
UNITED NATIONS ENVIRONMENT PROGRAMME
INTERNATIONAL LABOUR ORGANISATION
WORLD HEALTH ORGANIZATION
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 212
PRINCIPLES AND METHODS FOR ASSESSING ALLERGIC
HYPERSENSITIZATION ASSOCIATED WITH EXPOSURE
TO CHEMICALS
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Published under the joint sponsorship of the United Nations
Environment Programme, the International Labour Organisation, and the
World Health Organization, and produced within the framework of the
Inter-Organization Programme for the Sound Management of Chemicals.
World Health Organization
Geneva, 1999
The International Programme on Chemical Safety (IPCS), established in
1980, is a joint venture of the United Nations Environment Programme
(UNEP), the International Labour Organisation (ILO), and the World
Health Organization (WHO). The overall objectives of the IPCS are to
establish the scientific basis for assessment of the risk to human
health and the environment from exposure to chemicals, through
international peer review processes, as a prerequisite for the
promotion of chemical safety, and to provide technical assistance in
strengthening national capacities for the sound management of
chemicals.
The Inter-Organization Programme for the Sound Management of Chemicals
(IOMC) was established in 1995 by UNEP, ILO, the Food and Agriculture
Organization of the United Nations, WHO, the United Nations Industrial
Development Organization, the United Nations Institute for Training
and Research, and the Organisation for Economic Co-operation and
Development (Participating Organizations), following recommendations
made by the 1992 UN Conference on Environment and Development to
strengthen cooperation and increase coordination in the field of
chemical safety. The purpose of the IOMC is to promote coordination
of the policies and activities pursued by the Participating
Organizations, jointly or separately, to achieve the sound management
of chemicals in relation to human health and the environment.
WHO Library Cataloguing-in-Publication Data
Principles and methods for assessing allergic hypersensitization
associated with exposure to chemicals.
(Environmental health criteria ; 212)
1.Hypersensitivity - chemically induced 2.Immune tolerance
3.Autoimmunity - physiology 4.Immunologic tests
5.Environmental exposure 6.Occupational exposure 7.Risk
assessment - methods
I.International Programme on Chemical Safety II.Series
ISBN 92 4 157212 4 (NLM Classification: QW 900)
ISSN 0250-863X
The World Health Organization welcomes requests for permission to
reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made to
the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1999
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material in
this publication do not imply the expression of any opinion whatsoever
on the part of the Secretariat of the World Health Organization
concerning the legal status of any country, territory, city, or area
or of its authorities, or concerning the delimitation of its frontiers
or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar nature
that are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
PRINCIPLES AND METHODS FOR ASSESSING ALLERGIC HYPERSENSITIZATION
ASSOCIATED WITH EXPOSURE TO CHEMICALS
PREAMBLE
ABBREVIATIONS
PREFACE
1. THE IMMUNE SYSTEM
1.1. Introduction
1.1.1. Evolution and function of the adaptive immune
system
1.1.2. Immunosuppression, immunodeficiency and
autoimmunity
1.1.3. Allergy and allergic diseases
1.1.4. Conclusion
1.2. Physiology and components of the immune system
1.2.1. T-cells
1.2.1.1 Balancing the immune response
1.2.2. B-cells
1.2.3. Macrophages
1.2.4. Antigen-presenting cells
1.2.4.1 Co-stimulatory molecules in T-cell
activation
1.2.5. Adhesion molecules
1.2.6. Fc receptors
1.2.7. Polymorphonulear leukocytes
1.2.8. Cytotoxic lymphocytes
1.2.9. Mast cells
1.2.10. Basophils
1.2.11. Eosinophils
1.2.12. Complement components
1.2.13. Immunoglobulins
1.2.13.1 IgG
1.2.13.2 IgA
1.2.13.3 IgM
1.2.13.4 IgD
1.2.13.5 IgE
1.3. Immunotoxicology
1.4. Immunosuppression/immunodeficiency
1.4.1. Biological basis of
immunosuppression/immunodeficiency
1.4.2. Consequences of immunosuppression/immunodeficiency
1.5. Immunological tolerance
1.5.1. T-cell tolerance to self-antigens
1.5.2. B-cell tolerance to self antigens
1.5.3. Tolerance to non-self antigens
1.5.3.1 Scope
1.5.3.2 Mucosal defence against exogenous toxic
pressures
1.5.3.3 Induction of oral tolerance
1.5.3.4 Factors determining the development of
oral tolerance
1.5.3.5 Orally induced flare-up reactions and
desensitization
1.5.3.6 Mechanisms of tolerance
1.5.3.7 Conclusions
2. HYPERSENSITIVITY AND AUTOIMMUNITY -- OVERVIEW OF MECHANISMS
2.1. Classification of immune reactions
2.1.1. Type I hypersensitivity
2.1.1.1 Anaphylaxis
2.1.2. Type II hypersensitivity
2.1.3. Type III hypersensitivity -- immune complex
reaction
2.1.3.1 Arthus reaction
2.1.4. Type IV -- delayed-type hypersensitivity
2.1.4.1 Mechanisms of allergic contact
dermatitis
2.1.4.2 T-cell responses in chemically induced
pulmonary diseases
2.1.5. Type V stimulatory hypersensitivity
2.2. Regulation of hypersensitivity
2.2.1. Regulation of IgE synthesis by IL-4 and IFN-gamma
2.2.2. Eosinophilia and IL-5
2.2.3. The relationship between Th2 cells and type I
hypersensitivity
2.2.4. IL-12 drives the immune response towards Th1
2.2.5. IL-13, an interleukin-4-like cytokine
2.3. Autoimmune reactions
2.4. Possible mechanisms of autoimmune reactions
2.4.1. Release of anatomically sequestered antigens
2.4.2. The "cryptic self" hypothesis
2.4.3. The self-ignorance hypothesis
2.4.4. The molecular mimicry hypothesis
2.4.5. The "modified self" hypothesis
2.4.5.1 Hapten-induced antibody responses to
"modified self"
2.4.5.2 Hapten-induced autoantibodies that
recognize "self" proteins
2.4.6. Immunoregulatory disturbances
2.4.6.1 Errors in central or peripheral
tolerance
2.4.6.2 Polyclonal activators
2.5. Type I hypersensitivity diseases and allied disorders
2.5.1. Asthma
2.5.1.1 Definition
2.5.1.2 Airways inflammation and asthma
2.5.2. Occupational asthma
2.5.2.1 Occupational asthma and allergy
2.5.3. Atmospheric pollutants and asthma
2.5.4. Rhinitis
2.5.5. Atopic eczema
2.5.6. Urticaria
2.5.7. Gastrointestinal tract diseases: mechanisms of
food-induced symptoms
2.5.7.1 Non IgE-mediated food-sensitive
enteropathy
2.5.7.2 IgE-mediated food allergy
2.5.7.3 Role of gastrointestinal tract
physiology in food allergy
2.6. Type II hypersensitivity diseases
2.6.1. Drug-induced Type II reactivity
2.6.2. Transfusion reactions
2.6.3. Autoimmune haemolytic anaemia
2.6.4. Autoimmune thrombocytopenic purpura
2.6.5. Pemphigus and pemphigoid
2.6.6. Myasthenia gravis
2.7. Type III hypersensitivity diseases
2.7.1. Immune complex disease
2.7.2. Serum sickness
2.7.3. Allergic bronchopulmonary aspergillosis
2.7.4. Extrinsic allergic alveolitis
2.7.4.1 Farmer's lung
2.7.4.2 Bird-fancier's lung
2.8. Type IV hypersensitivity diseases
2.8.1. Chronic beryllium disease
2.8.2. Systemic autoimmune diseases
2.8.2.1 Systemic lupus erythematosus
2.8.2.2 Rheumatoid arthritis
2.8.2.3 Scleroderma
2.8.2.4 Sjögren's syndrome
2.8.2.5 Hashimoto's disease
3. FACTORS INFLUENCING ALLERGENICITY
3.1. Introduction
3.2. Inherent allergenicity
3.2.1. Inherent properties of chemicals inducing
autoimmunity
3.3. Exogenous factors affecting sensitization
3.3.1. Exposure
3.3.1.1 Magnitude of exposure
3.3.1.2 Frequency of exposure
3.3.1.3 Route of exposure
3.3.2. Atmospheric pollution
3.3.2.1 Tobacco smoke
3.3.2.2 Geographical factors
3.3.3. Metals
3.3.4. Detergents
3.4. Endogenous factors affecting sensitization
3.4.1. Genetic influence
3.4.1.1 Contact sensitization
3.4.1.2 IgE-related allergy
3.4.1.3 Other genetic factors
3.4.2. Tolerance
3.4.2.1 Orally induced flare-up reactions and
desensitization
3.4.2.2 Non-specific and specific mechanisms of
unresponsiveness
3.4.3. Underlying disease
3.4.4. Age
3.4.5. Diet
3.4.6. Gender
4. CLINICAL ASPECTS OF THE MOST IMPORTANT ALLERGIC DISEASES
4.1. Clinical aspects of allergic contact dermatitis
4.1.1. Introduction
4.1.2. Regional dermatitis
4.1.2.1 Hand eczema
4.1.2.2 Facial dermatitis
4.1.2.3 Other types of dermatitis
4.1.3. Special types of allergic contact reactions
4.1.3.1 Systemic contact dermatitis
4.1.3.2 Allergic photo-contact dermatitis
4.1.3.3 Non-eczematous reactions
4.1.3.4 Allergic contact urticaria
4.1.4. Allergic contact dermatitis as an occupational
disease
4.1.5. Diagnostic methods
4.1.5.1 Patch testing
4.1.5.2 In vitro testing
4.1.6. Assessment of exposure
4.1.7. Treatment and prevention of allergic contact
dermatitis
4.1.7.1 Primary prevention
4.1.7.2 Secondary prevention
4.1.7.3 Ways of preventing contact sensitization
4.1.8. Information needed for a preventative programme
4.2. Atopic eczema (atopic dermatitis)
4.2.1. Definition
4.2.2. Epidemiology of atopic eczema
4.2.3. Clinical manifestations and diagnostic criteria
4.2.3.1 Age-dependent clinical manifestations
4.2.3.2 Diagnosis of atopic eczema
4.2.3.3 Stigmata of the atopic constitution
4.2.3.4 Prognosis
4.2.4. Etiology
4.2.4.1 Genetic influence
4.2.5. Environmental provocation factors
4.2.6. Pathophysiology
4.2.6.1 Dry skin
4.2.6.2 Autonomic dysregulation
4.2.6.3 Cellular immunodeficiency
4.2.6.4 Increased IgE production
4.2.6.5 Psychosomatic aspects
4.2.7. Diagnostic approach
4.2.7.1 Medical history
4.2.7.2 Skin tests
4.2.7.3 Laboratory tests
4.2.7.4 Provocation tests
4.2.8. Therapeutic considerations
4.2.8.1 Avoidance of provocation factors
4.2.8.2 Basic dermatological therapy
4.2.8.3 Anti-inflammatory therapy
4.2.9. Conclusion
4.3. Allergic rhinitis and conjunctivitis
4.3.1. Introduction
4.3.2. Definition
4.3.3. Clinical manifestations
4.3.3.1 Seasonal allergic rhinitis and
conjunctivitis (hay fever, pollinosis)
4.3.3.2 Perennial allergic rhinitis and
conjunctivitis
4.3.3.3 Prognosis
4.3.4. Etiology
4.3.4.1 Allergic rhinitis and conjunctivitis
caused by contact with chemicals
4.3.5. Pathophysiology
4.3.6. Diagnostic techniques
4.3.6.1 Medical history
4.3.6.2 Clinical examination
4.3.6.3 Allergy testing
4.3.7. Therapeutic considerations
4.4. Clinical aspects of allergic asthma caused by contact with
chemicals
4.4.1. Introduction
4.4.2. Importance of occupational asthma
4.4.3. Chemical causes of occupational asthma
4.4.3.1 Isocyanates
4.4.3.2 Acid anhydrides
4.4.3.3 Complex platinum salts
4.4.4. Diagnosis of occupational asthma
4.4.4.1 Investigation of causes of occupational
asthma
4.4.4.2 Serial peak expiratory flow (PEF) rate
measurements
4.4.4.3 Immunological investigations
4.4.4.4 Inhalation challenge tests
4.4.5. Outcome of occupational asthma
4.4.6. Management and prevention of occupational asthma
4.5. Food allergy
4.5.1. Definitions
4.5.2. IgE-mediated food allergy
4.5.2.1 Oral allergy syndrome
4.5.2.2 Allergic reactions after ingestion of
food
4.5.2.3 Allergic reactions following skin
contact with food
4.5.3. Non-IgE-mediated immune reactions
4.5.3.1 Gluten-sensitive enteropathy (coeliac
disease)
4.5.4. Diagnosis of adverse food reactions
4.5.4.1 Case history and elimination diet
4.5.4.2 Skin tests
4.5.4.3 Specific serum IgE
4.5.4.4 IgG determination
4.5.4.5 Other in vitro tests
4.5.4.6 Oral challenge tests
4.5.5. Therapeutic considerations
4.5.6. Prevalence
4.5.6.1 Introduction
4.5.6.2 Children
4.5.6.3 Adults
4.5.6.4 Conclusions
4.6. Autoimmune diseases associated with drugs, chemicals and
environmental factors
4.6.1. Introduction
4.6.2. Systemic lupus erythematosus
4.6.3. Scleroderma: environmental and drug exposure
4.6.4. Silicone breast implants
4.6.5. Toxic oil syndrome
4.6.6. Eosinophilia-myalgia syndrome
4.6.7. Vinyl chloride disease (occupational
acro-o-steolysis)
4.6.8. Systemic vasculitis: environmental factors and
drugs
4.6.9. Conclusion
5. EPIDEMIOLOGY OF ASTHMA AND ALLERGIC DISEASE
5.1. Introduction
5.2. Definition and measurement of allergic disease
5.2.1. Asthma
5.2.1.1 Definition
5.2.1.2 Assessment
5.2.2. Rhinitis
5.2.3. Atopic dermatitis
5.2.3.1 Definition
5.2.3.2 Assessment
5.2.4. Skin-prick test and serum IgE
5.2.5. Allergic contact dermatitis
5.3. Asthma and atopy: prevalence rates and time trends in
prevalence rates
5.3.1. Europe
5.3.1.1 Prevalences
5.3.1.2 Time trends
5.3.2. Oceania
5.3.2.1 Prevalences
5.3.2.2 Time trends
5.3.3. Eastern Mediterranean
5.3.4. Africa
5.3.5. Asia
5.3.5.1 Prevalences
5.3.5.2 Time trends
5.3.6. North America
5.3.6.1 Prevalences
5.3.6.2 Time trends
5.3.7. The International Study of Asthma and Allergies in
Childhood
5.3.8. Conclusion
5.4. Age and gender distribution
5.5. Migration
5.6. Viral infection
5.7. Socioeconomic status
5.8. Occupational exposure
5.8.1. Chemicals with low relative molecular mass
5.8.1.1 Diisocyanates
5.8.1.2 Acrylates
5.8.1.3 Anhydrides
5.8.1.4 Solder flux
5.8.2. Metals
5.8.2.1 Cobalt
5.8.2.2 Metal-polishing industry
5.8.2.3 Aluminium
5.8.2.4 Platinum salts
5.8.3. Natural rubber latex
5.8.4. Flour
5.8.5. Animals
5.8.6. Other agents
5.9. Allergic contact dermatitis
5.9.1. Epidemiology of allergic contact dermatitis
5.9.1.1 Nickel
5.9.1.2 Chromates
5.9.1.3 Fragrances
5.9.1.4 Preservatives
5.9.1.5 Medicines
5.9.1.6 Plants and woods
5.9.2. Lack of a relationship between atopy and allergic
contact sensitization
5.10. Diet
5.10.1. Breast feeding
5.10.2. Sodium
5.10.3. Selenium
5.10.4. Vitamins and antioxidants
5.11. Number of siblings and crowding
5.12. Indoor environment
5.12.1. Tobacco smoke
5.12.2. Pets
5.12.3. Biocontaminants
5.12.3.1 House dust mites and insects
5.12.3.2 Moulds
5.12.4. Other indoor factors
5.13. Indoor and outdoor environmental factors
5.13.1. Nitrogen dioxide
5.13.2. Sulfur dioxide, acid aerosols and particulate
matter
5.13.3. Volatile organic compounds, formaldehyde and other
chemicals
5.14. Outdoor air pollution
5.14.1. Pollen and dust
5.14.2. Ozone
5.14.3. Motor vehicle emissions
5.15. Conclusions
6. HAZARD IDENTIFICATION: DEMONSTRATION OF ALLERGENICITY
6.1. Hazard and risk; allergy and toxicity
6.1.1. Testing allergic potential and toxicity testing
6.1.2. Databases and prior experience
6.2. Validation and quality assurance
6.3. Structure-activity relationships
6.3.1. Case-Multicase system
6.3.2. DEREK skin sensitization rulebase
6.3.3. SAR for respiratory hypersensitivity
6.4. Predictive testing in vivo
6.4.1. Testing for skin allergy
6.4.1.1 Testing in guinea-pigs
6.4.1.2 Testing in mice
6.4.1.3 Predictive testing for skin allergy in
humans
6.4.2. Testing for respiratory allergy
6.4.2.1 Guinea-pig model
6.4.2.2 Mouse IgE model
6.4.2.3 Rat model
6.4.2.4 Predictive testing for respiratory
allergy in humans
6.4.2.5 Cytokine fingerprinting
6.5. Testing for food allergy
6.6. In vitro approaches
6.7. Testing for autoimmunity
6.7.1. Popliteal lymph node assay
6.7.2. Animal models of autoimmune disease
6.8. Clues from general toxicity tests
7. RISK ASSESSMENT
7.1. Introduction
7.2. Risk assessment of allergy
7.3. Factors in risk assessment of allergy
7.4. Information aspects
7.4.1. No information about hazard
7.4.2. Scanty or no information about exposure
7.4.3. Unreliable or scanty information about risk
7.5. Conclusions
8. TERMINOLOGY
9. CONCLUSIONS
10. RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH
11. FURTHER RESEARCH
REFERENCES
CONCLUSIONS
CONCLUSIONES
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the criteria
monographs as accurately as possible without unduly delaying their
publication. In the interest of all users of the Environmental Health
Criteria monographs, readers are requested to communicate any errors
that may have occurred to the Director of the International Programme
on Chemical Safety, World Health Organization, Geneva, Switzerland, in
order that they may be included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Case postale
356, 1219 Châtelaine, Geneva, Switzerland (telephone no. + 41
22 - 9799111, fax no. + 41 22 - 7973460, E-mail irptc@unep.ch).
* * *
This publication was made possible by grant number
5 U01 ES02617-15 from the National Institute of Environmental Health
Sciences, National Institutes of Health, USA, and by financial support
from the European Commission.
Environmental Health Criteria
PREAMBLE
Objectives
In 1973 the WHO Environmental Health Criteria Programme was
initiated with the following objectives:
(i) to assess information on the relationship between exposure
to environmental pollutants and human health, and to provide
guidelines for setting exposure limits;
(ii) to identify new or potential pollutants;
(iii) to identify gaps in knowledge concerning the health effects
of pollutants;
(iv) to promote the harmonization of toxicological and
epidemiological methods in order to have internationally
comparable results.
The first Environmental Health Criteria (EHC) monograph, on
mercury, was published in 1976 and since that time an ever-increasing
number of assessments of chemicals and of physical effects have been
produced. In addition, many EHC monographs have been devoted to
evaluating toxicological methodology, e.g., for genetic, neurotoxic,
teratogenic and nephrotoxic effects. Other publications have been
concerned with epidemiological guidelines, evaluation of short-term
tests for carcinogens, biomarkers, effects on the elderly and so
forth.
Since its inauguration the EHC Programme has widened its scope,
and the importance of environmental effects, in addition to health
effects, has been increasingly emphasized in the total evaluation of
chemicals.
The original impetus for the Programme came from World Health
Assembly resolutions and the recommendations of the 1972 UN Conference
on the Human Environment. Subsequently the work became an integral
part of the International Programme on Chemical Safety (IPCS), a
cooperative programme of UNEP, ILO and WHO. In this manner, with the
strong support of the new partners, the importance of occupational
health and environmental effects was fully recognized. The EHC
monographs have become widely established, used and recognized
throughout the world.
The recommendations of the 1992 UN Conference on Environment and
Development and the subsequent establishment of the Intergovernmental
Forum on Chemical Safety with the priorities for action in the six
programme areas of Chapter 19, Agenda 21, all lend further weight to
the need for EHC assessments of the risks of chemicals.
Scope
The criteria monographs are intended to provide critical reviews
on the effect on human health and the environment of chemicals and of
combinations of chemicals and physical and biological agents. As
such, they include and review studies that are of direct relevance for
the evaluation. However, they do not describe every study carried
out. Worldwide data are used and are quoted from original studies,
not from abstracts or reviews. Both published and unpublished reports
are considered and it is incumbent on the authors to assess all the
articles cited in the references. Preference is always given to
published data. Unpublished data are only used when relevant
published data are absent or when they are pivotal to the risk
assessment. A detailed policy statement is available that describes
the procedures used for unpublished proprietary data so that this
information can be used in the evaluation without compromising its
confidential nature (WHO (1990) Revised Guidelines for the Preparation
of Environmental Health Criteria Monographs. PCS/90.69, Geneva, World
Health Organization).
In the evaluation of human health risks, sound human data,
whenever available, are preferred to animal data. Animal and in
vitro studies provide support and are used mainly to supply evidence
missing from human studies. It is mandatory that research on human
subjects is conducted in full accord with ethical principles,
including the provisions of the Helsinki Declaration.
The EHC monographs are intended to assist national and
international authorities in making risk assessments and subsequent
risk management decisions. They represent a thorough evaluation of
risks and are not, in any sense, recommendations for regulation or
standard setting. These latter are the exclusive purview of national
and regional governments.
Content
The layout of EHC monographs for chemicals is outlined below.
* Summary -- a review of the salient facts and the risk evaluation
of the chemical
* Identity -- physical and chemical properties, analytical methods
* Sources of exposure
* Environmental transport, distribution and transformation
* Environmental levels and human exposure
* Kinetics and metabolism in laboratory animals and humans
* Effects on laboratory mammals and in vitro test systems
* Effects on humans
* Effects on other organisms in the laboratory and field
* Evaluation of human health risks and effects on the environment
* Conclusions and recommendations for protection of human health
and the environment
* Further research
* Previous evaluations by international bodies, e.g., IARC, JECFA,
JMPR
Selection of chemicals
Since the inception of the EHC Programme, the IPCS has organized
meetings of scientists to establish lists of priority chemicals for
subsequent evaluation. Such meetings have been held in: Ispra, Italy,
1980; Oxford, United Kingdom, 1984; Berlin, Germany, 1987; and North
Carolina, USA, 1995. The selection of chemicals has been based on the
following criteria: the existence of scientific evidence that the
substance presents a hazard to human health and/or the environment;
the possible use, persistence, accumulation or degradation of the
substance shows that there may be significant human or environmental
exposure; the size and nature of populations at risk (both human and
other species) and risks for environment; international concern, i.e.
the substance is of major interest to several countries; adequate data
on the hazards are available.
If an EHC monograph is proposed for a chemical not on the
priority list, the IPCS Secretariat consults with the Cooperating
Organizations and all the Participating Institutions before embarking
on the preparation of the monograph.
Procedures
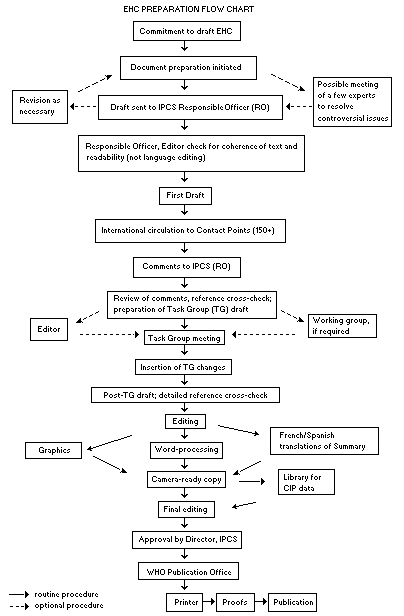
The order of procedures that result in the publication of an EHC
monograph is shown in the flow chart. A designated staff member of
IPCS, responsible for the scientific quality of the document, serves
as Responsible Officer (RO). The IPCS Editor is responsible for
layout and language. The first draft, prepared by consultants or,
more usually, staff from an IPCS Participating Institution, is based
initially on data provided from the International Register of
Potentially Toxic Chemicals, and reference data bases such as Medline
and Toxline.
The draft document, when received by the RO, may require an
initial review by a small panel of experts to determine its scientific
quality and objectivity. Once the RO finds the document acceptable as
a first draft, it is distributed, in its unedited form, to well over
150 EHC contact points throughout the world who are asked to comment
on its completeness and accuracy and, where necessary, provide
additional material. The contact points, usually designated by
governments, may be Participating Institutions, IPCS Focal Points, or
individual scientists known for their particular expertise. Generally
some four months are allowed before the comments are considered by the
RO and author(s). A second draft incorporating comments received and
approved by the Director, IPCS, is then distributed to Task Group
members, who carry out the peer review, at least six weeks before
their meeting.
The Task Group members serve as individual scientists, not as
representatives of any organization, government or industry. Their
function is to evaluate the accuracy, significance and relevance of
the information in the document and to assess the health and
environmental risks from exposure to the chemical. A summary and
recommendations for further research and improved safety aspects are
also required. The composition of the Task Group is dictated by the
range of expertise required for the subject of the meeting and by the
need for a balanced geographical distribution.
The three cooperating organizations of the IPCS recognize the
important role played by nongovernmental organizations.
Representatives from relevant national and international associations
may be invited to join the Task Group as observers. While observers
may provide a valuable contribution to the process, they can only
speak at the invitation of the Chairperson. Observers do not
participate in the final evaluation of the chemical; this is the sole
responsibility of the Task Group members. When the Task Group
considers it to be appropriate, it may meet in camera.
All individuals who as authors, consultants or advisers
participate in the preparation of the EHC monograph must, in addition
to serving in their personal capacity as scientists, inform the RO if
at any time a conflict of interest, whether actual or potential, could
be perceived in their work. They are required to sign a conflict of
interest statement. Such a procedure ensures the transparency and
probity of the process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, it then goes for language editing, reference checking, and
preparation of camera-ready copy. After approval by the Director,
IPCS, the monograph is submitted to the WHO Office of Publications for
printing. At this time a copy of the final draft is sent to the
Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern for
health or environmental effects of the agent because of greater
exposure; an appreciable time period has elapsed since the last
evaluation.
All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
 WHO TASK GROUP MEETING ON PRINCIPLES AND METHODS FOR ASSESSING
ALLERGIC HYPERSENSITIZATION ASSOCIATED WITH EXPOSURE TO CHEMICALS
Members
Professor V. Bencko, Institute of Hygiene and Epidemiology,
Charles University, Prague, Czech Republic
Dr K. Brockow, Clinic for Dermatology and Allergic Disease,
Biederstein Technical University, Munich, Germany
Professor A.D. Dayan, Department of Toxicology, Department of
Health, St Bartholomew's Hospital Medical College, London, United
Kingdom ( Chairman)
Dr D. D'Cruz, Department of Rheumatology, Royal London
Hospital, London, United Kingdom
Professor M. Eglite, Institute of Occupational and Environmental
Health, Medical Academy of Latvia, Riga, Latvia
Dr M.-A. Flyvholm, Department of Allergy and Irritation, National
Institute of Occupational Health, Copenhagen, Denmark
Dr J. Gergely, Department of Immunology, Lorand Eötvös
University, God, Hungary
Dr D. Germolec, National Toxicology Program, National Institute
of Environmental Health Sciences, Research Triangle Park, North
Carolina, USA ( Joint Rapporteur)
Dr H.S. Koren, National Health and Environmental Effects
Research Laboratory, US Environmental Protection Agency, Research
Triangle Park, North Carolina, USA
Dr M. Lovik, National Institute of Public Health, Oslo, Norway
( Joint Rapporteur)
Dr C. Madsen, Institute of Toxicology, Danish Veterinary and Food
Administration, Söborg, Denmark
Dr A. Penninks, Nutrition and Food Research Institute TNO, Zeist,
Netherlands
Professor R.J. Scheper, Institute of Pathology, Amsterdam,
Netherlands
Dr H. van Loveren, Laboratory for Pathology, National Institute of
Public Health and the Environment, Bilthoven, Netherlands
( Vice-Chairman)
Dr B.M.E. von Blomberg, Institute of Pathology, Amsterdam,
Netherlands
Dr J.G. Vos, National Institute of Public Health and the
Environment, Bilthoven, Netherlands
Secretariat
Dr E.M. Smith, International Programme on Chemical Safety,
World Health Organization, Geneva, Switzerland
Assisting the Secretariat
Dr H. Duhme, Institute for Epidemiology and Social Medicine,
Münster, Germany (8-10 September 1997)
Dr M. Kammüller, Rheinfelden, Germany (8-10 September 1997)
Professor M.H. Karol, Department of Environmental and Occupational
Health, University of Pittsburgh, Pittsburg, PA, USA (8-10 September
1997)
Dr I. Kimber, ZENECA Central Toxicology Laboratory, Alderley Park,
Cheshire, United Kingdom (11-12 September 1997)
Representatives of other Organizations
Dr D. Basketter, Unilever, Sharnbrook, Bedford, United Kingdom
(representing the European Centre for Ecotoxicology and Toxicology of
Chemicals)
Dr D. Metcalfe, Allergy and Immunology Institute, International
Life Sciences Institute, Washington DC, USA
Dr C. D'Ambrosio, Drug Allergy Unit, Catholic University of
Sacred Heart, Rome, Italy (representing the International Union of
Pharmacology).
ENVIRONMENTAL HEALTH CRITERIA ON PRINCIPLES AND METHODS FOR ASSESSING
ALLERGIC HYPERSENSITIZATION ASSOCIATED WITH EXPOSURE TO CHEMICALS
A WHO Task Group on Principles and Methods for Assessing Allergic
Hypersensitization Associated with Exposure to Chemicals met at the
National Institute of Public Health and the Environment, Bilthoven,
Netherlands from 8 to 12 September 1997. Dr E.M. Smith, IPCS, welcomed
the participants on behalf of Dr M. Mercier, Director of the IPCS, and
on behalf of the three IPCS cooperating organizations (UNEP/ILO/WHO).
The Group reviewed and revised the draft and made an evaluation of the
risks to human health and of allergic hypersensitization associated
with exposure to chemicals.
The main authors were
Professor A.D. Dayan, London, United Kingdom
Dr D. D'Cruz, London, United Kingdom
Dr H. Duhme, Münster, Germany
Dr M. Kammüller, Rheinfelden, Germany
Professor M.H. Karol, Pittsburgh, PA, USA
Professor U. Keil, Münster, Germany
Dr I. Kimber, Macclesfield, United Kingdom
Dr H.S. Koren, Research Triangle Park, NC, USA
Dr C. Madsen, Söborg, Denmark
Professor T. Menné, Hellerup, Denmark
Professor A.J. Newman Taylor, London, United Kingdom
Professor J. Ring, Munich, Germany
Professor R.J. Scheper, Amsterdam, Netherlands
Dr H. van Loveren, Bilthoven, Netherlands
Dr B.M.E. von Blomberg, Amsterdam, Netherlands
Professor B. Wüthrich, Zurich, Switzerland
Contributing authors were:
Dr D. Abeck, Munich, Germany
Dr D. Basketter, Sharnbrook, Bedford, United Kingdom
Dr K. Brockow, Munich, Germany
Dr D. Germolec, Research Triangle Park, NC, USA
Dr G. Hughes, London, United Kingdom
Dr M. Lovik, Oslo, Norway
Dr A. Penninks, Zeist, Netherlands
Dr T. Rustemeyer, Amsterdam, Netherlands
Dr E.M. Smith, Geneva, Switzerland
Dr M. Stender, Münster, Germany
Dr S.K. Weiland, Münster, Germany
Dr E.M. Smith and Dr P.G. Jenkins, both of the IPCS Central Unit,
were responsible for the scientific aspects of the monograph and for
the technical editing, respectively.
The efforts of all who helped in the preparation and finalization
of the monograph are gratefully acknowledged.
IPCS expresses its gratitude to the external reviewers who
provided comments and other relevant material, in particular to the
United Kingdom Department of Health, the US Environmental Protection
Agency, the European Centre for Ecotoxicology and Toxicology of
Chemicals (ECETOC), and to the Netherlands National Institute for
Public Health and the Environment (RIVM) for hosting the meeting.
Funds for the preparation, review and publication of this
monograph were generously provided by the US Environmental Protection
Agency, the Department of Toxicology, Department of Health, United
Kingdom, and the Netherlands National Institute for Public Health and
the Environment.
ABBREVIATIONS
APC antigen-presenting cell
COPD chronic obstructive pulmonary disease
DEREK deductive estimation of risk from existing knowledge
DTH delayed-type hypersensitivity
FcR Fc receptor
FEV1 forced expiratory volume in 1 second
FVC forced vital capacity
HIV human immunodeficiency virus
ICAM intercellular adhesion molecule
Ig immunoglobulin
IL interleukin
LAK lymphokine-activated killer
LC Langerhans cell
LPS lipopolysaccharide
MALTs mucosal-associated lymphoid tissues
MDR multiple drug resistance
NCAM neural cell adhesion molecule
NK natural killer
PAM pulmonary alveolar macrophage
PDGFR platelet-derived growth factor receptor
QSAR quantitative structure-activity relationship
SAR structure-activity relationship
SLE systemic lupus erythematosus
TCPA tetrachlorophthalic anhydride
TCR T-cell antigen receptor
TDI toluene diisocyanate
Th T helper
TNF tumour necrosis factor
PREFACE
Normal functioning of the immune system prevents serious
illnesses, such as infections and tumours. Immunotoxicology represents
abnormalities in the immune system produced by exposure to chemicals
and drugs. One consequence of dysfunction of the immune system is
partial or complete immunosuppression, resulting in reduced defences
against these conditions. This is often termed "immunotoxicity" and
the IPCS Environmental Health Criteria monograph 180: Principles and
Methods for Assessing Direct Immunotoxicity Associated with Exposure
to Chemicals (IPCS, 1996) provides an extensive review of the causes,
consequences and detection of this type of disorder.
Allergy is another type of adverse effect on health produced by
harmful immune responses following exposure to certain chemicals. The
initial exposure results in the state of allergic sensitization, in
which the immune system is primed to respond inappropriately on
subsequent exposure to the same agent, and allergy is the functional
disorder caused by that response. The best-known types of allergic
response affect the skin, i.e., allergic contact dermatitis and atopic
eczema, and the airways, i.e., asthma and allergic rhinitis, but any
tissue in the body may be affected.
Allergic responses usually occur to foreign antigens, although
self-antigens may sometimes be the targets of damaging immune
responses. This is known as autoimmunity and may occur because the
self-antigens have been modified by chemicals or because the latter
have adversely affected the control mechanisms that normally prevent
autoimmune reactions.
Both allergic and autoimmune disorders may be caused by the
responses of the immune system to substances of low (e.g., transition
metals and simple organic compounds) or high relative molecular mass
(e.g., proteins, including food components). The harmful reactions may
occur at the site of exposure or systemically. The genetic make-up of
the individual may be one predisposing factor.
Once developed, sensitization persists, sometimes for life, and
further exposure, even to a low concentration of the allergen, may
result in serious disease. After the chemical nature of the substance,
exposure (concentration, route, duration and frequency) is the most
important factor in the development of sensitization, as increased
exposure to allergens leads to increased risk of sensitization.
Allergic disorders represent major ill-health and economic loss to the
public and in the workplace. There are suggestions that pollution and
other environmental factors, such as lifestyle and smoking, may be
involved in the rising number of affected people in both developed and
developing countries.
The incidence of chemically induced autoimmune diseases is low,
but they represent important adverse consequences of the use of
certain medicines and, possibly, of exposure to various chemicals.
The structure and functional processes of the immune system and
the mechanisms of sensitization, allergic responses and autoimmunity
need to be considered in relation to the corresponding disorders and
chemicals known to produce them. This consideration will include
factors that affect the allergenicity of substances and the
development of sensitization and autoimmunity, such as the chemical
nature of allergens, special features of the causal exposures, and the
physiology of affected subjects.
Allergic disorders are important causes of ill-health at work and
in the community, and defining their epidemiology and the evaluation
of methods to study their occurrence are crucial. Hazard
identification and risk assessment are important if the incidence of
allergy and autoimmune disorders is to be contained or reduced. Test
methods for the prediction of some forms of sensitization and the risk
of disease following a given exposure are now available.
Allergic disorders of humans have been described for many years,
but the pace of advances in knowledge of the immune system means that
awareness and understanding of allergy and autoimmunity and their
consequences are increasing. Our understanding of allergy is
developing rapidly, and hypotheses about causes and mechanisms will
change as more is learnt about normal and abnormal functioning of the
immune system.
Because understanding of sensitization, allergy and autoimmunity
is still limited by the extent of knowledge of basic immunology there
is a need for fundamental and applied research in areas of the basic
mechanisms, detection and prevention of allergy.
1. THE IMMUNE SYSTEM
1.1 Introduction
The role of the immune system may be succinctly stated as the
"preservation of integrity". This system is responsible for
identifying what is "self" and what is "non-self". The great
complexity of the mammalian system is an indication of the importance,
as well as the difficulty, of this task. If the system fails to
recognize as non-self an infectious entity or the neoantigens
expressed by a newly arisen tumour, then the host is in danger of
rapidly succumbing to the unopposed invasion. Alternatively, if some
integral bodily tissue is not identified as self, then the host is in
danger of turning its considerable defensive abilities against the
tissue and an autoimmune disease is the result. The cost to the host
of these mistakes, made in either direction, may be quite high.
Therefore, an extremely complex array of organs, cells, soluble
factors and interactions has evolved to regulate this system and
minimize the frequency of either of the above-described errors. Recent
advances in cellular and molecular biology have dramatically increased
our understanding of the mammalian immune system. It is now possible
to study in detail biochemical and signal transduction pathways, as
well as the regulation of genes in lymphocytes, because of the novel
chemical and molecular probes that have been developed. Most
importantly, the identification and characterization of the cells,
cell surface receptors and cytokines that participate in the immune
response have enabled immunologists to produce transgenic and gene
"knockout" (disrupted target gene) mice, which will allow even more
in-depth study of critical elements in the immune response to
antigens. Along with the increased power of experimental immunology
has come the ability to study both the direct and indirect actions of
drugs and environmental chemicals (i.e., xenobiotics) on immunological
processes. Of particular importance are new insights regarding the
interactive role of the immune system with other organ systems such as
the nervous and endocrine systems. By way of mutual physical and
chemical communication between these organ systems, both direct and
indirect alteration of immunological function may occur through the
actions of xenobiotics.
1.1.1 Evolution and function of the adaptive immune system
Even the most primitive species of animals display some form of
immune system that enables identification of "non-self" and that
provides for some rudimentary host defence against environmental
challenges. With the emergence of the vertebrates, however, there is
seen the evolution of an adaptive immune system that has as its
primary physiological responsibility protection of the organism from
microbiological challenge and tumour development. The structure and
function of the immune system at the anatomical, biochemical and
functional levels are broadly comparable in all mammals.
Natural immunity is phylogenetically more ancient than the
adaptive immune response, but nevertheless is of critical importance
in providing resistance to infectious microorganisms, and the
nonspecific or innate immune system acts as a first line of defence.
Among the functions of the natural immune system is provision of a
physicochemical barrier at external surfaces in the skin and the
mucosal tissues of the gastrointestinal, reproductive and respiratory
tracts, and the physical elimination of bacteria by coughing,
sneezing, etc. The ability of these surfaces to renew themselves and
secrete antimicrobial agents such as fatty acids and lysozyme reduces
penetration by microbes. However, microbes that bypass these barriers
must be dealt with by other more advanced immunological mechanisms,
which can be either specific or nonspecific in nature. Cellular
elements of the natural immune system include natural killer (NK)
cells, mononuclear phagocytes, and eosinophil and neutrophil
polymorphonuclear cells. In addition, a complex series of plasma
proteins and glycoproteins together comprise the complement system,
which acts together with antibody in the elimination of bacteria, but
which can also be activated to provide natural immune function in the
absence of, or before, a specific immune response. The adaptive immune
system acts together with innate or natural immune mechanisms to
provide host resistance to infectious and malignant disease.
The adaptive immune system comprises organs, tissues, cells and
molecules that must act in concert to provide an integrated immune
response. The three cardinal characteristics of adaptive immunity are
memory, specificity and the capacity to distinguish between self and
non-self. Each of these characteristics are displayed by lymphocytes:
the main cellular vectors of adaptive immune responses. Immunological
memory is the ability to distinguish a foreign material as a previous
invader and to mount a greatly increased and lasting response to that
particular antigen. This process is the product of immunocompetent
cell cooperativity and allows for both amplification of the immune
response after repeated encounters with the same antigen
(immunization) and tolerance to self tissues. In contrast, nonspecific
or innate mechanisms do not possess individuality and do not lead to
memory.
Mature lymphocytes circulate throughout the body, between and
within lymphoid tissues. If a lymphocyte encounters a foreign antigen
in an appropriate form under suitable conditions then the cell becomes
activated and an immune response is initiated. The primary response
takes place in organized lymphoid tissues. It has been estimated that
in a normal adult human the immune system is capable of recognizing
and responding to many millions of antigens; even antigens that have
never been encountered previously, such as for instance new synthetic
chemicals. This enormous repertoire is provided by the clonal
diversity of lymphocytes; these cells being clonally distributed with
respect to antigen specificity. Thus, each clone of mature lymphocytes
differs one from another in terms of the antigenic structures that
will induce activation. Antigen recognition is effected via
specialized membrane receptors that have diversified among lymphocytes
during development of the immune system by a process of somatic
recombination of antigen receptor genes. It is the possession of these
receptors by lymphocytes that confers specificity to immune responses.
Recognition of antigen by lymphocytes in primary lymphoid tissues
results in rapid cellular activation and the stimulation of division
and differentiation. Division provides for a selective expansion in
numbers of those lymphocytes that are able to recognize and interact
with the inducing antigen. Selective clonal expansion forms the basis
of immunological memory. After first encounter with antigen,
responsive lymphocytes have increased in number such that if the
individual is exposed subsequently to the same antigenic material then
an accelerated and more aggressive response will be mounted. These are
the central events necessary for adaptive immunity and those that are
made use of in vaccination against infectious microorganisms.
All lymphocytes involved in adaptive immune responses interact
specifically with antigen, and they divide and differentiate in
response to antigenic challenge. These cells may be subdivided into
two main populations, T-lymphocytes and B-lymphocytes, that differ
with respect to their origins and development pathways, the way in
which antigen is recognized, and the effector cells into which they
ultimately differentiate. Both populations arise in the bone marrow
from primitive precursors, but thereafter follow discrete
developmental pathways. Cells committed to becoming T-lymphocytes
(pre-T-cells) require passage through and differentiation within the
thymus to achieve immunological maturity. The thymus serves also to
identify and destroy most of those T lymphocytes that display membrane
receptors which would permit interaction with self antigens. When they
leave the thymus the mature antigen-sensitive T-lymphocytes join the
recirculating pool.
Bone marrow derived B-cells also join the recirculating pool
where, with T-lymphocytes, they seek antigen for which they have
complementary membrane receptors. B-lymphocytes recognize antigen
usually in its native form. Activation triggers B-lymphocyte
differentiation and division. The end-cell of B-lymphocyte
differentiation is the plasma cell that possesses the synthetic and
secretory machinery to manufacture and export large amounts of
antibody. The antibody secreted by an individual plasma cell is of a
single specificity and matches identically the specificity of the
membrane receptor on the B-lymphocyte from which the plasma cell
differentiated. The purpose of antibody is essentially to form a
bridge between the inducing antigen and biological mechanisms that
serve to eliminate it. The interaction of antibody with antigen
facilitates the activation of complement (lysis of bacteria) and
phagocytosis by mononuclear phagocytes and neutrophils (intracellular
killing of bacteria) and results in the clearance of pathogenic
bacteria. The importance of B-lymphocytes and the antibodies that
derive from their activation is protection against extracellular
infection by bacteria and parasites.
The existence of T-lymphocytes was recognized for many years
before the true nature of their role in adaptive immune responses was
appreciated. Cell-mediated immune responses effected by T-lymphocyte
participate in host defence against all types of infectious organisms,
but of greatest evolutionary significance is immunity against viruses.
Humoral immunity effected by antibody is of relevance only in the
viraemic stage of viral infections. Viruses are obligate intracellular
parasites and once inside the infected host cell are protected from
antibody-mediated mechanisms.
The overall purpose of these host defence mechanisms is to
provide the organism with resistance to a challenging microbial
environment and to confer protection from the internal development of
non-self neoplasms or tumours. When normal immune function is absent
or compromised, the consequences for human health are serious.
Consideration of immunosuppression and immunodeficiency illustrates
the evolutionary importance of immune function.
1.1.2 Immunosuppression, immunodeficiency and autoimmunity
Active immune function is clearly beneficial for health, whereas
the consequences of a compromised immune system are adverse health
effects.
Immunodeficiency disorders can be congenital or acquired.
Congenital immunodeficiency is comparatively rare, but is frequently
very serious and can be fatal. Examples include a complete, or almost
complete, failure of the immune system to develop due to the absence
or aberrant maturation of lymphocyte or leukocyte progenitors,
resulting in severe combined immunodeficiency disease or reticular
dysgenesis. Without appropriate treatment these conditions are fatal,
children succumbing to overwhelming infection.
Acquired immunodeficiency can be secondary to malnutrition,
severe stress, treatment with immunosuppressive drugs or with cancer
chemotherapeutic agents, exposure to certain environmental chemicals
or infection, such as infection with the human immunodeficiency virus
(HIV), the cause of acquired immunodeficiency syndrome (AIDS). In all
instances immunosuppression is associated with reduced host resistance
and more persistent infection, often with unusual microorganisms that
are resisted well by immunocompetent individuals. Immunodeficiency is,
in addition, associated with an increased incidence of malignant
diseases that are known or suspected to be associated with oncogenic
viruses.
The benefits that derive from active immune function do not come
without a cost, however. While the adaptive immune system acts as a
"friend" in providing host defence, it may also act as a "foe", being
instrumental in the pathogenesis of certain diseases. The immune
system can, for instance, turn on the host if the fine discrimination
between self and non-self breaks down. The result is the development
of autoimmune responses and autoimmune disease. The mechanisms by
which autoimmunity develops are multifactorial, complex and remain
poorly understood. The majority of cases are idiopathic, although
diseases such as systemic sclerosis have been associated with organic
chemicals and silica.
1.1.3 Allergy and allergic diseases
Allergy may be defined as the adverse health effects resulting
from hypersensitivity caused by exposure to an exogenous antigen
(allergen) resulting in a marked increase in reactivity and
responsiveness to that particular antigen on subsequent exposure.
Allergy is not necessarily, or usually, the consequence of perturbed
immune function, but the result of an immune system response to an
antigen (in this case allergen) in such a way that a temporary or
long-lasting disease results. The immunological processes that are
involved in the development of allergic responses and allergic disease
are in principle and practice no different to those that provide
protective immunity and host resistance against potential pathogens.
Allergy normally develops in two phases. The first phase is
induced following initial encounter of the susceptible individual with
the allergen. A primary immune response is mounted that results in a
state of heightened responsiveness to that particular antigen
(specific sensitization). In immunological terms sensitization to an
allergen does not differ from immunization to a pathogenic
microorganism. Following second or subsequent exposure of the now
sensitized individual to the inducing allergen a more vigorous and
accelerated secondary immune response is provoked and it is at this
stage that adverse health effects are normally first recognized. The
aggressive secondary immune response against the allergen causes local
tissue disruption and inflammation that is recognized clinically as
allergic disease.
Individuals vary widely in terms of allergic responsiveness and
susceptibility to allergic disease. There are a number of factors of
importance here including opportunities for encounter with the
inducing allergen, the route, the dose or concentration of allergen,
extent and duration of exposure and genetic predisposition. The latter
is incompletely understood but clearly impacts significantly upon
susceptibility. Respiratory allergy (including hay fever and asthma)
to protein aeroallergens is associated frequently with atopy; a
genetic predisposition for increased production of IgE, the class of
antibody that causes respiratory hypersensitivity to proteins. In
addition, the immunological repertoire of individuals and the ability
of their immune system to recognize and respond to certain antigenic
structures will also influence susceptibility.
Allergic diseases are widespread and can be caused by allergens
encountered in the external environment, home or work. They range from
comparatively mild inflammatory responses localized to a single site
to systemic anaphylactic responses that may prove fatal. Allergic
disease, as well as representing an important and widespread health
problem, is also of great economic significance with respect to the
cost of health care and time lost from work. It has been recognized
that some forms of allergy are increasing in prevalence, compounding
the health impact of these diseases. The incidence of asthma, for
instance, has grown significantly in some developed countries, an
increase that may be attributable to changing allergen exposure
patterns, alterations in lifestyle, environmental pollution or to a
combination of all of these factors.
In the context of occupational and environmental health the two
most important allergic diseases caused by exposure to chemicals are
allergic contact dermatitis and respiratory hypersensitivity. The
former is very common and can be induced by industrial chemicals,
metals and natural products. Sensitization results from dermal
exposure of the susceptible individual to the inducing allergen.
Allergic contact dermatitis reactions are provoked subsequently when
the now sensitized individual is exposed for a second time to the
inducing allergen at the same or different skin site. Many hundreds of
contact allergens, varying enormously in potency, have been
identified.
Although from the occupational and environmental health
standpoint allergic contact dermatitis and respiratory
hypersensitivity represent the most important types of allergy induced
by chemicals, it should not be forgotten that exposure to xenobiotics
has been implicated in other forms of allergic disease. Certain drugs
are associated with systemic allergic reactions that are sometimes
reminiscent of autoimmune diseases. In addition, food components and
food additives are implicated in adverse reactions, which in some
cases take the form of an allergic response.
1.1.4 Conclusion
An active adaptive immune system is essential for health and
survival in a hostile microbiological environment. A price paid for
the host resistance provided by the immune system is that some immune
responses, often to benign antigens, result in the adverse health
effects of allergic disease.
1.2 Physiology and components of the immune system
Immunity refers to all those physiological mechanisms/processes
that enable an animal (i.e., the host) to recognize materials as
foreign to "self" and to neutralize, eliminate or metabolize them,
with or without injury to its own tissue. The immune system of higher
animals is therefore capable of distinguishing between self materials
from which they are constituted and "non-self" (i.e., those that are
foreign or antigenic). It probably evolved to confer a selective
advantage to organisms that could withstand colonization and microbial
invasion. The immune response must decipher sometimes quite subtle
differences between self and non-self, without error, to both provide
protection and avoid self-attack. Accomplishment of this selective
process requires the concerted action of a number of cell types.
Mammals have developed a highly complex, intertwined and redundant
system composed of layers of protective mechanisms to cope with more
sophisticated environmental threats.
The immune system comprises both lymphoid organs and specialized
cells. Erythrocytes, myeloid cells, megakaryocytes (which mature to
form platelets) and lymphocytes arise from a totipotent or pluripotent
stem cell in the yolk sac of the developing fetus and, later, the
fetal liver. In adult mammals, the stem cells are manufactured in the
bone marrow and progress via different pathways of differentiation to
become mature cells that may carry out specialized functions, such as
antibody production or phagocytosis (Abramson et al., 1977). The
thymus and bone marrow are the primary lymphoid organs that serve to
nurture the development of stem cells into mature effector cells.
Mature lymphocytes traffic to the secondary lymphoid organs, the lymph
nodes, spleen and mucosal-associated lymphoid tissues (MALTs), and
form immune-reactive units that respond vigorously to antigens. The
design of these secondary organs is such that the specialized
populations of lymphocytes reside in proximity, can interact with each
other, and can regulate the antigen-driven immune response required.
The lymph nodes, which are situated throughout the body, filter out
antigens draining from the peripheral bodily tissues. The spleen
monitors the blood and functions as a factory for red blood cell
turnover. The MALTs provide a frontline defence for microbes that are
ingested. Lymphocytes that reside in the spleen can, upon encountering
antigen, respond in situ or migrate to the site of infection via the
blood, colonizing a sensitized response unit in a local lymph node.
The virgin stem cell is believed to receive different maturational
stimuli in the microenvironment of the bone marrow, with stromal cell
contact and lymphokine exposure inducing entry into one of several
pathways of development. Functional lymphocytes are continuously
formed from stem cells and pass from the bone marrow through the
bloodstream to the lymphoid organs. The migratory pattern of the
lymphocyte determines its lifespan and behaviour, as described in
greater detail below for T-cells, B-cells and other immunocompetent
cells.
1.2.1 T-cells
Stem cells that enter the thymus gland, formed from the third and
fourth pharyngeal pouches in mammals, rapidly divide, acquire their
antigen specificity and are selectively deleted if they bear any
self-reactivity. The "educated" daughter cells, termed thymus-derived
or T-lymphocytes, then leave the thymus and travel to other lymphoid
tissues, persisting for weeks or even years. As stem cells pass
through the thymic subcapsular region, cortex and medulla, they
display plasma membrane-bound surface molecules that define their
function. It is possible to experimentally identify and isolate
subpopulations of T-lymphocytes by exploiting the differential
expression of these marker glycoproteins, using alloantisera or
monoclonal antibodies and immunostaining techniques. Murine
T-lymphocytes possess both the Thy-1 marker and the T-cell antigen
receptor (TCR)-CD3 complex, and fall into two major classes, either
T-helper/inducer cells expressing CD4 or T-suppressor/cytotoxic cells,
which display CD8.
Studies in inbred mice show that the T-cell antigen receptor only
recognizes antigen processed and presented on major histocompatibility
complex (MHC) molecules from the same thymic environment. MHC proteins
are products of the immune response (Ir) genes, which are primarily
responsible for tissue graft and organ transplantation rejection. In
general, CD4+ T-cells complex with antigen associated with MHC Class
I molecules, which are only found on certain cells of the immune
system, while CD8+ T-cells only see antigen when associated with MHC
Class I molecules, located on all nucleated cells. T-cell selection of
this type is termed positive and deletion of clones reactive to self
is termed negative selection (Zinkernagel & Doherty, 1975). Upon
contact with antigen, mature T-cells may either respond clonally in an
antigen-specific manner and initiate an immune response, or become
inactivated and eliminated in a process which is not well understood,
potentially leaving the animal unable to recognize the antigen. This
latter phenomenon is referred to as T-cell anergy.
The majority of lymphocytes in the peripheral blood and lymph
nodes and about one half of the cells in the spleen are T-cells.
Thymectomized animals or naturally occurring athymic or nude mice
(because they are also hairless) and children with Di George syndrome
are immunocompromised hosts that lack cell-mediated immune function
and responses to T-dependent antigens (Sell, 1987). The endocrine
function of the thymus has been recognized through partial recovery of
T-cell function in thymectomized animals given cell-free thymic
extracts, suggesting thymic hormones may, to some extent, replace
thymus-driven T-cell maturation (Law et al., 1968). However, the
thymic microenvironment appears necessary for proper selection and
differentiation of the T-cell repertoire. Imbalances in the function
of mature T-cell subpopulations may also occur clinically, as shown by
HIV infection of CD4+ T cells, resulting in decreased T-helper cell
levels (Stahl et al., 1982; Lane & Fauci, 1985), and systemic lupus
erythematosus in which lowered CD8+ T-suppressor cell activity is
thought to contribute to elevated antibody production and to
exacerbate the autoimmune state.
1.2.1.1 Balancing the immune response
It is clear that in the mouse most T-cells show predominant
production of two different sets of cytokines with pronounced, often
mutually exclusive, effects on different features of the immune
response (Romagnani, 1992a,b; Bloom et al., 1992; Mosmann & Sad,
1996). While some details of cytokine production are known to be
different in the human, they are generally similar to that in the
mouse. In brief, mouse Th1-cells produce IL-2, IFN-gamma and
lymphotoxin (LT), whereas Th2-cells produce IL-4, 5,6,9,10,13, as
shown in Table 1. Human Th1 and Th2 cells produce similar patterns,
although the synthesis of IL-2,6,10,13 is not as tightly restricted to
a single subset as in mouse T-cells. In the mouse Th1-cell (or Type I)
responses result in delayed-type hypersensitivity (DTH) reactions,
activation of macrophages to kill phagocytosed microorganisms, and in
IgG2a, rather than IgG1 and IgE, synthesis. Th2 (Type 2) responses
generate IgG1- and IgE-secreting cells, and eosinophilia. Notably,
Th2-derived IL-4 is an important switch factor for B cells to produce
the IgG1 and IgE immunoglobulin-isotypes. Th1- and Th2-cells arise
from a common lineage since they use the same T-cell receptor
repertoire, and naive precursor T-cells, not yet exhibiting either of
these cytokine profiles (Th0), can differentiate into both directions
(see also section 2.1.5). Although cytotoxic CD8+ T-cells often
secrete a Th1-like cytokine pattern, there is evidence for the
existence of Th2-like CD8+ T (Tc2) cells in humans and mice (Croft
et al., 1994; Mosmann & Sad, 1996). Type 2 cytokines such as IL-4
shift T cell differentiation away from the production of Type I
cytokines, whereas the Type I cytokine IFN-gamma is very potent in
preventing the development of Th2-cells.
Cytokines are soluble mediators synthesized by cells of the
immune system that bind to specific receptors or target cells and
modulate cell function in immunological reactions (Fig. 1). When
starting clonal expansion after antigen stimulation, T-cells develop
major cytokine profiles depending on the site of primary contact.
Along mucosal surfaces predominant local IL-4 release, possibly by
mast cells, basophils or locally residing T-cells, favours the
development of Th2-cells (Scott, 1993; Weiner et al., 1994; Mosmann
et al., 1996). In some individuals over-prone to IgE-switching, this
response may be excessive, leading to mucosal allergies, such as
respiratory hypersensitivity (see also chapter 4). The induction of
Type 2 T-cell responses after antigen introduction along mucosal
surfaces is probably further promoted by high local densities of
B-cells as compared to the skin compartment. B-cells are excellent
IL-10 producers, and antigen-presentation by B-cells is known to
favour Th2 responses (Eynon & Parker, 1992). In addition to the
archetype Type 2 cytokines, TGF-beta has also been associated with Th2
functions, but preferential production by either a Th2 subset, or a
distinct Th3 subset (Chen et al., 1994), is more likely to occur. As
mentioned above, TGF-beta plays the key role in immune suppression
along mucosal surfaces, e.g., by controlling several different
IFN-gamma-associated effector T-cell and macrophage functions
(Karpus & Swanborg, 1991; Oswald et al., 1992; Khoury et al., 1992;
Table 1. Cytokine production in the mouse
Cytokine
production T-cells Other cells
Th0 Th1 Th2 B-cell Macrophage NK-cell Mast cell Keratinocyte LC
IL-1 +alpha +beta
IL-2 + +
IFN-gamma + + +
LT (TNF-beta) + +
IL-3 + + + +
GM-CSF + + + +
TNF-alpha + + + + + +
IL-4 + + + +
IL-5 + +
IL-6 + + + + +
IL-10 + + + + + +
IL-12 + + +
IL-13 + + + + + +
Meade et al., 1992) and by maintaining epithelial cell layer integrity
(Planchon et al., 1994). Moreover, TGF-beta serves as a switch
factor for IgA production. To what extent T-cells preferentially
releasing TGF-beta may also contribute to mucosal tolerance to
IgE-inducing atopic allergens is still unclear. In sharp contrast,
along the skin route local release of IL-12 from, for instance,
macrophages and NK-cells stimulates the production of IFN-gamma by
T cells and facilitates predominant development of Th1 cells. Exposure
of the skin to exogenous antigenic substances, including contact
allergens, therefore preferentially induces specific Type 1,
pro-inflammatory T-cell responses.
1.2.2 B-cells
In contrast to T-lymphocyte maturation, the development of
lymphocytes capable of synthesizing and secreting antibody
(immunoglobulin) molecules in mammals is thought to occur in several
sites, including the bone marrow, spleen and MALTs. Because these
cells were first characterized in birds, which, unlike mammals,
possess a unique lymphoid organ, the bursa of Fabricius, and because
the precursors of these cells are formed in the bone marrow, these
cells have been termed B-lymphocytes. B-cells tend to reside for long
periods of time in the secondary lymphoid organs and form the lymphoid
follicles and germinal centres. Following activation by antigen or
antigen-activated T-helper cells (Noelle et al., 1990) and
lymphokines, B-cells proliferate and terminally differentiate to
antibody-producing plasma cells, which turn over rapidly and are
replenished by newly differentiated cells.
Like the T-cell antigen receptor (TCR)-CD3 complex, B-cells
express surface antigen-combining receptor molecules which are of
identical specificity to the immunoglobulins they synthesize and
secrete. The diversity of the natural world has necessitated a complex
series of molecular events in B-cell development designed to produce a
spectrum of immunoglobulins capable of protecting the organism. B-cell
maturation is marked by immunoglobulin gene rearrangements,
recombinations and somatic mutations, so that a relatively small
number of genes may efficiently produce a large number of antibody
specificities.
B-lymphocytes synthesize immunoglobulins of five different types:
IgM, IgG, IgA, IgD, and IgE. These proteins are composed of two
separate types of polypeptide chains joined by disulfide linkages,
termed the heavy and light chains because of differences in their
relative molecular masses (the heavy chains are about twice as large)
(see Fig. 2). Light chains are derived from either kappa or lambda
genes and combine with the five different heavy chains mu, gamma,
alpha, delta and epsilon (i.e., for the five different types of
immunoglobulin identified above). Enzymatic digestion of
immunoglobulin molecules yields fragments which indicate arrangement
in a Y-shaped structure, consisting of two arms containing the
WHO TASK GROUP MEETING ON PRINCIPLES AND METHODS FOR ASSESSING
ALLERGIC HYPERSENSITIZATION ASSOCIATED WITH EXPOSURE TO CHEMICALS
Members
Professor V. Bencko, Institute of Hygiene and Epidemiology,
Charles University, Prague, Czech Republic
Dr K. Brockow, Clinic for Dermatology and Allergic Disease,
Biederstein Technical University, Munich, Germany
Professor A.D. Dayan, Department of Toxicology, Department of
Health, St Bartholomew's Hospital Medical College, London, United
Kingdom ( Chairman)
Dr D. D'Cruz, Department of Rheumatology, Royal London
Hospital, London, United Kingdom
Professor M. Eglite, Institute of Occupational and Environmental
Health, Medical Academy of Latvia, Riga, Latvia
Dr M.-A. Flyvholm, Department of Allergy and Irritation, National
Institute of Occupational Health, Copenhagen, Denmark
Dr J. Gergely, Department of Immunology, Lorand Eötvös
University, God, Hungary
Dr D. Germolec, National Toxicology Program, National Institute
of Environmental Health Sciences, Research Triangle Park, North
Carolina, USA ( Joint Rapporteur)
Dr H.S. Koren, National Health and Environmental Effects
Research Laboratory, US Environmental Protection Agency, Research
Triangle Park, North Carolina, USA
Dr M. Lovik, National Institute of Public Health, Oslo, Norway
( Joint Rapporteur)
Dr C. Madsen, Institute of Toxicology, Danish Veterinary and Food
Administration, Söborg, Denmark
Dr A. Penninks, Nutrition and Food Research Institute TNO, Zeist,
Netherlands
Professor R.J. Scheper, Institute of Pathology, Amsterdam,
Netherlands
Dr H. van Loveren, Laboratory for Pathology, National Institute of
Public Health and the Environment, Bilthoven, Netherlands
( Vice-Chairman)
Dr B.M.E. von Blomberg, Institute of Pathology, Amsterdam,
Netherlands
Dr J.G. Vos, National Institute of Public Health and the
Environment, Bilthoven, Netherlands
Secretariat
Dr E.M. Smith, International Programme on Chemical Safety,
World Health Organization, Geneva, Switzerland
Assisting the Secretariat
Dr H. Duhme, Institute for Epidemiology and Social Medicine,
Münster, Germany (8-10 September 1997)
Dr M. Kammüller, Rheinfelden, Germany (8-10 September 1997)
Professor M.H. Karol, Department of Environmental and Occupational
Health, University of Pittsburgh, Pittsburg, PA, USA (8-10 September
1997)
Dr I. Kimber, ZENECA Central Toxicology Laboratory, Alderley Park,
Cheshire, United Kingdom (11-12 September 1997)
Representatives of other Organizations
Dr D. Basketter, Unilever, Sharnbrook, Bedford, United Kingdom
(representing the European Centre for Ecotoxicology and Toxicology of
Chemicals)
Dr D. Metcalfe, Allergy and Immunology Institute, International
Life Sciences Institute, Washington DC, USA
Dr C. D'Ambrosio, Drug Allergy Unit, Catholic University of
Sacred Heart, Rome, Italy (representing the International Union of
Pharmacology).
ENVIRONMENTAL HEALTH CRITERIA ON PRINCIPLES AND METHODS FOR ASSESSING
ALLERGIC HYPERSENSITIZATION ASSOCIATED WITH EXPOSURE TO CHEMICALS
A WHO Task Group on Principles and Methods for Assessing Allergic
Hypersensitization Associated with Exposure to Chemicals met at the
National Institute of Public Health and the Environment, Bilthoven,
Netherlands from 8 to 12 September 1997. Dr E.M. Smith, IPCS, welcomed
the participants on behalf of Dr M. Mercier, Director of the IPCS, and
on behalf of the three IPCS cooperating organizations (UNEP/ILO/WHO).
The Group reviewed and revised the draft and made an evaluation of the
risks to human health and of allergic hypersensitization associated
with exposure to chemicals.
The main authors were
Professor A.D. Dayan, London, United Kingdom
Dr D. D'Cruz, London, United Kingdom
Dr H. Duhme, Münster, Germany
Dr M. Kammüller, Rheinfelden, Germany
Professor M.H. Karol, Pittsburgh, PA, USA
Professor U. Keil, Münster, Germany
Dr I. Kimber, Macclesfield, United Kingdom
Dr H.S. Koren, Research Triangle Park, NC, USA
Dr C. Madsen, Söborg, Denmark
Professor T. Menné, Hellerup, Denmark
Professor A.J. Newman Taylor, London, United Kingdom
Professor J. Ring, Munich, Germany
Professor R.J. Scheper, Amsterdam, Netherlands
Dr H. van Loveren, Bilthoven, Netherlands
Dr B.M.E. von Blomberg, Amsterdam, Netherlands
Professor B. Wüthrich, Zurich, Switzerland
Contributing authors were:
Dr D. Abeck, Munich, Germany
Dr D. Basketter, Sharnbrook, Bedford, United Kingdom
Dr K. Brockow, Munich, Germany
Dr D. Germolec, Research Triangle Park, NC, USA
Dr G. Hughes, London, United Kingdom
Dr M. Lovik, Oslo, Norway
Dr A. Penninks, Zeist, Netherlands
Dr T. Rustemeyer, Amsterdam, Netherlands
Dr E.M. Smith, Geneva, Switzerland
Dr M. Stender, Münster, Germany
Dr S.K. Weiland, Münster, Germany
Dr E.M. Smith and Dr P.G. Jenkins, both of the IPCS Central Unit,
were responsible for the scientific aspects of the monograph and for
the technical editing, respectively.
The efforts of all who helped in the preparation and finalization
of the monograph are gratefully acknowledged.
IPCS expresses its gratitude to the external reviewers who
provided comments and other relevant material, in particular to the
United Kingdom Department of Health, the US Environmental Protection
Agency, the European Centre for Ecotoxicology and Toxicology of
Chemicals (ECETOC), and to the Netherlands National Institute for
Public Health and the Environment (RIVM) for hosting the meeting.
Funds for the preparation, review and publication of this
monograph were generously provided by the US Environmental Protection
Agency, the Department of Toxicology, Department of Health, United
Kingdom, and the Netherlands National Institute for Public Health and
the Environment.
ABBREVIATIONS
APC antigen-presenting cell
COPD chronic obstructive pulmonary disease
DEREK deductive estimation of risk from existing knowledge
DTH delayed-type hypersensitivity
FcR Fc receptor
FEV1 forced expiratory volume in 1 second
FVC forced vital capacity
HIV human immunodeficiency virus
ICAM intercellular adhesion molecule
Ig immunoglobulin
IL interleukin
LAK lymphokine-activated killer
LC Langerhans cell
LPS lipopolysaccharide
MALTs mucosal-associated lymphoid tissues
MDR multiple drug resistance
NCAM neural cell adhesion molecule
NK natural killer
PAM pulmonary alveolar macrophage
PDGFR platelet-derived growth factor receptor
QSAR quantitative structure-activity relationship
SAR structure-activity relationship
SLE systemic lupus erythematosus
TCPA tetrachlorophthalic anhydride
TCR T-cell antigen receptor
TDI toluene diisocyanate
Th T helper
TNF tumour necrosis factor
PREFACE
Normal functioning of the immune system prevents serious
illnesses, such as infections and tumours. Immunotoxicology represents
abnormalities in the immune system produced by exposure to chemicals
and drugs. One consequence of dysfunction of the immune system is
partial or complete immunosuppression, resulting in reduced defences
against these conditions. This is often termed "immunotoxicity" and
the IPCS Environmental Health Criteria monograph 180: Principles and
Methods for Assessing Direct Immunotoxicity Associated with Exposure
to Chemicals (IPCS, 1996) provides an extensive review of the causes,
consequences and detection of this type of disorder.
Allergy is another type of adverse effect on health produced by
harmful immune responses following exposure to certain chemicals. The
initial exposure results in the state of allergic sensitization, in
which the immune system is primed to respond inappropriately on
subsequent exposure to the same agent, and allergy is the functional
disorder caused by that response. The best-known types of allergic
response affect the skin, i.e., allergic contact dermatitis and atopic
eczema, and the airways, i.e., asthma and allergic rhinitis, but any
tissue in the body may be affected.
Allergic responses usually occur to foreign antigens, although
self-antigens may sometimes be the targets of damaging immune
responses. This is known as autoimmunity and may occur because the
self-antigens have been modified by chemicals or because the latter
have adversely affected the control mechanisms that normally prevent
autoimmune reactions.
Both allergic and autoimmune disorders may be caused by the
responses of the immune system to substances of low (e.g., transition
metals and simple organic compounds) or high relative molecular mass
(e.g., proteins, including food components). The harmful reactions may
occur at the site of exposure or systemically. The genetic make-up of
the individual may be one predisposing factor.
Once developed, sensitization persists, sometimes for life, and
further exposure, even to a low concentration of the allergen, may
result in serious disease. After the chemical nature of the substance,
exposure (concentration, route, duration and frequency) is the most
important factor in the development of sensitization, as increased
exposure to allergens leads to increased risk of sensitization.
Allergic disorders represent major ill-health and economic loss to the
public and in the workplace. There are suggestions that pollution and
other environmental factors, such as lifestyle and smoking, may be
involved in the rising number of affected people in both developed and
developing countries.
The incidence of chemically induced autoimmune diseases is low,
but they represent important adverse consequences of the use of
certain medicines and, possibly, of exposure to various chemicals.
The structure and functional processes of the immune system and
the mechanisms of sensitization, allergic responses and autoimmunity
need to be considered in relation to the corresponding disorders and
chemicals known to produce them. This consideration will include
factors that affect the allergenicity of substances and the
development of sensitization and autoimmunity, such as the chemical
nature of allergens, special features of the causal exposures, and the
physiology of affected subjects.
Allergic disorders are important causes of ill-health at work and
in the community, and defining their epidemiology and the evaluation
of methods to study their occurrence are crucial. Hazard
identification and risk assessment are important if the incidence of
allergy and autoimmune disorders is to be contained or reduced. Test
methods for the prediction of some forms of sensitization and the risk
of disease following a given exposure are now available.
Allergic disorders of humans have been described for many years,
but the pace of advances in knowledge of the immune system means that
awareness and understanding of allergy and autoimmunity and their
consequences are increasing. Our understanding of allergy is
developing rapidly, and hypotheses about causes and mechanisms will
change as more is learnt about normal and abnormal functioning of the
immune system.
Because understanding of sensitization, allergy and autoimmunity
is still limited by the extent of knowledge of basic immunology there
is a need for fundamental and applied research in areas of the basic
mechanisms, detection and prevention of allergy.
1. THE IMMUNE SYSTEM
1.1 Introduction
The role of the immune system may be succinctly stated as the
"preservation of integrity". This system is responsible for
identifying what is "self" and what is "non-self". The great
complexity of the mammalian system is an indication of the importance,
as well as the difficulty, of this task. If the system fails to
recognize as non-self an infectious entity or the neoantigens
expressed by a newly arisen tumour, then the host is in danger of
rapidly succumbing to the unopposed invasion. Alternatively, if some
integral bodily tissue is not identified as self, then the host is in
danger of turning its considerable defensive abilities against the
tissue and an autoimmune disease is the result. The cost to the host
of these mistakes, made in either direction, may be quite high.
Therefore, an extremely complex array of organs, cells, soluble
factors and interactions has evolved to regulate this system and
minimize the frequency of either of the above-described errors. Recent
advances in cellular and molecular biology have dramatically increased
our understanding of the mammalian immune system. It is now possible
to study in detail biochemical and signal transduction pathways, as
well as the regulation of genes in lymphocytes, because of the novel
chemical and molecular probes that have been developed. Most
importantly, the identification and characterization of the cells,
cell surface receptors and cytokines that participate in the immune
response have enabled immunologists to produce transgenic and gene
"knockout" (disrupted target gene) mice, which will allow even more
in-depth study of critical elements in the immune response to
antigens. Along with the increased power of experimental immunology
has come the ability to study both the direct and indirect actions of
drugs and environmental chemicals (i.e., xenobiotics) on immunological
processes. Of particular importance are new insights regarding the
interactive role of the immune system with other organ systems such as
the nervous and endocrine systems. By way of mutual physical and
chemical communication between these organ systems, both direct and
indirect alteration of immunological function may occur through the
actions of xenobiotics.
1.1.1 Evolution and function of the adaptive immune system
Even the most primitive species of animals display some form of
immune system that enables identification of "non-self" and that
provides for some rudimentary host defence against environmental
challenges. With the emergence of the vertebrates, however, there is
seen the evolution of an adaptive immune system that has as its
primary physiological responsibility protection of the organism from
microbiological challenge and tumour development. The structure and
function of the immune system at the anatomical, biochemical and
functional levels are broadly comparable in all mammals.
Natural immunity is phylogenetically more ancient than the
adaptive immune response, but nevertheless is of critical importance
in providing resistance to infectious microorganisms, and the
nonspecific or innate immune system acts as a first line of defence.
Among the functions of the natural immune system is provision of a
physicochemical barrier at external surfaces in the skin and the
mucosal tissues of the gastrointestinal, reproductive and respiratory
tracts, and the physical elimination of bacteria by coughing,
sneezing, etc. The ability of these surfaces to renew themselves and
secrete antimicrobial agents such as fatty acids and lysozyme reduces
penetration by microbes. However, microbes that bypass these barriers
must be dealt with by other more advanced immunological mechanisms,
which can be either specific or nonspecific in nature. Cellular
elements of the natural immune system include natural killer (NK)
cells, mononuclear phagocytes, and eosinophil and neutrophil
polymorphonuclear cells. In addition, a complex series of plasma
proteins and glycoproteins together comprise the complement system,
which acts together with antibody in the elimination of bacteria, but
which can also be activated to provide natural immune function in the
absence of, or before, a specific immune response. The adaptive immune
system acts together with innate or natural immune mechanisms to
provide host resistance to infectious and malignant disease.
The adaptive immune system comprises organs, tissues, cells and
molecules that must act in concert to provide an integrated immune
response. The three cardinal characteristics of adaptive immunity are
memory, specificity and the capacity to distinguish between self and
non-self. Each of these characteristics are displayed by lymphocytes:
the main cellular vectors of adaptive immune responses. Immunological
memory is the ability to distinguish a foreign material as a previous
invader and to mount a greatly increased and lasting response to that
particular antigen. This process is the product of immunocompetent
cell cooperativity and allows for both amplification of the immune
response after repeated encounters with the same antigen
(immunization) and tolerance to self tissues. In contrast, nonspecific
or innate mechanisms do not possess individuality and do not lead to
memory.
Mature lymphocytes circulate throughout the body, between and
within lymphoid tissues. If a lymphocyte encounters a foreign antigen
in an appropriate form under suitable conditions then the cell becomes
activated and an immune response is initiated. The primary response
takes place in organized lymphoid tissues. It has been estimated that
in a normal adult human the immune system is capable of recognizing
and responding to many millions of antigens; even antigens that have
never been encountered previously, such as for instance new synthetic
chemicals. This enormous repertoire is provided by the clonal
diversity of lymphocytes; these cells being clonally distributed with
respect to antigen specificity. Thus, each clone of mature lymphocytes
differs one from another in terms of the antigenic structures that
will induce activation. Antigen recognition is effected via
specialized membrane receptors that have diversified among lymphocytes
during development of the immune system by a process of somatic
recombination of antigen receptor genes. It is the possession of these
receptors by lymphocytes that confers specificity to immune responses.
Recognition of antigen by lymphocytes in primary lymphoid tissues
results in rapid cellular activation and the stimulation of division
and differentiation. Division provides for a selective expansion in
numbers of those lymphocytes that are able to recognize and interact
with the inducing antigen. Selective clonal expansion forms the basis
of immunological memory. After first encounter with antigen,
responsive lymphocytes have increased in number such that if the
individual is exposed subsequently to the same antigenic material then
an accelerated and more aggressive response will be mounted. These are
the central events necessary for adaptive immunity and those that are
made use of in vaccination against infectious microorganisms.
All lymphocytes involved in adaptive immune responses interact
specifically with antigen, and they divide and differentiate in
response to antigenic challenge. These cells may be subdivided into
two main populations, T-lymphocytes and B-lymphocytes, that differ
with respect to their origins and development pathways, the way in
which antigen is recognized, and the effector cells into which they
ultimately differentiate. Both populations arise in the bone marrow
from primitive precursors, but thereafter follow discrete
developmental pathways. Cells committed to becoming T-lymphocytes
(pre-T-cells) require passage through and differentiation within the
thymus to achieve immunological maturity. The thymus serves also to
identify and destroy most of those T lymphocytes that display membrane
receptors which would permit interaction with self antigens. When they
leave the thymus the mature antigen-sensitive T-lymphocytes join the
recirculating pool.
Bone marrow derived B-cells also join the recirculating pool
where, with T-lymphocytes, they seek antigen for which they have
complementary membrane receptors. B-lymphocytes recognize antigen
usually in its native form. Activation triggers B-lymphocyte
differentiation and division. The end-cell of B-lymphocyte
differentiation is the plasma cell that possesses the synthetic and
secretory machinery to manufacture and export large amounts of
antibody. The antibody secreted by an individual plasma cell is of a
single specificity and matches identically the specificity of the
membrane receptor on the B-lymphocyte from which the plasma cell
differentiated. The purpose of antibody is essentially to form a
bridge between the inducing antigen and biological mechanisms that
serve to eliminate it. The interaction of antibody with antigen
facilitates the activation of complement (lysis of bacteria) and
phagocytosis by mononuclear phagocytes and neutrophils (intracellular
killing of bacteria) and results in the clearance of pathogenic
bacteria. The importance of B-lymphocytes and the antibodies that
derive from their activation is protection against extracellular
infection by bacteria and parasites.
The existence of T-lymphocytes was recognized for many years
before the true nature of their role in adaptive immune responses was
appreciated. Cell-mediated immune responses effected by T-lymphocyte
participate in host defence against all types of infectious organisms,
but of greatest evolutionary significance is immunity against viruses.
Humoral immunity effected by antibody is of relevance only in the
viraemic stage of viral infections. Viruses are obligate intracellular
parasites and once inside the infected host cell are protected from
antibody-mediated mechanisms.
The overall purpose of these host defence mechanisms is to
provide the organism with resistance to a challenging microbial
environment and to confer protection from the internal development of
non-self neoplasms or tumours. When normal immune function is absent
or compromised, the consequences for human health are serious.
Consideration of immunosuppression and immunodeficiency illustrates
the evolutionary importance of immune function.
1.1.2 Immunosuppression, immunodeficiency and autoimmunity
Active immune function is clearly beneficial for health, whereas
the consequences of a compromised immune system are adverse health
effects.
Immunodeficiency disorders can be congenital or acquired.
Congenital immunodeficiency is comparatively rare, but is frequently
very serious and can be fatal. Examples include a complete, or almost
complete, failure of the immune system to develop due to the absence
or aberrant maturation of lymphocyte or leukocyte progenitors,
resulting in severe combined immunodeficiency disease or reticular
dysgenesis. Without appropriate treatment these conditions are fatal,
children succumbing to overwhelming infection.
Acquired immunodeficiency can be secondary to malnutrition,
severe stress, treatment with immunosuppressive drugs or with cancer
chemotherapeutic agents, exposure to certain environmental chemicals
or infection, such as infection with the human immunodeficiency virus
(HIV), the cause of acquired immunodeficiency syndrome (AIDS). In all
instances immunosuppression is associated with reduced host resistance
and more persistent infection, often with unusual microorganisms that
are resisted well by immunocompetent individuals. Immunodeficiency is,
in addition, associated with an increased incidence of malignant
diseases that are known or suspected to be associated with oncogenic
viruses.
The benefits that derive from active immune function do not come
without a cost, however. While the adaptive immune system acts as a
"friend" in providing host defence, it may also act as a "foe", being
instrumental in the pathogenesis of certain diseases. The immune
system can, for instance, turn on the host if the fine discrimination
between self and non-self breaks down. The result is the development
of autoimmune responses and autoimmune disease. The mechanisms by
which autoimmunity develops are multifactorial, complex and remain
poorly understood. The majority of cases are idiopathic, although
diseases such as systemic sclerosis have been associated with organic
chemicals and silica.
1.1.3 Allergy and allergic diseases
Allergy may be defined as the adverse health effects resulting
from hypersensitivity caused by exposure to an exogenous antigen
(allergen) resulting in a marked increase in reactivity and
responsiveness to that particular antigen on subsequent exposure.
Allergy is not necessarily, or usually, the consequence of perturbed
immune function, but the result of an immune system response to an
antigen (in this case allergen) in such a way that a temporary or
long-lasting disease results. The immunological processes that are
involved in the development of allergic responses and allergic disease
are in principle and practice no different to those that provide
protective immunity and host resistance against potential pathogens.
Allergy normally develops in two phases. The first phase is
induced following initial encounter of the susceptible individual with
the allergen. A primary immune response is mounted that results in a
state of heightened responsiveness to that particular antigen
(specific sensitization). In immunological terms sensitization to an
allergen does not differ from immunization to a pathogenic
microorganism. Following second or subsequent exposure of the now
sensitized individual to the inducing allergen a more vigorous and
accelerated secondary immune response is provoked and it is at this
stage that adverse health effects are normally first recognized. The
aggressive secondary immune response against the allergen causes local
tissue disruption and inflammation that is recognized clinically as
allergic disease.
Individuals vary widely in terms of allergic responsiveness and
susceptibility to allergic disease. There are a number of factors of
importance here including opportunities for encounter with the
inducing allergen, the route, the dose or concentration of allergen,
extent and duration of exposure and genetic predisposition. The latter
is incompletely understood but clearly impacts significantly upon
susceptibility. Respiratory allergy (including hay fever and asthma)
to protein aeroallergens is associated frequently with atopy; a
genetic predisposition for increased production of IgE, the class of
antibody that causes respiratory hypersensitivity to proteins. In
addition, the immunological repertoire of individuals and the ability
of their immune system to recognize and respond to certain antigenic
structures will also influence susceptibility.
Allergic diseases are widespread and can be caused by allergens
encountered in the external environment, home or work. They range from
comparatively mild inflammatory responses localized to a single site
to systemic anaphylactic responses that may prove fatal. Allergic
disease, as well as representing an important and widespread health
problem, is also of great economic significance with respect to the
cost of health care and time lost from work. It has been recognized
that some forms of allergy are increasing in prevalence, compounding
the health impact of these diseases. The incidence of asthma, for
instance, has grown significantly in some developed countries, an
increase that may be attributable to changing allergen exposure
patterns, alterations in lifestyle, environmental pollution or to a
combination of all of these factors.
In the context of occupational and environmental health the two
most important allergic diseases caused by exposure to chemicals are
allergic contact dermatitis and respiratory hypersensitivity. The
former is very common and can be induced by industrial chemicals,
metals and natural products. Sensitization results from dermal
exposure of the susceptible individual to the inducing allergen.
Allergic contact dermatitis reactions are provoked subsequently when
the now sensitized individual is exposed for a second time to the
inducing allergen at the same or different skin site. Many hundreds of
contact allergens, varying enormously in potency, have been
identified.
Although from the occupational and environmental health
standpoint allergic contact dermatitis and respiratory
hypersensitivity represent the most important types of allergy induced
by chemicals, it should not be forgotten that exposure to xenobiotics
has been implicated in other forms of allergic disease. Certain drugs
are associated with systemic allergic reactions that are sometimes
reminiscent of autoimmune diseases. In addition, food components and
food additives are implicated in adverse reactions, which in some
cases take the form of an allergic response.
1.1.4 Conclusion
An active adaptive immune system is essential for health and
survival in a hostile microbiological environment. A price paid for
the host resistance provided by the immune system is that some immune
responses, often to benign antigens, result in the adverse health
effects of allergic disease.
1.2 Physiology and components of the immune system
Immunity refers to all those physiological mechanisms/processes
that enable an animal (i.e., the host) to recognize materials as
foreign to "self" and to neutralize, eliminate or metabolize them,
with or without injury to its own tissue. The immune system of higher
animals is therefore capable of distinguishing between self materials
from which they are constituted and "non-self" (i.e., those that are
foreign or antigenic). It probably evolved to confer a selective
advantage to organisms that could withstand colonization and microbial
invasion. The immune response must decipher sometimes quite subtle
differences between self and non-self, without error, to both provide
protection and avoid self-attack. Accomplishment of this selective
process requires the concerted action of a number of cell types.
Mammals have developed a highly complex, intertwined and redundant
system composed of layers of protective mechanisms to cope with more
sophisticated environmental threats.
The immune system comprises both lymphoid organs and specialized
cells. Erythrocytes, myeloid cells, megakaryocytes (which mature to
form platelets) and lymphocytes arise from a totipotent or pluripotent
stem cell in the yolk sac of the developing fetus and, later, the
fetal liver. In adult mammals, the stem cells are manufactured in the
bone marrow and progress via different pathways of differentiation to
become mature cells that may carry out specialized functions, such as
antibody production or phagocytosis (Abramson et al., 1977). The
thymus and bone marrow are the primary lymphoid organs that serve to
nurture the development of stem cells into mature effector cells.
Mature lymphocytes traffic to the secondary lymphoid organs, the lymph
nodes, spleen and mucosal-associated lymphoid tissues (MALTs), and
form immune-reactive units that respond vigorously to antigens. The
design of these secondary organs is such that the specialized
populations of lymphocytes reside in proximity, can interact with each
other, and can regulate the antigen-driven immune response required.
The lymph nodes, which are situated throughout the body, filter out
antigens draining from the peripheral bodily tissues. The spleen
monitors the blood and functions as a factory for red blood cell
turnover. The MALTs provide a frontline defence for microbes that are
ingested. Lymphocytes that reside in the spleen can, upon encountering
antigen, respond in situ or migrate to the site of infection via the
blood, colonizing a sensitized response unit in a local lymph node.
The virgin stem cell is believed to receive different maturational
stimuli in the microenvironment of the bone marrow, with stromal cell
contact and lymphokine exposure inducing entry into one of several
pathways of development. Functional lymphocytes are continuously
formed from stem cells and pass from the bone marrow through the
bloodstream to the lymphoid organs. The migratory pattern of the
lymphocyte determines its lifespan and behaviour, as described in
greater detail below for T-cells, B-cells and other immunocompetent
cells.
1.2.1 T-cells
Stem cells that enter the thymus gland, formed from the third and
fourth pharyngeal pouches in mammals, rapidly divide, acquire their
antigen specificity and are selectively deleted if they bear any
self-reactivity. The "educated" daughter cells, termed thymus-derived
or T-lymphocytes, then leave the thymus and travel to other lymphoid
tissues, persisting for weeks or even years. As stem cells pass
through the thymic subcapsular region, cortex and medulla, they
display plasma membrane-bound surface molecules that define their
function. It is possible to experimentally identify and isolate
subpopulations of T-lymphocytes by exploiting the differential
expression of these marker glycoproteins, using alloantisera or
monoclonal antibodies and immunostaining techniques. Murine
T-lymphocytes possess both the Thy-1 marker and the T-cell antigen
receptor (TCR)-CD3 complex, and fall into two major classes, either
T-helper/inducer cells expressing CD4 or T-suppressor/cytotoxic cells,
which display CD8.
Studies in inbred mice show that the T-cell antigen receptor only
recognizes antigen processed and presented on major histocompatibility
complex (MHC) molecules from the same thymic environment. MHC proteins
are products of the immune response (Ir) genes, which are primarily
responsible for tissue graft and organ transplantation rejection. In
general, CD4+ T-cells complex with antigen associated with MHC Class
I molecules, which are only found on certain cells of the immune
system, while CD8+ T-cells only see antigen when associated with MHC
Class I molecules, located on all nucleated cells. T-cell selection of
this type is termed positive and deletion of clones reactive to self
is termed negative selection (Zinkernagel & Doherty, 1975). Upon
contact with antigen, mature T-cells may either respond clonally in an
antigen-specific manner and initiate an immune response, or become
inactivated and eliminated in a process which is not well understood,
potentially leaving the animal unable to recognize the antigen. This
latter phenomenon is referred to as T-cell anergy.
The majority of lymphocytes in the peripheral blood and lymph
nodes and about one half of the cells in the spleen are T-cells.
Thymectomized animals or naturally occurring athymic or nude mice
(because they are also hairless) and children with Di George syndrome
are immunocompromised hosts that lack cell-mediated immune function
and responses to T-dependent antigens (Sell, 1987). The endocrine
function of the thymus has been recognized through partial recovery of
T-cell function in thymectomized animals given cell-free thymic
extracts, suggesting thymic hormones may, to some extent, replace
thymus-driven T-cell maturation (Law et al., 1968). However, the
thymic microenvironment appears necessary for proper selection and
differentiation of the T-cell repertoire. Imbalances in the function
of mature T-cell subpopulations may also occur clinically, as shown by
HIV infection of CD4+ T cells, resulting in decreased T-helper cell
levels (Stahl et al., 1982; Lane & Fauci, 1985), and systemic lupus
erythematosus in which lowered CD8+ T-suppressor cell activity is
thought to contribute to elevated antibody production and to
exacerbate the autoimmune state.
1.2.1.1 Balancing the immune response
It is clear that in the mouse most T-cells show predominant
production of two different sets of cytokines with pronounced, often
mutually exclusive, effects on different features of the immune
response (Romagnani, 1992a,b; Bloom et al., 1992; Mosmann & Sad,
1996). While some details of cytokine production are known to be
different in the human, they are generally similar to that in the
mouse. In brief, mouse Th1-cells produce IL-2, IFN-gamma and
lymphotoxin (LT), whereas Th2-cells produce IL-4, 5,6,9,10,13, as
shown in Table 1. Human Th1 and Th2 cells produce similar patterns,
although the synthesis of IL-2,6,10,13 is not as tightly restricted to
a single subset as in mouse T-cells. In the mouse Th1-cell (or Type I)
responses result in delayed-type hypersensitivity (DTH) reactions,
activation of macrophages to kill phagocytosed microorganisms, and in
IgG2a, rather than IgG1 and IgE, synthesis. Th2 (Type 2) responses
generate IgG1- and IgE-secreting cells, and eosinophilia. Notably,
Th2-derived IL-4 is an important switch factor for B cells to produce
the IgG1 and IgE immunoglobulin-isotypes. Th1- and Th2-cells arise
from a common lineage since they use the same T-cell receptor
repertoire, and naive precursor T-cells, not yet exhibiting either of
these cytokine profiles (Th0), can differentiate into both directions
(see also section 2.1.5). Although cytotoxic CD8+ T-cells often
secrete a Th1-like cytokine pattern, there is evidence for the
existence of Th2-like CD8+ T (Tc2) cells in humans and mice (Croft
et al., 1994; Mosmann & Sad, 1996). Type 2 cytokines such as IL-4
shift T cell differentiation away from the production of Type I
cytokines, whereas the Type I cytokine IFN-gamma is very potent in
preventing the development of Th2-cells.
Cytokines are soluble mediators synthesized by cells of the
immune system that bind to specific receptors or target cells and
modulate cell function in immunological reactions (Fig. 1). When
starting clonal expansion after antigen stimulation, T-cells develop
major cytokine profiles depending on the site of primary contact.
Along mucosal surfaces predominant local IL-4 release, possibly by
mast cells, basophils or locally residing T-cells, favours the
development of Th2-cells (Scott, 1993; Weiner et al., 1994; Mosmann
et al., 1996). In some individuals over-prone to IgE-switching, this
response may be excessive, leading to mucosal allergies, such as
respiratory hypersensitivity (see also chapter 4). The induction of
Type 2 T-cell responses after antigen introduction along mucosal
surfaces is probably further promoted by high local densities of
B-cells as compared to the skin compartment. B-cells are excellent
IL-10 producers, and antigen-presentation by B-cells is known to
favour Th2 responses (Eynon & Parker, 1992). In addition to the
archetype Type 2 cytokines, TGF-beta has also been associated with Th2
functions, but preferential production by either a Th2 subset, or a
distinct Th3 subset (Chen et al., 1994), is more likely to occur. As
mentioned above, TGF-beta plays the key role in immune suppression
along mucosal surfaces, e.g., by controlling several different
IFN-gamma-associated effector T-cell and macrophage functions
(Karpus & Swanborg, 1991; Oswald et al., 1992; Khoury et al., 1992;
Table 1. Cytokine production in the mouse
Cytokine
production T-cells Other cells
Th0 Th1 Th2 B-cell Macrophage NK-cell Mast cell Keratinocyte LC
IL-1 +alpha +beta
IL-2 + +
IFN-gamma + + +
LT (TNF-beta) + +
IL-3 + + + +
GM-CSF + + + +
TNF-alpha + + + + + +
IL-4 + + + +
IL-5 + +
IL-6 + + + + +
IL-10 + + + + + +
IL-12 + + +
IL-13 + + + + + +
Meade et al., 1992) and by maintaining epithelial cell layer integrity
(Planchon et al., 1994). Moreover, TGF-beta serves as a switch
factor for IgA production. To what extent T-cells preferentially
releasing TGF-beta may also contribute to mucosal tolerance to
IgE-inducing atopic allergens is still unclear. In sharp contrast,
along the skin route local release of IL-12 from, for instance,
macrophages and NK-cells stimulates the production of IFN-gamma by
T cells and facilitates predominant development of Th1 cells. Exposure
of the skin to exogenous antigenic substances, including contact
allergens, therefore preferentially induces specific Type 1,
pro-inflammatory T-cell responses.
1.2.2 B-cells
In contrast to T-lymphocyte maturation, the development of
lymphocytes capable of synthesizing and secreting antibody
(immunoglobulin) molecules in mammals is thought to occur in several
sites, including the bone marrow, spleen and MALTs. Because these
cells were first characterized in birds, which, unlike mammals,
possess a unique lymphoid organ, the bursa of Fabricius, and because
the precursors of these cells are formed in the bone marrow, these
cells have been termed B-lymphocytes. B-cells tend to reside for long
periods of time in the secondary lymphoid organs and form the lymphoid
follicles and germinal centres. Following activation by antigen or
antigen-activated T-helper cells (Noelle et al., 1990) and
lymphokines, B-cells proliferate and terminally differentiate to
antibody-producing plasma cells, which turn over rapidly and are
replenished by newly differentiated cells.
Like the T-cell antigen receptor (TCR)-CD3 complex, B-cells
express surface antigen-combining receptor molecules which are of
identical specificity to the immunoglobulins they synthesize and
secrete. The diversity of the natural world has necessitated a complex
series of molecular events in B-cell development designed to produce a
spectrum of immunoglobulins capable of protecting the organism. B-cell
maturation is marked by immunoglobulin gene rearrangements,
recombinations and somatic mutations, so that a relatively small
number of genes may efficiently produce a large number of antibody
specificities.
B-lymphocytes synthesize immunoglobulins of five different types:
IgM, IgG, IgA, IgD, and IgE. These proteins are composed of two
separate types of polypeptide chains joined by disulfide linkages,
termed the heavy and light chains because of differences in their
relative molecular masses (the heavy chains are about twice as large)
(see Fig. 2). Light chains are derived from either kappa or lambda
genes and combine with the five different heavy chains mu, gamma,
alpha, delta and epsilon (i.e., for the five different types of
immunoglobulin identified above). Enzymatic digestion of
immunoglobulin molecules yields fragments which indicate arrangement
in a Y-shaped structure, consisting of two arms containing the
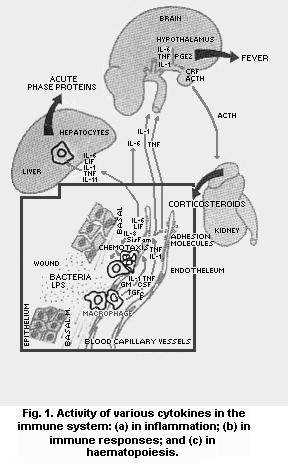
 antibody-combining sites for antigen, Fab fragments, and a tail region
(Fc) which is important for effector functions and regulation of
antibody responses. Surface immunoglobulin is predominantly of the IgM
and IgD types on naive B cells and secreted immunoglobulin may be
either IgM, IgG of four subclasses (1 to 4), IgA, or IgE. IgM is
primarily secreted early, in what is termed the primary antibody
response to antigen, with IgG constituting the later, secondary
response. Lymphokines such as IL-4 and TGF-beta induce heavy chain
class switching in B-cell antibody responses, leading to the
production of either IgGl and IgE, or IgA, respectively (Coffman
et al., 1986; Coffman et al., 1989). The nature of the antigen
encountered portends these lymphokine-mediated events. IgA-secreting
B-cells are predominant in the MALTs, while IgE is of central
importance in allergic reactions.
In addition to surface immunoglobulin, B-cells display receptors
for Fc regions of immunoglobulin molecules, MHC Class II molecules,
receptors for complement proteins, and the CD40 molecule which plays
an essential role in the contact between B- and T-cells. B-cells
appear to be comprised of two separate lineages, those that do and
those that do not express the surface marker CD5 (E32). CD5+ B-cells
comprise a small percentage of the splenic B-cell population, are more
prevalent in the peritoneal cavity of mice, and appear to be
long-lived, activated cells that differ from conventional B-cells in
their activational characteristics and capacity for self-renewal.
1.2.3 Macrophages
Stem cells also give rise to mononuclear phagocytes of the
myeloid series, of which the macrophage is the primary cell type.
Immature macrophages leave the bone marrow and are found in the
lymphoid organs, the liver, lungs, gastrointestinal tract, central
nervous system, serous cavities, bone, synovium and skin, and
differentiate within these sites. Macrophages are attracted to
microbes by the gradient of foreign molecules emanating from them, a
process called chemotaxis. Upon contact, the macrophage can engulf the
microbe, process and present the derived antigen via its MHC molecules
to T cells, and secrete cytokines (e.g., IL-1, TNF-alpha, IL-12),
degradative enzymes, complement components, reactive oxygen
intermediates and coagulation factors. Macrophages readily infiltrate
tumours and provide one mechanism of host defence against
malignancies.
1.2.4 Antigen-presenting cells
If an antigen penetrates the tissues it will be processed by
antigen-presenting cells (APCs) and transported to the draining lymph
nodes. Antigens that are encountered in the upper respiratory tract or
intestine are trapped by local mucosal-associated lymphoid tissues,
whereas antigens in the blood provoke a reaction in the spleen.
antibody-combining sites for antigen, Fab fragments, and a tail region
(Fc) which is important for effector functions and regulation of
antibody responses. Surface immunoglobulin is predominantly of the IgM
and IgD types on naive B cells and secreted immunoglobulin may be
either IgM, IgG of four subclasses (1 to 4), IgA, or IgE. IgM is
primarily secreted early, in what is termed the primary antibody
response to antigen, with IgG constituting the later, secondary
response. Lymphokines such as IL-4 and TGF-beta induce heavy chain
class switching in B-cell antibody responses, leading to the
production of either IgGl and IgE, or IgA, respectively (Coffman
et al., 1986; Coffman et al., 1989). The nature of the antigen
encountered portends these lymphokine-mediated events. IgA-secreting
B-cells are predominant in the MALTs, while IgE is of central
importance in allergic reactions.
In addition to surface immunoglobulin, B-cells display receptors
for Fc regions of immunoglobulin molecules, MHC Class II molecules,
receptors for complement proteins, and the CD40 molecule which plays
an essential role in the contact between B- and T-cells. B-cells
appear to be comprised of two separate lineages, those that do and
those that do not express the surface marker CD5 (E32). CD5+ B-cells
comprise a small percentage of the splenic B-cell population, are more
prevalent in the peritoneal cavity of mice, and appear to be
long-lived, activated cells that differ from conventional B-cells in
their activational characteristics and capacity for self-renewal.
1.2.3 Macrophages
Stem cells also give rise to mononuclear phagocytes of the
myeloid series, of which the macrophage is the primary cell type.
Immature macrophages leave the bone marrow and are found in the
lymphoid organs, the liver, lungs, gastrointestinal tract, central
nervous system, serous cavities, bone, synovium and skin, and
differentiate within these sites. Macrophages are attracted to
microbes by the gradient of foreign molecules emanating from them, a
process called chemotaxis. Upon contact, the macrophage can engulf the
microbe, process and present the derived antigen via its MHC molecules
to T cells, and secrete cytokines (e.g., IL-1, TNF-alpha, IL-12),
degradative enzymes, complement components, reactive oxygen
intermediates and coagulation factors. Macrophages readily infiltrate
tumours and provide one mechanism of host defence against
malignancies.
1.2.4 Antigen-presenting cells
If an antigen penetrates the tissues it will be processed by
antigen-presenting cells (APCs) and transported to the draining lymph
nodes. Antigens that are encountered in the upper respiratory tract or
intestine are trapped by local mucosal-associated lymphoid tissues,
whereas antigens in the blood provoke a reaction in the spleen.
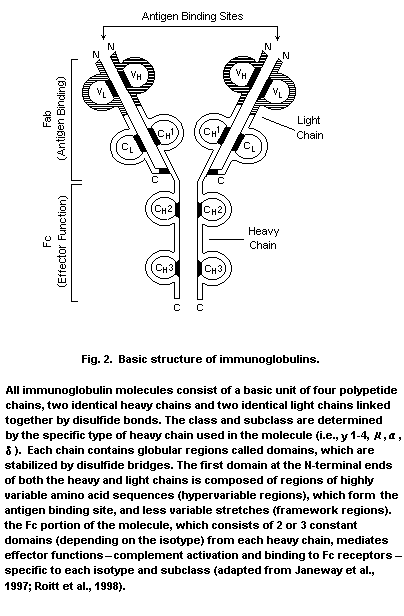 Macrophages in the liver will filter blood-borne antigens and degrade
them without producing an immune response, since they are not
strategically placed with respect to lymphoid tissue. Classically, it
has always been recognized that antigens draining into lymphoid tissue
are taken up by macrophages. They are then partially, if not
completely, broken down in the lysosomes; some may escape from the
cell in a soluble form to be taken up by other APCs and a fraction may
reappear at the surface either as a large fragment or as a processed
peptide associated with MHC Class II major histocompatibility
molecules. Although resting resident macrophages do not express MHC
Class II, antigens are usually encountered in the context of a
microbial infectious agent which can induce the expression of MHC
Class II by its adjuvant-like properties expressed through molecules
such as bacterial lipopolysaccharide (LPS). There is general agreement
that the APC must bear antigen on its surface for effective activation
of lymphocytes and ample evidence that antigen-pulsed macrophages can
stimulate specific T- and B-cells both in vitro and when injected
back in vivo. Some antigens, such as polymeric carbohydrates like
ficoll, cannot be degraded because the macrophages lack the enzymes
required; in these instances, specialized macrophages in the marginal
zone of the spleen or the lymph node subcapsular sinus, trap and
present the antigen to B-cells directly, apparently without any
processing or intervention from T-cells. Notwithstanding this
impressive account of the macrophage in antigen presentation, there is
one function where it is seemingly deficient, namely, the priming of
naive lymphocytes. Animals that have been depleted of macrophages by
selective uptake of liposomes containing the drug dichloromethylene
diphosphonate are as good as control animals with intact macrophages
in responding to T-dependent antigens. It must be concluded that cells
other than macrophages prime T-helper cells and it is generally
accepted that these belong to the group of dendritic cells.
Dendritic cells are large, motile, weakly phagocytic,
"professional" APCs that usually have several elongated pseudopodia.
Dendritic cells comprise about 2% of the cells in the secondary
lymphoid organs. They are localized strategically in the T-cell areas
of the lymph node (interdigitating dendritic cells). Interdigitating
cells express large amounts of MHC Class II molecules, and this
expression plays a pivotal role in the presentation and induction of
certain kinds of immune cells (such as Th 1) and the presentation of
antigen to CD4+ T-cells. Active follicular dendritic cells, although
not derived from haematopoietic stem cells, express high levels of
CD23 (an IgE Fc receptor) and C3 receptors, which allows them to trap
antigen-antibody complexes and present them to memory B-cells. Normal
skin contains a population of dendritic cells called Langerhans cells
that change their morphology to become interdigitating dendritic cells
within the T-cell areas of lymph nodes. Langerhans cells give the
immune system information regarding foreign substances that breach the
skin. Langerhans cells pick up skin-sensitizing antigens (e.g.,
antigens of the poison ivy plant) and migrate to the draining lymph
nodes. Langerhans cells are important in the delayed-type
hyper-sensitivity response known as contact dermatitis.
The need for physical linkage of hapten and carrier strongly
suggests that T-helper cells must recognize the carrier determinants
on the responding B-cell in order to provide the relevant accessory
stimulatory signals. However, since T-cells only recognize processed
membrane-bound antigen in association with MHC molecules, the T-helper
cells cannot recognize native antigen bound simply to the Ig-receptors
of the B-cell. Primed B-cells can present antigen to T-helper cells;
in fact, they work at much lower antigen concentrations than
conventional presenting cells because they can focus antigen through
their surface receptors. They must therefore be capable of processing
the antigen and the current view is that antigen bound to surface Ig
is internalized in endosomes, which then fuse with vesicles containing
MHC Class II molecules with their invariant chain. Processing of the
protein antigen then occurs and the resulting antigenic peptide is
then recycled to the surface in association with the Class II
molecules where it is available for recognition by specific T-helper
cells.
1.2.4.1 Co-stimulatory molecules in T-cell activation
Binding of the antigen/MHC-complex to the T-cell receptor
(Fig. 3) and co-receptors like CD4 and CD8 is not sufficient to
stimulate naive T-lymphocytes to proliferate and differentiate into
effector T-cells. For antigen-specific clonal expansion and
differentiation, a second, co-stimulatory signal is required. The same
cell that presents the specific antigen to the T-cell receptor must
deliver this co-stimulatory signal. The best-characterized
co-stimulatory moleculeson APCs are the so-called B7 molecules, B7.1
(CD80) and B7.2 (CD 86). Their receptor on T-cells is CD28; all three
molecules mentioned are members of the so-called immunoglobulin
superfamily. B7.2 is present on resting APCs, whereas B7.1 is
expressed predominantly on activated cells. It has been suggested that
B7.2 is of particular importance in the allergic immune response and
represents a potential therapeutic target (Robinson, 1998). However,
clear functional differences between B7.1 and B7.2 have not been
defined (Lenshow et al., 1996; Chambers & Allison, 1997).
On naive T-cells, CD28 is the only receptor for B7 molecules.
Activated T-cells, in contrast, also express another receptor for B7
called CTLA-4, which closely resembles CD28 but delivers a negative
signal to the T-cells (Chambers & Allison, 1997). Thus, binding of B7
to CTLA-4 will contribute to limiting or down-regulating the
proliferative response and T-cell production of IL-2.
Because of the requirement for co-stimulatory signals to obtain
productive antigenic stimulation of T-cells, only so-called
professional APCs, that is cells that are able to deliver proper
co-stimulation, can initiate a T-cell-dependent immune response. If
antigen binds to the T-cell receptor in the absence of proper
co-stimulation, the T-cell will not be activated but may instead
become refractory to activation, a state called anergy. In addition to
the co-stimulatory B7 molecules, a professional APC must also express
Macrophages in the liver will filter blood-borne antigens and degrade
them without producing an immune response, since they are not
strategically placed with respect to lymphoid tissue. Classically, it
has always been recognized that antigens draining into lymphoid tissue
are taken up by macrophages. They are then partially, if not
completely, broken down in the lysosomes; some may escape from the
cell in a soluble form to be taken up by other APCs and a fraction may
reappear at the surface either as a large fragment or as a processed
peptide associated with MHC Class II major histocompatibility
molecules. Although resting resident macrophages do not express MHC
Class II, antigens are usually encountered in the context of a
microbial infectious agent which can induce the expression of MHC
Class II by its adjuvant-like properties expressed through molecules
such as bacterial lipopolysaccharide (LPS). There is general agreement
that the APC must bear antigen on its surface for effective activation
of lymphocytes and ample evidence that antigen-pulsed macrophages can
stimulate specific T- and B-cells both in vitro and when injected
back in vivo. Some antigens, such as polymeric carbohydrates like
ficoll, cannot be degraded because the macrophages lack the enzymes
required; in these instances, specialized macrophages in the marginal
zone of the spleen or the lymph node subcapsular sinus, trap and
present the antigen to B-cells directly, apparently without any
processing or intervention from T-cells. Notwithstanding this
impressive account of the macrophage in antigen presentation, there is
one function where it is seemingly deficient, namely, the priming of
naive lymphocytes. Animals that have been depleted of macrophages by
selective uptake of liposomes containing the drug dichloromethylene
diphosphonate are as good as control animals with intact macrophages
in responding to T-dependent antigens. It must be concluded that cells
other than macrophages prime T-helper cells and it is generally
accepted that these belong to the group of dendritic cells.
Dendritic cells are large, motile, weakly phagocytic,
"professional" APCs that usually have several elongated pseudopodia.
Dendritic cells comprise about 2% of the cells in the secondary
lymphoid organs. They are localized strategically in the T-cell areas
of the lymph node (interdigitating dendritic cells). Interdigitating
cells express large amounts of MHC Class II molecules, and this
expression plays a pivotal role in the presentation and induction of
certain kinds of immune cells (such as Th 1) and the presentation of
antigen to CD4+ T-cells. Active follicular dendritic cells, although
not derived from haematopoietic stem cells, express high levels of
CD23 (an IgE Fc receptor) and C3 receptors, which allows them to trap
antigen-antibody complexes and present them to memory B-cells. Normal
skin contains a population of dendritic cells called Langerhans cells
that change their morphology to become interdigitating dendritic cells
within the T-cell areas of lymph nodes. Langerhans cells give the
immune system information regarding foreign substances that breach the
skin. Langerhans cells pick up skin-sensitizing antigens (e.g.,
antigens of the poison ivy plant) and migrate to the draining lymph
nodes. Langerhans cells are important in the delayed-type
hyper-sensitivity response known as contact dermatitis.
The need for physical linkage of hapten and carrier strongly
suggests that T-helper cells must recognize the carrier determinants
on the responding B-cell in order to provide the relevant accessory
stimulatory signals. However, since T-cells only recognize processed
membrane-bound antigen in association with MHC molecules, the T-helper
cells cannot recognize native antigen bound simply to the Ig-receptors
of the B-cell. Primed B-cells can present antigen to T-helper cells;
in fact, they work at much lower antigen concentrations than
conventional presenting cells because they can focus antigen through
their surface receptors. They must therefore be capable of processing
the antigen and the current view is that antigen bound to surface Ig
is internalized in endosomes, which then fuse with vesicles containing
MHC Class II molecules with their invariant chain. Processing of the
protein antigen then occurs and the resulting antigenic peptide is
then recycled to the surface in association with the Class II
molecules where it is available for recognition by specific T-helper
cells.
1.2.4.1 Co-stimulatory molecules in T-cell activation
Binding of the antigen/MHC-complex to the T-cell receptor
(Fig. 3) and co-receptors like CD4 and CD8 is not sufficient to
stimulate naive T-lymphocytes to proliferate and differentiate into
effector T-cells. For antigen-specific clonal expansion and
differentiation, a second, co-stimulatory signal is required. The same
cell that presents the specific antigen to the T-cell receptor must
deliver this co-stimulatory signal. The best-characterized
co-stimulatory moleculeson APCs are the so-called B7 molecules, B7.1
(CD80) and B7.2 (CD 86). Their receptor on T-cells is CD28; all three
molecules mentioned are members of the so-called immunoglobulin
superfamily. B7.2 is present on resting APCs, whereas B7.1 is
expressed predominantly on activated cells. It has been suggested that
B7.2 is of particular importance in the allergic immune response and
represents a potential therapeutic target (Robinson, 1998). However,
clear functional differences between B7.1 and B7.2 have not been
defined (Lenshow et al., 1996; Chambers & Allison, 1997).
On naive T-cells, CD28 is the only receptor for B7 molecules.
Activated T-cells, in contrast, also express another receptor for B7
called CTLA-4, which closely resembles CD28 but delivers a negative
signal to the T-cells (Chambers & Allison, 1997). Thus, binding of B7
to CTLA-4 will contribute to limiting or down-regulating the
proliferative response and T-cell production of IL-2.
Because of the requirement for co-stimulatory signals to obtain
productive antigenic stimulation of T-cells, only so-called
professional APCs, that is cells that are able to deliver proper
co-stimulation, can initiate a T-cell-dependent immune response. If
antigen binds to the T-cell receptor in the absence of proper
co-stimulation, the T-cell will not be activated but may instead
become refractory to activation, a state called anergy. In addition to
the co-stimulatory B7 molecules, a professional APC must also express
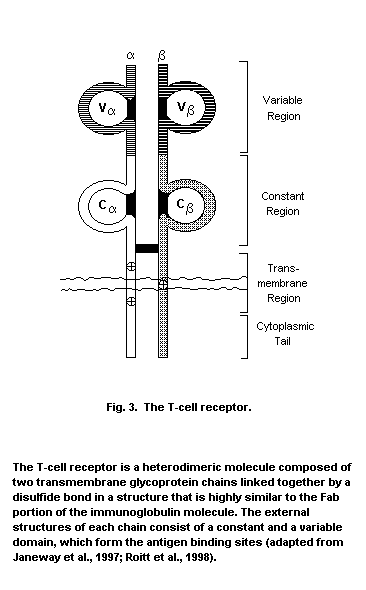 adhesion molecules like ICAM-1, ICAM-2 and LFA-3 and be able to
process antigen. There is evidence that different types of APCs differ
with regard to their co- stimulatory properties.
1.2.5 Adhesion molecules
Adhesive interactions of leukocytes with other immune cells or
with non-immune cells are central to the successful functioning of the
immune system. Such cell-cell interactions are mediated by different
types of accessory molecules which stabilize attachment, for instance
between T-cells and APCs, and which may provide (co-)stimulatory
signals upon triggering of the antigen receptor. These molecules are
also regularly used as identification markers for distinct leukocytes
subclasses or for their activational state (Schleimer & Bochner,
1998). Three families of such cell surface molecules have been
categorized:
(i) The immunoglobulin-gene superfamily includes the
antigen-specific receptors of B- and T-cells as well as the
CD4 and CD8 molecules and their respective ligands MHC Class
II and I; the adhesion molecules CD2, CD54, CD58 and CD102
also belong to this group.
(ii) The integrin family accounts for antigen-independent adhesion
between cells; their ligands are found on other leukocytes, on
endothelial cells and in the extracellular matrix; some
representative members of this family are CD11a/CD18,
CD11b/CD18, CD11c/CD18 (referring to the alpha/beta chains,
respectively) and the so-called very late activation (VLA-)
molecules on T-lymphocytes, which facilitate the migration of
these cells to peripheral inflammatory sites.
(iii) The third family, the selectins, can be expressed on
leukocytes (L-selectin) and endothelium (E-selectin). These
molecules play a role in the directed migration of lymphocytes
(for instance naive lymphocytes bind preferentially to the
high endothelial cells in the lymph nodes), neutrophils and
macrophages.
Table 2 shows the molecules facilitating the cellular contact
between APC and T-cells, and adhesion molecules playing a role in the
migration of leukocytes are shown in Table 3. Fig. 4 illustrates
antigen presentation and cell-cell contact.
1.2.6 Fc receptors
Fc receptors (FcR) are cell surface glycoproteins interacting
specifically with the Fc domains of different isotypes of
immunoglobulins (Ravetch, 1994, 1997; Gergely & Sarmay, 1996; Deo et
al., 1997; Vivier & Daeron 1997). FcRs are widely distributed on cells
of the immune system and mediate different effector responses. In
addition, they play an important role in the initiation of
immunocomplex-triggered inflammation and regulate the antibody
production of B-cells. Immunoglobulin-binding receptors, including the
high affinity receptor for IgE (Fc-epsilon-RI) on mast cells and
basophils, the high and low affinity receptors for IgG (Fc-gamma-RI,
Fc-gamma-RII and Fc-gamma-RIII) and the high affinity receptor for
IgA, belong to the immunoglobulin supergene family. The low affinity
Fc-epsilon-RII (CD32) is a lectin-like molecule (Table 4).
The ligand binding chains (alpha) of all Fc-gamma-Rs contain
extracellular parts comprising Ig-domains (Fc-gamma-RI has three, the
others two). The high affinity IgE-binding receptor (Fc-epsilon-RI) is
a tetrameric molecule containing one alpha, one beta and two gamma
chains. The IgE-binding site is located on the extracellular part of
the alpha chain. The beta chain has four transmembrane loops while the
dimeric gamma chains possess very long cytoplasmic tails.
Fc-gamma-RI, Fc-gamma-RIII and Fc-epsilon-RI belong to the family
of multisubunit immune recognition receptors (MIRRs), which are
characterized by a complex hetero-oligomeric structure in which ligand
binding and signal transducing functions are segregated into distinct
receptor substructures (Table 5).
1.2.7 Polymorphonuclear leukocytes
Polymorphonuclear leukocytes (PMNs) are myeloid phagocytic cells
important for the inflammatory responses of both specific and
nonspecific immunity. Polymorphonuclear leukocytes are also called
granulocytes because they contain granules composed of digestive
enzymes and bactericidal substances. The granulocyte progenitor can
develop into cells called either neutrophils, basophils/mast cells or
eosinophils, names which refer to the variable dye staining patterns
of their cytoplasm. These cells are also chemotactic and are attracted
by lymphokines released from lymphocytes in areas of infection. Like
macrophages, polymorphonuclear leukocytes participate in
antibody-dependent cell-mediated cytotoxicity (ADCC) reactions, in
which coating (opsonization) of microbial surfaces by specific
antibody enhances their recognition by cytotoxic or phagocytic
leukocytes.
1.2.8 Cytotoxic lymphocytes
Cytotoxic lymphocytes are defined by their capacity to recognize
and kill target cells. These cells fall into at least two different
populations, a) those that require recognition of MHC Class I
molecules for their activation, namely CD8+ T-cells, and b) those
that are silenced by recognition of these molecules, namely natural
killer (NK) cells, previously named "null cells" or large granular
lymphocytes (LGL). Cytotoxic CD8+ T-cells constitute the major
population of cytotoxic T lymphocytes (CTL) and are crucial for the
defence against intracellular, in particular viral, pathogens.
Peptides derived from such pathogens are processed into the endogenous
Table 2. Adhesion and (co-)stimulatory molecules mediating antigen presentation
to T-cells (modified from Janeway et al., 1997)
Adhesion molecules expressed on Ligand expressed on T-cell
antigen-presenting cell (APC)
Initial contact
between APC and T-cell CD58 (LFA-3) CD2
CD54(ICAM-1) } CD11a/CD18 (LFA-1)
CD102 (ICAM-2) }
CD11a/CD18 (LFA-1) CD50 (ICAM-3)
Antigen presentation and
T-cell activation antigenic peptide in MHC context TCR/CD3
MHC-Class II CD4
MHC-Class I CD8
CD80 (B7.1) } { CD28
CD86 (B7.2) } { CTLA-4
Table 3. Adhesion molecules mediating leukocyte migration (from Janeway et al., 1997)
Adhesion molecules Ligand on endothelium or
expressed on leukocyte extracellular matrix
Migration of naive T-cells
into lymphoid tissue CD62L (L-selectin) { CD34
{ GlyCAM-1
{ MadCAM-1 (Mucosae)
Migration of memory T-cells
into peripheral tissue CD11a/CD18(LFA-1) { CD54 (ICAM-1)
{ CD102 (ICAM-2)
Cutaneous lymphocyte CD62E (E-selectin)
antigen (CLA)
CD49d/CD29 (VLA-4) CD106 (VCAM-1)
CD49d/CD29 (VLA-5) fibronectin
Migration of neutrophil
and macrophages into
peripheral tissue sialyl-Lewis x moiety { CD62E (E-selectin)
{ CD62P (P-selectin)
CD11a/CD18 (LFA-1) { CD54 (ICAM-1)
{ CD102 (ICAM-2)
CD11b/CD18 (MAC-1) CD54 (ICAM-1)
adhesion molecules like ICAM-1, ICAM-2 and LFA-3 and be able to
process antigen. There is evidence that different types of APCs differ
with regard to their co- stimulatory properties.
1.2.5 Adhesion molecules
Adhesive interactions of leukocytes with other immune cells or
with non-immune cells are central to the successful functioning of the
immune system. Such cell-cell interactions are mediated by different
types of accessory molecules which stabilize attachment, for instance
between T-cells and APCs, and which may provide (co-)stimulatory
signals upon triggering of the antigen receptor. These molecules are
also regularly used as identification markers for distinct leukocytes
subclasses or for their activational state (Schleimer & Bochner,
1998). Three families of such cell surface molecules have been
categorized:
(i) The immunoglobulin-gene superfamily includes the
antigen-specific receptors of B- and T-cells as well as the
CD4 and CD8 molecules and their respective ligands MHC Class
II and I; the adhesion molecules CD2, CD54, CD58 and CD102
also belong to this group.
(ii) The integrin family accounts for antigen-independent adhesion
between cells; their ligands are found on other leukocytes, on
endothelial cells and in the extracellular matrix; some
representative members of this family are CD11a/CD18,
CD11b/CD18, CD11c/CD18 (referring to the alpha/beta chains,
respectively) and the so-called very late activation (VLA-)
molecules on T-lymphocytes, which facilitate the migration of
these cells to peripheral inflammatory sites.
(iii) The third family, the selectins, can be expressed on
leukocytes (L-selectin) and endothelium (E-selectin). These
molecules play a role in the directed migration of lymphocytes
(for instance naive lymphocytes bind preferentially to the
high endothelial cells in the lymph nodes), neutrophils and
macrophages.
Table 2 shows the molecules facilitating the cellular contact
between APC and T-cells, and adhesion molecules playing a role in the
migration of leukocytes are shown in Table 3. Fig. 4 illustrates
antigen presentation and cell-cell contact.
1.2.6 Fc receptors
Fc receptors (FcR) are cell surface glycoproteins interacting
specifically with the Fc domains of different isotypes of
immunoglobulins (Ravetch, 1994, 1997; Gergely & Sarmay, 1996; Deo et
al., 1997; Vivier & Daeron 1997). FcRs are widely distributed on cells
of the immune system and mediate different effector responses. In
addition, they play an important role in the initiation of
immunocomplex-triggered inflammation and regulate the antibody
production of B-cells. Immunoglobulin-binding receptors, including the
high affinity receptor for IgE (Fc-epsilon-RI) on mast cells and
basophils, the high and low affinity receptors for IgG (Fc-gamma-RI,
Fc-gamma-RII and Fc-gamma-RIII) and the high affinity receptor for
IgA, belong to the immunoglobulin supergene family. The low affinity
Fc-epsilon-RII (CD32) is a lectin-like molecule (Table 4).
The ligand binding chains (alpha) of all Fc-gamma-Rs contain
extracellular parts comprising Ig-domains (Fc-gamma-RI has three, the
others two). The high affinity IgE-binding receptor (Fc-epsilon-RI) is
a tetrameric molecule containing one alpha, one beta and two gamma
chains. The IgE-binding site is located on the extracellular part of
the alpha chain. The beta chain has four transmembrane loops while the
dimeric gamma chains possess very long cytoplasmic tails.
Fc-gamma-RI, Fc-gamma-RIII and Fc-epsilon-RI belong to the family
of multisubunit immune recognition receptors (MIRRs), which are
characterized by a complex hetero-oligomeric structure in which ligand
binding and signal transducing functions are segregated into distinct
receptor substructures (Table 5).
1.2.7 Polymorphonuclear leukocytes
Polymorphonuclear leukocytes (PMNs) are myeloid phagocytic cells
important for the inflammatory responses of both specific and
nonspecific immunity. Polymorphonuclear leukocytes are also called
granulocytes because they contain granules composed of digestive
enzymes and bactericidal substances. The granulocyte progenitor can
develop into cells called either neutrophils, basophils/mast cells or
eosinophils, names which refer to the variable dye staining patterns
of their cytoplasm. These cells are also chemotactic and are attracted
by lymphokines released from lymphocytes in areas of infection. Like
macrophages, polymorphonuclear leukocytes participate in
antibody-dependent cell-mediated cytotoxicity (ADCC) reactions, in
which coating (opsonization) of microbial surfaces by specific
antibody enhances their recognition by cytotoxic or phagocytic
leukocytes.
1.2.8 Cytotoxic lymphocytes
Cytotoxic lymphocytes are defined by their capacity to recognize
and kill target cells. These cells fall into at least two different
populations, a) those that require recognition of MHC Class I
molecules for their activation, namely CD8+ T-cells, and b) those
that are silenced by recognition of these molecules, namely natural
killer (NK) cells, previously named "null cells" or large granular
lymphocytes (LGL). Cytotoxic CD8+ T-cells constitute the major
population of cytotoxic T lymphocytes (CTL) and are crucial for the
defence against intracellular, in particular viral, pathogens.
Peptides derived from such pathogens are processed into the endogenous
Table 2. Adhesion and (co-)stimulatory molecules mediating antigen presentation
to T-cells (modified from Janeway et al., 1997)
Adhesion molecules expressed on Ligand expressed on T-cell
antigen-presenting cell (APC)
Initial contact
between APC and T-cell CD58 (LFA-3) CD2
CD54(ICAM-1) } CD11a/CD18 (LFA-1)
CD102 (ICAM-2) }
CD11a/CD18 (LFA-1) CD50 (ICAM-3)
Antigen presentation and
T-cell activation antigenic peptide in MHC context TCR/CD3
MHC-Class II CD4
MHC-Class I CD8
CD80 (B7.1) } { CD28
CD86 (B7.2) } { CTLA-4
Table 3. Adhesion molecules mediating leukocyte migration (from Janeway et al., 1997)
Adhesion molecules Ligand on endothelium or
expressed on leukocyte extracellular matrix
Migration of naive T-cells
into lymphoid tissue CD62L (L-selectin) { CD34
{ GlyCAM-1
{ MadCAM-1 (Mucosae)
Migration of memory T-cells
into peripheral tissue CD11a/CD18(LFA-1) { CD54 (ICAM-1)
{ CD102 (ICAM-2)
Cutaneous lymphocyte CD62E (E-selectin)
antigen (CLA)
CD49d/CD29 (VLA-4) CD106 (VCAM-1)
CD49d/CD29 (VLA-5) fibronectin
Migration of neutrophil
and macrophages into
peripheral tissue sialyl-Lewis x moiety { CD62E (E-selectin)
{ CD62P (P-selectin)
CD11a/CD18 (LFA-1) { CD54 (ICAM-1)
{ CD102 (ICAM-2)
CD11b/CD18 (MAC-1) CD54 (ICAM-1)
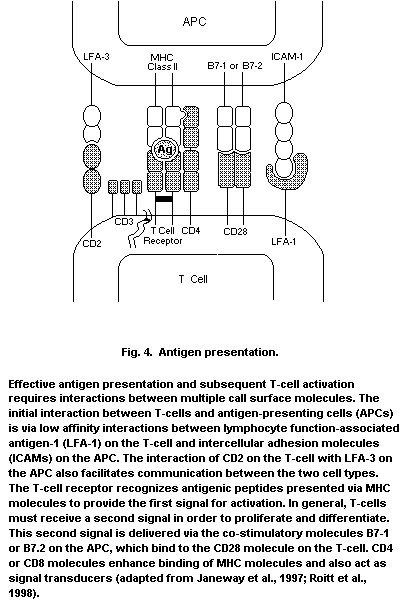 pathway of antigen presentation and exposed on the outer cell membrane
by Class I molecules. This complex is recognized by the T-cell
receptor, after which CTL-target cell binding is further stabilized by
CD8-Class I interaction. In contrast, NK cell-target cell recognition
is largely non-specific, but involves receptors recognizing disturbed
surface carbohydrates and an Fc receptor for IgG that can facilitate
antibody-dependent cell-mediated cytotoxicity (ADCC). NK cells are
unique in bearing distinct receptors which, when bound to MHC Class I
molecules, deliver signals interfering with their cytolytic activity.
For both types of cytotoxic lymphocytes the actual killing
process involves two major mechanisms, i.e., release of a membrane
pore-forming protein named perforin from granules, leading to osmotic
lysis of target cells, and release of lymphotoxin which activates
enzymes in the target cell to cleave DNA in the nucleus. The latter
process is also known as apoptosis. Most cytotoxic lymphocytes also
express a member of the tumour necrosis factor (TNF) superfamily,
i.e., Fas-ligand, mediating a third lytic mechanism for target cells
expressing the Fas antigen. The killing capacity of cytotoxic
lymphocytes is greatly enhanced by distinct cytokines, in particular
IL-2 and IL-12. Microscopically this is reflected by the appearance of
more prominent granules, e.g., in the so-called lymphokine-activated
killer (LAK) cells. Both major cytotoxic lymphocyte populations are
crucial to various phases of viral attack, but are not prominent in
causing allergic disorders. Nevertheless, contact allergens may
directly associate with surface-bound Class I molecules or enter the
cytoplasm of, for instance, Langerhans cells and associate with
peptides presented along the endogenous route of antigen presentation.
In this way, CD8+ T-cells may become involved in allergic contact
dermatitis reactions.
1.2.9 Mast cells
Mast cells are derived from precursors in the bone marrow that
migrate to specific tissue sites to mature. While they are found
throughout the body, they are most prominent in the skin, the upper
and lower respiratory tract, and the gastrointestinal tract (Tharp,
1990). In most organs mast cells tend to be concentrated around the
small blood vessels, the lymphatics, the nerves and the glandular
tissue (Tharp, 1990). These cells contain numerous cytoplasmic
granules that are enclosed by a bilayered membrane. There appear to be
two different populations of mast cells in humans, based on the
presence or absence of certain proteolytic enzymes, notably tryptase
and chymase (Tharp, 1990). Mast cells found in the skin and connective
tissue have both enzymes, while those in the alveoli, bronchial and
bronchiolar regions, and mucosa of the small bowel contain only
tryptase (Irani et al., 1986). However, both types of cells are
triggered in the same manner.
Table 4. Cellular distribution and binding properties of Fc-gamma receptors
Class CD Relative molecular mass Affinity (Ka) Expressiona Ig-bindingb
Fc-gamma-RI CD64 72 000 108-109 M-1 Mo, M hu, 3>1>4>>2
Fc-gamma-RII CD32 40 000 <107 M-1 Mo, N, Ba, Eo, Langerhans cell, B-cell hu, 3>1>>2,4
mu, 2b>>2a
Fc-gamma-RIII CD16 50 000-80 000 Thr, endothelial cells of the placenta
Fc-gamma-IIIa 3×107 M-1 Mo, M, LGL/NK, T-cell hu, 1=3>>2,4
mu, 1=3>>2,4
Fc-gamma-IIIb <107 M-1 N
a Mo = monocyte, M = macrophage, N = neutrophil granulocyte, Ba = basophil granulocyte, Eo = eosinophil granulocyte,
Thr = platelet, LGL = large granular lymphocyte, NK = natural killer cell
b hu = human, mu = murine
Table 5. Multisubunit immune recognition receptors (MIRRs) family
Receptor Ligand-binding subunit Signal transducing subunit
BCR
(B-cell
antigen receptor) mIg Ig-alpha (CD79a)
Ig-beta (CD79b)
TCR alpha-beta or gamma-delta CD3-gamma, delta and epsilon zeta-zeta or zeta-eta
(T-cell
antigen receptor)
Fc-epsilon-RI alpha-chain beta and gamma chain
Fc-gamma-RIIIa alpha-chain Fc-epsilon-RI-gamma-chain or TCR zeta-chain
Fc-gamma-RI alpha-chain Fc-epsilon-RI-gamma-chain
Mast cells may be activated by antigen-specific IgE bound to high
affinity receptors (Fc RI), antigen-specific IgE bound to low affinity
IgG receptors (Fc-epsilon-RII/III), or through complement receptors.
Following activation, most cells release preformed mediators such as
histamines and generate newly formed mediators such as TNF-alpha and
leukotriene C4 (LTC4) (Van Loveren et al., 1997). Both mast cells and
basophils arise from CD34 pluripotent stem cells. At what point the
cell lineages diverge is unknown, but mature mast cells depend on the
local production of C-kit ligand (stem cell factor) for their
survival. Basophils will not survive in the presence of stem cell
factors but do respond to IL-3.
1.2.10 Basophils
Basophils represent approximately 1% of the white blood cells in
peripheral blood. They have a half-life of about 3 days. They respond
to chemotactic stimulation and tend to accumulate in inflammatory
reactions. Basophils have high affinity IgE receptors as do mast
cells. Cross-linking of surface-bound IgE by a multivalent specific
allergen causes changes in the cell membrane and signal transduction
that result in the release of mediators from the cytoplasmic granules.
These preformed mediators include histamine, many other potent
mediators, and proteolytic enzymes (Tharp, 1990; Goust, 1993; Janeway
et al., 1997). Release of these substances from mast cells and
basophils is responsible for the early phase symptoms seen in allergic
reactions, which occur within 30 to 60 min after exposure to the
allergen. IL-4 synthesis and release occurs hours later. Release of
these basophil-derived mediators is believed to contribute to the late
phase allergic response. The clinical manifestations due to release of
both preformed and newly synthesized mediators from mast cells and
basophils vary from a localized skin reaction to a systemic response
known as anaphylaxis. Symptoms depend on variables such as route of
exposure, dosage and frequency of exposure (Marsh & Norman, 1988).
1.2.11 Eosinophils
Eosinophils represent 2-5% of the leukocytes. Polymorphonuclear
eosinophils resemble polymorphonuclear neutrophils, with the
difference that they contain large red granulations (eosin staining)
and refringent crystals, which may also be traced in the expectorates
of asthmatic patients (Charcot-Leyden crystals). Eosinophil counts are
increased, especially in allergic reactions, but they also act as a
defence against certain parasites, in chronic inflammatory phenomena,
and perhaps also in the defence against cancer. Like neutrophils, they
do not return to the bone marrow from which they originate, but are
eliminated via mucosal surfaces.
In the biphasic pattern of certain asthma attacks (an acute phase
followed, about 6 h later, by a late phase), eosinophils attracted to
the inflammatory zone during the late phase cause extensive
destruction of the bronchial mucosa. This is similar to the
destruction by eosinophils of certain parasites like schistosomes,
responsible for schistosomiasis.
1.2.12 Complement components
Protective immunity requires the interaction of the immune cell
types described above with secreted proteins found in the blood and
lymph. In addition to antibody and lymphokines, the complement
proteins represent a series of important protective substances
(Table 6). More than 20 of these proteins participate in reactions
that mediate lysis of foreign cells. Complement-mediated lysis of
bacterial cells, for example, can take place through two routes, the
classical pathway, which is catalysed by complexes of antibody
molecules, or the alternative pathway, which can be activated by the
antigen alone and by some immunoglobulins (Fig. 5). This results in
deposition of a membrane attack complex of complement proteins on the
surface of the microbial cell, leading to lysis. This process occurs
as a cascade of enzymatic cleavage reactions, yielding both the lytic
structure and production of biologically active components that induce
migration of lymphocytes and an inflammatory response.
1.2.13 Immunoglobulins
Table 7 summarizes the human immunoglobulin isotypes and their
concentrations in serum.
1.2.13.1 IgG
IgG represents 75-80% of the total Ig in humans. IgG2 and IgG4
cross the placental barrier. Thus, at birth, a baby temporarily
carries IgG of its mother, which lasts for 4-6 months.
IgG intervenes in infections by means of opsonization and it can
neutralize toxins. IgG appears especially following a secondary immune
response, i.e., after a second encounter with antigen. The secretion
of IgG is modulated by collaboration between B- and T-lymphocytes. IgG
is strongly opsonizing for macrophages and polymorphonuclear cells
possessing receptors for the Fc portion of IgG.
Antigenic analysis of IgG myelomas revealed further variation and
showed that they could be grouped into four isotypic subclasses now
termed IgG1, IgG2, IgG3 and IgG4. The differences all lie in the heavy
chains, which have been labelled gamma1, gamma2, gamma3 and gamma4,
respectively. These heavy chains show considerable homology and have
certain structures in common with each other -- those which react with
specific anti-antisera -- but each has one or more additional
structures characteristic of its own subclass arising from differences
in primary amino acid composition and in interchain disulfide
bridging. These give rise to differences in biological behaviour
(Table 8).
Table 6. Principal components of the complement system
Protein Relative molecular Concentration in Characterization
mass serum (µg/ml) and function
Early components
Classical pathway
C1q 410 000 70 consists of a
collagen-like and
a globular part; binds to the Fc part of Ig
C1r 85 000 50 serine protease; activates C1s
C1s 85 000 50 serine protease; activates C4-C2
C4 210 000 300 C4b binds to C2b
C2 110 000 25 serine protease; catalytical part of C4bC2ba
Lectin pathway
MBL (Mannose-binding 410 000 1 consists of a collagen-like and a carbohydrate part
lectin)
MASP1 (Mannose-binding
lectin associated
serine protease) 85 000 5 serine protease; activates MASP2
MASP2 85 000 5 serine protease; activates C4
Alternative pathway
Factor-D 25 000 1 serine protease; activates factor-B
Factor-B 93 000 200 serine protease; as the component of
C3bBba convertase activates C3
Properdin 220 000 25 stabilizes the C3bBba convertase
Table 6. (continued)
Protein Relative molecular Concentration in Characterization
mass serum (µg/ml) and function
Common component of
the various pathways
C3 190 000 1300 together with C3b, interacting with
C4b2ba and C3bBba forms C5-convertase;
fragment C3a is one of the anaphylatoxins
Terminal components
C5 190 000 70 fragment C5b binds C6; fragment C5a
is one of the anaphylatoxins
C6 120 000 60 binds C7
C7 110 000 55 binds C8
C8 150 000 55 binds C9
C9 70 000 60 its polymerized form is the MAC
(membrane attack complex)
a The MBL-MASP complex (which is structurally similar to the C1 complex) activates the complement system.
The carbohydrate-binding domain of MBL binds to the carbohydrate components of various microorganisms
and the MASP cleaves C4.
pathway of antigen presentation and exposed on the outer cell membrane
by Class I molecules. This complex is recognized by the T-cell
receptor, after which CTL-target cell binding is further stabilized by
CD8-Class I interaction. In contrast, NK cell-target cell recognition
is largely non-specific, but involves receptors recognizing disturbed
surface carbohydrates and an Fc receptor for IgG that can facilitate
antibody-dependent cell-mediated cytotoxicity (ADCC). NK cells are
unique in bearing distinct receptors which, when bound to MHC Class I
molecules, deliver signals interfering with their cytolytic activity.
For both types of cytotoxic lymphocytes the actual killing
process involves two major mechanisms, i.e., release of a membrane
pore-forming protein named perforin from granules, leading to osmotic
lysis of target cells, and release of lymphotoxin which activates
enzymes in the target cell to cleave DNA in the nucleus. The latter
process is also known as apoptosis. Most cytotoxic lymphocytes also
express a member of the tumour necrosis factor (TNF) superfamily,
i.e., Fas-ligand, mediating a third lytic mechanism for target cells
expressing the Fas antigen. The killing capacity of cytotoxic
lymphocytes is greatly enhanced by distinct cytokines, in particular
IL-2 and IL-12. Microscopically this is reflected by the appearance of
more prominent granules, e.g., in the so-called lymphokine-activated
killer (LAK) cells. Both major cytotoxic lymphocyte populations are
crucial to various phases of viral attack, but are not prominent in
causing allergic disorders. Nevertheless, contact allergens may
directly associate with surface-bound Class I molecules or enter the
cytoplasm of, for instance, Langerhans cells and associate with
peptides presented along the endogenous route of antigen presentation.
In this way, CD8+ T-cells may become involved in allergic contact
dermatitis reactions.
1.2.9 Mast cells
Mast cells are derived from precursors in the bone marrow that
migrate to specific tissue sites to mature. While they are found
throughout the body, they are most prominent in the skin, the upper
and lower respiratory tract, and the gastrointestinal tract (Tharp,
1990). In most organs mast cells tend to be concentrated around the
small blood vessels, the lymphatics, the nerves and the glandular
tissue (Tharp, 1990). These cells contain numerous cytoplasmic
granules that are enclosed by a bilayered membrane. There appear to be
two different populations of mast cells in humans, based on the
presence or absence of certain proteolytic enzymes, notably tryptase
and chymase (Tharp, 1990). Mast cells found in the skin and connective
tissue have both enzymes, while those in the alveoli, bronchial and
bronchiolar regions, and mucosa of the small bowel contain only
tryptase (Irani et al., 1986). However, both types of cells are
triggered in the same manner.
Table 4. Cellular distribution and binding properties of Fc-gamma receptors
Class CD Relative molecular mass Affinity (Ka) Expressiona Ig-bindingb
Fc-gamma-RI CD64 72 000 108-109 M-1 Mo, M hu, 3>1>4>>2
Fc-gamma-RII CD32 40 000 <107 M-1 Mo, N, Ba, Eo, Langerhans cell, B-cell hu, 3>1>>2,4
mu, 2b>>2a
Fc-gamma-RIII CD16 50 000-80 000 Thr, endothelial cells of the placenta
Fc-gamma-IIIa 3×107 M-1 Mo, M, LGL/NK, T-cell hu, 1=3>>2,4
mu, 1=3>>2,4
Fc-gamma-IIIb <107 M-1 N
a Mo = monocyte, M = macrophage, N = neutrophil granulocyte, Ba = basophil granulocyte, Eo = eosinophil granulocyte,
Thr = platelet, LGL = large granular lymphocyte, NK = natural killer cell
b hu = human, mu = murine
Table 5. Multisubunit immune recognition receptors (MIRRs) family
Receptor Ligand-binding subunit Signal transducing subunit
BCR
(B-cell
antigen receptor) mIg Ig-alpha (CD79a)
Ig-beta (CD79b)
TCR alpha-beta or gamma-delta CD3-gamma, delta and epsilon zeta-zeta or zeta-eta
(T-cell
antigen receptor)
Fc-epsilon-RI alpha-chain beta and gamma chain
Fc-gamma-RIIIa alpha-chain Fc-epsilon-RI-gamma-chain or TCR zeta-chain
Fc-gamma-RI alpha-chain Fc-epsilon-RI-gamma-chain
Mast cells may be activated by antigen-specific IgE bound to high
affinity receptors (Fc RI), antigen-specific IgE bound to low affinity
IgG receptors (Fc-epsilon-RII/III), or through complement receptors.
Following activation, most cells release preformed mediators such as
histamines and generate newly formed mediators such as TNF-alpha and
leukotriene C4 (LTC4) (Van Loveren et al., 1997). Both mast cells and
basophils arise from CD34 pluripotent stem cells. At what point the
cell lineages diverge is unknown, but mature mast cells depend on the
local production of C-kit ligand (stem cell factor) for their
survival. Basophils will not survive in the presence of stem cell
factors but do respond to IL-3.
1.2.10 Basophils
Basophils represent approximately 1% of the white blood cells in
peripheral blood. They have a half-life of about 3 days. They respond
to chemotactic stimulation and tend to accumulate in inflammatory
reactions. Basophils have high affinity IgE receptors as do mast
cells. Cross-linking of surface-bound IgE by a multivalent specific
allergen causes changes in the cell membrane and signal transduction
that result in the release of mediators from the cytoplasmic granules.
These preformed mediators include histamine, many other potent
mediators, and proteolytic enzymes (Tharp, 1990; Goust, 1993; Janeway
et al., 1997). Release of these substances from mast cells and
basophils is responsible for the early phase symptoms seen in allergic
reactions, which occur within 30 to 60 min after exposure to the
allergen. IL-4 synthesis and release occurs hours later. Release of
these basophil-derived mediators is believed to contribute to the late
phase allergic response. The clinical manifestations due to release of
both preformed and newly synthesized mediators from mast cells and
basophils vary from a localized skin reaction to a systemic response
known as anaphylaxis. Symptoms depend on variables such as route of
exposure, dosage and frequency of exposure (Marsh & Norman, 1988).
1.2.11 Eosinophils
Eosinophils represent 2-5% of the leukocytes. Polymorphonuclear
eosinophils resemble polymorphonuclear neutrophils, with the
difference that they contain large red granulations (eosin staining)
and refringent crystals, which may also be traced in the expectorates
of asthmatic patients (Charcot-Leyden crystals). Eosinophil counts are
increased, especially in allergic reactions, but they also act as a
defence against certain parasites, in chronic inflammatory phenomena,
and perhaps also in the defence against cancer. Like neutrophils, they
do not return to the bone marrow from which they originate, but are
eliminated via mucosal surfaces.
In the biphasic pattern of certain asthma attacks (an acute phase
followed, about 6 h later, by a late phase), eosinophils attracted to
the inflammatory zone during the late phase cause extensive
destruction of the bronchial mucosa. This is similar to the
destruction by eosinophils of certain parasites like schistosomes,
responsible for schistosomiasis.
1.2.12 Complement components
Protective immunity requires the interaction of the immune cell
types described above with secreted proteins found in the blood and
lymph. In addition to antibody and lymphokines, the complement
proteins represent a series of important protective substances
(Table 6). More than 20 of these proteins participate in reactions
that mediate lysis of foreign cells. Complement-mediated lysis of
bacterial cells, for example, can take place through two routes, the
classical pathway, which is catalysed by complexes of antibody
molecules, or the alternative pathway, which can be activated by the
antigen alone and by some immunoglobulins (Fig. 5). This results in
deposition of a membrane attack complex of complement proteins on the
surface of the microbial cell, leading to lysis. This process occurs
as a cascade of enzymatic cleavage reactions, yielding both the lytic
structure and production of biologically active components that induce
migration of lymphocytes and an inflammatory response.
1.2.13 Immunoglobulins
Table 7 summarizes the human immunoglobulin isotypes and their
concentrations in serum.
1.2.13.1 IgG
IgG represents 75-80% of the total Ig in humans. IgG2 and IgG4
cross the placental barrier. Thus, at birth, a baby temporarily
carries IgG of its mother, which lasts for 4-6 months.
IgG intervenes in infections by means of opsonization and it can
neutralize toxins. IgG appears especially following a secondary immune
response, i.e., after a second encounter with antigen. The secretion
of IgG is modulated by collaboration between B- and T-lymphocytes. IgG
is strongly opsonizing for macrophages and polymorphonuclear cells
possessing receptors for the Fc portion of IgG.
Antigenic analysis of IgG myelomas revealed further variation and
showed that they could be grouped into four isotypic subclasses now
termed IgG1, IgG2, IgG3 and IgG4. The differences all lie in the heavy
chains, which have been labelled gamma1, gamma2, gamma3 and gamma4,
respectively. These heavy chains show considerable homology and have
certain structures in common with each other -- those which react with
specific anti-antisera -- but each has one or more additional
structures characteristic of its own subclass arising from differences
in primary amino acid composition and in interchain disulfide
bridging. These give rise to differences in biological behaviour
(Table 8).
Table 6. Principal components of the complement system
Protein Relative molecular Concentration in Characterization
mass serum (µg/ml) and function
Early components
Classical pathway
C1q 410 000 70 consists of a
collagen-like and
a globular part; binds to the Fc part of Ig
C1r 85 000 50 serine protease; activates C1s
C1s 85 000 50 serine protease; activates C4-C2
C4 210 000 300 C4b binds to C2b
C2 110 000 25 serine protease; catalytical part of C4bC2ba
Lectin pathway
MBL (Mannose-binding 410 000 1 consists of a collagen-like and a carbohydrate part
lectin)
MASP1 (Mannose-binding
lectin associated
serine protease) 85 000 5 serine protease; activates MASP2
MASP2 85 000 5 serine protease; activates C4
Alternative pathway
Factor-D 25 000 1 serine protease; activates factor-B
Factor-B 93 000 200 serine protease; as the component of
C3bBba convertase activates C3
Properdin 220 000 25 stabilizes the C3bBba convertase
Table 6. (continued)
Protein Relative molecular Concentration in Characterization
mass serum (µg/ml) and function
Common component of
the various pathways
C3 190 000 1300 together with C3b, interacting with
C4b2ba and C3bBba forms C5-convertase;
fragment C3a is one of the anaphylatoxins
Terminal components
C5 190 000 70 fragment C5b binds C6; fragment C5a
is one of the anaphylatoxins
C6 120 000 60 binds C7
C7 110 000 55 binds C8
C8 150 000 55 binds C9
C9 70 000 60 its polymerized form is the MAC
(membrane attack complex)
a The MBL-MASP complex (which is structurally similar to the C1 complex) activates the complement system.
The carbohydrate-binding domain of MBL binds to the carbohydrate components of various microorganisms
and the MASP cleaves C4.
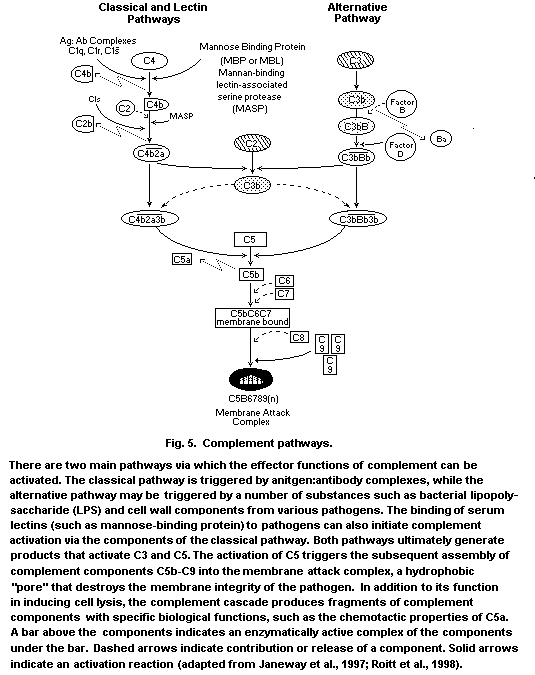 Table 7. Human immunoglobulin isotypes
Class Subclass H-chain Relative molecular mass Concentration in serum
(mg/ml)
IgA IgA1 alpha-1 150 000, 3.0
300 000,
400 000a
IgA2 alpha-2 150 000, 0.5
300 000,
400 000a
IgD - delta 180 000 trace
IgE - epsilon 190 000 trace
IgG IgG1 gamma-1 150 000 9.0
IgG2 gamma-2 150 000 3.0
IgG3 gamma-3 150 000 1.0
IgG4 gamma-4 150 000 0.5
IgM - mu 950 000b 1.5
a monomeric, dimeric, trimeric
b pentameric
1.2.13.2 IgA
IgA represents 15-20% of the human serum immunoglobulin pool,
where it occurs as a monomer of the regular immunoglobulin four-chain
unit, in contrast to secretory IgA, which mainly occurs in dimeric
form. The J chain which joins 2 IgA monomers facilitates the transfer
of the secretory component through cells. IgA is the predominant
immunoglobulin in seromucous secretions such as saliva, colostrum,
milk, and tracheobronchial and genitourinary secretions. Dimer
secretory IgA (sIgA), which may be of either of two subclasses (IgA1
or IgA2), but is mainly IgA2, is normally associated with yet another
protein, known as the secretory component. The bound secretory
component facilitates the transport of sIgA through the epithelial
cell layer(s) into the secretions and protects the antibody-dimer
against subsequent proteolytic attack. IgA2 predominates in secretions
since many microorganisms in the respiratory and gastrointestinal
tracts release proteases that cleave IgA1, but not IgA2. Next to IgA,
varying levels of IgE may be produced by locally residing plasma
cells, but the primary site of action of this antibody isotype is in
the sub-epithelial mucosal layers, e.g., in sensitizing locally
intruding protozoan parasites and worms for subsequent cytolytic
attack, notably by eosinophils. The secretory IgA (IgA-s) does not
opsonize. It fixes antigen via its variable part and forms unabsorbed
complexes. By capturing antigens, it prevents bacteria and viruses
from adhering to the mucous membrane, thereby preventing their
penetration into the organism.
The Fc fragment of IgA does not play any role, probably because
it is obstructed by the secretory component.
IgA deficiency is encountered in one of 700 individuals, causing
in such patients more frequent respiratory or gastrointestinal
infections. In case of IgA deficiency, IgM can take over. In severe
cases, there may be a simultaneous deficiency of both IgA and IgM.
1.2.13.3 IgM
IgM represents about 10% of immunoglobulins. IgM antibodies are
pentamers (5 units), the monomeric units being fixed by a J chain.
They are also known as macroglobulins or heavy globulins. IgM is the
first to appear in an immune response, and is the predominant antibody
isotype in the early phase of humoral immunity. As it has a short life
span, its presence points out to a recent infection (e.g., in
toxoplasmosis). Owing to its polyvalent structure, IgM can easily
produce agglutination and readily fixes complement. Because of its
large volume, it remains localized principally in blood. It does not
cross the placental barrier and is the first molecule to meet a viral
or microbial intruder in a blood vessel.
IgM antibodies tend to be of relatively low affinity as measured
against single determinants (haptens) but, because of their high
valency, they bind with considerable avidity to antigens with multiple
Table 8. The properties of human Ig isotypes
IgG1 IgG2 IgG3 IgG4 IgM IgA1 IgA2 IgD IgE
Complement activation,
classical pathway ++ + +++ - +++ - - - -
Complement activation,
alternative pathway - - - - - + - - -
Placental transfer + + - - - - -
Binding to macrophages
and other phagocytic cells + - + - - - - - +
High affinity binding to
mast cells and basophils - - - - - - - - +++
epitopes. For the same reason, these antibodies are extremely
efficient agglutinating and cytolytic agents and, since they appear
early in the response to infection and are largely confined to the
bloodstream, it is likely that they play a role of particular
importance in cases of bacteraemia. The isohaemagglutinins (anti-A,
anti-B) and many of the "natural" antibodies to microorganisms are
usually IgM; antibodies to the typhoid O antigen (endotoxin) and the
WR antibodies in syphilis are also found in this class. IgM appears to
precede other isotypes in the phylogeny of the immune response in
vertebrates.
Monomeric IgM (i.e., a single four-peptide unit), with a
hydrophobic sequence in the C-terminal end of the heavy chain to
anchor the molecule in the cell membrane, is the major antibody
receptor used by B-lymphocytes to recognize antigen.
1.2.13.4 IgD
This class was recognized through the discovery of a myeloma
protein that did not have the antigenic specificity of A or M,
although it reacted with antibodies to immunoglobulin light chains and
had the basic four-peptide structure. The hinge region is particularly
extended and, although protected to some degree by carbohydrate, it
may be this feature that makes IgD, among the different immunoglobulin
classes, uniquely susceptible to proteolytic degradation, and accounts
for its short half-life in plasma (2.8 days). It has been demonstrated
that nearly all the IgD is present, together with IgM, on the surface
of a proportion of B-lymphocytes where it seems likely that they may
operate as mutually interacting antigen receptors for the control of
lymphocyte activation and suppression. The greater susceptibility of
IgD to proteolysis on combination with antigen could well be
implicated in such a function.
1.2.13.5 IgE
The plasma level of IgE in normal individuals is low (Table 7).
The IgE level is commonly increased in patients suffering from Type I
allergies. It is a cytophilic Ig, i.e., it fixes to the surface of
certain cells, especially mast cells and basophils. It does not fix
complement. IgE occurs predominantly in perivascular tissues where
mast cells are localized. IgEs are responsible for Type I allergic
reactions. The binding of IgE with an antigen specific to this IgE on
the mast cell membrane provokes the release of mediators from mast
cell granules (degranulation)(see section 1.2.9).
IgE plays a major role in allergy, but it also appears to
intervene in the defence against parasites and perhaps also against
cancer cells. A high IgE level in apparently healthy babies has been
suggested as an accurate indicator of later allergic disorders (see
chapter 5).
IgE levels are particularly elevated in atopic eczema and in
intestinal parasitoses. Similarly elevated levels are also found in
certain myelomas and in disorders involving a long- or short-term
deficiency in T-lymphocytes, such as measles, infectious
mononucleosis, Hodgkin's disease, and dysglobulinaemia. Specific IgE
plays an important role in Type I allergies.
1.3 Immunotoxicology
Immunotoxicology may be defined as the scientific discipline
concerned with the adverse effects resulting from the interaction of
the immune system with xenobiotics. It includes the consequences of an
action (i.e., either suppression or enhancement) by a substance (or
its metabolite) on the immune system, as well as the immunological
response to such a substance (IPCS, 1996). A major focus of
immunotoxicology is the detection and evaluation of undesired effects
of substances by means of appropriate experiments. The prime concern
is to assess the importance of these interactions in regard to human
health. Toxic responses may occur when the immune system is the target
of chemical insults, resulting in altered immune function; this in
turn can result in decreased resistance to infection, certain forms of
neoplasia, or immune dysregulation or stimulation which exacerbates
allergy or autoimmunity. Alternatively, toxicity may arise when the
immune system responds to the antigenic specificity of the chemical as
part of a specific immune response (i.e., allergy or autoimmunity).
Certain drugs induce autoimmunity. The differentiation between direct
toxicity and toxicity due to an immune response to a compound is, to a
certain extent, artificial. Some compounds can exert a direct toxic
action on the immune system as well as altering the immune response.
Heavy metals like lead and mercury, for instance, manifest
immunosuppressive activity, hypersensitivity and autoimmunity.
1.4 Immunosuppression/immunodeficiency
1.4.1 Biological basis of immunosuppression/immunodeficiency
The occurrence of acquired immunodeficiency states was recognized
sporadically in scattered individuals during the 1960s and 1970s. In
the late 1970s and early 1980s, a new syndrome that spread rapidly
through certain groups was identified as a generalized type of
acquired immunodeficiency syndrome (AIDS). This disorder was found to
be due to a specific retrovirus that infects and destroys T helper
(Th) cells in humans (Fauci et al., 1991). These helper lymphocytes
have been identified in experimental studies as the key cells in the
recognition of antigen. Decrease in numbers of Th-cells leads to
impaired immune responses to a variety of infectious agents as well as
the occurrence of certain types of neoplasms. AIDS appears to result
from declining numbers of Th-cells with persistence of residual
populations of CD8+. Progression of AIDS is associated with
progressive loss of the Th-cells and an increased frequency of
infections by bacterial, fungal, viral and parasitic agents.
Other types of acquired immunodeficiency conditions have been
recognized and defined in the past two decades. Many have been related
to specific immunosuppressive drugs, chemotherapeutic agents and
certain chemicals (IPCS, 1996). The immunosuppressive effects of
xenobiotics in humans due to environmental exposure, when compared to
genetically determined immunodeficiency defects, do not reveal the
same degree of severity and persistence in the xenobiotic-related
immune defects as seen in the genetic disorders.
The dynamic nature of the immune system renders it especially
vulnerable to toxic influence. Reactions of lymphoid cells are
associated with gene amplification, transcription and translation.
Compounds that affect the processes of cell proliferation and
differentiation are especially immunotoxic. This applies in particular
to the rapidly dividing haematopoietic cells of the bone marrow and
thymocytes. Thus, the disappearance of lymphoid cells from bone
marrow, blood and tissue, and thymus weight may be the first and most
obvious signs of toxicity. Thymocytes are very susceptible to the
action of toxic compounds (Schuurman et al., 1992). It should be noted
that thymocyte depletion, suggestive of toxicity towards this cell
population, may actually be an indirect effect in cases where the cell
microenvironment is damaged and unable to support thymocyte growth.
The susceptibility of thymocytes to toxicity is related to the fragile
composition of these cells, especially cortical thymocytes, and to the
sensitive interactions between thymocytes and their microenvironment.
For instance, thymocytes are programmed to enter apoptosis when
activated during the physiological process of selection. The main
function of the thymus is T-cell (repertoire) generation during fetal
and early postnatal life. Its susceptibility to toxic compounds and
the subsequent effects on the cell-mediated immune system are most
prominent during this period of life. The skin, respiratory tract and
gastrointestinal tract together form an enormous surface that is in
close contact with the outside world, and they are potentially exposed
to a vast magnitude of microbial agents and potential toxicants. For
the respiratory tract, this is illustrated by human data on the
immunopathogenesis of lung diseases including asthma, fibrosis and
pulmonary infections. Examples of inhaled pollutants that may induce
these diseases are oxidant gases and particulates such as silica,
asbestos and coal dust.
The skin is an important target in immunotoxicology, as, for
instance, when there is contact with chemical allergens (Kimber &
Cumberbatch, 1992a,b) and UV-B irradiation (Goettsch et al., 1993).
The skin can respond to many xenobiotics by a specific immune response
(contact hypersensitivity) or by a non-specific inflammatory response
(contact irritancy); both responses are associated with the induction
of pro-inflammatory cytokines.
Drugs provide examples illustrating susceptibility to immunotoxic
effects. A number of cytostatic drugs are immunosuppressant. In
clinical medicine, cytostatic drugs used in cancer therapy often
produce bone marrow depression as a major side effect with increased
risk for infections as the result.
1.4.2 Consequences of immunosuppression/immunodeficiency
The major consequence of immunodeficiency or impaired immune
responsiveness is failure of protection of the host by antibody or
effector cells directed against specific target antigens. Antibody and
effector cells are essential for a protective effect against
infectious and toxic agents that can cause destructive tissue injury
and disseminated infections (Buckley, 1992). An impaired immune
response also limits the response to protective vaccines that normally
build adequate levels of cellular and antibody protection against
infectious agents. Selective impairment of immune responsiveness in
some instances may also lead to hypersensitivity states due to
dysregulation. This effect could also result in autoimmune disease by
promoting recognition of self-antigens, and hyperresponsiveness with
increased antibody and effector cell production (Bigazzi, 1988;
Broughton & Thrasher, 1988; Chandor, 1988). Increased potential for
the development of neoplasia and disseminated malignancies, especially
those of the lymphocytic tissues, may occur with impaired immune
surveillance (Radl et al., 1985; Byers et al., 1988).
The duration of immunodeficiency states might be transient or
long-lasting, depending on the severity and site of the specific
xenobiotic effect (Bekesi et al., 1987; Broughton & Thrasher, 1988).
The immune impairment that results from continued specific drug
therapy with immunosuppressive agents or human immunodeficiency virus
(HIV) infection are the only examples of long-lasting acquired
immunodeficiency in humans (Jenkins et al., 1988; Fauci et al., 1991).
Indeed, studies that have reported acquired deficiency of immune
function as a result of xenobiotics or radiation have shown the marked
capacity for self-restoring activity of the immune system, so that
once an offending agent has been cleared from the body the various
cellular components return to a normal state (Kishimoto & Hirano,
1984).
1.5 Immunological tolerance
Immunological tolerance refers to a state of non-responsiveness
that is specific for a particular antigen, and is induced by prior
exposure to that antigen. Tolerance can be induced to non-self
antigens, but the most important aspect of tolerance is
self-tolerance, which prevents the body from mounting an immune attack
against itself. The potential for attacking the body's own cells
arises because the immune system randomly generates a great diversity
of antigen-specific receptors, some of which will be self-reactive.
Cells bearing these receptors must be eliminated, either functionally
or physically.
1.5.1 T-cell tolerance to self-antigens
The thymus is central to the development of T-cells. Within the
thymus, T-cells develop from precursors that have not undergone
rearrangement of their T-cell antigen receptor (TCR) genes. In the
thymus, T-cells acquire the "education" that ensures that they respond
to antigens only in the context of molecules encoded by self major
histocompatibility complex (MHC) molecules. It is likely that
self-reactive T-cells are also dealt with and eliminated in the
thymus.
The high proliferative rate of thymocytes is paralleled by a
massive rate of cell death: the vast majority of T-cells, at the
double positive (CD4+ CD8+) stage, die within the thymus. Among the
factors that account for this are aberrant T-cell antigen receptor
(TCR) rearrangement, negative selection, and failure to be positively
selected. Positive selection occurs when T-cells, with some degree of
binding avidity for polymorphic regions of major histocompatibility
complex (MHC) molecules, are selected for survival. The MHC molecules
are encountered on thymic cortical epithelial cells, and binding is
presumed to protect the cells from programmed cell death. This
positive selection process ensures that the mature T-cell only
recognizes antigen (peptides) when associated with self-MHC molecules,
and so will be self-MHC restricted. Negative selection, on the other
hand, eliminates self-reactive T-cells, discarding those clones of
T-cells that are specifically reactive to self-antigens present
intrathymically.
The timing and precise localization of negative selection depends
on a variety of factors, including the accessibility of developing
T-cells to self-antigen, the combined avidity of the T-cell receptor
and accessory molecules, CD8 or CD4, for the self-MHC-self-peptide
complex, and the identity of the deleting cells. Elimination of
self-reactive cells is clearly a function of the thymic dendritic
cells or macrophages which are rich in MHC Class I and II molecules
and situated predominantly at the corticomedullary junction. Some
medullary or cortical epithelial cells may also impose negative
selection. Other cells involved in deletion may be the thymocytes
themselves. Specialized "veto" cells bearing self epitopes would
impart a negative signal, killing the self-reactive clone. Under
physiological conditions, veto signals occur when a T-cell with T-cell
receptors for self antigens binds to a veto cell. The veto cell is a
specialized T-cell expressing self epitopes. For the veto effect to
occur, the T-cell antigen receptor (TCR) has to bind to self antigen
in association with MHC Class I on the veto cell, while the CD8 of the
veto cell binds to MHC Class I on the T-cell. Once binding has
occurred, the T-cell is killed.
1.5.2 B-cell tolerance to self antigens
Production of high-affinity autoantibodies is T-cell dependent.
For this reason, and since the threshold of tolerance for T-cells is
lower than that for B-cells, the simplest explanation for
non-self-reactivity by B-cells is a lack of T-cell help. Nevertheless,
circumstances exist in which B-cells need to develop tolerance
directly. For example, there may be cross-reactive antigens on
microorganisms, which include both foreign T-cell-reactive epitopes
and other epitopes resembling self epitopes and capable of stimulating
B-cells (molecular mimicry). Such antigens could result in a vigorous
antibody response to self antigens. Furthermore, in contrast to T-cell
receptors, the immunoglobulin receptors on mature,
antigenically-stimulated B-cells can undergo hypermutation and may
acquire anti-self reactivities at this late stage. Tolerance must thus
be imposed on B-cells, both during their development and after
anti-genic stimulation in secondary lymphoid tissues.
The fate of self-reactive B-cells has been determined using
transgenic technology. The transgenic models showed that induction of
tolerance by self-antigens could lead to one of several end results.
The outcome depends on the affinity of the B-cell antigen receptor and
on the nature of the antigen it encounters, whether an integral
membrane protein, such as an MHC Class I molecule, or a soluble and
largely monomeric protein present in the circulation.
When B-cells encounter cell-membrane-associated self-antigens
capable of cross-linking Ig receptors on the B-cells with high
avidity, the B-cells are eliminated from lymphoid tissues. This type
of tolerance occurs whether the self-antigens are expressed on cells
in the bone marrow or elsewhere. In either case, the bone marrow
contains residual self-reactive B-cells, suggesting that immature
B-cells are less readily deleted than immature T-cells during the
early stages of differentiation.
If self-reactive B-cells are exposed to soluble antigen that is
largely monomeric (not capable of cross-linking receptors), then the
cells are not deleted from secondary lymphoid tissues, where they can
be found in normal numbers, but are rendered anergic. This effect only
occurs when the antigen is above a critical concentration threshold.
Anergy is associated with down-regulation of the membrane IgM
receptor. The maturation of the self-reactive B-cells is also arrested
in the follicular mantle zone and there is a striking reduction in
marginal zone B-cells with high levels of surface IgM. No evidence for
the activity of T-cells or of anti-idiotypic B-cells was found in
these transgenic models.
1.5.3 Tolerance to non-self antigens
1.5.3.1 Scope
Exposure to environmental and occupational allergens mainly takes
place along the skin and the mucosal surfaces lining the
gastrointestinal tract and the airways. Since no nutrients have to
pass the skin, skin barrier function simply focuses on exclusion of
exogenous molecules. Any macromolecule bypassing the skin epithelial
barrier is a potential health threat, and is subjected to
pro-inflammatory responses aimed at the most rapid destruction and/or
killing of the exogenous material. In sharp contrast, mucosal surfaces
along the gastrointestinal tract and the airways face a liquid or
moist environment which may contain valuable nutrient molecules, next
to a plethora of potentially toxic substances, including
microorganisms. Subtly balanced defence mechanisms have evolved,
therefore, along these mucosal surfaces to exclude microorganisms, and
to facilitate the entry of smaller nutrient molecules, such as
oligopeptides.
As a consequence, mucosal contacts with potential allergens may,
depending on the conditions, lead to either tolerance or
sensitization. The molecular and cell-biological characterization of
cytokines and adhesion molecules has led to better understanding of
the mechanisms involved in oral tolerance. There are primary,
non-immunological factors determining mucosal defence against
exogenous toxic pressures, including the roles of transmembrane
transporter molecules and TGF-beta in epithelial barrier function, as
well as alveolar macrophages and secretory IgA. The dichotomy between
Th1- and Th2-type immune responses in skin and mucosa, and the
supplementary role of TGF-beta are important.
1.5.3.2 Mucosal defence against exogenous toxic pressures
Distinct molecular mechanisms provide primary protection of
mucosal tissues against toxic pressure from exogenous toxic agents. If
these mechanisms fail, exogenous compounds penetrate the mucosa, reach
mucosal immunocytes, and induce undue immune reactivity. This leads to
local release of immunopharmacological mediators, such as
leukotrienes, further enhancing entry of xenobiotics by opening the
tight junctions. Studies, primarily aiming at elucidating mechanisms
of cytostatic drug-resistance in tumour cells, have shown the
existence of different molecular pumps mediating transmembrane
transport of potentially toxic molecules. Localization of these
molecules on the outer plasmacellular membrane contributes to the
efflux of exogenous toxic substrates from the cell interior to the
extracellular space, and localization on vesicle membranes contributes
to their loading into exocytotic vesicles, thus facilitating their
removal. While over-expression of these molecules on tumour cells
contributes to resistance to a vast array of cytostatic drugs
(multidrug resistance: MDR), the presence of such molecular pumps on
epithelial cells lining mucosal surfaces is thought to mediate a
primary barrier function to exogenous toxic pressure.
MDR-related proteins are abundantly present in various normal
tissues (Flens et al., 1996; Izquierdo et al., 1996). There,
MDR-related proteins represent physiological mechanisms of cellular
resistance to potentially toxic compounds. In normal tissues high
levels of these proteins can be observed on the luminal membranes of
epithelial cells lining mucosal surfaces chronically exposed to
xenobiotic agents, such as the respiratory epithelia in the trachea
and bronchi within the lung, and colonic epithelial cells. In the gut
they are thought to prevent too high intracellular concentrations of
potentially toxic molecules showing some degree of lipophilicity (van
der Valk et al., 1990; Weinstein et al., 1991). No regulatory
mechanisms have yet been defined determining to what extent MDR
molecules are expressed in mucosal lining cells. It is also still
unknown whether chronic inflammatory processes in the gastrointestinal
tract and airways might develop after failure of detoxifying
mechanisms similar to those mediating drug-resistance in tumour cells.
Mucosal epithelial barrier function is not only dependent on the
capacity of individual cells to resist uptake and passage of
potentially toxic molecules, but also on the integrity of the
epithelial cell layer(s). Important roles in maintaining this
integrity are played by two cytokines, IFN-gamma and TGF-beta
(Planchon et al., 1996). Of substances released by lymphocytes,
including those that reside in the mucosa, only IFN-gamma has been
reported to have a potent effect in reducing the barrier function of
epithelial monolayers in vitro (Madara & Stafford, 1989; Adams et
al., 1993). TGF-beta was found to enhance the integrity of epithelium
for normal homoeostasis (Derynck et al., 1988; Graycar et al., 1989;
Planchon et al., 1994). TGF-beta stimulates the synthesis of
extracellular matrix proteins (collagen, fibronectin) by up-regulating
their gene expression (Ignotz & Massague, 1986) and alters the
expression of integrins that act as receptors for these proteins,
thereby enhancing the cell's ability to bind them (Heine et al.,
1989). IFN-gamma and TGF-beta antagonism is most clearly revealed by
the striking ability of TGF-beta-1 to reduce the capacity of IFN-gamma
to disrupt epithelial barrier function (Planchon et al., 1996).
Another critical factor in the prevention of the potential
harmful entry of excessively large doses of antigens or microorganisms
into the mucosal tissues is the presence of IgA in the mucosal
secretions. IgA is highly efficient in complexing luminal antigenic
molecules and particles, thus reducing their chance of sneaking
through the epithelial barrier, and facilitates their uptake and
degradation by luminal phagocytes, e.g., pulmonary alveolar
macrophages (PAMs). The fact that TGF-beta is an important factor in
switching B-cell immunoglobulin synthesis to IgA production supports
the critical role of this cytokine in maintaining homoeostasis within
the mucosal tissues.
The maintenance of homoeostasis in the lungs requires particular
protection against environmental antigens. Chronic inflammatory immune
responses would be detrimental for these delicate tissues involved in
gas exchange. Highly active macrophages are present within the
alveolar spaces able to digest and eradicate exogenous antigens and
microorganisms, thus preventing these from even reaching the
epithelial barrier. Activation of PAMs is reflected by their
production of nitric oxide synthetase, leading to the local release of
nitric oxide, known as an effector molecule in macrophage-mediated
antimicrobial responses (Nussler & Billiar, 1993). Since nitric oxide
release is not a constitutive property of resident PAMs, effective
scavenging function requires a milieu of activating cytokines, such as
IFN-gamma, and the often synergistic cytokines IL-2 and TNF-alpha. On
the other hand, under steady state conditions, pro-inflammatory
processes are tightly controlled by lymphocytostatic signals generated
by the same resident PAMs. The mechanism(s) by which PAMs mediate
immune suppression, e.g., of T-cell proliferation, has been the
subject of much debate, and proposed mediators include prostaglandins
(Monick et al., 1987; Fireman et al., 1988), TGF-beta (Roth & Golub,
1993) and interleukin-1 receptor-antagonist (Moore et al., 1992).
TGF-beta has been identified as a most critical mediator in
suppressing local pro-inflammatory responses by its unique activity
in antagonizing IFN-gamma-induced macrophage activation (Bilyk & Holt,
1995).
1.5.3.3 Induction of oral tolerance
Chase (1946) confirmed that oral feeding of antigen could result
in a state of specific immunological unresponsiveness. Feeding contact
allergens to guinea-pigs made the animals refractory to subsequent
sensitization via the skin. Handling of antigen by the gut is
important in terms of both general and secretory immunity. The
induction of immunological unresponsiveness in humans by oral
ingestion of potential allergens was supported by the observation that
South American Indians ate poison ivy leaves in an attempt to prevent
contact sensitivity reactions to the plant (Dakin, 1982).
Systemic unresponsiveness after antigen feeding has been
described for a large variety of T-cell-dependent antigens, of which
the protein ovalbumin has been most extensively studied (reviewed in
Mowat, 1987). In addition, proteins such as bovine serum albumin
(Silverman et al., 1982; Domen et al., 1987), particulate
(erythrocyte-bound) antigens (Kagnoff, 1982; MacDonald, 1983;
Mattingly, 1984), inactivated viruses and bacteria (Stokes et al.,
1979) and autoimmune-related antigens (Thompson & Staines, 1990), as
well as contact allergens, have been shown to induce oral tolerance
(Asherson et al., 1977; Newby et al., 1980; Gautam et al., 1985).
Generally, T-cell-mediated delayed-type hypersensitivity responses and
IgE production are the types of immune responses to which tolerance
develops most readily. Persistent tolerance can be induced with
relatively low antigen doses (proteins: Heppel & Kilshaw, 1982;
Jarrett & Hall, 1984; contact allergens: Asherson et al., 1977; Polak,
1980; van Hoogstraten et al., 1992; Hariya et al., 1994). In sharp
contrast, local (secretory) IgA responses are generally unaffected
(Challacombe, 1983; Fuller et al., 1990). The apparent ability of the
intestinal immune system to prevent allergic hypersensitivity to
soluble, non-replicating antigens seems to be an important factor in
preventing enteropathies (Mowat, 1984, 1987; Mowat et al., 1986;
Challacombe & Tomasi, 1987). In contrast to potentially harmful,
pro-inflammatory DTH and IgE responses, the secretory IgA response
seems favourable. This immunoglobulin does not fix complement, nor
does it cause allergic reactions, whereas its release may rather
prevent enteropathies by inhibition of the entry of potentially
damaging molecules. Abrogation of oral tolerance to, for instance,
ovalbumin was found to lead to hypersensitivity responses in the
intestinal mucosa and gut-associated lymphoid tissues, resembling
those observed in food-sensitive enteropathies, e.g., coeliac disease.
Indeed, IgE and DTH responses are most frequently associated with
clinical food hypersensitivity.
1.5.3.4 Factors determining the development of oral tolerance
Several factors can play a role in the development of mucosal
tolerance, notably the nature of the antigens and the genetic
background, age and immune status of the individual. With regard to
the nature of the antigens, available experimental and clinical
evidence indicates that the ability of antigens to sensitize along the
skin route parallels the ability to induce tolerance upon mucosal
exposure. Thus, feeding of chemicals such as dinitrochlorobenze (DNCB)
and picryl chloride, which are strong sensitizers when first applied
to the skin, rapidly induces tolerance. Also nickel, which is amongst
the top ten of clinical contact sensitizing agents, is an effective
tolerance inducer in both experimental animals and humans (van
Hoogstraten, 1991, 1992, 1993). However, when the mucosal epithelial
barrier fails to prevent antigen passage, in particular the entry of
live viruses or bacteria, this may lead to priming for
pro-inflammatory immune responses rather than to the induction of
tolerance. The fact that such microorganisms are strong inducers of
local IL-12 and IFN-gamma release suggests that these cytokines could
play a role as antagonists for tolerance induction. Indeed, adequate
vaccination via the oral route can be achieved with live, attenuated
strains of microorganisms, e.g., with poliomyelitis vaccine (Stites &
Terr, 1991).
Essentially similar requirements for skin-sensitizing and
mucosal-tolerizing capacities of chemical allergens are also evident
from the apparent lack of major genetic influences on either of these
phenomena in outbred animals or humans. No or minimal genetic
restrictions have been found for the risk of developing contact
allergies to, for instance, nickel, nor for the induction of oral
tolerance to the same allergens. It would appear that the same
T-cell-receptor repertoire is being addressed under both conditions
but that, depending on the site of first encounter with the allergen,
sensitization or tolerance may ensue. On the other hand, inbred mouse
strains can show strong differences in their ability to develop
tolerance after protein feeding (Stokes et al., 1983 a,b; Tomasi et
al., 1983; Lamont et al., 1988). Noticeably, certain mouse strains
that are prone to autoimmune diseases fail to develop oral tolerance
to some proteins (Carr et al., 1985).
With regard to age, it was demonstrated in mice that ovalbumin
did not induce tolerance for either DTH or antibody responses during
the early postnatal period (1-2 days old), suggesting an increased
risk of allergic sensitization during infancy. The lack of tolerance
development in neonatal mice may be due to immaturity of the
intestinal immune system at birth in this species. The ability to
develop tolerance starts around day 4, but a transient defect in
tolerance induction occurs around the time of weaning (Strobel &
Ferguson, 1984; Hananan, 1990). Interestingly, clinical food
hypersensitivities in human infants often develop around the time of
weaning. This may be directly related to the physiological and dietary
changes associated with weaning, when large numbers of new antigens
are introduced. At the other end of the time scale, in ageing
individuals reduced abilities to develop new hypersensitivities and
tolerance are observed.
1.5.3.5 Orally induced flare-up reactions and desensitization
Considering the immune status of individuals, strong and
long-lasting oral tolerance can only be achieved in naive individuals,
i.e., those who have not been previously exposed to the antigen via
the skin. In mice, a single feed of ovalbumin was reported to suppress
fully subsequent systemic immune responses, and this state of
tolerance persisted for up to two years. In contrast, in primed
animals tolerance is hard to induce but partial and transient
unresponsiveness (desensitization) may eventually develop after
prolonged feeding of the antigen. Similar results have been obtained
in guinea-pig studies with various different chemical allergens,
including dinitrochlorobenzene (Polak, 1980), nickel (van Hoogstraten,
1994) and amlexanol (Hariya et al., 1994). Unfortunately, essentially
similar results have been obtained in early clinical trials aiming at
the treatment of autoimmune diseases, e.g., rheumatoid arthritis and
multiple sclerosis, by oral administration of relevant auto-antigens
(Weiner et al., 1994). Another problem with oral tolerance induction
in previously sensitized individuals arises owing to the tendency of
former inflammatory sites to re-inflame (flare-up reaction). Local
flare-up reactions confirm a previous sensitization process, and are
probably due to allergen-specific effector T-cells, which can persist
for periods up to several months at former inflammatory sites (Scheper
et al., 1983).
Two distinct features of immunocyte maturation may explain the
seemingly insurmountable differences between immunological responses
in naive and primed individuals, involving changes in expression
patterns of cellular adhesion/homing molecules, and lymphocyte
maturation features. First, a qualitative distinction exists between
naive (difficult to stimulate/afferently acting) cells and
effector/memory cells (easy to stimulate/efferently acting). In
contrast to naive lymphocytes, which only are activated by allergen
(modified-self constituents) if presented by professional dendritic
(e.g., Langerhans) cells, their progeny, known as effector/memory
lymphocytes, can also be stimulated by other cell types presenting
allergen-modified MHC Class II molecules, e.g., monocytes, endothelial
cells and B-cells. Effector/memory cells display increased numbers of
intercellular adhesion molecules (ICAMs), allowing for more
promiscuous cellular interactions. Amongst these, the most prominent
ICAMs are the CD28 and LFA-1 molecules, with B7-1/2 and ICAM-1 as
their respective ligands on APCs. Also, priming of T-cells leads to
the loss of homing receptors, such as L-selectin, which facilitate
interactions with high endothelial venules in peripheral lymph nodes.
Apparently, after sensitization T-cells are less capable of
recirculating through the lymphoid organs, but gain in ability to
migrate into the peripheral tissues. Indeed, interactions with
endothelia within inflamed skin are facilitated by the enhanced
expression of ICAMs like the cutaneous lymphocyte-associated antigen
CLA. Thus, effector/memory T-cells largely distribute over the
peripheral tissues where conditions may be insufficient to convey
effective tolerogenic signals. The second problem in inducing
tolerance in previously primed individuals relates directly to the
actual mechanism(s) of oral tolerance.
1.5.3.6 Mechanisms of tolerance
As discussed above, a preliminary factor contributing to
immunological non-responsiveness and/or lack of hypersensitivity
reactions at mucosal surfaces is the epithelial barrier function,
preventing entry of potentially harmful allergens. Obviously, from an
immunological point of view, this is a null-event and does not have
implications for subsequent encounters with the same allergen. Also as
discussed above, TGF-beta, a cytokine locally produced by epithelial
cells and immunocytes, plays a pivotal role in maintaining epithelial
barrier integrity. Importantly, the same cytokine also has broad
non-specific immunosuppressive functions, for example, antagonizing
phagocytic effector cell functions of pulmonary alveolar macrophages.
Similarly, other immunosuppressive cytokines may be locally released
from epithelial cells and may act in concert with TGF-beta to
down-regulate immune effector functions, such as epithelial
cell-derived P15E-related factors which show sequence homology with
retroviral envelope proteins (Oostendorp et al., 1993).
In contrast, specific immunological tolerance depends on
decreased responsiveness of specific B- or T-cells, or release of
immunosuppressive mediators from these cells after specific challenge.
Exposure to high doses of antigens may induce clonal deletion or
anergy of specific B- or T-cells by induction of apoptosis or
antigen-receptor down-regulation (Jones et al., 1990; Schönrich et
al., 1991; Ohashi et al., 1991; Melamed & Friedman, 1993).
Generally, ligation of the T-cell antigen receptor (TCR) in the
absence of appropriate co-stimulatory signals results in T-cell
non-responsiveness, not only in Th1- but also in Th2- cells. Human
CD4+ Th2-clones specific for the house dust mite allergen Der p I
can be rendered non-responsive to subsequent Der p I challenges by
incubating them with Der p I-derived peptides, representing the
relevant minimal T-cell activation-inducing epitopes, in the absence
of professional APC (Yssel et al., 1992). The anergized Th2-cells also
failed to produce cytokines (including IL-4 and IL-13) and failed to
provide help for B-cell IgE synthesis. The mechanisms underlying this
T-cell unresponsiveness have not yet been determined. Although these
cells cannot be activated through their T-cell antigen receptor (TCR),
they proliferate well in response to IL-2, or following activation by
Ca++ ionophore and the phorbol ester 12-O-tetradecanoylphorbol
13-acetate (TPA), suggesting that TCR activation or signalling
pathways immediately downstream of the TCR are disturbed.
Interestingly, the anergized Th2 cells expressed normal levels of
CD40 ligand, but their lack of help for B-cell IgE synthesis could not
be restored by exogenous IL-4 or IL-13, suggesting that in addition to
CD40L-CD40 interactions, other molecules are required for initiating
productive T- and B-cell interactions resulting in Ig isotype
production. It is likely that these molecules are down-regulated in
anergic T-cells. Peptide-induced Th2 cell tolerance and inhibition of
T-cell help for IgE synthesis may provide the basis for successful
immunotherapy in allergy. This anergy-based type of tolerance is
generally short-lived, since (functionally) deleted lymphocytes are
gradually replenished by newly arising clones in the bone marrow and
thymus and, in experimental animal models, cannot be transferred to
naive recipients, since these still contain a fully functional
repertoire, compensating for any missing clones. On the other hand,
mucosal contacts of naive individuals with relatively low amounts of
antigens, such as can be the case with environmental or occupational
exposure to chemical sensitizers, frequently induces a long-lasting
state of specific tolerance. Transfer of lymphoid cells, in particular
T-cells, from orally tolerized animals to syngeneic naive recipients
prevents their capacity to subsequently mount immune responses to the
same allergen, revealing the existence of so-called regulatory or
suppressor T-cells (Polak et al., 1980; van Hoogstraten et al., 1992,
1994; Weiner et al., 1994).
Although "professional" suppressor T-cells may not exist (Bloom
et al., 1992; Arnon & Teitelbaum, 1993), available data support the
possible development of specific regulatory T-cells that suppress
distinct immune functions. Depending on the experimental models, such
regulatory T-cells can belong to either or both the CD4+ or CD8+
subsets (Bloom et al., 1992). Regulatory T-cells may exert their
suppressive actions through different pathways, including the shedding
of TCR-alpha chains or hapten-binding TCR, through anti-idiotypic
reactivities, or through IL-2/cytokine consumption from the milieu
(Bloom et al., 1992; Fairchild et al., 1993; Kuchroo et al., 1995).
There is evidence that regulatory T-cells most often exert their role,
after antigen-specific activation, by releasing distinct cytokines
antagonizing specific effector T-cell functions (see section 1.2.1).
When starting clonal expansion after antigen-stimulation, T-cells
develop major cytokine profiles depending on the site of primary
contact (see section 1.2.1.1). For potential mechanism(s) of oral
tolerance T-cell subsets producing mutually suppressive cytokines can
be regarded as suppressor, or, better, as regulatory cells, depending
on the functions tested. Considering overt inflammatory reactions as
being most harmful to the individual and the primary cause of mucosal
hypersensitivities, Th2-cells and putative TGF-beta producing Th3-cells
are the most obvious candidates to mediate oral tolerance to proteins
and chemical allergens.
1.5.3.7 Conclusions
Although the phenomenon of oral tolerance has been known for over
a century the research on cellular resistance molecules, T-cell
cytokine patterns and cellular adhesion molecules has opened promising
avenues for further research on mechanisms and therapeutic options.
Clearly, the skin-versus-mucosa routing hypothesis discussed above
leaves many questions unanswered, such as the question of why some
chemicals may elicit strong Th2 responses and IgE antibody production
even when applied to the skin, without apparent reduction of delayed
allergic reactivity (Dearman et al., 1991). The preliminary
understanding of regulatory mechanisms in allergic contact dermatitis
has not yet led to further therapeutic progress. So far, no methods of
permanent desensitization have been devised. Nevertheless, the way in
which T-cells specifically recognize distinct allergens, as well as
how these and other inflammatory cells interact to generate
inflammation, is beginning to be understood. Defined cellular
interaction molecules and mediators provide promising targets for
anti-inflammatory drugs. Obviously, drugs found to be effective in
preventing severe T-cell-mediated conditions, e.g., rejection of a
vital organ graft, should be carefully evaluated before their use in
allergic skin disease is considered.
2. HYPERSENSITIVITY AND AUTOIMMUNITY - OVERVIEW OF MECHANISMS
Numerous environmental chemicals have the ability to produce a
hypersensitivity response. Although hypersensitivity diseases are
common, affecting millions of people, the incidence associated with
environmental pollutants or occupational exposure is largely unknown.
The characteristic that distinguishes allergic responses from immune
mechanisms involved in host defence is the nature of the reaction,
which often leads to tissue damage. Chemically induced
hypersensitivities usually fall into two responses distinguished not
only mechanistically but temporally: (1) immediate hypersensitivity,
which is mediated by immunoglobulin, most commonly IgE, and is
manifested within minutes of exposure to an allergen, and (2)
delayed-type hypersensitivity (DTH), a cell-mediated response that
occurs within 24-48 h. The type of immediate hypersensitivity response
elicited (anaphylactic, cytotoxic, Arthus or immune complex) depends
on the interaction of a sensitizing antigen or structurally related
compound with antibody. Delayed-type hypersensitivity responses are
characterized by T-lymphocytes bearing antigen-specific receptors
which, on contact with macrophage-associated antigen, respond by
secreting cytokines that mediate the delayed-type hypersensitivity
response. Almost any organ can be targeted by hypersensitivity
reactions, including the gastrointestinal tract, blood elements and
vessels, joints, kidneys, central nervous system and thyroid, although
the skin and lung, respectively, are the most common targets.
Various risk factors are involved in producing allergic
sensitization and influencing its severity. For instance, in the case
of aeroallergens, exposure can play a role in the primary
sensitization, in the development of symptomatic allergic disease, and
in the frequency and severity of acute symptomatic episodes. Other
risk factors include genetic predisposition, and age at the time of
the primary exposure.
Exposure to enzymes (mainly proteases) used in detergents have
also been associated with respiratory sensitization and symptoms.
Though sensitization is due to more than one factor, magnitude of
exposure has been demonstrated as a critical factor in the control of
primary sensitization to enzyme-containing detergents (Sarlo et al.,
1997).
Environmental factors have been suggested to contribute to the
prevalence of allergic diseases by modulating the allergen load
required for the sensitization as well as for the exacerbation and
intensity of allergic symptoms (Ollier & Davies, 1994).
2.1 Classification of immune reactions
Gell & Coombs (1963) classified immune reactions into four basic
types. Since then knowledge of immune reactions has increased and the
frequent overlaps between the different types must be stressed. This
classification is still very useful but the physiopathological reality
is frequently more complex.
The four major types of hypersensitivity according to Gell &
Coombs (1963) are:
Type I anaphylactic, immediate reaction
Type II cytotoxic reaction
Type III immune complex reaction
Type IV delayed or cell-mediated reaction
Sometimes a fifth type of hypersensitivity is added, i.e., Type V
stimulatory hypersensitivity (Roitt et al., 1998). In addition,
certain allergic diseases can be expressions of two or more types of
hypersensitivity.
The sections below review the mechanistic basis for phenomena and
diseases associated with each type of hypersensitivity.
2.1.1 Type I hypersensitivity
The distinguishing feature of Type I hypersensitivity is the
short time lag, usually seconds to minutes, between exposure to
antigen and the onset of clinical symptoms. The key reactant in Type I
or immediate sensitivity reactions is IgE (see Fig. 6). Antigens that
trigger formation of IgE are called atopic antigens, or allergens
(Marsh & Norman, 1988). Atopy refers to an inherited tendency to
respond to naturally occurring inhaled and ingested allergens with
continual production of IgE (Terr, 1994a). Patients who exhibit
allergic or immediate hypersensitivity reactions typically produce
antigen-specific IgE in response to a small concentration of antigen
(Atkinson & Platts-Mills, 1988). IgE levels appear to depend on the
interaction of both genetic and environmental factors.
Prausnitz & Kustner (1921) showed that a serum factor was
responsible for Type I reactions. This type of reaction is known as
passive cutaneous anaphylaxis. It occurs when serum is transferred
from an allergic individual to a non-allergic individual, and then the
second individual is challenged with specific antigen. This experiment
was conducted in 1921 but it was not until 1966 that the serum factor
responsible, namely IgE, was identified (Ishizaka & Ishizaka, 1966).
IgE is primarily synthesized in the lymphoid tissue of the respiratory
and gastrointestinal tracts. The regulation of IgE production appears
to be a function of T-cells. Th2 cytokines, in particular IL-4 and
IL-13 are essential for IgE synthesis, i.e., for the final
differentiation and isotype switch of the IgE-producing B-cells,
committing particular B-cells to IgE production (Goust, 1993). IL-2,
IL-5 and IL-6 also play a role, probably as sequential growth and
differentiation factors that select for IgE synthesis (Tharp, 1990).
Once an individual has become sensitized, the IgE produced
spreads throughout the body and binds in the peripheral tissues to
mast cells and basophils via the high affinity receptor for
Table 7. Human immunoglobulin isotypes
Class Subclass H-chain Relative molecular mass Concentration in serum
(mg/ml)
IgA IgA1 alpha-1 150 000, 3.0
300 000,
400 000a
IgA2 alpha-2 150 000, 0.5
300 000,
400 000a
IgD - delta 180 000 trace
IgE - epsilon 190 000 trace
IgG IgG1 gamma-1 150 000 9.0
IgG2 gamma-2 150 000 3.0
IgG3 gamma-3 150 000 1.0
IgG4 gamma-4 150 000 0.5
IgM - mu 950 000b 1.5
a monomeric, dimeric, trimeric
b pentameric
1.2.13.2 IgA
IgA represents 15-20% of the human serum immunoglobulin pool,
where it occurs as a monomer of the regular immunoglobulin four-chain
unit, in contrast to secretory IgA, which mainly occurs in dimeric
form. The J chain which joins 2 IgA monomers facilitates the transfer
of the secretory component through cells. IgA is the predominant
immunoglobulin in seromucous secretions such as saliva, colostrum,
milk, and tracheobronchial and genitourinary secretions. Dimer
secretory IgA (sIgA), which may be of either of two subclasses (IgA1
or IgA2), but is mainly IgA2, is normally associated with yet another
protein, known as the secretory component. The bound secretory
component facilitates the transport of sIgA through the epithelial
cell layer(s) into the secretions and protects the antibody-dimer
against subsequent proteolytic attack. IgA2 predominates in secretions
since many microorganisms in the respiratory and gastrointestinal
tracts release proteases that cleave IgA1, but not IgA2. Next to IgA,
varying levels of IgE may be produced by locally residing plasma
cells, but the primary site of action of this antibody isotype is in
the sub-epithelial mucosal layers, e.g., in sensitizing locally
intruding protozoan parasites and worms for subsequent cytolytic
attack, notably by eosinophils. The secretory IgA (IgA-s) does not
opsonize. It fixes antigen via its variable part and forms unabsorbed
complexes. By capturing antigens, it prevents bacteria and viruses
from adhering to the mucous membrane, thereby preventing their
penetration into the organism.
The Fc fragment of IgA does not play any role, probably because
it is obstructed by the secretory component.
IgA deficiency is encountered in one of 700 individuals, causing
in such patients more frequent respiratory or gastrointestinal
infections. In case of IgA deficiency, IgM can take over. In severe
cases, there may be a simultaneous deficiency of both IgA and IgM.
1.2.13.3 IgM
IgM represents about 10% of immunoglobulins. IgM antibodies are
pentamers (5 units), the monomeric units being fixed by a J chain.
They are also known as macroglobulins or heavy globulins. IgM is the
first to appear in an immune response, and is the predominant antibody
isotype in the early phase of humoral immunity. As it has a short life
span, its presence points out to a recent infection (e.g., in
toxoplasmosis). Owing to its polyvalent structure, IgM can easily
produce agglutination and readily fixes complement. Because of its
large volume, it remains localized principally in blood. It does not
cross the placental barrier and is the first molecule to meet a viral
or microbial intruder in a blood vessel.
IgM antibodies tend to be of relatively low affinity as measured
against single determinants (haptens) but, because of their high
valency, they bind with considerable avidity to antigens with multiple
Table 8. The properties of human Ig isotypes
IgG1 IgG2 IgG3 IgG4 IgM IgA1 IgA2 IgD IgE
Complement activation,
classical pathway ++ + +++ - +++ - - - -
Complement activation,
alternative pathway - - - - - + - - -
Placental transfer + + - - - - -
Binding to macrophages
and other phagocytic cells + - + - - - - - +
High affinity binding to
mast cells and basophils - - - - - - - - +++
epitopes. For the same reason, these antibodies are extremely
efficient agglutinating and cytolytic agents and, since they appear
early in the response to infection and are largely confined to the
bloodstream, it is likely that they play a role of particular
importance in cases of bacteraemia. The isohaemagglutinins (anti-A,
anti-B) and many of the "natural" antibodies to microorganisms are
usually IgM; antibodies to the typhoid O antigen (endotoxin) and the
WR antibodies in syphilis are also found in this class. IgM appears to
precede other isotypes in the phylogeny of the immune response in
vertebrates.
Monomeric IgM (i.e., a single four-peptide unit), with a
hydrophobic sequence in the C-terminal end of the heavy chain to
anchor the molecule in the cell membrane, is the major antibody
receptor used by B-lymphocytes to recognize antigen.
1.2.13.4 IgD
This class was recognized through the discovery of a myeloma
protein that did not have the antigenic specificity of A or M,
although it reacted with antibodies to immunoglobulin light chains and
had the basic four-peptide structure. The hinge region is particularly
extended and, although protected to some degree by carbohydrate, it
may be this feature that makes IgD, among the different immunoglobulin
classes, uniquely susceptible to proteolytic degradation, and accounts
for its short half-life in plasma (2.8 days). It has been demonstrated
that nearly all the IgD is present, together with IgM, on the surface
of a proportion of B-lymphocytes where it seems likely that they may
operate as mutually interacting antigen receptors for the control of
lymphocyte activation and suppression. The greater susceptibility of
IgD to proteolysis on combination with antigen could well be
implicated in such a function.
1.2.13.5 IgE
The plasma level of IgE in normal individuals is low (Table 7).
The IgE level is commonly increased in patients suffering from Type I
allergies. It is a cytophilic Ig, i.e., it fixes to the surface of
certain cells, especially mast cells and basophils. It does not fix
complement. IgE occurs predominantly in perivascular tissues where
mast cells are localized. IgEs are responsible for Type I allergic
reactions. The binding of IgE with an antigen specific to this IgE on
the mast cell membrane provokes the release of mediators from mast
cell granules (degranulation)(see section 1.2.9).
IgE plays a major role in allergy, but it also appears to
intervene in the defence against parasites and perhaps also against
cancer cells. A high IgE level in apparently healthy babies has been
suggested as an accurate indicator of later allergic disorders (see
chapter 5).
IgE levels are particularly elevated in atopic eczema and in
intestinal parasitoses. Similarly elevated levels are also found in
certain myelomas and in disorders involving a long- or short-term
deficiency in T-lymphocytes, such as measles, infectious
mononucleosis, Hodgkin's disease, and dysglobulinaemia. Specific IgE
plays an important role in Type I allergies.
1.3 Immunotoxicology
Immunotoxicology may be defined as the scientific discipline
concerned with the adverse effects resulting from the interaction of
the immune system with xenobiotics. It includes the consequences of an
action (i.e., either suppression or enhancement) by a substance (or
its metabolite) on the immune system, as well as the immunological
response to such a substance (IPCS, 1996). A major focus of
immunotoxicology is the detection and evaluation of undesired effects
of substances by means of appropriate experiments. The prime concern
is to assess the importance of these interactions in regard to human
health. Toxic responses may occur when the immune system is the target
of chemical insults, resulting in altered immune function; this in
turn can result in decreased resistance to infection, certain forms of
neoplasia, or immune dysregulation or stimulation which exacerbates
allergy or autoimmunity. Alternatively, toxicity may arise when the
immune system responds to the antigenic specificity of the chemical as
part of a specific immune response (i.e., allergy or autoimmunity).
Certain drugs induce autoimmunity. The differentiation between direct
toxicity and toxicity due to an immune response to a compound is, to a
certain extent, artificial. Some compounds can exert a direct toxic
action on the immune system as well as altering the immune response.
Heavy metals like lead and mercury, for instance, manifest
immunosuppressive activity, hypersensitivity and autoimmunity.
1.4 Immunosuppression/immunodeficiency
1.4.1 Biological basis of immunosuppression/immunodeficiency
The occurrence of acquired immunodeficiency states was recognized
sporadically in scattered individuals during the 1960s and 1970s. In
the late 1970s and early 1980s, a new syndrome that spread rapidly
through certain groups was identified as a generalized type of
acquired immunodeficiency syndrome (AIDS). This disorder was found to
be due to a specific retrovirus that infects and destroys T helper
(Th) cells in humans (Fauci et al., 1991). These helper lymphocytes
have been identified in experimental studies as the key cells in the
recognition of antigen. Decrease in numbers of Th-cells leads to
impaired immune responses to a variety of infectious agents as well as
the occurrence of certain types of neoplasms. AIDS appears to result
from declining numbers of Th-cells with persistence of residual
populations of CD8+. Progression of AIDS is associated with
progressive loss of the Th-cells and an increased frequency of
infections by bacterial, fungal, viral and parasitic agents.
Other types of acquired immunodeficiency conditions have been
recognized and defined in the past two decades. Many have been related
to specific immunosuppressive drugs, chemotherapeutic agents and
certain chemicals (IPCS, 1996). The immunosuppressive effects of
xenobiotics in humans due to environmental exposure, when compared to
genetically determined immunodeficiency defects, do not reveal the
same degree of severity and persistence in the xenobiotic-related
immune defects as seen in the genetic disorders.
The dynamic nature of the immune system renders it especially
vulnerable to toxic influence. Reactions of lymphoid cells are
associated with gene amplification, transcription and translation.
Compounds that affect the processes of cell proliferation and
differentiation are especially immunotoxic. This applies in particular
to the rapidly dividing haematopoietic cells of the bone marrow and
thymocytes. Thus, the disappearance of lymphoid cells from bone
marrow, blood and tissue, and thymus weight may be the first and most
obvious signs of toxicity. Thymocytes are very susceptible to the
action of toxic compounds (Schuurman et al., 1992). It should be noted
that thymocyte depletion, suggestive of toxicity towards this cell
population, may actually be an indirect effect in cases where the cell
microenvironment is damaged and unable to support thymocyte growth.
The susceptibility of thymocytes to toxicity is related to the fragile
composition of these cells, especially cortical thymocytes, and to the
sensitive interactions between thymocytes and their microenvironment.
For instance, thymocytes are programmed to enter apoptosis when
activated during the physiological process of selection. The main
function of the thymus is T-cell (repertoire) generation during fetal
and early postnatal life. Its susceptibility to toxic compounds and
the subsequent effects on the cell-mediated immune system are most
prominent during this period of life. The skin, respiratory tract and
gastrointestinal tract together form an enormous surface that is in
close contact with the outside world, and they are potentially exposed
to a vast magnitude of microbial agents and potential toxicants. For
the respiratory tract, this is illustrated by human data on the
immunopathogenesis of lung diseases including asthma, fibrosis and
pulmonary infections. Examples of inhaled pollutants that may induce
these diseases are oxidant gases and particulates such as silica,
asbestos and coal dust.
The skin is an important target in immunotoxicology, as, for
instance, when there is contact with chemical allergens (Kimber &
Cumberbatch, 1992a,b) and UV-B irradiation (Goettsch et al., 1993).
The skin can respond to many xenobiotics by a specific immune response
(contact hypersensitivity) or by a non-specific inflammatory response
(contact irritancy); both responses are associated with the induction
of pro-inflammatory cytokines.
Drugs provide examples illustrating susceptibility to immunotoxic
effects. A number of cytostatic drugs are immunosuppressant. In
clinical medicine, cytostatic drugs used in cancer therapy often
produce bone marrow depression as a major side effect with increased
risk for infections as the result.
1.4.2 Consequences of immunosuppression/immunodeficiency
The major consequence of immunodeficiency or impaired immune
responsiveness is failure of protection of the host by antibody or
effector cells directed against specific target antigens. Antibody and
effector cells are essential for a protective effect against
infectious and toxic agents that can cause destructive tissue injury
and disseminated infections (Buckley, 1992). An impaired immune
response also limits the response to protective vaccines that normally
build adequate levels of cellular and antibody protection against
infectious agents. Selective impairment of immune responsiveness in
some instances may also lead to hypersensitivity states due to
dysregulation. This effect could also result in autoimmune disease by
promoting recognition of self-antigens, and hyperresponsiveness with
increased antibody and effector cell production (Bigazzi, 1988;
Broughton & Thrasher, 1988; Chandor, 1988). Increased potential for
the development of neoplasia and disseminated malignancies, especially
those of the lymphocytic tissues, may occur with impaired immune
surveillance (Radl et al., 1985; Byers et al., 1988).
The duration of immunodeficiency states might be transient or
long-lasting, depending on the severity and site of the specific
xenobiotic effect (Bekesi et al., 1987; Broughton & Thrasher, 1988).
The immune impairment that results from continued specific drug
therapy with immunosuppressive agents or human immunodeficiency virus
(HIV) infection are the only examples of long-lasting acquired
immunodeficiency in humans (Jenkins et al., 1988; Fauci et al., 1991).
Indeed, studies that have reported acquired deficiency of immune
function as a result of xenobiotics or radiation have shown the marked
capacity for self-restoring activity of the immune system, so that
once an offending agent has been cleared from the body the various
cellular components return to a normal state (Kishimoto & Hirano,
1984).
1.5 Immunological tolerance
Immunological tolerance refers to a state of non-responsiveness
that is specific for a particular antigen, and is induced by prior
exposure to that antigen. Tolerance can be induced to non-self
antigens, but the most important aspect of tolerance is
self-tolerance, which prevents the body from mounting an immune attack
against itself. The potential for attacking the body's own cells
arises because the immune system randomly generates a great diversity
of antigen-specific receptors, some of which will be self-reactive.
Cells bearing these receptors must be eliminated, either functionally
or physically.
1.5.1 T-cell tolerance to self-antigens
The thymus is central to the development of T-cells. Within the
thymus, T-cells develop from precursors that have not undergone
rearrangement of their T-cell antigen receptor (TCR) genes. In the
thymus, T-cells acquire the "education" that ensures that they respond
to antigens only in the context of molecules encoded by self major
histocompatibility complex (MHC) molecules. It is likely that
self-reactive T-cells are also dealt with and eliminated in the
thymus.
The high proliferative rate of thymocytes is paralleled by a
massive rate of cell death: the vast majority of T-cells, at the
double positive (CD4+ CD8+) stage, die within the thymus. Among the
factors that account for this are aberrant T-cell antigen receptor
(TCR) rearrangement, negative selection, and failure to be positively
selected. Positive selection occurs when T-cells, with some degree of
binding avidity for polymorphic regions of major histocompatibility
complex (MHC) molecules, are selected for survival. The MHC molecules
are encountered on thymic cortical epithelial cells, and binding is
presumed to protect the cells from programmed cell death. This
positive selection process ensures that the mature T-cell only
recognizes antigen (peptides) when associated with self-MHC molecules,
and so will be self-MHC restricted. Negative selection, on the other
hand, eliminates self-reactive T-cells, discarding those clones of
T-cells that are specifically reactive to self-antigens present
intrathymically.
The timing and precise localization of negative selection depends
on a variety of factors, including the accessibility of developing
T-cells to self-antigen, the combined avidity of the T-cell receptor
and accessory molecules, CD8 or CD4, for the self-MHC-self-peptide
complex, and the identity of the deleting cells. Elimination of
self-reactive cells is clearly a function of the thymic dendritic
cells or macrophages which are rich in MHC Class I and II molecules
and situated predominantly at the corticomedullary junction. Some
medullary or cortical epithelial cells may also impose negative
selection. Other cells involved in deletion may be the thymocytes
themselves. Specialized "veto" cells bearing self epitopes would
impart a negative signal, killing the self-reactive clone. Under
physiological conditions, veto signals occur when a T-cell with T-cell
receptors for self antigens binds to a veto cell. The veto cell is a
specialized T-cell expressing self epitopes. For the veto effect to
occur, the T-cell antigen receptor (TCR) has to bind to self antigen
in association with MHC Class I on the veto cell, while the CD8 of the
veto cell binds to MHC Class I on the T-cell. Once binding has
occurred, the T-cell is killed.
1.5.2 B-cell tolerance to self antigens
Production of high-affinity autoantibodies is T-cell dependent.
For this reason, and since the threshold of tolerance for T-cells is
lower than that for B-cells, the simplest explanation for
non-self-reactivity by B-cells is a lack of T-cell help. Nevertheless,
circumstances exist in which B-cells need to develop tolerance
directly. For example, there may be cross-reactive antigens on
microorganisms, which include both foreign T-cell-reactive epitopes
and other epitopes resembling self epitopes and capable of stimulating
B-cells (molecular mimicry). Such antigens could result in a vigorous
antibody response to self antigens. Furthermore, in contrast to T-cell
receptors, the immunoglobulin receptors on mature,
antigenically-stimulated B-cells can undergo hypermutation and may
acquire anti-self reactivities at this late stage. Tolerance must thus
be imposed on B-cells, both during their development and after
anti-genic stimulation in secondary lymphoid tissues.
The fate of self-reactive B-cells has been determined using
transgenic technology. The transgenic models showed that induction of
tolerance by self-antigens could lead to one of several end results.
The outcome depends on the affinity of the B-cell antigen receptor and
on the nature of the antigen it encounters, whether an integral
membrane protein, such as an MHC Class I molecule, or a soluble and
largely monomeric protein present in the circulation.
When B-cells encounter cell-membrane-associated self-antigens
capable of cross-linking Ig receptors on the B-cells with high
avidity, the B-cells are eliminated from lymphoid tissues. This type
of tolerance occurs whether the self-antigens are expressed on cells
in the bone marrow or elsewhere. In either case, the bone marrow
contains residual self-reactive B-cells, suggesting that immature
B-cells are less readily deleted than immature T-cells during the
early stages of differentiation.
If self-reactive B-cells are exposed to soluble antigen that is
largely monomeric (not capable of cross-linking receptors), then the
cells are not deleted from secondary lymphoid tissues, where they can
be found in normal numbers, but are rendered anergic. This effect only
occurs when the antigen is above a critical concentration threshold.
Anergy is associated with down-regulation of the membrane IgM
receptor. The maturation of the self-reactive B-cells is also arrested
in the follicular mantle zone and there is a striking reduction in
marginal zone B-cells with high levels of surface IgM. No evidence for
the activity of T-cells or of anti-idiotypic B-cells was found in
these transgenic models.
1.5.3 Tolerance to non-self antigens
1.5.3.1 Scope
Exposure to environmental and occupational allergens mainly takes
place along the skin and the mucosal surfaces lining the
gastrointestinal tract and the airways. Since no nutrients have to
pass the skin, skin barrier function simply focuses on exclusion of
exogenous molecules. Any macromolecule bypassing the skin epithelial
barrier is a potential health threat, and is subjected to
pro-inflammatory responses aimed at the most rapid destruction and/or
killing of the exogenous material. In sharp contrast, mucosal surfaces
along the gastrointestinal tract and the airways face a liquid or
moist environment which may contain valuable nutrient molecules, next
to a plethora of potentially toxic substances, including
microorganisms. Subtly balanced defence mechanisms have evolved,
therefore, along these mucosal surfaces to exclude microorganisms, and
to facilitate the entry of smaller nutrient molecules, such as
oligopeptides.
As a consequence, mucosal contacts with potential allergens may,
depending on the conditions, lead to either tolerance or
sensitization. The molecular and cell-biological characterization of
cytokines and adhesion molecules has led to better understanding of
the mechanisms involved in oral tolerance. There are primary,
non-immunological factors determining mucosal defence against
exogenous toxic pressures, including the roles of transmembrane
transporter molecules and TGF-beta in epithelial barrier function, as
well as alveolar macrophages and secretory IgA. The dichotomy between
Th1- and Th2-type immune responses in skin and mucosa, and the
supplementary role of TGF-beta are important.
1.5.3.2 Mucosal defence against exogenous toxic pressures
Distinct molecular mechanisms provide primary protection of
mucosal tissues against toxic pressure from exogenous toxic agents. If
these mechanisms fail, exogenous compounds penetrate the mucosa, reach
mucosal immunocytes, and induce undue immune reactivity. This leads to
local release of immunopharmacological mediators, such as
leukotrienes, further enhancing entry of xenobiotics by opening the
tight junctions. Studies, primarily aiming at elucidating mechanisms
of cytostatic drug-resistance in tumour cells, have shown the
existence of different molecular pumps mediating transmembrane
transport of potentially toxic molecules. Localization of these
molecules on the outer plasmacellular membrane contributes to the
efflux of exogenous toxic substrates from the cell interior to the
extracellular space, and localization on vesicle membranes contributes
to their loading into exocytotic vesicles, thus facilitating their
removal. While over-expression of these molecules on tumour cells
contributes to resistance to a vast array of cytostatic drugs
(multidrug resistance: MDR), the presence of such molecular pumps on
epithelial cells lining mucosal surfaces is thought to mediate a
primary barrier function to exogenous toxic pressure.
MDR-related proteins are abundantly present in various normal
tissues (Flens et al., 1996; Izquierdo et al., 1996). There,
MDR-related proteins represent physiological mechanisms of cellular
resistance to potentially toxic compounds. In normal tissues high
levels of these proteins can be observed on the luminal membranes of
epithelial cells lining mucosal surfaces chronically exposed to
xenobiotic agents, such as the respiratory epithelia in the trachea
and bronchi within the lung, and colonic epithelial cells. In the gut
they are thought to prevent too high intracellular concentrations of
potentially toxic molecules showing some degree of lipophilicity (van
der Valk et al., 1990; Weinstein et al., 1991). No regulatory
mechanisms have yet been defined determining to what extent MDR
molecules are expressed in mucosal lining cells. It is also still
unknown whether chronic inflammatory processes in the gastrointestinal
tract and airways might develop after failure of detoxifying
mechanisms similar to those mediating drug-resistance in tumour cells.
Mucosal epithelial barrier function is not only dependent on the
capacity of individual cells to resist uptake and passage of
potentially toxic molecules, but also on the integrity of the
epithelial cell layer(s). Important roles in maintaining this
integrity are played by two cytokines, IFN-gamma and TGF-beta
(Planchon et al., 1996). Of substances released by lymphocytes,
including those that reside in the mucosa, only IFN-gamma has been
reported to have a potent effect in reducing the barrier function of
epithelial monolayers in vitro (Madara & Stafford, 1989; Adams et
al., 1993). TGF-beta was found to enhance the integrity of epithelium
for normal homoeostasis (Derynck et al., 1988; Graycar et al., 1989;
Planchon et al., 1994). TGF-beta stimulates the synthesis of
extracellular matrix proteins (collagen, fibronectin) by up-regulating
their gene expression (Ignotz & Massague, 1986) and alters the
expression of integrins that act as receptors for these proteins,
thereby enhancing the cell's ability to bind them (Heine et al.,
1989). IFN-gamma and TGF-beta antagonism is most clearly revealed by
the striking ability of TGF-beta-1 to reduce the capacity of IFN-gamma
to disrupt epithelial barrier function (Planchon et al., 1996).
Another critical factor in the prevention of the potential
harmful entry of excessively large doses of antigens or microorganisms
into the mucosal tissues is the presence of IgA in the mucosal
secretions. IgA is highly efficient in complexing luminal antigenic
molecules and particles, thus reducing their chance of sneaking
through the epithelial barrier, and facilitates their uptake and
degradation by luminal phagocytes, e.g., pulmonary alveolar
macrophages (PAMs). The fact that TGF-beta is an important factor in
switching B-cell immunoglobulin synthesis to IgA production supports
the critical role of this cytokine in maintaining homoeostasis within
the mucosal tissues.
The maintenance of homoeostasis in the lungs requires particular
protection against environmental antigens. Chronic inflammatory immune
responses would be detrimental for these delicate tissues involved in
gas exchange. Highly active macrophages are present within the
alveolar spaces able to digest and eradicate exogenous antigens and
microorganisms, thus preventing these from even reaching the
epithelial barrier. Activation of PAMs is reflected by their
production of nitric oxide synthetase, leading to the local release of
nitric oxide, known as an effector molecule in macrophage-mediated
antimicrobial responses (Nussler & Billiar, 1993). Since nitric oxide
release is not a constitutive property of resident PAMs, effective
scavenging function requires a milieu of activating cytokines, such as
IFN-gamma, and the often synergistic cytokines IL-2 and TNF-alpha. On
the other hand, under steady state conditions, pro-inflammatory
processes are tightly controlled by lymphocytostatic signals generated
by the same resident PAMs. The mechanism(s) by which PAMs mediate
immune suppression, e.g., of T-cell proliferation, has been the
subject of much debate, and proposed mediators include prostaglandins
(Monick et al., 1987; Fireman et al., 1988), TGF-beta (Roth & Golub,
1993) and interleukin-1 receptor-antagonist (Moore et al., 1992).
TGF-beta has been identified as a most critical mediator in
suppressing local pro-inflammatory responses by its unique activity
in antagonizing IFN-gamma-induced macrophage activation (Bilyk & Holt,
1995).
1.5.3.3 Induction of oral tolerance
Chase (1946) confirmed that oral feeding of antigen could result
in a state of specific immunological unresponsiveness. Feeding contact
allergens to guinea-pigs made the animals refractory to subsequent
sensitization via the skin. Handling of antigen by the gut is
important in terms of both general and secretory immunity. The
induction of immunological unresponsiveness in humans by oral
ingestion of potential allergens was supported by the observation that
South American Indians ate poison ivy leaves in an attempt to prevent
contact sensitivity reactions to the plant (Dakin, 1982).
Systemic unresponsiveness after antigen feeding has been
described for a large variety of T-cell-dependent antigens, of which
the protein ovalbumin has been most extensively studied (reviewed in
Mowat, 1987). In addition, proteins such as bovine serum albumin
(Silverman et al., 1982; Domen et al., 1987), particulate
(erythrocyte-bound) antigens (Kagnoff, 1982; MacDonald, 1983;
Mattingly, 1984), inactivated viruses and bacteria (Stokes et al.,
1979) and autoimmune-related antigens (Thompson & Staines, 1990), as
well as contact allergens, have been shown to induce oral tolerance
(Asherson et al., 1977; Newby et al., 1980; Gautam et al., 1985).
Generally, T-cell-mediated delayed-type hypersensitivity responses and
IgE production are the types of immune responses to which tolerance
develops most readily. Persistent tolerance can be induced with
relatively low antigen doses (proteins: Heppel & Kilshaw, 1982;
Jarrett & Hall, 1984; contact allergens: Asherson et al., 1977; Polak,
1980; van Hoogstraten et al., 1992; Hariya et al., 1994). In sharp
contrast, local (secretory) IgA responses are generally unaffected
(Challacombe, 1983; Fuller et al., 1990). The apparent ability of the
intestinal immune system to prevent allergic hypersensitivity to
soluble, non-replicating antigens seems to be an important factor in
preventing enteropathies (Mowat, 1984, 1987; Mowat et al., 1986;
Challacombe & Tomasi, 1987). In contrast to potentially harmful,
pro-inflammatory DTH and IgE responses, the secretory IgA response
seems favourable. This immunoglobulin does not fix complement, nor
does it cause allergic reactions, whereas its release may rather
prevent enteropathies by inhibition of the entry of potentially
damaging molecules. Abrogation of oral tolerance to, for instance,
ovalbumin was found to lead to hypersensitivity responses in the
intestinal mucosa and gut-associated lymphoid tissues, resembling
those observed in food-sensitive enteropathies, e.g., coeliac disease.
Indeed, IgE and DTH responses are most frequently associated with
clinical food hypersensitivity.
1.5.3.4 Factors determining the development of oral tolerance
Several factors can play a role in the development of mucosal
tolerance, notably the nature of the antigens and the genetic
background, age and immune status of the individual. With regard to
the nature of the antigens, available experimental and clinical
evidence indicates that the ability of antigens to sensitize along the
skin route parallels the ability to induce tolerance upon mucosal
exposure. Thus, feeding of chemicals such as dinitrochlorobenze (DNCB)
and picryl chloride, which are strong sensitizers when first applied
to the skin, rapidly induces tolerance. Also nickel, which is amongst
the top ten of clinical contact sensitizing agents, is an effective
tolerance inducer in both experimental animals and humans (van
Hoogstraten, 1991, 1992, 1993). However, when the mucosal epithelial
barrier fails to prevent antigen passage, in particular the entry of
live viruses or bacteria, this may lead to priming for
pro-inflammatory immune responses rather than to the induction of
tolerance. The fact that such microorganisms are strong inducers of
local IL-12 and IFN-gamma release suggests that these cytokines could
play a role as antagonists for tolerance induction. Indeed, adequate
vaccination via the oral route can be achieved with live, attenuated
strains of microorganisms, e.g., with poliomyelitis vaccine (Stites &
Terr, 1991).
Essentially similar requirements for skin-sensitizing and
mucosal-tolerizing capacities of chemical allergens are also evident
from the apparent lack of major genetic influences on either of these
phenomena in outbred animals or humans. No or minimal genetic
restrictions have been found for the risk of developing contact
allergies to, for instance, nickel, nor for the induction of oral
tolerance to the same allergens. It would appear that the same
T-cell-receptor repertoire is being addressed under both conditions
but that, depending on the site of first encounter with the allergen,
sensitization or tolerance may ensue. On the other hand, inbred mouse
strains can show strong differences in their ability to develop
tolerance after protein feeding (Stokes et al., 1983 a,b; Tomasi et
al., 1983; Lamont et al., 1988). Noticeably, certain mouse strains
that are prone to autoimmune diseases fail to develop oral tolerance
to some proteins (Carr et al., 1985).
With regard to age, it was demonstrated in mice that ovalbumin
did not induce tolerance for either DTH or antibody responses during
the early postnatal period (1-2 days old), suggesting an increased
risk of allergic sensitization during infancy. The lack of tolerance
development in neonatal mice may be due to immaturity of the
intestinal immune system at birth in this species. The ability to
develop tolerance starts around day 4, but a transient defect in
tolerance induction occurs around the time of weaning (Strobel &
Ferguson, 1984; Hananan, 1990). Interestingly, clinical food
hypersensitivities in human infants often develop around the time of
weaning. This may be directly related to the physiological and dietary
changes associated with weaning, when large numbers of new antigens
are introduced. At the other end of the time scale, in ageing
individuals reduced abilities to develop new hypersensitivities and
tolerance are observed.
1.5.3.5 Orally induced flare-up reactions and desensitization
Considering the immune status of individuals, strong and
long-lasting oral tolerance can only be achieved in naive individuals,
i.e., those who have not been previously exposed to the antigen via
the skin. In mice, a single feed of ovalbumin was reported to suppress
fully subsequent systemic immune responses, and this state of
tolerance persisted for up to two years. In contrast, in primed
animals tolerance is hard to induce but partial and transient
unresponsiveness (desensitization) may eventually develop after
prolonged feeding of the antigen. Similar results have been obtained
in guinea-pig studies with various different chemical allergens,
including dinitrochlorobenzene (Polak, 1980), nickel (van Hoogstraten,
1994) and amlexanol (Hariya et al., 1994). Unfortunately, essentially
similar results have been obtained in early clinical trials aiming at
the treatment of autoimmune diseases, e.g., rheumatoid arthritis and
multiple sclerosis, by oral administration of relevant auto-antigens
(Weiner et al., 1994). Another problem with oral tolerance induction
in previously sensitized individuals arises owing to the tendency of
former inflammatory sites to re-inflame (flare-up reaction). Local
flare-up reactions confirm a previous sensitization process, and are
probably due to allergen-specific effector T-cells, which can persist
for periods up to several months at former inflammatory sites (Scheper
et al., 1983).
Two distinct features of immunocyte maturation may explain the
seemingly insurmountable differences between immunological responses
in naive and primed individuals, involving changes in expression
patterns of cellular adhesion/homing molecules, and lymphocyte
maturation features. First, a qualitative distinction exists between
naive (difficult to stimulate/afferently acting) cells and
effector/memory cells (easy to stimulate/efferently acting). In
contrast to naive lymphocytes, which only are activated by allergen
(modified-self constituents) if presented by professional dendritic
(e.g., Langerhans) cells, their progeny, known as effector/memory
lymphocytes, can also be stimulated by other cell types presenting
allergen-modified MHC Class II molecules, e.g., monocytes, endothelial
cells and B-cells. Effector/memory cells display increased numbers of
intercellular adhesion molecules (ICAMs), allowing for more
promiscuous cellular interactions. Amongst these, the most prominent
ICAMs are the CD28 and LFA-1 molecules, with B7-1/2 and ICAM-1 as
their respective ligands on APCs. Also, priming of T-cells leads to
the loss of homing receptors, such as L-selectin, which facilitate
interactions with high endothelial venules in peripheral lymph nodes.
Apparently, after sensitization T-cells are less capable of
recirculating through the lymphoid organs, but gain in ability to
migrate into the peripheral tissues. Indeed, interactions with
endothelia within inflamed skin are facilitated by the enhanced
expression of ICAMs like the cutaneous lymphocyte-associated antigen
CLA. Thus, effector/memory T-cells largely distribute over the
peripheral tissues where conditions may be insufficient to convey
effective tolerogenic signals. The second problem in inducing
tolerance in previously primed individuals relates directly to the
actual mechanism(s) of oral tolerance.
1.5.3.6 Mechanisms of tolerance
As discussed above, a preliminary factor contributing to
immunological non-responsiveness and/or lack of hypersensitivity
reactions at mucosal surfaces is the epithelial barrier function,
preventing entry of potentially harmful allergens. Obviously, from an
immunological point of view, this is a null-event and does not have
implications for subsequent encounters with the same allergen. Also as
discussed above, TGF-beta, a cytokine locally produced by epithelial
cells and immunocytes, plays a pivotal role in maintaining epithelial
barrier integrity. Importantly, the same cytokine also has broad
non-specific immunosuppressive functions, for example, antagonizing
phagocytic effector cell functions of pulmonary alveolar macrophages.
Similarly, other immunosuppressive cytokines may be locally released
from epithelial cells and may act in concert with TGF-beta to
down-regulate immune effector functions, such as epithelial
cell-derived P15E-related factors which show sequence homology with
retroviral envelope proteins (Oostendorp et al., 1993).
In contrast, specific immunological tolerance depends on
decreased responsiveness of specific B- or T-cells, or release of
immunosuppressive mediators from these cells after specific challenge.
Exposure to high doses of antigens may induce clonal deletion or
anergy of specific B- or T-cells by induction of apoptosis or
antigen-receptor down-regulation (Jones et al., 1990; Schönrich et
al., 1991; Ohashi et al., 1991; Melamed & Friedman, 1993).
Generally, ligation of the T-cell antigen receptor (TCR) in the
absence of appropriate co-stimulatory signals results in T-cell
non-responsiveness, not only in Th1- but also in Th2- cells. Human
CD4+ Th2-clones specific for the house dust mite allergen Der p I
can be rendered non-responsive to subsequent Der p I challenges by
incubating them with Der p I-derived peptides, representing the
relevant minimal T-cell activation-inducing epitopes, in the absence
of professional APC (Yssel et al., 1992). The anergized Th2-cells also
failed to produce cytokines (including IL-4 and IL-13) and failed to
provide help for B-cell IgE synthesis. The mechanisms underlying this
T-cell unresponsiveness have not yet been determined. Although these
cells cannot be activated through their T-cell antigen receptor (TCR),
they proliferate well in response to IL-2, or following activation by
Ca++ ionophore and the phorbol ester 12-O-tetradecanoylphorbol
13-acetate (TPA), suggesting that TCR activation or signalling
pathways immediately downstream of the TCR are disturbed.
Interestingly, the anergized Th2 cells expressed normal levels of
CD40 ligand, but their lack of help for B-cell IgE synthesis could not
be restored by exogenous IL-4 or IL-13, suggesting that in addition to
CD40L-CD40 interactions, other molecules are required for initiating
productive T- and B-cell interactions resulting in Ig isotype
production. It is likely that these molecules are down-regulated in
anergic T-cells. Peptide-induced Th2 cell tolerance and inhibition of
T-cell help for IgE synthesis may provide the basis for successful
immunotherapy in allergy. This anergy-based type of tolerance is
generally short-lived, since (functionally) deleted lymphocytes are
gradually replenished by newly arising clones in the bone marrow and
thymus and, in experimental animal models, cannot be transferred to
naive recipients, since these still contain a fully functional
repertoire, compensating for any missing clones. On the other hand,
mucosal contacts of naive individuals with relatively low amounts of
antigens, such as can be the case with environmental or occupational
exposure to chemical sensitizers, frequently induces a long-lasting
state of specific tolerance. Transfer of lymphoid cells, in particular
T-cells, from orally tolerized animals to syngeneic naive recipients
prevents their capacity to subsequently mount immune responses to the
same allergen, revealing the existence of so-called regulatory or
suppressor T-cells (Polak et al., 1980; van Hoogstraten et al., 1992,
1994; Weiner et al., 1994).
Although "professional" suppressor T-cells may not exist (Bloom
et al., 1992; Arnon & Teitelbaum, 1993), available data support the
possible development of specific regulatory T-cells that suppress
distinct immune functions. Depending on the experimental models, such
regulatory T-cells can belong to either or both the CD4+ or CD8+
subsets (Bloom et al., 1992). Regulatory T-cells may exert their
suppressive actions through different pathways, including the shedding
of TCR-alpha chains or hapten-binding TCR, through anti-idiotypic
reactivities, or through IL-2/cytokine consumption from the milieu
(Bloom et al., 1992; Fairchild et al., 1993; Kuchroo et al., 1995).
There is evidence that regulatory T-cells most often exert their role,
after antigen-specific activation, by releasing distinct cytokines
antagonizing specific effector T-cell functions (see section 1.2.1).
When starting clonal expansion after antigen-stimulation, T-cells
develop major cytokine profiles depending on the site of primary
contact (see section 1.2.1.1). For potential mechanism(s) of oral
tolerance T-cell subsets producing mutually suppressive cytokines can
be regarded as suppressor, or, better, as regulatory cells, depending
on the functions tested. Considering overt inflammatory reactions as
being most harmful to the individual and the primary cause of mucosal
hypersensitivities, Th2-cells and putative TGF-beta producing Th3-cells
are the most obvious candidates to mediate oral tolerance to proteins
and chemical allergens.
1.5.3.7 Conclusions
Although the phenomenon of oral tolerance has been known for over
a century the research on cellular resistance molecules, T-cell
cytokine patterns and cellular adhesion molecules has opened promising
avenues for further research on mechanisms and therapeutic options.
Clearly, the skin-versus-mucosa routing hypothesis discussed above
leaves many questions unanswered, such as the question of why some
chemicals may elicit strong Th2 responses and IgE antibody production
even when applied to the skin, without apparent reduction of delayed
allergic reactivity (Dearman et al., 1991). The preliminary
understanding of regulatory mechanisms in allergic contact dermatitis
has not yet led to further therapeutic progress. So far, no methods of
permanent desensitization have been devised. Nevertheless, the way in
which T-cells specifically recognize distinct allergens, as well as
how these and other inflammatory cells interact to generate
inflammation, is beginning to be understood. Defined cellular
interaction molecules and mediators provide promising targets for
anti-inflammatory drugs. Obviously, drugs found to be effective in
preventing severe T-cell-mediated conditions, e.g., rejection of a
vital organ graft, should be carefully evaluated before their use in
allergic skin disease is considered.
2. HYPERSENSITIVITY AND AUTOIMMUNITY - OVERVIEW OF MECHANISMS
Numerous environmental chemicals have the ability to produce a
hypersensitivity response. Although hypersensitivity diseases are
common, affecting millions of people, the incidence associated with
environmental pollutants or occupational exposure is largely unknown.
The characteristic that distinguishes allergic responses from immune
mechanisms involved in host defence is the nature of the reaction,
which often leads to tissue damage. Chemically induced
hypersensitivities usually fall into two responses distinguished not
only mechanistically but temporally: (1) immediate hypersensitivity,
which is mediated by immunoglobulin, most commonly IgE, and is
manifested within minutes of exposure to an allergen, and (2)
delayed-type hypersensitivity (DTH), a cell-mediated response that
occurs within 24-48 h. The type of immediate hypersensitivity response
elicited (anaphylactic, cytotoxic, Arthus or immune complex) depends
on the interaction of a sensitizing antigen or structurally related
compound with antibody. Delayed-type hypersensitivity responses are
characterized by T-lymphocytes bearing antigen-specific receptors
which, on contact with macrophage-associated antigen, respond by
secreting cytokines that mediate the delayed-type hypersensitivity
response. Almost any organ can be targeted by hypersensitivity
reactions, including the gastrointestinal tract, blood elements and
vessels, joints, kidneys, central nervous system and thyroid, although
the skin and lung, respectively, are the most common targets.
Various risk factors are involved in producing allergic
sensitization and influencing its severity. For instance, in the case
of aeroallergens, exposure can play a role in the primary
sensitization, in the development of symptomatic allergic disease, and
in the frequency and severity of acute symptomatic episodes. Other
risk factors include genetic predisposition, and age at the time of
the primary exposure.
Exposure to enzymes (mainly proteases) used in detergents have
also been associated with respiratory sensitization and symptoms.
Though sensitization is due to more than one factor, magnitude of
exposure has been demonstrated as a critical factor in the control of
primary sensitization to enzyme-containing detergents (Sarlo et al.,
1997).
Environmental factors have been suggested to contribute to the
prevalence of allergic diseases by modulating the allergen load
required for the sensitization as well as for the exacerbation and
intensity of allergic symptoms (Ollier & Davies, 1994).
2.1 Classification of immune reactions
Gell & Coombs (1963) classified immune reactions into four basic
types. Since then knowledge of immune reactions has increased and the
frequent overlaps between the different types must be stressed. This
classification is still very useful but the physiopathological reality
is frequently more complex.
The four major types of hypersensitivity according to Gell &
Coombs (1963) are:
Type I anaphylactic, immediate reaction
Type II cytotoxic reaction
Type III immune complex reaction
Type IV delayed or cell-mediated reaction
Sometimes a fifth type of hypersensitivity is added, i.e., Type V
stimulatory hypersensitivity (Roitt et al., 1998). In addition,
certain allergic diseases can be expressions of two or more types of
hypersensitivity.
The sections below review the mechanistic basis for phenomena and
diseases associated with each type of hypersensitivity.
2.1.1 Type I hypersensitivity
The distinguishing feature of Type I hypersensitivity is the
short time lag, usually seconds to minutes, between exposure to
antigen and the onset of clinical symptoms. The key reactant in Type I
or immediate sensitivity reactions is IgE (see Fig. 6). Antigens that
trigger formation of IgE are called atopic antigens, or allergens
(Marsh & Norman, 1988). Atopy refers to an inherited tendency to
respond to naturally occurring inhaled and ingested allergens with
continual production of IgE (Terr, 1994a). Patients who exhibit
allergic or immediate hypersensitivity reactions typically produce
antigen-specific IgE in response to a small concentration of antigen
(Atkinson & Platts-Mills, 1988). IgE levels appear to depend on the
interaction of both genetic and environmental factors.
Prausnitz & Kustner (1921) showed that a serum factor was
responsible for Type I reactions. This type of reaction is known as
passive cutaneous anaphylaxis. It occurs when serum is transferred
from an allergic individual to a non-allergic individual, and then the
second individual is challenged with specific antigen. This experiment
was conducted in 1921 but it was not until 1966 that the serum factor
responsible, namely IgE, was identified (Ishizaka & Ishizaka, 1966).
IgE is primarily synthesized in the lymphoid tissue of the respiratory
and gastrointestinal tracts. The regulation of IgE production appears
to be a function of T-cells. Th2 cytokines, in particular IL-4 and
IL-13 are essential for IgE synthesis, i.e., for the final
differentiation and isotype switch of the IgE-producing B-cells,
committing particular B-cells to IgE production (Goust, 1993). IL-2,
IL-5 and IL-6 also play a role, probably as sequential growth and
differentiation factors that select for IgE synthesis (Tharp, 1990).
Once an individual has become sensitized, the IgE produced
spreads throughout the body and binds in the peripheral tissues to
mast cells and basophils via the high affinity receptor for
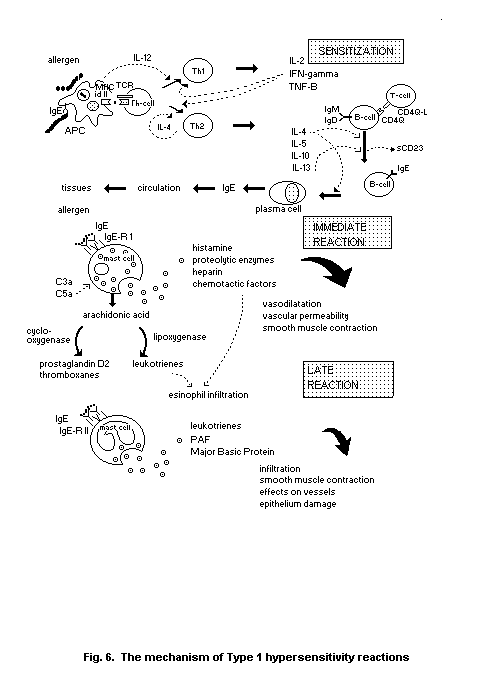 IgE (Fc-epsilon-RI). Upon contact with the allergen, the IgE molecules
will be cross-linked and the cells will release their granules
supplying the tissue with histamine, proteolytic enzymes, heparin and
chemotactic factors for eosinophils, neutrophils and monocytes. These
mediators induce vasodilatation, increased vascular permeability and
smooth muscle contraction and lead to an "immediate reaction", which
becomes clinically manifest within 20 min as a typical "wheal and
flare" in the skin or as bronchoconstriction in the respiratory tract.
At the same time, from the cell membrane new mediators, such as
prostaglandin-D2, thromboxanes and leukotrienes are being generated.
Together with the now attracted and activated eosinophils (which
produce platelet activating factor and major basic protein), these
mediators cause further infiltration, smooth muscle contraction,
mucosal oedema and damage of the epithelial cells, resulting in the so
called "late phase" reaction (12-24 h after challenge). Like the
immediate reaction the late phase responses can be observed both in
the skin and in the respiratory tract.
While actual antibody synthesis is regulated by the action of
cytokines, the tendency to respond to specific allergens appears to be
linked to inheritance of certain MHC genes. Various HLA class II
antigens seem to be associated with a high response to individual
allergens (Goust, 1993). As an example, individuals who possess the
HLA antigens B7 and DR2 are more likely to respond to a specific
ragweed antigen (Goust, 1993). The nature of this association is
unclear at this time.
2.1.1.1 Anaphylaxis
Anaphylaxis is the most severe type of allergic response, as it
involves multiple organs and may be fatal. Anaphylactic reactions are
typically triggered by glycoproteins or large polypeptides. Smaller
molecules, such as penicillin, are haptens that may become immunogenic
by combining with host cells or proteins. Typical agents that induce
anaphylaxis include venom from insects in the Hymenoptera family,
drugs such as penicillin, and foods such as seafood or egg albumin
(Widmann, 1989).
Allergic reaction to allergens (e.g., in food, venom) that result
in systemic anaphylaxis are, in the vast majority of instances,
believed to be mediated by allergen-specific IgE bound to high
affinity IgE receptors (Fc-epsilon-RI) on the surfaces of basophils
and mast cells. As described earlier, the subsequent activation of
basophils/mast cells results in the release (e.g., histamines) and
generation (e.g., leukotrienes) of potent chemical mediators of
anaphylaxis.
2.1.2 Type II hypersensitivity
Type II hypersensitivity reactions are caused by IgG and IgM
antibodies directed towards cell surface antigens. These antigens may
be altered self-antigens or heteroantigens. Such antibodies, bound to
the cell membrane, can activate inflammatory phagocytes by Fc receptor
triggering. These phagocytes will then try to kill or to inactivate
their target as they would kill a microorganism. If they are unable to
phagocytose the whole cell, they will cause cell damage by secreting
oxygen radicals and by generating inflammatory mediators such as
arachidonic acid metabolites (prostaglandins and leukotrienes) from
their cell membrane.
Moreover, cell-bound antibodies activate the complement system.
The presence of C3b on the cell membrane, in addition to the
immunoglobulin, facilitates phagocytosis, whereas the further
complement cascade will induce membrane perforation and cell lysis.
Together, these reactions result in destruction of antibody-coated
cells and thus in cytopenia or in considerable tissue damage.
Not only granulocytes and macrophages are able to kill
antibody-coated cells. Specialized large granular non-B,
non-T-lymphoid cells, called natural killer (NK) cells, also bear Fc
receptors (CD16) and are capable of killing antibody-coated target
cells. NK cell-mediated killing is achieved by the release of
cytoplasmic granules containing perforin and granzymes. This process
is called antibody-dependent cell-mediated cytotoxicity (ADCC) and,
although not yet recognized at the time of Gell & Coombs (1963), it
should strictly be considered as a Type II effector mechanism. ADCC
reactions have been well established in vitro to tumour antigens and
viral proteins, but their precise role in host defence and
hypersensitivity reactions is still not completely understood.
2.1.3 Type III hypersensitivity -- immune complex reaction
Type III hypersensitivity reactions are similar to Type II
reactions in that IgG or IgM is involved and that destruction is
complement-mediated. However, in the case of Type III diseases, the
antigen is soluble. When soluble antigen combines with antibody,
complexes are formed that precipitate out of the serum. These
complexes deposit in the tissues and bind complement, causing damage
to the particular tissue. Deposition of antigen-antibody complexes is
influenced by the relative concentration of both components. If a
large excess of antigen is present, sites on antibody molecules become
filled before cross-links can be formed. In antibody excess, a lattice
cannot be formed due to the relative sparsity of antigenic determinant
sites. The small complexes that result in either of the above cases
remain suspended or may pass directly into the urine. Precipitating
complexes, on the other hand, occur in mild antigen excess, and these
are the ones most likely to deposit in the tissues. Sites where this
typically occurs include the glomerular basement membrane, vascular
endothelium, joint linings, and pulmonary alveolar membranes (Roitt et
al., 1998).
Complement binds to these complexes in the tissues, causing the
release of mediators that increase vasodilation and vasopermeability,
attract macrophages and neutrophils, and enhance binding of phagocytic
cells by means of C3b deposited in the tissues. If the target cells
are large and cannot be engulfed for phagocytosis to take place,
granule and lysosomal contents are released by a process known as
exocytosis (Roitt et al., 1998). This results in the damage to host
tissue that is typified by Type III reactions.
2.1.3.1 Arthus reaction
The classic example of a Type III reaction is the Arthus
reaction, a local necrotic lesion resulting from a local
antigen-antibody reaction produced by intradermal injection of an
antigen into a previously sensitized animal. This reaction is
characterized by erythema and oedema, peaks within 3 to 8 h, and is
followed by a haemorrhagic necrotic lesion that ulcerates. The
inflammatory response is due to antigen-antibody combination and
subsequent formation of immune complexes that deposit in small dermal
blood vessels. Complement is fixed, attracting neutrophils and causing
aggregation of platelets. Activation of complement is, in fact,
essential for the Arthus reaction, as the C3a and C5a generated
activated mast cells to release permeability factors, with the
consequent localization of immune complexes along the endothelial cell
basement membrane (Terr, 1994b). The Arthus reaction is rare in
humans.
2.1.4 Type IV -- delayed-type hypersensitivity
Type IV reactions were originally described by Gell & Coombs
(1963) as those skin reactions which take more than 12 h to develop
after antigen application. The classical Type IV reaction is the
tuberculin reaction, which reaches its maximum 24-72 h after the
intradermal injection of mycobacterial extracts. This delayed type
skin reaction to intradermally injected protein is characterized by a
pronounced induration reflecting a dense mononuclear cell infiltrate.
Since it became clear that antigen-specific T-cells are
responsible for these reactions, the term Type IV reactivity has been
used not only in relation to delayed-type hypersensitivity (DTH)
reactions in the skin, but also to T-cell-mediated inflammatory
reactions in other tissues. In addition, other T-cell-mediated
reactions, such as those to infectious agents or tumour antigens,
which are rather protective than hypersensitive, are regularly
described as Type IV reactions.
Although CD8+ T-cells have been shown in some experimental
animal models to transfer DTH, generally CD4+ T-cells are held
responsible for DTH responsiveness. The majority of antigen specific
T-cells cloned from DTH reaction sites are of the CD4+ subset.
Increased frequencies of antigen-specific CD4+ T-cells can also be
detected in the circulation of sensitized individuals. Such memory
T-cells show enhanced expression of adhesion molecules, which
facilitates their recirculation through the peripheral tissues. So,
whereas priming of naive T-cells takes place in the lymph nodes
draining the area of antigen contact, the secondary DTH response of
memory T-cells rather takes place in the peripheral tissues at the
site of antigen contact.
Here they may encounter the antigens for which they were
originally sensitized. The T-cells do not recognize the whole antigen
or conformational epitopes as antibodies do, but they recognize small
peptides derived from these antigens after processing by
antigen-presenting cells (APC). MHC class II molecules bind these
peptides already within the intracellular vesicles and present them
subsequently on the APC membrane to helper T-cells (Fig. 5). If the
memory T-cells recognize the peptide in its MHC class II context, the
cells become activated and produce a characteristic set of cytokines.
In the DTH reaction that now develops, predominantly mononuclear
cells are attracted from the circulation and contribute to the local
inflammatory reaction. An essential chemokine found to play a role in
the early accumulation of leukocytes at the DTH reaction site is IL-8
(Larsen et al., 1995), whereas RANTES (Regulated in Activation Normal
T-cells Expressed and Secreted), produced by endothelial cells, was
shown to attract preferentially macrophages and CD4+ T-cells to the
DTH reaction (Marfaing-Koka et al., 1995). In addition to a number of
different chemokines, IFN-gamma, TNF-alpha and LT (lymphotoxin) are
produced in the DTH reaction (Tsicopoulos et al., 1992). These are
typical Th1 effector cytokines, which are either directly cytotoxic
for pathogens or indirectly by activating the macrophage bactericidal
mechanism. Together, the cytokine cascade during this secondary
response shows an extreme amplification power, as illustrated by
experimental studies in which measurable oedema could be triggered by
only one specific T-cell (Marchal et al., 1982). Therefore, DTH
reactions are mediated by Th1 cells, the most prominent cytokines
being IL-2, LT and IFN-gamma. Indeed, at the site of DTH reactions
these cytokines can be detected (Tsicopoulos et al., 1992). It should
be realized, however, that the immune response is always the resultant
of a Th1-Th2 balance and that this delicate balance can be influenced
by several external factors, such as drugs, hormones, infections and
altered antigen exposure. Chronic antigen stimulation, for instance,
may induce a shift away from Th1, DTH-associated immunity towards a
Th2 response (Kitagaki et al., 1995; Mosmann & Sad, 1996). Th2
cytokines, such as IL-4, IL-5 and IL-10, rather help to induce
antibody responses, particularly IgE. In chronic infectious disease
indeed high levels of antibodies can be detected while DTH reactivity
is waning ( Mycobacteria, Trichophyton). In the human system, a
Th1 to Th2 shift correlating with a clinical conversion from disease
resistance to susceptibility and disease progression has been shown
in Leishmania, Candida, Mycobacteria and HIV infection
(Mosmann & Sad, 1996).
2.1.4.1 Mechanisms of allergic contact dermatitis
a) Sensitization
In allergic contact dermatitis, Type IV reactivity is raised
against small, chemically reactive environmental agents that enter the
body via the skin. In the skin, epidermal dendritic Langerhans cells
(LC), bearing large numbers of class II molecules (HLA-DR, -DP and
-DQ) on their cell membrane, are the primary allergen-presenting
cells. They form a contiguous network in which agents penetrating the
skin are efficiently trapped. Langerhans cells stem from the bone
marrow, but their continuous presence in the epidermis is at least
partly maintained by local proliferation (Czernielewski & Demarchez,
1987; Breathnach, 1988).
Upon penetration through the epidermis, contact allergens readily
bind to a plethora of skin constituents. Whereas most allergens bind
spontaneously, some need metabolic conversion (Anderson et al., 1995)
or photoinduced activation before they bind. The latter allergens are
called contact photoallergens (White, 1992).
Only those allergens that modify the Langerhans cell MHC Class II
molecules can eventually sensitize T-cells; this occurs either by
direct binding to the MHC Class II molecules and to peptides within
their grooves or by uptake and processing of haptenized proteins
followed by presentation of the derived peptides in the MHC Class II
molecules of the antigen-presenting cells. It has been shown that in
individuals allergic to nickel some nickel-specific T-cell clones
recognize unprocessed nickel, bound to the MHC Class II molecules of
fixed antigen-presenting cells, whereas other nickel-specific T-cell
clones are dependent on viable antigen-presenting cells for
processing, most likely of preformed nickel-protein conjugates (Moulon
et al., 1995).
In a similar way, MHC Class I peptides may become modified by the
allergen, triggering class I restricted, CD8-positive T-cell clones.
Notably the size of the allergic metal ions is much smaller than the
peptides to which they bind in the groove. In some instances different
metallic allergens modify the MHC Class II peptides in a very similar
way. T-cell clones then show complete cross reactivity between the
metals, for instance between nickel and palladium, or between nickel
and copper (Pistoor et al., 1995). Clinical signs of cross reactivity
of nickel with other related metals, such as cobalt, however, most
probably result from concomitant sensitization by exposure to metal
alloys.
The majority of chemically reactive allergen, however, binds
covalently to distinct amino acids, thus forming haptenized proteins.
Usually, haptens, like picryl- or penicilloyl- are much larger than
metal allergens, and hapten-specific T-cell responses, i.e.,
independent of the carrier protein, can be observed.
Upon exposure of the skin to chemical contact sensitizing agents,
cytokine production in the epidermis by both keratinocytes and
Langerhans cells is immediately up-regulated, thereby initiating the
process of Langerhans cell maturation and migration.
The first cytokine to be up-regulated, within 15 min after
allergen application, is IL1-beta, produced by Langerhans cells. This
up-regulation was found to be allergen-specific, just like the
subsequent production of IL-1-alpha, and of the chemokines IP-10
(IFN-gamma-inducible protein 10) and MIP-2 (macrophage inflammatory
protein-2) by keratinocytes, and could not be detected upon irritant
application. TNF-alpha up-regulation in keratinocytes, on the other
hand, appeared to be a less allergen-specific event. The most relevant
cytokines for Langerhans cell maturation and egress are GM-CSF, IL-1
and TNF-alpha, while IL-10, which is also produced by keratinocytes,
but at a later stage, may serve as a down-regulatory molecule for
Langerhans cell maturation. (Heufler et al., 1988; Kimber &
Cumberbatch, 1992a,b; Enk & Katz, 1995). Interestingly, IL-1 and
TNF-alpha were found to down-regulate the membrane expression on
Langerhans cells of E-cadherin, a molecule that mediates
Langerhans-cell-keratinocyte adhesion. Thus, Langerhans cells with
allergen-modified MHC Class II molecules leave the epidermis and
migrate via the dermis and lymphatics to the draining lymph nodes,
where they settle within the paracortical areas. Indeed increased
numbers of dendritic cells appear in the regional lymph nodes around
24 h after hapten application (Kimber et al., 1990). Whereas resident
epidermal Langerhans cells are still relatively inefficient
antigen-presenting cells, once they have arrived in the lymph nodes
they have matured into fully active antigen-presenting dendritic cells
and are capable of stimulating even naive unprimed T-cells. Naive
cells express, in contrast to memory cells, low levels of cellular
adhesion molecules (CAM) and therefore require optimally functioning
antigen-presenting cells for stimulation. Matured Langerhans cells or
dendritic cells (DCs) have an increased expression of MHC Class II,
ICAM-1 and B7 molecules, allowing for optimal T-cell triggering
(Steinman et al., 1995); in addition the intricate structure of the
paracortical area offers an appropriate environment for this
sensitization process to take place. Naive T-cells, again in contrast
to memory cells, recirculate preferentially through the peripheral
lymphoid organs, rather than through the tissues, due to the
expression of distinct adhesion molecules (L-selectin) that recognize
the high endothelial venules in the lymph nodes. The probability of
hitting unprimed specific T-cells is thus increased.
When successful triggering and subsequent proliferation of
allergen-specific T-cells have taken place, the lymphocyte progeny
will leave the lymph nodes to join the recirculating pool of
lymphocytes. The frequency of specific cells in the circulation can
thus be increased from around 1:100 000 to 1:1000-10 000 and the
individual has now become "sensitized".
b) Elicitation of allergic contact dermatitis
Upon re-exposure to contact sensitizing agents, specific
recirculating memory T-cells present in the skin immediately recognize
the allergen modified MHC Class II molecules on the Langerhans cell
membranes. The probability that the allergen is indeed found by
specific memory T-cells is largely increased by the expression of
organ-specific interaction molecules on the T-cell surface. The
cutaneous lymphocyte-associated antigen (CLA), recognized by the
monoclonal antibody HECA-452, is present on a small subpopulation
(approximately 16%) of peripheral blood T-cells which preferentially
recirculates via the skin. Here the endothelial adhesion molecule
E-selectin acts as a vascular addressin for the skin-homing memory
T-cells (Picker et al., 1993; Bos & Kapsenberg, 1993). As described in
the section on Type IV - delayed-type hypersensitivity (section
2.1.4), mainly CD4-positive allergen-specific cells thus enter the
skin. Since memory cells have relatively low stimulation thresholds,
they can be triggered by less efficient antigen-presenting cells, like
the local resident Langerhans cells. The T-cells will now initiate a
Th1-type cytokine cascade, which eventually leads after 24-72 h to the
typical delayed-type contact allergic reaction. Because the reaction
takes place in superficial layers of the skin, erythema and blistering
are characteristic features, in contrast to the tuberculin DTH where
induration is most pronounced.
The challenge reaction in allergic contact dermatitis resolves
spontaneously within one week. It is therefore commonly used as a
primary diagnostic test in allergic contact dermatitis. To this end,
low non-toxic dosages of allergen are generally applied onto the skin
under an occlusive patch to allow for maximal skin penetration. The
main drawbacks of such an in vivo skin test procedure are the
potential sensitization and boosting by such an intense allergen
contact. Indeed it was shown experimentally in guinea-pigs that even
one epicutaneous application of allergen could direct the immune
response towards (still subclinical) sensitization, as shown by a
failure of subsequent tolerance induction (Van Hoogstraten et al.,
1994). Also clinically, occasional sensitization by epicutaneous skin
tests can be observed. For this reason much effort has been put in the
development of in vitro diagnostic procedures in allergic contact
dermatitis (Von Blomberg et al., 1990).
Up to now, in vitro assays in allergic contact dermatitis have
been successful for relatively non-toxic water-soluble allergens, such
as metal salts. For other allergens, occasionally positive results are
obtained by pre-pulsing antigen-presenting cells or proteins or by
using special solvents. So, despite the fact that most of our
knowledge of the pathogenesis of human allergic contact dermatitis is
due to in vitro experiments with blood from allergic patients, for
routine assessment of allergic contact dermatitis these assays are
still too complicated.
Repeated contact with low dosages of allergen, as typically
occurs for most contact allergens, may lead to continuous triggering
of Type IV reactivity in the skin and thus to an allergic contact
dermatitis (Scheper & Von Blomberg, 1992). The dermatitis only
disappears when the allergen is entirely eliminated from the
environment.
Even if the reaction is clinically healed, allergen-specific
T-cells may persist in the skin for up to several months. Thus,
locally increased allergen-specific hyperreactivity, either detectable
through accelerated "retest" reactivity (peaking at 6-8 h) or flare-up
reactivity after allergen entry from the circulation, may be observed
for several months at former allergic contact dermatitis reaction
sites (Scheper et al., 1983; Yamashita et al., 1989). The presence of
specific T-cells at former eczematous sites can thus be maintained by
low dosages of inhaled or ingested allergen, in the absence of
allergenic skin contacts.
Repeated contact with relatively high dosages of allergen, on the
other hand, may result in a local desensitization. The initial
erythematous reaction gradually decreases. Such a local
hypo-responsiveness of the skin, which is known as "hardening" in
occupational contact dermatitis, is largely reversed after a period of
allergen restrain. However, also systemically, DTH reactivity
decreases upon repeated allergen application. This decrease in DTH is
associated with increased antibody responses and a shift towards
immediate-type hypersensitivity, reflecting a shift from Th1 to Th2
reactivity (Boerrigter & Scheper, 1987; Kitagaki et al., 1995).
It appears, therefore, that although exposure of the skin to
exogenous antigens generally results in Th1 responses, the
micro-environment in chronically inflamed tissues, rather than the
site of allergen exposure or the nature of the allergen, determines
the type of immune reaction.
2.1.4.2 T-cell responses in chemically induced pulmonary diseases
Asthma is a chronic pulmonary inflammatory disease associated
with bronchial hyperreactivity. In the majority of asthma cases a
clear association exists with atopic IgE-mediated hypersensitivity,
involving relatively large protein allergens. Here T-cells dominate in
the late phase and the chronic reaction. The pivotal role of T-cells
in chronic asthma is stressed by the finding of activated T-cells in
the bronchial mucosa and the effectiveness of T-cell immunosuppressive
drugs. In particular, the number of activated CD4-positive cells was
found to correlate with the numbers of eosinophils in the
bronchoalveolar lavage (BAL) and with disease severity (Walker et al.,
1991). The T-cells, present in the bronchial mucosae and in the lavage
fluid, were shown to produce predominantly Th2 cytokines, in
particular IL-5, a cytokine known to activate eosinophils (Corrigan &
Kay, 1992). Therefore, although T-cell-mediated immunity is clearly
playing a role, Type IV reactivity, mediated by Th1 cells, does not
seem to be involved in this type of asthma.
Of particular interest is the hypersensitivity pneumonitis
induced by environmental small chemical allergens. Such allergens are
known to cause DTH when applied to the skin. Occasionally these
allergens induce, in addition or as first manifestation, asthmatic
disease upon inhalation. It could be questioned whether these
allergens, in contrast to the atopic protein allergens, would induce
Type IV reactivity.
Experimentally it has been shown in mice that Type IV
hypersensitivity to small chemical allergens, such as picryl chloride,
can indeed induce lung disease upon intranasal application (Garssen et
al., 1991, 1994).
Contact allergens that have been reported to induce asthma
include formaldehyde, platinum salts, nickel, cobalt and chromium
(Nordman et al., 1985; Estlander et al., 1993; Cirla, 1994;
Park et al., 1994; Merget et al., 1996). In a number of cases this
asthmatic disease could be associated with the presence of circulatory
IgG to the causative allergen and positive bronchial provocation
tests.
Trimellitic anhydride, phthalic anhydride and toluene
diisocyanate are reactive chemicals behaving primarily as respiratory
allergens, causing asthmatic disease and pulmonary irritation. The
immune reactions leading to asthmatic disease are quite variable; the
role of clear-cut Type IV reactivity is uncertain.
2.1.5 Type V stimulatory hypersensitivity
Stimulatory hypersensitivity occurs when antibodies binding to a
cell surface molecule cause inappropriate stimulation of the cell.
Normal feedback inhibition will then fail. An example is Graves'
disease (exophthalmic goitre), in which autoantibodies to the
thyroid-stimulating hormone receptor on thyroid cells stimulate the
production of excessive amounts of thyroid hormone, resulting in
disease.
2.2 Regulation of hypersensitivity
In 1986, the existence of two CD4+ Th-cell subsets was
discovered in mice, and they were designated Th1 and Th2. Their
identification has greatly improved understanding of the regulation of
immune effector functions, not least on Type I and Type IV
hypersensitivity responses. These Th subsets are defined by the
patterns of cytokines that they produce, which leads to strikingly
different T-cell functions (Table 9). Broadly speaking, Th2-cells are
more efficient B-cell helpers, especially in the production of IgE
antibody, whereas Th1-cells mediate DTH reactions. In addition, they
cross-regulate by producing mutually antagonistic cytokines. Their
specific function and characteristics in rodents and humans have not
yet been clearly established (Muraille & Leo, 1998).
Table 9. Characteristics of Th1- and Th2-associated immunitya in vivo
(modified from Röcken et al., 1996)
Characteristics Th1 Th2
IFN-gamma high variable, frequently low
IL-2 high variable, frequently low
IL-4 low/negative high
major mode of action DTH reactions eosinophil-associated
(cellular immunity) cytotoxicity
complement-binding non-complement-binding
antibodies and IgE antibodies and IgE
protective effects against intracellular against
microorganisms and extracellular parasites
tumours
harmful effects contact hypersensitivity atopic diseases
tissue-specific autoimmunity immunoglobulin-mediated
allergic encephalitis autoimmunity
juvenile diabetes bullous autoimmune
rheumatoid arthritis diseases
thyroiditis sclerosing diseases ?
uveitis
a Th1 and Th2 immunity characterizes T-cell populations, not single T-cells
In addition to Th1- and Th2-cells, additional cytokine production
phenotypes of CD4+ cells exist. They are, however, characterized
less thoroughly. Most resting T-cells mainly produce IL-2 on first
contact with antigen, and differentiate within a few days into cells
producing multiple cytokines, such as IL-4 and IFN-gamma. In addition
to Th1- and Th2-cells, the existence of undefined precursor cells has
been suggested. These precursor cells (IL-2 producing) are the virgin
Th cells, producing only or predominantly IL-2, and Th0 cells are in
the process of differentiation, producing cytokines of both Th1 type
(such as IL-2 and IFN-gamma) and Th2 type (such as IL-4, IL-5 and
IL-10). The pathways of differentiation from the precursor cells are,
however, unclear. In addition, it is unknown whether there is a single
common precursor cell or whether precursor cells are already committed
to a particular cytokine pattern before exposure to antigen (the
cytokine production profiles of Th-cell subsets in the mouse are shown
in Table 1). In conclusion, it is believed that Th1- and Th2-cells
represent the most differentiated populations of the CD4+ phenotype
that develop following prolonged exposure to antigen or following
stimulation by potent immunogens.
At least two mechanisms can influence the selective
differentiation of Th-cell subsets. Firstly, the cytokines that are
present during differentiation, in particular IFN-gamma, IL-4 and
IL-12, may greatly influence the type of Th that will be generated.
IF-gamma augments development of Th-type responses and IL-4 promotes
differentiation of Th-cells (Romagnani, 1992a). Secondly, the type of
APC is thought to influence the characteristics of immune responses.
Upon activation, Th2 cells express p39 on their surface, which
interacts with CD40 on the surface of B-cells. The interactions of p39
with CD40 and of T-cell antigen receptor (TCR) with antigen and MHC
Class II together lead to production of IL-4, IL-5 and IL-6 by Th2
cells, stimulating B-cells to antibody production. Th1 cells, on the
other hand, may interact with macrophages. A pair of cell surface
molecules analogous to p39/CD40 have not as yet been identified.
However, the interaction of Th1 cells with macrophages leads to
IFN-gamma production by Th1 cells, stimulating macrophages to produce
monokines.
The difference in APCs, macrophage versus B-cell, that
preferentially activates Th1 or Th2 suggests differences in antigen
requirements for activation, e.g., large particulate antigens
requiring phagocytosis for Th1 and low antigen concentration for Th2.
Whereas moderate concentrations of antigen preferentially activate
Th1, extremely high concentrations are believed to inhibit Th1 and
select for Th2 responses (Pfeiffer et al., 1991).
If Th1 and Th2 clones are stimulated by immobilized anti-CD3 (in
the absence of APC), both types produce their respective cytokine
pattern. The proliferative responses are, however, very different.
Whereas Th2 clones exhibit good proliferative responses, Th1 not only
fail to do so, but are even rendered incapable of proliferating in
response to exogenously added IL-2 (Williams & Unanue, 1990; Williams
et al., 1990). These Th1 clones are in a state of anergy or tolerance
(Schwartz & Weiss, 1990).
IFN-gamma inhibits the proliferation of Th2 responding to either
IL-2 or IL-4, but does not inhibit Th1. IL-10 inhibits the synthesis
of cytokines by Th1 cells, and, although growth factor requirement is
not affected, the reduction in IL-2 synthesis can lead to decreased
proliferation. It has been shown in in vitro human systems that
IL-10 can suppress the antigen-presenting capacity of monocytes and
dendritic cells by down-regulation of MHC Class II. IL-10 had no
effect on the antigen-presenting capacity of B-cells or
down-regulation of their MHC Class II. These results suggest a
mechanism for the general observation that macrophages/dendritic cells
preferentially stimulate Th1, whereas B-cells preferentially stimulate
Th2.
IL-2 is a T-cell growth factor (TCGF) that mediates autocrine
proliferation of Th1, whereas the TCGF IL-4 mediates autocrine
proliferation of Th2. Interestingly, it has been shown that IL-4 is
the major TCGF produced by T-cells from lymphoid organs that drain
mucosal tissues, whereas IL-2 is the major TCGF produced by T-cells
from other lymphoid organs (Daynes et al., 1990b). Involvement of
dehydroepiandosterone in this site/tissue-specific control on
lymphokine production was suggested (Daynes et al., 1990a).
Dihydrotestosterone and 1,2,5-dihydroxyvitamin D3 also change the
cytokine production pattern of T-cells.
In humans, a predominant fraction of CD4+ T-cell clones was
found to produce IL-2, IL-4 and IFN-gamma, although the quantities
varied considerably. Bearing in mind the findings in mice (see above),
it was thought that unrestricted profiles are mainly a property of
T-cells that are not yet committed to a certain differentiation
pathway. Consequently, functional heterogeneity of CD4+ cells
should most likely be found in chronically stimulated responders.
Kapsenberg et al. (1991) studied two categories of patients, those
with nickel hypersensitivity, an example of Type IV hypersensitivity,
and those with house dust mite ( Dermatophagoides pteronysinnus
(Dp))hypersensitivity, an example of Type I hypersensitivity.
Most house dust mite-specific T-cell clones from peripheral blood
(Wierenga et al., 1990) and lesional skin biopsies of house dust
mite-allergic patients show a Th2-like production profile. House dust
mite-specific clones from atopic patients induce IgE production (see
also below). It was shown that this production is dependent on a high
IL-4/IFN-gamma ratio, and is not dependent on the origin of B-cells.
Only IgE specific to house dust mite (and not, for instance, IgE
specific to tetanus toxoid or Candida albicans) was elevated in
atopic house dust mite-allergic patients.
The majority of allergen-specific human T-cell clones produce
IL-4 and IL-5, but not IFN-gamma. Virtually all T-cell clones specific
for bacterial components, which were derived from the same patients,
was found to produce large amounts of IL-2 and IFN-gamma, and few
produced IL-4 and/or IL-5 (Wierenga et al., 1990; Parronchi et al.,
1991). In a subsequent study, antigen-specific T-cell clones were
derived for the bacterial antigen purified protein derivate (PPD) from
Mycobacterium tuberculosis and for the helminth antigen Toxicara
canis excretory-secretory (TES). Most PPD-specific clones produced
IL-2 and IFN-gamma, but not IL-4 and IL-5, whereas most TES-specific
clones produced IL-4 and IL-5, but not IL-2 and IFN-gamma. This study
shows that in the course of natural immunization certain infectious agents
preferentially expand T-cell subsets. PPD expands Th1, parallelling
the (Th1-mediated) tuberculin DTH, whereas TES expands Th2,
parallelling the (Th2-mediated) parasite infection.
In a large series of human T-cell clones, all Th1 clones were
found to lyse EBV-transformed autologous B-cells pulsed with the
specific antigen, and the decrease of Ig production correlated with
the lytic activity of Th1 clones against autologous antigen-presenting
B-cell targets (Romagnani, 1991). This suggests an important mechanism
for down-regulation of antibody responses in vivo.
Almost all nickel-specific T-cell clones produce TNF-alpha,
GM-CSF, IL-2 and high levels of IFN-gamma, but low or undetectable
levels of IL-4 and IL-5, thus resembling Th1 cells. Nickel induces DTH
in the skin of allergic patients. Since IFN-gamma is an important
mediator for DTH, IFN-gamma may be essential to DTH. However, no clear
difference in cytokine production profile between allergic patients
and control individuals was found.
2.2.1 Regulation of IgE synthesis by IL-4 and IFN-gamma
Atopy is associated with enhanced serum titres of
allergen-specific IgE. The production of IgE is heightened and
sustained by B-cells in atopic patients. IL-2 secreted by Th cells is
necessary for the production of all isotypes of immunoglobulins
(Kapsenberg et al., 1991). Activated B-cells are induced by IL-4 to
undergo immunoglobulin heavy-chain rearrangements to the
epsilon-constant region, resulting in synthesis of IgE (Coffman et
al., 1986). So far, IL-4 can mediate this isotype switch, which is
blocked very efficiently by IFN-gamma (Romagnani, 1991). IFN-gamma
induces switching to gamma-2a (Coffman et al., 1986). IL-4 and
IFN-gamma are produced by Th2 and Th1 cells, respectively; a response
that involves mainly Th2 cells should produce a large amount of IgE,
whereas responses involving mainly Th1 cells, such as DTH reactions,
should be non-permissive for IgE production. In vivo experiments
have confirmed these predictions. IL-4-deficient mice lack IgE and
IgG1 responses (Kuhn et al., 1991), whereas transgenic mice
constitutively producing IL-4 show elevated serum IgE levels.
Injection of mice with anti-IgD antibodies results in a strong
stimulation of both B- and T-cell populations, leading to polyclonal
antibody production and very high IgE levels. Anti-IL-4 antibodies
dramatically reduce IgE levels after anti-IgD immunization, whereas
anti-IFN-gamma antibodies elevate IgE levels even further. Similarly,
administration of IFN-gamma results in considerable inhibition of the
IgE response. Because the anti-IgD immunization leads to a response
that involves high levels of Th2 cytokines, all of these results are
consistent with the effects of IL-4 and IFN-gamma on IgE synthesis as
defined by in vitro model systems. Similar correlations between
Th2-like responses and high IgE production are seen during several
parasite infections.
2.2.2 Eosinophilia and IL-5
Many parasitic infections induce high levels of circulating
eosinophils. Because IL-5 has been implicated as a major growth and
differentiation factor for eosinophils the association of IgE and
eosinophilia may be explained by the association of IL-4 and IL-5 as
products of Th2-cells (Gulbenkian et al., 1992). Supporting evidence
has been provided by experiments in vivo, in which administration of
anti-IL-5 during a strong anti-parasitic immune response completely
abrogated eosinophilia (Coffman et al., 1989), and from studies of
transgenic mice that express high levels of IL-5. The major
abnormality in these animals is the presence of extremely high levels
of eosinophils in the blood and various lymphoid organs. Patients with
filaria-induced eosinophilia exhibit a significantly greater frequency
of IL-5-producing T-cells than uninfected individuals.
2.2.3 The relationship between Th2 cells and type I hypersensitivity
In mice, in addition to enhancing IgE production via IL-4, Th2
cells also influence other features of allergic reactions. Firstly,
IL-3, IL-4 and IL-10 are mast cell growth factors that act in synergy,
at least in vitro, and secondly, IL-5 induces the proliferation and
differentiation of eosinophils in vitro and in vivo (Coffman,
1989; Sanderson, 1990). In addition, IL-3 and IL-4 have been shown to
enhance the secretory function of murine mast cells. So, Th2-cell
activation not only increases the level of IgE synthesized, but also
potentially increases the number of IgE-binding cells that will
degranulate in response to allergen challenge.
Mast cells and basophils produce IL-4. It has been hypothesized
that IL-4 produced by these cells induces the development of Th2
cells, and that these cells in turn produce IL-4. In addition, mast
cells are an important source of IL-5.
2.2.4 IL-12 drives the immune response towards Th1
The pivotal role of the cytokine IL-12 in the differentiation of
Th-cells towards Th1 is evident from both in vitro and in vivo
studies (Scott, 1993). IL-12 is produced by T-cells, B-cells,
macrophages and dendritic cells and stimulates the production of
IFN-gamma from T-cells and NK-cells. IL-12 enhances Th1-cell expansion
in cell lines from atopic patients (Manetti et al., 1993). The
presence of IL-12 during primary stimulation of naive CD4+ cells skews
the response in the direction of Th1 differentiation. These data
suggest that IL-12 may be the IL-4 equivalent for the differentiation
of Th1-cells. IL-10 has been shown to inhibit lymphocyte IFN-gamma
production by suppressing IL-12 synthesis in accessory cells. A
variety of pathogens that are associated with Th1 development have
been shown to induce IL-12 production (Scott, 1993).
2.2.5 IL-13, an interleukin-4-like cytokine
Information on cytokine IL-13 is based on limited information
about its activities in vitro. As it shares biological activities
with IL-4, these activities will, however, be briefly discussed. IL-13
is produced by activated T-cells. The activities in vitro of IL-13
are similar to those of IL-4, with two major exceptions. Firstly,
IL-13 does not act on T-cells and secondly, IL-13 does not act on
murine B-cells (Zurawski & de Vries, 1994). Similarly to IL-4 and
IL-10, IL-13 inhibits the production by LPS-stimulated monocytes of
proinflammatory cytokines, chemokines and haematopoietic growth
factors. In contrast to IL-10, however, IL-13 up-regulates the
antigen-presenting capacity of monocytes. Similarly to IL-4, IL-13
inhibits transcription of IFN-gamma and both alpha- and beta-chains of
IL-12. Thus, IL-13 may (like IL-4) suppress the development of
Th1- cells through down-regulation of IFN-gamma and IL-12 production
by monocytes, favouring the generation of Th2 cells. Also in the
mouse, IL-13 inhibits production of proinflammatory cytokines and
expression of IL-12 alpha- and beta-chain mRNA. Murine IL-13 does not
affect macrophage antigen-presenting capacity. Similarly to IL-4,
IL-13 acts on human B-cells in inducing class switch to production of
IgG4 and IgE and inducing CD23 surface expression (Punnonen et al.,
1993; Punnonen & de Vries, 1994). Following activation of T-cells,
IL-13 is produced earlier and for much longer periods than IL-4 (Yssel
et al., 1994). Thus, IL-13 may play an important role in the
regulation of enhanced IgE synthesis in allergic patients. In contrast
to IL-4, murine and human IL-13 do not induce IgE synthesis in murine
B-cells. Importantly, this may restrict the use of mice as an animal
model for allergy.
In summary, IL-13 may favour development of Th2-cells, consistent
with the induction of IgG4 and IgE synthesis. Determination of the
actual role of IL-13 requires more information on the biological
effects in vivo.
2.3 Autoimmune reactions
A wide spectrum of human and animal diseases appears to be wholly
or partially attributable to autoimmune reactions. Despite the
extensive growth of information relating to the mechanisms of
self-tolerance (see section 1.5), the understanding of the mechanisms
leading to pathogenic autoimmunity is still fragmentary and incomplete
(Theofilopoulos, 1995a).
Important issues that need to be resolved in this context
concern: (i) the nature of the inciting antigens (self, neo-self,
foreign); (ii) the definition of the criteria by which a disease can
be termed autoimmune; (iii) the principles that govern the spectrum
and extent of an autoimmune response; (iv) the mechanisms by which
spontaneous remissions and exacerbations of autoimmune diseases occur;
(v) the nature of environmental factors that initiate/precipitate
autoimmune reactions; (vi) the structural and other characteristics
that differentiate pathogenic from non-pathogenic autoantibodies and
T-cells; and (vii) the identity of the genes that predispose or
accelerate autoimmunity, as well as their mechanism of action
(Theofilopoulos, 1995a).
The most urgent of these questions concerns the nature of the
inciting antigen. Although autoimmune disorders are often defined and
diagnosed by the presence of autoantibodies (Osterland, 1994), it
should be noted that (a) autoantibodies may indeed be the actual
pathogenic agents of disease (e.g., autoimmune haemolytic anaemia,
pemphigus, and myasthenia gravis; see sections 2.6.3, 2.6.5 and
2.6.6), (b) they may arise as a consequence of another disease process
(e.g., organ-specific autoantibodies due to tissue damage to those
organs), or (c) they may merely mark, like footprints, the presence of
the etiological agent while not themselves causing disease (Naparstek
& Plotz, 1993; Theofilopoulos, 1995a). The latter possibility is
complicated by the fact that determinants recognized by the
autoantibody and the prerequisite Th-cell may reside on different
molecules within a supramolecular complex (Theofilopoulos, 1995a). For
example, for many years, it was believed that native nDNA itself was
the immunogen for anti-nDNA antibodies, but efforts to induce such
autoantibodies by immunization with nDNA have generally been
unsuccessful. It has been suggested that for anti-nDNA antibody
induction, the scenario may involve intermolecular help, via the
binding of nucleosomes or other protein-DNA complexes to anti-DNA
idiotype-displaying B-cells, followed by processing of the protein and
presentation to the corresponding Th-cells (Theofilopoulos, 1995a). In
this connection it is of interest that in systemic autoimmune diseases
autoantibodies frequently appear to be directed against the entire set
of polypeptides associated with discrete supramolecular cellular
entities, such as the nucleosome particle or the nucleocytoplasmic
ribonucleoprotein particles (see Table 10).
It has become clear that T-cells are primary players in the
initiation and perpetuation of spontaneous (Theofilopoulos & Dixon,
1985; Singer & Theofilopoulos, 1990) as well as induced systemic
autoimmune disorders (Druet, 1989; Goldman et al., 1991). Many immune
responses seem to be functionally dominated either by Th1 or Th2
cytokines. Therefore, the Th1-Th2 balance during immune reactions in
vivo significantly determines the outcome of immunopathological
processes (Röcken et al., 1996). Whereas organ-specific autoimmune
disease are predominantly mediated by IFN-gamma-producing Th1-cells,
IL-4-producing Th2-cells are involved in immunoglobulin-mediated
autoimmune diseases such as systemic lupus erythematosus (SLE)
(Goldman et al., 1991; Röcken et al., 1996) (Table 9).
Table 10. Examples of autoantigens in organ-specific and systemic autoimmune diseasesa
Organ/cell/nucleus Target antigens Diagnosis
Organ-specific autoimmune diseases
Pancreatic islet cells glutamic acid decarboxylase 65 insulin-dependent diabetes mellitus
glutamic acid decarboxylase 67
tyrosine phosphatase IA-2
tyrosine phosphatase IA-2b
Adrenal cortex 21-hydroxylase Addison's disease
Leydig cells, testes, granulosa cytochrome side-chain cleavage hypogonadism
theca enzyme
Ovary 17a-hydroxylase hypogonadism
Gastric parietal cell H+/K+-ATPase pernicious anaemia
intrinsic factor
Thyroid epithelium thyroid peroxidase autoimmune thyroid diseases
thyroglobulin
thyroid-stimulating hormone (TSH)
TSH-receptor
triiodothyronine
thyroxine
Hepatocyte CYP 2D6 (LKM-1) chronic active hepatitis
halothane-induced hepatitis
Melanocyte tyrosinase vitiligo
Parathyroid calcium-sensing receptor autoimmune parathyroidism
Table 10. (continued)
Organ/cell/nucleus Target antigens Diagnosis
Systemic autoimmune diseases
Native DNA DNA backbone systemic lupus erythematosus (SLE)-renal
ss-DNA nucleotides SLE and other connective tissue diseases
Nucleoprotein DNA histone SLE - central nervous system,
histone 1 H1, 2A, 2B, 3, 4 renal - drug-induced SLE
histone 2 H3 connective tissue disease
Sm SnRNP SLE
Nuclear RNP non Sm SnRNP mixed connective tissues disease, SLE
Ribosomal RNP phosphoproteins SLE
Scl-70 topoisomerase 1 scleroderma
Centromere kinetochore CRESTb, Raynaud's syndrome
SS-A (Ro) RNP SLE-cutaneous, photosensitivity
SS-B (La) RNA-pol protein Sicca syndrome, SLE, neonatal lupus
Cardiolipin phospholipid SLE - thrombosis, cytopenia
PM-1 protein complex myositis, scleroderma
Jo-1 histidyl tRNA synthesis myositis
Mi-2 dermatomyositis
Table 10. (continued)
Organ/cell/nucleus Target antigens Diagnosis
PCNA cyclin SLE
Ku protein on terminal chromosome SLE
nucleolar RNA-pol 1, RNA
fibrillaren scleroderma, drug-induced connective
tissue disease
Nuclear membrane laminins scleroderma, SLE
a modified from Osterland (1994) and Song et al. (1996a); responses encompass both Th1 and Th2 responses
and involve both Th1 (IFN-alpha) and Th2 (IL-4)
b CREST (calcinosis, Raynaud's phenomenon, oesophageal involvement, sclerodactyly and telangiectasia)
The major non-mutually exclusive etiological concepts of
autoimmune disorders have been reviewed (Theofilopoulos, 1995a,b) and
are summarized in Table 11.
Table 11. Possible mechanisms of autoimmune reactions
(modified from Theofilopoulos, 1995a,b)
Release of anatomically sequestered antigens
The "cryptic self" hypothesis
The self-ignorance hypothesis
The molecular mimicry hypothesis
The "modified self" hypothesis
Immunoregulatory disturbances
Errors in central or peripheral tolerance
Polyclonal activators
2.4 Possible mechanisms of autoimmune reactions
2.4.1 Release of anatomically sequestered antigens
In general, antigens associated with peripheral tissues,
especially those sequestered behind anatomic barriers, may not come
into contact with the developing T-cell repertoire, and, therefore,
tolerance may be unnecessary for such antigens. Induction of
organ-specific autoimmune disease following contact with antigens of
such so-called "immunologically privileged" sites has been well
documented, as exemplified by the development of ophthalmia following
eye injury and orchitis following vasectomy.
Data have also clearly established that antigens associated with
peripheral tissues can cause tolerance, and therefore loss of
susceptibility to tissue-specific autoimmune diseases, when
experimentally introduced into the thymus. Intrathymic injection of
pancreatic islet cells can prevent autoimmune diabetes in the
BioBreeding (BB) rat (Posselt et al., 1992) and the non-obese diabetic
(NOD) mouse (Gerling et al., 1992). Tissue trauma alone may not be
sufficient to elicit a conventional self-directed immunological
response. Tissue-trophic pathogens, such as viruses, may be important
in inducing the initial damage that results not only in availability
of previously sequestered antigens but also in the production of
co-stimulatory factors necessary for the immune response.
2.4.2 The "cryptic self" hypothesis
A corollary hypothesis for the mechanism of induction of
pathogenic autoimmune responses addresses molecular, rather than
anatomic, sequestration and relates to the presence of cryptic
self-determinants. Each self-protein presents only a small minority of
dominant determinants, which are involved in negative selection during
thymic maturation and development of tolerance of the organism to
them. Because of many constraints to peptide presentation, only a few
peptide stretches of a given protein antigen are presented to the
T-cell repertoire, namely those that have the highest affinity to the
MHC-binding site and are present at a sufficient concentration. These
peptides are the so-called dominant antigenic determinants. It is
important to realize that, because antigen-presenting cells cannot
distinguish "self" and "non-self" proteins, foreign and "self"
peptides are presented indiscriminately (Bloksma et al., 1995). The
subsequent immune responses, however, are diametrically opposed to
each other. Whereas foreign peptide sequences, in general, induce
"stimulatory" T-cell responses, the dominantly presented "self"
sequences induce "inhibitory" T-cell responses through
peptide-specific thymic cell deletion during development of the T-cell
repertoire and/or induction of specific tolerance or anergy in the
established peripheral T-cell repertoire. The poorly displayed
majority of subdominant/cryptic determinants, constituting the
"cryptic self", do not induce tolerance and, therefore, a large cohort
of potentially "self"-reactive T-cells exists. The presentation of
cryptic "self" peptides, however, can be up-regulated under certain
conditions (Lehmann et al., 1993). Evidence for the role of cryptic
determinants in the pathogenesis of autoimmunity has been provided in
the non-obese diabetic mouse (NOD) model (Kaufman et al., 1993; Tisch
et al., 1993), but the exact mechanisms of these immune responses are
not fully known. One suggestion is that pathogens such as viruses may
provide the initial stimulus through increased presentation of the
subdominant determinant, either by molecular mimicry (see below)
and/or by interferon-induced up-regulation of gene-expression,
including genes for antigen-presenting MHC molecules (Theofilopoulos,
1995a).
Processing of chemically altered "self" proteins may result in
the presentation of cryptic, thus potentially T-cell-activating,
self-peptides by creation of new binding sites with high affinity to
MHC molecules or modification/preventing of the physiological
intracellular protein degradation (Bloksma et al., 1995). Expression
of "cryptic self" peptides of nucleolar proteins appears to be a
decisive step in the pathogenesis of HgCl2-induced formation of
anti-nucleolar autoantibodies in mice (Kubicka-Muranyi et al., 1996).
2.4.3 The self-ignorance hypothesis
Evidence suggests that mature resting T-cells specific for
extrathymic antigens presented by non-professional antigen-presenting
cells (other than dendritic cells and macrophages) are induced to
undergo anergy because of the absence of appropriate "second signals"
or "co-stimulatory" factors (Theofilopoulos, 1995a). An alternative
possibility is that there is no induction of anergy, but that the
mature T-cells are unable to receive appropriate signals and/or help.
This would result in T-cells simply ignoring such antigens and
remaining quiescent. It follows that, if adequate antigen presentation
and co-stimulation occurs through professional antigen-presenting
cells, then these self-reactive but quiescent cells may be activated
and cause tissue damage (Theofilopoulos, 1995a).
2.4.4 The molecular mimicry hypothesis
Molecular mimicry is defined by homology in a linear amino acid
sequence between "self" molecules and foreign molecules. The above
theories of cryptic or ignored "self" are compatible with the
molecular mimicry hypothesis of autoimmunity, particularly as it
pertains to infectious agents. Closely related or identical peptides
are often found in unrelated proteins. Thus, many peptide fragments of
infectious agents are homologous with host proteins. Among microbial
antigens implicated in autoimmunity induced by molecular mimicry, heat
shock proteins (hsp), found in virtually all life forms, have received
prime attention (Minowada & Welch, 1995). Comparisons of the amino
acid sequence of hsp60 with the entire database of known human
sequences revealed that 86 human peptides have similar regions to
hsp60 and, of these, 19 are known disease-associated autoantigens
(Jones et al., 1993). However, the importance of mimicry to the
pathogenesis of spontaneous autoimmune disease is uncertain, as it is
unclear why immunological responses to hsp, which are expressed in
every cell, could lead to organ-specific autoimmune diseases.
2.4.5 The "modified self" hypothesis
This theory suggests that autoimmunity may arise as a result of
an immune response against modified "self" determinants ("neo-self"
determinants), which may be particularly relevant for chemical-induced
autoimmune responses. Drugs, their metabolites or other haptenic
chemicals may bind to "self" determinants. A number of possibilities
should be considered.
2.4.5.1 Hapten-induced antibody responses to "modified self"
In such reactions the hapten conjugates to "self" and forms an
integral component of the determinant that is recognized by the
antibody. In this mechanism hapten-specific T-cells provide cognate
help to the B-cell that is then induced to synthesize antibodies which
recognize the hapten-modified but not the native form of the "self"
protein. Therefore, these reactions against a particular hapten are
not truly autoimmune in nature. Penicillin, quinidine, halothane (Gut
et al., 1995), and tienilic acid are good examples of compounds that
can induce antibody responses to hapten-modified "self".
2.4.5.2 Hapten-induced autoantibodies that recognize "self" proteins
In their native form these can be considered true autoimmune
responses, since the determinant that is recognized by antibody does
not incorporate a drug-derived determinant. However, the determinant
that is recognized by the Th-cells that promote the B-cell response
may be drug-derived. The following theories have been put forward:
a) Drugs might break tolerance by binding to "self" macromolecules,
thereby creating new determinants that could be recognized
by T-cells. T-cells recognizing this new determinant would
clonally expand and go on to provide help for B cells that
recognize adjacent autoantigens on the same drug "self"
conjugate. These in turn would clonally expand and differentiate
into autoantibody-producing plasma cells (Fig. 7). In this way
the normal process of suppression that operates through either
clonal or functional deletion of Th-cells is effectively bypassed
(Allison, 1989). A considerable body of experimental evidence,
largely from work with mice, supports this concept.
Administration of arsenilic acid-conjugated autologous
thyroglobulin or dinitrophenylated autologous immunoglobulin to
mice has been found to lead to breakdown of tolerance and
elicitation of autoantibodies to these self-proteins (Weigle,
1965; Iverson, 1970). Furthermore, mice sensitized to
p-aminobenzoic acid (PAB) and then administered PAB-conjugated
isologous red blood cells developed a T-cell-dependent antibody
response to their own red blood cells, with consequent haemolytic
anaemia (Yamashita et al., 1976).
b) It has been postulated that a drug or metabolite might
interact chemically with "self"-MHC molecules on
antigen-presenting cells (macrophages or B-cells) in such a way
that they appear as "non-self" to T-cells. These T-cells,
following clonal expansion, would then provide help
indiscriminately to all B-cells carrying the drug-modified
"self"-MHC molecule. Assuming that the drug modifies MHC
molecules without regard for the antigen specificity of the
B-cell, the resulting cognate T-/B-cell interaction would lead to
polyclonal B-cell activation and induction of synthesis of
antibodies of multiple, including "anti-self", specificities
(Gleichmann et al., 1984, 1989). This mechanism would be
analogous to a graft-versus-host (GVH) reaction. Indeed,
experiments with mercuric chloride (Pelletier et al., 1994),
D-penicillamine (Tournade et al., 1990) and gold salts (Schuhmann
et al., 1990) in Brown Norway rats or particular strains of mice
led to immune disregulatory changes (elevated immunoglobulin
levels, particularly IgE, induction of autoantibodies to the
glomerular basement membrane, DNA, IgG, collagen and nuclear and
nucleolar proteins) resembling those seen in graft-versus-host
(GVH) disease (Gleichmann et al., 1984, 1989; Goldman et al.,
1991; Bloksma et al., 1995). The elevations in IgE, IgG1 and IL-4
in mercury chloride-treated susceptible mice and rats implicate
the Th2 subset in this response (Goldman et al., 1991).
IgE (Fc-epsilon-RI). Upon contact with the allergen, the IgE molecules
will be cross-linked and the cells will release their granules
supplying the tissue with histamine, proteolytic enzymes, heparin and
chemotactic factors for eosinophils, neutrophils and monocytes. These
mediators induce vasodilatation, increased vascular permeability and
smooth muscle contraction and lead to an "immediate reaction", which
becomes clinically manifest within 20 min as a typical "wheal and
flare" in the skin or as bronchoconstriction in the respiratory tract.
At the same time, from the cell membrane new mediators, such as
prostaglandin-D2, thromboxanes and leukotrienes are being generated.
Together with the now attracted and activated eosinophils (which
produce platelet activating factor and major basic protein), these
mediators cause further infiltration, smooth muscle contraction,
mucosal oedema and damage of the epithelial cells, resulting in the so
called "late phase" reaction (12-24 h after challenge). Like the
immediate reaction the late phase responses can be observed both in
the skin and in the respiratory tract.
While actual antibody synthesis is regulated by the action of
cytokines, the tendency to respond to specific allergens appears to be
linked to inheritance of certain MHC genes. Various HLA class II
antigens seem to be associated with a high response to individual
allergens (Goust, 1993). As an example, individuals who possess the
HLA antigens B7 and DR2 are more likely to respond to a specific
ragweed antigen (Goust, 1993). The nature of this association is
unclear at this time.
2.1.1.1 Anaphylaxis
Anaphylaxis is the most severe type of allergic response, as it
involves multiple organs and may be fatal. Anaphylactic reactions are
typically triggered by glycoproteins or large polypeptides. Smaller
molecules, such as penicillin, are haptens that may become immunogenic
by combining with host cells or proteins. Typical agents that induce
anaphylaxis include venom from insects in the Hymenoptera family,
drugs such as penicillin, and foods such as seafood or egg albumin
(Widmann, 1989).
Allergic reaction to allergens (e.g., in food, venom) that result
in systemic anaphylaxis are, in the vast majority of instances,
believed to be mediated by allergen-specific IgE bound to high
affinity IgE receptors (Fc-epsilon-RI) on the surfaces of basophils
and mast cells. As described earlier, the subsequent activation of
basophils/mast cells results in the release (e.g., histamines) and
generation (e.g., leukotrienes) of potent chemical mediators of
anaphylaxis.
2.1.2 Type II hypersensitivity
Type II hypersensitivity reactions are caused by IgG and IgM
antibodies directed towards cell surface antigens. These antigens may
be altered self-antigens or heteroantigens. Such antibodies, bound to
the cell membrane, can activate inflammatory phagocytes by Fc receptor
triggering. These phagocytes will then try to kill or to inactivate
their target as they would kill a microorganism. If they are unable to
phagocytose the whole cell, they will cause cell damage by secreting
oxygen radicals and by generating inflammatory mediators such as
arachidonic acid metabolites (prostaglandins and leukotrienes) from
their cell membrane.
Moreover, cell-bound antibodies activate the complement system.
The presence of C3b on the cell membrane, in addition to the
immunoglobulin, facilitates phagocytosis, whereas the further
complement cascade will induce membrane perforation and cell lysis.
Together, these reactions result in destruction of antibody-coated
cells and thus in cytopenia or in considerable tissue damage.
Not only granulocytes and macrophages are able to kill
antibody-coated cells. Specialized large granular non-B,
non-T-lymphoid cells, called natural killer (NK) cells, also bear Fc
receptors (CD16) and are capable of killing antibody-coated target
cells. NK cell-mediated killing is achieved by the release of
cytoplasmic granules containing perforin and granzymes. This process
is called antibody-dependent cell-mediated cytotoxicity (ADCC) and,
although not yet recognized at the time of Gell & Coombs (1963), it
should strictly be considered as a Type II effector mechanism. ADCC
reactions have been well established in vitro to tumour antigens and
viral proteins, but their precise role in host defence and
hypersensitivity reactions is still not completely understood.
2.1.3 Type III hypersensitivity -- immune complex reaction
Type III hypersensitivity reactions are similar to Type II
reactions in that IgG or IgM is involved and that destruction is
complement-mediated. However, in the case of Type III diseases, the
antigen is soluble. When soluble antigen combines with antibody,
complexes are formed that precipitate out of the serum. These
complexes deposit in the tissues and bind complement, causing damage
to the particular tissue. Deposition of antigen-antibody complexes is
influenced by the relative concentration of both components. If a
large excess of antigen is present, sites on antibody molecules become
filled before cross-links can be formed. In antibody excess, a lattice
cannot be formed due to the relative sparsity of antigenic determinant
sites. The small complexes that result in either of the above cases
remain suspended or may pass directly into the urine. Precipitating
complexes, on the other hand, occur in mild antigen excess, and these
are the ones most likely to deposit in the tissues. Sites where this
typically occurs include the glomerular basement membrane, vascular
endothelium, joint linings, and pulmonary alveolar membranes (Roitt et
al., 1998).
Complement binds to these complexes in the tissues, causing the
release of mediators that increase vasodilation and vasopermeability,
attract macrophages and neutrophils, and enhance binding of phagocytic
cells by means of C3b deposited in the tissues. If the target cells
are large and cannot be engulfed for phagocytosis to take place,
granule and lysosomal contents are released by a process known as
exocytosis (Roitt et al., 1998). This results in the damage to host
tissue that is typified by Type III reactions.
2.1.3.1 Arthus reaction
The classic example of a Type III reaction is the Arthus
reaction, a local necrotic lesion resulting from a local
antigen-antibody reaction produced by intradermal injection of an
antigen into a previously sensitized animal. This reaction is
characterized by erythema and oedema, peaks within 3 to 8 h, and is
followed by a haemorrhagic necrotic lesion that ulcerates. The
inflammatory response is due to antigen-antibody combination and
subsequent formation of immune complexes that deposit in small dermal
blood vessels. Complement is fixed, attracting neutrophils and causing
aggregation of platelets. Activation of complement is, in fact,
essential for the Arthus reaction, as the C3a and C5a generated
activated mast cells to release permeability factors, with the
consequent localization of immune complexes along the endothelial cell
basement membrane (Terr, 1994b). The Arthus reaction is rare in
humans.
2.1.4 Type IV -- delayed-type hypersensitivity
Type IV reactions were originally described by Gell & Coombs
(1963) as those skin reactions which take more than 12 h to develop
after antigen application. The classical Type IV reaction is the
tuberculin reaction, which reaches its maximum 24-72 h after the
intradermal injection of mycobacterial extracts. This delayed type
skin reaction to intradermally injected protein is characterized by a
pronounced induration reflecting a dense mononuclear cell infiltrate.
Since it became clear that antigen-specific T-cells are
responsible for these reactions, the term Type IV reactivity has been
used not only in relation to delayed-type hypersensitivity (DTH)
reactions in the skin, but also to T-cell-mediated inflammatory
reactions in other tissues. In addition, other T-cell-mediated
reactions, such as those to infectious agents or tumour antigens,
which are rather protective than hypersensitive, are regularly
described as Type IV reactions.
Although CD8+ T-cells have been shown in some experimental
animal models to transfer DTH, generally CD4+ T-cells are held
responsible for DTH responsiveness. The majority of antigen specific
T-cells cloned from DTH reaction sites are of the CD4+ subset.
Increased frequencies of antigen-specific CD4+ T-cells can also be
detected in the circulation of sensitized individuals. Such memory
T-cells show enhanced expression of adhesion molecules, which
facilitates their recirculation through the peripheral tissues. So,
whereas priming of naive T-cells takes place in the lymph nodes
draining the area of antigen contact, the secondary DTH response of
memory T-cells rather takes place in the peripheral tissues at the
site of antigen contact.
Here they may encounter the antigens for which they were
originally sensitized. The T-cells do not recognize the whole antigen
or conformational epitopes as antibodies do, but they recognize small
peptides derived from these antigens after processing by
antigen-presenting cells (APC). MHC class II molecules bind these
peptides already within the intracellular vesicles and present them
subsequently on the APC membrane to helper T-cells (Fig. 5). If the
memory T-cells recognize the peptide in its MHC class II context, the
cells become activated and produce a characteristic set of cytokines.
In the DTH reaction that now develops, predominantly mononuclear
cells are attracted from the circulation and contribute to the local
inflammatory reaction. An essential chemokine found to play a role in
the early accumulation of leukocytes at the DTH reaction site is IL-8
(Larsen et al., 1995), whereas RANTES (Regulated in Activation Normal
T-cells Expressed and Secreted), produced by endothelial cells, was
shown to attract preferentially macrophages and CD4+ T-cells to the
DTH reaction (Marfaing-Koka et al., 1995). In addition to a number of
different chemokines, IFN-gamma, TNF-alpha and LT (lymphotoxin) are
produced in the DTH reaction (Tsicopoulos et al., 1992). These are
typical Th1 effector cytokines, which are either directly cytotoxic
for pathogens or indirectly by activating the macrophage bactericidal
mechanism. Together, the cytokine cascade during this secondary
response shows an extreme amplification power, as illustrated by
experimental studies in which measurable oedema could be triggered by
only one specific T-cell (Marchal et al., 1982). Therefore, DTH
reactions are mediated by Th1 cells, the most prominent cytokines
being IL-2, LT and IFN-gamma. Indeed, at the site of DTH reactions
these cytokines can be detected (Tsicopoulos et al., 1992). It should
be realized, however, that the immune response is always the resultant
of a Th1-Th2 balance and that this delicate balance can be influenced
by several external factors, such as drugs, hormones, infections and
altered antigen exposure. Chronic antigen stimulation, for instance,
may induce a shift away from Th1, DTH-associated immunity towards a
Th2 response (Kitagaki et al., 1995; Mosmann & Sad, 1996). Th2
cytokines, such as IL-4, IL-5 and IL-10, rather help to induce
antibody responses, particularly IgE. In chronic infectious disease
indeed high levels of antibodies can be detected while DTH reactivity
is waning ( Mycobacteria, Trichophyton). In the human system, a
Th1 to Th2 shift correlating with a clinical conversion from disease
resistance to susceptibility and disease progression has been shown
in Leishmania, Candida, Mycobacteria and HIV infection
(Mosmann & Sad, 1996).
2.1.4.1 Mechanisms of allergic contact dermatitis
a) Sensitization
In allergic contact dermatitis, Type IV reactivity is raised
against small, chemically reactive environmental agents that enter the
body via the skin. In the skin, epidermal dendritic Langerhans cells
(LC), bearing large numbers of class II molecules (HLA-DR, -DP and
-DQ) on their cell membrane, are the primary allergen-presenting
cells. They form a contiguous network in which agents penetrating the
skin are efficiently trapped. Langerhans cells stem from the bone
marrow, but their continuous presence in the epidermis is at least
partly maintained by local proliferation (Czernielewski & Demarchez,
1987; Breathnach, 1988).
Upon penetration through the epidermis, contact allergens readily
bind to a plethora of skin constituents. Whereas most allergens bind
spontaneously, some need metabolic conversion (Anderson et al., 1995)
or photoinduced activation before they bind. The latter allergens are
called contact photoallergens (White, 1992).
Only those allergens that modify the Langerhans cell MHC Class II
molecules can eventually sensitize T-cells; this occurs either by
direct binding to the MHC Class II molecules and to peptides within
their grooves or by uptake and processing of haptenized proteins
followed by presentation of the derived peptides in the MHC Class II
molecules of the antigen-presenting cells. It has been shown that in
individuals allergic to nickel some nickel-specific T-cell clones
recognize unprocessed nickel, bound to the MHC Class II molecules of
fixed antigen-presenting cells, whereas other nickel-specific T-cell
clones are dependent on viable antigen-presenting cells for
processing, most likely of preformed nickel-protein conjugates (Moulon
et al., 1995).
In a similar way, MHC Class I peptides may become modified by the
allergen, triggering class I restricted, CD8-positive T-cell clones.
Notably the size of the allergic metal ions is much smaller than the
peptides to which they bind in the groove. In some instances different
metallic allergens modify the MHC Class II peptides in a very similar
way. T-cell clones then show complete cross reactivity between the
metals, for instance between nickel and palladium, or between nickel
and copper (Pistoor et al., 1995). Clinical signs of cross reactivity
of nickel with other related metals, such as cobalt, however, most
probably result from concomitant sensitization by exposure to metal
alloys.
The majority of chemically reactive allergen, however, binds
covalently to distinct amino acids, thus forming haptenized proteins.
Usually, haptens, like picryl- or penicilloyl- are much larger than
metal allergens, and hapten-specific T-cell responses, i.e.,
independent of the carrier protein, can be observed.
Upon exposure of the skin to chemical contact sensitizing agents,
cytokine production in the epidermis by both keratinocytes and
Langerhans cells is immediately up-regulated, thereby initiating the
process of Langerhans cell maturation and migration.
The first cytokine to be up-regulated, within 15 min after
allergen application, is IL1-beta, produced by Langerhans cells. This
up-regulation was found to be allergen-specific, just like the
subsequent production of IL-1-alpha, and of the chemokines IP-10
(IFN-gamma-inducible protein 10) and MIP-2 (macrophage inflammatory
protein-2) by keratinocytes, and could not be detected upon irritant
application. TNF-alpha up-regulation in keratinocytes, on the other
hand, appeared to be a less allergen-specific event. The most relevant
cytokines for Langerhans cell maturation and egress are GM-CSF, IL-1
and TNF-alpha, while IL-10, which is also produced by keratinocytes,
but at a later stage, may serve as a down-regulatory molecule for
Langerhans cell maturation. (Heufler et al., 1988; Kimber &
Cumberbatch, 1992a,b; Enk & Katz, 1995). Interestingly, IL-1 and
TNF-alpha were found to down-regulate the membrane expression on
Langerhans cells of E-cadherin, a molecule that mediates
Langerhans-cell-keratinocyte adhesion. Thus, Langerhans cells with
allergen-modified MHC Class II molecules leave the epidermis and
migrate via the dermis and lymphatics to the draining lymph nodes,
where they settle within the paracortical areas. Indeed increased
numbers of dendritic cells appear in the regional lymph nodes around
24 h after hapten application (Kimber et al., 1990). Whereas resident
epidermal Langerhans cells are still relatively inefficient
antigen-presenting cells, once they have arrived in the lymph nodes
they have matured into fully active antigen-presenting dendritic cells
and are capable of stimulating even naive unprimed T-cells. Naive
cells express, in contrast to memory cells, low levels of cellular
adhesion molecules (CAM) and therefore require optimally functioning
antigen-presenting cells for stimulation. Matured Langerhans cells or
dendritic cells (DCs) have an increased expression of MHC Class II,
ICAM-1 and B7 molecules, allowing for optimal T-cell triggering
(Steinman et al., 1995); in addition the intricate structure of the
paracortical area offers an appropriate environment for this
sensitization process to take place. Naive T-cells, again in contrast
to memory cells, recirculate preferentially through the peripheral
lymphoid organs, rather than through the tissues, due to the
expression of distinct adhesion molecules (L-selectin) that recognize
the high endothelial venules in the lymph nodes. The probability of
hitting unprimed specific T-cells is thus increased.
When successful triggering and subsequent proliferation of
allergen-specific T-cells have taken place, the lymphocyte progeny
will leave the lymph nodes to join the recirculating pool of
lymphocytes. The frequency of specific cells in the circulation can
thus be increased from around 1:100 000 to 1:1000-10 000 and the
individual has now become "sensitized".
b) Elicitation of allergic contact dermatitis
Upon re-exposure to contact sensitizing agents, specific
recirculating memory T-cells present in the skin immediately recognize
the allergen modified MHC Class II molecules on the Langerhans cell
membranes. The probability that the allergen is indeed found by
specific memory T-cells is largely increased by the expression of
organ-specific interaction molecules on the T-cell surface. The
cutaneous lymphocyte-associated antigen (CLA), recognized by the
monoclonal antibody HECA-452, is present on a small subpopulation
(approximately 16%) of peripheral blood T-cells which preferentially
recirculates via the skin. Here the endothelial adhesion molecule
E-selectin acts as a vascular addressin for the skin-homing memory
T-cells (Picker et al., 1993; Bos & Kapsenberg, 1993). As described in
the section on Type IV - delayed-type hypersensitivity (section
2.1.4), mainly CD4-positive allergen-specific cells thus enter the
skin. Since memory cells have relatively low stimulation thresholds,
they can be triggered by less efficient antigen-presenting cells, like
the local resident Langerhans cells. The T-cells will now initiate a
Th1-type cytokine cascade, which eventually leads after 24-72 h to the
typical delayed-type contact allergic reaction. Because the reaction
takes place in superficial layers of the skin, erythema and blistering
are characteristic features, in contrast to the tuberculin DTH where
induration is most pronounced.
The challenge reaction in allergic contact dermatitis resolves
spontaneously within one week. It is therefore commonly used as a
primary diagnostic test in allergic contact dermatitis. To this end,
low non-toxic dosages of allergen are generally applied onto the skin
under an occlusive patch to allow for maximal skin penetration. The
main drawbacks of such an in vivo skin test procedure are the
potential sensitization and boosting by such an intense allergen
contact. Indeed it was shown experimentally in guinea-pigs that even
one epicutaneous application of allergen could direct the immune
response towards (still subclinical) sensitization, as shown by a
failure of subsequent tolerance induction (Van Hoogstraten et al.,
1994). Also clinically, occasional sensitization by epicutaneous skin
tests can be observed. For this reason much effort has been put in the
development of in vitro diagnostic procedures in allergic contact
dermatitis (Von Blomberg et al., 1990).
Up to now, in vitro assays in allergic contact dermatitis have
been successful for relatively non-toxic water-soluble allergens, such
as metal salts. For other allergens, occasionally positive results are
obtained by pre-pulsing antigen-presenting cells or proteins or by
using special solvents. So, despite the fact that most of our
knowledge of the pathogenesis of human allergic contact dermatitis is
due to in vitro experiments with blood from allergic patients, for
routine assessment of allergic contact dermatitis these assays are
still too complicated.
Repeated contact with low dosages of allergen, as typically
occurs for most contact allergens, may lead to continuous triggering
of Type IV reactivity in the skin and thus to an allergic contact
dermatitis (Scheper & Von Blomberg, 1992). The dermatitis only
disappears when the allergen is entirely eliminated from the
environment.
Even if the reaction is clinically healed, allergen-specific
T-cells may persist in the skin for up to several months. Thus,
locally increased allergen-specific hyperreactivity, either detectable
through accelerated "retest" reactivity (peaking at 6-8 h) or flare-up
reactivity after allergen entry from the circulation, may be observed
for several months at former allergic contact dermatitis reaction
sites (Scheper et al., 1983; Yamashita et al., 1989). The presence of
specific T-cells at former eczematous sites can thus be maintained by
low dosages of inhaled or ingested allergen, in the absence of
allergenic skin contacts.
Repeated contact with relatively high dosages of allergen, on the
other hand, may result in a local desensitization. The initial
erythematous reaction gradually decreases. Such a local
hypo-responsiveness of the skin, which is known as "hardening" in
occupational contact dermatitis, is largely reversed after a period of
allergen restrain. However, also systemically, DTH reactivity
decreases upon repeated allergen application. This decrease in DTH is
associated with increased antibody responses and a shift towards
immediate-type hypersensitivity, reflecting a shift from Th1 to Th2
reactivity (Boerrigter & Scheper, 1987; Kitagaki et al., 1995).
It appears, therefore, that although exposure of the skin to
exogenous antigens generally results in Th1 responses, the
micro-environment in chronically inflamed tissues, rather than the
site of allergen exposure or the nature of the allergen, determines
the type of immune reaction.
2.1.4.2 T-cell responses in chemically induced pulmonary diseases
Asthma is a chronic pulmonary inflammatory disease associated
with bronchial hyperreactivity. In the majority of asthma cases a
clear association exists with atopic IgE-mediated hypersensitivity,
involving relatively large protein allergens. Here T-cells dominate in
the late phase and the chronic reaction. The pivotal role of T-cells
in chronic asthma is stressed by the finding of activated T-cells in
the bronchial mucosa and the effectiveness of T-cell immunosuppressive
drugs. In particular, the number of activated CD4-positive cells was
found to correlate with the numbers of eosinophils in the
bronchoalveolar lavage (BAL) and with disease severity (Walker et al.,
1991). The T-cells, present in the bronchial mucosae and in the lavage
fluid, were shown to produce predominantly Th2 cytokines, in
particular IL-5, a cytokine known to activate eosinophils (Corrigan &
Kay, 1992). Therefore, although T-cell-mediated immunity is clearly
playing a role, Type IV reactivity, mediated by Th1 cells, does not
seem to be involved in this type of asthma.
Of particular interest is the hypersensitivity pneumonitis
induced by environmental small chemical allergens. Such allergens are
known to cause DTH when applied to the skin. Occasionally these
allergens induce, in addition or as first manifestation, asthmatic
disease upon inhalation. It could be questioned whether these
allergens, in contrast to the atopic protein allergens, would induce
Type IV reactivity.
Experimentally it has been shown in mice that Type IV
hypersensitivity to small chemical allergens, such as picryl chloride,
can indeed induce lung disease upon intranasal application (Garssen et
al., 1991, 1994).
Contact allergens that have been reported to induce asthma
include formaldehyde, platinum salts, nickel, cobalt and chromium
(Nordman et al., 1985; Estlander et al., 1993; Cirla, 1994;
Park et al., 1994; Merget et al., 1996). In a number of cases this
asthmatic disease could be associated with the presence of circulatory
IgG to the causative allergen and positive bronchial provocation
tests.
Trimellitic anhydride, phthalic anhydride and toluene
diisocyanate are reactive chemicals behaving primarily as respiratory
allergens, causing asthmatic disease and pulmonary irritation. The
immune reactions leading to asthmatic disease are quite variable; the
role of clear-cut Type IV reactivity is uncertain.
2.1.5 Type V stimulatory hypersensitivity
Stimulatory hypersensitivity occurs when antibodies binding to a
cell surface molecule cause inappropriate stimulation of the cell.
Normal feedback inhibition will then fail. An example is Graves'
disease (exophthalmic goitre), in which autoantibodies to the
thyroid-stimulating hormone receptor on thyroid cells stimulate the
production of excessive amounts of thyroid hormone, resulting in
disease.
2.2 Regulation of hypersensitivity
In 1986, the existence of two CD4+ Th-cell subsets was
discovered in mice, and they were designated Th1 and Th2. Their
identification has greatly improved understanding of the regulation of
immune effector functions, not least on Type I and Type IV
hypersensitivity responses. These Th subsets are defined by the
patterns of cytokines that they produce, which leads to strikingly
different T-cell functions (Table 9). Broadly speaking, Th2-cells are
more efficient B-cell helpers, especially in the production of IgE
antibody, whereas Th1-cells mediate DTH reactions. In addition, they
cross-regulate by producing mutually antagonistic cytokines. Their
specific function and characteristics in rodents and humans have not
yet been clearly established (Muraille & Leo, 1998).
Table 9. Characteristics of Th1- and Th2-associated immunitya in vivo
(modified from Röcken et al., 1996)
Characteristics Th1 Th2
IFN-gamma high variable, frequently low
IL-2 high variable, frequently low
IL-4 low/negative high
major mode of action DTH reactions eosinophil-associated
(cellular immunity) cytotoxicity
complement-binding non-complement-binding
antibodies and IgE antibodies and IgE
protective effects against intracellular against
microorganisms and extracellular parasites
tumours
harmful effects contact hypersensitivity atopic diseases
tissue-specific autoimmunity immunoglobulin-mediated
allergic encephalitis autoimmunity
juvenile diabetes bullous autoimmune
rheumatoid arthritis diseases
thyroiditis sclerosing diseases ?
uveitis
a Th1 and Th2 immunity characterizes T-cell populations, not single T-cells
In addition to Th1- and Th2-cells, additional cytokine production
phenotypes of CD4+ cells exist. They are, however, characterized
less thoroughly. Most resting T-cells mainly produce IL-2 on first
contact with antigen, and differentiate within a few days into cells
producing multiple cytokines, such as IL-4 and IFN-gamma. In addition
to Th1- and Th2-cells, the existence of undefined precursor cells has
been suggested. These precursor cells (IL-2 producing) are the virgin
Th cells, producing only or predominantly IL-2, and Th0 cells are in
the process of differentiation, producing cytokines of both Th1 type
(such as IL-2 and IFN-gamma) and Th2 type (such as IL-4, IL-5 and
IL-10). The pathways of differentiation from the precursor cells are,
however, unclear. In addition, it is unknown whether there is a single
common precursor cell or whether precursor cells are already committed
to a particular cytokine pattern before exposure to antigen (the
cytokine production profiles of Th-cell subsets in the mouse are shown
in Table 1). In conclusion, it is believed that Th1- and Th2-cells
represent the most differentiated populations of the CD4+ phenotype
that develop following prolonged exposure to antigen or following
stimulation by potent immunogens.
At least two mechanisms can influence the selective
differentiation of Th-cell subsets. Firstly, the cytokines that are
present during differentiation, in particular IFN-gamma, IL-4 and
IL-12, may greatly influence the type of Th that will be generated.
IF-gamma augments development of Th-type responses and IL-4 promotes
differentiation of Th-cells (Romagnani, 1992a). Secondly, the type of
APC is thought to influence the characteristics of immune responses.
Upon activation, Th2 cells express p39 on their surface, which
interacts with CD40 on the surface of B-cells. The interactions of p39
with CD40 and of T-cell antigen receptor (TCR) with antigen and MHC
Class II together lead to production of IL-4, IL-5 and IL-6 by Th2
cells, stimulating B-cells to antibody production. Th1 cells, on the
other hand, may interact with macrophages. A pair of cell surface
molecules analogous to p39/CD40 have not as yet been identified.
However, the interaction of Th1 cells with macrophages leads to
IFN-gamma production by Th1 cells, stimulating macrophages to produce
monokines.
The difference in APCs, macrophage versus B-cell, that
preferentially activates Th1 or Th2 suggests differences in antigen
requirements for activation, e.g., large particulate antigens
requiring phagocytosis for Th1 and low antigen concentration for Th2.
Whereas moderate concentrations of antigen preferentially activate
Th1, extremely high concentrations are believed to inhibit Th1 and
select for Th2 responses (Pfeiffer et al., 1991).
If Th1 and Th2 clones are stimulated by immobilized anti-CD3 (in
the absence of APC), both types produce their respective cytokine
pattern. The proliferative responses are, however, very different.
Whereas Th2 clones exhibit good proliferative responses, Th1 not only
fail to do so, but are even rendered incapable of proliferating in
response to exogenously added IL-2 (Williams & Unanue, 1990; Williams
et al., 1990). These Th1 clones are in a state of anergy or tolerance
(Schwartz & Weiss, 1990).
IFN-gamma inhibits the proliferation of Th2 responding to either
IL-2 or IL-4, but does not inhibit Th1. IL-10 inhibits the synthesis
of cytokines by Th1 cells, and, although growth factor requirement is
not affected, the reduction in IL-2 synthesis can lead to decreased
proliferation. It has been shown in in vitro human systems that
IL-10 can suppress the antigen-presenting capacity of monocytes and
dendritic cells by down-regulation of MHC Class II. IL-10 had no
effect on the antigen-presenting capacity of B-cells or
down-regulation of their MHC Class II. These results suggest a
mechanism for the general observation that macrophages/dendritic cells
preferentially stimulate Th1, whereas B-cells preferentially stimulate
Th2.
IL-2 is a T-cell growth factor (TCGF) that mediates autocrine
proliferation of Th1, whereas the TCGF IL-4 mediates autocrine
proliferation of Th2. Interestingly, it has been shown that IL-4 is
the major TCGF produced by T-cells from lymphoid organs that drain
mucosal tissues, whereas IL-2 is the major TCGF produced by T-cells
from other lymphoid organs (Daynes et al., 1990b). Involvement of
dehydroepiandosterone in this site/tissue-specific control on
lymphokine production was suggested (Daynes et al., 1990a).
Dihydrotestosterone and 1,2,5-dihydroxyvitamin D3 also change the
cytokine production pattern of T-cells.
In humans, a predominant fraction of CD4+ T-cell clones was
found to produce IL-2, IL-4 and IFN-gamma, although the quantities
varied considerably. Bearing in mind the findings in mice (see above),
it was thought that unrestricted profiles are mainly a property of
T-cells that are not yet committed to a certain differentiation
pathway. Consequently, functional heterogeneity of CD4+ cells
should most likely be found in chronically stimulated responders.
Kapsenberg et al. (1991) studied two categories of patients, those
with nickel hypersensitivity, an example of Type IV hypersensitivity,
and those with house dust mite ( Dermatophagoides pteronysinnus
(Dp))hypersensitivity, an example of Type I hypersensitivity.
Most house dust mite-specific T-cell clones from peripheral blood
(Wierenga et al., 1990) and lesional skin biopsies of house dust
mite-allergic patients show a Th2-like production profile. House dust
mite-specific clones from atopic patients induce IgE production (see
also below). It was shown that this production is dependent on a high
IL-4/IFN-gamma ratio, and is not dependent on the origin of B-cells.
Only IgE specific to house dust mite (and not, for instance, IgE
specific to tetanus toxoid or Candida albicans) was elevated in
atopic house dust mite-allergic patients.
The majority of allergen-specific human T-cell clones produce
IL-4 and IL-5, but not IFN-gamma. Virtually all T-cell clones specific
for bacterial components, which were derived from the same patients,
was found to produce large amounts of IL-2 and IFN-gamma, and few
produced IL-4 and/or IL-5 (Wierenga et al., 1990; Parronchi et al.,
1991). In a subsequent study, antigen-specific T-cell clones were
derived for the bacterial antigen purified protein derivate (PPD) from
Mycobacterium tuberculosis and for the helminth antigen Toxicara
canis excretory-secretory (TES). Most PPD-specific clones produced
IL-2 and IFN-gamma, but not IL-4 and IL-5, whereas most TES-specific
clones produced IL-4 and IL-5, but not IL-2 and IFN-gamma. This study
shows that in the course of natural immunization certain infectious agents
preferentially expand T-cell subsets. PPD expands Th1, parallelling
the (Th1-mediated) tuberculin DTH, whereas TES expands Th2,
parallelling the (Th2-mediated) parasite infection.
In a large series of human T-cell clones, all Th1 clones were
found to lyse EBV-transformed autologous B-cells pulsed with the
specific antigen, and the decrease of Ig production correlated with
the lytic activity of Th1 clones against autologous antigen-presenting
B-cell targets (Romagnani, 1991). This suggests an important mechanism
for down-regulation of antibody responses in vivo.
Almost all nickel-specific T-cell clones produce TNF-alpha,
GM-CSF, IL-2 and high levels of IFN-gamma, but low or undetectable
levels of IL-4 and IL-5, thus resembling Th1 cells. Nickel induces DTH
in the skin of allergic patients. Since IFN-gamma is an important
mediator for DTH, IFN-gamma may be essential to DTH. However, no clear
difference in cytokine production profile between allergic patients
and control individuals was found.
2.2.1 Regulation of IgE synthesis by IL-4 and IFN-gamma
Atopy is associated with enhanced serum titres of
allergen-specific IgE. The production of IgE is heightened and
sustained by B-cells in atopic patients. IL-2 secreted by Th cells is
necessary for the production of all isotypes of immunoglobulins
(Kapsenberg et al., 1991). Activated B-cells are induced by IL-4 to
undergo immunoglobulin heavy-chain rearrangements to the
epsilon-constant region, resulting in synthesis of IgE (Coffman et
al., 1986). So far, IL-4 can mediate this isotype switch, which is
blocked very efficiently by IFN-gamma (Romagnani, 1991). IFN-gamma
induces switching to gamma-2a (Coffman et al., 1986). IL-4 and
IFN-gamma are produced by Th2 and Th1 cells, respectively; a response
that involves mainly Th2 cells should produce a large amount of IgE,
whereas responses involving mainly Th1 cells, such as DTH reactions,
should be non-permissive for IgE production. In vivo experiments
have confirmed these predictions. IL-4-deficient mice lack IgE and
IgG1 responses (Kuhn et al., 1991), whereas transgenic mice
constitutively producing IL-4 show elevated serum IgE levels.
Injection of mice with anti-IgD antibodies results in a strong
stimulation of both B- and T-cell populations, leading to polyclonal
antibody production and very high IgE levels. Anti-IL-4 antibodies
dramatically reduce IgE levels after anti-IgD immunization, whereas
anti-IFN-gamma antibodies elevate IgE levels even further. Similarly,
administration of IFN-gamma results in considerable inhibition of the
IgE response. Because the anti-IgD immunization leads to a response
that involves high levels of Th2 cytokines, all of these results are
consistent with the effects of IL-4 and IFN-gamma on IgE synthesis as
defined by in vitro model systems. Similar correlations between
Th2-like responses and high IgE production are seen during several
parasite infections.
2.2.2 Eosinophilia and IL-5
Many parasitic infections induce high levels of circulating
eosinophils. Because IL-5 has been implicated as a major growth and
differentiation factor for eosinophils the association of IgE and
eosinophilia may be explained by the association of IL-4 and IL-5 as
products of Th2-cells (Gulbenkian et al., 1992). Supporting evidence
has been provided by experiments in vivo, in which administration of
anti-IL-5 during a strong anti-parasitic immune response completely
abrogated eosinophilia (Coffman et al., 1989), and from studies of
transgenic mice that express high levels of IL-5. The major
abnormality in these animals is the presence of extremely high levels
of eosinophils in the blood and various lymphoid organs. Patients with
filaria-induced eosinophilia exhibit a significantly greater frequency
of IL-5-producing T-cells than uninfected individuals.
2.2.3 The relationship between Th2 cells and type I hypersensitivity
In mice, in addition to enhancing IgE production via IL-4, Th2
cells also influence other features of allergic reactions. Firstly,
IL-3, IL-4 and IL-10 are mast cell growth factors that act in synergy,
at least in vitro, and secondly, IL-5 induces the proliferation and
differentiation of eosinophils in vitro and in vivo (Coffman,
1989; Sanderson, 1990). In addition, IL-3 and IL-4 have been shown to
enhance the secretory function of murine mast cells. So, Th2-cell
activation not only increases the level of IgE synthesized, but also
potentially increases the number of IgE-binding cells that will
degranulate in response to allergen challenge.
Mast cells and basophils produce IL-4. It has been hypothesized
that IL-4 produced by these cells induces the development of Th2
cells, and that these cells in turn produce IL-4. In addition, mast
cells are an important source of IL-5.
2.2.4 IL-12 drives the immune response towards Th1
The pivotal role of the cytokine IL-12 in the differentiation of
Th-cells towards Th1 is evident from both in vitro and in vivo
studies (Scott, 1993). IL-12 is produced by T-cells, B-cells,
macrophages and dendritic cells and stimulates the production of
IFN-gamma from T-cells and NK-cells. IL-12 enhances Th1-cell expansion
in cell lines from atopic patients (Manetti et al., 1993). The
presence of IL-12 during primary stimulation of naive CD4+ cells skews
the response in the direction of Th1 differentiation. These data
suggest that IL-12 may be the IL-4 equivalent for the differentiation
of Th1-cells. IL-10 has been shown to inhibit lymphocyte IFN-gamma
production by suppressing IL-12 synthesis in accessory cells. A
variety of pathogens that are associated with Th1 development have
been shown to induce IL-12 production (Scott, 1993).
2.2.5 IL-13, an interleukin-4-like cytokine
Information on cytokine IL-13 is based on limited information
about its activities in vitro. As it shares biological activities
with IL-4, these activities will, however, be briefly discussed. IL-13
is produced by activated T-cells. The activities in vitro of IL-13
are similar to those of IL-4, with two major exceptions. Firstly,
IL-13 does not act on T-cells and secondly, IL-13 does not act on
murine B-cells (Zurawski & de Vries, 1994). Similarly to IL-4 and
IL-10, IL-13 inhibits the production by LPS-stimulated monocytes of
proinflammatory cytokines, chemokines and haematopoietic growth
factors. In contrast to IL-10, however, IL-13 up-regulates the
antigen-presenting capacity of monocytes. Similarly to IL-4, IL-13
inhibits transcription of IFN-gamma and both alpha- and beta-chains of
IL-12. Thus, IL-13 may (like IL-4) suppress the development of
Th1- cells through down-regulation of IFN-gamma and IL-12 production
by monocytes, favouring the generation of Th2 cells. Also in the
mouse, IL-13 inhibits production of proinflammatory cytokines and
expression of IL-12 alpha- and beta-chain mRNA. Murine IL-13 does not
affect macrophage antigen-presenting capacity. Similarly to IL-4,
IL-13 acts on human B-cells in inducing class switch to production of
IgG4 and IgE and inducing CD23 surface expression (Punnonen et al.,
1993; Punnonen & de Vries, 1994). Following activation of T-cells,
IL-13 is produced earlier and for much longer periods than IL-4 (Yssel
et al., 1994). Thus, IL-13 may play an important role in the
regulation of enhanced IgE synthesis in allergic patients. In contrast
to IL-4, murine and human IL-13 do not induce IgE synthesis in murine
B-cells. Importantly, this may restrict the use of mice as an animal
model for allergy.
In summary, IL-13 may favour development of Th2-cells, consistent
with the induction of IgG4 and IgE synthesis. Determination of the
actual role of IL-13 requires more information on the biological
effects in vivo.
2.3 Autoimmune reactions
A wide spectrum of human and animal diseases appears to be wholly
or partially attributable to autoimmune reactions. Despite the
extensive growth of information relating to the mechanisms of
self-tolerance (see section 1.5), the understanding of the mechanisms
leading to pathogenic autoimmunity is still fragmentary and incomplete
(Theofilopoulos, 1995a).
Important issues that need to be resolved in this context
concern: (i) the nature of the inciting antigens (self, neo-self,
foreign); (ii) the definition of the criteria by which a disease can
be termed autoimmune; (iii) the principles that govern the spectrum
and extent of an autoimmune response; (iv) the mechanisms by which
spontaneous remissions and exacerbations of autoimmune diseases occur;
(v) the nature of environmental factors that initiate/precipitate
autoimmune reactions; (vi) the structural and other characteristics
that differentiate pathogenic from non-pathogenic autoantibodies and
T-cells; and (vii) the identity of the genes that predispose or
accelerate autoimmunity, as well as their mechanism of action
(Theofilopoulos, 1995a).
The most urgent of these questions concerns the nature of the
inciting antigen. Although autoimmune disorders are often defined and
diagnosed by the presence of autoantibodies (Osterland, 1994), it
should be noted that (a) autoantibodies may indeed be the actual
pathogenic agents of disease (e.g., autoimmune haemolytic anaemia,
pemphigus, and myasthenia gravis; see sections 2.6.3, 2.6.5 and
2.6.6), (b) they may arise as a consequence of another disease process
(e.g., organ-specific autoantibodies due to tissue damage to those
organs), or (c) they may merely mark, like footprints, the presence of
the etiological agent while not themselves causing disease (Naparstek
& Plotz, 1993; Theofilopoulos, 1995a). The latter possibility is
complicated by the fact that determinants recognized by the
autoantibody and the prerequisite Th-cell may reside on different
molecules within a supramolecular complex (Theofilopoulos, 1995a). For
example, for many years, it was believed that native nDNA itself was
the immunogen for anti-nDNA antibodies, but efforts to induce such
autoantibodies by immunization with nDNA have generally been
unsuccessful. It has been suggested that for anti-nDNA antibody
induction, the scenario may involve intermolecular help, via the
binding of nucleosomes or other protein-DNA complexes to anti-DNA
idiotype-displaying B-cells, followed by processing of the protein and
presentation to the corresponding Th-cells (Theofilopoulos, 1995a). In
this connection it is of interest that in systemic autoimmune diseases
autoantibodies frequently appear to be directed against the entire set
of polypeptides associated with discrete supramolecular cellular
entities, such as the nucleosome particle or the nucleocytoplasmic
ribonucleoprotein particles (see Table 10).
It has become clear that T-cells are primary players in the
initiation and perpetuation of spontaneous (Theofilopoulos & Dixon,
1985; Singer & Theofilopoulos, 1990) as well as induced systemic
autoimmune disorders (Druet, 1989; Goldman et al., 1991). Many immune
responses seem to be functionally dominated either by Th1 or Th2
cytokines. Therefore, the Th1-Th2 balance during immune reactions in
vivo significantly determines the outcome of immunopathological
processes (Röcken et al., 1996). Whereas organ-specific autoimmune
disease are predominantly mediated by IFN-gamma-producing Th1-cells,
IL-4-producing Th2-cells are involved in immunoglobulin-mediated
autoimmune diseases such as systemic lupus erythematosus (SLE)
(Goldman et al., 1991; Röcken et al., 1996) (Table 9).
Table 10. Examples of autoantigens in organ-specific and systemic autoimmune diseasesa
Organ/cell/nucleus Target antigens Diagnosis
Organ-specific autoimmune diseases
Pancreatic islet cells glutamic acid decarboxylase 65 insulin-dependent diabetes mellitus
glutamic acid decarboxylase 67
tyrosine phosphatase IA-2
tyrosine phosphatase IA-2b
Adrenal cortex 21-hydroxylase Addison's disease
Leydig cells, testes, granulosa cytochrome side-chain cleavage hypogonadism
theca enzyme
Ovary 17a-hydroxylase hypogonadism
Gastric parietal cell H+/K+-ATPase pernicious anaemia
intrinsic factor
Thyroid epithelium thyroid peroxidase autoimmune thyroid diseases
thyroglobulin
thyroid-stimulating hormone (TSH)
TSH-receptor
triiodothyronine
thyroxine
Hepatocyte CYP 2D6 (LKM-1) chronic active hepatitis
halothane-induced hepatitis
Melanocyte tyrosinase vitiligo
Parathyroid calcium-sensing receptor autoimmune parathyroidism
Table 10. (continued)
Organ/cell/nucleus Target antigens Diagnosis
Systemic autoimmune diseases
Native DNA DNA backbone systemic lupus erythematosus (SLE)-renal
ss-DNA nucleotides SLE and other connective tissue diseases
Nucleoprotein DNA histone SLE - central nervous system,
histone 1 H1, 2A, 2B, 3, 4 renal - drug-induced SLE
histone 2 H3 connective tissue disease
Sm SnRNP SLE
Nuclear RNP non Sm SnRNP mixed connective tissues disease, SLE
Ribosomal RNP phosphoproteins SLE
Scl-70 topoisomerase 1 scleroderma
Centromere kinetochore CRESTb, Raynaud's syndrome
SS-A (Ro) RNP SLE-cutaneous, photosensitivity
SS-B (La) RNA-pol protein Sicca syndrome, SLE, neonatal lupus
Cardiolipin phospholipid SLE - thrombosis, cytopenia
PM-1 protein complex myositis, scleroderma
Jo-1 histidyl tRNA synthesis myositis
Mi-2 dermatomyositis
Table 10. (continued)
Organ/cell/nucleus Target antigens Diagnosis
PCNA cyclin SLE
Ku protein on terminal chromosome SLE
nucleolar RNA-pol 1, RNA
fibrillaren scleroderma, drug-induced connective
tissue disease
Nuclear membrane laminins scleroderma, SLE
a modified from Osterland (1994) and Song et al. (1996a); responses encompass both Th1 and Th2 responses
and involve both Th1 (IFN-alpha) and Th2 (IL-4)
b CREST (calcinosis, Raynaud's phenomenon, oesophageal involvement, sclerodactyly and telangiectasia)
The major non-mutually exclusive etiological concepts of
autoimmune disorders have been reviewed (Theofilopoulos, 1995a,b) and
are summarized in Table 11.
Table 11. Possible mechanisms of autoimmune reactions
(modified from Theofilopoulos, 1995a,b)
Release of anatomically sequestered antigens
The "cryptic self" hypothesis
The self-ignorance hypothesis
The molecular mimicry hypothesis
The "modified self" hypothesis
Immunoregulatory disturbances
Errors in central or peripheral tolerance
Polyclonal activators
2.4 Possible mechanisms of autoimmune reactions
2.4.1 Release of anatomically sequestered antigens
In general, antigens associated with peripheral tissues,
especially those sequestered behind anatomic barriers, may not come
into contact with the developing T-cell repertoire, and, therefore,
tolerance may be unnecessary for such antigens. Induction of
organ-specific autoimmune disease following contact with antigens of
such so-called "immunologically privileged" sites has been well
documented, as exemplified by the development of ophthalmia following
eye injury and orchitis following vasectomy.
Data have also clearly established that antigens associated with
peripheral tissues can cause tolerance, and therefore loss of
susceptibility to tissue-specific autoimmune diseases, when
experimentally introduced into the thymus. Intrathymic injection of
pancreatic islet cells can prevent autoimmune diabetes in the
BioBreeding (BB) rat (Posselt et al., 1992) and the non-obese diabetic
(NOD) mouse (Gerling et al., 1992). Tissue trauma alone may not be
sufficient to elicit a conventional self-directed immunological
response. Tissue-trophic pathogens, such as viruses, may be important
in inducing the initial damage that results not only in availability
of previously sequestered antigens but also in the production of
co-stimulatory factors necessary for the immune response.
2.4.2 The "cryptic self" hypothesis
A corollary hypothesis for the mechanism of induction of
pathogenic autoimmune responses addresses molecular, rather than
anatomic, sequestration and relates to the presence of cryptic
self-determinants. Each self-protein presents only a small minority of
dominant determinants, which are involved in negative selection during
thymic maturation and development of tolerance of the organism to
them. Because of many constraints to peptide presentation, only a few
peptide stretches of a given protein antigen are presented to the
T-cell repertoire, namely those that have the highest affinity to the
MHC-binding site and are present at a sufficient concentration. These
peptides are the so-called dominant antigenic determinants. It is
important to realize that, because antigen-presenting cells cannot
distinguish "self" and "non-self" proteins, foreign and "self"
peptides are presented indiscriminately (Bloksma et al., 1995). The
subsequent immune responses, however, are diametrically opposed to
each other. Whereas foreign peptide sequences, in general, induce
"stimulatory" T-cell responses, the dominantly presented "self"
sequences induce "inhibitory" T-cell responses through
peptide-specific thymic cell deletion during development of the T-cell
repertoire and/or induction of specific tolerance or anergy in the
established peripheral T-cell repertoire. The poorly displayed
majority of subdominant/cryptic determinants, constituting the
"cryptic self", do not induce tolerance and, therefore, a large cohort
of potentially "self"-reactive T-cells exists. The presentation of
cryptic "self" peptides, however, can be up-regulated under certain
conditions (Lehmann et al., 1993). Evidence for the role of cryptic
determinants in the pathogenesis of autoimmunity has been provided in
the non-obese diabetic mouse (NOD) model (Kaufman et al., 1993; Tisch
et al., 1993), but the exact mechanisms of these immune responses are
not fully known. One suggestion is that pathogens such as viruses may
provide the initial stimulus through increased presentation of the
subdominant determinant, either by molecular mimicry (see below)
and/or by interferon-induced up-regulation of gene-expression,
including genes for antigen-presenting MHC molecules (Theofilopoulos,
1995a).
Processing of chemically altered "self" proteins may result in
the presentation of cryptic, thus potentially T-cell-activating,
self-peptides by creation of new binding sites with high affinity to
MHC molecules or modification/preventing of the physiological
intracellular protein degradation (Bloksma et al., 1995). Expression
of "cryptic self" peptides of nucleolar proteins appears to be a
decisive step in the pathogenesis of HgCl2-induced formation of
anti-nucleolar autoantibodies in mice (Kubicka-Muranyi et al., 1996).
2.4.3 The self-ignorance hypothesis
Evidence suggests that mature resting T-cells specific for
extrathymic antigens presented by non-professional antigen-presenting
cells (other than dendritic cells and macrophages) are induced to
undergo anergy because of the absence of appropriate "second signals"
or "co-stimulatory" factors (Theofilopoulos, 1995a). An alternative
possibility is that there is no induction of anergy, but that the
mature T-cells are unable to receive appropriate signals and/or help.
This would result in T-cells simply ignoring such antigens and
remaining quiescent. It follows that, if adequate antigen presentation
and co-stimulation occurs through professional antigen-presenting
cells, then these self-reactive but quiescent cells may be activated
and cause tissue damage (Theofilopoulos, 1995a).
2.4.4 The molecular mimicry hypothesis
Molecular mimicry is defined by homology in a linear amino acid
sequence between "self" molecules and foreign molecules. The above
theories of cryptic or ignored "self" are compatible with the
molecular mimicry hypothesis of autoimmunity, particularly as it
pertains to infectious agents. Closely related or identical peptides
are often found in unrelated proteins. Thus, many peptide fragments of
infectious agents are homologous with host proteins. Among microbial
antigens implicated in autoimmunity induced by molecular mimicry, heat
shock proteins (hsp), found in virtually all life forms, have received
prime attention (Minowada & Welch, 1995). Comparisons of the amino
acid sequence of hsp60 with the entire database of known human
sequences revealed that 86 human peptides have similar regions to
hsp60 and, of these, 19 are known disease-associated autoantigens
(Jones et al., 1993). However, the importance of mimicry to the
pathogenesis of spontaneous autoimmune disease is uncertain, as it is
unclear why immunological responses to hsp, which are expressed in
every cell, could lead to organ-specific autoimmune diseases.
2.4.5 The "modified self" hypothesis
This theory suggests that autoimmunity may arise as a result of
an immune response against modified "self" determinants ("neo-self"
determinants), which may be particularly relevant for chemical-induced
autoimmune responses. Drugs, their metabolites or other haptenic
chemicals may bind to "self" determinants. A number of possibilities
should be considered.
2.4.5.1 Hapten-induced antibody responses to "modified self"
In such reactions the hapten conjugates to "self" and forms an
integral component of the determinant that is recognized by the
antibody. In this mechanism hapten-specific T-cells provide cognate
help to the B-cell that is then induced to synthesize antibodies which
recognize the hapten-modified but not the native form of the "self"
protein. Therefore, these reactions against a particular hapten are
not truly autoimmune in nature. Penicillin, quinidine, halothane (Gut
et al., 1995), and tienilic acid are good examples of compounds that
can induce antibody responses to hapten-modified "self".
2.4.5.2 Hapten-induced autoantibodies that recognize "self" proteins
In their native form these can be considered true autoimmune
responses, since the determinant that is recognized by antibody does
not incorporate a drug-derived determinant. However, the determinant
that is recognized by the Th-cells that promote the B-cell response
may be drug-derived. The following theories have been put forward:
a) Drugs might break tolerance by binding to "self" macromolecules,
thereby creating new determinants that could be recognized
by T-cells. T-cells recognizing this new determinant would
clonally expand and go on to provide help for B cells that
recognize adjacent autoantigens on the same drug "self"
conjugate. These in turn would clonally expand and differentiate
into autoantibody-producing plasma cells (Fig. 7). In this way
the normal process of suppression that operates through either
clonal or functional deletion of Th-cells is effectively bypassed
(Allison, 1989). A considerable body of experimental evidence,
largely from work with mice, supports this concept.
Administration of arsenilic acid-conjugated autologous
thyroglobulin or dinitrophenylated autologous immunoglobulin to
mice has been found to lead to breakdown of tolerance and
elicitation of autoantibodies to these self-proteins (Weigle,
1965; Iverson, 1970). Furthermore, mice sensitized to
p-aminobenzoic acid (PAB) and then administered PAB-conjugated
isologous red blood cells developed a T-cell-dependent antibody
response to their own red blood cells, with consequent haemolytic
anaemia (Yamashita et al., 1976).
b) It has been postulated that a drug or metabolite might
interact chemically with "self"-MHC molecules on
antigen-presenting cells (macrophages or B-cells) in such a way
that they appear as "non-self" to T-cells. These T-cells,
following clonal expansion, would then provide help
indiscriminately to all B-cells carrying the drug-modified
"self"-MHC molecule. Assuming that the drug modifies MHC
molecules without regard for the antigen specificity of the
B-cell, the resulting cognate T-/B-cell interaction would lead to
polyclonal B-cell activation and induction of synthesis of
antibodies of multiple, including "anti-self", specificities
(Gleichmann et al., 1984, 1989). This mechanism would be
analogous to a graft-versus-host (GVH) reaction. Indeed,
experiments with mercuric chloride (Pelletier et al., 1994),
D-penicillamine (Tournade et al., 1990) and gold salts (Schuhmann
et al., 1990) in Brown Norway rats or particular strains of mice
led to immune disregulatory changes (elevated immunoglobulin
levels, particularly IgE, induction of autoantibodies to the
glomerular basement membrane, DNA, IgG, collagen and nuclear and
nucleolar proteins) resembling those seen in graft-versus-host
(GVH) disease (Gleichmann et al., 1984, 1989; Goldman et al.,
1991; Bloksma et al., 1995). The elevations in IgE, IgG1 and IL-4
in mercury chloride-treated susceptible mice and rats implicate
the Th2 subset in this response (Goldman et al., 1991).
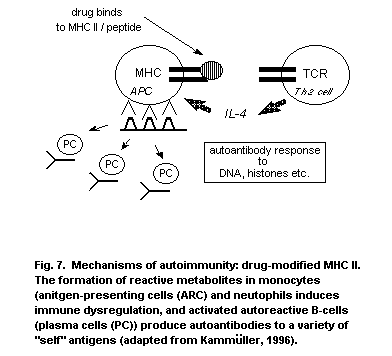 In order to become antigenic to T-cells, haptens need to
bind to carrier proteins and it has been discussed whether
or not T-cells may require covalent modification of MHC
molecules for hapten recognition. Several studies investigating
trinitrophenol- and gold-hapten formation have pointed to a major
role of hapten-modified MHC-associated peptides as
T-cell-antigenic structures (Martin & Weltzien, 1994; Sinigaglia,
1994; Weltzien et al., 1996).
c) Another theory of drug-induced autoimmunity suggests that
certain drugs or chemicals might induce, or protect from
suppression, populations of T-cells that recognize unmodified
"self" MHC. This would be analogous to graft-versus-host
reactions, but the difference with the aforementioned mechanism
would be that the chemical's effect is targeted at the T-cell
rather than the B-cell. In the Brown Norway (BN) rat model of
autoimmunity induced by D-penicillamine, gold and mercuric
chloride, autoreactive T-cells that recognize unmodified "self"
MHC Class II molecules on normal B-cells have been reported,
rather than T-cells that recognize chemically modified "self"
(Pelletier et al., 1994). This supports the concept that several
compounds might induce autoreactivity by modifying T-cells rather
than B-cells.
2.4.6 Immunoregulatory disturbances
2.4.6.1 Errors in central or peripheral tolerance
Errors in central or peripheral tolerance at the T- or B-cell
level have also been suggested as causes for autoimmunity.
The association between development of immunodeficiency, benign
or neoplastic lymphoproliferation and autoimmune diseases,
particularly in the context of thymic abnormalities, is well known
(Fudenberg, 1966). It has been observed upon immunosuppressive
treatment, among others with cyclophosphamide and cyclosporin A. The
reversibility of lymphoproliferative lesions upon withdrawal of the
immunosuppressive drug therapy suggests a causal relationship (Starzl
et al., 1984). Studies in rodents have provided more solid evidence of
the relationship between the development of autoimmune disease and
induced disturbance of thymic function (Sakaguchi & Sakaguchi, 1989,
1990; Barrett et al., 1995). Notably, cyclosporin A, which is
successfully used in the prevention of transplant rejection and
treatment of various autoimmune diseases in humans, has been shown to
interfere with the deletion of T-cells recognizing autoantigens in the
thymic medulla and to cause organ-specific and systemic autoimmune
disease under specific conditions. This occurs when cyclosporin A is
given to neonates (Sakaguchi & Sakaguchi, 1989), but not to older
animals (Hess & Fischer, 1989), and to bone marrow transplant
recipients that received a high dose of irradiation prior to
transplantation (Glazier et al., 1983; Hess & Fischer, 1989). The
development of autoimmune disease under these conditions has been
attributed to the absence of an established regulatory peripheral
T-cell repertoire. Because cyclosporin A may interfere at different
levels of immunological tolerance, autoreactive T-cells leaving the
thymus as a consequence of cyclosporin A treatment may not be
functionally inactivated in the periphery (Prud'Homme et al., 1991).
However, a study using the bone marrow transplant model in different
mouse strains suggested the involvement of other mechanisms, because
effects of cyclosporin-A on T-cell deletion did not correlate with
development of autoimmune effects (Bryson et al., 1991). The study
suggested a polyfactorial etiology of cyclosporin-A-induced autoimmune
disease and may explain why autoimmune side-effects have been observed
only rarely in cyclosporin A-treated human bone marrow transplant
patients (Jones et al., 1989).
Patients with primary immunodefiency, especially various B-cell
deficiencies, are known to have a high incidence of autoimmune disease
(Rosen, 1987). For example, selective IgA deficiency is associated
with systemic autoimmune diseases, such as systemic lupus
erythematosus (SLE) (Cleland & Bell, 1978; Rosen, 1987). Moreover,
drugs with a documented ability to cause systemic autoimmune
disorders, i.e., diphenylhydantoin (Seager et al., 1975) and
D-penicillamine, have been shown to reduce secretory and/or serum IgA
levels. However, the relationship between IgA deficiency and
susceptibility to autoimmune disease is not known. It is most likely
influenced by other factors as well, since the prevalence of selective
IgA deficiency in a normal population is much higher (1 in 700) than
the prevalence of systemic autoimmune disease.
Both cyclosporin A and diphenylhydantoin have immunosuppressive
activities and affect the thymus. Although neonatal exposure
experiments with diphenylhydantoin have been performed (Chapman &
Roberts, 1984; Kohler et al., 1987), autoimmune side effects have not
been reported. This may be related to the different intrathymic
targets of both compounds. Cyclosporin A is thought to disturb
thymocyte differentiation by affecting interdigitating and epithelial
cells (Schuurman et al., 1992), while diphenylhydantoin affects the
more immature cortical thymocytes probably by a
glucocorticoid-mediated effect. As pointed out by Schuurman et al.
(1992), such differences in intrathymic targets may have different
consequences for immune function ranging from immunodeficiency to
autoimmune disorders. It illustrates the complex relationship between
immunodeficiency, lymphoproliferation and autoimmune effects and the
difficulty of immunotoxicological hazard identification (chapter 6)
and risk assessment (chapter 7).
2.4.6.2 Polyclonal activators
Polyclonal B- and/or T-cell activation has been considered a
contributing or initiating mechanism of autoimmunity, particularly in
systemic diseases. Although exogenous polyclonal B-cell activators
(i.e., lipopolysaccharide) may exacerbate or precipitate SLE, they
appear to be insufficient in themselves (Hang et al., 1985;
Theofilopoulos, 1995a).
Polyclonal T-cell activation in autoimmune disease is exemplified
in graft-versus-host (GVH)-induced autoimmunity, where alloreactive
donor T-cells initiate recipient B-cell differentiation into
antibody-secreting cells, particularly those recognizing polymeric
"self" antigens (Gleichmann et al., 1989; Goldman et al., 1991;
Bloksma et al., 1995). It has been suggested that in this model, as
well as in some models of chemically induced systemic autoimmunity,
there is a predominant engagement of Th2-cells that promote the
humoral response (IL-4 hyperproduction) (Goldman et al., 1991).
Polyclonal stimulation of a large set of T-cells by bacterial/viral
superantigens is another possible scenario. T-cells that react with
MHC Class II-bound superantigens on B-cells may mutually stimulate
superantigen-displaying B-cells, thereby leading to production of
polyclonal immunoglobulins and, in some instances, autoantibodies
(Friedman et al., 1991).
The development of autoimmune reactions as outlined above is only
the first step in the production of autoimmune disease. Multiple
mechanisms can lead to the same overall clinical manifestations both
in organ-specific and in systemic autoimmune syndromes, and therefore,
expectations for a single etiological explanation appears unrealistic.
For organ-specific autoimmune diseases, the most straightforward
explanation to emerge is the concept that these diseases are caused by
otherwise conventional immunological responses against self-antigens
for which T-cell tolerance is normally not established (i.e., anatomic
sequestration, inadequate presentation due to the cryptic nature of
the self-determinant, and/or lack of co-stimulatory factors). With
regard to systemic autoimmune diseases such as SLE, the situation is
less clear, but neither exogenous polyclonal B- or T-cell activators
nor immunoregulatory disturbances appear to provide satisfactory
explanations. Physical, chemical and infectious assaults may
precipitate heterogenous syndromes such as SLE, characterized by an
almost all-encompassing autoimmune response against a vast array of
mostly dissimilar self-antigens, possibly mediated by the engagement
of a large set of non-tolerant T-cells that recognize diverse
self-peptides displayed on MHC molecules.
2.5 Type I hypersensitivity diseases and allied disorders
Allergy and atopy have become synonymous for the same set of
hypersensitivity disorders, several of which commonly occur in the
same individual. They comprise predisposition to develop IgE-mediated
immediate (Type I) hypersensitivity responses to common environmental
antigens, in part genetically mediated and manifested as eczema,
rhinitis, conjunctivitis and asthma.
Allergic diseases which are considered to result from Type I
(immediate) hypersensitivity reactions are shown in Table 12.
Table 12. Examples of Type I hypersensitivity and reaction sites
Disease Reaction site
Urticaria Skin
Atopic eczema Skin
Angioedema Skin or mucous membranes
Asthma Respiratory tract
Rhinitis Respiratory tract
Conjunctivitis Conjunctiva
Anaphylaxis } Variable, including skin,
Insect venom allergy } gastrointestinal tract,
Food allergy } respiratory system,
Drug allergy } cardiovascular system or
} generalized
The ability of protein antigens encountered in the environment or
workplace to cause IgE antibody-mediated rhinitis and asthma is now
well established. Thus, for example, a variety of pollens is known to
cause seasonal hay fever in susceptible individuals. It is now
apparent that certain chemicals are able to induce similar symptoms in
a proportion of exposed individuals (Butcher & Salvaggio, 1986; Karol,
1992). Among chemicals of small relative molecular mass known to cause
respiratory allergy in humans are: acid anhydrides such as phthalic
anhydride, tetrachlorophthalic anhydride, hexahydrophthalic anhydride
and trimellitic anhydride (Bernstein et al., 1982a, 1984; Moller et
al., 1985); certain isocyanates including toluene diisocyanate,
diphenylmethane-4,4'diisocyanate and hexamethylene diisocyanate
(Tanser et al., 1973; Zamit-Tabona et al., 1983; Keskinen et al.,
1988); some reactive dyes (Alanko et al., 1978); and platinum salts
(Biagini et al., 1985). A number of chemicals induce hypersensitivity
disorders that have features similar to Type I hypersensitivity
reactions but do not easily fall within the classification of Gell &
Coombs (1963).
The characteristics of respiratory allergic hypersensitivity to
chemicals are of specific pulmonary reactions usually induced only in
a minority of the exposed population and which are provoked by
atmospheric concentrations of the allergen that were previously
tolerable and that fail to cause symptoms in non-sensitized
individuals. Thus, it has been found that with toluene diisocyanate an
asthmatic response can be caused by atmospheric concentrations of the
chemical far below those that are necessary to induce irritant effects
(Newman Taylor, 1988).
Allergic respiratory hypersensitivity induced by chemicals may be
of immediate- and/or late-onset. An obligatory universal role for IgE
antibody in the pathogenesis of chemical respiratory allergen is
uncertain, not least because many symptomatic individuals lack
detectable IgE for the relevant allergen. In some cases it may be that
insufficiently sensitive or inappropriate methods have been employed
for detection of IgE antibody. Nonetheless, it is possible that
T-lymphocytes and cell-mediated immune responses may also effect
respiratory hypersensitivity reactions to chemicals. There is
generally a latent period between the onset of exposure and the
appearance of respiratory symptoms. In the case of certain
diisocyanates, asthma has been found to develop within a few months.
In other instances, however, there may be a latent period of several
years. While this is almost certainly the case for protein respiratory
allergens, there is no a priori reason to suppose that provocation
of the immune responses necessary for respiratory sensitization to
chemical allergens will result only from exposure via the respiratory
tract. Indeed, there is evidence that occupational respiratory
sensitization may be caused by dermal exposure to chemical allergens
following industrial spillage or splashing (Karol, 1986).
Allergic respiratory hypersensitivity, by definition, results
from the induction of a specific immunological response. While there
is no doubt that the acute onset of respiratory symptoms associated
with hypersensitivity to protein aeroallergens is due to
homocytotropic (primarily IgE) antibody, the nature of the immune
responses responsible for chemical respiratory allergy is still
controversial. Although IgE specific for all recognized chemical
respiratory allergens has been found, and despite a clear association
for some chemical allergens between the presence of specific IgE
antibody and the development of respiratory symptoms, a clear link
between allergic responses and serum IgE antibody has, in some
instances (notably with some diisocyanates), failed to emerge. It is
nevertheless the case that the induction of acute-onset
hypersensitivity reactions in the respiratory tract is usually
considered as being dependent upon IgE antibody and the elicitation of
classical immediate-type hypersensitivity responses.
In the light of present uncertainties, perhaps the most realistic
conclusion that can be drawn is that in many, but perhaps not all,
cases the development of chemically induced respiratory allergy is
dependent upon IgE antibody and the elicitation of immediate-type
hypersensitivity reactions in the respiratory tract. It is possible,
however, that in some instances respiratory hypersensitivity to
chemical allergens results from the action of T-lymphocytes operating
independently of IgE antibody. Irrespective of a putative
IgE-independent cell-mediated immune mechanism for the induction of
chemical respiratory hypersensitivity, it now appears likely that
T-lymphocytes play an important role in late phase reactions and in
the pathogenesis of chronic bronchial inflammation.
2.5.1 Asthma
2.5.1.1 Definition
Asthma is a respiratory disease that eludes easy definition. It
is characterized by variable airflow limitation due to bronchial
responsiveness and often by inflammatory changes in the airways.
Asthma has been classified as intrinsic or extrinsic; extrinsic asthma
is provoked by sensitivity to a foreign substance, including
idiosyncratic drug rections, while intrinsic asthma is characterized
by reactivity to non-allergic factors, such as infection and physical
and/or psychological stimuli (Barbee, 1987). However, this
classification is considered artificial because the clinical signs of
both types of asthma are similar.
The US National Institutes of Health (NIH, 1991) published a
consensus definition that included the following characteristics:
airway obstruction that is reversible (but not completely so in some
patients) either spontaneously or with treatment; airway inflammation,
and increased airway responsiveness to a variety of stimuli. In
practice, and especially in epidemiological surveys, it has been
diagnosed from the replies to questionnaires that have focused on such
symptoms as episodic wheezing and shortness of breath (see section
5.2.1.2b). Asthma is distinguished from chronic obstructive pulmonary
disease (COPD), i.e., chronic bronchitis and emphysema, by the
prominent reversibility of the airways obstruction.
The term reactive airways dysfunction syndrome (RADS) was coined
to refer to persistent asthma after high-level irritant exposure
(Brooks et al., 1985), but the term irritant-induced asthma is just as
suitable. To prevent unnecessary confusion, the use of terms other
than asthma should be avoided.
The prevalence of asthma has been increasing in a number of
countries in recent years (Buist & Vollmer, 1990; Strachan, 1995;
ISAAC, 1998). Although some of the increase may be the result of a
change in diagnostic classification and increased reporting, a true
increase in disease prevalence is likely. The causes of this increase
are currently unknown, but environmental pollution is one potential
contributory factor.
Allergy is associated with asthma. Up to 80% of patients with
asthma have positive immediate reactions to skin-prick testing with a
battery of common aeroallergens (Nelson, 1985), although this
percentage probably over-represents the importance of allergy in
asthma. Whereas allergy clearly plays a primary role in childhood
asthma, many adults with asthma do not appear to be sensitized to
specific aeroallergens. This observation provided the basis for the
traditional characterization of the disease into two major types:
i) extrinsic asthma (with sensitization to specific aeroallergens) and
ii) intrinsic asthma (without specific sensitization).
There is a genetic component to the risk of developing asthma.
Children with one asthmatic parent have an increased risk of
developing the disease themselves, and when both parents are
asthmatic, the risk is even higher. A parental history of atopy also
increases the risk. Up to 40% of the population is atopic: however,
many sensitized people do not develop asthma or asymptomatic airway
hyperresponsiveness (Witt et al., 1986). Thus allergy alone does not
explain the development of persistent asthma, although continuous or
recurrent exposure to allergen may serve to sustain asthma in a
genetically susceptible subpopulation.
Infections aggravate asthma and elicit exacerbations of the
disease (Johnston et al., 1995). When it comes to the role of viral
infections in the induction of the disease, evidence is conflicting
(Martinez, 1995). There is evidence that some infections, in
particular respiratory syncytial virus (RSV), may predispose for the
development of asthma (Sigurs et al., 1995). On the other hand, there
is increasing evidence that childhood infections may protect against
the development of allergy and allergic diseases, including asthma
(Holt, 1996; Shaheen et al., 1996; Shirakawa et al., 1997; Matricardi
et al., 1997). Some anecdotal evidence and small studies suggested
that childhood vaccination may increase the prevalence of asthma and
allergy (Kemp et al., 1997).
2.5.1.2 Airways inflammation and asthma
Over the past decade airway inflammation has emerged as an
important feature of clinical asthma. It has long been known from
autopsy studies of patients that die from status asthmaticus that
airway inflammation is present in such patients. The use of
fibre-optic bronchoscopy to obtain bronchoalveolar lavage and
bronchial-mucosal biopsy specimens has allowed the study of patients
with less severe asthma. Airway inflammation is clearly present in
these patients as well. Asthmatic airways are characterized by: (a)
infiltration with inflammatory cells, especially eosinophils; (b)
oedema; and (c) loss of epithelial integrity. Airflow obstruction in
asthma is believed to be the result of changes associated with airway
inflammation, mucus production and bronchoconstriction. Airway
inflammation is believed to play an important role in the genesis of
airway hyperresponsiveness in asthma (Holgate et al., 1987).
Much of the research on mechanisms that mediate airway
inflammation in asthma has focused on allergen-induced responses.
Inhalation of allergen in a specifically sensitized patient with
asthma will trigger rapid-onset but self-limited bronchoconstriction,
called the early response. In 30 to 50% of extrinsic asthmatic
subjects undergoing an allergen inhalation challenge, a delayed
reaction will occur 4 to 8 h later, called the late response (O'Byrne
et al., 1987). The late response is characterized by persistent
airflow obstruction, airway inflammation and airway
hyperresponsiveness (Cartier et al., 1982). Mast cell degranulation
and release of mediators such as histamine are believed to be
responsible for the immediate response (Liu et al., 1990). The role of
the mast cell in the genesis of the late response is more
controversial, but the release of chemoattractant substances such as
leukotrienes and cytokines (i.e., interleukins: IL-3, IL-4 and IL-5)
may be involved in the influx of neutrophils and eosinophils into the
airway epithelium, which is a key feature of this response. The
infiltration of the airway wall with eosinophils is also a key feature
of the late response (Metzger et al., 1987; Djukanovic et al., 1990).
The number of Th2-cells in the airway epithelium appears to be higher
in patients with allergy-related asthma and may be responsible for the
maintenance of chronic airway inflammation (Ollerenshaw & Woolcock,
1992). The Th2-cells are involved in the release of cytokines that may
activate both mast cells (IL-3 and IL-4) and eosinophils (IL-5). The
eosinophil can release proteins (e.g., major basic protein,
eosinophilic cationic protein, eosinophilic peroxidase or
eosinophil-derived neurotoxin), lipid mediators, oxygen radicals and
enzymes that can cause epithelial injury.
2.5.2 Occupational asthma
Occupational asthma induced by protein allergens is invariably
associated with atopy and with the presence of specific IgE antibody.
In contrast, occupational asthma induced by chemical allergens is not
restricted to atopic individuals and is not always associated with the
presence of demonstrable IgE antibody. For both forms of asthma the
inflammatory response in the respiratory tract is similar and
characterized by T-lymphocyte and eosinophil infiltration.
The immunopathology of occupational asthma has the characteristic
features of airway smooth muscle contraction, oedema, and fluid
accumulation, resulting presumably from the local release by mast
cells of inflammatory mediators such as histamine and leukotrienes.
Alternatively, it has been hypothesized that, in some instances of
chemically induced respiratory allergic hypersensitivity, the initial
inflammatory response results from a chronic cell-mediated immune
mechanism operating independently, or in the absence, of IgE antibody
(Corrigan & Kay, 1992). Chronic inflammation is recognized as playing
an important role in asthma and is associated with infiltration of the
bronchial mucosa with inflammatory cells, mucus production, the
destruction and sloughing of airway epithelial cells, and
subepithelial fibrosis secondary to collagen deposition (Roche et al.,
1989; Beasley et al., 1989). Of particular importance in the
development of bronchial mucosal inflammation and injury is the
eosinophil, acting in concert with infiltrating T-lymphocytes (Beasley
et al., 1989; Gleich, 1990). While the exact role of eosinophils in
the development of bronchial hyperreactivity has yet to be
established, there is no doubt that the eosinophilia associated with
allergen-induced respiratory reactions is influenced markedly by
cytokines and, in particular, by IL-5 (Chand et al., 1992; Gulbenkian
et al., 1992; Iwami et al., 1993). A role for T- lymphocytes in asthma
begs questions regarding the nature of allergen handling in the
respiratory tract and the characteristics of local antigen-presenting
cells. In the context of primary sensitization following inhalation
exposure to the inducing allergen, it is likely that the network of
dendritic cells found within the airway epithelium is of vital
importance (Holt et al., 1990; Schon-Hegrad et al., 1991).
2.5.2.1 Occupational asthma and allergy
Hypersensitivity-induced occupational asthma (see also section
4.3.3) fulfils the criteria for an acquired specific hypersensitivity
response:
a) It occurs in only a proportion -- usually a minority -- of
those exposed to the allergen.
b) It develops only after an initial symptom-free period of
exposure ranging from days even up to several years.
c) In those who develop asthma, airway responses (both
reduction in calibre and induction of hyperresponsiveness to
non-specific stimuli) are provoked by inhalation of the specific
agent in concentrations that were previously tolerable and that
do not provoke similar responses in others equally exposed.
These characteristics have stimulated a search for evidence of a
specific immunological response to the causes of occupational asthma,
both proteins and chemicals of low relative molecular mass. Attention
has been directed towards the identification of specific IgE and IgG
antibodies. In general, IgE and IgG4 have been found in exposed
populations to be associated with disease and total specific IgG with
exposure. For example, specific IgE and IgG4 were associated with
asthma and IgG with exposure to acid anhydride workers (Forster et
al., 1988).
Studies have suggested a central role for the T-lymphocyte and in
particular the Th2-lymphocyte in the development of the eosinophilic
bronchitis characteristic of asthma. Evidence for the involvement of
T-lymphocytes in occupational asthma was found in nine patients with
isocyanate-induced asthma who had activated T-lymphocytes and
eosinophils in bronchial biopsy specimens (Bentley et al., 1991).
Nonetheless, the IgE antibody-mast cell interaction is probably an
important associated response dependent upon Th2-lymphocyte
stimulation, and specific IgE remains a valuable marker of the
immunological response associated with asthma caused by several agents
inhaled at work.
Specific IgE has been identified in the sera of patients with
asthma caused by some low relative molecular mass chemicals,
particularly acid anhydrides (Newman Taylor et al., 1987) and reactive
dyes (Luczynska & Topping, 1986), but not others, notably isocyanates.
In a study to examine the determinants of allergenicity of low
relative molecular mass chemicals, the properties of two beta lactam
antibiotics were compared: clavulanic acid, which is not allergenic;
and a carbapenam MM2283, which can cause asthma and stimulate IgE
antibody production in man. The characteristics identified as relevant
to allergenicity were (a) reactivity with body proteins; (b) hapten of
single chemical structure and (c) stability of the conjugate formed
(Davies et al., 1977).
Specific IgE antibody has been identified in only some 15% of
cases of isocyanate-induced asthma. This may reflect the difficulties
of working with reactive chemicals in in vitro systems or failure to
prepare the relevant in vivo chemical-protein conjugate for the
in vitro test.
Duration and intensity of exposure are the major factors
contributing to the development of occupational asthma in populations
exposed to its causes. Additional factors such as atopy and tobacco
smoking may also contribute. The importance of these factors varies
for different causes of the disease. However, for no cause do they
adequately explain the development of the disease in the minority who
develop it. In part this may reflect the limited knowledge of
exposures experienced but probably also suggests other important,
including genetic (such as HLA haplotype), determinants.
2.5.3 Atmospheric pollutants and asthma
There is evidence that air pollutants are involved in
exacerbating asthma (Vos et al., 1996). Evidence from laboratory
studies suggests that certain air pollutants have the potential to
stimulate bronchoconstriction or airways inflammation (see also
chapter 5.) Exposure to SO2 is associated with chest tightness and
bronchoconstriction, with the concentration required to induce a
response being dependent upon the degree of hyperresponsiveness. It
may be that the effects of SO2 will be augmented in the presence of
other pollutants. It has been reported that the sensitivity of mild
asthmatics to SO2 is increased by prior exposure to ozone (O3).
Ozone is a prototype oxidant pollutant that reacts rapidly with tissue
components. It is formed by photochemical reactions involving oxides
of nitrogen and organic molecules and occurs with other photochemical
oxidants and fine particles in the complex mixture called "smog".
Bates & Sizto (1987) studied hospital admissions in Southern
Ontario, Canada, an area with a population of seven million people,
and observed an association between rates of admissions for asthmatic
subjects during the summer season and ambient air levels of both O3
and suspended sulfates. However, the study design could not separate
the 03 effects from concomitant effects of acidic aerosol and SO2.
Thurston et al. (1992a,b) found strong associations between elevated
summer "haze" pollution (H+, sulfate, O3) and increased asthma (and
total respiratory) admissions to hospitals in Buffalo and New York
City, USA, especially in 1988 when air pollution was severe. However,
the specific role of O3 as opposed to H+ was less clear.
Controlled (environmental chamber) human exposure studies have
clearly demonstrated that some healthy young adults and children
respond to O3 exposure (at levels occurring in ambient air) with
irritative cough and substernal chest pain on inspiration and
decrements in FVC and FEV1 (Koren et al., 1989; Folinsbee, 1992).
When exercising outdoors in summer such individuals show decrements in
FEV1 that are consistent with the observed ambient air O3 levels.
Controlled exposure to similar levels of ozone has also been shown to
cause an inflammatory response of the respiratory tract in all species
that have been studied including humans (Lippmann, 1989). The use of
bronchoalveolar lavage (BAL) as a research tool has afforded the
opportunity to sample lung and lower airways after exposure to O3 and
to ascertain the extent and course of inflammation and its
constitutive elements. The BAL studies (Devlin et al., 1991) have
clearly demonstrated that O3, even at very low concentration, causes
increases in numbers of neutrophils, and a variety of other
constituents of BAL fluid, some with potential inflammatory properties
such as prostaglandin E2, fibronectin, elastase and IL-6.
Inflammation was also detected in the upper airways of O3-exposed
subjects as shown by an increase in neutrophils and other inflammatory
indicators in the nasal lavage (NAL) fluid (Koren et al., 1990).
Interestingly, both NAL fluid and BAL fluid from non-asthmatic
subjects exposed to O3 have been shown to contain the mast cell
marker tryptase. This and another study (Bascom et al., 1990)
suggested that O3-induced inflammation may share certain features of
the response observed in subjects with allergic rhinitis challenged
with allergen.
A study demonstrated that asthmatic subjects exposed to low
levels of O3 (0.16 ppm) for 7.6 h while performing moderate exercise
showed more respiratory symptoms and greater decrements in FEV1 than
did similarly exposed non-asthmatics (Ball et al., 1993).
The concept of influencing the asthmatic response by combining
exposure to O3 with specific allergen challenge has created interest
in the potential "indirect" effects of O3 exposure. In one study,
individuals with allergic rhinitis were initially exposed to clean air
or 0.5 ppm O3 for 4 h (Bascom et al., 1990). The high level of
exposure to O3 did not enhance the subsequent acute response to
antigen in the nose under these experimental conditions. A study by
Molfino et al. (1991) examined the effect of pre-exposure to O3 (0.12
ppm for one hour at rest) on the subsequent airway response to inhaled
ragweed or grass pollen antigen in seven subjects with allergic
asthma. They reported O3-induced increases in bronchial
responsiveness to specific allergen challenge. Preliminary data from
studies currently conducted examining the effects of pre-exposure to
O3 (0.4 ppm for 2 h at rest) followed by a specific allergen nasal
challenge in asthmatics sensitive to house dust mite suggest that the
O3 pre-exposure caused a significant decrease in the dose of allergen
needed to induce symptoms (Peden et al., 1994). Eosinophil influx and
increase in eosinophil cationic protein were observed 4 h after nasal
allergen challenge following both O3 and clean air pre-exposure.
These changes were more dramatic following O3 pre-exposure although
the mean allergen dose was smaller.
The health relevance of oxides of nitrogen, and in particulate
NO2, has attracted some interest since the gas is present not only
outdoors but also indoors. A number of studies suggest mild effects of
NO2 in asthmatics at concentrations less than 1 ppm but others have
not found responses at levels up to 4 ppm.
Particulate air pollutants, especially fine particles derived
from combustion sources, are also of interest although there have been
few controlled exposure studies outside those involving acid aerosols.
Bioaerosols, to which an asthmatic is sensitized, are well known to
exacerbate asthma. Epidemiological studies describing the increase in
mortality associated with particulate matter (PM) provide provocative
evidence for adverse pulmonary health effects associated with
particulate pollution (Dockery et al., 1993, 1994; Brunekreef et al.,
1995; Pope et al., 1995). The association between PM and acute
mortality and morbidity has been demonstrated most strongly with
elderly people who have chronic cardiopulmonary disease (Pope et al.,
1992; Burnett et al., 1995; Schwartz & Morris, 1995). Experimental
studies with diesel exhaust particles show that they increase IL-4 and
specific IgE production, and exacerbate the response to allergen in
allergic individuals (Diaz-Sanchez et al., 1997). Studies in mice have
demonstrated that diesel exhaust particles facilitate the induction of
allergy (Takafuji et al., 1987; Lovik et al., 1997). Chemicals
adsorbed to the diesel exhaust particles, as well as carbon particles
with very little chemicals on them appear to enhance the allergic
immune response (Diaz-Sanchez et al., 1997, 1999; Lovik et al., 1997).
Environmental air pollutants including tobacco smoke may affect
the prevalence and/or severity of asthma in several different ways. In
hyperresponsive airways, pollutants may act as triggers of asthmatic
reactions without the presence of the specific allergen.
Alternatively, a pollutant could induce or increase airway
inflammation and, as a result, cause airway hyperreactivity that
persists after exposure has ceased. Some pollutants may have a direct
toxic effect on the respiratory epithelium leading to inflammation,
airway hyperreactivity and the appearance of asthma-like symptoms in
previously non-asthmatic individuals. Lastly, there are certain
pollutants that may have the ability to augment or modify immune
responses to inhaled antigens or to enhance the severity of reactions
elicited in the respiratory tract following inhalation exposure of the
sensitized individual to the inducing allergen.
2.5.4 Rhinitis
Rhinitis frequently, but not invariably, occurs in atopic
diseases. Similarities and differences between rhinitis and asthma are
considered below.
Allergic responses of the nasal mucosa cause an orchestrated set
of responses. The acute allergic reaction occurs within minutes and is
manifested as rhinorrhoea, pruritus and sneezing, and congestion, due
(respectively) to increased vascular permeability, sensory nerve
stimulation, and vasodilation with sinusoidal pooling plus oedema
formation. These responses are due to mediators released from the
mucosal mast cells, and histamine is a major participant.
Following this acute response is the slower development of the
late phase allergic reaction which is manifested by congestion and
hyperirritability and is due to cellular infiltration with
eosinophils, neutrophils and some basophils. There is interest in
whether lymphocytes also participate in this reaction, but the data
are not clear as yet.
Of the cells that participate in rhinitis, mast cells,
neutrophils, eosinophils and lymphocytes may all be important. Mast
cells initiate the response through the release of the mediators of
anaphylaxis. Work also indicates that mast cells generate a number of
cytokines (generally thought of as lymphocyte products, but clearly
generated by activated mast cells as well). These products include
IL-3, IL-4, IL-5, IL-6 and TNF. Neutrophils are the first cells to
infiltrate areas undergoing allergic reactions. The role of the
neutrophil in allergy is not clear. However, neutrophils appear to be
necessary for the development of increased airway hyperactivity in
animal models of asthma. Neutrophils also release factors that
activate mast cells (neutrophil-derived histamine releasing factor),
and the influx of neutrophils occurs simultaneously with recrudescent
histamine release in the late phase reaction. Eosinophils have
received a lot of attention, as they are the hallmark of allergic
inflammation. Eosinophils infiltrate areas more slowly than do
neutrophils, but persist much longer. The eosinophil can cause
epithelial denudation, mucus secretion and histamine release. Both
eosinophil and neutrophil infiltrates are inhibited by
corticosteroids.
Interest has focused on the possible contribution by lymphocytes
to the late-phase reaction. After mast cell activation, about 10% of
the superficial lymphocytes express the IL-2 receptor, indicating
their activation. There are suggestions that some cytokines are
released during this time period, either from mast cells or
lymphocytes.
2.5.5 Atopic eczema
In atopic eczema, the patient is much troubled by itching skin;
there is a history of chronic or chronically relapsing dermatitis,
worst on the flexures, which are excoriated and lichenified, and there
is a family or personal history of atopy. This is the typical picture
of atopic eczema, though some of the features may be absent (Hanifin &
Rajka, 1980). In any discussion of pathogenesis, family history is
important because atopic eczema is part of the atopic syndrome that
includes genetically determined phenotypes such as extrinsic bronchial
asthma, allergic rhinitis, allergic conjunctivitis and
gastrointestinal allergy. Important laboratory indices are blood and
tissue eosinophilia and antigen-specific IgE bound to mast cells in
skin (intracutaneous challenge) or peripheral blood
(radioallergosorbence assays). The Wiscott-Aldrich syndrome and
hyper-IgE syndrome, which can closely resemble atopic eczema, are
usually distinguishable by the associated life-threatening infections.
The clinical course of atopic eczema is unpredictable. Sometimes
it remits in childhood, but occasional patients have recurrences
throughout life. Some patients (or their parents) are convinced that
exacerbations are related to stress and/or exposure to environmental
antigens such as food or animals. Secondary skin infection by
Staphylococcus aureus, herpes simplex virus, varicella virus and,
possibly, fungal infections can lead to severe exacerbations. Finally,
autonomic nervous system disturbances and changes in fatty acid
metabolism and phosphodiesterase activity have been implicated.
Despite the development of numerous theories, the pathophysiology
of eczema is still remarkably little understood. Researchers are
currently focusing on Langerhans cells, which are thought to be
involved in eczema, because these cells possess abundant receptors for
IgE. Once in contact with allergen distributed after ingestion or
following direct skin contact, Langerhans cells present the allergen
to T-lymphocytes. They may also be directly stimulated to produce
inflammatory cytokines, which are responsible for eczematous lesions.
Atopic eczema is often accompanied by very high IgE levels. In babies,
an elevated IgE level is taken as a reliable predictive sign for the
development of asthma and/or hay fever in later life.
The relation between cell-mediated immunity and IgE in atopic
eczema was first established by Bruijnzeel-Koomen et al. (1986) who
identified the presence of IgE on Langerhans cells in atopic eczema.
It is now evident that this binding of IgE is the result of the
presence of the high-affinity receptor for IgE on these Langerhans
cells (Bieber & Ring, 1992). Langerhans cells and other
antigen-presenting cells in skin also express low-affinity Fc
receptors that efficiently bind allergen-precomplexed IgE. The
functional consequence of the expression of these Fc receptors for IgE
on antigen-presenting cells in skin is that the local response to
minute quantities of allergens in the skin is amplified. By
facilitated antigen-processing, only minute quantities of allergens
are needed to be presented to T-cells, because the
IgE-receptor-allergen complex aids processing and subsequent
presentation up to a 1000-fold (Van der Heijden et al., 1993).
Therefore, the onset of atopic eczema as an expression of atopic
allergy may result from an interplay between the degrees of expression
of one or more Fc receptor types, the serum concentration of
allergen-specific IgE, and the number of skin-infiltrating T-cells
specific for that allergen and, of course, exposure to the allergen.
Atopic syndrome is genetically determined. When both parents have
atopic disease of the same sort, their child has a risk of around 70%
of developing a similar phenotype. If parents have different atopic
diseases, the incidence of atopic disease in a child is 30% (Björksten
& Kjellmann, 1987). With asthmatics as probands in molecular genetic
studies, a gene predisposing to atopy has been found on chromosome I
Iql3 (Cookson et al., 1989), possibly coding for the beta subunit of
high-affinity IgE Type I Fc receptor (Sandford et al., 1993). However,
the genetic mapping of atopy is far from simple. For example, the
increasing prevalence of atopic eczema in the past three decades
(Williams, 1992) is difficult to explain on the basis of genetics
alone. Furthermore, a maternal pattern of inheritance has been found
(Cookson et al., 1992), which might be due to paternal genomic
imprinting or to maternal modification of developing immune responses
in utero or via breast milk. Linkage of atopy with a gene on I Iql3
could not be shown when patients with atopic eczema were taken as
probands. Thus more than one gene seems to be involved.
Environmental factors, such as exposure to allergens, are thought
to be involved in the phenotypic expression of atopic eczema. For
example, the presence of a strong atopic background has been
associated with enhanced protective responses to helminthic infections
(Lynch et al., 1998). However, a precise understanding of the
environmental factors that determine whether or not the atopic
genotype is expressed as an atopic phenotype is lacking.
2.5.6 Urticaria
Urticaria (hives, nettle rash) may be defined as an eruption of
short-lived red oedematous swellings of the skin, associated with
itching. The relative incidence of the different types of urticaria
and angioedema in the general population is unknown.
Urticaria usually involves degranulation of mast cells and
release of histamine. Many different elicitors have to be considered.
Allergy due to a reaction between a specific antigen and a mast
cell-fixed IgE antibody is only one mechanism. Pseudo-allergic
reactions, toxic effects and viral infections play a major role.
Acute urticaria resolves within a period of six weeks. If it
persists, it is called chronic urticaria. Wheals may be circular,
polycyclic or figured. If subcutaneous extension occurs, angioedema is
present. Although, like urticaria, angioedema may occur anywhere, the
genitalia, eyelids, lips and mucous membranes are especially common
sites. Itching is almost always present in patients with urticaria but
is inconsistent in angioedema. The duration of urticarial wheals is
usually 3 to 4 h, but angioedema lesions may last much longer.
Skin previously involved by wheals or angioedema looks entirely
normal apart from occasional purpura or other signs of trauma due to
scratching. The mucous membranes are frequently involved including the
tongue, soft palate and pharynx. Although discomfort and breathing
difficulty may occur, fatalities are almost exclusively associated
with hereditary angioedema. Acute urticaria may be associated with
systemic anaphylactic symptoms (wheezing, dyspnoea, syncope, abdominal
pain, vomiting). Occasionally acute urticaria may merge into serum
sickness, arthritis, fever, proteinuria). Common causes of allergic
acute urticaria include ingestion of penicillin, shellfish, soft fruit
and nuts.
Urticaria of immunological origin may arise rapidly (often less
than 60 min) at the site of contact of the skin or mucous membranes
with a specific substance.
Contact urticaria may also be of non-immunological origin, and
there are frequent instances in which the mechanism is uncertain. When
an immune mechanism is involved, the final common pathway is probably
the same. Contact urticaria of immunological origin involves
IgE-mediated hypersensitivity as indicated by a positive
radioallergosorbent test (RAST). In non-immunological examples, the
offending substance may evoke histamine release directly from
cutaneous mast cells. Such substances include ammonium persulfate
(Mahzoon et al., 1977), dimethyl sulfoxide (Odom & Maibach, 1976) and
cinnamaldehyde (Kirton, 1978); however, several other mechanisms are
also involved.
Immunological contact urticaria is more frequent in atopic
subjects. These patients often give a history of acute oedema of the
lips or buccal mucous membrane after ingestion of food items such as
fish, egg or nuts. In common with other types of allergy, healthy
control subjects are negative on skin testing. The offending allergen
is usually a high relative molecular mass substance and skin testing
is rarely positive in completely normal skin. Open and closed patch
tests and closed patch tests on lightly abraded skin (scratch-patch
tests) should be performed. The diagnosis is confirmed by a positive
radioallergosorbent test (RAST).
Non-immunological contact urticaria may be elicited in healthy
asymptomatic individuals, with the triggering substance frequently
being of low relative molecular mass, and contact reactions may be
elicitable in clinically normal skin. The danger of such generalized
reactions should be borne in mind before skin testing is performed.
2.5.7 Gastrointestinal tract diseases: mechanisms of food-induced
symptoms
2.5.7.1 Non IgE-mediated food-sensitive enteropathy
Slow onset gastrointestinal symptoms are described in children,
especially in relation to ingestion of cow's milk. The clinical
features are chronic diarrhoea and failure to thrive. The pathological
lesion found in the small intestine is crypt hyperplastic villous
atrophy of variable severity. The lesions are often patchy. There is
an increased expression of the markers of T-cell activation on the
T-cells of the lamina propria, and it is likely that a cell-mediated
reaction in the lamina propria is the basis of the abnormality,
although IgE involvement has also been described (Walker-Smith, 1992).
Nagata et al. (1995) suggested that activated CD4 cells in the lamina
propria of the small intestinal mucosa may contribute to the mucosal
damage, probably by releasing cytokines.
2.5.7.2 IgE-mediated food allergy
Food allergic patients often describe itching and tingling of the
mouth and throat as the first immediate symptoms of an allergic food
reactions. In addition papules/blisters on the mucosa and swelling of
the lips can be seen. These symptoms occur as a result of direct
contact between the allergen and the mucosa of the mouth and throat.
The concentration of mast cells is very high in the oropharyngeal
mucosa and the symptoms are probably caused by degranulation of
mucosal mast cells bearing specific IgE towards the offending allergen
(Pastorello et al., 1995).
Symptoms like nausea, vomiting, abdominal pains, loose stools and
gas production are described in connection with immediate allergic
reactions. In a direct challenge of the gastric mucosa using a
gastrofibrescope, Romanski (1987, 1989) found gastric changes within
5-20 min of contact with the introduced food. The macroscopic changes
were: pale mucosa, oedema, punctate haemorrhage, hyperperistalsis,
hypersecretion, erythema. Microscopic examination revealed oedema,
hyperaemia, capillary haemorrhage, eosinophilic infiltration and
inflammation.
The underlying mechanisms of IgE-mediated gastrointestinal
symptoms are a result of degranulation of intestinal mast cells with
release of mediators that act directly on the epithelium, endothelium
or muscle indirectly through nerves and mesenchymal cells. The result
is altered gastric acid secretion, ion transport, mucus production,
gut barrier function, and motility (Crowe & Perdue, 1992).
2.5.7.3 Role of gastrointestinal tract physiology in food allergy
Many elements of the gastrointestinal tract physiology influence
the ultimate allergenicity of food proteins. These include the pH,
digestive enzymes, bile, peristalsis, transit time, bacterial
fermentation, and the intestinal barrier function, permeability, and
absorption. Several food allergens or allergenic determinants were
reported to be relatively resistant to acid denaturation and
proteolytic digestion (Elsayed & Apold, 1977; Schwartz et al., 1980;
Kurisaki et al., 1981; Metcalfe, 1985; Taylor, 1986; Taylor, 1992;
Kortekangas-Savolainen et al., 1993). Unfortunately, insufficient
information is available on possible differences in susceptibility to
acid denaturation and gastrointestinal digestion between strongly
allergenic food proteins and proteins that possess weak or virtually
no allergenic potential. Attempts have also been made to correlate the
susceptibility to enzymatic breakdown of cow's milk proteins, their
intestinal permeability and allergenic properties (Taylor, 1986;
Marcon-Genty et al., 1989; Savilahti & Kuitunen, 1992). The important
role of digestion with respect to food protein allergenicity was
clearly demonstrated in mice showing that pre-feeding of an
endopeptidase inhibitor (aprotinin) to mice resulted in an inhibition
of normally expected oral tolerance induction by protein feeding
(Hanson et al., 1993). An abnormal digestive breakdown of proteins may
also be of importance, since intragastric administration more easily
results in anaphylactic sensitization as compared to ad libitum
feedings, generally resulting in tolerance induction, as has been
shown in rodents (Knippels et al., 1997). However, as digestion of
food proteins is part of the normal sequence of events following
consumption of food, it is likely that food allergic patients become
sensitized to digested allergens rather than to the native proteins.
Enzymatically digested food allergens may show the same, more, or less
binding to specific IgE from patients (Haddad et al., 1979; Schwartz
et al., 1980).
The intestinal barrier function, permeability, and absorption are
also hardly, or not, taken into account in the evaluation of the
allergenicity of food proteins. Knowledge of the intestinal uptake of
specific protein antigens and their fragments may provide some
additional information in the evaluation of the potential
allergenicity of protein products. There is evidence of limited
macromolecular exclusion by the epithelial barrier (Seifert et al.,
1974, 1977; Gardner, 1988; Teichberg, 1990).
2.6 Type II hypersensitivity diseases
Pathogenic Type II reactions may occur towards autoantigens,
alloantigens (in blood transfusions), infective agents and drugs or
chemicals, as described above. As shown in Table 13, these immune
reactions may cause corresponding disorders, i.e., autoimmune
diseases, transplantation/transfusion reactions or drug-induced
haemolytic reactions. As an illustration of Type II reaction-induced
diseases, three autoimmune disorders that are also inducible by drugs,
i.e., haemolytic reactions, pemphigus and myasthenia gravis, will be
dealt with in more detail.
2.6.1 Drug-induced Type II reactivity
Some drugs or their metabolites are chemically reactive agents
that readily bind to cells and tissues. Such drugs, present on the
cell membrane of blood cells, are obvious targets for pathogenic Type
II reactivity.
The most frequent allergic reaction occurs with penicillin and
its relatives. Benzylpenicillin is a small molecule with a relative
molecular mass of 372.47 and with a highly reactive beta-lactam ring,
which may bind to amino groups on proteins (carrier), forming covalent
conjugates. The thus formed penicilloyl hapten is considered as the
major determinant in penicillin allergy. Although penicillin is able
Table 13. Clinical disease due to Type II hypersensitivity reactions
Antigen Disease Symptoms
Autoantigens glomerular basement membrane Goodpasture's syndrome vasculitis, renal failure
epidermal desmosomes (desmoglein-3) pemphigus vulgaris skin blistering (intra-epidermal)
epidermal hemidesmosomes on bullous pemphigoid skin blistering (subepidermal)
basal keratinocytes
acetylcholine receptor myasthenia gravis striated muscle weakness
Rhesus antigen autoimmune haemolytic destruction of red cells, anaemia
anaemia
platelet integrin gpIIb:IIIa autoimmune thrombocytopenia abnormal bleeding
purpura
Alloantigens donor red cell antigens delayed haemolytic destruction of transfused red cells
transfusion reaction
Infective agents Streptococcal cell wall antigens acute rheumatic fever arthritis, myocarditis
cross-reacting with cardiac muscle
Klebsiella antigens cross-reacting ankylosing spondylitis (?) arthritis involving the spine
with HLA-B27
Drugs, chemicals penicillins, cephalosporins drug-specific haemolytic lysis of hapten-coated red cells
trimellitic anhydride anaemia
to induce all types of hypersensitivity reactions (IgE-, immune
complex- or T-cell-mediated), haemolytic anaemia with
penicillin-specific IgG antibodies reacting with penicillin-coated
erythrocytes is a typical example of Type II reactivity.
Interestingly, the specificity of drug-induced antibodies is
often much broader than would be expected from the penicillin example.
Ultimately, drugs trigger Type II reactivity without being involved in
the final destructive reaction. In addition to hapten-specific
antibodies, drugs can induce antibodies to metabolites, to
drug-carrier combinations or to the carrier alone, resulting in
clear-cut autoimmune reactivity. D-penicillamine is a classical
example of a drug inducing autoimmunity, but chemicals such as mercury
and gold are also able to induce autoimmunity.
The mechanism by which drugs can induce autoantibodies is shown
in detail in Fig. 6. By presenting the hapten in or on their MHC-class
II molecules, autoreactive B-cells, which are normally present at very
low frequencies without being activated, can trigger hapten-specific
T-cells to help them (the B-cells) differentiate into
antibody-producing plasma cells. Although the induction of the disease
is drug-dependent, the Type II effector reaction towards autologous
targets may be drug-independent. Hence, in this case the induced
autoimmune disease would continue after the exposure to the drug had
ceased.
2.6.2 Transfusion reactions
Transfusion reactions are examples of the cellular destruction
that results from antibody combining with heteroantigens. There are at
least 21 blood group systems, with more than 600 antigens within these
systems. Some antigens are stronger than others and are more likely to
stimulate antibody production. Certain antibodies are produced
naturally with no prior exposure to red blood cells, while other
antibodies are only produced after contact with cells carrying that
antigen.
The ABO blood groups are of primary importance in considering
transfusions. Anti-A and anti-B antibodies are so-called naturally
occurring antibodies. Individuals do not form such antibodies to their
own red blood cells. Thus, an individual who has Type A blood would
have anti-B in the serum, and a person with Type B blood has anti-A
antibodies. An individual with Type O blood has both anti-A and anti-B
in the serum, as O cells have neither of these two antigens.
If a patient is given blood for which antibodies are already
present, a transfusion reaction occurs. This can range from acute
massive intravascular haemolysis to a small decrease in red blood cell
survival. Acute haemolytic transfusion reactions may occur within
minutes or hours after transfusion of incompatible blood.
Delayed haemolytic reactions occur 4 to 10 days following a
transfusion and are due to a secondary response to the antigen.
Antibody-coated red blood cells are removed extravascularly, in the
spleen or in the liver, and the patient may experience a mild fever
and anaemia.
Haemolytic disease of the newborn appears in infants whose
mothers have been sensitized by exposure to fetal blood cells carrying
antigens, commonly of the Rhesus family, that differ from their own.
The mother makes IgG antibodies in response, and these cross the
placenta to cause destruction of fetal red cells. A common antigen
involved is the Rhesus D antigen
2.6.3 Autoimmune haemolytic anaemia
Drugs account for about 12% of the autoimmune haemolytic anaemia.
They can cause haemolysis by three different mechanisms: by acting as
a hapten, by inducing a classical autoimmune haemolytic anaemia, or by
forming immune complexes with antibodies that can be adsorbed by the
patient's red cells, the "innocent bystanders" (Fig. 8).
Antigen-presenting cells phagocytose and process haptenized
cells, such as erythrocytes. In addition, free drug molecules may bind
to MHC-class II or to peptides within the groove. The hapten is thus
presented by MHC-class II molecules to the T-cell receptor (TCR)
of T helper cells. Hapten-specific T-cells now proliferate and
differentiate, so that they can either attack haptenized cells (The
cells causing Type IV reactivity, not shown) or can help nearby
B-cells to produce antibodies (in particular The cells).
B-cells ingest and process the antigens to which their
immunoglobulin receptors bind and present peptides, including
haptenized peptides, derived from these antigens in their MHC-class II
molecules. Thus B-cells may trigger hapten-specific T-cells by
presenting haptenized peptides. Alternatively, external drug molecules
can bind to peptides in the MHC-class II groove. Differentiation of
B-cells is dependent on adjacent T-cells providing membrane signals
(to CD40, not shown) and the growth factors IL-4 and IL-6.
In drug-specific haemolytic anaemia (on the left of Fig. 8),
drug-specific B-cells present the drug to drug-specific T-cells.
Mutual activation of T- and B-cells now induces the B-cell to become a
plasma cell, producing drug-specific antibodies. Eventually these
antibodies lead to destruction of haptenized erythrocytes, while
normal cells remain intact.
In drug-induced autoimmune haemolytic anaemia (on the right of
Fig. 8), an autoreactive B-cell ingests and processes erythrocyte
membranes, including the haptenized parts. Normally, autoreactive
B cells exist but do not become activated by lack of appropriate
stimulating T-cells. If drug-specific T-cells are present, however,
In order to become antigenic to T-cells, haptens need to
bind to carrier proteins and it has been discussed whether
or not T-cells may require covalent modification of MHC
molecules for hapten recognition. Several studies investigating
trinitrophenol- and gold-hapten formation have pointed to a major
role of hapten-modified MHC-associated peptides as
T-cell-antigenic structures (Martin & Weltzien, 1994; Sinigaglia,
1994; Weltzien et al., 1996).
c) Another theory of drug-induced autoimmunity suggests that
certain drugs or chemicals might induce, or protect from
suppression, populations of T-cells that recognize unmodified
"self" MHC. This would be analogous to graft-versus-host
reactions, but the difference with the aforementioned mechanism
would be that the chemical's effect is targeted at the T-cell
rather than the B-cell. In the Brown Norway (BN) rat model of
autoimmunity induced by D-penicillamine, gold and mercuric
chloride, autoreactive T-cells that recognize unmodified "self"
MHC Class II molecules on normal B-cells have been reported,
rather than T-cells that recognize chemically modified "self"
(Pelletier et al., 1994). This supports the concept that several
compounds might induce autoreactivity by modifying T-cells rather
than B-cells.
2.4.6 Immunoregulatory disturbances
2.4.6.1 Errors in central or peripheral tolerance
Errors in central or peripheral tolerance at the T- or B-cell
level have also been suggested as causes for autoimmunity.
The association between development of immunodeficiency, benign
or neoplastic lymphoproliferation and autoimmune diseases,
particularly in the context of thymic abnormalities, is well known
(Fudenberg, 1966). It has been observed upon immunosuppressive
treatment, among others with cyclophosphamide and cyclosporin A. The
reversibility of lymphoproliferative lesions upon withdrawal of the
immunosuppressive drug therapy suggests a causal relationship (Starzl
et al., 1984). Studies in rodents have provided more solid evidence of
the relationship between the development of autoimmune disease and
induced disturbance of thymic function (Sakaguchi & Sakaguchi, 1989,
1990; Barrett et al., 1995). Notably, cyclosporin A, which is
successfully used in the prevention of transplant rejection and
treatment of various autoimmune diseases in humans, has been shown to
interfere with the deletion of T-cells recognizing autoantigens in the
thymic medulla and to cause organ-specific and systemic autoimmune
disease under specific conditions. This occurs when cyclosporin A is
given to neonates (Sakaguchi & Sakaguchi, 1989), but not to older
animals (Hess & Fischer, 1989), and to bone marrow transplant
recipients that received a high dose of irradiation prior to
transplantation (Glazier et al., 1983; Hess & Fischer, 1989). The
development of autoimmune disease under these conditions has been
attributed to the absence of an established regulatory peripheral
T-cell repertoire. Because cyclosporin A may interfere at different
levels of immunological tolerance, autoreactive T-cells leaving the
thymus as a consequence of cyclosporin A treatment may not be
functionally inactivated in the periphery (Prud'Homme et al., 1991).
However, a study using the bone marrow transplant model in different
mouse strains suggested the involvement of other mechanisms, because
effects of cyclosporin-A on T-cell deletion did not correlate with
development of autoimmune effects (Bryson et al., 1991). The study
suggested a polyfactorial etiology of cyclosporin-A-induced autoimmune
disease and may explain why autoimmune side-effects have been observed
only rarely in cyclosporin A-treated human bone marrow transplant
patients (Jones et al., 1989).
Patients with primary immunodefiency, especially various B-cell
deficiencies, are known to have a high incidence of autoimmune disease
(Rosen, 1987). For example, selective IgA deficiency is associated
with systemic autoimmune diseases, such as systemic lupus
erythematosus (SLE) (Cleland & Bell, 1978; Rosen, 1987). Moreover,
drugs with a documented ability to cause systemic autoimmune
disorders, i.e., diphenylhydantoin (Seager et al., 1975) and
D-penicillamine, have been shown to reduce secretory and/or serum IgA
levels. However, the relationship between IgA deficiency and
susceptibility to autoimmune disease is not known. It is most likely
influenced by other factors as well, since the prevalence of selective
IgA deficiency in a normal population is much higher (1 in 700) than
the prevalence of systemic autoimmune disease.
Both cyclosporin A and diphenylhydantoin have immunosuppressive
activities and affect the thymus. Although neonatal exposure
experiments with diphenylhydantoin have been performed (Chapman &
Roberts, 1984; Kohler et al., 1987), autoimmune side effects have not
been reported. This may be related to the different intrathymic
targets of both compounds. Cyclosporin A is thought to disturb
thymocyte differentiation by affecting interdigitating and epithelial
cells (Schuurman et al., 1992), while diphenylhydantoin affects the
more immature cortical thymocytes probably by a
glucocorticoid-mediated effect. As pointed out by Schuurman et al.
(1992), such differences in intrathymic targets may have different
consequences for immune function ranging from immunodeficiency to
autoimmune disorders. It illustrates the complex relationship between
immunodeficiency, lymphoproliferation and autoimmune effects and the
difficulty of immunotoxicological hazard identification (chapter 6)
and risk assessment (chapter 7).
2.4.6.2 Polyclonal activators
Polyclonal B- and/or T-cell activation has been considered a
contributing or initiating mechanism of autoimmunity, particularly in
systemic diseases. Although exogenous polyclonal B-cell activators
(i.e., lipopolysaccharide) may exacerbate or precipitate SLE, they
appear to be insufficient in themselves (Hang et al., 1985;
Theofilopoulos, 1995a).
Polyclonal T-cell activation in autoimmune disease is exemplified
in graft-versus-host (GVH)-induced autoimmunity, where alloreactive
donor T-cells initiate recipient B-cell differentiation into
antibody-secreting cells, particularly those recognizing polymeric
"self" antigens (Gleichmann et al., 1989; Goldman et al., 1991;
Bloksma et al., 1995). It has been suggested that in this model, as
well as in some models of chemically induced systemic autoimmunity,
there is a predominant engagement of Th2-cells that promote the
humoral response (IL-4 hyperproduction) (Goldman et al., 1991).
Polyclonal stimulation of a large set of T-cells by bacterial/viral
superantigens is another possible scenario. T-cells that react with
MHC Class II-bound superantigens on B-cells may mutually stimulate
superantigen-displaying B-cells, thereby leading to production of
polyclonal immunoglobulins and, in some instances, autoantibodies
(Friedman et al., 1991).
The development of autoimmune reactions as outlined above is only
the first step in the production of autoimmune disease. Multiple
mechanisms can lead to the same overall clinical manifestations both
in organ-specific and in systemic autoimmune syndromes, and therefore,
expectations for a single etiological explanation appears unrealistic.
For organ-specific autoimmune diseases, the most straightforward
explanation to emerge is the concept that these diseases are caused by
otherwise conventional immunological responses against self-antigens
for which T-cell tolerance is normally not established (i.e., anatomic
sequestration, inadequate presentation due to the cryptic nature of
the self-determinant, and/or lack of co-stimulatory factors). With
regard to systemic autoimmune diseases such as SLE, the situation is
less clear, but neither exogenous polyclonal B- or T-cell activators
nor immunoregulatory disturbances appear to provide satisfactory
explanations. Physical, chemical and infectious assaults may
precipitate heterogenous syndromes such as SLE, characterized by an
almost all-encompassing autoimmune response against a vast array of
mostly dissimilar self-antigens, possibly mediated by the engagement
of a large set of non-tolerant T-cells that recognize diverse
self-peptides displayed on MHC molecules.
2.5 Type I hypersensitivity diseases and allied disorders
Allergy and atopy have become synonymous for the same set of
hypersensitivity disorders, several of which commonly occur in the
same individual. They comprise predisposition to develop IgE-mediated
immediate (Type I) hypersensitivity responses to common environmental
antigens, in part genetically mediated and manifested as eczema,
rhinitis, conjunctivitis and asthma.
Allergic diseases which are considered to result from Type I
(immediate) hypersensitivity reactions are shown in Table 12.
Table 12. Examples of Type I hypersensitivity and reaction sites
Disease Reaction site
Urticaria Skin
Atopic eczema Skin
Angioedema Skin or mucous membranes
Asthma Respiratory tract
Rhinitis Respiratory tract
Conjunctivitis Conjunctiva
Anaphylaxis } Variable, including skin,
Insect venom allergy } gastrointestinal tract,
Food allergy } respiratory system,
Drug allergy } cardiovascular system or
} generalized
The ability of protein antigens encountered in the environment or
workplace to cause IgE antibody-mediated rhinitis and asthma is now
well established. Thus, for example, a variety of pollens is known to
cause seasonal hay fever in susceptible individuals. It is now
apparent that certain chemicals are able to induce similar symptoms in
a proportion of exposed individuals (Butcher & Salvaggio, 1986; Karol,
1992). Among chemicals of small relative molecular mass known to cause
respiratory allergy in humans are: acid anhydrides such as phthalic
anhydride, tetrachlorophthalic anhydride, hexahydrophthalic anhydride
and trimellitic anhydride (Bernstein et al., 1982a, 1984; Moller et
al., 1985); certain isocyanates including toluene diisocyanate,
diphenylmethane-4,4'diisocyanate and hexamethylene diisocyanate
(Tanser et al., 1973; Zamit-Tabona et al., 1983; Keskinen et al.,
1988); some reactive dyes (Alanko et al., 1978); and platinum salts
(Biagini et al., 1985). A number of chemicals induce hypersensitivity
disorders that have features similar to Type I hypersensitivity
reactions but do not easily fall within the classification of Gell &
Coombs (1963).
The characteristics of respiratory allergic hypersensitivity to
chemicals are of specific pulmonary reactions usually induced only in
a minority of the exposed population and which are provoked by
atmospheric concentrations of the allergen that were previously
tolerable and that fail to cause symptoms in non-sensitized
individuals. Thus, it has been found that with toluene diisocyanate an
asthmatic response can be caused by atmospheric concentrations of the
chemical far below those that are necessary to induce irritant effects
(Newman Taylor, 1988).
Allergic respiratory hypersensitivity induced by chemicals may be
of immediate- and/or late-onset. An obligatory universal role for IgE
antibody in the pathogenesis of chemical respiratory allergen is
uncertain, not least because many symptomatic individuals lack
detectable IgE for the relevant allergen. In some cases it may be that
insufficiently sensitive or inappropriate methods have been employed
for detection of IgE antibody. Nonetheless, it is possible that
T-lymphocytes and cell-mediated immune responses may also effect
respiratory hypersensitivity reactions to chemicals. There is
generally a latent period between the onset of exposure and the
appearance of respiratory symptoms. In the case of certain
diisocyanates, asthma has been found to develop within a few months.
In other instances, however, there may be a latent period of several
years. While this is almost certainly the case for protein respiratory
allergens, there is no a priori reason to suppose that provocation
of the immune responses necessary for respiratory sensitization to
chemical allergens will result only from exposure via the respiratory
tract. Indeed, there is evidence that occupational respiratory
sensitization may be caused by dermal exposure to chemical allergens
following industrial spillage or splashing (Karol, 1986).
Allergic respiratory hypersensitivity, by definition, results
from the induction of a specific immunological response. While there
is no doubt that the acute onset of respiratory symptoms associated
with hypersensitivity to protein aeroallergens is due to
homocytotropic (primarily IgE) antibody, the nature of the immune
responses responsible for chemical respiratory allergy is still
controversial. Although IgE specific for all recognized chemical
respiratory allergens has been found, and despite a clear association
for some chemical allergens between the presence of specific IgE
antibody and the development of respiratory symptoms, a clear link
between allergic responses and serum IgE antibody has, in some
instances (notably with some diisocyanates), failed to emerge. It is
nevertheless the case that the induction of acute-onset
hypersensitivity reactions in the respiratory tract is usually
considered as being dependent upon IgE antibody and the elicitation of
classical immediate-type hypersensitivity responses.
In the light of present uncertainties, perhaps the most realistic
conclusion that can be drawn is that in many, but perhaps not all,
cases the development of chemically induced respiratory allergy is
dependent upon IgE antibody and the elicitation of immediate-type
hypersensitivity reactions in the respiratory tract. It is possible,
however, that in some instances respiratory hypersensitivity to
chemical allergens results from the action of T-lymphocytes operating
independently of IgE antibody. Irrespective of a putative
IgE-independent cell-mediated immune mechanism for the induction of
chemical respiratory hypersensitivity, it now appears likely that
T-lymphocytes play an important role in late phase reactions and in
the pathogenesis of chronic bronchial inflammation.
2.5.1 Asthma
2.5.1.1 Definition
Asthma is a respiratory disease that eludes easy definition. It
is characterized by variable airflow limitation due to bronchial
responsiveness and often by inflammatory changes in the airways.
Asthma has been classified as intrinsic or extrinsic; extrinsic asthma
is provoked by sensitivity to a foreign substance, including
idiosyncratic drug rections, while intrinsic asthma is characterized
by reactivity to non-allergic factors, such as infection and physical
and/or psychological stimuli (Barbee, 1987). However, this
classification is considered artificial because the clinical signs of
both types of asthma are similar.
The US National Institutes of Health (NIH, 1991) published a
consensus definition that included the following characteristics:
airway obstruction that is reversible (but not completely so in some
patients) either spontaneously or with treatment; airway inflammation,
and increased airway responsiveness to a variety of stimuli. In
practice, and especially in epidemiological surveys, it has been
diagnosed from the replies to questionnaires that have focused on such
symptoms as episodic wheezing and shortness of breath (see section
5.2.1.2b). Asthma is distinguished from chronic obstructive pulmonary
disease (COPD), i.e., chronic bronchitis and emphysema, by the
prominent reversibility of the airways obstruction.
The term reactive airways dysfunction syndrome (RADS) was coined
to refer to persistent asthma after high-level irritant exposure
(Brooks et al., 1985), but the term irritant-induced asthma is just as
suitable. To prevent unnecessary confusion, the use of terms other
than asthma should be avoided.
The prevalence of asthma has been increasing in a number of
countries in recent years (Buist & Vollmer, 1990; Strachan, 1995;
ISAAC, 1998). Although some of the increase may be the result of a
change in diagnostic classification and increased reporting, a true
increase in disease prevalence is likely. The causes of this increase
are currently unknown, but environmental pollution is one potential
contributory factor.
Allergy is associated with asthma. Up to 80% of patients with
asthma have positive immediate reactions to skin-prick testing with a
battery of common aeroallergens (Nelson, 1985), although this
percentage probably over-represents the importance of allergy in
asthma. Whereas allergy clearly plays a primary role in childhood
asthma, many adults with asthma do not appear to be sensitized to
specific aeroallergens. This observation provided the basis for the
traditional characterization of the disease into two major types:
i) extrinsic asthma (with sensitization to specific aeroallergens) and
ii) intrinsic asthma (without specific sensitization).
There is a genetic component to the risk of developing asthma.
Children with one asthmatic parent have an increased risk of
developing the disease themselves, and when both parents are
asthmatic, the risk is even higher. A parental history of atopy also
increases the risk. Up to 40% of the population is atopic: however,
many sensitized people do not develop asthma or asymptomatic airway
hyperresponsiveness (Witt et al., 1986). Thus allergy alone does not
explain the development of persistent asthma, although continuous or
recurrent exposure to allergen may serve to sustain asthma in a
genetically susceptible subpopulation.
Infections aggravate asthma and elicit exacerbations of the
disease (Johnston et al., 1995). When it comes to the role of viral
infections in the induction of the disease, evidence is conflicting
(Martinez, 1995). There is evidence that some infections, in
particular respiratory syncytial virus (RSV), may predispose for the
development of asthma (Sigurs et al., 1995). On the other hand, there
is increasing evidence that childhood infections may protect against
the development of allergy and allergic diseases, including asthma
(Holt, 1996; Shaheen et al., 1996; Shirakawa et al., 1997; Matricardi
et al., 1997). Some anecdotal evidence and small studies suggested
that childhood vaccination may increase the prevalence of asthma and
allergy (Kemp et al., 1997).
2.5.1.2 Airways inflammation and asthma
Over the past decade airway inflammation has emerged as an
important feature of clinical asthma. It has long been known from
autopsy studies of patients that die from status asthmaticus that
airway inflammation is present in such patients. The use of
fibre-optic bronchoscopy to obtain bronchoalveolar lavage and
bronchial-mucosal biopsy specimens has allowed the study of patients
with less severe asthma. Airway inflammation is clearly present in
these patients as well. Asthmatic airways are characterized by: (a)
infiltration with inflammatory cells, especially eosinophils; (b)
oedema; and (c) loss of epithelial integrity. Airflow obstruction in
asthma is believed to be the result of changes associated with airway
inflammation, mucus production and bronchoconstriction. Airway
inflammation is believed to play an important role in the genesis of
airway hyperresponsiveness in asthma (Holgate et al., 1987).
Much of the research on mechanisms that mediate airway
inflammation in asthma has focused on allergen-induced responses.
Inhalation of allergen in a specifically sensitized patient with
asthma will trigger rapid-onset but self-limited bronchoconstriction,
called the early response. In 30 to 50% of extrinsic asthmatic
subjects undergoing an allergen inhalation challenge, a delayed
reaction will occur 4 to 8 h later, called the late response (O'Byrne
et al., 1987). The late response is characterized by persistent
airflow obstruction, airway inflammation and airway
hyperresponsiveness (Cartier et al., 1982). Mast cell degranulation
and release of mediators such as histamine are believed to be
responsible for the immediate response (Liu et al., 1990). The role of
the mast cell in the genesis of the late response is more
controversial, but the release of chemoattractant substances such as
leukotrienes and cytokines (i.e., interleukins: IL-3, IL-4 and IL-5)
may be involved in the influx of neutrophils and eosinophils into the
airway epithelium, which is a key feature of this response. The
infiltration of the airway wall with eosinophils is also a key feature
of the late response (Metzger et al., 1987; Djukanovic et al., 1990).
The number of Th2-cells in the airway epithelium appears to be higher
in patients with allergy-related asthma and may be responsible for the
maintenance of chronic airway inflammation (Ollerenshaw & Woolcock,
1992). The Th2-cells are involved in the release of cytokines that may
activate both mast cells (IL-3 and IL-4) and eosinophils (IL-5). The
eosinophil can release proteins (e.g., major basic protein,
eosinophilic cationic protein, eosinophilic peroxidase or
eosinophil-derived neurotoxin), lipid mediators, oxygen radicals and
enzymes that can cause epithelial injury.
2.5.2 Occupational asthma
Occupational asthma induced by protein allergens is invariably
associated with atopy and with the presence of specific IgE antibody.
In contrast, occupational asthma induced by chemical allergens is not
restricted to atopic individuals and is not always associated with the
presence of demonstrable IgE antibody. For both forms of asthma the
inflammatory response in the respiratory tract is similar and
characterized by T-lymphocyte and eosinophil infiltration.
The immunopathology of occupational asthma has the characteristic
features of airway smooth muscle contraction, oedema, and fluid
accumulation, resulting presumably from the local release by mast
cells of inflammatory mediators such as histamine and leukotrienes.
Alternatively, it has been hypothesized that, in some instances of
chemically induced respiratory allergic hypersensitivity, the initial
inflammatory response results from a chronic cell-mediated immune
mechanism operating independently, or in the absence, of IgE antibody
(Corrigan & Kay, 1992). Chronic inflammation is recognized as playing
an important role in asthma and is associated with infiltration of the
bronchial mucosa with inflammatory cells, mucus production, the
destruction and sloughing of airway epithelial cells, and
subepithelial fibrosis secondary to collagen deposition (Roche et al.,
1989; Beasley et al., 1989). Of particular importance in the
development of bronchial mucosal inflammation and injury is the
eosinophil, acting in concert with infiltrating T-lymphocytes (Beasley
et al., 1989; Gleich, 1990). While the exact role of eosinophils in
the development of bronchial hyperreactivity has yet to be
established, there is no doubt that the eosinophilia associated with
allergen-induced respiratory reactions is influenced markedly by
cytokines and, in particular, by IL-5 (Chand et al., 1992; Gulbenkian
et al., 1992; Iwami et al., 1993). A role for T- lymphocytes in asthma
begs questions regarding the nature of allergen handling in the
respiratory tract and the characteristics of local antigen-presenting
cells. In the context of primary sensitization following inhalation
exposure to the inducing allergen, it is likely that the network of
dendritic cells found within the airway epithelium is of vital
importance (Holt et al., 1990; Schon-Hegrad et al., 1991).
2.5.2.1 Occupational asthma and allergy
Hypersensitivity-induced occupational asthma (see also section
4.3.3) fulfils the criteria for an acquired specific hypersensitivity
response:
a) It occurs in only a proportion -- usually a minority -- of
those exposed to the allergen.
b) It develops only after an initial symptom-free period of
exposure ranging from days even up to several years.
c) In those who develop asthma, airway responses (both
reduction in calibre and induction of hyperresponsiveness to
non-specific stimuli) are provoked by inhalation of the specific
agent in concentrations that were previously tolerable and that
do not provoke similar responses in others equally exposed.
These characteristics have stimulated a search for evidence of a
specific immunological response to the causes of occupational asthma,
both proteins and chemicals of low relative molecular mass. Attention
has been directed towards the identification of specific IgE and IgG
antibodies. In general, IgE and IgG4 have been found in exposed
populations to be associated with disease and total specific IgG with
exposure. For example, specific IgE and IgG4 were associated with
asthma and IgG with exposure to acid anhydride workers (Forster et
al., 1988).
Studies have suggested a central role for the T-lymphocyte and in
particular the Th2-lymphocyte in the development of the eosinophilic
bronchitis characteristic of asthma. Evidence for the involvement of
T-lymphocytes in occupational asthma was found in nine patients with
isocyanate-induced asthma who had activated T-lymphocytes and
eosinophils in bronchial biopsy specimens (Bentley et al., 1991).
Nonetheless, the IgE antibody-mast cell interaction is probably an
important associated response dependent upon Th2-lymphocyte
stimulation, and specific IgE remains a valuable marker of the
immunological response associated with asthma caused by several agents
inhaled at work.
Specific IgE has been identified in the sera of patients with
asthma caused by some low relative molecular mass chemicals,
particularly acid anhydrides (Newman Taylor et al., 1987) and reactive
dyes (Luczynska & Topping, 1986), but not others, notably isocyanates.
In a study to examine the determinants of allergenicity of low
relative molecular mass chemicals, the properties of two beta lactam
antibiotics were compared: clavulanic acid, which is not allergenic;
and a carbapenam MM2283, which can cause asthma and stimulate IgE
antibody production in man. The characteristics identified as relevant
to allergenicity were (a) reactivity with body proteins; (b) hapten of
single chemical structure and (c) stability of the conjugate formed
(Davies et al., 1977).
Specific IgE antibody has been identified in only some 15% of
cases of isocyanate-induced asthma. This may reflect the difficulties
of working with reactive chemicals in in vitro systems or failure to
prepare the relevant in vivo chemical-protein conjugate for the
in vitro test.
Duration and intensity of exposure are the major factors
contributing to the development of occupational asthma in populations
exposed to its causes. Additional factors such as atopy and tobacco
smoking may also contribute. The importance of these factors varies
for different causes of the disease. However, for no cause do they
adequately explain the development of the disease in the minority who
develop it. In part this may reflect the limited knowledge of
exposures experienced but probably also suggests other important,
including genetic (such as HLA haplotype), determinants.
2.5.3 Atmospheric pollutants and asthma
There is evidence that air pollutants are involved in
exacerbating asthma (Vos et al., 1996). Evidence from laboratory
studies suggests that certain air pollutants have the potential to
stimulate bronchoconstriction or airways inflammation (see also
chapter 5.) Exposure to SO2 is associated with chest tightness and
bronchoconstriction, with the concentration required to induce a
response being dependent upon the degree of hyperresponsiveness. It
may be that the effects of SO2 will be augmented in the presence of
other pollutants. It has been reported that the sensitivity of mild
asthmatics to SO2 is increased by prior exposure to ozone (O3).
Ozone is a prototype oxidant pollutant that reacts rapidly with tissue
components. It is formed by photochemical reactions involving oxides
of nitrogen and organic molecules and occurs with other photochemical
oxidants and fine particles in the complex mixture called "smog".
Bates & Sizto (1987) studied hospital admissions in Southern
Ontario, Canada, an area with a population of seven million people,
and observed an association between rates of admissions for asthmatic
subjects during the summer season and ambient air levels of both O3
and suspended sulfates. However, the study design could not separate
the 03 effects from concomitant effects of acidic aerosol and SO2.
Thurston et al. (1992a,b) found strong associations between elevated
summer "haze" pollution (H+, sulfate, O3) and increased asthma (and
total respiratory) admissions to hospitals in Buffalo and New York
City, USA, especially in 1988 when air pollution was severe. However,
the specific role of O3 as opposed to H+ was less clear.
Controlled (environmental chamber) human exposure studies have
clearly demonstrated that some healthy young adults and children
respond to O3 exposure (at levels occurring in ambient air) with
irritative cough and substernal chest pain on inspiration and
decrements in FVC and FEV1 (Koren et al., 1989; Folinsbee, 1992).
When exercising outdoors in summer such individuals show decrements in
FEV1 that are consistent with the observed ambient air O3 levels.
Controlled exposure to similar levels of ozone has also been shown to
cause an inflammatory response of the respiratory tract in all species
that have been studied including humans (Lippmann, 1989). The use of
bronchoalveolar lavage (BAL) as a research tool has afforded the
opportunity to sample lung and lower airways after exposure to O3 and
to ascertain the extent and course of inflammation and its
constitutive elements. The BAL studies (Devlin et al., 1991) have
clearly demonstrated that O3, even at very low concentration, causes
increases in numbers of neutrophils, and a variety of other
constituents of BAL fluid, some with potential inflammatory properties
such as prostaglandin E2, fibronectin, elastase and IL-6.
Inflammation was also detected in the upper airways of O3-exposed
subjects as shown by an increase in neutrophils and other inflammatory
indicators in the nasal lavage (NAL) fluid (Koren et al., 1990).
Interestingly, both NAL fluid and BAL fluid from non-asthmatic
subjects exposed to O3 have been shown to contain the mast cell
marker tryptase. This and another study (Bascom et al., 1990)
suggested that O3-induced inflammation may share certain features of
the response observed in subjects with allergic rhinitis challenged
with allergen.
A study demonstrated that asthmatic subjects exposed to low
levels of O3 (0.16 ppm) for 7.6 h while performing moderate exercise
showed more respiratory symptoms and greater decrements in FEV1 than
did similarly exposed non-asthmatics (Ball et al., 1993).
The concept of influencing the asthmatic response by combining
exposure to O3 with specific allergen challenge has created interest
in the potential "indirect" effects of O3 exposure. In one study,
individuals with allergic rhinitis were initially exposed to clean air
or 0.5 ppm O3 for 4 h (Bascom et al., 1990). The high level of
exposure to O3 did not enhance the subsequent acute response to
antigen in the nose under these experimental conditions. A study by
Molfino et al. (1991) examined the effect of pre-exposure to O3 (0.12
ppm for one hour at rest) on the subsequent airway response to inhaled
ragweed or grass pollen antigen in seven subjects with allergic
asthma. They reported O3-induced increases in bronchial
responsiveness to specific allergen challenge. Preliminary data from
studies currently conducted examining the effects of pre-exposure to
O3 (0.4 ppm for 2 h at rest) followed by a specific allergen nasal
challenge in asthmatics sensitive to house dust mite suggest that the
O3 pre-exposure caused a significant decrease in the dose of allergen
needed to induce symptoms (Peden et al., 1994). Eosinophil influx and
increase in eosinophil cationic protein were observed 4 h after nasal
allergen challenge following both O3 and clean air pre-exposure.
These changes were more dramatic following O3 pre-exposure although
the mean allergen dose was smaller.
The health relevance of oxides of nitrogen, and in particulate
NO2, has attracted some interest since the gas is present not only
outdoors but also indoors. A number of studies suggest mild effects of
NO2 in asthmatics at concentrations less than 1 ppm but others have
not found responses at levels up to 4 ppm.
Particulate air pollutants, especially fine particles derived
from combustion sources, are also of interest although there have been
few controlled exposure studies outside those involving acid aerosols.
Bioaerosols, to which an asthmatic is sensitized, are well known to
exacerbate asthma. Epidemiological studies describing the increase in
mortality associated with particulate matter (PM) provide provocative
evidence for adverse pulmonary health effects associated with
particulate pollution (Dockery et al., 1993, 1994; Brunekreef et al.,
1995; Pope et al., 1995). The association between PM and acute
mortality and morbidity has been demonstrated most strongly with
elderly people who have chronic cardiopulmonary disease (Pope et al.,
1992; Burnett et al., 1995; Schwartz & Morris, 1995). Experimental
studies with diesel exhaust particles show that they increase IL-4 and
specific IgE production, and exacerbate the response to allergen in
allergic individuals (Diaz-Sanchez et al., 1997). Studies in mice have
demonstrated that diesel exhaust particles facilitate the induction of
allergy (Takafuji et al., 1987; Lovik et al., 1997). Chemicals
adsorbed to the diesel exhaust particles, as well as carbon particles
with very little chemicals on them appear to enhance the allergic
immune response (Diaz-Sanchez et al., 1997, 1999; Lovik et al., 1997).
Environmental air pollutants including tobacco smoke may affect
the prevalence and/or severity of asthma in several different ways. In
hyperresponsive airways, pollutants may act as triggers of asthmatic
reactions without the presence of the specific allergen.
Alternatively, a pollutant could induce or increase airway
inflammation and, as a result, cause airway hyperreactivity that
persists after exposure has ceased. Some pollutants may have a direct
toxic effect on the respiratory epithelium leading to inflammation,
airway hyperreactivity and the appearance of asthma-like symptoms in
previously non-asthmatic individuals. Lastly, there are certain
pollutants that may have the ability to augment or modify immune
responses to inhaled antigens or to enhance the severity of reactions
elicited in the respiratory tract following inhalation exposure of the
sensitized individual to the inducing allergen.
2.5.4 Rhinitis
Rhinitis frequently, but not invariably, occurs in atopic
diseases. Similarities and differences between rhinitis and asthma are
considered below.
Allergic responses of the nasal mucosa cause an orchestrated set
of responses. The acute allergic reaction occurs within minutes and is
manifested as rhinorrhoea, pruritus and sneezing, and congestion, due
(respectively) to increased vascular permeability, sensory nerve
stimulation, and vasodilation with sinusoidal pooling plus oedema
formation. These responses are due to mediators released from the
mucosal mast cells, and histamine is a major participant.
Following this acute response is the slower development of the
late phase allergic reaction which is manifested by congestion and
hyperirritability and is due to cellular infiltration with
eosinophils, neutrophils and some basophils. There is interest in
whether lymphocytes also participate in this reaction, but the data
are not clear as yet.
Of the cells that participate in rhinitis, mast cells,
neutrophils, eosinophils and lymphocytes may all be important. Mast
cells initiate the response through the release of the mediators of
anaphylaxis. Work also indicates that mast cells generate a number of
cytokines (generally thought of as lymphocyte products, but clearly
generated by activated mast cells as well). These products include
IL-3, IL-4, IL-5, IL-6 and TNF. Neutrophils are the first cells to
infiltrate areas undergoing allergic reactions. The role of the
neutrophil in allergy is not clear. However, neutrophils appear to be
necessary for the development of increased airway hyperactivity in
animal models of asthma. Neutrophils also release factors that
activate mast cells (neutrophil-derived histamine releasing factor),
and the influx of neutrophils occurs simultaneously with recrudescent
histamine release in the late phase reaction. Eosinophils have
received a lot of attention, as they are the hallmark of allergic
inflammation. Eosinophils infiltrate areas more slowly than do
neutrophils, but persist much longer. The eosinophil can cause
epithelial denudation, mucus secretion and histamine release. Both
eosinophil and neutrophil infiltrates are inhibited by
corticosteroids.
Interest has focused on the possible contribution by lymphocytes
to the late-phase reaction. After mast cell activation, about 10% of
the superficial lymphocytes express the IL-2 receptor, indicating
their activation. There are suggestions that some cytokines are
released during this time period, either from mast cells or
lymphocytes.
2.5.5 Atopic eczema
In atopic eczema, the patient is much troubled by itching skin;
there is a history of chronic or chronically relapsing dermatitis,
worst on the flexures, which are excoriated and lichenified, and there
is a family or personal history of atopy. This is the typical picture
of atopic eczema, though some of the features may be absent (Hanifin &
Rajka, 1980). In any discussion of pathogenesis, family history is
important because atopic eczema is part of the atopic syndrome that
includes genetically determined phenotypes such as extrinsic bronchial
asthma, allergic rhinitis, allergic conjunctivitis and
gastrointestinal allergy. Important laboratory indices are blood and
tissue eosinophilia and antigen-specific IgE bound to mast cells in
skin (intracutaneous challenge) or peripheral blood
(radioallergosorbence assays). The Wiscott-Aldrich syndrome and
hyper-IgE syndrome, which can closely resemble atopic eczema, are
usually distinguishable by the associated life-threatening infections.
The clinical course of atopic eczema is unpredictable. Sometimes
it remits in childhood, but occasional patients have recurrences
throughout life. Some patients (or their parents) are convinced that
exacerbations are related to stress and/or exposure to environmental
antigens such as food or animals. Secondary skin infection by
Staphylococcus aureus, herpes simplex virus, varicella virus and,
possibly, fungal infections can lead to severe exacerbations. Finally,
autonomic nervous system disturbances and changes in fatty acid
metabolism and phosphodiesterase activity have been implicated.
Despite the development of numerous theories, the pathophysiology
of eczema is still remarkably little understood. Researchers are
currently focusing on Langerhans cells, which are thought to be
involved in eczema, because these cells possess abundant receptors for
IgE. Once in contact with allergen distributed after ingestion or
following direct skin contact, Langerhans cells present the allergen
to T-lymphocytes. They may also be directly stimulated to produce
inflammatory cytokines, which are responsible for eczematous lesions.
Atopic eczema is often accompanied by very high IgE levels. In babies,
an elevated IgE level is taken as a reliable predictive sign for the
development of asthma and/or hay fever in later life.
The relation between cell-mediated immunity and IgE in atopic
eczema was first established by Bruijnzeel-Koomen et al. (1986) who
identified the presence of IgE on Langerhans cells in atopic eczema.
It is now evident that this binding of IgE is the result of the
presence of the high-affinity receptor for IgE on these Langerhans
cells (Bieber & Ring, 1992). Langerhans cells and other
antigen-presenting cells in skin also express low-affinity Fc
receptors that efficiently bind allergen-precomplexed IgE. The
functional consequence of the expression of these Fc receptors for IgE
on antigen-presenting cells in skin is that the local response to
minute quantities of allergens in the skin is amplified. By
facilitated antigen-processing, only minute quantities of allergens
are needed to be presented to T-cells, because the
IgE-receptor-allergen complex aids processing and subsequent
presentation up to a 1000-fold (Van der Heijden et al., 1993).
Therefore, the onset of atopic eczema as an expression of atopic
allergy may result from an interplay between the degrees of expression
of one or more Fc receptor types, the serum concentration of
allergen-specific IgE, and the number of skin-infiltrating T-cells
specific for that allergen and, of course, exposure to the allergen.
Atopic syndrome is genetically determined. When both parents have
atopic disease of the same sort, their child has a risk of around 70%
of developing a similar phenotype. If parents have different atopic
diseases, the incidence of atopic disease in a child is 30% (Björksten
& Kjellmann, 1987). With asthmatics as probands in molecular genetic
studies, a gene predisposing to atopy has been found on chromosome I
Iql3 (Cookson et al., 1989), possibly coding for the beta subunit of
high-affinity IgE Type I Fc receptor (Sandford et al., 1993). However,
the genetic mapping of atopy is far from simple. For example, the
increasing prevalence of atopic eczema in the past three decades
(Williams, 1992) is difficult to explain on the basis of genetics
alone. Furthermore, a maternal pattern of inheritance has been found
(Cookson et al., 1992), which might be due to paternal genomic
imprinting or to maternal modification of developing immune responses
in utero or via breast milk. Linkage of atopy with a gene on I Iql3
could not be shown when patients with atopic eczema were taken as
probands. Thus more than one gene seems to be involved.
Environmental factors, such as exposure to allergens, are thought
to be involved in the phenotypic expression of atopic eczema. For
example, the presence of a strong atopic background has been
associated with enhanced protective responses to helminthic infections
(Lynch et al., 1998). However, a precise understanding of the
environmental factors that determine whether or not the atopic
genotype is expressed as an atopic phenotype is lacking.
2.5.6 Urticaria
Urticaria (hives, nettle rash) may be defined as an eruption of
short-lived red oedematous swellings of the skin, associated with
itching. The relative incidence of the different types of urticaria
and angioedema in the general population is unknown.
Urticaria usually involves degranulation of mast cells and
release of histamine. Many different elicitors have to be considered.
Allergy due to a reaction between a specific antigen and a mast
cell-fixed IgE antibody is only one mechanism. Pseudo-allergic
reactions, toxic effects and viral infections play a major role.
Acute urticaria resolves within a period of six weeks. If it
persists, it is called chronic urticaria. Wheals may be circular,
polycyclic or figured. If subcutaneous extension occurs, angioedema is
present. Although, like urticaria, angioedema may occur anywhere, the
genitalia, eyelids, lips and mucous membranes are especially common
sites. Itching is almost always present in patients with urticaria but
is inconsistent in angioedema. The duration of urticarial wheals is
usually 3 to 4 h, but angioedema lesions may last much longer.
Skin previously involved by wheals or angioedema looks entirely
normal apart from occasional purpura or other signs of trauma due to
scratching. The mucous membranes are frequently involved including the
tongue, soft palate and pharynx. Although discomfort and breathing
difficulty may occur, fatalities are almost exclusively associated
with hereditary angioedema. Acute urticaria may be associated with
systemic anaphylactic symptoms (wheezing, dyspnoea, syncope, abdominal
pain, vomiting). Occasionally acute urticaria may merge into serum
sickness, arthritis, fever, proteinuria). Common causes of allergic
acute urticaria include ingestion of penicillin, shellfish, soft fruit
and nuts.
Urticaria of immunological origin may arise rapidly (often less
than 60 min) at the site of contact of the skin or mucous membranes
with a specific substance.
Contact urticaria may also be of non-immunological origin, and
there are frequent instances in which the mechanism is uncertain. When
an immune mechanism is involved, the final common pathway is probably
the same. Contact urticaria of immunological origin involves
IgE-mediated hypersensitivity as indicated by a positive
radioallergosorbent test (RAST). In non-immunological examples, the
offending substance may evoke histamine release directly from
cutaneous mast cells. Such substances include ammonium persulfate
(Mahzoon et al., 1977), dimethyl sulfoxide (Odom & Maibach, 1976) and
cinnamaldehyde (Kirton, 1978); however, several other mechanisms are
also involved.
Immunological contact urticaria is more frequent in atopic
subjects. These patients often give a history of acute oedema of the
lips or buccal mucous membrane after ingestion of food items such as
fish, egg or nuts. In common with other types of allergy, healthy
control subjects are negative on skin testing. The offending allergen
is usually a high relative molecular mass substance and skin testing
is rarely positive in completely normal skin. Open and closed patch
tests and closed patch tests on lightly abraded skin (scratch-patch
tests) should be performed. The diagnosis is confirmed by a positive
radioallergosorbent test (RAST).
Non-immunological contact urticaria may be elicited in healthy
asymptomatic individuals, with the triggering substance frequently
being of low relative molecular mass, and contact reactions may be
elicitable in clinically normal skin. The danger of such generalized
reactions should be borne in mind before skin testing is performed.
2.5.7 Gastrointestinal tract diseases: mechanisms of food-induced
symptoms
2.5.7.1 Non IgE-mediated food-sensitive enteropathy
Slow onset gastrointestinal symptoms are described in children,
especially in relation to ingestion of cow's milk. The clinical
features are chronic diarrhoea and failure to thrive. The pathological
lesion found in the small intestine is crypt hyperplastic villous
atrophy of variable severity. The lesions are often patchy. There is
an increased expression of the markers of T-cell activation on the
T-cells of the lamina propria, and it is likely that a cell-mediated
reaction in the lamina propria is the basis of the abnormality,
although IgE involvement has also been described (Walker-Smith, 1992).
Nagata et al. (1995) suggested that activated CD4 cells in the lamina
propria of the small intestinal mucosa may contribute to the mucosal
damage, probably by releasing cytokines.
2.5.7.2 IgE-mediated food allergy
Food allergic patients often describe itching and tingling of the
mouth and throat as the first immediate symptoms of an allergic food
reactions. In addition papules/blisters on the mucosa and swelling of
the lips can be seen. These symptoms occur as a result of direct
contact between the allergen and the mucosa of the mouth and throat.
The concentration of mast cells is very high in the oropharyngeal
mucosa and the symptoms are probably caused by degranulation of
mucosal mast cells bearing specific IgE towards the offending allergen
(Pastorello et al., 1995).
Symptoms like nausea, vomiting, abdominal pains, loose stools and
gas production are described in connection with immediate allergic
reactions. In a direct challenge of the gastric mucosa using a
gastrofibrescope, Romanski (1987, 1989) found gastric changes within
5-20 min of contact with the introduced food. The macroscopic changes
were: pale mucosa, oedema, punctate haemorrhage, hyperperistalsis,
hypersecretion, erythema. Microscopic examination revealed oedema,
hyperaemia, capillary haemorrhage, eosinophilic infiltration and
inflammation.
The underlying mechanisms of IgE-mediated gastrointestinal
symptoms are a result of degranulation of intestinal mast cells with
release of mediators that act directly on the epithelium, endothelium
or muscle indirectly through nerves and mesenchymal cells. The result
is altered gastric acid secretion, ion transport, mucus production,
gut barrier function, and motility (Crowe & Perdue, 1992).
2.5.7.3 Role of gastrointestinal tract physiology in food allergy
Many elements of the gastrointestinal tract physiology influence
the ultimate allergenicity of food proteins. These include the pH,
digestive enzymes, bile, peristalsis, transit time, bacterial
fermentation, and the intestinal barrier function, permeability, and
absorption. Several food allergens or allergenic determinants were
reported to be relatively resistant to acid denaturation and
proteolytic digestion (Elsayed & Apold, 1977; Schwartz et al., 1980;
Kurisaki et al., 1981; Metcalfe, 1985; Taylor, 1986; Taylor, 1992;
Kortekangas-Savolainen et al., 1993). Unfortunately, insufficient
information is available on possible differences in susceptibility to
acid denaturation and gastrointestinal digestion between strongly
allergenic food proteins and proteins that possess weak or virtually
no allergenic potential. Attempts have also been made to correlate the
susceptibility to enzymatic breakdown of cow's milk proteins, their
intestinal permeability and allergenic properties (Taylor, 1986;
Marcon-Genty et al., 1989; Savilahti & Kuitunen, 1992). The important
role of digestion with respect to food protein allergenicity was
clearly demonstrated in mice showing that pre-feeding of an
endopeptidase inhibitor (aprotinin) to mice resulted in an inhibition
of normally expected oral tolerance induction by protein feeding
(Hanson et al., 1993). An abnormal digestive breakdown of proteins may
also be of importance, since intragastric administration more easily
results in anaphylactic sensitization as compared to ad libitum
feedings, generally resulting in tolerance induction, as has been
shown in rodents (Knippels et al., 1997). However, as digestion of
food proteins is part of the normal sequence of events following
consumption of food, it is likely that food allergic patients become
sensitized to digested allergens rather than to the native proteins.
Enzymatically digested food allergens may show the same, more, or less
binding to specific IgE from patients (Haddad et al., 1979; Schwartz
et al., 1980).
The intestinal barrier function, permeability, and absorption are
also hardly, or not, taken into account in the evaluation of the
allergenicity of food proteins. Knowledge of the intestinal uptake of
specific protein antigens and their fragments may provide some
additional information in the evaluation of the potential
allergenicity of protein products. There is evidence of limited
macromolecular exclusion by the epithelial barrier (Seifert et al.,
1974, 1977; Gardner, 1988; Teichberg, 1990).
2.6 Type II hypersensitivity diseases
Pathogenic Type II reactions may occur towards autoantigens,
alloantigens (in blood transfusions), infective agents and drugs or
chemicals, as described above. As shown in Table 13, these immune
reactions may cause corresponding disorders, i.e., autoimmune
diseases, transplantation/transfusion reactions or drug-induced
haemolytic reactions. As an illustration of Type II reaction-induced
diseases, three autoimmune disorders that are also inducible by drugs,
i.e., haemolytic reactions, pemphigus and myasthenia gravis, will be
dealt with in more detail.
2.6.1 Drug-induced Type II reactivity
Some drugs or their metabolites are chemically reactive agents
that readily bind to cells and tissues. Such drugs, present on the
cell membrane of blood cells, are obvious targets for pathogenic Type
II reactivity.
The most frequent allergic reaction occurs with penicillin and
its relatives. Benzylpenicillin is a small molecule with a relative
molecular mass of 372.47 and with a highly reactive beta-lactam ring,
which may bind to amino groups on proteins (carrier), forming covalent
conjugates. The thus formed penicilloyl hapten is considered as the
major determinant in penicillin allergy. Although penicillin is able
Table 13. Clinical disease due to Type II hypersensitivity reactions
Antigen Disease Symptoms
Autoantigens glomerular basement membrane Goodpasture's syndrome vasculitis, renal failure
epidermal desmosomes (desmoglein-3) pemphigus vulgaris skin blistering (intra-epidermal)
epidermal hemidesmosomes on bullous pemphigoid skin blistering (subepidermal)
basal keratinocytes
acetylcholine receptor myasthenia gravis striated muscle weakness
Rhesus antigen autoimmune haemolytic destruction of red cells, anaemia
anaemia
platelet integrin gpIIb:IIIa autoimmune thrombocytopenia abnormal bleeding
purpura
Alloantigens donor red cell antigens delayed haemolytic destruction of transfused red cells
transfusion reaction
Infective agents Streptococcal cell wall antigens acute rheumatic fever arthritis, myocarditis
cross-reacting with cardiac muscle
Klebsiella antigens cross-reacting ankylosing spondylitis (?) arthritis involving the spine
with HLA-B27
Drugs, chemicals penicillins, cephalosporins drug-specific haemolytic lysis of hapten-coated red cells
trimellitic anhydride anaemia
to induce all types of hypersensitivity reactions (IgE-, immune
complex- or T-cell-mediated), haemolytic anaemia with
penicillin-specific IgG antibodies reacting with penicillin-coated
erythrocytes is a typical example of Type II reactivity.
Interestingly, the specificity of drug-induced antibodies is
often much broader than would be expected from the penicillin example.
Ultimately, drugs trigger Type II reactivity without being involved in
the final destructive reaction. In addition to hapten-specific
antibodies, drugs can induce antibodies to metabolites, to
drug-carrier combinations or to the carrier alone, resulting in
clear-cut autoimmune reactivity. D-penicillamine is a classical
example of a drug inducing autoimmunity, but chemicals such as mercury
and gold are also able to induce autoimmunity.
The mechanism by which drugs can induce autoantibodies is shown
in detail in Fig. 6. By presenting the hapten in or on their MHC-class
II molecules, autoreactive B-cells, which are normally present at very
low frequencies without being activated, can trigger hapten-specific
T-cells to help them (the B-cells) differentiate into
antibody-producing plasma cells. Although the induction of the disease
is drug-dependent, the Type II effector reaction towards autologous
targets may be drug-independent. Hence, in this case the induced
autoimmune disease would continue after the exposure to the drug had
ceased.
2.6.2 Transfusion reactions
Transfusion reactions are examples of the cellular destruction
that results from antibody combining with heteroantigens. There are at
least 21 blood group systems, with more than 600 antigens within these
systems. Some antigens are stronger than others and are more likely to
stimulate antibody production. Certain antibodies are produced
naturally with no prior exposure to red blood cells, while other
antibodies are only produced after contact with cells carrying that
antigen.
The ABO blood groups are of primary importance in considering
transfusions. Anti-A and anti-B antibodies are so-called naturally
occurring antibodies. Individuals do not form such antibodies to their
own red blood cells. Thus, an individual who has Type A blood would
have anti-B in the serum, and a person with Type B blood has anti-A
antibodies. An individual with Type O blood has both anti-A and anti-B
in the serum, as O cells have neither of these two antigens.
If a patient is given blood for which antibodies are already
present, a transfusion reaction occurs. This can range from acute
massive intravascular haemolysis to a small decrease in red blood cell
survival. Acute haemolytic transfusion reactions may occur within
minutes or hours after transfusion of incompatible blood.
Delayed haemolytic reactions occur 4 to 10 days following a
transfusion and are due to a secondary response to the antigen.
Antibody-coated red blood cells are removed extravascularly, in the
spleen or in the liver, and the patient may experience a mild fever
and anaemia.
Haemolytic disease of the newborn appears in infants whose
mothers have been sensitized by exposure to fetal blood cells carrying
antigens, commonly of the Rhesus family, that differ from their own.
The mother makes IgG antibodies in response, and these cross the
placenta to cause destruction of fetal red cells. A common antigen
involved is the Rhesus D antigen
2.6.3 Autoimmune haemolytic anaemia
Drugs account for about 12% of the autoimmune haemolytic anaemia.
They can cause haemolysis by three different mechanisms: by acting as
a hapten, by inducing a classical autoimmune haemolytic anaemia, or by
forming immune complexes with antibodies that can be adsorbed by the
patient's red cells, the "innocent bystanders" (Fig. 8).
Antigen-presenting cells phagocytose and process haptenized
cells, such as erythrocytes. In addition, free drug molecules may bind
to MHC-class II or to peptides within the groove. The hapten is thus
presented by MHC-class II molecules to the T-cell receptor (TCR)
of T helper cells. Hapten-specific T-cells now proliferate and
differentiate, so that they can either attack haptenized cells (The
cells causing Type IV reactivity, not shown) or can help nearby
B-cells to produce antibodies (in particular The cells).
B-cells ingest and process the antigens to which their
immunoglobulin receptors bind and present peptides, including
haptenized peptides, derived from these antigens in their MHC-class II
molecules. Thus B-cells may trigger hapten-specific T-cells by
presenting haptenized peptides. Alternatively, external drug molecules
can bind to peptides in the MHC-class II groove. Differentiation of
B-cells is dependent on adjacent T-cells providing membrane signals
(to CD40, not shown) and the growth factors IL-4 and IL-6.
In drug-specific haemolytic anaemia (on the left of Fig. 8),
drug-specific B-cells present the drug to drug-specific T-cells.
Mutual activation of T- and B-cells now induces the B-cell to become a
plasma cell, producing drug-specific antibodies. Eventually these
antibodies lead to destruction of haptenized erythrocytes, while
normal cells remain intact.
In drug-induced autoimmune haemolytic anaemia (on the right of
Fig. 8), an autoreactive B-cell ingests and processes erythrocyte
membranes, including the haptenized parts. Normally, autoreactive
B cells exist but do not become activated by lack of appropriate
stimulating T-cells. If drug-specific T-cells are present, however,
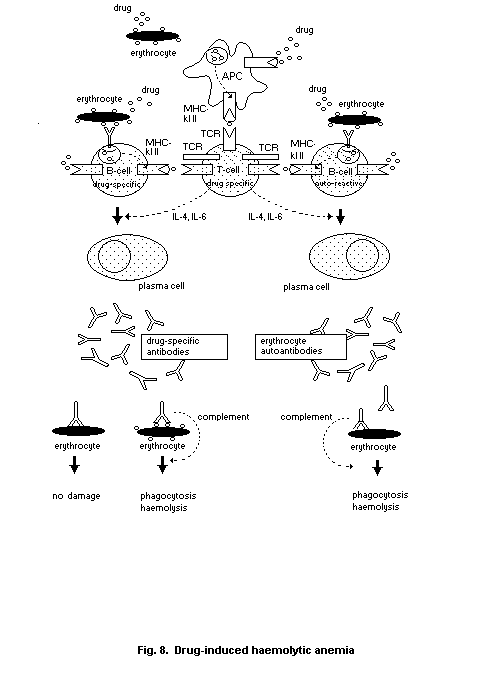 these B-cells, presenting haptenized peptides in at least some of
their class II molecules, may become activated and differentiate into
autoantibody-producing plasma cells. These antibodies may induce
haemolysis of all erythrocytes in a drug-independent manner.
The hapten type of autoimmune haemolytic anaemia is caused by the
presence of drug-specific antibodies. These antibodies may be partly
considered as autoimmune since the combination of drug and autologous
carrier forms the actual target of the antibodies. When drugs, like
penicillin, bind covalently to red blood cells, these drug-specific
antibodies bind to the cells and induce their elimination by
phagocytosis in the spleen. The induction of high titres of penicillin
IgG antibodies typically occurs upon intramuscular administration,
rather than upon intravenous penicillin therapy. On the other hand,
relatively high intravenous doses of penicillin are required to make
the erythrocytes susceptible to immune-mediated haemolysis. Thus, most
patients with penicillin-induced haemolytic anaemia have received
large doses of drug over a protracted period. After discontinuation of
therapy the haemolysis quickly resolves and the antiglobulin test
becomes negative within weeks.
Some drugs appear to be able to induce true autoimmune haemolytic
anaemia, with Rhesus antigens as the most common targets of the red
cell antibodies. It is conceivable that the same auto-specificities
are found in drug-induced and idiopathic autoimmune haemolytic
anaemia, because the "normally" present (but silent) autoreactive
B-cells become activated upon haptenization of the autoantigens, as
shown in Fig. 8. As in drug-induced pemphigus and myasthenia, the drug
itself does not seem to be involved in the destructive autoimmune
reaction. While several drugs (Table 14) have been reported to provoke
red cell autoantibodies, alpha-methyldopa is the best studied example.
Only after prolonged therapy anti-red cell autoantibodies (IgG
anti-Rh) are formed. Upon withdrawal of the drug the antibody titres
usually decline and haemolysis ceases. Alpha-Methyldopa not only
induces red cell antibodies, but also antinuclear factors, rheumatoid
factors and gastric mucosa antibodies.
Table 14. Summary of drugs causing the different types of
immune or autoimmune haemolytic anaemia (from: Foerster, 1993)
Type of drug-induced (A)IHA Drugs
Drug- or hapten-specific IHA penicillin, cephalosporins, tetracycline
Autoimmune IHA alpha-methyldopa, levodopa, mefanamic acid,
procainamide
Immune complex mediated IHA stibophen, p-aminosalicylic acid, chlorambucil,
("innocent bystander" type) quinidine, quinine, phenacetin, sulfonamides,
isoniazid, rifampicin, etc.
If soluble drug-specific antibodies are present, they may form
immune complexes with administered drugs and fix complement. The
complexes are then adsorbed by erythrocytes and thrombocytes resulting
in lysis or clearance of these "innocent bystanders". Strictly
speaking, this haemolysis is caused by Type III reactivity. The
mechanism of adsorption, however, is not completely understood. It is
clear that it does not simply involve Fc receptors, since the F(ab)2
domain of the antibodies, in particular, adheres to the target cells.
Low-affinity attachment of drugs to the cells seem to make them more
susceptible for complex binding, and specific red cell antigens also
seem to be involved. Perhaps the bystanders are less innocent than
initially thought, and Type II reactivity combines here effectively
with Type III reactivity. Many different drugs, usually of low
relative molecular mass, are able to induce this type of haemolysis
(Table 14) (Foerster, 1993).
2.6.4 Autoimmune thrombocytopenic purpura
Autoimmune or idiopathic thrombocytopenic purpura (ITP) is
another example of a Type II reaction involving destruction of
self-antigens. This disease is characterized by shortened platelet
survival and the presence of antibody bound to platelets. It can be
classified as acute, intermittent or chronic, depending on the
severity and frequency of the symptoms. Acute ITP occurs mainly in
children following an upper respiratory viral illness (Karpatkin,
1988). The disease lasts an average of 1 to 2 months. Intermittent ITP
may occur in a child or an adult. It is characterized by episodes
where the platelet count drops, followed by periods where the count is
normal. Chronic ITP is seen in adults, and it may last for years, or
indefinitely (Karpatkin, 1988).
ITP can be drug-induced by the following: quinidine, quinine,
sulfonamides, p-aminosalicylic acid, phenytoin and sedormid (no
longer used). The drug acts as a hapten and adheres to the surface of
the platelets. This type of ITP is reversed when the drug is
withdrawn.
2.6.5 Pemphigus and pemphigoid
Type II reactions in the skin may cause different types of
blistering diseases depending on the antigen (location) to which the
autoantibodies are directed.
Antibodies towards desmosomal antigens induce intra-epidermal
blistering (acantholysis), leading to pemphigus, which is a
potentially fatal disease. The presence of these intra-epidermal
antibodies can be shown by direct immunofluorescence of perilesional
skin and provides the main diagnostic parameter. Most patients also
have circulating antibodies with titres reflecting disease activity.
Since removal of these antibodies by plasmapheresis reduces disease
activity and transfer of positive sera to mouse and monkeys can induce
pemphigus-like lesions, it is believed that the anti-desmosomal
antibodies are the causative agent of the clinical lesions in
pemphigus.
Investigations have unravelled the different desmosomal molecules
serving as autoantigens in pemphigus. The most important antigens in
pemphigus are the desmogleins; these are transmembrane glycoproteins,
which are members of the cadherin gene superfamily. Desmogleins bear
the same calcium-binding motifs as other cadherins do, and calcium
appears to be essential for the formation of the conformational
epitopes that are recognized by pemphigus sera (Amagi et al., 1995).
Interestingly, the two clinical variants of the disease, pemphigus
vulgaris and pemphigus foliaceus, develop antibodies to different
desmogleins, i.e., desmoglein-3 and desmoglein-1, respectively. The
differential expression of these two desmogleins in the upper and
lower epidermis could explain the different levels of acantholysis
seen in the two pemphigus variants (Shimizu et al., 1995).
Drugs may play a precipitating role in pemphigus.
Penicillamine-D, thiopronine, ampicillin, rifampicin, phenylbutazone,
captopril, pyrazolon, enalapril and piroxicam can all induce
pemphigus. It would appear that the presence of certain chemically
reactive groups in the drugs, in addition to the pemphigus
susceptibility genes in the patient (HLA-DRB1*0402 or DQB1*0503;
Matzner et al., 1995; Wucherpfennig et al., 1995), predispose for
drug-induced pemphigus. Sulfhydryl groups (-SH), present in
D-penicillamine, and active amide groups (-CO-N-), typically present
in enalapril, are held responsible for the acantholytic effects in
human skin cultures. In the group of penicillin and cephalosporins,
this active amide group is probably more important for the induction
of pemphigus than the sulfhydryl group (Wolf & Brenner, 1994).
The mechanism by which the drugs induce pemphigus is still not
completely understood. It is clear, however, that in addition to the
direct acantholytic effects, which can be observed in human skin
cultures in vitro, an autoimmune reaction is being induced. The
resulting autoantibodies appear to have the same antigenic
specificity, i.e., to desmoglein-1 and desmoglein-3, as in idiopathic
pemphigus patients (Korman et al., 1991). Together these findings
would be in line with involvement of the same mechanism of
drug-induced autoimmunity as described for autoimmune haemolytic
anaemia.
Antibodies towards antigens present in the lamina lucida of the
basement membrane cause a less severe type of blistering disease,
called bullous pemphigoid. Direct immunofluorescence of perilesional
skin reveals the presence of autoantibodies along the dermo-epidermal
junction. Concordantly, sub-epidermal blisters are being formed.
The antigens in bullous pemphigoid have been identified as
transmembrane proteins of 180 000 and 230 000 relative molecular mass,
present in the hemidesmosomes of the basal keratinocytes (Korman,
1995). These hemidesmosomes are believed to play a role in the
epidermal-dermal adhesion. Also in bullous pemphigoid autoantibodies
with the same specificity can be detected in the circulation, but
their titres do not correlate with disease activity.
The precipitating role of drugs for bullous pemphigoid is not
well established, although the disease may occasionally follow drug
ingestion (e.g., after furosemide).
2.6.6 Myasthenia gravis
Myasthenia gravis is an autoimmune disease that is mediated by
IgG antibodies directed to the acetylcholine receptors in the
postsynaptic membrane of the muscle (Vincent et al., 1995). The number
of receptors can be considerably reduced by complement-mediated lysis
and accelerated internalization. Additionally, the residual receptors
may be blocked by autoantibodies directed to the acetylcholine binding
site, thus leading to further impairment of the transmission from
nerve to muscle. As a consequence, the disease is characterized by
weakness and fatigue of the striated muscles. In some patients only
few muscles are affected; a well-known localized form of the disease
is ocular myasthenia (Weinberg et al., 1994).
In young patients with myasthenia gravis (40-50% of patients,
usually female) the thymus is an important site of autoantibody
production and T-cell activation. Within the hyperplastic thymus,
formation of lymphoid follicles can be observed, with germinal centres
surrounded by T-cells. The acetylcholine receptor antigens are here
presented to the immune system by muscle-like myoid cells, which bear
MHC-class II molecules. As therapeutic treatment, in addition to
immunosuppression, thymectomy is beneficial in these patients, since a
substantial source of both antigen and antibody-producing plasma cells
is thus removed. On the other hand, in late onset (usually male)
patients (15-20%), the thymus is rather atrophic and autoantibody
production by thymic cells is relatively low. In another minority of
patients (15-20%) thymoma may develop. In this last group of patients,
autoantibodies to striated muscles are typically found in addition to
the acetylcholine receptor autoantibodies.
Like pemphigus, myasthenia gravis can be induced by a number of
drugs. D-penicillamine, used for treatment of rheumatoid arthritis,
has been most frequently reported as a trigger for myasthenia gravis.
A few other drugs are suspected of inducing myasthenia gravis; among
them are thiopronin and chloroquin.
Drug-induced myasthenia is characterized by frequent involvement
of facial and oropharyngeal muscles (Bonnet et al., 1995). The disease
seldom generalizes or results in thymoma. Autoantibodies to
acetylcholine receptors are measurable in the circulation in the
majority of patients (approximately 80%), whereas almost half of them
have blocking antibodies. Similar frequencies of these antibodies are
found in idiopathic myasthenia gravis (Morel et al., 1991). The
autoantibodies disappear upon discontinuation of the drug, and full
recovery may be obtained within a few months.
2.7 Type III hypersensitivity diseases
2.7.1 Immune complex disease
Immune complexes are formed every time antibody meets antigen.
Generally they are removed effectively by the reticuloendothelial
system but occasionally their formation can lead to a hypersensitivity
reaction. Diseases resulting from immune-complex formation can be
placed broadly into three groups.
a) The combined effects of a low-grade persistent infection
(such as occurs with alpha-haemolytic Streptococcus viridans or
staphylococcal infective endocarditis, or with a parasite such as
Plasmodium vivax, or in viral hepatitis), together with a weak
antibody response, leads to chronic immune-complex formation with
the eventual deposition of complexes in the tissues.
b) Immune complex disease is a frequent complication of
auto-immune disease where the continued production of
autoantibody to a self-antigen leads to prolonged immune-complex
formation. The mononuclear phagocyte, erythrocyte and complement
systems (which are responsible for the removal of complexes)
become overloaded and the complexes are deposited in the tissues,
such as occurs in systemic lupus erythematosus (SLE).
c) Immune complexes may be formed at multiple sites, such as in
the lungs following repeated inhalation of antigenic materials
from moulds, plants or animals. This is exemplified in Farmer's
lung and Pigeon fancier's lung, where there are circulating
antibodies to the actinomycete fungi found in mouldy hay or to
pigeon antigens. Both diseases are forms of extrinsic allergic
alveolitis, and they only occur after repeated exposure to the
antigen. The antibodies induced by these antigens are primarily
IgG, rather than IgE, as in immediate (Type I) hypersensitivity
reactions. When antigen again enters the body by inhalation,
local immune complexes are formed in the alveoli leading to
inflammation. Precipitating antibodies to the inhaled
actinomycete antigens are found in the sera of 90% of patients
with Farmer's lung, but since they are also found in some people
with no disease, and are absent from some sufferers, it seems
that other factors are also involved, including Type IV
hypersensitivity reactions.
The sites of immune-complex deposition are partly determined by
the localization of the antigen in the tissues and partly by how
circulating complexes become deposited.
Immune complexes trigger a variety of inflammatory processes.
They can interact with the complement system leading to the generation
of C3a and C5a (anaphylatoxins), which cause the release of vasoactive
amines from mast cells and basophils, thus increasing vascular
permeability. These anaphylatoxins are also chemotactic for
polymorphs. Cytokines released from macrophages, particularly
TNF-alpha and IL-1, are also important in localized immune-complex
diseases, such as alveolitis, through a mechanism involving neutrophil
recruitment. Platelets can also interact with immune complexes,
through their Fc receptors, leading to aggregation and microthrombus
formation and hence a further increase in vascular permeability due to
the release of vasoactive amines. Platelets are a rich source of
growth factors, and release of these may contribute to the cellular
proliferation found in immune-complex diseases such as
glomerulonephritis and rheumatoid arthritis.
The attracted polymorphs attempt to ingest the complexes, but in
the case of tissue-trapped complexes this is difficult and the
phagocytes are therefore likely to release their lysosomal enzymes to
the exterior, causing tissue damage. If simply released into the blood
or tissue fluids, these lysosomal enzymes are unlikely to cause much
inflammation, because they are rapidly neutralized by serum enzyme
inhibitors. But if the phagocyte applies itself closely to the
tissue-trapped complexes through Fc binding, then serum inhibitors are
excluded and the enzymes may damage the underlying tissue. A classic
example of this type of inflammatory response is the Arthus reaction
(see section 2.1.3.1).
2.7.2 Serum sickness
Serum sickness is a Type III reaction that is seen in humans,
although not as frequently as it used to be. Serum sickness results
from passive immunization with animal anti-serum used to treat such
infections as tetanus and gangrene, usually horse or bovine
anti-serum. Approximately 50% of the individuals who receive a single
injection develop the disease (Barrett, 1988). Generalized symptoms
appear about 1 to 2 weeks after injection of the animal serum and
include headache, nausea, vomiting, joint pain and lymphadenopathy.
Recovery takes between 7 and 30 days (Terr, 1994b).
In this disease, the sensitizing and the shock-producing dose of
antigen are one and the same, as antibodies develop while antigen
still present. High levels of antibody form immune complexes that
deposit in the tissues. Usually this is a benign and self-limiting
disease, but previous exposure to animal serum can cause
cardiovascular collapse upon re-exposure (Terr, 1994b). Antibiotic use
has diminished the need for this type of therapy.
2.7.3 Allergic bronchopulmonary aspergillosis
Allergic bronchopulmonary aspergillosis (ABPA) is a syndrome
characterized by respiratory and constitutional symptoms caused by
hypersensitivity reactions to fungal antigens of Aspergillus
fumigatus. Allergic bronchopulmonary aspergillosis is characterized
by episodic wheezing, pulmonary infiltrates, eosinophilia in sputum
and blood, markedly elevated serum IgE levels, positive immediate and
late skin tests to A. fumigatus, serum precipitating antibody to
Aspergillus, and sputum containing brown plugs or flakes. Not all of
these changes may be present during active disease and a diagnosis of
allergic bronchopulmonary aspergillosis is usually considered when
asthma is complicated by radiographic or clinical evidence of
recurrent pneumonic infiltrates, bronchiectasis or pulmonary fibrosis.
A variety of disease-related immunological alterations have been
reported in allergic bronchopulmonary aspergillosis. Antigen extracts
of A. fumigatus, A. niger and A. clavatus have been shown to
activate the alternative complement pathway in fresh human serum from
healthy humans. Total serum IgE is elevated in most, but not all,
instances of allergic bronchopulmonary aspergillosis and
Aspergillus-specific IgE is substantially increased as measured by
radioimmunoassay.
The immune pathogenesis of allergic bronchopulmonary
aspergillosis is thought to involve direct activation of complement by
Aspergillus antigen, IgE-antibody production with subsequent release
of vasoactive amines from mast cells, as well as IgG-antibody
production and deposition of antigen-antibody complexes in the
broncho-alveolar tree. Local deposition of immune complexes may
activate the complement pathway and generate chemotactic factors for
polymorphonuclear leucocytes in peripheral blood and produce a
resultant immune complex-initiated Arthus-type reaction in lung
tissues. Also "late phase" eosinophil-mediated IgE-dependent reactions
have been suggested to be involved in the pathogenesis of the disease.
2.7.4 Extrinsic allergic alveolitis
Extrinsic allergic alveolitis (EAA) is usually defined in
pathological terms as a granulomatous inflammatory reaction which
predominantly involves the gas-exchanging parts of the lung and which
is the outcome of a specific immunological response to an inhaled
substance. The vast majority of reported cases have been caused by
inhaled organic dusts, but a few cases have been attributed to inhaled
isocyanates, particularly diphenyl methane diisocyanate (MDI) but also
hexamethylene diisocyanate (HDI) and toluene diisocyanate (TDI). No
reported case has been validated by biopsy evidence of the
characteristic pathological appearances; cases have been identified on
the basis of:
a) Characteristic clinical history;
b) Changes on chest radiograph;
c) Pattern of functional change following controlled isocyanate
inhalation;
d) Proportions of cells received at bronchoalveolar lavage.
Typically, patients present with a history of recurrent episodes
of breathlessness associated with systemic symptoms of fever, malaise
and chills. A few (but only a minority of reported cases) have had
abnormal chest radiographs.
In the majority of cases the diagnosis has been made by the
response to inhalation testing or the pattern of cells recovered at
bronchoalveolar lavage. Inhalation testing provoked the changes of an
"alveolar reaction" with proportionate reduction in forced expiratory
volume in 1 second (FEV1), forced vital capacity (FVC) and in
transfer factor (TLCO) accompanied by a neutrophil leucocytosis and
fever. The cells recovered at bronchoalveolar lavage have, as is
characteristic of EAA, shown an increase in the proportion of
lymphocytes, on occasion by more than 50%.
In some cases IgG antibody to a human serum albumin conjugate of
the relevant isocyanate - MDI-HSA, TDI-HSA and HDI-HSA - has been
identified in serum. The outcome of EAA caused by isocyanates has been
little reported, but most cases, even if showing significant
functional impairment at the time of diagnosis, would seem to have no
permanent residual disability after avoidance of isocyanate exposure.
2.7.4.1 Farmer's lung
Farmer's lung affects workers who handle mouldy hay or grain. It
originates from poor conditions of storage, involving high dust levels
and humidity. Microorganisms responsible for Farmer's lung are moulds,
above all Micropolyspora faeni, and Thermoactinomyces vulgaris.
Specific precipitins are found in the blood, especially antibodies of
the IgG class. This disease is classified as a Type III
hypersensitivity. Better storage and work practices reduce the
incidence.
Sensitization takes some time to occur. Clinically, patients
suffer respiratory distress accompanied by fever appearing 8-10 h
after handling mouldy hay, straw or grain and presenting with fever,
shivering, chest pains, lassitude, sweating, headaches and coughing,
sometimes accompanied by haemoptysis. Fine auscultatory chest
crepitations may be present. In typical forms, the chest X-ray shows
miliary infiltrates and micronodules. Later, pulmonary fibrosis
appears progressively when the disease reaches a chronic stage. There
is also impairment in alveolar gas diffusion (so called restrictive
syndrome) and, in the most advanced cases, an alveolar-capillary
block, which leads to chronic pulmonary heart failure.
2.7.4.2 Bird-fancier's lung
Bird fancier's lung is another disease of the same type, found
especially among pigeon breeders, but also in those handling other
birds. The disease is due to the development of precipitating
antibodies against serum proteins of relevant avian species, e.g.,
pigeons, parrots, chickens, pheasants and turkeys.
2.8 Type IV hypersensitivity diseases
Although cell-mediated immunity has fully developed in
vertebrates for their benefit by facilitating effective eradication of
microorganisms and abnormal cells, T-cell mediated reactions can,
under certain conditions, also cause disease (Table 15). Although
allergic contact dermatitis probably represents the most common T-cell
mediated disease, a few other pathological conditions are briefly
reviewed here.
Table 15. Pathology caused by Type IV hypersensitivity
Type IV induced disease Antigens, chemicals
Allergic contact dermatitis low relative molecular mass chemicals, drugs
Protein contact dermatitis proteins
Granulomatous disease mycobacterial antigens, beryllium
Autoimmune disease, e.g., autoantigens, e.g., pancreatic islet antigens
diabetes mellitus Type I
Hypersensitivity pneumonitis toluene diisocyanate, beryllium, heavy metals
Protein contact dermatitis is another example of Type IV
hypersensitivity.
One of the more serious complications of Type IV hypersensitivity
is the formation of granulomata. In general, T-cell immunity to
infectious agents confers a long-lasting state of protective immunity.
Macrophages, activated by the T-cell cytokines, can attack the
pathogen and should be considered as important effector cells here. If
the microorganisms are not readily killed and degraded, however,
macrophages may become "frustrated". They fuse to form multi-nucleated
giant cells or develop into large macrophages ("epitheloid" cells).
Together these cells can form new structures, so-called granulomata,
in which the macrophages with the foreign material are being isolated
from the environment by a layer of surrounding T-cells producing
cytokines and fibroblasts. The expansion and outgrowth of new
granulomata, especially at vulnerable sites, may cause considerable
tissue damage and loss of function.
In general, granuloma formation occurs when Type IV reactivity is
directed towards persistent indigestible antigens. A number of
organisms can induce granulomatous disease: Mycobacterium
tuberculosis and M. leprae, Treponema pallidum, Schistosoma,
and Yersinia enterocolitica. Importantly, a number of exogenous,
non-infectious agents can also evoke granulomatous reactions.
T-cell-mediated immune reactions may also cause disease when the
T-cell response is directed to autologous tissue. The crucial role of
T-cells, for instance, in the breakdown of the insulin-producing
beta-cells of the pancreatic Islets, leading to insulin-dependent
diabetes mellitus, is well established.
Other autoimmune diseases can sometimes be precipitated by Type
IV reactions evoked by completely unrelated antigens. Psoriasis may be
an example of an autoimmune disease that could be triggered by contact
allergens. How exactly these chemicals trigger the disease is not
completely clear. It is, however, most likely that any damage to the
skin, either toxic, physical or immunological, that recruits
sufficient lymphocytes from the circulation to include some
auto-reactive, keratin-specific T-cells will trigger a local response
in susceptible patients, resulting in a psoriatic lesion.
T-cell-mediated immunity plays a crucial role in the pathogenesis
of some lung diseases. Environmental organic chemicals like toluene
diisocyanate and trimellitic anhydride, but also inorganic compounds
as chromium and nickel are known sometimes to cause pulmonary disease.
The extent to which Type IV-mediated immune responses are involved in
these disorders is discussed in section 2.1.4.2.
Allergic contact dermatitis is considered to be the most frequent
pathological manifestation of Type IV reactivity. In allergic contact
dermatitis, T-cells are sensitized to proteins, environmental agents
and chemicals, entering the body via the skin. Repeated exposure to
such chemicals results in persistent eczematous inflammatory reactions
at the site of allergen contact. Although allergic contact dermatitis
can be regarded as a prototype of delayed-type hypersensitivity, the
sensitization process for chemical contact allergens, which already
starts in the most superficial layers of the skin, is very special.
The mechanism by which chemicals induce and elicit hypersensitivity
reactions in the skin will, therefore, be described in more detail.
2.8.1 Chronic beryllium disease
Chronic beryllium disease is a systemic disorder with primary
manifestations in the lungs. The pathogenic beryllium compounds
include metallic beryllium, beryllium alloys and beryllium oxide fume
(IARC, 1993). Inhalation of low levels of beryllium dusts or salts
over months to years is associated with a chronic interstitial
pulmonary granulomatous disorder clinically similar to sarcoidosis
(Freiman & Hardy, 1970; Jones Williams, 1988; Williams, 1989). The
skin manifestations of beryllium disease consist of contact dermatitis
and subcutaneous granuloma formation with occasional ulceration.
The concept that the granulomas of chronic beryllium disease are
T-cell-mediated immune granulomas is supported by the observations
that:
a) beryllium (i.e., the antigen) persists in the lung for long
periods (Jones Williams & Wallach, 1989);
b) large numbers of T-cells and non-caseating granulomas are
present in the lung (Williams, 1989);
c) in response to beryllium salts, lung and blood T-cells
proliferate and release lymphokines in vitro, a parameter also
used diagnostically to distinguish beryllium disease from
sarcoidosis (Williams & Williams, 1982; Rossman et al., 1988;
Newman & Kreiss, 1992; Newman et al., 1994);
d) intradermal administration of beryllium salts induces a
local granulomatous response in these individuals.
In chronic beryllium disease, the lung T-cell population is
predominantly of the CD4+ phenotype (Rossman et al., 1988; Saltini et
al., 1989). These CD4+ T-cells, compared to blood T-cells from the
same individual or compared to T-cells from normal individuals,
exhibit increased proliferation in response to beryllium (Rossman et
al., 1988; Saltini et al., 1989). The T-cells are activated,
expressing HLA class II molecules and IL-2R and releasing IL-2
(Pinkston et al., 1984; Saltini et al., 1989). Furthermore, the
beryllium-induced lung T-cell proliferation is Class II-restricted.
Chronic beryllium disease is strongly associated with HLA-DPB1 *0201,
and all beryllium-specific (BeSO2) T-cell clones have been shown to
be restricted by this allele.
Analysis of T-cell lines and T-cell clones of individuals with
this disease has confirmed that the beryllium-induced response is
antigen- specific and that all the responder cells are CD4+ T-cells
(Saltini et al., 1989).
Thus, from the information available, it appears that chronic
beryllium disease is a classic example of an immune granuloma host
response. Why an element like beryllium should do this is not clear,
but two not mutually exclusive hypotheses could explain it. Firstly,
it is likely that most disease is caused by dusts of beryllium metal
or salts, so that the particulate forms a nidus around which
macrophages ingest, allowing the beryllium to be slowly released.
Secondly, soluble beryllium salts interact with proteins, such that
the beryllium becomes an immunogenic hapten in the context of the
protein.
In an epidemiological study of groups exposed to the combustion
products of coal containing a high concentration of beryllium, Bencko
et al. (1980) found elevated levels of IgG and IgA and increased
concentrations of autoantibodies (anti-nuclear and anti-mitochondrial
antibodies).
2.8.2 Systemic autoimmune diseases
Several organ-specific autoimmune diseases such as pemphigus and
pemphigoid (section 2.6.5) and myasthenia gravis (section 2.6.6) have
been discussed above. Many of the major rheumatological disorders are
autoimmune in nature. Although systemic lupus erythematosus (SLE) can
be ranked under Type III immune complex disorders, for other
autoimmune diseases this categorization is less clear-cut.
2.8.2.1 Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is a chronic systemic
inflammatory disease that follows a course of alternating
exacerbations and remissions. Multiple organ system involvement
characteristically occurs during periods of disease activity (Fye &
Sack, 1991) (see also section 4.6.2). The disease predominantly
affects women (female to male ratio of 9:1) of childbearing age;
however, the age at onset ranges from 2 to 90 years. It is more
prevalent among non-whites than Caucasians. Family studies have
demonstrated a genetic susceptibility to the development of SLE.
Autoantibody formation in SLE is partially genetically determined:
patients with HLA-DR2 are more likely to produce anti-dsDNA
antibodies, those with HLA-DR3 produce anti-SS-A and anti-SS-B
antibodies, and those with HLA-DR4 and HLA-DR5 produce anti-Sm and
anti-RNP antibodies. Reduced serum complement and the presence of
autoantibodies to double-stranded (ds) DNA are hallmarks of active
SLE, distinguishing this entity from other lupus variants. Antibodies
to single-stranded DNA and particularly against histone proteins are
characteristic of some drug-induced forms of SLE, such as
procainamide-induced lupus (Rubin, 1989; Rubin et al., 1995).
Although in most cases the etiology of SLE is unknown, a wide
variety of medicinal and environmental agents have been associated
with the elicitation of SLE at low incidence in susceptible
individuals (Kammüller et al., 1989a; Adams & Hess, 1991; Uetrecht,
1992).
2.8.2.2 Rheumatoid arthritis
Rheumatoid arthritis is a chronic, recurrent, systemic
inflammatory disease primarily involving the joints (Fye & Sack,
1991). It affects 1-3% of people in the USA, with a female to male
ratio of 3:1. Constitutional symptoms include malaise, fever and
weight loss. The disease characteristically begins in the small joints
of the hands and feet and progresses in a centripetal and symmetrical
fashion. Elderly patients may present with more proximal large-joint
involvement and deformities are common. Extra-articular manifestations
such as vasculitis, atrophy of skin and muscle, lymphadenopathy,
splenomegaly and leucopenia, are characteristic of rheumatoid
arthritis and often cause significant morbidity.
The cause of the unusual immune responses and subsequent
inflammation in rheumatoid arthritis is unknown. HLA-D4 and HLA-DR4
occur in approximately 70% of patients with rheumatoid arthritis. Some
patients who are negative for HLA-D4 and HLA-DR4 carry the HLA-DR1
gene. It is possible that these and perhaps other genetic determinants
impart susceptibility to an unidentified environmental factor, such as
a virus, that initiates the disease process. The most important
serological finding is the elevated rheumatoid factor titre, present
in over 75% of patients (Fye & Sack, 1991).
2.8.2.3 Scleroderma
Scleroderma or progressive systemic sclerosis is a disease of
unknown cause characterized by abnormally increased collagen
deposition in the skin (Fye & Sack, 1991) (see also section 4.6.3).
The course is usually slowly progressive and chronically disabling,
but it can be rapidly progressive and fatal because of involvement of
internal organs. It usually begins in the third or fourth decade of
life. Children are occasionally affected. The prevalence of the
disease is 4-12.5 cases per million population. Women are affected
twice as often as men, and there is no racial predisposition.
Scleroderma is a manifestation of various diseases, many of them
autoimmune (Alarcon-Segovia, 1985). Two primary forms of scleroderma
exist: localized or systemic. The systemic form, progressive systemic
sclerosis (PSS), has in turn two variants: the diffuse and the CREST
syndrome (acronym for Calcinosis, Raynaud's phenomenon, Esophageal
involvement, Sclerodactyly and Telangectasia). Autoantibodies to DNA
topoisomerase I (scl 70) and centromere may be useful serological
markers for these respective diseases. The occurrence of progressive
systemic sclerosis with features previously considered characteristic
of SLE, rheumatoid arthritis, dermatomyositis and Sjögren's syndrome
and associated with high titres of antibodies to nuclear
ribonucleoprotein has been termed mixed connective tissue disease
(MCTD) (Alarcon-Segovia, 1985). In contrast to other autoimmune
diseases, cellular infiltration in scleroderma is minimal or absent in
all organs except the synovium, where impressive collections of
lymphocytes and plasma cells can be seen. Unfortunately, research on
the pathogenesis of scleroderma is severely hampered by the absence of
an animal model.
It has been suggested that certain chemicals may be associated
with some forms of scleroderma, e.g., tri- and perchloroethylene
(Sparrow, 1977; Saihan et al., 1978; Flindt-Hansen & Isager, 1987),
vinyl chloride (Lange et al., 1974; Ward et al., 1976; Black et al.,
1983), silicone (Rose & Potter, 1995) and epoxy resins (Yamakage et
al., 1980).
2.8.2.4 Sjögren's syndrome
Sjögren's syndrome is a chronic inflammatory disease of unknown
cause characterized by diminished lacrimal and salivary gland
secretion resulting in keratoconjunctivitis sicca and xerostomia (Fye
& Sack, 1991). There is a dryness of the eyes, mouth, nose, trachea,
bronchi, vagina and skin. In one-third of the patients, the disease
occurs as a primary pathological entity (primary Sjögren's syndrome).
In the remaining patients, it occurs in association with rheumatoid
arthritis or other connective tissue disorders such as SLE. Ninety
percent of patients with Sjögren's syndrome are female. Although the
mean age at onset is 50 years, the disease also occurs in children.
Patients with Sjögren's syndrome have an abnormal immunological
response to one or more unidentified antigens characterized by
excessive B-cell and plasma cell activity, manifested by polyclonal
hypergammaglobulinaemia and the production of rheumatoid factor,
antinuclear factors, including antibodies to SS(A) and SS(B),
cryoglobulins, and anti-salivary duct antibodies. Both B- and
Th-lymphocytes and plasma cells infiltrate involved tissues. No single
immunological test is diagnostic for Sjögren's syndrome, although a
spectrum of nonspecific immunological abnormalities occurs in these
patients. Histological demonstration of lymphocytic infiltration in a
biopsy specimen taken from the minor labial salivary gland is the most
specific and sensitive diagnostic test for Sjögren's syndrome (Fye &
Sack, 1991).
2.8.2.5 Hashimoto's disease
Hashimoto's disease, autoimmune thyroiditis, is the classical
example from which much of the knowledge of autoimmune disorders has
come (Gell et al., 1975; Roitt et al., 1998).
Antibodies are formed to several antigens in follicular cells of
the thyroid, including specific domains of thyroglobulin, thyroid
peroxidase and certain surface receptors. Delayed-type cellular
hypersensitization also occurs. The consequence is often initial
stimulation of the thyroid, followed after a variable period by
progressive destruction of the follicular cells, infiltration by
lymphocytes and plasma cells, often containing germinal centres, and
eventual fibrosis. The clinical disease, which is much more common in
women than in men, may be marked by initial thyrotoxicosis, which is
invariably followed by progressive hypothyroidism and myxoedema.
Thyroid autoantibodies and variable lymphocytic infiltration are
common in many other autoimmune diseases, so other tissues and organs
may also be affected and antibodies against these are frequently
found.
The cause of Hashimoto's disease is rarely known but it may
sometimes follow an overt viral infection of the thyroid and it has
been associated with high exposure to iodine.
Thyroid autoantibodies of several types are found in many
apparently healthy individuals and are common in patients suffering
from other autoimmune diseases.
3. FACTORS INFLUENCING ALLERGENICITY
3.1 Introduction
Allergens can be defined as antigens that give rise to allergy
(Sherrill et al., 1994). The molecular properties that distinguish an
allergen from an antigen are not known, but certain features appear to
be associated with allergens. Induction of allergic responses is
highly dependent upon a number of exogenous, as well as endogenous,
factors.
3.2 Inherent allergenicity
Most allergens are proteins. The structurally known allergens
from pollen, mammals, insects and foods are all proteins (or
glycoproteins) with a relative molecular mass of 10 000-40 000 (King
et al., 1995). Regarding IgE-mediated allergy, it is known that the
IgE antibodies are not formed to an entire allergen, but rather to
certain epitopes on the molecule. IgE binding sites are referred to as
B-cell epitopes. For a protein to be allergenic, it must be
multivalent, expressing more than one B-cell epitope. This allows
antigen to bind to more than one IgE molecule on the surface of a mast
cell or basophil, and induce these cells to generate and release
mediators that initiate the allergic reaction.
B-cell epitopes usually involve 12-15 linear amino acids,
although these epitopes may be non-contiguous. In the latter
situations, tertiary folding of the molecule provides the epitopes
(i.e., conformational epitopes). Allergens must also exhibit T-cell
epitopes, the 6-8 amino acid fragments presented to T-cells by
antigen-presenting cells such as macrophages. This interaction is
necessary to initiate the process of antigen-specific IgE synthesis.
Factors such as "foreignness", size and charge influence
allergenicity and sensitization. Allergenic proteins do not possess
physicochemical properties that distinguish them from non-allergenic
proteins. Foreignness refers to the concept of "non-self". In general,
the more foreign the substance, the greater is its immunogenicity. The
relationship of foreignness to allergenicity is not known. The larger
the antigen, the more likely it is to contain epitopes.
Compounds with a relative molecular mass smaller than 1000
typically are not immunogenic; those with relative molecular mass
between 1000 and 6000 may or may not be immunogenic, whereas those
with a relative molecular mass greater than 6000 are generally
immunogenic (Benjamini & Leskowitz, 1991). Allergens with small
relative molecular mass are termed "haptens". Such chemicals are
believed to couple to macromolecules to become immunogenic. In
general, the nature or identity of macromolecular "carriers" is not
known.
Certain inorganic chemicals are particularly potent sensitizers
on exposure of the skin, e.g., nickel- and platinum-containing
compounds, and, in some instances, of the respiratory tract (Rycroft
et al., 1995; Vos et al., 1996). Cross-reactivity has been observed
between allergic sensitization to nickel and chromium salts, and
between platinum, palladium and related elements.
Physicochemical complexity of a compound also favours
immunogenicity, whereas homopolymers of amino acids, such as
polylysine, are usually poor immunogens. When the complexity is
increased, i.e., by attachment of moieties that of themselves are not
immunogenic, the entire molecule becomes immunogenic (Landsteiner &
Rostenberg, 1939). For example, attachment of dinitrophenol to
polylysine renders the structure immunogenic, (Benjamini & Leskowitz,
1991).
Certain physical and chemical characteristics appear to be
associated with allergens. Protein allergens tend to possess
biological activity. Haptens tend to have chemical reactivity (or are
metabolized into reactive compounds); contact allergens are often
lipophilic. Such factors might have functional importance by
facilitating access of the allergen to the immune system, and by
interfering with regulatory mechanisms of the immune response. For
example, many protein allergens have been shown to possess enzymatic
activity (Stewart, 1994). The house dust mite allergen Der p I is a
serine protease (Chua et al., 1988). There is evidence that the
proteolytic activity enhances penetration of the allergen through the
mucosa (Herbert et al., 1995) and stimulates the synthesis and release
of the Th2-associated allergy-promoting cytokine IL-4 from mast cells
and basophils (Machado et al., 1996). Furthermore, it has been shown
that Der p I selectively cleaves the lymphocyte surface membrane
molecules CD23 (Hewitt et al., 1995; Schulz et al., 1997) and
CD25-alpha subunit (Schulz et al., 1998) and releases them into the
fluids surrounding the cells. Whereas the low-affinity IgE receptor
CD23 on the cell surface mediates negative feedback on IgE synthesis,
released soluble CD23 promotes IgE synthesis. Thus, the enzymatic
cleavage of CD23 by Der p I will enhance the synthesis of IgE, a key
mediator molecule in allergy. Furthermore, cleavage of the IL-2
receptor CD25-alpha subunit will strongly inhibit the proliferative
response and production of IFN-gamma in Th1-cells. Consequently, the
immune response to Der p I, and possibly other protein antigens
simultaneously presented to the immune system, will be biased towards
Th2-cells and an allergic response.
Many of the respiratory chemical allergens possess distinctive
functionalities that are thought to endow the chemical with
allergenicity. Studies have been undertaken of structural features and
physicochemical properties associated with respiratory allergens, and
structure-activity relationship (SAR) models have been developed
(Graham et al., 1997). Such factors include transport parameters,
electron density and chemical reactivities. These models, as well as
SAR models of allergic contact dermatitis, are discussed in chapter 6.
The ability of the immune system to recognize and distinguish
specific spatial regions (epitopes) on molecules has resulted in the
development of reagents and methodology to map these epitopes on
molecules such as drugs, proteins and microorganisms (Saint-Remy,
1997). The immune system can distinguish between structures that are
almost identical, i.e., that differ from one another by a single amino
acid substitution, or by a conformational change. Epitope mapping is
performed by generating panels of antibodies of known specificity.
Examples of the use of such antibodies are: a) in physiology to
identify structures that allow molecules to interact with their
receptor, b) in pathology to identify particular T- or B-cell epitopes
on antigens, c) in design of vaccines to either increase efficacy or
stimulate certain types of responses, such as T-cell responses, d) in
microbiology to aid in typing microorganisms.
3.2.1 Inherent properties of chemicals inducing autoimmunity
A variety of medicinal drugs with a relative molecular mass of
less than 1000 can elicit systemic hypersensitivity reactions and
autoimmune disorders in susceptible individuals at low incidence
(Adams & Hess, 1991). Chemical agents, drugs in particular, with a
documented potential to induce autoimmune disorders such as SLE,
belong to different chemical classes. These include, among others,
derivatives of aromatic amines, hydrazines, hydantoins, thioureylenes,
oxazolidinediones, succinimides, dibenzazepines, phenothiaines,
sulfoamides, pyrazolines, amino acids (Kammüller et al., 1989a; Adams
& Hess 1991; Uetrecht, 1992), amines (Nilsson & Kristofferson, 1989),
halothane (Gut et al., 1995), mercuric chloride (Pelletier et al.,
1994), gold preparations (Sinigaglia, 1994), occupational or
environmental chemicals such as tri- and perchloroethylene (Sparrow,
1977; Saihan et al., 1978) and vinyl chloride (Ward et al., 1976;
Black et al., 1983) (see also section 4.4). Environmental nitrophenols
have been suggested to be able to elicit or perpetuate human
autoimmune disorders (Lauer, 1990). Many of these compounds are
heterocyclic and contain at least one aromatic group, suggesting that
particular chemical entities may favour induction of immune
dysregulation.
From a pharmacological point of view, the majority of autoimmune
disease-inducing drugs are beta-adrenergic-receptor-blocking
compounds, drugs acting on the central nervous system (CNS),
anti-thyroid agents and anti-infective agents. In view of the tight
functional connectivity between immune, nervous and endocrine systems,
which is at least partially effected by shared receptors and mediators
among the systems, it is possible that CNS drugs modulate immune
responses by acting at these receptors or inducing common mediators.
Lupus-inducing compounds have the capacity to be oxidized by the
extracellular myeloperoxidase-H2O2 system of activated neutrophils,
despite their chemical and pharmacological heterogeneity (Uetrecht,
1992; Jiang et al., 1994). Despite this substrate promiscuity of
myeloperoxidase, analogues of lupus-inducing drugs with blocked or
missing functional groups such as -NH2, -NHNH2-, -SH, -Cl or OHC3
are not metabolized by myeloperoxidase (Jiang et al., 1994).
In order to become antigenic to T-cells, haptens must bind
carrier proteins, and whether or not T-cells may require covalent
modification of MHC molecules for hapten recognition is a matter of
debate. Investigation of mechanisms of allergic and autoimmune
reactions has pointed to a major role of trinitrophenol- and
gold-hapten-modified MHC-associated peptides as T-cell-antigenic
structures (Martin & Weltzien, 1994; Sinigaglia, 1994; Weltzien et
al., 1996).
3.3 Exogenous factors affecting sensitization
3.3.1 Exposure
3.3.1.1 Magnitude of exposure
The development of sensitization and the responses in individuals
depend upon the frequency and intensity of acute symptomatic episodes
(Friedmann et al., 1983; Ollier & Davies 1994). Clinical and
experimental evidence indicates that exposure concentration is of
critical importance for the development and exacerbation of allergy.
For dermal and respiratory sensitization, in animal and human studies,
the dose-response concept has been shown to operate at both the
induction and elicitation phases of sensitivity.
The role of dose in induction of contact sensitization has been
demonstrated in animal models, including guinea-pigs and mice (Chan et
al., 1983; Stadler & Karol, 1985). Data revealed a relationship
between the amount of chemical applied epicutaneously to the animals
and both the severity of the ensuing reaction and the percentage of
animals responding. In both species, and with all chemicals tested, a
no-effect concentration was also observed.
Both the induction and elicitation phases of respiratory
sensitization have been shown to be under the influence of the dose
(concentration) of allergen. With protein allergens, sensitization to
detergent enzymes was found to diminish as the workplace atmospheric
levels of the enzyme dust were reduced (Juniper et al., 1977). With
chemical allergens, clinical studies have indicated an association of
episodic high (accidental) exposure with development of sensitization
(Brooks, 1982). In a study of isocyanate workers, a relationship was
found between the number of spills and the percentage of workers
displaying symptoms of allergic disease (asthma, bronchitis and
decreased pulmonary function). With Western red cedar, an association
was also noted between workplace exposure and either the incidence of
pulmonary sensitization to the wood dust or the prevalence of
occupational asthma (Brooks, 1982). A further indication of the
importance of exposure concentration on sensitization is the reported
decrease in the number of cases of toluene diisocyanate (TDI)
sensitization coincident with the lowering of the permissible
occupational exposure levels (Karol, 1992).
Animal studies have established more precisely the relationship
between the exposure concentration, the elicitation concentration, and
development of respiratory sensitivity (Karol, 1994 a,b). Once again,
the concentration of inhaled allergen was shown to be a prime factor
controlling the development of sensitivity (Karol, 1983). Exposure of
guinea-pigs to monitored concentrations of TDI vapour resulted in
development of pulmonary sensitization only when the exposure
concentration was > 0.25 ppm (> 1.8 mg/m3) (Karol, 1983).
Exposure to lesser concentrations, even for extended periods of time,
did not result in sensitization. Both a threshold concentration and a
no-effect concentration were observed, suggesting the existence of a
safe level of exposure for this potent allergenic chemical (Karol,
1986).
A threshold concentration for sensitization to the allergenic
proteolytic enzyme, subtilisin, was also noted in animal studies
(Thorne et al., 1986). Groups of guinea-pigs were exposed to
atmospheres containing increased concentrations of the enzyme for
15 min per day on each of 5 consecutive days. Sensitivity developed in
animals exposed to the high concentrations but not in those exposed to
the lesser ones. Even long-term exposure of animals to the lower
concentrations failed to produce sensitization, although the animals
had received a cumulative exposure comparable to that which regularly
induced sensitivity when given over 5 days. This enzyme is believed to
be a particularly potent allergen and has a threshold limit value of
0.06 mg/m3. Clinically, workplace sensitization to the enzyme has
been dramatically reduced by lowering workplace exposures, and by
changing the formulation of the allergen to make it less readily
airborne (Juniper et al., 1977; Thorne et al., 1986; Sarlo & Karol,
1994).
3.3.1.2 Frequency of exposure
Increased frequency of inhalation exposure to allergen increased
the sensitization rate (Karol, 1986). However, studies clearly
demonstrated the importance of the exposure concentration exceeding a
threshold level for the chemicals. Repeated inhalation exposure of
guinea-pigs to sub-threshold concentrations of subtilisin (Thorne et
al., 1986) or TDI (Karol, 1983) failed to sensitize the animals,
whereas the same total exposure given over a shorter time span
consistently resulted in sensitization. Long-term sub-threshold
exposure to TDI resulted in neither respiratory sensitization nor
production of specific antibodies (Karol, 1983).
Clinically, chronic low-level exposure has been implicated in the
development of respiratory allergy to some airborne chemicals, notably
TDI (Karol, 1986). However, at that time the ability to measure low
concentrations of TDI was limited. Long sampling periods were often
required which eliminated the possibility of detecting sporadic high
TDI concentrations (Karol, 1986). As a result, in such studies no
conclusion can be drawn regarding the development of sensitization as
a result of repeated low-level exposure.
The influence of chronic low-level exposure to detergent enzymes
on the development of occupational sensitization to these enzymes has
been studied (Juniper et al., 1977). Using skin prick tests as an
indication of sensitization, conversion to skin test positivity was
observed following 20 months of employment for both high- and
low-exposure groups. A reduction in the dust levels in the workplace
was coincident with a decreased conversion rate (Juniper et al.,
1977).
In the platinum industry, respiratory sensitization to soluble
platinum salts has occurred under conditions where exposure is below
the official workplace limit. Maynard et al. (1997) examined the
possibility that high short-term exposures might be responsible but
found there was no evidence for this. In a cross-sectional study of
respiratory and dermal sensitization to platinum salts in a population
of precious metals refinery workers, skin reactivity was found in
workers exposed to permissible levels of platinum salts and was
associated with respiratory and dermal sensitization, but not with
atopic status (Baker et al., 1990). Merget et al. (1994), in a study
of platinum refinery workers, found that in workers who developed
immediate-type asthma caused by platinum salts both nonspecific and
specific bronchial responsiveness did not decrease after removal from
exposure.
Repeated exposure of guinea-pigs to contact allergens resulted in
reduced local reactions (Boerrigter et al., 1987) with eventual
diminution such that the skin reactions were almost non-existent.
However, the state of unresponsiveness disappeared upon
discontinuation of the repeated allergen exposures.
In humans, repeated exposure may also down-regulate the local
inflammatory response in the skin. This phenomenon is termed
"hardening". However, the individual remains sensitized. By contrast,
repeated systemic exposure could also "desensitize". This effect is
thought to be due to the high total dose administered.
3.3.1.3 Route of exposure
The route of exposure has an influence on the outcome of exposure
to an allergen. In general, exposure by the inhalation or dermal route
favours sensitization, whereas exposure by the oral route favours
tolerance (unresponsiveness ). Immunological unresponsiveness can be
induced in animals by non-cutaneous exposure. Induction of "tolerance"
in humans to nickel as a result of exposure to nickel-releasing
orthodontic braces during early age has been suggested (Van
Hoogstraten et al., 1991).
Systemic unresponsiveness after ingestion of antigen has now been
described for a large variety of T-cell-dependent antigens (Mowat,
1987). Proteins such as ovalbumin and bovine serum albumin (Silverman
et al., 1982; Domen et al., 1987), particulate (erythrocyte-bound)
antigens (Kagnoff, 1982; MacDonald, 1983; Mattingly, 1984),
inactivated viruses and bacteria (Stokes et al., 1979; Rubin et al.,
1981), autoimmune-related antigens (Thompson & Staines, 1990), as well
as contact allergens, have been reported to induce oral tolerance
(Asherson et al., 1977; Newby et al., 1980; Gautam et al., 1985).
Generally, T-cell-mediated delayed-type hypersensitivity responses and
IgE production are the types of immune responses most readily
tolerized. Persistent tolerance can be induced with relatively low
antigen doses of proteins (Heppel & Kilshaw, 1982; Jarrett, 1984;
Jarrett & Hall, 1984) and contact allergens (Asherson et al., 1977;
Polak 1980; van Hoogstraten et al., 1992; Hariya et al., 1994). The
apparent ability of the intestinal immune system to prevent allergic
hypersensitivity to soluble, non-replicating antigens seems an
important pathway to prevent enteropathies (Challacombe & Tomasi,
1987; Mowat, 1984, 1987). Abrogation of oral tolerance to, for
instance, ovalbumin was found to lead to hypersensitivity responses in
the intestinal mucosa and gut-associated lymphoid tissues, resembling
those observed in food-sensitive enteropathies, e.g., coeliac disease
(see section 1.5.1.3).
If mucosal cells in the respiratory tract are the site of initial
exposure, the result is frequently production of IgA and IgE
antibodies and predisposition to Type I allergic reactions. Initial
exposure of mucosal cells in the gastrointestinal tract may have the
same effect but often produces tolerance. By contrast, skin exposure
favours Type IV sensitization. It appears that the route of first
encounter with the chemical allergen determines whether the outcome is
sensitization or unresponsiveness.
Once an individual is sensitized via the skin, subsequent oral
exposure does not tolerize, but might contribute to further
sensitization by boosting the ongoing immune response. It is even
possible to induce systemic allergic reaction via the oral route in
skin-sensitized individuals. Overall, all of these factors are
dependent upon the nature of the allergen.
3.3.2 Atmospheric pollution
The effect of indoor and outdoor air pollution on allergic
disease has received considerable attention. Environmental pollutants
have been reported to contribute to the prevalence of allergic
disease, the precipitation of allergic symptoms, and their intensity
(Ollier & Davies, 1994). Both epidemiological and experimental studies
have demonstrated that a variety of atmospheric substances (including
sulfur dioxide (SO)2, nitrogen dioxide (NO2), ozone (O3) and
particles) influence the induction and elicitation phases of the
allergic response. Effects have included adjuvant activity for
allergen-specific IgE production, modulation of mediator release from
inflammatory cells, and irritant effects on effector organs of the
allergic response (Behrendt et al., 1995) (see sections 5.13 and
5.14).
The question of whether environmental factors may be involved in
the observed increase in the prevalence of allergy is a matter of
controversy (Ring et al., 1995b; Behrendt et al., 1995; Vos et al.,
1996). There is no doubt that pollutants such as suspended particles,
automobile exhaust, ozone, sulfur dioxide and nitric oxides can be
measured in rather high concentrations in the air of many countries
that show an increasing prevalence of atopic diseases. However, some
of these pollutants, like sulfur dioxide, have shown a decrease in air
concentrations during the last decades. In a controlled prospective
trial comparing different living areas with various degrees of air
pollution in western and eastern Germany, striking differences were
shown with regard to the prevalence of respiratory atopic diseases,
with higher values for western compared to eastern Germany (von
Mutius, 1992; Schlipköter et al., 1992; Behrendt et al., 1993, 1996;
Ring et al., 1995). In contrast to atopic respiratory diseases, there
was a trend to higher prevalence rates of atopic eczema in eastern
Germany. In the same study there was evidence of an increased risk of
developing atopic eczema after exposure to natural allergens as well
as air pollutants from outdoor and indoor sources (Ring et al., 1995;
Krämer et al., 1996; Schäfer et al., 1996).
The mechanisms by which air pollutants influence allergic
reactions are not clear. Some pollutants may have a direct toxic
effect on the respiratory epithelium leading to inflammation, airway
hyperreactivity and the appearance of asthma-like symptoms in
previously non-asthmatic individuals. In cell systems, certain
pollutants have been shown to modulate degranulation and histamine
release from basophils (Ring et al., 1995). Polychlorinated biphenyls
enhance eicosanoid production by granulocytes and platelets (Raulf &
Konig, 1991). Certain pollutants may have the ability to augment or
modify immune responses to inhaled antigens or to enhance the severity
of reactions elicited in the respiratory tract following inhalation
exposure of the sensitized individual to the inducing allergen.
High concentrations of air pollutants can have irritant effects
and aggravate the symptoms of allergic respiratory and skin diseases
(Ring et al., 1995; Behrendt et al., 1996). Laboratory studies suggest
that certain air pollutants have the potential to stimulate
broncho-constriction and airway inflammation. Exposure to SO2 is
associated with chest tightness and bronchoconstriction, the
concentration required to induce a response being dependent upon the
degree of hyperresponsiveness of the individual. The effects of SO2
may be augmented in the presence of other pollutants. It has been
reported, for instance, that the sensitivity of mild asthmatics to
SO2 is increased by prior exposure to O3. Ozone has been
investigated extensively and has been found to cause bronchial
hyperresponsiveness. In controlled clinical exposure studies,
researchers have demonstrated that asthmatics are more responsive to
O3 than normal people (Ball et al., 1993; WHO, in press). Exposure of
asthmatics to O3 for 1 h caused an increase in airway responsiveness
to inhaled allergen. The proportion of cynomologous monkeys that
developed asthma and a positive skin test after inhalation of complex
platinum salts was increased in those animals that inhaled O3
concurrently (Biagini et al., 1986). The health relevance of oxides of
nitrogen, and in particular NO2, has attracted some interest since
the gas is present both outdoors and indoors. Some studies have
suggested mild effects of NO2 in asthmatics at concentrations of less
than 1 ppm (< 1.88 mg/m3); others have not found responses at levels
up to 4 ppm (7.52 mg/m3). Particulate air pollutants, especially fine
particles derived from combustion sources, are also of interest
although there have been few controlled exposure studies apart from
those involving acid aerosols.
Bioaerosols to which an asthmatic is sensitized are well known to
exacerbate asthma. Epidemiological studies describing the increase in
mortality associated with inhaled particulate matter (PM-10) provide
provocative evidence for adverse pulmonary health effects associated
with particulate pollution. The association between particulate matter
and acute mortality and morbidity has been demonstrated most strongly
with elderly people who have chronic cardiopulmonary disease
(Thurston, 1996).
Studies have demonstrated an effect on allergic disease from
substances adsorbed to airborne particles. Such substances were found
to release histamine from human basophils and had a priming effect on
anti-IgE-induced release of histamine and LTC4 (Behrendt et al.,
1995). These in vitro studies indicated that particle-adherent
substances interfere with cells involved in inflammatory processes.
There is evidence of an interaction between pollen and air
pollutants. Pollen grains in polluted areas have been shown to be
loaded with particles including heavy metals, such as lead, cadmium
and mercury. In vitro, these pollen grains were found to have
altered surface features and increased ability to release cytosolic
allergenic proteins (Behrendt et al., 1991).
3.3.2.1 Tobacco smoke
Passive exposure to tobacco smoke is a risk factor for childhood
asthma (Seaton et al., 1994; Becher et al., 1996). Studies to detect a
possible association between passive smoke and allergic disease in
adults are much more difficult to design. Asthmatic patients
frequently report exposure to passive smoke. In children, there is
evidence that tobacco smoke increases the risk for development of
wheezy bronchitis and asthma.
Tobacco smoking is associated with an increased risk of
developing IgE antibodies and asthma. The mechanism of this effect of
tobacco smoke is unknown, but may be a result of injury to the
respiratory mucosa. Several studies have indicated that subjects who
smoke cigarettes have higher IgE levels (Zummo & Karol, 1996).
Specific IgE antibody or an immediate skin test response was found to
be 4-5 times more frequent in smokers exposed to tetrachlorophthalic
acid (TCPA) and ammonium hexachloroplatinate. Initially smokers had
IgE levels similar to those of controls, but, with age, IgE levels in
smokers did not decline at the same rate as they did in the
non-smokers (Sherrill et al., 1994). This may provide an explanation
for the difference in IgE values observed in adult smokers. Moreover,
a relationship was noted between the number of cigarettes smoked and
the IgE level, suggesting causality. In female smokers, there was a
trend toward increased IgE at older ages (i.e., > 50 years).
Passive smoking has been found to be a risk factor for
development of sensitization in children (Halken et al., 1995). The
association does not necessarily imply an allergic mechanism, rather
the association can be a result of direct irritation and inflammation
of the respiratory tract. In children with atopic predisposition, a
significant correlation was found between exposure to tobacco smoke
and wheezing/persistent wheezy bronchitis. A prospective study of 94
asthmatic children found significantly more asthma symptoms in those
exposed to maternal tobacco smoke. A retrospective study with 199
children with asthma found acute exacerbations of asthma increased
with exposure to tobacco smoke. In children with past or present
atopic dermatitis, asthma was found more frequently in cases where the
mother smoked cigarettes (Halken et al., 1995).
3.3.2.2 Geographical factors
Exposure to airborne allergens, notably pollens, depends on
location, climate and time of year (Emberlin, 1994). Certain types of
air pollution reduce the amount of pollen produced, but they can also
render the proteins on pollen more allergenic (Ruffin et al., 1986).
3.3.3 Metals
Nickel is a frequent cause of contact sensitization, having a
sensitization rate of 15-50% in experimental studies. Most cases of
nickel allergy can be attributed to exposure to nickel alloys in close
skin contact, which release high concentrations of nickel when exposed
to sweat. Similarly, chromate dermatitis often relates to exposure to
hexavalent chromate in wet cement (Andersen et al., 1995).
Investigations of monozygotic female twins, where one or both were
nickel sensitive, have shown that only the twin with a history of
contact dermatitis by nickel alloy exposures gives a positive
diagnostic patch test to nickel (Menné & Holm, 1983). In vitro
diagnostic testing failed to demonstrate subclinical nickel
sensitization in family members of nickel-sensitive individuals
(Silvennoinen-Kassinen, 1981).
3.3.4 Detergents
Reports of respiratory allergic reactions in workers involved in
large-scale production of enzyme-containing detergents suggest that
the detergent component may contribute to the sensitization to the
enzyme component. The symptoms of rhinitis and/or asthma suggested a
Type I sensitization. Experimental studies in guinea-pigs, using
either inhalation or intratracheal dosing, indicated that detergents
and proteolytic enzymes enhance sensitization to allergenic proteins
(Ritz et al., 1993; Sarlo et al., 1997) when sensitization was
assessed by production of allergic antibody and respiratory responses
to allergen challenge.
3.4 Endogenous factors affecting sensitization
3.4.1 Genetic influence
3.4.1.1 Contact sensitization
Although significant genetic influences on contact sensitization
have been reported, lack of reproducibility and smallness of these
effects suggest their minor importance, as compared to exposure, in
clinical contact sensitization. A few studies utilizing different
inbred mice and guinea-pig strains noted differences in sensitization
rates for some contact allergens (Parker et al., 1975; Andersen &
Maibach, 1985). In humans, a well-controlled family study indicated
that experimental contact sensitization in children was greater when
both parents could be sensitized by the same substance compared to
children where only one parent could be sensitized (Walker et al.,
1967). A population-based twin study focusing on nickel allergy found
a significant genetic effect for the risk of developing this contact
sensitivity (Menné & Holm, 1983). However, twin studies, using other
designs, have failed to show such an association. Also, studies on
frequencies of HLA genes in contact hypersensitive individuals have
not revealed consistent patterns (Menné & Holm, 1986). Comparisons
between frequencies of sensitization in different ethnic populations,
e.g., for nickel in black and Caucasian groups, revealed either
similar or different rates, depending on the study designs (Menné &
Wilkinson, 1995).
Histamine releasibility from mast cells and basophils is a
critical event in many allergic disorders. In twin studies, this event
(which is related to the quantity of IgE present on the cells) was
shown to be under genetic control (Bonini et al., 1994).
Products of HLA class II genes are involved in allergen
presentation by antigen-presenting cells. Since these genes are highly
polymorphic, different HLA genes represent risk factors for
development of allergic asthma. Increased responsiveness to the
ragweed allergen Ra 5 was found to be associated with the HLA gene DR
2/DW 2.
There is evidence for a genetic contribution to sensitization to
some allergens of low relative molecular mass.
3.4.1.2 IgE-related allergy
One of the characteristic features of atopy is the production of
IgE in an exuberant and prolonged fashion to common largely innocuous
environmental allergens, such as house dust mites and pollen. Most
atopics are allergic to more than one common environmental allergen
and this introduces the concept that the causation of atopy occurs at
a variety of levels: generalized hyper-IgE responsiveness; IgE
response to specific allergens or epitopes; clinical disease
expression (Hopkins, 1997).
The genetics of production of total serum IgE have been studied.
In such studies consideration has to be given to the following
factors, since each has been shown to affect IgE levels: allergic
exposure, parasitic infection, age, sex and smoking. A correlation was
found between the total serum IgE of parents and children, suggesting
the involvement of one or more genes (Sherrill et al., 1994). However,
agreement on the model of inheritance is lacking. Linkage of loci for
total serum IgE and BHR to chromosome 5q has been reported (Sherrill
et a1., 1994). Mapping of this area of the chromosome will be
important for further progress. Total serum IgE appears to be under
strong genetic control (Bonini et al., l994), even in the presence of
environmental factors such as smoking. A gene for IgE response with
maternal inheritance was identified at chromosome 11q (Cookson et al.,
1989). High levels of IgE in cord blood appear to be a strong
indicator of subsequent development of atopic disease.
The genetic factors that determine the specificity of the
IgE-mediated response are thought to be independent of those governing
total serum IgE and may be linked to the human leucocyte antigen (HLA)
complex (Sibbald, 1997). Products of HLA Class II genes are involved
in allergen presentation by antigen-presenting cells. HLA Class II
genes are highly polymorphic. Different HLA genes represent risk
factors for the development of asthma associated with sensitization to
allergens. Increased responsiveness to ragweed antigen (Ra5) was found
to be associated with HLADR2/DW2, and response to ryegrass (Lol pI and
Lol pII) with HLADR23 and DR5 (Marsh, 1990). Environmental factors,
such as the quality, intensity, route and duration of allergen
exposure appear to be more relevant than genetic factors in causing
allergic reaction to specific allergens (Bonini et al., 1994).
Twin studies have suggested polyfactorial control of allergy
variables such as serum levels of total IgE and IgG4, mediator release
from inflammatory cells, and target organ response. Clinical data from
32 monozygotic and 71 dizygotic twin pairs yielded a concordance for
allergic disease of 50.0% of monozygotic pairs (16/32) and 35.2% of
dizygotic pairs (25/71). The difference was not statistically
significant (Cockcroft, 1988). Histamine releasibility from mast cells
and basophils is a crucial event in allergic disorders. In twin
studies, this event (which is related to the quantity of IgE present
on the cells) was shown to be under genetic control (Bonini et al.,
l994).
Development of respiratory allergies to small relative molecular
mass chemicals, i.e., relative molecular mass less than 5000, such as
isocyanates and acid anhydrides has not been found to be associated
with atopy (Chan-Yeung, 1995), although atopy has been shown to be a
risk factor for development of respiratory symptoms to some chemical
allergens, such as hexachloroplatinate (Dally et al., 1980).
Regarding low molecular mass, or chemical allergens, an
association between sensitization to acid anhydrides and HLA-DR3
haplotype has been reported (Young et al., 1993). An association of
HLA class II alleles and isocyanate asthma was detected (Bignon et
al., 1994). Twenty-eight patients with isocyanate-induced asthma (as
documented by positive inhalation challenge) were compared with 16
exposed individuals with no history of the disease. HLA DQB1*0503 and
allelic combination DQB1*0201/0301 were associated with susceptibility
to asthma. Conversely, allele DQB1*0501 and the
DQA1*0101-DQB1*0501-DR1 haplotype conferred protection in exposed
healthy subjects. No significant difference was detected in the
distribution of HLA Class II alleles and/or haplotypes among the
immediate, late or dual responders to TDI. These results are
consistent with the hypothesis that immune mechanisms are involved in
isocyanate asthma and that specific genetic factors may increase or
decrease the risk of development of isocyanate asthma in exposed
individuals.
3.4.1.3 Other genetic factors
Another factor that may contribute to susceptibility, or
resistance, to sensitization relates to genes that control production
of IL-4, a pleotropic cytokine that influences the development of both
Th- and B-lymphocytes, the induction of Class II MHC antigens and
immunoglobulin class switching from IgM to IgE. Genes for IL-3, IL-4,
IL-5 and GM-CSF have been identified on chromosome 5 (Van Lee Uwen et
al., l989). The IL-4 gene, as well as genes that regulate its
expression, appear to be prime candidates for predisposition to atopy
since there are reports that cells isolated from atopic individuals
have the ability to overexpress the IL-4 gene relative to those from
non-atopic individuals. In addition, the human IL-4 proximal promoter
exists in multiple allelic forms, with one of the alleles having a
markedly enhanced promoter activity (Song et al., l996b). This finding
suggests a gene target to screen for genetic predisposition for atopy.
3.4.2 Tolerance
Allergenicity of a given compound may be strongly reduced in
individuals who previously developed immunological tolerance. This has
been frequently seen when the primary contacts with the allergen were
at mucosal surfaces, e.g., by its presence in food. Principles and
mechanisms of immunological hyporesponsiveness and tolerance have been
dealt with in detail above (see section 1.5).
3.4.2.1 Orally induced flare-up reactions and desensitization
Strong and long-lasting oral tolerance can only be achieved in
naive individuals, i.e., those who have not been previously exposed to
the antigen via the skin. In mice, a single feed of ovalbumin was
reported to fully suppress subsequent systemic immune responses, with
this state of tolerance persisting for up to 2 years. In contrast,
tolerance is hard to induce in primed animals but partial and
transient unresponsiveness ("desensitization") may develop after
prolonged feeding of the antigen. Similar results have been obtained
in guinea-pigs with various chemical allergens, including
dinitrochlorobenzene (DNCB) (Polak, 1980), nickel (van Hoogstraten,
1994) and amlexanol (Hariya et al., 1994). Unfortunately, essentially
similar results have been obtained in clinical trials aiming at the
treatment of autoimmune diseases, e.g., rheumatoid arthritis and
multiple sclerosis, by oral administration of putative autoantigens
(Weiner et al., 1994). Another problem with oral tolerance induction
in previously sensitized individuals arises from the tendency of
former inflammatory sites to re-inflame ("flare-up reactions"). These
reactions are likely to be due to allergen-specific effector T-cells,
which can persist for periods of several months at former inflammatory
sites (Scheper et al., 1983).
The differences between immunological responses in naive and
primed individuals may reflect changes in expression of cellular
adhesion/homing molecules and lymphocyte maturation. A qualitative
distinction exists between (difficult to stimulate/afferently acting)
naive and (easy to stimulate/efferently acting) effector/memory cells.
In contrast to naive lymphocytes, which only are activated by allergen
(modified self constituents) if presented by professional dendritic,
e.g., Langerhans cells, their progeny, known as effector/memory
lymphocytes, can also be stimulated by other cell types presenting
allergen-modified MHC class II-molecules, e.g., monocytes, endothelial
cells and B-cells. Clearly, effector/memory cells display increased
numbers of cellular adhesion molecules (CAMs), allowing for more
promiscuous cellular interactions. Amongst these, the most prominent
CAMs are the CD28 and LFA-1 molecules, with B7.1 and B7.2 and ICAM-1
as their respective ligands on APC. In addition, priming of T-cells
leads to the loss of homing receptors, such as L-selectin, which
facilitate interactions with high endothelial venules in peripheral
lymph nodes. Apparently, after sensitization, T-cells are less capable
of recirculating through the lymphoid organs, but gain ability to
migrate into the peripheral tissues. Interactions with endothelia
within inflamed skin are facilitated by the enhanced expression of
CAMs, such as the cutaneous lymphocyte-associated antigen CLA, and
effector/memory T-cells largely distribute over the peripheral
tissues, where conditions may be insufficient to convey effective
tolerogenic signals.
3.4.2.2 Non-specific and specific mechanisms of unresponsiveness
A preliminary factor contributing to non-responsiveness and/or
lack of hypersensitivity reactions at mucosal surfaces is the
epithelial barrier function, preventing entry of potentially harmful
allergens. Obviously, from an immunological point of view, this is a
"null-event", and does not have implications to subsequent encounters
with the same allergen. TGF-beta, a cytokine locally produced by
epithelial cells and immunocytes, plays a pivotal role in maintaining
epithelial barrier integrity. Importantly, the same cytokine also has
broad nonspecific immunosuppressive functions, e.g., by antagonizing
phagocytic effector cell functions of pulmonary alveolar macrophages.
Similarly, other immunosuppressive cytokines may be locally released
from epithelial cells and may act in concert with TGF-beta to
down-regulate immune effector functions, such as epithelial
cell-derived P15E-related factors which show sequence homology with
retroviral envelope proteins (Oostendorp et al., 1993).
In contrast, specific immunological tolerance depends on
decreased responsiveness of specific B- or T- cells, or release of
immunosuppressive mediators from these cells after specific challenge.
So far, no methods of permanent desensitization have been devised.
Nevertheless, how T-cells specifically recognize distinct allergens,
and how these and other inflammatory cells interact to generate
inflammation, is beginning to be understood. Exposure to high doses of
antigens may induce clonal deletion or anergy of specific B- or
T-cells by induction of apoptosis or antigen-receptor down-regulation
(Jones et al., 1990; Schönrich et al., 1991; Ohashi et al., 1991;
Melamed & Friedman, 1993).
As IL-4 and IL-13 direct IgE isotype switching, one way to
intervene in allergen-specific IgE synthesis and to inhibit or prevent
IgE-mediated allergic disease is to inhibit IL-4 and IL-13 production
by allergen-specific Th2-cells. In addition to TCR engagement by
peptide MHC complexes, optimal T-cell activation and proliferation
generally requires co-stimulatory signals provided by interaction
between CD28 or CTLA-4 on T-cells and their ligands CD80 or CD86 on
professional APC. Ligation of the TCR in the absence of these
co-stimulatory signals can result in T-cell non-responsiveness. Human
CD4+ Th2 clones specific for the house dust mite allergen Der p I
can be rendered non-responsive to subsequent Der p I challenges by
incubating them with Der p I-derived peptides, representing the
relevant minimal T-cell activation inducing epitopes, in the absence
of professional APC (Yssel et al., 1994). The mechanisms underlying
this T-cell unresponsiveness have not yet been determined. Although
these cells cannot be activated through their TCR, they proliferate
well in response to IL-2 or following activation by Ca++ ionophore
and TPA, suggesting that TCR activation or signalling pathways
immediately downstream of the TCR are disturbed.
This type of tolerance is generally short-lasting, since
(functionally) deleted lymphocytes are gradually replenished by newly
arising clones in the bone marrow and thymus and, in experimental
animal models, cannot be transferred to naive recipients, since these
still contain a fully functional repertoire, compensating for any
missing clones. On the other hand, mucosal contacts of naive
individuals with relatively low amounts of antigens, such as can be
the case with environmental or occupational exposure to chemical
sensitizers, frequently induce a long-lasting state of specific
tolerance. Transfer of lymphoid cells, in particular T-cells, from
orally tolerized animals to syngeneic naive recipients prevents their
capacity to subsequently mount immune responses to the same allergen,
revealing the existence of so-called T-regulator or suppressor cells
(Polak et al., 1980; van Hoogstraten et al., 1992, 1994; Weiner et
al., 1994).
Although "professional" suppressor T-cells may not exist (Bloom
et al., 1992; Arnon & Teitelbaum, 1993) available data support the
development of specific "regulatory" T-cells that suppress distinct
immune functions. Depending on the experimental models, such
regulatory T-cells can belong to either or both the CD4+ or CD8+
subsets (Bloom et al., 1992). Evidence is accumulating that regulatory
T-cells most often exert their role, after antigen-specific
activation, by releasing distinct cytokines antagonizing distinct
effector T-cell functions.
3.4.3 Underlying disease
There is ample evidence that underlying diseases are able to
influence the susceptibility of individuals to develop allergy. Both
the induction and the manifestation of allergy may be affected.
Conditions that promote sensitization include ongoing
inflammatory reactions at the site of allergen contact. It has, for
instance, been described that late-phase reactions of the respiratory
tract and the associated state of hyperresponsiveness, may facilitate
sensitization (priming) to other allergens (Connell, 1969). At skin
sites, a pre-existing eczema provides a risk factor for acquiring
contact sensitization. The most important factor here is probably the
local disturbance of the skin barrier, allowing for an increased
penetration of allergen. The fact that all components for an immune
response (cytokines, T-cells) have already been attracted to the site
of allergen contact may, however, additionally contribute to this
increased risk for new sensitization.
The most important diseases affecting the hosts' immune
responsiveness, and thus allergic responsiveness, include infectious
disease, neoplastic disease and immune deficiencies. The relation
between infection and the development of allergic disease is quite
complex. On one hand, respiratory viral infections are believed to
contribute to the exacerbation of asthmatic disease (Busse, 1990).
However, from clinical and epidemiological studies it would appear
that under certain conditions viral infections can also protect
against asthma. These studies include the observation of incidental
spontaneous remission of asthma during hepatitis, fever or measles, as
well as the finding of a general inverse relationship between
infections and asthma or atopy (Matricardi, 1997; Serafini, 1997). In
line with such a "protective" role it is believed that natural
infections during early childhood would prevent the development of
atopic disease later on, presumably by activation of the Th1
lymphocytes through IFN-gamma (Serafini, 1997). Reduction in family
size and increased hygiene could thus contribute to the increased
frequency of atopic disease in developed countries. Interestingly,
infectious diseases, which are known to be associated with a
predominant Th2 immune responsivenesses, like parasite infections, do
not seem to favour the development of atopic disease (Bell, 1996). In
contrast, people suffering from severe parasite infection may have
less severe reactions to other allergens, due to competition of IgE at
the Fc epsilon receptor level on mast cells. Also in HIV-positive
patients, where Th2 responses may become dominant, no clear evidence
has been obtained for enhanced atopic sensitization, although allergic
manifestations are frequently observed in these patients.
Conditions that suppress allergic reactions have been extensively
described, since contact sensitization has been applied as a method
for immune status determination in different patient groups. It is a
well-known fact that in clinical conditions associated with general
immune suppression and anergy, such as malnutrition, immunosuppressive
treatment, malignancies and severe physical trauma, Type IV reactivity
to recall antigens as well as primary sensitization to contact
allergens like dinitrochlorobenzene can be dramatically impaired.
Finally, it should be noted that certain immunological
conditions, such as those found in some immunodeficiency diseases,
e.g., in the Wiskott-Aldrich syndrome, may predispose for the
development of atopic eczema. Atopic disease is also commonly seen in
IgA deficiency.
3.4.4 Age
Childhood asthma is becoming more common and doubled in the
United Kingdom, New Zealand and Australia between 1970 and 1990.
Because of their greater activity and their developing lungs, children
may be more susceptible to sensitization as well as to adverse effects
of irritants (Zummo & Karol, 1996).
The ability to become sensitized to dinitrochlorobenzene has been
shown to be largely unchanged with age. Patch testing with Rhus
oleoresins in subjects with a history of poison ivy sensitization
showed diminished responses in the elderly (Lejman et al., 1984).
However, exposure differences as a function of age must always be
considered (Menné & Wilkinson, 1995).
IgE levels change with age. Peak levels occur in the first or
second decades of life. A longitudinal study of more than 2000
subjects conducted over a 20-year period found no gender difference in
total IgE (Sherrill et al., 1994). Both sexes had their highest IgE
levels as children. Levels fell gradually up to around age 40 and
thereafter remained constant.
3.4.5 Diet
To explain the observed increase in incidence of allergy and
asthma during the last two decades, it has been suggested that a
change in host resistance to allergy may have occurred (Seaton et al.,
1994). A change in the diet in several Western countries has been
documented. Specifically, a 20-50% fall in consumption of fresh fruits
and vegetables has been noted. Since these foods are sources of
antioxidants such as vitamin C and beta-carotene, decreased consumption,
together with that of red meat and fresh fish, would mean less
ubiquinone and fewer cofactors (such as zinc and copper) for
antioxidant defence (see section 5.10).
3.4.6 Gender
In general, women appear to have greater immune capability than
men (Menné & Wilkinson, 1995). Animal and human studies have indicated
a greater incidence of autoimmune disease in women compared with men,
as well as higher IgG and IgM levels. Women have also been reported to
produce greater cell-mediated immune responses.
In a large, controlled study, men were found more susceptible to
sensitization by dinitrochlorobenzene than women (Walker et al.,
1967). However, women were more readily sensitized to
p-aminodiphenyl aniline than were men (Walker et al., 1967). In
these studies, the issue of previous exposure to the chemical, and
therefore greater susceptibility, could not be dismissed. This factor
may also explain greater female sensitization in clinical patch tests
with nickel and cobalt. Male and female sensitization rates obtained
by maximization testing were comparable (Leyden & Kligman, 1977).
In a study of the influence of sex hormones on sensitization,
response to dinitrochlorobenzene was enhanced in women receiving oral
contraceptive hormones (Rea, 1979)
4. CLINICAL ASPECTS OF THE MOST IMPORTANT ALLERGIC DISEASES
Allergic diseases give rise to symptoms in many different organ
systems and involve many different medical disciplines. The most
important allergic diseases comprise allergic contact dermatitis,
atopic eczema, allergic rhinitis and conjunctivitis, asthma and food
allergy, and autoimmune diseases associated with chemicals.
4.1 Clinical aspects of allergic contact dermatitis
4.1.1 Introduction
Like the mucous membranes and the gut, the skin is an advanced
part of the immune system. Together with the skin barrier, the immune
system defends the body surface against microorganisms. Skin contact
with small molecules (haptens) tends to induce cellular-mediated
contact sensitization. The consequence of this contact sensitization
is allergic contact dermatitis. If the same molecules are given orally
before cutaneous contact, they may induce persistent immunological
tolerance. Allergic contact dermatitis is a common disease and the
prevalence at any given time varies between 2-4% (Fig. 9, 10, 11).
Allergic contact dermatitis of the hands has particularly important
implications for society as prolonged sick leave is common.
Most contact allergens are small molecules with a relative
molecular mass below 6000. Contact sensitization is not inborn but is
always a consequence of earlier cutaneous contact. Contact
sensitization is considered to be life-long, but might become weaker
if exposure is avoided. Contact sensitized individuals are at risk of
developing the skin disease allergic contact dermatitis if re-exposed
to the specific chemical. The term dermatitis is used synonymously
with eczema and describes either an acute skin disease with redness,
oedema and vesicles (water blisters) or a more chronic type with
hyperkeratosis, fissures and scaling. The most important differential
diagnosis of contact dermatitis is psoriasis, dermatophytosis, and
scabies. IgE-mediated immunological contact urticaria is covered
briefly.
4.1.2 Regional dermatitis
4.1.2.1 Hand eczema
Epidemiological studies including 20 000 individuals representing
the general population showed a one-year prevalence of hand eczema of
10% (Meding, 1990); 20% of cases were classified as caused by contact
allergy. The average duration was 12.8 years and 22% had periods of
sick leave. Allergic contact dermatitis on the hands is therefore both
a common disease and costly for the society, and it can imply
significant socioeconomic consequences for the individual.
these B-cells, presenting haptenized peptides in at least some of
their class II molecules, may become activated and differentiate into
autoantibody-producing plasma cells. These antibodies may induce
haemolysis of all erythrocytes in a drug-independent manner.
The hapten type of autoimmune haemolytic anaemia is caused by the
presence of drug-specific antibodies. These antibodies may be partly
considered as autoimmune since the combination of drug and autologous
carrier forms the actual target of the antibodies. When drugs, like
penicillin, bind covalently to red blood cells, these drug-specific
antibodies bind to the cells and induce their elimination by
phagocytosis in the spleen. The induction of high titres of penicillin
IgG antibodies typically occurs upon intramuscular administration,
rather than upon intravenous penicillin therapy. On the other hand,
relatively high intravenous doses of penicillin are required to make
the erythrocytes susceptible to immune-mediated haemolysis. Thus, most
patients with penicillin-induced haemolytic anaemia have received
large doses of drug over a protracted period. After discontinuation of
therapy the haemolysis quickly resolves and the antiglobulin test
becomes negative within weeks.
Some drugs appear to be able to induce true autoimmune haemolytic
anaemia, with Rhesus antigens as the most common targets of the red
cell antibodies. It is conceivable that the same auto-specificities
are found in drug-induced and idiopathic autoimmune haemolytic
anaemia, because the "normally" present (but silent) autoreactive
B-cells become activated upon haptenization of the autoantigens, as
shown in Fig. 8. As in drug-induced pemphigus and myasthenia, the drug
itself does not seem to be involved in the destructive autoimmune
reaction. While several drugs (Table 14) have been reported to provoke
red cell autoantibodies, alpha-methyldopa is the best studied example.
Only after prolonged therapy anti-red cell autoantibodies (IgG
anti-Rh) are formed. Upon withdrawal of the drug the antibody titres
usually decline and haemolysis ceases. Alpha-Methyldopa not only
induces red cell antibodies, but also antinuclear factors, rheumatoid
factors and gastric mucosa antibodies.
Table 14. Summary of drugs causing the different types of
immune or autoimmune haemolytic anaemia (from: Foerster, 1993)
Type of drug-induced (A)IHA Drugs
Drug- or hapten-specific IHA penicillin, cephalosporins, tetracycline
Autoimmune IHA alpha-methyldopa, levodopa, mefanamic acid,
procainamide
Immune complex mediated IHA stibophen, p-aminosalicylic acid, chlorambucil,
("innocent bystander" type) quinidine, quinine, phenacetin, sulfonamides,
isoniazid, rifampicin, etc.
If soluble drug-specific antibodies are present, they may form
immune complexes with administered drugs and fix complement. The
complexes are then adsorbed by erythrocytes and thrombocytes resulting
in lysis or clearance of these "innocent bystanders". Strictly
speaking, this haemolysis is caused by Type III reactivity. The
mechanism of adsorption, however, is not completely understood. It is
clear that it does not simply involve Fc receptors, since the F(ab)2
domain of the antibodies, in particular, adheres to the target cells.
Low-affinity attachment of drugs to the cells seem to make them more
susceptible for complex binding, and specific red cell antigens also
seem to be involved. Perhaps the bystanders are less innocent than
initially thought, and Type II reactivity combines here effectively
with Type III reactivity. Many different drugs, usually of low
relative molecular mass, are able to induce this type of haemolysis
(Table 14) (Foerster, 1993).
2.6.4 Autoimmune thrombocytopenic purpura
Autoimmune or idiopathic thrombocytopenic purpura (ITP) is
another example of a Type II reaction involving destruction of
self-antigens. This disease is characterized by shortened platelet
survival and the presence of antibody bound to platelets. It can be
classified as acute, intermittent or chronic, depending on the
severity and frequency of the symptoms. Acute ITP occurs mainly in
children following an upper respiratory viral illness (Karpatkin,
1988). The disease lasts an average of 1 to 2 months. Intermittent ITP
may occur in a child or an adult. It is characterized by episodes
where the platelet count drops, followed by periods where the count is
normal. Chronic ITP is seen in adults, and it may last for years, or
indefinitely (Karpatkin, 1988).
ITP can be drug-induced by the following: quinidine, quinine,
sulfonamides, p-aminosalicylic acid, phenytoin and sedormid (no
longer used). The drug acts as a hapten and adheres to the surface of
the platelets. This type of ITP is reversed when the drug is
withdrawn.
2.6.5 Pemphigus and pemphigoid
Type II reactions in the skin may cause different types of
blistering diseases depending on the antigen (location) to which the
autoantibodies are directed.
Antibodies towards desmosomal antigens induce intra-epidermal
blistering (acantholysis), leading to pemphigus, which is a
potentially fatal disease. The presence of these intra-epidermal
antibodies can be shown by direct immunofluorescence of perilesional
skin and provides the main diagnostic parameter. Most patients also
have circulating antibodies with titres reflecting disease activity.
Since removal of these antibodies by plasmapheresis reduces disease
activity and transfer of positive sera to mouse and monkeys can induce
pemphigus-like lesions, it is believed that the anti-desmosomal
antibodies are the causative agent of the clinical lesions in
pemphigus.
Investigations have unravelled the different desmosomal molecules
serving as autoantigens in pemphigus. The most important antigens in
pemphigus are the desmogleins; these are transmembrane glycoproteins,
which are members of the cadherin gene superfamily. Desmogleins bear
the same calcium-binding motifs as other cadherins do, and calcium
appears to be essential for the formation of the conformational
epitopes that are recognized by pemphigus sera (Amagi et al., 1995).
Interestingly, the two clinical variants of the disease, pemphigus
vulgaris and pemphigus foliaceus, develop antibodies to different
desmogleins, i.e., desmoglein-3 and desmoglein-1, respectively. The
differential expression of these two desmogleins in the upper and
lower epidermis could explain the different levels of acantholysis
seen in the two pemphigus variants (Shimizu et al., 1995).
Drugs may play a precipitating role in pemphigus.
Penicillamine-D, thiopronine, ampicillin, rifampicin, phenylbutazone,
captopril, pyrazolon, enalapril and piroxicam can all induce
pemphigus. It would appear that the presence of certain chemically
reactive groups in the drugs, in addition to the pemphigus
susceptibility genes in the patient (HLA-DRB1*0402 or DQB1*0503;
Matzner et al., 1995; Wucherpfennig et al., 1995), predispose for
drug-induced pemphigus. Sulfhydryl groups (-SH), present in
D-penicillamine, and active amide groups (-CO-N-), typically present
in enalapril, are held responsible for the acantholytic effects in
human skin cultures. In the group of penicillin and cephalosporins,
this active amide group is probably more important for the induction
of pemphigus than the sulfhydryl group (Wolf & Brenner, 1994).
The mechanism by which the drugs induce pemphigus is still not
completely understood. It is clear, however, that in addition to the
direct acantholytic effects, which can be observed in human skin
cultures in vitro, an autoimmune reaction is being induced. The
resulting autoantibodies appear to have the same antigenic
specificity, i.e., to desmoglein-1 and desmoglein-3, as in idiopathic
pemphigus patients (Korman et al., 1991). Together these findings
would be in line with involvement of the same mechanism of
drug-induced autoimmunity as described for autoimmune haemolytic
anaemia.
Antibodies towards antigens present in the lamina lucida of the
basement membrane cause a less severe type of blistering disease,
called bullous pemphigoid. Direct immunofluorescence of perilesional
skin reveals the presence of autoantibodies along the dermo-epidermal
junction. Concordantly, sub-epidermal blisters are being formed.
The antigens in bullous pemphigoid have been identified as
transmembrane proteins of 180 000 and 230 000 relative molecular mass,
present in the hemidesmosomes of the basal keratinocytes (Korman,
1995). These hemidesmosomes are believed to play a role in the
epidermal-dermal adhesion. Also in bullous pemphigoid autoantibodies
with the same specificity can be detected in the circulation, but
their titres do not correlate with disease activity.
The precipitating role of drugs for bullous pemphigoid is not
well established, although the disease may occasionally follow drug
ingestion (e.g., after furosemide).
2.6.6 Myasthenia gravis
Myasthenia gravis is an autoimmune disease that is mediated by
IgG antibodies directed to the acetylcholine receptors in the
postsynaptic membrane of the muscle (Vincent et al., 1995). The number
of receptors can be considerably reduced by complement-mediated lysis
and accelerated internalization. Additionally, the residual receptors
may be blocked by autoantibodies directed to the acetylcholine binding
site, thus leading to further impairment of the transmission from
nerve to muscle. As a consequence, the disease is characterized by
weakness and fatigue of the striated muscles. In some patients only
few muscles are affected; a well-known localized form of the disease
is ocular myasthenia (Weinberg et al., 1994).
In young patients with myasthenia gravis (40-50% of patients,
usually female) the thymus is an important site of autoantibody
production and T-cell activation. Within the hyperplastic thymus,
formation of lymphoid follicles can be observed, with germinal centres
surrounded by T-cells. The acetylcholine receptor antigens are here
presented to the immune system by muscle-like myoid cells, which bear
MHC-class II molecules. As therapeutic treatment, in addition to
immunosuppression, thymectomy is beneficial in these patients, since a
substantial source of both antigen and antibody-producing plasma cells
is thus removed. On the other hand, in late onset (usually male)
patients (15-20%), the thymus is rather atrophic and autoantibody
production by thymic cells is relatively low. In another minority of
patients (15-20%) thymoma may develop. In this last group of patients,
autoantibodies to striated muscles are typically found in addition to
the acetylcholine receptor autoantibodies.
Like pemphigus, myasthenia gravis can be induced by a number of
drugs. D-penicillamine, used for treatment of rheumatoid arthritis,
has been most frequently reported as a trigger for myasthenia gravis.
A few other drugs are suspected of inducing myasthenia gravis; among
them are thiopronin and chloroquin.
Drug-induced myasthenia is characterized by frequent involvement
of facial and oropharyngeal muscles (Bonnet et al., 1995). The disease
seldom generalizes or results in thymoma. Autoantibodies to
acetylcholine receptors are measurable in the circulation in the
majority of patients (approximately 80%), whereas almost half of them
have blocking antibodies. Similar frequencies of these antibodies are
found in idiopathic myasthenia gravis (Morel et al., 1991). The
autoantibodies disappear upon discontinuation of the drug, and full
recovery may be obtained within a few months.
2.7 Type III hypersensitivity diseases
2.7.1 Immune complex disease
Immune complexes are formed every time antibody meets antigen.
Generally they are removed effectively by the reticuloendothelial
system but occasionally their formation can lead to a hypersensitivity
reaction. Diseases resulting from immune-complex formation can be
placed broadly into three groups.
a) The combined effects of a low-grade persistent infection
(such as occurs with alpha-haemolytic Streptococcus viridans or
staphylococcal infective endocarditis, or with a parasite such as
Plasmodium vivax, or in viral hepatitis), together with a weak
antibody response, leads to chronic immune-complex formation with
the eventual deposition of complexes in the tissues.
b) Immune complex disease is a frequent complication of
auto-immune disease where the continued production of
autoantibody to a self-antigen leads to prolonged immune-complex
formation. The mononuclear phagocyte, erythrocyte and complement
systems (which are responsible for the removal of complexes)
become overloaded and the complexes are deposited in the tissues,
such as occurs in systemic lupus erythematosus (SLE).
c) Immune complexes may be formed at multiple sites, such as in
the lungs following repeated inhalation of antigenic materials
from moulds, plants or animals. This is exemplified in Farmer's
lung and Pigeon fancier's lung, where there are circulating
antibodies to the actinomycete fungi found in mouldy hay or to
pigeon antigens. Both diseases are forms of extrinsic allergic
alveolitis, and they only occur after repeated exposure to the
antigen. The antibodies induced by these antigens are primarily
IgG, rather than IgE, as in immediate (Type I) hypersensitivity
reactions. When antigen again enters the body by inhalation,
local immune complexes are formed in the alveoli leading to
inflammation. Precipitating antibodies to the inhaled
actinomycete antigens are found in the sera of 90% of patients
with Farmer's lung, but since they are also found in some people
with no disease, and are absent from some sufferers, it seems
that other factors are also involved, including Type IV
hypersensitivity reactions.
The sites of immune-complex deposition are partly determined by
the localization of the antigen in the tissues and partly by how
circulating complexes become deposited.
Immune complexes trigger a variety of inflammatory processes.
They can interact with the complement system leading to the generation
of C3a and C5a (anaphylatoxins), which cause the release of vasoactive
amines from mast cells and basophils, thus increasing vascular
permeability. These anaphylatoxins are also chemotactic for
polymorphs. Cytokines released from macrophages, particularly
TNF-alpha and IL-1, are also important in localized immune-complex
diseases, such as alveolitis, through a mechanism involving neutrophil
recruitment. Platelets can also interact with immune complexes,
through their Fc receptors, leading to aggregation and microthrombus
formation and hence a further increase in vascular permeability due to
the release of vasoactive amines. Platelets are a rich source of
growth factors, and release of these may contribute to the cellular
proliferation found in immune-complex diseases such as
glomerulonephritis and rheumatoid arthritis.
The attracted polymorphs attempt to ingest the complexes, but in
the case of tissue-trapped complexes this is difficult and the
phagocytes are therefore likely to release their lysosomal enzymes to
the exterior, causing tissue damage. If simply released into the blood
or tissue fluids, these lysosomal enzymes are unlikely to cause much
inflammation, because they are rapidly neutralized by serum enzyme
inhibitors. But if the phagocyte applies itself closely to the
tissue-trapped complexes through Fc binding, then serum inhibitors are
excluded and the enzymes may damage the underlying tissue. A classic
example of this type of inflammatory response is the Arthus reaction
(see section 2.1.3.1).
2.7.2 Serum sickness
Serum sickness is a Type III reaction that is seen in humans,
although not as frequently as it used to be. Serum sickness results
from passive immunization with animal anti-serum used to treat such
infections as tetanus and gangrene, usually horse or bovine
anti-serum. Approximately 50% of the individuals who receive a single
injection develop the disease (Barrett, 1988). Generalized symptoms
appear about 1 to 2 weeks after injection of the animal serum and
include headache, nausea, vomiting, joint pain and lymphadenopathy.
Recovery takes between 7 and 30 days (Terr, 1994b).
In this disease, the sensitizing and the shock-producing dose of
antigen are one and the same, as antibodies develop while antigen
still present. High levels of antibody form immune complexes that
deposit in the tissues. Usually this is a benign and self-limiting
disease, but previous exposure to animal serum can cause
cardiovascular collapse upon re-exposure (Terr, 1994b). Antibiotic use
has diminished the need for this type of therapy.
2.7.3 Allergic bronchopulmonary aspergillosis
Allergic bronchopulmonary aspergillosis (ABPA) is a syndrome
characterized by respiratory and constitutional symptoms caused by
hypersensitivity reactions to fungal antigens of Aspergillus
fumigatus. Allergic bronchopulmonary aspergillosis is characterized
by episodic wheezing, pulmonary infiltrates, eosinophilia in sputum
and blood, markedly elevated serum IgE levels, positive immediate and
late skin tests to A. fumigatus, serum precipitating antibody to
Aspergillus, and sputum containing brown plugs or flakes. Not all of
these changes may be present during active disease and a diagnosis of
allergic bronchopulmonary aspergillosis is usually considered when
asthma is complicated by radiographic or clinical evidence of
recurrent pneumonic infiltrates, bronchiectasis or pulmonary fibrosis.
A variety of disease-related immunological alterations have been
reported in allergic bronchopulmonary aspergillosis. Antigen extracts
of A. fumigatus, A. niger and A. clavatus have been shown to
activate the alternative complement pathway in fresh human serum from
healthy humans. Total serum IgE is elevated in most, but not all,
instances of allergic bronchopulmonary aspergillosis and
Aspergillus-specific IgE is substantially increased as measured by
radioimmunoassay.
The immune pathogenesis of allergic bronchopulmonary
aspergillosis is thought to involve direct activation of complement by
Aspergillus antigen, IgE-antibody production with subsequent release
of vasoactive amines from mast cells, as well as IgG-antibody
production and deposition of antigen-antibody complexes in the
broncho-alveolar tree. Local deposition of immune complexes may
activate the complement pathway and generate chemotactic factors for
polymorphonuclear leucocytes in peripheral blood and produce a
resultant immune complex-initiated Arthus-type reaction in lung
tissues. Also "late phase" eosinophil-mediated IgE-dependent reactions
have been suggested to be involved in the pathogenesis of the disease.
2.7.4 Extrinsic allergic alveolitis
Extrinsic allergic alveolitis (EAA) is usually defined in
pathological terms as a granulomatous inflammatory reaction which
predominantly involves the gas-exchanging parts of the lung and which
is the outcome of a specific immunological response to an inhaled
substance. The vast majority of reported cases have been caused by
inhaled organic dusts, but a few cases have been attributed to inhaled
isocyanates, particularly diphenyl methane diisocyanate (MDI) but also
hexamethylene diisocyanate (HDI) and toluene diisocyanate (TDI). No
reported case has been validated by biopsy evidence of the
characteristic pathological appearances; cases have been identified on
the basis of:
a) Characteristic clinical history;
b) Changes on chest radiograph;
c) Pattern of functional change following controlled isocyanate
inhalation;
d) Proportions of cells received at bronchoalveolar lavage.
Typically, patients present with a history of recurrent episodes
of breathlessness associated with systemic symptoms of fever, malaise
and chills. A few (but only a minority of reported cases) have had
abnormal chest radiographs.
In the majority of cases the diagnosis has been made by the
response to inhalation testing or the pattern of cells recovered at
bronchoalveolar lavage. Inhalation testing provoked the changes of an
"alveolar reaction" with proportionate reduction in forced expiratory
volume in 1 second (FEV1), forced vital capacity (FVC) and in
transfer factor (TLCO) accompanied by a neutrophil leucocytosis and
fever. The cells recovered at bronchoalveolar lavage have, as is
characteristic of EAA, shown an increase in the proportion of
lymphocytes, on occasion by more than 50%.
In some cases IgG antibody to a human serum albumin conjugate of
the relevant isocyanate - MDI-HSA, TDI-HSA and HDI-HSA - has been
identified in serum. The outcome of EAA caused by isocyanates has been
little reported, but most cases, even if showing significant
functional impairment at the time of diagnosis, would seem to have no
permanent residual disability after avoidance of isocyanate exposure.
2.7.4.1 Farmer's lung
Farmer's lung affects workers who handle mouldy hay or grain. It
originates from poor conditions of storage, involving high dust levels
and humidity. Microorganisms responsible for Farmer's lung are moulds,
above all Micropolyspora faeni, and Thermoactinomyces vulgaris.
Specific precipitins are found in the blood, especially antibodies of
the IgG class. This disease is classified as a Type III
hypersensitivity. Better storage and work practices reduce the
incidence.
Sensitization takes some time to occur. Clinically, patients
suffer respiratory distress accompanied by fever appearing 8-10 h
after handling mouldy hay, straw or grain and presenting with fever,
shivering, chest pains, lassitude, sweating, headaches and coughing,
sometimes accompanied by haemoptysis. Fine auscultatory chest
crepitations may be present. In typical forms, the chest X-ray shows
miliary infiltrates and micronodules. Later, pulmonary fibrosis
appears progressively when the disease reaches a chronic stage. There
is also impairment in alveolar gas diffusion (so called restrictive
syndrome) and, in the most advanced cases, an alveolar-capillary
block, which leads to chronic pulmonary heart failure.
2.7.4.2 Bird-fancier's lung
Bird fancier's lung is another disease of the same type, found
especially among pigeon breeders, but also in those handling other
birds. The disease is due to the development of precipitating
antibodies against serum proteins of relevant avian species, e.g.,
pigeons, parrots, chickens, pheasants and turkeys.
2.8 Type IV hypersensitivity diseases
Although cell-mediated immunity has fully developed in
vertebrates for their benefit by facilitating effective eradication of
microorganisms and abnormal cells, T-cell mediated reactions can,
under certain conditions, also cause disease (Table 15). Although
allergic contact dermatitis probably represents the most common T-cell
mediated disease, a few other pathological conditions are briefly
reviewed here.
Table 15. Pathology caused by Type IV hypersensitivity
Type IV induced disease Antigens, chemicals
Allergic contact dermatitis low relative molecular mass chemicals, drugs
Protein contact dermatitis proteins
Granulomatous disease mycobacterial antigens, beryllium
Autoimmune disease, e.g., autoantigens, e.g., pancreatic islet antigens
diabetes mellitus Type I
Hypersensitivity pneumonitis toluene diisocyanate, beryllium, heavy metals
Protein contact dermatitis is another example of Type IV
hypersensitivity.
One of the more serious complications of Type IV hypersensitivity
is the formation of granulomata. In general, T-cell immunity to
infectious agents confers a long-lasting state of protective immunity.
Macrophages, activated by the T-cell cytokines, can attack the
pathogen and should be considered as important effector cells here. If
the microorganisms are not readily killed and degraded, however,
macrophages may become "frustrated". They fuse to form multi-nucleated
giant cells or develop into large macrophages ("epitheloid" cells).
Together these cells can form new structures, so-called granulomata,
in which the macrophages with the foreign material are being isolated
from the environment by a layer of surrounding T-cells producing
cytokines and fibroblasts. The expansion and outgrowth of new
granulomata, especially at vulnerable sites, may cause considerable
tissue damage and loss of function.
In general, granuloma formation occurs when Type IV reactivity is
directed towards persistent indigestible antigens. A number of
organisms can induce granulomatous disease: Mycobacterium
tuberculosis and M. leprae, Treponema pallidum, Schistosoma,
and Yersinia enterocolitica. Importantly, a number of exogenous,
non-infectious agents can also evoke granulomatous reactions.
T-cell-mediated immune reactions may also cause disease when the
T-cell response is directed to autologous tissue. The crucial role of
T-cells, for instance, in the breakdown of the insulin-producing
beta-cells of the pancreatic Islets, leading to insulin-dependent
diabetes mellitus, is well established.
Other autoimmune diseases can sometimes be precipitated by Type
IV reactions evoked by completely unrelated antigens. Psoriasis may be
an example of an autoimmune disease that could be triggered by contact
allergens. How exactly these chemicals trigger the disease is not
completely clear. It is, however, most likely that any damage to the
skin, either toxic, physical or immunological, that recruits
sufficient lymphocytes from the circulation to include some
auto-reactive, keratin-specific T-cells will trigger a local response
in susceptible patients, resulting in a psoriatic lesion.
T-cell-mediated immunity plays a crucial role in the pathogenesis
of some lung diseases. Environmental organic chemicals like toluene
diisocyanate and trimellitic anhydride, but also inorganic compounds
as chromium and nickel are known sometimes to cause pulmonary disease.
The extent to which Type IV-mediated immune responses are involved in
these disorders is discussed in section 2.1.4.2.
Allergic contact dermatitis is considered to be the most frequent
pathological manifestation of Type IV reactivity. In allergic contact
dermatitis, T-cells are sensitized to proteins, environmental agents
and chemicals, entering the body via the skin. Repeated exposure to
such chemicals results in persistent eczematous inflammatory reactions
at the site of allergen contact. Although allergic contact dermatitis
can be regarded as a prototype of delayed-type hypersensitivity, the
sensitization process for chemical contact allergens, which already
starts in the most superficial layers of the skin, is very special.
The mechanism by which chemicals induce and elicit hypersensitivity
reactions in the skin will, therefore, be described in more detail.
2.8.1 Chronic beryllium disease
Chronic beryllium disease is a systemic disorder with primary
manifestations in the lungs. The pathogenic beryllium compounds
include metallic beryllium, beryllium alloys and beryllium oxide fume
(IARC, 1993). Inhalation of low levels of beryllium dusts or salts
over months to years is associated with a chronic interstitial
pulmonary granulomatous disorder clinically similar to sarcoidosis
(Freiman & Hardy, 1970; Jones Williams, 1988; Williams, 1989). The
skin manifestations of beryllium disease consist of contact dermatitis
and subcutaneous granuloma formation with occasional ulceration.
The concept that the granulomas of chronic beryllium disease are
T-cell-mediated immune granulomas is supported by the observations
that:
a) beryllium (i.e., the antigen) persists in the lung for long
periods (Jones Williams & Wallach, 1989);
b) large numbers of T-cells and non-caseating granulomas are
present in the lung (Williams, 1989);
c) in response to beryllium salts, lung and blood T-cells
proliferate and release lymphokines in vitro, a parameter also
used diagnostically to distinguish beryllium disease from
sarcoidosis (Williams & Williams, 1982; Rossman et al., 1988;
Newman & Kreiss, 1992; Newman et al., 1994);
d) intradermal administration of beryllium salts induces a
local granulomatous response in these individuals.
In chronic beryllium disease, the lung T-cell population is
predominantly of the CD4+ phenotype (Rossman et al., 1988; Saltini et
al., 1989). These CD4+ T-cells, compared to blood T-cells from the
same individual or compared to T-cells from normal individuals,
exhibit increased proliferation in response to beryllium (Rossman et
al., 1988; Saltini et al., 1989). The T-cells are activated,
expressing HLA class II molecules and IL-2R and releasing IL-2
(Pinkston et al., 1984; Saltini et al., 1989). Furthermore, the
beryllium-induced lung T-cell proliferation is Class II-restricted.
Chronic beryllium disease is strongly associated with HLA-DPB1 *0201,
and all beryllium-specific (BeSO2) T-cell clones have been shown to
be restricted by this allele.
Analysis of T-cell lines and T-cell clones of individuals with
this disease has confirmed that the beryllium-induced response is
antigen- specific and that all the responder cells are CD4+ T-cells
(Saltini et al., 1989).
Thus, from the information available, it appears that chronic
beryllium disease is a classic example of an immune granuloma host
response. Why an element like beryllium should do this is not clear,
but two not mutually exclusive hypotheses could explain it. Firstly,
it is likely that most disease is caused by dusts of beryllium metal
or salts, so that the particulate forms a nidus around which
macrophages ingest, allowing the beryllium to be slowly released.
Secondly, soluble beryllium salts interact with proteins, such that
the beryllium becomes an immunogenic hapten in the context of the
protein.
In an epidemiological study of groups exposed to the combustion
products of coal containing a high concentration of beryllium, Bencko
et al. (1980) found elevated levels of IgG and IgA and increased
concentrations of autoantibodies (anti-nuclear and anti-mitochondrial
antibodies).
2.8.2 Systemic autoimmune diseases
Several organ-specific autoimmune diseases such as pemphigus and
pemphigoid (section 2.6.5) and myasthenia gravis (section 2.6.6) have
been discussed above. Many of the major rheumatological disorders are
autoimmune in nature. Although systemic lupus erythematosus (SLE) can
be ranked under Type III immune complex disorders, for other
autoimmune diseases this categorization is less clear-cut.
2.8.2.1 Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is a chronic systemic
inflammatory disease that follows a course of alternating
exacerbations and remissions. Multiple organ system involvement
characteristically occurs during periods of disease activity (Fye &
Sack, 1991) (see also section 4.6.2). The disease predominantly
affects women (female to male ratio of 9:1) of childbearing age;
however, the age at onset ranges from 2 to 90 years. It is more
prevalent among non-whites than Caucasians. Family studies have
demonstrated a genetic susceptibility to the development of SLE.
Autoantibody formation in SLE is partially genetically determined:
patients with HLA-DR2 are more likely to produce anti-dsDNA
antibodies, those with HLA-DR3 produce anti-SS-A and anti-SS-B
antibodies, and those with HLA-DR4 and HLA-DR5 produce anti-Sm and
anti-RNP antibodies. Reduced serum complement and the presence of
autoantibodies to double-stranded (ds) DNA are hallmarks of active
SLE, distinguishing this entity from other lupus variants. Antibodies
to single-stranded DNA and particularly against histone proteins are
characteristic of some drug-induced forms of SLE, such as
procainamide-induced lupus (Rubin, 1989; Rubin et al., 1995).
Although in most cases the etiology of SLE is unknown, a wide
variety of medicinal and environmental agents have been associated
with the elicitation of SLE at low incidence in susceptible
individuals (Kammüller et al., 1989a; Adams & Hess, 1991; Uetrecht,
1992).
2.8.2.2 Rheumatoid arthritis
Rheumatoid arthritis is a chronic, recurrent, systemic
inflammatory disease primarily involving the joints (Fye & Sack,
1991). It affects 1-3% of people in the USA, with a female to male
ratio of 3:1. Constitutional symptoms include malaise, fever and
weight loss. The disease characteristically begins in the small joints
of the hands and feet and progresses in a centripetal and symmetrical
fashion. Elderly patients may present with more proximal large-joint
involvement and deformities are common. Extra-articular manifestations
such as vasculitis, atrophy of skin and muscle, lymphadenopathy,
splenomegaly and leucopenia, are characteristic of rheumatoid
arthritis and often cause significant morbidity.
The cause of the unusual immune responses and subsequent
inflammation in rheumatoid arthritis is unknown. HLA-D4 and HLA-DR4
occur in approximately 70% of patients with rheumatoid arthritis. Some
patients who are negative for HLA-D4 and HLA-DR4 carry the HLA-DR1
gene. It is possible that these and perhaps other genetic determinants
impart susceptibility to an unidentified environmental factor, such as
a virus, that initiates the disease process. The most important
serological finding is the elevated rheumatoid factor titre, present
in over 75% of patients (Fye & Sack, 1991).
2.8.2.3 Scleroderma
Scleroderma or progressive systemic sclerosis is a disease of
unknown cause characterized by abnormally increased collagen
deposition in the skin (Fye & Sack, 1991) (see also section 4.6.3).
The course is usually slowly progressive and chronically disabling,
but it can be rapidly progressive and fatal because of involvement of
internal organs. It usually begins in the third or fourth decade of
life. Children are occasionally affected. The prevalence of the
disease is 4-12.5 cases per million population. Women are affected
twice as often as men, and there is no racial predisposition.
Scleroderma is a manifestation of various diseases, many of them
autoimmune (Alarcon-Segovia, 1985). Two primary forms of scleroderma
exist: localized or systemic. The systemic form, progressive systemic
sclerosis (PSS), has in turn two variants: the diffuse and the CREST
syndrome (acronym for Calcinosis, Raynaud's phenomenon, Esophageal
involvement, Sclerodactyly and Telangectasia). Autoantibodies to DNA
topoisomerase I (scl 70) and centromere may be useful serological
markers for these respective diseases. The occurrence of progressive
systemic sclerosis with features previously considered characteristic
of SLE, rheumatoid arthritis, dermatomyositis and Sjögren's syndrome
and associated with high titres of antibodies to nuclear
ribonucleoprotein has been termed mixed connective tissue disease
(MCTD) (Alarcon-Segovia, 1985). In contrast to other autoimmune
diseases, cellular infiltration in scleroderma is minimal or absent in
all organs except the synovium, where impressive collections of
lymphocytes and plasma cells can be seen. Unfortunately, research on
the pathogenesis of scleroderma is severely hampered by the absence of
an animal model.
It has been suggested that certain chemicals may be associated
with some forms of scleroderma, e.g., tri- and perchloroethylene
(Sparrow, 1977; Saihan et al., 1978; Flindt-Hansen & Isager, 1987),
vinyl chloride (Lange et al., 1974; Ward et al., 1976; Black et al.,
1983), silicone (Rose & Potter, 1995) and epoxy resins (Yamakage et
al., 1980).
2.8.2.4 Sjögren's syndrome
Sjögren's syndrome is a chronic inflammatory disease of unknown
cause characterized by diminished lacrimal and salivary gland
secretion resulting in keratoconjunctivitis sicca and xerostomia (Fye
& Sack, 1991). There is a dryness of the eyes, mouth, nose, trachea,
bronchi, vagina and skin. In one-third of the patients, the disease
occurs as a primary pathological entity (primary Sjögren's syndrome).
In the remaining patients, it occurs in association with rheumatoid
arthritis or other connective tissue disorders such as SLE. Ninety
percent of patients with Sjögren's syndrome are female. Although the
mean age at onset is 50 years, the disease also occurs in children.
Patients with Sjögren's syndrome have an abnormal immunological
response to one or more unidentified antigens characterized by
excessive B-cell and plasma cell activity, manifested by polyclonal
hypergammaglobulinaemia and the production of rheumatoid factor,
antinuclear factors, including antibodies to SS(A) and SS(B),
cryoglobulins, and anti-salivary duct antibodies. Both B- and
Th-lymphocytes and plasma cells infiltrate involved tissues. No single
immunological test is diagnostic for Sjögren's syndrome, although a
spectrum of nonspecific immunological abnormalities occurs in these
patients. Histological demonstration of lymphocytic infiltration in a
biopsy specimen taken from the minor labial salivary gland is the most
specific and sensitive diagnostic test for Sjögren's syndrome (Fye &
Sack, 1991).
2.8.2.5 Hashimoto's disease
Hashimoto's disease, autoimmune thyroiditis, is the classical
example from which much of the knowledge of autoimmune disorders has
come (Gell et al., 1975; Roitt et al., 1998).
Antibodies are formed to several antigens in follicular cells of
the thyroid, including specific domains of thyroglobulin, thyroid
peroxidase and certain surface receptors. Delayed-type cellular
hypersensitization also occurs. The consequence is often initial
stimulation of the thyroid, followed after a variable period by
progressive destruction of the follicular cells, infiltration by
lymphocytes and plasma cells, often containing germinal centres, and
eventual fibrosis. The clinical disease, which is much more common in
women than in men, may be marked by initial thyrotoxicosis, which is
invariably followed by progressive hypothyroidism and myxoedema.
Thyroid autoantibodies and variable lymphocytic infiltration are
common in many other autoimmune diseases, so other tissues and organs
may also be affected and antibodies against these are frequently
found.
The cause of Hashimoto's disease is rarely known but it may
sometimes follow an overt viral infection of the thyroid and it has
been associated with high exposure to iodine.
Thyroid autoantibodies of several types are found in many
apparently healthy individuals and are common in patients suffering
from other autoimmune diseases.
3. FACTORS INFLUENCING ALLERGENICITY
3.1 Introduction
Allergens can be defined as antigens that give rise to allergy
(Sherrill et al., 1994). The molecular properties that distinguish an
allergen from an antigen are not known, but certain features appear to
be associated with allergens. Induction of allergic responses is
highly dependent upon a number of exogenous, as well as endogenous,
factors.
3.2 Inherent allergenicity
Most allergens are proteins. The structurally known allergens
from pollen, mammals, insects and foods are all proteins (or
glycoproteins) with a relative molecular mass of 10 000-40 000 (King
et al., 1995). Regarding IgE-mediated allergy, it is known that the
IgE antibodies are not formed to an entire allergen, but rather to
certain epitopes on the molecule. IgE binding sites are referred to as
B-cell epitopes. For a protein to be allergenic, it must be
multivalent, expressing more than one B-cell epitope. This allows
antigen to bind to more than one IgE molecule on the surface of a mast
cell or basophil, and induce these cells to generate and release
mediators that initiate the allergic reaction.
B-cell epitopes usually involve 12-15 linear amino acids,
although these epitopes may be non-contiguous. In the latter
situations, tertiary folding of the molecule provides the epitopes
(i.e., conformational epitopes). Allergens must also exhibit T-cell
epitopes, the 6-8 amino acid fragments presented to T-cells by
antigen-presenting cells such as macrophages. This interaction is
necessary to initiate the process of antigen-specific IgE synthesis.
Factors such as "foreignness", size and charge influence
allergenicity and sensitization. Allergenic proteins do not possess
physicochemical properties that distinguish them from non-allergenic
proteins. Foreignness refers to the concept of "non-self". In general,
the more foreign the substance, the greater is its immunogenicity. The
relationship of foreignness to allergenicity is not known. The larger
the antigen, the more likely it is to contain epitopes.
Compounds with a relative molecular mass smaller than 1000
typically are not immunogenic; those with relative molecular mass
between 1000 and 6000 may or may not be immunogenic, whereas those
with a relative molecular mass greater than 6000 are generally
immunogenic (Benjamini & Leskowitz, 1991). Allergens with small
relative molecular mass are termed "haptens". Such chemicals are
believed to couple to macromolecules to become immunogenic. In
general, the nature or identity of macromolecular "carriers" is not
known.
Certain inorganic chemicals are particularly potent sensitizers
on exposure of the skin, e.g., nickel- and platinum-containing
compounds, and, in some instances, of the respiratory tract (Rycroft
et al., 1995; Vos et al., 1996). Cross-reactivity has been observed
between allergic sensitization to nickel and chromium salts, and
between platinum, palladium and related elements.
Physicochemical complexity of a compound also favours
immunogenicity, whereas homopolymers of amino acids, such as
polylysine, are usually poor immunogens. When the complexity is
increased, i.e., by attachment of moieties that of themselves are not
immunogenic, the entire molecule becomes immunogenic (Landsteiner &
Rostenberg, 1939). For example, attachment of dinitrophenol to
polylysine renders the structure immunogenic, (Benjamini & Leskowitz,
1991).
Certain physical and chemical characteristics appear to be
associated with allergens. Protein allergens tend to possess
biological activity. Haptens tend to have chemical reactivity (or are
metabolized into reactive compounds); contact allergens are often
lipophilic. Such factors might have functional importance by
facilitating access of the allergen to the immune system, and by
interfering with regulatory mechanisms of the immune response. For
example, many protein allergens have been shown to possess enzymatic
activity (Stewart, 1994). The house dust mite allergen Der p I is a
serine protease (Chua et al., 1988). There is evidence that the
proteolytic activity enhances penetration of the allergen through the
mucosa (Herbert et al., 1995) and stimulates the synthesis and release
of the Th2-associated allergy-promoting cytokine IL-4 from mast cells
and basophils (Machado et al., 1996). Furthermore, it has been shown
that Der p I selectively cleaves the lymphocyte surface membrane
molecules CD23 (Hewitt et al., 1995; Schulz et al., 1997) and
CD25-alpha subunit (Schulz et al., 1998) and releases them into the
fluids surrounding the cells. Whereas the low-affinity IgE receptor
CD23 on the cell surface mediates negative feedback on IgE synthesis,
released soluble CD23 promotes IgE synthesis. Thus, the enzymatic
cleavage of CD23 by Der p I will enhance the synthesis of IgE, a key
mediator molecule in allergy. Furthermore, cleavage of the IL-2
receptor CD25-alpha subunit will strongly inhibit the proliferative
response and production of IFN-gamma in Th1-cells. Consequently, the
immune response to Der p I, and possibly other protein antigens
simultaneously presented to the immune system, will be biased towards
Th2-cells and an allergic response.
Many of the respiratory chemical allergens possess distinctive
functionalities that are thought to endow the chemical with
allergenicity. Studies have been undertaken of structural features and
physicochemical properties associated with respiratory allergens, and
structure-activity relationship (SAR) models have been developed
(Graham et al., 1997). Such factors include transport parameters,
electron density and chemical reactivities. These models, as well as
SAR models of allergic contact dermatitis, are discussed in chapter 6.
The ability of the immune system to recognize and distinguish
specific spatial regions (epitopes) on molecules has resulted in the
development of reagents and methodology to map these epitopes on
molecules such as drugs, proteins and microorganisms (Saint-Remy,
1997). The immune system can distinguish between structures that are
almost identical, i.e., that differ from one another by a single amino
acid substitution, or by a conformational change. Epitope mapping is
performed by generating panels of antibodies of known specificity.
Examples of the use of such antibodies are: a) in physiology to
identify structures that allow molecules to interact with their
receptor, b) in pathology to identify particular T- or B-cell epitopes
on antigens, c) in design of vaccines to either increase efficacy or
stimulate certain types of responses, such as T-cell responses, d) in
microbiology to aid in typing microorganisms.
3.2.1 Inherent properties of chemicals inducing autoimmunity
A variety of medicinal drugs with a relative molecular mass of
less than 1000 can elicit systemic hypersensitivity reactions and
autoimmune disorders in susceptible individuals at low incidence
(Adams & Hess, 1991). Chemical agents, drugs in particular, with a
documented potential to induce autoimmune disorders such as SLE,
belong to different chemical classes. These include, among others,
derivatives of aromatic amines, hydrazines, hydantoins, thioureylenes,
oxazolidinediones, succinimides, dibenzazepines, phenothiaines,
sulfoamides, pyrazolines, amino acids (Kammüller et al., 1989a; Adams
& Hess 1991; Uetrecht, 1992), amines (Nilsson & Kristofferson, 1989),
halothane (Gut et al., 1995), mercuric chloride (Pelletier et al.,
1994), gold preparations (Sinigaglia, 1994), occupational or
environmental chemicals such as tri- and perchloroethylene (Sparrow,
1977; Saihan et al., 1978) and vinyl chloride (Ward et al., 1976;
Black et al., 1983) (see also section 4.4). Environmental nitrophenols
have been suggested to be able to elicit or perpetuate human
autoimmune disorders (Lauer, 1990). Many of these compounds are
heterocyclic and contain at least one aromatic group, suggesting that
particular chemical entities may favour induction of immune
dysregulation.
From a pharmacological point of view, the majority of autoimmune
disease-inducing drugs are beta-adrenergic-receptor-blocking
compounds, drugs acting on the central nervous system (CNS),
anti-thyroid agents and anti-infective agents. In view of the tight
functional connectivity between immune, nervous and endocrine systems,
which is at least partially effected by shared receptors and mediators
among the systems, it is possible that CNS drugs modulate immune
responses by acting at these receptors or inducing common mediators.
Lupus-inducing compounds have the capacity to be oxidized by the
extracellular myeloperoxidase-H2O2 system of activated neutrophils,
despite their chemical and pharmacological heterogeneity (Uetrecht,
1992; Jiang et al., 1994). Despite this substrate promiscuity of
myeloperoxidase, analogues of lupus-inducing drugs with blocked or
missing functional groups such as -NH2, -NHNH2-, -SH, -Cl or OHC3
are not metabolized by myeloperoxidase (Jiang et al., 1994).
In order to become antigenic to T-cells, haptens must bind
carrier proteins, and whether or not T-cells may require covalent
modification of MHC molecules for hapten recognition is a matter of
debate. Investigation of mechanisms of allergic and autoimmune
reactions has pointed to a major role of trinitrophenol- and
gold-hapten-modified MHC-associated peptides as T-cell-antigenic
structures (Martin & Weltzien, 1994; Sinigaglia, 1994; Weltzien et
al., 1996).
3.3 Exogenous factors affecting sensitization
3.3.1 Exposure
3.3.1.1 Magnitude of exposure
The development of sensitization and the responses in individuals
depend upon the frequency and intensity of acute symptomatic episodes
(Friedmann et al., 1983; Ollier & Davies 1994). Clinical and
experimental evidence indicates that exposure concentration is of
critical importance for the development and exacerbation of allergy.
For dermal and respiratory sensitization, in animal and human studies,
the dose-response concept has been shown to operate at both the
induction and elicitation phases of sensitivity.
The role of dose in induction of contact sensitization has been
demonstrated in animal models, including guinea-pigs and mice (Chan et
al., 1983; Stadler & Karol, 1985). Data revealed a relationship
between the amount of chemical applied epicutaneously to the animals
and both the severity of the ensuing reaction and the percentage of
animals responding. In both species, and with all chemicals tested, a
no-effect concentration was also observed.
Both the induction and elicitation phases of respiratory
sensitization have been shown to be under the influence of the dose
(concentration) of allergen. With protein allergens, sensitization to
detergent enzymes was found to diminish as the workplace atmospheric
levels of the enzyme dust were reduced (Juniper et al., 1977). With
chemical allergens, clinical studies have indicated an association of
episodic high (accidental) exposure with development of sensitization
(Brooks, 1982). In a study of isocyanate workers, a relationship was
found between the number of spills and the percentage of workers
displaying symptoms of allergic disease (asthma, bronchitis and
decreased pulmonary function). With Western red cedar, an association
was also noted between workplace exposure and either the incidence of
pulmonary sensitization to the wood dust or the prevalence of
occupational asthma (Brooks, 1982). A further indication of the
importance of exposure concentration on sensitization is the reported
decrease in the number of cases of toluene diisocyanate (TDI)
sensitization coincident with the lowering of the permissible
occupational exposure levels (Karol, 1992).
Animal studies have established more precisely the relationship
between the exposure concentration, the elicitation concentration, and
development of respiratory sensitivity (Karol, 1994 a,b). Once again,
the concentration of inhaled allergen was shown to be a prime factor
controlling the development of sensitivity (Karol, 1983). Exposure of
guinea-pigs to monitored concentrations of TDI vapour resulted in
development of pulmonary sensitization only when the exposure
concentration was > 0.25 ppm (> 1.8 mg/m3) (Karol, 1983).
Exposure to lesser concentrations, even for extended periods of time,
did not result in sensitization. Both a threshold concentration and a
no-effect concentration were observed, suggesting the existence of a
safe level of exposure for this potent allergenic chemical (Karol,
1986).
A threshold concentration for sensitization to the allergenic
proteolytic enzyme, subtilisin, was also noted in animal studies
(Thorne et al., 1986). Groups of guinea-pigs were exposed to
atmospheres containing increased concentrations of the enzyme for
15 min per day on each of 5 consecutive days. Sensitivity developed in
animals exposed to the high concentrations but not in those exposed to
the lesser ones. Even long-term exposure of animals to the lower
concentrations failed to produce sensitization, although the animals
had received a cumulative exposure comparable to that which regularly
induced sensitivity when given over 5 days. This enzyme is believed to
be a particularly potent allergen and has a threshold limit value of
0.06 mg/m3. Clinically, workplace sensitization to the enzyme has
been dramatically reduced by lowering workplace exposures, and by
changing the formulation of the allergen to make it less readily
airborne (Juniper et al., 1977; Thorne et al., 1986; Sarlo & Karol,
1994).
3.3.1.2 Frequency of exposure
Increased frequency of inhalation exposure to allergen increased
the sensitization rate (Karol, 1986). However, studies clearly
demonstrated the importance of the exposure concentration exceeding a
threshold level for the chemicals. Repeated inhalation exposure of
guinea-pigs to sub-threshold concentrations of subtilisin (Thorne et
al., 1986) or TDI (Karol, 1983) failed to sensitize the animals,
whereas the same total exposure given over a shorter time span
consistently resulted in sensitization. Long-term sub-threshold
exposure to TDI resulted in neither respiratory sensitization nor
production of specific antibodies (Karol, 1983).
Clinically, chronic low-level exposure has been implicated in the
development of respiratory allergy to some airborne chemicals, notably
TDI (Karol, 1986). However, at that time the ability to measure low
concentrations of TDI was limited. Long sampling periods were often
required which eliminated the possibility of detecting sporadic high
TDI concentrations (Karol, 1986). As a result, in such studies no
conclusion can be drawn regarding the development of sensitization as
a result of repeated low-level exposure.
The influence of chronic low-level exposure to detergent enzymes
on the development of occupational sensitization to these enzymes has
been studied (Juniper et al., 1977). Using skin prick tests as an
indication of sensitization, conversion to skin test positivity was
observed following 20 months of employment for both high- and
low-exposure groups. A reduction in the dust levels in the workplace
was coincident with a decreased conversion rate (Juniper et al.,
1977).
In the platinum industry, respiratory sensitization to soluble
platinum salts has occurred under conditions where exposure is below
the official workplace limit. Maynard et al. (1997) examined the
possibility that high short-term exposures might be responsible but
found there was no evidence for this. In a cross-sectional study of
respiratory and dermal sensitization to platinum salts in a population
of precious metals refinery workers, skin reactivity was found in
workers exposed to permissible levels of platinum salts and was
associated with respiratory and dermal sensitization, but not with
atopic status (Baker et al., 1990). Merget et al. (1994), in a study
of platinum refinery workers, found that in workers who developed
immediate-type asthma caused by platinum salts both nonspecific and
specific bronchial responsiveness did not decrease after removal from
exposure.
Repeated exposure of guinea-pigs to contact allergens resulted in
reduced local reactions (Boerrigter et al., 1987) with eventual
diminution such that the skin reactions were almost non-existent.
However, the state of unresponsiveness disappeared upon
discontinuation of the repeated allergen exposures.
In humans, repeated exposure may also down-regulate the local
inflammatory response in the skin. This phenomenon is termed
"hardening". However, the individual remains sensitized. By contrast,
repeated systemic exposure could also "desensitize". This effect is
thought to be due to the high total dose administered.
3.3.1.3 Route of exposure
The route of exposure has an influence on the outcome of exposure
to an allergen. In general, exposure by the inhalation or dermal route
favours sensitization, whereas exposure by the oral route favours
tolerance (unresponsiveness ). Immunological unresponsiveness can be
induced in animals by non-cutaneous exposure. Induction of "tolerance"
in humans to nickel as a result of exposure to nickel-releasing
orthodontic braces during early age has been suggested (Van
Hoogstraten et al., 1991).
Systemic unresponsiveness after ingestion of antigen has now been
described for a large variety of T-cell-dependent antigens (Mowat,
1987). Proteins such as ovalbumin and bovine serum albumin (Silverman
et al., 1982; Domen et al., 1987), particulate (erythrocyte-bound)
antigens (Kagnoff, 1982; MacDonald, 1983; Mattingly, 1984),
inactivated viruses and bacteria (Stokes et al., 1979; Rubin et al.,
1981), autoimmune-related antigens (Thompson & Staines, 1990), as well
as contact allergens, have been reported to induce oral tolerance
(Asherson et al., 1977; Newby et al., 1980; Gautam et al., 1985).
Generally, T-cell-mediated delayed-type hypersensitivity responses and
IgE production are the types of immune responses most readily
tolerized. Persistent tolerance can be induced with relatively low
antigen doses of proteins (Heppel & Kilshaw, 1982; Jarrett, 1984;
Jarrett & Hall, 1984) and contact allergens (Asherson et al., 1977;
Polak 1980; van Hoogstraten et al., 1992; Hariya et al., 1994). The
apparent ability of the intestinal immune system to prevent allergic
hypersensitivity to soluble, non-replicating antigens seems an
important pathway to prevent enteropathies (Challacombe & Tomasi,
1987; Mowat, 1984, 1987). Abrogation of oral tolerance to, for
instance, ovalbumin was found to lead to hypersensitivity responses in
the intestinal mucosa and gut-associated lymphoid tissues, resembling
those observed in food-sensitive enteropathies, e.g., coeliac disease
(see section 1.5.1.3).
If mucosal cells in the respiratory tract are the site of initial
exposure, the result is frequently production of IgA and IgE
antibodies and predisposition to Type I allergic reactions. Initial
exposure of mucosal cells in the gastrointestinal tract may have the
same effect but often produces tolerance. By contrast, skin exposure
favours Type IV sensitization. It appears that the route of first
encounter with the chemical allergen determines whether the outcome is
sensitization or unresponsiveness.
Once an individual is sensitized via the skin, subsequent oral
exposure does not tolerize, but might contribute to further
sensitization by boosting the ongoing immune response. It is even
possible to induce systemic allergic reaction via the oral route in
skin-sensitized individuals. Overall, all of these factors are
dependent upon the nature of the allergen.
3.3.2 Atmospheric pollution
The effect of indoor and outdoor air pollution on allergic
disease has received considerable attention. Environmental pollutants
have been reported to contribute to the prevalence of allergic
disease, the precipitation of allergic symptoms, and their intensity
(Ollier & Davies, 1994). Both epidemiological and experimental studies
have demonstrated that a variety of atmospheric substances (including
sulfur dioxide (SO)2, nitrogen dioxide (NO2), ozone (O3) and
particles) influence the induction and elicitation phases of the
allergic response. Effects have included adjuvant activity for
allergen-specific IgE production, modulation of mediator release from
inflammatory cells, and irritant effects on effector organs of the
allergic response (Behrendt et al., 1995) (see sections 5.13 and
5.14).
The question of whether environmental factors may be involved in
the observed increase in the prevalence of allergy is a matter of
controversy (Ring et al., 1995b; Behrendt et al., 1995; Vos et al.,
1996). There is no doubt that pollutants such as suspended particles,
automobile exhaust, ozone, sulfur dioxide and nitric oxides can be
measured in rather high concentrations in the air of many countries
that show an increasing prevalence of atopic diseases. However, some
of these pollutants, like sulfur dioxide, have shown a decrease in air
concentrations during the last decades. In a controlled prospective
trial comparing different living areas with various degrees of air
pollution in western and eastern Germany, striking differences were
shown with regard to the prevalence of respiratory atopic diseases,
with higher values for western compared to eastern Germany (von
Mutius, 1992; Schlipköter et al., 1992; Behrendt et al., 1993, 1996;
Ring et al., 1995). In contrast to atopic respiratory diseases, there
was a trend to higher prevalence rates of atopic eczema in eastern
Germany. In the same study there was evidence of an increased risk of
developing atopic eczema after exposure to natural allergens as well
as air pollutants from outdoor and indoor sources (Ring et al., 1995;
Krämer et al., 1996; Schäfer et al., 1996).
The mechanisms by which air pollutants influence allergic
reactions are not clear. Some pollutants may have a direct toxic
effect on the respiratory epithelium leading to inflammation, airway
hyperreactivity and the appearance of asthma-like symptoms in
previously non-asthmatic individuals. In cell systems, certain
pollutants have been shown to modulate degranulation and histamine
release from basophils (Ring et al., 1995). Polychlorinated biphenyls
enhance eicosanoid production by granulocytes and platelets (Raulf &
Konig, 1991). Certain pollutants may have the ability to augment or
modify immune responses to inhaled antigens or to enhance the severity
of reactions elicited in the respiratory tract following inhalation
exposure of the sensitized individual to the inducing allergen.
High concentrations of air pollutants can have irritant effects
and aggravate the symptoms of allergic respiratory and skin diseases
(Ring et al., 1995; Behrendt et al., 1996). Laboratory studies suggest
that certain air pollutants have the potential to stimulate
broncho-constriction and airway inflammation. Exposure to SO2 is
associated with chest tightness and bronchoconstriction, the
concentration required to induce a response being dependent upon the
degree of hyperresponsiveness of the individual. The effects of SO2
may be augmented in the presence of other pollutants. It has been
reported, for instance, that the sensitivity of mild asthmatics to
SO2 is increased by prior exposure to O3. Ozone has been
investigated extensively and has been found to cause bronchial
hyperresponsiveness. In controlled clinical exposure studies,
researchers have demonstrated that asthmatics are more responsive to
O3 than normal people (Ball et al., 1993; WHO, in press). Exposure of
asthmatics to O3 for 1 h caused an increase in airway responsiveness
to inhaled allergen. The proportion of cynomologous monkeys that
developed asthma and a positive skin test after inhalation of complex
platinum salts was increased in those animals that inhaled O3
concurrently (Biagini et al., 1986). The health relevance of oxides of
nitrogen, and in particular NO2, has attracted some interest since
the gas is present both outdoors and indoors. Some studies have
suggested mild effects of NO2 in asthmatics at concentrations of less
than 1 ppm (< 1.88 mg/m3); others have not found responses at levels
up to 4 ppm (7.52 mg/m3). Particulate air pollutants, especially fine
particles derived from combustion sources, are also of interest
although there have been few controlled exposure studies apart from
those involving acid aerosols.
Bioaerosols to which an asthmatic is sensitized are well known to
exacerbate asthma. Epidemiological studies describing the increase in
mortality associated with inhaled particulate matter (PM-10) provide
provocative evidence for adverse pulmonary health effects associated
with particulate pollution. The association between particulate matter
and acute mortality and morbidity has been demonstrated most strongly
with elderly people who have chronic cardiopulmonary disease
(Thurston, 1996).
Studies have demonstrated an effect on allergic disease from
substances adsorbed to airborne particles. Such substances were found
to release histamine from human basophils and had a priming effect on
anti-IgE-induced release of histamine and LTC4 (Behrendt et al.,
1995). These in vitro studies indicated that particle-adherent
substances interfere with cells involved in inflammatory processes.
There is evidence of an interaction between pollen and air
pollutants. Pollen grains in polluted areas have been shown to be
loaded with particles including heavy metals, such as lead, cadmium
and mercury. In vitro, these pollen grains were found to have
altered surface features and increased ability to release cytosolic
allergenic proteins (Behrendt et al., 1991).
3.3.2.1 Tobacco smoke
Passive exposure to tobacco smoke is a risk factor for childhood
asthma (Seaton et al., 1994; Becher et al., 1996). Studies to detect a
possible association between passive smoke and allergic disease in
adults are much more difficult to design. Asthmatic patients
frequently report exposure to passive smoke. In children, there is
evidence that tobacco smoke increases the risk for development of
wheezy bronchitis and asthma.
Tobacco smoking is associated with an increased risk of
developing IgE antibodies and asthma. The mechanism of this effect of
tobacco smoke is unknown, but may be a result of injury to the
respiratory mucosa. Several studies have indicated that subjects who
smoke cigarettes have higher IgE levels (Zummo & Karol, 1996).
Specific IgE antibody or an immediate skin test response was found to
be 4-5 times more frequent in smokers exposed to tetrachlorophthalic
acid (TCPA) and ammonium hexachloroplatinate. Initially smokers had
IgE levels similar to those of controls, but, with age, IgE levels in
smokers did not decline at the same rate as they did in the
non-smokers (Sherrill et al., 1994). This may provide an explanation
for the difference in IgE values observed in adult smokers. Moreover,
a relationship was noted between the number of cigarettes smoked and
the IgE level, suggesting causality. In female smokers, there was a
trend toward increased IgE at older ages (i.e., > 50 years).
Passive smoking has been found to be a risk factor for
development of sensitization in children (Halken et al., 1995). The
association does not necessarily imply an allergic mechanism, rather
the association can be a result of direct irritation and inflammation
of the respiratory tract. In children with atopic predisposition, a
significant correlation was found between exposure to tobacco smoke
and wheezing/persistent wheezy bronchitis. A prospective study of 94
asthmatic children found significantly more asthma symptoms in those
exposed to maternal tobacco smoke. A retrospective study with 199
children with asthma found acute exacerbations of asthma increased
with exposure to tobacco smoke. In children with past or present
atopic dermatitis, asthma was found more frequently in cases where the
mother smoked cigarettes (Halken et al., 1995).
3.3.2.2 Geographical factors
Exposure to airborne allergens, notably pollens, depends on
location, climate and time of year (Emberlin, 1994). Certain types of
air pollution reduce the amount of pollen produced, but they can also
render the proteins on pollen more allergenic (Ruffin et al., 1986).
3.3.3 Metals
Nickel is a frequent cause of contact sensitization, having a
sensitization rate of 15-50% in experimental studies. Most cases of
nickel allergy can be attributed to exposure to nickel alloys in close
skin contact, which release high concentrations of nickel when exposed
to sweat. Similarly, chromate dermatitis often relates to exposure to
hexavalent chromate in wet cement (Andersen et al., 1995).
Investigations of monozygotic female twins, where one or both were
nickel sensitive, have shown that only the twin with a history of
contact dermatitis by nickel alloy exposures gives a positive
diagnostic patch test to nickel (Menné & Holm, 1983). In vitro
diagnostic testing failed to demonstrate subclinical nickel
sensitization in family members of nickel-sensitive individuals
(Silvennoinen-Kassinen, 1981).
3.3.4 Detergents
Reports of respiratory allergic reactions in workers involved in
large-scale production of enzyme-containing detergents suggest that
the detergent component may contribute to the sensitization to the
enzyme component. The symptoms of rhinitis and/or asthma suggested a
Type I sensitization. Experimental studies in guinea-pigs, using
either inhalation or intratracheal dosing, indicated that detergents
and proteolytic enzymes enhance sensitization to allergenic proteins
(Ritz et al., 1993; Sarlo et al., 1997) when sensitization was
assessed by production of allergic antibody and respiratory responses
to allergen challenge.
3.4 Endogenous factors affecting sensitization
3.4.1 Genetic influence
3.4.1.1 Contact sensitization
Although significant genetic influences on contact sensitization
have been reported, lack of reproducibility and smallness of these
effects suggest their minor importance, as compared to exposure, in
clinical contact sensitization. A few studies utilizing different
inbred mice and guinea-pig strains noted differences in sensitization
rates for some contact allergens (Parker et al., 1975; Andersen &
Maibach, 1985). In humans, a well-controlled family study indicated
that experimental contact sensitization in children was greater when
both parents could be sensitized by the same substance compared to
children where only one parent could be sensitized (Walker et al.,
1967). A population-based twin study focusing on nickel allergy found
a significant genetic effect for the risk of developing this contact
sensitivity (Menné & Holm, 1983). However, twin studies, using other
designs, have failed to show such an association. Also, studies on
frequencies of HLA genes in contact hypersensitive individuals have
not revealed consistent patterns (Menné & Holm, 1986). Comparisons
between frequencies of sensitization in different ethnic populations,
e.g., for nickel in black and Caucasian groups, revealed either
similar or different rates, depending on the study designs (Menné &
Wilkinson, 1995).
Histamine releasibility from mast cells and basophils is a
critical event in many allergic disorders. In twin studies, this event
(which is related to the quantity of IgE present on the cells) was
shown to be under genetic control (Bonini et al., 1994).
Products of HLA class II genes are involved in allergen
presentation by antigen-presenting cells. Since these genes are highly
polymorphic, different HLA genes represent risk factors for
development of allergic asthma. Increased responsiveness to the
ragweed allergen Ra 5 was found to be associated with the HLA gene DR
2/DW 2.
There is evidence for a genetic contribution to sensitization to
some allergens of low relative molecular mass.
3.4.1.2 IgE-related allergy
One of the characteristic features of atopy is the production of
IgE in an exuberant and prolonged fashion to common largely innocuous
environmental allergens, such as house dust mites and pollen. Most
atopics are allergic to more than one common environmental allergen
and this introduces the concept that the causation of atopy occurs at
a variety of levels: generalized hyper-IgE responsiveness; IgE
response to specific allergens or epitopes; clinical disease
expression (Hopkins, 1997).
The genetics of production of total serum IgE have been studied.
In such studies consideration has to be given to the following
factors, since each has been shown to affect IgE levels: allergic
exposure, parasitic infection, age, sex and smoking. A correlation was
found between the total serum IgE of parents and children, suggesting
the involvement of one or more genes (Sherrill et al., 1994). However,
agreement on the model of inheritance is lacking. Linkage of loci for
total serum IgE and BHR to chromosome 5q has been reported (Sherrill
et a1., 1994). Mapping of this area of the chromosome will be
important for further progress. Total serum IgE appears to be under
strong genetic control (Bonini et al., l994), even in the presence of
environmental factors such as smoking. A gene for IgE response with
maternal inheritance was identified at chromosome 11q (Cookson et al.,
1989). High levels of IgE in cord blood appear to be a strong
indicator of subsequent development of atopic disease.
The genetic factors that determine the specificity of the
IgE-mediated response are thought to be independent of those governing
total serum IgE and may be linked to the human leucocyte antigen (HLA)
complex (Sibbald, 1997). Products of HLA Class II genes are involved
in allergen presentation by antigen-presenting cells. HLA Class II
genes are highly polymorphic. Different HLA genes represent risk
factors for the development of asthma associated with sensitization to
allergens. Increased responsiveness to ragweed antigen (Ra5) was found
to be associated with HLADR2/DW2, and response to ryegrass (Lol pI and
Lol pII) with HLADR23 and DR5 (Marsh, 1990). Environmental factors,
such as the quality, intensity, route and duration of allergen
exposure appear to be more relevant than genetic factors in causing
allergic reaction to specific allergens (Bonini et al., 1994).
Twin studies have suggested polyfactorial control of allergy
variables such as serum levels of total IgE and IgG4, mediator release
from inflammatory cells, and target organ response. Clinical data from
32 monozygotic and 71 dizygotic twin pairs yielded a concordance for
allergic disease of 50.0% of monozygotic pairs (16/32) and 35.2% of
dizygotic pairs (25/71). The difference was not statistically
significant (Cockcroft, 1988). Histamine releasibility from mast cells
and basophils is a crucial event in allergic disorders. In twin
studies, this event (which is related to the quantity of IgE present
on the cells) was shown to be under genetic control (Bonini et al.,
l994).
Development of respiratory allergies to small relative molecular
mass chemicals, i.e., relative molecular mass less than 5000, such as
isocyanates and acid anhydrides has not been found to be associated
with atopy (Chan-Yeung, 1995), although atopy has been shown to be a
risk factor for development of respiratory symptoms to some chemical
allergens, such as hexachloroplatinate (Dally et al., 1980).
Regarding low molecular mass, or chemical allergens, an
association between sensitization to acid anhydrides and HLA-DR3
haplotype has been reported (Young et al., 1993). An association of
HLA class II alleles and isocyanate asthma was detected (Bignon et
al., 1994). Twenty-eight patients with isocyanate-induced asthma (as
documented by positive inhalation challenge) were compared with 16
exposed individuals with no history of the disease. HLA DQB1*0503 and
allelic combination DQB1*0201/0301 were associated with susceptibility
to asthma. Conversely, allele DQB1*0501 and the
DQA1*0101-DQB1*0501-DR1 haplotype conferred protection in exposed
healthy subjects. No significant difference was detected in the
distribution of HLA Class II alleles and/or haplotypes among the
immediate, late or dual responders to TDI. These results are
consistent with the hypothesis that immune mechanisms are involved in
isocyanate asthma and that specific genetic factors may increase or
decrease the risk of development of isocyanate asthma in exposed
individuals.
3.4.1.3 Other genetic factors
Another factor that may contribute to susceptibility, or
resistance, to sensitization relates to genes that control production
of IL-4, a pleotropic cytokine that influences the development of both
Th- and B-lymphocytes, the induction of Class II MHC antigens and
immunoglobulin class switching from IgM to IgE. Genes for IL-3, IL-4,
IL-5 and GM-CSF have been identified on chromosome 5 (Van Lee Uwen et
al., l989). The IL-4 gene, as well as genes that regulate its
expression, appear to be prime candidates for predisposition to atopy
since there are reports that cells isolated from atopic individuals
have the ability to overexpress the IL-4 gene relative to those from
non-atopic individuals. In addition, the human IL-4 proximal promoter
exists in multiple allelic forms, with one of the alleles having a
markedly enhanced promoter activity (Song et al., l996b). This finding
suggests a gene target to screen for genetic predisposition for atopy.
3.4.2 Tolerance
Allergenicity of a given compound may be strongly reduced in
individuals who previously developed immunological tolerance. This has
been frequently seen when the primary contacts with the allergen were
at mucosal surfaces, e.g., by its presence in food. Principles and
mechanisms of immunological hyporesponsiveness and tolerance have been
dealt with in detail above (see section 1.5).
3.4.2.1 Orally induced flare-up reactions and desensitization
Strong and long-lasting oral tolerance can only be achieved in
naive individuals, i.e., those who have not been previously exposed to
the antigen via the skin. In mice, a single feed of ovalbumin was
reported to fully suppress subsequent systemic immune responses, with
this state of tolerance persisting for up to 2 years. In contrast,
tolerance is hard to induce in primed animals but partial and
transient unresponsiveness ("desensitization") may develop after
prolonged feeding of the antigen. Similar results have been obtained
in guinea-pigs with various chemical allergens, including
dinitrochlorobenzene (DNCB) (Polak, 1980), nickel (van Hoogstraten,
1994) and amlexanol (Hariya et al., 1994). Unfortunately, essentially
similar results have been obtained in clinical trials aiming at the
treatment of autoimmune diseases, e.g., rheumatoid arthritis and
multiple sclerosis, by oral administration of putative autoantigens
(Weiner et al., 1994). Another problem with oral tolerance induction
in previously sensitized individuals arises from the tendency of
former inflammatory sites to re-inflame ("flare-up reactions"). These
reactions are likely to be due to allergen-specific effector T-cells,
which can persist for periods of several months at former inflammatory
sites (Scheper et al., 1983).
The differences between immunological responses in naive and
primed individuals may reflect changes in expression of cellular
adhesion/homing molecules and lymphocyte maturation. A qualitative
distinction exists between (difficult to stimulate/afferently acting)
naive and (easy to stimulate/efferently acting) effector/memory cells.
In contrast to naive lymphocytes, which only are activated by allergen
(modified self constituents) if presented by professional dendritic,
e.g., Langerhans cells, their progeny, known as effector/memory
lymphocytes, can also be stimulated by other cell types presenting
allergen-modified MHC class II-molecules, e.g., monocytes, endothelial
cells and B-cells. Clearly, effector/memory cells display increased
numbers of cellular adhesion molecules (CAMs), allowing for more
promiscuous cellular interactions. Amongst these, the most prominent
CAMs are the CD28 and LFA-1 molecules, with B7.1 and B7.2 and ICAM-1
as their respective ligands on APC. In addition, priming of T-cells
leads to the loss of homing receptors, such as L-selectin, which
facilitate interactions with high endothelial venules in peripheral
lymph nodes. Apparently, after sensitization, T-cells are less capable
of recirculating through the lymphoid organs, but gain ability to
migrate into the peripheral tissues. Interactions with endothelia
within inflamed skin are facilitated by the enhanced expression of
CAMs, such as the cutaneous lymphocyte-associated antigen CLA, and
effector/memory T-cells largely distribute over the peripheral
tissues, where conditions may be insufficient to convey effective
tolerogenic signals.
3.4.2.2 Non-specific and specific mechanisms of unresponsiveness
A preliminary factor contributing to non-responsiveness and/or
lack of hypersensitivity reactions at mucosal surfaces is the
epithelial barrier function, preventing entry of potentially harmful
allergens. Obviously, from an immunological point of view, this is a
"null-event", and does not have implications to subsequent encounters
with the same allergen. TGF-beta, a cytokine locally produced by
epithelial cells and immunocytes, plays a pivotal role in maintaining
epithelial barrier integrity. Importantly, the same cytokine also has
broad nonspecific immunosuppressive functions, e.g., by antagonizing
phagocytic effector cell functions of pulmonary alveolar macrophages.
Similarly, other immunosuppressive cytokines may be locally released
from epithelial cells and may act in concert with TGF-beta to
down-regulate immune effector functions, such as epithelial
cell-derived P15E-related factors which show sequence homology with
retroviral envelope proteins (Oostendorp et al., 1993).
In contrast, specific immunological tolerance depends on
decreased responsiveness of specific B- or T- cells, or release of
immunosuppressive mediators from these cells after specific challenge.
So far, no methods of permanent desensitization have been devised.
Nevertheless, how T-cells specifically recognize distinct allergens,
and how these and other inflammatory cells interact to generate
inflammation, is beginning to be understood. Exposure to high doses of
antigens may induce clonal deletion or anergy of specific B- or
T-cells by induction of apoptosis or antigen-receptor down-regulation
(Jones et al., 1990; Schönrich et al., 1991; Ohashi et al., 1991;
Melamed & Friedman, 1993).
As IL-4 and IL-13 direct IgE isotype switching, one way to
intervene in allergen-specific IgE synthesis and to inhibit or prevent
IgE-mediated allergic disease is to inhibit IL-4 and IL-13 production
by allergen-specific Th2-cells. In addition to TCR engagement by
peptide MHC complexes, optimal T-cell activation and proliferation
generally requires co-stimulatory signals provided by interaction
between CD28 or CTLA-4 on T-cells and their ligands CD80 or CD86 on
professional APC. Ligation of the TCR in the absence of these
co-stimulatory signals can result in T-cell non-responsiveness. Human
CD4+ Th2 clones specific for the house dust mite allergen Der p I
can be rendered non-responsive to subsequent Der p I challenges by
incubating them with Der p I-derived peptides, representing the
relevant minimal T-cell activation inducing epitopes, in the absence
of professional APC (Yssel et al., 1994). The mechanisms underlying
this T-cell unresponsiveness have not yet been determined. Although
these cells cannot be activated through their TCR, they proliferate
well in response to IL-2 or following activation by Ca++ ionophore
and TPA, suggesting that TCR activation or signalling pathways
immediately downstream of the TCR are disturbed.
This type of tolerance is generally short-lasting, since
(functionally) deleted lymphocytes are gradually replenished by newly
arising clones in the bone marrow and thymus and, in experimental
animal models, cannot be transferred to naive recipients, since these
still contain a fully functional repertoire, compensating for any
missing clones. On the other hand, mucosal contacts of naive
individuals with relatively low amounts of antigens, such as can be
the case with environmental or occupational exposure to chemical
sensitizers, frequently induce a long-lasting state of specific
tolerance. Transfer of lymphoid cells, in particular T-cells, from
orally tolerized animals to syngeneic naive recipients prevents their
capacity to subsequently mount immune responses to the same allergen,
revealing the existence of so-called T-regulator or suppressor cells
(Polak et al., 1980; van Hoogstraten et al., 1992, 1994; Weiner et
al., 1994).
Although "professional" suppressor T-cells may not exist (Bloom
et al., 1992; Arnon & Teitelbaum, 1993) available data support the
development of specific "regulatory" T-cells that suppress distinct
immune functions. Depending on the experimental models, such
regulatory T-cells can belong to either or both the CD4+ or CD8+
subsets (Bloom et al., 1992). Evidence is accumulating that regulatory
T-cells most often exert their role, after antigen-specific
activation, by releasing distinct cytokines antagonizing distinct
effector T-cell functions.
3.4.3 Underlying disease
There is ample evidence that underlying diseases are able to
influence the susceptibility of individuals to develop allergy. Both
the induction and the manifestation of allergy may be affected.
Conditions that promote sensitization include ongoing
inflammatory reactions at the site of allergen contact. It has, for
instance, been described that late-phase reactions of the respiratory
tract and the associated state of hyperresponsiveness, may facilitate
sensitization (priming) to other allergens (Connell, 1969). At skin
sites, a pre-existing eczema provides a risk factor for acquiring
contact sensitization. The most important factor here is probably the
local disturbance of the skin barrier, allowing for an increased
penetration of allergen. The fact that all components for an immune
response (cytokines, T-cells) have already been attracted to the site
of allergen contact may, however, additionally contribute to this
increased risk for new sensitization.
The most important diseases affecting the hosts' immune
responsiveness, and thus allergic responsiveness, include infectious
disease, neoplastic disease and immune deficiencies. The relation
between infection and the development of allergic disease is quite
complex. On one hand, respiratory viral infections are believed to
contribute to the exacerbation of asthmatic disease (Busse, 1990).
However, from clinical and epidemiological studies it would appear
that under certain conditions viral infections can also protect
against asthma. These studies include the observation of incidental
spontaneous remission of asthma during hepatitis, fever or measles, as
well as the finding of a general inverse relationship between
infections and asthma or atopy (Matricardi, 1997; Serafini, 1997). In
line with such a "protective" role it is believed that natural
infections during early childhood would prevent the development of
atopic disease later on, presumably by activation of the Th1
lymphocytes through IFN-gamma (Serafini, 1997). Reduction in family
size and increased hygiene could thus contribute to the increased
frequency of atopic disease in developed countries. Interestingly,
infectious diseases, which are known to be associated with a
predominant Th2 immune responsivenesses, like parasite infections, do
not seem to favour the development of atopic disease (Bell, 1996). In
contrast, people suffering from severe parasite infection may have
less severe reactions to other allergens, due to competition of IgE at
the Fc epsilon receptor level on mast cells. Also in HIV-positive
patients, where Th2 responses may become dominant, no clear evidence
has been obtained for enhanced atopic sensitization, although allergic
manifestations are frequently observed in these patients.
Conditions that suppress allergic reactions have been extensively
described, since contact sensitization has been applied as a method
for immune status determination in different patient groups. It is a
well-known fact that in clinical conditions associated with general
immune suppression and anergy, such as malnutrition, immunosuppressive
treatment, malignancies and severe physical trauma, Type IV reactivity
to recall antigens as well as primary sensitization to contact
allergens like dinitrochlorobenzene can be dramatically impaired.
Finally, it should be noted that certain immunological
conditions, such as those found in some immunodeficiency diseases,
e.g., in the Wiskott-Aldrich syndrome, may predispose for the
development of atopic eczema. Atopic disease is also commonly seen in
IgA deficiency.
3.4.4 Age
Childhood asthma is becoming more common and doubled in the
United Kingdom, New Zealand and Australia between 1970 and 1990.
Because of their greater activity and their developing lungs, children
may be more susceptible to sensitization as well as to adverse effects
of irritants (Zummo & Karol, 1996).
The ability to become sensitized to dinitrochlorobenzene has been
shown to be largely unchanged with age. Patch testing with Rhus
oleoresins in subjects with a history of poison ivy sensitization
showed diminished responses in the elderly (Lejman et al., 1984).
However, exposure differences as a function of age must always be
considered (Menné & Wilkinson, 1995).
IgE levels change with age. Peak levels occur in the first or
second decades of life. A longitudinal study of more than 2000
subjects conducted over a 20-year period found no gender difference in
total IgE (Sherrill et al., 1994). Both sexes had their highest IgE
levels as children. Levels fell gradually up to around age 40 and
thereafter remained constant.
3.4.5 Diet
To explain the observed increase in incidence of allergy and
asthma during the last two decades, it has been suggested that a
change in host resistance to allergy may have occurred (Seaton et al.,
1994). A change in the diet in several Western countries has been
documented. Specifically, a 20-50% fall in consumption of fresh fruits
and vegetables has been noted. Since these foods are sources of
antioxidants such as vitamin C and beta-carotene, decreased consumption,
together with that of red meat and fresh fish, would mean less
ubiquinone and fewer cofactors (such as zinc and copper) for
antioxidant defence (see section 5.10).
3.4.6 Gender
In general, women appear to have greater immune capability than
men (Menné & Wilkinson, 1995). Animal and human studies have indicated
a greater incidence of autoimmune disease in women compared with men,
as well as higher IgG and IgM levels. Women have also been reported to
produce greater cell-mediated immune responses.
In a large, controlled study, men were found more susceptible to
sensitization by dinitrochlorobenzene than women (Walker et al.,
1967). However, women were more readily sensitized to
p-aminodiphenyl aniline than were men (Walker et al., 1967). In
these studies, the issue of previous exposure to the chemical, and
therefore greater susceptibility, could not be dismissed. This factor
may also explain greater female sensitization in clinical patch tests
with nickel and cobalt. Male and female sensitization rates obtained
by maximization testing were comparable (Leyden & Kligman, 1977).
In a study of the influence of sex hormones on sensitization,
response to dinitrochlorobenzene was enhanced in women receiving oral
contraceptive hormones (Rea, 1979)
4. CLINICAL ASPECTS OF THE MOST IMPORTANT ALLERGIC DISEASES
Allergic diseases give rise to symptoms in many different organ
systems and involve many different medical disciplines. The most
important allergic diseases comprise allergic contact dermatitis,
atopic eczema, allergic rhinitis and conjunctivitis, asthma and food
allergy, and autoimmune diseases associated with chemicals.
4.1 Clinical aspects of allergic contact dermatitis
4.1.1 Introduction
Like the mucous membranes and the gut, the skin is an advanced
part of the immune system. Together with the skin barrier, the immune
system defends the body surface against microorganisms. Skin contact
with small molecules (haptens) tends to induce cellular-mediated
contact sensitization. The consequence of this contact sensitization
is allergic contact dermatitis. If the same molecules are given orally
before cutaneous contact, they may induce persistent immunological
tolerance. Allergic contact dermatitis is a common disease and the
prevalence at any given time varies between 2-4% (Fig. 9, 10, 11).
Allergic contact dermatitis of the hands has particularly important
implications for society as prolonged sick leave is common.
Most contact allergens are small molecules with a relative
molecular mass below 6000. Contact sensitization is not inborn but is
always a consequence of earlier cutaneous contact. Contact
sensitization is considered to be life-long, but might become weaker
if exposure is avoided. Contact sensitized individuals are at risk of
developing the skin disease allergic contact dermatitis if re-exposed
to the specific chemical. The term dermatitis is used synonymously
with eczema and describes either an acute skin disease with redness,
oedema and vesicles (water blisters) or a more chronic type with
hyperkeratosis, fissures and scaling. The most important differential
diagnosis of contact dermatitis is psoriasis, dermatophytosis, and
scabies. IgE-mediated immunological contact urticaria is covered
briefly.
4.1.2 Regional dermatitis
4.1.2.1 Hand eczema
Epidemiological studies including 20 000 individuals representing
the general population showed a one-year prevalence of hand eczema of
10% (Meding, 1990); 20% of cases were classified as caused by contact
allergy. The average duration was 12.8 years and 22% had periods of
sick leave. Allergic contact dermatitis on the hands is therefore both
a common disease and costly for the society, and it can imply
significant socioeconomic consequences for the individual.
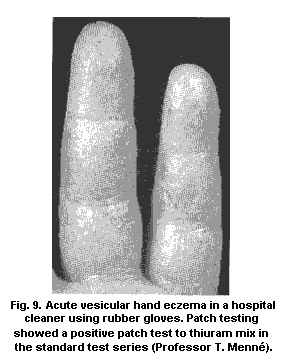

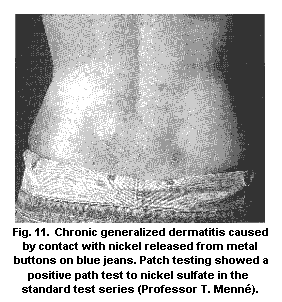 In a survey of 564 cases of permanent disability caused by skin
diseases, 222 of the 564 were caused by allergic contact dermatitis of
the hands (Menné & Bachmann, 1979).
Frequent causes of allergic hand eczema are nickel, chromate,
rubber additives (Fig. 9) preservatives, and fragrances (Menné &
Maibach, 1993). It can be acute or chronic, and it can be located on
either the dorsal or volar surfaces, or only on the fingers. It can
also present as a diffuse dermatitis. Spread to the face and forearms
is common.
4.1.2.2 Facial dermatitis
The face is second to the hands in the frequency of allergic
contact dermatitis. The exposure can be direct to airborne allergens
or indirect by contact with allergens transferred from the hands to
the face. Acute allergic contact dermatitis in the face is often
dramatic with severe oedema particularly of the eyelid regions.
Chronic cases frequently show patchy dermatitis even if the allergen
is uniformly spread on the face. Cosmetics, particularly fragrances,
are the most common causes of facial dermatitis. Allergic contact
dermatitis from medicaments (e.g., eye drops) and airborne
occupational dermatitis are seen. Severe oedema of the eyelids is a
common pattern of plant dermatitis. Facial dermatitis causes distress
to the individual because of pain, itching and disfiguration.
4.1.2.3 Other types of dermatitis
Stasis eczema and leg ulcers are a common disease among the
elderly as complications of arterial and venous insufficiency and
arteriosclerotic heart disease. Stasis eczema is a consequence of skin
malnutrition and can be followed by chronic ulceration. Both entities
are treated with topical medicaments such as emollients, steroids,
antiseptics and antibiotics. These compounds generally do not have a
high sensitizing capacity, but because they are used on damaged skin
under occlusion for prolonged periods, contact sensitivity is not
uncommon. Patch testing is routinely recommended in the work-up of leg
ulcer and leg eczema patients. On average 50% of these patients have a
positive patch test of actual or past relevance.
Intertriginous areas such as the axillae, external ear and
perianal area are also frequent sites of primary sensitization from
topically used medicaments and fragrances because of the natural
occlusion.
Shoe dermatitis is located in the skin area in direct contact
with the offending material, most frequently chromate-tanned leather,
rubber and glues (Podmore, 1995).
Allergic contact dermatitis from textiles gives a characteristic
clinical pattern with dermatitis in areas where textiles are in close
contact with the skin on the trunk and extremities. The offending
sensitizers are textile dyes and formaldehyde-releasing textile resins
(Fowler et al., 1992).
4.1.3 Special types of allergic contact reactions
4.1.3.1 Systemic contact dermatitis
Systemic contact dermatitis can be seen in primary contact
sensitized individuals when they are later exposed systemically to the
chemical (or drug) either orally, intravenously, by inhalation or by
transcutaneous absorption (Menné et al., 1994). The clinical symptoms
can either be erythematous flare in areas with earlier contact
dermatitis or a combination of symptoms including vesicular hand
eczema and inflammatory skin reaction in the flexural and genital
area. The explanation for the flare reaction is probably specific
sensitized lymphocytes persisting at the site of earlier allergic
contact dermatitis areas. The mechanism behind the other type of
reactions is speculative. Histologically this widespread reaction does
not have the picture of contact dermatitis but frequently presents the
picture of a lymphocytic vasculitis. The pathogenesis may be
circulating immune complexes or a general reaction to released
cytokines.
Systemic contact dermatitis is mostly seen in patients sensitized
to topically used medicaments when they are systemically treated with
the medicament or a cross-reacting medicament. Systemic contact
dermatitis has been described for a large number of substances.
4.1.3.2 Allergic photo-contact dermatitis
Most substances that cause photo-contact allergy are halogenated
aromatic hydrocarbons or sunscreen agents (White, 1995). The
combination of light, predominantly ultraviolet (UV), and the specific
chemical make the complete hapten. Clinical allergic photo-contact
dermatitis will therefore present a dermatitis (often severe) in
sun-exposed areas. This will typically be on the face, the forearms or
the dorsal aspects of the hands. In cases where photo-contact allergy
is suspected, patch testing is performed in duplicate and one site is
exposed to UVA. If a positive patch test only appears on the
UV-exposed site, photoallergy is likely.
4.1.3.3 Non-eczematous reactions
Allergic contact sensitivity in the skin can give rise to
clinical reaction patterns other than dermatitis (Goh, 1995). These
types of reactions are rare and to only a few chemicals. Even if these
patients have a clinical reaction type other than dermatitis, they
frequently have a positive patch test with the usual eczematous
morphology. The most common types of non-eczematous contact reactions
are erythema multiforme and lichen planus. Erythema multiforme-like
reactions are caused by contact with plant allergens and the lichen
planus type by contact with photographic chemicals.
4.1.3.4 Allergic contact urticaria
Contact urticaria is an immediate wheal reaction in the skin
caused by vasodilatation, with subsequent oedema. Contact urticaria
can either be allergic or non-allergic. In the non-allergic types
chemical causes a degranulation of the mast cells without involvement
of the immune system. The allergic types are mediated via IgE bound to
specific receptors on the mast cells and basophil lymphocytes in the
skin. The clinical types are similar with urticaria localized at the
contact site. Generalized anaphylactic reactions are rare. Both
organic and inorganic substances have now been described as causes of
allergic contact urticaria (Amin et al., 1996).
Contact urticaria is a frequent occupational disease among
individuals handling animals and animal products. Allergic contact
urticaria from proteins in rubber latex is a frequent and troublesome
problem among workers, particularly health personnel, due to
widespread use of rubber gloves (Taylor & Praditsuwan, 1996; NIOSH,
1997). A sensitization frequency of 2.8 to 10.7% has been reported in
health personnel (Turjanmaa, 1996). Individuals occupationally
sensitized to rubber latex proteins can develop anaphylactic reactions
if exposed to rubber gloves as patients.
4.1.4 Allergic contact dermatitis as an occupational disease
Occupational skin diseases are defined as skin diseases either
wholly or partly caused by the patient's occupation (Rycroft, 1995).
The epidemiology of occupational skin diseases, which mostly comprise
contact dermatitis of the hands, is known from population and
cross-sectional studies of specific occupational groups. Information
from centralized notification systems exists in some countries, but
the quality of data can be questioned. In particular, the problem of
under-reporting is difficult to quantify.
Skin diseases comprise between 20 and 40% of all occupational
diseases, depending on geographical area. Approximately one-third is
caused by allergic contact dermatitis and the rest mainly by irritant
dermatitis. The principal occupational contact sensitizing chemicals
are listed in Table 16. Not unexpectedly there is an overlap between
exposure to chemicals in occupational and domestic environments (see
section 4.1 and Table 19). The common high-risk occupations for
allergic contact dermatitis, modified from Rycroft (1995), are given
in Table 17 (Flyvholm et al., 1996). The prevalence of occupational
contact dermatitis in these occupations varies from a few percent up
to 15% (Rycroft, 1995).
Table 16. Main allergens related to occupational exposure
(from Flyvholm et al., 1996)
Allergens Sources of exposure
Acrylates adhesives; bone cement; dental products; UV-curing lacquers, etc.
Amines hardeners/curing agents for epoxy resin
Chromate cement; leather; pigments
Cobalt paints/lacquers
Colophony adhesives; dental products; paper; tin solder, etc.
Epoxy resin adhesives; paints; electric insulation
Formaldehyde disinfectants; preservatives; laboratory chemicals; formaldehyde resins;
funeral service
Formaldehyde releasers metal working fluids; paints; adhesives
Formaldehyde resins adhesives; paints/lacquers; impregnated textiles and paper; inks
Isocyanates adhesives; paints; fillings; polyurethane foams
Medicaments human and animal health care workers
Nickel coins; nickel plated objects; contaminated oils, etc.
Paraphenylenediamine hair dyes; rubber additive
Plastics/resins adhesives; paints; fillings, containers, etc.
Preservatives water-based products: metal working fluids; paints; adhesives;
cleaning agents; cosmetics; polishes; skin protection creams; process water, etc.
Rubber additives rubber gloves; rubber tubing; washers, etc.
Table 17. High-risk occupations for allergic contact dermatitis
Adhesives/plastics workers Horticulturalists
Agriculturalists Leather tanners
Cement casters Painters
Construction workers Pharmaceutical/chemical workers
Glass workers Rubber workers
Graphic workers Textile workers
Hairdressers Tilers
Health care workers Wood workers
It is difficult to give exact data concerning the costs of
occupational allergic contact dermatitis, as the compensation
regulation differs significantly from one country to another. However,
in the United Kingdom in 1996 it was estimated that 84 000 people had
occupational contact dermatitis, and 132 000 working days were lost
with a cost to employers of 20 million pounds per year (HSE, 1996).
4.1.5 Diagnostic methods
4.1.5.1 Patch testing
The aim of patch testing is to diagnose contact sensitization to
environmental chemicals. The patch test was introduced in 1896 by the
Swiss dermatologist Jadahsson (Wahlberg, 1995). The technology is a
biological test where contact allergy is proved by re-exposing the
skin to the specific chemical under occlusion on a skin area of 0.5
cm2 on the upper back for 2 days. A positive test is a reproduction
of the clinical disease showing redness, infiltration and eventual
vesicles. Standardization has taken place, particularly influenced by
the Scandinavian and later the International Contact Dermatitis
Research Group (ICDRG). The test should only be performed using
standardized test materials. All patients are primarily tested with
the Standard series including the most frequent sensitizing chemicals
such as metals, preservatives, fragrances, rubber additives and
topically used medicaments. Testing is frequently supplemented with
substances present in the patient's private or occupational
environments. Specially trained staff are necessary to obtain high
quality outcome of the procedure.
Sensitization can be quantified according to the degree of
positive patch test reaction (+ to +++), patch test concentration
threshold defined by dilution series, and finally by the "Use test".
In the latter test the individual is exposed to the chemical
simulating normal use.
The outcome of patch testing defines whether contact allergy is
present or not. Quantification of allergy combined with quantitative
exposure data is the basis for individual and general risk assessment
(Flyvholm et al., 1996).
The frequency of positive patch test reactions in the general
population (Nielsen & Menné, 1992) and in eczema patients tested at a
dermatological clinic in the same area of greater Copenhagen, Denmark,
is shown in Table 18. The allergens causing positive reactions most
frequently in eczema patients were nickel, fragrance mix, cobalt
chloride, colophony and balsam of Peru. For the general population,
nickel and thiomersal were the most common causes of positive patch
test reactions. Contact sensitization is generally more frequent among
patients investigated at dermatological centres than it is in the
general population.
4.1.5.2 In vitro testing
Several attempts have been made to develop in vitro methods for
testing contact sensitization (von Blomberg et al., 1990; McMillan &
Burrows, 1995). Yet, logistical and technical complexities, including
allergen toxicities, and the generally low frequencies of circulating
allergen-specific T-effector-memory cells, mean that currently
available methods are not appropriate for routine clinical use.
Nevertheless, in vitro tests, in particular the lymphocyte
proliferation test (LPT), using patient-derived white blood cell
samples, can be of considerable value in answering specific scientific
questions, e.g., on the involvement of allergen-specific T-cells or on
potential cross-reactivity patterns between allergens (Bruynzeel et
al., 1985; Pistoor et al., 1995).
4.1.6 Assessment of exposure
To establish the diagnosis of allergic contact dermatitis, the
outcome of patch testing needs to be combined with a detailed exposure
history (Flyvholm et al., 1996). Both domestic and work-related
exposures need to be elucidated. Factory visits are valuable but
rarely done (Rycroft, 1995). The most common contact allergens are
metals, preservatives, rubber additives, perfumes and medicaments. The
main sources of exposure to contact allergens can be divided into
groups of substances, products or use categories. Exposure to
allergens occurs under many circumstances, such as occupational,
domestic work, hobby and leisure time activities, topical medicaments,
cosmetics, personal care products, clothing and shoes. Examples of
such allergens are listed in Table 19 (Flyvholm et al., 1996). For
examples of occupational exposure, see Table 16 (section 4.1.4).
Exposure data can be obtained from databases, product labelling or
chemical analysis, and by contact with manufacturers and suppliers.
The prognosis for the individual patient depends upon the quality of
diagnostic patch testing and the ability to prevent contact of the
patient with the allergen.
Table 18. Comparison of frequencies of positive patch test reactions
in the general population (Nielsen & Menné, 1992) and in eczema patients at a
dermatological clinic in the same area of greater Copenhagen in 1990a
Test substance General populationb Dermatological clinicc
(% positive of tested) (% positive of tested)
Men Women Total Men Women Total
(n=279) (n=288) (n=567) (n=262) (n=410) (n=672)
Potassium dichromate 0.7 0.3 0.5 1.9 2.7 2.4
Neomycin sulfate 0.0 0.0 0.0 3.4 3.7 3.6
Thiuram mixture 0.7 0.3 0.5 4.6 2.7 3.4
p-Phenylenediamine 0.0 0.0 0.0 1.9 2.7 2.4
Cobalt chloride 0.7 1.4 1.1 2.3 2.7 2.5
Benzocaine - - NT 0.4 0.7 0.6
Caine(R) (local 0.0 0.0 0.0 - - NT
anaesthetic) mix
Formaldehyded - - NT 1.9 2.2 2.1
Colophony 0.4 1.0 0.7 4.6 5.4 5.1
Quinoline mix 0.4 0.3 0.4 1.9 0.5 1.0
Balsam of Peru 0.7 1.4 1.1 3.4 5.4 4.6
PPD black rubber mix 0.4 0.0 0.2 1.2 0.0 0.5
Wool alcohols 0.4 0.0 0.2 1.2 1.7 1.5
Mercapto mix 0.7 0.0 0.4 1.2 0.2 0.6
Epoxy resin 0.4 0.7 0.5 0.8 0.2 0.5
Paraben mix 0.4 0.3 0.4 0.8 0.2 0.5
p-tert-Butylphenol 1.1 1.0 1.1 0.4 1.2 0.9
formaldehyde resin
Fragrance mix 1.1 1.0 1.1 6.1 7.1 6.7
Ethylenediamine 0.4 0.0 0.2 0.8 0.7 0.7
dihydrochloridee
Quaternium 15 0.4 0.0 0.2 0.0 0.0 0.0
Nickel sulfate 2.2 11.1 6.7 4.2 16.1 11.5
MCI/MI 0.4 1.0 0.7 0.4 0.7 0.6
(chloro-methyl- and
methyl-isothiazolinone)
Table 18. (continued)
Test substance General populationb Dermatological clinicc
(% positive of tested) (% positive of tested)
Men Women Total Men Women Total
(n=279) (n=288) (n=567) (n=262) (n=410) (n=672)
Mercaptobenzothiazole 0.4 0.0 0.2 1.2 0.2 0.6
Priminf - - NT 0.4 1.5 1.0
Thiomersalg 3.6 3.1 3.4 - - NT
Carba mixh 0.7 0.0 0.4 - - NT
a Menné, unpublished (personal communication by T. Menné to the IPCS, 1997)
b Patch tested with the ready-to-apply TRUE test, Pharmacia (Sweden)
c Test substances from Hermal (Germany)
d Formaldehyde not included in TRUE test at the time of study
e Ethylenediamine dihydrochloride excluded from the European Standard series as of August 1992
f Primin not included in the TRUE test at the time of study
g Thiomersal not included in the European Standard series
h Carba mix excluded from the European Standard series as of January 1989
Table 19. Main allergens related to non-occupational exposure
Allergens Sources of exposure
Domestic work
Chromium leather; footwear
Colophony shoe polish; crayons; plasticine; paper
Flowers/plants gardening; house plants
Nickel nickel-plated objects
Plastics/resins adhesives; paints; containers
Preservatives cleaning agents; polishes; personal care products
Rubber additives gloves; other rubber objects
Wood repairs; handicraft
Hobbies and leisure time activities
Chromium leather; footwear
Colophony adhesive tapes; plasticine; paper; violin bow resin
crayons; artists' paints; textiles
Dyes/pigments gardening; house plants
Flowers/plants textile resins; preservative in various products
Formaldehyde nickel-plated objects
Nickel adhesives; paints; containers
Plastics/resins paints; personal care products
Preservatives gloves; sports equipment
Rubber additives handicrafts
Woods
Cosmetics and personal care products
Colophony mascara.
Dyes hair dyes; miscellaneous cosmetics
Fragrances
Glyceryl thioglycolate permanent waving
Lanolin
Table 19 (cont'd)
Allergens Sources of exposure
Paraphenylenediamine hair dyes; creams; lotions; shampoos;
liquid soap, etc. (i.e., most cosmetic
and personal care products)
Preservatives, e.g.,
formaldehyde releasers,
isothiazolines parabens
UV filters sunscreens
Topical medicaments
Antibiotics
Antihistamines
Antimicrobials
Balsams
Benzocaine
Colophony
Ethylenediamine
Formaldehyde releasers
Lanolin
Parabens
Preservatives
Tars
4.1.7 Treatment and prevention of allergic contact dermatitis
The treatment of allergic contact dermatitis requires medical
intervention. It usually involves the controlled use of emollients or
corticosteroids as well as prevention of further exposure to the
offending allergen (Wilkinson, 1995). A distinction is usually made
between primary prevention, focusing on the induction of contact
sensitization, and secondary prevention, focusing on the eliciting of
contact sensitization. In many instances the preventive measures for
the two different types overlap.
4.1.7.1 Primary prevention
In the 1960s an epidemic of contact dermatitis from dish-washing
products occurred in Scandinavia. The epidemic was resolved by the
concerted action of dermatologists and manufacturers. Extensive
chemical analysis combined with animal predictive testing, identified
highly sensitizing sultones to be present in some products (Magnusson
& Gilje, 1973; Ritz et al., 1975). It was determined that these
specific chemicals occurred as an impurity in the manufacturing
process, when temperature control was not strictly maintained. The
evaluation of the problem led to a solution, and there have been no
recurrences.
There are examples of exposure to hapten concentrations being
legally regulated in an attempt to prevent contact sensitization
(Hjorth & Menné, 1990). There is a complex European Union regulation
on cosmetic products, forbidding certain substances and regulating
others, i.e., preservatives, by a concentration limit (Council of the
European Communities, 1976).
Since the 1950s, chromate in cement has been know to be one of
the main causes of allergic chromate dermatitis among construction
workers. At the start of the 1980s the Scandinavian countries added
ferrosulfate at a low concentration to cement to reduce the hexavalent
chromate to trivalent chromate. The idea of this initiative was that
the trivalent chromate is not absorbed, or only to a minor degree,
through human skin, and therefore the risk of primary sensitization
from this salt is significantly less than from hexavalent chromate.
Epidemiological studies on construction sites performed at the
beginning of the 1980s and at the end of the 1980s in Denmark,
strongly suggest that this measure has been successful, as the
frequency of allergic chromate dermatitis has been reduced in Denmark
(Avnstorp, 1992).
Nickel is a common contact allergen on a global scale. This
allergy is caused by intimate skin contact with metal alloys,
releasing nickel when exposed to human sweat. Under simulated use
conditions, some alloys release high amounts and other alloys low
amounts of nickel (Lidén et al., 1996). Based on such research, some
Scandinavian countries have introduced regulations and quality
criteria for nickel alloys intended to be in prolonged skin contact.
It is believed that such measures might reduce significantly the
frequency of nickel allergy in the population. Regulation of nickel
exposure along similar lines has been adopted within the European
Union (Council of the European Communities, 1994).
In considering different glove materials to protect against skin
irritation and mechanical skin damage, it should be noted that most
small sensitizing chemicals rapidly penetrate most rubber and plastic
gloves, and appropriate gloves should therefore be used (Estlander &
Jolanki, 1988; Mellström et al., 1989; Roed-Petersen, 1989).
There is no method of predicting an individual propensity to
contact sensitization to a given chemical. When patch testing with
strong sensitizing chemicals is performed, active sensitization from
the test cannot completely be excluded. Pre-employment testing is
therefore not a method of preventing contact sensitization.
4.1.7.2 Secondary prevention
The cornerstones of the secondary prevention of allergic contact
dermatitis (elicitation of contact dermatitis) are based on sufficient
diagnostic procedures and patient information systems. The
availability of standardized patch test materials is essential.
Furthermore, it is crucial that it is possible for the doctor to
inform the patient where exposure to the specific allergen can be
expected. Of course, it is even more crucial that the patient is able
to understand and use the information over the following years to
identify the allergen in the home and occupational environments. It
seems obvious that this type of diagnostic follow-up will work, but it
has only been evaluated in a limited number of studies. Edman (1988)
found that the prognosis for patients sensitive to topical medicaments
depended upon whether the patients were able to follow the doctor's
advice on the occurrence of sensitizers in different products. Later
studies have shown that patients with contact allergy to formaldehyde
often continued to be exposed to formaldehyde (Cronin, 1991; Flyvholm
& Menné, 1992). When a careful investigation was made, formaldehyde
exposure could be demonstrated in nearly all the patients which seemed
to be decisive for the prognosis of their hand eczema (Flyvholm,
1997).
4.1.7.3 Ways of preventing contact sensitization
The following ways of preventing contact sensitization have been
suggested.
a) replacement of certain chemicals or particular products;
b) regulation of exposure (concentration) to sensitizing
chemicals, either general or in specific products, or during
particular work processes;
c) optimal diagnostic and information systems; education of
either groups or individuals;
d) individual oriented preventive methods; gloves, barrier
creams, protective clothing.
The problems of contact sensitization have been identified over
many years, and different types of preventive measures have been
tried. Some have been successful, but a number of chemicals still give
problems to a significant number of people. Different strategies
should be considered, whether it concerns common environmental
chemicals or chemicals with rare specific exposures. Chemicals
frequently used in both the domestic and occupational environment need
to be regulated by society, either with suggestion of replacement or
regulation of the exposure concentration. For rare chemicals it is
often sufficient to focus on specific occupational processes and to
educate the exposed individuals in no-touch techniques or introduce
individually oriented preventive measures.
4.1.8 Information needed for a preventative programme
The prevention of allergic contact dermatitis should be based on
preventing sensitization and, subsequently, on avoiding sufficient
exposure to elicit a response in a person already sensitized. This
requires information on the following aspects.
a) Occurrence of sensitizing substances
Products used at work or domestically should be labelled to
indicate the presence of substances capable of causing sensitization
and their concentrations, so that the user may take appropriate
precautions.
If there are suitable alternatives there may be no need to use a
sensitizing agent.
At present, the potential of new substances to cause
sensitization is determined from the results of tests on animals or
sometimes on humans (Rycroft, 1995), after databases have been
searched for relevant published information. Structure-activity
relationships should be assessed and may give valuable indications of
sensitizing potential for substances of a similar structure to known
contact allergens.
Comprehensive information about the composition of products and
the allergenic activity of their ingredients should be collected in
each country and be made available to health care professionals and
users. This should include the results of surveys of standardized
patch testing of humans so that trends in allergic sensitization can
be followed.
b) Avoiding or minimizing exposure
Induction of sensitization and eliciting an allergic disorder
both follow dose-response relationships, albeit at very different
concentrations.
It is important to minimize initial exposure to sensitizing
agents by restricting their availability or, if they cannot be
avoided, by minimizing exposure. Exposure can be minimized by ensuring
adequate ventilation and using personal protective equipment
appropriate to the work situation or in the home, e.g., gloves, masks,
etc. (see also chapter 7).
4.2 Atopic eczema (atopic dermatitis)
4.2.1 Definition
Atopic eczema or atopic dermatitis is a chronic pruritic
inflammatory skin disease characterized by a typical age-related
distribution and skin morphology (Figs. 12, 13). The diagnosis of
atopic eczema is based primarily on clinical grounds and the patient's
history (Hanifin, 1983; Rajka, 1990). Onset at an early age, pruritus
and excoriation, chronic or chronic relapsing course for more than
6 weeks, age-related eczematous morphology and distribution, as well
as a positive family history for atopic diseases (allergic bronchial
asthma, allergic rhinitis and conjunctivitis or atopic eczema), form
the most striking criteria. Together with allergic rhinitis and
conjunctivitis and bronchial asthma, atopic eczema forms the classical
triad of atopic diseases (Rajka, 1990; Ruzicka et al., 1991). Atopy
can be defined as "familial hypersensitivity of skin and mucous
membranes against environmental substances associated with increased
IgE production and/or nonspecific reactivity" (Ring, 1991). This
underlines two components held to be responsible for induction of this
disease. Although it is genetically determined, environmental
influences may play a role. During the last century many synonyms for
atopic eczema/atopic dermatitis have been evolved, e.g.,
neurodermatitis, prurigo Besnier, endogenous eczema and diffuse
neurodermatitis (Ring, 1991). The diagnosis of this skin disease is
based on clinical criteria, family history and/or demonstration of
IgE-mediated sensitization.
4.2.2 Epidemiology of atopic eczema
Atopic eczema is a common disease among children and adults. In
the 1950s the frequency of eczema was estimated to be between 1.1 and
3.1% (Walker & Warin, 1956). In the 1980s and 1990s the frequency of
atopic eczema was found to be up to 25% on the basis of questionnaires
(Bakke et al., 1990) and up to 9.7% for dermatologically examined
cohorts (Varjonen et al., 1992; Schäfer & Ring, 1997). Epidemiological
studies on the prevalence of atopic eczema in Germany were conducted
with questionnaire, physical and dermatological examination including
allergy tests. In 1989-1991 8.3% of 988 Bavarian school children aged
In a survey of 564 cases of permanent disability caused by skin
diseases, 222 of the 564 were caused by allergic contact dermatitis of
the hands (Menné & Bachmann, 1979).
Frequent causes of allergic hand eczema are nickel, chromate,
rubber additives (Fig. 9) preservatives, and fragrances (Menné &
Maibach, 1993). It can be acute or chronic, and it can be located on
either the dorsal or volar surfaces, or only on the fingers. It can
also present as a diffuse dermatitis. Spread to the face and forearms
is common.
4.1.2.2 Facial dermatitis
The face is second to the hands in the frequency of allergic
contact dermatitis. The exposure can be direct to airborne allergens
or indirect by contact with allergens transferred from the hands to
the face. Acute allergic contact dermatitis in the face is often
dramatic with severe oedema particularly of the eyelid regions.
Chronic cases frequently show patchy dermatitis even if the allergen
is uniformly spread on the face. Cosmetics, particularly fragrances,
are the most common causes of facial dermatitis. Allergic contact
dermatitis from medicaments (e.g., eye drops) and airborne
occupational dermatitis are seen. Severe oedema of the eyelids is a
common pattern of plant dermatitis. Facial dermatitis causes distress
to the individual because of pain, itching and disfiguration.
4.1.2.3 Other types of dermatitis
Stasis eczema and leg ulcers are a common disease among the
elderly as complications of arterial and venous insufficiency and
arteriosclerotic heart disease. Stasis eczema is a consequence of skin
malnutrition and can be followed by chronic ulceration. Both entities
are treated with topical medicaments such as emollients, steroids,
antiseptics and antibiotics. These compounds generally do not have a
high sensitizing capacity, but because they are used on damaged skin
under occlusion for prolonged periods, contact sensitivity is not
uncommon. Patch testing is routinely recommended in the work-up of leg
ulcer and leg eczema patients. On average 50% of these patients have a
positive patch test of actual or past relevance.
Intertriginous areas such as the axillae, external ear and
perianal area are also frequent sites of primary sensitization from
topically used medicaments and fragrances because of the natural
occlusion.
Shoe dermatitis is located in the skin area in direct contact
with the offending material, most frequently chromate-tanned leather,
rubber and glues (Podmore, 1995).
Allergic contact dermatitis from textiles gives a characteristic
clinical pattern with dermatitis in areas where textiles are in close
contact with the skin on the trunk and extremities. The offending
sensitizers are textile dyes and formaldehyde-releasing textile resins
(Fowler et al., 1992).
4.1.3 Special types of allergic contact reactions
4.1.3.1 Systemic contact dermatitis
Systemic contact dermatitis can be seen in primary contact
sensitized individuals when they are later exposed systemically to the
chemical (or drug) either orally, intravenously, by inhalation or by
transcutaneous absorption (Menné et al., 1994). The clinical symptoms
can either be erythematous flare in areas with earlier contact
dermatitis or a combination of symptoms including vesicular hand
eczema and inflammatory skin reaction in the flexural and genital
area. The explanation for the flare reaction is probably specific
sensitized lymphocytes persisting at the site of earlier allergic
contact dermatitis areas. The mechanism behind the other type of
reactions is speculative. Histologically this widespread reaction does
not have the picture of contact dermatitis but frequently presents the
picture of a lymphocytic vasculitis. The pathogenesis may be
circulating immune complexes or a general reaction to released
cytokines.
Systemic contact dermatitis is mostly seen in patients sensitized
to topically used medicaments when they are systemically treated with
the medicament or a cross-reacting medicament. Systemic contact
dermatitis has been described for a large number of substances.
4.1.3.2 Allergic photo-contact dermatitis
Most substances that cause photo-contact allergy are halogenated
aromatic hydrocarbons or sunscreen agents (White, 1995). The
combination of light, predominantly ultraviolet (UV), and the specific
chemical make the complete hapten. Clinical allergic photo-contact
dermatitis will therefore present a dermatitis (often severe) in
sun-exposed areas. This will typically be on the face, the forearms or
the dorsal aspects of the hands. In cases where photo-contact allergy
is suspected, patch testing is performed in duplicate and one site is
exposed to UVA. If a positive patch test only appears on the
UV-exposed site, photoallergy is likely.
4.1.3.3 Non-eczematous reactions
Allergic contact sensitivity in the skin can give rise to
clinical reaction patterns other than dermatitis (Goh, 1995). These
types of reactions are rare and to only a few chemicals. Even if these
patients have a clinical reaction type other than dermatitis, they
frequently have a positive patch test with the usual eczematous
morphology. The most common types of non-eczematous contact reactions
are erythema multiforme and lichen planus. Erythema multiforme-like
reactions are caused by contact with plant allergens and the lichen
planus type by contact with photographic chemicals.
4.1.3.4 Allergic contact urticaria
Contact urticaria is an immediate wheal reaction in the skin
caused by vasodilatation, with subsequent oedema. Contact urticaria
can either be allergic or non-allergic. In the non-allergic types
chemical causes a degranulation of the mast cells without involvement
of the immune system. The allergic types are mediated via IgE bound to
specific receptors on the mast cells and basophil lymphocytes in the
skin. The clinical types are similar with urticaria localized at the
contact site. Generalized anaphylactic reactions are rare. Both
organic and inorganic substances have now been described as causes of
allergic contact urticaria (Amin et al., 1996).
Contact urticaria is a frequent occupational disease among
individuals handling animals and animal products. Allergic contact
urticaria from proteins in rubber latex is a frequent and troublesome
problem among workers, particularly health personnel, due to
widespread use of rubber gloves (Taylor & Praditsuwan, 1996; NIOSH,
1997). A sensitization frequency of 2.8 to 10.7% has been reported in
health personnel (Turjanmaa, 1996). Individuals occupationally
sensitized to rubber latex proteins can develop anaphylactic reactions
if exposed to rubber gloves as patients.
4.1.4 Allergic contact dermatitis as an occupational disease
Occupational skin diseases are defined as skin diseases either
wholly or partly caused by the patient's occupation (Rycroft, 1995).
The epidemiology of occupational skin diseases, which mostly comprise
contact dermatitis of the hands, is known from population and
cross-sectional studies of specific occupational groups. Information
from centralized notification systems exists in some countries, but
the quality of data can be questioned. In particular, the problem of
under-reporting is difficult to quantify.
Skin diseases comprise between 20 and 40% of all occupational
diseases, depending on geographical area. Approximately one-third is
caused by allergic contact dermatitis and the rest mainly by irritant
dermatitis. The principal occupational contact sensitizing chemicals
are listed in Table 16. Not unexpectedly there is an overlap between
exposure to chemicals in occupational and domestic environments (see
section 4.1 and Table 19). The common high-risk occupations for
allergic contact dermatitis, modified from Rycroft (1995), are given
in Table 17 (Flyvholm et al., 1996). The prevalence of occupational
contact dermatitis in these occupations varies from a few percent up
to 15% (Rycroft, 1995).
Table 16. Main allergens related to occupational exposure
(from Flyvholm et al., 1996)
Allergens Sources of exposure
Acrylates adhesives; bone cement; dental products; UV-curing lacquers, etc.
Amines hardeners/curing agents for epoxy resin
Chromate cement; leather; pigments
Cobalt paints/lacquers
Colophony adhesives; dental products; paper; tin solder, etc.
Epoxy resin adhesives; paints; electric insulation
Formaldehyde disinfectants; preservatives; laboratory chemicals; formaldehyde resins;
funeral service
Formaldehyde releasers metal working fluids; paints; adhesives
Formaldehyde resins adhesives; paints/lacquers; impregnated textiles and paper; inks
Isocyanates adhesives; paints; fillings; polyurethane foams
Medicaments human and animal health care workers
Nickel coins; nickel plated objects; contaminated oils, etc.
Paraphenylenediamine hair dyes; rubber additive
Plastics/resins adhesives; paints; fillings, containers, etc.
Preservatives water-based products: metal working fluids; paints; adhesives;
cleaning agents; cosmetics; polishes; skin protection creams; process water, etc.
Rubber additives rubber gloves; rubber tubing; washers, etc.
Table 17. High-risk occupations for allergic contact dermatitis
Adhesives/plastics workers Horticulturalists
Agriculturalists Leather tanners
Cement casters Painters
Construction workers Pharmaceutical/chemical workers
Glass workers Rubber workers
Graphic workers Textile workers
Hairdressers Tilers
Health care workers Wood workers
It is difficult to give exact data concerning the costs of
occupational allergic contact dermatitis, as the compensation
regulation differs significantly from one country to another. However,
in the United Kingdom in 1996 it was estimated that 84 000 people had
occupational contact dermatitis, and 132 000 working days were lost
with a cost to employers of 20 million pounds per year (HSE, 1996).
4.1.5 Diagnostic methods
4.1.5.1 Patch testing
The aim of patch testing is to diagnose contact sensitization to
environmental chemicals. The patch test was introduced in 1896 by the
Swiss dermatologist Jadahsson (Wahlberg, 1995). The technology is a
biological test where contact allergy is proved by re-exposing the
skin to the specific chemical under occlusion on a skin area of 0.5
cm2 on the upper back for 2 days. A positive test is a reproduction
of the clinical disease showing redness, infiltration and eventual
vesicles. Standardization has taken place, particularly influenced by
the Scandinavian and later the International Contact Dermatitis
Research Group (ICDRG). The test should only be performed using
standardized test materials. All patients are primarily tested with
the Standard series including the most frequent sensitizing chemicals
such as metals, preservatives, fragrances, rubber additives and
topically used medicaments. Testing is frequently supplemented with
substances present in the patient's private or occupational
environments. Specially trained staff are necessary to obtain high
quality outcome of the procedure.
Sensitization can be quantified according to the degree of
positive patch test reaction (+ to +++), patch test concentration
threshold defined by dilution series, and finally by the "Use test".
In the latter test the individual is exposed to the chemical
simulating normal use.
The outcome of patch testing defines whether contact allergy is
present or not. Quantification of allergy combined with quantitative
exposure data is the basis for individual and general risk assessment
(Flyvholm et al., 1996).
The frequency of positive patch test reactions in the general
population (Nielsen & Menné, 1992) and in eczema patients tested at a
dermatological clinic in the same area of greater Copenhagen, Denmark,
is shown in Table 18. The allergens causing positive reactions most
frequently in eczema patients were nickel, fragrance mix, cobalt
chloride, colophony and balsam of Peru. For the general population,
nickel and thiomersal were the most common causes of positive patch
test reactions. Contact sensitization is generally more frequent among
patients investigated at dermatological centres than it is in the
general population.
4.1.5.2 In vitro testing
Several attempts have been made to develop in vitro methods for
testing contact sensitization (von Blomberg et al., 1990; McMillan &
Burrows, 1995). Yet, logistical and technical complexities, including
allergen toxicities, and the generally low frequencies of circulating
allergen-specific T-effector-memory cells, mean that currently
available methods are not appropriate for routine clinical use.
Nevertheless, in vitro tests, in particular the lymphocyte
proliferation test (LPT), using patient-derived white blood cell
samples, can be of considerable value in answering specific scientific
questions, e.g., on the involvement of allergen-specific T-cells or on
potential cross-reactivity patterns between allergens (Bruynzeel et
al., 1985; Pistoor et al., 1995).
4.1.6 Assessment of exposure
To establish the diagnosis of allergic contact dermatitis, the
outcome of patch testing needs to be combined with a detailed exposure
history (Flyvholm et al., 1996). Both domestic and work-related
exposures need to be elucidated. Factory visits are valuable but
rarely done (Rycroft, 1995). The most common contact allergens are
metals, preservatives, rubber additives, perfumes and medicaments. The
main sources of exposure to contact allergens can be divided into
groups of substances, products or use categories. Exposure to
allergens occurs under many circumstances, such as occupational,
domestic work, hobby and leisure time activities, topical medicaments,
cosmetics, personal care products, clothing and shoes. Examples of
such allergens are listed in Table 19 (Flyvholm et al., 1996). For
examples of occupational exposure, see Table 16 (section 4.1.4).
Exposure data can be obtained from databases, product labelling or
chemical analysis, and by contact with manufacturers and suppliers.
The prognosis for the individual patient depends upon the quality of
diagnostic patch testing and the ability to prevent contact of the
patient with the allergen.
Table 18. Comparison of frequencies of positive patch test reactions
in the general population (Nielsen & Menné, 1992) and in eczema patients at a
dermatological clinic in the same area of greater Copenhagen in 1990a
Test substance General populationb Dermatological clinicc
(% positive of tested) (% positive of tested)
Men Women Total Men Women Total
(n=279) (n=288) (n=567) (n=262) (n=410) (n=672)
Potassium dichromate 0.7 0.3 0.5 1.9 2.7 2.4
Neomycin sulfate 0.0 0.0 0.0 3.4 3.7 3.6
Thiuram mixture 0.7 0.3 0.5 4.6 2.7 3.4
p-Phenylenediamine 0.0 0.0 0.0 1.9 2.7 2.4
Cobalt chloride 0.7 1.4 1.1 2.3 2.7 2.5
Benzocaine - - NT 0.4 0.7 0.6
Caine(R) (local 0.0 0.0 0.0 - - NT
anaesthetic) mix
Formaldehyded - - NT 1.9 2.2 2.1
Colophony 0.4 1.0 0.7 4.6 5.4 5.1
Quinoline mix 0.4 0.3 0.4 1.9 0.5 1.0
Balsam of Peru 0.7 1.4 1.1 3.4 5.4 4.6
PPD black rubber mix 0.4 0.0 0.2 1.2 0.0 0.5
Wool alcohols 0.4 0.0 0.2 1.2 1.7 1.5
Mercapto mix 0.7 0.0 0.4 1.2 0.2 0.6
Epoxy resin 0.4 0.7 0.5 0.8 0.2 0.5
Paraben mix 0.4 0.3 0.4 0.8 0.2 0.5
p-tert-Butylphenol 1.1 1.0 1.1 0.4 1.2 0.9
formaldehyde resin
Fragrance mix 1.1 1.0 1.1 6.1 7.1 6.7
Ethylenediamine 0.4 0.0 0.2 0.8 0.7 0.7
dihydrochloridee
Quaternium 15 0.4 0.0 0.2 0.0 0.0 0.0
Nickel sulfate 2.2 11.1 6.7 4.2 16.1 11.5
MCI/MI 0.4 1.0 0.7 0.4 0.7 0.6
(chloro-methyl- and
methyl-isothiazolinone)
Table 18. (continued)
Test substance General populationb Dermatological clinicc
(% positive of tested) (% positive of tested)
Men Women Total Men Women Total
(n=279) (n=288) (n=567) (n=262) (n=410) (n=672)
Mercaptobenzothiazole 0.4 0.0 0.2 1.2 0.2 0.6
Priminf - - NT 0.4 1.5 1.0
Thiomersalg 3.6 3.1 3.4 - - NT
Carba mixh 0.7 0.0 0.4 - - NT
a Menné, unpublished (personal communication by T. Menné to the IPCS, 1997)
b Patch tested with the ready-to-apply TRUE test, Pharmacia (Sweden)
c Test substances from Hermal (Germany)
d Formaldehyde not included in TRUE test at the time of study
e Ethylenediamine dihydrochloride excluded from the European Standard series as of August 1992
f Primin not included in the TRUE test at the time of study
g Thiomersal not included in the European Standard series
h Carba mix excluded from the European Standard series as of January 1989
Table 19. Main allergens related to non-occupational exposure
Allergens Sources of exposure
Domestic work
Chromium leather; footwear
Colophony shoe polish; crayons; plasticine; paper
Flowers/plants gardening; house plants
Nickel nickel-plated objects
Plastics/resins adhesives; paints; containers
Preservatives cleaning agents; polishes; personal care products
Rubber additives gloves; other rubber objects
Wood repairs; handicraft
Hobbies and leisure time activities
Chromium leather; footwear
Colophony adhesive tapes; plasticine; paper; violin bow resin
crayons; artists' paints; textiles
Dyes/pigments gardening; house plants
Flowers/plants textile resins; preservative in various products
Formaldehyde nickel-plated objects
Nickel adhesives; paints; containers
Plastics/resins paints; personal care products
Preservatives gloves; sports equipment
Rubber additives handicrafts
Woods
Cosmetics and personal care products
Colophony mascara.
Dyes hair dyes; miscellaneous cosmetics
Fragrances
Glyceryl thioglycolate permanent waving
Lanolin
Table 19 (cont'd)
Allergens Sources of exposure
Paraphenylenediamine hair dyes; creams; lotions; shampoos;
liquid soap, etc. (i.e., most cosmetic
and personal care products)
Preservatives, e.g.,
formaldehyde releasers,
isothiazolines parabens
UV filters sunscreens
Topical medicaments
Antibiotics
Antihistamines
Antimicrobials
Balsams
Benzocaine
Colophony
Ethylenediamine
Formaldehyde releasers
Lanolin
Parabens
Preservatives
Tars
4.1.7 Treatment and prevention of allergic contact dermatitis
The treatment of allergic contact dermatitis requires medical
intervention. It usually involves the controlled use of emollients or
corticosteroids as well as prevention of further exposure to the
offending allergen (Wilkinson, 1995). A distinction is usually made
between primary prevention, focusing on the induction of contact
sensitization, and secondary prevention, focusing on the eliciting of
contact sensitization. In many instances the preventive measures for
the two different types overlap.
4.1.7.1 Primary prevention
In the 1960s an epidemic of contact dermatitis from dish-washing
products occurred in Scandinavia. The epidemic was resolved by the
concerted action of dermatologists and manufacturers. Extensive
chemical analysis combined with animal predictive testing, identified
highly sensitizing sultones to be present in some products (Magnusson
& Gilje, 1973; Ritz et al., 1975). It was determined that these
specific chemicals occurred as an impurity in the manufacturing
process, when temperature control was not strictly maintained. The
evaluation of the problem led to a solution, and there have been no
recurrences.
There are examples of exposure to hapten concentrations being
legally regulated in an attempt to prevent contact sensitization
(Hjorth & Menné, 1990). There is a complex European Union regulation
on cosmetic products, forbidding certain substances and regulating
others, i.e., preservatives, by a concentration limit (Council of the
European Communities, 1976).
Since the 1950s, chromate in cement has been know to be one of
the main causes of allergic chromate dermatitis among construction
workers. At the start of the 1980s the Scandinavian countries added
ferrosulfate at a low concentration to cement to reduce the hexavalent
chromate to trivalent chromate. The idea of this initiative was that
the trivalent chromate is not absorbed, or only to a minor degree,
through human skin, and therefore the risk of primary sensitization
from this salt is significantly less than from hexavalent chromate.
Epidemiological studies on construction sites performed at the
beginning of the 1980s and at the end of the 1980s in Denmark,
strongly suggest that this measure has been successful, as the
frequency of allergic chromate dermatitis has been reduced in Denmark
(Avnstorp, 1992).
Nickel is a common contact allergen on a global scale. This
allergy is caused by intimate skin contact with metal alloys,
releasing nickel when exposed to human sweat. Under simulated use
conditions, some alloys release high amounts and other alloys low
amounts of nickel (Lidén et al., 1996). Based on such research, some
Scandinavian countries have introduced regulations and quality
criteria for nickel alloys intended to be in prolonged skin contact.
It is believed that such measures might reduce significantly the
frequency of nickel allergy in the population. Regulation of nickel
exposure along similar lines has been adopted within the European
Union (Council of the European Communities, 1994).
In considering different glove materials to protect against skin
irritation and mechanical skin damage, it should be noted that most
small sensitizing chemicals rapidly penetrate most rubber and plastic
gloves, and appropriate gloves should therefore be used (Estlander &
Jolanki, 1988; Mellström et al., 1989; Roed-Petersen, 1989).
There is no method of predicting an individual propensity to
contact sensitization to a given chemical. When patch testing with
strong sensitizing chemicals is performed, active sensitization from
the test cannot completely be excluded. Pre-employment testing is
therefore not a method of preventing contact sensitization.
4.1.7.2 Secondary prevention
The cornerstones of the secondary prevention of allergic contact
dermatitis (elicitation of contact dermatitis) are based on sufficient
diagnostic procedures and patient information systems. The
availability of standardized patch test materials is essential.
Furthermore, it is crucial that it is possible for the doctor to
inform the patient where exposure to the specific allergen can be
expected. Of course, it is even more crucial that the patient is able
to understand and use the information over the following years to
identify the allergen in the home and occupational environments. It
seems obvious that this type of diagnostic follow-up will work, but it
has only been evaluated in a limited number of studies. Edman (1988)
found that the prognosis for patients sensitive to topical medicaments
depended upon whether the patients were able to follow the doctor's
advice on the occurrence of sensitizers in different products. Later
studies have shown that patients with contact allergy to formaldehyde
often continued to be exposed to formaldehyde (Cronin, 1991; Flyvholm
& Menné, 1992). When a careful investigation was made, formaldehyde
exposure could be demonstrated in nearly all the patients which seemed
to be decisive for the prognosis of their hand eczema (Flyvholm,
1997).
4.1.7.3 Ways of preventing contact sensitization
The following ways of preventing contact sensitization have been
suggested.
a) replacement of certain chemicals or particular products;
b) regulation of exposure (concentration) to sensitizing
chemicals, either general or in specific products, or during
particular work processes;
c) optimal diagnostic and information systems; education of
either groups or individuals;
d) individual oriented preventive methods; gloves, barrier
creams, protective clothing.
The problems of contact sensitization have been identified over
many years, and different types of preventive measures have been
tried. Some have been successful, but a number of chemicals still give
problems to a significant number of people. Different strategies
should be considered, whether it concerns common environmental
chemicals or chemicals with rare specific exposures. Chemicals
frequently used in both the domestic and occupational environment need
to be regulated by society, either with suggestion of replacement or
regulation of the exposure concentration. For rare chemicals it is
often sufficient to focus on specific occupational processes and to
educate the exposed individuals in no-touch techniques or introduce
individually oriented preventive measures.
4.1.8 Information needed for a preventative programme
The prevention of allergic contact dermatitis should be based on
preventing sensitization and, subsequently, on avoiding sufficient
exposure to elicit a response in a person already sensitized. This
requires information on the following aspects.
a) Occurrence of sensitizing substances
Products used at work or domestically should be labelled to
indicate the presence of substances capable of causing sensitization
and their concentrations, so that the user may take appropriate
precautions.
If there are suitable alternatives there may be no need to use a
sensitizing agent.
At present, the potential of new substances to cause
sensitization is determined from the results of tests on animals or
sometimes on humans (Rycroft, 1995), after databases have been
searched for relevant published information. Structure-activity
relationships should be assessed and may give valuable indications of
sensitizing potential for substances of a similar structure to known
contact allergens.
Comprehensive information about the composition of products and
the allergenic activity of their ingredients should be collected in
each country and be made available to health care professionals and
users. This should include the results of surveys of standardized
patch testing of humans so that trends in allergic sensitization can
be followed.
b) Avoiding or minimizing exposure
Induction of sensitization and eliciting an allergic disorder
both follow dose-response relationships, albeit at very different
concentrations.
It is important to minimize initial exposure to sensitizing
agents by restricting their availability or, if they cannot be
avoided, by minimizing exposure. Exposure can be minimized by ensuring
adequate ventilation and using personal protective equipment
appropriate to the work situation or in the home, e.g., gloves, masks,
etc. (see also chapter 7).
4.2 Atopic eczema (atopic dermatitis)
4.2.1 Definition
Atopic eczema or atopic dermatitis is a chronic pruritic
inflammatory skin disease characterized by a typical age-related
distribution and skin morphology (Figs. 12, 13). The diagnosis of
atopic eczema is based primarily on clinical grounds and the patient's
history (Hanifin, 1983; Rajka, 1990). Onset at an early age, pruritus
and excoriation, chronic or chronic relapsing course for more than
6 weeks, age-related eczematous morphology and distribution, as well
as a positive family history for atopic diseases (allergic bronchial
asthma, allergic rhinitis and conjunctivitis or atopic eczema), form
the most striking criteria. Together with allergic rhinitis and
conjunctivitis and bronchial asthma, atopic eczema forms the classical
triad of atopic diseases (Rajka, 1990; Ruzicka et al., 1991). Atopy
can be defined as "familial hypersensitivity of skin and mucous
membranes against environmental substances associated with increased
IgE production and/or nonspecific reactivity" (Ring, 1991). This
underlines two components held to be responsible for induction of this
disease. Although it is genetically determined, environmental
influences may play a role. During the last century many synonyms for
atopic eczema/atopic dermatitis have been evolved, e.g.,
neurodermatitis, prurigo Besnier, endogenous eczema and diffuse
neurodermatitis (Ring, 1991). The diagnosis of this skin disease is
based on clinical criteria, family history and/or demonstration of
IgE-mediated sensitization.
4.2.2 Epidemiology of atopic eczema
Atopic eczema is a common disease among children and adults. In
the 1950s the frequency of eczema was estimated to be between 1.1 and
3.1% (Walker & Warin, 1956). In the 1980s and 1990s the frequency of
atopic eczema was found to be up to 25% on the basis of questionnaires
(Bakke et al., 1990) and up to 9.7% for dermatologically examined
cohorts (Varjonen et al., 1992; Schäfer & Ring, 1997). Epidemiological
studies on the prevalence of atopic eczema in Germany were conducted
with questionnaire, physical and dermatological examination including
allergy tests. In 1989-1991 8.3% of 988 Bavarian school children aged

 5 to 6 years suffered from atopic eczema (Schäfer et al., 1994), and
in a study comparing eastern and western German areas in 1991 atopic
eczema was diagnosed in 12.9% of 1086 pre-school children (Ring et
al., 1995; Krämer et al., 1996; Schäfer & Ring, 1997). In studies in
the United Kingdom, Denmark and Switzerland, the same methodological
analyses were applied for a longer time interval to obtain figures on
the changes in frequency of atopic eczema. These studies showed a
dramatic increase in the prevalence of atopic eczema (Schäfer & Ring,
1995). In the United Kingdom the figures for the prevalence of atopic
eczema were 5.1% in 1946, 5.3% in 1964, 7.3% in 1958, and around 12%
for 1970-1989. Similarly, in Denmark the prevalence in 1964-1969 was
3.2% compared with 11.2% for 1970-1979. In Switzerland there was an
increase from 2.2% in 1968 to 2.8% in 1981.
4.2.3 Clinical manifestations and diagnostic criteria
4.2.3.1 Age-dependent clinical manifestations
In most patients with atopic eczema, the disease begins in
infancy between 3 and 12 months of age (Hill & Sulzberger, 1935) as an
erythematous, squamous or papulo-vesiculous inflammation, which may
worsen to the point of exudation. It is often found on the face, the
extremities (especially extensor aspects) and finally the trunk.
Oozing and crusted lesions can often be found on the scalp (cradle
cap). More and more, itching becomes an essential feature; the infant
may be irritable, restless and tries to scratch the affected areas
(after 3rd month of life). The course is chronically persistent or
relapsing. Later, between 2 and 5 years, the appearance of the lesions
changes. They become nummular and infiltrated. The localization
changes and affects flexures of popliteal and antecubital fossae, the
nape of the neck and the backs of the hands and feet. In severe cases
there may be an involvement of the entire skin surface. Dry skin
becomes another characteristic feature especially in the adult phase
and creates itching followed by scratching. This may lead to severe
excoriation with nodule formation and perpetuation of the inflammatory
reaction ("Prurigo Besnier"). Chronic inflammation produces thickening
(lichenification) of the skin, especially in flexural regions.
4.2.3.2 Diagnosis of atopic eczema
Many diagnostic systems have tried to collect reliable criteria
for this disease. The features listed by Hanifin & Rajka (1980) are
those referred to most often in the literature. A combination of a
number of major and minor criteria allows the establishment of the
diagnosis. A more simple selection of criteria for practical purposes
has been proposed (Ring, 1991). Williams et al. (1994a) proposed a new
arrangement of diagnostic criteria, primarily for epidemiological
studies. However, it must be kept in mind that all these diagnostic
systems have their drawbacks in this heterogenous disease. Clinical
criteria, as well as the patient's history and presence of
IgE-mediated sensitizations must be considered together and are the
mainstay for establishing the diagnosis. However, minimal forms exist
and sometimes do not meet the required criteria. Papular or nodular
variants as well as localized forms (e.g., exfoliating cheilitis,
infra-auricular rhagades, nipple eczema, finger pad or toe eczema)
constitute minimal expressions of this disease (Wüthrich, 1991).
Typical eczematous lesions may not only be triggered by IgE-mediated
allergic reactions in patients with a positive family history of
atopy, but can also be triggered by food additives. In most patients,
establishing the diagnosis is not too difficult. In selected cases,
clinical findings, history and IgE-mediated sensitization have to be
regarded critically and all important differential diagnoses have to
be ruled out thoroughly.
4.2.3.3 Stigmata of the atopic constitution
The diagnosis of atopic eczema often depends on further
additional features. Stigmata of atopic constitution are prevalent in
many patients with atopic eczema, although they are not specific for
this disease. Dry skin, hyperlinearity of palms and soles,
intraorbital fold, white dermographism, facial pallor, orbital
darkening, low hairline and thinning of the lateral portions of the
eyebrow are found more often in this group of patients (Przybilla,
1991). They are typical constitutional markers, which may add another
clue in establishing the diagnosis (Ring, 1988).
4.2.3.4 Prognosis
Variability and chronic relapses are characteristics of the
course of atopic eczema. Atopic eczema most frequently begins during
infancy (Hanifin, 1983; Rajka, 1990). In about two-thirds of infants
with atopic eczema, the disease clears during childhood. In the
remaining patients it persists into adult life. Minimal forms and
stigmata of the disease often remain throughout life (Vickers, 1991).
Sometimes atopic eczema starts only in adulthood. A definite prognosis
about the course of an individual patient cannot be made; there is
controversy about prognostic factors (Vickers, 1991).
4.2.4 Etiology
The manifestation of atopic eczema is subject to a multifactorial
genetic predisposition as well as to environmental provocation
factors.
4.2.4.1 Genetic influence
There is no doubt about the existence of a genetic component
favouring the manifestation of atopic eczema (Schnyder, 1960; Küster
et al., 1990). Twin studies show a concordance in homozygous twins of
83 and 86%, compared to 28 and 21% in heterozygous twins (Niermann,
1964; Schultz-Larsen, 1991). The chance of developing atopic eczema
depends on the family history of atopy. Whereas about 10-15% of
children without a family history of atopy develop atopic eczema, with
a positive history of one parent the risk rises to 25-30% and, with a
positive history of both parents, to 50-75% (Schultz-Larsen et al.,
1986; Björksten & Kjellman, 1987).
4.2.5 Environmental provocation factors
The activity of atopic eczema can be influenced by a large number
of environmental provocation factors (Table 20). These can either act
specifically in the sense of individual hypersensitivity, primarily
IgE-mediated allergy or, more often, as unspecific provocation factors
irritating the skin or affecting emotional status. The question of the
possible involvement of environmental atmospheric pollution in the
increase in the prevalence of atopic eczema remains controversial (see
section 3.3.2).
4.2.6 Pathophysiology
Although knowledge concerning components of the immune system and
inflammatory responses in patients with atopic eczema has increased
widely in recent decades, the pathophysiology of atopic eczema also
remains controversial (Marchionini, 1960; Rajka, 1990, 1996).
4.2.6.1 Dry skin
Dry or rough skin is a major feature of skin alteration in
patients with atopic eczema. Although a number of studies have
investigated the pathophysiology of dry skin, there is no consensus
(Melnik & Plewig, 1991; Lindskov & Holmer, 1992). An attractive
hypothesis is that even clinically non-inflamed "dry skin" shows
histologically a mild inflammatory infiltrate, and this is supported
by skin biopsies in atopic patients (Uehara, 1991). There seems to be
an intimate relation between dry skin, irritability and itch (Rajka,
1990; Ruzicka et al., 1991).
4.2.6.2 Autonomic dysregulation
In addition to immunological abnormalities, signs of
dysregulation of the autonomic nervous system have been described
(Szentivanyi, 1968; Ring et al., 1988; Ring & Thomas, 1989; Hanifin,
1993). Elevated phosphodiesterase activity in mononuclear leukocytes
seems to correlate with increased IgE production and vasoactive
mediator secretion (Butler et al., 1983; Cooper et al., 1985).
4.2.6.3 Cellular immunodeficiency
First described by Kaposi in 1895, patients with atopic eczema
are more susceptible to infection with viruses (e.g., Herpes
simplex, Human papilloma) and bacteria (especially Staphylococcus
aureus) (Kaposi, 1895). Earlier reports about decreased frequencies
of allergic contact sensitization in atopic eczema are contradicted by
Table 20. Important environmental provocation factors in atopic eczema
(adapted from Ring et al., 1996)
Unspecific provocation factors:
Irritants
Microbial skin colonization or infection
e.g., Staphylococcus aureus
Pityrosporum ovale
Herpes simplex (Eczema herpeticum)
Psychological stress, emotional factors
Specific provocation factors (individual hypersensitivity):
IgE-mediated allergy
e.g., Food
House dust mite
Animal dander
Pollen
Microbial colonisation?
Contact allergy
Pseudo-allergy (idiosyncrasy) and intolerance
e.g., preservatives in foods
citrus fruits
others who claim that the tendency to develop contact allergy is
increased (Rajka, 1990). However, Enders et al. (1988) reported that
the prevalence of positive patch test reactions for contact allergy in
patients with atopic eczema was almost equal to that of patients with
allergic contact dermatitis.
4.2.6.4 Increased IgE production
Serum IgE levels are elevated in the majority of patients with
atopic eczema (Ogawa et al., 1971). They tend to correlate with the
extent and severity of the disease (Johansson & Juhlin, 1970;
Wüthrich, 1975). Specific antibodies can be measured against common
environmental allergens (Rajka, 1990; Ruzicka et al., 1991). Although
often the clinical significance of these antibodies is lacking, in
some patients eczematous skin responses can be provoked by
aeroallergens (grass pollen, house dust mite or animal dander), a
procedure that has been called "atopy patch test" (Reitamo et al.,
1986; Adinoff et al., 1988; Ramb-Lindhauer et al., 1990; Ring et al.,
1991a,b; Platts-Mills et al. 1991; Vieluf et al., 1993). IgE
antibodies to foods are frequently found in patients and may induce
urticaria as well as eczematous reactions. Well-controlled clinical
trials showed that in a high number of patients with atopic eczema,
skin lesions were exacerbated after specific oral provocation with
certain foods in double-blind studies (Sampson & Albergo, 1984). Apart
from aeroallergens and foods, microbial allergens ( Staphylococcus
aureus, Pityrosporum ovale) might play a role. Chronic colonization
of atopic skin could provide a continuing cause of allergen
stimulation (Leyden et al., 1974; Ring et al., 1992, 1995; Neuber et
al., 1995; Kröger et al., 1995). After allergen stimulation of
IgE-bearing mast cells or basophils, the released vasoactive mediators
(such as histamine, eicanosoids, etc.) might induce itching, and also
eczema via a late-phase reaction (Dorsch & Ring, 1981). Langerhans
cells in the epidermis express high affinity receptors for IgE as well
as CD23 and IgE binding-protein (Bieber & Ring, 1992). Allergen
contact might result in the generation of Th2-helper cells, a subset
producing IL-4 and IL-5, thereby maintaining the allergic
inflammation. Also other cell types might be involved in the
inflammatory process; lymphocytes might act directly through
cytokines, and eosinophils through release of pro-inflammatory
mediators (Jakob et al., 1991; Kapp, 1995).
4.2.6.5 Psychosomatic aspects
It is well known from clinical experience that psychological and
emotional factors can greatly influence the clinical course of this
skin disease (Borelli & Schnyder, 1962; Jordan & Whitlock, 1972, 1974;
Ring et al., 1986; Cotterill, 1991). There is no convincing evidence
that psychological factors per se are the primary cause for atopic
eczema; however, it is clear that psychological factors may influence
existing eczematous lesions or even trigger new exacerbations of
eczema in many patients (Rajka, 1990; Ruzicka et al., 1991). For
children, the family situation, e.g., the interaction between parents
and the affected child, seems to be of particular importance (Ring et
al., 1976; Niebel, 1995).
4.2.7 Diagnostic approach
In atopic eczema diagnosis not only comprises the identification
of the disease but should also focus on individual provoking factors
able to trigger disease activity (Ring et al., 1991a,b; Morren et al.,
1994). Each patient may be susceptible to an individual set of
provocation factors. Often, exacerbations can be prevented or the skin
condition can be directly improved by avoidance of these factors (Ring
et al., 1996). Diagnostic procedures used are intended to reveal
provocation factors for the individual patient. Specific provocation
of atopic eczema often is the result of an individual
hypersensitivity. Although diagnostic tests normally differ from the
natural exposure with allergens, they provide useful information in
the hands of a trained allergist (Ring, 1988). Allergy diagnosis is
based on the four foundations: the patient's history, skin tests,
in vitro (laboratory) tests and provocation tests.
4.2.7.1 Medical history
The patient's history forms the backbone of allergy diagnosis.
Often the patient notices associations between disease activity and
specific conditions or actions (e.g., intake of foods, seasonal or
daily variations, contact with animals, heavy pollen emission). These
observations are very valuable in revealing individual provocation
factors. On the other hand, positive allergy tests must always be
verified for their clinical significance for the patient's disease by
comparing them with the history.
4.2.7.2 Skin tests
Skin test methods are divided into percutaneous (skin-prick,
intradermal) tests and epicutaneous (patch) tests (American Medical
Association, 1987a). Percutaneous tests search for immediate-type
IgE-mediated hypersensitivity and are especially indicated in atopic
eczema. The skin-prick test (prick puncture test) has gained the
widest acceptance because of its high convenience and safety (Dreborg,
1989). A drop of the test extract is placed on the volar surface of
the forearm and the solution is introduced into the epidermis with a
disposable hypodermic needle. After 15 min the reactions are graded in
relation to the erythema and wheal that are induced. In intradermal
testing 0.02 to 0.05 ml of the test extract is injected intradermally
with a syringe. Scratch tests (applying the extract to a superficial
scratch) and rub tests (rubbing of the skin with native allergen) are
other variants applied only for special indications. Because of the
danger of producing anaphylactic reactions these tests should be
performed only by trained allergists with experience in emergency
treatment. In patients with atopic dermatitis, percutaneous tests are
widely used for the detection of hypersensitivity against
environmental aeroallergens and foods (Ring, 1988).
Epicutaneous tests primarily focus on the detection of contact
allergy by cell-mediated immunity. The extract is put in an aluminum
chamber and fixed onto the skin of the patient for 48 h. The test
reaction is graded after 48 and 72 h. An eczematous response is
regarded as positive. Since it has been shown that in patients with
atopic eczema, eczematous skin responses can be elicited by epidermal
application with aeroallergens (especially the house dust mite),
epicutaneous testing with the atopy-patch test is gaining wide
acceptance (Adinoff et al., 1988; Vieluf et al., 1990; Darsow et al.,
1995). Although the definite mechanism is still unknown, this test
might fill the gap between IgE-mediated hypersensitivity and an
eczematous response.
4.2.7.3 Laboratory tests
In the serum of patients with atopic eczema, hypersensitivity can
be detected by laboratory methods. In atopic eczema the most important
hypersensitivity reactions are thought to be IgE-mediated. IgE
antibodies can be determined by binding to an allergen in a solid
phase and radioactive, enzymatic or fluorometric labelling (Ring,
1988). Specific antibodies against environmental allergens are
detected by the RAST (Radio-Allergo-Sorbent Test) and expressed
semi-quantitatively in different classes. Positive reactions must be
interpreted with regard to their clinical relevance (Pastorello et
al., 1989).
4.2.7.4 Provocation tests
Oral provocation tests and elimination diets are often necessary
for the evaluation of the clinical relevance of a suspected food
hypersensitivity (Przybilla & Ring, 1990). Also, allergy-like symptoms
to food additives, medications, etc., may be produced by
non-IgE-mediated mechanisms ("pseudo-allergy") (Vieluf et al., 1990).
In these cases elimination diets and provocation tests are performed.
Foods unlikely to produce adverse reactions can be screened by
elimination diets or open challenges. Oral provocation by double-blind
placebo-controlled food challenges is regarded as the "gold" standard
for the diagnosis of food allergies (Sampson, 1983; Bruijnzeel-Koomen
et al., 1995). However, there are pitfalls and problems with this
procedure (Bindslev-Jensen, 1994a).
4.2.8 Therapeutic considerations
The disease can be effectively controlled by a combination of
avoidance procedures, basic dermatological therapy and
anti-inflammatory therapy for exacerbations (Przybilla et al., 1994;
Ring et al., 1996). However, the patient has to accept that there is
no simple therapy allowing permanent cure. The integration and active
cooperation of the patient in the therapeutic concept ("patient
management") is a prerequisite for an effective therapy. In atopic
eczema, diagnostic and therapeutic approaches are intimately
connected.
4.2.8.1 Avoidance of provocation factors
During the first year of life food allergies are frequent. Later,
sensitization to aeroallergens becomes more important (Guillet &
Guillet, 1992). Food allergies were found in 63% of children with
extensive atopic eczema (Sampson, 1982).
Eggs, cow's milk, wheat, seafood and nuts present the most
important food allergens. Citrus fruits and preservatives in foods
often affect patients via non-allergic mechanisms (Przybilla & Ring,
1990). Individual provocation factors (hypersensitivity) have to be
revealed by allergological diagnostic procedures. Therapy consists in
the elimination of the relevant allergens from the diet. If extensive
interventions are planned, the help of a dietitian is needed.
Controversy exists about the value of prophylactic dietary
manipulations. Exclusive breast feeding for six months, maternal
avoidance of allergens during lactation, and delay of solid food
feeding seem to have a protective influence in postponing or avoiding
atopic eczema (Kajosaari & Saarinen, 1983; Arshad et al., 1992;
Saarinen & Kajorsaari, 1995).
Sensitization to aeroallergens is frequently found in older
children and in adults. As shown by atopy patch tests, in some
patients direct contact with house dust mite allergen, animal dander
and pollen on intact skin results in eczematous skin lesions (Ring et
al., 1991a,b; Darsow et al., 1995). In the case of a suspected allergy
to house dust mites, reduction procedures should include encasing of
bedding with impermeable synthetic material and removal of carpets and
upholstered furniture (Platts-Mills & Chapman, 1987; Platts-Mills et
al., 1991; Lau et al., 1995). When allergy to animal dander is shown,
contact with the animal must be avoided. In case of exacerbation of
atopic eczema due to aeroallergens, rehabilitation in
aeroallergen-poor climates (sea level or high altitude mountains) has
been recommended (Borelli, 1981). In patients with severe atopic
eczema without adequate improvement of skin condition despite therapy,
additional contact allergy should be suspected and excluded by
epicutaneous (patch) testing.
Furthermore, there are various nonspecific provocation factors
influencing the disease activity in patients with atopic eczema. The
skin of these patients is highly susceptible to irritants, such as
wool, coarse fabrics, soap, detergents, frequent bathing,
disinfectants, wet working conditions and others. Patients need to be
educated about avoidance of these factors (Ring et al., 1996).
Chronic microbial colonization of the skin (e.g.,
Staphylococcus aureus, Pityrosporum ovale) and superinfection are
possible additional provocation factors and should be treated (Cooper,
1994). Psychological factors such as stress are well-known triggering
factors for a subgroup of patients. In these patients, psychosomatic
intervention has been proven successful and psychosomatic approaches
should be supported (Cotterill, 1991; Ehlers et al., 1995).
4.2.8.2 Basic dermatological therapy
In patients with atopic eczema there is a defective skin barrier
against exogenous substances (Ruzicka et al., 1991; Schöpf et al.,
1995). Regular basic therapy with emollients with or without addition
of moisturizers and bath oils is needed for the treatment of the
irritable dry skin to prevent the itch/scratch cycle.
4.2.8.3 Anti-inflammatory therapy
Recurrent relapses are a characteristic feature of atopic eczema.
Anti-inflammatory therapy of exacerbations is aimed to control
effectively disease activity and permit a return to basic
dermatological therapy as soon as possible. Topical corticosteroids
are the drugs of choice for acute exacerbations.
4.2.9 Conclusion
Atopic eczema is one of the most common skin diseases in many
countries of the world with an increasing prevalence. Prevalence rates
range between 10 and 20% of school children. Owing to the immense
suffering caused by the skin disfigurement and the often unbearable
itching, as well as the large number of people affected, it presents a
major health problem. The role of allergy in this skin disease has
been controversial but it has been shown that in the majority of
patients, allergic reactions -- preferentially by IgE-mediated
sensitization -- seem to play a clinically relevant role in eliciting
and maintaining eczematous skin lesions.
4.3 Allergic rhinitis and conjunctivitis
4.3.1 Introduction
Allergic reactions can occur in the respiratory tract and ocular
conjunctiva. In the respiratory tract allergic reactions occur in:
a) the upper respiratory tract predominantly involving the
nose - rhinitis;
b) bronchial airways - asthma;
c) gas exchanging parts of the lung - extrinsic allergic
alveolitis.
Allergic reactions in the nose and airways are characterized by
mucosal infiltration with eosinophils and T-lymphocytes, diseases now
considered to be the manifestation of a local Th2-lymphocyte-dependant
eosinophilic inflammation. In contrast, extrinsic alveolitis is
characterized by granulomata and mononuclear cell inflammation within
alveoli, centred upon bronchioles; the disease is considered to be the
manifestation of a local Th1-dependant granulomatous inflammation.
Both patterns of reaction are predominantly induced by agents
suspended in the air, such as dust or fume particulates, aerosol
droplets or vapour, inhaled into the respiratory tract. In general
larger particles will be deposited and soluble chemicals dissolved in
the upper respiratory tract; smaller particles (<5 µm aerodynamic
diameter) and insoluble chemicals can penetrate into the gas
exchanging parts of the lung.
4.3.2 Definition
Allergic rhinitis and conjunctivitis are common allergic
inflammatory conditions induced by hypersensitivity to environmental
allergens affecting the nasal (rhinitis) and/or conjunctival mucosa
(conjunctivitis) (Mygind, 1986, 1989). Rhinitis, characterized by one
or more of the symptoms of nasal congestion, rhinorrhea, sneezing and
itching, is defined as the inflammation of the lining of the nose
(International Rhinitis Management Working Group, 1994). The symptoms
of allergic conjunctivitis consist of redness, lachrymation, itching
and burning of the conjunctiva (Ring, 1991). There is an increased
likelihood of the development of asthma in these patients.
4.3.3 Clinical manifestations
4.3.3.1 Seasonal allergic rhinitis and conjunctivitis (hay fever,
pollinosis)
Seasonal allergic rhinitis and conjunctivitis consists of
paroxysms of sneezing, nasal itching, nasal congestion and rhinorrhea
(Druce, 1993; Mygind, 1986). In severe cases the conjunctiva and
mucous membranes of the Eustachian tube, middle ear and paranasal
sinuses also may be involved. In these cases additional symptoms
usually present with low-grade itching, lacrimation, burning,
stinging, photophobia, redness and watery discharge, as well as ear
fullness, ear popping and pressure over the cheeks and the forehead.
This may be complicated by malaise, weakness and fatigue. Symptoms
typically show a periodic distribution manifesting at individual time
intervals during the pollen seasons of tree, grass and weed pollen
between spring and autumn months. About 20% of patients have asthmatic
symptoms as well (Smith, 1983). Food allergy, often manifesting as
"oral allergy syndrome" due to cross-reacting allergens, may also be
present (see section 4.5.2).
4.3.3.2 Perennial allergic rhinitis and conjunctivitis
In perennial allergic rhinitis and conjunctivitis, indoor
allergens are the main cause of symptoms, which are similar to those
of seasonal allergic rhinitis and conjunctivitis although nasal
blockage is more pronounced and itching of the eyes is a common
problem. Among the indoor allergens, house dust mites, cockroaches,
animal dander and moulds are important. The chronic and persistent
symptoms can present as a "permanent cold" and may be accompanied by
secondary complaints, such as mouth breathing, snoring and sinusitis
(Lucente, 1989). Occupational hypersensitivity to an airborne allergen
at the workplace may lead to symptoms only during the week with a
disease-free interval at weekends, for example in laboratory animal
workers.
4.3.3.3 Prognosis
The peak prevalence of allergic rhinitis and conjunctivitis is in
adolescents and young adults. The first manifestations of seasonal
allergic rhinitis and conjunctivitis develop before 20 years of age in
most patients. After 30 years of age, disease severity usually
moderates and is only occasionally a problem in the elderly. Repeated
exposure to allergens may cause nasal hyperreactivity also to other
allergens, thus broadening the spectrum of hypersensitivity (Connell,
1969). A proportion of patients will develop asthma in the course of
their disease (Evans, 1993).
4.3.4 Etiology
Symptoms of allergic rhinitis and conjunctivitis are provoked by
environmental aeroallergens. Typical seasonal allergens are tree
pollen in the spring, grass pollen in the early and mid summer and
weed pollen in the late summer. In temperate climates of the Northern
hemisphere the most important tree pollens derive from birch, alder
and hazel; among grass pollens timothy and ryegrass, and among weed
pollens mugwort and ragweed are the most important. However, regional
differences are also of importance, e.g., cedar pollen in Japan,
parietaria pollen in the Mediterranean area and ragweed pollen in the
USA being the most important allergens. Sometimes mould spores, e.g.,
Cladosporium and Alternaria, cause symptoms during summer and
autumn months. In perennial allergic rhinitis and conjunctivitis
mainly indoor allergens present in the environment throughout the year
are relevant triggers. The house dust mites Dermatophagoides
pteronyssinus and Dermatophagoides farinae, in Southern countries
Bloomia tropicalis, animal dander from horses, cats, dogs and other
pets, cockroaches, and moulds such as Aspergillus species are the
most important allergens.
Epidemiological studies indicate a significant increase in the
prevalence of allergic rhinitis and conjunctivitis. There is evidence
that outdoor air pollution plays a role in the increasing morbidity
from allergic rhinitis and conjunctivitis. The disease seems to be
more common in urban than in rural areas (Broder et al., 1974a,b).
There is evidence that air pollutants may interact directly with
pollen with a possible impact on allergenicity (Behrendt et al.,
1992).
Beside exogenous factors, the association of allergic rhinitis
and conjunctivitis with other atopic diseases, such as atopic eczema
or asthma and a positive family history for atopy clearly demonstrates
the genetically determined susceptibility (Coca & Cooke, 1923).
4.3.4.1 Allergic rhinitis and conjunctivitis caused by contact with
chemicals
Allergic rhinitis and conjunctivitis caused by contact with
chemicals is less common than by contact with proteins. The prevalence
is unknown. The scope of the problem is probably underestimated
because of diagnostic failure (Mygind, 1986). The majority of cases
reported in the literature are in association with occupational
diseases. Upper respiratory tract hypersensitivity involving the nose
often coexists with asthma, conjunctivitis, bronchitis, and
occasionally with contact dermatitis, allergic alveolitis or fever.
Occupational chemicals may be haptens, allergens, mediator- releasing
or pharmacological agents and irritants. Eliciting agents that
sometimes are shown to induce an immediate-type IgE-mediated
hypersensitivity include anhydrides, metallic salts, dyes,
diisocyanates and antibiotics. In many, but not all, workers with
trimellitic acid-induced rhinitis and asthma, specific IgE antibodies
and positive skin tests can be found, suggesting Type I and Type III
allergic mechanisms (Bernstein et al., 1982a). In isocyanate workers
with rhinitis, conjunctivitis, asthma, bronchitis, chronic obstructive
lung disease, cutaneous reactions or fever, 26% had positive
skin-prick tests and in 14% specific IgE antibodies could be detected
after conjugation of isocyanates with serum albumin (Baur et al.,
1984). In the majority of cases with occupational rhinitis,
conjunctivitis and asthma caused by platinum salts, a Type I
hypersensitivity was proved by skin tests, in vitro histamine
release and passive cutaneous anaphylaxis (Schultze-Werninghaus et
al., 1978). In textile workers exposed to reactive dyes, who had
respiratory complaints, skin-prick tests and patch tests were positive
(Alanko et al., 1978; Estlander, 1988). It is thought that these small
molecule chemicals are haptens that combine with proteins to form
antigenic determinants.
Symptoms caused by chemicals may also be due to a contact-allergy
and delayed-type hypersensitivity. This applies more often for ocular
allergy. Rubbing of the eyes after handling detergents or other
chemicals may provoke a contact conjunctivitis. Positive patch tests
are found to chemicals such as antibiotics, thiomersal, benzalkonium
chloride, solutions for contact lenses, and metallic salts. In these
cases a Type IV hypersensitivity seems to be the primary allergic
mechanism.
However, allergies have to be differentiated from toxic and
irritative mechanisms. Strongly toxic chemicals may elicit symptoms by
directly damaging the mucosa after single contact. Milder irritants,
such as sulfur dioxide, urea formaldehyde, detergents, solvents or
dusts may cause hyperreactivity after repeated (cumulative) contact.
Exposure to cotton defoliants causes asthma, rhinitis and
conjunctivitis, which is thought to be a result of direct histamine
release. It is important to note that there is often an overlap
between allergic and irritative processes. Hyperreactivity to
irritants occurs predominantly after repeated contact in patients with
pre-existing atopic diseases, with or without an allergic basis.
Chemicals are often not only irritants but also allergens.
4.3.5 Pathophysiology
Allergens transported by the air come into contact with the
mucosal surface. Contact with mast cells or basophils leads to
IgE-dependent activation and degranulation of mast cells. Preformed
mediators stored in the granules (e.g., histamine, tryptase) are
released rapidly and elicit immediate symptoms. Other mediators are
eluted slowly (e.g., heparin) or are synthesized de novo (e.g.,
prostaglandins, leukotrienes) (Bachert et al., 1995). Afferent nerve
stimulation may provoke an axon reflex, and the release of
neuropeptides (substance P, tachykinins) may amplify this reaction
(Barnes et al., 1991). Mediators that are released slowly induce a
late- phase reaction after 6 to 12 h, which results in local
accumulation of inflammatory cells including CD4+ T-lymphocytes,
eosinophils, basophils and neutrophils (Dvoracek et al., 1984). These
cells and mast cells release cytokines and proteins (e.g., eosinophil
basic proteins) that perpetuate the reaction (Bachert et al., 1995).
Inflammatory cytokines (e.g., IL-4) may selectively recruit
eosinophils by increasing the expression of adhesion molecules on the
vascular endothelium (VCAM-1, ICAM-1).
The late-phase reaction results in an increased
hyper-responsiveness, which may be specific for an allergen
("priming") or nonspecific to a variety of irritant triggers (Connell,
1969).
4.3.6 Diagnostic techniques
Diagnostic techniques are applied for differential diagnosis and
verification of a definite diagnosis. The patient's history, physical
examination with rhinoscopy and allergy testing represent the basic,
readily accessible diagnostic techniques. Rhinomanometry with
assessment of nasal resistance and nonspecific provocation tests
demonstrating hyperreactivity of the nasal mucosa are also often used
for evaluation of clinical relevance (International Rhinitis
Management Working Group, 1994).
4.3.6.1 Medical history
A careful history of seasonal and/or perennial symptoms provoked
by specific exogenous factors is most important for the diagnosis of
allergic rhinitis and conjunctivitis. The conditions that precipitate
or aggravate symptoms should be asked for in detail. In particular,
the presence of allergens in the patient's environment and the
possible causal relationship to the symptoms should be evaluated.
Exposure factors, such as contact with air pollutants, automobile
exhaust emissions or detergents, a history of atopic diseases and the
family history provide further important information. The severity of
the disease may be estimated by the frequency, distribution and
severity of symptoms. Standardized questionnaires are useful in
obtaining detailed information.
4.3.6.2 Clinical examination
Special devices are usually unnecessary for examination of the
eyes, whereas rhinoscopy is obligatory for the examination of the
nose. The use of indirect laryngoscopy and full endoscopic
ear-nose-throat examination are not mandatory, but may be of value in
special patients (International Rhinitis Management Working Group,
1994). The nasal mucosa is usually reddened, oedematous and produces
large quantities of a clear mucous discharge. The periorbital tissues
may be oedematous. Cyanosis, conjunctival injection, increased
lacrimation and mucous discharge of the eyes are further symptoms. The
quality and quantity of the secretions should be noted.
4.3.6.3 Allergy testing
Immediate hypersensitivity skin tests (skin-prick test,
intracutaneous test) are the primary diagnostic tool, skin-prick tests
being the method of choice for the majority of cases (Dreborg, 1989;
Ring, 1991). Skin testing with commercially available aeroallergens
generally has a high reliability. The number of skin tests that should
be performed is confined to a few common environmental allergens
tested routinely but should be extended specifically if the individual
patient's history indicates a role of other allergens.
The determination of total serum IgE is of limited value for this
disease but tests for specific IgE antibodies (e.g., RAST) are useful.
Positive results of the skin-prick test and determination of specific
IgE antibodies (sensitizations) should always be evaluated in
combination with the patient's history. Nasal and conjunctival
challenges with commercially available allergens should be used
whenever the clinical relevance of a sensitization to an allergen
cannot otherwise be estimated. However, there is no universally
accepted standard for this technique. As all in vivo tests are
potentially dangerous, with the risk of anaphylaxis, tests should be
carried out only by personnel trained in cardio-pulmonary
resuscitation.
4.3.7 Therapeutic considerations
The therapeutic repertoire of antiallergic therapy includes
environmental control to minimize exposure to the allergen responsible
for provoking symptoms, symptomatic medications, and immunotherapy
under strict medical supervision (Druce, 1993).
4.4 Clinical aspects of allergic asthma caused by contact with
chemicals
4.4.1 Introduction
Asthma is by far the most frequently reported outcome of
an allergic respiratory reaction to inhaled chemicals, primarily
occurring as the consequence of exposures experienced at work, i.e.,
occupational asthma. Allergic rhinitis is generally caused by the same
agents and may occur in isolation or in association with asthma.
4.4.2 Importance of occupational asthma
The contribution of occupational causes to the prevalence of
asthma in the community is not generally known. Estimates in different
countries have varied between 2% and 15% but their basis is not
secure. In Spain, occupational causes accounted for between 1 in 15
and 1 in 20 of cases of asthma in young Spanish adults aged between 20
and 44 years. Information in the United Kingdom is limited to the
numbers awarded compensation and the number of cases reported to
voluntary surveillance schemes, both of which are likely to
underestimate the true frequency of the disease.
In the United Kingdom a surveillance scheme for work-related
diseases (SWORD) with voluntary reporting of new cases of occupational
lung disease by respiratory and occupational physicians reported 2101
new cases in 1989 of which 554 (26%) were asthma. The agents most
frequently reported to cause occupational asthma were isocyanates,
which accounted for 22% of cases, and grain, wood dusts and laboratory
animals, which together accounted for a further 17% of cases. The
annual incidence rate for occupational asthma in the working
population was estimated to be 22 per million. The highest rates in
the occupational groups occurred primarily among those encountering
chemicals at work, i.e., coach and spray painters, chemical
processors, plastics making and processing, metal making and treating,
and welders (Table 21).
The incidence reported in this survey is lower than that reported
in Finland by Meredith & Nordman (1996); Finland is one of the few
countries where occupational lung diseases are registered. The
incidence in 1981 of occupational asthma in Finland was estimated to
be 71 per million (compared to the rate in the United Kingdom of 22
per million). However, within the United Kingdom there was
considerable regional variation in reported rates, and the area of
highest incidence, West Midlands Metropolitan Area, had a rate of 63
per million, similar to the reported incidence in Finland. Meredith &
Nordman (1996) suggested that the differences in regional rates might
at least in part be due to differences in ascertainment and reporting,
and that the true incidence of occupational asthma in the United
Kingdom was three or more times that reported.
4.4.3 Chemical causes of occupational asthma
Many different chemicals encountered at work can stimulate a
hypersensitivity response and cause asthma. The more prevalent causes
are shown in Table 22.
4.4.3.1 Isocyanates
Diisocyanates are bifunctional molecules used commercially to
polymerize polyglycol and polyhydroxyl (polyols) compounds to form
polyurethanes. Because each diisocyanate molecule has two reactive
isocyanate (NCO) groups, they link adjacent polyols to form a
three-dimensional lattice. Isocyanates also react with water to evolve
carbon dioxide, a reaction exploited in the manufacture of flexible
polyurethane foam. The urethane reaction is exothermic and the heat
generated sufficient to evaporate diisocyanates with high vapour
pressures, such as toluene diisocyanate (TDI) and hexamethylene
diisocyanate (HDI). Diphenyl methane diisocyanate (MDI) and
naphthalene diisocyanate (NDI), whose vapour pressures are lower,
evaporate in significant amounts when heat is applied.
It is estimated that approximately 5% of workers regularly
exposed to TDI develop asthma, which may be manifested as immediate
and/or late onset responses. TDI can act as a direct irritant, can
stimulate nerve reflexes, and, in the minority of patients, elicit an
IgE antibody response and occasionally an IgG response (Baur &
Fruhmann 1981; Baur et al., 1994). In addition, persistent activation
of T-cells and continuous expression of pro-inflammatory cytokines
seems to maintain a state of chronic inflammation (Maestrelli et al.,
1995).
Polyurethanes have widespread applications, and exposure to
isocyanates occurs in many different occupations. These include the
manufacture of flexible and rigid polyurethane foam, the application
of two part polyurethane paints by brush and by spray painting, and in
flexible packaging production where isocyanates are used in inks and
as laminating adhesives.
Table 21. Incidence of occupational asthma in high-risk occupational
groups reported to the United Kingdom Surveillance of Work-Related
and Occupational Respiratory Disease Project (SWORD) in 1989
(Meredith, 1993)
Occupational Group Cases Population Incidence/106/year
Coach and spray painters 35 54 737 639
Chemical processors 31 73 189 424
Bakers 29 70 839 409
Plastics making and processing 27 66 005 409
Metal making and treating 14 56 270 249
Laboratory technician and 26 127 478 204
assistant
Welders/solderers electronic 35 220 068 159
assemblers
Working population 22
Table 22. Examples of occupational chemical respiratory allergens associated
with positive bronchial provocation challenges
(adapted from Karol et al., 1996)
Isocyanates Dyes
Diphenylamine-4,4'-diisocyanate (MDI) Brilliant orange GR
Hexamethylene diisocyanate (HDI) Carminic acid
Isophorone diisocyanate (IPDI) Reactive orange 3R
Naphthalene-1,5-diisocyanate Rifafix red BBN
Toluene 2,4-diisocyanate (2,4 TDI) Rifazol black GR
Toluene 2,6-diisocyanate (2,6 TDI)
Amines Acid anhydrides
Dimethyl ethanolamine Phthalic anhydride
Ethanolamine Tetrachlorophthalic anhydride
Ethylenediamine Trimellitic anhydride
Triethylenetetramine
Others
Abietic acid Glutaraldehyde
6-Aminopenicillanic acid Iso-nonanoyl sulfonate oxybenzene
7-Aminocephalosporanic acid Methyl-2-cyanoacrylate
Ampicillin alpha-Methyldopa
Azocarbonamide Phenylglycine acid chloride
Table 22 (continued)
Isocyanates Dyes
2-(n-Benzyl-N-tert-butylamino)4'-hydroxy Piperacillin
3'-hydroxymethylacetophenone diacetate Piperazine
Benzylpenicillin Plicatic acid
Cephalexin Spiramycin
Chlorhexidine Styrene
Complex platinum salts
Ethyl cyanoacrylate Tylosin
Natural rubber latex
Inhaled isocyanates have been reported to cause four different
respiratory reactions:
a) Toxic bronchitis and asthma caused by isocyanate inhalation
at toxic concentrations. Exposure to TDI at an atmospheric
concentration of 0.5 ppm (3.6 mg/m3) causes irritation of
mucosal surfaces - eyes, nose and throat (Henschler et al.,
1962). Persistent asthma and reactive airways dysfunction
syndrome (RADS) has been reported following a single inhalation
of TDI at toxic concentrations (Luo et al., 1990).
b) Bronchial asthma caused by sensitization to isocyanates.
c) Accelerated decline of forced expiratory volume in 1 second
(FEV1). The rate of decline of FEV1 in an isocyanate
manufacturing plant workforce was similar in non-smokers with
high cumulative exposures to toluene diisocyanate (TDI) to the
rate observed in smokers in both the high- and low-exposure
groups. The rate in non-smokers with low cumulative exposure was
not different from that expected for control non-smokers. No
additive effect of TDI with smoking was observed (Diem et al.,
1982).
d) Extrinsic allergic alveolitis, which has been reported
particularly in workers exposed to MDI (Zeiss et al., 1980) and
also to HDI (Malo et al., 1983).
Of the four reactions, bronchial asthma caused by
hypersensitivity to isocyanates has been the most frequently reported
and is the most important both in terms of prevalence and morbidity.
TDI and MDI have been the most widely used isocyanates and are the
major causes of asthma, although, with its increasing use in spray
paints, HDI is becoming a more prevalent cause. A study of workers
employed at a new TDI manufacturing plant identified 12 workers (4% of
the total workforce) who had developed asthma during a 5-year period,
with 9 developing it in the first year of employment. The average
exposure to TDI monitored by paper tapes was 0.002 ppm (14 µg/m3)
(Weill et al., 1981). Half of the cases had been exposed to spills;
six were maintenance workers, one was a laboratory worker and only
five were process workers. A cross-sectional study of a steel coating
plant, where TDI had been introduced into the process some years
before, identified 21 cases of asthma out of a total of 221, which was
probably an underestimate of the true number of cases (Venables et
al., 1985a).
Inhalation challenge tests with TDI have shown that asthmatic
responses may be provoked in sensitized workers by very low
atmospheric concentrations, as low as 0.001 ppm (7 µg/m3) (O'Brien et
al., 1979). Late asthmatic responses provoked by isocyanates are
associated with the development of an increase in nonspecific airway
responsiveness (Durham et al., 1987), and cells recovered from
bronchoalveolar lavage during a late asthmatic reaction provoked by
TDI have an increased proportion of neutrophils, identifying an
inflammatory response in the airways provoked by TDI (Fabbri et al.,
1987).
4.4.3.2 Acid anhydrides
Acid anhydrides are low relative molecular mass chemicals used
industrially as curing agents in the production of epoxy and alkyd
resins and in the manufacture of the plasticizer dioctyl phthalate.
Epoxy and alkyd resins have widespread applications as paints,
plastics and adhesives. Six acid anhydrides, i.e., phthalic anhydride
(PA) (Maccia et al., 1976), trimellitic anhydride (TMA) (Fawcett et
al., 1977; Zeiss et al., 1977), tetrachlorophthalic anhydride (TCPA)
(Howe et al., 1983), maleic anhydride (MA) (Durham et al., 1987;
Topping et al., 1986), hexahydrophthalic anhydride (Moller et al.,
1985) and himic anhydride (Bernstein et al., 1984), have been reported
to cause occupational asthma. Inhalation tests with the causal acid
anhydride provoked asthmatic responses, and specific IgE or IgG
antibodies, or both, to the specific anhydride conjugated to human
serum albumin were identified in the sera of the great majority of
cases, although this was less frequent with maleic than with the other
anhydrides. Zeiss et al. (1977) suggested that four separate clinical
syndromes were caused by TMA, for which they proposed separate
immunological mechanisms: i) toxic airway irritation; ii) immediate
IgE-mediated rhinitis and asthma; iii) IgG-mediated late asthma with
systemic symptoms ("TMA flu"); iv) pulmonary haemorrhage-haemolytic
anaemia syndrome as the outcome of antibody binding to circulating red
blood cells and to pulmonary vascular cells. The distinction between
"immediate" and "late" asthmatic reactions with influenza-like
symptoms and their different pattern of immunological response has not
been consistently observed by other investigators. It seems more
likely that asthma caused by acid anhydrides, including TMA, may be
associated with specific IgE or IgG, or both, although specific IgE
and IgG4 seem to be more associated with asthma and IgG with exposure
(Forster et al., 1988). A relationship between HLA DR3 and the
development of specific IgE to TMA and possibly tetrachlorophthalic
anhydride (TCPA), but not phthalic anhydride (PA), has been reported
(Young et al., 1995). The pulmonary haemorrhage haemolytic anaemia
syndrome is real but rare. It has been reported in individuals exposed
to hot trimellitic anhydride fume and may be the outcome of a toxic
reaction to inhalation of trimellitic anhydride at very high
concentration rather than a hypersensitivity reaction.
4.4.3.3 Complex platinum salts
The complex platinum salt ammonium hexachloroplatinate is an
essential intermediate in the refining of platinum, a corrosion
resistant metal used as a catalyst and in jewellery. Allergy to
platinum salts in refinery workers was first reported in 1945 (Hunter
et al., 1945). Subsequently, inhalation of ammonium
hexachloroplatinate was shown to provoke asthmatic responses and to
elicit immediate skin test responses in sensitized individuals (Pepys
et al., 1972).
The incidence of occupational allergy in the platinum refining
industry was high in United Kingdom in the mid 1970s. In a cohort
study of 91 workers who entered employment in a platinum refinery in
the two years 1973 and 1974 (Venables et al., 1989), 22 developed
respiratory symptoms and an immediate skin test response to ammonium
hexachloroplatinate. The risk was greatest in the first year of
employment and smoking was more important than atopy as a predictor of
developing a positive skin test reaction.
4.4.4 Diagnosis of occupational asthma
Accurate and early diagnosis of cases of occupational asthma is
important. Remission of respiratory symptoms and restoration of normal
lung function, including nonspecific airway responsiveness, can follow
avoidance of exposure to the specific initiating cause. Furthermore,
chronic asthma is more likely to develop in those who remain exposed
to the initiating cause after the onset of symptoms. However,
avoidance of exposure frequently requires a change of employment.
Accurate diagnosis is also essential if those whose asthma is not
occupationally caused are to avoid being advised unnecessarily to
change or leave their work.
The diagnosis of occupational asthma requires:
a) differentiation of asthma from other causes of respiratory
symptoms, in particular chronic airflow limitation and
hyperventilation;
b) differentiation of occupational cause from non-occupational
asthma;
c) differentiation of asthma initiated by an agent inhaled at
work from pre-existing or incidental asthma exacerbated by
nonspecific irritants, such as sulfur dioxide and cold air,
inhaled at work.
The diagnosis of occupational asthma is usually suggested by the
history. It commonly occurs in an individual exposed at work to an
agent recognised to cause occupational asthma and only develops after
an initial symptom-free period when the patient has been exposed
without symptoms to concentrations in air that now provoke asthma.
Respiratory symptoms occur during the working week, may increase in
severity as the week progresses, and improve during absences from
work, at weekends or during holidays. The patient may also be aware of
others who have developed similar respiratory symptoms at the place of
work.
Nonspecific stimuli provoke asthmatic reactions that usually
occur within minutes of exposure to them and resolve within 1-2 h of
avoidance of exposure. Where work-related respiratory symptoms are due
to the provocation of asthma by a respiratory irritant encountered at
work, the onset of asthma will often have preceded initial exposure to
the irritant and the severity of asthma does not significantly improve
when away from work. Nonspecific irritants such as organic solvents,
which often have a characteristic and unpleasant smell, may also
provoke a hyperventilation response, when difficulty with breathing is
associated with symptoms that are consequences of a low pCO2, such as
tingling of the fingers, headaches and dizziness.
4.4.4.1 Investigation of causes of occupational asthma
In the majority of cases a confident diagnosis of occupational
asthma induced by an inhaled chemical can be made from knowledge of
exposure at work to a recognised cause of occupational asthma and a
characteristic history. Where possible, these should be supported by
objective evidence from serial measurements of peak expiratory flow
(PEF) or immunological tests or both. Inhalation testing is reserved
for occasions when the results of these investigations do not provide
an adequate basis for advice about future employment.
4.4.4.2 Serial peak expiratory flow (PEF) rate measurements
Asthma can be attributed with confidence to an agent inhaled at
work where exposure to it in the work place reproducibly provokes
airway narrowing. Repeated measurements of airway calibre, most
conveniently made as PEF rates, need to be made during a period long
enough to allow observation of the consistency of any changes and
their relationship to periods at work. Measurements need to be made
repeatedly during each day for a period of several weeks and the
patient can take measurements and record the results. Self-recording
of PEF measurements is now widely used. Patients are lent a peak flow
meter and asked to record the best of 3 measurements of PEF made every
2 h from waking to sleeping over a period of one month in the first
instance. To allow sufficient time for lung function to recover from
exposure to an agent inhaled at work, it is helpful if the month
includes a period away from work which is longer than a weekend,
ideally a one or two week holiday. Self-recording requires patient
compliance and honesty. The measurements may be conveniently
summarized to show the maximum, minimum and mean peak flow
measurements for each day and differences between periods at and away
from work observed. This method of patient investigation has proved,
in the hands of those experienced in its use, to be reliable and a
relatively sensitive and specific index of occupational asthma.
4.4.4.3 Immunological investigations
The demonstration of specific IgE antibody is a helpful, but not
conclusive, criterion to establish the diagnosis of occupational
asthma or rhinitis, because the presence of specific IgE antibody is
not unique to clinically allergic individuals. Antigen-specific IgE is
rare (5-15% of clinical cases). The presence of specific IgE antibody
can be detected by in vivo tests such as skin-prick test and in
vitro tests such as radioallargosorbent test (RAST) or enzyme-linked
immunosorbent assay (ELISA). Other in vitro tests, such as histamine
release from basophils, are less standardised. When allergens are not
available, histamine release from basophils can be an alternative to
direct measurement of IgE.
The application of immunological tests in the investigation of
occupational asthma caused by inhaled chemicals has widened with the
preparation of hapten protein conjugates suitable for immunological
testing (e.g., acid anhydride and reactive dye-human serum albumin
conjugates) and the development of reliable methods for identification
of specific IgE antibody in serum. Complex platinum salts such as
ammonium hexochloroplatinate can elicit immediate skin-prick test
responses without the need for conjugation to human serum albumin.
The value of such tests in the diagnosis of occupational asthma
depends upon their sensitivity and specificity in populations exposed
to the particular cause. An immediate skin-prick test response and
specific IgE antibody identified by RAST to conjugates of the acid
anhydride tetrachlorophthalic anhydride (TCPA) with human serum
albumin (Howe et al., 1983) were found to be associated with cases of
asthma in exposed populations and not simply a reflection of exposure.
4.4.4.4 Inhalation challenge tests
Inhalation tests are rarely undertaken because they are
potentially hazardous. Inhalation tests with occupational agents
should only be undertaken by those experienced in conducting them and
who have adequate hospital facilities for continuous monitoring of
patients for 24 h after each test.
There are 4 major indications for inhalation challenge testing in
the diagnosis of occupational asthma:
a) Where the agent thought to be responsible for causing asthma
has not previously been reliably shown to do so.
b) Where an individual with occupational asthma is exposed at
work to more than one potential cause, and his future employment
depends on knowledge of which one is responsible.
c) Where asthma is of such severity that further uncontrolled
exposure in the work environment is not justifiable.
d) Where the diagnosis of occupational asthma remains in doubt
after other appropriate investigations, including serial peak
expiratory flow rate (PEF) and immunological tests, have been
completed.
The aim in an occupational-type inhalation challenge test is to
expose the individual under single blind conditions to the putative
cause of the asthma in circumstances which resemble as closely as
possible the conditions of the exposure at work. Wherever possible,
atmospheric concentrations of the inhaled agent should be based on
knowledge of the concentrations experienced at work, and the physical
conditions of exposure, (e.g., size of dust particles, whether vapour
or aerosol, and temperatures to which the materials are heated),
should be similar to those encountered at work.
Measurements of airway responses provoked by inhalation challenge
tests should ideally include measurements of both changes in airway
calibre and in nonspecific airway responsiveness. Changes in airway
calibre are most conveniently measured by regular measurements of
forced expiratory volume in 1 second (FEV1) and forced vital capacity
(FVC) or PEF rate before and at regular intervals after the test, for
at least 24 h. Changes in airway responsiveness can be made by
estimating the concentration of inhaled histamine or metacholine that
provokes a 20% fall in FEV1 (PC20) before the test and at 3 and 24 h
after the test. The changes in airway calibre and nonspecific
responsiveness observed are compared to those following a control
challenge test, each test being made on a separate day.
The patterns of change in airway calibre provoked by inhalation
testing are distinguished by their time of onset and duration.
Immediate responses occur within minutes and resolve spontaneously
within 1-2 h. Such reactions can be provoked by both allergic (e.g.,
grass pollen) and non-allergic (e.g., inhaled histamine or sulfur
dioxide) stimuli. The response depends upon the concentration of the
provoking agent and the degree of pre-existing nonspecific airway
responsiveness. Lone immediate responses (i.e., an immediate response
not followed by a late response) are not usually associated with an
increase in nonspecific airway responsiveness. Late responses develop
one or more hours after the inhalation test exposure, usually after
some 3-4 h, and may persist for 24-36 h. Unlike the immediate
response, late responses are often associated with an increase in
nonspecific responsiveness which can be identified 3 h post test prior
to the onset of the late asthmatic response and less reliably at 24 h
after the test (Durham et al., 1987).
A dual response is an immediate response followed by a late
response. Recurrent nocturnal responses may be provoked by a single
inhalation test exposure with asthmatic responses occurring during
several successive nights with partial or complete remission during
the intervening days. Such responses are almost certainly a
manifestation of a provoked increase in nonspecific airway
responsiveness. The question to be answered from the results of an
inhalation test is whether or not in the individual case the
particular agent inhaled at work has induced asthma. The most reliable
means of answering this question is to determine whether or not
inhalation of the specific agent at concentrations to which exposure
occurs at work reproducibly provokes a non-immediate asthmatic
response and increases nonspecific airway responsiveness. In such
cases the specific agent can be considered to be the inducing cause in
that particular individual. Nonspecific irritants may provoke
immediate responses in individuals with hyper-responsive airways, but
do not provoke either an increase in nonspecific airway responsiveness
or a late asthmatic reaction. Agents that induce specific IgE
antibody, however, may provoke lone asthmatic responses; in these
cases inferences from the inhalation test result should take the
immunological test into account.
4.4.5 Outcome of occupational asthma
Asthma induced by an agent inhaled at work may become chronic,
persisting for several years, if not indefinitely, after avoidance of
exposure to its initiating cause. This seems particularly, although
not exclusively, to occur with asthma caused by low relative molecular
mass chemicals. Asthma caused by isocyanates and acid anhydrides has
been reported to have persisted in over half of cases. Six cases of
asthma caused by the acid anhydride tetrachlorophthalic anhydride
(TCPA) were followed up 4 years after avoidance of exposure: all had
chronic respiratory symptoms consistent with persistent airway
hyper-responsiveness and a measurable histamine PC20 was present in
the five in whom it was assessed. The rate of decline of specific IgE
to a TCPA-human serum albumin conjugate during the period of avoidance
of exposure was parallel in all 6 subjects and exponential with a
half-time of one year, making it very improbable their continuing
asthma was caused by further, albeit inadvertent, exposure (Venables
et al., 1987).
Chronic asthma in these cases is likely to be a manifestation of
persistent airway inflammation which, although initiated by the agent
inhaled at work, persists in its absence. Ten patients with
TDI-induced asthma, who had continuing respiratory symptoms and airway
hyper-responsiveness, were investigated for 4-40 months from their
last exposure. Bronchial biopsies obtained from eight patients showed
basement membrane thickening with infiltration of the mucosa by
eosinophils, lymphocytes and neutrophils. In four patients in whom
airway responsiveness had not improved, the proportion of eosinophils
in fluid recovered by BAL was increased, whereas this was the case in
only one of five patients whose airway responsiveness had improved
(Paggiaro et al., 1990).
4.4.6 Management and prevention of occupational asthma
Reduction in the incidence of occupational asthma will follow
adequate control of exposure to its causes (Table 22). Substitution of
a different paint for one containing TDI halted an epidemic of asthma
in a steel coating plant (Venables et al., 1985a). Measures to secure
control of exposure to the majority of causes of occupational asthma
have, however, been impeded by lack of knowledge of the nature of
exposure-response relationships for sensitizing chemicals.
The development of control measures that will significantly
reduce the incidence of occupational asthma requires examination of
exposure-response relationships and the effects of interventions in
longitudinal studies.
Management advice for patients with occupational asthma has been
greatly influenced by the results of studies of outcome of
occupational asthma which have found evidence of continuing asthma and
airway hyper-responsiveness despite many years avoidance of exposure
to the initiating cause, and particularly studies, such as those of
azodicarbonamide workers, that have identified a relationship between
the duration of symptomatic exposure and the risk of chronic asthma.
The importance of accurate identification of the specific cause cannot
be over-emphasised. Avoidance of exposure often involves relocation or
change of employment. Misdiagnosis of occupational asthma can be as
hazardous for individual patients as missing the diagnosis because of
the implications for employment.
Rigorous prevention of exposure is a key to the prevention of
occupational asthma. Patients who develop occupational asthma in whom
a specific cause is identified should be advised to avoid further
exposure to the cause of their asthma. This seems particularly
important where low relative molecular mass chemicals, such as
isocyanates, plicatic acid or acid anhydrides, are the cause as these
seem particularly, although not exclusively, associated with the
development of chronic asthma and airway hyper-responsiveness.
However, environmental changes, unless they involve substitution,
are often not able to reduce exposures sufficiently to prevent
continuing airway responses in sensitized individuals. Venables &
Newman Taylor (1990) examined the relationship between the
concentration of TCPA in air and the provocation of asthmatic
responses in inhalation challenge tests in 4 sensitized individuals.
They observed a log-linear relationship between the magnitude of late
asthmatic responses and TCPA concentration, which passed through the
origin, suggesting no threshold.
When an individual remains in employment exposed to the cause of
the asthma, either directly or indirectly, the effectiveness of
relocation or of respiratory protection needs to be monitored. This
can be conveniently done by serial self recordings of peak flow to
determine whether or not asthma persists and, if so, whether or not it
is work related.
4.5 Food allergy
4.5.1 Definitions
Food allergy is an adverse reaction to food occurring in
susceptible individuals, which is mediated by a classical immune
mechanism specific for the food in question. Therefore, the "true"
allergic reaction to a food is caused by an over-sensitive reaction of
the body's immune system (in essence, "immunity gone wrong"). Food
intolerances are all non-immune-mediated adverse reactions to food.
The subgroups of food intolerance are enzymatic (resulting from an
enzymatic defect, e.g., lactase deficiency), pharmacological
(depending on the direct effect of certain substances found in foods,
e.g., caffeine) and undefined food intolerance (Bruijnzeel-Koomen et
al., 1995) (Fig. 14). Food intolerances will not be described further
since, as far as is known, they do not involve immune mechanisms.
The foods of animal origin that most commonly cause allergic
reactions are milk, eggs, cod fish and shrimps. Those of plant origin
that most often cause allergic reactions are peanuts, soybeans,
celery, apple, hazelnuts and wheat.
In food allergy the best established mechanism is the presence of
IgE antibodies against the offending food (Type I allergy). Other
immune mechanisms may be involved in other clinical patterns of food
allergy. For example, Type III and Type IV mechanisms may play a role
in the genesis of food protein-induced enterocolitis of newborns and
infants.
A subgroup of patients with contact allergy (T-cell mediated) to
nickel or balsam of Peru can develop cutaneous symptoms (haematogenous
contact eczema) to double-blind, placebo-controlled food challenge
(DBPCFC) (Veien et al., 1987; Menné & Maibach, 1991). Foods with very
high amounts of nickel or containing natural flavours, cross-reacting
with balsam of Peru, may provoke skin symptoms.
In the following text only adverse food reactions with a proven
involvement of the immune system are covered.
4.5.2 IgE-mediated food allergy
Clinical manifestations of IgE-mediated food allergy can remain
localized at the site of the primary direct contact, i.e., the mouth
and throat (oral allergy syndrome) or the gastrointestinal tract
(isolated gastrointestinal food allergy). However, after ingestion
of the specific food, food-allergic patients often exhibit symptoms in
various organs: the skin, the respiratory tract, the gastrointestinal
tract and the cardiovascular system (systemic anaphylaxis) are the
most frequently affected. Usually, the patients show involvement of
two or more organ systems. Among 402 patients with a systemic
IgE-mediated allergy to one or more specific foods -- the oral allergy
syndrome was not included -- diagnosed over a 10-year period at the
Allergy Unit in the Zurich University Hospital, the affected organ was
most often the skin (46%), followed by the respiratory tract (25%),
the gastrointestinal tract (20%) and the cardiovascular system
(10%)(Wüthrich, 1993). Twenty percent of the food allergic patients
had skin symptoms exclusively, 11% had isolated gastrointestinal
manifestations and 8% isolated respiratory symptoms. In only 7% of all
cases was food allergy responsible for a chronic condition such as
chronic urticaria, perennial asthma or gastroenterocolitis.
4.5.2.1 Oral allergy syndrome
Mainly patients with pollinosis may describe, spontaneously or by
exact questioning, itching of the lips, mouth, palate and throat
immediately after intake of some fresh foods, namely fruits and
vegetables. Hoarseness and/or swelling of the lips, tongue, uvula and
larynx can occur infrequently. Oral allergy syndrome must be carefully
differentiated from the beginning of a generalized anaphylactic
reaction in which itching of the mouth and throat may be the first
symptoms. These symptoms were described in the USA in
ragweed-sensitive patients (6.2% in one series) after ingestion of
melon and banana (Anderson et al., 1970; Ross et al., 1991).
Cross-reactivity of pollen-sensitized subjects to various food
allergens can occur (Fig. 15). Most patients allergic to birch pollen
react to apples, hazelnuts and numerous other vegetables and fruits
from the Rosaceae family such as cherries, peaches, pears and almonds
(Eriksson et al., 1982; Dreborg & Foucard, 1983; Pauli et al., 1992) ;
mugwort pollen sensitive patients may react to celery root (celeriac)
and spices, the so called mugwort-celery-spices-syndrome (Wüthrich &
Hofer, 1984; Wüthrich et al., 1990). Grass pollen allergic patients
may react to cereals or tomatoes (de Martino et al., 1988). It has
been shown by prick, RAST studies and RAST inhibition experiments that
a celery thermolabile allergen seems to be involved in
celery-birch-pollen allergic patients whereas a thermostable allergen
is involved in celery-mugwort-allergic patients (Wüthrich et al.,
1990). Patients with chestnut and banana oral allergy syndrome may be
sensitized to natural rubber latex (M'Raihi et al., 1991). Many
patients are able to identify the offending fruit or vegetable.
5 to 6 years suffered from atopic eczema (Schäfer et al., 1994), and
in a study comparing eastern and western German areas in 1991 atopic
eczema was diagnosed in 12.9% of 1086 pre-school children (Ring et
al., 1995; Krämer et al., 1996; Schäfer & Ring, 1997). In studies in
the United Kingdom, Denmark and Switzerland, the same methodological
analyses were applied for a longer time interval to obtain figures on
the changes in frequency of atopic eczema. These studies showed a
dramatic increase in the prevalence of atopic eczema (Schäfer & Ring,
1995). In the United Kingdom the figures for the prevalence of atopic
eczema were 5.1% in 1946, 5.3% in 1964, 7.3% in 1958, and around 12%
for 1970-1989. Similarly, in Denmark the prevalence in 1964-1969 was
3.2% compared with 11.2% for 1970-1979. In Switzerland there was an
increase from 2.2% in 1968 to 2.8% in 1981.
4.2.3 Clinical manifestations and diagnostic criteria
4.2.3.1 Age-dependent clinical manifestations
In most patients with atopic eczema, the disease begins in
infancy between 3 and 12 months of age (Hill & Sulzberger, 1935) as an
erythematous, squamous or papulo-vesiculous inflammation, which may
worsen to the point of exudation. It is often found on the face, the
extremities (especially extensor aspects) and finally the trunk.
Oozing and crusted lesions can often be found on the scalp (cradle
cap). More and more, itching becomes an essential feature; the infant
may be irritable, restless and tries to scratch the affected areas
(after 3rd month of life). The course is chronically persistent or
relapsing. Later, between 2 and 5 years, the appearance of the lesions
changes. They become nummular and infiltrated. The localization
changes and affects flexures of popliteal and antecubital fossae, the
nape of the neck and the backs of the hands and feet. In severe cases
there may be an involvement of the entire skin surface. Dry skin
becomes another characteristic feature especially in the adult phase
and creates itching followed by scratching. This may lead to severe
excoriation with nodule formation and perpetuation of the inflammatory
reaction ("Prurigo Besnier"). Chronic inflammation produces thickening
(lichenification) of the skin, especially in flexural regions.
4.2.3.2 Diagnosis of atopic eczema
Many diagnostic systems have tried to collect reliable criteria
for this disease. The features listed by Hanifin & Rajka (1980) are
those referred to most often in the literature. A combination of a
number of major and minor criteria allows the establishment of the
diagnosis. A more simple selection of criteria for practical purposes
has been proposed (Ring, 1991). Williams et al. (1994a) proposed a new
arrangement of diagnostic criteria, primarily for epidemiological
studies. However, it must be kept in mind that all these diagnostic
systems have their drawbacks in this heterogenous disease. Clinical
criteria, as well as the patient's history and presence of
IgE-mediated sensitizations must be considered together and are the
mainstay for establishing the diagnosis. However, minimal forms exist
and sometimes do not meet the required criteria. Papular or nodular
variants as well as localized forms (e.g., exfoliating cheilitis,
infra-auricular rhagades, nipple eczema, finger pad or toe eczema)
constitute minimal expressions of this disease (Wüthrich, 1991).
Typical eczematous lesions may not only be triggered by IgE-mediated
allergic reactions in patients with a positive family history of
atopy, but can also be triggered by food additives. In most patients,
establishing the diagnosis is not too difficult. In selected cases,
clinical findings, history and IgE-mediated sensitization have to be
regarded critically and all important differential diagnoses have to
be ruled out thoroughly.
4.2.3.3 Stigmata of the atopic constitution
The diagnosis of atopic eczema often depends on further
additional features. Stigmata of atopic constitution are prevalent in
many patients with atopic eczema, although they are not specific for
this disease. Dry skin, hyperlinearity of palms and soles,
intraorbital fold, white dermographism, facial pallor, orbital
darkening, low hairline and thinning of the lateral portions of the
eyebrow are found more often in this group of patients (Przybilla,
1991). They are typical constitutional markers, which may add another
clue in establishing the diagnosis (Ring, 1988).
4.2.3.4 Prognosis
Variability and chronic relapses are characteristics of the
course of atopic eczema. Atopic eczema most frequently begins during
infancy (Hanifin, 1983; Rajka, 1990). In about two-thirds of infants
with atopic eczema, the disease clears during childhood. In the
remaining patients it persists into adult life. Minimal forms and
stigmata of the disease often remain throughout life (Vickers, 1991).
Sometimes atopic eczema starts only in adulthood. A definite prognosis
about the course of an individual patient cannot be made; there is
controversy about prognostic factors (Vickers, 1991).
4.2.4 Etiology
The manifestation of atopic eczema is subject to a multifactorial
genetic predisposition as well as to environmental provocation
factors.
4.2.4.1 Genetic influence
There is no doubt about the existence of a genetic component
favouring the manifestation of atopic eczema (Schnyder, 1960; Küster
et al., 1990). Twin studies show a concordance in homozygous twins of
83 and 86%, compared to 28 and 21% in heterozygous twins (Niermann,
1964; Schultz-Larsen, 1991). The chance of developing atopic eczema
depends on the family history of atopy. Whereas about 10-15% of
children without a family history of atopy develop atopic eczema, with
a positive history of one parent the risk rises to 25-30% and, with a
positive history of both parents, to 50-75% (Schultz-Larsen et al.,
1986; Björksten & Kjellman, 1987).
4.2.5 Environmental provocation factors
The activity of atopic eczema can be influenced by a large number
of environmental provocation factors (Table 20). These can either act
specifically in the sense of individual hypersensitivity, primarily
IgE-mediated allergy or, more often, as unspecific provocation factors
irritating the skin or affecting emotional status. The question of the
possible involvement of environmental atmospheric pollution in the
increase in the prevalence of atopic eczema remains controversial (see
section 3.3.2).
4.2.6 Pathophysiology
Although knowledge concerning components of the immune system and
inflammatory responses in patients with atopic eczema has increased
widely in recent decades, the pathophysiology of atopic eczema also
remains controversial (Marchionini, 1960; Rajka, 1990, 1996).
4.2.6.1 Dry skin
Dry or rough skin is a major feature of skin alteration in
patients with atopic eczema. Although a number of studies have
investigated the pathophysiology of dry skin, there is no consensus
(Melnik & Plewig, 1991; Lindskov & Holmer, 1992). An attractive
hypothesis is that even clinically non-inflamed "dry skin" shows
histologically a mild inflammatory infiltrate, and this is supported
by skin biopsies in atopic patients (Uehara, 1991). There seems to be
an intimate relation between dry skin, irritability and itch (Rajka,
1990; Ruzicka et al., 1991).
4.2.6.2 Autonomic dysregulation
In addition to immunological abnormalities, signs of
dysregulation of the autonomic nervous system have been described
(Szentivanyi, 1968; Ring et al., 1988; Ring & Thomas, 1989; Hanifin,
1993). Elevated phosphodiesterase activity in mononuclear leukocytes
seems to correlate with increased IgE production and vasoactive
mediator secretion (Butler et al., 1983; Cooper et al., 1985).
4.2.6.3 Cellular immunodeficiency
First described by Kaposi in 1895, patients with atopic eczema
are more susceptible to infection with viruses (e.g., Herpes
simplex, Human papilloma) and bacteria (especially Staphylococcus
aureus) (Kaposi, 1895). Earlier reports about decreased frequencies
of allergic contact sensitization in atopic eczema are contradicted by
Table 20. Important environmental provocation factors in atopic eczema
(adapted from Ring et al., 1996)
Unspecific provocation factors:
Irritants
Microbial skin colonization or infection
e.g., Staphylococcus aureus
Pityrosporum ovale
Herpes simplex (Eczema herpeticum)
Psychological stress, emotional factors
Specific provocation factors (individual hypersensitivity):
IgE-mediated allergy
e.g., Food
House dust mite
Animal dander
Pollen
Microbial colonisation?
Contact allergy
Pseudo-allergy (idiosyncrasy) and intolerance
e.g., preservatives in foods
citrus fruits
others who claim that the tendency to develop contact allergy is
increased (Rajka, 1990). However, Enders et al. (1988) reported that
the prevalence of positive patch test reactions for contact allergy in
patients with atopic eczema was almost equal to that of patients with
allergic contact dermatitis.
4.2.6.4 Increased IgE production
Serum IgE levels are elevated in the majority of patients with
atopic eczema (Ogawa et al., 1971). They tend to correlate with the
extent and severity of the disease (Johansson & Juhlin, 1970;
Wüthrich, 1975). Specific antibodies can be measured against common
environmental allergens (Rajka, 1990; Ruzicka et al., 1991). Although
often the clinical significance of these antibodies is lacking, in
some patients eczematous skin responses can be provoked by
aeroallergens (grass pollen, house dust mite or animal dander), a
procedure that has been called "atopy patch test" (Reitamo et al.,
1986; Adinoff et al., 1988; Ramb-Lindhauer et al., 1990; Ring et al.,
1991a,b; Platts-Mills et al. 1991; Vieluf et al., 1993). IgE
antibodies to foods are frequently found in patients and may induce
urticaria as well as eczematous reactions. Well-controlled clinical
trials showed that in a high number of patients with atopic eczema,
skin lesions were exacerbated after specific oral provocation with
certain foods in double-blind studies (Sampson & Albergo, 1984). Apart
from aeroallergens and foods, microbial allergens ( Staphylococcus
aureus, Pityrosporum ovale) might play a role. Chronic colonization
of atopic skin could provide a continuing cause of allergen
stimulation (Leyden et al., 1974; Ring et al., 1992, 1995; Neuber et
al., 1995; Kröger et al., 1995). After allergen stimulation of
IgE-bearing mast cells or basophils, the released vasoactive mediators
(such as histamine, eicanosoids, etc.) might induce itching, and also
eczema via a late-phase reaction (Dorsch & Ring, 1981). Langerhans
cells in the epidermis express high affinity receptors for IgE as well
as CD23 and IgE binding-protein (Bieber & Ring, 1992). Allergen
contact might result in the generation of Th2-helper cells, a subset
producing IL-4 and IL-5, thereby maintaining the allergic
inflammation. Also other cell types might be involved in the
inflammatory process; lymphocytes might act directly through
cytokines, and eosinophils through release of pro-inflammatory
mediators (Jakob et al., 1991; Kapp, 1995).
4.2.6.5 Psychosomatic aspects
It is well known from clinical experience that psychological and
emotional factors can greatly influence the clinical course of this
skin disease (Borelli & Schnyder, 1962; Jordan & Whitlock, 1972, 1974;
Ring et al., 1986; Cotterill, 1991). There is no convincing evidence
that psychological factors per se are the primary cause for atopic
eczema; however, it is clear that psychological factors may influence
existing eczematous lesions or even trigger new exacerbations of
eczema in many patients (Rajka, 1990; Ruzicka et al., 1991). For
children, the family situation, e.g., the interaction between parents
and the affected child, seems to be of particular importance (Ring et
al., 1976; Niebel, 1995).
4.2.7 Diagnostic approach
In atopic eczema diagnosis not only comprises the identification
of the disease but should also focus on individual provoking factors
able to trigger disease activity (Ring et al., 1991a,b; Morren et al.,
1994). Each patient may be susceptible to an individual set of
provocation factors. Often, exacerbations can be prevented or the skin
condition can be directly improved by avoidance of these factors (Ring
et al., 1996). Diagnostic procedures used are intended to reveal
provocation factors for the individual patient. Specific provocation
of atopic eczema often is the result of an individual
hypersensitivity. Although diagnostic tests normally differ from the
natural exposure with allergens, they provide useful information in
the hands of a trained allergist (Ring, 1988). Allergy diagnosis is
based on the four foundations: the patient's history, skin tests,
in vitro (laboratory) tests and provocation tests.
4.2.7.1 Medical history
The patient's history forms the backbone of allergy diagnosis.
Often the patient notices associations between disease activity and
specific conditions or actions (e.g., intake of foods, seasonal or
daily variations, contact with animals, heavy pollen emission). These
observations are very valuable in revealing individual provocation
factors. On the other hand, positive allergy tests must always be
verified for their clinical significance for the patient's disease by
comparing them with the history.
4.2.7.2 Skin tests
Skin test methods are divided into percutaneous (skin-prick,
intradermal) tests and epicutaneous (patch) tests (American Medical
Association, 1987a). Percutaneous tests search for immediate-type
IgE-mediated hypersensitivity and are especially indicated in atopic
eczema. The skin-prick test (prick puncture test) has gained the
widest acceptance because of its high convenience and safety (Dreborg,
1989). A drop of the test extract is placed on the volar surface of
the forearm and the solution is introduced into the epidermis with a
disposable hypodermic needle. After 15 min the reactions are graded in
relation to the erythema and wheal that are induced. In intradermal
testing 0.02 to 0.05 ml of the test extract is injected intradermally
with a syringe. Scratch tests (applying the extract to a superficial
scratch) and rub tests (rubbing of the skin with native allergen) are
other variants applied only for special indications. Because of the
danger of producing anaphylactic reactions these tests should be
performed only by trained allergists with experience in emergency
treatment. In patients with atopic dermatitis, percutaneous tests are
widely used for the detection of hypersensitivity against
environmental aeroallergens and foods (Ring, 1988).
Epicutaneous tests primarily focus on the detection of contact
allergy by cell-mediated immunity. The extract is put in an aluminum
chamber and fixed onto the skin of the patient for 48 h. The test
reaction is graded after 48 and 72 h. An eczematous response is
regarded as positive. Since it has been shown that in patients with
atopic eczema, eczematous skin responses can be elicited by epidermal
application with aeroallergens (especially the house dust mite),
epicutaneous testing with the atopy-patch test is gaining wide
acceptance (Adinoff et al., 1988; Vieluf et al., 1990; Darsow et al.,
1995). Although the definite mechanism is still unknown, this test
might fill the gap between IgE-mediated hypersensitivity and an
eczematous response.
4.2.7.3 Laboratory tests
In the serum of patients with atopic eczema, hypersensitivity can
be detected by laboratory methods. In atopic eczema the most important
hypersensitivity reactions are thought to be IgE-mediated. IgE
antibodies can be determined by binding to an allergen in a solid
phase and radioactive, enzymatic or fluorometric labelling (Ring,
1988). Specific antibodies against environmental allergens are
detected by the RAST (Radio-Allergo-Sorbent Test) and expressed
semi-quantitatively in different classes. Positive reactions must be
interpreted with regard to their clinical relevance (Pastorello et
al., 1989).
4.2.7.4 Provocation tests
Oral provocation tests and elimination diets are often necessary
for the evaluation of the clinical relevance of a suspected food
hypersensitivity (Przybilla & Ring, 1990). Also, allergy-like symptoms
to food additives, medications, etc., may be produced by
non-IgE-mediated mechanisms ("pseudo-allergy") (Vieluf et al., 1990).
In these cases elimination diets and provocation tests are performed.
Foods unlikely to produce adverse reactions can be screened by
elimination diets or open challenges. Oral provocation by double-blind
placebo-controlled food challenges is regarded as the "gold" standard
for the diagnosis of food allergies (Sampson, 1983; Bruijnzeel-Koomen
et al., 1995). However, there are pitfalls and problems with this
procedure (Bindslev-Jensen, 1994a).
4.2.8 Therapeutic considerations
The disease can be effectively controlled by a combination of
avoidance procedures, basic dermatological therapy and
anti-inflammatory therapy for exacerbations (Przybilla et al., 1994;
Ring et al., 1996). However, the patient has to accept that there is
no simple therapy allowing permanent cure. The integration and active
cooperation of the patient in the therapeutic concept ("patient
management") is a prerequisite for an effective therapy. In atopic
eczema, diagnostic and therapeutic approaches are intimately
connected.
4.2.8.1 Avoidance of provocation factors
During the first year of life food allergies are frequent. Later,
sensitization to aeroallergens becomes more important (Guillet &
Guillet, 1992). Food allergies were found in 63% of children with
extensive atopic eczema (Sampson, 1982).
Eggs, cow's milk, wheat, seafood and nuts present the most
important food allergens. Citrus fruits and preservatives in foods
often affect patients via non-allergic mechanisms (Przybilla & Ring,
1990). Individual provocation factors (hypersensitivity) have to be
revealed by allergological diagnostic procedures. Therapy consists in
the elimination of the relevant allergens from the diet. If extensive
interventions are planned, the help of a dietitian is needed.
Controversy exists about the value of prophylactic dietary
manipulations. Exclusive breast feeding for six months, maternal
avoidance of allergens during lactation, and delay of solid food
feeding seem to have a protective influence in postponing or avoiding
atopic eczema (Kajosaari & Saarinen, 1983; Arshad et al., 1992;
Saarinen & Kajorsaari, 1995).
Sensitization to aeroallergens is frequently found in older
children and in adults. As shown by atopy patch tests, in some
patients direct contact with house dust mite allergen, animal dander
and pollen on intact skin results in eczematous skin lesions (Ring et
al., 1991a,b; Darsow et al., 1995). In the case of a suspected allergy
to house dust mites, reduction procedures should include encasing of
bedding with impermeable synthetic material and removal of carpets and
upholstered furniture (Platts-Mills & Chapman, 1987; Platts-Mills et
al., 1991; Lau et al., 1995). When allergy to animal dander is shown,
contact with the animal must be avoided. In case of exacerbation of
atopic eczema due to aeroallergens, rehabilitation in
aeroallergen-poor climates (sea level or high altitude mountains) has
been recommended (Borelli, 1981). In patients with severe atopic
eczema without adequate improvement of skin condition despite therapy,
additional contact allergy should be suspected and excluded by
epicutaneous (patch) testing.
Furthermore, there are various nonspecific provocation factors
influencing the disease activity in patients with atopic eczema. The
skin of these patients is highly susceptible to irritants, such as
wool, coarse fabrics, soap, detergents, frequent bathing,
disinfectants, wet working conditions and others. Patients need to be
educated about avoidance of these factors (Ring et al., 1996).
Chronic microbial colonization of the skin (e.g.,
Staphylococcus aureus, Pityrosporum ovale) and superinfection are
possible additional provocation factors and should be treated (Cooper,
1994). Psychological factors such as stress are well-known triggering
factors for a subgroup of patients. In these patients, psychosomatic
intervention has been proven successful and psychosomatic approaches
should be supported (Cotterill, 1991; Ehlers et al., 1995).
4.2.8.2 Basic dermatological therapy
In patients with atopic eczema there is a defective skin barrier
against exogenous substances (Ruzicka et al., 1991; Schöpf et al.,
1995). Regular basic therapy with emollients with or without addition
of moisturizers and bath oils is needed for the treatment of the
irritable dry skin to prevent the itch/scratch cycle.
4.2.8.3 Anti-inflammatory therapy
Recurrent relapses are a characteristic feature of atopic eczema.
Anti-inflammatory therapy of exacerbations is aimed to control
effectively disease activity and permit a return to basic
dermatological therapy as soon as possible. Topical corticosteroids
are the drugs of choice for acute exacerbations.
4.2.9 Conclusion
Atopic eczema is one of the most common skin diseases in many
countries of the world with an increasing prevalence. Prevalence rates
range between 10 and 20% of school children. Owing to the immense
suffering caused by the skin disfigurement and the often unbearable
itching, as well as the large number of people affected, it presents a
major health problem. The role of allergy in this skin disease has
been controversial but it has been shown that in the majority of
patients, allergic reactions -- preferentially by IgE-mediated
sensitization -- seem to play a clinically relevant role in eliciting
and maintaining eczematous skin lesions.
4.3 Allergic rhinitis and conjunctivitis
4.3.1 Introduction
Allergic reactions can occur in the respiratory tract and ocular
conjunctiva. In the respiratory tract allergic reactions occur in:
a) the upper respiratory tract predominantly involving the
nose - rhinitis;
b) bronchial airways - asthma;
c) gas exchanging parts of the lung - extrinsic allergic
alveolitis.
Allergic reactions in the nose and airways are characterized by
mucosal infiltration with eosinophils and T-lymphocytes, diseases now
considered to be the manifestation of a local Th2-lymphocyte-dependant
eosinophilic inflammation. In contrast, extrinsic alveolitis is
characterized by granulomata and mononuclear cell inflammation within
alveoli, centred upon bronchioles; the disease is considered to be the
manifestation of a local Th1-dependant granulomatous inflammation.
Both patterns of reaction are predominantly induced by agents
suspended in the air, such as dust or fume particulates, aerosol
droplets or vapour, inhaled into the respiratory tract. In general
larger particles will be deposited and soluble chemicals dissolved in
the upper respiratory tract; smaller particles (<5 µm aerodynamic
diameter) and insoluble chemicals can penetrate into the gas
exchanging parts of the lung.
4.3.2 Definition
Allergic rhinitis and conjunctivitis are common allergic
inflammatory conditions induced by hypersensitivity to environmental
allergens affecting the nasal (rhinitis) and/or conjunctival mucosa
(conjunctivitis) (Mygind, 1986, 1989). Rhinitis, characterized by one
or more of the symptoms of nasal congestion, rhinorrhea, sneezing and
itching, is defined as the inflammation of the lining of the nose
(International Rhinitis Management Working Group, 1994). The symptoms
of allergic conjunctivitis consist of redness, lachrymation, itching
and burning of the conjunctiva (Ring, 1991). There is an increased
likelihood of the development of asthma in these patients.
4.3.3 Clinical manifestations
4.3.3.1 Seasonal allergic rhinitis and conjunctivitis (hay fever,
pollinosis)
Seasonal allergic rhinitis and conjunctivitis consists of
paroxysms of sneezing, nasal itching, nasal congestion and rhinorrhea
(Druce, 1993; Mygind, 1986). In severe cases the conjunctiva and
mucous membranes of the Eustachian tube, middle ear and paranasal
sinuses also may be involved. In these cases additional symptoms
usually present with low-grade itching, lacrimation, burning,
stinging, photophobia, redness and watery discharge, as well as ear
fullness, ear popping and pressure over the cheeks and the forehead.
This may be complicated by malaise, weakness and fatigue. Symptoms
typically show a periodic distribution manifesting at individual time
intervals during the pollen seasons of tree, grass and weed pollen
between spring and autumn months. About 20% of patients have asthmatic
symptoms as well (Smith, 1983). Food allergy, often manifesting as
"oral allergy syndrome" due to cross-reacting allergens, may also be
present (see section 4.5.2).
4.3.3.2 Perennial allergic rhinitis and conjunctivitis
In perennial allergic rhinitis and conjunctivitis, indoor
allergens are the main cause of symptoms, which are similar to those
of seasonal allergic rhinitis and conjunctivitis although nasal
blockage is more pronounced and itching of the eyes is a common
problem. Among the indoor allergens, house dust mites, cockroaches,
animal dander and moulds are important. The chronic and persistent
symptoms can present as a "permanent cold" and may be accompanied by
secondary complaints, such as mouth breathing, snoring and sinusitis
(Lucente, 1989). Occupational hypersensitivity to an airborne allergen
at the workplace may lead to symptoms only during the week with a
disease-free interval at weekends, for example in laboratory animal
workers.
4.3.3.3 Prognosis
The peak prevalence of allergic rhinitis and conjunctivitis is in
adolescents and young adults. The first manifestations of seasonal
allergic rhinitis and conjunctivitis develop before 20 years of age in
most patients. After 30 years of age, disease severity usually
moderates and is only occasionally a problem in the elderly. Repeated
exposure to allergens may cause nasal hyperreactivity also to other
allergens, thus broadening the spectrum of hypersensitivity (Connell,
1969). A proportion of patients will develop asthma in the course of
their disease (Evans, 1993).
4.3.4 Etiology
Symptoms of allergic rhinitis and conjunctivitis are provoked by
environmental aeroallergens. Typical seasonal allergens are tree
pollen in the spring, grass pollen in the early and mid summer and
weed pollen in the late summer. In temperate climates of the Northern
hemisphere the most important tree pollens derive from birch, alder
and hazel; among grass pollens timothy and ryegrass, and among weed
pollens mugwort and ragweed are the most important. However, regional
differences are also of importance, e.g., cedar pollen in Japan,
parietaria pollen in the Mediterranean area and ragweed pollen in the
USA being the most important allergens. Sometimes mould spores, e.g.,
Cladosporium and Alternaria, cause symptoms during summer and
autumn months. In perennial allergic rhinitis and conjunctivitis
mainly indoor allergens present in the environment throughout the year
are relevant triggers. The house dust mites Dermatophagoides
pteronyssinus and Dermatophagoides farinae, in Southern countries
Bloomia tropicalis, animal dander from horses, cats, dogs and other
pets, cockroaches, and moulds such as Aspergillus species are the
most important allergens.
Epidemiological studies indicate a significant increase in the
prevalence of allergic rhinitis and conjunctivitis. There is evidence
that outdoor air pollution plays a role in the increasing morbidity
from allergic rhinitis and conjunctivitis. The disease seems to be
more common in urban than in rural areas (Broder et al., 1974a,b).
There is evidence that air pollutants may interact directly with
pollen with a possible impact on allergenicity (Behrendt et al.,
1992).
Beside exogenous factors, the association of allergic rhinitis
and conjunctivitis with other atopic diseases, such as atopic eczema
or asthma and a positive family history for atopy clearly demonstrates
the genetically determined susceptibility (Coca & Cooke, 1923).
4.3.4.1 Allergic rhinitis and conjunctivitis caused by contact with
chemicals
Allergic rhinitis and conjunctivitis caused by contact with
chemicals is less common than by contact with proteins. The prevalence
is unknown. The scope of the problem is probably underestimated
because of diagnostic failure (Mygind, 1986). The majority of cases
reported in the literature are in association with occupational
diseases. Upper respiratory tract hypersensitivity involving the nose
often coexists with asthma, conjunctivitis, bronchitis, and
occasionally with contact dermatitis, allergic alveolitis or fever.
Occupational chemicals may be haptens, allergens, mediator- releasing
or pharmacological agents and irritants. Eliciting agents that
sometimes are shown to induce an immediate-type IgE-mediated
hypersensitivity include anhydrides, metallic salts, dyes,
diisocyanates and antibiotics. In many, but not all, workers with
trimellitic acid-induced rhinitis and asthma, specific IgE antibodies
and positive skin tests can be found, suggesting Type I and Type III
allergic mechanisms (Bernstein et al., 1982a). In isocyanate workers
with rhinitis, conjunctivitis, asthma, bronchitis, chronic obstructive
lung disease, cutaneous reactions or fever, 26% had positive
skin-prick tests and in 14% specific IgE antibodies could be detected
after conjugation of isocyanates with serum albumin (Baur et al.,
1984). In the majority of cases with occupational rhinitis,
conjunctivitis and asthma caused by platinum salts, a Type I
hypersensitivity was proved by skin tests, in vitro histamine
release and passive cutaneous anaphylaxis (Schultze-Werninghaus et
al., 1978). In textile workers exposed to reactive dyes, who had
respiratory complaints, skin-prick tests and patch tests were positive
(Alanko et al., 1978; Estlander, 1988). It is thought that these small
molecule chemicals are haptens that combine with proteins to form
antigenic determinants.
Symptoms caused by chemicals may also be due to a contact-allergy
and delayed-type hypersensitivity. This applies more often for ocular
allergy. Rubbing of the eyes after handling detergents or other
chemicals may provoke a contact conjunctivitis. Positive patch tests
are found to chemicals such as antibiotics, thiomersal, benzalkonium
chloride, solutions for contact lenses, and metallic salts. In these
cases a Type IV hypersensitivity seems to be the primary allergic
mechanism.
However, allergies have to be differentiated from toxic and
irritative mechanisms. Strongly toxic chemicals may elicit symptoms by
directly damaging the mucosa after single contact. Milder irritants,
such as sulfur dioxide, urea formaldehyde, detergents, solvents or
dusts may cause hyperreactivity after repeated (cumulative) contact.
Exposure to cotton defoliants causes asthma, rhinitis and
conjunctivitis, which is thought to be a result of direct histamine
release. It is important to note that there is often an overlap
between allergic and irritative processes. Hyperreactivity to
irritants occurs predominantly after repeated contact in patients with
pre-existing atopic diseases, with or without an allergic basis.
Chemicals are often not only irritants but also allergens.
4.3.5 Pathophysiology
Allergens transported by the air come into contact with the
mucosal surface. Contact with mast cells or basophils leads to
IgE-dependent activation and degranulation of mast cells. Preformed
mediators stored in the granules (e.g., histamine, tryptase) are
released rapidly and elicit immediate symptoms. Other mediators are
eluted slowly (e.g., heparin) or are synthesized de novo (e.g.,
prostaglandins, leukotrienes) (Bachert et al., 1995). Afferent nerve
stimulation may provoke an axon reflex, and the release of
neuropeptides (substance P, tachykinins) may amplify this reaction
(Barnes et al., 1991). Mediators that are released slowly induce a
late- phase reaction after 6 to 12 h, which results in local
accumulation of inflammatory cells including CD4+ T-lymphocytes,
eosinophils, basophils and neutrophils (Dvoracek et al., 1984). These
cells and mast cells release cytokines and proteins (e.g., eosinophil
basic proteins) that perpetuate the reaction (Bachert et al., 1995).
Inflammatory cytokines (e.g., IL-4) may selectively recruit
eosinophils by increasing the expression of adhesion molecules on the
vascular endothelium (VCAM-1, ICAM-1).
The late-phase reaction results in an increased
hyper-responsiveness, which may be specific for an allergen
("priming") or nonspecific to a variety of irritant triggers (Connell,
1969).
4.3.6 Diagnostic techniques
Diagnostic techniques are applied for differential diagnosis and
verification of a definite diagnosis. The patient's history, physical
examination with rhinoscopy and allergy testing represent the basic,
readily accessible diagnostic techniques. Rhinomanometry with
assessment of nasal resistance and nonspecific provocation tests
demonstrating hyperreactivity of the nasal mucosa are also often used
for evaluation of clinical relevance (International Rhinitis
Management Working Group, 1994).
4.3.6.1 Medical history
A careful history of seasonal and/or perennial symptoms provoked
by specific exogenous factors is most important for the diagnosis of
allergic rhinitis and conjunctivitis. The conditions that precipitate
or aggravate symptoms should be asked for in detail. In particular,
the presence of allergens in the patient's environment and the
possible causal relationship to the symptoms should be evaluated.
Exposure factors, such as contact with air pollutants, automobile
exhaust emissions or detergents, a history of atopic diseases and the
family history provide further important information. The severity of
the disease may be estimated by the frequency, distribution and
severity of symptoms. Standardized questionnaires are useful in
obtaining detailed information.
4.3.6.2 Clinical examination
Special devices are usually unnecessary for examination of the
eyes, whereas rhinoscopy is obligatory for the examination of the
nose. The use of indirect laryngoscopy and full endoscopic
ear-nose-throat examination are not mandatory, but may be of value in
special patients (International Rhinitis Management Working Group,
1994). The nasal mucosa is usually reddened, oedematous and produces
large quantities of a clear mucous discharge. The periorbital tissues
may be oedematous. Cyanosis, conjunctival injection, increased
lacrimation and mucous discharge of the eyes are further symptoms. The
quality and quantity of the secretions should be noted.
4.3.6.3 Allergy testing
Immediate hypersensitivity skin tests (skin-prick test,
intracutaneous test) are the primary diagnostic tool, skin-prick tests
being the method of choice for the majority of cases (Dreborg, 1989;
Ring, 1991). Skin testing with commercially available aeroallergens
generally has a high reliability. The number of skin tests that should
be performed is confined to a few common environmental allergens
tested routinely but should be extended specifically if the individual
patient's history indicates a role of other allergens.
The determination of total serum IgE is of limited value for this
disease but tests for specific IgE antibodies (e.g., RAST) are useful.
Positive results of the skin-prick test and determination of specific
IgE antibodies (sensitizations) should always be evaluated in
combination with the patient's history. Nasal and conjunctival
challenges with commercially available allergens should be used
whenever the clinical relevance of a sensitization to an allergen
cannot otherwise be estimated. However, there is no universally
accepted standard for this technique. As all in vivo tests are
potentially dangerous, with the risk of anaphylaxis, tests should be
carried out only by personnel trained in cardio-pulmonary
resuscitation.
4.3.7 Therapeutic considerations
The therapeutic repertoire of antiallergic therapy includes
environmental control to minimize exposure to the allergen responsible
for provoking symptoms, symptomatic medications, and immunotherapy
under strict medical supervision (Druce, 1993).
4.4 Clinical aspects of allergic asthma caused by contact with
chemicals
4.4.1 Introduction
Asthma is by far the most frequently reported outcome of
an allergic respiratory reaction to inhaled chemicals, primarily
occurring as the consequence of exposures experienced at work, i.e.,
occupational asthma. Allergic rhinitis is generally caused by the same
agents and may occur in isolation or in association with asthma.
4.4.2 Importance of occupational asthma
The contribution of occupational causes to the prevalence of
asthma in the community is not generally known. Estimates in different
countries have varied between 2% and 15% but their basis is not
secure. In Spain, occupational causes accounted for between 1 in 15
and 1 in 20 of cases of asthma in young Spanish adults aged between 20
and 44 years. Information in the United Kingdom is limited to the
numbers awarded compensation and the number of cases reported to
voluntary surveillance schemes, both of which are likely to
underestimate the true frequency of the disease.
In the United Kingdom a surveillance scheme for work-related
diseases (SWORD) with voluntary reporting of new cases of occupational
lung disease by respiratory and occupational physicians reported 2101
new cases in 1989 of which 554 (26%) were asthma. The agents most
frequently reported to cause occupational asthma were isocyanates,
which accounted for 22% of cases, and grain, wood dusts and laboratory
animals, which together accounted for a further 17% of cases. The
annual incidence rate for occupational asthma in the working
population was estimated to be 22 per million. The highest rates in
the occupational groups occurred primarily among those encountering
chemicals at work, i.e., coach and spray painters, chemical
processors, plastics making and processing, metal making and treating,
and welders (Table 21).
The incidence reported in this survey is lower than that reported
in Finland by Meredith & Nordman (1996); Finland is one of the few
countries where occupational lung diseases are registered. The
incidence in 1981 of occupational asthma in Finland was estimated to
be 71 per million (compared to the rate in the United Kingdom of 22
per million). However, within the United Kingdom there was
considerable regional variation in reported rates, and the area of
highest incidence, West Midlands Metropolitan Area, had a rate of 63
per million, similar to the reported incidence in Finland. Meredith &
Nordman (1996) suggested that the differences in regional rates might
at least in part be due to differences in ascertainment and reporting,
and that the true incidence of occupational asthma in the United
Kingdom was three or more times that reported.
4.4.3 Chemical causes of occupational asthma
Many different chemicals encountered at work can stimulate a
hypersensitivity response and cause asthma. The more prevalent causes
are shown in Table 22.
4.4.3.1 Isocyanates
Diisocyanates are bifunctional molecules used commercially to
polymerize polyglycol and polyhydroxyl (polyols) compounds to form
polyurethanes. Because each diisocyanate molecule has two reactive
isocyanate (NCO) groups, they link adjacent polyols to form a
three-dimensional lattice. Isocyanates also react with water to evolve
carbon dioxide, a reaction exploited in the manufacture of flexible
polyurethane foam. The urethane reaction is exothermic and the heat
generated sufficient to evaporate diisocyanates with high vapour
pressures, such as toluene diisocyanate (TDI) and hexamethylene
diisocyanate (HDI). Diphenyl methane diisocyanate (MDI) and
naphthalene diisocyanate (NDI), whose vapour pressures are lower,
evaporate in significant amounts when heat is applied.
It is estimated that approximately 5% of workers regularly
exposed to TDI develop asthma, which may be manifested as immediate
and/or late onset responses. TDI can act as a direct irritant, can
stimulate nerve reflexes, and, in the minority of patients, elicit an
IgE antibody response and occasionally an IgG response (Baur &
Fruhmann 1981; Baur et al., 1994). In addition, persistent activation
of T-cells and continuous expression of pro-inflammatory cytokines
seems to maintain a state of chronic inflammation (Maestrelli et al.,
1995).
Polyurethanes have widespread applications, and exposure to
isocyanates occurs in many different occupations. These include the
manufacture of flexible and rigid polyurethane foam, the application
of two part polyurethane paints by brush and by spray painting, and in
flexible packaging production where isocyanates are used in inks and
as laminating adhesives.
Table 21. Incidence of occupational asthma in high-risk occupational
groups reported to the United Kingdom Surveillance of Work-Related
and Occupational Respiratory Disease Project (SWORD) in 1989
(Meredith, 1993)
Occupational Group Cases Population Incidence/106/year
Coach and spray painters 35 54 737 639
Chemical processors 31 73 189 424
Bakers 29 70 839 409
Plastics making and processing 27 66 005 409
Metal making and treating 14 56 270 249
Laboratory technician and 26 127 478 204
assistant
Welders/solderers electronic 35 220 068 159
assemblers
Working population 22
Table 22. Examples of occupational chemical respiratory allergens associated
with positive bronchial provocation challenges
(adapted from Karol et al., 1996)
Isocyanates Dyes
Diphenylamine-4,4'-diisocyanate (MDI) Brilliant orange GR
Hexamethylene diisocyanate (HDI) Carminic acid
Isophorone diisocyanate (IPDI) Reactive orange 3R
Naphthalene-1,5-diisocyanate Rifafix red BBN
Toluene 2,4-diisocyanate (2,4 TDI) Rifazol black GR
Toluene 2,6-diisocyanate (2,6 TDI)
Amines Acid anhydrides
Dimethyl ethanolamine Phthalic anhydride
Ethanolamine Tetrachlorophthalic anhydride
Ethylenediamine Trimellitic anhydride
Triethylenetetramine
Others
Abietic acid Glutaraldehyde
6-Aminopenicillanic acid Iso-nonanoyl sulfonate oxybenzene
7-Aminocephalosporanic acid Methyl-2-cyanoacrylate
Ampicillin alpha-Methyldopa
Azocarbonamide Phenylglycine acid chloride
Table 22 (continued)
Isocyanates Dyes
2-(n-Benzyl-N-tert-butylamino)4'-hydroxy Piperacillin
3'-hydroxymethylacetophenone diacetate Piperazine
Benzylpenicillin Plicatic acid
Cephalexin Spiramycin
Chlorhexidine Styrene
Complex platinum salts
Ethyl cyanoacrylate Tylosin
Natural rubber latex
Inhaled isocyanates have been reported to cause four different
respiratory reactions:
a) Toxic bronchitis and asthma caused by isocyanate inhalation
at toxic concentrations. Exposure to TDI at an atmospheric
concentration of 0.5 ppm (3.6 mg/m3) causes irritation of
mucosal surfaces - eyes, nose and throat (Henschler et al.,
1962). Persistent asthma and reactive airways dysfunction
syndrome (RADS) has been reported following a single inhalation
of TDI at toxic concentrations (Luo et al., 1990).
b) Bronchial asthma caused by sensitization to isocyanates.
c) Accelerated decline of forced expiratory volume in 1 second
(FEV1). The rate of decline of FEV1 in an isocyanate
manufacturing plant workforce was similar in non-smokers with
high cumulative exposures to toluene diisocyanate (TDI) to the
rate observed in smokers in both the high- and low-exposure
groups. The rate in non-smokers with low cumulative exposure was
not different from that expected for control non-smokers. No
additive effect of TDI with smoking was observed (Diem et al.,
1982).
d) Extrinsic allergic alveolitis, which has been reported
particularly in workers exposed to MDI (Zeiss et al., 1980) and
also to HDI (Malo et al., 1983).
Of the four reactions, bronchial asthma caused by
hypersensitivity to isocyanates has been the most frequently reported
and is the most important both in terms of prevalence and morbidity.
TDI and MDI have been the most widely used isocyanates and are the
major causes of asthma, although, with its increasing use in spray
paints, HDI is becoming a more prevalent cause. A study of workers
employed at a new TDI manufacturing plant identified 12 workers (4% of
the total workforce) who had developed asthma during a 5-year period,
with 9 developing it in the first year of employment. The average
exposure to TDI monitored by paper tapes was 0.002 ppm (14 µg/m3)
(Weill et al., 1981). Half of the cases had been exposed to spills;
six were maintenance workers, one was a laboratory worker and only
five were process workers. A cross-sectional study of a steel coating
plant, where TDI had been introduced into the process some years
before, identified 21 cases of asthma out of a total of 221, which was
probably an underestimate of the true number of cases (Venables et
al., 1985a).
Inhalation challenge tests with TDI have shown that asthmatic
responses may be provoked in sensitized workers by very low
atmospheric concentrations, as low as 0.001 ppm (7 µg/m3) (O'Brien et
al., 1979). Late asthmatic responses provoked by isocyanates are
associated with the development of an increase in nonspecific airway
responsiveness (Durham et al., 1987), and cells recovered from
bronchoalveolar lavage during a late asthmatic reaction provoked by
TDI have an increased proportion of neutrophils, identifying an
inflammatory response in the airways provoked by TDI (Fabbri et al.,
1987).
4.4.3.2 Acid anhydrides
Acid anhydrides are low relative molecular mass chemicals used
industrially as curing agents in the production of epoxy and alkyd
resins and in the manufacture of the plasticizer dioctyl phthalate.
Epoxy and alkyd resins have widespread applications as paints,
plastics and adhesives. Six acid anhydrides, i.e., phthalic anhydride
(PA) (Maccia et al., 1976), trimellitic anhydride (TMA) (Fawcett et
al., 1977; Zeiss et al., 1977), tetrachlorophthalic anhydride (TCPA)
(Howe et al., 1983), maleic anhydride (MA) (Durham et al., 1987;
Topping et al., 1986), hexahydrophthalic anhydride (Moller et al.,
1985) and himic anhydride (Bernstein et al., 1984), have been reported
to cause occupational asthma. Inhalation tests with the causal acid
anhydride provoked asthmatic responses, and specific IgE or IgG
antibodies, or both, to the specific anhydride conjugated to human
serum albumin were identified in the sera of the great majority of
cases, although this was less frequent with maleic than with the other
anhydrides. Zeiss et al. (1977) suggested that four separate clinical
syndromes were caused by TMA, for which they proposed separate
immunological mechanisms: i) toxic airway irritation; ii) immediate
IgE-mediated rhinitis and asthma; iii) IgG-mediated late asthma with
systemic symptoms ("TMA flu"); iv) pulmonary haemorrhage-haemolytic
anaemia syndrome as the outcome of antibody binding to circulating red
blood cells and to pulmonary vascular cells. The distinction between
"immediate" and "late" asthmatic reactions with influenza-like
symptoms and their different pattern of immunological response has not
been consistently observed by other investigators. It seems more
likely that asthma caused by acid anhydrides, including TMA, may be
associated with specific IgE or IgG, or both, although specific IgE
and IgG4 seem to be more associated with asthma and IgG with exposure
(Forster et al., 1988). A relationship between HLA DR3 and the
development of specific IgE to TMA and possibly tetrachlorophthalic
anhydride (TCPA), but not phthalic anhydride (PA), has been reported
(Young et al., 1995). The pulmonary haemorrhage haemolytic anaemia
syndrome is real but rare. It has been reported in individuals exposed
to hot trimellitic anhydride fume and may be the outcome of a toxic
reaction to inhalation of trimellitic anhydride at very high
concentration rather than a hypersensitivity reaction.
4.4.3.3 Complex platinum salts
The complex platinum salt ammonium hexachloroplatinate is an
essential intermediate in the refining of platinum, a corrosion
resistant metal used as a catalyst and in jewellery. Allergy to
platinum salts in refinery workers was first reported in 1945 (Hunter
et al., 1945). Subsequently, inhalation of ammonium
hexachloroplatinate was shown to provoke asthmatic responses and to
elicit immediate skin test responses in sensitized individuals (Pepys
et al., 1972).
The incidence of occupational allergy in the platinum refining
industry was high in United Kingdom in the mid 1970s. In a cohort
study of 91 workers who entered employment in a platinum refinery in
the two years 1973 and 1974 (Venables et al., 1989), 22 developed
respiratory symptoms and an immediate skin test response to ammonium
hexachloroplatinate. The risk was greatest in the first year of
employment and smoking was more important than atopy as a predictor of
developing a positive skin test reaction.
4.4.4 Diagnosis of occupational asthma
Accurate and early diagnosis of cases of occupational asthma is
important. Remission of respiratory symptoms and restoration of normal
lung function, including nonspecific airway responsiveness, can follow
avoidance of exposure to the specific initiating cause. Furthermore,
chronic asthma is more likely to develop in those who remain exposed
to the initiating cause after the onset of symptoms. However,
avoidance of exposure frequently requires a change of employment.
Accurate diagnosis is also essential if those whose asthma is not
occupationally caused are to avoid being advised unnecessarily to
change or leave their work.
The diagnosis of occupational asthma requires:
a) differentiation of asthma from other causes of respiratory
symptoms, in particular chronic airflow limitation and
hyperventilation;
b) differentiation of occupational cause from non-occupational
asthma;
c) differentiation of asthma initiated by an agent inhaled at
work from pre-existing or incidental asthma exacerbated by
nonspecific irritants, such as sulfur dioxide and cold air,
inhaled at work.
The diagnosis of occupational asthma is usually suggested by the
history. It commonly occurs in an individual exposed at work to an
agent recognised to cause occupational asthma and only develops after
an initial symptom-free period when the patient has been exposed
without symptoms to concentrations in air that now provoke asthma.
Respiratory symptoms occur during the working week, may increase in
severity as the week progresses, and improve during absences from
work, at weekends or during holidays. The patient may also be aware of
others who have developed similar respiratory symptoms at the place of
work.
Nonspecific stimuli provoke asthmatic reactions that usually
occur within minutes of exposure to them and resolve within 1-2 h of
avoidance of exposure. Where work-related respiratory symptoms are due
to the provocation of asthma by a respiratory irritant encountered at
work, the onset of asthma will often have preceded initial exposure to
the irritant and the severity of asthma does not significantly improve
when away from work. Nonspecific irritants such as organic solvents,
which often have a characteristic and unpleasant smell, may also
provoke a hyperventilation response, when difficulty with breathing is
associated with symptoms that are consequences of a low pCO2, such as
tingling of the fingers, headaches and dizziness.
4.4.4.1 Investigation of causes of occupational asthma
In the majority of cases a confident diagnosis of occupational
asthma induced by an inhaled chemical can be made from knowledge of
exposure at work to a recognised cause of occupational asthma and a
characteristic history. Where possible, these should be supported by
objective evidence from serial measurements of peak expiratory flow
(PEF) or immunological tests or both. Inhalation testing is reserved
for occasions when the results of these investigations do not provide
an adequate basis for advice about future employment.
4.4.4.2 Serial peak expiratory flow (PEF) rate measurements
Asthma can be attributed with confidence to an agent inhaled at
work where exposure to it in the work place reproducibly provokes
airway narrowing. Repeated measurements of airway calibre, most
conveniently made as PEF rates, need to be made during a period long
enough to allow observation of the consistency of any changes and
their relationship to periods at work. Measurements need to be made
repeatedly during each day for a period of several weeks and the
patient can take measurements and record the results. Self-recording
of PEF measurements is now widely used. Patients are lent a peak flow
meter and asked to record the best of 3 measurements of PEF made every
2 h from waking to sleeping over a period of one month in the first
instance. To allow sufficient time for lung function to recover from
exposure to an agent inhaled at work, it is helpful if the month
includes a period away from work which is longer than a weekend,
ideally a one or two week holiday. Self-recording requires patient
compliance and honesty. The measurements may be conveniently
summarized to show the maximum, minimum and mean peak flow
measurements for each day and differences between periods at and away
from work observed. This method of patient investigation has proved,
in the hands of those experienced in its use, to be reliable and a
relatively sensitive and specific index of occupational asthma.
4.4.4.3 Immunological investigations
The demonstration of specific IgE antibody is a helpful, but not
conclusive, criterion to establish the diagnosis of occupational
asthma or rhinitis, because the presence of specific IgE antibody is
not unique to clinically allergic individuals. Antigen-specific IgE is
rare (5-15% of clinical cases). The presence of specific IgE antibody
can be detected by in vivo tests such as skin-prick test and in
vitro tests such as radioallargosorbent test (RAST) or enzyme-linked
immunosorbent assay (ELISA). Other in vitro tests, such as histamine
release from basophils, are less standardised. When allergens are not
available, histamine release from basophils can be an alternative to
direct measurement of IgE.
The application of immunological tests in the investigation of
occupational asthma caused by inhaled chemicals has widened with the
preparation of hapten protein conjugates suitable for immunological
testing (e.g., acid anhydride and reactive dye-human serum albumin
conjugates) and the development of reliable methods for identification
of specific IgE antibody in serum. Complex platinum salts such as
ammonium hexochloroplatinate can elicit immediate skin-prick test
responses without the need for conjugation to human serum albumin.
The value of such tests in the diagnosis of occupational asthma
depends upon their sensitivity and specificity in populations exposed
to the particular cause. An immediate skin-prick test response and
specific IgE antibody identified by RAST to conjugates of the acid
anhydride tetrachlorophthalic anhydride (TCPA) with human serum
albumin (Howe et al., 1983) were found to be associated with cases of
asthma in exposed populations and not simply a reflection of exposure.
4.4.4.4 Inhalation challenge tests
Inhalation tests are rarely undertaken because they are
potentially hazardous. Inhalation tests with occupational agents
should only be undertaken by those experienced in conducting them and
who have adequate hospital facilities for continuous monitoring of
patients for 24 h after each test.
There are 4 major indications for inhalation challenge testing in
the diagnosis of occupational asthma:
a) Where the agent thought to be responsible for causing asthma
has not previously been reliably shown to do so.
b) Where an individual with occupational asthma is exposed at
work to more than one potential cause, and his future employment
depends on knowledge of which one is responsible.
c) Where asthma is of such severity that further uncontrolled
exposure in the work environment is not justifiable.
d) Where the diagnosis of occupational asthma remains in doubt
after other appropriate investigations, including serial peak
expiratory flow rate (PEF) and immunological tests, have been
completed.
The aim in an occupational-type inhalation challenge test is to
expose the individual under single blind conditions to the putative
cause of the asthma in circumstances which resemble as closely as
possible the conditions of the exposure at work. Wherever possible,
atmospheric concentrations of the inhaled agent should be based on
knowledge of the concentrations experienced at work, and the physical
conditions of exposure, (e.g., size of dust particles, whether vapour
or aerosol, and temperatures to which the materials are heated),
should be similar to those encountered at work.
Measurements of airway responses provoked by inhalation challenge
tests should ideally include measurements of both changes in airway
calibre and in nonspecific airway responsiveness. Changes in airway
calibre are most conveniently measured by regular measurements of
forced expiratory volume in 1 second (FEV1) and forced vital capacity
(FVC) or PEF rate before and at regular intervals after the test, for
at least 24 h. Changes in airway responsiveness can be made by
estimating the concentration of inhaled histamine or metacholine that
provokes a 20% fall in FEV1 (PC20) before the test and at 3 and 24 h
after the test. The changes in airway calibre and nonspecific
responsiveness observed are compared to those following a control
challenge test, each test being made on a separate day.
The patterns of change in airway calibre provoked by inhalation
testing are distinguished by their time of onset and duration.
Immediate responses occur within minutes and resolve spontaneously
within 1-2 h. Such reactions can be provoked by both allergic (e.g.,
grass pollen) and non-allergic (e.g., inhaled histamine or sulfur
dioxide) stimuli. The response depends upon the concentration of the
provoking agent and the degree of pre-existing nonspecific airway
responsiveness. Lone immediate responses (i.e., an immediate response
not followed by a late response) are not usually associated with an
increase in nonspecific airway responsiveness. Late responses develop
one or more hours after the inhalation test exposure, usually after
some 3-4 h, and may persist for 24-36 h. Unlike the immediate
response, late responses are often associated with an increase in
nonspecific responsiveness which can be identified 3 h post test prior
to the onset of the late asthmatic response and less reliably at 24 h
after the test (Durham et al., 1987).
A dual response is an immediate response followed by a late
response. Recurrent nocturnal responses may be provoked by a single
inhalation test exposure with asthmatic responses occurring during
several successive nights with partial or complete remission during
the intervening days. Such responses are almost certainly a
manifestation of a provoked increase in nonspecific airway
responsiveness. The question to be answered from the results of an
inhalation test is whether or not in the individual case the
particular agent inhaled at work has induced asthma. The most reliable
means of answering this question is to determine whether or not
inhalation of the specific agent at concentrations to which exposure
occurs at work reproducibly provokes a non-immediate asthmatic
response and increases nonspecific airway responsiveness. In such
cases the specific agent can be considered to be the inducing cause in
that particular individual. Nonspecific irritants may provoke
immediate responses in individuals with hyper-responsive airways, but
do not provoke either an increase in nonspecific airway responsiveness
or a late asthmatic reaction. Agents that induce specific IgE
antibody, however, may provoke lone asthmatic responses; in these
cases inferences from the inhalation test result should take the
immunological test into account.
4.4.5 Outcome of occupational asthma
Asthma induced by an agent inhaled at work may become chronic,
persisting for several years, if not indefinitely, after avoidance of
exposure to its initiating cause. This seems particularly, although
not exclusively, to occur with asthma caused by low relative molecular
mass chemicals. Asthma caused by isocyanates and acid anhydrides has
been reported to have persisted in over half of cases. Six cases of
asthma caused by the acid anhydride tetrachlorophthalic anhydride
(TCPA) were followed up 4 years after avoidance of exposure: all had
chronic respiratory symptoms consistent with persistent airway
hyper-responsiveness and a measurable histamine PC20 was present in
the five in whom it was assessed. The rate of decline of specific IgE
to a TCPA-human serum albumin conjugate during the period of avoidance
of exposure was parallel in all 6 subjects and exponential with a
half-time of one year, making it very improbable their continuing
asthma was caused by further, albeit inadvertent, exposure (Venables
et al., 1987).
Chronic asthma in these cases is likely to be a manifestation of
persistent airway inflammation which, although initiated by the agent
inhaled at work, persists in its absence. Ten patients with
TDI-induced asthma, who had continuing respiratory symptoms and airway
hyper-responsiveness, were investigated for 4-40 months from their
last exposure. Bronchial biopsies obtained from eight patients showed
basement membrane thickening with infiltration of the mucosa by
eosinophils, lymphocytes and neutrophils. In four patients in whom
airway responsiveness had not improved, the proportion of eosinophils
in fluid recovered by BAL was increased, whereas this was the case in
only one of five patients whose airway responsiveness had improved
(Paggiaro et al., 1990).
4.4.6 Management and prevention of occupational asthma
Reduction in the incidence of occupational asthma will follow
adequate control of exposure to its causes (Table 22). Substitution of
a different paint for one containing TDI halted an epidemic of asthma
in a steel coating plant (Venables et al., 1985a). Measures to secure
control of exposure to the majority of causes of occupational asthma
have, however, been impeded by lack of knowledge of the nature of
exposure-response relationships for sensitizing chemicals.
The development of control measures that will significantly
reduce the incidence of occupational asthma requires examination of
exposure-response relationships and the effects of interventions in
longitudinal studies.
Management advice for patients with occupational asthma has been
greatly influenced by the results of studies of outcome of
occupational asthma which have found evidence of continuing asthma and
airway hyper-responsiveness despite many years avoidance of exposure
to the initiating cause, and particularly studies, such as those of
azodicarbonamide workers, that have identified a relationship between
the duration of symptomatic exposure and the risk of chronic asthma.
The importance of accurate identification of the specific cause cannot
be over-emphasised. Avoidance of exposure often involves relocation or
change of employment. Misdiagnosis of occupational asthma can be as
hazardous for individual patients as missing the diagnosis because of
the implications for employment.
Rigorous prevention of exposure is a key to the prevention of
occupational asthma. Patients who develop occupational asthma in whom
a specific cause is identified should be advised to avoid further
exposure to the cause of their asthma. This seems particularly
important where low relative molecular mass chemicals, such as
isocyanates, plicatic acid or acid anhydrides, are the cause as these
seem particularly, although not exclusively, associated with the
development of chronic asthma and airway hyper-responsiveness.
However, environmental changes, unless they involve substitution,
are often not able to reduce exposures sufficiently to prevent
continuing airway responses in sensitized individuals. Venables &
Newman Taylor (1990) examined the relationship between the
concentration of TCPA in air and the provocation of asthmatic
responses in inhalation challenge tests in 4 sensitized individuals.
They observed a log-linear relationship between the magnitude of late
asthmatic responses and TCPA concentration, which passed through the
origin, suggesting no threshold.
When an individual remains in employment exposed to the cause of
the asthma, either directly or indirectly, the effectiveness of
relocation or of respiratory protection needs to be monitored. This
can be conveniently done by serial self recordings of peak flow to
determine whether or not asthma persists and, if so, whether or not it
is work related.
4.5 Food allergy
4.5.1 Definitions
Food allergy is an adverse reaction to food occurring in
susceptible individuals, which is mediated by a classical immune
mechanism specific for the food in question. Therefore, the "true"
allergic reaction to a food is caused by an over-sensitive reaction of
the body's immune system (in essence, "immunity gone wrong"). Food
intolerances are all non-immune-mediated adverse reactions to food.
The subgroups of food intolerance are enzymatic (resulting from an
enzymatic defect, e.g., lactase deficiency), pharmacological
(depending on the direct effect of certain substances found in foods,
e.g., caffeine) and undefined food intolerance (Bruijnzeel-Koomen et
al., 1995) (Fig. 14). Food intolerances will not be described further
since, as far as is known, they do not involve immune mechanisms.
The foods of animal origin that most commonly cause allergic
reactions are milk, eggs, cod fish and shrimps. Those of plant origin
that most often cause allergic reactions are peanuts, soybeans,
celery, apple, hazelnuts and wheat.
In food allergy the best established mechanism is the presence of
IgE antibodies against the offending food (Type I allergy). Other
immune mechanisms may be involved in other clinical patterns of food
allergy. For example, Type III and Type IV mechanisms may play a role
in the genesis of food protein-induced enterocolitis of newborns and
infants.
A subgroup of patients with contact allergy (T-cell mediated) to
nickel or balsam of Peru can develop cutaneous symptoms (haematogenous
contact eczema) to double-blind, placebo-controlled food challenge
(DBPCFC) (Veien et al., 1987; Menné & Maibach, 1991). Foods with very
high amounts of nickel or containing natural flavours, cross-reacting
with balsam of Peru, may provoke skin symptoms.
In the following text only adverse food reactions with a proven
involvement of the immune system are covered.
4.5.2 IgE-mediated food allergy
Clinical manifestations of IgE-mediated food allergy can remain
localized at the site of the primary direct contact, i.e., the mouth
and throat (oral allergy syndrome) or the gastrointestinal tract
(isolated gastrointestinal food allergy). However, after ingestion
of the specific food, food-allergic patients often exhibit symptoms in
various organs: the skin, the respiratory tract, the gastrointestinal
tract and the cardiovascular system (systemic anaphylaxis) are the
most frequently affected. Usually, the patients show involvement of
two or more organ systems. Among 402 patients with a systemic
IgE-mediated allergy to one or more specific foods -- the oral allergy
syndrome was not included -- diagnosed over a 10-year period at the
Allergy Unit in the Zurich University Hospital, the affected organ was
most often the skin (46%), followed by the respiratory tract (25%),
the gastrointestinal tract (20%) and the cardiovascular system
(10%)(Wüthrich, 1993). Twenty percent of the food allergic patients
had skin symptoms exclusively, 11% had isolated gastrointestinal
manifestations and 8% isolated respiratory symptoms. In only 7% of all
cases was food allergy responsible for a chronic condition such as
chronic urticaria, perennial asthma or gastroenterocolitis.
4.5.2.1 Oral allergy syndrome
Mainly patients with pollinosis may describe, spontaneously or by
exact questioning, itching of the lips, mouth, palate and throat
immediately after intake of some fresh foods, namely fruits and
vegetables. Hoarseness and/or swelling of the lips, tongue, uvula and
larynx can occur infrequently. Oral allergy syndrome must be carefully
differentiated from the beginning of a generalized anaphylactic
reaction in which itching of the mouth and throat may be the first
symptoms. These symptoms were described in the USA in
ragweed-sensitive patients (6.2% in one series) after ingestion of
melon and banana (Anderson et al., 1970; Ross et al., 1991).
Cross-reactivity of pollen-sensitized subjects to various food
allergens can occur (Fig. 15). Most patients allergic to birch pollen
react to apples, hazelnuts and numerous other vegetables and fruits
from the Rosaceae family such as cherries, peaches, pears and almonds
(Eriksson et al., 1982; Dreborg & Foucard, 1983; Pauli et al., 1992) ;
mugwort pollen sensitive patients may react to celery root (celeriac)
and spices, the so called mugwort-celery-spices-syndrome (Wüthrich &
Hofer, 1984; Wüthrich et al., 1990). Grass pollen allergic patients
may react to cereals or tomatoes (de Martino et al., 1988). It has
been shown by prick, RAST studies and RAST inhibition experiments that
a celery thermolabile allergen seems to be involved in
celery-birch-pollen allergic patients whereas a thermostable allergen
is involved in celery-mugwort-allergic patients (Wüthrich et al.,
1990). Patients with chestnut and banana oral allergy syndrome may be
sensitized to natural rubber latex (M'Raihi et al., 1991). Many
patients are able to identify the offending fruit or vegetable.
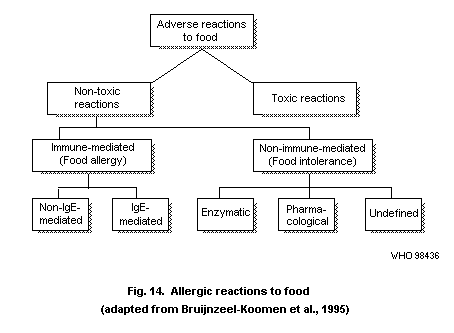 Cooking often destroys the reactivity (Wüthrich et al., 1990). Shared
epitopes and cross-reactive IgE antibodies are the most probable
explanation for the observed clinical symptoms in cases of association
between pollen allergy and food hypersensitivity (Calkhoven et al.,
1987).
4.5.2.2 Allergic reactions after ingestion of food
The main symptoms of gastrointestinal food allergy are vomiting,
nausea, diarrhoea and abdominal pains (colics or cramps) (Anderson,
1981; Atkins et al., 1985a,b). Skin reactions include local or
generalized pruritus, flush, urticaria, angioedema, morbilliform
exanthema and flare-up of atopic dermatitis. The symptoms of the upper
and the lower respiratory tract are rhinitis (sneezing, pruritus of
the nose, nasal stuffiness, nasal obstruction), larynx oedema, cough,
wheezing and bronchial asthma. Itching, redness and watering eyes
(conjunctivitis) is often associated with the above symptoms, but can
also appear as an isolated manifestation of food allergy.
Systemic anaphylaxis (cardiovascular collapse) always involves
other organ symptoms, e.g., of the gastrointestinal tract, the skin or
the respiratory tract. A particular subtype is food-dependent
exercise-induced anaphylaxis (Maulitz et al., 1979).
4.5.2.3 Allergic reactions following skin contact with food
Urticarial lesions can be provoked by contact with certain foods,
such as fish, shrimps, flour and pork meat (Maibach, 1976). Chronic
contact with food may induce protein contact dermatitis in food
handlers (Hjorth & Roed-Petersen, 1976).
4.5.3 Non-IgE-mediated immune reactions
Immune complexes with IgG antibodies and milk antigen inducing
complement-mediated damage have been suggested to induce Heiner's
syndrome and haemorrhagic gastroenteritis in childhood (Heiner et al.,
1962; Gryboski, 1967). Other non-IgE-mediated immune reactions to food
include gluten-sensitive enteropathy (coeliac disease), food-induced
colitis, and cutaneous allergic vasculitis (purpura).
4.5.3.1 Gluten-sensitive enteropathy (coeliac disease)
Gluten sensitive enteropathy or coeliac disease occurs in
susceptible individuals. It is characterized by damage to the small
intestinal mucosa with symptoms of malabsorption (Kagnoff, 1992). The
prevalence of coeliac disease has been estimated to be 0.2-0.5% with
considerable geographical variation (Logan, 1992).
Affected individuals develop specific immunological reactions to
gliadin, a protein that is a major alcohol-soluble fraction of gluten,
present in wheat, rye, barley and oat. Humoral cell-mediated immunity
and genetic factors seem to be involved in the pathogenesis.
The peak incidence of symptoms is in infancy after the
introduction of cereals, and a second peak occurs during the third
decade. In children it is often a semi-acute disease. The symptoms of
coeliac disease in children are recurrent abdominal pain, loose
stools, anorexia, short stature and delayed puberty. Symptoms of
malabsorption are unexplained nutritional deficiencies such as iron
and folic acid deficiency, anaemia and rickets. Dental enamel
hypoplasia and recurrent aphthae are also associated with coeliac
disease.
Patients with coeliac disease must avoid gliadins and related
proteins for life. As little as 100 mg gliadin has been described as
causing intestinal damage.
4.5.4 Diagnosis of adverse food reactions
4.5.4.1 Case history and elimination diet
The diagnosis of food allergy or food intolerance is the result
of a careful case history and clinical examination, and of several
in vitro and in vivo diagnostic procedures. A record of food
intake and its relation to clinical symptoms usually provides useful
information in the case of acute reactions, occurring from a few
minutes to a few hours after ingestion. History is, however, less
reliable when symptoms are chronic and caused by food that generally
is consumed daily, such as milk, egg, wheat and meat or by additives.
An elimination diet based on rice, potatoes and mineral water over a
week is useful in patients with chronic symptoms when a food allergy
to other foods, hidden allergens, spices or additives are suspected.
If there is no evident improvement of the symptoms after the diet
period, the role of food is practically excluded. In case of suspicion
of an enzymatic intolerance, appropriate laboratory tests must be
performed, as well as careful gastrointestinal examinations including
mucosal biopsies in the case of a chronic gastrointestinal disease.
4.5.4.2 Skin tests
Besides the case history, skin testing with various methods
(skin-prick tests with commercial glycerol extracts, prick-prick or
scratch tests with raw food or intracutaneous tests with aqueous
extracts) with a panel of routine or selected food is the normal
screening procedure for diagnosing food allergy (Metcalfe & Sampson,
1990; Sampson & Metcalfe, 1992). However, the allergen source is of
major importance and the standardization of commercial food extracts
is not optimal. When reviewing the literature on sensitivity and
specificity of skin-prick tests, as compared with the outcome of
double-blind placebo-controlled food challenge (DBPCFC), widely
variable results are found, depending on the allergen source
(commercial extracts) and the different food items (Sampson & Albergo,
1984; Atkins et al., 1985a,b; Pastorello et al., 1989). Using fresh or
raw food, e.g., raw milk, apple, celery, carrot, increases the
sensitivity of skin-prick tests (Ortolani et al., 1989). A typical
Cooking often destroys the reactivity (Wüthrich et al., 1990). Shared
epitopes and cross-reactive IgE antibodies are the most probable
explanation for the observed clinical symptoms in cases of association
between pollen allergy and food hypersensitivity (Calkhoven et al.,
1987).
4.5.2.2 Allergic reactions after ingestion of food
The main symptoms of gastrointestinal food allergy are vomiting,
nausea, diarrhoea and abdominal pains (colics or cramps) (Anderson,
1981; Atkins et al., 1985a,b). Skin reactions include local or
generalized pruritus, flush, urticaria, angioedema, morbilliform
exanthema and flare-up of atopic dermatitis. The symptoms of the upper
and the lower respiratory tract are rhinitis (sneezing, pruritus of
the nose, nasal stuffiness, nasal obstruction), larynx oedema, cough,
wheezing and bronchial asthma. Itching, redness and watering eyes
(conjunctivitis) is often associated with the above symptoms, but can
also appear as an isolated manifestation of food allergy.
Systemic anaphylaxis (cardiovascular collapse) always involves
other organ symptoms, e.g., of the gastrointestinal tract, the skin or
the respiratory tract. A particular subtype is food-dependent
exercise-induced anaphylaxis (Maulitz et al., 1979).
4.5.2.3 Allergic reactions following skin contact with food
Urticarial lesions can be provoked by contact with certain foods,
such as fish, shrimps, flour and pork meat (Maibach, 1976). Chronic
contact with food may induce protein contact dermatitis in food
handlers (Hjorth & Roed-Petersen, 1976).
4.5.3 Non-IgE-mediated immune reactions
Immune complexes with IgG antibodies and milk antigen inducing
complement-mediated damage have been suggested to induce Heiner's
syndrome and haemorrhagic gastroenteritis in childhood (Heiner et al.,
1962; Gryboski, 1967). Other non-IgE-mediated immune reactions to food
include gluten-sensitive enteropathy (coeliac disease), food-induced
colitis, and cutaneous allergic vasculitis (purpura).
4.5.3.1 Gluten-sensitive enteropathy (coeliac disease)
Gluten sensitive enteropathy or coeliac disease occurs in
susceptible individuals. It is characterized by damage to the small
intestinal mucosa with symptoms of malabsorption (Kagnoff, 1992). The
prevalence of coeliac disease has been estimated to be 0.2-0.5% with
considerable geographical variation (Logan, 1992).
Affected individuals develop specific immunological reactions to
gliadin, a protein that is a major alcohol-soluble fraction of gluten,
present in wheat, rye, barley and oat. Humoral cell-mediated immunity
and genetic factors seem to be involved in the pathogenesis.
The peak incidence of symptoms is in infancy after the
introduction of cereals, and a second peak occurs during the third
decade. In children it is often a semi-acute disease. The symptoms of
coeliac disease in children are recurrent abdominal pain, loose
stools, anorexia, short stature and delayed puberty. Symptoms of
malabsorption are unexplained nutritional deficiencies such as iron
and folic acid deficiency, anaemia and rickets. Dental enamel
hypoplasia and recurrent aphthae are also associated with coeliac
disease.
Patients with coeliac disease must avoid gliadins and related
proteins for life. As little as 100 mg gliadin has been described as
causing intestinal damage.
4.5.4 Diagnosis of adverse food reactions
4.5.4.1 Case history and elimination diet
The diagnosis of food allergy or food intolerance is the result
of a careful case history and clinical examination, and of several
in vitro and in vivo diagnostic procedures. A record of food
intake and its relation to clinical symptoms usually provides useful
information in the case of acute reactions, occurring from a few
minutes to a few hours after ingestion. History is, however, less
reliable when symptoms are chronic and caused by food that generally
is consumed daily, such as milk, egg, wheat and meat or by additives.
An elimination diet based on rice, potatoes and mineral water over a
week is useful in patients with chronic symptoms when a food allergy
to other foods, hidden allergens, spices or additives are suspected.
If there is no evident improvement of the symptoms after the diet
period, the role of food is practically excluded. In case of suspicion
of an enzymatic intolerance, appropriate laboratory tests must be
performed, as well as careful gastrointestinal examinations including
mucosal biopsies in the case of a chronic gastrointestinal disease.
4.5.4.2 Skin tests
Besides the case history, skin testing with various methods
(skin-prick tests with commercial glycerol extracts, prick-prick or
scratch tests with raw food or intracutaneous tests with aqueous
extracts) with a panel of routine or selected food is the normal
screening procedure for diagnosing food allergy (Metcalfe & Sampson,
1990; Sampson & Metcalfe, 1992). However, the allergen source is of
major importance and the standardization of commercial food extracts
is not optimal. When reviewing the literature on sensitivity and
specificity of skin-prick tests, as compared with the outcome of
double-blind placebo-controlled food challenge (DBPCFC), widely
variable results are found, depending on the allergen source
(commercial extracts) and the different food items (Sampson & Albergo,
1984; Atkins et al., 1985a,b; Pastorello et al., 1989). Using fresh or
raw food, e.g., raw milk, apple, celery, carrot, increases the
sensitivity of skin-prick tests (Ortolani et al., 1989). A typical
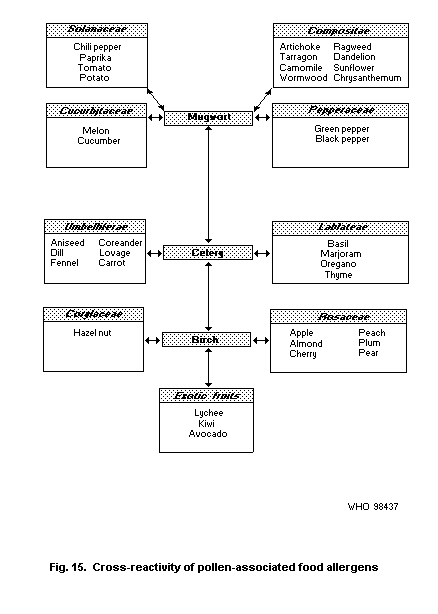 case history (e.g., oral allergy syndrome) or a severe,
life-threatening immediate Type I reaction after ingestion of a
defined food (e.g., apple or peanut), supported by a clear positive
specific skin test with the suspected food, establishes the diagnosis
(Metcalfe & Sampson, 1990). The demonstration of IgE antibodies
towards the culprit food underlines the IgE-mediated pathogenesis
(Johansson et al., 1984).
4.5.4.3 Specific serum IgE
The determination of food-specific serum IgE antibodies with
different techniques (e.g., RAST = Radio-Allergo-Sorbent-Test, ELISA =
Enzyme-Linked Immuno-Sorbent-Assay, FEIA = Fluoro-Enzyme Immuno-Assay)
has become a routine diagnostic tool in many Allergy Centres and among
practitioners (Johansson et al., 1984). However, a positive IgE
determination as well as a positive skin test do not mean actual food
allergy, but only a food sensitization. Cross-reactive IgE antibodies
to inhaled allergens (birch or mugwort pollen, cat dander, cow
epidermis) (Aalberse et al., 1981; Calkhoven et al., 1987) or to
botanically related food (e.g., legumes and peanut, soy and chickpea)
can be observed in negative challenge tests (Bernhisel-Broadbent &
Sampson 1989; Bernhisel-Broadbent et al., 1989). On the other hand,
some food allergic patients who show positive results in skin tests
have negative results in RAST. Like skin testing, the diagnostic value
of IgE determination is hampered by the lack of standardized food
extracts. Also the cut-off limit of IgE determination, i.e., the
minimal value in kU/litre or class, that should be considered
clinically relevant is still a matter of debate.
4.5.4.4 IgG determination
Specific IgG antibodies against food (Wüthrich, 1990) can be
found in many different physiological and pathological conditions.
Their determination does not prove the existence of a clinically
relevant immune reaction (Dannaeus & Inganäs, 1981).
4.5.4.5 Other in vitro tests
The Histamine Release Test (HRT) (Clinton et al., 1986) and the
Cellular Allergen Stimulation Test (CAST) (de Weck et al., 1993),
which determine the histamine or the sulfidoleucotrienes release from
basophil leucocytes in the peripheral blood, are of considerable
scientific interest but are too complicated and time-consuming for
daily routine use.
4.5.4.6 Oral challenge tests
At present, double-blind, placebo-controlled, food challenge
(DBPCFC) is considered as the "gold standard" for diagnosis of adverse
reactions to foods (Bernstein et al., 1982b; Bock et al., 1988).
Although several procedures for performing DBPCFC have been developed,
their application in normal clinical practice is hampered by their
heavy demands on resources and time.
The use of DBPCFC is necessary to assess objectively food
allergy/food intolerance, because several studies have demonstrated a
tremendous discrepancy between subjective perception of food
allergy/food intolerance and the results of DBPCFC. However, there are
difficulties in conducting studies of this nature in large population
samples. For example, challenge tests are dangerous with the risk of
severe anaphylactic reactions in the case of positive skin-prick tests
and/or positive serum IgE (RAST/CAP) against the offending food.
Moreover, there may be several potential hazards in the procedure and
in the interpretation of food challenges (Bindslev-Jensen et al.,
1994b), namely:
a) The nature of the food being tested (raw, freeze-dried,
cooked, food allergen extract).
b) The amount of food necessary to provoke objective signs.
c) DBPCFC may bypass important sites, e.g., mouth when using
capsules.
d) Additive or synergistic effect of multiple
hypersensitivities (e.g., concomitant sensitization to spices or
inhaled allergens such as pollen) or trigger factors (e.g.,
drugs, aspirin, alcohol, exercise, stress).
e) If the patient only reports subjective symptoms, e.g., itch,
headache, it may be necessary to increase the number of challenge
tests (three active and three placebo provocations) to avoid
false positive reactions and to obtain statistical significance
(Young et al., 1994).
In clinical practice it is often convenient to apply an open or a
single-blind food challenge: a negative test is of high predictive
value. A positive test, with the presence of objective signs (e.g.,
urticaria, asthma) or laboratory markers, increase of serum histamine,
tryptase or ECP (Eosinophil Cationic Protein) levels after challenge,
is usually sufficient to verify the diagnosis.
4.5.5 Therapeutic considerations
The only proven therapy for food allergy is to avoid the allergen
causing the disease. To be able to avoid, for instance, wheat flour,
milk or egg in the daily diet, professional advice is necessary both
to avoid hidden allergens in processed food and to ensure that the
diet is nutritionally adequate.
4.5.6 Prevalence
4.5.6.1 Introduction
It is possible to make more or less well-documented estimates of
the prevalence of different adverse reactions to foods. Frequency and
duration of breast feeding, eating habits, and flora (e.g., birch
trees) are factors influencing the prevalence. It is not known whether
the prevalence of food allergy is increasing. The prevalence of pollen
rhinitis is increasing (Wüthrich et al., 1995), and it is possible
that the prevalence of pollen-related food allergies may also be
rising.
4.5.6.2 Children
In many countries cow's milk is the first food allergen newborns
meet. The prevalence of cow's milk allergy is 2-5% in one-year-old
babies (Jakobsson & Lindberg, 1979; Bock, 1987; Host et al., 1988;
Hill & Hosking, 1997). This figure declines rapidly during the first
three years of life (Host & Halken, 1990).
The overall prevalence of food allergy is somewhat higher with a
maximum around one year. Bock (1987) followed a cohort of North
American children from birth to their third birthday; 7.7% had adverse
reactions to food not including fruit and fruit juices and 85% of the
adverse reactions were found during the first year of life. By the
third birthday less than 1% had adverse food reactions. The cumulative
prevalence of food allergy/intolerance diagnosed by elimination and
open challenge in a group of unselected Danish infants 18 months of
age was 6%. In the same study the prevalence in high-risk infants was
17% (Halken et al., 1992).
In a British study of a birth cohort of children age 4 years,
0.7% had allergy to peanuts or tree nuts and 0.5% had allergy to
peanuts alone. These figures were based on skin-prick tests and
positive history (Tariq et al., 1996).
The estimated prevalence of hypersensitivity reactions in
Australia to common foods in infancy and childhood is: cows's milk 2%,
egg 3.2%, and peanut 1.9%. Based on food challenge studies from
Australia, Japan, Indonesia, Malaysia and the Philippines, milk and
egg are the commonest food allergens. Soy, wheat and peanut
hypersensitivity are next commonest, but rice allergy is rare (Hill,
1997).
In a study of allergic children, Crespo et al. (1995) found that
allergy to cow's milk, egg and fish predominantly began before the
second year of age, whereas allergy to fruit, legumes and vegetables
predominantly began after the second year. This is in accordance with
the findings in a Danish study (Saval et al., 1993) where in older
children an increased prevalence of allergic pollen rhinitis was
followed by an increase in oral itch, the symptom characteristic for
allergic reactions to fruit and vegetables. In the 14-16 year old the
prevalence of oral itch was 2-2.9%.
4.5.6.3 Adults
a) IgE-mediated allergy
Allergic reaction to foods that cross-react with pollen is
probably the most common food allergic reaction in adults, at least in
countries where tree pollen allergy is common. In a study of Swedish
medical students, Foucard (1991) found that 78% of the students with
allergic rhinitis and positive skin test to birch pollen reported
clinical sensitivity to nuts and/or apples or other fruits. In another
Swedish study 70% of the patients with birch pollen allergy and 19% of
the patients with allergy to other pollen had food-related symptoms
(Eriksson et al., 1982). These estimates were mainly based on
self-reported symptoms.
In Switzerland approximately 10% (9.1-11.2%) of the adult
population currently suffers from hay fever. The prevalence of
sensitization measured by skin-prick test, but not necessarily
followed by clinical symptoms, is 12.7% for grass pollen and 7.9% for
birch pollen (Wüthrich et al., 1995). Other reports of the prevalence
of pollen rhinitis vary from 2-15% depending on, among other things,
the age group studied (Sibbald, 1993).
Applying Swedish figures for pollen-related reactions to the
Swiss data, taking into account the prick test results, approximately
50% of hay fever patients would have adverse reactions to
pollen-related foods, suggesting a prevalence of 3-5% in the adult
population. In a study by Kremser (1989) only about 10% of subjects
allergic to birch pollen had more severe reactions than oral itch.
Combining this with the Swiss figure gives a prevalence of
pollen-related reactions other than oral itch of approximately 0.5%.
In adults, the prevalence of allergy to non-pollen-related food
allergens such as milk, egg, shrimp, meat is probably about 0.1%. In a
study by Wüthrich (1993) of 402 patients with food allergy,
approximately 75% of the positive reactions were caused by
pollen-related food and 25% of the reactions were caused by
non-pollen-related food. Patients where the only food-related symptom
was itching of the mouth were not included in the study.
In some patients IgE allergy to natural rubber latex causes
allergic reactions to ingested banana, avocado, chestnut, etc. (Blanco
et al., 1994). The prevalence of natural rubber latex allergy in
European health-care workers screened with skin-prick tests has ranged
from 2.8 to 10.7%. About half of patients allergic to natural rubber
latex have experienced symptoms after eating banana (Turjanmaa et al.,
1996). In a study by Beezhold et al. (1996) 36% of a group of patients
allergic to natural rubber latex had clinical reactions to related
foods. The majority of these patients had anaphylaxis.
In the Netherlands an attempt has been made to estimate the total
prevalence of adverse reactions to foods and additives in adults. The
study used double-blind placebo-controlled food challenge (Niestijl
Jansen et al., 1994) and 12.4% of the studied population reported food
allergy or intolerance to specific food. Food groups mentioned most
frequently were fruit, chocolate and vegetables. The prevalence of
food allergy or intolerance in the adult Dutch population was
estimated to be 2.4%. If the positive reactions to food additives,
including menthol and glucose, is disregarded, the estimated
prevalence of food allergy/intolerance is 1.2%.
In a British study (Young et al., 1994) approximately 20% of a
survey population reported adverse reactions to foods. Eight foods
commonly perceived to cause sensitivity were canned or made into candy
bars and used for double-blind placebo-controlled challenge. The foods
were: cow's milk, hen's egg, wheat, soya, citrus fruits (orange),
fish/shellfish (prawn), nuts (peanut, brazil nut, walnut and hazel
nut) and chocolate. These eight foods accounted for 49.3% of reactions
reported in the study questionnaire. Adding some of the persons with
severe reactions and estimating the number of theoretically positive
in the non-challenged groups, the estimated prevalence of food
allergy/intolerance to the eight foods was 1.4-1.8%. As the eight
foods used for challenge accounted for approximately 50% of reported
reactions, the total prevalence estimate must be 3-4%.
b) Contact allergens
In dermally sensitized subjects, ingestion of the contact
allergen may cause skin flare reactions or other symptoms, for
instance, from the gastrointestinal tract. It has been reported that
10% of Danish women have contact allergy caused by nickel (Menné &
Holm, 1983). Up to 10% of these may benefit from a nickel-restricted
diet (Veien et al., 1993). The prevalence of systemic reactions via
the food from other contact allergens is not known. The chemicals of
fragrances and of food flavours, natural or synthetic, are often
identical. Veien et al. (1987) found an effect of a diet low in
flavours in patients having a flare of dermatitis after oral challenge
with balsam of Peru.
4.5.6.4 Conclusions
Milk and egg seems to be the most common food allergens in
infants and children worldwide. Peanuts are also reported to be common
food allergens in the United Kingdom, USA, Australia and some Asian
countries. The overall prevalence of food allergy/intolerance in young
children may be around ó-8%. This figure declines before 3 years of
age. In older children and young adults the prevalence of allergy to
pollen-related fruits, nuts and vegetables increases.
The results from the epidemiological studies, combined with the
knowledge on pollen and latex cross-reactions, systemic reactions to
contact allergens, and coeliac disease (prevalence estimate 0.2-0.5%),
point to an estimated prevalence of food allergy in the adult
population of 3-5%, giving that some of the reactions may be
coincident. These figures are based on the existing prevalence data,
which are mainly European. It is not yet known whether prevalence data
in the rest of the world are comparable.
4.6 Autoimmune diseases associated with drugs, chemicals and
environmental factors
4.6.1 Introduction
The autoimmune connective tissue diseases include conditions such
as systemic lupus erythematosus (SLE), systemic sclerosis, Sjögren's
syndrome, rheumatoid arthritis and the systemic vasculitides such as
Wegener's granulomatosis, Churg Strauss syndrome and polyarteritis
nodosa. The etiology of these conditions remains largely unknown but
there is a consensus that several factors may be important, including
genetic, ethnic and hormonal; many of these conditions have a female
preponderance (Table 23). Some of these conditions may also have an
environmental component to their pathogenesis and this particularly
applies to systemic sclerosis. Many drugs have been associated with
the development of SLE and cutaneous vasculitis. There has been some
interest in the possible development of connective tissue diseases
associated with silicone breast implants.
4.6.2 Systemic lupus erythematosus
SLE is predominantly a disease of young women (9:1 female: male
ratio) and is commoner amongst certain ethnic groups such as
Afro-Caribbeans and Orientals (Beeson 1994). Clinically, the disease
manifestations include arthritis, serositis, photosensitivity, oral
ulceration, malar rashes, recurrent thromboses, glomerulonephritis and
central nervous system involvement, e.g., epilepsy, psychoses.
Serologically, SLE is characterized by autoantibody production to
nuclear components such as anti-nuclear antibodies, antibodies to double
stranded DNA and antibodies to extractable nuclear antigens.
Table 23. Age and sex associations of some autoimmune diseases
(adapted from Beeson, 1994)
Age % Females
1. Autoimmune diseases that may
appear during childhood
Systemic lupus erythematosus 2-12 70
Dermatomyositis 1-15 70
Table 23. (continued)
Age % Females
Sydenham's chorea 5-15 65
Rheumatoid arthritis 1-10 65
Rheumatic fever 5-15 50
Thrombocytopenic purpura 2-5 50
Polyglandular syndromes 2-15 50
Bullous pemphigoid 1-15 50
Diabetes mellitus 2-15 45
Henoch-Schonlein purpura 2-5 40
Post-streptococcal nephritis 5-15 35
2. Autoimmune diseases that usually
appear during early adult life
Systemic lupus erythematosus 15-40 90
Erythema nodosum 15-30 90
Takayasu's arteritis 10-30 85
Myasthenia gravis 20-30 75
Thrombocytopenic purpura 15-45 75
Addison's disease 20-50 75
Rheumatoid arthritis 20-40 65
Multiple sclerosis 20-35 60
Sarcoidosis 20-40 55
Ulcerative colitis 15-40 50
Erythema multiforme 20-40 45
IgA nephropathy 10-30 35
Polyarteritis nodosa 20-50 30
Ankylosing spondylitis 20-30 25
Goodpasture's syndrome 15-35 15
Ankylosing spondylitis 20-30 25
Goodpasture's syndrome 15-35 15
Thromboangiitis obliterans 20-40 5
3. Autoimmune diseases that usually appear during mature adult life
Sjogren's syndrome 40-60 95
Primary biliary cirrhosis 30-75 90
Hashimoto's thyroiditis 30-50 85
Thyrotoxicosis 30-50 85
Scleroderma 30-50 80
Chronic active hepatitis 30-50 65
Polymyositis/dermatomyositis 40-60 55
Polychondritis 40-60 50
Pemphigus vulgaris 50-80 50
Wegener's granulomatosis 10-80 50
Henoch-Schonlein purpura 30-65 45
Table 23. (continued)
Age % Females
Membranous nephropathy 30-60 35
Amyotrophic lateral sclerosis 40-70 35
Tabes dorsalis 30-50 20
4. Autoimmune diseases that usually
appear late in life
Giant cell arteritis/polymyalgia 50-90 65
Pernicious anemia 40-80 60
Bullous pemphigoid 60-75 50
Rapidly progressive glomerulonephritis 50-70 45
Myasthenia gravis 50-80 40
Fibrosing alveolitis 40-70 35
The precise etiology of SLE remains obscure but some environmental
factors are known to exacerbate the disease. One of the most important
of these is ultraviolet light. In particular UV-B in the 295 to 305 nm
range is known to be toxic to SLE patients (McGrath et al., 1994).
Furthermore, patients with antibodies to Ro (SSA) are sensitive to UV
light, which provokes a photosensitive skin rash and may be followed by
a generalized disease flare (Gilliam & Sontheimer, 1982). Being in a
lower socioeconomic group may also increase the risk for the development
of glomerulonephritis in patients with SLE (McAlindon et al., 1993),
although the reasons for this remain obscure.
Many patients with SLE are allergic to a variety of substances
including drugs and chemicals, and a case-control study has supported
this clinical observation (Sequeira et al., 1993). Patients with lupus
and particularly those with Sjögren's syndrome appear to be sensitive to
cotrimoxazole and other sulfonamide-containing drugs. Oral contraceptive
pills, especially those with high oestrogen doses, may provoke flares of
SLE, and these agents should generally be avoided (Jungers et al.,
1982). The progesterone-only pill is associated with fewer flares.
Table 24. Drugs associated with the development of
systemic lupus erythematosus
Drugs commonly inducing systemic lupus erythematosus
Debrisoquine
Hydralazine
Quinidine
Procainamide
Drugs with good evidence for inducing systemic lupus erythematosus
Carbamazepine
Chlorpromazine
Hydrazine
Isoniazid
Minocycline
Methyldopa
Penicillamine
Phenytoin
The majority of cases of SLE are idiopathic but certain drugs are
known to cause SLE in genetically predisposed individuals. Of the 70 or
so drugs reported to be associated with drug-induced lupus, some of
which are shown in Table 24, only procainamide and hydralazine have been
studied in detail. Clinically, drug-induced lupus is similar to
idiopathic lupus, but there are one or two striking differences. For
example, serositis and pleuro-pulmonary involvement is much commoner in
drug-induced lupus, whereas renal disease and central nervous system
disease is less common in comparison to idiopathic lupus (Yung et al.,
1995). Serologically, anti-nuclear antibodies and antibodies to both
single-stranded and double-stranded DNA are found in both drug-induced
and idiopathic lupus, but it is uncommon to find very high levels of
anti-DNA antibodies in drug-induced lupus. Antibodies directed against
certain components of histone are thought to be characteristic of
drug-induced lupus. Although anti-histone antibodies may commonly be
found in idiopathic lupus, they react to the H1 and H2B subunits of
histone. In drug-induced lupus, the specificity appears to be against
the H2A-H2B dimer in procainamide-induced lupus, or against H3 and H4 in
hydralazine-induced lupus (Burlingame & Rubin, 1991).
Idiopathic lupus erythematosus is associated with HLA DR2 or DR3,
at least in Caucasian patients. One of the best clues towards an
explanation for a genetic predisposition to drug-induced lupus comes
from the study of the acetylator status of these patients. "Slow
acetylators" are homozygous for this gene and have low levels of
N-acetyltransferase in the liver. Both procainamide and hydralazine
are metabolized by this pathway and slow acetylators have a higher risk
of developing drug-induced lupus following exposure to these drugs
(Strandberg et al., 1976; Sonnhag et al., 1979).
The exact mechanisms by which drugs can induce lupus remain unknown
and a number of possibilities are being considered. It may be that the
ability of both procainamide and hydralazine to bind polynucleotides
in vitro may render DNA and/or histones antigenic (Dubroff & Reid,
1980; Tomura & van Lancker, 1988). A more specific mechanism has been
suggested whereby drugs interfere with the normal process of methylation
of DNA. Following DNA replication, cytosine residues are methylated at
the 5-position by the enzyme DNA methyltransferase. Failure of
methylation of regulatory sequences is associated with gene expression,
whereas methylation is associated with suppression of gene
transcription. Thus, DNA methylation is a mechanism regulating gene
expression (Yung et al., 1995). Studies have shown that
procainamide- and hydralazine-treated human T-cells show evidence of
hypomethylation (Scheinbart et al., 1991). In particular, procainamide
is capable of reversibly inhibiting T-cell DNA methyltransferase in a
dose-dependent manner (Scheinbart et al., 1991). Furthermore, these
T-cells become autoreactive in response to procainamide and hydralazine
(Yung et al., 1995). These autoreactive T-cells are capable of inducing
B-cells to differentiate into IgG-secreting cells without the addition
of antigen or mitogen, thus providing a mechanism for polyclonal B-cell
activation that is seen in both idiopathic and procainamide-induced
lupus (Richardson et al., 1990).
Experiments have shown that UV light is also capable of inhibiting
T-cell DNA methylation, increasing LFA-1 expression and inducing
autoreactivity (Richardson et al., 1994). Thus, drugs such as
procainamide and hydralazine and environmental factors such as UV light
may induce T-cell autoreactivity by inhibiting T-cell DNA
methyltransferase, thereby altering T-cell gene expression and making
autoimmunity more likely.
4.6.3 Scleroderma: environmental and drug exposure
Scleroderma (progressive systemic sclerosis) is a multisystem
connective tissue disease of unknown etiology. The prevalence ranges
from 47.9 to 290 per million among females, with an estimated incidence
of between 3.6 and 16 per million per year (Silman et al., 1996). It is
commoner in women (the sex ratio ranges from 3:1 to 8:1) and some ethnic
minorities such as Afro-Caribbeans and is characterized by widespread,
diffuse sclerosis affecting the peripheral vasculature, skin,
gastrointestinal tract, heart and muscle. Raynaud's phenomenon is a very
common early feature, and pulmonary and renal involvement may be serious
and life-threatening. Serologically, antibodies to topoisomerase I
(anti-Scl 70) and RNA polymerase III may predict prognosis in terms of
more extensive disease.
Scleroderma-like conditions (pseudoscleroderma) and scleroderma
have been associated with a variety of chemical and environmental agents
(Silman & Hochberg, 1996) (Table 25).
4.6.4 Silicone breast implants
Two studies have reported possible interactions between silicone
breast implants and the immune system. Tenenbaum et al. (1997) showed
the existence of a relationship between the level of anti-polymer
antibodies in the serum and the severity of clinical complications
(related to connective tissue) in silicone breast implant recipients.
Although these antibodies were not directed against silicone polymers
this might be the first objective marker to be used as a diagnostic
feature in silicone breast implant patients. Smalley et al. (1995, 1997)
reported on a lymphocytic response to silica (silicon dioxide) similar
to that for silicone (polysiloxane polymer), a silicon derivative, and
present in silicone breast implant material, in silicone breast implant
patients and their children.
Table 25. Some agents associated with scleroderma
Organic chemicals:
Toluene
Benzene
Xylene
Aromatic mixes e.g., white spirit
Vinyl chloride
Trichloroethylene
Perchloroethylene
Naphtha-n-hexane
Epoxy resins
Metaphenylenediamine
Urea formaldehyde foam insulation
Drugs:
Bleomycin
Carbidopa
L-5-hydroxytryptophan
Table 25. (continued)
Drugs:
Pentazocine
Cocaine
Diethyl proprion
Fenfluramine HCl
However, several thorough reviews of the subject, including a
case-control study (Sanchez-Guerrero et al., 1994, 1995; Gabriel et
al. 1994; Hochberg et al., 1996), concluded that there was little or
no association between silicone breast implants and either connective
tissue diseases or a unique arthralgia/myalgia/fibromyalgia syndrome.
They suggested that the connective tissue diseases that had been
reported were most likely to have been idiopathic in origin.
In a large retrospective cohort study, the Women's Health Study
sent questionnaires to 395 543 women and found 10 830 women who
reported breast implants (Hennekens et al., 1996). This study showed a
small but statistically significant increased risk of the combined
end-point of any connective tissue disease in patients with breast
implants, with a relative risk of 1.24 (95% confidence intervals
1.08-1.41, P = 0.0015). Of the specific connective tissue diseases
studied, scleroderma was associated with a relative risk of 1.89 (95%
confidence intervals 0.98-3.45) but this was not significant
( P = 0.06). For patients with scleroderma with implants less than 4
years, the relative risk was significant: 2.68 (95% confidence
intervals 1.11-6.51, P = 0.029) The precise nature of the implants
was not ascertained so it is not clear how many of the implants were
silicone gel filled. Another possibility for bias is the publicity
surrounding breast implants, which may have led to a degree of
over-reporting amongst the cases.
Other studies have supported the epidemiological view that there
is no association between connective tissue disease and silicone
breast implants (Nyren et al., 1998; Edworthy et al., 1998). Both were
very large cohort studies that compared patients who had either
undergone breast reduction (Nyren et al., 1998) or non-implant
cosmetic surgery (Edworthy et al., 1998). Neither found an excess of
any connective tissue disease among patients with silicone implants. A
United Kingdom review (DOH, 1998) came to the same conclusion.
Overall, apart from one retrospective study, there is no clear
evidence that patients with silicone breast implants have any risk of
developing a connective tissue disease but the area remains
controversial.
4.6.5 Toxic oil syndrome
This epidemic started in May 1981 when large numbers of patients
in the Madrid industrial area suffered acute respiratory illnesses
that did not respond to antibiotics. The etiological agent was
identified as being rapeseed oil that had been denatured with 2%
aniline for industrial use but sold illegally by itinerant vendors for
domestic food use. Initially 20 688 cases were recorded with 835
deaths -- a cumulative mortality of 4%, but over the first 2 years of
the epidemic the cumulative mortality was 2.3%. Oleyl-anilide proved
to be an excellent marker for case-related oil specimens although the
precise nature of the etiological agent has never been described. It
has proved impossible to consistently replicate the syndrome in animal
studies.
Various clinical features have been described (Kammüller et al.,
1984; Philen & Posada, 1993) including severe myalgia, arthralgia,
Raynaud's phenomenon, scleroderma-like skin changes, carpal tunnel
syndrome, joint contracture, pulmonary hypertension and peripheral
neuropathy. The most consistent laboratory features were eosinophilia
and high IgE counts and low levels of antibodies such as antinuclear
antibodies. HLA DR3-DR4 was associated with the chronic phase of the
disease. Histologically, the most consistent finding was a vascular
lesion characterized by intimal proliferation with fibrosis, vascular
occlusion and thrombosis.
4.6.6 Eosinophilia-myalgia syndrome
This epidemic was first identified in October 1989 in New Mexico
(Hertzman et al., 1990). The report was followed by a case-control
study that firmly linked the consumption of possibly contaminated
L-tryptophan from one source with the eosinophilia-myalgia syndrome
(Eidson et al., 1990). L-tryptophan was widely used as a
non-prescription food supplement by health conscious individuals for a
wide variety of minor ailments. The source of the epidemic was traced
to a single manufacturer (Philen & Posada, 1993).
The vast majority of cases occurred in the USA. The majority of
these patients were white middle-class middle-aged females, probably
reflecting the pattern of use of health supplements rather than any
innate risk factor. The mortality rate was reported as 2.7% (37 deaths
among 1370 known cases) (Philen & Posada, 1993).
The clinical features resembled those of toxic oil syndrome,
although scleroderma-like changes and Raynaud's phenemenon were not
reported (Kaufman et al., 1991; Philen & Posada, 1993). These authors
also noted that HLA DR4 was associated with an increased risk of
chronic disease. Laboratory investigations consistently showed an
eosinophilia early in the disease course, although this diminished
spontaneously even when the patients continued to be ill (Philen &
Posada, 1993). The main factor in treatment was the avoidance of
further ingestion of L-tryptophan. Glucocorticoids were used widely
and helped the myalgias and reduced the eosinophil count, but there
was often a recurrence of symptoms on stopping the steroid treatment,
and progression to chronic disease was not altered.
4.6.7 Vinyl chloride disease (occupational acro-osteolysis)
Vinyl chloride (CH2=CHCl) is a combustible colourless gas at
room temperature that is used in the manufacture of a variety of
plastics. Several methods can be used to polymerize the gas to make
polyvinyl chloride (PVC). In the mid-1960s a new syndrome affecting
workers involved in polymerizing vinyl chloride was recognised (Wilson
et al., 1967). These patients developed paraesthesia of the fingers,
cold sensitivity, Raynaud's phenomenon, pseudoclubbing of the fingers,
skin oedema and thickening of the fingers, hands and forearms, and
chest X-ray changes (Veltman et al., 1975). The risk of development of
symptoms was related to cumulative exposures over time and work
practices but was not related to handling the finished PVC product,
and in Wilson's (1967) study it occurred in less than 3% of exposed
individuals.
An increased prevalence of HLA DR3 and DR3/B8 haplotypes has been
noted in patients with vinyl chloride disease (Black et al., 1983).
Vinyl chloride is a cause of non-cirrhotic portal hypertension and
angiosarcoma of the liver.
The skin changes of vinyl chloride disease resemble morphoea
clinically and histologically, and vascular changes were often present
with luminal narrowing of the digital arteries and subtotal occlusion
of these vessels (Veltman et al., 1975). The most dramatic
radiological change is acro-osteolysis seen in the terminal phalanges
of the fingers; a transverse lytic band is seen across the distal
phalangeal shaft. Ward et al. (1976) reported immunological
abnormalities in vinyl chloride disease including polyclonal increases
of IgG, cryoglobulins, evidence of complement activation and low titre
anti-nuclear antibodies. Vascular endothelial, medial and sub-intimal
deposits of IgG, C3, C4 and fibrin/fibrinogen were seen on histology
of small and medium-sized arterioles. Reduced T-cell and modestly
increased B-cell numbers were also observed (Ward et al., 1976).
4.6.8 Systemic vasculitis: environmental factors and drugs
Various drugs are associated with hypersensitivity reactions and
the most common mechanism is an immune-complex-mediated vasculitis
(Dubost et al., 1991). Drugs account for approximately 10-20% of
dermal vasculitis and this figure is on the increase. The cutaneous
lesions most commonly seen include palpable purpura, although
urticarial lesions may be seen in 10% (Dubost et al., 1991). They
usually occur symmetrically on the lower limbs extending to the thighs
and buttocks (Mullick et al., 1979). In Mullick's series of 30
patients, 19 had disseminated vasculitis with other organ involvement,
including renal disease, synovitis, pleuropulmonary and cardiovascular
disease with coronary vessel vasculitis and cardiac failure. More than
80% of patients had constitutional features such as fatigue, malaise
and fever.
4.6.9 Conclusion
The vast majority of the autoimmune connective tissue diseases
have no known etiological agents. Certain drugs, occupational
exposures, and UV radiation have been shown either to exacerbate a
known autoimmune disease or, occasionally, to trigger the onset of a
syndrome that closely resembles one of the established diseases.
5. EPIDEMIOLOGY OF ASTHMA AND ALLERGIC DISEASE
5.1 Introduction
Asthma and allergies, such as atopic diseases (i.e., bronchial
asthma, allergic rhinitis, atopic dermatitis) and allergic contact
dermatitis, are common medical problems.
Asthma, allergic rhinitis and atopic dermatitis are conditions
that have a variety of clinical similarities and epidemiological
connections (Montgomery-Smith, 1983). Asthma has frequently been
classified as of allergic or non-allergic origin. Different atopic
conditions often occur together in the same person. A family history
of atopic disorders has consistently been found to be an important
predisposing factor for the development of atopic diseases (Croner &
Kjellman, 1990). However, epidemiological studies suggest that
exogenous factors also play an important role in their etiology
(Newman Taylor, 1995). There have been epidemiological studies on
geographical variation and time trends in prevalence rates of these
disorders, and knowledge about the effects of a variety of
environmental factors, including outdoor exposures, indoor exposures,
diet and occupational exposures, is increasing.
5.2 Definition and measurement of allergic disease
5.2.1 Asthma
5.2.1.1 Definition
Asthma is a respiratory disease that is not well defined. It is
characterized by variable airflow limitation due to bronchial
hyper-responsiveness and often by inflammatory changes in the airways
(see section 2.5.1)
5.2.1.2 Assessment
a) Questionnaires
In epidemiological studies many different questionnaires have
been used to investigate in populations the prevalence of asthma
symptoms such as wheezing and shortness of breath, as well as
diagnosed asthma. These include the MRC (United Kingdom Medical
Research Council), the ECSC (European Coal and Steel Community), the
ATS-DLD (American Thoracic Society and the Division of Lung Disease),
and the IUATLD (International Union against Tuberculosis and Lung
Disease) questionnaires (Toren et al., 1993).
When questionnaires are used in epidemiological studies to
compare prevalence rates between populations, findings may be
influenced by differences in language, interpretations of the concept
of wheeze in different communities, or the general awareness of the
disease in the community (Strachan et al., 1990). Nevertheless, the
IUATLD questionnaire has been shown to provide valid and comparable
data even when translated (Burney et al., 1989a). A similar written
questionnaire was developed for the International Study of Asthma and
Allergies in Childhood (ISAAC) (Pearce et al., 1993; Asher et al.,
1995; Shaw et al., 1995). In addition, a video questionnaire depicting
(in five scenes) adolescents with different symptoms of asthma has
been developed to overcome problems with the translation of
questionnaires (Asher et al., 1995). The sensitivity and specificity
for predicting bronchial hyperresponsiveness of the video
questionnaire was similar to the respective questions in the IUATLD
questionnaire (Shaw et al., 1992a,b; Shaw et al., 1995). By providing
data relatively free from biases due to language, culture, literacy or
interviewing techniques, the video questionnaire may prove
particularly useful for comparisons of prevalence and severity of
asthma in different populations (Asher et al., 1995).
b) Bronchial hyperresponsiveness
In epidemiological settings, a major limitation of using
bronchial hyperresponsiveness (BHR), measured by challenge, as a gold
standard for the definition of asthma is that a considerable
proportion of subjects with bronchial hyperresponsiveness report no
respiratory symptoms (Sterk et al., 1993). Thus, bronchial
hyperresponsiveness cannot be used synonymously as a diagnosis of
asthma (Sterk et al., 1993). The thresholds used to define bronchial
hyperresponsiveness -- usually a fall in the forced expiratory volume
in one second (FEV1) of more than 15% or 20%, or a fall in peak
expiratory flow rate (PEF) of more than 10% -- were chosen
arbitrarily. Various methods including exercise tests and inhalation
of metacholine, histamine or cold air have been described to measure
bronchial hyperresponsiveness (Sterk et al., 1993). It is now well
recognized that a change in osmolarity of the periciliary fluid is a
potent stimulus to airway narrowing and may be a common cause for
provoking an attack of asthma (Anderson & Smith, 1991). This has led
to the use of hypertonic saline aerosols to document bronchial
hyperresponsiveness in epidemiological studies (Riedler et al., 1994).
However, a negative response to any of these provocation methods does
not exclude asthma. In general, measuring bronchial
hyperresponsiveness in epidemiological studies has a moderate
specificity and a relatively low positive predictive value (Sterk et
al., 1993).
5.2.2 Rhinitis
There are no clear-cut criteria for defining hay fever and
perennial rhinitis. Rhinitis is frequently underdiagnosed and
misdiagnosed (Sibbald & Rink, 1991a,b). Patients are generally
classified according to the suspected etiology of their conditions.
Rhinitis is labelled "allergic" when a causal allergen can be
identified, otherwise it is labelled "non-allergic". Subjects with
seasonal symptoms are twice as likely to be labelled as having
allergic rhinitis by their doctors. The usual complaints of allergic
rhinitis are "summer flu", series of sneezing and a stuffy nose. The
classification is based primarily on nasal smears and skin-prick tests
compared with the patient's history (Weeke, 1987). Perennial allergic
rhinitis in patients who are allergic to pets is easy to diagnose if
symptoms such as sneezing and itchy eyes occur immediately after
contact with pets. If the symptoms are caused, for instance, by mites,
it is more difficult to obtain a clear-cut medical history. Symptoms
of patients with non-allergic rhinitis are often different from those
with allergic causes. The main symptoms in non-allergic cases are
stuffy nose and loss of sense of smell, while in the allergic type,
sneezing and watery secretion are more prominent. A standardized
questionnaire for assessing the prevalence of rhinitis in children in
epidemiological studies has been developed (Asher et al., 1995).
5.2.3 Atopic dermatitis
5.2.3.1 Definition
Atopic dermatitis is a disease that is difficult to define,
because of the frequently subtle clinical manifestations, the lack of
an identifying laboratory marker, and the lack of a distinguishing
primary lesion. Generally, atopic dermatitis is a chronic cutaneous
inflammatory disease with a strong tendency of patients to overproduce
IgE (Hanifin, 1987).
5.2.3.2 Assessment
The absence of diagnostic criteria for atopic dermatitis prompted
Hanifin & Lobitz, (1977) and Hanifin & Rajka (1980) to suggest major
and minor diagnostic criteria for atopic dermatitis based on clinical
features. Disadvantages of these criteria were that many of them had
no precise definition, that some were very infrequent, and that others
were nonspecific. A minimum set of diagnostic criteria for atopic
dermatitis was derived by the United Kingdom working party composed of
dermatologists, family practitioners, paediatricians and
epidemiologists (Williams et al., 1994a). These criteria included a
history of flexural involvement, a history of dry skin, the onset
under the age of 2 years, a personal history of asthma, a history of
pruritic skin condition, and visible flexural dermatitis. The criteria
were used for assessing the prevalence of atopic dermatitis in
children in an international multiple cross-sectional study (Asher et
al., 1995). The European Task Force (ETFAD, 1993) on atopic dermatitis
has developed a scoring index for the severity of atopic dermatitis,
which is based on a combination of symptoms such as erythema,
oedema/papulation, oozing/crusts, excoriation and lichenification.
Nevertheless, more objective tests, such as raised IgE and/or
skin-prick test positivity, remain important tools for epidemiological
research.
5.2.4 Skin-prick test and serum IgE
Skin-prick testing is commonly used to assess allergic
sensitization in epidemiological studies. The test is easy to perform;
serious local or systemic reactions occur only very rarely, and
different test devices are available. However, results of different
studies are often not comparable due to differences in allergen
concentrations, type of needles used for pricking, and criteria for
defining a positive reaction (Nelson et al., 1993). The most common
criterion is that the diameter of the allergen wheal be >2-3 mm after
subtraction of the reaction to the negative control (Pepys, 1994).
Even well-standardized skin-prick tests may be subject to measurement
error arising from different field workers and variations in the
degree of skin reactivity in different racial groups or under
different environmental conditions. Measurements of total and specific
serum IgE may therefore provide valuable additional information on the
atopic susceptibility and of the atopic status of an individual. Total
serum IgE, however, may also be raised in association with other
conditions, such as parasitic infections.
5.2.5 Allergic contact dermatitis
Allergic contact dermatitis is the condition in which contact
with haptens induces cell-mediated contact sensitization.
Patch testing is used to diagnose allergic contact dermatitis.
The test should be performed by a skilled operator using standardized
test materials and also with substances present in the patient's
domestic or occupational environment which are considered to be
possible sensitizers.
5.3 Asthma and atopy: prevalence rates and time trends in prevalence
rates
Asthma is the most common single chronic disease in childhood.
Most of the prevalence studies on asthma have therefore been conducted
in children and adolescents. Many of these studies have also
determined the prevalence of rhinitis and atopic dermatitis. In the
last 20 years prevalence estimates have been reported from many
different geographical regions in all five continents. Several studies
were repeated after a number of years applying the same methods at
different points in time, thereby providing information on time trends
in prevalence. Estimates on asthma prevalence were mostly derived by
questionnaires, often in combination with a lung function test and
determination of bronchial hyperresponsiveness. In addition to
questionnaire data, prevalence rates of atopic sensitization were
assessed by skin-prick tests and, to a lesser extent, by determination
of total or specific IgE. The overview given below is not intended to
be comprehensive, but will give some insight into the geographical
variation and recent time trends. The comparability of prevalence
estimates between study centres, however, is limited as most studies
applied different methods.
5.3.1 Europe
5.3.1.1 Prevalences
Between 1989 and 1992 a parental questionnaire, a skin-prick test
and a cold air challenge test were administered to 9- to 11-year-old
children in western and eastern Germany. Atopic sensitization was more
frequent in western German children living in Munich (5.9%), compared
to children living in Leipzig and Halle in eastern Germany (3.9%).
Bronchial hyperresponsiveness was also more prevalent in western
Germany (8.3%) than in eastern Germany (5.5%) (von Mutius et al.,
1994b). Hay fever and rhinitis were reported less often in Leipzig
than in Munich (2.4% and 16.6% compared to 8.6% and 19.7%), whereas
bronchitis was more prevalent in Leipzig. In contrast to atopic
respiratory disease, the prevalences for atopic eczema were similar in
the two study areas (von Mutius et al., 1992).
The occurrence of allergic diseases was studied during 1979 and
1980 on the basis of a questionnaire sent to the parents of 20 000
children 7, 10 and 14 years of age in three parts of Sweden with
different climatic conditions (Aberg et al., 1989). The prevalence of
asthma was significantly higher in the northern part of the country,
and this higher prevalence could not be explained by other factors
than by the cold and dry climate. In Viborg, Denmark, the frequency of
rhinitis was 10.5%, of atopic eczema 7%, of urticaria 3.2%, and of
asthma 4.5% among 5- to 16-year-old school children studied in 1990
(Saval et al., 1993). Asthma and rhinitis were more frequent among
boys, while atopic eczema was more frequent among girls. For both
sexes, the frequency of rhinitis increased during their years at
school, while the frequency of skin symptoms decreased.
In a general practice in London, symptoms, atopic state and
medical history were compared among 16- to 65-year-old patients with
seasonal and perennial rhinitis (Sibbald & Rink, 1991b). The
prevalence of rhinitis was 24%, 3% had seasonal symptoms only and 13%
had perennial symptoms only. Distinguishing between atopic and
non-atopic subjects by skin-prick testing with five common allergens
revealed that subjects with seasonal rhinitis were more likely to be
atopic. Moreover, subjects with seasonal rhinitis were also more
likely to have eczema and to have a family history of hay fever.
Rates of reported eczema during early childhood have been studied
by health visitor interview in three national cohorts of children born
in England, Scotland and Wales in 1946 (at age 6 years), 1958 (at age
7 years), 1970 (at age 5 years) (Taylor et al., 1984). The reported
rates in the three birth cohorts were 5.1%, 7.3% and 12.2%.
5.3.1.2 Time trends
From 1926 to 1961 the prevalence of asthma in Finnish men
registered through the defence forces statistics ranged between 0.02
and 0.08%, whereas from 1961 to 1989 the prevalence rose from 0.29 to
1.79% (Haahtela et al., 1990).
In the United Kingdom several studies were conducted in different
epidemiological settings to estimate the prevalence and time trends in
the prevalence of allergic disorders. The different settings comprised
patients of general practices (Fleming & Crombie, 1987; Sibbald &
Rink, 1991a,b), national birth cohorts (Taylor et al., 1984),
representative samples of school children in England (Burney et al.,
1990), in South Wales (Burr et al., 1989), in the London borough of
Croydon (Anderson et al., 1983; Anderson et al., 1994), in Aberdeen
(Ninan & Russell, 1992), in the South Thames region (Barbee et al.,
1987), and hospital admission statistics (Strachan & Anderson, 1992;
Anderson, 1989). The latter showed between 1978 and 1985 an increase
in the number of hospital admissions because of asthma in infants up
to 4 years old (186%) and in 5- to 16-year-olds (56%) in the South
West Thames region (Anderson, 1989). Changes in mode of referral,
severity on admission and readmission ratio were also explored, but
little evidence was found for a reduction in severity or change in
readmission rate since 1978. These findings contrast with findings of
two identical surveys that were conducted in 1978 and 1991 to explore
prevalence changes and use of medical services (Strachan & Anderson,
1992). These data showed a substantial increase in self-referral
together with an increase in readmission. A comparison of two surveys
of morbidity carried out in 1970-1971 and 1981-1982 in general
practices in England and Wales showed an increase in prevalence rates
of asthma in men from 8.8 to 15.9% (Fleming & Crombie, 1987).
Between 1973 and 1989 the lifetime prevalence of wheezing among
12-year-old children in South Wales increased from 17 to 22%, a
history of asthma rose from 6 to 12%, and current asthma from 4 to 9%
(Burr et al., 1989). Increases occurred also in the frequency of
history of eczema (5 to 16%) and of hay fever (9 to 15%), while
wheezing not attributable to asthma remained constant. Increasing
prevalences of asthma were also observed between 1973 and 1986 in
English school children (relative increase in boys of 6.9% and in
girls of 12.8%), which could not simply be explained by changes in
diagnostic fashion (Burney et al., 1990).
In Aberdeen, Scotland, the prevalence of wheeze among 8- to
13-year-old school children rose from 10.4% in 1964 to 19.8% in 1989,
and shortness of breath rose from 5.4 to 10.0%. Wheeze and shortness
of breath were more prevalent in boys than in girls. Asthma rose from
4.1 to 10.2%, hay fever from 3.2 to 11.9% and eczema from 5.3 to 12%
(Ninan & Russell, 1992). Between 1978 and 1991 significant relative
increases in the 12-month prevalence rates of attacks of wheezing and
of asthma were found in the London borough of Croydon (Anderson et
al., 1994). Results from a repeated cross-sectional study performed in
1991-1992 and 1995-1996 in 10-year-old children in Leipzig, Germany,
showed a rise in hay fever and atopic sensitization. However, no
increase in the prevalence of asthma or bronchial hyperresponsiveness
was observed over the 4-year period (von Mutius et al., 1998).
5.3.2 Oceania
5.3.2.1 Prevalences
Several cross-sectional surveys were performed in Australian and
New Zealand school children to assess the prevalence of respiratory
symptoms and bronchial hyperresponsiveness. A comparison of 769
children living in Wagga Wagga (inland Australia), and 718 children
living in Belmont (coastal Australia) carried out in 1982, showed that
respiratory symptoms, asthma, bronchial hyperresponsiveness, hay fever
and atopy were all more common in the dry inland area than in the
humid coastal area. In both areas 38% of the children were atopic
(Britton et al., 1986).
The prevalence of wheeze in Melbourne school children was 23.1%
for 7-year-olds, 21.7% for 12-year-olds, and 18.6% for 15-year-olds.
History of wheeze was more common for boys than for girls at age 7
years, but not at age 15 years. A history of asthma among 7-year-olds
was reported for 46% of the children in 1990 compared to 19.1% in 1964
(Robertson et al., 1991).
When comparing school children living in New Zealand with school
children living in South Wales in 1990, the prevalence of a history of
asthma at any time was higher in New Zealand (17%) than in South Wales
(12%) (Barry et al., 1991). Wheeze ever and wheeze brought on by
running were also higher in New Zealand than in South Wales. The sex
ratio of asthmatic and wheezy children was similar. In a comparison of
12- to 15-year-old school children living in five regions in four
countries, the prevalence of wheezing during the last 12 months, as
assessed by both a written and a video questionnaire, was similar in
West Sussex, England (29% and 30%), Wellington, New Zealand (28% and
36%), Adelaide, Australia (29% and 37%), and Sydney, Australia (30%
and 40%), and was lower in Bochum, Germany (20% and 27%) (Pearce et
al., 1993). One year prevalence of severe wheezing limiting speech was
greater in Wellington (11%), Adelaide (10%) and Sydney (13%) than in
West Sussex (7%) and Bochum (6%). In addition, the one year prevalence
of frequent attacks of wheezing, frequent nocturnal wheezing, and
doctor diagnosed asthma were higher in Australia and New Zealand than
in the European centres.
5.3.2.2 Time trends
Between 1982 and 1984 the prevalence of bronchial
hyper-responsiveness assessed by histamine challenge test among 2363
Australian children was 17.9% (Salome et al., 1987). The prevalence of
respiratory symptoms, bronchial hyperresponsiveness, severity of
bronchial hyperresponsiveness and bronchial hyperresponsiveness
combined with symptoms was compared between children living in
Auckland, New Zealand and two locations in Australia: Wagga Wagga in
the inland and Belmont on the coast (Asher et al., 1988). The
prevalences were similar in Auckland and Wagga Wagga, but lower in
Belmont. When the same study was repeated 10 years later with
Australian 8-10 year olds, the prevalence of wheeze had increased from
10.4% in 1982 to 27.6% in 1992 in Belmont and from 15.5 to 23.1% in
Wagga Wagga. Bronchial hyperresponsiveness increased twofold up to
19.8% in Belmont and 1.4 fold up to 18.1% in Wagga Wagga (Peat et al.,
1994). The prevalence of atopy remained unchanged. The reported asthma
or wheeze in a Maori population rose from 26.2% in 1975 to 34% in 1989
(Shaw et al., 1990).
5.3.3 Eastern Mediterranean
The prevalence of asthma was studied in Israeli adolescents by
means of computerized medical draft records of conscripts aged 17-18
years who were born over a 9-year period and examined up to the end of
1989 (Laor et al., 1993). Asthma was more prevalent in males than in
females and the prevalence of asthma increased over time. Risk of
asthma was higher for subjects of Western origin and lowest for those
of African origin. By area of residence the risk for asthma was
highest in coastal and lowest in inland regions.
5.3.4 Africa
In Africa, 694 children from a Cape Town township and 671 from a
rural area in Transkei performed an exercise tolerance test. Of the
children living in the urban area 3.2% had asthma compared to 0.14%
living in a rural district (Van Niekerk et al., 1979). The prevalence
of reversible airway obstruction after running for 6 min was studied
in 7- to 9-year-old school children living in three areas in Zimbabwe.
Prevalence of airway obstruction was 5.8% in northern Harare, an urban
area with more people of higher socioeconomic status, 3.1% in southern
Harare and 0.1% in Wedza, both more rural areas. Prevalence in white
children living in northern Harare was similar to the prevalence in
black children in that region, (5.3% and 5.9%, respectively) (Keeley
et al., 1991).
5.3.5 Asia
5.3.5.1 Prevalences
In 1992, the prevalences of hay fever, eczema and wheeze were
estimated by parental questionnaire in secondary school students, ages
12 to 19 years, in the three south-east Asian cities Hong Kong, Kota
Kinabalu and San Bu (Leung & Ho, 1994). The prevalences for hay fever
were 15.7%, 11.2% and 2.1%, for eczema 20.1%, 7.6% and 7.2%, and for
wheeze 11.6%, 8.2% and 1.9%, in the respective cities. Skin test
reactivity to one of five common allergens was common and present in
49.0-63.9% of subjects. In Singapore, allergic rhinitis was studied in
a cross-sectional population-based study of 2868 adults, aged 20-74
years (Ng & Tan, 1994b). Allergic rhinitis was reported by 4.5% of
subjects.
5.3.5.2 Time trends
Two surveys in 7- to 15-year-old school children in Taipei,
Taiwan, were conducted in 1974 and 1985 (Hsieh & Shen, 1988). The
prevalence of childhood asthma increased from 1.3% in 1974 to 5.1% in
1985, with boys showing higher rates in both studies.
5.3.6 North America
5.3.6.1 Prevalences
Significant variation in asthma mortality between different
geographical regions was reported. Regions with elevated rates
included the central plain states and three large urban metropolitan
areas: Chicago IL, New York NY and Phoenix AZ (Weiss et al., 1989).
5.3.6.2 Time trends
Asthma morbidity in Indian children and adults in Saskatchewan
was registered using hospitalization data from 1970 to 1989. Asthma
hospitalization was higher among boys than girls at age 0-4 years,
but this was reversed at the ages of 15-34 and 35-64 years.
Hospitalization for asthma had significantly increased for the age
groups 0-4 and 35-64 years. Increases of asthma morbidity in recent
years were also observed (Senthilselvan & Habbick, 1995).
The reported prevalence of ever having asthma increased among
6- to 11-year-old children between the first (1971 to 1974) and second
(1976 to 1980) US National Health and Nutrition Examination Surveys
(NHANES) from 4.8 to 7.6% (Gergen et al., 1988). Asthma was more
common in blacks than in whites, in boys than in girls, and in urban
than in rural areas.
Changes in the asthma prevalence between 1981 and 1988 were
studied in the US National Health Interview Survey (NHIS) in 0- to
17-year-old children (Weitzman et al., 1992). The prevalence of
parent-reported childhood asthma increased from 3.1 to 4.3%. Trends
towards a lower rate of hospitalization and better overall health
status of the asthmatics from 1981 to 88 were reported. The overall
prevalence increased by 40%, which was mainly due to a prevalence
increase in white children. However, the prevalence was still higher
in blacks. There was no evidence of an increase in severity of asthma.
An example of the uncertainties in collecting and interpreting
epidemiological data is provided by consideration of the age-adjusted
death rate for asthma in the USA, which increased by 40% between 1982
and 1991. The increase was higher in females (59%) than in males
(34%). The death rates were consistently higher in blacks than in
whites. Prevalence rates for self-reported asthma increased also by
42%, with a much greater increase in females (82%) than in males
(29%). Death rate and self-reported asthma increased but
hospitalization rates remained stable. This was possibly due to an
improved outpatient treatment and changed billing practices reflecting
changes in the classification of cases of asthma under other
diagnostic categories. Ethnic differences in self-reported morbidity
may potentially be explained by accessibility to health services and
socioeconomic factors (CDC, 1995). This is an example of the
difficulties in collecting and interpreting epidemiological data.
5.3.7 The International Study of Asthma and Allergies in Childhood
The International Study of Asthma and Allergies in Childhood
(ISAAC) was initiated to maximized the value of epidemiological
research into asthma and allergic disease by establishing a
standardized methodology and facilitating international collaboration
(Asher et al., 1995). Its specific aims are:
a) to describe the prevalence and severity of asthma, rhinitis and
eczema in children living in different centres, and to make
comparisons within and between countries;
b) to obtain baseline measures for assessment of future trends in
the prevalence and severity of these diseases;
c) to provide a framework for further etiological research into
genetic, lifestyle, environmental and medical care factors
affecting these diseases.
The first phase of the collaborative studies was completed by 155
centres in 56 countries and included more than 720 000 participants
(Strachan et al., 1997). The core questionnaires used were designed to
assess the prevalence and severity of asthma, allergic rhinitis and
eczema in children. A 20-fold difference in the 12-month-period
prevalence of wheezing in 13- to 14-year-old school children (range
1.6-36.8%) and an 8-fold variation between the 10th and 90th
percentiles (3.9-30.6%) were observed (ISAAC Steering Committee, 1998)
(Fig. 16). The 12-month-period prevalence of symptoms of allergic
rhinitis in 13- to 14-year-olds varied from 1.4 to 39.7% with a
four-fold variation seen between the 10th and 90th percentiles
(4.9-21.0%) (Strachan et al., 1997; ISAAC Steering Committee, 1998).
There was also a high variability in the 12-month-period prevalences
of atopic eczema (defined as flexural dermatitis), which varied from
0.3 to 20.5% (ISAAC Steering Committee, 1998).
5.3.8 Conclusion
Despite the differences in methodology between epidemiological
studies conducted in the last 20-30 years, there is evidence that the
prevalence of asthma and other atopic disorders is increasing in many
countries (e.g., Burr et al., 1989; Burney et al., 1990; Ninan &
Russell, 1992) (Fig. 17).
A change in genetic susceptibility of populations to the
manifestation of these diseases is unlikely to explain the observed
time trend. However, although the studies suggest an increase in
prevalence, this may partly reflect changes in diagnostic fashion and
in public awareness of these diseases. The scientific evidence for
increases in the prevalence of asthma in children and young adults
since 1970, for example, is still weak because the measures used are
susceptible to bias (Magnus & Jaakkola, 1997). Such methodological
problems can only be minimized if a methodology using standardized
questions on disease severity, including objective measurements, is
applied (see also section 5.15).
The ISAAC study in which such a standardized methodology was
applied found a high worldwide variation in the prevalence and
severity of asthma, rhinitis and eczema. In addition, countries such
as Australia and New Zealand that have high prevalence rates of asthma
in the ISAAC study are similar to those countries, including the
United Kingom, with the highest prevalence rates for asthma symptoms
in adults in the European Community Respiratory Health Survey (ECRHS)
(Burney et al., 1996) and countries such as India or Algeria are in
the lowest quartile for asthma prevalence in both studies. Together
with the observed increases in the prevalences of these diseases in
several countries during relatively short time periods of one or two
decades, this variability in prevalence rates probably is a reflection
of different lifestyle and environmental factors which seem to play an
important role in the etiology of allergic diseases.
There are worldwide differences in the distribution of these
diseases. Among the exogenous factors that have been suggested are
changes in allergen exposure, outdoor and indoor pollutants, cigarette
smoke, work place exposure, personal hygiene and changes in diet.
5.4 Age and gender distribution
The natural history of asthma is not well understood (Martinez et
al., 1995). Many infants have episodes of wheezing associated with
viral infections soon after birth and during the first years of life.
However, the majority of these children have transient conditions
(Martinez et al., 1995). The period prevalence of wheezing has a peak
before the age of 10 years (Anderson et al., 1992). During infancy
boys are more often affected than girls, but this difference seems to
disappear during and after adolescence (Barbee, 1987; Anderson et al.,
1992). Several studies have shown a higher prevalence of asthma
symptoms and bronchial hyperresponsiveness (BHR) in female than in
male adolescents (Shaw et al., 1991; Anderson et al., 1992; Pearce et
al., 1993; Riedler et al., 1994).
Hay fever has a median age of onset of around 15 years. The
prevalence has a peak in people aged 16-24 years. For perennial
rhinitis the median age of onset is around 20 years and the prevalence
reaches a peak in 20-30 year olds (Broder et al., 1974; Schachter &
Higgins, 1976; Viner & Jackman, 1976; Sibbald & Rink, 1991a,b). A
number of studies reported a slightly higher prevalence of allergic
rhinitis in males than in females (Weeke, 1987). Atopic dermatitis
case history (e.g., oral allergy syndrome) or a severe,
life-threatening immediate Type I reaction after ingestion of a
defined food (e.g., apple or peanut), supported by a clear positive
specific skin test with the suspected food, establishes the diagnosis
(Metcalfe & Sampson, 1990). The demonstration of IgE antibodies
towards the culprit food underlines the IgE-mediated pathogenesis
(Johansson et al., 1984).
4.5.4.3 Specific serum IgE
The determination of food-specific serum IgE antibodies with
different techniques (e.g., RAST = Radio-Allergo-Sorbent-Test, ELISA =
Enzyme-Linked Immuno-Sorbent-Assay, FEIA = Fluoro-Enzyme Immuno-Assay)
has become a routine diagnostic tool in many Allergy Centres and among
practitioners (Johansson et al., 1984). However, a positive IgE
determination as well as a positive skin test do not mean actual food
allergy, but only a food sensitization. Cross-reactive IgE antibodies
to inhaled allergens (birch or mugwort pollen, cat dander, cow
epidermis) (Aalberse et al., 1981; Calkhoven et al., 1987) or to
botanically related food (e.g., legumes and peanut, soy and chickpea)
can be observed in negative challenge tests (Bernhisel-Broadbent &
Sampson 1989; Bernhisel-Broadbent et al., 1989). On the other hand,
some food allergic patients who show positive results in skin tests
have negative results in RAST. Like skin testing, the diagnostic value
of IgE determination is hampered by the lack of standardized food
extracts. Also the cut-off limit of IgE determination, i.e., the
minimal value in kU/litre or class, that should be considered
clinically relevant is still a matter of debate.
4.5.4.4 IgG determination
Specific IgG antibodies against food (Wüthrich, 1990) can be
found in many different physiological and pathological conditions.
Their determination does not prove the existence of a clinically
relevant immune reaction (Dannaeus & Inganäs, 1981).
4.5.4.5 Other in vitro tests
The Histamine Release Test (HRT) (Clinton et al., 1986) and the
Cellular Allergen Stimulation Test (CAST) (de Weck et al., 1993),
which determine the histamine or the sulfidoleucotrienes release from
basophil leucocytes in the peripheral blood, are of considerable
scientific interest but are too complicated and time-consuming for
daily routine use.
4.5.4.6 Oral challenge tests
At present, double-blind, placebo-controlled, food challenge
(DBPCFC) is considered as the "gold standard" for diagnosis of adverse
reactions to foods (Bernstein et al., 1982b; Bock et al., 1988).
Although several procedures for performing DBPCFC have been developed,
their application in normal clinical practice is hampered by their
heavy demands on resources and time.
The use of DBPCFC is necessary to assess objectively food
allergy/food intolerance, because several studies have demonstrated a
tremendous discrepancy between subjective perception of food
allergy/food intolerance and the results of DBPCFC. However, there are
difficulties in conducting studies of this nature in large population
samples. For example, challenge tests are dangerous with the risk of
severe anaphylactic reactions in the case of positive skin-prick tests
and/or positive serum IgE (RAST/CAP) against the offending food.
Moreover, there may be several potential hazards in the procedure and
in the interpretation of food challenges (Bindslev-Jensen et al.,
1994b), namely:
a) The nature of the food being tested (raw, freeze-dried,
cooked, food allergen extract).
b) The amount of food necessary to provoke objective signs.
c) DBPCFC may bypass important sites, e.g., mouth when using
capsules.
d) Additive or synergistic effect of multiple
hypersensitivities (e.g., concomitant sensitization to spices or
inhaled allergens such as pollen) or trigger factors (e.g.,
drugs, aspirin, alcohol, exercise, stress).
e) If the patient only reports subjective symptoms, e.g., itch,
headache, it may be necessary to increase the number of challenge
tests (three active and three placebo provocations) to avoid
false positive reactions and to obtain statistical significance
(Young et al., 1994).
In clinical practice it is often convenient to apply an open or a
single-blind food challenge: a negative test is of high predictive
value. A positive test, with the presence of objective signs (e.g.,
urticaria, asthma) or laboratory markers, increase of serum histamine,
tryptase or ECP (Eosinophil Cationic Protein) levels after challenge,
is usually sufficient to verify the diagnosis.
4.5.5 Therapeutic considerations
The only proven therapy for food allergy is to avoid the allergen
causing the disease. To be able to avoid, for instance, wheat flour,
milk or egg in the daily diet, professional advice is necessary both
to avoid hidden allergens in processed food and to ensure that the
diet is nutritionally adequate.
4.5.6 Prevalence
4.5.6.1 Introduction
It is possible to make more or less well-documented estimates of
the prevalence of different adverse reactions to foods. Frequency and
duration of breast feeding, eating habits, and flora (e.g., birch
trees) are factors influencing the prevalence. It is not known whether
the prevalence of food allergy is increasing. The prevalence of pollen
rhinitis is increasing (Wüthrich et al., 1995), and it is possible
that the prevalence of pollen-related food allergies may also be
rising.
4.5.6.2 Children
In many countries cow's milk is the first food allergen newborns
meet. The prevalence of cow's milk allergy is 2-5% in one-year-old
babies (Jakobsson & Lindberg, 1979; Bock, 1987; Host et al., 1988;
Hill & Hosking, 1997). This figure declines rapidly during the first
three years of life (Host & Halken, 1990).
The overall prevalence of food allergy is somewhat higher with a
maximum around one year. Bock (1987) followed a cohort of North
American children from birth to their third birthday; 7.7% had adverse
reactions to food not including fruit and fruit juices and 85% of the
adverse reactions were found during the first year of life. By the
third birthday less than 1% had adverse food reactions. The cumulative
prevalence of food allergy/intolerance diagnosed by elimination and
open challenge in a group of unselected Danish infants 18 months of
age was 6%. In the same study the prevalence in high-risk infants was
17% (Halken et al., 1992).
In a British study of a birth cohort of children age 4 years,
0.7% had allergy to peanuts or tree nuts and 0.5% had allergy to
peanuts alone. These figures were based on skin-prick tests and
positive history (Tariq et al., 1996).
The estimated prevalence of hypersensitivity reactions in
Australia to common foods in infancy and childhood is: cows's milk 2%,
egg 3.2%, and peanut 1.9%. Based on food challenge studies from
Australia, Japan, Indonesia, Malaysia and the Philippines, milk and
egg are the commonest food allergens. Soy, wheat and peanut
hypersensitivity are next commonest, but rice allergy is rare (Hill,
1997).
In a study of allergic children, Crespo et al. (1995) found that
allergy to cow's milk, egg and fish predominantly began before the
second year of age, whereas allergy to fruit, legumes and vegetables
predominantly began after the second year. This is in accordance with
the findings in a Danish study (Saval et al., 1993) where in older
children an increased prevalence of allergic pollen rhinitis was
followed by an increase in oral itch, the symptom characteristic for
allergic reactions to fruit and vegetables. In the 14-16 year old the
prevalence of oral itch was 2-2.9%.
4.5.6.3 Adults
a) IgE-mediated allergy
Allergic reaction to foods that cross-react with pollen is
probably the most common food allergic reaction in adults, at least in
countries where tree pollen allergy is common. In a study of Swedish
medical students, Foucard (1991) found that 78% of the students with
allergic rhinitis and positive skin test to birch pollen reported
clinical sensitivity to nuts and/or apples or other fruits. In another
Swedish study 70% of the patients with birch pollen allergy and 19% of
the patients with allergy to other pollen had food-related symptoms
(Eriksson et al., 1982). These estimates were mainly based on
self-reported symptoms.
In Switzerland approximately 10% (9.1-11.2%) of the adult
population currently suffers from hay fever. The prevalence of
sensitization measured by skin-prick test, but not necessarily
followed by clinical symptoms, is 12.7% for grass pollen and 7.9% for
birch pollen (Wüthrich et al., 1995). Other reports of the prevalence
of pollen rhinitis vary from 2-15% depending on, among other things,
the age group studied (Sibbald, 1993).
Applying Swedish figures for pollen-related reactions to the
Swiss data, taking into account the prick test results, approximately
50% of hay fever patients would have adverse reactions to
pollen-related foods, suggesting a prevalence of 3-5% in the adult
population. In a study by Kremser (1989) only about 10% of subjects
allergic to birch pollen had more severe reactions than oral itch.
Combining this with the Swiss figure gives a prevalence of
pollen-related reactions other than oral itch of approximately 0.5%.
In adults, the prevalence of allergy to non-pollen-related food
allergens such as milk, egg, shrimp, meat is probably about 0.1%. In a
study by Wüthrich (1993) of 402 patients with food allergy,
approximately 75% of the positive reactions were caused by
pollen-related food and 25% of the reactions were caused by
non-pollen-related food. Patients where the only food-related symptom
was itching of the mouth were not included in the study.
In some patients IgE allergy to natural rubber latex causes
allergic reactions to ingested banana, avocado, chestnut, etc. (Blanco
et al., 1994). The prevalence of natural rubber latex allergy in
European health-care workers screened with skin-prick tests has ranged
from 2.8 to 10.7%. About half of patients allergic to natural rubber
latex have experienced symptoms after eating banana (Turjanmaa et al.,
1996). In a study by Beezhold et al. (1996) 36% of a group of patients
allergic to natural rubber latex had clinical reactions to related
foods. The majority of these patients had anaphylaxis.
In the Netherlands an attempt has been made to estimate the total
prevalence of adverse reactions to foods and additives in adults. The
study used double-blind placebo-controlled food challenge (Niestijl
Jansen et al., 1994) and 12.4% of the studied population reported food
allergy or intolerance to specific food. Food groups mentioned most
frequently were fruit, chocolate and vegetables. The prevalence of
food allergy or intolerance in the adult Dutch population was
estimated to be 2.4%. If the positive reactions to food additives,
including menthol and glucose, is disregarded, the estimated
prevalence of food allergy/intolerance is 1.2%.
In a British study (Young et al., 1994) approximately 20% of a
survey population reported adverse reactions to foods. Eight foods
commonly perceived to cause sensitivity were canned or made into candy
bars and used for double-blind placebo-controlled challenge. The foods
were: cow's milk, hen's egg, wheat, soya, citrus fruits (orange),
fish/shellfish (prawn), nuts (peanut, brazil nut, walnut and hazel
nut) and chocolate. These eight foods accounted for 49.3% of reactions
reported in the study questionnaire. Adding some of the persons with
severe reactions and estimating the number of theoretically positive
in the non-challenged groups, the estimated prevalence of food
allergy/intolerance to the eight foods was 1.4-1.8%. As the eight
foods used for challenge accounted for approximately 50% of reported
reactions, the total prevalence estimate must be 3-4%.
b) Contact allergens
In dermally sensitized subjects, ingestion of the contact
allergen may cause skin flare reactions or other symptoms, for
instance, from the gastrointestinal tract. It has been reported that
10% of Danish women have contact allergy caused by nickel (Menné &
Holm, 1983). Up to 10% of these may benefit from a nickel-restricted
diet (Veien et al., 1993). The prevalence of systemic reactions via
the food from other contact allergens is not known. The chemicals of
fragrances and of food flavours, natural or synthetic, are often
identical. Veien et al. (1987) found an effect of a diet low in
flavours in patients having a flare of dermatitis after oral challenge
with balsam of Peru.
4.5.6.4 Conclusions
Milk and egg seems to be the most common food allergens in
infants and children worldwide. Peanuts are also reported to be common
food allergens in the United Kingdom, USA, Australia and some Asian
countries. The overall prevalence of food allergy/intolerance in young
children may be around ó-8%. This figure declines before 3 years of
age. In older children and young adults the prevalence of allergy to
pollen-related fruits, nuts and vegetables increases.
The results from the epidemiological studies, combined with the
knowledge on pollen and latex cross-reactions, systemic reactions to
contact allergens, and coeliac disease (prevalence estimate 0.2-0.5%),
point to an estimated prevalence of food allergy in the adult
population of 3-5%, giving that some of the reactions may be
coincident. These figures are based on the existing prevalence data,
which are mainly European. It is not yet known whether prevalence data
in the rest of the world are comparable.
4.6 Autoimmune diseases associated with drugs, chemicals and
environmental factors
4.6.1 Introduction
The autoimmune connective tissue diseases include conditions such
as systemic lupus erythematosus (SLE), systemic sclerosis, Sjögren's
syndrome, rheumatoid arthritis and the systemic vasculitides such as
Wegener's granulomatosis, Churg Strauss syndrome and polyarteritis
nodosa. The etiology of these conditions remains largely unknown but
there is a consensus that several factors may be important, including
genetic, ethnic and hormonal; many of these conditions have a female
preponderance (Table 23). Some of these conditions may also have an
environmental component to their pathogenesis and this particularly
applies to systemic sclerosis. Many drugs have been associated with
the development of SLE and cutaneous vasculitis. There has been some
interest in the possible development of connective tissue diseases
associated with silicone breast implants.
4.6.2 Systemic lupus erythematosus
SLE is predominantly a disease of young women (9:1 female: male
ratio) and is commoner amongst certain ethnic groups such as
Afro-Caribbeans and Orientals (Beeson 1994). Clinically, the disease
manifestations include arthritis, serositis, photosensitivity, oral
ulceration, malar rashes, recurrent thromboses, glomerulonephritis and
central nervous system involvement, e.g., epilepsy, psychoses.
Serologically, SLE is characterized by autoantibody production to
nuclear components such as anti-nuclear antibodies, antibodies to double
stranded DNA and antibodies to extractable nuclear antigens.
Table 23. Age and sex associations of some autoimmune diseases
(adapted from Beeson, 1994)
Age % Females
1. Autoimmune diseases that may
appear during childhood
Systemic lupus erythematosus 2-12 70
Dermatomyositis 1-15 70
Table 23. (continued)
Age % Females
Sydenham's chorea 5-15 65
Rheumatoid arthritis 1-10 65
Rheumatic fever 5-15 50
Thrombocytopenic purpura 2-5 50
Polyglandular syndromes 2-15 50
Bullous pemphigoid 1-15 50
Diabetes mellitus 2-15 45
Henoch-Schonlein purpura 2-5 40
Post-streptococcal nephritis 5-15 35
2. Autoimmune diseases that usually
appear during early adult life
Systemic lupus erythematosus 15-40 90
Erythema nodosum 15-30 90
Takayasu's arteritis 10-30 85
Myasthenia gravis 20-30 75
Thrombocytopenic purpura 15-45 75
Addison's disease 20-50 75
Rheumatoid arthritis 20-40 65
Multiple sclerosis 20-35 60
Sarcoidosis 20-40 55
Ulcerative colitis 15-40 50
Erythema multiforme 20-40 45
IgA nephropathy 10-30 35
Polyarteritis nodosa 20-50 30
Ankylosing spondylitis 20-30 25
Goodpasture's syndrome 15-35 15
Ankylosing spondylitis 20-30 25
Goodpasture's syndrome 15-35 15
Thromboangiitis obliterans 20-40 5
3. Autoimmune diseases that usually appear during mature adult life
Sjogren's syndrome 40-60 95
Primary biliary cirrhosis 30-75 90
Hashimoto's thyroiditis 30-50 85
Thyrotoxicosis 30-50 85
Scleroderma 30-50 80
Chronic active hepatitis 30-50 65
Polymyositis/dermatomyositis 40-60 55
Polychondritis 40-60 50
Pemphigus vulgaris 50-80 50
Wegener's granulomatosis 10-80 50
Henoch-Schonlein purpura 30-65 45
Table 23. (continued)
Age % Females
Membranous nephropathy 30-60 35
Amyotrophic lateral sclerosis 40-70 35
Tabes dorsalis 30-50 20
4. Autoimmune diseases that usually
appear late in life
Giant cell arteritis/polymyalgia 50-90 65
Pernicious anemia 40-80 60
Bullous pemphigoid 60-75 50
Rapidly progressive glomerulonephritis 50-70 45
Myasthenia gravis 50-80 40
Fibrosing alveolitis 40-70 35
The precise etiology of SLE remains obscure but some environmental
factors are known to exacerbate the disease. One of the most important
of these is ultraviolet light. In particular UV-B in the 295 to 305 nm
range is known to be toxic to SLE patients (McGrath et al., 1994).
Furthermore, patients with antibodies to Ro (SSA) are sensitive to UV
light, which provokes a photosensitive skin rash and may be followed by
a generalized disease flare (Gilliam & Sontheimer, 1982). Being in a
lower socioeconomic group may also increase the risk for the development
of glomerulonephritis in patients with SLE (McAlindon et al., 1993),
although the reasons for this remain obscure.
Many patients with SLE are allergic to a variety of substances
including drugs and chemicals, and a case-control study has supported
this clinical observation (Sequeira et al., 1993). Patients with lupus
and particularly those with Sjögren's syndrome appear to be sensitive to
cotrimoxazole and other sulfonamide-containing drugs. Oral contraceptive
pills, especially those with high oestrogen doses, may provoke flares of
SLE, and these agents should generally be avoided (Jungers et al.,
1982). The progesterone-only pill is associated with fewer flares.
Table 24. Drugs associated with the development of
systemic lupus erythematosus
Drugs commonly inducing systemic lupus erythematosus
Debrisoquine
Hydralazine
Quinidine
Procainamide
Drugs with good evidence for inducing systemic lupus erythematosus
Carbamazepine
Chlorpromazine
Hydrazine
Isoniazid
Minocycline
Methyldopa
Penicillamine
Phenytoin
The majority of cases of SLE are idiopathic but certain drugs are
known to cause SLE in genetically predisposed individuals. Of the 70 or
so drugs reported to be associated with drug-induced lupus, some of
which are shown in Table 24, only procainamide and hydralazine have been
studied in detail. Clinically, drug-induced lupus is similar to
idiopathic lupus, but there are one or two striking differences. For
example, serositis and pleuro-pulmonary involvement is much commoner in
drug-induced lupus, whereas renal disease and central nervous system
disease is less common in comparison to idiopathic lupus (Yung et al.,
1995). Serologically, anti-nuclear antibodies and antibodies to both
single-stranded and double-stranded DNA are found in both drug-induced
and idiopathic lupus, but it is uncommon to find very high levels of
anti-DNA antibodies in drug-induced lupus. Antibodies directed against
certain components of histone are thought to be characteristic of
drug-induced lupus. Although anti-histone antibodies may commonly be
found in idiopathic lupus, they react to the H1 and H2B subunits of
histone. In drug-induced lupus, the specificity appears to be against
the H2A-H2B dimer in procainamide-induced lupus, or against H3 and H4 in
hydralazine-induced lupus (Burlingame & Rubin, 1991).
Idiopathic lupus erythematosus is associated with HLA DR2 or DR3,
at least in Caucasian patients. One of the best clues towards an
explanation for a genetic predisposition to drug-induced lupus comes
from the study of the acetylator status of these patients. "Slow
acetylators" are homozygous for this gene and have low levels of
N-acetyltransferase in the liver. Both procainamide and hydralazine
are metabolized by this pathway and slow acetylators have a higher risk
of developing drug-induced lupus following exposure to these drugs
(Strandberg et al., 1976; Sonnhag et al., 1979).
The exact mechanisms by which drugs can induce lupus remain unknown
and a number of possibilities are being considered. It may be that the
ability of both procainamide and hydralazine to bind polynucleotides
in vitro may render DNA and/or histones antigenic (Dubroff & Reid,
1980; Tomura & van Lancker, 1988). A more specific mechanism has been
suggested whereby drugs interfere with the normal process of methylation
of DNA. Following DNA replication, cytosine residues are methylated at
the 5-position by the enzyme DNA methyltransferase. Failure of
methylation of regulatory sequences is associated with gene expression,
whereas methylation is associated with suppression of gene
transcription. Thus, DNA methylation is a mechanism regulating gene
expression (Yung et al., 1995). Studies have shown that
procainamide- and hydralazine-treated human T-cells show evidence of
hypomethylation (Scheinbart et al., 1991). In particular, procainamide
is capable of reversibly inhibiting T-cell DNA methyltransferase in a
dose-dependent manner (Scheinbart et al., 1991). Furthermore, these
T-cells become autoreactive in response to procainamide and hydralazine
(Yung et al., 1995). These autoreactive T-cells are capable of inducing
B-cells to differentiate into IgG-secreting cells without the addition
of antigen or mitogen, thus providing a mechanism for polyclonal B-cell
activation that is seen in both idiopathic and procainamide-induced
lupus (Richardson et al., 1990).
Experiments have shown that UV light is also capable of inhibiting
T-cell DNA methylation, increasing LFA-1 expression and inducing
autoreactivity (Richardson et al., 1994). Thus, drugs such as
procainamide and hydralazine and environmental factors such as UV light
may induce T-cell autoreactivity by inhibiting T-cell DNA
methyltransferase, thereby altering T-cell gene expression and making
autoimmunity more likely.
4.6.3 Scleroderma: environmental and drug exposure
Scleroderma (progressive systemic sclerosis) is a multisystem
connective tissue disease of unknown etiology. The prevalence ranges
from 47.9 to 290 per million among females, with an estimated incidence
of between 3.6 and 16 per million per year (Silman et al., 1996). It is
commoner in women (the sex ratio ranges from 3:1 to 8:1) and some ethnic
minorities such as Afro-Caribbeans and is characterized by widespread,
diffuse sclerosis affecting the peripheral vasculature, skin,
gastrointestinal tract, heart and muscle. Raynaud's phenomenon is a very
common early feature, and pulmonary and renal involvement may be serious
and life-threatening. Serologically, antibodies to topoisomerase I
(anti-Scl 70) and RNA polymerase III may predict prognosis in terms of
more extensive disease.
Scleroderma-like conditions (pseudoscleroderma) and scleroderma
have been associated with a variety of chemical and environmental agents
(Silman & Hochberg, 1996) (Table 25).
4.6.4 Silicone breast implants
Two studies have reported possible interactions between silicone
breast implants and the immune system. Tenenbaum et al. (1997) showed
the existence of a relationship between the level of anti-polymer
antibodies in the serum and the severity of clinical complications
(related to connective tissue) in silicone breast implant recipients.
Although these antibodies were not directed against silicone polymers
this might be the first objective marker to be used as a diagnostic
feature in silicone breast implant patients. Smalley et al. (1995, 1997)
reported on a lymphocytic response to silica (silicon dioxide) similar
to that for silicone (polysiloxane polymer), a silicon derivative, and
present in silicone breast implant material, in silicone breast implant
patients and their children.
Table 25. Some agents associated with scleroderma
Organic chemicals:
Toluene
Benzene
Xylene
Aromatic mixes e.g., white spirit
Vinyl chloride
Trichloroethylene
Perchloroethylene
Naphtha-n-hexane
Epoxy resins
Metaphenylenediamine
Urea formaldehyde foam insulation
Drugs:
Bleomycin
Carbidopa
L-5-hydroxytryptophan
Table 25. (continued)
Drugs:
Pentazocine
Cocaine
Diethyl proprion
Fenfluramine HCl
However, several thorough reviews of the subject, including a
case-control study (Sanchez-Guerrero et al., 1994, 1995; Gabriel et
al. 1994; Hochberg et al., 1996), concluded that there was little or
no association between silicone breast implants and either connective
tissue diseases or a unique arthralgia/myalgia/fibromyalgia syndrome.
They suggested that the connective tissue diseases that had been
reported were most likely to have been idiopathic in origin.
In a large retrospective cohort study, the Women's Health Study
sent questionnaires to 395 543 women and found 10 830 women who
reported breast implants (Hennekens et al., 1996). This study showed a
small but statistically significant increased risk of the combined
end-point of any connective tissue disease in patients with breast
implants, with a relative risk of 1.24 (95% confidence intervals
1.08-1.41, P = 0.0015). Of the specific connective tissue diseases
studied, scleroderma was associated with a relative risk of 1.89 (95%
confidence intervals 0.98-3.45) but this was not significant
( P = 0.06). For patients with scleroderma with implants less than 4
years, the relative risk was significant: 2.68 (95% confidence
intervals 1.11-6.51, P = 0.029) The precise nature of the implants
was not ascertained so it is not clear how many of the implants were
silicone gel filled. Another possibility for bias is the publicity
surrounding breast implants, which may have led to a degree of
over-reporting amongst the cases.
Other studies have supported the epidemiological view that there
is no association between connective tissue disease and silicone
breast implants (Nyren et al., 1998; Edworthy et al., 1998). Both were
very large cohort studies that compared patients who had either
undergone breast reduction (Nyren et al., 1998) or non-implant
cosmetic surgery (Edworthy et al., 1998). Neither found an excess of
any connective tissue disease among patients with silicone implants. A
United Kingdom review (DOH, 1998) came to the same conclusion.
Overall, apart from one retrospective study, there is no clear
evidence that patients with silicone breast implants have any risk of
developing a connective tissue disease but the area remains
controversial.
4.6.5 Toxic oil syndrome
This epidemic started in May 1981 when large numbers of patients
in the Madrid industrial area suffered acute respiratory illnesses
that did not respond to antibiotics. The etiological agent was
identified as being rapeseed oil that had been denatured with 2%
aniline for industrial use but sold illegally by itinerant vendors for
domestic food use. Initially 20 688 cases were recorded with 835
deaths -- a cumulative mortality of 4%, but over the first 2 years of
the epidemic the cumulative mortality was 2.3%. Oleyl-anilide proved
to be an excellent marker for case-related oil specimens although the
precise nature of the etiological agent has never been described. It
has proved impossible to consistently replicate the syndrome in animal
studies.
Various clinical features have been described (Kammüller et al.,
1984; Philen & Posada, 1993) including severe myalgia, arthralgia,
Raynaud's phenomenon, scleroderma-like skin changes, carpal tunnel
syndrome, joint contracture, pulmonary hypertension and peripheral
neuropathy. The most consistent laboratory features were eosinophilia
and high IgE counts and low levels of antibodies such as antinuclear
antibodies. HLA DR3-DR4 was associated with the chronic phase of the
disease. Histologically, the most consistent finding was a vascular
lesion characterized by intimal proliferation with fibrosis, vascular
occlusion and thrombosis.
4.6.6 Eosinophilia-myalgia syndrome
This epidemic was first identified in October 1989 in New Mexico
(Hertzman et al., 1990). The report was followed by a case-control
study that firmly linked the consumption of possibly contaminated
L-tryptophan from one source with the eosinophilia-myalgia syndrome
(Eidson et al., 1990). L-tryptophan was widely used as a
non-prescription food supplement by health conscious individuals for a
wide variety of minor ailments. The source of the epidemic was traced
to a single manufacturer (Philen & Posada, 1993).
The vast majority of cases occurred in the USA. The majority of
these patients were white middle-class middle-aged females, probably
reflecting the pattern of use of health supplements rather than any
innate risk factor. The mortality rate was reported as 2.7% (37 deaths
among 1370 known cases) (Philen & Posada, 1993).
The clinical features resembled those of toxic oil syndrome,
although scleroderma-like changes and Raynaud's phenemenon were not
reported (Kaufman et al., 1991; Philen & Posada, 1993). These authors
also noted that HLA DR4 was associated with an increased risk of
chronic disease. Laboratory investigations consistently showed an
eosinophilia early in the disease course, although this diminished
spontaneously even when the patients continued to be ill (Philen &
Posada, 1993). The main factor in treatment was the avoidance of
further ingestion of L-tryptophan. Glucocorticoids were used widely
and helped the myalgias and reduced the eosinophil count, but there
was often a recurrence of symptoms on stopping the steroid treatment,
and progression to chronic disease was not altered.
4.6.7 Vinyl chloride disease (occupational acro-osteolysis)
Vinyl chloride (CH2=CHCl) is a combustible colourless gas at
room temperature that is used in the manufacture of a variety of
plastics. Several methods can be used to polymerize the gas to make
polyvinyl chloride (PVC). In the mid-1960s a new syndrome affecting
workers involved in polymerizing vinyl chloride was recognised (Wilson
et al., 1967). These patients developed paraesthesia of the fingers,
cold sensitivity, Raynaud's phenomenon, pseudoclubbing of the fingers,
skin oedema and thickening of the fingers, hands and forearms, and
chest X-ray changes (Veltman et al., 1975). The risk of development of
symptoms was related to cumulative exposures over time and work
practices but was not related to handling the finished PVC product,
and in Wilson's (1967) study it occurred in less than 3% of exposed
individuals.
An increased prevalence of HLA DR3 and DR3/B8 haplotypes has been
noted in patients with vinyl chloride disease (Black et al., 1983).
Vinyl chloride is a cause of non-cirrhotic portal hypertension and
angiosarcoma of the liver.
The skin changes of vinyl chloride disease resemble morphoea
clinically and histologically, and vascular changes were often present
with luminal narrowing of the digital arteries and subtotal occlusion
of these vessels (Veltman et al., 1975). The most dramatic
radiological change is acro-osteolysis seen in the terminal phalanges
of the fingers; a transverse lytic band is seen across the distal
phalangeal shaft. Ward et al. (1976) reported immunological
abnormalities in vinyl chloride disease including polyclonal increases
of IgG, cryoglobulins, evidence of complement activation and low titre
anti-nuclear antibodies. Vascular endothelial, medial and sub-intimal
deposits of IgG, C3, C4 and fibrin/fibrinogen were seen on histology
of small and medium-sized arterioles. Reduced T-cell and modestly
increased B-cell numbers were also observed (Ward et al., 1976).
4.6.8 Systemic vasculitis: environmental factors and drugs
Various drugs are associated with hypersensitivity reactions and
the most common mechanism is an immune-complex-mediated vasculitis
(Dubost et al., 1991). Drugs account for approximately 10-20% of
dermal vasculitis and this figure is on the increase. The cutaneous
lesions most commonly seen include palpable purpura, although
urticarial lesions may be seen in 10% (Dubost et al., 1991). They
usually occur symmetrically on the lower limbs extending to the thighs
and buttocks (Mullick et al., 1979). In Mullick's series of 30
patients, 19 had disseminated vasculitis with other organ involvement,
including renal disease, synovitis, pleuropulmonary and cardiovascular
disease with coronary vessel vasculitis and cardiac failure. More than
80% of patients had constitutional features such as fatigue, malaise
and fever.
4.6.9 Conclusion
The vast majority of the autoimmune connective tissue diseases
have no known etiological agents. Certain drugs, occupational
exposures, and UV radiation have been shown either to exacerbate a
known autoimmune disease or, occasionally, to trigger the onset of a
syndrome that closely resembles one of the established diseases.
5. EPIDEMIOLOGY OF ASTHMA AND ALLERGIC DISEASE
5.1 Introduction
Asthma and allergies, such as atopic diseases (i.e., bronchial
asthma, allergic rhinitis, atopic dermatitis) and allergic contact
dermatitis, are common medical problems.
Asthma, allergic rhinitis and atopic dermatitis are conditions
that have a variety of clinical similarities and epidemiological
connections (Montgomery-Smith, 1983). Asthma has frequently been
classified as of allergic or non-allergic origin. Different atopic
conditions often occur together in the same person. A family history
of atopic disorders has consistently been found to be an important
predisposing factor for the development of atopic diseases (Croner &
Kjellman, 1990). However, epidemiological studies suggest that
exogenous factors also play an important role in their etiology
(Newman Taylor, 1995). There have been epidemiological studies on
geographical variation and time trends in prevalence rates of these
disorders, and knowledge about the effects of a variety of
environmental factors, including outdoor exposures, indoor exposures,
diet and occupational exposures, is increasing.
5.2 Definition and measurement of allergic disease
5.2.1 Asthma
5.2.1.1 Definition
Asthma is a respiratory disease that is not well defined. It is
characterized by variable airflow limitation due to bronchial
hyper-responsiveness and often by inflammatory changes in the airways
(see section 2.5.1)
5.2.1.2 Assessment
a) Questionnaires
In epidemiological studies many different questionnaires have
been used to investigate in populations the prevalence of asthma
symptoms such as wheezing and shortness of breath, as well as
diagnosed asthma. These include the MRC (United Kingdom Medical
Research Council), the ECSC (European Coal and Steel Community), the
ATS-DLD (American Thoracic Society and the Division of Lung Disease),
and the IUATLD (International Union against Tuberculosis and Lung
Disease) questionnaires (Toren et al., 1993).
When questionnaires are used in epidemiological studies to
compare prevalence rates between populations, findings may be
influenced by differences in language, interpretations of the concept
of wheeze in different communities, or the general awareness of the
disease in the community (Strachan et al., 1990). Nevertheless, the
IUATLD questionnaire has been shown to provide valid and comparable
data even when translated (Burney et al., 1989a). A similar written
questionnaire was developed for the International Study of Asthma and
Allergies in Childhood (ISAAC) (Pearce et al., 1993; Asher et al.,
1995; Shaw et al., 1995). In addition, a video questionnaire depicting
(in five scenes) adolescents with different symptoms of asthma has
been developed to overcome problems with the translation of
questionnaires (Asher et al., 1995). The sensitivity and specificity
for predicting bronchial hyperresponsiveness of the video
questionnaire was similar to the respective questions in the IUATLD
questionnaire (Shaw et al., 1992a,b; Shaw et al., 1995). By providing
data relatively free from biases due to language, culture, literacy or
interviewing techniques, the video questionnaire may prove
particularly useful for comparisons of prevalence and severity of
asthma in different populations (Asher et al., 1995).
b) Bronchial hyperresponsiveness
In epidemiological settings, a major limitation of using
bronchial hyperresponsiveness (BHR), measured by challenge, as a gold
standard for the definition of asthma is that a considerable
proportion of subjects with bronchial hyperresponsiveness report no
respiratory symptoms (Sterk et al., 1993). Thus, bronchial
hyperresponsiveness cannot be used synonymously as a diagnosis of
asthma (Sterk et al., 1993). The thresholds used to define bronchial
hyperresponsiveness -- usually a fall in the forced expiratory volume
in one second (FEV1) of more than 15% or 20%, or a fall in peak
expiratory flow rate (PEF) of more than 10% -- were chosen
arbitrarily. Various methods including exercise tests and inhalation
of metacholine, histamine or cold air have been described to measure
bronchial hyperresponsiveness (Sterk et al., 1993). It is now well
recognized that a change in osmolarity of the periciliary fluid is a
potent stimulus to airway narrowing and may be a common cause for
provoking an attack of asthma (Anderson & Smith, 1991). This has led
to the use of hypertonic saline aerosols to document bronchial
hyperresponsiveness in epidemiological studies (Riedler et al., 1994).
However, a negative response to any of these provocation methods does
not exclude asthma. In general, measuring bronchial
hyperresponsiveness in epidemiological studies has a moderate
specificity and a relatively low positive predictive value (Sterk et
al., 1993).
5.2.2 Rhinitis
There are no clear-cut criteria for defining hay fever and
perennial rhinitis. Rhinitis is frequently underdiagnosed and
misdiagnosed (Sibbald & Rink, 1991a,b). Patients are generally
classified according to the suspected etiology of their conditions.
Rhinitis is labelled "allergic" when a causal allergen can be
identified, otherwise it is labelled "non-allergic". Subjects with
seasonal symptoms are twice as likely to be labelled as having
allergic rhinitis by their doctors. The usual complaints of allergic
rhinitis are "summer flu", series of sneezing and a stuffy nose. The
classification is based primarily on nasal smears and skin-prick tests
compared with the patient's history (Weeke, 1987). Perennial allergic
rhinitis in patients who are allergic to pets is easy to diagnose if
symptoms such as sneezing and itchy eyes occur immediately after
contact with pets. If the symptoms are caused, for instance, by mites,
it is more difficult to obtain a clear-cut medical history. Symptoms
of patients with non-allergic rhinitis are often different from those
with allergic causes. The main symptoms in non-allergic cases are
stuffy nose and loss of sense of smell, while in the allergic type,
sneezing and watery secretion are more prominent. A standardized
questionnaire for assessing the prevalence of rhinitis in children in
epidemiological studies has been developed (Asher et al., 1995).
5.2.3 Atopic dermatitis
5.2.3.1 Definition
Atopic dermatitis is a disease that is difficult to define,
because of the frequently subtle clinical manifestations, the lack of
an identifying laboratory marker, and the lack of a distinguishing
primary lesion. Generally, atopic dermatitis is a chronic cutaneous
inflammatory disease with a strong tendency of patients to overproduce
IgE (Hanifin, 1987).
5.2.3.2 Assessment
The absence of diagnostic criteria for atopic dermatitis prompted
Hanifin & Lobitz, (1977) and Hanifin & Rajka (1980) to suggest major
and minor diagnostic criteria for atopic dermatitis based on clinical
features. Disadvantages of these criteria were that many of them had
no precise definition, that some were very infrequent, and that others
were nonspecific. A minimum set of diagnostic criteria for atopic
dermatitis was derived by the United Kingdom working party composed of
dermatologists, family practitioners, paediatricians and
epidemiologists (Williams et al., 1994a). These criteria included a
history of flexural involvement, a history of dry skin, the onset
under the age of 2 years, a personal history of asthma, a history of
pruritic skin condition, and visible flexural dermatitis. The criteria
were used for assessing the prevalence of atopic dermatitis in
children in an international multiple cross-sectional study (Asher et
al., 1995). The European Task Force (ETFAD, 1993) on atopic dermatitis
has developed a scoring index for the severity of atopic dermatitis,
which is based on a combination of symptoms such as erythema,
oedema/papulation, oozing/crusts, excoriation and lichenification.
Nevertheless, more objective tests, such as raised IgE and/or
skin-prick test positivity, remain important tools for epidemiological
research.
5.2.4 Skin-prick test and serum IgE
Skin-prick testing is commonly used to assess allergic
sensitization in epidemiological studies. The test is easy to perform;
serious local or systemic reactions occur only very rarely, and
different test devices are available. However, results of different
studies are often not comparable due to differences in allergen
concentrations, type of needles used for pricking, and criteria for
defining a positive reaction (Nelson et al., 1993). The most common
criterion is that the diameter of the allergen wheal be >2-3 mm after
subtraction of the reaction to the negative control (Pepys, 1994).
Even well-standardized skin-prick tests may be subject to measurement
error arising from different field workers and variations in the
degree of skin reactivity in different racial groups or under
different environmental conditions. Measurements of total and specific
serum IgE may therefore provide valuable additional information on the
atopic susceptibility and of the atopic status of an individual. Total
serum IgE, however, may also be raised in association with other
conditions, such as parasitic infections.
5.2.5 Allergic contact dermatitis
Allergic contact dermatitis is the condition in which contact
with haptens induces cell-mediated contact sensitization.
Patch testing is used to diagnose allergic contact dermatitis.
The test should be performed by a skilled operator using standardized
test materials and also with substances present in the patient's
domestic or occupational environment which are considered to be
possible sensitizers.
5.3 Asthma and atopy: prevalence rates and time trends in prevalence
rates
Asthma is the most common single chronic disease in childhood.
Most of the prevalence studies on asthma have therefore been conducted
in children and adolescents. Many of these studies have also
determined the prevalence of rhinitis and atopic dermatitis. In the
last 20 years prevalence estimates have been reported from many
different geographical regions in all five continents. Several studies
were repeated after a number of years applying the same methods at
different points in time, thereby providing information on time trends
in prevalence. Estimates on asthma prevalence were mostly derived by
questionnaires, often in combination with a lung function test and
determination of bronchial hyperresponsiveness. In addition to
questionnaire data, prevalence rates of atopic sensitization were
assessed by skin-prick tests and, to a lesser extent, by determination
of total or specific IgE. The overview given below is not intended to
be comprehensive, but will give some insight into the geographical
variation and recent time trends. The comparability of prevalence
estimates between study centres, however, is limited as most studies
applied different methods.
5.3.1 Europe
5.3.1.1 Prevalences
Between 1989 and 1992 a parental questionnaire, a skin-prick test
and a cold air challenge test were administered to 9- to 11-year-old
children in western and eastern Germany. Atopic sensitization was more
frequent in western German children living in Munich (5.9%), compared
to children living in Leipzig and Halle in eastern Germany (3.9%).
Bronchial hyperresponsiveness was also more prevalent in western
Germany (8.3%) than in eastern Germany (5.5%) (von Mutius et al.,
1994b). Hay fever and rhinitis were reported less often in Leipzig
than in Munich (2.4% and 16.6% compared to 8.6% and 19.7%), whereas
bronchitis was more prevalent in Leipzig. In contrast to atopic
respiratory disease, the prevalences for atopic eczema were similar in
the two study areas (von Mutius et al., 1992).
The occurrence of allergic diseases was studied during 1979 and
1980 on the basis of a questionnaire sent to the parents of 20 000
children 7, 10 and 14 years of age in three parts of Sweden with
different climatic conditions (Aberg et al., 1989). The prevalence of
asthma was significantly higher in the northern part of the country,
and this higher prevalence could not be explained by other factors
than by the cold and dry climate. In Viborg, Denmark, the frequency of
rhinitis was 10.5%, of atopic eczema 7%, of urticaria 3.2%, and of
asthma 4.5% among 5- to 16-year-old school children studied in 1990
(Saval et al., 1993). Asthma and rhinitis were more frequent among
boys, while atopic eczema was more frequent among girls. For both
sexes, the frequency of rhinitis increased during their years at
school, while the frequency of skin symptoms decreased.
In a general practice in London, symptoms, atopic state and
medical history were compared among 16- to 65-year-old patients with
seasonal and perennial rhinitis (Sibbald & Rink, 1991b). The
prevalence of rhinitis was 24%, 3% had seasonal symptoms only and 13%
had perennial symptoms only. Distinguishing between atopic and
non-atopic subjects by skin-prick testing with five common allergens
revealed that subjects with seasonal rhinitis were more likely to be
atopic. Moreover, subjects with seasonal rhinitis were also more
likely to have eczema and to have a family history of hay fever.
Rates of reported eczema during early childhood have been studied
by health visitor interview in three national cohorts of children born
in England, Scotland and Wales in 1946 (at age 6 years), 1958 (at age
7 years), 1970 (at age 5 years) (Taylor et al., 1984). The reported
rates in the three birth cohorts were 5.1%, 7.3% and 12.2%.
5.3.1.2 Time trends
From 1926 to 1961 the prevalence of asthma in Finnish men
registered through the defence forces statistics ranged between 0.02
and 0.08%, whereas from 1961 to 1989 the prevalence rose from 0.29 to
1.79% (Haahtela et al., 1990).
In the United Kingdom several studies were conducted in different
epidemiological settings to estimate the prevalence and time trends in
the prevalence of allergic disorders. The different settings comprised
patients of general practices (Fleming & Crombie, 1987; Sibbald &
Rink, 1991a,b), national birth cohorts (Taylor et al., 1984),
representative samples of school children in England (Burney et al.,
1990), in South Wales (Burr et al., 1989), in the London borough of
Croydon (Anderson et al., 1983; Anderson et al., 1994), in Aberdeen
(Ninan & Russell, 1992), in the South Thames region (Barbee et al.,
1987), and hospital admission statistics (Strachan & Anderson, 1992;
Anderson, 1989). The latter showed between 1978 and 1985 an increase
in the number of hospital admissions because of asthma in infants up
to 4 years old (186%) and in 5- to 16-year-olds (56%) in the South
West Thames region (Anderson, 1989). Changes in mode of referral,
severity on admission and readmission ratio were also explored, but
little evidence was found for a reduction in severity or change in
readmission rate since 1978. These findings contrast with findings of
two identical surveys that were conducted in 1978 and 1991 to explore
prevalence changes and use of medical services (Strachan & Anderson,
1992). These data showed a substantial increase in self-referral
together with an increase in readmission. A comparison of two surveys
of morbidity carried out in 1970-1971 and 1981-1982 in general
practices in England and Wales showed an increase in prevalence rates
of asthma in men from 8.8 to 15.9% (Fleming & Crombie, 1987).
Between 1973 and 1989 the lifetime prevalence of wheezing among
12-year-old children in South Wales increased from 17 to 22%, a
history of asthma rose from 6 to 12%, and current asthma from 4 to 9%
(Burr et al., 1989). Increases occurred also in the frequency of
history of eczema (5 to 16%) and of hay fever (9 to 15%), while
wheezing not attributable to asthma remained constant. Increasing
prevalences of asthma were also observed between 1973 and 1986 in
English school children (relative increase in boys of 6.9% and in
girls of 12.8%), which could not simply be explained by changes in
diagnostic fashion (Burney et al., 1990).
In Aberdeen, Scotland, the prevalence of wheeze among 8- to
13-year-old school children rose from 10.4% in 1964 to 19.8% in 1989,
and shortness of breath rose from 5.4 to 10.0%. Wheeze and shortness
of breath were more prevalent in boys than in girls. Asthma rose from
4.1 to 10.2%, hay fever from 3.2 to 11.9% and eczema from 5.3 to 12%
(Ninan & Russell, 1992). Between 1978 and 1991 significant relative
increases in the 12-month prevalence rates of attacks of wheezing and
of asthma were found in the London borough of Croydon (Anderson et
al., 1994). Results from a repeated cross-sectional study performed in
1991-1992 and 1995-1996 in 10-year-old children in Leipzig, Germany,
showed a rise in hay fever and atopic sensitization. However, no
increase in the prevalence of asthma or bronchial hyperresponsiveness
was observed over the 4-year period (von Mutius et al., 1998).
5.3.2 Oceania
5.3.2.1 Prevalences
Several cross-sectional surveys were performed in Australian and
New Zealand school children to assess the prevalence of respiratory
symptoms and bronchial hyperresponsiveness. A comparison of 769
children living in Wagga Wagga (inland Australia), and 718 children
living in Belmont (coastal Australia) carried out in 1982, showed that
respiratory symptoms, asthma, bronchial hyperresponsiveness, hay fever
and atopy were all more common in the dry inland area than in the
humid coastal area. In both areas 38% of the children were atopic
(Britton et al., 1986).
The prevalence of wheeze in Melbourne school children was 23.1%
for 7-year-olds, 21.7% for 12-year-olds, and 18.6% for 15-year-olds.
History of wheeze was more common for boys than for girls at age 7
years, but not at age 15 years. A history of asthma among 7-year-olds
was reported for 46% of the children in 1990 compared to 19.1% in 1964
(Robertson et al., 1991).
When comparing school children living in New Zealand with school
children living in South Wales in 1990, the prevalence of a history of
asthma at any time was higher in New Zealand (17%) than in South Wales
(12%) (Barry et al., 1991). Wheeze ever and wheeze brought on by
running were also higher in New Zealand than in South Wales. The sex
ratio of asthmatic and wheezy children was similar. In a comparison of
12- to 15-year-old school children living in five regions in four
countries, the prevalence of wheezing during the last 12 months, as
assessed by both a written and a video questionnaire, was similar in
West Sussex, England (29% and 30%), Wellington, New Zealand (28% and
36%), Adelaide, Australia (29% and 37%), and Sydney, Australia (30%
and 40%), and was lower in Bochum, Germany (20% and 27%) (Pearce et
al., 1993). One year prevalence of severe wheezing limiting speech was
greater in Wellington (11%), Adelaide (10%) and Sydney (13%) than in
West Sussex (7%) and Bochum (6%). In addition, the one year prevalence
of frequent attacks of wheezing, frequent nocturnal wheezing, and
doctor diagnosed asthma were higher in Australia and New Zealand than
in the European centres.
5.3.2.2 Time trends
Between 1982 and 1984 the prevalence of bronchial
hyper-responsiveness assessed by histamine challenge test among 2363
Australian children was 17.9% (Salome et al., 1987). The prevalence of
respiratory symptoms, bronchial hyperresponsiveness, severity of
bronchial hyperresponsiveness and bronchial hyperresponsiveness
combined with symptoms was compared between children living in
Auckland, New Zealand and two locations in Australia: Wagga Wagga in
the inland and Belmont on the coast (Asher et al., 1988). The
prevalences were similar in Auckland and Wagga Wagga, but lower in
Belmont. When the same study was repeated 10 years later with
Australian 8-10 year olds, the prevalence of wheeze had increased from
10.4% in 1982 to 27.6% in 1992 in Belmont and from 15.5 to 23.1% in
Wagga Wagga. Bronchial hyperresponsiveness increased twofold up to
19.8% in Belmont and 1.4 fold up to 18.1% in Wagga Wagga (Peat et al.,
1994). The prevalence of atopy remained unchanged. The reported asthma
or wheeze in a Maori population rose from 26.2% in 1975 to 34% in 1989
(Shaw et al., 1990).
5.3.3 Eastern Mediterranean
The prevalence of asthma was studied in Israeli adolescents by
means of computerized medical draft records of conscripts aged 17-18
years who were born over a 9-year period and examined up to the end of
1989 (Laor et al., 1993). Asthma was more prevalent in males than in
females and the prevalence of asthma increased over time. Risk of
asthma was higher for subjects of Western origin and lowest for those
of African origin. By area of residence the risk for asthma was
highest in coastal and lowest in inland regions.
5.3.4 Africa
In Africa, 694 children from a Cape Town township and 671 from a
rural area in Transkei performed an exercise tolerance test. Of the
children living in the urban area 3.2% had asthma compared to 0.14%
living in a rural district (Van Niekerk et al., 1979). The prevalence
of reversible airway obstruction after running for 6 min was studied
in 7- to 9-year-old school children living in three areas in Zimbabwe.
Prevalence of airway obstruction was 5.8% in northern Harare, an urban
area with more people of higher socioeconomic status, 3.1% in southern
Harare and 0.1% in Wedza, both more rural areas. Prevalence in white
children living in northern Harare was similar to the prevalence in
black children in that region, (5.3% and 5.9%, respectively) (Keeley
et al., 1991).
5.3.5 Asia
5.3.5.1 Prevalences
In 1992, the prevalences of hay fever, eczema and wheeze were
estimated by parental questionnaire in secondary school students, ages
12 to 19 years, in the three south-east Asian cities Hong Kong, Kota
Kinabalu and San Bu (Leung & Ho, 1994). The prevalences for hay fever
were 15.7%, 11.2% and 2.1%, for eczema 20.1%, 7.6% and 7.2%, and for
wheeze 11.6%, 8.2% and 1.9%, in the respective cities. Skin test
reactivity to one of five common allergens was common and present in
49.0-63.9% of subjects. In Singapore, allergic rhinitis was studied in
a cross-sectional population-based study of 2868 adults, aged 20-74
years (Ng & Tan, 1994b). Allergic rhinitis was reported by 4.5% of
subjects.
5.3.5.2 Time trends
Two surveys in 7- to 15-year-old school children in Taipei,
Taiwan, were conducted in 1974 and 1985 (Hsieh & Shen, 1988). The
prevalence of childhood asthma increased from 1.3% in 1974 to 5.1% in
1985, with boys showing higher rates in both studies.
5.3.6 North America
5.3.6.1 Prevalences
Significant variation in asthma mortality between different
geographical regions was reported. Regions with elevated rates
included the central plain states and three large urban metropolitan
areas: Chicago IL, New York NY and Phoenix AZ (Weiss et al., 1989).
5.3.6.2 Time trends
Asthma morbidity in Indian children and adults in Saskatchewan
was registered using hospitalization data from 1970 to 1989. Asthma
hospitalization was higher among boys than girls at age 0-4 years,
but this was reversed at the ages of 15-34 and 35-64 years.
Hospitalization for asthma had significantly increased for the age
groups 0-4 and 35-64 years. Increases of asthma morbidity in recent
years were also observed (Senthilselvan & Habbick, 1995).
The reported prevalence of ever having asthma increased among
6- to 11-year-old children between the first (1971 to 1974) and second
(1976 to 1980) US National Health and Nutrition Examination Surveys
(NHANES) from 4.8 to 7.6% (Gergen et al., 1988). Asthma was more
common in blacks than in whites, in boys than in girls, and in urban
than in rural areas.
Changes in the asthma prevalence between 1981 and 1988 were
studied in the US National Health Interview Survey (NHIS) in 0- to
17-year-old children (Weitzman et al., 1992). The prevalence of
parent-reported childhood asthma increased from 3.1 to 4.3%. Trends
towards a lower rate of hospitalization and better overall health
status of the asthmatics from 1981 to 88 were reported. The overall
prevalence increased by 40%, which was mainly due to a prevalence
increase in white children. However, the prevalence was still higher
in blacks. There was no evidence of an increase in severity of asthma.
An example of the uncertainties in collecting and interpreting
epidemiological data is provided by consideration of the age-adjusted
death rate for asthma in the USA, which increased by 40% between 1982
and 1991. The increase was higher in females (59%) than in males
(34%). The death rates were consistently higher in blacks than in
whites. Prevalence rates for self-reported asthma increased also by
42%, with a much greater increase in females (82%) than in males
(29%). Death rate and self-reported asthma increased but
hospitalization rates remained stable. This was possibly due to an
improved outpatient treatment and changed billing practices reflecting
changes in the classification of cases of asthma under other
diagnostic categories. Ethnic differences in self-reported morbidity
may potentially be explained by accessibility to health services and
socioeconomic factors (CDC, 1995). This is an example of the
difficulties in collecting and interpreting epidemiological data.
5.3.7 The International Study of Asthma and Allergies in Childhood
The International Study of Asthma and Allergies in Childhood
(ISAAC) was initiated to maximized the value of epidemiological
research into asthma and allergic disease by establishing a
standardized methodology and facilitating international collaboration
(Asher et al., 1995). Its specific aims are:
a) to describe the prevalence and severity of asthma, rhinitis and
eczema in children living in different centres, and to make
comparisons within and between countries;
b) to obtain baseline measures for assessment of future trends in
the prevalence and severity of these diseases;
c) to provide a framework for further etiological research into
genetic, lifestyle, environmental and medical care factors
affecting these diseases.
The first phase of the collaborative studies was completed by 155
centres in 56 countries and included more than 720 000 participants
(Strachan et al., 1997). The core questionnaires used were designed to
assess the prevalence and severity of asthma, allergic rhinitis and
eczema in children. A 20-fold difference in the 12-month-period
prevalence of wheezing in 13- to 14-year-old school children (range
1.6-36.8%) and an 8-fold variation between the 10th and 90th
percentiles (3.9-30.6%) were observed (ISAAC Steering Committee, 1998)
(Fig. 16). The 12-month-period prevalence of symptoms of allergic
rhinitis in 13- to 14-year-olds varied from 1.4 to 39.7% with a
four-fold variation seen between the 10th and 90th percentiles
(4.9-21.0%) (Strachan et al., 1997; ISAAC Steering Committee, 1998).
There was also a high variability in the 12-month-period prevalences
of atopic eczema (defined as flexural dermatitis), which varied from
0.3 to 20.5% (ISAAC Steering Committee, 1998).
5.3.8 Conclusion
Despite the differences in methodology between epidemiological
studies conducted in the last 20-30 years, there is evidence that the
prevalence of asthma and other atopic disorders is increasing in many
countries (e.g., Burr et al., 1989; Burney et al., 1990; Ninan &
Russell, 1992) (Fig. 17).
A change in genetic susceptibility of populations to the
manifestation of these diseases is unlikely to explain the observed
time trend. However, although the studies suggest an increase in
prevalence, this may partly reflect changes in diagnostic fashion and
in public awareness of these diseases. The scientific evidence for
increases in the prevalence of asthma in children and young adults
since 1970, for example, is still weak because the measures used are
susceptible to bias (Magnus & Jaakkola, 1997). Such methodological
problems can only be minimized if a methodology using standardized
questions on disease severity, including objective measurements, is
applied (see also section 5.15).
The ISAAC study in which such a standardized methodology was
applied found a high worldwide variation in the prevalence and
severity of asthma, rhinitis and eczema. In addition, countries such
as Australia and New Zealand that have high prevalence rates of asthma
in the ISAAC study are similar to those countries, including the
United Kingom, with the highest prevalence rates for asthma symptoms
in adults in the European Community Respiratory Health Survey (ECRHS)
(Burney et al., 1996) and countries such as India or Algeria are in
the lowest quartile for asthma prevalence in both studies. Together
with the observed increases in the prevalences of these diseases in
several countries during relatively short time periods of one or two
decades, this variability in prevalence rates probably is a reflection
of different lifestyle and environmental factors which seem to play an
important role in the etiology of allergic diseases.
There are worldwide differences in the distribution of these
diseases. Among the exogenous factors that have been suggested are
changes in allergen exposure, outdoor and indoor pollutants, cigarette
smoke, work place exposure, personal hygiene and changes in diet.
5.4 Age and gender distribution
The natural history of asthma is not well understood (Martinez et
al., 1995). Many infants have episodes of wheezing associated with
viral infections soon after birth and during the first years of life.
However, the majority of these children have transient conditions
(Martinez et al., 1995). The period prevalence of wheezing has a peak
before the age of 10 years (Anderson et al., 1992). During infancy
boys are more often affected than girls, but this difference seems to
disappear during and after adolescence (Barbee, 1987; Anderson et al.,
1992). Several studies have shown a higher prevalence of asthma
symptoms and bronchial hyperresponsiveness (BHR) in female than in
male adolescents (Shaw et al., 1991; Anderson et al., 1992; Pearce et
al., 1993; Riedler et al., 1994).
Hay fever has a median age of onset of around 15 years. The
prevalence has a peak in people aged 16-24 years. For perennial
rhinitis the median age of onset is around 20 years and the prevalence
reaches a peak in 20-30 year olds (Broder et al., 1974; Schachter &
Higgins, 1976; Viner & Jackman, 1976; Sibbald & Rink, 1991a,b). A
number of studies reported a slightly higher prevalence of allergic
rhinitis in males than in females (Weeke, 1987). Atopic dermatitis
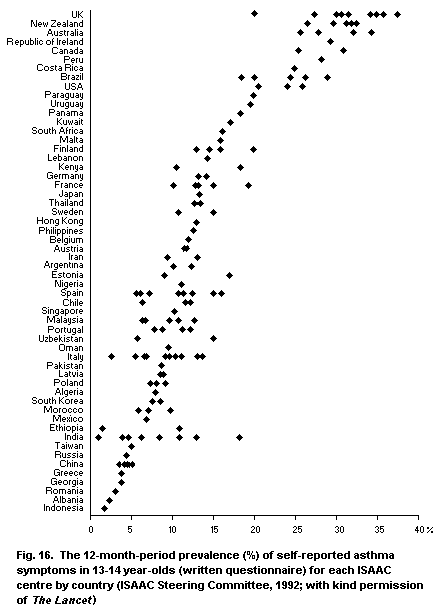
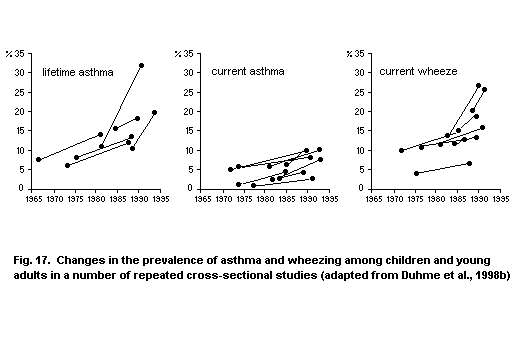 starts typically during the first year of life (Hanifin, 1987). The
onset of the disease is before the age of 7 in 60 to 90% of the cases
(Schultz Larsen & Hanifin, 1992). The prevalence appears to be higher
among females than among males (Schultz Larsen & Hanifin, 1992;
Schultz Larsen, 1993).
5.5 Migration
Migration studies have suggested that the environment has a
strong effect on the development of atopic disorders. Analyses of
interregional migrants in the United Kingdom showed that the regional
variation in cohorts, aged 5-7 years, was primarily related to the
region of current residence, and not to the region of birth (Strachan
et al., 1990). Ethnic group differences in the prevalence of atopic
dermatitis were studied in London school children. It appeared that,
compared to white children, London-born black Caribbean children were
at increased risk of atopic dermatitis (Williams et al., 1995a).
Striking differences in prevalence rates of asthma were found in
young urban and rural Xhosa children (Van Niekerk et al., 1979). In
this society, asthma was found mainly in urban communities and there
appears to be a striking absence of asthma in the rural environment.
Whether this is due to alterations in lifestyle or the possibility
that the rural traditional way of life exerts a protective effect in
the prevention of asthma is not clear.
The prevalence of asthma in Tokelauan children has been studied
in two environments, in Tokelau and in New Zealand (Waite et al.,
1980). Prevalence of asthma assessed by an interview of the mothers
was much higher among those children who were examined in New Zealand
than among those examined in Tokelau. Furthermore, for the children
examined in New Zealand, there was no significant difference in the
asthma prevalence between those children born in New Zealand and those
born in Tokelau.
Asthma in children and adolescents living in the New Guinea
Highlands was extremely uncommon in the sixties and early seventies.
In 58% of the observed asthma cases the onset of the disease was not
before the age of 30 years. Multiple sensitivities as assessed by
skin-prick test were common and did not diminish with increasing age
at onset. One possible explanation was that the degree of exposure to
allergens is high enough to cause sensitization, but not high enough
to cause lower respiratory symptoms until prolonged exposure in a
perhaps especially sensitive person has taken place (Anderson, 1974).
The prevalence of asthma among adults but not children living in the
Eastern highlands of Papua New Guinea has risen drastically between
1975 and 1985. Allergy to house dust mite appeared to be a significant
feature in the disease pathogenesis, and it is likely that this is
associated with modifications to traditional life style due to the
introduction of blankets and changes in sleeping habits, which promote
a more fertile environment for growth and multiplication of mites
(Dowse et al., 1985).
5.6 Viral infection
Findings of different studies suggest that viral infections in
early life may play a part in the prevention of allergic sensitization
(Martinez, 1994). Interesting observations with regard to a possibly
preventive role of viral infections in the development of asthma were
made in the population of Tristan da Cunha. The prevalence of asthma
among these islanders had earlier been reported to be one of the
highest in the world (Mantle & Pepys, 1974). After the evacuation from
Tristan da Cunha because of a volcanic eruption immunoassays in blood
samples of the islanders revealed a low prevalence of serum antibodies
against common viruses. During the years of evacuation, when the
islanders lived in the United Kingdom, a high incidence of respiratory
infection was observed. Thus, this population with a high prevalence
of asthma and a high prevalence of allergic sensitization had a very
low incidence of respiratory infections while living on the remote
island, and became heavily infected after being exposed to respiratory
viruses that they had probably rarely encountered before. Similar
observations were made in the Western Carolina islands where the
prevalence of asthmatic symptoms among children was very high, but
viral infections presumably very uncommon (Martinez, 1994). A study
from the United Kingdom found an inverse association between hay fever
and the number of older siblings (Strachan, 1989; Strachan, 1995).
Factors directly or indirectly related to the number of siblings may
decrease the susceptibility of children to become atopic. Again,
infections acquired during early childhood were proposed to be
protective against allergic sensitization. It has been speculated that
declining family size may in part contribute to the increasing
prevalence of atopic diseases reported in Western countries over the
past few decades, because of a lower chance of infection by older
siblings. In children from eastern and western Germany, allergic
sensitization as assessed by skin test reactivity was also inversely
associated with the number of siblings (von Mutius et al., 1994b).
5.7 Socioeconomic status
Hay fever has been recognized as a complaint of the more affluent
classes since the 19th century. A study of adults in south London
found little difference in the prevalence of rhinitis symptoms or
skin-prick reactions by social class, but a greater use of the label
"hay fever" by doctors for patients of higher socioeconomic status
(Sibbald & Rink, 1991a).
Eczema is more prevalent in British school children of higher
socioeconomic status than in those of lower status. Exposures
associated with social class are probably at least as important as
genetic factors in the expression of childhood eczema (Williams et
al., 1994b). The authors suggest that most or even all of the reported
changes in the prevalence of atopic dermatitis are due to a secular
trend in diagnosis. Population studies have used a range of different
methods and definitions for atopic dermatitis ranging from
questionnaire-based recall of eczema as a child or parental recall to
health visitors' and general practitioners' records. It is likely that
the term eczema is heavily biased by social class (allergy being a
more acceptable term in higher social classes) and with time (eczema
was a less acceptable label in earlier years due to connotations of
uncleanliness and arthropod infestation).
Hospital admissions for eczema have fallen over the last 20
years, but such in-patient data are misleading as much of this
reduction is probably due to the success of treatment with topical
corticosteroid preparations (Williams, 1992).
The prevalence of wheeze varied little by socioeconomic group in
an investigation of 5472 children, aged 5-17 years, in the United
Kingdom (Strachan et al., 1994). However, marked trends of severity
towards increased morbidity in poorer families were observed.
Diagnostic labelling and drug treatment of wheezy children did not
differ substantially with socioeconomic status. Thus, a degree of
socioeconomic equality existed in the process of medical care for
childhood asthma in the United Kingdom (Strachan et al., 1994;
Strachan, 1996).
No effect of socioeconomic status on the prevalence of asthma was
noted in the first and second US National Health and Nutrition
Examination Surveys (NHANES) (Gergen et al., 1988). In the Auckland
region, 1050 children aged 8-9 years were examined by parental
questionnaire and histamine inhalation challenge (Mitchell et al.,
1989). There was no relationship between socioeconomic status and
asthma diagnosis, bronchial hyperresponsiveness, or any combination of
bronchial hyperresponsiveness with symptoms or diagnosis. The relative
importance of socioeconomic status and several other factors in the
etiology of wheezing illness in the first 5 years and on the
persistence of this illness at the age of 16 years was studied in over
15 000 children born in the United Kingdom during one week of April
1970. Persistence of wheeze at age 16 years was related to high social
status (Lewis et al., 1995).
During a study of the prevalence of asthma and bronchitis in
Sydney school children, some social and environmental factors were
documented to ascertain if these affected the prevalence of either of
these diseases (Peat et al., 1980). No consistent relationship was
found between social class and lung disease with the exception of
increased prevalence of asthma in boys and girls of higher
socioeconomic status. Differential access and utilization of medical
care by the poor and rich may contribute to differences in asthma
prevalence. The relationship of socioeconomic status to various
indicators of asthma was studied in Canada in the context of universal
access to medical care. As compared with children from the most
advantaged homes, children from the least advantaged homes were more
likely to present exercise-induced bronchospasm, while there was no
excess of reported wheeze or diagnosed asthma. This result was
interpreted as indicating that there are unidentified environmental
factors that contribute to the excess asthma morbidity in children
(Ernst et al., 1995). In general, the association of socioeconomic
status with hay fever and eczema seems to be more consistent than with
asthma and respiratory symptoms.
5.8 Occupational exposure
Many prevalence studies have been conducted among workers in
high-risk occupations. Asthmatic workers may differ with regard to the
frequency of attacks, to the occurrence and to the onset of airway
obstruction. Diagnostic guidelines for occupational asthma were
internationally proposed in the USA and in Europe (Cartier et al.,
1989; Burge, 1989; EAACI, 1992). The diagnosis of occupational asthma
is based both on the clinical signs of the disease and the
demonstration of the occurrence of a recognized allergen in the
patient's workplace. Objective tests, such as skin testing, spirometry
and serial measurements of the peak expiratory flow, should be
performed to ascertain the occupational nature of the disorder (see
also section 4.4.4). Other diagnostic tools in epidemiological surveys
are standardized questionnaires inquiring information on work-related
symptoms. Typical symptoms of occupational asthma are symptoms
comprising difficulty in breathing, chest tightness, wheezing, a
period of initial exposure of 2 weeks or longer before the first onset
of symptoms, or evidence of airflow obstruction, and improvement of
symptoms when the subject is not working for days or longer.
Surveillance programs in the United Kingdom and Canada indicate
that occupational asthma is the most frequently reported occupational
lung disease accounting for 26 to 52% of the reports (Chan-Yeung &
Malo, 1995a). The proportion of asthma attributable to occupational
exposure is not known. Estimates range from 2 to 15%. The role of
occupational exposure is difficult to ascertain, because: a)
occupational asthma is still poorly recognized; b) affected workers
are scattered through many small workplaces often employing few
workers; c) methods of confirming work relationships are often rather
crude; d) there is no good reporting system of occupational asthma in
many countries; e) there are very few screening or surveillance
programmes for this condition.
Owing to such methodological problems it is often not possible to
give unbiased estimates of the true prevalence and incidence of
occupational asthma in populations if official government statistics
such as disabling benefit awards or worker's compensation boards are
used and comparisons within and between countries may be distorted
(Meredith & Nordman, 1996).
Occupational asthma falls into two categories: a) pre-existing
asthma that is aggravated by irritant or physical stimuli in the
workplace and b) asthma that is specifically induced by sensitization
to a workplace chemical (Jarvis et al., 1996). About 250 agents that
can give rise to occupational asthma are recognized (Chan-Yeung &
Malo, 1995a). Isocyanates are responsible for the most common form of
the disease, i.e., occupational asthma with latency. Occupational
asthma without latency follows exposure to high concentration of
irritant gases, fumes or chemicals; chlorine and ammonia are the most
common agents (Chan-Yeung & Malo, 1995a,b). Some substances may give
rise to asthma by inducing specific IgE antibodies. These allergens
are mostly substances of high relative molecular mass (>5000) such as
proteins in the urine of laboratory rats. Others are compounds of low
relative molecular mass, such as complex halogenated platinum salts or
acid anhydrides; these agents act as haptens and combine with a body
protein to form a complete antigen (Venables & Chan-Yeung, 1997). It
has been proposed that certain chemical allergens may cause
sensitization of the respiratory tract via an IgE-independent
mechanism. However, it is possible that new or technically refined IgE
measurements may reveal a much greater association between IgE
antibodies and chemical respiratory sensitization than is presently
assumed (Kimber & Wilks, 1995). Intermittent exposure to high levels
of an occupational agent is associated with a higher risk of
development of work-related asthma than a steady exposure to lower
concentrations, typical for isocyanate exposure.
Not all subjects develop occupational asthma under the same
exposure conditions. Various host markers (genetically determined) and
factors (acquired) have been incriminated in occupational asthma. In
general, atopy appears to be an important risk factor for occupational
asthma due to compounds of high relative molecular mass, such as
enzyme detergents or exposure to laboratory animals (Chang-Yeung,
1990). In a survey of workers exposed to flour in bakeries or mills,
the relation with symptoms was independent of atopic status (Cullinan
et al., 1994). Atopy has little predictive value in occupational
asthma due to chemicals of low relative molecular mass, and routine
screening for atopy in high-risk workplaces may not be justified
(Chan-Yeung & Malo, 1995b).
The effect of smoking on occupational asthma is not clear and
appears to be dependent on the type of occupational agent. When the
agent induces asthma by producing specific IgE antibodies, cigarette
smoking may enhance sensitization. However, cigarette smoking was not
associated with increased work-related asthmatic symptoms in workers
exposed to detergent enzymes, laboratory animals or colophony
(Chan-Yeung, 1990; Chan-Yeung & Malo, 1995a,b).
As part of the European Community Respiratory Health Survey
(ECRHS), the risk of occupational asthma has been estimated in a
random sample of 2646 young Spanish adults aged 20-44 years. Depending
on the definition of asthma, 2.6-6.7% of asthma cases were
attributable to occupational exposures (Kogevinas et al., 1996).
Incidence rates for occupational asthma for 1992 were estimated to be
approximately 153 cases per million workers in Finland (Meredith &
Nordman, 1996). Incidence rates reported for the United Kingdom in the
Surveillance of Work-related and Occupational Respiratory Disease
Project (SWORD) between 1989 and 1991 showed a high risk of
occupational asthma among paint sprayers, chemical and food
processors, laboratory staff, plastics and metal treatment workers,
and in welders and electronic assemblers (Meredith, 1993). For 1993
the incidence was estimated to be 37 per million working persons per
year (Meredith & Nordman, 1996). The most frequent agents causing
asthma among the organic agents were flour, grain, hay, wood dust and
laboratory animals; among the chemical agents were isocyanates and
glutaraldehyde, and some miscellaneous compounds such as solder,
colophony, glues and resins (Meredith & McDonald, 1994).
In the UK SWORD project there is a high level of national
coverage by chest physicians participating in the surveillance project
and estimates of the working population at risk are available, so that
incidence rates can be calculated by age, gender, region and
occupation. However, there is underestimation, because some patients
are seen only by general practitioners. Questions remain regarding
diagnostic accuracy and etiology.
In the following sections, studies on specific occupational
exposures in relation to occupational atopic diseases, with emphasis
on occupational asthma, are reviewed. The selection of exposures is
based on findings of the SWORD project and the availability of
epidemiological data.
5.8.1 Chemicals with low relative molecular mass
A total of 314 cases of occupational asthma were diagnosed at the
Institute of Occupational Health in Helsinki, Finland during the
period 1987 to 1990 (Savonius et al., 1993). By far the most common
causes of occupational asthma were low relative molecular mass
chemicals such as the isocyanates (76 cases), followed by formaldehyde
(18 ases), epoxy resin and epoxy resin hardeners (17 cases), and
cyanoacrylates (6 cases).
5.8.1.1 Diisocyanates
The main occupational hazards caused by polyurethane chemicals
are asthma and rhinitis, but contact dermatitis and urticaria may also
develop (Estlander et al., 1992). Exposures to toluene diisocyanate
were studied for effects on respiratory health of workers in two
plants manufacturing polyurethane foams (Jones et al., 1992).
Intensive personal monitoring was performed to characterized job
exposures. Initial questionnaire and spirometry data were obtained in
386 workers. Multiple regression analyses showed significant adverse
effects of cumulative toluene diisocyanate exposure on airway
responsiveness. According to Diller (1987), the incidence of
isocyanate asthma reported from different studies varies between 0 and
25%. Reasons for differences in observed incidence are intensity of
isocyanate exposure, criteria for diagnosis, mode of calculation,
sensitizing capacity of different isocyanates, individual
predisposition and confounding factors. No geographical or ethnic
difference was observed.
5.8.1.2 Acrylates
Acrylate monomers are used in a variety of industrial fields.
Their industrial use is increasing, since they have many features that
make them superior to formerly used chemicals (Savonius et al., 1993).
Contact sensitization is a well-known adverse health effect of
exposure to acrylates, but they may also cause respiratory symptoms.
The main acrylic compounds currently in use are acrylates,
cyanoacrylates and methacrylates. While methacrylates are well-known
contact sensitizers, cyanoacrylates have caused only few cases of
contact allergy. Acrylates may also have other harmful health effects.
Hand and finger symptoms and paraesthesiae have been reported among
dental personnel preparing acrylates with their hands. The domestic
use of acrylates is limited and the sensitizing problem is mainly
occupational, but probably occurs in many different industries.
Cyanoacrylates are used mostly as a component of a high strength glue
used for joining different materials. Methacrylates are used as
adhesives, as dental and orthopaedic fillings, as material for
protheses and as an embedding material for different purposes,
including histological preparations.
5.8.1.3 Anhydrides
Methylhexahydrophthalic anhydride (MHHPA) and
methyltetrahydrophthalic anhydride (MTHPA) are dicarboxylic anhydrides
used as hardeners for epoxy resins. MHHPA and MTHPA typically require
an elevated curing temperature (50-200 °C), which facilitates escape
of anhydride vapours. Anhydrides are low relative molecular mass
chemicals that have been reported to cause immunologically mediated
respiratory diseases (Tarvainen et al., 1995). Contact urticaria and
other skin symptoms have also been described. Some anhydrides, e.g.,
phthalic anhydride, have caused generalized urticaria, in connection
with respiratory symptoms, after high exposure.
5.8.1.4 Solder flux
Questionnaires and lung function measurements were administered
to 104 electronic workers in the USA who soldered printed circuits
boards. Symptoms of eye, throat and nose irritation occurred in nearly
half of the group. Lower respiratory tract symptoms, including cough,
phlegm production and wheezing, also occurred with increased
frequency, compared with reported rates among a general population
sample (Greaves et al., 1984).
5.8.2 Metals
5.8.2.1 Cobalt
Several clinical and experimental findings point to cobalt as a
main sensitizer and causal agent of hard metal asthma (Nemery et al.,
1992; Swennen et al., 1993; Cirla, 1994; Lauwerys & Lison, 1994).
Clinical features have been clearly identified by bronchial
provocation tests. IgE and IgG antibodies with cobalt specificity have
been demonstrated. Clinically, the ability of cobalt to induce delayed
hypersensitivity is well known for contact dermatitis (Cirla, 1994).
Occupational exposure to cobalt occurs mainly by inhalation in various
industries and occupations involved in the production and processing
of metal and various cobalt-containing alloys and salts. The potential
for exposure to cobalt is particularly important during the production
of cobalt powder, the production and processing and use of hard
metals, the polishing of diamonds with cobalt-containing disks, the
use of pigments and dryers containing cobalt salts and the processing
of cobalt alloys. In a cross-sectional survey of 194 workers from 10
diamond polishing workshops and 59 workers from 3 other workshops, a
questionnaire was administered and spirometry was performed to assess
whether exposure to cobalt was associated with respiratory impairments
(Nemery et al., 1992). Spirometry showed significantly lower indices
of ventilatory function in the group with the highest exposure to
cobalt. These differences were not due to differences in smoking
habits.
5.8.2.2 Metal-polishing industry
A comparative study of spirometric measurements in 104 polishers
and 90 unexposed controls was carried out in 25 brass and steel ware
polishing industries in Moradabad, India (Rastogi et al., 1992). The
polishing process generates dust, containing fine particles of emery
and metal, which are mainly composed of copper and zinc and constantly
inhaled by the polishers. Of the polishers 58.6% had one or more
respiratory symptoms as compared to 25.5% of the controls.
Occupational asthma was found to be confined to polishers, 4.8% being
affected. The polishers exhibited significantly greater reduction in
various lung function parameters over the work shift, which was larger
in smokers than in non-smokers. The duration of exposure was directly
correlated with acute fall in lung function.
5.8.2.3 Aluminium
Occupational asthma is a major respiratory health problem within
the primary aluminium industry (O'Donnell, 1995). In Australia and New
Zealand the incidence of occupational asthma in primary aluminium
smelting varies between smelters, estimates ranging from 0 to 2%.
Cases showed no association between the frequency of the symptoms or
the severity of bronchial hyperresponsiveness and a family history of
asthma, atopic skin test, tobacco smoking or age. Current evidence
suggests that occupational asthma in the aluminium industry is
irritant induced and caused by inhalation exposure to gaseous or
particulate fluoride compounds.
5.8.2.4 Platinum salts
Nonspecific and specific bronchial hyperresponsiveness in
immediate-type asthma caused by platinum salts did not cease after
removal from exposure (Merget et al., 1994) (see also section
4.3.4.1).
5.8.3 Natural rubber latex
Populations at increased risk of developing natural rubber latex
hypersensitivity include health care workers, rubber industry workers
and subjects undergoing multiple surgical procedures, especially
children with spina bifida and urogenital abnormalities. Prevalence
figures for natural rubber latex allergy in studies using skin-prick
tests range from 2.9 to 17% among hospital employees and are around
11% among glove-manufacturing workers (Vandenplas, 1995). Natural
rubber latex allergy has been demonstrated in 32 to 50% of the
children with spina bifida by skin-prick test or serological testing.
The prevalence of sensitization to natural rubber latex in the general
population ranged from 0 to 9% according to the atopic status of the
populations under study (Turjanmaa, 1994; Vandenplas, 1995). The
observed rise in incidence of sensitization to natural rubber latex
during the last decade is probably related to the increased use of
natural rubber latex devices as a protective barrier against
infections. Other possible determinant factors include increased
recognition of natural rubber latex allergy by exposed workers and
clinicians, changes in manufacturing methods, and discontinuation of
steam sterilization.
Complications were thought to be limited to contact dermatitis
due to irritation and sensitivity to certain rubber additives, and
only few reports of phenomena consistent with natural rubber latex
sensitivity were published before 1984. A possible explanation for the
abrupt rise in the incidence of natural rubber latex sensitivity is a
change in rubber manufacturing or rubber processing. There is a lack
of epidemiological studies observing the longitudinal trends in
prevalence and natural history of natural rubber latex allergy. The
risk for natural rubber latex allergy among health care workers
appears to vary with the frequency and intensity of exposure.
Cross-reactivity between natural rubber latex and certain foods,
particularly banana, avocado, chestnut and other fruits such as kiwi,
papaya and passion fruit, have been described (Charous, 1994).
A questionnaire and skin-prick tests with natural rubber latex
and common inhalant allergens were administered to hospital personnel
(201 nurses, 50 members of the cleaning staff, 38 laboratory
technicians), of whom 4.7% showed skin reactivity to latex. Among
those with a negative skin test to natural rubber latex no one had a
history of occupational asthma. For the latex-sensitive subjects
(N=12), a histamine challenge test was performed, which showed
bronchial hyperresponsiveness in all subjects. Seven of the twelve
developed significant bronchial response to a challenge test with
latex gloves. The overall prevalence of occupational asthma due to
natural rubber latex was estimated as 2.5% (Vandenplas et al., 1995).
A skin-prick test was performed on 77 surgeons and nurses with an
allergen solution made from latex gloves and rubber latex catheters.
In addition, subjects answered a questionnaire on the history of hand
dermatitis, atopic eczema, rhinitis or asthma, and symptoms when using
natural rubber latex gloves. The prevalence of relevant immediate
allergy assessed by skin-prick test was 5.2%. The reliability in
detecting sensitized persons was limited. The agent used previously in
skin-prick testing to demonstrate natural rubber latex sensitization
gave unreliable results; the use of a standardized reagent is
necessary (Cormio et al., 1993).
Among 512 hospital employees 7.4% of the doctors and 5.6% of the
nurses working in operational units were allergic to natural rubber
latex. The frequency was lower in non-operating units and among
laboratory personnel (Turjanmaa, 1987).
5.8.4 Flour
Occupational respiratory diseases are common among bakers
(Keskinen et al., 1978; Meredith, 1993; Reijula & Patterson, 1994; De
Zotti et al., 1994; Lauwerys & Lison, 1994). In Finland, for example,
the mean annual incidence of occupational respiratory diseases was 31
per 100 000 among the general work force compared to 374 per 100 000
among bakery workers (Reijula & Patterson, 1994). The annual incidence
of occupational asthma as reported to the SWORD project in the United
Kingdom was 334 per million for bakery workers as compared to 658 per
million for paint sprayers or 175 per million for electronic
assemblers (Meredith, 1993). A substantial prevalence of wheat flour
allergy was found in bakers and pastry cooks (Armentia et al., 1990).
Not only wheat allergens, but also alpha-amylase must be considered as
the causative agent (De Zotti et al., 1994). Other allergens such as
storage mites are suspected to play a role in the development of
occupational asthma (Tee et al., 1992a,b). Findings from the initial
cross-sectional phase of a cohort study of employees exposed to flour
in bakeries or mills showed that subjects without previous exposure to
flour expressed related symptoms especially to flour aeroallergen
(Cullinan et al., 1994). Forty-four male workers exposed to flour and
164 unexposed controls were examined by using personal samplers
measuring inspirable dust concentrations. The proportion of subjects
with one or more symptoms and with bronchial hyperresponsiveness was
significantly greater among workers exposed to flour than among
controls. The conclusion was drawn that despite exposure to relatively
low concentration levels of inspirable flour dust, subjects working in
the baking industry are at risk of developing both respiratory
symptoms and airway hyperresponsiveness (Bohadana et al., 1994).
5.8.5 Animals
The occurrence of respiratory disease was studied in 257 active
veterinarians and 100 control subjects who had no occupational animal
contact. Asthma was significantly more prevalent in veterinarians than
in controls (Lutsky et al., 1985). Selected indicators of allergy and
atopy were studied to determine predictors of laboratory animal
allergy in a prospective study of laboratory technicians. Although the
prevalence of atopy and allergic symptoms had increased in exposed
technicians after the follow-up period of 2 years, this was also found
in an unexposed matched control group, and there were no significant
differences between the groups in any measured variable at follow-up
(Renstrom et al., 1994).
5.8.6 Other agents
The association between occupational exposure to low levels of
airway irritants and bronchial responsiveness to histamine was
assessed in 688 male workers of synthetic fibre plants (Kremer et al.,
1995). According to job titles, exposure status was grouped into 7
categories: 1) reference group, 2) white collars, 3) SO2, HCl,
SO42-, 4) polyester vapour, 5) oil mist and oil vapour,
6) polyamide and polyester vapour, 7) multiple exposures. A higher
prevalence of airway responsiveness was associated with history of
allergy and respiratory symptoms. A slight trend was seen for subjects
with more than 5 years of exposure to polyester vapour and oil mist
and oil vapour towards a higher prevalence of bronchial
hyperresponsiveness. No overall association could be demonstrated.
5.9 Allergic contact dermatitis
5.9.1 Epidemiology of allergic contact dermatitis
Only a few studies have investigated the frequency of allergic
contact dermatitis in the general population (Coenraads & Smit, 1995).
Nielsen & Menné (1992) studied the distribution of allergic contact
sensitization in an unselected sample of 793 individuals. Of these,
567 participants were patch tested with the standard series
(TRUE-test). This series included the most common contact sensitizers,
such as metals, fragrances, preservatives and medicaments. It has been
found that 50-80% (depending on patient selection) of all cases of
allergic contact dermatitis will be diagnosed using this series (Menné
et al., 1992). Among the 567 volunteers were 15.2% who positively
reacted to one or more of the substances included in the test series.
Multiple contact sensitizations were thus observed more commonly than
expected (Nielsen & Menné, 1992). Patients evaluated at patch test
clinics often have multiple contact allergies. In experimental studies
individuals with one contact allergy were found to be more easily
sensitized to a second one (Friedmann, 1990). In the clinical
situation the causes of multiple sensitivities are difficult to
evaluate, because factors such as genetic susceptibility, broken skin
barrier and multiple exposures are mixed up. Multiple contact
sensitivities are typically seen in individuals with long-lasting
chronic dermatitis.
In the following paragraphs, specific exposures closely related
to allergic contact dermatitis are discussed.
5.9.1.1 Nickel
Nickel-containing metal alloys and nickel-plated surfaces release
free nickel ions when in direct contact with human sweat. The typical
nickel dermatitis is therefore located beneath metal items, such as
buttons, jewellery, suspenders, glasses and similar objects. Among the
567 participants of an unselected sample of 793 subjects were 11.1%
women and 2.2% men affected with a nickel allergy (Nielsen & Menné,
1993) (see Table 18; section 4.1.5.1). Individuals primarily
sensitized from consumer items might, at a later stage, develop
occupational nickel dermatitis when occupationally exposed to this
metal. Tools and equipment used in different jobs by workers such as
carpenters, electricians, painters and plumbers were found to release
nickel (Lidén et al., 1996).
5.9.1.2 Chromates
Chromium salts, but not metallic chromium, are sensitizing. The
hexavalent salts are the main sensitizers as they penetrate the skin
more easily than the trivalent chromate (Gammelgaard et al., 1992).
Chromate was found to be a cause of occupational hand eczema among
employees in the construction industry, mainly because of the presence
of hexavalent chromate in wet cement (Irvine et al., 1994). The
irritating capacity of cement (abrasive and high pH), combined with
the potent allergen hexavalent chromate, caused occupational hand
eczema in up to 10% of the workers. Addition of ferrosulfate to cement
reduces hexavalent chromate to the trivalent state which has a minimal
bioavailability. Introduction of ferrosulfate addition to cement in
the Scandinavian countries has significantly reduced occupational hand
eczema (Avnstorp, 1992).
5.9.1.3 Fragrances
Allergic contact dermatitis can also be caused by fragrances
(Johansen & Menné, 1995; Johansen et al., 1996a, 1997; Scheinman,
1996). In a study by Nielsen & Menné (1992), 1%-2% of the 567
participants were affected by fragrance allergy (Table 18). Geier &
Schnuch (1996) found a frequency of fragrance allergy of 8 to 17%
among eczema patients.
5.9.1.4 Preservatives
Preservatives are widely used in water-based cosmetics,
households, and industrial products. Preservatives have antimicrobial
effect against a wide range of microorganisms. The most widely used
preservatives in cosmetics are the parabens, formaldehyde and
formaldehyde releasing substances (quaternium 15, diazolidinyl urea,
imidazolidinyl urea) and the isothiazolones (Andersen & Rycroft,
1991). Formaldehyde and isothiazolones are also frequently used in
industrial products. In a British study, the frequency of contact
allergy in dermatology patients varied between 1 and 3% for different
preservatives (Jacobs et al., 1995). In another study the prevalence
of contact allergy to formaldehyde in eczema patients was as high as
8% (Flyvholm et al., 1997).
5.9.1.5 Medicines
The frequency of allergic contact dermatitis to topical medicines
varies considerably from country to country, and even within regions,
depending upon the availability and product preference by local
prescribing doctors. Most cases of allergic contact dermatitis to
medicines are caused by substances having doubtful documented effect
or which can be replaced by other medicines (Angelini, 1995).
Systematic studies have shown that 1-3% of eczema patients are
contact-sensitized to topically used steroids (Dooms-Goossens &
Morren, 1992; Lauerma, 1992). In addition, traditional remedies, such
as balsam of Peru and Propolis, are well-known sensitizers (Li, 1995).
5.9.1.6 Plants and woods
Plants and woods contain a diversity of strong and weak allergens
(Ducombs & Schmidt, 1995). Allergic contact dermatitis to plants and
woods usually presents itself with acute oedematous bullous lesions
that spread to skin areas distant from the primary contact. Airborne
and dustborne patterns can be seen with severe facial dermatitis and
flexural dermatitis. In North America poison ivy and poison oak are
important plants because of their high content of urushiols, which are
potent allergens (Kligman, 1958a,b). These allergens are also present
in plants and trees in Asia and Australia. Allergic contact dermatitis
from these plants represents an occupational health problem for
outdoor workers.
The Compositae family comprises 13 000 species. It includes
decorative flowers such as chrysanthemums and dahlias, but also a
number of common weeds and salads. Some cases of Compositae
dermatitis present an airborne pattern often with a photo-aggravated
facial dermatitis. This type of dermatitis is caused by different
weeds in Europe, North America and Australia. Another variant of the
disease has been recognized, giving a high morbidity and some
mortality, in rural areas in India. The disease has been termed
parthenium dermatitis after the offending plants ( Parthenium
hysteropherous) (Lonkar et al., 1974). The main allergens in
Compositae plants are different sesquiterpene lactones. In
consecutive patch-tested eczema patients in Northern Europe 1-3%
reacted to the sesquiterpene lactone mix (Ducombs et al., 1990). Most
of these were hobby gardeners with severe hand eczema, but there were
some cases among professional gardeners. The development of the
sesquiterpene lactone mix illustrated how new diagnostic technologies
can change the understanding of allergic skin diseases. The
development of these techniques for routine testing has dramatically
changed the prognosis for these patients, as the correlation between
plant contact and their hand eczema was unrecognised earlier.
5.9.2 Lack of a relationship between atopy and allergic contact
sensitization
Atopic hospital employees, who performed wet work, had a similar
prevalence of contact allergy (22%) to that of their non-atopic
colleagues (21%) (Lammintausta et al., 1982). In a hospital-based case
series, the prevalence of contact allergy against ingredients of
topical medications was common in subjects with atopic dermatitis.
However, the sensitization rate to multiple other substances was low
among these patients (Lammintausta et al., 1992). In another hospital
based-study of patients with hand eczema, participants with past or
present atopic disease showed a positive patch test reaction in a
significantly lower proportion than non-atopic individuals.
Furthermore, of all participants with a history of atopy, 22% had
developed allergic contact dermatitis, while the corresponding figure
for non-atopics was 45% (Rystedt, 1985). The reason for the apparently
lower prevalence of allergic contact dermatitis in atopics might be an
abnormal function of T-lymphocytes in atopic patients (Menne et al.,
1987; Hanifin & Chan, 1995). However, a population-based study in
Norwegian school children indicated a higher rate of contact allergies
in atopic compared to non-atopic participants (Dotterud & Falk, 1994;
Dotterud & Falk, 1995).
5.10 Diet
It has been hypothesized that changes in food habits are related
to the increased prevalence of atopic diseases. Seaton et al. (1994)
suggested that the increase in atopic disorders in the United Kingdom
may in part be a result of a change in diet. Between 1961 and 1985 the
average weekly consumption of fresh fruit, green vegetables, potatoes,
fresh fish and red meat was reduced in Britain. The authors argue that
a reduced dietary intake of natural antioxidants is related to a
higher susceptibility to oxidant attack and airway inflammation. The
food groups that were consumed less frequently between 1961 and 1985
are main sources of antioxidants, such as vitamin C and beta-carotene,
ubiquinone, and cofactors for antioxidant defence mechanisms, such as
selenium, zinc and copper.
The relationship between certain food groups and the risk of
asthma was recently studied by Hodge et al. (1996). In this study,
diet was assessed in 574 children by a detailed food frequency
questionnaire including 200 food items, which was related to airway
disease defined by respiratory symptoms or airway responsiveness to
exercise. Children who ate fresh, oily fish had a significantly
reduced risk of current asthma. No other food groups or nutrients were
significantly associated with either an increased or reduced risk of
current asthma (Hodge et al., 1996). The results of a cross-sectional
study in Leipzig, Germany, indicated that a change towards a higher
consumption of margarine was positively associated with hay fever; in
turn, changes in the consumption of butter showed an inverse
association with hay fever and atopic sensitization (von Mutius et
al., 1998).
Adverse reactions to food are a commonly encountered condition,
especially in infancy and early childhood. The highest prevalence
occurs between 1.5 and 3 years of age when in this age group as many
as 25% have been reported to have adverse reactions to food (Björksten
& Kjellman, 1987). However, only a minority of these reactions depend
on immunological mechanisms (i.e., allergy) (Björksten & Kjellman,
1987). Epidemiological studies on the most important dietary factors
in relation to atopic diseases are reviewed below.
5.10.1 Breast feeding
The prophylaxis of atopy has been sought by elimination diets and
by other preventive measures (Saarinen & Kajosaari, 1995). The role of
breast feeding and/or avoidance of formulas based on cows' milk in
early infancy has been the focus of much controversy (Anonymous,
1982). Small amounts of protein ingested by the mother are secreted
unchanged into breast milk. Potentially allergenic food ingested by
the mother may thereby be transferred to the infant and cause
sensitization. For this reason, maternal dietary restriction during
lactation has been recommended (Hattevig et al., 1989). A prospective,
long-term follow-up study from infancy to early adulthood indicated
that breast feeding can protect against development of atopic disease
(Saarinen & Kajosaari, 1995). Differences between the infant feeding
groups were identified for atopic eczema, food allergy and respiratory
allergy. Breast feeding for longer than 1 month without other milk
supplements offers prophylaxis against food allergy at 3 years of age
and also against respiratory allergy at age 17 years. Six months of
breast feeding was required to prevent eczema during the first 3 years
and possibly also to prevent substantial atopy in adolescence. Thus in
this study, breast feeding seemed to confer long-term protection
against allergic sensitization.
In a trial to examine the effect of different feeding patterns on
the incidence of atopic disease in newborns with a family history of
atopy, the incidence of atopic symptoms in the control group of
infants was 27% for those with a single relative with atopy and 50%
for infants with a biparental history of atopy (Bardare et al., 1993).
Breast feeding effectively reduced the incidence of atopic symptoms
when the mothers complied with prescribed dietary restrictions. This
may indicate that dietary restrictions are especially important in
infants with biparental history of atopy.
The stated reasons for discouraging the premature introduction of
solid food include the possible risk of excessive weight gain,
vulnerability of the gut to infection, and increased susceptibility to
the development of allergic disease. The incidence of gastrointestinal
illness, wheeze, and nappy dermatitis was not found to be related to
early introduction of solid food feeding. There was a significant but
rather small increase in respiratory illness at a particular age among
infants given solids early. The incidence of eczema was increased in
those infants who received solids at 8-12 weeks of age (Forsyth et
al., 1993).
Risk of atopy has been associated with a high cord-blood IgE
(Kjellman & Croner, 1984). Therefore, many studies have focused on
investigating those infants with high cord-blood IgE because
development of an atopic disorder is more likely in these children. A
dual approach of allergen avoidance, focusing on foods (breast milk
and extensively hydrolysed formulas) and aero-allergens (treatment of
bed- and living-room with acaricides), in comparison with controls who
did not undergo any intervention, was beneficial in selected high-risk
infants (Hide et al., 1994). Avoidance of potent food allergens in
early life may increase the threshold for sensitization in those
high-risk infants. Whether sensitization has been avoided or merely
deferred has yet to be proved. Reduced exposure of infants to
allergens in food and in house dust lowered the frequency of allergic
disorders in the first year of life for high-risk children,
pre-natally randomized to a prophylactic or control group (Arshad et
al., 1992). A similar result was found in an outpatient follow-up of
777 infants with a very low birth weight. Comparison after random
assignment to early diet of human milk versus cows' milk-based
pre-term or term formula revealed an increased risk of atopy for early
exposure to cows' milk (Lucas et al., 1990).
5.10.2 Sodium
Asthma mortality is related geographically to sales of table salt
and both epidemiological and experimental evidence suggest that a high
dietary sodium intake may increase airway responsiveness. Studies have
shown that a low-sodium diet contributes to a decrease in symptoms in
children with severe asthma. Further investigations have presented
ecological, observational and experimental evidence supporting a
relation between salt intake and airway responsiveness (Tribe et al.,
1994). Regional data from England and Wales showed a strong
correlation between table salt purchases and asthma mortality for
adult men and children of both sexes, but not for adult women. Asthma
mortality in women was found to be more closely related to other
factors (Burney, 1987).
As part of a wider survey on asthma, 138 men living in two
Hampshire villages in England underwent a bronchial histamine
challenge test and had their 24-h urinary excretion of sodium
measured. Bronchial reactivity was strongly related to 24-h excretion
of sodium, suggesting that a high-sodium diet may enhance bronchial
reactivity (Burney et al., 1986). A study on the effect of dietary
sodium intake on the airway response to histamine supports the
hypothesis that a high-sodium diet increases bronchial reactivity in
men but not in women and suggests that moderate restriction of sodium
intake in asthmatic men would reduce bronchial reactivity (Burney et
al., 1989b).
A controlled cross-over study of 14 asthmatics found that high
salt intake worsened forced expiratory volume in one second (FEV1),
peak expiratory flow rate (PEF) and symptoms (Medici & Vetter, 1991).
This was interpreted as indicating a generalized dysfunction of
cellular sodium regulation and providing an explanation for the
salt-sensitivity of the asthmatics. In contrast, another study
(Britton et al., 1994) found no support for the hypothesis that a high
dietary sodium intake is a risk factor for airway hyperreactivity or
atopic disease in the general adult population. No relationship either
between Na+ and K+ intake assessed by a 7-day recall and bronchial
hyperresponsiveness or chronic respiratory symptoms was found in a
sample of 205 subjects (Zoia et al., 1995).
5.10.3 Selenium
There seems to be an association between reduced serum selenium
concentration and lowered activity of the selenium-dependent enzyme
glutathione peroxide (GSH-Px) (Hasselmark et al., 1993). The aim of a
double-blind randomized study in 24 patients suffering from asthma was
to investigate whether selenium (Se) supplementation in asthmatic
patients increases GSH-Px activity and possibly brings about clinical
improvement in the Se-supplemented group as compared to the placebo
group. In the Se-supplemented group there were significant increases
in serum Se and platelet GSH-Px activity after intervention, while no
significant changes in these parameters could be observed in the
placebo group. Although there were no significant changes in lung
function measures in the Se-supplemented or placebo group, a
statistically significant clinical improvement was observed in the
Se-supplemented group. There are several possible mechanisms whereby a
reduced selenium status with associated lower GSH-Px activity may
contribute to the pathogenesis of asthma. Further research into the
role of selenium and GSH-Px is required (Beasley et al., 1991).
Selenium concentrations and GSH-Px activity were lower in 56
asthmatic subjects compared to 59 control subjects (Flatt et al.,
1990). In both the asthmatic and control groups, the mean whole blood
selenium concentrations and GSH-Px activity were generally higher in
those who reported having eczema or rhinitis or had positive responses
to skin-prick tests. These findings are consistent with the hypothesis
that low selenium concentrations may play a role in the pathogenesis
of asthma in New Zealand.
5.10.4 Vitamins and antioxidants
Data from the Nurses Health Study (Troisi et al., 1995), a
prospective investigation of major chronic diseases, suggests that
anti-oxidant supplementation and intake of various fats during
adulthood are not important determinants of asthma, although vitamin E
from diet may have a modest protective effect. An effect of vitamin E
on the inflammatory process seems plausible. The relation between lung
function and dietary intake of the antioxidant vitamins C and E in the
general population was investigated in a cross-sectional survey of a
random sample of adults of Nottingham, England (Britton et al., 1995).
After adjustment for gender, height, skin-prick test result and
smoking, lung function parameters were significantly and independently
related to the mean daily intake of vitamin C. These data support the
hypothesis that lung function in the general population is related to
antioxidant intake and that these vitamins may play a role in
protecting against the development of chronic obstructive pulmonary
disease. The generalizability of these study results, however, may be
limited due to a low participation rate of less than 60%. In the
Zutphen study, fruit intake was inversely related to the incidence of
chronic nonspecific lung diseases (Miedema et al., 1993). No
association was observed with intake of several antioxidants. The
second US National Health and Nutrition Examination Survey (NHANES)
reported an inverse association between wheezing and serum vitamin C
and the serum zinc/copper ratio. These data suggest that several
dietary constituents may influence the occurrence of respiratory
symptoms in adults, independently of cigarette smoking (Schwartz &
Weiss, 1990). The relationship between ventilatory function and winter
fresh fruit consumption was studied in a random sample of British
adults (Strachan et al., 1991). These findings suggest that
antioxidant and other actions of vitamin C may protect against
pulmonary emphysema and may reduce bronchorestrictor responses to
environmental pollutants.
5.11 Number of siblings and crowding
Factors playing a role in the prevalence of allergic disease in
industrialized countries might be associated with the generally
improved standard of living (Williams et al., 1994b). Along with this
change goes a smaller family size and lower number of children in
families. Studies have observed a decrease in the prevalence of
allergic rhinitis, eczema (Strachan, 1989, 1996)) and asthma symptoms
(Shaw et al., 1994) with an increase in the number of older siblings.
In addition, it was shown that the prevalence of atopic sensitization
decreases with an increasing number of siblings (von Mutius et al.,
1994a), and a strong inverse relation was found between atopic
sensitization and the number of persons per room in households
(Braback et al., 1995). Viral or bacterial cross-infections,
especially in early life, which may occur more frequently in larger
families, have been discussed to have a protective role against atopic
disease by preventing proliferation of Th2-lymphocytes (Romagnani,
1992b; Holt, 1994; Martinez, 1994). Helminth infestations have also
been suggested to have a protective effect against allergy development
(Williams, 1992) but studies in populations with a high prevalence of
asthma indicated no such beneficial effect (Mantle & Pepys, 1974;
Martinez, 1994).
5.12 Indoor environment
One of the most important changes in lifestyle during the past
decades in industrialized countries is the change in indoor climate
mainly due to modern building and furnishing materials, better
insulation, wall-to-wall carpeting, increased indoor temperature and
less ventilation. As a consequence of these factors and behavioural
changes, such as indoor pet keeping, many pollutants and allergens can
accumulate within dwellings. This may lead to higher sensitization
rates to indoor allergens and consequently to an increased incidence
of allergic diseases like asthma. This is especially relevant because
people in industrialized countries spend up to 90% of their time
indoors (Platts-Mills, 1994; Berglund et al., 1994). A variety of
indoor and outdoor sources contribute to the indoor pollution burden
and might play a role in the development or exacerbation of allergic
disease. Important indoor sources are respirable particles from stoves
and tobacco smoke; combustion products from cookers, ovens and
heaters; formaldehyde from foam insulation, chipboards, furniture and
fabrics; volatile organic compounds (VOCs) and other chemicals from
paints, sprays fabrics, and combustion; biological material from
animal sources such as dust mites, cats, etc., or from fungi, bacteria
and pollen (Angle, 1988; Karol, 1991). Ventilation, humidity and
temperature of dwellings also have an important influence on indoor
concentrations of many pollutants and allergens (Brunekreef et al.,
1989; Beggs & Curson, 1995). For many indoor factors, there is still a
lack of good epidemiological studies.
5.12.1 Tobacco smoke
Passive exposure to tobacco smoke is an important risk factor for
childhood asthma and wheezing, particularly when the child's mother
smokes. Many studies have reported a higher risk of asthma or
bronchial hyperresponsiveness among children exposed to tobacco smoke
(Ware et al., 1984; Dekker et al., 1991; Martinez et al., 1992;
Forastiere et al., 1992) and younger children are particularly prone
to these side effects (Arshad et al., 1993; Stoddard & Miller, 1995;
Platts-Mills et al., 1995). Children of smoking parents have also
increased reactivity to allergens as assessed by skin-prick test
(Martinez et al., 1988; Braback et al., 1995). Furthermore, children
whose mothers smoked during pregnancy show increased IgE-levels in
cord blood and increased risk of infant allergy (Magnusson, 1986).
Therefore, the increasing prevalence of smoking in women of
childbearing age may have contributed to the increase in atopic
disease in children (Burney et al., 1990).
Although a number of studies in adults have shown an association
between active smoking and asthma, others have failed to find such an
association. Thus, the evidence is not conclusive and the effect may
be only small (Platts-Mills et al., 1995). Active smoking can increase
total IgE serum concentration and ex-smokers show a decline in serum
IgE concentration after cessation of smoking (Burrows et al., 1981).
Studies on occupational allergies showed that the frequency of IgE
sensitization and asthma is higher in cigarette smokers (Anonymous
1985; Venables et al., 1985b). Little is known about the role of
active and passive smoking on allergic rhinitis, and studies show, if
any, only small effects (Ng & Tan, 1994a,b; Strachan, 1995; Tsunoda et
al., 1995).
In summary, smoking is known as a respiratory irritant and as an
important source for indoor pollution. However, although many studies
suggested that smoking plays a role in the etiology of asthma, there
is no clear evidence from the available data to indict smoking as the
driving force for the observed increases in asthma morbidity and
mortality (Weiss et al., 1993). Nevertheless, even if the relative
effect of active and passive smoking may be small, it can still have a
major impact if large parts of populations are exposed (attributable
risk) (Stoddard & Miller, 1995).
5.12.2 Pets
Many animals like cats, dogs, rodents, rabbits and cage birds
live within or in close proximity to homes. About one third to one
half of houses in the USA have a mammalian pet (Colloff et al., 1992;
Ledford, 1994). There is strong evidence that exposure to a number of
animal allergens can lead to primary sensitization and an increased
risk of developing allergic disease (Colloff et al., 1992). The most
important pet allergens are those from cats and dogs (Sears et al.,
1989; von Mutius et al., 1994b; Ledford, 1994; Strachan & Carey,
1995). Even after permanent removal of a cat from the home it may take
several months before the concentration of allergens in domestic dust
falls (Wood et al., 1992; Colloff et al., 1992; Munir et al., 1994b).
Pet owners visiting a home with no pets can bring in pet allergens on
their clothes, and rub it off during their visit, resulting in a
considerable amount of pet allergens in the visited flat (Munir et
al., 1993; Munir et al., 1994a,b).
5.12.3 Biocontaminants
5.12.3.1 House dust mites and insects
House dust mites are an important source of indoor allergens.
Most frequently detected species are Dermatophagoides pteronyssinus,
Dermatophagoides farina, Euroglyphus maynei and Blomia
tropicalis (Colloff et al., 1992; Ledford, 1994). The mites have a
narrow optimal temperature range for growth from 18°C to 27°C
(Ledford, 1994). Because dust mites depend on ambient humidity they
grow poorly in dry or high altitude climates. Mite allergens can be
measured directly by ELISA. Indirect measurements of mite allergens
rely on guanine quantification in house dust. However, this method has
been shown to be of limited clinical value since a considerable amount
of guanine may originate from non-house-dust mite sources (Hallas et
al., 1993). Sensitization to dust mites seems to play an important
role in the relation between damp homes and childhood respiratory
symptoms (Verhoeff et al., 1995). High mite allergen exposure has been
reported to increase the risk of sensitization in atopic children (Lau
et al., 1989) and exposure in early childhood has been shown to be a
determinant of subsequent asthma (Sporik et al., 1990). Sensitization
to Dermatophagoides pteronyssinus and other allergens was addressed
in a study performed shortly after the fall of the Berlin wall on
9- to 11-year-old children in eastern and western Germany. Skin-prick
testing showed sensitization rates of 10.3% to Dermatophagoides
pteronyssinus (wheal reaction >3mm) in western Germany and of
4.2% in eastern Germany, indicating that environmental and lifestyle
factors may have an influence on sensitization to common allergens
(von Mutius, 1994b).
Cockroaches are also a substantial indoor allergen source.
Sensitization has been shown to be a risk factor for emergency room
visits in asthmatics (Pollart et al., 1989). Exposure to high levels
of cockroach allergens and allergy against cockroaches was found to
explain much of the asthma-related health problems in inner-city
children from the USA (Rosenstreich et al., 1997). Exposure to
cockroaches has also been associated with allergic rhinitis (Ng & Tan,
1994b). Few data on sensitization to cockroaches exist for central
Europe (Colloff et al., 1992), but the available data suggest that the
prevalence of sensitization to cockroaches is low (Mosimann et al.,
1992; Munir et al., 1994a). Other allergens from insects include
moths, crickets, midges, locusts, beetles and various flies (Ledford,
1994).
5.12.3.2 Moulds
The most common indoor moulds responsible for allergies are
Aspergillus species, Cladosporium, and Penicillium (Ledford,
1994). Moulds are responsive to temperature, humidity and substrate
moisture level (Beggs & Curson, 1995). Damp housing has frequently
been shown to be a risk factor for respiratory symptoms in children
and adults (Brunekreef et al., 1989; Dekker et al., 1991; Brunekreef,
1992) and may play a role in the sensitization to mould allergens
(Verhoeff et al., 1995). However, it has been suggested that the
observed association between damp housing and childhood asthma may be
partly due to parental over-reporting (Strachan, 1988). Other possible
indoor sources of fungal allergens include air conditioning equipment,
humidifiers, degrading organic materials, and soil used for indoor
plants; these sources can also be important for allergens from
bacteria or protozoa (Dekker et al., 1991; Ledford, 1994).
5.12.4 Other indoor factors
A large variety of everyday rubber products (e.g., balloons, baby
pacifiers, sports equipment, adhesives, etc.) contain natural latex,
which may be a source of sensitization and a factor in allergic
disease expression. Airborne leaf parts of the popular indoor plant
Ficus benjamina (weeping fig) also contain latex particles (Axelsson
et al., 1990; Bircher et al., 1993). It is not yet known whether
natural latex, which is a well known occupational allergen
(Vandenplas, 1995) (see also section 5.8.3) has any impact on allergic
disease on a population level. Contrary to expectation, non-feather
bedding, especially foam pillows, have been suggested to be a possible
determinant for symptoms of asthma in adolescents (Strachan & Carey,
1995).
5.13 Indoor and outdoor environmental factors
5.13.1 Nitrogen dioxide
Nitrogen dioxide (NO2) is a strong oxidant. Indoor sources are
cigarette smoke, gas and oil heaters or cookers, which can result in a
high indoor concentration (Angle, 1988; Wardlaw, 1993). Main outdoor
sources are combustion of fossil fuels in motor vehicles and power
generation (Wardlaw, 1993). Combustion processes also generate a
mixture of NO2 and nitric oxide (NO), and formation of indoor nitrous
acid (HNO2) has also been demonstrated and associated with adverse
respiratory effects (Samet et al., 1993). Experimental exposure and
several epidemiological studies are inconclusive concerning the effect
of NO2 on lung function (Ware et al., 1984; Angle, 1988; Neas et al.,
1991; Gorski & Tarkowski, 1992; Samet et al., 1993). The current
understanding of NO2 is that it can decrease pulmonary function in
asthmatics, but its overall role in the development of allergic
disease is not clear (Angle, 1988).
5.13.2 Sulfur dioxide, acid aerosols and particulate matter
The sources of these major outdoor air pollutants are mainly
combustion of fossil fuels, like coal and oil, wood, and certain
industrial processes. Sulfur dioxide (SO2) in water forms sulfurous
acid (Gorski & Tarkowski, 1992). The indoor concentration of SO2 is
raised if unvented combustion devices for kerosene are used or when
cigarettes are smoked (Angle, 1988). Several studies on asthmatics
have shown that SO2 can provoke bronchoconstriction and asthma-like
symptoms even at low levels, especially during exercise and mouth
breathing (Linn et al., 1983a,b). Animal studies indicated an
enhancing effect of SO2 on sensitization to allergens (Riedel et al.,
1988).
Acid aerosols are comprised mainly of sulfuric acid and ammonium
bisulfate (Wardlaw, 1993), which has also been shown to cause an
increase in respiratory symptoms and a decrease in lung function
(Koenig et al., 1983; Hackney et al., 1989). Acid aerosols can also
comprise nitric acid, hydrochloric acid and hydroxymethansulfonic
acid. Outdoor airborne acidity is associated with daily respiratory
symptoms in asthmatics (Ostro et al., 1991).
Particulate matter (e.g., dust, dirt and smoke) is a major source
of air pollution and a complex and varying mixture of substances.
Sources are motor vehicle emissions, such as exhaust fumes and abraded
tyre fragments (Williams et al., 1995b), factory and utility
smokestacks, residential wood burning, construction activity, mining,
agricultural tilling, open burning, wind blown dust, fires, etc.
Several studies have suggested an association between particulate
matter (PM) exposure and asthmatic symptoms (Dockery et al., 1989; Xu
& Wang, 1993; Dockery & Pope, 1994; Abbey et al., 1995; Brunekreef et
al., 1995). Particles of a diameter <10 µm (PM10) are especially
important for respiratory disease because they are readily inhaled
deep into the lungs (CDC, 1994). Among them fine particles of a
diameter <2.5 µm and ultrafine particles (<0.1 µm) may be most
important (Peters et al., 1997). Animal studies indicated that diesel
exhaust particles may increase the risk of sensitization against
allergens (Muranaka et al., 1986; Takafuji et al., 1987; Fujimaki et
al., 1994).
5.13.3 Volatile organic compounds, formaldehyde and other chemicals
Volatile organic compounds (VOCs) comprise a broad spectrum of
substances including benzene, toluene, xylenes and aldehydes (Angle,
1988; Becher et al., 1996). VOCs are emitted by a large number of
materials and only few subgroups have been investigated for their
potential role in allergy. The total indoor amount of VOCs and several
sub-types like toluene and terpenes have been associated with
asthmatic symptoms (Norback et al., 1995). Outdoor sources of VOCs
have also been found to be related to increased rates of chronic
respiratory symptoms characteristic of reactive airways (Ware et al.,
1993). Another important VOC is formaldehyde, which is ubiquitous in
the human environment. Important indoor sources are smoking,
particle-boards, foam insulation and textiles (Imbus, 1985; Koenig,
1988). Formaldehyde exposure has been related to asthma and other
allergies (Imbus, 1985; Angle, 1988; Wjst et al., 1994; Norback et
al., 1995), but evidence that formaldehyde exposure can cause allergic
diseases in the airways is limited (Becher et al., 1996). Other
chemicals such as ethylenediamine and isocyanate may also contribute
to the allergic disease burden in the general community (Ledford,
1994), but population-based data showing an association between
allergies and indoor contamination by these pollutants are not
available. The increasing use and diversity of household cleaning
materials during the past decades have also been implicated in the
expression of allergic diseases (Williams, 1992).
5.14 Outdoor air pollution
A link between outdoor air pollution and the increased prevalence
of respiratory allergies such as asthma and allergic rhinitis has been
suspected for some time. However, whether atmospheric air pollution
can cause respiratory and other allergies is still not clear (Wardlaw,
1993). Contamination of the air with plant pollen is a major natural
source of air pollution. Important causes of outdoor air pollution are
burning fuels such as coal, oil and wood, smelting of ores, and other
industrial processes. Natural sources like volcanoes play only a small
role. Motor vehicle tail-pipe emissions are a major contributor of
several pollutants, such as diesel particles, oxides of nitrogen
(NOX), carbon monoxide and other airborne particles (Bascom, 1996).
The increase of allergic respiratory diseases coincided with a
decrease in many outdoor air pollutants like SO2 and total suspended
particles (TSP) (Weiss et al., 1993), whereas air pollution from motor
vehicle emissions (e.g., O3, NOX) increased during the same period
(Newman Taylor, 1995). Motor vehicles are also a major source of fine
suspended particles (see section 2.5.3). Most outdoor air pollutants
provoke more or less severe adverse effects in asthmatics, and
exposure to multiple pollutants may cause synergistic effects (Koenig
et al., 1990). A number of experimental animal studies indicate an
association between allergy and air pollution (Osebold et al., 1980;
Biagini et al., 1986; Muranaka et al., 1986; Takafuji et al., 1987;
Riedel et al., 1988; Takafuji et al., 1989; Suzuki et al., 1993;
Fujimaki et al., 1994). It is also assumed that pollutants can enhance
the allergenicity of common allergens like pollen (Ishizaki et al.,
1987; Behrendt et al., 1991; Gorski & Tarkowski, 1992). However, the
observation that the sensitization rate to common allergens in cities
in eastern Germany with previously high levels of traditional air
pollutants such as SO2 or particulate matter was low in comparison to
that of western Germany cities argues against the hypothesis that
traditional air pollution from industrial production and household
coal burning is the major factor driving the changes in morbidity
patterns of allergic disease (von Mutius et al., 1992; von Mutius et
al., 1994b). Similarly, a Swiss study reported effects of moderate
average air pollution concentration from PM10, NO2 and SO2 on
respiratory symptoms, such as chronic or nocturnal dry cough and
bronchitis, but not between air pollution and asthmatic and allergic
symptoms or diseases in children (Braun-Fahrländer et al., 1997).
These results are in close agreement with findings from the US Six
Cities Study (Dockery et al., 1989). In addition, the findings of the
ISAAC study do not provide support for an association between air
pollution and childhood wheezing; for example, countries with low
degrees of ambient air pollution such as New Zealand were among those
with the highest prevalence of asthma symptoms (ISAAC Steering
Committee, 1998). Likewise, recent results from the Pollution Effects
on Asthmatic Children in Europe (PEACE) project found only little
overall adverse effect of ambient air pollutants (e.g., PM10, black
smoke, SO2 and NO2) on respiratory health in children (PEACE, 1998).
5.14.1 Pollen and dust
Natural sources of air contamination are plant pollen from grass,
ragweed, trees, etc. (Schutz-Kiss et al., 1995), moulds (Dawson &
Mitchell, 1990), and natural particulate matter such as dust or dirt
(CDC, 1994) which can cause symptoms of asthma and allergic rhinitis.
Their role as a causal factor for the development of these diseases,
however, is not clear. Air pollution due to natural factors like
pollen or dust cannot easily explain the observed increase in allergic
diseases in many regions because humans have always had intense
outdoor contact with these substances. Several asthma outbreaks in
populations have been shown to be related to man-made airborne
allergen pollution. Examples are castor bean dust in USA, South Africa
and Brazil (Anto, 1995), and soybean dust which was released during
unloading of soybeans in the city harbour of Barcelona (Anto et al.,
1989).
5.14.2 Ozone
A major source of ozone (O3) is motor vehicle exhaust. O3 is a
main constituent of photochemical smog, and its formation requires
NOX and reactive organic compounds as precursors and ultraviolet
radiation (Gorski & Tarkowski, 1992; Wardlaw, 1993; Beggs & Curson,
1995). It is the most ubiquitous air pollutant in the USA (Koenig,
1995). Although O3 is mainly an outdoor pollutant, it is also present
in low concentrations in the indoor environment (Koren, 1995). Many
studies have shown that O3 has an aggravating effect in asthmatics
and can reduce lung function (Krzyzanowski et al., 1992; Gorski &
Tarkowski, 1992; Wardlaw, 1993; Koenig, 1995; Koren & Bromberg, 1995).
Unlike SO2 there seems to be no marked difference in acute responses
in asthmatics and non-asthmatics (Koenig et al., 1988). Animal models
suggest that O3, like other air pollutants, may increase
sensitization to allergens (Osebold et al., 1980; Sears et al., 1989).
Ambient levels of O3 may also have a synergistic effect with pollen
in the causation of allergic rhinitis (Bascom et al., 1990). However,
only a few epidemiological investigations have been performed on a
population level to evaluate the effect of ambient O3 on asthma and
allergies (e.g., Braun-Fahrländer et al., 1997) and further
investigations are needed to study the effect of O3 on these
disorders under real-life exposure conditions (Magnussen et al.,
1998).
5.14.3 Motor vehicle emissions
Motor vehicle traffic has increased dramatically in many
countries (Utell et al., 1994) and it has been speculated that this
increase may play a role in the observed changes of the prevalence of
allergies. A number of occupational studies have shown an association
between exposure to motor vehicle exhausts and adverse effects on
respiratory symptoms and lung function (Gamble et al., 1987; Evans et
al., 1988a; Ulfvarson & Alexandersson, 1990; Wade & Newman, 1993;
Raaschou Nielsen et al., 1995). Others, however, have failed to find
such an association (Speizer & Ferris, 1973; Tollerud et al., 1983;
Ames et al., 1984). An association between allergic sensitization and
components of motor vehicle exhaust fumes has been shown in various
animal studies (Osebold et al., 1980; Muranaka et al., 1986; Takafuji
et al., 1987; Riedel et al., 1988; Suzuki et al., 1993; Fujimaki et
al., 1994; Lovik et al., 1997). A number of experimental studies
suggested also an association between allergic disease and traffic
pollution (Molfino et al., 1991; Braun Fahrländer et al., 1994;
Devalia et al., 1994). Several epidemiological studies observed a
relationship between exposure to motor vehicle traffic at residence
and morbidity from respiratory and allergic disorders in adults
(Yokoyama et al., 1985; Ishizaki et al., 1987; Nitta et al., 1993) and
in children (Wjst et al., 1993; Edwards et al., 1994; Weiland et al.,
1994; Keil et al., 1996; Oosterlee et al., 1996; Duhme et al., 1996;
Brunekreef et al., 1997; Duhme et al., 1998a). Air pollution from
motor vehicle traffic derives also from mechanical abrasion of tyres,
which contain potentially allergenic latex particles (Williams et al.,
1995b). However, evidence that exposure to motor vehicle traffic can
cause asthma or allergies is not conclusive.
5.15 Conclusions
Asthma and allergic disorders represent a substantial burden not
only on the affected individuals but also on health care resources in
many countries. The costs of asthma are partly due to uncontrolled
disease, and are likely to rise as its prevalence and severity
increase (Barnes et al., 1996). One approach to reduce costs would be
to improve disease control. Another approach would be to reduce the
prevalence of asthma by preventive measures, and this would be
accompanied by a reduction in the costs of treatment and care (Peat,
1996). Environmental factors that have changed in the last decades
appear to be largely responsible for the observed increase in the
prevalence of asthma and allergic disease in many countries. The
determinants of these changes need to be identified in order to design
interventions that can reverse these trends. Such prevention
strategies in the field of asthma and allergies can aim at high-risk
groups or at populations as a whole (Rose, 1985). It is important to
note that even if a preventive measure offers little to each
individual it can bring large benefits to the community into which the
preventive measure is introduced (prevention paradox) (Rose, 1985).
It is often impossible to blame a single culprit within a complex
mixture of behaviours and exposures (such as indoor or outdoor air
pollution) for observed adverse health outcomes, and the studied risk
factor might also have different effects in the presence of other
factors (Greenland, 1993). Because randomized assignment of
individuals to certain exposures is impractical, if not unethical, in
environmental epidemiology, researchers rely mainly on data from
non-experimental studies with well-known inherent methodological
shortcomings (Rothman, 1993). Depending on the hypotheses being
studied, a number of epidemiological research strategies are
available. The applications, strengths and weaknesses of different
studies have been described (e.g., Hennekens & Buring, 1987; Rothman,
1993; Morgenstern & Thomas, 1993). Interpretation of (epidemiological)
study results has to consider random errors of estimation (due to
chance) and systematic errors or bias. Important sources of bias
(selection bias, information bias and confounding) may hamper the
validity of observed results. Therefore, possible systematic errors
should be considered already at the planning stage of any study and
measures should be implemented to minimize or control bias. It is also
crucial at the planning stage of a study to perform statistical power
calculations to evaluate how many study participants are needed to
assure a given probability of detecting a true effect of a given
magnitude.
Epidemiological studies applying accurate exposure and disease
measurements and taking into account important covariates, confounders
and effect modifiers are needed (Hatch & Thomas, 1993; Prentice &
Thomas, 1993). They can make an important contribution through
regulatory decisions in public health in the difficult field of risk
assessment for complex exposures.
Besides other measures, occupational sentinel health events
(Mullan & Murthy, 1991) and structure-activity research (Jarvis et
al., 1996; Graham et al., 1997) may be useful tools to evaluate the
sensitizing capacity of a chemical substance in populations. Lists of
well-known allergens categorized according to potency and degree of
exposure are available (Kayser & Schlede, 1995), and criteria for
classification of sensitizing substances in the environment have been
defined. Furthermore, by comparing different populations with
different grades of exposures, major disease determinants can be
uncovered that would otherwise remain undetected, if the suspected
risk factors show only little variation within a single population
(Rose, 1985). For comparison reasons, data should be collected and
analysed in a standardized way wherever possible. Using such
strategies, major health determinants have been successfully described
and studied in other fields, such as cardiovascular diseases and
cancer. It is an essential prerequisite of such internationally
conducted studies to obtain the health outcome and exposure data in a
standardized way. New epidemiological initiatives investigating
determinants of asthma and allergies need to incorporate these
principles, as is the case with the ongoing International Study of
Asthma and Allergies in Childhood (ISAAC) (Asher et al., 1995; ISAAC
Steering Committee, 1998).
6. HAZARD IDENTIFICATION: DEMONSTRATION OF ALLERGENICITY
6.1 Hazard and risk; allergy and toxicity
The conventional scheme for chemical risk assessment for human
health protection follows the sequence:
a) Hazard identification; what is the potential of the substance to
cause harm (sensitization and provocation of an allergic
reaction), and what is the dose-response relationship?
b) Exposure assessment
c) Risk assessment: what is the likelihood of eliciting an allergic
reaction in humans at the relevant level of exposure; are there
any groups of increased susceptibility?
d) Risk characterization: non-scientific consideration of the risk
weighed against the benefit of using the substance resulting in
the decision to ban or limit exposure (NAS, 1983, 1993).
Hazard identification, therefore, comprises procedures to
determine the potential of a substance to induce allergy or elicit
allergic reactions and the relationship between those properties and
the circumstances of exposure.
The prediction may be based on theoretical considerations of
chemical structure, possibly on in vitro experimental results, on
in vivo animal data, and on prior observations in humans. If it
depends on the results of laboratory studies and not on clinical
observations, the prediction, as is common to toxicity tests, must
take account of species differences in metabolism, responsivity, dose
(exposure), and the adequacy of validation of the experimental system.
Testing allergenic potential requires study of selected
immunological effects and differs from conventional toxicity testing
in the nature and content of its procedures, which are focused on
responses of the immune system and not on general screening for
changes in all body systems. In both types of testing, however, there
will be some form of relation between dose (exposure) and effect, as
the capacity of a substance to produce effects, its potency, will be
represented by the dose (exposure) required to produce sensitization
(or toxicity). A strong sensitizer will require only a small dose,
whereas a less potent compound will require a higher dose, or multiple
exposures. Unlike conventional toxicity, further exposure of a
sensitized animal (or man) will elicit a harmful allergic reaction
after a much smaller dose than that required for sensitization,
although there will still be a graduation of the severity and nature
of the hypersensitivity reaction, for example ranging from slight
bronchoconstriction to fatal bronchospasm or anaphylaxis after
respiratory challenge.
An additional important difference between conventional toxicity
and allergy is that allergic sensitization (the induced state of
hyperreactivity to a substance) normally persists for a long time,
even for life, whereas for many toxic responses a state of lasting
responsiveness is not induced. It is possible for different types of
hypersensitization and provocation to be effective in the same organ
but it is also possible for the route of sensitization and response to
subsequent challenge to differ, e.g., sensitization via the skin and
subsequent asthma on inhalation exposure.
6.1.1 Testing allergic potential and toxicity testing
Much testing to identify toxic hazard is done during industrial
development of substances for purposes ranging from new medicines or
consumer products to industrial intermediates or pesticides. There are
well-developed regimes of accepted procedures done under controlled
circumstances applicable to each intended use, and the results are
used for regulatory purposes to control risk. Most of these procedures
are not directed at revealing effects involving the immune system,
although indirect indications may sometimes be obtained that can
arouse suspicion that the immune system may have been affected.
Certain specialized procedures are also conducted, based on
consideration of the way in which humans may be exposed (e.g., in the
skin or by inhalation), which experience has shown can reveal certain
types of sensitizing and allergenic potential. The latter types of
test are the most important in the present context. The procedures and
their value and limitations are discussed here.
Laboratory tests, especially in vivo procedures, should be done
in such a way as to minimize the need for experimental animals and
scarce human and technical resources, and every attempt is made to
extract as much information as possible from the work that is done.
That includes considerable attention paid to quality assurance of
tests.
Accordingly, the testing of a new substance will follow a
sequence, moving from theoretical to practical procedures. Only those
techniques focused on the immune system are noted here, but it must be
realized that product development and occupational safety needs
require many other studies, too.
6.1.2 Databases and prior experience
A preliminary search should always be made for any information
about experimental or clinical findings about the immunological
consequences of exposure to the substance. This may include special
consideration of any groups considered to be particularly susceptible,
for example, because of pre-existing disease.
6.2 Validation and quality assurance
Obviously, the ideal situation would be that predictive tests
yield easy-to-interpret outcomes and no false negatives or false
positives, and that the tests will always give the same results,
regardless of when or where they are carried out. In order to achieve
this goal, validation of tests is required. Validation should occur at
two levels: first at the level of technical quality and second at the
level of specificity and sensitivity of the assay. It is important
that potential methods are carefully evaluated for interlaboratory
reproducibility and transferability, and for their ability to predict
an in vivo end-point. Owing to the complexity of the immune system,
it is quite likely that predictive assays for the capacity of
chemicals to induce skin, respiratory or food allergy, or autoimmunity
will not always function adequately to show the absence or presence of
such activity (Kammüller, 1996). Therefore, the outcome of these tests
should always be evaluated with great care.
6.3 Structure-activity relationships
Structure-activity models are directed towards a fuller
understanding of the relationship between chemical structure and
physicochemical properties and skin-sensitizing activity, with the
objective of deriving ideally quantitative structure-activity
relationships (QSAR). In this context, parameters that appear to be of
particular importance are protein reactivity and lipophilicity
associated with the capacity to penetrate into the viable epidermis
(Basketter & Roberts, 1990; Barratt et al., 1994a). The correlation of
the protein reactivity of chemicals with their skin-sensitization
potential is well established (e.g., Dupuis & Benezra, 1982), and it
is accepted that if a chemical is capable of reacting with a protein,
either directly or after appropriate (bio)-chemical transformation,
then it has the potential to be a contact allergen, assuming of course
that it can locate in the appropriate epidermal compartment.
It is not within the scope of this monograph to give a
comprehensive description of all the available models. A common
feature of many of these models is that their development was based
upon the mechanism of sensitization, i.e., the absorption of the
chemical sensitizer through the skin, followed by its covalent
modification of a skin-associated protein. Each of the existing
structure-activity relationship (SAR) models proposes structural
alerts, i.e., moieties associated with sensitizing activity. In all
cases, the structural alerts comprise electrophilic moieties, or
moieties that can be metabolized into electrophilic fragments
(proelectrophiles). For example, Benezra et al. (1985) developed a
hierarchical index of structures believed to be associated with
allergic contact dermatitis that contained amines, ketones, metals,
nitrogen-containing heterocycles, and oxygen-containing heterocycles,
among others. Barratt et al. (1994a,b) developed a list of structural
alerts based on the requirement for protein reactivity that included
alkylating, acylating, and arylating agents, electrophiles, thiol
exchange compounds, and free radical generators. Many of the existing
SAR systems have incorporated physicochemical considerations (Roberts
& Basketter, 1990a,b; Basketter et al., 1992; Ashby et al., 1995). The
mathematical model described by Roberts & Basketter (1990a,b) for
alkyl transfer agents has incorporated both a rate constant for
reaction of a chemical with a nucleophile, and a lipophilicity factor
(log P). Each of these models has been successful in predicting the
activity of moderate to strong contact sensitizers. Barratt et al.
(1994b) reported the greatest success in predicting active sensitizing
potential (98%). A smaller database using similar alerts (Payne &
Walsh, 1994) had a much poorer positive predictive ability of 57%. The
classification model of allergic contact dermatitis used by Hostynek
et al. (1996) made use of several parameters and multiple regression
to predict 79% of the active sensitizers and 88% of the inactive
chemicals. The relative alkylation index (RAI) is useful only in the
prediction of a homologous series of chemicals (Roberts & Basketter,
1990a,b). The model used by Benezra et al. (1985) did not specify
whether a validation was attempted, therefore predictive ability is
not known.
6.3.1 Case-Multicase system
The Case-Multicase system is not dependent upon a particular
mechanism of sensitization and has shown ability to predict activity
of weak sensitizers (Graham et al., 1996). The model operates by
fragmenting chemicals in the database into substructures containing
two or more heavy (non-hydrogen) atoms. It then identifies those
fragments that are statistically associated with active chemicals and
uses such fragments as structural alerts for prediction of test
chemicals.
The database for this model was derived from reports of animal
and human studies and consists of more than 1000 chemicals (Graham et
al., 1996). The model identified 49 structural alerts of allergic
contact dermatitis. The major ones were: (a) a nitrogen double-bonded
to a carbon or a nitrogen; (b) substituted aromatic structures; (c)
thiol- and disulfide-containing fragments; and (d) electrophilic
moieties. The model has been evaluated by testing its ability to
predict correctly the activity of chemicals for which there is
evidence of sensitization ability. The concordance between predictions
by the model and established evidence of sensitization was 90% (Graham
et al., 1996).
6.3.2 DEREK skin sensitization rulebase
A historical database (Cronin & Basketter, 1994) containing
results of about 300 guinea-pig maximization tests (Magnusson &
Kligman, 1970a,b), carried out over a number of years according to a
single protocol on defined single substances, was used to derive a set
of structural alerts for skin sensitization. The approach employed was
to group the substances, where possible, according to their most
likely mechanism of reaction with skin proteins. Where no mechanism
could be clearly identified, structural alerts were derived for groups
of chemicals with similar functional groups. This process initially
resulted in the production of around 40 structure-activity rules
(Barratt et al., 1994a), now increased to over 50. These were
incorporated into the expert system Deductive Estimation of Risk from
Existing Knowledge (DEREK) (Sanderson & Earnshaw, 1991; Ridings et
al., 1996). DEREK contains both a controlling programme and a chemical
rulebase. The chemical rulebase consists of descriptions of molecular
structural alerts, which correlate with specific toxicological
end-points.
Whilst details of the biology of skin sensitization are only
partly understood, it is now widely accepted that the ability to react
with a nucleophile, either directly or after appropriate metabolism,
is a prerequisite for the large majority of skin sensitizers. However,
the potential of a chemical to act as a contact allergen is further
modulated by its ability to penetrate the stratum corneum and
partition into the epidermal compartment of skin; this is apparent
from a number of QSAR studies (Roberts & Basketter, 1990a,b; Basketter
et al., 1992) in which skin-sensitization potential was found to
depend crucially on physicochemical parameters such as the log
octanol/water partition coefficient (log Pow). These parameters have
also been found to be equally important determinants of percutaneous
absorption (Flynn, 1990), with higher log Pow values, i.e., greater
lipophilicity, broadly leading to greater permeability. In QSAR
studies of skin permeability, the in vitro human skin permeability
coefficient has also been shown to decrease with increasing relative
molecular mass (Flynn, 1990) or molecular volume (Barratt, 1995). The
skin-sensitization potential of a series of substituted phenyl
benzoates was found to depend on log Pow and relative molecular
volume in the same way (Barratt et al., 1994c). The logical
consequence is that two chemicals may contain the same structural
alert (i.e., be reactive, presumably by the same mechanism), but one
will be a skin sensitizer because it can penetrate the skin whilst the
other will not be a skin sensitizer because its skin permeability is
too low, e.g., N-methyl- N-nitrosourea and streptozotocin (Ashby et
al., 1995).
6.3.3 SAR for respiratory hypersensitivity
An initial SAR model for respiratory-sensitizing chemicals has
been described (Karol et al., 1996). The model is based on the
Case-Multicase system and the database was derived from a critical
review of the published clinical literature. Criteria for inclusion of
published data included a decrement in pulmonary function resulting
from inhalation challenge with a non-irritating concentration of the
chemical.
In all, 39 respiratory chemical allergens were identified from
the literature search, all being obtained from human studies. Among
the chemicals were diisocyanates, acid anhydrides, antibiotics and
dyes. Since the model requires a data set of inactive chemicals, and
such chemicals could not be found in the literature, chemicals that
were inactive as dermal sensitizers were assumed to be inactive as
respiratory sensitizers as well, and were added to the respiratory
model as "inactive" chemicals.
The model identified structural alerts including the isocyanate
functionality, amines and aromatic fragments. When respiratory
sensitizers were compared with dermal sensitizers for both structural
alerts and physicochemical characteristics, differences were noted.
Among the physicochemical properties, respiratory chemicals had higher
mean relative molecular mass and greater water solubility when
compared with dermal sensitizers (Karol et al., 1996). However, the
discrimination of dermal and respiratory sensitizers remains
problematic.
6.4 Predictive testing in vivo
6.4.1 Testing for skin allergy
6.4.1.1 Testing in guinea-pigs
The guinea-pig was for many years the animal of choice for
experimental studies of contact sensitization, and several test
methods were developed in this species. The Draize test was developed
over 50 years ago (Draize et al., 1944) and was widely used, but this
is no longer the case and it has been superseded. Currently, the best
known and most widely applied are the Buehler test (Buehler, 1965),
the guinea-pig maximization test (Magnusson & Kligman, 1970a,b), and
the guinea-pig optimization test (Maurer et al., 1975), and have
formed the basis of hazard assessment for many years. Both the
Magnusson and the Buehler test are recommended according to an OECD
guideline (OECD, 1992). While these tests differ with respect to
procedural details, they are in principle similar. Guinea-pigs are
exposed to the test material or to the relevant vehicle. In the
Buehler test, both induction and challenge exposures are done
topically; in this test, false negatives are frequently observed. The
test was improved by occluded application of the test compound. In the
guinea-pig maximization test, induction is produced by intradermal and
occluded epidermal exposure, and in the optimization test induction is
done by intradermal exposure and challenge by intradermal and occluded
epidermal exposure. Adjuvant is employed also to augment the induction
of the immune responses. For induction, concentrations of up to 5% or
a maximum non-irritant concentration are used for intradermal
injections, and up to 25 % for epidermal application. Some time after
induction exposure, test and control animals are challenged at a
distant site with a sub-irritant concentration of the chemical, which
is generally lower than the concentration used for induction.
Challenge-induced inflammatory reactions, measured as a function of
erythema and/or oedema, are recorded 24 and 48 h later. Classification
of sensitizing activity is based usually upon the percentage of test
animals that display macroscopically detectable challenge reactions.
Any compound inducing at least 30% positive animals in an adjuvant
test is labelled as a sensitizer; in the case of a non-adjuvant test,
15% is sufficient for classification as a sensitizer.
Of the available guinea-pig test methods, the guinea-pig
maximization test is generally selected when the aim is to identify
the weakest of skin sensitizers. However, the method is not well
suited to the estimation of relative sensitizing potency, because of
the requirement for the use of intradermal injections of test material
and Freund's complete adjuvant (FCA) (Basketter et al., 1996). The
Buehler test is easier to use because the mode of application is
epicutaneous occlusive treatment for both induction and elicitation
(Chan et al., 1983).
Although guinea-pig test methods, such as the Buehler test and
the guinea-pig maximization test, have been in use for more than 25
years, there has not been extensive examination of their sensitivity
and specificity in comparison to what is known about human skin
sensitization. Both the tests, when conducted properly, offer
sufficient sensitivity to detect many known human skin sensitizers
(Wahlberg & Boman, 1985; Basketter et al., 1996). However, there has
been very little assessment on the specificity of these methods.
It should be noted that these guinea-pig methods are not well
suited to the identification of protein allergens. In general, the
maximization of exposure involved in these skin sensitization tests
will result simply in a large immune response. This will mask any
differential allergic response.
6.4.1.2 Testing in mice
Increased understanding of the cellular and molecular mechanisms
associated with contact allergy have derived largely from experimental
investigations in the mouse. Two different types of tests to predict
the capacity of chemicals to induce skin allergy have been developed.
One is the mouse ear swelling test (MEST), which, like the guinea-pig
methods described above, is based upon the evaluation of
challenge-induced reactions in previously sensitized animals (Gad et
al., 1986). In this test, mice sensitized by a comparatively rigorous
regime (the intradermal injection of adjuvant followed by the daily
application of the test material, for 4 consecutive days, to
tape-stripped skin) are challenged on one ear with the test compound
and on the contralateral ear with vehicle alone. Sensitizing potential
is evaluated by consideration of both the degree of oedema (ear
swelling) induced and the percentage of animals displaying a reaction
(Thorne et al., 1991).
The second test developed in mice is the local lymph node assay
(Kimber et al., 1989). In contrast to the mouse ear swelling test and
guinea-pig assays, activity here is measured as a function of events
occurring during the induction, rather than elicitation, phase of
contact sensitization. Mice are treated daily, for 3 consecutive days,
on the dorsum of both ears with the test material or with an equal
volume of vehicle alone. Proliferative activity in draining lymph
nodes (measured by the incorporation in situ of radiolabelled
thymidine) is evaluated 5 days following the initiation of exposure.
The local lymph node assay has been the subject of extensive
comparisons with guinea-pig methods (Kimber et al., 1994; Basketter et
al., 1996), and offers significant advantages compared with the
available guinea-pig test methods. Important among these is the fact
that there is an objective read-out. The assay has been evaluated by
collaborative trials, and an OECD (Organisation for Economic
Co-operation and Development) test guideline issued in 1992 states
that the local lymph node assay (or the MEST) can now be used as a
first stage in an assessment of skin-sensitizing activity (OECD, 1992)
If a positive result is seen, then a test substance may be designated
a potential sensitizer and it may not be necessary to conduct a
further guinea-pig test. However, if a negative result is seen, a
guinea-pig test must be conducted subsequently.
6.4.1.3 Predictive testing for skin allergy in humans
There are various skin test procedures for the diagnosis of
several types of contact dermatitis (see also chapter 4). Basically,
predictive tests in humans for skin allergy are similar to diagnostic
tests for contact sensitization, but the aims are different. For
diagnostic tests, the aim is determining sensitization to chemicals to
which there was a prior exposure, and avoiding new sensitizations
because of the procedure. For predictive testing in humans the aim is
to show the sensitizing capacity in individuals that have not been
exposed to the chemical previously.
Predictive testing in humans generally requires multiple
occlusive patches for induction of sensitization (10 patches, 48 h
each, same site) followed by a 2-week rest period and then challenge
(48 h) with a patch at a new skin site (Marzulli & Maibach, 1973,
1996). There are a number of variations in these procedures, including
the use of provocative chemical agents such as sodium lauryl sulfate
(Kligman, 1966c), special skin preparation such as stripping (Spier &
Sixt, 1955) or freezing (Epstein et al., 1963), special patches
(Magnusson & Hersle, 1965), high concentrations at induction (Marzulli
& Maibach, 1974), and 25-200 test subjects (Draize et al., 1944;
Kligman, 1966b). It is not entirely clear, however, how useful these
variations are and what the limitations are under the use conditions
because validation has not kept pace with the use of the different
approaches. Furthermore, predictive tests are often performed on a
single chemical entity, whereas ultimate use may occur as part of a
multicomponent formulation in a marketed product, where the vehicle
and associated ingredients may influence the outcome. It is essential
that the test has sufficient statistical power to provide appropriate
protection for the population at risk. This is illustrated by the
mathematical considerations of Henderson & Riley (1945) in their
classical paper on extrapolating data from a small test population to
large numbers of users. Briefly stated, there may be no skin reactions
in a test population of 200 random subjects, yet as many as 15 of
every 1000 of the general population (95% confidence), or up to 22 of
every 1000 may react (99% confidence). If the test group is reduced to
100 subjects, up to 30 of every 1000 of the general population may
react (95% confidence). Conversely, when 1 of 200 subjects in a test
population becomes sensitized, a test population of 10 000 subjects
might show from 1 to 275 sensitized, with 95% confidence.
Prospective tests of skin sensitization using human volunteers
should always be conducted in accordance with ethical principles.
6.4.2 Testing for respiratory allergy
Most of the animal models that are used for studying specific
respiratory hypersensitivity were developed using allergens with high
relative molecular mass, notably proteins. Very few animal models have
been developed as predictive tests for hazard identification and risk
assessment in the area of chemical-induced respiratory allergy. The
majority of these models are based upon antibody-mediated events. The
models differ with regard to the following aspects: the animal species
utilized, the route of administration of the agent, the protocol for
both induction and elicitation of responses, type of response
measured, and judgment of significant response.
6.4.2.1 Guinea-pig model
The guinea-pig has been used for decades for the study of
anaphylactic shock and pulmonary hypersensitivity (Sarlo & Karol,
1994). The guinea-pig is similar to humans in that the lung is a major
shock organ for anaphylactic responses to antigens. The guinea-pig
responds to histamine and can experience both immediate-onset and
late-onset responses. Airway hyperreactivity and eosinophil influx and
inflammation can also be demonstrated in this animal species.
Mechanistic studies have been hampered by the lack of reagents needed
to identify cells and mediators in respiratory allergy. In addition,
the major anaphylactic antibody is IgG1a, whereas it is IgE in other
rodent species and in humans. The in vivo passive cutaneous
anaphylaxis (PCA) assay was used to measure IgG1a antibody responses,
but ELISA methods have now been developed that have eliminated some of
the variability seen with the PCA.
The guinea-pig inhalation model focuses on identifying chemical
sensitizers by measurement of the response, or elicitation phase, of
sensitization. In contrast with the mouse IgE test, the model does not
depend upon a preconceived mechanism of sensitization. Rather, it
functions by reproducing the characteristics that typify the
hypersensitivity reactions, i.e., the physiological response of the
airways and the pulmonary inflammation. Measurement of specific
antibody formation provides ancillary evidence of the response. The
method has been successfully used to distinguish low relative
molecular mass contact sensitizers from respiratory sensitizers. The
model utilizes inhalation as the route of exposure for both the
sensitization phase and the elicitation phase of the response. A
variation of the method is the use of intratracheal administration of
the agent (Sarlo & Karol, 1994). The model has the capacity to assess
immediate-onset responses (IAR), as well as late-onset responses
(LAR). The latter is possible since minimal restraint of animals is
used. A dynamic air supply and passive detection devices allow
continuous 24-h monitoring of respiratory function of animals. The
ability to detect late-onset responses makes the model particularly
appropriate for evaluation of chemical allergy, where late-onset
responses are a frequent occurrence.
The advantages of the guinea-pig model for chemical sensitization
include: use of inhalation as the relevant route of exposure;
generation of atmospheres of reactive chemicals; measurement of
physiological responses including immediate-onset responses,
late-onset responses, fever and hyperreactive airways; measurement of
specific antibody production; and histopathological evaluation of
pulmonary tissue. Disadvantages of the model are the cost, the time
involved, the need for specialized facilities, and the employment of
guinea-pigs. The latter is a disadvantage in that IgGl rather than IgE
is the major class of cytophilic hypersensitivity antibody in
guinea-pigs.
Variations of the guinea-pig model have been developed to
optimize the response, to monitor additional respiratory parameters,
or to simplify the procedure (Briatico-Vangosa et al., 1994). Such
variations include single or repeated intradermal administration of
free chemical, elicitation with a multivalent chemical-adducted
protein, and measurement of flow-volume loops, respiratory minute
volume, inspiratory and expiratory time, and peak respiratory flow
rates. Using chemical-protein adducts for elicitation avoids the
possible development of airway hyperreactivity due to chemical
irritation (analogous to the reactive airways syndrome in humans).
Using the guinea-pig model, differences were readily apparent
between sensitization to allergens of high relative molecular mass
(HRMM) versus those of low relative molecular mass (LRMM). Responses
to ovalbumin and to bacterial subtilisin (HRMM allergens) consisted of
severe immediate-onset responses in 90-100% of animals and late-onset
responses in 50% of animals (Thorne & Karol, 1989). By contrast,
sensitization to diphenylmethane-4,4-diisocyanate (MDI), a LRMM
allergen, consisted predominantly of late-onset responses (Karol &
Thorne, 1988). This finding reflects the human experience where
late-onset responses are the most frequently observed responses to
LRMM allergens.
The model has been validated in two ways. Firstly, it has been
established in several laboratories (Pauluhn & Eben, 1991; Sarlo &
Clark, 1992; Stadler & Loveless, 1992; Warren et al., 1993; Sarlo &
Karol, 1994) and responses of animals to inhaled toluene diisocyanate
have been reproduced. This confirms the robustness of the model.
Secondly, the model has been found to distinguish pulmonary from
dermal chemical sensitizers, and from non-sensitizers. For example,
inhalation of toluene diisocyanate (Karol, 1983) and
diphenylmethane-4,4-diisocyanate (Karol & Thorne, 1988) by animals
resulted in pulmonary sensitization, whereas similar exposure to
formaldehyde (Lee et al., 1984) and hydrogenated
diphenylmethane-4,4-diisocyanate (Karol & Thorne, 1988), two
recognized contact sensitizers, resulted in dermal sensitivity.
Further validation with additional classes of chemicals is needed to
generate confidence that information will be applicable to human
disease.
It should be emphasized that the measurement of respiratory
responses induced by chemical respiratory allergens is technically
demanding, and consequently conflicting results have been reported.
For example, it has proven difficult to induce robust respiratory
responses in animals sensitized to the potent human respiratory
allergen diphenylmethane-4,4-diisocyanate. Pauluhn & Mohr (1994)
reported that a proportion of animals sensitized to
diphenylmethane-4,4-diisocyanate by inhalation responded to challenge
with inhaled diphenylmethane-4,4-diisocyanate, while those sensitized
by intradermal injection did not. The converse finding was reported by
Rattray et al. (1994).
Although the guinea-pig can be used for testing the capacity of
chemicals to induce respiratory allergy, it needs further validation
in terms of predictive value. However, it should be noted that it can
also be used to test proteins for their ability to stimulate the
production of anaphylactic antibody (Blaikie et al., 1995; Sarlo et
al., 1997). Such information may be of value in assessing the relative
allergenic potency of proteins.
6.4.2.2 Mouse IgE model
A mouse inhalation model to study airway responses to sensitizing
chemicals has been developed (Garssen et al., 1991), but has not yet
been used with a wide range of chemicals associated with respiratory
allergy. The mouse IgE test currently represents the furthest
developed systematic approach to the prediction of respiratory allergy
in the mouse. It does not evaluate actual airway responses, but is
based on the notion of the nature of immune responses elicited in mice
by chemical allergens and of the qualitative differences in immune
responses provoked by contact and respiratory sensitizers as they are
generally found.
Topical administration to mice of chemical respiratory allergens
stimulated a substantial increase in the serum concentration of total
IgE, a response not seen with contact allergens considered to lack the
ability to cause sensitization of the respiratory tract (Dearman &
Kimber, 1991, 1992). Observations suggested that it might be possible
to identify chemical respiratory sensitizers as a function of induced
changes in serum IgE concentration; the advantage of this approach
being that measurement of a serum protein, rather than of
hapten-specific antibody, is required. This forms the basis of the
mouse IgE test.
Investigations have suggested that the mouse IgE test may provide
a useful method for the prospective identification of chemical
respiratory allergens, a conclusion that is supported by recent
studies in an independent laboratory (Potter & Wederbrand, 1995). It
must be emphasized, however, that to date the assay has been evaluated
only with a limited number of chemicals, most of the analyses have
been performed in a single laboratory, and the mechanistic basis of
the model has not been established.
Respiratory allergic responses, associated with increased
reactivity of airways, were observed in mice that were topically
sensitized to and intranasally challenged with picryl chloride, a
Th1-type immune-response-inducing chemical (Garssen et al., 1991). In
addition, it has been shown that IgE-deficient mice undergo
anaphylaxis (Oettigen et al., 1994). Other investigators have noted a
decrease, rather than an increase, in serum IgE following exposure of
mice to toluene diisocyanate (TDI) (Satoh et al., 1995). For this
reason, actual testing of lung function in vivo after sensitization
and challenge with chemicals known to sensitize, yet unable to produce
IgE responses, as can be done in mice (Garssen et al., 1991) as well
as in the guinea-pig (section 6.4.2.1), seems prudent. Moreover,
standardization of the methodology is necessary, including dosages
appropriate for testing, optimal time for repeated administration of
the chemical and for obtaining sera, and clarification of the meaning
of "elevated" IgE titre. Validation of the method is still needed.
6.4.2.3 Rat model
A rat bioassay has been developed by Pauluhn (1996). Nose-only
exposure for 1 or 2 weeks, using non-irritant concentrations as judged
in short-term pilot experiments, is carried out, upon which lung
functions are tested and biochemical and morphological signs of
effects in the airways are determined. The model needs further
validation to evaluate whether it is suitable for the prediction of
respiratory sensitization. A rat model to study respiratory syndromes,
including the IgE-mediated allergic responses, in addition to anaemia
and haemolysis in workers exposed to trimellitic anhydride (Zeiss et
al., 1977), was used by Leach et al. (1987). In this model, animals
are subjected to single or multiple exposures at several
concentrations of trimellitic anhydride dust and at selected time
points are challenged with a single exposure of trimellitic anhydride
dust. The lungs are evaluated for haemorrhage and serum is tested for
IgG antibody specific for trimellitic anhydride. The model has also
been applied for other types of anhydrides.
6.4.2.4 Predictive testing for respiratory allergy in humans
For obvious reasons, predictive testing for respiratory
sensitization is not done in humans. Occasionally, case reports may
serve as an adequate hazard identification but not as a risk estimate,
because data on route and extent of exposure, and on the "population
at risk" are usually lacking. In the absence of case reports, it is
not possible to conclude that there exists no potential for
sensitization.
6.4.2.5 Cytokine fingerprinting
Contact and respiratory chemical allergens provoke in mice
qualitatively different immune responses suggestive of divergent
Th-cell activation and characterized by different patterns of cytokine
production. Chronic exposure of mice, over a 13-day period, to
trimellitic anhydride was found to result in the production of high
levels of mitogen-inducible IL-4 and another Th2-cell cytokine
interleukin 10 (IL-10) by draining lymph node cells, but only low
levels of IFN-gamma. In contrast, treatment of mice under the same
conditions of exposure with oxazolone (a potent contact allergen)
caused the production by draining lymph node cells of only
comparatively low levels of IL-4 and IL-10, but high concentrations of
IFN-gamma (Dearman et al., 1995). Similar selective cytokine secretion
profiles have been recorded following exposure of mice to other
contact and respiratory chemical allergens (Dearman, 1996). These data
raise the question of whether it might be possible to monitor the
sensitizing properties of chemicals as a function of induced cytokine
production profiles. Evidence suggests that this is the case, and the
value of cytokine fingerprinting in the routine identification and
classification of chemical allergens is being explored currently.
6.5 Testing for food allergy
Despite the availability of several methods to study antigenic
and allergenic properties of protein products, current testing
possibilities to investigate the allergenicity of food proteins at a
pre-market stage are very poor. Validated methods with a high
sensitivity have not yet been developed. However, some strategies can
be followed to obtain additional relevant information apart from the
information obtained from the currently applied assays; for instance,
taking account of the role of the gastrointestinal tract physiology.
The major adverse reactions to food constituents that involve the
immune system are to proteins; non-proteins may cause food intolerance
that does not involve the immune system, and hence are non-allergic
reactions that do not fall within the scope of this monograph.
Most proteins encountered by the immune system can produce immune
responses. To determine the antigenicity of proteins, several assays
are operational. However, these assays are based on parenteral
application of the test proteins to laboratory animals. For food
allergy research, three rodent species have frequently been used: the
mouse, the guinea-pig and the rat. In many studies, sensitization was
performed parenterally or passively and effects of enteral challenges
were subsequently studied (Bloch & Walker, 1981; Freier et al., 1985;
Granato & Piguet, 1986; Pahud et al., 1988; Miller & Nicklin, 1988;
Turner et al., 1988, 1990; Curtis et al., 1990). In addition, effects
of challenges have frequently been investigated in in vitro studies
with intestinal tissue or with, for instance, ligated gut (Roberts et
al., 1981; Baird et al., 1984; Lake et al., 1984; Catto-Smith et al.,
1989a,b). The guinea-pig is the most regular test species. In general,
any protein that may be recognized as an antigen (foreign protein)
will induce a humoral immune response upon injection and will most
likely give a positive testing result in rodent assays. Although the
information from antigenicity assays may be of major relevance, in
that they will provide information on the quality and vigour of the
response, it must always be recognized that such assays only provide
information in the species examined. Whether a protein has a high or
low potency of inducing food allergic reactions in (susceptible)
humans cannot be concluded or predicted based only on the results of
parenteral antigenicity assays.
In addition to in vivo antigenicity assays, several
(combinations of) physicochemical and immunochemical analyses are used
routinely to detect antigenicity.
Determination of allergenic proteins or fragments that are able
to cause activation of mast cells and basophils is possible using in
vitro mast cell or basophil degranulation tests. For these assays,
mast cells or basophils are loaded with antigen-specific cytophilic
antibodies using serum from sensitized humans or test animals. The
cells are subsequently incubated with the antigen or test product, and
degranulation of the cells can then be determined. A well-validated
in vivo counterpart for the detection of mast cell activation is the
Passive Cutaneous Anaphylaxis (PCA) test, in which sera from
sensitized animals are injected subcutaneously in unsensitized
animals. Possible cytophilic antibodies present in the sera are bound
by the receptors on the mast cells in the skin, and bridging of the
antigen-specific antibodies on the mast cells by injected antigen
induces mast cell activation -- the resulting reaction is detectable
by various methods. In the Active Systemic Anaphylaxis (ASA) test,
which is also a validated anaphylaxis test, actively sensitized
animals are injected with the test substance intravenously and several
parameters are recorded to determine the systemic anaphylactic
response as a measure for the degree of sensitization.
In the evaluation of the potential allergenicity of food
products, clinical assays such as skin-prick tests or challenge
procedures may also be used. For instance, these assays are applicable
in the evaluation of the residual allergenicity of hypoallergenic
products, in the evaluation of cross-allergenicity, or in the
evaluation of the possible allergenicity of food products derived from
biotechnologically derived crops in which a gene from a known
allergenic source species has been introduced. However, the use of
patients in such assays for non-diagnostic purposes requires careful
ethical consideration. Since many of the questions may also be
addressed by performing in vitro assays such as immunochemical
analyses, the use of sera from patients is always preferable to
intentional exposure of humans.
6.6 In vitro approaches
Another approach for the detection of potential sensitizing
capacity that would not rely on in vivo animal or human testing is
directed towards the construction of in vitro experimental models
that reflect accurately some pivotal event during skin sensitization.
Accurate modelling of the immune system in vitro is not possible
without considerable difficulty. Nevertheless advances have been made.
Wass & Belin (1990) and Gauggel et al. (1993) used biochemical
techniques to examine the ability of low relative molecular mass
chemicals to act as haptens by combining them with a model protein or
a polypeptide. Gauggel et al. (1993) showed that their test correctly
identified 12 out of 14 known human allergenic haptens and 23 out of
24 non-allergenic low relative molecular mass chemicals. Neither
method can detect sensitizers that must be metabolized to form a
hapten.
6.7 Testing for autoimmunity
Although there are currently no predictive assays developed and
validated to identify in the early phases of toxicity testing the
potential of chemicals to induce autoimmune responses, it should be
noted that assays to identify contact sensitizers (Kimber et al.,
1994; Vial & Descotes, 1994) might be helpful to identify systemic
sensitizers. Clinical signs of systemic immune-mediated side-effects
usually become manifest only during advanced clinical development of
drugs. The conditions used in routine preclinical toxicological
screening are obviously not optimal for the detection of the
immune-dysregulating potential of drugs and chemicals (e.g., small
animal number, use of outbred animal strains, dynamics of disease
development versus snapshot determinations, lack of predictive
parameters). An economically and practically relevant question
concerning screening studies is whether actual evidence of an agent's
ability to induce manifest hypersensitivity or autoimmune disease
should be and can be obtained, or whether (preferably short-term)
assays not measuring the actual clinical end-points can be
sufficiently predictive in this respect.
In vitro tests for biological effects of sensitizers or
chemicals able to induce autoimmunity are at present in their infancy.
It should be emphasized that there are two major limitations: (a) it
is so far impossible to completely reproduce in vitro the complex
microenvironment in which immune responses are initiated in vivo;
and (b) some immune reactions are elicited not by native xenobiotics
but by their metabolic products generated in vivo.
6.7.1 Popliteal lymph node assay
At present this appears to be the only method available to
examine the ability of chemicals to induce an autoimmune response, but
the usefulness of the results in risk assessment remains to be
demonstrated. Based on the hypothesis that chemicals may elicit
autoimmune disorders by a mechanism resembling graft-versus-host
reactions, an existing graft-versus-host assay, the popliteal lymph
node assay, has been modified to study chemical-induced immune
reactions (Bloksma et al., 1995). Using the popliteal lymph node
assay, many drugs known to occasionally induce immune-mediated
systemic side-effects in humans were shown to trigger significant
reactions in mice and rats (Gleichmann, 1981; Gleichmann et al., 1983,
1989; Hurtenbach et al., 1987; Kammüller & Seinen, 1988;
Stiller-Winkler et al., 1988; Kammüller, 1989a; Thomas et al., 1989,
1991; de Backer et al., 1990; Verdier et al., 1990; Katsutani &
Shionoya, 1992; Krzystyniak et al., 1992; Bloksma et al., 1995).
Effects observed were very similar to those induced during a local
graft-versus-host reaction in the popliteal lymph node assay (de
Bakker et al., 1990). Immunogenetic studies in mice with
diphenylhydantoin (Bloksma et al., 1988) and D-penicillamine
(Hurtenbach et al., 1987) have indicated that the extent of popliteal
lymph node enlargement is controlled by major histocompatibility
complex (MHC (H-2)) as well as non-major histocompatibility complex
genes. Furthermore, the popliteal lymph node assay was able to
discriminate between structurally closely related compounds, for
example chemical congeners of D-penicillamine (Hurtenbach et al.,
1987), diphenylhydantoin (Kammüller & Seinen, 1988) and zimeldine
(Thomas et al., 1989, 1991). Thus, the direct popliteal lymph node
assay seems to be a versatile tool for recognizing T-cell-activating
drugs and chemicals, including autoimmunogenic chemicals, but it also
produces false-negative results (Bloksma et al., 1995). With the
adoptive transfer popliteal lymph node assay, sensitized cells are
used as probes to detect the formation in vivo of immunogenic
metabolites of low relative molecular mass chemicals (Kubicka-Muranyi
et al., 1993; Bloksma et al., 1995). However, further mechanistic
studies and interlaboratory validation is required before either
variant of the assay can be recommended for routine use in the
preclinical toxicity screening (Verdier et al., 1997).
6.7.2 Animal models of autoimmune disease
Three basic types of animal models may be employed to identify
the potential of drugs or chemicals to induce systemic
hypersensitivity or autoimmune responses: (a) genetically predisposed
animals; (b) autoimmunization; and (c) organic or chemically induced
(Table 26). In each type of model the development and severity of
symptoms is multifactorial, in that the disease state can be
influenced by age, hormonal and/or environmental factors. In addition
there is a tendency for more than one autoimmune disorder to occur in
a number of individual models. Nevertheless, a number of syndromes
similar to that observed in humans can be mimicked in animal models.
The genetically predisposed models, whether naturally occurring,
transgenic or knockout based, tend to be the most reliable and
therefore have been more commonly employed in autoimmunity research
(Lo, 1996). In this type of model, mild to severe syndromes
spontaneously develop, usually due to specific MHC allele mutations
encoding class II molecules and often inducing function abnormalities
of the CD4+ Th-cell (Theofilopoulos, 1995a,b). The most common
genetic models include several MRL mouse variants for autoimmune
thyroiditis, arthritis and lupus, several variants of New Zealand
Brown mice (NZB), New Zealand White mice (NZW) and BXSB mice for
lupus, and the non-obese diabetic (NOD) mouse and BioBreeding (BB) rat
for insulin-dependent diabetes mellitus (Cohen & Miller, 1994).
Autoimmunization with purified self-antigens can elicit a
specific autoimmune response, particular when adjuvants are
administered in conjunction with self-proteins. A frequently used
model of this type, experimental autoimmune encephalomyelitis, is
induced by immunization of rodents with myelin basic protein. The
resulting pathology is a CD4+ T-cell-mediated autoimmune disease
characterized by central nervous system perivascular lymphocyte
infiltration and destruction of the myelin nerve sheath with resultant
paralysis, similar to that observed in patients with multiple
sclerosis (Constantinescu et al., 1998). Additional models included
immunization with thyroglobulin to simulate Hashimoto's thyroiditis,
and the injection of type II collagen to induce rheumatoid arthritis
(Wick et al., 1974; Durie et al., 1994).
In experimental models, foreign substances are used to induce the
autoimmune disease state. These include chemicals, drugs and
biological substances such as bacterial or viral antigens. Brown
Norway (BN) rats injected with non-toxic amounts of mercuric chloride
which produce no signs of overt toxicity develop an immunologically
mediated disease characterized by a T-cell-dependent polyclonal B-cell
activation (Pelletier et al., 1994). These animals demonstrate
increases in serum IgE and the production of autoantibodies to a
number of proteins including DNA, laminin, collagen IV and other
components of the glomerular basement membrane. Proteinuria and
Table 26. Experimental models for autoimmune diseasesa
Autoimmune disease Classification
Genetically predisposed strainsb Autoimmunizationc Biological/chemical induction
Autoimmune thyroiditis Murphy-Roths lymphoma (MRL) mouse, Thyroglobulin - EAT
(Hashimoto's and Graves') BioBreeding (BB) rat, (mouse, rat)
Obese strain (OS) chicken
Insulin-dependent Non-obese diabetic (NOD) mouse, Streptozotocin (STZ) (mouse)
Diabetes mellitus BioBreeding (BB) rat,
Diseases resistant BioBreeding
(DRBB) rat,
Brown Norway rat
Myasthenia gravis Acetylcholine receptor Penicillamine (mouse, rat)
- EAMG (mouse, rat)
Multiple sclerosis Myelin basic protein
EAE (mouse, rat, chicken)
Rheumatoid arthritis Murphy-Roths lymphoma CFA + Type II collagen Streptococcal cell-wall (rat)
(MRL)/lpr mouse, (mouse, rat, monkey)
Severe combined immunodeficient CFA + TB hsp (mouse, rat)
(SCID) mouse,
Human leukocyte antigen
B27 (HLA B27) transgenic rat
Systemic lupus Murphy-Roths lymphoma CFA + antiDNA antibodies Mercury (mouse, rat, monkey)
erythematosus MRL +/+ mouse, (mouse, rat) Penicillamine (mouse, rat)
Murphy-Roths lymphoma MRL/lpr Procainamide (mouse, rat)
mouse,
Murphy-Roths lymphoma
(MRL)-mp-lpr/lpr mouse,
New Zealand White (NZW) 2410 mouse,
New Zealand Black (NZB) mouse/
New Zealand White (NZW) mouse
Table 26. (continued)
Autoimmune disease Classification
Genetically predisposed strainsb Autoimmunizationc Biological/chemical induction
Systemic sclerosis Tight skin (TSK) mouse,
(Scleroderma)
a Adapted from Cohen & Miller (1994); Farine (1997); Bigazzi (1997)
b lpr = lymphoproliferation
c CFA = Complete Freunds Adjuvant
nephrotic syndrome similar to that observed in humans are also
observed. The disease is characterized by a glomerulonephropathy
evolving in two phases: (a) a linear anti-GBM antibody deposition
along the glomerular capillary pattern, and (b) a change to a granular
pattern of immunofluorescence with the appearance of immune complex
deposits (Pelletier et al., 1994).
6.8 Clues from general toxicity tests
The conditions used in conventional systemic toxicological
screening are not designed for the detection of the potential of drugs
and chemicals to induce allergy or autoimmunity, as general toxicity
screening does not include specialized tests required for this
purpose. In more recent guidelines for general toxicity screening,
attention is given to the immune system, especially with the purpose
of detecting direct toxicity to components of the immune system. This
pertains for instance to the updated OECD guideline for 28-day oral
toxicity testing (Koeter, 1995). According to this guideline, lymphoid
organs are weighed and lymphoid tissues are examined microscopically.
In addition to morphological examinations of lymphoid organs, a
selection of non-lymphoid tissues (e.g., blood vessels, renal
glomeruli, synovial membranes, thyroid, skin, liver and lungs) should
be investigated. Tissue damage, protein (immune) complex deposits,
and/or inflammatory cell infiltrates in these tissues may indicate the
induction of hypersensitivity or autoimmune phenomena.
It is common practice in toxicological pathology to rely most on
statistically significant differences in incidence between test groups
and controls. However, individual variability can be high, especially
regarding hypersensitivity and autoimmune phenomena (Holt & Sedgwick,
1987; Kammüller et al., 1989). Therefore, in studies with a limited
number of animals per group, a change in only a single or a few test
animals may have considerable biological relevance. This is
particularly true in outbred animals.
In addition to morphological examinations in routine
toxicological studies, measurements of some immunologically relevant
serum parameters can provide important information about
antibody-mediated responses. Parameters may comprise levels of total
immunoglobulins and of various immunoglobulin (sub)classes, immune
complexes, and some commonly observed autoantibodies, e.g.,
anti-nuclear antibodies (ANAs), anti-histone, and anti-single-stranded
deoxyribonucleic acid (denatured; ssDNA) autoantibodies. Some of these
autoantibodies proved to be useful in the diagnosis of
procainamide-induced systemic lupus erythematosus in humans as shown
by Rubin et al. (1995). Measurement of these parameters at suitably
chosen intervals, especially during subchronic and chronic exposure,
may obviate the snapshot nature of the histopathological examinations
and will give a reflection of cellular and humoral immune function in
time. With regard to autoantibody measurements it is important to
measure both total immunoglobulin classes and subclasses. This can be
illustrated by murine models of spontaneous systemic lupus
erythematosus in which a switch from IgM to IgG autoantibodies could
be associated with development of overt disease. Also, data on
drug-induced systemic lupus erythematosus in humans point in this
direction. Anti-nuclear antibodies in patients with
procainamide-induced systemic lupus erythematosus appeared to be of
the IgG class (in particular IgG1 and IgG3), whereas IgM anti-nuclear
antibodies are predominant in asymptomatic users of the drug.
It has been suggested that parameters of the immune system are
included in conventional chronic toxicity testing and also in
reproductive toxicity testing and evaluation, since the developing
immune system may be particularly vulnerable to immune dysregulating
effects (Hollady & Luster, 1996). For example, a newly developed
immunomodulating agent was found to induce thyroiditis and significant
antithyroglobulin autoantibodies in a 6-month and a 2-year rat study
(Verdier et al., 1997).
7. RISK ASSESSMENT
7.1 Introduction
Risk assessment for human health protection is the final stage in
evaluating the likelihood that potential adverse health effects will
manifest themselves in humans.
7.2 Risk assessment of allergy
Effective risk assessment requires an appreciation of the
potential of a chemical or drug to induce an allergic response, or
elicit an allergic reaction, and its potency. Potency here is
considered as the amount of chemical necessary to induce an allergic
response or to elicit an allergic reaction. Risk assessment demands
also an understanding of the conditions of human exposure, i.e., the
extent, duration and route(s) of likely exposure.
With the assembly of the available data completed, risk assessors
must decide whether sufficient data are available to proceed. It may
be necessary to produce a preliminary or provisional risk assessment
irrespective of the availability of full documentation. However, at
this stage, following comparison of what is available with minimal
data sets, consideration is normally given to further searching,
further requests for information to industry, and the use of modelling
or default values to fill data gaps. A decision is then made on
whether to proceed.
Risk assessment of allergy is complicated by the fact that there
exist two related, but nevertheless independent considerations.
Allergic disease almost always occurs in two phases. During the first
phase the previously non-sensitized individual is exposed to
sufficient allergen in such a way that an allergic response is induced
which results in sensitization. This is the induction phase. An
allergic reaction will be elicited if this now sensitized individual
is exposed again to the same allergen under conditions where a
secondary, more aggressive allergic response is provoked. This is
known as the elicitation phase. It is important to appreciate that
these phases usually differ with respect to the exposure conditions
required. The induction of allergic sensitization in a previously
naive individual almost invariably requires exposure to concentrations
of the allergen that are greater than those necessary to elicit a
response in a sensitized subject. Allergic reactions can be provoked
in sensitized individuals with small amounts of the inducing allergen
that are without effect in non-sensitized populations. It is possible
also that sensitization may be achieved from exposure via a route that
is different from that needed to elicit a response. Thus, there is
some evidence that dermal exposure to certain chemical allergens may
stimulate the quality of immune response necessary to cause
sensitization of the respiratory tract. The elicitation of pulmonary
responses will subsequently be provoked following inhalation exposure
of the now sensitized individual to the relevant chemical.
Taken together it is clear that risk assessment for chemically
induced allergic disease has two components: (a) the likelihood that a
chemical will induce sensitization in a previously unsensitized
individual; and (b) the conditions under which a chemical will provoke
allergic reactions in those who are already sensitized.
Information on risk, plus suggestions about any group of
increased susceptibility, is used for risk evaluation, that is the
process of deciding for medical, ethical, economic and legal reasons
whether the identified risk will be "acceptable" and under what
conditions, e.g., restricted availability of the substance in special
circumstances or wider availability with a warning label and advice
about protective measures. The decision may also be made to ban the
use of a material deemed to carry too large a risk in relation to
anticipated benefit from its use. These decisions reflect
socioeconomic and political factors as well as medical and scientific
conclusions, and so lie outside the purpose of this monograph.
7.3 Factors in risk assessment of allergy
Risk assessment requires answers to the following questions:
* Who will be exposed?
How many people: Is there any information to suggest particular
susceptibility (or resistance) to allergic hypersensitivity, e.g.,
genetic, nutritional or other factors?
* Circumstances of the exposure
Is it an initial sensitizing exposure or a subsequent provocative
exposure that may elicit an allergic reaction?
* Extent and duration of exposure
This defines the "load" of the allergen. Most of the available
evidence suggests that it is the peak concentration of exposure rather
than the cumulative delivered dose that is critical in determining the
extent to which sensitization will develop. The same principle
probably applies also to the elicitation of allergic reactions in
sensitized individuals.
* Nature of exposure
Exposure to chemical allergens occurs frequently in the context
of mixtures and formulations. An important consideration is whether
other components of the mixture will influence the ability of the
chemical allergen to induce sensitization or elicit a reaction.
* Route
If it is the first exposure, is it by a route that may sensitize
people, usually via inhalation or skin contact, or by ingestion, which
is much less likely to do so? If an individual is already sensitized,
is it via the route by which he has already been sensitized, in which
case an allergic response may occur? Or, will it be by another route,
which may not necessarily evoke an immunological response?
* Evidence of hazard
What is the nature and quality, i.e., the "strength", of the
laboratory information showing the sensitizing potential of the
substance? Has it come from laboratory tests of proven value, from
chance observations during some other type of experiment, or is it
based on prediction from structure-activity relationships? In each of
these, are the data qualitative or quantitative?
* Prior evidence of risk
Are there already results from humans, either as tests in a
clinical laboratory setting, or following exposure in the real world
of work or home, showing that people have been sensitized and how, and
the consequences of subsequent exposure by any route?
Taking all this information together, each aspect of which has
already been discussed in this monograph, should suggest the totality
of the risk under a given set of circumstances, in other words, the
likelihood that a given exposure to a substance will sensitize people,
or evoke an allergic response in them, perhaps the proportion
affected, and the nature of that response (including clinical feature
of the disorder and their severity). That represents the ideal. In
practice, the available information may be incomplete, and skilled
judgement is required to extrapolate from whatever is known to suggest
what may happen in practice.
7.4 Information aspects
The three commonest deficiencies in knowledge are discussed
below.
7.4.1 No information about hazard
For a novel substance, a novel use, or because hypersensitivity
may not previously have been recognised, there will be no information
from humans. In that case, prediction can only be based upon the
chemical structure of the substance, the results of appropriate
laboratory tests, and assumptions about exposure of people.
Such an extrapolation based solely on laboratory-defined hazard
is the usual situation faced by the toxicologist, occupational health
specialist and regulator in dealing with a new substance.
As discussed earlier in this monograph, many of the laboratory
procedures available to identify hazard are well proven and some are
valuable in assessing relative potency for skin sensitizers. Their
quantitative predictivity, sensitivity and specificity are difficult
to state, but they probably give about 80% correct negative results,
i.e., sensitization is unlikely (Kimber & Maurer, 1996) and about 60%
correct positive predictions, i.e., skin sensitization is likely to
occur.
For respiratory tract sensitization, far fewer results are
available, but a positive finding in any of the types of experiment
considered here should be regarded as a powerful indicator of this
type of hazard. The strength of a negative finding is less certain,
because there is little published information.
True hypersensitivity to oral antigens is still poorly
understood, and the more common but more fickle "food intolerance"
remains clouded by uncertainties. For a novel substance in a food, it
is not yet possible to predict the likelihood that it will cause true
allergy or intolerance if consumed. Known allergens and the other
materials that sometimes cause disorders when eaten can be indicated
by reference to published clinical information, which should also
point both to the likely frequency of the harmful response, and
whether it is affected by ethnic or by other physiological factors,
the methods of preparation and cooking, and the matrix in which a
putative food allergen is presented, because the complex nature of the
mixtures that are foods does affect the risk of sensitization and
elicitation of reactions to materials in the diet.
The best predictor at present of the risk of true immunologically
mediated hypersensitivity to a food component is probably
demonstration of the presence of antibodies to known potent allergens,
such as peanut (groundnut) protein.
Predicting the risk of autoimmunity, too, can only be done by
demonstrating the presence of a known antigen in an appropriate
matrix, or, with less certainty, by demonstrating a very close
structural analogy to a known cause of an autoimmune response.
7.4.2 Scanty or no information about exposure
It is sometimes difficult to envisage exactly how a substance
will be used in the workplace or at home, or how it may be
disseminated via the environmental media. In both of these instances
the prediction of risk must carry some uncertainty.
The problem of predicting exposure is well shown by the
demonstration of an increased incidence of asthmatic attacks in the
population of the city of Barcelona during large-scale transhipment of
soy beans in the docks. That appears to have released a sufficient
quantity of an allergen into the air for it to be disseminated and to
expose the entire population under certain meteorological conditions,
resulting in allergic asthma in so many people as to have a serious
effect on the health of the community.
7.4.3 Unreliable or scanty information about risk
This is a particular problem because of the propensity of
clinicians to report unusual or unique examples of disorders and not
to describe negative findings. It may be difficult, therefore, to gain
a balanced or critical view of the allergenic effects of substances
from much of the clinical literature. It can provide an indication of
suspicion, but large, carefully constructed series of patients or
cautious surveillance results are rare.
At a different level, but well able to provide helpful
information, are national occupational health statistics on
appropriate industrial diseases, particularly those for which a
pension has been awarded. For many reasons those series are unlikely
to provide unbiased information about incidence or prevalence, because
of the inevitable bias when legal liability may be involved, but they
can act as broad indicators of risk, and even of the allergenic
potential of particular substances (Drever, 1995).
Databases of consumer products used by the general population are
very limited and may be close to capricious in their content, but they
should be consulted when possible.
Overall, therefore, the risk assessor has a very difficult task
in the general field of allergic hypersensitivity. At present,
laboratory tests may indicate the potential for a hazard, and
searching consideration may suggest many of the features about
exposure. From that a risk to humans may be predicted in qualitative
terms, at least for the common route of domestic and occupational
exposure of the skin. The prediction of respiratory tract allergy
remains less certain, and the likelihood of effects following
ingestion, or of an autoimmune disorder, can very rarely be predicted.
Many predictive test methods serve simply to identify the
inherent potential of a chemical to induce allergy but provide no
indication of the potency with which it will do so. One problem is
that some methods do not incorporate a dose-response analysis. The
other issue is that some tests measure activity as the frequency of
responses rather than the severity of responses. The need is to have
available information on potency defined as the quantity of chemical
necessary to induce sensitization (or to elicit a reaction). Some
newer test methods are beginning to address this issue and are
providing information about allergic potency relative to index
allergens. Such comparisons have been of value in setting safe
occupational exposure levels and minimizing the risk of allergic
disease.
As in any form of toxic reaction, "dose" is important, in that
initial sensitization requires at least a certain minimum exposure
(concentration of allergen, its local availability at the site of
administration, and the duration of contact). In someone already
sensitized, the likelihood of producing a clinical disorder and its
severity are also related to dose, although, by definition, the
quantity of allergen required to produce an effect is very much
smaller than that associated with a conventional toxic action. This
aspect of the extent or intensity of dose (=exposure) is more
important at the practical level of preventing sensitization or
protecting the sensitized individual, i.e., risk management, but it
should not ignored at the risk assessment stage.
A further difficulty experienced by the risk assessor in this
area is that of individual susceptibility. Genotype does play some
role in the propensity of people to develop clinical disorders due to
hypersensitivity, as shown, for example, by the familial nature of
atopy. There is also good clinical evidence, however, to show that the
inheritance of eczematous skin hypersensitivity, or of asthmatic
hypersensitivity to common domestic allergens, involves many powerful
factors in addition to genetic constitution. Although there is no a
priori reason to believe that humans lack "immune response" genes
for hypersensitivity as pragmatically defined in animals, their
importance remains uncertain. Some chemically related autoimmune
diseases though are associated with certain "immune response"
genotypes. For example, HLA DR3 and DR3/B8 haplotypes are associated
with vinyl chloride disease (Black et al., 1983).
Skin sensitization risk assessment is not a highly prescriptive
process that should always be followed in the same way (Calvin, 1992).
On the contrary, what is necessary is that it is carried out
thoroughly to the point where the risk has been adequately assessed.
In some circumstances, this point may be reached quite quickly and
with minimal expenditure of time and effort. In other cases,
substantial and sustained effort is required. For example, in the
situation where exposure to the contact allergen is essentially zero,
then even for highly potent contact allergens, there is no need to go
further with a risk assessment because allergic contact dermatitis
will not occur. Furthermore, if the exposure is sufficiently low, it
may not be necessary to know precisely the potency of a contact
allergen. Simply the knowledge that it is not a very strong allergen
may be sufficient to permit a proper conclusion of the risk
assessment. Another situation where risk assessment may be relatively
simple is the replacement of an ingredient with another of the same or
similar type (e.g., an alternative supplier of a raw material). In
such a case, and where the risk is already known to be very low, all
that may be necessary is to confirm that the specification of the new
source of raw material is the same. Alternatively, data that provide
evidence that the relative sensitization potential of the old and new
materials is similar may suffice. In contrast, even where the
intrinsic sensitization potential is very low, if skin contact is
sufficiently intense and prolonged, then sensitization may occur. An
example of this is the situation where medicines are applied
continuously to skin, often damaged and/or inflamed skin, under
occlusion. A prime example is found with stasis ulcers, where a
variety of medicines and chemicals with negligible sensitization
potential, such as cetostearyl alcohol and paraben esters, quite
frequently cause allergic contact dermatitis.
The comments above on skin sensitization all presuppose a good
knowledge of the relative sensitizing potency of the substance under
consideration. Such data may be obtained by examination of the skin
sensitization potential of the substance in guinea-pig methods
referred to earlier in this monograph. Critical to this type of
analysis is data from the same method at the same testing institution
with suitable benchmark substances. These may well need to be selected
on a case by case basis in the light of the risk assessment that needs
to be made. Alternatively, use made be made of the local lymph node
assay, the results of which can be interpreted to yield an objective
estimate of relative sensitizing potency (Kimber & Basketter, 1997).
To determine what represents a safe threshold for a sensitizer is
a complex matter (Basketter et al., 1997). The key point is that the
threshold depends on the method used to determine it. Thresholds in
animal models my differ substantially from those for humans even in
situations where the pattern of exposure is similar. However, where it
is possible to match the type of exposure in the test in humans to
that which is expected in practice, then such data may be interpreted
directly to humans (Johansen et al., 1996b).
7.5 Conclusions
The scientific, practical and clinical uncertainties that affect
allergic hypersensitization make the task of the risk assessor
particularly difficult. In practice, it may be reasonable in such
assessments to favour the fail-safe approach by emphasising the
"precautionary principle", namely that if there is even a suspicion of
a risk, exposure should be minimized and preferably be entirely
prevented. That will have a considerable influence on the development
of new substances, on devising new uses for existing materials, and on
instituting controls over exposure in the home and safe working
practices, because an assessment that suggests a risk of allergic
sensitization must lead at once to an appropriate decision about risk
management.
8. TERMINOLOGY
Adhesion molecules. Molecules, belonging mainly to the
immuno-globulin or integrin superfamily of molecules (e.g., LFA-1,
ICAM-1), expressed on the membrane of various cells of the immune
system. Interactions with each other as receptors and corresponding
ligands facilitate cooperation (cross-talk) of cells, signal
transduction and information transfer between cells.
Adjuvant. A material that enhances immune response to substances in
a non-antigen-specific manner.
Allergen. An antigen that provokes allergy.
Allergic contact dermatitis. An inflammatory skin disease resulting
from allergic sensitization.
Allergy. Hypersensitivity caused by exposure to an exogenous
antigen (allergen) resulting in a marked increase in reactivity and
responsiveness to that antigen on subsequent exposure, resulting in
adverse health effects.
Allogenic. Term describing genetically different phenotypes in
different (non-inbred) individuals of the same species.
Anaphylaxis/anaphylactic reaction. A local or systemic immediate
hypersensitivity reaction initiated by mediators released after
immunological stimulation. Symptoms can be a drop in blood pressure
related to vascular permeability and vascular dilatation, and
obstruction of airways related to smooth muscle
contraction/bronchoconstriction.
Anergy. Lack of immune responsiveness (usually defined as lack of
response to common recall antigens).
Antibody. An immunoglobulin produced by activated B-cells and
plasma cells after exposure to an antigen with specificity for the
inducing antigen.
Antibody-dependent cell-mediated cytotoxicity (ADCC). Lysis of
various target cells coated with antibody by Fc receptor-bearing
killer cells, including large granular lymphocytes (NK cells),
neutro-phils, eosinophils and mononuclear phagocytes.
Antigen. Any compound recognized by antigen-receptor-bearing
lymphocytes. Antigens induce immune responses or tolerance. Antigens
inducing immune responses only with the help of T-cells are
T-dependent antigens, while those that do not need T-help are
T-independent antigens. All immunogens are antigens but not all
antigens are necessarily immunogens (see also immunogens).
Antigen-presenting cells. Cells expressing MHC gene products, with
the capacity to process and present antigen. Macrophages, dendritic
cells, B-lymphocytes and Langerhans cells are termed professional or
constitutive antigen-presenting cells. However, other cells (such as
endothelial cells) can acquire the ability to present antigen in
certain pathological conditions.
Antigen processing and presentation. Protein antigens are processed
(cleaved by enzymes) in various compartments of antigen-presenting
cells. The immunogenic peptides interact with the binding sites of MHC
class II products (exogenous antigens) or with those in MHC class I
products (endogenous antigens, including viruses). The processed
antigen-MHC complex is recognized by the antigen receptor complex of
T-lymphocytes.
Antigenic determinant. A single antigenic site (epitope) usually
exposed on the surface of a complex antigen. Epitopes are recognized
by antigen-receptors on T- or B-cells (T-cell epitopes or B-cell
epitopes).
Anti-nuclear antibody. Antibody directed to nuclear antigen. These
antibodies can have various specificities (e.g., to single- or
double-stranded DNA, or to histone proteins). These antibodies are
frequently observed in patients with rheumatoid arthritis,
scleroderma, Sjögren's syndrome, systemic lupus erythematosus, and
mixed connective tissue disease. Also called anti-nuclear factor
(ANF).
Arthus reaction (Gell and Coombs Type III reaction). Inflammatory
response, generally evoked in skin, that is induced by immune
complexes formed after injection of antigen in an individual
containing antibodies.
Asthma. A respiratory disorder characterized by variable air flow
limitation. Most cases are associated with bronchial
hyperresponsiveness and chronic inflammatory changes in the airways.
Atopic dermatitis. Inflammation of the skin in atopic individuals.
The term is broader than atopic eczema (see also atopic eczema).
Atopic eczema. Chronic skin disease, often localized on flexural
surfaces, in individuals with propensity to develop IgE-mediated
allergy. The term describes eczema occurring in atopic individuals and
does not imply mechanisms (see also eczema).
Atopy. A genetic predisposition toward development of IgE-mediated
immediate hypersensitivity reactions against common environmental
antigens.
Autoimmune disease. A disease involving immune responses against
self antigens, resulting in pathological change.
Autoimmunity. Responses against self (autologous) antigens.
B-lymphocytes. Bone-marrow-derived lymphocytes, expressing an
antigen-receptor complex composed of membrane-bound immunoglobulin
(mIg) and associated molecular chains. B-cell receptors interact with
epitopes directly (no MHC restriction). Activated B-lymphocytes
produce antibody and are efficient antigen-presenting cells. They are
the precursors of plasma cells.
Basophil. A circulating granular leukocyte having prominent
cytoplasmic granules when stained with dyes that indicate a basic pH.
The granules contain histamine and sulfated mucopolysaccharides. After
binding of antigen to membrane-bound IgE via Fc-epsilon-RI receptors,
they release histamine, platelet activating factor (PAF) and
leukotrienes, and other inflammatory mediators.
Bronchoalveolar lavage. Harvesting of cells and fluid from the
lung, commonly by bronchoscopy and lavage.
Bronchoprovocation. Use of inhaled triggers (cold air, histamine,
methacholine, allergen, etc.) to assess the responsivity of the
airways.
Carrier. An immunogenic macromolecule (usually protein) to which a
hapten is attached, allowing the hapten to be immunogenic.
CD. A molecular marker on a cell surface that may be used
operationally to define phenotype, origin and activation state of the
cell.
CD3. A molecule composed of five polypeptide chains associated with
the heterodimer T-cell receptor (TCR), forming the T-cell receptor
complex (TCR/ CD3); CD3 transduces the activating signals when antigen
binds to the TCR.
CD4. A cell surface antigen belonging to the immunoglobulin
superfamily of molecules. Marker of T helper cells. As an adhesion
molecule, it interacts with the non-polymorphic part of MHC class II
gene product.
CD8. A cell surface molecule belonging to the immunoglobulin
superfamily of molecules. Marker of suppressor and cytotoxic T-cells.
As an adhesion molecule, it interacts with the MHC class I gene
product.
CD16. Low-affinity Fc-gamma receptor (Fc-gamma-RIII) expressed
mainly on NK cells, granulocytes and macrophages, mediating ADCC.
CD23. Low-affinity Fc-epsilon receptor induced by IL-4 and
expressed on activated B-cells and macrophages.
Cell-mediated or cellular response. A specific immune response in
which T-lymphocytes mediate the effects, either through the release of
cytokines or through cytotoxicity.
Class I MHC gene products. Antigens encoded by the MHC class I
genes are expressed on all nucleated cells. They present
antigen-derived peptides of endogenous origin.
Class II MHC gene products. Antigens encoded by the MHC class II
genes are expressed on antigen-presenting cells. They present
antigen-derived peptides of endogenous origin.
Clonal anergy. A form of self tolerance developing as a consequence
of negative selection during the thymic selection processes. Clones of
thymocytes whose antigen receptors (TCR) bind with high affinity to
self antigens in association with MHC molecules are inactivated.
Complement system. A group of serum proteins with the capacity to
interact with each other when activated. The chain reaction of the
activated complement components results in formation of a lytic
complex and several biologically active peptides of low relative
molecular mass (anaphylatoxins). The system can be activated by
antigen-antibody complexes (classical pathway) and by other
components, e.g., bacteria (alternative pathway). As an effector
mechanism of the humoral immune response, the activated complement
system facilitates opsonization, phagocytosis and lysis of cellular
antigens.
Contact sensitivity. A state of immunological sensitization in
which an eczematous epidermal reaction may occur when a hapten is
applied to the skin of a sensitized individual. (see allergic contact
dermatitis).
Contact urticaria. Urticaria provoked by contact with inducing
agents (see urticaria).
Cross-reactivity. The ability of an antibody or a T-cell, specific
for one antigen, to react with a second antigen; a measure of
relatedness between two antigenic substances, and/or polyspecificity
of the antibody molecule (e.g., some rheumatoid factors), or of the
T-cell receptor.
Cytokines. Group of substances (biologically active peptides),
mainly synthesized by lymphocytes (lymphokines) or monocytes/
macrophages (monokines), that modulate the function of cells in
immunological reactions; cytokines include interleukins. Some
cytokines (pleotrophic cytokines) have a broad spectrum of biological
actions, including: neuromodulation, growth factor activity and
proinflammatory activity (see also interleukins).
Cytotoxic T-cell (cytolytic T-cell)(CTL). A subpopulation of T-cells
with the capacity to lyse target cells displaying a determinant in
association with MHC gene products, recognized by its antigen receptor
complex (TCR/CD3).
Delayed-type hypersensitivity (DTH) (Gell and Coombs Type IV
reaction). A form of T-cell-mediated immunity in which the ultimate
effector cell is the activated mononuclear phagocyte (macrophage); the
response of DTH appears fully over 24 to 48 h. Previous exposure is
required. Examples include response to Mycobacterium tuberculosis
(tuberculin test) and contact dermatitis.
Dendritic cell. A cell type characterized by extended cytoplasmic
protrusions and a high expression of adhesion molecules and Class II
MHC gene products effecting antigen presentation to specific
lymphocytes (see also Langerhans' cell).
Dermatitis. Inflammatory skin disease showing redness, swelling,
infiltration, scaling and sometimes vesicles and blisters.
Desensitization. Generally transient state of specific
non-reactivity in previously sensitized individual, resulting from
repeated antigen exposures.
Eczema. A dermatitis characterized by non-contagious inflammation
of skin with typical clinical (itch, erythema, papules, seropapules,
vesicles, squames, crusts, lichenification) and dermatohistological
(spongiosis, acanthosis, parakeratosis, lymphocytic infiltration)
findings. Often due to sensitization.
Elicitation. Production of a cell-mediated or antibody-mediated
allergic response by exposure of a sensitized individual to an
allergen.
Endocytosis. The uptake by a cell of a substance from the
environment by invagination of its plasma membrane; it includes both
phagocytosis mediated by receptors and pinocytosis.
Enzyme-linked immunosorbent assay (ELISA). An assay in which an
enzyme is linked to an antibody and a labelled substance is used to
measure the activity of bound enzyme and, hence, the amount of bound
antibody. With a fixed amount of immobilized antigen, the amount of
labelled antibody bound decreases as the concentration of unlabelled
antigen is increased, allowing quantification of unlabelled antigen
(competitive ELISA). With a fixed amount of one immobilized antibody,
the binding of a second, labelled antibody increases as the
concentration of antigen increases, allowing quantification of antigen
(sandwich ELISA).
Eosinophil. A circulating granular leukocyte having prominent
granules that stain specifically by eosin and containing numerous
lysosomes. Eosinophils are important effector cells in immune
reactions to antigens that induce high levels of IgE antibodies (e.g.,
parasites). Eosinophils are also abundant at sites of immediate
hypersensitivity reactions.
Epidemiology. The study of the distribution and determinants of
health-related states or events in specified populations, and the
application of this knowledge to manage health problems.
Epitope. A single antigenic determinant.
Fc receptors. Receptors expressed on a wide range of cells,
interacting with the Fc portion of immunoglobulins belonging to
various isotypes. Membrane-bound Fc receptors mediate different
effector functions (endocytosis, antibody-dependenT-cellular
cytotoxicity (ADCC)) and induce mediator release. Both the
membrane-bound and soluble forms of Fc receptors regulate antibody
production of B-cells.
Forced expiratory volume in 1 second (FEV1). Physiological
measurement of the volume of air expired in one second with a maximal
respiratory effort.
Forced ventilatory capacity (FVC). The physiological measurement of
lung volume associated with a complete respiratory effort.
Glomerulonephropathy. Disease of the glomeruli, which may show
either thickening of the basement membrane -- membranous
glomerulopathy associated with IgG deposits -- due to the accretion of
proteins, or "minimal change glomerulopathy", in which there is
functional damage but little structural change by light microscopy.
Hapten. A non-immunogenic compound of low relative molecular mass
which becomes immunogenic after conjugation with a carrier protein or
cell and in this form induces immune responses. Antibodies, but not
T-cells, can bind the hapten alone in the absence of carrier.
Helper T-cell. A functional subpopulation of T-cells (expressing
CD4 antigen) that help to generate cytotoxic T-cells and cooperate
with B-cells in the production of an antibody response. Helper T-cells
recognize antigen in association with MHC class II gene products.
Depending on their capacity to produce various cytokines one can
functionally differentiate Th1 (IL-2 and IFN gamma producing) and Th2
(IL-3, IL-4 and IL-6 producing) cells.
Human leukocyte antigen (HLA). The major human histocompatibility
complex situated on chromosome 6. Human HLA-A, -B and -C (resembling
mouse H-2K, D and L) are class I MHC molecules, whereas HLA DP, -DQ
and -DR (resembling mouse I-A and I-E) are class II MHC molecules.
Humoral immune response. An immune response in which specific
antibodies induce the effector functions (such as phagocytosis and
activation of the complement system).
Hyperreactivity. An abnormally increased response to a stimulus.
Hypersensitivity. Abnormally increased immunologically mediated
response to a stimulus. Sometimes used loosely for any increased
response (see also hypersusceptibility, hyperreactivity)
Hypersensitivity pneumonitis (HPS) / extrinsic allergic alveolitis
(Gell and Coombes Type III reaction). An immune-mediated
inflammatory disease of the lung parenchyma caused by exposure to an
inhaled chemical allergen or organic dust.
Hypersusceptibility. Adverse effects in an individual occurring
under exposure conditions that result in no effects in the great
majority of the population or an individual exhibiting exaggerated
effects in comparison with the great majority of those showing some
adverse effects.
IgE-binding Fc receptors. The high-affinity IgE-binding
Fc-epsilon-R type I is expressed on mast-cells and basophils.
Interacts with IgE antibodies with high affinity. The cross-linking of
these receptors results in release of mediators (such as histamine).
The receptor is composed of alpha, beta and gamma chains; the alpha
chain contains the IgE binding site, while the gamma chain is
responsible for signal transfer. The low-affinity IgE binding Fc
receptor (CD23) is expressed on B-cells, its soluble (truncated) form
is generated by proteolytic cleavage and regulates IgE production of
B-cells.
Immediate-type hypersensitivity (Gell and Coombs Type I reaction).
A form of antibody mediated immunity that takes place in minutes to
hours after the administration of antigen. Previous exposure is
required. An example is allergic rhinitis to pollen antigen.
Immunodeficiency. Defects in one or more components of the immune
system resulting in inability to eliminate or neutralize non-self
antigens. Congenital or primary immunodeficiencies are genetic or due
to developmental disorders (such as congenital thymic aplasia).
Acquired or secondary immunodeficiencies develop as a consequence of
malnutrition, malignancies, immunosuppressive compounds, radiation or
infection of immunocompetent T-cells with human immunodeficiency virus
(HIV). Defects of the nonspecific defence system may also result in
immunodeficiency.
Immunogen. A substance capable of eliciting a specific immune
response manifested by the formation of specific antibodies and/or
specifically committed lymphocytes. To induce an antibody response an
immunogen must possess structurally and functionally distinct
determinants for activation of B-cells and T-cells.
Immunoglobulin (Ig). Immunity-conferring portion of the plasma- or
serum-gammaglobulins. Various isotypes (classes and subclasses) of
immunoglobulins have a common core structure of two identical light
(L) and two identical heavy (H) polypeptide chains, which contain
repeating homologous units folded in common globular motifs (Ig
domains). The amino acid sequences of the N-terminal domains are
variable (V domains), in contrast to the more conserved constant
regions (C domains). The V domains contain the
complementarity-determining regions (CDRS) forming the antigen-binding
sites, whereas the C domains trigger several effector functions of the
immune system (see also antibody).
Immunoglobulin gene superfamily. Genes encoding proteins containing
one or more Ig domains (homology units) that are homologous to either
Ig V or C domains. Cell surface and soluble molecules mediating
recognition, adhesion or binding functions in and outside the immune
system, derived from the same precursor, belong to this family of
molecules (e.g., Ig, TCR, MHC Class I and II, CD4, CD8, Fc-gamma-R,
NCAM, PDGFR).
Incidence (epidemiological). The number of new cases of disease in
a defined population during a specified period of time.
Interleukin. Immunoregulatory proteins, also designated as
lymphokines, monokines or cytokines. General features are: low
relative molecular mass (<80 000) and frequently glycosylated;
regulate immune cell function and inflammation by binding to specific
cell surface receptors; transient and local production; act in
paracrine, autocrine or endocrine manner, with stimulatory or blocking
effect on growth/differentiation; very potent, function at picomolar
concentrations. Interleukins represent an extensive series of
mediators with a wide range of overlapping functions. Other mediators
in this series are c-kit ligand, interferons, tumour necrosis
factor, transforming growth factor, and a family of low relative
molecular mass mediators, called chemokines.
Intolerance. Non-immunologically mediated adverse reactions. In
food intolerances these may be due to pharmacological properties of
food constituents, metabolic disorders or responses of unknown
etiology.
Langerhans' cells. Bone-marrow-derived epidermal cells with a
dendritic morphology, expressing CD1 marker in humans and containing
the cytoplasmic organelle, called the Birbeck granule. They express
Class II MHC antigen and are capable of antigen presentation (see also
dendritic cells).
Lymphocyte. Bone-marrow-derived cell with little cytoplasm, with
the ability to migrate and exchange between the circulation and
tissues, to home to sites of antigen exposure, and to be held back at
these sites. The only cells that specifically recognize and respond to
antigens (mainly with the help of accessory cells). Lymphocytes
consist of various subsets differing in their function and products
(e.g., B-lymphocytes, helper-T-cells, cytolytic T-cells).
Macrophage. Mononuclear cells derived from monocytes residing in
tissues. Activated by different stimuli they may appear in various
forms such as epitheloid cells and multinucleate giant cells.
Macrophages found in different organs and connective tissues have been
named according the specific locations, e.g., as microglia, alveolar
macrophages or Kupffer cells. Macrophages may function as
antigen-presenting cells, effector cells of cell-mediated immunity,
and phagocytes eliminating opsonized antigens.
Major basic protein. A small basic arginine-rich peptide (pI 10.9,
relative molecular mass of 13 800) in the granules of eosinophils that
kills helminths and protozoa.
Major histocompatibility complex (MHC). A cluster of genes encoding
cell surface antigens that are polymorphic within a species and have a
crucial function in signalling between lymphocytes and cells
expressing antigen and in recognition of self.
Mast cell. Tissue bound mononuclear granular cells with staining
affinity for basic dyes at low pH. The specific granules contain
mediators of allergic inflammation, e.g., histamine. Upon stimulation
with antigen via membrane-bound IgE antibodies, they release preformed
and newly generated mediators. Two types of mast cells exist.
Tryptase-containing T-mast cells are mainly associated with mucosal
epithelial ceils. Chymase-containing TC mast cells are long-living
connective tissue cells.
Mitogen. A substance that causes cells to synthesize DNA and
proliferate without acting as an antigen.
Monocyte. Bone-marrow-derived mononuclear phagocytic leukocyte, with
bean-shaped nucleus and fine granular cytoplasm containing lysosomes,
phagocytic vacuoles and cytoskeletal filaments. Once transported to
tissues they develop into macrophages.
Natural killer (NK) cell. A subset of lymphocytes found in blood
and some lymphoid tissues, derived from the bone marrow and appearing
as large granular lymphocytes (LGL). NK cells possess the capacity to
kill certain tumour cells or virus-infected normal cells. The killing
is not induced by specific antigen and is not restricted by MHC
molecules.
Nephropathy. Disease of the kidney that may involve either or both
the glomeruli (specialized structures where blood is filtered) and the
renal tubules (connected structures where the composition of the
filtrate is greatly modified in accordance with the physiological
needs of the body).
Nephrotic syndrome. A clinical disease in which damage to glomeruli
has caused leaky filtration, resulting in major loss of protein from
the body.
Neutrophil (polymorphonuclear leukocyte). Granular leukocytes
having a nucleus with three to five lobes and fine cytoplasmic
granules stainable by neutral dyes. The cells have properties of
chemotaxis, adherence to immune complexes, and phagocytosis. The cells
are involved in a variety of inflammatory processes including
late-phase allergic reactions.
Occupational asthma. Asthma caused by a sensitizing agent present
in the workplace, usually after a period of asymptomatic exposure.
Opsonization. Coating of antigens with antibody and/or complement
components. The interaction of opsonized complexes with Fc- or
complement-receptors facilitates their uptake by the receptor-bearing
phagocytic cells.
Oral tolerance. Orally induced and immune-mediated
non-responsiveness.
Peak expiratory flow rate (PEFR). A physiological measure of the
maximum air flow.
Plasma cell. A terminally differentiated B-lymphocyte with little
or no capacity for mitotic division, that can synthesize and secrete
antibody. Plasma cells have eccentric nuclei, abundant cytoplasm and
distinct perinuclear haloes. The cytoplasm contains dense rough
endoplasmic reticulum and a large Golgi complex.
Platelet activating factor. Low relative molecular mass
phospholipid generated from alkyl phospholipids in mast cells,
basophilic and neutrophilic granulocytes, and monocytes-macrophages,
which mediates microthrombus formation of platelets in
hypersensitivity reactions.
Prevalence (epidemiology). The number of cases of disease occurring
in a given population at a designated time.
Prevention of allergy. Primary prevention is the control of the
exposures inducing allergy. Secondary prevention is the control of
exposure of sensitized individuals.
Pseudo-allergy. Non-immunological "hypersensitivity" with clinical
symptoms and signs mimicking those of allergic diseases.
Radioallergosorbent test (RAST). A solid-phase radioimmunoassay for
detecting IgE antibody specific for a particular antigen.
Rate (epidemiology). The frequency with which an event occurs in a
defined population.
Reactive airways dysfunction syndrome (RADS). A syndrome
characterized by reversible airflow limitation and complicating
bronchial hyperresponsiveness induced by acute exposure to high
concentrations of non-sensitizing irritant gases at work.
Sensitization. Induction of specialized immunological memory in an
individual by exposure to an allergen.
Stem cell. Pluripotent cells, representing 0.01% of bone marrow
cells, having the capacity for self renewal, and committed to
differentiate along particular lineages, e.g., erythroid,
megakaryocytic, granulocytic, monocytic and lymphocytic. Cytokines
stimulate the proliferation and maturation of distinct precursors.
Suppressor T-lymphoctye. A subpopulation of T-lymphocytes that
inhibits the activation phase of immune responses. They are CD8+, and
their growth and differentiation may be regulated by CD4+ cells.
Tolerance. Persistent condition of specific immunological
unresponsiveness, resulting from previous non-sensitizing exposure to
the antigen.
Urticaria. Transient eruption of skin characterized by erythematous
or oedematous swelling (wheal) of the dermis or subcutaneous tissue.
9. CONCLUSIONS
Allergy is an important, world-wide, health problem. It affects a
substantial proportion of the population and all age groups. For
reasons that are poorly understood, the overall frequency of allergy
is increasing.
The various and complex interactions between chemicals, drugs and
proteins, the immune system and the target organ(s) that lead to the
manifestation of allergic hypersensitivity and autoimmunity are
reviewed in this monograph. Extensive research into these topics
continues, so new developments are anticipated. The multiplicity of
endogenous and exogenous factors that have an impact upon allergy are
considered. Exogenous factors, including allergens themselves, as well
as infection, air pollution and life style (e.g., tobacco smoking) are
all of importance. In addition, genetic predisposition to a particular
allergic disorder is an important determinant of reactivity.
The epidemiology of allergy demonstrates the widespread nature of
this group of disorders and, as a consequence, highlights the need for
proper attention to the identification of allergic hazards and their
assessment in the implementation of appropriate risk management
strategies.
Methods of hazard identification for skin sensitizers are well
established. They have still to be standardized for respiratory
sensitizers and are not yet available for other types of allergen.
Techniques to measure the potency of skin sensitizers are being
applied for the purpose of risk assessment.
Once allergic hazards have been well characterized, risk
assessment, risk management and risk communication are critical
elements to reduce the incidence of allergic disorders. Risk
assessment requires that the hazard, of known potency, is evaluated in
the light of the nature and extent of exposure. Risk management
measures, such as control of exposure and product labelling, must be
implemented when the risk assessment indicates the need.
More is being learned of the physicochemical and immunological
features of food allergens, which may eventually be of predictive
value.
The complexity of the mechanisms of allergic disorders makes it
difficult at present to suggest in vitro predictive methods of
general applicability, although application of structure-activity
relationships deserves further consideration.
10. RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH
a) Effective strategies to prevent allergy should be employed, based
on good information about allergens in products and the
environment. Control of exposure should be the basis for
preventing or minimizing the occurrence of allergic disease.
b) There is urgent need to determine the cause of the increased
frequency of allergy.
c) Methods of surveillance should be instituted to define the
frequency of allergies of different types.
d) The measurement of exposure of individuals and of populations may
be difficult, but adequate assessment is essential to any
analysis of the association between exposure and effect. The
specific nature of immune responses represents a unique type of
biomarker in studying past exposure, for example, by the use of
skin patch or prick testing, or assay of IgE antibodies to detect
sensitization by specific allergens.
e) Worker surveillance systems, the quality of medical examination
of workers and education of workers exposed to chemicals should
be improved in order to reveal occupational allergic diseases at
an early stage. Relatively simple notification schemes for
occupational disorders and post-marketing surveillance of
medicines provide economic screening and alerting systems for
allergic diseases. Monitoring these disorders in the workplace is
particularly valuable because exposure there is likely to be
greater than anywhere else.
f) The efficacy and value of primary and secondary prevention and
intervention strategies should be assessed at intervals using
validated epidemiological techniques.
g) For allergic contact dermatitis, the available in vivo
predictive models are of proven value for antigens of low
relative molecular mass. The need is to discover how best they
can be used to show the potency of allergens.
h) For allergic disorders of the respiratory tract, available in
vivo test methods for substances of low relative molecular mass
are promising, but their predictive value and specificity need to
be substantiated. For protein allergens, some animal test methods
are being developed, but they require further evaluation using
substances of known allergenic potential in humans.
i) It is important that information about the presence of allergens
in products (e.g., food) be readily available, for instance in
databases and on product labels, so that regulators and
individuals can adopt appropriate precautions.
j) The main method for avoiding occupational chemically induced
autoimmune disease is the control of exposure. Pre-exposure
assessment of those exposed to chemicals of immunomodulating
potential should be considered in order to document any
pre-existing features of connective tissue diseases. Strict
adherence to guidelines to avoid or minimize exposure is advised,
including the use of good occupational hygiene practices. Other
risk factors such as smoking should be minimized and regular
occupational medical examinations should be considered.
k) There is a need for investigation of the quantitative
relationships between immune responses induced by chemicals and
the severity of allergic reactions.
l) Determination of the minimal exposure and the duration of it
required to cause sensitization or elicit an allergic response is
important in controlling allergic disease.
m) It is important to devise standard strategies for the clinical
investigation and diagnosis of allergy and to apply them
internationally to examine the causes and incidence of allergic
disorders. This will require the production and availability of
standardized extracts of biological allergens of controlled
potency, as well as regular consideration of the components of
standard series of allergens used in testing.
n) Public health authorities, health professionals and government
agencies should consider how to estimate the human and economic
costs to individuals and society of allergic diseases.
o) Public health authorities, health professionals, the public and
especially the workforce would benefit from better information
about the occurrence, causes, clinical manifestations and
consequences of different types of allergy.
11. FURTHER RESEARCH
a) The adjuvant effect of environmental factors, such as pollutants,
particulates, tobacco smoke and UV radiation, on the induction
and elicitation of allergic responses to other chemicals should
be investigated. The influence of other forms of toxicity,
including direct immunotoxicity, should also be studied.
b) The influence of formulation and mixtures on the induction and
elicitation of chemical allergy needs to be explored.
c) The relevance of route of exposure to chemicals to the
development of allergic disease, particularly allergic
sensitization of the respiratory tract, needs to be investigated.
d) The relevance of route of exposure to the development of
tolerance should be explored.
e) The role of IgE antibody and other immune effector mechanisms in
the development of chemical respiratory allergy needs to be
clarified.
f) Understanding of the cellular mechanisms responsible for the
induction of allergy, such as how allergens are processed and
presented to T-cells, is necessary to facilitate the development
of predictive in vitro tests.
g) Improved, reliable, sensitive, specific and robust biomarkers of
exposure to allergens are needed. Ideally, they should be
non-invasive and suitable for field use. Similarly, better
techniques for dosimetry of allergens are needed.
h) The basis for the difference in the response of asthmatics and
non-asthmatics to airborne pollutants needs exploration.
i) It is important to understand the relative contributions of
lifestyle, nutrition, pollution and change in the pattern of
childhood infection and immunization to the development of
allergy.
j) Mechanisms of sensitization to food allergens need further
research. In addition, more needs to be known about the
occurrence and clinical importance of cross-reactivity of complex
allergens, especially those of natural origin, such as pollen and
food components.
k) It is necessary to identify the properties that control the
allergenic characteristics and potency of proteins, including the
reasons why they induce transient or persistent food allergy.
l) In the case of autoimmune disease, predictive methods need to be
developed and validated.
REFERENCES
Aalberse RC, Koshte V, & Clemens JG (1981) Immunoglobulin E antibodies
that cross react with vegetable foods, pollen and hymenoptera venom. J
Allergy Clin Immunol, 68: 356-364.
Abbey DE, Hwang BL, Burchette RJ, Vancuren T, & Mills PK (1995)
Estimated long-term ambient concentrations of PM10 and development of
respiratory symptoms in a nonsmoking population. Arch Environ Health,
50(2): 139-152.
Aberg N, Engstrom I, & Lindberg U (1989) Allergic diseases in Swedish
school children. Acta Paediatr Scand, 78: 246-252.
Abramson S, Miller RG, & Phillips RA (1977) The identification in
adult bone marrow of pluripotent and restricted stem cells of the
myeloid and lymphoid systems. J Exp Med, 145: 1567-1579.
Adams LE & Hess EV (1991) Drug-related Lupus: Incidence, mechanisms
and clinical implications. Drug Saf, 6: 431-449.
Adams RB, Planchon SM, & Roche JK (1993) Interferon-gamma modulation
of epithelial barrier function: time course, reversibility, and site
of cytokine binding. J Immunol, 150: 2356-2363.
Adinoff AD, Telez P, & Clark RAF (1988) Atopic dermatitis and
aeroallergen contact sensitivity. J Allergy Clin Immunol, 81: 736-742.
Alanko K, Keskinen H, Björkstén F, & Ojanen S (1978) Immediate-type
hypersensitivity to reactive dyes. Clin Allergy, 8: 25-31.
Alarcon-Segovia D (1985) Scleroderma. In: Rose NR & Mackay IR ed. The
autoimmune diseases. New York, London, San Diego, Academic Press, pp
119-143.
Allison AC (1989) Theories of self tolerance and autoimmunity. In:
Kammüller M, Bloksma N, & Seinen W ed. Autoimmunity and toxicology:
Immune disregulation induced by drugs and chemicals. Amsterdam,
Oxford, New York, Elsevier Science Publishers, pp 67-115.
Amagi M, Ishii K, Hashimoto T, Gamou S, Shimizu N, & Nishikawa T
(1995) Conformational epitopes of pemphigus antigens (Dsg1 and Dsg3)
are calcium dependent and glycosylation dependent. J Invest Dermatol,
105: 243-247.
American Medical Association (1987a) Council of Scientific Affairs,
American Medical Association, Report I, Part I, of the Allergy Panel
(1987): in vivo diagnostic testing and immunotherapy for allergy. J
Am Med Assoc, 258: 1363-1367.
American Medical Association (1987b) Council of Scientific Affairs,
American Medical Association, Report I, Part II, of the Allergy Panel
(1987): in vitro testing for allergy. J Am Med Assoc, 258:
1639-1643.
Ames RG, Hall DS, & Reger RB (1984) Chronic respiratory effects of
exposure to diesel emissions in coal mines. Arch Environ Health, 39:
389-394.
Amin S, Lahti A, & Maibach HI (1996) Contact urticaria and the contact
urticaria syndrome. In: Marzulli FN & Maibach HI ed.
Dermatotoxicology, 5th ed. London, Taylor and Francis, pp 485-505.
Andersen KE & Maibach HI (1985) Guinea pig sensitization assays. In:
Andersen KE & Maibach HI ed. Contact allergy: Predictive tests in
guinea pigs. Basel, Karger, pp 263-290.
Andersen KE & Rycroft RJG (1991) Recommended patch test concentrations
for preservatives, biocides and antimicrobials. Contact Dermatitis,
25: 1-18.
Andersen KE, Burrows D, & White IR (1995) Allergens from the standard
series. In: Rycroft RJG, Menné T, Frosch PJ, & Benezra C ed. Textbook
of contact dermatitis, 2nd ed. Berlin, Heidelberg, New York, Springer
Verlag, pp 416-457.
Anderson HR (1974) The epidemiological and allergic features of asthma
in the New Guinea Highlands. Clin Allergy, 4: 171-183.
Anderson HR (1989) Increase in hospital admissions for childhood
asthma: trends in referral, severity, and readmissions from 1970 to
1985 in a health region of the United Kingdom. Thorax, 44: 614-619.
Anderson JA (1991) The clinical spectrum of food allergy in adults.
Clin Exp Allergy, 21(suppl 1): 304-314.
Anderson SD & Smith CM (1991) Osmotic challenges in the assessment of
bronchial hyperresponsiveness. Am Rev Respir Dis, 143(3/2): S43-S46.
Anderson LB, Dreyfuss EM, Logan U, Johnstone E, & Glaser J (1970)
Melon and banana sensitivity coincident with ragweed pollinosis. J
Allergy Clin. Immunol, 45: 310-319.
Anderson HR, Bailey PA, Cooper JS, Palmer JC, & West S (1983)
Morbidity and school absence caused by asthma and wheezing illness.
Arch Dis Child, 58: 777-784.
Anderson HR, Pottier AC, & Strachan DP (1992) Asthma from birth to age
23: incidence and relation to prior and concurrent atopic disease.
Thorax, 47: 537-542.
Anderson HR, Butland BK, & Strachan DP (1994) Trends in prevalence and
severity of childhood asthma. Br Med J, 308: 1600-1604.
Anderson C, Hehr A, Robbins R, Hasan R, Athar M, Mukhtar H, & Elmets
CA (1995) Metabolic requirements for induction of contact sensitivity
to immunotoxic polyaromatic hydrocarbons. J Immunol,
155(7): 3530-3537.
Angelini G (1995) Allergens related to specific exposures: Topical
drugs. In: Rycroft RJG, Menné T, Frosch PJ, & Benezra C ed. Textbook
of contact dermatitis, 2nd ed. Berlin, Heidelberg, New York, Springer
Verlag, pp 477-498.
Angle CR (1988) Indoor air pollutants. Adv Pediatr, 35: 239-280.
Anonymous (1982) Breast feeding and eczema/asthma. Lancet, 1(8277):
910-911.
Anonymous (1985) Smoking, occupation, and allergic lung disease.
Lancet, 1(8435): 965.
Anto JM (1995) Asthma outbreaks: an opportunity for research? Thorax,
50(3): 220-222.
Anto JM, Sunyer J, Rodriguez Roisin R, Suarez Cervera M, & Vazquez L
(1989) Community outbreaks of asthma associated with inhalation of
soybean dust. Toxicoepidemiological Committee. New Engl J Med, 320:
1097-1102.
Armentia A, Martin Santos JM, Quintero A, Fernandez A, Barber D,
Alonso E, & Gil I (1990) Bakers' asthma: Prevalence and evaluation of
immunotherapy with a wheat flour extract. Ann Allergy, 65(4): 265-272.
Arnon R & Teitelbaum D (1993) On the existence of suppressor cells.
Int Arch Allergy Immunol, 100: 2-7.
Arshad SH, Matthews S, Gant C, & Hide DW (1992) Effect of allergen
avoidance on development of allergic disorders in infancy. Lancet,
339: 1493-1497.
Arshad SH, Stevens M, & Hide DW (1993) The effect of genetic and
environmental factors on the prevalence of allergic disorders at the
age of two years. Clin Exp Allergy, 23: 504-511.
Ashby J, Basketter DA, Paton D, & Kimber I (1995) Structure activity
relationships in skin sensitizing sensitization using the murine local
lymph node assay. Toxicology, 103: 177-194.
Asher MI, Pattemore PK, Harrison AC, Mitchell EA, Rea HH, Stewart AW,
& Woolcock AJ (1988) International comparison of the prevalence of
asthma symptoms and bronchial hyperresponsiveness. Am Rev Respir Dis,
138: 524-529.
Asher MI, Keil U, Anderson HR, Beasley R, Crane J, Martinez F,
Mitchell EA, Pearce N, Sibbald B, Stewart AW, Strachan D, Weiland SK,
& Williams HC (1995) International study of asthma and allergies in
childhood (ISAAC): Rationale and methods. Eur Respir J, 8(3): 483-491.
Asherson GL, Zembala M, Perera MACC, Mayhew B, & Thomas WR (1977)
Production of immunity and unresponsiveness in the mouse by feeding
contact sensitizing agents and the role of suppressor cells in the
Peyers's patches, mesenteric lymph nodes and other lymphoid tissue.
Cell Immunol, 33: 145-155.
Atkins FM, Steinberg SS, & Metcalfe DD (1985a) Evaluation of immediate
adverse reactions to foods in adult patients: I. Correlation of
demographic, laboratory, and prick skin test data with response to
controlled oral food challenge. J Allergy Clin Immunol, 75: 348-355.
Atkins FM, Steinberg SS, & Metcalfe DD (1985b) Evaluation of immediate
adverse reactions to foods in adult patients: II. A detailed analysis
of reaction patterns during oral food challenge. J Allergy Clin
Immunol, 75: 356-355.
Atkinson NF Jr & Platts-Mills TAE (1988) IgE and atopic disease. In:
Immunological diseases, 4th ed. Boston, Massachusetts, Little Brown,
vol 2, pp 1009-1026.
Avnstorp C (1992) Cement eczema: An epidemiological intervention
study. Acta Dermatol Venereol, 179(suppl): 1-22.
Axelsson IG, Johansson SG, Larsson PH, & Zetterstrom O (1990)
Characterization of allergenic components in sap extract from the
weeping fig (Ficus benjamina). Int Arch Allergy Appl Immunol, 91(2):
130-135.
Bachert C, Hauser U, Prem B, Rudack C, & Ganzer U (1995)
Proinflammatory cytokines in allergic rhinitis. Eur Arch
Otorhinolaryngol, 252(Suppl 1): 155-214.
Baird AW, Coombs RRA, McLaughlan P, & Cuthbert AW (1984) Immediate
hypersensitivity reactions to cow milk proteins in isolated epithelium
from ileum of milk-drinking guinea-pigs: Comparison with colonic
epithelia. Int Arch Allergy Appl Immunol, 75: 255-263.
Baker DB, Gann PH, Brooks SM, Gallagher J, & Bernstein IL (1990)
Cross-sensitization study of platinum salts sensitization among
precious metals refinery workers. Am J Ind Med, 18: 653-664.
Bakke P, Gulsvikk A, & Eide GE (1990) Hay fever, eczema and urticaria
in southwest Norway. Allergy, 45: 515-522.
Ball B, Horstman D, Folinsbee L, Gerrity T, DeWitt P, Abdul-Salaam S,
& Brown J (1993) Pulmonary responses in asthmatics performing light
exercise in clean air(air) and 0.16 ppm ozone (O3). Am Rev Respir
Dis, 147: A640 (abstract).
Barbee RA (1987) The epidemiology of asthma. Monogr Allergy, 21:
21-41.
Barbee RA, Kaltenborn W, Lebowitz MD, & Burrows B (1987) Longitudinal
changes in allergen skin test reactivity in a community population
sample. J Allergy Clin Immunol, 79(1): 16-24.
Bardare M, Vaccari A, Allievi E, Brunelli L, Coco F, de Gaspari GC, &
Flauto U (1993) Influence of dietary manipulation on incidence of
atopic disease in infants at risk. Ann Allergy, 71(4): 366-371.
Barnes PJ, Baranjuk J, & Belvisi MG (1991) Neuropeptides in the
respiratory tract. Am Rev Respir Dis, 144: 1187-1198.
Barnes PJ, Jonsson B, & Klim JB (1996) The costs of asthma. Eur Respir
J, 9: 636-642.
Barratt MD (1995) Quantitative structure activity relationships for
skin permeability. Toxicol in vitro, 9: 27-37.
Barratt MD, Basketter DA, Chamberlain M, Payne MP, Admans GD, &
Langowski JJ (1994a) Development of an expert system rule base for
identifying contact allergens. Toxicol in vitro, 8: 837-839.
Barratt MD, Basketter DA, Chamberlain M, Admans GD, & Langowski JJ
(1994b) An expert system rule base for identifying contact allergens.
Toxicol in vitro, 8: 1053-1060.
Barratt MD, Basketter DA, & Roberts DW (1994c) Skin sensitization
structure-activity relationships for phenyl benzoates. Toxicol in
vitro, 8: 823-826.
Barrett JT (1988) Textbook of immunology, 5th ed. St. Louis, Missouri,
C.V. Mosby & Co.
Barrett SP, Toh BH, Alderuccio F, van Driel I, & Gleeson PA (1995)
Organ-specific autoimmunity induced by adult thymectomy and
cyclophosphamide-induced lymphopenia. Eur J Immunol, 25: 238-244.
Barry DM, Burr ML, & Limb ES (1991) Prevalence of asthma among 12 year
old children in New Zealand and South Wales: a comparative survey.
Thorax, 46: 405-409.
Bascom R (1996) Environmental factors and respiratory
hypersensitivity: the Americas. Toxicol Lett, 86(2-3): 115-130.
Bascom R, Naclerio RM, Fitzgerald TK, Kagey Sobotka A, & Proud D
(1990) Effect of ozone inhalation on the response to nasal challenge
with antigen of allergic subjects. Am Rev Respir Dis, 142(3): 594-601.
Basketter DA & Roberts DW (1990) Structure activity relationships in
contact allergy. Int J Cosmet Sci, 12: 81-90.
Basketter DA, Roberts DW, Cronin M, & Scholes EW (1992) The value of
the local lymph node assay in quantitative structure-activity
investigations. Contact Dermatitis, 27: 137-142.
Basketter DA, Gerberich GF, Kimber I, & Loveless SE (1996) The local
lymph node assay -- a valuable alternative to currently accepted skin
sensitisation tests. Food Chem Toxicol, 34: 651-660.
Basketter DA, Cookman G, Gerberich GF, Hamaide N, & Potokar M (1997)
Skin sensitisation thresholds: Determination in predictive models.
Food Chem Toxicol, 35: 417-425.
Bates DV & Sizto R (1987) Air pollution and hospital admissions in
southern Ontario: The acid summer haze effect. Environ Res, 43:
317-331.
Baur V & Fruhmann G (1981) Specific IgE antibodies in patients with
isocyanate asthma. Chest, 80(suppl): 73S-76S.
Baur X, Dewair M, & Fruhmann G (1984) Detection of immunologically
sensitised isocyanate workers by RAST and intracutaneous skin tests. J
Allergy Clin Immunol, 73: 610-618.
Baur X, Marek W, Ammon J, Czuppon AB, Marczynski B, Raulf-Heimsoth M,
Roemmelt H, & Fruhman G (1994) Respiratory and other hazards of
isocyanates. Int Arch Occup Environ Health, 66: 141-152.
Beasley R, Roche WR, Roberts JA, & Holgate ST (1989) Cellular events
in the bronchi in mild asthma and after bronchial provocation. Am Rev
Respir Dis, 139: 806-817.
Beasley R, Thomson C, & Pearce N (1991) Selenium, glutathione
peroxidase and asthma. Clin Exp Allergy, 21(2): 157-159.
Becher R, Hongslo JK, Jantunen J, & Dybing E (1996) Environmental
chemicals relevant for respiratory hypersensitivity: the indoor
environment. Toxicol Lett, 86: 155-162.
Beeson PB (1994) age and sex associations of 40 autoimmune diseases.
Am J Med, 96: 457-462.
Beezhold DH, Sussman GL, Liss GM, & Chang N-S (1996) Latex allergy can
induce clinical reactions to specific foods. Clin Exp Allergy, 26:
416-422.
Beggs PJ & Curson PH (1995) An integrated environmental asthma model.
Arch Environ Health, 50(2): 87-94.
Behrendt H, Friedrich KH, Kainka-Stänicke E, Darsow U, Becker WM, &
Tomingas R (1991) New trends in allergy. In: Ring J & Pryzbilla B ed.
Allergens and pollutants in the air: A complex interaction. Berlin,
Heidelberg, New York, Springer Verlag, pp 467-478.
Behrendt H, Becker W, Friedrichs K, Darsow U, & Tomingas R (1992)
Interaction between aeroallergens and airborne particulate matter. Int
Arch Allergy Immunol, 99: 425-428.
Behrendt H, Krämer U, Dolgner R, Hinrichs J, Willer H, Hagenbeck H, &
Schlipköter W (1993) Elevated levels of total IgE in East German
children: Atopy, parasites or pollutants? Allergo J, 2: 31-40.
Behrendt H, Friedrich KH, Krämer U, Hitzfeld B, Becker WM, & Ring J
(1996) The role of indoor and outdoor air pollution in allergic
diseases. In: Vos J, Younes M, & Smith E ed. Allergic
hypersensitivities induced by chemicals. Boca Raton, Florida, CRC
Press, pp 173-182.
Bekesi JG, Roboz JP, Fischbein A, & Mason P (1987) Immunotoxicology:
Environmental contamination by polybrominated biphenyls and immune
dysfunction among residents of the State of Michigan. Cancer Detect
Prev, 1(suppl): 29-37.
Bell RG (1996) IgE, allergies and helminth parasites: a new
perspective on an old conundrum. Immunol Cell Biol, 74: 337-345.
Bencko V, Vasilieva EV, & Symon K (1980) Immunological aspects of
exposure to emissions from burning coal of high beryllium content.
Environ Res, 22(2): 439-449.
Benezra C, Sigman CC, Perry LR, Helmes CT, & Maibach HI (1985) A
systematic search for structure-activity relationships of skin contact
sensitizers: methodology. J Invest Dermatol, 85: 351-356.
Benjamini E & Leskowitz S (1991) Immunology: A short course. New York,
Wiley-Liss.
Bentley AM, Maestrelli P, Fabbri LM, Menz G, Storz Chr, Bradley B,
Jeffrey PK, Durham SR, & Kay AB (1991) Immunohistology of the
bronchial mucosa in occupational, intrinsic and extrinsic asthma. J
Allergy Clin Immunol, 87: 246 (abstract).
Berglund M, Braback L, Bylin G, Jonson JO, & Vahter M (1994) Personal
NO2 exposure monitoring shows high exposure among ice-skating
schoolchildren. Arch Environ Health, 49: 17-24.
Bernhisel-Broadbent J & Sampson HA (1989) Cross-allergenicity in the
legume botanical family in children with food hypersensitivity. J
Allergy Clin Immunol, 83: 435-440.
Bernhisel-Broadbent J, Taylor S, & Sampson HA (1989)
Cross-allergenicity in the legume botanical family in children with
food hypersensitivity: II. Laboratory correlates. J Allergy Clin
Immunol, 84: 701-709.
Bernstein DI, Patterson R, & Zeiss CR (1982a) Clinical and immunologic
evaluation of trimellitic anhydride- and phthalic anhydride-exposed
workers using a questionnaire and comparative analysis of enzyme
linked immunosorbent and radioimmunoassay studies. J Allergy Clin
Immunol, 69: 311-318.
Bernstein M, Day JH, & Welsh A (1982b) Double-blind food challenge in
the diagnosis of food sensitivity in the adult. J Allergy Clin
Immunol, 70: 205-211.
Bernstein DI, Gallagher JA, D'Souza L, & Bernstein IL (1984)
Heterogeneity of specific IgE responses in workers sensitised to acid
anhydride compounds. J Allergy Clin Immunol, 74: 794-801.
Biagini RE, Bernstein IL, Gallagher JS, Moorman WJ, Brooks S, & Gann
PH (1985) The diversity of reaginic immune responses to platinum and
palladium salts. J. Allergy Clin Immunol, 76(6): 794-802.
Biagini RE, Moorman WJ, Lewis TR, & Bernstein IL (1986) Ozone
enhancement of platinum asthma in a primate model. Annu Rev Respir
Dis, 134: 719-725.
Bieber TH & Ring J (1992) In vivo modulation of the high-affinity
receptor for IgE (Fc-epsilon-RI) on human epidermal Langerhans cells.
Int Arch Allergy Immunol, 99: 204-207.
Bigazzi PE (1988) Autoimmunity induced by chemicals. J Toxicol Clin
Toxicol, 26: 125-156.
Bigazzi PE (1997) Autoimmunity caused by xenobiotics. Toxicology, 119:
1-21.
Bignon JS, Aron Y, Ju LY, Kopferschmitt MC, Garnier R, Mapp C, Fabbri
LM, Pauli G, Lockhart A, Charron D, & Swierczewski E (1994) HLA class
II alleles in isocyanate-induced asthma. Am J Respir Crit Care Med,
149: 71-75.
Bilyk N & Holt PG (1995) Cytokine modulation of the immunosuppressive
phenotype of pulmonary alveolar macrophage populations. Immunology,
86: 231-237.
Bindslev-Jensen C, Hansen TK, Norgaard A, Vestergaard H, & Lars K
(1994a) New controversial techniques in the diagnosis of food
hypersensitivity. In: Johansson SGO ed. Progress in allergy and
clinical immunology. Toronto, Bern, Hans Huber, vol 3, pp 468-274.
Bindslev-Jensen C, Skov P, Madsen F, & Poulsen LK (1994b) Food allergy
and food intolerance - what is the difference? Ann Allergy, 72:
317-320.
Bircher AJ, Wüthrich B, Langauer S, & Schmid P (1993) [Ficus
benjamina, a perennial inhalation allergen of increasing importance.]
Schweiz Med Wochenschr, 123(22): 1153-1159 (in German).
Björksten B & Kjellman NIM (1987) Perinatal factors influencing the
development of allergy. Clin Rev Allergy, 5: 339-347.
Black CM, Welsh KI, Walker AE, Bernstein RM, Catoggio LJ, McGregor AR,
& Lloyd Jones JK (1983) Genetic susceptibility to scleroderma-like
syndrome induced by vinyl chloride. Lancet, 1: 53-55.
Blaikie L, Basketter DA, & Morrow T (1995) Experience with a guinea
pig model for the assessment of respiratory allergens. Hum Exp
Toxicol, 14: 73.
Blanco C, Carrillo T, Castillo R, Quiralte J, & Cuevas M (1994)
Avocado hypersensitivity. Allergy, 49: 454-459.
Bloch KJ & Walker WA (1981) Effect of locally induced intestinal
anaphylaxis on the uptake of a bystander antigen. J Allergy Clin
Immunol, 67: 312-316.
Bloksma N, Kammüller ME, Punt P, & Seinen W (1988) Strain-dependence
of primary popliteal lymph node reactions to subcutaneous injection of
diphenylhydantoin in mice. In: Kammüller ME ed. A toxicological
approach to chemical-induced autoimmunity. Utrecht, The Netherlands,
University of Utrecht, pp 79-92 (PhD Thesis).
Bloksma N, Kubicka-Muranyi M, Schuppe H-C, Gleichmann E, & Gleichmann
H (1995) Predictive immunotoxicological test systems: Suitability of
the popliteal lymph node assay in mice and rats. Crit Rev Toxicol, 25:
369-396.
Bloom BR, Salgame P, & Diamons B (1992) Revisiting and revising
suppressor T cells. Immunol Today, 13(4): 131-136.
Bock SA (1987) Prospective appraisal of complaints of adverse
reactions to foods in children during the first 3 years of life.
Pediatrics, 79(5): 683-688.
Bock SA, Sampson HA, Atkins FM, Zeiger RS, Lehrer S, Sachs M, Bush RK,
& Metcalfe DD (1988) Double-blind, placebo-controlled food challenge
(DBPCFC) as an office procedure: a manual. J Allergy Clin Immunol, 82:
986-997.
Boerrigter GH & Scheper RJ (1987) Local and systemic desensitization
induced by repeated epicutaneous hapten application. J Invest
Dermatol, 88(1): 3-7.
Bohadana AB, Massin N, Wild P, Kolopp MN, & Toamain JP (1994)
Respiratory symptoms and airway responsiveness in apparently healthy
workers exposed to flour dust. Eur Respir J, 7(6): 1070-1076.
Bonini S, Magrini L, Rotiroti G, Ronchetti MP, & Onotati P (1994)
Genetic and environmental factors in the changing incidence of
allergy. Allergy, 49: 6-14.
Bonnet M, Angibaud G, Cantagrel A, Montastruc JL, & Janet M (1995)
Myasthénie induite par la tiopronine au cours du traitement de la
polyarthrite rhumatoïde. Rev Neurol, 151: 67-68.
Borelli S (1981) [Dermatological indications for climatherapy in the
high altitude of Davos (1560 m and higher.] In: Borelli S & Düngemann
H ed. [Allergology and dermatology.] Frankfurt, Germany, IMP-Verlag,
pp 564-660 (in German).
Borelli S & Schnyder UW (1962) [Constitutional dermatitis without
atopy.] In: Miescher G & Storck H ed. [Dermatitis: I. Handbook of
treatment.] Berlin, Heidelberg, New York, Springer Verlag, vol 1/1, pp
254-319 (in German).
Bos JD & Kapsenberg ML (1993) The skin immune system: progress in
cutaneous biology. Immunol Today, 14: 75-78.
Braback L, Breborowicz A, Julge K, Knutsson A, Riikjarv MA, Vasar M, &
Bjorksten B (1995) Risk factors for respiratory symptoms and atopic
sensitisation in the Baltic area. Arch Dis Child, 72(6): 487-493.
Braun Fahrländer C, Kunzli N, Domenighetti G, Carell CF, & Ackermann
Liebrich U (1994) Acute effects of ambient ozone on respiratory
function of Swiss schoolchildren after a 10-minute heavy exercise.
Pediatr Pulmonol, 17: 169-177.
Braun-Fahrländer C, Vuille JC, Sennhauser FH, Neu U, Künzle T, Grize
L, Gassner M, Minder C, Schindler C, Varonier HS, & Wüthrich B (1997)
Respiratory health and long-term exposure to air pollutants in Swiss
schoolchildren -- SCARPOL Team: Swiss study on childhood allergy and
respiratory symptoms with respect to air pollution, climate and
pollen. Am J Respir Crit Care Med, 155: 1042-1049.
Breathnach SM (1988) The Langerhans cell: Centenary review. Br J
Dermatol, 119(4): 463-469.
Briatico-Vangosa G, Braun CLJ, Cookman G, Hofmann T, Kimber I,
Loveless SE, Morrow T, Pauluhn J, Sorensen T, & Niessen HJ (1994)
Respiratory allergy: Hazard identification and risk assessment. Fundam
Appl Toxicol, 23: 145-158.
Britton WJ, Woolcock AJ, Peat JK, Sedgwick CJ, Lloyd DM, & Leeder SR
(1986) Prevalence of bronchial hyperresponsiveness in children: the
relationship between asthma and skin reactivity to allergens in two
communities. Int J Epidemiol, 15(2): 202-209.
Britton J, Pavord I, Richards K, Knox A, Wisniewski A, Weiss S, &
Tattersfield A (1994) Dietary sodium intake and the risk of airway
hyperreactivity in a random adult population. Thorax, 49(9): 875-880.
Britton JR, Pavord ID, Richards KA, Knox AJ, Wisniewski AF, Lewis SA,
Tattersfield AE, & Weiss ST (1995) Dietary antioxidant vitamin intake
and lung function in the general population. Am J Respir Crit Care
Med, 151(5): 1383-1387.
Broder I, Higgins MW, Mathews KP, & Keller JB (1974a) Epidemiology of
asthma and allergic rhinitis in a total community, Tecumseh, Michigan.
J Allergy Clin Immunol, 53: 127-138.
Broder I, Higgins MW, Matthews KP, & Keeler JB (1974b) Epidemiology of
asthma and allergic rhinitis in a total community, Tecumseh, Michigan:
3. Second survey of the community. J Allergy Clin Immunol, 53:
127-138.
Brooks SM (1982) The evaluation of occupational airways disease in the
laboratory and workplace. J Allergy Clin Immunol, 70: 56-66.
Broughton A & Thrasher JD (1988) Antibodies and altered cell-mediated
immunity in formaldehyde-exposed humans. Comments Toxicol, 2: 155-174.
Bruijnzeel-Koomen C, Van Wichen DF, Toonstra J, Berrens L, &
Bruijnzeel PL (1986) The presence of IgE molecules on epidermal
Langerhans cells in patients with atopic dermatitis. Arch Dermatol
Res, 278(3): 199-205.
Bruijnzeel-Koomen C, Ortolani C, Aas K, Bindslev-Jensen C, Björkstén
B, Moneret-Vautrin D, & Wüthrich B (1995) Position paper of the
European Academy of Allergy and Clinical Immunology on adverse
reactions to food: Adverse reactions to food. Allergy, 50: 623-635.
Brunekreef B (1992) Damp housing and adult respiratory symptoms.
Allergy, 47(5): 498-502.
Brunekreef B, Dockery DW, Speizer FE, Ware JH, Spengler JD, & Ferris
BG (1989) Home dampness and respiratory morbidity in children. Am Rev
Respir Dis, 140: 1363-1367.
Brunekreef B, Dockery DW, & Krzyzanowski M (1995) Epidemiologic
studies on short-term effects of low levels of major ambient air
pollution components. Environ Health Perspect; 103(Suppl 2): 3-13.
Brunekreef B, Janssen NA, de Hartog J, Harssema H, Knape M, & van
Vliet P (1997) Air pollution from truck traffic and lung function in
children living near motorways. Epidemiology, 8(3): 298-303.
Bruynzeel DP, von Blomberg-van der Flier M, Scheper RJ, van Ketel WG,
& de Haan P (1985) Penicillin allergy and the relevance of
epicutaneous tests. Dermatologica, 171: 429-434.
Bryson JS, Caywood BE, & Kaplan AM (1991) Relationship of cyclosporine
A-mediated inhibition of clonal deletion and development of syngeneic
graft-versus-host disease. J Immunol, 147: 391-397.
Buckley RH (1992) Primary immunodeficiency diseases. In: Wyngaarden J
& Smith L ed. Cecil's textbook of medicine. Philadelphia,
Pennsylvania, W.B. Saunders Company, pp 1446-1453.
Buehler EV (1965) Delayed contact hypersensitivity in the guinea pig.
Arch Dermatol, 5(91): 171-177.
Buist AS & Vollmer WM (1990) Reflections on the rise in asthma
morbidity and mortality. J Am Med Assoc, 264: 1719-1720.
Burge PS (1989) Diagnosis of occupational asthma. Clin Exp Allergy,
19(6): 649-652.
Burlingame RW & Rubin RL (1991) Drug-induced anti-histone
autoantibodies display two patterns of reactivity with substructures
of chromatin. J Clin Invest, 88: 680-690.
Burney P (1987) A diet rich in sodium may potentiate asthma:
Epidemiologic evidence for a new hypothesis. Chest, 91: 143S-148S.
Burney PG, Britton JR, Chinn S, Tattersfield AE, Platt HS, Papacosta
AO, & Kelson MC (1986) Response to inhaled histamine and 24 hour
sodium excretion. Br Med J Clin Res Ed, 292: 1483-1486.
Burney PG, Laitinen LA, Perdrizet S, Huckauf H, Tattersfield AE, Chinn
S, Poisson N, Heeren A, Britton JR, & Jones T (1989a) Validity and
repeatability of the IUATLD (1984) bronchial symptoms questionnaire:
An international comparison. Eur Respir J, 2(10): 940-945.
Burney PG, Neild JE, Twort CH, Chinn S, Jones TD, Mitchell WD, Bateman
C, & Cameron IR (1989b) Effect of changing dietary sodium on the
airway response to histamine. Thorax, 44: 36-41.
Burney PG, Chinn S & Rana RJ (1990) Has the prevalence of asthma
increased in children? Evidence from the national study of health and
growth 1973-86. Br Med J, 300: 1306-1310.
Burney P, Chinn S, Jarvis D, Luczynska C, & Lai E (1996) Variations in
the prevalence of respiratory symptoms, self-reported asthma attacks,
and use of asthma medication in the European Community respiratory
health survey (ECRHS). Eur Respir J, 9: 687-695.
Burnett RT, Dales R, Krewski D, Vincent R, Dann T, & Brook JR (1995)
Associations between ambient particulate sulphate and admissions to
Ontario hospitals for cardiac and respiratory diseases. Am J
Epidemiol, 142: 15-22.
Burr ML, Butland BK, King S, & Vaughan Williams E (1989) Changes in
asthma prevalence: Two surveys 15 years apart. Arch Dis Child 1989,
64: 1452-1456.
Burrows B, Halonen M, Barbee RA, & Lebowitz MD (1981) The relationship
of serum immunoglobulin E to cigarette smoking. Am Rev Respir Dis,
124(5): 523-525.
Busse WW (1990) Respiratory infections: their role in airway
responsiveness and the pathogenesis of asthma. J Allergy Clin Immunol,
85: 671-683.
Butcher BT & Salvaggio JE (1986) Occupational asthma. J Allergy Clin
Immunol, 78(4): 547-556.
Butler JM, Chan SC, Stevens S, & Hanifin JM (1983) Increased leukocyte
histamine release with elevated cyclic AMP-phosphodiesterase activity
in atopic dermatitis. J Allergy Clin Immunol, 71: 490-497.
Byers VS, Levin AS, Ozonoff DM, & Baldwin RW (1988) Association
between clinical symptoms and lymphocyte abnormalities in a population
with chronic domestic exposure to industrial solvent contaminated
domestic water supply and a high incidence of leukaemia. Cancer
Immunol Immunother, 27: 77-81.
Calkhoven P, Aalbers M, Koshte L, Pos O, Oei HD, & Aalberse RC (1987)
Cross-reactivity among birch pollen, vegetables and fruits as detected
by IgE antibodies is due to at least three distinct cross-reactive
structures. Allergy, 42: 382-390.
Calvin G (1992) Risk management case history -- detergents. In:
Richardson ML ed. Risk management of chemicals. London, Royal Society
of Chemistry, pp 120-136.
Carr RI, Etheridge PD, & Tilley D (1985) Failure of oral tolerance in
NZB/W mice is antigen dependent and parallels antibody patterns in
human systemic lupus erythematosus (SLE). Fed Proc, 44: 1542
(abstract).
Cartier A, Thompson NC, Frith PA, Roberts R, & Hargreave FE (1982)
Allergen-induced increase in bronchial responsiveness to histamine:
relationship to the late asthmatic response and change in airway
caliber. J Allergy Clin Immunol, 70: 170-177.
Cartier A, Bernstein IL, Burge PS, Cohn JR, Fabbri LM, Hargreave FE,
Malo JL, McKay RT, & Salvaggio JE (1989) Guidelines for
bronchoprovocation on the investigation of occupational asthma: Report
of the Subcommittee on Bronchoprovocation for Occupational Asthma. J
Allergy Clin Immunol, 84(5/2): 823-829.
Catto-Smith AG, Patrick MK, Hardin JA, & Gall DG (1989a) Intestinal
anaphylaxis in the rat: Mediators responsible for the ion transport
abnormalities. Agents Actions, 28: 185-191.
Catto-Smith AG, Patrick MK, Scott RB, Davidson JS, & Gall DG (1989b)
Gastric response to mucosal IgE-mediated reactions. Am J Physiol, 257:
G704 (abstract).
CDC (US Center for Disease Control) (1994) Populations at risk from
particulate air pollution -- United States, 1992. Morbid Mortal Wkly
Rep, 43: 290-293.
CDC (US Center for Disease Control) (1995) Asthma--United States,
1982-1992. Morbid Mortal Wkly Rep, 43: 952-955.
Challacombe SJ (1983) Salivary antibodies and systemic tolerance in
mice after oral immunization with bacterial antigens. Ann NY Acad Sci,
409: 177-193.
Challacombe SJ & Tomasi TB (1987) Oral tolerance. In: Brostoff J &
Challacombe SJ ed. Food allergy and intolerance, Section F: Model
systems of antigen handling. Philadelphia, Pennsylvania, Ballière
Tindall, pp 255-268.
Chambers CA & Allison JP (1997) Co-stimulation in T cell responses.
Curr Opin Immunol, 9: 396-404.
Chan PK, Baldwin RC, Parsons RD, Moss JN, Stiratelle R, Smith JM, &
Hayes AW (1983) Kathon biocide: Manifestation of delayed contact
dermatitis in guinea pigs is dependent on the concentrations for
induction and challenge. J Invest Dermatol, 81: 409-411.
Chan-Yeung M (1990) Occupational asthma. Chest, 98(suppl 5):
148S-161S.
Chan-Yeung M (1995) Occupational Asthma. Environ Health Perspect,
103(suppl 6): 249-252.
Chan-Yeung M & Malo JL (1995a) Occupational asthma. N Engl J Med,
333(2): 107-112.
Chan Yeung M & Malo JL (1995b) Epidemiology of occupational asthma.
In: Asthma and rhinitis. Oxford, London, Boston, Scientific
Publications, pp 44-57.
Chan-Yeung M, Malo JL, Busse W, & Holgate ST ed. (1995) Asthma and
rhinitis: Epidemiology of occupational asthma. Oxford, London, Boston,
Blackwell Scientific Publications, pp 44-57.
Chand N, Harrison JE, Rooney S, Pillar J, Jakubicki R, Nolan K,
Diamantis W, & Sofia RD (1992) Anti-IL-5 monoclonal antibody inhibits
allergic late phase bronchial eosinophilia in guinea pigs: a
therapeutic approach. Eur J Pharmacol, 211: 121-123.
Chandor SB (1988) Autoimmune phenomena in lymphoid malignancies. Clin
Lab Med, 8: 373-384.
Chapman JR & Roberts DW (1984) Humoral immune dysfunction as a result
of prenatal exposure to diphenylhydantoin: correlation with the
occurrence of physical defects. Teratology, 30: 107-117.
Charous BL (1994) The puzzle of latex allergy: some answers, still
more questions. Ann Allergy, 73(4): 277-281.
Chase MW (1946) Inhibition of experimental drug allergy by prior
feeding of the sensitizing agent. Proc Soc Exp Biol Med, 61: 257-259.
Chen Y, Kuchroo VK, Inobe J, Hafler DA, & Weiner HL (1994) Regulatory
T cell clones induced by oral tolerance: Suppression of autoimmune
encephalomyelitis. Science, 265: 1237-1240.
Chua KY, Stewart GA, Thomas WR, Simpson RJ, Dilworth RJ, Plozza TM, &
Turner KJ (1988) Sequence analysis of cDNA coding for a major house
dust mite allergen, Der p 1. J Exp Med, 167: 175-182.
Cirla AM (1994) Cobalt-related asthma: clinical and immunological
aspects. Sci Total Environ, 150(1-3): 85-94.
Cleland LG & Bell DA (1978) The occurrence of systemic lupus
erythematosus in two kindreds in association with severe selective IgA
deficiency. J Rheumatol, 5: 288-293.
Clinton PM, Kemeny DM, Amlot P, Urbanek R, & Lessof MH (1986)
Histamine release from peripheral blood leucocytes in egg-allergic
patients. Clin Allergy, 16: 345-354.
Coca AF & Cooke RA (1923) On the classification of the phenomena of
hypersensitiveness. J Immunol, 8: 163-182.
Cockcroft DW (1988) Allergens. In: Barnes PJ, Rodger IW, & Thomson NC
ed. Asthma: Basic mechanisms and clinical management. New York,
London, San Diego, Academic Press, pp 445-464.
Coenraads PJ & Smit J (1995) Epidemiology. In: Rycroft RJG, Menné T,
Frosch PJ, & Benezra C ed. Textbook of contact dermatitis, 2nd ed.
Berlin, Heidelberg, New York, Springer Verlag, pp 133-150.
Coffman RL, O'Hara J, Bond MW, Carty J, Zlotnik A, & Paul WE (1986)
B-cell stimulatory factor-1 enhances the IgE response of
lipopolysaccharide activated B-cells. J Immunol, 136(12): 4538-4541.
Coffman RL, Lebman D, & Shrader B (1989) Transforming growth
factor-beta specifically enhances IgA production by
lipopolysaccharide-stimulated murine B-lymphocytes. J Exp Med, 170:
1039-1044.
Cohen IR & Miller A ed. (1994) Autoimmune disease models. New York,
London, San Diego, Academic Press.
Colloff MJ, Ayres J, Carswell F, Howarth PH, Merrett TG, Mitchell EB,
Walshaw MJ, Warner JO, Warner JA, & Woodcock AA (1992) The control of
allergens of dust mites and domestic pets: A position paper. Clin Exp
Allergy, 22(suppl 2): 1-28.
Conell JT (1969) Quantitative intranasal pollen challenges: III. The
priming effect in allergic rhinitis. J Allergy, 43: 33-44.
Constantinescu CS, Hilliard B, Fujioka T, Bhopale MK, Calida D, &
Rostami AM (1998) Pathogenesis of neuroimmunologic diseases. Immunol
Res, 17: 217-227.
Cookson WOCM, Sharp PA, Faux JA, & Hopkin JM (1989) Linkage between
immunoglobulin E responses underlying asthma and rhinitis and
chromosome 11q. Lancet, 1: 1292-1295.
Cookson WOCM, Young RP, Sandford AJ, Moffatt MF, Shirakawa T, Sharp
PA, Faux JA, Julier C, Le Souef PN, Lathrop GM, & Hopkin JM (1992)
Maternal inheritance of atopic IgE responsiveness on chromosome 11q.
Lancet, 340: 381-384.
Cooper KD (1994) Atopic dermatitis: recent trends in pathogenesis and
therapy. J Invest Dermatol, 102: 128-137.
Cooper KD, Kang K, Chan SC, & Hanifin JM (1985) Phosphodiesterase
inhibition by Ro20-1724 reduces hyper-IgE synthesis by atopic
dermatitis cells in vitro. J Invest Dermatol, 804: 477-482
Cormio L, Turjanmaa K, Talja M, Andersson LC, & Ruutu M (1993)
Toxicity and immediate allergenicity of latex gloves. Clin Exp
Allergy, 23(7): 618-623.
Corrigan CJ & Kay AB (1992) T cells and eosinophils in the
pathogenesis of asthma. Immunol Today, 13: 501-507.
Cotterill JA (1991) Psychosomatic approaches in the treatment of
atopic eczema. In: Ruzicka T, Ring J, & Przybilla B ed. Handbook of
atopic eczema. Berlin, Heidelberg, New York, Springer Verlag, pp
459-465.
Crespo JF, Pascual C, Burks AW, Helms RM, & Esteban MM (1995)
Frequency of food allergy in a pediatric population from Spain.
Pediatr Allergy Immunol, 6: 39-43.
Croner S & Kjellman NI (1990) Development of atopic disease in
relation to family history and cord blood IgE levels. Pediatr Allergy
Immunol, 1: 14-20.
Cronin E (1991) Formaldehyde is a significant allergen in women with
hand eczema. Contact Dermatitis, 25: 276-283.
Cronin MTD & Basketter DA (1994) Multivariate QSAR analysis of a skin
sensitization database: SAR and QSAR. Environ Res, 2: 1-21.
Crowe SE & Perdue MH (1992) Gastrointestinal food hypersensitivity:
Basic mechanisms of pathophysiology. Gastroenterology, 103: 1075-1095.
Cullinan P, Lowson D, Nieuwenhuijsen MJ, Sandiford C, Tee RD, Venables
KM, McDonald JC, & Newman Taylor AJ (1994) Work related symptoms,
sensitisation, and estimated exposure in workers not previously
exposed to flour. Occup Environ Med, 51(9): 579-583.
Curtis GH, Patrick MK, Catto-Smith AG, & Gall DG (1990) Intestinal
anaphylaxis in the rat: Effect of chronic antigen exposure.
Gastroenterology, 98: 1558-1566.
Czernielewski JM & Demarchez M (1987) Further evidence for the
self-reproducing capacity of Langerhans cells in human skin. J Invest
Dermatol, 88: 17-20.
Dakin R (1982) Remarks on cutaneous affection, produced by certain
poisonous vegetables. Am J Med Sci, 4: 98-100.
Dally MB, Hunter JV, Hughes EG, Stewart M, & Newman Taylor AJ (1980)
Hypersensitivity to platinum salts: a population study. Am Rev Respir
Dis, 12(4): 120 (abstract).
Dannaeus A & Inganäs M (1981) A follow-up study of children with food
allergy: Clinical course in relation to serum IgE-antibody levels to
milk, egg and fish. Clin Allergy, 11: 533-539.
Darsow U, Vieluf D, & Ring J (1995) Atopy patch test with different
vehicles and allergen concentrations: an approach to standardisation.
J. Allergy Clin Immunol, 95: 677-684.
Davies RJ, Butcher BR, O'Neil CE, & Salvaggio JE (1977) The in vitro
effect of toluene di-isocyanate on lymphocyte cyclic adenosine
monophosphate production by isoproterenol, prostaglandin and
histamine. J Allergy Clin Immunol, 60: 223-229.
Dawson KP & Mitchell EA (1990) Asthma in New Zealand children. J
Asthma, 27: 291-297.
Daynes RA, Dudley DJ, & Araneo BA (1990) Regulation of murine
lymphokine production in vivo. II. Dehydroepiandrosterone is a
natural enhancer of interleukin synthesis by helper T cells. Eur J
Immunol, 20(49): 793-802.
Dearman RJ & Kimber I (1991) Differential stimulation of immune
function by respiratory and contact chemical allergens. Immunology,
72: 563-570.
Dearman RJ & Kimber I (1992) Divergent immune responses to respiratory
and contact chemical allergens: antibody elicited by phthalic
anhydride and oxazolone. Clin Exp Allergy, 22: 241-250.
Dearman RJ, Hegarty JM, & Kimber I (1991) Inhalation exposure of mice
to trimellitic anhydride induces both IgG and IgE anti-hapten
antibody. Int Arch Allergy Appl Immunol, 95: 70-76.
Dearman RJ, Basketter DA, & Kimber I (1995) Differential cytokine
production following chronic exposure of mice to chemical respiratory
and contact allergens. Immunology, 86: 545-550.
Dearman RJ, Basketter DA, & Kimber I (1996) Characterization of
chemical allergens as a function of divergent cytokine secretion
profiles induced in mice. Toxicol Appl Pharmacol, 138: 308-316.
De Bakker JM, Kammüller ME, Muller ESM, Lam AW, Seinen W, & Bloksma N
(1990) Kinetics and morphology of chemically induced popliteal lymph
node reactions compared with antigen-, mitogen-, and
graft-versus-host-reaction-induced responses. Virchows Arch B Cell
Pathol, 58: 279-287.
Dekker C, Dales R, Bartlett S, Brunekreef B, & Zwanenburg H (1991)
Childhood asthma and the indoor environment. Chest, 100: 922-926.
De Martino M, Novembre E, & Cozza G (1988) Sensitivity to tomato and
peanut allergens in children monosensitized to grass pollen. Allergy,
43: 206-213.
Deo YM, Graziano RF, Repp R, & van de Winkel JGJ (1997) Clinical
significance of IgG Fc receptors and Fc-gamma-directed
immunotherapies. Immunol Today, 18: 127-135.
Derynck R, Lindquist PB, Lee A, Wen D, Tamm T, Graycar JL, Rhee L,
Mason AJ, Miller DA, Coffey RJ, Moses HL, & Chen EY (1988) A new type
of transforming growth factor-beta. EMBO J, 7: 3737-3743.
Devalia JL, Rusznak C, Herdman MJ, Trigg CJ, Tarraf H, & Davies RJ
(1994) Effect of nitrogen dioxide and sulphur dioxide on airway
response of mild asthmatic patients to allergen inhalation. Lancet,
344: 1668-1671.
Devlin RB, McDonnell WF, Mann R, Becker S, House DE, Schreinemachers
D, & Koren HS (1991) Exposure of humans to ambient levels of ozone for
6.6 hours causes cellular and biochemical changes in the lung. Am J
Respir Cell Mol Biol, 4: 72-81.
De Weck AL, Stadler BM, Urwyler A, Wehner HU, & Bühlmann RP (1993)
Cellular allergen stimulation test (CAST) -- A new dimension in
allergy diagnostic. ACI News, 5: 9.
De Zotti R, Larese F, Bovenzi M, Negro C, & Molinari S (1994) Allergic
airway disease in Italian bakers and pastry makers. Occup Environ Med,
51(8): 548-552.
Diaz-Sanchez D, Tsien A, Fleming J, & Saxon A (1997) Combined diesel
exhaust particulate and ragweed allergen challenge markedly enhances
human in vivo nasal ragweed-specific IgE and skews cytokine
production to a T helper cell 2-type pattern. J Immunol, 158:
2406-2413.
Diaz-Sanchez D, Tsien A, Fleming J, & Saxon A (1999) Effect of topical
fluticasone propionate on the mucosal allergic response induced by
ragweed allergen and diesel exhaust particle challenge. Clin Immunol,
90(3): 313-322.
Diem JE, John RN, Hendrick DJ, Glindmeyer HW, & Dharmarajan V (1982)
Five year longitudinal study of workers employed in a new toluene
di-isocyanate manufacturing plant. Am Rev Respir Dis, 126(3): 420-428.
Diller WF (1987) Facts and fallacies involved in the epidemiology of
isocyanate asthma. Bull Eur Physiopathol Respir, 23(6): 551-553.
Djukanovic R, Wilson J, Britten K, Wilson SJ, Walls AF, Roche WR,
Howarth PH, & Holgate ST (1990) Quantitation of mast cells and
eosinophils in the bronchial mucosa of symptomatic atopic individuals
and healthy control subjects using immunohistochemistry. Am Rev Respir
Dis, 142: 863-871.
Dockery DW & Pope CA (1994) Acute respiratory effects of particulate
air pollution. Annu Rev Public Health, 15: 107-132.
Dockery DW, Speizer FE, Stram DO, Ware JH, Spengler JD, & Ferris BGJ
(1989) Effects of inhalable particles on respiratory health of
children. Am Rev Respir Dis, 139: 587-594.
Dockery DW, Pope CA III, Xu X, Spengler JD, Ware JH, Fay ME, Ferris BG
Jr, & Speizer FE (1993) An association between air pollution and
mortality in six US cities. N Engl J Med, 329: 1753-1759.
DOH (1998) Report of the Silicone Gel Breast Implants Independent
Review Group. London, Department of Health, pp 1-36.
Domen PL, Muckerheide A, & Michael JG III (1987) Abrogation of oral
tolerance. J Immunol, 139: 3195-3198.
Dooms-Goossens A & Morren M (1992) Results of routine patch testing
with corticosteroids in 2073 patients. Contact Dermatitis, 26:
182-191.
Dorsch W & Ring J (1981) Induction of late cutaneous reactions by skin
blister fluid from allergen-tested and normal skin. J Allergy Clin
Immunol, 67: 117-123.
Dotterud LK & Falk ES (1994) Metal allergy in north Norwegian
schoolchildren and its relationship with ear piercing and atopy.
Contact Dermatitis, 31(5): 308-313.
Dotterud LK & Falk ES (1995) Contact allergy in relation to hand
eczema and atopic diseases in north Norwegian schoolchildren.
Acta-Paediatr, 84(4): 402-406.
Dowse GK, Turner KJ, Stewart GA, Alpers MP, & Woolcock AJ (1985) The
association between dermatophagoides mites and the increasing
prevalence of asthma in village communities within the Papua New
Guinea highlands. J Allergy Clin Immunol, 75(1): 75-83.
Draize JH, Woodard G, & Calvery HD (1944) Methods for the study of
irritation and toxicity of substances applied topically to the skin
and mucous membranes. J Pharmacol Exp Ther, 83: 377-390.
Dreborg S (1989) Skin tests used for standardization of allergenic
preparation. In: Dreborg S ed. Skin tests used in type I allergy
testing: Position paper prepared by the Subcommittee on Skin Tests of
the European Academy of Allergology and Clinical Immunology. Allergy,
44(suppl 10): 13-51.
Dreborg S & Foucard T (1983) Allergy to apple, carrot and potato in
children with birch pollen allergy. Allergy, 38: 167-172.
Drever F ed. (1995) Occupational health -- Decennial supplement: The
Registrar General`s decennial supplement for England and Wales.
London, Office of Population Censuses and Surveys, Health and Safety
Executive, 374 pp (Series DS No. 10).
Druce HM (1993) Allergic and nonallergic rhinitis. In: Middleton E,
Reed Cellis EF, Adkinson NF, Junginger JW, & Busse WW ed. Allergy:
Principles and practice. St. Louis, Missouri, C.V. Mosby & Co., pp
1433-1453.
Druet P (1989) Contributions of immunological reactions to
nephrotoxicity. Toxicol Lett, 46: 55-64.
Dubost J-J, Souteyrand P, & Sauvezie B (1991) Drug-induced
vasculitides. Clin Rheumatol, 5: 119-138.
Dubroff LM & Reid RJ Jr (1980) Hydralazine-pyrimidine interactions may
explain hydralazine induced lupus erythematosus. Science, 208:
404-406.
Ducombs G & Schmidt R (1995) Plant and plant products. In: Rycroft
RJG, Menné T, Frosch PJ, & Benezra C ed. Textbook of contact
dermatitis, 2nd ed. Berlin, Heidelberg, New York, Springer Verlag, pp
589-634.
Ducombs G, Benezra C, Talaga P, Andersen KE, Burrows D, Camarasa JG,
Dooms-Goossens A, Lachapelle J-M, Frosch P, Menné T, Rycroft R, White
IR, & Wilkinson JD (1990) Patch testing with sesquiterpene lactone
mix: A marker for contact allergy to Compositae and certain other
plants -- A multicenter study of the E.E.C.D.R.G., 1989. Contact
Dermatitis, 22: 249-253.
Duhme H, Weiland SK, Keil U, Kraemer B, Schmid M, Stender M, &
Chambless L (1996) The association between self-reported symptoms of
asthma and allergic rhinitis and self-reported traffic density on
street of residence in adolescents. Epidemiology, 7: 578-582.
Duhme H, Weiland SK, Rudolph P, Wienke A, Kramer A, & Keil U (1998a)
Asthma and allergies among children in West and East Germany: a
comparison between Münster and Greifswald using the ISAAC phase I
protocol. Eur Respir J, 11: 840-847.
Duhme H, Weiland SK, & Keil U (1998b) Epidemiological analyses of the
relationship between environmental pollution and asthma. Toxicol Lett,
102-103: 307-316.
Dupuis G & Benezra C (1982) Contact dermatitis to simple chemicals: A
molecular approach. New York, Marcel Dekker.
Durham SR, Graneek BJ, Hawkins R, & Newman Taylor AJ (1987) The
temporal relationship between increases in airway responsiveness to
histamine and late asthmatic responses induced by occupational agents.
J Allergy Clin Immunol, 79: 398-406.
Durie FH, Fava RA, & Noelle RJ (1994) Collagen-induced arthritis as a
model of rheumatoid arthritis. Clin Immunol Immunopathol,
73(1): 11-18.
Dvoracek JE, Yunginger JW, Kern EB, Hyatt RE, & Gleich GJ (1984)
Induction of nasal late-phase reaction by insufflation of ragweed
pollen extract. J Allergy Clin Immunol, 73: 363-368.
EAACI (European Academy of Allergology and Clinical Immunology) (1992)
Subcommittee on Occupational Allergy -- Guidelines for the diagnosis
of occupational asthma. Clin Exp Allergy, 22(1): 103-108.
Edman B (1988) The usefulness of detailed information to patients with
contact allergy. Contact Dermatitis, 19: 43-48.
Edwards J, Walters S, & Griffiths RK (1994) Hospital admissions for
asthma in preschool children: Relationship to major roads in
Birmingham, United Kingdom. Arch Environ Health, 49: 223-227.
Edworthy SM, Martin L, Barr SG, Birdsell DC, Brant RF, & Fritzler MJ
(1998) A clinical study of the relationship between silicone breast
implants and connective tissue disease. J Rheumatol, 25(2): 254-260.
Ehlers A, Stangier U, & Gieler U (1995) Treatment of atopic
dermatitis: A comparison of psychological and dermatological
approaches to relapse prevention. J Consult Clin Psychol, 63(4):
624-635.
Eidson M, Philen RM, Sewell CM, Voorhees R, & Kilbourne EM (1990)
L-tryptophan and eosinophilia-myalgia syndrome in New Mexico. Lancet;
335(8690): 645-648.
Elsayed S & Apold J (1977) Allergenic structure of allergen M from
cod: II. Allergenicity of the limited tryptic hydrolysis peptides of
fragment TM2. Int Arch Allergy Appl Immunol, 54(2): 171-175.
Emberlin J (1994) The effects of patterns in climate and pollen
abundance on allergy. Allergy, 49: 15-20.
Enders F, Przybilla B, Ring J, Burg G, & Braun-Falco O (1988)
[Epicutaneous testing with standard batteries of allergens: Results
from 12,026 patients.] Hautarzt, 39: 779-786 (in German).
Enk AH & Katz SI (1995) Contact sensitivity as a model for T cell
activation in skin. J Invest Dermatol, 105: 80S-83S.
Epstein WL, Kligman AM, & Senecal IP (1963) Role of regional lymph
nodes in contact sensitization. Arch Dermatol, 88: 789-792.
Eriksson NE, Formgren H, & Svenonius E (1982) Food hypersensitivity in
patients with pollen allergy in Sweden. Allergy, 37(3): 437-443.
Ernst P, Demissie K, Joseph L, Locher U, & Becklake MR (1995)
Socioeconomic status and indicators of asthma in children. Am J Respir
Crit Care Med, 152(2): 570-575.
Estlander T & Jolanki R (1988) How to protect the hands. Dermatol
Clin, 6: 105-114.
Estlander T, Keskinen H, Jolanki R, & Kanerva L (1992) Occupational
dermatitis from exposure to polyurethane chemicals. Contact
Dermatitis, 27(3): 161-165.
Estlander T, Kanerva L, Tupasela O, Keskinen H, & Jolanki R (1993)
Immediate and delayed allergy to nickel with contact urticaria,
rhinitis, asthma and contact dermatitis. Clin Exp Allergy, 23:
306-310.
ETFAD (1993) Severity scoring of atopic dermatitis: the SCORAD index.
Consensus Report of the European Task Force on Atopic Dermatitis.
Dermatology, 186: 23-31.
Council of the European Communities (1976) Council directive of 27
July 1976 on the approximation of the laws of the Member States
relating to cosmetic products (76/768/EEC). Off J Eur Communities,
L-262: 169-183.
Council of the European Communities (1994) European Parliament and
Council directive 94/27/EC of 30 June 1994: Nickel. Off J Eur
Communities, L 18811-2.
Evans R (1993) Epidemiology and natural history of asthma, allergic
rhinitis and atopic dermatitis (eczema). In: Middleton E, Reed Cellis
EF, Adkinson NF, Junginger JW, & Busse WW ed. Allergy: Principles and
practice. St. Louis, Missouri, C.V. Mosby & Co., pp 1109-1136.
Evans RG, Webb K, Homan S, & Ayres SM (1988a) Cross-sectional and
longitudinal changes in pulmonary function associated with automobile
pollution among bridge and tunnel officers. Am J Ind Med, 14: 25-36.
Evans RG, Webb KB, Knutsen AP, Roodman ST, Roberts DW, Bagby JR,
Garrett WA, & Andrews JS (1988b) A medical follow-up of the health
effects of long-term exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin.
Arch Environ Health, 43: 273-278.
Eynon BE & Parker DC (1992) Small B cells as antigen-presenting cells
in the induction of tolerance to soluble protein antigens. J Exp Med,
175: 131-138.
Fabbri LM, Boschetto P, Zocca E, Melani G, Pivirotto F, & Plebani M
(1987) Bronchoalveolar neutrophilia during late asthmatic reactions
induced by toluene di-isocyanate. Am Rev Respir Dis, 136: 36-42.
Fairchild RL, Palmer E, & Moorhead JW (1993) Production of
DNP-specific/Class I MHC-restricted suppressor molecules is linked to
the expression of T cell receptor alpha- and beta-chain genes. J
Immunol, 150(1): 67-77.
Farine J-C (1997) Animal models in autoimmune disease in
immunotoxicity assessment. Toxicology, 119: 29-35.
Fauci A, Schnittman SM, & Poli G (1991) Immunopathologenic mechanisms
in human immunodeficiency virus (HIV) infection. Ann Intern Med, 114:
678-693.
Fawcett IW, Newman Taylor AJ, & Pepys J (1977) Asthma due to inhaled
chemical agents - epoxy resin systems containing phthalic anhydride,
trimellitic anhydride and triethylene tetramine. Clin Allergy, 7:
1-14.
Fireman E, Ben Efraim S, Grief J, Alguetti A, Ayalon D, & Topilsky M
(1988) Correlation between prostaglandin E2 production and suppressor
activity of alveolar macrophages from patients with interstitial lung
diseases. Immunol Lett, 18(2): 159-165.
Flatt A, Pearce N, Thomson CD, Sears MR, Robinson MF, & Beasley R
(1990) Reduced selenium in asthmatic subjects in New Zealand. Thorax,
45: 95-99.
Fleming DM & Crombie DL (1987) Prevalence of asthma and hay fever in
England and Wales. Br Med J Clin Res Ed, 294(6567): 279-283.
Flens MJ, Zaman GJR, Van der Valk P, Izquierdo MA, Schroeijers AB,
Scheffer GL, van der Groep P, De Haas M, Meijer CJLM, & Scheper RJ
(1996) Tissue distribution of the multidrug resistance protein. Am J
Pathol, 148: 1237-1247.
Flindt-Hansen H & Isager H (1987) Scleroderma after occupational
exposure to trichloroethylene and trichloroethane. Acta Dermatol
Venereol, 67: 263-264.
Flynn GL (1990) Physicochemical determinants of skin absorption. In:
Gerrity TR & Henry CJ ed. Principles of route-to-route extrapolation
for risk assessment. Amsterdam, Oxford, New York, Elsevier Science
Publishers, pp 93-127.
Flyvholm M-A (1997) Formaldehyde exposure at the workplace and in the
environment. Allergologie, 20(5): 225-231.
Flyvholm M-A & Menné T (1992) Allergic contact dermatitis from
formaldehyde: A case study focusing on sources of formaldehyde
exposure. Contact Dermatitis, 27: 27-37.
Flyvholm M-A, Menné T, & Maibach HI (1996) Skin allergy: Exposures and
dose-response relationships. In: Vos JG, Younes M, & Smith E ed.
Allergic hypersensitization induced by chemicals: Recommendations for
prevention. Boca Raton, Florida, CRC Press, pp 261-285.
Flyvholm M-A, Hall BM, Agner T, Tiedemann E, Greenhill P, Vanderveken
W, Freeberg FE, & Menné T (1997) Threshold for occluded formaldehyde
patch test in formaldehyde-sensitive patients. Contact Dermatitis, 36:
26-33.
Foerster J (1993) Autoimmune hemolytic anemias. In: Lee GR, Bithell
TC, Foerster J, Athens JW, & Lukens JN ed. Wintrobe's clinical
haematology, 9th ed. London, Lea and Febiger, pp 1170-1196.
Folinsbee LJ (1992) Human health effects of air pollution. Environ.
Health Perspect, 100: 45-56.
Forastiere F, Corbo GM, Michelozzi P, Pistelli R, Agabiti N, Brancato
G, Ciappi G, & Perucci CA (1992) Effects of environment and passive
smoking on the respiratory health of children. Int J Epidemiol, 21(1):
66-73.
Forster H, Topping M, & Newman Taylor AJ (1988) Specific IgG and IgG4
antibody to tetrachlorophthalic anhydride. Allergy Proc, 9: 296.
Forsyth JS, Ogston SA, Clark A, Florey CD, & Howie PW (1993) Relation
between early introduction of solid food to infants and their weight
and illnesses during the first two years of life. Br Med J, 306(6892):
1572-1576.
Foucard T (1991) Allergy and allergy-like symptoms in 1,050 medical
students. Allergy, 46: 20-26.
Fowler JF, Skinner SM, & Belsito DV (1992) Allergic contact dermatitis
from formaldehyde resins in permanent press clothing: An
underdiagnosed cause of generalized dermatitis. J Am Acad Dermatol,
27: 962-968.
Freier S, Eran M, & Goldstein R (1985) The effect of immediate type
gastrointestinal allergic reactions on brush border enzymes and gut
morphology in the rat. Pediatr Res, 19: 456-459.
Freiman DE & Hardy HL (1970) Beryllium disease: The relation of
pulmonary pathology to clinical course and prognosis based on a study
of 130 cases from the US Beryllium Case Registry. Hum Pathol,
1: 25-44.
Friedmann PS (1990) The immunology of allergic contact dermatitis: The
DNCB story. Adv Dermatol, 5: 175-196.
Friedmann PS, Moss C, Shuster S, & Simpson JM (1983) Quantitative
relationships between sensitizing dose of DNCB and reactivity in
normal subjects. Clin Exp Immunol, 53: 706-710.
Friedman SM, Posnett DN, Tumang JR, Cole BC, & Crow K (1991) A
potential role for microbial superantigens in the pathogenesis of
systemic autoimmune disease. Arthritis Rheum, 34: 468-480.
Fudenberg HH (1966) Immunological deficiency, autoimmune disease, and
lymphoma: observations, implications, and speculations. Arthritis
Rheum, 9: 464-472.
Fujimaki H, Nohara O, Ichinose T, Watanabe N, & Saito S (1994) IL-4
production in mediastinal lymph node cells in mice intratracheally
instilled with diesel exhaust particulates and antigen. Toxicology,
92: 261-268.
Fuller KA, Pearl D, & Whitacre CC (1990) Oral tolerance in
experimental autoimmune encephalomyelitis: Serum and salivary antibody
responses. J Neuroimmunol, 28: 14-26.
Fye KH & Sack KE (1991) Rheumatic diseases. In: Stites DP & Terr AI
ed. Basic and clinical immunology, 7th ed. Norwalk, San Mateo,
Appleton & Lange, pp 438-463.
Gabriel SE, O`Fallon WM, Kurland LT, Berad CM, Woods JE, & Melton LJ
(1994) Risk of connective-tissue diseases and other disorders after
breast implantation. N Engl J Med, 330(24): 1697-1702.
Gad SC, Dunn BJ, Dobbs DW, Reilly C, & Walsh RD (1986) Development and
validation of an alternative dermal sensitization test : The mouse ear
swelling test (MEST). Toxicol Appl Pharmacol, 84: 93-114.
Gamble J, Jones W, & Minshall S (1987) Epidemiological-environmental
study of diesel bus garage workers: chronic effects of diesel exhaust
on the respiratory system. Environ Res, 44: 6-17.
Gammelgaard B, Fullerton F, Avnstorp C, & Menné T (1992) Permeation of
chromium salts through human skin in vitro. Contact Dermatitis, 27:
302-310.
Gardner MLG (1988) Gastrointestinal absorption of intact proteins.
Annu Rev Nutr, 8: 329-350.
Garssen J, Nijkamp FP, Van der Vliet H, & Van Loveren H (1991) T-cell
mediated induction of airway hyperreactivity in mice. Am Rev Resp Dis,
144: 931-938.
Garssen J, Nijkamp FP, Van de Vliet H, & Van Loveren H (1994) A role
for cellular immunity in the induction of airway hyperresposiveness
induced by small molecular weight compounds. Toxicol Lett,
72: 151-154.
Gauggel DL, Sarlo K, & Asquith TN (1993) A proposed screen for
evaluating low-molecular-weight chemicals as potential respiratory
allergens. J Appl Toxicol, 13(5): 307-313.
Gautam SC, Chikkala NF, & Battisto JR (1985) Orally induced tolerance
generates an efferently acting suppressor T cell and an acceptor T
cell that together downregulate contact sensitivity. J Immunol, 135:
2975-2983.
Geier J & Schnuch A (1996) No cross-sensitization between MCI/MI,
benzisothiazolinone and octylisothiazolinone. Contact Dermatitis,
34(2): 148-149.
Gell PGH & Coombs RRA (1963) Clinical aspects of immunology. Oxford,
London, Boston, Blackwell Scientific Publications.
Gell PGH, Coombs RRA, & Lachman R (1975) Clinical aspects of
immunology, 3rd ed. Oxford, London, Boston, Blackwell Scientific
Publications.
Gergely J & Sarmay G (1996) Fc-gamma-RII-mediated regulation of human
B cells. Scand J Immunol, 44: 1-10.
Gergen PJ, Mullally DI, & Evans R (1988) National survey of prevalence
of asthma among children in the United States, 1976 to 1980.
Pediatrics, 81: 1-7.
Gerling I, Serreze DV, Christianson S, & Leiter E (1992) Intrathymic
islet cell transplantation reduces beta-cell autoimmunity and prevents
diabetes in NOD/Lt mice. Diabetes, 41: 1672-1676.
Gilliam JN & Sontheimer RD (1982) Skin manifestations of SLE. Clin
Rheum Dis, 8: 207-218.
Glazier A, Tutschka PJ, Farmer ER, & Santos GW (1983)
Graft-versus-host disease in cyclosporine A-treated rats after
syngeneic and autologous bone marrow reconstitution. J Exp Med, 158:
1-8.
Gleich GJ (1990) The eosinophil and bronchial asthma: Current
understanding. J Allergy Clin Immunol, 85(2): 422-436.
Gleichmann H (1981) Studies on the mechanism of drug sensitization:
T-cell-dependent popliteal lymph node reaction to diphenylhydantoin.
Clin Immunol Immunopathol, 18: 203-211.
Gleichmann HIK, Pals ST, & Radaszkiewicz T (1983) T-cell-dependent
B-cell proliferation and activation induced by administration of the
drug diphenylhydantoin to mice. Hematol Oncol, 1: 165-176.
Gleichmann E, Pals ST, Rolink AG, Radaszkiewicz T, & Gleichmann H
(1984) Graft-versus-host reactions: clues to the etiopathology of a
spectrum of immunological diseases. Immunol Today, 5: 324-332.
Gleichmann E, Vohr H-W, Stringer C, Nuyens J, & Gleichmann H (1989)
Testing the sensitization of T cells to chemicals: From murine
graft-versus-host (GVH) reactions to chemical-induced GVH-like
immunological diseases. In: Kammüller ME, Bloksma N, & Seinen W ed.
Autoimmunity and toxicology. Amsterdam, Oxford, New York, Elsevier
Science Publishers, pp 363-390.
Goettsch W, Garssen J, De Gruijl FR, & Van Loveren H (1993) UV-B and
the immune system: a review with special emphasis on T cell-mediated
immunity. Thymus, 21: 93-114.
Goh C (1995) Non eczematous contact reactions. In: Rycroft RJG, Menné
T, Frosch PJ, & Benezra C ed. Textbook of contact dermatitis, 2nd ed.
Berlin, Heidelberg, New York, Springer Verlag, pp 221-233.
Goldman M, Druet P, & Gleichmann E (1991) TH2 cells in systemic
autoimmunity: insights from allogeneic diseases and chemically-induced
autoimmunity. Immunol Today, 12: 223-227.
Gorski P & Tarkowski M (1992) Non specific environmental factors and
asthma development. Pol J Occup Med Environ Health, 5: 227-236.
Goust JM (1993) Immediate hypersensitivity. Immunol Ser, 58: 343-359.
Graham C, Gealy R, Macina OT, Karol MH, & Rosenkranz HS (1996) QSAR
for allergic contact dermatitis. Quant Struct-Activ Relat, 15: 1-7.
Graham C, Rosenkranz HS, & Karol MH (1997) Structure-activity model of
chemicals that cause human respiratory sensitization. Regul Toxicol
Pharmacol, 26: 296-306.
Granato DA & Piguet PF (1986) A mouse monoclonal IgE antibody anti
bovine milk beta-lactoglobulin allows studies of allergy in the
gastrointestinal tract. Clin Exp Immunol, 63(3): 703-710.
Graycar JL, Miller DA, Arrick BA, Lyons RM, Moses HL, & Derynck R
(1989) Human transforming growth factor-beta-3: recombinant
expression, purification, and biologic activities in comparison with
transforming growth factors beta 1 and beta 2. Mol Endocrinol,
3: 1977-1986.
Greaves IA, Wegman DH, Smith TJ, & Spiegelman DL (1984) Respiratory
effects of two types of solder flux used in the electronics industry.
J Occup Med, 26(2): 81-85.
Greenland S (1993) Basic problems in interaction assessment. Environ
Health Perspect, 101(suppl): 59-66.
Gryboski JD (1967) Gastrointestinal milk allergy in infants.
Pediatrics, 40: 354-362.
Guillet GG & Guillet MH (1992) Natural history of sensitizations in
atopic dermatitis. Arch Dermatol, 128: 187-192.
Gulbenkian AR, Egan RW, Fernandez X, Jones H, Kreutner W, Kung T,
Payvandi F, Sullivan L, Zurcher JA, & Watnik AS (1992) Interleukin-5
modulates eosinophil accumulation in allergic guinea pig lung. Am Rev
Respir Dis, 146: 263-266.
Gut J, Christen U, Frey N, Koch V, & Stoffler D (1995) Molecular
mimicry in halothane hepatitis: biochemical and structural
characterization of lipoylated autoantigens. Toxicology, 97: 199-224.
Haahtela T, Lindholm H, Bjorksten F, Koskenvuo K, & Laitinen LA (1990)
Prevalence of asthma in Finnish young men. Br Med J, 301: 266-268.
Hackney JD, Linn WS, & Avol EL (1989) Acid fog: Effects on respiratory
function and symptoms in healthy and asthmatic volunteers. Environ
Health Perspect, 79: 159-162.
Haddad ZH, Kalra V, & Verma S (1979) IgE antibodies to peptic and
peptic-tryptic digests of beta-lactoglobulin: significance in food
hypersensitivity. Ann Allergy, 42: 368-371.
Halken S, Host A, Hansen LG, & Osterballe O (1992) Effect of an
allergy prevention programme on incidence of atopic symptoms in
infancy. Allergy, 47: 545-553.
Halken S, Host A, Nilsson L, & Taudorf E (1995) Passive smoking as a
risk factor for development of obstructive respiratory disease and
allergic sensitization. Allergy, 50(2): 97-105.
Hallas TE, Yi X, & Schou C (1993) Does guanine concentration in
house-dust samples reflect house-dust mite exposure? Allergy, 48(5):
303-305.
Hananan D (1990) Transgenic mouse models of self-tolerance and
autoreactivity by the immune system. Annu Rev Cell Biol, 6: 493-537.
Hang L, Aguado MT, Dixon FJ, & Theofilopoulos AN (1985) Induction of
severe autoimmune disease in normal mice by simultaneous action of
multiple immunostimulators. J Exp Med, 161: 423-428.
Hanifin JM (1983) Atopic dermatitis and other endogenous eczemas.
Semin Dermatol, 2: 1-44.
Hanifin JM (1987) Epidemiology of atopic dermatitis. Monogr Allergy,
21: 116-131.
Hanifin JM (1993) Atopic dermatitis. In: Middleton E, Reed Cellis EF,
Adkinson NF, Junginger JW, & Busse WW ed. Allergy: Principles and
practice. St. Louis, Missouri, C.V. Mosby & Co., pp 1581-1604.
Hanifin JM & Chan SC (1995) Monocyte phosphodiesterase abnormalities
and dysregulation of lymphocyte function in atopic dermatitis. J
Invest Dermatol, 105(suppl 1): 84S-88S.
Hanifin JM & Lobitz WC Jr (1977) Newer concepts of atopic dermatitis.
Arch Dermatol, 113(5): 663-670.
Hanifin JM & Rajka G (1980) Diagnostic features of atopic dermatitis.
Acta Dermatol Venereol, 92(suppl): 44-47.
Hanson DG, Roy MJ, Green GM, & Miller SD (1993) Inhibition of
orally-induced immune tolerance in mice by prefeeding an endopeptidase
inhibitor. Region Immunol, 5: 76-84.
Hariya T, Ikezawa Z, Aihara M, Kitamura K, Osawa J, & Nakajima H
(1994) Allergenicity and tolerogenicity of amlexanox in the guinea
pig. Contact Dermatitis, 31: 31-36.
Hasselmark L, Malmgren R, Zetterstrom O, & Unge G (1993) Selenium
supplementation in intrinsic asthma. Allergy, 48(1): 30-36.
Hatch M & Thomas D (1993) Measurement issues in environmental
epidemiology. Environ Health Perspect, 101(Suppl 4): 49-57.
Hattevig G, Kjellman B, Sigurs N, Bjorksten B, & Kjellman NI (1989)
Effect of maternal avoidance of eggs, cow's milk and fish during
lactation upon allergic manifestations in infants. Clin Exp Allergy,
19(1): 27-32.
Heine J, Ignotz RA, Hemler ME, Crouse C, & Massague J (1989)
Regulation of cell adhesion receptors by transforming growth
factor-beta. Concomitant regulation of integrins that share a common
beta-1 subunit. J Biol Chem, 264: 380-388.
Heiner DC, Sears JW, & Kniker WT (1962) Multiple precipitins to cow's
milk chronic respiratory disease: A syndrome including poor growth,
gastrointestinal symptoms, evidence of allergy, iron deficiency anemia
and pulmonary hemosiderosis. Am J Dis Child, 103: 634-651.
Henderson CR & Riley EC (1945) Certain statistical considerations in
patch testing. J Invest Dermatol, 6: 227-232.
Hennekens LIM & Buring JE (1987) Epidemiology in medicine. Boston,
Toronto, Little, Brown & Co., 400 pp.
Hennekens LIM, Cook NR, Hebert PR, Karlson EW, LaMotte F, Manson JE, &
Buring JE (1996) Self-reported breast implants and connective tissue
diseases in female health professionals. J Am Med Assoc, 275: 616-621.
Henschler D, Assman W, & Meyer K (1962) [The toxicology of toluene
diisocyanate.] Arch Toxicol, 19: 364-387 (in German).
Heppel LMJ & Kilshaw PJ (1982) Immune responses of guinea pigs to
dietary protein: I. Induction of tolerance by feeding with ovalbumin.
Int Arch Allergy Appl Immunol, 68: 54-59.
Herbert CA, King CM, Ring PC, Holgate ST, Stewart GA, Thompson PJ, &
Robinson C (1995) Augmentation of permeability in the bronchial
epithelium by the house dust mite allergen Der p 1. Am J Respir Cell
Mol Biol, 12: 369-378.
Hertzman PA, Blevins WL, Mayer J, Greenfield B, Ting M, & Gleich GJ
(1990) Association of the eosinophilia-myalgia syndrome with the
ingestion of tryptophan. N Eng J Med, 322(130): 869-873.
Hess AD & Fischer AC (1989) Immune mechanisms in cyclosporine-induced
syngeneic graft-versus- host disease. Transplantation, 48: 895-900.
Heufler C, Koch F, & Schuler G (1988) Granulocyte/macrophage
colony-stimulating factor and interleukin-1 mediate the maturation of
murine epidermal Langerhans cells into potent immunostimulatory
dendritic cells. J Exp Med, 167: 700-705.
Hewitt CRA, Brown AP, Hart BJ, & Pritchard DI (1995) A major house
dust mite allergen disrupts the immunoglobulin E network by
selectively cleaving CD23: innate protection by antiproteases. J Exp
Med, 182: 1537-1544.
Hide DW, Matthews S, Matthews L, Stevens M, Ridout S, Twiselton R,
Gant C, & Arshad SH (1994) Effect of allergen avoidance in infancy on
allergic manifestations at age two years. J Allergy Clin Immunol,
93(5): 842-846.
Hill DJ & Hosking CS (1997) Emerging disease profiles in infants and
young children with food allergy. Pediatr Allergy Immunol, 8(suppl
10): 21-26.
Hill LW & Sulzberger MB (1935) Evolution of atopic dermatitis. Arch
Dermatol Syph, 32: 451.
Hjorth N & Menné T (1990) Prevention of allergic contact
sensitization: a historical perspective. In: Menné T & Maibach HI ed.
Exogenous dermatoses. Boca Raton, Florida, CRC Press, chapter 30.
Hjorth N & Roed-Petersen J (1976) Occupational protein contact
dermatitis in food handlers. Contact Dermatitis, 2(1): 28-42.
Hochberg MC, Perlmutter DL, Medsger TA Jr, Nguyen K, Steen V, Weisman
MH, White B, & Wigley FM (1996) Lack of association between
augmentation mammoplasty and systemic sclerosis (scleroderma).
Arthritis Rheum, 39: 1125-1131.
Hodge L, Salome CM, Peat JK, Haby MM, Xuan W, & Woolcock AJ (1996)
Consumption of oily fish and childhood asthma risk. Med J Aust,
164(3): 137-140.
Holgate ST, Beasley R, & Twentyman OP (1987) The pathogenesis and
significance of bronchial hyperresponsiveness in airways disease. Clin
Sci, 73(6): 561-572.
Hollady SD & Luster MI (1996) Alterations in fetal thymic and liver
hematopoietic cells as indicators of exposure to developmental
immunotoxicants. Environ Health Perspect, 104(suppl 4): 809-813.
Holt PG (1994) A potential vaccine strategy for asthma and allied
atopic diseases during early childhood. Lancet, 344: 456-458.
Holt PG (1996) Infections and the development of allergy. Toxicol
Lett, 86: 205-210.
Holt PG & Sedgwick JD (1987) Suppression of IgE responses following
antigen inhalation: A natural homeostatic mechanism which limits
sensitization to aeroallergens. Immunol Today, 60(1): 97-102.
Holt PG, Schon-Hegrad MA, & McMenamin PG (1990) Dendritic cells in the
respiratory tract. Int. Rev Immunol, 6(2-3): 139-149.
Hopkins JM (1997) Genetics of atopy. In: Kay AB ed. Allergy and
allergic diseases. Oxford, London, Boston, Blackwell Scientific
Publications, pp 1187-1195.
Host A & Halken S (1990) A prospective study of cow milk allergy in
Danish infants during the first 3 years of life. Allergy, 45: 587.
Host A, Husby S, & Osterballe O (1988) A prospective study of cow's
milk allergy in exclusively breast-fed infants: Incidence,
pathogenetic role of inadvertent exposure to cow's milk formula, and
characterisation of bovine milk protein in human milk. Acta Paediatr
Scand, 77(5): 663-670.
Hostynek JJ, Magee PS, & Maibach HI (1996) QSAR predictive of contact
allergy: scope and limitations. Curr Probl Dermatol, 14: 59-106.
Howe W, Venables KM, Topping MD, Dally MB, Hawkins R, Law SJ, & Newman
Taylor AJ (1983) Tetrachlorophthalic anhydride asthma: evidence for
specific IgE antibody. J Allergy Clin Immunol, 71: 5-11.
HSE (1996) Good health is good business: Employers guide
-- Work-related dermatitis. London, United Kingdom, Health and Safety
Executive, 3 pp.
Hsieh KH & Shen JJ (1988) Prevalence of childhood asthma in Taipei,
Taiwan, and other Asian Pacific countries. J Asthma, 25(2): 73-82.
Hunter D, Milton R, & Perry KMA (1945) Asthma caused by the complex
salts of platinum. Br J Ind Med, 2: 92-98.
Hurtenbach U, Gleichmann H, Nagata N, & Gleichmann E (1987) Immunity
to D-penicillamine: genetic, cellular, and chemical requirements for
induction of popliteal lymph node enlargement in the mouse. J Immunol,
139: 411-416.
IARC (1993) Beryllium, cadmium, mercury, and exposures in the glass
industry. Lyon, International Agency for Research on Cancer, pp 88-89
(IARC Monographs on the Evaluation of Carcinogenic Risks to Humans,
Volume 58).
Ignotz RA & Massague J (1986) Transforming growth factor-beta
stimulates the expression of fibronectin and collagen and their
incorporation into the extracellular matrix. J Biol Chem, 261(9):
4337-4345.
Imbus HR (1985) Clinical evaluation of patients with complaints
related to formaldehyde exposure. J Allergy Clin Immunol, 76(6):
831-840.
International Rhinitis Management Working Group (1994) International
consensus report on the diagnosis and management of rhinitis. Allergy,
49(suppl 19): 1-34.
ISAAC (International Study of Asthma and Allergies in Childhood)
(1998) ISAAC Steering Committee -- Worldwide variation in the
prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and
atopic eczema. Lancet, 351: 1225-1232.
IPCS (1996) Environmental health criteria 180: Principles and methods
for assessing direct immunotoxicity associated with exposure to
chemicals. Geneva, World Health Organization, International Programme
on Chemical Safety, 390 pp.
Irani AA, Schechter NM, Craig SS, DeBlois G, & Schwartz LB (1986) Two
types of human mast cells that have distinct neutral protease
compositions. Proc Natl Acad Sci (USA), 83: 4464-4468.
Irvine C, Pugh CE, Hansen EJ, & Rycroft RJG (1994) Cement dermatitis
in underground workers during construction of the Channel Tunnel.
Occup Med, 44: 17-23.
Ishizaka K & Ishizaka T (1966) Physicochemical properties of reginic
antibody: 1. Association of reaginic activity with an immunoglobulin
other than gammaA- or gammaG-globulin. J Allergy, 37(3): 169-185.
Ishizaki T, Koizumi K, Ikemori R, Ishiyama Y, & Kushibiki E (1987)
Studies of prevalence of Japanese cedar pollinosis among the residents
in a densely cultivated area. Ann Allergy, 58: 265-270.
Iverson GM (1970) Ability of CBA mice to produce anti-idiotypic sera
to 5563 myeloma protein. Nature (Lond), 227: 273-274.
Iwami T, Nagai H, Tsuruoka N, & Koda A (1993) Effect of murine
recombinant interleukin-5 on bronchial reactivity in guinea-pigs. Clin
Exp Allergy, 23(1): 32-38.
Izquierdo MA, Scheffer GL, Flens MJ, Giaccone G, Broxterman HJ, Van
der Valk P, Meijer CJLM, & Scheper RJ (1996) Broad distribution of the
multidrug resistance -- related Vault Lung Resistance Protein in
normal human tissues and tumors. Am J Pathol, 148: 877-887.
Jacobs MC, White IR, Rycroft RJG, & Taub N (1995) Patch testing with
preservatives at St. John's from 1982 to 1993. Contact Dermatitis, 33:
247-255.
Jakob T, Hermann K, & Ring J (1991) Eosinophil cationic protein in
atopic eczema. Arch Dermatol Res, 283: 5-6.
Jakobsson I & Lindberg T (1979) A prospective study of cow's milk
protein intolerance in Swedish infants. Acta Pædiatr Scand, 68:
853-859.
Janeway CA, Travers P, Hunt S, & Walport M (1997) Immunobiology: The
immune system in health and disease. Edinburgh, Churchill Livingstone,
400 pp.
Jarrett EEE (1984) Immunoregulation of IgE responses: the role of the
gut in perspective. Ann Allergy, 53: 550-556.
Jarrett EEE & Hall E (1984) The development of IgE suppressive
immunocompetence in young animals: influence of exposure to antigen in
the presence or absence of maternal immunity. Immunology, 53: 365-373.
Jarrett EEE, Haig DM, McDougall W, & McNulty E (1976) Rat IgE
production: II. Primary and booster reaginic antibody responses
following intradermal or oral immunization. Immunology, 30: 671-677.
Jarvis J, Agius R, & Sawyer L (1996) Odds on for asthma. Chem Br, 32:
51-53.
Jenkins MK, Schwartz RH, & Pardoll DM (1988) Effects of cyclosporine A
on T-cell development and clonal deletion. Science, 24: 1655-1658.
Jiang X, Khursigara G, & Rubin RL (1994) Transformation of
lupus-inducing drugs to cytotoxic products by activated neutrophils.
Science, 266: 810-813.
Johansen J & Menné T (1995) The fragrance mix and its constituents: a
14-year material. Contact Dermatitis, 32: 18-23.
Johansen J, Rastogi SC, & Menné T (1996a) Exposure to selected
fragrance materials. A case study of fragrance-mix-positive eczema
patients. Contact Dermatitis, 34: 106-110.
Johansen JD, Andersen KE, & Menné T (1996b) Quantitative aspects of
isoeugenol contact allergy assessed by use and patch tests. Contact
Dermatitis, 34: 414-418.
Johansen JD, Rastogi SC, Andersen KE, & Menné T (1997) Content and
reactivity to product perfumes in fragrance mix positive and negative
eczema patients: A study of perfumes used in toiletries and skin-care
products. Contact Dermatitis, 36(6): 291-296.
Johansson SGO & Juhlin L (1970) Immunoglobulin-E in "healed" atopic
dermatitis and after treatment with corticosteroids and azathioprine.
Br J Dermatol, 82: 10-13.
Johansson SGO, Dannaeus A, & Lilja G (1984) The relevance of anti-food
antibodies for the diagnosis of food allergy. Ann Allergy, 53: 665.
Johnston SL, Pattemore PK, Sanderson G, Smith S, Lampe F, Josephs L,
Symington P, O`Toole S, Myint SH, & Tyrrell DAJ (1995) Community study
of role of viral infections in exacerbations of asthma in 9-11 year
old children. Br Med J, 310: 1225-1229.
Jones RJ, Vogelsang GB, Hess AD, Farmer ER, Mann RB, Geller RB,
Piantadosi S, & Santos GW (1989) Induction of graft-versus-host
disease after autologous bone marrow transplantation. Lancet, 1:
754-757.
Jones LA, Chin LT, Longo DL, & Kruisbeek AM (1990) Peripheral clonal
elimination of functional T cells. Science, 250: 1726-1729.
Jones RN, Rando RJ, Glindmeyer HW, Foster TA, Hughes JM, O'Neil CE, &
Weill H (1992) Abnormal lung function in polyurethane foam producers:
Weak relationship to toluene diisocyanate exposures. Am Rev Respir
Dis, 146(4): 871-877.
Jones DB, Coulson AFW, & Duff GW (1993) Sequence homologies between
hsp60 and autoantigens. Immunol Today, 14: 115-118.
Jones Williams W (1988) Beryllium disease. Postgrad Med J, 64:
511-516.
Jones Williams W & Wallach ER (1989) Laser probe mass spectrometry
(LAMMS) analysis of beryllium, sarcoidosis and other granulomatous
diseases. Sarcoidosis, 6: 111-117.
Jordan JM & Whitlock FA (1972) Emotions and the skin: the conditioning
of scratch responses in cases of atopic dermatitis. Br J Dermatol,
86(6): 574-585.
Jordan JM & Whitlock FA (1974) Atopic dermatitis, anxiety and
conditioned scratch responses. J Psychosom Res, 18(5): 297-299.
Jungers P, Dougados M, Pelissier C, Kutten F, Tron F, Lesavre P, &
Bach JF (1982) Influence of oral contraceptive therapy on the activity
of systemic lupus erythematosus. Arthritis Rheum, 25: 618-623.
Juniper CP, How MJ, Goodwin BFJ, & Kinshott AK (1977) Bacillus
subtilis enzymes: a seven year clinical, epidemiological and
immunological study of an industrial allergen. J Soc Occup Med, 27:
3-12.
Kagnoff MF (1982) Oral tolerance. Ann NY Acad Sci, 392: 248-265.
Kagnoff MF (1992) Celiac disease: A gastrointestinal disease with
environmental, genetic, and immunologic components. Gastr Clin North
Am, 21(2): 405-425.
Kajosaari M & Saarinen UM (1983) Prophylaxis of atopic disease by six
month's total solid food elimination. Acta Paediatr Scand, 72:
411-414.
Kammüller ME (1996) Drug-induced autoimmune disorders. Drug Inf J, 30:
293-299.
Kammüller ME & Seinen W (1988) Structural requirements for hydantoins
and 2-thiohydantoins to induce lymphoproliferative popliteal lymph
node reactions in the mouse. Int J Immunopharmacol, 10: 997-1010.
Kammüller ME, Penninks AH, & Seinen W (1984) Spanish toxic oil
syndrome is a chemically induced GVHD-like epidemic. Lancet, 1:
1174-1175.
Kammüller ME, Bloksma N, & Seinen W ed. (1989a) Autoimmunity and
toxicology: Immune disregulation induced by drugs and chemicals.
Amsterdam, Oxford, New York, Elsevier Science Publishers, pp 3-34.
Kammüller ME, Thomas C, De Bakker JM, Bloksma N, & Seinen W (1989b)
The popliteal lymph node assay in mice to screen for the immune
disregulating potential of chemicals -- a preliminary study. Int J
Immunopharmacol, 11: 293-300.
Kaposi M (1895) Pathology and treatment of diseases of the skin, 4th
ed. New York, William Wood.
Kapp A (1995) [Atopic dermatitis: The skin manifestations.] Allergo J,
4: 229-238 (in German).
Kapsenberg ML, Wierenga, EA, Bos JD, & Jansen HM (1991) Functional
subsets of allergen-reactive human CD4+ T cells. Immunol Today, 12:
392-395.
Karol MH (1983) Concentration-dependent immunologic response to
toluene diisocyanate (TDI) following inhalation exposure. Toxicol Appl
Pharmacol, 68: 229-241.
Karol MH (1986) Respiratory effects of inhaled isocyanates. CRC Crit
Rev Toxicol, 16: 349-380.
Karol MH (1991) Allergic reactions to indoor air pollutants. Environ
Health Perspect, 95: 45-51.
Karol MH (1992) Occupational asthma and allergic reactions to inhaled
compounds. In: Miller K, Turk J, & Nicklin S ed. Principles and
practice of immunotoxicology. Oxford, London, Boston, Blackwell
Scientific Publications, pp 228-241.
Karol MH (1993) Concentration-dependent immunologic response to
toluene diisocyanate (TDI) following inhalation exposure. Toxicol Appl
Pharmacol, 68: 229.
Karol MH (1994a) Occupational asthma pig predictive tests for
respiratory allergy. In: Dean JH, Luster MI, Munson AE, & Kimber I ed.
Immunotoxicology and immunopharmacology, 2nd ed. New York, Raven
Press, pp 703-720.
Karol MH (1994b) Animal models of occupational asthma. Eur Respir J,
7(3): 555-568.
Karol MH & Thorne PS (1988) Pulmonary hypersensitivity and
hyperreactivity: implications for assessing allergic responses. In:
Gardner DE, Crapo JD, & Massaro EJ ed. Toxicology of the lung. New
York, Raven Press, p 427.
Karol MH, Graham C, Gealy R, Macina OT, Sussman N, & Rosenkranz HS
(1996) Structure- activity relationships and computer-assisted
analysis of respiratory sensitization potential. Toxicol Lett, 86:
187-191.
Karpatkin S (1988) Immunological platelet disorders. In: Immunological
diseases, 4th ed. Boston, Little Brown, vol 2, pp 1631-1662.
Karpus WJ & Swanborg RH (1991) CD4+ suppressor cells inhibit the
function of effector cells of experimental autoimmune
encephalomyelitis through a mechanism involving transforming growth
factor-beta. J Immunol, 146: 1163-1168.
Katsutani N & Shionoya H (1992) Popliteal lymph node enlargement
induced by procainamide. Int J Immunopharmacol, 14: 681-686.
Kaufman LD, Gruber BL, & Gregersen PK (1991) Clinical follow-up and
immunogenetic studies of 32 patients with eosinophilia-myalgia
syndrome. Lancet, 337: 1071-1074.
Kaufman DL, Clare-Salzler M, Tian J, Forsthuber T, Ting GSP, Robinson
P, Atkinson MA, Sercarz EE, Tobin AJ, & Lehmann PV (1993) Spontaneous
loss of T-cell tolerance to glutamic acid decarboxylase in murine
insulin-dependent diabetes. Nature (Lond), 366: 69-72.
Kayser D & Schlede E (1995) [Chemicals and contact allergy -- a
summary evaluation.] Berlin, Federal Institute for Consumer's Health
Protection and Veterinary Medicine (BGVV) (in German).
Keeley DJ, Neill P, & Gallivan S (1991) Comparison of the prevalence
of reversible airways obstruction in rural and urban Zimbabwean
children. Thorax, 46: 549-553.
Keil U, Weiland SK, Duhme H, & Chambless L (1996) The international
study of asthma and allergies in childhood (ISAAC): Objectives and
methods -- Results from German ISAAC centres concerning traffic
density and wheezing and allergic rhinitis. Toxicol Lett, 86: 99-103.
Kemp T, Pearce N, Fitzharris P, Crane J, Fergusson D, St. George I,
Wickens K, & Beasley R (1997) Is infant immunization a risk factor for
childhood asthma or allergy? Epidemiology, 8: 678-680.
Keskinen H, Alanko K, & Saarinen L (1978) Occupational asthma in
Finland. Clin Allergy, 8(6): 569-579.
Keskinen H, Tupasela O, Tiikkainen U, & Nordman H (1988) Experience of
specific in IgE asthma due to diisocyanates. Clin Allergy, 18(6):
597-604.
Khoury SJ, Hancock WW, & Weiner HL (1992) Oral tolerance to myelin
basic protein and natural recovery from experimental autoimmune
encephalomyelitis are associated with downregulation of inflammatory
cytokines and differential upregulation of transforming growth factor
beta, interleukin 4 and prostaglandin E expression in the brain. J Exp
Med, 176: 1355-1364.
Kimber I & Basketter DA (1997) Contact sensitisation: a new approach
to risk assessment. Hum Ecol Risk Assess, 3: 385-395.
Kimber I & Cumberbatch M (1992a) Dendritic cells and cutaneous immune
responses to chemical allergens. Toxicol Appl Pharmacol, 117: 137-146.
Kimber I & Cumberbatch M (1992b) Stimulation of Langerhans cell
migration by tumour necrosis factor alpha. J Invest Dermatol, 99:
48S-50S.
Kimber I & Maurer T ed. (1996) Toxicology of contact hypersensitivity.
London, Taylor & Francis, 170 pp.
Kimber I & Wilks MF (1995) Chemical respiratory allergy: Toxicological
and occupational health issues. Hum Exp Toxicol, 14(9): 735-736.
Kimber I, Hilton J, & Weisenberger C (1989) The murine local lymph
node assay for identification of contact allergens: a preliminary
evaluation of in situ measurement of lymphocyte proliferation. Contact
Dermatitis, 21: 215.
Kimber I, Kinnaird A, Peters SW, & Mitchell JA (1990) Correlation
between lymphocyte proliferative responses and dendritic cell
migration in regional lymph nodes following skin painting with contact
sensitizing agents. Int Arch Allerg Appl Immunol, 93: 47-53.
Kimber I, Dearman RJ, Scholes EW, & Basketter DA (1994) The local
lymph node assay developments and applications. Toxicology, 93: 13-31.
King TP, Hoffman D, Lowenstein H, Marsh DG, Platts-Mills TAE, & Thomas
W (1995) Allergen nomenclature. J Allergy Clin Immunol, 96: 5-14.
Kirton V (1978) Contact urticaria and cinnamic aldehyde. Contact
Dermatitis, 4(6): 374-375.
Kishimoto T & Hirano T (1984) beta-Lymphocyte activation,
proliferation, and immunoglobulin secretion. In: Paul WE ed.
Fundamental immunology, 2nd ed. New York, Raven Press, pp 385-411.
Kitagaki H, Fujisawa S, Watanabe K, Hayakawa K, & Shiohara T (1995)
Immediate type hypersensitivity response followed by a late reaction
is induced by repeated epicutaneous application of contact sensitizing
agents in mice. J Invest Dermatol, 105: 749-755.
Kjellman NI & Croner S (1984) Cord blood IgE determination for allergy
prediction -- a follow-up to seven years of age in 1,651 children. Ann
Allergy, 53(2): 167-171.
Kligman AM (1958a) Poison ivy (Rhus) dermatitis. Arch Dermatol, 77:
159-180.
Kligman AM (1958b) Hyposensitization against Rhus dermatitis. Arch
Dermatol, 78: 47-72.
Kligman AM (1966a) The identification of contact allergies by human
assay: II. Factors influencing the induction and measurement of
allergic contact dermatitis. J Invest Dermatol, 47: 375-393.
Kligman AM (1966b) The identification of contact allergens by human
assay: III. The maximization test -- A procedure for screening and
rating contact sensitizers. J Invest Dermatol, 47: 393-409.
Kligman AM (1966c) The SLS provocative patch test in allergic
sensitization. J Invest Dermatol, 46: 573-595.
Knippels LM, Penninks AH, Spanhaak S, & Houben GF (1998) Oral
sensitization to food proteins: a Brown Norway rat model. Clin Exp
Allergy, 28(3): 368-375.
Koenig JQ (1988) Indoor and outdoor pollutants and the upper
respiratory tract. J Allergy Clin Immunol, 81: 1055-1059.
Koenig JQ (1995) Effect of ozone on respiratory responses in subjects
with asthma. Environ Health Perspect, 103(suppl 2): 103-105.
Koenig JQ, Pierson WE, & Horike M (1983) The effects of inhaled
sulfuric acid on pulmonary function in adolescent asthmatics. Am Rev
Respir Dis, 128(2): 221-225.
Koenig JQ, Pierson WE, Covert DS, Marshall SG, Morgan MS, & van Belle
G (1988) The effects of ozone and nitrogen dioxide on lung function in
healthy and asthmatic adolescents. Res Rep Health Eff Inst, 14: 5-24.
Koenig JQ, Covert DS, Hanley QS, van Belle G, & Pierson WE (1990)
Prior exposure to ozone potentiates subsequent response to sulfur
dioxide in adolescent asthmatic subjects. Am Rev Respir Dis, 141:
377-380.
Koëter HBWM (1995) International harmonisation of immunotoxicity
testing. Hum Exp Toxicol, 14: 151-154.
Kogevinas M, Anto J, Soriano JB, Tobias A, & Burney P (1996) The risk
of asthma attributable to occupational exposures. Ann Resp Crit Care
Med, 154: 137-143.
Kohler C, Jeanvoine G, Pierrez J, Olive D, & Gerard H (1987)
Modifications of the thymus and splenic thymic dependent zones after
in utero exposure to phenytoin: Qualitative and quantitative analysis
in C3H mice. Dev Pharmacol Therap, 10: 405-412.
Koren HS (1995) Associations between criteria air pollutants and
asthma. Environ Health Perspect, 103(suppl 6): 235-242.
Koren HS & Bromberg PA (1995) Respiratory responses of asthmatics to
ozone. Int Arch Allergy Immunol, 107: 236-238.
Koren HS, Devlin RB, Graham DE, Mann R, McGee MP, Horstman DH, Kozumbo
WJ, Becker S, House DE, McDonnel WF, & Bromberg PA (1989)
Ozone-induced inflammation in the lower airways of human subjects. Am
Rev Respir Dis, 139: 407-415.
Koren HS, Hatch GE, & Graham DE (1990) Nasal lavage as a tool in
assessing acute inflammation in response to inhaled pollutants.
Toxicology, 60: 15-25.
Korman NJ (1995) In situ bound antibodies eluted from the skin of
patients with bullous pemphigoid are preferentially directed against
the 230-kD bullous pemphigoid antigen. J Invest Dermatol,
105: 824-830.
Korman NJ, Eyre RW, Zone J, & Stanley JR (1991) Drug-induced
pemphigus: autoantibodies directed against the pemphigus antigen
complexes are present in penicillamine and captopril-induced
pemphigus. J Invest Dermatol, 96: 273-276.
Kortekangas-Savolainen O, Savolainen J, & Einarsson R (1993)
Gastrointestinal stability of baker's yeast allergens: an in vitro
study. Clin Exp Allergy, 23(7): 578-590.
Krämer U, Behrendt H, & Ring J (1996) Air pollution as a risk factor
for allergy: The East-West German experience. In: Ring J, Behrendt H,
& Vieluf D ed. New trends in allergy. Berlin, Heidelberg, New York,
Springer Verlag, vol IV, pp 25-35.
Kremer AM, Pal TM, Schouten JP, & Rijcken B (1995) Airway
hyperresponsiveness in workers exposed to low levels of irritants. Eur
Respir J, 8(1): 53-61.
Kremser M (1989) [Food allergies -- oropharyngeal reactions.] Wien Med
Wochenschr, 139(6/7): 135-139.
Kröger S, Neuber K, Gruseck E, Ring J, & Abeck D (1995) Pityrosporum
ovale extracts increase interleukin-4, interleukin-10 and IgE
synthesis in patients with atopic eczema. Acta Dermatol Venereol, 75:
357-360.
Krzystyniak K, Brouland J-P, Panayi G, Patriarca C, Verdier F,
Descotes J, & Revillard J-P (1992) Activation of CD4+ and CD8+
lymphocyte subsets by streptozotocin in murine popliteal lymph node
(PLN) test. J Autoimmun, 5: 183-197.
Krzyzanowski M, Quackenboss JJ, & Lebowitz MD (1992) Relation of peak
expiratory flow rates and symptoms to ambient ozone. Arch Environ
Health, 47(2): 107-115.
Kubicka-Muranyi M, Goebels R, Goebel C, Uetrecht J, & Gleichmann E
(1993) T lymphocytes ignore procainamide, but respond to its reactive
metabolites in peritoneal cells: demonstration by the adoptive
transfer popliteal lymph node assay. Toxicol Appl Pharmacol, 122:
88-94.
Kubicka-Muranyi M, Griem P, Lübben B, Rottmann N, Lührmann R, &
Gleichmann E (1996) Mercuric chloride-induced autoimmunity in mice
involves an upregulated presentation of altered and unaltered
nucleolar self antigen. Int Arch Allergy Immunol, 108: 1-10.
Kuchroo VK, Byrne MC, Greenfield E, Whitters MJ, Nalefsky EA, Rao A,
Collins M, & Dorf ME (1995) Transfection of TCR alpha-chains into
suppressor and T helper cell hybridomas. J Immunol, 154: 5030-5038.
Kuhn R, Rajewsky K, & Muller W (1991) Generation and analysis of
interleukin-4 deficient mice. Science, 254(5032): 707-710.
Kurisaki J, Konishi Y, Kaminogawa S, & Yamauchi K (1981) Studies on
the allergenic structure of hen ovomucoid by chemical and enzymatic
fragmentation. Agric Biol Chem, 45: 879.
Küster W, Petersen M, Christophers E, Goos M, & Sterry W (1990) A
family study of atopic dermatitis. Arch Dermatol Res, 282: 98-102.
Lake AM, Kagey-Sobotka A, Jakubowicz T, & Lichtenstein LM (1984)
Histamine release in acute anaphylactic enteropathy of the rat. J
Immunol, 133(3): 1529-1534.
Lammintausta K, Kalimo K, & Havu VK (1982) Contact allergy in atopics
who perform wet work in hospital. Derm Beruf Umwelt, 30(6): 184-188.
Lammintausta K, Kalimo K, & Fagerlund VL (1992) Patch test reactions
in atopic patients. Contact Dermatitis, 26(4): 234-240.
Lamont AG, Mowat A McI, Browning MJ, & Parrott DMV (1988) Genetic
control of oral tolerance to ovalbumin in mice. Immunology, 63:
737-739.
Landsteiner K & Rostenberg A Jr (1939) Individual differences in
susceptibility to eczematous sensitization with simple chemical
substances. J Invest Dermatol, 2: 25-29.
Lane HC & Fauci AS (1985) Immunologic abnormalities in the acquired
immunodeficiency syndrome. Annu Rev Immunol, 3: 477-500.
Lange CE, Jühe S, Stein G, & Veltman G (1974) [The so-called vinyl
chloride disease -- an occupational scleroderma?] Int Arch Arbeitsmed,
32: 1-32 (in German).
Laor A, Cohen L, & Danon YL (1993) Effects of time, sex, ethnic
origin, and area of residence on prevalence of asthma in Israeli
adolescents. Br Med J, 307: 841-844.
Larsen CG, Thomsen MK, Gesser B, Deleuran BW, & Thestrup-Pedersen K
(1995) The delayed type hypersensitivity reaction is dependent on
IL-8. Inhibition of a tuberculin skin reaction by an anti-IL-8
monoclonal antibody. J Immunol, 155(4S): 2151-2157.
Lau S, Falkenhorst G, Weber A, Werthmann I, Lind P, Buettner Goetz P,
& Wahn U (1989) High mite-allergen exposure increases the risk of
sensitization in atopic children and young adults. J Allergy Clin
Immunol, 84: 718-725.
Lau S, Ehnert B, Cremer B, Nasert S, Buettner P, Czarnetzki BM, & Wahn
U (1995) [Reduction of house dust mite exposure in sensitized patients
with atopic eczema.] Allergo J, 4: 432-437 (in German).
Lauer K (1990) Environmental nitrophenols and autoimmunity (Letter to
the Editor). Mol Immunol, 27: 697-698.
Lauerma AI (1992) Contact hypersensitivity to glucocorticosteroids. Am
J Contact Dermatitis, 3: 112-132.
Lauwerys R & Lison D (1994) Health risks associated with cobalt
exposure -- an overview. Sci Total Environ, 150(1-3): 1-6.
Law LW, Goldstein AL, & White A (1968) Influence of thymosin on
immunological competence of lymphoid cells from thymectosized mice.
Nature (Lond), 219(161): 1391-1392.
Leach CL, Hatoum NS, Ratajcak HV, Zeiss CR, Roger JC, & Garvin PJ
(1987) Pathologic and immunologic response to inhaled trimellitic
anhydride in rats. Toxicol Appl Pharmacol, 87: 67-80.
Ledford DK (1994) Indoor allergens. J Allergy Clin Immunol, 94:
327-334.
Lee HK, Alarie Y, & Karol MH (1984) Induction of formaldehyde
sensitivity in guinea pigs. Toxicol Appl Pharmacol, 75: 147-155.
Lehmann PV, Secarz EE, Fortshuber T, Dayan CM, & Gammon G (1993)
Determinant spreading and the dynamics of the autoimmune T cell
repertoire. Immunol Today, 14: 203-208.
Lejman E, Stoudemayer T, Grove G, & Kligman AM (1984) Age differences
in poison ivy dermatitis. Contact Dermatitis, 11: 163-167.
Lenshow DJ, Walunas TL, & Bluestone JA (1996) CD28/B7 system of T cell
co-stimulation. Annu Rev Immunol, 14: 233-258.
Leung R & Ho P (1994) Asthma, allergy, and atopy in three south-east
Asian populations. Thorax, 49: 1205-1210.
Lewis S, Richards D, Bynner J, Butler N, & Britton J (1995)
Prospective study of risk factors for early and persistent wheezing in
childhood. Eur Respir J, 8(3): 349-356.
Leyden JJ & Kligman AM (1977) Allergic contact dermatitis: Sex
differences. Contact Dermatitis, 3: 333-336.
Leyden JE, Marples RR, & Kligman AM (1974) Staphylococcus aureus in
the lesions of atopic dermatitis. Br J Dermatol, 90(5): 525-530.
Li LF (1995) A clinical and patch test study of contact dermatitis
from traditional Chinese medical materials. Contact Dermatitis, 33:
392-395.
Lidén C, Menné T, & Burrows D (1996) Nickel-containing alloys and
platings and their ability to cause dermatitis. Br J Dermatol, 134:
193-198.
Lindskov R & Holmer G (1992) Polyunsaturated fatty acid in plasma, red
blood cells and mononuclear phospholipids in patients with atopic
dermatitis. Allergy, 47: 517-521.
Linn WS, Shamoo DA, Spier CE, Valencia LM, Anzar UT, Venet TG, &
Hackney JD (1983a) Respiratory effects of 0.75 ppm sulfur dioxide in
exercising asthmatics: influence of upper-respiratory defenses.
Environ Res, 30: 340-348.
Linn WS, Venet TG, Shamoo DA, Valencia LM, Anzar UT, Spier CE, &
Hackney JD (1983b) Respiratory effects of sulfur dioxide in heavily
exercising asthmatics: A dose-response study. Am Rev Respir Dis, 127:
278-283.
Lippmann M (1989) Health effects of ozone: a critical review. J Air
Pollut Control Assoc, 39: 672-695.
Liu MC, Bleecker ER, Lichtenstein LM, Kagey-Sobotka A, Niv Y, Mclemore
TL, Permutt S, Proud D, & Hubbard WC (1990) Evidence for elevated
levels of histamine, prostaglandin D2, and other bronchoconstricting
prostaglandins in the airways of subjects with mild asthma. Am Rev
Respir Dis, 142: 126-132.
Lo D (1996) Animal models of human disease -- Transgenic and knockout
models of autoimmunity: building a better disease? Clin Immunol
Immunopathol, 79(2): 96-104.
Logan RFA (1992) In: Auricchio S & Visakorpi JK ed. Problems and
pitfalls in epidemiological studies of coeliac disease, common food
intolerances: 1. Epidemiology of coeliac disease. Basel, Karger, p 14.
Lonkar A, Mitchell JC, & Calnan CD (1974) Contact dermatitis from
parthenium hysterophorus. Trans St. John's Hosp Derm Soc, 60: 43-53.
Lovik M, Hogseth A-K, Gaarder PI, Hagemann R, & Eide I (1997) Diesel
exhaust particles and carbon black have adjuvant activity on the local
lymph node response and systemic IgE production to ovalbumin.
Toxicology, 121: 165-178.
Lucas A, Brooke OG, Morley R, Cole TJ, & Bamford MF (1990) Early diet
of preterm infants and development of allergic or atopic disease:
randomised prospective study. Br Med J, 300(6728): 837-840.
Lucente FE (1989) Rhinitis and nasal obstruction. Otolaryngol Clin
North Am, 22: 307-318.
Luczynska CM & Topping MD (1986) Specific IgE antibodies to reactive
dye-albumin conjugates. J Immunol Methods, 95: 177-186.
Luo J-C, Nelsen KG, & Fischbein A (1990) Persistent reactive airways
dysfunction syndrome after exposure to toluene di-isocyanate. Br J Ind
Med, 47: 239-241.
Lutsky I, Baum GL, Teichtahl H, Mazar A, Aizer F, & Bar Sela S (1985)
Occupational respiratory disease in veterinarians. Ann Allergy, 55(2):
153-156.
Lynch NR, Hagel IA, Palenque ME, Di Prisco MC, Escudero JE, Corao LA,
Sandia JA, Ferreira LJ, Botto C, Perez M, & Le Souef PN (1998)
Relationship between helminthic infection and IgE response in atopic
and nonatopic children in a tropical environment. J Allergy Clin
Immunol, 101(2): 217-221.
McAlindon T, Gianotta L, Taub N, D'Cruz D, & Hughes GRV (1993)
Environmental factors predicting nephritis in systemic lupus
erythematosus. Ann Rheum Dis, 52: 720-724.
Maccia CA, Berstein IL, Emmett EA, & Brookes SSM (1976) In vitro
demonstration of specific IgE in phthalic anhydride sensitivity. Am
Rev Respir Dis, 113: 701-704.
MacDonald TT (1983) Immunosuppression caused by antigen feeding: II.
Suppressor T cells mask Peyer's patch B cell priming to orally
administered antigen. Eur J Immunol, 13: 138-142.
McGrath H Jr, Bell JM, & Haycock JW (1994) Fluorescent light activates
the immunomodulator cis-urocanic acid in vitro: Implications for
patients with systemic lupus erythematosus. Ann Rheum Dis, 53(6):
396-399.
Machado DC, Horton D, Harrop R, Peachell PT, & Helm BA (1996)
Potential allergens stimulate the release of mediators of the allergic
response from cells of mast cell lineage in the absence of
sensitization with antigen-specific IgE. Eur J Immunol, 26: 2972-2980.
McMillan C & Burrows D (1995) In vitro testing in contact
hypersensitivity. In: Rycroft RJG, Menné T, Frosch PJ, & Benezra C ed.
Textbook of contact dermatitis, 2nd ed. Berlin, Heidelberg, New York,
Springer Verlag, pp 306-318.
Madara JL & Stafford J (1989) Interferon-gamma directly affects
barrier function of cultured intestinal epithelial monolayers. J Clin
Invest, 83(2): 724-727.
Maestrelli P, Di Stefano A, Occari P, Turato G, Milani G, Pivirotto F,
Mapp CE, Fabbri LM, & Saetta M (1995) Cytokines in the airway mucosa
of subjects with asthma induced by toluene diisocyanate. Am J Resp
Crit Care Med, 151: 607-612.
Magnus P & Jaakkola JJ (1997) Secular trend in the occurrence of
asthma among children and young adults: critical appraisal of repeated
cross sectional surveys. Br Med J, 314: 1795-1799
Magnusson CG (1986) Maternal smoking influences cord serum IgE and IgD
levels and increases the risk for subsequent infant allergy. J Allergy
Clin Immunol, 78: 898-904.
Magnusson B & Gilje O (1973) Allergic contact dermatitis from a
dish-washing liquid containing laurylethersulphate. Acta Dermatol
Venereol, 53: 136-140.
Magnusson B & Hersle K (1965) Patch test methods. Acta Dermatol
Venereol, 45: 123-128.
Magnusson B & Kligman AM (1970a) Allergic contact dermatitis in the
guinea pig. Springfield, Charles C Thomas, pp 3-120.
Magnusson B & Kligman AM (1970b) The identification of contact
allergens by animal assay: The guinea pig maximisation test. J Invest
Dermatol, 52: 268-276.
Magnussen H, Holz O, & Jörres RA (1998) Asthma and the environment: Is
ozone really important? Eur Respir Rev, 8: 141-144.
Mahzoon S, Yamamoto S, & Greaves MW (1977) Response of skin to
ammonium persulphate. Acta Dermatol Venereol, 57(2): 125-126.
Maibach H (1976) Immediate hypersensitivity in hand dermatitis. Arch
Dermatol, 112(9): 1289-1291.
Malo J-L, Ouimet G, Cartier A, Lebitz D, & Ziess CR (1983) Combined
alveolitis and asthma due to hexamethylene di-isocyanate (HDI) with
evidence of crossed respiratory and immunologic reactivities to
diphenylmethane di-isocyanate MDI. J Allergy Clin Immunol,
72: 413-419.
Manetti R, Parronchi P, Giudizi MG, Piccinni MP, Maggi E, Trinchieri
G, & Romagnani S (1993) Natural killer cell stimulatory factor
(interleukin 12 [IL-12]) induces T helper type 1 (Th1)-specific immune
responses and inhibits the development of IL-4 producing Th cells. J
Exp Med, 177(4): 1199-1204.
Mantle J & Pepys J (1974) Asthma amongst Tristan da Cunha islanders.
Clin Allergy, 4: 161-170.
Marchal G, Seman M, Milon G, Truffa-Bacchi P, & Zilberfarb V (1982)
Local adoptive transfer of skin delayed type hypersensitivity
initiated by a single T lymphocyte. J Immunol, 129(3): 954-958.
Marchionini A (1960) [New studies of constitutional neurodermatitis.]
In: Marchionini A & Röckl M ed. [Dermatology and venereology.] Berlin,
Heidelberg, New York, Springer Verlag, vol 3, pp 42-46 (in German).
Marcon-Genty D, Tomé D, Dumontier AM, Kheroua O, & Desjeux JF (1989)
Permeability of milk protein antigens across the intestinal
epithelium in vitro. Reprod Nutr Dev, 29(6): 717-723.
Marfaing-Koka A, Devergne O, Gorgone G, Portier A, Schall TJ, Galanaud
P, & Emilie D (1995) Regulation of the production of RANTES chemokine
by endothelial cells -- Synergistic induction by IFN-gamma plus
TNF-alpha and inhibition by IL-4 and IL-13. J Immunol, 154(suppl 4):
1870-1878.
Marsh DG (1990) Immunogenetic and immunochemical factors determining
immune responsiveness to allergens: studies in unrelated subjects. In:
Marsh DG & Blumenthal M ed. Genetic and environmental factors in
clinical allergy. Minneapolis, University of Minneapolis Press, pp
97-120.
Marsh DG & Norman PS (1988) Antigens that cause atopic disease. In:
Immunological diseases, 4th ed. Boston, Little Brown, vol 2, pp
981-1008.
Martin S & Weltzien HU (1994) T cell recognition of haptens, a
molecular view. Int Arch Allergy Immunol, 104: 10-16.
Martinez FD (1994) Role of viral infections in the inception of asthma
and allergies during childhood: could they be protective? Thorax, 49:
1189-1191.
Martinez FD (1995) Viral infections and the development of asthma. Am
J Respir Crit Care Med, 151: 1644-1648.
Martinez FD, Antognoni G, Macri F, Bonci E, Midulla F, De Castro G, &
Ronchetti R (1988) Parental smoking enhances bronchial responsiveness
in nine-year-old children. Am Rev Respir Dis, 138(3): 518-523.
Martinez FD, Cline M, & Burrows B (1992) Increased incidence of asthma
in children of smoking mothers. Pediatrics, 89: 21-26.
Martinez FD, Wright AL, Taussig LM, Holberg CJ, Halonen M, & Morgan WJ
(1995) Asthma and wheezing in the first six years of life. The Group
Health Medical Associates. N Engl J Med, 332: 133-138.
Marzulli FN & Maibach HI (1973) Antimicrobials: Experimental contact
sensitization in man. J Soc Cosmet Chem, 24: 399-421.
Marzulli FN & Maibach HI (1974) The use of graded concentrations in
studying skin sensitizers: experimental contact sensitization in man.
Food Cosmet Toxicol, 12: 219-227.
Marzulli FN & Maibach HI (1996) Test methods for allergic contact
dermatitis in humans.
In: Marzulli FN & Maibach HI ed. Dermatotoxicology, 5th ed.
Washington, New York, London, Hemisphere Publishing Corporation, pp
477-482.
Matricardi PM (1997) Infections preventing atopy: Facts and new
questions. Allergy, 52: 879-882.
Matricardi PM, Rosmini F, Ferrigno L, Nasion R, Rapicetta M, Chionne
P, Stroffolini T, Pasquini P, & D`Amelio R (1997) Cross sectional
retrospective study of the prevalence of atopy among Italian military
students with antibodies against hepatitis A virus. Br Med J, 314:
999-1003.
Mattingly JA (1984) Immunologic suppression after oral administration
of antigen: III. Activation of suppressor-inducer cells in the Peyer's
patches. Cell Immunol, 86: 46-52.
Matzner Y, Erlich HA, Brautbar C, Sanilevitch A, Landau M, Brenner S,
& Friedmann A (1995) Identical HLA class II alleles predispose to
drug-triggered and idiopathic pemphigus vulgaris. Acta Dermatol
Venereol, 75: 12-14.
Maulitz RM, Pratt DS, & Schocket AL (1979) Exercise-induced
anaphylactic reaction to shellfish. J Allergy Clin Immunol, 63(6):
433-434.
Maurer T, Thomann P, Weirich EG, & Hess R (1975) The optimization test
in the guinea pig: A method for the predictive evaluation of the
contact allergenicity of chemicals. Agents Actions, 5: 174-179.
Maynard AD, Northage C, Hemingway M, & Bradley SD (1997) Measurement
of short-term exposure to airborne soluble platinum in the platinum
industry. Ann Occup Hyg, 41(1): 77-94.
Meade R, Askenase PW, Geba GP, Neddermann K, Jacoby RO, & Pasternak RD
(1992) Transforming growth factor-beta 1 inhibits murine immediate and
delayed type hypersensitivity. J Immunol, 149(2): 521-528.
Medici TC & Vetter W (1991) [Bronchial asthma and kitchen salt.]
Schweiz Med Wochenschr, 121(14): 501-508 (in German).
Meding B (1990) Epidemiology of hand eczema in an industrial city.
Acta Dermatol Venereol, 153(suppl): 2-43.
Melamed D & Friedman A (1993) Direct evidence for anergy in T
lymphocytes tolerized by oral administration of ovalbumin. Eur J
Immunol, 23: 935-942.
Mellström G, Lindahl G, & Wahlberg J (1989) DAISY: reference database
on protective gloves. Semin Dermatol, 8: 75-79.
Melnik B & Plewig G (1991) Atopic dermatitis and disturbances of
essential fatty acid in prostaglandin E metabolism. J Am Acad
Dermatol, 25: 859-860.
Menné T & Bachmann E (1979) Permanent disability from skin diseases.
Dermatosen, 27: 37-42.
Menné T & Holm NV (1983) Nickel allergy in a female twin population.
Int J Dermatol, 22: 22-28.
Menné T & Holm NV (1986) Gene susceptibility in human allergic contact
sensitization. Semin Dermatol, 5: 301-306.
Menné T & Maibach HI (1991) Systemic contact-type dermatitis. In:
Marzulli FN & Maibach HI ed. Dermatotoxicology. Washington, New York,
London, Hemisphere Publishing Corporation, p 453.
Menné T & Maibach HI ed. (1994) Hand eczema. Boca Raton, Florida, CRC
Press, 334 pp.
Menné T & Wilkinson JD (1995) Individual predisposition to contact
dermatitis. In: Rycroft RJG, Menné T, Frosch PJ, & Benezra C ed.
Textbook of contact dermatitis. Berlin, Heidelberg, New York, Springer
Verlag, pp 123-130.
Menné T, Christophersen J, & Maibach HI (1987) Epidemiology of
allergic contact sensitization. Monogr Allergy, 21: 132-161.
Menné T, Dooms-Goossens A, Wahlberg JE, White I, & Wilkinson J (1992)
How large a proportion of contact sensitivities are diagnosed with the
European standard series. Contact Dermatitis, 26: 201-202.
Menné T, Veien N, Sjolin KE, & Maibach HI (1994) Systemic contact
dermatitis. Am J Contact Dermatitis, 5: 1-12.
Meredith S (1993) Reported incidence of occupational asthma in the
United Kingdom, 1989-90. J Epidemiol Community Health, 47(6): 459-463.
Meredith SK & McDonald JC (1994) Work-related respiratory disease in
the United Kingdom, 1989-1992: Report on the SWORD project. Occup Med,
44(4): 183-189.
Meredith S & Nordman H (1996) Occupational asthma: measures of
frequency from four countries. Thorax, 51(4): 435-440.
Merget R, Reineke M, Rueckmann A, Bergmann EM, & Schultze Werninghaus
G (1994) Nonspecific and specific bronchial responsiveness in
occupational asthma caused by platinum salts after allergen avoidance.
Am J Respir Crit Care Med, 150(4): 1146-1149.
Merget R, Dierkes A, Rueckmann A, Bergmann EM, & Schultze Werninghaus
G (1996) Absence of relationship between degree of nonspecific and
specific bronchial responsiveness in occupational asthma due to
platinum salts. Eur Respir J, 9(2): 211-216.
Metcalfe DD (1985) Food allergens. Clin Rev Allergy, 3(3): 331-349.
Metcalfe DD & Sampson HA (1990) Workshop on experimental methodology
for clinical studies of adverse reactions to foods and food additives.
J Allergy Clin Immunol, 86: 421-442.
Metzger WJ, Zavala D, Richerson HB, Mosely P, Iwamota P, Monick M,
Sjoerdsma K, & Hunninghake GW (1987) Local allergen challenge and
bronchoalveolar lavage of allergic asthmatic lungs. Am Rev Respir Dis,
135: 433-440.
Miedema I, Feskens EJ, Heederik D, & Kromhout D (1993) Dietary
determinants of long-term incidence of chronic nonspecific lung
diseases: The zutphen study. Am J Epidemiol, 138(1): 37-45.
Miller K & Nicklin S (1988) Mechanisms of food intolerances:
development and use of experimental animal models. In: Walker R &
Quattrucci E ed. Nutritional and toxicological aspects of food
processing. London, New York, Philadelphia, Taylor & Francis, pp
351-364.
Minowada G & Welch WJ (1995) Clinical implications of the stress
response. J Clin Invest, 95: 3-12.
Mitchell EA, Stewart AW, Pattemore PK, Asher MI, Harrison AC, & Rea HH
(1989) Socioeconomic status in childhood asthma. Int J Epidemiol,
18(4): 888-890.
Molfino NA, Wright SC, Katz I, Tarlo S, Silverman F, McClean PA,
Szalai JP, Raizenne M, Slutsky AS, & Zamel N (1991) Effect of low
concentrations of ozone on inhaled allergen responses in asthmatic
subjects. Lancet, 338: 199-203.
Moller DR, Gallagher JS, Benstein DI, Wilcox TG, Burroughs HE, &
Bernstein IL (1985) Detection of IgE mediated respiratory
sensitisation in workers exposed to hexahydrophthalic anhydride. J
Allergy Clin Immunol, 75: 663-672.
Monick M, Glazier J, & Hunninghake GW (1987) Human alveolar
macrophages suppress interleukin 1 activity via the secretion of
prostaglandin E2. Am Rev Respir Dis, 135: 72-77.
Montgomery-Smith J (1983) Epidemiology and natural history of asthma,
allergic rhinitis and atopic dermatitis (eczema). In: Allergy:
Principles and practice. St Louis, Missouri, C.V. Mosby & Co., pp
771-803.
Montgomery-Smith J, Middleton E, Reed CE, Ellis EF, Adkinson NF, &
Yunginger JW ed. (1983) Allergy: Principles and practice
-- Epidemiology and natural history of asthma, allergic rhinitis and
atopic dermatitis (eczema). St Louis, Missouri, C.V. Mosby & Co., pp
771-803.
Moore SA, Strieter RM, Rolfe MW, Standiford TJ, Burdick MD, & Kunkel
SL (1992) Expression and regulation of human alveolar
macrophage-derived interleukin-1 receptor antagonist. Am J Respir Cell
Mol Biol, 6(6): 569-575.
Morel E, Feuillet-Fieux MN, Vernet-der Garabedian B, Raimond F,
D'Anglejan J, Bataille R, Sany J, & Bach JF (1991) Autoantibodies in
D-penicillamine-induced myasthenia gravis: a comparison with
idiopathic myasthenia and rheumatoid arthritis. Clin Immunol
Immunopathol, 58: 318-330.
Morgenstern H & Thomas D (1993) Principles of study design in
environmental epidemiology. Environ Health Perspect, 101(suppl 4):
23-28.
Morren MA, Przybilla B, Bamelis M, Heykants B, Reynaers A, & Degreef H
(1994) Atopic dermatitis: triggering factors. J Am Acad Dermatol, 31:
467-473.
Mosimann B, Peitrequin R, Blanc C, & Pecoud A (1992) Allergie aux
blattes (cafards) dans une population suisse souffrant d'asthme et de
rhinite chroniques. Schweiz Med Wochenschr, 122(34): 1245-1248.
Mosmann TR & Sad S (1996) The expanding universe of T-cell subsets:
Th1, Th2 and more. Immunol Today, 17: 138-146.
Moulon C, Vollmer J, & Weltzien HU (1995) Characterization of
processing requirements and metal cross-reactivities in T cell clones
from patients with allergic contact dermatitis to nickel. Eur J
Immunol, 25: 3308-3315.
Mowatt AMcI (1984) The immunopathogenesis of food-sensitive
enteropathies. In: Newly TJ & Stokes CR ed. Local immune responses of
the gut. Boca Raton, Florida, CRC Press, pp 199-225.
Mowatt AMcI (1987) The regulation of immune responses to dietary
protein antigens. Immunol Today, 8: 93-98.
Mowatt AMcI, Lamont AG, Strobel S, & Mackenzie S (1986) The role of
antigen processing and suppressor T cells in immune responses to
dietary proteins in mice. Adv Exp Med Biol, 216A: 709-720.
M'Raihi L, Charpin D, Pons A, Bongrande P, & Vervloet D (1991)
Cross-reactivity between latex and banana. J Allergy Clin Immunol, 87:
129-130.
Mullan RJ & Murthy LI (1991) Occupational sentinel health events: An
up-dated list for physician recognition and public health
surveillance. Am J Ind Med, 19(6): 775-799.
Mullick FG, McAllister HA, Wagner BM, & Fenoglio JJ (1979) Drug
related vasculitis: Clinicopathologic correlations in 30 patients. Hum
Pathol, 10: 313-325.
Munir AK, Einarsson R, Schou C, & Dreborg SK (1993) Allergens in
school dust: I. The amount of the major cat (Fel d I) and dog (Can f
I) allergens in dust from Swedish schools is high enough to probably
cause perennial symptoms in most children with asthma who are
sensitized to cat and dog. J Allergy Clin Immunol, 91(5): 1067-1074.
Munir AK, Einarsson R, & Dreborg SK (1994a) Indirect contact with pets
can confound the effect of cleaning procedures for reduction of animal
allergen levels in house dust. Pediatr Allergy Immunol, 5(1): 32-39.
Munir AK, Bjorksten B, Einarsson R, Schou C, Ekstrand Tobin A, Warner
A, & Kjellman NI (1994b) Cat (Fel d I), dog (Can f I), and cockroach
allergens in homes of asthmatic children from three climatic zones in
Sweden. Allergy, 49(7): 508-516.
Muraille E & Leo O (1998) Revisiting the Th1/Th2 paradigm. Scand J
Immunol, 47: 1-9.
Muranaka M, Suzuki S, Koizumi K, Takafuji S, Miyamoto T, Ikemori R, &
Tokiwa H (1986) Adjuvant activity of diesel-exhaust particulates for
the production of IgE antibody in mice. J Allergy Clin Immunol, 77:
616-623.
Mygind N (1986) Essential allergy, 1st ed. Oxford, London, Boston,
Blackwell Scientific Publications, 480 pp.
Mygind N (1989) Nasal allergy, 2nd ed. Oxford, London, Boston,
Blackwell Scientific Publications, pp 219-223.
Nagata S, Yamashiro Y, Ohtuska Y, Shioya T, Oguchi S, Shimizu T, &
Maeta M (1995) Quantitative analysis and immunohistochemical studies
on small intestinal mucosa of food-sensitive enteropathy. J Pediatr
Gastroenterol Nutr, 20: 44-48.
Naparstek Y & Plotz PH (1993) The role of autoantibodies in autoimmune
disease. Annu Rev Immunol, 11: 79-104.
NAS (National Academy of Science) (1983) Committee on the
Institutional Means for Assessment of Risks to Public Health -- Risk
assessment in the Federal Government: Managing the process.
Washington, DC, National Academy Press, pp 1-50.
NAS (National Academy of Science) (1993) Committee on Risk Assessment
Methodology, Board on Environmental Studies and Toxicology -- Issues
in risk assessment. Washington, DC, National Academy Press, 356 pp.
Neas LM, Dockery DW, Ware JH, Spengler JD, Speizer FE, & Ferris BG Jr
(1991) Association of indoor nitrogen dioxide with respiratory
symptoms and pulmonary function in children. Am J Epidemiol, 134(2):
204-219.
Nelson HS (1985) The atopic diseases. Ann Allergy, 55: 441-447.
Nelson HS, Rosloniec DM, McCall LI, & Ikle D (1993) Comparative
performance of five commercial prick skin test devices. J Allergy Clin
Immunol, 92(5): 750-756.
Nemery B, Casier P, Roosels D, Lahaye D, & Demedts M (1992) Survey of
cobalt exposure and respiratory health in diamond polishers. Am Rev
Respir Dis, 145(3): 610-616.
Newly TJ, Stokes CR, & Bourne FJ (1980) Effects of feeding bacterial
lipopolysaccharide and dectran sulphate on the development of oral
tolerance to contact sensitizing agents. Immunology, 41: 617-621.
Newman LS & Kreiss K (1992) Nonoccupational beryllium disease
masquerading as sarcoidosis: identification by blood lymphocyte
proliferative response to beryllium. Am Rev Resp Dis, 145: 1212-1214.
Newman LS, Bobka C, Schumacher B, Daniloff E, Zhen B, Mroz MM, & King
TE Jr (1994) Compartmentalized immune response reflects clinical
severity of beryllium disease. Am J Resp Crit Care Med, 150: 135-142.
Newman Taylor AJ (1988) Occupational asthma. Postgrad Med J,
64: 505-510.
Newman Taylor AJ (1995) Environmental determinants of asthma. Lancet,
345(8945): 296-299.
Newman Taylor AJ, Venables KM, Durham S, Graneek BJ, & Topping MD
(1987) Acid anhydrides and asthma. Int Arch Allergy Appl Immunol, 82:
435-439.
Ng TP & Tan WC (1994a) Epidemiology of chronic (perennial) rhinitis in
Singapore: prevalence estimates, demographic variation and clinical
allergic presentation. Ann Acad Med Singap, 23: 83-88.
Ng TP & Tan WC (1994b) Epidemiology of allergic rhinitis and its
associated risk factors in Singapore. Int J Epidemiol, 23(3): 553-558.
Niebel G (1995) [Behavioural medicine of chronic skin disease.] Bern,
Göttingen, Toronto, Seattle, Hogrefe & Huber Publishers.
Nielsen NH & Menné T (1992) Allergic contact sensitization in an
unselected Danish population -- the Glostrup allergy study, Denmark.
Acta Dermatol Venereol, 72: 456-460.
Nielsen NH & Menné T (1993) Nickel sensitization and ear piercing in
an unselected Danish population -- The Glostrup allergy study,
Denmark. Contact Dermatitis, 29: 16-21.
Niermann H (1964) [Dermatology in twins.] Berlin, Heidelberg, New
York, Springer Verlag, 108 pp.
Niestijl Jansen JJ, Kardinaal AFM, Huijbers G, Vlieg-Boerstra BJ,
Martens BPM, & Ockhuizen T (1994) Prevalence of food allergy and
intolerance in the adult Dutch population. J Allergy Clin Immunol, 93:
446-456.
NIH (National Institutes of Health) (1991) National Asthma Education
Program Expert Panel -- Guidelines for the diagnosis and management of
asthma. Bethesda, Maryland, National Heart, Lung and Blood Institute,
72 pp (NIH Publication No. 91-3042A).
Nilsson B & Kristofferson A (1989) Zimelidine: Febrile reaction and
peripheral neuropathy. In: Kammüller ME, Bloksma N, & Seinen W ed.
Autoimmunity and toxicology. Amsterdam, Oxford, New York, Elsevier
Science Publishers, pp 183-214.
Ninan TK & Russell G (1992) Respiratory symptoms and atopy in Aberdeen
schoolchildren: Evidence from two surveys 25 years apart. Br Med J,
304: 873-875.
NIOSH (1997) NIOSH alert: Preventing allergic reactions to natural
rubber latex in the workplace. Cincinnati, Ohio, National Institute
for Occupational Safety and Health, 11 pp.
Nitta H, Sato T, Nakai S, Maeda K, Aoki S, & Ono M (1993) Respiratory
health associated with exposure to automobile exhaust. I. Results of
cross-sectional studies in 1979, 1982, and 1983. Arch Environ Health,
48: 53-58.
Noelle RJ, Daum J, Bartlett WC, McCann J, & Shepherd DM (1990) Cognate
interactions between helper T-cells and B-cells. V. Reconstitution of
helper T-cell function using purified plasma membranes from activated
Th1 and Th2 helper T-cells and lymphokines. J Immunol, 146(4):
1118-1124.
Norback D, Bjornsson E, Janson C, Widstrom J, & Boman G (1995)
Asthmatic symptoms and volatile organic compounds, formaldehyde, and
carbon dioxide in dwellings. Occup Environ Med, 52(6): 388-395.
Nordman H, Keskinen H, & Tuppurainen M (1985) Formaldehyde asthma
-- rare or overlooked? J Allergy Clin Immunol, 75: 91-99.
Nussler AK & Billiar TR (1993) Inflammation, immunoregulation, and
inducible nitric oxide synthase. J Leuk Biol, 54(2): 171-178.
Nyren O, Yin L, Josefsson S, McLaughlin JK, Blot WJ, Engqvist M,
Hakelius L, Boice JD Jr, & Adami HO (1998) Risk of connective tissue
disease and related disorders among women with breast implants: a
nation-wide retrospective cohort study in Sweden. Br Med J, 316(7129):
417-422.
O'Brien IM, Harries MG, Burge PS, & Pepys J (1979) Toluene
di-isocyanate induced asthma. Reactions to TDI, MDI, HDI and
histamine. Clin Allergy, 19: 1-6.
O'Byrne PM, Dolovich J, & Hargreave FE (1987) Late asthmatic
responses. Am Rev Respir Dis, 134: 740-751.
Odom RB & Maibach HI (1976) Contact urticaria: a different contact
dermatitis. Cutis, 18(5): 672-676.
O'Donnell TV (1995) Asthma and respiratory problems -- a review. Sci
Total Environ, 163(1-3): 137-145.
OECD (1992) Guideline 406 for testing chemicals. Paris, Organisation
for Economic Cooperation and Development.
Oettigen HC, Martin TR, Wynshaw-Boris A, Deng C, Drazen JM, & Leder P
(1994) Active anaphylaxis in IgE-deficient mice. Nature (Lond), 370:
367-370.
Ogawa M, Berger PA, McIntyre OR, & Clendenning WE (1971) IgE in atopic
dermatitis. Arch Dermatol, 103: 575.
Ohashi PS, Oehnen S, Buerki K, Pircher H, Ohashi CT, Odermatt B,
Malissen B, Zinkernagel RM, & Hengartner H (1991) Ablation of
"tolerance" and induction of diabetes by virus infection in viral
antigen transgenic mice. Cell, 65: 305-317.
Ollerenshaw S & Woolcock AJ (1992) Characteristic of inflammation in
biopsies from large airways of subjects with asthma and subjects with
chronic airflow limitation. Am Rev Respir Dis, 145: 922-927.
Ollier S & Davies RJ (1994) A report of the relationship between
allergen load, primary immunologic sensitization and the expression of
allergic disease. Respir Med, 88: 407-415.
Oostendorp RAJ, Meijer CJLM, & Scheper RJ (1993) Immunosuppression by
retroviral-envelope-related peptides, and their role in non-retroviral
human disease. Crit Rev Oncol Hematol, 14: 189-206.
Oosterlee A, Drijver M, Lebret E, & Brunekreef B (1996) Chronic
respiratory symptoms in children and adults living along streets with
high traffic density. Occup Environ Med, 53: 241-247.
Ortolani C, Ispano M, Pastorello EA, Ansaloni R, & Magri GC (1989)
Comparison of results of skin prick tests (with fresh foods and
commercial food extracts) and RAST in 100 patients with oral allergy
syndrome. J Allergy Clin Immunol, 83: 683-690.
Osebold JW, Gershwin LJ, & Zee YC (1980) Studies on the enhancement of
allergic lung sensitization by inhalation of ozone and sulfuric acid
aerosol. J Environ Pathol Toxicol, 3: 221-234.
Osterland CK (1994) Laboratory diagnosis and monitoring in chronic
systemic autoimmune diseases. Clin Chem, 40(11B): 2146-2153.
Ostro BD, Lipsett MJ, Wiener MB, & Selner JC (1991) Asthmatic
responses to airborne acid aerosols. Am J Public Health, 81(6):
694-702.
Oswald IP, Gazzinelli RT, Sher A, & James SL (1992) IL-10 synergizes
with IL-4 and transforming growth factor-beta to inhibit macrophage
cytotoxic activity. J Immunol, 148(11): 3578-3582.
Paggiaro P, Bacci E, Paoletti P, Bernard P, Dente FL, Marchetti G,
Talini D, Menconi GF, & Giuntini C (1990) Bronchoalveolar lavage and
morphology of the airways after cessation of exposure in asthmatic
subjects sensitised to toluene di-isocyanate. Chest, 98(3): 536-542.
Park HS, Yu HJ, & Jung KS (1994) Occupational asthma caused by
chromium. Clin Exp Allergy, 24: 676-681.
Parker D, Sommer G, & Turk JL (1975) Variation in guinea pig
responsiveness. Cell Immunol, 18: 233-238.
Parronchi P, Macchia D, Piccini M-P, Biswas P, Simonelli C, Maggi E,
Ricci M, Ansari A, & Romagnani S (1991) Allergen-and bacterial
antigen-specific T-cell clones established from atopic donors show a
different profile of cytokine production. Proc Natl Acad Sci (USA),
88: 4538-4542.
Pastorello E, Stocchi L, Bigi A, Pravettoni V, Schilke ML, Valente D,
& Zanussi C (1989) Value and limits of diagnostic tests in food
hypersensitivity. Allergy, 44(suppl): 151-158.
Pastorello EA, Incorvaia C, & Ortolani C (1995) The mouth and pharynx.
In: Ortolani C ed. Atlas of mechanisms in adverse reactions to food.
Allergy, 50(20): 41-44.
Pauli G, de Blay F, Bessort JC, & Dietermann A (1992) The association
between respiratory allergies and food hypersensitivities. ACI News,
4: 43-47.
Pauluhn J (1996) Predictive testing for respiratory sensitisation.
Toxicol Lett, 86(2-3): 177-185.
Pauluhn J & Eben A (1991) Validation of a non-invasive technique to
assess immediate or delayed onset of airway hypersensitivity in
guinea-pigs. J Appl Toxicol, 11: 423-431.
Pauluhn J & Mohr U (1994) Assessment of respiratory hypersensitivity
in guinea pigs sensitized to diphenylmethane-4,4-diisocyanate (MDI)
and challenged with MDI acetylcholine or MDI albumin conjugate.
Toxicology, 92: 53-74.
Payne MP & Walsh PT (1994) Structure-activity relationships for skin
sensitization potential: development of structural alerts for use in
knowledge-based toxicity prediction systems. J Chem Inf Comput Sci,
34(1): 154-161.
PEACE(1998) Study of pollution effects on asthmatic children in Europe
(PEACE). Eur Respir Rev, 8: 1-130.
Pearce N, Weiland S, Keil U, Langridge P, Anderson HR, Strachan D,
Bauman A, Young L, Gluyas P, Ruffin D, Crane J, & Beasley R (1993)
Self-reported prevalence of asthma symptoms in children in Australia,
England, Germany and New Zealand: an international comparison using
the ISAAC protocol. Eur Respir J, 6: 1455-1461.
Peat JK (1996) Prevention of asthma. Eur Respir J, 9: 1545-1555.
Peat J., Woolcock A, Leeder SR, & Blackburn CR (1980) Asthma and
bronchitis in Sydney schoolchildren: II. The effect of social factors
and smoking on prevalence. Am J Epidemiol, 111(6): 728-735.
Peat J., van den Berg RH, Green WF, Mellis CM, Leeder SR & Woolcock A
(1994) Changing prevalence of asthma in Australian children. Br Med J,
308: 1591-1596.
Peden DB, Carter J, Dailey LA, & Devlin R (1994) The effect of ozone
on house dust mite allergen-induced nasal inflammation in asthmatics.
Am J Respir Crit Care Med, 149: A154 (abstract).
Pelletier L, Castedo M, Bellon B, & Druet P (1994) Mercury and
autoimmunity. In: Dean JH, Luster MI, Munson AE, & Kimber I ed.
Immunotoxicology and immunopharmacology, 2nd ed. New York, Raven
Press, pp 539-552.
Pepys J (1994) "Atopy": a study in definition. Allergy, 49(6):
397-399.
Pepys J, Pickering CAC, & Hughes EG (1972) Asthma due to inhaled
chemical agents -- complex salts of platinum. Clin Allergy, 2:
391-396.
Peters A, Dockery DW, Heinrich J, & Watchman HE (1997) Short-term
effects of particulate air pollution on respiratory morbidity in
asthmatic children. Eur Resp J, 10(4): 872-879.
Pfeiffer C, Murray J, Madri J, & Bottomly K (1991) Selective
activation of Th1- and Th2-like cells in vivo: response to human
collagen IV. Immunol Rev, 123: 65-84.
Philen RM & Posada M (1993) Toxic oil syndrome and
eosinophilia-myalgia syndrome: May 8-10, 1991, World Health
Organization Meeting report. Semin Arthritis Rheum, 23: 104-124.
Picker LJ, Treer JR, Ferguson-Darnell B, Collins PA, Bergstresser PR,
& Terstappen LWMM (1993) Control of lymphocyte recirculation in man:
II. Differential regulation of the cutaneous lymphocyte associated
antigen, a tissue-selective homing receptor for skin-homing T cells. J
Immunol, 150: 1122-1136.
Pinkston P, Bitterman PB, & Crystal RG (1984) Interleukin-2 in the
alveolitis of beryllium-induced lung disease. Am Rev Respir Dis,
129(411): A161 (abstract).
Pistoor FAM., Kapsenberg ML, Bos JD, Meinardi MMHM, von Blomberg BME,
& Scheper RJ (1995) Cross-reactivity of human nickel-reactive T
lymphocyte clones with copper and palladium. J Invest Dermatol, 105:
92-95.
Planchon SM, Martins CAP, Guerrant RL, & Roche J. (1994) Regulation of
intestinal epithelial barrier function by TGF-beta-1. J Immunol, 153:
5730-5739.
Platts-Mills TA (1994) How environment affects patients with allergic
disease: indoor allergens and asthma. Ann Allergy, 72: 381-384.
Platts-Mills TA & Chapman MD (1987) Dust mites: immunology, allergic
disease, and environmental control. J Allergy Clin Immunol, 80:
755-775.
Platts-Mills TAE, Chapman MD, Mitchell B, Heymann PW, & Deuell B
(1991) Role of inhalant allergens in atopic eczema. In: Ruzicka T,
Ring J, & Przybilla B ed. Handbook of atopic eczema. Berlin,
Heidelberg, New York, Springer Verlag, pp 192-203.
Platt-Mills TA, Sporik RB, Chapman MD, & Heymann PW (1995) The role of
indoor allergens in asthma. Allergy, 50(suppl 22): 5-12.
Podmore P (1995) Shoes. In: Rycroft RJG, Menné T, Frosch PJ, & Benezra
C ed. Textbook of contact dermatitis, 2nd ed. Berlin, Heidelberg, New
York, Springer Verlag, pp 516-527.
Polak L (1980) Immunological aspects of contact sensitivity: An
experimental study. Monogr Allergy, 15: 1-170.
Pollart SM, Chapman MD, Fiocco GP, Rose G, & Platts-Mills TA (1989)
Epidemiology of acute asthma: IgE antibodies to common inhalant
allergens as a risk factor for emergency room visits. J Allergy Clin
Immunol, 83: 875-882.
Pope CA III, Schwarz J, & Ransom MR (1992) Daily mortality and PM10
pollution in Utah Valley. Arch Environ Health, 47: 211-217.
Pope CA III, Bates DV, & Raizenne ME (1995) Health effects of
particulate air pollution: Time for reassessment. Environ Health
Perspect, 103(5): 472-480.
Posselt AM, Barker CF, Friedman AL, & Naji A (1992) Prevention of
autoimmune diabetes in the BB rat by intrathymic islet transplantation
at birth. Science, 256: 1321-1324.
Potter DW & Wederbrand KS (1995) Total IgE antibody in BALB/c mice
after dermal exposure to chemicals. Fundam Appl Toxicol, 26: 127-135.
Prausnitz C & Kustner H (1921) [Studies concerning sensitivity.] Zent.
bl Bakteriol I Orig, 86: 160-169
Prentice RL & Thomas D (1993) Methodologic research needs in
environmental epidemiology: Data analysis. Environ Health Perspect,
101(suppl 4): 39-48.
Prud'Homme GJ, Parfrey NA, & Vanier LE (1991) Cyclosporine-induced
autoimmunity and immune hyperreactivity. Autoimmunity, 9: 345-356.
Przybilla B & Ring J (1990) Food allergy and atopic eczema. Semin
Dermatol, 9: 220-225.
Przybilla B, Eberlein-König B, & Rueff F (1994) Practical management
of atopic eczema. Lancet, 343: 1342-1346.
Punnonen J & de Vries JE (1994) IL-13 induces proliferation, Ig
isotype switching, and Ig synthesis by immature human fetal B cells. J
Immunol, 152: 1094-1102.
Punnonen J, Aversa G, Cocks BG, McKenzie AN, Menon S, Zurawski G, de
Waal Malefyt R, & de Vries JE (1993) Interleukin-13 induces
interleukin 4-independent IgG4 and IgE synthesis and CD23 expression
by human B cells. Proc Natl Acad Sci (USA), 90(8): 3730-3734.
Raaschou Nielsen O, Nielsen ML, & Gehl J (1995) Traffic-related air
pollution: exposure and health effects in Copenhagen street cleaners
and cemetery workers. Arch Environ Health, 50(3): 207-213.
Radl J, Valentijn RM, Haaijman JJ, & Paul LC (1985) Monoclonal
gamma-pathies in patients undergoing immunosuppressive treatment after
renal transplantation. Clin Immunol Immunopathol, 37: 98-102.
Rajka G (1990) Essential aspects of atopic eczema. Berlin, Heidelberg,
New York, Springer Verlag, 261 pp.
Rastogi SK, Gupta BN, Husain T, Mathur N, Pangtey BS, & Garg N (1992)
Respiratory symptoms and ventilatory capacity in metal polishers. Hum
Exp Toxicol, 11(6): 466-472.
Rattray NJ, Botham PA, Hext PM, Woodcock DR, Fielding I, Dearman RJ, &
Kimber I (1994) Induction of respiratory hypersensitivity to
diphenylmethane-4,4-diisocyanate in guinea pigs. Influence of route of
exposure. Toxicology, 88: 15-30.
Raulf M & Konig W (1991) Modulation of leukotriene generation from
human polymorphonuclear granulocytes by polychlorinated biphenyls
(PCB). Immunology, 73(4): 485-490.
Ravetch JV (1994) Fc receptors: rubor redux. Cell, 78: 553-560
Ravetch JV (1997) Fc receptors. Curr Opin Immunol, 9: 121-125.
Rea TH (1979) Quantitative enhancement of dinitrochlorobenzene
responsivity in women receiving oral contraceptives. Arch Dermatol,
115(3): 361-362.
Reijula K & Patterson R (1994) Occupational allergies in Finland in
1981-91. Allergy Proc, 15(3): 163-168.
Reitamo S, Visa K, Kahonen K, Kayhko K, Stubb S, & Salo OP (1986)
Eczematous reactions in atopic patients caused by epicutaneous testing
with inhalant allergens. Br J Dermatol, 114: 303-309.
Renstrom A, Malmberg P, Larsson K, Sundblad BM, & Larsson PH (1994)
Prospective study of laboratory-animal allergy: factors predisposing
to sensitization and development of allergic symptoms. Allergy, 49(7):
548-552.
Richardson BC, Liebling MR, & Hudson JL (1990) CD4+ cells treated with
DNA methylation inhibitors induce autologous B cell differentiation.
Clin Immunol Immunopathol, 55: 368-381.
Richardson B, Powers D, Hooper F, Yung RL, & O'Rourke K (1994)
Lymphocyte function-associated antigen 1 overexpression and T cell
autoreactivity. Arthritis Rheum, 37: 1363-1372.
Ridings JE, Barrat MD, Cary R, Earnshaw CG, Eggington CE, Ellis MK,
Judson PN, Langowski JJ, Marchant CA, Payne MP, Watson WP, & Yih TD
(1996) Computer prediction of possible toxic action from chemical
structure: an update on the DEREK system. Toxicology, 106: 267-279.
Riedel F, Kramer M, Scheibenbogen C, & Rieger CH (1988) Effects of
SO2 exposure on allergic sensitization in the guinea pig. J Allergy
Clin Immunol, 82: 527-534.
Riedler J, Reade T, Dalton M, Holst D, & Robertson C (1994) Hypertonic
saline challenge in an epidemiologic survey of asthma in children. Am
J Respir Crit Care Med, 150: 1632-1639.
Ring J (1988) [Applied allergology], 2nd ed. Munich, MMW Medizin
Verlag GmbH, pp 85-86 (in German).
Ring J (1991) Atopy: Conditions, disease or syndrome? In: Ruzicka T,
Ring J, & Przybilla B ed. Handbook of atopic eczema. Berlin,
Heidelberg, New York, Springer Verlag, pp 3-8.
Ring J & Thomas P (1989) Histamine and atopic eczema. Acta Dermatol
Venereol, 144: 70-77.
Ring J, Sedlmeier F, von der Helm D, Mayr T, Walz U, Ibel H, Przybilla
B, Reimann HJ, & Dorsch W (1988) Histamine and allergic disease. In:
Ring J & Burg G ed. New trends in allergy. Berlin, Heidelberg, New
York, Springer Verlag, pp 44-77.
Ring J, Bieber T, Vieluf D, Kunz B, & Przybilla B (1991a) Atopic
eczema, Langerhans cells and allergy. Int Arch Allergy Immunol, 94:
194-201.
Ring J, Ruzicka T, & Przybilla B (1991b) The pathophysiology of atopic
eczema. In: Ruzicka T, Ring J, & Przybilla B ed. Handbook of atopic
eczema. Berlin, Heidelberg, New York, Springer Verlag, pp 330-335.
Ring J, Abeck D, & Neuber K (1992) Atopic eczema: role of
microorganisms on the skin surface. Allergy, 47: 265-269.
Ring J, Behrendt H, Schäfer T, Vieluf D, & Krämer U (1995) Impact of
air pollution on allergic diseases: Clinical and epidemiologic
studies. In: Johansson SGO ed. Progress in allergy and clinical
immunology. Bern, Göttingen, Toronto, Seattle, Hogrefe & Huber
Publishers, vol 3, pp 174-179.
Ring J, Brockow K, & Abeck D (1996) The therapeutic concept of patient
management in atopic eczema. Allergy, 51: 206-215.
Ritz HL, Conner DS, & Sauter ED (1975) Contact sensitization of
guinea-pigs with unsaturated and halogenated sultones. Contact
Dermatitis, 1: 349-359.
Ritz HL, Evans BL, Bruce RD, Fletcher ER, Fisher GL, & Sarlo K (1993)
Respiratory and immunological responses of guinea pigs to
enzyme-containing detergents: A comparison of intratracheal and
inhalation modes of exposure. Fundam Appl Toxicol, 21: 31-37.
Roberts DW & Basketter DA (1990a) A quantitative structure
activity/dose relationship for contact allergenic potential of alkyl
group transfer agents. Toxicology in vitro, 4(4/5): 686-687.
Roberts DW & Basketter DA (1990b) A quantitative structure
activity/dose response relationship for contact allergic potential of
alkyl group transfer agents. Contact Dermatitis, 23: 331-335.
Roberts SA, Reinhardt MC, Paganelli R, & Levinsky RJ (1981) Specific
antigen exclusion and non-specific facilitation of antigen entry
across the gut in rats allergic to food proteins. Clin Exp Immunol,
45: 131-136.
Robertson CF, Heycock E, Bishop J, Nolan T, Olinsky A, & Phelan PD
(1991) Prevalence of asthma in Melbourne schoolchildren: Changes over
26 years. Br Med J, 302: 1116-1118.
Robinson DS (1998) T cell co-stimulation: a potential therapeutic
target in asthma? Clin Exp Allergy, 28: 788-790.
Roche WR, Beasley R, Williams JH, & Holgate ST (1989) Subepithelial
fibrosis in the bronchi of asthmatics. Lancet, 1: 520-523.
Röcken M, Racke M, & Shevach EM (1996) IL-4-induced immune deviation
as antigen-specific therapy for inflammatory autoimmune disease.
Immunol Today, 17: 225-231.
Roed-Petersen J (1989) A new glove material protective against epoxy
and acrylate monomer. In: Frosch PJ, Dooms-Goossens A, Lachapelle J-M,
Rycroft RJG, & Scheper RJ ed. Current topics in contact dermatitis.
Berlin, Heidelberg, New York, Springer Verlag, pp 603-606.
Roitt IM, Brostoff J, & Male DK ed. (1998) Immunology, 5 th ed. St.
Louis, Missouri, C.V. Mosby & Co., 424 pp.
Romagnani S (1991) Human Th1 and Th2 subsets: doubt no more.
Immmunol Today, 12(8): 256-257.
Romagnani S (1992a) Induction of Th1 and Th2 responses: A key role for
the "natural" immune response? Immunol Today, 13(10): 379-381.
Romagnani S (1992b) Human TH1 and TH2 subsets: Regulation of
differentiation and role in protection and immunopathology. Int Arch
Allergy Immunol, 98: 279-285.
Romanski B (1987) The pathology of food allergy studied by gastric
allergen challenge. In: Brostoff J & Challacombe SJ ed. Food allergy
and intolerance. London, Baillière Tindall, pp 917-931.
Romanski B (1989) The pathology of food allergy studied by gastric
allergen challenge. In: Paul WE ed. Fundamental immunology. New York,
Raven Press, p 41.
Rose G (1985) Sick individuals and sick populations. Int J Epidemiol,
14: 32-38.
Rose NR & Potter M (1995) The silicone controversy: towards a
resolution. Immunol Today, 16: 459-460.
Rosen FS (1987) Autoimmunity and immunodeficiency disease. In: Evered
D & Whelan J ed. Autoimmunity and autoimmune disease. New York,
Chichester, Brisbane, Toronto, John Wiley & Sons, pp 135-148 (Ciba
Foundation Symposium No. 129).
Rosenstreich DL, Eggleston P, Kattan M, Baker D, Slavin RG, Gergen P,
Mitchell H, McNiff-Mortimer K, Lynn H, Ownby D, & Malveaux F (1997)
The role of cockroach allergy and exposure to cockroach allergen in
causing morbidity among inner-city children with asthma. N Engl J Med,
336(19): 1356-1363.
Ross B, McCullough J, & Ownby DR (1991) Evidence of allergenic
cross-reactivity between banana and watermelon. J Allergy Clin
Immunol, 87: 274 (abstract).
Rossman MD, Kern JA, Elias JA, Cullen MR, Epsein PE, Preuss OP,
Markham TN, & Daniele RP (1988) Proliferative response of
bronchoalveolar lymphocytes to beryllium. Ann Intern Med, 108:
687-693.
Roth MD & Golub SH (1993) Human pulmonary macrophages utilize
prostaglandins and transforming growth factor beta 1 to suppress
lymphocyte activation. J Leuk Biol, 53(4): 366-371.
Rothman KJ (1993) Methodologic frontiers in environmental
epidemiology. Environ Health Perspect, 101(suppl 4): 19-21.
Rubin RL (1989) Autoimmune reactions induced by procainamide and
hydralazine. In: Kammüller M, Bloksma N, & Seinen W ed. Autoimmunity
and toxicology: Immune disregulation induced by drugs and chemicals.
Amsterdam, Oxford, New York, Elsevier Science Publishers, pp 119-150.
Rubin RL, Burlingame RW, Arnott JE, Totoritis MC, McNally EM, &
Johnson AD (1995) IgG but not other classes of anti-[(H2A-H2B)-DNA] is
an early sign of procainamide-induced lupus. J Immunol, 154:
2483-2493.
Ruffin J, Liu MYG, Sessions R, Banerjee S, & Banerjee UC (1986)
Effects of certain atmospheric pollutants (SO2, NO2, CO) on the
soluble amino acids, molecular weight and antigenicity of some
airborne pollen grains. Cytobios, 46: 119-129.
Ruzicka T, Ring J, & Przybilla B ed. (1991) Handbook of atopic eczema.
Berlin, Heidelberg, New York, Springer Verlag, 481 pp.
Rycroft RJG (1995) Occupational contact dermatitis. In: Rycroft RJG,
Menné T, Frosch PJ, & Benezra C ed. Textbook of contact dermatitis,
2nd ed. Berlin, Heidelberg, New York, Springer Verlag, pp 343-400.
Rycroft RJG, Menné T, Frosch PJ, & Benezra C ed. (1995) Textbook of
contact dermatitis, 2nd ed. Berlin, Heidelberg, New York, Springer
Verlag, 400 pp.
Rystedt I (1985) Atopic background in patients with occupational hand
eczema. Contact Dermatitis, 12(5): 247-254.
Saarinen UM & Kajosaari M (1995) Breastfeeding as prophylaxis against
atopic disease: Prospective follow-up study until 17 years old.
Lancet, 346(8982): 1065-1069.
Saihan EM, Burton JL, & Heaton KW (1978) A new syndrome with
pigmentation, scleroderma, gynaecomastia, Raynaud's phenomenon and
peripheral neuropathy. Br J Dermatol, 99: 437-440.
Saint-Remy J-MR (1997) Epitope mapping, a new method for biological
evaluation and immunotoxicology. Toxicology, 119: 77-81.
Sakaguchi S & Sakaguchi N (1989) Organ-specific autoimmune disease
induced in mice by elimination of T cell subsets: V. Neonatal
administration of cyclosporine A causes autoimmune disease. J Immunol,
142: 471-480.
Sakaguchi S & Sakaguchi N (1990) Thymus and autoimmunity: capacity of
the normal thymus to produce pathogenic self-reactive T cells and
conditions required for their induction of autoimmune disease. J Exp
Med, 172: 537-545.
Salome CM, Peat J., Britton WJ, & Woolcock A (1987) Bronchial
hyperresponsiveness in two populations of Australian schoolchildren:
I. Relation to respiratory symptoms and diagnosed asthma. Clin
Allergy, 17: 271-281.
Saltini C, Winestock K, Kirby M, Pinkston P, & Crysal RG (1989)
Maintenance of the alveolitis in patients with chronic beryllium
disease by beryllium-specific helper T-cells. N Engl J Med, 320:
1103-1109.
Samet JM, Lambert WE, Skipper BJ, Cushing AH, Hunt WC, Young SA,
McLaren LC, Schwab M, & Spengler JD (1993) Nitrogen dioxide and
respiratory illnesses in infants. Am Rev Respir Dis, 148: 1258-1265.
Sampson HA (1982) The immunopathogenic role of food hypersensitivity
in atopic dermatitis. Acta Dermatol Venereol, 176(suppl): 34-37.
Sampson HA (1983) Role of immediate food hypersensitivity in the
pathogenesis of atopic dermatitis. J Allergy Clin Immunol, 71:
473-480.
Sampson HA & Albergo R (1984) Comparison of results of skin tests,
RAST, and double-blind, placebo-controlled food challenges in children
with atopic dermatitis. J Allergy Clin Immunol, 74: 26-33.
Sampson HA & Metcalfe DD (1992) Food allergies. J Am Med Assoc,
268(20): 2840-2844.
Sanchez-Guerrero J, Schur PH, Sergent JS, & Liang MH (1994) Silicone
breast implants and rheumatic diseases. Arthritis Rheum, 37: 158-168.
Sanchez-Guerrero J, Colditz GA, Karlson EW, Hunter DJ, Speizer FE, &
Liang MH (1995) Silicone breast implants and the risk of
connective-tissue diseases and symptoms. N Engl J Med, 332(25):
1666-1670.
Sanderson CJ (1990) Eosinophil differentiation factor (interleukin-5).
Immunol Ser, 49: 231-256.
Sanderson DM & Earnshaw CG (1991) Computer prediction of possible
toxic action from chemical structure; The DEREK system. Hum Exp
Toxicol, 10: 261-273.
Sandford A, Shirakawa T, Moffatt MF, Daniels SE, Ra C, Faux JA, Young
RP, Nakamura Y, Lathrop GM, Cookson WOCM, & Hopkin JM (1993)
Localization of atopy and beta subunit of high-affinity IgE receptor
(Fc-epsilon-R1) on chromosome 11q. Lancet, 341: 332-334.
Sarlo K & Clark ED (1992) A tier approach for evaluating the
respiratory allergenicity of low molecular weight chemicals. Fundam
Appl Toxicol, 18: 107-114.
Sarlo K & Karol MH (1994) Guinea pig predictive tests for respiratory
allergy. In: Dean JH, Luster MI, Munson AE, & Kimber I ed.
Immunotoxicology and immunopharmacology, 2nd ed. New York, Raven
Press, pp 703-720.
Sarlo K, Ritz HL, Fletcher ER, Schrotel KR, & Clark ED (1997)
Proteolytic detergent enzymes enhance the allergic antibody responses
of guinea pigs to nonproteolytic detergent enzymes in a mixture:
implications for occupational exposure. J Allergy Clin Immunol, 100:
480-487.
Satoh T, Kramarik JA, Tollerud DJ, & Karol MH (1995) A murine model
for assessing the respiratory hypersensitivity potential of chemical
allergens. Toxicol Lett, 78: 57-66.
Saval P, Fuglsang G, Madsen C, & Osterballe O (1993) Prevalence of
atopic disease among Danish school children. J Pediatr Allergy
Immunol, 4: 117-122.
Savilahti E & Kuitunen M (1992) Allergenicity of cow milk proteins. J
Pediatr, 121: S12-S20.
Savonius B, Keskinen H, Tuppurainen M, & Kanerva L (1993) Occupational
respiratory disease caused by acrylates. Clin Exp Allergy, 23(5):
416-424.
Schachter J & Higgins MW (1976) Median age at onset of asthma and
allergic rhinitis in Tecumseh, Michigan. J Allergy Clin Immunol,
57(4): 342-351.
Schäfer T & Ring J (1997) Epidemiology of allergic diseases. Allergy,
52(suppl 38): 14-22.
Schäfer T, Przybilla B, Überla K, Pöschl C, & Ring J (1994) Frequency
of atopic diseases in children and parental predisposition -- results
of an epidemiological survey. ACI News, 2(suppl): 170.
Scheinbart LS, Johnson MA, Gross LA, Edelstein SR, & Richardson BC
(1991) Procainamide inhibits DNA methyltransferase in a human T cell
line. J Rheumatol, 18: 530-534.
Scheinman PL (1996) Allergic contact dermatitis to fragrances: A
review. Am J Contact Dermatitis, 7: 65-76.
Scheper RJ & von Blomberg BME (1992) Cellular mechanisms in allergic
contact dermatitis. In: Rycroft RJG, Menné T, Frosch PJ, & Benezra C
ed. Textbook of contact dermatitis. Berlin, Heidelberg, New York,
Springer Verlag, pp 11-27.
Scheper RJ, von Blomberg M, Boerrigter GH, Bruynzeel DP,
von Dinther-Janssen ACHM, & Vos A (1983) Induction of immunological
memory in the skin. Role of local T cell retention. Clin Exp Immunol,
51: 141-148.
Schleimer RP & Bochner BS (1998) The role of adhesion molecules in
allergic inflammation and their suitability as targets of antiallergic
therapy. Clin Exp Allergy, 28(suppl 39): 15-23.
Schlipköter HW, Krämer U, Behrendt H, Dolgner R, Stiller-Winkler R,
Ring J, & Willer HJ (1992) Impact of air pollution on children's
health: Results from Saxony-Anhalt and Saxony as compared to
Northrhine-Westphalia. In: Health and ecological effects -- Critical
issues in the global environment. Pittsburgh, Pennsylvania, Air and
Waste Management Association, vol 5 (Publication IU-A 2103).
Schnyder UW (1960) [Neurodermitis, asthma, rhinitis: A genetic
allergological study.] Basel, Karger, 106 pp.
Schon-Hegrad MA, Oliver J, McMenamin PG, & Holt PG (1991) Studies on
the density, distribution and surface phenotype of intraepithelial
class II MHC antigen (Ia)-bearing dendritic cells (DC) in the
conducting airways. J Exp Med, 173(6): 1345-1356.
Schönrich G, Kalinke U, Momburg F, Malissen M, Schmitt-Verhulst AM,
Malissen B, Hämmerling GJ, & Arnold B (1991) Down-regulation of T cell
receptors on self-reactive T cells as a novel mechanism for
extrathymic tolerance induction. Cell, 65: 293-304.
Schöpf E, Mueller JM, & Ostermann T (1995) [Significance of adjuvant
basis therapy in chronic relapsing skin disease.] Hautarzt, 44:
451-454 (in German).
Schuhmann D, Kubicka-Muranyi M, Mirtschewa J, Gunther J, Kind P, &
Gleichmann E (1990) Adverse reactions to gold: I. Chronic treatment
with a Au(I) drug sensitizes mouse T cells not to Au(I) but to Au(III)
and induces autoantibody formation. J Immunol, 145: 2132-2139.
Schulz O, Sutton BJ, Beavil RL, Shi J, Sewell HF, Gould HJ, Laing P, &
Shakib F (1997) Cleavage of the low-affinity receptor for human IgE
(CD23) by a mite cysteine protease: nature of the cleaved fragment in
relation to the structure and function of CD23. Eur J Immunol, 27:
584-588.
Schulz O, Sewell HF, & Shakib F (1998) Proteolytic cleavage of CD25,
the a subunit of the human T cell interleukin 2 receptor, by Der p 1,
a major mite allergen with cysteine protease activity. J Exp Med, 18:
271-275.
Schultz Larsen F (1991) Genetic aspects of atopic eczema. In: Ruzicka
T, Ring J, & Przybilla B ed. Handbook of atopic eczema. Berlin,
Heidelberg, New York, Springer Verlag, pp 15-26.
Schultz-Larsen F (1993) The epidemiology of atopic dermatitis. Monogr
Allergy, 31: 9-28.
Schultz Larsen F & Hanifin JM (1992) Secular change in the occurrence
of atopic dermatitis. Acta Dermatol Venereol, 176(suppl): 7-12.
Schultz-Larsen F, Holm NV, & Hennigsen K (1986) Atopic dermatitis: A
genetic-epidemiology study in a population-based twin sample. J Am
Assoc Dermatol, 15: 487-494.
Schultze-Werninghaus G, Roesch A, Wilhelms OH, Gonsior E, & Meir Sydow
J (1978) [Bronchial asthma due to occupational allergy of immediate
type (I) to platinum salts.] Dtsch Med Wochenschr, 103: 972-975 (in
German).
Schutz-Kiss D, Popp W, Wagner C, Reiser K, Havelec L, & Zwick H (1995)
[Sensitization to inhaled allergens in the Vienna population.] Wien
Klin Wochenschr, 107(11): 331-335 (in German).
Schuurman H-J, Van Loveren H, Rozing J, & Vos JG (1992) Chemicals
trophic for the thymus: risk for immunodeficiency and autoimmunity.
Int J Immunopharmac, 14: 369-375.
Schwartz J & Morris R (1995) Air pollution and hospital admissions for
cardiovascular disease in Detroit, Michigan. Am J Epidemiol, 142:
23-35.
Schwartz J & Weiss ST (1990) Dietary factors and their relation to
respiratory symptoms -- The second national health and nutrition
examination survey. Am J Epidemiol, 132(1): 67-76.
Schwartz HR, Nerurkar LS, Spies JR, Scanlon RT, & Bellanti JA (1980)
Milk hypersensitivity: RAST studies using new antigens generated by
pepsin hydrolysis of beta-lactoglobulin. Ann Allergy, 45: 242-245.
Scott P (1993) IL-12: Initiation cytokine for cell-mediated immunity.
Science, 260(5107): 496-497.
Seager J, Jamison DL, Wilson J, Hayward AR, & Soothill JF (1975) IgA
deficiency, epilepsy, and phenytoin treatment. Lancet, II: 632-635.
Sears MR, Herbison GP, Holdaway MD, Hewitt CJ, Flannery EM, & Silva PA
(1989) The relative risks of sensitivity to grass pollen, house dust
mite and cat dander in the development of childhood asthma. Clin Exp
Allergy, 19: 419-424.
Seaton A, Godden DJ, & Brown K (1994) Increase in asthma: a more toxic
environment or a more susceptible population? Thorax, 49: 171-174.
Seifert J, Ring J, & Brendel W (1974) Prolongation of skin allografts
after oral application of ALS in rats. Nature (Lond), 249(459): 776.
Seifert J, Ring J, Steininger J, & Brendel W (1977) Influence of the
immune response on the absorption of protein from the gut. Nutr Metab,
21(Suppl 1): 256-258.
Sell S (1987) Immune deficiency states. In: Immunology,
immunopathology, and immunity. Amsterdam, Oxford, New York, Elsevier
Science Publishers, 617 pp.
Senthilselvan A & Habbick BF (1995) Increased asthma hospitalizations
among registered Indian children and adults in Saskatchewan,
1970-1989. J Clin Epidemiol, 48(10): 1277-1283.
Sequeira J, Cesic D, Keser G, Bukelica M, Karanagnostis S, Khamashta
MA, & Hughes GRV (1993) Allergic disorders in systemic lupus
erythematosus. Lupus, 2: 187-192.
Serafini U (1997) Do infections protect against asthma and atopy?
Allergy, 52(9): 955-957.
Shaheen SO, Aaby P, Hall A, Barker DJP, Heyes CB, Shiell AW, &
Goudiaby A (1996) Measles and atopy in Guinea-Bissau. Lancet, 347:
1792-1796.
Shaw RA, Crane J, O'Donnell TV, Porteous LE, & Coleman ED (1990)
Increasing asthma prevalence in a rural New Zealand adolescent
population: 1975-89. Arch Dis Child, 65: 1319-1323.
Shaw RA, Crane J, & O'Donnell TV (1991) Asthma symptoms, bronchial
hyperresponsiveness and atopy in a Maori and European adolescent
population. NZ Med J, 104: 175-179.
Shaw RA, Crane J, O'Donnell TV, Lewis ME, Stewart B, & Beasley R
(1992a) The use of a videotaped questionnaire for studying asthma
prevalence: A pilot study among New Zealand adolescents. Med J Aust,
157(5): 311-314.
Shaw RA, Crane J, Pearce N, Burgess CD, Bremner P, Woodman K, &
Beasley R (1992b) Comparison of a video questionnaire with the IUATLD
written questionnaire for measuring asthma prevalence. Clin Exp
Allergy, 22: 561-568.
Shaw R, Woodman K, Crane J, Moyes C, Kennedy J, & Pearce N (1994) Risk
factors for asthma symptoms in Kawerau children. NZ Med J, 107:
387-391.
Shaw R, Woodman K, Ayson M, Dibdin S, Winkelmann R, Crane J, Beasley
R, & Pearce N (1995) Measuring the prevalence of bronchial
hyper-responsiveness in children. Int J Epidemiol, 24(3): 597-602.
Sherrill DL, Halonen M, & Burrows B (1994) Relationships between total
serum IgE, atopy, and smoking: a twenty-year follow-up analysis. J
Allergy Clin Immunol, 94: 954-962.
Shimizu H, Masunaga T, Ishiko A, Kikuchi A, Hashimoto T, & Nishikawa T
(1995) Pemphigus vulgaris and Pemphigus foilaceus sera show an
inversely graded pattern to extracellular regions of desmosomes in
different layers of human epidermis. J Invest Dermatol, 105: 153-159.
Shirakawa T, Enomoto T, Shimazu S, & Hopkin JM (1997) The inverse
association between tuberculin responses and atopic disorder. Science,
275: 77-79.
Sibbald B (1993) Epidemiology of allergic rhinitis. In: Burr ML ed.
Epidemiology of clinical allergy. Basel, Karger, pp 61-79 (Monograph
on Allergy Series).
Sibbald B (1997) Familial inheritance of asthma and allergy. In: Kay
AB ed. Allergy and allergic diseases. Oxford, London, Boston,
Blackwell Scientific Publications, pp 1177-1186.
Sibbald B & Rink E (1991a) Labelling of rhinitis and hayfever by
doctors. Thorax, 46: 378-381.
Sibbald B & Rink E (1991b) Epidemiology of seasonal and perennial
rhinitis: Clinical presentation and medical history. Thorax, 46:
895-901.
Sigurs N, Bjarnson R, Sigurbergsson F, Kjellman B, & Björkstén (1995)
Asthma and immunoglobulin E antibodies after respiratory syncitial
virus bronchiolitis: a prospective cohort study with matched controls.
Pediatrics, 95: 500-505.
Silman A & Hochberg MC (1996) Occupational and environmental
influences on scleroderma. Rheum Dis Clin North Am, 22(4): 737-749.
Silman A, Black CM, & Welsh KL (1996) Epidemiology, demographics,
genetics. In: Clements PJ & Furst DE ed. Systemic sclerosis.
Baltimore, Maryland, Williams and Wilkins, pp 23-49.
Silvennoinen-Kassinen S (1981) The specificity of nickel sulphate
reaction in vitro: A family study and a study of chromium-allergic
subjects. Scand J Immunol, 13: 231-235.
Silverman GA, Peri BA, & Rothberg RM (1982) Systemic antibody
responses of different species following ingestion of soluble protein
antigens. Dev Comp Immunol, 6: 747-752.
Singer PA & Theofilopoulos AN (1990) T-cell receptor Vb repertoire
expression in murine models of SLE. Immunol Rev, 118: 103-127.
Sinigaglia F (1994) The molecular basis of metal recognition by T
cells. J Invest Dermatol, 102: 398-401.
Smalley DL, Shanklin DR, Hall MF, Stevens MV, & Hanissian A (1995)
Immunologic stimulation of T lymphocytes by silica after use of
silicone mammary implants. FASEB J, 9: 424-427.
Smalley DL, Levine JJ, Shanklin DR, Hall MF, & Stevens MV (1997)
Lymphocyte response to silica among offspring of silicone breast
implant recipients. Immunobiology, 196: 567-574.
Smith IM (1983) Epidemiology and natural history of asthma, allergic
rhinitis and atopic dermatitis (eczema). In: Middleton E, Reed Cellis
EF, Adkinson NF, Junginger JW, & Busse WW ed. Allergy: Principles and
practice. St. Louis, Missouri, C.V. Mosby & Co., pp 771-804.
Song YH, Li Y, & Maclaren NK (1996) The nature of autoantigens
targeted in autoimmune endocrine diseases. Immunol Today, 17: 232-237.
Song Z, Casolaro V, Chen R, Georas SN, Monos D, & Ono SJ (1996)
Polymorphic nucleotides within the human IL-4 promoter that mediate
over expression of the gene. J Immunol, 156: 424-429.
Sonnhag C, Karlsson E, & Hed J (1979) Procainamide induced lupus
erythematosus like syndrome in relation to acetylator phenotype and
plasma levels of procainamide. Acta Med Scand, 206: 245-251.
Sparrow GP (1977) A connective tissue disorder similar to vinyl
chloride disease in a patient exposed to perchlorethylene. Clin Exp
Dermatol, 2: 17-22.
Speizer FE & Ferris BGJ (1973) Exposure to automobile exhaust: I.
Prevalence of respiratory symptoms and disease. Arch Environ Health,
26: 313-318.
Spier HW & Sixt I (1955) [Investigation of the dependence of eczema on
the thickness of the stratum corneum.] Hautarzt, 6: 152-159 (in
German).
Sporik R, Holgate ST, Platts Mills TA, & Cogswell JJ (1990) Exposure
to house-dust mite allergen (Der p I) and the development of asthma in
childhood: A prospective study. N Engl J Med, 323: 502-507.
Stadler JC & Karol MH (1985) Use of dose-response data to compare the
skin sensitizing abilities of dicyclohexylmethane-4,4'-diisocyanante
and picryl chloride in two animal species. Toxicol Appl Pharmacol, 78:
445-450.
Stadler JC & Loveless SE (1992) Guinea pigs exhibit extended latency
period for the development of sensitivity to an amine. Toxicologist,
12: 44 (abstract).
Stahl RE, Friedman-Kien A, Dubin R, Marmor M, & Zolla-Pazner S (1982)
Immunologic abnormalities in homosexual men. Am J Med, 73(2): 171-178.
Starzl TE, Nalesnik MA, Porter KA, Ho M, Iwatsuki S, Griffith BP,
Rosenthal JT, Hakala TR, Shaw BW Jr, Hardesty RL, Atchinson RW, Jaffe
R, & Bahnson HT (1984) Reversibility of lymphomas and
lymphoproliferative lesions developing under cyclosporin-steroid
therapy. Lancet, 8377: 583-587.
Steinman R, Hoffman L, & Pope M (1995) Maturation and migration of
cutaneous dendritic cells. J Invest Dermatol, 105: 2S-7S.
Sterk PJ, Fabbri LM, Quanjer PH, Cockcroft DW, O'Byrne PM, Anderson
SD, Juniper EF, & Malo JL (1993) Airway responsiveness: Standardized
challenge testing with pharmacological, physical and sensitizing
stimuli in adults. Report of Working Party on Standardization of Lung
Function Tests, European Community for Steel and Coal. Official
Statement of the European Respiratory Society. Eur Respir J,
16(suppl): 53-83.
Stewart GA (1994) Molecular biology of allergens. In: Busse WW &
Holgate ST ed. Asthma and rhinitis. Oxford, London, Boston, Blackwell
Scientific Publications, pp 898-932.
Stiller-Winkler R, Radaszkiewicz T, & Gleichmann E (1988)
Immunopathological signs in mice treated with mercury compounds-I.
Identification by the popliteal lymph node assay of responder and
non-responder strains. Int J Immunopharmacol, 10: 475-484.
Stites DP & Terr AI (1991) Basic and clinical immunology. London,
Appleton & Lange, p 797.
Stoddard JJ & Miller T (1995) Impact of parental smoking on the
prevalence of wheezing respiratory illness in children. Am J
Epidemiol, 141: 96-102.
Stokes CR, Newly JR, Huntley JH, Patel D, & Bourne FJ (1979) The
immune response of mice to bacterial antigens given by mouth.
Immunology, 38: 497-502.
Stokes CR, Newly TJ, & Bourne FJ (1983a) The influence of oral
immunization on local and systemic immune response to heterologous
antigens. Clin Exp Immunol, 52: 399-406.
Stokes CR, Swarbrick ET, & Soothill JF (1983b) Genetic differences in
immune exclusion and partial tolerance to ingested antigens. Clin Exp
Immunol, 52(3): 678-684.
Strachan DP (1988) Damp housing and childhood asthma: validation of
reporting of symptoms. Br Med J, 297: 1223-1226.
Strachan DP (1989) Hay fever, hygiene, and household size. Br Med J,
299: 1259-1260.
Strachan DP (1995) Epidemiology of hay fever: towards a community
diagnosis. Clin Exp Allergy, 25(4): 296-303.
Strachan DP (1996) Socioeconomic factors and the development of
allergy. Toxicol Lett, 86: 199-203.
Strachan DP & Anderson HR (1992) Trends in hospital admission rates
for asthma in children. Br Med J, 304: 819-820.
Strachan DP & Carey IM (1995) Home environment and severe asthma in
adolescents: a population based case-control study. Br Med J, 311:
1053-1056.
Strachan DP, Golding J, & Anderson HR (1990) Regional variations in
wheezing illness in British children: effect of migration during early
childhood. J Epidemiol Community Health, 44: 231-236.
Strachan DP, Cox BD, Erzinclioglu SW, Walters DE, & Whichelow MJ
(1991)Ventilatory function and winter fresh fruit consumption in a
random sample of British adults. Thorax, 46: 624-629.
Strachan DP, Anderson HR, Limb ES, O'Neill A, & Wells N (1994) A
national survey of asthma prevalence, severity, and treatment in Great
Britain. Arch Dis Child, 70: 174-178.
Strachan DP, Sibbald B, Weiland SK, Ait-Khaled N, Anabwani G, Anderson
HR, Asher MI, Beasley R, Bjorksten B, Burr M, Clayton T, Crane J,
Ellwood P, Keil U, Lai C, Mallol J, Martinez F, Mitchell E, Montefort
S, Pearce N, Robertson C, Shah J, Stewart A, von Mutius E, & Williams
H (1997) Worldwide variations in prevalence of symptoms of allergic
rhinoconjunctivitis in children: the international study of asthma and
allergies in childhood (ISAAC). Pediatr Allergy Immunol, 8: 161-176.
Strandberg I, Boman G, Hassler L, & Sjoqvist F (1976) Acetylator
phenotype in patients with hydralazine induced lupoid syndrome. Acta
Med Scand, 200: 367-371.
Strobel S & Ferguson A (1984) Immune responses to fed protein antigens
in mice: III. Systemic tolerance or priming is related to age at which
antigen is first encountered. Pediatr Res, 18: 588-594.
Suzuki T, Kanoh T, Kanbayashi M, Todome Y, & Ohkuni H (1993) The
adjuvant activity of pyrene in diesel exhaust on IgE antibody
production in mice. Jpn J Allergol, 42: 963-968.
Swennen B, Buchet JP, Stanescu D, Lison D, & Lauwerys R (1993)
Epidemiological survey of workers exposed to cobalt oxides, cobalt
salts, and cobalt metal. Br J Ind Med, 50(9): 835-842.
Szentivanyi A (1968) The beta adrenergic theory of the atopic
abnormality in asthma. J Allergy, 42: 203.
Takafuji S, Suzuki S, Koizumi K, Tadokoro K, Miyamoto T, Ikemori R, &
Muranaka M (1987) Diesel-exhaust particulates inoculated by the
intranasal route have an adjuvant activity for IgE production in mice.
J Allergy Clin Immunol, 79: 639-645.
Takafuji S, Suzuki S, Koizumi K, Tadokoro K, Ohashi H, Muranaka M, &
Miyamoto T (1989) Enhancing effect of suspended particulate matter on
the IgE antibody production in mice. Int Arch Allergy Appl Immunol,
90: 1-7.
Tanser AR, Bourke MP, & Blandford AG (1973) Isocyanate asthma:
respiratory symptoms caused by diphenylmethane di-isocyanate. Thorax,
28(5): 596-600.
Tariq SM, Stevens M, Matthews S, Ridout S, Twiselton R, & Hide DW
(1996) Cohort study of peanut and tree nut sensitisation by the age of
4 years. Br Med J, 313: 514-517.
Tarvainen K, Jolanki R, Estlander T, Tupasela O, Pfaffli P, & Kanerva
L (1995) Immunologic contact urticaria due to airborne
methylhexahydrophthalic and methyltetrahydrophthalic anhydrides.
Contact Dermatitis, 32(4): 204-209.
Taylor SL (1986) Immunologic and allergic properties of cow's milk
proteins in humans. J Food Protect, 49: 239-250.
Taylor SL (1992) Chemistry and detection of food allergens. Food
Technol, 46: 146-152.
Taylor JS & Praditsuwan P (1996) Latex allergy. Arch Dermatol, 132:
265-271.
Taylor B, Wadsworth J, Wadsworth M, & Peckham C (1984) Changes in the
reported prevalence of childhood eczema since the 1939-45 war. Lancet,
2: 1255-1257
Tee RD, Gordon DJ, Gordon S, Crook B, Nunn A, Musk AW, Venables KM, &
Taylor A (1992a) Immune response to flour and dust mites in a United
Kingdom bakery. Br J Ind Med, 49(8): 581-587.
Tee RD, Gordon DJ, van Hage Hamsten M, Gordon S, Nunn A, Johansson SG,
& Taylor A (1992b) Comparison of allergic responses to dust mites in
UK bakery workers and Swedish farmers. Clin Exp Allergy, 22(2):
233-239.
Teichberg S, Isolauri E, Wapnir RA, Roberts B, & Lifshitz F (1990)
Development of the neonatal rat small intestinal barrier to
nonspecific macromolecular absorption: Effect of early weaning to
artificial diets. Pediatr Res, 28(1): 31-37.
Tenenbaum SA, Rice JC, Espinoza LR, Cuéllar ML, Plymale DR, Sander DM,
Williamson LL, Haislip AM, Gluck OS, Tesser JRP, Nogy L, Stribrny Km,
Bevan JA, & Garry RF (1997) Use of antipolymer assay in recipients of
silicone breast implants. Lancet, 349: 449-454.
Terr AI (1994a) The atopic diseases. In: Stites DP, Terr AI, & Parslow
TG ed. Basic and clinical immunology, 8th ed. East Norwalk,
Connecticut, Appleton & Lange, pp 27-346.
Terr AI (1994b) Immune-complex allergic disease. In: Stites DP, Terr
AI, & Parslow TG ed. Basic and clinical immunology, 8th ed. East
Norwalk, Connecticut, Appleton & Lange, pp 357-362.
Tharp MD (1990) Immunodermatology. IgE and immediate hypersensitivity.
Dermatol Clin North Am, 8(4): 619-631.
Theofilopoulos AN (1995a) The basis of autoimmunity: Part I.
Mechanisms of aberrant self-recognition. Immunol Today, 16: 90-98.
Theofilopoulos AN (1995b) The basis of autoimmunity: Part II. Genetic
predisposition. Immunol Today, 16: 150-158.
Theofilopoulos AN & Dixon FJ (1985) Murine models of systemic lupus
erythematosus. Adv Immunol, 37: 269-390.
Thomas C, Groten J, Kammüller ME, De Bakker JM, Seinen W, & Bloksma N
(1989) Popliteal lymph node reactions in mice induced by the drug
zimeldine. Int J Immunopharmacol, 11: 693-702.
Thomas C, Lippe W, Seinen W, & Bloksma N (1991) Popliteal lymph node
enlargement and antibody production in the mouse induced by drugs
affecting monoamine levels in the brain. Int J Immunopharmacol, 13:
621-629.
Thompson HSG & Staines NA (1990) Could specific oral tolerance be a
therapy for autoimmune disease? Immunol Today, 11: 398-399.
Thorne PS & Karol MH (1989) Association of fever with late-onset
pulmonary hypersensitivity responses in the guinea pig. Toxicol Appl
Pharmacol, 100: 247-258.
Thorne PS, Hillebrand J, Magreni C, Riley EJ, & Karol MH (1986)
Experimental sensitization to subtilisin: I. Production of
immediate- and late-onset pulmonary reactions. Toxicol Appl Pharmacol,
86: 112-123.
Thorne PS, Hawk C, Kaliszewski SD, & Gurney PD (1991) The noninvasive
mouse ear swelling assay. I. Refinements for detecting weak contact
sensitizers. Fundam Appl Toxicol, 17: 790-806.
Thurston GD (1996) A critical review of PM10-mortality time-series
studies. J Expo Anal Environ Epidemiol, 6(1): 3-21.
Thurston GD, Ito K, Kinney PL, & Lippmann M (1992a) A multi-year study
of air pollution and respiratory hospital admissions in three New York
State metropolitan areas: Results for 1988 and 1989 summers. J Expo
Anal Environ Epidemiol, 2: 429-450.
Thurston G, D'Souza N, Lippmann M, Bartoszek M, & Fine I (1992b)
Associations between summer haze air pollution and asthma
exacerbations: A pilot camp study. Am Rev Respir Dis, 145: A429
(abstract).
Tisch R, Yang XD, Singer SM, Liblau LS, Fugger L, & McDevitt HO (1993)
Immune response to glutamic acid decarboxylase correlates with
insulitis in non-obese diabetic mice. Nature (Lond), 366: 72-75.
Tollerud DJ, Weiss ST, Elting E, Speizer FE, & Ferris B (1983) The
health effects of automobile exhaust. VI. Relationship of respiratory
symptoms and pulmonary function in tunnel and turnpike workers. Arch
Environ Health, 38: 334-340.
Tomasi TB, Barr WG, Challacombe SJ, & Curran G (1983) Oral tolerance
and accessory-cell function of Peyer's patches. Ann NY Acad Sci, 409:
145-163.
Tomura T & van Lancker JL (1988) Procainamide -- DNA interaction. J
Rheumatol, 15: 59-64.
Topping MD, Venables KM, Luczynska CM, Howe W, & Newman Taylor A
(1986) Specificity of the human IgE response to inhaled acid
anhydride. J Allergy Clin Immunol, 77: 834-842.
Toren K, Brisman J, & Jarvholm B (1993) Asthma and asthma-like
symptoms in adults assessed by questionnaires: A literature review.
Chest, 104(2): 600-608.
Tournade H, Pelletier L, Pasquier R, Vial MC, Mandet C, & Druet P
(1990) D-penicillamine- induced autoimmunity in Brown Norway rats:
Similarities with HgCl2-induced autoimmunity. J Immunol, 144:
2985-2991.
Tribe RM, Barton JR, Poston L, & Burney PG (1994) Dietary sodium
intake, airway responsiveness, and cellular sodium transport. Am J
Respir Crit Care Med, 149(6): 1426-1433.
Troisi RJ, Willett WC, Weiss ST, Trichopoulos D, Rosner B, & Speizer
FE (1995) A prospective study of diet and adult-onset asthma. Am J
Respir Crit Care Med, 151(5): 1401-1408.
Tsicopoulos A, Hamid Q, Varney V, Ying S, Moqbel R, Durham SR, & Kay
AB (1992) Preferential messenger RNA expression of Th1-type cells
(IFN-gamma+, IL2+) in classical delayed-type (tuberculin)
hypersensitivity reactions in human skin. J Immunol, 148(7):
2058-2061.
Tsunoda K, Ohta Y, Shinogami M, & Soda Y (1995) Does passive smoking
affect the incidence of nasal allergies? Am J Public Health, 85(7):
1019-1020.
Turjanmaa K (1987) Incidence of immediate allergy to latex gloves in
hospital personnel. Contact Dermatitis, 17(5): 270-275.
Turjanmaa K (1994) Allergy to natural rubber latex: a growing problem.
Ann Med, 26: 297-300.
Turjanmaa K, Alenius H, Mäkinen-Kiljunen S, Reunala T, & Palosuo T
(1996) Natural rubber latex allergy. Allergy, 51: 593-602.
Turner MW, Boulton P, Shields JG, Strobel S, Gibson S, Miller HRP, &
Levinsky RJ (1988) Intestinal hypersensitivity reactions in the rat:
I. Uptake of intact protein, permeability to sugars and their
correlation with mucosal mast-cell activation. Immunology, 63(1):
119-124.
Turner MW, Barnett GE, & Strobel S (1990) Mucosal mast cell activation
patterns in the rat following repeated feeding of antigen. Clin Exp
Allergy, 20(4): 421-427.
Uehara M (1991) Dry skin and inflammation. In: Ruzicka T, Ring J, &
Przybilla B ed. Handbook of atopic eczema. Berlin, Heidelberg, New
York, Springer Verlag, pp 84-89.
Uetrecht JP (1992) The role of leukocyte-generated reactive
metabolites in the pathogenesis of idiosyncratic drug reactions. Drug
Metab Rev, 24: 299-366.
Ulfvarson U & Alexandersson R (1990) Reduction in adverse effect on
pulmonary function after exposure to filtered diesel exhaust. Am J Ind
Med, 17: 341-347.
Utell MJ, Warren J, & Sawyer RF (1994) Public health risks from motor
vehicle emissions. Annu Rev Public Health, 15: 157-178.
Vandenplas O (1995) Occupational asthma caused by natural rubber
latex. Eur Respir J, 8: 1957-1965.
Vandenplas O, Delwiche JP, Evrard G, Aimont P, van der Brempt X,
Jamart J, & Delaunois L (1995) Prevalence of occupational asthma due
to latex among hospital personnel. Am J Respir Crit Care Med, 151(1):
54-60.
Van der Heijden FL, Van Neerven RJJ, Van Katwijk M, Bos JD, &
Kapsenberg ML (1993) Serum IgE-facilitated allergen presentation in
atopic disease. J Immunol, 150: 3643-3650.
Van der Valk P, van Kalken CK, Ketelaars H, Broxterman HJ, Kuiper CM,
Tsuruo T, Lankelma J, Meijer CJLM, Pinedo HM, & Scheper RJ (1990)
Distribution of multi-drug resistance-associated P-glycoprotein in
normal and neoplastic human tissues -- Analysis with 3 monoclonal
antibodies recognizing different epitopes of the P-Glycoprotein
molecule. Ann Oncol, 1: 56-64.
Van Hoogstraten IMW, Boden D, von Blomberg BME, Kraal G, & Scheper RJ
(1992) Persistent immune tolerance to nickel and chromium by oral
administration prior to cutaneous sensitization. J Invest Dermatol,
99: 608-616.
Van Hoogstraten IMW, Boos C, Boden D, von Blomberg BME, Scheper RJ, &
Kraal G (1993) Oral induction of tolerance to nickel sensitization in
mice. J Invest Dermatol, 101: 26-31.
Van Hoogstraten IMW, von Blomberg BME, Boden D, Kraal G, & Scheper RJ
(1994) Non-sensitizing percutaneous skin contacts prevent subsequent
induction of immune tolerance. J Invest Dermatol, 102: 80-83.
Van Lee Uwen BH, Martinsom ME, Webb GC, & Young IG (1989) Molecular
organization of the cytokine gene cluster, involving the human IL-3,
IL-4, IL-5, and GM-CSF genes on human chromosome 5. Blood, 73:
1142-1148.
Van Loveren H, Redegeld F, Matsuda H, Buckley T, Teppema JS, & Garssen
J (1997) Mast cells. In: Bos JD ed. Skin immune system (SIS), 2nd ed.
Boca Raton, Florida, CRC Press, pp 160-184.
Van Niekerk CH, Weinberg EG, Shore SC, Heese HV, & Van Schalkwyk J
(1979) Prevalence of asthma: A comparative study of urban and rural
Xhosa children. Clin Allergy, 9: 319-324.
Varjonen E, Kalimo K, Lammintausta K, & Terho P (1992) Prevalence of
atopic disorders among adolescents in Turku, Finland. Allergy, 47:
243-248.
Veien NK, Hattel T, Justese O, & Norholm A (1987) Dietary restrictions
in the treatment of adult patients with eczema. Contact Dermatitis,
17: 223-228.
Veien NK, Hattel T, & Laurberg G (1993) Low nickel diet: An open
prospective trial. J Am Acad Dermatol, 29: 1002-1007.
Veltman C, Lange CE, Juhe S, Stein G, & Bachner V (1975) Clinical
manifestations and course of vinyl chloride disease. Proc NY Acad Sci,
246: 6-17.
Venables KM & Chan Yeung M (1997) Occupational asthma. Lancet,
349(9063): 1465-1469.
Venables KM & Newman Taylor A (1990) Exposure-response relationships
in tetrachlorophthalic anhydride asthma. J Allergy Clin Immunol, 85:
55-58.
Venables KM, Dally MB, Burge PS, Pickering CAC, & Newman Taylor A
(1985a) Occupational asthma in a steel coating plant. Br J Ind Med,
42: 517-524.
Venables KM, Topping MD, Howe W, Luczynska CM, Hawkins R, & Newman
Taylor A (1985b) Interaction of smoking and atopy in producing
specific IgE antibody against a hapten protein conjugate. Br Med J,
290: 201-204.
Venables KM, Topping MD, Nunn A, Howe W, & Newman Taylor A (1987)
Immunologic and functional consequences of chemical
(tetrachlorophthalic anhydride) induced asthma after 4 years of
avoidance of exposure. J Allergy Clin Immunol, 80: 212-218.
Venables KM, Dally MB, Nunn A, Stevens JF, Stephens R, Farrer N,
Hunter JV, Stewart M, Hughes EG, & Newman Taylor A (1989) Smoking and
occupational allergy in a platinum refinery. Br Med J, 299: 939-942.
Verdier F, Virat M, & Descotes J (1990) Applicability of the popliteal
lymph node assay in the Brown-Norway rat. Immunopharmacol
Immunotoxicol, 12: 669-677.
Verdier F, Patriarca C, & Descotes J (1997) Autoantibodies in
conventional toxicity testing. Toxicology, 119: 51-58.
Verhoeff AP, van Strien RT, van Wijnen JH, & Brunekreef B (1995) Damp
housing and childhood respiratory symptoms: the role of sensitization
to dust mites and molds. Am J Epidemiol, 141: 103-110.
Vial T & Descotes J (1994) Contact sensitization assays in guinea
pigs: are they predictive of the potential for systemic allergic
reactions? Toxicology, 93: 63-75.
Vickers CFH (1991) Natural history of atopic eczema. In: Ruzicka T,
Ring J, & Przybilla B ed. Handbook of atopic eczema. Berlin,
Heidelberg, New York, Springer Verlag, pp 80-83.
Vieluf D, Przybilla B, Traenkner I, & Ring J (1990) Oral provocation
with food additives in atopic eczema. J Allergy Clin Immunol, 85: 206
(abstract).
Vieluf D, Kunz B, Bieber T, Przybilla B, & Ring J (1993) "Atopy patch
test" with aeroallergens in patients with atopic eczema. Allergo J, 2:
9-12
Vincent A, Roberts M, Willison H, Lang B, & Newsom-Davis J (1995)
Autoantibodies, neurotoxins and the nervous system. J Physiol, 89(3):
129-136.
Viner AS & Jackman N (1976) Retrospective survey of 1271 patients
diagnosed as perennial rhinitis. Clin Allergy, 6(3): 251-259.
Vivier E & Daeron M (1997) Immunoreceptor tyrosine-based inhibition
motifs. Immunol Today, 18: 286-291.
Von Blomberg BME, Bruynzeel DP, & Scheper RJ (1990) Advances in
mechanisms of allergic contact dermatitis: in vitro and in vivo
research. In: Marzulli FN & Maibach HI ed. Dermatotoxicology, 4th ed.
Washington, New York, London, Hemisphere Publishing Corporation, pp
255-362.
Von Mutius E, Fritzsch C, Weiland SK, Roll G, & Magnussen H (1992)
Prevalence of asthma and allergic disorders among children in united
Germany: A descriptive comparison. Br Med J, 305: 1395-1399.
Von Mutius E, Martinez FD, Fritzsch C, Nicolai T, Roell G, & Thiemann
HH (1994a) Prevalence of asthma and atopy in two areas of West and
East Germany. Am J Respir Crit Care Med, 149: 358-364.
Von Mutius E, Martinez FD, Fritzsch C, Nicolai T, Reitmeir P, &
Thiemann HH (1994b) Skin test reactivity and number of siblings. Br
Med J, 308: 692-695.
Von Mutius E, Weiland SK, Fritzsch C, Duhme H, & Keil U (1998)
Increasing prevalence of hay fever and atopy among children in
Leipzig, East Germany. Lancet, 351: 862-866.
Vos JG, Younes M, & Smith E ed. (1996) Allergic hypersensitivities
induced by chemicals: Recommendations for prevention. Boca Raton,
Florida, CRC Press, 348 pp.
Vreeburg KJJ, Wilkinson JD, & Scheper RJ (1991) Reduced frequency of
nickel allergy upon oral nickel contact at an early age. Clin Exp
Immunol, 85: 441-445.
Wade JF 3rd & Newman LS (1993) Diesel asthma: Reactive airways disease
following overexposure to locomotive exhaust. J Occup Med, 35(2):
149-154.
Wahlberg JE (1995) Patch testing. In: Rycroft RJG, Menné T, Frosch PJ,
& Benezra C ed. Textbook of contact dermatitis, 2nd ed. Berlin,
Heidelberg, New York, Springer Verlag, pp 241-265.
Wahlberg JE & Boman A (1985) Guinea pig maximisation test. Curr Probl
Dermatol, 14: 59-106.
Waite DA, Eyles EF, Tonkin SL, & O'Donnell TV (1980) Asthma prevalence
in Tokelauan children in two environments. Clin Allergy, 10: 71-75.
Walker RB & Warin RP (1956) The incidence of eczema in early
childhood. Br J Dermatol, 68: 182-183.
Walker FB, Smith PD, & Maibach HI (1967) Genetic factors in human
allergic contact dermatitis. Int Arch Allergy, 32: 453-462.
Walker C, Kaegi MK, Braun MD, & Blaser K (1991) Activated T cells and
eosinophilia in bronchoalveolar lavages from subjects with asthma
correlated with disease severity. J Allergy Clin Immunol, 88: 935-942.
Walker-Smith JA (1992) Immunology of gastrointestinal food allergy in
infancy and early childhood. In: MacDonald TT ed. Immunology of
gastrointestinal disease. Dodrecht, Kluwer Academic Publishers, pp
61-73.
Ward AM, Udnoon S, Watkins J, Walker AE, & Darke CS (1976)
Immunological mechanisms in the pathogenesis of vinyl chloride
disease. Br Med J, 1: 936-938.
Wardlaw A (1993) The role of air pollution in asthma. Clin Exp
Allergy, 23: 81-96.
Ware JH, Dockery DW, Spiro A 3rd, Speizer FE, & Ferris BG Jr (1984)
Passive smoking, gas cooking, and respiratory health of children
living in six cities. Am Rev Respir Dis, 129(3): 366-374.
Ware JH, Spengler JD, Neas LM, Samet JM, Wagner GR, Coultas D,
Ozkaynak H, & Schwab M (1993) Respiratory and irritant health effects
of ambient volatile organic compounds: The Kanawha County health
study. Am J Epidemiol, 137(12): 1287-1301.
Warren DL, Shiotsuka RN, Sangha GK, & Lyon JP (1993) Comparison of
respiratory sensitization responses to 2,4 and 2,6 toluene
diisocyanate. Toxicologist, 13: 41 (abstract).
Wass U & Belin L (1990) An in vitro method for predicting
sensitising properties of inhaled chemicals. Scand J Work Environ
Health, 16(3): 208-214.
Weeke ER (1987) Epidemiology of hay fever and perennial allergic
rhinitis. Monogr Allergy, 21: 1-20.
Weigle WO (1965) The production of thyroiditis and antibody following
injection of unaltered thyroglobulin into rabbits previously
stimulated with altered thyroglobulin. J Exp Med, 122: 1049-1062.
Weiland SK, Mundt KA, Rückmann A, & Keil U (1994) Self-reported
wheezing and allergic rhinitis in children and traffic density on
street of residence. Ann Epidemiol, 4: 243-247.
Weill H, Butcher B, & Charmarajan V (1981) Respiratory and immunologic
evaluation of isocyanate exposure in a new manufacturing plant.
Cincinnati, Ohio, National Institute of Occupational Safety and Health
(NIOSH Technical Report; DHHS (NIOSH) Publication No. 81-125).
Weinberg DA, Lesser RL, & Vollmer TL (1994) Ocular myasthenia: A
protean disorder. Surv Ophthalmol, 39: 169-210.
Weiner HL, Friedman A, Miller A, Khoury SJ, Al-Sabbagh A, Santos L,
Sayegh M, Nussenblatt RB, Trentham DE, & Hafler DA (1994) Oral
tolerance: Immunologic mechanisms and treatment of animal and human
organ-specific autoimmune diseases by oral administration of
autoantigens. Annu Rev Immunol, 12: 809-837.
Weinstein RS, Grogan TM, Kuszak JR, Jakate SM, Kluskens LF, & Coon JS
(1991) Multidrug resistance gene product (P-glycoprotein) in normal
tissue and tumors. St. Louis, Missouri, Mosby Year Book, Inc., pp
207-234 (Advances in Pathology and Laboratory Medicine Series).
Weiss KB, Wagener DK, Aberg N, Engstrom I, & Lindberg U (1989)
Geographic variations in US asthma mortality: Small-area analyses of
excess mortality, 1981-1985 -- Allergic diseases in Swedish school
children. Am J Epidemiol, 78(2): 246-252.
Weiss KB, Gergen PJ, & Wagener DK (1993) Breathing better or wheezing
worse? The changing epidemiology of asthma morbidity and mortality.
Annu Rev Public Health, 14: 491-513.
Weitzman M, Gortmaker SL, Sobol AM, & Perrin JM (1992) Recent trends
in the prevalence and severity of childhood asthma. J Am Med Assoc,
268: 2673-2677.
Weltzien HU, Moulon C, Martin S, Padovan E, Hartmann U, & Kohler J
(1996) T cell immune responses to haptens: Structural models for
allergic and autoimmune reactions. Toxicology, 107: 141-151.
White IR (1995) Phototoxic and photoallergic reactions. In: Rycroft
RJG, Menné T, Frosch PJ, & Benezra C ed. Textbook of contact
dermatitis. Berlin, Heidelberg, New York, Springer Verlag, pp 75-91.
WHO (in press) Air quality guidelines. Copenhagen, World Health
Organization, Regional Office for Europe.
Wick G, Sundick RS, & Albini B (1974) The obese strain (OS) of
chickens: An animal model with spontaneous autoimmune thyroiditis.
Clin Immunol Immunopathol, 3: 272-300.
Widmann FK (1989) An introduction to clinical immunology.
Philadelphia, Pennsylvania, F.A. Davis, 424 pp.
Wierenga EA, Smoek M, de Groot C, Chrétien I, Bos JD, Jansen HM, &
Kapsemberg ML (1990) Evidence for compartmentalization of functional
subsets of CD4+ T lymphocytes in atopic patients. J Immunol, 144:
4651-4656.
Wilkinson JD (1995) The management of contact dermatitis. In: Rycroft
RJG, Menné T, Frosch PJ, & Benezra C ed. Textbook of contact
dermatitis, 2nd ed. Berlin, Heidelberg, New York, Springer Verlag, pp
660-683.
Williams WJ (1989) Beryllium workers -- sarcoidosis or chronic
beryllium disease. Sarcoidosis, 6(suppl): 34-35.
Williams HC (1992) Is the prevalence of atopic dermatitis increasing?
Clin Exp Dermatol, 17: 385-391.
Williams IR & Unanue ER (1990) Costimulatory requirements of murine
Th1 clones: The role of accessory cell-derived signals in response to
anti-CD3 antibody. J Immunol, 145(1): 85-93.
Williams WR & Williams WJ (1982) Development of beryllium lymphocyte
transformation tests in chronic beryllium disease. Int Arch Allergy
Appl Immunol, 67: 175-180.
Williams ME, Lichtman AH, & Abbas AK (1990) Anti-CD3 antibody induces
unresponsiveness to IL-2 in Th1 clones but not in Th2 clones. J
Immunol, 144(4): 1208-1214.
Williams HC, Burney PG, Hay RJ, Archer CB, Shipley MJ, Hunter JJ,
Bingham EA, Finlay AY, Pembroke AC, Graham Brown RA, Atherton DA,
Lewis-Jones MS, Holden CA, Harper JI, Champion RH, Poyner TF, Launer
J, & David TJ (1994a) The UK Working Party's diagnostic criteria for
atopic dermatitis: I. Derivation of a minimum set of discriminators
for atopic dermatitis. Br J Dermatol, 131(3): 383-396.
Williams HC, Strachan DP, & Hay RJ (1994b) Childhood eczema: disease
of the advantaged? Br Med J, 308: 1132-1135.
Williams HC, Pembroke AC, Forsdyke H, Boodoo G, Hay RJ, & Burney PG
(1995a) London-born black Caribbean children are at increased risk of
atopic dermatitis. J Am Acad Dermatol, 32(2/1): 212-217.
Williams PB, Buhr MP, Weber RW, Volz MA, Koepke JW, & Selner JC
(1995b) Latex allergen in respirable particulate air pollution. J
Allergy Clin Immunol, 95(1/1): 88-95.
Witt C, Stuckey MS, Woolcock A, & Dawkins RL (1986) Positive allergy
prick tests associated with bronchial histamine responsiveness in an
unselected population. J Allergy Clin Immunol, 77: 698-702.
Wjst M, Reitmeir P, Dold S, Wulff A, Nicolai T, von Loeffelholz
Colberg EF, & von Mutius E (1993) Road traffic and adverse effects on
respiratory health in children. Br Med J, 307: 596-600.
Wjst M, Heinrich J, Liu P, Dold S, Wassmer G, Merkel G, Huelsse C, &
Watchman HE (1994) Indoor factors and IgE levels in children. Allergy,
49: 766-771.
Wolf R & Brenner S (1994) An active amide group in the molecule of
drugs that induce pemphigus: a casual or causal relationship?
Dermatology, 189: 1-4.
Wood RA, Mudd KE, & Eggleston PA (1992) The distribution of cat and
dust mite allergens on wall surfaces. J Allergy Clin Immunol, 89(1/1):
126-130.
Wucherpfennig KW, Yu B, Monos DS, Argyris E, Karr RW, Ahmed AR, &
Strominger JL (1995) Structural basis for major histocompatibility
complex (MHC)-linked susceptibility to autoimmunity: charged residues
of a single MHC binding pocket confer selective presentation of
self-peptides in pemphigus vulgaris. Proc Natl Acad Sci (USA), 92(25):
11935-11939.
Wüthrich B (1975) [Immunopathology of constitutional neurodermatitis.]
Bern, Göttingen, Toronto, Seattle, Hogrefe & Huber Publishers, 178 pp
(in German).
Wüthrich B (1990) [Do food allergies of Type III exist?] Allergologie,
13: 371 (in German).
Wüthrich B (1991) Minimal forms of atopic eczema. In: Ruzicka T, Ring
J, & Przybilla B ed. Handbook of atopic eczema. Berlin, Heidelberg,
New York, Springer Verlag, pp 46-53.
Wüthrich B (1993) [Food allergies: Frequency of symptoms and the
allergy-eliciting foods in 402 patients.] Allergologie, 16: 280-287
(in German).
Wüthrich B & Hofer T (1984) [Food allergy: The "celery-mugwort-spices
syndrome" association with mango allergy?] Dtsch Med Wochenschr,
109(250): 981-986 (in German).
Wüthrich B, Stäger J, & Johansson SGO (1990) Celery allergy associated
with birch and mugwort pollinosis. Allergy, 45(80): 566-571.
Wüthrich B, Schindler C, Leuenberger P, Ackermann-Liebrich U, &
SAPALDIA Team (1995) Prevalence of atopy and pollinosis in the adult
population of Switzerland (SAPALDIA-study). Int Arch Allergy Immunol,
106: 149.
Xu X & Wang L (1993) Association of indoor and outdoor particulate
level with chronic respiratory illness. Am Rev Respir Dis, 148(6/1):
1516-1522.
Yamakage A, Ishikawa H, Saito Y, & Hattori A (1980) Occupational
scleroderma-like disorder occurring in men engaged in the
polymerization of epoxy resins. Dermatologica, 161: 33-44.
Yamashita U, Takami T, Hamasaka T, & Kitigawa M (1976) The role of
hapten-reactive T-lymphocytes in the induction of autoimmunity in
mice: II. Termination of self tolerance to erythrocytes by
immunization with hapten-isologous erythrocytes. Cell Immunol, 25:
32-40.
Yamashita N, Natsuaki M, & Sagami S (1989) Flare-up reaction on murine
contact hypersensitivity: I. Description of an experimental model:
rechallenge system. Immunology, 67(3): 365-369.
Yokoyama Y, Nitta H, Maeda K, & Aoki S (1985) What interaction does
indoor nitrogen dioxide have on the effect of the automobile exhaust.
Tokai J Exp Clin Med, 10: 379-384.
Young RP, Barker R, Cookson WOCM, & Newman Taylor A (1993) HLA-DR and
DP antigen frequencies and acid anhydride sensitization. Am Rev Respir
Dis, 147(suppl): A113 (abstract).
Young E, Stoneham MD, Petruckevitch A, Barton J, & Rana R (1994) A
population study of food intolerance. Lancet, 343: 1127-1130.
Young RP, Barker RD, Pile KD, Cookson WOCM, & Newman Taylor A (1995)
The association of HLA-DR3 with specific IgE to inhaled acid
anhydrides. Am J Resp Crit Care Med, 151: 219-221.
Yssel H, Johnson KE, Schneider PV, Wideman J, Terr A, Kastelein R, &
de Vries JE (1992) T cell activation inducing epitopes of the house
dust mite allergen Der p I -- Induction of a restricted cytokine
production profile of Der p l-specific T cell clones upon
antigen-specific activation. J Immunol, 148: 738-745.
Yssel H, Fasler S, Lamb J, & de Vries JE (1994) Induction of
non-responsiveness in human allergen-specific type 2 T helper cells.
Curr Opin Immunol, 6: 847-852.
Yung RL, Quddus J, Crisp CE, Johnson KJ, & Richardson BC (1995)
Mechanisms of drug induced lupus: I. Cloned Th2 cells modified with
DNA methylation inhibitors in vitro cause autoimmunity in vivo. J
Immunol, 154: 3025-3035.
Zeiss CR, Patterson R, Pruzansky JJ, Miller MM, Resonberg M, & Levitz
D (1977) Trimellitic anhydride induced airways syndrome: Clinical and
immunologic studies. J Allergy Clin Immunol, 60: 96-103.
Zeiss CR, Kanellaks TM, Bellone TD, Levitz D, Pruzansky JJ, &
Patterson R (1980) Immunoglobulin E mediated asthma and
hyper-sensitivity pneumonitis with precipitating antihapten antibodies
due to diphenylmethane di-isocyanate (MDI) exposure. J Allergy Clin
Immunol, 65: 347-352.
Zinkernagel RM & Doherty PC (1975) H-2 compatibility requirement for
T-cell mediated lysis of target cells infected with lymphocytic
choriomeningitis virus -- Different cytotoxic T-cell specifities are
associated with structures coded for H-2K or H-2D. J Exp Med, 141(6):
1427-1436.
Zoia MC, Fanfulla F, Bruschi C, Basso O, De Marco R, Casali L, &
Cerveri I (1995) Chronic respiratory symptoms, bronchial
responsiveness and dietary sodium and potassium: a population-based
study. Monaldi Arch Chest Dis, 50(2): 104-108.
Zummo SM & Karol MH (1996) Indoor air pollution: Acute adverse health
effects and host susceptibility. Environ Health Sci, Jan/Feb: 25-29.
Zurawski G & de Vries JE (1994) Interleukin 13, an interleukin 4-like
cytokine that acts on monocytes and B cells, but not on T cells.
Immunol Today, 15(1): 19-26.
1. CONCLUSIONS
L'allergie constitue un problème de santé à l'échelle mondiale.
Elle touche une proportion importante de la population et toutes les
tranches d'âge. Pour des raisons mal connues, sa fréquence globale est
en augmentation.
La présente monographie fait le point des interactions diverses
et complexes entre produits chimiques, médicaments, système
immunitaire et organes cibles, interactions qui aboutissent à des
manifestations d'hypersensibilité allergique et à des troubles
d'origine autoimmune. D'importantes recherches sont menées dans tous
ces domaines et l'on peut donc s'attendre à de nouvelles découvertes.
Les études portent sur les facteurs endogènes et exogènes multiples
qui interviennent dans l'allergie. Les facteurs exogènes ont tous leur
importance, qu'il s'agisse des allergènes eux-mêmes ou encore des
maladies infectieuses, de la pollution de l'air et du mode de vie (par
exemple, le tabagisme). Par ailleurs, les prédispositions génétiques à
un trouble allergique donné déterminent en grande partie la réactivité
du sujet.
L'épidémiologie de l'allergie montre que cet ensemble
d'affections est très répandu et que, par conséquent, il importe
d'accorder l'attention voulue à l'identification du risque allergique
et à son évaluation pour être à même de mettre en oeuvre des
stratégies de prise en charge adaptées au risque.
Les méthodes d'identification des substances produisant une
sensibilisation cutanée sont bien connues. Elles restent à normaliser
dans le cas des sensibilisants respiratoires et font encore défaut
pour les autres types d'allergènes. On utilise les techniques de
mesure de l'activité des sensibilisants cutanés pour l'évaluation du
risque.
Une fois le risque allergique bien caractérisé, l'évaluation de
ce risque et sa prise en charge sont d'une importance primordiale pour
réduire l'incidence des troubles allergiques. Pour évaluer le risque,
il faut déterminer l'activité allergisante de l'agent en cause en
fonction de la nature et de l'intensité de l'exposition. Si
l'évaluation en fait ressortir la nécessité, il faudra alors prendre
en charge ce risque en mettant en oeuvre un certain nombre de mesures
consistant par exemple à limiter l'exposition et à étiqueter le
produit.
Les progrès accomplis dans la connaissance des caractéristiques
physicochimiques et immunologiques des allergènes alimentaires
pourraient déboucher sur la mise au point d'indicateurs prédictifs.
En raison de la complexité des mécanismes qui sont à la base des
troubles allergiques, il est encore difficile de proposer des tests
prédictifs in vitro qui soient d'une portée générale, mais
l'application des relation structure-activité mérite d'être étudiée de
manière plus approfondie.
2. RECOMMANDATIONS POUR LA PROTECTION DE LA SANTÉ HUMAINE
a) Il convient de mettre en oeuvre des stratégies efficaces
pour prévenir les allergies, fondées sur des informations fiables
relatives aux agents en cause et à l'environnement. L'action en
vue de prévenir ou de réduire au minimum la fréquence des
allergies doit reposer sur la limitation de l'exposition.
b) Elucider la cause de la fréquence accrue des allergie est
devenu d'une urgente nécessité.
c) Il faut mettre en oeuvre des méthodes de surveillance afin
de déterminer la fréquence des divers types d'allergie.
d) Il peut s'avérer difficile de mesurer l'exposition des
individus et des populations, mais une évaluation correcte est
néanmoins nécessaire à toute analyse de la relation
effet-exposition. Du fait de sa spécificité, la réponse
immunitaire constitue un biomarqueur unique en son genre pour
étudier les antécédents d'exposition, par exemple par apposition
d'un timbre cutané, par intradermoréaction ou par titrage des
anticorps IgE pour mettre en évidence une sensibilisation par
des allergènes déterminés.
e) Il faut améliorer les systèmes de surveillance sanitaire des
travailleurs, la qualité des visites médicales effectuées dans le
cadre de la médecine du travail ainsi que l'information de ceux
qui sont exposés à des produits chimiques, afin de mettre en
évidence le plus tôt possible toute maladie allergique d'origine
professionnelle. La surveillance de ces affections sur le lieu de
travail est particulièrement importante, du fait que l'exposition
y est vraisemblablement plus intense qu'ailleurs.
f) Il faut évaluer périodiquement l'efficacité et l'intérêt de
la prévention primaire et secondaire, de même que celle des
stratégies d'intervention en utilisant des techniques
d'évaluation épidémiologique dûment validées.
g) Dans le cas de la dermatite de contact d'origine allergique,
les modèles prédictifs in vivo dont on dispose à l'heure
actuelle ont fait la preuve de leur valeur pour les antigènes de
faible masse moléculaire relative. Il faut maintenant trouver le
moyen de les utiliser au mieux pour mesurer l'activité des
allergènes.
h) Dans le cas des troubles allergiques intéressant les voies
respiratoires, les tests in vivo relatifs aux substances de
faible masse moléculaire relative sont prometteurs, mais il faut
encore s'assurer de leur valeur prédictive et de leur
spécificité. Dans le cas des allergènes de nature protéique, on
est en train de mettre au point des méthodes d'épreuve, mais il
faudra encore les évaluer de manière plus approfondie sur des
substances dont on connaît le pouvoir allergénique chez l'Homme.
i) Il importe que les services de contrôle et les consommateurs
puissent s'informer facilement de la présence éventuelle
d'allergènes dans certains produits (par ex. les denrées
alimentaires) en consultant par exemple des bases de données ou
grâce à un étiquetage approprié, et soient de la sorte en mesure
de prendre les précautions voulues.
j) Le principal moyen dont on dispose pour éviter les
affections autoimmunes provoquées par des produits chimiques sur
le lieu de travail consiste à limiter l'exposition. Il convient
d'envisager un examen préalable des personnes qui vont être
exposées à des produits chimiques à action immunomodulatrice pour
rechercher tout signe de maladie affectant le tissu conjonctif.
Il est souhaitable que les consignes visant à éviter ou à réduire
l'exposition soient scrupuleusement suivies, notamment en ce qui
concerne les règles d'hygiène et sécurité. Il faut aussi réduire
au maximum les autres facteurs de risque, comme le tabagisme, et
envisager des contrôles médicaux réguliers dans le cadre de la
médecine du travail.
k) Il est nécessaire d'étudier les relations quantitatives
entre la réponse immunitaire suscitée par une exposition à des
produits chimiques et la gravité des réactions allergiques.
l) Il est également important de déterminer, pour lutter contre
ce type de maladies, quelles sont l'intensité et la durée
minimales d'exposition nécessaires pour provoquer une
sensibilisation ou déclencher une réaction allergique.
m) Il importe de mettre au point des stratégies normalisées
pour l'examen clinique et le diagnostic des allergies et de les
mettre en oeuvre à l'échelle internationale afin de déterminer
les causes et l'incidence des maladies d'origine allergique. A
cet effet, il faudra produire et mettre à disposition des
extraits normalisés d'allergènes biologiques d'activité contrôlée
et procéder périodiquement à l'examen des produits qui entreront
dans la composition des batteries normalisées d'allergènes
destinées aux divers tests.
n) Les autorités en charge de la santé publique, les membres
des professions de santé et les organismes publics devront
réfléchir aux moyens d'évaluer le coût humain et économique des
maladies allergiques pour l'individu et la société.
o) Les autorités en charge de la santé publique, les membres
des professions de santé, le public en général et les
travailleurs en particulier tireront avantage d'une meilleure
information sur la fréquence, les causes, les manifestations
cliniques et les conséquences des différents types d'allergie.
1. CONCLUSIONES
La alergia es un importante problema de salud de alcance mundial.
Afecta a una proporción sustancial de la población y a todos los
grupos de edad. Por razones poco conocidas, la frecuencia general de
la alergia está en aumento.
En esta monografía se examinan las interacciones diversas y
complejas entre productos químicos, medicamentos y proteínas, el
sistema inmunitario y el(los) órgano(s) diana que dan lugar a la
manifestación de la hipersensibilidad alérgica y la autoinmunidad.
Estos temas son objeto de una investigación extensa continua, de
manera que se prevén nuevos descubrimientos. Se examina la
multiplicidad de factores endógenos y exógenos que tienen
repercusiones sobre la alergia. Los factores exógenos, como los
alergenos mismos, las infecciones, la contaminación del aire y el modo
de vida (por ejemplo, el tabaquismo) son todos ellos importantes.
Además, la predisposición genética a un trastorno alérgico específico
es un determinante importante de la reactividad.
La epidemiología de la alergia demuestra el carácter extendido de
este grupo de trastornos y, como consecuencia, pone de relieve la
necesidad de prestar suficiente atención a la identificación de los
riesgos de alergia y la evaluación de éstos en la aplicación de
estrategias apropiadas de gestión de riesgos.
Hay métodos de reconocida eficacia para la determinación de
riesgos en relación con los sensibilizadores de la piel. Hace falta
normalizar los aplicables a los sensibilizadores a nivel respiratorio
y aún no se dispone de métodos aplicables a otros tipos de alergenos.
En la evaluación de riesgos se están aplicando técnicas de
determinación de la potencia de los sensibilizadores de la piel.
Una vez que los riesgos de alergia se han caracterizado bien, la
evaluación de riesgos, la gestión de riesgos y la comunicación sobre
los riesgos son elementos decisivos para reducir la incidencia de los
trastornos alérgicos. La evaluación de riesgos requiere que el riesgo,
de una potencia conocida, se evalúe atendiendo a la naturaleza y la
magnitud de la exposición. Cuando la evaluación de riesgos indica que
es necesario, se deben aplicar medidas de gestión de los riesgos,
tales como el control de la exposición y el etiquetado de productos.
Se están adqduiriendo más conocimientos sobre las características
fisicoquímicas e inmunológicas de alergenos alimentarios, que con el
tiempo podrán llegar a tener valor predictivo.
Dada la complejidad de los mecanismos de los trastornos
alérgicos, por el momento resulta difícil proponer métodos preditivos
in vitro de aplicabilidad general; no obstante, la aplicación de las
relaciones entre actividad y estructura merece mayor consideración.
2. RECOMENDACIONES PARA LA PROTECCION DE LA SALUD HUMANA
a) Para prevenir la alergia se deben aplicar estrategias
eficaces basadas en buena información acerca de los alergenos
presentes en los productos y el medio ambiente. El control de la
exposición debe ser la base de la prevención o la reducción al
mínimo de la aparición de enfermedades alérgicas.
b) Hay una necesidad urgente de determinar las causas del
aumento de la frecuencia de la alergia.
c) Se deben adoptar métodos de vigilancia para definir la
frecuencia de alergias de diferentes tipos.
d) Puede ser difícil medir la exposición de los individuos y
las poblaciones, pero una evaluación adecuada es esencial para
cualquier análisis de la asociación entre la exposición y el
efecto. La especificidad de las respuestas inmunitarias
representa un tipo único de marcador biológico a la hora de
estudiar la exposición pasada, por ejemplo mediante la
utilización de pruebas epicutáneas o de puntura, o la valoración
de los anticuerpos de IgE para detectar la sensibilización por
alergenos específicos.
e) Se deben mejorar los sistemas de vigilancia de los
- trabajadores, la calidad del examen médico de éstos y la
educación de los expuestos a productos químicos para detectar
enfermedades alérgicas ocupacionales en un estadio inicial. Los
sistemas relativamente sencillos de notificación de los
trastornos ocupacionales y la vigilancia posterior a la
comercialización de los medicamentos constituyen formas
económicas de reconocimiento y alerta en relación con las
enfermedades alérgicas. La vigilancia de estos trastornos en el
lugar de trabajo es particularmente valiosa porque es allí donde
la exposición probablemente sea mayor que en cualquier otro
lugar.
f) La eficacia y el valor de las estrategias de prevención
primaria y secundaria e intervención deben evaluarse a intervalos
que utilizan técnicas epidemiológicas validadas.
g) Con respecto a la dermatitis de contacto alérgica, los
modelos preditivos in vivo disponibles tienen un valor
comprobado para los antígenos de masa molecular relativa baja. Es
necesario descubrir la mejor manera de utilizarlos para
determinar la potencia de los alergenos.
h) Con respecto a los trastornos alérgicos de las vías
respiratorias, los métodos de prueba in vivo disponibles para
las sustancias de masa molecular relativa baja son prometedores,
pero es necesario comprobar su valor predictivo y su
especificidad. Para los alergenos proteicos, están
desarrollándose algunos métodos de prueba en animales, pero es
preciso someterlos a evaluaciones ulteriores utilizando
sustancias de potencial alergénico conocido en los seres humanos.
i) Es importante que la información acerca de la presencia de
alergenos en los productos (por ejemplo alimentos) esté
fácilmente disponible, por ejemplo en bases de datos y en las
etiquetas de los productos, para que las instancias reguladoras y
los individuos puedan tomar precauciones apropiadas.
j) El método principal para prevenir enfermedades
autoinmunitarias ocupacionales inducidas químicamente es el
control de la exposición. Se debe considerar la evaluación previa
a la exposición de los expuestos a los productos químicos con
potencial inmunomodelador para documentar cualquier
característica preexistente de morbilidad del tejido conectivo.
Se recomienda una adhesión estricta a las normas para evitar o
reducir al mínimo la exposición, inclusive la utilización de
buenas prácticas de higiene ocupacional. Se deben reducir al
mínimo otros factores de riesgo, como el consumo de tabaco y se
debe considerar la posibilidad de efectuar exámenes médicos
ocupacionales regulares.
k) Es necesario investigar las relaciones cuantitativas
existentes entre las respuestas inmunitarias inducido por los
productos químicos y la gravedad de las reacciones alérgicas.
l) Para controlar las enfermedades alérgicas es importante
determinar la exposición mínima y su duración necesaria para
causar sensibilización o producir una respuesta alérgica.
m) Es importante idear estrategias normalizadas de
investigación y diagnóstico clínicos de la alergia y aplicarlas
internacionalmente para examinar las causas y la incidencia de
los trastornos alérgicos. Ello requerirá la producción y la
disponibilidad de extractos normalizados de alergenos biológicos
de potencia controlada, así como el examen regular de los
componentes de series normalizadas de alergenos utilizados en las
pruebas.
n) Las autoridades de salud pública, los profesionales de la
salud y las dependencias gubernamentales deben examinar la manera
de calcular los costos humanos y económicos de las enfermedades
alérgicas para los individuos y la sociedad.
o) Las autoridades de salud pública, los profesionales de la
salud, el público y, especialmente, los trabajadores necesitan
mejor información acerca de la frecuencia, las causas, las
manifestaciones clínicas y las consecuencias de diferentes tipos
de alergia.
starts typically during the first year of life (Hanifin, 1987). The
onset of the disease is before the age of 7 in 60 to 90% of the cases
(Schultz Larsen & Hanifin, 1992). The prevalence appears to be higher
among females than among males (Schultz Larsen & Hanifin, 1992;
Schultz Larsen, 1993).
5.5 Migration
Migration studies have suggested that the environment has a
strong effect on the development of atopic disorders. Analyses of
interregional migrants in the United Kingdom showed that the regional
variation in cohorts, aged 5-7 years, was primarily related to the
region of current residence, and not to the region of birth (Strachan
et al., 1990). Ethnic group differences in the prevalence of atopic
dermatitis were studied in London school children. It appeared that,
compared to white children, London-born black Caribbean children were
at increased risk of atopic dermatitis (Williams et al., 1995a).
Striking differences in prevalence rates of asthma were found in
young urban and rural Xhosa children (Van Niekerk et al., 1979). In
this society, asthma was found mainly in urban communities and there
appears to be a striking absence of asthma in the rural environment.
Whether this is due to alterations in lifestyle or the possibility
that the rural traditional way of life exerts a protective effect in
the prevention of asthma is not clear.
The prevalence of asthma in Tokelauan children has been studied
in two environments, in Tokelau and in New Zealand (Waite et al.,
1980). Prevalence of asthma assessed by an interview of the mothers
was much higher among those children who were examined in New Zealand
than among those examined in Tokelau. Furthermore, for the children
examined in New Zealand, there was no significant difference in the
asthma prevalence between those children born in New Zealand and those
born in Tokelau.
Asthma in children and adolescents living in the New Guinea
Highlands was extremely uncommon in the sixties and early seventies.
In 58% of the observed asthma cases the onset of the disease was not
before the age of 30 years. Multiple sensitivities as assessed by
skin-prick test were common and did not diminish with increasing age
at onset. One possible explanation was that the degree of exposure to
allergens is high enough to cause sensitization, but not high enough
to cause lower respiratory symptoms until prolonged exposure in a
perhaps especially sensitive person has taken place (Anderson, 1974).
The prevalence of asthma among adults but not children living in the
Eastern highlands of Papua New Guinea has risen drastically between
1975 and 1985. Allergy to house dust mite appeared to be a significant
feature in the disease pathogenesis, and it is likely that this is
associated with modifications to traditional life style due to the
introduction of blankets and changes in sleeping habits, which promote
a more fertile environment for growth and multiplication of mites
(Dowse et al., 1985).
5.6 Viral infection
Findings of different studies suggest that viral infections in
early life may play a part in the prevention of allergic sensitization
(Martinez, 1994). Interesting observations with regard to a possibly
preventive role of viral infections in the development of asthma were
made in the population of Tristan da Cunha. The prevalence of asthma
among these islanders had earlier been reported to be one of the
highest in the world (Mantle & Pepys, 1974). After the evacuation from
Tristan da Cunha because of a volcanic eruption immunoassays in blood
samples of the islanders revealed a low prevalence of serum antibodies
against common viruses. During the years of evacuation, when the
islanders lived in the United Kingdom, a high incidence of respiratory
infection was observed. Thus, this population with a high prevalence
of asthma and a high prevalence of allergic sensitization had a very
low incidence of respiratory infections while living on the remote
island, and became heavily infected after being exposed to respiratory
viruses that they had probably rarely encountered before. Similar
observations were made in the Western Carolina islands where the
prevalence of asthmatic symptoms among children was very high, but
viral infections presumably very uncommon (Martinez, 1994). A study
from the United Kingdom found an inverse association between hay fever
and the number of older siblings (Strachan, 1989; Strachan, 1995).
Factors directly or indirectly related to the number of siblings may
decrease the susceptibility of children to become atopic. Again,
infections acquired during early childhood were proposed to be
protective against allergic sensitization. It has been speculated that
declining family size may in part contribute to the increasing
prevalence of atopic diseases reported in Western countries over the
past few decades, because of a lower chance of infection by older
siblings. In children from eastern and western Germany, allergic
sensitization as assessed by skin test reactivity was also inversely
associated with the number of siblings (von Mutius et al., 1994b).
5.7 Socioeconomic status
Hay fever has been recognized as a complaint of the more affluent
classes since the 19th century. A study of adults in south London
found little difference in the prevalence of rhinitis symptoms or
skin-prick reactions by social class, but a greater use of the label
"hay fever" by doctors for patients of higher socioeconomic status
(Sibbald & Rink, 1991a).
Eczema is more prevalent in British school children of higher
socioeconomic status than in those of lower status. Exposures
associated with social class are probably at least as important as
genetic factors in the expression of childhood eczema (Williams et
al., 1994b). The authors suggest that most or even all of the reported
changes in the prevalence of atopic dermatitis are due to a secular
trend in diagnosis. Population studies have used a range of different
methods and definitions for atopic dermatitis ranging from
questionnaire-based recall of eczema as a child or parental recall to
health visitors' and general practitioners' records. It is likely that
the term eczema is heavily biased by social class (allergy being a
more acceptable term in higher social classes) and with time (eczema
was a less acceptable label in earlier years due to connotations of
uncleanliness and arthropod infestation).
Hospital admissions for eczema have fallen over the last 20
years, but such in-patient data are misleading as much of this
reduction is probably due to the success of treatment with topical
corticosteroid preparations (Williams, 1992).
The prevalence of wheeze varied little by socioeconomic group in
an investigation of 5472 children, aged 5-17 years, in the United
Kingdom (Strachan et al., 1994). However, marked trends of severity
towards increased morbidity in poorer families were observed.
Diagnostic labelling and drug treatment of wheezy children did not
differ substantially with socioeconomic status. Thus, a degree of
socioeconomic equality existed in the process of medical care for
childhood asthma in the United Kingdom (Strachan et al., 1994;
Strachan, 1996).
No effect of socioeconomic status on the prevalence of asthma was
noted in the first and second US National Health and Nutrition
Examination Surveys (NHANES) (Gergen et al., 1988). In the Auckland
region, 1050 children aged 8-9 years were examined by parental
questionnaire and histamine inhalation challenge (Mitchell et al.,
1989). There was no relationship between socioeconomic status and
asthma diagnosis, bronchial hyperresponsiveness, or any combination of
bronchial hyperresponsiveness with symptoms or diagnosis. The relative
importance of socioeconomic status and several other factors in the
etiology of wheezing illness in the first 5 years and on the
persistence of this illness at the age of 16 years was studied in over
15 000 children born in the United Kingdom during one week of April
1970. Persistence of wheeze at age 16 years was related to high social
status (Lewis et al., 1995).
During a study of the prevalence of asthma and bronchitis in
Sydney school children, some social and environmental factors were
documented to ascertain if these affected the prevalence of either of
these diseases (Peat et al., 1980). No consistent relationship was
found between social class and lung disease with the exception of
increased prevalence of asthma in boys and girls of higher
socioeconomic status. Differential access and utilization of medical
care by the poor and rich may contribute to differences in asthma
prevalence. The relationship of socioeconomic status to various
indicators of asthma was studied in Canada in the context of universal
access to medical care. As compared with children from the most
advantaged homes, children from the least advantaged homes were more
likely to present exercise-induced bronchospasm, while there was no
excess of reported wheeze or diagnosed asthma. This result was
interpreted as indicating that there are unidentified environmental
factors that contribute to the excess asthma morbidity in children
(Ernst et al., 1995). In general, the association of socioeconomic
status with hay fever and eczema seems to be more consistent than with
asthma and respiratory symptoms.
5.8 Occupational exposure
Many prevalence studies have been conducted among workers in
high-risk occupations. Asthmatic workers may differ with regard to the
frequency of attacks, to the occurrence and to the onset of airway
obstruction. Diagnostic guidelines for occupational asthma were
internationally proposed in the USA and in Europe (Cartier et al.,
1989; Burge, 1989; EAACI, 1992). The diagnosis of occupational asthma
is based both on the clinical signs of the disease and the
demonstration of the occurrence of a recognized allergen in the
patient's workplace. Objective tests, such as skin testing, spirometry
and serial measurements of the peak expiratory flow, should be
performed to ascertain the occupational nature of the disorder (see
also section 4.4.4). Other diagnostic tools in epidemiological surveys
are standardized questionnaires inquiring information on work-related
symptoms. Typical symptoms of occupational asthma are symptoms
comprising difficulty in breathing, chest tightness, wheezing, a
period of initial exposure of 2 weeks or longer before the first onset
of symptoms, or evidence of airflow obstruction, and improvement of
symptoms when the subject is not working for days or longer.
Surveillance programs in the United Kingdom and Canada indicate
that occupational asthma is the most frequently reported occupational
lung disease accounting for 26 to 52% of the reports (Chan-Yeung &
Malo, 1995a). The proportion of asthma attributable to occupational
exposure is not known. Estimates range from 2 to 15%. The role of
occupational exposure is difficult to ascertain, because: a)
occupational asthma is still poorly recognized; b) affected workers
are scattered through many small workplaces often employing few
workers; c) methods of confirming work relationships are often rather
crude; d) there is no good reporting system of occupational asthma in
many countries; e) there are very few screening or surveillance
programmes for this condition.
Owing to such methodological problems it is often not possible to
give unbiased estimates of the true prevalence and incidence of
occupational asthma in populations if official government statistics
such as disabling benefit awards or worker's compensation boards are
used and comparisons within and between countries may be distorted
(Meredith & Nordman, 1996).
Occupational asthma falls into two categories: a) pre-existing
asthma that is aggravated by irritant or physical stimuli in the
workplace and b) asthma that is specifically induced by sensitization
to a workplace chemical (Jarvis et al., 1996). About 250 agents that
can give rise to occupational asthma are recognized (Chan-Yeung &
Malo, 1995a). Isocyanates are responsible for the most common form of
the disease, i.e., occupational asthma with latency. Occupational
asthma without latency follows exposure to high concentration of
irritant gases, fumes or chemicals; chlorine and ammonia are the most
common agents (Chan-Yeung & Malo, 1995a,b). Some substances may give
rise to asthma by inducing specific IgE antibodies. These allergens
are mostly substances of high relative molecular mass (>5000) such as
proteins in the urine of laboratory rats. Others are compounds of low
relative molecular mass, such as complex halogenated platinum salts or
acid anhydrides; these agents act as haptens and combine with a body
protein to form a complete antigen (Venables & Chan-Yeung, 1997). It
has been proposed that certain chemical allergens may cause
sensitization of the respiratory tract via an IgE-independent
mechanism. However, it is possible that new or technically refined IgE
measurements may reveal a much greater association between IgE
antibodies and chemical respiratory sensitization than is presently
assumed (Kimber & Wilks, 1995). Intermittent exposure to high levels
of an occupational agent is associated with a higher risk of
development of work-related asthma than a steady exposure to lower
concentrations, typical for isocyanate exposure.
Not all subjects develop occupational asthma under the same
exposure conditions. Various host markers (genetically determined) and
factors (acquired) have been incriminated in occupational asthma. In
general, atopy appears to be an important risk factor for occupational
asthma due to compounds of high relative molecular mass, such as
enzyme detergents or exposure to laboratory animals (Chang-Yeung,
1990). In a survey of workers exposed to flour in bakeries or mills,
the relation with symptoms was independent of atopic status (Cullinan
et al., 1994). Atopy has little predictive value in occupational
asthma due to chemicals of low relative molecular mass, and routine
screening for atopy in high-risk workplaces may not be justified
(Chan-Yeung & Malo, 1995b).
The effect of smoking on occupational asthma is not clear and
appears to be dependent on the type of occupational agent. When the
agent induces asthma by producing specific IgE antibodies, cigarette
smoking may enhance sensitization. However, cigarette smoking was not
associated with increased work-related asthmatic symptoms in workers
exposed to detergent enzymes, laboratory animals or colophony
(Chan-Yeung, 1990; Chan-Yeung & Malo, 1995a,b).
As part of the European Community Respiratory Health Survey
(ECRHS), the risk of occupational asthma has been estimated in a
random sample of 2646 young Spanish adults aged 20-44 years. Depending
on the definition of asthma, 2.6-6.7% of asthma cases were
attributable to occupational exposures (Kogevinas et al., 1996).
Incidence rates for occupational asthma for 1992 were estimated to be
approximately 153 cases per million workers in Finland (Meredith &
Nordman, 1996). Incidence rates reported for the United Kingdom in the
Surveillance of Work-related and Occupational Respiratory Disease
Project (SWORD) between 1989 and 1991 showed a high risk of
occupational asthma among paint sprayers, chemical and food
processors, laboratory staff, plastics and metal treatment workers,
and in welders and electronic assemblers (Meredith, 1993). For 1993
the incidence was estimated to be 37 per million working persons per
year (Meredith & Nordman, 1996). The most frequent agents causing
asthma among the organic agents were flour, grain, hay, wood dust and
laboratory animals; among the chemical agents were isocyanates and
glutaraldehyde, and some miscellaneous compounds such as solder,
colophony, glues and resins (Meredith & McDonald, 1994).
In the UK SWORD project there is a high level of national
coverage by chest physicians participating in the surveillance project
and estimates of the working population at risk are available, so that
incidence rates can be calculated by age, gender, region and
occupation. However, there is underestimation, because some patients
are seen only by general practitioners. Questions remain regarding
diagnostic accuracy and etiology.
In the following sections, studies on specific occupational
exposures in relation to occupational atopic diseases, with emphasis
on occupational asthma, are reviewed. The selection of exposures is
based on findings of the SWORD project and the availability of
epidemiological data.
5.8.1 Chemicals with low relative molecular mass
A total of 314 cases of occupational asthma were diagnosed at the
Institute of Occupational Health in Helsinki, Finland during the
period 1987 to 1990 (Savonius et al., 1993). By far the most common
causes of occupational asthma were low relative molecular mass
chemicals such as the isocyanates (76 cases), followed by formaldehyde
(18 ases), epoxy resin and epoxy resin hardeners (17 cases), and
cyanoacrylates (6 cases).
5.8.1.1 Diisocyanates
The main occupational hazards caused by polyurethane chemicals
are asthma and rhinitis, but contact dermatitis and urticaria may also
develop (Estlander et al., 1992). Exposures to toluene diisocyanate
were studied for effects on respiratory health of workers in two
plants manufacturing polyurethane foams (Jones et al., 1992).
Intensive personal monitoring was performed to characterized job
exposures. Initial questionnaire and spirometry data were obtained in
386 workers. Multiple regression analyses showed significant adverse
effects of cumulative toluene diisocyanate exposure on airway
responsiveness. According to Diller (1987), the incidence of
isocyanate asthma reported from different studies varies between 0 and
25%. Reasons for differences in observed incidence are intensity of
isocyanate exposure, criteria for diagnosis, mode of calculation,
sensitizing capacity of different isocyanates, individual
predisposition and confounding factors. No geographical or ethnic
difference was observed.
5.8.1.2 Acrylates
Acrylate monomers are used in a variety of industrial fields.
Their industrial use is increasing, since they have many features that
make them superior to formerly used chemicals (Savonius et al., 1993).
Contact sensitization is a well-known adverse health effect of
exposure to acrylates, but they may also cause respiratory symptoms.
The main acrylic compounds currently in use are acrylates,
cyanoacrylates and methacrylates. While methacrylates are well-known
contact sensitizers, cyanoacrylates have caused only few cases of
contact allergy. Acrylates may also have other harmful health effects.
Hand and finger symptoms and paraesthesiae have been reported among
dental personnel preparing acrylates with their hands. The domestic
use of acrylates is limited and the sensitizing problem is mainly
occupational, but probably occurs in many different industries.
Cyanoacrylates are used mostly as a component of a high strength glue
used for joining different materials. Methacrylates are used as
adhesives, as dental and orthopaedic fillings, as material for
protheses and as an embedding material for different purposes,
including histological preparations.
5.8.1.3 Anhydrides
Methylhexahydrophthalic anhydride (MHHPA) and
methyltetrahydrophthalic anhydride (MTHPA) are dicarboxylic anhydrides
used as hardeners for epoxy resins. MHHPA and MTHPA typically require
an elevated curing temperature (50-200 °C), which facilitates escape
of anhydride vapours. Anhydrides are low relative molecular mass
chemicals that have been reported to cause immunologically mediated
respiratory diseases (Tarvainen et al., 1995). Contact urticaria and
other skin symptoms have also been described. Some anhydrides, e.g.,
phthalic anhydride, have caused generalized urticaria, in connection
with respiratory symptoms, after high exposure.
5.8.1.4 Solder flux
Questionnaires and lung function measurements were administered
to 104 electronic workers in the USA who soldered printed circuits
boards. Symptoms of eye, throat and nose irritation occurred in nearly
half of the group. Lower respiratory tract symptoms, including cough,
phlegm production and wheezing, also occurred with increased
frequency, compared with reported rates among a general population
sample (Greaves et al., 1984).
5.8.2 Metals
5.8.2.1 Cobalt
Several clinical and experimental findings point to cobalt as a
main sensitizer and causal agent of hard metal asthma (Nemery et al.,
1992; Swennen et al., 1993; Cirla, 1994; Lauwerys & Lison, 1994).
Clinical features have been clearly identified by bronchial
provocation tests. IgE and IgG antibodies with cobalt specificity have
been demonstrated. Clinically, the ability of cobalt to induce delayed
hypersensitivity is well known for contact dermatitis (Cirla, 1994).
Occupational exposure to cobalt occurs mainly by inhalation in various
industries and occupations involved in the production and processing
of metal and various cobalt-containing alloys and salts. The potential
for exposure to cobalt is particularly important during the production
of cobalt powder, the production and processing and use of hard
metals, the polishing of diamonds with cobalt-containing disks, the
use of pigments and dryers containing cobalt salts and the processing
of cobalt alloys. In a cross-sectional survey of 194 workers from 10
diamond polishing workshops and 59 workers from 3 other workshops, a
questionnaire was administered and spirometry was performed to assess
whether exposure to cobalt was associated with respiratory impairments
(Nemery et al., 1992). Spirometry showed significantly lower indices
of ventilatory function in the group with the highest exposure to
cobalt. These differences were not due to differences in smoking
habits.
5.8.2.2 Metal-polishing industry
A comparative study of spirometric measurements in 104 polishers
and 90 unexposed controls was carried out in 25 brass and steel ware
polishing industries in Moradabad, India (Rastogi et al., 1992). The
polishing process generates dust, containing fine particles of emery
and metal, which are mainly composed of copper and zinc and constantly
inhaled by the polishers. Of the polishers 58.6% had one or more
respiratory symptoms as compared to 25.5% of the controls.
Occupational asthma was found to be confined to polishers, 4.8% being
affected. The polishers exhibited significantly greater reduction in
various lung function parameters over the work shift, which was larger
in smokers than in non-smokers. The duration of exposure was directly
correlated with acute fall in lung function.
5.8.2.3 Aluminium
Occupational asthma is a major respiratory health problem within
the primary aluminium industry (O'Donnell, 1995). In Australia and New
Zealand the incidence of occupational asthma in primary aluminium
smelting varies between smelters, estimates ranging from 0 to 2%.
Cases showed no association between the frequency of the symptoms or
the severity of bronchial hyperresponsiveness and a family history of
asthma, atopic skin test, tobacco smoking or age. Current evidence
suggests that occupational asthma in the aluminium industry is
irritant induced and caused by inhalation exposure to gaseous or
particulate fluoride compounds.
5.8.2.4 Platinum salts
Nonspecific and specific bronchial hyperresponsiveness in
immediate-type asthma caused by platinum salts did not cease after
removal from exposure (Merget et al., 1994) (see also section
4.3.4.1).
5.8.3 Natural rubber latex
Populations at increased risk of developing natural rubber latex
hypersensitivity include health care workers, rubber industry workers
and subjects undergoing multiple surgical procedures, especially
children with spina bifida and urogenital abnormalities. Prevalence
figures for natural rubber latex allergy in studies using skin-prick
tests range from 2.9 to 17% among hospital employees and are around
11% among glove-manufacturing workers (Vandenplas, 1995). Natural
rubber latex allergy has been demonstrated in 32 to 50% of the
children with spina bifida by skin-prick test or serological testing.
The prevalence of sensitization to natural rubber latex in the general
population ranged from 0 to 9% according to the atopic status of the
populations under study (Turjanmaa, 1994; Vandenplas, 1995). The
observed rise in incidence of sensitization to natural rubber latex
during the last decade is probably related to the increased use of
natural rubber latex devices as a protective barrier against
infections. Other possible determinant factors include increased
recognition of natural rubber latex allergy by exposed workers and
clinicians, changes in manufacturing methods, and discontinuation of
steam sterilization.
Complications were thought to be limited to contact dermatitis
due to irritation and sensitivity to certain rubber additives, and
only few reports of phenomena consistent with natural rubber latex
sensitivity were published before 1984. A possible explanation for the
abrupt rise in the incidence of natural rubber latex sensitivity is a
change in rubber manufacturing or rubber processing. There is a lack
of epidemiological studies observing the longitudinal trends in
prevalence and natural history of natural rubber latex allergy. The
risk for natural rubber latex allergy among health care workers
appears to vary with the frequency and intensity of exposure.
Cross-reactivity between natural rubber latex and certain foods,
particularly banana, avocado, chestnut and other fruits such as kiwi,
papaya and passion fruit, have been described (Charous, 1994).
A questionnaire and skin-prick tests with natural rubber latex
and common inhalant allergens were administered to hospital personnel
(201 nurses, 50 members of the cleaning staff, 38 laboratory
technicians), of whom 4.7% showed skin reactivity to latex. Among
those with a negative skin test to natural rubber latex no one had a
history of occupational asthma. For the latex-sensitive subjects
(N=12), a histamine challenge test was performed, which showed
bronchial hyperresponsiveness in all subjects. Seven of the twelve
developed significant bronchial response to a challenge test with
latex gloves. The overall prevalence of occupational asthma due to
natural rubber latex was estimated as 2.5% (Vandenplas et al., 1995).
A skin-prick test was performed on 77 surgeons and nurses with an
allergen solution made from latex gloves and rubber latex catheters.
In addition, subjects answered a questionnaire on the history of hand
dermatitis, atopic eczema, rhinitis or asthma, and symptoms when using
natural rubber latex gloves. The prevalence of relevant immediate
allergy assessed by skin-prick test was 5.2%. The reliability in
detecting sensitized persons was limited. The agent used previously in
skin-prick testing to demonstrate natural rubber latex sensitization
gave unreliable results; the use of a standardized reagent is
necessary (Cormio et al., 1993).
Among 512 hospital employees 7.4% of the doctors and 5.6% of the
nurses working in operational units were allergic to natural rubber
latex. The frequency was lower in non-operating units and among
laboratory personnel (Turjanmaa, 1987).
5.8.4 Flour
Occupational respiratory diseases are common among bakers
(Keskinen et al., 1978; Meredith, 1993; Reijula & Patterson, 1994; De
Zotti et al., 1994; Lauwerys & Lison, 1994). In Finland, for example,
the mean annual incidence of occupational respiratory diseases was 31
per 100 000 among the general work force compared to 374 per 100 000
among bakery workers (Reijula & Patterson, 1994). The annual incidence
of occupational asthma as reported to the SWORD project in the United
Kingdom was 334 per million for bakery workers as compared to 658 per
million for paint sprayers or 175 per million for electronic
assemblers (Meredith, 1993). A substantial prevalence of wheat flour
allergy was found in bakers and pastry cooks (Armentia et al., 1990).
Not only wheat allergens, but also alpha-amylase must be considered as
the causative agent (De Zotti et al., 1994). Other allergens such as
storage mites are suspected to play a role in the development of
occupational asthma (Tee et al., 1992a,b). Findings from the initial
cross-sectional phase of a cohort study of employees exposed to flour
in bakeries or mills showed that subjects without previous exposure to
flour expressed related symptoms especially to flour aeroallergen
(Cullinan et al., 1994). Forty-four male workers exposed to flour and
164 unexposed controls were examined by using personal samplers
measuring inspirable dust concentrations. The proportion of subjects
with one or more symptoms and with bronchial hyperresponsiveness was
significantly greater among workers exposed to flour than among
controls. The conclusion was drawn that despite exposure to relatively
low concentration levels of inspirable flour dust, subjects working in
the baking industry are at risk of developing both respiratory
symptoms and airway hyperresponsiveness (Bohadana et al., 1994).
5.8.5 Animals
The occurrence of respiratory disease was studied in 257 active
veterinarians and 100 control subjects who had no occupational animal
contact. Asthma was significantly more prevalent in veterinarians than
in controls (Lutsky et al., 1985). Selected indicators of allergy and
atopy were studied to determine predictors of laboratory animal
allergy in a prospective study of laboratory technicians. Although the
prevalence of atopy and allergic symptoms had increased in exposed
technicians after the follow-up period of 2 years, this was also found
in an unexposed matched control group, and there were no significant
differences between the groups in any measured variable at follow-up
(Renstrom et al., 1994).
5.8.6 Other agents
The association between occupational exposure to low levels of
airway irritants and bronchial responsiveness to histamine was
assessed in 688 male workers of synthetic fibre plants (Kremer et al.,
1995). According to job titles, exposure status was grouped into 7
categories: 1) reference group, 2) white collars, 3) SO2, HCl,
SO42-, 4) polyester vapour, 5) oil mist and oil vapour,
6) polyamide and polyester vapour, 7) multiple exposures. A higher
prevalence of airway responsiveness was associated with history of
allergy and respiratory symptoms. A slight trend was seen for subjects
with more than 5 years of exposure to polyester vapour and oil mist
and oil vapour towards a higher prevalence of bronchial
hyperresponsiveness. No overall association could be demonstrated.
5.9 Allergic contact dermatitis
5.9.1 Epidemiology of allergic contact dermatitis
Only a few studies have investigated the frequency of allergic
contact dermatitis in the general population (Coenraads & Smit, 1995).
Nielsen & Menné (1992) studied the distribution of allergic contact
sensitization in an unselected sample of 793 individuals. Of these,
567 participants were patch tested with the standard series
(TRUE-test). This series included the most common contact sensitizers,
such as metals, fragrances, preservatives and medicaments. It has been
found that 50-80% (depending on patient selection) of all cases of
allergic contact dermatitis will be diagnosed using this series (Menné
et al., 1992). Among the 567 volunteers were 15.2% who positively
reacted to one or more of the substances included in the test series.
Multiple contact sensitizations were thus observed more commonly than
expected (Nielsen & Menné, 1992). Patients evaluated at patch test
clinics often have multiple contact allergies. In experimental studies
individuals with one contact allergy were found to be more easily
sensitized to a second one (Friedmann, 1990). In the clinical
situation the causes of multiple sensitivities are difficult to
evaluate, because factors such as genetic susceptibility, broken skin
barrier and multiple exposures are mixed up. Multiple contact
sensitivities are typically seen in individuals with long-lasting
chronic dermatitis.
In the following paragraphs, specific exposures closely related
to allergic contact dermatitis are discussed.
5.9.1.1 Nickel
Nickel-containing metal alloys and nickel-plated surfaces release
free nickel ions when in direct contact with human sweat. The typical
nickel dermatitis is therefore located beneath metal items, such as
buttons, jewellery, suspenders, glasses and similar objects. Among the
567 participants of an unselected sample of 793 subjects were 11.1%
women and 2.2% men affected with a nickel allergy (Nielsen & Menné,
1993) (see Table 18; section 4.1.5.1). Individuals primarily
sensitized from consumer items might, at a later stage, develop
occupational nickel dermatitis when occupationally exposed to this
metal. Tools and equipment used in different jobs by workers such as
carpenters, electricians, painters and plumbers were found to release
nickel (Lidén et al., 1996).
5.9.1.2 Chromates
Chromium salts, but not metallic chromium, are sensitizing. The
hexavalent salts are the main sensitizers as they penetrate the skin
more easily than the trivalent chromate (Gammelgaard et al., 1992).
Chromate was found to be a cause of occupational hand eczema among
employees in the construction industry, mainly because of the presence
of hexavalent chromate in wet cement (Irvine et al., 1994). The
irritating capacity of cement (abrasive and high pH), combined with
the potent allergen hexavalent chromate, caused occupational hand
eczema in up to 10% of the workers. Addition of ferrosulfate to cement
reduces hexavalent chromate to the trivalent state which has a minimal
bioavailability. Introduction of ferrosulfate addition to cement in
the Scandinavian countries has significantly reduced occupational hand
eczema (Avnstorp, 1992).
5.9.1.3 Fragrances
Allergic contact dermatitis can also be caused by fragrances
(Johansen & Menné, 1995; Johansen et al., 1996a, 1997; Scheinman,
1996). In a study by Nielsen & Menné (1992), 1%-2% of the 567
participants were affected by fragrance allergy (Table 18). Geier &
Schnuch (1996) found a frequency of fragrance allergy of 8 to 17%
among eczema patients.
5.9.1.4 Preservatives
Preservatives are widely used in water-based cosmetics,
households, and industrial products. Preservatives have antimicrobial
effect against a wide range of microorganisms. The most widely used
preservatives in cosmetics are the parabens, formaldehyde and
formaldehyde releasing substances (quaternium 15, diazolidinyl urea,
imidazolidinyl urea) and the isothiazolones (Andersen & Rycroft,
1991). Formaldehyde and isothiazolones are also frequently used in
industrial products. In a British study, the frequency of contact
allergy in dermatology patients varied between 1 and 3% for different
preservatives (Jacobs et al., 1995). In another study the prevalence
of contact allergy to formaldehyde in eczema patients was as high as
8% (Flyvholm et al., 1997).
5.9.1.5 Medicines
The frequency of allergic contact dermatitis to topical medicines
varies considerably from country to country, and even within regions,
depending upon the availability and product preference by local
prescribing doctors. Most cases of allergic contact dermatitis to
medicines are caused by substances having doubtful documented effect
or which can be replaced by other medicines (Angelini, 1995).
Systematic studies have shown that 1-3% of eczema patients are
contact-sensitized to topically used steroids (Dooms-Goossens &
Morren, 1992; Lauerma, 1992). In addition, traditional remedies, such
as balsam of Peru and Propolis, are well-known sensitizers (Li, 1995).
5.9.1.6 Plants and woods
Plants and woods contain a diversity of strong and weak allergens
(Ducombs & Schmidt, 1995). Allergic contact dermatitis to plants and
woods usually presents itself with acute oedematous bullous lesions
that spread to skin areas distant from the primary contact. Airborne
and dustborne patterns can be seen with severe facial dermatitis and
flexural dermatitis. In North America poison ivy and poison oak are
important plants because of their high content of urushiols, which are
potent allergens (Kligman, 1958a,b). These allergens are also present
in plants and trees in Asia and Australia. Allergic contact dermatitis
from these plants represents an occupational health problem for
outdoor workers.
The Compositae family comprises 13 000 species. It includes
decorative flowers such as chrysanthemums and dahlias, but also a
number of common weeds and salads. Some cases of Compositae
dermatitis present an airborne pattern often with a photo-aggravated
facial dermatitis. This type of dermatitis is caused by different
weeds in Europe, North America and Australia. Another variant of the
disease has been recognized, giving a high morbidity and some
mortality, in rural areas in India. The disease has been termed
parthenium dermatitis after the offending plants ( Parthenium
hysteropherous) (Lonkar et al., 1974). The main allergens in
Compositae plants are different sesquiterpene lactones. In
consecutive patch-tested eczema patients in Northern Europe 1-3%
reacted to the sesquiterpene lactone mix (Ducombs et al., 1990). Most
of these were hobby gardeners with severe hand eczema, but there were
some cases among professional gardeners. The development of the
sesquiterpene lactone mix illustrated how new diagnostic technologies
can change the understanding of allergic skin diseases. The
development of these techniques for routine testing has dramatically
changed the prognosis for these patients, as the correlation between
plant contact and their hand eczema was unrecognised earlier.
5.9.2 Lack of a relationship between atopy and allergic contact
sensitization
Atopic hospital employees, who performed wet work, had a similar
prevalence of contact allergy (22%) to that of their non-atopic
colleagues (21%) (Lammintausta et al., 1982). In a hospital-based case
series, the prevalence of contact allergy against ingredients of
topical medications was common in subjects with atopic dermatitis.
However, the sensitization rate to multiple other substances was low
among these patients (Lammintausta et al., 1992). In another hospital
based-study of patients with hand eczema, participants with past or
present atopic disease showed a positive patch test reaction in a
significantly lower proportion than non-atopic individuals.
Furthermore, of all participants with a history of atopy, 22% had
developed allergic contact dermatitis, while the corresponding figure
for non-atopics was 45% (Rystedt, 1985). The reason for the apparently
lower prevalence of allergic contact dermatitis in atopics might be an
abnormal function of T-lymphocytes in atopic patients (Menne et al.,
1987; Hanifin & Chan, 1995). However, a population-based study in
Norwegian school children indicated a higher rate of contact allergies
in atopic compared to non-atopic participants (Dotterud & Falk, 1994;
Dotterud & Falk, 1995).
5.10 Diet
It has been hypothesized that changes in food habits are related
to the increased prevalence of atopic diseases. Seaton et al. (1994)
suggested that the increase in atopic disorders in the United Kingdom
may in part be a result of a change in diet. Between 1961 and 1985 the
average weekly consumption of fresh fruit, green vegetables, potatoes,
fresh fish and red meat was reduced in Britain. The authors argue that
a reduced dietary intake of natural antioxidants is related to a
higher susceptibility to oxidant attack and airway inflammation. The
food groups that were consumed less frequently between 1961 and 1985
are main sources of antioxidants, such as vitamin C and beta-carotene,
ubiquinone, and cofactors for antioxidant defence mechanisms, such as
selenium, zinc and copper.
The relationship between certain food groups and the risk of
asthma was recently studied by Hodge et al. (1996). In this study,
diet was assessed in 574 children by a detailed food frequency
questionnaire including 200 food items, which was related to airway
disease defined by respiratory symptoms or airway responsiveness to
exercise. Children who ate fresh, oily fish had a significantly
reduced risk of current asthma. No other food groups or nutrients were
significantly associated with either an increased or reduced risk of
current asthma (Hodge et al., 1996). The results of a cross-sectional
study in Leipzig, Germany, indicated that a change towards a higher
consumption of margarine was positively associated with hay fever; in
turn, changes in the consumption of butter showed an inverse
association with hay fever and atopic sensitization (von Mutius et
al., 1998).
Adverse reactions to food are a commonly encountered condition,
especially in infancy and early childhood. The highest prevalence
occurs between 1.5 and 3 years of age when in this age group as many
as 25% have been reported to have adverse reactions to food (Björksten
& Kjellman, 1987). However, only a minority of these reactions depend
on immunological mechanisms (i.e., allergy) (Björksten & Kjellman,
1987). Epidemiological studies on the most important dietary factors
in relation to atopic diseases are reviewed below.
5.10.1 Breast feeding
The prophylaxis of atopy has been sought by elimination diets and
by other preventive measures (Saarinen & Kajosaari, 1995). The role of
breast feeding and/or avoidance of formulas based on cows' milk in
early infancy has been the focus of much controversy (Anonymous,
1982). Small amounts of protein ingested by the mother are secreted
unchanged into breast milk. Potentially allergenic food ingested by
the mother may thereby be transferred to the infant and cause
sensitization. For this reason, maternal dietary restriction during
lactation has been recommended (Hattevig et al., 1989). A prospective,
long-term follow-up study from infancy to early adulthood indicated
that breast feeding can protect against development of atopic disease
(Saarinen & Kajosaari, 1995). Differences between the infant feeding
groups were identified for atopic eczema, food allergy and respiratory
allergy. Breast feeding for longer than 1 month without other milk
supplements offers prophylaxis against food allergy at 3 years of age
and also against respiratory allergy at age 17 years. Six months of
breast feeding was required to prevent eczema during the first 3 years
and possibly also to prevent substantial atopy in adolescence. Thus in
this study, breast feeding seemed to confer long-term protection
against allergic sensitization.
In a trial to examine the effect of different feeding patterns on
the incidence of atopic disease in newborns with a family history of
atopy, the incidence of atopic symptoms in the control group of
infants was 27% for those with a single relative with atopy and 50%
for infants with a biparental history of atopy (Bardare et al., 1993).
Breast feeding effectively reduced the incidence of atopic symptoms
when the mothers complied with prescribed dietary restrictions. This
may indicate that dietary restrictions are especially important in
infants with biparental history of atopy.
The stated reasons for discouraging the premature introduction of
solid food include the possible risk of excessive weight gain,
vulnerability of the gut to infection, and increased susceptibility to
the development of allergic disease. The incidence of gastrointestinal
illness, wheeze, and nappy dermatitis was not found to be related to
early introduction of solid food feeding. There was a significant but
rather small increase in respiratory illness at a particular age among
infants given solids early. The incidence of eczema was increased in
those infants who received solids at 8-12 weeks of age (Forsyth et
al., 1993).
Risk of atopy has been associated with a high cord-blood IgE
(Kjellman & Croner, 1984). Therefore, many studies have focused on
investigating those infants with high cord-blood IgE because
development of an atopic disorder is more likely in these children. A
dual approach of allergen avoidance, focusing on foods (breast milk
and extensively hydrolysed formulas) and aero-allergens (treatment of
bed- and living-room with acaricides), in comparison with controls who
did not undergo any intervention, was beneficial in selected high-risk
infants (Hide et al., 1994). Avoidance of potent food allergens in
early life may increase the threshold for sensitization in those
high-risk infants. Whether sensitization has been avoided or merely
deferred has yet to be proved. Reduced exposure of infants to
allergens in food and in house dust lowered the frequency of allergic
disorders in the first year of life for high-risk children,
pre-natally randomized to a prophylactic or control group (Arshad et
al., 1992). A similar result was found in an outpatient follow-up of
777 infants with a very low birth weight. Comparison after random
assignment to early diet of human milk versus cows' milk-based
pre-term or term formula revealed an increased risk of atopy for early
exposure to cows' milk (Lucas et al., 1990).
5.10.2 Sodium
Asthma mortality is related geographically to sales of table salt
and both epidemiological and experimental evidence suggest that a high
dietary sodium intake may increase airway responsiveness. Studies have
shown that a low-sodium diet contributes to a decrease in symptoms in
children with severe asthma. Further investigations have presented
ecological, observational and experimental evidence supporting a
relation between salt intake and airway responsiveness (Tribe et al.,
1994). Regional data from England and Wales showed a strong
correlation between table salt purchases and asthma mortality for
adult men and children of both sexes, but not for adult women. Asthma
mortality in women was found to be more closely related to other
factors (Burney, 1987).
As part of a wider survey on asthma, 138 men living in two
Hampshire villages in England underwent a bronchial histamine
challenge test and had their 24-h urinary excretion of sodium
measured. Bronchial reactivity was strongly related to 24-h excretion
of sodium, suggesting that a high-sodium diet may enhance bronchial
reactivity (Burney et al., 1986). A study on the effect of dietary
sodium intake on the airway response to histamine supports the
hypothesis that a high-sodium diet increases bronchial reactivity in
men but not in women and suggests that moderate restriction of sodium
intake in asthmatic men would reduce bronchial reactivity (Burney et
al., 1989b).
A controlled cross-over study of 14 asthmatics found that high
salt intake worsened forced expiratory volume in one second (FEV1),
peak expiratory flow rate (PEF) and symptoms (Medici & Vetter, 1991).
This was interpreted as indicating a generalized dysfunction of
cellular sodium regulation and providing an explanation for the
salt-sensitivity of the asthmatics. In contrast, another study
(Britton et al., 1994) found no support for the hypothesis that a high
dietary sodium intake is a risk factor for airway hyperreactivity or
atopic disease in the general adult population. No relationship either
between Na+ and K+ intake assessed by a 7-day recall and bronchial
hyperresponsiveness or chronic respiratory symptoms was found in a
sample of 205 subjects (Zoia et al., 1995).
5.10.3 Selenium
There seems to be an association between reduced serum selenium
concentration and lowered activity of the selenium-dependent enzyme
glutathione peroxide (GSH-Px) (Hasselmark et al., 1993). The aim of a
double-blind randomized study in 24 patients suffering from asthma was
to investigate whether selenium (Se) supplementation in asthmatic
patients increases GSH-Px activity and possibly brings about clinical
improvement in the Se-supplemented group as compared to the placebo
group. In the Se-supplemented group there were significant increases
in serum Se and platelet GSH-Px activity after intervention, while no
significant changes in these parameters could be observed in the
placebo group. Although there were no significant changes in lung
function measures in the Se-supplemented or placebo group, a
statistically significant clinical improvement was observed in the
Se-supplemented group. There are several possible mechanisms whereby a
reduced selenium status with associated lower GSH-Px activity may
contribute to the pathogenesis of asthma. Further research into the
role of selenium and GSH-Px is required (Beasley et al., 1991).
Selenium concentrations and GSH-Px activity were lower in 56
asthmatic subjects compared to 59 control subjects (Flatt et al.,
1990). In both the asthmatic and control groups, the mean whole blood
selenium concentrations and GSH-Px activity were generally higher in
those who reported having eczema or rhinitis or had positive responses
to skin-prick tests. These findings are consistent with the hypothesis
that low selenium concentrations may play a role in the pathogenesis
of asthma in New Zealand.
5.10.4 Vitamins and antioxidants
Data from the Nurses Health Study (Troisi et al., 1995), a
prospective investigation of major chronic diseases, suggests that
anti-oxidant supplementation and intake of various fats during
adulthood are not important determinants of asthma, although vitamin E
from diet may have a modest protective effect. An effect of vitamin E
on the inflammatory process seems plausible. The relation between lung
function and dietary intake of the antioxidant vitamins C and E in the
general population was investigated in a cross-sectional survey of a
random sample of adults of Nottingham, England (Britton et al., 1995).
After adjustment for gender, height, skin-prick test result and
smoking, lung function parameters were significantly and independently
related to the mean daily intake of vitamin C. These data support the
hypothesis that lung function in the general population is related to
antioxidant intake and that these vitamins may play a role in
protecting against the development of chronic obstructive pulmonary
disease. The generalizability of these study results, however, may be
limited due to a low participation rate of less than 60%. In the
Zutphen study, fruit intake was inversely related to the incidence of
chronic nonspecific lung diseases (Miedema et al., 1993). No
association was observed with intake of several antioxidants. The
second US National Health and Nutrition Examination Survey (NHANES)
reported an inverse association between wheezing and serum vitamin C
and the serum zinc/copper ratio. These data suggest that several
dietary constituents may influence the occurrence of respiratory
symptoms in adults, independently of cigarette smoking (Schwartz &
Weiss, 1990). The relationship between ventilatory function and winter
fresh fruit consumption was studied in a random sample of British
adults (Strachan et al., 1991). These findings suggest that
antioxidant and other actions of vitamin C may protect against
pulmonary emphysema and may reduce bronchorestrictor responses to
environmental pollutants.
5.11 Number of siblings and crowding
Factors playing a role in the prevalence of allergic disease in
industrialized countries might be associated with the generally
improved standard of living (Williams et al., 1994b). Along with this
change goes a smaller family size and lower number of children in
families. Studies have observed a decrease in the prevalence of
allergic rhinitis, eczema (Strachan, 1989, 1996)) and asthma symptoms
(Shaw et al., 1994) with an increase in the number of older siblings.
In addition, it was shown that the prevalence of atopic sensitization
decreases with an increasing number of siblings (von Mutius et al.,
1994a), and a strong inverse relation was found between atopic
sensitization and the number of persons per room in households
(Braback et al., 1995). Viral or bacterial cross-infections,
especially in early life, which may occur more frequently in larger
families, have been discussed to have a protective role against atopic
disease by preventing proliferation of Th2-lymphocytes (Romagnani,
1992b; Holt, 1994; Martinez, 1994). Helminth infestations have also
been suggested to have a protective effect against allergy development
(Williams, 1992) but studies in populations with a high prevalence of
asthma indicated no such beneficial effect (Mantle & Pepys, 1974;
Martinez, 1994).
5.12 Indoor environment
One of the most important changes in lifestyle during the past
decades in industrialized countries is the change in indoor climate
mainly due to modern building and furnishing materials, better
insulation, wall-to-wall carpeting, increased indoor temperature and
less ventilation. As a consequence of these factors and behavioural
changes, such as indoor pet keeping, many pollutants and allergens can
accumulate within dwellings. This may lead to higher sensitization
rates to indoor allergens and consequently to an increased incidence
of allergic diseases like asthma. This is especially relevant because
people in industrialized countries spend up to 90% of their time
indoors (Platts-Mills, 1994; Berglund et al., 1994). A variety of
indoor and outdoor sources contribute to the indoor pollution burden
and might play a role in the development or exacerbation of allergic
disease. Important indoor sources are respirable particles from stoves
and tobacco smoke; combustion products from cookers, ovens and
heaters; formaldehyde from foam insulation, chipboards, furniture and
fabrics; volatile organic compounds (VOCs) and other chemicals from
paints, sprays fabrics, and combustion; biological material from
animal sources such as dust mites, cats, etc., or from fungi, bacteria
and pollen (Angle, 1988; Karol, 1991). Ventilation, humidity and
temperature of dwellings also have an important influence on indoor
concentrations of many pollutants and allergens (Brunekreef et al.,
1989; Beggs & Curson, 1995). For many indoor factors, there is still a
lack of good epidemiological studies.
5.12.1 Tobacco smoke
Passive exposure to tobacco smoke is an important risk factor for
childhood asthma and wheezing, particularly when the child's mother
smokes. Many studies have reported a higher risk of asthma or
bronchial hyperresponsiveness among children exposed to tobacco smoke
(Ware et al., 1984; Dekker et al., 1991; Martinez et al., 1992;
Forastiere et al., 1992) and younger children are particularly prone
to these side effects (Arshad et al., 1993; Stoddard & Miller, 1995;
Platts-Mills et al., 1995). Children of smoking parents have also
increased reactivity to allergens as assessed by skin-prick test
(Martinez et al., 1988; Braback et al., 1995). Furthermore, children
whose mothers smoked during pregnancy show increased IgE-levels in
cord blood and increased risk of infant allergy (Magnusson, 1986).
Therefore, the increasing prevalence of smoking in women of
childbearing age may have contributed to the increase in atopic
disease in children (Burney et al., 1990).
Although a number of studies in adults have shown an association
between active smoking and asthma, others have failed to find such an
association. Thus, the evidence is not conclusive and the effect may
be only small (Platts-Mills et al., 1995). Active smoking can increase
total IgE serum concentration and ex-smokers show a decline in serum
IgE concentration after cessation of smoking (Burrows et al., 1981).
Studies on occupational allergies showed that the frequency of IgE
sensitization and asthma is higher in cigarette smokers (Anonymous
1985; Venables et al., 1985b). Little is known about the role of
active and passive smoking on allergic rhinitis, and studies show, if
any, only small effects (Ng & Tan, 1994a,b; Strachan, 1995; Tsunoda et
al., 1995).
In summary, smoking is known as a respiratory irritant and as an
important source for indoor pollution. However, although many studies
suggested that smoking plays a role in the etiology of asthma, there
is no clear evidence from the available data to indict smoking as the
driving force for the observed increases in asthma morbidity and
mortality (Weiss et al., 1993). Nevertheless, even if the relative
effect of active and passive smoking may be small, it can still have a
major impact if large parts of populations are exposed (attributable
risk) (Stoddard & Miller, 1995).
5.12.2 Pets
Many animals like cats, dogs, rodents, rabbits and cage birds
live within or in close proximity to homes. About one third to one
half of houses in the USA have a mammalian pet (Colloff et al., 1992;
Ledford, 1994). There is strong evidence that exposure to a number of
animal allergens can lead to primary sensitization and an increased
risk of developing allergic disease (Colloff et al., 1992). The most
important pet allergens are those from cats and dogs (Sears et al.,
1989; von Mutius et al., 1994b; Ledford, 1994; Strachan & Carey,
1995). Even after permanent removal of a cat from the home it may take
several months before the concentration of allergens in domestic dust
falls (Wood et al., 1992; Colloff et al., 1992; Munir et al., 1994b).
Pet owners visiting a home with no pets can bring in pet allergens on
their clothes, and rub it off during their visit, resulting in a
considerable amount of pet allergens in the visited flat (Munir et
al., 1993; Munir et al., 1994a,b).
5.12.3 Biocontaminants
5.12.3.1 House dust mites and insects
House dust mites are an important source of indoor allergens.
Most frequently detected species are Dermatophagoides pteronyssinus,
Dermatophagoides farina, Euroglyphus maynei and Blomia
tropicalis (Colloff et al., 1992; Ledford, 1994). The mites have a
narrow optimal temperature range for growth from 18°C to 27°C
(Ledford, 1994). Because dust mites depend on ambient humidity they
grow poorly in dry or high altitude climates. Mite allergens can be
measured directly by ELISA. Indirect measurements of mite allergens
rely on guanine quantification in house dust. However, this method has
been shown to be of limited clinical value since a considerable amount
of guanine may originate from non-house-dust mite sources (Hallas et
al., 1993). Sensitization to dust mites seems to play an important
role in the relation between damp homes and childhood respiratory
symptoms (Verhoeff et al., 1995). High mite allergen exposure has been
reported to increase the risk of sensitization in atopic children (Lau
et al., 1989) and exposure in early childhood has been shown to be a
determinant of subsequent asthma (Sporik et al., 1990). Sensitization
to Dermatophagoides pteronyssinus and other allergens was addressed
in a study performed shortly after the fall of the Berlin wall on
9- to 11-year-old children in eastern and western Germany. Skin-prick
testing showed sensitization rates of 10.3% to Dermatophagoides
pteronyssinus (wheal reaction >3mm) in western Germany and of
4.2% in eastern Germany, indicating that environmental and lifestyle
factors may have an influence on sensitization to common allergens
(von Mutius, 1994b).
Cockroaches are also a substantial indoor allergen source.
Sensitization has been shown to be a risk factor for emergency room
visits in asthmatics (Pollart et al., 1989). Exposure to high levels
of cockroach allergens and allergy against cockroaches was found to
explain much of the asthma-related health problems in inner-city
children from the USA (Rosenstreich et al., 1997). Exposure to
cockroaches has also been associated with allergic rhinitis (Ng & Tan,
1994b). Few data on sensitization to cockroaches exist for central
Europe (Colloff et al., 1992), but the available data suggest that the
prevalence of sensitization to cockroaches is low (Mosimann et al.,
1992; Munir et al., 1994a). Other allergens from insects include
moths, crickets, midges, locusts, beetles and various flies (Ledford,
1994).
5.12.3.2 Moulds
The most common indoor moulds responsible for allergies are
Aspergillus species, Cladosporium, and Penicillium (Ledford,
1994). Moulds are responsive to temperature, humidity and substrate
moisture level (Beggs & Curson, 1995). Damp housing has frequently
been shown to be a risk factor for respiratory symptoms in children
and adults (Brunekreef et al., 1989; Dekker et al., 1991; Brunekreef,
1992) and may play a role in the sensitization to mould allergens
(Verhoeff et al., 1995). However, it has been suggested that the
observed association between damp housing and childhood asthma may be
partly due to parental over-reporting (Strachan, 1988). Other possible
indoor sources of fungal allergens include air conditioning equipment,
humidifiers, degrading organic materials, and soil used for indoor
plants; these sources can also be important for allergens from
bacteria or protozoa (Dekker et al., 1991; Ledford, 1994).
5.12.4 Other indoor factors
A large variety of everyday rubber products (e.g., balloons, baby
pacifiers, sports equipment, adhesives, etc.) contain natural latex,
which may be a source of sensitization and a factor in allergic
disease expression. Airborne leaf parts of the popular indoor plant
Ficus benjamina (weeping fig) also contain latex particles (Axelsson
et al., 1990; Bircher et al., 1993). It is not yet known whether
natural latex, which is a well known occupational allergen
(Vandenplas, 1995) (see also section 5.8.3) has any impact on allergic
disease on a population level. Contrary to expectation, non-feather
bedding, especially foam pillows, have been suggested to be a possible
determinant for symptoms of asthma in adolescents (Strachan & Carey,
1995).
5.13 Indoor and outdoor environmental factors
5.13.1 Nitrogen dioxide
Nitrogen dioxide (NO2) is a strong oxidant. Indoor sources are
cigarette smoke, gas and oil heaters or cookers, which can result in a
high indoor concentration (Angle, 1988; Wardlaw, 1993). Main outdoor
sources are combustion of fossil fuels in motor vehicles and power
generation (Wardlaw, 1993). Combustion processes also generate a
mixture of NO2 and nitric oxide (NO), and formation of indoor nitrous
acid (HNO2) has also been demonstrated and associated with adverse
respiratory effects (Samet et al., 1993). Experimental exposure and
several epidemiological studies are inconclusive concerning the effect
of NO2 on lung function (Ware et al., 1984; Angle, 1988; Neas et al.,
1991; Gorski & Tarkowski, 1992; Samet et al., 1993). The current
understanding of NO2 is that it can decrease pulmonary function in
asthmatics, but its overall role in the development of allergic
disease is not clear (Angle, 1988).
5.13.2 Sulfur dioxide, acid aerosols and particulate matter
The sources of these major outdoor air pollutants are mainly
combustion of fossil fuels, like coal and oil, wood, and certain
industrial processes. Sulfur dioxide (SO2) in water forms sulfurous
acid (Gorski & Tarkowski, 1992). The indoor concentration of SO2 is
raised if unvented combustion devices for kerosene are used or when
cigarettes are smoked (Angle, 1988). Several studies on asthmatics
have shown that SO2 can provoke bronchoconstriction and asthma-like
symptoms even at low levels, especially during exercise and mouth
breathing (Linn et al., 1983a,b). Animal studies indicated an
enhancing effect of SO2 on sensitization to allergens (Riedel et al.,
1988).
Acid aerosols are comprised mainly of sulfuric acid and ammonium
bisulfate (Wardlaw, 1993), which has also been shown to cause an
increase in respiratory symptoms and a decrease in lung function
(Koenig et al., 1983; Hackney et al., 1989). Acid aerosols can also
comprise nitric acid, hydrochloric acid and hydroxymethansulfonic
acid. Outdoor airborne acidity is associated with daily respiratory
symptoms in asthmatics (Ostro et al., 1991).
Particulate matter (e.g., dust, dirt and smoke) is a major source
of air pollution and a complex and varying mixture of substances.
Sources are motor vehicle emissions, such as exhaust fumes and abraded
tyre fragments (Williams et al., 1995b), factory and utility
smokestacks, residential wood burning, construction activity, mining,
agricultural tilling, open burning, wind blown dust, fires, etc.
Several studies have suggested an association between particulate
matter (PM) exposure and asthmatic symptoms (Dockery et al., 1989; Xu
& Wang, 1993; Dockery & Pope, 1994; Abbey et al., 1995; Brunekreef et
al., 1995). Particles of a diameter <10 µm (PM10) are especially
important for respiratory disease because they are readily inhaled
deep into the lungs (CDC, 1994). Among them fine particles of a
diameter <2.5 µm and ultrafine particles (<0.1 µm) may be most
important (Peters et al., 1997). Animal studies indicated that diesel
exhaust particles may increase the risk of sensitization against
allergens (Muranaka et al., 1986; Takafuji et al., 1987; Fujimaki et
al., 1994).
5.13.3 Volatile organic compounds, formaldehyde and other chemicals
Volatile organic compounds (VOCs) comprise a broad spectrum of
substances including benzene, toluene, xylenes and aldehydes (Angle,
1988; Becher et al., 1996). VOCs are emitted by a large number of
materials and only few subgroups have been investigated for their
potential role in allergy. The total indoor amount of VOCs and several
sub-types like toluene and terpenes have been associated with
asthmatic symptoms (Norback et al., 1995). Outdoor sources of VOCs
have also been found to be related to increased rates of chronic
respiratory symptoms characteristic of reactive airways (Ware et al.,
1993). Another important VOC is formaldehyde, which is ubiquitous in
the human environment. Important indoor sources are smoking,
particle-boards, foam insulation and textiles (Imbus, 1985; Koenig,
1988). Formaldehyde exposure has been related to asthma and other
allergies (Imbus, 1985; Angle, 1988; Wjst et al., 1994; Norback et
al., 1995), but evidence that formaldehyde exposure can cause allergic
diseases in the airways is limited (Becher et al., 1996). Other
chemicals such as ethylenediamine and isocyanate may also contribute
to the allergic disease burden in the general community (Ledford,
1994), but population-based data showing an association between
allergies and indoor contamination by these pollutants are not
available. The increasing use and diversity of household cleaning
materials during the past decades have also been implicated in the
expression of allergic diseases (Williams, 1992).
5.14 Outdoor air pollution
A link between outdoor air pollution and the increased prevalence
of respiratory allergies such as asthma and allergic rhinitis has been
suspected for some time. However, whether atmospheric air pollution
can cause respiratory and other allergies is still not clear (Wardlaw,
1993). Contamination of the air with plant pollen is a major natural
source of air pollution. Important causes of outdoor air pollution are
burning fuels such as coal, oil and wood, smelting of ores, and other
industrial processes. Natural sources like volcanoes play only a small
role. Motor vehicle tail-pipe emissions are a major contributor of
several pollutants, such as diesel particles, oxides of nitrogen
(NOX), carbon monoxide and other airborne particles (Bascom, 1996).
The increase of allergic respiratory diseases coincided with a
decrease in many outdoor air pollutants like SO2 and total suspended
particles (TSP) (Weiss et al., 1993), whereas air pollution from motor
vehicle emissions (e.g., O3, NOX) increased during the same period
(Newman Taylor, 1995). Motor vehicles are also a major source of fine
suspended particles (see section 2.5.3). Most outdoor air pollutants
provoke more or less severe adverse effects in asthmatics, and
exposure to multiple pollutants may cause synergistic effects (Koenig
et al., 1990). A number of experimental animal studies indicate an
association between allergy and air pollution (Osebold et al., 1980;
Biagini et al., 1986; Muranaka et al., 1986; Takafuji et al., 1987;
Riedel et al., 1988; Takafuji et al., 1989; Suzuki et al., 1993;
Fujimaki et al., 1994). It is also assumed that pollutants can enhance
the allergenicity of common allergens like pollen (Ishizaki et al.,
1987; Behrendt et al., 1991; Gorski & Tarkowski, 1992). However, the
observation that the sensitization rate to common allergens in cities
in eastern Germany with previously high levels of traditional air
pollutants such as SO2 or particulate matter was low in comparison to
that of western Germany cities argues against the hypothesis that
traditional air pollution from industrial production and household
coal burning is the major factor driving the changes in morbidity
patterns of allergic disease (von Mutius et al., 1992; von Mutius et
al., 1994b). Similarly, a Swiss study reported effects of moderate
average air pollution concentration from PM10, NO2 and SO2 on
respiratory symptoms, such as chronic or nocturnal dry cough and
bronchitis, but not between air pollution and asthmatic and allergic
symptoms or diseases in children (Braun-Fahrländer et al., 1997).
These results are in close agreement with findings from the US Six
Cities Study (Dockery et al., 1989). In addition, the findings of the
ISAAC study do not provide support for an association between air
pollution and childhood wheezing; for example, countries with low
degrees of ambient air pollution such as New Zealand were among those
with the highest prevalence of asthma symptoms (ISAAC Steering
Committee, 1998). Likewise, recent results from the Pollution Effects
on Asthmatic Children in Europe (PEACE) project found only little
overall adverse effect of ambient air pollutants (e.g., PM10, black
smoke, SO2 and NO2) on respiratory health in children (PEACE, 1998).
5.14.1 Pollen and dust
Natural sources of air contamination are plant pollen from grass,
ragweed, trees, etc. (Schutz-Kiss et al., 1995), moulds (Dawson &
Mitchell, 1990), and natural particulate matter such as dust or dirt
(CDC, 1994) which can cause symptoms of asthma and allergic rhinitis.
Their role as a causal factor for the development of these diseases,
however, is not clear. Air pollution due to natural factors like
pollen or dust cannot easily explain the observed increase in allergic
diseases in many regions because humans have always had intense
outdoor contact with these substances. Several asthma outbreaks in
populations have been shown to be related to man-made airborne
allergen pollution. Examples are castor bean dust in USA, South Africa
and Brazil (Anto, 1995), and soybean dust which was released during
unloading of soybeans in the city harbour of Barcelona (Anto et al.,
1989).
5.14.2 Ozone
A major source of ozone (O3) is motor vehicle exhaust. O3 is a
main constituent of photochemical smog, and its formation requires
NOX and reactive organic compounds as precursors and ultraviolet
radiation (Gorski & Tarkowski, 1992; Wardlaw, 1993; Beggs & Curson,
1995). It is the most ubiquitous air pollutant in the USA (Koenig,
1995). Although O3 is mainly an outdoor pollutant, it is also present
in low concentrations in the indoor environment (Koren, 1995). Many
studies have shown that O3 has an aggravating effect in asthmatics
and can reduce lung function (Krzyzanowski et al., 1992; Gorski &
Tarkowski, 1992; Wardlaw, 1993; Koenig, 1995; Koren & Bromberg, 1995).
Unlike SO2 there seems to be no marked difference in acute responses
in asthmatics and non-asthmatics (Koenig et al., 1988). Animal models
suggest that O3, like other air pollutants, may increase
sensitization to allergens (Osebold et al., 1980; Sears et al., 1989).
Ambient levels of O3 may also have a synergistic effect with pollen
in the causation of allergic rhinitis (Bascom et al., 1990). However,
only a few epidemiological investigations have been performed on a
population level to evaluate the effect of ambient O3 on asthma and
allergies (e.g., Braun-Fahrländer et al., 1997) and further
investigations are needed to study the effect of O3 on these
disorders under real-life exposure conditions (Magnussen et al.,
1998).
5.14.3 Motor vehicle emissions
Motor vehicle traffic has increased dramatically in many
countries (Utell et al., 1994) and it has been speculated that this
increase may play a role in the observed changes of the prevalence of
allergies. A number of occupational studies have shown an association
between exposure to motor vehicle exhausts and adverse effects on
respiratory symptoms and lung function (Gamble et al., 1987; Evans et
al., 1988a; Ulfvarson & Alexandersson, 1990; Wade & Newman, 1993;
Raaschou Nielsen et al., 1995). Others, however, have failed to find
such an association (Speizer & Ferris, 1973; Tollerud et al., 1983;
Ames et al., 1984). An association between allergic sensitization and
components of motor vehicle exhaust fumes has been shown in various
animal studies (Osebold et al., 1980; Muranaka et al., 1986; Takafuji
et al., 1987; Riedel et al., 1988; Suzuki et al., 1993; Fujimaki et
al., 1994; Lovik et al., 1997). A number of experimental studies
suggested also an association between allergic disease and traffic
pollution (Molfino et al., 1991; Braun Fahrländer et al., 1994;
Devalia et al., 1994). Several epidemiological studies observed a
relationship between exposure to motor vehicle traffic at residence
and morbidity from respiratory and allergic disorders in adults
(Yokoyama et al., 1985; Ishizaki et al., 1987; Nitta et al., 1993) and
in children (Wjst et al., 1993; Edwards et al., 1994; Weiland et al.,
1994; Keil et al., 1996; Oosterlee et al., 1996; Duhme et al., 1996;
Brunekreef et al., 1997; Duhme et al., 1998a). Air pollution from
motor vehicle traffic derives also from mechanical abrasion of tyres,
which contain potentially allergenic latex particles (Williams et al.,
1995b). However, evidence that exposure to motor vehicle traffic can
cause asthma or allergies is not conclusive.
5.15 Conclusions
Asthma and allergic disorders represent a substantial burden not
only on the affected individuals but also on health care resources in
many countries. The costs of asthma are partly due to uncontrolled
disease, and are likely to rise as its prevalence and severity
increase (Barnes et al., 1996). One approach to reduce costs would be
to improve disease control. Another approach would be to reduce the
prevalence of asthma by preventive measures, and this would be
accompanied by a reduction in the costs of treatment and care (Peat,
1996). Environmental factors that have changed in the last decades
appear to be largely responsible for the observed increase in the
prevalence of asthma and allergic disease in many countries. The
determinants of these changes need to be identified in order to design
interventions that can reverse these trends. Such prevention
strategies in the field of asthma and allergies can aim at high-risk
groups or at populations as a whole (Rose, 1985). It is important to
note that even if a preventive measure offers little to each
individual it can bring large benefits to the community into which the
preventive measure is introduced (prevention paradox) (Rose, 1985).
It is often impossible to blame a single culprit within a complex
mixture of behaviours and exposures (such as indoor or outdoor air
pollution) for observed adverse health outcomes, and the studied risk
factor might also have different effects in the presence of other
factors (Greenland, 1993). Because randomized assignment of
individuals to certain exposures is impractical, if not unethical, in
environmental epidemiology, researchers rely mainly on data from
non-experimental studies with well-known inherent methodological
shortcomings (Rothman, 1993). Depending on the hypotheses being
studied, a number of epidemiological research strategies are
available. The applications, strengths and weaknesses of different
studies have been described (e.g., Hennekens & Buring, 1987; Rothman,
1993; Morgenstern & Thomas, 1993). Interpretation of (epidemiological)
study results has to consider random errors of estimation (due to
chance) and systematic errors or bias. Important sources of bias
(selection bias, information bias and confounding) may hamper the
validity of observed results. Therefore, possible systematic errors
should be considered already at the planning stage of any study and
measures should be implemented to minimize or control bias. It is also
crucial at the planning stage of a study to perform statistical power
calculations to evaluate how many study participants are needed to
assure a given probability of detecting a true effect of a given
magnitude.
Epidemiological studies applying accurate exposure and disease
measurements and taking into account important covariates, confounders
and effect modifiers are needed (Hatch & Thomas, 1993; Prentice &
Thomas, 1993). They can make an important contribution through
regulatory decisions in public health in the difficult field of risk
assessment for complex exposures.
Besides other measures, occupational sentinel health events
(Mullan & Murthy, 1991) and structure-activity research (Jarvis et
al., 1996; Graham et al., 1997) may be useful tools to evaluate the
sensitizing capacity of a chemical substance in populations. Lists of
well-known allergens categorized according to potency and degree of
exposure are available (Kayser & Schlede, 1995), and criteria for
classification of sensitizing substances in the environment have been
defined. Furthermore, by comparing different populations with
different grades of exposures, major disease determinants can be
uncovered that would otherwise remain undetected, if the suspected
risk factors show only little variation within a single population
(Rose, 1985). For comparison reasons, data should be collected and
analysed in a standardized way wherever possible. Using such
strategies, major health determinants have been successfully described
and studied in other fields, such as cardiovascular diseases and
cancer. It is an essential prerequisite of such internationally
conducted studies to obtain the health outcome and exposure data in a
standardized way. New epidemiological initiatives investigating
determinants of asthma and allergies need to incorporate these
principles, as is the case with the ongoing International Study of
Asthma and Allergies in Childhood (ISAAC) (Asher et al., 1995; ISAAC
Steering Committee, 1998).
6. HAZARD IDENTIFICATION: DEMONSTRATION OF ALLERGENICITY
6.1 Hazard and risk; allergy and toxicity
The conventional scheme for chemical risk assessment for human
health protection follows the sequence:
a) Hazard identification; what is the potential of the substance to
cause harm (sensitization and provocation of an allergic
reaction), and what is the dose-response relationship?
b) Exposure assessment
c) Risk assessment: what is the likelihood of eliciting an allergic
reaction in humans at the relevant level of exposure; are there
any groups of increased susceptibility?
d) Risk characterization: non-scientific consideration of the risk
weighed against the benefit of using the substance resulting in
the decision to ban or limit exposure (NAS, 1983, 1993).
Hazard identification, therefore, comprises procedures to
determine the potential of a substance to induce allergy or elicit
allergic reactions and the relationship between those properties and
the circumstances of exposure.
The prediction may be based on theoretical considerations of
chemical structure, possibly on in vitro experimental results, on
in vivo animal data, and on prior observations in humans. If it
depends on the results of laboratory studies and not on clinical
observations, the prediction, as is common to toxicity tests, must
take account of species differences in metabolism, responsivity, dose
(exposure), and the adequacy of validation of the experimental system.
Testing allergenic potential requires study of selected
immunological effects and differs from conventional toxicity testing
in the nature and content of its procedures, which are focused on
responses of the immune system and not on general screening for
changes in all body systems. In both types of testing, however, there
will be some form of relation between dose (exposure) and effect, as
the capacity of a substance to produce effects, its potency, will be
represented by the dose (exposure) required to produce sensitization
(or toxicity). A strong sensitizer will require only a small dose,
whereas a less potent compound will require a higher dose, or multiple
exposures. Unlike conventional toxicity, further exposure of a
sensitized animal (or man) will elicit a harmful allergic reaction
after a much smaller dose than that required for sensitization,
although there will still be a graduation of the severity and nature
of the hypersensitivity reaction, for example ranging from slight
bronchoconstriction to fatal bronchospasm or anaphylaxis after
respiratory challenge.
An additional important difference between conventional toxicity
and allergy is that allergic sensitization (the induced state of
hyperreactivity to a substance) normally persists for a long time,
even for life, whereas for many toxic responses a state of lasting
responsiveness is not induced. It is possible for different types of
hypersensitization and provocation to be effective in the same organ
but it is also possible for the route of sensitization and response to
subsequent challenge to differ, e.g., sensitization via the skin and
subsequent asthma on inhalation exposure.
6.1.1 Testing allergic potential and toxicity testing
Much testing to identify toxic hazard is done during industrial
development of substances for purposes ranging from new medicines or
consumer products to industrial intermediates or pesticides. There are
well-developed regimes of accepted procedures done under controlled
circumstances applicable to each intended use, and the results are
used for regulatory purposes to control risk. Most of these procedures
are not directed at revealing effects involving the immune system,
although indirect indications may sometimes be obtained that can
arouse suspicion that the immune system may have been affected.
Certain specialized procedures are also conducted, based on
consideration of the way in which humans may be exposed (e.g., in the
skin or by inhalation), which experience has shown can reveal certain
types of sensitizing and allergenic potential. The latter types of
test are the most important in the present context. The procedures and
their value and limitations are discussed here.
Laboratory tests, especially in vivo procedures, should be done
in such a way as to minimize the need for experimental animals and
scarce human and technical resources, and every attempt is made to
extract as much information as possible from the work that is done.
That includes considerable attention paid to quality assurance of
tests.
Accordingly, the testing of a new substance will follow a
sequence, moving from theoretical to practical procedures. Only those
techniques focused on the immune system are noted here, but it must be
realized that product development and occupational safety needs
require many other studies, too.
6.1.2 Databases and prior experience
A preliminary search should always be made for any information
about experimental or clinical findings about the immunological
consequences of exposure to the substance. This may include special
consideration of any groups considered to be particularly susceptible,
for example, because of pre-existing disease.
6.2 Validation and quality assurance
Obviously, the ideal situation would be that predictive tests
yield easy-to-interpret outcomes and no false negatives or false
positives, and that the tests will always give the same results,
regardless of when or where they are carried out. In order to achieve
this goal, validation of tests is required. Validation should occur at
two levels: first at the level of technical quality and second at the
level of specificity and sensitivity of the assay. It is important
that potential methods are carefully evaluated for interlaboratory
reproducibility and transferability, and for their ability to predict
an in vivo end-point. Owing to the complexity of the immune system,
it is quite likely that predictive assays for the capacity of
chemicals to induce skin, respiratory or food allergy, or autoimmunity
will not always function adequately to show the absence or presence of
such activity (Kammüller, 1996). Therefore, the outcome of these tests
should always be evaluated with great care.
6.3 Structure-activity relationships
Structure-activity models are directed towards a fuller
understanding of the relationship between chemical structure and
physicochemical properties and skin-sensitizing activity, with the
objective of deriving ideally quantitative structure-activity
relationships (QSAR). In this context, parameters that appear to be of
particular importance are protein reactivity and lipophilicity
associated with the capacity to penetrate into the viable epidermis
(Basketter & Roberts, 1990; Barratt et al., 1994a). The correlation of
the protein reactivity of chemicals with their skin-sensitization
potential is well established (e.g., Dupuis & Benezra, 1982), and it
is accepted that if a chemical is capable of reacting with a protein,
either directly or after appropriate (bio)-chemical transformation,
then it has the potential to be a contact allergen, assuming of course
that it can locate in the appropriate epidermal compartment.
It is not within the scope of this monograph to give a
comprehensive description of all the available models. A common
feature of many of these models is that their development was based
upon the mechanism of sensitization, i.e., the absorption of the
chemical sensitizer through the skin, followed by its covalent
modification of a skin-associated protein. Each of the existing
structure-activity relationship (SAR) models proposes structural
alerts, i.e., moieties associated with sensitizing activity. In all
cases, the structural alerts comprise electrophilic moieties, or
moieties that can be metabolized into electrophilic fragments
(proelectrophiles). For example, Benezra et al. (1985) developed a
hierarchical index of structures believed to be associated with
allergic contact dermatitis that contained amines, ketones, metals,
nitrogen-containing heterocycles, and oxygen-containing heterocycles,
among others. Barratt et al. (1994a,b) developed a list of structural
alerts based on the requirement for protein reactivity that included
alkylating, acylating, and arylating agents, electrophiles, thiol
exchange compounds, and free radical generators. Many of the existing
SAR systems have incorporated physicochemical considerations (Roberts
& Basketter, 1990a,b; Basketter et al., 1992; Ashby et al., 1995). The
mathematical model described by Roberts & Basketter (1990a,b) for
alkyl transfer agents has incorporated both a rate constant for
reaction of a chemical with a nucleophile, and a lipophilicity factor
(log P). Each of these models has been successful in predicting the
activity of moderate to strong contact sensitizers. Barratt et al.
(1994b) reported the greatest success in predicting active sensitizing
potential (98%). A smaller database using similar alerts (Payne &
Walsh, 1994) had a much poorer positive predictive ability of 57%. The
classification model of allergic contact dermatitis used by Hostynek
et al. (1996) made use of several parameters and multiple regression
to predict 79% of the active sensitizers and 88% of the inactive
chemicals. The relative alkylation index (RAI) is useful only in the
prediction of a homologous series of chemicals (Roberts & Basketter,
1990a,b). The model used by Benezra et al. (1985) did not specify
whether a validation was attempted, therefore predictive ability is
not known.
6.3.1 Case-Multicase system
The Case-Multicase system is not dependent upon a particular
mechanism of sensitization and has shown ability to predict activity
of weak sensitizers (Graham et al., 1996). The model operates by
fragmenting chemicals in the database into substructures containing
two or more heavy (non-hydrogen) atoms. It then identifies those
fragments that are statistically associated with active chemicals and
uses such fragments as structural alerts for prediction of test
chemicals.
The database for this model was derived from reports of animal
and human studies and consists of more than 1000 chemicals (Graham et
al., 1996). The model identified 49 structural alerts of allergic
contact dermatitis. The major ones were: (a) a nitrogen double-bonded
to a carbon or a nitrogen; (b) substituted aromatic structures; (c)
thiol- and disulfide-containing fragments; and (d) electrophilic
moieties. The model has been evaluated by testing its ability to
predict correctly the activity of chemicals for which there is
evidence of sensitization ability. The concordance between predictions
by the model and established evidence of sensitization was 90% (Graham
et al., 1996).
6.3.2 DEREK skin sensitization rulebase
A historical database (Cronin & Basketter, 1994) containing
results of about 300 guinea-pig maximization tests (Magnusson &
Kligman, 1970a,b), carried out over a number of years according to a
single protocol on defined single substances, was used to derive a set
of structural alerts for skin sensitization. The approach employed was
to group the substances, where possible, according to their most
likely mechanism of reaction with skin proteins. Where no mechanism
could be clearly identified, structural alerts were derived for groups
of chemicals with similar functional groups. This process initially
resulted in the production of around 40 structure-activity rules
(Barratt et al., 1994a), now increased to over 50. These were
incorporated into the expert system Deductive Estimation of Risk from
Existing Knowledge (DEREK) (Sanderson & Earnshaw, 1991; Ridings et
al., 1996). DEREK contains both a controlling programme and a chemical
rulebase. The chemical rulebase consists of descriptions of molecular
structural alerts, which correlate with specific toxicological
end-points.
Whilst details of the biology of skin sensitization are only
partly understood, it is now widely accepted that the ability to react
with a nucleophile, either directly or after appropriate metabolism,
is a prerequisite for the large majority of skin sensitizers. However,
the potential of a chemical to act as a contact allergen is further
modulated by its ability to penetrate the stratum corneum and
partition into the epidermal compartment of skin; this is apparent
from a number of QSAR studies (Roberts & Basketter, 1990a,b; Basketter
et al., 1992) in which skin-sensitization potential was found to
depend crucially on physicochemical parameters such as the log
octanol/water partition coefficient (log Pow). These parameters have
also been found to be equally important determinants of percutaneous
absorption (Flynn, 1990), with higher log Pow values, i.e., greater
lipophilicity, broadly leading to greater permeability. In QSAR
studies of skin permeability, the in vitro human skin permeability
coefficient has also been shown to decrease with increasing relative
molecular mass (Flynn, 1990) or molecular volume (Barratt, 1995). The
skin-sensitization potential of a series of substituted phenyl
benzoates was found to depend on log Pow and relative molecular
volume in the same way (Barratt et al., 1994c). The logical
consequence is that two chemicals may contain the same structural
alert (i.e., be reactive, presumably by the same mechanism), but one
will be a skin sensitizer because it can penetrate the skin whilst the
other will not be a skin sensitizer because its skin permeability is
too low, e.g., N-methyl- N-nitrosourea and streptozotocin (Ashby et
al., 1995).
6.3.3 SAR for respiratory hypersensitivity
An initial SAR model for respiratory-sensitizing chemicals has
been described (Karol et al., 1996). The model is based on the
Case-Multicase system and the database was derived from a critical
review of the published clinical literature. Criteria for inclusion of
published data included a decrement in pulmonary function resulting
from inhalation challenge with a non-irritating concentration of the
chemical.
In all, 39 respiratory chemical allergens were identified from
the literature search, all being obtained from human studies. Among
the chemicals were diisocyanates, acid anhydrides, antibiotics and
dyes. Since the model requires a data set of inactive chemicals, and
such chemicals could not be found in the literature, chemicals that
were inactive as dermal sensitizers were assumed to be inactive as
respiratory sensitizers as well, and were added to the respiratory
model as "inactive" chemicals.
The model identified structural alerts including the isocyanate
functionality, amines and aromatic fragments. When respiratory
sensitizers were compared with dermal sensitizers for both structural
alerts and physicochemical characteristics, differences were noted.
Among the physicochemical properties, respiratory chemicals had higher
mean relative molecular mass and greater water solubility when
compared with dermal sensitizers (Karol et al., 1996). However, the
discrimination of dermal and respiratory sensitizers remains
problematic.
6.4 Predictive testing in vivo
6.4.1 Testing for skin allergy
6.4.1.1 Testing in guinea-pigs
The guinea-pig was for many years the animal of choice for
experimental studies of contact sensitization, and several test
methods were developed in this species. The Draize test was developed
over 50 years ago (Draize et al., 1944) and was widely used, but this
is no longer the case and it has been superseded. Currently, the best
known and most widely applied are the Buehler test (Buehler, 1965),
the guinea-pig maximization test (Magnusson & Kligman, 1970a,b), and
the guinea-pig optimization test (Maurer et al., 1975), and have
formed the basis of hazard assessment for many years. Both the
Magnusson and the Buehler test are recommended according to an OECD
guideline (OECD, 1992). While these tests differ with respect to
procedural details, they are in principle similar. Guinea-pigs are
exposed to the test material or to the relevant vehicle. In the
Buehler test, both induction and challenge exposures are done
topically; in this test, false negatives are frequently observed. The
test was improved by occluded application of the test compound. In the
guinea-pig maximization test, induction is produced by intradermal and
occluded epidermal exposure, and in the optimization test induction is
done by intradermal exposure and challenge by intradermal and occluded
epidermal exposure. Adjuvant is employed also to augment the induction
of the immune responses. For induction, concentrations of up to 5% or
a maximum non-irritant concentration are used for intradermal
injections, and up to 25 % for epidermal application. Some time after
induction exposure, test and control animals are challenged at a
distant site with a sub-irritant concentration of the chemical, which
is generally lower than the concentration used for induction.
Challenge-induced inflammatory reactions, measured as a function of
erythema and/or oedema, are recorded 24 and 48 h later. Classification
of sensitizing activity is based usually upon the percentage of test
animals that display macroscopically detectable challenge reactions.
Any compound inducing at least 30% positive animals in an adjuvant
test is labelled as a sensitizer; in the case of a non-adjuvant test,
15% is sufficient for classification as a sensitizer.
Of the available guinea-pig test methods, the guinea-pig
maximization test is generally selected when the aim is to identify
the weakest of skin sensitizers. However, the method is not well
suited to the estimation of relative sensitizing potency, because of
the requirement for the use of intradermal injections of test material
and Freund's complete adjuvant (FCA) (Basketter et al., 1996). The
Buehler test is easier to use because the mode of application is
epicutaneous occlusive treatment for both induction and elicitation
(Chan et al., 1983).
Although guinea-pig test methods, such as the Buehler test and
the guinea-pig maximization test, have been in use for more than 25
years, there has not been extensive examination of their sensitivity
and specificity in comparison to what is known about human skin
sensitization. Both the tests, when conducted properly, offer
sufficient sensitivity to detect many known human skin sensitizers
(Wahlberg & Boman, 1985; Basketter et al., 1996). However, there has
been very little assessment on the specificity of these methods.
It should be noted that these guinea-pig methods are not well
suited to the identification of protein allergens. In general, the
maximization of exposure involved in these skin sensitization tests
will result simply in a large immune response. This will mask any
differential allergic response.
6.4.1.2 Testing in mice
Increased understanding of the cellular and molecular mechanisms
associated with contact allergy have derived largely from experimental
investigations in the mouse. Two different types of tests to predict
the capacity of chemicals to induce skin allergy have been developed.
One is the mouse ear swelling test (MEST), which, like the guinea-pig
methods described above, is based upon the evaluation of
challenge-induced reactions in previously sensitized animals (Gad et
al., 1986). In this test, mice sensitized by a comparatively rigorous
regime (the intradermal injection of adjuvant followed by the daily
application of the test material, for 4 consecutive days, to
tape-stripped skin) are challenged on one ear with the test compound
and on the contralateral ear with vehicle alone. Sensitizing potential
is evaluated by consideration of both the degree of oedema (ear
swelling) induced and the percentage of animals displaying a reaction
(Thorne et al., 1991).
The second test developed in mice is the local lymph node assay
(Kimber et al., 1989). In contrast to the mouse ear swelling test and
guinea-pig assays, activity here is measured as a function of events
occurring during the induction, rather than elicitation, phase of
contact sensitization. Mice are treated daily, for 3 consecutive days,
on the dorsum of both ears with the test material or with an equal
volume of vehicle alone. Proliferative activity in draining lymph
nodes (measured by the incorporation in situ of radiolabelled
thymidine) is evaluated 5 days following the initiation of exposure.
The local lymph node assay has been the subject of extensive
comparisons with guinea-pig methods (Kimber et al., 1994; Basketter et
al., 1996), and offers significant advantages compared with the
available guinea-pig test methods. Important among these is the fact
that there is an objective read-out. The assay has been evaluated by
collaborative trials, and an OECD (Organisation for Economic
Co-operation and Development) test guideline issued in 1992 states
that the local lymph node assay (or the MEST) can now be used as a
first stage in an assessment of skin-sensitizing activity (OECD, 1992)
If a positive result is seen, then a test substance may be designated
a potential sensitizer and it may not be necessary to conduct a
further guinea-pig test. However, if a negative result is seen, a
guinea-pig test must be conducted subsequently.
6.4.1.3 Predictive testing for skin allergy in humans
There are various skin test procedures for the diagnosis of
several types of contact dermatitis (see also chapter 4). Basically,
predictive tests in humans for skin allergy are similar to diagnostic
tests for contact sensitization, but the aims are different. For
diagnostic tests, the aim is determining sensitization to chemicals to
which there was a prior exposure, and avoiding new sensitizations
because of the procedure. For predictive testing in humans the aim is
to show the sensitizing capacity in individuals that have not been
exposed to the chemical previously.
Predictive testing in humans generally requires multiple
occlusive patches for induction of sensitization (10 patches, 48 h
each, same site) followed by a 2-week rest period and then challenge
(48 h) with a patch at a new skin site (Marzulli & Maibach, 1973,
1996). There are a number of variations in these procedures, including
the use of provocative chemical agents such as sodium lauryl sulfate
(Kligman, 1966c), special skin preparation such as stripping (Spier &
Sixt, 1955) or freezing (Epstein et al., 1963), special patches
(Magnusson & Hersle, 1965), high concentrations at induction (Marzulli
& Maibach, 1974), and 25-200 test subjects (Draize et al., 1944;
Kligman, 1966b). It is not entirely clear, however, how useful these
variations are and what the limitations are under the use conditions
because validation has not kept pace with the use of the different
approaches. Furthermore, predictive tests are often performed on a
single chemical entity, whereas ultimate use may occur as part of a
multicomponent formulation in a marketed product, where the vehicle
and associated ingredients may influence the outcome. It is essential
that the test has sufficient statistical power to provide appropriate
protection for the population at risk. This is illustrated by the
mathematical considerations of Henderson & Riley (1945) in their
classical paper on extrapolating data from a small test population to
large numbers of users. Briefly stated, there may be no skin reactions
in a test population of 200 random subjects, yet as many as 15 of
every 1000 of the general population (95% confidence), or up to 22 of
every 1000 may react (99% confidence). If the test group is reduced to
100 subjects, up to 30 of every 1000 of the general population may
react (95% confidence). Conversely, when 1 of 200 subjects in a test
population becomes sensitized, a test population of 10 000 subjects
might show from 1 to 275 sensitized, with 95% confidence.
Prospective tests of skin sensitization using human volunteers
should always be conducted in accordance with ethical principles.
6.4.2 Testing for respiratory allergy
Most of the animal models that are used for studying specific
respiratory hypersensitivity were developed using allergens with high
relative molecular mass, notably proteins. Very few animal models have
been developed as predictive tests for hazard identification and risk
assessment in the area of chemical-induced respiratory allergy. The
majority of these models are based upon antibody-mediated events. The
models differ with regard to the following aspects: the animal species
utilized, the route of administration of the agent, the protocol for
both induction and elicitation of responses, type of response
measured, and judgment of significant response.
6.4.2.1 Guinea-pig model
The guinea-pig has been used for decades for the study of
anaphylactic shock and pulmonary hypersensitivity (Sarlo & Karol,
1994). The guinea-pig is similar to humans in that the lung is a major
shock organ for anaphylactic responses to antigens. The guinea-pig
responds to histamine and can experience both immediate-onset and
late-onset responses. Airway hyperreactivity and eosinophil influx and
inflammation can also be demonstrated in this animal species.
Mechanistic studies have been hampered by the lack of reagents needed
to identify cells and mediators in respiratory allergy. In addition,
the major anaphylactic antibody is IgG1a, whereas it is IgE in other
rodent species and in humans. The in vivo passive cutaneous
anaphylaxis (PCA) assay was used to measure IgG1a antibody responses,
but ELISA methods have now been developed that have eliminated some of
the variability seen with the PCA.
The guinea-pig inhalation model focuses on identifying chemical
sensitizers by measurement of the response, or elicitation phase, of
sensitization. In contrast with the mouse IgE test, the model does not
depend upon a preconceived mechanism of sensitization. Rather, it
functions by reproducing the characteristics that typify the
hypersensitivity reactions, i.e., the physiological response of the
airways and the pulmonary inflammation. Measurement of specific
antibody formation provides ancillary evidence of the response. The
method has been successfully used to distinguish low relative
molecular mass contact sensitizers from respiratory sensitizers. The
model utilizes inhalation as the route of exposure for both the
sensitization phase and the elicitation phase of the response. A
variation of the method is the use of intratracheal administration of
the agent (Sarlo & Karol, 1994). The model has the capacity to assess
immediate-onset responses (IAR), as well as late-onset responses
(LAR). The latter is possible since minimal restraint of animals is
used. A dynamic air supply and passive detection devices allow
continuous 24-h monitoring of respiratory function of animals. The
ability to detect late-onset responses makes the model particularly
appropriate for evaluation of chemical allergy, where late-onset
responses are a frequent occurrence.
The advantages of the guinea-pig model for chemical sensitization
include: use of inhalation as the relevant route of exposure;
generation of atmospheres of reactive chemicals; measurement of
physiological responses including immediate-onset responses,
late-onset responses, fever and hyperreactive airways; measurement of
specific antibody production; and histopathological evaluation of
pulmonary tissue. Disadvantages of the model are the cost, the time
involved, the need for specialized facilities, and the employment of
guinea-pigs. The latter is a disadvantage in that IgGl rather than IgE
is the major class of cytophilic hypersensitivity antibody in
guinea-pigs.
Variations of the guinea-pig model have been developed to
optimize the response, to monitor additional respiratory parameters,
or to simplify the procedure (Briatico-Vangosa et al., 1994). Such
variations include single or repeated intradermal administration of
free chemical, elicitation with a multivalent chemical-adducted
protein, and measurement of flow-volume loops, respiratory minute
volume, inspiratory and expiratory time, and peak respiratory flow
rates. Using chemical-protein adducts for elicitation avoids the
possible development of airway hyperreactivity due to chemical
irritation (analogous to the reactive airways syndrome in humans).
Using the guinea-pig model, differences were readily apparent
between sensitization to allergens of high relative molecular mass
(HRMM) versus those of low relative molecular mass (LRMM). Responses
to ovalbumin and to bacterial subtilisin (HRMM allergens) consisted of
severe immediate-onset responses in 90-100% of animals and late-onset
responses in 50% of animals (Thorne & Karol, 1989). By contrast,
sensitization to diphenylmethane-4,4-diisocyanate (MDI), a LRMM
allergen, consisted predominantly of late-onset responses (Karol &
Thorne, 1988). This finding reflects the human experience where
late-onset responses are the most frequently observed responses to
LRMM allergens.
The model has been validated in two ways. Firstly, it has been
established in several laboratories (Pauluhn & Eben, 1991; Sarlo &
Clark, 1992; Stadler & Loveless, 1992; Warren et al., 1993; Sarlo &
Karol, 1994) and responses of animals to inhaled toluene diisocyanate
have been reproduced. This confirms the robustness of the model.
Secondly, the model has been found to distinguish pulmonary from
dermal chemical sensitizers, and from non-sensitizers. For example,
inhalation of toluene diisocyanate (Karol, 1983) and
diphenylmethane-4,4-diisocyanate (Karol & Thorne, 1988) by animals
resulted in pulmonary sensitization, whereas similar exposure to
formaldehyde (Lee et al., 1984) and hydrogenated
diphenylmethane-4,4-diisocyanate (Karol & Thorne, 1988), two
recognized contact sensitizers, resulted in dermal sensitivity.
Further validation with additional classes of chemicals is needed to
generate confidence that information will be applicable to human
disease.
It should be emphasized that the measurement of respiratory
responses induced by chemical respiratory allergens is technically
demanding, and consequently conflicting results have been reported.
For example, it has proven difficult to induce robust respiratory
responses in animals sensitized to the potent human respiratory
allergen diphenylmethane-4,4-diisocyanate. Pauluhn & Mohr (1994)
reported that a proportion of animals sensitized to
diphenylmethane-4,4-diisocyanate by inhalation responded to challenge
with inhaled diphenylmethane-4,4-diisocyanate, while those sensitized
by intradermal injection did not. The converse finding was reported by
Rattray et al. (1994).
Although the guinea-pig can be used for testing the capacity of
chemicals to induce respiratory allergy, it needs further validation
in terms of predictive value. However, it should be noted that it can
also be used to test proteins for their ability to stimulate the
production of anaphylactic antibody (Blaikie et al., 1995; Sarlo et
al., 1997). Such information may be of value in assessing the relative
allergenic potency of proteins.
6.4.2.2 Mouse IgE model
A mouse inhalation model to study airway responses to sensitizing
chemicals has been developed (Garssen et al., 1991), but has not yet
been used with a wide range of chemicals associated with respiratory
allergy. The mouse IgE test currently represents the furthest
developed systematic approach to the prediction of respiratory allergy
in the mouse. It does not evaluate actual airway responses, but is
based on the notion of the nature of immune responses elicited in mice
by chemical allergens and of the qualitative differences in immune
responses provoked by contact and respiratory sensitizers as they are
generally found.
Topical administration to mice of chemical respiratory allergens
stimulated a substantial increase in the serum concentration of total
IgE, a response not seen with contact allergens considered to lack the
ability to cause sensitization of the respiratory tract (Dearman &
Kimber, 1991, 1992). Observations suggested that it might be possible
to identify chemical respiratory sensitizers as a function of induced
changes in serum IgE concentration; the advantage of this approach
being that measurement of a serum protein, rather than of
hapten-specific antibody, is required. This forms the basis of the
mouse IgE test.
Investigations have suggested that the mouse IgE test may provide
a useful method for the prospective identification of chemical
respiratory allergens, a conclusion that is supported by recent
studies in an independent laboratory (Potter & Wederbrand, 1995). It
must be emphasized, however, that to date the assay has been evaluated
only with a limited number of chemicals, most of the analyses have
been performed in a single laboratory, and the mechanistic basis of
the model has not been established.
Respiratory allergic responses, associated with increased
reactivity of airways, were observed in mice that were topically
sensitized to and intranasally challenged with picryl chloride, a
Th1-type immune-response-inducing chemical (Garssen et al., 1991). In
addition, it has been shown that IgE-deficient mice undergo
anaphylaxis (Oettigen et al., 1994). Other investigators have noted a
decrease, rather than an increase, in serum IgE following exposure of
mice to toluene diisocyanate (TDI) (Satoh et al., 1995). For this
reason, actual testing of lung function in vivo after sensitization
and challenge with chemicals known to sensitize, yet unable to produce
IgE responses, as can be done in mice (Garssen et al., 1991) as well
as in the guinea-pig (section 6.4.2.1), seems prudent. Moreover,
standardization of the methodology is necessary, including dosages
appropriate for testing, optimal time for repeated administration of
the chemical and for obtaining sera, and clarification of the meaning
of "elevated" IgE titre. Validation of the method is still needed.
6.4.2.3 Rat model
A rat bioassay has been developed by Pauluhn (1996). Nose-only
exposure for 1 or 2 weeks, using non-irritant concentrations as judged
in short-term pilot experiments, is carried out, upon which lung
functions are tested and biochemical and morphological signs of
effects in the airways are determined. The model needs further
validation to evaluate whether it is suitable for the prediction of
respiratory sensitization. A rat model to study respiratory syndromes,
including the IgE-mediated allergic responses, in addition to anaemia
and haemolysis in workers exposed to trimellitic anhydride (Zeiss et
al., 1977), was used by Leach et al. (1987). In this model, animals
are subjected to single or multiple exposures at several
concentrations of trimellitic anhydride dust and at selected time
points are challenged with a single exposure of trimellitic anhydride
dust. The lungs are evaluated for haemorrhage and serum is tested for
IgG antibody specific for trimellitic anhydride. The model has also
been applied for other types of anhydrides.
6.4.2.4 Predictive testing for respiratory allergy in humans
For obvious reasons, predictive testing for respiratory
sensitization is not done in humans. Occasionally, case reports may
serve as an adequate hazard identification but not as a risk estimate,
because data on route and extent of exposure, and on the "population
at risk" are usually lacking. In the absence of case reports, it is
not possible to conclude that there exists no potential for
sensitization.
6.4.2.5 Cytokine fingerprinting
Contact and respiratory chemical allergens provoke in mice
qualitatively different immune responses suggestive of divergent
Th-cell activation and characterized by different patterns of cytokine
production. Chronic exposure of mice, over a 13-day period, to
trimellitic anhydride was found to result in the production of high
levels of mitogen-inducible IL-4 and another Th2-cell cytokine
interleukin 10 (IL-10) by draining lymph node cells, but only low
levels of IFN-gamma. In contrast, treatment of mice under the same
conditions of exposure with oxazolone (a potent contact allergen)
caused the production by draining lymph node cells of only
comparatively low levels of IL-4 and IL-10, but high concentrations of
IFN-gamma (Dearman et al., 1995). Similar selective cytokine secretion
profiles have been recorded following exposure of mice to other
contact and respiratory chemical allergens (Dearman, 1996). These data
raise the question of whether it might be possible to monitor the
sensitizing properties of chemicals as a function of induced cytokine
production profiles. Evidence suggests that this is the case, and the
value of cytokine fingerprinting in the routine identification and
classification of chemical allergens is being explored currently.
6.5 Testing for food allergy
Despite the availability of several methods to study antigenic
and allergenic properties of protein products, current testing
possibilities to investigate the allergenicity of food proteins at a
pre-market stage are very poor. Validated methods with a high
sensitivity have not yet been developed. However, some strategies can
be followed to obtain additional relevant information apart from the
information obtained from the currently applied assays; for instance,
taking account of the role of the gastrointestinal tract physiology.
The major adverse reactions to food constituents that involve the
immune system are to proteins; non-proteins may cause food intolerance
that does not involve the immune system, and hence are non-allergic
reactions that do not fall within the scope of this monograph.
Most proteins encountered by the immune system can produce immune
responses. To determine the antigenicity of proteins, several assays
are operational. However, these assays are based on parenteral
application of the test proteins to laboratory animals. For food
allergy research, three rodent species have frequently been used: the
mouse, the guinea-pig and the rat. In many studies, sensitization was
performed parenterally or passively and effects of enteral challenges
were subsequently studied (Bloch & Walker, 1981; Freier et al., 1985;
Granato & Piguet, 1986; Pahud et al., 1988; Miller & Nicklin, 1988;
Turner et al., 1988, 1990; Curtis et al., 1990). In addition, effects
of challenges have frequently been investigated in in vitro studies
with intestinal tissue or with, for instance, ligated gut (Roberts et
al., 1981; Baird et al., 1984; Lake et al., 1984; Catto-Smith et al.,
1989a,b). The guinea-pig is the most regular test species. In general,
any protein that may be recognized as an antigen (foreign protein)
will induce a humoral immune response upon injection and will most
likely give a positive testing result in rodent assays. Although the
information from antigenicity assays may be of major relevance, in
that they will provide information on the quality and vigour of the
response, it must always be recognized that such assays only provide
information in the species examined. Whether a protein has a high or
low potency of inducing food allergic reactions in (susceptible)
humans cannot be concluded or predicted based only on the results of
parenteral antigenicity assays.
In addition to in vivo antigenicity assays, several
(combinations of) physicochemical and immunochemical analyses are used
routinely to detect antigenicity.
Determination of allergenic proteins or fragments that are able
to cause activation of mast cells and basophils is possible using in
vitro mast cell or basophil degranulation tests. For these assays,
mast cells or basophils are loaded with antigen-specific cytophilic
antibodies using serum from sensitized humans or test animals. The
cells are subsequently incubated with the antigen or test product, and
degranulation of the cells can then be determined. A well-validated
in vivo counterpart for the detection of mast cell activation is the
Passive Cutaneous Anaphylaxis (PCA) test, in which sera from
sensitized animals are injected subcutaneously in unsensitized
animals. Possible cytophilic antibodies present in the sera are bound
by the receptors on the mast cells in the skin, and bridging of the
antigen-specific antibodies on the mast cells by injected antigen
induces mast cell activation -- the resulting reaction is detectable
by various methods. In the Active Systemic Anaphylaxis (ASA) test,
which is also a validated anaphylaxis test, actively sensitized
animals are injected with the test substance intravenously and several
parameters are recorded to determine the systemic anaphylactic
response as a measure for the degree of sensitization.
In the evaluation of the potential allergenicity of food
products, clinical assays such as skin-prick tests or challenge
procedures may also be used. For instance, these assays are applicable
in the evaluation of the residual allergenicity of hypoallergenic
products, in the evaluation of cross-allergenicity, or in the
evaluation of the possible allergenicity of food products derived from
biotechnologically derived crops in which a gene from a known
allergenic source species has been introduced. However, the use of
patients in such assays for non-diagnostic purposes requires careful
ethical consideration. Since many of the questions may also be
addressed by performing in vitro assays such as immunochemical
analyses, the use of sera from patients is always preferable to
intentional exposure of humans.
6.6 In vitro approaches
Another approach for the detection of potential sensitizing
capacity that would not rely on in vivo animal or human testing is
directed towards the construction of in vitro experimental models
that reflect accurately some pivotal event during skin sensitization.
Accurate modelling of the immune system in vitro is not possible
without considerable difficulty. Nevertheless advances have been made.
Wass & Belin (1990) and Gauggel et al. (1993) used biochemical
techniques to examine the ability of low relative molecular mass
chemicals to act as haptens by combining them with a model protein or
a polypeptide. Gauggel et al. (1993) showed that their test correctly
identified 12 out of 14 known human allergenic haptens and 23 out of
24 non-allergenic low relative molecular mass chemicals. Neither
method can detect sensitizers that must be metabolized to form a
hapten.
6.7 Testing for autoimmunity
Although there are currently no predictive assays developed and
validated to identify in the early phases of toxicity testing the
potential of chemicals to induce autoimmune responses, it should be
noted that assays to identify contact sensitizers (Kimber et al.,
1994; Vial & Descotes, 1994) might be helpful to identify systemic
sensitizers. Clinical signs of systemic immune-mediated side-effects
usually become manifest only during advanced clinical development of
drugs. The conditions used in routine preclinical toxicological
screening are obviously not optimal for the detection of the
immune-dysregulating potential of drugs and chemicals (e.g., small
animal number, use of outbred animal strains, dynamics of disease
development versus snapshot determinations, lack of predictive
parameters). An economically and practically relevant question
concerning screening studies is whether actual evidence of an agent's
ability to induce manifest hypersensitivity or autoimmune disease
should be and can be obtained, or whether (preferably short-term)
assays not measuring the actual clinical end-points can be
sufficiently predictive in this respect.
In vitro tests for biological effects of sensitizers or
chemicals able to induce autoimmunity are at present in their infancy.
It should be emphasized that there are two major limitations: (a) it
is so far impossible to completely reproduce in vitro the complex
microenvironment in which immune responses are initiated in vivo;
and (b) some immune reactions are elicited not by native xenobiotics
but by their metabolic products generated in vivo.
6.7.1 Popliteal lymph node assay
At present this appears to be the only method available to
examine the ability of chemicals to induce an autoimmune response, but
the usefulness of the results in risk assessment remains to be
demonstrated. Based on the hypothesis that chemicals may elicit
autoimmune disorders by a mechanism resembling graft-versus-host
reactions, an existing graft-versus-host assay, the popliteal lymph
node assay, has been modified to study chemical-induced immune
reactions (Bloksma et al., 1995). Using the popliteal lymph node
assay, many drugs known to occasionally induce immune-mediated
systemic side-effects in humans were shown to trigger significant
reactions in mice and rats (Gleichmann, 1981; Gleichmann et al., 1983,
1989; Hurtenbach et al., 1987; Kammüller & Seinen, 1988;
Stiller-Winkler et al., 1988; Kammüller, 1989a; Thomas et al., 1989,
1991; de Backer et al., 1990; Verdier et al., 1990; Katsutani &
Shionoya, 1992; Krzystyniak et al., 1992; Bloksma et al., 1995).
Effects observed were very similar to those induced during a local
graft-versus-host reaction in the popliteal lymph node assay (de
Bakker et al., 1990). Immunogenetic studies in mice with
diphenylhydantoin (Bloksma et al., 1988) and D-penicillamine
(Hurtenbach et al., 1987) have indicated that the extent of popliteal
lymph node enlargement is controlled by major histocompatibility
complex (MHC (H-2)) as well as non-major histocompatibility complex
genes. Furthermore, the popliteal lymph node assay was able to
discriminate between structurally closely related compounds, for
example chemical congeners of D-penicillamine (Hurtenbach et al.,
1987), diphenylhydantoin (Kammüller & Seinen, 1988) and zimeldine
(Thomas et al., 1989, 1991). Thus, the direct popliteal lymph node
assay seems to be a versatile tool for recognizing T-cell-activating
drugs and chemicals, including autoimmunogenic chemicals, but it also
produces false-negative results (Bloksma et al., 1995). With the
adoptive transfer popliteal lymph node assay, sensitized cells are
used as probes to detect the formation in vivo of immunogenic
metabolites of low relative molecular mass chemicals (Kubicka-Muranyi
et al., 1993; Bloksma et al., 1995). However, further mechanistic
studies and interlaboratory validation is required before either
variant of the assay can be recommended for routine use in the
preclinical toxicity screening (Verdier et al., 1997).
6.7.2 Animal models of autoimmune disease
Three basic types of animal models may be employed to identify
the potential of drugs or chemicals to induce systemic
hypersensitivity or autoimmune responses: (a) genetically predisposed
animals; (b) autoimmunization; and (c) organic or chemically induced
(Table 26). In each type of model the development and severity of
symptoms is multifactorial, in that the disease state can be
influenced by age, hormonal and/or environmental factors. In addition
there is a tendency for more than one autoimmune disorder to occur in
a number of individual models. Nevertheless, a number of syndromes
similar to that observed in humans can be mimicked in animal models.
The genetically predisposed models, whether naturally occurring,
transgenic or knockout based, tend to be the most reliable and
therefore have been more commonly employed in autoimmunity research
(Lo, 1996). In this type of model, mild to severe syndromes
spontaneously develop, usually due to specific MHC allele mutations
encoding class II molecules and often inducing function abnormalities
of the CD4+ Th-cell (Theofilopoulos, 1995a,b). The most common
genetic models include several MRL mouse variants for autoimmune
thyroiditis, arthritis and lupus, several variants of New Zealand
Brown mice (NZB), New Zealand White mice (NZW) and BXSB mice for
lupus, and the non-obese diabetic (NOD) mouse and BioBreeding (BB) rat
for insulin-dependent diabetes mellitus (Cohen & Miller, 1994).
Autoimmunization with purified self-antigens can elicit a
specific autoimmune response, particular when adjuvants are
administered in conjunction with self-proteins. A frequently used
model of this type, experimental autoimmune encephalomyelitis, is
induced by immunization of rodents with myelin basic protein. The
resulting pathology is a CD4+ T-cell-mediated autoimmune disease
characterized by central nervous system perivascular lymphocyte
infiltration and destruction of the myelin nerve sheath with resultant
paralysis, similar to that observed in patients with multiple
sclerosis (Constantinescu et al., 1998). Additional models included
immunization with thyroglobulin to simulate Hashimoto's thyroiditis,
and the injection of type II collagen to induce rheumatoid arthritis
(Wick et al., 1974; Durie et al., 1994).
In experimental models, foreign substances are used to induce the
autoimmune disease state. These include chemicals, drugs and
biological substances such as bacterial or viral antigens. Brown
Norway (BN) rats injected with non-toxic amounts of mercuric chloride
which produce no signs of overt toxicity develop an immunologically
mediated disease characterized by a T-cell-dependent polyclonal B-cell
activation (Pelletier et al., 1994). These animals demonstrate
increases in serum IgE and the production of autoantibodies to a
number of proteins including DNA, laminin, collagen IV and other
components of the glomerular basement membrane. Proteinuria and
Table 26. Experimental models for autoimmune diseasesa
Autoimmune disease Classification
Genetically predisposed strainsb Autoimmunizationc Biological/chemical induction
Autoimmune thyroiditis Murphy-Roths lymphoma (MRL) mouse, Thyroglobulin - EAT
(Hashimoto's and Graves') BioBreeding (BB) rat, (mouse, rat)
Obese strain (OS) chicken
Insulin-dependent Non-obese diabetic (NOD) mouse, Streptozotocin (STZ) (mouse)
Diabetes mellitus BioBreeding (BB) rat,
Diseases resistant BioBreeding
(DRBB) rat,
Brown Norway rat
Myasthenia gravis Acetylcholine receptor Penicillamine (mouse, rat)
- EAMG (mouse, rat)
Multiple sclerosis Myelin basic protein
EAE (mouse, rat, chicken)
Rheumatoid arthritis Murphy-Roths lymphoma CFA + Type II collagen Streptococcal cell-wall (rat)
(MRL)/lpr mouse, (mouse, rat, monkey)
Severe combined immunodeficient CFA + TB hsp (mouse, rat)
(SCID) mouse,
Human leukocyte antigen
B27 (HLA B27) transgenic rat
Systemic lupus Murphy-Roths lymphoma CFA + antiDNA antibodies Mercury (mouse, rat, monkey)
erythematosus MRL +/+ mouse, (mouse, rat) Penicillamine (mouse, rat)
Murphy-Roths lymphoma MRL/lpr Procainamide (mouse, rat)
mouse,
Murphy-Roths lymphoma
(MRL)-mp-lpr/lpr mouse,
New Zealand White (NZW) 2410 mouse,
New Zealand Black (NZB) mouse/
New Zealand White (NZW) mouse
Table 26. (continued)
Autoimmune disease Classification
Genetically predisposed strainsb Autoimmunizationc Biological/chemical induction
Systemic sclerosis Tight skin (TSK) mouse,
(Scleroderma)
a Adapted from Cohen & Miller (1994); Farine (1997); Bigazzi (1997)
b lpr = lymphoproliferation
c CFA = Complete Freunds Adjuvant
nephrotic syndrome similar to that observed in humans are also
observed. The disease is characterized by a glomerulonephropathy
evolving in two phases: (a) a linear anti-GBM antibody deposition
along the glomerular capillary pattern, and (b) a change to a granular
pattern of immunofluorescence with the appearance of immune complex
deposits (Pelletier et al., 1994).
6.8 Clues from general toxicity tests
The conditions used in conventional systemic toxicological
screening are not designed for the detection of the potential of drugs
and chemicals to induce allergy or autoimmunity, as general toxicity
screening does not include specialized tests required for this
purpose. In more recent guidelines for general toxicity screening,
attention is given to the immune system, especially with the purpose
of detecting direct toxicity to components of the immune system. This
pertains for instance to the updated OECD guideline for 28-day oral
toxicity testing (Koeter, 1995). According to this guideline, lymphoid
organs are weighed and lymphoid tissues are examined microscopically.
In addition to morphological examinations of lymphoid organs, a
selection of non-lymphoid tissues (e.g., blood vessels, renal
glomeruli, synovial membranes, thyroid, skin, liver and lungs) should
be investigated. Tissue damage, protein (immune) complex deposits,
and/or inflammatory cell infiltrates in these tissues may indicate the
induction of hypersensitivity or autoimmune phenomena.
It is common practice in toxicological pathology to rely most on
statistically significant differences in incidence between test groups
and controls. However, individual variability can be high, especially
regarding hypersensitivity and autoimmune phenomena (Holt & Sedgwick,
1987; Kammüller et al., 1989). Therefore, in studies with a limited
number of animals per group, a change in only a single or a few test
animals may have considerable biological relevance. This is
particularly true in outbred animals.
In addition to morphological examinations in routine
toxicological studies, measurements of some immunologically relevant
serum parameters can provide important information about
antibody-mediated responses. Parameters may comprise levels of total
immunoglobulins and of various immunoglobulin (sub)classes, immune
complexes, and some commonly observed autoantibodies, e.g.,
anti-nuclear antibodies (ANAs), anti-histone, and anti-single-stranded
deoxyribonucleic acid (denatured; ssDNA) autoantibodies. Some of these
autoantibodies proved to be useful in the diagnosis of
procainamide-induced systemic lupus erythematosus in humans as shown
by Rubin et al. (1995). Measurement of these parameters at suitably
chosen intervals, especially during subchronic and chronic exposure,
may obviate the snapshot nature of the histopathological examinations
and will give a reflection of cellular and humoral immune function in
time. With regard to autoantibody measurements it is important to
measure both total immunoglobulin classes and subclasses. This can be
illustrated by murine models of spontaneous systemic lupus
erythematosus in which a switch from IgM to IgG autoantibodies could
be associated with development of overt disease. Also, data on
drug-induced systemic lupus erythematosus in humans point in this
direction. Anti-nuclear antibodies in patients with
procainamide-induced systemic lupus erythematosus appeared to be of
the IgG class (in particular IgG1 and IgG3), whereas IgM anti-nuclear
antibodies are predominant in asymptomatic users of the drug.
It has been suggested that parameters of the immune system are
included in conventional chronic toxicity testing and also in
reproductive toxicity testing and evaluation, since the developing
immune system may be particularly vulnerable to immune dysregulating
effects (Hollady & Luster, 1996). For example, a newly developed
immunomodulating agent was found to induce thyroiditis and significant
antithyroglobulin autoantibodies in a 6-month and a 2-year rat study
(Verdier et al., 1997).
7. RISK ASSESSMENT
7.1 Introduction
Risk assessment for human health protection is the final stage in
evaluating the likelihood that potential adverse health effects will
manifest themselves in humans.
7.2 Risk assessment of allergy
Effective risk assessment requires an appreciation of the
potential of a chemical or drug to induce an allergic response, or
elicit an allergic reaction, and its potency. Potency here is
considered as the amount of chemical necessary to induce an allergic
response or to elicit an allergic reaction. Risk assessment demands
also an understanding of the conditions of human exposure, i.e., the
extent, duration and route(s) of likely exposure.
With the assembly of the available data completed, risk assessors
must decide whether sufficient data are available to proceed. It may
be necessary to produce a preliminary or provisional risk assessment
irrespective of the availability of full documentation. However, at
this stage, following comparison of what is available with minimal
data sets, consideration is normally given to further searching,
further requests for information to industry, and the use of modelling
or default values to fill data gaps. A decision is then made on
whether to proceed.
Risk assessment of allergy is complicated by the fact that there
exist two related, but nevertheless independent considerations.
Allergic disease almost always occurs in two phases. During the first
phase the previously non-sensitized individual is exposed to
sufficient allergen in such a way that an allergic response is induced
which results in sensitization. This is the induction phase. An
allergic reaction will be elicited if this now sensitized individual
is exposed again to the same allergen under conditions where a
secondary, more aggressive allergic response is provoked. This is
known as the elicitation phase. It is important to appreciate that
these phases usually differ with respect to the exposure conditions
required. The induction of allergic sensitization in a previously
naive individual almost invariably requires exposure to concentrations
of the allergen that are greater than those necessary to elicit a
response in a sensitized subject. Allergic reactions can be provoked
in sensitized individuals with small amounts of the inducing allergen
that are without effect in non-sensitized populations. It is possible
also that sensitization may be achieved from exposure via a route that
is different from that needed to elicit a response. Thus, there is
some evidence that dermal exposure to certain chemical allergens may
stimulate the quality of immune response necessary to cause
sensitization of the respiratory tract. The elicitation of pulmonary
responses will subsequently be provoked following inhalation exposure
of the now sensitized individual to the relevant chemical.
Taken together it is clear that risk assessment for chemically
induced allergic disease has two components: (a) the likelihood that a
chemical will induce sensitization in a previously unsensitized
individual; and (b) the conditions under which a chemical will provoke
allergic reactions in those who are already sensitized.
Information on risk, plus suggestions about any group of
increased susceptibility, is used for risk evaluation, that is the
process of deciding for medical, ethical, economic and legal reasons
whether the identified risk will be "acceptable" and under what
conditions, e.g., restricted availability of the substance in special
circumstances or wider availability with a warning label and advice
about protective measures. The decision may also be made to ban the
use of a material deemed to carry too large a risk in relation to
anticipated benefit from its use. These decisions reflect
socioeconomic and political factors as well as medical and scientific
conclusions, and so lie outside the purpose of this monograph.
7.3 Factors in risk assessment of allergy
Risk assessment requires answers to the following questions:
* Who will be exposed?
How many people: Is there any information to suggest particular
susceptibility (or resistance) to allergic hypersensitivity, e.g.,
genetic, nutritional or other factors?
* Circumstances of the exposure
Is it an initial sensitizing exposure or a subsequent provocative
exposure that may elicit an allergic reaction?
* Extent and duration of exposure
This defines the "load" of the allergen. Most of the available
evidence suggests that it is the peak concentration of exposure rather
than the cumulative delivered dose that is critical in determining the
extent to which sensitization will develop. The same principle
probably applies also to the elicitation of allergic reactions in
sensitized individuals.
* Nature of exposure
Exposure to chemical allergens occurs frequently in the context
of mixtures and formulations. An important consideration is whether
other components of the mixture will influence the ability of the
chemical allergen to induce sensitization or elicit a reaction.
* Route
If it is the first exposure, is it by a route that may sensitize
people, usually via inhalation or skin contact, or by ingestion, which
is much less likely to do so? If an individual is already sensitized,
is it via the route by which he has already been sensitized, in which
case an allergic response may occur? Or, will it be by another route,
which may not necessarily evoke an immunological response?
* Evidence of hazard
What is the nature and quality, i.e., the "strength", of the
laboratory information showing the sensitizing potential of the
substance? Has it come from laboratory tests of proven value, from
chance observations during some other type of experiment, or is it
based on prediction from structure-activity relationships? In each of
these, are the data qualitative or quantitative?
* Prior evidence of risk
Are there already results from humans, either as tests in a
clinical laboratory setting, or following exposure in the real world
of work or home, showing that people have been sensitized and how, and
the consequences of subsequent exposure by any route?
Taking all this information together, each aspect of which has
already been discussed in this monograph, should suggest the totality
of the risk under a given set of circumstances, in other words, the
likelihood that a given exposure to a substance will sensitize people,
or evoke an allergic response in them, perhaps the proportion
affected, and the nature of that response (including clinical feature
of the disorder and their severity). That represents the ideal. In
practice, the available information may be incomplete, and skilled
judgement is required to extrapolate from whatever is known to suggest
what may happen in practice.
7.4 Information aspects
The three commonest deficiencies in knowledge are discussed
below.
7.4.1 No information about hazard
For a novel substance, a novel use, or because hypersensitivity
may not previously have been recognised, there will be no information
from humans. In that case, prediction can only be based upon the
chemical structure of the substance, the results of appropriate
laboratory tests, and assumptions about exposure of people.
Such an extrapolation based solely on laboratory-defined hazard
is the usual situation faced by the toxicologist, occupational health
specialist and regulator in dealing with a new substance.
As discussed earlier in this monograph, many of the laboratory
procedures available to identify hazard are well proven and some are
valuable in assessing relative potency for skin sensitizers. Their
quantitative predictivity, sensitivity and specificity are difficult
to state, but they probably give about 80% correct negative results,
i.e., sensitization is unlikely (Kimber & Maurer, 1996) and about 60%
correct positive predictions, i.e., skin sensitization is likely to
occur.
For respiratory tract sensitization, far fewer results are
available, but a positive finding in any of the types of experiment
considered here should be regarded as a powerful indicator of this
type of hazard. The strength of a negative finding is less certain,
because there is little published information.
True hypersensitivity to oral antigens is still poorly
understood, and the more common but more fickle "food intolerance"
remains clouded by uncertainties. For a novel substance in a food, it
is not yet possible to predict the likelihood that it will cause true
allergy or intolerance if consumed. Known allergens and the other
materials that sometimes cause disorders when eaten can be indicated
by reference to published clinical information, which should also
point both to the likely frequency of the harmful response, and
whether it is affected by ethnic or by other physiological factors,
the methods of preparation and cooking, and the matrix in which a
putative food allergen is presented, because the complex nature of the
mixtures that are foods does affect the risk of sensitization and
elicitation of reactions to materials in the diet.
The best predictor at present of the risk of true immunologically
mediated hypersensitivity to a food component is probably
demonstration of the presence of antibodies to known potent allergens,
such as peanut (groundnut) protein.
Predicting the risk of autoimmunity, too, can only be done by
demonstrating the presence of a known antigen in an appropriate
matrix, or, with less certainty, by demonstrating a very close
structural analogy to a known cause of an autoimmune response.
7.4.2 Scanty or no information about exposure
It is sometimes difficult to envisage exactly how a substance
will be used in the workplace or at home, or how it may be
disseminated via the environmental media. In both of these instances
the prediction of risk must carry some uncertainty.
The problem of predicting exposure is well shown by the
demonstration of an increased incidence of asthmatic attacks in the
population of the city of Barcelona during large-scale transhipment of
soy beans in the docks. That appears to have released a sufficient
quantity of an allergen into the air for it to be disseminated and to
expose the entire population under certain meteorological conditions,
resulting in allergic asthma in so many people as to have a serious
effect on the health of the community.
7.4.3 Unreliable or scanty information about risk
This is a particular problem because of the propensity of
clinicians to report unusual or unique examples of disorders and not
to describe negative findings. It may be difficult, therefore, to gain
a balanced or critical view of the allergenic effects of substances
from much of the clinical literature. It can provide an indication of
suspicion, but large, carefully constructed series of patients or
cautious surveillance results are rare.
At a different level, but well able to provide helpful
information, are national occupational health statistics on
appropriate industrial diseases, particularly those for which a
pension has been awarded. For many reasons those series are unlikely
to provide unbiased information about incidence or prevalence, because
of the inevitable bias when legal liability may be involved, but they
can act as broad indicators of risk, and even of the allergenic
potential of particular substances (Drever, 1995).
Databases of consumer products used by the general population are
very limited and may be close to capricious in their content, but they
should be consulted when possible.
Overall, therefore, the risk assessor has a very difficult task
in the general field of allergic hypersensitivity. At present,
laboratory tests may indicate the potential for a hazard, and
searching consideration may suggest many of the features about
exposure. From that a risk to humans may be predicted in qualitative
terms, at least for the common route of domestic and occupational
exposure of the skin. The prediction of respiratory tract allergy
remains less certain, and the likelihood of effects following
ingestion, or of an autoimmune disorder, can very rarely be predicted.
Many predictive test methods serve simply to identify the
inherent potential of a chemical to induce allergy but provide no
indication of the potency with which it will do so. One problem is
that some methods do not incorporate a dose-response analysis. The
other issue is that some tests measure activity as the frequency of
responses rather than the severity of responses. The need is to have
available information on potency defined as the quantity of chemical
necessary to induce sensitization (or to elicit a reaction). Some
newer test methods are beginning to address this issue and are
providing information about allergic potency relative to index
allergens. Such comparisons have been of value in setting safe
occupational exposure levels and minimizing the risk of allergic
disease.
As in any form of toxic reaction, "dose" is important, in that
initial sensitization requires at least a certain minimum exposure
(concentration of allergen, its local availability at the site of
administration, and the duration of contact). In someone already
sensitized, the likelihood of producing a clinical disorder and its
severity are also related to dose, although, by definition, the
quantity of allergen required to produce an effect is very much
smaller than that associated with a conventional toxic action. This
aspect of the extent or intensity of dose (=exposure) is more
important at the practical level of preventing sensitization or
protecting the sensitized individual, i.e., risk management, but it
should not ignored at the risk assessment stage.
A further difficulty experienced by the risk assessor in this
area is that of individual susceptibility. Genotype does play some
role in the propensity of people to develop clinical disorders due to
hypersensitivity, as shown, for example, by the familial nature of
atopy. There is also good clinical evidence, however, to show that the
inheritance of eczematous skin hypersensitivity, or of asthmatic
hypersensitivity to common domestic allergens, involves many powerful
factors in addition to genetic constitution. Although there is no a
priori reason to believe that humans lack "immune response" genes
for hypersensitivity as pragmatically defined in animals, their
importance remains uncertain. Some chemically related autoimmune
diseases though are associated with certain "immune response"
genotypes. For example, HLA DR3 and DR3/B8 haplotypes are associated
with vinyl chloride disease (Black et al., 1983).
Skin sensitization risk assessment is not a highly prescriptive
process that should always be followed in the same way (Calvin, 1992).
On the contrary, what is necessary is that it is carried out
thoroughly to the point where the risk has been adequately assessed.
In some circumstances, this point may be reached quite quickly and
with minimal expenditure of time and effort. In other cases,
substantial and sustained effort is required. For example, in the
situation where exposure to the contact allergen is essentially zero,
then even for highly potent contact allergens, there is no need to go
further with a risk assessment because allergic contact dermatitis
will not occur. Furthermore, if the exposure is sufficiently low, it
may not be necessary to know precisely the potency of a contact
allergen. Simply the knowledge that it is not a very strong allergen
may be sufficient to permit a proper conclusion of the risk
assessment. Another situation where risk assessment may be relatively
simple is the replacement of an ingredient with another of the same or
similar type (e.g., an alternative supplier of a raw material). In
such a case, and where the risk is already known to be very low, all
that may be necessary is to confirm that the specification of the new
source of raw material is the same. Alternatively, data that provide
evidence that the relative sensitization potential of the old and new
materials is similar may suffice. In contrast, even where the
intrinsic sensitization potential is very low, if skin contact is
sufficiently intense and prolonged, then sensitization may occur. An
example of this is the situation where medicines are applied
continuously to skin, often damaged and/or inflamed skin, under
occlusion. A prime example is found with stasis ulcers, where a
variety of medicines and chemicals with negligible sensitization
potential, such as cetostearyl alcohol and paraben esters, quite
frequently cause allergic contact dermatitis.
The comments above on skin sensitization all presuppose a good
knowledge of the relative sensitizing potency of the substance under
consideration. Such data may be obtained by examination of the skin
sensitization potential of the substance in guinea-pig methods
referred to earlier in this monograph. Critical to this type of
analysis is data from the same method at the same testing institution
with suitable benchmark substances. These may well need to be selected
on a case by case basis in the light of the risk assessment that needs
to be made. Alternatively, use made be made of the local lymph node
assay, the results of which can be interpreted to yield an objective
estimate of relative sensitizing potency (Kimber & Basketter, 1997).
To determine what represents a safe threshold for a sensitizer is
a complex matter (Basketter et al., 1997). The key point is that the
threshold depends on the method used to determine it. Thresholds in
animal models my differ substantially from those for humans even in
situations where the pattern of exposure is similar. However, where it
is possible to match the type of exposure in the test in humans to
that which is expected in practice, then such data may be interpreted
directly to humans (Johansen et al., 1996b).
7.5 Conclusions
The scientific, practical and clinical uncertainties that affect
allergic hypersensitization make the task of the risk assessor
particularly difficult. In practice, it may be reasonable in such
assessments to favour the fail-safe approach by emphasising the
"precautionary principle", namely that if there is even a suspicion of
a risk, exposure should be minimized and preferably be entirely
prevented. That will have a considerable influence on the development
of new substances, on devising new uses for existing materials, and on
instituting controls over exposure in the home and safe working
practices, because an assessment that suggests a risk of allergic
sensitization must lead at once to an appropriate decision about risk
management.
8. TERMINOLOGY
Adhesion molecules. Molecules, belonging mainly to the
immuno-globulin or integrin superfamily of molecules (e.g., LFA-1,
ICAM-1), expressed on the membrane of various cells of the immune
system. Interactions with each other as receptors and corresponding
ligands facilitate cooperation (cross-talk) of cells, signal
transduction and information transfer between cells.
Adjuvant. A material that enhances immune response to substances in
a non-antigen-specific manner.
Allergen. An antigen that provokes allergy.
Allergic contact dermatitis. An inflammatory skin disease resulting
from allergic sensitization.
Allergy. Hypersensitivity caused by exposure to an exogenous
antigen (allergen) resulting in a marked increase in reactivity and
responsiveness to that antigen on subsequent exposure, resulting in
adverse health effects.
Allogenic. Term describing genetically different phenotypes in
different (non-inbred) individuals of the same species.
Anaphylaxis/anaphylactic reaction. A local or systemic immediate
hypersensitivity reaction initiated by mediators released after
immunological stimulation. Symptoms can be a drop in blood pressure
related to vascular permeability and vascular dilatation, and
obstruction of airways related to smooth muscle
contraction/bronchoconstriction.
Anergy. Lack of immune responsiveness (usually defined as lack of
response to common recall antigens).
Antibody. An immunoglobulin produced by activated B-cells and
plasma cells after exposure to an antigen with specificity for the
inducing antigen.
Antibody-dependent cell-mediated cytotoxicity (ADCC). Lysis of
various target cells coated with antibody by Fc receptor-bearing
killer cells, including large granular lymphocytes (NK cells),
neutro-phils, eosinophils and mononuclear phagocytes.
Antigen. Any compound recognized by antigen-receptor-bearing
lymphocytes. Antigens induce immune responses or tolerance. Antigens
inducing immune responses only with the help of T-cells are
T-dependent antigens, while those that do not need T-help are
T-independent antigens. All immunogens are antigens but not all
antigens are necessarily immunogens (see also immunogens).
Antigen-presenting cells. Cells expressing MHC gene products, with
the capacity to process and present antigen. Macrophages, dendritic
cells, B-lymphocytes and Langerhans cells are termed professional or
constitutive antigen-presenting cells. However, other cells (such as
endothelial cells) can acquire the ability to present antigen in
certain pathological conditions.
Antigen processing and presentation. Protein antigens are processed
(cleaved by enzymes) in various compartments of antigen-presenting
cells. The immunogenic peptides interact with the binding sites of MHC
class II products (exogenous antigens) or with those in MHC class I
products (endogenous antigens, including viruses). The processed
antigen-MHC complex is recognized by the antigen receptor complex of
T-lymphocytes.
Antigenic determinant. A single antigenic site (epitope) usually
exposed on the surface of a complex antigen. Epitopes are recognized
by antigen-receptors on T- or B-cells (T-cell epitopes or B-cell
epitopes).
Anti-nuclear antibody. Antibody directed to nuclear antigen. These
antibodies can have various specificities (e.g., to single- or
double-stranded DNA, or to histone proteins). These antibodies are
frequently observed in patients with rheumatoid arthritis,
scleroderma, Sjögren's syndrome, systemic lupus erythematosus, and
mixed connective tissue disease. Also called anti-nuclear factor
(ANF).
Arthus reaction (Gell and Coombs Type III reaction). Inflammatory
response, generally evoked in skin, that is induced by immune
complexes formed after injection of antigen in an individual
containing antibodies.
Asthma. A respiratory disorder characterized by variable air flow
limitation. Most cases are associated with bronchial
hyperresponsiveness and chronic inflammatory changes in the airways.
Atopic dermatitis. Inflammation of the skin in atopic individuals.
The term is broader than atopic eczema (see also atopic eczema).
Atopic eczema. Chronic skin disease, often localized on flexural
surfaces, in individuals with propensity to develop IgE-mediated
allergy. The term describes eczema occurring in atopic individuals and
does not imply mechanisms (see also eczema).
Atopy. A genetic predisposition toward development of IgE-mediated
immediate hypersensitivity reactions against common environmental
antigens.
Autoimmune disease. A disease involving immune responses against
self antigens, resulting in pathological change.
Autoimmunity. Responses against self (autologous) antigens.
B-lymphocytes. Bone-marrow-derived lymphocytes, expressing an
antigen-receptor complex composed of membrane-bound immunoglobulin
(mIg) and associated molecular chains. B-cell receptors interact with
epitopes directly (no MHC restriction). Activated B-lymphocytes
produce antibody and are efficient antigen-presenting cells. They are
the precursors of plasma cells.
Basophil. A circulating granular leukocyte having prominent
cytoplasmic granules when stained with dyes that indicate a basic pH.
The granules contain histamine and sulfated mucopolysaccharides. After
binding of antigen to membrane-bound IgE via Fc-epsilon-RI receptors,
they release histamine, platelet activating factor (PAF) and
leukotrienes, and other inflammatory mediators.
Bronchoalveolar lavage. Harvesting of cells and fluid from the
lung, commonly by bronchoscopy and lavage.
Bronchoprovocation. Use of inhaled triggers (cold air, histamine,
methacholine, allergen, etc.) to assess the responsivity of the
airways.
Carrier. An immunogenic macromolecule (usually protein) to which a
hapten is attached, allowing the hapten to be immunogenic.
CD. A molecular marker on a cell surface that may be used
operationally to define phenotype, origin and activation state of the
cell.
CD3. A molecule composed of five polypeptide chains associated with
the heterodimer T-cell receptor (TCR), forming the T-cell receptor
complex (TCR/ CD3); CD3 transduces the activating signals when antigen
binds to the TCR.
CD4. A cell surface antigen belonging to the immunoglobulin
superfamily of molecules. Marker of T helper cells. As an adhesion
molecule, it interacts with the non-polymorphic part of MHC class II
gene product.
CD8. A cell surface molecule belonging to the immunoglobulin
superfamily of molecules. Marker of suppressor and cytotoxic T-cells.
As an adhesion molecule, it interacts with the MHC class I gene
product.
CD16. Low-affinity Fc-gamma receptor (Fc-gamma-RIII) expressed
mainly on NK cells, granulocytes and macrophages, mediating ADCC.
CD23. Low-affinity Fc-epsilon receptor induced by IL-4 and
expressed on activated B-cells and macrophages.
Cell-mediated or cellular response. A specific immune response in
which T-lymphocytes mediate the effects, either through the release of
cytokines or through cytotoxicity.
Class I MHC gene products. Antigens encoded by the MHC class I
genes are expressed on all nucleated cells. They present
antigen-derived peptides of endogenous origin.
Class II MHC gene products. Antigens encoded by the MHC class II
genes are expressed on antigen-presenting cells. They present
antigen-derived peptides of endogenous origin.
Clonal anergy. A form of self tolerance developing as a consequence
of negative selection during the thymic selection processes. Clones of
thymocytes whose antigen receptors (TCR) bind with high affinity to
self antigens in association with MHC molecules are inactivated.
Complement system. A group of serum proteins with the capacity to
interact with each other when activated. The chain reaction of the
activated complement components results in formation of a lytic
complex and several biologically active peptides of low relative
molecular mass (anaphylatoxins). The system can be activated by
antigen-antibody complexes (classical pathway) and by other
components, e.g., bacteria (alternative pathway). As an effector
mechanism of the humoral immune response, the activated complement
system facilitates opsonization, phagocytosis and lysis of cellular
antigens.
Contact sensitivity. A state of immunological sensitization in
which an eczematous epidermal reaction may occur when a hapten is
applied to the skin of a sensitized individual. (see allergic contact
dermatitis).
Contact urticaria. Urticaria provoked by contact with inducing
agents (see urticaria).
Cross-reactivity. The ability of an antibody or a T-cell, specific
for one antigen, to react with a second antigen; a measure of
relatedness between two antigenic substances, and/or polyspecificity
of the antibody molecule (e.g., some rheumatoid factors), or of the
T-cell receptor.
Cytokines. Group of substances (biologically active peptides),
mainly synthesized by lymphocytes (lymphokines) or monocytes/
macrophages (monokines), that modulate the function of cells in
immunological reactions; cytokines include interleukins. Some
cytokines (pleotrophic cytokines) have a broad spectrum of biological
actions, including: neuromodulation, growth factor activity and
proinflammatory activity (see also interleukins).
Cytotoxic T-cell (cytolytic T-cell)(CTL). A subpopulation of T-cells
with the capacity to lyse target cells displaying a determinant in
association with MHC gene products, recognized by its antigen receptor
complex (TCR/CD3).
Delayed-type hypersensitivity (DTH) (Gell and Coombs Type IV
reaction). A form of T-cell-mediated immunity in which the ultimate
effector cell is the activated mononuclear phagocyte (macrophage); the
response of DTH appears fully over 24 to 48 h. Previous exposure is
required. Examples include response to Mycobacterium tuberculosis
(tuberculin test) and contact dermatitis.
Dendritic cell. A cell type characterized by extended cytoplasmic
protrusions and a high expression of adhesion molecules and Class II
MHC gene products effecting antigen presentation to specific
lymphocytes (see also Langerhans' cell).
Dermatitis. Inflammatory skin disease showing redness, swelling,
infiltration, scaling and sometimes vesicles and blisters.
Desensitization. Generally transient state of specific
non-reactivity in previously sensitized individual, resulting from
repeated antigen exposures.
Eczema. A dermatitis characterized by non-contagious inflammation
of skin with typical clinical (itch, erythema, papules, seropapules,
vesicles, squames, crusts, lichenification) and dermatohistological
(spongiosis, acanthosis, parakeratosis, lymphocytic infiltration)
findings. Often due to sensitization.
Elicitation. Production of a cell-mediated or antibody-mediated
allergic response by exposure of a sensitized individual to an
allergen.
Endocytosis. The uptake by a cell of a substance from the
environment by invagination of its plasma membrane; it includes both
phagocytosis mediated by receptors and pinocytosis.
Enzyme-linked immunosorbent assay (ELISA). An assay in which an
enzyme is linked to an antibody and a labelled substance is used to
measure the activity of bound enzyme and, hence, the amount of bound
antibody. With a fixed amount of immobilized antigen, the amount of
labelled antibody bound decreases as the concentration of unlabelled
antigen is increased, allowing quantification of unlabelled antigen
(competitive ELISA). With a fixed amount of one immobilized antibody,
the binding of a second, labelled antibody increases as the
concentration of antigen increases, allowing quantification of antigen
(sandwich ELISA).
Eosinophil. A circulating granular leukocyte having prominent
granules that stain specifically by eosin and containing numerous
lysosomes. Eosinophils are important effector cells in immune
reactions to antigens that induce high levels of IgE antibodies (e.g.,
parasites). Eosinophils are also abundant at sites of immediate
hypersensitivity reactions.
Epidemiology. The study of the distribution and determinants of
health-related states or events in specified populations, and the
application of this knowledge to manage health problems.
Epitope. A single antigenic determinant.
Fc receptors. Receptors expressed on a wide range of cells,
interacting with the Fc portion of immunoglobulins belonging to
various isotypes. Membrane-bound Fc receptors mediate different
effector functions (endocytosis, antibody-dependenT-cellular
cytotoxicity (ADCC)) and induce mediator release. Both the
membrane-bound and soluble forms of Fc receptors regulate antibody
production of B-cells.
Forced expiratory volume in 1 second (FEV1). Physiological
measurement of the volume of air expired in one second with a maximal
respiratory effort.
Forced ventilatory capacity (FVC). The physiological measurement of
lung volume associated with a complete respiratory effort.
Glomerulonephropathy. Disease of the glomeruli, which may show
either thickening of the basement membrane -- membranous
glomerulopathy associated with IgG deposits -- due to the accretion of
proteins, or "minimal change glomerulopathy", in which there is
functional damage but little structural change by light microscopy.
Hapten. A non-immunogenic compound of low relative molecular mass
which becomes immunogenic after conjugation with a carrier protein or
cell and in this form induces immune responses. Antibodies, but not
T-cells, can bind the hapten alone in the absence of carrier.
Helper T-cell. A functional subpopulation of T-cells (expressing
CD4 antigen) that help to generate cytotoxic T-cells and cooperate
with B-cells in the production of an antibody response. Helper T-cells
recognize antigen in association with MHC class II gene products.
Depending on their capacity to produce various cytokines one can
functionally differentiate Th1 (IL-2 and IFN gamma producing) and Th2
(IL-3, IL-4 and IL-6 producing) cells.
Human leukocyte antigen (HLA). The major human histocompatibility
complex situated on chromosome 6. Human HLA-A, -B and -C (resembling
mouse H-2K, D and L) are class I MHC molecules, whereas HLA DP, -DQ
and -DR (resembling mouse I-A and I-E) are class II MHC molecules.
Humoral immune response. An immune response in which specific
antibodies induce the effector functions (such as phagocytosis and
activation of the complement system).
Hyperreactivity. An abnormally increased response to a stimulus.
Hypersensitivity. Abnormally increased immunologically mediated
response to a stimulus. Sometimes used loosely for any increased
response (see also hypersusceptibility, hyperreactivity)
Hypersensitivity pneumonitis (HPS) / extrinsic allergic alveolitis
(Gell and Coombes Type III reaction). An immune-mediated
inflammatory disease of the lung parenchyma caused by exposure to an
inhaled chemical allergen or organic dust.
Hypersusceptibility. Adverse effects in an individual occurring
under exposure conditions that result in no effects in the great
majority of the population or an individual exhibiting exaggerated
effects in comparison with the great majority of those showing some
adverse effects.
IgE-binding Fc receptors. The high-affinity IgE-binding
Fc-epsilon-R type I is expressed on mast-cells and basophils.
Interacts with IgE antibodies with high affinity. The cross-linking of
these receptors results in release of mediators (such as histamine).
The receptor is composed of alpha, beta and gamma chains; the alpha
chain contains the IgE binding site, while the gamma chain is
responsible for signal transfer. The low-affinity IgE binding Fc
receptor (CD23) is expressed on B-cells, its soluble (truncated) form
is generated by proteolytic cleavage and regulates IgE production of
B-cells.
Immediate-type hypersensitivity (Gell and Coombs Type I reaction).
A form of antibody mediated immunity that takes place in minutes to
hours after the administration of antigen. Previous exposure is
required. An example is allergic rhinitis to pollen antigen.
Immunodeficiency. Defects in one or more components of the immune
system resulting in inability to eliminate or neutralize non-self
antigens. Congenital or primary immunodeficiencies are genetic or due
to developmental disorders (such as congenital thymic aplasia).
Acquired or secondary immunodeficiencies develop as a consequence of
malnutrition, malignancies, immunosuppressive compounds, radiation or
infection of immunocompetent T-cells with human immunodeficiency virus
(HIV). Defects of the nonspecific defence system may also result in
immunodeficiency.
Immunogen. A substance capable of eliciting a specific immune
response manifested by the formation of specific antibodies and/or
specifically committed lymphocytes. To induce an antibody response an
immunogen must possess structurally and functionally distinct
determinants for activation of B-cells and T-cells.
Immunoglobulin (Ig). Immunity-conferring portion of the plasma- or
serum-gammaglobulins. Various isotypes (classes and subclasses) of
immunoglobulins have a common core structure of two identical light
(L) and two identical heavy (H) polypeptide chains, which contain
repeating homologous units folded in common globular motifs (Ig
domains). The amino acid sequences of the N-terminal domains are
variable (V domains), in contrast to the more conserved constant
regions (C domains). The V domains contain the
complementarity-determining regions (CDRS) forming the antigen-binding
sites, whereas the C domains trigger several effector functions of the
immune system (see also antibody).
Immunoglobulin gene superfamily. Genes encoding proteins containing
one or more Ig domains (homology units) that are homologous to either
Ig V or C domains. Cell surface and soluble molecules mediating
recognition, adhesion or binding functions in and outside the immune
system, derived from the same precursor, belong to this family of
molecules (e.g., Ig, TCR, MHC Class I and II, CD4, CD8, Fc-gamma-R,
NCAM, PDGFR).
Incidence (epidemiological). The number of new cases of disease in
a defined population during a specified period of time.
Interleukin. Immunoregulatory proteins, also designated as
lymphokines, monokines or cytokines. General features are: low
relative molecular mass (<80 000) and frequently glycosylated;
regulate immune cell function and inflammation by binding to specific
cell surface receptors; transient and local production; act in
paracrine, autocrine or endocrine manner, with stimulatory or blocking
effect on growth/differentiation; very potent, function at picomolar
concentrations. Interleukins represent an extensive series of
mediators with a wide range of overlapping functions. Other mediators
in this series are c-kit ligand, interferons, tumour necrosis
factor, transforming growth factor, and a family of low relative
molecular mass mediators, called chemokines.
Intolerance. Non-immunologically mediated adverse reactions. In
food intolerances these may be due to pharmacological properties of
food constituents, metabolic disorders or responses of unknown
etiology.
Langerhans' cells. Bone-marrow-derived epidermal cells with a
dendritic morphology, expressing CD1 marker in humans and containing
the cytoplasmic organelle, called the Birbeck granule. They express
Class II MHC antigen and are capable of antigen presentation (see also
dendritic cells).
Lymphocyte. Bone-marrow-derived cell with little cytoplasm, with
the ability to migrate and exchange between the circulation and
tissues, to home to sites of antigen exposure, and to be held back at
these sites. The only cells that specifically recognize and respond to
antigens (mainly with the help of accessory cells). Lymphocytes
consist of various subsets differing in their function and products
(e.g., B-lymphocytes, helper-T-cells, cytolytic T-cells).
Macrophage. Mononuclear cells derived from monocytes residing in
tissues. Activated by different stimuli they may appear in various
forms such as epitheloid cells and multinucleate giant cells.
Macrophages found in different organs and connective tissues have been
named according the specific locations, e.g., as microglia, alveolar
macrophages or Kupffer cells. Macrophages may function as
antigen-presenting cells, effector cells of cell-mediated immunity,
and phagocytes eliminating opsonized antigens.
Major basic protein. A small basic arginine-rich peptide (pI 10.9,
relative molecular mass of 13 800) in the granules of eosinophils that
kills helminths and protozoa.
Major histocompatibility complex (MHC). A cluster of genes encoding
cell surface antigens that are polymorphic within a species and have a
crucial function in signalling between lymphocytes and cells
expressing antigen and in recognition of self.
Mast cell. Tissue bound mononuclear granular cells with staining
affinity for basic dyes at low pH. The specific granules contain
mediators of allergic inflammation, e.g., histamine. Upon stimulation
with antigen via membrane-bound IgE antibodies, they release preformed
and newly generated mediators. Two types of mast cells exist.
Tryptase-containing T-mast cells are mainly associated with mucosal
epithelial ceils. Chymase-containing TC mast cells are long-living
connective tissue cells.
Mitogen. A substance that causes cells to synthesize DNA and
proliferate without acting as an antigen.
Monocyte. Bone-marrow-derived mononuclear phagocytic leukocyte, with
bean-shaped nucleus and fine granular cytoplasm containing lysosomes,
phagocytic vacuoles and cytoskeletal filaments. Once transported to
tissues they develop into macrophages.
Natural killer (NK) cell. A subset of lymphocytes found in blood
and some lymphoid tissues, derived from the bone marrow and appearing
as large granular lymphocytes (LGL). NK cells possess the capacity to
kill certain tumour cells or virus-infected normal cells. The killing
is not induced by specific antigen and is not restricted by MHC
molecules.
Nephropathy. Disease of the kidney that may involve either or both
the glomeruli (specialized structures where blood is filtered) and the
renal tubules (connected structures where the composition of the
filtrate is greatly modified in accordance with the physiological
needs of the body).
Nephrotic syndrome. A clinical disease in which damage to glomeruli
has caused leaky filtration, resulting in major loss of protein from
the body.
Neutrophil (polymorphonuclear leukocyte). Granular leukocytes
having a nucleus with three to five lobes and fine cytoplasmic
granules stainable by neutral dyes. The cells have properties of
chemotaxis, adherence to immune complexes, and phagocytosis. The cells
are involved in a variety of inflammatory processes including
late-phase allergic reactions.
Occupational asthma. Asthma caused by a sensitizing agent present
in the workplace, usually after a period of asymptomatic exposure.
Opsonization. Coating of antigens with antibody and/or complement
components. The interaction of opsonized complexes with Fc- or
complement-receptors facilitates their uptake by the receptor-bearing
phagocytic cells.
Oral tolerance. Orally induced and immune-mediated
non-responsiveness.
Peak expiratory flow rate (PEFR). A physiological measure of the
maximum air flow.
Plasma cell. A terminally differentiated B-lymphocyte with little
or no capacity for mitotic division, that can synthesize and secrete
antibody. Plasma cells have eccentric nuclei, abundant cytoplasm and
distinct perinuclear haloes. The cytoplasm contains dense rough
endoplasmic reticulum and a large Golgi complex.
Platelet activating factor. Low relative molecular mass
phospholipid generated from alkyl phospholipids in mast cells,
basophilic and neutrophilic granulocytes, and monocytes-macrophages,
which mediates microthrombus formation of platelets in
hypersensitivity reactions.
Prevalence (epidemiology). The number of cases of disease occurring
in a given population at a designated time.
Prevention of allergy. Primary prevention is the control of the
exposures inducing allergy. Secondary prevention is the control of
exposure of sensitized individuals.
Pseudo-allergy. Non-immunological "hypersensitivity" with clinical
symptoms and signs mimicking those of allergic diseases.
Radioallergosorbent test (RAST). A solid-phase radioimmunoassay for
detecting IgE antibody specific for a particular antigen.
Rate (epidemiology). The frequency with which an event occurs in a
defined population.
Reactive airways dysfunction syndrome (RADS). A syndrome
characterized by reversible airflow limitation and complicating
bronchial hyperresponsiveness induced by acute exposure to high
concentrations of non-sensitizing irritant gases at work.
Sensitization. Induction of specialized immunological memory in an
individual by exposure to an allergen.
Stem cell. Pluripotent cells, representing 0.01% of bone marrow
cells, having the capacity for self renewal, and committed to
differentiate along particular lineages, e.g., erythroid,
megakaryocytic, granulocytic, monocytic and lymphocytic. Cytokines
stimulate the proliferation and maturation of distinct precursors.
Suppressor T-lymphoctye. A subpopulation of T-lymphocytes that
inhibits the activation phase of immune responses. They are CD8+, and
their growth and differentiation may be regulated by CD4+ cells.
Tolerance. Persistent condition of specific immunological
unresponsiveness, resulting from previous non-sensitizing exposure to
the antigen.
Urticaria. Transient eruption of skin characterized by erythematous
or oedematous swelling (wheal) of the dermis or subcutaneous tissue.
9. CONCLUSIONS
Allergy is an important, world-wide, health problem. It affects a
substantial proportion of the population and all age groups. For
reasons that are poorly understood, the overall frequency of allergy
is increasing.
The various and complex interactions between chemicals, drugs and
proteins, the immune system and the target organ(s) that lead to the
manifestation of allergic hypersensitivity and autoimmunity are
reviewed in this monograph. Extensive research into these topics
continues, so new developments are anticipated. The multiplicity of
endogenous and exogenous factors that have an impact upon allergy are
considered. Exogenous factors, including allergens themselves, as well
as infection, air pollution and life style (e.g., tobacco smoking) are
all of importance. In addition, genetic predisposition to a particular
allergic disorder is an important determinant of reactivity.
The epidemiology of allergy demonstrates the widespread nature of
this group of disorders and, as a consequence, highlights the need for
proper attention to the identification of allergic hazards and their
assessment in the implementation of appropriate risk management
strategies.
Methods of hazard identification for skin sensitizers are well
established. They have still to be standardized for respiratory
sensitizers and are not yet available for other types of allergen.
Techniques to measure the potency of skin sensitizers are being
applied for the purpose of risk assessment.
Once allergic hazards have been well characterized, risk
assessment, risk management and risk communication are critical
elements to reduce the incidence of allergic disorders. Risk
assessment requires that the hazard, of known potency, is evaluated in
the light of the nature and extent of exposure. Risk management
measures, such as control of exposure and product labelling, must be
implemented when the risk assessment indicates the need.
More is being learned of the physicochemical and immunological
features of food allergens, which may eventually be of predictive
value.
The complexity of the mechanisms of allergic disorders makes it
difficult at present to suggest in vitro predictive methods of
general applicability, although application of structure-activity
relationships deserves further consideration.
10. RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH
a) Effective strategies to prevent allergy should be employed, based
on good information about allergens in products and the
environment. Control of exposure should be the basis for
preventing or minimizing the occurrence of allergic disease.
b) There is urgent need to determine the cause of the increased
frequency of allergy.
c) Methods of surveillance should be instituted to define the
frequency of allergies of different types.
d) The measurement of exposure of individuals and of populations may
be difficult, but adequate assessment is essential to any
analysis of the association between exposure and effect. The
specific nature of immune responses represents a unique type of
biomarker in studying past exposure, for example, by the use of
skin patch or prick testing, or assay of IgE antibodies to detect
sensitization by specific allergens.
e) Worker surveillance systems, the quality of medical examination
of workers and education of workers exposed to chemicals should
be improved in order to reveal occupational allergic diseases at
an early stage. Relatively simple notification schemes for
occupational disorders and post-marketing surveillance of
medicines provide economic screening and alerting systems for
allergic diseases. Monitoring these disorders in the workplace is
particularly valuable because exposure there is likely to be
greater than anywhere else.
f) The efficacy and value of primary and secondary prevention and
intervention strategies should be assessed at intervals using
validated epidemiological techniques.
g) For allergic contact dermatitis, the available in vivo
predictive models are of proven value for antigens of low
relative molecular mass. The need is to discover how best they
can be used to show the potency of allergens.
h) For allergic disorders of the respiratory tract, available in
vivo test methods for substances of low relative molecular mass
are promising, but their predictive value and specificity need to
be substantiated. For protein allergens, some animal test methods
are being developed, but they require further evaluation using
substances of known allergenic potential in humans.
i) It is important that information about the presence of allergens
in products (e.g., food) be readily available, for instance in
databases and on product labels, so that regulators and
individuals can adopt appropriate precautions.
j) The main method for avoiding occupational chemically induced
autoimmune disease is the control of exposure. Pre-exposure
assessment of those exposed to chemicals of immunomodulating
potential should be considered in order to document any
pre-existing features of connective tissue diseases. Strict
adherence to guidelines to avoid or minimize exposure is advised,
including the use of good occupational hygiene practices. Other
risk factors such as smoking should be minimized and regular
occupational medical examinations should be considered.
k) There is a need for investigation of the quantitative
relationships between immune responses induced by chemicals and
the severity of allergic reactions.
l) Determination of the minimal exposure and the duration of it
required to cause sensitization or elicit an allergic response is
important in controlling allergic disease.
m) It is important to devise standard strategies for the clinical
investigation and diagnosis of allergy and to apply them
internationally to examine the causes and incidence of allergic
disorders. This will require the production and availability of
standardized extracts of biological allergens of controlled
potency, as well as regular consideration of the components of
standard series of allergens used in testing.
n) Public health authorities, health professionals and government
agencies should consider how to estimate the human and economic
costs to individuals and society of allergic diseases.
o) Public health authorities, health professionals, the public and
especially the workforce would benefit from better information
about the occurrence, causes, clinical manifestations and
consequences of different types of allergy.
11. FURTHER RESEARCH
a) The adjuvant effect of environmental factors, such as pollutants,
particulates, tobacco smoke and UV radiation, on the induction
and elicitation of allergic responses to other chemicals should
be investigated. The influence of other forms of toxicity,
including direct immunotoxicity, should also be studied.
b) The influence of formulation and mixtures on the induction and
elicitation of chemical allergy needs to be explored.
c) The relevance of route of exposure to chemicals to the
development of allergic disease, particularly allergic
sensitization of the respiratory tract, needs to be investigated.
d) The relevance of route of exposure to the development of
tolerance should be explored.
e) The role of IgE antibody and other immune effector mechanisms in
the development of chemical respiratory allergy needs to be
clarified.
f) Understanding of the cellular mechanisms responsible for the
induction of allergy, such as how allergens are processed and
presented to T-cells, is necessary to facilitate the development
of predictive in vitro tests.
g) Improved, reliable, sensitive, specific and robust biomarkers of
exposure to allergens are needed. Ideally, they should be
non-invasive and suitable for field use. Similarly, better
techniques for dosimetry of allergens are needed.
h) The basis for the difference in the response of asthmatics and
non-asthmatics to airborne pollutants needs exploration.
i) It is important to understand the relative contributions of
lifestyle, nutrition, pollution and change in the pattern of
childhood infection and immunization to the development of
allergy.
j) Mechanisms of sensitization to food allergens need further
research. In addition, more needs to be known about the
occurrence and clinical importance of cross-reactivity of complex
allergens, especially those of natural origin, such as pollen and
food components.
k) It is necessary to identify the properties that control the
allergenic characteristics and potency of proteins, including the
reasons why they induce transient or persistent food allergy.
l) In the case of autoimmune disease, predictive methods need to be
developed and validated.
REFERENCES
Aalberse RC, Koshte V, & Clemens JG (1981) Immunoglobulin E antibodies
that cross react with vegetable foods, pollen and hymenoptera venom. J
Allergy Clin Immunol, 68: 356-364.
Abbey DE, Hwang BL, Burchette RJ, Vancuren T, & Mills PK (1995)
Estimated long-term ambient concentrations of PM10 and development of
respiratory symptoms in a nonsmoking population. Arch Environ Health,
50(2): 139-152.
Aberg N, Engstrom I, & Lindberg U (1989) Allergic diseases in Swedish
school children. Acta Paediatr Scand, 78: 246-252.
Abramson S, Miller RG, & Phillips RA (1977) The identification in
adult bone marrow of pluripotent and restricted stem cells of the
myeloid and lymphoid systems. J Exp Med, 145: 1567-1579.
Adams LE & Hess EV (1991) Drug-related Lupus: Incidence, mechanisms
and clinical implications. Drug Saf, 6: 431-449.
Adams RB, Planchon SM, & Roche JK (1993) Interferon-gamma modulation
of epithelial barrier function: time course, reversibility, and site
of cytokine binding. J Immunol, 150: 2356-2363.
Adinoff AD, Telez P, & Clark RAF (1988) Atopic dermatitis and
aeroallergen contact sensitivity. J Allergy Clin Immunol, 81: 736-742.
Alanko K, Keskinen H, Björkstén F, & Ojanen S (1978) Immediate-type
hypersensitivity to reactive dyes. Clin Allergy, 8: 25-31.
Alarcon-Segovia D (1985) Scleroderma. In: Rose NR & Mackay IR ed. The
autoimmune diseases. New York, London, San Diego, Academic Press, pp
119-143.
Allison AC (1989) Theories of self tolerance and autoimmunity. In:
Kammüller M, Bloksma N, & Seinen W ed. Autoimmunity and toxicology:
Immune disregulation induced by drugs and chemicals. Amsterdam,
Oxford, New York, Elsevier Science Publishers, pp 67-115.
Amagi M, Ishii K, Hashimoto T, Gamou S, Shimizu N, & Nishikawa T
(1995) Conformational epitopes of pemphigus antigens (Dsg1 and Dsg3)
are calcium dependent and glycosylation dependent. J Invest Dermatol,
105: 243-247.
American Medical Association (1987a) Council of Scientific Affairs,
American Medical Association, Report I, Part I, of the Allergy Panel
(1987): in vivo diagnostic testing and immunotherapy for allergy. J
Am Med Assoc, 258: 1363-1367.
American Medical Association (1987b) Council of Scientific Affairs,
American Medical Association, Report I, Part II, of the Allergy Panel
(1987): in vitro testing for allergy. J Am Med Assoc, 258:
1639-1643.
Ames RG, Hall DS, & Reger RB (1984) Chronic respiratory effects of
exposure to diesel emissions in coal mines. Arch Environ Health, 39:
389-394.
Amin S, Lahti A, & Maibach HI (1996) Contact urticaria and the contact
urticaria syndrome. In: Marzulli FN & Maibach HI ed.
Dermatotoxicology, 5th ed. London, Taylor and Francis, pp 485-505.
Andersen KE & Maibach HI (1985) Guinea pig sensitization assays. In:
Andersen KE & Maibach HI ed. Contact allergy: Predictive tests in
guinea pigs. Basel, Karger, pp 263-290.
Andersen KE & Rycroft RJG (1991) Recommended patch test concentrations
for preservatives, biocides and antimicrobials. Contact Dermatitis,
25: 1-18.
Andersen KE, Burrows D, & White IR (1995) Allergens from the standard
series. In: Rycroft RJG, Menné T, Frosch PJ, & Benezra C ed. Textbook
of contact dermatitis, 2nd ed. Berlin, Heidelberg, New York, Springer
Verlag, pp 416-457.
Anderson HR (1974) The epidemiological and allergic features of asthma
in the New Guinea Highlands. Clin Allergy, 4: 171-183.
Anderson HR (1989) Increase in hospital admissions for childhood
asthma: trends in referral, severity, and readmissions from 1970 to
1985 in a health region of the United Kingdom. Thorax, 44: 614-619.
Anderson JA (1991) The clinical spectrum of food allergy in adults.
Clin Exp Allergy, 21(suppl 1): 304-314.
Anderson SD & Smith CM (1991) Osmotic challenges in the assessment of
bronchial hyperresponsiveness. Am Rev Respir Dis, 143(3/2): S43-S46.
Anderson LB, Dreyfuss EM, Logan U, Johnstone E, & Glaser J (1970)
Melon and banana sensitivity coincident with ragweed pollinosis. J
Allergy Clin. Immunol, 45: 310-319.
Anderson HR, Bailey PA, Cooper JS, Palmer JC, & West S (1983)
Morbidity and school absence caused by asthma and wheezing illness.
Arch Dis Child, 58: 777-784.
Anderson HR, Pottier AC, & Strachan DP (1992) Asthma from birth to age
23: incidence and relation to prior and concurrent atopic disease.
Thorax, 47: 537-542.
Anderson HR, Butland BK, & Strachan DP (1994) Trends in prevalence and
severity of childhood asthma. Br Med J, 308: 1600-1604.
Anderson C, Hehr A, Robbins R, Hasan R, Athar M, Mukhtar H, & Elmets
CA (1995) Metabolic requirements for induction of contact sensitivity
to immunotoxic polyaromatic hydrocarbons. J Immunol,
155(7): 3530-3537.
Angelini G (1995) Allergens related to specific exposures: Topical
drugs. In: Rycroft RJG, Menné T, Frosch PJ, & Benezra C ed. Textbook
of contact dermatitis, 2nd ed. Berlin, Heidelberg, New York, Springer
Verlag, pp 477-498.
Angle CR (1988) Indoor air pollutants. Adv Pediatr, 35: 239-280.
Anonymous (1982) Breast feeding and eczema/asthma. Lancet, 1(8277):
910-911.
Anonymous (1985) Smoking, occupation, and allergic lung disease.
Lancet, 1(8435): 965.
Anto JM (1995) Asthma outbreaks: an opportunity for research? Thorax,
50(3): 220-222.
Anto JM, Sunyer J, Rodriguez Roisin R, Suarez Cervera M, & Vazquez L
(1989) Community outbreaks of asthma associated with inhalation of
soybean dust. Toxicoepidemiological Committee. New Engl J Med, 320:
1097-1102.
Armentia A, Martin Santos JM, Quintero A, Fernandez A, Barber D,
Alonso E, & Gil I (1990) Bakers' asthma: Prevalence and evaluation of
immunotherapy with a wheat flour extract. Ann Allergy, 65(4): 265-272.
Arnon R & Teitelbaum D (1993) On the existence of suppressor cells.
Int Arch Allergy Immunol, 100: 2-7.
Arshad SH, Matthews S, Gant C, & Hide DW (1992) Effect of allergen
avoidance on development of allergic disorders in infancy. Lancet,
339: 1493-1497.
Arshad SH, Stevens M, & Hide DW (1993) The effect of genetic and
environmental factors on the prevalence of allergic disorders at the
age of two years. Clin Exp Allergy, 23: 504-511.
Ashby J, Basketter DA, Paton D, & Kimber I (1995) Structure activity
relationships in skin sensitizing sensitization using the murine local
lymph node assay. Toxicology, 103: 177-194.
Asher MI, Pattemore PK, Harrison AC, Mitchell EA, Rea HH, Stewart AW,
& Woolcock AJ (1988) International comparison of the prevalence of
asthma symptoms and bronchial hyperresponsiveness. Am Rev Respir Dis,
138: 524-529.
Asher MI, Keil U, Anderson HR, Beasley R, Crane J, Martinez F,
Mitchell EA, Pearce N, Sibbald B, Stewart AW, Strachan D, Weiland SK,
& Williams HC (1995) International study of asthma and allergies in
childhood (ISAAC): Rationale and methods. Eur Respir J, 8(3): 483-491.
Asherson GL, Zembala M, Perera MACC, Mayhew B, & Thomas WR (1977)
Production of immunity and unresponsiveness in the mouse by feeding
contact sensitizing agents and the role of suppressor cells in the
Peyers's patches, mesenteric lymph nodes and other lymphoid tissue.
Cell Immunol, 33: 145-155.
Atkins FM, Steinberg SS, & Metcalfe DD (1985a) Evaluation of immediate
adverse reactions to foods in adult patients: I. Correlation of
demographic, laboratory, and prick skin test data with response to
controlled oral food challenge. J Allergy Clin Immunol, 75: 348-355.
Atkins FM, Steinberg SS, & Metcalfe DD (1985b) Evaluation of immediate
adverse reactions to foods in adult patients: II. A detailed analysis
of reaction patterns during oral food challenge. J Allergy Clin
Immunol, 75: 356-355.
Atkinson NF Jr & Platts-Mills TAE (1988) IgE and atopic disease. In:
Immunological diseases, 4th ed. Boston, Massachusetts, Little Brown,
vol 2, pp 1009-1026.
Avnstorp C (1992) Cement eczema: An epidemiological intervention
study. Acta Dermatol Venereol, 179(suppl): 1-22.
Axelsson IG, Johansson SG, Larsson PH, & Zetterstrom O (1990)
Characterization of allergenic components in sap extract from the
weeping fig (Ficus benjamina). Int Arch Allergy Appl Immunol, 91(2):
130-135.
Bachert C, Hauser U, Prem B, Rudack C, & Ganzer U (1995)
Proinflammatory cytokines in allergic rhinitis. Eur Arch
Otorhinolaryngol, 252(Suppl 1): 155-214.
Baird AW, Coombs RRA, McLaughlan P, & Cuthbert AW (1984) Immediate
hypersensitivity reactions to cow milk proteins in isolated epithelium
from ileum of milk-drinking guinea-pigs: Comparison with colonic
epithelia. Int Arch Allergy Appl Immunol, 75: 255-263.
Baker DB, Gann PH, Brooks SM, Gallagher J, & Bernstein IL (1990)
Cross-sensitization study of platinum salts sensitization among
precious metals refinery workers. Am J Ind Med, 18: 653-664.
Bakke P, Gulsvikk A, & Eide GE (1990) Hay fever, eczema and urticaria
in southwest Norway. Allergy, 45: 515-522.
Ball B, Horstman D, Folinsbee L, Gerrity T, DeWitt P, Abdul-Salaam S,
& Brown J (1993) Pulmonary responses in asthmatics performing light
exercise in clean air(air) and 0.16 ppm ozone (O3). Am Rev Respir
Dis, 147: A640 (abstract).
Barbee RA (1987) The epidemiology of asthma. Monogr Allergy, 21:
21-41.
Barbee RA, Kaltenborn W, Lebowitz MD, & Burrows B (1987) Longitudinal
changes in allergen skin test reactivity in a community population
sample. J Allergy Clin Immunol, 79(1): 16-24.
Bardare M, Vaccari A, Allievi E, Brunelli L, Coco F, de Gaspari GC, &
Flauto U (1993) Influence of dietary manipulation on incidence of
atopic disease in infants at risk. Ann Allergy, 71(4): 366-371.
Barnes PJ, Baranjuk J, & Belvisi MG (1991) Neuropeptides in the
respiratory tract. Am Rev Respir Dis, 144: 1187-1198.
Barnes PJ, Jonsson B, & Klim JB (1996) The costs of asthma. Eur Respir
J, 9: 636-642.
Barratt MD (1995) Quantitative structure activity relationships for
skin permeability. Toxicol in vitro, 9: 27-37.
Barratt MD, Basketter DA, Chamberlain M, Payne MP, Admans GD, &
Langowski JJ (1994a) Development of an expert system rule base for
identifying contact allergens. Toxicol in vitro, 8: 837-839.
Barratt MD, Basketter DA, Chamberlain M, Admans GD, & Langowski JJ
(1994b) An expert system rule base for identifying contact allergens.
Toxicol in vitro, 8: 1053-1060.
Barratt MD, Basketter DA, & Roberts DW (1994c) Skin sensitization
structure-activity relationships for phenyl benzoates. Toxicol in
vitro, 8: 823-826.
Barrett JT (1988) Textbook of immunology, 5th ed. St. Louis, Missouri,
C.V. Mosby & Co.
Barrett SP, Toh BH, Alderuccio F, van Driel I, & Gleeson PA (1995)
Organ-specific autoimmunity induced by adult thymectomy and
cyclophosphamide-induced lymphopenia. Eur J Immunol, 25: 238-244.
Barry DM, Burr ML, & Limb ES (1991) Prevalence of asthma among 12 year
old children in New Zealand and South Wales: a comparative survey.
Thorax, 46: 405-409.
Bascom R (1996) Environmental factors and respiratory
hypersensitivity: the Americas. Toxicol Lett, 86(2-3): 115-130.
Bascom R, Naclerio RM, Fitzgerald TK, Kagey Sobotka A, & Proud D
(1990) Effect of ozone inhalation on the response to nasal challenge
with antigen of allergic subjects. Am Rev Respir Dis, 142(3): 594-601.
Basketter DA & Roberts DW (1990) Structure activity relationships in
contact allergy. Int J Cosmet Sci, 12: 81-90.
Basketter DA, Roberts DW, Cronin M, & Scholes EW (1992) The value of
the local lymph node assay in quantitative structure-activity
investigations. Contact Dermatitis, 27: 137-142.
Basketter DA, Gerberich GF, Kimber I, & Loveless SE (1996) The local
lymph node assay -- a valuable alternative to currently accepted skin
sensitisation tests. Food Chem Toxicol, 34: 651-660.
Basketter DA, Cookman G, Gerberich GF, Hamaide N, & Potokar M (1997)
Skin sensitisation thresholds: Determination in predictive models.
Food Chem Toxicol, 35: 417-425.
Bates DV & Sizto R (1987) Air pollution and hospital admissions in
southern Ontario: The acid summer haze effect. Environ Res, 43:
317-331.
Baur V & Fruhmann G (1981) Specific IgE antibodies in patients with
isocyanate asthma. Chest, 80(suppl): 73S-76S.
Baur X, Dewair M, & Fruhmann G (1984) Detection of immunologically
sensitised isocyanate workers by RAST and intracutaneous skin tests. J
Allergy Clin Immunol, 73: 610-618.
Baur X, Marek W, Ammon J, Czuppon AB, Marczynski B, Raulf-Heimsoth M,
Roemmelt H, & Fruhman G (1994) Respiratory and other hazards of
isocyanates. Int Arch Occup Environ Health, 66: 141-152.
Beasley R, Roche WR, Roberts JA, & Holgate ST (1989) Cellular events
in the bronchi in mild asthma and after bronchial provocation. Am Rev
Respir Dis, 139: 806-817.
Beasley R, Thomson C, & Pearce N (1991) Selenium, glutathione
peroxidase and asthma. Clin Exp Allergy, 21(2): 157-159.
Becher R, Hongslo JK, Jantunen J, & Dybing E (1996) Environmental
chemicals relevant for respiratory hypersensitivity: the indoor
environment. Toxicol Lett, 86: 155-162.
Beeson PB (1994) age and sex associations of 40 autoimmune diseases.
Am J Med, 96: 457-462.
Beezhold DH, Sussman GL, Liss GM, & Chang N-S (1996) Latex allergy can
induce clinical reactions to specific foods. Clin Exp Allergy, 26:
416-422.
Beggs PJ & Curson PH (1995) An integrated environmental asthma model.
Arch Environ Health, 50(2): 87-94.
Behrendt H, Friedrich KH, Kainka-Stänicke E, Darsow U, Becker WM, &
Tomingas R (1991) New trends in allergy. In: Ring J & Pryzbilla B ed.
Allergens and pollutants in the air: A complex interaction. Berlin,
Heidelberg, New York, Springer Verlag, pp 467-478.
Behrendt H, Becker W, Friedrichs K, Darsow U, & Tomingas R (1992)
Interaction between aeroallergens and airborne particulate matter. Int
Arch Allergy Immunol, 99: 425-428.
Behrendt H, Krämer U, Dolgner R, Hinrichs J, Willer H, Hagenbeck H, &
Schlipköter W (1993) Elevated levels of total IgE in East German
children: Atopy, parasites or pollutants? Allergo J, 2: 31-40.
Behrendt H, Friedrich KH, Krämer U, Hitzfeld B, Becker WM, & Ring J
(1996) The role of indoor and outdoor air pollution in allergic
diseases. In: Vos J, Younes M, & Smith E ed. Allergic
hypersensitivities induced by chemicals. Boca Raton, Florida, CRC
Press, pp 173-182.
Bekesi JG, Roboz JP, Fischbein A, & Mason P (1987) Immunotoxicology:
Environmental contamination by polybrominated biphenyls and immune
dysfunction among residents of the State of Michigan. Cancer Detect
Prev, 1(suppl): 29-37.
Bell RG (1996) IgE, allergies and helminth parasites: a new
perspective on an old conundrum. Immunol Cell Biol, 74: 337-345.
Bencko V, Vasilieva EV, & Symon K (1980) Immunological aspects of
exposure to emissions from burning coal of high beryllium content.
Environ Res, 22(2): 439-449.
Benezra C, Sigman CC, Perry LR, Helmes CT, & Maibach HI (1985) A
systematic search for structure-activity relationships of skin contact
sensitizers: methodology. J Invest Dermatol, 85: 351-356.
Benjamini E & Leskowitz S (1991) Immunology: A short course. New York,
Wiley-Liss.
Bentley AM, Maestrelli P, Fabbri LM, Menz G, Storz Chr, Bradley B,
Jeffrey PK, Durham SR, & Kay AB (1991) Immunohistology of the
bronchial mucosa in occupational, intrinsic and extrinsic asthma. J
Allergy Clin Immunol, 87: 246 (abstract).
Berglund M, Braback L, Bylin G, Jonson JO, & Vahter M (1994) Personal
NO2 exposure monitoring shows high exposure among ice-skating
schoolchildren. Arch Environ Health, 49: 17-24.
Bernhisel-Broadbent J & Sampson HA (1989) Cross-allergenicity in the
legume botanical family in children with food hypersensitivity. J
Allergy Clin Immunol, 83: 435-440.
Bernhisel-Broadbent J, Taylor S, & Sampson HA (1989)
Cross-allergenicity in the legume botanical family in children with
food hypersensitivity: II. Laboratory correlates. J Allergy Clin
Immunol, 84: 701-709.
Bernstein DI, Patterson R, & Zeiss CR (1982a) Clinical and immunologic
evaluation of trimellitic anhydride- and phthalic anhydride-exposed
workers using a questionnaire and comparative analysis of enzyme
linked immunosorbent and radioimmunoassay studies. J Allergy Clin
Immunol, 69: 311-318.
Bernstein M, Day JH, & Welsh A (1982b) Double-blind food challenge in
the diagnosis of food sensitivity in the adult. J Allergy Clin
Immunol, 70: 205-211.
Bernstein DI, Gallagher JA, D'Souza L, & Bernstein IL (1984)
Heterogeneity of specific IgE responses in workers sensitised to acid
anhydride compounds. J Allergy Clin Immunol, 74: 794-801.
Biagini RE, Bernstein IL, Gallagher JS, Moorman WJ, Brooks S, & Gann
PH (1985) The diversity of reaginic immune responses to platinum and
palladium salts. J. Allergy Clin Immunol, 76(6): 794-802.
Biagini RE, Moorman WJ, Lewis TR, & Bernstein IL (1986) Ozone
enhancement of platinum asthma in a primate model. Annu Rev Respir
Dis, 134: 719-725.
Bieber TH & Ring J (1992) In vivo modulation of the high-affinity
receptor for IgE (Fc-epsilon-RI) on human epidermal Langerhans cells.
Int Arch Allergy Immunol, 99: 204-207.
Bigazzi PE (1988) Autoimmunity induced by chemicals. J Toxicol Clin
Toxicol, 26: 125-156.
Bigazzi PE (1997) Autoimmunity caused by xenobiotics. Toxicology, 119:
1-21.
Bignon JS, Aron Y, Ju LY, Kopferschmitt MC, Garnier R, Mapp C, Fabbri
LM, Pauli G, Lockhart A, Charron D, & Swierczewski E (1994) HLA class
II alleles in isocyanate-induced asthma. Am J Respir Crit Care Med,
149: 71-75.
Bilyk N & Holt PG (1995) Cytokine modulation of the immunosuppressive
phenotype of pulmonary alveolar macrophage populations. Immunology,
86: 231-237.
Bindslev-Jensen C, Hansen TK, Norgaard A, Vestergaard H, & Lars K
(1994a) New controversial techniques in the diagnosis of food
hypersensitivity. In: Johansson SGO ed. Progress in allergy and
clinical immunology. Toronto, Bern, Hans Huber, vol 3, pp 468-274.
Bindslev-Jensen C, Skov P, Madsen F, & Poulsen LK (1994b) Food allergy
and food intolerance - what is the difference? Ann Allergy, 72:
317-320.
Bircher AJ, Wüthrich B, Langauer S, & Schmid P (1993) [Ficus
benjamina, a perennial inhalation allergen of increasing importance.]
Schweiz Med Wochenschr, 123(22): 1153-1159 (in German).
Björksten B & Kjellman NIM (1987) Perinatal factors influencing the
development of allergy. Clin Rev Allergy, 5: 339-347.
Black CM, Welsh KI, Walker AE, Bernstein RM, Catoggio LJ, McGregor AR,
& Lloyd Jones JK (1983) Genetic susceptibility to scleroderma-like
syndrome induced by vinyl chloride. Lancet, 1: 53-55.
Blaikie L, Basketter DA, & Morrow T (1995) Experience with a guinea
pig model for the assessment of respiratory allergens. Hum Exp
Toxicol, 14: 73.
Blanco C, Carrillo T, Castillo R, Quiralte J, & Cuevas M (1994)
Avocado hypersensitivity. Allergy, 49: 454-459.
Bloch KJ & Walker WA (1981) Effect of locally induced intestinal
anaphylaxis on the uptake of a bystander antigen. J Allergy Clin
Immunol, 67: 312-316.
Bloksma N, Kammüller ME, Punt P, & Seinen W (1988) Strain-dependence
of primary popliteal lymph node reactions to subcutaneous injection of
diphenylhydantoin in mice. In: Kammüller ME ed. A toxicological
approach to chemical-induced autoimmunity. Utrecht, The Netherlands,
University of Utrecht, pp 79-92 (PhD Thesis).
Bloksma N, Kubicka-Muranyi M, Schuppe H-C, Gleichmann E, & Gleichmann
H (1995) Predictive immunotoxicological test systems: Suitability of
the popliteal lymph node assay in mice and rats. Crit Rev Toxicol, 25:
369-396.
Bloom BR, Salgame P, & Diamons B (1992) Revisiting and revising
suppressor T cells. Immunol Today, 13(4): 131-136.
Bock SA (1987) Prospective appraisal of complaints of adverse
reactions to foods in children during the first 3 years of life.
Pediatrics, 79(5): 683-688.
Bock SA, Sampson HA, Atkins FM, Zeiger RS, Lehrer S, Sachs M, Bush RK,
& Metcalfe DD (1988) Double-blind, placebo-controlled food challenge
(DBPCFC) as an office procedure: a manual. J Allergy Clin Immunol, 82:
986-997.
Boerrigter GH & Scheper RJ (1987) Local and systemic desensitization
induced by repeated epicutaneous hapten application. J Invest
Dermatol, 88(1): 3-7.
Bohadana AB, Massin N, Wild P, Kolopp MN, & Toamain JP (1994)
Respiratory symptoms and airway responsiveness in apparently healthy
workers exposed to flour dust. Eur Respir J, 7(6): 1070-1076.
Bonini S, Magrini L, Rotiroti G, Ronchetti MP, & Onotati P (1994)
Genetic and environmental factors in the changing incidence of
allergy. Allergy, 49: 6-14.
Bonnet M, Angibaud G, Cantagrel A, Montastruc JL, & Janet M (1995)
Myasthénie induite par la tiopronine au cours du traitement de la
polyarthrite rhumatoïde. Rev Neurol, 151: 67-68.
Borelli S (1981) [Dermatological indications for climatherapy in the
high altitude of Davos (1560 m and higher.] In: Borelli S & Düngemann
H ed. [Allergology and dermatology.] Frankfurt, Germany, IMP-Verlag,
pp 564-660 (in German).
Borelli S & Schnyder UW (1962) [Constitutional dermatitis without
atopy.] In: Miescher G & Storck H ed. [Dermatitis: I. Handbook of
treatment.] Berlin, Heidelberg, New York, Springer Verlag, vol 1/1, pp
254-319 (in German).
Bos JD & Kapsenberg ML (1993) The skin immune system: progress in
cutaneous biology. Immunol Today, 14: 75-78.
Braback L, Breborowicz A, Julge K, Knutsson A, Riikjarv MA, Vasar M, &
Bjorksten B (1995) Risk factors for respiratory symptoms and atopic
sensitisation in the Baltic area. Arch Dis Child, 72(6): 487-493.
Braun Fahrländer C, Kunzli N, Domenighetti G, Carell CF, & Ackermann
Liebrich U (1994) Acute effects of ambient ozone on respiratory
function of Swiss schoolchildren after a 10-minute heavy exercise.
Pediatr Pulmonol, 17: 169-177.
Braun-Fahrländer C, Vuille JC, Sennhauser FH, Neu U, Künzle T, Grize
L, Gassner M, Minder C, Schindler C, Varonier HS, & Wüthrich B (1997)
Respiratory health and long-term exposure to air pollutants in Swiss
schoolchildren -- SCARPOL Team: Swiss study on childhood allergy and
respiratory symptoms with respect to air pollution, climate and
pollen. Am J Respir Crit Care Med, 155: 1042-1049.
Breathnach SM (1988) The Langerhans cell: Centenary review. Br J
Dermatol, 119(4): 463-469.
Briatico-Vangosa G, Braun CLJ, Cookman G, Hofmann T, Kimber I,
Loveless SE, Morrow T, Pauluhn J, Sorensen T, & Niessen HJ (1994)
Respiratory allergy: Hazard identification and risk assessment. Fundam
Appl Toxicol, 23: 145-158.
Britton WJ, Woolcock AJ, Peat JK, Sedgwick CJ, Lloyd DM, & Leeder SR
(1986) Prevalence of bronchial hyperresponsiveness in children: the
relationship between asthma and skin reactivity to allergens in two
communities. Int J Epidemiol, 15(2): 202-209.
Britton J, Pavord I, Richards K, Knox A, Wisniewski A, Weiss S, &
Tattersfield A (1994) Dietary sodium intake and the risk of airway
hyperreactivity in a random adult population. Thorax, 49(9): 875-880.
Britton JR, Pavord ID, Richards KA, Knox AJ, Wisniewski AF, Lewis SA,
Tattersfield AE, & Weiss ST (1995) Dietary antioxidant vitamin intake
and lung function in the general population. Am J Respir Crit Care
Med, 151(5): 1383-1387.
Broder I, Higgins MW, Mathews KP, & Keller JB (1974a) Epidemiology of
asthma and allergic rhinitis in a total community, Tecumseh, Michigan.
J Allergy Clin Immunol, 53: 127-138.
Broder I, Higgins MW, Matthews KP, & Keeler JB (1974b) Epidemiology of
asthma and allergic rhinitis in a total community, Tecumseh, Michigan:
3. Second survey of the community. J Allergy Clin Immunol, 53:
127-138.
Brooks SM (1982) The evaluation of occupational airways disease in the
laboratory and workplace. J Allergy Clin Immunol, 70: 56-66.
Broughton A & Thrasher JD (1988) Antibodies and altered cell-mediated
immunity in formaldehyde-exposed humans. Comments Toxicol, 2: 155-174.
Bruijnzeel-Koomen C, Van Wichen DF, Toonstra J, Berrens L, &
Bruijnzeel PL (1986) The presence of IgE molecules on epidermal
Langerhans cells in patients with atopic dermatitis. Arch Dermatol
Res, 278(3): 199-205.
Bruijnzeel-Koomen C, Ortolani C, Aas K, Bindslev-Jensen C, Björkstén
B, Moneret-Vautrin D, & Wüthrich B (1995) Position paper of the
European Academy of Allergy and Clinical Immunology on adverse
reactions to food: Adverse reactions to food. Allergy, 50: 623-635.
Brunekreef B (1992) Damp housing and adult respiratory symptoms.
Allergy, 47(5): 498-502.
Brunekreef B, Dockery DW, Speizer FE, Ware JH, Spengler JD, & Ferris
BG (1989) Home dampness and respiratory morbidity in children. Am Rev
Respir Dis, 140: 1363-1367.
Brunekreef B, Dockery DW, & Krzyzanowski M (1995) Epidemiologic
studies on short-term effects of low levels of major ambient air
pollution components. Environ Health Perspect; 103(Suppl 2): 3-13.
Brunekreef B, Janssen NA, de Hartog J, Harssema H, Knape M, & van
Vliet P (1997) Air pollution from truck traffic and lung function in
children living near motorways. Epidemiology, 8(3): 298-303.
Bruynzeel DP, von Blomberg-van der Flier M, Scheper RJ, van Ketel WG,
& de Haan P (1985) Penicillin allergy and the relevance of
epicutaneous tests. Dermatologica, 171: 429-434.
Bryson JS, Caywood BE, & Kaplan AM (1991) Relationship of cyclosporine
A-mediated inhibition of clonal deletion and development of syngeneic
graft-versus-host disease. J Immunol, 147: 391-397.
Buckley RH (1992) Primary immunodeficiency diseases. In: Wyngaarden J
& Smith L ed. Cecil's textbook of medicine. Philadelphia,
Pennsylvania, W.B. Saunders Company, pp 1446-1453.
Buehler EV (1965) Delayed contact hypersensitivity in the guinea pig.
Arch Dermatol, 5(91): 171-177.
Buist AS & Vollmer WM (1990) Reflections on the rise in asthma
morbidity and mortality. J Am Med Assoc, 264: 1719-1720.
Burge PS (1989) Diagnosis of occupational asthma. Clin Exp Allergy,
19(6): 649-652.
Burlingame RW & Rubin RL (1991) Drug-induced anti-histone
autoantibodies display two patterns of reactivity with substructures
of chromatin. J Clin Invest, 88: 680-690.
Burney P (1987) A diet rich in sodium may potentiate asthma:
Epidemiologic evidence for a new hypothesis. Chest, 91: 143S-148S.
Burney PG, Britton JR, Chinn S, Tattersfield AE, Platt HS, Papacosta
AO, & Kelson MC (1986) Response to inhaled histamine and 24 hour
sodium excretion. Br Med J Clin Res Ed, 292: 1483-1486.
Burney PG, Laitinen LA, Perdrizet S, Huckauf H, Tattersfield AE, Chinn
S, Poisson N, Heeren A, Britton JR, & Jones T (1989a) Validity and
repeatability of the IUATLD (1984) bronchial symptoms questionnaire:
An international comparison. Eur Respir J, 2(10): 940-945.
Burney PG, Neild JE, Twort CH, Chinn S, Jones TD, Mitchell WD, Bateman
C, & Cameron IR (1989b) Effect of changing dietary sodium on the
airway response to histamine. Thorax, 44: 36-41.
Burney PG, Chinn S & Rana RJ (1990) Has the prevalence of asthma
increased in children? Evidence from the national study of health and
growth 1973-86. Br Med J, 300: 1306-1310.
Burney P, Chinn S, Jarvis D, Luczynska C, & Lai E (1996) Variations in
the prevalence of respiratory symptoms, self-reported asthma attacks,
and use of asthma medication in the European Community respiratory
health survey (ECRHS). Eur Respir J, 9: 687-695.
Burnett RT, Dales R, Krewski D, Vincent R, Dann T, & Brook JR (1995)
Associations between ambient particulate sulphate and admissions to
Ontario hospitals for cardiac and respiratory diseases. Am J
Epidemiol, 142: 15-22.
Burr ML, Butland BK, King S, & Vaughan Williams E (1989) Changes in
asthma prevalence: Two surveys 15 years apart. Arch Dis Child 1989,
64: 1452-1456.
Burrows B, Halonen M, Barbee RA, & Lebowitz MD (1981) The relationship
of serum immunoglobulin E to cigarette smoking. Am Rev Respir Dis,
124(5): 523-525.
Busse WW (1990) Respiratory infections: their role in airway
responsiveness and the pathogenesis of asthma. J Allergy Clin Immunol,
85: 671-683.
Butcher BT & Salvaggio JE (1986) Occupational asthma. J Allergy Clin
Immunol, 78(4): 547-556.
Butler JM, Chan SC, Stevens S, & Hanifin JM (1983) Increased leukocyte
histamine release with elevated cyclic AMP-phosphodiesterase activity
in atopic dermatitis. J Allergy Clin Immunol, 71: 490-497.
Byers VS, Levin AS, Ozonoff DM, & Baldwin RW (1988) Association
between clinical symptoms and lymphocyte abnormalities in a population
with chronic domestic exposure to industrial solvent contaminated
domestic water supply and a high incidence of leukaemia. Cancer
Immunol Immunother, 27: 77-81.
Calkhoven P, Aalbers M, Koshte L, Pos O, Oei HD, & Aalberse RC (1987)
Cross-reactivity among birch pollen, vegetables and fruits as detected
by IgE antibodies is due to at least three distinct cross-reactive
structures. Allergy, 42: 382-390.
Calvin G (1992) Risk management case history -- detergents. In:
Richardson ML ed. Risk management of chemicals. London, Royal Society
of Chemistry, pp 120-136.
Carr RI, Etheridge PD, & Tilley D (1985) Failure of oral tolerance in
NZB/W mice is antigen dependent and parallels antibody patterns in
human systemic lupus erythematosus (SLE). Fed Proc, 44: 1542
(abstract).
Cartier A, Thompson NC, Frith PA, Roberts R, & Hargreave FE (1982)
Allergen-induced increase in bronchial responsiveness to histamine:
relationship to the late asthmatic response and change in airway
caliber. J Allergy Clin Immunol, 70: 170-177.
Cartier A, Bernstein IL, Burge PS, Cohn JR, Fabbri LM, Hargreave FE,
Malo JL, McKay RT, & Salvaggio JE (1989) Guidelines for
bronchoprovocation on the investigation of occupational asthma: Report
of the Subcommittee on Bronchoprovocation for Occupational Asthma. J
Allergy Clin Immunol, 84(5/2): 823-829.
Catto-Smith AG, Patrick MK, Hardin JA, & Gall DG (1989a) Intestinal
anaphylaxis in the rat: Mediators responsible for the ion transport
abnormalities. Agents Actions, 28: 185-191.
Catto-Smith AG, Patrick MK, Scott RB, Davidson JS, & Gall DG (1989b)
Gastric response to mucosal IgE-mediated reactions. Am J Physiol, 257:
G704 (abstract).
CDC (US Center for Disease Control) (1994) Populations at risk from
particulate air pollution -- United States, 1992. Morbid Mortal Wkly
Rep, 43: 290-293.
CDC (US Center for Disease Control) (1995) Asthma--United States,
1982-1992. Morbid Mortal Wkly Rep, 43: 952-955.
Challacombe SJ (1983) Salivary antibodies and systemic tolerance in
mice after oral immunization with bacterial antigens. Ann NY Acad Sci,
409: 177-193.
Challacombe SJ & Tomasi TB (1987) Oral tolerance. In: Brostoff J &
Challacombe SJ ed. Food allergy and intolerance, Section F: Model
systems of antigen handling. Philadelphia, Pennsylvania, Ballière
Tindall, pp 255-268.
Chambers CA & Allison JP (1997) Co-stimulation in T cell responses.
Curr Opin Immunol, 9: 396-404.
Chan PK, Baldwin RC, Parsons RD, Moss JN, Stiratelle R, Smith JM, &
Hayes AW (1983) Kathon biocide: Manifestation of delayed contact
dermatitis in guinea pigs is dependent on the concentrations for
induction and challenge. J Invest Dermatol, 81: 409-411.
Chan-Yeung M (1990) Occupational asthma. Chest, 98(suppl 5):
148S-161S.
Chan-Yeung M (1995) Occupational Asthma. Environ Health Perspect,
103(suppl 6): 249-252.
Chan-Yeung M & Malo JL (1995a) Occupational asthma. N Engl J Med,
333(2): 107-112.
Chan Yeung M & Malo JL (1995b) Epidemiology of occupational asthma.
In: Asthma and rhinitis. Oxford, London, Boston, Scientific
Publications, pp 44-57.
Chan-Yeung M, Malo JL, Busse W, & Holgate ST ed. (1995) Asthma and
rhinitis: Epidemiology of occupational asthma. Oxford, London, Boston,
Blackwell Scientific Publications, pp 44-57.
Chand N, Harrison JE, Rooney S, Pillar J, Jakubicki R, Nolan K,
Diamantis W, & Sofia RD (1992) Anti-IL-5 monoclonal antibody inhibits
allergic late phase bronchial eosinophilia in guinea pigs: a
therapeutic approach. Eur J Pharmacol, 211: 121-123.
Chandor SB (1988) Autoimmune phenomena in lymphoid malignancies. Clin
Lab Med, 8: 373-384.
Chapman JR & Roberts DW (1984) Humoral immune dysfunction as a result
of prenatal exposure to diphenylhydantoin: correlation with the
occurrence of physical defects. Teratology, 30: 107-117.
Charous BL (1994) The puzzle of latex allergy: some answers, still
more questions. Ann Allergy, 73(4): 277-281.
Chase MW (1946) Inhibition of experimental drug allergy by prior
feeding of the sensitizing agent. Proc Soc Exp Biol Med, 61: 257-259.
Chen Y, Kuchroo VK, Inobe J, Hafler DA, & Weiner HL (1994) Regulatory
T cell clones induced by oral tolerance: Suppression of autoimmune
encephalomyelitis. Science, 265: 1237-1240.
Chua KY, Stewart GA, Thomas WR, Simpson RJ, Dilworth RJ, Plozza TM, &
Turner KJ (1988) Sequence analysis of cDNA coding for a major house
dust mite allergen, Der p 1. J Exp Med, 167: 175-182.
Cirla AM (1994) Cobalt-related asthma: clinical and immunological
aspects. Sci Total Environ, 150(1-3): 85-94.
Cleland LG & Bell DA (1978) The occurrence of systemic lupus
erythematosus in two kindreds in association with severe selective IgA
deficiency. J Rheumatol, 5: 288-293.
Clinton PM, Kemeny DM, Amlot P, Urbanek R, & Lessof MH (1986)
Histamine release from peripheral blood leucocytes in egg-allergic
patients. Clin Allergy, 16: 345-354.
Coca AF & Cooke RA (1923) On the classification of the phenomena of
hypersensitiveness. J Immunol, 8: 163-182.
Cockcroft DW (1988) Allergens. In: Barnes PJ, Rodger IW, & Thomson NC
ed. Asthma: Basic mechanisms and clinical management. New York,
London, San Diego, Academic Press, pp 445-464.
Coenraads PJ & Smit J (1995) Epidemiology. In: Rycroft RJG, Menné T,
Frosch PJ, & Benezra C ed. Textbook of contact dermatitis, 2nd ed.
Berlin, Heidelberg, New York, Springer Verlag, pp 133-150.
Coffman RL, O'Hara J, Bond MW, Carty J, Zlotnik A, & Paul WE (1986)
B-cell stimulatory factor-1 enhances the IgE response of
lipopolysaccharide activated B-cells. J Immunol, 136(12): 4538-4541.
Coffman RL, Lebman D, & Shrader B (1989) Transforming growth
factor-beta specifically enhances IgA production by
lipopolysaccharide-stimulated murine B-lymphocytes. J Exp Med, 170:
1039-1044.
Cohen IR & Miller A ed. (1994) Autoimmune disease models. New York,
London, San Diego, Academic Press.
Colloff MJ, Ayres J, Carswell F, Howarth PH, Merrett TG, Mitchell EB,
Walshaw MJ, Warner JO, Warner JA, & Woodcock AA (1992) The control of
allergens of dust mites and domestic pets: A position paper. Clin Exp
Allergy, 22(suppl 2): 1-28.
Conell JT (1969) Quantitative intranasal pollen challenges: III. The
priming effect in allergic rhinitis. J Allergy, 43: 33-44.
Constantinescu CS, Hilliard B, Fujioka T, Bhopale MK, Calida D, &
Rostami AM (1998) Pathogenesis of neuroimmunologic diseases. Immunol
Res, 17: 217-227.
Cookson WOCM, Sharp PA, Faux JA, & Hopkin JM (1989) Linkage between
immunoglobulin E responses underlying asthma and rhinitis and
chromosome 11q. Lancet, 1: 1292-1295.
Cookson WOCM, Young RP, Sandford AJ, Moffatt MF, Shirakawa T, Sharp
PA, Faux JA, Julier C, Le Souef PN, Lathrop GM, & Hopkin JM (1992)
Maternal inheritance of atopic IgE responsiveness on chromosome 11q.
Lancet, 340: 381-384.
Cooper KD (1994) Atopic dermatitis: recent trends in pathogenesis and
therapy. J Invest Dermatol, 102: 128-137.
Cooper KD, Kang K, Chan SC, & Hanifin JM (1985) Phosphodiesterase
inhibition by Ro20-1724 reduces hyper-IgE synthesis by atopic
dermatitis cells in vitro. J Invest Dermatol, 804: 477-482
Cormio L, Turjanmaa K, Talja M, Andersson LC, & Ruutu M (1993)
Toxicity and immediate allergenicity of latex gloves. Clin Exp
Allergy, 23(7): 618-623.
Corrigan CJ & Kay AB (1992) T cells and eosinophils in the
pathogenesis of asthma. Immunol Today, 13: 501-507.
Cotterill JA (1991) Psychosomatic approaches in the treatment of
atopic eczema. In: Ruzicka T, Ring J, & Przybilla B ed. Handbook of
atopic eczema. Berlin, Heidelberg, New York, Springer Verlag, pp
459-465.
Crespo JF, Pascual C, Burks AW, Helms RM, & Esteban MM (1995)
Frequency of food allergy in a pediatric population from Spain.
Pediatr Allergy Immunol, 6: 39-43.
Croner S & Kjellman NI (1990) Development of atopic disease in
relation to family history and cord blood IgE levels. Pediatr Allergy
Immunol, 1: 14-20.
Cronin E (1991) Formaldehyde is a significant allergen in women with
hand eczema. Contact Dermatitis, 25: 276-283.
Cronin MTD & Basketter DA (1994) Multivariate QSAR analysis of a skin
sensitization database: SAR and QSAR. Environ Res, 2: 1-21.
Crowe SE & Perdue MH (1992) Gastrointestinal food hypersensitivity:
Basic mechanisms of pathophysiology. Gastroenterology, 103: 1075-1095.
Cullinan P, Lowson D, Nieuwenhuijsen MJ, Sandiford C, Tee RD, Venables
KM, McDonald JC, & Newman Taylor AJ (1994) Work related symptoms,
sensitisation, and estimated exposure in workers not previously
exposed to flour. Occup Environ Med, 51(9): 579-583.
Curtis GH, Patrick MK, Catto-Smith AG, & Gall DG (1990) Intestinal
anaphylaxis in the rat: Effect of chronic antigen exposure.
Gastroenterology, 98: 1558-1566.
Czernielewski JM & Demarchez M (1987) Further evidence for the
self-reproducing capacity of Langerhans cells in human skin. J Invest
Dermatol, 88: 17-20.
Dakin R (1982) Remarks on cutaneous affection, produced by certain
poisonous vegetables. Am J Med Sci, 4: 98-100.
Dally MB, Hunter JV, Hughes EG, Stewart M, & Newman Taylor AJ (1980)
Hypersensitivity to platinum salts: a population study. Am Rev Respir
Dis, 12(4): 120 (abstract).
Dannaeus A & Inganäs M (1981) A follow-up study of children with food
allergy: Clinical course in relation to serum IgE-antibody levels to
milk, egg and fish. Clin Allergy, 11: 533-539.
Darsow U, Vieluf D, & Ring J (1995) Atopy patch test with different
vehicles and allergen concentrations: an approach to standardisation.
J. Allergy Clin Immunol, 95: 677-684.
Davies RJ, Butcher BR, O'Neil CE, & Salvaggio JE (1977) The in vitro
effect of toluene di-isocyanate on lymphocyte cyclic adenosine
monophosphate production by isoproterenol, prostaglandin and
histamine. J Allergy Clin Immunol, 60: 223-229.
Dawson KP & Mitchell EA (1990) Asthma in New Zealand children. J
Asthma, 27: 291-297.
Daynes RA, Dudley DJ, & Araneo BA (1990) Regulation of murine
lymphokine production in vivo. II. Dehydroepiandrosterone is a
natural enhancer of interleukin synthesis by helper T cells. Eur J
Immunol, 20(49): 793-802.
Dearman RJ & Kimber I (1991) Differential stimulation of immune
function by respiratory and contact chemical allergens. Immunology,
72: 563-570.
Dearman RJ & Kimber I (1992) Divergent immune responses to respiratory
and contact chemical allergens: antibody elicited by phthalic
anhydride and oxazolone. Clin Exp Allergy, 22: 241-250.
Dearman RJ, Hegarty JM, & Kimber I (1991) Inhalation exposure of mice
to trimellitic anhydride induces both IgG and IgE anti-hapten
antibody. Int Arch Allergy Appl Immunol, 95: 70-76.
Dearman RJ, Basketter DA, & Kimber I (1995) Differential cytokine
production following chronic exposure of mice to chemical respiratory
and contact allergens. Immunology, 86: 545-550.
Dearman RJ, Basketter DA, & Kimber I (1996) Characterization of
chemical allergens as a function of divergent cytokine secretion
profiles induced in mice. Toxicol Appl Pharmacol, 138: 308-316.
De Bakker JM, Kammüller ME, Muller ESM, Lam AW, Seinen W, & Bloksma N
(1990) Kinetics and morphology of chemically induced popliteal lymph
node reactions compared with antigen-, mitogen-, and
graft-versus-host-reaction-induced responses. Virchows Arch B Cell
Pathol, 58: 279-287.
Dekker C, Dales R, Bartlett S, Brunekreef B, & Zwanenburg H (1991)
Childhood asthma and the indoor environment. Chest, 100: 922-926.
De Martino M, Novembre E, & Cozza G (1988) Sensitivity to tomato and
peanut allergens in children monosensitized to grass pollen. Allergy,
43: 206-213.
Deo YM, Graziano RF, Repp R, & van de Winkel JGJ (1997) Clinical
significance of IgG Fc receptors and Fc-gamma-directed
immunotherapies. Immunol Today, 18: 127-135.
Derynck R, Lindquist PB, Lee A, Wen D, Tamm T, Graycar JL, Rhee L,
Mason AJ, Miller DA, Coffey RJ, Moses HL, & Chen EY (1988) A new type
of transforming growth factor-beta. EMBO J, 7: 3737-3743.
Devalia JL, Rusznak C, Herdman MJ, Trigg CJ, Tarraf H, & Davies RJ
(1994) Effect of nitrogen dioxide and sulphur dioxide on airway
response of mild asthmatic patients to allergen inhalation. Lancet,
344: 1668-1671.
Devlin RB, McDonnell WF, Mann R, Becker S, House DE, Schreinemachers
D, & Koren HS (1991) Exposure of humans to ambient levels of ozone for
6.6 hours causes cellular and biochemical changes in the lung. Am J
Respir Cell Mol Biol, 4: 72-81.
De Weck AL, Stadler BM, Urwyler A, Wehner HU, & Bühlmann RP (1993)
Cellular allergen stimulation test (CAST) -- A new dimension in
allergy diagnostic. ACI News, 5: 9.
De Zotti R, Larese F, Bovenzi M, Negro C, & Molinari S (1994) Allergic
airway disease in Italian bakers and pastry makers. Occup Environ Med,
51(8): 548-552.
Diaz-Sanchez D, Tsien A, Fleming J, & Saxon A (1997) Combined diesel
exhaust particulate and ragweed allergen challenge markedly enhances
human in vivo nasal ragweed-specific IgE and skews cytokine
production to a T helper cell 2-type pattern. J Immunol, 158:
2406-2413.
Diaz-Sanchez D, Tsien A, Fleming J, & Saxon A (1999) Effect of topical
fluticasone propionate on the mucosal allergic response induced by
ragweed allergen and diesel exhaust particle challenge. Clin Immunol,
90(3): 313-322.
Diem JE, John RN, Hendrick DJ, Glindmeyer HW, & Dharmarajan V (1982)
Five year longitudinal study of workers employed in a new toluene
di-isocyanate manufacturing plant. Am Rev Respir Dis, 126(3): 420-428.
Diller WF (1987) Facts and fallacies involved in the epidemiology of
isocyanate asthma. Bull Eur Physiopathol Respir, 23(6): 551-553.
Djukanovic R, Wilson J, Britten K, Wilson SJ, Walls AF, Roche WR,
Howarth PH, & Holgate ST (1990) Quantitation of mast cells and
eosinophils in the bronchial mucosa of symptomatic atopic individuals
and healthy control subjects using immunohistochemistry. Am Rev Respir
Dis, 142: 863-871.
Dockery DW & Pope CA (1994) Acute respiratory effects of particulate
air pollution. Annu Rev Public Health, 15: 107-132.
Dockery DW, Speizer FE, Stram DO, Ware JH, Spengler JD, & Ferris BGJ
(1989) Effects of inhalable particles on respiratory health of
children. Am Rev Respir Dis, 139: 587-594.
Dockery DW, Pope CA III, Xu X, Spengler JD, Ware JH, Fay ME, Ferris BG
Jr, & Speizer FE (1993) An association between air pollution and
mortality in six US cities. N Engl J Med, 329: 1753-1759.
DOH (1998) Report of the Silicone Gel Breast Implants Independent
Review Group. London, Department of Health, pp 1-36.
Domen PL, Muckerheide A, & Michael JG III (1987) Abrogation of oral
tolerance. J Immunol, 139: 3195-3198.
Dooms-Goossens A & Morren M (1992) Results of routine patch testing
with corticosteroids in 2073 patients. Contact Dermatitis, 26:
182-191.
Dorsch W & Ring J (1981) Induction of late cutaneous reactions by skin
blister fluid from allergen-tested and normal skin. J Allergy Clin
Immunol, 67: 117-123.
Dotterud LK & Falk ES (1994) Metal allergy in north Norwegian
schoolchildren and its relationship with ear piercing and atopy.
Contact Dermatitis, 31(5): 308-313.
Dotterud LK & Falk ES (1995) Contact allergy in relation to hand
eczema and atopic diseases in north Norwegian schoolchildren.
Acta-Paediatr, 84(4): 402-406.
Dowse GK, Turner KJ, Stewart GA, Alpers MP, & Woolcock AJ (1985) The
association between dermatophagoides mites and the increasing
prevalence of asthma in village communities within the Papua New
Guinea highlands. J Allergy Clin Immunol, 75(1): 75-83.
Draize JH, Woodard G, & Calvery HD (1944) Methods for the study of
irritation and toxicity of substances applied topically to the skin
and mucous membranes. J Pharmacol Exp Ther, 83: 377-390.
Dreborg S (1989) Skin tests used for standardization of allergenic
preparation. In: Dreborg S ed. Skin tests used in type I allergy
testing: Position paper prepared by the Subcommittee on Skin Tests of
the European Academy of Allergology and Clinical Immunology. Allergy,
44(suppl 10): 13-51.
Dreborg S & Foucard T (1983) Allergy to apple, carrot and potato in
children with birch pollen allergy. Allergy, 38: 167-172.
Drever F ed. (1995) Occupational health -- Decennial supplement: The
Registrar General`s decennial supplement for England and Wales.
London, Office of Population Censuses and Surveys, Health and Safety
Executive, 374 pp (Series DS No. 10).
Druce HM (1993) Allergic and nonallergic rhinitis. In: Middleton E,
Reed Cellis EF, Adkinson NF, Junginger JW, & Busse WW ed. Allergy:
Principles and practice. St. Louis, Missouri, C.V. Mosby & Co., pp
1433-1453.
Druet P (1989) Contributions of immunological reactions to
nephrotoxicity. Toxicol Lett, 46: 55-64.
Dubost J-J, Souteyrand P, & Sauvezie B (1991) Drug-induced
vasculitides. Clin Rheumatol, 5: 119-138.
Dubroff LM & Reid RJ Jr (1980) Hydralazine-pyrimidine interactions may
explain hydralazine induced lupus erythematosus. Science, 208:
404-406.
Ducombs G & Schmidt R (1995) Plant and plant products. In: Rycroft
RJG, Menné T, Frosch PJ, & Benezra C ed. Textbook of contact
dermatitis, 2nd ed. Berlin, Heidelberg, New York, Springer Verlag, pp
589-634.
Ducombs G, Benezra C, Talaga P, Andersen KE, Burrows D, Camarasa JG,
Dooms-Goossens A, Lachapelle J-M, Frosch P, Menné T, Rycroft R, White
IR, & Wilkinson JD (1990) Patch testing with sesquiterpene lactone
mix: A marker for contact allergy to Compositae and certain other
plants -- A multicenter study of the E.E.C.D.R.G., 1989. Contact
Dermatitis, 22: 249-253.
Duhme H, Weiland SK, Keil U, Kraemer B, Schmid M, Stender M, &
Chambless L (1996) The association between self-reported symptoms of
asthma and allergic rhinitis and self-reported traffic density on
street of residence in adolescents. Epidemiology, 7: 578-582.
Duhme H, Weiland SK, Rudolph P, Wienke A, Kramer A, & Keil U (1998a)
Asthma and allergies among children in West and East Germany: a
comparison between Münster and Greifswald using the ISAAC phase I
protocol. Eur Respir J, 11: 840-847.
Duhme H, Weiland SK, & Keil U (1998b) Epidemiological analyses of the
relationship between environmental pollution and asthma. Toxicol Lett,
102-103: 307-316.
Dupuis G & Benezra C (1982) Contact dermatitis to simple chemicals: A
molecular approach. New York, Marcel Dekker.
Durham SR, Graneek BJ, Hawkins R, & Newman Taylor AJ (1987) The
temporal relationship between increases in airway responsiveness to
histamine and late asthmatic responses induced by occupational agents.
J Allergy Clin Immunol, 79: 398-406.
Durie FH, Fava RA, & Noelle RJ (1994) Collagen-induced arthritis as a
model of rheumatoid arthritis. Clin Immunol Immunopathol,
73(1): 11-18.
Dvoracek JE, Yunginger JW, Kern EB, Hyatt RE, & Gleich GJ (1984)
Induction of nasal late-phase reaction by insufflation of ragweed
pollen extract. J Allergy Clin Immunol, 73: 363-368.
EAACI (European Academy of Allergology and Clinical Immunology) (1992)
Subcommittee on Occupational Allergy -- Guidelines for the diagnosis
of occupational asthma. Clin Exp Allergy, 22(1): 103-108.
Edman B (1988) The usefulness of detailed information to patients with
contact allergy. Contact Dermatitis, 19: 43-48.
Edwards J, Walters S, & Griffiths RK (1994) Hospital admissions for
asthma in preschool children: Relationship to major roads in
Birmingham, United Kingdom. Arch Environ Health, 49: 223-227.
Edworthy SM, Martin L, Barr SG, Birdsell DC, Brant RF, & Fritzler MJ
(1998) A clinical study of the relationship between silicone breast
implants and connective tissue disease. J Rheumatol, 25(2): 254-260.
Ehlers A, Stangier U, & Gieler U (1995) Treatment of atopic
dermatitis: A comparison of psychological and dermatological
approaches to relapse prevention. J Consult Clin Psychol, 63(4):
624-635.
Eidson M, Philen RM, Sewell CM, Voorhees R, & Kilbourne EM (1990)
L-tryptophan and eosinophilia-myalgia syndrome in New Mexico. Lancet;
335(8690): 645-648.
Elsayed S & Apold J (1977) Allergenic structure of allergen M from
cod: II. Allergenicity of the limited tryptic hydrolysis peptides of
fragment TM2. Int Arch Allergy Appl Immunol, 54(2): 171-175.
Emberlin J (1994) The effects of patterns in climate and pollen
abundance on allergy. Allergy, 49: 15-20.
Enders F, Przybilla B, Ring J, Burg G, & Braun-Falco O (1988)
[Epicutaneous testing with standard batteries of allergens: Results
from 12,026 patients.] Hautarzt, 39: 779-786 (in German).
Enk AH & Katz SI (1995) Contact sensitivity as a model for T cell
activation in skin. J Invest Dermatol, 105: 80S-83S.
Epstein WL, Kligman AM, & Senecal IP (1963) Role of regional lymph
nodes in contact sensitization. Arch Dermatol, 88: 789-792.
Eriksson NE, Formgren H, & Svenonius E (1982) Food hypersensitivity in
patients with pollen allergy in Sweden. Allergy, 37(3): 437-443.
Ernst P, Demissie K, Joseph L, Locher U, & Becklake MR (1995)
Socioeconomic status and indicators of asthma in children. Am J Respir
Crit Care Med, 152(2): 570-575.
Estlander T & Jolanki R (1988) How to protect the hands. Dermatol
Clin, 6: 105-114.
Estlander T, Keskinen H, Jolanki R, & Kanerva L (1992) Occupational
dermatitis from exposure to polyurethane chemicals. Contact
Dermatitis, 27(3): 161-165.
Estlander T, Kanerva L, Tupasela O, Keskinen H, & Jolanki R (1993)
Immediate and delayed allergy to nickel with contact urticaria,
rhinitis, asthma and contact dermatitis. Clin Exp Allergy, 23:
306-310.
ETFAD (1993) Severity scoring of atopic dermatitis: the SCORAD index.
Consensus Report of the European Task Force on Atopic Dermatitis.
Dermatology, 186: 23-31.
Council of the European Communities (1976) Council directive of 27
July 1976 on the approximation of the laws of the Member States
relating to cosmetic products (76/768/EEC). Off J Eur Communities,
L-262: 169-183.
Council of the European Communities (1994) European Parliament and
Council directive 94/27/EC of 30 June 1994: Nickel. Off J Eur
Communities, L 18811-2.
Evans R (1993) Epidemiology and natural history of asthma, allergic
rhinitis and atopic dermatitis (eczema). In: Middleton E, Reed Cellis
EF, Adkinson NF, Junginger JW, & Busse WW ed. Allergy: Principles and
practice. St. Louis, Missouri, C.V. Mosby & Co., pp 1109-1136.
Evans RG, Webb K, Homan S, & Ayres SM (1988a) Cross-sectional and
longitudinal changes in pulmonary function associated with automobile
pollution among bridge and tunnel officers. Am J Ind Med, 14: 25-36.
Evans RG, Webb KB, Knutsen AP, Roodman ST, Roberts DW, Bagby JR,
Garrett WA, & Andrews JS (1988b) A medical follow-up of the health
effects of long-term exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin.
Arch Environ Health, 43: 273-278.
Eynon BE & Parker DC (1992) Small B cells as antigen-presenting cells
in the induction of tolerance to soluble protein antigens. J Exp Med,
175: 131-138.
Fabbri LM, Boschetto P, Zocca E, Melani G, Pivirotto F, & Plebani M
(1987) Bronchoalveolar neutrophilia during late asthmatic reactions
induced by toluene di-isocyanate. Am Rev Respir Dis, 136: 36-42.
Fairchild RL, Palmer E, & Moorhead JW (1993) Production of
DNP-specific/Class I MHC-restricted suppressor molecules is linked to
the expression of T cell receptor alpha- and beta-chain genes. J
Immunol, 150(1): 67-77.
Farine J-C (1997) Animal models in autoimmune disease in
immunotoxicity assessment. Toxicology, 119: 29-35.
Fauci A, Schnittman SM, & Poli G (1991) Immunopathologenic mechanisms
in human immunodeficiency virus (HIV) infection. Ann Intern Med, 114:
678-693.
Fawcett IW, Newman Taylor AJ, & Pepys J (1977) Asthma due to inhaled
chemical agents - epoxy resin systems containing phthalic anhydride,
trimellitic anhydride and triethylene tetramine. Clin Allergy, 7:
1-14.
Fireman E, Ben Efraim S, Grief J, Alguetti A, Ayalon D, & Topilsky M
(1988) Correlation between prostaglandin E2 production and suppressor
activity of alveolar macrophages from patients with interstitial lung
diseases. Immunol Lett, 18(2): 159-165.
Flatt A, Pearce N, Thomson CD, Sears MR, Robinson MF, & Beasley R
(1990) Reduced selenium in asthmatic subjects in New Zealand. Thorax,
45: 95-99.
Fleming DM & Crombie DL (1987) Prevalence of asthma and hay fever in
England and Wales. Br Med J Clin Res Ed, 294(6567): 279-283.
Flens MJ, Zaman GJR, Van der Valk P, Izquierdo MA, Schroeijers AB,
Scheffer GL, van der Groep P, De Haas M, Meijer CJLM, & Scheper RJ
(1996) Tissue distribution of the multidrug resistance protein. Am J
Pathol, 148: 1237-1247.
Flindt-Hansen H & Isager H (1987) Scleroderma after occupational
exposure to trichloroethylene and trichloroethane. Acta Dermatol
Venereol, 67: 263-264.
Flynn GL (1990) Physicochemical determinants of skin absorption. In:
Gerrity TR & Henry CJ ed. Principles of route-to-route extrapolation
for risk assessment. Amsterdam, Oxford, New York, Elsevier Science
Publishers, pp 93-127.
Flyvholm M-A (1997) Formaldehyde exposure at the workplace and in the
environment. Allergologie, 20(5): 225-231.
Flyvholm M-A & Menné T (1992) Allergic contact dermatitis from
formaldehyde: A case study focusing on sources of formaldehyde
exposure. Contact Dermatitis, 27: 27-37.
Flyvholm M-A, Menné T, & Maibach HI (1996) Skin allergy: Exposures and
dose-response relationships. In: Vos JG, Younes M, & Smith E ed.
Allergic hypersensitization induced by chemicals: Recommendations for
prevention. Boca Raton, Florida, CRC Press, pp 261-285.
Flyvholm M-A, Hall BM, Agner T, Tiedemann E, Greenhill P, Vanderveken
W, Freeberg FE, & Menné T (1997) Threshold for occluded formaldehyde
patch test in formaldehyde-sensitive patients. Contact Dermatitis, 36:
26-33.
Foerster J (1993) Autoimmune hemolytic anemias. In: Lee GR, Bithell
TC, Foerster J, Athens JW, & Lukens JN ed. Wintrobe's clinical
haematology, 9th ed. London, Lea and Febiger, pp 1170-1196.
Folinsbee LJ (1992) Human health effects of air pollution. Environ.
Health Perspect, 100: 45-56.
Forastiere F, Corbo GM, Michelozzi P, Pistelli R, Agabiti N, Brancato
G, Ciappi G, & Perucci CA (1992) Effects of environment and passive
smoking on the respiratory health of children. Int J Epidemiol, 21(1):
66-73.
Forster H, Topping M, & Newman Taylor AJ (1988) Specific IgG and IgG4
antibody to tetrachlorophthalic anhydride. Allergy Proc, 9: 296.
Forsyth JS, Ogston SA, Clark A, Florey CD, & Howie PW (1993) Relation
between early introduction of solid food to infants and their weight
and illnesses during the first two years of life. Br Med J, 306(6892):
1572-1576.
Foucard T (1991) Allergy and allergy-like symptoms in 1,050 medical
students. Allergy, 46: 20-26.
Fowler JF, Skinner SM, & Belsito DV (1992) Allergic contact dermatitis
from formaldehyde resins in permanent press clothing: An
underdiagnosed cause of generalized dermatitis. J Am Acad Dermatol,
27: 962-968.
Freier S, Eran M, & Goldstein R (1985) The effect of immediate type
gastrointestinal allergic reactions on brush border enzymes and gut
morphology in the rat. Pediatr Res, 19: 456-459.
Freiman DE & Hardy HL (1970) Beryllium disease: The relation of
pulmonary pathology to clinical course and prognosis based on a study
of 130 cases from the US Beryllium Case Registry. Hum Pathol,
1: 25-44.
Friedmann PS (1990) The immunology of allergic contact dermatitis: The
DNCB story. Adv Dermatol, 5: 175-196.
Friedmann PS, Moss C, Shuster S, & Simpson JM (1983) Quantitative
relationships between sensitizing dose of DNCB and reactivity in
normal subjects. Clin Exp Immunol, 53: 706-710.
Friedman SM, Posnett DN, Tumang JR, Cole BC, & Crow K (1991) A
potential role for microbial superantigens in the pathogenesis of
systemic autoimmune disease. Arthritis Rheum, 34: 468-480.
Fudenberg HH (1966) Immunological deficiency, autoimmune disease, and
lymphoma: observations, implications, and speculations. Arthritis
Rheum, 9: 464-472.
Fujimaki H, Nohara O, Ichinose T, Watanabe N, & Saito S (1994) IL-4
production in mediastinal lymph node cells in mice intratracheally
instilled with diesel exhaust particulates and antigen. Toxicology,
92: 261-268.
Fuller KA, Pearl D, & Whitacre CC (1990) Oral tolerance in
experimental autoimmune encephalomyelitis: Serum and salivary antibody
responses. J Neuroimmunol, 28: 14-26.
Fye KH & Sack KE (1991) Rheumatic diseases. In: Stites DP & Terr AI
ed. Basic and clinical immunology, 7th ed. Norwalk, San Mateo,
Appleton & Lange, pp 438-463.
Gabriel SE, O`Fallon WM, Kurland LT, Berad CM, Woods JE, & Melton LJ
(1994) Risk of connective-tissue diseases and other disorders after
breast implantation. N Engl J Med, 330(24): 1697-1702.
Gad SC, Dunn BJ, Dobbs DW, Reilly C, & Walsh RD (1986) Development and
validation of an alternative dermal sensitization test : The mouse ear
swelling test (MEST). Toxicol Appl Pharmacol, 84: 93-114.
Gamble J, Jones W, & Minshall S (1987) Epidemiological-environmental
study of diesel bus garage workers: chronic effects of diesel exhaust
on the respiratory system. Environ Res, 44: 6-17.
Gammelgaard B, Fullerton F, Avnstorp C, & Menné T (1992) Permeation of
chromium salts through human skin in vitro. Contact Dermatitis, 27:
302-310.
Gardner MLG (1988) Gastrointestinal absorption of intact proteins.
Annu Rev Nutr, 8: 329-350.
Garssen J, Nijkamp FP, Van der Vliet H, & Van Loveren H (1991) T-cell
mediated induction of airway hyperreactivity in mice. Am Rev Resp Dis,
144: 931-938.
Garssen J, Nijkamp FP, Van de Vliet H, & Van Loveren H (1994) A role
for cellular immunity in the induction of airway hyperresposiveness
induced by small molecular weight compounds. Toxicol Lett,
72: 151-154.
Gauggel DL, Sarlo K, & Asquith TN (1993) A proposed screen for
evaluating low-molecular-weight chemicals as potential respiratory
allergens. J Appl Toxicol, 13(5): 307-313.
Gautam SC, Chikkala NF, & Battisto JR (1985) Orally induced tolerance
generates an efferently acting suppressor T cell and an acceptor T
cell that together downregulate contact sensitivity. J Immunol, 135:
2975-2983.
Geier J & Schnuch A (1996) No cross-sensitization between MCI/MI,
benzisothiazolinone and octylisothiazolinone. Contact Dermatitis,
34(2): 148-149.
Gell PGH & Coombs RRA (1963) Clinical aspects of immunology. Oxford,
London, Boston, Blackwell Scientific Publications.
Gell PGH, Coombs RRA, & Lachman R (1975) Clinical aspects of
immunology, 3rd ed. Oxford, London, Boston, Blackwell Scientific
Publications.
Gergely J & Sarmay G (1996) Fc-gamma-RII-mediated regulation of human
B cells. Scand J Immunol, 44: 1-10.
Gergen PJ, Mullally DI, & Evans R (1988) National survey of prevalence
of asthma among children in the United States, 1976 to 1980.
Pediatrics, 81: 1-7.
Gerling I, Serreze DV, Christianson S, & Leiter E (1992) Intrathymic
islet cell transplantation reduces beta-cell autoimmunity and prevents
diabetes in NOD/Lt mice. Diabetes, 41: 1672-1676.
Gilliam JN & Sontheimer RD (1982) Skin manifestations of SLE. Clin
Rheum Dis, 8: 207-218.
Glazier A, Tutschka PJ, Farmer ER, & Santos GW (1983)
Graft-versus-host disease in cyclosporine A-treated rats after
syngeneic and autologous bone marrow reconstitution. J Exp Med, 158:
1-8.
Gleich GJ (1990) The eosinophil and bronchial asthma: Current
understanding. J Allergy Clin Immunol, 85(2): 422-436.
Gleichmann H (1981) Studies on the mechanism of drug sensitization:
T-cell-dependent popliteal lymph node reaction to diphenylhydantoin.
Clin Immunol Immunopathol, 18: 203-211.
Gleichmann HIK, Pals ST, & Radaszkiewicz T (1983) T-cell-dependent
B-cell proliferation and activation induced by administration of the
drug diphenylhydantoin to mice. Hematol Oncol, 1: 165-176.
Gleichmann E, Pals ST, Rolink AG, Radaszkiewicz T, & Gleichmann H
(1984) Graft-versus-host reactions: clues to the etiopathology of a
spectrum of immunological diseases. Immunol Today, 5: 324-332.
Gleichmann E, Vohr H-W, Stringer C, Nuyens J, & Gleichmann H (1989)
Testing the sensitization of T cells to chemicals: From murine
graft-versus-host (GVH) reactions to chemical-induced GVH-like
immunological diseases. In: Kammüller ME, Bloksma N, & Seinen W ed.
Autoimmunity and toxicology. Amsterdam, Oxford, New York, Elsevier
Science Publishers, pp 363-390.
Goettsch W, Garssen J, De Gruijl FR, & Van Loveren H (1993) UV-B and
the immune system: a review with special emphasis on T cell-mediated
immunity. Thymus, 21: 93-114.
Goh C (1995) Non eczematous contact reactions. In: Rycroft RJG, Menné
T, Frosch PJ, & Benezra C ed. Textbook of contact dermatitis, 2nd ed.
Berlin, Heidelberg, New York, Springer Verlag, pp 221-233.
Goldman M, Druet P, & Gleichmann E (1991) TH2 cells in systemic
autoimmunity: insights from allogeneic diseases and chemically-induced
autoimmunity. Immunol Today, 12: 223-227.
Gorski P & Tarkowski M (1992) Non specific environmental factors and
asthma development. Pol J Occup Med Environ Health, 5: 227-236.
Goust JM (1993) Immediate hypersensitivity. Immunol Ser, 58: 343-359.
Graham C, Gealy R, Macina OT, Karol MH, & Rosenkranz HS (1996) QSAR
for allergic contact dermatitis. Quant Struct-Activ Relat, 15: 1-7.
Graham C, Rosenkranz HS, & Karol MH (1997) Structure-activity model of
chemicals that cause human respiratory sensitization. Regul Toxicol
Pharmacol, 26: 296-306.
Granato DA & Piguet PF (1986) A mouse monoclonal IgE antibody anti
bovine milk beta-lactoglobulin allows studies of allergy in the
gastrointestinal tract. Clin Exp Immunol, 63(3): 703-710.
Graycar JL, Miller DA, Arrick BA, Lyons RM, Moses HL, & Derynck R
(1989) Human transforming growth factor-beta-3: recombinant
expression, purification, and biologic activities in comparison with
transforming growth factors beta 1 and beta 2. Mol Endocrinol,
3: 1977-1986.
Greaves IA, Wegman DH, Smith TJ, & Spiegelman DL (1984) Respiratory
effects of two types of solder flux used in the electronics industry.
J Occup Med, 26(2): 81-85.
Greenland S (1993) Basic problems in interaction assessment. Environ
Health Perspect, 101(suppl): 59-66.
Gryboski JD (1967) Gastrointestinal milk allergy in infants.
Pediatrics, 40: 354-362.
Guillet GG & Guillet MH (1992) Natural history of sensitizations in
atopic dermatitis. Arch Dermatol, 128: 187-192.
Gulbenkian AR, Egan RW, Fernandez X, Jones H, Kreutner W, Kung T,
Payvandi F, Sullivan L, Zurcher JA, & Watnik AS (1992) Interleukin-5
modulates eosinophil accumulation in allergic guinea pig lung. Am Rev
Respir Dis, 146: 263-266.
Gut J, Christen U, Frey N, Koch V, & Stoffler D (1995) Molecular
mimicry in halothane hepatitis: biochemical and structural
characterization of lipoylated autoantigens. Toxicology, 97: 199-224.
Haahtela T, Lindholm H, Bjorksten F, Koskenvuo K, & Laitinen LA (1990)
Prevalence of asthma in Finnish young men. Br Med J, 301: 266-268.
Hackney JD, Linn WS, & Avol EL (1989) Acid fog: Effects on respiratory
function and symptoms in healthy and asthmatic volunteers. Environ
Health Perspect, 79: 159-162.
Haddad ZH, Kalra V, & Verma S (1979) IgE antibodies to peptic and
peptic-tryptic digests of beta-lactoglobulin: significance in food
hypersensitivity. Ann Allergy, 42: 368-371.
Halken S, Host A, Hansen LG, & Osterballe O (1992) Effect of an
allergy prevention programme on incidence of atopic symptoms in
infancy. Allergy, 47: 545-553.
Halken S, Host A, Nilsson L, & Taudorf E (1995) Passive smoking as a
risk factor for development of obstructive respiratory disease and
allergic sensitization. Allergy, 50(2): 97-105.
Hallas TE, Yi X, & Schou C (1993) Does guanine concentration in
house-dust samples reflect house-dust mite exposure? Allergy, 48(5):
303-305.
Hananan D (1990) Transgenic mouse models of self-tolerance and
autoreactivity by the immune system. Annu Rev Cell Biol, 6: 493-537.
Hang L, Aguado MT, Dixon FJ, & Theofilopoulos AN (1985) Induction of
severe autoimmune disease in normal mice by simultaneous action of
multiple immunostimulators. J Exp Med, 161: 423-428.
Hanifin JM (1983) Atopic dermatitis and other endogenous eczemas.
Semin Dermatol, 2: 1-44.
Hanifin JM (1987) Epidemiology of atopic dermatitis. Monogr Allergy,
21: 116-131.
Hanifin JM (1993) Atopic dermatitis. In: Middleton E, Reed Cellis EF,
Adkinson NF, Junginger JW, & Busse WW ed. Allergy: Principles and
practice. St. Louis, Missouri, C.V. Mosby & Co., pp 1581-1604.
Hanifin JM & Chan SC (1995) Monocyte phosphodiesterase abnormalities
and dysregulation of lymphocyte function in atopic dermatitis. J
Invest Dermatol, 105(suppl 1): 84S-88S.
Hanifin JM & Lobitz WC Jr (1977) Newer concepts of atopic dermatitis.
Arch Dermatol, 113(5): 663-670.
Hanifin JM & Rajka G (1980) Diagnostic features of atopic dermatitis.
Acta Dermatol Venereol, 92(suppl): 44-47.
Hanson DG, Roy MJ, Green GM, & Miller SD (1993) Inhibition of
orally-induced immune tolerance in mice by prefeeding an endopeptidase
inhibitor. Region Immunol, 5: 76-84.
Hariya T, Ikezawa Z, Aihara M, Kitamura K, Osawa J, & Nakajima H
(1994) Allergenicity and tolerogenicity of amlexanox in the guinea
pig. Contact Dermatitis, 31: 31-36.
Hasselmark L, Malmgren R, Zetterstrom O, & Unge G (1993) Selenium
supplementation in intrinsic asthma. Allergy, 48(1): 30-36.
Hatch M & Thomas D (1993) Measurement issues in environmental
epidemiology. Environ Health Perspect, 101(Suppl 4): 49-57.
Hattevig G, Kjellman B, Sigurs N, Bjorksten B, & Kjellman NI (1989)
Effect of maternal avoidance of eggs, cow's milk and fish during
lactation upon allergic manifestations in infants. Clin Exp Allergy,
19(1): 27-32.
Heine J, Ignotz RA, Hemler ME, Crouse C, & Massague J (1989)
Regulation of cell adhesion receptors by transforming growth
factor-beta. Concomitant regulation of integrins that share a common
beta-1 subunit. J Biol Chem, 264: 380-388.
Heiner DC, Sears JW, & Kniker WT (1962) Multiple precipitins to cow's
milk chronic respiratory disease: A syndrome including poor growth,
gastrointestinal symptoms, evidence of allergy, iron deficiency anemia
and pulmonary hemosiderosis. Am J Dis Child, 103: 634-651.
Henderson CR & Riley EC (1945) Certain statistical considerations in
patch testing. J Invest Dermatol, 6: 227-232.
Hennekens LIM & Buring JE (1987) Epidemiology in medicine. Boston,
Toronto, Little, Brown & Co., 400 pp.
Hennekens LIM, Cook NR, Hebert PR, Karlson EW, LaMotte F, Manson JE, &
Buring JE (1996) Self-reported breast implants and connective tissue
diseases in female health professionals. J Am Med Assoc, 275: 616-621.
Henschler D, Assman W, & Meyer K (1962) [The toxicology of toluene
diisocyanate.] Arch Toxicol, 19: 364-387 (in German).
Heppel LMJ & Kilshaw PJ (1982) Immune responses of guinea pigs to
dietary protein: I. Induction of tolerance by feeding with ovalbumin.
Int Arch Allergy Appl Immunol, 68: 54-59.
Herbert CA, King CM, Ring PC, Holgate ST, Stewart GA, Thompson PJ, &
Robinson C (1995) Augmentation of permeability in the bronchial
epithelium by the house dust mite allergen Der p 1. Am J Respir Cell
Mol Biol, 12: 369-378.
Hertzman PA, Blevins WL, Mayer J, Greenfield B, Ting M, & Gleich GJ
(1990) Association of the eosinophilia-myalgia syndrome with the
ingestion of tryptophan. N Eng J Med, 322(130): 869-873.
Hess AD & Fischer AC (1989) Immune mechanisms in cyclosporine-induced
syngeneic graft-versus- host disease. Transplantation, 48: 895-900.
Heufler C, Koch F, & Schuler G (1988) Granulocyte/macrophage
colony-stimulating factor and interleukin-1 mediate the maturation of
murine epidermal Langerhans cells into potent immunostimulatory
dendritic cells. J Exp Med, 167: 700-705.
Hewitt CRA, Brown AP, Hart BJ, & Pritchard DI (1995) A major house
dust mite allergen disrupts the immunoglobulin E network by
selectively cleaving CD23: innate protection by antiproteases. J Exp
Med, 182: 1537-1544.
Hide DW, Matthews S, Matthews L, Stevens M, Ridout S, Twiselton R,
Gant C, & Arshad SH (1994) Effect of allergen avoidance in infancy on
allergic manifestations at age two years. J Allergy Clin Immunol,
93(5): 842-846.
Hill DJ & Hosking CS (1997) Emerging disease profiles in infants and
young children with food allergy. Pediatr Allergy Immunol, 8(suppl
10): 21-26.
Hill LW & Sulzberger MB (1935) Evolution of atopic dermatitis. Arch
Dermatol Syph, 32: 451.
Hjorth N & Menné T (1990) Prevention of allergic contact
sensitization: a historical perspective. In: Menné T & Maibach HI ed.
Exogenous dermatoses. Boca Raton, Florida, CRC Press, chapter 30.
Hjorth N & Roed-Petersen J (1976) Occupational protein contact
dermatitis in food handlers. Contact Dermatitis, 2(1): 28-42.
Hochberg MC, Perlmutter DL, Medsger TA Jr, Nguyen K, Steen V, Weisman
MH, White B, & Wigley FM (1996) Lack of association between
augmentation mammoplasty and systemic sclerosis (scleroderma).
Arthritis Rheum, 39: 1125-1131.
Hodge L, Salome CM, Peat JK, Haby MM, Xuan W, & Woolcock AJ (1996)
Consumption of oily fish and childhood asthma risk. Med J Aust,
164(3): 137-140.
Holgate ST, Beasley R, & Twentyman OP (1987) The pathogenesis and
significance of bronchial hyperresponsiveness in airways disease. Clin
Sci, 73(6): 561-572.
Hollady SD & Luster MI (1996) Alterations in fetal thymic and liver
hematopoietic cells as indicators of exposure to developmental
immunotoxicants. Environ Health Perspect, 104(suppl 4): 809-813.
Holt PG (1994) A potential vaccine strategy for asthma and allied
atopic diseases during early childhood. Lancet, 344: 456-458.
Holt PG (1996) Infections and the development of allergy. Toxicol
Lett, 86: 205-210.
Holt PG & Sedgwick JD (1987) Suppression of IgE responses following
antigen inhalation: A natural homeostatic mechanism which limits
sensitization to aeroallergens. Immunol Today, 60(1): 97-102.
Holt PG, Schon-Hegrad MA, & McMenamin PG (1990) Dendritic cells in the
respiratory tract. Int. Rev Immunol, 6(2-3): 139-149.
Hopkins JM (1997) Genetics of atopy. In: Kay AB ed. Allergy and
allergic diseases. Oxford, London, Boston, Blackwell Scientific
Publications, pp 1187-1195.
Host A & Halken S (1990) A prospective study of cow milk allergy in
Danish infants during the first 3 years of life. Allergy, 45: 587.
Host A, Husby S, & Osterballe O (1988) A prospective study of cow's
milk allergy in exclusively breast-fed infants: Incidence,
pathogenetic role of inadvertent exposure to cow's milk formula, and
characterisation of bovine milk protein in human milk. Acta Paediatr
Scand, 77(5): 663-670.
Hostynek JJ, Magee PS, & Maibach HI (1996) QSAR predictive of contact
allergy: scope and limitations. Curr Probl Dermatol, 14: 59-106.
Howe W, Venables KM, Topping MD, Dally MB, Hawkins R, Law SJ, & Newman
Taylor AJ (1983) Tetrachlorophthalic anhydride asthma: evidence for
specific IgE antibody. J Allergy Clin Immunol, 71: 5-11.
HSE (1996) Good health is good business: Employers guide
-- Work-related dermatitis. London, United Kingdom, Health and Safety
Executive, 3 pp.
Hsieh KH & Shen JJ (1988) Prevalence of childhood asthma in Taipei,
Taiwan, and other Asian Pacific countries. J Asthma, 25(2): 73-82.
Hunter D, Milton R, & Perry KMA (1945) Asthma caused by the complex
salts of platinum. Br J Ind Med, 2: 92-98.
Hurtenbach U, Gleichmann H, Nagata N, & Gleichmann E (1987) Immunity
to D-penicillamine: genetic, cellular, and chemical requirements for
induction of popliteal lymph node enlargement in the mouse. J Immunol,
139: 411-416.
IARC (1993) Beryllium, cadmium, mercury, and exposures in the glass
industry. Lyon, International Agency for Research on Cancer, pp 88-89
(IARC Monographs on the Evaluation of Carcinogenic Risks to Humans,
Volume 58).
Ignotz RA & Massague J (1986) Transforming growth factor-beta
stimulates the expression of fibronectin and collagen and their
incorporation into the extracellular matrix. J Biol Chem, 261(9):
4337-4345.
Imbus HR (1985) Clinical evaluation of patients with complaints
related to formaldehyde exposure. J Allergy Clin Immunol, 76(6):
831-840.
International Rhinitis Management Working Group (1994) International
consensus report on the diagnosis and management of rhinitis. Allergy,
49(suppl 19): 1-34.
ISAAC (International Study of Asthma and Allergies in Childhood)
(1998) ISAAC Steering Committee -- Worldwide variation in the
prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and
atopic eczema. Lancet, 351: 1225-1232.
IPCS (1996) Environmental health criteria 180: Principles and methods
for assessing direct immunotoxicity associated with exposure to
chemicals. Geneva, World Health Organization, International Programme
on Chemical Safety, 390 pp.
Irani AA, Schechter NM, Craig SS, DeBlois G, & Schwartz LB (1986) Two
types of human mast cells that have distinct neutral protease
compositions. Proc Natl Acad Sci (USA), 83: 4464-4468.
Irvine C, Pugh CE, Hansen EJ, & Rycroft RJG (1994) Cement dermatitis
in underground workers during construction of the Channel Tunnel.
Occup Med, 44: 17-23.
Ishizaka K & Ishizaka T (1966) Physicochemical properties of reginic
antibody: 1. Association of reaginic activity with an immunoglobulin
other than gammaA- or gammaG-globulin. J Allergy, 37(3): 169-185.
Ishizaki T, Koizumi K, Ikemori R, Ishiyama Y, & Kushibiki E (1987)
Studies of prevalence of Japanese cedar pollinosis among the residents
in a densely cultivated area. Ann Allergy, 58: 265-270.
Iverson GM (1970) Ability of CBA mice to produce anti-idiotypic sera
to 5563 myeloma protein. Nature (Lond), 227: 273-274.
Iwami T, Nagai H, Tsuruoka N, & Koda A (1993) Effect of murine
recombinant interleukin-5 on bronchial reactivity in guinea-pigs. Clin
Exp Allergy, 23(1): 32-38.
Izquierdo MA, Scheffer GL, Flens MJ, Giaccone G, Broxterman HJ, Van
der Valk P, Meijer CJLM, & Scheper RJ (1996) Broad distribution of the
multidrug resistance -- related Vault Lung Resistance Protein in
normal human tissues and tumors. Am J Pathol, 148: 877-887.
Jacobs MC, White IR, Rycroft RJG, & Taub N (1995) Patch testing with
preservatives at St. John's from 1982 to 1993. Contact Dermatitis, 33:
247-255.
Jakob T, Hermann K, & Ring J (1991) Eosinophil cationic protein in
atopic eczema. Arch Dermatol Res, 283: 5-6.
Jakobsson I & Lindberg T (1979) A prospective study of cow's milk
protein intolerance in Swedish infants. Acta Pædiatr Scand, 68:
853-859.
Janeway CA, Travers P, Hunt S, & Walport M (1997) Immunobiology: The
immune system in health and disease. Edinburgh, Churchill Livingstone,
400 pp.
Jarrett EEE (1984) Immunoregulation of IgE responses: the role of the
gut in perspective. Ann Allergy, 53: 550-556.
Jarrett EEE & Hall E (1984) The development of IgE suppressive
immunocompetence in young animals: influence of exposure to antigen in
the presence or absence of maternal immunity. Immunology, 53: 365-373.
Jarrett EEE, Haig DM, McDougall W, & McNulty E (1976) Rat IgE
production: II. Primary and booster reaginic antibody responses
following intradermal or oral immunization. Immunology, 30: 671-677.
Jarvis J, Agius R, & Sawyer L (1996) Odds on for asthma. Chem Br, 32:
51-53.
Jenkins MK, Schwartz RH, & Pardoll DM (1988) Effects of cyclosporine A
on T-cell development and clonal deletion. Science, 24: 1655-1658.
Jiang X, Khursigara G, & Rubin RL (1994) Transformation of
lupus-inducing drugs to cytotoxic products by activated neutrophils.
Science, 266: 810-813.
Johansen J & Menné T (1995) The fragrance mix and its constituents: a
14-year material. Contact Dermatitis, 32: 18-23.
Johansen J, Rastogi SC, & Menné T (1996a) Exposure to selected
fragrance materials. A case study of fragrance-mix-positive eczema
patients. Contact Dermatitis, 34: 106-110.
Johansen JD, Andersen KE, & Menné T (1996b) Quantitative aspects of
isoeugenol contact allergy assessed by use and patch tests. Contact
Dermatitis, 34: 414-418.
Johansen JD, Rastogi SC, Andersen KE, & Menné T (1997) Content and
reactivity to product perfumes in fragrance mix positive and negative
eczema patients: A study of perfumes used in toiletries and skin-care
products. Contact Dermatitis, 36(6): 291-296.
Johansson SGO & Juhlin L (1970) Immunoglobulin-E in "healed" atopic
dermatitis and after treatment with corticosteroids and azathioprine.
Br J Dermatol, 82: 10-13.
Johansson SGO, Dannaeus A, & Lilja G (1984) The relevance of anti-food
antibodies for the diagnosis of food allergy. Ann Allergy, 53: 665.
Johnston SL, Pattemore PK, Sanderson G, Smith S, Lampe F, Josephs L,
Symington P, O`Toole S, Myint SH, & Tyrrell DAJ (1995) Community study
of role of viral infections in exacerbations of asthma in 9-11 year
old children. Br Med J, 310: 1225-1229.
Jones RJ, Vogelsang GB, Hess AD, Farmer ER, Mann RB, Geller RB,
Piantadosi S, & Santos GW (1989) Induction of graft-versus-host
disease after autologous bone marrow transplantation. Lancet, 1:
754-757.
Jones LA, Chin LT, Longo DL, & Kruisbeek AM (1990) Peripheral clonal
elimination of functional T cells. Science, 250: 1726-1729.
Jones RN, Rando RJ, Glindmeyer HW, Foster TA, Hughes JM, O'Neil CE, &
Weill H (1992) Abnormal lung function in polyurethane foam producers:
Weak relationship to toluene diisocyanate exposures. Am Rev Respir
Dis, 146(4): 871-877.
Jones DB, Coulson AFW, & Duff GW (1993) Sequence homologies between
hsp60 and autoantigens. Immunol Today, 14: 115-118.
Jones Williams W (1988) Beryllium disease. Postgrad Med J, 64:
511-516.
Jones Williams W & Wallach ER (1989) Laser probe mass spectrometry
(LAMMS) analysis of beryllium, sarcoidosis and other granulomatous
diseases. Sarcoidosis, 6: 111-117.
Jordan JM & Whitlock FA (1972) Emotions and the skin: the conditioning
of scratch responses in cases of atopic dermatitis. Br J Dermatol,
86(6): 574-585.
Jordan JM & Whitlock FA (1974) Atopic dermatitis, anxiety and
conditioned scratch responses. J Psychosom Res, 18(5): 297-299.
Jungers P, Dougados M, Pelissier C, Kutten F, Tron F, Lesavre P, &
Bach JF (1982) Influence of oral contraceptive therapy on the activity
of systemic lupus erythematosus. Arthritis Rheum, 25: 618-623.
Juniper CP, How MJ, Goodwin BFJ, & Kinshott AK (1977) Bacillus
subtilis enzymes: a seven year clinical, epidemiological and
immunological study of an industrial allergen. J Soc Occup Med, 27:
3-12.
Kagnoff MF (1982) Oral tolerance. Ann NY Acad Sci, 392: 248-265.
Kagnoff MF (1992) Celiac disease: A gastrointestinal disease with
environmental, genetic, and immunologic components. Gastr Clin North
Am, 21(2): 405-425.
Kajosaari M & Saarinen UM (1983) Prophylaxis of atopic disease by six
month's total solid food elimination. Acta Paediatr Scand, 72:
411-414.
Kammüller ME (1996) Drug-induced autoimmune disorders. Drug Inf J, 30:
293-299.
Kammüller ME & Seinen W (1988) Structural requirements for hydantoins
and 2-thiohydantoins to induce lymphoproliferative popliteal lymph
node reactions in the mouse. Int J Immunopharmacol, 10: 997-1010.
Kammüller ME, Penninks AH, & Seinen W (1984) Spanish toxic oil
syndrome is a chemically induced GVHD-like epidemic. Lancet, 1:
1174-1175.
Kammüller ME, Bloksma N, & Seinen W ed. (1989a) Autoimmunity and
toxicology: Immune disregulation induced by drugs and chemicals.
Amsterdam, Oxford, New York, Elsevier Science Publishers, pp 3-34.
Kammüller ME, Thomas C, De Bakker JM, Bloksma N, & Seinen W (1989b)
The popliteal lymph node assay in mice to screen for the immune
disregulating potential of chemicals -- a preliminary study. Int J
Immunopharmacol, 11: 293-300.
Kaposi M (1895) Pathology and treatment of diseases of the skin, 4th
ed. New York, William Wood.
Kapp A (1995) [Atopic dermatitis: The skin manifestations.] Allergo J,
4: 229-238 (in German).
Kapsenberg ML, Wierenga, EA, Bos JD, & Jansen HM (1991) Functional
subsets of allergen-reactive human CD4+ T cells. Immunol Today, 12:
392-395.
Karol MH (1983) Concentration-dependent immunologic response to
toluene diisocyanate (TDI) following inhalation exposure. Toxicol Appl
Pharmacol, 68: 229-241.
Karol MH (1986) Respiratory effects of inhaled isocyanates. CRC Crit
Rev Toxicol, 16: 349-380.
Karol MH (1991) Allergic reactions to indoor air pollutants. Environ
Health Perspect, 95: 45-51.
Karol MH (1992) Occupational asthma and allergic reactions to inhaled
compounds. In: Miller K, Turk J, & Nicklin S ed. Principles and
practice of immunotoxicology. Oxford, London, Boston, Blackwell
Scientific Publications, pp 228-241.
Karol MH (1993) Concentration-dependent immunologic response to
toluene diisocyanate (TDI) following inhalation exposure. Toxicol Appl
Pharmacol, 68: 229.
Karol MH (1994a) Occupational asthma pig predictive tests for
respiratory allergy. In: Dean JH, Luster MI, Munson AE, & Kimber I ed.
Immunotoxicology and immunopharmacology, 2nd ed. New York, Raven
Press, pp 703-720.
Karol MH (1994b) Animal models of occupational asthma. Eur Respir J,
7(3): 555-568.
Karol MH & Thorne PS (1988) Pulmonary hypersensitivity and
hyperreactivity: implications for assessing allergic responses. In:
Gardner DE, Crapo JD, & Massaro EJ ed. Toxicology of the lung. New
York, Raven Press, p 427.
Karol MH, Graham C, Gealy R, Macina OT, Sussman N, & Rosenkranz HS
(1996) Structure- activity relationships and computer-assisted
analysis of respiratory sensitization potential. Toxicol Lett, 86:
187-191.
Karpatkin S (1988) Immunological platelet disorders. In: Immunological
diseases, 4th ed. Boston, Little Brown, vol 2, pp 1631-1662.
Karpus WJ & Swanborg RH (1991) CD4+ suppressor cells inhibit the
function of effector cells of experimental autoimmune
encephalomyelitis through a mechanism involving transforming growth
factor-beta. J Immunol, 146: 1163-1168.
Katsutani N & Shionoya H (1992) Popliteal lymph node enlargement
induced by procainamide. Int J Immunopharmacol, 14: 681-686.
Kaufman LD, Gruber BL, & Gregersen PK (1991) Clinical follow-up and
immunogenetic studies of 32 patients with eosinophilia-myalgia
syndrome. Lancet, 337: 1071-1074.
Kaufman DL, Clare-Salzler M, Tian J, Forsthuber T, Ting GSP, Robinson
P, Atkinson MA, Sercarz EE, Tobin AJ, & Lehmann PV (1993) Spontaneous
loss of T-cell tolerance to glutamic acid decarboxylase in murine
insulin-dependent diabetes. Nature (Lond), 366: 69-72.
Kayser D & Schlede E (1995) [Chemicals and contact allergy -- a
summary evaluation.] Berlin, Federal Institute for Consumer's Health
Protection and Veterinary Medicine (BGVV) (in German).
Keeley DJ, Neill P, & Gallivan S (1991) Comparison of the prevalence
of reversible airways obstruction in rural and urban Zimbabwean
children. Thorax, 46: 549-553.
Keil U, Weiland SK, Duhme H, & Chambless L (1996) The international
study of asthma and allergies in childhood (ISAAC): Objectives and
methods -- Results from German ISAAC centres concerning traffic
density and wheezing and allergic rhinitis. Toxicol Lett, 86: 99-103.
Kemp T, Pearce N, Fitzharris P, Crane J, Fergusson D, St. George I,
Wickens K, & Beasley R (1997) Is infant immunization a risk factor for
childhood asthma or allergy? Epidemiology, 8: 678-680.
Keskinen H, Alanko K, & Saarinen L (1978) Occupational asthma in
Finland. Clin Allergy, 8(6): 569-579.
Keskinen H, Tupasela O, Tiikkainen U, & Nordman H (1988) Experience of
specific in IgE asthma due to diisocyanates. Clin Allergy, 18(6):
597-604.
Khoury SJ, Hancock WW, & Weiner HL (1992) Oral tolerance to myelin
basic protein and natural recovery from experimental autoimmune
encephalomyelitis are associated with downregulation of inflammatory
cytokines and differential upregulation of transforming growth factor
beta, interleukin 4 and prostaglandin E expression in the brain. J Exp
Med, 176: 1355-1364.
Kimber I & Basketter DA (1997) Contact sensitisation: a new approach
to risk assessment. Hum Ecol Risk Assess, 3: 385-395.
Kimber I & Cumberbatch M (1992a) Dendritic cells and cutaneous immune
responses to chemical allergens. Toxicol Appl Pharmacol, 117: 137-146.
Kimber I & Cumberbatch M (1992b) Stimulation of Langerhans cell
migration by tumour necrosis factor alpha. J Invest Dermatol, 99:
48S-50S.
Kimber I & Maurer T ed. (1996) Toxicology of contact hypersensitivity.
London, Taylor & Francis, 170 pp.
Kimber I & Wilks MF (1995) Chemical respiratory allergy: Toxicological
and occupational health issues. Hum Exp Toxicol, 14(9): 735-736.
Kimber I, Hilton J, & Weisenberger C (1989) The murine local lymph
node assay for identification of contact allergens: a preliminary
evaluation of in situ measurement of lymphocyte proliferation. Contact
Dermatitis, 21: 215.
Kimber I, Kinnaird A, Peters SW, & Mitchell JA (1990) Correlation
between lymphocyte proliferative responses and dendritic cell
migration in regional lymph nodes following skin painting with contact
sensitizing agents. Int Arch Allerg Appl Immunol, 93: 47-53.
Kimber I, Dearman RJ, Scholes EW, & Basketter DA (1994) The local
lymph node assay developments and applications. Toxicology, 93: 13-31.
King TP, Hoffman D, Lowenstein H, Marsh DG, Platts-Mills TAE, & Thomas
W (1995) Allergen nomenclature. J Allergy Clin Immunol, 96: 5-14.
Kirton V (1978) Contact urticaria and cinnamic aldehyde. Contact
Dermatitis, 4(6): 374-375.
Kishimoto T & Hirano T (1984) beta-Lymphocyte activation,
proliferation, and immunoglobulin secretion. In: Paul WE ed.
Fundamental immunology, 2nd ed. New York, Raven Press, pp 385-411.
Kitagaki H, Fujisawa S, Watanabe K, Hayakawa K, & Shiohara T (1995)
Immediate type hypersensitivity response followed by a late reaction
is induced by repeated epicutaneous application of contact sensitizing
agents in mice. J Invest Dermatol, 105: 749-755.
Kjellman NI & Croner S (1984) Cord blood IgE determination for allergy
prediction -- a follow-up to seven years of age in 1,651 children. Ann
Allergy, 53(2): 167-171.
Kligman AM (1958a) Poison ivy (Rhus) dermatitis. Arch Dermatol, 77:
159-180.
Kligman AM (1958b) Hyposensitization against Rhus dermatitis. Arch
Dermatol, 78: 47-72.
Kligman AM (1966a) The identification of contact allergies by human
assay: II. Factors influencing the induction and measurement of
allergic contact dermatitis. J Invest Dermatol, 47: 375-393.
Kligman AM (1966b) The identification of contact allergens by human
assay: III. The maximization test -- A procedure for screening and
rating contact sensitizers. J Invest Dermatol, 47: 393-409.
Kligman AM (1966c) The SLS provocative patch test in allergic
sensitization. J Invest Dermatol, 46: 573-595.
Knippels LM, Penninks AH, Spanhaak S, & Houben GF (1998) Oral
sensitization to food proteins: a Brown Norway rat model. Clin Exp
Allergy, 28(3): 368-375.
Koenig JQ (1988) Indoor and outdoor pollutants and the upper
respiratory tract. J Allergy Clin Immunol, 81: 1055-1059.
Koenig JQ (1995) Effect of ozone on respiratory responses in subjects
with asthma. Environ Health Perspect, 103(suppl 2): 103-105.
Koenig JQ, Pierson WE, & Horike M (1983) The effects of inhaled
sulfuric acid on pulmonary function in adolescent asthmatics. Am Rev
Respir Dis, 128(2): 221-225.
Koenig JQ, Pierson WE, Covert DS, Marshall SG, Morgan MS, & van Belle
G (1988) The effects of ozone and nitrogen dioxide on lung function in
healthy and asthmatic adolescents. Res Rep Health Eff Inst, 14: 5-24.
Koenig JQ, Covert DS, Hanley QS, van Belle G, & Pierson WE (1990)
Prior exposure to ozone potentiates subsequent response to sulfur
dioxide in adolescent asthmatic subjects. Am Rev Respir Dis, 141:
377-380.
Koëter HBWM (1995) International harmonisation of immunotoxicity
testing. Hum Exp Toxicol, 14: 151-154.
Kogevinas M, Anto J, Soriano JB, Tobias A, & Burney P (1996) The risk
of asthma attributable to occupational exposures. Ann Resp Crit Care
Med, 154: 137-143.
Kohler C, Jeanvoine G, Pierrez J, Olive D, & Gerard H (1987)
Modifications of the thymus and splenic thymic dependent zones after
in utero exposure to phenytoin: Qualitative and quantitative analysis
in C3H mice. Dev Pharmacol Therap, 10: 405-412.
Koren HS (1995) Associations between criteria air pollutants and
asthma. Environ Health Perspect, 103(suppl 6): 235-242.
Koren HS & Bromberg PA (1995) Respiratory responses of asthmatics to
ozone. Int Arch Allergy Immunol, 107: 236-238.
Koren HS, Devlin RB, Graham DE, Mann R, McGee MP, Horstman DH, Kozumbo
WJ, Becker S, House DE, McDonnel WF, & Bromberg PA (1989)
Ozone-induced inflammation in the lower airways of human subjects. Am
Rev Respir Dis, 139: 407-415.
Koren HS, Hatch GE, & Graham DE (1990) Nasal lavage as a tool in
assessing acute inflammation in response to inhaled pollutants.
Toxicology, 60: 15-25.
Korman NJ (1995) In situ bound antibodies eluted from the skin of
patients with bullous pemphigoid are preferentially directed against
the 230-kD bullous pemphigoid antigen. J Invest Dermatol,
105: 824-830.
Korman NJ, Eyre RW, Zone J, & Stanley JR (1991) Drug-induced
pemphigus: autoantibodies directed against the pemphigus antigen
complexes are present in penicillamine and captopril-induced
pemphigus. J Invest Dermatol, 96: 273-276.
Kortekangas-Savolainen O, Savolainen J, & Einarsson R (1993)
Gastrointestinal stability of baker's yeast allergens: an in vitro
study. Clin Exp Allergy, 23(7): 578-590.
Krämer U, Behrendt H, & Ring J (1996) Air pollution as a risk factor
for allergy: The East-West German experience. In: Ring J, Behrendt H,
& Vieluf D ed. New trends in allergy. Berlin, Heidelberg, New York,
Springer Verlag, vol IV, pp 25-35.
Kremer AM, Pal TM, Schouten JP, & Rijcken B (1995) Airway
hyperresponsiveness in workers exposed to low levels of irritants. Eur
Respir J, 8(1): 53-61.
Kremser M (1989) [Food allergies -- oropharyngeal reactions.] Wien Med
Wochenschr, 139(6/7): 135-139.
Kröger S, Neuber K, Gruseck E, Ring J, & Abeck D (1995) Pityrosporum
ovale extracts increase interleukin-4, interleukin-10 and IgE
synthesis in patients with atopic eczema. Acta Dermatol Venereol, 75:
357-360.
Krzystyniak K, Brouland J-P, Panayi G, Patriarca C, Verdier F,
Descotes J, & Revillard J-P (1992) Activation of CD4+ and CD8+
lymphocyte subsets by streptozotocin in murine popliteal lymph node
(PLN) test. J Autoimmun, 5: 183-197.
Krzyzanowski M, Quackenboss JJ, & Lebowitz MD (1992) Relation of peak
expiratory flow rates and symptoms to ambient ozone. Arch Environ
Health, 47(2): 107-115.
Kubicka-Muranyi M, Goebels R, Goebel C, Uetrecht J, & Gleichmann E
(1993) T lymphocytes ignore procainamide, but respond to its reactive
metabolites in peritoneal cells: demonstration by the adoptive
transfer popliteal lymph node assay. Toxicol Appl Pharmacol, 122:
88-94.
Kubicka-Muranyi M, Griem P, Lübben B, Rottmann N, Lührmann R, &
Gleichmann E (1996) Mercuric chloride-induced autoimmunity in mice
involves an upregulated presentation of altered and unaltered
nucleolar self antigen. Int Arch Allergy Immunol, 108: 1-10.
Kuchroo VK, Byrne MC, Greenfield E, Whitters MJ, Nalefsky EA, Rao A,
Collins M, & Dorf ME (1995) Transfection of TCR alpha-chains into
suppressor and T helper cell hybridomas. J Immunol, 154: 5030-5038.
Kuhn R, Rajewsky K, & Muller W (1991) Generation and analysis of
interleukin-4 deficient mice. Science, 254(5032): 707-710.
Kurisaki J, Konishi Y, Kaminogawa S, & Yamauchi K (1981) Studies on
the allergenic structure of hen ovomucoid by chemical and enzymatic
fragmentation. Agric Biol Chem, 45: 879.
Küster W, Petersen M, Christophers E, Goos M, & Sterry W (1990) A
family study of atopic dermatitis. Arch Dermatol Res, 282: 98-102.
Lake AM, Kagey-Sobotka A, Jakubowicz T, & Lichtenstein LM (1984)
Histamine release in acute anaphylactic enteropathy of the rat. J
Immunol, 133(3): 1529-1534.
Lammintausta K, Kalimo K, & Havu VK (1982) Contact allergy in atopics
who perform wet work in hospital. Derm Beruf Umwelt, 30(6): 184-188.
Lammintausta K, Kalimo K, & Fagerlund VL (1992) Patch test reactions
in atopic patients. Contact Dermatitis, 26(4): 234-240.
Lamont AG, Mowat A McI, Browning MJ, & Parrott DMV (1988) Genetic
control of oral tolerance to ovalbumin in mice. Immunology, 63:
737-739.
Landsteiner K & Rostenberg A Jr (1939) Individual differences in
susceptibility to eczematous sensitization with simple chemical
substances. J Invest Dermatol, 2: 25-29.
Lane HC & Fauci AS (1985) Immunologic abnormalities in the acquired
immunodeficiency syndrome. Annu Rev Immunol, 3: 477-500.
Lange CE, Jühe S, Stein G, & Veltman G (1974) [The so-called vinyl
chloride disease -- an occupational scleroderma?] Int Arch Arbeitsmed,
32: 1-32 (in German).
Laor A, Cohen L, & Danon YL (1993) Effects of time, sex, ethnic
origin, and area of residence on prevalence of asthma in Israeli
adolescents. Br Med J, 307: 841-844.
Larsen CG, Thomsen MK, Gesser B, Deleuran BW, & Thestrup-Pedersen K
(1995) The delayed type hypersensitivity reaction is dependent on
IL-8. Inhibition of a tuberculin skin reaction by an anti-IL-8
monoclonal antibody. J Immunol, 155(4S): 2151-2157.
Lau S, Falkenhorst G, Weber A, Werthmann I, Lind P, Buettner Goetz P,
& Wahn U (1989) High mite-allergen exposure increases the risk of
sensitization in atopic children and young adults. J Allergy Clin
Immunol, 84: 718-725.
Lau S, Ehnert B, Cremer B, Nasert S, Buettner P, Czarnetzki BM, & Wahn
U (1995) [Reduction of house dust mite exposure in sensitized patients
with atopic eczema.] Allergo J, 4: 432-437 (in German).
Lauer K (1990) Environmental nitrophenols and autoimmunity (Letter to
the Editor). Mol Immunol, 27: 697-698.
Lauerma AI (1992) Contact hypersensitivity to glucocorticosteroids. Am
J Contact Dermatitis, 3: 112-132.
Lauwerys R & Lison D (1994) Health risks associated with cobalt
exposure -- an overview. Sci Total Environ, 150(1-3): 1-6.
Law LW, Goldstein AL, & White A (1968) Influence of thymosin on
immunological competence of lymphoid cells from thymectosized mice.
Nature (Lond), 219(161): 1391-1392.
Leach CL, Hatoum NS, Ratajcak HV, Zeiss CR, Roger JC, & Garvin PJ
(1987) Pathologic and immunologic response to inhaled trimellitic
anhydride in rats. Toxicol Appl Pharmacol, 87: 67-80.
Ledford DK (1994) Indoor allergens. J Allergy Clin Immunol, 94:
327-334.
Lee HK, Alarie Y, & Karol MH (1984) Induction of formaldehyde
sensitivity in guinea pigs. Toxicol Appl Pharmacol, 75: 147-155.
Lehmann PV, Secarz EE, Fortshuber T, Dayan CM, & Gammon G (1993)
Determinant spreading and the dynamics of the autoimmune T cell
repertoire. Immunol Today, 14: 203-208.
Lejman E, Stoudemayer T, Grove G, & Kligman AM (1984) Age differences
in poison ivy dermatitis. Contact Dermatitis, 11: 163-167.
Lenshow DJ, Walunas TL, & Bluestone JA (1996) CD28/B7 system of T cell
co-stimulation. Annu Rev Immunol, 14: 233-258.
Leung R & Ho P (1994) Asthma, allergy, and atopy in three south-east
Asian populations. Thorax, 49: 1205-1210.
Lewis S, Richards D, Bynner J, Butler N, & Britton J (1995)
Prospective study of risk factors for early and persistent wheezing in
childhood. Eur Respir J, 8(3): 349-356.
Leyden JJ & Kligman AM (1977) Allergic contact dermatitis: Sex
differences. Contact Dermatitis, 3: 333-336.
Leyden JE, Marples RR, & Kligman AM (1974) Staphylococcus aureus in
the lesions of atopic dermatitis. Br J Dermatol, 90(5): 525-530.
Li LF (1995) A clinical and patch test study of contact dermatitis
from traditional Chinese medical materials. Contact Dermatitis, 33:
392-395.
Lidén C, Menné T, & Burrows D (1996) Nickel-containing alloys and
platings and their ability to cause dermatitis. Br J Dermatol, 134:
193-198.
Lindskov R & Holmer G (1992) Polyunsaturated fatty acid in plasma, red
blood cells and mononuclear phospholipids in patients with atopic
dermatitis. Allergy, 47: 517-521.
Linn WS, Shamoo DA, Spier CE, Valencia LM, Anzar UT, Venet TG, &
Hackney JD (1983a) Respiratory effects of 0.75 ppm sulfur dioxide in
exercising asthmatics: influence of upper-respiratory defenses.
Environ Res, 30: 340-348.
Linn WS, Venet TG, Shamoo DA, Valencia LM, Anzar UT, Spier CE, &
Hackney JD (1983b) Respiratory effects of sulfur dioxide in heavily
exercising asthmatics: A dose-response study. Am Rev Respir Dis, 127:
278-283.
Lippmann M (1989) Health effects of ozone: a critical review. J Air
Pollut Control Assoc, 39: 672-695.
Liu MC, Bleecker ER, Lichtenstein LM, Kagey-Sobotka A, Niv Y, Mclemore
TL, Permutt S, Proud D, & Hubbard WC (1990) Evidence for elevated
levels of histamine, prostaglandin D2, and other bronchoconstricting
prostaglandins in the airways of subjects with mild asthma. Am Rev
Respir Dis, 142: 126-132.
Lo D (1996) Animal models of human disease -- Transgenic and knockout
models of autoimmunity: building a better disease? Clin Immunol
Immunopathol, 79(2): 96-104.
Logan RFA (1992) In: Auricchio S & Visakorpi JK ed. Problems and
pitfalls in epidemiological studies of coeliac disease, common food
intolerances: 1. Epidemiology of coeliac disease. Basel, Karger, p 14.
Lonkar A, Mitchell JC, & Calnan CD (1974) Contact dermatitis from
parthenium hysterophorus. Trans St. John's Hosp Derm Soc, 60: 43-53.
Lovik M, Hogseth A-K, Gaarder PI, Hagemann R, & Eide I (1997) Diesel
exhaust particles and carbon black have adjuvant activity on the local
lymph node response and systemic IgE production to ovalbumin.
Toxicology, 121: 165-178.
Lucas A, Brooke OG, Morley R, Cole TJ, & Bamford MF (1990) Early diet
of preterm infants and development of allergic or atopic disease:
randomised prospective study. Br Med J, 300(6728): 837-840.
Lucente FE (1989) Rhinitis and nasal obstruction. Otolaryngol Clin
North Am, 22: 307-318.
Luczynska CM & Topping MD (1986) Specific IgE antibodies to reactive
dye-albumin conjugates. J Immunol Methods, 95: 177-186.
Luo J-C, Nelsen KG, & Fischbein A (1990) Persistent reactive airways
dysfunction syndrome after exposure to toluene di-isocyanate. Br J Ind
Med, 47: 239-241.
Lutsky I, Baum GL, Teichtahl H, Mazar A, Aizer F, & Bar Sela S (1985)
Occupational respiratory disease in veterinarians. Ann Allergy, 55(2):
153-156.
Lynch NR, Hagel IA, Palenque ME, Di Prisco MC, Escudero JE, Corao LA,
Sandia JA, Ferreira LJ, Botto C, Perez M, & Le Souef PN (1998)
Relationship between helminthic infection and IgE response in atopic
and nonatopic children in a tropical environment. J Allergy Clin
Immunol, 101(2): 217-221.
McAlindon T, Gianotta L, Taub N, D'Cruz D, & Hughes GRV (1993)
Environmental factors predicting nephritis in systemic lupus
erythematosus. Ann Rheum Dis, 52: 720-724.
Maccia CA, Berstein IL, Emmett EA, & Brookes SSM (1976) In vitro
demonstration of specific IgE in phthalic anhydride sensitivity. Am
Rev Respir Dis, 113: 701-704.
MacDonald TT (1983) Immunosuppression caused by antigen feeding: II.
Suppressor T cells mask Peyer's patch B cell priming to orally
administered antigen. Eur J Immunol, 13: 138-142.
McGrath H Jr, Bell JM, & Haycock JW (1994) Fluorescent light activates
the immunomodulator cis-urocanic acid in vitro: Implications for
patients with systemic lupus erythematosus. Ann Rheum Dis, 53(6):
396-399.
Machado DC, Horton D, Harrop R, Peachell PT, & Helm BA (1996)
Potential allergens stimulate the release of mediators of the allergic
response from cells of mast cell lineage in the absence of
sensitization with antigen-specific IgE. Eur J Immunol, 26: 2972-2980.
McMillan C & Burrows D (1995) In vitro testing in contact
hypersensitivity. In: Rycroft RJG, Menné T, Frosch PJ, & Benezra C ed.
Textbook of contact dermatitis, 2nd ed. Berlin, Heidelberg, New York,
Springer Verlag, pp 306-318.
Madara JL & Stafford J (1989) Interferon-gamma directly affects
barrier function of cultured intestinal epithelial monolayers. J Clin
Invest, 83(2): 724-727.
Maestrelli P, Di Stefano A, Occari P, Turato G, Milani G, Pivirotto F,
Mapp CE, Fabbri LM, & Saetta M (1995) Cytokines in the airway mucosa
of subjects with asthma induced by toluene diisocyanate. Am J Resp
Crit Care Med, 151: 607-612.
Magnus P & Jaakkola JJ (1997) Secular trend in the occurrence of
asthma among children and young adults: critical appraisal of repeated
cross sectional surveys. Br Med J, 314: 1795-1799
Magnusson CG (1986) Maternal smoking influences cord serum IgE and IgD
levels and increases the risk for subsequent infant allergy. J Allergy
Clin Immunol, 78: 898-904.
Magnusson B & Gilje O (1973) Allergic contact dermatitis from a
dish-washing liquid containing laurylethersulphate. Acta Dermatol
Venereol, 53: 136-140.
Magnusson B & Hersle K (1965) Patch test methods. Acta Dermatol
Venereol, 45: 123-128.
Magnusson B & Kligman AM (1970a) Allergic contact dermatitis in the
guinea pig. Springfield, Charles C Thomas, pp 3-120.
Magnusson B & Kligman AM (1970b) The identification of contact
allergens by animal assay: The guinea pig maximisation test. J Invest
Dermatol, 52: 268-276.
Magnussen H, Holz O, & Jörres RA (1998) Asthma and the environment: Is
ozone really important? Eur Respir Rev, 8: 141-144.
Mahzoon S, Yamamoto S, & Greaves MW (1977) Response of skin to
ammonium persulphate. Acta Dermatol Venereol, 57(2): 125-126.
Maibach H (1976) Immediate hypersensitivity in hand dermatitis. Arch
Dermatol, 112(9): 1289-1291.
Malo J-L, Ouimet G, Cartier A, Lebitz D, & Ziess CR (1983) Combined
alveolitis and asthma due to hexamethylene di-isocyanate (HDI) with
evidence of crossed respiratory and immunologic reactivities to
diphenylmethane di-isocyanate MDI. J Allergy Clin Immunol,
72: 413-419.
Manetti R, Parronchi P, Giudizi MG, Piccinni MP, Maggi E, Trinchieri
G, & Romagnani S (1993) Natural killer cell stimulatory factor
(interleukin 12 [IL-12]) induces T helper type 1 (Th1)-specific immune
responses and inhibits the development of IL-4 producing Th cells. J
Exp Med, 177(4): 1199-1204.
Mantle J & Pepys J (1974) Asthma amongst Tristan da Cunha islanders.
Clin Allergy, 4: 161-170.
Marchal G, Seman M, Milon G, Truffa-Bacchi P, & Zilberfarb V (1982)
Local adoptive transfer of skin delayed type hypersensitivity
initiated by a single T lymphocyte. J Immunol, 129(3): 954-958.
Marchionini A (1960) [New studies of constitutional neurodermatitis.]
In: Marchionini A & Röckl M ed. [Dermatology and venereology.] Berlin,
Heidelberg, New York, Springer Verlag, vol 3, pp 42-46 (in German).
Marcon-Genty D, Tomé D, Dumontier AM, Kheroua O, & Desjeux JF (1989)
Permeability of milk protein antigens across the intestinal
epithelium in vitro. Reprod Nutr Dev, 29(6): 717-723.
Marfaing-Koka A, Devergne O, Gorgone G, Portier A, Schall TJ, Galanaud
P, & Emilie D (1995) Regulation of the production of RANTES chemokine
by endothelial cells -- Synergistic induction by IFN-gamma plus
TNF-alpha and inhibition by IL-4 and IL-13. J Immunol, 154(suppl 4):
1870-1878.
Marsh DG (1990) Immunogenetic and immunochemical factors determining
immune responsiveness to allergens: studies in unrelated subjects. In:
Marsh DG & Blumenthal M ed. Genetic and environmental factors in
clinical allergy. Minneapolis, University of Minneapolis Press, pp
97-120.
Marsh DG & Norman PS (1988) Antigens that cause atopic disease. In:
Immunological diseases, 4th ed. Boston, Little Brown, vol 2, pp
981-1008.
Martin S & Weltzien HU (1994) T cell recognition of haptens, a
molecular view. Int Arch Allergy Immunol, 104: 10-16.
Martinez FD (1994) Role of viral infections in the inception of asthma
and allergies during childhood: could they be protective? Thorax, 49:
1189-1191.
Martinez FD (1995) Viral infections and the development of asthma. Am
J Respir Crit Care Med, 151: 1644-1648.
Martinez FD, Antognoni G, Macri F, Bonci E, Midulla F, De Castro G, &
Ronchetti R (1988) Parental smoking enhances bronchial responsiveness
in nine-year-old children. Am Rev Respir Dis, 138(3): 518-523.
Martinez FD, Cline M, & Burrows B (1992) Increased incidence of asthma
in children of smoking mothers. Pediatrics, 89: 21-26.
Martinez FD, Wright AL, Taussig LM, Holberg CJ, Halonen M, & Morgan WJ
(1995) Asthma and wheezing in the first six years of life. The Group
Health Medical Associates. N Engl J Med, 332: 133-138.
Marzulli FN & Maibach HI (1973) Antimicrobials: Experimental contact
sensitization in man. J Soc Cosmet Chem, 24: 399-421.
Marzulli FN & Maibach HI (1974) The use of graded concentrations in
studying skin sensitizers: experimental contact sensitization in man.
Food Cosmet Toxicol, 12: 219-227.
Marzulli FN & Maibach HI (1996) Test methods for allergic contact
dermatitis in humans.
In: Marzulli FN & Maibach HI ed. Dermatotoxicology, 5th ed.
Washington, New York, London, Hemisphere Publishing Corporation, pp
477-482.
Matricardi PM (1997) Infections preventing atopy: Facts and new
questions. Allergy, 52: 879-882.
Matricardi PM, Rosmini F, Ferrigno L, Nasion R, Rapicetta M, Chionne
P, Stroffolini T, Pasquini P, & D`Amelio R (1997) Cross sectional
retrospective study of the prevalence of atopy among Italian military
students with antibodies against hepatitis A virus. Br Med J, 314:
999-1003.
Mattingly JA (1984) Immunologic suppression after oral administration
of antigen: III. Activation of suppressor-inducer cells in the Peyer's
patches. Cell Immunol, 86: 46-52.
Matzner Y, Erlich HA, Brautbar C, Sanilevitch A, Landau M, Brenner S,
& Friedmann A (1995) Identical HLA class II alleles predispose to
drug-triggered and idiopathic pemphigus vulgaris. Acta Dermatol
Venereol, 75: 12-14.
Maulitz RM, Pratt DS, & Schocket AL (1979) Exercise-induced
anaphylactic reaction to shellfish. J Allergy Clin Immunol, 63(6):
433-434.
Maurer T, Thomann P, Weirich EG, & Hess R (1975) The optimization test
in the guinea pig: A method for the predictive evaluation of the
contact allergenicity of chemicals. Agents Actions, 5: 174-179.
Maynard AD, Northage C, Hemingway M, & Bradley SD (1997) Measurement
of short-term exposure to airborne soluble platinum in the platinum
industry. Ann Occup Hyg, 41(1): 77-94.
Meade R, Askenase PW, Geba GP, Neddermann K, Jacoby RO, & Pasternak RD
(1992) Transforming growth factor-beta 1 inhibits murine immediate and
delayed type hypersensitivity. J Immunol, 149(2): 521-528.
Medici TC & Vetter W (1991) [Bronchial asthma and kitchen salt.]
Schweiz Med Wochenschr, 121(14): 501-508 (in German).
Meding B (1990) Epidemiology of hand eczema in an industrial city.
Acta Dermatol Venereol, 153(suppl): 2-43.
Melamed D & Friedman A (1993) Direct evidence for anergy in T
lymphocytes tolerized by oral administration of ovalbumin. Eur J
Immunol, 23: 935-942.
Mellström G, Lindahl G, & Wahlberg J (1989) DAISY: reference database
on protective gloves. Semin Dermatol, 8: 75-79.
Melnik B & Plewig G (1991) Atopic dermatitis and disturbances of
essential fatty acid in prostaglandin E metabolism. J Am Acad
Dermatol, 25: 859-860.
Menné T & Bachmann E (1979) Permanent disability from skin diseases.
Dermatosen, 27: 37-42.
Menné T & Holm NV (1983) Nickel allergy in a female twin population.
Int J Dermatol, 22: 22-28.
Menné T & Holm NV (1986) Gene susceptibility in human allergic contact
sensitization. Semin Dermatol, 5: 301-306.
Menné T & Maibach HI (1991) Systemic contact-type dermatitis. In:
Marzulli FN & Maibach HI ed. Dermatotoxicology. Washington, New York,
London, Hemisphere Publishing Corporation, p 453.
Menné T & Maibach HI ed. (1994) Hand eczema. Boca Raton, Florida, CRC
Press, 334 pp.
Menné T & Wilkinson JD (1995) Individual predisposition to contact
dermatitis. In: Rycroft RJG, Menné T, Frosch PJ, & Benezra C ed.
Textbook of contact dermatitis. Berlin, Heidelberg, New York, Springer
Verlag, pp 123-130.
Menné T, Christophersen J, & Maibach HI (1987) Epidemiology of
allergic contact sensitization. Monogr Allergy, 21: 132-161.
Menné T, Dooms-Goossens A, Wahlberg JE, White I, & Wilkinson J (1992)
How large a proportion of contact sensitivities are diagnosed with the
European standard series. Contact Dermatitis, 26: 201-202.
Menné T, Veien N, Sjolin KE, & Maibach HI (1994) Systemic contact
dermatitis. Am J Contact Dermatitis, 5: 1-12.
Meredith S (1993) Reported incidence of occupational asthma in the
United Kingdom, 1989-90. J Epidemiol Community Health, 47(6): 459-463.
Meredith SK & McDonald JC (1994) Work-related respiratory disease in
the United Kingdom, 1989-1992: Report on the SWORD project. Occup Med,
44(4): 183-189.
Meredith S & Nordman H (1996) Occupational asthma: measures of
frequency from four countries. Thorax, 51(4): 435-440.
Merget R, Reineke M, Rueckmann A, Bergmann EM, & Schultze Werninghaus
G (1994) Nonspecific and specific bronchial responsiveness in
occupational asthma caused by platinum salts after allergen avoidance.
Am J Respir Crit Care Med, 150(4): 1146-1149.
Merget R, Dierkes A, Rueckmann A, Bergmann EM, & Schultze Werninghaus
G (1996) Absence of relationship between degree of nonspecific and
specific bronchial responsiveness in occupational asthma due to
platinum salts. Eur Respir J, 9(2): 211-216.
Metcalfe DD (1985) Food allergens. Clin Rev Allergy, 3(3): 331-349.
Metcalfe DD & Sampson HA (1990) Workshop on experimental methodology
for clinical studies of adverse reactions to foods and food additives.
J Allergy Clin Immunol, 86: 421-442.
Metzger WJ, Zavala D, Richerson HB, Mosely P, Iwamota P, Monick M,
Sjoerdsma K, & Hunninghake GW (1987) Local allergen challenge and
bronchoalveolar lavage of allergic asthmatic lungs. Am Rev Respir Dis,
135: 433-440.
Miedema I, Feskens EJ, Heederik D, & Kromhout D (1993) Dietary
determinants of long-term incidence of chronic nonspecific lung
diseases: The zutphen study. Am J Epidemiol, 138(1): 37-45.
Miller K & Nicklin S (1988) Mechanisms of food intolerances:
development and use of experimental animal models. In: Walker R &
Quattrucci E ed. Nutritional and toxicological aspects of food
processing. London, New York, Philadelphia, Taylor & Francis, pp
351-364.
Minowada G & Welch WJ (1995) Clinical implications of the stress
response. J Clin Invest, 95: 3-12.
Mitchell EA, Stewart AW, Pattemore PK, Asher MI, Harrison AC, & Rea HH
(1989) Socioeconomic status in childhood asthma. Int J Epidemiol,
18(4): 888-890.
Molfino NA, Wright SC, Katz I, Tarlo S, Silverman F, McClean PA,
Szalai JP, Raizenne M, Slutsky AS, & Zamel N (1991) Effect of low
concentrations of ozone on inhaled allergen responses in asthmatic
subjects. Lancet, 338: 199-203.
Moller DR, Gallagher JS, Benstein DI, Wilcox TG, Burroughs HE, &
Bernstein IL (1985) Detection of IgE mediated respiratory
sensitisation in workers exposed to hexahydrophthalic anhydride. J
Allergy Clin Immunol, 75: 663-672.
Monick M, Glazier J, & Hunninghake GW (1987) Human alveolar
macrophages suppress interleukin 1 activity via the secretion of
prostaglandin E2. Am Rev Respir Dis, 135: 72-77.
Montgomery-Smith J (1983) Epidemiology and natural history of asthma,
allergic rhinitis and atopic dermatitis (eczema). In: Allergy:
Principles and practice. St Louis, Missouri, C.V. Mosby & Co., pp
771-803.
Montgomery-Smith J, Middleton E, Reed CE, Ellis EF, Adkinson NF, &
Yunginger JW ed. (1983) Allergy: Principles and practice
-- Epidemiology and natural history of asthma, allergic rhinitis and
atopic dermatitis (eczema). St Louis, Missouri, C.V. Mosby & Co., pp
771-803.
Moore SA, Strieter RM, Rolfe MW, Standiford TJ, Burdick MD, & Kunkel
SL (1992) Expression and regulation of human alveolar
macrophage-derived interleukin-1 receptor antagonist. Am J Respir Cell
Mol Biol, 6(6): 569-575.
Morel E, Feuillet-Fieux MN, Vernet-der Garabedian B, Raimond F,
D'Anglejan J, Bataille R, Sany J, & Bach JF (1991) Autoantibodies in
D-penicillamine-induced myasthenia gravis: a comparison with
idiopathic myasthenia and rheumatoid arthritis. Clin Immunol
Immunopathol, 58: 318-330.
Morgenstern H & Thomas D (1993) Principles of study design in
environmental epidemiology. Environ Health Perspect, 101(suppl 4):
23-28.
Morren MA, Przybilla B, Bamelis M, Heykants B, Reynaers A, & Degreef H
(1994) Atopic dermatitis: triggering factors. J Am Acad Dermatol, 31:
467-473.
Mosimann B, Peitrequin R, Blanc C, & Pecoud A (1992) Allergie aux
blattes (cafards) dans une population suisse souffrant d'asthme et de
rhinite chroniques. Schweiz Med Wochenschr, 122(34): 1245-1248.
Mosmann TR & Sad S (1996) The expanding universe of T-cell subsets:
Th1, Th2 and more. Immunol Today, 17: 138-146.
Moulon C, Vollmer J, & Weltzien HU (1995) Characterization of
processing requirements and metal cross-reactivities in T cell clones
from patients with allergic contact dermatitis to nickel. Eur J
Immunol, 25: 3308-3315.
Mowatt AMcI (1984) The immunopathogenesis of food-sensitive
enteropathies. In: Newly TJ & Stokes CR ed. Local immune responses of
the gut. Boca Raton, Florida, CRC Press, pp 199-225.
Mowatt AMcI (1987) The regulation of immune responses to dietary
protein antigens. Immunol Today, 8: 93-98.
Mowatt AMcI, Lamont AG, Strobel S, & Mackenzie S (1986) The role of
antigen processing and suppressor T cells in immune responses to
dietary proteins in mice. Adv Exp Med Biol, 216A: 709-720.
M'Raihi L, Charpin D, Pons A, Bongrande P, & Vervloet D (1991)
Cross-reactivity between latex and banana. J Allergy Clin Immunol, 87:
129-130.
Mullan RJ & Murthy LI (1991) Occupational sentinel health events: An
up-dated list for physician recognition and public health
surveillance. Am J Ind Med, 19(6): 775-799.
Mullick FG, McAllister HA, Wagner BM, & Fenoglio JJ (1979) Drug
related vasculitis: Clinicopathologic correlations in 30 patients. Hum
Pathol, 10: 313-325.
Munir AK, Einarsson R, Schou C, & Dreborg SK (1993) Allergens in
school dust: I. The amount of the major cat (Fel d I) and dog (Can f
I) allergens in dust from Swedish schools is high enough to probably
cause perennial symptoms in most children with asthma who are
sensitized to cat and dog. J Allergy Clin Immunol, 91(5): 1067-1074.
Munir AK, Einarsson R, & Dreborg SK (1994a) Indirect contact with pets
can confound the effect of cleaning procedures for reduction of animal
allergen levels in house dust. Pediatr Allergy Immunol, 5(1): 32-39.
Munir AK, Bjorksten B, Einarsson R, Schou C, Ekstrand Tobin A, Warner
A, & Kjellman NI (1994b) Cat (Fel d I), dog (Can f I), and cockroach
allergens in homes of asthmatic children from three climatic zones in
Sweden. Allergy, 49(7): 508-516.
Muraille E & Leo O (1998) Revisiting the Th1/Th2 paradigm. Scand J
Immunol, 47: 1-9.
Muranaka M, Suzuki S, Koizumi K, Takafuji S, Miyamoto T, Ikemori R, &
Tokiwa H (1986) Adjuvant activity of diesel-exhaust particulates for
the production of IgE antibody in mice. J Allergy Clin Immunol, 77:
616-623.
Mygind N (1986) Essential allergy, 1st ed. Oxford, London, Boston,
Blackwell Scientific Publications, 480 pp.
Mygind N (1989) Nasal allergy, 2nd ed. Oxford, London, Boston,
Blackwell Scientific Publications, pp 219-223.
Nagata S, Yamashiro Y, Ohtuska Y, Shioya T, Oguchi S, Shimizu T, &
Maeta M (1995) Quantitative analysis and immunohistochemical studies
on small intestinal mucosa of food-sensitive enteropathy. J Pediatr
Gastroenterol Nutr, 20: 44-48.
Naparstek Y & Plotz PH (1993) The role of autoantibodies in autoimmune
disease. Annu Rev Immunol, 11: 79-104.
NAS (National Academy of Science) (1983) Committee on the
Institutional Means for Assessment of Risks to Public Health -- Risk
assessment in the Federal Government: Managing the process.
Washington, DC, National Academy Press, pp 1-50.
NAS (National Academy of Science) (1993) Committee on Risk Assessment
Methodology, Board on Environmental Studies and Toxicology -- Issues
in risk assessment. Washington, DC, National Academy Press, 356 pp.
Neas LM, Dockery DW, Ware JH, Spengler JD, Speizer FE, & Ferris BG Jr
(1991) Association of indoor nitrogen dioxide with respiratory
symptoms and pulmonary function in children. Am J Epidemiol, 134(2):
204-219.
Nelson HS (1985) The atopic diseases. Ann Allergy, 55: 441-447.
Nelson HS, Rosloniec DM, McCall LI, & Ikle D (1993) Comparative
performance of five commercial prick skin test devices. J Allergy Clin
Immunol, 92(5): 750-756.
Nemery B, Casier P, Roosels D, Lahaye D, & Demedts M (1992) Survey of
cobalt exposure and respiratory health in diamond polishers. Am Rev
Respir Dis, 145(3): 610-616.
Newly TJ, Stokes CR, & Bourne FJ (1980) Effects of feeding bacterial
lipopolysaccharide and dectran sulphate on the development of oral
tolerance to contact sensitizing agents. Immunology, 41: 617-621.
Newman LS & Kreiss K (1992) Nonoccupational beryllium disease
masquerading as sarcoidosis: identification by blood lymphocyte
proliferative response to beryllium. Am Rev Resp Dis, 145: 1212-1214.
Newman LS, Bobka C, Schumacher B, Daniloff E, Zhen B, Mroz MM, & King
TE Jr (1994) Compartmentalized immune response reflects clinical
severity of beryllium disease. Am J Resp Crit Care Med, 150: 135-142.
Newman Taylor AJ (1988) Occupational asthma. Postgrad Med J,
64: 505-510.
Newman Taylor AJ (1995) Environmental determinants of asthma. Lancet,
345(8945): 296-299.
Newman Taylor AJ, Venables KM, Durham S, Graneek BJ, & Topping MD
(1987) Acid anhydrides and asthma. Int Arch Allergy Appl Immunol, 82:
435-439.
Ng TP & Tan WC (1994a) Epidemiology of chronic (perennial) rhinitis in
Singapore: prevalence estimates, demographic variation and clinical
allergic presentation. Ann Acad Med Singap, 23: 83-88.
Ng TP & Tan WC (1994b) Epidemiology of allergic rhinitis and its
associated risk factors in Singapore. Int J Epidemiol, 23(3): 553-558.
Niebel G (1995) [Behavioural medicine of chronic skin disease.] Bern,
Göttingen, Toronto, Seattle, Hogrefe & Huber Publishers.
Nielsen NH & Menné T (1992) Allergic contact sensitization in an
unselected Danish population -- the Glostrup allergy study, Denmark.
Acta Dermatol Venereol, 72: 456-460.
Nielsen NH & Menné T (1993) Nickel sensitization and ear piercing in
an unselected Danish population -- The Glostrup allergy study,
Denmark. Contact Dermatitis, 29: 16-21.
Niermann H (1964) [Dermatology in twins.] Berlin, Heidelberg, New
York, Springer Verlag, 108 pp.
Niestijl Jansen JJ, Kardinaal AFM, Huijbers G, Vlieg-Boerstra BJ,
Martens BPM, & Ockhuizen T (1994) Prevalence of food allergy and
intolerance in the adult Dutch population. J Allergy Clin Immunol, 93:
446-456.
NIH (National Institutes of Health) (1991) National Asthma Education
Program Expert Panel -- Guidelines for the diagnosis and management of
asthma. Bethesda, Maryland, National Heart, Lung and Blood Institute,
72 pp (NIH Publication No. 91-3042A).
Nilsson B & Kristofferson A (1989) Zimelidine: Febrile reaction and
peripheral neuropathy. In: Kammüller ME, Bloksma N, & Seinen W ed.
Autoimmunity and toxicology. Amsterdam, Oxford, New York, Elsevier
Science Publishers, pp 183-214.
Ninan TK & Russell G (1992) Respiratory symptoms and atopy in Aberdeen
schoolchildren: Evidence from two surveys 25 years apart. Br Med J,
304: 873-875.
NIOSH (1997) NIOSH alert: Preventing allergic reactions to natural
rubber latex in the workplace. Cincinnati, Ohio, National Institute
for Occupational Safety and Health, 11 pp.
Nitta H, Sato T, Nakai S, Maeda K, Aoki S, & Ono M (1993) Respiratory
health associated with exposure to automobile exhaust. I. Results of
cross-sectional studies in 1979, 1982, and 1983. Arch Environ Health,
48: 53-58.
Noelle RJ, Daum J, Bartlett WC, McCann J, & Shepherd DM (1990) Cognate
interactions between helper T-cells and B-cells. V. Reconstitution of
helper T-cell function using purified plasma membranes from activated
Th1 and Th2 helper T-cells and lymphokines. J Immunol, 146(4):
1118-1124.
Norback D, Bjornsson E, Janson C, Widstrom J, & Boman G (1995)
Asthmatic symptoms and volatile organic compounds, formaldehyde, and
carbon dioxide in dwellings. Occup Environ Med, 52(6): 388-395.
Nordman H, Keskinen H, & Tuppurainen M (1985) Formaldehyde asthma
-- rare or overlooked? J Allergy Clin Immunol, 75: 91-99.
Nussler AK & Billiar TR (1993) Inflammation, immunoregulation, and
inducible nitric oxide synthase. J Leuk Biol, 54(2): 171-178.
Nyren O, Yin L, Josefsson S, McLaughlin JK, Blot WJ, Engqvist M,
Hakelius L, Boice JD Jr, & Adami HO (1998) Risk of connective tissue
disease and related disorders among women with breast implants: a
nation-wide retrospective cohort study in Sweden. Br Med J, 316(7129):
417-422.
O'Brien IM, Harries MG, Burge PS, & Pepys J (1979) Toluene
di-isocyanate induced asthma. Reactions to TDI, MDI, HDI and
histamine. Clin Allergy, 19: 1-6.
O'Byrne PM, Dolovich J, & Hargreave FE (1987) Late asthmatic
responses. Am Rev Respir Dis, 134: 740-751.
Odom RB & Maibach HI (1976) Contact urticaria: a different contact
dermatitis. Cutis, 18(5): 672-676.
O'Donnell TV (1995) Asthma and respiratory problems -- a review. Sci
Total Environ, 163(1-3): 137-145.
OECD (1992) Guideline 406 for testing chemicals. Paris, Organisation
for Economic Cooperation and Development.
Oettigen HC, Martin TR, Wynshaw-Boris A, Deng C, Drazen JM, & Leder P
(1994) Active anaphylaxis in IgE-deficient mice. Nature (Lond), 370:
367-370.
Ogawa M, Berger PA, McIntyre OR, & Clendenning WE (1971) IgE in atopic
dermatitis. Arch Dermatol, 103: 575.
Ohashi PS, Oehnen S, Buerki K, Pircher H, Ohashi CT, Odermatt B,
Malissen B, Zinkernagel RM, & Hengartner H (1991) Ablation of
"tolerance" and induction of diabetes by virus infection in viral
antigen transgenic mice. Cell, 65: 305-317.
Ollerenshaw S & Woolcock AJ (1992) Characteristic of inflammation in
biopsies from large airways of subjects with asthma and subjects with
chronic airflow limitation. Am Rev Respir Dis, 145: 922-927.
Ollier S & Davies RJ (1994) A report of the relationship between
allergen load, primary immunologic sensitization and the expression of
allergic disease. Respir Med, 88: 407-415.
Oostendorp RAJ, Meijer CJLM, & Scheper RJ (1993) Immunosuppression by
retroviral-envelope-related peptides, and their role in non-retroviral
human disease. Crit Rev Oncol Hematol, 14: 189-206.
Oosterlee A, Drijver M, Lebret E, & Brunekreef B (1996) Chronic
respiratory symptoms in children and adults living along streets with
high traffic density. Occup Environ Med, 53: 241-247.
Ortolani C, Ispano M, Pastorello EA, Ansaloni R, & Magri GC (1989)
Comparison of results of skin prick tests (with fresh foods and
commercial food extracts) and RAST in 100 patients with oral allergy
syndrome. J Allergy Clin Immunol, 83: 683-690.
Osebold JW, Gershwin LJ, & Zee YC (1980) Studies on the enhancement of
allergic lung sensitization by inhalation of ozone and sulfuric acid
aerosol. J Environ Pathol Toxicol, 3: 221-234.
Osterland CK (1994) Laboratory diagnosis and monitoring in chronic
systemic autoimmune diseases. Clin Chem, 40(11B): 2146-2153.
Ostro BD, Lipsett MJ, Wiener MB, & Selner JC (1991) Asthmatic
responses to airborne acid aerosols. Am J Public Health, 81(6):
694-702.
Oswald IP, Gazzinelli RT, Sher A, & James SL (1992) IL-10 synergizes
with IL-4 and transforming growth factor-beta to inhibit macrophage
cytotoxic activity. J Immunol, 148(11): 3578-3582.
Paggiaro P, Bacci E, Paoletti P, Bernard P, Dente FL, Marchetti G,
Talini D, Menconi GF, & Giuntini C (1990) Bronchoalveolar lavage and
morphology of the airways after cessation of exposure in asthmatic
subjects sensitised to toluene di-isocyanate. Chest, 98(3): 536-542.
Park HS, Yu HJ, & Jung KS (1994) Occupational asthma caused by
chromium. Clin Exp Allergy, 24: 676-681.
Parker D, Sommer G, & Turk JL (1975) Variation in guinea pig
responsiveness. Cell Immunol, 18: 233-238.
Parronchi P, Macchia D, Piccini M-P, Biswas P, Simonelli C, Maggi E,
Ricci M, Ansari A, & Romagnani S (1991) Allergen-and bacterial
antigen-specific T-cell clones established from atopic donors show a
different profile of cytokine production. Proc Natl Acad Sci (USA),
88: 4538-4542.
Pastorello E, Stocchi L, Bigi A, Pravettoni V, Schilke ML, Valente D,
& Zanussi C (1989) Value and limits of diagnostic tests in food
hypersensitivity. Allergy, 44(suppl): 151-158.
Pastorello EA, Incorvaia C, & Ortolani C (1995) The mouth and pharynx.
In: Ortolani C ed. Atlas of mechanisms in adverse reactions to food.
Allergy, 50(20): 41-44.
Pauli G, de Blay F, Bessort JC, & Dietermann A (1992) The association
between respiratory allergies and food hypersensitivities. ACI News,
4: 43-47.
Pauluhn J (1996) Predictive testing for respiratory sensitisation.
Toxicol Lett, 86(2-3): 177-185.
Pauluhn J & Eben A (1991) Validation of a non-invasive technique to
assess immediate or delayed onset of airway hypersensitivity in
guinea-pigs. J Appl Toxicol, 11: 423-431.
Pauluhn J & Mohr U (1994) Assessment of respiratory hypersensitivity
in guinea pigs sensitized to diphenylmethane-4,4-diisocyanate (MDI)
and challenged with MDI acetylcholine or MDI albumin conjugate.
Toxicology, 92: 53-74.
Payne MP & Walsh PT (1994) Structure-activity relationships for skin
sensitization potential: development of structural alerts for use in
knowledge-based toxicity prediction systems. J Chem Inf Comput Sci,
34(1): 154-161.
PEACE(1998) Study of pollution effects on asthmatic children in Europe
(PEACE). Eur Respir Rev, 8: 1-130.
Pearce N, Weiland S, Keil U, Langridge P, Anderson HR, Strachan D,
Bauman A, Young L, Gluyas P, Ruffin D, Crane J, & Beasley R (1993)
Self-reported prevalence of asthma symptoms in children in Australia,
England, Germany and New Zealand: an international comparison using
the ISAAC protocol. Eur Respir J, 6: 1455-1461.
Peat JK (1996) Prevention of asthma. Eur Respir J, 9: 1545-1555.
Peat J., Woolcock A, Leeder SR, & Blackburn CR (1980) Asthma and
bronchitis in Sydney schoolchildren: II. The effect of social factors
and smoking on prevalence. Am J Epidemiol, 111(6): 728-735.
Peat J., van den Berg RH, Green WF, Mellis CM, Leeder SR & Woolcock A
(1994) Changing prevalence of asthma in Australian children. Br Med J,
308: 1591-1596.
Peden DB, Carter J, Dailey LA, & Devlin R (1994) The effect of ozone
on house dust mite allergen-induced nasal inflammation in asthmatics.
Am J Respir Crit Care Med, 149: A154 (abstract).
Pelletier L, Castedo M, Bellon B, & Druet P (1994) Mercury and
autoimmunity. In: Dean JH, Luster MI, Munson AE, & Kimber I ed.
Immunotoxicology and immunopharmacology, 2nd ed. New York, Raven
Press, pp 539-552.
Pepys J (1994) "Atopy": a study in definition. Allergy, 49(6):
397-399.
Pepys J, Pickering CAC, & Hughes EG (1972) Asthma due to inhaled
chemical agents -- complex salts of platinum. Clin Allergy, 2:
391-396.
Peters A, Dockery DW, Heinrich J, & Watchman HE (1997) Short-term
effects of particulate air pollution on respiratory morbidity in
asthmatic children. Eur Resp J, 10(4): 872-879.
Pfeiffer C, Murray J, Madri J, & Bottomly K (1991) Selective
activation of Th1- and Th2-like cells in vivo: response to human
collagen IV. Immunol Rev, 123: 65-84.
Philen RM & Posada M (1993) Toxic oil syndrome and
eosinophilia-myalgia syndrome: May 8-10, 1991, World Health
Organization Meeting report. Semin Arthritis Rheum, 23: 104-124.
Picker LJ, Treer JR, Ferguson-Darnell B, Collins PA, Bergstresser PR,
& Terstappen LWMM (1993) Control of lymphocyte recirculation in man:
II. Differential regulation of the cutaneous lymphocyte associated
antigen, a tissue-selective homing receptor for skin-homing T cells. J
Immunol, 150: 1122-1136.
Pinkston P, Bitterman PB, & Crystal RG (1984) Interleukin-2 in the
alveolitis of beryllium-induced lung disease. Am Rev Respir Dis,
129(411): A161 (abstract).
Pistoor FAM., Kapsenberg ML, Bos JD, Meinardi MMHM, von Blomberg BME,
& Scheper RJ (1995) Cross-reactivity of human nickel-reactive T
lymphocyte clones with copper and palladium. J Invest Dermatol, 105:
92-95.
Planchon SM, Martins CAP, Guerrant RL, & Roche J. (1994) Regulation of
intestinal epithelial barrier function by TGF-beta-1. J Immunol, 153:
5730-5739.
Platts-Mills TA (1994) How environment affects patients with allergic
disease: indoor allergens and asthma. Ann Allergy, 72: 381-384.
Platts-Mills TA & Chapman MD (1987) Dust mites: immunology, allergic
disease, and environmental control. J Allergy Clin Immunol, 80:
755-775.
Platts-Mills TAE, Chapman MD, Mitchell B, Heymann PW, & Deuell B
(1991) Role of inhalant allergens in atopic eczema. In: Ruzicka T,
Ring J, & Przybilla B ed. Handbook of atopic eczema. Berlin,
Heidelberg, New York, Springer Verlag, pp 192-203.
Platt-Mills TA, Sporik RB, Chapman MD, & Heymann PW (1995) The role of
indoor allergens in asthma. Allergy, 50(suppl 22): 5-12.
Podmore P (1995) Shoes. In: Rycroft RJG, Menné T, Frosch PJ, & Benezra
C ed. Textbook of contact dermatitis, 2nd ed. Berlin, Heidelberg, New
York, Springer Verlag, pp 516-527.
Polak L (1980) Immunological aspects of contact sensitivity: An
experimental study. Monogr Allergy, 15: 1-170.
Pollart SM, Chapman MD, Fiocco GP, Rose G, & Platts-Mills TA (1989)
Epidemiology of acute asthma: IgE antibodies to common inhalant
allergens as a risk factor for emergency room visits. J Allergy Clin
Immunol, 83: 875-882.
Pope CA III, Schwarz J, & Ransom MR (1992) Daily mortality and PM10
pollution in Utah Valley. Arch Environ Health, 47: 211-217.
Pope CA III, Bates DV, & Raizenne ME (1995) Health effects of
particulate air pollution: Time for reassessment. Environ Health
Perspect, 103(5): 472-480.
Posselt AM, Barker CF, Friedman AL, & Naji A (1992) Prevention of
autoimmune diabetes in the BB rat by intrathymic islet transplantation
at birth. Science, 256: 1321-1324.
Potter DW & Wederbrand KS (1995) Total IgE antibody in BALB/c mice
after dermal exposure to chemicals. Fundam Appl Toxicol, 26: 127-135.
Prausnitz C & Kustner H (1921) [Studies concerning sensitivity.] Zent.
bl Bakteriol I Orig, 86: 160-169
Prentice RL & Thomas D (1993) Methodologic research needs in
environmental epidemiology: Data analysis. Environ Health Perspect,
101(suppl 4): 39-48.
Prud'Homme GJ, Parfrey NA, & Vanier LE (1991) Cyclosporine-induced
autoimmunity and immune hyperreactivity. Autoimmunity, 9: 345-356.
Przybilla B & Ring J (1990) Food allergy and atopic eczema. Semin
Dermatol, 9: 220-225.
Przybilla B, Eberlein-König B, & Rueff F (1994) Practical management
of atopic eczema. Lancet, 343: 1342-1346.
Punnonen J & de Vries JE (1994) IL-13 induces proliferation, Ig
isotype switching, and Ig synthesis by immature human fetal B cells. J
Immunol, 152: 1094-1102.
Punnonen J, Aversa G, Cocks BG, McKenzie AN, Menon S, Zurawski G, de
Waal Malefyt R, & de Vries JE (1993) Interleukin-13 induces
interleukin 4-independent IgG4 and IgE synthesis and CD23 expression
by human B cells. Proc Natl Acad Sci (USA), 90(8): 3730-3734.
Raaschou Nielsen O, Nielsen ML, & Gehl J (1995) Traffic-related air
pollution: exposure and health effects in Copenhagen street cleaners
and cemetery workers. Arch Environ Health, 50(3): 207-213.
Radl J, Valentijn RM, Haaijman JJ, & Paul LC (1985) Monoclonal
gamma-pathies in patients undergoing immunosuppressive treatment after
renal transplantation. Clin Immunol Immunopathol, 37: 98-102.
Rajka G (1990) Essential aspects of atopic eczema. Berlin, Heidelberg,
New York, Springer Verlag, 261 pp.
Rastogi SK, Gupta BN, Husain T, Mathur N, Pangtey BS, & Garg N (1992)
Respiratory symptoms and ventilatory capacity in metal polishers. Hum
Exp Toxicol, 11(6): 466-472.
Rattray NJ, Botham PA, Hext PM, Woodcock DR, Fielding I, Dearman RJ, &
Kimber I (1994) Induction of respiratory hypersensitivity to
diphenylmethane-4,4-diisocyanate in guinea pigs. Influence of route of
exposure. Toxicology, 88: 15-30.
Raulf M & Konig W (1991) Modulation of leukotriene generation from
human polymorphonuclear granulocytes by polychlorinated biphenyls
(PCB). Immunology, 73(4): 485-490.
Ravetch JV (1994) Fc receptors: rubor redux. Cell, 78: 553-560
Ravetch JV (1997) Fc receptors. Curr Opin Immunol, 9: 121-125.
Rea TH (1979) Quantitative enhancement of dinitrochlorobenzene
responsivity in women receiving oral contraceptives. Arch Dermatol,
115(3): 361-362.
Reijula K & Patterson R (1994) Occupational allergies in Finland in
1981-91. Allergy Proc, 15(3): 163-168.
Reitamo S, Visa K, Kahonen K, Kayhko K, Stubb S, & Salo OP (1986)
Eczematous reactions in atopic patients caused by epicutaneous testing
with inhalant allergens. Br J Dermatol, 114: 303-309.
Renstrom A, Malmberg P, Larsson K, Sundblad BM, & Larsson PH (1994)
Prospective study of laboratory-animal allergy: factors predisposing
to sensitization and development of allergic symptoms. Allergy, 49(7):
548-552.
Richardson BC, Liebling MR, & Hudson JL (1990) CD4+ cells treated with
DNA methylation inhibitors induce autologous B cell differentiation.
Clin Immunol Immunopathol, 55: 368-381.
Richardson B, Powers D, Hooper F, Yung RL, & O'Rourke K (1994)
Lymphocyte function-associated antigen 1 overexpression and T cell
autoreactivity. Arthritis Rheum, 37: 1363-1372.
Ridings JE, Barrat MD, Cary R, Earnshaw CG, Eggington CE, Ellis MK,
Judson PN, Langowski JJ, Marchant CA, Payne MP, Watson WP, & Yih TD
(1996) Computer prediction of possible toxic action from chemical
structure: an update on the DEREK system. Toxicology, 106: 267-279.
Riedel F, Kramer M, Scheibenbogen C, & Rieger CH (1988) Effects of
SO2 exposure on allergic sensitization in the guinea pig. J Allergy
Clin Immunol, 82: 527-534.
Riedler J, Reade T, Dalton M, Holst D, & Robertson C (1994) Hypertonic
saline challenge in an epidemiologic survey of asthma in children. Am
J Respir Crit Care Med, 150: 1632-1639.
Ring J (1988) [Applied allergology], 2nd ed. Munich, MMW Medizin
Verlag GmbH, pp 85-86 (in German).
Ring J (1991) Atopy: Conditions, disease or syndrome? In: Ruzicka T,
Ring J, & Przybilla B ed. Handbook of atopic eczema. Berlin,
Heidelberg, New York, Springer Verlag, pp 3-8.
Ring J & Thomas P (1989) Histamine and atopic eczema. Acta Dermatol
Venereol, 144: 70-77.
Ring J, Sedlmeier F, von der Helm D, Mayr T, Walz U, Ibel H, Przybilla
B, Reimann HJ, & Dorsch W (1988) Histamine and allergic disease. In:
Ring J & Burg G ed. New trends in allergy. Berlin, Heidelberg, New
York, Springer Verlag, pp 44-77.
Ring J, Bieber T, Vieluf D, Kunz B, & Przybilla B (1991a) Atopic
eczema, Langerhans cells and allergy. Int Arch Allergy Immunol, 94:
194-201.
Ring J, Ruzicka T, & Przybilla B (1991b) The pathophysiology of atopic
eczema. In: Ruzicka T, Ring J, & Przybilla B ed. Handbook of atopic
eczema. Berlin, Heidelberg, New York, Springer Verlag, pp 330-335.
Ring J, Abeck D, & Neuber K (1992) Atopic eczema: role of
microorganisms on the skin surface. Allergy, 47: 265-269.
Ring J, Behrendt H, Schäfer T, Vieluf D, & Krämer U (1995) Impact of
air pollution on allergic diseases: Clinical and epidemiologic
studies. In: Johansson SGO ed. Progress in allergy and clinical
immunology. Bern, Göttingen, Toronto, Seattle, Hogrefe & Huber
Publishers, vol 3, pp 174-179.
Ring J, Brockow K, & Abeck D (1996) The therapeutic concept of patient
management in atopic eczema. Allergy, 51: 206-215.
Ritz HL, Conner DS, & Sauter ED (1975) Contact sensitization of
guinea-pigs with unsaturated and halogenated sultones. Contact
Dermatitis, 1: 349-359.
Ritz HL, Evans BL, Bruce RD, Fletcher ER, Fisher GL, & Sarlo K (1993)
Respiratory and immunological responses of guinea pigs to
enzyme-containing detergents: A comparison of intratracheal and
inhalation modes of exposure. Fundam Appl Toxicol, 21: 31-37.
Roberts DW & Basketter DA (1990a) A quantitative structure
activity/dose relationship for contact allergenic potential of alkyl
group transfer agents. Toxicology in vitro, 4(4/5): 686-687.
Roberts DW & Basketter DA (1990b) A quantitative structure
activity/dose response relationship for contact allergic potential of
alkyl group transfer agents. Contact Dermatitis, 23: 331-335.
Roberts SA, Reinhardt MC, Paganelli R, & Levinsky RJ (1981) Specific
antigen exclusion and non-specific facilitation of antigen entry
across the gut in rats allergic to food proteins. Clin Exp Immunol,
45: 131-136.
Robertson CF, Heycock E, Bishop J, Nolan T, Olinsky A, & Phelan PD
(1991) Prevalence of asthma in Melbourne schoolchildren: Changes over
26 years. Br Med J, 302: 1116-1118.
Robinson DS (1998) T cell co-stimulation: a potential therapeutic
target in asthma? Clin Exp Allergy, 28: 788-790.
Roche WR, Beasley R, Williams JH, & Holgate ST (1989) Subepithelial
fibrosis in the bronchi of asthmatics. Lancet, 1: 520-523.
Röcken M, Racke M, & Shevach EM (1996) IL-4-induced immune deviation
as antigen-specific therapy for inflammatory autoimmune disease.
Immunol Today, 17: 225-231.
Roed-Petersen J (1989) A new glove material protective against epoxy
and acrylate monomer. In: Frosch PJ, Dooms-Goossens A, Lachapelle J-M,
Rycroft RJG, & Scheper RJ ed. Current topics in contact dermatitis.
Berlin, Heidelberg, New York, Springer Verlag, pp 603-606.
Roitt IM, Brostoff J, & Male DK ed. (1998) Immunology, 5 th ed. St.
Louis, Missouri, C.V. Mosby & Co., 424 pp.
Romagnani S (1991) Human Th1 and Th2 subsets: doubt no more.
Immmunol Today, 12(8): 256-257.
Romagnani S (1992a) Induction of Th1 and Th2 responses: A key role for
the "natural" immune response? Immunol Today, 13(10): 379-381.
Romagnani S (1992b) Human TH1 and TH2 subsets: Regulation of
differentiation and role in protection and immunopathology. Int Arch
Allergy Immunol, 98: 279-285.
Romanski B (1987) The pathology of food allergy studied by gastric
allergen challenge. In: Brostoff J & Challacombe SJ ed. Food allergy
and intolerance. London, Baillière Tindall, pp 917-931.
Romanski B (1989) The pathology of food allergy studied by gastric
allergen challenge. In: Paul WE ed. Fundamental immunology. New York,
Raven Press, p 41.
Rose G (1985) Sick individuals and sick populations. Int J Epidemiol,
14: 32-38.
Rose NR & Potter M (1995) The silicone controversy: towards a
resolution. Immunol Today, 16: 459-460.
Rosen FS (1987) Autoimmunity and immunodeficiency disease. In: Evered
D & Whelan J ed. Autoimmunity and autoimmune disease. New York,
Chichester, Brisbane, Toronto, John Wiley & Sons, pp 135-148 (Ciba
Foundation Symposium No. 129).
Rosenstreich DL, Eggleston P, Kattan M, Baker D, Slavin RG, Gergen P,
Mitchell H, McNiff-Mortimer K, Lynn H, Ownby D, & Malveaux F (1997)
The role of cockroach allergy and exposure to cockroach allergen in
causing morbidity among inner-city children with asthma. N Engl J Med,
336(19): 1356-1363.
Ross B, McCullough J, & Ownby DR (1991) Evidence of allergenic
cross-reactivity between banana and watermelon. J Allergy Clin
Immunol, 87: 274 (abstract).
Rossman MD, Kern JA, Elias JA, Cullen MR, Epsein PE, Preuss OP,
Markham TN, & Daniele RP (1988) Proliferative response of
bronchoalveolar lymphocytes to beryllium. Ann Intern Med, 108:
687-693.
Roth MD & Golub SH (1993) Human pulmonary macrophages utilize
prostaglandins and transforming growth factor beta 1 to suppress
lymphocyte activation. J Leuk Biol, 53(4): 366-371.
Rothman KJ (1993) Methodologic frontiers in environmental
epidemiology. Environ Health Perspect, 101(suppl 4): 19-21.
Rubin RL (1989) Autoimmune reactions induced by procainamide and
hydralazine. In: Kammüller M, Bloksma N, & Seinen W ed. Autoimmunity
and toxicology: Immune disregulation induced by drugs and chemicals.
Amsterdam, Oxford, New York, Elsevier Science Publishers, pp 119-150.
Rubin RL, Burlingame RW, Arnott JE, Totoritis MC, McNally EM, &
Johnson AD (1995) IgG but not other classes of anti-[(H2A-H2B)-DNA] is
an early sign of procainamide-induced lupus. J Immunol, 154:
2483-2493.
Ruffin J, Liu MYG, Sessions R, Banerjee S, & Banerjee UC (1986)
Effects of certain atmospheric pollutants (SO2, NO2, CO) on the
soluble amino acids, molecular weight and antigenicity of some
airborne pollen grains. Cytobios, 46: 119-129.
Ruzicka T, Ring J, & Przybilla B ed. (1991) Handbook of atopic eczema.
Berlin, Heidelberg, New York, Springer Verlag, 481 pp.
Rycroft RJG (1995) Occupational contact dermatitis. In: Rycroft RJG,
Menné T, Frosch PJ, & Benezra C ed. Textbook of contact dermatitis,
2nd ed. Berlin, Heidelberg, New York, Springer Verlag, pp 343-400.
Rycroft RJG, Menné T, Frosch PJ, & Benezra C ed. (1995) Textbook of
contact dermatitis, 2nd ed. Berlin, Heidelberg, New York, Springer
Verlag, 400 pp.
Rystedt I (1985) Atopic background in patients with occupational hand
eczema. Contact Dermatitis, 12(5): 247-254.
Saarinen UM & Kajosaari M (1995) Breastfeeding as prophylaxis against
atopic disease: Prospective follow-up study until 17 years old.
Lancet, 346(8982): 1065-1069.
Saihan EM, Burton JL, & Heaton KW (1978) A new syndrome with
pigmentation, scleroderma, gynaecomastia, Raynaud's phenomenon and
peripheral neuropathy. Br J Dermatol, 99: 437-440.
Saint-Remy J-MR (1997) Epitope mapping, a new method for biological
evaluation and immunotoxicology. Toxicology, 119: 77-81.
Sakaguchi S & Sakaguchi N (1989) Organ-specific autoimmune disease
induced in mice by elimination of T cell subsets: V. Neonatal
administration of cyclosporine A causes autoimmune disease. J Immunol,
142: 471-480.
Sakaguchi S & Sakaguchi N (1990) Thymus and autoimmunity: capacity of
the normal thymus to produce pathogenic self-reactive T cells and
conditions required for their induction of autoimmune disease. J Exp
Med, 172: 537-545.
Salome CM, Peat J., Britton WJ, & Woolcock A (1987) Bronchial
hyperresponsiveness in two populations of Australian schoolchildren:
I. Relation to respiratory symptoms and diagnosed asthma. Clin
Allergy, 17: 271-281.
Saltini C, Winestock K, Kirby M, Pinkston P, & Crysal RG (1989)
Maintenance of the alveolitis in patients with chronic beryllium
disease by beryllium-specific helper T-cells. N Engl J Med, 320:
1103-1109.
Samet JM, Lambert WE, Skipper BJ, Cushing AH, Hunt WC, Young SA,
McLaren LC, Schwab M, & Spengler JD (1993) Nitrogen dioxide and
respiratory illnesses in infants. Am Rev Respir Dis, 148: 1258-1265.
Sampson HA (1982) The immunopathogenic role of food hypersensitivity
in atopic dermatitis. Acta Dermatol Venereol, 176(suppl): 34-37.
Sampson HA (1983) Role of immediate food hypersensitivity in the
pathogenesis of atopic dermatitis. J Allergy Clin Immunol, 71:
473-480.
Sampson HA & Albergo R (1984) Comparison of results of skin tests,
RAST, and double-blind, placebo-controlled food challenges in children
with atopic dermatitis. J Allergy Clin Immunol, 74: 26-33.
Sampson HA & Metcalfe DD (1992) Food allergies. J Am Med Assoc,
268(20): 2840-2844.
Sanchez-Guerrero J, Schur PH, Sergent JS, & Liang MH (1994) Silicone
breast implants and rheumatic diseases. Arthritis Rheum, 37: 158-168.
Sanchez-Guerrero J, Colditz GA, Karlson EW, Hunter DJ, Speizer FE, &
Liang MH (1995) Silicone breast implants and the risk of
connective-tissue diseases and symptoms. N Engl J Med, 332(25):
1666-1670.
Sanderson CJ (1990) Eosinophil differentiation factor (interleukin-5).
Immunol Ser, 49: 231-256.
Sanderson DM & Earnshaw CG (1991) Computer prediction of possible
toxic action from chemical structure; The DEREK system. Hum Exp
Toxicol, 10: 261-273.
Sandford A, Shirakawa T, Moffatt MF, Daniels SE, Ra C, Faux JA, Young
RP, Nakamura Y, Lathrop GM, Cookson WOCM, & Hopkin JM (1993)
Localization of atopy and beta subunit of high-affinity IgE receptor
(Fc-epsilon-R1) on chromosome 11q. Lancet, 341: 332-334.
Sarlo K & Clark ED (1992) A tier approach for evaluating the
respiratory allergenicity of low molecular weight chemicals. Fundam
Appl Toxicol, 18: 107-114.
Sarlo K & Karol MH (1994) Guinea pig predictive tests for respiratory
allergy. In: Dean JH, Luster MI, Munson AE, & Kimber I ed.
Immunotoxicology and immunopharmacology, 2nd ed. New York, Raven
Press, pp 703-720.
Sarlo K, Ritz HL, Fletcher ER, Schrotel KR, & Clark ED (1997)
Proteolytic detergent enzymes enhance the allergic antibody responses
of guinea pigs to nonproteolytic detergent enzymes in a mixture:
implications for occupational exposure. J Allergy Clin Immunol, 100:
480-487.
Satoh T, Kramarik JA, Tollerud DJ, & Karol MH (1995) A murine model
for assessing the respiratory hypersensitivity potential of chemical
allergens. Toxicol Lett, 78: 57-66.
Saval P, Fuglsang G, Madsen C, & Osterballe O (1993) Prevalence of
atopic disease among Danish school children. J Pediatr Allergy
Immunol, 4: 117-122.
Savilahti E & Kuitunen M (1992) Allergenicity of cow milk proteins. J
Pediatr, 121: S12-S20.
Savonius B, Keskinen H, Tuppurainen M, & Kanerva L (1993) Occupational
respiratory disease caused by acrylates. Clin Exp Allergy, 23(5):
416-424.
Schachter J & Higgins MW (1976) Median age at onset of asthma and
allergic rhinitis in Tecumseh, Michigan. J Allergy Clin Immunol,
57(4): 342-351.
Schäfer T & Ring J (1997) Epidemiology of allergic diseases. Allergy,
52(suppl 38): 14-22.
Schäfer T, Przybilla B, Überla K, Pöschl C, & Ring J (1994) Frequency
of atopic diseases in children and parental predisposition -- results
of an epidemiological survey. ACI News, 2(suppl): 170.
Scheinbart LS, Johnson MA, Gross LA, Edelstein SR, & Richardson BC
(1991) Procainamide inhibits DNA methyltransferase in a human T cell
line. J Rheumatol, 18: 530-534.
Scheinman PL (1996) Allergic contact dermatitis to fragrances: A
review. Am J Contact Dermatitis, 7: 65-76.
Scheper RJ & von Blomberg BME (1992) Cellular mechanisms in allergic
contact dermatitis. In: Rycroft RJG, Menné T, Frosch PJ, & Benezra C
ed. Textbook of contact dermatitis. Berlin, Heidelberg, New York,
Springer Verlag, pp 11-27.
Scheper RJ, von Blomberg M, Boerrigter GH, Bruynzeel DP,
von Dinther-Janssen ACHM, & Vos A (1983) Induction of immunological
memory in the skin. Role of local T cell retention. Clin Exp Immunol,
51: 141-148.
Schleimer RP & Bochner BS (1998) The role of adhesion molecules in
allergic inflammation and their suitability as targets of antiallergic
therapy. Clin Exp Allergy, 28(suppl 39): 15-23.
Schlipköter HW, Krämer U, Behrendt H, Dolgner R, Stiller-Winkler R,
Ring J, & Willer HJ (1992) Impact of air pollution on children's
health: Results from Saxony-Anhalt and Saxony as compared to
Northrhine-Westphalia. In: Health and ecological effects -- Critical
issues in the global environment. Pittsburgh, Pennsylvania, Air and
Waste Management Association, vol 5 (Publication IU-A 2103).
Schnyder UW (1960) [Neurodermitis, asthma, rhinitis: A genetic
allergological study.] Basel, Karger, 106 pp.
Schon-Hegrad MA, Oliver J, McMenamin PG, & Holt PG (1991) Studies on
the density, distribution and surface phenotype of intraepithelial
class II MHC antigen (Ia)-bearing dendritic cells (DC) in the
conducting airways. J Exp Med, 173(6): 1345-1356.
Schönrich G, Kalinke U, Momburg F, Malissen M, Schmitt-Verhulst AM,
Malissen B, Hämmerling GJ, & Arnold B (1991) Down-regulation of T cell
receptors on self-reactive T cells as a novel mechanism for
extrathymic tolerance induction. Cell, 65: 293-304.
Schöpf E, Mueller JM, & Ostermann T (1995) [Significance of adjuvant
basis therapy in chronic relapsing skin disease.] Hautarzt, 44:
451-454 (in German).
Schuhmann D, Kubicka-Muranyi M, Mirtschewa J, Gunther J, Kind P, &
Gleichmann E (1990) Adverse reactions to gold: I. Chronic treatment
with a Au(I) drug sensitizes mouse T cells not to Au(I) but to Au(III)
and induces autoantibody formation. J Immunol, 145: 2132-2139.
Schulz O, Sutton BJ, Beavil RL, Shi J, Sewell HF, Gould HJ, Laing P, &
Shakib F (1997) Cleavage of the low-affinity receptor for human IgE
(CD23) by a mite cysteine protease: nature of the cleaved fragment in
relation to the structure and function of CD23. Eur J Immunol, 27:
584-588.
Schulz O, Sewell HF, & Shakib F (1998) Proteolytic cleavage of CD25,
the a subunit of the human T cell interleukin 2 receptor, by Der p 1,
a major mite allergen with cysteine protease activity. J Exp Med, 18:
271-275.
Schultz Larsen F (1991) Genetic aspects of atopic eczema. In: Ruzicka
T, Ring J, & Przybilla B ed. Handbook of atopic eczema. Berlin,
Heidelberg, New York, Springer Verlag, pp 15-26.
Schultz-Larsen F (1993) The epidemiology of atopic dermatitis. Monogr
Allergy, 31: 9-28.
Schultz Larsen F & Hanifin JM (1992) Secular change in the occurrence
of atopic dermatitis. Acta Dermatol Venereol, 176(suppl): 7-12.
Schultz-Larsen F, Holm NV, & Hennigsen K (1986) Atopic dermatitis: A
genetic-epidemiology study in a population-based twin sample. J Am
Assoc Dermatol, 15: 487-494.
Schultze-Werninghaus G, Roesch A, Wilhelms OH, Gonsior E, & Meir Sydow
J (1978) [Bronchial asthma due to occupational allergy of immediate
type (I) to platinum salts.] Dtsch Med Wochenschr, 103: 972-975 (in
German).
Schutz-Kiss D, Popp W, Wagner C, Reiser K, Havelec L, & Zwick H (1995)
[Sensitization to inhaled allergens in the Vienna population.] Wien
Klin Wochenschr, 107(11): 331-335 (in German).
Schuurman H-J, Van Loveren H, Rozing J, & Vos JG (1992) Chemicals
trophic for the thymus: risk for immunodeficiency and autoimmunity.
Int J Immunopharmac, 14: 369-375.
Schwartz J & Morris R (1995) Air pollution and hospital admissions for
cardiovascular disease in Detroit, Michigan. Am J Epidemiol, 142:
23-35.
Schwartz J & Weiss ST (1990) Dietary factors and their relation to
respiratory symptoms -- The second national health and nutrition
examination survey. Am J Epidemiol, 132(1): 67-76.
Schwartz HR, Nerurkar LS, Spies JR, Scanlon RT, & Bellanti JA (1980)
Milk hypersensitivity: RAST studies using new antigens generated by
pepsin hydrolysis of beta-lactoglobulin. Ann Allergy, 45: 242-245.
Scott P (1993) IL-12: Initiation cytokine for cell-mediated immunity.
Science, 260(5107): 496-497.
Seager J, Jamison DL, Wilson J, Hayward AR, & Soothill JF (1975) IgA
deficiency, epilepsy, and phenytoin treatment. Lancet, II: 632-635.
Sears MR, Herbison GP, Holdaway MD, Hewitt CJ, Flannery EM, & Silva PA
(1989) The relative risks of sensitivity to grass pollen, house dust
mite and cat dander in the development of childhood asthma. Clin Exp
Allergy, 19: 419-424.
Seaton A, Godden DJ, & Brown K (1994) Increase in asthma: a more toxic
environment or a more susceptible population? Thorax, 49: 171-174.
Seifert J, Ring J, & Brendel W (1974) Prolongation of skin allografts
after oral application of ALS in rats. Nature (Lond), 249(459): 776.
Seifert J, Ring J, Steininger J, & Brendel W (1977) Influence of the
immune response on the absorption of protein from the gut. Nutr Metab,
21(Suppl 1): 256-258.
Sell S (1987) Immune deficiency states. In: Immunology,
immunopathology, and immunity. Amsterdam, Oxford, New York, Elsevier
Science Publishers, 617 pp.
Senthilselvan A & Habbick BF (1995) Increased asthma hospitalizations
among registered Indian children and adults in Saskatchewan,
1970-1989. J Clin Epidemiol, 48(10): 1277-1283.
Sequeira J, Cesic D, Keser G, Bukelica M, Karanagnostis S, Khamashta
MA, & Hughes GRV (1993) Allergic disorders in systemic lupus
erythematosus. Lupus, 2: 187-192.
Serafini U (1997) Do infections protect against asthma and atopy?
Allergy, 52(9): 955-957.
Shaheen SO, Aaby P, Hall A, Barker DJP, Heyes CB, Shiell AW, &
Goudiaby A (1996) Measles and atopy in Guinea-Bissau. Lancet, 347:
1792-1796.
Shaw RA, Crane J, O'Donnell TV, Porteous LE, & Coleman ED (1990)
Increasing asthma prevalence in a rural New Zealand adolescent
population: 1975-89. Arch Dis Child, 65: 1319-1323.
Shaw RA, Crane J, & O'Donnell TV (1991) Asthma symptoms, bronchial
hyperresponsiveness and atopy in a Maori and European adolescent
population. NZ Med J, 104: 175-179.
Shaw RA, Crane J, O'Donnell TV, Lewis ME, Stewart B, & Beasley R
(1992a) The use of a videotaped questionnaire for studying asthma
prevalence: A pilot study among New Zealand adolescents. Med J Aust,
157(5): 311-314.
Shaw RA, Crane J, Pearce N, Burgess CD, Bremner P, Woodman K, &
Beasley R (1992b) Comparison of a video questionnaire with the IUATLD
written questionnaire for measuring asthma prevalence. Clin Exp
Allergy, 22: 561-568.
Shaw R, Woodman K, Crane J, Moyes C, Kennedy J, & Pearce N (1994) Risk
factors for asthma symptoms in Kawerau children. NZ Med J, 107:
387-391.
Shaw R, Woodman K, Ayson M, Dibdin S, Winkelmann R, Crane J, Beasley
R, & Pearce N (1995) Measuring the prevalence of bronchial
hyper-responsiveness in children. Int J Epidemiol, 24(3): 597-602.
Sherrill DL, Halonen M, & Burrows B (1994) Relationships between total
serum IgE, atopy, and smoking: a twenty-year follow-up analysis. J
Allergy Clin Immunol, 94: 954-962.
Shimizu H, Masunaga T, Ishiko A, Kikuchi A, Hashimoto T, & Nishikawa T
(1995) Pemphigus vulgaris and Pemphigus foilaceus sera show an
inversely graded pattern to extracellular regions of desmosomes in
different layers of human epidermis. J Invest Dermatol, 105: 153-159.
Shirakawa T, Enomoto T, Shimazu S, & Hopkin JM (1997) The inverse
association between tuberculin responses and atopic disorder. Science,
275: 77-79.
Sibbald B (1993) Epidemiology of allergic rhinitis. In: Burr ML ed.
Epidemiology of clinical allergy. Basel, Karger, pp 61-79 (Monograph
on Allergy Series).
Sibbald B (1997) Familial inheritance of asthma and allergy. In: Kay
AB ed. Allergy and allergic diseases. Oxford, London, Boston,
Blackwell Scientific Publications, pp 1177-1186.
Sibbald B & Rink E (1991a) Labelling of rhinitis and hayfever by
doctors. Thorax, 46: 378-381.
Sibbald B & Rink E (1991b) Epidemiology of seasonal and perennial
rhinitis: Clinical presentation and medical history. Thorax, 46:
895-901.
Sigurs N, Bjarnson R, Sigurbergsson F, Kjellman B, & Björkstén (1995)
Asthma and immunoglobulin E antibodies after respiratory syncitial
virus bronchiolitis: a prospective cohort study with matched controls.
Pediatrics, 95: 500-505.
Silman A & Hochberg MC (1996) Occupational and environmental
influences on scleroderma. Rheum Dis Clin North Am, 22(4): 737-749.
Silman A, Black CM, & Welsh KL (1996) Epidemiology, demographics,
genetics. In: Clements PJ & Furst DE ed. Systemic sclerosis.
Baltimore, Maryland, Williams and Wilkins, pp 23-49.
Silvennoinen-Kassinen S (1981) The specificity of nickel sulphate
reaction in vitro: A family study and a study of chromium-allergic
subjects. Scand J Immunol, 13: 231-235.
Silverman GA, Peri BA, & Rothberg RM (1982) Systemic antibody
responses of different species following ingestion of soluble protein
antigens. Dev Comp Immunol, 6: 747-752.
Singer PA & Theofilopoulos AN (1990) T-cell receptor Vb repertoire
expression in murine models of SLE. Immunol Rev, 118: 103-127.
Sinigaglia F (1994) The molecular basis of metal recognition by T
cells. J Invest Dermatol, 102: 398-401.
Smalley DL, Shanklin DR, Hall MF, Stevens MV, & Hanissian A (1995)
Immunologic stimulation of T lymphocytes by silica after use of
silicone mammary implants. FASEB J, 9: 424-427.
Smalley DL, Levine JJ, Shanklin DR, Hall MF, & Stevens MV (1997)
Lymphocyte response to silica among offspring of silicone breast
implant recipients. Immunobiology, 196: 567-574.
Smith IM (1983) Epidemiology and natural history of asthma, allergic
rhinitis and atopic dermatitis (eczema). In: Middleton E, Reed Cellis
EF, Adkinson NF, Junginger JW, & Busse WW ed. Allergy: Principles and
practice. St. Louis, Missouri, C.V. Mosby & Co., pp 771-804.
Song YH, Li Y, & Maclaren NK (1996) The nature of autoantigens
targeted in autoimmune endocrine diseases. Immunol Today, 17: 232-237.
Song Z, Casolaro V, Chen R, Georas SN, Monos D, & Ono SJ (1996)
Polymorphic nucleotides within the human IL-4 promoter that mediate
over expression of the gene. J Immunol, 156: 424-429.
Sonnhag C, Karlsson E, & Hed J (1979) Procainamide induced lupus
erythematosus like syndrome in relation to acetylator phenotype and
plasma levels of procainamide. Acta Med Scand, 206: 245-251.
Sparrow GP (1977) A connective tissue disorder similar to vinyl
chloride disease in a patient exposed to perchlorethylene. Clin Exp
Dermatol, 2: 17-22.
Speizer FE & Ferris BGJ (1973) Exposure to automobile exhaust: I.
Prevalence of respiratory symptoms and disease. Arch Environ Health,
26: 313-318.
Spier HW & Sixt I (1955) [Investigation of the dependence of eczema on
the thickness of the stratum corneum.] Hautarzt, 6: 152-159 (in
German).
Sporik R, Holgate ST, Platts Mills TA, & Cogswell JJ (1990) Exposure
to house-dust mite allergen (Der p I) and the development of asthma in
childhood: A prospective study. N Engl J Med, 323: 502-507.
Stadler JC & Karol MH (1985) Use of dose-response data to compare the
skin sensitizing abilities of dicyclohexylmethane-4,4'-diisocyanante
and picryl chloride in two animal species. Toxicol Appl Pharmacol, 78:
445-450.
Stadler JC & Loveless SE (1992) Guinea pigs exhibit extended latency
period for the development of sensitivity to an amine. Toxicologist,
12: 44 (abstract).
Stahl RE, Friedman-Kien A, Dubin R, Marmor M, & Zolla-Pazner S (1982)
Immunologic abnormalities in homosexual men. Am J Med, 73(2): 171-178.
Starzl TE, Nalesnik MA, Porter KA, Ho M, Iwatsuki S, Griffith BP,
Rosenthal JT, Hakala TR, Shaw BW Jr, Hardesty RL, Atchinson RW, Jaffe
R, & Bahnson HT (1984) Reversibility of lymphomas and
lymphoproliferative lesions developing under cyclosporin-steroid
therapy. Lancet, 8377: 583-587.
Steinman R, Hoffman L, & Pope M (1995) Maturation and migration of
cutaneous dendritic cells. J Invest Dermatol, 105: 2S-7S.
Sterk PJ, Fabbri LM, Quanjer PH, Cockcroft DW, O'Byrne PM, Anderson
SD, Juniper EF, & Malo JL (1993) Airway responsiveness: Standardized
challenge testing with pharmacological, physical and sensitizing
stimuli in adults. Report of Working Party on Standardization of Lung
Function Tests, European Community for Steel and Coal. Official
Statement of the European Respiratory Society. Eur Respir J,
16(suppl): 53-83.
Stewart GA (1994) Molecular biology of allergens. In: Busse WW &
Holgate ST ed. Asthma and rhinitis. Oxford, London, Boston, Blackwell
Scientific Publications, pp 898-932.
Stiller-Winkler R, Radaszkiewicz T, & Gleichmann E (1988)
Immunopathological signs in mice treated with mercury compounds-I.
Identification by the popliteal lymph node assay of responder and
non-responder strains. Int J Immunopharmacol, 10: 475-484.
Stites DP & Terr AI (1991) Basic and clinical immunology. London,
Appleton & Lange, p 797.
Stoddard JJ & Miller T (1995) Impact of parental smoking on the
prevalence of wheezing respiratory illness in children. Am J
Epidemiol, 141: 96-102.
Stokes CR, Newly JR, Huntley JH, Patel D, & Bourne FJ (1979) The
immune response of mice to bacterial antigens given by mouth.
Immunology, 38: 497-502.
Stokes CR, Newly TJ, & Bourne FJ (1983a) The influence of oral
immunization on local and systemic immune response to heterologous
antigens. Clin Exp Immunol, 52: 399-406.
Stokes CR, Swarbrick ET, & Soothill JF (1983b) Genetic differences in
immune exclusion and partial tolerance to ingested antigens. Clin Exp
Immunol, 52(3): 678-684.
Strachan DP (1988) Damp housing and childhood asthma: validation of
reporting of symptoms. Br Med J, 297: 1223-1226.
Strachan DP (1989) Hay fever, hygiene, and household size. Br Med J,
299: 1259-1260.
Strachan DP (1995) Epidemiology of hay fever: towards a community
diagnosis. Clin Exp Allergy, 25(4): 296-303.
Strachan DP (1996) Socioeconomic factors and the development of
allergy. Toxicol Lett, 86: 199-203.
Strachan DP & Anderson HR (1992) Trends in hospital admission rates
for asthma in children. Br Med J, 304: 819-820.
Strachan DP & Carey IM (1995) Home environment and severe asthma in
adolescents: a population based case-control study. Br Med J, 311:
1053-1056.
Strachan DP, Golding J, & Anderson HR (1990) Regional variations in
wheezing illness in British children: effect of migration during early
childhood. J Epidemiol Community Health, 44: 231-236.
Strachan DP, Cox BD, Erzinclioglu SW, Walters DE, & Whichelow MJ
(1991)Ventilatory function and winter fresh fruit consumption in a
random sample of British adults. Thorax, 46: 624-629.
Strachan DP, Anderson HR, Limb ES, O'Neill A, & Wells N (1994) A
national survey of asthma prevalence, severity, and treatment in Great
Britain. Arch Dis Child, 70: 174-178.
Strachan DP, Sibbald B, Weiland SK, Ait-Khaled N, Anabwani G, Anderson
HR, Asher MI, Beasley R, Bjorksten B, Burr M, Clayton T, Crane J,
Ellwood P, Keil U, Lai C, Mallol J, Martinez F, Mitchell E, Montefort
S, Pearce N, Robertson C, Shah J, Stewart A, von Mutius E, & Williams
H (1997) Worldwide variations in prevalence of symptoms of allergic
rhinoconjunctivitis in children: the international study of asthma and
allergies in childhood (ISAAC). Pediatr Allergy Immunol, 8: 161-176.
Strandberg I, Boman G, Hassler L, & Sjoqvist F (1976) Acetylator
phenotype in patients with hydralazine induced lupoid syndrome. Acta
Med Scand, 200: 367-371.
Strobel S & Ferguson A (1984) Immune responses to fed protein antigens
in mice: III. Systemic tolerance or priming is related to age at which
antigen is first encountered. Pediatr Res, 18: 588-594.
Suzuki T, Kanoh T, Kanbayashi M, Todome Y, & Ohkuni H (1993) The
adjuvant activity of pyrene in diesel exhaust on IgE antibody
production in mice. Jpn J Allergol, 42: 963-968.
Swennen B, Buchet JP, Stanescu D, Lison D, & Lauwerys R (1993)
Epidemiological survey of workers exposed to cobalt oxides, cobalt
salts, and cobalt metal. Br J Ind Med, 50(9): 835-842.
Szentivanyi A (1968) The beta adrenergic theory of the atopic
abnormality in asthma. J Allergy, 42: 203.
Takafuji S, Suzuki S, Koizumi K, Tadokoro K, Miyamoto T, Ikemori R, &
Muranaka M (1987) Diesel-exhaust particulates inoculated by the
intranasal route have an adjuvant activity for IgE production in mice.
J Allergy Clin Immunol, 79: 639-645.
Takafuji S, Suzuki S, Koizumi K, Tadokoro K, Ohashi H, Muranaka M, &
Miyamoto T (1989) Enhancing effect of suspended particulate matter on
the IgE antibody production in mice. Int Arch Allergy Appl Immunol,
90: 1-7.
Tanser AR, Bourke MP, & Blandford AG (1973) Isocyanate asthma:
respiratory symptoms caused by diphenylmethane di-isocyanate. Thorax,
28(5): 596-600.
Tariq SM, Stevens M, Matthews S, Ridout S, Twiselton R, & Hide DW
(1996) Cohort study of peanut and tree nut sensitisation by the age of
4 years. Br Med J, 313: 514-517.
Tarvainen K, Jolanki R, Estlander T, Tupasela O, Pfaffli P, & Kanerva
L (1995) Immunologic contact urticaria due to airborne
methylhexahydrophthalic and methyltetrahydrophthalic anhydrides.
Contact Dermatitis, 32(4): 204-209.
Taylor SL (1986) Immunologic and allergic properties of cow's milk
proteins in humans. J Food Protect, 49: 239-250.
Taylor SL (1992) Chemistry and detection of food allergens. Food
Technol, 46: 146-152.
Taylor JS & Praditsuwan P (1996) Latex allergy. Arch Dermatol, 132:
265-271.
Taylor B, Wadsworth J, Wadsworth M, & Peckham C (1984) Changes in the
reported prevalence of childhood eczema since the 1939-45 war. Lancet,
2: 1255-1257
Tee RD, Gordon DJ, Gordon S, Crook B, Nunn A, Musk AW, Venables KM, &
Taylor A (1992a) Immune response to flour and dust mites in a United
Kingdom bakery. Br J Ind Med, 49(8): 581-587.
Tee RD, Gordon DJ, van Hage Hamsten M, Gordon S, Nunn A, Johansson SG,
& Taylor A (1992b) Comparison of allergic responses to dust mites in
UK bakery workers and Swedish farmers. Clin Exp Allergy, 22(2):
233-239.
Teichberg S, Isolauri E, Wapnir RA, Roberts B, & Lifshitz F (1990)
Development of the neonatal rat small intestinal barrier to
nonspecific macromolecular absorption: Effect of early weaning to
artificial diets. Pediatr Res, 28(1): 31-37.
Tenenbaum SA, Rice JC, Espinoza LR, Cuéllar ML, Plymale DR, Sander DM,
Williamson LL, Haislip AM, Gluck OS, Tesser JRP, Nogy L, Stribrny Km,
Bevan JA, & Garry RF (1997) Use of antipolymer assay in recipients of
silicone breast implants. Lancet, 349: 449-454.
Terr AI (1994a) The atopic diseases. In: Stites DP, Terr AI, & Parslow
TG ed. Basic and clinical immunology, 8th ed. East Norwalk,
Connecticut, Appleton & Lange, pp 27-346.
Terr AI (1994b) Immune-complex allergic disease. In: Stites DP, Terr
AI, & Parslow TG ed. Basic and clinical immunology, 8th ed. East
Norwalk, Connecticut, Appleton & Lange, pp 357-362.
Tharp MD (1990) Immunodermatology. IgE and immediate hypersensitivity.
Dermatol Clin North Am, 8(4): 619-631.
Theofilopoulos AN (1995a) The basis of autoimmunity: Part I.
Mechanisms of aberrant self-recognition. Immunol Today, 16: 90-98.
Theofilopoulos AN (1995b) The basis of autoimmunity: Part II. Genetic
predisposition. Immunol Today, 16: 150-158.
Theofilopoulos AN & Dixon FJ (1985) Murine models of systemic lupus
erythematosus. Adv Immunol, 37: 269-390.
Thomas C, Groten J, Kammüller ME, De Bakker JM, Seinen W, & Bloksma N
(1989) Popliteal lymph node reactions in mice induced by the drug
zimeldine. Int J Immunopharmacol, 11: 693-702.
Thomas C, Lippe W, Seinen W, & Bloksma N (1991) Popliteal lymph node
enlargement and antibody production in the mouse induced by drugs
affecting monoamine levels in the brain. Int J Immunopharmacol, 13:
621-629.
Thompson HSG & Staines NA (1990) Could specific oral tolerance be a
therapy for autoimmune disease? Immunol Today, 11: 398-399.
Thorne PS & Karol MH (1989) Association of fever with late-onset
pulmonary hypersensitivity responses in the guinea pig. Toxicol Appl
Pharmacol, 100: 247-258.
Thorne PS, Hillebrand J, Magreni C, Riley EJ, & Karol MH (1986)
Experimental sensitization to subtilisin: I. Production of
immediate- and late-onset pulmonary reactions. Toxicol Appl Pharmacol,
86: 112-123.
Thorne PS, Hawk C, Kaliszewski SD, & Gurney PD (1991) The noninvasive
mouse ear swelling assay. I. Refinements for detecting weak contact
sensitizers. Fundam Appl Toxicol, 17: 790-806.
Thurston GD (1996) A critical review of PM10-mortality time-series
studies. J Expo Anal Environ Epidemiol, 6(1): 3-21.
Thurston GD, Ito K, Kinney PL, & Lippmann M (1992a) A multi-year study
of air pollution and respiratory hospital admissions in three New York
State metropolitan areas: Results for 1988 and 1989 summers. J Expo
Anal Environ Epidemiol, 2: 429-450.
Thurston G, D'Souza N, Lippmann M, Bartoszek M, & Fine I (1992b)
Associations between summer haze air pollution and asthma
exacerbations: A pilot camp study. Am Rev Respir Dis, 145: A429
(abstract).
Tisch R, Yang XD, Singer SM, Liblau LS, Fugger L, & McDevitt HO (1993)
Immune response to glutamic acid decarboxylase correlates with
insulitis in non-obese diabetic mice. Nature (Lond), 366: 72-75.
Tollerud DJ, Weiss ST, Elting E, Speizer FE, & Ferris B (1983) The
health effects of automobile exhaust. VI. Relationship of respiratory
symptoms and pulmonary function in tunnel and turnpike workers. Arch
Environ Health, 38: 334-340.
Tomasi TB, Barr WG, Challacombe SJ, & Curran G (1983) Oral tolerance
and accessory-cell function of Peyer's patches. Ann NY Acad Sci, 409:
145-163.
Tomura T & van Lancker JL (1988) Procainamide -- DNA interaction. J
Rheumatol, 15: 59-64.
Topping MD, Venables KM, Luczynska CM, Howe W, & Newman Taylor A
(1986) Specificity of the human IgE response to inhaled acid
anhydride. J Allergy Clin Immunol, 77: 834-842.
Toren K, Brisman J, & Jarvholm B (1993) Asthma and asthma-like
symptoms in adults assessed by questionnaires: A literature review.
Chest, 104(2): 600-608.
Tournade H, Pelletier L, Pasquier R, Vial MC, Mandet C, & Druet P
(1990) D-penicillamine- induced autoimmunity in Brown Norway rats:
Similarities with HgCl2-induced autoimmunity. J Immunol, 144:
2985-2991.
Tribe RM, Barton JR, Poston L, & Burney PG (1994) Dietary sodium
intake, airway responsiveness, and cellular sodium transport. Am J
Respir Crit Care Med, 149(6): 1426-1433.
Troisi RJ, Willett WC, Weiss ST, Trichopoulos D, Rosner B, & Speizer
FE (1995) A prospective study of diet and adult-onset asthma. Am J
Respir Crit Care Med, 151(5): 1401-1408.
Tsicopoulos A, Hamid Q, Varney V, Ying S, Moqbel R, Durham SR, & Kay
AB (1992) Preferential messenger RNA expression of Th1-type cells
(IFN-gamma+, IL2+) in classical delayed-type (tuberculin)
hypersensitivity reactions in human skin. J Immunol, 148(7):
2058-2061.
Tsunoda K, Ohta Y, Shinogami M, & Soda Y (1995) Does passive smoking
affect the incidence of nasal allergies? Am J Public Health, 85(7):
1019-1020.
Turjanmaa K (1987) Incidence of immediate allergy to latex gloves in
hospital personnel. Contact Dermatitis, 17(5): 270-275.
Turjanmaa K (1994) Allergy to natural rubber latex: a growing problem.
Ann Med, 26: 297-300.
Turjanmaa K, Alenius H, Mäkinen-Kiljunen S, Reunala T, & Palosuo T
(1996) Natural rubber latex allergy. Allergy, 51: 593-602.
Turner MW, Boulton P, Shields JG, Strobel S, Gibson S, Miller HRP, &
Levinsky RJ (1988) Intestinal hypersensitivity reactions in the rat:
I. Uptake of intact protein, permeability to sugars and their
correlation with mucosal mast-cell activation. Immunology, 63(1):
119-124.
Turner MW, Barnett GE, & Strobel S (1990) Mucosal mast cell activation
patterns in the rat following repeated feeding of antigen. Clin Exp
Allergy, 20(4): 421-427.
Uehara M (1991) Dry skin and inflammation. In: Ruzicka T, Ring J, &
Przybilla B ed. Handbook of atopic eczema. Berlin, Heidelberg, New
York, Springer Verlag, pp 84-89.
Uetrecht JP (1992) The role of leukocyte-generated reactive
metabolites in the pathogenesis of idiosyncratic drug reactions. Drug
Metab Rev, 24: 299-366.
Ulfvarson U & Alexandersson R (1990) Reduction in adverse effect on
pulmonary function after exposure to filtered diesel exhaust. Am J Ind
Med, 17: 341-347.
Utell MJ, Warren J, & Sawyer RF (1994) Public health risks from motor
vehicle emissions. Annu Rev Public Health, 15: 157-178.
Vandenplas O (1995) Occupational asthma caused by natural rubber
latex. Eur Respir J, 8: 1957-1965.
Vandenplas O, Delwiche JP, Evrard G, Aimont P, van der Brempt X,
Jamart J, & Delaunois L (1995) Prevalence of occupational asthma due
to latex among hospital personnel. Am J Respir Crit Care Med, 151(1):
54-60.
Van der Heijden FL, Van Neerven RJJ, Van Katwijk M, Bos JD, &
Kapsenberg ML (1993) Serum IgE-facilitated allergen presentation in
atopic disease. J Immunol, 150: 3643-3650.
Van der Valk P, van Kalken CK, Ketelaars H, Broxterman HJ, Kuiper CM,
Tsuruo T, Lankelma J, Meijer CJLM, Pinedo HM, & Scheper RJ (1990)
Distribution of multi-drug resistance-associated P-glycoprotein in
normal and neoplastic human tissues -- Analysis with 3 monoclonal
antibodies recognizing different epitopes of the P-Glycoprotein
molecule. Ann Oncol, 1: 56-64.
Van Hoogstraten IMW, Boden D, von Blomberg BME, Kraal G, & Scheper RJ
(1992) Persistent immune tolerance to nickel and chromium by oral
administration prior to cutaneous sensitization. J Invest Dermatol,
99: 608-616.
Van Hoogstraten IMW, Boos C, Boden D, von Blomberg BME, Scheper RJ, &
Kraal G (1993) Oral induction of tolerance to nickel sensitization in
mice. J Invest Dermatol, 101: 26-31.
Van Hoogstraten IMW, von Blomberg BME, Boden D, Kraal G, & Scheper RJ
(1994) Non-sensitizing percutaneous skin contacts prevent subsequent
induction of immune tolerance. J Invest Dermatol, 102: 80-83.
Van Lee Uwen BH, Martinsom ME, Webb GC, & Young IG (1989) Molecular
organization of the cytokine gene cluster, involving the human IL-3,
IL-4, IL-5, and GM-CSF genes on human chromosome 5. Blood, 73:
1142-1148.
Van Loveren H, Redegeld F, Matsuda H, Buckley T, Teppema JS, & Garssen
J (1997) Mast cells. In: Bos JD ed. Skin immune system (SIS), 2nd ed.
Boca Raton, Florida, CRC Press, pp 160-184.
Van Niekerk CH, Weinberg EG, Shore SC, Heese HV, & Van Schalkwyk J
(1979) Prevalence of asthma: A comparative study of urban and rural
Xhosa children. Clin Allergy, 9: 319-324.
Varjonen E, Kalimo K, Lammintausta K, & Terho P (1992) Prevalence of
atopic disorders among adolescents in Turku, Finland. Allergy, 47:
243-248.
Veien NK, Hattel T, Justese O, & Norholm A (1987) Dietary restrictions
in the treatment of adult patients with eczema. Contact Dermatitis,
17: 223-228.
Veien NK, Hattel T, & Laurberg G (1993) Low nickel diet: An open
prospective trial. J Am Acad Dermatol, 29: 1002-1007.
Veltman C, Lange CE, Juhe S, Stein G, & Bachner V (1975) Clinical
manifestations and course of vinyl chloride disease. Proc NY Acad Sci,
246: 6-17.
Venables KM & Chan Yeung M (1997) Occupational asthma. Lancet,
349(9063): 1465-1469.
Venables KM & Newman Taylor A (1990) Exposure-response relationships
in tetrachlorophthalic anhydride asthma. J Allergy Clin Immunol, 85:
55-58.
Venables KM, Dally MB, Burge PS, Pickering CAC, & Newman Taylor A
(1985a) Occupational asthma in a steel coating plant. Br J Ind Med,
42: 517-524.
Venables KM, Topping MD, Howe W, Luczynska CM, Hawkins R, & Newman
Taylor A (1985b) Interaction of smoking and atopy in producing
specific IgE antibody against a hapten protein conjugate. Br Med J,
290: 201-204.
Venables KM, Topping MD, Nunn A, Howe W, & Newman Taylor A (1987)
Immunologic and functional consequences of chemical
(tetrachlorophthalic anhydride) induced asthma after 4 years of
avoidance of exposure. J Allergy Clin Immunol, 80: 212-218.
Venables KM, Dally MB, Nunn A, Stevens JF, Stephens R, Farrer N,
Hunter JV, Stewart M, Hughes EG, & Newman Taylor A (1989) Smoking and
occupational allergy in a platinum refinery. Br Med J, 299: 939-942.
Verdier F, Virat M, & Descotes J (1990) Applicability of the popliteal
lymph node assay in the Brown-Norway rat. Immunopharmacol
Immunotoxicol, 12: 669-677.
Verdier F, Patriarca C, & Descotes J (1997) Autoantibodies in
conventional toxicity testing. Toxicology, 119: 51-58.
Verhoeff AP, van Strien RT, van Wijnen JH, & Brunekreef B (1995) Damp
housing and childhood respiratory symptoms: the role of sensitization
to dust mites and molds. Am J Epidemiol, 141: 103-110.
Vial T & Descotes J (1994) Contact sensitization assays in guinea
pigs: are they predictive of the potential for systemic allergic
reactions? Toxicology, 93: 63-75.
Vickers CFH (1991) Natural history of atopic eczema. In: Ruzicka T,
Ring J, & Przybilla B ed. Handbook of atopic eczema. Berlin,
Heidelberg, New York, Springer Verlag, pp 80-83.
Vieluf D, Przybilla B, Traenkner I, & Ring J (1990) Oral provocation
with food additives in atopic eczema. J Allergy Clin Immunol, 85: 206
(abstract).
Vieluf D, Kunz B, Bieber T, Przybilla B, & Ring J (1993) "Atopy patch
test" with aeroallergens in patients with atopic eczema. Allergo J, 2:
9-12
Vincent A, Roberts M, Willison H, Lang B, & Newsom-Davis J (1995)
Autoantibodies, neurotoxins and the nervous system. J Physiol, 89(3):
129-136.
Viner AS & Jackman N (1976) Retrospective survey of 1271 patients
diagnosed as perennial rhinitis. Clin Allergy, 6(3): 251-259.
Vivier E & Daeron M (1997) Immunoreceptor tyrosine-based inhibition
motifs. Immunol Today, 18: 286-291.
Von Blomberg BME, Bruynzeel DP, & Scheper RJ (1990) Advances in
mechanisms of allergic contact dermatitis: in vitro and in vivo
research. In: Marzulli FN & Maibach HI ed. Dermatotoxicology, 4th ed.
Washington, New York, London, Hemisphere Publishing Corporation, pp
255-362.
Von Mutius E, Fritzsch C, Weiland SK, Roll G, & Magnussen H (1992)
Prevalence of asthma and allergic disorders among children in united
Germany: A descriptive comparison. Br Med J, 305: 1395-1399.
Von Mutius E, Martinez FD, Fritzsch C, Nicolai T, Roell G, & Thiemann
HH (1994a) Prevalence of asthma and atopy in two areas of West and
East Germany. Am J Respir Crit Care Med, 149: 358-364.
Von Mutius E, Martinez FD, Fritzsch C, Nicolai T, Reitmeir P, &
Thiemann HH (1994b) Skin test reactivity and number of siblings. Br
Med J, 308: 692-695.
Von Mutius E, Weiland SK, Fritzsch C, Duhme H, & Keil U (1998)
Increasing prevalence of hay fever and atopy among children in
Leipzig, East Germany. Lancet, 351: 862-866.
Vos JG, Younes M, & Smith E ed. (1996) Allergic hypersensitivities
induced by chemicals: Recommendations for prevention. Boca Raton,
Florida, CRC Press, 348 pp.
Vreeburg KJJ, Wilkinson JD, & Scheper RJ (1991) Reduced frequency of
nickel allergy upon oral nickel contact at an early age. Clin Exp
Immunol, 85: 441-445.
Wade JF 3rd & Newman LS (1993) Diesel asthma: Reactive airways disease
following overexposure to locomotive exhaust. J Occup Med, 35(2):
149-154.
Wahlberg JE (1995) Patch testing. In: Rycroft RJG, Menné T, Frosch PJ,
& Benezra C ed. Textbook of contact dermatitis, 2nd ed. Berlin,
Heidelberg, New York, Springer Verlag, pp 241-265.
Wahlberg JE & Boman A (1985) Guinea pig maximisation test. Curr Probl
Dermatol, 14: 59-106.
Waite DA, Eyles EF, Tonkin SL, & O'Donnell TV (1980) Asthma prevalence
in Tokelauan children in two environments. Clin Allergy, 10: 71-75.
Walker RB & Warin RP (1956) The incidence of eczema in early
childhood. Br J Dermatol, 68: 182-183.
Walker FB, Smith PD, & Maibach HI (1967) Genetic factors in human
allergic contact dermatitis. Int Arch Allergy, 32: 453-462.
Walker C, Kaegi MK, Braun MD, & Blaser K (1991) Activated T cells and
eosinophilia in bronchoalveolar lavages from subjects with asthma
correlated with disease severity. J Allergy Clin Immunol, 88: 935-942.
Walker-Smith JA (1992) Immunology of gastrointestinal food allergy in
infancy and early childhood. In: MacDonald TT ed. Immunology of
gastrointestinal disease. Dodrecht, Kluwer Academic Publishers, pp
61-73.
Ward AM, Udnoon S, Watkins J, Walker AE, & Darke CS (1976)
Immunological mechanisms in the pathogenesis of vinyl chloride
disease. Br Med J, 1: 936-938.
Wardlaw A (1993) The role of air pollution in asthma. Clin Exp
Allergy, 23: 81-96.
Ware JH, Dockery DW, Spiro A 3rd, Speizer FE, & Ferris BG Jr (1984)
Passive smoking, gas cooking, and respiratory health of children
living in six cities. Am Rev Respir Dis, 129(3): 366-374.
Ware JH, Spengler JD, Neas LM, Samet JM, Wagner GR, Coultas D,
Ozkaynak H, & Schwab M (1993) Respiratory and irritant health effects
of ambient volatile organic compounds: The Kanawha County health
study. Am J Epidemiol, 137(12): 1287-1301.
Warren DL, Shiotsuka RN, Sangha GK, & Lyon JP (1993) Comparison of
respiratory sensitization responses to 2,4 and 2,6 toluene
diisocyanate. Toxicologist, 13: 41 (abstract).
Wass U & Belin L (1990) An in vitro method for predicting
sensitising properties of inhaled chemicals. Scand J Work Environ
Health, 16(3): 208-214.
Weeke ER (1987) Epidemiology of hay fever and perennial allergic
rhinitis. Monogr Allergy, 21: 1-20.
Weigle WO (1965) The production of thyroiditis and antibody following
injection of unaltered thyroglobulin into rabbits previously
stimulated with altered thyroglobulin. J Exp Med, 122: 1049-1062.
Weiland SK, Mundt KA, Rückmann A, & Keil U (1994) Self-reported
wheezing and allergic rhinitis in children and traffic density on
street of residence. Ann Epidemiol, 4: 243-247.
Weill H, Butcher B, & Charmarajan V (1981) Respiratory and immunologic
evaluation of isocyanate exposure in a new manufacturing plant.
Cincinnati, Ohio, National Institute of Occupational Safety and Health
(NIOSH Technical Report; DHHS (NIOSH) Publication No. 81-125).
Weinberg DA, Lesser RL, & Vollmer TL (1994) Ocular myasthenia: A
protean disorder. Surv Ophthalmol, 39: 169-210.
Weiner HL, Friedman A, Miller A, Khoury SJ, Al-Sabbagh A, Santos L,
Sayegh M, Nussenblatt RB, Trentham DE, & Hafler DA (1994) Oral
tolerance: Immunologic mechanisms and treatment of animal and human
organ-specific autoimmune diseases by oral administration of
autoantigens. Annu Rev Immunol, 12: 809-837.
Weinstein RS, Grogan TM, Kuszak JR, Jakate SM, Kluskens LF, & Coon JS
(1991) Multidrug resistance gene product (P-glycoprotein) in normal
tissue and tumors. St. Louis, Missouri, Mosby Year Book, Inc., pp
207-234 (Advances in Pathology and Laboratory Medicine Series).
Weiss KB, Wagener DK, Aberg N, Engstrom I, & Lindberg U (1989)
Geographic variations in US asthma mortality: Small-area analyses of
excess mortality, 1981-1985 -- Allergic diseases in Swedish school
children. Am J Epidemiol, 78(2): 246-252.
Weiss KB, Gergen PJ, & Wagener DK (1993) Breathing better or wheezing
worse? The changing epidemiology of asthma morbidity and mortality.
Annu Rev Public Health, 14: 491-513.
Weitzman M, Gortmaker SL, Sobol AM, & Perrin JM (1992) Recent trends
in the prevalence and severity of childhood asthma. J Am Med Assoc,
268: 2673-2677.
Weltzien HU, Moulon C, Martin S, Padovan E, Hartmann U, & Kohler J
(1996) T cell immune responses to haptens: Structural models for
allergic and autoimmune reactions. Toxicology, 107: 141-151.
White IR (1995) Phototoxic and photoallergic reactions. In: Rycroft
RJG, Menné T, Frosch PJ, & Benezra C ed. Textbook of contact
dermatitis. Berlin, Heidelberg, New York, Springer Verlag, pp 75-91.
WHO (in press) Air quality guidelines. Copenhagen, World Health
Organization, Regional Office for Europe.
Wick G, Sundick RS, & Albini B (1974) The obese strain (OS) of
chickens: An animal model with spontaneous autoimmune thyroiditis.
Clin Immunol Immunopathol, 3: 272-300.
Widmann FK (1989) An introduction to clinical immunology.
Philadelphia, Pennsylvania, F.A. Davis, 424 pp.
Wierenga EA, Smoek M, de Groot C, Chrétien I, Bos JD, Jansen HM, &
Kapsemberg ML (1990) Evidence for compartmentalization of functional
subsets of CD4+ T lymphocytes in atopic patients. J Immunol, 144:
4651-4656.
Wilkinson JD (1995) The management of contact dermatitis. In: Rycroft
RJG, Menné T, Frosch PJ, & Benezra C ed. Textbook of contact
dermatitis, 2nd ed. Berlin, Heidelberg, New York, Springer Verlag, pp
660-683.
Williams WJ (1989) Beryllium workers -- sarcoidosis or chronic
beryllium disease. Sarcoidosis, 6(suppl): 34-35.
Williams HC (1992) Is the prevalence of atopic dermatitis increasing?
Clin Exp Dermatol, 17: 385-391.
Williams IR & Unanue ER (1990) Costimulatory requirements of murine
Th1 clones: The role of accessory cell-derived signals in response to
anti-CD3 antibody. J Immunol, 145(1): 85-93.
Williams WR & Williams WJ (1982) Development of beryllium lymphocyte
transformation tests in chronic beryllium disease. Int Arch Allergy
Appl Immunol, 67: 175-180.
Williams ME, Lichtman AH, & Abbas AK (1990) Anti-CD3 antibody induces
unresponsiveness to IL-2 in Th1 clones but not in Th2 clones. J
Immunol, 144(4): 1208-1214.
Williams HC, Burney PG, Hay RJ, Archer CB, Shipley MJ, Hunter JJ,
Bingham EA, Finlay AY, Pembroke AC, Graham Brown RA, Atherton DA,
Lewis-Jones MS, Holden CA, Harper JI, Champion RH, Poyner TF, Launer
J, & David TJ (1994a) The UK Working Party's diagnostic criteria for
atopic dermatitis: I. Derivation of a minimum set of discriminators
for atopic dermatitis. Br J Dermatol, 131(3): 383-396.
Williams HC, Strachan DP, & Hay RJ (1994b) Childhood eczema: disease
of the advantaged? Br Med J, 308: 1132-1135.
Williams HC, Pembroke AC, Forsdyke H, Boodoo G, Hay RJ, & Burney PG
(1995a) London-born black Caribbean children are at increased risk of
atopic dermatitis. J Am Acad Dermatol, 32(2/1): 212-217.
Williams PB, Buhr MP, Weber RW, Volz MA, Koepke JW, & Selner JC
(1995b) Latex allergen in respirable particulate air pollution. J
Allergy Clin Immunol, 95(1/1): 88-95.
Witt C, Stuckey MS, Woolcock A, & Dawkins RL (1986) Positive allergy
prick tests associated with bronchial histamine responsiveness in an
unselected population. J Allergy Clin Immunol, 77: 698-702.
Wjst M, Reitmeir P, Dold S, Wulff A, Nicolai T, von Loeffelholz
Colberg EF, & von Mutius E (1993) Road traffic and adverse effects on
respiratory health in children. Br Med J, 307: 596-600.
Wjst M, Heinrich J, Liu P, Dold S, Wassmer G, Merkel G, Huelsse C, &
Watchman HE (1994) Indoor factors and IgE levels in children. Allergy,
49: 766-771.
Wolf R & Brenner S (1994) An active amide group in the molecule of
drugs that induce pemphigus: a casual or causal relationship?
Dermatology, 189: 1-4.
Wood RA, Mudd KE, & Eggleston PA (1992) The distribution of cat and
dust mite allergens on wall surfaces. J Allergy Clin Immunol, 89(1/1):
126-130.
Wucherpfennig KW, Yu B, Monos DS, Argyris E, Karr RW, Ahmed AR, &
Strominger JL (1995) Structural basis for major histocompatibility
complex (MHC)-linked susceptibility to autoimmunity: charged residues
of a single MHC binding pocket confer selective presentation of
self-peptides in pemphigus vulgaris. Proc Natl Acad Sci (USA), 92(25):
11935-11939.
Wüthrich B (1975) [Immunopathology of constitutional neurodermatitis.]
Bern, Göttingen, Toronto, Seattle, Hogrefe & Huber Publishers, 178 pp
(in German).
Wüthrich B (1990) [Do food allergies of Type III exist?] Allergologie,
13: 371 (in German).
Wüthrich B (1991) Minimal forms of atopic eczema. In: Ruzicka T, Ring
J, & Przybilla B ed. Handbook of atopic eczema. Berlin, Heidelberg,
New York, Springer Verlag, pp 46-53.
Wüthrich B (1993) [Food allergies: Frequency of symptoms and the
allergy-eliciting foods in 402 patients.] Allergologie, 16: 280-287
(in German).
Wüthrich B & Hofer T (1984) [Food allergy: The "celery-mugwort-spices
syndrome" association with mango allergy?] Dtsch Med Wochenschr,
109(250): 981-986 (in German).
Wüthrich B, Stäger J, & Johansson SGO (1990) Celery allergy associated
with birch and mugwort pollinosis. Allergy, 45(80): 566-571.
Wüthrich B, Schindler C, Leuenberger P, Ackermann-Liebrich U, &
SAPALDIA Team (1995) Prevalence of atopy and pollinosis in the adult
population of Switzerland (SAPALDIA-study). Int Arch Allergy Immunol,
106: 149.
Xu X & Wang L (1993) Association of indoor and outdoor particulate
level with chronic respiratory illness. Am Rev Respir Dis, 148(6/1):
1516-1522.
Yamakage A, Ishikawa H, Saito Y, & Hattori A (1980) Occupational
scleroderma-like disorder occurring in men engaged in the
polymerization of epoxy resins. Dermatologica, 161: 33-44.
Yamashita U, Takami T, Hamasaka T, & Kitigawa M (1976) The role of
hapten-reactive T-lymphocytes in the induction of autoimmunity in
mice: II. Termination of self tolerance to erythrocytes by
immunization with hapten-isologous erythrocytes. Cell Immunol, 25:
32-40.
Yamashita N, Natsuaki M, & Sagami S (1989) Flare-up reaction on murine
contact hypersensitivity: I. Description of an experimental model:
rechallenge system. Immunology, 67(3): 365-369.
Yokoyama Y, Nitta H, Maeda K, & Aoki S (1985) What interaction does
indoor nitrogen dioxide have on the effect of the automobile exhaust.
Tokai J Exp Clin Med, 10: 379-384.
Young RP, Barker R, Cookson WOCM, & Newman Taylor A (1993) HLA-DR and
DP antigen frequencies and acid anhydride sensitization. Am Rev Respir
Dis, 147(suppl): A113 (abstract).
Young E, Stoneham MD, Petruckevitch A, Barton J, & Rana R (1994) A
population study of food intolerance. Lancet, 343: 1127-1130.
Young RP, Barker RD, Pile KD, Cookson WOCM, & Newman Taylor A (1995)
The association of HLA-DR3 with specific IgE to inhaled acid
anhydrides. Am J Resp Crit Care Med, 151: 219-221.
Yssel H, Johnson KE, Schneider PV, Wideman J, Terr A, Kastelein R, &
de Vries JE (1992) T cell activation inducing epitopes of the house
dust mite allergen Der p I -- Induction of a restricted cytokine
production profile of Der p l-specific T cell clones upon
antigen-specific activation. J Immunol, 148: 738-745.
Yssel H, Fasler S, Lamb J, & de Vries JE (1994) Induction of
non-responsiveness in human allergen-specific type 2 T helper cells.
Curr Opin Immunol, 6: 847-852.
Yung RL, Quddus J, Crisp CE, Johnson KJ, & Richardson BC (1995)
Mechanisms of drug induced lupus: I. Cloned Th2 cells modified with
DNA methylation inhibitors in vitro cause autoimmunity in vivo. J
Immunol, 154: 3025-3035.
Zeiss CR, Patterson R, Pruzansky JJ, Miller MM, Resonberg M, & Levitz
D (1977) Trimellitic anhydride induced airways syndrome: Clinical and
immunologic studies. J Allergy Clin Immunol, 60: 96-103.
Zeiss CR, Kanellaks TM, Bellone TD, Levitz D, Pruzansky JJ, &
Patterson R (1980) Immunoglobulin E mediated asthma and
hyper-sensitivity pneumonitis with precipitating antihapten antibodies
due to diphenylmethane di-isocyanate (MDI) exposure. J Allergy Clin
Immunol, 65: 347-352.
Zinkernagel RM & Doherty PC (1975) H-2 compatibility requirement for
T-cell mediated lysis of target cells infected with lymphocytic
choriomeningitis virus -- Different cytotoxic T-cell specifities are
associated with structures coded for H-2K or H-2D. J Exp Med, 141(6):
1427-1436.
Zoia MC, Fanfulla F, Bruschi C, Basso O, De Marco R, Casali L, &
Cerveri I (1995) Chronic respiratory symptoms, bronchial
responsiveness and dietary sodium and potassium: a population-based
study. Monaldi Arch Chest Dis, 50(2): 104-108.
Zummo SM & Karol MH (1996) Indoor air pollution: Acute adverse health
effects and host susceptibility. Environ Health Sci, Jan/Feb: 25-29.
Zurawski G & de Vries JE (1994) Interleukin 13, an interleukin 4-like
cytokine that acts on monocytes and B cells, but not on T cells.
Immunol Today, 15(1): 19-26.
1. CONCLUSIONS
L'allergie constitue un problème de santé à l'échelle mondiale.
Elle touche une proportion importante de la population et toutes les
tranches d'âge. Pour des raisons mal connues, sa fréquence globale est
en augmentation.
La présente monographie fait le point des interactions diverses
et complexes entre produits chimiques, médicaments, système
immunitaire et organes cibles, interactions qui aboutissent à des
manifestations d'hypersensibilité allergique et à des troubles
d'origine autoimmune. D'importantes recherches sont menées dans tous
ces domaines et l'on peut donc s'attendre à de nouvelles découvertes.
Les études portent sur les facteurs endogènes et exogènes multiples
qui interviennent dans l'allergie. Les facteurs exogènes ont tous leur
importance, qu'il s'agisse des allergènes eux-mêmes ou encore des
maladies infectieuses, de la pollution de l'air et du mode de vie (par
exemple, le tabagisme). Par ailleurs, les prédispositions génétiques à
un trouble allergique donné déterminent en grande partie la réactivité
du sujet.
L'épidémiologie de l'allergie montre que cet ensemble
d'affections est très répandu et que, par conséquent, il importe
d'accorder l'attention voulue à l'identification du risque allergique
et à son évaluation pour être à même de mettre en oeuvre des
stratégies de prise en charge adaptées au risque.
Les méthodes d'identification des substances produisant une
sensibilisation cutanée sont bien connues. Elles restent à normaliser
dans le cas des sensibilisants respiratoires et font encore défaut
pour les autres types d'allergènes. On utilise les techniques de
mesure de l'activité des sensibilisants cutanés pour l'évaluation du
risque.
Une fois le risque allergique bien caractérisé, l'évaluation de
ce risque et sa prise en charge sont d'une importance primordiale pour
réduire l'incidence des troubles allergiques. Pour évaluer le risque,
il faut déterminer l'activité allergisante de l'agent en cause en
fonction de la nature et de l'intensité de l'exposition. Si
l'évaluation en fait ressortir la nécessité, il faudra alors prendre
en charge ce risque en mettant en oeuvre un certain nombre de mesures
consistant par exemple à limiter l'exposition et à étiqueter le
produit.
Les progrès accomplis dans la connaissance des caractéristiques
physicochimiques et immunologiques des allergènes alimentaires
pourraient déboucher sur la mise au point d'indicateurs prédictifs.
En raison de la complexité des mécanismes qui sont à la base des
troubles allergiques, il est encore difficile de proposer des tests
prédictifs in vitro qui soient d'une portée générale, mais
l'application des relation structure-activité mérite d'être étudiée de
manière plus approfondie.
2. RECOMMANDATIONS POUR LA PROTECTION DE LA SANTÉ HUMAINE
a) Il convient de mettre en oeuvre des stratégies efficaces
pour prévenir les allergies, fondées sur des informations fiables
relatives aux agents en cause et à l'environnement. L'action en
vue de prévenir ou de réduire au minimum la fréquence des
allergies doit reposer sur la limitation de l'exposition.
b) Elucider la cause de la fréquence accrue des allergie est
devenu d'une urgente nécessité.
c) Il faut mettre en oeuvre des méthodes de surveillance afin
de déterminer la fréquence des divers types d'allergie.
d) Il peut s'avérer difficile de mesurer l'exposition des
individus et des populations, mais une évaluation correcte est
néanmoins nécessaire à toute analyse de la relation
effet-exposition. Du fait de sa spécificité, la réponse
immunitaire constitue un biomarqueur unique en son genre pour
étudier les antécédents d'exposition, par exemple par apposition
d'un timbre cutané, par intradermoréaction ou par titrage des
anticorps IgE pour mettre en évidence une sensibilisation par
des allergènes déterminés.
e) Il faut améliorer les systèmes de surveillance sanitaire des
travailleurs, la qualité des visites médicales effectuées dans le
cadre de la médecine du travail ainsi que l'information de ceux
qui sont exposés à des produits chimiques, afin de mettre en
évidence le plus tôt possible toute maladie allergique d'origine
professionnelle. La surveillance de ces affections sur le lieu de
travail est particulièrement importante, du fait que l'exposition
y est vraisemblablement plus intense qu'ailleurs.
f) Il faut évaluer périodiquement l'efficacité et l'intérêt de
la prévention primaire et secondaire, de même que celle des
stratégies d'intervention en utilisant des techniques
d'évaluation épidémiologique dûment validées.
g) Dans le cas de la dermatite de contact d'origine allergique,
les modèles prédictifs in vivo dont on dispose à l'heure
actuelle ont fait la preuve de leur valeur pour les antigènes de
faible masse moléculaire relative. Il faut maintenant trouver le
moyen de les utiliser au mieux pour mesurer l'activité des
allergènes.
h) Dans le cas des troubles allergiques intéressant les voies
respiratoires, les tests in vivo relatifs aux substances de
faible masse moléculaire relative sont prometteurs, mais il faut
encore s'assurer de leur valeur prédictive et de leur
spécificité. Dans le cas des allergènes de nature protéique, on
est en train de mettre au point des méthodes d'épreuve, mais il
faudra encore les évaluer de manière plus approfondie sur des
substances dont on connaît le pouvoir allergénique chez l'Homme.
i) Il importe que les services de contrôle et les consommateurs
puissent s'informer facilement de la présence éventuelle
d'allergènes dans certains produits (par ex. les denrées
alimentaires) en consultant par exemple des bases de données ou
grâce à un étiquetage approprié, et soient de la sorte en mesure
de prendre les précautions voulues.
j) Le principal moyen dont on dispose pour éviter les
affections autoimmunes provoquées par des produits chimiques sur
le lieu de travail consiste à limiter l'exposition. Il convient
d'envisager un examen préalable des personnes qui vont être
exposées à des produits chimiques à action immunomodulatrice pour
rechercher tout signe de maladie affectant le tissu conjonctif.
Il est souhaitable que les consignes visant à éviter ou à réduire
l'exposition soient scrupuleusement suivies, notamment en ce qui
concerne les règles d'hygiène et sécurité. Il faut aussi réduire
au maximum les autres facteurs de risque, comme le tabagisme, et
envisager des contrôles médicaux réguliers dans le cadre de la
médecine du travail.
k) Il est nécessaire d'étudier les relations quantitatives
entre la réponse immunitaire suscitée par une exposition à des
produits chimiques et la gravité des réactions allergiques.
l) Il est également important de déterminer, pour lutter contre
ce type de maladies, quelles sont l'intensité et la durée
minimales d'exposition nécessaires pour provoquer une
sensibilisation ou déclencher une réaction allergique.
m) Il importe de mettre au point des stratégies normalisées
pour l'examen clinique et le diagnostic des allergies et de les
mettre en oeuvre à l'échelle internationale afin de déterminer
les causes et l'incidence des maladies d'origine allergique. A
cet effet, il faudra produire et mettre à disposition des
extraits normalisés d'allergènes biologiques d'activité contrôlée
et procéder périodiquement à l'examen des produits qui entreront
dans la composition des batteries normalisées d'allergènes
destinées aux divers tests.
n) Les autorités en charge de la santé publique, les membres
des professions de santé et les organismes publics devront
réfléchir aux moyens d'évaluer le coût humain et économique des
maladies allergiques pour l'individu et la société.
o) Les autorités en charge de la santé publique, les membres
des professions de santé, le public en général et les
travailleurs en particulier tireront avantage d'une meilleure
information sur la fréquence, les causes, les manifestations
cliniques et les conséquences des différents types d'allergie.
1. CONCLUSIONES
La alergia es un importante problema de salud de alcance mundial.
Afecta a una proporción sustancial de la población y a todos los
grupos de edad. Por razones poco conocidas, la frecuencia general de
la alergia está en aumento.
En esta monografía se examinan las interacciones diversas y
complejas entre productos químicos, medicamentos y proteínas, el
sistema inmunitario y el(los) órgano(s) diana que dan lugar a la
manifestación de la hipersensibilidad alérgica y la autoinmunidad.
Estos temas son objeto de una investigación extensa continua, de
manera que se prevén nuevos descubrimientos. Se examina la
multiplicidad de factores endógenos y exógenos que tienen
repercusiones sobre la alergia. Los factores exógenos, como los
alergenos mismos, las infecciones, la contaminación del aire y el modo
de vida (por ejemplo, el tabaquismo) son todos ellos importantes.
Además, la predisposición genética a un trastorno alérgico específico
es un determinante importante de la reactividad.
La epidemiología de la alergia demuestra el carácter extendido de
este grupo de trastornos y, como consecuencia, pone de relieve la
necesidad de prestar suficiente atención a la identificación de los
riesgos de alergia y la evaluación de éstos en la aplicación de
estrategias apropiadas de gestión de riesgos.
Hay métodos de reconocida eficacia para la determinación de
riesgos en relación con los sensibilizadores de la piel. Hace falta
normalizar los aplicables a los sensibilizadores a nivel respiratorio
y aún no se dispone de métodos aplicables a otros tipos de alergenos.
En la evaluación de riesgos se están aplicando técnicas de
determinación de la potencia de los sensibilizadores de la piel.
Una vez que los riesgos de alergia se han caracterizado bien, la
evaluación de riesgos, la gestión de riesgos y la comunicación sobre
los riesgos son elementos decisivos para reducir la incidencia de los
trastornos alérgicos. La evaluación de riesgos requiere que el riesgo,
de una potencia conocida, se evalúe atendiendo a la naturaleza y la
magnitud de la exposición. Cuando la evaluación de riesgos indica que
es necesario, se deben aplicar medidas de gestión de los riesgos,
tales como el control de la exposición y el etiquetado de productos.
Se están adqduiriendo más conocimientos sobre las características
fisicoquímicas e inmunológicas de alergenos alimentarios, que con el
tiempo podrán llegar a tener valor predictivo.
Dada la complejidad de los mecanismos de los trastornos
alérgicos, por el momento resulta difícil proponer métodos preditivos
in vitro de aplicabilidad general; no obstante, la aplicación de las
relaciones entre actividad y estructura merece mayor consideración.
2. RECOMENDACIONES PARA LA PROTECCION DE LA SALUD HUMANA
a) Para prevenir la alergia se deben aplicar estrategias
eficaces basadas en buena información acerca de los alergenos
presentes en los productos y el medio ambiente. El control de la
exposición debe ser la base de la prevención o la reducción al
mínimo de la aparición de enfermedades alérgicas.
b) Hay una necesidad urgente de determinar las causas del
aumento de la frecuencia de la alergia.
c) Se deben adoptar métodos de vigilancia para definir la
frecuencia de alergias de diferentes tipos.
d) Puede ser difícil medir la exposición de los individuos y
las poblaciones, pero una evaluación adecuada es esencial para
cualquier análisis de la asociación entre la exposición y el
efecto. La especificidad de las respuestas inmunitarias
representa un tipo único de marcador biológico a la hora de
estudiar la exposición pasada, por ejemplo mediante la
utilización de pruebas epicutáneas o de puntura, o la valoración
de los anticuerpos de IgE para detectar la sensibilización por
alergenos específicos.
e) Se deben mejorar los sistemas de vigilancia de los
- trabajadores, la calidad del examen médico de éstos y la
educación de los expuestos a productos químicos para detectar
enfermedades alérgicas ocupacionales en un estadio inicial. Los
sistemas relativamente sencillos de notificación de los
trastornos ocupacionales y la vigilancia posterior a la
comercialización de los medicamentos constituyen formas
económicas de reconocimiento y alerta en relación con las
enfermedades alérgicas. La vigilancia de estos trastornos en el
lugar de trabajo es particularmente valiosa porque es allí donde
la exposición probablemente sea mayor que en cualquier otro
lugar.
f) La eficacia y el valor de las estrategias de prevención
primaria y secundaria e intervención deben evaluarse a intervalos
que utilizan técnicas epidemiológicas validadas.
g) Con respecto a la dermatitis de contacto alérgica, los
modelos preditivos in vivo disponibles tienen un valor
comprobado para los antígenos de masa molecular relativa baja. Es
necesario descubrir la mejor manera de utilizarlos para
determinar la potencia de los alergenos.
h) Con respecto a los trastornos alérgicos de las vías
respiratorias, los métodos de prueba in vivo disponibles para
las sustancias de masa molecular relativa baja son prometedores,
pero es necesario comprobar su valor predictivo y su
especificidad. Para los alergenos proteicos, están
desarrollándose algunos métodos de prueba en animales, pero es
preciso someterlos a evaluaciones ulteriores utilizando
sustancias de potencial alergénico conocido en los seres humanos.
i) Es importante que la información acerca de la presencia de
alergenos en los productos (por ejemplo alimentos) esté
fácilmente disponible, por ejemplo en bases de datos y en las
etiquetas de los productos, para que las instancias reguladoras y
los individuos puedan tomar precauciones apropiadas.
j) El método principal para prevenir enfermedades
autoinmunitarias ocupacionales inducidas químicamente es el
control de la exposición. Se debe considerar la evaluación previa
a la exposición de los expuestos a los productos químicos con
potencial inmunomodelador para documentar cualquier
característica preexistente de morbilidad del tejido conectivo.
Se recomienda una adhesión estricta a las normas para evitar o
reducir al mínimo la exposición, inclusive la utilización de
buenas prácticas de higiene ocupacional. Se deben reducir al
mínimo otros factores de riesgo, como el consumo de tabaco y se
debe considerar la posibilidad de efectuar exámenes médicos
ocupacionales regulares.
k) Es necesario investigar las relaciones cuantitativas
existentes entre las respuestas inmunitarias inducido por los
productos químicos y la gravedad de las reacciones alérgicas.
l) Para controlar las enfermedades alérgicas es importante
determinar la exposición mínima y su duración necesaria para
causar sensibilización o producir una respuesta alérgica.
m) Es importante idear estrategias normalizadas de
investigación y diagnóstico clínicos de la alergia y aplicarlas
internacionalmente para examinar las causas y la incidencia de
los trastornos alérgicos. Ello requerirá la producción y la
disponibilidad de extractos normalizados de alergenos biológicos
de potencia controlada, así como el examen regular de los
componentes de series normalizadas de alergenos utilizados en las
pruebas.
n) Las autoridades de salud pública, los profesionales de la
salud y las dependencias gubernamentales deben examinar la manera
de calcular los costos humanos y económicos de las enfermedades
alérgicas para los individuos y la sociedad.
o) Las autoridades de salud pública, los profesionales de la
salud, el público y, especialmente, los trabajadores necesitan
mejor información acerca de la frecuencia, las causas, las
manifestaciones clínicas y las consecuencias de diferentes tipos
de alergia.