
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organization or the World Health Organization.
Environmental Health Criteria 229
First draft prepared by Drs J. Kielhorn, U. Wahnschaffe and I. Mangelsdorf, Fraunhofer Institute of Toxicology and Aerosol Research, Hanover, Germany
Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organization and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals.
World Health Organization
Geneva, 2003
The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organization (ILO) and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals.
The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and Agriculture Organization of the United Nations, WHO, the United Nations Industrial Development Organization, the United Nations Institute for Training and Research and the Organisation for Economic Co-operation and Development (Participating Organizations), following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase coordination in the field of chemical safety. The purpose of the IOMC is to promote coordination of the policies and activities pursued by the Participating Organizations, jointly or separately, to achieve the sound management of chemicals in relation to human health and the environment.
WHO Library Cataloguing-in-Publication Data
Selected nitro- and nitro-oxy-polycyclic aromatic hydrocarbons.
(Environmental health criteria ; 229)
1.Polycyclic hydrocarbons, Aromatic - toxicity 2.Polycyclic hydrocarbons,
Aromatic - adverse effects 3.Environmental exposure 4.Risk assessment
I.International Programme for Chemical Safety II.Series
ISBN 92 4 157229 9 (NLM classification: QD 341.H9)
ISSN 0250-863X
The World Health Organization welcomes requests for permission to reproduce or translate its publications, in part or in full. Applications and enquiries should be addressed to the Office of Publications, World Health Organization, Geneva, Switzerland, which will be glad to provide the latest information on any changes made to the text, plans for new editions, and reprints and translations already available.
©World Health Organization 2003
Publications of the World Health Organization enjoy copyright protection in accordance with the provisions of Protocol 2 of the Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries.
The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters.
ENVIRONMENTAL HEALTH CRITERIA FOR
SELECTED NITRO- AND NITRO-OXY-POLYCYCLIC AROMATIC HYDROCARBONS
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the criteria monographs as accurately as possible without unduly delaying their publication. In the interest of all users of the Environmental Health Criteria monographs, readers are requested to communicate any errors that may have occurred to the Director of the International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland, in order that they may be included in corrigenda.
* * *
The WHO Environmental Health Criteria Programme is financially supported by the US Environmental Protection Agency, European Commission, German Federal Ministry of the Environment, Nature Conservation, and Nuclear Safety, and Japanese Ministry of Health, Labour and Welfare.
Environmental Health Criteria
Objectives
In 1973, the WHO Environmental Health Criteria Programme was initiated with the following objectives:
The first Environmental Health Criteria (EHC) monograph, on mercury, was published in 1976, and since that time an ever-increasing number of assessments of chemicals and of physical effects have been produced. In addition, many EHC monographs have been devoted to evaluating toxicological methodology, e.g. for genetic, neurotoxic, teratogenic and nephrotoxic effects. Other publications have been concerned with epidemiological guidelines, evaluation of short-term tests for carcinogens, biomarkers, effects on the elderly and so forth.
Since its inauguration, the EHC Programme has widened its scope, and the importance of environmental effects, in addition to health effects, has been increasingly emphasized in the total evaluation of chemicals.
The original impetus for the Programme came from World Health Assembly resolutions and the recommendations of the 1972 UN Conference on the Human Environment. Subsequently the work became an integral part of the International Programme on Chemical Safety (IPCS), a cooperative programme of UNEP, ILO and WHO. In this manner, with the strong support of the new partners, the importance of occupational health and environmental effects was fully recognized. The EHC monographs have become widely established, used and recognized throughout the world.
The recommendations of the 1992 UN Conference on Environment and Development and the subsequent establishment of the Intergovernmental Forum on Chemical Safety with the priorities for action in the six programme areas of Chapter 19, Agenda 21, all lend further weight to the need for EHC assessments of the risks of chemicals.
Scope
The criteria monographs are intended to provide critical reviews on the effects on human health and the environment of chemicals and of combinations of chemicals and physical and biological agents. As such, they include and review studies that are of direct relevance for the evaluation. However, they do not describe every study carried out. Worldwide data are used and are quoted from original studies, not from abstracts or reviews. Both published and unpublished reports are considered, and it is incumbent on the authors to assess all the articles cited in the references. Preference is always given to published data. Unpublished data are used only when relevant published data are absent or when they are pivotal to the risk assessment. A detailed policy statement is available that describes the procedures used for unpublished proprietary data so that this information can be used in the evaluation without compromising its confidential nature (WHO (1990) Revised Guidelines for the Preparation of Environmental Health Criteria Monographs. PCS/90.69, Geneva, World Health Organization).
In the evaluation of human health risks, sound human data, whenever available, are preferred to animal data. Animal and in vitro studies provide support and are used mainly to supply evidence missing from human studies. It is mandatory that research on human subjects is conducted in full accord with ethical principles, including the provisions of the Helsinki Declaration.
The EHC monographs are intended to assist national and international authorities in making risk assessments and subsequent risk management decisions. They represent a thorough evaluation of risks and are not, in any sense, recommendations for regulation or standard setting. These latter are the exclusive purview of national and regional governments.
Content
The layout of EHC monographs for chemicals is outlined below.
Selection of chemicals
Since the inception of the EHC Programme, the IPCS has organized meetings of scientists to establish lists of priority chemicals for subsequent evaluation. Such meetings have been held in Ispra, Italy, 1980; Oxford, United Kingdom, 1984; Berlin, Germany, 1987; and North Carolina, USA, 1995. The selection of chemicals has been based on the following criteria: the existence of scientific evidence that the substance presents a hazard to human health and/or the environment; the possible use, persistence, accumulation or degradation of the substance shows that there may be significant human or environmental exposure; the size and nature of populations at risk (both human and other species) and risks for environment; international concern, i.e., the substance is of major interest to several countries; adequate data on the hazards are available.
If an EHC monograph is proposed for a chemical not on the priority list, the IPCS Secretariat consults with the Cooperating Organizations and all the Participating Institutions before embarking on the preparation of the monograph.
Procedures
The order of procedures that result in the publication of an EHC monograph is shown in the flow chart. A designated staff member of IPCS, responsible for the scientific quality of the document, serves as Responsible Officer (RO). The IPCS Editor is responsible for layout and language. The first draft, prepared by consultants or, more usually, staff from an IPCS Participating Institution, is based on extensive literature searches from reference databases such as Medline and Toxline.
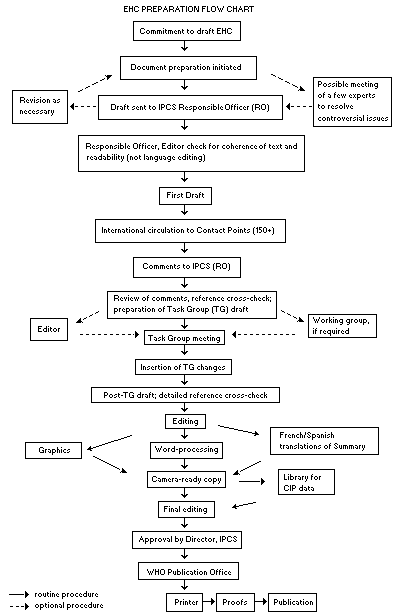
EHC PREPARATION FLOW CHART
The draft document, when received by the RO, may require an initial review by a small panel of experts to determine its scientific quality and objectivity. Once the RO finds the document acceptable as a first draft, it is distributed, in its unedited form, to well over 150 EHC contact points throughout the world who are asked to comment on its completeness and accuracy and, where necessary, provide additional material. The contact points, usually designated by governments, may be Participating Institutions, IPCS Focal Points or individual scientists known for their particular expertise. Generally some four months are allowed before the comments are considered by the RO and author(s). A second draft incorporating comments received and approved by the Director, IPCS, is then distributed to Task Group members, who carry out the peer review, at least six weeks before their meeting.
The Task Group members serve as individual scientists, not as representatives of any organization, government or industry. Their function is to evaluate the accuracy, significance and relevance of the information in the document and to assess the health and environmental risks from exposure to the chemical. A summary and recommendations for further research and improved safety aspects are also required. The composition of the Task Group is dictated by the range of expertise required for the subject of the meeting and by the need for a balanced geographical distribution.The three cooperating organizations of the IPCS recognize the important role played by nongovernmental organizations. Representatives from relevant national and international associations may be invited to join the Task Group as observers. Although observers may provide a valuable contribution to the process, they can speak only at the invitation of the Chairperson. Observers do not participate in the final evaluation of the chemical; this is the sole responsibility of the Task Group members. When the Task Group considers it to be appropriate, it may meet in camera.
All individuals who as authors, consultants or advisers participate in the preparation of the EHC monograph must, in addition to serving in their personal capacity as scientists, inform the RO if at any time a conflict of interest, whether actual or potential, could be perceived in their work. They are required to sign a conflict of interest statement. Such a procedure ensures the transparency and probity of the process.
When the Task Group has completed its review and the RO is satisfied as to the scientific correctness and completeness of the document, it then goes for language editing, reference checking and preparation of camera-ready copy. After approval by the Director, IPCS, the monograph is submitted to the WHO Office of Publications for printing. At this time, a copy of the final draft is sent to the Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the updating of an EHC monograph: new data are available that would substantially change the evaluation; there is public concern for health or environmental effects of the agent because of greater exposure; an appreciable time period has elapsed since the last evaluation.
All Participating Institutions are informed, through the EHC progress report, of the authors and institutions proposed for the drafting of the documents. A comprehensive file of all comments received on drafts of each EHC monograph is maintained and is available on request. The Chairpersons of Task Groups are briefed before each meeting on their role and responsibility in ensuring that these rules are followed.
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR SELECTED NITRO- AND NITRO-OXY-POLYCYCLIC AROMATIC HYDROCARBONS
Members
Professor D. Anderson, Department of Biomedical Sciences, University of Bradford, Bradford, West Yorkshire, United Kingdom (Chairperson)
Professor J. Arey, Air Pollution Research Center, University of California, Riverside, California, USA
Dr R.P. Bos, Department of Pharmacology & Toxicology, UMC St. Radboud, University of Nijmegen, Nijmegen, The Netherlands
Dr A. Cecinato, Istituto sull’Inquinamento Atmosferico-CNR, CP10 Monterotondo Stazione, Rome, Italy
Dr K. El-Bayoumy, Division of Cancer Etiology & Prevention, American Health Foundation, Valhalla, New York, USA (Vice-Chairperson)
Dr P.C. Howard, Division of Biochemical Toxicology, National Center for Toxicological Research, Jefferson, Arkansas, USA (Co-Rapporteur)
Dr J. Kielhorn, Chemical Risk Assessment, Fraunhofer Institute of Toxicology and Aerosol Research, Hanover, Germany (Co-Rapporteur)
Professor M. Kirsch-Volders, Laboratory of Cell Genetics, Free University of Brussels, Brussels, Belgium
Dr I. Mangelsdorf, Chemical Risk Assessment, Fraunhofer Institute of Toxicology and Aerosol Research, Hanover, Germany
Dr S. Pavittranon, Toxicology and Environmental Laboratory, National Institute of Health, Department of Medical Sciences, Ministry of Public Health, Nontaburi, Thailand
Dr H. Tokiwa, Department of Environmental Health Science, Kyushu Women’s University, Kitakyushu, Japan
Dr U. Wahnschaffe, Consultant, Uetze, Germany
Professor Z. Yuxin, Institute of Occupational Medicine, Chinese Academy of Preventive Medicine, Beijing, People’s Republic of China
Secretariat
Mr T. Ehara, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
Mrs P. Harlley, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR SELECTED NITRO- AND NITRO-OXY-POLYCYCLIC AROMATIC HYDROCARBONS
The first and second drafts of this monograph were prepared by the authors, Drs J. Kielhorn, U. Wahnschaffe and I. Mangelsdorf.
A WHO Task Group on Environmental Health Criteria for Selected Nitro- and Nitro-oxy-Polycyclic Aromatic Hydrocarbons met at the Fraunhofer Institute of Toxicology and Aerosol Research, in Hanover, Germany, on 26–30 November 2001. The group reviewed the draft and the peer review comments, revised the draft and made an evaluation of the risks for human health and environment from exposure to selected nitro- and nitro-oxy-polycyclic aromatic hydrocarbons.
Dr P. Jenkins and Mr T. Ehara of the IPCS central unit were responsible for the scientific aspects of the monograph, and Ms. Marla Sheffer was responsible for the technical editing.
The efforts of all, especially the Fraunhofer Institute of Toxicology and Aerosol Research, which helped in the preparation and finalization of the monograph, are gratefully acknowledged.
|
BaP |
benzo[a]pyrene |
|
bw |
body weight |
|
CAS |
Chemical Abstracts Service |
|
cDNA |
complementary (or copy) DNA |
|
CHO |
Chinese hamster ovary |
|
CYP |
cytochrome P450 |
|
D2 |
no. 2 diesel fuel |
|
dA |
deoxyadenosine |
|
dA-C8-2-AP |
N-(deoxyadenosin-8-yl)-2-aminopyrene |
|
DCM |
dichloromethane |
|
dG |
deoxyguanosine |
|
dG-C8-AAF |
N-(deoxyguanosin-8-yl)-2-acetylaminofluorene |
|
dG-C8-AF |
N-(deoxyguanosin-8-yl)-2-aminofluorene |
|
dG-C8-1-amino-6-NP |
N-(deoxyguanosin-8-yl)-1-amino-6-nitropyrene |
|
dG-C8-1-amino-8-NP |
N-(deoxyguanosin-8-yl)-1-amino-8-nitropyrene |
|
dG-C8-AP |
N-(deoxyguanosin-8-yl)-1-aminopyrene |
|
dG-C8-2-AP |
N-(deoxyguanosin-8-yl)-2-aminopyrene |
|
dG-C8-4-AP |
N-(deoxyguanosin-8-yl)-4-aminopyrene |
|
dG-N2-AAF |
C3-(deoxyguanosin-N2-yl)-2-acetylaminofluorene |
|
dG-1-nitroBaP-DE |
10-(deoxyguanosin-N2-yl)-7,8,9-trihydroxy-7,8,9,10-tetrahydro-1-nitrobenzo[a]pyrene |
|
dG-3-nitroBaP-DE |
10-(deoxyguanosin-N2-yl)-7,8,9-trihydroxy-7,8,9,10-tetrahydro-3-nitrobenzo[a]pyrene |
|
6-dG-N2-1-aminoBaP |
6-(deoxyguanosin-N2-yl)-1-aminobenzo[a]pyrene |
|
6-dG-N2-3-aminoBaP |
6-(deoxyguanosin-N2-yl)-3-aminobenzo[a]pyrene |
|
dI |
deoxyinosine |
|
DMSO |
dimethyl sulfoxide |
|
DNA |
deoxyribonucleic acid |
|
DNP |
dinitropyrene |
|
EC |
electron capture |
|
EC50 |
median effective concentration |
|
ECD |
electron capture detector |
|
ED50 |
median effective dose |
|
EHC |
Environmental Health Criteria monograph |
|
EI |
electron impact |
|
ELISA |
enzyme-linked immunosorbent assay |
|
EPA |
Environmental Protection Agency (USA) |
|
FAO |
Food and Agriculture Organization of the United Nations |
|
FTP |
Federal Test Procedure (USA) |
|
GC |
gas chromatography |
|
GPC |
semipreparative gel permeation chromatography |
|
GST |
glutathione S-transferase |
|
Hb |
haemoglobin |
|
HCFC-22 |
chlorodifluoromethane |
|
HDD |
heavy-duty diesel |
|
HPLC |
high-performance liquid chromatography |
|
IARC |
International Agency for Research on Cancer |
|
ILO |
International Labour Organization |
|
i.m. |
intramuscular |
|
i.p. |
intraperitoneal |
|
IPCS |
International Programme on Chemical Safety |
|
i.v. |
intravenous |
|
JECFA |
Joint FAO/WHO Expert Committee on Food Additives |
|
JMPR |
Joint FAO/WHO Meeting on Pesticide Residues |
|
Koc |
organic carbon/water partition coefficient |
|
Kow |
n-octanol/water partition coefficient |
|
LC50 |
median lethal concentration |
|
LC100 |
lethal concentration for 100% of test organisms |
|
LD50 |
median lethal dose |
|
LOAEL |
lowest-observed-adverse-effect level |
|
LOEL |
lowest-observed-effect level |
|
LPG |
liquefied petroleum gas |
|
MA |
metabolic activation |
|
MN |
micronucleus induction |
|
MS |
mass spectrometry |
|
MS/MS |
tandem mass spectrometry |
|
MTD |
maximum tolerated dose |
|
MW |
molecular weight (relative molecular mass) |
|
NADH |
nicotinamide adenine dinucleotide |
|
NADPH |
reduced nicotinamide adenine dinucleotide |
|
NAT |
N-acetyltransferase |
|
NB |
nitrobenzanthrone |
|
nd |
not detected |
|
NICI |
negative ion chemical ionization |
|
NIST |
National Institute of Standards and Technology (USA) |
|
nitroPAH |
nitro-polycyclic aromatic hydrocarbon |
|
NMR |
nuclear magnetic resonance |
|
NOAEL |
no-observed-adverse-effect level |
|
NPD |
nitrogen–phosphorus detector |
|
NP-LC |
normal-phase high-performance liquid chromatography |
|
NPR |
NADPH-cytochrome P450 reductase |
|
OCC |
oxidation catalytic converter |
|
OECD |
Organisation for Economic Co-operation and Development |
|
OH-AAF |
N-acetyl-2-aminofluoren-x-ol |
|
OH-2-NF |
2-nitrofluorenol |
|
PAH |
polycyclic aromatic hydrocarbon |
|
PCB |
polychlorinated biphenyl |
|
PEF |
potency equivalency factor |
|
PICI |
positive ion chemical ionization |
|
PM2.5 |
particulate matter <2.5 µm in diameter |
|
PM10 |
particulate matter <10 µm in diameter |
|
p.o. |
per os |
|
ppb |
part per billion |
|
ppm |
part per million |
|
ppt |
part per trillion |
|
PUF |
polyurethane foam |
|
RO |
Responsible Officer |
|
RP-LC |
reversed-phase high-performance liquid chromatography |
|
s.c. |
subcutaneous |
|
SCE |
sister chromatid exchange |
|
SEOM |
solvent extractable organic matter |
|
SOF |
soluble organic fraction |
|
SPE |
solid-phase extraction |
|
SRM |
Standard Reference Material |
|
TA98AT+ |
N-hydroxyarylamine O-acetyltransferase-overproducing S. typhimurium strain |
|
TA98NR+ |
nitroreductase-overproducing S. typhimurium strain |
|
TA98AT– |
N-hydroxyarylamine O-acetyltransferase-deficient S. typhimurium strain |
|
TA98NR– |
nitroreductase-deficient S. typhimurium strain |
|
TEA |
thermal energy analyser |
|
TID |
thermionic detector |
|
TLC |
thin-layer chromatography |
|
UDS |
unscheduled DNA synthesis |
|
UN |
United Nations |
|
UNEP |
United Nations Environment Programme |
|
UV |
ultraviolet |
|
WHO |
World Health Organization |
|
XOC |
extractable organic component |
1.1 Identity, physical and chemical properties, and analytical methods
Nitro-polycyclic aromatic hydrocarbons (nitroPAHs) are derivatives of polycyclic aromatic hydrocarbons (PAHs), which contain two or more fused aromatic rings made of carbon and hydrogen atoms. NitroPAHs occur in the environment as a mixture together with parent PAHs and hundreds of other organic compounds. NitroPAHs are usually present in much smaller quantities than PAHs.
NitroPAHs in the environment occur either in the vapour phase or adsorbed to particulate matter. NitroPAHs are insoluble or sparingly soluble in water but mostly soluble in organic solvents.
The sampling of nitroPAHs is similar to that of PAHs. Ambient air is sampled by collecting particulate matter on special filters by means of high-volume samplers. Vapour-phase nitroPAHs are commonly collected on solid sorbents such as polyurethane foam.
Solvent extraction is followed by cleanup using liquid chromatography with silica gel or alumina, high-performance liquid chromatography (HPLC) or solid-phase extraction. The nitroPAH fraction must be separated from the PAH fraction and oxygenated PAH fraction by HPLC on silica. Methods used for the separation and detection of nitroPAHs include gas chromatography with a variety of detectors, HPLC with fluorescence, chemiluminescence or electrochemical detector, and mass spectrometric techniques. Analysis is dependent on the standards available.
Another approach to analysis of complex mixtures is bioassay-directed chemical analysis, where mutagenically active fractions are bioassayed and characterized until the major class or specific compounds potentially responsible for the mutagenicity are identified. The use of bacterial tester strains selectively sensitive to nitroarenes has led to the identification of nitroPAHs as potent mutagens in complex mixtures from diverse sources. Synthetic standards are required for this type of analysis.
The nitroketone 3-nitrobenzanthrone and nitrolactones, such as 2- and 4-nitrodibenzopyranone, are nitro-oxy compounds, which have been detected together with nitroPAHs and are analysed by similar methods.
1.2 Sources of human and environmental exposure
NitroPAHs originate primarily as direct or indirect products of incomplete combustion. Only a few nitroPAHs are produced industrially; commercially produced nitronaphthalenes and 5-nitroacenaphthene, for example, are used primarily as chemical intermediates.
NitroPAHs originate from PAHs (generally adsorbed on particu-late matter and themselves products of incomplete combustion) by at least two distinct processes: (1) through nitration during combustion processes (e.g., in vehicle exhaust, particularly diesel, but also gasoline and aircraft emissions; industrial emissions; domestic residential heating/cooking; wood burning) and (2) through atmospheric formation from PAHs by either gas-phase reactions — daytime hydroxyl radical addition to the PAH followed by reaction with nitrogen dioxide and loss of a water molecule and nighttime nitrate radical addition to the PAH followed by reaction with nitrogen dioxide and loss of nitric acid — or heterogeneous gas–particle interaction of parent PAHs adsorbed onto particles with nitrating agents.
The distribution of nitroPAH isomers in samples of ambient air has been found to be significantly different from that in direct emissions from combustion. 2-Nitrofluoranthene and 2-nitropyrene are ubiquitous components of particulate matter, although they are not directly emitted from most combustion sources. The nitroPAH profile, or the relative quantities of certain "marker" PAHs, is a pointer to the source of formation of the nitroPAH. The most abundant nitro isomers of pyrene, fluorene and fluoranthene observed in diesel exhaust are 1-nitropyrene, 2-nitrofluorene and 3-nitrofluoranthene, whereas the isomers formed from the hydroxyl radical reactions of these PAHs are 2-nitropyrene, 3-nitrofluorene and 2-nitrofluoranthene.
The majority of ambient nitroPAHs are now thought to be formed in the atmosphere from the gas-phase reactions of PAHs with four rings or less.
Many mono- and some di- and trinitroPAH isomers have been identified and quantified in various samples of diesel exhaust, 1-nitropyrene usually being the most abundant. 1-Nitropyrene is the "marker" nitroPAH for diesel exhaust, and its presence in ambient air samples is a sign of pollution by diesel vehicle traffic. Diesel fuel, engine types and catalytic traps are continually being modified, so the various studies of nitroPAHs in diesel exhaust cannot be directly compared. In general, the mass emission of particles, emissions of particle-bound PAHs and nitroPAHs, and mutagenic activity levels generally decreased with the use of either particulate traps or catalytic converters.
The concentration of 1-nitropyrene was much less in gasoline exhaust particles than in diesel exhaust particles, but the concentrations of 1,3-, 1,6- and 1,8-dinitropyrenes were found to be almost the same in gasoline and diesel exhaust particles.
There is evidence of the presence of nitroPAHs in jet aeroplane exhaust.
NitroPAHs have been detected in the emissions of kerosene heaters, fuel gas and liquefied petroleum gas (LPG) burners, which are used in many countries for heating and cooking at home.
3-Nitrobenzanthrone has been detected in diesel exhaust particulate and in urban air samples. 2-Nitrodibenzopyranone and 4-nitrodibenzopyranone as well as nitropyrene lactones have been observed in ambient particulate matter.
1.3 Environmental transport, distribution and transformation
NitroPAHs can be transported in the vapour phase or adsorbed onto particulate matter. Those with liquid-phase vapour pressures greater than 10–4 Pa at ambient air temperature (i.e., two- to four-ring PAHs and two-ring nitroPAHs) will exist at least partially in the gas phase.
Owing to their low aqueous solubility or insolubility, nitroPAHs are not expected to be transported in water. Data available give high values for sorption coefficients (log Koc), suggesting that nitroPAHs, similar to PAHs, adsorb onto soil and sediments. Leaching into groundwater is thought to be negligible. Some nitroPAHs may be slowly biodegradable under certain conditions.
The values for the n-octanol/water partition coefficient (log Kow) range from 2.5 for 1-nitronaphthalene to 6.3 for 3-nitroperylene, suggesting a potential for bioaccumulation. There were no data available on biomagnification.
Many anaerobic and aerobic bacteria reduce nitroPAHs to mutagenic aminoPAHs. Nitroreduction by intestinal microflora plays a major role in the metabolism of nitroPAHs in mammals. Although a wide variety of bacteria, fungi and algae have been shown to degrade the parent PAHs containing two to five rings, nitro-substituted PAHs are only slowly degraded by indigenous microorganisms and may persist in soils and sediments. The recalcitrance of high molecular weight nitroPAHs is due in part to the strong adsorption to soil organic matter, low solubility, large molecular size and the polar character of the nitro group.
Time course studies in microcosms showed that 1-nitropyrene was degraded slowly under aerobic and anaerobic conditions in estuarine sediments.
Sphingomonas paucimobilis strain EPA 505 (a soil bacterium capable of utilizing fluoranthene as the sole source of carbon and energy) biodegraded 1-nitropyrene to 48.6% after 6 h.
The filamentous fungus Cunninghamella elegans has been shown to oxidatively metabolize, via a cytochrome P450 monooxygenase, a number of nitroPAHs (1-nitropyrene, 2-nitrofluorene, 2- and 3-nitrofluoranthene, 6-nitrochrysene, 1-nitrobenzo[e]pyrene and 6-nitrobenzo[a]pyrene) to products that are less mutagenic than the nitroPAHs themselves.
A plant cell culture derived from alligator weed (Alternanthera philoxeroides) detoxified 1-nitropyrene and 1,3-, 1,6- and 1,8-dinitropyrene, all direct-acting mutagens, when incubated with them, as shown by mutagenicity response in the Salmonella typhimurium TA98 assay.
The photolysis of nitroPAHs has been studied under varied conditions of irradiation. The rate of photolysis depends not only on the conditions of irradiation but also on whether the nitroPAH is in the gaseous phase (e.g., 1-and 2-nitronaphthalene), in solution (type of solvent) or bound to solids/particles. In the latter case, the type and age of the particle seem to influence the photochemistry of the respective nitroPAH. The rate of photodecomposition, identification of photolytic products and resulting loss or gain of metabolic activity as determined by the S. typhimurium assay have been the main end-points studied.
Calculated atmospheric lifetimes of nitroPAHs due to photolysis and gas-phase reactions with hydroxyl and nitrate radicals and with ozone under atmospheric conditions show that the dominant loss process for nitroPAHs (e.g., 1- and 2-nitronaphthalene) is photolysis. Particle oxidation of nitroPAHs by ozone may be the main loss process at night.
1.4 Environmental levels and human exposure
NitroPAHs that have been detected in ambient air include 1- and 2-nitronaphthalene and methylnitronaphthalenes (predominantly in the vapour phase), 2-nitrofluorene, 9-nitroanthracene, 9-nitrophenanthrene, 2-, 3- and 8-nitrofluoranthene, 1- and 2-nitropyrene, 1,3-, 1,6- and 1,8-dinitropyrene and 6-nitrochrysene.
At remote and forest sites, nitroPAHs were either not detected or detected in the low picogram per cubic metre range (e.g., 17 pg/m3 for 2-nitrofluoranthene; 4 pg/m3 for 1-nitropyrene). The concentration of nitroPAHs in the atmosphere of urban regions depends on the season, the type of heating used and the number and regulation of traffic vehicles. Reported levels in air do not usually exceed 1 ng/m3, although maxima of up to 13 ng/m3 have been reported.
Various studies have been performed monitoring certain isomeric nitroPAHs. Investigators have concentrated on the nitroPAHs that seem to be of quantitative/environmental (e.g., nitroPAHs of relative molecular mass 247: 1-nitropyrene, 2-nitropyrene, 2-nitrofluoranthene) or carcinogenic (e.g., 1-nitropyrene, 2-nitrofluorene, dinitropyrenes) importance.
Studies of daytime/nighttime concentrations of specific isomeric nitroPAHs in certain regions (in particular California, USA) and parallel environmental chamber studies have led to an understanding of the atmospheric formation of certain nitroPAHs (2-nitrofluoranthene and 2-nitropyrene). Concurrent studies of certain nitroPAHs (1-nitropyrene, dinitropyrenes) and traffic volume have confirmed that traffic emission is a source of nitroPAHs.
Most seasonal studies show higher winter/spring concentrations of marker nitroPAHs, which parallels the use of domestic heating, although this is not always the case.
As nitroPAHs have been detected in the emissions of kerosene heaters, fuel gas and LPG burners used for heating and cooking at home, as well as in the fumes of cooking oils, there is therefore a potential indoor exposure to nitroPAHs in poorly ventilated conditions.
Concentrations of polyaromatic compounds, including nitroPAHs, were measured in a study of indoor and outdoor air levels associated with 33 homes located in two US cities: Columbus, Ohio, and Azusa, California. The overall levels were much higher in homes occupied by smokers, but the use of natural gas heating and cooking appliances also appeared to increase the nitroPAH levels slightly.
1-Nitropyrene (4.2–25 600 ng/litre) was detected in 36 of 55 samples of wastewater from oil–water separating tanks of gasoline stations and in used crankcase oil.
1- and 2-nitronaphthalene and 1,3- and 1,5-dinitronaphthalene were detected in river water in Japan at concentrations of 1.3, 11.7, 1.7 and 3.2 ng/litre, respectively. In another water sample, 1-nitropyrene was identified.
There are only limited data on the presence of nitroPAHs in samples of soil, sewage sludge, sediment and incinerator ash (e.g., for 1-nitropyrene, 0.03–0.8 µg/kg dry weight in soil, 0.68 µg/kg in sewage sludge, 25.2 µg/kg in sediment and <0.01–0.89 mg/kg in incinerator ash).
With the exception of spices, smoked and grilled foods and peanuts, the concentrations of nitroPAHs in foods are below 5 µg/kg.
In a study in the United Kingdom, foodstuffs were monitored for the presence of 9-nitroanthracene and 1-nitropyrene. Twenty-five out of 28 foods contained no detectable levels of these nitroPAHs. 9-Nitroanthracene was tentatively identified in peated malt, at 0.9 µg/kg, and 1-nitropyrene in two samples of tea leaves, at 1.7 and 0.17 µg/kg.
Another survey of nitroPAH levels in various foods in Austria showed mostly detectable levels of 2-nitrofluorene, 1-nitropyrene and 2-nitronaphthalene. The highest concentrations were found in spices, smoked foods and teas, in particular Mate tea, which is roasted. NitroPAHs were also detected in vegetables and fruits, probably due to atmospheric pollution.
1-Nitropyrene was detected in grilled corn, mackerel and (in considerable amounts) pork and yakitori (grilled chicken) grilled with sauce (up to 43 ng/g).
In 1980, studies showed that extracts of selected xerographic toners and paper photocopies were mutagenic. The fraction of the carbon black B responsible for 80% of the mutagenicity contained 1-nitropyrene, 1,3-, 1,6- and 1,8-dinitropyrene, 1,3,6-trinitropyrene and 1,3,6,8-tetranitropyrene. As a result of this finding, the manufacturers modified the production of carbon black B, substantially reducing the levels of nitropyrenes.
Occupational exposure to nitroPAHs has been demonstrated in workplaces associated with the use of diesel engines. For example, concentrations of 1-nitropyrene in air were measured in various workplaces associated with the use of diesel engines. The highest levels (42 ng/m3) reported were determined in the breathing zones of the underground workers (drivers of diesel-powered excavators) at an oil shale mine in Estonia.
1.5 Kinetics and metabolism in laboratory animals and humans
1-Nitropyrene and 2-nitrofluorene administered by various routes are rapidly absorbed, and the resulting metabolites are conjugated and excreted. Radiolabelled 1-nitropyrene was found to be widely distributed in the body of rats and mice following administration by all routes. Other nitroPAHs have not been as well studied.
The metabolism of nitroPAHs is complex. It seems that there are at least five metabolic activation pathways through which mutations can be induced by nitroPAHs in bacterial and mammalian systems and/or through which DNA binding occurs. These are 1) nitroreduction; 2) nitroreduction followed by esterification (in particular acetylation); 3) ring oxidation; 4) ring oxidation and nitroreduction; and 5) ring oxidation and nitroreduction followed by esterification. In bacteria, nitroreduction seems to be the major metabolic pathway, whereas the fungus Cunninghamella elegans is an example of a species in which nitroPAHs are metabolized by ring oxidation.
Nitroreduction of nitroPAHs in vivo probably occurs mainly by bacteria in the intestinal tract. In oxidative metabolism, the first step is transformation to phase I primary metabolites such as epoxides, phenols and dihydrodiols, and then to secondary metabolites, such as diol epoxides, tetrahydrotetrols and phenol epoxides. In mammalian systems, the phase I metabolites are then conjugated with glutathione, sulfate or glucuronic acid to form phase II metabolites, which are more polar and water-soluble than the parent hydrocarbons. On reaching the intestine, the conjugated metabolites can be deconjugated by the intestinal microflora and absorbed, entering enterohepatic circulation. Nitroreduction and N-acetylation can occur, resulting in the excretion in urine and faeces of metabolites such as acetylaminopyrenols after 1-nitropyrene administration.
Different cytochrome P450 enzymes may be involved in the metabolism of a specific nitroPAH, and these may differ in the related isomers, resulting in possibly different kinetics and pathways. Cytochrome P450 enzymes responsible for the metabolism of nitroPAHs may vary between species and in different target organs and in different cell types within target organs.
All nitroPAHs do not follow the same activation pathways. Some are mutagenic when reduced to an arylhydroxylamine (e.g., 1-nitropyrene is metabolized mainly by hydroxylation of the aromatic moiety, followed by nitroreduction and N-acetylation); others (e.g., 1,8- and 1,6-dinitropyrene) are reduced to the arylhydroxylamine and then require further O-esterification (in particular O-acetylation) to an acyloxy ester for mutagenicity. Some may be mutagenic only after activation by oxidation to reactive epoxides or dihydrodiol epoxides (as possibly in 6-nitrobenzo[a]pyrene, similar to benzo[a]pyrene, or BaP). The main DNA adducts detected with nitroPAHs in vivo and in vitro are C8-substituted deoxyguanosine adducts; however, N2-substituted deoxyguanosine and C8-substituted deoxyadenosine derivatives have also been detected and may predominate in nitroPAHs with greater hydrocarbon character (e.g., 3-nitrobenzo[a]pyrene and 6-nitrochrysene). DNA adducts of dinitropyrenes are formed only via nitroreduction, presumably owing to the high electron deficiency in the aromatic rings caused by the presence of two nitro groups. The DNA adducts resulting from the nitroreduction of nitroPAHs are better characterized than those arising from oxidative metabolism, although the latter may be of more importance in mammalian metabolism.
1.6 Effects on laboratory mammals and in vitro test systems
Only six nitroPAHs have been tested for acute toxicity. In rats, an LD50 of 86 mg/kg of body weight (kg bw) after intraperitoneal (i.p.) application was reported for 1-nitronaphthalene; in mice, an oral LD50 of 1300 mg/kg bw was reported for 2-nitronaphthalene. In further studies on both substances, systemic effects on the target organs lung and liver were observed after single high doses; however, 2-nitronaphthalene seemed to be less toxic than 1-nitronaphthalene. 5-Nitroacenaphthene at an i.p. dose of 1700 mg/kg bw was lethal to all treated rats. For 2-nitrofluorene, an oral LD50 of 1600 mg/kg bw in mice was reported, whereas gavaging with up to 5000 mg 1-nitropyrene/kg bw resulted in no observable toxic effects. Local inflammation and ulceration were seen in rats after subcutaneous (s.c.) injection of 8 mg 3-nitrofluoranthene/kg bw.
Data on systemic or local non-neoplastic effects caused by short-term or long-term treatment with nitroPAHs are limited, as the end-point of most studies has been carcinogenicity. In most cases, non-neoplastic toxic effects were observed at doses at which carcinogenic responses are also manifested. Systemic non-neoplastic toxic effects, such as reduced body weight or increased mortality, appeared presumably independently of carcinogenic effects in feeding studies with 5-nitroacenaphthene at a dose level of 500 mg/kg bw per day (rat) or 40 mg/kg bw per day (mice) and with 2-nitrofluorene at a dose of 25 mg/kg bw per day (rat). Medium-term exposure via inhalation to 1-nitropyrene resulted in metaplasia of the upper respiratory tract at concentrations of >0.5 mg/m3.
No data are available on skin and eye irritation, sensitization or reproductive toxicity.
Data on genotoxicity in vitro are available on 95 nitroPAHs; for 74 nitroPAHs, however, only one or two end-points, mainly in bacterial test systems, were investigated. A sufficient database, including eukaryotic test systems, has been found only with 21 nitroPAHs. Most of these substances (67 out of 95) showed positive results, but the results were derived from a small database. Clearly positive results were obtained for 19 nitroPAHs, and questionable results for 8 nitroPAHs. With none of the nitroPAHs were clearly negative results obtained.
For 86 nitroPAHs, data on the S. typhimurium microsome test are available. In contrast to the parent PAHs, most nitroPAHs were clearly more effective in the Salmonella microsome test without metabolic activation. There are five nitroPAHs that showed exceptionally high mutagenic potency (>100 000 revertants/nmol) in this test system: 3,7- and 3,9-dinitrofluoranthene, 1,6- and 1,8-dinitropyrene, and 3,6-dinitrobenzo[a]pyrene.
Bacterial nitroreductase and acetyltransferase are involved in the metabolic activation of nitroPAHs, but not all nitroPAHs follow the same metabolic activation pathways. Furthermore, there is no uniform mutagenic effect of the different nitroPAHs, as they produce both frameshift and base pair substitutions in the S. typhimurium microsome test. There is evidence that nitroPAHs with nitro groups perpendicular to the aromatic ring are not as mutagenic as isomers having parallel nitro orientation.
Data on the in vivo genotoxicity of nitroPAHs are available for 15 nitroPAHs. All nitroPAHs that gave positive results in vivo were also positive in vitro. Four nitroPAHs that were positive in in vitro genotoxicity tests revealed inconsistent or inconclusive genotoxicity (2-nitronaphthalene, 5-nitroacenaphthene and 3-nitrofluoranthene) or negative genotoxicity (2,7-dinitrofluorene; limited validity) results in vivo.
3-Nitrobenzanthrone, like 1,6- and 1,8-dinitropyrene, is highly mutagenic in bacteria through nitroreduction and O-esterification. 3-Nitrobenzanthrone is also an effective gene mutagen and causes micronuclei formation in human cells in vitro and in mice in vivo.
2-Nitrodibenzopyranone was reported to be highly mutagenic in the S. typhimurium microsome test in strain TA98 (–S9), being more mutagenic than 2-nitrofluorene and 1-nitropyrene. 1- and 3-nitropyrene lactones have been found to be highly mutagenic in the S. typhimurium microsome test.
Studies on the in vitro genotoxicity of 2-nitrodibenzopyranone in forward mutation assays using two human B-lymphoblastoid cell lines are conflicting. Nitropyrene lactones were found to induce mutations at the tk and hprt loci in both cell lines. Further, they induced kinetochore-positive and -negative micronuclei in the CREST modified micronucleus assay, which detects chromosomal loss and breakage events.
Data on carcinogenic effects are available for 28 nitroPAHs. Although inhalation is the main exposure route in humans, no long-term inhalation study on any nitroPAH is available. Most studies examined the carcinogenic effects of nitroPAHs by oral administration, topical application, pulmonary implantation or intratracheal administration.
Owing to the limitations in experimental design, none of the negative studies confirmed the absence of carcinogenic effects in animals. However, results showed carcinogenic effects in experimental animals for 5-nitroacenaphthene, 2-nitrofluorene, 3-nitrofluoranthene, 3,7- and 3,9-dinitrofluoranthene, 1- and 4-nitropyrene, 1,3-, 1,6- and 1,8-dinitropyrene and 6-nitrochrysene. Some carcinogenic effects in experimental animals were observed for 2-nitropyrene, 7-nitrobenz[a]anthracene, 2- and 6-nitrobenzo[a]pyrene, 3,6-dinitrobenzo[a]pyrene, 7-nitrodibenz[a,h]anthracene and 3-nitroperylene. For the remaining 10 nitroPAHs tested, not enough data were available with which to evaluate their carcinogenicity in experimental animals.
Besides local effects at the site of injection, nitroPAHs induced mainly systemic tumours in mammary tissue, lung, liver and the haematopoietic system. 6-Nitrochrysene appears to be the most carcinogenic of the nitroPAHs considered here. With systemic effects after s.c. or i.p. injection, 1-nitropyrene was more carcinogenic than the dinitropyrenes. The carcinogenicity of 1-nitropyrene and dinitropyrenes varies, depending on the route of administration.
Nitrated benzo[a]pyrenes are generally less potent carcinogens than the parent compound BaP. However, the mono- or dinitrated pyrenes are more carcinogenic than pyrene. Similar results were presented for 3-nitroperylene compared with perylene and for 6-nitrochrysene compared with chrysene; with local effects after dermal exposure, however, 6-nitrochrysene was less active than chrysene.
Data were available on carcinogenic effects of some metabolites of 2-nitrofluorene, 1-nitropyrene and 6-nitrochrysene. Comparing 2-nitrofluorene with its metabolites in rats, the highest carcinogenic potency was shown by 2-acetylaminofluorene. 1-Nitropyrene was significantly more carcinogenic after oral application in rats than either 1-nitrosopyrene or 1-aminopyrene. In contrast, 1-nitrosopyrene induced a higher incidence of liver tumours in mice after i.p. application than 1-nitropyrene; no effects were observed with ring hydroxylated metabolites. 6-Nitrosochrysene and 6-aminochrysene were inactive, in contrast to the ring hydroxylated metabolites, which showed carcinogenic activity in the liver similar to that of the parent compound 6-nitrochrysene; this indicates that the metabolic activation of 6-nitrochrysene occurs by ring oxidation and/or a combination of ring oxidation and nitroreduction.
There are no reports on the effects of individual nitroPAHs on humans. As would be expected, since nitroPAHs occur in complex mixtures in the atmosphere and exhaust, the exact contribution of nitroPAHs to the adverse health consequences of exposure to polluted atmospheres and to exhaust cannot be elucidated.
At present, investigations on the effects of nitroPAHs on human health are being carried out using biomarkers of exposure. Several reports have described the development of methods for and provided data on the evaluation of 1-nitropyrene as a biomarker for occupational exposure to diesel exhaust. Urinary metabolites of PAHs and nitroPAHs were determined in the urine of diesel mechanics using the enzyme-linked immunosorbent assay (ELISA). In another study, metabolites of 1-nitropyrene (namely, N-acetyl-1-aminopyren-6-ol and N-acetyl-1-aminopyren-8-ol) were measured in the urine of workers in a shipping department. Several studies have focused on measuring the haemoglobin and plasma adducts of metabolites of 1-nitropyrene and other nitroPAHs and may provide appropriate biomarkers in future molecular epidemiological investigations.
1.8 Effects on other organisms in the laboratory and field
Data on the acute toxicity of nitroPAHs to aquatic organisms are available only for 1-nitronaphthalene. An LC50 (96 h) of 9.0 mg/litre was reported for the fathead minnow (Pimephales promelas). Furthermore, this nitroPAH inhibited the growth of the ciliate Tetrahymena pyriformis, with an EC50 (60 h) of 17.3 mg/litre.
Some studies have been concerned with the effect of nitroPAHs on the metabolism of some aquatic species — for example, the subcellular and tissue distribution of two- and one-electron NAD(P)H-dependent nitroreductase activity in marine invertebrates from three phyla: mussel (Mytilus edulis), crab (Carcinus maenas) and starfish (Asteria rubens). NADPH-dependent two-electron nitroreductase activity, occurring only under anaerobic conditions, was detected in the microsomal and cytosolic fractions of the major digestive tissues of mussel (digestive gland) and crab, but not in the gills of either species. 1-Aminopyrene was the only metabolite identified. No activity was detectable in the pyloric caeca or stomach region of the starfish. NAD(P)H-dependent one-electron nitroreduction was present in all subcellular fractions of the major digestive tissues of the three species.
In the presence of calf thymus DNA, adducts derived from 1-nitropyrene were detected in vitro using hepatic S9 fractions prepared from fish. The ability of 1-nitropyrene to form DNA adducts was also established in vivo using brown trout (Salmo trutta) and turbot (Scophthalmus maximus). These DNA adducts were comparable to those obtained in Wistar rats treated with 1-nitropyrene.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL METHODS
Nitro-polycyclic aromatic hydrocarbons (nitroPAHs) are derivatives of polycyclic aromatic hydrocarbons (PAHs), which contain two or more fused aromatic rings made of carbon and hydrogen atoms, formed as a result of incomplete combustion (see IPCS, 1998). NitroPAHs occur in the environment as a mixture together with parent PAHs and hundreds of other organic compounds (see chapter 3). NitroPAHs are usually present in smaller quantities (by 2 orders of magnitude) than PAHs.
Interest was focused on nitroPAHs in the early 1980s as correlations were found between the presence of nitroPAHs in diesel exhaust and environmental extracts and mutagenic activity. A large number of groups of nitro, oxy and mixed nitro-oxy compounds eluted together in the mutagenic fractions. Analytical methods were developed to separate and identify these compounds and to specify their isometric composition, as the biological action of these compounds also depends on their stereospecificity (see chapters 6 and 7). As it would be impossible to evaluate all these compounds in one document, a decision was made to include mono- and dinitroPAHs (2–5 rings) but, in general, not methylated or hydroxylated nitroPAHs. Some nitro-oxyPAHs are also included: nitroketones (3-nitrobenzanthrone) and selected nitrolactones (e.g., the nitrophenanthrene lactones: 2- and 4-nitrodibenzopyranone [2- and 4-nitro-6H-dibenzo[b,d]pyran-6-one] and nitropyrene lactones), which have recently been shown to be present in the extracts of the polar fractions of diesel exhaust and airborne particulates.
The nomenclature, molecular formula, relative molecular mass and Chemical Abstracts Service (CAS) number of selected nitroPAHs and nitro-oxyPAHs are given in Table 1. The structural formulas of some selected nitroPAHs and nitro-oxyPAHs are shown in Figure 1.
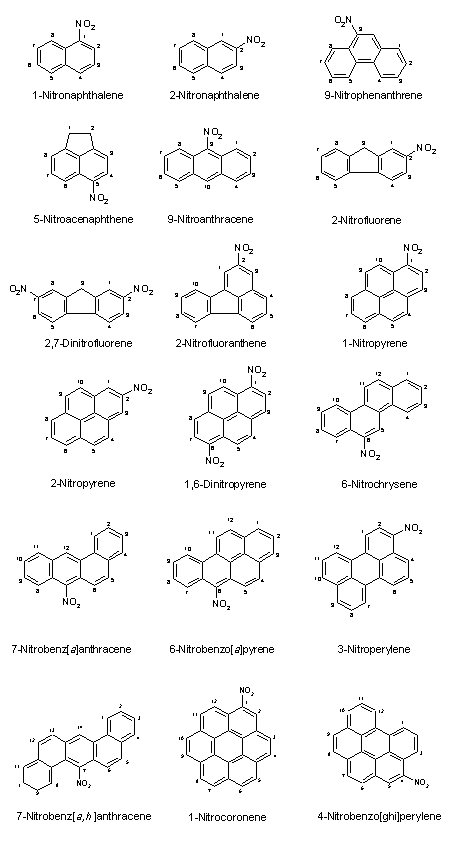
Fig. 1. Structural formulas of some nitroPAHs and some nitro-oxyPAHs.
Table 1. Nomenclature, molecular formulas, relative molecular mass and CAS numbers of selected nitroPAHs and their oxygen-containing derivatives
|
Parent PAHs |
Nitro derivative |
Molecular formula |
Relative molecular mass |
CAS number |
|
Two-ring PAHs |
||||
|
Naphthalene |
1-Nitronaphthalene |
C10H7NO2 |
173.17 |
|
|
2-Nitronaphthalene |
" |
" |
|
|
|
1,3-Dinitronaphthalene |
C10H6N2O4 |
218.17 |
|
|
|
1,5-Dinitronaphthalene |
" |
" |
|
|
|
1,8-Dinitronaphthalene |
" |
" |
|
|
|
2,7-Dinitronaphthalene |
" |
" |
|
|
|
2,3,5-Trinitronaphthalene |
C10H5N3O6 |
263.17 |
|
|
|
1,3,6,8-Tetranitronaphthalene |
C10H4N4O8 |
308.16 |
|
|
Three-ring PAHs |
||||
|
Acenaphthene |
3-Nitroacenaphthene |
C12H9NO2 |
199.21 |
|
|
5-Nitroacenaphthene |
" |
" |
|
|
|
Fluorene |
1-Nitrofluorene |
C13H9NO2 |
211.22 |
|
|
2-Nitrofluorene |
" |
" |
|
|
|
3-Nitrofluorene |
" |
" |
|
|
|
4-Nitrofluorene |
" |
" |
|
|
|
2,7-Dinitrofluorene |
C13H8N2O4 |
256.22 |
|
|
|
Anthracene |
2-Nitroanthracene |
C14H9NO2 |
223.23 |
|
|
9-Nitroanthracene |
" |
" |
|
|
|
9,10-Dinitroanthracene |
C14H8N2O4 |
268.23 |
|
|
|
Phenanthrene |
2-Nitrophenanthrene |
C14H9NO2 |
223.23 |
|
|
9-Nitrophenanthrene |
" |
" |
|
|
|
2,6-Dinitrophenanthrene |
C14H8N2O4 |
268.23 |
||
|
Four-ring PAHs |
||||
|
Fluoranthene |
1-Nitrofluoranthene |
C16H9NO2 |
247.25 |
|
|
2-Nitrofluoranthene |
" |
" |
|
|
|
3-Nitrofluoranthene |
" |
" |
|
|
|
7-Nitrofluoranthene |
" |
" |
|
|
|
8-Nitrofluoranthene |
" |
" |
|
|
|
1,2-Dinitrofluoranthene |
C16H8N2O4 |
292.25 |
|
|
|
2,3-Dinitrofluoranthene |
" |
" |
|
|
|
2,4-Dinitrofluoranthene |
" |
" |
|
|
|
2,5-Dinitrofluoranthene |
" |
" |
|
|
|
3,4-Dinitrofluoranthene |
" |
" |
||
|
3,7-Dinitrofluoranthene |
" |
" |
|
|
|
3,9-Dinitrofluoranthene |
" |
" |
|
|
|
1,2,4-Trinitrofluoranthene |
C16H7N3O6 |
337.25 |
|
|
|
1,2,5-Trinitrofluoranthene |
" |
" |
|
|
|
2,3,5-Trinitrofluoranthene |
" |
" |
|
|
|
Pyrene |
1-Nitropyrene |
C16H9NO2 |
247.25 |
|
|
2-Nitropyrene |
" |
" |
|
|
|
4-Nitropyrene |
" |
" |
|
|
|
1,3-Dinitropyrene |
C16H8N2O4 |
292.25 |
|
|
|
1,6-Dinitropyrene |
" |
" |
|
|
|
1,8-Dinitropyrene |
" |
" |
|
|
|
1,3,6-Trinitropyrene |
C16H7N3O6 |
337.25 |
|
|
|
1,3,6,8-Tetranitropyrene |
C16H6N4O8 |
382.24 |
|
|
|
Benz[a]anthracene |
7-Nitrobenz[a]anthracene |
C18H11NO2 |
273.29 |
|
|
Chrysene |
2-Nitrochrysene |
C18H11NO2 |
273.29 |
|
|
5-Nitrochrysene |
" |
" |
|
|
|
6-Nitrochrysene |
" |
" |
|
|
|
Five-ring PAHs |
||||
|
Benzo[e]fluoranthene |
3-Nitrobenzo[e]fluoranthene |
C20H11NO2 |
297.31 |
|
|
Benzo[a]pyrene |
1-Nitrobenzo[a]pyrene |
C20H11NO2 |
297.31 |
70021-997 |
|
3-Nitrobenzo[a]pyrene |
" |
" |
|
|
|
6-Nitrobenzo[a]pyrene |
" |
" |
|
|
|
3,6-Dinitrobenzo[a]pyrene |
C20H10N2O4 |
342.31 |
|
|
|
Benzo[e]pyrene |
1-Nitrobenzo[e]pyrene |
C20H11NO2 |
297.31 |
|
|
3-Nitrobenzo[e]pyrene |
" |
" |
|
|
|
Perylene |
3-Nitroperylene |
C20H11NO2 |
297.31 |
|
|
Dibenz[a,h]anthracene |
7-Nitrodibenz[a,h]anthracene |
C22 H13 NO2 |
323.4 |
|
|
Six-ring PAHs |
||||
|
Benzo[ghi]perylene |
4-Nitrobenzo[ghi]perylene |
C22H11NO2 |
321.34 |
|
|
7-Nitrobenzo[ghi]perylene |
" |
" |
||
|
Coronene |
1-Nitrocoronene |
C24H11NO2 |
345.36 |
|
|
Nitro-oxyPAHs |
||||
|
3-Nitrobenzanthrone |
C17H9NO3 |
275.26 |
1711-34-9 |
|
|
2-Nitrobenzopyranone |
C13H7NO4 |
241.20 |
|
|
|
3-Nitrobenzopyranone |
C13H7NO4 |
241.20 |
|
|
|
4-Nitrobenzopyranone |
C13H7NO4 |
241.20 |
|
|
|
Nitropyrene lactones |
C15H7NO4 |
265.22 |
2.2 Physical and chemical properties
At ambient temperatures, nitroPAHs are yellowish to orange solids that tend to sublime (White, 1985). NitroPAHs in the environment occur in the vapour phase or are absorbed and/or adsorbed to particulate matter, depending upon their vapour pressure and the ambient conditions. Two- to four-ring nitroPAHs are present partially in the vapour phase under certain conditions; for example, 1-nitronaphthalene (in certain climates) occurs mainly in the vapour phase (Arey et al., 1987), whereas 2-nitrofluorene occurs equally in both vapour and particulate phases, and 1-nitropyrene occurs in the particulate phase (Schuetzle & Frazier, 1986).
NitroPAHs are insoluble or sparingly soluble in water but are mostly soluble in organic solvents such as acetone, benzene, dimethyl sulfoxide (DMSO) and methylene chloride.
Table 2 gives details of some environmentally relevant physical and chemical properties of nitroPAHs together with those of the parent PAHs (see IPCS, 1998). Nitrodibenzopyranones have a lower vapour pressure than nitroPAHs and therefore are to be found predominantly in the particulate phase.
Table 2. Physical and chemical properties of nitroPAHs and their parent PAHsa
|
Parent PAHs; |
Melting point (°C) |
Boiling point (°C) at 101.3 kPa |
Vapour pressure |
Solubility in water at 25 °C (mg/litre) |
Henry’s law constant at 25 °C (kPa·m3/mol) |
Log Kowb |
Log Kocb |
|
Two-ring PAHs |
|||||||
|
Naphthalene |
81 |
218 |
10.4 |
31.7 |
4.9 × 10–2 |
3.4 |
|
|
1-Nitro- |
58–61.5 (56.5c) |
330; 314 sublimesd |
0.0154 (20 °C)d 3.2 × 10–2 e |
34f |
6.1 × 10–1 e |
2.50c |
3.02 |
|
2-Nitro- |
74–79 (76c) |
304e |
3.2 × 10–2 e |
26f |
6.1 × 10–1 e |
2.78c |
3.09 |
|
1,3-Dinitro- |
144–149 |
2.83g,h |
|||||
|
1,5-Dinitro- |
215–219 |
2.58g,h |
|||||
|
1,8-Dinitro- |
171–172 |
2.52g,h |
|||||
|
2,7-Dinitro- |
234 |
||||||
|
1,3,6,8-Tetranitro- |
195 |
2.29g |
|||||
|
Three-ring PAHs |
|||||||
|
Acenaphthene |
95 |
279 |
2.9 × 10–1 |
3.93 |
1.5 × 10–2 |
3.92 |
|
|
3-Nitro- |
151 |
||||||
|
5-Nitro- |
102 |
3.36c |
|||||
|
Fluorene |
115 |
295 |
8 × 10–2 |
1.98 |
1.0 × 10–2 |
4.18 |
|
|
1-Nitro- |
326 |
9.7 × 10–5 |
0.28 |
7.2 × 10–2 |
3.76e |
||
|
2-Nitro- |
154–158 (158c) |
326 |
9.7 × 10–5 |
0.216 |
9.5 × 10–2 |
4.08c |
3.16e |
|
3-Nitro- |
105–106 |
326 |
9.7 × 10–5 |
0.28 |
7.2 × 10–2 |
3.76e |
|
|
4-Nitro- |
75–76 |
326 |
9.7 × 10–5 |
0.28 |
7.2 × 10–2 |
3.76e |
|
|
2,7-Dinitro- |
334 |
3.35g,h |
|||||
|
Anthracene |
216 |
342 |
8 × 10–4 |
0.073 |
7.3 × 10–2 |
4.5 |
|
|
2-Nitro- |
172 |
4.23g |
|||||
|
9-Nitro- |
141–146 (146c) |
4.16c |
4.69j |
||||
|
9,10-Dinitro- |
263, 310 |
4.10c |
|||||
|
Phenanthrene |
100.5 |
340 |
1.6 × 10–2 |
1.29 |
|||
|
2-Nitro- |
119–120 |
4.23g |
|||||
|
9-Nitro- |
116–117 |
||||||
|
2,7-Dinitro- |
|||||||
|
Four-ring PAHs |
|||||||
|
Fluoranthene |
108.8 |
375 |
1.2 × 10–3 |
0.26 |
6.5 × 10–4 (20 °C) |
5.22 |
|
|
1-Nitro- |
4.69g |
||||||
|
2-Nitro- |
420e |
9.9 × 10–7 e |
0.019e |
1.3 × 10–2 e |
4.48e |
||
|
3-Nitro- |
156–162 (166c) |
5.15 |
|||||
|
7-Nitro- |
144–145 |
420e |
9.9 × 10–7 e |
0.017e |
1.4 × 10–2 e |
4.69g |
4.48e |
|
8-Nitro- |
158–164 |
420e |
9.9 × 10–7 e |
0.017e |
1.4 × 10–2 e |
4.69g |
4.48e |
|
3,4-Dinitro- |
279–280k |
||||||
|
3,7-Dinitro- |
203–204k |
||||||
|
3,9-Dinitro- |
275–276 |
||||||
|
Pyrene |
150.4 |
393 |
6.0 × 10–4 |
0.135 |
1.1 × 10–3 |
5.18 |
|
|
1-Nitro- |
151–152 (153c) |
472e |
4.4 × 10–6 e |
0.017e |
6.4 × 10–2 e |
5.29c |
4.48e |
|
2-Nitro- |
197–199 |
472e |
4.4 × 10–6 e |
0.021e |
6.4 × 10–2 e |
3.53e |
|
|
4-Nitro- |
190–192 |
472e |
4.4 × 10–6 e |
0.017e |
6.4 × 10–2 e |
4.48e |
|
|
1,3-Dinitro- |
295–297 |
4.44g |
|||||
|
1,6-Dinitro- |
309–310 |
4.44g |
|||||
|
1,8-Dinitro- |
299–300 |
4.44g |
|||||
|
1,3,6-Trinitro- |
4.18g |
||||||
|
1,3,6,8-Tetranitro- |
335 |
3.92g |
|||||
|
Benz[a]-anthracene |
160.7 |
400 |
2.8 × 10–5 |
0.014 |
5.61 |
||
|
7-Nitro- |
161–162c |
5.34c |
|||||
|
Chrysene |
253.8 |
448 |
8.4 × 10–5 |
0.002 |
5.91 |
||
|
2-Nitro- |
|||||||
|
5-Nitro- |
|||||||
|
6-Nitro- |
208c |
5.41c |
|||||
|
Five-ring PAHs |
|||||||
|
Benzo[a]pyrene |
178.1 |
496 |
7.3 × 10–7 |
0.004 |
3.4 × 10–5 (20 °C) |
6.50 |
|
|
1-Nitro- |
|||||||
|
3-Nitro- |
|||||||
|
6-Nitro- |
260c |
567e |
0.012e |
6.13c |
5.66e |
||
|
3,6-Dinitro- |
|||||||
|
Benzo[e]pyrene |
178.7 |
493 |
7.4 × 10–7 |
0.005 |
6.44 |
||
|
1-Nitro- |
5.87g |
||||||
|
Perylene |
277.5 |
503 |
0.0004 |
5.3 |
|||
|
3-Nitro- |
209c |
6.34c |
|||||
|
Six-ring PAHs |
|||||||
|
Coronene |
439 |
525 |
2 × 10–10 |
0.000 14 |
5.4 |
||
|
1-Nitro- |
|||||||
|
Benzo[ghi]-perylene |
278.3 |
545 |
1.4 × 10–8 |
0.000 26 |
2.7 × 10–5 (20 °C) |
7.10 |
|
|
4-Nitro- |
|||||||
|
7-Nitro- |
|||||||
|
Nitro-oxyPAHs |
|||||||
|
3-Nitrobenzanthrone |
256–257l |
||||||
|
2-Nitrodibenzo[b,d]-pyranone |
368e |
2.5 × 10–6 e |
128e,f |
2.9 × 10–6 e |
2.04e |
||
|
4-Nitrodibenzo[b,d]-pyranone |
368e |
2.5 × 10–6 e |
128e,f |
2.9 × 10–6 e |
2.04e |
|
a |
The data for parent PAHs are from IPCS (1998). The data for nitroPAHs are from White (1985), unless stated otherwise. |
|
b |
Log Kow = octanol/water partition coefficient; log Koc = sorption coefficient. |
|
c |
From Karcher et al. (1991). |
|
d |
From IUCLID dataset on 1-nitronaphthalene, 2000. |
|
e |
From Yaffe et al. (2001). |
|
f |
From Al-Bashir et al. (1994). |
|
g |
From Compadre et al. (1990). |
|
h |
Experimental values. |
|
i |
From Sinks et al. (1997). |
|
j |
From Nielsen et al. (1997). |
|
k |
From Nakagawa et al. (1987). |
|
l |
From Suzuki et al. (1997). |
Atmospheric concentrations of nitroPAHs are usually expressed as micrograms, nanograms or picograms per cubic metre. At 25 °C and 101.3 kPa, the conversion factors for a compound of given molecular mass are obtained as follows:
ppb = µg/m3 × 24.45/relative molecular mass
µg/m3 = ppb × relative molecular mass/24.45
where ppb is parts per billion, and one billion is 109.
For example, for 1-nitropyrene, 1 ppb = 10.1 µg/m3, and 1 µg/m3 = 0.099 ppb.
A direct analysis of nitroPAHs from environmental sources is not possible, as all environmental matrices are very complex. The samples often contain thousands of combustion products, including parent PAHs and other closely related derivatives (in particular oxygenated PAHs such as aldehydes, ketones and carboxylic acids), which tend to co-elute with nitroPAHs under a variety of liquid and gas chromatographic conditions and are present at concentrations 1 or 2 orders of magnitude higher than those of the nitro-substituted compounds (Schuetzle, 1983; Vincenti et al., 1996; see Table 3). An extensive sample cleanup and prefractionation of the sample are tedious but necessary prerequisites for trace analysis of nitroPAHs.
Isomer-specific identification is necessary, as the biological activity depends on the position of the nitro substituent. The source of the nitroPAH (e.g., from combustion or from atmospheric reactions; see chapter 3) may determine the isomeric specificity of the nitroPAH.
Although 1- and 2-nitronaphthalene are expected to be found predominantly in the gas phase, other semivolatile nitroPAHs will be distributed between the particulate and gas phases, depending upon the ambient temperature. Thus, many ambient measurements will underestimate the total nitroPAHs present unless both gas- and particulate-associated species have been measured.
Analysis is hindered by a lack of adequate instrumental sensitivity or selectivity and limited availability of native and isotope-labelled standards (Chiu & Miles, 1996).
Bioassay-directed fractionation (usually using the Ames test or modifications of this with specific Salmonella typhimurium strains) and subsequent chemical characterization have been used for the identification of nitroPAHs in a number of complex mixtures (see section 2.4.5).
As most nitroPAHs are mutagenic, special precautions should be taken, even at ultratrace concentrations.
Table 3. Relative concentrations of various PAH compounds and PAH derivatives in the non-polar and moderately polar fractions of a diesel particle extract, showing only the nitroPAH fraction in detaila
|
Compound |
Fraction concentration |
||
|
Non-polar fractions |
1000 |
||
|
Total PAHs, hydrocarbons, alkylbenzenes |
1000 |
||
|
Moderately polar fractions |
1000 |
||
|
PAH ketones |
147 |
||
|
PAH carboxyaldehydes |
122.2 |
||
|
PAH acid anhydrides |
54.1 |
||
|
HydroxyPAHs |
113.1 |
||
|
PAH quinones |
71.3 |
||
|
NitroPAHs |
2.9 |
||
|
Nitrofluorenes |
0.34 |
||
|
Nitro(anthracenes and phenanthrenes) |
0.71 |
||
|
Nitrofluoranthenes |
0.05 |
||
|
Nitropyrenes |
1.5 |
||
|
Methyl nitro(pyrenes and fluoranthenes) |
0.25 |
||
|
Other oxygenated PAHs |
83.4 |
||
|
PAH carry-over phthalates, hydrocarbon contaminants |
340 |
||
a Adapted from Schuetzle (1983).
It should be noted that many nitroPAHs, in particular 9-nitro-anthracene, are unstable in the presence of light; therefore, reduced light conditions should be used (see chapter 4).
Details of methods used for the analysis of nitroPAHs in different matrices are given in Table 4. Reviews on the analysis of nitroPAHs are given by White (1985), Vincenti et al. (1996), CONCAWE (1998) and Hayakawa (2000).
Table 4. Analysis of nitroPAHsa
|
Sample type |
Extraction |
Cleanup |
Analysis |
Detector |
Detection limitb |
References |
|
|
Air |
|||||||
|
Ambient air |
Soxhlet (DCM) |
NP-LC |
GC |
MS in SIM |
Arey et al. (1987) |
||
|
Reaction chamber study |
Soxhlet (DCM) |
NP-LC |
GC |
MS in SIM |
Atkinson et al. (1987a) |
||
|
Ambient air |
Ultrasonication (DCM) |
Extensive, including |
GC |
FPD; NPD; MS EI |
Fernández & Bayona (1992) |
||
|
Air particulate |
Ultrasonication (DCM) |
Silica gel |
HPLC |
Electrochemical |
Galceran & Moyano (1993) |
||
|
Air particulate |
Ultrasonication (benzene/ethanol) |
NP-LC |
HPLC (with on-line reduction) |
CL |
0.3–5 fmol |
Murahashi & Hayakawa (1997) |
|
|
Air particulate |
Ultrasonication (DCM) |
Silica/cyclohexane |
HPLC switching technique |
FL |
20 fmol |
Zühlke et al. (1998) |
|
|
Diesel exhaust |
|||||||
|
Diesel |
Soxhlet |
NP-LC |
GC |
NPD (FL) |
Schuetzle & Perez (1983) |
||
|
Diesel particulate |
Soxhlet (DCM) |
NP-LC |
GC |
NPD |
0.5 ppm |
Paputa-Peck et al. (1983) |
|
|
Diesel or air particulates |
Soxhlet (DCM) |
Extensive; also |
GC |
FID |
Niles & Tan (1989) |
||
|
Diesel or air particulates |
Ultrasonication (acetone) |
SPE and on-line reduction |
GC |
MS EI |
1 ng/g |
Scheepers et al. (1994a) |
|
|
Air and diesel particulates |
Soxhlet (DCM) |
NP-LC |
LC |
Electrochemical FL |
60 pg |
MacCrehan et al. (1988) |
|
|
Diesel or air particulates and gaseous |
DCM |
SPE |
HRGC |
HRMS |
low ng/g (diesel particulate); pg/m3 range |
Chiu & Miles (1996) |
|
|
Vehicle or air particulates |
Ultrasonication (benzene/ethanol) |
Precolumn reduction |
HPLC (column switching) |
CL with on-line reduction |
Hayakawa et al. (1999a) |
||
|
Water |
|||||||
|
River water |
DCM; florisil column |
GC |
MS |
Takahashi et al. (1995) |
|||
|
River water |
Concentration by blue chitin column; methanol |
GC |
MS |
Nagai et al. (1999) |
|||
|
Wastewater |
Fractionation into diethyl ether-soluble neutral, acidic and basic fractions |
|
HPLC |
UV and FL |
|
Manabe et al. (1984) |
|
|
Crankcase oil |
Extraction with methanol, concentration and dissolution in water; then as above |
HPLC |
UV and FL |
Manabe et al. (1984) |
|||
|
Soil, sewage sludge, sediment |
|||||||
|
Urban dust, residual of incinerator, soil |
Soxhlet (acetonitrile) |
SPE |
GC |
NPD |
Librando et al. (1993) |
||
|
Sewage sludge |
Dimethyl formamide |
LSE or SPE |
GC |
FID; GC-MS |
Bodzek & Janoszka (1995) |
||
|
River sediment |
Ether; sodium hydroxide or |
Silica gel column; reduced/acylated |
GC |
ECD |
Sato et al. (1985) |
||
|
Soil |
Methanol |
Silica gel column; hexane/benzene/ |
Analytical ODS column for HPLC |
Reduction to amino derivatives; fluorescence |
Dinitropyrenes 0.7–4 pg |
Watanabe et al. (1999) |
|
|
Soil |
Methanol/acetone |
SPE; HPLC |
GC |
MS EI or NICI |
30 ng/kg dry weight |
Niederer (1998) |
|
|
Biological samples |
|||||||
|
Rat tissue |
Homogenate treated with acetonitrile; evaporated under nitrogen; residue dissolved in methanol; extraction with Blue Rayon |
HPLC; on-line reduction |
Fluorescence |
van Bekkum et al. (1999) |
|||
|
Human lung specimens |
Ultrasonication (DCM) |
Bioassay-directed chemical analysis, HPLC, GC |
MS |
Tokiwa et al. (1993a, 1998a) |
|||
|
Food and beverages |
|||||||
|
Various food samples |
Homogenization; acetonitrile; hexane |
Silica gel column; DCM |
GC |
TEA |
12 pg |
Dennis et al. (1984) |
|
|
Various food samples |
GC |
MS |
5 pg |
Schlemitz & Pfannhauser (1996a) |
|||
|
Grilled and smoked food samples |
HPLC; on-line reduction |
Fluorescence |
Schlemitz & Pfannhauser (1996b) |
||||
|
Fish, meat products and cheese |
HPLC; on-line reduction |
Fluorescence |
50 ng/kg |
Dafflon et al. (2000) |
|||
|
a |
Abbreviations: |
|||
|
CL |
chemiluminescence |
MS |
mass spectrometry |
|
|
DCM |
dichloromethane |
MS EI |
mass spectrometer electron impact mode |
|
|
ECD |
electron capture detector |
NICI |
negative-ion chemical ionization |
|
|
ELISA |
enzyme-linked immunosorbent assay |
NPD |
nitrogen–phosphorus detection |
|
|
FID |
flame ionization detector |
NP-LC |
normal-phase high-performance liquid chromatography |
|
|
FL |
fluorescence detection |
ODS |
octadecylsilyl |
|
|
FPD |
flame photometric detection |
ppm |
parts per million |
|
|
GC |
gas chromatography |
RP-LC |
reversed-phase high-performance liquid chromatography |
|
|
HPLC |
high-performance liquid chromatography |
SIM |
selective ion monitoring |
|
|
HRGC |
high-resolution gas chromatography |
SPE |
solid-phase microextraction |
|
|
HRMS |
high-resolution mass spectrometry |
TEA |
thermal energy analyser |
|
|
LC |
liquid chromatography |
UV |
ultraviolet |
|
|
LSE |
adsorption column chromatography on silica gel |
|||
|
b |
The percent recovery was not given in most cases. |
|||
Methods of collecting air particulates include a) Mega sampler with 50% cut-off point of 20 µm and typically 6-h sampling periods (used in Claremont, California, USA); b) an electrostatic precipitator with an impact stage designed for a 15-µm cut-off (used in Aurskog, Norway); and c) specially designed filter baghouses collected over a period of a year (National Institute of Standards and Technology [NIST] Standard Reference Materials [SRMs]) (Ramdahl et al., 1986). A 10-µm size-selective inlet (for particulate matter less than 10 µm in diameter, or PM10) sampler is also reported (Nishioka & Lewtas, 1992) .
For investigations into the formation and photochemistry of nitroPAHs, three different collection media for ambient air sampling have been used: Tenax-GC solid adsorbent, polyurethane foam (PUF) and filters for high-volume sampling (Arey et al., 1991).
Three methods for the sampling of the semivolatile phase of diesel exhaust were compared: cryogenic sampling, adsorbent sampler with XAD-2 and PUF. The PUF technique gave the highest recovery of PAHs and mutagenic activity. The three sampling techniques for the semivolatile phase resulted in extracts with different chemical composition, different mutagenic potency and different mutagenicity profiles (Westerholm et al., 1991).
In general, vapour-phase constituents are collected on solid sorbents such as XAD and PUF, and particles are collected on Teflon-impregnated glass fibre filters (see Table 4).
For sampling of diesel exhaust particulates, a dilution tunnel method is mainly used (Hayakawa, 2000). The exhaust is diluted by the filtered air in the tunnel to simulate the real road conditions, and an aliquot is sampled on the filter. Gaseous substances are trapped in PUF. The sampling and analytical methods are reviewed by Levsen (1988).
Blue cotton bearing covalently linked copper phthalocyanine trisulfonates as a ligand adsorbs polyaromatic compounds and preconcentrates several nitroPAHs in water (Hayatsu, 1992).
Extraction of filter and PUF samples can be carried out by dichloromethane (DCM) in a Soxhlet apparatus for 16 h, the Soxhlet body being loosely covered in aluminium foil to exclude light (Chiu & Miles, 1996). DCM was found to be the most efficient solvent for extraction of mutagenic compounds from diesel particles (Montreuil et al., 1992).
Toluene as solvent has also been reported (Spitzer, 1993; Vincenti et al., 1996). Supercritical fluid extraction of nitroPAHs from diesel exhaust particulate matter using carbon dioxide–chlorodifluoromethane (HCFC-22) or carbon dioxide–toluene has been demonstrated (Paschke et al., 1992).
Soil sample extraction has been described using the Soxhlet device using 1:1 (v/v) toluene:methanol (Vincenti et al., 1996) or 5% ethanol in toluene (Spitzer, 1993).
Various procedures for fractionation of particulate extracts have been described:
From open-column liquid chromatography, four main fractions are obtained by eluting with different solvents (in parentheses): aliphatics (hexane), aromatics (hexane/benzene), moderately polar (DCM) and highly polar (methanol) (Lewtas, 1988). The nitroPAHs are to be found in the moderately polar fraction, but together with a number of oxy derivatives (e.g., aldehydes, ketones, quinones), which may interfere with them. Therefore, a more effective fractionation is achieved by NP-LC, but here the nitroPAHs are not collected in a single fraction but are distributed in several fractions (Vincenti et al., 1996). Another approach to separate nitroPAHs from hydroxyPAHs uses a sequential cleanup with silica and alumina (Moyano & Galceran, 1997). Nitrated and hydroxylated PAHs extracted from air particulates could be fractionated from other micropollutants by semipreparative packed-column supercritical fluid chromatography on silica gel (Medvedovici et al., 1998).
Figure 2 shows a schematic diagram of an analytical method used for the cleanup and separation of particulate organic matter (Ciccioli et al., 1996).
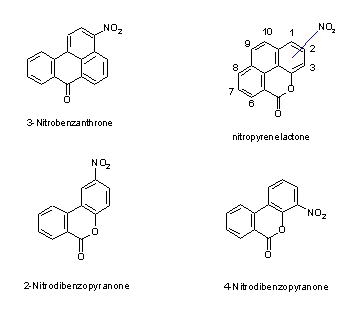
|
Fig. 2. Schematic diagram of the analytical method for the cleanup and separation of particulate organic matter (Ciccioli et al., 1996). DCM = dichloromethane; DMSO = dimethyl sulfoxide; GC-FID = gas chromatography with flame ionization detection; GC-MS = gas chromatography/mass spectrometry; HPLC = high-performance liquid chromatography. |
The most frequently used techniques for the detection of nitroPAHs are (Vincenti et al., 1996):
A thin-layer chromatography method using plates coated with a silica gel layer has been developed for analysing nitroPAHs. DCM and DCM with n-hexane and methanol were used for the mobile phases. The chromatograms developed using a mixture of n-hexane–DCM (1:1 v/v) were observed under ultraviolet (UV) light before and after being sprayed with a reducing agent (sodium borohydride dissolved in methanol and copper chloride solution). Light at lambda = 254 nm induced green fluorescence for 1-nitropyrene and 1,3-dinitropyrene only and a violet colour for the remaining compounds. Carbon disulfide quenched fluorescence (observed at lambdaexc = 365 nm) for 1-nitronaphthalene, 9-nitroanthracene, 1-nitropyrene and 1,3-dinitropyrene only (Tyrpien, 1993; Tyrpien et al., 1997 ). Janoszka et al. (1997) used acetonitrile/water as the mobile phase. The method is suggested as a simple and quick method for identifying the above nitroPAHs in airborne particulate matter after separation in moderately polar fractions by column chromatography on silica gel. Isomeric nitroPAHs cannot be separated.
More recent developments in this field include a) selective detection of several nitroPAHs by using time-of-flight MS (Bentz et al., 1995; Dotter et al., 1996; Bezabeh et al., 1997); b) a method using supercritical fluid extraction and on-line multidimensional chromatographic methods (NP-LC coupled to a high-resolution GC) and ion trap detector MS (Lewis et al., 1995a,b; Feilberg et al., 2001); and c) particle beam liquid chromatography–MS with NICI mode (Bonfanti et al., 1996). High-resolution NICI is reported to be at least 20 times more sensitive than the low-resolution NICI or EI for determination of nitroPAHs in air samples (Chiu & Miles, 1996). A method has been developed involving the derivatization of nitroPAHs to their corresponding fluorinated derivatives, followed by GC-ECD analysis. The sensitivity of the method is an order of magnitude higher than that of direct GC-ECD analysis of nitroPAHs themselves. This method is suitable for routine monitoring of nitroPAHs in air samples (Jinhui & Lee, 2001).
Owing to small differences in GC retention times for 2- and 3-nitrofluoranthene, the 2-nitrofluoranthene present in some samples was incorrectly reported as 3-nitrofluoranthene in earlier reports. After reanalysis, the original reports were corrected (Ramdahl et al., 1986; Nishioka et al., 1988). Use of more selective stationary phases enabled better separation of isomeric pairs (Ciccioli et al., 1988). For the best separation of all of the nitrofluoranthene and nitropyrene isomers, the use of a DB13 (or SP30-type) column is recommended (A. Cecinato, personal communication, 2002).
Sampling gas-phase PAHs from environmental chambers onto Tenax adsorbent under conditions not typical of ambient atmospheres (e.g., 19 mg nitrogen dioxide/m3, 44 mg nitrogen pentoxide/m3) can lead to artificial formation of nitro derivatives via reactions with the Tenax adsorbent (Zielinska et al., 1986a).
The chemical analysis of trace levels of organic mutagens in ambient air is more complex than comparable analyses of emissions from specific sources. Source-emitted pollutants account for only part of the whole ambient air sample. Aging of air samples introduces unknown, highly variable chemical and meteorological factors. Further, the concentrations of pollutants are much lower in ambient air than at the sources (Greenberg et al., 1993).
The procedures followed for investigating nitro-oxyPAHs in emissions and ambient air are, in general, the same as those adopted for nitroPAHs.
Bioassay-directed fractionation closely coupled to chemical characterization has been developed as a method of determining nitroPAHs in complex mixtures (see Figure 3) (Schuetzle & Lewtas, 1986; Lewtas, 1988; Lewtas & Nishioka, 1990; Legzdins et al., 1995; Enya et al., 1997). In this approach, the complex mixture is fractionated, and each fraction is bioassayed; the mutagenic activity for each (HPLC) fraction is plotted in a manner analogous to a conventional chromatogram, and the plot is referred to as a mutagram (mutagenicity profile = mutagram).
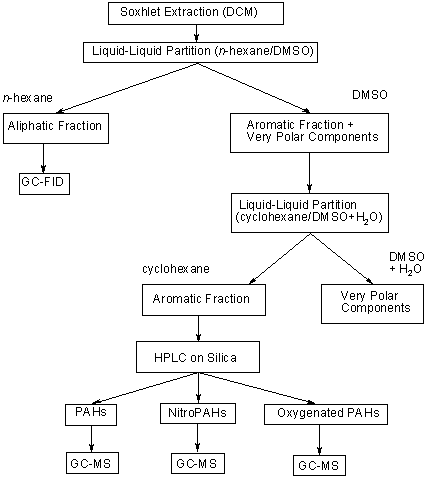
Fig. 3. Bioassay-directed chemical analysis scheme for the determination of air particulate matter. The numbers under each fraction represent the percent distribution of mass and mutagenicity in Salmonella tester strains (in parentheses); adapted from Schuetzle & Lewtas (1986) and Lewtas et al. (1990a).
Mutagenically active fractions are further fractionated, bioassayed and characterized until the major class of compounds or specific com-pounds potentially responsible for the mutagenicity are identified. Use of bacterial tester strains selectively sensitive to nitroarenes has led to the identification of nitroPAHs as potent mutagens in complex mixtures from diverse sources.
Non-polar fractions of an extract of diesel particulates (SRM 1650) accounted for less than 2–3% of the mutagenicity in the total extract. The distribution of mutagenicity in the moderately polar and polar fractions was dependent on the source sample (Schuetzle & Lewtas, 1986).
Studies using this bioassay-directed fractionation and chemical characterization (see also section 7.5.5) include, for example, studies of diesel exhaust extracts (Schuetzle et al., 1981; Nishioka et al., 1982; Claxton et al., 1992; Legzdins et al., 1994; Enya et al., 1997; Hayakawa et al., 1997), xerographic toners (Rosenkranz et al., 1980), cigarette smoke (Kier et al., 1974), ambient atmospheric particles (Nishioka et al., 1988; Lewtas et al., 1990a; Arey et al., 1992; Casellas et al., 1995; Hayakawa et al., 1995b) and the metabolites of 1-nitropyrene (Lewtas et al., 1990b).
For example, Casellas et al. (1995) made a detailed chemical analysis of mutagenic fractions using bioassay-directed chemical analysis in urban airborne particulate matter in Barcelona, Spain. The first fractionation of the solvent (DCM) extractable organic matter was achieved by semipreparative gel permeation chromatography (GPC). The second fractionation was achieved with NP-LC. The collected fractions were tested for mutagenicity using the S. typhimurium microsome assay with strains TA98, TA98NR– and TA98AT– (for more details, see chapter 7). The chemical characterization of mutagenic fractions was carried out by an extensive application of capillary GC-MS in the EI and NICI modes. Those fractions exhibiting the highest levels of mutagenicity were subjected to a third level of fractionation by reversed-phase HPLC (RP-LC) and analysed by GC-MS. Two sampling sites in Barcelona were monitored during 1990. Samples (24 h) of air particulate matter over periods of 1 week per season were processed.
The direct mutagenicity in the fractions NP-LC 3 (and 4) isolated from the GPC-2 fraction of airborne particulate matter collected in Barcelona (autumn 1990) seemed to be accounted for by nitrated arenes, 9-nitroanthracene, 2-nitrofluoranthene and 2-nitropyrene. 6-Nitrobenzo[a]pyrene in polar fraction 5 needed application of metabolic activation (+S9) for mutagenicity. In order to evaluate the contribution of nitro derivatives to the total mutagenicity, the NP-LC fractions were tested against TA98NR– and TA98AT–. Generally, a remarkable decrease in mutagenic activity was observed in all fractions; these decreases were more apparent in fractions NP-LC3 to NP-LC7, thus suggesting a significant contribution of nitroarenes to the mutagenicity of these fractions of medium and high polarity. Subfraction NP-LC2 was fractionated further. The expected composition was PAHs, but two nitro derivatives, 2-nitrofluoranthene and 1-nitropyrene, were identified in subfraction RP-LC3, which may be responsible for the high direct-acting mutagenic activity observed in this fraction and in the NP-LC2 fraction (Casellas et al., 1995).
NitroPAHs originate primarily as direct or indirect products of incomplete combustion. Only a few nitroPAHs are produced industrially (e.g., nitronaphthalenes and 5-nitroacenaphthene).
Treatment of naphthalene with mixed sulfuric/nitric acids at 60 °C yields 95% 1-nitronaphthalene and 5% 2-nitronaphthalene, together with traces of dinitronaphthalene and dinaphthol. Higher temperatures (80–100 °C) result in a mixture of 1,5- and 1,8-dinitronaphthalene in about a 2:3 ratio (Booth, 1991). The dinitro isomers can then be further separated for specific uses.
Nitronaphthalenes are produced in Germany and Japan. The production capacity of dinitronaphthalenes in Japan was 1200 tonnes per year (no year given; Booth, 1991).
1-Nitronaphthalene is used almost exclusively for catalytic reduction to 1-naphthylamine. Further uses, such as use as a deblooming agent for petroleum and oils and as a component in the formulation of explosives, are of historical interest only (Booth, 1991).
1,5-Dinitronaphthalene is an intermediate in the production of naphthazarin (5,8-dihydroxy-1,4-naphthoquinone) and 1,5-naphthalenediamine, which is mainly converted to naphthalene 1,5-diisocyanate. It is further used as a sensitizing agent for ammonium nitrate explosives (Booth, 1991).
1,8-Dinitronaphthalene is catalytically hydrogenated to 1,8-naphthalenediamine for use mainly as a colorant intermediate for naphthperinones (Booth, 1991).
5-Nitroacenaphthene is reported to be an intermediate in the synthesis of naphthalimide dyes that are used as fluorescent whitening agents and photochemical agents (Yahagi et al., 1975; IARC, 1978).
NitroPAHs in the environment originate from direct emissions from combustion sources and nitration of PAHs, primarily in the atmosphere.
The nitroPAHs emitted from combustion sources are nitroPAHs that would be formed through electrophilic nitration (e.g., 1-nitropyrene and 2-nitrofluorene; see section 3.2.1). NitroPAHs have been observed in vehicle exhaust (particularly diesel), industrial emissions and emissions from domestic residential heating/cooking and wood burning.
NitroPAHs are formed in the atmosphere from PAHs by the following reactions:
These processes are described in more detail in section 3.2.2. Other less important pathways, which are briefly mentioned here, include:
The distribution of nitroPAH isomers in samples of ambient air has been found to be significantly different from that in direct emissions from combustion (compare Table 5 and chapter 5). For example, 2-nitrofluoranthene and 2-nitropyrene are ubiquitous components of particulate matter that have been detected in urban, suburban, forest and remote areas located in Europe, America, Asia and Antarctica (Ciccioli et al., 1996), although they are not directly emitted from most combustion sources (see chapter 5). The nitroPAH profile, or the relative quantities of certain "marker" PAHs, is a pointer to the source of formation of nitroPAHs — for example, markers of direct emissions from combustion, in particular diesel exhaust, are 1-nitropyrene and 2-nitrofluorene, whereas the presence of 2-nitrofluoranthene and 2-nitropyrene points to atmospheric transformation.
Table 5. NitroPAHs detected in diesel emissionsa,b
|
Parent PAH; |
Concentration (ppm = µg/g, unless otherwise stated) |
||||||||||||||
|
a) |
b) |
c) |
d) |
e) |
f) |
g) |
h) |
i) |
j) |
k) |
l) |
m) |
n) |
o) |
|
|
Naphthalene |
|||||||||||||||
|
1-Nitro- |
X (nd) |
X |
X |
0.7 |
0.88 |
0.013 |
|||||||||
|
2-Nitro- |
nd |
X |
X |
0.02 |
0.039 |
||||||||||
|
1,3-Dinitro- |
X |
X |
|||||||||||||
|
1,5-Dinitro- |
X (nd) |
X |
X |
||||||||||||
|
1,8-Dinitro- |
X (nd) |
||||||||||||||
|
Acenaphthene |
|||||||||||||||
|
3-Nitro- |
X* |
||||||||||||||
|
Fluorene |
|||||||||||||||
|
1-Nitro- |
5.91* |
0.08* |
|||||||||||||
|
2-Nitro- |
X |
X |
X |
nd |
0.11 |
4.1 |
<0.01 |
0.27 |
0.001 |
X |
27 |
8.77 |
0.99 |
nd |
|
|
2,5-Dinitro- |
X (nd) |
||||||||||||||
|
2,7-Dinitro- |
X (nd) |
0.13 |
|||||||||||||
|
Anthracene |
|||||||||||||||
|
1-Nitro- |
X* |
300* |
4.6 |
X* |
|||||||||||
|
2-Nitro- |
X |
X |
10.1 |
||||||||||||
|
9-Nitro- |
X |
X |
X |
2.8 |
6.02 |
10 |
1.19 |
1.36 |
7 |
63 |
1.00 |
||||
|
Phenanthrene |
|||||||||||||||
|
1-Nitro- |
0.5 |
||||||||||||||
|
2-Nitro- |
X |
X |
X |
16.5* |
1.8* |
||||||||||
|
3-Nitro- |
9.3 |
||||||||||||||
|
4-Nitro- |
0.7 |
||||||||||||||
|
9-Nitro- |
2.3 |
0.37 |
0.18 |
0.27 |
|||||||||||
|
Fluoranthene |
|||||||||||||||
|
1-Nitro- |
X (nd) |
X |
4.1 |
X* |
|||||||||||
|
2-Nitro- |
nd |
X |
0.06 |
||||||||||||
|
3-Nitro- |
X (nd) |
X |
1 |
10 |
0.33 |
1.34 |
1.47 |
<1 |
10 |
0.06 |
|||||
|
7-Nitro- |
X (nd) |
X |
1.6 |
||||||||||||
|
8-Nitro- |
X (nd) |
X |
2.0 |
0.25 |
|||||||||||
|
Pyrene |
|||||||||||||||
|
1-Nitro- |
75 |
X |
X |
590 |
2.84 |
43 |
18; 19.6* |
20 |
16.6; 18.0* |
16.4 |
19 |
450 |
0.2 (+1,3-DNP) |
3.43 |
6.49 |
|
2-Nitro- |
nd |
||||||||||||||
|
4-Nitro- |
nd |
0.07 |
0.04 |
||||||||||||
|
1,3-Dinitro- |
0.30 |
X |
0.6 |
<0.1 |
0.58 |
0.6 |
with 7-NP |
0.12 all DNP |
0.17 |
||||||
|
1,6-Dinitro- |
0.40 |
0.6 |
<0.1 |
0.64 |
1.39 |
0.37 |
0.04 |
||||||||
|
1,8-Dinitro- |
0.53 |
0.4 |
<0.1 |
0.49 |
1.55 |
0.07 |
|||||||||
|
Benz[a]anthracene |
|||||||||||||||
|
7-Nitro- |
nd |
X |
8* |
0.45 |
2.8 |
1.99 |
1.62 |
<1 |
10 |
0.61 |
|||||
|
Chrysene |
|||||||||||||||
|
1-Nitro- |
X (nd) |
0.8* |
2.9* |
||||||||||||
|
5-Nitro- |
4.6 |
||||||||||||||
|
6-Nitro- |
X |
0.04 |
0.87 |
0.782 |
X |
4 |
0.08 |
0.10 |
|||||||
|
Benzo[a]pyrene |
|||||||||||||||
|
1-Nitro- |
1- + 3- |
10* |
1.2* |
1.4* |
|||||||||||
|
6-Nitro- |
X |
X |
5.8 |
1.4 |
1.6 |
0.641 |
X |
0.12 |
0.30 |
||||||
|
Benzo[e]pyrene |
|||||||||||||||
|
1-Nitro- |
0.83 |
||||||||||||||
|
3-Nitro- |
2.2 |
||||||||||||||
|
Perylene |
|||||||||||||||
|
3-Nitro- |
X |
X |
0.64 |
||||||||||||
|
a |
E = extract; P = particulate; DNP = dinitropyrene; 7-NP = 7-nitropyrene; X = detected; * = mixture of isomers; X (nd) = detected by GC/nitrogen–phosphorus detector (NPD) (0.5 µg/g) but not by GC-MS (detection limit 5 µg/g); nd = not detected by either. |
|
|
b |
Samples were as follows: |
|
|
a) |
Light-duty diesel particulate extract (Paputa-Peck et al., 1983). |
|
|
b) |
Levsen (1988); Schilhabel & Levsen (1989). Thirty-eight nitroPAHs were detected (including methyl-nitroPAHs and mixed isomers, not noted in the table). Identification by comparison with reference compounds or relative retention times reported by Paputa-Peck et al. (1983). |
|
|
c) |
Hartung et al. (1984). Exhaust from light-duty diesel test engine. |
|
|
d) |
Concentration in extract from VW Rabbit diesel (Nishioka et al., 1982, 1983). |
|
|
e) |
Yu et al. (1984). |
|
|
f) |
Campbell & Lee (1984); g/g light-duty diesel particulate extract (US Environmental Protection Agency [EPA] recalculated concentrations from mg/g extract to g/g particle using a value of 44% for extractable material). |
|
|
g) |
Diesel particulate SRM 1650 (Chiu & Miles, 1996). |
|
|
h) |
Diesel particulate SRM 1650 (MacCrehan et al., 1988). |
|
|
i) |
Diesel extract SRM 1975 (Chiu & Miles, 1996). |
|
|
j) |
SRM 1975 diesel particle extract (DCM extract of diesel particulate matter collected from an industrial diesel-powered forklift) (NIST, 2000) (SRMs are analytical reference samples for quality control, not implied as representative of environmental diesel samples). |
|
|
k) |
Heavy-duty diesel particulate matter (SRM 1650) (Niles & Tan, 1989). |
|
|
l) |
NitroPAHs extracted from bus soot (Paschke et al., 1992). Bus soot contained much higher concentrations of 1-nitropyrene than standard diesel sample SRM 1650 (450 compared with 20 µg/g); extraction by carbon dioxide–HCFC-22. |
|
|
m) |
Particulate nitroPAH concentration; 1988 Cummins LTA engine operated under baseline conditions (no trap) at US EPA steady-state engine mode 9; mean of n = 3; vapour-phase exhaust was >0.025 µg/m3. Details also given with mode 11; with and without ceramic particle trap and with copper fuel additive (Harvey et al., 1994). |
|
|
n) |
µg/km US Federal Test Procedure 72 (FTP-72) cycle with hot start, four-cylinder engine, light diesel vehicle (n = 3–6) (Scheepers & Bos, 1992a). |
|
|
o) |
pmol/mg; particulate emission from 1995 diesel engine vehicle, idling engine (Hayakawa et al., 1997). |
|
The majority of ambient nitroPAHs are now thought to be formed in the atmosphere from the gas-phase reactions of PAHs with four rings or less (Atkinson & Arey, 1994).
1) Qualitative and quantitative studies of nitroPAHs in diesel exhaust
NitroPAHs have been detected in particulate exhaust emissions of motor vehicles, in particular diesel exhaust emissions, together with hundreds of other organic compounds (see Table 3; Schuetzle, 1983). Interest was focused on nitroPAHs in the early 1980s because correlations were found between the presence of nitroPAHs in diesel exhaust (and environmental extracts) and mutagenic activity in Salmonella typhimurium (see chapter 7). A large number of groups of nitro, oxy and mixed nitro-oxy compounds eluted together in the mutagenic fractions. Analytical methods were developed to separate and identify as many mononitro- and dinitroPAHs in diesel exhaust as technically possible, first as isomer groups and then using isomer-specific identification (e.g., Schuetzle et al., 1981, 1982; Newton et al., 1982; Xu et al., 1982; Henderson et al., 1983; Paputa-Peck et al., 1983; Campbell & Lee, 1984; Levsen, 1988; Niles & Tan, 1989; Schilhabel & Levsen, 1989; Chiu & Miles, 1996; see also Table 5). The number of nitroPAHs quantified is generally limited due to lack of standards. Investigators have used different samples of diesel exhaust as well as different analytical methods. Further, the concentration of nitroPAHs adsorbed on diesel particulate varies substantially from sample to sample (Levsen, 1988). It is therefore difficult to compare the various nitroPAH profiles. Usually, 1-nitropyrene is the predominant component, and concentrations of 7–165 µg/g particulate have been reported (Levsen, 1988). However, 1-nitropyrene is not always the dominating substance. Especially in heavy-duty diesel, 2-nitrofluorene may exceed 1-nitropyrene by a factor of 1.8–15, with an average of 9.8. When 1-nitropyrene is the dominant substance, a 2-nitrofluorene concentration equal to 15% of that of 1-nitropyrene is common (Beije & Möller, 1988a).
Some studies have focused on measuring concentrations of specific nitroPAHs — for example, 1-nitropyrene as a marker of nitroPAH formation (see below), 2-nitrofluorene (Beije & Möller, 1988a; Möller et al., 1993a) or dinitroPAHs, in particular 1,3-, 1,6- and 1,8-dinitropyrenes, which have been found to be more mutagenic (although not necessarily more carcinogenic; see chapter 7) than mononitroPAHs (Pederson et al., 1984; Hayakawa et al., 1992, 1994) (see Table 6). Dinitropyrenes are formed from the further nitration of 1-nitropyrene and are present at only about 1% of its concentration (Schuetzle & Frazier, 1986). Mononitrofluoranthene and mononitropyrenes were separated and identified in diesel extracts (Ciccioli et al., 1988).
Table 6. Concentrations of 1-nitropyrene and 1,3-, 1,6- and 1,8-dinitropyrene in diesel particulate and gasoline exhausta
|
NitroPAH |
Concentration (µg/g) |
||||||
|
a) |
b) |
c) |
d) |
e) |
f) |
g) |
|
|
1-Nitropyrene |
3.9 |
3.9–116 |
13 |
37 |
0.32 |
0.44 |
2.1 (<5) |
|
1,3-Dinitropyrene |
<0.005 |
0.07 |
0.08 |
0.05 |
0.06 |
||
|
1,6-Dinitropyrene |
0.033 |
0.04–4.47 |
0.07 |
0.15 |
0.06 |
0.12 |
|
|
1,8-Dinitropyrene |
0.013 |
0.04–6.24 |
0.06 |
0.23 |
0.08 |
0.10 |
|
|
a |
Samples were as follows: |
|
|
a) |
From 1978 Opel diesel (Gibson, 1982, 1983). |
|
|
b) |
Range of nitropyrenes detected in diesel particulate from five different motors (1979–1983) under different driving speeds and cycles and collected by different methods (Pederson et al., 1984). |
|
|
c) |
Mean from seven diesel vehicles (1983–1991) (Hayakawa et al., 1994). |
|
|
d) |
Diesel exhaust from 1980 Isuzu truck (Sera et al., 1994). |
|
|
e) |
Gasoline exhaust from 1985 Toyota automobile (Sera et al., 1994). |
|
|
f) |
Exhaust from eight gasoline vehicles (year not given) (Hayakawa et al., 1994). |
|
|
g) |
On-road emission factors for 1-nitropyrene of 0.49 versus <0.03 µg/km (2.1 versus <5 µg/g, by mass of the extracted particulate matter) were given for heavy-duty diesel trucks and spark ignition gasoline light-duty passenger cars, respectively (Gorse et al., 1983). |
|
2) Formation of nitroPAHs and comparison of emissions
NitroPAHs are formed in vehicle engines from the reactions of PAHs with nitrating species that are provided by the conversion of nitrogen and oxygen at high temperatures in the combustion chamber (Scheepers & Bos, 1992a).
Studies compare the emissions of nitroPAHs from various engines (Henderson et al., 1983), under differing driving conditions (different speeds, loads, etc.) (Schuetzle & Perez, 1983; Draper, 1986; Lorber & Mollenhauer, 1989; Veigl et al., 1994), with and without an oxidation catalytic converter (Johnson et al., 1994; Mitchell et al., 1994; Pataky et al., 1994), with and without catalysed or non-catalysed particulate traps (Westerholm et al., 1986; Bagley et al., 1993; Harvey et al., 1994; Johnson et al., 1994) and using a variety of fuels and/or additives (Harvey et al., 1994; Johnson et al., 1994).
1-Nitropyrene concentrations in exhaust (given as µg/g dust) from heavy-duty trucks during simulated driving cycles are as follows: suburban, 1.94; urban, 3.39; and motorway, 1.94 and 2.73 (Scheepers et al., 1994a). 1-Nitropyrene emission rates in exhaust emissions from a heavy-duty diesel vehicle during transient driving conditions were 1.6 µg/km in the particulate and <0.05 µg/km in the semivolatile phase (Westerholm et al., 1991). Light-duty trucks with oxidation catalysts emitted 1-nitropyrene at 0.36 µg/km with the US Federal Test Procedure 75 (FTP-75) driving cycle and 0.32 and 0.22 µg/km with the European driving cycle (cold and hot start, respectively; Scheepers et al., 1994a).
In a multivariate analysis of exhaust emissions, 10 different fuels were combusted using two different types of heavy-duty diesel engines. The levels of 1-nitropyrene in the exhaust from the different fuels ranged from 0.07 to 2.17 µg/km in one engine and from 0.32 to 7.19 µg/km in the other. It was not found that one fuel gave a high 1-nitropyrene emission in both engine types. There was a negative correlation between 1-nitropyrene and nitrogen oxides and pyrene content, indicating the formation of 1-nitropyrene from the reaction between nitrogen oxides and pyrene (Sjögren et al., 1996).
Studies using low-sulfur fuel showed that, independent of the type of engine and exhaust after-treatment device, the primary effect of reducing sulfur content in diesel fuel was to substantially decrease the sulfate emissions and the number of respirable particles (Bagley et al., 1996). The total particulate matter emissions were not significantly diminished. However, because sulfate particles are so small, decreases in the emissions of these particles do not significantly diminish the emissions of total particulate matter. Using low-sulfur fuel had no significant effect on emissions of semivolatile organic compounds in the vapour phase or of soluble organic compounds in the particulate phase. However, the emission of hydrocarbon gases, the emission of some PAHs in the particulate and vapour phases and the mutagenicity of particulate-phase fractions were significantly elevated under some operating conditions, which may have been due to differences in the hydrocarbon composition of the low-sulfur fuels compared with that of conventional, high-sulfur fuels. This study did not measure nitroPAHs but should be relevant for these compounds (Bagley et al., 1996).
Particulate-associated and vapour-phase emissions of 2-nitrofluorene, 1,3-, 1,6- and 1,8-dinitropyrene, 1-nitropyrene, 3-nitrofluoranthene, 7-nitrobenz[a]anthracene and 6-nitrochrysene were measured with a low-sulfur (0.01% by mass sulfur) fuel and a particulate trap at steady-state mode 9 (Johnson et al., 1994; Table 7). Relatively large amounts of nitroPAHs were found in the vapour phase. In contrast to other studies involving diesel exhausts, it is not clear why the dinitropyrene concentrations that were detected are so high relative to the 1-nitropyrene concentration (see also Harvey et al., 1994, in Table 5). The use of a low-sulfur fuel with the particulate trap is known to alter particle size distributions and partitioning of PAHs (see Table 8).
Table 7. Particulate-associated (SOF) and vapour-phase (XOC) nitroPAH emissions with a low-sulfur fuel and a
particulate trap at steady-state mode 9a
|
Phaseb |
Mean nitroPAH level (ng/m3) |
|||||||
|
2-Nitro- |
1,6/1,8-Dinitropyrene |
1,3-Dinitropyrene |
1-Nitropyrene |
3-Nitro- |
7-Nitro- |
6-Nitrochrysene |
||
|
Baseline |
SOF |
420 |
550 |
420 |
120 |
1020 |
<24 |
<21 |
|
XOC |
<25 |
830 |
82 |
77 |
<38 |
10 |
140 |
|
|
Trap |
SOF |
920 |
1200 |
100 |
≤16 |
25 |
≤20 |
<12 |
|
XOC |
<16 |
1300 |
240 |
130 |
<24 |
170 |
38 |
|
a From Johnson et al. (1994).
b SOF = soluble organic fraction; XOC = extractable organic component.
Table 8. Changes in PAH emissions with a non-catalysed particulate trapa
|
Fuel type |
Particulate-associated PAH (%) |
Vapour-phase PAH (%) |
Overall PAH (%) |
|
Commercial No. 2 (0.32% sulfur) |
–25 |
+15 |
+2 |
|
"Low sulfur" (0.01% sulfur) |
–65 |
+140 |
+18 |
a From CONCAWE (1998).
Low-sulfur no. 2 diesel fuel and 100% soy methyl ester biodiesel fuel were tested with and without an oxidation catalytic converter over a light-duty transient test cycle (Bagley et al., 1998). Of the nitroPAH compounds analysed (1-nitropyrene, 2-nitrofluorene, 6-nitrochrysene and 1,3- and 1,6-dinitropyrenes), only 1-nitropyrene was found in quantifiable levels in all particle-associated samples (although 1,3-dinitropyrene may also be present, as it co-elutes with 1-nitropyrene in the method used in this study). The use of the oxidation catalytic converter with low-sulfur no. 2 diesel fuel reduced 1-nitropyrene by 66%, but this was not significant.
From CONCAWE’s review of studies on PAHs in automotive exhaust emissions, it can be said that, in general, after-treatment systems can substantially decrease PAH emissions. Diesel oxidation catalysts may be more effective in reducing the vapour-phase PAHs, whereas particulate traps seem to deal more effectively with PAHs condensed onto particulate matter. The few data on the effects of after-treatment (e.g., catalysts) on nitrated PAHs and the associated mutagenicity of the exhaust are variable, and the results of different studies can be contradictory (CONCAWE, 1998).
However, there does seem to be increasing evidence in recent studies that nitroPAHs, in particular in volatile and semivolatile fractions, are still emitted in diesel exhaust emissions. For example, in a study by Sharp (2000), all of the PAH and nitroPAH compounds investigated were present in the exhaust of all three engines tested (Cummins N14, DDC Series 50 and Cummins B5.9) when operated on a special batch of diesel fuel, blended to meet the stringent specifications required by the US Clean Air Act, at levels well above the US Environmental Protection Agency (EPA) required threshold of 0.5 ng/hp-h (Table 9). Further, Sharp (2000) showed that emissions of PAH and nitroPAH compounds were substantially lower with biodiesel than with conventional diesel fuel. This is not unexpected, when considering that the biodiesel contains no aromatics and no PAH compounds. The catalyst-out nitroPAH data presented an unexpected trend, in that catalyst-out nitroPAH levels were significantly higher than engine-out levels. This trend was most evident with the lighter nitroPAH compounds.
Table 9. PAHs or nitroPAHs measured in exhaust from different types of engine with diesel fuel with and without an oxidation catalysta
|
PAH or nitroPAH |
Mass in exhaust (ng/hp-h) |
||||
|
Cummins N14 |
DDC 50 |
Cummins B5.9 |
|||
|
No catalyst |
No catalyst |
Catalyst |
No catalyst |
Catalyst |
|
|
Benzo[a]pyrene |
1435 |
1614 |
226 |
723 |
241 |
|
2-Nitrofluorene |
123 |
88 |
90 |
257 |
478 |
|
1-Nitropyrene |
82 |
83 |
76 |
210 |
2171 |
|
7-Nitrobenz[a]anthracene |
1.9 |
1.4 |
1.4 |
34 |
60 |
|
6-Nitrochrysene |
0.8 |
0.8 |
5.8 |
11 |
56 |
|
6-Nitrobenz[a]pyrene |
2.5 |
11.1 |
4.2 |
6.1 |
6.8 |
a From Sharp (2000).
Diesel fuel, engine types and catalytic traps/converters are continually being modified, so the various studies of nitroPAHs in diesel exhaust cannot be directly compared. A detailed survey of nitroPAHs in exhaust emissions under various conditions is beyond the scope of this document.
Although diesel use is likely to increase during the coming years (Lloyd & Cackette, 2001), a relatively small number of tests have been conducted to determine the gaseous, semivolatile and particulate organic matter in diesel fuel and exhaust. Very little is known about how this composition changes with different operating conditions and the introduction of new technologies. Further understanding will require significant improvements in the analytical methods and procedures used in emissions testing of diesel engines (Chow, 2001).
3) Diesel engine oil
New diesel engine oil did not contain 1-nitropyrene at a detection limit of 0.1 mg/litre; after a vehicle travel distance of 9000 km, however, 0.5 mg/litre was detected in the used oil. The presence of 1-nitropyrene in diesel engine oil points to another source of environmental contamination (e.g., used engine oil; Jensen et al., 1986).
Concentrations of some nitroPAHs detected in the exhaust particulate from mufflers of gasoline engines were much lower than those from diesel engines — for example, for 1-nitropyrene (0.16 versus 27.7 µg/g tar, respectively) and 2-nitrofluorene (0.16 versus 5.52 µg/g tar, respectively) (Handa et al., 1983). These differences were not as pronounced in a comparative study of 1-nitropyrene in the soluble organic fraction (SOF) of particles from exhaust emissions of in-use gasoline- and diesel-powered passenger cars under simulated driving conditions (mean 27.4 versus 53 µg/g, respectively) (Tejada et al., 1986).
Particulate-associated 1-nitropyrene was emitted at a rate of 0.03–0.05 µg/km driving distance from two three-way catalyst-equipped light-duty gasoline-fuelled vehicles using the US FTP-75 driving cycle under three different driving conditions — cold transient, stabilized and hot transient (Westerholm et al., 1996). (Three-way catalysts for gasoline exhaust seem to be effective in reducing both vapour- and particulate-phase PAHs [CONCAWE, 1998].)
Although the concentration of 1-nitropyrene, for example, was less in gasoline particles than in diesel particles, the concentrations of 1,3-, 1,6- and 1,8-dinitropyrene were found to be almost the same (Hayakawa et al., 1994; Sera et al., 1994; see Table 6).
Studies on the mutagenicity of aircraft (jet aeroplane) particulate extracts suggested the presence of nitroarenes, but no analytical determination was made (McCartney et al., 1986). The mutagenicity derived from idling aircraft was greater than that collected across an active runway. This is in agreement with the finding that particulates from idling aircraft are much richer in PAHs than those collected during simulated landings and takeoffs (Robertson et al., 1980). PAHs emitted during simulated idling are greatly enriched with respect to three- and four-ring structures. The majority of organic pollutants in airports come from idling aircraft. Takeoffs contribute only 1–2% to the total burden (Gelinas & Fan, 1979). In simulation experiments, the particulate-adsorbed PAHs constitute less than 1% of the total PAHs emitted by aircraft, the remainder residing in the gaseous phase (Robertson et al., 1980).
NitroPAHs have been detected in the emissions of kerosene heaters, fuel gas and liquefied petroleum gas (LPG) burners, which are used in many countries (e.g., Japan, China, Taiwan; also mobile homes in USA) for heating and cooking at home (Tokiwa et al., 1985, 1990a; Kinouchi et al., 1988).
In a study on indoor air concentrations in mobile homes with kerosene heaters, 1-nitronaphthalene, 2- or 3-nitrofluoranthene and 1-nitropyrene were found in the particulate phase (dinitropyrenes were below the detection limit). In the semivolatile organic fraction, from these nitroPAHs, only 1-nitronaphthalene was detected (naphthalene itself was present at a concentration more than 1000-fold higher than the 1-nitronapthalene concentration) (Mumford et al., 1991).
Fume samples from three different commercial cooking oils frequently used in Taiwan were collected and analysed. As well as several PAHs (benzo[a]pyrene [BaP], benz[a]anthracene and dibenz[a,h]anthracene), two nitroPAHs were also identified. Concentrations of 1-nitropyrene and 1,3-dinitropyrene were, respectively, 1.1 and 0.9 µg/m3 in fumes from lard oil, 2.9 and 3.4 µg/m3 from soybean oil, and 1.5 and 0.4 µg/m3 from peanut oil (Wu et al., 1998). (The incidence of lung cancer in Chinese women is relatively high and is thought to be associated with cooking practices [Ko et al., 1997].)
Williams et al. (1986) reported significant levels of 18 different species of nitroPAHs in diesel extract but did not find any in coke oven mains, roofing tar vapour or cigarette smoke condensate at a detection level of <50 pg. The lack of 1-nitropyrene, 1-nitronaphthalene and 6-nitrochrysene in mainstream cigarette smoke was also shown by El-Bayoumy et al. (1985).
In another study, coke oven emission extractable organic matter was found to contain 3-nitrophenanthrene and 1-nitropyrene (66 and 27 ng/mg, respectively) in the slightly polar fraction and 9-nitroanthracene and 3-nitrophenanthrene (78 and 23 ng/mg, respectively) in the acidic fraction. However, 9-nitrophenanthrene, 3-nitrofluoranthene, 6-nitrochrysene and 6-nitrobenzopyrene were not detectable. In vitro mammalian cell cultures were used to determine whether DNA adducts derived from individual nitroPAHs or from organic extracts of coke oven emissions can be detected. Using the 32P-postlabelling method, 4-nitropyrene, 6-nitrochrysene and 3-nitrofluoranthene were reported to cause 10–100 DNA adducts per 108 nucleotides. When the extractable organic matter was used, the results suggest that nitroPAH adducts (detected by 32P-postlabelling) were present and may contribute to the genotoxicity of coke oven emissions (Topinka et al., 1998).
Nitroarenes were found to be an important contributor to the mutagenic activity of the emissions from municipal waste incinerators (DeMarini et al., 1996; see also section 7.5.5.7). 1-Nitropyrene was identified in coal fly ash (Harris et al., 1984). NitroPAHs, in particular 2-nitrofluoranthene and 2-nitropyrene, have been detected in the stack emissions of a plant manufacturing carbon electrodes (Ciccioli et al., 1988, 1989).
The gas-phase formation of nitroPAHs in the atmosphere was first proposed by Pitts et al. (1985a) to explain the unexpected presence of 2-nitrofluoranthene and 2-nitropyrene in particulate organic matter sampled in the Los Angeles basin in California, USA (see chapter 5). These two nitroPAHs have not been identified in diesel exhaust or other combustion products. The occurrence of these nitroPAHs in different locations of southern California (Arey et al., 1987; Atkinson et al., 1987a; Zielinska et al., 1989a) and Europe (Nielsen & Ramdahl, 1986; Ramdahl et al., 1986; Ciccioli et al., 1989; Feilberg et al., 2001) as well as in forest and remote areas of America and Asia (Ciccioli et al., 1996; see chapter 5 and Table 19) showed their ubiquitous presence. Further studies showing differences in daytime/nighttime concentrations of certain nitroPAHs under various climatic conditions (see Figure 6 in chapter 5) provided further support for the gas-phase formation of nitroPAHs.
A further confirmation of the atmospheric origin of 2-nitrofluoranthene is the finding of its absence in tunnel air (Queensway, Birmingham, United Kingdom) and its abundant presence in the ambient atmosphere of this city (Dimashki et al., 2000; see also Table 10).
Table 10. Measurements of nitroPAHs in the atmosphere showing distribution in particulate and vapour phasesa
|
Compound |
Particulate (ng/m3) |
Vapour (ng/m3) |
||
|
Mean |
Range |
Mean |
Range |
|
|
1-Nitronaphthalene |
<0.61x10–4 |
0.089 |
0.033–0.207 |
|
|
2-Nitronaphthalene |
<1.4x10–4 |
0.067 |
0.027–0.176 |
|
|
9-Nitroanthracene |
0.130 |
0.034–0.520 |
0.057 |
0.014–0.177 |
|
1-Nitropyrene |
0.090 |
0.019–0.204 |
<0.22x10–4 |
|
|
2-Nitrofluoranthene |
0.221 |
0.046–0.586 |
<0.21x10–4 |
|
|
7-Nitrobenz[a]anthracene |
0.033 |
0.011–0.059 |
<0.74x10–4 |
|
a From Dimashki et al. (2000); Birmingham, United Kingdom (November 1995 – February 1996).
Two-ring nitroPAHs are present mainly in the vapour phase; under some climatic conditions, other nitroPAHs are also present in the vapour phase. For example, according to Arey et al. (1987) in a study in California, the four-ring PAHs fluoranthene and pyrene were mainly (>90%) present in the gas phase; even at 0 °C, 30% and 70%, respectively, were measured in the gas phase and were therefore available for reaction. However, the nitrofluoranthenes and nitropyrenes exist almost exclusively in the particulate phase. 2-Nitrofluoranthene and 2-nitropyrene formed in the gas phase by hydroxyl-initiated reactions were observed to condense on airborne particles immediately, so that these particles were not detected in the gas phase (Fan et al., 1995). Ambient data from California suggest that the nitrofluorenes are distributed between the gas and particulate phases (Helmig et al., 1992c). Table 10 shows the concentration of selected nitroPAHs measured in vapour and particulate phases in ambient air.
Laboratory studies were carried out to understand the formation of nitroPAHs in ambient air (e.g., Arey et al., 1986, 1989a, 1990; Zielinska et al., 1989b; Atkinson et al., 1990a,b). From these studies, it was concluded that gas-phase daytime hydroxyl radical- and nighttime nitrate radical-initiated reactions of simple volatile and semivolatile PAHs do form nitroPAH derivatives (Atkinson, 1990).
Both daytime hydroxyl radical-initiated reactions and nighttime nitrate radical-initiated reactions have been shown to produce ambient nitroPAHs. For references summarizing the atmospheric formation of nitroPAHs, see Atkinson & Arey (1994) and Arey (1998).
The photolysis of ozone in the troposphere results in the formation of the hydroxyl radical. The hydroxyl radical-initiated reactions of PAHs lead to the formation of nitroPAHs in low yields (5% or less; see Table 11) — for example, 2-nitrofluoranthene (Arey et al., 1986; Zielinska et al., 1986b; Atkinson et al., 1990a), 2-nitropyrene (Arey et al., 1986; Atkinson et al., 1990a) and 3-nitrofluorene (Helmig et al., 1992c). The proposed mechanism involves hydroxyl radical reaction with the gaseous PAH, followed by nitrogen dioxide addition at the free radical site. Although this reaction occurs in competition with the reaction with oxygen, nitroPAH formation is preferred in the presence of sufficient nitrogen dioxide. The resulting nitroPAH products having a relatively low vapour pressure may then condense out on the surface of ambient particles (Atkinson & Arey, 1994).
This hydroxyl radical-initiated mechanism could also explain the formation of volatile nitroarenes such as 1- and 2-nitronaphthalene from gaseous naphthalene (Atkinson et al., 1987b, 1990a). Ambient measurements indicate that the atmospheric reaction products of the two-ring PAHs, such as nitronaphthalenes, remain predominantly in the gas phase. Phenanthrene is more abundant in ambient air than anthracene, fluoranthene and pyrene. Helmig et al. (1992a,b) observed nitrophenanthrenes at only very low yields (# 1%), although other authors (Wilson et al., 1995) found that 9-nitrophenanthrene was the second most abundant nitroPAH after 1-nitronaphthalene (see Figure 6 in chapter 5).
In ambient air, in contrast to environmental chambers, the nitrate radical is formed from the reaction of nitrogen dioxide with ozone. Concentrations of nitrate radical are low during daylight hours because of the rapid photolysis of the nitrate radical (with a photolysis lifetime at solar noon of approximately 5 s) and the rapid reactions of nitric oxide with ozone and nitrate with nitric oxide (Atkinson et al., 1992). At night, however, in the absence of nitric oxide, the concentrations of the nitrate radical and nitrogen pentoxide increase (Atkinson et al., 1986). Average nitrate radical concentrations in the lower troposphere over continental areas during nighttime hours have been estimated as 5 × 108 molecules/cm3 (~20 ppt), but are lower over marine areas (Atkinson & Arey, 1994). In the dark, nitrate radicals react in the gas phase with PAHs to form nitro derivates in significant yield (see Table 11) (Pitts et al., 1985b,c,d; Sweetman et al., 1986; Zielinska et al., 1986b; Arey et al., 1989b; Atkinson et al., 1990a,b; Inazu et al., 1996, 1997).
Table 11. NitroPAHs formed from the gas-phase reactions of PAHs known to be present in ambient air with hydroxyl radicals and nitrate radicals (both in the presence of nitrogen dioxide) and their yieldsa
|
PAH |
Daytime reactions: |
Nighttime reactions: |
|
Initiation by hydroxyl radical followed by reaction with nitrogen dioxide |
Reaction with nitrate radical |
|
|
Naphthalene |
1-Nitronaphthalene (0.3%) |
1-Nitronaphthalene (17%) |
|
1-Methyl-naphthalene |
All 1-methylnitronaphthalene isomers except 1-methyl-2-nitronaphthalene (~0.4%) |
All 1-methylnitronaphthalene isomers (~30%) |
|
2-Methyl-naphthalene |
All 2-methylnitronaphthalene isomers except 2-methyl-1- and 2-methyl-3-nitronaphthalene (~0.2%) |
All 2-methylnitronaphthalene isomers (~30%) |
|
Acenaphthene |
5-Nitroacenaphthene |
4-Nitroacenaphthene (40%)b |
|
Acenaphthylene |
4-Nitroacenaphthylene (2%) |
No nitro isomers formed |
|
Fluorene |
3-Nitrofluorene (~1.4%) |
|
|
Phenanthrene |
Two nitro isomers (not 9-nitrophenanthrene) in trace yields |
Four nitro isomers (including 9-nitrophenanthrene) in trace yields |
|
Anthracenec |
1-Nitroanthracene, low yield |
1-Nitroanthracene, low yield |
|
Pyrene |
2-Nitropyrene (~0.5%) |
4-Nitropyrene (~0.06%) |
|
Fluoranthene |
2-Nitrofluoranthene (~3%) |
2-Nitrofluoranthene (~24%) |
|
a |
From Arey (1998); Atkinson & Arey (1994). |
|
b |
Yields for the nitrate radical addition pathway to the fused aromatic rings (Arey et al., 1989a). |
|
c |
9-Nitroanthracene was observed in both the hydroxyl and nitrate radical reactions, but may not be a product of these reactions, because it is also formed from exposure to nitrogen dioxide/nitric acid. |
The proposed mechanism of reaction of naphthalene in nitrogen pentoxide–nitrate–nitrogen dioxide–air mixtures occurs by the initial addition of the nitrate radical to the aromatic rings to form a nitratocyclohexadienyl-type radical, which then either decomposes to reactants or reacts exclusively with nitrogen dioxide (Atkinson & Arey, 1994).
3-Nitrobenzanthrone was found by bioassay-directed fractionation of diesel particulates (0.6–6.6 µg/g, depending on load; Enya et al., 1997). 3-Nitrobenzanthrone was also detected in airborne particle extracts from urban samples taken in autumn/winter during the day (nd–5.2 pg/m3) and night (7.7–11.5 pg/m3) (Enya et al., 1997). The day/night differences could be the result of different emissions or different meteorological conditions.
Mutagenic nitropyrene lactones (El-Bayoumy & Hecht, 1986) were identified in environmental chamber simulations of atmospheric reactions of pyrene (Sasaki et al., 1995).
2- and 4-nitrodibenzopyranone were identified in the products of the gas-phase hydroxyl radical-initiated reaction of phenanthrene (Helmig et al., 1992a,b).
The transport and distribution of nitroPAHs depend on their physicochemical characteristics (see chapter 2), but data for nitroPAHs are scarce. Their behaviour in the environment is expected to be similar to that of the parent PAHs (see IPCS, 1998), for which there is more information.
NitroPAHs are either formed in the atmosphere from PAHs or emitted directly into the atmosphere during combustion processes (see chapter 3). They can be transported in the vapour phase or adsorbed onto particulate matter. Those with liquid-phase vapour pressures greater than approximately 10–4 Pa at ambient air temperature (i.e., two- to four-ring PAHs and two-ring nitroPAHs) will exist at least partially in the gas phase (Atkinson & Arey, 1994). NitroPAHs having a relatively low vapour pressure will condense out on the surface of ambient particles. Based on ambient measurements, 1- and 2-nitronaphthalene are expected to be found predominantly in the gas phase (Arey et al., 1987). This has also been shown for 1-nitronaphthalene in diesel exhaust samples (Feilberg et al., 1999). Concentrations of a number of PAHs and nitroPAHs were measured (Arey et al., 1987; Atkinson & Arey, 1994), and it was shown that at daytime temperatures in, for example, California, USA, the four-ring PAHs fluoranthene and pyrene are mainly (>90%) present in the gas phase; even at 0 °C, 30% and 70%, respectively, were measured in the gas phase and were therefore available for radical-initiated reactions. There is currently no clear understanding of the partitioning of PAHs between the gas and particulate phases or of the size distribution and mass of the particulates in exhaust gases (CONCAWE, 1998). This is equally true for ambient air, in particular for nitroPAHs.
Just as PAHs are ubiquitous in the environment, so are nitroPAHs. This has been shown in particular for nitroPAHs that are formed not by combustion but by atmospheric transformation (e.g., 2-nitrofluoranthene and 2-nitropyrene), which have been found in different types of airsheds throughout the world (Ciccioli et al., 1995, 1996; see also chapters 3 and 5).
In recent monitoring investigations in downtown Rome, Italy, 1-nitropyrene, a marker indicative of direct emissions, was found not only in the coarse fraction (2.5–10 µm) of atmospheric particulates, but also in the fine fraction (0.01–2.5 µm [PM2.5]), whereas 2-nitrofluoranthene of photochemical origin was mostly found in the fine particulate fraction (Cecinato et al., 1999). 1-Nitropyrene and 1,3-, 1,6- and 1,8-dinitropyrene monitored in Kanazawa, Japan, were found almost exclusively in the particulate fraction <1.1 µm (Hayakawa et al., 1999b). It should be noted that particles with diameters below 2.1 µm can reach terminal bronchi and alveoli (Cecinato et al., 1999).
Owing to their low aqueous solubility or insolubility, nitroPAHs are not expected to accumulate in the hydrosphere. However, considering the low Henry’s law constants (see Table 2 in chapter 2), nitroPAHs present in the hydrosphere are not expected to be transferred significantly to the gas phase.
Although data are scarce, the sorption coefficients (log Koc) for nitroPAHs are high, indicating that nitroPAHs adsorb strongly to the organic fraction of soils and sediments. Leaching into groundwater is therefore thought to be negligible.
The affinity of nitroPAHs for organic phases is much higher than that for water. The n-octanol/water partition coefficients (log Kow) range from 2.5 for 1-nitronaphthalene to 6.3 for 3-nitroperylene (see Table 2), indicating a potential for bioaccumulation. Bioaccumulation of 2-nitrofluorene by Daphnia magna was reported to follow first-order kinetics (Gang & Xiaobai, 1994). A bioconcentration factor of 170 was reported for daphnia exposed to a 2-nitrofluorene concentration of 0.124 mg/litre for up to 8 h.
There were no data on biomagnification.
The diverse metabolic pathways for the microbial metabolism of nitroPAHs are summarized in Figure 4.
Less is known about the metabolism of nitroPAHs by aquatic and terrestrial microorganisms than for the parent PAHs. Although a wide variety of bacteria, fungi and algae have been shown to degrade the parent PAHs containing two to five rings, nitro-substituted PAHs are only slowly degraded by indigenous microorganisms and may persist in soils and sediments. The recalcitrance of high molecular weight nitroPAHs is due in part to the strong adsorption to soil organic matter, low solubility, large molecular size and the hydrophilic character of the nitro group (Cerniglia & Somerville, 1995).
The stability of 1-nitropyrene and 1,6-dinitropyrene was studied in four samples of water (sea, unpolluted river, polluted river and pond water) and filtrates of various soil suspensions with and without 0.1% peptone (Tahara et al., 1995). The mutagenicity decreased rapidly when 1-nitropyrene and 1,6-dinitropyrene were incubated at 30 °C, but not when the test solutions had been autoclaved. Mutagenicity attributed to 1-nitropyrene (3 µg/ml) decreased by 50% in 1.95–3.55 days for water samples and in 0.56–2.37 days for soil filtrate depending on the content of microflora in the test solutions. 1-Aminopyrene was detected as a degradation product of 1-nitropyrene. Mutagenicity attributed to 1,6-dinitropyrene (10 µg/ml) decreased by 50% in 0.53–2.15 days for water samples and in 0.50–0.61 days for soil filtrate.
Time course studies in microcosms showed that 1-nitropyrene was degraded slowly under aerobic and anaerobic conditions in estuarine sediments. Less than 1% had been converted to 14CO2 after 8 weeks of aerobic incubation. Addition of 1-nitropyrene to anaerobic sediments resulted in no 14CO2 evolution, but the 1-nitropyrene was reduced to 1-aminopyrene. The low mineralization of 1-nitropyrene compared with that of the parent compound pyrene could be due to the nitro substituent in the C1 position decreasing the enzymatic oxidation (Cerniglia & Somerville, 1995).
A bacterium isolated from sediments chronically exposed to petrogenic hydrocarbons mineralized 1-nitropyrene and 6-nitrochrysene only to a small extent (12.3% and 2%) compared with non-nitrated PAHs after 10 days of incubation (Heitkamp & Cerniglia, 1988). In pure culture, this bacterium, Mycobacterium sp. strain Pyr-1, was found to metabolize nitroPAHs by both oxidative and reductive pathways. In media with pyrene, the cells oxidized up to 20% of the added 1-nitropyrene to 1-nitro-cis-9,10- and 1-nitro-cis-4,5-dihydrodiols (Heitkamp et al., 1991). However, cells that had been grown in media without pyrene did not produce dihydrodiols, but reduced up to 70% of the 1-nitropyrene to aminopyrene. Further, extracts from cells that had been grown without pyrene activated 1-nitropyrene, 1,3- and 1,6-dinitropyrene and 6-nitrochrysene to DNA-damaging products, as shown in Salmonella typhimurium tester strains and by the umu test (Rafii et al., 1994).
Sphingomonas paucimobilis strain EPA 505 (a soil bacterium capable of utilizing fluoranthene as the sole source of carbon and energy) biodegraded 1-nitropyrene to 48.6% after 6 h (Ye et al., 1996).
The filamentous fungus Cunninghamella elegans has been shown to metabolize a number of nitroPAHs via oxidation pathways to products that are, in general, less mutagenic than the nitroPAHs themselves. The nitroPAH is initially oxidized via a cytochrome P450 monooxygenase to arene oxides, which isomerize to form phenols (hydroxyl derivatives) or are enzymatically hydrated to form trans-dihydrodiols. The phenols can be subsequently conjugated with sulfate, glucose, xylose or glucuronic acid to form detoxified products (Cerniglia & Somerville, 1995) (Table 12 and Figure 4). The metabolites formed are similar to those oxidative metabolites found in rat microsomes and in vivo studies (see chapter 6); however, whereas the trans-dihydrodiol metabolites in rat liver microsomes are predominantly in the R,R configuration, metabolism by C. elegans produces trans-dihydrodiol metabolites in the S,S absolute configuration.
These studies with the fungus Cunninghamella elegans have been extended to include comparison of the biotransformation of nitroPAHs with that of their parent PAHs. For example, comparison of the metabolism pattern between 1-nitrobenzo[e]pyrene and its parent PAH, benzo[e]pyrene, indicates that the nitro group at the C1 position of benzo[e]pyrene drastically altered the regioselectivity of the metabolism (Pothuluri et al., 1999a).
The fungal biotransformation of a mixture of 2- and 3-nitrofluoranthenes was similar to that of the individual nitrofluoranthenes; however, the mammalian system (rat liver microsomes) showed differences in the regioselectivity of nitrofluoranthene at positions C4, C5, C8 and C9 (Pothuluri et al., 1998a).
A plant cell culture (100 mg/ml wet weight) derived from alligator weed (Alternanthera philoxeroides) detoxified 1-nitropyrene and 1,3-, 1,6- and 1,8-dinitropyrene, all direct-acting mutagens, when incubated with them, as shown by mutagenicity response in the Salmonella typhimurium TA98 assay (Shane et al., 1993).
Table 12. Biotransformation of nitroPAHs by the fungus Cunninghamella elegans (48-h incubation)
|
NitroPAH |
Metabolites (major metabolites are in bold) |
Reference |
|
1-Nitropyrene |
Glucoside conjugates of 1-nitropyren-6-ol and 1-nitropyren-8-ol |
Cerniglia et al. (1985) |
|
2-Nitrofluoranthene |
Sulfate conjugates of 2-nitrofluoranthen-8-ol and 2-nitrofluoranthen-9-ol |
Pothuluri et al. (1998a) |
|
3-Nitrofluoranthene |
Sulfate conjugates of 3-nitrofluoranthen-8-ol and 3-nitrofluoranthen-9-ol |
Pothuluri et al. (1994) |
|
6-Nitrochrysene |
6-Nitrochrysene-1-sulfate |
Pothuluri et al. (1998b) |
|
1-Nitrobenzo[e]pyrene |
Sulfate and glucoside conjugates of |
Pothuluri et al. (1999a) |
|
6-Nitrobenzo[a]pyrene |
Glucoside conjugates of |
Millner et al. (1986); Cerniglia & Somerville (1995) |
|
2-Nitrofluorene |
2-nitrofluoren-9-ol, 2-nitro-9-fluorenone, |
Pothuluri et al. (1996) |
|
9-Nitroanthracene |
Phenol and dihydrodiol derivatives |
Pothuluri et al. (1999b) |
The biotransformation of aquatic species is discussed in chapter 9.
The photolysis of nitroPAHs has been studied under varied conditions of irradiation (see Table 13). The rate of photolysis depends not only on the conditions of irradiation but also on whether the nitroPAH is in the gaseous stage (e.g., 1-and 2-nitronaphthalene), in solution (type of solvent) or bound to solids or particles. In the latter case, the type and age of the particle seem to influence the photochemistry of the respective nitroPAH. The rate of photodecomposition, identification of photolytic products and the resulting loss or gain of mutagenic activity as determined by the Salmonella typhimurium assay have been the main end-points studied. Decomposition products include quinones, hydroxy-nitroPAHs and hydroxyPAHs (see Table 13). Although most studies show that the mutagenic activity of decomposition products was less than that of the original nitroPAH, some results show an increase.
Table 13. Rates of photolysis of nitroPAHs in different media and comparison of the mutagenic activity of the products
|
NitroPAH |
Media |
Source of light |
Major product |
Mutagenic activitya |
Rate of photolysis |
Reference |
|
1-Nitronaphthalene |
Gaseous |
Sunlight |
1,4-Naphthoquinone |
Atmospheric lifetime = 1.7 h |
Atkinson et al. (1989) |
|
|
1- and 2-nitro-naphthalenes |
Gaseous |
Sunlight |
0.5 h and 11 h, respectively |
Feilberg et al. (1999) |
||
|
1- and 2-nitro-naphthalenes |
2-Propanol |
320–418 nm for 2 h |
No degradation |
Stärk et al. (1985) |
||
|
3-Nitrofluoranthene |
DMSO |
Less |
12.5 days |
Holloway et al. (1987) |
||
|
2-Nitrofluorene |
DMSO |
Cool-white light or artificial sunlight; up to 5 h |
More |
White et al. (1985) |
||
|
9-Nitroanthracene |
Acetone and silica gel |
Mainly 9,10-anthraquinone (via iminoxyl radical) |
Chapman et al. (1966); Pitts et al. (1978) |
|||
|
9-Nitroanthracene |
Hexane |
Anthraquinone |
Transformed to 40% after a 1-day exposure to light |
Schlemitz & Pfannhauser (1997) |
||
|
1-Nitropyrene |
Solid |
Sunlight |
Pyrenol, pyrene quinone, pyrenediol |
Less |
Biphasic; half-times = 14 h and 533 h |
Benson et al. (1985) |
|
1-Nitropyrene |
Methanol |
>300 nm in presence of oxygen |
Pyren-1-ol (88%) and 2-nitropyren-1-ol (7%) |
95% transformation after 2.5 h |
van den Braken-van Leersum et al. (1987) |
|
|
1-Nitropyrene |
Benzene |
Sunlight |
Nitropyren-9-ol |
Less |
Rapid, 10% after 1 h; 53% after 10 h |
Koizumi et al. (1994) |
|
1-Nitropyrene |
Soot |
Sunlight |
Nitropyren-9-ol |
Less |
Slow, 42% in 40 days |
Koizumi et al. (1994) |
|
1-Nitropyrene |
DMSO |
Light |
Less |
1.2 days |
Holloway et al. (1987) |
|
|
1-Nitropyrene |
Silica |
Light |
Less |
6 days |
Holloway et al. (1987) |
|
|
1-Nitropyrene |
2-Propanol |
320–418 nm for 90 min |
Almost total loss |
Rapid |
Stärk et al. (1985) |
|
|
1-Nitropyrene |
DMSO |
Fluorescent sunlamps; 4 and 24 h |
Pyrene quinone, pyrenol, 1-nitropyrenol |
Less |
Yang et al. (1994) |
|
|
2-Nitropyrene |
Methanol |
>300 nm in presence of oxygen |
Very stable |
Only 19% conversion after 7.5 h |
van den Braken-van Leersum et al. (1987) |
|
|
4-Nitropyrene |
Methanol |
>300 nm in presence of oxygen |
Pyrene (9%) and traces of other products |
van den Braken-van Leersum et al. (1987) |
||
|
1,8-Dinitropyrene |
DMSO |
Light |
1-Nitropyren-8-ol |
0.7 days |
Holloway et al. (1987) |
|
|
1,8-Dinitropyrene |
Silica |
Light |
1-Nitropyren-8-ol |
5.7 days |
Holloway et al. (1987) |
|
|
1,8-Dinitropyrene |
Silica |
Less |
>20 days |
Holloway et al. (1987) |
||
|
7-Nitro-benz[a]-anthracene |
DMSO |
Cool-white light or artificial sunlight; up to 5 h |
More |
White et al. (1985) |
||
|
7-Nitrodibenzo[a,h]-anthracene |
DMSO |
Fluorescent sunlamps; 4, 24 and 48 h |
More at 4 h, then decreasing |
Yang et al. (1994) |
||
|
9-Nitrodibenzo[a,c]-anthracene |
DMSO |
Fluorescent sunlamps; 4, 24 and 48 h |
More at 4 h, then decreasingb |
Yang et al. (1994) |
||
|
1-Nitrobenzo[a]pyrene |
DMSO |
Fluorescent sunlamps; 4, 24 and 48 h |
Less |
Yang et al. (1994) |
||
|
6-Nitrobenzo[a]pyrene |
Silica gel |
Benzo[a]pyrene quinones: 1,6-, 3,6- and 6,12- isomers |
Pitts (1983) |
|||
|
6-Nitrobenzo[a]pyrene |
DMSO |
Cool-white light or artificial sunlight; up to 5 h |
More |
White et al. (1985) |
||
|
6-Nitrobenzo[a]pyrene |
In solution |
UV light |
Benzo[a]pyrene-3,6-quinone via benzo[a]pyren-6-oxyl radical |
Ioki (1977) |
||
|
3,6-Dinitrobenzo[a]-pyrene |
UV light at 312 nm |
3-Nitrobenzo[a]-pyrene-6-quinone |
Sera et al. (1991) |
|||
|
3-Nitrobenzo[e]pyrene |
DMSO |
Cool-white light or artificial sunlight; up to 5 h |
More |
White et al. (1985) |
a Compared with original nitroPAH.
b Also increasing cytotoxicity with length of exposure time.
1) Photolysis of gaseous nitroPAHs
Feilberg et al. (1999) showed that 1-nitronaphthalene photolyses faster than 2-nitronaphthalene. Using a large outdoor smog chamber facility, the gas-phase photolysis rates were determined to be 0.07 × kNO2 and 0.005 × kNO2 for 1- and 2-nitronaphthalene, respectively (Feilberg et al., 1999). Using an average kNO2 of 5.2 × 10–3/s, the lifetimes of these two nitroPAHs with respect to photolysis were calculated to be 0.5 and 11 h, respectively. Therefore, gas-phase photolysis is the major degradation pathway for 1-nitronaphthalene; for 2-nitronaphthalene, other pathways (such as reaction with hydroxyl radicals; see Tables 14 and 15) may be important (Feilberg et al., 1999).
Table 14. Room temperature rate constants, k, for the gas-phase reactions of hydroxyl radicals, nitrate radicals and ozone with nitronaphthalenesa
|
Nitronaphthalene |
k (cm3/molecule per second) for reaction with |
||
|
Hydroxyl (OH)b |
Nitrate |
Ozone |
|
|
1-Nitronaphthalene |
5.4 x 10–12 |
3.0 x 10–29 [NO2]b |
<6 x 10–19 |
|
2-Nitronaphthalene |
5.6 x 10–12 |
2.7 x 10–29 [NO2]b |
<6 x 10–19 |
a Adapted from Atkinson & Arey (1994).
b Atkinson (1991).
Table 15. Calculated atmospheric lifetimes of nitronaphthalenes due to photolysis and gas-phase reactions with hydroxyl and nitrate radicals and with ozone
|
Nitronaphthalenes |
Atmospheric lifetimes due to reaction with |
|||
|
Hydroxyl (OH) |
Nitrate (NO3) |
Ozone |
Photolysis |
|
|
1-Nitronaphthalene |
2.7 daysa |
18 yearsb |
>28 daysc |
0.5 hd |
|
2-Nitronaphthalene |
2.6 daysa |
20 yearsb |
>28 daysc |
11 hd |
|
a |
From Atkinson & Arey (1994) for a 12-h daytime average hydroxyl radical concentration of 1.6 x 106 molecules/cm3 (Prinn et al., 1992). |
|
b |
From Atkinson & Arey (1994) for a 12-h average nighttime nitrate radical concentration of 5 x 108 molecules/cm3 (Atkinson, 1991) and nitrogen dioxide concentration of 2.4 x 1011 molecules/cm3. |
|
c |
From Atkinson & Arey (1994) for a 24-h average ozone concentration of 7 x 1011 molecules/cm3 (Logan, 1985). |
|
d |
From Feilberg et al. (1999) using an average 12-h daytime nitrogen dioxide photolysis rate kNO2 = 5.2 x 10–3/s. |
2) Photolysis of PAHs in solution or on solids
Studies comparing the photodecomposition of nitroPAHs on solids or in solution show that decomposition rates or times are much longer on solids (see Table 13; e.g., with 1-nitropyrene [Koizumi et al., 1994]; 1-nitropyrene, 1,8-dinitropyrene, 3-nitrofluoranthene [Holloway et al., 1987]; see also section 3) below on photostability of nitroPAHs on particles).
Several authors have studied the photolysis of isomeric or other groups of nitroPAHs and compared their rates of decomposition and their mutagenicities with their differences in chemical structure (see Table 13).
Some nitroPAHs can be readily decomposed when exposed to light both in solution or on particles to form quinones and possibly phenolic derivatives (Pitts, 1983). The reactions are complex and depend on the presence or absence of air, the type of solvent and the wavelength of light used (Chapman et al., 1966). A mechanistic theory was suggested using the example of 9-nitroanthracene. Illumination in acetone resulted in rearrangement into a nitrite, followed by dissociation into nitric oxide and a phenoxy-type radical and ultimately anthraquinone (Chapman et al., 1966). 9-Nitroanthracene forms 9,10-anthraquinone on irradiation both in solution and on silica gel (Pitts et al., 1978).
UV light-induced oxidation of 6-nitrobenzo[a]pyrene resulted in the formation of benzo[a]pyrene-3,6-quinone (Ioki, 1977). 3,6-Dinitrobenzo[a]pyrene was readily decomposed by UV radiation at 312 nm to 3-nitrobenzo[a]pyrene-6-quinone (Sera et al., 1991).
6-Nitrobenzo[a]pyrene on silica gel photolyses rapidly to BaP quinones (1,6-, 3,6- and 6,12- isomers); the 1- and 3- isomers are more stable (Pitts, 1983). (According to Chapman’s hypothesis, the 6-nitro- isomer with two peri-hydrogens should be less stable than the 1- and 3- isomers with only one peri-hydrogen.)
A 1-day exposure of 9-nitroanthracene to light resulted in 40% conversion to anthraquinone (Schlemitz & Pfannhauser, 1997).
The influence of the nitro group on the aromatic B -system of pyrene has been studied by comparing the spectroscopic and photochemical properties of 1-, 2- and 4-nitropyrene (van den Braken-van Leersum et al., 1987). Whereas the UV and mass spectra of 1- and 4-nitropyrene show an interaction normal for nitro-aromatic compounds, 2-nitropyrene shows a lack of interaction, reflected in a UV spectrum very similar to that of pyrene and a mass spectrum with a low abundance of [M–NO]. The photochemical behaviour of the three compounds is governed by the degree of interaction. In the presence of oxygen, 1-nitropyrene shows the nitro-nitrite rearrangement, leading to 1-hydroxypyrene (pyren-1-ol) (88%) and 1-hydroxy-2-nitropyrene (2-nitropyren-1-ol) (7%); under anaerobic conditions, equimolar amounts of 1-nitrosopyrene and 2-nitropyren-1-ol are formed. The photochemical products of 4-nitropyrene in air are pyrene (9%) and unstable products that react with the solvent. 1-Nitropyrene was more reactive than 4-nitropyrene. 2-Nitropyrene is very stable under photochemical conditions due to lack of interaction. After irradiation of a solution of 2-nitropyrene 3 times longer than needed for 95% conversion of 1-nitropyrene, 81% of the starting material was recovered.
Yang et al. (1994) studied the photodecomposition of four sets of isomeric nitroPAHs (total of 10 nitroPAHs) in DMSO for 4, 24 and 48 h under fluorescent sunlamps. Decomposition products were multiple — e.g., quinones and/or hydroxy-nitroPAHs. The order of ease of photodecomposition for the different isomeric groups was as follows (see also Table 16):
6-nitro-BaP > 1-nitro-BaP $ 3-nitro-BaP
1-nitropyrene > 4-nitropyrene > 2-nitropyrene
9-nitroanthracene > 2-nitroanthracene
7-nitrodibenzo[a,h]anthracene = 9-nitrodibenzo[a,c]anthracene
Table 16. Comparison of the decomposition rates of the isomeric nitroPAHs after exposure in DMSO to fluorescent sunlampsa
|
Compound |
Nitro orientation |
Degree of decomposition |
|
6-Nitrobenzo[a]pyrene |
Perpendicular |
+ + + + + + |
|
1-Nitrobenzo[a]pyrene |
Parallel |
+ + + |
|
3-Nitrobenzo[a]pyrene |
Parallel |
+ + + |
|
1-Nitropyrene |
Parallel |
+ + + |
|
4-Nitropyrene |
Parallel |
+ + |
|
2-Nitropyrene |
Parallel |
+ |
|
9-Nitroanthracene |
Perpendicular |
+ + + + |
|
2-Nitroanthracene |
Parallel |
+ + |
|
7-Nitrodibenzo[a,h]anthracene |
Perpendicular |
+ + + + |
|
9-Nitrodibenzo[a,c]anthracene |
Perpendicular |
+ + + + |
a From Yang et al. (1994).
It appears that those nitroPAHs having a perpendicular orientation decompose faster than those with a parallel orientation (see Table 16).
1-Nitropyrene, 2-nitropyrene, 2-nitrofluoranthene, 3-nitrofluoranthene, 9-nitroanthracene and 6-nitrobenzo[a]pyrene were irradiated in cyclohexane solutions (Feilberg & Nielsen, 2000). In the absence of co-solutes, the photodegradation of nitroPAHs is strongly dependent on the orientation of the nitro group. In 9-nitroanthracene and 6-nitrobenzo[a]pyrene, the nitro group adapts an approximately perpendicular orientation relative to the aromatic plane, and, as expected, both compounds decayed very quickly, with less than 1% remaining after 15 min. 1-Nitropyrene decayed moderately, whereas 2-nitrofluorene, 2-nitrofluoranthene and 3-nitrofluoranthene were stable towards photolysis.
The mutagenicity of the photodecomposition products of 1-nitropyrene (a moderate direct-acting mutagen), 1-nitrobenzo[a]pyrene (a potent direct-acting mutagen), 7-nitrodibenzo[a,h]anthracene and 9-nitrodibenzo[a,c]anthracene (both non-direct-acting mutagens) were tested using the Salmonella typhimurium microsome assay with TA98 without metabolic activation (Yang et al., 1994). Most studies show that the mutagenic activity of decomposition products was less than that of the original nitroPAHs (Benson et al., 1985; Koizumi et al., 1994; Yang et al., 1994) (see Table 13). In contrast, following 3- or 5-h exposures in DMSO, 6-nitrobenzo[a]pyrene, 7-nitrobenz[a]anthracene, 3-nitrobenzo[e]pyrene and 2-nitrofluorene were found to have elevated mutagenicities in the S. typhimurium microsome assay when exposed to sunlight (White et al., 1985). Irradiated 6-nitrobenzo[a]pyrene was about 15 times more mutagenic than 7-nitrobenz[a]anthracene.
3) Photostability of nitroPAHs on particles
NitroPAHs that are particle-associated under atmospheric conditions may be fully or partially protected from photolysis (Atkinson & Arey, 1994).
In a chamber study, the stability of nitroPAHs (1-nitropyrene, 7-nitrobenz[a]anthracene and 6-nitrobenzo[a]pyrene) on airborne, fresh diesel soot particles was assessed (Kamens et al., 1994). They degraded very rapidly in the sunlight; the half-lives for the nitropyrenes were around 2.5 h. 6-Nitrobenzo[a]pyrene degraded more rapidly, with a half-life of 30 min. However, nitroPAHs associated with aerosolized diesel soot (SRM 1650) did not decay when exposed to sunlight (no further details given). 1-Nitropyrene adsorbed to coal fly ash was resistant to photodecomposition (Holder et al., 1994).
NitroPAHs, in spite of structural differences, all showed similar decay rates on diesel soot particles or wood smoke, indicating that other chemicals associated with the diesel particles somehow affect the photooxidation of nitroPAHs in sunlight and that the effect of the structure is not a dominant factor, as it is with individual nitroPAHs studied (Fan et al., 1996a; Feilberg & Nielsen, 2000, 2001).
1) Gaseous nitroPAHs
Transformations of 1- and 2-nitronaphthalene with hydroxyl and nitrate radicals and ozone were studied, and the rate constants for the gas-phase reactions are given in Table 14.
The reactions of nitrate radicals and ozone with gaseous nitroPAHs (see Table 15) appear to be of negligible importance as atmospheric loss processes (Atkinson et al., 1989). Reactions of gaseous nitroPAHs with hydroxyl radicals are probably of secondary importance to photolysis (e.g., for 1-nitronaphthalene), although recent studies with 2-nitronaphthalene suggest that this loss process may be as important as photolysis (Feilberg et al., 1999).
2) NitroPAHs on particles
No physical or chemical loss processes were found when 1-nitropyrene was coated on glass and Teflon filters exposed to 100 ppb of nitric acid-free nitrogen dioxide, ozone and sulfur dioxide (Grosjean et al., 1983). In contrast, 1,3-, 1,6- and 1,8-dinitropyrene were detected when 1-nitropyrene-coated particles on a filter surface were exposed to 260 mg/m3 of nitrogen dioxide (Lee et al., 1989).
Oxidation of 1-nitropyrene adsorbed on silica gel with dimethyldioxirane led to the formation of 1-nitropyrene-4,5-oxide and 1-nitropyrene-9,10-oxide, in a ratio 74:26. When the adsorbed 1-nitropyrene was exposed to the gas-phase ozonolysis of tetramethylethylene, the same oxides were produced in the ratio 72:28. Reaction of 1-nitropyrene with ozone alone did not lead to oxide formation (Murray & Singh, 1998).
Degradation of nitroPAHs was observed in the presence of both ozone and nitrogen dioxide plus ozone on real soot particles studied in an outdoor smog chamber in both cool and warm temperatures (between 2 and 20 °C) (Fan et al., 1996b) (see Table 17). The degradation rate constants of 1- and 2-nitropyrene and 2-, 3- and 8-nitrofluoranthene with ozone ranged from 0.0015 to 0.0025/ppm per minute.
Table 17. Particle nitroPAH atmospheric half-lives of 2-nitrofluoranthene and 1-nitropyrene from photolysis and dark heterogeneous reactions with ozonea,b
|
Compound |
Photolysis rate constant (min–1) |
O3 rate constant |
Half-life in sunlight |
Half-life with 0.2 ppm O3 (h) |
|
2-Nitrofluoranthene |
0.013 |
0.002 |
1 |
29 |
|
1-Nitropyrene |
0.020 |
0.002 |
0.6 |
29 |
|
a |
Data from Fan et al. (1995, 1996b). |
|
b |
NitroPAH photolysis rate constants are derived from an average nitrogen dioxide photolysis rate of kNO2 = 0.5/min; an ozone concentration of 0.2 ppm (1 ppm = 1.96 mg/m3) is assumed, which represents a moderately to highly polluted atmosphere. |
The rate constant of particle nitroPAHs with nitrogen dioxide–nitrate–nitrogen pentoxide was less than 0.001/ppm per minute. Particle oxidation of nitroPAHs by ozone does not appear to be as important as photodegradation of nitroPAHs in daytime, but it may be the main loss process at night.
NitroPAHs can be formed as direct or indirect products of incomplete combustion. Formation of nitroPAHs from diesel and gasoline exhaust is discussed in chapter 3. NitroPAHs can be expected in urban air as a result of traffic and domestic heating. However, nitroPAHs can also be formed indirectly from nitration of PAHs transported in ambient air to other sites (see chapter 3).
Some studies have monitored one sample to identify as many nitroPAHs as possible (Table 18). NitroPAHs that have been detected in ambient air include 1- and 2-nitronaphthalene, all 14 methylnitronaphthalene isomers (predominantly in the vapour phase), 2-nitrofluorene, 9-nitroanthracene, 9-nitrophenanthrene, 2-, 3- and 8-nitrofluoranthene, 1- and 2-nitropyrene, 1,3-, 1,6- and 1,8-dinitropyrene, 6-nitrochrysene, 7-nitrobenz[a]anthracene and 1- and 2-nitrotriphenylene (Ishii et al., 2000, 2001). Also observed in low concentrations (relative to 2-nitrofluoranthene) were 7-nitrofluoranthene, 4-nitropyrene and nitroacephenanthrylene isomers. Other nitroPAHs have been cited in trace quantities by one author only (see Table 18). However, the importance of the nitroPAHs depends not only on their relative concentrations in air but also on their mutagenicity; dinitropyrenes and 3,6-dinitrobenzo[a]pyrene, for example, are present in relatively low concentrations but have a high mutagenicity (see below and chapter 7).
Ambient air monitoring of nitroPAHs in many parts of the world has revealed the unexpected presence in airborne particles of 2-nitrofluoranthene and 2-nitropyrene at levels equal to or higher than those found for 1-nitropyrene (see Table 19). 2-Nitrofluoranthene and 2-nitropyrene are not usually found as a result of combustion processes, but are formed by atmospheric reactions (see chapter 3).
Table 18. NitroPAHs detected in ambient aira
|
Parent PAH; nitro derivative |
Concentration (ng/m3)b,c |
|||||||||||||||
|
a) |
b) |
c) |
d) |
e) |
f) |
g) |
h) |
i) |
j) |
k) |
l) |
m) |
n) |
|||
|
P |
P |
V+P |
V+P |
V+P |
V |
P |
P |
V+P |
P |
P |
V+P |
P |
||||
|
Naphthalene |
||||||||||||||||
|
1-Nitro- |
m |
2.7 |
5.7 |
0.10 |
4.7 |
0.39 |
0.90 |
0.352 |
0.39 |
|||||||
|
2-Nitro- |
2.6 |
3.1 |
0.17 |
2.3 |
0.55 |
0.55 |
0.065 |
0.21 |
||||||||
|
1,3-Dinitro- |
0.12 |
0.23 |
||||||||||||||
|
1,5-Dinitro- |
0.06 |
0.029 |
||||||||||||||
|
1,8-Dinitro- |
0.017 |
|||||||||||||||
|
Epsilon-Methylnitro- |
X |
X |
X |
6.0 |
0.005 |
|||||||||||
|
Acenaphthene |
||||||||||||||||
|
5-Nitro- |
0.11 |
|||||||||||||||
|
Fluorene |
||||||||||||||||
|
2-Nitro- |
0.05 |
0.21 |
0.37 |
|||||||||||||
|
2,5-Dinitro- |
0.19 |
|||||||||||||||
|
2,7-Dinitro- |
1.5 |
0.095 |
||||||||||||||
|
Anthracene |
||||||||||||||||
|
9-Nitro- |
X |
0.11 |
0.25 |
0.11 |
0.03 |
0.04 |
0.17 |
0.029 |
||||||||
|
Phenanthrene |
||||||||||||||||
|
9-Nitro- |
0.29 |
0.105 |
||||||||||||||
|
Fluoranthene |
||||||||||||||||
|
2-Nitro- |
X |
X |
0.35 |
2.0 |
0.14 |
X |
0.15 |
0.16 |
0.017 |
0.039 |
0.065 |
|||||
|
3-Nitro- |
np |
np |
np |
0.2–0.8 |
0.13 |
0.013 |
||||||||||
|
7-Nitro- |
X |
|||||||||||||||
|
8-Nitro- |
X |
0.003 |
0.004 |
0.003 |
X |
|||||||||||
|
3,7-Dinitro- |
0.005 |
|||||||||||||||
|
3,9-Dinitro- |
0.004 |
|||||||||||||||
|
Pyrene |
||||||||||||||||
|
1-Nitro- |
X |
X |
0.015 |
0.015 |
0.026 |
0.009 |
0.14 |
0.04 |
1.7–1.9 |
0.068 |
0.004 |
0.009 |
0.13 |
|||
|
2-Nitro- |
X |
0.014 |
0.032 |
0.030 |
X |
0.008 |
||||||||||
|
4-Nitro- |
X |
|||||||||||||||
|
1,3-Dinitro- |
0.01 |
|||||||||||||||
|
1,6-Dinitro |
0.008 |
|||||||||||||||
|
Benz[a]anthracene |
||||||||||||||||
|
7-Nitro- |
0.014 |
|||||||||||||||
|
10-Nitro- |
0.014 |
|||||||||||||||
|
Chrysene |
||||||||||||||||
|
6-Nitro- |
0.27 |
0.8–1.5 |
0.0002 |
|||||||||||||
|
Benzo[a]pyrene |
||||||||||||||||
|
6-Nitro- |
t |
X |
0.27 |
np |
||||||||||||
|
Total nitroPAHs |
5.8 |
11.1 |
0.59 |
13.0 |
1.4 |
2.22 |
0.60 |
1.5 |
||||||||
|
a |
See also Tables 19 and 20 and Figures 5 and 6. |
|
|
b |
P = particulate; V = vapour; t = trace; m = monoisomers (specific isomers not identified); np = not present in detectable amounts; X = detected (no quantification). It should be noted that there is some historical misidentification of certain isomers due to lack of standards (e.g., nitrochrysenes and nitrofluoranthenes). |
|
|
c |
Samples were as follows: |
|
|
a) |
Urban air particles from St. Louis, Missouri, USA, NIST SRM (Ramdahl et al., 1982); 2-nitrofluoranthene originally reported as 3-nitrofluoranthene. |
|
|
b) |
Torrance, California, USA. Analysis by GC and negative ion atmospheric pressure ionization MS, by which means small peaks of 4-nitropyrene and 7-nitrofluoranthene could be detected (Korfmacher et al., 1987). |
|
|
c) |
Glendora, California, USA, daytime composite sample (Atkinson et al., 1988). |
|
|
d) |
Glendora, California, USA, nighttime composite sample (Atkinson et al., 1988). |
|
|
e) |
Concord, California, USA, daytime composite sample (Atkinson et al., 1988). |
|
|
f) |
Redlands, California, USA, nighttime sample (Gupta, 1995). |
|
|
g) |
ng/m3; mean of 30 samples of airborne particulate matter from rural area 30 km west of Copenhagen, Denmark, February to April 1982 (Nielsen et al., 1984). Original publication gives 3-nitrofluoranthene, but this was reanalysed and given as 2-nitrofluoranthene (Ramdahl et al., 1986). |
|
|
h) |
Tokyo air (Matsushita & Iida, 1986); only methylnitronaphthalene reported was 2-methyl-1-nitronaphthalene. |
|
|
i) |
Fresno, California, USA, composite daytime sample; five samples, December 1990 – January 1991 (Hunt & Maisel, 1995); see Figure 6. |
|
|
j) |
Suburban, Bayreuth, Germany, in 1983 (Garner et al., 1986). 3-Nitrofluoranthene reported as present, likely 2- + 3-nitrofluoranthene. No nitroPAHs in snow sample near motorway. |
|
|
k) |
Air sample from Sapporo, Japan, in 1989 (Tokiwa et al., 1990a). |
|
|
l) |
Aerosol from Amazon forest (Alta Floresta) near biomass burning (Vasconcellos et al., 1998; see also Figure 6). |
|
|
m) |
ng/m3; vapour + particulate phase; Houston, Texas, USA, annual average; see also seasonal pattern (Wilson et al., 1995). |
|
|
n) |
Assay of 19 independent 48-h air samplings from three sampling sites in downtown urban environment (Florence, Italy), January–February 1993; 3-nitrofluoranthene reported as present, likely 2- + 3-nitrofluoranthene (Berlincioni et al., 1995). |
|
Table 19. Average concentrations of MW 247 nitroPAHs as represented by 2-nitrofluoranthene (2-NFL), 1-nitropyrene (1-NP)
and 2-nitropyrene (2-NP) detected in tropospheric samplesa,b
|
Source |
Site |
Year |
Season |
Concentration (pg/m3) |
Reference |
|||||
|
Total PAHs |
BaP |
2-NFL |
1-NP |
2-NP |
2-NFL/ |
|||||
|
Urban |
Milan (Italy) |
1990 |
Winter |
240 |
30 |
4 |
8 |
Ciccioli et al. (1993) |
||
|
Urban |
Milan (Italy) |
1990 |
Summer |
170 |
20 |
nr |
8.5 |
Ciccioli et al. (1993) |
||
|
Urban |
Barcelona (Spain) |
1989–1990 |
Year |
50 000 |
3500 |
120 |
30 |
20 |
4 |
Bayona et al. (1994) |
|
Urban |
Hamilton (Canada) |
1990–1991 |
Spring/ summer |
2000 |
6 |
12 |
4 |
0.5 |
Legzdins et al. (1995) |
|
|
Downtown |
Kanazawa (Japan), Site 1 |
1994 |
Summer |
nr |
nr |
59 |
32 |
4 |
1.8 |
Murahashi & Hayakawa (1997) |
|
Suburban |
Kanazawa (Japan), Site 2 |
1994 |
Summer |
nr |
nr |
16 |
10 |
2 |
1.6 |
Murahashi & Hayakawa (1997) |
|
Urban |
Damascus (Syria) |
1994 |
Winter |
4500 |
230 |
120 |
nr |
1.9 |
Dimashki et al. (1996) |
|
|
Urban |
Milan, Viale Marche (Italy) |
1991 |
Winter |
48 600 |
2850 |
960 |
140 |
nr |
6.8 |
Cecinato et al. (1998) |
|
Urban |
Milan, Brera Tower (Italy) |
1993 |
Winter |
70 200 |
1350 |
1730 |
590 |
nr |
2.9 |
Cecinato et al. (1998) |
|
Urban |
Milan (Italy) |
1991–1993 |
Winter |
nr |
nr |
1140 |
220 |
300 |
5.2 |
Ciccioli et al. (1995) |
|
Urban |
Rome (Italy) |
1991–1993 |
Winter |
36 000 |
1600 |
250 |
80 |
nr |
3.1 |
Cecinato et al. (1998) |
|
Urban |
Rome (Italy) |
1991–1993 |
Summer |
nr |
nr |
470 |
70 |
70 |
6.7 |
Ciccioli et al. (1995) |
|
Urban |
Sao Paulo (Brazil) |
1991–1993 |
Winter |
nr |
nr |
139 |
16 |
42 |
8.7 |
Ciccioli et al. (1995) |
|
Urban |
Athens (Greece) |
1996 |
Year |
23 900 |
610 |
90 |
40 |
60 |
2.3 |
Marino et al. (2000) |
|
Urban |
St. Louis (USA) |
Year |
560c |
160c |
60c |
3.5 |
Ramdahl et al. (1986) |
|||
|
Remote |
Svalbard Island (Norway) |
1998–1999 |
Year |
36 |
2 |
13 |
8 |
0.16 |
Cecinato et al. (2000) |
|
|
Urban industrial |
Bab Ezzouar (Algeria) |
1999 |
Summer |
2000 |
1500 |
0 |
n.ev. |
– |
Yassaa et al. (2001a) |
|
|
Landfill |
Oued Smar (Algeria) |
1998–1999 |
Summer |
3900 |
310 |
80 |
10 |
37 |
Yassaa et al. (2001b) |
|
|
Urban |
Algiers (Algeria) |
1999 |
Winter |
2300 |
450 |
140 |
20 |
3.2 |
Yassaa et al. (2001b) |
|
|
Urban |
Glendora (USA) |
1986 |
Summer |
240 |
630 |
16 |
19 |
~40 |
Atkinson et al. (1988) |
|
|
Biomass burning |
Yuba City (USA) |
1986 |
Autumn |
200 |
130 |
8 |
8 |
16 |
Atkinson et al. (1988) |
|
|
Industrial |
Concord (USA) |
1986–1987 |
Winter |
4400 |
290 |
30 |
50 |
10 |
Atkinson et al. (1988) |
|
|
Wood smoke |
Mammoth Lakes (USA) |
1987 |
Winter |
6200 |
29 |
8 |
3 |
3.6 |
Atkinson et al. (1988) |
|
|
Oil production |
Oildale (USA) |
1987 |
Summer |
490 |
28 |
7 |
1 |
4 |
Atkinson et al. (1988) |
|
|
Urban |
Reseda (USA) |
1987 |
Summer |
290 |
150 |
8 |
13 |
~19 |
Atkinson et al. (1988) |
|
|
Remote |
Pt. Arguello (USA) |
1987 |
Summer |
nd |
5 |
0.5 |
0.3 |
10 |
Atkinson et al. (1988) |
|
|
Remote |
San Nicolas Island (USA) |
1987 |
Summer |
nd |
2 |
3 |
nd |
0.67 |
Atkinson et al. (1988) |
|
|
Urban |
Washington (USA) |
Year |
600c |
200c |
50c |
3 |
Ramdahl et al. (1986) |
|||
|
Urban |
Riversided (USA) |
Autumn |
nr |
nr |
193 |
22 |
8 |
8.8 |
Pitts et al. (1985d) |
|
|
Suburban |
Montelibretti (Italy) |
1991–1993 |
Year |
nr |
nr |
87 |
12 |
16 |
7.3 |
Ciccioli et al. (1995) |
|
Suburban |
Madrid (Spain) |
1991–1993 |
Autumn |
nr |
nr |
70 |
10 |
20 |
7 |
Ciccioli et al. (1995) |
|
Suburban |
Torranced (USA) |
Winter |
nr |
nr |
310 |
32 |
30 |
9.7 |
Arey et al. (1987) |
|
|
Suburban |
Glendorad (USA) |
nr |
nr |
270 |
nr |
12 |
Arey et al. (1988b) |
|||
|
Suburban |
Claremontd (USA) |
Autumn |
nr |
nr |
408 |
16 |
3 |
25.5 |
Zielinska et al. (1989a) |
|
|
Suburban |
Claremont (USA) |
1985 |
Autumn |
nr |
nr |
2800c |
360c |
80c |
7 |
Ramdahl et al. (1986) |
|
Rural residential |
Aurskog (Norway) |
1984 |
Winter |
nr |
nr |
560c |
150c |
170c |
3.7 |
Ramdahl et al. (1986) |
|
Forest areas |
Castelporziano (Italy) |
1992 |
Winter |
1560 |
140 |
70 |
3 |
14 |
23 |
Ciccioli et al. (1995) |
|
Forest areas |
Castelporziano (Italy) |
1993 |
Summer |
600 |
10 |
25 |
nd |
5 |
>25 |
Ciccioli et al. (1995) |
|
Forest areas |
Alta Floresta (Brazil) |
1993 |
Winter, spring |
2550 |
160 |
15 |
2 |
80 |
7.5 |
Ciccioli et al. (1995) |
|
Forest areas |
Storkow (Germany) |
1991 |
Summer |
430 |
10 |
3 |
nd |
nd |
>3 |
Ciccioli et al. (1995) |
|
Remote |
CNR Pyramid (Nepal) |
1991 |
Autumn |
130 |
3 |
3 |
nd |
nd |
>3 |
Ciccioli et al. (1995) |
|
Remote |
Terra Nova Bay (Antarctica) |
1991 |
Summer |
3 |
nd |
nd |
nd |
nd |
Ciccioli et al. (1995) |
|
|
a |
Adapted from Ciccioli et al. (1995, 1996). |
|
b |
nr = not reported; nd = not detected; Year (under "Season") = mean for year; n.ev. = 2-NP could not be evaluated since an interference co-eluted. |
|
c |
Units of ng/g; this is a NIST SRM of ambient particles. |
|
d |
California, USA. |
Table 20. Concentrations of benzo[a]pyrene and selected nitroPAHs in urban air from Teplice and Prachatice, Czech Republica
|
Compound |
Concentration (ng/m3) |
||||
|
Teplice, |
Teplice |
Teplice |
Prachatice |
Prachatice |
|
|
BaP |
6.05 |
0.57 |
4.76 |
3.68 |
0.41 |
|
9-Nitroanthracene |
0.018 |
0.002 |
0.033 |
0.005 |
0.002 |
|
3-Nitrofluorantheneb |
0.090 |
0.009 |
0.139 |
0.013 |
0.003 |
|
1-Nitropyrene |
0.055 |
0.008 |
0.048 |
not given |
0.005 |
a From Lenicek et al. (2001).
b Probably 2- plus 3-nitrofluoranthene.
From the results of these broad monitoring studies, other investigators have concentrated on the nitroPAHs that seem to be of quantitative/environmental (e.g., nitroPAHs of relative molecular mass 247 [MW 247]: 1-nitropyrene, 2-nitropyrene, 2-nitrofluoranthene; Table 19; Figures 5 and 6), 1-nitropyrene, 9-nitroanthracene, 3-nitrofluoranthracene (together with many PAHs in the Teplice, Czech Republic, program, Table 20) or carcinogenic (e.g., 1-nitropyrene, dinitropyrenes; Table 21) importance. As noted in Table 20, in many studies the report of only the 3- isomer of fluoranthene may in fact reflect a single GC peak in which 2- and 3-nitrofluoranthene co-eluted. Where both isomers have not been discussed, caution must be used in interpreting the data, for example, regarding the sources of the nitrofluoranthene isomers.
Table 21. Monitoring studies on dinitropyrenesa
|
Location |
Date |
BaP |
1-Nitropyrene |
1,3-Dinitropyrene |
1,6-Dinitropyrene |
1,8-Dinitropyrene |
Reference |
|
|
Shopping mall, Fukuoka, Japan |
Winter 1992 |
15 pg/m3 |
9.5 pg/m3 |
7.5 pg/m3 |
Maeda et al. (1994) |
|||
|
Kanazawa, Japan |
1989–1992 |
173 pg/m3 |
0.64 pg/m3 |
1.17 pg/m3 |
1.05 pg/m3 |
Hayakawa et al. (1995a) |
||
|
Kanazawa, Japan |
Downtown |
July 1993–February 1994 |
2495±1260 pg/m3 |
222±101 pg/m3 |
1.3±0.8 pg/m3 |
1.0±0.6 pg/m3 |
1.4±0.7 pg/m3 |
Murahashi et al. (1995) |
|
Location not given |
15–134 pg/m3 |
nd–4.7 pg/m3 |
0.3–8.7 pg/m3 |
nd–6.6 pg/m3 |
Tanabe et al. (1986) |
|||
|
Location not given |
190–1600 ng/g |
nd–56 ng/g |
5–105 ng/g |
nd–79 ng/g |
Tanabe et al. (1986) |
|||
|
Fukuoka, Japan |
140 ng/g |
12 ng/g |
10 ng/g |
25 ng/g |
Sera et al. (1994) |
|||
|
Bermuda |
Remote |
August 1992 |
280 ng/g particulate |
520 ng/g |
8.1 ng/g |
3.5 ng/g |
Gibson (1986) |
|
|
Bermuda |
Remote |
January/ February 1983 |
320 ng/g |
720 ng/g |
8.3 ng/g |
4.4 ng/g |
Gibson (1986) |
|
|
Delaware (USA) |
Rural |
July/August 1982 |
570 ng/g |
540 ng/g |
4.9 ng/g |
2.4 ng/g |
Gibson (1986) |
|
|
Warren (USA) |
Suburban |
December 1982 |
6500 ng/g |
360 ng/g |
<6 ng/g |
<6 ng/g |
Gibson (1986) |
|
|
Warren (USA) |
Suburban |
June 1984 |
4100 ng/g |
350 ng/g |
4.6 ng/g |
2.1 ng/g |
Gibson (1986) |
|
|
Detroit (USA) |
Urban |
June–August 1981 |
7800 ng/g |
220 ng/g |
3.6 ng/g |
2.5 ng/g |
Gibson (1986) |
|
|
River Rouge (USA) |
Industrial |
July 1982 |
4900 ng/g |
590 ng/g |
46 ng/g |
13.1 ng/g |
Gibson (1986) |
|
|
Dearborn (USA) |
Industrial |
June–August 1980 |
66 000 ng/g |
150 ng/g |
41 ng/g |
20 ng/g |
Gibson (1986) |
|
|
February–April 1983 |
14 000 ng/g |
Gibson (1986) |
a nd = not detected.
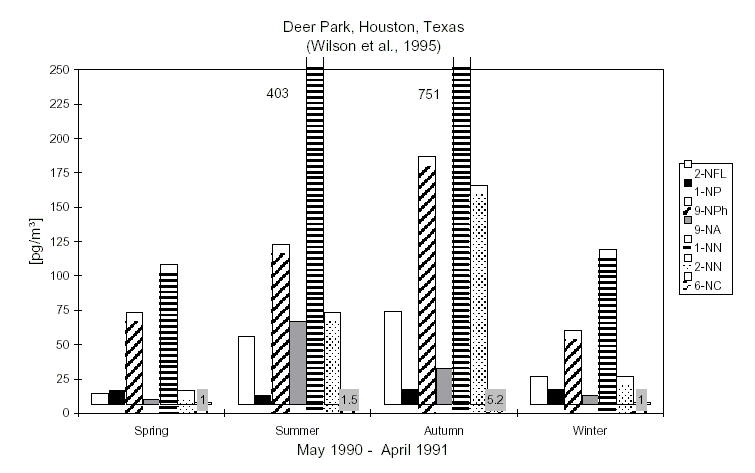
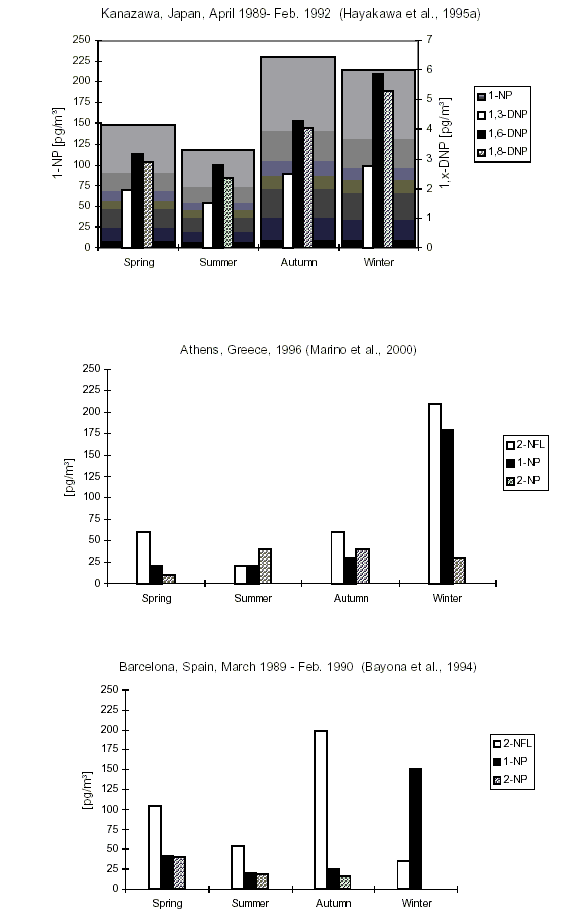
Fig. 5. Seasonal studies of selected nitroPAHs in ambient air. 2-NFL = 2-nitrofluoranthene; 1-NP = 1-nitropyrene; 2-NP = 2-nitropyrene; 1,3-DNP = 1,3-dinitropyrene; 1,6-DNP = 1,6-dinitropyrene; 1,8-DNP = 1,8-dinitropyrene; 9-NPh = 9-nitrophenanthrene; 9-NA = 9-nitroanthracene; 1-NN = 1-nitronaphthalene; 2-NN = 2-nitronaphthalene; 6-NC = 6-nitrochrysene.
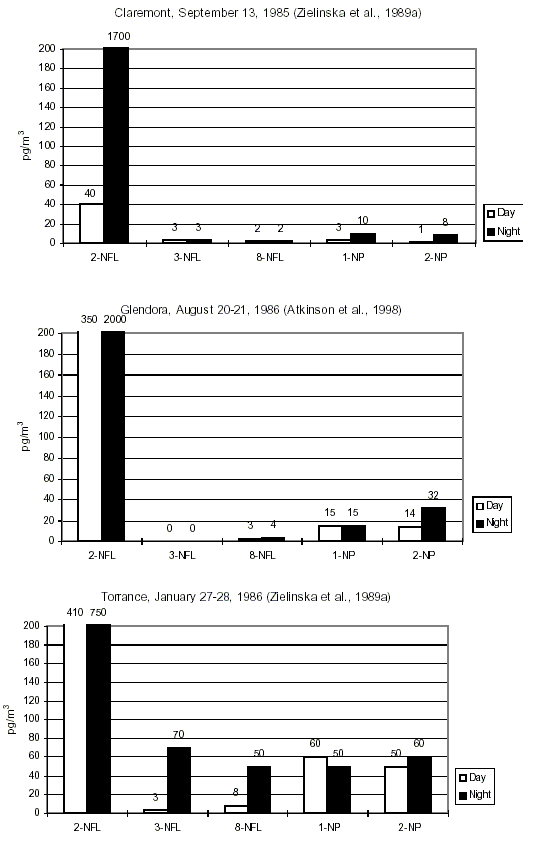
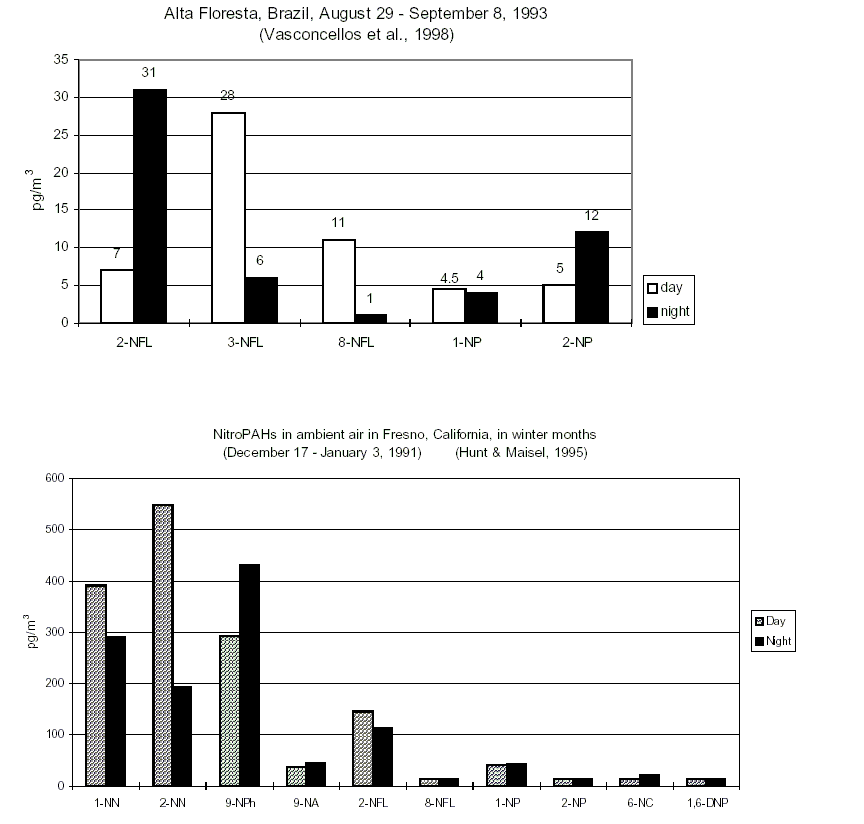
Fig. 6. Diurnal differences in MW 247 compounds. 2-NFL = 2-nitrofluoranthene; 3-NFL = 3-nitrofluoranthene; 8-NFL = 8-nitrofluoranthene; 1-NP = 1-nitropyrene; 2-NP = 2-nitropyrene; 1-NN = 1-nitronaphthalene; 2-NN = 2-nitronaphthalene; 9-NPh = 9-nitrophenanthrene; 9-NA = 9-nitroanthracene; 6-NC = 6-nitrochrysene; 1,6-DNP = 1,6-dinitropyrene.
3,6-Dinitrobenzo[a]pyrene was detected in airborne particulates in Santiago, Chile, in the winter of 1988–1989 at concentrations of 10 ng/g of total particulate (0.002 ng/m3 of air) (Sera et al., 1991). Although the concentration was usually low, the mutagenicity of the chemicals accounted for about 40% of the total activity of the crude extracts.
The nitroarene fraction of MW 247 is of interest because in diesel exhaust, about 10% of direct mutagenicity is associated with this fraction, comprising nitrofluoranthene and nitropyrene isomers (in particular 1-nitropyrene and 3- and 8-nitrofluoranthene in diesel exhaust) (Salmeen et al., 1984). Furthermore, 2-nitrofluoranthene and 2-nitropyrene, which are formed from atmospheric reactions, also have a relative molecular mass of 247.
Table 19 also compares the concentrations of total PAHs, BaP and nitroPAHs of MW 247. From the data available, it can be seen that the concentration of BaP always exceeds that of 1-nitropyrene (ranging from 2 to 100 times more). 2-Nitrofluoranthene concentrations usually exceed those of 1-nitropyrene (by 1–40 times). Because 2-nitrofluoranthene is formed from gas-phase radical-initiated reactions of fluoranthene and 1-nitropyrene is emitted in combustion sources, the high 2-nitrofluoranthene/1-nitropyrene ratio suggests the importance of atmospheric nitroPAH formation. Additionally, the very high nitronaphthalene and methylnitronaphthalene concentrations in Table 18 when vapour and particles have been measured also reflect the significance of the atmospheric formation of nitroPAHs (Atkinson et al., 1988; Gupta et al., 1996; Arey, 1998).
1) Abundance of volatile and particulate-bound nitroPAHs in ambient air
2-Nitrofluoranthene is usually the most abundant MW 247 nitroPAH observed in ambient particulate organic matter extracts; however, in the ambient air samples where vapour-phase nitroPAHs have been measured (see Table 19), the more volatile nitronaphthalenes and methylnitronaphthalenes (two-ring PAHs) were most abundant (Arey et al., 1987). From seasonal studies at Houston, Texas, USA, it was found that 1-nitronaphthalene was always the most prevalent nitroPAH, followed by 9-nitrophenanthrene (three-ring PAH), 2-nitronaphthalene and 2-nitrofluoranthene (four-ring PAH) and 9-nitroanthracene (three-ring PAH) (Wilson et al., 1995; see Figure 5). The concentrations of all these compounds were several times higher in summer and autumn than in winter and spring. In comparison, 1-nitropyrene (four-ring PAH) was present in comparatively small amounts, but more in autumn and winter. Although 1- and 2-nitronaphthalene are expected to be found dominantly in the gas phase, the other semivolatile nitroPAHs will be distributed between the particle and gas phases, depending upon the ambient temperature. Thus, many ambient measurements will underestimate the total nitroPAHs present unless both gas- and particle-associated species have been measured. Hunt & Maisel (1995) also reported 1-nitronaphthalene, 2-nitronaphthalene and 9-nitrophenanthrene as the most abundant nitroPAHs (see Figure 6).
At seven sites throughout California, USA (Atkinson et al., 1988), the nitronaphthalenes were the most abundant nitroPAHs; at Glendora (traffic impacted), Yuba City (biomass burning and traffic), Oildale (oil production) and Reseda (traffic), the nitronaphthalene concentrations exceeded those of BaP.
2) Studies on ambient particulate matter: worldwide surveys
Remote sites
There are some reports of the determination of nitroPAHs at remote and forest sites: Storkow (Germany), CNR Pyramid (Nepal), Terra Nova Bay (Antarctica) and Alta Floresta (Brazil) (see Table 19; Ciccioli et al., 1995, 1996). Recently, nitronaphthalenes were reported to be the most abundant nitroPAHs in Antarctica (1–200 fg/m3 range) (Vincenti et al., 2001).
NitroPAHs were either not detected or in the low picogram per cubic metre range (e.g., 2-nitrofluoranthene, 17 pg/m3; 1-nitropyrene, 4 pg/m3). NitroPAHs were determined in the aerosol from the Amazon forest (Alta Floresta), but the author notes that it was near biomass burning (Vasconcellos et al., 1998).
Urban sites
Reports of urban and suburban concentrations of nitroPAHs are given in Tables 18 and 19. The levels depend, for example, on the climatic conditions, the number and regulation of traffic vehicles and the type of heating used. For example, concentrations of nitroPAHs (9-nitroanthracene, 2-nitrofluoranthene and 1-nitropyrene) taken during the same time period (January to March 1994) were 2–4 times higher in Damascus, Syria, than in Birmingham, United Kingdom, possibly due to insufficient burning of fuel oil employed for domestic heating and older cars in the Syrian capital (Dimashki et al., 1996).
Even in urban areas, the concentrations of 2-nitrofluoranthene have been found to be greater than those of 1-nitropyrene (see Table 19), with some values exceeding 1 ng/m3. Maximum values of 2-nitrofluoranthene and 1-nitropyrene were reported in Milan (4540 and 1070 pg/m3, respectively). This is in the range of reported concentrations of BaP in particulate matter. The concentration of total PAHs is 2 orders of magnitude higher. In Glendora, California, USA, 2-nitrofluoranthene reached 2000 pg/m3 when the corresponding BaP concentration was 320 pg/m3 (Atkinson et al., 1988).
2-Nitrofluoranthene and 1-nitropyrene concentrations were monitored daily for 4 weeks (February/March 1991) in Milan and Rome, Italy, and compared with BaP and "total" PAH levels (Cecinato et al., 1998). Concentrations were, respectively, 0.95 and 0.25 ng/m3 in Milan compared with 0.25 and 0.08 ng/m3 in Rome. Together with the results of previous studies, it was shown that mean winter levels were higher than summer levels. In Milan, the concentrations of these two nitroPAHs increased from 1990 to 1993.
The outdoor background concentration of 1-nitropyrene measured in and around Southampton, United Kingdom, was usually below 10 pg/m3, but occasionally a higher value was observed (Scheepers et al., 1999).
Some studies on urban atmospheric particulates have concentrated on monitoring 2-nitrofluorene, which, together with 1-nitropyrene, is associated with diesel emissions. Concentrations of nitroPAHs in particulate samples in the summer in six different sites in the industrially developed region of Upper Silesia in Poland were 90–290 pg/m3 for 2-nitrofluorene and up to 350 pg/m3 for 1-nitropyrene (and/or 3-nitrofluoranthene) (Warzecha, 1993). In studies on levels of 2-nitrofluorene in various cities, the following concentrations were measured: 24–50 pg/m3 (Tokyo, Japan), 50–700 pg/m3 (Beijing, China) and 170–4190 pg/m3 (Berlin, Germany) (Moriske et al., 1984; Beije & Möller, 1988a).
3) Seasonal differences
Seasonal studies on certain selected nitroPAHs (Bayona et al., 1994; Hayakawa et al., 1995a; Wilson et al., 1995; Murahashi et al., 1999; Marino et al., 2000) (see Figure 5) show that the concentrations of 1-nitropyrene and dinitropyrenes (from combustion sources) in ambient air particulates are usually higher in winter than in the other months. In contrast, in most studies, levels of 2-nitrofluoranthene and 2-nitropyrene (atmospheric transformation) are less in winter months than in the warmer seasons. This is also the case for the more volatile nitroPAHs (e.g., 1-nitronaphthalene, 2-nitronaphthalene, 9-nitrophenanthrene and 9-nitroanthracene) (Wilson et al., 1995; see Figure 5). The concentrations of all these compounds were several times higher in summer and autumn than in winter and spring.
Table 20 shows the concentrations of BaP and some nitroPAHs measured in a large study of semivolatile and volatile compounds at two sites in the Czech Republic during the years 1993–1994 and 1996–1997. Higher concentrations were found in winter than in summer due to the higher fuel consumption in winter. Although coal combustion was replaced in this region by gas for heating purposes in the years 1994–1995, it was found that similar distribution profiles and concentrations of PAHs were in the air in Teplice in winter 1993 and 1996 (Lenicek et al., 2001).
4) Diurnal differences
Daytime and nighttime studies on MW 247 nitroPAHs in ambient air collected at three sites (Claremont, Torrance and Glendora) situated in the Los Angeles basin in California, USA (Arey et al., 1987, 1988b; Zielinska et al., 1989b), showed increases of certain nitroPAHs during the nighttime (Figure 6; Table 18). From studies of PAHs (and nitroPAHs) in simulated atmospheres, it has been proposed that PAHs react with gaseous co-pollutants in the atmosphere to produce these nitroPAHs. Whereas the nitration of particle-bound PAHs with nitrogen dioxide/nitric acid and nitrogen pentoxide is probably not significant under most atmospheric conditions, this may not be the case in airsheds that have high nitrogen dioxide and nighttime nitrogen pentoxide concentrations. Daytime hydroxyl radical-initiated reactions of PAHs lead to the formation of nitroPAHs in low yields. For a discussion of the atmospheric formation of nitroPAHs, see chapter 3. The high nighttime concentrations of 2-nitrofluoranthene (and 1- and 2-nitronaphthalene and certain methylnitronaphthalenes) are attributed to nighttime gas-phase reactions of the parent PAH with the nitrate radical.
Composite samples (vapour-phase and particulate portions) of a number of chemical compounds, including nitroPAHs, were monitored in Fresno, California, USA, during the winter months. Elevated retene (1-methyl-7-isopropylphenanthrene) and enhanced nocturnal PAH (in particular naphthalene) concentrations suggested the strong influence of residential wood burning. Photochemical reactions involving these parent compounds and hydroxyl radicals resulted in elevated concentrations of 1- and 2-naphthalene (Hunt & Maisel, 1995; see Figure 6).
In a study on the chemical composition of aerosol in the Amazon, it could be seen from diurnal studies that the nitroPAHs 2-nitrofluoranthene and 2-nitropyrene were formed from nighttime photochemical reactions. The comparatively high levels of daytime 3- and 8-nitrofluoranthene were thought to be due to direct emissions (flaming and smouldering) from forest fires (Vasconcellos et al., 1998; see Figure 6).
Diurnal studies in which 1-nitropyrene and dinitropyrene levels in ambient air in downtown Kanazawa, Japan (sampling period July 1993 to February 1994), were compared with BaP concentrations and traffic volume found the nitroPAH levels to be high in the morning and evening and low from midnight to early morning, indicating that the major contributor to the dinitropyrenes and 1-nitropyrene was traffic emission (Hayakawa et al., 1995a, 1999b; Murahashi et al., 1995; see Figure 7). Similar studies monitoring 2-nitrofluoranthene, 1-, 2- and 4-nitropyrene and 6-nitrochrysene (4–7 January 1995) showed the highest concentrations of 4-nitropyrene and 6-nitrochrysene around 10 a.m. to 2 p.m. (comparable to 1-nitropyrene); however, the highest concentrations of 2-nitrofluoranthene and 2-nitropyrene were observed about 6 h later, around 6 p.m. to 8 p.m. (Murahashi et al., 1999).
Diurnal and seasonal (summer/winter) atmospheric concentrations of 1-nitropyrene and dinitropyrenes were recorded in three cities in Japan: Kanazawa, Sapporo and Tokyo (Kakimoto et al., 2000, 2001; see Table 22 and also Table 21). The concentrations tended to be higher in winter than in summer and higher in daytime than at night.
Table 22. Mean atmospheric concentrations of 1-nitropyrene and dinitropyrenes in three Japanese cities in 1995a
|
City |
Season |
Day/night |
1-Nitropyrene |
Dinitropyrenesb |
|
Kanazawa |
Winter |
Day |
59.2 |
0.91 |
|
Night |
38.3 |
0.62 |
||
|
Summer |
Day |
26.7 |
0.38 |
|
|
Night |
11.3 |
0.28 |
||
|
Sapporo |
Winter |
Day |
413 |
5.7 |
|
Night |
197 |
2.6 |
||
|
Summer |
Day |
206 |
2.4 |
|
|
Night |
149 |
2.3 |
||
|
Tokyo |
Winter |
Day |
163 |
2.0 |
|
Night |
120 |
2.2 |
||
|
Summer |
Day |
130 |
2.3 |
|
|
Night |
75.3 |
2.9 |
a From Kakimoto et al. (2000, 2001).
b Dinitropyrenes = 1,6-dinitropyrene + 1,8-dinitropyrene +1,3-dinitropyrene.
Daytime and nighttime variations in concentrations of 2-nitrofluoranthene, 1-nitropyrene and 2-nitropyrene, together with aerosols, ozone and nitrated species, were recorded in downtown Milan, Italy, in September 1990 and February to March 1991 (Ciccioli et al., 1993).
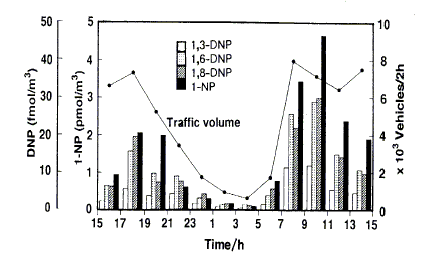
Fig. 7. Diurnal studies of 1-nitropyrene (1-NP) and 1,3-, 1,6- and 1,8-dinitropyrene (1,3-, 1,6- and 1,8-DNP) in downtown Kanazawa, Japan, July to February 1994 (from Hayakawa et al., 1999b), compared with traffic volume, indicating that the major contributor to the dinitropyrenes and 1-nitropyrene was traffic emissions.
Hydroxyl radical-initiated reactions were assumed to be responsible for the formation of 2-nitrofluoranthene and 2-nitropyrene. Since 2-nitrofluoranthene in nighttime hours was at concentrations much higher than those of 2-nitropyrene, it was supposed that reaction with nitrate also occurred.
Thus, combustion emissions, the occurrence of atmospheric formation reactions (daytime hydroxyl radical reactions and nighttime nitrate radical reactions) and meteorological and atmospheric dynamics will all influence the measured concentrations of nitroPAHs.
5) Nitro-oxyPAHs
2- and 4-nitrodibenzopyranones were observed in an ambient particulate extract collected from Riverside, California, USA, and accounted for ~20% of the total mutagenic activity (Helmig et al., 1992a). It should be noted that the microsuspension modification of the Ames assay was used. 2-Nitrodibenzopyranone has an unusually high response in this assay. 2- and 4-nitrodibenzopyranone were also detected in other ambient air samples from California (0.05–1 and <0.02–0.21 ng/m3 for the 2- and 4- isomers, respectively) and in an urban dust sample (NIST SRM 1649) from Washington, D.C. (0.82 and 0.52 µg/g, respectively; see Table 23), but with much lower concentrations in diesel particulate material (~0.2 and <0.1 µg/g, respectively), suggesting that nitrodibenzopyranones are formed in the atmosphere (Helmig et al., 1992b). 3-Nitrodibenzopyranone has not been reported in ambient air samples.
Table 23. 2- and 4-nitrodibenzopyranone concentrations in atmospheric samples from Los Angeles, California, USA, in urban dust (SRM 1649) and diesel exhaust particles (SRM 1650) compared with MW 247 nitroPAHsa
|
Compound |
Concentrations in the Los Angeles basin (ng/m3) |
Concentration (µg/g particles) |
|
|
SRM 1649 |
SRM 1650 |
||
|
2-Nitrodibenzopyranone |
0.05–1 |
0.82 ± 0.15 |
~0.2 |
|
4-Nitrodibenzopyranone |
<0.02–0.21 |
0.52 ±0.09 |
<0.1 |
|
2-Nitrofluoranthene |
0.03–2.0b |
0.60c |
|
|
1-Nitropyrene |
0.003–0.06b |
0.06d |
19 ± 2d |
|
2-Nitropyrene |
0.001–0.06b |
0.27 |
|
|
9-Nitroanthracene |
0.2 |
10 |
|
|
7-Nitrobenz[a]anthracene |
3 |
||
|
6-Nitrobenzo[a]pyrene |
1.6 |
||
a Adapted from Helmig et al. (1992b).
b From Arey et al. (1987); Zielinska et al. (1989a).
c From Ramdahl et al. (1986).
d From May et al. (1992).
NitroPAHs have been detected in the emissions of kerosene heaters, fuel gas and LPG burners (city gas and heavy oil) used for heating and cooking at home, as well as in the fumes of cooking oils (see chapter 3). In poorly ventilated conditions, there is therefore a potential for indoor exposure to nitroPAHs.
Concentrations of polyaromatic compounds, including nitroPAHs, were measured in a study of indoor and outdoor air levels associated with 33 homes located in two US cities, Columbus, Ohio, and Azusa, California (Wilson et al., 1991). The overall levels were much higher in homes occupied by smokers, but the use of natural gas heating and cooking appliances also appeared to increase the nitroPAH levels slightly (Table 24). Concentrations were up to 0.28 ng/m3 for 1-nitropyrene and from 0.006 to 0.20 ng/m3 for 2-nitrofluoranthene. The average outdoor level for 2-nitrofluoranthene was 0.06 ng/m3. Concentrations of 1-nitropyrene were detected at <1.0–8.6 pg/m3 in indoor air in Southampton, United Kingdom (Scheepers et al., 1999).
Table 24. Typical nitroPAH concentrations in homes averaged over three locations: living room, bedroom and kitchena
|
NitroPAH |
Concentrations (ng/m3) |
||
|
Homes with smokers |
Homes with non-smokers |
||
|
with gas heating and gas stove |
with electric heating and electric stove |
with gas heating and gas stove |
|
|
1-Nitropyrene |
0.044 |
0.020 |
0.021 |
|
2-Nitrofluoranthene |
0.052 |
0.012 |
0.022 |
a From Wilson et al. (1991).
1-Nitropyrene was detected in water samples from the Yodo River, Kyoto, Japan, which is used as a source of drinking-water. No quantitative data were given (Ohe, 1996).
1- and 2-nitronaphthalene and 1,3- and 1,5-dinitronaphthalene were detected in river water in Japan at concentrations of 1.3, 11.7, 1.7 and 3.2 ng/litre, respectively (Takahashi et al., 1995).
The following nitroPAHs were not detected in the Nagura River in Japan: 1-nitropyrene (<20 ng/litre), 3-nitrofluoranthene (<20 ng/litre), 1,3-, 1,6- and 1,8-dinitropyrene (<200 ng/litre) and 2,7-dinitrofluorene (<200 ng/litre) (Nagai et al., 1999).
1-Nitropyrene (4.2–25 600 ng/litre) was detected in 36 of 55 samples of wastewater from oil/water separating tanks of gasoline stations and in used crankcase oil (Manabe et al., 1984). The 1-nitropyrene accounted for 0.3–58.5% of the total mutagenicity of the neutral fractions. Nineteen samples did not contain any 1-nitropyrene.
1-Nitropyrene and nitrofluoranthene isomers as well as nitronaphthalene, nitrofluorene and methylated derivatives were detected together with other PAHs in samples of sewage sludge in Upper Silesia, Poland (Bodzek & Janoszka, 1995; Bodzek et al., 1997) (levels not given). 1-Nitropyrene was identified in sewage sludge at 0.68 µg/kg dry weight (Fernández et al., 1992).
In samples of surface soil from the city of Basel, Switzerland, nitroPAHs (mainly 3-nitrofluoranthene and 1-nitropyrene) were found in concentrations between 0.03 and 0.8 µg/kg dry weight. Levels of oxyPAHs and parent PAHs were 102- to 104-fold higher (Niederer, 1998).
1,3-, 1,6- and 1,8-dinitropyrene (0.012–3.27, 0.014–5.59 and 0.013–6.80 µg/kg dry weight, respectively) were detected in all 110 samples of non-agricultural soil collected from five geographically different areas in Japan between November 1996 and March 1997 (Watanabe et al., 2000).
2-Nitrofluorene, 2,7-dinitrofluorene and 1-nitropyrene (1.5, 3.8 and 25.2 µg/kg sediment, respectively) were detected in sediments in the bed of the Suimon River. The direct-acting mutagenic activity that had been observed in sediment samples from this river was consistent with the mutagenicity of these nitroPAHs, in particular 1-nitropyrene (Sato et al., 1985).
Coastal sediments collected offshore from Barcelona, Spain, were fractionated and characterized using bioassay-directed chemical analysis (Fernández et al., 1992). 6-Nitrochrysene, 6-nitrobenzo[a]pyrene and nitrobenzofluoranthenes were identified at concentrations of 0.52, 0.34 and 0.26 µg/kg dry weight, respectively, in sediment from urban littoral stations.
Librando et al. (1993) reported concentrations of selected nitroPAHs of up to 5000 µg/kg in incinerator ash. The highest values were given by 1-nitronaphthalene (1.59–2.86 mg/kg), 2-nitronaphthalene (1.06–3.46 mg/kg) and 9-nitroanthracene (0.96–4.84 mg/kg). Values for 1-nitropyrene were <0.01–0.89 mg/kg.
5.1.4.1 Food
Foodstuffs were monitored for the presence of 9-nitroanthracene and 1-nitropyrene in the United Kingdom (Dennis et al., 1984). Of the 28 samples analysed, 25 contained no detectable levels of these nitroPAHs. 9-Nitroanthracene was tentatively identified in peated malt at 0.9 µg/kg and 1-nitropyrene in two samples of tea leaf at 1.7 and 0.17 µg/kg.
1-Nitropyrene was detected in grilled corn, mackerel and (in considerable amounts) pork and yakitori (grilled chicken) grilled with sauce (up to 43 ng/g) (Ohnishi et al., 1986). The authors concluded that the formation of 1-nitropyrene is due to pyrene produced by the incomplete combustion of fat in the chicken, its nitration at acidic pH by nitrogen dioxide emitted by the burning of cooking gas and some components of the marinating sauce.
Various foods were analysed for the presence of 2-nitrofluorene, 1-nitropyrene and 2-nitronaphthalene (Schlemitz & Pfannhauser, 1996a; Table 25). These nitroPAHs were detected in most samples, the highest concentrations being found in spices and smoked food, but also in vegetables and fruits, probably due to atmospheric pollution. In a parallel study using another analytical method, the presence of nitroPAHs was monitored in cheese as well as in meats and fish prepared by grilling or roasting (Schlemitz & Pfannhauser, 1996b; see Table 25).
Table 25. Occurrence of nitroPAHs in food samples
|
Sample |
Measurea |
Concentrationsb (µg/kg) |
Referencec |
|||
|
1-Nitronaphthalene |
2-Nitronaphthalene |
2-Nitrofluorene |
1-Nitropyrene |
|||
|
Vegetable, fruits, nuts |
||||||
|
Lettuce |
n. sp. |
– |
<0.2 |
1.6 |
<0.2 |
Schlemitz & Pfannhauser (1996a) |
|
Parsley |
n. sp. |
– |
<0.2 |
0.8 |
1.7 |
Schlemitz & Pfannhauser (1996a) |
|
Carrot |
n. sp. |
– |
nd (0.2) |
0.9 |
0.4 |
Schlemitz & Pfannhauser (1996a) |
|
Apple |
n. sp. |
– |
nd (0.2) |
0.8 |
0.2 |
Schlemitz & Pfannhauser (1996a) |
|
Peanuts |
n. sp. |
– |
nd (0.2) |
16.6 |
<0.5 |
Schlemitz & Pfannhauser (1996a) |
|
Spices |
||||||
|
Paprika |
n. sp. |
– |
7.8 |
26.4 |
9.3 |
Schlemitz & Pfannhauser (1996a) |
|
Marjoram |
n. sp. |
– |
3.6 |
23.1 |
14.1 |
Schlemitz & Pfannhauser (1996a) |
|
Caraway |
n. sp. |
– |
3.1 |
350.7 |
10.9 |
Schlemitz & Pfannhauser (1996a) |
|
Oils |
||||||
|
Olive oil |
n. sp. |
– |
<0.2 |
0.8 |
0.6 |
Schlemitz & Pfannhauser (1996a) |
|
Milk products |
||||||
|
Alp-cheese I |
mean (3) |
nd |
– |
1.1 |
nd |
Schlemitz & Pfannhauser (1996b) |
|
Alp-cheese II |
mean (3) |
nd |
– |
0.3 |
nd |
Schlemitz & Pfannhauser (1996b) |
|
Smoked cheese |
mean (3) |
0.6 |
– |
0.5 |
nd |
Schlemitz & Pfannhauser (1996b) |
|
Fish |
||||||
|
Grilled fish |
mean (3) |
0.2 |
– |
0.3 |
nd |
Schlemitz & Pfannhauser (1996b) |
|
Grilled mackerel |
0.45 |
Ohnishi et al. (1986) |
||||
|
Smoked fish |
mean (3) |
0.3 |
– |
0.2 |
nd |
Schlemitz & Pfannhauser (1996b) |
|
Meat |
||||||
|
Grilled meat (pork) |
n. sp. |
– |
0.1 |
2.0 |
1.0 |
Schlemitz & Pfannhauser (1996a) |
|
Grilled sausages |
n. sp. |
|
<0.2 |
0.2 |
0.8 |
Schlemitz & Pfannhauser (1996a) |
|
mean (3) |
nd |
|
0.1 |
|
Schlemitz & Pfannhauser (1996b) |
|
|
Smoked meat |
n. sp. |
– |
10.2 |
2.0 |
2.2 |
Schlemitz & Pfannhauser (1996a) |
|
Smoked sausages |
n. sp. |
– |
8.4 |
19.6 |
4.2 |
Schlemitz & Pfannhauser (1996a) |
|
Roasted meat (pork) |
mean (3) |
nd |
– |
0.5 |
0.3 |
Schlemitz & Pfannhauser (1996b) |
|
Roasted turkey |
mean (3) |
nd |
– |
0.3 |
nd |
Schlemitz & Pfannhauser (1996b) |
|
Grilled chicken |
range (10) |
– |
– |
– |
0.4–11 |
Kinouchi et al. (1986a) |
|
Grilled chicken |
up to 43 |
Ohnishi et al. (1986) |
||||
|
a |
In parentheses: number of samples; n.sp.= not specified. |
|
b |
nd = not detected (detection limit in parentheses, if given); – = no data available. |
|
c |
Schlemitz & Pfannhauser (1996a, 1996b) studies conducted in Austria; Kinouchi et al. (1986a) and Ohnishi et al. (1986) studies conducted in Japan. |
In another study (Ziegler et al., 1999), concentrations of 2-nitrofluorene and 1- and 2-nitronaphthalene in a variety of fruits and vegetables (apple, grapes, red pepper, broccoli, kohlrabi and cauliflower) were below the detection limit (5 µg/kg).
To study the effect of cooking on nitroPAHs, cauliflower and broccoli samples were artificially contaminated with a solution of 2-nitrofluorene and 1- and 2-nitronaphthalene and cooked for 20 min. No more than 4% of the nitroPAHs transferred into the boiling water. The nitronaphthalenes volatilized to a great extent, but 2-nitrofluorene remained almost quantitatively on the solid parts.
The same solution of three nitroPAHs was dropped onto the peel of apples. After 18 h, it was found that almost the whole content remained in the peel fraction, only a small amount of the nitronaphthalenes being found in the edible interior part. Washing the contaminated apples in hot water did not remove any nitroPAHs from the peel. Similar results were found with kohlrabi. Therefore, nitroPAHs remain on the surface of fruits and vegetables. In the environment, however, the nitroPAHs are attached to particles and are not applied directly to the surface of plants, so washing the fruit with hot water should remove the particulate matter and the nitroPAHs; peeling fruit is the best way to reduce potential exposure to nitroPAHs (Ziegler et al., 1999).
NitroPAHs (2-nitrofluorene, 1-nitropyrene and 1- and 2-nitronaphthalene) were found in smoked foods from the market (n = 92), such as fish (n = 69) and meat products (n = 14). No traces of such compounds could be detected in smoked cheese (n = 9) after discarding the cheese rind (Dafflon et al., 2000).
Surveys of nitroPAH levels in various foods showed that substantial concentrations of nitroPAHs were present in tea and coffee (Schlemitz & Pfannhauser, 1996a). In a further study (using supercritical fluid extraction), samples of a variety of teas were investigated for their nitroPAH and PAH content (Schlemitz & Pfannhauser, 1997; see Table 26). Six nitroPAHs were measured. Very high concentrations of nitroPAHs (128 µg/kg) and PAHs (7536 µg/kg) were found in Mate tea, which is roasted with combustion fumes to gain its unique aroma. Other teas had concentrations of around 20 µg/kg for the six nitroPAHs, with 2-nitrofluorene and 9-nitroanthracene having the highest levels. Although nitroPAHs are usually not very soluble in water (see chapter 2), the authors found that up to 25% of the nitroPAH concentration could be measured in the tea water. It is possible that other components of the tea act as co-solvents, thereby increasing the solubility of nitroPAHs in the tea water. In all tea samples, the PAH content was always much higher than that of nitroPAHs. The presence of nitroPAHs and PAHs in tea probably originates from several sources, including technological processes used during the preparation of tea, such as roasting and drying, and atmospheric pollution.
Table 26. Occurrence of nitroPAHs in tea leaves and coffee beansa
|
Sample (n = 3) |
Concentrationb (µg/kg) |
|||||||
|
1-NN |
2-NN |
2-NF |
1-NP |
9-NA |
3-NFL |
Total nitroPAHs |
Total PAHs |
|
|
Assam tea |
nd |
0.58 |
6.45 |
2.32 |
5.21 |
4.36 |
18.92 |
28.56 |
|
Earl Grey tea |
0.90 |
1.38 |
9.77 |
7.75 |
4.17 |
1.38 |
25.35 |
313.77 |
|
Ceylon tea |
0.39 |
0.67 |
0.61 |
1.54 |
16.64 |
1.73 |
21.58 |
79.7 |
|
Darjeeling tea |
13.07 |
1.43 |
nd |
4.00 |
2.5 |
0.98 |
21.98 |
775.67 |
|
Mate tea (roasted) |
0.47 |
4.09 |
20.04 |
37.89 |
22.19 |
43.04 |
127.72 |
7536.33 |
|
Mate tea (green) |
nd |
1.52 |
5.32 |
0.80 |
1.81 |
1.21 |
10.66 |
6475.9 |
|
Formosa Sencha (green tea) |
nd |
1.05 |
9.27 |
3.10 |
3.06 |
3.66 |
20.14 |
549.76 |
|
Nettle leaf tea |
0.25 |
0.18 |
10.02 |
1.96 |
12.6 |
3.71 |
28.72 |
97.77 |
|
Peppermint tea |
0.46 |
0.85 |
16.41 |
3.79 |
3.94 |
1.85 |
27.30 |
140.39 |
|
Fennel tea (instant) |
nd |
0.41 |
0.09 |
nd |
0.51 |
nd |
1.01 |
13.41 |
|
Fruit tea (instant) |
0.82 |
0.22 |
0.56 |
0.55 |
0.60 |
0.28 |
3.03 |
17.53 |
|
Teac |
1.5 |
5.3 |
nd |
|||||
|
Coffeec |
4.0 |
30.1 |
2.4 |
|||||
|
a |
From Schlemitz & Pfannhauser (1997). |
|
b |
1-NN = 1-nitronaphthalene; 2-NN = 2-nitronaphthalene; 2-NF = 2-nitrofluorene; 1-NP = 1-nitropyrene; 9-NA = 9-nitroanthracene; 3-NFL = 3-nitrofluoranthene; nd = not detected. |
|
c |
From Schlemitz & Pfannhauser (1996b); in this study, only three nitroPAHs were determined. |
NitroPAHs are found in the ground coffee bean, so filtered coffee would probably not contain nitroPAHs at such levels. Drinking instant coffee would lead to the direct ingestion of the nitroPAHs (Schlemitz & Pfannhauser, 1996a).
Extracts of selected xerographic toners and paper photocopies were found to be mutagenic (Löfroth et al., 1980). The fraction of the carbon black B responsible for 80% of the mutagenicity contained 1-nitropyrene, 1,3-, 1,6- and 1,8-dinitropyrene, 1,3,6-trinitropyrene and 1,3,6,8-tetranitropyrene. As a result of this finding, the manufacturers modified the production of carbon black B, substantially reducing the levels of nitropyrenes (Rosenkranz et al., 1980).
Although "parent" PAHs have been detected in tobacco smoke (IARC, 1986), there are no reports of nitroPAHs in cigarette smoke condensate. 1-Nitronaphthalene (<10 ng/cigarette), 1-nitropyrene (<10 ng/cigarette) and 6-nitrochrysene (<1 ng/cigarette) were not detected in the mainstream cigarette smoke of three different types of cigarette (El-Bayoumy et al., 1985). No 1-nitropyrene was detected in the tar extracts of five commercial brands of cigarettes at a limit of determination of 30 pg/cigarette (Scheepers et al., 2001).
NitroPAHs have been found to be ubiquitous in ambient air and in particular in air polluted by vehicular traffic. Indoor air exposure from kerosene heaters and from cooking oils has been described. NitroPAHs are found in certain foods, in particular grilled foods (see section 5.1.4.1). Drinking-water is not likely to contain nitroPAHs, as they are sparingly soluble or insoluble in water.
NitroPAHs have been detected in samples of resected lung tissue from patients in Japan (Tokiwa et al., 1993a, 1998a,b; Sera, 1998; Tokiwa & Sera, 2000) (see Table 27; section 8.1). The lung specimens were surgically resected from 293 patients with carcinoma and 63 patients with tuberculosis without carcinoma as controls during the periods 1961–1962 (time of heavy air pollution in Japan) and 1991–1996 (time of regulated air pollution in Japan). The ages of the patients ranged from 28 to 68 years. Lung specimens collected in this study originated from patients, including smokers and non-smokers, who had lived in the Fukuoka prefecture in Japan for over 20 years.
Table 27. Concentrations of PAHs and nitroPAHs in lung tissuesa
|
Compound |
Concentration of compound (pg/g of dry weight) |
|||
|
With carcinomab |
Controlb |
With carcinomac |
Controlc |
|
|
1-Nitropyrene |
30.8 ± 11.4 |
27 ± 14.8 |
19.7 ± 10.5* |
18.7 ± 10.2 |
|
1,3-Dinitropyrene |
4 ± 0.7 |
4.2 ± 0.35 |
3.5 ± 0.14 |
2.6 ± 0.17 |
|
1,6-Dinitropyrene |
5.1 ± 2.3 |
5 ± 2.8 |
4.82 ± 1.69 |
4.6 ± 1.8 |
|
1,8-Dinitropyrene |
6.7 ± 2.6 |
6.2 ± 2.7 |
6.26 ± 1.76 |
6.1 ± 2.1 |
|
2-Nitrofluoranthene |
43.2 ± 19.7 |
43.5 ± 2.5 |
38.6 ± 17.2 |
34.7 ± 15.4 |
|
3-Nitrofluoranthene |
32.6 ± 12.8 |
33.6 ± 15.2 |
39.1 ± 14.2 |
36.5 ± 13.8 |
|
Benzo[a]pyrene |
341 ± 210 |
330 ± 107 |
196 ± 114* |
178 ± 118 |
|
Pyrene |
583 ± 220 |
539 ± 356 |
510 ± 286 |
462 ± 115 |
|
Fluoranthene |
701 ± 356 |
673 ± 421 |
605 ± 370* |
586 ± 136 |
a From Tokiwa et al. (1998a); * P < 0.05.
b Periods 1961–1962 (time of heavy air pollution in Japan); samples from patients with lung carcinoma and, as control, from patients with tuberculosis but without carcinoma.
c Periods 1991–1996 (time of regulated air pollution in Japan); samples from patients with lung carcinoma and, as control, from patients with tuberculosis but without carcinoma.
Occupational exposure to nitroPAHs is expected in workplaces associated with the use of diesel engines. Underground workers (drivers of diesel-powered excavators) at an oil shale mine in Estonia were subjects in a study for the evaluation of 1-nitropyrene as a biomarker of exposure. The concentrations of 1-nitropyrene associated with respirable particles as determined in the breathing zones of the workers ranged up to 42 ng/m3 (mean 2.5 ng/m3) (Scheepers et al., 2002; see Table 28).
Table 28. Concentration of particle-associated 1-nitropyrene (pg/m3) in the breathing zones of drivers of diesel-powered excavators
(main study and pilot study)a
|
Surface |
Underground |
|||||||
|
No. of measurements |
Geometric mean (pg/m3) |
Median (pg/m3) |
Range (pg/m3) |
No. of measurements |
Geometric mean (pg/m3) |
Median (pg/m3) |
Range (pg/m3) |
|
|
Main study |
42b |
85 |
83 |
4–2166 |
50 |
637 |
694 |
29–5031 |
|
Pilot study (first shift) |
9 |
9 |
6 |
3–50 |
10 |
2483 |
2021 |
602–42 190 |
|
Pilot study (second shift) |
9 |
45 |
34 |
12–686 |
10 |
984 |
1179 |
134–3455 |
|
a |
From Scheepers et al. (2002). |
|
b |
1-Nitropyrene could not be quantified in seven workers because the internal standard peak area was too low. Air sampling failed in one worker. |
Air concentrations of 1-nitropyrene were measured in various workplaces associated with the use of diesel engines (Scheepers et al., 1994a; see Table 29). The highest mean level (1 ng/m3) was reported for forklift truck drivers at an aluminium rolling factory.
Table 29. Air concentrations of 1-nitropyrene as determined from extracts of total suspended particulate matter collected at workplaces associated with the use of diesel enginesa
|
Facility |
Sources of diesel exhaust |
No. of samples (n) |
1-Nitropyrene (mean)b (ng/m3) |
|
River vessel |
Ship’s engine |
3 |
0.05 |
|
Repair shop for trains |
Train engines |
4 |
0.32 |
|
Army driving lessons |
Armoured cars |
2 |
0.01 |
|
Flower auction |
Trucks |
2 |
0.08 |
|
Farming |
Tractor |
1 |
nd |
|
Gardening |
Passing traffic |
1 |
0.04 |
|
Airport platform |
Platform vehiclesc |
3 |
0.04 |
|
Concrete manufacturing |
Forklift trucks |
2 |
0.66 |
|
Chemical plant |
Forklift trucks |
4 |
0.27 |
|
Aluminium rolling |
Forklift trucks |
4 |
1.06 |
|
Galvanization workshop |
Forklift trucks |
4 |
0.10 |
|
Grass verge maintenance |
Lawn mowers |
1 |
0.007 |
|
River vessel |
Ship’s aggregate |
1 |
0.79 |
a From Scheepers (1994); Scheepers et al. (1994a); sample volumes 135–600 m3.
b Using gas chromatography–high-resolution mass spectrometry detection; nd = not detected.
c Lift platforms, power supplies, trucks, pull-offs.
In a more detailed report, 1-nitropyrene levels were determined in stationary samples of total suspended particulate matter collected on two consecutive workdays in January in a repair shop for train engines (Scheepers et al., 1994b). Air concentrations of particulate-associated 1-nitropyrene ranged from non-detectable to 5.6 ng/m3. In spot urine samples, urinary metabolites of PAHs and nitroPAHs were determined using enzyme-linked immunosorbent assay (ELISA), based on an antibody with high affinity to 1-aminopyrene. In urine samples of three non-smoking diesel mechanics, both cumulative and average excretion of urinary metabolites over 48 and 72 h were significantly enhanced (P < 0.05) compared with the excreted levels in urine samples from two office clerks. However, in a further study in May, this could not be confirmed (Scheepers et al., 1995a). Spot urine samples were collected during two consecutive workdays in January and May from a group of 30 railroad workers. Three job categories were involved: a) diesel mechanics exposed indoors to exhaust derived from running diesel engines in a repair shop for diesel trains; b) electrical engineers working outdoors and exposed to low levels of emissions derived from remote sources of incomplete combustion; and c) office workers. Levels of 1-aminopyrene equivalents were very similar in subjects working indoors and outdoors, regardless of whether they were smokers or non-smokers. It is possible that the significant effect found in the January study was due to the higher number of diesel engines being repaired or a lack of ventilation compared with the study in May, when windows, for example, were left open (Scheepers, 1994; Scheepers et al., 1995a,b, 1999, 2001).
For further studies on biomonitoring, see chapter 8.
The metabolism of nitroPAHs in vivo, with the exception of 1-nitropyrene and 2-nitrofluorene, has not been well studied. However, from the results on these two compounds, it can be expected that, in general, nitroPAHs administered by various routes are rapidly absorbed and metabolized, mainly by ring oxidation and nitroreduction, followed by conjugation and excretion of the metabolites formed, mainly in faeces and urine. Radiolabelled 1-nitropyrene was found to be widely distributed in the body of rats and mice after administration by all routes. For recent reviews on the metabolism and activation of nitroPAHs, see Fu (1990), Beland & Marques (1994), Fu & Herrenos-Saenz (1999) and Purohit & Basu (2000).
NitroPAHs constitute a complex group of chemicals showing different metabolisms. For a particular nitroPAH, there may be several metabolic pathways, often depending on the route of administration. Intestinal microflora seem to play a role in metabolism through nitroreduction and/or deconjugation. Deconjugation can enhance enterohepatic circulation. For most of the compounds studied, the metabolic pathways are not clearly understood.
While metabolism of PAHs involves oxidation and subsequent hydrolysis and/or conjugation reactions (IPCS, 1998), the metabolism of nitroPAHs is even more complex. It seems that there are at least five metabolic activation pathways through which mutations can be induced by nitroPAHs in bacterial and mammalian systems and/or through which DNA binding occurs. These are:
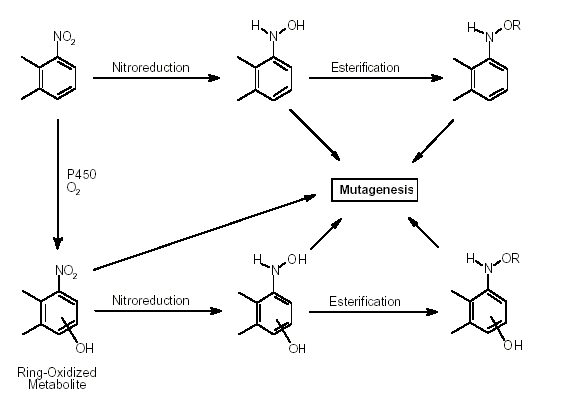
Fig. 8. Metabolic activation pathways of nitroPAHs leading to mutation (Fu, 1990).
In bacteria, nitroreduction seems to be the major metabolic pathway, whereas the fungus Cunninghamella elegans is an example of a species in which nitroPAHs are metabolized by ring oxidation (see chapter 4).
Studies into the enzymes involved in metabolic pathways — in particular those of cytochrome P450 — have shown that different P450 enzymes may be involved in metabolism of a specific nitroPAH (Chae et al., 1993); that these may differ in the related isomers, resulting in possibly different kinetics and pathways (Chae et al., 1999a); and that P450 enzymes responsible for the metabolism of nitroPAHs may vary from species to species and in different target organs (Howard et al., 1988, 1990; Silvers et al., 1992, 1994; Guengerich et al., 1999).
The nitroreduction of a nitroPAH may involve one- and/or two-electron transfers and result in the formation of the corresponding nitrosoPAH, further reduction to the N-hydroxyaminoPAH and final reduction to the aminoPAH. Both bacterial and mammalian enzymatic systems are capable of reductively metabolizing nitroPAHs under anaerobic or hypoxic incubation conditions (see also chapter 4). Nitroreduction of nitroPAHs in vivo probably occurs mainly by bacteria in the intestinal tract. In mammalian cells, nitroreduction is catalysed by a variety of enzymes, including cytosolic aldehyde oxidase (Tatsumi et al., 1986), xanthine oxidase (Howard & Beland, 1982; Bauer & Howard, 1990, 1991), DT-diaphorase or microsomal NADPH cytochrome P450 reductase (Djuric et al., 1986a). These enzymes are found in a variety of tissues; for example, xanthine oxidase occurs in liver, intestinal mucosa and mammary glands and is also present in the milk and colostrum of most mammals (Howard et al., 1995).
In oxidative metabolism, the first step is transformation to phase I primary metabolites, such as epoxides, phenols and dihydrodiols, and then to secondary metabolites, such as diol epoxides, tetrahydrotetrols and phenol epoxides. In mammalian systems, the phase I metabolites are then conjugated with glutathione, sulfate or glucuronic acid to form phase II metabolites, which are more polar and water soluble than the parent hydrocarbons. On reaching the intestine, the conjugated metabolites can be deconjugated by the intestinal microflora and absorbed, entering the enterohepatic recirculation. Nitroreduction and N-acetylation can occur, resulting in the excretion in urine and faeces of metabolites such as, for example, acetylaminopyrenols after 1-nitropyrene administration.
Acetylation is an important conjugation reaction whereby water solubility is reduced. N-Acetyltransferases are found in the liver and in the intestine. Arylhydroxyamines can be acetylated at either the -NH or -OH of the -NHOH moiety, resulting in either an arylhydroxyamic acid or an acetoxyarylamine.
NitroPAHs do not follow the same activation pathways. Mutagenic activation can occur by deacetylation (to arylhydroxyamines) or O-acetylation (e.g., 1,8- and 1,6-dinitropyrene) and intramolecular acyl transfer to acetoxyarylamines with the production of electrophilic nitrenium ion (-N+), which binds to DNA. Some nitroPAHs (e.g., 6-nitrobenzo[a]pyrene) may be mutagenic only after activation by oxidation to reactive epoxides or dihydrodiol epoxides, similarly to BaP.
The main DNA adducts detected with nitroPAHs in vivo and in vitro are N-(deoxyguanosin-8-yl) (C8-substituted deoxyguanosine [dG]) derivatives, but N2-substituted dG and C8-substituted deoxyadenosine (dA) derivatives have also been detected and may predominate in certain nitroPAHs with greater hydrocarbon character (e.g., 3-nitrobenzo[a]pyrene and 6-nitrochrysene). DNA adducts of dinitropyrenes are formed only via nitroreduction, presumably owing to the high electron deficiency in the aromatic rings caused by the presence of two nitro groups. The DNA adducts resulting from the nitroreduction of nitroPAHs are better characterized than those arising from oxidative metabolism, although the latter may be of more importance in mammalian metabolism. There is recent evidence for oxidative metabolism, e.g., in the genotoxicity of the atmospheric reaction product 2-nitronaphthalene in human lymphoblastoid cell lines (Grosovsky et al., 1999; Sasaki et al., 1999).
In this document, it is not possible to present the results of all the studies on the metabolism of nitroPAHs because of the amount of data and the number of different compounds. Most studies on the metabolism of nitroPAHs have concentrated on 1-nitropyrene and, to a lesser extent, 2-nitrofluorene, as they are the most abundant nitroPAHs in diesel exhaust. These are given in sections 6.2 and 6.4, respectively. An overview of the metabolism, metabolic activation and DNA binding of isomeric and some individual nitroPAHs (sections 6.3, 6.5–6.9) is then given to illustrate how the three-dimensional structure of individual nitroPAHs, in particular the orientation of the nitro functional group, seems to determine the biological activity of individual nitroPAHs (reviewed by Fu et al., 1988a; Hart et al., 1988; Fu, 1990). For recent reviews on the metabolism and activation of nitroPAHs, refer to the first paragraph of this chapter.
Early peak levels of radioactivity observed in blood and tissue homogenates indicated a rapid (within 3 h) absorption from the gastrointestinal tract after intragastric administration of [14C]1-nitropyrene (single dose, 27.6 µCi, 750 mg/kg bw) to male Cpb:WU (Wistar) rats (van Bekkum et al., 1999).
After gavage administration of radiolabelled 1-nitropyrene to rats, rapid absorption was found after 30 min (Sun et al., 1983) and 1 h (Bond et al., 1986). Binding of 1-nitropyrene to particles reduced absorption significantly (see section 6.2.2).
No data were available on dermal absorption.
Radioactivity was found to be widely distributed in tissues of rats after intragastric administration of 14C- or 3H-labelled 1-nitropyrene, being found mainly in the liver and kidney, but also in the gastrointestinal tract, bladder, adipose tissue, brain, lung and heart (Kinouchi et al., 1986b).
Carbon-14 was widely distributed in tissues of male F344/Crl rats 1 h after nose-only exposure to 50 and 490 ng [14C]1-nitropyrene/litre air or [14C]1-nitropyrene (650 ng/litre) coated on diesel exhaust particles. Highest concentrations of 14C were found in the respiratory tract (nasal turbinates, trachea, lung), kidneys, liver and urinary bladder. Lungs of rats exposed to the diesel particles contained nearly 5 times (148 versus 29 pmol/g lung) more 14C than lungs exposed to aerosols without particles 1 h after exposure and 80-fold (80 versus 1 pmol/g lung) 94 h after exposure, demonstrating that particle association of nitropyrene significantly alters the biological fate of inhaled nitropyrene (Bond et al., 1986). Concentrations of diesel soot in the exposure chamber ranged from 3.7 to 6.1 µg/litre air. The activity median diffusion equivalent diameter of aerosols of [14C]1-nitropyrene coated on diesel exhaust particles was 0.22 µm.
Once 1-nitropyrene becomes available in the bloodstream, it is rapidly metabolized, followed by conjugation and rapid elimination of the metabolites formed. Evidence for pathways of metabolism has come from in vivo studies on these metabolites in rodents, including studies with germ-free animals (see section 6.2.3.5 and Table 30) and studies on bile-cannulated rats (see Tables 30 and 31), as well as from in vitro studies (El-Bayoumy & Hecht, 1983; Ball & King, 1985; Howard et al., 1985; King & Lewtas, 1993).
Table 30. Metabolites formed in in vivo studies (either unconjugated 1-nitropyrene metabolites or aglycons, after incubation with,
for example, aryl sulfatase or β-glucuronidase)a,b
|
Metabolite |
I |
II |
IIIa germ-free |
IIIb |
IIIc |
IV |
Va |
Vb |
Vc |
VI |
VII |
|
i.p. |
i.v. |
oral |
oral |
oral |
inhal. |
oral |
oral |
oral |
oral |
oral |
|
|
1-Nitropyren-3-olc |
U, F |
Bil |
F,U |
Bil |
B |
nd |
U |
P, Li, Ki, Lu |
|||
|
1-Nitropyren-6- and/or -8-ol |
U, F |
Bil |
F,U |
Bil |
U |
B |
x |
x |
U |
P, Li, Ki, Lu |
|
|
Aminopyrenols |
Bil |
||||||||||
|
N-Acetyl-1-aminopyren-3-ol |
U, F |
F |
Bil |
nd |
|||||||
|
N-Acetyl-1-aminopyren-6-ol |
U ,F |
U, F |
Bil |
U, F |
U |
P, Li, Ki, |
|||||
|
N-Acetyl-1-aminopyren-8-ol |
U, F |
U, F |
Bil |
U, F |
U |
P Li, Ki, |
|||||
|
trans-4,5-Dihydro-4,5-dihydroxy-1-nitropyrene (1-nitropyrene-4,5-dihydrodiol) |
U, F |
Bil |
F,U |
Bil |
B |
x |
P Li, Ki, |
||||
|
N-Acetyl-1-aminopyrene |
U, F |
F |
nd |
x |
nd |
||||||
|
1-Aminopyrene |
U, F |
F |
F |
B |
x |
x |
U |
P Li, Ki, Lu |
|||
|
1-Nitropyrene |
U, F |
F |
nd |
P, Li, Ki, Lu |
|
a |
i.p. = intraperitoneal; i.v. = intravenous; inhal. = inhalation; P = in plasma; B = in blood; U = in urine; F = in faeces; Bil = biliary glucuronide conjugates; Li = liver, Ki = kidney; Lu = lung ; nd = not detected; x = whole tissue; bold type, main metabolites. |
|
|
b |
Studies were as follows: |
|
|
I) |
Intraperitoneal administration of [14C]1-nitropyrene to CD rats (Ball et al., 1984a). |
|
|
II) |
Intravenous administration of [4,5,9,10-3H]1-nitropyrene to bile-cannulated rats resulted in rapid appearance of radioactivity in the bile with maximum concentration of radioactivity being observed between 0 and 1 h. Thirty per cent was excreted in 4 h. By comparison, following oral administration of [4,5,9,10-3H]1-nitropyrene, the highest concentration of radioactivity in the bile was detected between 2 and 4 h, and only 10% of the dose was found in the bile within 12 h. Nearly all the radioactivity in the bile was due to conjugated 1-nitropyrene metabolites (Howard et al., 1985). |
|
|
IIIa) |
Major faecal metabolites in germ-free rats following oral administration of [3H]1-nitropyrene (48 h). The same metabolites were found in the urine as their glucuronide conjugates. 1-Nitropyrene was found in the faeces (Kinouchi et al., 1986b). Similar results were found using [4,5,9,10-14C ]1-nitropyrene (120 h) (El-Bayoumy et al., 1984a). |
|
|
IIIb) |
Major faecal metabolites in conventional rats following oral administration of [3H]1-nitropyrene (48 h). Substantial nitroreduction and N-acetylation occurred (Kinouchi et al., 1986b). Similar results were found using [4,5,9,10-14C]1-nitropyrene (120 h) (El-Bayoumy et al., 1984a). |
|
|
IIIc) |
As above using bile-cannulated rats (Kinouchi et al., 1986b). Nearly all the metabolites in the bile were conjugated. With time (24–48 h), the proportion of nitrated metabolites decreased, while the proportion of aminopyrenols and acetylaminopyrenols increased. |
|
|
IV) |
Rats exposed to 50 and 490 ng [14C]1-nitropyrene/litre air, or [14C]1-nitropyrene (650 ng/litre) coated on diesel exhaust particles (Bond et al., 1986). Large quantities of unmetabolized 1-nitropyrene (presumably cleared from upper respiratory tract and cleared unmetabolized and unabsorbed via gastrointestinal tract). Lungs: unmetabolized nitropyrene, unconjugated acetylaminopyrenols, small quantities of polar metabolites. Liver and kidneys: polar nitropyrene metabolites. |
|
|
Va) |
HPLC analysis of organic extractable metabolites (24 h) from blood samples of female C57B1/6N mice administered [4,5,9,10-3H]1-nitropyrene by gavage (Howard et al., 1995). |
|
|
Vb) |
C57B1/6N mouse fetal homogenates. 1-Aminopyrene was detected in the amniotic fluid, and 0.7% of the administered dose passed the placenta (Howard et al., 1995). |
|
|
Vc) |
Neonatal homogenate from nursing mice 12 h after mothers administered 1-nitropyrene by gavage. Each neonate received about 0.1% of the administered dose (Howard et al., 1995). |
|
|
VI) |
Single oral dose of 20 mg of diesel exhaust particle sample to rats. Approximately 13% of the 1-nitropyrene present on the diesel exhaust particle sample was excreted in urine and identified as 1-nitropyrene metabolites (van Bekkum et al., 1998). |
|
|
VII) |
Metabolites present following intragastric administration of [14C]1-nitropyrene to male Wistar rats (van Bekkum et al., 1999). |
|
|
c |
Also called 3-hydroxy-1-nitropyrene. |
|
1-Nitropyrene undergoes extensive metabolism both on the pyrene moiety and on the nitro function of the molecule (see Figure 9). The metabolism of 1-nitropyrene occurs 1) through cytochrome P450-mediated ring C-oxidation to epoxides, with subsequent rearrangement to nitropyrenols and conjugation or hydration to dihydrodiols, or 2) through nitroreduction in one- or two-electron steps to form 1-nitrosopyrene, N-hydroxy-1-aminopyrene or 1-aminopyrene with or without subsequent acetylation, or 3) a combination of ring oxidation and nitroreduction followed by acetylation. The complex biotransformation is reflected in the variety of metabolites present in plasma and tissue homogenate (see Figures 10–12) and in the variety of adducts formed with blood proteins (van Bekkum et al., 1999) and with DNA (El-Bayoumy et al., 1994a) (see section 6.2.5).
The metabolites of 1-nitropyrene are highly conjugated in vivo. After an i.p. administration of [14C]1-nitropyrene to CD rats, >95% of the urinary 14C eluted with the solvent front (Ball et al., 1984a). Incubation of excreted metabolites in the urine with specific deconjugating enzymes β-glucuronidase and sulfatase reduced this (see Figure 10), but still over 50% of the radioactivity was to be detected in the solvent front, so that many metabolites have not yet been identified.
Urinary metabolites that in general have been observed in in vivo studies with rats and mice (following hydrolysis of conjugates) are 1-nitropyren-3-ol, 1-nitropyren-6-ol and 1-nitropyren-8-ol (also referred to as 3-, 6- and 8-hydroxy-1-nitropyrene), N-acetyl-1-aminopyren-3-ol, N-acetyl-1-aminopyren-6-ol and N-acetyl-1-aminopyren-8-ol (also referred to as 3-, 6- and 8-hydroxy-N-acetylaminopyrene), 1-nitropyrene-trans-dihydrodiols (mainly trans-4,5-dihydro-4,5-dihydroxy-1-nitropyrene, also called 1-nitropyrene-4,5-dihydrodiol), N-acetyl-1-aminopyrene and 1-aminopyrene, and in some studies unmetabolized 1-nitropyrene (Ball et al., 1984a; El-Bayoumy & Hecht, 1984a; Howard et al., 1985, 1995; Kinouchi et al., 1986b; van Bekkum et al., 1999). In all studies, the main metabolite identified (total up to 48 h) in urine of rats administered 1-nitropyrene was N-acetyl-1-aminopyren-6-ol, which (e.g., in the study by Ball et al., 1984a) accounted for about 20% of the total radioactivity observed in the urine and is the predominant source of mutagenic activity from this source in the urine. 1-Aminopyrene is hardly measurable in the urine but is found as the major metabolite in faeces (Ball et al., 1984a; Kinouchi et al., 1986b; see Figures 10 and 11). The major metabolites in the bile from bile-cannulated rats were 1-nitropyren-3-ol, 1-nitropyren-6/8-ol and trans-4,5-dihydro-4,5-dihydroxy-1-nitropyrene (Kinouchi et al., 1986b; Figure 11). An overview of the studies into the metabolites formed during the metabolism of 1-nitropyrene is given in Table 30.
The nature of the metabolites (biliary, urinary and faecal) changes with time. The early metabolites are predominantly primary oxidative products — phenols and dihydrodiol(s). Ball et al. (1984a) measured metabolites after i.p. injection after 4 h and found that 1-nitropyren-3-ol, 1-nitropyren-6/8-ol and trans-4,5-dihydro-4,5-dihydroxy-1-nitropyrene were the major metabolites (Figure 10). The major metabolites in plasma, liver and kidneys after 3 and 6 h were likewise 1-nitropyren-6/8-ol (van Bekkum et al., 1999; see Figure 12). At later time points, the major products (N-acetyl-1-aminopyren-6-ol, N-acetyl-1-aminopyren-8-ol and N-acetyl-1-aminopyren-3-ol) have undergone ring oxidation, nitroreduction and also N-acetylation (see Figures 9–12).
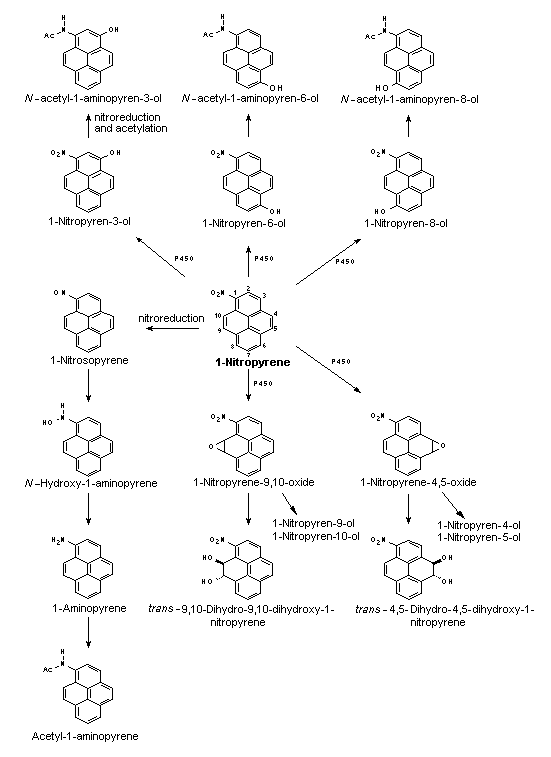
Fig. 9. Scheme of metabolism of 1-nitropyrene in vivo (from Howard et al., 1995).
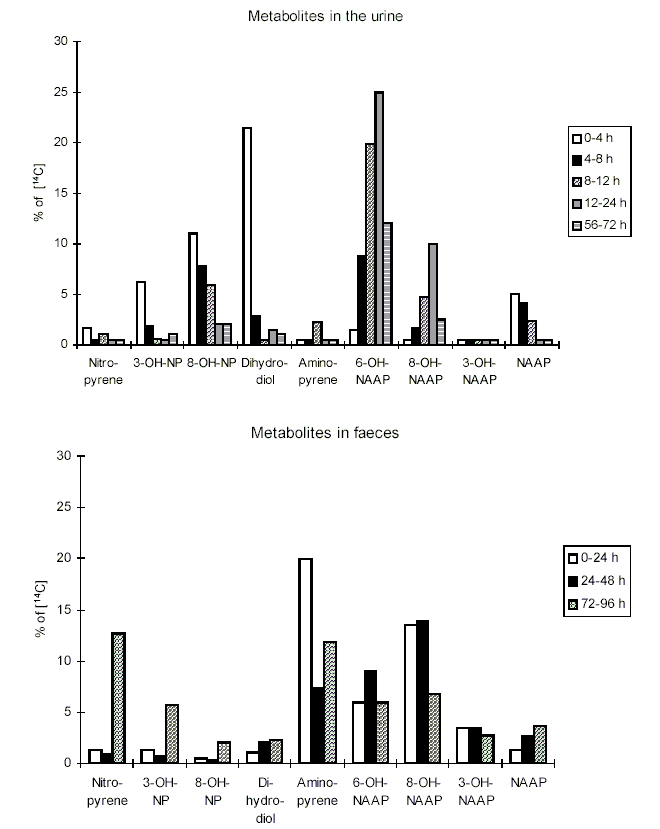
Fig. 10. The metabolites of [14C]1-nitropyrene in the urine and faeces of rats at specified times after i.p. administration (Ball et al., 1984a). Deconjugating enzymes: β-glucuronidase and sulfatase; % of urinary [14C] on solvent front = 52, 72, 64, 58 and 79% for the given time points; 3-OH-NP = 1-nitropyren-3-ol; 8-OH-NP = 1-nitropyren-8-ol; 6-OH-NAAP = N-acetyl-1-aminopyren-6-ol; 8-OH-NAAP = N-acetyl-1-aminopyren-8-ol; 3-OH-NAAP = N-acetyl-1-aminopyren-3-ol; NAAP = N-acetyl-1-aminopyrene.
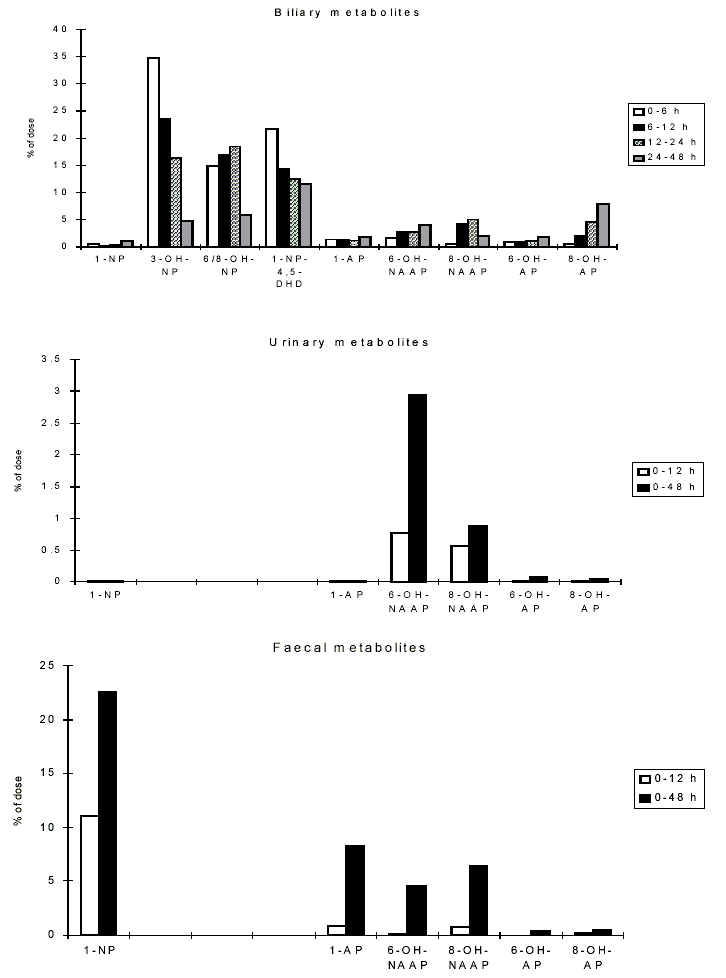
Fig. 11. Metabolites excreted from rats at given times after oral exposure to [³H]1-nitropyrene (percentage of diethylether-soluble material after β-glucuronidase treatment) (Kinouchi et al., 1986b). 1-NP = 1-nitropyrene; 3-OH-NP = 1-nitropyren-3-ol; 6/8-OH-NP = 1-nitropyren-6/8-ol; 1-NP-4,5-DHD = trans-4,5-dihydro-4,5-dihydroxy-1-nitropyrene; 1-AP = 1-aminopyrene; 6-OH-NAAP = N-acetyl-1-aminopyren-6-ol; 8-OH-NAAP = N-acetyl-1-aminopyren-8-ol; 6-OH-AP = 1-aminopyren-6-ol; 8-OH-AP = 1-aminopyren-8-ol.
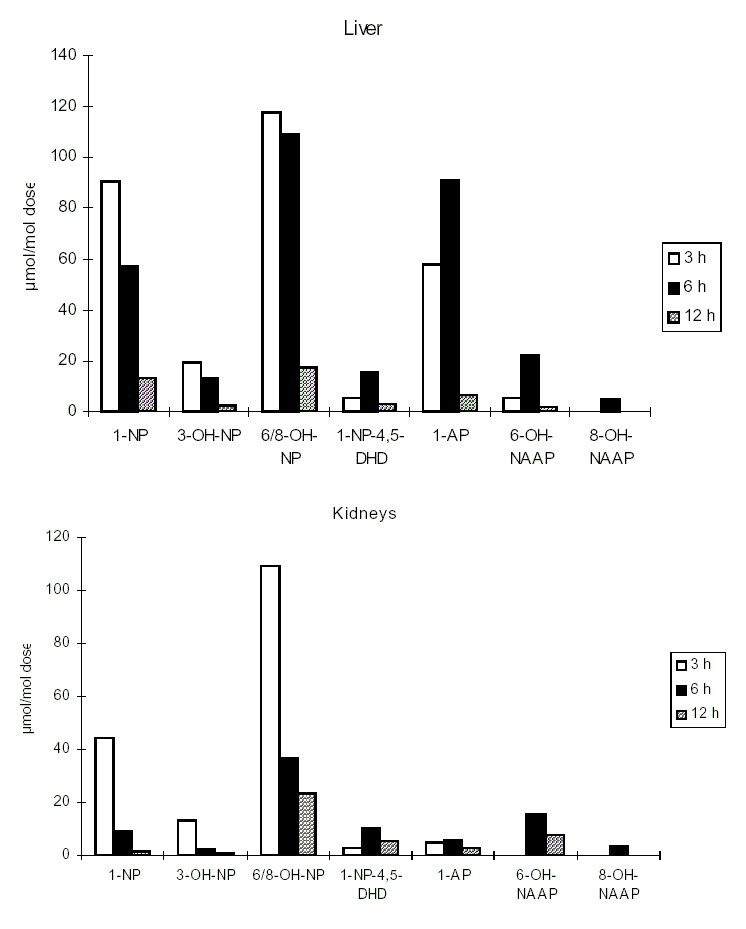
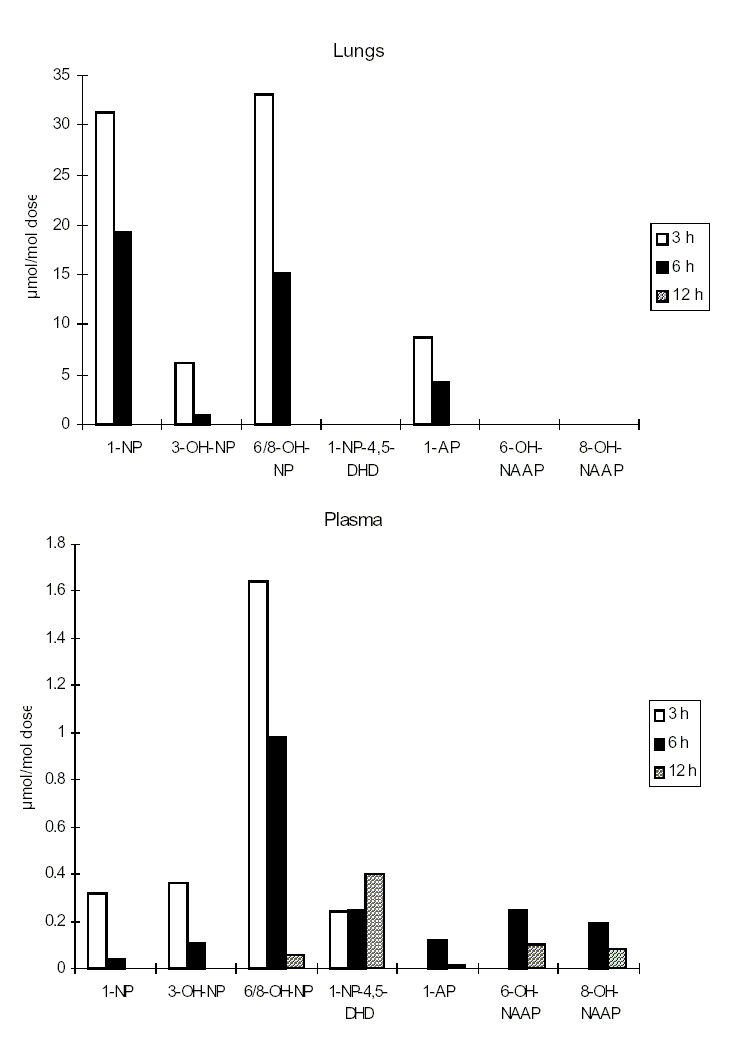
Fig. 12. Metabolites excreted after oral application to Wistar rats after given times; single dose of [14C]1-nitropyrene; 27.6 µCi, 750 mg/kg bw (van Bekkum et al., 1999). 1-NP = 1-nitropyrene; 3-OH-NP = 1-nitropyren-3-ol; 6/8-OH-NP = 1-nitropyren-6/8-ol; 1-NP-4,5-DHD = trans-4,5-dihydro-4,5-dihydroxy-1-nitropyrene; 1-AP = 1-aminopyrene; 6-OH-NAAP = N-acetyl-1-aminopyren-6-ol; 8-OH-NAAP = N-acetyl-1-aminopyren-8-ol.
The C-oxidative metabolism of 1-nitropyrene in different species is catalysed by different cytochrome P450 isoenzymes: in rabbit by CYP2C3 (Howard et al., 1988) and in rat by CYP2B1 and CYP2C (Silvers et al., 1994). The CYP3A subfamily (in particular CYP3A3 and CYP3A4) seems to be the enzymes involved in human metabolism of 1-nitropyrene (Howard et al., 1990; Silvers et al., 1992; Chae et al., 1999a).
Cytochrome P450 can catalyse the direct formation of three phenols (1-nitropyren-3-ol, 1-nitropyren-6-ol and 1-nitropyren-8-ol). These phenols are further conjugated to sulfate and glucuronide derivatives. However, in vitro studies showed that human liver enzymes preferentially produce metabolites with the -OH function situated at the 3- position, whereas 6- and 8- isomers are the main metabolites produced by rat liver enzymes (Howard et al., 1990; Silvers et al., 1992; Chae et al., 1999a). 1-Nitropyren-3-ol is considerably more mutagenic in Salmonella typhimurium than 1-nitropyren-6-ol and 1-nitropyren-8-ol, the mutagenicity of the phenols presumably resulting from nitroreduction to hydroxyamine derivatives (Ball et al., 1984a; Consolo et al., 1989).
Alternatively, the C-oxidation of 1-nitropyrene by cytochrome P450 can result in the formation of two arene K-region oxides, 1-nitropyrene-4,5-oxide and 1-nitropyrene-9,10-oxide. The K-region oxides are mutagenic in S. typhimurium either with or without exogeneous activating enzymes (Smith et al., 1990a; Beland, 1991). The K-region oxides can be hydrolysed by epoxide hydrolase to the corresponding K-region trans-dihydrodiols or can rearrange to form four K-region phenols (1-nitropyren-4-ol, 1-nitropyren-5-ol, 1-nitropyren-9-ol or 1-nitropyren-10-ol). Mutagenicity of K-region derivatives of 1-nitropyrene was reported (El-Bayoumy & Hecht, 1986).
Species differences were found in both activation of 1-nitropyrene to 1-nitropyrene oxides and inactivation of 1-nitropyrene oxides by epoxide hydration and glutathione conjugation in hepatic subcellular fractions. For example, 1-nitropyrene-4,5-oxide-producing activity was highest in guinea-pig and dog, followed in order by hamster, rat, human and mouse; 1-nitropyrene-9,10-oxide-producing activity was highest in hamster, followed in order by guinea-pig, rat, dog, mouse and human; glutathione conjugation of 1-nitropyrene oxides was higher in rodents than in human and dog. There was also a wide degree of interindividual variations in these activities (Kataoka et al., 1991).
In S. typhimurium, 1-nitropyrene is mutagenic through nitroreduction to the corresponding nitroso derivative, then to the hydroxyamino derivative, which has been shown to form a C8-guanyl adduct (Howard et al., 1983a). This adduct is also responsible for the mutagenicity of 1-nitropyrene in Chinese hamster ovary (CHO) cells (Heflich et al., 1986a) and cultured human diploid fibroblasts (Howard et al., 1983b; Beland et al., 1986). The reductive metabolism of 1-nitropyrene by rat liver microsomes gave 1-aminopyrene as the sole metabolite (Saito et al., 1984a).
Human metabolism of 1-nitropyrene has been studied in vitro. Metabolites derived from nitroreduction (1-aminopyrene) as well as ring oxidation (trans-dihydriols and phenols) were detected in the human hepatoma cell line HepG2 treated with 1-nitropyrene (Eddy et al., 1987; Silvers et al., 1994, 1997). In contrast, human hepatic microsomal incubations of 1-nitropyrene yielded only oxidized products (Silvers et al., 1992; Chae et al., 1999a).
Studies have shown that human red blood cells may also possess the metabolic competence to reduce 1-nitropyrene (Belisario et al., 1996).
Metabolism of nitroPAHs by intestinal microflora has been demonstrated both in vitro and in vivo (Cerniglia & Somerville, 1995). Intestinal microflora have been shown to be important in hydrolysing conjugated metabolites (e.g., via β-glucuronidase activity) and thereby facilitating enterohepatic recirculation of aglycones as well as playing a role in nitroreduction (Morotomi et al., 1985; Cerniglia & Somerville, 1995).
Studies using germ-free animals (intestinal microflora were absent) treated with 1-nitropyrene showed that the predominant metabolites formed were primarily ring-hydroxylated derivatives of 1-nitropyrene, whereas conventional animals used both oxidative and reductive pathways to form aminopyrene and acetylated and ring-hydroxylated metabolites. Further, the excreta collected from germ-free animals were less mutagenic than those from conventional animals. Following oral administration, the radioactivity passed through the conventional rats much more rapidly than through the germ-free rats (El-Bayoumy et al., 1983, 1984a; Kinouchi et al., 1986b). In studies on the major end metabolite N-acetyl-1-aminopyren-6-ol, which is excreted mainly as its β-glucuronidase conjugate, it was shown using germ-free animals that intestinal microflora are vital not only for nitroreduction but also for the hydrolysis of glucuronides released for enterohepatic recirculation (Ball et al., 1991). Scheepers et al. (1994d; ref #2 from Bos) studied haemoglobin (Hb) adduct formation after intragastric administration of 2-nitrofluorene (and 2-aminofluorene) in germ-free rats compared with rats equipped with a bacterial flora derived from human or rat faeces. After administration of 2-nitrofluorene, no Hb adducts could be detected in germ-free rats. Rats with a rat or human microflora showed low levels of Hb adducts, less than 1% of the Hb adduct levels obtained after administration of the same dose of 2-aminofluorene.
These results, together with those on pure and mixed cultures of intestinal flora from humans and rodents (see Cerniglia & Somerville, 1995), which reduced 1-nitropyrene to 1-aminopyrene, indicate the important role of intestinal flora in the bioactivation of 1-nitropyrene (as a model for nitroPAHs in general).
However, the intestinal microflora are not separate from the host metabolic processes, and the synergistic metabolic interactions between enzymes of the gut mucosa, hepatic tissue and microflora are important for the metabolic activation of nitroPAHs. The extent of nitroreduction of nitroPAHs to the corresponding aryl amines by cultures of intestinal microflora depends upon the geometric structure of the nitroPAH, the position of the nitro substituent, the source of intestinal microflora and diet. Generally, the nitroreductive capacities of mixed populations of intestinal microflora are greater than those observed in pure culture (Cerniglia & Somerville, 1995).
It has been suggested that in vivo, intestinal microflora are involved in the biotransformation of 1-nitropyrene to 1-aminopyrene (El-Bayoumy et al., 1983; Howard et al., 1983c; Ayres et al., 1985). The upper intestinal tract has, however, been shown to contain a low capacity of nitroreductase activity compared with the lower intestinal tract (Kinouchi et al., 1993). Data from a recent study in rats after intragastric administration of 1-nitropyrene (van Bekkum et al., 1999) imply that 1-nitropyrene is hardly reduced by the intestinal microflora in the rat prior to absorption from the upper intestinal tract and transportation to the liver. Ring hydroxylation in the liver initially seems to be the predominant route of biotransformation, whereas nitroreduction in the liver occurs to a small extent. v Nitroreduction seems to play a significant role in the enterohepatic recirculation only following biliary excretion and deconjugation, explaining the presence of reduced and acetylated metabolites in plasma, urine and tissue homogenates at later time points (van Bekkum et al., 1999).
From the profile of the metabolites, it seems that the main metabolic pathway for 1-nitropyrene is absorption, hydroxylation of the aromatic moiety and conjugation in the liver. After passing via the bile to the intestine, it is deconjugated, reduced (intestinal microflora) and reabsorbed. N-Acetylation of amino derivatives of 1-nitropyrene in vivo appears to occur primarily in the liver (Kinouchi et al., 1986b). Excretion of metabolites is via urine, but the unabsorbed metabolites are reduced to 1-aminopyrene and excreted in the faeces.
After oral or i.p. administration of [4,5,9,10-3H]1-nitropyrene to female C57B1/6N mice, rapid distribution and elimination of the 1-nitropyrene and its metabolites were observed. In both routes, the radioactivity in the serum rose to maximum levels between 6 and 12 h. The elimination kinetics was described as a two-component curve with half-lives for the rapid component and for the slower phase, respectively, of 0.5 and 3 days for the i.p. route and 0.3 and 1.8 days for animals treated orally (Howard et al., 1995).
Radioactivity (14C) was cleared rapidly from all tissues of male F344/Crl rats in a biphasic manner after inhalation exposure to 50 and 490 ng [14C]1-nitropyrene/litre (0.05 and 0.49 mg/m3) air or [14C]1-nitropyrene (650 ng/litre [0.65 mg/m3] air) coated on diesel exhaust particles. The short-term half-life of 14C in the lungs, liver and kidneys was 1, 3 and 0.5 h, respectively, and the long-term half-life was 40, 35 and 120 h, respectively (Bond et al., 1986).
1-Nitro[4,5,9,10-14C]pyrene, alone, co-administered with or vapour-coated onto aerosolized diesel particles, was administered to Sprague-Dawley rats by intratracheal instillation (Ball et al., 1986). After 24 h, the pattern of excretion was similar in all three regimens, the majority of the radioactivity being in the gastrointestinal tract. The pure compound resulted in the most extensive clearance of 14C from the lung and faster movement of 14C along the gut. The urines analysed by HPLC were similar to each other (and to urines from rats injected i.p.) in terms of the distribution of metabolites.
In a nose-only inhalation study, the elimination half-life of 1-nitropyrene in the lungs was about 1 h for rats exposed to 8 mg/m3 and 6 h for rats exposed to 50 mg/m3 (NTP, 1996). Lung burdens of 1-nitropyrene in rats exposed to 8 mg/m3 remained the same for the 13-week duration; however, lung burdens in rats exposed to 50 mg/m3 increased with time, indicating that the rats were unable to clear the 1-nitropyrene between exposures. The half-life of 1-nitropyrene in the plasma of rats was about 1 h (NTP, 1996).
Following intragastric and i.p. administration and following inhalation of 1-nitropyrene or 1-nitropyrene coated on diesel exhaust particles, the majority (50–60%) of the administered dose has been shown to be excreted in the faeces, whereas urine contained about 15–20% of the dose (see Table 31).
Table 31. Excretion pattern for 1-nitropyrene in vivo
|
Route of administration |
Radiolabel |
Dose |
Rats |
Time |
% of dose in faeces |
% of dose in urine |
% of dose in bile |
Reference |
|
Oral |
3H |
n.g. |
Female Fischer-344 |
96 |
46a |
18a |
Dutcher & Sun (1983) |
|
|
i.p. |
14C |
10 mg/kg bw |
Female Fischer-344 |
24 |
15 |
30 |
Dutcher et al. (1985) |
|
|
Oral |
14C |
10 mg/kg bw |
Female Fischer-344 |
48 |
35 |
55 |
Dutcher et al. (1985) |
|
|
i.p. |
14C |
10 mg/kg bw |
Not given |
24 |
40 |
15 |
Ball et al. (1984a) |
|
|
i.p. |
14C |
6 µmol |
Male Sprague-Dawley |
24 |
40–60 |
20–30 |
Ball & King (1985) |
|
|
Oral; coated on diesel particles |
14C |
380 µg/g; 5 mg/rat |
Sprague-Dawley |
24 |
40–60 |
20–30 |
Ball & King (1985) |
|
|
Intratracheal instillation; coated on diesel particles |
14C |
380 µg/g; 5 mg/rat |
24 |
40–60 |
20–30 |
Ball & King (1985) |
||
|
Gavage |
14C |
100 mg/kg bw |
Male F344 |
48 |
41 |
16 |
El-Bayoumy & Hecht (1984a) |
|
|
Gavageb |
14C |
100 mg/kg bw |
Male F344b |
72 |
6 |
37 |
||
|
Oral |
14C |
10 µg/kg bw |
Male F344/Crl |
c |
Bond et al. (1986) |
|||
|
i.v. |
14C |
10 µg/kg bw |
c |
|||||
|
Oral; coated on diesel particles |
14C |
10 µg/kg bw |
c |
|||||
|
Inhalation (nose only) |
14C |
70–1000 ng/litre |
c |
|||||
|
Inhalation; coated on diesel particles |
14C |
50–1100 ng/litre |
c |
|||||
|
Oral (gavage) |
3H |
30 µmol (700 µg) |
Male Wistar |
24 |
52 |
18 |
Kinouchi et al. (1986b) |
|
|
Oral (gavage) |
3H |
30 µmol (700 µg) |
Male Wistar (germ-free) |
24 |
9 |
4 |
||
|
i.v. |
3H |
0.3, 1.2 µmol (71, 286 µg) |
Male F344 |
24 |
3, 12 |
17, 7 |
Medinsky et al. (1985) |
|
|
i.v.b |
3H |
0.3, 1.2 µmol |
Male F344b |
24 |
8 |
80, 60 |
||
|
Intragastric |
14C |
750 mg/kg bw |
Male Cpb:WU |
50–60 |
15–20 |
van Bekkum et al. (1999) |
||
|
i.p. |
3H |
24 mg/kg bw |
Female CD |
24 |
3.2 |
6.5 |
Chae et al. (1997) |
|
a |
Greater than 36% was converted into the volatile form, presumably 3H2O. |
|
b |
Bile duct-cannulated rats. |
|
c |
Excretion in faeces was the dominant route of elimination. In general, quantities of 14C excreted were approximately 1.5–2 times greater than those measured in urine. Elimination half-lives for 14C in urine for different concentrations and different exposure modes ranged from 13 to 20 h. For faeces, elimination half-lives for 14C ranged from 15 to 21 h (Bond et al., 1986). |
Elimination half-lives for 14C in urine for different concentrations and different exposure modes ranged from 13 to 20 h. For faeces, elimination half-lives for 14C ranged from 15 to 21 h (Bond et al., 1986).
In biliary excretion, metabolites are excreted directly from the liver into the bile and thus into the small intestine without first entering the bloodstream for excretion by the kidneys. In the small intestine, if conditions are favourable, reabsorption can occur, resulting in an enterohepatic recirculation.
In studies with 1-nitropyrene using bile duct-cannulated rats, biliary excretion accounted for 37% of an intragastric dose after 72 h in one study (El-Bayoumy & Hecht, 1984a) and for 60–80% after 24 h in an intravenous (i.v.) study (Medinsky et al., 1985) (see Table 31). It was shown using germ-free animals that intestinal microflora are vital for the hydrolysis of glucuronides, enabling enterohepatic recirculation (Ball et al., 1991). Enterohepatic recirculation following biliary excretion seems to be an important route of excretion, at least in the rat (Howard et al., 1985; Medinsky et al., 1985; Kinouchi et al., 1990).
Reactive metabolites have been shown to bind to macromolecules, such as DNA and blood proteins, in in vitro and in vivo studies (Howard & Beland, 1982; Howard et al., 1983a; Djuric et al., 1986b; Roy et al., 1989; Smith et al., 1990b; El-Bayoumy et al., 1994a,b,c).
Hb binding to 1-nitropyrene, 2-nitronaphthalene and 2-nitrofluorene administered orally to male SD rats was found to be significantly lower than Hb binding to the corresponding amines. Reactive metabolites of 1-nitropyrene bound abundantly to the plasma proteins (Suzuki et al., 1989).
Reactive metabolites of 1-nitropyrene were reported to bind to Hb of both male and female F344 rats at a level of 0.08% of the dose in a dose-related manner. 1-Nitropyrene–Hb adducts appeared to be stable and accumulated with long-term exposure. After administration of a single dose of 1-nitropyrene, the half-life for clearance of Hb-associated radioactivity was 1.6 days. It seemed that reactive metabolites of 1-nitropyrene bind to the haem rather than to the globin moiety (El-Bayoumy et al., 1994a,c).
Van Bekkum et al. (1997) developed a method for determining Hb adducts following oral administration to rats. Low levels of Hb adducts were found 24 h after administration of a single dose of 1-nitropyrene. This method was used for biomonitoring studies (see below).
Van Bekkum et al. (1999) found that with intragastric administration of [14C]1-nitropyrene, levels of radioactivity associated with plasma proteins were approximately 4 times higher than the radioactivity associated with Hb (401.0 and 84.1 pmol/g protein per µmol 1-nitropyrene per kg bw, respectively, at 24 h). There were no indications of reactive metabolites of 1-nitropyrene binding to the haem of the Hb.
Aryl nitrenium ions generated by nitro reduction or K-region nitropyrene epoxides generated by ring oxidation can react with DNA, forming adducts (for review on DNA adducts from nitroPAHs, see Beland & Marques, 1994). In bacteria and in in vitro studies, the predominant DNA adduct derived from 1-nitropyrene is N-(deoxyguanosin-8-yl)-1-aminopyrene (dG-C8-AP), formed via the nitroreduction pathway. In vivo experiments with rats and mice have shown the presence of dG-C8-AP and, in smaller amounts, further adducts that have not yet been identified, formed via oxidative metabolic pathways (El-Bayoumy et al., 1994a,c; see Table 32 and also Tables 33 and 34 for some other nitroPAHs).
Table 32. DNA adducts in vivo after exposure to 1-, 2- and 4-nitropyrene (or their metabolites)a
|
NitroPAH |
Species; strain; sex |
Route |
Organ |
N or O |
Adducts identified |
Reference |
|
1-Nitropyrene |
Rat; Wistar; m |
i.p. |
Liver |
N |
dG-C8-AP |
Hashimoto & Shudo (1985) |
|
1-Nitropyrene |
Rat; Wistar; f |
i.p. |
Liver, kidney, mammary gland |
N |
dG-C8-AP |
Stanton et al. (1985) |
|
1-Nitropyrene |
Mouse (newborn) |
i.p. |
Liver, lung |
N |
dG-C8-AP |
El-Bayoumy et al. (1988a) |
|
1-Nitropyrene |
Mouse; B6C3F1; m |
i.t. |
Lung, liver, kidney |
N |
dG-C8-AP, major adduct; further unidentified adducts |
Mitchell (1988a) |
|
1-Nitropyrene |
Rat; Sprague-Dawley; f |
Gavage |
Liver, mammary fat pads |
N/O |
dG-C8-AP, a minor adduct; further unidentified adducts |
Roy et al. (1989) |
|
1-Nitropyrene |
Mouse; B6C3F1; m |
Inhalation |
Lung |
N |
dG-C8-AP |
Bond et al. (1990) |
|
1-Nitropyrene |
Rat; CD; f |
s.c. |
Injection site |
N |
dG-C8-AP |
Smith et al. (1990b) |
|
Mammary gland |
dG-C8-AP |
|||||
|
1-Nitropyrene |
Mouse; CD-1; m (newborn) |
s.c. |
Liver |
N |
dG-C8-AP |
Smith et al. (1990b) |
|
1-Nitropyrene |
Mouse; A/J; m |
i.p. |
Lung |
N |
dG-C8-AP |
Smith et al. (1990b) |
|
1-Nitropyrene |
Rat; Sprague-Dawley; f |
Mammary gland |
N |
dG-C8-AP |
Herreno-Saenz et al. (1995) |
|
|
1-Nitropyrene |
Rat; Sprague-Dawley; f |
Gavage |
Liver |
N |
dG-C8-AP |
El-Bayoumy et al. (1994c) |
|
1-Nitropyrene-4,5-oxide |
Mouse; ICR; m |
Gavage |
Lower intestinal mucosa |
O |
Three different adducts, unidentified |
Kinouchi et al. (1993) |
|
1-Nitropyrene-9,10-oxide |
Mouse; ICR; m |
Gavage |
Lower intestinal mucosa |
O |
Three different adducts, unidentified |
Kinouchi et al. (1993) |
|
[3H]2-Nitropyrene |
Rat; Sprague-Dawley; f |
Gavage |
Liver, kidney, mammary gland |
Multiple adducts; minor: dG-C8-2-AP and dA-C8-2-AP |
Upadhyaya et al. (1992) |
|
|
[3H]2-Nitropyrene |
Rat; CD; f |
i.p. |
Liver, mammary gland |
No adducts found |
Chae et al. (1997) |
|
|
[3H]4-Nitropyrene |
Rat; CD; f |
i.p. |
Liver, mammary gland |
N |
Four radioactive peaks: two unstable adducts decomposed to pyrene-4,5-dione, a tentative identification of dI adduct |
Chae et al. (1997, 1999b) |
|
a |
m = male; f = female; i.p. = intraperitoneal; i.t. = intratracheal instillation; s.c. = subcutaneous; N = nitroreduction pathway; O = oxidation pathway; major adduct given in bold type; dG-C8-AP = N-(deoxyguanosin-8-yl)-1-aminopyrene; dG-C8-2-AP = N-(deoxyguanosin-8-yl)-2-aminopyrene; dA-C8-2-AP = N-(deoxyadenosin-8-yl)-2-aminopyrene. |
Table 33. DNA adducts from 2-nitrofluorenea
|
Animal |
Route |
Organ |
Main adduct |
Others |
Reference |
|
|
Rat, Wistar; m |
Oral |
Liver |
Unidentified (80%) |
dG-C8-AF (15%) |
Mulder et al. (1990) |
|
|
Rat, Wistar; m |
Oral |
Liver |
Unidentified |
dG-C8-AF |
Wierckx et al. (1991) |
|
|
Rat, Wistar; m |
i.v. |
With catheterized bile ducts |
Same unidentified as above |
Wierckx et al. (1991) |
||
|
Rat, Wistar; m |
i.p. |
Liver |
dG-C8-AF |
dG-C8-AAF |
Möller et al. (1993b) |
|
|
Rat, Wistar; m |
Gavage |
Liver |
dG-C8-AF |
dG-C8-AAF |
Möller & Zeisig (1993) |
|
|
Rat, AGUS; f |
Gavage |
Liver (kidney, lung, heart) |
dG-C8-AF (95%) |
Möller et al. (1994) |
||
|
Rat, AGUS; f |
Gavage |
Germ-free |
Liver |
Unidentified |
Möller et al. (1994) |
|
|
Rat, Wistar; f |
Gavage |
Germ-free |
Liver |
Unidentified |
dG-C8-AF |
Scheepers et al. (1994c) |
|
Rat, Wistar; f |
Gavage |
Germ-free + rat microflora |
Liver |
Unidentified |
dG-C8-AF |
Scheepers et al. (1994c) |
|
Rat, Wistar; m |
Diet |
Forestomach (liver, kidney) |
dG-C8-AF |
2–4 unidentified |
Cui et al. (1995) |
|
|
Rat, Wistar; m |
Diet |
Forestomach, liver, kidney (spleen) |
dG-C8-AF, dG-C8-AAF, 2 unidentified |
Cui et al. (1999) |
|
a |
m = male; f = female; i.v. = intravenous; i.p. = intraperitoneal; dG-C8-AF = N-(deoxyguanosin-8-yl)-2-aminofluorene; dG-C8-AAF = N-(deoxyguanosin-8-yl)-2-acetylaminofluorene; dG-N2-AAF = C3-(deoxyguanosin-N2-yl)-2-acetylaminofluorene. |
Table 34. DNA adducts of nitroPAHs other than 1-nitropyrene and 2-nitrofluorenea
|
NitroPAH |
Animal |
Route |
Organ |
N or O |
Major DNA adducts |
Reference |
|
2-Nitrofluoranthene |
Neonatal B6C3F1 mice |
N |
dG-C8-2-aminofluoranthene |
Herreno-Saenz et al. (1994) |
||
|
3-Nitrofluoranthene |
In vitro |
N |
dG-C8-3-aminofluoranthene |
Dietrich et al. (1988) |
||
|
1-Nitrobenzo[a]pyrene |
In vitro |
O |
dG-1-nitroBaP trans-7,8-diol anti-9,10-epoxide |
Fu et al. (1997) |
||
|
1-Nitrobenzo[a]pyrene |
In vitro |
|
|
N |
6-dG-N2-1-aminoBaP |
Fu et al. (1997) |
|
3-Nitrobenzo[a]pyrene |
In vitro |
O |
dG-3-nitroBaP trans-7,8-diol anti-9,10-epoxide |
|||
|
3-Nitrobenzo[a]pyrene |
In vitro |
N |
6-dG-N2-3-aminoBaP |
Herreno-Saenz et al. (1993) |
||
|
1,6-Dinitropyrene |
Male CD-1 preweanling mice |
i.p. |
Liver |
N |
dG-C8-1-amino-6-NP |
Delclos et al. (1987b) |
|
1,6-Dinitropyrene |
Male Sprague-Dawley rats |
i.p. |
Bladder, liver, mammary gland, kidney, lung |
N |
dG-C8-1-amino-6-NP |
Djuric et al. (1988) |
|
1,6-Dinitropyrene |
Rats |
Lung |
N |
dG-C8-1-amino-6-NP |
Smith et al. (1993); Beland et al. (1994) |
|
|
1,6-Dinitropyrene |
Male F344 rats |
Lung, liver |
N |
dG-C8-1-amino-6-NP |
Beland et al. (1994) |
|
|
1,6-Dinitropyrene |
Male B6C3F1 mice |
Liver |
N |
dG-C8-1-amino-6-NP |
Howard & Beland (1994) |
|
|
1,8-Dinitropyrene |
Rat |
Mesentery, mammary glands |
N |
dG-C8-1-amino-8-NP |
Heflich et al. (1986b) |
|
|
6-Nitrochrysene |
In vitro and in bacterial systems |
N |
N-(dG-8-yl)-6-aminochrysene; N-(dI-8-yl)-6-aminochrysene; 5-(dG-N2-yl)-6-aminochrysene |
Delclos et al. (1987b) |
||
|
6-Nitrochrysene |
Preweanling CD-1 mice |
i.p. |
Lung |
O/N |
dG with 1,2-DHD-6-aminochrysene-3,4-epoxide |
Delclos et al. (1988); Li et al. (1993, 1994) |
|
6-Nitrochrysene |
Female CD rat |
i.p. |
Lung, liver, mammary gland and colon |
O/N |
dG with 1,2-DHD-6-aminochrysene-3,4-epoxide |
Chae et al. (1996) |
|
N |
5-(dG-N2-yl)-6-aminochrysene |
|
a |
i.p. = intraperitoneal; N = nitroreductive pathway; O = oxidative pathway; O/N = oxidative or reductive; dG-C8-2-aminofluoranthene = N-(deoxyguanosin-8-yl)-2-aminofluoranthene; dG-C8-3-aminofluoranthene = N-(deoxyguanosin-8-yl)-3-aminofluoranthene; dG-1-nitroBaP trans-7,8-diol anti-9,10-epoxide = deoxyguanosine-1-nitrobenzo[a]pyrene trans-7,8-diol anti-9,10-epoxide; dG-3-nitroBaP trans-7,8-diol anti-9,10-epoxide = deoxyguanosine-3-nitrobenzo[a]pyrene trans-7,8-diol anti-9,10-epoxide; 6-dG-N2-1-aminoBaP = 6-(deoxyguanosin-N2-yl)-1-aminobenzo[a]pyrene; 6-dG-N2-3-aminoBaP = 6-(deoxyguanosin-N2-yl)-3-aminobenzo[a]pyrene; dG-C8-1-amino-6-NP = N-(deoxyguanosin-8-yl)-1-amino-6-nitropyrene; dG-C8-1-amino-8-NP = N-(deoxyguanosin-8-yl)-1-amino-8-nitropyrene; N-(dG-8-yl)-6-aminochrysene = N-(deoxyguanosin-8-yl)-6-aminochrysene; N-(dI-8-yl)-6-aminochrysene = N-(deoxyinosin-8-yl)-6-aminochrysene; 5-(dG-N2-yl)-6-aminochrysene = 5-(deoxyguanosin-N2-yl)-6-aminochrysene; 1,2-DHD-6-aminochrysene-3,4-epoxide = trans-1,2-dihydro-1,2-dihydroxy-6-aminochrysene-3,4-epoxide. |
1) Reduction pathway of 1-nitropyrene and formation of dG-C8-AP in in vitro studies
Early in vitro studies, such as the detection of reduced metabolites (e.g., 1-aminopyrene and N-acetyl-1-aminopyrene; Messier et al., 1981) and decreased mutagenic activity in nitroreductase-deficient strains of S. typhimurium (Mermelstein et al., 1981; Rosenkranz et al., 1981), showed that the reduction of 1-nitropyrene to N-hydroxy-1-aminopyrene was associated with the induction of mutations. N-Hydroxy-1-aminopyrene has been shown to undergo an acid-catalysed reaction with DNA to yield the C8-substituted dG adduct dG-C8-AP (see Figure 13) (Howard et al., 1983a). This adduct is the predominant product formed from 1-nitropyrene and from 1-nitrosopyrene, its reduced derivative, in S. typhimurium TA1538 (Howard et al., 1983a; Heflich et al., 1985a,b), CHO cells (Heflich et al., 1985a, 1986a; Thornton-Manning et al., 1991a,b), Chinese hamster lung fibroblasts (Edwards et al., 1986a) and human diploid fibroblasts (Beland et al., 1986; Patton et al., 1986).
Evidence that dG-C8-AP is probably the premutagenic lesion comes from the observation that mutations are induced primarily at G:C base pairs in the lambda cI gene of an Escherichia coli uvr– lysogen (Stanton et al., 1988), in pBR322 introduced into E. coli (Melchior et al., 1990), in pZ189 replicating in human embryonic kidney cells (Yang et al., 1988; Maher et al., 1990) and in the hprt gene of CHO cells (Newton et al., 1992). dG-C8-AP causes a –2 deletion of a G:C or C:G pair within a CGCGCGCG hot-spot sequence upstream of the hisD3052 mutation in S. typhimurium strain TA98 (Bell et al., 1991; Malia et al., 1996). It could be shown in a comparative study using S. typhimurium strain TA98NR (which does not produce a nitroreductase), strain TA98 and strain YG1021 (which contains a plasmid carrying the nitroreductase-encoding gene) that nitroreductase activity, DNA adduct level and mutagenicity were strongly correlated with each other and that the higher the adduct level, the higher the level of mutagenicity (Arimochi et al., 1998).
Ring oxidation, which is a major metabolic pathway for 1-nitropyrene in vivo, results in the formation of metabolites such as phenols, dihydrodiols and K-region epoxides, which are more mutagenic than 1-nitropyrene (El-Bayoumy & Hecht, 1986). In vitro studies reacting calf thymus DNA with 1-nitropyrene-4,5-epoxide yielded three major adducts, identified as diastereomers, two with a trans configuration at the C4–C5 bond and one cis, resulting from the addition of the exocyclic N2 of dG to the C5 benzylic position of the epoxide (Roy et al., 1991; see Figure 13). These were probably the same adducts as those described in a similar experiment (Smith et al., 1990b). DNA adducts with characteristics of ring oxidation have also been detected in CHO cells incubated with 1-nitropyrene in the presence of an exogeneous microsomal preparation from liver (S9) (Thornton-Manning et al., 1991a).
The role of nitroreduction and C-oxidation in DNA adduct formation was investigated in HepG2 cells (Silvers et al., 1997). Cytochrome P450s were induced with differing concentrations of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). As the P450s were induced, the metabolism of 1-nitropyrene in the HepG2 switched from nitroreduction (i.e., formation of 1-aminopyrene) to C-oxidation (i.e., formation of pyrenols). The metabolic switch from nitroreduction to C-oxidation was accompanied by a decrease in DNA adduct formation, suggesting that generation of C-oxidized metabolites in HepG2 does not result in DNA adduct formation.
2) DNA adducts in in vivo studies
Metabolites indicative of ring oxidation and nitroreduction have also been detected in vivo in experimental animals. In mammalian systems, nitroreductase enzymes typically exhibit low levels of activity (Peterson et al., 1979). dG-C8-AP has also been detected in vivo in mice and rats treated with 1-nitropyrene or 1-nitrosopyrene (see Table 32), but it represents only a small proportion of the total molecular binding. Additional adducts were found in most cases, but none of these has been identified fully. For example, Kinouchi et al. (1992) studied DNA formation in the liver of B6C3F1 mice after administration of 1-nitropyrene and found that the major adduct was not dG-C8-AP; however, the major 32P-labelled spots migrated to the same position as the in vitro DNA adduct spots of K-region epoxides of 1-nitropyrene. In a separate investigation, similar findings were reported (El-Bayoumy et al., 1994a,c).
Further activation of oxidized metabolites of 1-nitropyrene has been suggested (Ball et al., 1996). 1-Nitropyren-6-ol can be activated to K-region epoxidation. 1-Acetamidopyren-6-ol undergoes several steps, possibly including both deacetylation and N-oxidation, yielding an arylnitrenium ion, as well as K-region oxidation, yielding an epoxide as active intermediate. DNA adducts derived from these activated compounds may be formed in tissues susceptible to 1-nitropyrene.
For approaches to biomonitoring of persons exposed to nitroPAHs, in particular 1-nitropyrene as a marker for diesel exhaust exposure, see chapter 8.
4-Nitropyrene seems to differ from 1- and 2-nitropyrene concerning elimination and excretion patterns, metabolites and DNA adducts (see below). This may account for 4-nitropyrene being the most carcinogenic of the nitropyrenes in rat mammary gland.
The data on faecal and urinary excretion of 2- and 4-nitropyrene after intragastric gavage suggest differences between the excretion patterns of 4-nitropyrene compared with 1- and 2-nitropyrene (Upadhyaya et al., 1992, 1994; see Table 35). The results of comparative studies via the i.p. route are inconclusive owing to the low excretion rate reported (Chae et al., 1997).
Table 35. Excretion pattern for 2- and 4-nitropyrene compared with that of 1-nitropyrene
|
NitroPAH |
Route |
Radio-label |
Rats |
Time (h) |
Faeces (% of dose) |
Urine (% of dose) |
Main metabolites in urine and faeces |
Reference |
|
1-Nitropyrene |
All |
24 |
50–60 |
15–20 |
See Table 30 |
See Table 30 |
||
|
2-Nitropyrene |
Intragastric |
14C |
Male F344 |
48 |
60 |
10 |
N-Acetyl-2-aminopyren-6-ol |
Upadhyaya et al. (1992) |
|
4-Nitropyrene |
Intragastric |
3H |
Female Sprague-Dawley |
48 and |
30 |
40 |
4-Aminopyrene, N-acetyl-4-aminopyren-9(10)-ol + many unknown metabolites |
Upadhyaya et al. (1994) |
|
1-Nitropyrene |
i.p. |
3H |
Female CD |
24 |
3.2 |
6.5 |
Chae et al. (1997) |
|
|
2-Nitropyrene |
i.p. |
3H |
Female CD |
24 |
2.2 |
6.2 |
Chae et al. (1997) |
|
|
4-Nitropyrene |
i.p. |
3H |
Female CD |
24 |
0.8 |
2.1 |
Chae et al. (1997) |
Analogous to 1-nitropyrene, N-acetyl-2-aminopyren-6-ol was the main metabolite detected in urine and faeces (2% and 20% of the dose, respectively) after intragastric gavage of 2-nitropyrene in rats (Upadhyaya et al., 1992). In contrast, after 4-nitropyrene administration, 4-aminopyrene and N-acetyl-4-aminopyren-9(10)-ol were the major metabolites, but at much lower yields. There were many unknown metabolites, including possibly 4-nitropyrene-9,10-dione (Upadhyaya et al., 1994). In a comparative study on 1-, 2- and 4- nitropyrene administered i.p., metabolites derived from nitroreduction and ring oxidation pathways (acetylaminopyrenes and their phenolic derivatives) were found in all three cases, although a quantitative analysis was not possible as a result of the low levels detected (Chae et al., 1997).
A comparative study on 1-, 2- and 4-nitropyrene in human hepatic and pulmonary microsomes showed some differences in their metabolism (Chae et al., 1999a). Human hepatic samples were competent in metabolizing 1-, 2- and 4-nitropyrene. With human pulmonary microsomal samples, similar patterns were obtained, but at much lower levels. Ring-oxidized metabolites (phenols and trans-dihydrodiols) were produced from all three isomers. However, the reductive metabolism leading to the formation of aminopyrene was evident only with 4-nitropyrene, which is the most potent carcinogen of the three.
Whereas most of the hepatic microsomal metabolism of 1- and 4-nitropyrene could be attributed to CYP3A4, none of the P450 enzymes tested seemed to be involved in the human hepatic microsomal metabolism of 2-nitropyrene (Chae et al., 1999a). Thus, the role of specific human P450 enzymes depends upon the position of the nitro group.
The major DNA adduct found in S. typhimurium (Howard et al., 1983a), cells in culture (Heflich et al., 1986a; Silvers et al., 1997) and rodents treated with 1-nitropyrene (see Table 32) is dG-C8-AP. With 2-nitropyrene, both dG and, to a lesser extent, dA adducts derived from nitroreduction [N-(deoxyguanosin-8-yl)-2-aminopyrene (dG-C8-2-AP) and N-(deoxyadenosin-8-yl)-2-aminopyrene (dA-C8-2-AP)] were formed in S. typhimurium (Yu et al., 1991) and upon incubation of 2-nitropyrene with DNA in the presence of rat hepatic microsomes (Fu et al., 1991).
DNA binding with 2-nitropyrene has been studied in vivo in female rats administered a single dose by gavage (Upadhyaya et al., 1992). The two adducts, dG-C8-2-AP and dA-C8-2-AP, formed by nitroreduction were detected in liver, mammary gland and kidney DNA, but were only a small percentage of the total DNA hydrolysate products (see Table 32).
In a comparative study, after i.p. treatment with [3H]1-, 2- or 4-nitropyrene, binding to rat mammary gland DNA was 0.6, 0.3 and 2.1 pmol/mg DNA, respectively. Only 4-nitropyrene yielded multiple putative DNA adducts (Chae et al., 1997). HPLC analysis yielded four radioactive peaks, which were found to co-elute with standards derived from the nitroreduction of 4-nitropyrene. One peak was identified as pyrene-4,5-dione and was formed from decomposition of two putative DNA–4-nitropyrene adducts. Another was tentatively identified as a deoxyinosine (dI) adduct. None of the peaks co-eluted with the major DNA adducts derived from 4-nitropyrene-9,10-epoxide, a ring-oxidized metabolite, or with the adduct standard N-(deoxyguanosin-8-yl)-4-aminopyrene (dG-C8-4-AP) (Chae et al., 1999b).
Although earlier studies inferred that the metabolism and excretion patterns of 2-nitrofluorene differed greatly from those of 1-nitropyrene, it seems with increasing data that there are many similarities. Whereas it was postulated that 2-nitrofluorene is metabolized in vivo via reduction to 2-aminofluorene (via reductase) in the intestine (and liver) followed by acetylation to N-acetyl-2-aminofluorene and then hydroxylation (Möller et al., 1985), this now appears to be only the case for one pathway after oral administration. Other studies show that the metabolic pathway via hydroxylated nitrofluorenes is similar to that of 1-nitropyrene (see Figure 14). Hydroxylated nitrofluorenes are more mutagenic in Salmonella than 2-nitrofluorene alone (Möller et al., 1988).
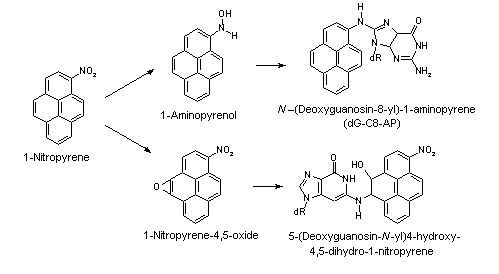
Fig. 13. Formation of DNA adduct from 1-nitropyrene; oxidative pathways and DNA adduct formation in vitro
(adapted from Beland & Marques, 1994).
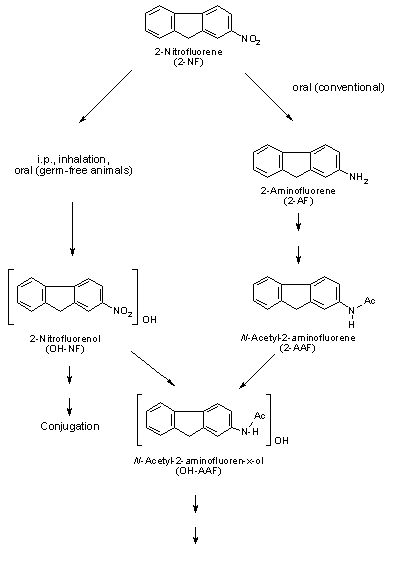
Fig. 14. Metabolism of 2-nitrofluorene in vivo
(adapted from Möller et al., 1985, 1987a,b, 1994).
2-Nitrofluorene is readily absorbed, distributed and excreted after administration by all routes. Data on excretion studies are given in Table 36. The data are contradictory, with some studies showing a higher percentage of the dose in urine than in faeces and vice versa. Some studies show that, similar to 1-nitropyrene, about 20–30% of the dose is excreted in the urine after 24 h. Excretion of metabolites was accompanied by an excretion of mutagenicity. (In both urine and faeces, the direct-acting mutagenicity [–S9] dominated over mutagenicity with S9 [Möller et al., 1987a, 1988, 1994]. Forty-three per cent of the total mutagenicity [and ~75% of the direct-acting mutagenicity] in the urine of rats after an oral dose of 2-nitro[9-14C]fluorene is due to free [unconjugated] metabolites: mostly N-acetylaminofluorene and some nitrofluorenols [Möller et al., 1987a].)
Table 36. Excretion pattern and metabolites for 2-nitrofluorene in vivoa
|
Route |
Radio-label |
Dose |
Rats |
Time (h) |
Faeces |
Urine |
Bile |
Main metabolites in the urine and bile |
Reference |
|
Oral, intragastric |
14C |
Single dose, 5 mg/rat |
Sprague-Dawley |
24 |
2–9 |
31–35 |
7- and 5-OH-AAF; minor: 1-, 3-, 8-, 9-OH-AAF; 2-NF not observed |
Möller et al. (1985, 1987a) |
|
|
Oral, intragastric |
14C |
Single dose, 5 mg/rat |
AGUS conventional rats |
24 |
17 |
22 |
7- and 5-OH-AAF |
Möller et al. (1988) |
|
|
Oral, intragastric |
Single dose, 5 mg/rat |
Germ-free |
24 |
8 |
18 |
9-OH-NF and 2-NF; |
Möller et al. (1988) |
||
|
Oral |
14C |
Not given |
Male Wistar |
24 |
Not given |
20 |
OH-NF and 2-AAF |
Mulder et al. (1990) |
|
|
i.v. |
3H and 14C |
6 µmol/kg bw |
Male Wistar |
2 |
40 |
In bile, glucuronide conjugate of 9-OH-NF |
Mulder et al. (1990) |
||
|
i.p. |
None |
1 mmol/kg bw |
Male Sprague-Dawley |
Up to 72; maximum at 24 |
Five hydroxylated 2-nitrofluorenes |
Castaneda-Acosta et al. (1997) |
|
a |
i.v. = intravenous; i.p. = intraperitoneal; 2-NF = 2-nitrofluorene; 2-AAF = 2-acetylaminofluorene; x-OH-AAFs = N-acetylaminofluoren-x-ols; x-OH-NF = nitrofluoren-x-ols. |
The pharmacokinetics of 2-nitrofluorene elimination from the blood after i.v. administration was studied in rats with cannulated bile ducts. 2-Nitrofluorene was found to be removed from the blood in a biphasic manner, with t½ values of 2.5 min and 2.5 h. Biliary excretion was likewise biphasic, with t½ values of 9 min and 1 h. After 2 h, about 40% of the dose had been excreted in the bile. The major metabolite was the glucuronide conjugate of 2-nitrofluoren-9-ol. The great difference between the high biliary excretion of 2-nitrofluorene metabolites (40% in 2 h) and the rather low excretion of the compound and its metabolites in urine (20% in 24 h) is possibly due to reabsorption of 2-nitrofluorene or its metabolites from the gut and subsequent enterohepatic recirculation (Mulder et al., 1990).
In contrast to former studies using oral administration (see below), the time-dependent metabolism of 2-nitrofluorene after i.p. administration to rats (Castaneda-Acosta et al., 1997) showed that N-acetylaminofluorenols, 2-acetylaminofluorene and 2-aminofluorene were not detected in the urine using HPLC or 1H-nuclear magnetic resonance (NMR); radioactive labelling was not used. After hydrolysis with β-glucuronidase/arylsulfatase, five 2-nitrofluorenols were(isolated by HPLC and identified by a combination of data from NMR, UV, and MS but not radioactive labelling) identified: trans-6,9-dihydro-6,9-dihydroxy-2-nitrofluorene [trans-6,9-dihydro-2-nitrofluoren-6,9-diol], 2-nitrofluoren-6-ol, 2-nitrofluoren-7-ol, 2-nitrofluoren-8-ol and 2-nitrofluoren-9-ol. Two conjugated metabolites were identified as 6- and 7-[(2-nitrofluoren-6-ol and 2-nitrofluoren-7-ol sulfate esters)oxy]-2-nitrofluorene. These latter conjugates were present as a mixture and were probably sulfates. They were by far the main metabolites and had a maximum at 12 h (the first time point given).
After an oral dose to rats, 2-nitrofluorene was metabolized to unconjugated hydroxy metabolites of 2-acetylaminofluorene compounds, mainly the 7- and 5-hydroxyl metabolites and, to a lesser extent, the 9-, 8-, 3- and 1-hydroxy metabolites and N-acetyl-2-amino(x)fluorenol (Möller et al., 1985). In another study, N-acetyl-2-aminofluoren-9-ol and, to a lesser extent, N-acetyl-2-aminofluoren-7-ol were observed as abundant urinary metabolites after 2-nitrofluorene intragastric administration. 2-Nitrofluorene phenols were also detected (Scheepers et al., 1994c). In contrast, in germ-free animals, 2-acetylaminofluorenols are hardly detected in urine or faeces after a single oral dose of 2-nitrofluorene; instead, 2-nitrofluorene phenols are mainly detected (Scheepers et al., 1994c).
After administration of a single oral dose of 2-nitrofluorene to germ-free and conventional rats, radioactivity with associated mutagenic activity was rapidly excreted in both urine and faeces. The mutagenicity in the excreta from germ-free animals exceeded that from conventional animals (in contrast to findings with 1-nitropyrene). Nitrofluorenols were associated with this high direct-acting mutagenicity (Möller et al., 1988). It is suggested that the microflora are responsible for reducing the nitrofluorenols (or 2-nitrofluorene). After oral administration to conventional rats, another metabolic route (similar to that for 1-nitropyrene) results in the formation of hydroxylated nitrofluorenes (2-nitrofluorenols), which could play a role in the carcinogenicity of 2-nitrofluorene, since the tumour patterns for 2-nitrofluorene differ from those for N-acetylaminofluorene (Miller et al., 1955; Cui et al., 1995; see chapter 7). 2-Nitrofluoren-7-ol and 2-nitrofluoren-9-ol (but not 2-nitrofluoren-5-ol) were shown to induce similar DNA adducts and preneoplastic liver lesions, but in smaller quantities than with 2-nitrofluorene (Cui et al., 1996).
The formation of 2-nitrofluorenols has also been shown in inhalation studies (in isolated, perfused lung and liver; Möller et al., 1987b) as well as in germ-free animals (Möller et al., 1988) and after induction of the cytochrome P450 system in vivo with β-naphthoflavone, resulting in an increase in the mutagenicity of urine and faeces in induced animals (Mφller et al., 1987a). A β-naphthoflavone-inducible microsomal enzyme, most likely CYP1A1, catalyses the hydroxylation of nitrofluorene both in the 9- position and in other positions (Törnquist et al., 1990). It seems that inhaled 2-nitrofluorene is metabolized by the lung to 2-nitrofluorenols or transported to the liver as nitrofluorene and then ring hydroxylated. The liver conjugates the 2-nitrofluorenols and excretes them as glucuronides via the bile. In the intestine, however, the 2-nitrofluorenols may be liberated by the action of β-glucuronidase (Mφller et al., 1994).
The in vivo metabolism of 2-nitrofluorene, an environmental pollutant, and 2-aminofluorene and its alkylated derivatives, 2-formylaminofluorene and N-acetyl-2-aminofluorene, was examined in rat and dog (Ueda et al., 2001a). 2-Nitrofluoren-7-ol, 2-nitrofluoren-5-ol, 2-aminofluorene, N-acetyl-2-aminofluorene, N-formyl-2-aminofluorene, 2-aminofluoren-5-ol, 2-aminofluoren-7-ol, N-acetyl-2-aminofluoren-5-ol, N-acetyl-2-aminofluoren-7-ol, N-formyl-2-aminofluoren-5-ol and N-formyl-2-aminofluoren-7-ol were identified as urinary and faecal metabolites of nitrofluorene in rat and dog. Acetylaminofluorene and its hydroxylated derivatives were detected as major metabolites of nitrofluorene in rat, but formylaminofluorene and its hydroxylated derivatives were mainly excreted in dogs.
DNA adduct formation was studied in the liver, kidney, spleen and stomach after oral administration of 2-nitrofluorene in rats (Cui et al., 1999). Four major DNA adducts were induced. DNA adduct D co-migrating with N-(deoxyguanosin-8-yl)-2-aminofluorene (dG-C8-AF) had previously been identified after oral administration of 2-nitrofluorene (Wierckx et al., 1991; Möller & Zeisig, 1993; Cui et al., 1995). DNA adduct C co-migrated with C3-(deoxyguanosin-N2-yl)-2-acetylaminofluorene (dG-N2-AAF), whereas the other two (A and B) did not co-migrate with any of the adduct standards used and could not be identified. The four DNA–2-nitrofluorene adducts showed different kinetics of formation and persistence, which may play different roles in 2-nitrofluorene-induced tumour formation (Cui et al., 1999). In the forestomach, after 10 days, there was a high level of only adduct D (dG-C8-AF), but the amount of DNA adducts after 11 months could not be analysed due to multiple tumours. In the liver, the amount of dG-C8-AF decreased with time. The unidentified adducts A and B were the major adducts after 11 months of 2-nitrofluorene feeding. All adducts, in particular A and C, persisted long after the withdrawal of 2-nitrofluorene (Cui et al., 1999).
Other studies on DNA adducts after 2-nitrofluorene administration are summarized in Table 33. Almost all studies after oral administration show the presence of unidentified DNA adducts that do not elute with known DNA adducts found after oral administration of 2-aminofluorene or N-acetyl-2-aminofluorenol (i.e., dG-C8-AF, N-(deoxyguanosin-8-yl)-2-acetylaminofluorene [dG-C8-AAF], dG-N2-AAF; e.g., Scheepers et al., 1994c). The metabolites 2-nitrofluoren-7-ol and 2-nitrofluoren-9-ol (but not 2-nitrofluoren-5-ol) were shown to induce DNA adducts that were similar to those unidentified DNA adducts after 2-nitrofluorene administration (Cui et al., 1996), suggesting that these DNA adducts are formed via an oxidative pathway. Möller et al. (1987a) had previously shown that cytochrome P450 induction dramatically increases the excretion of hydroxylated nitrofluorenes, indicating the involvement of epoxides.
Germ-free animals administered 2-nitrofluorene by gavage had lower levels of DNA adducts than conventional animals, suggesting that the microflora plays an important role in the formation of DNA adducts. Although DNA adducts were detected in the liver, they have not been identified (Möller et al., 1994; Scheepers et al., 1994c).
After oral administration of 2-nitrofluorene, Hb adduct levels were low, and no Hb adducts were detected in the blood in the absence of microflora (Scheepers et al., 1994c,d).
Dinitropyrenes are detected at much lower concentrations than 1-nitropyrene; however, these compounds, in particular 1,6- and 1,8-dinitropyrene, are exceedingly potent bacterial mutagens and in most studies are more tumorigenic than 1-nitropyrene (see chapter 7).
The metabolic activation of dinitropyrenes occurs by reduction of one nitro group to yield N-hydroxy-1-amino-x-nitropyrene, where x is 3, 6 or 8, depending on the original compound. These N-hydroxyarylamine intermediates can undergo acid-catalysed DNA binding or, in contrast to 1-nitropyrene, which is only N-acetylated, can be converted into highly reactive O-acetyl metabolites by bacterial (Orr et al., 1985) and mammalian (Djuric et al., 1985) transacetylases (Beland & Marques, 1994). This activation pathway has been shown to be responsible for their extreme mutagenicity in Salmonella (Fu, 1990; Beland, 1991). In rat liver cytosolic incubations, 1-nitropyrene and 1,3-dinitropyrene were reduced to a much lesser extent than 1,6- or 1,8-dinitropyrene, which suggests that there may be fundamental differences in the reduction pathways between these nitroPAHs (Djuric et al., 1986a; Fu, 1990).
Following a single dose of 1,8-dinitropyrene, only one adduct, identified as N-(deoxyguanosin-8-yl)-1-amino-8-nitropyrene (dG-C8-1-amino-8-NP), was detected in mesentery and in mammary glands of rats (Heflich et al., 1986b) (see Table 34 and Figure 15).
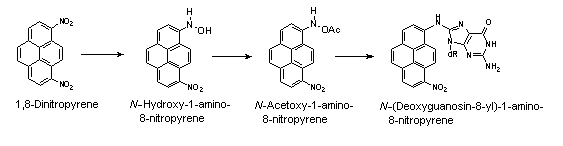
Fig. 15. Metabolic activation pathway and DNA adduct formation of 1,8-dinitropyrene (from Beland & Marques, 1994).
Similarly, a single DNA adduct, N-(deoxyguanosin-8-yl)-1-amino-6-nitropyrene (dG-C8-1-amino-6-NP), has been found in the livers of newborn mice (Delclos et al., 1987a) and in the mammary glands (Djuric et al., 1988) and lungs of rats (Smith et al., 1993) treated with 1,6-dinitropyrene. After administration by gavage, measurable DNA adducts (probably dG-C8-1-amino-6-NP) were detected only in intestinal mucosa and urinary bladder; after i.p. injection, higher levels of the DNA adduct were found, mostly in bladder, white blood cells and lung, but only a lower adduct level was found in liver (Wolff et al., 1993). In male F344 rats after direct pulmonary instillation of 1,6-dinitropyrene, the major DNA adduct in target (lung) and surrogate (liver, white blood cells and spleen lymphocytes) tissues was dG-C8-1-amino-6-NP (Beland et al., 1994). Nitroreduction as well as factors such as O-acetylation seem to be important in determining the extent of DNA binding by 1,6- and 1,8-dinitropyrene in vivo (Beland, 1989).
The relationship between induction of DNA adducts and gene mutations was analysed in F344 rats given 1,6-dinitropyrene directly to the lungs by implantation. A dose-dependent increase of adducts was found in spleen lymphocytes but not in the lung. In parallel, a significant increase of gene mutation frequency (hprt locus) was detected in spleen T-lymphocytes (Smith et al., 1993). Similar studies (Beland et al., 1994; Beland, 1995) compared the adducts in lung, liver and lymphocytes and gene mutations in spleen T-lymphocytes. These findings indicate that concentrations of 1,6-dinitropyrene that produce a dose-dependent induction of lung tumours also result in a dose-dependent formation of DNA adducts and induction of lymphocyte mutations, but that the dose–response curves for DNA binding and mutations are different. These results suggest that T-lymphocyte mutations may be a more sensitive and longer-lived biomarker than DNA adducts for assessing previous exposures to nitroPAHs.
Sequencing of DNA amplification products from 20 1,6-dinitropyrene-induced lung tumours identified five mutations in K-ras codon 12 (four GGT to TGT transversions and one GGT to GAT transition), but not K-ras codons 13 or 61; and mutations in p53 exons 5–8 (eight substitutions at G:C base pairs and one deletion) in 9 of 20 tumour samples. The mutations identified in the dinitropyrene-induced lung tumours and TGr T-lymphocytes are consistent with the formation of dG adducts by 1,6-dinitropyrene (Smith et al., 1997).
The i.p. administration of 100 nmol of 1,6-dinitropyrene to B6C3F1 mice resulted in the detection of 0.46 ± 0.05 fmol of DNA adducts per microgram of DNA. The co-administration of a 25-fold molar excess of 1-nitropyrene (but not 2.5-fold) increased the 1,6-dinitropyrene DNA adduct level to 0.59 ± 0.07 fmol/µg DNA. Conversely, co-administration of 25-fold molar excess of pyrene resulted in a significant decrease in 1,6-dinitropyrene DNA adducts to 0.34 ± 0.04 fmol/µg DNA. While there has been no follow-up on these observations, they do suggest that the metabolic activation of 1,6-dinitropyrene, when existing as part of a complex mixture (e.g., inhaled pollution or particulates), might be affected by the more highly abundant mononitroPAHs or PAHs (see also section 7.5.5 on complex mixtures).
The orientation of the nitro group may be the reason for the differences in mutagenicity and tumorigenicity of the mononitro derivatives of the potent carcinogenic BaP (see chapter 7). While 1- and 3-nitrobenzo[a]pyrene have their nitro group preferentially adopting a parallel (coplanar) orientation, 6-nitrobenzo[a]pyrene has its nitro group in a perpendicular orientation.
Very few data were found on in vivo studies on excretion patterns or metabolites after application of nitrobenzo[a]pyrene. Most studies available are in vitro studies.
As a comparison, BaP requires metabolic activation and is metabolized by a number of oxidative pathways; it is also activated via "bay-region" vicinal diol epoxides (e.g., 7,8-dihydrodiol-9,10-epoxide) to form DNA adducts (IPCS, 1998).
Isomeric nitrobenzo[a]pyrenes are activated to DNA-damaging and mutagenic derivatives by nitroreduction, ring oxidation or a combination of these two pathways. Comparison of metabolic patterns between BaP and 1-, 3- and 6-nitrobenzo[a]pyrene indicates that nitro orientation may affect the regioselectivity of the cytochrome P450 isozymes (Wang et al., 1988).
For 2-nitrobenzo[a]pyrene, the principal metabolic activation pathway using mouse or rat liver microsomes is similar to that of BaP, via ring oxidation to the corresponding 7,8- and 9,10-dihydrodiols. 2- Nitrobenzo[a]pyrene and both of its dihydrodiols are potent mutagens with and without S9 activation. 2-Nitrobenzo[a]pyrene, however, showed higher mutagenic activity in the presence of S9 than in the absence of S9. Collectively, these results suggest that ring oxidation of 2-nitrobenzo[a]pyrene to the bay-region diol epoxide, 2-nitrobenzo[a]pyrene trans-7,8-dihydro-9,10-epoxide, is the principal route of metabolic activation (Von Tungeln et al., 1994a).
Incubation of 3-nitrobenzo[a]pyrene with rat liver microsomes under aerobic conditions yielded 3-nitrobenzo[a]pyrene trans-7,8-dihydrodiol and 3-nitrobenzo[a]pyrene trans-9,10-dihydrodiol as major metabolites, which give 3-nitrobenzo[a]pyrene trans-7,8-diol anti-9,10-epoxide. In contrast, metabolism of 3-nitrobenzo[a]pyrene under hypoxic conditions yielded 3-aminobenzo[a]pyrene, presumably via 3-nitroso- and N-hydroxy-3-aminobenzo[a]pyrene (Chou et al., 1985; Wang et al., 1988). Analogous metabolites were found with 1-nitrobenzo[a]pyrene (Chou et al., 1986).
The S9-mediated mutagenicity of 1- and 3-nitrobenzo[a]pyrene seems to result from binding of 1- and 3-nitrobenzo[a]pyrene trans-7,8-diol anti-9,10-epoxide metabolites to DNA and the subsequent formation of 10-(deoxyguanosin-N2-yl)-7,8,9-trihydroxy-7,8,9,10-tetrahydro-1-nitrobenzo[a]pyrene (dG-1-nitroBaP-DE) and 10-(deoxyguanosin-N2-yl)-7,8,9-trihydroxy-7,8,9,10-tetrahydro-3-nitrobenzo[a]pyrene (dG-3-nitroBaP-DE), respectively. In contrast, the direct-acting mutagenicity of 1- and 3-nitrobenzo[a]pyrene is due to the reaction of N-hydroxyamino derivatives with DNA, which produces the corresponding 6-(deoxyguanosin-N2-yl)-1-aminobenzo[a]pyrene (6-dG-N2-1-amino-BaP) and 6-(deoxyguanosin-N2-yl)-3-aminobenzo[a]pyrene (6-dG-N2-3-amino-BaP) adducts (Fu et al., 1997; Fu & Herreno-Saenz, 1999) (see Table 34).
The aerobic metabolism of 6-nitrobenzo[a]pyrene by rat liver microsomes generated 6-nitrobenzo[a]pyren-3-ol as the major metabolite and 6-nitrobenzo[a]pyren-6-ol and quinones as minor products (Fu et al., 1982).
Whereas 3- and 8-nitrofluoranthene and 3,7- and 3,9-dinitrofluoranthene have been detected in diesel exhaust, 2-nitrofluoranthene has been found to be one of the most abundant nitroPAHs in ambient particulate matter in most locations not associated with traffic pollution (see chapter 5). It is formed from the parent fluoranthene by gas-phase hydroxyl radical- or nitrate radical-initiated reactions (see chapter 3).
The oxidative metabolism of 3-nitrofluoranthene by liver microsomes and cytosols of several mammalian species has been reported to yield various phenolic derivatives, the major metabolites from microsomes isolated from Sprague-Dawley rat and C57B16 mouse being 3-nitrofluoranthen-8-ol and 3-nitrofluoranthen-9-ol (Howard et al., 1988). After i.p. administration of [14C]3-nitrofluoranthene, urine was found to contain 15–20% and faeces about 30% of the dose eliminated within the first 24 h after dosing. Analysis of the metabolic fractions after hydrolysis with β-glucuronidase and sulfatase indicated that positions 4, 8 and 9 were major sites of oxidation. Four major metabolites were tentatively identified as 3-acetamidofluoranthen-1-ol, 3-aminofluoranthene-4,9-quinone and 4-hydroxyfluoranthene-3,8-quinone (from the oxidation of 3-aminofluoranthen-4,9-diol and 3-aminofluoranthen-4-ol-8,9-quinone) and 3-aminofluoranthen-4-ol-8,9-quinone (Gold et al., 1996).
Studies using rat lung cytosol suggest that the mononitrofluoranthenes can be metabolized in the lung by both nitroreductive and oxidative pathways. The absence of any significant oxidative metabolism of 3,9-dinitrofluoranthene suggests that the major pathway for its activation may be nitroreduction followed by O-acetylation. Nitroreduction of 3,9-dinitrofluoranthene occurred at rates twice that of 3-nitrofluoranthene, while both 8- and 2-nitrofluoranthene were metabolized more slowly (Mitchell et al., 1993).
Rat hepatic enzymes catalyse both reductive and oxidative nitrofluoranthene metabolism in vitro. Under aerobic conditions, hydroxylation of the aromatic ring is the main pathway, whereas in the absence of oxygen, only reduction of the nitro group occurs. However, it seems that the isomeric position of the nitro group and the coplanar or perpendicular conformation of the nitro group with respect to the plane of aromatic rings have an influence on the biological activity of the nitrofluoranthenes. The isomers 1-, 7- and 8-nitrofluoranthene were better substrates of rat hepatic microsomal hydrolases than 3-nitrofluoranthene, which, along with 1-nitrofluoranthene, was the preferred substrate for microsomal and cytosolic rat liver nitroreductases. Further, 1- and 3-nitrofluoranthene isomer oxidative metabolism was mediated to a larger extent by phenobarbital-induced rat liver microsomes, whereas 7- and 8-nitrofluoranthene isomer activation was greater in liver microsomes from 3-methylcholanthrene-induced rats (Belisario et al., 1990).
Results from studies of 2-nitrofluoranthene with S. typhimurium TA98 suspension cultures and from neonatal mice administered 2-nitrofluoranthene suggest that 2-nitrofluoranthene is metabolically activated to N-hydroxy-2-aminofluoranthene via nitroreduction, and the resulting DNA adduct [N-(deoxyguanosin-8-yl)-2-aminofluoranthene] is responsible, at least in part, for the mutagenic properties of this compound (Herreno-Saenz et al., 1992, 1994). The major adduct formed in vitro from 3-nitrofluoranthene in the presence of xanthine oxidase and DNA is similarly N-(deoxyguanosin-8-yl)-3-aminofluoranthene. No evidence for other adducts was found (Dietrich et al., 1988; see Table 34).
2-Nitroanthracene contains a nitro group that is coplanar or nearly coplanar with the aromatic ring, whereas 9-nitroanthracene has a nitro group that is perpendicular or nearly so to the aromatic moiety, so that steric interactions with adjacent protons are minimized. This difference in structure is hypothesized to be the basis for differences in metabolism and biological activity of the two nitroPAHs (Fu, 1990).
2-Nitroanthracene was reduced to 2-aminoanthracene in vitro under hypoxic conditions (3% oxygen), but the 9- isomer was not nitro reduced, even with a lower oxygen concentration (0.2%). Aerobic metabolism of 2-nitroanthracene by liver microsomes of rats (after induction with 3-methylcholanthrene) produced 2-nitroanthracene trans-5,6-dihydrodiol and 2-nitroanthracene trans-7,8-dihydrodiol, which were further metabolized mainly to 2-nitroanthracene 5,6,7,8-tetrahydro-trans-diols (dihydrodiol ketones). Under similar conditions, aerobic metabolism of 9-nitroanthracene via 9-nitroanthracene trans-3,4-dihydrodiol yielded 9-nitroanthracene 1,2,3,4-tetrahydrotetrol as the principal metabolite (Fu et al., 1985a, 1986).
Although 6-nitrochrysene is present in the environment in rather low levels, research into this nitroPAH has been based on its remarkable carcinogenic activity in the newborn mouse lung adenoma assay, being the most potent nitroPAH tested in this model (see chapter 7). Figure 16 describes some of the main pathways of 6-nitrochrysene metabolism.
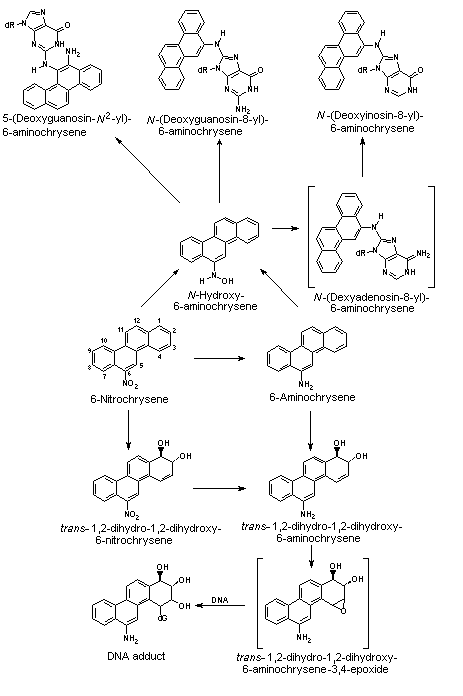
Fig. 16. Metabolic activation pathways and DNA adducts of 6-nitrochrysene (Beland & Marques, 1994).
In rats injected with [3,4,9,10-3H]6-nitrochrysene i.p., after 24 h, only 1.3% of the dose was excreted in the urine and 23% in the faeces (compare with excretion of 1-nitropyrene in Table 31). The extent of metabolism in rats was extremely limited, with 6-nitrochrysene being the major component found in the faeces after 24 h, accounting for 98% of the radioactivity. There is evidence of both reductive and aromatic ring oxidation pathways. The major faecal and urinary metabolite was 6-aminochrysene. Other metabolites observed in the faeces were trans-1,2-dihydro-1,2-dihydroxy-6-nitrochrysene and chrysene-5,6-quinone; further urinary metabolites were trans-1,2-dihydro-1,2-dihydroxy-6-nitrochrysene and trans-9,10-dihydro-9,10-dihydroxy-6-nitrochrysene (Chae et al., 1996). Oral administration of 6-nitrochrysene to female CD rats was also performed, and similar results were obtained (Boyiri et al., 2000). Metabolites detected in extracts from whole animals (mice) after dosing with [3,4,9,10-3H]6-nitrochrysene i.p. were likewise 6-aminochrysene, trans-1,2-dihydro-1,2-dihydroxy-6-nitrochrysene and trans-9,10-dihydro-9,10-dihydroxy-6-nitrochrysene (Delclos et al., 1988).
Studies in mice and in vitro assays have indicated that 6-nitrochrysene can be activated by two major pathways (Figure 16; Li et al., 1994). The major pathway seems to proceed by a combination of ring oxidation and nitroreduction via the formation of the proximate tumorigen trans-1,2-dihydro-1,2-dihydroxy-6-aminochrysene to yield a single major DNA adduct, which has not been rigorously characterized but appears to involve the formation of trans-1,2-dihydro-1,2-dihydroxy-6-aminochrysene-3,4-epoxide with dG (Delclos et al., 1988; Li et al., 1993, 1994). This pathway seems to be the major contributor to the formation of DNA adducts in preweanling mice (Delclos et al., 1987a, 1988; El-Bayoumy et al., 1989a) and rats in vivo (Chae et al., 1996; see Table 34).
Adducts derived from simple nitroreduction of 6-nitrochrysene differ from those obtained from incubating trans-1,2-dihydro-1,2-dihydroxy-6-nitrochrysene in vitro with calf thymus DNA (Krzeminski et al., 2000).
Oxidation to trans-1,2-dihydro-1,2-dihydroxy-6-nitrochrysene is catalysed by CYP1A2 in human liver and CYP1A1 in human lung; reduction to aminochrysene is catalysed by CYP3A4 (Chae et al., 1993). Results of Chen et al. (2000) suggest that 6-nitrochrysene is an inducer of human CYP1A1, and the induction occurs at a transcriptional level in HepG2 cells.
A second pathway has been observed in isolated rat hepatocytes treated with 6-nitrochrysene, involving the formation of N-hydroxy-6-aminochrysene by simple nitroreduction, yielding three major adducts: N-(deoxyguanosin-8-yl)-6-aminochrysene, N-(deoxyinosin-8-yl)-6-aminochrysene, and 5-(deoxyguanosin-N2-yl)-6-aminochrysene (Delclos et al., 1987b). The dI adduct probably results from the oxidative deamination of the corresponding dA adduct.
1-, 2- and 3-BaP, 1- and 3-nitrobenzo[e]pyrene, 2- and 3-nitrofluoranthene and 9-nitrodibenz[a,c]anthracene were tested for tumorigenicity in the neonatal male B6C3F1 mouse with 6-nitrochrysene as positive control (see chapter 7). K- and H-ras mutations were analysed in liver tumours of the treated mice and were found to occur mainly at the first base of K-ras codon 133, resulting in GGC CGC transversion (von Tungeln et al., 1999b). The results indicate that liver tumours from mice treated with nitroPAHs possess ras mutations typical of PAHs and their derivatives rather than H-ras mutations, which are typical for those produced by arylamines (nitroreduction pathway). This result is consistent with the theory that positive responses for nitroPAHs in the neonatal B6C3F1 mouse bioassay are due to metabolism via ring oxidation.
CGC transversion (von Tungeln et al., 1999b). The results indicate that liver tumours from mice treated with nitroPAHs possess ras mutations typical of PAHs and their derivatives rather than H-ras mutations, which are typical for those produced by arylamines (nitroreduction pathway). This result is consistent with the theory that positive responses for nitroPAHs in the neonatal B6C3F1 mouse bioassay are due to metabolism via ring oxidation.
Sixty-four (88%) of 73 analysed lung adenomas and all 15 analysed adenocarcinomas from newborn CD-1 mice injected with 6-nitrochrysene i.p. had a K-ras mutation in codon 12, 13 or 61. All of the mutations in codons 12 and 13 involved a G:C base pair (Li et al., 1994).
The Ah receptor-regulated enzymes CYP1A+, CYP1A2 and CYP1B1 are especially important for the activation of nitroPAHs. The induction of these enzymes by smoking and other exposures will cause a high activating capacity in a fraction of exposed individuals. Many of the enzymes involved (cytochrome P450s, N-acetyltransferases, glutathione S-transferase µ1 [GSTµ1], etc.) show high interindividual variation in humans due to genetic polymorphism (for more information, see section 8.3.2.2).
There are few studies on the acute toxicity of nitroPAHs (Table 37). Only studies on acute lung and liver toxicity of 1- and 2-nitronaphthalene after parenteral application have been reported in any detail.
Table 37. Acute toxicity of nitroPAHs
|
Substance |
Species |
Sex |
Route |
LD50 (mg/kg bw) |
Referencea |
|
1-Nitro-naphthalene |
Sprague-Dawley |
Male |
i.p. |
86 |
Johnson et al. (1984) |
|
2-Nitro-naphthalene |
Swiss-Webster mice |
Male |
Oral |
1300 |
Simmon et al. (1979) |
|
2-Nitrofluorene |
Swiss-Webster mice |
Male |
Oral |
1600 |
Simmon et al. (1979) |
|
1-Nitropyrene |
F344 rats |
Male or female |
Gavage |
>5000 |
Marshall et al. (1982) |
a Cited studies not conducted according to current guidelines.
Johnson et al. (1984) reported respiratory distress in male Sprague-Dawley rats (n = 4–23 per group) within 24 h after a single i.p. injection of 25–200 mg 1-nitronaphthalene/kg bw (ED50 = 60 mg/kg bw). Rats showed laboured breathing and pronounced gasping (severe form). Histopathology revealed acute necrosis of non-ciliated bronchiolar epithelial cells and inflammation. Furthermore, authors reported centrilobular liver necrosis at 100 mg/kg bw, which was most pronounced 72 h after injection. At the time of sacrifice, the relative liver weight was also significantly elevated. The authors estimated an LD50 of 86 mg/kg bw after i.p. application. Further details on lung and liver toxicity are given in section 7.7.
Acute toxicity in male mice after oral application was determined by Simmon et al. (1979). The authors reported an LD50 of 1300 mg/kg bw at a post-exposure observation period of 24 h (no further data).
Details on lung and liver toxicity are given in section 7.7.
Morita et al. (1997) reported an LD100 of approximately 1700 mg/kg bw in male and female rats after i.p. injection. Rats died 48–72 h after treatment (no further data available).
An LD50 of 1600 mg/kg bw at a post-exposure observation period of 24 h was reported in male mice after oral application of 2-nitrofluorene (no further data) (Simmon et al., 1979).
Male rats receiving intrapulmonary implants at a dose of 50 µg per rat showed reduced body weight (Horikawa et al., 1991).
In a dose range-finding study, no observable toxic effects and no histopathological alterations were reported in female F344 rats (n = 1 per dose) that were gavaged with 500–5000 mg/kg bw and sacrificed 4 days after treatment. Similar results were noted after 5000 mg/kg bw (two males and two females) and post-exposure observation periods up to 14 days (Marshall et al., 1982).
There are very few (if any) reports on just the non-neoplastic effects of nitroPAHs, but details can be obtained from studies available on the carcinogenicity of nitroPAHs after repeated administration (see section 7.6). In most carcinogenicity studies, non-neoplastic effects such as increased mortality or reduced body weight are due to tumour formation. In this section, those studies are presented in which non-neoplastic toxic effects that are presumably independent of carcinogenic effects appeared or the specified dose resulted in neither carcinogenic effects nor other toxic effects. However, there seems to be no underlying pattern, and it is impossible to generalize about these effects. Details on experimental design and results are described in Tables 38 and 39. Studies with limited validity (see footnotes for Table 39) are not discussed in this section. The criteria that were considered include (a) toxic effects that were statistically significant, (b) dose–response and (c) clear positive results where statistics were not given.
The Task Group evaluated the strength of each of the in vivo studies based on the following criteria: (a) numbers of animals in each group, (b) duration of study, (c) adequate dosing (e.g., maximum tolerated dose [MTD] in negative studies) and (d) appropriateness of controls. Specific comments on the strengths and weaknesses of the studies are included in Table 39.
In chronic feeding studies on mice and rats, up to 160 or 90 mg/kg bw per day, respectively, for 78 weeks plus latency period resulted in no obvious toxic effects (NCI, 1978a).
In a subchronic intermittent feeding study on rats, a dose of approximately 500 mg/kg bw per day resulted in reduced body weight and increased mortality in females but not in males (Takemura et al., 1974).
In male and female mice, lipidosis of the liver, liver tumour formation and calcification of the renal papilla were observed after exposure via the diet for 78 weeks at doses >40 mg/kg bw per day. The authors also noted reduced survival (NCI, 1978b).
Cui et al. (1995) reported reduced body weight during the exposure period of 11 months in a feeding study on rats at a dose level of about 25 mg/kg bw per day. No effect was noted at <10 mg/kg bw per day.
In a nose-only inhalation study, F344 rats (40 males and 40 females per group) were exposed to 0 or 7.5 mg 1-nitropyrene/m3 for 2 h per day, 5 days per week, for 4 weeks. One day, 2 weeks, 6 months or 12 months after the exposure period, groups of three males and three females were sacrified for histopathology of lung and nasal cavity. There was no effect on body or lung weight, and histopathology revealed no significant lesion. In additional immunization experiments, no increase in the number of antibody-forming cells in the lung-associated lymph nodes was noted (Wolff et al., 1988).
In contrast to the study of Wolff et al. (1988), adverse effects at much lower concentrations were observed in a nose-only study on F344 rats after 13 weeks of exposure (NTP, 1996; for details, see Table 38). Even at the low dose of 0.51 mg/m3, squamous metaplasia of the epiglottis was reported (see also section 7.7).
Repeated administration to rats via gavage (3 times weekly for 4 weeks) at a dose of approximately 2.5 mg/kg bw showed no alteration in body weight and survival (King, 1988).
No effect on body or organ weight was reported in rats after a single intrapulmonary implantation of 1.5 mg per rat (Maeda et al., 1986) or i.p. injection of approximately 17 mg/kg bw, 3 times weekly for 4 weeks (Imaida et al., 1991b).
Table 38. Short-term studies on toxicity and preneoplastic lesions induced by nitroPAHsa
|
NitroPAH; purity |
Species; strain; number per group, sex, ageb |
Route and dose |
Treatment duration; study duration |
Type of lesion: incidence in male and female dose groups |
Results |
Non-neoplastic effects; remarks |
Reference |
|
2-Nitro-fluorene; n.g. |
Rat; Wistar; n.g., 6 weeks |
Gavage |
Four applications on consecutive days and two 2 and 4 days after two-thirds hepatectomy on day 5; 7 weeks |
gamma-Glutamyl transferase-positive foci (preneoplastic changes) volume in % of liver volume: |
+ |
No data on toxicity |
Möller et al. (1989) |
|
|
Rat; Wistar; 3 m, 6–8 weeks |
Gavage |
Single application; 6 weeks |
gamma -Glutamyl transferase-positive foci per cm3 in the liver: |
+ |
No data on toxicity |
Möller et al. (1993b) |
|
2-Nitro-fluorene; n.g. |
Rat; Wistar; n.g., m, 6 weeks |
i.p. |
Single application; 6 weeks |
gamma -Glutamyl transferase-positive foci per cm3 in the liver: |
+ |
No data on toxicity |
Möller et al. (1989) |
|
|
Rat; Wistar; 3 m, 6–8 weeks |
i.p. |
Single application; 6 weeks |
gamma -Glutamyl transferase-positive foci per cm3 in the liver: |
+ |
No data on toxicity |
Möller et al. (1993b) |
|
2-Nitrofluorene; |
Rat; Wistar; 5–7 m, 6–8 weeks |
i.p. |
Single application; 6 weeks |
gamma -Glutamyl transferase-positive foci per cm3 in the liver: |
+ |
No data on toxicity |
Möller et al. (1993b) |
|
2,7-Dinitro-fluorene; |
Rat; Wistar; 3 m, 6–8 weeks |
Gavage |
Single application; 6 weeks |
gamma -Glutamyl transferase-positive foci per cm3 in the liver: |
+ |
No data on toxicity |
Möller et al. (1993b) |
|
2,7-Dinitro-fluorene; |
Rat; Wistar; 3 m, 6–8 weeks |
i.p. |
Single application; 6 weeks |
gamma -Glutamyl transferase-positive foci per cm3 in the liver: |
+ |
No data on toxicity |
Möller et al. (1993b) |
|
1-Nitropyrene; |
Rat; Fischer 344; 6–20 m, |
Gavage |
6 days; 6 weeks |
gamma -Glutamyl transferase-positive foci per cm3 in the liver: |
+ |
No data on toxicity |
Denda et al. (1989) |
|
1-Nitropyrene; |
Rat; F344/N; 7 weeks |
Inhalation |
13 weeks; 13 weeks |
Epiglottis squamous metaplasia #: |
+ |
No effect on body weight except slight decrease in high-dose males, increased relative and absolute liver weights in m at >2 mg/m3; haematology and clinical chemistry revealed no treatment-related effects; only non-neoplastic effects but short observation period; nose-only exposure to aerosol; NOAEL for m 0.51 mg/m3; LOAEL for f 0.51 mg/m3 |
NTP (1996) |
|
a |
n.g. = not given; m = male; f = female; # = no data on statistical evaluation; TS = test substance; p.o. = per os; NOAEL = no-observed-adverse-effect level; LOAEL = lowest-observed-adverse-effect level; significant compared with control, * (P < 0.05), ** (P < 0.01), *** (P < 0.001). |
|
b |
Age of animals at start of the experiment. |
Table 39. Long-term toxicity and carcinogenicity of nitroPAHsa
|
NitroPAH and purity |
Species; strain; number per group per sex; ageb |
Route and dose |
Treatment duration; study duration |
Type of lesion: incidence in male and female dose groups |
Resultc |
Non-neoplastic effects; remarks |
References |
|
1-Nitronaphthalene |
|||||||
|
Technical grade, impurity not specified |
Mouse; B6C3F1; |
Diet, 0, 0.06, 0.12% (~0, 80, 160 mg/kg bw per day) |
78 weeks; 96–98 weeks |
No significant effects with any tumour type |
(–) |
No clinical abnormalities; no effect on survival, but body weight reduced in m and f (no statistics); increase in incidences of non-neoplastic lesions not compound related; complete histopathology at termination; statistical evaluation with all animals surviving 52 weeks; reduced body weight also in low-dose control compared with high-dose control; high dose presumably not the MTD |
NCI (1978a) |
|
Technical grade, impurity not specified |
Rat; F344; 50 m, 50 f (separate high-dose control 25 m and 25 f); |
Diet, 0, 0.05% for 12 weeks then 0.06% for 66 weeks, 0.18% (~0, 25/30, 90 mg/kg bw per day) |
78 weeks; 107–109 weeks |
No significant effects with any tumour type |
(–) |
No treatment-related clinical abnormalities; no effect on survival; body weight decreased in high-dose group compared with concurrent control but not with low-dose control (no statistics); no compound-related increase in non-neoplastic lesions; complete histopathology at termination; statistical evaluation with all animals surviving 52 weeks; high dose presumably not the MTD |
NCI (1978a) |
|
2-Nitronaphthalene |
|||||||
|
Purified |
Rhesus monkey; n.g.; 1 f; n.g. |
Oral gelatin capsules, 121 mg/kg bw twice daily, 6 days per week |
54 months; |
Papilloma in urinary bladder: |
(+) |
No data on toxicity; four papillomas observed; no further macroscopic lesions detected; no control |
Conzelman et al. (1970) |
|
n.g. |
Mouse; n.g.; |
n.g. |
Single implantation; |
Bladder carcinomas: |
(–) |
No effect on survival (no further data); cholesterol pellet containing the TS implanted into the bladder; only mice surviving longer than 175 days evaluated |
Bryan et al. (1964) |
|
5-Nitroacenaphthene |
|||||||
|
n.g. |
Rat; Wistar; |
Diet, 0, 1% |
4 months, 2 interruptions, each of 3 weeks after 1 and 2 months of exposure; 282–500 days |
Rhabdomyosarcomas #: |
+ |
Reduced body weight and survival also without tumour formation; only 12 treated rats lived more than 200 days; 29 controls survived >500 days |
Takemura et al. (1974) |
|
n.g. |
Rat; Wistar; |
Diet, 1% (~500 mg/kg bw per day) |
6 months; |
No malignant tumours (no further data) |
(–) |
No effects on survival (>500 days) or on body weight; no control data |
Takemura et al. (1974) |
|
Fairly high purity (no further data) |
Rat; F344; |
Diet, 0 (two control groups), 0.12, 0.24% (~0, 60, 120 mg/kg bw per day) |
78 weeks (high-dose m 70 weeks); control 108–110 weeks, low dose 100 weeks, high-dose m 70 weeks (f 87 weeks) |
Lung adenomas/carcinomas: |
+ |
Dose-dependent reduction in body weight in m and f (no statistics); dose-dependent increase in mortality in m and f (high tumour incidence); histopathology revealed no compound-related increase in non-neoplastic lesions; concurrent high-dose and low-dose control groups |
NCI (1978b) |
|
Fairly high purity (no further data) |
Mouse; B6C3F1; |
Diet, 0, 0.06 (51 weeks) and then 0.03, 0.12% (~0, 80/40, 160 mg/kg bw per day) |
78 weeks; |
Hepatocellular carcinomas: |
+ |
Dose-dependent depression in body weight in m but not in f; reduced survival (not tumour related) in m and f; treatment-related fatty metamorphosis of the liver (low- and high-dose m, high-dose f) and calcification of renal papilla (high-dose f, low- and high-dose m) |
NCI (1978b) |
|
n.g. |
Syrian golden hamster; n.g.; |
Diet, 0, 1% |
6 months; |
Cholangiomas #: |
+ |
In f, reduced body weight compared with control (no further data); reduced survival, 13 f and 7 m survived 270 days (no data on control) |
Takemura et al. (1974) |
|
2-Nitrofluorene |
|||||||
|
n.g. |
Rat; Holtzman; |
Diet, 0, 342 mg/kg diet |
8 months; |
Mammary gland tumours #: |
(+) |
No effect on survival (no data on body weight); metabolites 2-aminofluorene and 2-acetylaminofluorene were more active |
Miller et al. (1955) |
|
n.g. |
Rat; Holtzman; |
Diet, 0, 342 mg/kg diet |
12 months; |
Forestomach squamous cell carcinomas #: |
+ |
No effect on health or survival up to the time of tumour formation (no further data); glandular portion of the forestomach not involved in tumour formation |
Miller et al. (1955) |
|
n.g. |
Rat; Minnesota; |
Diet, 0, 0.05% |
23 weeks; |
Mammary adenocarcinomas #: |
(–) |
No effect on survival; reduced body weight; small number of animals |
Morris et al. (1950) |
|
95.3% |
Rat; Wistar; |
Diet, 0, 0.24, 0.95, 2.37 mmol/kg diet |
11 months; |
Liver hepatocellular carcinomas #: |
+ |
During exposure period, reduced body weight in the high-dose group; after exposure period, dose-dependent increase in mortality due to tumour formation in mid- and high-dose group |
Cui et al. (1995) |
|
98% |
Mouse; Sencar; 20 f; |
Dermal, 0, 50, 150 µg/mouse |
Single application; |
A few skin tumours arose, but effect was not significant concerning tumours per animal (no data on incidence) |
(–) |
No effect on body weight; initiation/promotion study; TS as initiator; vehicle acetone; 6 days after initiation was complete, 2 µg tetradecanoylphorbol acetate applied twice weekly for 13 weeks (promotion) |
Möller et al. (1993c) |
|
98% |
Mouse; Sencar; 25 f; |
Dermal, 0, 500, 1500 µg/mouse |
Single application; |
A few skin tumours arose, but effect was not significant concerning tumours per animal (no data on incidence) |
(–) |
Final body weight significantly decreased in both TS treatment groups; TS without tetradecanoylphorbol acetate treatment increased body weight; initiation/promotion study; TS as initiator; vehicle acetone; 5 days after initiation was complete, 5 µg tetradecanoylphorbol acetate applied twice weekly for 19 weeks (promotion) |
Möller et al. (1993c) |
|
n.g. |
Rat; Minnesota; |
Dermal, 3 or, after 6 months, 6 drops of 0 or 2% thrice weekly |
80 weeks; |
Total tumours #: |
(+) |
Slightly reduced survival, no effect on body weight; no skin tumours induced; TS dissolved in acetone; total dose 69 mg/rat; small number of animals |
Morris et al. (1950) |
|
>98% |
Rat; Sprague-Dawley; 15 f; 50 days |
Intramammary, 0 or 2.04 µmol/injection, see remarks |
Single application; |
Mammary gland tumour (malignant tumours): 10(2)/15, 5(1)/15; total number of tumours (malignant tumours): 12(2)/15; 8(1)/15 |
(–) |
No effect on body weight gain; vehicle DMSO; each of eight glands injected underneath the nipples; high incidence in vehicle control |
Malejka-Giganti et al. (1999) |
|
2,5-Dinitrofluorene |
|||||||
|
n.g. |
Rat; n.g.; |
Diet, 0 or 1.62 mmol/kg diet |
8 months; |
Mammary gland tumours #: |
(+) |
Survivors at termination: m 9/12, 6/8; f 11/12, 6/8 (no further data); autopsy included ear duct, mammary gland and abdominal and thoracic organs; only histopathology of tumours |
Miller et al. (1962) |
|
2,7-Dinitrofluorene |
|||||||
|
n.g. |
Rat; n.g.; |
Diet, 0 or 1.62 mmol/kg diet |
8 months; |
Malignant mammary gland tumours #: |
(+) |
Survivors at termination: m 9/12, 4/8; f 11/12, 0/8 (no further data); autopsy included ear duct, mammary gland and abdominal and thoracic organs; only histopathology of tumours |
Miller et al. (1962) |
|
>98% |
Rat; Sprague-Dawley; 15 f; 50 days |
Intramammary, 0 or 2.04 µmol/injection, see remarks |
Single application; |
Mammary gland tumour (malignant tumours): 10(2)/15, 14(14)/15; total number of tumours (malignant tumours): 12(2)/15; 33(27)/15*** |
(+) |
No effect on body weight gain; vehicle DMSO; each of eight glands injected underneath the nipples; high incidence in vehicle control |
Malejka-Giganti et al. (1999) |
|
2-Nitrofluoranthene |
|||||||
|
>99% |
Mouse; B6C3F1; |
i.p., total dose 0, 400 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose on days 1, 8 and 15, respectively; |
Liver adenomas: 2/24, 8/23; liver carcinomas: 1/24, 3/23; lung tumours: 0/24, 0/23 |
(–) |
Mortality: 0%, 4%; no effects on body weight; solvent DMSO; only liver and lung tumours studied |
von Tungeln et al. (1994b, 1999b) |
|
3-Nitrofluoranthene |
|||||||
|
99.99% |
Rat; F344/DuCrj; |
Intrapulmonary implants, 0, 1000 µg/rat |
Single implantation; |
Lung squamous cell carcinomas #: |
(–) |
Reduced body weight (but not significant) in treated rats; survival not reduced; further lung lesions also in controls; TS in a mixture of beeswax–tricaprylin |
Horikawa et al. (1991) |
|
Purified (no further data) |
Rat; n.g.; 20 m; n.g. |
Intrapulmonary implants, 0, 1000 µg/rat |
Single implantation; |
Lung tumours #: |
(–) |
No data on toxicity; TS in a mixture of beeswax–tricaprylin |
Tokiwa et al. (1990b) |
|
>99% |
Rat; F344/DuCrj; |
s.c., 0, 2 mg/rat twice weekly |
7.5 weeks; |
Tumours at injection site: |
+ |
Reversible inflammation and ulcerations at the injection site after treatment with TS; decreased body weight gain; solvent DMSO |
Ohgaki et al. (1982) |
|
>99.9% |
Mouse; BLU:Ha; |
i.p., total dose 0, 63, 315 µg/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; |
total no. of mice with lung tumours: 20/192, 15/53***, 16/52**; |
+ |
No data on toxicity; solvent DMSO; lung tumours studied; sexes not separately evaluated for statistical purposes |
Busby et al. (1989) |
|
>99% |
Mouse; B6C3F1; |
i.p., total dose 0, 400 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose on days 1, 8 and 15, respectively; |
Liver adenomas: 2/24, 7/24; liver carcinomas: 1/24, 1/24; lung tumours: 0/24, 0/24 |
(–) |
Mortality: 0%, 0%; no effects on body weight; solvent DMSO; only liver and lung tumours studied |
Von Tungeln et al. (1994b, 1999b) |
|
3,7-Dinitrofluoranthene |
|||||||
|
Purified by recrystallization |
Rat; F344/DuCrj; |
s.c., 0, 50 µg/rat twice weekly |
10 weeks; |
Local subcutaneous tumours #: |
+ |
<10% of treated rats alive at termination (control 100%); TS dissolved in DMSO |
Tokiwa et al. (1987a) |
|
99.90% |
Rat; F344/DuCrj; |
Intrapulmonary implants, 0, 200 µg/rat |
Single implantation; |
Lung tumours #: |
+ |
Reduced body weight and survival in treated rats, presumably due to tumour formation; TS in a mixture of beeswax–tricaprylin |
Tokiwa et al. (1990b); Horikawa et al. (1991) |
|
3,9-Dinitrofluoranthene |
|||||||
|
Purified by recrystallization |
Rat; F344/DuCrj; |
s.c., 0, 50 µg/rat twice weekly |
10 weeks; |
Subcutaneous tumours #: |
+ |
All treated rats died or were sacrificed moribund at weeks 15–36 (survival in control 100% at week 50) due to local tumour formation; TS dissolved in DMSO |
Tokiwa et al. (1987a) |
|
99.98% |
Rat; F344/DuCrj; |
Intrapulmonary implants, 0, 50, 100, 200 µg/rat |
Single implantation; |
Lung tumours #: |
+ |
Reduced body weight in treated rats (dose related); reduced survival in high-dose group; effects in low-dose group presumably not related to tumour formation; TS in a mixture of beeswax–tricaprylin |
Horikawa et al. (1991) |
|
1-Nitropyrene |
|||||||
|
>99.9% |
Rat; CD; 35–36 f; weanling |
Gavage, 0, 10 µmol (2.5 mg)/kg bw, thrice weekly |
4 weeks; |
No significant carcinogenic effects |
(–) |
No effects on body weight and survival; solvent DMSO |
King (1988); Imaida et al. (1991a) |
|
>99.9% |
Rat; CD; 30 f; 30 days |
Gavage, 0, 50 µmol/rat (42–125 mg/kg bw), once weekly |
8 weeks; |
Mammary fibroadenomas: |
+ |
No effect on body weight and survival; vehicle trioctanoin |
El-Bayoumy et al. (1995) |
|
>99.9%, absence of dinitropyrenes |
Rat; Sprague-Dawley; 22–36 m and 24–31 f; |
Gavage, 0, 100, 250 µmol/kg bw (0, 25, 62 mg/kg bw), once weekly |
16 weeks; |
Mammary adenocarcinomas: |
+ |
No effect on body weight, but reduced survival in both treatment groups, presumably due to tumour formation; vehicle trioctanoin; metabolites 1-nitrosopyrene and 1-aminopyrene were less carcinogenic |
El-Bayoumy et al. (1988b) |
|
>99% |
Mouse; Crl/CD-1 (ICR)BR; |
Dermal, 0, 0.1 mg/mouse per day |
10 days; |
No significant effect on skin tumour formation |
(–) |
No data on toxicity; initiation/promotion study; TS as initiator; vehicle acetone; 10 days after initiation was complete, 2.5 µg tetradecanoylphorbol acetate applied thrice weekly for 25 weeks (promotion) |
El-Bayoumy et al. (1982) |
|
99.5% |
Mouse; |
Dermal, 0, 0.03, 0.1, 0.3, 1, 3 mg/mouse |
Single application except high dose (2 treatments, no further information); |
No significant tumour initiating activity in contrast to the positive control BaP (0.05 mg/mouse) |
(–) |
No data on toxicity; initiation/promotion study; vehicle acetone; 1 week after initiation was complete, 2 µg tetradecanoylphorbol acetate applied twice weekly for 30 weeks (promotion) |
Nesnow et al. (1984) |
|
98% (0.008% 1,3-dinitropyrene, 0.6% 1,6- and 1,8-dinitropy-rene, 1.3% pyrene) |
Syrian golden hamster; n.g.; |
Intratracheal instillation, 0, 2 mg/hamster, once weekly |
15 weeks; |
No significant effects on lung or trachea tumour formation in contrast to the positive control BaP (same dose) |
(–) |
24 (control 16) hamsters alive after treatment period (pneumonia during exposure period), no effect on survival; vehicle phosphate buffer; animals observed during entire life span |
Yamamoto et al. (1987) |
|
99.9% |
Rat; F344/DuCrj; |
Intrapulmonary implants, 0, 1.5 mg/rat |
Single implantation; |
No lung tumour induction in contrast to the positive control 0.5 mg 3-methylcholanthrene (19/19) |
(–) |
No effect on body or organ weight (no data on survival); TS in a mixture of beeswax–tricaprylin |
Maeda et al. (1986) |
|
99.9% |
Mouse; BALB/c; 20 m; 6 weeks |
s.c., 0, 0.1 mg/mouse (~3.3 mg/kg bw), once weekly |
20 weeks; |
No significant carcinogenic effect in contrast to 1,6-dinitropyrene (same dose; subcutaneous tumours) |
(–) |
Slightly reduced survival, presumably not treatment related; no data on body weight; solvent DMSO; low number of mice |
Tokiwa et al. (1984, 1986) |
|
>99.75% |
Rat; F344/DuCrj; |
s.c., 0, 0.2, 2 mg/rat (0, 2, 20 mg/kg bw), twice weekly |
10 weeks; |
No tumours at injection site, no other carcinogenic effects |
(–) |
No effect on body weight and survival; solvent DMSO |
Ohgaki et al. (1985) |
|
Purified, contamination by dinitro-pyrenes <0.02% |
Rat; Sprague-Dawley CD; |
s.c., 0, 50, 100 µmol (12.5 or 25 mg)/kg bw, once weekly |
8 weeks; |
Tumours at injection site: m 0/28, 2/29, 10/31***; f 0/31, 3/31, 9/32** (mostly malignant fibrous histiosarcomas); mammary tumours: f 2/31, 7/31, 15/32*** (mostly adenocarcinomas and fibroadenomas) |
+ |
No significant effect on body weight or survival; no effect on liver and kidney weight, but spleen weight increased in m and f in high-dose group (marked haematopoiesis due to tumour formation); solvent DMSO |
Hirose et al. (1984) |
|
n.g. |
Rat; CD; 29–30 f; weanling |
s.c., 0, 100 µmol (25 mg)/kg bw, once weekly |
4 weeks; 87–90 weeks |
Mammary fibroadenomas: |
(+) |
No significant effect on body or organ weight (liver, kidney, spleen); no data on survival; solvent DMSO |
Imaida et al. (1991b) |
|
>99.9% |
Rat; F344; 55 f; newborn |
s.c., 0, 100 µmol (25 mg)/kg bw, once weekly |
8 weeks; |
Leukaemia: |
+ |
No effects on survival; no data on body weight; solvent DMSO |
King (1988); Imaida et al. (1995) |
|
>99.9% |
Rat; CD; 47–48 f; newborn |
s.c., 0, 100 µmol (25 mg)/kg bw, once weekly |
8 weeks; |
Mammary adenocarcinomas: |
+ |
No effects on survival; no data on body weight; solvent DMSO; no significant effects with the metabolites 1-nitropyren-3-ol, 1-nitropyren-6-ol and 1-nitropyren-8-ol |
King (1988); Imaida et al. (1995) |
|
>99.9% |
Rat; CD; control 40 f, treated 49 f; newborn |
s.c., 0, 2.5–10 µmol/kg bw (0.6–2.5 mg/kg bw) (see remarks), once weekly |
8 weeks; |
Mammary adenocarcinomas: |
+ |
No effects on survival; solvent DMSO; 1st injection 2.5 µmol, 2nd and 3rd 5 µmol, 4th–8th 10 µmol/kg bw |
King (1988); Imaida et al. (1995) |
|
>99.99%, no dinitropyrenes detected |
Mouse, A/J; |
i.p., 0 or 93 mg/kg bw, thrice weekly |
6 weeks (total of 17 injections); |
Incidence of lung tumours significantly increased (P < 0.0001, control 1/23, no further data) |
(+) |
No data on toxicity; solvent DMSO; no definitive carcinogenicity test with this mouse strain |
Bai et al. (1998) |
|
>99%, absence of dinitropyrenes |
Mouse, A/J; 15–16 m and 12–16 f; 6–8 weeks |
i.p., not specified, but total dose 0, 175, 525, 1575 mg/kg bw, injections thrice weekly |
6 weeks; 24 weeks |
Lung tumours: |
(+) |
No data on toxicity; vehicle trioctanoin; the strain used in this study is sensitive to lung tumour development and, on occasion, conflicts with standard 2-year studies |
El-Bayoumy & Hecht (1983); El-Bayoumy et al. (1984b) |
|
>99% |
Mouse; CD-1; |
i.p., total dose 0, 700, 2800 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; 12 months |
Liver carcinomas: |
+ |
54–85% survived to weaning (no further data on toxicity); vehicle DMSO; two vehicle-treated control groups; tumour incidence from weaning to month 12 recorded; metabolite 1-nitrosopyrene was more effective |
Wislocki et al. (1986) |
|
>99.9% |
Mouse; BLU:Ha; |
i.p., total dose 0, 21, 105 µg/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; days 1–15; 26 weeks |
No effects on lung tumour formation |
(–) |
No data on toxicity; solvent DMSO; only lung tumours studied |
Busby et al. (1989) |
|
99.5% |
Mouse; SENCAR; |
i.p., 0, 1, 2, 4, 6, 8 mg/mouse (~0, 33, 66, 132, 200, 264 mg/kg bw) |
Single application; |
No significant tumour initiating activity in contrast to the positive control BaP |
(–) |
No data on toxicity; initiation/promotion study; vehicle corn oil; 1 week after initiation, 2 µg tetradecanoylphorbol acetate applied dermally twice weekly for 30 weeks (promotion) |
Nesnow et al. (1984) |
|
>99.9% |
Rat; CD; 36 f; weanling |
i.p., 0, 10 µmol/kg bw (2.5 mg/kg bw), thrice weekly |
4 weeks; |
Mammary tumours: |
+ |
No effects on body weight or survival; solvent DMSO |
King (1988); Imaida et al. (1991a) |
|
n.g. |
Rat; CD; 29 f; weanling |
i.p., 0, 67 µmol (17 mg)/kg bw, thrice weekly (total dose 119 µmol/rat) |
4 weeks; |
No significant carcinogenic effects also with the metabolites (same dose) N-hydroxy-N-acetyl-1-aminopyrene and N-acetyl-1-aminopyrene |
(–) |
No significant effect on body or organ weight (liver, kidney, spleen); no altered liver foci; no data on survival; solvent DMSO |
Imaida et al. (1991b) |
|
n.g. |
Rat; CD; 29–30 f; weanling |
i.p., 0, 100 µmol (25 mg)/kg bw, once weekly |
4 weeks; |
Mammary adenocarcinomas: |
+ |
No significant effect on body or organ weight (liver, kidney, spleen); no data on survival; solvent DMSO |
Imaida et al. (1991b) |
|
Mixture of 1-nitropyrene and small amounts of 1,3-, 1,6- and 1,8-dinitropyrene |
|||||||
|
0.11% 1,3-dinitropyrene, 0.27% 1,6-dinitropyrene, 0.23% 1,8-dinitropyrene |
Rat; F-344/Jcl; |
Gavage, 0, 5, 10, 20 mg/kg bw, twice weekly |
55 weeks; |
Mammary tumours: |
+ |
No significant effect on body weight, but dose-related decrease in survival from week 70 on associated with tumour formation; no effects on organ weight (brain, heart, liver, spleen, kidney, adrenal, uterus, ovary, thyroid); vehicle olive oil |
Odagiri et al. (1986) |
|
>99%; traces of dinitro-pyrenes |
Rat; F344/DuCrj; |
s.c., 0, 2 mg/rat twice weekly |
10 weeks; |
Tumours at injection site: |
+ |
Decreased body weight gain (no data on survival) and reversible inflammation/ulcerations at the injection site in TS-treated rats; |
Ohgaki et al. (1982) |
|
n.g. |
Mouse; B6C3F1; |
i.p., 0, 20, 40, 50, 100 mg/kg bw |
Single injection; |
Significant (P < 0.05) induction of lung adenomas and adenocarcinomas (no further data) |
(+) |
Transplacental carcinogenesis studied; pregnant mice exposed; frequent abortions after injection of nitropyrene mixture; data from abstract, no further information available |
Odagiri et al. (1993) |
|
2-Nitropyrene |
|||||||
|
n.g. |
Rat; CD; 28 f; weanling |
Intramammary, 0, 2 µmol/injection, see remarks |
Single application; |
No effect on mammary tumour formation |
(–) |
No data on body or organ weight and survival; solvent DMSO; dose injected into tissue underlying each of the three thoracic nipples (left and right) |
Imaida et al. (1991b) |
|
n.g. |
Rat; CD; 28–29 f; weanling |
i.p., 0, 67 µmol (17 mg)/kg bw, thrice weekly (total dose 119 µmol/rat) |
4 weeks; |
Leukaemia/lymphoma: |
+ |
No significant effect on body or organ weight (liver, kidney, spleen); no altered liver foci; solvent DMSO |
Imaida et al. (1991b) |
|
4-Nitropyrene |
|||||||
|
Purified by HPLC |
Rat; CD; control 47 f, treated 27 f; |
s.c., 0, 100 µmol (25 mg)/kg bw, once weekly |
8 weeks; |
Mammary tumours: f 17/47, 20/27**; malignant fibrous histiocytomas: f 0/47, 10/27***; |
+ |
No effects on body weight during treatment period, but reduced survival; solvent DMSO |
King (1988); Imaida et al. (1995) |
|
>99% |
Mouse; CD-1; |
i.p., total dose 0, 2800 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; 12 months |
Liver carcinomas: |
+ |
54–85% survived to weaning (no further data on toxicity); vehicle DMSO; tumour incidence from weaning to month 12 recorded |
Wislocki et al. (1986) |
|
Purified by recrystallization |
Rat; CD; 29 f; weanling |
i.p., 0, 67 µmol (17 mg)/kg bw, thrice weekly (total dose 119 µmol/rat) |
4 weeks; |
Mammary adenocarcinomas: |
+ |
No significant effect on body or organ weight (liver, kidney, spleen); altered liver foci in 1 treated rat; no data on survival; solvent DMSO |
Imaida et al. (1991b) |
|
Purified by recrystallization |
Rat; CD; 28 f; weanling |
Intramammary, 0, 2 µmol/injection, see remarks |
Single application; |
Mammary adenocarcinomas: |
+ |
No data on body or organ weight and survival; solvent DMSO; dose injected into tissue underlying each of the three thoracic nipples (left and right) |
Imaida et al. (1991b) |
|
>99.8% |
Rat; CD; 30 f; 30 days |
Intramammary, 0, 2.04 µmol/injection, see remarks |
Single application; |
Mammary adenocarcinomas: |
+ |
No effects on body weight and survival; solvent DMSO; dose injected into tissue underlying each of the three thoracic nipples and three inguinal nipples (only left side, right side DMSO injection) |
El-Bayoumy et al. (1993) |
|
Mixture of 1,3-, 1,6- and 1,8-dinitropyrene |
|||||||
|
20.26% 1,3-dinitropyrene, 39.26% 1,6-dinitropyrene, 39.51% 1,8-dinitropyrene (>99%) |
Mouse; |
Dermal, total dose 0, 0.05, 0.1, 0.5, 1, 2 mg/mouse |
Single application (low dose) up to 2 applications per day for 5 days; 30 weeks |
Skin papillomas: |
+ |
No data on toxicity; initiation/promotion study; vehicle acetone/DMSO; 1 week after initiation was complete, 2 µg tetradecanoylphorbol acetate applied twice weekly for 30 weeks (promotion) |
Nesnow et al. (1984) |
|
1,3-Dinitropyrene |
|||||||
|
>99% |
Rat; CD; 35 f; |
Gavage, 0, 10 µmol (2.9 mg)/kg bw, thrice weekly |
4 weeks; |
No significant carcinogenic effects |
(–) |
No effects on body weight and survival; solvent DMSO |
King (1988); Imaida et al. (1991a) |
|
>99.9% |
Mouse; BALB/c; 13–18 m; 6 weeks |
s.c., 0, 50 µg/mouse (1.7 mg/kg bw), once weekly |
20 weeks; |
No significant carcinogenic effects in contrast to the positive control BaP (same dose) |
(–) |
No effect on survival (no data on body weight); solvent DMSO |
Tokiwa et al. (1986); Otofuji et al. (1987) |
|
>99% |
Rat; CD; 40–43 f; newborn |
s.c., 0, 2.5–10 µmol (0.7–3 mg)/kg bw (see remarks), once weekly |
8 weeks; |
Malignant fibrous histiocytomas: |
+ |
No effect on survival (no further data); solvent DMSO; 1st injection 2.5 µmol, 2nd and 3rd 5 µmol, 4th–8th 10 µmol/kg bw |
King (1988); Imaida et al. (1995) |
|
0.6% 1,6 dinitropyrene, 0.4% 1,8-dinitropyrene and 0.05% other nitropyrenes |
Rat; F344/DuCrj; |
s.c., 0, 0.2 mg/rat (~1 mg/kg bw), twice weekly |
10 weeks; |
Sarcomas at the site of injection #: |
+ |
No effect on body weight gain; in treated rats, reversible ulcer or scar formation at the site of injection (reversible); reduced survival due to local tumour development; solvent DMSO |
Ohgaki et al. (1984) |
|
>99% |
Mouse; CD-1; |
i.p., total dose 0, 200 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; 12 months |
No significant carcinogenic effects |
(–) |
54–85% survived to weaning (no further data on toxicity); tumour incidence from weaning to month 12 recorded; vehicle DMSO |
Wislocki et al. (1986) |
|
>99% |
Rat; CD; 31–36 f; weanling |
i.p., 0, 10 µmol (2.9 mg)/kg bw, thrice weekly |
4 weeks; 76–78 weeks |
Mammary tumours: f 7/31, 19/36** (9 adeno-carcinomas, 12 fibroadenomas and 1 adenoma) |
+ |
No effects on survival and body weight; solvent DMSO |
King (1988); Imaida et al. (1991a) |
|
1,6-Dinitropyrene |
|||||||
|
>99% |
Rat; CD; 36 f; weanling |
Gavage, 0, 10 µmol (3 mg)/kg bw, thrice weekly |
4 weeks; |
Mammary adenocarcinomas: |
(–) |
No effects on body weight or survival; solvent DMSO |
King (1988); Imaida et al. (1991a) |
|
>99.9% |
Syrian golden hamster; n.g.; |
Intratracheal instillation, 0, 0.5 mg/hamster (~5 mg/kg bw), once weekly |
26 weeks; |
Lung adenocarcinomas #: |
+ |
Reduced survival due to tumour formation; no data on body weight; TS suspended in 0.2 ml saline |
Takayama et al. (1985) |
|
99.9% |
Rat; F344/DuCrj; 28–31 m; 10–11 weeks |
Intrapulmonary implant, 0, 0.15 mg/rat |
Single implantation; |
Lung tumours: m 0/31, 23/28** (21 squamous cell carcinomas, 2 undifferentiated carcinomas) |
+ |
Reduced body and liver weight (no data on survival); TS in a mixture of beeswax–tricaprylin |
Maeda et al. (1986) |
|
99.8% |
Rat; F344/NSlc; |
Intrapulmonary implant, 0, 0.003, 0.01, 0.03, 0.1, 0.15 mg/rat |
Single implantation; |
Lung tumours: m 0/40, 0/39, 4/30, 13/31, 22/26, 6/9 (mainly undifferentiated neoplasm), highly significant dose–response relationship (no further data) |
+ |
No effect on body weight, but at >0.03 mg/rat, significantly reduced survival due to tumour formation; TS in a mixture of beeswax–tricaprylin |
Iwagawa et al. (1989); Tokiwa et al. (1990b) |
|
>99.9% |
Mouse; BALB/c; 20 m; 6 weeks |
s.c., 0, 0.1 mg/mouse, once weekly |
20 weeks; |
Malignant fibrous histiocytomas at the site of injection: |
+ |
Reduced survival presumably due to tumour formation; no data on body weight; solvent DMSO |
Tokiwa et al. (1984, 1986) |
|
Purity checked by HPLC (no further data) |
Rat; F344/DuCrj; |
s.c., 0, 0.2 mg/rat twice weekly |
10 weeks; |
Sarcomas at the site of injection #: |
+ |
No effect on body weight; reduced survival due to tumour development; solvent DMSO |
Ohgaki et al. (1985) |
|
>99% |
Rat; CD; 40–46 f; newborn |
s.c., 0, 2.5–10 µmol (0.7–3 mg)/kg bw (see remarks), once weekly |
8 weeks; |
Malignant fibrous histiocytomas: |
+ |
Survival reduced (149 versus 495 days) due to tumour development; enlarged liver and spleen (leukaemia); solvent DMSO; 1st injection 2.5 µmol, 2nd and 3rd 5 µmol, 4th–8th 10 µmol/kg bw |
King (1988); Imaida et al. (1995) |
|
>99% |
Mouse; CD-1; |
i.p., total dose 0, 200 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; |
Liver carcinomas: |
+ |
54–85% survived to weaning (no further data on toxicity); vehicle DMSO; tumour incidence from weaning to month 12 recorded |
Wislocki et al. (1986) |
|
>99% |
Rat; CD; control 31 f, treated 23 f; weanling |
i.p., 0, 10 µmol (2.9 mg)/kg bw, thrice weekly |
4 weeks; |
Malignant fibrous histiocytomas: |
+ |
No effect on body weight, but average survival 135 days versus 535 days in controls due to tumour formation in the peritoneal cavity; solvent DMSO |
King (1988); Imaida et al. (1991a) |
|
1,8-Dinitropyrene |
|||||||
|
>99% |
Rat; CD; 36 f; weanling |
Gavage, 0, 10 µmol (2.9 mg)/kg bw, thrice weekly |
4 weeks; |
Mammary tumours: |
+ |
No effects on body weight and survival; solvent DMSO |
King (1988); Imaida et al. (1991a) |
|
0.6% 1,6-dinitropyrene, 0.4% 1,3-dinitropyrene and 0.05% other nitropyrenes |
Rat; F344/DuCrj; |
s.c., 0, 0.2 mg/rat, twice weekly |
10 weeks; |
Sarcomas at the site of injection #: |
+ |
No effect on body weight gain; in treated rats, reversible ulcer or scar formation at the site of injection; reduced survival due to tumour development; solvent DMSO |
Ohgaki et al. (1984) |
|
>99.9% |
Mouse; BALB/c; 13–15 m; 6 weeks |
s.c., 0, 50 µg/mouse, once weekly |
20 weeks; |
Local subcutaneous tumours: |
+ |
No effect on survival (no data on body weight); solvent DMSO |
Tokiwa et al. (1986); Otofuji et al. (1987) |
|
0.4% 1,3-dinitropyrene (no further data) |
Rat; |
s.c., 0, 2, 20 µg/rat twice weekly |
10 weeks; |
Sarcomas at the site of injection #: |
+ |
No effect on body weight; reduced survival due to local tumour development; solvent DMSO |
Ohgaki et al. (1985) |
|
>99% |
Rat; CD; 37–40 f; newborn |
s.c., 0, 2.5–10 µmol (0.7–3 mg)/kg bw (see remarks), once weekly |
8 weeks; |
Malignant fibrous histiocytomas: |
+ |
Survival reduced (163 versus 495 days) due to tumour development; enlarged liver and spleen (leukaemia); solvent DMSO; 1st injection 2.5 µmol, 2nd and 3rd 5 µmol, 4th–8th 10 µmol/kg bw |
King (1988); Imaida et al. (1995) |
|
>99% |
Mouse; CD-1; |
i.p., total dose 0, 200 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; 12 months |
No significant carcinogenic effects |
(–) |
54–85% survived to weaning (no further data on toxicity); vehicle DMSO; tumour incidence from weaning to month 12 recorded |
Wislocki et al. (1986) |
|
>99% |
Rat; CD; 31–33 f; weanling |
i.p., 0, 10 µmol (2.9 mg)/kg bw, thrice weekly |
4 weeks; |
Malignant fibrous histiocytomas: |
+ |
No effect on body weight, but average survival 236 days versus 535 days in controls due to local tumour formation; solvent DMSO |
King (1988); Imaida et al. (1991a) |
|
7-Nitrobenz[a]anthracene |
|||||||
|
>99% |
Mouse; CD-1; |
i.p., total dose 0, 2800 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at day 1, 8 and 15 injected; 12 months |
Liver tumours: |
+ |
54–85% survived to weaning (no further data on toxicity); vehicle DMSO; tumour incidence from weaning to month 12 recorded; parent PAH was more effective |
Wislocki et al. (1986) |
|
6-Nitrochrysene |
|||||||
|
>99% |
Mouse; Crl/CD-1 (ICR)BR; 20 f; 50–55 days |
Dermal, 0, 0.1 mg/mouse per day |
10 days; 25 weeks |
Skin tumours: |
+ |
No data on toxicity; initiation/promotion study; 10 days after initiation, 2.5 µg tetradecanoylphorbol acetate applied thrice weekly for 25 weeks (promotion); vehicle acetone |
El-Bayoumy et al. (1982) |
|
>99.8% |
Rat, CD; 30 f; |
Intramammary, 0, 2.04 µmol/injection, see remarks |
Single application; |
Mammary adenocarcinomas: |
+ |
No effects on body weight and survival; solvent DMSO; dose injected into tissue underlying each of the three thoracic nipples and three inguinal nipples (only left side, right side DMSO injection) |
El-Bayoumy et al. (1993) |
|
>99.9% |
Mouse; BLU:Ha; control 91 m and 101 f, treated 26 m and 22 f; newborn |
i.p., total dose 0, 7.7 µg/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; 26 weeks |
Total no. of mice with lung tumours 20/192, 32/48***; also significant increase in no. of lung tumours/mouse |
+ |
No data on toxicity; solvent DMSO; lung tumours studied; no statistical evaluation for each sex; parent PAH revealed no carcinogenic effect |
Busby et al. (1989) |
|
>99% |
Mouse; CD-1; |
i.p., total dose 0, 700, 2800 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; 12 months |
Liver adenomas and carcinomas: |
+ |
54–85% survived to weaning, except high-dose group (25–30%); death occurred mainly between last injection and weaning (no further data); vehicle DMSO; two vehicle-treated control groups; tumour incidence from weaning to month 12 recorded; the parent PAH was less effective |
Wislocki et al. (1986) |
|
Purity checked by different methods (no further data) |
Mouse; BLU:Ha; 22–23 m and 15–29 f; newborn |
i.p., total dose 0, 38.5, 189 µg/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; 26 weeks |
Lung tumours #: |
+ |
A few mice died 1st 3 months (no further data on toxicity); solvent DMSO; mainly lung tumours studied; no statistical evaluation for each sex |
Busby et al. (1985) |
|
Purity checked (no further data) |
Mouse, Swiss-Webster BLU-Ha; 23–38 m and 24–46 f; newborn |
i.p., total dose 0, 100, 700 nmol/mouse (0, 27.3, 191 µg/mouse) |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; 32 weeks |
Lung tumours: |
+ |
No data on toxicity; solvent DMSO; liver and lung tumours studied; similar results with the metabolites trans-1,2-dihydro-1,2-dihydroxy-6-nitrochrysene and trans-1,2-dihydro-1,2-dihydroxy-6-aminochrysene; 6-nitroso- and 6-aminochysene not effective (equimolar doses) |
El-Bayoumy et al. (1989a) |
|
>99.5% |
Mouse, Crj:CD-1(ICR); |
i.p., total dose 0, 1.4 µmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; 24 weeks |
Lung adenomas: |
+ |
No data on toxicity; solvent DMSO |
Imaida et al. (1992) |
|
n.g. |
Mouse; CD-1; |
i.p., total dose 0, 0.25 µmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; 12 months |
Liver tumours #: |
+ |
No data on toxicity; solvent DMSO; only liver tumours evaluated; sequence analysis of mutations performed |
Manjanatha et al. (1996) |
|
>99% |
Mouse; CD-1; |
i.p., total dose 0, 100 or 400 nmol/mouse |
3 applications on days 1, 8 and 15, respectively; |
Liver adenomas: 1/20, 15/16***, 11/16***; liver carcinomas: 2/20, 14/16***, 15/16***; lung adenomas: 4/20, 16/16***, 16/16***; lung carcinomas: 0/20, 3/16, 10/16*** |
+ |
Mortality not compound related (no further data) |
von Tungeln et al. (1999a) |
|
n.g. |
Mouse; B6C3F1; |
i.p., total dose 0, 0.3 µmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; 12 months |
Liver tumours #: |
+ |
No data on toxicity; solvent DMSO; only liver tumours evaluated; sequence analysis of mutations performed |
Manjanatha et al. (1996) |
|
Purified by recrystallization |
Mouse; B6C; |
i.p., 0, 2.2 µmol/mouse (20 mg/kg bw), every 2nd week |
6 weeks; |
Lung adenomas: |
+ |
No data on toxicity; solvent DMSO; transgenic mice used, DNA analysis performed |
Ogawa et al. (1996) |
|
>99% |
Mouse; B6C3F1; 18–23 m; newborn |
i.p., total dose 0, 400 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; 12 months |
Liver adenomas: 2/18, 19/19*** |
+ |
No data on toxicity; solvent DMSO; caloric restriction inhibits tumour formation |
Fu et al. (1994a) |
|
>99% |
Mouse; B6C3F1; 18–23 m; 8 days |
i.p., total dose 0, 400 nmol/mouse |
2 applications: 3/7 and 4/7 of the total dose at days 8 and 15 injected; 12 months |
Liver adenomas: 2/18, 21/21*** |
+ |
No data on toxicity; solvent DMSO; caloric restriction inhibits tumour formation |
Fu et al. (1994a) |
|
>99% |
Mouse, B6C3F1; |
i.p., total dose 0, 400 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose on days 1, 8 and 15, respectively; |
Liver adenomas: 2/19, 19/19***; liver carcinomas: 0/19, 14/19***; lung tumours: 1/19, 11/19** |
+ |
Mortality: 0%, 21%; no effects on body weight; solvent DMSO; only liver and lung tumours studied |
von Tungeln et al. (1994b, 1999b) |
|
>99% |
Mouse, B6C3F1; |
i.p., total dose 0, 400 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose on days 1, 8 and 15, respectively; |
Liver adenomas: 2/24, 5/22; liver carcinomas: 1/24, 20/22***; lung tumours: 0/24, 13/22*** |
+ |
Mortality: 0%, 8%; no effects on body weight; solvent DMSO; only liver and lung tumours studied |
von Tungeln et al. (1999b) |
|
n.g. |
Mouse; C57BL/6; 15–19 m; 8 days |
i.p., 0, 7.8, 15.7 mg/kg bw |
2 applications at days 8 and 15; 12 months |
Liver tumour incidence: 0/17, 15/18*, 16/18* |
+ |
No data on toxicity; solvent DMSO; further test groups (sacrificed after 7 months after exposure) indicate that a deficiency in the p53 suppressor gene does not accelerate tumorigenesis |
Dass et al. (1999) |
|
>99.5% |
Rat; Crj:CD (SD); control 40 m and 29 f, treated 31 m and 32 f; newborn |
i.p., total dose 0 or 14.8 µmol/rat |
5 applications: 1/37, 2/37, 4/37, 10/37, 20/37 of the total dose at days 1, 8, 15, 22, 29 injected; |
Colon adenocarcinomas: |
+ |
No data on toxicity; solvent DMSO |
Imaida et al. (1992) |
|
1-Nitrobenzo[a]pyrene |
|||||||
|
Purified (no further data) |
Rat; F344/DuC1; |
s.c., 0, 0.5, 1, 2 mg/rat (~0, 2.5, 5, 10 mg/kg bw) |
Single injection; |
Local subcutaneous tumours: |
(–) |
No effect on body weight (no data on mortality); TS in beeswax–tricaprylin suspended; positive control |
Horikawa et al. (1998) |
|
>99% |
Mouse; B6C3F1; |
i.p., total dose 0, 400 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose on days 1, 8 and 15, respectively; |
Liver adenomas: 2/19, 6/23; liver carcinomas: 0/19, 0/23; lung tumours: 1/19, 0/23 |
(–) |
Mortality: 0%, 4%; no effects on body weight; solvent DMSO; only liver and lung tumours studied; carcinogenic effects not significant |
von Tungeln et al. (1994b, 1999b) |
|
>99% |
Mouse; CD-1; |
i.p., total dose 0, 100 nmol/mouse |
3 applications on days 1, 8 and 15; |
No significant carcinogenic effects in liver and lung |
(–) |
Mortality not compound related (no further data); low dose tested; metabolite 1-nitrobenzo[a]pyrene-trans-7,8-dihydrodiol also negative |
von Tungeln et al. (1999a) |
|
2-Nitrobenzo[a]pyrene |
|||||||
|
>99% |
Mouse, B6C3F1; |
i.p., total dose 0, 400 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose on days 1, 8 and 15, respectively; |
Liver adenomas: 2/19, 21/21***; liver carcinomas: 0/19, 15/21***; lung tumours: 1/19, 4/21 |
+ |
Mortality: 0%, 9%; no effects on body weight; solvent DMSO; only liver and lung tumours studied |
von Tungeln et al. (1994b, 1999b) |
|
3-Nitrobenzo[a]pyrene |
|||||||
|
Purified (no further data) |
Rat; F344/DuC1; |
s.c., 0, 0.5, 1, 2 mg/rat (~0, 2.5, 5, 10 mg/kg bw) |
Single injection; |
Local subcutaneous tumours: |
(–) |
No effect on body weight (no data on mortality) |
Horikawa et al. (1998) |
|
>99% |
Mouse; B6C3F1; |
i.p., total dose 0, 400 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose on days 1, 8 and 15, respectively; |
Liver adenomas: 2/19, 5/24; liver carcinomas: 0/19, 1/24; lung tumours: 1/19, 0/24 |
(–) |
Mortality: 0%, 0%; no effects on body weight; solvent DMSO; only liver and lung tumours studied |
von Tungeln et al. (1994b, 1999b) |
|
>99% |
Mousel CD-1; |
i.p., total dose 0, 100 nmol/mouse |
3 applications on days 1, 8 and 15; 12 months |
No significant carcinogenic effects in liver and lung |
(–) |
Mortality not compound related (no further data); low dose tested; metabolite 3-nitrobenzo[a]pyrene-trans-7,8-dihydrodiol also negative |
von Tungeln et al. (1999a) |
|
6-Nitrobenzo[a]pyrene |
|||||||
|
>99% |
Mouse; Crl/CD-1 (ICR)BR; 20 f; 50–55 days |
Dermal, 0, 0.1 mg/mouse per day |
10 days; 25 weeks |
No significant effect on skin tumour formation |
(–) |
No data on toxicity; initiation/promotion study; 10 days after initiation was complete, 2.5 µg tetradecanoylphorbol acetate applied thrice weekly for 25 weeks (promotion); vehicle acetone |
El-Bayoumy et al. (1982) |
|
>99% |
Mouse; CD-1; |
i.p., total dose 0, 560 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; 12 months |
Liver tumours: |
+ |
54–85% survived to weaning (no further data on toxicity); vehicle DMSO; tumour incidence from weaning to month 12 recorded; parent PAH was more effective |
Wislocki et al. (1986) |
|
>99.9% |
Mouse; BLU:Ha; control 91 m and 101 f, treated 23–24 m and 26–30 f; |
i.p., total dose 0, 14, 70 µg/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; 26 weeks |
No effects on lung tumour formation in contrast to parent PAH (equimolar dose) |
(–) |
No data on toxicity; solvent DMSO; only lung tumours studied |
Busby et al. (1989) |
|
1-Nitrobenzo[e]pyrene |
|||||||
|
>99% |
Mouse; B6C3F1; |
i.p., total dose 0, 400 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose on days 1, 8 and 15, respectively; |
Liver adenomas: 2/19, 4/24; liver carcinomas: 0/19, 1/24; lung tumours: 1/19, 0/24 |
(–) |
Mortality: 0%, 0%; no effects on body weight; solvent DMSO; only liver and lung tumours studied |
von Tungeln et al. (1994b, 1999b) |
|
3-Nitrobenzo[e]pyrene |
|||||||
|
>99% |
Mouse, B6C3F1; |
i.p., total dose 0, 400 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose on days 1, 8 and 15, respectively; |
Liver adenomas: 2/19, 4/24; liver carcinomas: 0/19, 0/24; lung tumours: 1/19, 1/24 |
(–) |
Mortality: 0%, 0%; no effects on body weight; solvent DMSO; only liver and lung tumours studied |
von Tungeln et al. (1994b, 1999b) |
|
1,6-Dinitrobenzo[a]pyrene |
|||||||
|
n.g. |
Rat; F344/DuCrj; |
s.c., 0, 8, 40, 200, 1000 µg/rat |
Single injection; |
No subcutaneous tumours at the site of injection in contrast to the parent PAH |
(–) |
No data on toxicity; TS dissolved in a mixture of equal volumes beeswax and tricaprylin |
Horikawa et al. (1993); Tokiwa et al. (1994) |
|
99.98% |
Rat; F344/DuC1; |
s.c., 0, 8, 40, 200, 1000 µg/rat (~0, 0.04, 0.2, 1, 5 mg/kg bw) |
Single injection; |
Subcutaneous malignant fibrous histiocytoma: |
(–) |
No effect on body weight (no data on mortality); TS suspended in beeswax–tricaprylin; positive control |
Horikawa et al. (1998) |
|
3,6-Dinitrobenzo[a]pyrene |
|||||||
|
n.g. |
Rat; F344/DuCrj; 20–21 m; 6 weeks |
s.c., 0, 8, 40, 200, 1000 µg/rat |
Single injection; |
Subcutaneous malignant fibrous histiocytomas at the site of injection #: 0/20, 1/21, 5/21, 8/21, 14/20 |
+ |
No data on toxicity; TS dissolved in a mixture of equal volumes beeswax and tricaprylin |
Horikawa et al. (1993); Tokiwa et al. (1994) |
|
99.99% |
Rat; F344/DuC1; |
s.c., 0, 8, 40, 200, 1000 µg/rat (~0, 0.04, 0.2, 1, 5 mg/kg bw) |
Single injection; |
Subcutaneous malignant fibrous histiocytoma: |
+ |
Dose-dependent reduction in body weight in all treatment groups, presumably due to tumour formation (no data on mortality); |
Horikawa et al. (1998) |
|
7-Nitrodibenz[a,h]anthracene |
|||||||
|
n.g. |
Mouse, B6C3F1; |
i.p., total dose 0, 400 nmol/mouse |
3 applications on days 1, 8 and 15; n.g. |
Increased incidence of liver tumours (no further data); parent PAH more active (100% liver tumour incidence) |
(+) |
Solvent DMSO; only liver tumours studied; data from abstract, no further information available |
von Tungeln et al. (1994b) |
|
n.g. |
Mouse, B6C3F1; |
i.p., total dose 0, 400 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose at days 1, 8 and 15 injected; 12 months |
Liver adenomas: 2/24, 12/24**; |
+ |
No treatment-related effect on mortality or body weight; solvent DMSO; parent PAH more effective |
Fu et al. (1998b) |
|
9-Nitrodibenz[a,c]anthracene |
|||||||
|
>99% |
Mouse, B6C3F1; |
i.p., total dose 0, 400 nmol/mouse |
3 applications: 1/7, 2/7 and 4/7 of the total dose on days 1, 8 and 15, respectively; |
Liver adenomas: 2/24, 2/24; liver carcinomas: 1/24, 0/24; lung tumours: 0/24, 0/24 |
(–) |
Mortality: 0%, 0%; no effects on body weight; solvent DMSO; only liver and lung tumours studied |
von Tungeln et al. (1994b, 1999b) |
|
3-Nitroperylene |
|||||||
|
>99% |
Mouse; Crl/CD-1 (ICR)BR; |
Dermal, 0, 0.1 mg/mouse per day |
10 days; 25 weeks |
Local skin tumours: |
+ |
No data on toxicity; initiation/promotion study; 10 days after initiation was complete, 2.5 µg tetradecanoylphorbol acetate applied thrice weekly for 25 weeks (promotion); vehicle acetone |
El-Bayoumy et al. (1982) |
|
a |
All studies used vehicle-treated controls unless otherwise stated; m = male; f = female; DMSO = dimethyl sulfoxide; # = no data on statistical evaluation; MTD: maximum tolerated dose; n.g. = not given; TS: test substance; significant compared with control, * (P < 0.05), ** (P < 0.01), *** (P < 0.001); n.g.= data not given. |
|
b |
Age of animals at start of the experiment. |
|
c |
The Task Group evaluated the strength of each of the in vivo studies based on the following criteria: a) numbers of animals in each group, b) duration of study, c) adequate dosing (e.g., MTD in negative studies) and d) appropriateness of controls. Specific comments on the strengths and weaknesses of the studies are included in the table. The Task Group recognized that some of the studies did not report statistical evaluations, and this is noted in the table: |
|
+, study positive and acceptable to Task Group (data are not from an abstract, more than 10 animals per group per sex, and included an appropriate control) for assessment of carcinogenicity |
|
|
(+), study claimed positive results; however, the Task Group considered the data "less than acceptable" for assessment (data from abstract, less than 10 animals per group per sex and/or no control) |
|
|
–, although none of the studies met this criterion, study negative and acceptable to Task Group (data are from manuscript, >25 surviving (2 years) animals per group per sex, and included a control) for assessment of carcinogenicity |
|
|
(–), study claimed negative results; however, the Task Group considered the data "less than acceptable" for assessment (data from abstract, insufficient number of animals per group per sex and/or no control). |
No effect on survival or body weight was observed in rats after repeated s.c. application (twice weekly for 10 weeks) of 20 mg/kg bw (Ohgaki et al., 1985).
Rats treated via gavage with about 3 mg/kg bw 3 times weekly for 4 weeks showed no effects on body weight or survival (King, 1988).
Local effects preceding tumour formation were noted in rats after repeated s.c. injection of 0.2 mg per animal. Rats showed ulcer and scar formation at the site of injection (Ohgaki et al., 1984).
Imaida et al. (1991a) reported no effects on body weight or survival in a gavage study on rats. Rats were treated 3 times per week for 4 weeks at a dose level of 3 mg/kg bw.
Ulcer and scar formation at the site of injection were noted in rats after repeated s.c. injection of 0.2 mg per animal (Ohgaki et al., 1984).
In the mouse newborn assay (Wislocki et al., 1986), the total dose of 2.8 µmol per mouse resulted in increased mortality shortly after the exposure period.
Subcutaneous bolus injection of 2.5–10 mg/kg bw did not affect body weight gain in rats (Horikawa et al., 1998).
In the same study (Horikawa et al., 1998), s.c. bolus injection of 0.04–5 mg/kg bw also resulted in no changes in body weight gain in rats.
No studies are available.
No studies are available.
The Task Group was aware that the calculations for mutagenic potency could only be estimated and were not exact, due to the following shortcomings: (1) uncontrolled interlaboratory variation among protocols; (2) absence of information about the mutability of the bacterial strains used in several laboratories (lack of use of different positive controls); and (3) differences in the purity of the nitroPAH (e.g., contamination by dinitroPAHs). The Task Group recognized that, historically, contamination of a mononitroPAH with small amounts of dinitroPAHs has dramatically affected the reported mutagenicity of the particular mononitroPAH (for discussion, see Rosenkranz & Mermelstein, 1983; Tokiwa & Ohnishi, 1986). For instance, the average mutagenicity of 1-nitropyrene (690 revertants/nmol in TA98; Table 40) is approximately 400-fold less than the mutagenicity of 1,6- or 1,8-dinitropyrene (average approximately 235 000 revertants/nmol; Table 40). Therefore, considering the mutagenicity of highly purified 1-nitropyrene was reported as 490 revertants/nmol in Tokiwa & Ohnishi (1986) and that for the 1,6- and 1,8-dinitropyrene averaged 216 000 revertants/nmol in TA98, the mutagenicity of 1400 revertants/nmol for 1-nitropyrene in TA98 reported by Ho et al. (1981) could be due to as little as 0.4% contamination with dinitropyrenes. Historically, the use of strains TA98NR– (nitroreductase deficient) and TA98AT– (O-acetyltransferase deficient) has allowed an assessment of the relative level of contamination.
Table 40. Summary of mutagenic potency of nitroPAHs in Salmonella microsome assay
|
Substancea (referencesb) |
Revertants per nmol nitroPAH in Salmonella typhimurium strains with and without metabolic activation (MA)c |
Predominant type of histidine mutatione |
|||||||||
|
TA98d |
TA100d |
TA1535d |
TA1537d |
TA1538d |
|||||||
|
–MA |
+MA |
–MA |
+MA |
–MA |
+MA |
–MA |
+MA |
–MA |
+MA |
||
|
1-Nitronaphthalene (a–l) |
0–1.0f |
0.1–2.7 |
0.8–9.9 |
0.55–6.3 |
0–0.05 |
0.04–0.07 |
0–<0.01 |
0 |
0.05–0.24 |
0.13 |
base pair substitution |
|
2-Nitronaphthalene (b–f, h, j, m–z) |
0.12–1.0g |
0.09–0.69 |
1.1–8.7 |
1.1–4.4 |
0.3–3.7 |
|
0.03–0.15 |
0.02–0.4 |
|
base pair substitution |
|
|
1,3-Dinitronaphthalene (h, s) |
0.89–0.9 |
6.7–7.3 |
0.15–3.1 |
0.7–0.76 |
base pair substitution |
||||||
|
1,5-Dinitronaphthalene (h, k, s, aa) |
2.2–4.0 |
|
4.7–14 |
|
0h |
|
base pair substitution |
||||
|
1,8-Dinitronaphthalene (h, k, aa) |
4.4–7.9 |
|
|
|
0h |
0h |
base pair substitution |
||||
|
2,3,5-Trinitronaphthalene (bb, cc) |
32–65 |
|
|
|
|
|
frameshift |
||||
|
1,3,6,8-Tetranitronaphthalene (h, cc) |
0.24–30 |
|
|
|
|
|
_* i |
||||
|
3-Nitroacenaphthene (l) |
|
|
<0.01 |
|
|
base pair substitution |
|||||
|
5-Nitroacenaphthene (a, g, k, l, r, dd–hh) |
1.6–30 |
4.3–25 |
2.0–40 |
14–71 |
<0.01–0.6 |
0.27–0.32 |
0.1–1.54 |
1.1–1.2 |
0.78–8.3 |
6.1–27 |
_* i |
|
2-Nitrofluorene (e–g, j–l, o, r, t, u–x, z, aa, ii, jj, ll–tt, vv–zz) |
2.8–430 |
6–72 |
4.6–200 |
7.5–35 |
0–0.01 |
0.04–1 |
6.5–270 |
15–35 |
_* i |
||
|
3-Nitrofluorene (yy) |
7.4 |
||||||||||
|
2,5-Dinitrofluorene (g, xx) |
1600–2500 |
|
|
|
|
frameshift |
|||||
|
2,7-Dinitrofluorene (g, k, ll, pp, qq, xx, A) |
470–5200 |
38–2600 |
6–174 |
14–90 |
0h |
290–310 |
350–3000 |
frameshift |
|||
|
2-Nitroanthracene (j, B) |
890–1600 |
|
|
|
|
_* i |
|||||
|
9-Nitroanthracene (g, k, p, qq, ss, tt, C, D, F) |
0.01–2.7 |
0.07–2.0 |
0.89–3.6 |
0.89–3.0 |
base pair substitution |
||||||
|
9,10-Dinitroanthracene (ff) |
0 |
<1 |
_* i |
||||||||
|
1-Nitrophenanthrene (G) |
110 |
330 |
base pair substitution |
||||||||
|
2-Nitrophenanthrene (j, ll) |
130–330 |
|
|
|
frameshift |
||||||
|
3-Nitrophenanthrene (G) |
330 |
620 |
_* i |
||||||||
|
9-Nitrophenanthrene (ll, vv, D, G) |
<0.45–290 |
|
<0.89–981 |
1.6–22 |
base pair substitution |
||||||
|
1,5-Dinitrophenanthrene (G) |
4 |
5 |
_* i |
||||||||
|
1,6-Dinitrophenanthrene (G) |
120 |
1200 |
base pair substitution |
||||||||
|
1,10-Dinitrophenanthrene (G) |
0.8 |
2 |
base pair substitution |
||||||||
|
2,6-Dinitrophenanthrene (G, H) |
730 |
1800 |
base pair substitution |
||||||||
|
2,7-Dinitrophenanthrene (ll) |
3900 |
_* i |
|||||||||
|
2,9-Dinitrophenanthrene (G) |
3 |
590 |
base pair substitution |
||||||||
|
2,10-Dinitrophenanthrene (G) |
120 |
240 |
base pair substitution |
||||||||
|
3,5-Dinitrophenanthrene (G) |
70 |
240 |
base pair substitution |
||||||||
|
3,6-Dinitrophenanthrene (G) |
90 |
2100 |
base pair substitution |
||||||||
|
3,10-Dinitrophenanthrene (G) |
110 |
1400 |
base pair substitution |
||||||||
|
4,9-Dinitrophenanthrene (G) |
1 |
2 |
base pair substitution |
||||||||
|
4,10-Dinitrophenanthrene (G) |
5.3 |
160 |
base pair substitution |
||||||||
|
1,5,9-Trinitrophenanthrene (G) |
170 |
110 |
_* i |
||||||||
|
1,5,10-Trinitrophenanthrene (G) |
14 |
4 |
frameshift |
||||||||
|
1,6,9-Trinitrophenanthrene (G) |
360 |
720 |
base pair substitution |
||||||||
|
1,7,9-Trinitrophenanthrene (G) |
1000 |
1300 |
_* i |
||||||||
|
2,5,10-Trinitrophenanthrene (G) |
63 |
370 |
base pair substitution |
||||||||
|
2,6,9-Trinitrophenanthrene (G) |
180 |
1200 |
base pair substitution |
||||||||
|
3,5,10-Trinitrophenanthrene (G) |
50 |
44 |
_* i |
||||||||
|
3,6,9-Trinitrophenanthrene (G) |
530 |
1300 |
base pair substitution |
||||||||
|
1-Nitrofluoranthene (l, gg, D, I) |
74–540 |
|
124–988 |
<0.1 |
|
|
_* i |
||||
|
2-Nitrofluoranthene (zz, I–L) |
61–1000 |
89–760 |
_* i |
||||||||
|
3-Nitrofluoranthene (f, k, l, aa, qq, vv, ww, D, H–P) |
1400–14 000 |
31–590 |
747–5800 |
31–640 |
<1–25 |
|
46–980 |
|
810–4500 |
12–338 |
frameshift |
|
7-Nitrofluoranthene (l, gg, D, I) |
4–544 |
|
123–990 |
<0.1 |
|
|
base pair substitution |
||||
|
8-Nitrofluoranthene (l, D, I, L, M) |
2200–18 000 |
|
400–750 |
|
<25 |
<123–210 |
9900–14 000 |
frameshift |
|||
|
1,2-Dinitrofluoranthene (I, L) |
1000–1300 |
180–190 |
_* i |
||||||||
|
1,3-Dinitrofluoranthene (I) |
2600 |
470 |
_* i |
||||||||
|
2,3-Dinitrofluoranthene (L) |
420 |
80 |
_* i |
||||||||
|
2,4-Dinitrofluoranthene (L) |
6000 |
550 |
_* i |
||||||||
|
2,5-Dinitrofluoranthene (L) |
210 |
540 |
_* i |
||||||||
|
3,4-Dinitrofluoranthene (H, O–Q) |
4100–4500 |
|
3200–3300 |
|
0h |
0h |
|
|
440–610 |
|
_* i |
|
3,7-Dinitrofluoranthene (H, O, P) |
|
|
25 000–31 000 |
|
0h |
0h |
4900–6800 |
|
13 000–19 000 |
|
frameshift |
|
3,9-Dinitrofluoranthene (H, O, P) |
|
|
|
|
0h |
0h |
2800–3200 |
|
|
|
frameshift |
|
1,2,4-Trinitrofluoranthene (L) |
3600 |
140 |
_* i |
||||||||
|
1,2,5-Trinitrofluoranthene (L) |
1400 |
69 |
_* i |
||||||||
|
2,3,5-Trinitrofluoranthene (L) |
2700 |
720 |
_* i |
||||||||
|
1-Nitropyrene (d–f, k, o, p, q, s, v–z, aa, ll, qq, ss, vv, ww, B, D, E, H, J, M, N, P, R–Z, AA–OO) |
50–4400 |
17–1700 |
41–910 |
29–98 |
0h |
0h |
1–500 |
70–1200 |
5–87 |
frameshift |
|
|
2-Nitropyrene (ll, oo, pp, D) |
1800–2800 |
|
390–740 |
|
0h |
|
frameshift |
||||
|
4-Nitropyrene (d, H, QQ) |
2500–2700 |
|
frameshift |
||||||||
|
1,3-Dinitropyrene (e, f, q, s, aa, ll, qq, vv, H, M, P, S, FF, HH–KK, RR, SS) |
29 000–16 0000 |
|
8300–37 000 |
|
0h |
3000–13 000 |
11 000–79 000 |
frameshift |
|||
|
1,6-Dinitropyrene (e, f, k, q, s, vv, aa, B, H, M, P, S, U, W, Z, EE, FF, HH–KK, MM, RR–TT) |
25 000–250 000 |
380–37 000 |
4300–33 000 |
61–1300 |
0h |
9200–33 000 |
12 000–61 000 |
frameshift |
|||
|
1,8-Dinitropyrene (e, f, k, q, s, w–z, aa, nn, qq, rr, vv, zz, H, M, P, S, W, Z, BB, FF, HH–KK, UU, RR, SS) |
55 000–2 000 000 |
120–77 000 |
5500– |
42–4400 |
0h |
12 000–21 000 |
9900–35 000 |
frameshift |
|||
|
2,7-Dinitropyrene (ll) |
38 000 |
_* i |
|||||||||
|
1,3,6-Trinitropyrene (e, f, s, vv, FF, HH–KK) |
17 000–240 000 |
1100–40 000 |
0h |
|
16 000–20 000 |
frameshift |
|||||
|
1,3,6,8-Tetranitropyrene (f, q, s, vv, FF, HH–KK) |
3200–84 000 |
100–13 000 |
0h |
|
|
frameshift |
|||||
|
7-Nitrobenz[a]anthracene (D, QQ, VV) |
<1 |
|
0.55–<1 |
|
base pair substitution |
||||||
|
2-Nitrochrysene (gg, PP) |
|
27–450 |
|
100–350 |
frameshift |
||||||
|
5-Nitrochrysenej (D) |
<0.55 |
2.7 |
2.7 |
5.5 |
base pair substitution |
||||||
|
6-Nitrochrysene (d, k, qq, vv, ww, D, VV–XX) |
5–270 |
12–110 |
38–270 |
40–410 |
|
|
base pair substitution |
||||
|
3-Nitrobenzo(k)-fluoranthene (l) |
580 |
990 |
<1 |
35 |
150 |
_* i |
|||||
|
7-Nitrobenzo(k)-fluoranthene (l) |
<0.01 |
<0.01 |
<0.01 |
<0.01 |
<0.01 |
_* i |
|||||
|
1-Nitrobenzo[a]pyrene (d, rr, H, QQ, YY, ZZ, aaa, bbb) |
73–840 |
150–920 |
34–2380 |
|
_* i |
||||||
|
2-Nitrobenzo[a]pyrene (PP) |
2500 |
610 |
190 |
720 |
frameshift |
||||||
|
3-Nitrobenzo[a]pyrene (d, rr, H, QQ, YY, aaa–fff) |
93–1900 |
250–861 |
28–3100 |
26–41 |
_* i |
||||||
|
6-Nitrobenzo[a]pyrene (k, rr–tt, D–F, VV, YY, bbb, ggg, hhh) |
<1–68 |
30–440 |
<1–59 |
19–440 |
|
|
_* i |
||||
|
1-Nitrobenzo[e]pyrene (D, Y, iii) |
1.3–39 |
11–45 |
|
|
<1.5 |
|
|
_* i |
|||
|
3-Nitrobenzo[e]pyrene (D, Y, QQ) |
890–3100 |
|
|
|
<1.5 |
<30 |
<30 |
frameshift |
|||
|
4-Nitrobenzo[e]pyrene (d) |
980 |
300 |
frameshift |
||||||||
|
1,3-Dinitrobenzo[e]pyrene (d) |
8500 |
_* i |
|||||||||
|
1,6-Dinitrobenzo[e]pyrene (Y) |
98 |
92 |
_* i |
||||||||
|
1,8-Dinitrobenzo[e]pyrene (Y) |
0h |
0h |
_* i |
||||||||
|
3,6-Dinitrobenzo[a]-pyrene (H) |
140 000 |
_* i |
|||||||||
|
3-Nitrodibenz[a,h]-anthracene (PP) |
320 |
960 |
<1 |
560 |
_* i |
||||||
|
7-Nitrodibenz[a,h]-anthracene (d) |
<1 |
<1 |
_* i |
||||||||
|
9-Nitrodibenz[a,c]-anthracene (d) |
<1 |
<1 |
_* i |
||||||||
|
3-Nitroperylene (p, tt, D, E, bbb, ggg) |
4.4–30 |
260–3600 |
|
890–1000 |
0h |
|
|
110–500 |
_* i |
||
|
3,6-Dinitroperylene (bbb) |
1500 |
1600 |
270 |
890 |
_* i |
||||||
|
3,7-Dinitroperylene (bbb) |
1600 |
<68 |
240 |
<34 |
frameshift |
||||||
|
1-Nitrocoronene (D) |
2.8 |
6.9 |
1.4 |
3.5 |
_* i |
||||||
|
4-Nitrobenzo[ghi]-perylene (gg) |
1900 |
49 |
frameshift |
||||||||
|
7-Nitrobenzo[ghi]-perylene (gg) |
<0.6 |
<0.6 |
_* i |
||||||||
|
a |
Substances with adequate database (>5 data with at least one strain, at least 3 data with the strain showing the maximal mutagenic activity) marked by bold-typed substance name. |
|
b |
Studies used for calculation of means are as follows: |
|
a) Dunkel et al. (1985); b) El-Bayoumy et al. (1981); c) Gupta et al. (1996); d) Jung et al. (1991); e) Klopman et al. (1984); f) Massaro et al. (1983); g) Matsuda (1981); h) McCoy et al. (1981a) ; i) Mortelmans et al. (1987); j) Scribner et al. (1979); k) Tokiwa et al. (1981b); l) Vance & Levin (1984); m) De Flora (1979); n) De Flora et al. (1984); o) Hagiwara et al. (1993); p) Ho et al. (1981); q) Löfroth (1981); r) McCann et al. (1975); s) Mermelstein et al. (1982); t) Rosenkranz & Poirier (1979); u) Simmon (1979a); v) Wang et al. (1980); w) Watanabe et al. (1989); x) Watanabe et al. (1990) ; y) Watanabe et al. (1993) ; z) Yamada et al. (1997); aa) Tokiwa et al. (1985); bb) Sorenson et al. (1983); cc) Whong & Edwards (1984); dd) McCoy et al. (1983a); ee) McCoy et al. (1983b); ff) Möller et al. (1985); gg) Rosenkranz & Mermelstein (1983); hh) Yahagi et al. (1975); ii) Castellino et al. (1978); jj) Cui et al. (1996); kk) Edenharder & Tang (1997); ll) Hirayama et al. (1988); mm) Hughes et al. (1997); nn) Jurado et al. (1994); oo) LaVoie et al. (1981); pp) McCoy et al. (1981b); qq) Pederson & Siak (1981); rr) Pitts et al. (1984); ss) Pitts et al. (1982); tt) Pitts (1983); uu) Schleibinger et al. (1989) ; vv) Sugimura & Takayama (1983); ww) Tokiwa et al. (1981a); xx) Vance et al. (1987); yy) Watanabe et al. (1997a); zz) Zielinska et al. (1987); A) Levin et al. (1979); B) Fu et al. (1986); C) Fu et al. (1985a); D) Greibrokk et al. (1984); E) Pitts et al. (1978); F) Wang et al. (1978); G) Sera et al. (1996); H) Tokiwa et al. (1994); I) Shane et al.. (1991); J) Ball et al. (1994); K) Ball et al. (1995); L) Zielinska et al. (1988) ; M) Ball & Young (1992); N) Sangaiah et al. (1996) ; O) Nakagawa et al. (1987); P) Tokiwa et al. (1986); Q) Horikawa et al. (1987); R) Ball et al. (1984b); S) Crebelli et al. (1995); T) DeMarini et al. (1996); U) El-Bayoumy & Hecht (1986); V) El-Bayoumy & Hecht (1983); W) Fifer et al. (1986a); X) Fifer et al. (1986b); Y) Fu et al. (1989); Z) González de Mejía et al. (1998); AA) Harris et al. (1984); BB) Heflich et al. (1985a); CC) King et al. (1984); DD) Lee et al. (1994); EE) Manabe et al. (1985); FF) McCoy et al. (1985a); GG) McCoy et al. (1984); HH) Mermelstein et al. (1981); JJ) Nakayasu et al. (1982); KK) Rosenkranz et al. (1980); LL) Taylor et al. (1995); MM) Tokiwa et al. (1984); NN) Urios et al. (1994); OO) Yu et al. (1991); PP) Yu et al. (1992); QQ) Fu et al. (1985b); RR) Shah et al. (1991); SS) Shane & Winston (1997); TT) Ashby et al. (1983); UU) Rosenkranz et al. (1982); VV) Fu et al. (1988b); WW) El-Bayoumy & Hecht (1984b); XX) El-Bayoumy et al. (1989b); YY) Chou et al. (1984); ZZ) Chou et al. (1986); aaa) Hass et al. (1986a); bbb) Löfroth et al. (1984); ccc) Chou et al. (1985); ddd) Fu et al. (1997); eee) Hass et al. (1984); fff) Heflich et al. (1989); ggg) Anderson et al. (1987); hhh) Fu et al. (1982); iii) Fu et al. (1988c). |
|
|
c |
Study results revealing no mutagenicity were not included (exception see below) if the strain was presumably not tested up to cytotoxicity; study results with no explicitness (e.g., < or > 1) not used for calculation of the mean; MA = metabolic activation (rodent liver microsomes). |
|
d |
In each of these columns, the following values are presented (separated in data with or without MA): a) range of mutagenic potency in revertants per nmol (2 or more data available), b) mean number of revertants per nmol and c) in parentheses the number n of data used for calculation of the mean. |
|
e |
Bold-typed letters indicate clearly more revertants (difference at least 10-fold) in strains detecting base pair substitutions (TA100, TA1535) relative to strains detecting frameshift mutations (TA98, TA1537, TA1538) or the reversal (comparison in dependence on metabolic activation); a difference of at least 2-fold is marked by normal-typed letters. |
|
f |
30 revertants/nmol after 90-min preincubation. |
|
g |
680 revertants/nmol after 90-min preincubation. |
|
h |
Presumably not tested up to cytotoxicity (included in the table if no other data were available). |
|
i |
_* = less than 2-fold increase. |
|
j |
Structural assignment not given. |
|
k |
In further studies, no mutagenicity was observed without MA, but the substance was not tested up to cytotoxicity. |
Details on the genotoxicity of nitroPAHs in vitro are presented in four tables. The results of the numerous studies on the Salmonella microsome assay (Maron & Ames, 1983) with nitroPAHs are summarized in Table 40. Data on the genotoxicity of nitroPAHs in other bacterial systems are documented in Table 41. Data on eukaryotic test systems, including human cells, are presented in Tables 42 and 43 (genotoxicity) and 44 (cell transformation). A summary of all data on genotoxicity in vitro is given in Table 45. The final evaluation of genotoxicity and a scaling of the mutagenic potency in the Salmonella microsome assay are presented in Table 46.
Table 41. Genotoxicity of nitroPAHs in bacteria, other than the Salmonella microsome test
|
Substance |
Test type |
Bacteriaa |
Resultsb |
Reference |
|
|
–MAc |
+MAc |
||||
|
1-Nitronaphthalene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
Reverse mutation assay |
E. coli WP2uvrA (trp+ reversion) |
– |
± |
Dunkel et al. (1985) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (umu gene expression) |
(±) |
n.g. |
Nakamura et al. (1987) |
|
|
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
– |
(±) |
Mersch-Sundermann et al. (1991, 1992) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 |
+d |
n.g. |
Oda et al. (1992, 1993) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 |
+ |
n.g. |
Schmid et al. (1997) |
|
|
2-Nitronaphthalene |
DNA damage/repair (rec assay) |
E. coli polA– (DNA repair deficient) or polA+ (proficient) |
+ |
n.g. |
Rosenkranz & Poirier (1979) |
|
DNA damage/repair (rec assay) |
E. coli WP67 (DNA repair deficient) or WP2 (proficient) |
+ |
+ |
De Flora et al. (1984) |
|
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (umu gene expression) |
+ |
n.g. |
Nakamura et al. (1987) |
|
|
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
+ |
(±) |
Mersch-Sundermann et al. (1991, 1992) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 |
+d |
n.g. |
Oda et al. (1992, 1993) |
|
|
1,3-Dinitronaphthalene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
1,5-Dinitronaphthalene |
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
– |
+ |
Mersch-Sundermann et al. (1991, 1992) |
|
DNA damage (prophage induction) |
E. coli (induction of prophage in lysogenic strain B/r.WP2(lambda), indicator strain SR714) |
+ |
n.g. |
Rossmann et al. (1991) |
|
|
2,7-Dinitronaphthalene |
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
+ |
(±) |
Mersch-Sundermann et al. (1991, 1992) |
|
2,3,5-Trinitronaphthalene |
Reverse mutation assay |
E. coli WP2uvrA (trp+ reversion) |
+ |
n.g. |
Sorenson et al. (1983) |
|
5-Nitroacenaphthene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
Reverse mutation assay |
E. coli WP2uvrA (trp+ reversion) |
(±) |
+ |
McCoy et al. (1983b) |
|
|
Reverse mutation assay |
E. coli WP2uvrA (trp+ reversion) |
– |
+ |
Dunkel et al. (1985) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 |
+d |
n.g. |
Oda et al. (1992, 1993) |
|
|
2-Nitrofluorene |
DNA cell binding |
E. coli Q13 |
+ |
+e |
Kubinski et al. (1981) |
|
DNA damage (K12 inductest) |
E. coli (induction of prophage in lysogenic strain GY5027, indicator strain GY4015) |
– |
– |
Mamber et al. (1984) |
|
|
DNA damage (prophage induction) |
E. coli K12 (lysogen; induction of prophage lambdaclts857) |
+ |
+e |
Ho & Ho (1981) |
|
|
DNA damage (prophage induction) |
E. coli (induction of prophage in lysogenic strain B/r.WP2(lambda), indicator strain SR714) |
+ |
n.g. |
Rossmann et al. (1991) |
|
|
DNA damage/repair (rec assay) |
E. coli polA– (DNA repair deficient) or polA+ (proficient) |
+ |
n.g. |
Rosenkranz & Poirier (1979) |
|
|
DNA damage/repair (rec assay) |
E. coli WP100 (DNA repair deficient) or WP2 (proficient) |
– |
+ |
McCarroll et al. (1981a) |
|
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
– |
+ |
McCarroll et al. (1981b) |
|
|
DNA damage/repair (rec assay) |
E. coli WP100 (DNA repair deficient) or WP2 (proficient) |
+ |
n.g. |
Doudney et al. (1981) |
|
|
DNA damage/repair (rec assay) |
E. coli JC (DNA repair deficient) or AB1157 (proficient) |
n.g. |
± |
Suter & Jaeger (1982) |
|
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
n.g. |
+ |
Suter & Jaeger (1982) |
|
|
DNA damage/repair (rec assay) |
E. coli WP100 (DNA repair deficient) or WP2 (proficient) |
+ |
n.g. |
Mamber et al. (1983, 1984) |
|
|
Forward mutation assay |
S. typhimurium TA1537, 100, 98 (resistance to 8-azaguanine) |
– |
n.g. |
Castellino et al. (1978) |
|
|
Forward mutation assay |
S. typhimurium TA1538, 1535 (resistance to 8-azaguanine) |
+ |
n.g. |
Castellino et al. (1978) |
|
|
Forward mutation assay |
E. coli WP2 (resistance to L-azetidine-2-carboxylic acid) |
+ |
n.g. |
Mitchell (1980) |
|
|
Forward mutation assay |
S. typhimurium BA8 (resistance to L-arabinose) |
+ |
n.g. |
Ruiz-Rubio et al. (1984) |
|
|
Forward mutation assay |
S. typhimurium SV50 (resistance to arabinose) |
– |
n.g. |
Xu et al. (1984) |
|
|
Forward mutation assay |
E. coli WP2 or CM891 (resistance to L-azetidine-2-carboxylic acid) |
(±) |
n.g. |
Mitchell & Gilbert (1985) |
|
|
Forward mutation assay |
S. typhimurium BA3 or BA9 (resistance to L-arabinose) |
+ |
n.g. |
Hera & Pueyo (1986) |
|
|
Reverse mutation assay |
E. coli WP2uvrA (trp reversion) |
+ |
n.g. |
McCoy et al. (1981b) |
|
|
Reverse mutation assay |
E. coli CM891 (trp reversion) |
+ |
n.g. |
Mitchell & Gilbert (1984) |
|
|
Reverse mutation assay |
S. typhimurium BA3, 8 (reversion to histidine auxotrophy) |
+ |
n.g. |
Ruiz-Rubio et al. (1984) |
|
|
Reverse mutation assay |
E. coli WP2 or CM891 (trp reversion, positive only in CM891) |
+ |
n.g. |
Mitchell & Gilbert (1985) |
|
|
Reverse mutation assay |
S. typhimurium TA1538 with plasmid pYG8031, pYG8011, pSE117, pKM101, pBR322 |
+ |
n.g. |
Nohmi et al. (1995) |
|
|
Reverse mutation assay |
E. coli MX100 (reversion of arginine auxotrophy) |
+ |
n.g. |
Kranendonk et al. (1996) |
|
|
Reverse mutation assay |
E. coli strains (reversion to Lac+; induction of frameshifts enhanced in strains with higher acetyltransferase activity) |
+ |
n.g. |
Hoffmann et al. (2001) |
|
|
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
n.g. |
+ |
Ohta et al. (1984) |
|
|
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
+ |
n.g. |
Quillardet et al. (1985) |
|
|
SOS DNA repair (SOS chromotest) |
E. coli K12 PQ37 |
+ |
n.g. |
Mamber et al. (1986) |
|
|
SOS DNA repair (SOS chromotest) |
E. coli K12 PQ37 |
+ |
+ |
Marzin et al. (1986) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (umu gene expression) |
+ |
n.g. |
Nakamura et al. (1987) |
|
|
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
– |
– |
Schleibinger et al. (1989) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 |
+ |
+ |
Schleibinger et al. (1989) |
|
|
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
+ |
(±) |
Mersch-Sundermann et al. (1991, 1992) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (umu gene expression) |
+d |
n.g. |
Oda et al. (1992, 1993, 1996) |
|
|
2,7-Dinitrofluorene |
Reverse mutation assay |
E. coli WP2uvrA (trp+ reversion, no data on cytotoxicity) |
– |
n.g. |
McCoy et al. (1981b) |
|
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
+ |
+e |
Mersch-Sundermann et al. (1991, 1992) |
|
|
9-Nitroanthracene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
– |
– |
Mersch-Sundermann et al. (1991, 1992) |
|
|
2-Nitrofluoranthene |
DNA damage |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Nakagawa et al. (1987) |
|
|
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
+ |
+ |
Schleibinger et al. (1989) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 |
+ |
+f |
Schleibinger et al. (1989) |
|
|
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
+ |
+e |
Mersch-Sundermann et al. (1991, 1992) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 |
+d |
n.g. |
Oda et al. (1992, 1993) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (MA by human CYP1A1, CYP1A2 or CYP1B1 co-expressed with NPR) |
+ |
+f |
Yamazaki et al. (2000) |
|
|
3-Nitrofluoranthene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1986) |
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002; NM2009 & 3009 (acetyltransferase activity |
+ |
n.g. |
Shimada et al. (1994) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (MA by human CYP1A1, CYP1A2 or CYP1B1 co-expressed with NPR) |
+ |
+e |
Yamazaki et al. (2000) |
|
|
8-Nitrofluoranthene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
3,4-Dinitrofluoranthene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Nakagawa et al. (1987) |
|
3,7-Dinitrofluoranthene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1986) |
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Nakagawa et al. (1987) |
|
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 |
+d |
n.g. |
Oda et al. (1992, 1993) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002; NM2009 and 3009 (acetyltransferase activity |
+ |
n.g. |
Shimada et al. (1994) |
|
|
3,9-Dinitrofluoranthene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1986) |
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Nakagawa et al. (1987) |
|
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 |
+d |
n.g. |
Oda et al. (1992, 1993) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002; NM2009 and 3009 (acetyltransferase activity |
+ |
n.g. |
Shimada et al. (1994) |
|
|
1-Nitropyrene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1986) |
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Horikawa et al. (1986) |
|
|
Forward mutation assay |
S. typhimurium TM677 (resistance to 8-azaguanine) |
+ |
+e |
Busby et al. (1994a) |
|
|
Reverse mutation assay |
E. coli WP2uvrA (trp+ reversion) |
– |
n.g. |
Mermelstein et al. (1981) |
|
|
Reverse mutation assay |
E. coli WP2uvrA (trp+ reversion) |
(±) |
n.g. |
Tokiwa et al. (1984) |
|
|
Reverse mutation assay |
E. coli WP2uvrA, 3001-04 (trp+ reversion; positive only in 3003 and 3004g) |
(±) |
n.g. |
McCoy et al. (1985b) |
|
|
Reverse mutation assay |
S. typhimurium TA1538 with plasmid pYG8031, pYG8011, pSE117, pKM101, pBR322 |
+ |
n.g. |
Nohmi et al. (1995) |
|
|
SOS DNA repair (SOS umu test) |
E. coli PQ37 |
+ |
n.g. |
Ohta et al. (1984) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (umu gene expression) |
+ |
n.g. |
Nakamura et al. (1987) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 |
(±) |
(±) |
Schleibinger et al. (1989) |
|
|
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
+ |
+f |
Schleibinger et al. (1989) |
|
|
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
+ |
+e |
Mersch-Sundermann et al. (1991, 1992) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (umu gene expression) |
+ |
n.g. |
Fretwurst & Ahlf (1996) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (MA by human CYP1A1, CYP1A2 or CYP1B1 co-expressed with NPR) |
+ |
+e |
Yamazaki et al. (2000) |
|
|
2-Nitropyrene |
Forward mutation assay |
S. typhimurium TM677 (resistance to 8-azaguanine) |
– |
+ |
Busby et al. (1994a) |
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (MA by CYP1B1 or CYP1A1, not CYP1A2) |
n.g. |
+ |
Shimada et al. (1996) |
|
|
4-Nitropyrene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Horikawa et al. (1986) |
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
|
Forward mutation assay |
S. typhimurium TM677 (resistance to 8-azaguanine) |
+ |
+e |
Busby et al. (1994a) |
|
|
1,3-Dinitropyrene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Horikawa et al. (1986) |
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1986) |
|
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
|
Forward mutation assay |
S. typhimurium TM677 (resistance to 8-azaguanine) |
+ |
+e |
Busby et al. (1994a) |
|
|
Reverse mutation assay |
E. coli WP2uvrA (trp+ reversion) |
– |
n.g. |
Mermelstein et al. (1981) |
|
|
Reverse mutation assay |
E. coli WP2uvrA, 3001-04 (trp+ reversion; positive only in 3003 and 3004g) |
+ |
n.g. |
McCoy et al. (1985b) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (umu gene expression) |
+ |
n.g. |
Nakamura et al. (1987) |
|
|
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
+ |
+e |
Mersch-Sundermann et al. (1991, 1992) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 |
+d |
n.g. |
Oda et al. (1992, 1993) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002; NM2009 and 3009 (acetyltransferase activity |
+ |
n.g. |
Shimada et al. (1994) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (umu gene expression) |
+ |
+e |
Shane & Winston (1997) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (MA by human CYP1A1, CYP1A2 or CYP1B1 co-expressed with NPR) |
+ |
+e |
Yamazaki et al. (2000) |
|
|
1,6-Dinitropyrene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Horikawa et al. (1986) |
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1986) |
|
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
|
Forward mutation assay |
S. typhimurium TM677 (resistance to 8-azaguanine) |
+ |
+e |
Busby et al. (1994a) |
|
|
Reverse mutation assay |
E. coli WP2uvrA (trp+ reversion) |
– |
n.g. |
Mermelstein et al. (1981) |
|
|
Reverse mutation assay |
E. coli WP2uvrA (trp+ reversion) |
+ |
n.g. |
Tokiwa et al. (1984) |
|
|
Reverse mutation assay |
E. coli WP2uvrA, 3001-04 (trp+ reversion; positive only in 3003 and 3004g) |
+ |
n.g. |
McCoy et al. (1985b) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (umu gene expression) |
+ |
n.g. |
Nakamura et al. (1987) |
|
|
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
+ |
+e |
Mersch-Sundermann et al. (1991, 1992) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 |
+d |
n.g. |
Oda et al. (1992, 1993) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002; NM2009 and 3009 (acetyltransferase activity |
+ |
n.g. |
Shimada et al. (1994) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (umu gene expression) |
+ |
+e |
Shane & Winston (1997) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (MA by human CYP1A1, CYP1A2 or CYP1B1 co-expressed with NPR) |
+ |
+e |
Yamazaki et al. (2000) |
|
|
1,8-Dinitropyrene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Horikawa et al. (1986) |
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1986) |
|
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
|
Forward mutation assay |
S. typhimurium TM677 (resistance to 8-azaguanine; mutagenicity proportional to DNA binding) |
+ |
n.g. |
Sanders et al. (1983) |
|
|
Forward mutation assay |
S. typhimurium BA15 (resistance to L-arabinose) |
+ |
n.g. |
Jurado et al. (1993) |
|
|
Forward mutation assay |
S. typhimurium TM677 (resistance to 8-azaguanine) |
+ |
+e |
Busby et al. (1994a) |
|
|
Forward mutation assay |
S. typhimurium BA14 (resistance to L-arabinose) |
+ |
n.g. |
Jurado et al. (1994) |
|
|
Reverse mutation assay |
E. coli WP2uvrA (trp+ reversion) |
– |
n.g. |
Mermelstein et al. (1981) |
|
|
Reverse mutation assay |
E. coli WP2uvrA, 3001-04 (trp+ reversion; positive only in 3003 and 3004g) |
+ |
n.g. |
McCoy et al. (1985b) |
|
|
Reverse mutation assay |
S. typhimurium TA1538 with plasmid pYG8031, pYG8011, pSE117, pKM101, pBR322 |
+ |
n.g. |
Nohmi et al. (1995) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (umu gene expression) |
+ |
n.g. |
Nakamura et al. (1987) |
|
|
SOS DNA repair (SOS chromotest) |
E. coli PQ37 |
+ |
+e |
Mersch-Sundermann et al. (1991, 1992) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 |
+d |
n.g. |
Oda et al. (1992, 1993) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (umu gene expression) |
+ |
+e |
Shane & Winston (1997) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (MA by human CYP1A1, CYP1A2 or CYP1B1 co-expressed with NPR) |
+ |
+e |
Yamazaki et al. (2000) |
|
|
1,3,6-Trinitropyrene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Horikawa et al. (1986) |
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
|
Reverse mutation assay |
E. coli WP2uvrA (trp+ reversion) |
+ |
n.g. |
Mermelstein et al. (1981) |
|
|
1,3,6,8-Tetranitropyrene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Horikawa et al. (1986) |
|
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
|
Reverse mutation assay |
E. coli WP2uvrA (trp+ reversion) |
– |
n.g. |
Mermelstein et al. (1981) |
|
|
7-Nitrobenz[a]anthracene |
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (MA by human CYP1A1, CYP1A2 or CYP1B1 co-expressed with NPR) |
– |
+ |
Yamazaki et al. (2000) |
|
6-Nitrochrysene |
DNA damage/repair (rec assay) |
B. subtilis M45 (DNA repair deficient) or H17 (proficient) |
+ |
n.g. |
Tokiwa et al. (1987b) |
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (MA by CYP1B1, not CYP1A1 or CYP1A2) |
n.g. |
+ |
Shimada et al. (1996) |
|
|
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (MA by human CYP1A1, CYP1A2 or CYP1B1 co-expressed with NPR) |
+ |
+e |
Yamazaki et al. (2000) |
|
|
6-Nitrobenzo[a]pyrene |
SOS DNA repair (SOS umu test) |
S. typhimurium TA1535 pSK1002 (MA by human CYP1A1, CYP1A2 or CYP1B1 co-expressed with NPR) |
– |
+ |
Yamazaki et al. (2000) |
|
a |
trp = tryptophan; NPR = NADPH-cytochrome P450 reductase; |
|
|
b |
The results in Table 41 are derived as follows: |
|
|
+ |
is a clear positive result, also given as + or positive by the authors of the publication. |
|
|
(±) |
is a weak positive result, also classified by the authors of the publication as ± or ?. |
|
|
(+) |
given where there were weak positive results with dose–response. |
|
|
± |
given when the result was inconclusive, which means that the result was negative, but testing was not performed up to cytotoxic concentrations. Further ± was given for weak positive effects without dose–response or without sufficient documentation for further assessment. |
|
|
– |
is a negative result. |
|
|
c |
MA = metabolic activation (preparation of liver microsomes [S9-mix] from rodent species); n.g. = not given. |
|
|
d |
Highly sensitive in strains having high O-acetyltransferase and nitroreductase activities. |
|
|
e |
Reduced by metabolic activation. |
|
|
f |
Increased by metabolic activation. |
|
|
g |
Positive results only in E. coli strains that have increased permeability to large molecules and DNA repair deficiency. |
|
The results in Tables 41–44 are derived as follows:
|
+ |
is a clear positive result, also given as + or positive by the authors of the publication; |
|
(±) |
is a weak positive result, also classified by the authors of the publication as ± or ?; |
|
(+) |
is a weak positive result with dose–response; |
|
± |
is an inconclusive result, which means that the result was negative, but testing was not performed up to cytotoxic concentrations; further ± was given for weak positive effects without dose–response or without sufficient documentation for further assessment; and |
|
– |
represents a negative result. |
The majority of the studies on the genotoxicity of nitroPAHs (more than 100 studies on a total of 91 nitroPAHs) have used the Salmonella typhimurium microsome assay with a standardized test protocol using the strains TA98, TA100, TA1535, TA1537 or TA1538. For conciseness, the details of each individual study are not given here but are compiled in Table 40. A summary of the data on the Salmonella microsome assay is presented in Table 45.
From the extensive data, it is possible to compare the mutagenic potency of nitroPAHs in Tables 40 and 46. In analogy to other publications (Tokiwa et al., 1981b, 1993b; Rosenkranz & Mermelstein, 1983; Purohit & Basu, 2000), the mutagenic potency of the examined nitroPAHs in the Salmonella microsome assay is determined in this documentation by the number of revertants per nanomole for each S. typhimurium strain (see Table 40). For different doses, the number of revertants per nanomole has been calculated and the number of spontaneous revertants subtracted. The dose resulting in the maximum number of revertants per nanomole is used for further calculation and assessment. Only those studies that fulfilled the following minimum requirements were documented (and mutagenic potencies determined): at least three doses tested with the corresponding strain, negative control, spontaneous reversion rate at least doubled, and graphical or tabulated presentation of results. Less valid studies were also documented if the database of the nitroPAH was small (<4 studies). Data from studies in which the authors calculated the mutagenic potency by more sophisticated methods (e.g., calculated from the linear regression analysis of the data) were also added to the data pool without change, except for conversion to the unit revertants per nanomole where necessary.
For most nitroPAHs, only a few data are available for one or more of the S. typhimurium strains (74 out of 91 nitroPAHs; see Table 40). Only 17 nitroPAHs revealed a large database on mutagenicity in the Salmonella microsome assay (>5 tests with at least one strain; bold type substance name in Table 40). There are five nitroPAHs that showed exceptionally high mutagenic potency (>100 000 revertants/nmol, highest scale in Table 40) in this test system: 3,7-, and 3,9-dinitrofluoranthene, 1,6- and 1,8-dinitropyrene, and 3,6-dinitrobenzo[a]pyrene (shown by ++++++ in Table 46). A somewhat lower potency was seen with 8-nitrofluoranthene, 1,3-dinitropyrene, 1,3,6-trinitropyrene and 1,3,6,8-tetranitropyrene, where the limit 100 000 revertants/nmol was not reached but >10 000 was (second highest scale in Table 40; shown by +++++ in Table 46).
Most nitroPAHs showed medium mutagenic potency (24 nitroPAHs marked in Table 46 with +++ [>100, <1000 revertants/nmol] or 29 nitroPAHs marked by ++++ [>1000, <10 000 revertants/nmol]). Eleven nitroPAHs reached the second lowest scale (marked by ++ [>10, <100 revertants/nmol]).
Eleven out of 91 nitroPAHs showed only a weak mutagenic potency relative to other nitroPAHs (lowest scale in Table 46, <10 revertants/nmol, marked by +): 1- and 2-nitronaphthalene, 1,3- and 1,5-dinitronaphthalene, 3-nitrofluorene, 9-nitroanthracene, 1,5-, 4,9- and 1,10-dinitrophenanthrene, 5-nitrochrysene and 1-nitrocoronene. In seven nitroPAHs, the mutagenic potency could not be calculated. At least with 1- and 2-nitronaphthalene, the weak mutagenic effect is influenced by the volatility (see also section 2), leading consequently to a short period of exposure of the bacteria to the substance. Gupta et al. (1996) reported an increase in the mutagenic activity of about 1 (1-nitronaphthalene) or 2 (2-nitronaphthalene) orders of magnitude by prolongation of the preincubation time to 90 min. With none of the nitroPAHs listed in Table 40 were clearly negative results obtained in the Salmonella microsome assay. Further, the validity of negative results is limited, for example, if the substance was not tested up to cytotoxicity (see 1,8-dinitrobenzo[e]pyrene in Table 40).
For comparison, it should be noted that the mutagenicity of BaP with exogenous metabolic activation is ~2.3 revertants/nmol, and no mutagenic activity is shown in the absence of metabolic activation.
Various strategies have been used to investigate the metabolism and "mutagenic activation pathways" leading to histidine reversion in bacteria. As well as through comparison of the mutagenic effect of individual nitroPAHs using the Ames Salmonella typhimurium reversion assay, other attempts include:
These and other studies have shown that all nitroPAHs do not follow the same metabolic and activation pathways.
1) The effect of metabolic activation on mutagenicity
The effect of metabolic activation in the Salmonella microsome assay can be assessed by comparing the maximum mutagenic potency with and without exogenous metabolic activation in the same strain (see Table 40 and summary in Table 46). For this, comparative data are available on 48 nitroPAHs (see column "mutagenic potency" in Table 46). No data are available on 43 nitroPAHs; 41 were tested only without and 2 only with metabolic activation in the same strain. It should be noted that parent PAHs need metabolic activation for mutagenic effects in S. typhimurium (IPCS, 1998).
Most nitroPAHs (35 out of 48), including those with exceptionally high mutagenic potency (e.g., dinitropyrenes), were clearly more effective in the Salmonella microsome assay without metabolic activation (marked in Table 46 by –S9) or exhibited a moderate difference (10 nitroPAHs, marked by –S9). This indicates that mutagenicity shown in the Salmonella microsome assay was induced mainly by the enzymes in the bacteria itself and not by the added mammalian activation system. Furthermore, there is evidence that with some nitroPAHs, the metabolic activation system inhibited mutagenicity — for example, see 3,7- and 3,9-dinitrofluoranthene in Table 40.
Only 3 out of these 48 nitroPAHs induced clearly more reverse mutations with metabolic activation (marked by +S9 in Table 46): 7-nitrobenz[a]anthracene, 6-nitrobenzo[a]pyrene and 3-nitroperylene. Metabolic activation of 6-nitrobenzo[a]pyrene (Fu et al., 1982) and 3-nitroperylene (Anderson et al., 1987) was investigated in more detail. In contrast to most other nitroPAHs, where bacterial nitroreduction is the first step in metabolic activation, for these nitroPAHs it is suggested that predominantly a ring oxidation by mixed-function oxidase enzymes is involved, followed by further bacterial metabolism to form the ultimate mutagen (Anderson et al., 1987). For comparison, most parent PAHs are also metabolized by a ring oxidation (IPCS, 1998).
With 10 nitroPAHs, the mutagenic potency was higher with metabolic activation but in the same order of magnitude as without (marked by normal type +S9 in Table 46), indicating a) activation by both bacterial enzymes and mammalian microsomal enzymes or b) that S9-mix has no inhibitory effect on mutagenicity.
It should be pointed out that the higher mutagenic activity (marked by +S9 in Table 46) of exogenous metabolic activation with mammalian microsomes is predominantly observed in nitroPAHs with five aromatic rings.
2) Activation of nitroPAHs in strains with altered enzyme activity
For several nitroPAHs (n = 17), a large database on the standard tester strains of S. typhimurium is available (bold type substance name in Table 40); for some of these nitroPAHs, additional studies have been carried out using nitroreductase- or O-acetyltransferase-deficient strains as well as strains overproducing both enzymes. Examples of these results are shown in Figure 17. The direct mutagenic activity of most nitroPAHs in the Salmonella reversion test is a consequence of reduction of the nitro group to a hydroxylamine, which is catalysed by bacterial nitroreductase (Rosenkranz & Mermelstein, 1983). This nitroreduction and the binding of the corresponding electrophilic arylnitrenium ion (R-NH+) to DNA are discussed as one of several possible metabolic activation pathways in S. typhimurium (Fu, 1990; Ball et al., 1995).
The use of a nitroreductase-deficient strain (marked with NR– in Figure 17) revealed a clear decrease (difference at least 5-fold) in mutagenic activity compared with the standard tester strain showing the maximal mutagenic potency for three nitroPAHs: 2-nitronaphthalene (McCoy et al., 1981a), 2,7-dinitrofluorene (McCoy et al., 1981b) and 1,3-dinitropyrene (Crebelli et al., 1995). A less clear decrease (only 2- to 5-fold) was seen with 5-nitroacenaphthene (McCoy et al., 1983a), 3-nitrofluoranthene (Tokiwa et al., 1986) and 1-nitropyrene (Figure 17). No data for direct comparison between the standard tester strain with maximal response and the corresponding nitroreductase-deficient strain are available with 2-nitrofluorene, but the decreasing effect was obvious when comparing TA98 versus TA98NR– (see Figure 17).
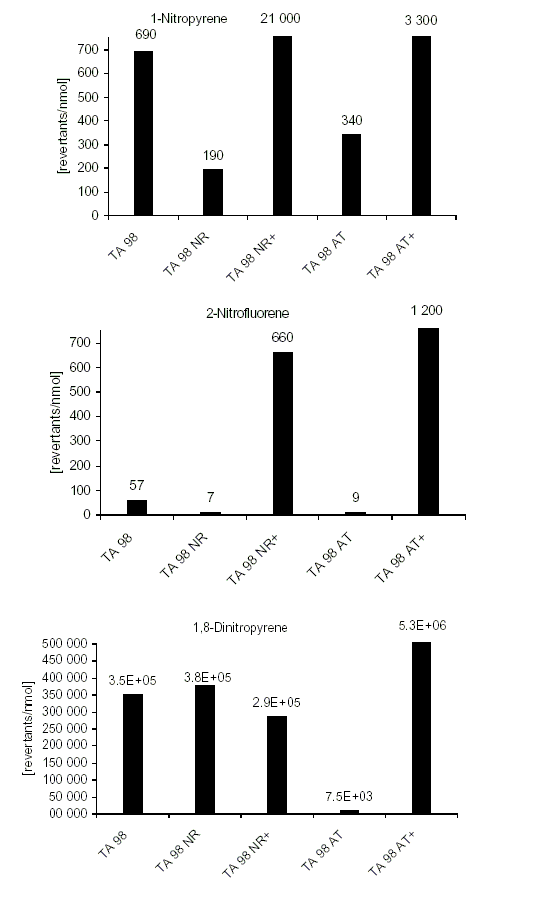
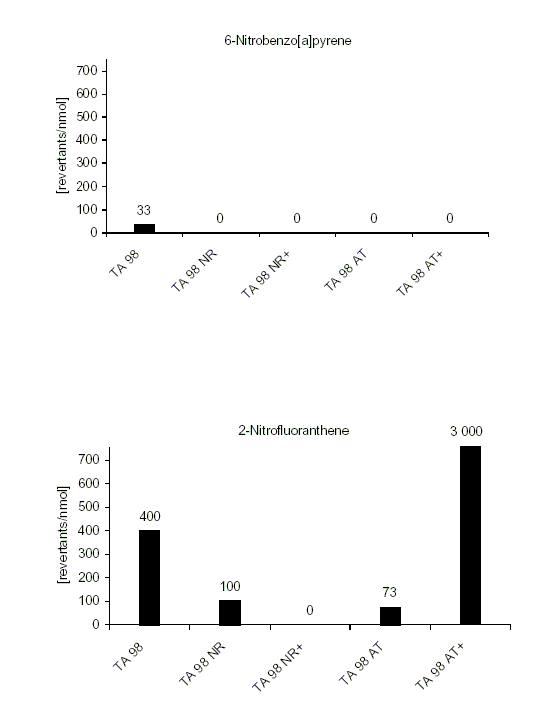
Fig. 17. Examples of mutagenicity studies on nitroPAHs using nitroreductase- or O-acetyltransferase-deficient strains as well as strains overproducing both enzymes. All experiments without metabolic activation system.
NR– = nitroreductase-deficient strain; NR+ = nitroreductase-overproducing strain; AT– = O-acetyltransferase-deficient strain; AT+ = O-acetyltransferase-overproducing strain.
Furthermore, using TA98 with high nitroreductase activity (e.g., TA98NR+ in Figure 17), these nitroPAHs showed an elevated mutagenic effect compared with the corresponding standard tester strain — for example, 2-nitronaphthalene (4-fold increase; Hagiwara et al., 1993), 2-nitrofluorene (12-fold; Figure 17) and 1-nitropyrene (30-fold; see Figure 17) (no further data available for comparison between the standard tester strain with maximal response and the corresponding strain with high nitroreductase activity).
Overall, these examples show that nitroreduction is an important mutagenic activation pathway in S. typhimurium.
Nitroreductases and O-acetyltransferases have been shown to be important enzymes in the mutagenic activation of nitroPAHs in bacteria. It is postulated that after reduction of the nitro group to hydroxylamine, this proximate mutagen requires further activation to form the ultimate mutagen (an arylnitrenium ion). For some nitroPAHs, this activation step occurs through O-acetylation of the hydroxylamine (McCoy et al., 1985a; Fu, 1990; Ball et al., 1994, 1995).
There are a limited number of nitroPAHs with available data on O-acetyltransferase-overproducing and -deficient strains. Several nitroPAHs showed a decrease in mutagenicity with O-acetyltransferase-deficient strains (e.g., TA98AT–) as well as with nitroreductase-deficient strains. For example, pronounced decreases were seen with 5-nitroacenaphthene (McCoy et al., 1983a), 3-nitrofluoranthene (Shane et al., 1991) and 1,3-dinitropyrene (Crebelli et al., 1995), whereas the decreases were not as pronounced with 2-nitrofluorene (see Figure 17), 2,7-dinitrofluorene (Hirayama et al., 1988) and 1-nitropyrene (see Figure 17).
Some nitroPAHs showed a clear increase in mutagenic potency (about 1 order of magnitude) relative to the standard tester strain in strains overproducing O-acetyltransferase (marked by AT+ in Figure 17) — for example, 2-nitrofluorene (20-fold increase; Figure 17), 3-nitrofluoranthene (20-fold; Ball et al., 1994; Sangaiah et al., 1996), 1,6-dinitropyrene (30-fold; Tokiwa et al., 1985, 1986, 1994) and 1,8-dinitropyrene (15-fold; see Figure 17). For these nitroPAHs, O-acetylation seems to be a mutagenic activation pathway in S. typhimurium.
For 1,6- and 1,8-dinitropyrene, no significant difference between nitroreductase-deficient strains, standard tester strains and nitroreductase-overproducing strains were seen (similar results with both nitroPAHs; data on 1,8-dinitropyrene, see Figure 17); with these two nitroPAHs, however, a clear decrease in mutagenic activity compared with the standard tester strain was obtained with O-acetyltransferase-deficient (AT–) strains (see Figure 17). Together with data on strains overproducing O-acetyltransferase (see above), this suggests that 1,6- and 1,8-dinitropyrene are examples of nitroPAHs that do not depend on nitroreductase for expressing maximal direct mutagenicity in S. typhimurium (Rosenkranz & Mermelstein, 1983). In accordance with this activation mechanism, 1,8-dinitropyrene was highly mutagenic in the O-acetyltransferase-overproducing strain (highest measured mutagenic potency, 15-fold increase relative to TA98), but no increasing effect was detected in the nitroreductase-overproducing strain (see Figure 17).
6-Nitrobenzo[a]pyrene (see Figure 17) is an example of a nitroPAH that needed an exogenous mammalian metabolic activation system for mutagenic effects in S. typhimurium. The mutagenic potency was only moderately increased in nitroreductase- and O-acetyltransferase-deficient strains compared with the standard tester strain.
Additional data on mechanisms of mutagenicity are presented in chapter 6.
3) Use of tester strains to determine frameshift versus base pair substitution
Data on standard tester strains recommended for general mutagenesis screening by Ames et al. (1975) are documented in Table 40. Strains TA1535 and TA100 detect mutagens that cause base pair substitutions, primarily at the G:C base pair in the hisG gene, and the other three strains detect various frameshift mutagens (Maron & Ames, 1983). Base pair substitutions induced by exposure to nitroPAHs were in general not detected by TA1535 but by its R-factor derivative TA100 (see Table 40) containing plasmid pKM101, which enhances an error-prone DNA repair and increases spontaneous and chemically induced mutations.
NitroPAHs that clearly induced more base pair substitutions than frameshift mutations (difference 10-fold or more) as well as nitroPAHs inducing more frameshift mutations (mainly detected by TA98, a strain also including plasmid pKM101) are tabulated in Table 40 (right column; marked by bold type letters). Six nitroPAHs clearly induced more base pair substitutions than frameshift mutations, and seven nitroPAHs were clearly more effective in strains detecting frameshift mutations. A difference of at least 2-fold is marked (more base pair substitutions, 21 nitroPAHs; more frameshift mutations, 16 nitroPAHs) also in Table 40 (normal type letters in the right column). No clear differences were observed with the remaining nitroPAHs, or a comparison was not possible (only one strain tested, 14 nitroPAHs).
There is a tendency in nitroPAHs with two or three aromatic rings towards more base pair substitutions and in nitroPAHs with three or four rings towards more frameshift mutations. For example, nitrated naphthalenes and phenanthrenes induced more base pair substitutions, and nitrated pyrenes induced more frameshift mutations.
Watanabe et al. (1997b) examined the mutational specificity of 12 nitroPAHs in six S. typhimurium strains (TA7001–7006) that reverted only by one specific base substitution. All tested nitroPAHs induced TA AT, CG AT and CG GC transversions and GC AT transition in his genes. AT GC transition and TA GC transversion were weakly or not induced by the tested nitroPAHs.
It is suggested that frameshift mutations induced by nitroPAHs are not due to simple intercalation but rather to DNA adduct formation (Rosenkranz & Mermelstein, 1983; see also chapter 6), mainly at the C8 position of guanine (Rosenkranz et al., 1985). The most frequent frameshift mutation in S. typhimurium TA98 induced, for example, by 1-nitropyrene is a –2 deletion of a G:C or C:G base pair within a CGCGCGCG hot-spot sequence of the hisD3052 mutation (Bell et al., 1991).
1) Number of aromatic hydrocarbon rings in nitroPAHs
There seems to be some relationship between mutagenic potency and the number of aromatic rings within the nitroPAH molecule.
In Table 46, nitrated (mono-, di-, tri-, tetra-) naphthalenes (two rings) showed only low mutagenic potency (two lowest scales in column "mutagenic potency"; <100 revertants/nmol; marked by + and ++).
NitroPAHs with three rings (acenaphthene, fluorene, anthracene, phenanthrene) revealed a low mutagenic potency (10 out of 32 examined nitroPAHs) or, in most cases (22 out of 32 investigated nitroPAHs), a medium mutagenic potency (two medium scales; >100, but <10 000 revertants/nmol; marked by +++ and ++++ in Table 46). The two highest scales (>10 000 revertants/nmol) were not reached in this group of nitroPAHs.
In contrast, 9 out of 30 examined nitroPAHs with four rings (fluoranthene, pyrene, benz[a]anthracene, chrysene) showed a high mutagenic potency (two highest scales in Table 46, marked by +++++ and ++++++), 19 a medium mutagenic potency and only 2 a low mutagenic potency. Vance & Levin (1984) also examined the effect of the number and arrangement of aromatic nuclei in nitroPAHs on mutagenicity in the Salmonella microsome assay. They presented evidence for highest activity in nitroPAHs with four aromatic rings and an overall length of three rings.
This tendency towards a higher mutagenic potency with increasing number of rings is not followed for nitroPAHs with five rings (e.g., benzo[x]fluoranthenes, benzo[x]pyrenes, perylene). However, 12 out of 15 nitroPAHs in Table 46 with available data revealed medium mutagenic potency, 1 nitroPAH high mutagenic potency and only 2 nitroPAHs low mutagenic potency.
Data on nitroPAHs with six or more rings (e.g., coronene) are not sufficient for assessment of a correlation (Table 46).
2) Number of nitro groups
There are several examples in Table 46 revealing an increased mutagenic potency for dinitration compared with mononitration of PAHs. Although influenced by the orientation of the nitro groups (see below), a second nitro group elevated mutagenic potency in fluorenes, phenanthrenes, fluoranthenes, pyrenes and benzo[a]pyrenes. The highest scale for mutagenic potency was observed in the dinitrated PAHs 3,7- and 3,9-dinitrofluoranthene, 1,6- and 1,8-dinitropyrene and 3,6-dinitrobenzo[a]pyrene.
There is no case for a further increase in mutagenic potency with a third or a fourth nitro group. Comparing, for example, the mononitrated pyrenes with 1,3,6-trinitro- and 1,3,6,8-tetranitropyrene, the mutagenic potency increased with further nitro groups, although the potency of the dinitropyrenes is not reached (Table 46).
3) Orientation of nitro group
The orientation of the nitro group is an important structural factor in determining the mutagenic activity of nitroPAHs. Comparing experimental results on direct mutagenic activity of nitroPAHs in the Salmonella microsome assay, it is suggested that nitroPAHs with their nitro group oriented perpendicular or nearly perpendicular to the plane of the aromatic rings generally exhibit weak or no mutagenicity (Fu et al., 1985b, 1988b, 1989, 1997; Jung et al., 1991). The decreased mutagenicity of nitroPAHs with nitro groups perpendicular to the aromatic ring system is thought to be due to their inability to fit into the active site of the bacterial nitroreductases because of steric interactions (Fu et al., 1985b).
In a recent study on 22 different nitrated phenanthrenes (Sera et al., 1996), three dinitrophenanthrenes showed weak mutagenic potency: 1,5-, 1,10- and 4,9-dinitrophenanthrene (Tables 40 and 46). In these phenanthrene derivatives, substituents were oriented perpendicular or nearly perpendicular to the aromatic system. In contrast, dinitrophenanthrenes with substituents showing a dihedral angle (calculated by the authors) below 10 degrees exhibited high mutagenic activity — for example, 3,6-dinitrophenanthrene. Similar results were presented with mononitrated phenanthrenes (highest mutagenicity with 3-nitrophenanthrene, revealing the lowest dihydral angle) and trinitrated phenanthrenes (lowest mutagenic potency in 1,5,10-trinitrophenanthrene, in which all three substituents reached a dihydral angle above 50 degrees) (Sera et al., 1996).
Further studies suggest that the nitroPAHs with the nitro group situated at the longest axis of the molecule exhibited the highest mutagenicity in S. typhimurium (Vance & Levin, 1984; Hirayama et al., 1988; Yu et al., 1992). For example, 2-nitropyrene has a higher mutagenic potency than 1- or 4-nitropyrene, and 2-nitrochrysene revealed a higher mutagenicity than 6-nitrochrysene. This conclusion is in accord with the hypothesis above, since nitroPAHs with nitro substituents oriented perpendicular to the plane of aromatic rings in general have their nitro group situated either at the shortest axis of the molecule or near the shortest axis with weak mutagenic activity (Yu et al., 1992).
Klopman et al. (1984) presented evidence for an inverse linear relationship between the calculated energies of the lowest unoccupied molecular orbital (LUMO) and the logarithm of the direct-acting mutagenicity of nitroPAHs in Salmonella strains. In a recent study on 11 dinitrophenanthrene derivatives (Sera et al., 1996), it could be shown that nitroPAHs with no nitro substituent perpendicular to the aromatic ring have a lower LUMO energy level.
Furthermore, structurally similar nitroPAHs showed a positive correlation between reduction potential measured by polarographic methods and direct mutagenicity (Jung et al., 1991).
The available data on the genotoxicity of nitroPAHs in bacterial test systems other than the Salmonella microsome assay are presented in Table 41. The literature is focused on two end-points: gene mutation and DNA damage/repair. Although the database is smaller than that on the Salmonella microsome assay (91 nitroPAHs), data on 27 nitroPAHs were given (see Table 41 and summary in Table 45).
With three exceptions, gene mutation assays (data on 13 nitroPAHs) in Table 41 confirmed the positive results in the Salmonella microsome assay (see Table 45). Exclusively positive results were obtained with DNA damage/repair assays on 26 nitroPAHs (Tables 41 and 45).
The effect of metabolic activation in the Salmonella microsome assay is discussed in detail in section 7.5.1.3. Bacterial systems other than the Salmonella microsome assay did not need metabolic activation systems (see Tables 41 and 45). In those studies where it was possible to compare genotoxic activities with and without exogenous metabolic activation, it was found that most nitroPAHs showed genotoxic effects irrespective of the presence of metabolic activation (11 nitroPAHs) and seven nitroPAHs were more active without, indicating the important role of bacterial enzymes in the metabolic activation of nitroPAHs in vitro.
Genotoxicity of nitroPAHs in eukaryotic cells, including fungi, plants and mammalian cells, is presented in Table 42, and genotoxicity of nitroPAHs in human cells is shown in Table 43 (summary of data in Table 45). These data are available for 28 nitroPAHs. Most of these nitroPAHs have been studied in more detail because they were detected in diesel particulate extracts (IARC, 1989) and were discussed as possible contributors to the carcinogenic risk of diesel exhaust. Twenty-five of the 28 nitroPAHs showing positive results in bacterial systems gave predominantly positive results in eukaryotic test systems, with the exception of 1,3,6,8-tetranitropyrene (two negative end-points), 9-nitroanthracene (one positive and one negative end-point) and 7-nitrobenz[a]anthracene (one inconclusive result; see Table 45).
Table 42. In vitro genotoxicity of nitroPAHs in eukaryotic cells, excluding human cells (see Table 43)
|
Substance |
End-pointa |
Tested cell type (remarks) |
Resultsb |
Reference |
|
|
–MAc |
+MAc |
||||
|
1-Nitronaphthalene |
GM |
Mouse lymphoma cells L5178Y (TK test) |
–d |
n.g. |
Shelby & Stasiewicz (1984) |
|
GM |
Chinese hamster lung cells V79 (MA by co-culture of rat hepatocytes, HPRT test) |
± |
± |
Boyes et al. (1991) |
|
|
GM |
Chinese hamster lung cells V79 (MA by co-culture of hamster hepatocytes, HPRT test) |
± |
+ |
Boyes et al. (1991) |
|
|
SCE |
Cultured mammalian cells (no details available) |
–d |
n.g. |
Shelby & Stasiewicz (1984) |
|
|
SCE |
Chinese hamster lung cells V79 (MA by co-culture with rat or hamster hepatocytes) |
– |
+ |
Boyes et al. (1991) |
|
|
CA |
Cultured mammalian cells (no details available) |
+d |
n.g. |
Shelby & Stasiewicz (1984) |
|
|
2-Nitronaphthalene |
MR |
S. cerevisiae D3 |
+ |
+ |
Simmon (1979b) |
|
UDS |
Primary rat hepatocytes |
– |
Mori et al. (1987) |
||
|
UDS |
Primary mouse hepatocytes |
– |
Mori et al. (1987) |
||
|
1,8-Dinitronaphthalene |
UDS |
Primary mouse hepatocytes |
– |
Mori et al. (1987) |
|
|
UDS |
Primary rat hepatocytes |
+ |
Mori et al. (1987) |
||
|
5-Nitroacenaphthene |
UDS |
Primary mouse hepatocytes |
+ |
Mori et al. (1987) |
|
|
CA |
Chinese hamster lung cells CHL |
+ |
+e |
Matsuoka et al. (1991) |
|
|
1-Nitrofluorene |
GM |
Mouse lymphoma cells L5178Y (TK test) |
+ |
n.g. |
Amacher et al. (1979) |
|
2-Nitrofluorene |
GM |
A. nidulans haploid strain35 (forward mutation; resistance to 8-azaguanine) |
– |
n.g. |
Bignami et al. (1982) |
|
GM |
A. nidulans haploid strain35 (forward mutation; induction of methA1 suppressors) |
– |
n.g. |
Bignami et al. (1982) |
|
|
MR |
S. cerevisiae D3 |
+ |
+f |
Simmon (1979b) |
|
|
GC |
S. cerevisiae D4 |
– |
n.g. |
Mitchell (1980) |
|
|
GM |
Tradescantia clone 4430 (plant cuttings exposed by inflorescence immersion) |
+ |
n.g. |
Schairer & Sautkulis (1982) |
|
|
GM |
Mouse lymphoma cells L5178Y (TK test) |
+ |
n.g. |
Oberly et al. (1984) |
|
|
GM |
Chinese hamster ovary cells (CHO, HPRT test) |
– |
+ |
Oberly et al. (1990) |
|
|
GM |
Mouse lymphoma cells L5178Y (TK test) |
+ |
n.g. |
Oberly et al. (1996) |
|
|
GM |
Chinese hamster V79 cells (cells with different enzyme activities tested: CYP1A2, CYP2C9 or CYP3A4 with and without endogenous acetyltransferase; HPRT test) |
– |
Kappers et al. (2000) |
||
|
UDS |
Primary rat hepatocytes |
– |
Probst et al. (1981) |
||
|
UDS |
Primary rat hepatocytes |
+ |
Mori et al. (1987) |
||
|
UDS |
Primary mouse hepatocytes |
+ |
Mori et al. (1987) |
||
|
2-Nitrofluorene |
SSB |
Alveolar type II cells, Clara cells or macrophages isolated from rabbit lung (alkaline elution assay, tested up to cytotoxicity) |
– |
n.g. |
Becher et al. (1993) |
|
VE |
C3H2K cells (no further data) infected with mouse leukaemia virus |
(±) |
n.g. |
Yoshikura et al. (1979) |
|
|
SCE |
Chinese hamster ovary (CHO) cells |
+ |
+e |
Nachtman & Wolff (1982) |
|
|
SCE |
Chinese hamster lung cells V79 |
– |
+ |
Schleibinger et al. (1989) |
|
|
CA |
Chinese hamster lung cells CHL |
+ |
+e |
Matsuoka et al. (1991) |
|
|
MN |
Chinese hamster lung cells V79 |
+ |
n.g. |
Glatt et al. (1990) |
|
|
MN |
Rat intestinal cells IEC-17/-18 (metabolically competent) |
+ |
Glatt et al. (1990) |
||
|
MN |
Embryonal human liver cells |
+ |
Glatt et al. (1990) |
||
|
MN |
Mouse BALB/c-3T3 cells |
+ |
n.g. |
Gu et al. (1992) |
|
|
MN |
Chinese hamster CHL cells (sublines expressing bacterial or human N-acetyltransferases more effective than parent CHL cells) |
+ |
n.g. |
Watanabe et al. (1994) |
|
|
3-Nitrofluorene |
CA |
Chinese hamster lung cells CHL |
+ |
+e |
Matsuoka et al. (1991) |
|
4-Nitrofluorene |
CA |
Chinese hamster lung cells CHL |
+ |
+e |
Matsuoka et al. (1991) |
|
2,7-Dinitrofluorene |
UDS |
Primary rat hepatocytes |
+ |
Probst et al. (1981) |
|
|
UDS |
Primary rat hepatocytes |
+ |
Mori et al. (1987) |
||
|
UDS |
Primary mouse hepatocytes |
+ |
Mori et al. (1987) |
||
|
9-Nitroanthracene |
UDS |
Primary rat hepatocytes |
– |
Mori et al. (1987) |
|
|
UDS |
Primary mouse hepatocytes |
– |
Mori et al. (1987) |
||
|
3-Nitrofluoranthene |
GM |
Chinese hamster lung cells V79 (HPRT test) |
– |
+g |
Berry et al. (1985) |
|
UDS |
Primary rat hepatocytes |
+ |
Mori et al. (1987) |
||
|
UDS |
Primary mouse hepatocytes |
+ |
Mori et al. (1987) |
||
|
SCE |
Chinese hamster lung cells V79 |
– |
+ |
Schleibinger et al. (1989) |
|
|
8-Nitrofluoranthene |
GM |
Chinese hamster lung cells V79 (HPRT test) |
– |
+g |
Berry et al. (1985) |
|
3,7-Dinitrofluoranthene |
GM |
Chinese hamster lung cells V79 (HPRT test) |
n.g. |
+ |
Tokiwa et al. (1988) |
|
CA |
Chinese hamster lung cells CHL (no induction of polyploid cells) |
+ |
(±) |
Matsuoka et al. (1993) |
|
|
3,9-Dinitrofluoranthene |
GM |
Chinese hamster lung cells V79 (HPRT test) |
n.g. |
+ |
Tokiwa et al. (1988) |
|
CA |
Chinese hamster lung cells CHL (no induction of polyploid cells) |
+ |
(±) |
Matsuoka et al. (1993) |
|
|
1-Nitropyrene |
GC |
S. cerevisiae D4 (trp locus; cells exposed in suspension or in plates) |
– |
n.g. |
McCoy et al. (1983c) |
|
GC |
S. cerevisiae D4 (trp locus) |
–h |
n.g. |
McCoy et al. (1984) |
|
|
GM |
Heterozygous soybean strain T219 (seeds exposed, mutated spots evaluated, MA by rat S9-mix; no effect with pyrene) |
+ |
– |
Katoh et al. (1994) |
|
|
GM |
Chinese hamster ovary cells (CHO, HPRT test) |
+ |
+e |
Marshall et al. (1982) |
|
|
GM |
Chinese hamster lung cells (maximum concentration of 20 µg/ml not cytotoxic, DT tested) |
– |
– |
Nakayasu et al. (1982) |
|
|
GM |
Chinese hamster ovary cells (CHO, HPRT test) |
+ |
+e |
Li & Dutcher (1983) |
|
|
GM |
Chinese hamster lung cells (not tested up to cytotoxicity, DT tested) |
– |
n.g. |
Sugimura & Takayama (1983) |
|
|
GM |
Chinese hamster lung cells V79 (MA by Syrian hamster embryo cells, NA test) |
– |
– |
Takayama et al. (1983) |
|
|
GM |
Chinese hamster lung cells V79 (MA by co-cultivated rat hepatocytes, HPRT test) |
– |
+ |
Ball et al. (1985) |
|
|
GM |
Chinese hamster lung cells V79 (HPRT test) |
(±) |
+ |
Berry et al. (1985) |
|
|
GM |
Chinese hamster ovary cells (not tested up to cytotoxicity; high activity of the metabolite 1-nitrosopyrene, HPRT test) |
– |
n.g. |
Fifer et al. (1986a) |
|
|
GM |
Chinese hamster ovary cells (CHO; not tested up to cytotoxicity, metabolites mutagenic without MA, HPRT test) |
– |
n.g. |
Heflich et al. (1985a, 1986a) |
|
|
GM |
Chinese hamster ovary cells (CHO; no data on cytotoxicity, metabolite 1-nitrosopyrene mutagenic without MA, HPRT test) |
– |
n.g. |
Heflich et al. (1986c) |
|
|
GM |
Chinese hamster ovary cells (CHO, HPRT test) |
– |
+e |
Heflich et al. (1990) |
|
|
GM |
Chinese hamster CHO-K1 (DNA repair proficient, HPRT test) |
– |
n.g. |
Thornton-Manning et al. (1991a) |
|
|
GM |
Chinese hamster CHO-UV5 (DNA repair deficient, HPRT test) |
+ |
n.g. |
Thornton-Manning et al. (1991a) |
|
|
GM |
Chinese hamster CHO-K1 (DNA repair proficient, HPRT test) |
– |
+ |
Thornton-Manning et al. (1991b) |
|
|
GM |
Chinese hamster CHO-UV5 (DNA repair deficient; more effective at anaerobic conditions, HPRT test) |
– |
+ |
Thornton-Manning et al. (1991b) |
|
|
GM |
Chinese hamster V79 cells (cells with different enzyme activities tested: CYP1A2 or CYP3A4 with and without endogenous acetyltransferase; HPRT test; positive with CYP1A2 plus acetyltransferase activity) |
+ |
Kappers et al. (2000) |
||
|
GM |
Mouse NIH/3T3 cells (cells with two different enzyme activities tested: a) only acetyltransferase or b) CYP1A2; HPRT test; positive only with CYP1A2) |
+ |
Kappers et al. (2000) |
||
|
UDS |
Primary rat hepatocytes (higher potency than in hamster hepatocytes) |
+ |
Kornbrust & Barfknecht (1984) |
||
|
UDS |
Primary Syrian golden hamster hepatocytes (see above) |
+ |
Kornbrust & Barfknecht (1984) |
||
|
UDS |
Primary rat tracheal epithelial cells |
+ |
n.g. |
Doolittle & Butterworth (1984) |
|
|
UDS |
Clara cells isolated from rabbit lung (primary cell culture) |
+ |
n.g. |
Haugen et al. (1986) |
|
|
UDS |
Alveolar type-II cells isolated from rabbit lung (primary cell culture) |
– |
n.g. |
Haugen et al. (1986) |
|
|
UDS |
Primary rat hepatocytes |
+ |
Mori et al. (1987) |
||
|
UDS |
Primary mouse hepatocytes |
+ |
Mori et al. (1987) |
||
|
SSB |
Chinese hamster lung cells V79 (alkaline elution assay) |
+ |
n.g. |
Saito et al. (1984b) |
|
|
SSB |
Rat hepatoma cell line H4-II-E (alkaline elution assay) |
+ |
Möller & Thorgeirsson (1985) |
||
|
SSB |
Primary mouse hepatocytes (alkaline elution assay) |
+ |
Möller & Thorgeirsson (1985) |
||
|
SSB |
Chinese hamster lung fibroblasts Don |
+ |
+ |
Edwards et al. (1986b) |
|
|
SSB |
Alveolar type II cells, Clara cells or macrophages isolated from rabbit lung (alkaline elution assay, tested up to cytotoxicity) |
– |
n.g. |
Becher et al. (1993) |
|
|
VE |
Rat fibroblasts H3 transformed by ts-a mutant of polyoma virus |
+ |
+e |
Lambert & Weinstein (1987) |
|
|
SCE |
Chinese hamster lung cells V79 (abstract) |
+ |
n.g. |
Heidemann & Miltenburger (1983) |
|
|
SCE |
Chinese hamster lung cells V79 |
– |
+ |
Schleibinger et al. (1989) |
|
|
CA |
Chinese hamster pulmonary cell line Don:Wg3h |
+ |
n.g. |
Lafi & Parry (1987) |
|
|
CA |
Chinese hamster lung cells CHL |
–i |
+ |
Matsuoka et al. (1991) |
|
|
2-Nitropyrene |
MN |
Isolated rodent hepatocytes (abstract) |
+ |
Müller et al. (1995) |
|
|
1,3-Dinitropyrene |
GC |
S. cerevisiae D4 (trp locus; cells exposed in suspension or in plates) |
– |
n.g. |
McCoy et al. (1983c) |
|
GM |
Heterozygous soybean strain T219 (seeds exposed, mutated spots evaluated, MA by rat S9-mix) |
+ |
+ |
Katoh et al. (1994) |
|
|
GM |
Chinese hamster lung cells (DT tested) |
+ |
n.g. |
Nakayasu et al. (1982) |
|
|
GM |
Chinese hamster ovary cells (CHO; mutagenicity increased with low amounts of MA system, HPRT test) |
+ |
+f |
Li & Dutcher (1983) |
|
|
GM |
Chinese hamster lung cells (DT tested) |
+ |
n.g. |
Sugimura & Takayama (1983) |
|
|
GM |
Chinese hamster lung cells V79 (MA by Syrian hamster embryo cells, NA test) |
(±) |
+ |
Takayama et al. (1983) |
|
|
GM |
Chinese hamster lung cells V79 (inhibition by haemin, NA test) |
+ |
n.g. |
Katoh et al. (1984) |
|
|
UDS |
Primary rat hepatocytes |
+ |
Mori et al. (1987) |
||
|
UDS |
Primary mouse hepatocytes |
+ |
Mori et al. (1987) |
||
|
SSB |
Primary mouse hepatocytes (alkaline elution assay) |
(±) |
Möller & Thorgeirsson (1985) |
||
|
VE |
Rat fibroblasts H3 transformed by ts-a mutant of polyoma virus |
+ |
n.g. |
Lambert & Weinstein (1987) |
|
|
CA |
Chinese hamster lung cells CHL |
+ |
+f |
Matsuoka et al. (1991) |
|
|
MN |
mouse hepatoma cells BWI-J (not tested up to cytotoxicity) |
– |
Roscher & Wiebel (1992) |
||
|
MN |
Rat hepatoma cells 5L (not tested up to cytotoxicity) |
– |
Roscher & Wiebel (1992) |
||
|
MN |
Rat hepatoma cells H4IIEC3/G- |
+ |
Roscher & Wiebel (1992) |
||
|
MN |
Chinese hamster lung cells V79 |
+ |
n.g. |
Roscher & Wiebel (1992) |
|
|
MN |
Rat fibroblastoid cells 208F (not tested up to cytotoxicity) |
– |
n.g. |
Roscher & Wiebel (1992) |
|
|
1,6-Dinitropyrene |
GC |
S. cerevisiae JD1 (trp and his loci) |
+j |
n.g. |
Wilcox & Parry (1981) |
|
GC |
S. cerevisiae JD1 (trp and his loci) |
+k |
+e |
Wilcox et al. (1982) |
|
|
GC |
S. cerevisiae D4 (trp locus; cells exposed in suspension or in plates) |
– |
n.g. |
McCoy et al. (1983c) |
|
|
GM |
Heterozygous soybean strain T219 (seeds exposed, mutated spots evaluated, MA by rat S9-mix) |
+ |
+ |
Katoh et al. (1994) |
|
|
GM |
Chinese hamster lung cells (DT tested) |
+ |
+f |
Nakayasu et al. (1982) |
|
|
GM |
Chinese hamster ovary cells (CHO, HPRT test) |
+ |
+f |
Li & Dutcher (1983) |
|
|
GM |
Chinese hamster lung cells (DT tested) |
+ |
n.g. |
Sugimura & Takayama (1983) |
|
|
GM |
Chinese hamster lung cells V79 (inhibition by haemin, NA test) |
+ |
n.g. |
Katoh et al. (1984) |
|
|
GM |
Chinese hamster ovary cells (CHO, HPRT test) |
+ |
n.g. |
Edgar & Brooker (1985) |
|
|
GM |
Chinese hamster ovary cells (CHO; higher activity of the metabolite 1-nitro-6-nitrosopyrene, HPRT test) |
(±) |
n.g. |
Fifer et al. (1986a) |
|
|
UDS |
Primary rat hepatocytes (higher potency than the positive control acetylaminofluorene) |
+ |
Butterworth et al. (1983) |
||
|
UDS |
Primary rat tracheal epithelial cells |
+ |
n.g. |
Doolittle & Butterworth (1984) |
|
|
UDS |
Primary rat spermatocytes and spermatids (no cytotoxicity) |
– |
n.g. |
Working & Butterworth (1984) |
|
|
UDS |
Clara cells isolated from rabbit lung (primary cell culture) |
+ |
n.g. |
Haugen et al. (1986) |
|
|
UDS |
Alveolar type-II cells isolated from rabbit lung (primary cell culture) |
+ |
n.g. |
Haugen et al. (1986) |
|
|
UDS |
Primary rat hepatocytes |
+ |
Mori et al. (1987) |
||
|
UDS |
Primary mouse hepatocytes |
+ |
Mori et al. (1987) |
||
|
SSB |
Chinese hamster lung cells V79 (alkaline elution assay) |
+ |
n.g. |
Saito et al. (1984b) |
|
|
SSB |
Primary mouse hepatocytes (alkaline elution assay) |
(±) |
Möller & Thorgeirsson (1985) |
||
|
SSB |
Rat hepatoma cell line H4-II-E (alkaline elution assay) |
– |
Möller & Thorgeirsson (1985) |
||
|
DCL |
Rat hepatoma cell line H4-II-E |
– |
Möller & Thorgeirsson (1985) |
||
|
VE |
Rat fibroblasts H3 transformed by ts-a mutant of polyoma virus |
+ |
n.g. |
Lambert & Weinstein (1987) |
|
|
SCE |
Chinese hamster ovary cells (CHO) |
+ |
n.g. |
Edgar & Brooker (1985) |
|
|
CA |
Rat liver epithelial cells RL4 (cells metabolically competent) |
+ |
Danford et al. (1982) |
||
|
CA |
Rat liver epithelial cell line RL4 (cells metabolically competent) |
+ |
Wilcox et al. (1982) |
||
|
CA |
Chinese hamster liver cell line |
+ |
Danford et al. (1983) |
||
|
CA |
Primary Chinese hamster liver cells (abstract) |
+ |
Danford et al. (1983) |
||
|
CA |
Chinese hamster ovary cells CHO |
+ |
n.g. |
Edgar & Brooker (1985) |
|
|
CA |
Chinese hamster lung cells CHL |
+ |
+f |
Matsuoka et al. (1991) |
|
|
MN |
Mouse hepatoma cells BWI-J (not tested up to cytotoxicity) |
– |
Roscher & Wiebel (1992) |
||
|
MN |
Rat hepatoma cells 5L (not tested up to cytotoxicity) |
– |
Roscher & Wiebel (1992) |
||
|
MN |
Rat hepatoma cells H4IIEC3/G- |
+ |
Roscher & Wiebel (1992) |
||
|
MN |
Chinese hamster lung cells V79 |
+ |
n.g. |
Roscher & Wiebel (1992) |
|
|
MN |
Rat fibroblastoid cells 208F |
+ |
n.g. |
Roscher & Wiebel (1992) |
|
|
1,8-Dinitropyrene |
GC |
S. cerevisiae JD1 (trp and his locus) |
+j |
n.g. |
Wilcox & Parry (1981) |
|
GC |
S. cerevisiae JD1 (trp and his locus) |
+k |
+e |
Wilcox et al. (1982) |
|
|
GC |
S. cerevisiae D4 (trp locus; cells exposed in suspension or in plates) |
– |
n.g. |
McCoy et al. (1983c) |
|
|
GM |
Heterozygous soybean strain T219 (seeds exposed, mutated spots evaluated, MA by rat S9-mix) |
+ |
+ |
Katoh et al. (1994) |
|
|
GM |
Mouse lymphoma cells L5178Y (TG, AC, OUA or MT tested) |
+ |
n.g. |
Cole et al. (1982); Arlett (1984) |
|
|
GM |
Chinese hamster lung cells (DT tested) |
+ |
n.g. |
Nakayasu et al. (1982) |
|
|
GM |
Chinese hamster ovary cells (CHO, HPRT test) |
+ |
+f |
Li & Dutcher (1983) |
|
|
GM |
Chinese hamster lung cells (DT tested) |
+ |
n.g. |
Sugimura & Takayama (1983) |
|
|
GM |
Chinese hamster lung cells V79 (MA by Syrian hamster embryo cells, NA test) |
+ |
+e |
Takayama et al. (1983) |
|
|
GM |
Chinese hamster lung cells V79 (inhibition by haemin, NA test) |
+ |
n.g. |
Katoh et al. (1984) |
|
|
GM |
mouse lymphoma cells L5178Y (TK test) |
+ |
n.g. |
Edgar (1985) |
|
|
GM |
Chinese hamster ovary cells (CHO, HPRT test) |
+ |
n.g. |
Edgar & Brooker (1985) |
|
|
GM |
Chinese hamster ovary cells (CHO; higher activity of the metabolite 1-nitro-8-nitrosopyrene, HPRT test) |
+ |
n.g. |
Fifer et al. (1986a) |
|
|
GM |
Chinese hamster ovary cells (CHO; no data on cytotoxicity, metabolite 1-nitro-8-nitrosopyrene more mutagenic, HPRT test) |
+ |
n.g. |
Heflich et al. (1986c) |
|
|
GM |
Chinese hamster ovary cells (CHO, HPRT test) |
+ |
n.g. |
O’Donovan (1990) |
|
|
GM |
Chinese hamster lung cells V79 (HPRT test) |
(±) |
n.g. |
O’Donovan (1990) |
|
|
UDS |
Clara cells isolated from rabbit lung (primary cell culture) |
+ |
n.g. |
Haugen et al. (1986) |
|
|
UDS |
Alveolar type-II cells isolated from rabbit lung (primary cell culture) |
+ |
n.g. |
Haugen et al. (1986) |
|
|
UDS |
Primary rat hepatocytes |
+ |
Mori et al. (1987) |
||
|
UDS |
Primary mouse hepatocytes |
+ |
Mori et al. (1987) |
||
|
SSB |
Chinese hamster lung cells V79 (alkaline elution assay) |
+ |
n.g. |
Saito et al. (1984b) |
|
|
SSB |
Primary mouse hepatocytes (alkaline elution assay) |
(±) |
Möller & Thorgeirsson (1985) |
||
|
SSB |
Rat hepatoma cell line H4-II-E (alkaline elution assay) |
+ |
Möller & Thorgeirsson (1985) |
||
|
DCL |
Rat hepatoma cell line H4-II-E |
– |
Möller & Thorgeirsson (1985) |
||
|
VE |
Rat fibroblasts H3 transformed by ts-a mutant of polyoma virus |
+ |
n.g. |
Lambert & Weinstein (1987) |
|
|
SCE |
Chinese hamster ovary cells (CHO) |
+ |
+e |
Nachtman & Wolff (1982) |
|
|
SCE |
Chinese hamster ovary cells (CHO) |
+ |
n.g. |
Edgar & Brooker (1985) |
|
|
CA |
Rat liver epithelial cells RL4 (cells metabolically competent) |
+ |
Danford et al. (1982) |
||
|
CA |
Rat liver epithelial cell line RL4 (cells metabolically competent) |
+ |
Wilcox et al. (1982) |
||
|
CA |
Chinese hamster ovary cells CHO |
+ |
n.g. |
Edgar & Brooker (1985) |
|
|
CA |
Chinese hamster lung cells CHL |
+ |
+f |
Matsuoka et al. (1991) |
|
|
MN |
Chinese hamster CHL cells (sublines expressing bacterial or human N-acetyltransferases more effective than parent CHL cells) |
+ |
n.g. |
Watanabe et al. (1994) |
|
|
1,3,6-Trinitropyrene |
GC |
S. cerevisiae D4 (trp locus; cells exposed in suspension or in plates) |
– |
n.g. |
McCoy et al. (1983c) |
|
GM |
Chinese hamster lung cells (DT tested) |
+ |
n.g. |
Nakayasu et al. (1982) |
|
|
GM |
Chinese hamster ovary cells (CHO, HPRT test) |
+ |
+f |
Li & Dutcher (1983) |
|
|
GM |
Chinese hamster lung cells (DT tested) |
+ |
n.g. |
Sugimura & Takayama (1983) |
|
|
SSB |
Primary mouse hepatocytes (alkaline elution assay) |
(±) |
Möller & Thorgeirsson (1985) |
||
|
VE |
Rat fibroblasts H3 transformed by ts-a mutant of polyoma virus |
+ |
n.g. |
Lambert & Weinstein (1987) |
|
|
1,3,6,8-Tetranitropyrene |
GC |
S. cerevisiae D4 (trp locus; cells exposed in suspension or in plates) |
– |
n.g. |
McCoy et al. (1983c) |
|
GM |
Chinese hamster lung cells (maximum concentration of 20 µg/ml not cytotoxic, DT tested) |
– |
n.g. |
Nakayasu et al. (1982) |
|
|
GM |
Chinese hamster lung cells (DT tested) |
– |
n.g. |
Sugimura & Takayama (1983) |
|
|
SSB |
Primary mouse hepatocytes (alkaline elution assay) |
(±) |
Möller & Thorgeirsson (1985) |
||
|
VE |
Rat fibroblasts H3 transformed by ts-a mutant of polyoma virus |
+ |
n.g. |
Lambert & Weinstein (1987) |
|
|
6-Nitrochrysene |
GM |
Chinese hamster CHO-K1 cells (DNA repair proficient, HPRT test) |
– |
(±) |
Delclos & Heflich (1992) |
|
GM |
Chinese hamster CHO-UV5 cells (DNA repair deficient, HPRT test) |
(±) |
+ |
Delclos & Heflich (1992) |
|
|
1-Nitrobenzo[a]pyrene |
GM |
Chinese hamster ovary cells (CHO, HPRT test) |
– |
n.g. |
Chou et al. (1984) |
|
GM |
Chinese hamster ovary cells (CHO; MA by hepatocytes; no cytotoxicity; abstract, HPRT test) |
– |
– |
Hass et al. (1984) |
|
|
GM |
Chinese hamster ovary cells (CHO; MA by S9 [positive result] or MA by rat hepatocytes [negative results], HPRT test) |
– |
+ |
Hass et al. (1986b) |
|
|
GM |
Chinese hamster ovary cells CHO-K1-BH4 (the trans-7,8-dihydrodiol metabolite revealed higher mutagenicity, HPRT test) |
– |
+ |
Thornton-Manning et al. (1988) |
|
|
GM |
Chinese hamster ovary cells CHO-K1-BH4 (higher mutagenicity of the metabolite 1-nitrosobenzo[a]pyrene, HPRT test) |
– |
n.g. |
Heflich et al. (1989) |
|
|
6-Nitrobenzo[a]pyrene |
GM |
Chinese hamster ovary cells (CHO, HPRT test) |
+ |
n.g. |
Chou et al. (1984) |
|
GM |
Chinese hamster ovary cells (CHO; MA by hepatocytes; abstract, HPRT test) |
(±) |
(±) |
Hass et al. (1984) |
|
|
GM |
Chinese hamster ovary cells (CHO; MA by S9 [positive result] or MA by rat hepatocytes [negative results], HPRT test) |
– |
(±) |
Hass et al. (1986b) |
|
|
GM |
Chinese hamster ovary cells CHO-K1-BH4 (higher mutagenicity of the metabolite 6-nitrosobenzo[a]pyrene, HPRT test) |
– |
n.g. |
Heflich et al. (1989) |
|
|
a |
End-points are as follows: |
|
|
CA = chromosomal aberrations; DCL = DNA cross-links; GC = gene conversion; GM = gene mutation; HPRT = mutation at the hypoxanthine–guanine phosphoribosyl-transferase locus; TK = mutation at the thymidine kinase locus; NA = mutation at the NA+/K+ ATPase locus; AG = resistance to 8-azaguanine; TG = resistance to 6-thioguanine; MT = resistance to methotrexate; OUA = resistance to ouabain; DT = resistance to diphtheria toxin; AC = resistance to 1-β-D-arabinofuranosyl cytosine; MN = induction of micronuclei; MR = mitotic recombination; SCE = sister chromatid exchange; SSB = single-strand breaks of DNA; UDS = unscheduled DNA synthesis; VE = enhanced viral DNA synthesis. |
||
|
b |
The results in Table 42 are derived as follows: |
|
|
+ |
is a clear positive result, also given as + or positive by the authors of the publication. |
|
|
(±) |
is a weak positive result, also classified by the authors of the publication as ± or ?. |
|
|
(+) |
given where there were weak positive results with dose–response. |
|
|
± |
given when the result was inconclusive, which means that the result was negative, but testing was not performed up to cytotoxic concentrations. Further ± was given for weak positive effects without dose–response or without sufficient documentation for further assessment. |
|
|
– |
is a negative result. |
|
|
c |
MA = metabolic activation system (rodent liver microsomes) or metabolically competent cells (e.g., isolated liver cells); n.g. = not given. |
|
|
d |
No information given on metabolic activation. |
|
|
e |
Increased by metabolic activation when compared with a positive response without metabolic activation. |
|
|
f |
Decreased by metabolic activation when compared with a positive response without metabolic activation. |
|
|
g |
S100 used (high nitroreductase activity). |
|
|
h |
Positive results with the metabolite 1-nitrosopyrene, authors’ comment: recombinogenic inactivity of nitropyrenes for yeast (see this table) is due to a deficiency in nitroreductase. |
|
|
i |
But polyploid cells induced. |
|
|
j |
Activity decreased with higher doses without effect on cytotoxicity. |
|
|
k |
Increased activity with nitrogen in the atmosphere (probably increased activity of nitroreductase), reduced in an oxygen atmosphere. |
|
Table 43. In vitro genotoxicity of nitroPAHs in human cells (see also Tables 47 and 48)
|
Substance |
End-pointa |
Tested cell typeb (remarks) |
Resultsc |
Reference |
|
|
–MAd |
+MAd |
||||
|
1-Nitronaphthalene |
GM |
Human lymphoblastoid cells MCL-5 (metabolically competent, TK or HPRT test) |
– |
Sasaki et al. (1996, 1997a,b); Grosovsky et al. (1999) |
|
|
SCE |
Cultured mammalian cells (no details available) |
–e |
n.g. |
Shelby & Stasiewicz (1984) |
|
|
CA |
Cultured mammalian cells (no details available) |
+e |
n.g. |
Shelby & Stasiewicz (1984) |
|
|
MN |
Human lymphoblastoid MCL-5 cells (metabolically competent) |
– |
Sasaki et al. (1996); Grosovsky et al. (1999) |
||
|
2-Nitronaphthalene |
GM |
Human lymphoblastoid cells MCL-5 (metabolically competent, TK test) |
± |
Sasaki et al. (1996, 1997a,b); Grosovsky et al. (1999) |
|
|
GM |
Human lymphoblastoid cells MCL-5 (metabolically competent, HPRT test) |
– |
Sasaki et al. (1996, 1997a,b); Grosovsky et al. (1999) |
||
|
GM |
Human lymphoblastoid cells AHH-1 1A1 (metabolically competent, TK test) |
+ |
Grosovsky et al. (1999) |
||
|
GM |
Human lymphoblastoid cells MCL-5 (metabolically competent, HPRT test) |
– |
Sasaki et al. (1996, 1997a,b); Grosovsky et al. (1999) |
||
|
GM |
Human lymphoblastoid cells AHH-1 1A1 (metabolically competent, HPRT test) |
+ |
Grosovsky et al. (1999) |
||
|
MN |
Human lymphoblastoid MCL-5 cells (metabolically competent) |
+ |
Sasaki et al. (1996, 1997a,b); Grosovsky et al. (1999) |
||
|
MN |
Human lymphoblastoid cells AHH-1 1A1 (metabolically competent) |
+ |
Grosovsky et al. (1999) |
||
|
2-Nitrofluorene |
GM |
Human lymphoblastoid cell line (metabolically competent, expresses CYP1A1, TK test) |
– |
Durant et al. (1996) |
|
|
IDS |
HeLa cells |
n.g. |
+ |
Painter & Howard (1982) |
|
|
MN |
Embryonal human liver cells |
+ |
Glatt et al. (1990) |
||
|
IDS |
HeLa cells |
n.g. |
+ |
Painter & Howard (1982) |
|
|
2,7-Dinitrofluorene |
UDS |
Primary human hepatocytes |
– |
Yoshimi et al. (1987) |
|
|
9-Nitroanthracene |
GM |
Human lymphoblastoid cell line (metabolically competent, express CYP1A1, TK test) |
+ |
Durant et al. (1996) |
|
|
2-Nitrofluoranthene |
GM |
Human lymphoblastoid cell line (metabolically competent, express CYP1A1, TK test) |
+ |
Durant et al. (1996) |
|
|
GM |
Human cell line MCL-5 (metabolically competent, TK test) |
– |
Busby et al. (1997) |
||
|
GM |
Human cell line h1A1v2 (metabolically competent, also CYP1A1, TK test) |
+ |
Busby et al. (1997) |
||
|
GM |
Human lymphoblastoid cell line (metabolically competent, express CYP1A1, TK test) |
+ |
Durant et al. (1996) |
||
|
3-Nitrofluoranthene |
GM |
Human lymphoblastoid cell line (metabolically competent, express CYP1A1, TK test) |
+ |
Durant et al. (1996) |
|
|
UDS |
Primary human hepatocytes |
– |
Yoshimi et al. (1987) |
||
|
1-Nitropyrene |
GM |
Human hepatoma HepG2 cells (metabolically competent, HPRT test) |
+ |
Eddy et al. (1985, 1987) |
|
|
GM |
Normal human diploid skin fibroblasts (HPRT test) |
+ |
n.g. |
Patton et al. (1986) |
|
|
GM |
Human diploid fibroblasts (DNA repair-deficient human xeroderma pigmentosum cells; more sensitive than normal cells, HPRT test) |
+ |
n.g. |
Patton et al. (1986) |
|
|
GM |
Human hepatoma HepG2 cells (metabolically competent, HPRT test) |
+ |
Eddy et al. (1985, 1987) |
||
|
GM |
Human lymphoblastoid cells MCL-5 (metabolically competent, TK test) |
+ |
Busby et al. (1994b) |
||
|
GM |
Human hepatoma HepG2 cells (metabolically competent, HPRT test) |
+ |
Silvers et al. (1994) |
||
|
GM |
Human lymphoblastoid cell line (metabolically competent, express CYP1A1, TK test) |
+ |
Durant et al. (1996) |
||
|
GM |
Human lymphoblastoid cells MCL-5 (metabolically competent, TK test) |
+ |
Busby et al. (1994b) |
||
|
UDS |
Primary human bronchus epithelial cells |
+ |
n.g. |
Sugimura & Takayama (1983) |
|
|
UDS |
Human hepatoma HepG2cells (metabolically competent) |
+ |
Eddy et al. (1985, 1987) |
||
|
UDS |
Primary human hepatocytes |
+ |
Yoshimi et al. (1987) |
||
|
UDS |
Human hepatoma HepG2cells (metabolically competent) |
+ |
Eddy et al. (1985, 1987) |
||
|
UDS |
Human hepatoma HepG2cells (metabolically competent) |
+ |
Silvers et al. (1994) |
||
|
2-Nitropyrene |
GM |
Human lymphoblastoid cell line (metabolically competent, express CYP1A1, TK test) |
– |
Durant et al. (1996) |
|
|
GM |
Human lymphoblastoid cells MCL-5 (metabolically competent, TK test) |
– |
Busby et al. (1994b) |
||
|
GM |
Human lymphoblastoid cell line (metabolically competent, express CYP1A1, TK test) |
– |
Durant et al. (1996) |
||
|
4-Nitropyrene |
GM |
Human lymphoblastoid cells MCL-5 (metabolically competent, TK test) |
+ |
Busby et al. (1994b) |
|
|
1,3-Dinitropyrene |
GM |
Human hepatoma HepG2 cells (HPRT test) |
+ |
Eddy et al. (1986) |
|
|
GM |
Human lymphoblastoid cells MCL-5 (metabolically competent, TK test) |
± |
Busby et al. (1994b) |
||
|
1,3-Dinitropyrene |
GM |
Human hepatoma HepG2 cells (metabolically competent, HPRT test) |
+ |
Silvers et al. (1994) |
|
|
GM |
Human lymphoblastoid cell line (metabolically competent, express CYP1A1, TK test) |
+ |
Durant et al. (1996) |
||
|
GM |
Human lymphoblastoid cells MCL-5 (metabolically competent, TK test) |
± |
Busby et al. (1994b) |
||
|
GM |
Human hepatoma HepG2 cells (HPRT test) |
+ |
Eddy et al. (1986) |
||
|
UDS |
Human hepatoma HepG2 cells |
+ |
Eddy et al. (1986) |
||
|
UDS |
Primary human hepatocytes |
+ |
Yoshimi et al. (1987) |
||
|
UDS |
Human hepatoma HepG2cells (metabolically competent) |
(±) |
Silvers et al. (1994) |
||
|
MN |
Human hepatoma cells HepG2 (not tested up to cytotoxicity) |
– |
Roscher & Wiebel (1992) |
||
|
1,6-Dinitropyrene |
GM |
Human hepatoma HepG2 cells (HPRT test) |
– |
Eddy et al. (1985) |
|
|
GM |
Human lymphoblastoid cells MCL-5 (metabolically competent, TK test) |
+ |
Busby et al. (1994b) |
||
|
GM |
Human hepatoma HepG2 cells (metabolically competent; no data on cytotoxicity, HPRT test) |
– |
Silvers et al. (1994) |
||
|
GM |
Human lymphoblastoid cell line (metabolically competent, express CYP1A1, TK test) |
+ |
Durant et al. (1996) |
||
|
GM |
Human lymphoblastoid cells MCL-5 (metabolically competent, TK test) |
+ |
Busby et al. (1994b) |
||
|
UDS |
Primary human hepatocytes (higher potency than the positive control acetylaminofluorene) |
+ |
Butterworth et al. (1983) |
||
|
UDS |
Primary human bronchial epithelial cells |
+ |
n.g. |
Sugimura & Takayama (1983) |
|
|
UDS |
Primary human bronchial epithelial cells (8 donors, positive in 3 out of 8) |
+ |
n.g. |
Doolittle et al. (1985) |
|
|
UDS |
Human hepatoma HepG2 cells (abstract) |
– |
Eddy et al. (1985) |
||
|
UDS |
Primary human hepatocytes |
+ |
Yoshimi et al. (1987) |
||
|
UDS |
Primary human hepatocytes (higher potency than the positive control acetylaminofluorene) |
+ |
Butterworth et al. (1983) |
||
|
UDS |
Primary human bronchial epithelial cells (8 donors, positive in 3 out of 8) |
+ |
n.g. |
Doolittle et al. (1985) |
|
|
UDS |
Human hepatoma HepG2 cells (metabolically competent; no data on cytotoxicity) |
– |
Silvers et al. (1994) |
||
|
CA |
Human fibroblast cell line HSBP |
n.g. |
+ |
Wilcox et al. (1982) |
|
|
MN |
Human hepatoma cells HepG2 (not tested up to cytotoxicity) |
– |
Roscher & Wiebel (1992) |
||
|
1,8-Dinitropyrene |
GM |
Human diploid lymphoblasts (abstract, HPRT test) |
+ |
n.g. |
Sanders et al. (1983) |
|
GM |
Human diploid lymphoblasts (abstract, NA test) |
+ |
n.g. |
Sanders et al. (1983) |
|
|
GM |
Human skin fibroblasts from normal or xeroderma pigmentosum patients (HPRT test) |
– |
n.g. |
Arlett (1984) |
|
|
GM |
Human hepatoma HepG2 cells (abstract, HPRT test) |
– |
Eddy et al. (1985) |
||
|
GM |
Human lymphoblastoid cells MCL-5 (metabolically competent, TK test) |
+ |
Busby et al. (1994b) |
||
|
GM |
Human hepatoma HepG2 cells (metabolically competent; no data on cytotoxicity, HPRT test) |
– |
Silvers et al. (1994) |
||
|
GM |
Human hepatoma HepG2 cells (abstract, HPRT test) |
– |
Eddy et al. (1985) |
||
|
GM |
Human lymphoblastoid cell line (metabolically competent, express CYP1A1, TK test) |
+ |
Durant et al. (1996) |
||
|
GM |
Human lymphoblastoid cells MCL-5 (metabolically competent, TK test) |
+ |
Busby et al. (1994b) |
||
|
GM |
Human skin fibroblasts from normal or xeroderma pigmentosum patients (HPRT test) |
– |
n.g. |
Arlett (1984) |
|
|
UDS |
Human hepatoma HepG2 cells (abstract) |
– |
Eddy et al. (1985) |
||
|
UDS |
Primary human hepatocytes |
+ |
Yoshimi et al. (1987) |
||
|
UDS |
Human hepatoma HepG2cells (metabolically competent, no data on cytotoxicity) |
– |
Silvers et al. (1994) |
||
|
CA |
Human fibroblast cell line HSBP |
n.g. |
+ |
Wilcox et al. (1982) |
|
|
MN |
Normal human skin fibroblasts (not tested up to cytotoxicity) |
– |
n.g. |
Arlett (1984) |
|
|
6-Nitrochrysene |
GM |
Human lymphoblastoid cell line AHH-1 (metabolically competent; metabolite 6-aminochrysene more competent, HPRT test) |
+ |
Morris et al. (1994) |
|
|
a |
End-points are as follows: |
|
|
CA = chromosomal aberrations; GM = gene mutation; HPRT = mutation at the hypoxanthine-guanine phosphoribosyl-transferase locus; TK = mutation at the thymidine kinase locus; NA = mutation at the Na+/K+ ATPase locus; IDS = inhibition of DNA synthesis; MN = induction of micronuclei; SCE = sister chromatid exchange; UDS = unscheduled DNA synthesis. |
||
|
b |
The cell line AHH-1 1A1 (also called h1A1v2) constitutively expresses CYP1A1 necessary for the metabolism of many promutagens. The cells of the MCL-5 cell line contain endogenous CYP1A1 activity and additionally two plasmids expressing cDNAs for epoxide hydrolase and CYP1A2, CYP2A6, CYP2E1 and CYP3A4. |
|
|
c |
The results in Table 43 are derived as follows: |
|
|
+ |
is a clear positive result, also given as + or positive by the authors of the publication. |
|
|
(±) |
is a weak positive result, also classified by the authors of the publication as ± or ?. |
|
|
(+) |
given where there were weak positive results with dose–response. |
|
|
± |
given when the result was inconclusive, which means that the result was negative, but testing was not performed up to cytotoxic concentrations. Further ± was given for weak positive effects without dose–response or without sufficient documentation for further assessment. |
|
|
– |
is a negative result. |
|
|
d |
MA = metabolic activation system (rodent liver microsomes) or metabolically competent cells; n.g. = not given. |
|
|
e |
No information given on metabolic activation. |
|
Data on the effects of metabolic activation on genotoxicity in eukaryotic cells are limited (see nitroPAHs in Table 45 with end-points investigated with and without metabolic activation, so showing entries in both lines, +MA and –MA). In most cases, these nitroPAHs resulted in genotoxic effects independent of metabolic activation. There is a tendency towards more negative or inconclusive results without metabolic activation (12 entries) compared with tests with metabolic activation (3 entries).
In Table 44, data on cell transformation in mammalian and human cells induced by nitroPAHs are tabulated. Studies on this end-point are available for 13 nitroPAHs. Positive results were obtained with 10 out of the 13 investigated nitroPAHs (see also summary in Table 45).
Table 44. Cell transformation in mammalian and human cells induced by nitroPAHs
|
Substance |
Cell (remarks) |
Resultsa |
Reference |
|
|
–MAb |
+MAb |
|||
|
1-Nitronaphthalene |
Mouse embryo cell line A31-1-13 clone of Balb/c-3T3 (LD50 464 µmol; tested up to cytotoxicity) |
± |
n.g. |
Matthews et al. (1993) |
|
2-Nitronaphthalene |
Primary Syrian hamster embryo cells (heterogenicity of cells, partial metabolically competent) |
+ |
n.g. |
Pienta (1980) |
|
2-Nitrofluorene |
Syrian hamster kidney cell BHK-21/cl13 and either human lung fibroblast cell line WI38 or human liver cells (no further details available) |
n.g. |
+ |
Purchase et al. (1978) |
|
Primary Syrian hamster embryo cells (heterogenicity of cells, MA by cultured hamster hepatocytes) |
– |
+ |
Poiley et al. (1979) |
|
|
Primary Syrian hamster embryo cells (heterogenicity of cells, MA by hamster S9-mix or cultured hamster hepatocytes) |
– |
+ |
Pienta (1980) |
|
|
Hamster BHK-21/Cl 13 cells |
+ |
+c |
Styles (1981) |
|
|
3-Nitrofluoranthene |
Syrian hamster embryo cells (heterogenicity of cells; target and X-irradiated feeder cells exposed) |
+ |
n.g. |
DiPaolo et al. (1983) |
|
1-Nitropyrene |
Syrian hamster embryo cells (heterogenicity of cells; target and X-irradiated feeder cells exposed) |
+ |
n.g. |
DiPaolo et al. (1983) |
|
Primary human diploid skin fibroblasts (effect only under anaerobic conditions; MA: mammalian nitroreductase) |
+ |
+c |
Howard et al. (1983b) |
|
|
Isolated rat tracheal epithelial cells |
– |
– |
Mitchell & Thomassen (1990) |
|
|
Mouse Balb 3T3 clone A31-1-1 cells |
+ |
n.g. |
Sheu et al. (1994) |
|
|
Isolated rat tracheal epithelial cells (cytotoxic effects, same results with parent PAH) |
(±) |
n.g. |
West & Rowland (1994) |
|
|
Isolated rat tracheal epithelial cells |
– |
n.g. |
Xiang et al. (1996); Ensell et al. (1998) |
|
|
2-Nitropyrene |
Mouse Balb 3T3 clone A31-1-1 cells |
+ |
n.g. |
Sheu et al. (1994) |
|
4-Nitropyrene |
Mouse Balb 3T3 clone A31-1-1 cells |
+ |
n.g. |
Sheu et al. (1994) |
|
4-Nitropyrene |
Isolated rat tracheal epithelial cells (cytotoxic effects, parent PAH less competent) |
(±) |
n.g. |
West & Rowland (1994) |
|
1,6-Dinitropyrene |
Isolated rat tracheal epithelial cells (cytotoxic effects, parent PAH less competent) |
+ |
n.g. |
West & Rowland (1994) |
|
1,8-Dinitropyrene |
Syrian hamster embryo cells (heterogenicity of cells; target and X-irradiated feeder cells exposed) |
+ |
n.g. |
DiPaolo et al. (1983) |
|
7-Nitrobenz[a]anthracene |
Mouse Balb 3T3 clone A31-1-1 cells |
± |
n.g. |
Sheu et al. (1994) |
|
6-Nitrochrysene |
Syrian hamster embryo cells (heterogenicity of cells; target and X-irradiated feeder cells exposed) |
+ |
n.g. |
DiPaolo et al. (1983) |
|
Mouse embryo cell line Balb 3T3clA31-1 (chrysene also negative results) |
– |
n.g. |
Sala et al. (1987) |
|
|
Mouse embryo fibroblast cell line C3H 10T1/2 (chrysene negative) |
(±) |
n.g. |
Sala et al. (1987) |
|
|
Syrian hamster embryo cells (heterogenicity of cells; target and X-irradiated feeder cells exposed; chrysene less effective) |
+ |
n.g. |
Sala et al. (1987) |
|
|
Isolated rat tracheal epithelial cells |
+ |
+c |
Mitchell & Thomassen (1990) |
|
|
Mouse Balb 3T3 clone A31-1-1 cells |
– |
n.g. |
Sheu et al. (1994) |
|
|
Isolated rat tracheal epithelial cells (cytotoxic effects, parent PAH less competent) |
+ |
n.g. |
West & Rowland (1994) |
|
|
6-Nitrobenzo[a]pyrene |
Syrian hamster embryo cells (heterogenicity of cells; target and X-irradiated feeder cells exposed; BaP more effective) |
+ |
n.g. |
DiPaolo et al. (1983) |
|
Primary human diploid skin fibroblasts (effect only under anaerobic conditions; MA: mammalian nitroreductase) |
+ |
+c |
Howard et al. (1983b) |
|
|
Syrian hamster embryo cells (heterogenicity of cells; target and X-irradiated feeder cells exposed; BaP more effective) |
+ |
n.g. |
Sala et al. (1987) |
|
|
Mouse embryo cell line Balb 3T3clA31-1 (clearly positive results with BaP) |
(±) |
n.g. |
Sala et al. (1987) |
|
|
Mouse embryo fibroblast cell line C3H 10T1/2 (BaP clearly positive) |
(+) |
n.g. |
Sala et al. (1987) |
|
|
Mouse Balb 3T3 clone A31-1-1 cells (clearly positive results with BaP) |
– |
n.g. |
Sheu et al. (1994) |
|
|
a |
The results in Table 44 are derived as follows: |
|
|
+ |
is a clear positive result, also given as + or positive by the authors of the publication. |
|
|
(±) |
is a weak positive result, also classified by the authors of the publication as ± or ?. |
|
|
(+) |
given where there were weak positive results with dose–response. |
|
|
± |
given when the result was inconclusive, which means that the result was negative, but testing was not performed up to cytotoxic concentrations. Further ± was given for weak positive effects without dose–response or without sufficient documentation for further assessment. |
|
|
– |
is a negative result. |
|
|
b |
MA = exogenous metabolic activation: microsomes, S9-mix or other metabolical activation systems (see remarks); n.g. = not given. |
|
|
c |
Increased by metabolic activation when compared with a positive response without metabolic activation. |
|
Table 45. Summary of genotoxicity in vitroa,b,c
|
Substance |
Bacteria |
Fungi |
Plants |
Mammalian cells |
Human cells |
|||||||||||||
|
GM-Ames |
GM other |
DD |
GM |
GC |
GM |
GM |
DD |
SCE |
CA |
MN |
CTd |
GM |
DD |
SCE |
CA |
MN |
CTd |
|
|
1-Nitronaphthalene –MA |
+ |
–e |
+ |
± |
– |
+ |
±e |
|||||||||||
|
1-Nitronaphthalene +MA |
+ |
±e |
(±)e |
+ |
+ |
–e |
–e |
|||||||||||
|
2-Nitronaphthalene –MA |
+ |
+ |
+ |
+ |
||||||||||||||
|
2-Nitronaphthalene +MA |
+ |
+ |
+ |
–2/2 |
+ |
+ |
||||||||||||
|
1,3-Dinitronaphthalene –MA |
+ |
+ |
||||||||||||||||
|
1,3-Dinitronaphthalene +MA |
||||||||||||||||||
|
1,5-Dinitronaphthalene –MA |
+ |
+ |
||||||||||||||||
|
1,5-Dinitronaphthalene +MA |
+ |
+ |
||||||||||||||||
|
1,8-Dinitronaphthalene –MA |
+ |
|||||||||||||||||
|
1,8-Dinitronaphthalene +MA |
+ |
+ |
||||||||||||||||
|
2,7-Dinitronaphthalene –MA |
+ |
|||||||||||||||||
|
2,7-Dinitronaphthalene +MA |
(±)e |
|||||||||||||||||
|
2,3,5-Trinitronaphthalene –MA |
+ |
+ |
||||||||||||||||
|
2,3,5-Trinitronaphthalene +MA |
+ |
|||||||||||||||||
|
1,3,6,8-Tetranitronaphthalene –MA |
+ |
|||||||||||||||||
|
1,3,6,8-Tetranitronaphthalene +MA |
+ |
|||||||||||||||||
|
3-Nitroacenaphthene –MA |
+ |
|||||||||||||||||
|
3-Nitroacenaphthene +MA |
||||||||||||||||||
|
5-Nitroacenaphthene –MA |
+ |
(±) |
+ |
|||||||||||||||
|
5-Nitroacenaphthene +MA |
+ |
+ |
+ |
|||||||||||||||
|
1-Nitrofluorene –MA |
+ |
|||||||||||||||||
|
1-Nitrofluorene +MA |
+ |
|||||||||||||||||
|
2-Nitrofluorene –MA |
+ |
+ |
+ |
–2/2 |
+ |
+ |
+ |
–e |
+ |
+ |
+ |
– |
||||||
|
2-Nitrofluorene +MA |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
–e |
||||||||
|
3-Nitrofluorene –MA |
+ |
+ |
||||||||||||||||
|
3-Nitrofluorene +MA |
+ |
|||||||||||||||||
|
4-Nitrofluorene –MA |
+ |
|||||||||||||||||
|
4-Nitrofluorene +MA |
+ |
|||||||||||||||||
|
2,5-Dinitrofluorene –MA |
+ |
|||||||||||||||||
|
2,5-Dinitrofluorene +MA |
+ |
|||||||||||||||||
|
2,7-Dinitrofluorene –MA |
+ |
–e |
+ |
|||||||||||||||
|
2,7-Dinitrofluorene +MA |
+ |
+ |
+ |
±e |
||||||||||||||
|
2-Nitroanthracene –MA |
+ |
|||||||||||||||||
|
2-Nitroanthracene +MA |
+ |
|||||||||||||||||
|
9-Nitroanthracene –MA |
+ |
+ |
||||||||||||||||
|
9-Nitroanthracene +MA |
+ |
–e |
–2/2 |
+ |
||||||||||||||
|
9,10-Dinitroanthracene –MA |
–e |
|||||||||||||||||
|
9,10-Dinitroanthracene +MA |
±e |
|||||||||||||||||
|
1-Nitrofluoranthene –MA |
+ |
|||||||||||||||||
|
1-Nitrofluoranthene +MA |
+ |
|||||||||||||||||
|
2-Nitrofluoranthene –MA |
+ |
+ |
||||||||||||||||
|
2-Nitrofluoranthene +MA |
+ |
+ |
+ |
|||||||||||||||
|
3-Nitrofluoranthene –MA |
+ |
+ |
–e |
–e |
+ |
|||||||||||||
|
3-Nitrofluoranthene +MA |
+ |
+ |
+ |
+ |
+ |
+ |
±e |
|||||||||||
|
7-Nitrofluoranthene –MA |
+ |
|||||||||||||||||
|
7-Nitrofluoranthene +MA |
+ |
|||||||||||||||||
|
8-Nitrofluoranthene –MA |
+ |
+ |
–e |
|||||||||||||||
|
8-Nitrofluoranthene +MA |
+ |
+ |
||||||||||||||||
|
1,2-Dinitrofluoranthene –MA |
+ |
|||||||||||||||||
|
1,2-Dinitrofluoranthene +MA |
+ |
|||||||||||||||||
|
1,3-Dinitrofluoranthene –MA |
+ |
|||||||||||||||||
|
1,3-Dinitrofluoranthene +MA |
+ |
|||||||||||||||||
|
2,3-Dinitrofluoran-thene –MA |
+ |
|||||||||||||||||
|
2,3-Dinitrofluoranthene +MA |
+ |
|||||||||||||||||
|
2,4-Dinitrofluoranthene –MA |
+ |
|||||||||||||||||
|
2,4-Dinitrofluoranthene +MA |
+ |
|||||||||||||||||
|
2,5-Dinitrofluoranthene –MA |
+ |
|||||||||||||||||
|
2,5-Dinitrofluoranthene +MA |
+ |
|||||||||||||||||
|
3,4-Dinitrofluoranthene –MA |
+ |
+ |
||||||||||||||||
|
3,4-Dinitrofluoranthene +MA |
+ |
|||||||||||||||||
|
3,7-Dinitrofluoranthene –MA |
+ |
+ |
+ |
|||||||||||||||
|
3,7-Dinitrofluoranthene +MA |
+ |
+ |
(±)e |
|||||||||||||||
|
3,9-Dinitrofluoranthene –MA |
+ |
+ |
+ |
|||||||||||||||
|
3,9-Dinitrofluoranthene +MA |
+ |
+ |
(±)e |
|||||||||||||||
|
1,2,4-Trinitrofluoranthene –MA |
+ |
|||||||||||||||||
|
1,2,4-Trinitrofluoranthene +MA |
+ |
|||||||||||||||||
|
1,2,5-Trinitrofluoranthene –MA |
+ |
|||||||||||||||||
|
1,2,5-Trinitrofluoranthene +MA |
+ |
|||||||||||||||||
|
2,3,5-Trinitrofluoranthene –MA |
+ |
|||||||||||||||||
|
2,3,5-Trinitrofluoranthene +MA |
+ |
|||||||||||||||||
|
1-Nitropyrene –MA |
+ |
+ |
+ |
–2/2 |
+ |
– |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
|||||
|
1-Nitropyrene +MA |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
–e |
+ |
+ |
+ |
|||||||
|
2-Nitropyrene –MA |
+ |
–e |
+ |
–2/2 |
||||||||||||||
|
2-Nitropyrene +MA |
+ |
+ |
+ |
+ |
||||||||||||||
|
4-Nitropyrene –MA |
+ |
+ |
+ |
+ |
+ |
|||||||||||||
|
4-Nitropyrene +MA |
+ |
|||||||||||||||||
|
1,3-Dinitropyrene –MA |
+ |
+ |
+ |
–e |
+ |
+ |
+ |
+ |
||||||||||
|
1,3-Dinitropyrene +MA |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
±e |
|||||||
|
1,6-Dinitropyrene –MA |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
||||||
|
1,6-Dinitropyrene +MA |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
± |
+ |
+ |
+ |
||||||
|
1,8-Dinitropyrene –MA |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
±e |
|||||
|
1,8-Dinitropyrene +MA |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
± |
+ |
||||||
|
2,7-Dinitropyrene –MA |
+ |
|||||||||||||||||
|
2,7-Dinitropyrene +MA |
||||||||||||||||||
|
1,3,6-Trinitropyrene –MA |
+ |
+ |
+ |
–e |
+ |
|||||||||||||
|
1,3,6-Trinitropyrene +MA |
+ |
(±)e |
||||||||||||||||
|
1,3,6,8-Tetranitropyrene –MA |
+ |
–e |
+ |
–e |
–2/2 |
|||||||||||||
|
1,3,6,8-Tetranitropyrene +MA |
||||||||||||||||||
|
7-Nitrobenz[a]anthracene –MA |
+ |
– |
± |
|||||||||||||||
|
7-Nitrobenz[a]anthracene +MA |
+ |
+ |
||||||||||||||||
|
2-Nitrochrysene –MA |
+ |
|||||||||||||||||
|
2-Nitrochrysene +MA |
+ |
|||||||||||||||||
|
5-Nitrochrysenef –MA |
+ |
|||||||||||||||||
|
5-Nitrochrysenef +MA |
+ |
|||||||||||||||||
|
6-Nitrochrysene –MA |
+ |
+ |
(±) |
+ |
||||||||||||||
|
6-Nitrochrysene +MA |
+ |
+ |
+ |
+ |
+ |
|||||||||||||
|
3-Nitrobenzo[k]-fluoranthene –MA |
+ |
|||||||||||||||||
|
3-Nitrobenzo[k]-fluoranthene +MA |
||||||||||||||||||
|
7-Nitrobenzo[k]-fluoranthene –MA |
±e |
|||||||||||||||||
|
7-Nitrobenzo[k]-fluoranthene +MA |
||||||||||||||||||
|
1-Nitrobenzo[a]pyrene –MA |
+ |
– |
||||||||||||||||
|
1-Nitrobenzo[a]pyrene +MA |
+ |
+ |
||||||||||||||||
|
2-Nitrobenzo[a]pyrene –MA |
+ |
|||||||||||||||||
|
2-Nitrobenzo[a]pyrene +MA |
+ |
|||||||||||||||||
|
3-Nitrobenzo[a]pyrene –MA |
+ |
– |
||||||||||||||||
|
3-Nitrobenzo[a]pyrene +MA |
+ |
+ |
||||||||||||||||
|
6-Nitrobenzo[a]pyrene –MA |
+ |
– |
± |
+ |
+ |
|||||||||||||
|
6-Nitrobenzo[a]pyrene +MA |
+ |
+ |
(±) |
+ |
||||||||||||||
|
1-Nitrobenzo[e]pyrene –MA |
+ |
|||||||||||||||||
|
1-Nitrobenzo[e]pyrene +MA |
+ |
|||||||||||||||||
|
3-Nitrobenzo[e]pyrene –MA |
+ |
|||||||||||||||||
|
3-Nitrobenzo[e]pyrene +MA |
+ |
|||||||||||||||||
|
4-Nitrobenzo[e]pyrene –MA |
+ |
|||||||||||||||||
|
4-Nitrobenzo[e]pyrene +MA |
||||||||||||||||||
|
1,3-Dinitrobenzo[e]pyrene –MA |
+ |
|||||||||||||||||
|
1,3-Dinitrobenzo[e]pyrene +MA |
||||||||||||||||||
|
1,6-Dinitrobenzo[e]pyrene –MA |
+ |
|||||||||||||||||
|
1,6-Dinitrobenzo[e]pyrene +MA |
+ |
|||||||||||||||||
|
1,8-Dinitrobenzo[e]pyrene –MA |
±e |
|||||||||||||||||
|
1,8-Dinitrobenzo[e]pyrene +MA |
±e |
|||||||||||||||||
|
3,6-Dinitrobenzo[a]pyrene –MA |
+ |
|||||||||||||||||
|
3,6-Dinitrobenzo[a]pyrene +MA |
||||||||||||||||||
|
3-Nitrodibenz[a,h]anthracene –MA |
± |
|||||||||||||||||
|
3-Nitrodibenz[a,h]anthracene +MA |
+ |
|||||||||||||||||
|
7-Nitrodibenz[a,h]anthracene –MA |
±e |
|||||||||||||||||
|
7-Nitrodibenz[a,h]anthracene +MA |
||||||||||||||||||
|
9-Nitrodibenz[a,c]anthracene –MA |
±e |
|||||||||||||||||
|
9-Nitrodibenz[a,c]-anthracene +MA |
||||||||||||||||||
|
3-Nitroperylene –MA |
+ |
|||||||||||||||||
|
3-Nitroperylene +MA |
+ |
|||||||||||||||||
|
3,6-Dinitroperylene –MA |
+ |
|||||||||||||||||
|
3,6-Dinitroperylene +MA |
+ |
|||||||||||||||||
|
3,7-Dinitroperylene –MA |
+ |
|||||||||||||||||
|
3,7-Dinitroperylene +MA |
+ |
|||||||||||||||||
|
1-Nitrocoronene –MA |
+ |
|||||||||||||||||
|
1-Nitrocoronene +MA |
+ |
|||||||||||||||||
|
4-Nitrobenzo[ghi]perylene –MA |
||||||||||||||||||
|
4-Nitrobenzo[ghi]perylene +MA |
+ |
|||||||||||||||||
|
7-Nitrobenzo[ghi]perylene –MA |
||||||||||||||||||
|
7-Nitrobenzo[ghi]perylene +MA |
±e |
|||||||||||||||||
|
a |
Data on 24 nitrated phenanthrenes were not presented in this table but in Tables 40 and 46. |
|
b |
CA = chromosomal aberration; CT = cell transformation; DD = DNA damage; GC = gene conversion and mitotic recombination; GM = gene mutation; GM-Ames = gene mutation in Salmonella microsome assay; GM other = gene mutation tests in bacteria other than the Salmonella microsome assay; MA = metabolic activation; MN = micronuclei; SCE = sister chromatid exchange. |
|
c |
+ = positive (>0% of presented study results are positive); – = negative (>50% of the presented study results are negative); on negative results, the proportion of negative assays at this end-point was indicated; (±) = marginal; ± = inconclusive. |
|
d |
Mechanisms other than genotoxicity might transform cells in this assay. |
|
e |
Only one study available (not marked with positive results). |
|
f |
Structural assignment of the test substance was not provided. |
Table 46. Overview of genotoxicity of nitroPAHs in vitro and in vivo compared with indications of carcinogenic effects of these compounds
|
Substance |
Genotoxicity in vitroa |
Genotoxicity in vivoa |
Carcinogenicitya,k |
||||||
|
Mutagenic potencyb,c |
nd |
Total end-pointse |
End-points in mammalian systemsf |
Resultg |
End-pointsh Drosophila |
End-pointsi rodents |
Resultj |
||
|
1-Nitronaphthalene |
+ (–S9) |
9 |
8/0/1/2 |
5/0/0/2 |
positive |
1/0/0/0 |
0 |
positive |
database insufficient |
|
2-Nitronaphthalene |
+ (–S9) |
9 |
6/0/0/1 |
4/0/0/1 |
positive |
0 |
2/0/0/1 |
inconclusive |
database insufficient |
|
1,3-Dinitronaphthalene |
+ |
2 |
2/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1,5-Dinitronaphthalene |
+ (–S9) |
3 |
2/0/0/0 |
0 |
positive |
1/0/0/0 |
0 |
positive |
n.d. |
|
1,8-Dinitronaphthalene |
++ (–S9) |
1 |
2/0/0/1 |
0 |
inconclusive |
n.d. |
n.d. |
||
|
2,7-Dinitronaphthalene |
n.d. |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
|||
|
2,3,5-Trinitronaphthalene |
++ (+S9) |
1 |
2/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1,3,6,8-Tetranitronaphthalene |
++ (–S9) |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
3-Nitroacenaphthene |
++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
5-Nitroacenaphthene |
++ (+S9) |
9 |
4/0/0/0 |
1/0/0/0 |
positive |
0 |
1/0/1/0 |
inconclusive |
positive |
|
1-Nitrofluorene |
n.d. |
1/0/0/0 |
1/0/0/0 |
positive |
n.d. |
n.d. |
|||
|
2-Nitrofluorene |
++ (–S9) |
10 |
12/0/0/2 |
6/0/0/1 |
positive |
6/1/0/1 |
1/0/1/0 |
positive |
positive |
|
3-Nitrofluorene |
+ |
1 |
2/0/0/0 |
1/0/0/0 |
positive |
n.d. |
n.d. |
||
|
4-Nitrofluorene |
n.d. |
1/0/0/0 |
1/0/0/0 |
positive |
n.d. |
n.d. |
|||
|
2,5-Dinitrofluorene |
++++ |
2 |
1/0/0/0 |
0 |
positive |
n.d. |
database insufficient |
||
|
2,7-Dinitrofluorene |
++++ |
7 |
5/0/1/1 |
2/0/1/0 |
positive |
0 |
1/0/0/1 |
(negative) |
database insufficient |
|
2-Nitroanthracene |
++++ |
2 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
9-Nitroanthracene |
+ (+S9) |
3 |
4/0/0/1 |
2/0/0/1 |
positive |
1/0/0/0 |
1/0/0/1 |
positive |
n.d. |
|
9,10-Dinitroanthracene |
n.d. |
1/0/1/0 |
0 |
inconclusive |
n.d. |
n.d. |
|||
|
1-Nitrophenanthrene |
+++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
2-Nitrophenanthrene |
+++ |
2 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
3-Nitrophenanthrene |
+++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
9-Nitrophenanthrene |
+++ (–S9) |
2 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1,5-Dinitrophenanthrene |
+ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1,6-Dinitrophenanthrene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1,10-Dinitrophenanthrene |
+ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
2,6-Dinitrophenan-threne |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
2,7-Dinitrophenanthrene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
2,9-Dinitrophenanthrene |
+++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
2,10-Dinitrophenanthrene |
+++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
3,5-Dinitrophenanthrene |
+++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
3,6-Dinitrophenanthrene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
3,10-Dinitrophenanthrene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
4,9-Dinitrophenanthrene |
+ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
4,10-Dinitrophenanthrene |
+++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1,5,9-Trinitrophenanthrene |
+++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1,5,10-Trinitrophenanthrene |
++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1,6,9-Trinitrophenanthrene |
+++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1,7,9-Trinitrophenanthrene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
2,5,10-Trinitrophenanthrene |
+++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
2,6,9-Trinitrophenanthrene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
3,5,10-Trinitrophenanthrene |
++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
3,6,9-Trinitrophenanthrene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1-Nitrofluoranthene |
+++ |
3 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
2-Nitrofluoranthene |
+++ (–S9) |
4 |
3/0/0/0 |
1/0/0/0 |
positive |
n.d. |
database insufficient |
||
|
3-Nitrofluoranthene |
++++ |
17 |
7/0/1/0 |
5/0/1/0 |
positive |
1/0/0/1 |
1/0/0/0 |
inconclusive |
positive |
|
7-Nitrofluoranthene |
+++ |
3 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
8-Nitrofluoranthene |
+++++ |
2 |
3/0/0/0 |
1/0/0/0 |
positive |
n.d. |
n.d. |
||
|
1,2-Dinitrofluoranthene |
++++ |
2 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1,3-Dinitrofluoranthene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
2,3-Dinitrofluoranthene |
+++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
2,4-Dinitrofluoranthene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
2,5-Dinitrofluoranthene |
+++ (+S9) |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
3,4-Dinitrofluoranthene |
++++ |
3 |
2/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
3,7-Dinitrofluoranthene |
++++++ |
2 |
4/0/0/0 |
2/0/0/0 |
positive |
0 |
1/0/0/0 |
positive |
positive |
|
3,9-Dinitrofluoranthene |
++++++ |
2 |
4/0/0/0 |
2/0/0/0 |
positive |
0 |
1/0/0/0 |
positive |
positive |
|
1,2,4-Trinitrofluoranthene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1,2,5-Trinitrofluoranthene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
2,3,5-Trinitrofluoranthene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1-Nitropyrene |
+++ (–S9) |
41 |
11/0/0/1 |
6/0/0/0 |
positive |
0 |
4/0/0/0 |
positive |
positive |
|
2-Nitropyrene |
++++ |
4 |
5/0/0/1 |
2/0/0/1 |
positive |
n.d. |
(positive) |
||
|
4-Nitropyrene |
++++ |
2 |
4/0/0/0 |
1/0/0/0 |
positive |
n.d. |
positive |
||
|
1,3-Dinitropyrene |
+++++ |
12 |
12/0/1/1 |
7/0/1/0 |
positive |
0 |
1/1/0/0 |
(positive) |
positive |
|
1,6-Dinitropyrene |
++++++ |
15 |
13/0/0/0 |
8/0/0/0 |
positive |
0 |
3/1/1/0 |
positive |
positive |
|
1,8-Dinitropyrene |
++++++ |
18 |
14/0/2/0 |
9/0/2/0 |
positive |
2/0/0/1 |
1/1/0/0 |
positive |
positive |
|
2,7-Dinitropyrene |
+++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1,3,6-Trinitropyrene |
+++++ |
6 |
6/1/0/1 |
2/1/0/0 |
positive |
n.d. |
n.d. |
||
|
1,3,6,8-Tetranitropyrene |
+++++ |
6 |
5/0/0/3 |
1/0/0/1 |
inconclusive |
n.d. |
n.d. |
||
|
7-Nitrobenz[a]-anthracene |
++ (+S9) |
1 |
2/0/0/0 |
0 |
positive |
n.d. |
(positive) |
||
|
2-Nitrochrysene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
5-Nitrochrysenel |
+ (+S9) |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
6-Nitrochrysene |
+++ (+S9) |
5 |
4/0/0/0 |
2/0/0/0 |
positive |
0 |
1/0/0/0 |
positive |
positive |
|
3-Nitrobenzo[k]-fluoranthene |
+++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
7-Nitrobenzo[k]-fluoranthene |
n.d. |
1/0/1/0 |
0 |
inconclusive |
n.d. |
n.d. |
|||
|
1-Nitrobenzo[a]pyrene |
+++ (–S9) |
4 |
2/0/0/0 |
1/0/0/0 |
positive |
n.d. |
database insufficient |
||
|
2-Nitrobenzo[a]pyrene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
(positive) |
||
|
3-Nitrobenzo[a]pyrene |
+++ (–S9) |
4 |
2/0/0/0 |
1/0/0/0 |
positive |
n.d. |
database insufficient |
||
|
6-Nitrobenzo[a]pyrene |
+++ (+S9) |
7 |
3/1/0/0 |
1/1/0/0 |
positive |
n.d. |
(positive) |
||
|
1-Nitrobenzo[e]pyrene |
++ (+S9) |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
database insufficient |
||
|
3-Nitrobenzo[e]pyrene |
++++ |
3 |
1/0/0/0 |
0 |
positive |
n.d. |
database insufficient |
||
|
4-Nitrobenzo[e]pyrene |
+++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1,3-Dinitrobenzo[e]-pyrene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1,6-Dinitrobenzo[a]-pyrene |
n.d. |
n.d. |
database insufficient |
||||||
|
1,6-Dinitrobenzo[e]-pyrene |
++ (–S9) |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1,8-Dinitrobenzo[e]-pyrene |
n.d. |
1/0/1/0 |
0 |
inconclusive |
n.d. |
n.d. |
|||
|
3,6-Dinitrobenzo[a]-pyrene |
++++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
(positive) |
||
|
3-Nitrodibenz[a,h]-anthracene |
+++ (+S9) |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
7-Nitrodibenz[a,h]-anthracene |
n.d. |
1/0/1/0 |
0 |
inconclusive |
n.d. |
(positive) |
|||
|
9-Nitrodibenz[a,c]-anthracene |
n.d. |
1/0/1/0 |
0 |
inconclusive |
n.d. |
database insufficient |
|||
|
3-Nitroperylene |
++++ |
4 |
1/0/0/0 |
0 |
positive |
n.d. |
(positive) |
||
|
3,6-Dinitroperylene |
++++ |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
3,7-Dinitroperylene |
++++(–S9) |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
1-Nitrocoronene |
+ (+S9) |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
4-Nitrobenzo[ghi]-perylene |
++++ (a) |
1 |
1/0/0/0 |
0 |
positive |
n.d. |
n.d. |
||
|
7-Nitrobenzo[ghi]-perylene |
n.d. |
1/0/1/0 |
0 |
inconclusive |
n.d. |
n.d. |
|||
|
a |
n.d. = not determined. |
|
b |
Maximum mean mutagenic potency of nitroPAHs in the Salmonella microsome assay presented in Table 40; + = <10 revertants/nmol; ++ = >0, <100 revertants/nmol; +++ = >00, <1000 revertants/nmol; ++++ = >000, <10 000 revertants/nmol; +++++ = >0 000, <100 000 revertants/nmol; ++++++ = >00 000 revertants/nmol. |
|
c |
The effects of metabolic activation in the Salmonella microsome assay are presented in parentheses; +S9 = mutagenic potency is maximal with metabolic activation, but the value is in the same order of magnitude as without metabolic activation; +S9 (bold typed) = difference is one or more orders of magnitude; –S9 = mutagenic potency is maximal without metabolic activation, but the value is in the same order of magnitude as with metabolic activation; –S9 (bold typed) = difference is one or more orders of magnitude; a = tested only with metabolic activation; no entry = tested only without metabolic activation. |
|
d |
Number of studies available for determination of the highest mutagenic potency in the Salmonella microsome assay. |
|
e |
End-points in bacterial and eukaryotic test systems; end-points corresponding to columns in Table 45 (cell transformation not included): total number of investigated end-points in Table 45 / number of end-points with weak positive results / number of end-points with inconclusive results / number of end-points with negative results. |
|
f |
End-points in mammalian test systems including human cells; end-points corresponding to columns in Table 45 (cell transformation not included): total number of investigated end-points in Table 45 / number of end-points with weak positive results / number of end-points with inconclusive results / number of end-points with negative results. |
|
g |
Normal type: limited database (data on fewer than three total end-points available, see Table 45) or inconclusive results, bold type: data on three or more total end-points available and majority of end-points positive, at least three end-points. |
|
h |
Number of investigated end-points in Table 50 (Genotoxicity of nitroPAHs to Drosophila melanogaster) / number of end-points with weak positive result / number of end-points with inconclusive results / number of end-points with negative results. |
|
i |
Number of investigated end-points in Table 49 (Genotoxicity of nitroPAHs in rodents) / number of end-points with weak positive result / number of end-points with inconclusive results / number of end-points with negative results. |
|
j |
Normal type: limited database (only data on one end-point available) or inconclusive results; bold type: data on two or more end-points available and majority of end-points positive; parentheses: limited validity. |
|
k |
Evaluation of carcinogenicity, see Table 52; (positive): limited evidence, only one positive study; positive in bold type: more than one positive study. |
|
l |
Structural assignment of the test substance was not provided. |
Some nitroPAHs are extremely mutagenic in bacteria. For example, when tested in S. typhimurium (strain TA98) in the absence of exogenous metabolizing enzymes (i.e., –S9), 1-nitropyrene, 1,3-dinitropyrene, 1,6-dinitropyrene and 1,8-dinitropyrene are reported to be 200, 63 000, 80 000 and 110 000 times more mutagenic than BaP (+S9) (see Table 47). This led to an earlier conclusion that nitroPAHs are among the most important mutagens in ambient aerosol samples. This acute sensitivity of S. typhimurium to nitroPAHs is attributed to the presence of native nitroreductase enzymes, which initiate the metabolism of nitroPAH to their ultimate mutagenic metabolites (arylhydroxylamines; Durant et al., 1996). Other bacterial assays were used as an alternative to reversion assays — e.g., S. typhimurium TM677 (a quantitative bacterial forward mutation assay, based on resistance to 8-azaguanine) — and it was found that, in general, these results were consistent with the results found in S. typhimurium TA98: that without metabolic activation, for example, the dinitropyrenes were 2–3 times more mutagenic than 1-nitropyrene, with an order of potency of 1,8-dinitropyrene > 1,6-dinitropyrene > 1-nitropyrene, and further that there is about a 1000-fold reduction in mutagenicity for the dinitropyrenes in the presence compared with the absence of metabolic activation (Busby et al., 1994a).
Table 47. Comparison of potency of gene mutation assays in human versus bacterial cells for nitroPAHs
|
Chemical |
S. typhimurium TA98 |
S. typhimurium TM677 |
Human MCL-5 cellsc |
Human |
|
Reversion |
Forward mutation |
Forward mutation |
Forward mutation |
|
|
BaP –S9 |
0 |
|||
|
BaP +S9 |
2.3 |
1 |
||
|
1-Nitropyrene |
453 |
19 000 |
9.1 |
0.025 |
|
1,6-Dinitropyrene |
184 000 |
3 200 000 |
0.8 |
0.28 |
|
1,8-Dinitropyrene |
254 000 |
9 400 000 |
1.5 |
0.046 |
a Rosenkranz & Mermelstein (1983).
b Busby et al. (1994a).
c Busby et al. (1994b) human B-lymphoblastoid cells.
d Durant et al. (1996) human B-lymphoblastoid cells. Comparison relative to BaP =1.
Further in vitro studies in mammalian cells have shown that these results in bacteria may be misleading, due to the high sensitivity of this bacterial assay. When a forward mutation assay based on human B-lymphoblastoid cells (cell line designated h1A1v2; constitutively expresses the cytochrome CYP1A1 necessary for the metabolism of many promutagens) was used, it was found that nitroPAHs as a group were less mutagenic than PAHs (see Table 47; see also Tables 43 and 48). The most active nitroPAH tested (1,6-dinitropyrene) had a minimum mutagen concentration ~3-fold lower than that of BaP (Durant et al., 1996). In a test on the mutagenicity of mono- and dinitropyrenes in another cell line (MCL-5 containing endogenous CYP1A1 and additionally two plasmids expressing cDNAs for four additional P450s: CYP1A2, CYP2A6, CYP2E1 and CYP3A4) of human B-lymphoblastoid cells, there was an 11-fold difference between the most potent (1,6-dinitropyrene) and 1-nitropyrene (Busby et al., 1994b; see Table 47).
For the specific examples in Table 47, these wide differences in sensitivity to nitroPAHs in prokaryotic and eukaryotic cells are likely due to interspecific differences in the types and concentrations of metabolizing enzymes present as well as in DNA repair or perhaps depend on other factors, such as the duration of treatment (Durant et al., 1996).
The mutagenicities of 1- and 2-nitronaphthalene compared with BaP have also been determined in forward mutation assays using MCL-5 and h1A1v2 (Grosovsky et al., 1999; see also Table 48). Mutagenicity of the compounds was determined at the heterozygous tk locus and the hemizygous hprt locus. Whereas BaP induced a significant increase in mutation frequency at the tk and hprt loci, 2-nitronaphthalene showed only a significant increase in mutation frequency at the tk locus and 1-nitronaphthalene at neither. In a further study, 2-nitronaphthalene showed no significant induction of mutants using L3 cells, which are isogenic with MCL-5 cells but are distinguished by the absence of transfected plasmids (Sasaki et al., 1999). Mutagenicity induced at the tk locus suggests that the genotoxicity of nitroPAHs in human cells requires oxidative metabolism.
Table 48. Comparison of 2-nitronaphthalene, 3-nitrobenzanthrone, 2-nitrodibenzopyranone and nitropyrene lactones in human B-lymphoblastoid cell lines MCL-5 and h1A1v2
|
Compound |
Concentration |
Induced mutation frequencya at tk and hprt loci at the highest concentration testedb,c |
|||
|
h1A1v2 |
MCL-5 |
||||
|
tk |
hprt |
tk |
hprt |
||
|
2-Nitronaphthalene |
462 |
96.7 |
0 |
83.6 |
0 |
|
2-Nitrodibenzopyranone |
4.15 |
16.7 |
0 |
7.2 |
0 |
|
Nitropyrene lactones |
3.77 |
37.6 |
14.3 |
27.1 |
46.4 |
|
3-Nitrobenzanthrone |
3.64 |
156.1 |
35.6 |
62.6 |
86.7 |
|
a |
Induced mutation frequency is defined as the number of mutants per 106 viable cells minus the number of background mutants per 106 viable cells. |
|
b |
Phousongphouang et al. (2000). |
|
c |
Grosovsky et al. (1999). |
Data on genotoxicity in vitro are available on 95 nitroPAHs (see Table 46); for 74 nitroPAHs, however, only one or two end-points, mainly in bacterial test systems, were investigated. Most of these substances (67 out of 74) showed positive results (marked in Table 46 by normal type "positive"); no conclusion could be drawn with eight nitroPAHs (marked in Table 46 with "inconclusive").
A sufficient database also including eukaryotic test systems has been found only with 21 nitroPAHs. In Table 46, 19 nitroPAHs with a clear genotoxic effect in vitro were marked by a bold type "positive," which means that three or more end-points in Table 45 are judged positive and at least one end-point in eukaryotic test systems showed positive results. With one nitroPAH (1,3,6,8-tetranitropyrene), inconsistent results were observed.
Genotoxicity studies of nitroPAHs in rodents in vivo are given in Table 49 and in Drosophila melanogaster in Table 50. The Task Group noted that several of the studies listed in Table 49 are of limited validity. Moreover, although inhalation is the main exposure route in humans, no (long-term) inhalation study on any nitroPAH is available. Exposure directly in the lung was performed by instillation for 1-nitropyrene or by implantation for 1,6-dinitropyrene. In both cases, genotoxic effects were observed, in particular gene mutations with 1,6-dinitropyrene.
Table 49. Genotoxicity of nitroPAHs in rodents in vivoa
|
Purity |
Species; strain; sex |
Test type |
Route; dose |
Test conditions; comments |
Resultb |
References |
|
2-Nitronaphthalene |
||||||
|
n.g. |
Mouse (5/group); Swiss-Webster; m |
Host-mediated mitotic recombination assay |
Gavage; 0 or 1300 mg/kg bw |
Mice injected i.p. with S. cerevisiae D3 and 4 h later yeast isolated from peritoneal fluid for further cultivation, vehicle DMSO; toxic effects |
– |
Simmon et al. (1979) |
|
n.g. |
Mouse (4–6/group); |
Host-mediated reverse gene mutation assay |
Gavage; 0 or 1300 mg/kg bw |
Mice injected i.p. with S. typhimurium TA1538 and 4 h later bacteria isolated from peritoneal fluid for further cultivation, vehicle DMSO; toxic effects |
– |
Simmon et al. (1979) |
|
n.g. |
Mouse (3/group); |
Host-mediated reverse gene mutation assay |
i.m. injection; 0 or 125 mg/kg bw |
Mice injected i.p. with S. typhimurium TA1538 or TA1530 and 4 h later bacteria isolated from peritoneal fluid for further cultivation, vehicle DMSO; |
+ |
Simmon et al. (1979) |
|
5-Nitroacenaphthene |
||||||
|
84.9% |
Mouse (5/group); |
Micronucleus test |
i.p. injection; 0, 250, 425, 500, 850, 1000 or 1700 mg/kg bw in m; 0, 212, 425, 850 or 1700 mg/kg bw in f |
Micronuclei in peripheral blood determined after 0, 24, 48, 72 h; mortality at high dose, no data on cytotoxicity, significant effects after 72 h in m in high-dose groups (marginal in f), judged by the authors as negative |
± |
Morita et al. (1997) |
|
84.9% |
Rat (3/group); Sprague-Dawley; m + f |
Micronucleus test |
i.p. injection; 425, 850 or 1700 mg/kg bw |
Micronuclei in peripheral blood determined after 0, 24, 48, 72 h; all rats died in high-dose group 24 h after injection; no data on cytotoxicity |
– |
Morita et al. (1997) |
|
2-Nitrofluorene |
||||||
|
n.g. |
Rat (2–3/group); F344; m |
Micronucleus test |
Gavage (2 equal doses 24 h apart); 0 or 125 mg/kg bw |
Micronuclei in liver and bone marrow of 4-week-old rats determined 24 h after the 2nd gavage, hepatocytes isolated by collagenase perfusion; negative results with bone marrow, but one dose |
+ |
Parton & Garriott (1997) |
|
n.g. |
Rat (5/group); |
Micronucleus test |
Gavage (4 equal doses 24 h apart); 0, 250, 500 or 1000 mg/kg bw |
Micronuclei in liver and bone marrow of 4-week-old rats determined 24 h after the 4th gavage, hepatocytes isolated from formalin-fixed liver; marginal results with bone marrow |
+ |
Parton & Garriott (1997) |
|
n.g. |
Rat (3–6/group); Wistar; m |
Unscheduled DNA synthesis in the liver |
Gavage; 0, 12.5, 25 or 50 mg/kg bw |
Hepatocytes examined after 12 or 24 h, vehicle corn oil; no data on toxicity, effect not dose dependent, TS less effective than the metabolite 2-acetylaminofluorene |
+ |
Beije & Möller (1988b) |
|
n.g. |
Rat (2–3/group); |
Unscheduled DNA synthesis in the liver |
Gavage; 0, 12.5 or 25 mg/kg bw |
Hepatocytes examined after 24 or 36 h, vehicle corn oil; no data on toxicity, this strain less sensitive than Wistar; TS less effective than the metabolite N-acetyl-2-aminofluorene (2-acetylaminofluorene) |
+ |
Beije & Möller (1988b) |
|
n.g. |
Mouse (3–14/group); |
Micronucleus test |
Injection (not specified); 0, 50, 100, 200 or 500 mg/kg bw |
Micronuclei determined in bone marrow cells after 48 h, vehicle corn oil/DMSO; no data on cytotoxicity |
–* |
Sakitani & Suzuki (1986) |
|
n.g. |
Mouse (6/group); ddY; m |
Micronucleus test |
i.p. injection; 0 or 80 mg/kg bw (one dose) |
Micronuclei determined in peripheral reticulocytes after 0, 24, 48, 72, 96 h, vehicle olive oil; no data on toxicity, effective positive control |
–* |
Murakami et al. (1996) |
|
n.g. |
Mouse (3/group); |
Host-mediated DNA repair assay |
i.v. injection; 0 or 105 mg/kg bw (one dose) |
Mice injected i.p. with E. coli bacterial mix, solvent DMSO, 2 h later organ homogenates prepared and survival of DNA repair-proficient and -deficient bacteria determined; no data on toxicity, no effect in any organ (liver, lung, spleen, kidney); positive results with BaP |
–* |
Heussen et al. (1990) |
|
n.g. |
Mouse (4–6/group); Swiss-Webster; m |
Host-mediated mitotic recombination assay |
Gavage; 0 or 1600 mg/kg bw |
Mice injected i.p. with S. cerevisiae D3 and 4 h later yeast isolated from peritoneal fluid for further cultivation, vehicle DMSO; toxic effects |
– |
Simmon et al. (1979) |
|
n.g. |
Mouse (3/group); |
Host-mediated reverse gene mutation assay |
i.m. injection; 0 or 125 mg/kg bw |
Mice injected i.p. with S. typhimurium TA1538 or TA1530 and 4 h later bacteria isolated from peritoneal fluid for further cultivation, vehicle DMSO; |
+ |
Simmon et al. (1979) |
|
n.g. |
Mouse (4–6/group); |
Host-mediated reverse gene mutation assay |
Gavage; 0 or 1600 mg/kg bw |
Mice injected i.p. with S. typhimurium TA1538 and 4 h later bacteria isolated from peritoneal fluid for further cultivation, vehicle DMSO; toxic effects |
(±) |
Simmon et al. (1979) |
|
n.g. |
Mouse (5/group); |
Induction of faulty differentiation in spermatozoa |
i.p. injection (once daily, 5 days); 250, 500, 750, 1000, 1250 or 1500 mg/kg bw |
5 weeks after the last dose, sperm examined for head abnormalities; lethal at high dose, sporadic increases in abnormalities |
(±) |
Topham (1980) |
|
n.g. |
Chinese hamster (2–3/dose); n.g.; f |
SCE test |
Gavage; 0, 125, 250 or 500 mg/kg bw |
Bone marrow cells prepared after 18 h; vehicle n.g.; no cytotoxicity, no mortality (no further data) |
+ |
Neal & Probst (1983) |
|
n.g. |
Chinese hamster (2–3/dose); n.g.; f |
SCE test |
i.p. injection; 0, 50, 100 or 200 mg/kg bw |
Bone marrow cells prepared after 18 h; vehicle n.g.; no cytotoxicity, no mortality (no further data) |
–* |
Neal & Probst (1983) |
|
2,7-Dinitrofluorene |
||||||
|
n.g. |
Mouse (6/group); ddY; m |
Micronucleus test |
i.p. injection; 0 or 80 mg/kg bw (one dose) |
Micronuclei determined in peripheral reticulocytes after 0, 24, 48, 72, 96 h, vehicle olive oil; no data on toxicity, effective positive control |
–* |
Murakami et al. (1996) |
|
9-Nitroanthracene |
||||||
|
n.g. |
Mouse (6/group); ddY; m |
Micronucleus test |
i.p. injection; 0 or 80 mg/kg bw (one dose) |
Micronuclei determined in peripheral reticulocytes after 0, 24, 48, 72, 96 h, vehicle olive oil; no data on toxicity, effective positive control |
–* |
Murakami et al. (1996) |
|
3-Nitrofluoranthene |
||||||
|
n.g. |
Mouse; ddY; n.g. |
Micronucleus test |
i.p. injection; 0, 40 or 80 mg/kg bw (2 doses) |
Micronuclei determined in bone marrow polychromatic erythrocytes after 24 h, vehicle olive oil; no data on toxicity or number of animals, effective positive control |
–* |
Tokiwa et al. (1993b) |
|
99.99% |
Mouse (6/group); ddY; m |
Micronucleus test |
i.p. injection; 0 or 80 mg/kg bw (one dose) |
Micronuclei determined in peripheral reticulocytes after 0, 24, 48, 72, 96 h, vehicle olive oil; no data on toxicity, effective positive control |
–* |
Murakami et al. (1996) |
|
n.g. |
Mouse (314/group); |
Micronucleus test |
Injection (not specified); 0, 50, 100, 200 or 500 mg/kg bw |
Micronuclei determined in bone marrow cells after 48 h, vehicle corn oil/DMSO |
+ |
Sakitani & Suzuki (1986) |
|
3,7-Dinitrofluoranthene |
||||||
|
n.g. |
Mouse; ddY; n.g. |
Micronucleus test |
i.p. injection; 0, 10, 20, 40, 80 or 160 mg/kg bw |
Micronuclei determined in bone marrow polychromatic erythrocytes after 24 h, vehicle olive oil; dose-dependent effect (10–80 mg/kg bw) |
+ |
Tokiwa et al. (1993b) |
|
3,9-Dinitrofluoranthene |
||||||
|
n.g. |
Mouse; ddY; n.g. |
Micronucleus test |
i.p. injection; 0, 10, 20, 40 or 80 mg/kg bw |
Micronuclei determined in bone marrow polychromatic erythrocytes after 24 h, vehicle olive oil; dose-dependent effect (10–40 mg/kg bw) |
+ |
Tokiwa et al. (1993b) |
|
1-Nitropyrene |
||||||
|
>98%, impurity pyrene |
Rat (3/dose); F344; f |
SCE test |
Gavage; 0, 500, 1500 or 5000 mg/kg bw |
Bone marrow cells prepared after 48 h; vehicle 2% gelatin; no toxic effects, dose-dependent effect |
+ |
Marshall et al. (1982) |
|
n.g. |
Rat (6/dose); CD; m |
SCE test |
Intratracheal instillation (3 doses in 24-h interval) |
SCE in lung cells determined; DNA adducts at lower doses, abstract, no further details |
+* |
Whong et al. (1993) |
|
n.g. |
Rat (6/dose); CD; m |
Micronucleus test |
Intratracheal instillation; 3 doses in 24-h interval |
Micronuclei in lung cells determined; DNA adducts at lower doses, abstract, no further details |
+* |
Whong et al. (1993) |
|
n.g. |
Rat; Sprague-Dawley; |
Cell transformation assay |
Intratracheal instillation; 15 or 30 mg/kg bw (3 times at 3-day intervals) |
Transformation determined in vitro after tracheal epithelial cell isolation; all transformed cell lines produced squamous cell carcinoma in nude mice |
+ |
Xiang et al. (1996, 1997); Ensell et al. (1999) |
|
n.g. |
Mouse (3–14/group); |
Micronucleus test |
Injection (not specified); 0, 50, 100 or 300 mg/kg bw |
Micronuclei determined in bone marrow cells after 48 h, vehicle corn oil/DMSO; no data on cytotoxicity |
–* |
Sakitani & Suzuki (1986) |
|
n.g. |
Mouse; B6C3F1; m |
DNA alkaline elution assay |
Intratracheal instillation; 25–100 mg/kg bw |
Strand breaks in lung DNA determined 2 h and 1 or 4 days after treatment; dose-dependent effect 2 h after treatment, DNA repair evident after 1 and 4 days (complete at low dose), TS more effective than BaP, data from abstract |
+* |
Mitchell (1985) |
|
>99%, <0.03% dinitropyrenes |
Mouse (4/group); |
DNA alkaline elution assay |
Intratracheal instillation; 0, 25, 50 or 75 mg/kg bw |
Single strand breaks in lung DNA determined 2 h and 1, 3, 7, 14 or 28 days after treatment, vehicle 0.2% gelatin, dose-dependent effect (maximum at 2 h), prelabelling of neonates with [3H]thymidine more effective than prelabelling of adults, also single strand break repair faster in mice prelabelled as neonates |
+ |
Mitchell (1988b) |
|
n.g. |
Chinese hamster; |
SCE test |
i.p. injection; 125 mg/kg bw |
Cytogenetic analysis in bone marrow cells; SCE rate increased by a factor of 3–4, abstract, no further details available |
+* |
Heidemann & Miltenburger (1983) |
|
n.g. |
Chinese hamster; |
Micronucleus test |
i.p. injection; 1000 mg/kg bw (or 2 doses each of 500 mg/kg at 24-h interval) |
Cytogenetic analysis in bone marrow cells; significant increase in micronucleus rate, abstract, no further details available |
+* |
Heidemann & Miltenburger (1983) |
|
1,3-Dinitropyrene |
||||||
|
>98% |
Mouse (5/group); |
Host-mediated reverse gene mutation assay |
Gavage; 0, 0.4, 1, 2 or 4 mg/kg bw |
S. typhimurium TA98 i.v. injected, vehicle DMSO, 1 h later liver removed, preparation of bacteria for cultivation; dose-dependent effect, binding to liver components reducing interaction with S. typhimurium |
(±) |
Shah et al. (1991) |
|
1,6-Dinitropyrene |
||||||
|
>99% |
Rat (1–3/group); |
Unscheduled DNA synthesis in the liver |
Gavage; 0 or 50 mg/kg bw (one dose) |
Hepatocytes examined after 2, 12 or 24 h, vehicle corn oil; no data on toxicity, positive results with 2-acetylaminofluorene (metabolite of 2-nitrofluorene) |
-* |
Butterworth et al. (1983) |
|
>99% |
Rat (2–3/group); |
Unscheduled DNA synthesis in spermatocytes |
Gavage; 0 or 50 mg/kg bw (one dose) |
Cells prepared after 2, 12 or 24 h, vehicle corn oil; no data on toxicity, positive control effective |
–* |
Working & Butterworth (1984) |
|
n.g. |
Rat (2/group); F344; m |
Gene mutation at the HPRT locus |
Implantation into the lung; 0, 30 or 100 µg TS |
Spleen lymphocytes isolated after 3, 9, 12, 15 or 21 weeks, T-lymphocytes cultivated in vitro and 6-thioguanine-resistant mutants determined; dose- and time-dependent (maximum at 21 weeks) effect, DNA binding in lung and spleen maximum at day 7 |
+ |
Smith et al. (1993) |
|
n.g. |
Rat (2/group); F344; m |
Gene mutation at the HPRT locus |
Implantation into the lung; 0, 30 or 100 µg TS |
Spleen lymphocytes isolated after 3, 9, 12, 15, 21, 27, 40 or 51 weeks, T-lymphocytes cultivated in vitro and 6-thioguanine-resistant mutants determined; dose- and time-dependent (maximum at 21–40 weeks) effect, DNA binding in lung, liver, blood and spleen maximum at day 3–7 |
+ |
Beland (1995); Smith et al. (1995) |
|
n.g. |
Rat (2/group); F344; m |
Gene mutation at the HPRT locus |
Implantation into the lung; 0, 0.3, 1, 3, 10, 30, 100 or 150 µg TS |
Spleen lymphocytes isolated after 21 weeks, T-lymphocytes cultivated in vitro and 6-thioguanine-resistant mutants determined; dose-dependent effect (1–100 µg), effect correlates with DNA binding in liver, lung and spleen |
+ |
Beland et al. (1994); Beland (1995); Smith et al. (1995) |
|
>98% |
Mouse (5/group); |
Host-mediated reverse gene mutation assay |
Gavage; 0, 0.4, 1, 2 or 4 mg/kg bw |
S. typhimurium TA98 i.v. injected, vehicle DMSO; 1 h later liver removed, preparation of bacteria for cultivation; dose-dependent effect, binding to liver components reducing interaction with S. typhimurium |
(±) |
Shah et al. (1991) |
|
1,8-Dinitropyrene |
||||||
|
>98% |
Mouse (5/group); |
Host-mediated reverse gene mutation assay |
Gavage; 0, 0.04, 0.1, 0.4, 1 or 4 mg/kg bw |
S. typhimurium TA98 i.v. injected, vehicle DMSO; 1 h later liver removed, preparation of bacteria for cultivation; dose-dependent effect, binding to liver components reducing interaction with S. typhimurium |
(±) |
Shah et al. (1991) |
|
6-Nitrochrysene |
||||||
|
n.g. |
Mouse (3–14/group); |
Micronucleus test |
Injection (not specified); 0, 50, 100, 200 or 500 mg/kg bw |
Micronuclei determined in bone marrow cells after 48 h, vehicle corn oil/DMSO |
+ |
Sakitani & Suzuki (1986) |
|
a |
f = females; m = males; n.g. = not given; SCE = sister chromatid exchange; TS = test substance; i.m. = intramuscular; HPRT = hypoxanthine-guanine phosphoribosyl-transferase. |
|
b |
+ = positive; (±) = marginally positive; ± = inconclusive; – = negative; results marked with an asterisk (*) are of limited validity; see comments in table. |
Table 50. Genotoxicity of nitroPAHs to Drosophila melanogaster sp. in vivoa
|
Purity |
Strain; sex |
Test type |
Test conditions; comments |
Result |
References |
|
1-Nitronaphthalene |
|||||
|
99.5% |
Canton-S; m |
Sex-linked recessive lethals test |
72 h after feeding a solution containing 0 or 25 mg TS/litre or 24 h after injection of 0 or 400 mg/litre (no data above volume), males mated to Basc females, presumptive lethals scored in F2 and F3; 13% (feeding) or 19% (injection) mortality |
– |
Valencia et al. (1985) |
|
n.g. |
UZ; m |
Somatic mutation/ recombination test |
Zeste-white eye assay, eye mosaicism examined, 2nd-instar larvae fed an instant medium containing 0, 0.5 or 1.0 mmol TS/litre until pupation, ~4000 eyes/group scored in adult m; no data on toxicity, dose-dependent effect; statistically not significant |
(±) |
Batiste-Alentorn et al. (1995) |
|
n.g. |
[(wi)4]; m |
Somatic mutation/ recombination test |
White ivory eye assay, eye mosaicism examined, 3rd-instar larvae fed an instant medium containing 0, 0.5 or 1.0 mmol TS/litre until pupation, 800–1422 eyes/group scored in adult m; no data on toxicity, dose-dependent effect; statistically not significant |
(±) |
Batiste-Alentorn et al. (1995) |
|
n.g. |
mwh (f) /flr³ (m); m + f |
Somatic mutation/ recombination test |
Wing spot assay, wing mosaicism examined, 3-day-old larvae fed an instant medium containing 0, 0.5, 1.0 or 2.0 mmol TS/litre until pupation, 48 wings/group scored; no data on toxicity, dose-dependent effect |
+ |
Batiste-Alentorn et al. (1995) |
|
n.g. |
mwh (m) /flr³ (f); m + f |
Somatic mutation/ recombination test |
Wing spot assay, wing mosaicism examined, 3-day-old larvae fed an instant medium containing 0, 1, 2 or 5 mmol TS/litre until pupation, 16–200 wings/group scored; tested up to toxicity, dose-dependent effect |
+ |
Delgado-Rodriguez et al. (1995) |
|
1,5-Dinitronaphthalene |
|||||
|
n.g. |
mwh (m) /flr³ (f); m + f |
Somatic mutation/ recombination test |
Wing spot assay, wing mosaicism examined, 3-day-old larvae fed an instant medium containing 0, 1, 10, 20, 50 or 100 mmol TS/litre until pupation, 18–200 wings/group scored; tested up to toxicity; no dose-dependent effect |
+ |
Delgado-Rodriguez et al. (1995) |
|
2-Nitrofluorene |
|||||
|
n.g. |
mwh (f) /flr³ (m); m + f |
Somatic mutation/ recombination test |
Wing spot assay, wing mosaicism examined, 3-day-old larvae fed a medium containing 0, 12.5 or 25 mmol TS/litre until pupation, 80–200 wings/group scored; no data on toxicity; positive only with twin spots |
± |
Graf et al. (1992) |
|
9-Nitroanthracene |
|||||
|
n.g. |
mwh (m) /flr³ (f); m + f |
Somatic mutation/ recombination test |
Wing spot assay, wing mosaicism examined, 3-day-old larvae fed an instant medium containing 0, 1, 5, 10 or 20 mmol TS/litre until pupation, 14–200 wings/group scored; tested up to toxicity, dose-dependent effect |
+ |
Delgado-Rodriguez et al. (1995) |
|
3-Nitrofluoranthene |
|||||
|
n.g. |
y (f) cross se h (m); m + f |
Mitotic recombination assay |
Eye mosaic assay, eye mosaicism examined, long-term exposure of larvae via food supplemented with 0 or 1 mmol TS/litre (solvent 3% ethanol), 868 (control) or 2296 eyes tested; highest dose tolerated |
– |
Vogel & Nivard (1993) |
|
1,8-Dinitropyrene |
|||||
|
n.g. |
mwh (f) /flr³ (m); m + f |
Somatic mutation/ recombination test |
Wing spot assay, wing mosaicism examined, 3-day-old larvae fed a medium containing 0 or 2 mmol TS/litre until pupation, 139 and 200 (control) wings/group scored; one dose, no data on toxicity |
– |
Graf et al. (1992) |
|
n.g. |
Rec– male × Rec+ female; |
DNA repair test |
3rd-instar larvae (~200 per dose) exposed to 0–10 nmol TS per g food until they eclosed, adult males (DNA repair deficient) and females (proficient) counted; decreased ratio male/female |
+ |
Obana & Nishimune (1993) |
a f = females; m = males; n.g. = not given; TS = test substance; + = positive; (±) = marginally positive; ± = inconclusive; – = negative.
Exposure by gavage was used for 2-nitronaphthalene, 2-nitrofluorene, 1-nitropyrene, 1,6-dinitropyrene and 1,8-dinitropyrene. Micronucleus induction was studied only for 2-nitrofluorene and showed a significant increase in liver and bone marrow. Sister chromatid exchange induction was positive for both studied nitroPAHs: 2-nitrofluorene and 1-nitropyrene. Unscheduled DNA synthesis (UDS) was positive for 2-nitrofluorene and negative for 1,6-dinitropyrene (limited validity of study). Host-mediated assays were performed, with negative results for 2-nitronaphthalene and positive results for 1,6-dinitropyrene.
Exposure by i.p. injection was used for different studies on eight nitroPAHs. However, most of the studies were of limited validity for different reasons (see Table 49) and gave negative results. Clear positive effects were observed only for 3,7-dinitrofluoranthene and 3,9-dinitrofluoranthene (micronucleus induction in bone marrow). Clear negative effects were reported for 5-nitroacenaphthalene (micronucleus induction in peripheral blood) and 2-nitrofluorene (host-mediated recombination assay).
Exposure by intramuscular (i.m.) injection gave a significant increase of reverse mutation in the host-mediated assay for 2-nitronaphthalene and 2-nitrofluorene. Injection (non-defined route) led to a significant increase of micronuclei in bone marrow for 3-nitrofluoranthene and 6-nitrochrysene.
Most of the tested nitroPAHs were positive in in vivo genotoxicity tests in somatic cells, and the data in Drosophila (Table 50) confirm that these compounds are in vivo somatic mutagens. There were no germ cell assays carried out in rodents, and in Drosophila, the single germ cell assay was negative.
Table 46 summarizes the studies on the genotoxicity of nitroPAHs in vivo given in Tables 49 and 50 and compares these data with the in vitro genotoxicity data.
Data on the genotoxicity of nitroPAHs in vivo are available on 15 nitroPAHs. Two or more end-points were investigated only on seven substances: 2-nitronaphthalene, 2-nitrofluorene, 9-nitroanthracene, 3-nitrofluoranthene, 1-nitropyrene and 1,6- and 1,8-dinitropyrene. Only two of these seven nitroPAHs revealed predominant positive results: 2-nitrofluorene and 1-nitropyrene (Tables 49 and 50, marked by bold type "positive" in Table 46). Inconsistent results were seen with 2-nitronaphthalene and 3-nitrofluoranthene. The other three nitroPAHs — 9-nitroanthracene and 1,6- and 1,8-dinitropyrene — were judged to be genotoxic in vivo, but the evidence is limited.
A limited database was found with eight nitroPAHs (one end-point studied; Tables 49 and 50; see also Table 46). In most cases (six nitroPAHs), the study on the investigated end-point revealed a positive result; with two nitroPAHs, the result was inconclusive.
All nitroPAHs that gave positive results in vivo (normal and bold type "positive" in Table 46) also revealed positive results in three or more in vitro end-points (bold type "positive"; Table 46). The exception is 1,5-dinitronaphthalene (only normal type "positive"; two end-points investigated in vitro, both positive).
Four nitroPAHs revealed inconclusive (2-nitronaphthalene, 5-nitroacenaphthene and 3-nitrofluoranthene) or negative (2,7-dinitrofluorene, limited validity) results on genotoxicity in vivo. 2-Nitronaphthalene and 3-nitrofluoranthene are investigated in detail in in vitro studies (seven and eight end-points, respectively), and both are clearly mutagenic (Table 46). Genotoxicity in vitro was also observed with 5-nitroacenaphthene and 2-nitrofluoranthene, although most positive results were seen in the bacterial test system and only one in mammalian cells (Table 45). Similar in vitro results are presented with 2,7-dinitrofluorene (Table 45).
1-Nitronaphthalene induced no sex-linked recessive lethal mutations, but did induce somatic mutation/recombinations in Drosophila. Data on mammalian test systems are not available. For comparison, a feeding study on mice and rats revealed no conclusive results on carcinogenicity. 1-Nitronaphthalene also induced gene mutations in bacteria (but showed low mutagenic potency) and mammalian cells, although negative results were obtained with human cells; in vitro results on bacteria and mammalian cells further indicated DNA damage (see Table 45).
Data on 2-nitronaphthalene are limited to the host-mediated assay, a test system with low sensitivity. Mutations of inoculated bacteria were induced after i.p. injection, but oral application revealed negative results, similar to the mitotic recombination assay. In vitro results on gene mutation in bacteria and human cells also revealed positive results (Table 45).
According to available in vitro results on gene mutation and DNA damage in bacteria (see Table 45), 1,5-dinitronaphthalene induced somatic mutation/recombination in Drosophila.
5-Nitroacenaphthene revealed in vivo negative or inconclusive results in the micronucleus test in two species; however, besides gene mutation in the Ames test, DNA damage was observed in bacteria and mammalian cells in vitro (Table 45).
In vivo, 2-nitrofluorene showed clastogenic activity in the micronucleus test with hepatocytes but no or marginal activity in the bone marrow. Sister chromatid exchange was increased in bone marrow cells after oral application. DNA damage was also evident in the liver. In vitro studies showed DNA-damaging effects in bacteria and mammalian cells (Table 45). Although 2-nitrofluorene induced mitotic recombination in yeast in vitro (see Table 45), this could not be confirmed in the host-mediated assay, whereas positive host-mediated reverse mutation assays are in accordance with in vitro results. In vivo, toxic doses reached testis tissue and induced weak effects on differentiation of spermatocytes. Data on gene mutation in vitro are contradictory (Table 45), and no data are available in mammals; the corresponding test in Drosophila was inconclusive. Overall, genotoxic activity of 2-nitrofluorene corresponds to the carcinogenic effects in animals.
No conclusion can be drawn from a single micronucleus test on 2,7-dinitrofluorene or 9-nitroanthracene, nitroPAHs with DNA-damaging activity in bacteria (both nitroPAHs) and mammalian cells (negative results with 9-nitroanthracene; see Table 45). 9-Nitroanthracene induced somatic mutation in insects and in vitro gene mutation in bacteria and human cells.
The carcinogen 3-nitrofluoranthene showed clastogenic activity in a valid micronucleus test but negative results in the Drosophila mitotic recombination assay. Data on gene mutation and DNA damage in vitro (see Table 45) were positive.
The positive micronucleus tests in vivo confirmed the in vitro (Table 45) clastogenic activity in mammalian cells and the DNA-damaging activity in bacteria of 3,7- and 3,9-dinitrofluoranthene.
1-Nitropyrene induced local effects in the respiratory system concerning DNA damage, clastogenicity, increased sister chromatid exchange and cell transformation. Systemic effects are inconclusive (micronucleus test) or positive (sister chromatid exchange). In vivo results on different genotoxicity end-points confirmed in vitro studies (Table 45).
Data on 1,3-dinitropyrene are limited for the host-mediated assay. Like the other dinitropyrenes (see below), 1,3-dinitropyrene is a carcinogen in animals and a potent mutagen in in vitro studies on bacteria (see Table 40), but only marginal effects were observed in vivo in a host-mediated assay with the same bacterial strain. The authors of the study (Shah et al., 1991) presented evidence that dinitropyrenes or their metabolites bind to tissue components of the host, reducing interaction with bacteria.
According to in vitro results, 1,6-dinitropyrene also induced gene mutation in rats. Negative results in in vivo studies on DNA damage are of limited validity, since evidence for systemic toxicity at the dose chosen has not been shown. DNA damage has been shown in different in vitro test systems (Table 45).
In addition to the weak positive result in a host-mediated assay (see also 1,3-dinitropyrene), 1,8-dinitropyrene induced DNA damage but no somatic mutation in Drosophila. Gene mutation as well as DNA-damaging effects were observed in several in vitro assays (Table 45).
In accordance with DNA-damaging activity in bacteria (Table 45), 6-nitrochrysene is genotoxic in the micronucleus test on mice.
A 32P post-labelling study was carried out to examine the adducts in DNA from human hepatoma HepG2 cells treated with 3-nitrobenzanthrone (Kawanishi et al., 2000). Two major and two minor spots were obtained. The two minor spots could be identified as N-acetyl-3-amino-2-(2'-deoxyguanosin-3',5'-bisphosphate-8-yl)-benzanthrone (identified in a previous in vitro study; Enya et al., 1998) and the respective dA adduct (Kawanishi et al., 2000).
Using the 32P post-labelling assay, 3-nitrobenzanthrone was found to bind covalently to calf thymus DNA after metabolic activation, forming multiple DNA adducts, all of which are reduction products, in vitro. Three of the seven adducts were detected in all three activation systems used; however, different amounts of individual spots were obtained in the different in vitro systems used (Bieler et al., 1999). Multiple DNA adducts were also detected in cultures of rat lung alveolar type II epithelial cells treated with 3-nitrobenzanthrone (Borlak et al., 2000).
Arlt et al. (2001) reported DNA adduct formation in rats after a single oral dose (via gavage) of 2 mg 3-nitrobenzanthrone/kg bw, using 32P post-labelling. Adducts were detectable in all organs, the highest levels being found in the small intestine (38 adducts/108 nucleotides), followed by stomach, kidney, liver, lung and bladder. There is evidence that the adducts are products derived from reduced metabolites covalently bound to dG (60%) and dA (40%).
The mutagenicity of 3-nitrobenzanthrone in the Ames S. typhimurium assay produced 208 000 revertants/nmol in strain TA98 (–S9) and 6 290 000 revertants/nmol in YG1024 (–S9) and compares favourably with the mutagenicity of 1,8-dinitropyrene. 3-Nitrobenzanthrone is the strongest mutagen so far reported for these compounds in this test. The 9- and 11-nitrobenzanthrone and 3,9- and 3,11-dinitrobenzanthrone isomers did not show this high mutagenicity (Enya et al., 1997).
3-Nitrobenzanthrone also induces micronuclei in mouse peripheral blood reticulocytes after i.p. administration (mean frequency of micronucleated reticulocytes = 0.64% at a dose of 25 mg/kg bw after 48 h), suggesting its potential genotoxicity to mammals, and is suspected to be a human carcinogen (Enya et al., 1997).
The genotoxic effects of 3-nitrobenzanthrone were determined in forward mutation assays using two human B-lymphoblastoid cell lines: the MCL-5 and h1A1v2 (Phousongphouang et al., 2000). The mutagenicity of the compounds was determined at the heterozygous tk locus and the hemizygous hprt locus and also in the CREST modified micronucleus assay, which detects chromosomal loss and breakage events. Results indicate that 3-nitrobenzanthrone is an effective human cell mutagen, significantly inducing mutations at the tk and hprt loci in both cell lines and inducing micronuclei in the h1A1v2 cell line (see Table 48).
2-, 3- and 4-nitrodibenzopyranone all formed multiple DNA adducts after incubation with xanthine oxidase and calf thymus DNA under anaerobic conditions. DNA adducts were detected in the liver DNA from rats treated with 2-nitrodibenzopyranone. The migration of these adducts was similar to that observed in the in vitro experiment. No significant increase in DNA adducts was detected in lung DNA; no further details were given (Watanabe et al., 1996).
2- and 4-nitrodibenzopyranone were found to be in the most mutagenic HPLC fraction of an ambient particulate extract collected from Riverside, California, USA, when these products were tested in the S. typhimurium microsuspension assay TA98 (–S9). 2- and 4-nitrodibenzopyranone gave activities of 58 600 and 480 revertants/nmol, respectively; the activity of the positive control mutagen, 2-nitrofluorene, was 6000 revertants/nmol (Arey et al., 1992; Helmig et al., 1992a).
2-, 3- and 4-nitrodibenzopyranones were tested for mutagenicity in the plate incorporation and microsuspension assays using S. typhimurium tester strains in the presence and absence of S9-mix. In both assays, all of the nitrodibenzopyranones showed mutagenicity in every strain. In the absence of S9-mix, TA98 was the strain most sensitive to the mutagenicity of nitrodibenzopyranones. The activity of nitrodibenzopyranones was reduced in TA98NR– and TA98AT–strains relative to TA98, suggesting that nitrodibenzopyranones cause frameshift mutations and that nitroreduction by "classical" reductase and acetylation are significant steps for their metabolic activation (Watanabe et al., 1995).
2-Nitrodibenzopyranone was tested with S. typhimurium TA98NR and was activated by each of the S9 fractions tested (from phenobarbital-, 3-methylcholanthrene-β-naphthoflavone- and PCB-treated and untreated rats). 2-Nitrodibenzopyranone was metabolically activated by NADH and xanthine oxidase, and this activation was found to be inhibited by allopurinol, indicating that cytosolic xanthine oxidase in rat liver S9 participates in the activation of 2-nitrodibenzopyranone (Watanabe et al., 1997a).
The mutagenicities of 2- and 3-nitrodibenzopyranone (and 2-nitrofluoranthene) were determined in a quantitative forward mutation assay with S. typhimurium TM677. 2-Nitrodibenzopyranone was non-mutagenic in the absence of S9 but was mutagenic in its presence. The converse result was found with 3-nitrodibenzopyranone. The minimal detectable mutagen concentration (in nmol/ml) for 2-nitrofluoranthene, 3-nitrodibenzopyranone and 2-nitrodibenzopyranone was 2.5, 16.9 and >415 (order of potency), respectively, and with S9, 1.2, 308 and 15.1, respectively (Busby et al., 1997).
2- and 3-nitrodibenzopyranone and 2-nitrofluoranthene were tested in two metabolically competent human cell lines (MCL-5 and h1A1v2) differing in their complement of cytochrome P450s and microsomal epoxide hydrolase. Neither 2-nitrodibenzopyranone nor 3-nitrodibenzopyranone was mutagenic at the tk locus in MCL-5 or h1A1v2 cells at up to 200 nmol/ml (Busby et al., 1997).
In contrast, in another study testing for mutagenicity in the MCL-5 human cell line, it was found that 2-nitrodibenzopyranone was mutagenic at the tk but not at the hprt locus, suggesting that the human cell mutagenicity of these compounds was exclusively attributable to loss of heterozygosity events not available at the hemizygous hprt locus (Sasaki et al., 1997a,b; Grosovsky et al., 1999).
The high mutagenicity in bacterial cells is in marked contrast to the results in human cells in vitro, but there were no data available on the effect of nitrodibenzopyranones in mammalian cells in vivo.
Two specific nitropyrene lactone isomers, 1-nitropyrene lactone and 3-nitropyrene lactone, have been found to be highly mutagenic in the S. typhimurium microsomal assay (El-Bayoumy & Hecht, 1986).
The mutagenicity of nitropyrene lactones was determined in forward mutation assays using MCL-5 and h1A1v2, and they were found to induce mutations at the tk and hprt loci in both cell lines. Further, they induced kinetochore-positive and -negative micronuclei in the CREST modified micronucleus assay, which detects chromosomal loss and breakage events (Phousongphouang et al., 2000).
Compounds chosen for their known presence and mutagenicity in ambient air samples (2-nitronaphthalene, 3-nitrobenzanthrone, 2-nitrodibenzopyranone and nitropyrene lactones) were compared in human B-lymphoblastoid cell lines MCL-5 and h1A1v2 (see Table 48).
In the MCL-5 cell line, both 3-nitrobenzanthrone and the nitropyrene lactones induced comparable mutation frequencies at hprt and tk, whereas they were both 3- to 4-fold more mutagenic at tk than at hprt in the h1A1v2 cell line. 3-Nitrobenzanthrone is 2- to 4-fold more potent than is the mixture of nitropyrene lactones and almost an order of magnitude more potent than 2-nitrodibenzopyranone (Phousongphouang et al., 2000). Previous studies with 2-nitronaphthalene showed induction only at the tk locus, showing that its genotoxicity is restricted to the induction of large-scale loss of heterozygosity events and micronuclei (Sasaki et al., 1997a,b, 1999). It is suggested that 3-nitrobenzanthrone and nitropyrene lactones may be capable of inducing a broader range of genetic alterations, including smaller-scale chromosomal changes and intragenic mutations (Phousongphouang et al., 2000).
Durant et al. (1996) tested two oxygen-containing nitroPAHs for mutagenicity at the tk locus in the human B-lymphoblastoid cell line h1A1v2. Whereas 2-nitrodibenzopyranone showed no mutagenic activity, 3-nitrodibenzopyranone was considered to be positive in this test system.
Data on the genotoxicity of nitroPAHs are summarized and related to carcinogenicity in Table 46.
Bioassay-directed chemical fractionation (see chapter 2) — e.g., using the S. typhimurium microsomal assay — is the test that has been mostly used for identifying classes of bioactive compounds and their mutagenic potential in complex environmental mixtures. The outcome of the bioassay-directed chemical analysis seems to depend on the biological end-point used (see below) (Hannigan et al., 1998).
Caution must be taken in the interpretation of Salmonella mutagenicity assays with complex mixtures.
Some nitroPAHs are extremely mutagenic in bacteria, in particular in Salmonella mutagenicity assays, but this does not necessarily parallel the mutagenicity in in vitro human cell systems or in in vivo studies or the carcinogenicity of the compound.
Another issue is the possible synergism or antagonism of combined nitroPAHs in mixtures compared with individual nitroPAHs. For example, mixtures of 1- and 3-nitrobenzo[a]pyrene showed 183% more TA98 revertants in the Ames test compared with equivalent amounts of the pure compounds (Thornton-Manning et al., 1989).
In a study on diesel exhaust particles extracted with benzene–ethanol (3:2 v/v) and separated into five fractions by silica gel column chromatography, it was found that the direct-acting mutagenic activity (assayed by the Ames test using S. typhimurium YG1024 strain without metabolic activation systems) of the five fractions was about 5 times greater than that of the crude extract, suggesting that activities in the fractions were suppressed in the crude extract (Hayakawa et al., 1997).
Eide & Johnsen (1998) investigated the possible interactions between mutagens in a mixture (organic extract of diesel exhaust particles) using nitroPAHs as an example. 1-Nitropyrene, 2-nitrofluorene and 1,8-dinitropyrene were added to the extract separately and in various combinations. The mixtures were tested for mutagenicity in the Ames assay using the TA98 strain of S. typhimurium. The three individual nitroPAHs and the diesel exhaust particle extract were found to act additively in the Ames test.
Nishioka et al. (1982) found that in diesel engine exhaust, nitropolycyclic aromatic compounds accounted for 20–25% of the bacterial mutagenic activity (without further enzymatic activation of the assay, i.e., –S9). Salmeen et al. (1984) found that mono- and dinitroPAHs accounted for 30–40% of bacterial mutagenic activity (–S9) of diesel engine exhaust particles.
In a diesel exhaust particle extract (benzene–ethanol), nitroPAHs were found mostly in fraction 4 (solvent DCM), which contained 61.5% of the total activity. Of this fraction, 53.1% of the activity was attributed to nitroPAHs, with the greatest contribution being from 1-nitropyrene and 1,3-, 1,6- and 1,8-dinitropyrene (Hayakawa et al., 1997).
Studies on ambient air showed that 1–8% of bacterial mutagenic activity (–S9) was due to nitroPAHs (Arey et al., 1988b).
It is generally accepted that most of the particulate mutagenicity is due to organics, which are more polar than nitroPAHs (Schuetzle & Lewtas, 1986; Nishioka et al., 1988; Lewtas et al., 1990a). In contrast, vapour-phase mutagenicity was highest in less polar fractions.
A comparison of the mutagenic activities of HPLC fractions of concurrently sampled vapour-phase and particulate organics shows that the ambient mutagenicity concentration in the vapour phase is comparable to that in the particulate matter (Harger et al., 1992). Using the Ames test, but with the modification of Kado et al. (1983) to increase sensitivity, Harger et al. (1992) found that the vapour-phase mutagens account for over 50% of the total. In agreement with previous reports (e.g., Arey et al., 1988b), about 10% of ambient particulate mutagenicity in the Ames test can be accounted for by nitrofluoranthenes and nitropyrenes, the dominant contributors to ambient particulate organic matter (Arey et al., 1988b). These nitroPAH isomers were largely formed from the atmospheric gas-phase reactions of the parent PAH rather than being directly emitted (Arey et al., 1986). Nitronaphthalenes were thought to account for about 13% of the mutagenicity in the fraction where the mutagenicity of the vapour phase was highest for these nitroPAHs.
1- and 2-nitronaphthalenes (and methylnitronaphthalenes) were identified and quantified in ambient vapour-phase samples collected in Redlands, California, USA, during moderate photochemical air pollution (Gupta et al., 1996). The mutagenic activities were determined using a microsuspension preincubation modification of the Ames test in strain TA98 without microsomal activation. The abundance of 1-nitronaphthalene was similar to that of 2-nitronaphthalene during the day but almost a factor of 2 higher at night. The calculated contributions of the nitronaphthalenes to the total vapour-phase mutagenic activity were about 18% for daytime and about 32% for nighttime.
Although the vapour phase may contribute substantially to the mutagenic potential (Nishioka et al., 1988; Tuominen et al., 1988; Harger et al., 1992; Greenberg et al., 1993), most studies on the bioassay-directed chemical analysis of genotoxic components in air have concentrated on urban airborne particulate matter.
DCM extracts isolated from urban airborne particulate matter in Barcelona, Spain, exhibited moderate mutagenicity against TA98, ranging from 20 to 109 revertants/m3 (Bayona et al., 1994). The application of metabolic activation (+S9) generally led to a slight decrease in mutagenicity (13–32%), indicating the predominance of direct-acting mutagens. In contrast, Garner et al. (1986) reported, after addition of S9-mix, at least a 2-fold increase in mutagenic activity in S. typhimurium TA98 and TA100 of DCM extracts isolated from suburban airborne particulate matter collected in Bayreuth, Germany.
Casellas et al. (1995) made a detailed chemical analysis of mutagenic fractions using bioassay-directed chemical analysis in urban airborne particulate matter in Barcelona (see chapter 2). The collected fractions were tested for mutagenicity using the Ames test with tester strains TA98, TA98NR– and TA98AT–. The direct mutagenicity in the fractions seemed to be accounted for by nitrated arenes, 9-nitroanthracene, 2-nitrofluoranthene and 2-nitropyrene. 6-Nitrobenzo[a]pyrene in polar fraction 5 needed application of metabolic activation (+S9) for mutagenicity. In order to evaluate the contribution of nitro derivatives to total mutagenicity, the fractions from the second fractionation were tested against TA98NR– and TA98AT–. Generally, a remarkable decrease in mutagenic activity was observed in all fractions, thus suggesting a significant contribution of nitroarenes to the mutagenicity of these fractions of medium and high polarity. Further subfractionation gave PAHs, but two nitro derivatives, 2-nitrofluoranthene and 1-nitropyrene, were identified, which may be responsible for the high direct-acting mutagenic activity observed in this fraction (Casellas et al., 1995).
Studies with the nitroreductase-deficient Salmonella strain TA98NR– indicated that, in urban samples from Helsinki and Lahti, Finland, particularly in winter, a considerable part of the mutagenicity detected in the Ames test was due to nitro-substituted compounds (Tuominen et al., 1988).
In investigations on the activity of mutagens in ambient airborne particles sampled in the Netherlands, seven distinct groups of mutagens were discerned; one group was identified as mononitrated PAHs. This group accounted for about 12% of the total effect in TA98 with a metabolic activation system and for about 24% of the total effect in TA98 without a metabolic activation system. These percentages were about 14 (with S9) and 13 (without S9) in TA100 (de Raat et al., 1994).
Complex mixtures extracted from air filters in downtown Mexico were assayed in the somatic mutation and recombination test in wings of Drosophila melanogaster using two different crosses, as well as in the Ames test using strain TA98 with and without S9 fraction (Delgado-Rodriguez et al., 1999). An extract of total suspended particles gave a higher response in Salmonella when no S9 fraction was added, and it was more active in the ST cross than in the HB cross in Drosophila. This sample also contained the highest amount of nitroPAHs (direct-acting mutagens).
In the benzene–ethanol extract of airborne particulate, the calculated mutagenic contribution of 1-nitropyrene and 1,3-, 1,6- and 1,8-dinitropyrene in the O-acetyltransferase-overproducing S. typhimurium strain YG1024 was 2.1, 2.5, 5 and 9%, respectively (Hayakawa et al., 1995b, 1997), assuming that the effect of coexisting compounds is negligible.
Studies on indoor air particulates in city and country houses indicated that in the absence of indoor sources (e.g., tobacco smoke, grilled meat, toast, cooking), the mutagenic activity of the particulate fraction in S. typhimurium TA98 is due to dusts penetrating from the outside. A contribution by nitroarenes of about 50% to this mutagenic activity is discussed. The particulate produced by indoor pollution sources resulted in varying mutagenic activities; the highest values were detected by pollution due to charcoal burning (Nardini et al., 1994).
A fine particulate matter composite sample from Los Angeles, California, USA (1993; 24-h sampling every 6 days at four locations) was tested for mutagenic activity at the thymidine locus in h1A1v2 (human) cells using a 72-h exposure (Hannigan et al., 1998; h1A1v2 cells are AHH-1 TK+/– cells bearing the plasmid pHSRAA, which contains two copies of the human CYP1A1 cDNA and confers resistance to 1-histidinol). This cell line has been shown to be sensitive to both PAHs and nitroPAHs (Durant et al., 1996) and has been used previously to investigate the seasonal and spatial variation of the human cell mutagenicity of fine organic aerosol in southern California (Hannigan et al., 1997). The subfractions containing unsubstituted polycyclic aromatic compounds were responsible for a considerable portion of the mutagenic potency of the whole atmospheric sample. The only nitroPAH quantified as being of importance was 2-nitrofluoranthene (Hannigan et al., 1998). Targeted compounds that were not found in any semipolar subfraction included nitronaphthalene isomers, nitrofluorene and dinitrofluorene isomers, nitropyrene, dinitropyrene and hydroxynitropyrene isomers, nitro- and dinitrobenzo[a]pyrene isomers and nitrofluorenone isomers.
A similar study was carried out with an organic extract of a standard reference sample (SRM 1649) using a bioassay-directed fractionation method; mutagenicity testing was done using the h1A1v2 cell line. NitroPAHs did not contribute significantly to the mutagenicity of this sample, because they were present at low concentrations and because they are not particularly mutagenic in h1A1v2 cells (Durant et al., 1998).
Studies on DNA adduct formation in mammalian cell cultures (primary cultures of rat hepatocytes) suggest that nitroPAHs contribute to the total genotoxicity of coke oven emissions (Topinka et al., 1998).
The mutagenic contributions of five selected nitroPAHs (1-, 2-, and 4-nitropyrene, 2-nitrofluoranthene and 6-nitrochrysene) at two sites (downtown and suburban areas of Kanazawa, Japan) were obtained by multiplying their mutagenic activity (revertants/pmol) by their concentration (pmol/m3) based on the yearly average (see Table 51). The value for 2-nitrofluoranthene was the largest at the suburban site and close to that of 1-nitropyrene at the downtown site, suggesting that the mutagenic contribution of 2-nitrofluoranthene may not be negligible (Murahashi et al., 1999) (see also chapter 5).
Table 51. Mutagenic activities in Salmonella typhimurium TA98 and mutagenic contributions of nitroPAHs
|
NitroPAH |
Mutagenic activity (revertants/pmol) |
Mutagenic contribution (103 revertants/m3) |
|
|
Site Ka |
Site Ta |
||
|
2-Nitrofluoranthene |
0.57 |
51 |
30 |
|
1-Nitropyrene |
0.48 |
62 |
7.2 |
|
2-Nitropyrene |
2.2 |
26 |
7.5 |
|
4-Nitropyrene |
3.7 |
7.0 |
2.1 |
|
6-Nitrochrysene |
0.25 |
6.0 |
1.9 |
a Site K is a downtown site; site T is a suburban site.
Nitroarenes were found to be an important contributor to the mutagenic activity of the emissions from municipal waste incinerators (DeMarini et al., 1996). Most (80–95%) of the mutagenic activity in the DCM-extractable particulate organics emitted by the municipal waste incinerators resided in the neutral/base fraction; however, the polar neutral fraction accounted for 12% of the direct-acting mutagenic activity. The mutagenic potencies of the whole extract and the various fractions were 4–15 times greater in the absence than in the presence of S9. Results with TA98 strains deficient in nitroreductase or transacetylase indicated that a majority of the direct-acting mutagenicity was due to nitroarenes. This was confirmed by bioassay-directed subfractionation of the neutral/base fraction. The results indicated that nitroarenes such as 1-nitropyrene that eluted in the neutral/base fraction accounted for at least 50% of the direct-acting mutagenicity and induced only a hotspot two-base deletion in the sequence (CG)4 in TA98. In contrast, most of the complex frameshifts induced by the whole extract were induced by nitroarenes other than 1-nitropyrene that were activated by transacetylation and that eluted in the polar neutral fraction.
Soils with low levels of mutagenic activities were exposed to traffic exhaust for 3–26 weeks. Increased mutagenic activity of solvent fractions was detected in the Salmonella microsome assay, the activity depending on exposure duration and depth of soil sampling. Although polar aromatics (including nitroarenes) contributed only 7.8% of total organic compounds in the soil, this fraction showed the major part of mutagenicity (55–65%). Studies with tester strains deficient in nitroreductase or overproducing nitroreductase or O-acetyltransferase indicate the major contribution of nitroarenes to the mutagenic activity of the soils. In in vitro studies on human lymphocyte, the fraction of polar aromatics induced a significant increase in sister chromatid exchanges. However, no increase in micronucleated polychromatic erythrocytes was observed in male mice after oral application of 2000 mg of this fraction per kilogram of body weight (Wesp et al., 2000).
Recently, there has been interest in the antimutagenic properties of nutrients in our diets, which may play protective roles in tumour promotion and progression. Some studies have concentrated on nitroPAHs, which are often present as pollutants in ambient air.
Carotenoids (carotenes and xanthophylls) and vitamin A effectively decreased the mutagenicity of nitroaromatics, including 1-nitropyrene, 2-nitrofluorene, 3-nitrofluoranthene, 1,6-dinitropyrene and 1,8-dinitropyrene, in the S. typhimurium strain TA98 and in its acetyl-CoA:N-hydroxyarylamine O-acetyltransferase-overproducing derivative YG1024 (González de MejR a et al., 1997, 1998; De Flora et al., 1999). It has been suggested that lutein may form an extracellular complex with, for example, 1-nitropyrene, which could limit the bioavailability of this compound to target cells (González de Mejía et al., 1997).
Diverse polysaccharides were tested for antimutagenic activities against 1-nitropyrene and 2-nitrofluorene with S. typhimurium strains TA98 and TA100. Although many compounds showed no effects, antimutagenic activity was found for xyloglucan and different pectins and pectin-like rhamnogalacturonans (Hensel & Meier, 1999).
Milk cultured with Bifidobacterium or Lactobacillus strains or uninoculated milk showed activity against the mutagenicity induced by 2-nitrofluorene in Salmonella tester strain TA98 (Cassand et al., 1994).
Tannic acid and its hydrolysed products, such as ellagic acid, gallic acid and propyl gallate, showed no antimutagenic activities against 2-nitrofluorene, 1-nitropyrene and 1,3-dinitropyrene using Ames Salmonella tester strains TA98 and TA100 (Chen & Chung, 2000).
Chili extract was tested for its effect on the mutagenicity of an urban air sample as well as 1-nitropyrene, 1,6-dinitropyrene or 1,8- dinitropyrene. The extract itself showed moderate mutagenic activity, and an additive or potentiation effect was noted with lower concentrations of chili and nitroPAHs or air extracts present. Only at the maximum concentrations of chili extract was there a decrease in the number of revertants. Chlorophyllin and β-carotene also showed antimutagenic effects against the nitroPAHs (Espinosa-Aguirre et al., 1993).
A detailed presentation of data on the carcinogenicity of nitroPAHs, including a conclusion for each test, is tabulated in Table 39. A summary of carcinogenicity data separated by administration routes as well as the final evaluation are given in Table 52. Whereas data on mutagenicity are available for numerous nitroPAHs, there are data on carcinogenic effects for only 28 nitroPAHs.
Although inhalation is the main exposure route in humans, no long-term inhalation study on any nitroPAH is available. One study on 1-nitropyrene with a 13-week duration showed preneoplastic lesions (see Table 38). Studies on intrapulmonary implants (done with five nitroPAHs) or intratracheal instillation (two nitroPAHs) are of limited validity in comparison with inhalation studies due to the non-physiological exposure routes (see Tables 39 and 52). However, 3,7- and 3,9-dinitrofluoranthene, as well as 1,6-dinitropyrene, revealed positive results with these application routes.
For eight nitroPAHs, data on oral exposure (gavage or via the diet) are available. Carcinogenic effects were observed with this administration route for 5-nitroacenaphthene, 2-nitrofluorene, 1-nitropyrene and 1,8-dinitropyrene (see Tables 39 and 52). Preneoplastic changes in the liver of rats were seen after oral exposure to 2-nitrofluorene or 2,7-dinitrofluorene (see Table 38).
The dermal exposure route has been investigated with five nitroPAHs (see Tables 39 and 52), mainly in initiation/promotion studies. Initiating activity was observed for 6-nitrochrysene and 3-nitroperylene.
In about two-thirds of all studies presented in Table 39, parenteral injection (s.c. or i.p.) was used in the carcinogenicity studies. Administration of the following 15 nitroPAHs by this application route resulted in carcinogenic effects: 3-nitrofluoranthene, 3,7- and 3,9-dinitrofluoranthene, 1-, 2- and 4-nitropyrene, 1,3-, 1,6- and 1,8-dinitropyrene, 7-nitrobenz[a]anthracene, 6-nitrochrysene, 2-nitrobenzo[a]pyrene, 6-nitrobenzo[a]pyrene, 3,6-dinitrobenzo[a]pyrene and 7-nitrodibenz[a,h]anthracene. The validity of these studies is limited compared with physiological exposure routes. Furthermore, in several studies, special study designs were used, such as short treatment duration and newborn or weanling animals at the start of treatment period (e.g., see mouse newborn assay, in Table 39; Wislocki et al., 1986).
The mouse newborn assay used in 16 nitroPAHs (see Tables 39 and 52) was evaluated by Fujii (1991). From this evaluation, it can be concluded that 31 out of 37 (83.8%) substances tested by the mouse newborn assay revealed similar carcinogenic or non-carcinogenic results when tested in either the adult mouse or the adult rat. Although the mouse newborn assay has some disadvantages (non-physiological administration route, metabolism in newborn different from that in adult, target tissue not extrapolative to humans), it is useful in screening for and predicting potential carcinogens (Fu et al., 1998a).
Local carcinogenic effects were also investigated using other application routes: injection into mammary tissue (2,7-dinitrofluorene, 2- and 4-nitropyrene, 6-nitrochrysene) and implantation into the bladder (2-nitronaphthalene; see Table 39). Carcinogenic effects were observed with 2,7-dinitrofluorene (limited validity), 4-nitropyrene and 6-nitrochrysene (El-Bayoumy et al., 1993).
Most studies described in Table 39 showed limitations concerning the study design — e.g., low number of animals, one dose, short exposure/study duration, etc. Due to these limitations, none of the negative studies confirmed the absence of carcinogenic effects in experimental animals.
For 10 nitroPAHs (1- and 2-nitronaphthalene, 2,7-dinitrofluorene, 2-nitrofluoranthene, 1- and 3-nitrobenzo[a]pyrene, 1- and 3-nitrobenzo[e]pyrene, 1,6-dinitrobenzo[a]pyrene, 9-nitrodibenz[a,c]anthracene; see Table 39), not enough data were available to show indications for carcinogenicity in experimental animals (see Table 52). Although the study on 1-nitronaphthalene (see Table 39) is performed with acceptable restrictions according to the study design proposed by the guidelines of the Organisation for Economic Co-operation and Development (OECD), none of the negative studies permitted an exclusion of carcinogenic effects, because the MTD was not reached.
Table 52. Summary and evaluation of carcinogenicity studies in animals
|
Substance |
Species |
Number of studiesa with positive (+) or negative (–) result, by administration route |
Carcinogenicityb |
|||||||||
|
Oral |
Dermal |
Subcutaneous |
Intraperitoneal |
Other |
||||||||
|
+ |
– |
+ |
– |
+ |
– |
+ |
– |
+ |
– |
|||
|
1-Nitronaphthalene |
Rat |
(1) |
Database insufficient |
|||||||||
|
Mouse |
(1) |
|||||||||||
|
2-Nitronaphthalene |
Monkey |
(1) |
Database insufficient |
|||||||||
|
Mouse |
(1c) |
|||||||||||
|
5-Nitroacenaphthene |
Rat |
2 |
(1) |
Positive |
||||||||
|
Mouse |
1 |
|||||||||||
|
Hamster |
1 |
|||||||||||
|
2-Nitrofluorene |
Rat |
2 |
|
|
|
|
|
|
|
|
(1d) |
Positive |
|
(1) |
(1) |
(1) |
|
|
|
|
|
|
|
||
|
Mouse |
|
|
|
(2) |
|
|
|
|
|
|
||
|
2,5-Dinitrofluorene |
Rat |
(1) |
Database insufficient |
|||||||||
|
2,7-Dinitrofluorene |
Rat |
(1) |
(1d) |
Database insufficient |
||||||||
|
3-Nitrofluoranthene |
Rat |
1 |
(2e) |
Positive |
||||||||
|
Mouse |
1 |
(1f) |
||||||||||
|
3,7-Dinitrofluoranthene |
Rat |
1 |
1e |
Positive |
||||||||
|
3,9-Dinitrofluoranthene |
Rat |
1 |
1e |
Positive |
||||||||
|
1-Nitropyrene |
Rat |
2 |
|
|
|
4 |
|
2 |
|
|
|
Positive |
|
|
(1) |
|
|
(1) |
(1) |
|
(1) |
|
(1e) |
|
|
|
Mouse |
|
|
|
|
|
|
1f |
|
|
|
|
|
|
|
|
|
(2) |
|
(1) |
(2) |
(2) |
|
|
|
|
Hamster |
(1g) |
|||||||||||
|
2-Nitropyrene |
Rat |
1 |
(1d) |
(Positive) |
||||||||
|
4-Nitropyrene |
Rat |
1 |
1 |
2d |
Positive |
|||||||
|
Mouse |
1f |
|||||||||||
|
1,3-Dinitropyrene |
Rat |
(1) |
2 |
1 |
Positive |
|||||||
|
Mouse |
(1) |
(1f) |
||||||||||
|
1,6-Dinitropyrene |
Rat |
(1) |
2 |
1 |
2e |
Positive |
||||||
|
Mouse |
1 |
1f |
||||||||||
|
Hamster |
1g |
|||||||||||
|
1,8-Dinitropyrene |
Rat |
1 |
3 |
1 |
Positive |
|||||||
|
Mouse |
1 |
(1f) |
||||||||||
|
7-Nitrobenz[a]-anthracene |
Mouse |
1 |
(Positive) |
|||||||||
|
6-Nitrochrysene |
Rat |
1f |
1d |
Positive |
||||||||
|
Mouse |
|
|
1 |
|
|
|
1 |
|
|
|
||
|
|
|
|
|
|
|
13f |
|
|
|
||
|
|
|
|
|
|
|
(1f) |
|
|
|
||
|
1-Nitrobenzo[a]pyrene |
Rat |
(1) |
Database insufficient |
|||||||||
|
Mouse |
(2f) |
|||||||||||
|
2-Nitrobenzo[a]pyrene |
Mouse |
1f |
(Positive) |
|||||||||
|
3-Nitrobenzo[a]pyrene |
Rat |
(1) |
Database insufficient |
|||||||||
|
Mouse |
(2f) |
|||||||||||
|
6-Nitrobenzo[a]pyrene |
Mouse |
(1) |
1f |
(1f) |
(Positive) |
|||||||
|
1-Nitrobenzo[e]pyrene |
Mouse |
(1f) |
Database insufficient |
|||||||||
|
3-Nitrobenzo[e]pyrene |
Mouse |
(1f) |
Database insufficient |
|||||||||
|
1,6-Dinitrobenzo[a]pyrene |
Rat |
(2) |
Database insufficient |
|||||||||
|
3,6-Dinitrobenzo[a]pyrene |
Rat |
2 |
(Positive)h |
|||||||||
|
7-Nitrodibenz[a,h]-anthracene |
Mouse |
|
|
|
|
|
|
1f |
|
|
|
(Positive) |
|
|
|
|
|
|
|
(1f) |
|
|
|
||
|
9-Nitrodibenz[a,c]-anthracene |
Mouse |
(1f) |
Database insufficient |
|||||||||
|
3-Nitroperylene |
Mouse |
1 |
(Positive) |
|||||||||
|
a |
Number without parentheses: study design sufficient for assessment; number in parentheses: study with limitations (data from abstract, fewer than 10 animals per group per sex and/or no control, studies with negative results not according to standard guidelines, positive studies with one dose low level of significance and high spontaneous incidence; for details, see Table 39. |
|
b |
Database insufficient: no valid study; (positive): only one positive study with study design sufficient for assessment; positive: more than one study with a study design sufficient for assessment. |
|
c |
Implantation into the bladder. |
|
d |
Injection into mammary tissue. |
|
e |
Intrapulmonary implants. |
|
f |
Newborn assay. |
|
g |
Intratracheal instillation. |
|
h |
Probably the same results are presented in two different publications. |
Some indication of carcinogenic effects (see Table 52) has been shown for seven nitroPAHs: 2-nitropyrene, 7-nitrobenz[a]anthracene, 2-nitrobenzo[a]pyrene, 6-nitrobenzo[a]pyrene, 3,6-dinitrobenzo[a]pyrene, 7-nitrodibenz[a,h]anthracene and 3-nitroperylene.
Although most of the positive studies had limitations in experimental design (see above) or insufficient documentation, the sum of experimental results showed indication of carcinogenic effects in experimental animals for the remaining 11 of the nitroPAHs tested (see Table 52): 5-nitroacenaphthene, 2-nitrofluorene, 3-nitrofluoranthene, 3,7-dinitrofluoranthene, 3,9-dinitrofluoranthene, 1-nitropyrene, 4-nitropyrene, 1,3-dinitropyrene, 1,6-dinitropyrene, 1,8-dinitropyrene and 6-nitrochrysene.
Numerous studies (22 studies in Table 39) have been performed with 1-nitropyrene. This is due to the fact that 1-nitropyrene has the highest concentration of all nitroPAHs in the nitroPAH fraction of diesel exhaust and to the resulting discussion of its possible carcinogenicity. Most of these studies revealed a study design sufficient for assessment of the carcinogenic activity (see Table 52), including the oral exposure route. In reviews on the carcinogenicity of nitroPAHs (IARC, 1989; Rippe & Pott, 1989), the authors discussed the carcinogenic activity of 1-nitropyrene as possible contamination of the test substance with dinitropyrenes and referred to the studies of Odagiri et al. (1986) (oral, systemic tumours; see Table 39) and Ohgaki et al. (1982, 1985) (s.c., local effects). In recent studies with higher purity of the test substance but similar study design and similar results (Hirose et al., 1984; El-Bayoumy et al., 1988b, 1995; see Table 39), it could, however, be shown that the carcinogenic effect is due to 1-nitropyrene itself.
NitroPAHs induced a variety of tumours at the site of application as well as distally in experimental animals. Tumour types observed after exposure to nitroPAHs are tabulated in Table 53 (for details on studies, see Table 39). Besides local effects at the site of injection, nitroPAHs induced mainly systemic tumours in mammary tissue, lung, liver and haematopoietic system.
Table 53. Type of tumours induced by nitroPAHs
|
Substance |
Species |
Routea |
Type of tumourb |
Local/ systemic effects |
References |
|
5-Nitroacenaphthene |
Rat |
Oral |
Mammary carcinoma |
Systemic |
Takemura et al. (1974); NCI (1978b) |
|
1-Nitropyrene |
Rat |
Oral |
Mammary tumours |
Systemic |
El-Bayoumy et al. (1988b, 1995) |
|
1,8-Dinitropyrene |
Rat |
Oral |
Mammary tumours |
Systemic |
King (1988) |
|
1-Nitropyrene |
Rat |
s.c. |
Mammary adenocarcinoma |
Systemic |
King (1988) |
|
4-Nitropyrene |
Rat |
s.c. |
Mammary tumours |
Systemic |
King (1988) |
|
1,3-Dinitropyrene |
Rat |
s.c. |
Mammary adenocarcinoma |
Systemic |
King (1988) |
|
1,8-Dinitropyrene |
Rat |
s.c. |
Mammary adenocarcinoma |
Systemic |
King (1988) |
|
1-Nitropyrene |
Rat |
i.p. |
Mammary tumours |
Systemic |
King (1988) |
|
4-Nitropyrene |
Rat |
i.p. |
Mammary adenocarcinoma |
Systemic |
Imaida et al. (1991b) |
|
1,3-Dinitropyrene |
Rat |
i.p. |
Mammary tumours |
Systemic |
King (1988) |
|
2,7-Dinitropyrene |
Rat |
Intramammary |
Mammary adenocarcinoma |
Local |
Malejka-Giganti et al. (1999) |
|
4-Nitropyrene |
Rat |
Intramammary |
Mammary adenocarcinoma |
Local |
Imaida et al. (1991b); El-Bayoumy et al. (1993) |
|
6-Nitrochrysene |
Rat |
Intramammary |
Mammary tumours |
Local |
El-Bayoumy et al. (1993) |
|
5-Nitroacenaphthene |
Rat |
Oral |
Lung adenoma/carcinoma |
Systemic |
NCI (1978b) |
|
1-Nitropyrene |
Rat |
Oral |
Lung tumours |
Systemic |
El-Bayoumy et al. (1988b) |
|
3,7-Dinitrofluoranthene |
Rat |
Intrapulmonary |
Lung tumours |
Local |
Horikawa et al. (1991) |
|
3,9-Dinitrofluoranthene |
Rat |
Intrapulmonary |
Lung tumours |
Local |
Horikawa et al. (1991) |
|
1,6-Dinitropyrene |
Rat |
Intrapulmonary |
Lung tumours |
Local |
Maeda et al. (1986); Iwagawa et al. (1989) |
|
1,6-Dinitropyrene |
Hamster |
i.tr. |
Lung adenocarcinoma |
Local |
Takayama et al. (1985) |
|
3-Nitrofluoranthene |
Mouse |
i.p.c |
Lung tumours |
Systemic |
Busby et al. (1989) |
|
4-Nitropyrene |
Mouse |
i.p.c |
Lung tumours |
Systemic |
Wislocki et al. (1986) |
|
6-Nitrochrysene |
Mouse |
i.p. |
Lung tumours |
Systemic |
Ogawa et al. (1996) |
|
6-Nitrochrysene |
Mouse |
i.p.c |
Lung tumours |
Systemic |
Wislocki et al. (1986); Busby et al. (1985, 1989); El-Bayoumy et al. (1989a); Imaida et al. (1992); Fu et al. (1994a); von Tungeln et al. (1999a) |
|
5-Nitroacenaphthene |
Mouse |
Oral |
Hepatocellular carcinoma |
Systemic |
NCI (1978b) |
|
5-Nitroacenaphthene |
Hamster |
Oral |
Cholangioma |
Systemic |
Takemura et al. (1974) |
|
2-Nitrofluorene |
Rat |
Oral |
Hepatocellular carcinoma |
Systemic |
Cui et al. (1995) |
|
1-Nitropyrene |
Mouse |
i.p.c |
Liver carcinoma |
Systemic |
Wislocki et al. (1986) |
|
4-Nitropyrene |
Mouse |
i.p.c |
Liver carcinoma |
Systemic |
Wislocki et al. (1986) |
|
1,6-Dinitropyrene |
Mouse |
i.p.c |
Liver carcinoma |
Systemic |
Wislocki et al. (1986) |
|
7-Nitrobenz[a]anthracene |
Mouse |
i.p.c |
Liver tumours |
Systemic |
Wislocki et al. (1986) |
|
6-Nitrochrysene |
Mouse |
i.p.c |
Liver tumours |
Systemic |
Wislocki et al. (1986); El-Bayoumy et al. (1989a); Fu et al. (1994a); Manjanatha et al. (1996); von Tungeln et al. (1999a) |
|
2-Nitrobenzo[a]pyrene |
Mouse |
i.p.c |
Liver tumours |
Systemic |
von Tungeln et al. (1999b) |
|
6-Nitrobenzo[a]pyrene |
Mouse |
i.p.c |
Liver tumours |
Systemic |
Wislocki et al. (1986) |
|
7-Nitrodibenz[a,h]-anthracene |
Mouse |
i.p.c |
Liver tumours |
Systemic |
von Tungeln et al. (1994b), data from abstract; Fu et al. (1998b) |
|
1-Nitropyrene |
Rat |
Oral |
Leukaemiad |
Systemic |
Odagiri et al. (1986) |
|
1,6-Dinitropyrene |
Hamster |
i.tr. |
Leukaemia |
Systemic |
Takayama et al. (1985) |
|
1-Nitropyrene |
Rat |
s.c. |
Leukaemia |
Systemic |
King (1988) |
|
4-Nitropyrene |
Rat |
s.c. |
Leukaemia |
Systemic |
King (1988) |
|
1,6-Dinitropyrene |
Rat |
s.c. |
Leukaemia |
Systemic |
King (1988) |
|
1,8-Dinitropyrene |
Rat |
s.c. |
Leukaemia |
Systemic |
King (1988) |
|
2-Nitropyrene |
Rat |
i.p. |
Leukaemia/lymphoma |
Systemic |
Imaida et al. (1991b) |
|
6-Nitrochrysene |
Mouse |
i.p.c |
Malignant lymphoma |
Systemic |
Wislocki et al. (1986) |
|
5-Nitroacenaphthene |
Rat |
Oral |
Clitoral gland carcinoma |
Systemic |
NCI (1978b) |
|
1-Nitropyrene |
Rat |
Oral |
Clitoral gland tumoursd |
Systemic |
Odagiri et al. (1986) |
|
2-Nitrofluorene |
Rat |
Oral |
Ear duct tumours |
Systemic |
Miller et al. (1955) |
|
5-Nitroacenaphthene |
Rat |
Oral |
Ear canal/skin carcinomas |
Systemic |
NCI (1978b) |
|
4-Nitropyrene |
Rat |
s.c. |
Zymbal gland carcinoma |
Systemic |
King (1988) |
|
5-Nitroacenaphthene |
Mouse |
Oral |
Ovarian tumour |
Systemic |
NCI (1978b) |
|
2-Nitrofluorene |
Rat |
Oral |
Kidney cortical carcinoma |
Systemic |
Cui et al. (1995) |
|
1-Nitropyrene |
Rat |
Oral |
Pancreas islet cell adenoma |
Systemic |
El-Bayoumy et al. (1988b) |
|
5-Nitroacenaphthene |
Rat |
Oral |
Small intestine adenocarcinoma |
Local |
Takemura et al. (1974) |
|
2-Nitrofluorene |
Rat |
Oral |
Forestomach squamous cell carcinoma |
Local |
Miller et al. (1955); Cui et al. (1995) |
|
6-Nitrochrysene |
Rat |
i.p. |
Colon tumours |
Local/ systemic |
Imaida et al. (1992) |
|
6-Nitrochrysene |
Mouse |
Dermal |
Skin tumours |
Local |
El-Bayoumy et al. (1982) |
|
3-Nitroperylene |
Mouse |
Dermal |
Skin tumours |
Local |
El-Bayoumy et al. (1982) |
|
3-Nitrofluoranthene |
Rat |
s.c. |
Malignant subcutaneous tumours |
Local |
Ohgaki et al. (1982) |
|
3,7-Dinitrofluoranthene |
Rat |
s.c. |
Malignant subcutaneous tumours |
Local |
Tokiwa et al. (1987a) |
|
3,9-Dinitrofluoranthene |
Rat |
s.c. |
Malignant subcutaneous tumours |
Local |
Tokiwa et al. (1987a) |
|
1-Nitropyrene |
Rat |
s.c. |
Malignant subcutaneous tumoursd |
Local |
Ohgaki et al. (1982); Hirose et al. (1984) |
|
4-Nitropyrene |
Rat |
s.c. |
Malignant subcutaneous tumours |
Local |
King (1988) |
|
1,3-Dinitropyrene |
Rat |
s.c. |
Malignant subcutaneous tumours |
Local |
Ohgaki et al. (1984); King (1988) |
|
1,6-Dinitropyrene |
Mouse |
s.c. |
Malignant subcutaneous tumours |
Local |
Tokiwa et al. (1986) |
|
1,6-Dinitropyrene |
Rat |
s.c. |
Malignant subcutaneous tumours |
Local |
Ohgaki et al. (1985); King (1988) |
|
1,8-Dinitropyrene |
Rat |
s.c. |
Malignant subcutaneous tumours |
Local |
Ohgaki et al. (1984, 1985); King (1988) |
|
1,8-Dinitropyrene |
Mouse |
s.c. |
Subcutaneous tumours |
Local |
Otofuji et al. (1987) |
|
3,6-Dinitrobenzo[a]pyrene |
Rat |
s.c. |
Malignant subcutaneous tumours |
Local |
Tokiwa et al. (1994); Horikawa et al. (1998) |
|
1,6-Dinitropyrene |
Rat |
i.p. |
Malignant fibrous histiocytoma |
Local |
King (1988) |
|
1,8-Dinitropyrene |
Rat |
i.p. |
Malignant fibrous histiocytoma |
Local |
King (1988) |
|
a |
Intrapulmonary = intrapulmonary implants; intramammary = injected into mammary tissue; i.tr. = intratracheal instillation. |
|
b |
Only data from studies sufficient for assessment (compare with Table 39). |
|
c |
Study using the newborn mouse assay (for details, see Table 39). |
|
d |
Effects probably due to impurities with dinitropyrenes. |
In view of the possible involvement of environmental carcinogenic nitroPAHs (in urban air, food, combustion) in breast cancer etiology (El-Bayoumy, 1992), it should be emphasized that several nitroPAHs increased the incidence of mammary tumours in rats (see Table 39): 5-nitroacenaphthene, 1-nitropyrene, 4-nitropyrene, 1,3-dinitropyrene and 1,8-dinitropyrene. Studies with limited validity (see Table 39) revealed indications of systemically induced mammary tumours also with 2-nitrofluorene, 1,6-dinitropyrene and 6-nitrochrysene. Local effects were observed with 2,7-dinitrofluorene and 6-nitrochrysene (see Table 53).
Exposure to dinitrofluoranthenes (rat) or 1,6-dinitropyrene (hamster/rat) via the respiratory tract led to local lung tumour formation. Systemic effects in the lung were observed with oral (rat: 5-nitroacenaphthene, 1-nitropyrene) or parenteral (mouse: 3-nitrofluoranthene, 4-nitropyrene, 6-nitrochrysene) application (see Table 53).
Ten of 28 nitroPAHs with available data on carcinogenicity induced systemic tumours in the liver of rodent species: eight nitroPAHs in mice after i.p. injection, 5-nitroacenaphthene in mouse and hamster after oral exposure, and 2-nitrofluorene in the rat after oral exposure (see Table 53).
Mononitrated pyrenes as well as 1,6- and 1,8-dinitropyrenes induced systemic effects on the haematopoietic system (leukaemia/ lymphoma) in rats after parenteral application; 1-nitropyrene induced these effects after oral exposure as well. Similar effects were observed with 6-nitrochrysene in mice and 1,6-dinitropyrene in hamters (see Table 53).
Other systemically induced tumour types are clitoral gland tumours in rats after oral exposure to 5-nitroacenaphthene or 1-nitroyrene. 5-Nitroacenaphthene also induced a high incidence of ear canal carcinomas; the effect of 4-nitropyrene reached only a low level of significance (see also Table 39). Ovarian tumours in mice were observed after oral exposure to 5-nitroacenaphthene. Worth mentioning are the kidney cortical carcinomas in rats after oral exposure to 2-nitrofluorene, since this tumour type reached a high incidence (see also Table 39). The biological significance of pancreas islet cell adenomas in rats is questionable, because the effect is not dose dependent (see also Table 39; also low level of significance).
Local carcinogenic effects in the intestinal tract of rats were observed after oral exposure to 2-nitrofluorene and 5-nitroacenaphthene. 6-Nitrochrysene and 3-nitroperylene revealed skin tumours in mice after dermal exposure. Local effects in rats (predominantly induction of malignant fibrous histiocytoma) were observed with nine nitroPAHs (see Table 53; for two nitroPAHs also in mice) after s.c. and/or i.p. injection. These local effects indicate metabolic activation of the nitroPAHs at the site of application.
Due to differences in study design (e.g., one dose versus long-term daily application, parenteral versus oral exposure, newborn versus adult animals, etc.), overall quantification and comparison of the carcinogenic potency are not possible. However, the carcinogenic potencies of several nitroPAHs can be compared because they were investigated with similar experimental design in the same laboratory. Results reported in these studies (see Table 39 for details on study design) concerning the carcinogenic potency are presented in Table 54. For comparison, available data on BaP have been added.
Table 54. Comparative studies on nitroPAHs
|
Species |
Route |
Local/systemic tumours |
Potency ranking order |
Reference |
|
Rat |
Intramammary injection |
Local |
2,7-dinitrofluorene > 2-nitrofluorene |
Malejka-Giganti et al. (1999) |
|
Rat |
s.c. |
Local |
1-nitropyrenea > 3-nitrofluoranthene |
Ohgaki et al. (1982) |
|
Rat |
Intrapulmonary implants |
Local |
3,9-dinitrofluoranthene > 3,7-dinitrofluoranthene > BaP > 3-nitrofluoranthene |
Horikawa et al. (1991) |
|
Rat |
s.c. |
Local |
3,7-dinitrofluoranthene = 3,9-dinitrofluoranthene |
Tokiwa et al. (1987a) |
|
Rat |
s.c. |
Systemic |
4-nitropyrene > 1-nitropyrene |
King (1988) |
|
Rat |
Intramammary injection |
Local |
4-nitropyrene > 1-nitropyrene = 2-nitropyrene |
Imaida et al. (1991b) |
|
Rat |
Oral |
Systemic (mammary) |
BaP > 1-nitropyrene |
El-Bayoumy et al. (1995) |
|
Rat |
Oral |
Systemic (mammary) |
1,8-dinitropyrene > 1,6-dinitropyrene > 1,3-dinitropyrene = 1-nitropyrene |
King (1988) |
|
Rat |
s.c. or i.p. |
Local |
1,6-dinitropyrene > 1,8-dinitropyrene > 1,3-dinitropyrene > 1-nitropyrene |
King (1988) |
|
Rat |
s.c. or i.p. |
Systemic (mammary) |
1-nitropyrene > 1,3-dinitropyrene > 1,8-dinitropyrene > 1,6-dinitropyrene |
King (1988) |
|
Mouse |
Dermal |
Local |
BaP > mixture of dinitropyrenes > 1-nitropyrene |
Nesnow et al. (1984) |
|
Rat |
Intrapulmonary implants |
Local |
1,6-dinitropyrene > 1-nitropyrene |
Maeda et al. (1986) |
|
Rat |
s.c. |
Local |
1,3-dinitropyrene = 1,8-dinitropyrene |
Ohgaki et al. (1984) |
|
Rat |
s.c. |
Local |
1,8-dinitropyrene > 1-nitropyrene |
Ohgaki et al. (1985) |
|
Mouse |
s.c. |
Local |
1,6-dinitropyrene > 1-nitropyrene |
Tokiwa et al. (1984, 1986) |
|
Mouse |
s.c. |
Local |
BaP > 1,8-dinitropyrene > 1,3-dinitropyrene |
Otofuji et al. (1987) |
|
Rat |
Intrapulmonary implants |
Local |
1,6-dinitropyrene > BaP |
Iwagawa et al. (1989) |
|
Mouse |
i.p.b |
Systemic |
1,6-dinitropyrene > 1,3-dinitropyrene > 1,8-dinitropyrene |
Wislocki et al. (1986) |
|
Mouse |
i.p.b |
Systemic |
6-nitrochrysene > 1,6-dinitropyrene |
Wislocki et al. (1986) |
|
Mouse |
i.p.b |
Systemic |
6-nitrochrysene > BaP > 4-nitropyrene > 6-nitrobenzo[a]pyrene > 1-nitropyrene > 7-nitrobenz[a]anthracene |
Wislocki et al. (1986) |
|
Rat |
Intramammary injection |
Local |
6-nitrochrysene > 4-nitropyrene |
El-Bayoumy et al. (1993) |
|
Mouse |
Dermal |
Local |
6-nitrochrysene > 3-nitroperylene > 1-nitropyrene |
El-Bayoumy et al. (1982) |
|
Mouse |
i.p.b |
Systemic |
6-nitrochrysene > BaP |
von Tungeln et al. (1999a) |
|
Mouse |
i.p.b |
Systemic |
6-nitrochrysene> 2-nitrobenzo[a]pyrene > 7-nitrodibenz[a,h]anthracene > 9-nitrodibenz[a,c]anthracene = 3-nitrobenzo[e]pyrene = 1-nitrobenzo[e]pyrene = 3-nitrobenzo[a]pyrene = 1-nitrobenzo[a]pyrene = 2-nitrofluoranthene = 3-nitrofluoranthene |
von Tungeln et al. (1994b; 1999b) |
|
Mouse |
i.p.b |
Systemic |
6-nitrochrysene > BaP > 3-nitrofluoranthene > 6-nitrobenzo[a]pyrene = 1-nitropyrene |
Busby et al. (1989) |
|
Mouse |
Dermal |
Local |
BaP > 6-nitrobenzo[a]pyrene |
El-Bayoumy et al. (1982) |
|
Rat |
s.c. |
Local |
3,6-dinitrobenzo[a]pyrene > BaP > 1,6-dinitrobenzo[a]pyrene |
Horikawa et al. (1998) |
|
Rat |
s.c. |
Local |
BaP > 3,6-dinitrobenzo[a]pyrene > 1,6-dinitrobenzo[a]pyrene |
Tokiwa et al. (1994) |
a Effect probably due to impurities with dinitropyrenes.
b Newborn mouse assay.
Due to the differences in the ranking order between study results on local and systemic carcinogenic effects, a synopsis of the ranking order including all tested nitroPAHs could not be given. However, the following conclusions can be drawn from Table 54:
The bay region diol epoxide derived from BaP was included in some studies for comparison with 6-nitrochrysene (Hecht et al., 1994).
Table 55. Comparison of dose-related incidence of liver tumours in the newborn mouse assaya
|
Substance |
Total dose per mouse (nmol [µg]) |
Dose per application (mg/kg bw)b |
Incidence of mice with liver tumoursc (%) |
Estimated dose (mg/kg bw per injection) inducing a liver tumour incidence of 25%d |
Reference |
|
6-Nitrochrysene |
100 (27) |
~4 |
94 |
1 |
von Tungeln et al. (1999a) |
|
2-Nitrobenzo[a]-pyrene |
400 (119) |
~17 |
100 |
4 |
von Tungeln et al. (1999b) |
|
1,6-Dinitropyrene |
200 (58) |
~8 |
32 |
6 |
Wislocki et al. (1986) |
|
7-Nitrodibenz[a,h]-anthracene |
400 (129) |
~18 |
58 |
8 |
Fu et al. (1998b) |
|
BaPe |
560 (141) |
~20 |
49 |
10 |
Wislocki et al. (1986) |
|
6-Nitrobenzo[a]-pyrene |
560 (166) |
~24 |
28 |
21 |
Wislocki et al. (1986) |
|
4-Nitropyrene |
2800 (692) |
~99 |
83 |
30 |
Wislocki et al. (1986) |
|
1-Nitropyrene |
2800 (692) |
~99 |
28 |
88 |
Wislocki et al. (1986) |
|
7-Nitrobenz[a]-anthracene |
2800 (765) |
~109 |
28 |
97 |
Wislocki et al. (1986) |
|
a |
Newborn mice assay: mice were injected i.p. with 1/7, 2/7 and 4/7 of the total dose at 1, 8 and 15 days of age, respectively; post-exposure observation period 12 months. All data are related to results in male mice, which are more sensitive in this assay. |
|
b |
On the assumption that the body weight was 1 g at day 1 postnatal. |
|
c |
Liver adenomas and carcinomas evaluated, spontaneous incidence not considered. |
|
d |
Calculated using the given dose and incidence and assuming linear dose–response relationship. |
|
e |
Benzo[a]pyrene added for comparison. |
In several studies tabulated in Table 39, the parent PAH was also tested with a study design that was similar to or the same as that used for nitroPAHs. The comparison of the carcinogenic activities of the nitroPAHs and the corresponding parent PAHs in these studies is presented in Table 56 (for details on study design, see Table 39).
No differences were observed between mononitrated fluoranthenes and the parent PAH fluoranthene or between 9-nitrodibenz[a,c]anthracene and the parent dibenz[a,c]anthracene (see Table 56).
Table 56. Comparison of nitroPAHs with parent PAHs
|
Species |
Route |
Local/ systemic tumours |
Potency ranking order |
Reference |
|
Mouse |
i.p.a |
Systemic |
fluoranthene = 2-nitrofluoranthene = 3-nitrofluoranthene |
von Tungeln et al. (1994b, 1999b) |
|
Mouse |
i.p.a |
Systemic |
3-nitrofluoranthene = fluoranthene |
Busby et al. (1989) |
|
Mouse |
i.p.a |
Systemic |
benz[a]anthracene > 7-nitrobenz[a]anthracene |
Wislocki et al. (1986) |
|
Mouse |
i.p.a |
Systemic |
1-nitropyrene = pyrene |
Busby et al. (1989) |
|
Mouse |
i.p.a |
Systemic |
4-nitropyrene > 1-nitropyrene > pyrene |
Wislocki et al. (1986) |
|
Mouse |
Dermal |
Local |
1-nitropyrene = pyrene |
El-Bayoumy et al. (1982) |
|
Mouse |
i.p.a |
Systemic |
1,6-dinitropyrene > 1,3-dinitropyrene > 1,8-dinitropyrene > pyrene |
Wislocki et al. (1986) |
|
Mouse |
i.p.a |
Systemic |
6-nitrochrysene > chrysene |
Wislocki et al. (1986) |
|
Mouse |
Dermal |
Local |
chrysene > 6-nitrochrysene |
El-Bayoumy et al. (1982) |
|
Mouse |
i.p.a |
Systemic |
6-nitrochrysene > chrysene |
Busby et al. (1989) |
|
Mouse |
i.p.a |
Systemic |
BaP > 6-nitrobenzo[a]pyrene |
Busby et al. (1989) |
|
Mouse |
i.p.a |
Systemic |
BaP > 6-nitrobenzo[a]pyrene |
Wislocki et al. (1986) |
|
Mouse |
Dermal |
Local |
BaP > 6-nitrobenzo[a]pyrene |
El-Bayoumy et al. (1982) |
|
Rat |
s.c. |
Local |
3,6-dinitrobenzo[a]pyrene > BaP > 1,6-dinitrobenzo[a]pyrene |
Horikawa et al. (1998) |
|
Rat |
s.c. |
Local |
BaP > 3,6-dinitrobenzo[a]pyrene > 1,6-dinitrobenzo[a]pyrene |
Tokiwa et al. (1994) |
|
Mouse |
i.p.a |
Systemic |
dibenz[a,c]anthracene = 9-nitrodibenz[a,c]anthracene |
von Tungeln et al. (1994b, 1999b) |
|
Mouse |
i.p.a |
Systemic |
dibenz[a,h]anthracene > 7-nitrodibenz[a,h]anthracene |
von Tungeln et al. (1994b) (data from abstract) |
|
Mouse |
i.p.a |
Systemic |
dibenz[a,h]anthracene > 7-nitrodibenz[a,h]anthracene |
Fu et al. (1998b) |
|
Mouse |
Dermal |
Local |
3-nitroperylene > perylene |
El-Bayoumy et al. (1982) |
|
a |
Newborn mouse assay. |
|
b |
Rosenkranz et al. (1980); LL) Taylor et al. (1995); MM) Tokiwa et al. (1984); NN) Urios et al. (1994); OO) Yu et al. (1991); PP) Yu et al. (1992); QQ) Fu et al. (1985b); RR) Shah et al. (1991); SS) Shane & Winston (1997); TT) Ashby et al. (1983); UU) Rosenkranz et al. (1982); VV) Fu et al. (1988b); WW) El-Bayoumy & Hecht (1984b); XX) El-Bayoumy et al. (1989b); YY) Chou et al. (1984); ZZ) Chou et al. (1986); aaa) Hass et al. (1986a); bbb) Löfroth et al. (1984); ccc) Chou et al. (1985); ddd) Fu et al. (1997); eee) Hass et al. (1984); fff) Heflich et al. (1989); ggg) Anderson et al. (1987); hhh) Fu et al. (1982); iii) Fu et al. (1988c). |
|
c |
Study results revealing no mutagenicity were not included (exception see below) if the strain was presumably not tested up to cytotoxicity; study results with no explicitness (e.g., < or > 1) not used for calculation of the mean; MA = metabolic activation (rodent liver microsomes). |
|
d |
In each of these columns, the following values are presented (separated in data with or without MA): a) range of mutagenic potency in revertants per nmol (2 or more data available), b) mean number of revertants per nmol and c) in parentheses the number n of data used for calculation of the mean. |
|
e |
Bold-typed letters indicate clearly more revertants (difference at least 10-fold) in strains detecting base pair substitutions (TA100, TA1535) relative to strains detecting frameshift mutations (TA98, TA1537, TA1538) or the reversal (comparison in dependence on metabolic activation); a difference of at least 2-fold is marked by normal-typed letters. |
|
f |
30 revertants/nmol after 90-min preincubation. |
|
g |
680 revertants/nmol after 90-min preincubation. |
|
h |
Presumably not tested up to cytotoxicity (included in the table if no other data were available). |
|
i |
_* = less than 2-fold increase. |
|
j |
Structural assignment not given. |
|
k |
In further studies, no mutagenicity was observed without MA, but the substance was not tested up to cytotoxicity. |
The mono- or dinitrated pyrenes are more carcinogenic than pyrene. Similar results were presented for 3-nitroperylene versus perylene and for 6-nitrochrysene versus chrysene; for local effects after dermal exposure, however, 6-nitrochrysene was less active than chrysene (see Table 56).
Data in Table 56 suggest that most mono- and dinitrated benzo[a]pyrenes are less carcinogenic than the parent BaP (see also Table 52), although there are contradictory results with 3,6-dinitrobenzo[a]pyrene. 2-Nitrobenzo[a]pyrene probably has a higher potency than BaP (see Table 55), but the direct comparison is not available.
Other examples for nitroPAHs with less activity than the parent PAH are 7-nitrobenz[a]anthracene versus benz[a]anthracene and 7-nitrodibenz[a,h]anthracene versus dibenz[a,h]anthracene (see Table 56). These alterations in carcinogenicity by addition of a nitro group to a PAH were discussed by Fu et al. (1994b). The authors suggested that the introduction of a nitro group to a PAH with a bay region such as BaP resulted in a weaker carcinogenicity of the corresponding nitroPAH if the nitro group adopts a perpendicular or nearly perpendicular orientation. The effect of nitro orientation on metabolism, mutagenesis and adduct formation is also presented in chapter 6.
Quantification and comparison of the carcinogenic potency are possible with studies using a similar study design. Data on several nitroPAHs are available for the newborn mouse assay (three i.p. injections on days 1, 7 and 15, respectively; post-exposure observation period 12 months; for details on study design, see Table 39). It is possible to rank these nitroPAHs (according to their potency) by calculating for each the estimated dose inducing a liver tumour incidence of 25% (see Table 55). 6-Nitrochrysene seems to be the most active nitroPAH in this assay. For comparison, data on BaP are included. The results of this ranking order are comparable with those in Table 54. Due to differences in study design and species, a similar quantification is problematical with other studies on the same or other administration routes.
A lifetime study has been started on F344 rats after intratracheal administration of 3-nitrobenzanthrone (in diesel exhaust) (Adachi et al., 1999).
Data on the carcinogenic effects of the metabolites of nitroPAHs are available for 2-nitrofluorene, 1-nitropyrene and 6-nitrochrysene. Metabolites were studied concurrently with the same study design and in equimolar doses (for details of study design and validity, see Table 39).
In a study on 2-nitrofluorene (Miller et al., 1955), the metabolite 2-aminofluorene induced a higher incidence of mammary tumours than 2-nitrofluorene itself, as well as ear duct tumours in females. The highest carcinogenic potency was shown by the metabolite 2-acetylaminofluorene, which caused small intestine tumours in male and female rats and liver tumours in male rats, neither of which were observed with 2-nitrofluorene itself.
El-Bayoumy et al. (1988b), on the other hand, presented data showing that the parent compound 1-nitropyrene was significantly more carcinogenic after oral application in rats than either 1-nitrosopyrene or 1-aminopyrene. In contrast, 1-nitrosopyrene induced a higher incidence of liver tumours in mice after i.p. application than 1-nitropyrene (Wislocki et al., 1986). No significant effects were observed with the metabolites 1-nitropyren-3-ol, 1-nitropyren-6-ol and 1-nitropyren-8-ol (King, 1988; Imaida et al., 1995) or with N-hydroxy-N-acetyl-1-aminopyrene and N-acetyl-1-aminopyrene (Imaida et al., 1991b).
El-Bayoumy et al. (1989a) studied the carcinogenic activity of 6-nitrochrysene and its metabolites in mice. In contrast to the parent compound 6-nitrochrysene, 6-nitrosochrysene and 6-aminochrysene were inactive at equimolar doses. The metabolites trans-1,2-dihydro-1,2-dihydroxy-6-nitrochrysene and trans-1,2-dihydro-1,2-dihydroxy-6-aminochrysene had carcinogenic activity comparable to that of 6-nitrochrysene; in the liver, trans-1,2-dihydro-1,2-dihydroxy-6-nitrochrysene induced more tumours per mouse than 6-nitrochrysene. These results indicated the metabolic activation of 6-nitrochrysene by ring oxidation.
1-Nitropyrene is often used as a marker for nitroPAHs, similar to the use of BaP as a marker for PAHs, in studies on diesel exhaust (see also chapter 3). In several studies on mice and rats (see Table 54), the carcinogenic activities of these two substances have been directly compared (oral, dermal and i.p. application). In all of these studies, BaP was clearly more carcinogenic than 1-nitropyrene. For example, Wislocki et al. (1986) showed that this difference was about 300% (see Table 55). Furthermore, with dermal exposure, BaP induced an incidence of 100% skin tumours at a dose of 0.05 mg per mouse, whereas 1-nitropyrene revealed no significant effect at doses up to 3 mg per mouse (Nesnow et al., 1984; Table 39).
Grimmer et al. (1987) studied the contribution of PAHs and nitroPAHs to the carcinogenic impact of diesel exhaust condensate. They separated the condensate by chromatographic methods into several fractions and implanted these mixtures into the lung of rats (vehicle beeswax/trioctanoin). Results are presented in Table 57. Findings from this study indicated that most of the carcinogenicity of diesel exhaust is provoked by PAHs consisting of four or more rings. A minor contribution was observed by the fraction of nitroPAHs. Furthermore, nitroPAHs revealed a low carcinogenic activity in comparison with BaP.
Although the discussion on effect mechanisms involved in cancer caused by diesel emissions is still in progress (Rosenkranz, 1996), it seems that the carcinogenicity of diesel exhaust condensates originates mainly from PAHs and not from nitroPAHs.
Table 57. Squamous cell lung carcinoma incidence from diesel exhaust condensate in ratsa,b
|
Implanted material |
Portion by weight (%) |
Dose (mg) |
Incidence (%) |
|
I) Hydrophilic fraction |
25 |
6.7 |
0/35 (0) |
|
II) Hydrophobic fraction |
75 |
20 |
5/35 (14) |
|
Vehicle control |
0 |
0/35 (0) |
|
|
Untreated control |
0 |
0/35 (0) |
|
|
BaP |
0.03 |
3/35 (9) |
|
a |
From Grimmer et al. (1987). |
|
b |
PAC = polycyclic aromatic compounds; no data about significance; mean survival 97–110 weeks; reduced survival (69 weeks) at the high-dose BaP. |
In Table 46, 11 nitroPAHs are tabulated that showed carcinogenic effects in more than one animal study (marked by a bold type "positive" under carcinogenicity). Data on the genotoxicity of nitroPAHs in vivo are available on 15 nitroPAHs, whereas two or more end-points were investigated on 7 substances, and only 2 of these 7 nitroPAHs revealed consistent positive results in vivo (also marked by the bold type "positive"; see Table 46). The database on genotoxicity in vivo is mainly not sufficient for detailed comparison with carcinogenicity data. Only two nitroPAHs, 2-nitrofluorene and 1-nitropyrene, gave consistent results on genotoxicity in vivo and carcinogenicity. Other carcinogenic nitroPAHs revealed positive in vivo data (3,7-dinitrofluoranthene, 3,9-dinitrofluoranthene, 1,3-, 1,6- and 1,8-dinitropyrene, 6-nitrochrysene) or inconsistent results in vivo (5-nitroacenaphthene, 3-nitrofluoranthene), but with a limited database. No data on genotoxicity in vivo are available with the carcinogen 4-nitropyrene.
More studies are available on genotoxicity in vitro than in vivo. All 11 clearly carcinogenic nitroPAHs (see Table 46) showed positive results in at least three genotoxic end-points in vitro (marked by a bold type "positive" in Table 46).
Six nitroPAHs showed some indication of carcinogenic effects in animals (marked by a normal type "(positive)" in Table 46), but no data are available on genotoxicity in vivo (Table 46). In vitro data revealed clearly positive results for 2-nitropyrene and 6-nitrobenzo[a]pyrene, positive results but with a limited database for 7-nitrobenz[a]anthracene, 3,6-dinitrobenzo[a]pyrene and 3-nitroperylene and inconclusive results for 7-nitrodibenz[a,h]anthracene (Table 46).
The carcinogenicity database was insufficient for 10 nitroPAHs (see Table 52). With the exception of 1- and 2-nitronaphthalene, which revealed positive or inconsistent results in vivo, no data are available on genotoxicity in vivo for these nitroPAHs (Table 46). However, data on genotoxicity in vitro showed positive results in three or more investigated end-points of 1- and 2-nitronaphthalene and 2-nitrofluoranthene; further, five nitroPAHs revealed positive results in vitro, although the database was limited (1-, 2- and 3-nitrobenzo[a]pyrene, 1- and 3-nitrobenzo[e]pyrene). Only inconclusive results are observed with 9-nitrodibenz[a,c]anthracene (Table 46). No data on genotoxicity in vitro and in vivo were available with 1,6-dinitrobenzo[a]pyrene.
Potency equivalency factors (PEFs) for PAHs for cancer induction have been derived by several authors (see also IPCS, 1998). Based on the limited carcinogenicity studies available (given in Table 39), the Office of Environmental Health Hazard Assessment of California, USA, has included nitroPAHs in its PEF scheme: BaP, 1.0 (index compound); 1,6-dinitropyrene and 6-nitrochrysene, 10; 1,8-dinitropyrene, 1.0; 1- and 4-nitropyrene, 0.1; 2-nitrofluorene, 0.01 (OEHHA, 1994).
The use of PEFs is disputed for many reasons (Collins et al., 1998): a) weak database; b) use of tests such as injection into newborn mouse lung; c) rounding to nearest factor of 10; d) the possibility that interactions of nitroPAHs are synergistic or antagonistic rather than being additive; and e) the possibility that some non-carcinogenic compounds potentiate the activity of carcinogenic nitroPAHs (or PAHs).
Ember et al. (2000) reported the increased expression of oncogenes and tumour suppressor genes in different organs of mice 24 h after i.p. injection of 30 µmol 1-nitropyrene/kg bw. Significant elevation was detected in the expression of the c-myc oncogene in spleen, bone marrow and lymph node. No significant change was observed in the expression of oncogenes of the ras gene family (N-ras, Ha-ras, Ki-ras) in lung, thymus, kidney, liver and spleen. However, the expression of the p53 suppressor gene exhibited an increase in the bone marrow and a moderate elevation in the spleen.
Forty-eight hours after i.p. injection of 30 µmol 1-nitropyrene/kg bw, no significantly elevated expression of c-myc oncogene or p53 tumour suppressor gene was detected in mice, but significant overexpression of the Ha-ras oncogene was observed in the lung (11.7-fold higher than vehicle control), liver (9.7-fold), spleen (7.7-fold), thymus (7.1-fold) and kidney (4.2-fold). This effect on the Ha-ras gene was more pronounced in males than in females (Pusztai et al., 1998).
In a short-term bioassay on the carcinogenicity of 6-nitrochrysene in 8-day-old mice (Dass et al., 1999; see Table 39 for details), no accelerated liver tumorigenesis was observed in mice deficient in the p53 suppressor gene in comparison with the wild-type mice.
Johnson et al. (1984) reported lung and liver toxicity of 1-nitronaphthalene after a single i.p. injection (see section 7.1). These results were confirmed by Sauer et al. (1995) in further studies on rats after a single i.p. injection of 100 mg 1-nitronaphthalene/kg bw.
Details on the dose-related epithelial toxicity of 1-nitronaphthalene in the rat lung were presented by Paige et al. (1997), 24 h after i.p. injection of 0, 25, 50, 100 or 150 mg/kg bw (n = 5–9 male Sprague-Dawley rats). At the lowest dose level, only non-ciliated (Clara) cells in the bronchioles were swollen and necrotic. With higher doses, damage to ciliated cells was also observed. The severity of lesions was dose dependent. At the high dose, the most severe lesion was observed in the trachea. Inflammation was noted only at the high dose levels. A dose-dependent increase in lung volume at >25 mg/kg bw was also reported. Re-evaluation of the same slides by transmission electron microscopy revealed injury in Clara cells of rats administered 25 and 50 mg/kg bw, including marked degeneration of mitochondria, swelling of perinuclear endoplasmic reticulum and increased opacity of the mitochondrial matrix. Active ciliogenesis was also observed at 50 mg/kg bw (Paige et al., 1998).
Cytotoxic effects on the epithelium of the lung were also reported in male mice after i.p. injection of 0.5–2 mmol/kg bw (86–346 mg/kg bw; Rasmussen et al., 1986).
Lung toxicity in experimental animals after in vivo administration has been confirmed by in vitro studies using rat lung slice cultures (Price et al., 1995).
Details on liver toxicity in rats after i.p. injection of 100 mg 1-nitronaphthalene/kg bw were reported by Sauer et al. (1997). Ultrastructural evaluation revealed necrosis of centrilobular hepatocytes 48 h after treatment with 1-nitronaphthalene, accompanied by elevated levels of alanine aminotransferase, aspartate aminotransferase and bilirubin in the serum.
1-Nitronaphthalene might be less toxic after oral exposure than after i.p. administration, since daily administration of up to 160 mg/kg bw via the diet revealed no clinical abnormalities in mice or rats (NCI, 1978a; see Table 39).
In comparison with 1-nitronaphthalene, the isomer 2-nitronaphthalene (Johnson et al., 1984) revealed no lung or liver toxicity in rats after a single i.p. dose of 100 mg/kg bw, using similar experimental design.
Twenty-four hours after i.p. injection of 200 mg/kg bw, 2-nitronaphthalene also induced histopathological alterations in the lung of male Sprague-Dawley rats, although respiratory distress syndrome was not observed (compare with similar studies on 1-nitronaphthalene). Pulmonary lesions were restricted to the bronchioles and consisted of epithelial cell necrosis, mild interstitial oedema and inflammation. Clinical chemistry data revealed hepatotoxicity, although no morphological changes were noted (Sauer & Sipes, 1995).
Similar results were presented in a comparative light and electron microscopic study on lung toxicity of nitronaphthalene isomers in i.p.-treated male Swiss-Webster mice (Rasmussen et al., 1986).
Squamous metaplasia of the epiglottis was observed in rats exposed via inhalation for 13 weeks (NTP, 1996; see Table 38). In females, this lesion occurred at an exposure concentration of >0.5 mg 1-nitropyrene/m3; in males, it was observed at >2 mg/m3. At much higher concentrations, squamous metaplasia was also observed in the bronchus of exposed male (>7.8 mg/m3) and female (>20 mg/m3) rats.
NitroPAHs, formed directly from diesel exhaust, heating stoves or other combustion processes, or formed through atmospheric transformation processes from PAHs, are ubiquitous atmospheric pollutants (see chapters 3 and 5).
Since the major route of exposure to nitroPAHs is through inhalation of complex mixtures (e.g., diesel exhaust, polluted urban air), the Task Group thought it appropriate to summarize the effects of diesel exhaust inhalation (for a more thorough treatise, see Scheepers & Bos, 1992b; IPCS, 1996; US EPA, 2000; Lloyd & Cackette, 2001; Sydbom et al., 2001). On the basis of available human and animal evidence, it is concluded that diesel exhaust can cause acute irritation (e.g., eye, throat, bronchial irritation), neurophysiological symptoms (e.g., light-headedness, nausea) and respiratory symptoms (cough and phlegm). There is also evidence for possible immunological effects and exacerbation of allergic responses to known allergens. Chronic animal inhalation studies show a spectrum of dose-dependent chronic inflammation and histopathological changes in the lung in several studies (US EPA, 2000).
Exposure to diesel exhaust by inhalation has the potential to induce cancer in humans and animals. There is considerable evidence demonstrating an association between diesel exhaust exposure and increased lung cancer risk among workers in different occupations. The human evidence, although strong, is not sufficient to allow a definite conclusion that diesel exhaust exposure is associated with lung cancer, due to confounding factors such as smoking and further to the lack of exact diesel exhaust exposure data for workers (HEI, 1995; US EPA, 2000). However, there is extensive evidence for the induction of lung cancer in the rat from long-term inhalation exposure to high concentrations of diesel exhaust and supporting evidence of carcinogenicity from exposure to diesel particulate matter and associated organic compounds in rats and mice by non-inhalation routes of exposure (IPCS, 1996, 1998; US EPA, 2000).
Diesel vapour and diesel-exhaust derived particulate matter extracts are genotoxic to bacterial and mammalian cell systems and can induce adverse chromosomal changes in animals. Elevated levels of DNA adducts have been associated with occupational exposure to diesel exhaust.
Due to the complexity of diesel exhaust, it is likely that some effects are caused by the gaseous components, whereas other effects relate to the particle content. Approximately 50–90% of the number of particles in diesel exhaust are in the ultrafine size range, with the majority of diesel particles ranging in size from 0.005 to 0.05 µm and the mode at about 0.02 µm. These are believed to be aerosol particles formed from exhaust constituents during cooling and to consist of sulfuric acid droplets, ash particles, condensed organic material and maybe carbon spherules. Although ultrafine diesel exhaust-derived particulate matter accounts for the majority of the number of particles, it makes up only 1–20% of the mass of diesel exhaust-derived particulate matter. Between 80% and 95% of the diesel particle mass is in the size range from 0.05 to 1.0 µm, with a mean diameter of about 0.02 µm (US EPA, 2000). These particles have a very large surface area per gram of mass, which make them excellent carriers for adsorbed inorganic and organic compounds. These particles are respirable and penetrate deep into the lungs, carrying these compounds with them (US EPA, 2000).
Recent epidemiological studies have associated mortality and respiratory morbidity with exposure to ambient concentrations of ultrafine particles, raising concern that diesel exhaust could contribute to or be the cause of the observed health effects.
There have been many developments in recent years concerning changes in engines, fuel (e.g., decreasing sulfur content), particle traps, etc., all of which have had an effect on emissions, on particle size and on the relationship between the vapour and particulate phases of organic chemicals (see also chapter 3; CONCAWE, 1998; US EPA, 2000).
Some organic compounds associated with the particles, in particular PAHs and nitroPAHs, are known to show genotoxic properties, and some compounds show carcinogenic properties. It is not certain whether PAHs and their nitro, oxy-alkylated or heterocyclic derivatives or possibly other compounds or the particles themselves are principally responsible for the effects of diesel exposure. Either the effects of gas-phase constituents on the carcinogenic properties of the particles and/or particle-associated organics have not been investigated or the findings have been inconclusive (Scheepers & Bos, 1992b).
As can be expected, as nitroPAHs occur in complex mixtures, there are no reports on effects on humans from individual nitroPAHs. It can be expected that some of the effects reported to be due to exposure to diesel exhaust (see IPCS, 1996) or PAHs (see IPCS, 1998) may be due partly to the nitroPAHs in the complex mixture. Mutagenicity studies on atmospheric samples containing nitroPAHs (see section 7.6.8) suggest that nitroPAHs are responsible for at least part of the total mutagenicity and therefore should be considered of importance in the study of the carcinogenicity of atmospheric pollutants.
There are no case reports specifically on the effects of nitroPAHs.
It is presumed that carcinogens present in human lungs contribute to the incidence of lung cancer. Most of the carcinogens are inhaled with particulate matter via the respiratory tract into the lung alveoli. NitroPAHs have been detected in samples of resected lung tissue from tuberculosis patients, with and without carcinoma, in the period 1991–1996, in Japan (Tokiwa et al., 1993a, 1998a,b; Sera, 1998) (see chapter 5 and Table 27). For 112 non-smoking lung cancer patients from whom lung specimens were collected, 5-year survival rates after the operation were determined on the basis of the nitroarene concentration at the resection time. Lung specimens were divided into higher and lower chemical concentration groups at the levels of 18 pg/g for 1-nitropyrene, 15 pg/g for 1,3-dinitropyrene and 35 pg/g for 3-nitrofluoranthene, and the results were statistically analysed by the Kaplan-Meier method. The hazard ratio significantly increased in the higher chemical concentration group if it was adjusted for age, gender and stage, and it also increased if it was adjusted for cell differentiation in addition to the other factors (Tokiwa et al., 1998a; Tokiwa & Sera, 2000).
Many workplaces have atmospheres with heavy loads of PAHs (see IPCS, 1998). In particular, workers exposed to diesel engine exhaust in the transport industry and in related occupations are exposed to nitroPAHs (section 5.3). 1-Nitropyrene has been used as a marker for exposure to nitroPAHs from diesel exhaust.
Although it is known that humans are exposed to nitroPAHs through their environment — e.g., diesel exhaust and cooking oil fumes — sensitive analytical methods for detection and quantification of nitroPAH adducts with protein and/or DNA or their metabolites in biological fluids are still being developed. First attempts were made with 1-nitropyrene, as this is the most abundant nitroPAH in numerous environmental sources (van Bekkum, 1999; Bos et al., 2000).
Biomonitoring studies in general appear to have a wide inter-individual variation (see below), and there is often a widely overlapping distribution of adduct concentrations in different exposure situations (e.g., occupational versus environmental), so that proving a cause–effect relationship in epidemiological studies is very difficult (Neumann et al., 1995b).
Biomonitoring for nitroPAHs has proved more difficult than expected due to the many possible metabolic pathways and low yield of multiple DNA adducts measured by 32P post-labelling (El-Bayoumy et al., 1994b,c; van Bekkum et al., 1999). Dinitropyrenes (in particular 1,6-dinitropyrene) have, as an alternative to 1-nitropyrene, also been suggested as biomarkers. Although they are present in much lower amounts (only 1% of that of 1-nitropyrene), they are more carcinogenic than 1-nitropyrene, and their DNA adducts are better characterized (Smith et al., 1993, 1995). A further development is the suggestion that T-lymphocyte mutations produced by the 1,6-dinitropyrene–DNA adducts may be more sensitive and longer-lived biomarkers than DNA adducts themselves in assessing previous exposures to nitroPAHs (e.g., from diesel exhaust) (Beland et al., 1994; Beland, 1995; Smith et al., 1995; see chapter 6).
Another approach is to use protein (albumin or Hb) adducts of nitroPAHs as biomarkers of exposure, as suggested by El-Bayoumy et al. (1994a,c). Development of sensitive analytical techniques has enabled the study of nitroPAH–Hb adducts as biomarkers of nitroPAH exposure in rats (van Bekkum et al., 1997) and in human occupational exposure groups: coke oven workers assigned to different job categories (Neumann et al., 1995a,b) and bus garage workers, as well as control groups having urban and rural exposure (Zwirner-Baier & Neumann, 1999). In the human biomarker studies, five nitroPAHs were selected — 1-nitropyrene, 2-nitrofluorene, 3-nitrofluoranthene, 9-nitrophenanthrene and 6-nitrochrysene — and methods were developed to determine the sulfinic acid-type Hb adducts that they form in vivo. (Hydrolysis of the Hb adducts yields the respective arylamines, which were analysed by GC-MS. The detection limit was 0.01–0.08 pmol/g Hb.) In the more recent study (Zwirner-Baier & Neumann, 1999), three exposure groups (high, medium, low) were chosen, assessed from analysis of 1-nitropyrene extracted from total suspended particulate matter in air samples from the chosen locations. Blood samples were analysed from 29 bus garage workers (occupationally exposed to diesel exhaust) and from 20 urban hospital workers and 14 rural council workers as controls. Hb adducts above the detection limit were found in most blood samples. The most abundant adducts were from 1-nitropyrene and 2-nitrofluorene, but there were no differences between the groups, suggesting that both are widespread environmental contaminants (Zwirner-Baier & Neumann, 1999).
A sensitive and selective method of detecting 1-nitropyrene metabolites in urine after diesel exhaust exposure has been investigated in rats, with the aim of developing the method for biomonitoring of human exposure. 1-Nitropyrenols were reduced to 1-aminopyrenols prior to derivatization with heptafluorobutyryl imidazole (van Bekkum et al., 1998; van Bekkum, 1999).
Occupational exposure to diesel exhaust was studied using 1-nitropyrene as a biomarker. Air samples collected at fixed locations in a large trading and distribution centre for mixed cargo contained 1-nitropyrene at concentrations ranging from 270 to 7850 pg/m3 and from 6.4 to 20.6 pg/m3 for indoor and outdoor locations, respectively. The N-acetyl derivatives of 1-aminopyren-6-ol and 1-aminopyren-8-ol were identified in urine, but concentrations were at the limit of detection. Hb and plasma adduct levels ranged from non-detectable to 3380 fg 1-aminopyrene/mg and to 107 fg 1-aminopyrene/mg, respectively, and did not correlate with airborne 1-nitropyrene concentrations (van Bekkum, 1999; Bos et al., 2000).
Biomonitoring of workers occupationally exposed to diesel exhaust was performed to determine their internal burden of diesel-associated aromatic compounds. Personal air sampling also allowed the determination of exposure of the miners at their workplace to several PAHs and nitroarenes. The urine of 18 underground salt miners was collected during and after their shift for 24 h. Nine miners were smokers. The urinary levels of 1-hydroxypyrene and hydroxylated phenanthrene metabolites were determined as biomarkers of PAH exposure, whereas urinary levels of 1-aminopyrene and 3-aminobenzanthrone were chosen to monitor exposure to specific nitroarenes from diesel exhaust, such as 1-nitropyrene and 3-nitrobenzanthrone. It was found that concentrations of 3-aminobenzanthrone (1–143 ng/24 h urine), determined for the first time in this study as a urinary metabolite of diesel exhaust exposure, were similar to 1-aminopyrene concentrations (2–200 ng/24 h urine). The excreted amounts of aromatic amines found as metabolites of the nitroarenes were about 5- to 10-fold higher, as one might expect from the levels determined by personal air sampling at the workplace of the individuals (Seidel et al., 2002).
On the basis of an existing antibody developed against 6-aminobenzo[a]pyrene, an immunochemical assay (ELISA) was developed for the detection of metabolites excreted in urine as a result of occupational exposure to PAHs and nitroPAHs. The method was validated in a study on the occupational exposure of 28 railroad workers (Scheepers et al., 1995b).
For a review of metabolic polymorphisms and susceptibility to cancer, see Vineis et al. (1999).
The C-oxidative metabolism of individual nitroPAHs in different species is catalysed by different cytochrome P450s. In contrast to rat and rabbit (see chapter 6), the CYP3A subfamily (in particular CYP3A3 and CYP3A4) seems to be the enzymes involved in human metabolism of 1-nitropyrene and 4-nitropyrene in HepG2 cells and hepatic microsome samples. A minor role of CYP1A2 has also been suggested (Howard et al., 1990; Silvers et al., 1992; Chae et al., 1999a). None of the P450 enzymes tested (CYP3A4, CYP1A2, CYP2E1, CYP2A6, CYP2D6 and CYP2C9) appeared to be involved in the oxidation of 2-nitropyrene. Nitroreduction, through CYP3A4, was observed only for 4-nitropyrene, not for 1-nitropyrene or 2-nitropyrene (Chae et al., 1999a).
6-Nitrochrysene induced CYP1A1 but not CYP1A2 in human hepatoma HepG2 cells. 6-Nitrochrysene was also able to induce pulmonary CYP1A1 in human lung carcinoma NCI-H322 cells (Chen et al., 2000).
The genotoxicities of four samples of diesel exhaust particle extracts and nine nitroarenes found in diesel exhaust particle extracts were investigated after activation catalysed by human cytochrome P450 family 1 enzymes co-expressed with NADPH-cytochrome P450 reductase (NPR) in Escherichia coli membranes. The diesel exhaust particle extract samples induced umu gene expression in Salmonella typhimurium TA1535/pSK1002 without any P450 system and were further activated by human CYP1B1/NPR membranes. Moderate activation of the diesel exhaust particle extract sample by CYP1A2/NPR membranes, but not by either CYP1A1/NPR or NPR membranes, was also observed. 1-Nitropyrene was strongly activated by human CYP1B1/NPR membranes. 1,8-Dinitropyrene was most highly activated by CYP1A1 and CYP1B1 systems for the three dinitropyrenes tested. In contrast, 1,3-dinitropyrene was inactivated by CYP1A1/NPR, CYP1A2/NPR and CYP1B1/NPR systems and slightly activated by NPR membranes. 2-Nitrofluoranthene and 3-nitrofluoranthene showed activities similar to that of 1-nitropyrene after bioactivation by CYP1B1/NPR membranes. However, the genotoxicities of 6-nitrochrysene, 7-nitrobenz[a]anthracene and 6-nitrobenzo[a]pyrene were all weak in this assay system (Yamazaki et al., 2000).
Both the HPRT assay and the Ames test did not show any involvement of CYP3A in the activation of 1-nitropyrene to a mutagenic metabolite. In addition, a clear involvement of CYP1A2 in the activation of 1-nitropyrene was demonstrated in both mutation assays using eukaryotic cells. However, no activation of 1-nitropyrene was seen in the eukaryotic cell lines when expressing only CYP1A2 or acetyltransferase. No clear involvement of cytochrome P450 could be demonstrated for activation of 2-nitrofluorene to a mutagenic metabolite (Kappers et al., 2000).
Of major importance in human biotransformation is the individual’s capacity to metabolize certain xenobiotics. As a result of interindividual differences in metabolic capacity (polymorphisms), persons are slow, intermediate or rapid metabolizers. Some important enzymes involved in the biotransformation of xenobiotics such as PAHs and nitroPAHs are polymorphic — e.g., some cytochrome P450s (CYP1A1, CYP1A2 and some CYP3A enzymes), NAT2 and GSTµ1.
CYP1A2 is a phase I enzyme involved in the biotransformation of, for example, arylamines to reactive N-hydroxyamines. This enzyme is induced by various environmental factors.
NAT2 activity is genetically determined and polymorphic. Fast and slow acetylators are distinguished based on their ability to metabolize, for example, amines to N-acetyl derivatives. Two major alleles at a single autosomal gene locus are involved in the production of the N-acetyltransferase enzyme. Since CYP1A2 and NAT2 both convert arylamines, both polymorphisms need to be considered when understanding the toxicokinetics of such compounds.
The distribution of GSTµ1 is also polymorphic; approximately 50% of the human population is GSTµ1 deficient, as a result of a homozygous deletion of the GSTµ1 gene (Ketterer et al., 1992). GSTµ is an important subfamily of enzymes largely responsible for conjugation of electrophilic compounds with glutathione. Among the substrates for GSTµ1 are metabolites of arylamines and BaP such as benzo[a]pyrene-4,5-oxide and the ultimate carcinogen benzo[a]pyrene-diolepoxide. Hence, glutathione conjugation can prevent binding of such reactive metabolites with DNA.
In a study on 1-nitropyrene as an exposure marker in a large trading and distribution centre for mixed cargo, blood protein adduct levels were not influenced by NAT2 phenotype, CYP1A2 phenotype or GST phenotype (van Bekkum, 1999).
The Task Group was made aware that a large study is in progress in the European Union, looking at miners and the association of polymorphisms with biomarker development (Scheepers et al., 2002).
The Task Group realized that the biotransformation and DNA damage studies could have been included in earlier chapters (e.g., chapter 4 or 6); however, the Task Group felt that these studies should be combined and considered in this chapter.
Schultz & Moulton (1985) reported an EC50 of 17.3 mg/litre for growth inhibition of the ciliate Tetrahymena pyriformis exposed to 1-nitronaphthalene in a static test system at 28 °C for 60 h. The 95% confidence interval was 14.72–21.26 mg/litre.
A 96-h LC50 value of 9.0 mg/litre was reported for the fathead minnow (Pimephales promelas) exposed to 1-nitronaphthalene in a static renewal test system (Curtis & Ward, 1981). The 95% confidence interval was 5.4–15 mg/litre. The test was carried out using water with a hardness of 30–35 mg calcium carbonate/litre, pH 7.2–7.9 and temperature of 22 ± 1 °C.
Lysak & Marcinek (1972) reported a 24-h LC100 value of 25 mg/litre for rainbow trout (Oncorhynchus mykiss) exposed to 1-nitronaphthalene in a static renewal test system at a temperature ranging from 16 to 21.5 °C. The corresponding 48-h LC0 concentration was 5 mg/litre. Mortality was reported in fish exposed to 7.5–15 mg/litre for 48 h.
9.1.2 Biotransformation studies in aquatic species
Post-mitochondrial supernatants (S9) of marine invertebrates from three phyla — mussel (Mytilus edulis), crab (Carcinus maenas) and starfish (Asteria rubens) — activated 1-nitropyrene to products that were mutagenic in S. typhimurium strain TA98NR (Marsh et al., 1992).
An NADPH-dependent two-electron nitroreductase activity, occurring only under anaerobic conditions, was detected in the microsomal and cytosolic fractions of the major digestive tissues of mussel (Mytilus edulis) (digestive gland) and crab (Carcinus maenas), but not in the gills of either species. 1-Aminopyrene was the only metabolite identified. No activity was detectable in the pyloric caeca or stomach region of the starfish (Asteria rubens). NAD(P)H-dependent one-electron nitroreduction was present in all subcellular fractions of the major digestive tissues of the three species (Hetherington et al., 1996).
1-Nitropyrene (8.3 mg/litre) was added to the water for goldfish (Carassius auratus). After 48 h, 1-aminopyrene, N-acetyl-1-aminopyrene and N-formyl-1-aminopyrene were detected in the water (Kitamura & Tatsumi, 1996), showing that goldfish can metabolize 1-nitropyrene via a nitroreduction pathway (Kitamura & Tatsumi, 1996).
In goldfish (Carassius auratus), 2-nitrofluorene is predominantly metabolized to and excreted as 2-aminofluorene and its acylated metabolites, but not as its hydroxylated metabolites (Ueda et al., 2001b).
1-Nitropyrene (100 µmol/litre) produced concentration-dependent increases in DNA strand breaks (using the "comet" assay) in isolated brown trout (Salmo trutta) hepatocytes incubated in vitro (17.1 ± 4.4 compared with control 3.7 ± 0.6), but no significant effects were found in blood cells (2.8 ± 0.4 compared with control 2.4 ± 0.4) (Mitchelmore & Chipman, 1998).
Isolated mussel (Mytilus edulis L.) digestive gland cells were analysed using the single-cell gel electrophoresis or "comet" assay (Mitchelmore et al., 1998a) to assess the ability of potential aquatic contaminants (e.g., BaP, 1-nitropyrene) to induce DNA strand breaks. There were significant concentration-dependent increases in the percentage of DNA in the comet tail (mean values ± standard deviation) for all doses compared with controls (P < 0.05) for BaP (up to 24.7 ± 5.1 at 100 µmol/litre) and 1-nitropyrene (up to 54.7 ± 5.0% at 200 µmol/litre). There was a decrease (P < 0.05) in viability (eosin Y exclusion) of exposed compared with control cells at 200 µmol/litre with BaP but not with 1-nitropyrene.
In a further study using mussel (Mytilus edulis L.) digestive gland, isolated cells were exposed in vitro to sub-cytotoxic concentrations (50 µmol/litre) of BaP and 1-nitropyrene for 1 h in the dark at 15 °C in the absence or presence of various cytochrome P450 inhibitors, antioxidant enzyme inhibitors, the free radical scavenger N,N-t-butyl-α-phenylnitrone and other modulators. DNA strand breakage was measured using the “comet” assay (Mitchelmore et al., 1998b). BaP-induced strand breakage was indicated to be cytochrome P450 catalysed and to occur via the production of BaP quinones. 1-Nitropyrene-induced strand breakage was indicated to occur via free radical mechanisms(s) (84% strand break inhibition by 50 mmol N,N-t-butyl-α-phenylnitrone/litre) and catalysis by different forms of cytochrome P450 than for BaP (61% strand break inhibition by 50 µmol α-naphthoflavone/litre, but none by clotrimazole at same concentration).
The ability of 1-nitropyrene to form DNA adducts in fish was investigated in vitro and in vivo using brown trout (Salmo trutta) and turbot (Scophthalmus maximus) and compared with that in Wistar rat (Mitchelmore et al., 1998c). Hepatic S9 fractions from brown trout, uninduced and induced with β-naphthoflavone, and from β-naphthoflavone-induced rat were incubated with calf thymus DNA and 1-nitropyrene. With all S9 fractions, the presence of three distinct 1-nitropyrene-related DNA adducts was detected using 32P post-labelling. Turbot, rat and brown trout (uninduced and induced with β-naphthoflavone) were dosed with 1-nitropyrene (i.p.; 100 mg/kg bw). Liver DNA from both turbot and rat exhibited a 1-nitropyrene-related adduct spot in a similar position to that seen in the incubations with S9 from rat and brown trout. The major DNA adducts in fish were consistent with the major 1-nitropyrene DNA adduct (dG-C8-AP), based on co-chromatography. However, in contrast to the in vitro studies, no 1-nitropyrene-related adducts were found in liver DNA from induced and uninduced brown trout, possibly reflecting the influence of detoxification systems.
No information on the effects of nitroPAHs on organisms in the field was identified in the literature.
Compounds in ambient air that have been implicated as being mutagenic/carcinogenic include nitroPAHs and, more recently, the nitro-oxy compounds — nitroketones (in particular 3-nitrobenzanthrone) and nitrolactones (in particular 2-nitrodibenzopyranone and nitropyrene lactones).
The Task Group was aware that changes in diesel fuel, engine technology, exhaust treatment and indoor heating may alter the relative concentration of nitroPAHs on the air particles, and the number and size of the particles may alter the bioavailability, and ultimately the impact, of the nitroPAHs.
There is increasing evidence in recent studies that nitroPAHs, in particular in volatile and semivolatile fractions, are still emitted in diesel exhaust emissions, even after use of catalysts, and their concentrations may in fact be increased by this process.
10.1.1.1 NitroPAHs
NitroPAHs are found in the environment from combustion source emissions or as the result of gas-phase radical-initiated atmospheric formation. NitroPAHs that have been detected in diesel exhaust include primarily 1-nitropyrene, 9-nitroanthracene, 3-nitrofluoranthene, 6-nitrochrysene, 7-nitrobenz[a]anthracene, 2-nitrofluorene and dinitropyrenes.
The highest levels of nitroPAHs in the general environment have been found in urban air. The major contributor to the concentrations of dinitropyrenes and 1-nitropyrene in ambient air is traffic emissions.
Some nitroPAHs, notably 2-nitrofluoranthene and 2-nitropyrene, which are not found in diesel exhaust, have been detected in urban, suburban, forest and remote areas. The ubiquitous occurrence of these nitroPAHs is probably due to their photochemical origin from gas-phase radical-initiated reaction of the parent PAHs and subsequent attachment of the nitroPAHs to carbon particles, which can be widely distributed in the atmosphere.
NitroPAHs that have been detected in ambient air include 1- and 2-nitronaphthalene and methylnitronaphthalenes (predominantly in the vapour phase), 2-nitrofluorene, 9-nitroanthracene, 9-nitrophenanthrene, 2-, 3- and 8-nitrofluoranthene, 1- and 2-nitropyrene, 1,3-, 1,6- and 1,8-dinitropyrene and 6-nitrochrysene.
From worldwide surveys of mononitropyrenes and fluoranthenes from a number of urban, suburban and remote areas, it can be seen that the concentrations of 2-nitrofluoranthene (atmospheric formation) in ambient particulate exceeds several-fold that of 1-nitropyrene (from combustion) in almost all studies.
Seasonal studies on selected nitroPAHs show that the concentrations of 1-nitropyrene and dinitropyrenes (from combustion sources) in ambient air particulate are usually higher in winter than in the other months. In contrast, in most studies, levels of 2-nitrofluoranthene and 2-nitropyrene (atmospheric transformation) are less in winter months than in the warmer seasons. When vapour- and particulate-phase nitroPAHs are monitored, it seems that the semivolatile nitroPAHs are the most predominant nitroPAHs.
It is difficult to give a comprehensive comparison of levels of the different nitroPAHs in rural and urban studies, as most studies have, for various reasons, concentrated on levels of a few selected nitroPAHs. The levels of individual nitroPAHs in ambient air vary considerably, depending on place of measurement, season and time of day. In general, however, levels of total nitroPAHs measured range from picograms to several nanograms per cubic metre.
NitroPAHs have been found in some food samples. Levels do not usually exceed 5 µg/kg, with the exception of spices, smoked food, tea (in particular Mate tea) and peanuts. NitroPAHs in vegetables and fruits are probably due to atmospheric pollution. The average daily intake of nitroPAHs is negligible compared with that for PAHs.
Air concentrations of 1-nitropyrene have been measured in various workplaces associated with the use of diesel engines. The exposure levels of nitroPAHs vary depending on occupation, and the highest levels found have been in underground mining (mean 2.5 ng/m3; maximum 42 ng/m3).
3-Nitrobenzanthrone was detected in diesel exhaust particulate (0.6–6.6 µg/g load) as well as in airborne particle extracts from urban samples (not detected to 12 pg/m3).
2-Nitrodibenzopyranone has been detected in ambient air (0.05–1 ng/m3) at about the same levels as 2-nitrofluoranthene, but at higher levels than 1- and 2-nitropyrene. 2-Nitrodibenzopyranone (0.8 µg/g) was also found in an urban dust sample, but much lower concentrations were found in diesel particulate material (0.2 µg/g), suggesting that nitrodibenzopyranones are formed in the atmosphere. Nitropyrene lactones have also been reported in ambient air.
1) Biomonitoring
Various reports have described the development of methods for and showed data on the evaluation of 1-nitropyrene as a biomarker for occupational exposure to diesel exhaust. 1-Nitropyrene has been measured in particulate matter as a marker for environmental exposure. Urinary metabolites of PAHs and nitroPAHs were determined in urine of diesel mechanics using an immunoassay (ELISA), and, in another study, metabolites of 1-nitropyrene (specifically N-acetyl-1-aminopyren-6-ol and N-acetyl-1-aminopyren-8-ol) were measured in urine of workers in a shipping department. Additional studies have focused on measuring the Hb and plasma adducts of metabolites of 1-nitropyrene and other nitroPAHs and may provide appropriate biomarkers in future molecular epidemiological investigations.
1-Nitropyrene and 2-nitrofluorene administered by various routes are rapidly absorbed and metabolized, and the metabolites are conjugated and excreted. Radiolabelled 1-nitropyrene was found to be widely distributed in the body of rats and mice after all routes of administration. Following intragastric and intraperitoneal administration and following inhalation of 1-nitropyrene or 1-nitropyrene coated on diesel exhaust particles, the majority, 50–60% of the administered dose, has been shown to be excreted in the faeces, whereas urine contained about 15–20% of the dose. In contrast, the major route of excretion of 2-nitrofluorene is the urine.
NitroPAHs constitute a complex group of chemicals showing different metabolic profiles. In mammals, there may be several metabolic pathways for a particular nitroPAH, often depending on the route of administration. Intestinal microflora play an important role in nitroreduction of nitroPAHs and in metabolism, by deconjugating metabolites, thereby enabling enterohepatic circulation. The metabolism of only a few nitroPAHs has been studied.
In vivo studies in mammals (e.g., 1-nitropyrene and 2-nitrofluorene) have shown that the metabolism of nitroPAHs occurs by both oxidative and reductive pathways, leading to several types of DNA adducts. Although the DNA adducts formed via nitroreduction pathways have mostly been identified and correspond to the DNA adducts found in in vitro studies (e.g., in particular the C8-substituted dG adduct), the DNA adducts resulting from oxidative pathways have not been thoroughly identified. There is increasing evidence to suggest that oxidative metabolic pathways are important in the biotransformation and possibly macromolecular adduct formation by nitroPAHs, supported by observations in vitro in human cells and in vivo in rats.
Using the 32P post-labelling assay, 3-nitrobenzanthrone was found to bind covalently to calf thymus DNA after metabolic activation, forming multiple DNA adducts in vitro, all of which are reduction products. Multiple DNA adducts were also detected in cultures of rat lung alveolar type II epithelial cells treated with 3-nitrobenzanthrone.
2-, 3- and 4-nitrodibenzopyranones all formed multiple DNA adducts after incubation with xanthine oxidase and calf thymus DNA under anaerobic conditions. DNA adducts were detected in the liver, but not the lungs, of rats treated with 2-nitrodibenzopyranone. The migration of these adducts was similar to that observed in the in vitro experiment.
The limited data indicate that nitroPAHs have a moderate to low acute toxicity. For example, the oral LD50 for 2-nitrofluorene in mice was 1600 mg/kg bw, whereas gavaging up to 5000 mg 1-nitropyrene/kg bw resulted in no observable toxic effects.
Oral administration of up to 160 mg 1-nitronaphthalene/kg bw via diet revealed no clinical abnormalities in mice and rats. In contrast, lung (and liver) toxicity has been reported after single i.p. injections of 100 mg 1-nitronaphthalene/kg bw and effects in non-ciliated cells in the bronchioles at concentrations as low as 25 mg/kg bw. In a 13-week inhalation study on rats conducted according to current acceptable standards, exposure to 1-nitropyrene resulted in histopathological effects in the upper respiratory tract, leading to a lowest-observed-effect level (LOEL) of 0.5 mg/m3.
There are few data on systemic or local non-neoplastic effects caused by short-term or long-term treatment with nitroPAHs. In most cases, non-neoplastic toxic effects were observed at doses at which carcinogenic responses are also manifested.
No data are available on skin and eye irritation, sensitization or reproductive toxicity.
There were no data available on the non-neoplastic effects of nitroketones and nitrolactones.
1) NitroPAH
Data on genotoxicity in vitro are available on 95 nitroPAHs (see Table 45), but only one or two end-points, mainly in bacterial test systems, were investigated for 74 nitroPAHs. A sufficient database, also including eukaryotic test systems, has been found only with 21 nitroPAHs. Sixty-seven of 95 nitroPAHs tested showed positive results, but these were derived from a small database. Clearly positive results were obtained for 19 nitroPAHs, and questionable results for 8 nitroPAHs. Clearly negative results were not obtained with any of the nitroPAHs.
For 86 nitroPAHs, data on the Salmonella typhimurium microsome test are available. In contrast to the parent PAHs, most nitroPAHs were clearly more effective in the S. typhimurium microsome test without metabolic activation. There are five nitroPAHs that showed exceptionally high mutagenic potency (>100 000 revertants/nmol) in this test system: 3,7- and 3,9-dinitrofluoranthene, 1,6- and 1,8-dinitropyrene and 3,6-dinitrobenzo[a]pyrene (see also nitroketones and nitrolactones below).
Bacterial nitroreductase and acetyltransferase are involved in the metabolic activation of the nitroPAHs, but not all nitroPAHs follow the same metabolic activation pathways. For different nitroPAHs, both frameshift and base pair substitutions have been reported in the S. typhimurium microsome test. There is evidence that nitroPAHs with nitro groups perpendicular to the aromatic ring are not as mutagenic as isomers having parallel nitro orientation.
Some nitroPAHs are extremely mutagenic in bacteria. This led to an earlier conclusion that nitroPAHs are among the most important mutagens in ambient aerosol samples. This sensitivity of S. typhimurium to nitroPAHs is attributed to the presence of native nitroreductase enzymes, which initiate the metabolism of nitroPAHs to their ultimate mutagenic metabolites (arylhydroxylamines). These results in bacteria may be misleading, as nitroPAHs as a group were found to be less mutagenic than PAHs in in vitro studies in human B-lymphoblastoid cells h1A1v2 and MCL-5. The most active nitroPAH tested (1,6-dinitropyrene) had a minimum mutagen concentration ~3-fold higher than that of BaP. However, these results in human cells must also be interpreted with caution, as they may underestimate the toxic potential of nitroPAHs; comparative carcinogenesis studies — e.g., the mouse newborn assay (Table 53) — show some nitroPAHs being more carcinogenic than BaP.
Data on the genotoxicity of nitroPAHs in vivo are available on 15 nitroPAHs. All nitroPAHs that gave positive results in vivo were also positive in vitro (Table 46). Four nitroPAHs that were positive in in vitro genotoxicity tests revealed inconsistent/inconclusive genotoxicity (2-nitronaphthalene, 5-nitroacenaphthene and 3-nitrofluoranthene) or negative genotoxicity (2,7-dinitrofluorene; limited validity) results in vivo.
Most of the tested nitroPAHs were positive in in vivo genotoxicity tests in somatic cells, and the data in Drosophila confirm that these compounds are in vivo somatic mutagens. There were no germ cell assays carried out in rodents, and the single germ cell assay in Drosophila was negative.
2) Nitrobenzanthrones
3-Nitrobenzanthrone, like 1,6- and 1,8-dinitropyrene, is highly mutagenic in bacteria through nitroreduction and O-esterification. 3-Nitrobenzanthrone is also an effective gene mutagen and causes micronuclei formation in human cells in vitro and in mice in vivo.
3) Nitrodibenzopyranones and nitrolactones
2-Nitrodibenzopyranone was reported to be highly mutagenic in the S. typhimurium microsome test in strain TA98 (–S9), being more mutagenic than 2-nitrofluorene and 1-nitropyrene.
1- and 3-nitropyrene lactones have been found to be highly mutagenic in the S. typhimurium microsome test.
Studies on the in vitro genotoxicity of 2-nitrodibenzopyranone in forward mutation assays using two human B-lymphoblastoid cell lines are conflicting. However, nitropyrene lactones were found to induce mutations at the tk and hprt loci in both cell lines. Further, they induced kinetochore-positive and -negative micronuclei in the CREST modified micronucleus assay, which detects chromosomal loss and breakage events.
4) Complex mixtures
Studies on the genotoxicity of individual nitroPAHs are necessary for an understanding of the mechanisms of toxicity, but studies on the mutagenicity of environmental samples, although much more complex, are needed to examine the effect of actual exposure conditions.
Most studies have used the S. typhimurium microsome test, although more recent studies have used Drosophila and human cell lines for mutagenicity testing. Another issue is the possible additivity, antagonism or synergism of combined nitroPAHs in mixtures compared with individual nitroPAHs. There may also be problems with collection and stability of samples and the type of nitroPAH.
5) Diesel engine exhaust
Earlier studies found that in diesel engine exhaust, nitroPAHs accounted for 20–25% of the bacterial mutagenic activity (without further enzymatic activation of the assay, i.e., –S9). Another study found that mono- and dinitroPAHs accounted for 30–40% of bacterial mutagenic activity (–S9) of diesel engine exhaust particles.
In a diesel exhaust particle extract (benzene–ethanol), nitroPAHs were found mostly in fraction 4 (solvent DCM), which contained 61.5% of the total activity. Of this fraction, 53.1% of the activity was attributed to nitroPAHs, with the greatest contribution being from 1-nitropyrene and 1,3-, 1,6- and 1,8-dinitropyrene.
Studies on ambient air showed that 1–8% of the mutagenic activity (–S9) in the S. typhimurium microsome test was due to nitroPAHs. In the benzene–ethanol extract of airborne particulate, the calculated mutagenic contributions of 1-nitropyrene and 1,3-, 1,6- and 1,8-dinitropyrene in the S. typhimurium YG1024 strain were 2.1, 2.5, 5 and 9%, respectively, assuming that the interaction between the compounds is negligible.
The 1-nitropyrene content in diesel exhaust particle samples correlates with the mutagenicity in four S. typhimurium strains.
6) Ambient air
Using a preincubation modification of the S. typhimurium microsome test, the vapour phase contributed substantially to the mutagenic potential of ambient air samples. The ambient mutagenicity concentration in the vapour phase was comparable with that in particulate matter. Nitronaphthalenes were thought to account for about 13% of the mutagenicity in the fraction where the mutagenicity of the vapour phase was highest for these nitroPAHs. About 10% of ambient particulate mutagenicity in the Ames test can be accounted for by nitrofluoranthenes and nitropyrenes, the dominant contributors to ambient particulate organic matter. These nitroPAH isomers were largely formed from the atmospheric gas-phase reactions of the parent PAH, rather than being directly emitted.
2- and 4-nitrodibenzopyranone were found in the most mutagenic HPLC fraction of ambient particle extracts collected from Riverside, California, USA, when tested in a preincubation modification of the S. typhimurium microsome test (TA98 [–S9]). This fraction accounted for ~20% of the total mutagenic activity, with the 2-nitro isomer contributing to the majority of this mutagenicity.
A human cell mutagenicity assay at the tk locus was recently used on samples from various sites in the Los Angeles, California, USA, area collected in 1993. This assay has previously been shown not to be very sensitive to nitroPAHs. 2-Nitrofluoranthene was the only nitroPAH in this study that significantly contributed to the mutagenicity.
1) NitroPAHs
Data on carcinogenic effects are available for 28 nitroPAHs (see Table 52). Although inhalation is the main exposure route in humans, no long-term inhalation study on any nitroPAH is available. Most studies examined the carcinogenic effects of nitroPAHs by oral, topical application, pulmonary implantation and intratracheal administration.
Owing to the limitations in experimental design, none of the negative studies confirmed the absence of carcinogenic effects in animals. However, results showed carcinogenic effects in experimental animals for 5-nitroacenaphthene, 2-nitrofluorene, 3-nitrofluoranthene, 3,7-dinitrofluoranthene, 3,9-dinitrofluoranthene, 1-nitropyrene, 4-nitropyrene, 1,3-dinitropyrene, 1,6-dinitropyrene, 1,8-dinitropyrene and 6-nitrochrysene. Some carcinogenic effects in experimental animals were observed for 2-nitropyrene, 7-nitrobenz[a]anthracene, 2-nitrobenzo[a]pyrene, 6-nitrobenzo[a]pyrene, 3,6-dinitrobenzo[a]pyrene, 7-nitrodibenz[a,h]anthracene and 3-nitroperylene. For the remaining 10 nitroPAHs tested, not enough data were available to evaluate the carcinogenicity in experimental animals.
Besides local effects at the site of injection, nitroPAHs induced mainly systemic tumours in mammary tissue, lung, liver and the haematopoietic system. 6-Nitrochrysene appears to be the most carcinogenic of the nitroPAHs considered here. With systemic effects after s.c. or i.p. injection, 1-nitropyrene was more carcinogenic than the dinitropyrenes. The carcinogenicity of 1-nitropyrene and dinitropyrenes varies, depending on the route of administration.
The substitution of the nitro group on the parent PAH does not alter the carcinogenicity and/or mutagenicity in a consistent manner (i.e., sometimes increases and sometimes decreases the effect). As examples, nitrated benzo[a]pyrenes were found to be generally less potent carcinogens than the parent compound BaP. However, the mono- or dinitrated pyrenes are more carcinogenic than pyrene, and 3-nitroperylene is more carcinogenic than perylene. 6-Nitrochrysene was more carcinogenic than chrysene after i.p. administration, but was less active with respect to local effects after dermal exposure.
2) Nitroketones and nitrolactones
There are no data on the carcinogenicity of these compounds.
Table 58 shows a summary of the exposure and effects of nitroPAHs that are probably of special relevance to health and the environment.
Table 58. Overview on exposure, genotoxicity of nitroPAHs in vitro and in vivo, and carcinogenic effects of selected nitroPAHs
Substance |
Human exposure |
Genotoxicity in vitro |
Genotoxicity in vivo |
Carcinogenicity |
||||
|
Ambient aira |
Diesel exhaust particlesb |
Resultc |
Resultd |
Indicatione |
Number of positive studiesf |
|||
|
Rat |
Mouse |
Hamster |
||||||
|
1-Nitronaphthalene |
+g,h |
+ |
Positive |
Positive |
Database insufficient |
|||
|
2-Nitronaphthalene |
+g,h |
+ |
Positive |
Inconclusive |
Database insufficient |
|||
|
5-Nitroacenaphthene |
Positive |
Inconclusive |
Positive |
2 |
1 |
1 |
||
|
2-Nitrofluorene |
+ |
+ |
Positive |
Positive |
Positive |
2 |
||
|
2,7-Dinitrofluorene |
+ |
+ |
Positive |
(Negative) |
Database insufficient |
|||
|
9-Nitroanthracene |
+ |
+ |
Positive |
Positive |
n.d. |
|||
|
9-Nitrophenanthrene |
+g |
+ |
Positive |
n.d. |
n.d. |
|||
|
2-Nitrofluoranthene |
+h |
Positive |
n.d. |
Database insufficient |
||||
|
3-Nitrofluoranthene |
+ |
+ |
Positive |
Inconclusive |
Positive |
1 |
1 |
|
|
3,7-Dinitrofluoranthene |
+ |
Positive |
Positive |
Positive |
2 |
|||
|
3,9-Dinitrofluoranthene |
+ |
Positive |
Positive |
Positive |
2 |
|||
|
1-Nitropyrene |
+ |
+ |
Positive |
Positive |
Positive |
8 |
1 |
|
|
2-Nitropyrene |
+h |
Positive |
n.d. |
(Positive) |
1 |
|||
|
4-Nitropyrene |
+ |
+ |
Positive |
n.d. |
Positive |
4 |
1 |
|
|
1,3-Dinitropyrene |
+ |
+ |
Positive |
(Positive) |
Positive |
3 |
||
|
1,6-Dinitropyrene |
+ |
+ |
Positive |
Positive |
Positive |
5 |
2 |
1 |
|
1,8-Dinitropyrene |
+ |
+ |
Positive |
Positive |
Positive |
5 |
1 |
|
|
7-Nitrobenz[a]anthracene |
+ |
+ |
Positive |
n.d. |
(Positive) |
1 |
||
|
6-Nitrochrysene |
+ |
+ |
Positive |
Positive |
Positive |
2 |
15 |
|
|
1-Nitrobenzo[a]pyrene |
Positive |
n.d. |
Database insufficient |
|||||
|
2-Nitrobenzo[a]pyrene |
Positive |
n.d. |
(Positive) |
1 |
||||
|
3-Nitrobenzo[a]pyrene |
Positive |
n.d. |
Database insufficient |
|||||
|
6-Nitrobenzo[a]pyrene |
+ |
+ |
Positive |
n.d. |
(Positive) |
1 |
||
|
1-Nitrobenzo[e]pyrene |
Positive |
n.d. |
Database insufficient |
|||||
|
3-Nitrobenzo[e]pyrene |
Positive |
n.d. |
Database insufficient |
|||||
|
1,6-Dinitrobenzo[a]pyrene |
Positive |
n.d. |
Database insufficient |
|||||
|
3,6-Dinitrobenzo[a]pyrene |
Positive |
n.d. |
(Positive) |
2i |
||||
|
7-Nitrodibenz[a,h]anthracene |
Inconclusive |
n.d. |
(Positive) |
1 |
||||
|
9-Nitrodibenz[a,c]anthracene |
Inconclusive |
n.d. |
Database insufficient |
|||||
|
3-Nitroperylene |
+ |
Positive |
n.d. |
(Positive) |
1 |
|||
|
3-Nitrobenzanthrone |
+ |
+ |
Positive |
Positive |
n.d. |
|||
|
2-Nitrodibenzopyranone |
+h |
+ |
Positive |
n.d. |
n.d. |
|||
|
a |
+ = nitroPAHs detected in ambient air. |
|
b |
+ = nitroPAHs detected in diesel exhaust particles. |
|
c |
Normal type: limited database (data on fewer than three end-points available) or inconsistent results; bold type: data on three or more end-points available and majority of end-points positive. |
|
d |
Normal type: limited database (only data on one end-point available) or inconsistent results; bold type: data on two or more end-points available and majority of end-points positive; parentheses: limited validity; n.d. = no data. |
|
e |
(Positive) = only one positive study with study design sufficient for assessment; n.d. = no data. |
|
f |
Number of carcinogenicity studies with positive results and experimental design sufficient for assessment; separated for different species; detailed data presented in Tables 39 and 52. |
|
g |
NitroPAHs predominant in the vapour phase. |
|
h |
NitroPAHs formed by tropospheric transformation. |
|
i |
Probably the same results are presented in two different publications. |
NitroPAHs are either formed in the atmosphere from PAHs or emitted directly into the atmosphere during combustion processes. They are transported in the vapour phase or adsorbed onto particulate matter. Those with liquid-phase vapour pressures greater than 10–4 Pa at ambient air temperature will exist at least partially in the gas phase — i.e., two- to four-ring PAHs and two-ring nitroPAHs.
Owing to their low aqueous solubility or insolubility, nitroPAHs are not expected to be transported in water. Data available give high values for log Koc, suggesting that nitroPAHs, similar to PAHs, adsorb onto soil and sediments. Leaching into groundwater is thought to be negligible. Some nitroPAHs may be slowly biodegradable under certain conditions.
The values for log Kow range from 2.5 for 1-nitronaphthalene to 6.3 for 3-nitroperylene, suggesting a potential for bioaccumulation. There were no data available on biomagnification.
Calculated atmospheric lifetimes of nitroPAHs due to photolysis and gas-phase reactions with hydroxyl and nitrate radicals and with ozone under atmospheric conditions show that the main degradation process for nitroPAHs (e.g., 1- and 2- nitronaphthalene) is photolysis. Particle oxidation of nitroPAHs by ozone may be the main degradation process at night.
Most studies reporting nitroPAH concentrations have focused on air samples. There are a few studies that indicate the presence of nitroPAHs in other environmental media, including water (ng/litre range) and sediment, soil and sewage sludge (µg/kg range).
Organisms living in water, sediment or soil may potentially be exposed to nitroPAHs.
Data on the acute toxicity of nitroPAHs to aquatic organisms are available only for 1-nitronaphthalene. An LC50 (96 h) of 9.0 mg/litre was reported for the fathead minnow (Pimephales promelas). Furthermore, this nitroPAH inhibited the growth of the ciliate Tetrahymena pyriformis, with an EC50 (60 h) of 17.3 mg/litre.
With 1-nitropyrene, DNA adducts were detected in vivo using brown trout (Salmo trutta) and turbot (Scophthalmus maximus) that were comparable to those obtained in Wistar rats.
The identification of all of the mutagenic compounds in urban air has not been achieved.
Most studies have concentrated on measuring concentrations of nitroPAHs on particulates and their levels of mutagenicity. Not enough data are available on the concentrations of nitroPAHs in, and mutagenicity of, the vapour phase.
There are not enough genotoxicity/carcinogenicity data on some nitroPAHs, such as the nitronaphthalenes, methylnitronaphthalenes, 2-nitrofluoranthene, 3-nitrobenzanthrone or nitrolactones.
The mutagenic responses of nitroPAHs in bacterial systems do not necessarily reflect those responses obtained in human cell lines or in vivo.
For certain nitroPAHs, there is increasing evidence for the role of oxidative metabolism in vivo, rather than nitroreductive biotransformation.
There is a lack of data on the biotransformation and genotoxicity (e.g., additivity, antagonism or synergism) of nitroPAHs when included in complex mixtures in which they exist (e.g., diesel and ambient particulates).
In addition, limited data are available on the toxic/genotoxic effects of nitroPAHs in target tissues of humans and animals and in human cell lines.
NitroPAHs, nitroketones and nitrolactones have been detected in ambient air and diesel exhaust.
Organisms living in water, sediment or soil may potentially be exposed to nitroPAHs. Some aquatic organisms are capable of metabolizing nitroPAHs to active intermediates that can damage DNA, and in certain cases nitroPAHs showed acutely toxic effects.
The nitroPAHs, nitroketones and nitrolactones listed in Table 58 are genotoxic. Many of the nitroPAHs are somatic mutagens in rodents and carcinogenic in more than one species. Even in light of a lack of human data, the overwhelming evidence supports a conclusion that the nitroPAHs are probably human carcinogens.
Reduce the overall concentration of PAHs in urban air, since they are a source of atmospheric nitroPAHs.
Reduce the level of nitroPAH emissions in diesel exhaust and other forms of combustion.
Improve the efficiency of exhaust catalysts and filters to remove PAHs and nitroPAHs.
Encourage development and implementation of less-polluting indoor heating sources.
Encourage power source development that does not require fossil fuel combustion.
Encourage improved industrial hygiene (ventilation, engine efficiency, personal protection) concerning fossil fuel engines.
Encourage increased examination by regulatory agencies of the occurrence of nitroPAHs in non-emission sources (e.g., Mate tea).
Improve communication on the risk of nitroPAHs by health agencies to industrial organizations (i.e., risk communication).
The following were identified by the Task Group as areas of basic research that required attention:
Table 59. Summary of previous evaluations of nitroPAHs by IARC
|
NitroPAH |
Evidence for carcinogenicity in experimental animals |
Evaluation in humans |
Overall evaluation |
IARC reference |
|
1-Nitronaphthalene |
Inadequate |
No data |
3 |
1989 |
|
2-Nitronaphthalene |
Inadequate |
No data |
3 |
1989 |
|
5-Nitroacenaphthene |
Sufficient |
No data |
2B |
1978; Suppl. 7 (1987) |
|
2-Nitrofluorene |
Sufficient |
No data |
2B |
1989 |
|
9-Nitroanthracene |
No data |
No data |
3 |
1984; Suppl. 7 (1987) |
|
3-Nitrofluoranthene |
Inadequate |
No data |
3 |
1984; Suppl. 7 (1987) |
|
3,7-Dinitrofluoranthene |
Sufficient |
Inadequate |
2B |
1989; revised 1996 |
|
3,9-Dinitrofluoranthene |
Sufficient |
Inadequate |
2B |
1989; revised 1996 |
|
1-Nitropyrene |
Sufficient |
No data |
2B |
1989 |
|
2-Nitropyrene |
Inadequate |
No data |
3 |
1989 |
|
4-Nitropyrene |
Sufficient |
No data |
2B |
1989 |
|
1,3-Dinitropyrene |
Limited |
No data |
3 |
1989 |
|
1,6-Dinitropyrene |
Sufficient |
No data |
2B |
1989 |
|
1,8-Dinitropyrene |
Sufficient |
No data |
2B |
1989 |
|
6-Nitrochrysene |
Sufficient |
No data |
2B |
1989 |
|
7-Nitrobenz[a]anthracene |
Limited |
No data |
3 |
1989 |
|
6-Nitrobenzopyrene |
Limited |
No data |
3 |
1989 |
|
3-Nitroperylene |
Inadequate |
No data |
3 |
1989 |
|
Diesel exhaust (whole) |
Sufficient |
Limited |
2A |
1989 |
a Group 2A: probably carcinogenic to humans; Group 2B: possibly carcinogenic to humans; Group 3: not classifiable as to its carcinogenicity to humans.
Adachi S, Kawamura K, Takemoto K, Suzuki H & Hisamatsu Y (1999) Carcinogenicity of 3-nitrobenzanthrone, a potent mutagen in diesel exhaust — preliminary results in F344 rats after intratracheal administration. In: Relationships between acute and chronic effects of air pollution. Washington, DC, International Life Sciences Institute.
Al-Bashir B, Hawari J, Leduc R & Samson R (1994) Behavior of nitrogen-substituted naphthalenes in flooded soil: Part 1. Sorption/desorption and biodegradation. Water Res, 28: 1817–1825.
Amacher DE, Paillet SC & Tu GN (1979) Utility of the mouse lymphoma L5178Y/TK assay for the detection of chemical mutagens. In: Hsie AW, O’Neill JP & McElheny VK ed. Mammalian cell mutagenesis. The maturation of test systems. New York, CSH Press, pp. 277–293 (Banbury Report No. 2).
Ames BN, McCann J & Yamasaki E (1975) Methods for detecting carcinogens and mutagens with the Salmonella/mammalian-microsome mutagenicity test. Mutat Res, 31: 347–364.
Anderson JG, McCalla DR, Bryant DW, McCarry BE & Bromke AM (1987) Metabolic activation of 3-nitroperylene in the Salmonella/S9 assay. Mutagenesis, 2: 279–285.
Arey J (1998) Atmospheric reactions of PAHs including formation of nitroarenes. In: Neilson AH ed. The handbook of environmental chemistry, vol. 3, part I, PAHs and related compounds. Berlin, Springer-Verlag, pp. 347–385.
Arey J, Zielinska B, Atkinson R, Winer AM, Ramdahl T & Pitts JN Jr (1986) The formation of nitro-PAH from the gas-phase reactions of fluoranthene and pyrene with the OH radical in the presence of NOx. Atmos Environ, 20: 2339–2345.
Arey J, Zielinska B, Atkinson R & Winer AM (1987) Polycyclic aromatic hydrocarbon and nitroarene concentrations in ambient air during a wintertime high-NO2 episode in the Los Angeles basin. Atmos Environ, 21: 1437–1444.
Arey J, Zielinska B, Atkinson R & Winer AM (1988a) Formation of nitroarenes during ambient high-volume sampling. Environ Sci Technol, 22: 457–462.
Arey J, Zielinska B, Harger WP, Atkinson R & Winer AM (1988b) The contribution of nitrofluoranthenes and nitropyrenes to the mutagenic activity of ambient particulate organic matter collected in Southern California. Mutat Res, 207: 45–51.
Arey J, Zielinska B, Atkinson R & Aschmann SM (1989a) Nitroarene products from the gas-phase reactions of volatile polycyclic aromatic hydrocarbons with the OH radical and N2O5. Int J Chem Kinet, 21: 775–799.
Arey J, Atkinson R, Zielinska B & McElroy PA (1989b) Diurnal concentrations of volatile polycyclic aromatic hydrocarbons and nitroarenes during a photochemical air pollution episode in Glendora, California. Environ Sci Technol, 23: 321–327.
Arey J, Atkinson R, Aschmann SM & Schuetzle D (1990) Experimental investigation of the atmospheric chemistry of 2-methyl-1-nitronaphthalene and a comparison of predicted nitroarene concentrations with ambient air data. Polycycl Aromat Comp, 1: 33–50.
Arey J, Zielinska B, Atkinson R & Winer AM (1991) Analysis of PAH and their nitro-derivatives in ambient air. In: Cooke M, Loening K & Merritt J ed. Polynuclear aromatic hydrocarbons: Measurement, means, and metabolism. 11th International Symposium on Polynuclear Aromatic Hydrocarbons, October 1987. Columbus, Ohio, Battelle Press, pp. 51–67.
Arey J, Harger WP, Helmig D & Atkinson R (1992) Bioassay-directed fractionation of mutagenic PAH atmospheric photooxidation products and ambient particulate extracts. Mutat Res, 281: 67–76.
Arimochi H, Kinouchi T, Kataoka K, Kuwahara T & Ohnishi Y (1998) Activation of 1-nitropyrene by nitroreductase increases the DNA adduct level and mutagenicity. J Med Invest, 44(3–4): 193–198.
Arlett CF (1984) Mutagenicity of 1,8-dinitropyrene in cultured mammalian cells. In: Rickert DE ed. Toxicity of nitroaromatic compounds. Washington, DC, Hemisphere, pp. 185–194.
Arlt V, Bieler C, Mier W, Wiessler M & Schmeiser H (2001) DNA adduct formation by the ubiquitous environmental contaminant 3-nitrobenzanthrone in rats determined by 32P-postlabeling. Int J Cancer, 93(3): 450–454.
Ashby J, Paton D, Wilcox P & Parry JM (1983) Synthesis of 1,6-diaminopyrene from 1,6-dinitropyrene and its S9 dependent mutagenicity to S. typimurium. Carcinogenesis, 4: 787–789.
Atkinson R (1990) Gas-phase tropospheric chemistry of organic compounds: a review. Atmos Environ Part A, 24: 1–42.
Atkinson R (1991) Kinetics and mechanisms of the gas-phase reactions of the NO3 radical with organic compounds. J Phys Chem Ref Data, 20: 449–507.
Atkinson R & Arey J (1994) Atmospheric chemistry of gas-phase polycyclic aromatic hydrocarbons: formation of atmospheric mutagens. Environ Health Perspect, 102(Suppl 4): 117–126.
Atkinson R, Winer AM & Pitts JN Jr (1986) Estimation of night-time N2O5 concentrations from ambient NO2 and NO3 radical concentrations and the role of N2O5 in night-time chemistry. Atmos Environ, 20: 331–339.
Atkinson R, Arey J, Zielinska B, Winer AM & Pitts JN Jr (1987a) The formation of nitropolycyclic hydrocarbons and their contribution to the mutagenicity of ambient air. In: Sandhu SS, DeMarini DM, Mass MJ, Moore MM & Mumford JS ed. Short-term bioassays in the analysis of complex environmental mixtures, V. New York, Plenum Press, pp. 291–309.
Atkinson R, Arey J, Zielinska B & Aschmann SM (1987b) Kinetics and products of the gas-phase reactions of OH radicals and N2O5 with naphthalene and biphenyl. Environ Sci Technol, 21: 1014–1022.
Atkinson R, Arey J, Winer AM, Zielinska B, Dinoff TM, Harger WP & McElroy PA (1988) A survey of ambient concentrations of selected polycyclic aromatic hydrocarbons (PAH) at various locations in California. Riverside, California, University of California, Statewide Air Pollution Research Center, May, 181 pp. (Report A5-185-32).
Atkinson R, Aschmann SM, Arey J, Zielinska B & Schuetzle D (1989) Gas-phase atmospheric chemistry of 1- and 2-nitronaphthalene and 1,4-naphthoquinone. Atmos Environ, 23: 2679–2690.
Atkinson R, Arey J, Zielinska B & Aschmann SM (1990a) Kinetics and nitro-products of the gas-phase OH and NO3 radical-initiated reactions of naphthalene-d8, fluoranthene-d10, and pyrene. Int J Chem Kinet, 22: 999–1014.
Atkinson R, Tuazon EC & Arey J (1990b) Reactions of naphthalene in N2O5–NO3–NO2–air mixtures. Int J Chem Kinet, 22: 1071–1082.
Atkinson R, Baulch DL, Cox RA, Hampson RF Jr, Kerr JA & Troe J (1992) Evaluated kinetic and photochemical data for atmospheric chemistry; supplement IV. J Phys Chem Ref Data, 21: 1125–1556.
Ayres PH, Sun JD & Bond JA (1985) Contribution of intestinal microfloral metabolism to the total macromolecular covalent binding of 1-nitropyrene in the lung and liver of the rat. Toxicology, 36: 263–273.273
Bagley ST, Gratz LD, Leddy DG & Johnson JH (1993) Characterization of particle- and vapor-phase organic fraction emissions from a heavy-duty diesel engine equipped with a particle trap and regeneration controls. Cambridge, Massachusetts, Health Effects Institute, pp. 1–135 (Research Report No. 56).
Bagley ST, Baumgard KJ, Gratz LD, Johnson JH & Leddy DG (1996) Characterization of fuel and aftertreatment device effects on diesel emissions. Prepared by the Department of Biological Sciences, Department of Mechanical Engineering and Engineering Mechanics, and Department of Chemistry, Michigan Technological University, Houghton, Michigan, for the Health Effects Institute, Cambridge, Massachusetts (Research Report No. 76).
Bagley ST, Gratz LD, Johnson JH & McDonald JF (1998) Effects of an oxidation catalytic converter and a biodiesel fuel on the chemical, mutagenic, and particle size characteristics of emissions from a diesel engine. Environ Sci Technol, 32: 1183–1191.
Bai F, Nakanishi Y, Takayama K, Pei XH, Tokiwa H & Hara N (1998) Ki-ras mutation and cell proliferation of lung lesions induced by 1-nitropyrene in A/J mice. Mol Carcinogen, 22: 258–264.
Ball JC & Young WC (1992) Evidence for a new class of mutagens in diesel particulate extracts. Environ Sci Technol, 26: 2181–2186.
Ball LM & King LC (1985) Metabolism, mutagenicity and activation of 1-nitropyrene in vivo and in vitro. Environ Int, 11: 355–362.
Ball LM, Kohan MJ, Inmon JP, Claxton LD & Lewtas J (1984a) Metabolism of 1-nitro[14C]pyrene in vivo in the rat and mutagenicity of urinary metabolites. Carcinogenesis, 5: 1557–1564.
Ball LM, Kohan MJ, Claxton LD & Lewtas J (1984b) Mutagenicity of derivatives and metabolites of 1-nitropyrene: activation by rat liver S9 and bacterial enzymes. Mutat Res, 138: 113–125.
Ball LM, Zacmanidis P & Salmeen IT (1985) The reduction of 1-nitropyrene to 1-aminopyrene does not correlate with the mutagenicity of 1-nitropyrene in V79 Chinese hamster cells. In: Cooke M & Dennis AJ ed. Polynuclear aromatic hydrocarbons: Mechanism, methods and metabolism. Columbus, Ohio, Battelle Press, pp. 113–120.
Ball LM, King LC, Jackson MA & Lewtas J (1986) In vivo metabolism, disposition and macromolecular binding of 1-nitro[14C]pyrene vapor-coated onto diesel particles. In: Cooke M & Dennis AJ ed. Polynuclear aromatic hydrocarbons: Chemistry, characterization and carcinogenesis; Ninth international symposium. Columbus, Ohio, Battelle Press, pp. 53–64.
Ball LM, Rafter JJ, Gustafsson JA, Gustafsson BE, Kohan MJ & Lewtas J (1991) Formation of mutagenic urinary metabolites from 1-nitropyrene in germ-free and conventional rats: role of the gut flora. Carcinogenesis, 12: 1–5.
Ball LM, Rosser-Duncan P & Boucher MN (1994) Oxidative and bacterial pathways in metabolic activation of nitrated polycyclic aromatic hydrocarbons. Polycycl Aromat Comp, 7: 35–42.
Ball LM, Stocking LM, Kohan MJ, Warren SH & Lewtas J (1995) Metabolic activation of the genotoxic environmental contaminants 2- and 3-nitrofluoranthene in variants of Salmonella typhimurium TA98. Mutagenesis, 10: 497–504.
Ball LM, Zhang J, Ramachandran P, Torchal MA & Sangaiah R (1996) Further activation of oxidised metabolites of 1-nitropyrene. Polycycl Aromat Comp, 10: 27–34.
Batiste-Alentorn M, Xamena N, Creus A & Marcos R (1995) Genotoxicity testing of five compounds in three Drosophila short-term somatic assays. Mutat Res, 341: 161–167.
Bauer SL & Howard PC (1990) The kinetics of 1-nitropyrene and 3-nitrofluoranthene metabolism using bovine liver xanthine oxidase. Cancer Lett, 54: 37–42.
Bauer SL & Howard PC (1991) Kinetics and cofactor requirements for the nitroreductive metabolism of 1-nitropyrene and 3-nitrofluoranthene by rabbit liver aldehyde oxidase. Carcinogenesis, 12(9): 1545–1549.
Bayona JM, Casellas M, Fernández P, Solanas AM & Albaigés J (1994) Sources and seasonal variability of mutagenic agents in the Barcelona city aerosol. Chemosphere, 29(3): 441–450.
Becher R, Laag M, Schwarze PE, Brunborg G, Soderlund EJ & Holme JA (1993) Chemically induced DNA damage in isolated rabbit lung cells. Mutat Res, 285: 303–311.
Beije B & Möller L (1988a) 2-Nitrofluorene and related compounds: Prevalence and biological effects. Mutat Res, 196: 177–209.
Beije B & Möller L (1988b) Correlation between induction of unscheduled DNA synthesis in the liver and excretion of mutagenic metabolites in the urine of rats exposed to the carcinogenic air pollutant 2-nitrofluorene. Carcinogenesis, 9: 1465–1470.
Beland FA (1989) DNA binding by 1-nitropyrene and dinitropyrenes in vitro and in vivo: Effects of nitroreductase induction. Prepared by Department of Biochemistry and Molecular Biology, University of Arkansas School for Medical Sciences, Little Rock, Arkansas, for Health Effects Institute, Cambridge, Massachusetts (Research Report No. 31).
Beland FA (1991) Role of ring oxidation in the metabolic activation of 1-nitropyrene. Prepared by Department of Biochemistry and Molecular Biology, University of Arkansas School for Medical Sciences, Little Rock, Arkansas, for Health Effects Institute, Cambridge, Massachusetts (HEI-RR-91/46; NTIS PB92-191295).
Beland FA (1995) DNA adduct formation and T lymphocyte mutation induction in F344 rats implanted with tumorigenic doses of 1,6-dinitropyrene. Prepared by Department of Biochemistry and Molecular Biology, University of Arkansas School for Medical Sciences, Little Rock, Arkansas, for Health Effects Institute, Cambridge, Massachusetts (Research Report No. 72; NTIS PB96-121843).
Beland FA & Marques MM (1994) DNA adducts of nitropolycyclic aromatic hydrocarbons. In: Hemmininki K, Dipple A, Shuker DEG, Kadlubar FF, Segerbäck D & Bartsch H ed. DNA adducts: Identification and biological significance. Lyon, International Agency for Research on Cancer, pp. 229–244.
Beland FA, Ribovich M, Howard PC, Heflich RH, Kurian P & Milo GE (1986) Cytotoxicity, cellular transformation and DNA adducts in normal human diploid fibroblasts exposed to 1-nitrosopyrene, a reduced derivative of the environmental contaminant, 1-nitropyrene. Carcinogenesis, 7: 1279–1283.
Beland FA, Fullerton NF, Smith BA & Heflich RH (1994) Formation of DNA adducts and induction of mutations in rats treated with tumorigenic doses of 1,6-dinitropyrene. Environ Health Perspect, 102: 185–189.
Belisario MA, Pecce R, Della Morte R, Arena AR, Cecinato A, Ciccioli P & Staiano N (1990) Characterization of oxidative and reductive metabolism in vitro of nitrofluoranthenes by rat liver enzymes. Carcinogenesis, 11(2): 213–218.
Belisario MA, Pecce R, Garofalo A, Sannolo N & Malorni A (1996) Erythrocyte enzymes catalyse 1-nitropyrene and 3-nitrofluoranthene nitroreduction. Toxicology, 108: 101–108.
Bell DA, Levine JG & DeMarini DM (1991) DNA sequence analysis of revertants of the hisD3052 allele of Salmonella typhimurium TA98 using the polymerase chain reaction and direct sequencing: Application to 1-nitropyrene-induced revertants. Mutat Res, 252: 35–44.
Benson JM, Brooks AL, Cheng YS, Henderson TR & White JE (1985) Environmental transformation of 1-nitropyrene on glass surfaces. Atmos Environ, 19: 1169–1174.
Bentz JWG, Goschnick J, Schuricht J, Ache HJ, Zehnpfennig J & Benninghoven A (1995) Analysis and classification of individual outdoor aerosol particles with SIMS time-of-flight mass spectrometry. Fresenius J Anal Chem, 353: 603–608.
Berlincioni M, Croce G, Ferri F, Iacovella N, La Rocca C, Lolini M, Megli A, Pupp M, Rizzi L, Turrio Baldassarri L & di Domenico A (1995) Priority organic microcontaminants in selected environmental and food matrices. Fresenius Environ Bull, 4(3): 169–174.
Berry DL, Schoofs GM & Vance WA (1985) Mutagenicity of nitrofluoranthenes, 3-aminofluoranthene and 1-nitropyrene in Chinese hamster V79 cells. Carcinogenesis, 6: 1403–1407.
Bezabeh D, Allen T, McCauley E, Kelly P & Jones A (1997) Negative ion laser desorption ionization time-of-flight mass spectrometry of nitrated polycyclic aromatic hydrocarbons. J Am Soc Mass Spectrom, 8: 630–636.
Bieler CA, Wiessler M, Erdinger L, Suzuki H, Enya T & Schmeiser HH (1999) DNA adduct formation from the mutagenic air pollutant 3-nitrobenzanthrone. Mutat Res, 439(2): 307–311.
Bignami M, Carere A, Conti G, Conti L, Crebelli R & Fabrizi M (1982) Evaluation of 2 different genetic markers for the detection of frameshift and missense mutagens in A. nidulans. Mutat Res, 97: 293–302.
Bodzek D & Janoszka B (1995) Determination of polycyclic aromatic hydrocarbons and their derivatives in sewage sludges in Upper Silesia, Poland. Vom Wasser, 84: 19–33.
Bodzek D, Janoszka B, Dobosz C, Warzecha L & Bodzek M (1997) Determination of polycyclic aromatic compounds and heavy metals in sludges from biological sewage treatment plants. J Chromatogr, 774: 177–192.
Bond JA, Sun JD, Medinsky MA, Jones RK & Yeh HC (1986) Deposition, metabolism, and excretion of 1-(14C)nitropyrene and 1-(14C)nitropyrene coated on diesel exhaust particles as influenced by exposure concentration. Toxicol Appl Pharmacol, 85: 102–117.
Bond JA, Mitchell CE, Mauderley JL & Wolff RKK (1990) Nitropolycyclic aromatic hydrocarbons and diesel exhaust: potential role of DNA binding in carcinogenicity. In: Howard PC, Hecht SS & Beland FA ed. Nitroarenes — Occurrence, metabolism, and biological impact. New York, Plenum Press, pp. 189–199.
Bonfanti L, Careri M, Mangia A, Manini P & Maspero M (1996) Simultaneous identification of different classes of hydrocarbons and determination of nitro-polycyclic aromatic hydrocarbons by means of particle beam liquid chromatography–mass spectrometry. J Chromatogr A, 728: 359–369.
Booth G (1991) Nitro compounds, aromatic. In: Elvers B, Hawkins S & Schulz G ed. Ullmann’s encyclopedia of industrial chemistry. Weinheim, VCH, pp. 411–455.
Borlak J, Hansen T, Yuan Z, Sikka HC, Kumar S, Schmidbauer S, Frank H, Jacob J & Seidel A (2000) Metabolism and DNA-binding of 3-nitrobenzanthrone in primary rat alveolar type II cells, in human fetal bronchial, rat epithelial and mesenchymal cell lines. Polycycl Aromat Comp, 21: 73–86.
Bos RP, van Bekkum YM & Scheepers PTJ (2000) Biomonitoring of diesel exhaust in exposed workers. In: Anderson D, Karakaya AE & Srám RJ ed. Human monitoring after environmental and occupational exposure to chemical and physical agents. Amsterdam, IOS Press, pp. 319–329.
Boyes BG, Rogers CG & Stapley R (1991) Genotoxicity of 1-nitronaphthalene in Chinese hamster V79 cells. Mutat Res, 259: 111–121.
Boyiri T, Desai D, Amin S, Rosa J & El-Bayoumy K (2000) Metabolism and DNA binding of the environmental mammary carcinogen 6-nitrochrysene (6-NC) in rats following oral administration. Proc Am Assoc Cancer Res, 41: 572.
Bryan GT, Brown RR & Price JM (1964) Mouse bladder carcinogenicity of certain tryptophan metabolites and other aromatic nitrogen compounds suspended in cholesterol. Cancer Res, 24: 596–602.
Busby WF, Garner RC, Chow FL, Martin CN, Stevens EK, Newberne PM & Wogan GN (1985) 6-Nitrochrysene is a potent tumorigen in newborn mice. Carcinogenesis, 6: 801–803.(sec 7)
Busby WF, Stevens EK, Martin CN, Chow FL & Garner RC (1989) Comparative lung tumorigenicity of parent and mononitro-polynuclear aromatic hydrocarbons in the BLU:Ha newborn mouse assay. Toxicol Appl Pharmacol, 99: 555–563.
Busby WF, Smith H, Bishop WW & Thilly WG (1994a) Mutagenicity of mono- and dinitropyrenes in the Salmonella typhimurium TM677 forward mutation assay. Mutat Res, 322: 221–232.
Busby WF, Penman BW & Crespi CL (1994b) Human cell mutagenicity of mono- and dinitropyrenes in metabolically competent MCL-5 cells. Mutat Res, 322: 233–242.
Busby WF, Smith H, Crespi CL, Penman BW & Lafleur AL (1997) Mutagenicity of the atmospheric transformation products 2-nitrofluoranthene and 2-nitrodibenzopyranone in Salmonella and human cell forward mutation assays. Mutat Res, 389(2–3): 261–270.
Butterworth BE, Earle LL, Strom D, Jirtle R & Michalopoulos G (1983) Induction of DNA repair in human and rat hepatocytes by 1,6-dinitropyrene. Mutat Res, 122: 73–80.
Campbell RM & Lee ML (1984) Capillary column gas chromatographic determination of nitro polycyclic aromatic compounds in particulate extracts. Anal Chem, 56: 1026–1030.
Casellas M, Fermandez P, Bayona JM & Solanas AM (1995) Bioassay-directed chemical analysis of genotoxic components in urban airborne particulate matter from Barcelona (Spain). Chemosphere, 30: 725–740.
Cassand P, Abdelali H, Bouley C, Denariaz G & Narbonne JF (1994) Inhibitory effect of dairy products on the mutagenicities of chemicals and dietary mutagens. J Dairy Res, 61: 545–552.
Castaneda-Acosta J, Shane BS & Winston GW (1997) Regioselective hydroxylations of 2-nitrofluorene in vivo: a nuclear magnetic resonance study. Chem Res Toxicol, 10(10): 1073–1079.
Castellino S, Lorenzetti R, Ravenna L & de Carneri I (1978) Forward mutation assays using bacteria and 8-azaguanine. Ecotoxicol Environ Saf, 2: 277–299.
Cecinato A, Ciccioli P, Brancaleoni E & Zagari M (1998) PAH and N-PAH in the urban atmosphere of Rome and Milan. Ann Chim, 88: 369–380.
Cecinato A, Marino F, Di Filippo P, Lepore L & Possanzini M (1999) Distribution of n-alkanes, polynuclear aromatic hydrocarbons and nitrated polynuclear aromatic hydrocarbons between the fine and coarse fractions of inhalable atmospheric particulates. J Chromatogr A, 846(1–2): 255–264.
Cecinato A, Mabilia R & Marino F (2000) Relevant organic components in ambient particulate matter collected at Svalbard Islands (Norway). Atmos Environ, 34: 5061–5066.
Cerniglia CE & Somerville CC (1995) Reductive metabolism of nitroaromatic and nitropolycyclic aromatic hydrocarbons. In: Spain JC ed. Biodegradation of nitroaromatic compounds. New York, Plenum Press, pp. 99–115.
Cerniglia CE, Freeman JP, White GL, Heflich RH & Miller DW (1985) Fungal metabolism and detoxification of the nitropolycyclic aromatic hydrocarbon 1-nitropyrene. Appl Environ Microbiol, 50: 649–655.
Chae YH, Yun C-H, Guengerich FP, Kadlubar FF & El-Bayoumy K (1993) Roles of human hepatic and pulmonary cytochrome P450 enzymes in the metabolism of the environmental carcinogen 6-nitrochrysene. Cancer Res, 53: 2028–2034.
Chae YH, Delclos KB, Blaydes B & El-Bayoumy K (1996) Metabolism and DNA binding of the environmental colon carcinogen 6-nitrochrysene in rats. Cancer Res, 56: 2052–2058.
Chae YH, Upadhyaya P, Ji BY, Fu PP & El-Bayoumy K (1997) Comparative metabolism and DNA binding of 1-, 2-, and 4-nitropyrene in rats. Mutat Res, 376: 21–28.
Chae YH, Thomas T, Guengerich FP, Fu PP & El-Bayoumi K (1999a) Comparative metabolism of 1-, 2-, and 4-nitropyrene by human hepatic and pulmonary microsomes. Cancer Res, 59(7): 1473–1480.
Chae YH, Ji BY, Lin JM, Fu PP, Cho BP & El-Bayoumy K (1999b) Nitroreduction of 4-nitropyrene is primarily responsible for DNA adduct formation in the mammary gland of female CD rats. Chem Res Toxicol, 12(2): 180–186...
Chapman OL, Heckert DC, Reasoner JW & Thackaberry SP (1966) Photochemical studies on 9-nitroanthracene. J Am Chem Soc, 88: 5550–5554.
Chen R, Chou M & Ueng T (2000) Induction of cytochrome P450 1A1 in human hepatoma HepG2 cells by 6-nitrochrysene. Toxicol Lett, 117(1–2): 69–77.
Chen SC & Chung KT (2000) Mutagenicity and antimutagenicity studies of tannic acid and its related compounds. Food Chem Toxicol, 38: 1–5.
Chiu C & Miles W (1996) An improved method for nitro-PAH analysis. Polycycl Aromat Comp, 9: 307–314.
Chou MW, Heflich RH, Casciano DA, Miller DW, Freeman JP, Evans FE & Fu PP (1984) Synthesis, spectral analysis, and mutagenicity of 1-, 3- and 6-nitrobenzo[a]pyrene. J Med Chem, 27: 1156–1161.
Chou MW, Heflich RH & Fu PP (1985) Multiple metabolic pathways for the mutagenic activation of 3-nitrobenzo[a]pyrene. Carcinogenesis, 6: 1235–1238.
Chou MW, Heflich RH & Fu PP (1986) Metabolism of 1-nitrobenzo[a]pyrene by rat liver microsomes to potent mutagenic metabolites. Carcinogenesis, 7: 1837–1844.
Chow J (2001) Diesel engines: environmental impact and control. J Air Waste Manage Assoc, 51(9): 1258–1270.
Ciccioli P, Cecinato A, Brancaleoni E & Liberti A (1988) Short capillary column for the separation and identification of mononitrofluoranthene and mononitropyrene isomers present in air and emission samples. J High Resol Chromatogr, 11: 306–312.
Ciccioli P, Cecinato A, Brancaleoni E, Draisci R & Liberti A (1989) Evaluation of nitrated polycyclic aromatic hydrocarbons in anthropogenic emission and air samples. Aerosol Sci Technol, 10: 296–310.
Ciccioli P, Cecinato A, Cabella R, Brancaleoni E & Buttini P (1993) The contribution of gas-phase reactions to the nitroarene fraction of molecular weight 247 present in carbon particles sampled in an urban area of northern Italy. Atmos Environ Part A, 27: 1261–1270.
Ciccioli P, Cecinato A, Brancaleoni E, Frattoni M, Zacchei P & de Castro Vasconcellos P (1995) The ubiquitous occurrence of nitro-PAH of photochemical origin in airborne particles. Ann Chim, 85: 455–469.
Ciccioli P, Cecinato A, Brancaleoni E, Frattoni M, Zacchei P, Miguel AH & de Castro Vasconcellos P (1996) Formation and transport of 2-nitrofluoranthene and 2-nitropyrene of photochemical origin in the troposphere. J Geophys Res, 101: 19567–19581.
Claxton L, Douglas G, Krewski D, Lewtas J, Matsushita H & Rosenkranz H (1992) Overview, conclusions, and recommendations of the IPCS collaborative study on complex mixtures. Mutat Res, 276: 61–80.
Cole J, Arlett CF, Lowe J & Bridges BA (1982) The mutagenic potency of 1,8-dinitropyrene in cultured mouse lymphoma cells. Mutat Res, 93: 213–220.
Collins JF, Brown JP, Alexeeff GV & Salmon AG (1998) Potency equivalency factors for some polycyclic aromatic hydrocarbons and polycyclic aromatic hydrocarbon derivatives. Regul Toxicol Pharmacol, 28: 45–54.
Compadre RLL, Debnath AK, Shusterman AJ & Hansch C (1990) LUMO energies and hydrophobicity as determinants of mutagenicity by nitroaromatic compounds in Salmonella typhimurium. Environ Mol Mutagen, 15: 44–55.
CONCAWE (1998) Polycyclic aromatic hydrocarbons in automotive exhaust emissions and fuels. Brussels, CONCAWE (98/55).
Consolo MC, Anders M & Howard PC (1989) Mutagenicity of the phenolic microsomal metabolites of 3-nitrofluoranthene and 1-nitropyrene in strains of Salmonella typhimurium. Mutat Res, 210: 263–269.
Conzelman GM, Moulton JE & Flanders LE (1970) Tumors in the urinary bladder of a monkey: induction with 2-nitronaphthalene. Gann (Jpn J Cancer Res), 61: 79–80.
Crebelli R, Conti L, Crochi B, Carere A, Bertoli C & Del-Giacomo N (1995) The effect of fuel composition on the mutagenicity of diesel engine exhaust. Mutat Res, 346: 167–172.
Cui XS, Torndal UB, Eriksson LC & Möller L (1995) Early formation of DNA adducts compared with tumor formation in a long-term tumor study in rats after administration of 2-nitrofluorene. Carcinogenesis, 16: 2135–2141.
Cui X-S, Bergman J & Möller L (1996) Preneoplastic lesions, DNA adduct formation and mutagenicity of 5-, 7- and 9-hydroxy-2-nitrofluorene, metabolites of the air pollutant 2-nitrofluorene. Mutat Res, 369: 147–155.
Cui XS, Eriksson LC & Möller L (1999) Formation and persistence of DNA adducts during and after a long-term administration of 2-nitrofluorene. Mutat Res, 442: 9–18.
Curtis MW & Ward CH (1981) Aquatic toxicity of forty industrial chemicals: Testing in support of hazardous substance spill prevention regulation. J Hydrol, 51: 359–367.
Dafflon O, Scheurer L, Koch H & Bosset JO (2000) Quantitation of nitrated polycyclic aromatic hydrocarbons in fish, meat products and cheese using liquid chromatography. Mitt Geb Lebensmittelunters Hyg, 91(2): 158–171.
Danford N, Wilcox N & Parry JM (1982) The clastogenic activity of dinitropyrenes in a rat-liver epithelial cell line. Mutat Res, 105: 349–355.
Danford N, Hogan L & Parry JM (1983) A modified hypotonic treatment to measure mitotic aneuploidy in a Chinese hamster primary cell line. Environ Mutagen, 5: 948.
Dass S, Bucci T, Heflich R & Casciano D (1999) Evaluation of the transgenic p53+/– mouse for detecting genotoxic liver carcinogens in a short-term bioassay. Cancer Lett, 143(1): 81–85.
De Flora S (1979) Metabolic activation and deactivation of mutagens and carcinogens. Ital J Biochem, 28: 81–103.
De Flora S, Zanacchi P, Camoirano A, Bennicelli C & Badolati C (1984) Genotoxic activity and potency of 135 compounds in the Ames reversion test and in a bacterial DNA-repair test. Mutat Res, 133: 161–198.
De Flora S, Bagnasco M & Vainio H (1999) Modulation of genotoxic and related effects by carotenoids and vitamin A in experimental models: mechanistic issues. Mutagenesis, 14(2): 153–172.
Delclos KB & Heflich RH (1992) Mutation induction and DNA adduct formation in Chinese hamster ovary cells treated with 6-nitrochrysene, 6-aminochrysene and their metabolites. Mutat Res, 279: 153–164.
Delclos KB, Walker RP, Dooley KL, Fu PP & Kadlubar FF (1987a) Carcinogen–DNA adduct formation in the lungs and livers of preweanling CD-1 male mice following administration of [3H]-6-nitrochrysene, [3H]-6-aminochrysene, and [3H]-1,6-dinitropyrene. Cancer Res, 47: 6272–6277.
Delclos KB, Miller DW, Lay JO Jr, Cascino DA, Walker RP, Fu PP & Kadlubar FF (1987b) Identification of C8-modified deoxyinosine and N2- and C8-modified deoxyguanosine as major products of the in vitro reaction of N-hydroxy-6-aminochrysene and 6-aminochrysene with DNA and the formation of these adducts in isolated rat hepatocytes treated with 6-nitrochrysene and 6-aminochrysene. Carcinogenesis, 8: 1703–1709.
Delclos KB, El-Bayoumy K, Hecht SS, Walker RP & Kadlubar FF (1988) Metabolism of the carcinogen [3H]6-nitrochrysene in the preweanling mouse: identification of 6-aminochrysene-1,2-dihydrodiol as the probable proximate carcinogenic metabolite. Carcinogenesis, 9(10): 1875–1884.
Delgado-Rodriguez A, Ortiz-Marttelo R, Graf U, Villalobos-Pietrini R & Gomez-Arroyo S (1995) Genotoxic activity of environmentally important polycyclic aromatic hydrocarbons and their nitro derivatives in the wing spot test of Drosophila melanogaster. Mutat Res, 341(4): 235–247.
Delgado-Rodriguez A, Ortiz-Marttelo R, Villalobos-Pietrini R, Gomez-Arroyo S & Graf U (1999) Genotoxicity of organic extracts of airborne particles in somatic cells of Drosophila melanogaster. Chemosphere, 39(1): 33–43.
DeMarini DM, Shelton ML & Bell DA (1996) Mutation spectra of chemical fractions of a complex mixture: role of nitroarenes in the mutagenic specificity of municipal waste incinerator emissions. Mutat Res, 349: 1–20.
Denda A, Tsutsumi M, Tsujiuchi T, Eimoto H, Konishi Y & Sato S (1989) Induction of rat liver γ-glutamyltranspeptidase-positive foci by oral administration of 1-nitropyrene. Cancer Lett, 45: 21–26.
Dennis MJ, Massey RC, McWeeny DJ & Knowles ME (1984) Estimation of nitropolycyclic aromatic hydrocarbons in foods. Food Addit Contam, 1: 29–37.
de Raat WK, Boers JP, Bakker GL, de Meijere FA, Hooimeijer A, Lohman PHM & Mohn GR (1994) Contribution of PAH and some of their nitrated derivatives to the mutagenicity of ambient airborne particles and coal fly ash. Sci Total Environ, 153: 7–28.
Dietrich AM, Guenat CR, Tomer KB & Ball LM (1988) Identification and characterization of the major DNA adduct formed chemically and in vitro from the environmental genotoxin 3-nitrofluoranthene. Carcinogenesis, 9: 2113–2119.
Dimashki M, Smith DJT & Harrison RM (1996) Urban levels of polycyclic aromatic hydrocarbons and nitro-PAH in atmospheric particles sampled from Birmingham, UK and Damascus, Syria. Polycycl Aromat Comp, 9: 201–208.
Dimashki M, Harrad S & Harrison RM (2000) Measurements of nitro-PAH in the atmospheres of two cities. Atmos Environ, 34: 2459–2469.
DiPaolo JA, DeMarinis AJ, Chow FL, Garner RC, Martin CN & Doniger J (1983) Nitration of carcinogenic and non-carcinogenic polycyclic aromatic hydrocarbons results in products able to induce transformation of Syrian hamster cells. Carcinogenesis, 3: 357–359.
Djuric Z, Fifer EK & Beland FA (1985) Acetyl coenzyme A-dependent binding of carcinogenic and mutagenic dinitropyrenes to DNA. Carcinogenesis, 6: 941–944.
Djuric Z, Potter DW, Heflich RH & Beland FA (1986a) Aerobic and anaerobic reduction of nitrated pyrenes in vitro. Chem-Biol Interact, 59: 309–324.
Djuric Z, Fifer E, Howard PC & Beland FA (1986b) Oxidative microsomal metabolism of 1-nitropyrene to DNA-binding of oxidized metabolites following nitroreduction. Carcinogenesis, 7: 1073–1079.
Djuric Z, Fifer EK, Yamazoe Y & Beland FA (1988) DNA binding by 1-nitropyrene and 1,6-dinitropyrene in vitro and in vivo: effects of nitroreductase induction. Carcinogenesis, 9(3): 357–364.
Doolittle DJ & Butterworth BE (1984) Assessment of chemically-induced DNA repair in rat tracheal epithelial cells. Carcinogenesis, 5: 773–779.
Doolittle DJ, Furlong JW & Butterworth BE (1985) Assessment of chemically-induced DNA repair in primary cultures of human bronchial epithelial cells. Toxicol Appl Pharmacol, 79: 28–38.
Dotter RN, Smith CH, Young MK, Ke PB, Jones AD, McCauley EM & Chang DP (1996) Laser desorption/ionization time-of-flight mass spectrometry of nitrated polycyclic aromatic hydrocarbons. Anal Chem, 68: 2319–2324.
Doudney CO, Franke MA & Rinaldi CN (1981) The DNA damage activity (DDA) assay and its application to river waters and diesel exhausts. Environ Int, 5: 293–297.
Draper W (1986) Quantitation of nitro- and dinitropolycyclic aromatic hydrocarbons in diesel exhaust particulate matter. Chemosphere, 15: 437–447.
Dunkel VC, Zeiger E, Brusiek D, McCoy E, McGregor D, Mortelmans K, Rosenkranz HS & Simmon VF (1985) Reproducibility of microbial mutagenicity assays: II. Testing of carcinogens and noncarcinogens in Salmonella typhimurium and Escherichia coli. Environ Mutagen, 7: 1–248.
Durant JL, Busby WF, Lafleur AL, Penman BW & Crespi CL (1996) Human cell mutagenicity of oxygenated, nitrated and unsubstituted polycyclic aromatic hydrocarbons associated with urban aerosols. Mutat Res, 371: 123–157.
Durant JL, Lafleur AL, Plummer EF, Taghizadeh K, Busby WF & Thilly WG (1998) Human lymphoblast mutagens in urban airborne particles. Environ Sci Technol, 32: 1894–1906.(sec 7)
Dutcher JS & Sun JD (1983) Metabolism and excretion of 1-nitropyrene in Fischer-344 rats. Fed Proc, 42: 914.
Dutcher JS, Sun JD, Bechtold WE & Unkefer CJ (1985) Excretion and metabolism of 1-nitropyrene in rats after oral or intraperitoneal administration. Fundam Appl Toxicol, 5: 287–296.
Eddy EP, Howard PC & Rosenkranz HS (1985) Metabolism, DNA repair and mutagenicity of 1-nitropyrene in the human hepatoma cell line HepG2. Proc Am Assoc Cancer Res, 26: 96.(sec 7)
Eddy EP, McCoy EC, Rosenkranz HS & Mermelstein R (1986) Dichotomy in the mutagenicity and genotoxicity of nitropyrenes: Apparent effect of the number of electrons involved in nitroreduction. Mutat Res, 161: 109–111.
Eddy EP, Howard PC, McCoy EC & Rosenkranz HS (1987) Mutagenicity, unscheduled DNA synthesis, and metabolism of 1-nitropyrene in the human hepatoma cell line HepG2. Cancer Res, 47: 3163–3168.
Edenharder R & Tang X (1997) Inhibition of the mutagenicity of 2-nitrofluorene, 3-nitrofluoranthene and 1-nitropyrene by flavonoids, coumarins, quinones and other phenolic compounds. Food Chem Toxicol, 35: 357–372.
Edgar DH (1985) The mutagenic potency of 4 agents at the thymidine kinase locus in mouse lymphoma L5178Y cells in vitro: Effects of exposure time. Mutat Res, 157: 199–204.
Edgar DH & Brooker PC (1985) Induction of 6-thioguanine resistance, chromosome aberrations and SCE by dinitropyrenes in Chinese hamster ovary cells in vitro. Mutat Res, 158: 209–215.
Edwards MJ, Batmanghelich S, Edwards S, Parry JM & Smith K (1986a) The induction of DNA adducts in mammalian cells exposed to 1-nitropyrene and its nitro-reduced derivatives. Mutagenesis, 1: 347–352.
Edwards MJ, Parry JM, Batmanghelich S & Smith K (1986b) Toxicity and DNA damage induced by 1-nitropyrene and its derivation in Chinese hamster lung fibroblasts. Mutat Res, 163: 81–89.
Eide I & Johnsen HG (1998) Mixture design and multivariate analysis in mixture research. Environ Health Perspect, 106(Suppl 6): 1373–1376.
El-Bayoumy K (1992) Environmental carcinogens that may be involved in human breast cancer etiology. Chem Res Toxicol, 5: 585–590.
El-Bayoumy K & Hecht SS (1983) Identification and mutagenicity of metabolites of 1-nitropyrene formed by rat liver. Cancer Res, 43: 3132–3137.
El-Bayoumy K & Hecht SS (1984a) Metabolism of 1-nitro[U-4,5,9,10-14C]pyrene in the F344 rat. Cancer Res, 44: 4317–4322.
El-Bayoumy K & Hecht SS (1984b) Identification of trans-1,2-dihydro-1,2-dihydroxy-6-nitrochrysene as a major mutagenic metabolite of 6-nitrochrysene. Cancer Res, 44: 3408–3413.
El-Bayoumy K & Hecht SS (1986) Mutagenicity of K-region derivates of 1-nitropyrene; remarkable activity of 1- and 3-nitro-5H-phenanthro[4,5-bcd]pyran-5-one. Mutat Res, 170: 31–40.
El-Bayoumy K, Lavoie EJ, Hecht SS, Fow EA & Hoffmann D (1981) The influence of methyl substitution on the mutagenicity of nitronaphthalenes and nitrobiphenyls. Mutat Res, 81: 143–153.
El-Bayoumy K, Hecht SS & Hoffmann D (1982) Comparative tumor initiating activity on mouse skin of 6-nitrobenzo[a]pyrene, 6-nitrochrysene, 3-nitroperylene, 1-nitropyrene and their parent hydrocarbons. Cancer Lett, 16: 333–337.
El-Bayoumy K, Sharma C, Louis YM, Reddy B & Hecht SS (1983) The role of intestinal microflora in the metabolic reduction of 1-nitropyrene to 1-aminopyrene in conventional and germfree rats and in humans. Cancer Lett, 19: 311–316.
El-Bayoumy K, Reddy B & Hecht SS (1984a) Identification of ring oxidized metabolites of 1-nitropyrene in the feces and urine of germfree F344 rats [short communication]. Carcinogenesis, 5: 1371–1373.
El-Bayoumy K, Hecht SS, Sackl T & Stoner GD (1984b) Tumorigenicity and metabolism of 1-nitropyrene in A/J mice. Carcinogenesis, 5: 1449–1452.
El-Bayoumy K, O’Donnel M, Hecht SS & Hoffmann D (1985) On the analysis of 1-nitronaphthalene, 1-nitropyrene and 6-nitrochrysene in cigarette smoke. Carcinogenesis, 6: 505–507.
El-Bayoumy K, Shiue GH & Hecht SS (1988a) Metabolism and DNA binding of 1-nitropyrene and 1-nitrosopyrene in newborn mice. Chem Res Toxicol, 1: 243–247.
El-Bayoumy K, Rivenson A, Johnson B, DiBello J, Little P & Hecht SS (1988b) Comparative tumorigenicity of 1-nitropyrene, 1-nitrosopyrene, and 1-aminopyrene administered by gavage to Sprague-Dawley rats. Cancer Res, 48: 4256–4260.
El-Bayoumy K, Shiue G-H & Hecht SS (1989a) Comparative tumorigenicity of 6-nitrochrysene and its metabolites in newborn mice. Carcinogenesis, 10: 369–372.
El-Bayoumy K, Delclos KB, Heflich RH, Walker R, Shiue G-H & Hecht SS (1989b) Mutagenicity, metabolism and DNA adduct formation of 6-nitrochrysene in Salmonella typhimurium. Mutagenesis, 4: 235–240.
El-Bayoumy K, Rivenson A, Upadhyaya P, Chae YH & Hecht SS (1993) Induction of mammary cancer by 6-nitrochrysene in female CD rats. Cancer Res, 53: 3719–3722.
El-Bayoumy K, Johnson B, Roy AK, Upadhyaya P, Partian S & Hecht SS (1994a) Development of methods to monitor exposure to 1-nitropyrene. Environ Health Perspect, 102(Suppl 6): 31–37.
El-Bayoumy K, Johnson B, Partian S, Upadhyaya P & Hecht SS (1994b) In vivo binding of 1-nitropyrene to albumin in the rat. Carcinogenesis, 15: 119–123.
El-Bayoumy K, Johnson BE, Roy AK, Upadhyaya P & Partian SJ (1994c) Biomonitoring of nitropolynuclear aromatic hydrocarbons via protein and DNA adducts. Cambridge, Massachusetts, Health Effects Institute, April, pp. 1–39 (Research Report No. 64).
El-Bayoumy K, Chae Y-H, Upadhyaya P, Rivenson A, Kurtzke C, Reddy B & Hecht SS (1995) Comparative tumorigenicity of benzo[a]pyrene, 1-nitropyrene and 2-amino-1-methyl-6-phenylimidazo-[4,5-b]pyridine administered by gavage to female CD rats. Carcinogenesis, 16: 431–434.
Ember I, Pusztai Z, Gyongyi Z & Kiss I (2000) 1-Nitropyrene induces elevated expression of oncogenes and tumor suppressor genes 24 hours after treatment in CBA/Ca mice. Anticancer Res, 20(3A): 1563–1566.
Ensell M-X, Whong W-Z, Heng Z-C, Nath J & Ong T (1998) In vitro and in vivo transformation in rat tracheal epithelial cells exposed to diesel emission particles and related compounds. Mutat Res, 412: 283–291.
Ensell M, Hubbs A, Zhou G, Battelli L, Nath J & Ong T (1999) Neoplastic potential of rat tracheal epithelial cell lines induced by 1-nitropyrene and dibenzo(a,i)pyrene. Mutat Res, 444(1): 193–199.
Enya T, Suzuki H, Watanabe T, Hisamatsu Y & Hisamatsu Y (1997) 3-Nitrobenzanthrone, a powerful bacterial mutagen and suspected human carcinogen found in diesel exhaust and airborne particulates. Environ Sci Technol, 31: 2772–2776.
Enya T, Kawanishi M, Suzuki H, Matsui S & Hisamatsu Y (1998) An unusual DNA adduct derived from the powerfully mutagenic environmental contaminant 3-nitrobenzanthrone. Chem Res Toxicol, 11(12): 1460–1467.
Espinosa-Aguirre JJ, Reyes RE, Rubio J, Ostrosky-Wegman P & Martinez G (1993) Mutagenic activity of urban air samples and its modulation by chili extracts. Mutat Res, 303: 55–61.
Fan Z, Chen D, Birla P & Kamens RM (1995) Modeling of nitro-polycyclic aromatic hydrocarbon formation and decay in the atmosphere. Atmos Environ, 29: 1171–1181.
Fan Z, Kamens RM, Hu J, Zhang J & McDow S (1996a) Photostability of nitro-polycyclic aromatic hydrocarbons on combustion soot particles in sunlight. Environ Sci Technol, 30(4): 1358–1364.
Fan Z, Kamens RM, Zhang J & Hu J (1996b) Ozone–nitrogen dioxide–NPAH heterogeneous soot particle reactions and modeling NPAH in the atmosphere. Environ Sci Technol, 30(9): 2821–2827.
Feilberg A & Nielsen T (2000) Effect of aerosol chemical composition on the photodegradation of nitro-polycyclic aromatic hydrocarbons. Environ Sci Technol, 34: 789–797..
Feilberg A & Nielsen T (2001) Photodegradation of nitro-PAHs in viscous organic media used as models of organic aerosols. Environ Sci Technol, 35: 108–113..
Feilberg A, Kamens RM, Strommen MR & Nielsen T (1999) Modeling the formation, decay, and partitioning of semivolatile nitro-polycyclic aromatic hydrocarbons (nitronaphthalenes) in the atmosphere. Atmos Environ, 33(8): 1231–1243.
Feilberg A, Poulsen MB, Nielsen T & Skow H (2001) Occurrence and sources of particulate nitro-polycyclic aromatic hydrocarbons in ambient air in Denmark. Atmos Environ, 35: 353–366.
Fernández P & Bayona JM (1992) Use of off-line gel permeation chromatography–normal-phase liquid chromatography for the determination of polycyclic aromatic compounds in environmental samples and standard reference materials (air particulate matter and marine sediment). J Chromatogr, 625: 141–149.
Fernández P, Grifoll M, Solanas AM, Bayona JM & Albaigés J (1992) Bioassay-directed chemical analysis of genotoxic components in coastal sediments. Environ Sci Technol, 26: 817–829.
Fifer EK, Heflich RH, Djuric Z, Howard PC & Beland FA (1986a) Synthesis and mutagenicity of 1-nitro-6-nitrosopyrene and 1-nitro-8-nitrosopyrene, potential intermediates in the metabolic activation of 1,6- and 1,8-dinitropyrene. Carcinogenesis, 7: 65–70.
Fifer EK, Howard PC, Heflich RH & Beland FA (1986b) Synthesis and mutagenicity of 1-nitropyrene 4,5-oxide and 1-nitropyrene 9,10-oxide, microsomal metabolites of 1-nitropyrene. Mutagenesis, 1: 433–438.
Fretwurst S & Ahlf W (1996) Modifikation des umu-Testes zum Nachweis gentoxischer und cytotoxischer Wirkungen von feststoffassoziierten Umweltchemikalien. [A modified umu-test for the detection of genotoxic and cytotoxic impacts of solid materials associated contaminants.] Vom Wasser, 86: 353–361.
Fu PP (1990) Metabolism of nitro-polycyclic aromatic hydrocarbons. Drug Metab Rev, 22: 209–268.
Fu PP & Herreno-Saenz D (1999) Nitro-polycyclic aromatic hydrocarbons: a class of genotoxic environmental pollutants. Environ Carcinogen Ecotoxicol Rev, 17(1): 1–43.
Fu PP, Chou MW, Yang SK, Beland FA, Kadlubar FF, Casciano DA, He RH & Evans FE (1982) Metabolism of the mutagenic environmental pollutant, 6-nitrobenzo[a]pyrene: Metabolic activation via ring oxidation. Biochem Biophys Res Commun, 105: 1037–1043.
Fu PP, von Tungeln LS & Chou MW (1985a) Metabolism of 9-nitroanthracene by rat liver microsomes: Identification and mutagenicity of metabolites. Carcinogenesis, 6: 753–757.
Fu PP, Chou MW, Miller DW, White GL, Heflich RH & Beland FA (1985b) The orientation of the nitro substituent predicts the direct-acting bacterial mutagenicity of nitrated polycyclic aromatic hydrocarbons. Mutat Res, 143: 173–181.
Fu PP, Heflich RH, von Tungeln LS, Yang DTC, Fifer EK & Beland FA (1986) Effect of the nitro group conformation on the rat liver microsomal metabolism and bacterial mutagenicity of 2- and 9-nitroanthracene. Carcinogenesis, 7: 1819–1827.
Fu PP, Chou MW & Beland FA (1988a) Effects of nitro substitution on the in vitro metabolic activation of polycyclic aromatic hydrocarbons. In: Yang SK & Silverman BD ed. Polycyclic aromatic hydrocarbon carcinogenesis: Structure–activity relationships. Boca Raton, Florida, CRC Press, pp. 37–65.
Fu PP, Heflich RH, Unruh IF, Shaikh AU, Wu YS, Lai CC & Lai JS (1988b) Relationships among direct-acting mutagenicity, nitro group orientation and polarographic reduction potential of 6-nitrobenzo[a]pyrene, 7-nitrobenz[a]anthracene and their derivatives. Mutat Res, 209: 115–122.
Fu PP, Heflich RH, von Tungeln LS, Miranda HZ & Evans FE (1988c) Microsomal metabolism of 1-nitrobenzo[e]pyrene to a highly mutagenic K-region dihydrodiol. Carcinogenesis, 9: 951–958.
Fu PP, Ni YC, Zhang YM, Heflich RH, Wang YK & Lai JS (1989) Effect of the orientation of nitro substituent on the bacterial mutagenicity of dinitrobenzo[e]pyrenes. Mutat Res, 225: 121–125.
Fu PP, Miller DW, von Tungeln LS, Lay JO Jr, Huang K, Jones L & Evans FE (1991) Formation of C8-modified deoxyguanosine and C8-modified deoxyadenosine as major DNA adducts from 2-nitropyrene metabolism mediated by rat and mouse liver microsomes and cytosols. Carcinogenesis, 12: 609–616.
Fu PP, Dooley KL, von Tungeln LS, Bucci T, Hart RW & Kadlubar FF (1994a) Caloric restriction profoundly inhibits liver tumor formation after initiation by 6-nitrochrysene in male mice. Carcinogenesis, 15: 159–161.
Fu PP, Herreno-Saenz D, von Tungeln LS, Lay JO, Wu YS, Lai JS & Evans FE (1994b) DNA adducts and carcinogenicity of nitro-polycyclic aromatic hydrocarbons. Environ Health Perspect, 102(Suppl 6): 177–183.
Fu PP, Qui FY, Jung H, von Tungeln LS, Zhan DJ, Lee MJ, Wu YS & Heflich RH (1997) Metabolism of isomeric nitrobenzo[a]pyrenes leading to DNA adducts and mutagenesis. Mutat Res, 376: 43–51.
Fu PP, von Tungeln L, Yi P, Xia Q, Casciano D, Flammang T & Kadlubar F (1998a) Neonatal mouse tumorigenicity bioassay. Drug Inf J, 32(3): 711–728.
Fu PP, von Tungeln LS, Chiu LH, Zhan DJ, Deck J, Bucci T & Wang JC (1998b) Structure, tumorigenicity, microsomal metabolism, and DNA binding of 7-nitrodibenz[a,h]anthracene. Chem Res Toxicol, 11(8): 937–945.
Fujii K (1991) Evaluation of the newborn mouse model for chemical tumorigenesis. Carcinogenesis, 12: 1409–1415.
Galceran MT & Moyano E (1993) Determination of oxygenated and nitro-substituted polycyclic aromatic hydrocarbons by HPLC and electrochemical detection. Talanta (London), 40: 615–621.
Gang Y & Xiaobai X (1994) Bioaccumulation and elimination of 2-nitrofluorene in Daphnia magna. Fresenius Environ Bull, 3: 75–79.
Garner RC, Stanton CA, Martin CN, Chow FL, Thomas W, Hubner D & Hermann R (1986) Bacterial mutagenicity and chemical analysis of polycyclic aromatic hydrocarbons and some nitro derivatives in environmental samples collected in West Germany. Environ Mutagen, 8(1): 109–117.
Gelinas CG & Fan HSL (1979) Reducing air pollutant emissions at airports by controlling aircraft ground operations. J Air Pollut Control Assoc, 29: 125–128.
Gibson TL (1982) Nitro derivates of polynuclear aromatic hydrocarbons in airborne and source particulate matter [preliminary communication]. Atmos Environ, 16: 2037–2040.
Gibson TL (1983) Sources of direct-acting nitroarene mutagens in airborne particulate matter. Mutat Res, 122: 115–121.
Gibson TL (1986) Sources of nitroaromatic mutagens in atmospheric polycyclic organic matter. J Air Pollut Control Assoc, 36: 1022–1025.
Glatt H, Gemperlein I, Setiabudi F, Platt KL & Oesch F (1990) Expression of xenobiotic-metabolizing enzymes in propagatable cell cultures and induction of micronuclei by 13 compounds. Mutagenesis, 5: 241–249.
Gold A, Sangaiah R, Inhof CJ, Zhang J & Ball LM (1996) Metabolic fate of the environmental mutagen 3-nitrofluoranthene in the rat. Polycycl Aromat Comp, 10: 19–26.
González de Mejía E, Loarca-Pina G & Ramos-Gomez M (1997) Antimutagenicity of xanthophylls present in Aztec marigold (Tagetes erecta) against 1-nitropyrene. Mutat Res, 389: 216–226.
González de Mejía E, Quintanar-Hernández JA & Loarca-Pina G (1998) Antimutagenic activity of carotenoids in green peppers against some nitroarenes. Mutat Res, 416(1–2): 11–19.
Gorse RA Jr, Riley TL, Ferris FC, Pero AM & Skewes LM (1983) 1-Nitropyrene concentration and bacterial mutagenicity in on-road vehicle emissions. Environ Sci Technol, 27: 198–202.
Graf U, Wild D & Würgler FE (1992) Genotoxicity of 2-amino-3-methylimidazol[4,5-f]quinoline(IQ) and related compounds in Drosophila. Mutagenesis, 7: 145–149.
Greenberg A, Lwo J, Atherholt T, Rosen R, Hartman T, Butler J & Louis J (1993) Bioassay-directed fractionation of organic compounds associated with airborne particulate matter: An interseasonal study. Atmos Environ Part A, 27: 1609–1626.
Greibrokk T, Löfroth G, Nilsson L, Toftgard R, Carlstedt-Duke J & Gustafsson JA (1984) Nitroarenes: mutagenicity in the Ames Salmonella/microsome assay and affinity to the TCDD-receptor protein. In: Rickert DE ed. Toxicity of nitroaromatic compounds. Washington, DC, Hemisphere, pp. 167–183.
Grimmer G, Brune H, Deutsch-Wenzel R, Dettbarn G, Jacob J, Naujack KW, Mohr U & Ernst H (1987) Contribution of polycyclic aromatic hydrocarbons and nitro-derivatives to the carcinogenic impact of diesel engine exhaust condensate evaluated by implantation into the lungs of rats. Cancer Lett, 37: 173–180.
Grosjean D, Harrison J & Fung K (1983) Exposure of 1-nitropyrene to gaseous atmospheric pollutants. Atmos Environ, 17(8): 1609–1612.
Grosovsky AJ, Sasaki JC, Arey J, Eastmond DA, Parks KK & Atkinson R (1999) Evaluation of the potential health effects of the atmospheric reaction products of polycyclic aromatic hydrocarbons. Cambridge, Massachusetts, Health Effects Institute, pp. 1–29 (Research Report No. 84).
Gu Z-W, Whong W-Z, Wallace WE & Ong T (1992) Induction of micronuclei in BALB/c-3T3 cells by selected chemicals and complex mixtures. Mutat Res, 279: 217–222.
Guengerich FP, Parikh A, Turesky RJ & Josephy PD (1999) Inter-individual differences in the metabolism of environmental toxicants: cytochrome P450 1a2 as a prototype. Mutat Res, 428(1,2): 115–124.
Gupta P (1995) The contribution of methylnitronaphthalenes to the vapor-phase mutagenicity observed in ambient air samples collected at Redlands, California. Masters’ thesis, University of California, Riverside, California.
Gupta P, Harger WP & Arey J (1996) The contribution of nitro- and methylnitro-naphthalenes to the vapor-phase mutagenicity of ambient air samples. Atmos Environ, 30: 3157–3166.
Hagiwara Y, Watanabe M, Oda Y, Sofuni T & Nohmi T (1993) Specificity and sensitivity of Salmonella typhimurium YG1041 and YG1042 strains possessing elevated levels of both nitroreductase and acetyltransferase activity. Mutat Res, 291: 171–180.
Handa T, Yamauchi T, Ohnishi M, Hisamatsu Y & Ishi T (1983) Detection and average content levels of carcinogenic and mutagenic compounds from the particulates on diesel and gasoline engine mufflers. Environ Int, 9: 335–341.
Hannigan M, Cass G, Penman B, Crespi C, Lafleur A, Busby W & Thilly W (1997) Human cell mutagens in Los Angeles air. Environ Sci Technol, 31: 438–447.
Hannigan M, Cass G, Penman B, Crespi C, Lafleur A, Busby W, Thilly W & Simoneit BT (1998) Bioassay-directed chemical analysis of Los Angeles airborne particulate matter using a human cell mutagenicity assay. Environ Sci Technol, 32(22): 3502–3514.
Harger P, Arey J & Atkinson R (1992) The mutagenicity of HPLC-separated vapor-phase and particulate organics in ambient air. Atmos Environ, 26A: 2463–2466.
Harris WR, Chess EK, Okamoto D, Remsen JF & Later DW (1984) Contribution of nitropyrene to the mutagenic activity of coal fly ash. Environ Mutagen, 6: 131–144.
Hart RW, Fu PP & Turturro A (1988) Nitro-polycyclic aromatic hydrocarbons: structural features, genotoxicity, and risk evaluation. In: Politzer P & Martin FJ ed. Chemical carcinogens: Activation, mechanisms, structural and electronic factors, and reactivity. Amsterdam, Elsevier, pp. 264–290.
Hartung A, Kraft J, Schulze J, Kieß H & Lies KH (1984) The identification of nitrated polycyclic aromatic hydrocarbons in diesel particulate extracts and their potential formation as artifacts during particulate collection. Chromatographia, 19: 269–273.
Harvey GD, Baumgard KJ, Johnson JH, Gratz LD, Bagley ST & Leddy DG (1994) Effects of a ceramic particle trap and copper fuel additive on heavy-duty diesel emissions. Warrendale, Pennsylvania, Society of Automotive Engineers, pp. 207–225 (SAE Paper SP-1053).
Hashimoto Y & Shudo K (1985) Modification of nucleic acids with 1-nitropyrene in the rat: Identification of the modified nucleic acid base. Gann (Jpn J Cancer Res), 76: 253–256.
Hass BS, Chou MW, Casciano DA, White GL, Fu PP & Heflich RH (1984) Differences in the mutagenicity response of Salmonella and of CHO cells to nitrobenzo[a]pyrenes. Environ Mutagen, 6: 417–418.
Hass BS, Heflich RH, Chou MW, White GL, Fu PP & Casciano DA (1986a) Hepatocyte-mediated mutagenicity of mononitrobenzo[a]pyrenes in Salmonella typhimurium strains. Mutat Res, 171: 123–129.
Hass BS, Heflich RH, Schol HM, Chou MW, Fu PP & Casciano DA (1986b) Mutagenicity of the mononitrobenzo[a]pyrenes in Chinese hamster ovary cells mediated by rat hepatocytes of liver S9. Carcinogenesis, 7: 681–684.
Haugen A, Aune T & Deilhaug T (1986) Nitropyrene-induced DNA repair in Clara cells and alveolar type-II cells isolated from rabbit lung. Mutat Res, 175: 259–262.
Hayakawa K (2000) Chromatographic methods for carcinogenic/mutagenic nitropolycyclic aromatic hydrocarbons. Biomed Chromatogr, 14(6): 397–405.
Hayakawa K, Butoh M & Miyazaki M (1992) Determination of dinitro- and nitropyrenes in emission particulates from diesel and gasoline engine vehicles by liquid chromatography with chemiluminescence detection after precolumn reduction. Anal Chim Acta, 266: 251–256.
Hayakawa K, Terai N, Suzuki K, Dinning PG, Yamada M & Miyazaki M (1993) Chromatographic determination method for 1-nitropyrene and its metabolites in biological samples with fluorescence detection after online reduction. Biomed Chromatogr, 7: 262–266.
Hayakawa K, Butoh M, Hirabayashi Y & Miyazaki M (1994) Determination of 1,3-, 1,6-, 1,8-dinitropyrenes and 1-nitropyrene in vehicle exhaust particulates. Jpn J Toxicol Environ Health, 40: 20–25.
Hayakawa K, Murahashi T, Butoh M & Miyazaki M (1995a) Determination of 1,3-, 1,6-, and 1,8-dinitropyrenes and 1-nitropyrene in urban air by high-performance liquid chromatography using chemiluminescence detection. Environ Sci Technol, 29: 928–932.
Hayakawa K, Kawaguchi Y, Murahashi T & Miyazaki M (1995b) Distributions of nitropyrenes and mutagenicity in airborne particulates collected with an Andersen sampler. Mutat Res, 348: 57–61.
Hayakawa K, Nakamura A, Terai N, Kizu R & Ando K (1997) Nitroarene concentrations and direct-acting mutagenicity of diesel exhaust particulates fractionated by silica-gel column chromatography. Chem Pharm Bull, 45: 1820–1822.
Hayakawa K, Murahashi T, Noji K & Kizu R (1999a) Column-switching high performance liquid chromatography for determining polycyclic aromatic hydrocarbons and nitropolycyclic aromatic hydrocarbons in environmental samples. Chromatogr J Sep Detect Sci, 20(3): 255–262.
Hayakawa K, Kizu R & Ando K (1999b) Study on atmospheric behavior and toxicity of carcinogenic nitroarenes by high-performance liquid chromatography using chemiluminescence detection. Chromatogr J Sep Detect Sci, 20(1): 37–43.
Hayatsu H (1992) Cellulose bearing covalently linked copper phthalocyanine trisulfonate as an adsorbent selective for polycyclic compounds and its use in studies of environmental mutagens and carcinogens. J Chromatogr, 497: 37–56.
Hecht SS, El-Bayoumy K, Rivenson A & Amin S (1994) Potent mammary carcinogenicity in female CD rats of a fjord region diol-epoxide of benzo[c]phenanthrene compared to a bay region diol-epoxide of benzo[a]pyrene. Cancer Res, 54: 21–24.
Heflich RH, Fifer EK, Djuric Z & Beland FA (1985a) DNA adduct formation and mutation induction by nitropyrenes in Salmonella and Chinese hamster ovary cells: relationships with nitroreduction and acetylation. Environ Health Perspect, 62: 135–143.
Heflich RH, Howard PC & Beland FA (1985b) 1-Nitrosopyrene: an intermediate in the metabolic activation of 1-nitropyrene to a mutagen in Salmonella typhimurium TA1538. Mutat Res, 149: 25–32.
Heflich RH, Fullerton NF & Beland FA (1986a) An examination of the weak mutagenic response of 1-nitropyrene in Chinese hamster ovary cells. Mutat Res, 161: 99–108.
Heflich RH, Djuric Z, Fifer EK, Cerniglia CE & Beland FA (1986b) Metabolism of dinitropyrenes to DNA-binding derivatives in vitro and in vivo. In: Ishinishi N, Koizumi A, McClellan RO & Stöber W ed. Carcinogenic and mutagenic effects of diesel engine exhaust. Amsterdam, Elsevier, pp. 185–197.
Heflich RH, Fifer EK, Djuric Z & Beland FA (1986c) Mutation induction and DNA adduct formation by 1,8-dinitropyrene in Chinese hamster ovary cells. In: Ramel C, Lambert B & Magnusson J ed. Genetic toxicology of environmental chemicals: Part A: Basic principles and mechanisms of action. Proceedings of the 4th international conference on environmental mutagens, Stockholm, Sweden, 24–28 June 1985. New York, Alan R. Liss, Inc., pp. 265–273.
Heflich RH, Unruh LE, Thornton-Manning JR, von Tungeln LS & Fu PP (1989) Mutagenicity of 1-, 3-, and 6-nitrobenzo[a]pyrene in Salmonella typhimurium and Chinese hamster ovary cells. Mutat Res, 225: 157–163.
Heflich RH, Thornton-Manning JR, Kinouchi T & Beland FA (1990) Mutagenicity of oxidized microsomal metabolites of 1-nitropyrene in Chinese hamster ovary cells. Mutagenesis, 5: 151–157.
HEI (1995) Executive summary 1995: Diesel exhaust: Critical analysis of emissions, exposure, and health effects. Boston, Massachusetts, Health Effects Institute (www.healtheffects.org).
Heidemann A & Miltenburger HG (1983) Investigations on the mutagenic activity of fractions from diesel exhaust particulate matter in mammalian cells in vivo and in vitro (Abstract No. 15). Mutat Res, 113: 339.
Heitkamp MA & Cerniglia CE (1988) Mineralization of polycyclic aromatic hydrocarbons by a bacterium isolated from sediments below an oil field. Appl Environ Microbiol, 54: 1612–1614.
Heitkamp MA, Freeman JP, Miller DW & Cerniglia CE (1991) Biodegradation of 1-nitropyrene. Arch Microbiol, 156(3): 223–230.
Helmig D, Arey J, Harger WP, Atkinson R & López-Cancio J (1992a) Formation of mutagenic nitrodibenzopyranones and their occurrence in ambient air. Environ Sci Technol, 26: 622–624.
Helmig D, Lopez-Cancio J, Arey J, Harger WP & Atkinson R (1992b) Quantification of ambient nitrodibenzopyranones: Further evidence for atmospheric mutagen formation. Environ Sci Technol, 26: 2207–2213.
Helmig D, Arey J, Atkinson R, Harger WP & McElroy PA (1992c) Products of the OH radical-initiated gas-phase reaction of fluorene in the presence of NOx. Atmos Environ, 26A: 1735–1745.
Henderson TR, Sun JD, Royer RE, Clark CR, Li AP, Harvey TM, Hunt DF, Fulford JE, Lovette AM & Davidson WR (1983) Triple-quadrupole mass spectrometry studies of nitroaromatic emissions from different diesel engines. Environ Sci Technol, 17: 443–449.
Hensel A & Meier K (1999) Pectins and xyloglucans exhibit antimutagenic activities against nitroaromatic compounds. Planta Med, 65(5): 395–399.
Hera C & Pueyo C (1986) Conditions for the optimal use of the L-arabinose-resistance mutagenesis test with Salmonella typhimurium. Mutagenesis, 1(4): 267–273.
Herreno-Saenz D, Evans FE, Heinze T, Lewtas J & Fu PP (1992) In vitro metabolism and DNA adduct formation from the mutagenic environmental contaminant 2-nitrofluoranthene. Chem Res Toxicol, 5: 863–869.
Herreno-Saenz D, Evans FE, Abian J & Fu PP (1993) Formation of 6-(deoxyguanosin-N2-yl)-3-aminobenzo[a]pyrene adduct from the mutagenic environmental contaminant 3-nitrobenzo[a]pyrene. Carcinogenesis, 14: 1065–1067.
Herreno-Saenz D, Heflich RH, von Tungeln LS, Lewtas J & Fu PP (1994) DNA adduct formation by 2-nitrofluoranthene in Salmonella typhimurium and neonatal B6C3F1 mice. Polycycl Aromat Comp, 6: 79–85.
Herreno-Saenz D, Evans FE, Beland FA & Fu PP (1995) Identification of two N2-deoxyguanosinyl DNA adducts upon nitroreduction of the environmental mutagen 1-nitropyrene. Chem Res Toxicol, 8: 269–277.
Hetherington LH, Livingstone DR & Walker CH (1996) Two- and one-electron dependent in vitro reductive metabolism of nitroaromatics by Mytilus edulis, Carcinus maenas and Asterias rubens. Comp Biochem Physiol, 113: 231–239.
Heussen GAH, Post JG & Alink GM (1990) Genotoxicity of benzo[a]pyrene, 2-nitrofluorene and airborne particulates in the DNA-repair host-mediated assay. Mutat Res, 241: 83–93.
Hirayama T, Watanabe T, Akita M, Shimomura S, Fujioka Y, Ozasa S & Fukui S (1988) Relationships between structure of nitrated arenes and their mutagenicity in Salmonella typhimurium; 2- and 2,7-nitro substituted fluorene, phenanthrene and pyrene. Mutat Res, 209: 67–74.
Hirose M, Lee MS, Wang CY & King CM (1984) Induction of rat mammary gland tumors by 1-nitropyrene, a recently recognized environmental mutagen. Cancer Res, 44: 1158–1162.
Ho CH, Clark BR, Guerin MR, Barkenbus BD, Rao TK & Epler JL (1981) Analytical and biological analyses of test materials from the synthetic fuel technologies. IV. Studies of chemical structure–mutagenic activity relationships of aromatic nitrogen compounds relevant to synfuels. Mutat Res, 85: 335–345.
Ho YL & Ho SK (1981) Screening of carcinogens with the prophage aclts857 induction test. Cancer Res, 41: 532–536.
Hoffmann GR, Janel-Bintz R & Fuchs RP (2001) Induction of -2 frameshift mutations by 2-nitrofluorene, N-hydroxyacetylaminofluorene, and N-2-acetylaminofluorene in reversion assays in Escherichia coli strains differing in permeability and acetyltransferase activity. Mutat Res, 493(1–2): 127–137.
Holder PS, Wehry EL & Mamantov G (1994) Photochemical transformation of 1-nitropyrene sorbed on coal fly ash fractions. Polycycl Aromat Comp, 4: 135–139.
Holloway MP, Biaglow MC, McCoy EC, Anders M, Rosenkranz HS & Howard PC (1987) Photochemical instability of 1-nitropyrene, 3-nitrofluoranthene, 1,8-dinitropyrene and their parent polycyclic aromatic hydrocarbons. Mutat Res, 187: 199–207.
Horikawa K, Sera N, Tokiwa H & Kada T (1986) Results of the rec-assay of nitropyrenes in the Bacillus subtilis test system. Mutat Res, 174: 89–92.
Horikawa K, Otofuji T, Nakagawa R, Sera N, Kuroda Y, Otsuka H & Tokiwa H (1987) Mutagenicity of dinitrofluoranthenes (DNFs) and their carcinogenicity in F344 rats. Mutat Res, 182: 360–361.
Horikawa K, Sera N, Otofuji T, Murakami K, Tokiwa H, Hwagawa M, Izumi K & Otsuka H (1991) Pulmonary carcinogenicity of 3,9- and 3,7-dinitrofluoranthene, 3-nitrofluoranthene and benzo(a)pyrene in F344 rats. Carcinogenesis, 12: 1003–1008.
Horikawa K, Sera N, Murakami K, Otofuji T, Tokiwa H, Izumi K & Otsuka H (1993) Carcinogenicity of dinitrobenzo[a]pyrenes and dinitrofluoranthenes in F344 rats. In: Dai Q, Armour MA & Zheng Q ed. Recent advances of chemistry and molecular biology in cancer research. Beijing, Science Press, pp. 319–325.
Horikawa K, Sera N, Murakami K, Sano N, Izumi K & Tokiwa H (1998) Comparative tumorigenicity of 1- and 3-nitrobenzo[a]pyrenes, and 3,6- and 1,6-dinitrobenzo[a]pyrenes in F344/DuCrj rats. Toxicol Lett, 98(1–2): 51–58.
Howard PC & Beland FA (1982) Xanthine oxidase catalyzed binding of 1-nitropyrene to DNA. Biochem Biophys Res Commun, 104: 727–732.
Howard PC & Beland FA (1994) The effects of copollutants on the metabolism and DNA binding of carcinogens. Cambridge, Massachusetts, Health Effects Institute, pp. 1–17 (Research Report No. 66).
Howard PC, Heflich RH, Evans FE & Beland FA (1983a) Formation of DNA adducts in vitro and in Salmonella typhimurium upon metabolic reduction of the environmental mutagen 1-nitropyrene. Cancer Res, 43: 2052–2058.
Howard PC, Gerrard JA, Milo GE, Fu PP, Beland FA & Kadlubar FF (1983b) Transformation of normal human skin fibroblasts by 1-nitropyrene and 6-nitrobenzo[a]pyrene. Carcinogenesis, 4: 353–355.
Howard PC, Beland FA & Cerniglia (1983c) Reduction of the carcinogen 1-nitropyrene to 1-aminopyrene by the rat intestinal bacteria. Carcinogenesis, 4: 985–990.
Howard PC, Flammang TJ & Beland FA (1985) Comparison of the in vitro and the in vivo hepatic metabolism of the carcinogen 1-nitropyrene. Carcinogenesis, 6: 243–249.
Howard PC, Mateescu GD & Consolo MC (1988) Liver microsomal metabolism of the environmental carcinogen 3-nitrofluoranthene. I. Phenolic metabolites. Carcinogenesis, 9: 911–917.
Howard PC, Aoyama T, Bauer SL, Gelboin HV & Gonzalez FJ (1990) The metabolism of 1-nitropyrene by human cytochromes P450. Carcinogenesis, 11: 1539–1542.
Howard PC, Consolo MC, Dooley KL & Beland FA (1995) Metabolism of 1-nitropyrene in mice: Transport across the placenta and mammary tissues. Chem-Biol Interact, 3: 309–325.
Hughes TJ, Lewtas J & Claxton LD (1997) Development of a standard reference material for diesel mutagenicity in the Salmonella plate incorporation assay. Mutat Res, 391: 243–258.
Hunt GT & Maisel BE (1995) Atmospheric concentrations of PCDDs/PCDFs, PAHs, and NO2-PAHs in Fresno, California, during winter months. Organ Comp, 24: 55–61.
IARC (1978) Some aromatic amines and related nitro compounds — Hair dyes, colouring agents and miscellaneous industrial chemicals. Lyon, International Agency for Research on Cancer, 400 pp. (Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 16).
IARC (1984) Polynuclear aromatic compounds, Part 2: Carbon blacks, mineral oils and some nitroarenes. Lyon, International Agency for Research on Cancer, 245 pp. (IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 33).
IARC (1986) Tobacco smoking. Lyon, International Agency for Research on Cancer, 139 pp. (IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 38).
IARC (1987) Overall evaluations in carcinogenicity: An updating of IARC monographs, volumes 1–42. Lyon, International Agency for Research on Cancer, 440 pp. (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Supplement 7).
IARC (1989) Diesel and gasoline exhausts and some nitro-compounds. Lyon, International Agency for Research on Cancer, 375 pp. (IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 46).
IARC (1996) 3,7-Dinitrofluoranthene and 3,9-dinitrofluoranthene. Lyon, International Agency for Research on Cancer, pp. 297–307 (IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 65).
Imaida K, Lee MS, Wang CY & King CM (1991a) Carcinogenicity of dinitropyrenes in the weanling female CD rat. Carcinogenesis, 12: 1187–1191.
Imaida K, Hirose M, Tay L, Lee MS, Wang CY & King CM (1991b) Cooperative carcinogenicities of 1-, 2-, and 4-nitropyrene and structurally related compounds in the female CD rat. Cancer Res, 51: 2902–2907.
Imaida K, Uneyama C, Ogasawara H, Hayashi S, Fukuhara K, Miyata N & Takahashi M (1992) Induction of colon adenocarcinomas in CD rats and lung adenomas in ICR mice by 6-nitrochrysene: comparison of carcinogenicity and aryl hydrocarbon hydroxylase induction in the target organs of each species. Cancer Res, 52: 1542–1545.
Imaida K, Lee M-S, Land SJ, Wang CY & King CM (1995) Carcinogenicity of nitropyrenes in the newborn female rat. Carcinogenesis, 16: 3027–3030.
Inazu K, Kobayashi T & Hisamatsu Y (1996) Formation of mutagenic nitrofluoranthenes in the gas–solid heterogeneous reaction of particle-associated fluoranthene in NO2–O3–O2 system. Chem Lett, 12: 1105–1106.
Inazu K, Kobayashi T & Hisamatsu Y (1997) Formation of 2-nitrofluoranthene in gas–solid heterogeneous photoreaction of fluoranthene supported on oxide particles in the presence of nitrogen dioxide. Chemosphere, 35: 607–622.
Ioki Y (1977) Aryloxyl radicals by photorearrangement of nitro-compounds. J Chem Soc Perkin Trans, II: 1240–1242.
IPCS (1996) Diesel fuel and exhaust emissions. Geneva, World Health Organization, International Programme on Chemical Safety, 389 pp. (Environmental Health Criteria 171).
IPCS (1998) Selected non-heterocyclic polycyclic aromatic hydrocarbons. Geneva, World Health Organization, International Programme on Chemical Safety, 883 pp. (Environmental Health Criteria 202).
Ishii S, Hisamatsu Y, Inazu K, Kadoi M & Aika K (2000) Ambient measurement of nitrotriphenylenes and possibility of nitrotriphenylene formation by atmospheric reaction. Environ Technol, 34: 1893–1899.
Ishii S, Hisamatsu Y, Inazu K & Aika K (2001) Environmental occurrence of nitrotriphenylene observed in airborne particulate matter. Chemosphere, 44: 681–690.
Iwagawa M, Maeda T, Izumi K, Otsuka H, Nishifuji K, Ohnishi Y & Aoki S (1989) Comparative dose–response study on the pulmonary carcinogenicity of 1,6-dinitropyrene and benzo(a)pyrene in F344 rats. Carcinogenesis, 10: 1285–1290.
Janoszka B, Bodzek D, Szotek A & Warzecha L (1997) HPLC and TLC identification of nitrogen polynuclear aromatic compounds in airborne particulate matter. J Planar Chromatogr Mod TLC, 10: 55–58.
Jensen TE, Richert JFO, Cleary AC, LaCouse DL & Gorse JRA (1986) 1-Nitropyrene in used diesel engine oil. J Air Pollut Control Assoc, 26: 1255–1256.
Jin Z & Rappaport SM (1983) Microbore liquid chromatography with electrochemical detection for determination of nitro-substituted polynuclear aromatic hydrocarbons in diesel soot. Anal Chem, 55: 1778–1781.
Jinhui X & Lee FC (2001) Analysis of nitrated polynuclear aromatic hydrocarbons. Chemosphere, 42(3): 245–250.
Johnson DE, Riley MGI & Cornish HH (1984) Acute target organ toxicity of 1-nitronaphthalene in the rat. J Appl Toxicol, 4: 253–257.
Johnson JH, Bagley ST, Gratz LD & Leddy DG (1994) A review of diesel particulate control technology and emissions effects — 1992 Horning memorial lecture. Warrendale, Pennsylvania, Society of Automotive Engineers (SAE Paper [SP-1020] 940233).
Jung II, Shaikh AU, Heflich RH & Fu PP (1991) Nitro group orientation, reduction potential and direct-acting mutagenicity of nitro-polycyclic aromatic hydrocarbons. Environ Mol Mutagen, 17: 169–180.
Jurado J, Alejandre-Duran E & Pueyo C (1993) Genetic differences between the standard Ames tester strains TA100 and TA98. Mutagenesis, 8: 527–532.
Jurado J, Alejandre DE & Pueyo C (1994) Mutagenicity testing in Salmonella typhimurium strains possessing both the His reversion and Ara forward mutation systems and different levels of classical nitroreductase or O-acetyltransferase activities. Environ Mol Mutagen, 23: 286–293.
Kado NY, Langley D & Eisenstadt E (1983) A simple modification of the Salmonella liquid-incubation assay increased sensitivity for detecting mutagens in human urine. Mutat Res, 121: 25–32.
Kakimoto H, Kitamura M, Matsumoto Y, Sakai S, Kanoh F, Murahashi T, Akutsu K, Kizu R & Hayakawa K (2000) Comparison of atmospheric polycyclic aromatic hydrocarbons and nitropolycyclic aromatic hydrocarbons in Kanazawa, Sapporo and Tokyo. J Health Sci, 46(1): 5–15.
Kakimoto H, Yokoe H, Matsumoto Y, Sakai S, Kanoh F, Murahashi M, Akutsu K, Toriba A, Kizu R & Hayakawa K (2001) Considerations of atmospheric behaviors of polycyclic aromatic hydrocarbons, nitropolycyclic aromatic hydrocarbons and inorganic pollutants based on their interrelationships. J Health Sci, 47(4): 385–393.
Kamens RM, Zhi-Hua F, Yao Y, Chen D, Chen S & Vartiainen M (1994) A methodology for modeling the formation and decay of nitro-PAH in the atmosphere. Chemosphere, 28: 1623–1632.
Kamiura T, Kawaraya T, Tanaka M & Nakadoi T (1991) Determination of 3-nitrofluoranthene and 1-nitropyrene in suspended particulate matter by liquid chromatography with fluorescence detection. Anal Chim Acta, 254: 27–31.
Kanoh T, Fukuda M, Hayami E, Kinouchi I, Nishifuji K & Ohnishi Y (1990) Nitro reaction in mice injected with pyrene during exposure to nitrogen dioxide. Mutat Res, 245: 1–4.
Kappers W, van Och F, de Groene E & Horbach G (2000) Comparison of three different in vitro mutation assays used for the investigation of cytochrome P450-mediated mutagenicity of nitro-polycyclic aromatic hydrocarbons. Mutat Res, 466(2): 143–159.
Karcher W, Devillers J, Garrigues P & Jacob J ed. (1991) Spectral atlas of polycyclic aromatic compounds: including information on aquatic toxicity, occurrence and biological activity. Dordrecht, Kluwer Academic Publishers.
Kataoka K, Kinouchi T & Ohnishi Y (1991) Species differences in metabolic activation and inactivation of 1-nitopyrene in the liver. Cancer Res, 51: 3919–3924.
Katoh Y, Takayama S & Shudo K (1984) Inhibition by hemin of dinitropyrene-induced mutagenesis in Chinese hamster V79 cells. Gann (Jpn J Cancer Res), 75: 574–577.
Katoh Y, Maekawa M & Sano Y (1994) Mutagenic effects of nitropyrenes in a soybean test system. Mutat Res, 320: 59–68.
Kawanishi M, Enya T, Suzuki H, Takebe H, Matsui S & Yagi T (2000) Postlabelling analysis of DNA adducts formed in human hepatoma cells treated with 3-nitrobenzanthrone. Mutat Res, 470(2): 133–139.
Ketterer B, Harris J, Talaska G, Meyer D, Pemble S, Taylor J, Lang N & Kadlubar F (1992) The human glutathione S-transferase supergene family, its polymorphism, and its effects on susceptibility to lung cancer. Environ Health Perspect, 98: 87–94.
Kier LD, Yamasaki E & Ames BN (1974) Detection of mutagenic activity in cigarette smoke condensates. Proc Natl Acad Sci USA, 71: 4159–4163.
King CM (1988) Metabolism and biological effects of nitropyrene and related compounds. Cambridge, Massachusetts, Health Effects Institute, pp. 1–22 (Research Report No. 16).
King LC & Lewtas J (1993) An evaluation of the comparative metabolism and kinetics of 1-nitropyrene by rabbit, rat, and hamster tracheal epithelial cells. Toxicol Appl Pharmacol, 122: 149–158.
King LC, Kohan MJ, Ball LM & Lewtas J (1984) Mutagenicity of 1-nitropyrene metabolites from lung S9. Cancer Lett, 22: 255–262.
Kinouchi T, Hideshi T & Ohnishi Y (1986a) Detection of 1-nitropyrene in Yakatori (grilled chicken). Mutat Res, 171: 105–113.
Kinouchi T, Morotomi M, Mutai M, Fifer EK, Beland FA & Ohnishi Y (1986b) Metabolism of 1-nitropyrene in germ-free and conventional rats. Gann (Jpn J Cancer Res), 77: 356–369.
Kinouchi T, Nishifuji K, Tsutsui H, Hoare SL & Ohnishi Y (1988) Mutagenicity and nitropyrene concentration of indoor air particulates exhausted from a kerosene heater. Gann (Jpn J Cancer Res), 79: 32–41.
Kinouchi T, Nishifuji K & Ohnishi Y (1990) Biliary excretion of glutathione conjugates of 4,5-epoxy-4,5-dihydro-1-nitropyrene and 9,10-epoxy-9,10-dihydro-1-nitropyrene in rats administered 1-nitropyrene orally and their further metabolism in the intestinal tract. Carcinogenesis, 11: 1381–1387.
Kinouchi T, Kataoka K, Miyanishi K, Akimoto S & Ohnishi Y (1992) Role of intestinal microflora in metabolism of glutathione conjugates of 1-nitropyrene 4,5-oxide and 1-nitropyrene 9,10-oxide. Tohoku J Exp Med, 168: 119–122.
Kinouchi T, Kataoka K, Miyanishi K, Akimoto S & Ohnishi Y (1993) Biological activities of the intestinal microflora in mice treated with antibiotics or untreated and the effects of the microflora on absorption and metabolic activation of orally administrated glutathione conjugates of K-region epoxides of 1-nitropyrene. Carcinogenesis, 14: 869–874.
Kitamura S & Tatsumi K (1996) Metabolism in vivo of 1-nitropyrene, an environmental pollutant, in fish. Biol Pharm Bull, 19: 1524–1526.
Klopman G, Tonucci DA, Holloway M & Rosenkranz HS (1984) Relationship between polarographic reduction potential and mutagenicity of nitroarenes. Mutat Res, 126: 139–144.
Ko YC, Lee CH, Chen MJ, Huang CC, Chang WY, Lin HJ, Wang HZ & Chang PY (1997) Risk factors for primary lung cancer among non-smoking women in Taiwan. Int J Epidemiol, 26(1): 24–31.
Koizumi A, Saitoh N, Suzuki T & Kamiyama S (1994) A novel compound, 9-hydroxy-1-nitropyrene, is a major photodegraded compound of 1-nitropyrene in the environment. Arch Environ Health, 49: 87–92.
Korfmacher WA, Rushing LG, Arey J, Zielinska B & Pitts JN Jr (1987) Identification of mononitropyrenes and mononitrofluoranthenes in air particulate matter via fused silica gas chromatography combined with negative ion atmospheric pressure ionization mass spectrometry. J High Resol Chromatogr Chromatogr Commun, 10: 641–646.
Kornbrust DJ & Barfknecht JR (1984) Comparison of rat and hamster hepatocyte primary culture/DNA repair assays. Environ Mutagen, 6: 1–11.
Kranendonk M, Pintado F, Mesquita P, Laires A, Vermeulen NPE & Rueff J (1996) MX100, a new Escherichia coli tester strain for use in genotoxicity studies. Mutagenesis, 11: 327–333.
Krzeminski J, Desai D, Lin J, Serebryany V, El-Bayoumy K & Amin S (2000) Synthesis of anti- 1,2-dihydroxy-3,4-epoxy-1,2,3,4-tetrahydro-6-nitrochrysene and its reaction with 2’-deoxyguanosine-5'-monophosphate, 2'-deoxyadenosine-5'-monophosphate, and calf thymus DNA in vitro. Chem Res Toxicol, 13(11):1143–1148.
Kubinski H, Gutzke GE & Kubinski ZO (1981) DNA-cell-binding (DCB) assay for suspected carcinogens and mutagens. Mutat Res, 89: 95–136.
LaCourse DL & Jensen TE (1986) Determination of 1-nitropyrene in extracts of vehicle particulate emissions. Anal Chem, 58: 1894–1895.
Lafi A & Parry JM (1987) Chromosome aberrations induced by nitro-, nitroso- and aminopyrenes in cultured Chinese hamster cells. Mutagenesis, 2: 23–26.
Lambert ME & Weinstein IB (1987) Nitropyrenes are inducers of polyoma viral DNA synthesis. Mutat Res, 183: 203–211.
LaVoie EJ, Goril A, Briggs G & Hoffmann D (1981) Mutagenicity of aminocarbazoles and nitrocarbazoles. Mutat Res, 90: 337–344.
Lee H, Cherng SH & Liu TY (1994) Bacterial mutagenicity and DNA adduct formation by binary mixtures of benzo[a]pyrene and 1-nitropyrene. Environ Mol Mutagen, 24: 229–234.
Lee PS, Gorski RA, Johnson JT & Soderholm SC (1989) Generation of diesel particles coated with polycyclic aromatic compounds — evidence suggesting that dinitropyrene formation is a collection artifact. J Aerosol Sci, 20: 627–637.
Legzdins AE, McCarry BE & Bryant DW (1994) Polycyclic aromatic compounds in Hamilton air: their mutagenicity, ambient concentrations and relationships with atmospheric pollutants. Polycycl Aromat Comp, 5: 157–165.
Legzdins AE, McCarry BE & Marvin CH (1995) Methodology for bioassay-directed fractionation studies of air particulate material and other complex environmental matrixes. Int J Environ Anal Chem, 60: 79–94.
Lenicek J, Sekyra M, Bednarkova K, Benes I & Sipek F (2001) Fractionation and chemical analysis of urban air particulate extracts. In: Sram RJ ed. Teplice program: Impact of air pollution on human health. Prague, Academia, pp. 87–96.
Levin DE, Barnes WS & Klekowski F (1979) Mutagenicity of fluorene derivatives: A proposed mechanism. Mutat Res, 63: 1–10.
Levsen K (1988) The analysis of diesel particulate. Fresenius Z Anal Chem, 331: 467–478.
Lewis AC, Askey SA, Robinson RE, Bartle KD & Pilling MJ (1995a) Identification of polycyclic aromatic compounds in urban air particulate extracts by on-line coupled LC-GC-ITD-MS. Anal Proc, 32: 297–300.
Lewis AC, Robinson RE, Bartle KD & Pilling MJ (1995b) Online coupled LC-GC-ITD/MS for the identification of alkylated, oxygenated, and nitrated polycyclic aromatic compounds in urban air particulate extracts. Environ Sci Technol, 29: 1977–1981.
Lewtas J (1988) Genotoxicity of complex mixtures: Strategies for the identification and comparative assessment of airborne mutagens and carcinogens from combustion sources. Fundam Appl Toxicol, 10: 571–589.
Lewtas J & Nishioka MG (1990) Nitroarenes: Their detection, mutagenicity and occurrence in the environment. In: Howard PC, Hecht SS & Beland FA ed. Nitroarenes: Occurrence, metabolism, and biological impact. New York, Plenum Press, pp. 61–72.
Lewtas J, Chuang J, Nishioka M & Peterson B (1990a) Bioassay-directed fractionation of the organic extract of SRM 1649 urban air particulate matter. Int J Environ Anal Chem, 39: 245–256.
Lewtas J, King LC, Williams K, Ball LM & DeMarini DM (1990b) Bioassay-directed fractionation of 1-nitropyrene metabolites: generation of mutagrams by coupling reverse-phase HPLC with micro-suspension mutagenicity assays. Mutagenesis, 5: 481–489.
Li AP & Dutcher JS (1983) Mutagenicity of mono-, di- and tri-nitropyrenes in Chinese hamster ovary cells. Mutat Res, 119: 387–392.
Li EE, Heflich RH & Delclos KB (1993) Trans-1,2-dihydro-1,2-dihydroxy-6-aminochrysene is metabolized to form a major adduct with deoxyguanosine and produces mutations in the hprt gene of Chinese hamster ovary cells at G:C basepairs. Carcinogenesis, 14: 2109–2114.
Li EE, Heflich RH, Bucci TJ, Manjanatha MG, Blaydes BS & Delclos KB (1994) Relationships of DNA adduct formation, K-ras activating mutations and tumorigenic activities of 6-nitrochrysene and its metabolites in the lungs of CD-1 mice. Carcinogenesis, 15: 1377–1385.
Liberti A, Ciccioli P, Cecinato A, Brancaleone E & Di Palo C (1984) Determination of nitrated-polyaromatic hydrocarbons (nitro-PAHs) in environmental samples by high resolution chromatographic techniques. J High Resol Chromatogr, 7: 389–397.
Librando V, Liberatori A & Fazzino SD (1993) The determination of nitro-polycyclic aromatic hydrocarbons in the ash of a sanitary waste incinerator. Polycycl Aromat Comp, 3: 587–594.
Lindskog A, Brorstrom-Lunden E, Alfheim I & Hagen I (1987) Chemical transformation of PAH on airborne particles by exposure to nitrogen dioxide during sampling: a comparison between two filter media. Sci Total Environ, 61: 51–58.
Lloyd A & Cackette T (2001) Diesel engines: environmental impact and control. J Air Waste Manage Assoc, 51(6): 809–847.
Löfroth G (1981) Comparison of the mutagenic activity in carbon particulate matter and in diesel and gasoline engine exhaust. In: Waters MD, Sandhu SS, Lewtas J, Claxton L & Nesnow S ed. Short-term bioassays in the analysis of complex environmental mixtures II. New York, Plenum Press, pp. 319–336.
Löfroth G, Hefner E, Alfheim I & Möller M (1980) Mutagenic activity in photocopies. Science, 209: 1037–1039.
Löfroth G, Toftgard R, Nilsson L, Agurell E & Gustafsson JA (1984) Short-term bioassays of nitro derivatives of benzo(a)pyrene and perylene. Carcinogenesis, 5: 925–930.
Logan JA (1985) Tropospheric ozone: seasonal behavior, trends, and anthropogenic influence. J Geophys Res, 90(10): 463–482.
Lorber KE & Mollenhauer K (1989) Emission und biologische Wirkung von N-haltigen polycyclischen Aromaten aus Dieselmotoren. Technische Universität Berlin, Berlin. Berlin, Umweltbundesamt, pp. 89–150.
Lysak A & Marcinek J (1972) Multiple toxic effect of simultaneous action of some chemical substances on fish. Rcozniki Nauk Rolniczych, 94(3): 53–63.
MacCrehan WA, May WE, Yang SD & Brenner BA (1988) Determination of nitro polynuclear aromatic hydrocarbons in air and diesel particulate matter using liquid chromatography with electrochemical and fluorescence detection. Anal Chem, 60: 194–199.
Maeda M, Tsukagoshi K, Murata M, Takagi M & Yamashita T (1994) Separation and determination of trace dinitropyrenes by means of off-line reduction–HPLC–chemiluminescence detection. Application to assessing atmospheric environment. Anal Sci, 10: 583–587.
Maeda T, Izumi T, Otsuka H, Manabe Y, Kinouchi T & Ohnishi Y (1986) Induction of squamous cell carcinoma in the rat lung by 1,6-dinitropyrene. J Natl Cancer Inst, 76: 693–701.
Maher VM, Mah MCM, Yang JL, Bhattacharyya NP & McCormick JJ (1990) Mutations and homologous recombination induced by N-substituted aryl compounds in mammalian cells. In: Howard PC, Hecht SS & Beland FA ed. Nitroarenes: occuurence, metabolism, and biological impact. New York, Plenum Press, pp. 149–156.
Malejka-Giganti D, Niehans GA, Reichert MA, Bennett KK & Bliss RL (1999) Potent carcinogenicity of 2,7-dinitrofluorene, an environmental pollutant, for the mammary gland of female Sprague-Dawley rats. Carcinogenesis, 20: 2017–1023.
Malia SA, Vyas RR & Basu AK (1996) Site-specific frame-shift mutagenesis by the 1-nitropyrene-DNA adduct N-(deoxyguanosin-8-yl)-1-aminopyrene located in the (CG)3 sequence: effects of SOS, proofreading, and mismatch repair. Biochemistry (Washington), 35: 4568–4577.
Mamber SW, Bryson V & Katz SE (1983) The Escherichia coli WP2/WP100 rec assay for detection of potential chemical carcinogens. Mutat Res, 119: 135–144.
Mamber SW, Bryson V & Katz SE (1984) Evaluation of the Escherichia coli K12 inductest for detection of potential carcinogens. Mutat Res, 130: 141–151.
Mamber SW, Okasinski WG, Pinter CD & Tunac JB (1986) The Escherichia coli K-12 SOS chromotest agar spot test for simple, rapid detection of genotoxic agents. Mutat Res, 171: 83–90.
Manabe Y, Kinouchi T, Wakisaka K, Tahara I & Ohnishi Y (1984) Mutagenic 1-nitropyrene in wastewater from oil–water separating tanks of gasoline stations and in used crankcase oil. Environ Mutagen, 6: 669–681.
Manabe Y, Kinouchi T & Ohnishi Y (1985) Identification and quantification of highly mutagenic nitrosacetoxypyrenes and nitrohydroxypyrenes in diesel-exhaust particles. Mutat Res, 158: 3–18.
Manjanatha MG, Li EE, Fu PP & Heflich RH (1996) H- and K-ras mutational profiles in chemically induced liver tumors from B6C3F-1 and CD-1 mice. J Toxicol Environ Health, 47: 195–208.
Marino F, Cecinato A & Siskos PA (2000) Nitro-PAH in ambient particulate matter in the atmosphere of Athens. Chemosphere, 40: 533–537.
Maron DM & Ames BN (1983) Revised methods for the Salmonella mutagenicity test. Mutat Res, 113: 173–215.
Marsh JW, Chipman JK & Livingstone DR (1992) Activation of xenobiotics to reactive and mutagenic products by the marine invertebrates Mytilus edulis, Carcinus maenus and Asterias rubens. Aquat Toxicol, 22: 115–128.
Marshall TC, Royer RE, Li AP, Kusewitt DF & Brooks AL (1982) Acute and genetic toxicity of 1-nitropyrene and its fate after single oral doses to rats. J Toxicol Environ Health, 10: 373–384.
Marzin DR, Olivier P & Vophi H (1986) Kinetic determination of enzymatic activity and modification of the metabolic activation system in the SOS chromotest. Mutat Res, 164: 353–359.
Massaro M, McCartney M, Rosenkranz EJ, Anders M, McCoy EC, Mermelstein R & Rosenkranz HS (1983) Evidence that nitroarene metabolites form mutagenic adducts with DNA-adenine as well as with DNA-guanine. Mutat Res, 122: 243–249.
Matsuda A (1981) Studies on the mutagenicity of nitro-aromatic compounds in environment. Acta Sch Med Univ Gifu, 29: 278–296.
Matsuoka A, Sofuni I, Miyata N & Ishidate MJ (1991) Clastogenicity of 1-nitropyrene, dinitropyrenes, fluorene and mononitrofluorenes in cultured Chinese hamster cells. Mutat Res, 259: 103–110.
Matsuoka A, Horikawa K, Yamazaki N, Sera N, Sofuni T & Tokiwa H (1993) Chromosomal aberrations induced in vitro by 3,7- and 3,9-dinitrofluoranthene. Mutat Res, 298: 255–259.
Matsushita H & Iida Y (1986) Application of capillary gas chromatography to environmental analysis. J High Resol Chromatogr, 9: 708–711.
Matthews EJ, Spalding JW & Tennant RW (1993) Transformation of BALB/c-3T3 cells: V. Transformation responses of 168 chemicals compared with mutagenicity in Salmonella and carcinogenicity in rodent bioassays. Environ Health Perspect, 101(Suppl 2): 347–482.
May WE, Benner BA, Wise, SA, Schuetzle D & Lewtas J (1992) Standard reference materials for chemical and biological studies of complex environmental samples. Mutat Res, 276: 11–22.
McCann J, Choi E, Yamasaki E & Ames BN (1975) Detection of carcinogens as mutagens in the Salmonella/microsome test: Assay of 300 chemicals. Part I. Proc Natl Acad Sci USA, 72: 5135–5139.
McCarroll NE, Piper CE & Keech BH (1981a) An E. coli microsuspension assay for the detection of DNA damage induced by direct-acting agents and promutagens. Environ Mutagen, 3: 429–444.
McCarroll NE, Keech BH & Piper CE (1981b) A microsuspension adaptation of the Bacillus subtilus "rec" assay. Environ Mutagen, 3: 607–616.
McCartney MA, Chatterjee BF, McCoy EC, Mortimer EAJ & Rosenkranz HS (1986) Airplane emissions: A source of mutagenic nitrated polycyclic aromatic hydrocarbons. Mutat Res, 171: 99–104.
McCoy EC, Rosenkranz EJ, Petrullo LA, Rosenkranz HS & Mermelstein R (1981a) Structural basis of the mutagenicity in bacteria of nitrated naphthalene and derivatives. Environ Mutagen, 3: 499–511.
McCoy EC, Rosenkranz EJ, Rosenkranz HS & Mermelstein R (1981b) Nitrated fluorene derivatives are potent frameshift mutagens. Mutat Res, 90: 11–20.
McCoy EC, DeMarco G, Rosenkranz EJ, Anders M, Rosenkranz HS & Mermelstein R (1983a) 5-Nitroacenaphthene: A newly recognized role for the nitro function in mutagenicity. Environ Mutagen, 5: 17–22.
McCoy EC, Rosenkranz EJ, Mermelstein R & Rosenkranz HS (1983b) Mutagenic specificity and the prediction of mechanisms and bioactivation pathways of genotoxicants: the mutagenicity of 5-nitroacenaphthene as an example. Mutat Res, 111: 61–68.
McCoy EC, Anders M, Rosenkranz HS & Mermelstein R (1983c) Apparent absence of recombinogenic activity of nitropyrenes for yeast. Mutat Res, 116: 119–127.
McCoy EC, Anders M, McCartney M, Howard PC, Beland PA & Rosenkranz HS (1984) The recombinogenic inactivity of the 1-nitropyrene for years is due to a deficiency in a functional nitroreductase. Mutat Res, 139: 115–118.
McCoy EC, Holloway M, Frierson M, Klopman G, Mermelstein R & Rosenkranz HS (1985a) Genetic and quantum chemical basis of the mutagenicity of nitroarenes for adenine–thymine base pairs. Mutat Res, 149: 311–319.
McCoy EC, Anders M, Rosenkranz HS & Mermelstein R (1985b) Mutagenicity of nitropyrenes for Escherichia coli: Requirement for increased cellular permeability. Mutat Res, 142: 163–167.
Medinsky MA, Shelton H, Bond JA & McClellan RO (1985) Biliary excretion and enterohepatic circulation of 1-nitropyrene metabolites in Fischer-344 rats. Biochem Pharmacol, 34: 2325–2330.
Medvedovici A, David F, Desmet G & Sandra P (1998) Fractionation of nitro and hydroxy polynuclear aromatic hydrocarbons from extracts of air particulates by supercritical fluid chromatography. J Microcolumn Sep, 10: 89–97.
Melchior WB, Marques MM, Smith BA & Beland FA (1990) Adduct distribution and mutations in DNA modified with 1-aminopyrene. Proc Am Assoc Cancer Res, 31: 93.
Mermelstein R, Kiriazides DK, Butler M, McCoy EC & Rosenkranz HS (1981) The extraordinary mutagenicity of nitropyrenes in bacteria. Mutat Res, 89: 187–196.
Mermelstein R, Rosenkranz HS & McCoy EC (1982) The microbial mutagenicity of nitroarenes. In: Tice RR, Costa DL & Schaich K ed. Genotoxic effects of airborne agents. New York, Plenum Press, pp. 369–396.
Mersch-Sundermann V, Kern S & Wintermann F (1991) Genotoxicity of nitrated polycyclic aromatic hydrocarbons and related structures on Escherichia coli PQ 37 (SOS chromotest). Environ Mol Mutagen, 18: 41–50.
Mersch-Sundermann V, Klopman G & Rosenkranz HS (1992) Structural requirements for the induction of the SOS repair in bacteria by nitrated polycyclic aromatic hydrocarbons and related chemicals. Mutat Res, 265: 61–73.
Messier F, Lu C, Andrews P, McCarry BE, Quilliam MA & McCalla DR (1981) Metabolism of 1-nitropyrene and formation of DNA adducts in Salmonella typhimurium. Carcinogenesis, 2: 1007–1011.
Miller E, Fletscher T, Margreth A & Miller J (1962) The carcinogenicities of derivatives of fluorene and biphenyl, fluoro derivatives as probes for active sites in 2-acetylaminofluorene. Cancer Res, 22: 1002–1014.
Miller JA, Sandin RB, Miller EC & Rusch HP (1955) The carcinogenicity of compounds related to 2-acetylaminofluorene. II. Variations in the bridges and the 2-substituent. Cancer Res, 15: 188–199.
Millner GC, Fu PP & Cerniglia CE (1986) Microbial transformation of 6-nitrobenzo[a]pyrene. J Toxicol Environ Health, 19: 519–530.
Mitchell CE (1985) Genotoxicity of 1-nitropyrene in mouse lung after intratracheal instillation. Fed Proc, 44: 924.
Mitchell CE (1988a) Formation of DNA adducts in mouse tissues after intratracheal instillation of 1-nitropyrene. Carcinogenesis, 9: 857–860.
Mitchell CE (1988b) Damage and repair of mouse lung DNA induced by 1-nitropyrene. Toxicol Lett, 42: 159–166.
Mitchell CE & Thomassen DG (1990) Cytotoxic and transformation responses of rat tracheal epithelial cells exposed to nitrated polycyclic aromatic hydrocarbons in culture. Carcinogenesis, 11: 155–158.
Mitchell CE, Bechthold WE & Belinsky SA (1993) Metabolism of nitrofluoranthenes by rat lung subcellular fractions. Carcinogenesis, 14: 1161–1166.
Mitchell I (1980) Forward mutation in Escherichia coli and gene conversion in Saccharomyces cerevisiae compared quantitatively with reversion in Salmonella typhimurium. Agents Actions, 10: 287–295.
Mitchell I & Gilbert PJ (1984) The effect of pretreatment of Escherichia coli CM891 with ethylenediaminetetraacetate on sensitivity to a variety of standard mutagens. Mutat Res, 140: 13–19.
Mitchell IG & Gilbert PJ (1985) An assessment of the importance of error-prone repair and point mutations to forward mutation to L-azetidine-2-carboxylic acid resistance in Escherichia coli. Mutat Res, 149: 303–310.
Mitchell K, Steere DE & Taylor JA (1994) Impact of diesel fuel aromatics on particulate, PAH and nitro-PAH emissions. Warrendale, Pennsylvania, Society of Automotive Engineers (SAE Paper 942053).
Mitchelmore CL & Chipman JK (1998) Detection of DNA strand breaks in brown trout (Salmo trutta) hepatocytes and blood cells using the single cell gel electrophoresis (comet) assay. Aquat Toxicol, 41(1–2): 161–182.
Mitchelmore CL, Birmelin C, Livingstone DR & Chipman JK (1998a) Detection of DNA strand breaks in isolated mussel (Mytilus edulis L.) digestive gland cells using the "comet" assay. Ecotoxicol Environ Saf, 41(1): 51–58.
Mitchelmore CL, Birmelin C, Chipman JK & Livingstone DR (1998b) Evidence for cytochrome P-450 catalysis and free radical involvement in the production of DNA strand breaks by benzo[a]pyrene and nitroaromatics in mussel (Mytilus edulis L.) digestive gland cells. Aquat Toxicol, 41(3): 193–212.
Mitchelmore CL, Livingstone DR & Chipman JK (1998c) Conversion of 1-nitropyrene by brown trout (Salmo trutta) and turbot (Scophthalamus maximus) to DNA adducts detected by 32P- postlabelling. Biomarkers, 3: 21–33.
Miyanishi K, Kinouchi T, Kataoka K, Kanoh T & Ohnishi Y (1996) In vivo formation of mutagens by intraperitoneal administration of polycyclic aromatic hydrocarbons in animals during exposure to nitrogen dioxide. Carcinogenesis, 17: 1483–1490.
Möller L & Zeisig M (1993) DNA adduct formation after oral administration of 2-nitrofluorene and N-acetyl-2-aminofluorene, analyzed by phosphorus-32 TLC and phosphorus-32 HPLC. Carcinogenesis, 14: 53–59.
Möller L, Nilsson L, Gustafsson JA & Rafter J (1985) Formation of mutagenic metabolites from 2-nitrofluorene, an air-pollutant, in the rat. Environ Int, 11: 363–368.
Möller L, Rafter J & Gustafsson JA (1987a) Metabolism of the carcinogenic air pollutant 2-nitrofluorene in the rat. Carcinogenesis, 8: 637–645.
Möller L, Törnquist S, Beije B, Rafter J, Toftgard R & Gustafsson JA (1987b) Metabolism of the carcinogenic air pollutant 2-nitrofluorene in the isolated perfused rat lung and liver. Carcinogenesis, 8: 1847–1852.
Möller L, Corrie M, Midtvedt T, Rafter J & Gustafsson JA (1988) The role of the intestinal microflora in the formation of mutagenic metabolites from the carcinogenic air pollutant 2-nitrofluorene. Carcinogenesis, 9: 823–830.
Möller L, Torndal UB, Eriksson LC & Gustafsson JA (1989) The air pollutant 2-nitrofluorene as initiator and promotor in a liver model for chemical carcinogenesis. Carcinogenesis, 10: 435–440.
Möller L, Lax I & Eriksson LC (1993a) Nitrated polycyclic aromatic hydrocarbons: A risk assessment for the Urban Citizen. Environ Health Perspect, 101(Suppl 3): 309–315.
Möller L, Cui XS, Torndal UB & Eriksson LC (1993b) Preneoplastic lesions and DNA adduct formation of the airborne genotoxic agents 2-nitrofluorene and 2,7-dinitrofluorene. Carcinogenesis, 14: 2627–2632.
Möller L, Zeisig M & Toftgaard R (1993c) Lack of initiating capacity of the genotoxic air pollutant 2-nitrofluorene in the mouse skin two-stage carcinogenesis system. Carcinogenesis, 14: 1723–1725.
Möller L, Zeisig M, Midtvedt T & Gustafsson JA (1994) Intestinal microflora enhances formation of DNA adducts following administration of 2-NF and 2-AAF. Carcinogenesis, 15: 857–861.
Möller ME & Thorgeirsson SS (1985) DNA damage induced by nitropyrenes in primary mouse hepatocytes and in rat H4-II-E hepatoma cells. Mutat Res, 151: 137–146.
Montreuil CN, Ball JC, Gorse JRA & Young WC (1992) Solvent extraction efficiencies of mutagenic components from diesel particles. Mutat Res, 282: 89–92.
Mori H, Sugie S, Yoshimi N, Kinouchi T & Oh Y (1987) Genotoxicity of a variety of nitroarenes and other nitro compounds in DNA-repair tests with rat and mouse hepatocytes. Mutat Res, 190: 159–167.
Moriske HJ, Block I & Rüden H (1984) Über polare organische Verbindungen im Stadtaerosol und deren mutagene Wirksamkeit im Salmonella typhimurium-Test nach Ames. Forum Städte Hyg, 35: 113–121.
Morita T, Asano N, Awogi T, Sasaki YF, Sato S, Shimada H, Sutou S, Suzuki T, Wakata A, Sofuni T & Hayashi M (1997) Evaluation of the rodent micronucleus assay in the screening of IARC carcinogens (groups 1, 2A and 2B). The summary report of the 6th collaborative study by CSGMT/JEMS NMS. Mutat Res, 389: 3–122.
Morotomi M, Nanno M, Watanabe T, Sakurai T & Mutai M (1985) Mutagenic activation of biliary metabolites of 1-nitropyrene by intestinal microflora. Mutat Res, 149: 171–178.
Morris HP, Dubnik CS & Johnson JM (1950) Studies of the carcinogenic action in the rat of 2-nitro-, 2-amino-, 2-acetylamino-, and 2-diacetyl-aminofluorene after ingestion and after painting. J Natl Cancer Inst, 10: 1201–1213.
Morris SM, Domon OE, Delclos KB, Chen JJ & Casciano DA (1994) Induction of mutations at the hypoxanthine phosphoribosyl transferase (HPRT) locus in AHH-1 human lymphoblastoid cells. Mutat Res, 310: 45–54.
Mortelmans K, Haworth S, Lawlor T, Speck W, Tainer B & Zeiger E (1987) Salmonella mutagenicity tests: II. Results from the testing of 270 chemicals. Environ Mutagen, 8(Suppl 7): 1–119.
Moyano E & Galceran MT (1997) Determination of oxy-, nitro- and hydroxy- polycyclic aromatic hydrocarbons in atmospheric aerosol samples. Quim Anal, 16(3): 159–164.
Mulder GJ, Wierckx FCJ, Wedzinga R & Meerman JHN (1990) Generation of reactive intermediates from 2-nitrofluorene that bind covalently to DNA, RNA and protein in vitro and in vivo. In: Howard PC, Hecht SS & Beland FA ed. Nitroarenes — Occurrence, metabolism, and biological impact. New York, Plenum Press, pp. 219–230.
Müller L, Müller-Tegethoff K, Hambach P, Schleicher R & Kasper P (1995) The hepatocyte micronucleus assay for the detection of genotoxic liver carcinogens. Environ Mol Mutagen, 25: 37.
Mumford JL, Williams RW, Walsh DB, Burton RM, Svensgaard DJ, Chuang JC, Houk VS & Lewtas J (1991) Indoor air pollutants from unvented kerosene heater emissions in mobile homes: Studies on particles, semivolatile organics, carbon monoxide, and mutagenicity. Environ Sci Technol, 25: 1732–1738.
Murahashi T & Hayakawa K (1997) A sensitive method for the determination of 6-nitrochrysene, 2-nitrofluoranthene and 1-, 2- and 4-nitropyrenes in airborne particulates using high-performance liquid chromatography with chemiluminescence detection. Anal Chim Acta, 343: 251–257.
Murahashi T, Miyazaki M, Kakizawa R, Yamagishi Y, Kitamura M & Hayakawa K (1995) Diurnal concentrations of 1,3-, 1,6-, 1,8-dinitropyrenes, 1-nitropyrene and benzo[a]pyrene in air in downtown Kanazawa and the contribution of diesel-engine vehicles. Jpn J Toxicol Environ Health, 41: 328–333.
Murahashi T, Kizu R, Kakimoto H, Toriba A & Hayakawa K (1999) 2-Nitrofluoranthene, 1-, 2- and 4-nitropyrenes and 6-nitrochrysene in diesel-engine exhaust and airborne particulates. J Health Sci, 45(5): 244–250.
Murakami K, Sera N & Horikawa K (1996) Evaluation of mutagenicity of complex mixture of nitroarenes using the mouse micronuclei test. Toxicol Environ Chem, 54: 161–165.
Murray RW & Singh M (1998) Oxidative transformations of 1-nitropyrene under simulated environmental conditions. Chem Res Toxicol, 11: 449–453.
Nachtman JP & Wolff S (1982) Activity of nitro-polynuclear aromatic hydrocarbons in the sister chromatid exchange assay with and without metabolic activation. Environ Mutagen, 4: 1–5.
Nagai A, Kano Y, Funasaka R & Nakamuro K (1999) A fundamental study on the characteristics of concentration using a blue chitin column for polycyclic aromatic hydrocarbons in water. J Health Sci, 45(3): 111–118.
Nakagawa R, Horikawa K, Sera N, Kodera Y & Tokiwa H (1987) Dinitrofluoranthene: induction, identification and gene mutation. Mutat Res, 191: 85–91.
Nakamura S-I, Oda Y, Shimada T, Oki I & Sugimoto K (1987) SOS-inducing activity of chemical carcinogens and mutagens in Salmonella typhimurium TA1535/pSK1002: Examination with 151 chemicals. Mutat Res, 192: 239–246.
Nakayasu M, Sakamoto H, Wakabayashi L, Te M, Su T & Rosenkranz HS (1982) Potent mutagenic activity of nitropyrenes on Chinese hamster lung cells with diphtheria toxin resistance as a selective marker. Carcinogenesis, 3: 917–922.
Nardini B, Granella M & Clonfero E (1994) Mutagens in indoor air particulate. Mutat Res 322: 193–202.
NCI (1978a) Bioassay of 1-nitronaphthalene for possible carcinogenicity. Bethesda, Maryland, National Cancer Institute (NCI-CG-TR 64; NIH 78-1314).
NCI (1978b) Bioassay of 5-nitroacenaphthene for possible carcinogenicity, CAS No.
Neal SB & Probst GS (1983) Chemically-induced sister-chromatid exchange in vivo in bone marrow of Chinese hamsters, an evaluation of 24 compounds. Mutat Res, 113: 33–43.
Nesnow S, Triplett LL & Slaga TJ (1984) Tumor initiating activities of 1-nitropyrene and its nitrated products in Sencar mice. Cancer Lett, 23: 1–8.
Neumann HG, van Dorp C & Zwirner-Baier I (1995a) The implications for risk assessment of measuring the relative contribution to exposure from occupation, environment and lifestyle: Hemoglobin adducts from amino- and nitro-arenes. Toxicol Lett, 82–83: 771–778.
Neumann H-G, Albrecht O, van Dorp C & Zwirner-Baier I (1995b) Macromolecular adducts caused by environmental chemicals. Clin Chem, 41: 1835–1840.
Newton DL, Erickson MD, Tomer KB, Pellizzari ED, Gentry P & Zweidinger RB (1982) Identification of nitroaromatics in diesel exhaust particulate using gas chromatography/negative ion chemical ionization mass spectrometry and other techniques. Environ Sci Technol, 16: 206–213.
Newton RK, Mittelstaedt RA, Manjanatha MG & Heflich RH (1992) DNA sequence analysis of 1-nitrosopyrene-induced mutations in the hprt gene of Chinese hamster ovary cells. Carcinogenesis, 13: 819–825.
Niederer M (1998) Determination of polycyclic aromatic hydrocarbons and substitutes (nitro-, oxy-PAHs) in urban soil and airborne particulate by GC-MS and NCI-MS/MS. Environ Sci Pollut Res, 5(4): 209–216.
Nielsen T (1983) Isolation of polycyclic aromatic hydrocarbons and nitro derivatives in complex mixtures by liquid chromatography. Anal Chem, 55: 286–290.
Nielsen T & Ramdahl T (1986) Determination of 2-nitrofluoranthene and 2-nitropyrene in ambient particulate matter: evidence for atmospheric reactions. Atmos Environ, 20: 1507–1509.
Nielsen T, Seitz B & Ramdahl T (1984) Occurrence of nitro-PAH in the atmosphere in a rural area. Atmos Environ, 18: 2159–2165.
Nielsen T, Siigur K, Helweg C, Jorgensen O, Hansen PE & Kirso U (1997) Sorption of polycyclic aromatic compounds to humic acid as studied by high-performance liquid chromatography. Environ Sci Technol, 31: 1102–1108.
Niles R & Tan YL (1989) Determination of polynuclear aromatic hydrocarbons and mononitrated derivatives in air and diesel particulates. Anal Chim Acta, 221: 53–63.
Nishioka MG & Lewtas J (1992) Quantification of nitro- and hydroxylated nitro-aromatic/polycyclic aromatic hydrocarbons in selected ambient air daytime winter samples. Atmos Environ, 26A(11): 2077–2087.
Nishioka MG, Petersen BA & Lewtas J (1982) Comparison of nitro-aromatic content and direct-acting mutagenicity of diesel emissions. In: Cooke M, Dennis AJ & Fisher GL ed. Polynuclear aromatic hydrocarbons: Physical and biological chemistry. Columbus, Ohio, Battelle Press, pp. 603–613.
Nishioka MG, Petersen B & Lewtas J (1983) Comparison of nitroaromatic content and direct-acting mutagenicity of passenger car engine emissions. In: Rondia D, Cooke M & Haroz RK ed. Mobile source emissions including polycyclic organic species. Dordrecht, D. Reidel, pp. 197–210.
Nishioka MG, Howard CC, Contos DA, Ball LM & Lewtas J (1988) Detection of hydroxylated nitro aromatic and hydroxylated nitro polycyclic aromatic compounds in an ambient air particulate extract using bioassay-directed fractionation. Environ Sci Technol, 22: 908–915.
NIST (2000) SRM 1975 diesel particle extract (dichloromethane extract of diesel particulate matter collected from an industrial diesel-powered forklift). Gaithersburg, Maryland, National Institute of Standards and Technology (http://srmcatalog.nist.gov/srmcatalog/tables/109-2.htm).
Nohmi T, Yamada M, Matsui M, Matsui K, Watanabe M & Sofuni T (1995) Involvement of umuDCST genes in nitropyrene-induced-CG frameshift mutagenesis at the repetitive CG sequence in the hisD3052 allele of Salmonella typhimurium. Mol Gen Genet, 247: 7–16.
NTP (1996) NTP technical report on toxicity studies of 1-nitropyrene. Administered by inhalation to F344/N rats. Research Triangle Park, North Carolina, US Department of Health and Human Services, National Toxicology Program, 62 pp. (Toxicity Report Series No. 34).
Obana H & Nishimune T (1993) Genotoxicity of environmental mutagens in a Drosophila DNA-repair test. Jpn J Toxicol Environ Health, 39: 577–581.
Oberly TJ, Bewsey BJ & Probst GS (1984) An evaluation of the L5178Y TK+/– mouse lymphoma forward mutation assay using 42 chemicals. Mutat Res, 125: 291–306.
Oberly TJ, Rexroat MA, Bewsey BJ, Richardson KK & Michaelis KC (1990) An evaluation of the CHO/HGPRT mutation assay involving suspension cultures and soft agar cloning: results for 33 chemicals. Environ Mol Mutagen, 16: 260–271.
Oberly TJ, Hoffman WP & Garriott ML (1996) An evaluation of the twofold rule for assessing a positive response in the L5178Y TK +/– mouse lymphoma assay. Mutat Res, 369: 221–232.
Oda Y, Shimada T, Watanabe M, Ishidate M & Nohmi T (1992) A sensitive umu test system for the detection of mutagenic nitroarenes in Salmonella typhimurium NM1011 having a high nitroreductase activity. Mutat Res, 272: 91–99.
Oda Y, Yamazaki H, Watanabe M, Nohmi T & Shimada T (1993) Highly sensitive umu test system for the detection of mutagenic nitroarenes in Salmonella typhimurium NM3009 having high O-acetyltransferase and nitroreductase activities. Environ Mol Mutagen, 21: 357–364.
Oda Y, Yamazaki H, Thier R, Ketterer B, Guengerich FP & Shimada T (1996) A new Salmonella typhimurium NM5004 strain expressing rat glutathione S-transferase 5-5: Use in detection of genotoxicity of dihaloalkanes using an SOS/umu test system. Carcinogenesis, 17: 297–302.
Odagiri Y, Adachi S, Katayama H, Matsushita H & Takemoto K (1986) Carcinogenic effects of a mixture of nitropyrenes in F344 rats following its repeated oral administrations. In: Ishinishi N, Koizumi A, McClellan RO & Stöber W ed. Carcinogenic and mutagenic effects of diesel engine exhaust. Amsterdam, Elsevier Science Publishers, pp. 291–307.
Odagiri Y, Katayama H, Matsushita H & Takemoto K (1993) [Transplacental carcinogenicity of a mixture of mono- and di-nitropyrenes in B6C3F1 mice.] Taiki Osen Gakkaishi, 28: 99–106 (in Japanese).
O’Donovan MR (1990) 1,8-Dinitropyrene: comparative mutagenicity in Chinese hamster V79 and CHO cells. Mutagenesis, 5: 275–277.
OEHHA (1994) Part B. Health assessment. In: Benzo[a]pyrene as a toxic air contaminant. Sacramento, California, California Environmental Protection Agency, Air Resources Board, Office of Environmental Health Hazard Assessment [cited in Collins et al., 1998].
Oehme M, Mano S & Stray HJ (1982) Determination of nitrated polycyclic hydrocarbons in aerosols using capillary gas chromatography combined with different electron capture detection methods. J High Resol Chromatogr, 5: 417–423.
Ogawa K, Imaida K, Masui T, Kawabe M, Hasegawa R, Kato K, Ito N & Shirai T (1996) Chemically induced lung and forestomach neoplasias in transgenic mice carry mutant forms of the human c-Ha-ras transgene. Carcinogenesis, 17: 341–345.
Ohe T (1984) Mutagenicity of photochemical reaction products of polycyclic aromatic hydrocarbons with nitrite. Sci Total Environ, 39: 161–175.
Ohe T (1996) Evaluation of SOS-inducing activity with an O-acetyltransferase-overexpressing strain Salmonella typhimurium NM2009 for municipal river water and the identification of 1-nitropyrene. Water Sci Technol, 33: 313–320.
Ohgaki H, Matsukura N, Morino K, Kawachi T, Sugimura T, Morita K, Tokiwa H & Hirota T (1982) Carcinogenicity in rats of the mutagenic compounds 1-nitropyrene and 3-nitrofluoranthene. Cancer Lett, 15: 1–7.
Ohgaki H, Negishi C, Wakabayashi K, Kusama K, Sato S & Sugimura T (1984) Induction of sarcomas in rats by subcutaneous injection of dinitropyrenes. Carcinogenesis, 5: 583–585.
Ohgaki H, Hasegawa H, Kato T, Negishi C, Sato S & Sugimura T (1985) Absence of carcinogenicity of 1-nitropyrene, correction of previous results, and new demonstration of carcinogenicity of 1,6-dinitropyrene in rats. Cancer Lett, 25: 239–245.
Ohnishi Y, Kinouchi T, Tsutsui H, Uejima M & Nishifuji K (1986) Mutagenic nitropyrenes in foods. In: Hayashi Y, Nagao M, Sugimura T, Takayama S, Tomatis L, Wattenberg LW & Wogan GN ed. Diet, nutrition and cancer. Tokyo, Japan Scientific Societies Press, pp. 107–118.
Ohta T, Nakamura N, Moriya M, Shirasu Y & Kada T (1984) The SOS-inducing activity of chemical mutagens in Escherichia coli. Mutat Res, 131: 101–109.
Orr JC, Bryant DW, McCalla DR & Quilliam MA (1985) Dinitropyrene-resistant Salmonella typhimurium are deficient in an acetyl-CoA acetyltransferase. Chem-Biol Interact, 54: 281–288.
Otofuji T, Horikawa K, Maeda T, Sano N, Izumi K, Otsuka H & Tokiwa H (1987) Tumorigenicity test of 1,3- and 1,8-dinitropyrene in BALB/c mice. J Natl Cancer Inst, 79: 185–188.
Paige R, Wong V & Plopper C (1997) Dose-related airway-selective epithelial toxicity of 1-nitronaphthalene in rats. Toxicol Appl Pharmacol, 147: 224–233.
Paige R, Wong V & Plopper C (1998) Acute ultrastructural changes in airway epithelium following exposure to 1-nitronaphthalene. Fed Am Soc Exp Biol J, 12(5): A786.
Painter RB & Howard R (1982) The HeLa DNA-synthesis inhibition test as a rapid screen for mutagenic carcinogens. Mutat Res, 92: 427–437.
Paputa-Peck MC, Marano RS, Schuetzle D, Riley TL, Hampton CV, Prater TJ, Skewes LM, Jensen TE, Ruehle PH, Bosch LC & Duncan WP (1983) Determination of nitrated polynuclear aromatic hydrocarbons in particulate extracts by capillary column gas chromatography with nitrogen selective detection. Anal Chem, 55: 1946–1954.
Parton JW & Garriott ML (1997) An evaluation of micronucleus induction in bone marrow and in hepatocytes isolated from collagenase perfused liver or from formalin-fixed liver using four-week-old rats treated with known clastogens. Environ Mol Mutagen, 29: 379–385.
Paschke T, Hawthorne SB, Miller DJ & Wenclawiak B (1992) Supercritical fluid extraction of nitrated polycyclic aromatic hydrocarbons and polycyclic aromatic hydrocarbons from diesel exhaust particulate matter. J Chromatogr, 609: 333–340.
Pataky GM, Baumgard KJ, Gratz LD, Bagley ST, Leddy DG & Johnson JH (1994) Effects of an oxidation catalytic converter on regulated and unregulated diesel emissions. Warrendale, Pennsylvania, Society of Automotive Engineers, pp. 129–144 (SAE Paper SP-1020).
Patton JD, Maher VM & McCormick JJ (1986) Cytotoxic and mutagenic effects of 1-nitropyrene and 1-nitrosopyrene in diploid human fibroblasts. Carcinogenesis, 7: 89–93.
Pederson TC & Siak JS (1981) The role of nitroaromatic compounds in the direct-acting mutagenicity of diesel particle extracts. J Appl Toxicol, 1: 54–60.
Pederson TC, Siak JS & Gibson TL (1984) Measurement of dinitropyrene isomers and mutagenic activity in diesel exhaust and airborne particulate. Warren, Michigan, General Motors Research Laboratories (GMR-4698).
Peterson FJ, Mason RP, Hovsepian J & Holtzman JL (1979) Oxygen-sensitive and -insensitive nitroreduction by Escherichia coli and rat hepatic microsomes. J Biol Chem, 254(10): 4009–4014.
Phousongphouang PT, Grosovsky AJ, Eastmond DA, Covarrubias M & Arey J (2000) The genotoxicity of 3-nitrobenzanthrone and the nitropyrene lactones in human lymphoblasts. Mutat Res, 472(1–2): 93–103.
Pienta RJ (1980) Evaluation and relevance of the Syrian hamster embryo cell system. In: Williams GM, Kroes R, Waaijers HW & van de Poll KW ed. The predictive value of short-term screening tests in carcinogenicity evaluation. Amsterdam, Elsevier, pp. 149–169.
Pitts JN Jr (1983) Formation and fate of gaseous and particulate mutagens and carcinogens in real and simulated atmospheres. Environ Health Perspect, 47: 115–140.
Pitts JN Jr, van Cauwenberghe KA, Grosjean D, Schmid JP, Fitz DR, Belser WLJ, Knudson GB & Hynds PM (1978) Atmospheric reactions of polycyclic aromatic hydrocarbons: Facile formation of mutagenic nitro derivatives. Science, 202: 515–519.
Pitts JN Jr, Lokensgard DM, Harger W, Fisher TS, Mejia V, Schuler JJ, Scorziell GM & Katzenstein YA (1982) Mutagens in diesel exhaust particulate identification and direct activities of 6-nitrobenzo(a)pyrene, 9-nitroanthracene, 1-nitropyrene and 5H-phenanthro(4,5-bcd)pyran-5-one. Mutat Res, 103: 241–249.
Pitts JN Jr, Zielinska B & Harger WP (1984) Isomeric mononitrobenzo[a]pyrenes: synthesis, identification and mutagenic activities. Mutat Res, 140: 81–85.
Pitts JN Jr, Sweetman JA, Zielinska B, Winer AM & Atkinson R (1985a) Determination of 2-nitrofluoranthene and 2-nitropyrene in ambient particulate organic matter: Evidence for atmospheric reactions. Atmos Environ, 19: 1601–1608.
Pitts JN Jr, Zielinska B, Sweetman JA, Atkinson R & Winer AM (1985b) Reactions of adsorbed pyrene and perylene with gaseous N2O5 under stimulated atmospheric conditions. Atmos Environ, 19: 911–915.
Pitts JN Jr, Sweetman JA, Zielinska B, Atkinson R, Wi AM & Harger WP (1985c) Formation of nitroarenes from the reaction of polycyclic aromatic hydrocarbons with dinitrogen pentaoxide. Environ Sci Technol, 19: 1115–1121.
Pitts JN Jr, Atkinson R, Sweetman JA & Zielinska B (1985d) The gas-phase reaction of naphthalene with N2O5 to form nitronaphthalenes. Atmos Environ, 19: 701–705.
Poiley JA, Raineri R & Pienta RJ (1979) Use of hamster hepatocytes to metabolize carcinogens in an in vitro bioassay. J Natl Cancer Inst, 63: 519–524.
Pothuluri JV, Evans FE, Heinze TM & Cerniglia CE (1994) Fungal metabolism of 3-nitrofluoranthene. J Toxicol Environ Health, 42: 209–218.
Pothuluri JV, Evans FE, Heinze TM, Fu PP & Cerniglia CE (1996) Fungal metabolism of 2-nitrofluorene. J Toxicol Environ Health, 47: 587–599.
Pothuluri JV, Doerge DR, Churchwell MI, Fu PP & Cerniglia CE (1998a) Fungal metabolism of nitrofluoranthenes. J Toxicol Environ Health, 53: 153–174.
Pothuluri JV, Sutherland JB, Freeman JP & Ce CE (1998b) Fungal biotransformation of 6-nitrochrysene. Appl Environ Microbiol, 64(8): 3106–3109.
Pothuluri JV, Freeman JP, Fu PP & Cerniglia CE (1999a) Biotransformation of 1-nitrobenzo[e]pyrene by the fungus Cunninghamella elegans. J Ind Microbiol Biotechnol, 22(1): 52–57.
Pothuluri JV, Freeman JP, Fu PP & Cerniglia CE (1999b) Fungal biotransformation of 9-nitroanthracene. Abstr Gen Meet Am Soc Microbiol, 99: 567.
Price RJ, Renwick AB, Wield PT, Beamand JA & Lake BG (1995) Toxicity of 3-methylindole, 1-nitronaphthalene and paraquat in precision-cut rat lung slices. Arch Toxicol, 69: 405–409.
Prinn R, Cunnold D, Simmonds P, Alyea F, Boldi R, Crawford A, Fraser P, Gutzler D, Hartley D, Rosen R & Rasmussen R (1992) Global average concentration and trend for hydroxyl radicals deduced from ALE/GAGE trichlorethane (methyl chloroform data for 1978–1990). J Geophys Res, 97(2): 445–461.
Probst GS, McMahon RE, Hill LE, Thompson CZ, Epp JK & Neal SB (1981) Chemically-induced unscheduled DNA synthesis in primary rat hepatocyte cultures: a comparison with bacterial mutagenicity using 218 compounds. Environ Mutagen, 3: 11–32.
Purchase IFH, Longstaff E, Ashby J, Styles JA, Anderson D, Lefevre PA & Westwood FR (1978) An evaluation of 6 short-term tests for detecting organic chemical carcinogens. Br J Cancer, 37: 873–903.
Purohit V & Basu AK (2000) Mutagenicity of nitroaromatic compounds. Chem Res Toxicol, 13(8): 673–692.
Pusztai Z, Selypes A & Ember I (1998) Short-term effects of 1-nitropyrene on chromosomes and on oncogene/tumor suppressor gene expression in vivo. Anticancer Res, 18: 4489–4492.
Quillardet P, de Bellecombe C & Hofnung M (1985) The SOS chromotest, a colorimetric bacterial assay for genotoxins: validation study with 83 compounds. Mutat Res, 147: 79–95.
Rafii F, Selby AL, Newton RK & Ce CE (1994) Reduction and mutagenic activation of nitroaromatic compounds by a Mycobacterium sp. Appl Environ Microbiol, 60: 4263–4267.
Ramdahl T & Urdal K (1982) Determination of nitrated polycyclic aromatic hydrocarbons by fused silica capillary gas chromatography/negative ion chemical ionization mass spectrometry. Anal Chem, 54: 2256–2260.
Ramdahl T, Becher G & Björseth A (1982) Nitrated polycyclic aromatic hydrocarbons in urban air particles. Environ Sci Technol, 16: 861–865.
Ramdahl T, Zielinska B, Arey J, Atkinson R, Winer AM & Pitts JN Jr (1986) Ubiquitous occurrence of 2-nitrofluoranthene and 2-nitropyrene in air. Nature, 321: 425–427.
Rasmussen RE, Do DH, Kim TS & Dearden LC (1986) Comparative cytotoxicity of naphthalene and its monomethyl- and mononitro-derivatives in the mouse lung. J Appl Toxicol, 6: 13–20.
Rippe R & Pott F (1989) Kanzerogenitätsuntersuchungen von Nitro-PAH (Nitroarenen) im Hinblick auf ihre Bedeutung für die krebserzeugende Wirkung von Dieselmotorabgas. In: Gesellschaft z. Förderung d. Lufthygiene und Silikoseforschung e. V. ed. Umwelthygiene Bd. 21. Medizinisches Institut für Umwelthygiene, Jahresbericht 1988/89. Düsseldorf, Stefan W. Albers, pp. 65–89.
Robbat A Jr, Corso NP & Doherty PJ (1986) Gas chromatographic chemiluminescent detection and evaluation of predictive models for identifying nitrated polycyclic aromatic hydrocarbons in a diesel fuel particulate extract. Anal Chem, 58: 2078–2084.
Robertson DJ, Groth RH & Blasko TJ (1980) Organic content of particulate matter in turbine engine exhaust. J Air Pollut Control Assoc, 30: 261–266.
Roscher E & Wiebel FJ (1992) Genotoxicity of 1,3- and 1,6-dinitropyrene: induction of micronuclei in a panel of mammalian test cell lines. Mutat Res, 278: 11–17.
Rosenkranz EJ, McCoy EC, Mermelstein R & Rosenkranz HS (1982) Evidence for the existence of distinct nitroreductases in Salmonella typhimurium: Roles in mutagenesis. Carcinogenesis, 3: 121–123.
Rosenkranz HS (1996) Mutagenic nitroarenes, diesel emissions, particulate-induced mutations and cancer: an essay on cancer-causation by a moving target. Mutat Res, 367: 65–72.
Rosenkranz HS & Mermelstein R (1983) Mutagenicity and genotoxicity of nitroarenes: All nitro-containing chemicals were not created equal. Mutat Res, 114: 217–267.
Rosenkranz HS & Poirier LA (1979) Evaluation of the mutagenicity and DNA-modifying activity of carcinogens and noncarcinogens in microbial systems. J Natl Cancer Inst, 62: 873–891.
Rosenkranz HS, McCoy EC, Sanders DR, Butler M, Kiriazides DK & Mermelstein R (1980) Nitropyrenes: Isolation, identification, and reduction of mutagenic impurities in carbon black and toners. Science, 209: 1039–1043.
Rosenkranz HS, McCoy EC, Mermelstein R & Speck WT (1981) A cautionary note on the use of nitroreductase-deficient strains of Salmonella typhimurium for the detection of nitroarenes as mutagens in complex mixtures including diesel exhausts. Mutat Res, 91(2): 103–105.
Rosenkranz HS, McCoy EC, Frierson M & Klopman G (1985) The role of DNA sequence and structure of the electrophile on the mutagenicity of nitroarenes and arylamine derivatives. Environ Mutagen, 7: 645–653.
Rossmann TG, Molina M, Meyer L, Boone P, Klein CB, Wang Z, Li F, Lin WC & Kinney PL (1991) Performance of 133 compounds in the lambda prophage induction endpoint of the microscreen assay and a comparison with S. typhimurium mutagenicity and rodent carcinogenicity assays. Mutat Res, 260: 349–367.
Roy AK, El-Bayoumy K & Hecht SS (1989) 32P-postlabeling analysis of 1-nitropyrene–DNA adducts in female Sprague-Dawley rats. Carcinogenesis, 10: 195–198.
Roy AK, Upadhyaya P, Evans FE & El-Bayoumy K (1991) Structural characterization of the major adducts formed by reaction of 4,5-epoxy-4,5-dihydro-1-nitropyrene with DNA. Carcinogenesis, 12: 577–581.
Ruiz-Rubio M, Hera C & Pueyo C (1984) Comparison of a forward and a reserve mutation assay in Salmonella typhimurium measuring L-arabinose resistance and histidine prototrophy. EMBO (Eur Mol Biol Org) J, 3: 1435–1440.
Saito K, Kamataki T & Kato R (1984a) Participation of cytochrome P-450 in reductive metabolism of 1-nitropyrene by rat liver microsomes. Cancer Res, 44: 3169–3173.
Saito K, Mita S, Kamataki T & Kato R (1984b) DNA single-strand breaks by nitropyrenes and related compounds in Chinese hamster V79 cells. Cancer Lett, 24: 121–127.
Sakitani T & Suzuki Y (1986) Mutagenic activities of air pollutants. Part 2. Mutagenic activities of air pollutants observed by micronucleus test. Tokyo Jikeikai Med J, 101: 259–266.
Sala M, Lasne C, Lu Y & Chouroulinkov I (1987) Morphological transformation in three mammalian cell systems following treatment with 6-nitrochrysene and 6-nitrobenzo(a)pyrene. Carcinogenesis, 8: 503–507.
Salmeen IT, Pero AM, Zator R, Schuetzle D & Riley TL (1984) Ames assay chromatograms and the identification of mutagens in diesel particle extracts. Environ Sci Technol, 18: 375–382.
Sanders DR, Temcharoen P & Thilly WG (1983) 1,8-Dinitropyrene mutagenicity in bacteria and human cells. Environ Mutagen, 5: 457.
Sangaiah R, Ramachadran P, Zhang J & Ball LM (1996) Synthesis and biological activity of metabolites of environmental nitroarenes. Polycycl Aromat Comp, 11: 283–290.
Sasaki J, Arey J & Harger W (1995) Formation of mutagens from the photooxidations of 2–4-ring PAH. Environ Sci Technol, 29: 1324–1335.
Sasaki J, Arey J & Atkinson DA (1996) Gene specific mutagenicity induced in human lymphoblasts by atmospheric reaction products of naphthalene. Environ Mol Mutagen, 27(Suppl): 57.
Sasaki JC, Arey J, Eastmond DA, Parks KK & Grosovsky AJ (1997a) Evidence for oxidative metabolism in the genotoxicity of 2-nitronaphthalene and 2-nitrodibenzopyranone. Environ Mol Mutagen, 28(Suppl): 44.
Sasaki JC, Arey J, Eastmond DA, Parks KK & Grosovsky AJ (1997b) Genotoxicity induced in human lymphoblasts by atmospheric reaction products of naphthalene and phenanthrene. Mutat Res, 393: 23–35.
Sasaki JC, Arey J, Eastmond DA, Parks KK, Phousongphouang PT & Grosovsky AJ (1999) Evidence for oxidative metabolism in the genotoxicity of the atmospheric reaction product 2-nitronaphthalene in human lymphoblastoid cell lines. Mutat Res, 445: 113–125.
Sato T, Kato K, Ose Y, Nagase H & Ishikawa T (1985) Nitroarenes in Suimon river sediment. Mutat Res, 157: 135–143.
Sauer J-M & Sipes IG (1995) Modulation of chemical-induced lung and liver toxicity by all-trans-retinol in the male Sprague-Dawley rat. Toxicology, 105: 237–249.
Sauer JM, Hooser SB & Sipes IG (1995) All-trans-retinol alteration of 1-nitronaphthalene induced pulmonary and hepatic injury by modulation of associated inflammatory responses in the male Sprague-Dawley rat. Toxicol Appl Pharmacol, 133: 139–149.
Sauer JM, Eversole RR, Lehmann CL, Johnson DE & Beuving LJ (1997) An ultrastructural evaluation of acute 1-nitronaphthalene induced hepatic and pulmonary toxicity in the rat. Toxicol Lett, 90: 19–27.
Schairer LA & Sautkulis RC (1982) Detection of ambient levels of mutagenic atmospheric pollutants with the higher plant Tradescantia. In: Klekowski EJ Jr ed. Environmental mutagenesis, carcinogenesis and plant biology. New York, Praeger, pp. 154–194.
Scheepers PTL (1994) Monitoring occupational exposure to diesel exhaust. Thesis, University of Nijmegen, Nijmegen, 238 pp.
Scheepers PTJ & Bos RP (1992a) Combustion of diesel fuel from a toxicological perspective. I. Origin of incomplete combustion products. Int Arch Occup Environ Health, 64: 149–161. (sec 3)
Scheepers PTJ & Bos RP (1992b) Combustion of diesel fuel from a toxicological perspective. II. Toxicity. Int Arch Occup Environ Health, 64:163–177.
Scheepers PTJ, Velders DD, Martens MHJ, Noordhoek J & Bos RP (1994a) Gas chromatographic–mass spectrometric determination of nitro polycyclic aromatic hydrocarbons in airborne particulate matter from workplace atmospheres contaminated with diesel exhaust. J Chromatogr A, 677: 107–121.
Scheepers PTJ, Thuis HJTM, Martens MHJ & Bos RP (1994b) Assessment of occupational exposure to diesel exhaust. The use of an immunoassay for the determination of urinary metabolites of nitroarenes and polycyclic aromatic hydrocarbons. Toxicol Lett, 72: 191–198.
Scheepers PTJ, Velders DD, Steenwinkel MJST, Van Delft JHM, Driessen W, Straetemans MME, Baan RA, Koopman JP, Noordhoek J & Bos RP (1994c) Role of the intestinal microflora in the formation of DNA and haemoglobin adducts in rats treated with 2-nitrofluorene and 2-aminofluorene by gavage. Carcinogenesis, 15: 1433–1441.
Scheepers PTJ, Straetemans MME, Koopman JP & Bos RP (1994d) Nitroreduction and formation of hemoglobin adducts in rats with a human intestinal microflora. Environ Health Perspect, 102(Suppl 6): 39–41.
Scheepers PTJ, Fijneman PHS, Beenakkers MFM, deLepper AJGM, Thuis HJTM, Stevens D, VanRooij JGM, Noordhoek J & Bos RP (1995a) Immunochemical detection of metabolites of parent and nitro polycyclic aromatic hydrocarbons in urine samples from persons occupationally exposed to diesel exhaust. Fresenius J Anal Chem, 351: 660–669.
Scheepers PTJ, Martens MHJ, Velders DD, Fijneman P, Van Kerkhoven M, Noordhoek J & Bos RP (1995b) 1-Nitropyrene as a marker for the mutagenicity of diesel exhaust-derived airborne particulate matter in workplace atmospheres. Environ Mol Mutagen, 25: 134–147.
Scheepers PTJ, Martens MHJ, Kimmel J-PF, Velders DD & Bos RP (1999) Determination of inhalation exposure to 1-nitropyrene in workplace atmospheres and urban dwellings. Polycycl Aromat Comp, 17: 267–275.
Scheepers PTJ, Anzion R & Bos RP (2001) GC–MS in occupational and environmental health risk assessment with some applications related to environmental and biological monitoring of 1-nitropyrene. In: Niessen WMA ed. Current practice of GC–MS. New York, Marcel Dekker, pp. 199–227.
Scheepers PTJ, Coggon D, Knudsen LE, Anzion R, Autrup H, Bogovski S, Bos RP, Dahmann D, Farmer P, Martin EA, Micka V, Muzyka V, Neumann HG, Poole J, Schmidt-Ott A, Seiler F, Volf J & Zwirner-Baier I (2002) BIOMarkers for Occupational Diesel exhaust Exposure Monitoring (BIOMODEM) — a study in underground mining. Toxicol Lett, 134: 305–317.
Schilhabel J & Levsen K (1989) Identification of nitrated polycyclic hydrocarbons in diesel particulate extracts by negative ion chemical ionization and tandem mass spectrometry. Fresenius Z Anal Chem, 333: 800–805.
Schleibinger H, Leberl C & Rüden H (1989) Nitrierte polyzyklische aromatische Kohlenwasserstoffe (Nitro-PAH)im Schwebestaub der Außenluft. 2. Mitteilung: Vergleich der Mutagenität von Nitro-PAH und Luftstaubextrakten im Ames-, SOS-Reparatur-Induktion- und SCE-Test. Zentralbl Hyg Umweltmed, 188: 421–438.
Schlemitz S & Pfannhauser W (1996a) Monitoring of nitropolycyclic aromatic hydrocarbons in food using gas chromatography. Z Lebensm-Unters Forsch, 203: 61–64.
Schlemitz S & Pfannhauser W (1996b) Analysis of nitro-PAHs in food matrices by on-line reduction and high performance liquid chromatography. Food Addit Contam, 13: 969–977.
Schlemitz S & Pfannhauser W (1997) Supercritical fluid extraction of mononitrated polycyclic aromatic hydrocarbons from tea — correlation with the PAH concentration. Z Lebensm-Unters Forsch, 205: 305–310.
Schmid C, Reifferscheid G, Zahn RK & Bachmann M (1997) Increase of sensitivity and validity of the SOS/umu-test after replacement of the beta-galactosidase reporter gene with luciferase. Mutat Res, 394: 9–16.
Schuetzle D (1983) Sampling of vehicle emissions for chemical analysis and biological testing. Environ Health Perspect, 47: 65–80.
Schuetzle D & Frazier JA (1986) Factors influencing the emission of vapor and particulate phase components from diesel engines. In: Ishinishi N, Koizumi A, McClellan RO & Stöber W ed. Carcinogenic and mutagenic effects of diesel engine exhaust. Amsterdam, Elsevier Science Publishers B.V., pp. 41–63.
Schuetzle D & Lewtas J (1986) Bioassay-directed chemical analysis in environmental research. Anal Chem, 58: 1060A–1075A.
Schuetzle D & Perez JM (1983) Factors influencing the emissions of nitrated-polynuclear aromatic hydrocarbons (nitro-PAH) from diesel engines. J Air Pollut Control Assoc, 33: 751–755.
Schuetzle D, Lee FSC, Prater TJ & Tejada SB (1981) The identification of polynuclear aromatic hydrocarbons (PAHs) derivatives in mutagenic fractions of diesel particulate extracts. Int J Environ Anal Chem, 9: 93–144.
Schuetzle D, Riley TL, Prater TJ, Harvey TM & Hunt DF (1982) Analysis of nitrated polycyclic aromatic hydrocarbons in diesel particulates. Anal Chem, 54: 265–271.
Schultz TW & Moulton BA (1985) Structure–activity relationships for nitrogen-containing aromatic molecules. Environ Toxicol Chem, 4: 353–359..
Scribner JD, Fisk SR & Scribner NK (1979) Mechanisms of action of carcinogenic aromatic amines: An investigation using mutagenesis in bacteria. Chem-Biol Interact, 26: 11–25.
Seidel A, Dahmann D, Krekeler H & Jacob J (2002) Biomonitoring of polycyclic aromatic compounds in the urine of mining workers occupationally exposed to diesel exhaust. Int J Hyg Environ Health, 204: 333–338.
Sera N (1998) Structure–activity relationship between mutagenicity and NO2 substitution of nitroarenes, and the presence of mutagens/carcinogens in excised lung cancer specimens from Japan and China. Environ Mutagen Res Commun, 20(2): 97–105.
Sera N, Kai M, Horikawa K, Fukuhara K, Miyata N & Tokiwa H (1991) Detection of 3,6-dinitrobenzo(a)pyrene in airborne particulates. Mutat Res, 263: 27–32.
Sera N, Fukuhara K, Miyata N & Tokiwa H (1994) Detection of nitro-azabenzo[a]pyrene derivatives in the semivolatile phase originating from airborne particulate matter, diesel and gasoline vehicles. Mutagenesis, 9: 47–52.
Sera N, Fukuhara K, Miyata N & Tokiwa H (1996) Mutagenicity of nitrophenanthrene derivatives for Salmonella typhimurium: effects of nitroreductase and acetyltransferase. Mutat Res, 349(1): 137–144.
Shah AB, Rowland IR & Combes RD (1991) Inhibition of dinitropyrene mutagenicity in vitro and in vivo using Salmonella typhimurium and the intrasanguinous host-mediated assay. Mutat Res, 253: 181–191. (sec 7)
Shane BS & Winston GW (1997) Activation and detoxification of dinitropyrenes by cytosol and microsomes from Aroclor-pretreated rats in the Ames and umu assays. Environ Mol Mutagen, 30: 303–311.
Shane BS, Squadrito GL, Church DF & Pryor WA (1991) Comparative mutagenicity of nitrofluoranthenes in Salmonella typhimurium TA98, TA98NR, and TA98/L8DNP6. Environ Mol Mutagen, 17: 130–138.
Shane BS, Many CR & Longstreth DJ (1993) Activation and detoxification of nitro-aromatic compounds by plant tissue culture cells. In: Gorsuch JW ed. Environmental toxicology and risk assessment, vol. 2. Symposium on Environmental Toxicology and Risk Assessment: Aquatic, Plant, and Terrestrial, Pittsburgh, Pennsylvania, 26–30 April 1992. Philadelphia, Pennsylvania, American Society for Testing and Materials, pp. 346–361 (ASTM Special Technical Publication 1216).
Sharp C (2000) The effect of biodiesel fuels on transient emissions from modern diesel engines, part II, unregulated emissions and chemical characterization. Warrendale, Pennsylvania, Society of Automotive Engineers (SAE Paper (sec2000-01-1968).
Shelby MD & Stasiewicz S (1984) Chemicals showing no evidence of carcinogenicity in long-term, two-species rodent studies: The need for short-term test data. Environ Mutagen, 6: 871–878.
Sheu CW, Dobras SN, Rodriguez I, Lee JK & Fu PP (1994) Transforming activity of selected polycyclic aromatic hydrocarbons and their nitro-derivatives in BALB/3T3 A31-1-1 cells. Food Chem Toxicol, 32: 611–615.
Shimada T, Oda Y, Yamazaki H, Mimura M & Guengerich FP (1994) SOS function test for studies of chemical carcinogenesis using Salmonella typhimurium. In: Adolph KW ed. Methods in molecular genetics, vol. 5, Gene and chromosome analysis, part C. San Diego, California, Academic Press, pp. 342–355.
Shimada T, Hayes CL, Yamazaki H, Amin S, Hecht SS, Gu FP & Sutter TR (1996) Activation of chemically diverse procarcinogens by human cytochrome P-450 1B1. Cancer Res, 56: 2979–2984.
Sigvardson KW & Birks JW (1984) Detection of nitro-polycyclic aromatic hydrocarbons in liquid chromatography by zinc reduction and peroxyoxalate chemiluminescence. J Chromatogr, 316: 507–518.
Silvers KJ, Chazinski T, McManus ME, Bauer SL, Gonzalez FJ, Gelboin HV, Maurel P & Howard PC (1992) Cytochrome P 450 3A4 (nifedipine oxidase) is responsible for the C-oxidative metabolism of 1-nitropyrene in human liver microsomal samples. Cancer Res, 52: 6237–6243.
Silvers KJ, Eddy EP, McCoy EC, Rosenkranz HS & Howard PC (1994) Pathways for the mutagenesis of 1-nitropyrene and dinitropyrenes in the human hepatoma cell line HepG2. Environ Health Perspect, 102(Suppl 6): 195–200.
Silvers KJ, Couch LH, Rorke EA & Howard PC (1997) Role of nitroreductases but not cytochromes P450 in the metabolic activation of 1-nitropyrene in the HepG2 human hepatoblastoma cell line. Biochem Pharmacol, 54: 927–936.
Simmon VE (1979a) In vitro mutagenicity assays of chemical carcinogens and related compounds with Salmonella typhimurium. J Natl Cancer Inst, 62: 893–899.
Simmon VF (1979b) In vitro assays for recombinogenic activity of chemical carcinogens and related compounds with Saccharomyces cerevisiae D3. J Natl Cancer Inst, 62: 901–909.
Simmon VF, Rosenkranz HS, Zeiger E & Poirier LA (1979) Mutagenic activity of chemical carcinogens and related compounds in the intraperitoneal host-mediated assay. J Natl Cancer Inst, 62: 911–918.
Sinks GD, Schultz TW & Hunter RS (1997) UVb-induced toxicity of PAHs: Effects of substituents and heteroatom substitution. Bull Environ Contam Toxicol, 59: 1–8.
Sjögren M, Li H, Rannug U & Westerholm R (1996) Multivariate analysis of exhaust emissions from heavy-duty diesel fuels. Environ Sci Technol, 30: 38–49.
Smith BA, Heflich RH, Ohnishi Y, Ohuchida A, Kinouchi T, Thornton-Manning JR & Beland FA (1990a) DNA adduct formation by 1-nitropyrene 4,5- and 9,10-oxide. In: Howard PC, Hecht SS & Beland FA ed. Nitroarenes — Occurrence, metabolism, and biological impact. New York, Plenum Press, pp. 181–187.
Smith BA, Korfmacher WA & Beland FA (1990b) DNA adduct formation in target tissues of Sprague-Dawley rats, CD-1 mice and A/J mice following tumorigenic doses of 1-nitropyrene. Carcinogenesis, 11: 1705–1710.
Smith BA, Fullerton NF, Aidoo A, Heflich RH & Beland FA (1993) DNA adduct formation in relation to lymphocyte mutations and lung tumor induction in F344 rats treated with the environmental pollutant 1,6-dinitropyrene. Environ Health Perspect, 99: 277–280.
Smith BA, Fullerton NF, Heflich RH & Beland FA (1995) DNA adduct formation and T-lymphocyte mutation induction in F334 rats implanted with tumorigenic doses of 1,6-dinitropyrene. Cancer Res, 55: 2316–2324.
Smith BA, Manjanatha MG, Pogribny IP, Mittelstaedt RA, Chen T, Fullerton NF, Beland FA & Heflich RH (1997) Analysis of mutations in the K-ras and p53 genes of lung tumors and in the hprt gene of 6-thioguanine-resistant T-lymphocytes from rats treated with 1,6-dinitropyrene. Mutat Res, 379: 61–68.
Sorenson WG, Whong WZ, Simpson JP, Brusick DJ & Ong TM (1983) Genotoxic properties of 2,4,7-trinitro-9-fluorenone. Mutat Res, 118: 167–176.
Spitzer TJ (1993) Selective clean-up for polynuclear aromatic compounds in airborne particles and soil. J Chromatogr, 643: 43–49.
Stanton CA, Chow FL, Phillips DH, Grover PL, Garner RC & Martin CN (1985) Evidence for N-(deoxyguanosin-8-yl)-1-aminopyrene as a major DNA adduct in female rats treated with 1-nitropyrene. Carcinogenesis, 6: 535–538.
Stanton CA, Garner RC & Martin CN (1988) The mutagenicity and DNA base sequence changes induced by 1-nitroso- and 1-nitropyrene in the cI gene of lambda prophage. Carcinogenesis, 9: 1153–1157.
Stärk G, Stauff J, Miltenburger HG & Stumm-Fischer I (1985) Photodecomposition of 1-nitropyrene and other direct-acting mutagens extracted from diesel-exhaust particulates. Mutat Res, 155: 27–33.
Stray H, Mano S, Mikalsen A & Oehme MJ (1984) Selective determination of substituted PAH in aerosols using liquid CO2-extraction, HPLC prefractionation on chemically activated silica, and HRGC combined with negative ion mass spectrometry. J High Resol Chromatogr, 7: 74–82.
Styles JA (1981) Use of BHK 21/CI 13 cells for chemical screening. In: Mishra N, Dunkel V & Mehlman M ed. Advances in modern environmental toxicology, vol. 1, Mammalian cell transformation by chemical carcinogens. Princeton Junction, New Jersey, Senate Press, pp. 85–131.
Sugimura T & Takayama S (1983) Biological actions of nitroarenes in short-term tests on Salmonella, cultured mammalian cells and cultured human tracheal tissues: possible basis for regulatory control. Environ Health Perspect, 47: 171–176.
Sun JD, Wolff RK, Aberman HM & McClellan RO (1983) Inhalation of 1-nitropyrene association with ultrafine insoluble particles or as a pure aerosol: A comparison of deposition and biological fate. Toxicol Appl Pharmacol, 69: 185–198.
Suter W & Jaeger I (1982) Comparative evaluation of different pairs of DNA repair-proficient bacterial tester strains for rapid detection of chemical mutagens and carcinogens. Mutat Res, 97: 1–18.
Suzuki J, Hagino T & Suzuki S (1987) Formation of 1-nitropyrene by photolysis of pyrene in water containing nitrite ion. Chemosphere, 16: 859.
Suzuki J, Meguro SI, Morita O, Hirayama S & Suzuki S (1989) Comparison of in vivo binding of aromatic nitro and amino compounds to rat hemoglobin. Biochem Pharmacol, 38: 3511–3519.
Suzuki H, Enya T & Hisamatsu Y (1997) Synthesis and characterization of some nitrobenzanthrones: Suspected new mutagens in atmospheric environment. Synthesis, 11: 1273–1276.
Sweetman AJ, Zielinska B, Atkinson R, Ramdahl T, Winer AM & Pitts JN Jr (1986) A possible formation pathway for the 2-nitrofluoranthene observed in ambient particulate organic matter. Atmos Environ, 20: 235–238.
Sydbom A, Blomberg A, Parnia S, Stenfors N, Sandstrom T & Dahlen S (2001) Health effects of diesel exhaust emissions. Eur Respir J, 17(4):733–746.
Tahara I, Kataoka K, Kinouchi T & Ohnishi Y (1995) Stability of 1-nitropyrene and 1,6-dinitropyrene in environmental water samples and soil suspensions. Mutat Res, 343: 109–119.
Takahashi Y, Nakagawa J, Hosokawa N, Asano M & Morita M (1995) Identification and determination of organic compounds in a river water. Kankyo Kagaku, 5: 207–214.
Takayama S, Tanaka M, Katoh Y, Terada M & Sugimura T (1983) Mutagenicity of nitropyrenes in Chinese hamster V79 cells. Gann (Jpn J Cancer Res), 74: 338–341.
Takayama S, Ishikawa T, Nakajima H & Sato S (1985) Lung carcinoma induction in Syrian golden hamsters by intratracheal instillation of 1,6-dinitropyrene. Gann (Jpn J Cancer Res), 76: 457–461.
Takemura N, Hashida C & Terasawa M (1974) Carcinogenic action of 5-nitroacenaphthene. Br J Cancer, 30: 481–483.
Tanabe K, Matsushita H, Kuo C-T & Imamiya S (1986) Determination of carcinogenic nitroarenes in airborne particulates by high performance liquid chromatography. J Jpn Soc Air Pollut, 21: 535–544.
Tatsumi K, Kitamura S & Narai N (1986) Reductive metabolism of aromatic nitro compounds including carcinogens by rabbit liver preparations. Cancer Res, 46: 1089–1093.
Taylor MS, Setzer RW & Demarini DM (1995) Examination of the additivity assumption using the spinal and standard Salmonella assays to evaluate binary combinations of mutagens. Mutat Res, 335: 1–14.
Tejada SB, Zweidinger RB & Sigsby JE Jr (1986) Fluorescence detection and identification of nitro derivates of polynuclear aromatic hydrocarbons by on-column catalytic reduction to aromatic amines. Anal Chem, 58: 1827–1834.
Thornton-Manning JR, Chou MW, Fu PP & Heflich RH (1988) Microsomal activation of 1- and 3-nitrobenzo[a]pyrene to mutagens in Chinese hamster ovary cells. Mutagenesis, 3: 233–237.
Thornton-Manning JR, Campbell WL, Hass BS, Chen JJ, Fu PP, Cerniglia CE & Heflich RH (1989) Role of nitroreduction in the synergistic mutational response induced by mixtures of 1- and 3-nitrobenzo[a]pyrene in Salmonella typhimurium. Environ Mol Mutagen, 13: 203–210.
Thornton-Manning JR, Smith BA, Beland FA & Heflich RH (1991a) Mutagenesis and DNA adduct formation by 1-nitropyrene in Chinese hamster ovary cells without exogenous metabolic activation. Toxicol Appl Pharmacol, 109: 538–546.
Thornton-Manning JR, Smith BA, Beland FA & Heflich RH (1991b) S9-mediated metabolism of 1-nitropyrene to a mutagen in Chinese hamster ovary cells by ring oxidation under aerobic conditions and by nitroreduction under anaerobic conditions. Carcinogenesis, 12: 2317–2323.
Tokiwa H & Ohnishi Y (1986) Mutagenicity and carcinogenicity of nitroarenes and their sources in the environment. CRC Crit Rev Toxicol, 17: 23–60.
Tokiwa H & Sera N (2000) Contribution of nitrated polycyclic aromatic hydrocarbons in diesel particles to human cancer induction. Polycycl Aromat Comp, 21: 231–245.
Tokiwa H, Nakagawa R, Morita K & Ohnishi Y (1981a) Mutagenicity of nitro derivatives induced by exposure of aromatic compounds to nitrogen dioxide. Mutat Res, 85: 195–205.
Tokiwa H, Nakagawa R & Ohnishi Y (1981b) Mutagenic assay of aromatic nitro compounds with Salmonella typhimurium. Mutat Res, 91: 321–325.
Tokiwa H, Otofuji T, Horikawa K, Kitamori S, Otsuka H, Manabe Y & Kinouchi T & Ohnishi Y (1984) 1,6-Dinitropyrene: Mutagenicity in Salmonella and carcinogenicity in BALB/c mice. J Natl Cancer Inst, 73: 1359–1363.
Tokiwa H, Nakagawa R & Horikawa K (1985) Mutagenic/carcinogenic agents in indoor pollutants: The dinitropyrenes generated by kerosene heaters and fuel gas and liquified petroleum gas burners. Mutat Res, 157: 39–47.
Tokiwa H, Otofuji T, Nakagawa R, Horikawa K, Maeda T, Sano N, Izumi K & Otsuka H (1986) Dinitro derivatives of pyrene and fluoranthene in diesel emission particulates and their tumorigenicity in mice and rats. In: Ishinishi N, Koizumi A, McClellan RO & Stöber W ed. Carcinogenic and mutagenic effects of diesel engine exhaust. Amsterdam, Elsevier, pp. 253–270.
Tokiwa H, Otofuji T, Horikawa K, Sera N, Nakagawa R, Maeda T, Sano N, Izumi K & Otsuka H (1987a) Induction of subcutaneous tumors in rats by 3,7- and 3,9-dinitrofluoranthene. Carcinogenesis, 8: 1919–1922
Tokiwa H, Nakagawa R, Horikawa K & Ohkubo A (1987b) The nature of the mutagenicity and carcinogenicity of nitrated, aromatic compounds in the environment. Environ Health Perspect, 73: 191–199.
Tokiwa H, Horikawa K, Omura H & Kuroda Y (1988) Mutagenicity in Chinese hamster V79 cells and induction of micronuclei in mice by nitrated fluoranthenes. Exp Oncol, 7: 33–37.
Tokiwa H, Sera N, Kai M, Horikawa K & Ohnishi Y (1990a) The role of nitroarenes in the mutagenicity of airborne particles indoors and outdoors. In: Waters MD, Daniel FB, Lewtas J, Moore MM & Nesnow S ed. Genetic toxicology of complex mixtures. New York, Plenum Press, pp. 165–172.
Tokiwa H, Horikawa K, Sera N, Izumi K, Iwagawa M, Otsuka H, Ohnishi Y, Nakashima A & Nakashima K (1990b) Carcinogenicity of dinitroarenes in rat lung. In: Howard PC, Hecht SS & Beland FA ed. Nitroarenes — Occurrence, metabolism, and biological impact. New York, Plenum Press, pp. 29–37.
Tokiwa H, Sera N, Horikawa K, Nakanishi Y & Shigematu N (1993a) The presence of mutagens/carcinogens in the excised lung and analysis of lung cancer induction. Carcinogenesis, 14: 1933–1938.
Tokiwa H, Horikawa K & Ohnishi Y (1993b) Genetic toxicology and carcinogenicity of mono- and dinitrofluoranthenes. Mutat Res, 297: 181–195.
Tokiwa H, Sera N, Nakashima A, Nakashima K, Nakanishi Y & Sigematu N (1994) Mutagenic and carcinogenic significance and the possible induction of lung cancer by nitro aromatic hydrocarbons in particulate pollutants. Environ Health Perspect, 102(Suppl 4): 107–110.
Tokiwa H, Nakanishi Y, Sera N, Hara N & Inuzuka S (1998a) Analysis of environmental carcinogens associated with the incidence of lung cancer. Toxicol Lett, 99(1): 33–41.
Tokiwa H, Sera N & Nakanishi Y (1998b) Biological action of environmental carcinogens associated with the incidence of lung cancer. Hum Exp Toxicol, 17: 51.
Tomkins BA, Brazell RS, Roth ME & Ostrum VH (1984) Isolation of mononitrated polycyclic aromatic hydrocarbons in particulate matter by liquid chromatography and determination by gas chromatography with the thermal energy analyzer. Anal Chem, 56: 781–786.
Topham JC (1980) Do induced sperm-head abnormalities in mice specifically identify mammalian mutagens rather than carcinogens? Mutat Res, 74: 379–387.
Topinka J, Schwarz L, Kiefer F, Wiebel F, Gajdos O, Vidova P, Dobias L, Fried M, Sram R & Wolff T (1998) DNA adduct formation in mammalian cell cultures by polycyclic aromatic hydrocarbons (PAH) and nitro-PAH in coke oven emission extract. Mutat Res, 419: 91–105.
Törnquist S, Möller L, Gabrielsson J, Gustafsson JA & Toftgard R (1990) 2-Nitrofluorene metabolism in the rat lung. Pharmacokinetic and metabolic effects of beta-naphthoflavone treatment. Carcinogenesis, 11(8): 1249–1254.
Tuominen J, Salomaa S, Pyysalo H, Skyttä E, Tikkanen L, Nurmela T, Sorsa M, Pohjola V, Sauri M & Himberg K (1988) Polynuclear aromatic compounds and genotoxicity in particulate and vapor phases of ambient air: effect of traffic, season, and meteorological conditions. Environ Sci Technol, 22: 1228–1234.
Tyrpien K (1993) TLC identification of some nitrogen derivatives of PAH in airborne particulate matter. J Planar Chromatogr Mod TLC, 6: 413–415.
Tyrpien K, Janoszka B & Bodzek D (1997) Application of planar chromatography to the analysis of polycyclic aromatic hydrocarbons and their derivatives in environmental samples. J Chromatogr A, 774: 111–120.
Ueda O, Kitamura S, Kubo R, Yano Y, Kanzaki Y, Fujimoto T, Tatsumi K & Ohta S (2001a) Metabolism of 2-nitrofluorene, 2-aminofluorene and 2-acylaminofluorene in rat and dog and the role of intestinal bacteria. Xenobiotica, 31(1): 33–49.
Ueda O, Kitamura S, Huruse Y, Saito T, Kanzaki Y, Yano Y, Tatsumi K & Ohta S (2001b) Metabolism of 2-nitrofluorene, an environmental pollutant, and 2-acetylaminofluorene in fish. Bull Environ Contam Toxicol, 66: 371–378.
Upadhyaya P, Roy AK, Fu PP & El-Bayoumy K (1992) Metabolism and DNA binding of 2-nitropyrene in the rat. Cancer Res, 52: 1176–1181.
Upadhyaya P, von Tungeln LS, Fu PP & El-Bayoumy K (1994) In vitro and in vivo metabolism of the carcinogen 4-nitropyrene. Chem Res Toxicol, 7: 690–695.
Urios A, Herrara G, Aleixandre V, Sommer S & Blanco M (1994) Mutability of Salmonella tester strains TA1538 (hisD3052) and Ta1535 (hisG46) containing the UmuD' and UmuC proteins of Escherichia coli. Environ Mol Mutagen, 23: 281–285.
US EPA (2000) Health assessment document for diesel exhaust. Washington, DC, US Environmental Protection Agency, National Center for Environmental Assessment, July, pp. 2-40–2-140 (EPA 600/8-90/057E).
Valencia R, Mason JM, Woodruff RC & Zimmering S (1985) Chemical mutagenesis testing in Drosophila. III. Results of 48 coded compounds tested for the National Toxicology Program. Environ Mutagen, 7: 325–348.
van Bekkum Y (1999) Biomonitoring of exposure to diesel exhaust: Development and application of techniques using 1-nitropyrene as a marker. Dissertation, University of Nijmegen, Nijmegen.
van Bekkum YM, Scheepers PTJ, van den Broek PHH, Velders DD, Noordhoek J & Bos RP (1997) Determination of hemoglobin adducts following oral administration of 1-nitropyrene to rats using gas chromatography–tandem mass spectrometry. J Chromatogr B, Biomed Sci Appl, 701: 19–28.
van Bekkum YM, van den Broek PH, Scheepers PT & Bos RP (1998) Sensitive and selective detection of urinary 1-nitropyrene metabolites following administration of a single intragastric dose of diesel exhaust particles (SRM 2975) to rats. Chem Res Toxicol, 11(11): 1382–1390.
van Bekkum YM, van den Broek PHH, Scheepers PTJ, Noordhoek J & Bos RP (1999) Biological fate of [14C]-1-nitropyrene in rats following intragastric administration. Chem-Biol Interact, 117(1): 15–33.
Vance WA & Levin DE (1984) Structural features of nitroaromatics that determine mutagenic activity in Salmonella typhimurium. Environ Mutagen, 6: 797–811.
Vance WA, Wang YY & Okamoto HS (1987) Disubstituted amino-, nitroso-, and nitrofluorenes: a physicochemical basis for structure–activity relationships in Salmonella typhimurium. Environ Mutagen, 9: 123–141.
van den Braken-van Leersum AM, Tintel C, van’t Zelfde M, Cornelisse J & Lugtenburg J (1987) Spectroscopic and photochemical properties of mononitropyrenes. Recl Trav Chim Pays-Bas, 106: 120–128.
Vasconcellos PC, Artaxo PE, Ciccioli P, Cecinato A, Brancalconi E & Frattoni M (1998) Chemical composition of aerosol collected in the Amazon forest. Quim Nova, 21: 385–393.
Veigl E, Posch W, Lindner W & Tritthart P (1994) Selective and sensitive analysis of 1-nitropyrene in diesel exhaust particulate extract by multidimensional HPLC. Chromatographia, 38: 199–206.
Vincenti M, Minero C, Pelizzetti E, Fontana M & De Maria R (1996) Sub-parts-per-billion determination of nitro-substituted polynuclear aromatic hydrocarbons in airborne particulate matter and soil by electron capture–tandem mass spectrometry. J Am Soc Mass Spectrom, 7: 1255–1265.
Vincenti M, Maurino V, Minero C & Pelizzetti E (2001) Detection of nitro-substituted polycyclic airborne particulate. Int J Environ Anal Chem, 79(4): 257–272.
Vineis P, Malats N, Lang M, d’Errico A, Caporaso N, Cuzick J & Boffetta P ed. (1999) Metabolic polymorphisms and susceptibility to cancer. Lyon, International Agency for Research on Cancer (IARC Scientific Publications No. 148).
Vogel EW & Nivard MJM (1993) Performance of 181 chemicals in a Drosophila assay predominantly monitoring interchromosomal mitotic recombination. Mutagenesis, 8: 57–81.
von Tungeln LS, Ewing DG, Weitkamp R, Cheng E, Herreno-Saenz D, Evans FE & Fu PP (1994a) Metabolic activation of the potent mutagen and tumorigen 2-nitrobenzo[a]pyrene. Polycycl Aromat Comp, 7: 91–98.
von Tungeln LS, Bucci T, Kadlubar FF & Fu PP (1994b) Tumorigenicity of nitro-polycyclic aromatic hydrocarbons in the neonatal B6C3F-1 male mouse. Proc Am Assoc Cancer Res Annu Meet, 35: 162.
von Tungeln LS, Xia Q, Bucci T, Heflich RH & Fu PP (1999a) Tumorigenicity and liver tumor ras-protooncogene mutations in CD-1 mice treated neonatally with 1- and 3-nitrobenzo[a]pyrene and their trans-7,8-dihydrodiol and aminobenzo[a]pyrene metabolites. Cancer Lett, 137(2): 137–143.
von Tungeln LS, Xia Q, Herreno-Saenz D, Bucci T, Heflich R & Fu P (1999b) Tumorigenicity of nitropolycyclic aromatic hydrocarbons in the neonatal B6C3F1 mouse bioassay and characterization of rash mutations in liver tumors from treated mice. Cancer Lett, 146(1): 1–7.
Wang CY, Lee M-S, King CM & Warner PO (1980) Evidence for nitroaromatics as direct acting mutagens of airborne particulates. Chemosphere, 9: 83–87.
Wang NY, Chou MW & Fu PP (1988) Microsomal metabolism of 3-nitrobenzo[a]pyrene: effect of the nitro substituent on the regioselective metabolism of benzo[a]pyrene. J Chin Biochem Soc, 17: 1–11.
Wang YY, Rappaport SM, Sawyer RF, Talcott RE & Wei ET (1978) Direct-acting mutagens in automobile exhaust. Cancer Lett, 5: 39–47.
Warzecha L (1993) Nitroarenes in airborne particulate organic extracts collected from upper Silesia — identification and quantitative analysis. Chem Anal (Warsaw), 38: 303–313.
Warzecha L (1996) Separation and analysis of nitroarenes from airborne particulate organic matter by HPLC and capillary GC–MS methods. J High Resol Chromatogr, 19: 639–642.
Watanabe M, Ishidate MJ & Nohmi T (1989) A sensitive method for the detection of mutagenic nitroarenes: construction of nitroreductase-overproducing derivatives of Salmonella typhimurium strains TA98 and TA100. Mutat Res, 216: 211–220.
Watanabe M, Ishidate MJ & Nohmi T (1990) Sensitive method for the detection of mutagenic nitroarenes and aromatic amines: new derivatives of Salmonella typhimurium tester strains possessing elevated O-acetyltransferase levels. Mutat Res, 234: 337–348.
Watanabe M, Sofuni T & Nohmi T (1993) Comparison of the sensitivity of Salmonella typhimurium strains YG1024 and YG1012 for detecting the mutagenicity of aromatic amines and nitroarenes. Mutat Res, 301: 7–12.
Watanabe M, Matsuoka A, Yamazaki N, Hayashi M, Deguchi T, Nohmi T & Sofuni T (1994) New sublines of Chinese hamster CHL stably expressing human NAT1 or NAT2 N-acetylransferases or Salmonella typhimurium O-acetyltransferase: comparison of the sensitivities to nitroarenes and aromatic amines using the in vitro micronucleus test. Cancer Res, 54: 1672–1677.
Watanabe T, Kohan MJ, Walsh D, Ball LM, DeMarini DM & Lewtas J (1995) Mutagenicity of nitrobenzopyranones in the Salmonella-plate incorporation and microsuspension assays. Mutat Res, 345(1–2): 1–9.
Watanabe T, Hikayama T & Lewtas J (1996) Mutagenicity and DNA adduct formation of air pollutants: nitrodibenzopyranone isomers and nitrodibenzofuran isomers. Jpn J Toxicol Environ Health, 42: 4.
Watanabe T, Kaji H, Takashima M, Kasai T, Lewtas J & Hirayama T (1997a) Metabolic activation of 2- and 3-nitrobenzopyranone isomers and related compounds by rat liver S9 and the effect of S9 on the mutational specificity of nitrodibenzopyranones. Mutat Res, 388(1): 67–78.
Watanabe T, Takashima M, Kasai T & Hirayama T (1997b) Comparison of the mutational specificity induced by environmental genotoxin nitrated polycyclic aromatic hydrocarbons in Salmonella typhimurium his genes. Mutat Res, 394(1–3): 103–112.
Watanabe T, Ishida S, Kishiji M, Takahashi Y, Furuta A, Kasai T, Wakabayashi K & Hirayama T (1999) High-performance liquid chromatography fluorescence determination of dinitropyrenes in soil after column chromatographic clean-up and on-line reduction. J Chromatogr A, 839(1/2): 41–48.
Watanabe T, Goto S, Matsumoto Y, Asanoma M, Hirayama T, Sera N, Takahashi Y, Endo O, Sakai S & Wakabayashi K (2000) Mutagenic activity of surface soil and quantification of 1,3-, 1,6-, and 1,8-dinitropyrene isomers in soil in Japan. Chem Res Toxicol, 13: 281–286.
Wesp H, Tang X & Edenharder R (2000) The influence of automobile exhausts on mutagenicity of soils: contamination with, fractionation, separation, and preliminary identification of mutagens in the Salmonella/reversion assay and effects of solvent fractions on the sister-chromatid exchanges in human lymphocyte cultures and in the in vivo mouse bone marrow micronucleus assay. Mutat Res, 472(1–2): 1–21.
West RW & Rowland KL (1994) In vitro transformation potential of N-polycyclic aromatic hydrocarbons in rat tracheal epithelial cells. Toxicol In Vitro, 8: 301–307.
Westerholm R, Alsberg T, Strandell M, Frommelin A, Grigoriadis V, Hantzaridou A, Maitra G, Winquist L, Rannug U, Egebäck KE & Bertilsson T (1986) Chemical analysis and biological testing of emissions from a heavy duty diesel truck with and without two different particulate traps. Warrendale, Pennsylvania, Society of Automotive Engineers, The Engineering Resource for Advancing Mobility, pp. 73–83 (SAE Technical Paper Series 860014).
Westerholm RN, Almen J, Li H, Rannug JU, Egeback KE & Gragg K (1991) Chemical and biological characterization of particulate-, semivolatile-, and gas-phase-associated compounds in diluted heavy-duty diesel exhausts: A comparison of three different semivolatile-phase samplers. Environ Sci Technol, 25: 332–338.
Westerholm R, Christensen A & Rosen A (1996) Regulated and unregulated exhaust emissions from two three-way catalyst equipped gasoline fuelled vehicles. Atmos Environ, 30: 3529–3536.
White CM (1985) Nitrated polycyclic aromatic hydrocarbons. Heidelberg, A. Huethig, pp. 1–376.
White GL, Fu PP & Heflich RH (1985) Effect of nitro substitution on the light-mediated mutagenicity of polycyclic aromatic hydrocarbons in Salmonella typhimurium TA98. Mutat Res, 144: 1–7.
Whong WZ & Edwards GS (1984) Genotoxic activity of nitro aromatic explosives and related compounds in Salmonella typhimurium. Mutat Res, 136: 209–215.
Whong WZ, Stewart J, Cutler D & Ong T (1993) Induction of DNA adducts, micronuclei and sister chromatid exchanges by three industrial PAHs in the rat lung cell system. Environ Mol Mutagen, 21(Suppl): 77.
Wierckx FCJ, Wedzinga R, Timmers-Reker AJM, Beland FA, Meerman JHN & Mulder GJ (1991) DNA adduct formation in the liver following administration of [3H]2-nitrofluorene to rats in vivo. Carcinogenesis, 12: 2053–2058.
Wilcox P & Parry JM (1981) The genetic activity of dinitropyrenes in yeast: Unusual dose response curves for induced mitotic gene conversion. Carcinogenesis, 2: 1201–1205.
Wilcox P, Danford N & Parry JM (1982) The genetic activity and metabolism of dinitropyrenes in eucaryotic cells. In: Sorsa M & Vainio H ed. Mutagens in our environment. New York, Alan R. Liss, pp. 249–258.
Williams R, Sparacino C, Petersen B, Bumgarner J, Jungers H & Lewtas J (1986) Comparative characterization of organic emissions from diesel particles, coke oven mains, roofing tar vapors and cigarette smoke condensate. Int J Environ Anal Chem, 26: 27–49.
Wilson NK, Chuang JC & Kuhlman MR (1991) Sampling polycyclic aromatic hydrocarbons and other semivolatile organic compounds in indoor air. Indoor Air, 4: 513–521.
Wilson NK, McCurdy TR & Chuang JC (1995) Concentrations and phase distributions of nitrated and oxygenated polycyclic aromatic hydrocarbons in ambient air. Atmos Environ, 29(19): 2575–2584.
Wislocki PG, Bagan ES, Lu AYH, Dooley KL, Fu PP, Han-Hsu H, Beland FA & Kadlubar FF (1986) Tumorigenicity of nitrated derivatives of pyrene, benz(a)anthracene, chrysene and benzo(a)pyrene in the newborn mouse assay. Carcinogenesis, 7: 1317–1322.
Wolff RK, Barr EB, Bond JA, Ei AF, Griffith WC, Hahn FF, Harkema JR, Henderson RF, Mitchell CE, Rothenberg SJ, Shopp GM & Sun JD (1988) Factors affecting possible carcinogenicity of inhaled nitropyrene aerosols. Prepared by Inhalation Toxicology Research Institute and Lovelace Biomedical and Environmental Research Institute, Albuquerque, New Mexico, for Health Effects Institute, National Toxicology Program, Cambridge, Massachusetts, pp. 1–41 (Research Report No. 19).
Wolff T, Bogan R, Wanders H & Wegenke M (1993) Biomonitoring of human exposure to carcinogenic nitroaromatic compounds: a pilot study on DNA adduct formation by 1,6-dinitropyrene in rats. Lyon, International Agency for Research on Cancer, pp. 195–199 (IARC Scientific Publications No. 124).
Working PK & Butterworth BE (1984) An assay to detect chemically induced DNA repair in the rat spermatocytes. Environ Mutagen, 6: 273–286.
Wu PF, Chiang TA, Wang LF, Chang CS & Ko YC (1998) Nitro-polycyclic aromatic hydrocarbon contents of fumes from heated cooking oils and prevention of mutagenicity by catechin. Mutat Res, 403(1–2): 29–34.
Xiang M, Whong WZ, Heng ZC, Nath J & Ong T (1996) In vivo and in vitro transformation in rat tracheal epithelial cells exposed to diesel emission particles and related compounds. Environ Mol Mutagen, 27(Suppl): 76.
Xiang M, Hubbs A, Zhou G, Nath J & Ong T (1997) The neoplastic potential of rat tracheal epithelial cell lines induced by dibenzo(a,i)pyrene and 1-nitropyrene. Environ Mol Mutagen, 28(Suppl): 58.
Xu J, Whong W-Z & Ong TM (1984) Validation of the Salmonella (SV50)/arabinose-resistant forward mutation assay system with 26 compounds. Mutat Res, 130: 79–86.
Xu XB, Nachtman JP, Jin ZL, Wei ET & Rappaport SM (1982) Isolation and identification of mutagenic nitro-PAH in diesel-exhaust particulates. Anal Chim Acta, 136: 163–174.
Yaffe D, Cohen Y, Arey J & Grosovsky A (2001) Multimedia analysis of PAHs and nitro-PAH daughter products in the Los Angeles basin. Risk Anal, 21(2): 275–294.(sec 2)
Yahagi T, Shimizu H, Nagao M, Takemura N & Sugimura T (1975) Mutagenicity of 5-nitroacenaphthene in Salmonella. Gann (Jpn J Cancer Res), 66: 581–582.
Yamada M, Espinosa-Aguirre JJ, Watanabe M, Matsui K, Sofuni T & Nohmi T (1997) Targeted disruption of the gene encoding the classical nitroreductase enzyme in Salmonella typhimurium Ames test strains TA1535 and TA1538. Mutat Res, 375: 9–17.
Yamamoto A, Inamasu T, Hisanaga A, Kitamori S & Ishinishi N (1987) Comparative study on the carcinogenicity of 1-nitropyrene and benzo(a)pyrene to the lung of Syrian golden hamsters induced by intermittent intratracheal instillations. J Jpn Soc Air Pollut, 22: 29–35.
Yamazaki H, Hatanaka N, Kizu R, Hayakawa K, Shimada N, Guengerich F, Nakajima M & Yokoi T (2000) Bioactivation of diesel exhaust particle extracts and their major nitrated polycyclic aromatic hydrocarbon components, 1-nitropyrene and dinitropyrenes, by human cytochromes P450 1A1, 1A2, and 1B1. Mutat Res, 472(1–2): 129–138.
Yang JL, Maher VM & McCormick JJ (1988) Kinds and spectrum of mutations induced by 1-nitrosopyrene adducts during plasmid replication in human cells. Mol Cell Biol, 8: 3364–3372.
Yang TC, Chou A, Chen E, Chiu L & Ni Y (1994) Photodecomposition of environmental nitro-polycyclic aromatic hydrocarbons. Polycycl Aromat Comp, 5: 201–208.
Yassaa N, Meklati B, Cecinato A & Marino F (2001a) Chemical characteristics of organic aerosol in Bab-Ezzouar (Algiers). Contribution of bituminous product manufacture. Chemosphere, 45: 315–322.
Yassaa N, Meklati B, Cecinato A & Marino F (2001b) Organic aerosols in urban and waste landfill of Algiers metropolitan area: occurrence and sources. Environ Sci Technol, 35: 306–311.
Ye D, Siddiqi MA, Maccubbin AE, Kumar S & Sikka HC (1996) Degradation of polynuclear aromatic hydrocarbons by Sphingomonas paucimobilis. Environ Sci Technol, 30: 136–142.
Yoshikura H, Kuchino T & Matsushimu T (1979) Carcinogenic chemicals enhance mouse leukemia virus infection in contact-inhibited culture: a new simple method of screening carcinogens. Cancer Lett, 7: 203.
Yoshimi N, Mori H, Sugie S, Iwata H, Kinouchi T & Ohnishi Y (1987) Genotoxicity of a variety of nitroarenes in DNA repair tests with human hepatocytes. Gann (Jpn J Cancer Res), 78: 807–813.
Yu S, Heflich RH, von Tungeln LS, El-Bayoumy K, Kadlubar FF & Fu PP (1991) Comparative direct-acting mutagenicity of 1- and 2-nitropyrene: evidence for 2-nitropyrene mutagenesis by both guanine and adenine adducts. Mutat Res, 250: 145–152.
Yu S, Herreno-Saenz D, Miller DW, Heflich RH, Kadlubar FF & Fu PP (1992) Mutagenicity of nitro-polycyclic aromatic hydrocarbons with the nitro substituent situated at the longest axis. Mutat Res, 283: 45–52.
Yu WC, Fine DH, Chiu KS & Biemann K (1984) Determination of nitrated polycyclic aromatic hydrocarbons in diesel particulates by gas chromatography with chemiluminescent detection. Anal Chem, 56: 1158–1162.
Ziegler W, Penalver LG, Preiss U & Wallnöfer PR (1999) Fate of nitropolycyclic aromatic hydrocarbons in artificially contaminated fruits and vegetables during food processing. Adv Food Sci, 21: 54–57.
Zielinska B, Arey J, Ramdahl T, Atkinson R & Winer AM (1986a) Potential for artifact formation during Tenax sampling of polycyclic aromatic hydrocarbons. J Chromatogr, 363: 382–386.
Zielinska B, Arey J, Atkinson R, Ramdahl T, Winer AM & Pitts JN Jr (1986b) Reaction of dinitrogen pentoxide with fluoranthene. J Am Chem Soc, 108: 4126–4132.
Zielinska B, Harger WP, Arey J, Winer AM, Haas RA & Hanson CV (1987) The mutagenicity of 2-nitrofluoranthene and its in vitro hepatic metabolites. Mutat Res, 190: 259–266.
Zielinska B, Arey J, Harger WP & Lee RWK (1988) Mutagenic activities of selected nitrofluoranthene derivatives in Salmonella typhimurium strains TA98, TA98NR and TA98/1,8-DNP6. Mutat Res, 206: 131–140.
Zielinska B, Arey J, Atkinson R & Winer AM (1989a) The nitroarenes of molecular weight 247 in ambient particulate samples collected in Southern California. Atmos Environ, 23: 223–229.
Zielinska B, Arey J, Atkinson R & McElroy PA (1989b) Formation of methylnitronaphthalenes from the gas-phase reactions of 1- and 2-methylnaphthalene with OH radicals and N2O5 and their occurrence in ambient air. Environ Sci Technol, 23: 723–729.
Zühlke J, Knopp D & Niessner R (1998) Determination of 1-nitropyrene with enzyme-linked immunosorbent assay versus high-performance column switching technique. J Chromatogr A, 807(2): 209–217.
Zwirner-Baier I & Neumann HG (1999) Polycyclic nitroarenes (nitro-PAHs) as biomarkers of exposure to diesel exhaust. Mutat Res, 441: 135–144.
Les hydrocarbures aromatiques polycycliques nitrés (nitro-HAP ou nitroarènes) sont des dérivés d’hydrocarbures aromatiques polycycliques (HAP) à noyaux condensés, c’est à dire contenant au moins deux cycles aromatiques accolés constitués d’atomes de carbone et d’hydrogène. Ils sont présents dans l’environnement sous forme de mélanges, à côté des HAP dont ils dérivent et de centaines d’autres composés organiques. Ils sont généralement beaucoup moins abondants que les HAP.
Dans l’environnement, ils sont présents soit en phase vapeur, soit adsorbés à la surface de particules. Ce sont des composés insolubles ou du moins peu solubles dans l’eau, mais pour la plupart solubles dans les solvants organiques.
Le prélèvement d’échantillons de nitro-HAP s’effectue comme pour les HAP. Pour les analyses portant sur l’air ambiant, on recueille les particules aéroportées sur des filtres spéciaux au moyen d’échantillonneurs de grand volume. Les nitro-HAP en phase vapeur sont habituellement recueillis sur un sorbant solide comme la mousse de polyuréthane.
Après une extraction par solvant, on procède à une purification par chromatographie en phase liquide sur gel de silice ou alumine, par chromatographie en phase liquide à haute performance (HPLC) ou encore par extraction en phase solide. Il faut séparer la fraction contenant les dérivés nitrés de celles qui contiennent les hydrocarbures aromatiques polycycliques et les HAP oxygénés par HPLC sur silice. Pour séparer et identifier les nitro-HAP, on a recours à la chromatographie en phase gazeuse avec divers types de détecteurs (détecteurs à fluorescence, à chimiluminescence, ou détecteurs électrochimiques) ou encore à la spectrométrie de masse. L’analyse est tributaire des étalons disponibles.
Dans le cas des mélanges complexes, on peut utiliser une autre méthode d’analyse qui repose sur les propriétés biologiques de ces composés : on soumet à des épreuves biologiques les fractions dotées de propriétés mutagènes et on les caractérise jusqu’à identification du composé ou du groupe de composés qui pourraient être responsables des effets mutagènes observés. En utilisant des souches bactériennes présentant une sensibilité sélective aux nitroarènes, on a pu identifier, grâce à leur forte mutagénicité, divers HAP nitrés présents dans des mélanges d’origines diverses. Ce type d’analyse nécessite un certain nombre d’étalons de synthèse.
Une nitrocétone aromatique, la 3-nitrobenzanthrone, ainsi que diverses nitrolactones comme la 2- et la 4-nitrodibenzopyranone sont des dérivés nitro-oxygénés que l’on a mis en évidence en même temps que les nitro-HAP et qui sont justiciables de méthodes d’analyses similaires.
Les nitro-HAP sont essentiellement des produits directs ou indirects d’une combustion incomplète. Seuls quelques-uns d’entre eux sont produits industriellement, comme les nitronaphtalènes et le 5-nitroacénaphtène que l’on utilise principalemement comme intermédiaires de synthèse.
Les nitro-HAP se forment à partir des HAP correspondants (qui sont généralement adsorbés sur des particules et résultent eux-même d’une combustion incomplète) selon au moins deux processus distincts : 1) par nitration au cours de la combustion (par ex.dans les gaz d’échappement de véhicules à moteur, notamment à moteur diesel, mais aussi dans ceux des moteurs à essence et des moteurs d’avion, dans les émissions de diverses industries, lors du chauffage ou de la cuisson des aliments au domicile ou encore lors de la combustion du bois) et 2) dans l’atmosphère, par des réactions en phase gazeuse - dans la journée, par addition de radicaux hydroxyles aux HAP suivie d’une attaque par le dioxyde d’azote et de la perte d’une molécule d’eau et au cours de la nuit, par addition de radicaux nitrate aux HAP suivie d’une attaque par le dioxyde d’azote et de l’élimination d’acide nitrique - ou encore, par une interaction hétérogène entre la phase gazeuse et les particules, au cours de laquelle des agents de nitration attaquent les HAP adsorbés sur les particules.
On a constaté que la distribution des nitro-HAP isomères dans des échantillons d’air ambiant était sensiblement différente de celle que l’on peut observer dans les émissions produites par la combustion. Le 2-nitrofluoranthène et le 2-nitropyrène sont omniprésents dans les matières particulaires, même s’ils ne proviennent pas directement des sources de combustion les plus fréquentes. Le « profil » des nitro-HAP ou l’abondance relative de certains HAP « marqueurs » renseigne sur l’origine de leur formation. Les isomères nitrés les plus abondants du pyrène, du fluorène et du fluoranthène que l’on trouve dans les gaz d’échappement des moteurs diesel sont le 1-nitropyrène, le 2-nitrofluorène et le 3-nitrofluoranthène, alors que les isomères qui se forment par l’action des radicaux hydroxyles sur ces HAP sont le 2-nitropyrène, le 3-nitrofluorène et le 2-nitrofluoranthène.
On pense maintenant que la majorité des nitro-HAP présents dans l’atmosphère se forment par nitration en phase gazeuse d’hydrocarbures aromatiques polycycliques contenant tout au plus quatre noyaux.
On a mis en évidence et dosé de nombreux isomères mononitrés et quelques isomères di- et trinitrés dans divers échantillons de gaz d’échappement de moteurs diesel, le 1-nitropyrène étant généralement le plus abondant. Le 1-nitropyrène est le nitro-HAP « marqueur » des gaz d’échappement de moteurs diesel et sa présence dans l’air ambiant trahit la présence d’une pollution par des véhicules dotés de tels moteurs. Comme le carburant diesel, les moteurs et les pots catalytiques sont sans cesse modifiés, il est impossible de procéder à une comparaison directe des diverses études consacrées aux nitro-HAP présents dans les gaz d’échappement des moteurs diesel. En règle générale, l’utilisation de pièges à particules ou de pots catalytiques entraîne une diminution des émissions massives de particules, des émissions de HAP et de nitro-HAP et du niveau de l’activité mutagène.
La concentration du 1-nitropyrène est beaucoup plus faible dans les particules émises par les moteurs à essence que dans celles qui sont émises par les moteurs diesel., mais on a constaté que la concentration du 1,3-, du 1,6- et du 1,8-dinitropyrène est pratiquement la même dans les deux cas.
On est fondé à penser que des nitro-HAP sont présents dans les gaz d’échappement des avions à réaction.
On a décelé la présence de nitro-HAP dans les émissions des poêles à kérosène, ainsi que dans celles des brûleurs à gaz et gaz de pétrole liquéfié (GPL), que l’on utilise dans de nombreux pays pour le chauffage et et la cuisine.
On a trouvé de la 3-nitrobenzanthrone dans les particules émises par les moteurs diesel ainsi que dans des échantillons d’air urbain. On a également mis en évidence la présence de 2-nitrodibenzopyranone, de 4-nitrodibenzopyranone et de nitropyrène-lactones dans des particules aéroportées présentes dans l’air ambiant.
3.1 Transport et distribution dans l’environnement
Les HAP et les nitro-HAP peuvent être transportés en phase vapeur ou adsorbés sur des particules. Ceux qui, à l’état liquide, ont une tension de vapeur supérieure à 10–4 Pa à la température ambiante (c’est-à-dire les HAP possédant deux à quatre noyaux aromatiques et les nitro-HAP à deux noyaux aromatiques) seront au moins partiellement présents dans la phase gazeuse.
En raison de leur faible solubilité ou de leur insolubilité dans l’eau, les nitro-HAP ne devraient pas être transportés par cette dernière. Selon les données disponibles, la valeur de leur coefficient de sorption (log Koc) est élevée, ce qui indique qu’à l’instar des HAP, les nitro-HAP sont adsorbés sur les sédiments et les particules du sol. Le passage dans les eaux souterraines par lessivage devrait être négligeable. Dans certaines conditions, quelques-uns d’entre eux peuvent subir une lente biodégradation.
La valeur du coefficient de partage n-octanol/eau (log Kow) va de 2,5 pour le 1-nitronaphtalène à 6,3 pour le 3-nitropérylène, ce qui donne à penser qu’une bioaccumulation est possible. On ne possède aucune donnée sur la bioamplification.
3.2 Biotransformation
Un grand nombre de bactéries aérobies et anaérobies réduisent les nitro-HAP en amino-HAP dotés de propriétés mutagènes.Chez les mammifères, la réduction du groupement nitro par la flore intestinale joue un rôle majeur dans le métabolisme des nitro-HAP. On a montré que si toutes sortes de bactéries, de champignons et d’algues sont capables de dégrader les HAP contenant de deux à cinq cycles, les dérivés nitrés correspondants ne sont que très lentement décomposés par les microorganismes indigènes et peuvent donc subsister dans le sol et les sédiments. Cette résistance des nitro-HAP de masse moléculaire élevée est due pour une part à leur forte adsorption aux matières organiques du sol, à leur faible solubilité, à la taille importante de leur molécule et au caractère polaire du groupement nitro.
Des études diachroniques effectuées sur un certain nombre de microcosmes ont montré que le 1-nitropyrène subissait une lente dégradation aérobie ou anaérobie dans les sédiments estuariels.
La souche EPA de Sphingomonas paucimobilis (une bactérie terricole capable d’utiliser le fluoranthène comme unique source de carbone et d’énergie) provoque une biodégradation du 1-nitropyrène qui le réduit en 6 h à 48,6 % de sa quantité initiale.
On a montré qu’en présence de Cunninghamella elegans, un champignon filamenteux, un certain nombre de nitro-HAP (1-nitropyrène, 2-nitrofluorène, 2- et 3-nitrofluoranthène, 6-nitrochrysène, 1-nitrobenzo[e]pyrène et 6-nitrobenzo[a]pyrène) subissent, sous l’action d’une monooxygénase du cytochrome P450, une métabolisation oxydative qui donne naissance à des produits moins mutagènes que les nitro-HAP eux-mêmes.
L’incubation de 1-nitropyrène et de 1,3-, 1,6- et 1,8-dinitropyrène avec une culture de cellules végétales provenant d’Alternanthera philoxeroides a détoxifié ces composés qui sont tous des mutagènes à action directe, comme l’a montré le test de mutagénicité d’Ames sur la souche TA98 de Salmonella typhimurium.
3.3 Dégradation abiotique
On a étudié la photolyse des nitro-HAP dans diverses conditions d’irradiation. La vitesse de photolyse dépend non seulement des conditions d’irradiation mais aussi de l’état dans lequel se trouve le composé : en phase gazeuse (par exemple dans le cas du 1- et du 2-nitronaphtalène), en solution (nature du solvant) ou lié à des matières solides ou à des particules. Dans ce dernier cas, la nature et l’âge des particules semblent avoir une influence sur la photochimie des nitro-HAP considérés. Les principaux points étudiés ont été la vitesse de photodécomposition, la nature des produits de photolyse et la perte ou le gain résultants d’activité mutagène déterminés par le test d’Ames sur S. typhimurium.
Le calcul de la durée de vie atmosphérique des nitro-HAP soumis à la photolyse et à des réactions en phase gazeuse avec des radicaux hydroxyle et nitrate ou encore avec l’ozone atmosphérique montre que le processus principal de destruction des nitro-HAP (comme le 1- et le 2-nitropyrène, par exemple) est la photolyse. La nuit, c’est principalement une oxydation par l’ozone des nitro-HAP portés par des particules qui semble être responsable de cette destruction.
Dans l’air ambiant, on a notamment décelé la présence des nitro-HAP suivants : 1- et 2-nitronaphtalène et méthylnitronaphtalènes (principalement dans la phase vapeur), 2-nitrofluorène, 9-nitroanthracène, 9-nitrophénanthrène, 2-, 3- et 8-nitrofluoranthène, 1- et 2-nitropyrène, 1,3-, 1,6- et 1,8-dinitropyrène et 6-nitrochrysène.
Dans les zones reculées ou les zones de forêt, les nitro-HAP se sont révélés soit non décelables, soit à la concentration de quelques picogrammes par mètre cube (par ex., 17 pg/m3 pour le 2-nitrofluoranthène et 4 pg/m3 pour le 1-nitropyrène). La concentration des nitro-HAP dans l’air des zones urbaines dépend de la saison, des moyens de chauffage utilisés ainsi que du nombre de véhicules en circulation et de la régulation du trafic. Les concentrations relevées dans l’air ne dépassent généralement pas 1 ng/m3, même si des valeurs maximales allant jusqu’à 13 ng/m3 ont été observées.
Diverses études ont été consacrées à la surveillance continue de certains nitro-HAP isomères. Les chercheurs se sont intéressés aux composés importants sur le plan quantitatif ou environnemental (par exemple ceux qui ont une masse moléculaire relative de 247 : 1-nitropyrène, 2-nitropyrène, 2-nitrofluoranthène,) ou qui ont des propriétés cancérogènes (1-nitropyrène, 2-nitrofluorène ou encore les dinitropyrènes, par ex.).
Des études portant sur les concentrations diurnes et nocturnes de certains nitro-HAP isomères dans diverses régions (notamment la Californie) et des études parallèles en chambre environnementale ont permis de comprendre comment se forment un certain nombre de nitro-HAP dans l’atmosphère (le 2-nitrofluoranthène et le 2-nitropyrène). D’autres études de ce type consacrées à la corrélation entre certains nitro-HAP (comme le 1-nitropyrène et les dinitropyrènes) et l’importance de la circulation automobile, ont confirmé que les émissions provenant du trafic routier sont une source de nitro-HAP.
La plupart des études consacrées à la variation saisonnière de la concentration montrent que celle des composés marqueurs augmente en hiver et au printemps, parallèlement à l’utilisation du chauffage domestique, sans toutefois que cette corrélation soit toujours observée.
4.1 Air intérieur
Comme on a décelé la présence de nitro-HAP dans les émissions des poêles à kérosène et des brûleurs à gaz de ville ou à GPL utilisés pour le chauffage ou la cuisine, ainsi que dans les fumées dégagées par les huiles de friture, il existe un risque d’exposition à ces composés dans les locaux mal ventilés.
Lors d’une étude sur l’air extérieur et sur l’air intérieur de 33 habitations situées dans deux villes des Etats-Unis (Columbus, dans l’Ohio et Azusa, en Californie), on a mesuré la concentration de composés aromatiques polycycliques et notamment de nitro-HAP. Dans l’ensemble, les concentrations se sont révélées beaucoup plus élevées dans les logements occupés par des fumeurs, mais on a également constaté que l’utilisation d’appareils de chauffage et de cuisine au gaz naturel était à l’origine d’une légère augmentation de la teneur en nitro-HAP.
On a trouvé du 1-nitropyrène (4,2 - 25 600 ng/litre) dans 36 échantillons d’eaux usées sur 55 prélevés dans des réservoirs de séparation huile-eau de stations-service ainsi que dans de l’huile de vidange de carter.
Dans des cours d’eau du Japon, on a mis en évidence la présence de 1- et de 2-nitronaphtalène ainsi que de 1,3- et de 1,5-dinitronaphtalène aux concentrations respectives de 1,3, 11,7, 1,7 et 3,2 ng/litre. Du 1-nitropyrène a été trouvé dans un autre échantillon.
On ne possède que des données limitées sur la présence de nitro-HAP dans des échantillons de sol, de boues d’égout, de sédiments et de cendres d’incinérateur (dans le cas du 1-nitropyrène par exemple, 0,03 - 0,8 μg/kg de poids sec dans le sol, 0,68 μg/kg dans des boues d’ιgout, 25,2 μg/kg dans des sιdiments et < 0,01 - 0,89 mg/kg dans des cendres d’incinérateur).
4.2 Aliments et boissons
A l’exception des épices, des produits alimentaires fumés ou grillés et des cacahuètes, la concentration des nitro-HAP dans les denrées alimentaires est inférieure à 5 μg/kg.
Dans une étude effectuée au Royaume-Uni, on a contrôlé des produits alimentaires à la recherche de 9-nitroanthracène et de 1-nitropyrène. Sur 28 produits analysés, 25 ne contenaient aucun de ces nitro-HAP en quantités décelables. Du 9-nitroanthracène a peut-être été mis en évidence à la concentration de 0,9 μg/kg dans du malt tourbeux, du 1-nitropyrène ayant par ailleurs été trouvé dans deux échantillons de feuilles de thé à la concentration respective de 1,7 et 0,17 μg/kg.
Lors d’une autre enquête menée en Autriche sur la teneur en nitro-HAP de divers produits alimentaires, on a constaté essentiellement la présence de 2-nitrofluorène, 1-nitropyrène et 2-nitronaphtalène en quantités décelables. Les concentrations les plus élevées ont été observées dans les épices, les produits fumés et diverses sortes de thé, notamment le Mate, qui est torréfié. On a également mis des nitro-HAP en évidence dans des fruits et des légumes, probablement contaminés par la pollution atmosphérique.
On a trouvé du 1-nitropyrène dans du maïs, du maquereau et (en très forte quantité) dans du porc grillés ainsi que dans du yakitori (poulet grillé), grillé avec de la sauce (jusqu’à 43 ng/kg).
4.3 Autres produits
En 1980, des études ont montré que des extraits de certaines encres xérographiques et de photocopies papier avaient des propriétés mutagènes. La fraction du noir de carbone B responsable à 80 % de cette activité mutagène contenait du 1-nitropyrène, du 1,3-, du 1,6- et du 1,8-dinitropyrène ainsi que du 1,3,6-trinitropyrène et du 1,3,6,8-tétranitropyrène. A la suite de cette découverte, les fabricants ont modifié le mode de production du noir de carbone, ce qui a permis de réduire sensiblement la teneur en nitro-HAP.
4.4 Exposition professionnelle
On a mis en évidence une exposition professionnelle aux nitro-HAP sur des lieux de travail où étaient utilisés des moteurs diesel. On a par exemple trouvé du 1-nitropyrène en quantités dosables dans l’air de divers lieux de travail où étaient utilisés ces moteurs. La concentration la plus forte (42 ng/m3) a été relevée en Estonie au niveau de la zone de respiration d’ouvriers mineurs qui conduisaient des excavatrices à moteur diesel dans une mine d’huile de schiste.
Administrés par différentes voies, le 1-nitropyrène et le 2-nitrofluorène sont rapidement résorbés et leurs métabolites sont excrétés après conjugaison. Après administration par toutes les voies possibles à des rats et à des souris, le 1-nitropyrène radiomarqué se distribue largement dans l’organisme. Les autres nitro-HAP n’ont pas été bien étudiés.
Les nitro-HAP ont un métabolisme complexe. Il semble qu’il existe au moins cinq voies d’activation métabolique par lesquelles ces composés sont capables de provoquer des mutations dans les systèmes bactériens et mammaliens et au cours desquelles ils se lient à l’ADN, à savoir : 1) la réduction du groupement nitro; 2) la réduction du groupement nitro suivie d’une esterification (notamment une acétylation) ; 3) une oxydation du noyau; 4) une oxydation du noyau et une réduction du groupement nitro; 5) une oxydation du noyau, une réduction du groupement nitro puis une esterification. Chez les bactéries, la réduction du groupement nitro semble constituer la voie métabolique principale, alors que le champignon Cunninghamella elegans est un exemple d’espèce métabolisant les nitro-HAP par oxydation du noyau.
In vivo, la réduction du groupement nitro des nitro-HAP est sans doute due principalement aux bactéries qui composent la flore intestinale. La première étape du métabolisme oxydatif est la formation des métabolites dits de la phase I (métabolites primaires), à savoir des époxydes, des phénols et des dihydrodiols. On passe ensuite aux métabolites secondaires, constitués de diols-époxydes, de tétrahydrodiols et phénols-époxydes. Dans les systèmes mammaliens, les métabolites de la phase I forment des glutathiono-, sulfo- ou glucuroconjugués pour donner les métabolites de la phase II, plus polaires et hydrosolubles que les hydrocarbures correspondants. Parvenus dans l’intestin, les métabolites conjugués peuvent subir une déconjugaison sous l’action de la flore intestinale et être absorbés pour passer dans le cycle entéro-hépatique. Il peut également y avoir réduction du groupement nitro et N-acétylation, puis, si c’est du 1-nitropyrène qui a été administré, excrétion dans les urines et les matières fécales de métabolites comme les acétylaminopyrénols.
Diverses enzymes du cytochrome P450 peuvent intervenir dans le métabolisme de certains nitro-HAP et les voies métaboliques de même que la cinétique peuvent varier selon les isomères. Les enzymes du cytochrome P450 responsables du métabolisme des nitro-HAP peuvent varier selon les espèces, les organes-cibles et les divers types de cellules présents dans ces organes.
Tous les nitro-HAP n’empruntent pas les mêmes voies d’activation métabolique. Certains se révèlent mutagènes une fois réduits en arylhydroxylamines (par ex. le 1-nitropyrène est principalement métabolisé par hydroxylation du reste aromatique, puis réduction du groupement nitré suivie d’une N-acétylation); d’autres (par ex. le 1,8- et le 1,6-dinitropyrène) doivent encore subir, après réduction en arylhydroxylamine, une O-esterification (notamment une O-acétylation) qui les transformera en esters alcoxy mutagènes. Pour d’autres encore, une oxydation en époxydes ou dihydrodiols-époxydes réactifs suffit à conférer des propriétés mutagènes (comme ce pourrait être le cas du 6-nitrobenzo[a]pyrène, analogue à cet égard au benzo[a]pyrène ou BaP). Les principaux adduits des nitro-HAP à l’ADN à avoir été détectés in vivo et in vitro sont des adduits à la désoxyguanosine C8-substitués. On a toutefois également mis en évidence la présence d’adduits à la désoxyguanosine N2-substitués ainsi que des adduits à l’adénosine C8-substitués, qui pourraient prédominer dans le cas des nitro-HAP dont les propriétés sont plus proches de celles des hydrocarbures (par ex. le 3-nitrobenzo[a]pyrène et le 6-nitrochrysène). Les adduits à l’ADN des dinitropyrènes ne se forment que par réduction du groupement nitro, vraisemblablement par suite du fort déficit en électrons des noyaux aromatiques dû à la présence des deux groupements nitro. Les adduits à l’ADN qui résultent de la réduction du groupement nitro des nitro-HAP sont mieux caractérisés que ceux qui proviennent du métabolisme oxydatif, encore que ces derniers puissent être plus importants chez les mammifères.
Seulement six nitro-HAP ont fait l’objet d’une analyse de leur toxicité aiguë. Chez le rat on a trouvé une DL50 de 86 mg/kg de poids corporel après injection de 1-nitronaphtalène par voie intrapéritonéale; chez la souris, on fait état d’une DL50 de 1300 mg/kg pc dans le cas du 2-nitronaphtalène. D’autres études consacrées à ces deux composés ont permis d’observer des effets généraux au niveau du poumon et du foie après administration d’une dose unique élevée; toutefois, le 2-nitronaphtalène s’est révélé moins toxique que le 1-nitronaphtalène. Une dose de 1700 mg/kg pc de 5-nitroacénaphtène administrée par voie intrapéritonéale a été mortelle pour tous les rats traités. Dans le cas du 2-nitrofluorène, on a obtenu une DL50 orale de 1600 mg/kg pc pour la souris, alors que l’administration par gavage de doses de 1-nitropyrène allant jusqu’à 5000 mg/kg pc n’a produit aucun effet toxique observable.Chez des rats qui avaient reçu une injection sous-cutanée de 8 mg de 3-nitrofluoranthène par kg de poids corporel, on a observé une inflammation et une ulcération locales.
Les données relatives aux effets non néoplasiques locaux ou généraux consécutifs à l’administration de nitro-HAP sur de courtes ou de longues périodes restent limitées car la plupart des études sont centrées sur la cancérogénicité de ces composés. Dans la plupart des cas, les doses qui provoquent des effets toxiques non néoplasiques perceptibles sont celles auxquelles se manifestent également des effets cancérogènes. Toutefois, des effets non néoplasiques généraux tels qu’une diminution du poids corporel et une augmentation de la mortalité, se sont manifestés, indépendamment semble-t-il, des effets cancérogènes, lors d’études au cours desquelles des rats et des souris ont reçu une alimentation contenant du 5-nitroacénaphtène à raison de 500 mg/kg pc par jour (rats) ou de 40 mg/kg pc par jour (souris) et du 2-nitrofluorène à raison de 25 mg/kg pc par jour (rats). Une exposition de durée moyenne à du 1-nitropyrène par la voie respiratoire a provoqué une métaplasie des voies respiratoires supérieures à une concentration >0,5 mg/m3.
On ne dispose d’aucune donnée sur l’irritation oculaire ou cutanée, ni sur la sensibilisation ou la toxicité génésique.
On possède des données sur la toxicité in vitro de 95 nitro-HAP; toutefois, pour 74 d’entre eux, seuls un ou deux points d’aboutissement des effets toxiques ont été étudiés, principalement sur des systèmes bactériens. On ne dispose d’une base de données suffisante, portant notamment sur des systèmes d’épreuve eucaryotes, que pour 21 nitro-HAP. Des résultats positifs ont été obtenus pour la plupart des composés (67 sur 95), mais les résultats sont tirés d’une petite base de données. Des résultats indiscutablement positifs n’ont été obtenus que pour 19 composés, les résultats étant discutables pour 8 autres. Aucun des nitro-HAP n’a donné de résultats clairement négatifs.
Les résultats du test d’Ames sur S. typhimurium sont connus pour 86 nitro-HAP. Contrairement aux HAP correspondants, les dérivés nitrés se révèlent nettement plus actifs dans le test sur salmonelles sans activation métabolique. Cinq d’entre eux présentent une activité mutagène exceptionnellement forte (>100 000 mutants réverses/nmol) dans ce test : le 3,7- et le 3,9-dinitrofluoranthène, le 1,6- et le 1,8-dinitropyrène et le 3,6-dinitrobenzo[a]pyrène.
La nitroréductase et l’acétyltransférase bactériennes interviennent dans l’activation métabolique des nitro-HAP, mais les voies d’activation métabolique ne sont pas identiques pour tous les composés. En outre, ces composés n’ont pas un effet mutagène uniforme car ils provoquent des mutations par décalage du cadre de lecture et par substitution de paires de bases dans le test d’Ames sur S. typhimurium. Il y a lieu de penser que les nitro-HAP dont les groupements nitro sont perpendiculaires au plan du noyau aromatique ne sont pas aussi mutagènes que ceux de leurs isomères dans lesquels ces groupements sont parallèles au plan du noyau.
On dispose de données sur la génotoxicité in vivo de 15 nitro-HAP. Tous les nitro-HAP qui donnent des résultats positifs in vivo en donnent également in vitro. Quatre de ces composés qui se sont révélés positifs in vitro on donné in vivo des résultats incohérents ou non concluants (2-nitronaphtalène, 5-nitroacénaphtène, 3-nitrofluoranthène) ou même négatifs (2,7-dinitrofluorène; validité limitée).
La 3-nitrobenzanthrone, comme le 1,6- et le 1,8-dinitropyrène, est fortement mutagène pour les bactéries, l’activation métabolique s’effectuant par réduction du groupement nitro et O-esterification. La 3-nitrobenzanthrone a également une forte activité mutagène au niveau génique et elle provoque la formation de micronoyaux dans les cellules humaines in vitro et les cellules murines in vivo.
On a fait état d’une forte mutagénicité de la 2-nitrodibenzopyranone dans le test d’Ames sur la souche TA98 (-S9) de S. typhimurium, supérieure à celle du 2-nitrofluorène et du 1-nitropyrène. La 1- et la 3-nitropyrène-lactone se sont également révélées très fortement mutagènes dans le test d’Ames sur S. typhimurium.
Avec la 2-nitrodibenzopyranone, on a obtenu des résultats contradictoires dans les tests de génotoxicité in vitro portant sur la production de mutations directes dans deux lignées de cellules lymphoblastoïdes B humaines. Les nitropyrènes-lactones se sont révélées capables de provoquer des mutations au niveau des locus tk et hprt dans les deux lignées. Elles ont en outre provoqué la formation de micronoyaux kinétochore-positifs et kinétochore-négatifs dans le test des micronoyaux modifié selon la méthode CREST, qui permet de déceler les pertes et les ruptures chromosomiques.
On possède des données de cancérogénicité concernant 28 nitro-HAP. Bien que l’inhalation soit la principale voie d’exposition chez l’Homme, il n’existe d’étude d’inhalation à long terme sur aucun nitro-HAP. La plupart des travaux portent sur les effets cancérogènes dus à l’administration de nitro-HAP par voie orale, application topique, implantation pulmonaire ou instillation intratrachéenne.
En raison d’insuffisances dans les protocoles expérimentaux, aucune des études ayant débouché sur des résultats négatifs ne permet de confirmer l’absence d’effets cancérogènes chez l’animal. D’ailleurs, des résultats témoignant d’effets cancérogènes ont été obtenus par expérimentation animale dans le cas des composés suivants : 5-nitroacénaphtène, 2-nitrofluorène, 3-nitrofluoranthène, 3,7- et 3,9-dinitrofluoranthène, 1- et 4-nitropyrène, 1,3-, 1,6 et 1,8-dinitropyrène et 6-nitrochrysène. Une certaine cancérogénicité ressort également de l’expérimentation animale dans le cas des composés suivants : 2-nitropyrène, 7-nitrobenz[a]anthracène, 2- et 6-nitrobenzo[a]pyrène, 3,6-dinitrobenzo[a]pyrène, 7-nitrodibenz[a,h]anthracène et 3-nitropérylène. En ce qui concerne les 10 nitro-HAP restants, on ne dispose pas de données suffisantes pour en évaluer la cancérogénicité chez les animaux de laboratoire.
Outre des effets locaux au point d’injection, les principaux effets cancérogènes généraux des nitro-HAP consistent en tumeurs des tissus mammaires, pulmonaires, hépatiques ou concernent le système hématopoïétique. De tous les nitro-HAP examinés ici, c’est le 6-nitrochrysène qui se révèle le plus cancérogène. Au regard de ses effets généraux après injection sous-cutanée ou intrapéritonéale, le 1-nitropyrène est plus cancérogène que les dinitropyrènes. La cancérogénicité du 1-nitropyrène et des dinitropyrènes varie selon la voie d’administration.
Les benzo[a]pyrènes nitrés sont généralement des cancérogènes moins puissants que le benzo[a]pyrène lui-même. En revanche, le mono- et les dinitropyrènes sont plus cancérogène que le pyrène. On a obtenu des résultats analogues dans le cas du 3-nitropérylène par rapport au pérylène et du 6-nitrochrysène par rapport au chrysène. Toutefois, le 6-nitrochrysène est moins actif que le chrysène relativement aux effets locaux consécutifs à une exposition de l’épiderme.
Il existe des données sur les effets cancérogènes de certains métabolites du 2-nitrofluorène, du 1-nitropyrène et du 6-nitrochrysène. En comparant, chez le rat, l’effet du 2-nitrofluorène à ceux de ses métabolites, on a constaté que le 2-acétylaminofluorène était le plus cancérogène. Le 1-nitropyrène s’est par contre révélé sensiblement plus cancérogène après administration par voie orale que le 1-nitrosopyrène ou le 1-aminopyrène. D’un autre côté, l’incidence des tumeurs du foie est plus forte après adminstration de 1-nitrosopyrène. Aucun effet n’a été observé dans le cas de métabolites hydroxylés sur le noyau. Le 6-nitrosochrysène et le 6-aminochrysène se sont montrés inactifs, contrairement aux métabolites hydroxylés sur le noyau, qui avaient une activité cancérogène sur les cellules hépatiques analogue à celle du 6-nitrochrysène. Cela montre que l’activation du 6-nitrochrysène se produit par oxydation du noyau ou par oxydation du noyau plus réduction du groupement nitro.
On ne dispose d’aucune publication concernant les effets de tel ou tel nitro-HAP sur l’Homme. Comme on peut s’y attendre du fait que les nitro-HAP sont présents sous la forme de mélanges complexes dans l’air ambiant et les gaz d’échappement, il n’est pas possible de connaître leur contribution exacte aux effets indésirables d’une exposition à une atmosphère polluée et aux gaz d’échappement.
Pour l’instant, l’étude des effets des nitro-HAP sur la santé humaine repose sur l’utilisation de biomarqueurs d’exposition. Plusieurs publications sont consacrées à l’élaboration de méthodes pour l’évaluation du 1-nitropyrène comme biomarqueur de l’exposition professionnelle aux gaz d’échappement des moteurs diesel et contiennent un certain nombre de données sur ce point. La méthode immunoenzymatique ELISA a été utilisée pour mettre en évidence la présence de métabolites de HAP et de nitro-HAP dans les urines de dieselistes. Dans une autre étude, on a dosé des métabolites du 1-nitropyrène (à savoir le N-acétyl-1-aminopyrène-6-ol et le N-acétyl-1-aminopyrène-8-ol) dans les urines de travailleurs d’un service d’expéditions. Plusieurs études ont été consacrées au dosage des adduits à l’hémoglobine et au plasma de métabolites du 1-nitropyrène et d’autres nitro-HAP et pourraient fournir des biomarqueurs appropriés pour les futures études d’épidémiologie moléculaire.
On ne possède de données sur la toxicité aiguë des nitro-HAP pour les organismes aquatiques que dans le cas du 1-nitronaphtalène. On a ainsi obtenu une CL50 à 96 h de 9,0 mg/litre pour le méné tête-de-boule (Pimephales promelas). Ce nitro-HAP inhibe également la croissance d’un cilié, Tetrahymena pyriformis, avec une CE50 à 60 h de 17,3 mg/litre.
Certaines études avaient pour objet l’effet des nitro-HAP sur le métabolisme de diverses espèces aquatiques-par exemple, sur la distribution infracellulaire et tissulaire de l’activité de la nitroréductase mono- et biélectronique NAD(P)H-dépendante chez les invertébrés marins appartenant à trois phylums : une moule (Mytilus edulis), un crabe (Carcinus maenas) et une étoile de mer, l’astérie rouge (Asteria rubens). On a mis en évidence une activité nitroréductase biélectronique NADPH-dépendante, présente uniquement en anaérobiose, dans les fractions microsomienne et cytosolique des principaux tissus digestifs de la moule (glande digestive) et du crabe, mais pas dans les branchies de ces deux espèces. Le seul métabolite a avoir été identifié était le 1-aminopyrène. Aucune activité nitroréductase n’était décelable dans les caeca pyloriques ou la région stomacale d’Asteria rubens. On a constaté qu’une nitroréduction monoélectronique NAD(P)H-dépendante se produisait dans toutes les fractions infracellulaires des principaux tissus digestifs des trois espèces.
En présence d’ADN de thymus de veau, on a mis en évidence in vitro des adduits du 1-nitropyrène à l’aide de fractions de S9 hépatique d’origine pisciaire. L’aptitude du 1-nitropyrène à former des adduits à l’ADN a également été démontrée in vivo sur la truite commune (Salmo trutta) et le turbot (Scophthalmus maximus). Ces adduits à l’ADN se sont révélés comparables à ceux obtenus sur des rats Wistar traités par du 1-nitropyrène.
Los hidrocarburos aromáticos policíclicos nitrogenados (nitroPAH) son derivados de los hidrocarburos aromáticos policíclicos (PAH), con dos o más anillos aromáticos fundidos formados por átomos de carbono y de hidrógeno. Estos compuestos se encuentran en la naturaleza mezclados con los PAH de origen y cientos de otros compuestos orgánicos. Suelen aparecer en cantidades muy inferiores a las de los PAH.
Los nitroPAH están en el medio ambiente en fase de vapor o adsorbidos en la materia particulada. Su solubilidad en agua es escasa o nula, pero en general son solubles en disolventes orgánicos.
El muestreo de los nitroPAH es semejante al de los PAH. Se toman muestras del aire recogiendo materia particulada sobre filtros especiales mediante muestradores de alto volumen. Los nitroPAH en fase de vapor se recogen normalmente en sorbentes sólidos, como la espuma de poliuretano.
La extracción con disolventes va seguida de un aclarado utilizando cromatografía líquida con gel de sílice o alúmina, cromatografía líquida de alto rendimiento o extracción en fase sólida. La fracción de los nitroPAH se debe separar de la fracción de los PAH y de la fracción de los PAH oxigenados mediante cromatografía líquida de alto rendimiento sobre sílice. Entre los métodos utilizados para la separación y detección de los nitroPAH cabe mencionar la cromatografía de gases con diversos detectores, la cromatografía líquida de alto rendimiento con fluorescencia, quimioluminiscencia o detector electroquímico y las técnicas de espectrometría de masas. El análisis depende de los patrones disponibles.
Otro método de análisis de mezclas complejas es el análisis químico orientado a la biovaloración, en el que se someten a biovaloración fracciones mutagénicamente activas y se caracterizan hasta que se identifican los compuestos principales o específicos que podrían provocar la mutagenicidad. La utilización de cepas bacterianas de comprobación con sensibilidad selectiva a los nitroarenos ha llevado a la identificación de los nitroPAH como mutágenos potentes en mezclas complejas de fuentes diversas. Para este tipo de análisis se necesitan patrones sintéticos.
La nitrocetona 3-nitrobenzantrona y las nitrolactonas, como la 2- y 4-nitrodibenzopiranona, son compuestos oxinitrogenados que se han detectado junto con los nitroPAH y se analizan utilizando métodos semejantes.
Los nitroPAH se forman fundamentalmente como productos directos o indirectos de la combustión incompleta. Sólo un pequeño número de nitroPAH procede de la industria; por ejemplo, los nitronaftalenos y el 5-nitroacenafteno producidos comercialmente se utilizan sobre todo como intermediarios químicos.
Los nitroPAH se forman a partir de los PAH (generalmente adsorbidos en la materia particulada y productos a su vez de la combustión incompleta) mediante dos procesos distintos como mínimo: 1) nitración durante los procesos de combustión (por ejemplo, en los gases de escape de los vehículos, en particular diesel, pero también de gasolina, y en las emisiones de las aeronaves; emisiones industriales; la calefacción/cocinas de las viviendas; la combustión de la madera); y 2) formación en la atmósfera a partir de los PAH, bien por reacciones en fase gaseosa - adición diurna de radicales hidroxilo a los PAH, seguida de reacción con el dióxido de nitrógeno y pérdida de una molécula de agua y adición nocturna de radicales nitrato a los PAH, seguida de reacción con el dióxido de nitrógeno y pérdida de ácido nítrico - o mediante una interacción heterogénea gas-partículas de los PAH de origen adsorbidos en las partículas con agentes nitrantes.
La distribución de los isómeros de los nitroPAH en las muestras de aire ambiente es muy diferente de la que se observa en las muestras obtenidas a partir de las emisiones directas de la combustión. El 2-nitrofluoranteno y el 2-nitropireno son componentes ubicuos de la materia particulada, aunque no los emitan de manera directa la mayor parte de las fuentes de combustión. El perfil de los nitroPAH, o las cantidades relativas de ciertos PAH «marcadores», son indicadores de su fuente de formación. Los nitroisómeros más abundantes del pireno, el fluoreno y el fluoranteno observados en los gases de escape de los motores diesel son el 1-nitropireno, el 2-nitrofluoreno y el 3-nitrofluoranteno, mientras que los isómeros resultantes de la reacción de los radicales hidroxilo de estos PAH son el 2-nitropireno, el 3-nitrofluoreno y el 2-nitrofluoranteno.
Ahora se considera que la mayoría de los nitroPAH se forman en la atmósfera por reacción en fase gaseosa de los PAH con cuatro anillos o menos.
Se han identificado y cuantificado muchos isómeros mono- y algunos di- y trinitroPAH en diversas muestras de gases de escape de motores diesel, siendo normalmente el 1-nitropireno el más abundante. El 1-nitropireno es el nitroPAH «marcador» para los gases de escape de los motores diesel y su presencia en las muestras de aire ambiente es un signo de contaminación a causa del tráfico de estos vehículos. El combustible diesel, los tipos de motores y los catalizadores están sufriendo continuas modificaciones, de manera que no es posible una comparación directa de los distintos estudios sobre la presencia de nitroPAH en los gases de escape de los motores diesel. En general, la emisión masiva de partículas, las emisiones de partículas ligadas a los PAH y los nitroPAH y los niveles de actividad mutagénica solían disminuir con el uso de interceptores de partículas o de convertidores catalíticos.
La concentración de 1-nitropireno era mucho menor en las partículas de los gases de escape de los motores de gasolina que en las de los diesel, pero se observó que las concentraciones de 1,3-, 1,6- y 1,8-dinitropirenos eran prácticamente iguales en las partículas de los gases de escape de los motores de gasolina y diesel.
No hay pruebas de la presencia de nitroPAH en los gases de escape de los aviones.
Se han detectado nitroPAH en las emisiones de los calefactores de queroseno y de los quemadores de gas combustible y gas licuado de petróleo que se utilizan en muchos países para la calefacción y para cocinar en las viviendas.
Se ha detectado 3-nitrobenzatrona en muestras de partículas de los gases de escape de los motores diesel y en las del aire urbano. Se han observado 2-nitrodibenzopiranona y 4-nitrodibenzopiranona, así como lactonas nitropirénicas en la materia particulada del medio ambiente.
3.1 Transporte y distribución en el medio ambiente
Los nitroPAH se pueden transportar en fase de vapor o adsorbidos en materia particulada. Los que tienen una presión de vapor en fase líquida superior a 10–4 Pa a la temperatura del aire ambiente (es decir, PAH de dos a cuatro anillos y nitroPAH de dos anillos) se encontrarán al menos parcialmente en la fase gaseosa.
Debido a su escasa o nula solubilidad en agua, no se prevé un transporte de los nitroPAH por este medio. Los datos disponibles muestran valores elevados para los coeficientes de sorción (log Koc), lo cual parece indicar que los nitroPAH, al igual que los PAH, se adsorben en el suelo y los sedimentos. Se considera que la lixiviación hacia el agua freática es insignificante. Algunos nitroPAH pueden sufrir una biodegradación lenta en determinadas condiciones.
Los valores del coeficiente de reparto n-octanol/agua (log Kow) oscilan entre 2,5 para el 1-nitronaftaleno y 6,3 para el 3-nitroperileno, lo que parece indicar una posible bioacumulación. No se dispone de datos sobre la bioamplificación.
3.2 Biotransformación
Numerosas bacterias anaerobias y aerobias reducen los nitroPAH a aminoPAH mutagénicos. La nitrorreducción por la microflora intestinal desempeña una función importante en el metabolismo de los nitroPAH en los mamíferos. Aunque se ha demostrado que hay una gran variedad de bacterias, hongos y algas que degradan los PAH de origen con dos a cinco anillos, la degradación de los PAH con sustituciones de nitrógeno por la flora endógena es un proceso lento. La resistencia de los nitroPAH de peso molecular alto se debe en parte a la fuerte adsorción a la materia orgánica del suelo, la baja solubilidad, el tamaño molecular elevado y el carácter polar del grupo nitrogenado.
Los estudios a lo largo del tiempo en distintos microcosmos pusieron de manifiesto que el 1-nitropireno se degradaba lentamente en condiciones aerobias y anaerobias en los sedimentos de los estuarios.
La cepa EPA 505 de Sphingomonas paucimobilis (bacteria del suelo capaz de utilizar fluoranteno como única fuente de carbono y energía) biodegradó el 1-nitropireno al 48,6% después de seis horas.
Se ha puesto de manifiesto que el hongo filamentoso Cunninghamella elegans metaboliza por vía oxidativa, mediante una citocromo P450 monooxigenasa, diversos nitroPAH (1-nitropireno, 2-nitrofluoreno, 2- y 3-nitrofluoranteno, 6-nitrocriseno, 1-nitrobenzo[e]pireno y 6-nitrobenzo[a]pireno) a productos que son menos mutagénicos que los propios nitroPAH.
Un cultivo de células vegetales obtenido a partir de la lagunilla (Alternanthera philoxeroides) destoxificó el 1-nitropireno y el 1,3-, 1,6- y 1,8-dinitropireno, mutágenos de acción directa, tras la incubación con ellos, como pone de manifiesto la respuesta a la mutagenicidad en la valoración con Salmonella typhimurium TA98.
3.3 Degradación abiótica
Se ha estudiado la fotolisis de los nitroPAH en diversas condiciones de irradiación. La velocidad de la fotolisis depende no sólo de las condiciones de irradiación, sino también de que el nitroPAH esté en fase gaseosa (por ejemplo, el 1- y 2-nitronaftaleno), en disolución (tipo de disolvente) o unido a sólidos/partículas. En el último caso, el tipo y la edad de la partícula parecen influir en la fotoquímica del respectivo nitroPAH. Los principales efectos finales estudiados son la velocidad de fotodescomposición, la identificación de los productos fotolíticos y el consiguiente aumento o disminución de la actividad metabólica determinada mediante la valoración con S. typhimurium.
La permanencia calculada en la atmósfera de los nitroPAH, debido a la fotolisis y a las reacciones en fase gaseosa con radicales hidroxilo y nitrato y con el ozono en condiciones atmosféricas, pone de manifiesto que el proceso de eliminación predominante de los nitroPAH (por ejemplo, el 1- y 2-nitronaftaleno) es la fotolisis. La oxidación de las partículas de nitroPAH por el ozono puede ser el principal proceso de eliminación durante la noche.
Entre los nitroPAH detectados en el aire ambiente cabe mencionar el 1- y 2-nitronaftaleno y los metilnitronaftalenos (sobre todo en fase de vapor), el 2-nitrofluoreno, el 9-nitroantraceno, el 9-nitrofenantreno, el 2-, 3- y 8-nitrofluoranteno, el 1- y 2-nitropireno, el 1,3-, 1,6- y 1,8-dinitropireno y el 6-nitrocriseno.
En lugares remotos y forestales no se detectaron nitroPAH o bien se obtuvo una concentración baja del orden de picogramos/m3 (por ejemplo, 17 pg/m3 para el 2-nitrofluoranteno; 4 pg/m3 para el 1-nitropireno). La concentración de nitroPAH en la atmósfera de las regiones urbanas depende de la estación, el tipo de calefacción utilizado y el número de vehículos y la regulación del tráfico. Los niveles en el aire notificados no suelen superar el valor de 1 ng/m3, aunque se han descrito máximos de hasta 13 ng/m3.
Se han realizado varios estudios mediante la vigilancia de ciertos isómeros de los nitroPAH. Los investigadores se han concentrado en los nitroPAH que parecen tener importancia cuantitativa/ambiental (por ejemplo, los nitroPAH con una masa molecular relativa de 247: 1-nitropireno, 2-nitropireno, 2-nitrofluoranteno) o carcinogénica (por ejemplo, el 1-nitropireno, el 2-nitrofluoreno, los dinitropirenos).
Los estudios de las concentraciones diurnas/nocturnas de los isómeros de nitroPAH específicos de ciertas regiones (en particular de California, Estados Unidos) y los estudios paralelos en cámaras climatizadas han permitido comprender la formación atmosférica de ciertos nitroPAH (el 2-nitrofluoranteno y el 2-nitropireno). En estudios simultáneos de ciertos nitroPAH (el 1-nitropireno y los dinitropirenos) y del volumen del tráfico se ha confirmado que éste es una fuente de emisión de nitroPAH.
La mayoría de los estudios estacionales ponen de manifiesto una mayor concentración de nitroPAH marcadores en invierno/primavera, debido al uso de la calefacción doméstica, aunque no siempre es así.
4.1 Aire de espacios cerrados
Dado que se han detectado nitroPAH en las emisiones de los calefactores de queroseno y de los quemadores de gas combustible y de gas licuado de petróleo que se utilizan en la calefacción y para cocinar en las viviendas, así como en el humo de los aceites de cocinar, hay por consiguiente una posible exposición a estos compuestos en los espacios cerrados en condiciones de escasa ventilación.
En un estudio del aire de espacios cerrados y abiertos en 33 viviendas situadas en dos ciudades de los Estados Unidos, Columbus (Ohio) y Azusa (California), se midieron las concentraciones de compuestos poliaromáticos, incluidos los nitroPAH. En general, los niveles fueron mucho más altos en las viviendas ocupadas por fumadores, pero la calefacción y los aparatos de cocinar de gas natural también parecían aumentar ligeramente su concentración.
Se detectó 1-nitropireno (4,2-25 600 ng/l) en 36 de 55 muestras de aguas residuales procedentes de depósitos de separación gasolina-agua de las estaciones de servicio y en el lubricante del cárter usado.
En agua de río del Japón se detectaron 1- y 2-nitronaftaleno y 1,3- y 1,5-dinitronaftaleno en concentraciones de 1,3, 11,7, 1,7 y 3,2 ng/litro, respectivamente. En otra muestra de agua se identificó el 1-nitropireno.
Sólo hay datos limitados sobre la presencia de nitroPAH en muestras de suelo, fangos cloacales, sedimentos y ceniza de incineradores (por ejemplo, para el 1-nitropireno, 0,03-0,8 µg/kg de peso seco en el suelo, 0,68 µg/kg en los fangos cloacales, 25,2 µg/kg en los sedimentos y < 0,01-0,89 mg/kg en la ceniza de los incineradores).
4.2 Alimentos y bebidas
Con la excepción de las especias, los alimentos ahumados y asados a la parrilla y los cacahuetes, las concentraciones de nitroPAH en los alimentos son inferiores a 5 µg/kg.
En un estudio realizado en el Reino Unido, se investigó la presencia de 9-nitroantraceno y 1-nitropireno en los productos alimenticios. De un total de 28 alimentos, 25 contenían niveles no detectables de estos nitroPAH. Se identificó provisionalmente 9-nitroantraceno en malta de turbera (0,9 µg/kg) y 1-nitropireno en dos muestras de hojas de té, con 1,7 y 0,17 µg/kg.
En otro estudio de los niveles de nitroPAH en diversos alimentos de Austria se pusieron de manifiesto concentraciones en su mayor parte detectables de 2-nitrofluoreno, 1-nitropireno y 2-nitronaftaleno. Las concentraciones más altas se encontraron en las especias, los alimentos ahumados y el té, en particular el té mate, que está tostado. También se detectaron nitroPAH en hortalizas y frutas, probablemente debido a la contaminación atmosférica.
Se detectó 1-nitropireno en el maíz asado a la parrilla, la caballa y (en cantidades considerables) la carne de cerdo y el yakitori (pollo a la parrilla) asado con salsa (hasta 43 ng/g).
4.3 Otros productos
En 1980, varios estudios pusieron de manifiesto que los extractos de determinados «toner» xerográficos y fotocopias de papel eran mutagénicos. La fracción del negro de carbón B responsable del 80% de la mutagenicidad contenía 1-nitropireno, 1,3- 1,6- y 1,8-dinitropireno, 1,3,6-trinitropireno y 1,3,6,8-tetranitropireno. Como resultado de este descubrimiento, los fabricantes modificaron la producción del negro de carbón B, reduciendo sustancialmente los niveles de nitropirenos.
4.4 Exposición profesional
Se ha demostrado la existencia de exposición profesional a los nitropirenos en lugares de trabajo relacionados con el uso de motores diesel. Por ejemplo, se midieron concentraciones de 1-nitropireno en el aire de diversos puestos de trabajo relacionados con el uso de motores diesel. Los niveles máximos notificados (42 ng/m3) se determinaron en zonas subterráneas de respiración de los trabajadores (conductores de excavadoras con motor diesel) en una mina de esquisto petrolífero de Estonia.
El 1-nitropireno y el 2-nitrofluoreno administrados por diversas vías se absorben con rapidez y los metabolitos resultantes se conjugan y se excretan. Tras la administración a ratas y ratones de 1-nitropireno radiomarcado por todas las vías se detectó su presencia ampliamente distribuido en el organismo. No se han realizado estudios tan completos de otros nitroPAH.
El metabolismo de los nitroPAH es complejo. Parece que hay por lo menos cinco vías de activación metabólica mediante las cuales pueden inducir mutaciones en sistemas bacterianos y de mamíferos y/o se produce unión al ADN. Son las siguientes: 1) nitrorreducción; 2) nitrorreducción seguida de esterificación (en particular acetilación); 3) oxidación de los anillos; 4) oxidación de los anillos y nitrorreducción; y 5) oxidación de los anillos y nitrorreducción seguida de esterificación. En bacterias, la principal vía metabólica parece ser la nitrorreducción, mientras que el hongo Cunninghamella elegans es un ejemplo de especie que metaboliza los nitroPAH mediante la oxidación de los anillos.
Probablemente se produce nitrorreducción in vivo de los nitroPAH por la acción de las bacterias del tracto intestinal. En el metabolismo oxidativo, el primer paso es la transformación a metabolitos primarios de la fase I, como epóxidos, fenoles y dihidrodioles, y luego a metabolitos secundarios, como epóxidos diólicos, tetrahidrotetroles y epóxidos fenólicos. En sistemas de mamíferos, los metabolitos de la fase I se conjugan luego con glutatión, sulfato o ácido glucurónico para formar metabolitos de la fase II, que son más polares y solubles en agua que los hidrocarburos originales. Al llegar al intestino, los metabolitos conjugados se pueden desconjugar mediante la flora intestinal y absorberse, pasando a la circulación enterohepática. Tras la administración de 1-nitropireno se puede producir nitrorreducción y N-acetilación, con la excreción en la orina y las heces de metabolitos como los acetilaminopirenoles.
En el metabolismo de un nitroPAH determinado pueden participar diferentes enzimas del citocromo P450 y dar lugar a distintos isómeros, con el resultado de cinéticas y vías metabólicas posiblemente diferentes. Las enzimas del citocromo P450 responsables del metabolismo de los nitroPAH pueden variar de una especie a otra y de un órgano destinatario a otro, así como en los distintos tipos de células de dichos órganos.
No todos los nitroPAH siguen las mismas vías de activación. Algunos son mutagénicos cuando se reducen a arilhidroxilamina (por ejemplo, el 1-nitropireno se metaboliza principalmente por hidroxilación del grupo aromático, seguida de nitrorreducción y N-acetilación); otros (por ejemplo, el 1,8- y el 1,6-dinitropireno) se reducen a arilhidroxilamina y luego requieren una ulterior O-esterificación (en particular O-acetilación) a un aciloxiéster para ser mutagénicos. Algunos pueden ser mutagénicos sólo tras la activación por oxidación a epóxidos reactivos o epóxidos de dihidrodiol (como ocurre posiblemente en el 6-nitrobenzo[a]pireno, semejante al benzo[a]pireno). Los aductos principales de ADN con nitroPAH detectados in vivo e in vitro son aductos de desoxiguanosina con una sustitución en C8; sin embargo, se han detectado derivados de la desoxiguanosina con una sustitución en N2 y de la desoxiadenosina con sustitución en C8 y pueden predominar en los nitroPAH con un carácter hidrocarbonado más fuerte (por ejemplo, el 3-nitrobenzo[a]pireno y el 6-nitrocriseno). Los aductos de ADN de los dinitropirenos sólo se forman por nitrorreducción, posiblemente debido a la elevada deficiencia de electrones en los anillos aromáticos a causa de la presencia de dos grupos nitrogenados. Se han caracterizado mejor los aductos de ADN derivados de la nitrorreducción de los nitroPAH que los procedentes del metabolismo oxidativo, aunque estos últimos pueden ser más importantes en el metabolismo de los mamíferos.
Sólo se han realizado pruebas de toxicidad aguda con seis nitroPAH. En ratas, se notificó una DL50 de 86 mg/kg de peso corporal tras la inyección intraperitoneal de 1-nitronaftaleno; en ratones, se informó de una DL50 de 1300 mg/kg de peso corporal tras la administración por vía oral de 2-nitronaftaleno. En ulteriores estudios con ambas sustancias se observaron efectos en los órganos destinatarios, pulmón e hígado, tras la administración de dosis elevadas únicas; sin embargo, el 2-nitronaftaleno parecía ser menos tóxico que el 1-nitronaftaleno. Una dosis intraperitoneal de 1700 mg /kg de peso corporal de 5-nitroacenafteno fue letal para todas las ratas tratadas. Para el 2-nitrofluoreno se notificó en ratones una DL50 por vía oral de 1600 mg/kg de peso corporal, mientras que la administración con sonda de hasta 5000 mg/kg de peso corporal de 1-nitropireno no tuvo efectos tóxicos observables. En ratas se observó inflamación y ulceración local tras la inyección subcutánea de 8 mg/kg de peso corporal de 3-nitrofluoranteno.
Los datos sobre los efectos no neoplásicos sistémicos o locales causados por el tratamiento prolongado o breve con nitroPAH son limitados, puesto que el efecto final de la mayor parte de los estudios ha sido la carcinogenicidad. En la mayoría de los casos se observaron efectos tóxicos no neoplásicos con dosis a las cuales también se manifestaban respuestas carcinogénicas. Los efectos tóxicos sistémicos no neoplásicos, como la reducción del peso corporal o el aumento de la mortalidad, posiblemente aparecieron con independencia de los efectos carcinogénicos en los estudios de alimentación con 5-nitroacenafteno en dosis de 500 mg/kg de peso corporal al día (ratas) o 40 mg/kg de peso corporal al día (ratones) y con 2-nitrofluoreno en dosis de 25 mg/kg de peso corporal al día (ratas). La exposición de duración media al 1-nitropireno por inhalación produjo metaplasia del tracto superior de las vías respiratorias en concentraciones >0,5 mg/m3.
No se dispone de datos sobre la irritación cutánea y ocular, la sensibilización o la toxicidad reproductiva.
Hay datos sobre la genotoxicidad in vitro de 95 nitroPAH; sin embargo, para 74 de ellos sólo se investigaron uno o dos efectos finales, principalmente en sistemas de prueba bacterianos. Sólo para 21 se ha encontrado una base de datos suficiente, incluidos sistemas de prueba eucarióticos. La mayoría de estas sustancias (67 de 95) mostraron resultados positivos, pero éstos se obtuvieron a partir de una base de datos pequeña. Se obtuvieron resultados claramente positivos para 19 nitroPAH, y dudosos para ocho. Con ninguno de ellos se obtuvieron resultados claramente negativos.
Hay datos relativos a la prueba microsomal en S. typhimurium para 86 nitroPAH. A diferencia de los PAH de origen, la mayor parte de los nitroPAH fueron claramente más efectivos en la prueba microsomal en Salmonella sin activación metabólica. Hay cinco nitroPAH que mostraron una potencia mutagénica excepcionalmente alta (>100 000 revertientes/nmol) en esta prueba: 3,7- y 3,9-dinitrofluoranteno, 1,6- y 1,8-dinitropireno y 3,6-dinitrobenzo[a]pireno.
En la activación metabólica de los nitroPAH intervienen la nitrorreductasa y la acetiltransferasa bacterianas, pero no todos ellos siguen las mismas vías de activación metabólica. Además, no hay un efecto mutagénico uniforme de los distintos nitroPAH, puesto que en la prueba microsomal en S. typhimurium producen tanto un cambio en el marco de lectura como sustituciones en los pares de bases. Hay pruebas de que los nitroPAH con grupos nitrogenados perpendiculares al anillo aromático no son tan mutagénicos como los isómeros con una orientación paralela de dicho grupo.
Hay datos sobre la genotoxicidad in vivo de 15 nitroPAH. Todos los que dieron resultado positivo in vivo también lo dieron in vitro. De los nitroPAH que dieron resultado positivo en las pruebas de genotoxicidad in vitro, cuatro mostraron una genotoxicidad contradictoria o nula (2-nitronaftaleno, 5-nitroacenafteno y 3-nitrofluoranteno) o dieron un resultado de genotoxicidad negativo (2,7-dinitrofluoreno; validez limitada) in vivo.
La 3-nitrobenzatrona, al igual que el 1,6- y el 1,8-dinitropireno, es muy mutagénica en bacterias mediante nitrorreducción y O-esterificación. La 3-nitrobenzatrona es también un mutágeno efectivo de genes y provoca la formación de micronúcleos en células humanas in vitro y en ratones in vivo.
Se informó de que la 2-nitrodibenzopiranona era muy mutagénica en la prueba microsomal en S. typhimurium con la cepa TA98 (-S9), siendo más mutagénica que el 2-nitrofluoreno y el 1-nitropireno. Se ha observado que la 1- y la 3-nitropirenolactonas son muy mutagénicas en la prueba microsomal en S. typhimurium.
Los resultados de los estudios sobre la genotoxicidad in vitro de la 2-nitrodibenzopiranona en valoraciones de mutaciones adaptativas utilizando dos líneas de células linfoblastoideas B humanas son contradictorios. Se observó que las nitropirenolactonas inducían mutaciones en los loci tk y hprt en ambas líneas de células. Además, inducían micronúcleos positivos y negativos en el cinetocoro en la valoración del micronúcleo modificado de CREST, que detecta pérdidas cromosomales y fenómenos de rotura.
Se dispone de datos sobre los efectos carcinogénicos para 28 nitroPAH. Aunque la inhalación es la vía principal de exposición en las personas, no hay ningún estudio de inhalación prolongada para ninguno de los nitroPAH. En la mayoría de los estudios se examinaron los efectos carcinogénicos de los nitroPAH mediante la administración oral, la aplicación cutánea, la implantación pulmonar o la administración intratraqueal.
Debido a limitaciones inherentes al diseño de los experimentos, ninguno de los estudios negativos confirmó la ausencia de efectos carcinogénicos en los animales. Sin embargo, los resultados pusieron de manifiesto efectos carcinogénicos en animales de experimentación con 5-nitroacenafteno, 2-nitrofluoreno, 3-nitrofluoranteno, 3,7- y 3,9-dinitrofluoranteno, 1- y 4-nitropireno, 1,3-, 1,6- y 1,8-dinitropireno y 6-nitrocriseno. Se observaron algunos efectos carcinogénicos en animales de experimentación con 2-nitropireno, 7-nitrodibenz[a]antraceno, 2- y 6-nitrobenzo[a]pireno, 3,6-dinitrobenzo[a]pireno, 7-nitrobenz[a,h]antraceno y 3-nitroperileno. Para el resto de los 10 nitroPAH sometidos a prueba no se disponía de datos suficientes con los cuales evaluar su carcinogenicidad en animales de experimentación.
Además de los efectos locales en el lugar de la inyección, los nitroPAH inducían principalmente tumores sistémicos en el tejido mamario, pulmonar y hepático y en el sistema hematopoyético. El 6-nitrocriseno parece ser el nitroPAH más carcinogénico de los examinados en el presente documento. El 1-nitropireno fue más carcinogénico que los dinitropirenos, con efectos sistémicos tras la inyección subcutánea o intraperitoneal. La carcinogenicidad del 1-nitropireno y de los dinitropirenos varía en función de la vía de administración.
Los nitrobenzo[a]pirenos suelen ser carcinógenos menos potentes que sus compuestos de origen. Sin embargo, los pirenos mono o dinitrados son más carcinogénicos que el pireno. Se presentaron resultados semejantes para el 3-nitroperileno en comparación con el perileno y para el 6-nitrocriseno en comparación con el criseno; sin embargo, con respecto a los efectos locales tras la exposición cutánea, el 6-nitrocriseno fue menos activo que el criseno.
Había datos disponibles sobre los efectos carcinogénicos de algunos metabolitos del 2-nitrofluoreno, el 1-nitropireno y el 6-nitrocriseno. Al comparar el 2-nitrofluoreno con sus metabolitos en ratas, se observó que el potencial carcinogénico más alto correspondía al 2-acetilaminofluoreno. Tras la administración oral en ratas, el 1-nitropireno fue considerablemente más carcinogénico que el 1-nitrosopireno o el 1-aminopireno. En cambio, el 1-nitrosopireno indujo tras la administración intraperitoneal una incidencia mayor de tumores hepáticos en ratones que el 1-nitropireno; no se observaron efectos con metabolitos de anillos hidroxilados. El 6-nitrosocriseno y el 6-aminocriseno fueron inactivos, a diferencia de los metabolitos de anillos hidroxilados, que mostraron en el hígado una actividad carcinogénica semejante a la del 6-nitrocriseno de origen; esto indica que la activación metabólica del 6-nitrocriseno se produce por oxidación de los anillos y/o una combinación de oxidación y nitrorreducción de los anillos.
No hay ningún informe sobre los efectos de los distintos nitroPAH en el ser humano. Como cabría esperar, puesto que los nitroPAH están presentes en mezclas complejas en la atmósfera y en los gases de escape, no se puede determinar la contribución exacta de los nitroPAH a las consecuencias adversas para la salud de la exposición a atmósferas contaminadas y a gases de escape.
En la actualidad se están llevando a cabo investigaciones sobre los efectos de los nitroPAH en la salud humana utilizando biomarcadores de la exposición. En varios informes se ha descrito la elaboración de métodos de evaluación del 1-nitropireno como biomarcador para la exposición ocupacional a los gases de escape de motores diesel y se han proporcionado datos al respecto. Se determinaron los metabolitos urinarios de los PAH y los nitroPAH en la orina de mecánicos de motores diesel utilizando la prueba de inmunosorción enzimática (ELISA). En otro estudio, se midieron los metabolitos del 1-nitropireno (a saber, N-acetil-1-aminopiren-6-ol y N-acetil-1-aminopiren-8-ol) en la orina de trabajadores de un departamento de transporte marítimo. Varios estudios se han concentrado en la medición de los aductos de los metabolitos del 1-nitropireno y otros nitroPAH en la hemoglobina y el plasma y pueden proporcionar biomarcadores apropiados para futuras investigaciones epidemiológicas moleculares.
Solamente hay datos disponibles sobre la toxicidad aguda de los nitroPAH en los organismos acuáticos para el 1-nitronaftaleno. Se notificó una CL50 (96 horas) de 9,0 mg/litro para Pimephales promelas. Además, este nitroPAH inhibió el crecimiento del ciliado Tetrahymena pyriformis, con una CE50 (60 horas) de 17,3 mg/litro.
Algunos estudios se han ocupado del efecto de los nitroPAH en el metabolismo de algunas especies acuáticas, por ejemplo la distribución subcelular y tisular de la actividad nitrorreductasa con dos y un electrones dependiente del NAD(P)H en invertebrados marinos de tres phyla: el mejillón (Mytilus edulis), el cangrejo (Carcinus maenas) y la estrella de mar (Asteria rubens). Se detectó actividad nitrorreductasa con dos electrones dependiente del NADPH, que se produce sólo en condiciones anaerobias, en las fracciones microsómica y citosólica de los principales tejidos digestivos del mejillón (glándula digestiva) y el cangrejo, pero no en las branquias de ninguna de las dos especies. El único metabolito identificado fue el 1-aminopireno. No se registró actividad detectable en la región de los ciegos pilóricos o el estómago de la estrella de mar. En todas las fracciones subcelulares de los principales tejidos digestivos de las tres especies había presente nitrorreducción de un electrón NAD(P)H-dependiente.
En presencia de ADN de timo de ternero se detectaron in vitro aductos derivados del 1-nitropireno utilizando fracciones S9 hepáticas preparadas a partir de peces. También se estableció la capacidad del 1-nitropireno para formar aductos de ADN in vivo utilizando trucha marina (Salmo trutta) y rodaballo (Scophthalmus maximus). Estos aductos de ADN eran comparables a los obtenidos en ratas Wistar tratadas con 1-nitropireno.
See Also:
Toxicological Abbreviations
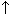 )
)