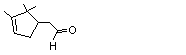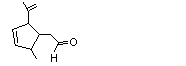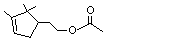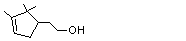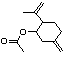First draft prepared by
Dr G.J.A. Speijers
Section Public Health of the Centre for Substances & Risk Assessment, National Institute of Public Health and Environmental Protection, Bilthoven, Netherlands
and Professor A. Renwick
Clinical Pharmacology Group, University of Southampton, Southampton, England
|
Application of the Procedure for the Safety Evaluation of Flavouring Agents |
The Committee evaluated a group of 26 flavouring agents that included alicyclic primary alcohols, aldehydes, acids and related esters (see Table 1), using the Procedure for the Safety Evaluation of Flavouring Agents (See Figure 1). The Committee had not evaluated any of these agents previously.
Table 1. Summary of results of safety evaluation of alicyclic primary alcohols, aldehydes, acids and related esters used as flavouring agentsa
|
Flavouring agent |
No. |
CAS No. and structure |
Steps A3 and B3b |
Step B4 |
Comments |
Conclusion based on current intake |
|
Structural class I |
|
|
|
|
|
|
|
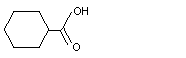
Cyclohexanecarboxylic acid |
961 |
98-89-5 |
A3 - No |
N/R |
See note 1 |
No safety concern |
|
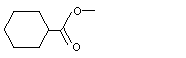
Methyl cyclohexanecarboxylate |
962 |
4630-82-4 |
A3 - No |
N/R |
See notes 1,2 |
No safety concern |
|
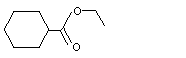
Ethyl cyclohexanecarboxylate |
963 |
3289-28-9 |
A3 - No |
N/R |
See notes 1,2 |
No safety concern |
|
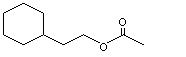
Cyclohexaneethyl acetate |
964 |
21722-83-8 |
A3 - No |
N/R |
See notes 2,3 |
No safety concern |
|
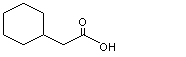
Cyclohexaneacetic acid |
965 |
5292-21-7 |
A3 - No |
N/R |
See note 3 |
No safety concern |
|
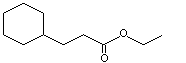
Ethyl cyclohexanepropionate |
966 |
10094-36-7 |
A3 - No |
N/R |
See notes 1,2 |
No safety concern |
|
2,2,3-Trimethylcyclopent-3-en-1-yl-acetaldehyde |
967 |
4501-58-0 |
A3 - No |
N/R |
See note 4 |
No safety concern |
|
cis- 5-Isopropenyl-cis-2-methylcyclopentan-1-carboxaldehyde |
968 |
55253-28-6 |
A3 - No |
N/R |
See note 4 |
No safety concern |
|
Campholene acetate |
969 |
36789-69-0 |
A3 - No |
N/R |
See notes 2,4 |
No safety concern |
|
alpha- Campholenic alcohol |
970 |
1901-38-8 |
A3 - No |
N/R |
See note 4 |
No safety concern |
|
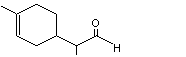
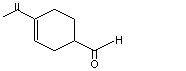
para- Menth-1-ene-9-al |
971 |
29548-14-9 |
A3 - No |
N/R |
See note 4 |
No safety concern |
|
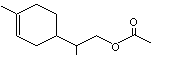
1-para-Menthen-9-yl-acetate |
972 |
17916-91-5 |
A3 - No |
N/R |
See notes 2,4 |
No safety concern |
|
para- Mentha-1,8-dien-7-al |
973 |
2111-75-3 |
A3 - No |
N/R |
See note 4 |
No safety concern |
|
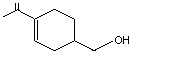
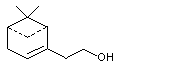
para- Mentha-1,8-dien-7-ol |
974 |
536-59-4 |
A3 - No |
N/R |
See note 4 |
No safety concern |
|
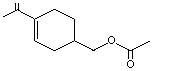
para- Mentha-1,8-dien-7-yl acetate |
975 |
15111-96-3 |
A3 - No |
N/R |
See notes 2,4 |
No safety concern |
|
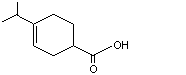
1,2,5,6-Tetrahydrocuminic acid |
976 |
71298-42-5 |
A3 - No |
N/R |
See note 1 |
No safety concern |
|
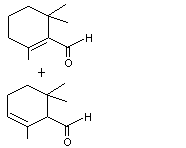
2,6,6-Trimethylcyclohexa-1,3-dienyl methanal |
977 |
116-26-7 |
B3 - No |
Yes. The NOELs of 120 mg/kg bw per day for perillyl alcohol in 90-day studies in rats and dogs and NOELs of 10 mg/kg bw per day for alpha-ionone and beta-ionone in a 90-da7 study in rats are > 2 million and > 200 000 times, respectively, the estimated intake of 2,6,6-trimethyl-cyclohexa-1,3-dienyl methanal when used as a flavouring agent. |
|
No safety concern |
|
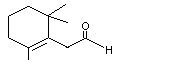
2,6,6-Trimethyl-1-cyclohexen-1-acetaldehyde |
978 |
472-66-2 |
A3 - No |
N/R |
See note 3. |
No safety concern |
|
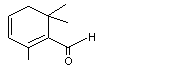
2,6,6-Trimethyl-1-cyclohexen-1-carboxaldehyde |
979 |
432-25-7 |
A3 - No |
N/R |
See note 1 |
No safety concern |
|
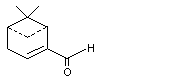
2-Formyl-6-6-dimethylbicyclo[3.1.1[hept-2-ene |
980 |
564-94-3 |
A3 - No |
N/R |
See note 4 |
No safety concern |
|
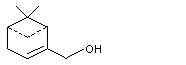
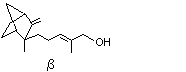
Myrtenol |
981 |
515-00-4 |
A3 - No |
N/R |
See note 4 |
No safety concern |
|
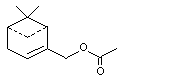
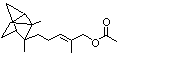
Myrtenyl acetate |
982 |
1079-01-2 |
A3 - No |
N/R |
See note 4 |
No safety concern |
|
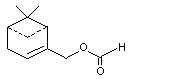
Myrtenal formate |
983 |
72928-52-0 |
A3 - No |
N/R |
See note 4 |
No safety concern |
|
Santalol (alpha and beta) |
984 |
115-71-9 |
A3 - No |
N/R |
See note 4 |
No safety concern |
|
|
77-42-9 |
|
|
|
|
|
Santalyl acetate |
985 |
1323-00-8 |
A3 - No |
N/R |
See note 4 |
No safety concern |
|
10-Hydroxymethylene-2-pinene |
986 |
128-50-7 |
A3 - No |
N/R |
See note 4 |
No safety concern |
|
CAS: Chemical Abstracts Service; ND: no intake data reported; N/R: not required for evaluation |
|
|
a |
Step 2: Except for No. 977, all the flavouring agents in this group were predicted to be metabolized to innocuous products. |
|
b |
The threshold for human intake for structural class I is 1800 µg/day. All intake values are expressed in µg/day. |
|
|
The combined intake of all flavouring agents in the group is 22 and 18 µg/person per day in Europe and the USA, respectively. |
|
Notes: |
|
|
1. |
Metabolized by beta-oxidation, aromatization and conjugation with glycine and glucuronic acid |
|
2. |
Metabolized by hydrolysis followed by oxidation and/or conjugation |
|
3. |
Metabolized by beta-oxidation and eliminated largely by metabolism via the citric acid cycle |
|
4. |
Metabolized largely by oxidation of the side-chain to the corresponding carboxylic acid, which is excreted unchanged and as conjugates |
Twenty of the 26 flavouring agents in this group are terpenoid alcohols, aldehydes or carboxylic acids or their related esters. Thirteen are common components of food and have been detected in vanilla, bourbon, rum, mango, rosemary, grapefruit juice, mandarin peel oil and blackberries (Maarse et al., 1999).
The total annual volume of the 26 flavouring agents in this group that is produced is approximately 170 kg in Europe (Insternational Organization of the Flavor Industry, 1995) and 130 kg in the USA (Lucas et al., 1999). None of the agents is produced in an annual volume greater than 55 kg. Most of the total annual volume in Europe is accounted for by (2,2,3-trimethylcyclopent-3-en-1-yl)acetaldehyde (No. 967; campholenic aldehyde), para-mentha-1,8-dien-7-al (No. 973; perillaldehyde), para-mentha-1,8-dien-7-ol (No. 974; perillyl alcohol), 2,6,6-trimethylcyclohexa-1,3-dienyl methanal (No. 977; safranal) and 2-formyl-6,6-dimethylbicyclo[3.1.1]hept-2-ene (No. 980; myrtenal). In the USA, approximately 91% of the total annual volume is accounted for by cyclohexanecarboxylic acid (No. 961), para-mentha-1,8-dien-7-al (No. 973; perillaldehyde), 2,6,6-trimethyl-1-cyclohexen-1-acetaldehyde (No. 978; beta-cyclocitral) and 2-formyl-6,6-dimethylbicyclo[3.1.1]hept-2-ene (No. 980; myrtenal). The estimated daily per capita intake of 11 of the substances in this group is 10 µg (see Table 2).
Table 2. Annual volumes of production of alicyclic primary alcohols, aldehydes, acids and related esters used as flavouring agents in Europe and the USA
|
Substance (No.) |
Most recent annual volume (kg)a |
Intakeb |
Annual volume in naturally occurring foods (kg)c |
Consumption ratiod |
|
|
µg/day |
µg/kg bw per day |
||||
|
Cyclohexanecarboxylic acid (961) |
|
|
|
|
|
|
Europe |
0,5 |
0.071 |
0.0012 |
– |
N/R |
|
USA |
32 |
4.2 |
0.071 |
|
|
|
Methyl cyclohexanecarboxylate (962) |
|
|
|
|
|
|
Europe |
0.6 |
0.086 |
0.0014 |
+ |
N/R |
|
USA |
0.1 |
0.013 |
0.0002 |
|
|
|
Ethyl cyclohexanecarboxylate (963) |
|
|
|
|
|
|
Europe |
ND |
|
|
+ |
N/R |
|
USA |
0.91e |
0.12 |
0.0020 |
|
|
|
Cyclohexaneethyl acetate (964) |
|
|
|
|
|
|
Europe |
8.0 |
1.1 |
0.019 |
– |
N/R |
|
USA |
ND |
|
|
|
|
|
Cyclohexaneacetic acid (965) |
|
|
|
|
|
|
Europe |
1.0 |
0.14 |
0.0024 |
– |
N/R |
|
USA |
2,7 |
0.36 |
0.0059 |
|
|
|
Ethyl cyclohexanepropionate (966) |
|
|
|
|
|
|
Europe |
ND |
|
|
– |
N/R |
|
USA |
0.9 |
0.12 |
0.0020 |
|
|
|
(2,2,3-Trimethylcyclopent-3-en-1-yl) acetaldehyde (967) |
|
|
|
|
|
|
Europe |
41 |
5.8 |
0.098 |
+ |
N/R |
|
USA |
ND |
|
|
|
|
|
cis-5-Isopropenyl-cis-2-methylcyclopentan-1-carboxaldehyde (968) |
|
|
|
|
|
|
Europe |
0.1 |
0.014 |
0.0002 |
– |
N/R |
|
USA |
ND |
|
|
|
|
|
Campholene acetate (969) |
|
|
|
|
|
|
Europe |
0.5 |
0.071 |
0.0012 |
– |
N/R |
|
USA |
ND |
|
|
|
|
|
alpha-Campholenic alcohol (970) |
|
|
|
|
|
|
Europe |
0.1 |
0.014 |
0.0002 |
+ |
N/R |
|
USA |
ND |
|
|
|
|
|
para-Menth-1-ene-9-al (971) |
|
|
|
|
|
|
Europe |
1.0 |
0.14 |
0.0024 |
5.8 |
5.8 |
|
USA |
ND |
ND |
ND |
|
|
|
1-para-Menthen-9-yl acetate (972) |
|
|
|
|
|
|
Europe |
7.0 |
1.0 |
0.017 |
+ |
N/R |
|
USA |
ND |
|
|
|
|
|
para-Mentha-1,8-dien-7-al (973) |
|
|
|
|
|
|
Europe |
17 |
2.4 |
0.040 |
110 |
0.07 |
|
USA |
11 |
1.5 |
0.025 |
|
|
|
para-Mentha-1,8-dien-7-ol (974) |
|
|
|
|
|
|
Europe |
13 |
1.8 |
0.031 |
+ |
N/R |
|
USA |
5.9 |
0.78 |
0.013 |
|
|
|
para-Mentha-1,8-dien-7-yl acetate (975) |
|
|
|
|
|
|
Europe |
2.9 |
0.41 |
0.069 |
1.2 |
000 |
|
USA |
0.5 |
0.066 |
0.0011 |
|
|
|
1,2,5,6-Tetrahydrocuminic acid (976) |
|
|
|
|
|
|
Europe |
0.1 |
0.014 |
0.0002 |
– |
N/R |
|
USA |
ND |
|
|
|
|
|
2,6,6-Trimethylcyclohexa-1,3-dienyl methanal (977) |
|
|
|
|
|
|
Europe |
29 |
4.1 |
0.069 |
75 |
0.03 |
|
USA |
0.5 |
0.066 |
0.0011 |
|
|
|
2,6,6-Trimethyl-1-cyclohexen-1-acetaldehyde (978) |
|
|
|
|
|
|
Europe |
2.0 |
0.28 |
0.0048 |
– |
N/R |
|
USA |
16 |
2.0 |
0.034 |
|
|
|
2,6,6-Trimethyl-1 and 2-cyclohexen-1-carboxaldehyde (979) |
|
|
|
|
|
|
Europe |
3.0 |
0.43 |
0.0071 |
74 |
25 |
|
USA |
ND |
|
|
|
|
|
2-Formyl-6,6-dimethylbicyclo[3.1.1] hept-2-ene (980) |
|
|
|
|
|
|
Europe |
33 |
4.2 |
0.078 |
+ |
N/R |
|
USA |
54 |
7.2 |
0.12 |
|
|
|
Myrtenol (981) |
|
|
|
|
|
|
Europe |
3.0 |
0.43 |
0.0071 |
+ |
N/R |
|
USA |
0.4 |
0.053 |
0.0009 |
|
|
|
Myrtenyl acetate (982) |
|
|
|
|
|
|
Europe |
3.0 |
0.43 |
0.0071 |
– |
N/R |
|
USA |
0.2 |
0.026 |
0.0004 |
|
|
|
2-Hydroxymethyl-6,6-dimethylbicyclo [3.1.1]hept-2-enyl formate (983) |
|
|
|
|
|
|
Europe |
2.5 |
0.36 |
0.0059 |
– |
N/R |
|
USA |
ND |
|
|
|
|
|
Santalol (alpha and beta) (984) |
|
|
|
|
|
|
Europe |
0.3 |
0.043 |
0.0007 |
– |
N/R |
|
USA |
ND |
|
|
|
|
|
Santalyl acetate (985) |
|
|
|
|
|
|
Europe |
ND |
|
|
– |
N/R |
|
USA |
0.1 |
0.013 |
0.0002 |
|
|
|
10-Hydroxymethylene-2-pinene (986) |
|
|
|
|
|
|
Europe |
ND |
|
|
– |
N/R |
|
USA |
0.1e |
0.013 |
0.0002 |
|
|
|
Total |
|
|
|
|
|
|
Europe |
170 |
|
|
|
|
|
USA |
130 |
|
|
|
|
|
ND, no data reported; N/R, not reported; +, reported to occur naturally in foods (Maarse et al., 1999) but no quantitative data available; –, not reported to occur naturally in foods |
|
|
a |
From International Organization of the Flavor Industry (1995) and Lucas et al. (1999) |
|
b |
Intake (µg/person per day) was calculated as follows: [(annual volume, kg) × (1 × 109 µg/kg)/(population × survey correction factor × 365 days)], where population (10%, ‘eaters only’) = 32 × 106 for Europe and 26 × 106 for the USA. The correction factor = 0.6 for Europe and 0.8 for the USA, representing the assumption that only 60% and 80% of the annual production volume of the flavour, resepctively, was reported in the poundage surveys. Intake (µg/kg bw per day) calculated as follows: [(µg/person per day)/body weight], where body weight = 60 kg. Slight variations may occur from rounding. |
|
c |
Quantitative data from Stofberg & Grundschober (1987) |
|
d |
Calculated as follows: (annual consumption in food, kg)/(most recently reported volume as a flavouring agent, kg) |
|
e |
Anticipated annual volume in the USA as reported by the Flavor and Extract Manufacturers Association |
The number of potential pathways of metabolism of alicyclic substances increases as the number and types of functional groups and ring substituents in the molecule increase. The cyclohexane derivatives (carboxylic acids and their related esters) in this group (Nos 961–966) can undergo beta-oxidation and subsequent ring cleavage and/or aromatization of the cyclohexane ring and/or can be excreted in urine as cyclohexane carboxylic acid and its glycine and glucuronide conjugates. If there is an even number of carbons in the side-chain, beta-oxidative cleavage leads to ring-opened oxygenated metabolites, primarily dicarboxylic acids, which are metabolized via the citric acid cycle. If there is an odd number of carbons in the side-chain, beta-oxidative cleavage leads to a cyclohexane carboxylic acid derivative that may subsequently undergo dehydrogenation to yield unsaturated derivatives. The two non-terpenoid substances containing a cyclohexene ring (Nos 978 and 979) would be metabolized and eliminated by similar pathways. The substance with two endocyclic double bonds, 2,6,6-trimethylcyclohexa-1,3-dienyl methanal (No. 977; safranal), is a structural analogue of dehydrodihydroionol (No. 397), which was evaluated by the Committee at its fifty-first meeting (Annex 1, reference 137), and, like that agent, cannot be predicted to be metabolized to innocuous products.
The flavouring agents with a cyclopentane or cyclopentene ring (Nos 967–970) also have an oxygenated side-chain, and this is predicted to be the main site of oxidative metabolism prior to excretion of more polar acid metabolites. The processes of side-chain and alicyclic ring oxidation were considered by the Committee at its fifty-first meeting (Annex 1, reference 137).
Monocyclic terpenoid primary alcohols (e.g., No. 974, perillyl alcohol) and aldehydes (e.g., Nos 971 and 973) would be oxidized to yield the corresponding carboxylic acid, while the esters (Nos 972 and 975) would undergo hydrolysis prior to oxidation. The acid metabolites of the above agents and 1,2,5,6-tetrahydrocuminic acid (No. 976) would be conjugated with glucuronic acid and excreted mainly in the urine. In a minor pathway, the aldehyde can be reduced to the alcohol and excreted as the glucuronic acid conjugate (Brewster et al., 1977a,b; Ishida et al., 1989; Haag & Gould, 1994). If an endocyclic double-bond is present, the flavouring agent or its metabolite could be reduced by the action of gut microflora. The acid metabolite may also undergo aromatization of the ring to yield a hippuric acid derivative (Ishida et al., 1989). Bicyclic and tricyclic compounds (e.g., Nos 980–986) would undergo similar metabolism to the monocyclic terpenoids and, in addition, may undergo ring oxidation.
Four agents in this group (Nos 973, 977, 979 and 980) contain an alpha,beta-unsaturated carbonyl group, which is a structural alert for toxicity. The Committee, at previous meetings, devoted considerable attention to the safety of flavouring agents containing this reactive moiety. The Committee concluded at its fifty-seventh meeting (Annex 1, reference 154) that the presence of protective processes in cells provides adequate detoxication capacity at the low doses associated with the use of compounds such as flavouring agents. These protective processes include conjugation with glutathione and reduction of the ketone to the corresponding alcohol (followed by conjugation of the alcohol with glucuronic acid).
|
Step 1 |
In applying the Procedure for the Safety Evaluation of Flavouring Agents, the Committee assigned all 26 agents to structural class I (Cramer et al., 1978). |
|
Step 2 |
The simple cyclohexane and cyclohexene derivatives (Nos 961–966, 978 and 979), the cyclopentane and cyclopentene compounds (Nos 967–970) and the simple mono-, bi- and tricyclic terpenoid derivatives (Nos 971–976 and 980–986) were predicted to be metabolized to innocuous products. The evaluation of these substances therefore proceeded via the A (left-hand) side of the decision-tree (see Figure 1). The remaining substance, 2,6,6-trimethylcyclohexa-1,3-dienyl methanal (No. 977; safranal), cannot be predicted to be metabolized to innocuous products and was therefore evaluated via the B (right-hand) side of the decision-tree. |
|
Step A3 |
The daily per capita intakes of each of the 25 flavouring agents evaluated at this step were below the daily human intake thresholds for structural class I (1800 µg per person), indicating that they present no safety concern when consumed as flavouring agents at current estimated levels. |
|
Step B3 |
The daily per capita intake of the monocyclic substance with two endocyclic double-bonds evaluated at this step, 2,6,6-trimethylcyclohexa-1,3-dienyl methanal (No. 977; safranal), was below the threshold for daily human intake of compounds of structural class I , and its evaluation therefore proceeded to step B4. |
|
Step B4 |
As the agent evaluated at this step, 2,6,6-trimethylcyclohexa-1,3-dienyl methanal (No. 977; safranal), is structurally related to perillyl alcohol (No. 974), data on the toxicity of perillyl alcohol were used to evaluate its safety. Perillyl alcohol given by intragastric gavage changed the weights of several organs in female rats when given at 400 mg/kg bw per day, but not at 120 mg/kg bw per day, in a 90-day study; changes in organ weights were not reported in male rats. Doses of 40, 120 and 400 mg/kg bw per day produced hyperexcitability and salivation, which the authors considered may have been due to its irritating properties (National Cancer Institute, 1996). A daily dose of 120 mg/kg bw was well tolerated by dogs in a 90-day study (National Cancer Institute, 1996). The daily intake of 2,6,6-trimethylcyclohexa-1,3-dienyl methanal (No. 977; safranal) is 4 µg/kg bw in Europe and 0.07 µg/kg bw in the USA. The margin of safety between these intakes and 120 mg/kg bw per day is > 2 000 000. The compound also shares structural similarities with alpha-ionone and beta-ionone (Nos 388 and 389), which were evaluated by the Committee at its fifty-first meeting (Annex 1, reference 137). The NOELs for these compounds were 10 mg/kg bw per day in a 90-day study in rats, providing a margin of safety of about 200 000. Therefore, 2,6,6-trimethylcyclohexa-1,3-dienyl methanal (No. 977; safranal) would not be a safety concern. |
In the unlikely event that all 26 flavouring agents were to be consumed concurrently on a daily basis, the estimated combined intake would not exceed the human intake threshold for structural class I. Therefore, the combined intake of the agents in this group would not represent a safety concern.
The Committee concluded that none of the 26 flavouring agents in this group of alicyclic primary alcohols, aldehydes, acids and related esters would present a safety concern at the current estimated levels of intake.
The background information summarizes key data relevant to the safety evaluation of 26 alicyclic substituted primary alcohols, aldehydes, acids and related esters used as flavouring agents. Six members of the group are carboxylic acids or related esters of cyclohexane. The esters are rapidly hydrolysed to their component alcohols and cyclohexane carboxylic acids. The cyclohexane carboxylic acids are simple alicyclic substances which are expected to participate in common routes of absorption, distribution and metabolic detoxication and have similar toxicological end-points.
The remaining 20 substances are mono- or bicyclic terpenes with alkyl ring substituents and containing a primary oxygenated group (alcohol, aldehyde or carboxylic acid) or a related ester functional group. Monocyclic terpenoid primary alcohols and monocyclic and bicyclic aldehydes, with alkyl ring substituents, are generally oxidized to the corresponding carboxylic acid, conjugated with glucuronic acid and excreted as urinary metabolites. Reduction of the aldehyde to the alcohol and excretion as glucuronide is a minor pathway. When there is an endocyclic double-bond, the metabolite could be reduced by the action of gut microflora after its excretion via the bile. If the terpene is monocyclic, the resulting acid metabolite may undergo aromatization of the ring to yield a benzoic acid derivative, which may be excreted as a glycine conjugate.
The total annual production volume of the 26 flavouring agents in this group is approximately 170 kg in Europe (Internal Organization of the Flavor Industry, 1995) and 120 kg in the USA (Lucas et al., 1999). The annual volume of use of none of the substances in this group is greater than 55 kg. The production volumes and intakes for each substance are reported in Table 2.
Alicyclic primary alcohols, aldehydes, acids and related esters have been detected mainly in fruits and related alcoholic beverages. They have been reported to be present in mango, rosemary, grapefruit juice, mandarin peel oil, blackberries, vanilla bourbon and rum (Maarse et al., 1999). As shown in Table 2, 13 of the substances in this group have been reported to occur naturally in foods. Quantitative data on natural occurrence and consumption ratios have been reported for para-menth-1-ene-9-al (No. 971), para-mentha-1,8-dien-7-al (No. 973), para-mentha-1,8-dien-7-yl acetate (No. 975), 2,6,6-trimethylcyclohexa-1,3-dienyl methanal (No. 977) and 2,6,6-trimethyl-1 and 2-cyclohexen-1-carboxaldehyde (No. 979). These values indicate that their intake results mainly from traditional foods (i.e. consumption ratio > 1; see Table 2) (Stofberg & Kirschman, 1985; Stofberg & Grundschober, 1987).
In general, aliphatic esters (Nos 962–964, 966, 969, 972, 975, 982, 983 and 985) are rapidly hydrolysed to their component alcohols and carboxylic acids. The hydrolysis is catalysed by classes of enzymes known as carboxylesterases (Heymann, 1980; Graffner-Nordberg et al., 1998), the most important of which are the beta-esterases. Although these enzymes are present in most mammalian tissues, they predominate in hepatocytes (Anders, 1989; Graffner-Nordberg et al., 1998). The substrate specificity of beta-carboxylesterase isoenzymes has been correlated with the structure of the alcohol and acid components (Heymann, 1980). Aliphatic esters used as flavour ingredients hydrolyse rapidly in liver homogenates, simulated pancreatic fluid, simulated gastric fluid and preparations of intestinal mucosa in vitro (Leegwater & van Straten, 1974; Butterworth et al., 1975; Grundschober, 1977; Longland & Gangolli, 1977; Junge & Heymann, 1979; Graffner-Nordberg et al., 1998).
Esters of cyclohexanecarboxylic acid (No. 961), cyclohexaneacetic acid (No. 965) and higher homologues are hydrolysed to the component alcohol and carboxylic acid (Ford & Moran, 1978). Methyl cyclohexanecarboxylate (No. 962) and ethyl cyclohexanecarboxylate (No. 963) were incubated separately with 50 ml of simulated gastric fluid at 37 °C, for 2, 4 or 6 h, and the amounts of unchanged ester and released acid were determined. The results showed approximately 20% hydrolysis of each ester in the gastric system. After a 5-h incubation in simulated intestinal fluid, 40% and 50% of methyl and ethyl cyclohexanecarboxylate were hydrolysed, respectively (Moran & Tyburcy, 1979). In a study of hydrolysis in vitro, 100% of ethyl cyclohexanepropionate (No. 966) was hydrolysed after a 2-h incubation in 5% pancreatin at 37 °C, as demonstrated by determination of the ester and the released alcohol (Grundschober, 1977; Leegwater & van Straten, 1974).
In an investigation of the hydrolysis of the structurally related aliphatic ester, cis/trans-1(7),8-para-menthadienyl acetate1, in rat liver homogenate, menthadienyl acetate was incubated in the homogenate at 37 °C for 15, 30, 60 or 240 min. Complete (100%) hydrolysis to cis/trans-1(7)-8-para-menthadien-2-ol was observed after 60 min, with 92% hydrolysis occurring within the first 15 min and 98% within 30 min (Salzer, 1998). The ready hydrolysis of aliphatic esters in simulated gastric and intestinal fluid supports the conclusion that the ester is hydrolysed extensively in vivo before absorption.
(i) Cyclohexyl derivatives
When sodium cyclohexanecarboxylate (No. 961) with a 14C-labelled ring was administered orally to male Wistar albino rats at a dose of 100 mg/kg bw, > 98% of the original dose was excreted in the urine as hippuric acid metabolites and glucuronic acid conjugates (hippurate, 3,4,5,6-tetrahydrohippurate, hexahydrohippurate, benzoyl- and cyclohexylcarbonylglucuronide). Less than 1% was excreted via the faeces or expired air (Brewster et al., 1977a).
Similar results were obtained when [14C]cyclohexanecarboxylate sodium salt was administered to male Wistar albino rats via a duodenal cannula at a dose of 0.5, 5.0, 35, 100 or 200 mg/kg bw. At the highest dose, 97% was recovered as five urinary (qualitatively the same as after oral dosing) and two biliary metabolites (glucuronide conjugates of cyclohexanecarboxylate and benzoate) within 7 h of dosing. As the dose increased, hippurate excretion decreased, glucuronide excretion increased, excretion of 3,4,5,6-tetrahydrohippurate and hexahydrohippurate in urine increased and biliary excretion increased. At the lowest dose (0.5 mg/kg bw), 95% of the original dose was excreted within the first 90 min (Table 3; Brewster et al., 1977a).
Table 3. Effect of dose on metabolism and excretionof sodium cyclohexanecarboxylate in rats
|
Dose (mg/kg bw) |
Excretion (% of dose after 7 h) |
|||||
|
Bile a |
Urine Benzoyl and cyclohexyl- carbonyl glucuronides |
Hippurate |
3,4,5,6-Tetra hydrohippurate |
Hexahydrohippurate |
Total |
|
|
200 |
8.0 |
18 |
48 |
12 |
10 |
97 |
|
100 |
7.7 |
14 |
56 |
9.9 |
10 |
97 |
|
35 |
3.9 |
6.9 |
70 |
8.6 |
6.1 |
96 |
|
5 |
3.8 |
2.4 |
75 |
9.2 |
6.6 |
97 |
|
0.5 |
3.0 |
1.4 |
81 |
8.1 |
4.9 |
98 |
a Almost entirely as glucuronide conjugates of cyclohexane-carboxylate and benzoate
Dose-dependent clearance of cyclohexanecarboxylic acid and a structural analogue 1-methyl-1-cyclohexanecarboxylic acid has been reported. Female Sprague-Dawley rats were given a single intravenous dose of cyclohexanecarboxylic acid at 67, 130 or 310 mg/kg bw or 1-methyl-1-cyclohexanecarboxylic acid at 74, 150 or 340 mg/kg bw. Blood and urine were collected for 24 and 48 h after dosing, respectively. 1-Methyl-1-cyclohexanecarboxylic acid was eliminated largely via renal excretion as both the parent compound and as a base-labile conjugate (probably the glucuronide), while cyclohexanecarboxylic acid was cleared mostly via other routes, presumably the pathways identified by Brewster et al. (1977a). The plasma and renal clearances of both compounds decreased with increasing dose, suggesting that the pathways by which they are eliminated from the body are saturable (Liu & Pollack, 1993). Further studies on the pharmacokinetics of these compounds indicated that biliary excretion of parent compound and glucuronide conjugate within 5 h accounted for 0.92 and 4.3% of the dose, respectively, for cyclohexanecarboxylic acid and 1.3 and 59% of the dose, respectively, for 1-methyl-1-cyclohexanecarboxylic acid. The data for cyclohexanecarboxylic acid are the most relevant for the substances evaluated in this group. The small amount of cyclohexane carboxylic acid excreted in the bile as the glucuronide conjugate suggests that enterohepatic circulation would be minimal (Liu et al., 1992).
(ii) Terpenoid primary alcohols, aldehydes and carboxylic acids
One of the substances in this group, para-mentha-1,8-dien-7-ol (i.e. perillyl alcohol; No. 974), has been studied for its anticarcinogenic potential in humans. Adult patients with various advanced malignancies were treated three times a day with oral doses of 800, 1600 or 2400 mg/m2, equivalent to approximately 20, 40 and 60 mg/kg bw. The plasma and urinary pharmacokinetics were investigated after the first dose and after 15 and 29 days of treatment. The parent drug was not detected in plasma. The peak plasma concentrations of the two main metabolites of perillyl alcohol were found at 1.5–3.5 h (perillic acid) and 3–5 h (dihydroperillic acid), and their half-lives on the basis of data for day 1 were approximately 2 h. About 9% of the two higher doses given on day 15 or 29 was recovered within 6 h of treatment, of which approximately 90% was perillic acid and 10% was dihydroperillic acid (Ripple et al., 1998).
Studies conducted in humans, dogs, and rats have shown that orally administered perillyl alcohol is rapidly absorbed and metabolized after ingestion. The compound was not detected in plasma samples taken 4 h after a single dose of 1000 mg/kg bw in sesame oil to female Wistar-Furth rats by gavage or after 3, 5 or 10 weeks’ administration at 2% in the diet.. The main metabolites in plasma were perillic acid, dihydroperillic acid and their methyl esters. The ratio of perillic acid to dihydroperillic acid after a single dose by gavage was about 14, whereas this ratio was 2–4 after dietary administration (Haag & Gould, 1994).
The pharmacokinetics of perillyl alcohol in plasma was studied in 28-day studies in rats and dogs. In rats given 600 mg/kg bw per day in two divided doses, the peak plasma concentration was found at 2 h, and the elimination half-time was 4–7 h. The peak concentration of dihydroperillic acid was found at 9 h; the plasma half-life was not given. In dogs given 600 mg/kg bw per day in three divided doses, the peak concentrations of both perillic acid and dihydroperillic acid were found at 2 h, and the elimination half-life of perillic acid was 2.2 h. The peak plasma concentrations of perillic acid were two- to threefold higher in dogs than in rats, whereas the peak concentrations of dihydroperillic acid were six- to sevenfold lower in dogs (National Cancer Institute, 1996).
The peak plasma levels of oxidized metabolites of perillyl alcohol (e.g. perillic acid and dihydroperillic acid) in one male and one female beagle dog given perillyl alcohol at 250 mg/kg bw by gavage were found 1 and 5 h after administration, respectively. The half-lives of perillic acid and dihydroperillic acid appeared to be 3.2 and 3.4 h, respectively. Analysis of blood specimens collected before dosing and 10 min to 48 h after dosing indicated the presence of the oxidized metabolites 10 min after administration. The parent compound, perillyl alcohol, was not detectable in plasma (Phillips et al., 1995).
On the basis of these studies, the cyclohexyl derivatives and terpenoid primary alcohols, aldehydes and carboxylic acids are expected to be rapidly metabolized and excreted, primarily as urinary metabolites.
(i) Cyclohexyl derivatives
Six of the 26 flavouring agents in this group have a cyclohexane ring substituent which contains a primary oxygenated functional group (Nos 961–966). The remaining 20 substances (Nos 967–986) contain a monocyclic or bicyclic terpene ring substituent. Regardless of the ring substituent, metabolism is governed mainly by the presence of a primary oxygenated functional group.
Studies on representative flavouring agents indicated that they are metabolized primarily by oxidation of the primary alcohol or aldehyde function to yield the corresponding carboxylic acid, or oxidation of the alkyl ring substituents to yield polyoxygenated polar metabolites, which are readily excreted. The metabolic options available to alicyclic substances increase as the number and types of functional groups and ring substituents in the molecule increase. If a primary alcohol, aldehyde or carboxylic acid function is present on an alkyl side-chain, the substance may undergo beta-oxidation and subsequent ring cleavage. If the number of carbons in the side-chain is even, beta-oxidative cleavage yields acyclic metabolites, primarily dicarboxylic acids, which presumably enter the citric acid cycle and are completely metabolized. If the number of carbons present in the side-chain is odd, beta-oxidative cleavage leads to aromatization and the production of cyclic metabolites which are excreted primarily in the urine.
Cyclohexanecarboxylic acid, which contains an odd number of carbons in its side-chain, undergoes ring dehydrogenation and aromatization to benzoic acid in vivo before its excretion as the glycine conjugate, hippuric acid (Williams, 1959) (see Figure 1).
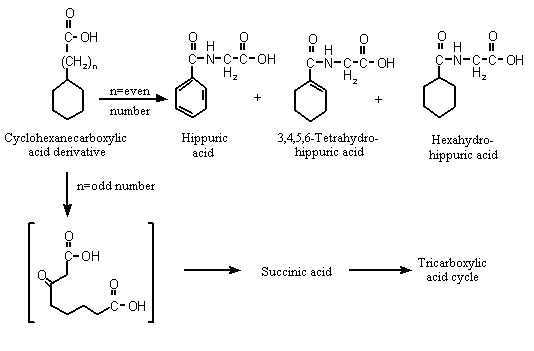
Figure 1. Metabolism of cyclohexanecarboxylic acid derivatives in mammals
Studies with [14C]cyclohexanecarboxylate sodium salt administered intraduo-denally to male Wistar albino rats showed decreasing hippurate excretion with increasing dose but increasing excretion of 3,4,5,6-tetrahydrohippurate and hexahydrohippurate and glucuronide in urine. Biliary excretion of the glucuronic acid conjugates of benzoic acid and cyclohexane carboxylic acid was reported to be dose-related. The authors suggested that the glycine conjugation pathway became saturated as the cyclohexanecarboxylic acid dose increased, resulting in increased excretion of the corresponding glucuronic acid conjugates (Brewster et al., 1977a). These results also confirm the aromatization of cyclohexanecarboxylic acid to yield benzoic acid in vivo (Williams, 1959; Brewster et al., 1977a). In a parallel experiment, male Wistar albino rats given sodium cyclohexanecarboxylic acid with a 14C-labelled ring orally at a dose of 100 mg/kg bw excreted > 98% of the original dose as the urinary metabolites hippurate, 3,4,5,6-tetrahydrohippurate, hexahydrohippurate and the glucuronide conjugates of benzoate and cyclohexanecarboxylate, and < 1% in faeces or expired air (Brewster et al., 1977a).
When 25 mg of [14C]cyclohexanecarboxylic acid were incubated with perfused rat liver preparations for 6 h, 16% cyclohexylcarbonyl-beta-D-glucuronide was excreted in the bile. The remainder was present in the perfusate as unchanged cyclohexane-carboxylic acid (10%), hippuric acid (50%), the glucuronic acid conjugate of cyclohexanecarboxylic acid (2–4%), hexahydrohippuric acid (2%), 3,4,5,6-tetrahydrohippuric acid (2%) and benzoic acid (1–2%). Unlike in the intact animal, no benzoyl glucuronide was detected. All five metabolites in the perfusate were detected by 0.5 h. After 2.5 h, the concentrations of cyclohexylcarbonyl glucuronide, hexahydrohippuric acid, tetrahydrohippuric acid and benzoic acid were at their maximum (10–12%, 4%, 4% and 2%, respectively). The amount of cyclohexyl-carbonyl glucuronide diminished dramatically during the final period of perfusion owing to excretion in the bile (Brewster et al., 1977b).
Two rabbits were given deuterated cyclohexanecarboxylic acid orally at a daily dose of 1000 mg per rabbit for 3 or 4 days, resulting in cumulative intakes of 3000 and 4000 mg per rabbit. Urine collected for 4 days during and after administration contained benzoic acid, representing 27% and 53% of the two doses, respectively. In the same study, two groups of twin male dogs were maintained on a diet containing 1000 mg of ring-deuterated cyclohexanecarboxylic acid per day, resulting in approximate intakes of 77 and 72 mg/kg bw per day for 8 days or 69 and 68 mg/kg bw per day for 4 days. At the end of treatment, urinary analysis revealed benzoic acid concentrations equivalent to 20% and 24% for dogs in the first group and 33% and 25% for those in the second. Also in the same study, four men and two women were given a single dose of 4000 mg of ring-deuterated cyclohexanecarboxylic acid; urine was collected from four persons for 3 days and from the remaining two for 3 days. The concentrations of deuterium-labelled benzoic acid in urine represented 55–91% of the administered dose (Bernhard & Caflisch-Weill, 1945).
As cyclohexaneacetic acid (No. 965) contains an even-numbered carbon side-chain, it is predicted to undergo beta-oxidation and subsequent ring cleavage to yield dicarboxylic acids, which can be completely oxidized via the citric acid cycle (see Figure 1; Williams, 1959). Succinic acid was identified as a urinary metabolite after subcutaneous injection of cyclohexaneacetic acid into a dog, suggesting that ring oxidation and cleavage had occurred. Unchanged cyclohexaneacetic acid was detected in the urine (Bernhard, 1937).
Cyclohexanepropionic acid undergoes oxidation to its homologue cyclohexanecarboxylic acid, which is subsequently aromatized to benzoic acid and excreted mainly as hippuric acid (Williams, 1959).
(ii) Terpenoid primary alcohols, aldehydes and carboxylic acids
In mammals, monocyclic terpenoid primary alcohols (e.g., perillyl alcohol) and monocyclic (e.g., perillaldehyde) and bicyclic (e.g., myrtenal) aldehydes which contain alkyl ring substituents are generally oxidized to the corresponding carboxylic acid, conjugated with glucuronic acid and excreted in urine. In a minor pathway, the aldehyde may be reduced to the alcohol and excreted as the glucuronide (Ishida et al., 1989; Haag & Gould, 1994). If an endocyclic double-bond is present, the metabolite can be reduced by the action of gut microflora, providing it reaches the lower intestinal tract, for example via biliary excretion. If the terpene is monocyclic (e.g., perillyl alcohol), the resulting acid metabolite can undergo aromatization of the ring to yield a benzoic acid derivative, which can be excreted as the glycine conjugate (see Figure 2; Ishida et al., 1989).
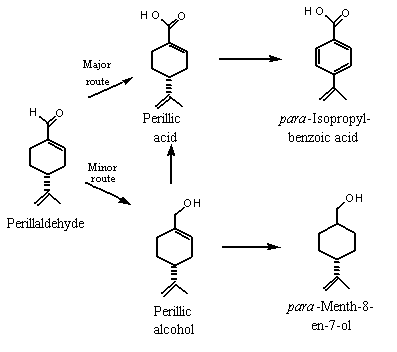
Figure 2. Proposed metabolism of perillaldehyde in mammals
The metabolic fate of the terpenoid aldehydes (Nos 967, 968, 971 and 977–979) can be predicted on the basis of known biotransformations of the representative aldehydes para-mentha-1,8-dien-7-al (i.e., perillaldehyde; No. 973) and 2-formyl-6,6-dimethylbicyclo[3.1.1]hept-2-ene (i.e., myrtenal, No. 980) (Ishida et al., 1989).
Six male rabbits weighing 2.5–3.0 kg were each given 2000 mg of para-mentha-1,8-dien-7-al (perillaldehyde, No. 973) orally, and their urine was collected for 3 consecutive days, pooled, extracted and separated into a neutral fraction (which contained 0.83 g of metabolites) and an acidic fraction which was methylated (which contained 4.6 g of metabolites). (–)-Perillyl alcohol and (–)-cis-shisool (i.e., cyclohexanemethanol, 4-(1-methylethenyl)- or para-menth-8-en-7-ol) represented 46% and 39% of the neutral metabolites, and perillic acid represented 57% of the acidic metabolites detected in the urine. para-Isopropylbenzoic acid was also detected in urine. These results indicate that perillaldehyde was oxidized mainly to para-mentha-1,8-dien-7-carboxylic acid (perillic acid), which was converted in part to para-isopropylbenzoic acid by aromatization of the cyclohexene ring and reduction of the isopropenyl double bond. To a lesser extent, perillaldehyde was reduced to perillyl alcohol, which can be selectively hydrogenated to yield para-mentha-8-en-7-ol (see Figure 2) (Ishida et al., 1989).
A similar study was performed in which six male rabbits were given a total dose of 12 000 mg of 2-formyl-6,6-dimethylbicyclo[3.1.1]hept-2-ene (i.e., (–)-myrtenal; No. 980). The dose was excreted in the urine as myrtenol, cis-10-pinanol (i.e., dihydromyrtenol), myrtenic acid and perillic acid. The urines from the rabbits were pooled, extracted and separated into a neutral fraction (which contained 0.64 g of metabolites) and an acidic fraction which was methylated (which contained 2.9 g of metabolites). Myrtenol and cis-10-pinalol represented 55 and 44% of the neutral extract, respectively, and myrtenic acid represented 76% of the acidic metabolites detected in urine. These results indicate that myrtenal was metabolized mainly to the corresponding carboxylic acid (myrtenic acid), which is presumably excreted as the glucuronic acid conjugate. The presence of perillic acid indicates some cleavage of the strained bicyclic ring. To a lesser extent, the aldehyde can either be reduced to myrtenol (No. 981), which is conjugated with glucuronic acid and excreted, or undergoes hydrogenation of the double-bond to yield dihydromyrtenol, probably via reduction by intestinal microflora (Ishida et al., 1989; Figure 3). Workers exposed to sawmill dust containing myrtenol excreted the glucuronic acid conjugate in their urine (Eriksson & Levin, 1990). Reduction of the aldehyde to the corresponding alcohol may be more important in the metabolism of sterically hindered aldehydes.
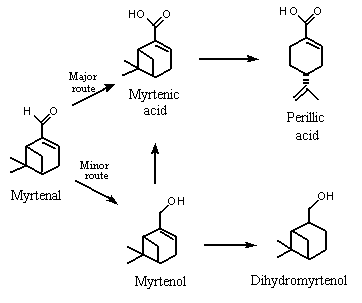
Figure 3. Proposed metabolism of myrtenal in rabbits
The metabolic fate of the terpenoid alcohols (Nos 970, 981 and 986) in this group is expected to be similar to that of the structurally related aldehydes, although there would probably be greater excretion of the alcohol glucuronide and less oxidation to the acid. Data on the biotransformation of para-mentha-1,8-dien-7-ol (i.e. perillyl alcohol, No. 974) and santalol (alpha and beta) (No. 984) support this conclusion (reference not given). Bicyclic and tricyclic compounds (e.g., Nos 980–986) would also undergo microsomal ring oxidation (Hold et al., 2002).
In female Wistar-Furth rats fed a diet of 2% perillyl alcohol for 3, 5 or 10 weeks, perillic acid and dihydroperillic acid were major metabolites in plasma, and the methyl esters of perillic acid and dihydroperillic acid were identified as minor metabolites. The authors concluded that the methyl esters were artefacts formed during processing of the urine. Unchanged perillyl alcohol was not detected. The same plasma metabolites were identified 4 h after female Wistar-Furth rats were given perillyl alcohol at a single dose of 1000 mg/kg bw in sesame oil by gavage. No trace of perillyl alcohol was found at any time, including 15 min after gavage. These results indicate that perillyl alcohol is rapidly metabolized and absorbed from the gastrointestinal tract. The presence of dihydroperillic acid indicated that the endocyclic alkene had been hydrogenated (Haag & Gould, 1994).
A study in male Wistar rats confirmed that the oxidation of perillyl alcohol to perillic acid involves perillaldehyde as an intermediate. The rats were given perillyl alcohol, perillaldehyde or perillic acid intravenously at 80 µmol/kg bw in dimethyl sulfoxide, representing doses of 12, 12 and 13 mg/kg bw, respectively, and urine and bile were collected for 2 h after administration. In all cases, the glucuronic acid conjugate of perillic acid was the predominant metabolite detected in urine and bile. The glucuronic acid conjugate of perillyl alcohol was also a major biliary metabolite after administration of perillyl alcohol. The authors concluded that 56% of the original dose had been oxidized to perillaldehyde within 2 h, followed by conversion to perillic acid and eventually excretion as a glucuronic acid conjugate (Boon et al., 2000).
If the cyclic terpene contains an alkyl substituent of greater chain length than C-3, the primary alcohol, aldehyde, acid or related ester may undergo beta-oxidation and cleavage of the side-chain to yield carboxylic acid metabolites of lower relatuve molecular mass. Elemental analysis of the urinary metabolites of rabbits given repeated oral doses of an alpha,beta-santalol mixture (a tricyclic primary alcohol containing a C-6 alkyl ring substituent; No. 984) showed that a metabolite was formed by loss of an isoprene (C-5) unit from santalol (Hildebrandt, 1902). Structurally related triterpenoid alcohols (farnesol) and ketones (geranylgeranylacetone) undergo beta-oxidation and cleavage to produce chain-shortened polar diacid fragments. For example, C-7, C-9 and C-11 diacid fragments (32%, 15% and 5.3%, respectively, of total radioactivity in urine) were identified in the urine of rats 24 h after an oral dose of [14C]geranylgeranylacetone at 125 mg/kg bw (Nishizawa et al., 1987).
The studies indicate that 19 of the 26 alicyclic substances are expected to be metabolized primarily by oxidation to yield carboxylic acid derivatives that are readily conjugated and excreted primarily in the urine. There are no adequate data on the metabolism of the remaining seven polycyclic terpenoid substances and also no adequate data to demonstrate that their metabolic pathway is similar to that of the monocyclic terpenoids.
Oral LD50 values have been reported for 16 of the 26 substances in this group. The values ranged from 890 to 5700 mg/kg bw for rats and > 1000 to 4000 mg/kg bw for mice, demonstrating that the oral acute toxicity of alicyclic primary alcohols, aldehydes, acids and related esters is low (Table 4).
Table 4. Studies of the acute toxicity of alicyclic primary alcohols, aldehydes, acids and related esters used as flavouring agents
|
No. |
Agent |
Species |
Sex1 |
Route |
LD50 |
Reference |
|
961 |
Cyclohexanecarboxylic acid |
Rat |
M,F |
Gavage |
3300 |
Terrell & Cooper (1978) |
|
M |
Gavage |
3200 |
||||
|
F |
Gavage |
3300 |
||||
|
961 |
Cyclohexanecarboxylic acid |
Rat |
M,F |
Gavage |
3300 |
Moran et al. (1980) |
|
962 |
Methylcyclohexanecarboxylate |
Rat |
M,F |
Gavage |
4000 |
Moran et al. (1980) |
|
963 |
Ethyl cyclohexanecarboxylate |
Rat |
M,F |
Gavage |
4000 |
Moran et al. (1980) |
|
964 |
Cyclohexaneethyl acetate |
Rat |
NR |
Oral |
3200 |
Wohl (1974) |
|
964 |
Cyclohexaneethyl acetate |
Rat |
NR |
Oral |
2200 |
Moreno (1978) |
|
967 |
2,2,3-Trimethylcyclopent-3-en-1-yl acetaldehyde |
Rat |
NR |
Oral |
4,300 |
British Industrial Biological Research Association (1976) |
|
967 |
2,2,3-Trimethylcyclopent-3-en-1-yl acetaldehyde |
Rat |
NR |
Oral |
4,100 |
Moreno (1978) |
|
969 |
Campholene acetate |
Rat |
M |
Gavage |
4600–5300 |
Piccirillo et al. (1979) |
|
F |
Gavage |
3000 |
||||
|
M, F |
Gavage |
3900 |
||||
|
970 |
alpha-Campholene alcohol |
Rat |
NR |
Gavage |
1000–2000 |
Levenstein (1982) |
|
973 |
para-Menth-1,8-dien-7-al |
Mouse |
M |
Gavage |
> 1000 |
Honda et al. (1986) |
|
973 |
para-Menth-1,8-dien-7-al |
Mouse |
NR |
Oral |
1700 |
Moreno (1978) |
|
974 |
para-Menth-1,8-dien-7-ol |
Rat |
NR |
Oral |
2,100 |
Moreno (1977) |
|
976 |
1,2,5,6-Tetrahydrocuminic acid |
Rat |
NR |
Gavage |
> 2500– |
Levenstein (1981) |
|
977 |
2,6,6-Trimethylcyclohexa-1,3-dienyl methanal |
Mouse |
M,F |
Oral |
3500 |
Pellmont (1967) |
|
977 |
2,6,6-Trimethylcyclohexa-1,3-dienyl methanal |
Rat |
NR |
Oral |
5700 |
Pellmont (1969) |
|
979 |
2,6,6-Trimethyl-1 and 2-cyclohexen-1-carboxaldehyde |
Mouse |
NR |
Oral |
4000 |
Pellmont (1973) |
|
980 |
2-Formyl-6,6-dimethylcicyclo[3.1.1]hept-2-ene |
Rat |
NR |
Oral |
2300 |
Moreno (1982) |
|
982 |
Myrtenyl acetate |
Rat |
M,F |
Gavage |
1000–2500 |
Kynoch & Lloyd (1977) |
|
982 |
Myrtenyl acetate |
Rat |
NR |
Oral |
2600 |
Moreno (1982) |
|
983 |
2-Hydroxy-6,6-dimethylbicyclo[3.1.1]hept-2-enyl formate |
Rat |
M,F |
Gavage |
> 5000 |
Moreno (1972) |
|
986 |
10-Hydroxymethylene-2 pinene |
Rat |
NR |
Oral |
890 |
Moreno (1977) |
M, male; F, female; NR, not reported
The results of short-term and long-term studies of toxicity with representative alicyclic primary alcohols, aldehydes, carboxylic acids and related esters are summarized in Table 5 and described below.
Table 5. Results of short-term and long-term studies of toxicity and carcinogenicity on alicyclic primary alcohols, aldehydes, acids, related esters and some related compounds used as flavouring agents
|
No. |
Substance |
Species; sex |
No. test groupsa/ |
Route |
Duration |
NOEL |
Reference |
|
967 |
(2,2,3-Trimethylcyclo-pent-3-en-1-yl)acetaldehyde |
Rat, MF |
1/16 per sex |
Gavage |
90 |
>12 |
British Industrial Biological Research Association (1976) |
|
974 |
para-Mentha-1,8-dien-7-ol |
Rat, MF |
3/NR |
Gavage |
28 |
200 |
National Cancer Institute (1996) |
|
974 |
para-Mentha-1,8-dien-7-ol |
Rat, MF |
3/NR |
Gavage |
90 |
120d |
National Cancer Institute (1996) |
|
|
|
|
|
|
|
< 40e |
|
|
974 |
para-Mentha-1,8-dien-7-ol |
Rat, F |
4/20 |
Diet |
105 |
1300 |
Haag & Gould (1994) |
|
974 |
para-Mentha-1,8-dien-7-ol |
Rat, F |
1/NR |
Diet |
14 |
> 2000 |
Ren & Gould (1998) |
|
974 |
para-Mentha-1,8-dien-7-ol |
Rat, F |
1/26 |
Diet |
70 |
> 2500 |
Haag & Gould (1994) |
|
974 |
para-Mentha-1,8-dien-7-ol |
Rat, M |
1/10 |
Diet |
133 |
> 2000 |
Mills et al. (1995) |
|
974 |
para-Mentha-1,8-dien-7-ol |
Rat, M |
2/36 |
Diet |
365 |
> 2000 |
Reddy et al. (1997) |
|
974 |
para-Mentha-1,8-dien-7-ol |
Hamster, M |
1/NR |
Diet |
42 |
> 1500 |
Stark et al. (1995) |
|
974 |
para-Mentha-1,8-dien-7-ol |
Hamster, M |
1/NR |
Diet |
28 |
> 4000 |
Burke et al. (1997) |
|
974 |
para-Mentha-1,8-dien-7-ol |
Dog, M/F |
3/NR |
Gelati capsule |
28 |
300 |
National Cancer Institute (1996) |
|
974 |
para-Mentha-1,8-dien-7-ol |
Dog, M/F |
3/NR |
Gelatin capsule |
90 |
120 |
National Cancer Institute (1996) |
M, male; F, female; NR, not reported
a Total number of test groups does not include control animals.
b Total number per test group includes both male and female animals.
c > indicates highest dose tested without adverse effect.
d Based on systemic effects
e Based on hyperexcitement and oral discharge
(i) Allyl ester of cyclohexanepropionic acid
Allyl cyclohexanepropionate was evaluated by the Committee at its forty-sixth meeting in a group of allyl esters (Annex 1, reference 122). The Committee concluded that allyl cyclohexanepropionate does not present a safety concern at present levels of intake. It is expected to hydrolyse in vivo to form allyl alcohol and cyclohexane-propionic acid, and the latter would undergo beta-oxidation to form cyclohexane-carboxylic acid (Williams, 1959).
Groups of five male and five female Osborne-Mendel rats were fed diets containing allyl cyclohexanepropionate at a concentration of 1000 or 2500 mg/kg, equivalent to 50 and 125 mg/kg bw per day, for 27–28 weeks or 1 year, respectively. A control group of 10 male and 10 female animals was included. The general health, food intake and weight of the rats were recorded weekly, with no significant difference found between test and control groups. Haematological examinations (haemoglobin, erythrocyte volume fraction, erythrocyte count, leukocyte count) conducted at months 3, 6 and 12 revealed normal values. At termination, the weights of the liver, kidneys, spleen, heart and testes showed no treatment-related changes. Macroscopic and microscopic examination of the liver, kidneys, spleen, heart, testes and remaining abdominal and thoracic viscera and a hind leg (for bone, bone marrow and muscle) revealed no changes that could be associated with administration of the test substance (Hagan et al., 1967).
(ii) (2,2,3-Trimethylcyclo-pent-3-en-1-yl)acetaldehyde (No. 967)
(2,2,3-Trimethylcyclo-pent-3-en-1-yl)acetaldehyde (No. 967) was administered in corn oil to groups of 16 Wistar rats of each sex by gavage for 90 days at a dose of 12 mg/kg bw per day. The vehicle control animals were given corn oil at 5 ml/kg bw per day. The rats were weighed for 6 days before treatment, on the first day of dosing and at various times throughout the study. Food intake was recorded at 3- or 4-day intervals throughout the study. Treated males showed slightly greater weight gain than controls due to greater food intake. The authors cited previous studies (Gaunt et al., 1971; Branton et al., 1972; Gaunt et al., 1974; Butterworth et al., 1975) to attribute the increased food intake to the strong flavour of the test substance. This food consumption pattern was less obvious in the female rats, but, on average, treated females consumed slightly more food than the controls. Haematological examination of eight animals of each sex per group at week 6 and of 16 of each sex per group at week 12 showed no differences in haemoglobin concentration, packed cell volume, erythrocyte or leukocyte count, differential leukocyte count or blood urea nitrogen between treated and control rats. A post-mortem examination was conducted in which the liver and kidneys were weighed and abnormalities noted, and the liver and kidneys of all rats and about 25 tissues from eight animals of each sex per group were examined microscopically. The only difference observed between test and control rats was thickened alveoli in the lungs of treated males. The authors considered that this effect was not due to the test substance (British Industrial Biological Research Association, 1976). The dose of 12 mg/kg bw per day that resulted in no adverse effects is at least 100 000 times the daily per capita intake (‘eaters only’) (see Table 2) of 0.073 µg/kg bw per day from use of (2,2,3-trimethyl-cyclo-pent-3-en-1-yl)acetaldehyde as a flavouring agent in Europe.
(iii) para-Mentha-1,8-dien-7-ol (perillyl alcohol; No. 974)
Numerous studies have shown that high dietary levels of perillyl alcohol given to experimental animals may be protective against known carcinogens. para-Mentha-1,8-dien-7-ol (i.e. perillyl alcohol) is a monoterpene that inhibits the growth of pancreatic, mammary and liver tumours. It has been used in animals as a chemotherapeutic agent for neuroblastoma, prostate and colon cancer and has possible chemotherapeutic applications for skin and lung cancer (Crowell, 1997; Belanger, 1998).
In preclinical studies, nude mice given perillyl alcohol by gavage at a dose of 1000 mg/kg bw in sesame oil had lost weight (3 g) by day 10 of treatment, and two of three mice died on day 13; seven of 10 nude mice bearing HT-29 xenografts that were given 1000 mg/kg bw in sesame oil by gavage died after two doses. Mice given 1800 mg/kg bw per day in two doses became lethargic after three days. Nude mice with PC-3 prostate tumor xenografts that were given 600 or 800 mg/kg bw per day showed early mortality, three of 10 mice at 600 mg/kg bw per day dying after four doses and six of 10 at 800 mg/kg bw per day dying after five doses (National Cancer Institute, 1996).
Fourteen-day range-finding studies were performed with perillyl alcohol in male Fischer 344 rats and male and female beagle dogs. The rats (number of groups and number per group not reported) received doses of 75–900 mg/kg bw per day in soya bean oil by gavage in three divided doses at an interval of 8 h. At the highest dose, the histological lesions included forestomach inflammation with hyperplasia and testicular degeneration. The dogs (number per group not given) received perillylalcohol at a dose of 60, 300, 600 or 1200 mg/kg bw per day in soya bean oil by gelatin capsule in three divided doses. Dose-related increases in the frequency of vomiting and soft or loose stools were observed. At 1200 mg/kg bw per day, leukocytosis and thrombocytosis were also seen (National Cancer Institute, 1996).
Female Wistar-Furth rats received a diet containing 2% perillyl alcohol, corresponding to about 2000 mg/kg per day (Food and Drug Administration, 1993), and pair-fed rats received the control diet of powdered laboratory chow. Two weeks after initiation of the perillyl alcohol diet, 10 µl of [14C]mevalonolactone were injected into the upper four abdominal mammary glands of each rat. Analyses of the mammary epithelial cells 3 h later showed that perillyl alcohol had inhibited coenzyme Q synthesis and protein prenylation in the mammary parenchyma by approximately 20%. Cholesterol synthesis was not affected. No observable signs of toxicity were reported. The authors suggested that inhibition of coenzyme Q synthesis could be involved in the anticancer action of perillyl alcohol (Ren & Gould, 1998). The dose of 2000 mg/kg bw per day that resulted in no adverse effects is 10 000 000 times the daily per capita intake (‘eaters only’) of 0.023 µg/kg bw per day from use of perillyl alcohol as a flavouring agent in Europe.
Twenty-eight-day studies were performed with perillyl alcohol in male and female Fischer 344 rats and male and female beagle dogs. The rats (number per group not reported) received 200, 600 or 1000 mg/kg bw per day in soya bean oil by gavage in two divided doses because of its irritating properties. Animals at the two higher doses showed 12–13% body-weight loss on days 4–10, increased activity of alanine aminotransferase (by 32–79% in females at 600 mg/kg bw per day and in both sexes at 1000 mg/kg bw per day), dose-related forestomach hyperplasia and testicular atrophy. Animals at 1000 mg/kg bw per day showed renal tubular degeneration (with rising blood urea nitrogen) and mortality (10% of males and 50% of females) concomittant with splenic atrophy (females only) and hepatocyte cytoplasmic vacuolization.
The dogs (number per group not reported) received 300, 600 or 1200 mg/kg bw per day in soya bean oil by gelatin capsule in three divided doses. One dog at 1200 mg/kg bw died on day 13 with signs of gastrointestinal distress after consistent low food consumption; mild bone marrow atrophy and lymph node and tissue atrophy were also found. Emesis and abnormal stools were seen at the two higher doses, with dose-related renal tubular degeneration, thymic atrophy and increased blood urea nitrogen values (National Cancer Institute, 1996).
In 90-day studies, perillyl alcohol in in soya bean oil was given at a dose of 40, 120 or 400 mg/kg bw per day by gavage to groups of Fischer 344 rats (number per group not reported) and by capsule to groups of beagle dogs (number per group not reported). The animals were given food and drinking-water ad libitum and monitored for survival, behaviour and haematological changes. In rats, no treatment-related deaths, abnormal haematological or clinical chemical findings or gross lesions were reported at any dose. Significant weight loss (10%) was seen at 400 mg/kg bw per day. The weights of the liver, kidney and lung were increased in females at this dose, but no microscopic changes were observed. The dose-related signs (in all treated groups) included hyperexcitement and a clear oral discharge. No deaths occurred among the dogs. The highest dose was associated with emesis (more severe in females) and mild anaemia. Prothrombin time, activated partial prothrombin time and the albumin:globulin ratio were increased significantly in females at the highest dose, possibly due to severe emesis. Gross necropsy revealed testicular atrophy in one male dog at this dose. The NOEL was 120 mg/kg bw per day (National Cancer Institute, 1996).
d-Limonene, the parent compound of perillyl alcohol, has been reported in published studies to induce hyaline droplet renal nephropathyin adult male rats. The protein reponsible for this lesion has been identified and found to be unique to certain strains. Although the protein is not present in humans, members of the same superfamily have been found; thus, it is possible that this effect may occur in humans. The National Cancer Institute (1996) suggested that renal nephropathy may be induced in rats of each sex at doses greater than 400 mg/kg bw per day.
Groups of 11 male Syrian golden hamsters aged 10 weeks which had been injected with PC-1 pancreatic carcinoma cells were given diets containing 0 or 3% perillyl alcohol, equivalent to 1500 mg/kg bw per day, immediately after palpation of tumours > 2 mm in diameter. After 3 weeks of treatment, perillyl alcohol had significantly reduced the tumour growth rate to less than half that of the controls. Complete regression of tumours was seen in 16% (5/31) of hamsters treated with perillyl alcohol and none of the controsl (0/25). The body-weight gain of the treated hamsters was normal, and no adverse microscopic effects were observed in the liver, kidney or pancreas (Stark et al., 1995).
In a similar study, groups pf 15 male Syrian golden hamsters received a control diet or a diet containing perillyl alcohol at a concentration of 40 000 mg/kg for 4 weeks and then 1 week later a subcutaneous injection of PC-1 hamster pancreatic ductal carcinoma cells. Tumour growth was inhibited by 50% in the group given perillyl acohol. The body weights of the animals were not affected, and the total plasma cholesterol concentrations were normal. Tumour growth was not inhibited at 20 000 mg/kg of diet (Burke et al., 1997).
To study the effect of perillyl alcohol on liver tumours, 21 male Fischer 344 rats were given drinking-water containing 50 ppm N-nitrosodiethylamine for 1 month. Two weeks later, 11 rats were fed a control diet of powdered food and 10 were fed powdered food containing 1% (w/w) perillyl alcohol for 1 week, followed by 2% (w/w) perillyl alcohol, equivalent to 2000 mg/kg bw per day (Food and Drug Administration, 1993), for 18 weeks. At the end of the study, the treated and untreated rats were killed and the livers removed, and total liver weight, tumour weight and number of tumours were recorded. The mean tumour mass per liver was 0.8 g in the treated and 7.0 g in the untreated animals, indicating a 10-fold decrease in the mean liver tumour mass of the perillyl alcohol-treated rats. No change in normal liver weight was observed. The authors determined that perillyl alcohol did not alter the proliferation rate of tumour cells but rather promoted apoptosis, thereby increasing the loss of tumour cells (a fivefold increase in apoptotic index in large tumours and a 10-fold increase in small tumours). The 10% decrease in body weight observed in the treated rats was attributed to a decrease in body fat (Mills et al., 1995).
In a long-term study designed to investigate the chemopreventive effects of perillyl alcohol on colon carcinogenesis, groups of 5-week-old male Fischer 344 rats were fed either a control diet (AIN-76A) or diets providing perillyl alcohol at approximately 1000 or 2000 mg/kg bw per day (Food and Drug Administration, 1993). At weeks 3 and 4 after introduction of the diet, groups of 36 rats intended for treatment with a carcinogen were given weekly subcutaneous injections of azoxymethane at 15 mg/kg bw. Control groups of 12 rats per group maintained on basal diet or a diet containing perillyl alcohol at 2000 mg/kg were injected with an equal volume of saline. The animals were maintained on the control and experimental diets for an additional 52 weeks after the last azoxymethane treatment. The general health of the animals was monitored daily, and body weights were recorded every 2 weeks for the first 10 weeks and then every 4 weeks. At the conclusion of the experiment, gross and histopathological examinations were conducted on the stomach, small intestine and large intestine.
The body weights of control animals maintained on the basal diet were similar to those fed the perillyl alcohol-supplemented diets. Gross examination of liver, kidneys, stomach, lungs and intestines at necroscopy revealed no gross changes in any of the three control groups. The body weights of the azoxymethane-treated rats on perillyl alcohol diets were comparable to those of controls throughout the experiment, and no signs of toxicity were reported at any dose. A significant decrease (p < 0.05) in the incidence and multiplicity of invasive adenocarcinoma of the colon was reported in the animals given 1000 mg/kg of diet, but no effect was seen on tumours in the small intestine. At 2000 mg/kg, the incidence and multiplicity of adenocarcinomas in the small intestine (specifically the invasive type) and the incidence of total (invasive plus noninvasive) adenocarcinomas in the colon were significantly decreased. Animals at both dietary concentrations showed a five- to sixfold increase in the apoptotic index (p < 0.0001) in colon tumours as compared with rats fed control diets. The authors suggested that perillyl alcohol-induced apoptosis inhibits tumour promotion in vivo (Reddy et al., 1997).
Studies on the related monoterpene limonene also give an indication of the potential preventive activity of perillyl alcohol against colon cancer and metastasis of established colon tumours. Limonene is metabolized to perillic acid and dihydroperillic acid, the same metabolites as perillyl alcohol. Two weeks before administration of a single dose of [3H]7,12-dimethylbenz[a]anthracene (DMBA) by gastric intubation, groups of six female Wistar-Furth rats were placed on either a control diet of laboratory chow or a diet containing 5% limonene (w/w in basal diet). Urine was collected for 24 h after intubation of DMBA, and the rats were then killed, blood was collected and DNA was extracted from the liver, lung, kidney and spleen. About 50% less DMBA-associated radioactivity was found in the tissues of the limonene-fed animals than in controls, although the circulating levels of DMBA-associated radioactivity did not differ from those of controls. A 2.3-fold increase in DMBA and/or DMBA-derived metabolites was reported in the urine of the limonene-fed animals. These results suggest that limonene induces modifications in DMBA metabolism, resulting in the formation of more water-soluble DMBA metabolites, which are excreted (Maltzman et al., 1991).
Limonene also significantly inhibited metastasis to the liver (80%) of colonic tumour cells implanted in the spleens of mice (Broitman et al., 1996; Belanger, 1998).
A series of studies was conducted on the effect of perillyl alcohol on rats with mammary tumours induced by DMBA at 50 mg/kg bw. In the first study, groups of 26 female Wistar-Furth rats were fed a diet containing 2.5% perillyl alcohol, equivalent to 2500 mg/kg bw per day (Food and Drug Administration, 1993), for 10 weeks after palpation of the first mammary tumour > 3 mm in diameter. After 3 weeks of treatment, 81% of the primary tumours detected in the perillyl alcohol-treated rats had regressed completely, as compared with 31% that regressed spontaneously in the pair-fed controls. The time required for complete regression was 3 and 9 weeks in the treated and control group, respectively. Perillyl alcohol also prevented the development of secondary tumours after the initial diet assignment. No signs of toxicity were reported. Slight weight loss due to initial food aversion was reported in the rats given the diet containing 2.5% perillyl alcohol. Increased weight gain was reported as food consumption increased, but the control weights were never achieved. Several animals given 3% perillyl alcohol in the diet in this study died (Haag & Gould, 1994).
In the second study, groups of 20 female Wistar-Furth rats were placed on diets containing 0, 0.5, 1, 1.5 or 2% perillyl alcohol for 15 weeks, after palpation of mammary tumours > 10 mm in diameter. The maximum daily intake of perillyl alcohol on the basis of food consumption was calculated to be 0, 410, 880, 1300 and 1600 mg/kg bw, respectively. A 25–75%, dose-related regression (partial plus complete) of primary tumours was observed in the treated rats. A level of 1% in the diet was required for significant (35%) complete regression. No spontaneous regression of primary tumours was observed in the control animals. No observable signs of toxicity were reported at any dose. In all treated groups, some food aversion was seen at the beginning of feeding, which disappeared within 2 weeks. Body-weight gain was slightly depressed at the highest dietary concentration. Interestingly the concentration of perillyl alcohol in the diet that blocked formation of subsequent tumours in rats (1%) was 10 times higher than the concentration (1000 mg/kg of diet) that was chemopreventive in the colon (Haag & Gould, 1994; National Cancer Institute, 1996).
No data were available for the six cyclohexane derivatives. Almost all the studies of monocyclic and polycyclic terpenoids were performed with para-mentha-1,8-dien-7-al (i.e., perillaldehyde), except for one assay for reverse mutation with 2,6,6-trimethylcyclohexa-1,3-dienyl methanal.
(i) In vitro
Alicyclic primary alcohols, aldehydes, acids and related esters: Two substances representative of the monocyclic and bicyclic terpenoid alcohols, aldehydes and carboxylic acids, namely 2,6,6-trimethylcyclohexa-1,3-dienyl methanal and para-mentha-1,8-dien-7-al (i.e., perillaldehyde), have been tested for genotoxicity. Negative results were reported in standard (Florin et al., 1980) assays for reverse mutation when Salmonella typhimurium strains TA98, TA100, TA1535 and TA1537 were incubated with 3 µmol of 2,6,6-trimethylcyclohexa-1,3-dienyl methanal/plate (No. 977) with and without metabolic activation. In modified assays for reverse mutation (Ishidate et al., 1984; Fujita et al., 1994), S. typhimurium strains TA92, TA94, TA97, TA98, TA100, TA102, TA1535, TA1537 and TA2637 were incubated with up to 1000 µg of para-mentha-1,8-dien-7-al (i.e., perillaldehyde) (No. 973), with and without metabolic activation. Negative results were reported (see Table 6).
Table 6. Studies of genotoxicity with alicyclic primary alcohols, aldehydes, acids and related esters used as flavouring agents
|
No. |
Agent |
End-point |
Test object |
Maximum concentration |
Results |
Reference |
|
In vitro |
|
|
|
|
|
|
|
973 |
para-Mentha-1,8-dien- 7-al |
DNA damage |
Bacillus subtilis M45 and H17 |
2.5 µl/disc |
Weak positive |
Yoo (1986) |
|
973 |
para-Mentha-1,8-dien- 7-al |
DNA damage |
Bacillus subtilis M45 and H17 |
0.63 µl |
Negative |
Kuroda et al. (1984) |
|
973 |
para-Mentha-1,8-dien- 7-al |
DNA damage |
Bacillus subtilis M45 and H17 |
1.25 µl |
Positive |
Kuroda et al. (1984) |
|
973 |
para-Mentha-1,8-dien- 7-al |
Reverse mutationa |
S. typhimurium TA97, TA102 |
100 µg/plate |
Negativeb |
Fujita et al. (1994) |
|
973 |
para-Mentha-1,8-dien- 7-al |
Reverse mutationa |
S. typhimurium TA92, TA1535, TA100, TA1537, TA94, TA98, TA2637 |
1000 µg/plate |
Negativeb |
Ishidate et al. (1984) |
|
977 |
2,6,6-Trimethylcyclohexa-1,3-dienyl methanal |
Reverse mutation |
S. typhimurium TA98, TA100, TA1535, TA1537 |
3 µmol/plate |
Negativeb |
Florin et al. (1980) |
|
973 |
para-Mentha-1,8-dien- 7-al |
DNA repair |
Escherichia coli PQ37 |
Not reported |
Negative |
Eder et al. (1993) |
|
973 |
para-Mentha-1,8-dien- 7-al |
Sister chromatid exchange |
Chinese hamster ovary cells |
200 µg/ml |
Positivec,d |
Tayama et al. (1990) |
|
973 |
para-Mentha-1,8-dien- 7-al |
Sister chromatid exchange |
Chinese hamster ovary cells |
300 µg/ml |
Positived,e |
Tayama et al. (1990) |
|
973 |
para-Mentha-1,8-dien- 7-al |
Chromosomal aberration |
Chinese hamster fibroblasts |
50 µg/ml |
Positivec |
Ishidate et al. (1984) |
|
973 |
para-Mentha-1,8-dien- 7-al |
Chromosomal aberration |
Chinese hamster ovary cells |
300 µg/ml |
Negatived,e,f |
Tayama et al. (1990) |
|
973 |
para-Mentha-1,8-dien- 7-al |
Chromosomal aberration |
Chinese hamster ovary cells |
200 µg/ml |
Negativec,d,f |
Tayama et al. (1990) |
|
973 |
para-Mentha-1,8-dien- 7-al |
Mutagenicity |
Chinese hamster ovary cells |
100 µg/ml |
Negativec,g |
Sasaki et al. (1990) |
|
973 |
para-Mentha-1,8-dien- 7-al |
Mutagenicity |
Human fetus cells (Rsa) |
0.025 µg/ml |
Positiveh |
Suzuki et al. (1990) |
|
973 |
para-Mentha-1,8-dien- 7-al |
Mutagenicity |
Human fetus cells (Rsa) |
0.010 µg/ml |
Negativeh |
Suzuki et al. (1990) |
|
973 |
para-Mentha-1,8-dien- 7-al |
Mutagenicity |
Human fetus cells (Rsa) |
200 ng/ml |
Positivei |
Suzuki & Suzuki (1994) |
|
973 |
para-Mentha-1,8-dien- 7-al |
Mutagenicity |
E. coli WP2 |
0.4 mg/plate |
Negative |
Yoo (1986) |
|
In vivo |
|
|
|
|
|
|
|
973 |
para-Mentha-1,8-dien- 7-al |
Micronucleus formation |
Mouse bone marrow erythrocytes |
600 mg/kg bw |
Negativej |
Hayashi et al. (1988) |
a Preincubation with exogenous metanolic system from rat liver
b Assay performed with and without metabolic activation
c Assay performed without metabolic activation
d Cytotoxic at 150 µg/ml
e Assay performed with metabolic activation
f Positive only at cytotoxic concentrations
g Cytotoxic at 12 µg/ml
h Cytotoxic at 0.025 µg/ml
i Cytotoxic at > 20 ng/ml
j Administered byintraperitoneal injection
para-Mentha-1,8-dien-7-al was considered to be weakly positive in an assay for DNA damage in Bacillus subtilis strains M45 (rec+) and H17 (rec–) at a concentration of 2.5 µl/disc (Yoo, 1986). Negative results were reported in a similar study at concentrations of 0.16–0.63 µl/plate, but positive results were found at higher concentrations (1.2–2.5 µl/plate) (Kuroda et al., 1984). Negative results were reported in tests in which para-mentha-1,8-dien-7-al was incubated with Escherichia coli WP2 cells at 0.05–0.4 mg/plate (Yoo, 1986).
The SOS Chromotest for DNA damage in genetically engineered E. coli PQ 37 gave negative results for para-mentha-1,8-dien-7-al (Eder et al., 1993). The maximum induction factor was calculated to be 1.0 (Positive results are considered to be significant if the maximum induction factor is at least 1.5). Standard assays for sister chromatid exchange and chromosomal aberrations with para-mentha-1,8-dien-7-al yielded equivocal results. An assay for sister chromatid exchange performed with and without metabolic activation in Chinese hamster ovary cells (CHO-K1), exposed for 3 h (expression time, 27 h) to concentrations of 50–300 and 50–100 µg, respectively, yielded positive results for genotoxicity; cytotoxicity was reported at 150 and 300 µg/ml without and with metabolic activation, respectively (Tayama et al., 1990). In another assay conducted in CHO-K1 cells, cytotoxicity was reported at concentrations > 12 µg/ml, and negative results were seen at 10 µg/ml. para-Mentha-1,8-dien-7-al may have enhanced N-methyl-N’-nitro-N-nitrosoguanine-induced ouabain-resistant mutations at 10 µg/ml (Sasaki et al., 1990). Chromosomal aberrations were reported to be induced by para-mentha-1,8-dien-7-al in Chinese hamster fibroblast cells after 24 and 48 h of exposure to concentrations of 40 and 50 µg/ml without metabolic activation. Polyploidy was observed in this study (Ishidate et al., 1984). No chromosomal aberrations were induced in CHO-K1 cells after 3 h of exposure (expression time, 27 h) to non-cytotoxic concentrations of 50–100 µg/ml without metabolic activation. At the cytotoxic concentration of 150 µg/ml, induction of chromosomal aberrations was observed without metabolic activation. With metabolic activation, concentrations of 50–200 µg/ml did not induce chromosomal aberrations, whereas the cytotoxic concentration of 300 µg/ml did (Tayama et al., 1990).
Studies of mutagenicity in human fetus cells (Rsa) exposed to a concentration of 0, 0.010, 0.015, 0.02 or 0.025 µg/ml gave negative results for para-mentha-1,8-dien-7-al at the lowest concentration and positive results at concentrations of 0.015 and 0.02 µg/ml. At the highest concentration of 0.025 µg/ml, the substance was cytotoxic (Suzuki et al., 1990). In another study with Rsa cells, mutagenicity, determined by ouabain resistance, was reported at concentrations of para-mentha-1,8-dien-7-al > 10 ng/ml, with apparent cytotoxicity at >20 ng/ml. In this study, mutagenicity was detected in K-ras codons at concentrations of 2–200 ng/ml (Suzuki & Suzuki, 1994).
(ii) In vivo
In an assay for micronucleus formation, groups of six male ddY mice aged 8 weeks were given a single intraperitoneal injection of para-mentha-1,8-dien-7-al at a dose of 0, 75, 150, 300 or 600 mg/kg bw in olive oil. A positive control group received mitomycin C. After 24 h, the mice were killed, and femoral bone-marrow cells were collected, fixed and stained with Giemsa. No indication of micronucleus induction was reported at any dose.
One group of five mice received intraperitoneal injections of 200 mg/kg bw per day for 4 days. No induction of micronuclei was observed 24 h after the last dose, and the percentage of polychromatic erythrocytes was not affected at any dose (Hayashi et al., 1988).
(iii) Conclusion
The relevance to human health of positive results in Chinese hamster ovary cells in vitro is questionable, for the following reasons:
2,6,6-Trimethylcyclohexa-1,3-dienyl methanal and para-mentha-1,8-dien-7-al did not induce reverse mutation. Mixed results were reported with para-mentha-1,8-dien-7-al in assays for DNA damage, sister chromatid exchange, chromosomal aberration and other geneotoxic end-points. As these substances are rapidly metabolized in vivo to compounds of lower toxicological potential, the Committee concluded that the monocyclic and bicyclic terpenes with alkyl ring substituents and containing an alcohol, aldehyde or carboxylic acid group would have little genotoxic potential in vivo.
A number of effects that did not contribute to the safety evaluation have been reported in studies on substances in this group. Increased hexobarbital sleeping time, lowered rectal temperature, diminished spontaneous locomotor activity and reduced acetic acid-induced writhing were reported in male ddY mice given a single oral dose of 100 mg or more of alpha- or beta-santalol (Okugawa et al., 1995).
A dose-related decrease in cell proliferation was reported in human and hamster pancreatic tumour cell lines treated with perillyl alcohol. A concentration-dependent increase in apoptosis was reported in rat phaeochromocytoma cells exposed to perillyl alcohol or perillaldehyde at a concentration of 500 and 200 mol/l, respectively (Boon et al., 2000).
The number of mutations induced by N-methyl-N’-nitro-N-nitrosoguanidine in CHO-K1 cells was reduced by treatment with perillaldehyde at 15 µg/ml (Sasaki et al., 1990). In the same study, perillaldehyde at 50 µg/ml showed immunosuppressive effects in mouse spleen cells.
A phase I clinical trial was conducted with perillyl alcohol in patients with advanced malignancies. The substance was administered orally in gelatin capsules, three times a day, at a dose of 800, 1600 or 2400 mg/m2; two, four and three patients were treated at these doses for 3 months or more. Episodes of gastrointestinal symptoms (such as nausea, unpleasant taste and satiety) and fatigue were reported. Two patients with ovarian cancer showed reversible granulocytopenia. There was no evidence of renal, hepatic or neurological toxicity (Ripple et al., 1998).
The phase I clinical trial was repeated with smaller, more frequent doses to reduce the gastrointestinal effects. Sixteen patients with advanced refractory malignancies were treated orally with perillyl alcohol in soya bean oil at a dose of 800, 1200 or 1600 mg/m2 four times a day. Mild gastrointestinal effects were observed (mainly at 1200 and 1600 mg/m2), and again there was no evidence of renal, hepatic or neurological toxicity (Ripple et al., 2000).
Nausea, vomiting, loss of appetite, weight loss, fatigue, short-term memory loss, slurred speech and loss of balance were reported in further phase I trials in which patients with cancer were given perillyl alcohol orally at a dose of 2100 or 2800 mg/m2. Similar signs of toxicity were not observed in the patients given a lower dose of 1600 mg/m2 (Belanger, 1998).
Anders, M.W. (1989) Biotransformation and bioactivation of xenobiotics by the kidney. In: Huston, D.H., Caldwell, J. & Paulson, G. D., eds, Intermediary Xenobiotic Metabolism in Animals, New York: Taylor & Francis, pp. 81–97.
Belanger, J.T. (1998) Perillyl alcohol: Applications in oncology. Altern. Med. Rev., 3, 448–457.
Bernhard, K. (1937) Metabolic hydrogenation of the cyclohexane ring. Hoppe-Seyler’s Z. Physiol. Chem., 248, 256–276.
Bernhard, K. & Caflisch-Weill, H. (1945) On the dehydrogenation of hexahydro-benzoic acid in the animal body. Helv. Chim. Acta, 28, 1697–1707.
Boon, P.J.M., van der Boon, D. & Mulder, G.J., (2000) Cytotoxicity and biotransformation of the anticancer drug perillyl alcohol in PC12 cells and in the rat. Toxicol. Appl. Pharmacol., 167, 55–62.
Bradley, M.O., Taylor, V.I., Armstrong, M.J. & Galloway, S.M. (1987) Relationships among cytotoxicity, lysosomal breakdown, chromosome aberrations, and DNA double-strand breaks. Mutat. Res., 189, 69–79.
Brantom, P.G., Gaunt, I.F., Grasso, P. & Lansdown, A.B.G. (1972) Short-term toxicity of tolualdehyde in rats. Food Cosmet. Toxicol., 10, 637–647.
Brewster, D., Jones, R.S. & Parke, D.V. (1977a) The metabolism of cyclohexanecarboxylic acid in the isolated perfused rat liver. Xenobiotica, 7, 601–609.
Brewster, D., Jones, R.S. & Parke, D.V. (1977b) The metabolism of cyclohexanecarboxylic acid in the rat. Biochem. J., 164, 595–600.
British Industrial Biological Research Association (1976) Short-term toxicity of samples TT171, TT172, TT173, and TT174 in rats. Unpublished report. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Broitman, S.A, Wilkinson, J., 4th, Cerda, S. & Branch, S.K. (1996) Effects of monoterpenes and mevinolin on murine colon tumor CT-26 in vitro and its hepatic ‘metastases’ in vivo. Adv. Exp. Med. Biol., 401, 111–130.
Brusick, D. (1986) Genotoxic effects in cultured mammalian cells produced by low pH treatment conditions and increased Ion concentrations. Environ. Mutag., 8, 879–886.
Burke, Y.D., Stark, M.J., Roach, S.L.., Sen, S.E. & Crowell, P.L (1997) Inhibition of pancreatic cancer growth by the dietary isopernoids farnesol and geraniol. Lipids, 32, 151–156.
Butterworth, K.B., Carponini, F:M.B., Gaunt, P.F., Grasso, P. & Lloyd, A.G. (1975) A New Approach to the Evaluation of the Safety of Flavouring Esters, Carshalton, Surrey: British Industrial Biological Research Association.
Cramer, G.M., Ford, R.A. & Hall, R.L. (1978) Estimation of toxic hazard—A decision tree approach. Food Cosmet. Toxicol., 16, 255–276.
Crowell, P.L. (1997) Monoterpenes in breast cancer chemoprevention. Breast Cancer Res. Treat., 46, 191–197.
Eder, E., Scheckenbach, S., Deininger, C. & Hoffman, C. (1993) The possible role of alpha,beta-unsaturated carbonyl compounds in mutagenesis and carcinogenesis. Toxicol. Lett., 67, 87–103.
Eriksson, K. & Levin, J.-O. (1990) Identification of cis- and trans- verbenol in human urine after occupational exposure to terpenes. Int. Arch. Occup. Environ. Health, 62, 379–383.
Florin, I., Rutberg, L., Curvall, M. & Enzell, C.R. (1980) Screening of tobacco smoke constituents for mutagenicity using the Ames’ test. Toxicology, 18, 219–232.
Food and Drug Administration (1993) Priority-based Assessment of Food Additives (PAFA) Database, Washington DC: Center for Food Safety and Applied Nutrition, p. 58.
Ford, D.M. & Moran, E.J. (1978) Preliminary indications of in vitro hydrolysis of two flavor chemical esters. Private communication. Submiited to WHO by the Flavor and Extract Manufacturers Association.
Fujita, H., Aoki, N. & Sasaki, M. (1994) Mutagenicity test of food additives with Salmonella typhirurium TA97 and TA102 (IX*). Ann. Rep. Tokyo Metr. Res. Lab. Public Health, 45, 191–199 (in Japanese; abstract and tables in English).
Gaunt, I.F., Agrelo, C.E., Colley, J., Lansdown, A.B.G. & Grasso, P. (1971) Short-term toxicity of isobornyl acetate in rats. Food Cosmet. Toxicol., 9, 355–366.
Gaunt, I.F., Mason, P.L., Hardy, J., Lansdown, A.B.G. & Gangolli, S.D. (1974) Short-term toxicity of methylpheylcarbinyl acetate in rats. Food Chem. Toxicol., 12, 185–194.
Graffner-Nordberg, M., Karin, S., Anders, T. & Anders, H. (1998) Synthesis and enzymie hydrolysis of esters, constituting simple models of soft drugs. Chem. Pharm. Bull., 46, 591–601.
Grundschober, F. (1977) Toxicological assessment of flavoring esters. Toxicology, 8, 387–390.
Haag, J.D. & Gould, M.N. (1994) Mammary carcinoma regression induced by perillyl alcohol, a hydroxylated analog of limonene. Cancer Chemother. Pharmacol., 34, 477–483.
Hagan, E.C., Hansen, W.H., Fitzhugh, O.G., Jenner, P.M., Jones, W.I. & Taylor, J.M. (1967) Food flavourings and compounds of related structure. II. Subacute and chronic toxicity. Food Cosmet. Toxicol., 5, 141–157.
Hayashi, M., Kishi, M., Sofuni, T. & Ishidate, M., Jr (1988) Micronucleus tests in mice on 39 food additives and eight miscellaneous chemicals. Food Chem. Toxicol., 26, 487–500.
He, L., Huanbiao, M., Hadisusilo, S., Qureshi, A.A. & Elson, C.E. (1997) Isoprenoids suppress the growth of murine B16 melanomas in vitro and in vivo. J. Nutr., 127, 668–674
Heck, J.D., Vollmuth, T.A., Cifone, M.A. Jagannath, D.R., Myhr, B. & Curren, R.D. (1989) An evaluation of food flavoring ingredients in a genetic toxicity screening battery. Toxicologist, 9, 206.
Heymann, E. (1980) Carboxlesterases and amidases. In: Jakoby, W.B., ed., Enzymatic Basics of Detoxication, 2nd Ed., New York: Academic Press, pp. 291–323.
Hildebrandt, H. (1902) On the behavior of carvone and santalol in the animal body. Hoppe Seyler’s Z. Physiol. Chem., 36, 452–461.
Hold, K.M., Sirisoma, N.S., Sparks, S.E. & Casida, J.E. (2002) Metabolism and mode of action of cis- and trans-3-pinanones (the active ingredients of hyssop oil). Xenobiotica, 32, 251–265.
Honda, G., Koeauka, Y., Kamisako, W. & Tabata, M. (1986) Isolation of sedative principles from Perilla frutescens. Chem. Pharm. Bull., 34, 1672–1677.
International Organization of the Flavor Industry (1995) European inquiry on volume of use. Unpublished report. Submitted to WHO by the Flavor and Extract Manufacturers Association
Ishida, T., Toyota, M. & Asakawa, Y.(1989) Terpenoid biotransformation in mammals. V. Metabolism of (+)-citronellal, (±)-citronellal, (±)-7-hydroxycitronellal, citral, (–)-perillaldehyde, (–)-myrtenal, cuminaldehyde, thujone, and (±)-carvone in rabbits. Xenobiotica, 19, 843–855.
Ishidate, M., Jr, Sofuni, T., Yoshikawa, K., Hayashi, M., Nohmi, T., Sawada, M. & Matsouoka, A. (1984) Primary mutagenicity screening of food additives currently used in Japan. Food Chem. Toxicol., 22, 623–636.
Junge, W. & Heymann, E. (1979) Characterization of the isoenzymes of pig-liver esterase. Eur. J. Biochem., 95, 519–525.
Kynoch, S.R. & Lloyd, G.K. (1977) Acute oral toxicity to rats of myrtenyl acetate. Unpublished report. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Kuroda, K., Setsuko, I., Young, S. & Takeji, Y. (1984) Rec-assay of food additives. Nippon Kosnu Eisei Zassni, 31, 277–281 (in Japanese; abstract and tables in English).
Leegwater, D.C. & van Straten, S. (1974) In vitro study on the hydrolysis of twenty-six organic esters by pancreatin. Unpublished report. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Levenstein, I. (1981) Oral toxicity testing of 1,2,5,6-tetrahydrocuminic acid in rats. Unpublished report. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Levenstein, I. (1982a) Acute oral, LD50 of alpha-campholenic alcohol in rats. Unpublished report. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Liu, M.-J. & Pollack, G.M. (1993) Pharmacokinetics and pharmacodynamics of valproate analogs in rats. II. Pharmacokinetics of octanoic acid, cyclohexanecarboxylic acid, and 1-methyl-1-cyclohexanecrboxylic acid. Biopharm. Drug Disposition, 14, 325–339.
Liu, M.-J., Brouwer, K.L.R. & Pollack, G.M. (1992) Pharmacokinetics and pharmacodynamics of valproate analogs in rats. III. Pharmacokinetics of valproic acid, cyclohexanecarboxylic acid, and 1-methyl-1-cyclohexanecarboxylic acid in the bile-exteriorized rat. Drug Metab. Disposition, 20, 810–815.
Longland, R.C. & Gangolli, S.D. (1977) The hydrolysis of flavouring esters by artificial gastrointestinal juices and rat tissue preparations. Toxicology, 8, 197–204.
Lucas, C.D., Putnam, J.M. & Hallagan, J.B. (1999) Flavor and Extract Manufacturers Association of the United States 1995 poundage and technical effects update survey. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Maarse, H., Visscher, C.A., Willemsens, L.C. & Boelens, M.H. (1999) Volatile Components in Food—Qualitative and Quantitative Data, Zeist: Centraal Instituut Voor Voedingsonderzioek TNO.
Maltzman, T.H., Christou, M., Gould, M.N. & Jefcoate, C.R. (1991) Effects of monoterpenoids on in vivo DMBA-DNA adduct formation and on phase I hepatic metabolising enzymes. Carcinogenesis, 12, 2081–2087.
Mills, J.J., Chari, R.S., Boyer, I.J., Gould, M.N. & Jirtle, R.L. (1995) Induction of apoptosis in liver tumors by the monoterpene perillyl alcohol. Cancer Res., 55, 979–983.
Moran, E.J., Esterday, O.D. & Oser, B.L. (1980) Acute oral toxicity of selected flavor chemicals. Drug Chem. Toxicol., 3, 249–258.
Moran, E.J. & Tyburcy, K.R. (1979) In vitro hydrolysis of flavoring esters in a simulated gastric system and a simulated intestinal system. Unpublished report. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Moreno, O.M. (1972) Acute toxicity studies in mice, rats and rabbits (2-hydroxymethyl-6,6-dimethylbicyclo[3.1.1]hept-2-enylformate). Unpublished report to RIFM. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Moreno, O.M. (1977) Acute oral toxicity in rats and dermal toxicity in rabbits (p-menth-1,8-dien-7-ol and 10-hydroxymethylene-2-pinene). Unpublished report to RIFM. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Moreno, O.M. (1978) Acute oral toxicity in rats and acute dermal toxicity in rabbits (cyclohexaneethyl acetate; 2,2,3-trimethylcyclo-pent-3-en-1-ylacetaldehyde; p-menth-1,8-dien-7-al). Unpublished report to RIFM. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Moreno, O.M. (1982) Acute oral toxicity in rats and acute dermal toxicity in rabbits (myrtenal; myrtenal acetate). Unpublished report to RIFM. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Naarden, Inc. (1974) In vitro study on the hydrolysis of twenty-six organic esters by pancreatin. Unpublished report. Submitted to WHO by the Flavor and Extract Manufacturers Association.
National Cancer Institute (1996) Clinical development plan: l—Perillyl alcohol. J. Cell. Biochem., 265, 137–148.
Nishizawa, Y., Abe, S., Yamada, K., Nakamura, T., Yamatsu, I. & Kinoshita, K. (1987) Identification of urinary and microsomal metabolites of geranylgeranylacetone in rats. Xenobiotica, 17, 575–584.
Okugawa, H., Ueda, R., Matsumoto, K., Kawanishi, K. & Kato, A. (1995) Effects of alpha-santalol and beta-santolol from sandalwood on the central nervous system in mice. Phytomedicine, 2, 119–126.
Ohta, T., Watanabe, M., Watanabe, K. & Shirasu, Y. (1986) Inhibitory effects of flavourings on mutagenesis induced by chemicals in bacteria. Food Chem. Toxicol., 24, 51–54.
Pellmont (1967) Acute toxicity in mice with 2,6,6-trimethylcyclohexa-1,3-dienyl methanal. Unpublished report. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Pellmont (1969) Acute toxicity in rats with 2,6,6-trimethylcyclohexa-1,3-dienyl methanal. Unpublished report. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Pellmont (1973) Acute oral toxicity in mice with cyclocitral. Unpublished report. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Phillips, L.R., Malspeis, L. & Supko, J.G. (1995) Pharmacokinetics of active drug metabolites after oral administration of perillyl alcohol, an investigational antineoplastic agent, to the dog. Drug Metab. Disposition, 23, 676–680.
Piccirillo, V.J., Leatherwood, L., Swidersky, P. & Shopp, G. (1979) Acute oral LD50 determination of campholene acetate in rats. Unpublished report. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Reddy, B.S., Wang, C.X., Samaha, H., Lubet, R., Steele, V.E., Kelloff, G.J. & Rao, C.V. (1997) Chemoprevention of colon carcinogenesis by dietary perillyl alcohol. Cancer Res., 57, 420–425.
Ren, Z. & Gould, M.N. (1998) Modulation of small G protein isoprenylation by anticancer monoterpenes in in situ mammary gland epithelial cells. Carcinogenesis, 19, 827–832.
Ripple, G.H., Gould, M.N.,Arzoomanian, R.Z., Alberti, D., Feierabend, C., Simon, K., Binger, K., Tutsch, K.D., Pomplun, M., Wahamaki, A., Marnocha, R., Wilding, G. & Bailey, H.H. (1998) Phase I clinical trial of perillyl alcohol administered daily. Clin. Cancer Res., 4, 1159–1164.
Ripple, G.H., Gould, M.N., Stewart, J.A., Tutsch, K.D., Arzoomanian, R.Z., Alberti, D., Feierabend, C., Pomplun, M., Wilding, G. & Bailey, H.H. (2000) Phase I clinical and pharmacokinetic study of perillyl alcohol administered four times a day. Clin. Cancer Res., 6, 390–396.
Salzer (1998) In vitro hydrolysis test. Unpublished report. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Sasaki, M., Yoshida, S., Ichikawa, H., Takano, I., Yasuda, I. & Akiyama, K. (1990) Cytological activities of perilla extracts. Ann. Rep. Tokyo Metr. Res. Lab. Public Health, 41, 275–281 (in Japanese; abstract and tables in English).
Stark, M.J., Burke, Y.D., McKinzie, J.H., Ayoubi, A.S. & Crowell, P.L. (1995) Chemotherapy of pancreatic cancer with the monoterpene perillyl alcohol. Cancer Lett., 96, 15–21.
Stofberg, J. & Grundschober, F. (1987) Consumption ratio and food predominance of flavoring materials. Perfumer Flavorist, 12, 27.
Stofberg, J. & Kirschman, J.C. (1985) The consumption ratio of flavoring materials: A mechanism for setting priorities for safety evaluation. Food Chem. Toxicol., 23, 857–860.
Suzuki, N. & Suzuki, H. (1994) Induction of K-ras codon 12 mutation by saccharin sodium with perillaldehyde-induced mutagenicity. Hen’igensei Shiken, 3, 155–160 (in Japanese; abstract and tables in English).
Suzuki, H., Ichikawa, H. & Sasaki, M. (1990) Mutagenicity of perillaldehyde in a human cell strain. Ann. Rep. Tokyo Metr. Res. Lab. Public Health, 41, 265–266 (in Japanese; abstract and tables in English).
Tayama, S., Ichikawa, H. & Sasaki, M. (1990) Effects of perillaldehyde on induction of chromosomal aberrations and sister-chromatid exchanges (SCEs) in CHO-KI-cells. Ann. Rep. Tokyo Metr. Res. Lab. Public Health, 41, 267–269 (in Japanese; abstract and tables in English).
Terrell & Cooper (1978) Unpublished report. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Williams, R.T. (1959) Detoxication Mechanisms. The Metabolism and Detoxication of Drugs, Toxic Substances, and Other Organic Compounds, 2nd Ed., London: Chapman & Hall, pp. 119–120.
Wohl, A. (1974) Acute oral and dermal toxicity studies (cyclohexyl ethyl acetate). Report to RIFM. Unpublished report. Submitted to WHO by the Flavor and Extract Manufacturers Association.
Yoo, Y.S. (1986) Mutagenic and antimutagenic activities of flavoring agents used in foodstuffs. J. Osaka City Med. Center, 34, 267–288 (in Japanese; abstract and tables in English).
Zajac-Kaye, M. & Ts’O, P.O.P. (1984) DNAase I encapsulated in liposomes can induce neoplastic transformation of Syrian hamster embryo cells in culture. Cell, 39, 427–437.
ENDNOTES:
See Also:
Toxicological Abbreviations