may be required if severely
poisoned patients are not responsive to the above measures (Hall et
al., 1986a). Experimental animal studies have suggested that
hyperbaric oxygen is both efficacious (Goldstein & Doull, 1971) and
not efficacious (Sheehy & Way, 1974) in reducing nitrite-induced
methaemoglobinaemia. Hyperbaric oxygen can be used to maintain
tissue oxygenation while exchange transfusion is prepared (Hall et
al., 1986a).
If the therapeutic administration of sodium nitrite in cyanide
poisoning produces excessive methaemoglobinaemia (Viana et al.,
1934; Berlin, 1970; Lasch & El Shawa, 1981; Feihl et al., 1982),
there is some controversy about appropriate treatment. The
administration of methylene blue has been recommended (Product
Information, 1984), and toluidine blue has also been used. However,
exchange transfusion rather than methylene or toluidine blue
administration has been suggested because conversion of
cyanmethaemoglobin back to normal haemoglobin could theoretically
release bound cyanide and worsen the poisoning (Rumack, 1987). This
controversy is unresolved at present.
7.10 Clinical Studies
No controlled human clinical trials have been performed to
compare the efficacy of various cyanide antidotes in acute human
poisoning.
7.11 Clinical Studies - Case Reports
In the absence of controlled clinical trials, anecdotal reports
of human poisoning treated with sodium nitrite are all that can be
evaluated. To compound the problem, only a single case of acute
cyanide poisoning treated solely with sodium nitrite has been
reported (Mota, 1933). All other reported patients were given more
than one antidote. Many patients have received a combination of
sodium nitrite and sodium thiosulfate, or amyl nitrite and sodium
nitrite plus sodium thiosulfate (Chen & Rose, 1952; Chen & Rose,
1956). In other cases, sodium nitrite has been administered in
various combinations with sodium thiosulfate, methylene blue,
dicobalt-EDTA (Kelocyanor [R]), and hydroxocobalamin (Motin et al.,
1970; Lutier et al., 1971; Yacoub et al., 1974). The clinical
efficacy of sodium nitrite used alone in acute cyanide poisoning is
therefore impossible to separate from its use in an antidotal
combination, and, judging from various animal studies showing
antidotal synergism between sodium nitrite and sodium thiosulfate
(Chen et al., 1934; Chen & Rose, 1952; Way et al., 1972), it should
probably not be used alone.
The first reported cases of human acute cyanide poisoning to be
treated with a combination of sodium nitrite and sodium thiosulfate
were described by Viana et al. (1934). Their first patient ingested
about 5 g of potassium cyanide and became comatose with a weak pulse
and respiratory distress, but recovered after being given 1500 mg of
sodium nitrite and 18 g of sodium thiosulfate. Their second patient
ingested about 2 g of potassium cyanide and developed coma,
respiratory distress, and convulsions, but recovered after being
given 750 mg of sodium nitrite and 12 g of sodium thiosulfate. No
cyanide assays were undertaken. Hypotension and cyanosis developed
in both cases (Viana et al., 1934).
The largest case series of acute cyanide poisoning treated with
the amyl nitrite/sodium nitrite/sodium thiosulfate antidote
combination was assembled by Chen & Rose (1952, 1956) comprising a
total of 49 patients. One patient who was moribund before being
administered these antidotes did not recover (Chen & Rose, 1952).
Only historical evidence of cyanide poisoning was available in these
cases; no cyanide assays were reported (Chen & Rose, 1952; Chen &
Rose, 1956).
Survival following acute cyanide poisoning from potassium and
sodium cyanide (De Busk & Seidl, 1969; Stewart, 1974; Feihl et al.,
1982; Peters et al., 1982; Wood, 1982; Litovitz et al., 1983; Wesson
et al., 1985; Hall et al., 1987), cyanogenic plants (Sayre &
Kaymakcalan, 1964; Rubino, 1978; Lasch & El Shawa, 1981; Shragg et
al., 1983), and laetrile (Moss et al., 1981; Beamer et al., 1983;
Hall et al., 1986b) has been reported. However, not all patients
with severe cyanide poisoning given the sodium nitrite/sodium
thiosulfate antidote combination have survived (Chen & Rose, 1952;
Braico et al., 1979; Litovitz et al., 1983). Patients with severe
acute cyanide poisoning (including coma, convulsions, and metabolic
acidosis) and blood cyanide levels, measured at various times after
exposure, ranging from 0.26 to 40 mg/l (10 to 1539 µmol/l) have
survived following the administration of sodium nitrite and sodium
thiosulfate in combination with supportive measures (Rubino, 1978;
Moss et al., 1981; Peters et al., 1982; Feihl et al, 1982; Litovitz
et al., 1983; Shragg et al., 1983; Wesson, et al., 1985; Hall et
al., 1987; Hall et al., 1986b). Patients poisoned with cyanide have
survived with supportive treatment only (Graham et al., 1977; Vogel
et al., 1981; Brivet et al., 1983), but the highest reported blood
cyanide level in a patient who survived without receiving specific
antidote treatment was 3.8 mg/l (147 µmol/l) (Edwards & Thomas,
1978). This suggests that patients with more severe cyanide
poisoning may have a better chance of recovery with both supportive
measures and specific antidotes than with supportive measures alone
(Hall & Rumack, 1986).
Administration of the sodium nitrite/sodium thiosulfate
antidote combination in five smoke inhalation victims with carbon
monoxide and cyanide poisoning and mean blood cyanide levels of
1.62 mg/l (62 µmol/l) has been reported (Hart et al., 1985). These
five patients also received hyperbaric oxygen therapy. There were
four survivors and one fatality in this series.
7.12 Summary of Evaluation
7.12.1 Indications
Sodium nitrite is indicated for use in acute cyanide poisoning.
It should not be used except where there is definite indication of
severe poisoning, such as loss of consciousness and deteriorating
vital functions. It is usually administered with sodium thiosulfate
and its administration may be preceded by the use of amyl nitrite.
7.12.2 Contraindications
Sodium nitrite should not be administered to asymptomatic
patients following exposure to cyanide. It should not be
administered to patients with smoke inhalation and combined carbon
monoxide and cyanide poisoning unless hyperbaric oxygen therapy is
available and such therapy has already been initiated.
G6DP-deficient individuals are theoretically at great risk from
nitrite therapy because of the likelihood of severe haemolysis,
although no such cases have been reported.
7.12.3 Advised route and dosage
Sodium nitrite is administered intravenously in an initial
adult dose of 300 mg (10 ml of a 3% solution) (Chen & Rose, 1952;
Hall & Rumack, 1986). While many authors recommend that this dose
be infused over a 5-min period, hypotension from the vasodilating
properties of sodium nitrite may occur. Hypotension may be avoided
by diluting sodium nitrite with normal saline and infusing the
medication over a 20-min period with frequent blood pressure
monitoring (Hall, 1986). The rate of infusion may be increased if
no hypotension occurs (Hall, 1986). Methaemoglobin levels should be
monitored to avoid excessive methaemoglobin induction (Berlin, 1970)
(see chapter 10). The paediatric dose for an average child,
frequently quoted in the literature and based on in vitro
experiments with canine blood, is 0.33 ml of a 3% solution per kg
body weight (about 10 mg/kg), administered intravenously with the
precautions noted above (Hall & Rumack, 1986). A pediatric dose of
approximately 0.2 ml/kg of a 3% solution has also been recommended
(Product Information, 1984). If there is reason to suspect anaemia,
tables devised to calculate a reduced dose, taking into account the
relatively lesser amount of haemoglobin present, should be consulted
(Berlin, 1970).
The adult dose may be lethal for a child. With a haemoglobin
level of 12 g/100 ml, only 10 mg/kg per body weight can be
administered immediately and 5 mg/kg repeated within 30 min if
necessary (Berlin, 1970). However, the calculation of Berlin was
based on a therapeutic level of methaemoglobinaemia of 15 mg/kg.
This may be too high because Weger (1968) observed severe
circulatory collapse in volunteers after 12 mg/kg was given
intravenously. In the same study, the administration of 4 mg/kg
(10 ml of a 3% solution NaNO2 solution, i.e., 300 mg) induced only
6% methaemoglobinaemia. As the adult dose of about 4 mg/kg has been
shown to be effective clinically, it may be safer to begin treatment
in children with sodium nitrite at 4 mg/kg (about 0.13 ml of a 3%
solution per kg body weight) and to administer additional sodium
nitrite only if excessive methaemoglobinaemia is not present and a
satisfactory clinical response has not occurred.
In both adults and children, another sodium nitrite dose of 50%
the initial amount administered may be repeated 30 min after the
first dose if there is inadequate clinical improvement (Hall &
Rumack, 1986).
7.12.4 Other consequential or supportive therapy
See section 1.10.
7.13 Model Information Sheet
7.13.1 Uses
Sodium nitrite is indicated for the treatment of acute cyanide
poisoning.
7.13.2 Dosage and route
The adult dose is 300 mg (10 ml of a 3% solution) infused
intravenously at the fastest rate possible without causing
hypotension (usually over 5 to 20 min). The initial paediatric dose
is 0.13-0.33 ml of a 3% solution per kg body weight (about
4-10 mg/kg). It is advisable to begin with lower doses in children
and increase to the desired effect. If anaemia is suspected,
standard tables should be consulted to calculate a reduced
paediatric sodium nitrite dose.
In both adults and children, another dose of one-half the
initial amount administered may be repeated 30 min after the first
dose if there is inadequate clinical improvement.
7.13.3 Precautions/contraindications
Excessive methaemoglobinaemia may occur, especially when doses
larger than those recommended are administered to children.
Hypotension may occur following rapid administration of sodium
nitrite, owing to its vasodilating properties. Blood pressure
should be monitored carefully during sodium nitrite administration,
and the infusion rate slowed if hypotension occurs.
Patients with smoke inhalation and combined carbon monoxide and
cyanide poisoning with elevated carboxyhaemoglobin levels should not
be given sodium nitrite unless treatment in a hyperbaric oxygen
chamber is available and such treatment has been initiated.
G6PD-deficient individuals are theoretically at great risk from
sodium nitrite therapy because of the likelihood of severe
haemolysis, although no such cases have been reported.
7.13.4 Adverse effects
Excessive methaemoglobinaemia may occur, especially with doses
exceeding those recommended. Hypotension may occur with rapid
intravenous infusion.
7.13.5 Use in pregnancy and lactation
There are no reported cases of the use of sodium nitrite during
pregnancy or lactation. Animal experiments indicate that some
sodium nitrite crosses the placenta and that fetal
methaemoglobinaemia may be induced. The risk to the fetus from
severe maternal cyanide poisoning would seem to be greater than the
risk of fetal methaemoglobin induction. No animal studies have
addressed the question of sodium nitrite excretion in breast milk or
its possible effects on the nursing infant.
7.13.6 Storage
Sodium nitrite should be stored at a controlled temperature of
15 to 30 °C (59 to 86 °F). The shelf-life is 5 years.
7.14 References
Aquanno JJ, Chan KM, & Dietzler DN (1981) Accidental poisoning of
two laboratory technologists with sodium nitrite. Clin Chem, 27:
1145-1146.
Asahina S, Friedman MA, Arnold E, Millar GN, Mishkin M, Bishop Y, &
Epstein SS (1971) Acute synergistic toxicity and hepatic necrosis
following oral administration of sodium nitrite and secondary amines
to mice. Cancer Res, 31: 1201-1205.
Bakshi SP, Fahey JL, & Pierce LE (1967) Sausage cyanosis - Acquired
methemoglobinemic nitrite poisoning. New Engl J Med, 277: 1072.
Baselt RC (1982) Cyanide and nitrite. In: Disposition of toxic drugs
and chemicals in man, 2nd ed. Davis, California, Biomedical
Publications, pp 209-214, 555-557.
Beamer WC, Shealy RM, & Prough DS (1983) Acute cyanide poisoning
from laetrile ingestion. Ann Emergency Med, 12: 449-451.
Becker CM (1985) The role of cyanide in fires. Vet Hum Toxicol, 27:
487-490.
Berlin AM (1970) The treatment of cyanide poisoning in children.
Pediatrics, 46: 793-796.
Braico KT, Humbert JR, Terplan KL, & Lehotay JM (1979) Laetrile
intoxication: Report of a fatal case. New Engl J Med, 300: 238-240.
Brivet F, Delfraissy JF, Duche M, Bertrand P, & Dormont J (1983)
Acute cyanide poisoning: Recovery with non-specific supportive
therapy. Intensive Care Med, 9: 33-35.
Burrows GE, Liu DHW, Isom GE, & Way JL (1982) Effect of antagonists
on the physiologic disposition of sodium cyanide. J Toxicol Environ
Health, 10: 181-189.
Chen KK & Rose CL (1952) Nitrite and thiosulfate therapy in cyanide
poisoning. J Am Med Assoc, 149: 113-119.
Chen KK & Rose CL (1956) Treatment of acute cyanide poisoning. J Am
Med Assoc, 162: 1154-1155.
Chen KK, Rose CL, & Clowes GHA (1933a) Methylene blue, nitrites, and
sodium thiosulphate against cyanide poisoning. Proc Soc Exp Biol
Med, 31 : 250-251.
Chen KK, Rose CL, & Clowes GHA (1933b) Potentiation of antidotal
action of sodium tetrathionate and sodium nitrite in cyanide
poisoning. Proc Soc Exp Biol Med, 31: 252-253.
Chen KK, Rose CL, & Clowes GHA (1934) Comparative values of several
antidotes in cyanide poisoning. Am J Med Sci, 188: 767-781.
Cohen MA & Guzzardi LJ (1984) A letter on the treatment of cyanide
poisoning. Vet Hum Toxicol, 26: 503-504.
De Busk RF & Seidl LG (1969) Attempted suicide by cyanide: A report
of two cases. California Med, 110: 394-396.
DiNapoli J, Hall AH, Orake R, & Rumack BH (1989) Cyanide and arsenic
poisoning by intravenous injection. Ann Emergency Med, 18: 308-311.
Edwards AC & Thomas ID (1978) Cyanide poisoning. Lancet, 1: 92-93.
Feihl F, Domenighetti G, & Perret Cl (1982) Intoxication massive au
cyanure avec évolution favorable. Schweiz Med Wochenschr, 112:
1280-1282.
Goldstein GM & Doull J (1971) Treatment of nitrite-induced
methemoglobinemia with hyperbaric oxygen. Proc Soc Exp Biol Med,
138: 137-139.
Gosselin RE, Smith RP, & Hodge HC ed. (1984) Nitrite. In: Clinical
toxicology of commercial products, 5th ed. Baltimore, Maryland,
Williams and Wilkins, pp 314-319.
Graham DL, Lamand D, Theodore J, & Robin ED (1977) Acute cyanide
poisoning complicated by lactic acidosis and pulmonary edema. Arch
Intern Med, 137: 1051-1055.
Grant WM (1986) Sodium nitrite. In: Toxicology of the eye, 3rd ed.
Springfield, Illinois, Charles C. Thomas, p 840.
Gruener N, Shuval HI, Behroozi K, Cohen S, & Shechter H (1973)
Methemoglobinemia induced by transplacental passage of nitrites in
rats. Bull Environ Contam Toxicol, 9: 44-48.
Hall AH (1986) Cyanide poisoning: Dealing with unexpected menace.
Emergency Med, 18: 919-205.
Hall AH & Rumack BH (1986) Clinical toxicology of cyanide. Ann
Emergency Med, 15: 1067-1074.
Hall AH, Kulig KW, & Rumack BH (1986a) Drug- and chemical-induced
methaemoglobinaemia: Clinical features and management. Med Toxicol,
1: 253-260.
Hall AH, Linden CH, Kulig KW, & Rumack BH (1986b) Cyanide poisoning
from laetrile ingestion: Role of nitrite therapy. Pediatrics, 78(2):
269-272.
Hall AH, Doutre W, Ludden T, Kulig K, & Rumack BH (1987)
Nitrite/thiosulfate treated acute cyanide poisoning: Estimated
kinetics after antidote. Clin Toxicol, 25: 121-133.
Hart GB, Strauss MB, Lennon PA, & Whitcraft DD (1985) Treatment of
smoke inhalation by hyperbaric oxygen. J Emergency Med, 3: 211-215.
Harvey SC (1980) Sodium nitrite. In: Osol A ed. Remington's
pharmaceutical science, 16th ed. Easton, Pennsylvania, Mack
Publishing Co., p 782.
Holmes RK & Way JL (1982) Mechanism of cyanide antagonism by sodium
nitrite. Pharmacologist, 24: 182 (Abstract).
Hug E (1932) L'intoxication par l'acide cyanhydrique. Action
antidote du bleu de méthylène, du nitrite de sodium et du sulfure de
sodium. C R Soc Biol (Paris), 111: 88-90.
Hug E (1933) Action combinée du nitrite de sodium dans le traitement
de l'intoxication cyanhydrique chez le lapin. C R Soc Biol (Paris),
114: 87-89.
Hug E & Marenzi AD (1933a) La fixation de l'acide cyanhydrique par
les hématies qui contiennent de la méthémoglobine. C R Soc Biol
(Paris), 114: 84-86.
Hug E & Marenzi AD (1933b) Mécanisme de l'action antidote du sodium
vis-à-vis de l'intoxication par l'acide cyanhydrique. C R Soc Biol
(Paris), 114: 86-87.
ITI (1985) Sodium nitrite. In: Toxic and hazardous industrial
chemicals safety manual. Tokyo, The International Technical
Information Institute, p 485.
Izmerov NF ed. (1982) IRPTC Scientific Reviews of Soviet Literature
on Toxicity and Hazards of Chemicals No. 22: Nitrites. Moscow,
Centre of International Projects, GKNT, 32 pp.
Jarvholm B, Lavenius B, & Sallsten G (1986) Cancer morbidity in
workers exposed to cutting fluids containing nitrites and amines. Br
J. Ind Med, 43: 563-565.
Jones J, McMullen MJ, & Dougherty J (1987) Toxic Smoke Inhalation:
cyanide poisoning in fire victims. Am J Emerg Med, 5: 318-321.
Korhonen A, Hemminki K, & Vainio H (1983) Toxicity of rubber
chemicals towards three-day chicken embryos. Scand J Work Environ
Health, 9: 115-119.
Kruszyna R, Kruszyna H, & Smith RP (1982) Comparison of
hydroxylamine, 4-dimethylaminophenol and nitrite protection against
cyanide poisoning in mice. Arch Toxicol, 49: 191-202.
Kruszyna H, Kruszyna R, & Smith RP (1985) Cyanide and sulfide
interact with nitrogenous compounds to influence the relaxation of
various smooth muscles. Proc Soc Exp Biol Med, 179: 44-49.
Kuroki T, Abbondandolo A, Drevon C, Huberman E, & Laval F (1980)
Mutagenesis assays with mammalian cells. In: Long-term and
short-term screening assays for carcinogens: A critical appraisal.
Lyons, International Agency for Research on Cancer, pp 107-133 (IARC
Monographs on the Evaluation of the Carcinogenic Risk of Chemicals
to Humans, Supplement 2).
Lasch EE & El Shawa R (1981) Multiple cases of cyanide poisoning by
apricot kernels in children from Gaza. Pediatrics, 68(1): 5-7.
Litovitz TL, Larkin RF, & Myers RAM (1983) Cyanide poisoning treated
with hyperbaric oxygen. Am J Emergency Med, 1: 94-101.
Lutier F, Dusoleil P, & De Montgros J (1971) Action de
l'hydroxocobalamine à dose massive dans l'intoxication aiguë au
cyanure. (A propos d'un cas). Arch Mal Prof, 32: 683-686.
MacDonald IA & Rao BG (1982) Nitrite-induced volatile fecal
mutagens: Some preliminary studies. Cancer, 49: 1405-1408.
MacQuiston TAC (1936) Fatal poisoning by sodium nitrite. Lancet, 2:
1153-1154.
Mansouri A (1985) Methemoglobinemia. Am J Med Sci, 298: 200-209.
Martindale (1989) In: Reynolds JEF ed. The extra pharmacopoeia, 29th
ed. London, The Pharmaceutical Press, p 854.
Mirvish SS, Salmasi S, Cohen SM, Patil K, & Mahboubi E (1983) Liver
and forestomach tumours and other forestomach lesions in rats
treated with morpholine and sodium nitrite, with and without sodium
ascorbate. J Natl Cancer Inst, 71: 81-85.
Moss M, Khalil N, & Gray J (1981) Deliberate self-poisoning with
laetrile. Can Med Assoc J, 125: 1126-1128.
Mota MM (1933) [On a case of intoxication by potassium cyanide
treated with sodium nitrite with survival.] Rev Med Rosario, 23:
670-674 (in Spanish).
Motin J, Bouletreau P, & Rouzioux JM (1970) Intoxication
cyanhydrique grave traitée avec succès par l'hydroxocobalamine. J
Med Strasbourg, 1: 717-722.
Musil J (1966) [The effects of chronic sodium nitrite intoxication
in rats.] Acta Biol Med Ger, 16: 388-394 (in German).
Padberg LR & Martin T (1939) Three fatal cases of sodium nitrite
poisoning. J Am Med Assoc, 113: 1733.
Paulet G & Menoret P (1954) Etude comparée de la sensibilité du sang
de l'homme (frais ou conservé) et du sang de chien à divers
méthémoglobinisants. C R Soc Biol (Paris), 148: 1014-1017.
Peters CG, Mundy JVB, & Rayner PR (1982) Acute cyanide poisoning:
The treatment of a suicide attempt. Anaesthesia, 37: 582-586.
Piantadosi CA, Sylvia AK, & Jobsis FF (1983) Cyanide-induced
cytochrome a,a3 oxidation-reduction responses in rat brain in vivo.
J. Clin Invest, 72: 1224-1233.
Product Information (1984) Product information for the cyanide
antidote package[R]. Indianapolis, Indiana, Eli Lilly & Co.
Rein H, Ristau O, Kupfer E, & Jung F (1968) [Nitrosohaemoglobin from
poisoning with sodium nitrite.] Folia Haematol, 90: 176-181 (in
German).
Rubino JM (1978) Cyanide toxicity: Report of a case. Connecticut
Poison Inf Bull, 3: 1-6.
Rumack BH ed. (1987) Cyanide poisoning management. In:
Poisindex[R]: Poison information system. Denver, Colorado,
Micromedex 52: 1-F4.
Sax NI (1984) Sodium nitrite. In: Dangerous properties of industrial
materials, 6th ed. New York, Van Nostrand Reinhold Co., pp
2442-2443.
Sayre JW & Kaymakcalan S (1964) Cyanide poisoning from apricot seeds
among children in central Turkey. New Engl J Med, 270: 1113-1115.
Schardein JL (1985) Chemically induced birth defects. New York,
Marcel Dekker, pp 74, 759, 810-824.
Sheehy MH & Way JL (1974) Nitrite intoxication: Protection with
methylene blue and oxygen. Toxicol Appl Pharmacol, 30: 221-226.
Shragg TA, Albertson TE, & Fischer CJ (1983) Cyanide poisoning after
bitter almond ingestion. West J. Med, 136: 65-69.
Standefer JC, Jones AM, Street E, & Inserra R (1979) Death
associated with nitrite ingestion: Report of a case. J Forensic Sci,
24: 678-771.
Stewart R (1974) Cyanide poisoning. Clin Toxicol, 7: 561-564.
Suarez KA & Bhonsle P (1978) Enhanced hepatotoxicity of carbon
tetrachloride following sodium nitrite pre-treatment. Arch Int
Pharmacodyn, 234: 329-334.
Surgenor DM (1970) Sodium nitrite USP. In: Osol E ed. Remington's
pharmaceutical sciences, 14th ed. Easton, Pennsylvania, Mack
Publishing Co., pp 846-847.
Ten Brink WAG, Wiezer JHA, Luijpen AFMG, Van Heijst ANP, Pikaar SA,
& Seldenrijk R (1982) Nitrate poisoning caused by food contaminated
with cooling fluid. J Toxicol Clin Toxicol, 19: 139-147.
Ten Eyck RP, Schaerdel AD, & Ottinger WE (1986) Comparison of
nitrite treatment and stroma-free methemoglobin solution as
antidotes for cyanide poisoning in a rat model. Clin Toxicol, 23:
477-487.
Tomoda A & Yoneyama Y (1979) On the relationship of hemoglobin
oxidation with the conformation of hemoglobin. Experientia (Basel),
35: 15-16.
United States Pharmacopeia (1985) Sodium nitrite and sodium nitrite
injection. In: United States Pharmacopoeia, 21st ed. Rockville,
Maryland, United States Pharmacopeial Convention, Inc., pp 974,
1186-1187, 1189-1190, 1206.
US NIOSH (1983) Registry of toxic effects of chemical substances
1981-1982. Atlanta, Georgia, National Institute of Occupational
Safety and Health, p 857.
Viana C, Cagnoli H, & Cedan J (1934) Action du nitrite de sodium
dans l'intoxication au cyanure. C R Soc Biol (Paris), 115:
1649-1651.
Vogel SN, Sultan TR, & Ten Eyck RP (1981) Cyanide poisoning. Clin
Toxicol, 18: 367-383.
Way JL, Gibbon SL, & Sheehy MH (1966) Effect of oxygen on cyanide
intoxication. I. Prophylactic protection. J Pharmacol Exp Ther, 153:
381-385.
Way JL, End E, Sheehy MH, De Miranda P, Feitknecht UF, Bachand R,
Gibbon SC, & Burrows GE (1972) Effects of oxygen on cyanide
intoxication. IV. Hyperbaric oxygen. Toxicol Appl Pharmacol, 22:
415-421.
Way JL, Sylvester D, Morgan RL, Isom GE, Burrows GE, Tamulinas CB, &
Way JL (1984) Recent perspectives on the toxicodynamic basis of
cyanid antagonism. Fundam Appl Toxicol, 4: S231-S239.
Weger N (1968) [Aminophenol as a cyanide antidote.] Arch Toxikol,
24: 49-50 (in German).
Weisburger JH, Horn CL, & Barnes WS (1983) Possible genotoxic
carcinogens in foods in relation to cancer causation. Semin Oncol,
10: 330-341.
Wesson DE, Foley R, Sabatini S, Wharton J, Kapusnik J, & Kurtzman NA
(1985) Treatment of acute cyanide intoxication with hemodialysis. Am
J Nephrol, 5: 121-126.
Windholz M, Budavari S, & Blumetti RF, ed. (1983) Sodium nitrite.
In: The Merck index, 10th ed. Rahway, New Jersey, Merck & Co., Inc.,
p 8480.
Wood GC (1982) Acute cyanide intoxication: Diagnosis and management.
Clin Toxicol Consultant, 4: 140-149.
Yacoub M, Faure J, Morena H, Vincent M, & Gaure H (1974)
L'intoxication cyanhydrique aiguë. Données actuelles sur le
métabolisme du cyanure et le traitement par hydroxocobalamine. J Eur
Toxicol, 7: 22-29.
8. 4-DIMETHYLAMINOPHENOL (4-DMAP)
8.1 Introduction
Cyanide has a special affinity for ferric ions, which are found
in cytochrome oxidase, the terminal oxidative respiratory enzyme in
mitochondria. Blood contains a substantial quantity of ferrous ions
within haemoglobin and it is possible to convert these readily to
ferric ions, for example, by the use of nitrites as suggested by
Chen et al. (1933). A disadvantage of the use of nitrites is the
fact that the concentration of methaemoglobin rises only slowly
after intravenous administration. In six experiments on volunteers,
using the dose of sodium nitrite suggested by Chen (4 mg/kg), a
methaemoglobin level of 6% resulted after 40 min (Kiese & Weger,
1969). This was considered to be too low a concentration of
methaemoglobin after too long a time. Experiments with several
aminophenols showed that the use of 4-dimethylaminophenol (4-DMAP)
resulted in rapid and controlled formation of methaemoglobin. An
intravenous dose of 3.25 mg/kg resulted in a methaemoglobin
concentration of 30% in 10 min, a methaemoglobin level of 15% being
reached within one min.
8.2 Name and Chemical Formula
Chemical name: Dimethyl( para)aminophenol
hydrochloride
Formula:
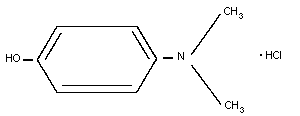 Total formula: C8H11 ON.HCl
Relative molecular mass: 173.5
Appearance: snow-white crystals
CAS number: 619-60-3
Manufacturer: Dr Franz Koehler Chemie GmbH,
Alsbach, Germany
Commercial name: 4-dimethylaminophenol (4-DMAP)
8.3 Physico-chemical Properties
Raw material: snow-white colourless crystals
Melting point: 145 °C + 1 °C
Solubility: very soluble in water. The
solution is oxidized by contact
with air and changes from being
colourless to black-brown
Optical properties:
IR-spectrum: peaks at (cm-1)
833: l,4-disubstituted aromatics
1240: phenols
1275: amine
1500: aromatics
2700: C-H aromatics
3200: N-H aromatics
UV data: maximum at 271 nm; extinction
coefficient 1255 l/mol
Acidity: no data available
pK (-N(CH3)2) = 6.15
pK (-OH) = 10.1
Stability in light: no data available
Thermal stability: no data available
Refractive index
and specific gravity: unknown
Weight on drying: no data available
Excipients and
pharmaceutical aids: unknown for the final product
although it may contain an
antioxidant such as sodium
pyrosulfate
8.4 Synthesis
Routes of synthesis: no information available
Manufacturing processes: no information available
8.5 Analytical Methods
8.5.1 Identity
To 5 ml of liquid from the ampoule, add one drop 30% hydrogen
peroxide. The solution will turn a deep violet colour within 5 min.
To 2.5 ml of liquid from the ampoule, add 0.5 ml 10% ferrichloride.
A pale-brown colour will appear immediately.
8.5.2 Quantification
AgNO3 titration: 2.5 ml liquid from the ampoule is diluted
in 40 ml water and, after the addition of 3 ml nitric acid
(2 mol/l), potentiometrically titrated with silver nitrate
(0.1 mol/l). 1 ml AgNO3 (0.1 mol/l) equiv. 17.365 mg
dimethylaminophenol hydrochloride.
HClO4 titration: 2.5 ml liquid from the ampoule is added to
40 ml of a 3% solution of mercury acetate in glacial acetic acid and
then potentiometrically titrated with perchloric acid (0.1 mol/l).
1 ml perchloric acid (0.1 mol/l) equiv. 17.365 mg
dimethylaminophenol hydrochloride.
8.5.3 Purity
Unknown
8.5.4 Methods for analysis of 4-DMAP in biological samples
Unknown.
8.6 Shelf-life
Since, in contact with air, 4-DMAP is readily oxidized and the
solution changes colour to black-brown, 4-DMAP must be stored in
opaque containers. An open ampoule cannot be kept. Coloured and
turbid solutions cannot be used. Correctly stored 4-DMAP can be
used after storage for up to 3 years. The influence of tropical
conditions is unknown.
8.7 General Properties
The use of 4-DMAP results in rapid formation of methaemoglobin.
An intravenous dose of 3.25 mg/kg resulted in a methaemoglobin
concentration of 30% within 10 min. Because there is a much larger
source of haemoglobin than there is of cytochrome oxidase,
cytochrome oxidase blocked by cyanide can be reactivated and its
role as an essential catalyst for tissue utilization of oxygen
restored.
8.8 Animal Studies
8.8.1 In Vitro Studies
In studies involving the addition of various aminophenols such
as 2-aminophenol, 2-amino-4-chlorophenol, 2-amino-4,6-
dichlorophenol, 2-amino-5-ethoxyphenol, 2-dimethylaminophenol,
4-aminophenol, 4-methylaminophenol, and 4-dimethylaminophenol to the
blood of mice, cats, dogs, cattle, and man, it has been shown that
4-dimethylaminophenol and 4-methylaminophenol produce consistent
amounts of methaemoglobin most rapidly. These studies also showed
that a concentration of 30-40% methaemoglobin was attained at widely
varying rates in different species (Kiese & Weger, 1969).
8.8.1.1 Metabolism of 4-DMAP in the liver
In a study by Eyer & Kampffmeyer (1978), nearly all the 4-DMAP
was conjugated in livers perfused with 4-DMAP (0.01 mmol/l)
(0.4 nmol/mg protein per min).
8.8.1.2 Red cell metabolism of 4-DMAP
4-DMAP rapidly catalyses methaemoglobin formation. The
reaction mechanism has been studied using purified human
haemoglobin. 4-DMAP transfers electrons catalytically from
ferrohaemoglobin to oxygen (Eyer et al., 1974; Eckert & Eyer, 1983)
and is thereby oxidized to the phenoxyl-radical and N,N-dimethyl-
quinoneimine (Eyer & Lengfelder, 1984). Both species are reduced to
ferrohaemoglobin with the formation of methaemoglobin.
Catalytic methaemoglobin formation is terminated by rapid
binding of oxidized 4-DMAP to reactive SH groups in haemoglobin and
thioether formation with reduced glutathione. Formation of
glutathione-S-conjugates with 4-DMAP has been shown to occur in
vitro in red cells (Eyer & Kiese, 1976), and in vivo in dogs
(Eyer & Gaber, 1978) and man (Jansco et al., 198l, Klimmek et al.,
1983). Kinetic studies of thioether formation by DMAP in the
presence of purified dog haemoglobin and glutathione in a reducing
system showed that S,S-(2-dimethylamino-5-hydroxy-1,3-phenylene)-
bis-gluthathione (di(GS)-DMAP) is formed initially. This thioether
is highly autoxidizable and the addition of another thiol gives
stable S,S,S(2-dimethylamino)-5-hydroxy-1,3,4-phenylene)-
tri-glutathione (tris-(GS)-DMAP). Despite its chemical stability,
it can be actively transported across the red blood cell membrane
(Eckert & Eyer, 1986).
4-DMAP is excreted in the urine mainly as the tris-cysteinyl
derivative, tris(Cys)-DMAP. This compound is presumed to be broken
down in the glutamyl cycle of the kidney (Meister, 1973), giving
rise to cysteinyl glycine and cysteinyl derivatives.
8.8.1.3 Toxic effects of 4-DMAP on erythrocytes
Studies have been undertaken to test the effect of 4-DMAP on
the osmotic resistance of human erythrocytes. The percentage
haemolysis did not differ from that of controls, even for an
incubation period of less than 20 h, at a 4-DMAP concentration of
1 mmol/l. However, the degree of haemolysis after 20 h was 14.5%
(±6.9%), compared with a control value of 2.7% (±0.4%), when
measured in 0.7% NaCl solution (Klimmek et al., 1983). Since the
haemolysis that occurred in the tests for osmotic resistance was
maximal after the same period as in in vivo studies, one may
assume a common lytic mechanism under in vitro and in vivo
conditions (Klimmek et al., 1983).
8.8.1.4 Toxic effects of 4-DMAP on isolated rat kidney tubules
Various toxic effects were observed when 4-DMAP was added to
suspensions of isolated rat kidney tubules, apparently as the result
of the irreversible binding of 4-DMAP to SH-containing compounds,
e.g., multiple enzyme inhibition, inhibition of gluconeogenesis,
decrease in glutathione, and membrane damage (Snizicz et al., 1979).
The results suggest that irreversible impairment of the integrity of
the cell membrane causes irreversible damage to tubular epithelial
cells, the resultant increase in permeability being followed by a
decrease in nucleotide content without marked effect on
gluconeogenesis or mitochondrial membrane integrity.
8.8.1.5 Oxygen saturation and methaemoglobin formation
From reports by Kiese (1974) and Eyer et al. (1979), it is
evident that quite small variations of pO2 can cause large
differences in the rate of methaemoglobin formation. At high
percentage oxyhaemoglobin levels, where the R-configuration of
haemoglobin prevails over the T-configuration, the rate of oxidation
is lower. At a 4-DMAP concentration of 0.032 mmol/l, increasing the
percentage saturation of haemoglobin of humans more than halved the
rate of methaemoglobin production (Marrs et al., 1982).
8.8.2 Pharmacokinetics
No data are available.
8.8.3 Pharmacodynamics
4-DMAP (3.25 mg/kg), given intravenously to dogs one min after
poisoning with a lethal dose of KCN (4 mg/kg), resulted in the
survival of all dogs. The maximum venous methaemoglobin content was
reached within 5-10 min and was 38.8% (±1.7%) of the total
haemoglobin. The injection of the same 4-DMAP dose intramuscularly
led to a maximum methaemoglobin level of 41.6% (±1.3%) after 30 min.
A methaemoglobin concentration of 35% was achieved within 5 min of
an intravenous dose of 3.25 mg/kg, 15 min of an intramuscular dose
of 3.25 mg/kg, and 30 min of an oral dose of 15 mg/kg (Klimmek et
al., 1983).
Bright & Marrs (1982) found that there was considerable
variation in the methaemoglobin levels produced in different dogs
using the same oral dose of 4-DMAP.
In experiments using different aminophenols, it was shown that
4-DMAP given intravenously rapidly produced consistent amounts of
methaemoglobin in the blood of different animals species. In mice
and rabbits, a rapid fall in the concentration of methaemoglobin was
observed after 2 min and after 20 min the concentration in these
species was less than 10%. In dogs and mice, however, the decrease
in methaemoglobin concentration was substantially less; after
20 min, the level was still above 30%. In contrast to 4-DMAP,
sodium nitrite caused increased methaemoglobin levels which lasted
longer than an hour in mice and rabbits (Kiese & Weger, 1969).
There was a marked increase in arterial pO2 after the
administration of 4-DMAP to dogs poisoned with cyanide, compared
with non-poisoned dogs. Presumably, part of the oxygen released
during the formation of methaemoglobin under hypoxic conditions can
be utilized by tissues (Klimmek et al., 1979a,b). In the same
experiments, an increase in respiratory volume and mean arterial
blood pressure, was observed together with a lessening of the
lactate-pyruvate ratio.
8.8.4 Toxicology
LD50 intravenous mouse: 50-70 mg/kg (Kiese & Weger, 1965;
Marrs et al., 1984)
LD50 oral mouse: 946 mg/kg (Marrs et al., 1984)
LD50 intravenous rat: 57 mg/kg (Kiese et al., 1975)
LD50 oral rat: 689-780 mg/kg (Marrs et al., 1984)
LD50 intravenous dog: 26 mg/kg (Weger, 1975)
In all five studies, the cause of death was inadequate oxygen
transport by the blood to the tissues because 85% of the haemoglobin
had been converted to methaemoglobin.
8.8.4.1 Nephrotoxicity
Some of the aminophenols, such as 4-aminophenol, produce kidney
lesions after intravenous administrations (Green et al., 1969;
Calder et al., 1971; Hinsberg & Treupel, 1984). The intravenous
injection of 100 mg 4-DMAP/kg, a dose nearly double the LD50 and
which oxidizes 60% of the haemoglobin to methaemoglobin, caused
renal damage in rats. Moderate to severe necrosis of the convoluted
tubules, with either few or no inflammatory cells, was found 24 h
after injection. The glomeruli were not affected and there was no
papillary damage. However, no tubular necrosis was detected with a
dose of 30 mg/kg. A high or low sodium diet did not noticeably
affect the nephrotoxicity of 4-DMAP (Kiese et al., l975).
A single oral dose of up to 25 mg 4-DMAP/kg did not result in
any macroscopic or histological abnormality in the gastrointestinal
tract, liver, or kidneys of rats killed up to 24 h after dosing. In
animals that had been kept for 7 days after dosing, no histological
abnormalities were found other than engorgement of the liver and
kidneys (Marrs et al., 1982).
8.8.4.2 Mutagenicity
A clear relationship between dose and mutation frequency was
shown by a mammalian cell assay employing V79 (Chinese hamster)
cells (Lee & Webber, 1983), despite a negative Ames test with and
without metabolic activation.
8.9 Volunteer Studies
In experiments by Weger (1968) on seven volunteers, an
intravenous dose of N,N-dimethyl- p-aminophenol (3.25 mg/kg)
resulted in a 15% methaemoglobin concentration within 1 min. After
10 min, the concentration was 30%. Other aminophenols tested gave
less favourable results. N-methyl- p-aminophenol had much the
same effect but only at an iv dose of 20 mg/kg. O-aminophenol
(30 mg/kg) administered intravenously resulted in 15%
methaemoglobinaemia after 12 min and 30% after 60 min. Comparison
was also made with sodium nitrite. When a dose of 4 mg/kg was given
intravenously to seven volunteers, the maximal methaemoglobin level
was 6%. This dose of sodium nitrite was that recommended by Chen et
al. (l933) and Chen & Rose (l952) and suggests that the antidotal
effect may be due to a mechanism other than methaemoglobin
formation.
In experiments by Klimmek et al. (1983) on volunteers a
methaemoglobin concentration of about 30% was reached within 5 min
after an intravenous dose of 3.25 mg 4-DMAP/kg (3 volunteers),
50 min after an intramuscular dose of 3.5 mg 4-DMAP/kg
(6 volunteers), and 30 min after an oral dose of 900 mg 4-DMAP
(5 volunteers).
8.9.1 Metabolism of 4-DMAP in the liver
The metabolic conversion of 4-DMAP to glucuronide, sulfate, and
thioether derivatives is likely to follow linear Michaelis-Menten
kinetics. The pattern of metabolites was studied in urine collected
over 24 hours from subjects treated with intravenous 4- DMAP
(3.25 mg/kg). An average of 68% of the dose was excreted as
metabolites in urine: 41% as glucuronide, 12% as sulfate, and 15%
as thioethers (Klimmek et al., 1983).
4-DMAP rapidly auto-oxidizes at pH values above 7 and is
transformed to a variety of degradation products (Eyer et al.,
1974). Thus partial degradation of 4-DMAP in the gastrointestinal
tract may account for the reduced total metabolite recovery
following oral administration compared with that following
intravenous administration.
8.9.2 Metabolism of 4-DMAP in erythrocytes
Rapid thioether formation following the intravenous
administration of 4-DMAP has been shown to occur in erythrocytes
where 4-DMAP is oxidized by oxyhaemoglobin to the corresponding
quinoneimine which yields adducts with red cell glutathione (Eyer &
Kiese, 1976). One of these, tris-(GS)-DMAP penetrates the red cell
membrane slowly and is excreted in the urine mainly as a
tris-cysteinyl derivative, tris-(Cys)-DMAP.
The 4-DMAP thioethers excreted in human urine (15.3% (±1.8%) of
the dose) are 1ikely to originate in the erythrocytes; this
underlines the importance of erythrocytes in 4-DMAP metabolism in
man.
8.9.3 Adverse effects
After 6-7 days, a phlebitis was observed in the antecubital
vein where 4-DMAP was infused (Klimmek et al., 1983). Following an
intramuscular injection of 4-DMAP, a slight pressure was felt after
5-10 min at the site of injection, slowly growing in intensity and
finally resulting in severe pain. Approximately 10 h after the
injection, shivering, sweating, and fever occurred. An oral 4-DMAP
dose of 300 mg was well tolerated, whereas 600 mg slowed mental
activity and caused tiredness, and 900 mg gave an effect of
dizziness, with headache and buzzing in the ears (Klimmek et al.,
1983).
After an intravenous injection of 4-DMAP (3.25 mg/kg), the
total bilirubin concentration increased by 140%, that of conjugated
bilirubin by 180% and that of iron by 200%. Within 24 h of an
intramuscular injection of 4-DMAP (3.5 mg/kg), the total bilirubin
increased by 270% and then declined rapidly, while the bilirubin
concentration rose by 120% and that of iron by 50% (Klimmek et al.,
1983).
After an oral dose of 900 mg 4-DMAP, total and conjugated
bilirubin levels increased by 170% and 80%, respectively, and the
iron concentration rose by 60%. The changes in bilirubin and iron
concentrations were the result of reduced erythrocyte survival
caused by 4-DMAP, which took about one day to develop. Since the
haemolysis that occurred in the tests for osmotic resistance was
maximal after the same period as in in vivo studies, one may
assume a common lytic mechanism both under in vitro and in vivo
conditions (Klimmek et al., 1983).
8.10 Clinical Studies
No data are available.
8.11 Clinical Studies - Case Reports
Table 6 summarizes clinical observations made in 19 cases of
cyanide poisoning treated by 4-DMAP. In most patients, there was a
long time interval between the occurrence of poisoning and the
administration of 4-DMAP. Patients with impaired vital function at
the time of treatment recovered. In six cases an overdose of 4-DMAP
was given, resulting in excessive methaemoglobinaemia, and in three
cases haemolysis was observed on the second day after administration
of the antidote. Even the recommended dose of 3.25 mg 4-DMAP/kg has
resulted in a methaemoglobin concentration of about 70% (van Dijk et
al., 1987).
8.12 Summary of Evaluation
8.12.1 Indications
4-DMAP should only be given to patients poisoned with cyanide
who are in a deep coma and who have dilated non-reactive pupils and
deteriorating cardio-respiratory function.
8.12.2 Recommended routes and dosage
An intravenous dose of 3.25 mg/kg body weight should be given
immediately. The intramuscular injection of 4-DMAP to patients in
shock due to cyanide poisoning cannot be recommended because
absorption is unpredictable.
8.12.3 Other consequential or supportive therapy
The administration of 4-DMAP should always be followed by that
of sodium thiosulfate (150-200 mg/kg intravenously) (see chapter 3).
Artificial respiration should be given if necessary.
Table 6. Clinical Observations in cyanide-poisoned patients treated by 4-DMAPa
Estimated Interval before Blood cyanide Interval before 4-DMAP dose, Additional Adverse Outcome Reference
cyanide first-aid concentration blood measurement interval before antidotes effects
ingested treatmentb (mg/l) usede administrationf
10 g oral 15 min ? 250 mg, 45 min CO2-EDTA recovery Daunderer et al. (1974)
? < 5 min 5-16.9 5 min 250 mg, 5 min CNS Jacobs (1984)
damage
? 1.5-2 h c 5.5 h 250 mg, > 5h recovery Von Clarmann (1987)
? 20 min NR NR 250 mg, 3.5h recovery Von Clarmann (1987)
? < 1h 1.9 > 24 h 250 mg, < 1 h CO2-EDTA met.Hb death Von Clarmann (1987)
900 mg 69-77&
? < 1 h ND 27 h 2 x 250 mg, CO2-EDTA death Von Clarmann (1987)
1 h and 27 h 7.5 mg
50-150 mg? < 1 h 1.7 20 min 250 mg, 20 min recovery Von Clarmann (1987)
? <1 h 9.2 measurement ?, < 20 min met.Hb death Von Clarmann (1987)
at autopsy 77%
10 g 2h d NR 250 mg, > 2h recovery Von Clarmann (1987)
? 20 min d NR 750 mg, 20 min death Von Clarmann (1987)
320 mg 8 min 0.24 > 1 h 2 x 250 mg, recovery Von Clarmann (1987)
10 min and 1 h
Table 6. Contd.
Estimated Interval before Blood cyanide Interval before 4-DMAP dose, Additional Adverse Outcome Reference
cyanide first-aid concentration blood measurement interval before antidotes effects
ingested treatmentb (mg/l) usede administrationf
? 10 min 70 30 min 2 x 250 mg, death Von Clarmann (1987)
10 min and 30 min
? 20 min 24 1.5 h 250 mg, 1.5j; death Von Clarmann (1987)
? ? 37 < 1 h 1250 mg haemolysis recovery Von Clarmann (1987)
1 g 1 h 6 > 1 h 500 mg, 1 h met. Hb recovery Von Clarmann (1987)
25 g ? 10.5 2.5 h 250 mg, > 3 h CO2-EDTA recovery Von Clarmann (1987)
300 mg
? 15 min 7.5 1 h 250 mg, 4 h NaNO2 met.Hb death Van Dijk et al. (1987)
? 15 min NR NR 1000 mg, 30 min hydroxo- met.Hb recovery Van Heijst et al. (1987)
cobalamin haemolysis
a met.Hb = methaemoblobin; ? = unknown; NR = not recorded; ND = not detectable.
b Interval between cyanide exposure and beginning of first-aid treatment.
c Cyanide was present in the expired air but no measurement of blood concentration was made.
d Positive in qualitative tests.
e Interval between cyanide exposure and measurement of blood cyanide concentration.
f Interval between cyanide exposure and commencement of 4-DMAP treatment.
8.12.4 Areas of use where there is insufficient information to make
recommendations
There is insufficient evidence regarding the efficacy of the
prophylactic use of oral 4-DMAP, e.g., with rescue personnel.
8.13 Model Information Sheet
8.13.1 Uses
4-DMAP is indicated for the treatment of severe cyanide
poisoning in patients who are in deep coma and who have dilated
non-reactive pupils and deteriorating cardio-respiratory function.
8.13.2 Dosage and route
An intravenous dose of 3.25 mg/kg body weight should be given.
8.13.3 Precautions/contraindications
There are differences in individual susceptibility, which may
result in an unacceptably high level of methaemoglobin after normal
therapeutic doses (van Dijk et al., 1987). Excess
methaemoglobinaemia may be corrected with either methylene or
toluidine blue, but this will result in the release of cyanide;
exchange transfusion is an alternative approach if this is
practicable. Determination of methaemoglobin will not allow the
amount of haemoglobin available for oxygen transport to be
calculated, because the cyanmethaemoglobin concentration will be
unknown and, at the present time, cannot be analysed.
Treatment with 4-DMAP is contraindicated in patients with G6PD
deficiency.
8.13.4 Adverse effects
Haemolysis is observed following an overdose. Mild headache,
dizziness, hyperventilation, cyanosis, and green/brown
discolouration of the urine have also been observed. Haemolysis may
occur even in people with normal erythrocytes given an appropriate
therapeutic dose.
8.13.5 Use in pregnancy and lactation
No information is available.
8.13.6 Storage
4-DMAP must be stored in opaque containers. The maximum
storage time is 3 years.
8.14 References
Bright JE & Marrs TC (1982) A comparison of the
methemoglobin-inducing activity of 4-DMAP and p-aminopropriophenone.
Toxicol Lett, 13(1-2): 81-86.
Calder IC, Funder CC, Green CR, Ham K, & Tange JD (1971) Comparative
nephrotoxicity of aspirin and phenacetin derivatives. Br Med J, 4:
518-521.
Chen KK & Rose CL (1952) Nitrite and thiosulfate therapy in cyanide
poisoning. J Am Med Assoc, 149: 113-119.
Chen KK, Rose CL, & Clowes GHA (1933) Amyl nitrite and cyanide
poisoning. J Am Med Assoc, 100: 1920-1922.
Daunderer H, Theml H, & Weger N (1974) [Treatment of prussic acid
poisoning with 4-dimethylaminophenol (4-DMAP).] Med Klin, 69:
1626-1631 (in German).
Eckert KG & Eyer P (1983) Differences in the reaction of isomeric
ortho- and para-aminophenols with hemoglobin. Biochem Pharmacol, 32:
1019-1027.
Eckert KG & Eyer P (1986) Formation and transport of xenobiotic
glutathion-S-conjugates in red cells. Biochem Pharmacol, 35:
325-329.
Eyer P & Gaber H (1978) Biotransformation of 4-dimethylaminophenol
in the dog. Biochem Pharmacol, 27: 2215-2221.
Eyer P & Kampffmeyer H (1978) Biotransformation of
4-dimethylaminophenol in the isolated perfused rat liver. Biochem
Pharmacol, 27: 2223-2228.
Eyer P & Kiese M (1976) Biotransformation of 4-dimethylaminophenol;
reaction with glutathion and some properties of the reaction
products. Chem-Biol Interact, 14: 165-178.
Eyer P & Lengfelder E (1984) Radical formation during autooxidation
of 4-dimethylaminophenol and some properties of the reaction
product. Biochem Pharmacol, 33: 1005-1013.
Eyer P, Kiese M, Lipowsky G, & Weger N (1974) Reactions of
4-dimethylaminophenol with hemoglobin, and autooxidation of
4-dimethylaminophenol. Chem-Biol Interact, 8: 41-59.
Eyer P, Herle H, Kiese M, & Kluin G (1979) Kinetics of
ferrihaemoglobin formation by some reducing agents and the role of
hydrogen peroxide. Mol Pharmacol, 11: 326-334.
Green CR, Ham KN, & Tange JD (1969) Kidney lesions induced in rats
by p-aminophenol. Br Med J, 1: 162-164.
Hinsberg O & Treupel G (1984) [The physiological effects of
p-aminophenol and some of its derivatives.] Naunyn-Schmiedebergs
Arch Exp Pathol Pharmakol, 33: 216-250 (in German).
Jacobs K (1984) [Report on experience with the administration of
4-DMAP in severe prussic acid poisoning. Consequences for medical
practice.] Zentralbl Arbeitsmed, 34: 274-277 (in German).
Jansco L, Szinicz L, & Eyer P (1981) Biotransformation of
4-dimethylaminophenol in man. Arch Toxicol, 47: 39-45.
Kiese M (1974) Methaemoglobinaemia, a comprehensive treatise.
Cleveland, Ohio, CRC Press, Inc.
Kiese M & Weger N (1965) The treatment of experimental cyanide
poisoning by hemoglobin formation. Arch Toxicol, 21: 89-100.
Kiese M & Weger N (1969) Formation of ferrihemoglobin with
aminophenols in the human for the treatment of cyanide poisoning.
Eur J Pharmacol, 7: 97-105.
Kiese M, Szinick L, Thiel N, & Weger N (1975) Ferrihemoglobin and
kidney lesions in rats produced by 4-aminophenol or
4-dimethylaminophenol. Arch Toxicol, 34: 337-340.
Klimmek R, Fladerer H, & Weger N (1979a) Circulation, respiration
and blood homeostasis in cyanide poisoned dogs after treatments with
4-aminophenol or cobalt compounds. Arch Toxicol, 42: 121-133.
Klimmek R, Fladerer H, Szinicz L, Weger N, & Kriese M (1979b)
Effects of 4-dimethylaminophenol and CO2 EDTA on circulation,
respiration, and blood homeostasis in dogs. Arch Toxicol, 42: 75-84.
Klimmek R, Krettek C, Szinicz L, Eyer R, & Weger N (1983) Effects of
biotransformation of 4-dimethylaminophenol in man and dogs. Arch
Toxicol, 53: 275-288.
Lee CG & Webber TD (1983) The mutagenicity of a cyanide antidote,
dimethylaminophenol, in Chinese hamster cells. Toxicol Lett, 16:
275-288.
Marrs TC, Bright JE, & Swanson DW (1982) The effects of prior
treatment with 4-dimethylaminophenol on animals experimentally
poisoned with hydrogen cyanide. Arch Toxicol, 51: 247-253.
Marrs TC, Scawin J, & Swanson DW (1984) The acute intravenous and
oral toxicity in mice, rats and guinea pigs of 4-DMAP and its
effects on haematological variables. Toxicology, 31: 165-173.
Meister A (1973) On the enzymology of amino acid transport. Science,
180: 33-39.
Szizicz L, Weger N, Schneiderhan W, & Kiese M (1979) Nephrotoxicity
of aminophenols: Effects of the 4-dimethylaminophenol on isolated
rat kidney tubules. Arch Toxicol, 42: 63-73.
Van Dijk A, Glerum JH, Van Heijst CNP, & Douze JMC (1987) Clinical
evaluation of the cyanide antagonist 4-DMAP in a lethal cyanide
poisoning case. Vet Hum Toxicol, 29(suppl 2): 38-39.
Van Heijst ANP, Douze JMC, Van Kesteren RG, Bergen JEAM, & Van Dijk
A (1987) Therapeutic problems in cyanide poisoning. Clin Toxicol,
25: 383-398.
Von Clarmann M (1987) 4-dimethylaminophenol. Communication at the
joint meeting of the EAPCC, CEC, IPCS and the National Poison
Control Centre in the Netherlands, Utrecht.
Weger N (1968) [Aminophenols as antidotes to prussic acid.] Arch
Toxikol, 24: 49-50 (in German).
Weger N (1975) [Cyanide poisoning and therapy.] Wehrmed Monatschr,
19(1): 6-11 (in German with English summary).
9. Methylene Blue and Toluidine Blue
9.1 Methylene Blue
9.1.1 Introduction
Methylene blue is a redox dye that has been used in clinical
medicine for approximately 100 years. Its present uses are based on
its tissue-staining properties and its oxidative-reductive capacity
(Bodansky & Gutman, 1947; Blass & Fung, 1976; Metz et al., 1976).
Administered locally it has been used for the detection of fistulae
and for the recognition of ruptured amniotic membranes in obstetrics
(Sparhr & Salisbury, 1980; Martindale, 1989; Windholz, 1983). It
may also be administered systemically to assist the detection of
endocrine tissue, e.g., on parathyroid glands or pancreatic adenoma
during an operation (Blass & Fung, 1976; Whitman et al., 1979;
Martindale, 1989).
Methylene blue has long been recognized as an effective
antidote for methaemoglobinaemia in man and in domestic animals
(Etteldorf, 1951; Beutler & Baluda, 1963; Burrows et al., 1977;
Gosselin et al., 1984). Injected systemically in low doses, its
reducing action is utilized in the treatment of toxic
methaemoglobinaemia.
Methylene blue, however, is relatively ineffective against
toxic methaemoglobinaemia in individuals with glucose-6-phosphate
dehydrogenase deficiency and in chlorate poisoning (Bodansky &
Gutman, 1947; Metz et al., 1976; Gosselin et al., 1984).
9.1.2 Name and chemical formula of antidote
3,7-bis(dimethylamino)-phenazothionium chloride trihydrate,
tetramethylthionine chloride trihydrate
Synonyms: Methylioninii chloridum, Methylenum caerulum, Azul
de Metileno, Swiss blue, Blu di metilene, Schultz No.
1038, CI Classic Blue 9.
Colour index no: 52015
Total formula: C8H11 ON.HCl
Relative molecular mass: 173.5
Appearance: snow-white crystals
CAS number: 619-60-3
Manufacturer: Dr Franz Koehler Chemie GmbH,
Alsbach, Germany
Commercial name: 4-dimethylaminophenol (4-DMAP)
8.3 Physico-chemical Properties
Raw material: snow-white colourless crystals
Melting point: 145 °C + 1 °C
Solubility: very soluble in water. The
solution is oxidized by contact
with air and changes from being
colourless to black-brown
Optical properties:
IR-spectrum: peaks at (cm-1)
833: l,4-disubstituted aromatics
1240: phenols
1275: amine
1500: aromatics
2700: C-H aromatics
3200: N-H aromatics
UV data: maximum at 271 nm; extinction
coefficient 1255 l/mol
Acidity: no data available
pK (-N(CH3)2) = 6.15
pK (-OH) = 10.1
Stability in light: no data available
Thermal stability: no data available
Refractive index
and specific gravity: unknown
Weight on drying: no data available
Excipients and
pharmaceutical aids: unknown for the final product
although it may contain an
antioxidant such as sodium
pyrosulfate
8.4 Synthesis
Routes of synthesis: no information available
Manufacturing processes: no information available
8.5 Analytical Methods
8.5.1 Identity
To 5 ml of liquid from the ampoule, add one drop 30% hydrogen
peroxide. The solution will turn a deep violet colour within 5 min.
To 2.5 ml of liquid from the ampoule, add 0.5 ml 10% ferrichloride.
A pale-brown colour will appear immediately.
8.5.2 Quantification
AgNO3 titration: 2.5 ml liquid from the ampoule is diluted
in 40 ml water and, after the addition of 3 ml nitric acid
(2 mol/l), potentiometrically titrated with silver nitrate
(0.1 mol/l). 1 ml AgNO3 (0.1 mol/l) equiv. 17.365 mg
dimethylaminophenol hydrochloride.
HClO4 titration: 2.5 ml liquid from the ampoule is added to
40 ml of a 3% solution of mercury acetate in glacial acetic acid and
then potentiometrically titrated with perchloric acid (0.1 mol/l).
1 ml perchloric acid (0.1 mol/l) equiv. 17.365 mg
dimethylaminophenol hydrochloride.
8.5.3 Purity
Unknown
8.5.4 Methods for analysis of 4-DMAP in biological samples
Unknown.
8.6 Shelf-life
Since, in contact with air, 4-DMAP is readily oxidized and the
solution changes colour to black-brown, 4-DMAP must be stored in
opaque containers. An open ampoule cannot be kept. Coloured and
turbid solutions cannot be used. Correctly stored 4-DMAP can be
used after storage for up to 3 years. The influence of tropical
conditions is unknown.
8.7 General Properties
The use of 4-DMAP results in rapid formation of methaemoglobin.
An intravenous dose of 3.25 mg/kg resulted in a methaemoglobin
concentration of 30% within 10 min. Because there is a much larger
source of haemoglobin than there is of cytochrome oxidase,
cytochrome oxidase blocked by cyanide can be reactivated and its
role as an essential catalyst for tissue utilization of oxygen
restored.
8.8 Animal Studies
8.8.1 In Vitro Studies
In studies involving the addition of various aminophenols such
as 2-aminophenol, 2-amino-4-chlorophenol, 2-amino-4,6-
dichlorophenol, 2-amino-5-ethoxyphenol, 2-dimethylaminophenol,
4-aminophenol, 4-methylaminophenol, and 4-dimethylaminophenol to the
blood of mice, cats, dogs, cattle, and man, it has been shown that
4-dimethylaminophenol and 4-methylaminophenol produce consistent
amounts of methaemoglobin most rapidly. These studies also showed
that a concentration of 30-40% methaemoglobin was attained at widely
varying rates in different species (Kiese & Weger, 1969).
8.8.1.1 Metabolism of 4-DMAP in the liver
In a study by Eyer & Kampffmeyer (1978), nearly all the 4-DMAP
was conjugated in livers perfused with 4-DMAP (0.01 mmol/l)
(0.4 nmol/mg protein per min).
8.8.1.2 Red cell metabolism of 4-DMAP
4-DMAP rapidly catalyses methaemoglobin formation. The
reaction mechanism has been studied using purified human
haemoglobin. 4-DMAP transfers electrons catalytically from
ferrohaemoglobin to oxygen (Eyer et al., 1974; Eckert & Eyer, 1983)
and is thereby oxidized to the phenoxyl-radical and N,N-dimethyl-
quinoneimine (Eyer & Lengfelder, 1984). Both species are reduced to
ferrohaemoglobin with the formation of methaemoglobin.
Catalytic methaemoglobin formation is terminated by rapid
binding of oxidized 4-DMAP to reactive SH groups in haemoglobin and
thioether formation with reduced glutathione. Formation of
glutathione-S-conjugates with 4-DMAP has been shown to occur in
vitro in red cells (Eyer & Kiese, 1976), and in vivo in dogs
(Eyer & Gaber, 1978) and man (Jansco et al., 198l, Klimmek et al.,
1983). Kinetic studies of thioether formation by DMAP in the
presence of purified dog haemoglobin and glutathione in a reducing
system showed that S,S-(2-dimethylamino-5-hydroxy-1,3-phenylene)-
bis-gluthathione (di(GS)-DMAP) is formed initially. This thioether
is highly autoxidizable and the addition of another thiol gives
stable S,S,S(2-dimethylamino)-5-hydroxy-1,3,4-phenylene)-
tri-glutathione (tris-(GS)-DMAP). Despite its chemical stability,
it can be actively transported across the red blood cell membrane
(Eckert & Eyer, 1986).
4-DMAP is excreted in the urine mainly as the tris-cysteinyl
derivative, tris(Cys)-DMAP. This compound is presumed to be broken
down in the glutamyl cycle of the kidney (Meister, 1973), giving
rise to cysteinyl glycine and cysteinyl derivatives.
8.8.1.3 Toxic effects of 4-DMAP on erythrocytes
Studies have been undertaken to test the effect of 4-DMAP on
the osmotic resistance of human erythrocytes. The percentage
haemolysis did not differ from that of controls, even for an
incubation period of less than 20 h, at a 4-DMAP concentration of
1 mmol/l. However, the degree of haemolysis after 20 h was 14.5%
(±6.9%), compared with a control value of 2.7% (±0.4%), when
measured in 0.7% NaCl solution (Klimmek et al., 1983). Since the
haemolysis that occurred in the tests for osmotic resistance was
maximal after the same period as in in vivo studies, one may
assume a common lytic mechanism under in vitro and in vivo
conditions (Klimmek et al., 1983).
8.8.1.4 Toxic effects of 4-DMAP on isolated rat kidney tubules
Various toxic effects were observed when 4-DMAP was added to
suspensions of isolated rat kidney tubules, apparently as the result
of the irreversible binding of 4-DMAP to SH-containing compounds,
e.g., multiple enzyme inhibition, inhibition of gluconeogenesis,
decrease in glutathione, and membrane damage (Snizicz et al., 1979).
The results suggest that irreversible impairment of the integrity of
the cell membrane causes irreversible damage to tubular epithelial
cells, the resultant increase in permeability being followed by a
decrease in nucleotide content without marked effect on
gluconeogenesis or mitochondrial membrane integrity.
8.8.1.5 Oxygen saturation and methaemoglobin formation
From reports by Kiese (1974) and Eyer et al. (1979), it is
evident that quite small variations of pO2 can cause large
differences in the rate of methaemoglobin formation. At high
percentage oxyhaemoglobin levels, where the R-configuration of
haemoglobin prevails over the T-configuration, the rate of oxidation
is lower. At a 4-DMAP concentration of 0.032 mmol/l, increasing the
percentage saturation of haemoglobin of humans more than halved the
rate of methaemoglobin production (Marrs et al., 1982).
8.8.2 Pharmacokinetics
No data are available.
8.8.3 Pharmacodynamics
4-DMAP (3.25 mg/kg), given intravenously to dogs one min after
poisoning with a lethal dose of KCN (4 mg/kg), resulted in the
survival of all dogs. The maximum venous methaemoglobin content was
reached within 5-10 min and was 38.8% (±1.7%) of the total
haemoglobin. The injection of the same 4-DMAP dose intramuscularly
led to a maximum methaemoglobin level of 41.6% (±1.3%) after 30 min.
A methaemoglobin concentration of 35% was achieved within 5 min of
an intravenous dose of 3.25 mg/kg, 15 min of an intramuscular dose
of 3.25 mg/kg, and 30 min of an oral dose of 15 mg/kg (Klimmek et
al., 1983).
Bright & Marrs (1982) found that there was considerable
variation in the methaemoglobin levels produced in different dogs
using the same oral dose of 4-DMAP.
In experiments using different aminophenols, it was shown that
4-DMAP given intravenously rapidly produced consistent amounts of
methaemoglobin in the blood of different animals species. In mice
and rabbits, a rapid fall in the concentration of methaemoglobin was
observed after 2 min and after 20 min the concentration in these
species was less than 10%. In dogs and mice, however, the decrease
in methaemoglobin concentration was substantially less; after
20 min, the level was still above 30%. In contrast to 4-DMAP,
sodium nitrite caused increased methaemoglobin levels which lasted
longer than an hour in mice and rabbits (Kiese & Weger, 1969).
There was a marked increase in arterial pO2 after the
administration of 4-DMAP to dogs poisoned with cyanide, compared
with non-poisoned dogs. Presumably, part of the oxygen released
during the formation of methaemoglobin under hypoxic conditions can
be utilized by tissues (Klimmek et al., 1979a,b). In the same
experiments, an increase in respiratory volume and mean arterial
blood pressure, was observed together with a lessening of the
lactate-pyruvate ratio.
8.8.4 Toxicology
LD50 intravenous mouse: 50-70 mg/kg (Kiese & Weger, 1965;
Marrs et al., 1984)
LD50 oral mouse: 946 mg/kg (Marrs et al., 1984)
LD50 intravenous rat: 57 mg/kg (Kiese et al., 1975)
LD50 oral rat: 689-780 mg/kg (Marrs et al., 1984)
LD50 intravenous dog: 26 mg/kg (Weger, 1975)
In all five studies, the cause of death was inadequate oxygen
transport by the blood to the tissues because 85% of the haemoglobin
had been converted to methaemoglobin.
8.8.4.1 Nephrotoxicity
Some of the aminophenols, such as 4-aminophenol, produce kidney
lesions after intravenous administrations (Green et al., 1969;
Calder et al., 1971; Hinsberg & Treupel, 1984). The intravenous
injection of 100 mg 4-DMAP/kg, a dose nearly double the LD50 and
which oxidizes 60% of the haemoglobin to methaemoglobin, caused
renal damage in rats. Moderate to severe necrosis of the convoluted
tubules, with either few or no inflammatory cells, was found 24 h
after injection. The glomeruli were not affected and there was no
papillary damage. However, no tubular necrosis was detected with a
dose of 30 mg/kg. A high or low sodium diet did not noticeably
affect the nephrotoxicity of 4-DMAP (Kiese et al., l975).
A single oral dose of up to 25 mg 4-DMAP/kg did not result in
any macroscopic or histological abnormality in the gastrointestinal
tract, liver, or kidneys of rats killed up to 24 h after dosing. In
animals that had been kept for 7 days after dosing, no histological
abnormalities were found other than engorgement of the liver and
kidneys (Marrs et al., 1982).
8.8.4.2 Mutagenicity
A clear relationship between dose and mutation frequency was
shown by a mammalian cell assay employing V79 (Chinese hamster)
cells (Lee & Webber, 1983), despite a negative Ames test with and
without metabolic activation.
8.9 Volunteer Studies
In experiments by Weger (1968) on seven volunteers, an
intravenous dose of N,N-dimethyl- p-aminophenol (3.25 mg/kg)
resulted in a 15% methaemoglobin concentration within 1 min. After
10 min, the concentration was 30%. Other aminophenols tested gave
less favourable results. N-methyl- p-aminophenol had much the
same effect but only at an iv dose of 20 mg/kg. O-aminophenol
(30 mg/kg) administered intravenously resulted in 15%
methaemoglobinaemia after 12 min and 30% after 60 min. Comparison
was also made with sodium nitrite. When a dose of 4 mg/kg was given
intravenously to seven volunteers, the maximal methaemoglobin level
was 6%. This dose of sodium nitrite was that recommended by Chen et
al. (l933) and Chen & Rose (l952) and suggests that the antidotal
effect may be due to a mechanism other than methaemoglobin
formation.
In experiments by Klimmek et al. (1983) on volunteers a
methaemoglobin concentration of about 30% was reached within 5 min
after an intravenous dose of 3.25 mg 4-DMAP/kg (3 volunteers),
50 min after an intramuscular dose of 3.5 mg 4-DMAP/kg
(6 volunteers), and 30 min after an oral dose of 900 mg 4-DMAP
(5 volunteers).
8.9.1 Metabolism of 4-DMAP in the liver
The metabolic conversion of 4-DMAP to glucuronide, sulfate, and
thioether derivatives is likely to follow linear Michaelis-Menten
kinetics. The pattern of metabolites was studied in urine collected
over 24 hours from subjects treated with intravenous 4- DMAP
(3.25 mg/kg). An average of 68% of the dose was excreted as
metabolites in urine: 41% as glucuronide, 12% as sulfate, and 15%
as thioethers (Klimmek et al., 1983).
4-DMAP rapidly auto-oxidizes at pH values above 7 and is
transformed to a variety of degradation products (Eyer et al.,
1974). Thus partial degradation of 4-DMAP in the gastrointestinal
tract may account for the reduced total metabolite recovery
following oral administration compared with that following
intravenous administration.
8.9.2 Metabolism of 4-DMAP in erythrocytes
Rapid thioether formation following the intravenous
administration of 4-DMAP has been shown to occur in erythrocytes
where 4-DMAP is oxidized by oxyhaemoglobin to the corresponding
quinoneimine which yields adducts with red cell glutathione (Eyer &
Kiese, 1976). One of these, tris-(GS)-DMAP penetrates the red cell
membrane slowly and is excreted in the urine mainly as a
tris-cysteinyl derivative, tris-(Cys)-DMAP.
The 4-DMAP thioethers excreted in human urine (15.3% (±1.8%) of
the dose) are 1ikely to originate in the erythrocytes; this
underlines the importance of erythrocytes in 4-DMAP metabolism in
man.
8.9.3 Adverse effects
After 6-7 days, a phlebitis was observed in the antecubital
vein where 4-DMAP was infused (Klimmek et al., 1983). Following an
intramuscular injection of 4-DMAP, a slight pressure was felt after
5-10 min at the site of injection, slowly growing in intensity and
finally resulting in severe pain. Approximately 10 h after the
injection, shivering, sweating, and fever occurred. An oral 4-DMAP
dose of 300 mg was well tolerated, whereas 600 mg slowed mental
activity and caused tiredness, and 900 mg gave an effect of
dizziness, with headache and buzzing in the ears (Klimmek et al.,
1983).
After an intravenous injection of 4-DMAP (3.25 mg/kg), the
total bilirubin concentration increased by 140%, that of conjugated
bilirubin by 180% and that of iron by 200%. Within 24 h of an
intramuscular injection of 4-DMAP (3.5 mg/kg), the total bilirubin
increased by 270% and then declined rapidly, while the bilirubin
concentration rose by 120% and that of iron by 50% (Klimmek et al.,
1983).
After an oral dose of 900 mg 4-DMAP, total and conjugated
bilirubin levels increased by 170% and 80%, respectively, and the
iron concentration rose by 60%. The changes in bilirubin and iron
concentrations were the result of reduced erythrocyte survival
caused by 4-DMAP, which took about one day to develop. Since the
haemolysis that occurred in the tests for osmotic resistance was
maximal after the same period as in in vivo studies, one may
assume a common lytic mechanism both under in vitro and in vivo
conditions (Klimmek et al., 1983).
8.10 Clinical Studies
No data are available.
8.11 Clinical Studies - Case Reports
Table 6 summarizes clinical observations made in 19 cases of
cyanide poisoning treated by 4-DMAP. In most patients, there was a
long time interval between the occurrence of poisoning and the
administration of 4-DMAP. Patients with impaired vital function at
the time of treatment recovered. In six cases an overdose of 4-DMAP
was given, resulting in excessive methaemoglobinaemia, and in three
cases haemolysis was observed on the second day after administration
of the antidote. Even the recommended dose of 3.25 mg 4-DMAP/kg has
resulted in a methaemoglobin concentration of about 70% (van Dijk et
al., 1987).
8.12 Summary of Evaluation
8.12.1 Indications
4-DMAP should only be given to patients poisoned with cyanide
who are in a deep coma and who have dilated non-reactive pupils and
deteriorating cardio-respiratory function.
8.12.2 Recommended routes and dosage
An intravenous dose of 3.25 mg/kg body weight should be given
immediately. The intramuscular injection of 4-DMAP to patients in
shock due to cyanide poisoning cannot be recommended because
absorption is unpredictable.
8.12.3 Other consequential or supportive therapy
The administration of 4-DMAP should always be followed by that
of sodium thiosulfate (150-200 mg/kg intravenously) (see chapter 3).
Artificial respiration should be given if necessary.
Table 6. Clinical Observations in cyanide-poisoned patients treated by 4-DMAPa
Estimated Interval before Blood cyanide Interval before 4-DMAP dose, Additional Adverse Outcome Reference
cyanide first-aid concentration blood measurement interval before antidotes effects
ingested treatmentb (mg/l) usede administrationf
10 g oral 15 min ? 250 mg, 45 min CO2-EDTA recovery Daunderer et al. (1974)
? < 5 min 5-16.9 5 min 250 mg, 5 min CNS Jacobs (1984)
damage
? 1.5-2 h c 5.5 h 250 mg, > 5h recovery Von Clarmann (1987)
? 20 min NR NR 250 mg, 3.5h recovery Von Clarmann (1987)
? < 1h 1.9 > 24 h 250 mg, < 1 h CO2-EDTA met.Hb death Von Clarmann (1987)
900 mg 69-77&
? < 1 h ND 27 h 2 x 250 mg, CO2-EDTA death Von Clarmann (1987)
1 h and 27 h 7.5 mg
50-150 mg? < 1 h 1.7 20 min 250 mg, 20 min recovery Von Clarmann (1987)
? <1 h 9.2 measurement ?, < 20 min met.Hb death Von Clarmann (1987)
at autopsy 77%
10 g 2h d NR 250 mg, > 2h recovery Von Clarmann (1987)
? 20 min d NR 750 mg, 20 min death Von Clarmann (1987)
320 mg 8 min 0.24 > 1 h 2 x 250 mg, recovery Von Clarmann (1987)
10 min and 1 h
Table 6. Contd.
Estimated Interval before Blood cyanide Interval before 4-DMAP dose, Additional Adverse Outcome Reference
cyanide first-aid concentration blood measurement interval before antidotes effects
ingested treatmentb (mg/l) usede administrationf
? 10 min 70 30 min 2 x 250 mg, death Von Clarmann (1987)
10 min and 30 min
? 20 min 24 1.5 h 250 mg, 1.5j; death Von Clarmann (1987)
? ? 37 < 1 h 1250 mg haemolysis recovery Von Clarmann (1987)
1 g 1 h 6 > 1 h 500 mg, 1 h met. Hb recovery Von Clarmann (1987)
25 g ? 10.5 2.5 h 250 mg, > 3 h CO2-EDTA recovery Von Clarmann (1987)
300 mg
? 15 min 7.5 1 h 250 mg, 4 h NaNO2 met.Hb death Van Dijk et al. (1987)
? 15 min NR NR 1000 mg, 30 min hydroxo- met.Hb recovery Van Heijst et al. (1987)
cobalamin haemolysis
a met.Hb = methaemoblobin; ? = unknown; NR = not recorded; ND = not detectable.
b Interval between cyanide exposure and beginning of first-aid treatment.
c Cyanide was present in the expired air but no measurement of blood concentration was made.
d Positive in qualitative tests.
e Interval between cyanide exposure and measurement of blood cyanide concentration.
f Interval between cyanide exposure and commencement of 4-DMAP treatment.
8.12.4 Areas of use where there is insufficient information to make
recommendations
There is insufficient evidence regarding the efficacy of the
prophylactic use of oral 4-DMAP, e.g., with rescue personnel.
8.13 Model Information Sheet
8.13.1 Uses
4-DMAP is indicated for the treatment of severe cyanide
poisoning in patients who are in deep coma and who have dilated
non-reactive pupils and deteriorating cardio-respiratory function.
8.13.2 Dosage and route
An intravenous dose of 3.25 mg/kg body weight should be given.
8.13.3 Precautions/contraindications
There are differences in individual susceptibility, which may
result in an unacceptably high level of methaemoglobin after normal
therapeutic doses (van Dijk et al., 1987). Excess
methaemoglobinaemia may be corrected with either methylene or
toluidine blue, but this will result in the release of cyanide;
exchange transfusion is an alternative approach if this is
practicable. Determination of methaemoglobin will not allow the
amount of haemoglobin available for oxygen transport to be
calculated, because the cyanmethaemoglobin concentration will be
unknown and, at the present time, cannot be analysed.
Treatment with 4-DMAP is contraindicated in patients with G6PD
deficiency.
8.13.4 Adverse effects
Haemolysis is observed following an overdose. Mild headache,
dizziness, hyperventilation, cyanosis, and green/brown
discolouration of the urine have also been observed. Haemolysis may
occur even in people with normal erythrocytes given an appropriate
therapeutic dose.
8.13.5 Use in pregnancy and lactation
No information is available.
8.13.6 Storage
4-DMAP must be stored in opaque containers. The maximum
storage time is 3 years.
8.14 References
Bright JE & Marrs TC (1982) A comparison of the
methemoglobin-inducing activity of 4-DMAP and p-aminopropriophenone.
Toxicol Lett, 13(1-2): 81-86.
Calder IC, Funder CC, Green CR, Ham K, & Tange JD (1971) Comparative
nephrotoxicity of aspirin and phenacetin derivatives. Br Med J, 4:
518-521.
Chen KK & Rose CL (1952) Nitrite and thiosulfate therapy in cyanide
poisoning. J Am Med Assoc, 149: 113-119.
Chen KK, Rose CL, & Clowes GHA (1933) Amyl nitrite and cyanide
poisoning. J Am Med Assoc, 100: 1920-1922.
Daunderer H, Theml H, & Weger N (1974) [Treatment of prussic acid
poisoning with 4-dimethylaminophenol (4-DMAP).] Med Klin, 69:
1626-1631 (in German).
Eckert KG & Eyer P (1983) Differences in the reaction of isomeric
ortho- and para-aminophenols with hemoglobin. Biochem Pharmacol, 32:
1019-1027.
Eckert KG & Eyer P (1986) Formation and transport of xenobiotic
glutathion-S-conjugates in red cells. Biochem Pharmacol, 35:
325-329.
Eyer P & Gaber H (1978) Biotransformation of 4-dimethylaminophenol
in the dog. Biochem Pharmacol, 27: 2215-2221.
Eyer P & Kampffmeyer H (1978) Biotransformation of
4-dimethylaminophenol in the isolated perfused rat liver. Biochem
Pharmacol, 27: 2223-2228.
Eyer P & Kiese M (1976) Biotransformation of 4-dimethylaminophenol;
reaction with glutathion and some properties of the reaction
products. Chem-Biol Interact, 14: 165-178.
Eyer P & Lengfelder E (1984) Radical formation during autooxidation
of 4-dimethylaminophenol and some properties of the reaction
product. Biochem Pharmacol, 33: 1005-1013.
Eyer P, Kiese M, Lipowsky G, & Weger N (1974) Reactions of
4-dimethylaminophenol with hemoglobin, and autooxidation of
4-dimethylaminophenol. Chem-Biol Interact, 8: 41-59.
Eyer P, Herle H, Kiese M, & Kluin G (1979) Kinetics of
ferrihaemoglobin formation by some reducing agents and the role of
hydrogen peroxide. Mol Pharmacol, 11: 326-334.
Green CR, Ham KN, & Tange JD (1969) Kidney lesions induced in rats
by p-aminophenol. Br Med J, 1: 162-164.
Hinsberg O & Treupel G (1984) [The physiological effects of
p-aminophenol and some of its derivatives.] Naunyn-Schmiedebergs
Arch Exp Pathol Pharmakol, 33: 216-250 (in German).
Jacobs K (1984) [Report on experience with the administration of
4-DMAP in severe prussic acid poisoning. Consequences for medical
practice.] Zentralbl Arbeitsmed, 34: 274-277 (in German).
Jansco L, Szinicz L, & Eyer P (1981) Biotransformation of
4-dimethylaminophenol in man. Arch Toxicol, 47: 39-45.
Kiese M (1974) Methaemoglobinaemia, a comprehensive treatise.
Cleveland, Ohio, CRC Press, Inc.
Kiese M & Weger N (1965) The treatment of experimental cyanide
poisoning by hemoglobin formation. Arch Toxicol, 21: 89-100.
Kiese M & Weger N (1969) Formation of ferrihemoglobin with
aminophenols in the human for the treatment of cyanide poisoning.
Eur J Pharmacol, 7: 97-105.
Kiese M, Szinick L, Thiel N, & Weger N (1975) Ferrihemoglobin and
kidney lesions in rats produced by 4-aminophenol or
4-dimethylaminophenol. Arch Toxicol, 34: 337-340.
Klimmek R, Fladerer H, & Weger N (1979a) Circulation, respiration
and blood homeostasis in cyanide poisoned dogs after treatments with
4-aminophenol or cobalt compounds. Arch Toxicol, 42: 121-133.
Klimmek R, Fladerer H, Szinicz L, Weger N, & Kriese M (1979b)
Effects of 4-dimethylaminophenol and CO2 EDTA on circulation,
respiration, and blood homeostasis in dogs. Arch Toxicol, 42: 75-84.
Klimmek R, Krettek C, Szinicz L, Eyer R, & Weger N (1983) Effects of
biotransformation of 4-dimethylaminophenol in man and dogs. Arch
Toxicol, 53: 275-288.
Lee CG & Webber TD (1983) The mutagenicity of a cyanide antidote,
dimethylaminophenol, in Chinese hamster cells. Toxicol Lett, 16:
275-288.
Marrs TC, Bright JE, & Swanson DW (1982) The effects of prior
treatment with 4-dimethylaminophenol on animals experimentally
poisoned with hydrogen cyanide. Arch Toxicol, 51: 247-253.
Marrs TC, Scawin J, & Swanson DW (1984) The acute intravenous and
oral toxicity in mice, rats and guinea pigs of 4-DMAP and its
effects on haematological variables. Toxicology, 31: 165-173.
Meister A (1973) On the enzymology of amino acid transport. Science,
180: 33-39.
Szizicz L, Weger N, Schneiderhan W, & Kiese M (1979) Nephrotoxicity
of aminophenols: Effects of the 4-dimethylaminophenol on isolated
rat kidney tubules. Arch Toxicol, 42: 63-73.
Van Dijk A, Glerum JH, Van Heijst CNP, & Douze JMC (1987) Clinical
evaluation of the cyanide antagonist 4-DMAP in a lethal cyanide
poisoning case. Vet Hum Toxicol, 29(suppl 2): 38-39.
Van Heijst ANP, Douze JMC, Van Kesteren RG, Bergen JEAM, & Van Dijk
A (1987) Therapeutic problems in cyanide poisoning. Clin Toxicol,
25: 383-398.
Von Clarmann M (1987) 4-dimethylaminophenol. Communication at the
joint meeting of the EAPCC, CEC, IPCS and the National Poison
Control Centre in the Netherlands, Utrecht.
Weger N (1968) [Aminophenols as antidotes to prussic acid.] Arch
Toxikol, 24: 49-50 (in German).
Weger N (1975) [Cyanide poisoning and therapy.] Wehrmed Monatschr,
19(1): 6-11 (in German with English summary).
9. Methylene Blue and Toluidine Blue
9.1 Methylene Blue
9.1.1 Introduction
Methylene blue is a redox dye that has been used in clinical
medicine for approximately 100 years. Its present uses are based on
its tissue-staining properties and its oxidative-reductive capacity
(Bodansky & Gutman, 1947; Blass & Fung, 1976; Metz et al., 1976).
Administered locally it has been used for the detection of fistulae
and for the recognition of ruptured amniotic membranes in obstetrics
(Sparhr & Salisbury, 1980; Martindale, 1989; Windholz, 1983). It
may also be administered systemically to assist the detection of
endocrine tissue, e.g., on parathyroid glands or pancreatic adenoma
during an operation (Blass & Fung, 1976; Whitman et al., 1979;
Martindale, 1989).
Methylene blue has long been recognized as an effective
antidote for methaemoglobinaemia in man and in domestic animals
(Etteldorf, 1951; Beutler & Baluda, 1963; Burrows et al., 1977;
Gosselin et al., 1984). Injected systemically in low doses, its
reducing action is utilized in the treatment of toxic
methaemoglobinaemia.
Methylene blue, however, is relatively ineffective against
toxic methaemoglobinaemia in individuals with glucose-6-phosphate
dehydrogenase deficiency and in chlorate poisoning (Bodansky &
Gutman, 1947; Metz et al., 1976; Gosselin et al., 1984).
9.1.2 Name and chemical formula of antidote
3,7-bis(dimethylamino)-phenazothionium chloride trihydrate,
tetramethylthionine chloride trihydrate
Synonyms: Methylioninii chloridum, Methylenum caerulum, Azul
de Metileno, Swiss blue, Blu di metilene, Schultz No.
1038, CI Classic Blue 9.
Colour index no: 52015
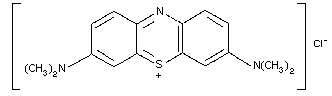 Molecular formula: C16H18C1N3S.3H2O (Martindale, 1989;
Windholz, 1983)
Relative molecular mass: 373.9
CAS number: 7220-79-3 (trihydrate) and 67-73-4 (anhydrous)
Commercial methylene blue is the double chloride of
tetramethylthionine and zinc and is not suitable for
medicinal use (Windholz, 1983).
9.1.3 Physico-chemical properties
Methylene blue is a dark green, almost odourless, hygroscopic
crystalline powder with a bronze-like lustre. It loses 8-18% of its
weight on drying. One gram dissolves in about 25 ml of water, in
about 65 ml alcohol, and in 450 ml of chloroform. Methylene blue is
insoluble in ether (Martindale, 1989; Windholz, 1983).
A 1% solution in water has a pH of 3 to 4.5. Solutions in
water are intensely blue-coloured and incompatible with caustic
alkalis, oxidising and reducing substances and iodides (Martindale,
1989).
Solutions are sterilised by autoclaving or by filtration and
should be stored in airtight containers (Windholz, 1983).
Data on melting and boiling points and on stability in light
are not available.
Excipients: water for injection.
9.1.4 Synthesis
First prepared by Caro in 1876, methylene blue is produced by
the so-called "thiosulfate process" in which a mixture of
dimethyl- p-phenylenediamine and dimethylaniline is oxidized,
usually with potassium dichromate, in the presence of sodium
thiosulfate and zinc chloride (Windholz, 1983).
9.1.5 Analytical methods
No information on analytical methods for methylene blue is
available.
9.1.6 Shelf-life
No data about specific conditions of temperature and humidity
are available, and the shelf-life is unknown. Methylene blue should
be stored in airtight containers.
9.1.7 General properties
Methylene blue functions as an intermediate in the transfer of
electrons from pyridine nucleotides to a suitable electron acceptor,
thereby stimulating the hexose monophosphate shunt (HMPS) pathway in
a variety of cell systems (Bodansky & Gutman, 1947; Beutler &
Baluda, 1963; Smith & Olson, 1973; Metz et al., 1976).
In the erythrocyte, methylene blue is reduced to leucomethylene
blue primarily by NADPH-dependent diaphorase (dihydrolipoamide
dehydrogenase). This diaphorase is reduced via oxidation of NADPH,
which in turn stimulates the HMPS, and leucomethylene blue transfers
electrons to an acceptor such as methaemoglobin (Bodansky & Gutman,
1947; Metz et al., 1976). This series of reactions can be used
clinically for the reduction of ferric to ferrous haem iron in
patients with acquired methaemoglobinaemia. The effectiveness of
methylene blue in reducing methaemoglobin is well established
(Etteldorf, 1951; Beutler & Baluda, 1963; Smith & Olson, 1973;
Harrison, 1977; Gosselin et al., 1984).
9.1.8 Animal studies
Several animal studies confirm that the administration of
methylene blue protects against death caused by
methaemoglobin-generating agents (Blass & Fung, 1976; Burrows et
al., 1977; Hrushesky et al., 1985). Burrows et al. (1977)
administered sodium nitrite (50 mg/kg) intervenously to four ewes.
This resulted in the formation of 70-80% methaemoglobinaemia (a
lethal level) within 45 min. The mortality associated with this
dose of sodium nitrite was successfully antagonized by the
administration of intravenous methylene blue (2.2 mg/kg) 30 min
later.
Pharmacological studies have been carried out in dogs and rats
(Blass & Fung, 1976). When intravenous boluses as large as 15 mg/kg
were given, the resultant data could be explained on the basis of a
one-compartment model with binding of methylene blue to tissues.
Few data on mutagenicity or teratogenicity testing are
available.
9.1.9 Volunteer studies
No data on volunteer studies are available.
9.1.10 Clinical studies
The clinical symptoms and signs associated with
methaemoglobinaemia depend on the percentage of haemoglobin oxidized
to the methaemoglobin form (Bodansky & Gutman, 1947). They consist
of greyish-blue cyanosis, without signs of cardiac or pulmonary
distress, which becomes apparent at about 15% methaemoglobinaemia.
Hypoxaemia may be accompanied by dyspnoea, dizziness, headache,
weakness, lethargy, and CNS depression, the severity of which will
increase with increasing concentrations of methaemoglobin.
An intense chocolate-brown-coloured blood and a central
cyanosis that does not respond to the administration of 100% oxygen
suggest methaemoglobinaemia (Smith & Olson, 1973; Gosselin et al.,
1984; Hall et al., 1986). Methaemoglobin levels may be directly
measured using a spectophotometric method (Hall et al., 1986).
Many authors agree that in asymptomatic patients, with
methaemoglobin levels of 30% or less, methylene blue administration
is not necessary (Bodansky & Gutman, 1947; Gosselin et al., 1984;
Hall et al., 1986).
9.1.11 Clinical studies - case reports
Ng et al. (1982) described a case of methaemoglobinaemia and
haemolysis resulting from the ingestion of paraquat by a 32-year-old
patient. He was administered methylene blue (1 mg/kg) intravenously,
with dramatic results: cyanosis was reversed and the patient became
alert and cooperative. The methaemoglobin level was 19.7% before
methylene blue was given and 1.6% after.
A single case of dapsone-induced methaemoglobinaemia was
treated by continuous methylene blue infusion (Berlin et al., 1985).
The patient recovered completely.
9.1.12 Summary of evaluation
9.1.12.1 Indications
Methylene blue is the agent of choice for accelerating the
reduction of methaemoglobin, at toxic levels, induced by aniline and
aminophenols, nitrites and nitrates, nitrobenzene, antimalarial
agents (chloroquine, primaquine), dapsone, local anaesthetics
(lidocaine, prilocaine, benzocaine), phenacetin, phenazopyridin, and
sulfamidines. It should be used with special caution in the
correction of excess methaemoglobinaemia induced by the treatment of
cyanide poisoning with nitrites.
9.1.12.2 Advised route and dosage
Methylene blue should be administered as an intravenous dose of
1-2 mg/kg body weight (0.1 to 0.2 ml/kg of a 1% solution), slowly
over 5 to 10 min. An improvement should be noted within 30-60 min
of administration. If cyanosis has not disappeared within 1 h, a
second dose should be given (Bodansky & Gutman, 1947; Etteldorf,
1951; Smith & Olson, 1973; Harrison, 1977; Hall et al., 1986).
9.1.12.3 Precautions and contraindications
Methylene blue should not be given by subcutaneous or
intrathecal injection because it causes necrotic abscess formation
(Whitman et al., 1979; Windholz, 1983; Gosselin et al., 1984). It
should be administered with caution in patients with severe renal
impairment (Windholz, 1983; Martindale, 1989; Gosselin et al.,
1984).
In patients with glucose-6-phosphate dehydrogenase deficiency
(who do not generate NADPH), methylene blue is likely to be
ineffective and has been reported to cause haemolytic anaemia
(Beutler & Baluda, 1963; Rosen et al., 1971; Whitman, et al., 1979;
Martindale, 1989).
Methylene blue should not be used in chlorate-induced
methaemoglobinaemia because it may enhance the toxicity of the
chlorate salts (Metz et al., 1976; Martindale, 1989).
9.1.12.4 Adverse effects
Nausea, abdominal and chest pain, headache, dizziness, mental
confusion and profuse sweating may occur following large intravenous
doses of methylene blue (Naidler et al., 1945; Martindale, 1989;
Windholz, 1983).
Whitman et al. (1979) described a 28-year-old patient who
received methylene blue (5 mg/kg) for the identification of an
insulin-secreting pancreatic adenoma. During surgery, the patient
developed methaemoglobin levels of up to 7.1%.
Sparhr & Salisbury (1980) reported a case where an
intra-amniotic injection of 10 mg of methylene blue was given in a
pregnant woman into the vitelline sac of one of two twins. On the
second day, the new-born baby developed a methaemoglobin level of
20%. Physical examination revealed a bluish-green skin colour and
respiratory distress and Heinz bodies were seen in the peripheral
blood. Other reports have stressed the neonatal morbidity related
to the use of the dye in obstetrics (Vincer et al., 1987).
Blass & Fung (1976) reported the case of a 4-year-old boy who
was given 1 g methylene blue intravenously during surgery. He
developed hypotension, tachycardia, and deep cyanosis and remained
intensely blue for several days.
Methylene blue may impart a blue-green colour to urine and
faeces (Martindale, 1989; Windholz, 1983).
9.1.12.5 Other consequential or supportive theory
See chapter 1.
9.1.13 Model information sheet
9.1.13.1 Uses
Methylene blue is used for the treatment of drug-induced, and
some forms of idiopathic, methaemoglobinaemia. It should be used
with great caution in the correction of nitrite-induced
methaemoglobinaemia arising from urgent treatment of cyanide
poisoning.
It is also used as a dye for the identification of fistulae and
glandular tissues.
9.1.13.2 Dosage and route of administration
A dose of 1-2 mg/kg (0.1-0.2 ml/kg of a 1% solution) should be
administered intravenously over 5-10 min.
9.1.13.3 Precautions and contraindications
Methylene blue may be administered when methaemoglobin levels
resulting from nitrite administration are more than 30-40%. It has
no value in the treatment of cyanide poisoning.
Regeneration of haemoglobin from methaemoglobin will release
cyanide back into the circulation.
9.1.13.4 Adverse effects
Large intravenous doses of methylene blue produce nausea,
abdominal and chest-pain, headache, dizziness, mental confusion, and
profuse sweating. Intravascular haemolysis, which may be
life-threatening, can also occur.
9.1.13.5 Use in pregnancy/lactation
Hyperbilirubinaemia and haemolysis may be seen in the neonate.
9.1.13.6 Storage
Methylene blue should be stored in airtight containers.
9.1.14 References
Berlin G, Brodin B, & Milden JO (1985) Acute dapsone intoxication: a
case treated with continuous infusion of methylene blue, forced
diuresis and plasma exchange. Clin Toxicol, 22: 537-548.
Beutler B & Baluda M (1963) Methemoglobin reduction: studies on the
interaction between cell populations and of the role of methylene
blue. Blood, 22: 323.
Blass N & Fung D (1976) Dyed but not dead - Methylene blue overdose.
Anesthesiology, 45: 458-459.
Bodansky O & Gutman H (1947) Treatment of methemoglobinemia. J
Pharmacol Exp Ther, 89: 45-46.
Burrows GM, Williams A, Davis A, & Way JI (1977) Antagonism of
nitrite toxicity by methylene blue or toluidine chloride. Proc West
Pharmacol Soc, 20: 439-443.
Etteldorf JN (1951) Methylene blue in treatment of methemoglobinemia
in premature infants caused by marking ink: 8 cases reported. J
Pediatr, 38: 24-27.
Gosselin RE, Smith HP, & Hodge HC ed. (1984) Nitrite. In: Clinical
toxicology of commercial products, 5th ed. Baltimore, Maryland,
Williams & Wilkins Co., pp III/314-III/319.
Hall AH, Konig MW, & Rumack BH (1986) Drug and chemical-induced
methemoglobinemia. Clinical features and treatment. Med Toxicol, 1:
253-260.
Harrison MR (1977) Toxic methemoglobinemia. A case of acute
nitrobenzene and aniline poisoning. Anaesthesia, 32: 270-272.
Hrushesky WJ, Olshefski R, Wood O, Meshnick S, & Eaton JW (1985)
Modifying intracellular redox balance: an approach to improving
therapeutic index. Lancet, 1: 565-567.
Martindale (1989) In: Reynolds JEF ed. The extra pharmacopoeia, 29th
ed. London, The Pharmaceutical Press, pp 843-844.
Metz, E, Balcerzak S, & Sacome A (1976) Mechanism of methylene blue
stimulation of the hexose-monophosphate shunt in erythrocytes. J
Clin Invest, 58(4): 797-802.
Naidler JE, Green H, & Rosenbaum A (1945) Intravenous injection of
methylene blue in man with reference to its toxic symptoms and
effect on the electrocardiogram. Am J. Med Sci, 188: 15-21.
Ng LL, Naik RB, & Polak A (1982) Paraquat ingestion with
methemoglobinemia treated with methylene blue. Br J Med, 284:
1445-1446.
Rosen PS, Johnson G, McGehee SG, & Beutler E (1971) Failure of
methylene blue treatment in toxic methemoglobinemia. Ann Intern Med,
75: 83-86.
Smith R & Olson M (1973) Drug-induced methemoglobinemia. Semin
Hematol, 10: 253-260.
Sparhr RC & Salisbury DJ (1980) Intra-amniotic injection of
methylene blue leading to methemoglobinemia in one of twins. Int J
Gynaecol Obstet, 17: 477-478.
Vincer MJ, Allen AC, Evans JR, Nwaesei C, & Stinson DA (1987)
Methylene-blue-induced hemolytic anemia in a neonate. Can Med Assoc
J, 136: 503-4.
Whitman JG, Taylor AR, & White JM (1979) Potential hazard of
methylene blue. Anaesthesia, 34: 181-182.
Windholz M ed. (1983) The Merck index: An encyclopedia of chemicals,
drugs and biologicals, 10th ed. Rahway, New Jersey, Merck and Co.,
Inc. pp 333-334.
9.2 Toluidine Blue
9.2.1 Introduction
Toluidine blue is one of a group of dyes that can be used to
treat methaemoglobinaemia. One such situation is where a
methaemoglobin-producing cyanide antidote has produced dangerously
high methaemoglobin levels. Additionally, toluidine blue has a
number of uses such as in vivo staining, which are not relevant to
the present monograph (Chobanian et al., 1987). It has also been
suggested as an antagonist to heparin (Deichmann & Gerarde, 1969).
9.2.2 Name and chemical formula of antidote
The Chemical Abstracts name of toluidine blue is
phenothiazin-5-ium-3-amino-7-(dimethylamino)-2-methyl chloride. The
Chemical Abstracts registration number is 92-31-9. Synonyms include
tolonium chloride and toluidinblau. The compound is listed in the
Colour Index as Basic Blue 17, chemical constitution number 52040
(Society of Dyers and Colourists, 1979), the supplier of the dye
being given as BASF, Ludwigshafen and Rhein, Germany.
9.2.3 Physico-chemical properties
The only supplier of the pharmaceutical preparation is Dr Franz
Kohler, Chemie GmbH, Neue Bergstrasse 3-7, Postfach 17, D-6146
Alsbach-Hahlein 1, Germany. It is supplied in packs containing 5
or 25 ampoules.
The 10-ml ampoules contain toluidine blue at a concentration of
40 mg/ml, i.e. 0.4 g/ampoule. No further information is available
on the constitution of the material.
The relative molecular mass is 305.85.
Toluidine blue has the following structure:
Molecular formula: C16H18C1N3S.3H2O (Martindale, 1989;
Windholz, 1983)
Relative molecular mass: 373.9
CAS number: 7220-79-3 (trihydrate) and 67-73-4 (anhydrous)
Commercial methylene blue is the double chloride of
tetramethylthionine and zinc and is not suitable for
medicinal use (Windholz, 1983).
9.1.3 Physico-chemical properties
Methylene blue is a dark green, almost odourless, hygroscopic
crystalline powder with a bronze-like lustre. It loses 8-18% of its
weight on drying. One gram dissolves in about 25 ml of water, in
about 65 ml alcohol, and in 450 ml of chloroform. Methylene blue is
insoluble in ether (Martindale, 1989; Windholz, 1983).
A 1% solution in water has a pH of 3 to 4.5. Solutions in
water are intensely blue-coloured and incompatible with caustic
alkalis, oxidising and reducing substances and iodides (Martindale,
1989).
Solutions are sterilised by autoclaving or by filtration and
should be stored in airtight containers (Windholz, 1983).
Data on melting and boiling points and on stability in light
are not available.
Excipients: water for injection.
9.1.4 Synthesis
First prepared by Caro in 1876, methylene blue is produced by
the so-called "thiosulfate process" in which a mixture of
dimethyl- p-phenylenediamine and dimethylaniline is oxidized,
usually with potassium dichromate, in the presence of sodium
thiosulfate and zinc chloride (Windholz, 1983).
9.1.5 Analytical methods
No information on analytical methods for methylene blue is
available.
9.1.6 Shelf-life
No data about specific conditions of temperature and humidity
are available, and the shelf-life is unknown. Methylene blue should
be stored in airtight containers.
9.1.7 General properties
Methylene blue functions as an intermediate in the transfer of
electrons from pyridine nucleotides to a suitable electron acceptor,
thereby stimulating the hexose monophosphate shunt (HMPS) pathway in
a variety of cell systems (Bodansky & Gutman, 1947; Beutler &
Baluda, 1963; Smith & Olson, 1973; Metz et al., 1976).
In the erythrocyte, methylene blue is reduced to leucomethylene
blue primarily by NADPH-dependent diaphorase (dihydrolipoamide
dehydrogenase). This diaphorase is reduced via oxidation of NADPH,
which in turn stimulates the HMPS, and leucomethylene blue transfers
electrons to an acceptor such as methaemoglobin (Bodansky & Gutman,
1947; Metz et al., 1976). This series of reactions can be used
clinically for the reduction of ferric to ferrous haem iron in
patients with acquired methaemoglobinaemia. The effectiveness of
methylene blue in reducing methaemoglobin is well established
(Etteldorf, 1951; Beutler & Baluda, 1963; Smith & Olson, 1973;
Harrison, 1977; Gosselin et al., 1984).
9.1.8 Animal studies
Several animal studies confirm that the administration of
methylene blue protects against death caused by
methaemoglobin-generating agents (Blass & Fung, 1976; Burrows et
al., 1977; Hrushesky et al., 1985). Burrows et al. (1977)
administered sodium nitrite (50 mg/kg) intervenously to four ewes.
This resulted in the formation of 70-80% methaemoglobinaemia (a
lethal level) within 45 min. The mortality associated with this
dose of sodium nitrite was successfully antagonized by the
administration of intravenous methylene blue (2.2 mg/kg) 30 min
later.
Pharmacological studies have been carried out in dogs and rats
(Blass & Fung, 1976). When intravenous boluses as large as 15 mg/kg
were given, the resultant data could be explained on the basis of a
one-compartment model with binding of methylene blue to tissues.
Few data on mutagenicity or teratogenicity testing are
available.
9.1.9 Volunteer studies
No data on volunteer studies are available.
9.1.10 Clinical studies
The clinical symptoms and signs associated with
methaemoglobinaemia depend on the percentage of haemoglobin oxidized
to the methaemoglobin form (Bodansky & Gutman, 1947). They consist
of greyish-blue cyanosis, without signs of cardiac or pulmonary
distress, which becomes apparent at about 15% methaemoglobinaemia.
Hypoxaemia may be accompanied by dyspnoea, dizziness, headache,
weakness, lethargy, and CNS depression, the severity of which will
increase with increasing concentrations of methaemoglobin.
An intense chocolate-brown-coloured blood and a central
cyanosis that does not respond to the administration of 100% oxygen
suggest methaemoglobinaemia (Smith & Olson, 1973; Gosselin et al.,
1984; Hall et al., 1986). Methaemoglobin levels may be directly
measured using a spectophotometric method (Hall et al., 1986).
Many authors agree that in asymptomatic patients, with
methaemoglobin levels of 30% or less, methylene blue administration
is not necessary (Bodansky & Gutman, 1947; Gosselin et al., 1984;
Hall et al., 1986).
9.1.11 Clinical studies - case reports
Ng et al. (1982) described a case of methaemoglobinaemia and
haemolysis resulting from the ingestion of paraquat by a 32-year-old
patient. He was administered methylene blue (1 mg/kg) intravenously,
with dramatic results: cyanosis was reversed and the patient became
alert and cooperative. The methaemoglobin level was 19.7% before
methylene blue was given and 1.6% after.
A single case of dapsone-induced methaemoglobinaemia was
treated by continuous methylene blue infusion (Berlin et al., 1985).
The patient recovered completely.
9.1.12 Summary of evaluation
9.1.12.1 Indications
Methylene blue is the agent of choice for accelerating the
reduction of methaemoglobin, at toxic levels, induced by aniline and
aminophenols, nitrites and nitrates, nitrobenzene, antimalarial
agents (chloroquine, primaquine), dapsone, local anaesthetics
(lidocaine, prilocaine, benzocaine), phenacetin, phenazopyridin, and
sulfamidines. It should be used with special caution in the
correction of excess methaemoglobinaemia induced by the treatment of
cyanide poisoning with nitrites.
9.1.12.2 Advised route and dosage
Methylene blue should be administered as an intravenous dose of
1-2 mg/kg body weight (0.1 to 0.2 ml/kg of a 1% solution), slowly
over 5 to 10 min. An improvement should be noted within 30-60 min
of administration. If cyanosis has not disappeared within 1 h, a
second dose should be given (Bodansky & Gutman, 1947; Etteldorf,
1951; Smith & Olson, 1973; Harrison, 1977; Hall et al., 1986).
9.1.12.3 Precautions and contraindications
Methylene blue should not be given by subcutaneous or
intrathecal injection because it causes necrotic abscess formation
(Whitman et al., 1979; Windholz, 1983; Gosselin et al., 1984). It
should be administered with caution in patients with severe renal
impairment (Windholz, 1983; Martindale, 1989; Gosselin et al.,
1984).
In patients with glucose-6-phosphate dehydrogenase deficiency
(who do not generate NADPH), methylene blue is likely to be
ineffective and has been reported to cause haemolytic anaemia
(Beutler & Baluda, 1963; Rosen et al., 1971; Whitman, et al., 1979;
Martindale, 1989).
Methylene blue should not be used in chlorate-induced
methaemoglobinaemia because it may enhance the toxicity of the
chlorate salts (Metz et al., 1976; Martindale, 1989).
9.1.12.4 Adverse effects
Nausea, abdominal and chest pain, headache, dizziness, mental
confusion and profuse sweating may occur following large intravenous
doses of methylene blue (Naidler et al., 1945; Martindale, 1989;
Windholz, 1983).
Whitman et al. (1979) described a 28-year-old patient who
received methylene blue (5 mg/kg) for the identification of an
insulin-secreting pancreatic adenoma. During surgery, the patient
developed methaemoglobin levels of up to 7.1%.
Sparhr & Salisbury (1980) reported a case where an
intra-amniotic injection of 10 mg of methylene blue was given in a
pregnant woman into the vitelline sac of one of two twins. On the
second day, the new-born baby developed a methaemoglobin level of
20%. Physical examination revealed a bluish-green skin colour and
respiratory distress and Heinz bodies were seen in the peripheral
blood. Other reports have stressed the neonatal morbidity related
to the use of the dye in obstetrics (Vincer et al., 1987).
Blass & Fung (1976) reported the case of a 4-year-old boy who
was given 1 g methylene blue intravenously during surgery. He
developed hypotension, tachycardia, and deep cyanosis and remained
intensely blue for several days.
Methylene blue may impart a blue-green colour to urine and
faeces (Martindale, 1989; Windholz, 1983).
9.1.12.5 Other consequential or supportive theory
See chapter 1.
9.1.13 Model information sheet
9.1.13.1 Uses
Methylene blue is used for the treatment of drug-induced, and
some forms of idiopathic, methaemoglobinaemia. It should be used
with great caution in the correction of nitrite-induced
methaemoglobinaemia arising from urgent treatment of cyanide
poisoning.
It is also used as a dye for the identification of fistulae and
glandular tissues.
9.1.13.2 Dosage and route of administration
A dose of 1-2 mg/kg (0.1-0.2 ml/kg of a 1% solution) should be
administered intravenously over 5-10 min.
9.1.13.3 Precautions and contraindications
Methylene blue may be administered when methaemoglobin levels
resulting from nitrite administration are more than 30-40%. It has
no value in the treatment of cyanide poisoning.
Regeneration of haemoglobin from methaemoglobin will release
cyanide back into the circulation.
9.1.13.4 Adverse effects
Large intravenous doses of methylene blue produce nausea,
abdominal and chest-pain, headache, dizziness, mental confusion, and
profuse sweating. Intravascular haemolysis, which may be
life-threatening, can also occur.
9.1.13.5 Use in pregnancy/lactation
Hyperbilirubinaemia and haemolysis may be seen in the neonate.
9.1.13.6 Storage
Methylene blue should be stored in airtight containers.
9.1.14 References
Berlin G, Brodin B, & Milden JO (1985) Acute dapsone intoxication: a
case treated with continuous infusion of methylene blue, forced
diuresis and plasma exchange. Clin Toxicol, 22: 537-548.
Beutler B & Baluda M (1963) Methemoglobin reduction: studies on the
interaction between cell populations and of the role of methylene
blue. Blood, 22: 323.
Blass N & Fung D (1976) Dyed but not dead - Methylene blue overdose.
Anesthesiology, 45: 458-459.
Bodansky O & Gutman H (1947) Treatment of methemoglobinemia. J
Pharmacol Exp Ther, 89: 45-46.
Burrows GM, Williams A, Davis A, & Way JI (1977) Antagonism of
nitrite toxicity by methylene blue or toluidine chloride. Proc West
Pharmacol Soc, 20: 439-443.
Etteldorf JN (1951) Methylene blue in treatment of methemoglobinemia
in premature infants caused by marking ink: 8 cases reported. J
Pediatr, 38: 24-27.
Gosselin RE, Smith HP, & Hodge HC ed. (1984) Nitrite. In: Clinical
toxicology of commercial products, 5th ed. Baltimore, Maryland,
Williams & Wilkins Co., pp III/314-III/319.
Hall AH, Konig MW, & Rumack BH (1986) Drug and chemical-induced
methemoglobinemia. Clinical features and treatment. Med Toxicol, 1:
253-260.
Harrison MR (1977) Toxic methemoglobinemia. A case of acute
nitrobenzene and aniline poisoning. Anaesthesia, 32: 270-272.
Hrushesky WJ, Olshefski R, Wood O, Meshnick S, & Eaton JW (1985)
Modifying intracellular redox balance: an approach to improving
therapeutic index. Lancet, 1: 565-567.
Martindale (1989) In: Reynolds JEF ed. The extra pharmacopoeia, 29th
ed. London, The Pharmaceutical Press, pp 843-844.
Metz, E, Balcerzak S, & Sacome A (1976) Mechanism of methylene blue
stimulation of the hexose-monophosphate shunt in erythrocytes. J
Clin Invest, 58(4): 797-802.
Naidler JE, Green H, & Rosenbaum A (1945) Intravenous injection of
methylene blue in man with reference to its toxic symptoms and
effect on the electrocardiogram. Am J. Med Sci, 188: 15-21.
Ng LL, Naik RB, & Polak A (1982) Paraquat ingestion with
methemoglobinemia treated with methylene blue. Br J Med, 284:
1445-1446.
Rosen PS, Johnson G, McGehee SG, & Beutler E (1971) Failure of
methylene blue treatment in toxic methemoglobinemia. Ann Intern Med,
75: 83-86.
Smith R & Olson M (1973) Drug-induced methemoglobinemia. Semin
Hematol, 10: 253-260.
Sparhr RC & Salisbury DJ (1980) Intra-amniotic injection of
methylene blue leading to methemoglobinemia in one of twins. Int J
Gynaecol Obstet, 17: 477-478.
Vincer MJ, Allen AC, Evans JR, Nwaesei C, & Stinson DA (1987)
Methylene-blue-induced hemolytic anemia in a neonate. Can Med Assoc
J, 136: 503-4.
Whitman JG, Taylor AR, & White JM (1979) Potential hazard of
methylene blue. Anaesthesia, 34: 181-182.
Windholz M ed. (1983) The Merck index: An encyclopedia of chemicals,
drugs and biologicals, 10th ed. Rahway, New Jersey, Merck and Co.,
Inc. pp 333-334.
9.2 Toluidine Blue
9.2.1 Introduction
Toluidine blue is one of a group of dyes that can be used to
treat methaemoglobinaemia. One such situation is where a
methaemoglobin-producing cyanide antidote has produced dangerously
high methaemoglobin levels. Additionally, toluidine blue has a
number of uses such as in vivo staining, which are not relevant to
the present monograph (Chobanian et al., 1987). It has also been
suggested as an antagonist to heparin (Deichmann & Gerarde, 1969).
9.2.2 Name and chemical formula of antidote
The Chemical Abstracts name of toluidine blue is
phenothiazin-5-ium-3-amino-7-(dimethylamino)-2-methyl chloride. The
Chemical Abstracts registration number is 92-31-9. Synonyms include
tolonium chloride and toluidinblau. The compound is listed in the
Colour Index as Basic Blue 17, chemical constitution number 52040
(Society of Dyers and Colourists, 1979), the supplier of the dye
being given as BASF, Ludwigshafen and Rhein, Germany.
9.2.3 Physico-chemical properties
The only supplier of the pharmaceutical preparation is Dr Franz
Kohler, Chemie GmbH, Neue Bergstrasse 3-7, Postfach 17, D-6146
Alsbach-Hahlein 1, Germany. It is supplied in packs containing 5
or 25 ampoules.
The 10-ml ampoules contain toluidine blue at a concentration of
40 mg/ml, i.e. 0.4 g/ampoule. No further information is available
on the constitution of the material.
The relative molecular mass is 305.85.
Toluidine blue has the following structure:
 9.2.4 Synthesis
No data are available.
9.2.5 Analysis
9.2.5.1 Analysis of methaemoglobin
Methaemoglobin may be estimated by the spectrophotometric
method of Evelyn & Malloy (1938) or using the IL 282 CO-oximeter
(Instrumentation Laboratories in UK, Ltd., Warrington, Cheshire,
United Kingdom.) It is very important to note that neither method
gives meaningful results in the presence of cyanide.
Toluidine blue may interfere with the estimation of
methaemoglobin by some methods (Smith, 1971) (see chapter 10).
9.2.6 Stability
No information on the stability of toluidine blue is available.
9.2.7 General properties
The mode of action of toluidine blue is similar to that of
methylene blue; it catalyses the transfer of electrons from the
pentose-phosphate pathway to methaemoglobin via NADPH-methaemoglobin
reductase (Kiese & Waller, 1951; Sass et al., 1969; Kiese et al.,
1972). It is likely that in individuals with glucose-6-
dehydrogenase deficiency or NADPH-methaemoglobin-reductase
deficiency the dyes would not be efficacious in reducing
methaemoglobin (Brewer & Tarlov, 1961; Sass et al., 1967).
Furthermore, certainly in vitro, and possibly in vivo, toluidine
blue will produce methaemoglobin from previously non-oxidized blood
(Kiese, 1945).
9.2.8 Animal studies
9.2.8.1 Pharmacokinetics
No data are available.
9.2.8.2 Pharmacodynamics
A number of studies have shown toluidine blue to be superior to
methylene blue in reducing methaemoglobin (Kiese, 1945; Friehoff &
Lobermann, 1952, 1953; Kiese et al., 1972). Kiese et al. (1972)
found that methaemoglobin produced by the intravenous injection of
dogs with 4-dimethylaminophenol was reduced 2-3 times more rapidly
by toluidine blue than by methylene blue. Burrows (1979) found that
toluidine blue given intravenously to sheep at a dose of 1.1 mg/kg
reduced methaemoglobin produced by sodium nitrite, although the rate
of reduction could be increased by the use of higher doses.
9.2.8.3 Toxicology
The intravenous LD50s values in mice, rats, and rabbits are,
respectively, 27.56, 28.93, and 13.44 mg/kg. Toxic signs observed
include increased respiration rate, convulsions, partial heart
block, and death due to respiratory and cardiac failure. Weekly
intravenous injections into rabbits at a dose one tenth the LD50
produced no change in haematological parameters, and histological
examination of a number of organs after 90 days revealed no
abnormality (Stolarsky & Haley, 1951).
Some early work on Drosophila melanogaster suggested that
toluidine blue might possess mutagenic properties (Landa et al.,
1965). Toluidine blue was reported by Au & Hsu (1979) to produce
extensive chromosome damage.
9.2.9 Volunteer studies
Kiese et al. (1972) reported that, in human volunteers,
toluidine blue reduced 4-dimethylaminophenol-induced methaemoglobin
about twice as rapidly as did methylene blue. No adverse effects
were reported with toluidine blue.
9.2.10 Clinical studies
No data are available.
9.2.11 Clinical studies - case reports
Few case reports exist in the literature and some of these
relate to the use of toluidine blue as a vital stain. A notable
case report of its use in toxic methaemoglobinaemia is that of
Büttner et al. (1967). Side-effects reported include nausea,
vomiting and leucocytosis (Deichmann & Gerarde, 1969).
9.2.12 Summary of evaluations
Toluidine blue has been recommended in a number of studies as
being preferable to methylene blue on the grounds of superior
efficacy (Daunderer, 1979, 1980; Marrs & Ballantyne, 1987).
Compared to methylene blue, there is little clinical experience and
this is confirmed by the paucity of case reports concerning the use
of toluidine blue. Furthermore, there is a lack of information
concerning the nature of the material and its storage
characteristics.
9.2.13 Model information sheet
9.2.13.1 Indications
Toluidine blue is indicated in methaemoglobinaemia, including
toxic methaemoglobinaemia; mild cases of methaemoglobinaemia,
though, would not normally require specific therapy. Toluidine blue
is not indicated in cyanosis due to any other cause. It would not
be effective in sulfhaemoglobinaemia and, on theoretical grounds,
would not be expected to be effecitve in the treatment of
methaemoglobinaemia in individuals with glucose-6-phosphate
dehydrogenase deficiency.
9.2.13.2 Side effects
Owing to the blue colour of the material, cyanosis may occur,
as may staining of the urine. Vomiting may occur following an
overdose.
9.2.13.3 Advised route and dose
The compound should be given intravenously at a dose of
2-4 mg/kg. This may be repeated after 30 min if the clinical
response has been inadequate. It is important to give the material
intravenously.
9.2.13.4 Use in pregnancy and children
No information is available and caution is advised in
administering toluidine blue to a pregnant woman because of
mutagenicity findings. Nevertheless, in life-threatening
methaemoglobinaemia, it would seem inadvisable to withhold
treatment.
9.2.13.5 Storage
No information is available at present.
9.2.14 References
Au W & Hsu TC (1979) Studies on the clastogenic effects of biologic
stains and dyes. Environ Mutagen, 1: 27-35.
Brewer GJ & Tarlov AR (1961) Studies on the mechanism of
primaquine-type hemolysis: the effect of methylene blue. Clin Res,
9: 65.
Burrows GE (1979) Methylene blue or tolonium chloride antagonism of
sodium nitrite induced methemoglobinemia. J Vet Clin Med, 2: 81-86.
Büttner H, Fortwich F, & Hansen HW (1967) [Treatment of a severe
case of nitrogensol poisoning with toluidine blue.] Verh Dtsch Ges
Inn Med, 72: 665-666 (in German).
Chobanian SJ, Cattau EL, Winters C, Johnson DA, Van Ness MM,
Miramadi A, Morwitz SL, & Colcher H (1987) In vivo staining with
toluidine blue as an adjunct to the endoscopic detection of
Barrett's esophagus. Gastrointest Endosc, 33: 99-101.
Daunderer M (1979) [Clinical experience with the antidote 4-DMAP, a
methaemoglobin component for the treatment of poisonings with
prussic acid and its salts, sulfuric acid, hydrazoic acid and its
salts.] University of Munich, Federal Republic of Germany
(Dissertation for appointment as Reader) (in German).
Daunderer M (1980) [Antidote therapy: toluidine blue in
methaemoglobinaemia.] Fortschr Med, 98: 462-464 (in German).
Deichmann WB & Gerarde HW (1969) Toxicology of drugs and chemicals.
New York, London, Academic Press, p 597.
Evelyn RA & Malloy HT (1938) Microdetermination of oxyhemoglobin,
methemoglobin and sulfhemoglobin in a single sample of blood. J.
Biol Chem, 126: 655-662.
Friehoff FJ & Lobermann KH (1952) [Toluidine blue in the treatment
of toxic haemoglobinaemias.] Ther Ggw, 91: 446 (in German).
Friehoff FJ & Lobermann KH (1953) [Dye therapy of toxic
haemoglobinaemias, bearing in mind the pathophysiology of such
poisonings.] Arzneimittelforschung, 3: 616 (in German).
Kiese M (1945) [Reduction of haemoglobin. IV.] Naunyn-Schmiedebergs
Arch Exp Pathol Pharmakol, 204: 288-312 (in German).
Kiese M & Waller HD (1951) [Reduction of haemoglobin and oxygen for
reversibly reducible dyes in red cells.] Naunyn-Schmiedebergs Arch
Pharmakol, 213: 44 (in German).
Kiese M, Lorcher W, Weger N, & Zierer A (1972) Comparative studies
on the effects of toluidine blue and methylene blue on the reduction
of ferrihemoglobin in man and dog. Eur J Clin Pharmacol, 4: 115-118.
Landa Z, Klouda P, & Pleskotova D (1965) In: Veleminsky J, Gichner
T, & Ridesova A ed. Induction of mutations and the mutation process.
Proceeding of a Symposium, Prague, 26-28 September, 1963. Prague,
Czechoslovak Academy of Sciences Publishing House, pp 115-122.
Marrs TC & Ballantyne B (1987) Clinical and experimental toxicology
of cyanides: an overview. In: Ballantyne B & Marrs TC ed. Clinical
and experimental toxicology of cyanides. Bristol, United Kingdom,
John Wright, pp 473-495.
Marrs TC, Bright JE, & Inns RH (1989) Methaemoglobin production and
reduction by methylene blue and the interaction of methylene blue
with sodium nitrite in vivo. Human Toxicol, 8: 359-364.
Sass MD, Caruso CJ, & Farhangi M (1967) TPNH-methemoglobin reductase
deficiency: a new red-cell enzyme defect. J Lab Clin Med, 70:
760-767.
Sass MD, Caruso CJ, & Axelrod DR (1969) Mechanism of the TPNH-linked
reduction of methemoglobin by methylene blue. Clin Chem Acta, 24:
77.
Smith RP (1971) Spectrophotometric determination of methemoglobin
and oxyhemoglobin in the presence of methylene blue. Clin Toxicol,
4: 273.
Society of Dyers and Colourists (1979) The colour index. Bradford,
United Kingdom, The Society of Dyers and Colourists.
Stolarsky F & Haley TJ (1951) Acute and chronic toxicity of
toluidine blue and neutral red. Fed Proc, 10: 337.
10. Analytical Methods for Cyanide Alone and in Combination with
Cyanide Antidotes in Blood
It is important to be sure of the diagnosis in every case of
poisoning, and this is particularly true of cyanide intoxication
where treatment with antidotes is often potentially hazardous. Fast
qualitative and quantitative methods for analysing cyanide in blood
are therefore necessary. The methods detailed below are approved
techniques for the detection and/or quantification of cyanide before
the administration of cyanide antidotes.
Measurement of whole blood free cyanide concentrations after
the administration of cyanide antidotes is not yet possible.
10.1 Qualitative Methods
10.1.1 Detection in blood with a detector tube (Bedside Test)
The time required, assuming that instrumentation and chemicals
are readily available, is 2-3 min.
10.1.1.1 Principle
Hydrocyanic acid is liberated from blood by acidification and
is then passed through a detector tube using a gas detector pump.
If hydrocyanic acid is present, the reactive zone of the tube
changes its colour from yellow to red as a result of the following
reaction:
HgCl2
(1) HCN --> HC1
(2) HCl + Methyl red --> red reaction product
A semiquantitative determination of the HCN concentration can
be made by means of a scale on the tube (M. von Clarmann, personal
communication, 1988).
10.1.1.2 Materials
Gas detector pump (Dräger a) (Fig. 5)
Detector tube "Hydrocyanic acid 2/a" (Dräger a) (Fig. 6)
Test tube height 6 cm, width 2 cm (Fig. 6)
Stopper with two drill-holes (Fig. 5)
10% sulfuric acid
specimen of venous blood, with or without an anticoagulant
a Drägerwerk AG, 2400 Lübeck Germany
9.2.4 Synthesis
No data are available.
9.2.5 Analysis
9.2.5.1 Analysis of methaemoglobin
Methaemoglobin may be estimated by the spectrophotometric
method of Evelyn & Malloy (1938) or using the IL 282 CO-oximeter
(Instrumentation Laboratories in UK, Ltd., Warrington, Cheshire,
United Kingdom.) It is very important to note that neither method
gives meaningful results in the presence of cyanide.
Toluidine blue may interfere with the estimation of
methaemoglobin by some methods (Smith, 1971) (see chapter 10).
9.2.6 Stability
No information on the stability of toluidine blue is available.
9.2.7 General properties
The mode of action of toluidine blue is similar to that of
methylene blue; it catalyses the transfer of electrons from the
pentose-phosphate pathway to methaemoglobin via NADPH-methaemoglobin
reductase (Kiese & Waller, 1951; Sass et al., 1969; Kiese et al.,
1972). It is likely that in individuals with glucose-6-
dehydrogenase deficiency or NADPH-methaemoglobin-reductase
deficiency the dyes would not be efficacious in reducing
methaemoglobin (Brewer & Tarlov, 1961; Sass et al., 1967).
Furthermore, certainly in vitro, and possibly in vivo, toluidine
blue will produce methaemoglobin from previously non-oxidized blood
(Kiese, 1945).
9.2.8 Animal studies
9.2.8.1 Pharmacokinetics
No data are available.
9.2.8.2 Pharmacodynamics
A number of studies have shown toluidine blue to be superior to
methylene blue in reducing methaemoglobin (Kiese, 1945; Friehoff &
Lobermann, 1952, 1953; Kiese et al., 1972). Kiese et al. (1972)
found that methaemoglobin produced by the intravenous injection of
dogs with 4-dimethylaminophenol was reduced 2-3 times more rapidly
by toluidine blue than by methylene blue. Burrows (1979) found that
toluidine blue given intravenously to sheep at a dose of 1.1 mg/kg
reduced methaemoglobin produced by sodium nitrite, although the rate
of reduction could be increased by the use of higher doses.
9.2.8.3 Toxicology
The intravenous LD50s values in mice, rats, and rabbits are,
respectively, 27.56, 28.93, and 13.44 mg/kg. Toxic signs observed
include increased respiration rate, convulsions, partial heart
block, and death due to respiratory and cardiac failure. Weekly
intravenous injections into rabbits at a dose one tenth the LD50
produced no change in haematological parameters, and histological
examination of a number of organs after 90 days revealed no
abnormality (Stolarsky & Haley, 1951).
Some early work on Drosophila melanogaster suggested that
toluidine blue might possess mutagenic properties (Landa et al.,
1965). Toluidine blue was reported by Au & Hsu (1979) to produce
extensive chromosome damage.
9.2.9 Volunteer studies
Kiese et al. (1972) reported that, in human volunteers,
toluidine blue reduced 4-dimethylaminophenol-induced methaemoglobin
about twice as rapidly as did methylene blue. No adverse effects
were reported with toluidine blue.
9.2.10 Clinical studies
No data are available.
9.2.11 Clinical studies - case reports
Few case reports exist in the literature and some of these
relate to the use of toluidine blue as a vital stain. A notable
case report of its use in toxic methaemoglobinaemia is that of
Büttner et al. (1967). Side-effects reported include nausea,
vomiting and leucocytosis (Deichmann & Gerarde, 1969).
9.2.12 Summary of evaluations
Toluidine blue has been recommended in a number of studies as
being preferable to methylene blue on the grounds of superior
efficacy (Daunderer, 1979, 1980; Marrs & Ballantyne, 1987).
Compared to methylene blue, there is little clinical experience and
this is confirmed by the paucity of case reports concerning the use
of toluidine blue. Furthermore, there is a lack of information
concerning the nature of the material and its storage
characteristics.
9.2.13 Model information sheet
9.2.13.1 Indications
Toluidine blue is indicated in methaemoglobinaemia, including
toxic methaemoglobinaemia; mild cases of methaemoglobinaemia,
though, would not normally require specific therapy. Toluidine blue
is not indicated in cyanosis due to any other cause. It would not
be effective in sulfhaemoglobinaemia and, on theoretical grounds,
would not be expected to be effecitve in the treatment of
methaemoglobinaemia in individuals with glucose-6-phosphate
dehydrogenase deficiency.
9.2.13.2 Side effects
Owing to the blue colour of the material, cyanosis may occur,
as may staining of the urine. Vomiting may occur following an
overdose.
9.2.13.3 Advised route and dose
The compound should be given intravenously at a dose of
2-4 mg/kg. This may be repeated after 30 min if the clinical
response has been inadequate. It is important to give the material
intravenously.
9.2.13.4 Use in pregnancy and children
No information is available and caution is advised in
administering toluidine blue to a pregnant woman because of
mutagenicity findings. Nevertheless, in life-threatening
methaemoglobinaemia, it would seem inadvisable to withhold
treatment.
9.2.13.5 Storage
No information is available at present.
9.2.14 References
Au W & Hsu TC (1979) Studies on the clastogenic effects of biologic
stains and dyes. Environ Mutagen, 1: 27-35.
Brewer GJ & Tarlov AR (1961) Studies on the mechanism of
primaquine-type hemolysis: the effect of methylene blue. Clin Res,
9: 65.
Burrows GE (1979) Methylene blue or tolonium chloride antagonism of
sodium nitrite induced methemoglobinemia. J Vet Clin Med, 2: 81-86.
Büttner H, Fortwich F, & Hansen HW (1967) [Treatment of a severe
case of nitrogensol poisoning with toluidine blue.] Verh Dtsch Ges
Inn Med, 72: 665-666 (in German).
Chobanian SJ, Cattau EL, Winters C, Johnson DA, Van Ness MM,
Miramadi A, Morwitz SL, & Colcher H (1987) In vivo staining with
toluidine blue as an adjunct to the endoscopic detection of
Barrett's esophagus. Gastrointest Endosc, 33: 99-101.
Daunderer M (1979) [Clinical experience with the antidote 4-DMAP, a
methaemoglobin component for the treatment of poisonings with
prussic acid and its salts, sulfuric acid, hydrazoic acid and its
salts.] University of Munich, Federal Republic of Germany
(Dissertation for appointment as Reader) (in German).
Daunderer M (1980) [Antidote therapy: toluidine blue in
methaemoglobinaemia.] Fortschr Med, 98: 462-464 (in German).
Deichmann WB & Gerarde HW (1969) Toxicology of drugs and chemicals.
New York, London, Academic Press, p 597.
Evelyn RA & Malloy HT (1938) Microdetermination of oxyhemoglobin,
methemoglobin and sulfhemoglobin in a single sample of blood. J.
Biol Chem, 126: 655-662.
Friehoff FJ & Lobermann KH (1952) [Toluidine blue in the treatment
of toxic haemoglobinaemias.] Ther Ggw, 91: 446 (in German).
Friehoff FJ & Lobermann KH (1953) [Dye therapy of toxic
haemoglobinaemias, bearing in mind the pathophysiology of such
poisonings.] Arzneimittelforschung, 3: 616 (in German).
Kiese M (1945) [Reduction of haemoglobin. IV.] Naunyn-Schmiedebergs
Arch Exp Pathol Pharmakol, 204: 288-312 (in German).
Kiese M & Waller HD (1951) [Reduction of haemoglobin and oxygen for
reversibly reducible dyes in red cells.] Naunyn-Schmiedebergs Arch
Pharmakol, 213: 44 (in German).
Kiese M, Lorcher W, Weger N, & Zierer A (1972) Comparative studies
on the effects of toluidine blue and methylene blue on the reduction
of ferrihemoglobin in man and dog. Eur J Clin Pharmacol, 4: 115-118.
Landa Z, Klouda P, & Pleskotova D (1965) In: Veleminsky J, Gichner
T, & Ridesova A ed. Induction of mutations and the mutation process.
Proceeding of a Symposium, Prague, 26-28 September, 1963. Prague,
Czechoslovak Academy of Sciences Publishing House, pp 115-122.
Marrs TC & Ballantyne B (1987) Clinical and experimental toxicology
of cyanides: an overview. In: Ballantyne B & Marrs TC ed. Clinical
and experimental toxicology of cyanides. Bristol, United Kingdom,
John Wright, pp 473-495.
Marrs TC, Bright JE, & Inns RH (1989) Methaemoglobin production and
reduction by methylene blue and the interaction of methylene blue
with sodium nitrite in vivo. Human Toxicol, 8: 359-364.
Sass MD, Caruso CJ, & Farhangi M (1967) TPNH-methemoglobin reductase
deficiency: a new red-cell enzyme defect. J Lab Clin Med, 70:
760-767.
Sass MD, Caruso CJ, & Axelrod DR (1969) Mechanism of the TPNH-linked
reduction of methemoglobin by methylene blue. Clin Chem Acta, 24:
77.
Smith RP (1971) Spectrophotometric determination of methemoglobin
and oxyhemoglobin in the presence of methylene blue. Clin Toxicol,
4: 273.
Society of Dyers and Colourists (1979) The colour index. Bradford,
United Kingdom, The Society of Dyers and Colourists.
Stolarsky F & Haley TJ (1951) Acute and chronic toxicity of
toluidine blue and neutral red. Fed Proc, 10: 337.
10. Analytical Methods for Cyanide Alone and in Combination with
Cyanide Antidotes in Blood
It is important to be sure of the diagnosis in every case of
poisoning, and this is particularly true of cyanide intoxication
where treatment with antidotes is often potentially hazardous. Fast
qualitative and quantitative methods for analysing cyanide in blood
are therefore necessary. The methods detailed below are approved
techniques for the detection and/or quantification of cyanide before
the administration of cyanide antidotes.
Measurement of whole blood free cyanide concentrations after
the administration of cyanide antidotes is not yet possible.
10.1 Qualitative Methods
10.1.1 Detection in blood with a detector tube (Bedside Test)
The time required, assuming that instrumentation and chemicals
are readily available, is 2-3 min.
10.1.1.1 Principle
Hydrocyanic acid is liberated from blood by acidification and
is then passed through a detector tube using a gas detector pump.
If hydrocyanic acid is present, the reactive zone of the tube
changes its colour from yellow to red as a result of the following
reaction:
HgCl2
(1) HCN --> HC1
(2) HCl + Methyl red --> red reaction product
A semiquantitative determination of the HCN concentration can
be made by means of a scale on the tube (M. von Clarmann, personal
communication, 1988).
10.1.1.2 Materials
Gas detector pump (Dräger a) (Fig. 5)
Detector tube "Hydrocyanic acid 2/a" (Dräger a) (Fig. 6)
Test tube height 6 cm, width 2 cm (Fig. 6)
Stopper with two drill-holes (Fig. 5)
10% sulfuric acid
specimen of venous blood, with or without an anticoagulant
a Drägerwerk AG, 2400 Lübeck Germany
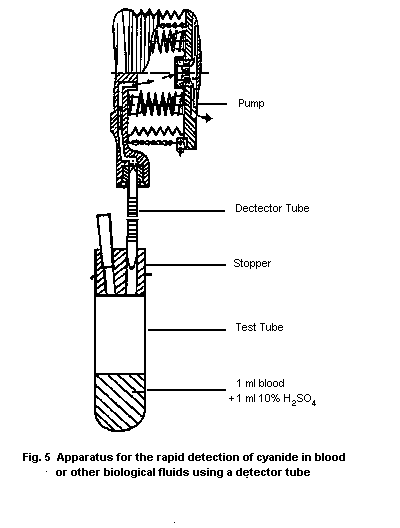
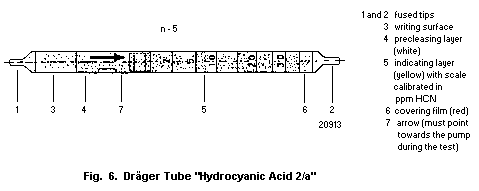 10.1.1.3 Procedure
Read carefully the operating instructions for the gas detector
pump and the detector tube.
Insert the detector tube into the free drill-hole of the
stopper and put the gas detector pump on the detector tube.
Place 1 ml blood and 1 ml 10% sulfuric acid into the test tube
and close it immediately with the stopper prepared as described
above. Shake the test tube cautiously, avoid splashing by all
means.
Compress the pump balk while closing the free end of the glass
tube with a finger. Then let the glass tube open while the balk of
the pump extends. Repeat the pumping 15 times.
Attention!
Take care not to suck some of the acid contents of the test
tube into the detector tube. If this happens, a false positive
result will be the consequence.
Judge the colour of the indicating layer of the detector tube
immediately after the pumping. If hydrocyanic acid is present, the
indicating layer turns becomes red. A very rough semiquantitative
determination of the HCN concentration can be made by means of the
scale on the tube. The detection limit in this test is 1 mg
CN-/l.
10.1.1.4 Specificity
The analysis is based on the reaction of hydrocyanic acid with
mercury salts: the hydrochloric acid thereby produced is
determined by methyl red. Specificity is ensured by the
precleansing layer. Acidic gases (HCl or SO2) have no influence
on the HCN reading, even if present in considerable excess. This
also applies to hydrogen sulfide.
Sodium azide gives a positive reaction.
10.1.2 Spot test
The time required, assuming that instrumentation and reagents
are readily available, is 2-3 min.
10.1.2.1 Principle
Hydrocyanic acid (HCN) is liberated from biological fluids by
acidification. The evolved HCN is passed through a filter paper
impregnated with an alkaline solution of palladium dimethylglyoxime
in which the palladium is a constituent of an inner-sphere complex
anion. Cyanide ions lead to a demasking of dimethylglyoxime, which
reacts with nickel (II) to produce red nickel-dimethylglyoxime
(Jakobs, 1984).
10.1.2.2 Equipment
Gas washing flask shown in Fig. 7 (measurements of the
flask: height 15 cm; width 3 cm).
Water bath
Centrifuge
Disposable syringes containing anticoagulant (e.g., K-EDTA
Monovetten R, Sarstedt Numbrecht, Germany)
Pipettes
10.1.2.3 Chemicals
Ethanol
Palladium(II) chloride
Potassium hydroxide
Ammonium chloride
Nickel(II) chloride. 6H2O
Concentrated sulfuric acid (96-98%)
Dimethylglyoxime (2,3-butanedione dioxime)
Antifoaming agent (e.g., silicon antifoaming emulsion
10.1.1.3 Procedure
Read carefully the operating instructions for the gas detector
pump and the detector tube.
Insert the detector tube into the free drill-hole of the
stopper and put the gas detector pump on the detector tube.
Place 1 ml blood and 1 ml 10% sulfuric acid into the test tube
and close it immediately with the stopper prepared as described
above. Shake the test tube cautiously, avoid splashing by all
means.
Compress the pump balk while closing the free end of the glass
tube with a finger. Then let the glass tube open while the balk of
the pump extends. Repeat the pumping 15 times.
Attention!
Take care not to suck some of the acid contents of the test
tube into the detector tube. If this happens, a false positive
result will be the consequence.
Judge the colour of the indicating layer of the detector tube
immediately after the pumping. If hydrocyanic acid is present, the
indicating layer turns becomes red. A very rough semiquantitative
determination of the HCN concentration can be made by means of the
scale on the tube. The detection limit in this test is 1 mg
CN-/l.
10.1.1.4 Specificity
The analysis is based on the reaction of hydrocyanic acid with
mercury salts: the hydrochloric acid thereby produced is
determined by methyl red. Specificity is ensured by the
precleansing layer. Acidic gases (HCl or SO2) have no influence
on the HCN reading, even if present in considerable excess. This
also applies to hydrogen sulfide.
Sodium azide gives a positive reaction.
10.1.2 Spot test
The time required, assuming that instrumentation and reagents
are readily available, is 2-3 min.
10.1.2.1 Principle
Hydrocyanic acid (HCN) is liberated from biological fluids by
acidification. The evolved HCN is passed through a filter paper
impregnated with an alkaline solution of palladium dimethylglyoxime
in which the palladium is a constituent of an inner-sphere complex
anion. Cyanide ions lead to a demasking of dimethylglyoxime, which
reacts with nickel (II) to produce red nickel-dimethylglyoxime
(Jakobs, 1984).
10.1.2.2 Equipment
Gas washing flask shown in Fig. 7 (measurements of the
flask: height 15 cm; width 3 cm).
Water bath
Centrifuge
Disposable syringes containing anticoagulant (e.g., K-EDTA
Monovetten R, Sarstedt Numbrecht, Germany)
Pipettes
10.1.2.3 Chemicals
Ethanol
Palladium(II) chloride
Potassium hydroxide
Ammonium chloride
Nickel(II) chloride. 6H2O
Concentrated sulfuric acid (96-98%)
Dimethylglyoxime (2,3-butanedione dioxime)
Antifoaming agent (e.g., silicon antifoaming emulsion
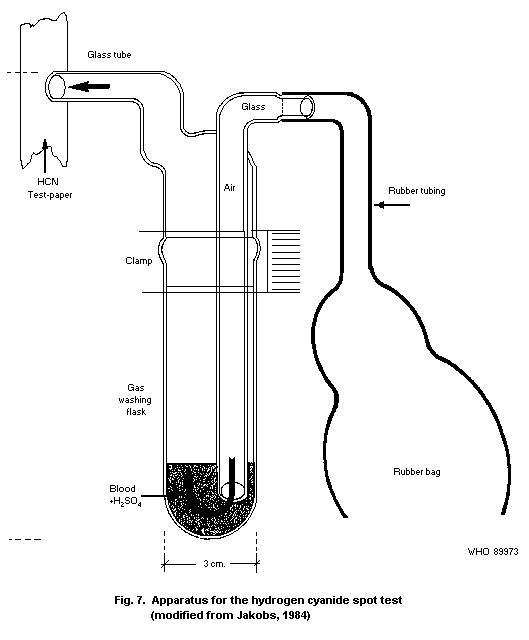 10.1.2.4. Reagents
50% Sulfuric acid
Hydrochloric acid (0.1 mol/l)
Potassium hydroxide (3 mol/l)
Alkali palladium dimethylglyoxime solution:
(a) Dissolve 50 mg palladium(II)chloride in 2.5 ml
hydrochloric acid (0.1 mol/l) by heating it in a water
bath for a little while.
(b) Dissolve 100 mg dimethylglyoxime in 10 ml ethanol.
(c) Add the palladium(II)chloride solution drop by drop to the
ethanolic solution of dimethylglyoxime.
(d) Centrifuge at 4000 g.
(e) Wash the residue with 10 ml distilled water and
centrifuge. Repeat this procedure three times.
(f) Dissolve the residue in 10 ml KOH (3mol/l) by heating it
in water bath for a little while.
(g) Allow to cool, centrifuge at 4000 g if necessary.
Nickel chloride solution (0.25 mol/l) saturated with ammonium
chloride.a
Dissolve 4 g ammonium chloride and 0.6 g NiCl2.6H2O in 10 ml
water.
All reagents are stable at 4 °C for at least one year.
10.1.2.5 Specimen collection
Blood is collected with disposable syringes which contain EDTA
as anticoagulant. If the syringes are not disposable, EDTA disodium
salt is added to a final concentration of 1 mg/ml blood and mixed
well. Other anticoagulants can be used as well.
10.1.2.6 Procedure
Filter paper is impregnated with the alkaline solution of
palladium dimethylglyoxime or a drop of this solution is placed on
filter paper.
a The presence of ammonium salts prevents the precipitation of
green Ni(OH)2
Into a gas washing flask (Fig. 7), 5 ml blood, 3-5 drops of the
antifoaming agent, and 5 drops of 50% sulfuric acid are placed.
The moist reagent paper is held on to the free opening of the
glass tube. Air is then blown through the flask, using the rubber
bag for 2 min. The part of the paper that was exposed to the stream
of gas is then spotted with the nickel ammonium chloride solution.
If HCN is present, a pink to red stain of nickel dimethylglyoxime
appears at once, the depth of the colour varying with the cyanide
content of the gas. The detection limit in this test is 1 mg
CN-/l.
10.1.2.7 Specificity
Bright yellow inner-sphere complex palladium dimethylglyoxime
(I) readily dissolves in caustic alkali to give a yellow solution of
alkaline palladium dimethylglyoxime (II) in which the palladium is a
constituent of an inner-sphere complex anion. The inner-sphere
complex bound dimethylglyoxime is masked in solutions of II, i.e. no
red Ni-dimethylglyoxime precipitate appears when Ni2+ ions are
added to the solution. Likewise, the palladium is masked against
practically all reagents that are normally characteristic for Pd2+
ions. Cyanide ions present an exception. If added in excess to the
yellow solutions of II, they cause immediate discharge of the
colour, and addition of nickel salt solutions (containing NH4Cl)
brings down red Ni-dimethylglyoxime.
Since the Ni-dimethylglyoxime reaction is highly sensitive,
cyanide ions can be detected through their demasking effect. (Feigl
& Feigl, 1949; Feigl et al., 1966). The reaction is specific to
HCN.
10.2 Quantitative Methods
10.2.1 Gas chromatographic head space technique
This procedure, described by Eben & Lewalter (1988), has been
slightly modified from that developed by McAuley & Reive (1983).
The gas chromatographic determination of hydrogen cyanide may be
carried out simply and rapidly with the head-space technique
described here. The use of a thermionic detector that is specific
for nitrogen makes it possible to obtain a sensitivity 10 times
greater than with a flame ionization detector.
The method described is highly accurate because practically no
sample treatment is required. This is confirmed by the precision
data presented here. Another advantage over alternative analytical
techniques is that gas chromatographic head space analysis is an
established part of the repertoire of most medical toxicological
laboratories.
10.2.1.1 Principle
Hydrogen cyanide gas is liberated by adding acid to
cyanide-containing blood and is then determined by means of gas
chromatography using head-space analysis. Fractionation may be
carried out on a packed capillary column. The cyanide is determined
using a thermionic nitrogen detector.
Calibration standards, which are mixed with whole blood, are
used for quantification. To evaluate the data, a calibration curve
is used in which peak height or peak area is plotted as a function
of the cyanide concentration in blood.
10.2.1.2 Equipment
* Gas chromatograph with thermionic nitrogen detector, and
chart recorder or integrator, if necessary with a
capillary injector
* Packed glass column: length, 2 m; inner diameter, 2.2 mm
* Column packing: Porapak Q 100-120 mesh
* Alternatively, a Duran (borosilicate) glass or quartz
capillary (length, 60 m; inner diameter, 0.3 mm) may be
used
* Stationary phase: SE 30 (Chrompack)a, chemically bonded,
film thickness 0.3 µm
* Gas-tight syringes (50 or 500 µl) for gas chromatography
* Bottles (5 ml) with serum caps fitted with PTFE-coated
septa, together with tools for sealing and opening
* Automatic pipettes, variable between 20 and 200 µl
* Syringe (100 µl) for gas chromatography
* Vortex mixer
* Centrifuge
* Transfer pipettes (1, 2, 4, and 10 ml)
* Volumetric flasks (10, 20, 50, and 100 ml)
* Disposable syringes containing anticoagulant (e.g., K-EDTA
MonovettenR, Sarstedt, Nümbrecht, Germany)
a Chompack, P.O. Box 8033, 4330
10.2.1.3 Chemicals
* Potassium cyanide, pa
* Sodium cyanide, pa
* Glacial acetic acid, pa
* Helium (99.999% purity) as carrier gas
* Hydrogen (99.90% purity)
* Synthetic air (80% purified nitrogen, 20% oxygen)
10.2.1.4 Solutions
* Physiological saline (9 g sodium chloride/l)
* Pooled stabilized whole blood or stabilized bovine blood
for the calibration standards
10.2.1.5 Calibration standards
(a) Starting solution
12.6 mg potassium cyanide (equivalent to 5.3 mg cyanide)
is weighed out exactly and transferred to a 50 ml
volumetric flask, which is then filled to the mark with
whole blood (106 mg cyanide/l blood).
(b) Stock solution
Starting solution (2 ml) is pipetted into a 20 ml
volumetric flask, which is then filled to the mark with
whole blood (10.6 mg cyanide/l blood). From this
solution, calibration standards within the concentration
range 0.2 - 4.2 mg cyanide/l blood are prepared by
dilution with whole blood as shown in the following table:
Volume of Final volume of Cyanide concentration
stock solution calibration standard of calibration standard
ml ml mg/l
4 10 4.24
2 10 2.12
1 10 1.06
1 20 0.53
1 50 0.212
The blood volume required for preparing the calibration
standards may be reduced to about 10 ml if the standards are
prepared directly in the bottles used for sample treatment. In this
case the procedure is as follows:
Stock solution A
Exactly 24.0 mg potassium cyanide (equivalent to 10 mg cyanide)
is transferred to a 100 ml volumetric flask, which is then filled to
the mark with physiological saline (100 mg cyanide/l).
Stock solution B
10 ml stock solution A is pipetted into a 100 ml volumetric
flask, which is then filled to the mark with physiological saline
(10 mg cyanide/l).
The calibration standards are prepared directly in the bottles
used for the sample treatment. Blood (1 ml) is pipetted into each
bottle and after adding the cyanide solution, the bottle is sealed
immediately. The difference in the volumes of the individual
standards does not reduce the reliability of the calibration for the
head-space analysis used here.
Volume of stock Volume of stock Cyanide concentration
solution A solution B of calibration standard
µl µl mg/l
- 20 0.2
- 40 0.4
- 50 0.5
10 - 1.0
20 - 2.0
30 - 3.0
50 - 5.0
10.2.1.6 Specimen collection and sample preparation
Blood is collected in disposable syringes containing an
anticoagulant. Immediately after collection, 1 ml of whole blood is
pipetted into a 5-ml bottle, which is sealed at once with a cap and
PTFE-coated septum.
Using a 100-µl syringe, 50 µl glacial acetic acid is injected
through the septum into the bottle to release the hydrogen cyanide.
The contents of the bottle are shaken for 30 seconds on a vortex
mixer and then centrifuged for 3 min at 3000 rpm. The bottle is
then incubated for 30 min at room temperature.
The blood samples containing cyanide should be processed within
30 min of collection, as losses can otherwise occur.
10.2.1.7 Operational parameters for gas chromatography
(a) Packed column
Column: Material: Glass
Length: 2 m
Inner diameter: 2.2 mm
Stationary phase: Porapak Q 100-120 mesh
Detector: Thermoionic nitrogen detector
Temperatures: Column: 120 °C
Injector: 220 °C
Detector: 300 °C
Carrier gas: Helium: 30 ml/min
Detector gases: Hydrogen: 3 ml/min
Synthetic air: 60 ml/min
Injected head-space 50 µl
Under these conditions, hydrogen cyanide has a retention
time of 1.5 min.
(b) Capillary column
Capillary column: Material: Quartz or Duran glass
Length: 50 m
Inner diameter: 0.3 mm
Stationary phase: SE 30 (chrompack) chemically
bonded, film thickness 0.3 mm
Detector: Thermionic specific detector (TSD)
Temperatures: Column: 80 °C
Injector: 200 °C
Detector: 300 °C
Split: 1:4
Carrier gas: Helium, 1.68 MPa; make up gas,
35 ml/min
Detector gases: Hydrogen: 1.45 MPa
(3.5-4.0 ml/min)
Synthetic air: 160 ml/min
Injected head-space 400 µl
Under these conditions, the retention time for hydrogen
cyanide is 3.1 min.
10.2.1.8 Analytical determination
50 µl (packed column) or 400 µl (capillary column) of the head
space above the prepared blood sample is injected into the gas
chromatograph under the conditions given in section 10.2.1.7.
10.2.1.9 Calibration
The calibration standards containing cyanide in blood are
prepared as described in section 10.2.1.5 and analysed. The
calibration curve is obtained by plotting the peak heights or areas
for the standards against the blood cyanide concentrations used (see
Figure 8). The linearity of the calibration curve has been tested up
to 6 mg/l.
10.1.2.4. Reagents
50% Sulfuric acid
Hydrochloric acid (0.1 mol/l)
Potassium hydroxide (3 mol/l)
Alkali palladium dimethylglyoxime solution:
(a) Dissolve 50 mg palladium(II)chloride in 2.5 ml
hydrochloric acid (0.1 mol/l) by heating it in a water
bath for a little while.
(b) Dissolve 100 mg dimethylglyoxime in 10 ml ethanol.
(c) Add the palladium(II)chloride solution drop by drop to the
ethanolic solution of dimethylglyoxime.
(d) Centrifuge at 4000 g.
(e) Wash the residue with 10 ml distilled water and
centrifuge. Repeat this procedure three times.
(f) Dissolve the residue in 10 ml KOH (3mol/l) by heating it
in water bath for a little while.
(g) Allow to cool, centrifuge at 4000 g if necessary.
Nickel chloride solution (0.25 mol/l) saturated with ammonium
chloride.a
Dissolve 4 g ammonium chloride and 0.6 g NiCl2.6H2O in 10 ml
water.
All reagents are stable at 4 °C for at least one year.
10.1.2.5 Specimen collection
Blood is collected with disposable syringes which contain EDTA
as anticoagulant. If the syringes are not disposable, EDTA disodium
salt is added to a final concentration of 1 mg/ml blood and mixed
well. Other anticoagulants can be used as well.
10.1.2.6 Procedure
Filter paper is impregnated with the alkaline solution of
palladium dimethylglyoxime or a drop of this solution is placed on
filter paper.
a The presence of ammonium salts prevents the precipitation of
green Ni(OH)2
Into a gas washing flask (Fig. 7), 5 ml blood, 3-5 drops of the
antifoaming agent, and 5 drops of 50% sulfuric acid are placed.
The moist reagent paper is held on to the free opening of the
glass tube. Air is then blown through the flask, using the rubber
bag for 2 min. The part of the paper that was exposed to the stream
of gas is then spotted with the nickel ammonium chloride solution.
If HCN is present, a pink to red stain of nickel dimethylglyoxime
appears at once, the depth of the colour varying with the cyanide
content of the gas. The detection limit in this test is 1 mg
CN-/l.
10.1.2.7 Specificity
Bright yellow inner-sphere complex palladium dimethylglyoxime
(I) readily dissolves in caustic alkali to give a yellow solution of
alkaline palladium dimethylglyoxime (II) in which the palladium is a
constituent of an inner-sphere complex anion. The inner-sphere
complex bound dimethylglyoxime is masked in solutions of II, i.e. no
red Ni-dimethylglyoxime precipitate appears when Ni2+ ions are
added to the solution. Likewise, the palladium is masked against
practically all reagents that are normally characteristic for Pd2+
ions. Cyanide ions present an exception. If added in excess to the
yellow solutions of II, they cause immediate discharge of the
colour, and addition of nickel salt solutions (containing NH4Cl)
brings down red Ni-dimethylglyoxime.
Since the Ni-dimethylglyoxime reaction is highly sensitive,
cyanide ions can be detected through their demasking effect. (Feigl
& Feigl, 1949; Feigl et al., 1966). The reaction is specific to
HCN.
10.2 Quantitative Methods
10.2.1 Gas chromatographic head space technique
This procedure, described by Eben & Lewalter (1988), has been
slightly modified from that developed by McAuley & Reive (1983).
The gas chromatographic determination of hydrogen cyanide may be
carried out simply and rapidly with the head-space technique
described here. The use of a thermionic detector that is specific
for nitrogen makes it possible to obtain a sensitivity 10 times
greater than with a flame ionization detector.
The method described is highly accurate because practically no
sample treatment is required. This is confirmed by the precision
data presented here. Another advantage over alternative analytical
techniques is that gas chromatographic head space analysis is an
established part of the repertoire of most medical toxicological
laboratories.
10.2.1.1 Principle
Hydrogen cyanide gas is liberated by adding acid to
cyanide-containing blood and is then determined by means of gas
chromatography using head-space analysis. Fractionation may be
carried out on a packed capillary column. The cyanide is determined
using a thermionic nitrogen detector.
Calibration standards, which are mixed with whole blood, are
used for quantification. To evaluate the data, a calibration curve
is used in which peak height or peak area is plotted as a function
of the cyanide concentration in blood.
10.2.1.2 Equipment
* Gas chromatograph with thermionic nitrogen detector, and
chart recorder or integrator, if necessary with a
capillary injector
* Packed glass column: length, 2 m; inner diameter, 2.2 mm
* Column packing: Porapak Q 100-120 mesh
* Alternatively, a Duran (borosilicate) glass or quartz
capillary (length, 60 m; inner diameter, 0.3 mm) may be
used
* Stationary phase: SE 30 (Chrompack)a, chemically bonded,
film thickness 0.3 µm
* Gas-tight syringes (50 or 500 µl) for gas chromatography
* Bottles (5 ml) with serum caps fitted with PTFE-coated
septa, together with tools for sealing and opening
* Automatic pipettes, variable between 20 and 200 µl
* Syringe (100 µl) for gas chromatography
* Vortex mixer
* Centrifuge
* Transfer pipettes (1, 2, 4, and 10 ml)
* Volumetric flasks (10, 20, 50, and 100 ml)
* Disposable syringes containing anticoagulant (e.g., K-EDTA
MonovettenR, Sarstedt, Nümbrecht, Germany)
a Chompack, P.O. Box 8033, 4330
10.2.1.3 Chemicals
* Potassium cyanide, pa
* Sodium cyanide, pa
* Glacial acetic acid, pa
* Helium (99.999% purity) as carrier gas
* Hydrogen (99.90% purity)
* Synthetic air (80% purified nitrogen, 20% oxygen)
10.2.1.4 Solutions
* Physiological saline (9 g sodium chloride/l)
* Pooled stabilized whole blood or stabilized bovine blood
for the calibration standards
10.2.1.5 Calibration standards
(a) Starting solution
12.6 mg potassium cyanide (equivalent to 5.3 mg cyanide)
is weighed out exactly and transferred to a 50 ml
volumetric flask, which is then filled to the mark with
whole blood (106 mg cyanide/l blood).
(b) Stock solution
Starting solution (2 ml) is pipetted into a 20 ml
volumetric flask, which is then filled to the mark with
whole blood (10.6 mg cyanide/l blood). From this
solution, calibration standards within the concentration
range 0.2 - 4.2 mg cyanide/l blood are prepared by
dilution with whole blood as shown in the following table:
Volume of Final volume of Cyanide concentration
stock solution calibration standard of calibration standard
ml ml mg/l
4 10 4.24
2 10 2.12
1 10 1.06
1 20 0.53
1 50 0.212
The blood volume required for preparing the calibration
standards may be reduced to about 10 ml if the standards are
prepared directly in the bottles used for sample treatment. In this
case the procedure is as follows:
Stock solution A
Exactly 24.0 mg potassium cyanide (equivalent to 10 mg cyanide)
is transferred to a 100 ml volumetric flask, which is then filled to
the mark with physiological saline (100 mg cyanide/l).
Stock solution B
10 ml stock solution A is pipetted into a 100 ml volumetric
flask, which is then filled to the mark with physiological saline
(10 mg cyanide/l).
The calibration standards are prepared directly in the bottles
used for the sample treatment. Blood (1 ml) is pipetted into each
bottle and after adding the cyanide solution, the bottle is sealed
immediately. The difference in the volumes of the individual
standards does not reduce the reliability of the calibration for the
head-space analysis used here.
Volume of stock Volume of stock Cyanide concentration
solution A solution B of calibration standard
µl µl mg/l
- 20 0.2
- 40 0.4
- 50 0.5
10 - 1.0
20 - 2.0
30 - 3.0
50 - 5.0
10.2.1.6 Specimen collection and sample preparation
Blood is collected in disposable syringes containing an
anticoagulant. Immediately after collection, 1 ml of whole blood is
pipetted into a 5-ml bottle, which is sealed at once with a cap and
PTFE-coated septum.
Using a 100-µl syringe, 50 µl glacial acetic acid is injected
through the septum into the bottle to release the hydrogen cyanide.
The contents of the bottle are shaken for 30 seconds on a vortex
mixer and then centrifuged for 3 min at 3000 rpm. The bottle is
then incubated for 30 min at room temperature.
The blood samples containing cyanide should be processed within
30 min of collection, as losses can otherwise occur.
10.2.1.7 Operational parameters for gas chromatography
(a) Packed column
Column: Material: Glass
Length: 2 m
Inner diameter: 2.2 mm
Stationary phase: Porapak Q 100-120 mesh
Detector: Thermoionic nitrogen detector
Temperatures: Column: 120 °C
Injector: 220 °C
Detector: 300 °C
Carrier gas: Helium: 30 ml/min
Detector gases: Hydrogen: 3 ml/min
Synthetic air: 60 ml/min
Injected head-space 50 µl
Under these conditions, hydrogen cyanide has a retention
time of 1.5 min.
(b) Capillary column
Capillary column: Material: Quartz or Duran glass
Length: 50 m
Inner diameter: 0.3 mm
Stationary phase: SE 30 (chrompack) chemically
bonded, film thickness 0.3 mm
Detector: Thermionic specific detector (TSD)
Temperatures: Column: 80 °C
Injector: 200 °C
Detector: 300 °C
Split: 1:4
Carrier gas: Helium, 1.68 MPa; make up gas,
35 ml/min
Detector gases: Hydrogen: 1.45 MPa
(3.5-4.0 ml/min)
Synthetic air: 160 ml/min
Injected head-space 400 µl
Under these conditions, the retention time for hydrogen
cyanide is 3.1 min.
10.2.1.8 Analytical determination
50 µl (packed column) or 400 µl (capillary column) of the head
space above the prepared blood sample is injected into the gas
chromatograph under the conditions given in section 10.2.1.7.
10.2.1.9 Calibration
The calibration standards containing cyanide in blood are
prepared as described in section 10.2.1.5 and analysed. The
calibration curve is obtained by plotting the peak heights or areas
for the standards against the blood cyanide concentrations used (see
Figure 8). The linearity of the calibration curve has been tested up
to 6 mg/l.
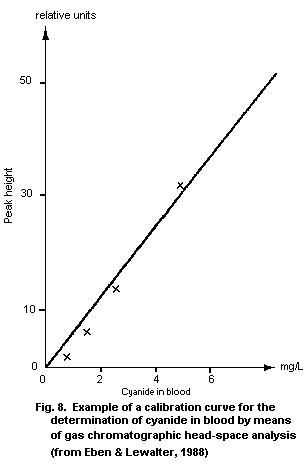 10.2.1.10 Calculation of the analytical result
From the peak height or area for the hydrogen cyanide in the
sample, the corresponding cyanide concentration in whole blood
(mg/l) is read off the calibration curve (see Figure 8).
10.2.1.11 Reliability of the method
The within-series precision and between-day precision were
determined using a packed or a capillary column and blood samples
with a defined cyanide content that were prepared as described in
section 10.2.1.5. The individual values are given in Tables 7 and
8.
Table 7. Within-series precision for the gas chromatographic
determination of cyanide in blood using a packed column
(from Eben & Lewalter, 1988)
n Cyanide in blood Sw
mg/l %
10 0.54 7.5
10 2.20 4.6
n = number of analysis performed
Sw = relative standard deviation derived from replicate
analyses of the same specimen
Table 8. Within-series precision and between-day precision for the
gas chromatographic determination of cyanide in blood
using a capillary column (from Eben & Lewalter, 1988)
Precision n Cyanide in blood s
(mg/l) %
Within-series 17 0.19 5.4
17 0.39 0.9
17 2.11 0.7
Between-day 17 0.19 14.5
17 0.39 5.5
17 2.11 6.7
n = number of analyses performed
s = relative standard deviation
Recovery experiments were carried out using spiked blood
samples. This procedure is the same as that used to prepare the
calibration curve and is thus of limited value. The results serve
only as an internal laboratory check. Recovery rates of r = 84-107%
were obtained for cyanide concentrations between 0.19 and 2.11 mg/l
using the capillary column.
Losses during sample treatment of between 25 and 40% were
revealed by comparison with a calibration curve established using
treated aqueous cyanide solution and the capillary column.
10.2.1.12 Detection limit
The concentration of cyanide that can be determined by this
method, using a packed gas chromatography column, is 0.07 mg/l. If
a capillary column is used, the detection limit is raised to 0.1 mg
cyanide/l blood, because with control blood a peak appears in the
position for hydrogen cyanide.
10.2.1.13 Specificity
The separation of the cyanide from interfering blood
constituents and its detection with the thermionic nitrogen detector
makes this procedure highly specific for cyanide. With the packed
column no interference was observed with ethanol, diethyl ether,
methanol, acetone, ethyl acetate, n-hexane, diisopropyl ether or
2-propanol (1 mg per analytical sample).
Blood or plasma samples from people who had not been exposed to
hydrogen cyanide or other cyanides yielded no peak in the position
for hydrogen cyanide when a packed column was used. With the
capillary column described here a peak was also observed for blood
samples from people not occupationally exposed to cyanides. The
peak, however, indicated a cyanide concentration below the detection
limit of 0.1 mg/l.
10.2.2 Microdiffusion technique
Microdiffusion analysis is admirably suited to the detection
and estimation of small quantities of volatile substances like HCN.
Conway (1950) pioneered the application of this method of analysis
in the determination of such substances as ammonia, alcohol, carbon
dioxide, volatile amines, and carbon monoxide. The Conway diffusion
dish has become a standard laboratory item.
Feldstein & Klendshoj (1954) adapted the technique to the
estimation of cyanide in biological materials.
10.2.2.1 Principle
Hydrocyanic acid is set free from the sample (blood, serum,
urine, stomach contents) by the addition of sulfuric acid and is
then absorbed into sodium hydroxide. The cyanide concentration is
measured colorimetrically in an aliquot of the alkaline solution
(Asmus & Garschagen, 1953).
10.2.2.2 Equipment
* Spectrophotometer (Vis)
* Conway microdiffusion cells (Fig. 9)a
* Volumetric flask 50 ml
* Test tubes
* Pipettes
10.2.1.10 Calculation of the analytical result
From the peak height or area for the hydrogen cyanide in the
sample, the corresponding cyanide concentration in whole blood
(mg/l) is read off the calibration curve (see Figure 8).
10.2.1.11 Reliability of the method
The within-series precision and between-day precision were
determined using a packed or a capillary column and blood samples
with a defined cyanide content that were prepared as described in
section 10.2.1.5. The individual values are given in Tables 7 and
8.
Table 7. Within-series precision for the gas chromatographic
determination of cyanide in blood using a packed column
(from Eben & Lewalter, 1988)
n Cyanide in blood Sw
mg/l %
10 0.54 7.5
10 2.20 4.6
n = number of analysis performed
Sw = relative standard deviation derived from replicate
analyses of the same specimen
Table 8. Within-series precision and between-day precision for the
gas chromatographic determination of cyanide in blood
using a capillary column (from Eben & Lewalter, 1988)
Precision n Cyanide in blood s
(mg/l) %
Within-series 17 0.19 5.4
17 0.39 0.9
17 2.11 0.7
Between-day 17 0.19 14.5
17 0.39 5.5
17 2.11 6.7
n = number of analyses performed
s = relative standard deviation
Recovery experiments were carried out using spiked blood
samples. This procedure is the same as that used to prepare the
calibration curve and is thus of limited value. The results serve
only as an internal laboratory check. Recovery rates of r = 84-107%
were obtained for cyanide concentrations between 0.19 and 2.11 mg/l
using the capillary column.
Losses during sample treatment of between 25 and 40% were
revealed by comparison with a calibration curve established using
treated aqueous cyanide solution and the capillary column.
10.2.1.12 Detection limit
The concentration of cyanide that can be determined by this
method, using a packed gas chromatography column, is 0.07 mg/l. If
a capillary column is used, the detection limit is raised to 0.1 mg
cyanide/l blood, because with control blood a peak appears in the
position for hydrogen cyanide.
10.2.1.13 Specificity
The separation of the cyanide from interfering blood
constituents and its detection with the thermionic nitrogen detector
makes this procedure highly specific for cyanide. With the packed
column no interference was observed with ethanol, diethyl ether,
methanol, acetone, ethyl acetate, n-hexane, diisopropyl ether or
2-propanol (1 mg per analytical sample).
Blood or plasma samples from people who had not been exposed to
hydrogen cyanide or other cyanides yielded no peak in the position
for hydrogen cyanide when a packed column was used. With the
capillary column described here a peak was also observed for blood
samples from people not occupationally exposed to cyanides. The
peak, however, indicated a cyanide concentration below the detection
limit of 0.1 mg/l.
10.2.2 Microdiffusion technique
Microdiffusion analysis is admirably suited to the detection
and estimation of small quantities of volatile substances like HCN.
Conway (1950) pioneered the application of this method of analysis
in the determination of such substances as ammonia, alcohol, carbon
dioxide, volatile amines, and carbon monoxide. The Conway diffusion
dish has become a standard laboratory item.
Feldstein & Klendshoj (1954) adapted the technique to the
estimation of cyanide in biological materials.
10.2.2.1 Principle
Hydrocyanic acid is set free from the sample (blood, serum,
urine, stomach contents) by the addition of sulfuric acid and is
then absorbed into sodium hydroxide. The cyanide concentration is
measured colorimetrically in an aliquot of the alkaline solution
(Asmus & Garschagen, 1953).
10.2.2.2 Equipment
* Spectrophotometer (Vis)
* Conway microdiffusion cells (Fig. 9)a
* Volumetric flask 50 ml
* Test tubes
* Pipettes
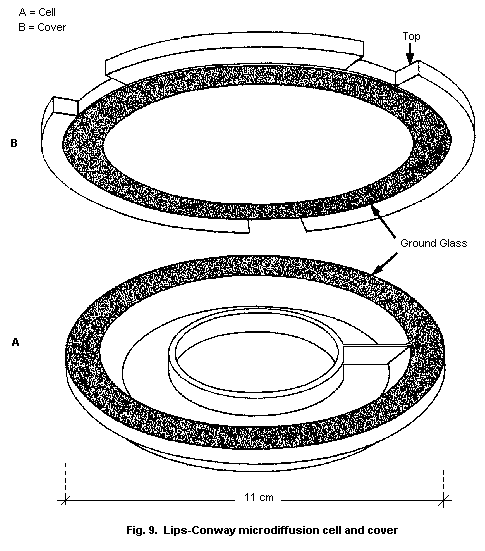 a Culture dishes, Lips-Conway type, Code no. 911313009, Glaswerke
Wertheim, Germany
10.2.2.3 Chemicals
* Potassium cyanide
* Hydrochloric acid (concentrated; 35-37%)
* Sulfuric acid (concentrated; 96-98%)
* Sodium hydroxide
* Sodium phosphate, monobasic (NaH2PO4.2H2O)
* Chloramine T ( N-chloro- p-toluenesulfonamide sodium salt
trihydrate)
* Barbituric acid (should be stored in a dissicator)
* Pyridine (should be stored in the dark)
* Silicone grease or other sealing agent
10.2.2.4 Solvents and reagents
* Sodium phosphate (1 mol/l)
* Sulfuric acid (10%)
* Sodium hydroxide (0.1 mol/l)
* Chloramine T (0.25%) (must be prepared fresh daily)
Pyridine-barbituric acid reagent
Barbituric acid (3 g), 15 ml pyridine, and 3 ml concentrated
hydrochloric acid are placed in a 50-ml volumetric flask. The
solution is mixed to dissolve the reagents, made up to 50 ml with
water, and filtered. This solution must be prepared fresh each time
it is used.
10.2.2.5 Calibration standards
Standard solutions of sodium cyanide containing 0.1 to 2.0 µg
of cyanide per ml of sodium hydroxide (0.1 mol/l) are prepared.
10.2.2.6 Specimen
Venous blood containing heparin or EDTA as anticoagulants,
serum, plasma, urine, or stomach content (or tissue slurry) may
serve as specimens. The samples should be analysed as soon as
possible.
10.2.2.7 Procedure
(a) Microdiffusion
A sample (2-4 ml) of the specimen is placed in the outer
compartment of the microdiffusion cell, and 3.3 ml of the
sodium hydroxide solution (0.1 mol/l) is pipetted into the
centre well of the unit. The ground-glass cover of the
unit is smeared with silicone grease or other sealing
agent and is placed on the unit so that a small portion of
the outer compartment remains uncovered. Then 3-4 drops
of 10% sulfuric acid are pipetted into the outer
compartment, and the lid is quickly moved into place to
cover the entire cell with an airtight seal. The unit is
then gently tilted and rotated to mix the fluids in the
outer compartment and allowed to stand for 3 h at 20-25 °C
for diffusion to be completed. At the end of that time
the lid is removed and an aliquot of the liquid in the
centre well is taken for analysis.
(b) Quantitative determination
1.0 ml of the absorbing solution in the centre well is
placed into each of three test tubes graduated at 10 ml.
A blank consisting of 1.0 ml of sodium hydroxide (0.1
mol/l) in a test tube is prepared. A 1-ml sample of a
calibration solution is used as a positive control.
To the blank, the control sample, and the unknown, 2.0 ml
of sodium phosphate solution (1 mol/l) and 1.0 ml of
chloramine T solution are added. After mixing and
allowing to stand for 2-3 min., 3.0 ml of the
pyridine-barbituric acid reagent is added. The contents
of the tubes are again mixed and allowed to stand for
10 min. The presence of a red colour indicates the
presence of cyanide. The optical density is determined in
a spectrophotometer at 580 nm with the blank set at zero
density.
(c) Calibration curve and calculation
With aliquots of the standard solutions of sodium cyanide
containing 0.1 to 2.0 µg of cyanide/ml of sodium hydroxide
(1 mol/l), the colour is developed as described above. A
calibration curve relating optical density and
concentration is obtained and the cyanide content of the
material being analysed is read from the curve.
10.2.2.8 Reliability of the method
For cyanide concentrations from 0.04 µg/ml to 0.38 µg/ml, the
within-run precision was ± 2.75% for blood and ± 5.56% for urine.
The recovery of cyanide added to biological samples is 96-102%.
10.2.2.9 Detection limit
The detection limit depends on the amount of the analysed
sample and of the NaOH (0.1 mol/l) for the absorption of the
released hydrocyanic acid. This limit can be lowered by reducing
the volume of sodium hydroxide in the centre well of the Conway unit
to 1.0 ml.
With 2 ml blood and 3.3 ml NaOH (0.1 mol/l), the detection
limit is 0.2 µg CN_ /ml blood.
10.2.2.10 Specificity
The separation of the cyanide from interfering blood
constituents by microdiffusion makes this procedure very specific.
Hydrogen sulfide and sulfur dioxide may interfere.
10.3 References
Asmus E & Garschagen H (1953) [The use of barbituric acid for the
photometric determination of cyanide and thiocyanate.] Z Anal Chem,
138: 414-422 (in German).
Conway EJ (1950) Microdiffusion analysis and volumetric error. New
York, Van Nostrand Reinhold Co.
Eben A & Lewalter J (1988) Cyanide determination in blood. In:
Angerer J & Schaller KH ed. Analysis of hazardous substances in
biological materials. Volume II: Methods for biological monitoring.
Weinheim, VCH-Verlags-gesellschaft.
Feigl F & Feigl HE (1949) On the reactivity of inner-complex-bonded
Palladium - a new specific test for cyanides in alkaline solutions.
Anal Chim Acta, 3: 300-309.
Feigl F, Anger V, & Oesper RE (1966) Spot tests in organic analysis,
7th ed. Amsterdam, Oxford, New York, Elsevier Science Publishers, pp
547-548.
Feldstein L & Klendshoj NC (1954) The determination of volatile
substances by microdiffusion analysis. J Forensic Sci, 2: 39-58.
Jakobs K (1984) [Report on experience with the administration of
4-DMAP in severe prussic acid poisoning. Consequences for medical
practice.] Zentralbl Arbeitsmed, 34: 274-277 (in German).
McAuley F & Reive DS (1983) Rapid quantitation of cyanide in blood
by gas chromatography. J Anal Toxicol, 7: 213-215.
a Culture dishes, Lips-Conway type, Code no. 911313009, Glaswerke
Wertheim, Germany
10.2.2.3 Chemicals
* Potassium cyanide
* Hydrochloric acid (concentrated; 35-37%)
* Sulfuric acid (concentrated; 96-98%)
* Sodium hydroxide
* Sodium phosphate, monobasic (NaH2PO4.2H2O)
* Chloramine T ( N-chloro- p-toluenesulfonamide sodium salt
trihydrate)
* Barbituric acid (should be stored in a dissicator)
* Pyridine (should be stored in the dark)
* Silicone grease or other sealing agent
10.2.2.4 Solvents and reagents
* Sodium phosphate (1 mol/l)
* Sulfuric acid (10%)
* Sodium hydroxide (0.1 mol/l)
* Chloramine T (0.25%) (must be prepared fresh daily)
Pyridine-barbituric acid reagent
Barbituric acid (3 g), 15 ml pyridine, and 3 ml concentrated
hydrochloric acid are placed in a 50-ml volumetric flask. The
solution is mixed to dissolve the reagents, made up to 50 ml with
water, and filtered. This solution must be prepared fresh each time
it is used.
10.2.2.5 Calibration standards
Standard solutions of sodium cyanide containing 0.1 to 2.0 µg
of cyanide per ml of sodium hydroxide (0.1 mol/l) are prepared.
10.2.2.6 Specimen
Venous blood containing heparin or EDTA as anticoagulants,
serum, plasma, urine, or stomach content (or tissue slurry) may
serve as specimens. The samples should be analysed as soon as
possible.
10.2.2.7 Procedure
(a) Microdiffusion
A sample (2-4 ml) of the specimen is placed in the outer
compartment of the microdiffusion cell, and 3.3 ml of the
sodium hydroxide solution (0.1 mol/l) is pipetted into the
centre well of the unit. The ground-glass cover of the
unit is smeared with silicone grease or other sealing
agent and is placed on the unit so that a small portion of
the outer compartment remains uncovered. Then 3-4 drops
of 10% sulfuric acid are pipetted into the outer
compartment, and the lid is quickly moved into place to
cover the entire cell with an airtight seal. The unit is
then gently tilted and rotated to mix the fluids in the
outer compartment and allowed to stand for 3 h at 20-25 °C
for diffusion to be completed. At the end of that time
the lid is removed and an aliquot of the liquid in the
centre well is taken for analysis.
(b) Quantitative determination
1.0 ml of the absorbing solution in the centre well is
placed into each of three test tubes graduated at 10 ml.
A blank consisting of 1.0 ml of sodium hydroxide (0.1
mol/l) in a test tube is prepared. A 1-ml sample of a
calibration solution is used as a positive control.
To the blank, the control sample, and the unknown, 2.0 ml
of sodium phosphate solution (1 mol/l) and 1.0 ml of
chloramine T solution are added. After mixing and
allowing to stand for 2-3 min., 3.0 ml of the
pyridine-barbituric acid reagent is added. The contents
of the tubes are again mixed and allowed to stand for
10 min. The presence of a red colour indicates the
presence of cyanide. The optical density is determined in
a spectrophotometer at 580 nm with the blank set at zero
density.
(c) Calibration curve and calculation
With aliquots of the standard solutions of sodium cyanide
containing 0.1 to 2.0 µg of cyanide/ml of sodium hydroxide
(1 mol/l), the colour is developed as described above. A
calibration curve relating optical density and
concentration is obtained and the cyanide content of the
material being analysed is read from the curve.
10.2.2.8 Reliability of the method
For cyanide concentrations from 0.04 µg/ml to 0.38 µg/ml, the
within-run precision was ± 2.75% for blood and ± 5.56% for urine.
The recovery of cyanide added to biological samples is 96-102%.
10.2.2.9 Detection limit
The detection limit depends on the amount of the analysed
sample and of the NaOH (0.1 mol/l) for the absorption of the
released hydrocyanic acid. This limit can be lowered by reducing
the volume of sodium hydroxide in the centre well of the Conway unit
to 1.0 ml.
With 2 ml blood and 3.3 ml NaOH (0.1 mol/l), the detection
limit is 0.2 µg CN_ /ml blood.
10.2.2.10 Specificity
The separation of the cyanide from interfering blood
constituents by microdiffusion makes this procedure very specific.
Hydrogen sulfide and sulfur dioxide may interfere.
10.3 References
Asmus E & Garschagen H (1953) [The use of barbituric acid for the
photometric determination of cyanide and thiocyanate.] Z Anal Chem,
138: 414-422 (in German).
Conway EJ (1950) Microdiffusion analysis and volumetric error. New
York, Van Nostrand Reinhold Co.
Eben A & Lewalter J (1988) Cyanide determination in blood. In:
Angerer J & Schaller KH ed. Analysis of hazardous substances in
biological materials. Volume II: Methods for biological monitoring.
Weinheim, VCH-Verlags-gesellschaft.
Feigl F & Feigl HE (1949) On the reactivity of inner-complex-bonded
Palladium - a new specific test for cyanides in alkaline solutions.
Anal Chim Acta, 3: 300-309.
Feigl F, Anger V, & Oesper RE (1966) Spot tests in organic analysis,
7th ed. Amsterdam, Oxford, New York, Elsevier Science Publishers, pp
547-548.
Feldstein L & Klendshoj NC (1954) The determination of volatile
substances by microdiffusion analysis. J Forensic Sci, 2: 39-58.
Jakobs K (1984) [Report on experience with the administration of
4-DMAP in severe prussic acid poisoning. Consequences for medical
practice.] Zentralbl Arbeitsmed, 34: 274-277 (in German).
McAuley F & Reive DS (1983) Rapid quantitation of cyanide in blood
by gas chromatography. J Anal Toxicol, 7: 213-215.