
UNITED NATIONS ENVIRONMENT PROGRAMME
INTERNATIONAL LABOUR ORGANISATION
WORLD HEALTH ORGANIZATION
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 196
Methanol
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Environmental Health Criteria 196
First draft prepared by Dr. L. Fishbein, Fairfax, Virginia, USA
Published under the joint sponsorship of the United Nations
Environment Programme, the International Labour Organisation, and the
World Health Organization, and produced within the framework of the
Inter-Organization Programme for the Sound Management of Chemicals.
World Health Organization
Geneva, 1997
The International Programme on Chemical Safety (IPCS) is a joint
venture of the United Nations Environment Programme, the International
Labour Organisation, and the World Health Organization. The main
objective of the IPCS is to carry out and disseminate evaluations of
the effects of chemicals on human health and the quality of the
environment. Supporting activities include the development of
epidemiological, experimental laboratory, and risk-assessment methods
that could produce internationally comparable results, and the
development of manpower in the field of toxicology. Other activities
carried out by the IPCS include the development of know-how for coping
with chemical accidents, coordination of laboratory testing and
epidemiological studies, and promotion of research on the mechanisms
of the biological action of chemicals.
WHO Library Cataloguing in Publication Data
Methanol.
(Environmental health criteria ; 196)
1.Alcohol, Methyl - toxicity 2.Alcohol, Methyl - adverse effects
3.Environmental exposure I.Series
ISBN 92 4 157196 9 (NLM Classification: QV 83)
ISSN 0250-863X
The World Health Organization welcomes requests for permission to
reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made to
the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1997
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved. The designations
employed and the presentation of the material in this publication do
not imply the expression of any opinion whatsoever on the part of the
Secretariat of the World Health Organization concerning the legal
status of any country, territory, city or area or of its authorities,
or concerning the delimitation of its frontiers or boundaries. The
mention of specific companies or of certain manufacturers' products
does not imply that they are endorsed or recommended by the World
Health Organization in preference to others of a similar nature that
are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR METHANOL
PREAMBLE
ABBREVIATIONS
1. SUMMARY
1.1. Identity, physical and chemical properties, analytical
methods
1.2. Sources of human exposure
1.3. Environmental levels and human exposure
1.4. Environmental distribution and transformation
1.5. Absorption, distribution, biotransformation and elimination
1.6. Effects on laboratory mammals and in vitro test systems
1.6.1. Systemic toxicity
1.6.2. Genotoxicity and carcinogenicity
1.6.3. Reproductive toxicity, embryotoxicity and
teratogenicity
1.7. Effects on humans
1.8. Effects on organisms in the environment
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL
METHODS
2.1. Identity
2.2. Physical and chemical properties
2.2.1. Physical properties
2.2.2. Chemical properties
2.3. Conversion factors
2.4. Analytical methods
2.4.1. Environmental samples
2.4.1.1 Methanol in air
2.4.1.2 Methanol in fuels
2.4.1.3 Methanol in fuel emissions
2.4.1.4 Methanol in sewage and aqueous solutions
2.4.1.5 Methanol in soils
2.4.2. Foods, beverages and consumer products
2.4.3. Biological materials
2.4.3.1 Methanol in exhaled air
2.4.3.2 Methanol in blood
2.4.3.3 Methanol in urine
2.4.3.4 Methanol in miscellaneous biological
tissues
2.4.3.5 Methanol metabolites in biological
fluids
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Anthropogenic sources
3.2.1. Production levels and processes
3.2.1.1 Production processes
3.2.1.2 Production figures
3.2.2. Uses
3.2.2.1 Use as feedstock for chemical syntheses
3.2.2.2 Use as fuel
3.2.2.3 Other uses
3.2.2.4 Losses into the environment
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1. Transport and distribution between media
4.2. Transformation
4.2.1. Biodegradation
4.2.1.1 Water and sewage sludge
4.2.1.2 Soils and sediments
4.2.2. Abiotic degradation
4.2.2.1 Water
4.2.2.2 Air
4.3.2. Bioconcentration
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Air
5.1.2. Water
5.1.3. Food
5.1.4. Tobacco smoke
5.2. Occupational exposure
5.3. General population
6. KINETICS AND METABOLISM IN LABORATORY ANIMALS AND HUMANS
6.1. Absorption
6.1.1. Inhalation
6.1.2. Oral
6.1.3. Dermal
6.2. Distribution
6.3. Metabolic transformation
6.4. Elimination and excretion
6.5. Modelling of pharmacokinetic and toxicokinetic data
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1. Single exposure
7.1.1. Non-primates
7.1.2. Non-human primates
7.2. Short-term exposure
7.2.1. Inhalation exposure
7.3. Long-term exposure
7.4. Skin and eye irritation; sensitization
7.5. Reproduction toxicity, embryotoxicity and teratogenicity
7.5.1. Reproductive toxicity (effects on fertility)
7.5.2. Developmental toxicity
7.5.3. Behavioural effects
7.5.4. In vitro studies
7.6. Mutagenicity and related end-points
7.6.1. In vitro studies
7.6.2. In vivo studies
7.7. Carcinogenicity
7.8. Special studies
7.8.1. Effects on hepatocytes
7.8.2. Toxic interactions
7.8.3. Studies with exhaust emissions from methanol-
fuelled engines
7.9. Mechanism of ocular toxicity
8. EFFECTS ON HUMANS
8.1. General population and occupational exposure
8.1.1. Acute toxicity
8.1.2. Clinical features of acute poisonings
8.1.3. Repeated or chronic exposure
8.1.4. Reproductive and developmental effects
8.1.5. Chromosomal and mutagenic effects
8.1.6. Carcinogenic effects
8.1.7. Sensitive sub-populations
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1. Aquatic organisms
9.1.1. Microorganisms
9.1.2. Algae
9.1.3. Aquatic invertebrates
9.1.4. Fish
9.2. Terrestrial organisms
9.2.1. Plants
10. EVALUATION OF EFFECTS ON HUMAN HEALTH AND THE ENVIRONMENT
10.1. Evaluation of human health risks
10.1.1. Exposure
10.1.2. Human health effects
10.1.3. Approaches to risk assessment
10.2. Evaluation of effects on the environment
11. RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH AND THE
ENVIRONMENT
11.1. Protection of human health
11.2. Protection of the environment
12. FURTHER RESEARCH
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
REFERENCES
RESUME
RESUMEN
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the criteria
monographs as accurately as possible without unduly delaying their
publication. In the interest of all users of the Environmental Health
Criteria monographs, readers are requested to communicate any errors
that may have occurred to the Director of the International Programme
on Chemical Safety, World Health Organization, Geneva, Switzerland, in
order that they may be included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Case postale
356, 1219 Châtelaine, Geneva, Switzerland (telephone no. + 41 22 -
9799111, fax no. + 41 22 - 7973460, E-mail irptc@unep.ch).
* * *
This publication was made possible by grant number 5 U01 ES02617-
15 from the National Institute of Environmental Health Sciences,
National Institutes of Health, USA, and by financial support from the
European Commission.
* * *
Financial support for this Task Group meeting was provided by the
United Kingdom Department of Health as part of its contributions to
the IPCS.
Environmental Health Criteria
PREAMBLE
Objectives
In 1973 the WHO Environmental Health Criteria Programme was
initiated with the following objectives:
(i) to assess information on the relationship between exposure to
environmental pollutants and human health, and to provide
guidelines for setting exposure limits;
(ii) to identify new or potential pollutants;
(iii) to identify gaps in knowledge concerning the health effects of
pollutants;
(iv) to promote the harmonization of toxicological and
epidemiological methods in order to have internationally
comparable results.
The first Environmental Health Criteria (EHC) monograph, on
mercury, was published in 1976 and since that time an ever-increasing
number of assessments of chemicals and of physical effects have been
produced. In addition, many EHC monographs have been devoted to
evaluating toxicological methodology, e.g., for genetic, neurotoxic,
teratogenic and nephrotoxic effects. Other publications have been
concerned with epidemiological guidelines, evaluation of short-term
tests for carcinogens, biomarkers, effects on the elderly and so
forth.
Since its inauguration the EHC Programme has widened its scope,
and the importance of environmental effects, in addition to health
effects, has been increasingly emphasized in the total evaluation of
chemicals.
The original impetus for the Programme came from World Health
Assembly resolutions and the recommendations of the 1972 UN Conference
on the Human Environment. Subsequently the work became an integral
part of the International Programme on Chemical Safety (IPCS), a
cooperative programme of UNEP, ILO and WHO. In this manner, with the
strong support of the new partners, the importance of occupational
health and environmental effects was fully recognized. The EHC
monographs have become widely established, used and recognized
throughout the world.
The recommendations of the 1992 UN Conference on Environment and
Development and the subsequent establishment of the Intergovernmental
Forum on Chemical Safety with the priorities for action in the six
programme areas of Chapter 19, Agenda 21, all lend further weight to
the need for EHC assessments of the risks of chemicals.
Scope
The criteria monographs are intended to provide critical reviews
on the effect on human health and the environment of chemicals and of
combinations of chemicals and physical and biological agents. As
such, they include and review studies that are of direct relevance for
the evaluation. However, they do not describe every study carried
out. Worldwide data are used and are quoted from original studies,
not from abstracts or reviews. Both published and unpublished reports
are considered and it is incumbent on the authors to assess all the
articles cited in the references. Preference is always given to
published data. Unpublished data are only used when relevant
published data are absent or when they are pivotal to the risk
assessment. A detailed policy statement is available that describes
the procedures used for unpublished proprietary data so that this
information can be used in the evaluation without compromising its
confidential nature (WHO (1990) Revised Guidelines for the Preparation
of Environmental Health Criteria Monographs. PCS/90.69, Geneva, World
Health Organization).
In the evaluation of human health risks, sound human data,
whenever available, are preferred to animal data. Animal and
in vitro studies provide support and are used mainly to supply
evidence missing from human studies. It is mandatory that research on
human subjects is conducted in full accord with ethical principles,
including the provisions of the Helsinki Declaration.
The EHC monographs are intended to assist national and
international authorities in making risk assessments and subsequent
risk management decisions. They represent a thorough evaluation of
risks and are not, in any sense, recommendations for regulation or
standard setting. These latter are the exclusive purview of national
and regional governments.
Content
The layout of EHC monographs for chemicals is outlined below.
* Summary - a review of the salient facts and the risk evaluation
of the chemical
* Identity - physical and chemical properties, analytical methods
* Sources of exposure
* Environmental transport, distribution and transformation
* Environmental levels and human exposure
* Kinetics and metabolism in laboratory animals and humans
* Effects on laboratory mammals and in vitro test systems
* Effects on humans
* Effects on other organisms in the laboratory and field
* Evaluation of human health risks and effects on the environment
* Conclusions and recommendations for protection of human health
and the environment
* Further research
* Previous evaluations by international bodies, e.g., IARC, JECFA,
JMPR
Selection of chemicals
Since the inception of the EHC Programme, the IPCS has organized
meetings of scientists to establish lists of priority chemicals for
subsequent evaluation. Such meetings have been held in: Ispra, Italy,
1980; Oxford, United Kingdom, 1984; Berlin, Germany, 1987; and North
Carolina, USA, 1995. The selection of chemicals has been based on the
following criteria: the existence of scientific evidence that the
substance presents a hazard to human health and/or the environment;
the possible use, persistence, accumulation or degradation of the
substance shows that there may be significant human or environmental
exposure; the size and nature of populations at risk (both human and
other species) and risks for environment; international concern, i.e.
the substance is of major interest to several countries; adequate data
on the hazards are available.
If an EHC monograph is proposed for a chemical not on the
priority list, the IPCS Secretariat consults with the Cooperating
Organizations and all the Participating Institutions before embarking
on the preparation of the monograph.
Procedures
The order of procedures that result in the publication of an EHC
monograph is shown in the flow chart. A designated staff member of
IPCS, responsible for the scientific quality of the document, serves
as Responsible Officer (RO). The IPCS Editor is responsible for
layout and language. The first draft, prepared by consultants or,
more usually, staff from an IPCS Participating Institution, is based
initially on data provided from the International Register of
Potentially Toxic Chemicals, and reference data bases such as Medline
and Toxline.
The draft document, when received by the RO, may require an
initial review by a small panel of experts to determine its scientific
quality and objectivity. Once the RO finds the document acceptable as
a first draft, it is distributed, in its unedited form, to well over
150 EHC contact points throughout the world who are asked to comment
on its completeness and accuracy and, where necessary, provide
additional material. The contact points, usually designated by
governments, may be Participating Institutions, IPCS Focal Points, or
individual scientists known for their particular expertise. Generally
some four months are allowed before the comments are considered by the
RO and author(s). A second draft incorporating comments received and
approved by the Director, IPCS, is then distributed to Task Group
members, who carry out the peer review, at least six weeks before
their meeting.
The Task Group members serve as individual scientists, not as
representatives of any organization, government or industry. Their
function is to evaluate the accuracy, significance and relevance of
the information in the document and to assess the health and
environmental risks from exposure to the chemical. A summary and
recommendations for further research and improved safety aspects are
also required. The composition of the Task Group is dictated by the
range of expertise required for the subject of the meeting and by the
need for a balanced geographical distribution.
The three cooperating organizations of the IPCS recognize
the important role played by nongovernmental organizations.
Representatives from relevant national and international associations
may be invited to join the Task Group as observers. While observers
may provide a valuable contribution to the process, they can only
speak at the invitation of the Chairperson. Observers do not
participate in the final evaluation of the chemical; this is the sole
responsibility of the Task Group members. When the Task Group
considers it to be appropriate, it may meet in camera.
All individuals who as authors, consultants or advisers
participate in the preparation of the EHC monograph must, in addition
to serving in their personal capacity as scientists, inform the RO if
at any time a conflict of interest, whether actual or potential, could
be perceived in their work. They are required to sign a conflict of
interest statement. Such a procedure ensures the transparency and
probity of the process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, it then goes for language editing, reference checking, and
preparation of camera-ready copy. After approval by the Director,
IPCS, the monograph is submitted to the WHO Office of Publications for
printing. At this time a copy of the final draft is sent to the
Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern for
health or environmental effects of the agent because of greater
exposure; an appreciable time period has elapsed since the last
evaluation.
All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
 WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR METHANOL
Members
Dr D. Anderson, British Industry Biological Research Association
(BIBRA) Toxicology International, Carshalton, Surrey, United
Kingdom
Dr S.A. Assimon, Contaminants Standards Monitoring and Projects
Branch, US Food and Drug Administration, Washington DC, USA
Dr H.B.S. Conacher, Bureau of Chemical Safety, Ottawa, Ontario,
Canada
Professor J. Eells, Department of Pharmacology and Toxicology,
Medical College of Wisconsin Milwaukee, USA (Chairman)
Mr J. Fawell, National Centre for Environmental Toxicology,
Marlow, Essex, United Kingdom
Dr L. Fishbein, Fairfax, Virginia, USA (Joint Rapporteur)
Dr K. McMartin, Department of Pharmacology and Therapeutics,
Louisiana State University Medical Center, Shreveport,
Louisiana, USA
Mr H. Malcolm, Institute of Terrestrial Ecology, Monks Wood,
Huntingdon, United Kingdom (Joint Rapporteur)
Dr H.B. Matthews, National Institute of Environmental Health
Sciences, Research Triangle Park, North Carolina, USA
Professor M. Piscator, Karolinska Institute, Stockholm, Sweden
(Vice-Chairman)
Dr G. Rosner, Merzhausen, Germany
Representatives of other Organizations
Professor K.R. Butterworth, BIBRA Toxicology International,
Carshalton, Surrey, United Kingdom (representing the
International Union of Toxicology)
Mr M.G. Penman, ICI Chemicals & Polymers Limited,
Middlesbrough, Cleveland, United Kingdom (representing the
European Centre for Ecotoxicology and Toxicology of
Chemicals)
Secretariat
Dr E. Smith, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland (Secretary)
Mr J.D. Wilbourn, Unit of Carcinogen Identification and
Evaluation, International Agency for Research on Cancer
(IARC), Lyon, France
ENVIRONMENTAL HEALTH CRITERIA FOR METHANOL
A WHO Task Group on Environmental Health Criteria for Methanol
met at the British Industrial Biological Research Association (BIBRA)
Toxicology International, Carshalton, Surrey, United Kingdom from 28
to 31 October 1996. Dr D. Anderson opened the meeting and welcomed
the participants on behalf of the host institute. Dr E. Smith, IPCS,
welcomed the participants on behalf of the Director, IPCS, and the
three IPCS cooperating organizations (UNEP/ILO/WHO). The Task Group
reviewed and revised the draft criteria monograph and made an
evaluation of the risks for human health and the environment from
exposure to methanol.
Dr L. Fishbein, Fairfax, Virginia, USA prepared the first draft
of this monograph. The second draft, incorporating comments received
following the circulation of the first draft to the IPCS Contact
Points for Environmental Health Criteria monographs, was also prepared
by Dr Fishbein.
Dr E.M. Smith and Dr P.G. Jenkins, both of the IPCS Central Unit,
were responsible for the overall scientific content and technical
editing, respectively.
The efforts of all who helped in the preparation and finalization
of the monograph are gratefully acknowledged.
ABBREVIATIONS
ATP adenosine triphosphate
BCF bioconcentration factor
BOD biochemical oxygen demand
COD chemical oxygen demand
CNS central nervous system
FID flame ionization detection
GC gas chromatography
MLD minimum lethal dose
MS mass spectrometry
MTBE methyl tertiary butyl ether
NAD nicotinamide adenine dinucleotide
NCAM neural cell adhesion molecule
NOAEL no-observed-adverse-effect level
THF tetrahydrofolate
TLV threshold limit value
1. SUMMARY
1.1 Identity, physical and chemical properties, analytical methods
Methanol is a clear, colourless, volatile flammable liquid with a
mild alcoholic odour when pure. It is miscible with water and many
organic solvents and forms many binary azeotropic mixtures.
Analytical methods, principally gas chromatography (GC) with
flame ionization detection (FID), are available for the determination
of methanol in various environmental media (air, water, soil and
sediments) and foods, as well as the determination of methanol and its
principal metabolite, formate, in body fluids and tissues. In addition
to GC-FID, enzymatic procedures with colorimetric end-points are
utilized for the determination of formate in blood, urine and tissues.
Determination of methanol in the workplace usually involves
collection and concentration on silica gel, followed by aqueous
extraction and GC-FID or GC-mass spectrometry analysis of the extract.
1.2 Sources of human exposure
Methanol occurs naturally in humans, animals and plants. It is a
natural constituent in blood, urine, saliva and expired air. A mean
urinary methanol level of 0.73 mg/litre (range 0.3-2.61 mg/litre) in
unexposed individuals and a range of 0.06 to 0.32 µg/litre in expired
air have been reported.
The two most important sources of background body burdens for
methanol and formate are diet and metabolic processes. Methanol is
available in the diet principally from fresh fruits and vegetables,
fruit juices (average 140 mg/litre, range 12 to 640 mg/litre),
fermented beverages (up to 1.5 g/litre) and diet foods (principally
soft drinks). The artificial sweetener aspartame is widely used and,
on hydrolysis, 10% (by weight) of the molecule is converted to free
methanol, which is available for absorption.
About 20 million tonnes of methanol were produced worldwide in
1991, principally by catalytic conversion of pressurized synthesis gas
(hydrogen, carbon dioxide and carbon monoxide). Worldwide capacity was
projected to rise to 30 million tonnes by 1995.
Methanol is used in the industrial production of many important
organic compounds, principally methyl tertiary butyl ether (MTBE),
formaldehyde, acetic acid, glycol methyl ethers, methylamine, methyl
halides and methyl methacrylate.
Methanol is a constituent of a large number of commercially
available solvents and consumer products including paints, shellacs,
varnishes, paint thinners, cleansing solutions, antifreeze solutions,
automotive windshield washer fluids and deicers, duplicating fluids,
denaturant for ethanol, and in hobby and craft adhesives. Potentially
large uses of methanol are in its direct use as a fuel, in gasoline
blends or as a gasoline extender. It should be noted that the highest
morbidity and mortality has been associated with deliberate or
accidental oral ingestion of methanol-containing mixtures.
Methanol has been identified in exhausts from both gasoline and
diesel engines and in tobacco smoke.
1.3 Environmental levels and human exposure
Emissions of methanol primarily occur from the miscellaneous
industrial and domestic solvent use, methanol production, end-product
manufacturing and bulk storage and handling losses.
Exposures to methanol can occur in occupational settings through
inhalation or dermal contact. Many national occupational health
exposure limits suggest that workers are protected from any adverse
effects if exposures do not exceed a time-weighted average of
260 mg/m3 (200 ppm) methanol for any 8-h day and for a 40-h working
week.
Current general population exposures through air are typically
10 000 times lower than occupational limits. The general population is
exposed to methanol in air at concentrations ranging from less than
0.001 mg/m3 (0.8 ppb) in rural air to nearly 0.04 mg/m3 (30 ppb) in
urban air.
Data on the occurrence of methanol in finished drinking-water is
limited, but methanol is frequently found in industrial effluents.
If the projected use of methanol as an alternate fuel or in
admixture with fuels increases significantly, it can be expected that
there will be widespread exposure to methanol via inhalation of
vapours from methanol-fuelled vehicles and/or siphoning or
percutaneous absorption of methanol fuels or blends.
1.4 Environmental distribution and transformation
Methanol is readily degraded in the environment by photo
oxidation and biodegradation processes. Half-lives of 7-18 days have
been reported for the atmospheric reaction of methanol with hydroxyl
radicals.
Many genera and strains of microorganisms are capable of using
methanol as a growth substrate. Methanol is readily degradable under
both aerobic and anaerobic conditions in a wide variety of
environmental media including fresh and salt water, sediments and
soils, ground water, aquifer material and industrial wastewater; 70%
of methanol in sewage systems is generally degraded within 5 days.
Methanol is a normal growth substrate for many soil
microorganisms, which are capable of completely degrading methanol to
carbon dioxide and water.
Methanol has a fairly low absorptive capacity on soils.
Bioconcentration in most organisms is low.
Methanol is of low toxicity to aquatic and terrestrial organisms,
and effects due to environmental exposure to methanol are unlikely to
be observed except in the case of a spill.
1.5 Absorption, distribution, biotransformation and elimination
Methanol is readily absorbed by inhalation, ingestion and dermal
exposure, and it is rapidly distributed to tissues according to the
distribution of body water. A small amount of methanol is excreted
unchanged by the lungs and kidneys.
Following ingestion, peak serum levels occur within 30-90 min,
and methanol is distributed throughout the body with a volume of
distribution of approximately 0.6 litre/kg.
Methanol is metabolized primarily in the liver by sequential
oxidative steps to formaldehyde, formic acid and carbon dioxide. The
initial step involves oxidation to formaldehyde by hepatic alcohol
dehydrogenase, which is a saturable rate-limiting process. The
relative affinity of alcohol dehydrogenase for ethanol and methanol is
approximately 20:1. In step 2, formaldehyde is oxidized by
formaldehyde dehydrogenase to formic acid/or formate depending on the
pH. In step 3, formic acid is detoxified to carbon dioxide by folate-
dependent reactions.
Elimination of methanol from the blood via the urine and exhaled
air and by metabolism appears to be slow in all species, especially
when compared to ethanol. Clearance proceeds with reported half-times
of 24 h or more with doses greater than 1 g/kg and half-times of
2.5-3 h for doses less than 0.1 g/kg. It is the rate of metabolic
detoxification, or removal of formate that is vastly different between
rodents and primates and is the basis for the dramatic differences in
methanol toxicity observed between rodents and primates.
1.6 Effects on laboratory mammals and in vitro test systems
1.6.1 Systemic toxicity
The acute and short-term toxicity of methanol varies greatly
between different species, toxicity being highest in species with a
relatively poor ability to metabolize formate. In such cases of poor
metabolism of formate, fatal methanol poisoning occurs as a result of
metabolic acidosis and neuronal toxicity, whereas, in animals that
readily metabolize formate, consequences of CNS depression (coma,
respiratory failure, etc.) are usually the cause of death. Sensitive
primate species (humans and monkeys) develop increased blood formate
concentrations following methanol exposure, while resistant rodents,
rabbits and dogs do not. Humans and non-human primates are uniquely
sensitive to the toxic effects of methanol. Overall methanol has a low
acute toxicity to non-primate animals. The LD50 values and minimal
lethal doses after oral exposure range from 7000 to 13 000 mg/kg in
the rat, mouse, rabbit and dog and from 2000 to 7000 mg/kg for the
monkey.
Rats exposed to levels of methanol up to 6500 mg/m3 (5000 ppm)
for 6 h/day, 5 days/week for 4 weeks, exhibited no exposure-related
effects except for increased discharges around the nose and eyes.
These were considered reflective of upper respiratory irritation.
Rats exposed to methanol vapour levels up to 13 000 mg/m3
(10 000 ppm) for 6 h/day, 5 days/week for 6 weeks, failed to
demonstrate pulmonary toxicity.
In the rabbit, methanol is a moderately irritant to the eye. It
was not skin-sensitizing in a modified maximization test.
Toxic effects found in methanol-exposed primates include
metabolic acidosis and ocular toxicity, effects that are not normally
found in folate-sufficient rodents. The differences in toxicity are
due to differences in the rate of metabolism of the methanol
metabolite formate. For instance, the clearance of formate from the
blood of exposed primates is at least 50% slower than for rodents.
Monkeys receiving methanol doses higher than 3000 mg/kg by gavage
demonstrated ataxia, weakness and lethargy within a few hours of
exposure. These signs tended to disappear within 24 h and were
followed by transient coma in some of the animals.
In monkeys exposed to methanol for 6 h/day for 5 days a week, 20
repeated exposures to 6500 mg/m3 (5000 ppm) methanol failed to elicit
ocular effects.
1.6.2 Genotoxicity and carcinogenicity
Methanol has given negative results for gene mutation in bacteria
and yeast assays, but it did induce chromosomal malsegregation in
Aspergillus. It did not induce sister chromatic exchanges in Chinese
hamster cells in vitro but caused significant increases in mutation
frequencies in L5178Y mouse lymphoma cells.
Methanol inhalation did not induce chromosomal damage in mice.
There is some evidence that oral or intraperitoneal administration
increased the incidence of chromosomal damage in mice.
There is no evidence from animal studies to suggest that methanol
is a carcinogen, although the lack of an appropriate animal model is
recognized.
1.6.3 Reproductive toxicity, embryotoxicity and teratogenicity
Conflicting results have been reported on the effects of
inhalation of methanol for up to six weeks on gonadotropin and
testosterone concentrations.
The inhalation of methanol by pregnant rodents throughout the
period of embryogenesis induces a wide range of concentration-
dependent teratogenic and embryolethal effects. Treatment-related
malformations, predominantly extra or rudimentary cervical ribs and
urinary or cardiovascular defects, were found in fetuses of rats
exposed 7 h/day for 7-15 days of gestation to 26 000 mg/m3
(20 000 ppm) methanol. Slight maternal toxicity was found at this
exposure level, and no adverse effects to the mother or offspring were
found in animals exposed to 6500 mg/m3 (5000 ppm), which was
interpreted as the no-observed-adverse-effect level (NOAEL) for this
test system.
Increased incidences of exencephaly and cleft palate were found
in the offspring of CD-1 mice exposed 7 h/day, on days 6-15 of
gestation, to methanol levels of 6500 mg/m3 (5000 ppm) or more. There
was increased embryo/fetal death at 9825 mg/m3 (7500 ppm) or more and
an increasing incidence of full-litter resorptions. Reduced fetal
weight was observed at 13 000 and 19 500 mg/m3 (10 000 or 15 000
ppm). The NOAEL for developmental toxicity was 1300 mg/m3 (1000 ppm)
methanol. There was no evidence of maternal toxicity at methanol
exposure levels below 9000 mg/m3 (7000 ppm).
When litters of pregnant CD-1 mice were given 4 g methanol/kg by
gavage, the incidences of adverse effects on resorption, external
defects including cleft palate, and fetal weight were similar to those
found in the 13 000 mg/m3 (10 000 ppm) inhalation exposure group,
presumably due to the greater rate of respiration of the mouse. The
mouse is more sensitive than the rat to developmental toxicity
resulting from inhaled methanol.
Transient neurological signs and reduced body weights were found
in CD-1 dams exposed to 19 500 mg/m3 (15 000 ppm) for 6 h/day
throughout organogenesis (gestational days 6-15). Fetal malformations
found at 13 000 and 19 500 mg/m3 (10 000 and 15 000 ppm) included
neural and ocular defects, cleft palate, hydronephrosis and limb
anomalies.
1.7 Effects on humans
Humans (and non-human primates) are uniquely sensitive to
methanol poisoning and the toxic effects in these species is
characterized by formic acidaemia, metabolic acidosis, ocular
toxicity, nervous system depression, blindness, coma and death. Nearly
all of the available information on methanol toxicity in humans
relates to the consequences of acute rather than chronic exposures. A
vast majority of poisonings involving methanol have occurred from
drinking adulterated beverages and from methanol-containing products.
Although ingestion dominates as the most frequent route of poisoning,
inhalation of high concentrations of methanol vapour and percutaneous
absorption of methanolic liquids are as effective as the oral route in
producing acute toxic effects. The most noted health consequence of
longer-term exposure to lower levels of methanol is a broad range of
ocular effects.
The toxic properties of methanol are based on factors that govern
both the conversion of methanol to formic acid and the subsequent
metabolism of formate to carbon dioxide in the folate pathway. The
toxicity is manifest if formate generation continues at a rate that
exceeds its rate of metabolism.
The lethal dose of methanol for humans is not known for certain.
The minimum lethal dose of methanol in the absence of medical
treatment is between 0.3 and 1 g/kg. The minimum dose causing
permanent visual defects is unknown.
The severity of the metabolic acidosis is variable and may not
correlate well with the amount of methanol ingested. The wide
interindividual variability of the toxic dose is a prominent feature
in acute methanol poisoning.
Two important determinants of human susceptibility to methanol
toxicity appear to be (1) concurrent ingestion of ethanol, which slows
the entrance of methanol into the metabolic pathway, and (2) hepatic
folate status, which governs the rate of formate detoxification.
The symptoms and signs of methanol poisoning, which may not
appear until after an asymptomatic period of about 12 to 24 h, include
visual disturbances, nausea, abdominal and muscle pain, dizziness,
weakness and disturbances of consciousness ranging from coma to clonic
seizures. Visual disturbances generally develop between 12 and 48 h
after methanol ingestion and range from mild photophobia and misty or
blurred vision to markedly reduced visual acuity and complete
blindness. In extreme cases death results. The principal clinical
feature is severe metabolic acidosis of the anion-gap type. The
acidosis is largely attributed to the formic acid produced when
methanol is metabolized.
The normal blood concentration of methanol from endogenous
sources is less than 0.5 mg/litre (0.02 mmol/litre), but dietary
sources may increase blood methanol levels. Generally, CNS effects
appear above blood methanol levels of 200 mg/litre (6 mmol/litre);
ocular symptoms appear above 500 mg/litre (16 mmol/litre), and
fatalities have occurred in untreated patients with initial methanol
levels in the range of 1500-2000 mg/litre (47-62 mmol/litre).
Acute inhalation of methanol vapour concentrations below
260 mg/m3 or ingestion of up to 20 mg methanol/kg by healthy or
moderately folate-deficient humans should not result in formate
accumulation above endogenous levels.
Visual disturbances of several types (blurring, constriction of
the visible field, changes in colour perception, and temporary or
permanent blindness) have been reported in workers who experienced
methanol air levels of about 1500 mg/m3 (1200 ppm) or more.
A widely used occupational exposure limit for methanol is
260 mg/m3 (200 ppm), which is designed to protect workers from any of
the effects of methanol-induced formic acid metabolic acidosis and
ocular and nervous system toxicity.
No other adverse effects of methanol have been reported in
humans except minor skin and eye irritation at exposures well above
260 mg/m3 (200 ppm).
1.8 Effects on organisms in the environment
LC50 values in aquatic organisms range from 1300 to
15 900 mg/litre for invertebrates (48-h and 96-h exposures), and
13 000 to 29 000 mg/litre for fish (96-h exposure).
Methanol is of low toxicity to aquatic organisms, and effects due
to environmental exposure to methanol are unlikely to be observed,
except in the case of a spill.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1 Identity
Chemical formula: CH3OH
Chemical structure: H
'
H - C - OH
'
H
Relative molecular mass: 32.04
CAS chemical name: methanol
CAS registry number: 67-56-1
RTECS number: PC 1400000
Synonyms: methyl alcohol, carbinol, wood
alcohol, wood spirits, wood
naphtha, Columbian spirits,
Manhattan spirits, colonial spirit,
hydroxymethane, methylol,
methylhydroxide,
monohydroxymethane, pyroxylic
spirit
Impurities in commercial methanol include acetone, acetaldehyde,
acetic acid and water.
2.2 Physical and chemical properties
2.2.1 Physical properties
Methanol is a colourless, volatile, flammable liquid with a mild
alcoholic odour when pure. However, the crude product may have a
repulsive pungent odour. Methanol is miscible with water, alcohols,
esters, ketones and most other solvents and forms many azeotropic
mixtures. It is only slightly soluble in fats and oils (Clayton &
Clayton, 1982; Windholz, 1983; Elvers et al., 1990).
Important physical constants and properties of methanol are
summarized in Table 1.
Table 1. Some physical properties of methanola
Appearance clear colourless liquid
Odour slight alcoholic when pure;
crude material pungent
Boiling point 64.7°C
Flash point 15.6°C (open cup)
12.2°C (closed cup)
Freezing point -97.68°C
Specific gravity 0.7915 (20/4°C)
0.7866 (25°C)
Vapour pressure
at 30°C 160 mmHg
at 20°C 92 mmHg
Henry's Law Constant (25°C) 1.35 x 10-4atm.m3/mole
Log P (octanol/water) -0.82; -0.77; -0.68
Partition constant -0.66; -0.64
Ignition temperature 470°C
Explosive limits in air lower 5.5
(% by volume) upper 44
Refractive index n20 1.3284
a Data from: Clayton & Clayton, 1982; Elvers et al., 1990;
Grayson, 1981; Howard, 1990; Windholz, 1983.
In the USA, sales grade methanol must normally meet the
following specifications:
methanol content (weight %) minimum 99.85
acetone and aldehydes (ppm) maximum 30
acid (as acetic acid) (ppm) maximum 30
water content (ppm) maximum 1.500
specific gravity (d2020) 0.7928
permanganate time, minimum 30
odour characteristic
distillation range at 101 kPa 1°C, must include
64.6°C
colour, platinum-cobalt scale, maximum 5
appearance clear-colourless
residual on evaporation, g/100 ml 0.001
carbonizable impurities, colour 30
platinum-cobalt scale, maximum 5
Grade AA differs in specifying an acetone maximum (20 ppm), a
minimum for ethanol (10 ppm), and in having a more stringent water
content specification (1.000 ppm, maximum) (Grayson, 1981).
2.2.2 Chemical properties
Methanol undergoes reactions that are typical of alcohols as a
chemical class. The reactions of particular industrial importance
include the following: dehydrogenation and oxidative dehydrogenation
over silver or molybdenum-iron oxide to form formaldehyde; the
acid-catalysed reaction with isobutylene to form methyl tertiary butyl
ether (MTBE); carbonylation to acetic acid catalysed by cobalt or
rhodium; esterification with organic acids and acid derivatives;
etherification; addition to unsaturated bonds and replacement of the
hydroxyl group (Grayson, 1981; Elvers et al., 1990).
2.3 Conversion factors
1 ppm = 1.31 mg/m3 (25°C, 1013hPa) 1 mmol/litre = 32 mg/litre
1 mg/m3 = 0.763 ppm (25°C, 1013hPa) 1 mg/litre =31.2 µmol/litre
(Adapted from Clayton & Clayton, 1982)
2.4 Analytical methods
Prior to the advent of sensitive gas chromatographic techniques,
the analysis of methanol in environmental, consumer and biological
samples was performed by procedures involving isolation of the
volatile alcohol and titrimetry. This was followed later by more
sensitive spectrophotometric methods based on the oxidation of
methanol to formaldehyde with potassium permanganate then reaction
with Schiff's reagent or rosaniline solution to produce an easily
recognizable and stable colour (Gettler, 1920; Boos, 1948; Skaug,
1956; Hindberg & Wieth, 1963; NIOSH, 1976).
The earliest procedures for the determination of methanol in
blood and urine were based on the initial distillation to isolate the
volatile alcohol (Gettler, 1920). Feldstein & Klendshog (1954)
determined methanol in biological fluids by initial microdiffusion
followed by oxidation to formaldehyde and subsequent reaction with
chromotropic acid (1,8-dihydroxy naphthalene-3,6-disulfonic acid). The
recovery ranged from 80 to 85% for less than 0.10 mg methanol. In the
procedure of Harger (1935), methanol was determined by oxidation with
bichromate to carbon dioxide and water followed by titration with a
mixture of ferrous sulfate and methyl orange. Jaselkis & Warriner
(1966) determined methanol in aqueous solution by titrimetry employing
xenon trioxide oxidation. Methanol was determined at a level of
0.03 mg with a relative standard deviation of 4%.
2.4.1 Environmental samples
The determination of methanol by primarily GC-FID procedures has
been frequently reported in ambient air, workplace air, fuels, fuel
emissions, sewage and aqueous solutions, soils, coal-gasification
condensate water and tobacco smoke.
The measurement of methanol in ambient and workplace air, usually
involves a preconcentration step in which the sample is passed through
a solid absorbent containing silica gel, Tenax GC, Porapak or
activated charcoal (NIOSH, 1976,1977,1984; CEC, 1988). It can also be
accomplished by on-column cryogenic trapping or can be analysed
directly. Direct reading infrared instruments with gas cuvettes can be
used for continuous monitoring of methanol in air (Lundberg, 1985).
2.4.1.1 Methanol in air
The use of absorption tubes to trap methanol from ambient and
workplace air with subsequent liquid or thermal desorption prior to
gas chromatographic analysis has been reported frequently. The US
National Institute of Occupational Safety and Health (NIOSH,
1977,1984) recommended the use of a glass tube (7 cm × 4 mm internal
diameter) containing two sections of 20-40 mesh silica gel separated
by a 2-mm portion of urethane foam (front=100 mg, back=50 mg). Water
is used to extract the methanol, which is separated on a 2 m × 2 mm
internal diameter glass column containing 60-80 mesh Tenax GC or the
equivalent using flame ionization detection (FID). The working range
is 25 to 900 mg/m3 (19 to 690 ppm) methanol for a 5-litre air sample.
The limit of detection has been reported to be 1.05 mg/m3 in a
3-litre air sample (NIOSH, 1976). At high concentrations of methanol
or at high relative humidity, a large silica gel tube is required
(700 mg silica gel front section). The injection, detector and column
temperatures are 200°C, 250-300°C and 80°C respectively. Positive
identification by mass spectrometry may be necessary in some cases,
and alternative gas chromatographic columns, e.g., SP-1000, SP-2100 or
FFAP, are also conformation aides.
Although GC-FID provides greater sensitivity than GC-MS, the
latter is generally considered more reliable for the measurement of
methanol in samples containing other alcohols or low molecular weight
oxygenates.Analysis of methanol in workplace air has been carried out
by head-space GC-FID using a column containing 15% Carbowax 1500 on
diatomaceous earth, 70-100 mesh operated at 100°C. The detection limit
was below 5 ml/m3 ( Heinrich & Angerer, 1982). Methanol in workplace
air was initially collected in silica gel tubes and the methanol
concentrations analysed by GC-FID equipped with a 50 m silica
capillary column containing Carbowax 20M. Additionally, methanol
vapour concentrations in the workplace have been analysed by a Miron-B
analyser with detection at a wavelength of 9.70 µm.
Methanol and other low molecular weight oxygenates have been
determined in ambient air by cryogradient sampling and two-dimensional
gas chromatography (Jonsson & Berg, 1983). Samples were initially
separated on a packed column (1,2,3-tris (2-cyanoethoxy)propane on
Chromosorb W-AW), then refocused on-line in a fused-silica capillary
cold trap, followed by on-line splitless reinjection onto a 50 m ×
0.3 mm internal diameter fused silica capillary column. The detection
limit for a typical oxygenate (3-methylbutanol) was 0.1 µg/m3 using a
3-litre sample. The detection limit for methanol was slightly higher.
Spectrophotometric methods have also been employed for the
determination of methanol in air. Aqueous potassium permanganate
acidified with phosphoric acid was used to absorb methanol from air
with the simultaneous oxidation to formaldehyde. After the addition of
p-aminoazobenzene and sulfur dioxide, the resulting pink dye was
determined spectrophotometrically at 505 nm. The limit of detection
was 5 µg methanol/ml air (Verma & Gupta, 1984).
Methanol from air was absorbed by acidified potassium
permanganate producing formaldehyde which on reaction with
4-nitroaniline produced a yellow dye determined spectroscopically at
395 nm (Upadhyay & Gupta, 1984).
Infrared spectrometry and infrared lasers have also been employed
for the determination of methanol in air (Diaz-Rueda et al., 1977;
Sweger & Travis, 1979). Methanol together with acetone, toluene and
ethyl acetate were recovered from 10 litres of air at a flow rate of
11 ml/min by passage through a tube containing 150 mg of activated
charcoal. The carbon disulfide extracts of the organic compounds were
determined by infrared at 1300 cm-1 using caesium bromide windows.
The minimum concentration of methanol detected quantitatively was
0.77 mg/m3 (0.60 ppm) and the minimum concentration required for
identification was 0.24 mg/m3 (0.18 ppm) (Diaz-Rueda et al., 1977).
Infrared lasers have been used to detect trace organic gases
including methanol. An air sample at 8 Tor was introduced to a
20-litre capacity sample cell, and laser radiation was detected
synchronously by a mercury-cadmium Te detector. The laser line
employed was P (34), the electric field was 1.40 kV/cm and the
measurement time was 2 min. The detection limit for methanol was
0.105 mg/m3 (0.08 ppm) (Sweger & Travis, 1979).
Methanol in the workplace can be measured by portable direct
reading instruments, real-time continuous monitoring systems and
passive dosimeters (NIOSH, 1976,1977,1984; Liesivouri & Savolainen,
1987; Kawai et al., 1990).
Kawai et al. (1990) described a personal diffusive badge type
that could absorb methanol vapour in linear relation to the exposure
duration up to 10 h and to exposure concentrations up to 1050 mg/m3
(800 ppm) the maximum duration and concentration tested respectively.
Additionally it was shown that the response to short-term peak
exposure was rapid enough and that no spontaneous desorption would
occur.
2.4.1.2 Methanol in fuels
Agarawal (1988) determined methanol quantitatively in commercial
gasoline via an initial extraction with ethylene glycol then by GC
utilizing a GB-1 fused silica capillary column (OV-1 equivalent, 60 m
× 0.32 mm internal diameter) and FID. The recovery of 4% methanol in
gasoline by this procedure was 95.4 ± 2.34% (SD).
In the procedure of Tackett (1987), gasoline samples were
injected directly on a Carbowax 20M column operated at 50°C for 3.0
min and then programmed to rise to 150°C at a rate of 10°C per min.
The calibration curve is linear up to 10% (v/v) methanol and the
detection limit was 0.2% employing a thermal conductivity detector.
Low molecular weight alcohols and MTBE were determined in
gasoline by GC-FID utilizing dual columns: 4.6 m × 3.2 mm o.d. column
packed with 30% m/m ethylene glycol succinate on Chromosorb P (85-100
mesh) and a 2.7 m × 3.2 mm o.d. stainless steel column packed with
Porapak P (80-100 mesh) operated at 150°C (Luke & Ray, 1984).
Gas chromatographic analyses of methanol, ethanol and tert-
butanol in gasoline have been reported by Pauls & McCoy (1981). The GC
column was 150 cm × 3 mm in o.d. stainless steel packed with Porapak R
(80-100 mesh) operated at 175°C and the injector and FID detector
temperatures were maintained at 250°C.
A direct liquid chromatographic method for the determination of
C1-C3 alcohols and water in gasoline-alcohol blends was described by
Zinbo (1984). The separation was performed on either one or two
microparticulate size-exclusion columns of ultrastyragel with toluene
as the mobile phase. The quantification of alcohols and water in the
effluent was achieved by a differential refractometer at 30°C. The
lower limits of detection for C1-C3 alcohols was 0.005 vol %. Methanol
in gasoline-alcohol blends has been determined by nuclear magnetic
resonance (Renzoni et al., 1985). The method takes advantage of a
window in the proton nuclear magnetic resonance spectrum of gasoline
that extends from a chemical shift of 2.8 to 6.8 ppm. Methanol was
quantified in gasoline by integration of the methyl singlet at
3.4 ppm. The method gave linear calibration curves in the range of
0-25% (v/v) methanol with a detection limit of less than 0.1%.
2.4.1.3 Methanol in fuel emissions
Methanol has been detected in motor vehicle emissions at levels
of 0.9 mg/m3 (0.69 ppm) and in ambient air by GC-FID utilizing a
360 cm × 0.27 cm internal diameter stainless steel column packed with
Porapak Q (50-80 mesh) operated at 150°C (Bellar & Sigsby, 1970).
Seizinger & Dimitriades (1972) determined methanol in simple
hydrocarbon fuel emissions utilizing GC with time-of-flight mass
spectrometry. The analytical procedure involved concentration of the
exhaust oxygenates drawn through a Chromosorb bed followed by GC-FID
initially on a 30 in by 1/4 in o.d. column packed with 10% 1,2,3-tris
(2-cyanoethoxy) propane (TCEP) programmed from -20°C to 110°C at
4°C/min. The second-stage column was a 45 m × 0.05 cm internal
diameter by 0.03 o.d Carbowax 20M support coated on tubular (SCOT)
column programmed from 60°C to 210°C at 10°C/min. The column effluent
was split for parallel detection with FID and mass spectrometry.
Methanol was found at levels of 0.1-0.8 mg/m3 (0.1-0.6 ppm) in
the exhaust of simple hydrocarbon fuels.
Methods for the quantification of evaporative emissions (running
losses, hot soak, diurnal and refuelling) from methanol-fuelled motor
vehicles (methanol/gasoline fuel mixtures of 100, 85, 50, 15 and 0%
methanol) have been described (Snow et al., 1989; Federal Register,
1989; Gabele & Knapp, 1993).
Methanol emissions from methanol-fuelled cars were determined by
GC employing a Quadrex 007 methyl silicone 50 m × 0.53 mm internal
diameter column with 5.0 µm film thickness. The separation was
affected isothermally at 75°C (limit of detection 0.25 µg/ml)
(Williams et al., 1990).
2.4.1.4 Methanol in sewage and aqueous solutions
Fox (1973) determined methanol at levels of 0.5-100 mg/litre
(0.5-100 ppm) in sewage or other aqueous solutions by GC-FID employing
a 0.5 m × 3.175 mm o.d. stainless steel column packed with Tenax GC
60/80 mesh and operated at 70°C isothermal.
C1-C4 alcohols in aqueous solution were determined
quantitatively by GC-FID using a 1 m × 0.32 cm stainless steel column
packed with 5% w/w Carbowax 20M on Chromosorb 101 (80-100 mesh) with a
column temperature of 65°C for methanol and ethanol and 100°C for n-
propanol and n-butanol (Sims, 1976).
Methanol and ethanol at the mg/litre level in aqueous solution
were determined by Komers & Sir (1976) utilizing a combination of
stripping and GC-FID technique. The alcohols were analysed as their
corresponding volatile nitrite on a 170 cm × 0.4 cm internal diameter
glass column containing Chromosorb 102 (80-120 mesh) operated at
104°C. Approximately 1 µg of the individual alcohol could be
determined in sample volumes of about 5 ml.
Mohr & King (1985) determined methanol in coal-gasification
condensate water by GC. Condensate water was injected directly on a 45
× 0.32 cm Porapak R column programmed from 80-200°C at 20°C/min.
A standard method for the analysis of methanol in raw, waste and
potable waters has been published by the UK Standing Committee of
Analysts (1982). The method is based on direct injection GC-FID using
a 2 m stainless steel column with 15% carbowax 1540 m chromosorb
W80-100 DMCS. The limit of detection is 0.11 mg/litre.
2.4.1.5 Methanol in soils
The biodegradation of methanol in gasolines by various soils was
determined by Novak et al. (1985). Methanol extracted in water (25%
v/v) was measured by direct injection GC-FID using a 2.1 m × 3 mm
stainless steel column packed with 0.2% Carbowax 1500 0n 80/100 mesh
Carbopak C at 120°C isothermal.
2.4.2 Foods, beverages and consumer products
Lund et al. (1981) determined methanol in orange and grapefruit
juice, fresh and canned, by GC-FID using a 1.5 × 3 mm column packed
with 50/80 mesh Porapak Q at 100°C with injector port and detector
block at 200°C.
Greizerstein (1981) utilized GC-FID and GC-MS for the analysis of
alcohols, aldehydes and esters in commercial beverages (beers, wines,
distilled spirits). Separations were carried out using a 3 m × 2 mm
internal diameter glass column packed with 30% Carbowax 20 M at 150°C.
A more satisfactory separation of methanol from the other congeners
was achieved using a 180-cm Porapak P column. Methanol was found at
levels of 6-27 mg/litre beer; 96-321 mg/litre in wines and
10-220 mg/litre in distilled spirits. Methanol in distilled liquors
and cordials has been determined by GC-FID (AOAC, 1990).
Rastogi (1993) analysed methanol content of 26 model and hobby
glues and found methanol in 12 of them by head-space GC-FID employing
capillary columns of different polarity. The polar GC column was a
Supelcowax 10, 60 m × 0.32 mm internal diameter; and the non-polar
column was a CP-Sil-5 CB, 50 m × 0.32 mm. The detection limit for
methanol was 20 mg/litre.
Methanol in wine vinegars was determined by GC-MS (Blanch et al.,
1992). Methanol with many other minor volatile components was
fractionated using a simultaneous distillation extraction technique
before GC analysis on a 4 m × 0.85 mm internal diameter micropacked
column coated with a mixture of Carbowax and bis-(2-ethylhexyl)-
sebecate (92:8), 4% on desilanized Volaspher A-2. The column
temperature was 60°C and the injector and FID detector were at 180°C.
2.4.3 Biological materials
A variety of primarily gas chromatographic methods have been
utilized for the determination of methanol in biological samples from
normal, poisoned and occupationally exposed individuals. Methanol
exposure has been measured in exhaled breath, blood and urine samples.
2.4.3.1 Methanol in exhaled air
Prior to analysis, expired air samples are normally collected in
sampling bags or glass containers or after preconcentration on Tenax
or other solid sorbents in adsorbent tubes and thermally desorbed, or
utilizing cryotraps (Franzblau et al., 1992a).
Free methanol has been detected and measured by GC in the expired
air of normal healthy humans with separations made on 1.52 m × 0.3 cm
columns filled with Anakrom ABS, 70-80 mesh coated with 2% N,N,-N,-N-
tetramethyl azeleamide and 8% behenyl alcohol at 86°C. The
concentration of methanol in nine subjects ranged from
0.06-0.32 µg/litre (Eriksen & Kulkarni, 1963). Methanol was only
infrequently detected in samples of human expired air and saliva by
Larsson (1965) employing GC-FID and a 1.75 mx 3.5 mm internal diameter
glass column containing polyethylene glycol (M=1500) 20% on Chromosorb
W.
Methanol in expired air and in head-space analysis of plasma was
determined as the nitrite ester utilizing GC-MS (Jones et al., 1983).
Condensed expired air samples were analysed on Porapak Q and the assay
of methanol nitrite ester was accomplished on a 2 m × 2 mm internal
diameter silanized glass column containing Tenax GC (30-60 mesh) at
60°C.
Krotosynski et al. (1977) analysed expired air from normal
healthy subjects using for sample preconcentration a 18 cm × 6 mm o.d.
stainless steel column containing Tenax GC. Sample analysis was
performed using GC-FID and a 91 m × 6 mm stainless steel column coated
with Emulphoron-870. Apart from methanol, 102 organic compounds were
detected.
Alveolar air of workers exposed to methanol was first collected
in gas sampling tubes and then analysed by GC-FID using a Porapak Q
(80-100 mesh) column at 150°C (Baumann & Angerer, 1979).
The detection of methanol and other endogenous compounds in
expired air by GC-FID with on-column concentration of sample and
separation on a 1.5 m × 3 mm o.d. stainless steel column packed with
Porapak Q, 80-100 mesh maintained at 35°C was described by Phillips &
Greenberg (1987).
The expired air of volunteer subjects exposed for periods of
about 90 min to atmospheres artificially contaminated with low levels
of methanol (ca. 130 mg/m3 (100 ppm)) was monitored during and
after the exposure using an atmospheric pressure ionization mass
spectrometer (API/MS) fitted with a direct breath analysis system
(Benoit et al., 1985).
A transportable Fourier Transform Infrared (FTIR) spectrometer
was utilized for the analysis of methanol vapour in alveolar and
ambient air in humans exposed to methanol vapour. The infrared
spectrum region used for methanol quantification was in the 950-1100
cm region. For the analysis of methanol in alveolar air with FTIR the
limit of detection for methanol was 0.4 mg/m3 (0.32 ppm), and for
methanol in ambient air the detection limit was 0.13 mg/m3 (0.1 ppm)
(Franzblau et al., 1992a).
2.4.3.2 Methanol in blood
A number of methods have been used to extract methanol from blood
prior to analysis including purge-and-trap, head-space analysis and
solvent extraction.
Baker et al. (1969) reported the simultaneous determination of
lower alcohols, acetone and acetaldehyde in blood by GC-FID utilizing
a 183 cm × 5 mm internal diameter column containing Porapak Q operated
at 100°C. The method did not require precipitation of protein prior to
analysis.
Methanol in whole blood and serum was analysed by GC-FID
employing 1.2 m and 1.8 m × 3 mm internal diameter glass columns
packed with 20% Hallcomid or 10% Carbowax on 60-80 mesh Diatopor TW
operated at 70°C (Mather & Assimos, 1965).
Blood serum was deproteinized and acetone and aliphatic alcohols
including methanol were determined by GC-FID using a pre-column of 3%
OV-1 on Gas Chrom Q and an analytical 30-m capillary column packed
with SPB-1 and operated at 35°C. Methanol and other alcohols were
separated in less than 3 min (Smith, 1984).
Methanol in deproteinized blood samples from occupationally
exposed workers was quantified by GC-FID employing a 1.8 m × 4 mm
internal diameter glass column packed with 60-80 mesh Carbopak B/5%
Carbowax 20M at 60°C. The detection limit for methanol was about
0.4 µg/ml (Lee et al., 1992).
Methanol in blood of occupationally exposed workers was
determined by head-space GC-FID utilizing a column containing 15%
Carbowax 1599 on diatomaceous earth, 70-80 mesh and operated at 70°C.
The detection limit was 0.6 mg/litre (Heinrich & Angerer, 1982).
The simultaneous determination of methanol, ethanol, acetone,
isopropanol and ethylene glycol in plasma by GC-FID was accomplished
using a 180 cm × 4 mm internal diameter glass column packed with
Porapak Q, 50-80 mesh. The column temperature was programmed from
199-210°C at 2°C/min, and the injection port and detector temperatures
were 210°C and 240°C respectively. The detection limit for methanol
was 0.1 nmol/ml. The procedure was recommended for methanol and
ethylene glycol intoxication cases (Cheung & Lin, 1987).
Methanol in blood from occupationally exposed workers was
determined directly without further pretreatment by GC-FID using a 4 m
× 3 mm glass column packed with 10% SBS 100 on Shimalite TPA, 60-80
mesh. The detector and oven were heated at 180°C and 60°C,
respectively (Kawai et al., 1991a).
Head-space GC-FID on methanol in blood from workers exposed at
sub-occupational exposure limits was reported by Kawai et al. (1992).
A 30 m × 0.53 mm capillary column coated with 1.0 um DB-Wax was used
with the injection port and detector heated at 200°C and the oven
temperature kept at 40°C for 1 min after the injection and then
elevated at a rate of 5°C/min to 110°C for 15 min. The detection limit
for methanol in blood was 100 µg/litre.
Leaf & Zatman (1952) utilized a colorimetric procedure for the
determination of methanol in air as well as in the blood and urine of
occupationally exposed workers in a methanol synthesis plant. The
procedure involved acid permanganate oxidation of methanol to
formaldehyde, which was then determined with a modified Schiff's
reagent. Concentrations of methanol up to 150 mg/litre were determined
to within 3%.
Determination of methanol in patients with acute methanol
poisoning was accomplished with a colorimetric procedure following
permanganate oxidation to formaldehyde and the subsequent reaction
with chromotropic acid (1,8-dihydroxy naphthalene 3,6-disulfonic
acid). Quantitative recovery of 100% was found for methanol following
the analysis of 3 ml of plasma, which required 45 min (Hindberg &
Wieth, 1963).
Accumulation of methanol in blood was detected in alcoholic
subjects during a 10-15 day period of chronic alcohol intake using
GC-FID and a 1.8 m column packed with Porapak Q, 80-100 mesh, or
Chromosorb 101 operated at 140°C (Majchrowicz & Mendelson, 1971). The
identity of methanol was also confirmed chemically using the
specificity of the colour reaction between permanganate and
formaldehyde.
Head-space GC was used to determine the concentrations of
methanol and ethanol in blood samples from 519 individuals suspected
of drinking and driving in Sweden. Methanol was determined in whole
blood without prior dilution with an internal standard. Carbopack C
(0.2% Carbowax 1500) was used as the stationary phase and the oven
temperature was 80°C (Jones & Lowinger, 1988).
Methanol in whole blood of poisoned patients was determined
without pretreatment by GC-FID using a 1800 mm × 4 mm internal
diameter glass column packed with 80-100 mesh Carbopack C/0.2% CW 1500
operated at 80°C; the detector temperature was 120°C (Jacobsen et al.,
1982a).
Serum methanol concentrations in men after oral administration of
the sweetening agent aspartame were determined by GC-MS utilizing a
fused silica capillary column 26 m × 0.22 mm internal diameter of
CPWAX 57 CB operated at 50°C isothermally (Davoli et al., 1986).
Methanol and formate in blood and urine of rats administered
methanol intravenously was determined by HPLC employing a REZEX-ROA-
organic acid column (300 mm × 7.8 mm internal diameter) and a
similarly packed pre-column (50 mm × 4.6 mm internal diameter). The
mobile phase was 0.043 N sulfuric acid with 10% acetonitrile at a flow
rate of 1 ml/min (Horton et al., 1992).
Methanol in serum has also been determined by high-field (500
MHZ) proton nuclear magnetic resonance at the 3.39 singlet peak. For
serum containing 20-500 mg of added methanol/litre, peak area was a
linear function of concentration (r=0.998). This NMR technique
permitted the determination of methanol and acetone in blood serum at
a level of less than 1mM (Bock, 1982).
Pollack & Kawagoe (1991) determined methanol in deproteinized
whole blood of rats by capillary GC-FID with direct column injection
utilizing a 15 m × 0.54 mm internal diameter fused silica capillary
column coated with Carbowax and operated at 35°C. The limit of
detection was 2 µg/ml.
2.4.3.3 Methanol in urine
Sedivec et al. (1981) determined methanol in urine in five
volunteers exposed to methanol vapour for 8 h. Head-space GC-FID was
used with a 120 cm × 3 mm column packed with Chromosorb 102, 60-80
mesh at 120°C. The detection limit of methanol was 0.1 mg/litre. The
methanol content in urine of 20 subjects occupationally exposed to
methanol was determined by head-space GC-FID utilizing a column
containing Porapak QS, 80-100 mesh and operated at 130°C. The
detection limit was 0.6 mg/litre (Heinrich & Angerer, 1982).
Methanol in the urine of exposed workers was determined by
head-space GC-FID using a 4.1 m × 3.2 mm glass column containing 10%
SBS-100 on Shimalite TPA, 60-80 mesh. The oven and injection port
temperatures were 60°C and 180°C respectively. The limit of detection
for methanol in urine was 0.1 mg/litre (Kawai et al., 1991b, 1992).
Urinary methanol as a measure of occupational exposure was
determined by GC-FID utilizing a 2 m glass column packed with Porapak
Q, 80-100 mesh. The detection limit for methanol was 0.32 mg/litre
(Liesivouri & Savolainen, 1987).
Urine concentrations of methanol in volunteers who had ingested
small amounts of methanol was determined by head-space GC-FID using
Tenax GC as the column packing (Ferry et al., 1980).
2.4.3.4 Methanol in miscellaneous biological tissues
Methanol and other alcohols have been determined in tissue
homogenates either per se or as their nitrite esters by GC-FID
employing a 1.8 m × 6 mm o.d. glass column packed with Chromosorb 101
operated at 145°C. The sensitivity was 8 µg per g of tissue (Gessner,
1970).
2.4.3.5 Methanol metabolites in biological fluids
The principal metabolite of methanol in humans and monkeys is
formate and it has been shown that accumulation of blood formate at
higher levels of methanol exposure coincides with the development of
metabolic acidosis and visual system toxicities (Clay et al., 1975;
McMartin et al., 1975; Baumbach et al., 1977; Tephly, 1991). Formate
is an endogenous product of single carbon metabolism and is normally
found in the urine of healthy individuals.
Formate has been analysed in blood and urine samples primarily by
enzymatic methods with a colorimetric or fluorimetric end-point or by
derivatization followed by analysis by GC-FID. Formate in plasma has
also been determined by isotachophoresis (Sejersted et al., 1983).
Ferry et al. (1980) measured formic acid as an ethyl ester formed
by the treatment of urine with 30% sulfuric acid in ethanol. The
samples were analysed by head-space GC-FID on a column packed with 10%
silar 10C on Chrom Q.
The analysis of formic acid in blood was performed via an initial
transformation of formic acid by concentrated sulfuric acid into water
and carbon monoxide, the latter being reduced to methane on a
catalytic column and analysed directly by GC-FID (Angerer & Lehnert,
1977; Baumann & Angerer, 1979; Heinrich & Angerer, 1982).
Urinary formic acid was determined after the methylation of the
acid and its conversion to N,N-dimethylformamide with GC-FID equipped
with a 50-m silica capillary column containing Carbowax 20M liquid
phase. The detection limit was 2.3 mg/litre (Liesivouri & Savolainen,
1987).
Franzblau et al. (1992b) found that urinary formic acid in
specimens collected 16 h following cessation of methanol exposure and
analysed by head-space GC-FID may not be an appropriate approach to
assess methanol exposure biologically.
Enzymatic methods for the determination of formate are based
primarily on the enzyme-catalysed conversion of formate to carbon
dioxide in the presence of nicotinamide adenine dinucleotide (NAD),
generating NADH as the other reaction product. NADH formation can be
subsequently measured directly or reacted in a coupled reaction to
generate a fluorescent or coloured complex.
A specific assay for formic acid in body fluids based on the
reaction of formate with bacterial formate dehydrogenase coupled to a
diaphorase-catalysed reduction of the non-fluorescent dye resazurin to
the fluorescent substance resorufin was reported by Makar et al.
(1975) and Makar & Tephly (1982). This permitted the accurate
determination of about 6 mg formate/litre blood at excitation
wavelength of 565 nm and an emission wavelength of 590 nm (Makar et
al., 1975; Makar & Tephly, 1982).
A serum formate enzymic assay based on modifications of the
formate dehydrogenase (FDH)-diaphorase procedure using NAD-diaphorase-
iodonitrotetrazolium violet to develop a red-coloured complex, which
is measured at 500 nm, was described by Grady & Osterloh (1986). The
calibration curve was linear over the formate range of 0 to
400 mg/litre.
Formate in plasma was determined by Lee et al. (1992) employing
an enzymatic procedure (Grady & Osterloh, 1986; Buttery & Chamberlin,
1988) and measured spectrophotometrically at 510 nm. The detection
limit was about 3 µg/ml.
Lee et al. (1992) determined that formate associated with acute
methanol toxicity in humans does not accumulate in blood when
atmospheric methanol exposure concentrations are below the
occupational threshold limit value of 260 mg/m3 (200 ppm) for 6 h in
exposed healthy volunteers.
d'Alessandro et al. (1994) found that serum and urine formate
determinations were not sensitive biological markers of methanol
exposure at the threshold limit value (TLV) in human volunteers.
Formate in serum was analysed by the enzymatic-colorimetric procedure
of Grady & Osterloh (1986). The sensitivity of the method was
0.5 mg/litre of formate in serum.
Buttery & Chamberlin (1988) developed an enzymatic method for the
determination of abnormal levels of formate in plasma requiring no
deproteinization and utilizing a stable colour reagent consisting of
phenazine methosulfate, p-iodonitrotetrazolium and NAD to produce a
stable red formazan colour. The precision at 1.0 and 5.0 mmol/litre
formate was 2.9% and 1.7%, respectively, within-day and 5.5% and 2.3%,
respectively, between days.
Urinary formic acid was determined using formate dehydrogenase
(FDH) in the presence of NAD. The detection limit was 0.5 mg/litre.
The normal formic acid excretion in urine is between 2.0 and
30 mg/litre (Triebig & Schaller, 1980).
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Methanol occurs naturally in humans, animals and plants (Axelrod
& Daly, 1965; CEC, 1988). It is a natural constituent of blood, urine
and saliva (Leaf & Zatman, 1952) and expired air (Erikssen & Kulkarni,
1963; Larsson, 1965; Krotosynski et al., 1979; Jones et al., 1990),
and has also been found in mother's milk (Pellizzari et al., 1982).
Humans have a background body burden of 0.5 mg/kg body weight (Kavet
& Nauss, 1990).
Levels of methanol in expired air are reported to range from 0.06
to 0.49 µg/litre (46-377 ppb) (Eriksen & Kulkarni, 1963). Methanol has
been detected in the expired air of normal, healthy non-smoking
subjects at a mean level of 0.5 ng/litre (Krotosynski et al., 1979).
It is believed that dietary sources are only partial contributors
to the total body pool of methanol (Stegink et al., 1981). It has been
suggested that methanol is formed by the activities of the intestinal
microflora or by other enzymatic processes (Axelrod & Daly, 1965). The
methanol-forming enzyme was shown to be protein carboxylmethylase, an
enzyme that methylates the carboxyl groups of proteins (Kim, 1973;
Morin & Liss, 1973).
Natural emission sources of methanol include volcanic gasses,
vegetation, microbes and insects (Owens et al., 1969; Holzer et al.,
1977; Graedel et al., 1986). Isidorov et al. (1985) identified
methanol emissions of evergreen cyprus in the forests of Northern
Europe and Asia. Methanol was identified as one of the volatile
components emitted by alfalfa (Owens et al., 1969) and it is formed
during biological decomposition of biological wastes, sewage and
sludges (US EPA, 1975; Howard, 1990; Nielsen et al., 1993).
3.2 Anthropogenic sources
The major anthropogenic sources of methanol include its
production, storage and use, principally its use as a solvent, as a
chemical intermediate, in the production of glycol ethers, and in the
manufacture of charcoal, and exhaust from vehicle engines (US EPA,
1976a,b, 1980a,b; CEC, 1988).
3.2.1 Production levels and processes
3.2.1.1 Production processes
The earliest important source of methanol ("wood alcohol") was
the dry distillation of wood at about 350°C, which was employed from
around 1830 to 1930. In countries where wood is plentiful and wood
products form an important industry, methanol is still obtained by
this procedure (ILO, 1983).
In 1880, about 1.5 million litres of wood alcohol were produced
in the USA while in 1910 the amount had increased to over 3 million
litres (Tyson & Schoenberg, 1914). However methanol produced from wood
contained more contaminants, primarily acetone, acetic acid and allyl
alcohol, than the chemical-grade methanol currently available
(Grayson, 1981; Elvers et al., 1990). Methanol was also produced as
one of the products of the non-catalytic oxidation of hydrocarbons (a
procedure discontinued in the USA in 1973), and as a by-product of
Fischer-Tropsch synthesis, which is no longer industrially important
(Grayson, 1981).
Modern industrial scale methanol production is based exclusively
on the catalytic conversion of pressurized synthesis gas (hydrogen,
carbon monoxide and carbon dioxide) in the presence of metallic
heterogenous catalysts. All carbonaceous materials such as coal, coke,
natural gas, petroleum and fractions obtained from petroleum (asphalt,
gasoline, gaseous compounds) can be employed as starting materials for
synthesis gas production (Grayson, 1981; Elvers et al., 1990).
The required synthesis pressure is dependant upon the activity of
the particular metallic catalyst employed, with copper-containing zinc
oxide-alumina catalysts being the most effective in industrial
methanol plants (Elvers et al., 1990). By convention the processes are
classified according to the pressure used: low-pressure processes,
50-100 atmospheres; medium-pressure processes, 100-250 atmospheres;
and high-pressure processes, 250-350 atmospheres. Low-pressure
technology is the most widely employed globally and accounted for 55%
of the USA methanol capacity in 1980 (Grayson, 1981).
Almost all the methanol produced in the USA is made from natural
gas. This is steam reformed to produce synthesis gas, which is
converted to methanol by low-pressure processes. A small amount of
methanol is obtained as a by-product from the oxidation of butane to
produce acetic acid and from the destructive distillation of wood to
produce charcoal (Grayson, 1981; Elvers et al., 1990).
The composition of methanol obtained directly from synthesis
without any purification or with only partial purification varies
according to the synthesis (e.g., pressure, catalyst, feedstock). The
principal impurities include 5-20% (by volume) water, higher alcohols
(principally ethanol), methyl formate and higher esters, and smaller
amounts of ethers and aldehydes (Grayson, 1981; Elvers et al., 1990).
Methanol is purified by distillation, the complexity required
depending on the desired methanol purity and the purity of the crude
methanol (Grayson, 1981; Elvers et al., 1990).
Natural gas, petroleum residues and naphtha accounted for 90% of
worldwide methanol capacity in 1980, miscellaneous off-gas sources
constituting the remaining 10%. Natural gas alone accounted for 70%,
petroleum residues 15%, and naphtha 5% (Grayson, 1981). Natural gas
feedstock accounted for 75% in the USA and 70% of global capacity in
1980. Methanol produced from residual oil accounted for approximately
15% of USA and worldwide capacity in 1980, while naphtha and coal
feedstocks accounted for approximately 5% and 2%, respectively, of
worldwide methanol capacity in 1980 (Grayson, 1981). About 90% of the
global methanol capacity is currently based on natural gas (SRI,
1992).
The production of methanol from coal, being independent of oil
and natural gas supplies, is noted to be an attractive alternative
feed stock in some quarters (Grayson, 1981; CEC, 1988). Newer
approaches to the production of methanol that have been suggested
include the catalytic conversion from carbon dioxide and hydrogen
avoiding conventional steam reforming (Rotman, 1994a) and the direct
catalytic conversion of methane to methanol (Rotman, 1994b).
3.2.1.2 Production figures
As shown in Table 2, worldwide annual capacity for methanol
production has increased over the past decades from approximately 15 ×
106 tonnes in 1979 (Grayson, 1981) to 21 × 106 tonnes in 1989
(Elvers et al., 1990) and more than 22.1 × 106 tonnes in the
beginning of 1991 (SRI, 1992). Worldwide demand was projected to rise
further to about 25.8 × 106 tonnes in 1994 (Anon., 1991; Nielsen et
al., 1993) and 30.1 × 106 tonnes in 1995 (SRI, 1992). The data
available do not allow capacity and production figures to be compared;
however, it is assumed that approximately 80% of production capacity
is utilized (Fiedler et al., 1990).
The USA and Canada are the largest methanol-producing countries.
About 85% of Canada's production is exported to the USA, Japan and
Europe (Heath, 1991). In Western Europe, Germany, the Netherlands and
the United Kingdom are the major methanol-producing countries,
accounting for 7%, 3% and over 2% of the world capacity, respectively
(SRI, 1992). The production of methanol in Germany in 1991 and 1992
amounted to 715 000 and 770 000 tonnes respectively.
The annual capacity in Eastern Europe was estimated to be 5.8 ×
106 tonnes in 1987. The production in the former USSR was 3.28 × 106
tonnes and 3.21 × 106 tonnes in 1987 and 1988, respectively (Rippen,
1990).
Table 2. Methanol production or production capacity (× 106 tonnes per year) from 1978 to 1995
Year World-wide USA Canada Western Japan Capacity/ Reference
Europe production
1978 12 3.4 3 1 capacity Grayson (1981)
production
1979 15 4.05 3.45 1.35 capacity Grayson (1981)
1980 2.5 production CEC (1988)
1981 8 production CEC (1988)
1983 15.9 5.52 (33%) 1.75 (11%) 2.53 1.27 (8%) capacity SRI (1992)
production CEC (1988)
1988 1.91 production Anderson (1993)
1989 21 capacity Elvers et al. (1990)
19 production
1990 22.3 capacity Anon. (1991);
Nielsen et al. (1993)
1991 22.1 4.42 (20%) 2.21 (10%) 2.65 (12%)a 0.22 (1%) capacity SRI (1992)
1991 2.22 0.077 production Anderson (1993)
1992 2.15 0.034 production Anderson (1993)
1992 3.66 2.15 production Reisch (1994)
1993 4.78 production Reisch (1994)
1995 30.1 capacity SRI (1992)
a Only Germany, the Netherlands and the United Kingdom.
The figures in Table 2 indicate a major shift in methanol
production from the developed countries to the developing areas. In
fact, the methanol industry underwent large structural changes during
the 1980s as a result of the discovery of large natural gas fields in
remote regions having little demand for natural gas themselves. Since
methanol production is a very suitable alternative for marketing
natural gases, a number of methanol production plants for export were
built or proposed to be built in Asia (Bahrein, Oman, Qatar, Saudi
Arabia, Indonesia, Malaysia), South America (Chile, Mexico,
Venezuela), the Caribbean (Trinidad) and in New Zealand and Norway
(Fiedler et al., 1990; SRI, 1992). The largest single train plant
based on this concept came on stream in southern Chile in 1988 with an
annual output of 750 000 tonnes (Fiedler et al., 1990).
Future trends in methanol production and demand are being driven
to a large extent by increasing demand for methyl tertiary butyl ether
(MTBE), which is used in gasoline blending as an octane enhancer and
to reduce carbon monoxide emissions (Anon., 1991; Morris, 1993;
Nielsen et al., 1993).
3.2.2 Uses
During the 1890s, the market for methanol (then better known as
wood alcohol) increased as a commercial product and as a solvent for
use in the workplace. It was included in many consumer products such
as witch hazel, Jamaica ginger, vanilla extract and perfumes (Wood &
Buller, 1904). The most notorious use of wood alcohol was and
continues to be as an adulterant in alcoholic beverages, which has led
to large-scale episodes of poisonings since 1900 (Bennett et al.,
1953; Kane et al., 1968).
Historically, in terms of commercial usage, about half of all
methanol produced has been used to produce formaldehyde. Other earlier
large-volume chemicals based on methanol include acetic acid, dimethyl
terephthalate, glycol methyl ethers, methyl halides, methylamines,
methyl acrylate and various solvent uses (Grayson, 1981; CEC, 1988;
Elvers et al., 1990; Nielsen et al., 1993).
3.2.2.1 Use as feedstock for chemical syntheses
Approximately 70% of the methanol produced worldwide is used as
feedstock for chemical syntheses. As shown in Table 3, formaldehyde,
methyl tertiary butyl ether (MTBE), acetic acid, methyl methacrylate,
and dimethyl terephthalate are, in order of importance, the main
chemicals produced from methanol. Methyl halides produced from
methanol include methyl chloride, methylene chloride and chloroform.
Nearly all the formaldehyde manufactured worldwide is produced by
oxidation of methanol with atmospheric oxygen. The annual formaldehyde
production was projected to increase at a rate of 3%, but because
other bulk products have higher growth rates, its relative importance
with respect to methanol use has decreased (Elvers et al., 1990;
Fiedler et al., 1990).
Table 3. Use pattern for methanol (as a percentage of production) according to region and year
Global Global USA USA Japan Western Europe Brazil
1979 1988 1973 1985 n.g. 1985 n.g.
Use for synthesis of:
formaldehyde 52 40 39 30 47 50 60
MTBE 4 20 8 - 5 -
acetic acid 6 9 3.4 12 10 5 -
dimethyl terephthalate 4 6.1 4 1 4 16
methyl methacrylate 4 3.7 4 6 3 2
methyl halides 8a 6.1 9 3 6 -
methyl amines 3.3 4 2 4 9
glycol methyl ethers 1.1
Direct use
solvent 10 6 6 2
fuel 6 - 5 -
Miscellaneous 14 16.9 13 25 12 11
Referenceb [1] [2] [3] [4] [4] [4] [4]
a together with methyl amines production
b Reference: [1] Kennedy & Shanks (1981); [2] Elvers et al. (1990); [3] US EPA (1980a); [4] Rippen (1990)
n.g. = year not given
MTBE has become an important octane-enhancing blending component
in gasoline, particularly in the USA where the Clean Air Act
Amendments of 1990 have prompted further steps toward reducing
emissions from motor vehicles by changing the formulations of
gasoline. This is achieved by using so-called oxygenated fuel, i.e.
fuel containing at least 2% oxygen by weight in the form of
oxygenates, but less benzene and other aromatic compounds than
conventional fuel (Health Effects Institute, 1996). MTBE is produced
by reacting methanol with isobutene in acid ion exchangers. In 1987,
MTBE (production of 1.6 × 106 tonnes) ranked 32nd among the top 50
chemicals produced in the USA (Scholz et al., 1990). In 1993, 11 ×
106 tonnes were produced, ranking MTBE ninth of the top 50 chemicals
(Reisch, 1994).
Acetic acid is produced by carbonylation of methanol with carbon
monoxide. Annual growth rates of 6% have been estimated (Fiedler et
al., 1990).
Methanol is present in a broad variety of commercial and consumer
products including shellacs, paints, varnishes, mixed solvents in
duplicating machines (95% concentration or greater), antifreeze and
gasoline deicers (generally containing 35-95% methanol), windshield
washer fluid (contains 35-90% methanol), cleansing solutions
(containing around 5% methanol), model and hobby glues and adhesives,
and Sterno ("canned heat") containing 4% methanol (Posner, 1975; US
EPA, 1980a; CEC, 1988; ATSDR, 1993).
Methanol is also used in the denitrification of wastewater,
sewage treatment application (carbon source for bacteria to aid in the
anaerobic conversion of nitrates to nitrogen and carbon dioxide), as a
substrate for fermentation production of animal feed protein (single
cell protein), as a hydrate inhibitor in natural gas, and in the
methanolysis of polyethylene terephthalate (PET) from recycled plastic
wastes (Posner, 1975; US EPA, 1980a; Kennedy & Shanks, 1981; ATSDR,
1993).
3.2.2.2 Use as fuel
Methanol is a potential substitute for petroleum. It can be
directly used in fuel as a replacement for gasoline in gasoline and
diesel blends. Methanol is in favour over conventional fuels because
of its lower ozone-forming potential, lower emissions of some
pollutants, particularly benzene and polycyclic aromatic hydrocarbons
and sulfur compounds, and low evaporative emissions. On the other
hand, the possibility of higher formaldehyde emissions, its higher
acute toxicity and, at present, lower cost-efficiency favour
conventional fuels (CONCAWE, 1995).
For use in gasoline engines, pure methanol (so-called M100 fuel)
or mixtures of 3, 15 and 85% methanol with conventional petroleum
products (M3, M15, M85) are most common. In diesel engines methanol
cannot be used as an exclusive fuel because of its low cetane number
that would impose proper ignition. Therefore, methanol is injected
into the cylinder after ignition of the conventional diesel fuel
(Fiedler et al., 1990).
3.2.2.3 Other uses
Methanol is used in refrigeration systems, e.g., in ethylene
plants, and as an antifreeze in heating and cooling circuits. However,
its use as an engine antifreeze has been replaced by glycol-based
products. Methanol is added to natural gas at the pumping stations of
pipelines to prevent formation of gas hydrates at low temperature and
can be recycled after removal of water. Methanol is also used as an
absorption agent in gas scrubbers to remove, for example, carbon
dioxide and hydrogen sulfide. According to Table 3, large amounts of
methanol are used as a solvent. Pure methanol is not usually used
alone as a solvent, but is included in solvent mixtures (Fiedler et
al., 1990).
3.2.2.4 Losses into the environment
Given the high production volume, widespread use and physical and
chemical properties of methanol, there is a very high potential for
large amounts of methanol to be released to the environment,
principally to air (US EPA, 1976a,b, 1980a,b, 1994; Nielsen et al.,
1993). Emissions of methanol primarily occur from miscellaneous
solvent usage, methanol production, end-product manufacturing, and
bulk storage and handling losses. The largest source of emissions of
methanol is the miscellaneous solvent use category.
US EPA (1980b) estimated emission factors for the release of
methanol and volatile organic compounds (VOC) from the low-pressure
synthesis of methanol from natural gas in a model plant with a
capacity of 450 000 tonnes/year. The process and capacity were typical
of those built in the late 1970s. The overall emission factors were
estimated to be: uncontrolled emissions, 1.56 kg methanol/tonne
produced; controlled emissions, 0.14 kg methanol/tonne produced
(Nielsen et al., 1993).
It was estimated that about 1% of the methanol used in the
production of formaldehyde would be released to the environment during
the production process by which formaldehyde is produced by either a
metallic silver-catalyst process or a metal oxide-catalyst process (US
EPA, 1976a; 1980b). In the oxidation-dehydrogenation process with
metallic silver catalyst, 0.89 kg methanol/tonne of 39% (by weight)
formaldehyde solution was released principally from the product
absorber vents, and 1.24 kg methanol/tonne from the fractionator
vents. The production of formaldehyde using the catalytic oxidation,
metal oxide catalyst process resulted in the release of 1.93 kg
methanol/tonne of 37% formaldehyde solution with emissions from the
absorber vent (US EPA, 1980b).
US EPA (1994) reported that methanol was the most released
chemical to the environment (air, water and land) based on the 1992
Toxic Release Inventory which utilized 81 016 individual chemical
reports from a total of 23 630 facilities (approximately 65% of
facilities reporting). The air, water and land releases of methanol
totalled 1.09 × 105 tonnes, consisting of 1.53 × 104 tonnes of
fugitive or non-point air emissions, 72 956 tonnes of stack or point
air emissions, 7444 tonnes of surface water discharges and 15 095
tonnes released to land. Additionally, 1.283 × 104 tonnes were
transferred via underground injection.
Methanol had the largest off-site transfers (51 672 tonnes) to
publicly owned treatment works (POTWs) in 1992. During the same
period, methanol ranked third largest of the Toxic Release Inventory
Chemicals with off-site transfers for treatment. The total transfers
to treatment were 18 098 tonnes, consisting of 4 tonnes for
solidification, 10 295 tonnes for incineration/thermal treatment, 1971
tonnes of incineration/insignificant fuel value; 5311 tonnes for
wastewater treatment and 147 tonnes to waste broker-waste treatment. A
total of 493 980 tonnes of methanol was treated, consisting of 260 875
tonnes treated on-site and 197 400 tonnes off-site. A total of 1510
tonnes of methanol was released to land, primarily to on-site
landfills (US EPA, 1994).
The total amount of methanol release in Canada in 1993 was
306 222 tonnes distributed as follows: air, 15 326; water, 14 248;
underground, 819 and land, 205 (Ministry of Supply & Services Canada,
1993).
Tail pipe emissions as well as evaporative emissions are
monitored by a number of agencies. Emissions and air quality modelling
results have been reported from methanol/gasoline blends in prototype
flexible/variable fuelled vehicles (US EPA, 1991; Auto/Oil Air Quality
Research Program, 1992, 1994). Motor vehicle emissions are affected in
various ways by the use of methanol fuels in production flexible/
variable fuel vehicles. Higher molecular weight hydrocarbons are
reduced and carbon monoxide is reduced under some circumstances, while
increases in methanol and formaldehyde can occur (US EPA, 1991).
Methanol has been found in significant amounts in the exhaust
from gasoline-powered vehicles as well as in diesel exhausts. Methanol
was measured at levels of 100-226 mg/kg in the exhaust emissions from
non-catalyst vehicles fuelled with isobutane/methanol/gasoline
(2/15/83; M-15). Methanol emissions from a light-duty diesel vehicle
fuelled with 95% methanol were one order of magnitude higher
(3.4 g/kg) (Jonsson et al., 1985).
Chang & Rudy (1990) reported methanol emission factors for
vehicles fuelled by M-85 (85% methanol + 15% gasoline) and M-100 (100%
methanol) in the USA. For M-85-fuelled vehicles, factors were 0.156-
0.7 g methanol/mile driven in exhaust emissions and 0.055-0.25 g
methanol/mile driven in evaporative emissions. For M-100 fuelled
vehicles, they were 0.5 g methanol/mile driven in exhaust emissions
and 0.072-0.134 g methanol/mile driven in evaporative emissions.
Methanol was found at levels of 130-800 µg/m3 (0.1 to 0.6 ppm)
in the exhaust from nine hydrocarbon test fuels, e.g., iso-octane,
iso-octene, benzene, 2-methyl-2-butene, toluene, o-xylene,
benzene/ n-pentane, toluene/ n-pentane and iso-octane/toluene/
iso-octene (Seizinger & Dimitriades, 1972).
Methanol, formaldehyde and hydrocarbon emissions from methanol-
fuelled cars were reported by Williams et al. (1990). The variable
methanol-fuelled vehicles using fuel mixtures of 100, 85, 50, 15 and
0% methanol and a dedicated methanol vehicle all gave similar emission
patterns. The organic composition of the exhaust was 85-90% methanol,
5-7% formaldehyde and 3-9% hydrocarbons.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
Methanol is released into the environment from both natural and
man-made sources, the latter being the most significant. Methanol
is released predominantly from its production and use as a solvent
in industrial processes (in extraction, washing, drying and
recrystallization operations), and to a lesser degree from a variety
of industrial processes and domestic uses (US EPA, 1980a,b; Graedel et
al., 1986; CEC, 1988; Howard, 1990; Nielsen et al., 1993).
Methanol volatilization half-lives of 5.3 and 2.6 days have been
estimated for a model river (1 m deep) and an environmental pond,
respectively (Howard, 1990).
Methanol is expected to exist almost entirely in the vapour phase
in the ambient atmosphere, based on its vapour pressure (Eisenreich
et al., 1981; Graedel et al., 1986). Because of methanol's water
solubility, rain would be expected to physically remove some methanol
from the air (US EPA, 1980a,b; Snider & Dawson, 1985).
Methanol has been found in the atmosphere (Graedel et al.,
1986). It can be the product of atmospheric alkane chemistry with
concentrations as high as 131 µg/m3 (100 ppb) being found. Methanol
is expected to become an important additional trace gas in the
atmosphere due to its projected increased use as an alternative fuel
to gasoline or in a gasoline blend (CEC, 1988; Chang & Rudy, 1990).
The miscibility of methanol in water and its low octanol/water
partition coefficient suggest high mobility in soil. Lkke (1984)
studied the adsorption of methanol onto three soil types at 6°C. The
soils tested comprised two sandy soils (organic matter contents of
0.09 and 0.1%), and a clay soil (organic matter content of 0.22%).
Methanol solutions with concentrations of 0.1, 1.0, 9 and 90 mg/litre
were used in 1-h exposure studies. Adsorption coefficients for all
soil methanol concentrations and soil types ranged from 0.13 to 0.61,
indicating methanol has a low adsorptive capacity on these soils.
However Nielsen et al. (1993) suggested that the soils used in the
Lkke (1984) study had low organic matter contents compared to typical
agricultural surface soil which can have organic matter contents of 1
to 2%, and up to 5% in some soils. A soil containing a typical amount
of organic matter might therefore be expected to retain methanol and
prevent it from reaching the subsoil.
Additionally, the relatively high vapour pressure and low
adsorptive capacity suggests significant evaporation from dry
surfaces.
4.2 Transformation
4.2.1 Biodegradation
Methanol is readily biodegradable in soil and sediments, both
under aerobic and anaerobic conditions. A large number of strains/
genera of microorganisms have been identified as capable of using
methanol as a growth substrate (Hanson, 1980; Braun & Stolp,
1985; Nielsen et al., 1993). These include Pseudomonas sp.,
Methylobacterium organophilium; Hyphomicrobium sp., Desulfovibrio;
Streptomyces sp., Rhodopseudomonas acidophilia; Paracoccus
denitrificans; Microcyclus aquaticus; Thiobacillus novellus;
Micrococcus denitrificans; Achromobacter 1L (isolated from activated
sewage sludge) and Mycobacterium 50 (isolated from activated sewage
sludge). Most microorganisms possess the enzyme alcohol dehydrogenase
which is necessary for methanol oxidation. The methanogen,
Methanosarcine barkeri can grow on and produce methane from methanol
(Hippe et al., 1979).
The following genera of methanol-oxidizing yeasts have been
reported: Pichia; Saccharomyces; Hansenula; Rhodotorula; Kloechera;
Candida; Torulopsis (Stensel et al., 1973; Hanson, 1980; Nielsen et
al., 1993). Okpokwasili & Amanchukwu (1988) isolated Candida sp.
from Niger Delta sediment which utilized methanol as a growth
substrate.
4.2.1.1 Water and sewage sludge
In a closed bottle test, according to OECD guideline 301D,
methanol was found to be readily biodegradable with 99% COD removal
after the test period of 30 days (Hüls AG, 1978). In another closed
bottle test using unadapted inoculum from domestic sewage the
degradation of methanol at concentrations of 3, 7 or 10 mg/litre in
both freshwater (settled domestic wastewater) and synthetic seawater
incubated for a maximum of 20 days under aerobic conditions was
studied by Price et al. (1974). Methanol was readily degraded in both
inocula at all concentrations with average disappearance of methanol
as follows: a) after 5 days, 76% bio-oxidation in fresh water and 69%
in salt water; b) after 10 days, 88% bio-oxidation in fresh water and
84% in salt water; c) after 15 days, 91% bio-oxidation in fresh water
and 85% in salt water and d) after 20 days, 95% bio-oxidation in fresh
water and 97% in salt water.
Matsui et al. (1988) studied the biodegradability of methanol in
a batch reactor using activated sludge from an industrial wastewater
treatment plant which was acclimatized to the wastewater originating
from a petrochemical complex in Japan. Methanol at an initial
concentration of 100 mg/litre and an acclimation period of 1 day was
found to be highly biodegradable with 91% COD removal and 92% TOC
removal achieved.
Incubation of 0.05 mg methanol/litre for 5 days in activated
sludge from a municipal sewage plant resulted in the degradation of
37% of the methanol (Freitag et al., 1985). Hatfield (1957) found that
at a feed rate of 333 or 500 mg/litre, methanol was virtually
completely oxidized (with a major portion of the BOD and COD removed)
by acclimated microorganisms within 6 h in a settled domestic sewage
inoculum.
The microbial metabolism of methanol in a model activated sludge
system monitored by Swain & Somerville (1978) revealed that methanol
was not broken down when added transiently (0.23% by volume) to the
system operating with a retention time of 11 h. However adaptation of
the sludge in such a system to 0.1% by volume occurred over a period
of several days. After 2 days acclimation, about 50% of the methanol
was utilized, and after 6 days acclimation more than 80% of the
methanol had been metabolized. There were no apparent toxic effects
caused by the addition of methanol (0.1% by volume) to the sludge
prior to and after adaptation to methanol.
The anaerobic treatment of wastes containing methanol and higher
alcohols (approximately 50:50 mix) was studied by Lettinga et al.
(1981). In batch and continuous experiments using an inoculum
consisting of sugar beet waste and active anaerobic sludge, the
breakdown of methanol began within a few days while the breakdown of
higher alcohols occurred immediately depending on the initial load of
waste applied.
Denitrification is facilitated by heterotrophic and autotrophic
bacteria. Heterotrophic bacteria require a carbon source for their
growth and cell metabolism which can be supplied by methanol (Stensel
et al., 1973; Nyberg et al., 1992; Jansen et al., 1993; Upton, 1993).
Bacteria such as the organisms of the genera Pseudomonas,
Micrococcus, Achromobacter, Spirillum, and Bacillus reduce
nitrate, nitrogen oxide and nitrous oxide under anaerobic conditions.
The addition of methanol to promote denitrification has been suggested
in situations where nitrate accumulates, and methanol has been
added as an economic exogenous organic carbon source to increase
denitrification (Stensel et al., 1973; Nyberg et al., 1992; Jansen et
al., 1993; Upton, 1993).
At a wastewater treatment plant in Malmo, Sweden, complete
denitrification was obtained after approximately one month at 10°C
after methanol was added for denitrification. Microscopic examination
revealed a growing population of budding and/or appendaged bacteria,
presumably Hyphomicrobrium spp. reaching a stable maximum at the
time when optimal nitrate removal occurred (Nyberg et al., 1992)
Upton (1993) described a pilot study in the United Kingdom
indicating that denitrification in deep-bed sand filters is a feasible
technology utilizing methanol addition. Nitrogen removals greater than
70% were possible at winter sewage temperatures.
Several other laboratory studies using a variety of methodologies
have demonstrated the rapid biodegradation of methanol by sewage
organisms. These show degradation of between 66 and 95%, and usually
greater than 80%, within five days (Kempa, 1976; Hüls AG, 1978; Matsui
et al., 1988).
4.2.1.2 Soils and sediments
Methanol is biodegradable in soils and sediments, both under
aerobic and anaerobic conditions. Methanol is a normal growth
substrate for many soil microorganisms, which are capable of
completely mineralizing methanol to carbon monoxide and water (CEC,
1988; Howard, 1990; Howard et al., 1991; Nielsen et al., 1993).
Methanol at concentrations of up to 1000 mg/litre was degraded to
non-measurable amounts within a year or less in subsurface soil and
ground water sites in Pennsylvania, New York and Virginia (USA)
believed to be previously uncontaminated. Complete degradation of
100 g methanol/litre occurred in less than 30 days in one aerobic soil
sample from a Pennsylvania site (Novak et al., 1985).
Scheunert et al. (1987) monitored the formation of 14CO2 from
labelled methanol in aerobic and anaerobic suspended soil and found
methanol to be readily degradable after 5 days incubation at 35°C.
Rates and patterns of biodegradation of methanol in surface and
subsurface soils from eight sites in New York, Pennsylvania and
Virginia in static microcosms under anaerobic conditions were
evaluated by Hickman & Novak (1989) and Hickman et al. (1989). The
rates of methanol degradation varied considerably between sites, but
the soils could be characterized into two general types, namely fast
soils, in which degradation rates were high and rates were increased
by addition of nitrate and sulfate, and slow soils, in which
biodegradation rates were low and decreased by the addition of nitrate
or sulfate and inhibition of sulfate increased degradation rates.
Biodegradation rates in subsurface soils were generally within the
range of 0.5-1.1 mg/litre per day and indicated that no acclimation
period was required. Biodegradation rates were calculated and used to
estimate a half-life of between 58 and 263 days for methanol in these
soils (Hickman et al., 1989).
Compared to other substrates studied, e.g., acetate,
trimethylamine and methylamine, methanol (at concentrations less than
3 µM) was degraded relatively slowly mainly to carbon dioxide,
principally via sulfite-reducing organisms, and could not be
considered a significant in situ precursor in surface sediments of
an intertidal zone in Maine, USA (King et al., 1983).
Methanol was found to be an important substrate for methanogenic
bacteria in anaerobic sediments (highly reduced and containing methane
and hydrogen sulfide), collected from a salt marsh located in
San Francisco Bay, California. The sediments were homogenized
anaerobically with San Francisco Bay water and 310-340 µmol methanol/
flask, resulting in 83-91% conversion to methane, carbon dioxide and
water after 3 days (Oremland et al., 1982).
A sulfate-reducing bacterium of the genus Desulfovibrio, which
is capable of degrading methanol after growth on pyruvate, malate or
fumarate, completely converted anaerobic samples of 14C-methanol to
carbon dioxide. However the 14C-label was not used as a carbon source
by the bacterium and was not assimilated into cellular material (Braun
& Stolp, 1985).
4.2.2 Abiotic degradation
4.2.2.1 Water
In a 5-day experiment, 14C-labelled methanol applied to
soil-water suspensions under both aerobic and anaerobic conditions
yielded 53.4 and 46.3% 14CO2, respectively (Scheunert et al., 1987).
Half-lives of 5.1 years and 46.6 days for the photooxidation of
methanol in water have been reported based on the measured rate data
for the reaction with hydroxyl radicals in aqueous solutions (Howard
et al., 1991). A bimolecular reaction rate constant of 8.5 × 10-13
cm3/molecule per second for the reaction of methanol and hydroxyl
radicals in water has been reported by Lemaire et al. (1982).
The rate constant for the reaction of methanol with hydroxyl
radicals in aqueous solution is approximately 1 × 109 litre/mol per
second (Gurten et al., 1984). If the hydroxyl radical concentration of
sunlit natural water is assumed to be 1 × 10-17 mol/litre (Mill et
al., 1980), the half-life of methanol would be approximately 2.2 years
(Howard, 1990).
Sediment and clay suspensions did not photo-catalyse the
degradation of methanol in aqueous solution during ultraviolet
irradiation at 300 nm. However, the addition of semi-conductor powders
such as titanium dioxide led to large increases in the yield of
formaldehyde upon irradiation, in contrast to the small amounts of
formaldehyde formed from the irradiation of 10% aqueous methanol
(Oliver et al., 1979).
Hustert et al. (1981) reported that methanol in aqueous solution
was stable when exposed to sunlight. Alcohols are generally resistant
to environmental aqueous hydrolysis (Lyman et al., 1982; Howard,
1990).
4.2.2.2 Air
Methanol reacts in the atmosphere with oxidizing species (Barnes
et al., 1982; Lemaire et al., 1982; Whitbeck, 1983; Graedel et al.,
1986; Montgomery, 1991; Nielsen et al., 1993; US EPA, 1994).
The atmospheric lifetime of methanol has been estimated to be 20
days based on the reaction of compounds with the hydroxyl radical,
and assuming a hydroxyl free radical concentration of 5 × 105
radicals/cm3 (Graedel et al., 1986). Methanol half-lives of 8.4 days
(US EPA, 1979), 8.0 days (Lemaire et al., 1982) and 7.3 days (Barnes
et al., 1982) have also been reported based on reactions at 300°K and
equations reported in Lyman et al. (1984) and Resenblatt (1990).
Gusten et al. (1984) reported that at 300 °K and atmospheric pressure,
an average hydroxyl concentration of 1 × 106 molecules/cm3 and a
reaction rate constant of 0.95 × 10-12 cm3 /mol per sec, the half-
life of methanol was 8.4 days.
Reaction of methanol with nitrogen dioxide in a smog chamber
yielded methyl nitrite and nitric acid and the surface reaction of
methanol and nitrogen dioxide was enhanced under ultraviolet light
(Akimoto & Takagi, 1986). The reaction of methanol with nitrogen
dioxide may be the major source of methyl nitrite found in polluted
atmospheres (Takagi et al., 1986; Howard, 1990). Only 4.1% of the
methanol applied to silica gel was degraded when irradiated for 17 h
at wavelengths greater than 290 nm (Freitag et al., 1985).
4.2.3 Bioconcentration
Bioconcentration factors (BCFs) of methanol experimentally
measured in aquatic organisms using a log kow value for methanol of
-0.77 and correlation equations reported in Lyman et al. (1990) ranged
from 0.01-0.51 (Nielsen et al., 1993). Based on the octanol/water
partition coefficient of -0.77, the BCF value for methanol was
estimated to be 0.2 (Howard, 1990).
Freitag et al. (1985) reported a BCF of < 10 (wet weight basis)
for the golden ide (Leuciscus idus melanotus) after 3 days exposure
to 0.05 mg methanol/litre.
Gluth et al. (1985) proposed a BCF of about 1 for the carp
Cyprinus carpio exposed to 14C-methanol for up to 72 h. The amount
of radioactivity was measured in the liver, kidneys, intestine,
muscle, blood and gills of carp exposed to methanol at 5 mg/litre. The
initial uptake of methanol into the different tissue types was the
same after 24 h and levels remained constant for over 72 h in the
liver, kidneys, gills and intestines, but dropped slightly in the
blood and muscle.
Geyer et al. (1984) calculated a BCF of 28 400 (dry weight.
basis) for the green alga Chlorella fusca exposed to 0.05 mg/litre
14C-labelled methanol for 24 h at a temperature of 20-25°C with 16 h
illumination and with agitation. Nielsen et al. (1993) suggested that
this high bioconcentration factor is anomalous compared to those for
other aquatic organisms. It may be due to the fact that methanol is
metabolized by the algae, and the 14C-label, which is measured to
calculate the BCF value, is incorporated into the algae in metabolic
forms other than methanol.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
Methanol was detected at mean ambient concentrations of 10 and
3 µg/m3 (7.9 and 2.6 ppb) at Tucson, Arizona, USA, and two remote
Arizona locations, respectively, during monitoring in 1982 of air
pollutants in the USA (Snider & Dawson, 1985). It was also detected in
rural air in Alabama (Holzer et al., 1977). Methanol was detected at
concentrations of 0.65-1.8 µg/m3 (0.5-1.2 ppb) (average 0.77 ppb
methanol plus ethanol) in Arctic air from Point Barrow, Alaska, in
September 1967 (Cavanaugh et al., 1969).
Urban air levels of methanol in the range of 10.5-131 µg/m3
(8-100 ppb) have been reported (Graedel et al., 1986). Jonsson et al.
(1985) reported significant amounts of methanol (0.59-94 µg/m3;
0.45-72 ppb) at dense traffic sites in Stockholm, Sweden. Average
ambient methanol concentrations of 5-30 µg/m3 (3.83-26.7 ppb) were
detected at five sites in and around Stockholm.
In 1994, methanol was listed as one of the 189 hazardous air
pollutants (HAPs) under the Clean Air Act Amendment of 1990, Title III
in the USA (Kelly et al., 1994). In a US EPA (1993) summary, median
methanol levels of 6-60 µg/m3 were found in 52 samples from three
locations (Boston, Houston, and Lima, Ohio) in the USA.
5.1.2 Water
Data on the occurrence of methanol in water, particularly
finished drinking-water, is limited. Methanol was identified in water
at 24 locations in the USA during the period 1974-1976 (US EPA,
1976b). The frequency of occurrence was as follows: finished drinking-
water, 12; effluents from chemical plants, 6; effluents from sewage
treatment, 4; effluents from paper production, 1; and effluents from
latex production, 1.
Methanol was detected in the USA at a mean level of 22 µg/litre
in rainwater collected during a thunderstorm in Arizona in 1982
(Snider & Dawson, 1985).
Methanol at levels of 17-80 mg/litre (17-80 ppm) was detected in
wastewater effluents from a speciality chemicals manufacturing
facility in Massachusetts, USA, but none was detected in associated
river water or sediments (Jungclaus et al., 1978). A concentration of
42.4 mg/litre were found in a leachate from the Love Canal in Niagara
Falls, New York (Venkataraman et al., 1984). Methanol at a level of
1050 mg/litre was detected in condensate waters discharged from a coal
gasification plant at North Dakota, USA (Mohr & King, 1985).
5.1.3 Food
Dietary methanol can arise in large part from fresh fruits and
vegetables where it occurs as the free alcohol, methyl esters of fatty
acids or methoxy group on polysaccharides such as pectin (Kirchner &
Miller, 1957; Casey et al., 1963; Self et al., 1963; Lund et al.,
1981; Stegink et al., 1981; Monte, 1984).
The methanol content of fresh and canned fruit juices
(principally orange and grapefruit juices) varies considerably and may
range from 1-43 mg/litre (Kirchner & Miller, 1957), 10-80 mg/litre
(Lund et al., 1981; Monte, 1984) and 12-640 mg/litre with an average
of 140 mg/litre (Francot & Geoffroy, 1956; Monte, 1984). Methanol
evolved during the cooking of high pectin foods (Casey et al., 1963)
has been accounted for in the volatile fraction during boiling and is
quickly lost to the atmosphere (Self et al., 1963). However entrapment
of the volatiles during canning is possible and probably accounts for
the elevated methanol levels of certain fruits and vegetables during
this process (Lund et al., 1981).
Fermented distilled beverages can contain high levels of
methanol, with some neutral spirits having as much as 1.5 g/litre
(Francot & Geoffroy, 1956). Methanol was found at levels of
6-27 mg/litre in beer, 96-321 mg/litre in wines and 10-220 mg/litre
in distilled spirits (Greizerstein, 1981). The methanol content in
representative beverage alcohol varied between 40 and 55 mg/litre
bourbon. This value is comparable with those reported by the
distillers. The concentration of methanol in 50% grain alcohol was
found to be approximately 1 mg/litre (Majchrowicz & Mendelson,
1971).The presence of methanol in distilled spirits is directly linked
to the pectin content of the raw materials. During the process of
making fruit spirits, pectic substances contained in different parts
of the fruit undergo degradation by pectin methylases, which can lead
to the formation of significant quantities of methanol (Bindler et
al., 1988). Concentrations of methanol permitted in brandies in the
USA, Canada and Italy range from 6-7 g/litre ethanol (Bindler et al.,
1988).
Methanol has been identified in the volatile fraction of sherry
wine vinegars (Blanch et al., 1992), lemon, orange and lime extracts,
distilled liquors and cordials (AOAC, 1980, 1990).
Methanol has been identified as a volatile component of dried
legumes with reported levels of 1.5-7.9 mg/kg in beans, 3.6 mg/kg in
split peas and 4.4 mg/kg in lentils (Lovegren et al., 1979). Methanol
has also been reported (no levels stated) in roasted filberts (Kinlin
et al., 1972) and baked potatoes (Coleman et al., 1981). It has been
detected in low-boiling volatile fractions of cooked foods, including
Brussels sprouts, carrots, celery, corn, onion, parsnip, peas and
potatoes (Self et al., 1963).
Humans can also ingest varying amounts of methanol in foods and
or drugs isolated or recrystallized from methanol, e.g., methanol is
used as an extraction solvent for spice oleoresins and hops (Lewis,
1989). Additionally, certain foods and drugs, consumed or administered
as their methyl ester, can release methanol during their metabolism
and excretion (Stegink et al., 1981; Davoli et al., 1986). For
example, 10% of the sweetening agent aspartame (L-aspartyl-L-
phenylalanine methyl ester) hydrolyzes in the gastrointestinal tract
to become free methanol. Carbonated beverages contain about 555 mg
aspartame/litre (Medinsky & Dorman, 1994), equivalent to approximately
56 mg methanol per litre.
The amount of methanol present in an average serving of beverage
sweetened by aspartame alone is considerably less than in the same
volume of many fruit and vegetable juices. For instance, tomato juice
will result in 6 times the amount of methanol exposure than
consumption of an equivalent volume of aspartame sweetened beverage
(Wucherpfennig et al., 1983).
Exposure to several industrial compounds such as methanol,
formaldehyde and acetone may contribute to increasing amounts of
formate in the body (Boeniger, 1987). Formate is present in blood at
background or endogenous levels that range from 0.07 to 0.4 mM.
Although it is essential for survival, an excess of formate, which
often occurs after intake of large doses of methanol, can cause severe
toxicity and even death (Medinsky & Dorman, 1994).
Ingestion of formate can arise from such foods as honey, fruit
syrups and roasted coffee as well as from its use as a food
preservative. Formate is also produced as a by-product of several
metabolic pathways including histidine and tryptophan degradation
(Stegink et al., 1983).
The possible utility of formic acid as a biomarker for
occupational exposure to methanol has been investigated (Angerer &
Lehnert, 1977; Baumann & Angerer, 1979; Ferry et al., 1980; Heinrich &
Angerer, 1982; Liesivouri & Savolainen, 1987; Franzblau et al., 1992b;
Lee et al., 1992; d'Alessandro et al., 1994).
5.1.4 Tobacco smoke
Methanol at levels of 180 µg/cigarette has been detected in the
vapour phase in mainstream smoke (Norman, 1977; Guerin et al., 1987).
It has been reported to represent about 2% by weight of the mainstream
smoke organic phase and particulate matter (Dube & Green, 1982).
5.2 Occupational exposure
US NIOSH (1976) estimated that 175 000 workers in the USA are
potentially exposed to methanol. As stated in Clayton & Clayton
(1982), the US Department of Labor reported that 72 occupations
involve exposure to methanol. Estimates derived from the NIOSH
1972/1974 National Occupational Hazard Survey and 1982-1983 National
Occupational Exposure Survey indicate that approximately 1-2 million
workers in the USA are potentially exposed to methanol (Howard, 1990).
In a 1978-1982 survey of solvent products associated with USA
industrial workplace exposure, methanol was identified in 9.8% of 275
solvent samples collected. The products represented solvent classes
such as thinners, degreasers, paints, inks and adhesives (Howard,
1990).Workplace concentrations in the range of 29-108 mg/m3 were
found during production of "fused collars" (Greenberg et al., 1938).
No signs or symptoms of methanol intoxication were observed.
In the vicinity of "spirit" duplicator machines operated with
methanol-based duplicator fluids, methanol concentrations of between
475 to 4000 mg/m3 were found in the breathing zone. Teacher aides and
clerical workers exposed to these concentrations experienced typical
symptoms of methanol intoxication (Kingsley & Hirsch, 1955; NIOSH,
1981; Frederick et al., 1984).
In a Japanese factory producing canned fuel containing mainly
methanol, air levels of methanol were high (Kawai et al., 1991b). A
mean geometric concentration of 600 mg/m3 (459 ppm) with a geometric
standard derivation of 4.1 was found in the breathing zone of 22
production workers (8-h sampling). This resulted in high blood and
urine levels of methanol (see section 8.1.3 for further details).
In a chemical plant, 30-min workplace concentrations ranged from
about 49 to 303 mg/m3 during the course of a shift, with a geometric
mean of 129 mg/m3. After an 8-h exposure, average methanol blood and
urine levels of 8.9 ± 14.7 and 21.8 ± 20.0 mg/litre and a mean formic
acid urine level of 29.9 ± 28.6 mg/litre were found (Heinrich &
Angerer, 1982).
Increases in blood and urine methanol and formate levels can be
measured in humans exposed to methanol vapours in the workplace at
concentrations below the ACGIH threshold limit value (TLV) of 260
mg/m3 (200 ppm). The recommended limit of 260 mg/m3 for methanol was
first proposed by Cook (1945), based on previous studies of Sayers et
al. (1942) who observed no symptoms in dogs exposed daily (7
days/week) for 379 days at concentrations between 590 and 655 mg/m3
(450 and 500 ppm). Printing office and chemical workers exposed to
approximately 130 mg/m3 (100 ppm) during the workshift exhibited a
1.5- to 2.5-fold increase in blood and urinary formate and a 15- to
20-fold increase in blood and urinary methanol at the end of the
workday, whereas unexposed workers did not exhibit an increase in
their blood and urinary methanol or formate levels (Baumann & Angerer,
1979; Heinrich & Angerer, 1982).
5.3 General population
Humans are routinely exposed to methanol from both the diet and
natural metabolic processes. Sedivec et al. (1981) reported a mean
blood methanol level of 0.73 mg/litre in 31 unexposed subjects (range:
0.32-2.61 mg/litre). Eriksen & Kulkarni (1963) measured a mean level
of 0.25 µg/litre in the expired air of 9 "normal" people (range:
0.06-0.45 µg/litre).
Methanol is available from the ingestion of dietary fruits and
vegetables, from the consumption of fruit juices and fermentation
beverages, and from the use of the synthetic sweetener aspartame,
which on hydrolysis yields 10% of its weight as free methanol, which
is available for absorption. Estimates of intakes of methanol from
these sources vary considerably. Consuming a 354 ml carbonated
beverage is approximately equivalent to a methanol intake of 20 mg.
Excluding exposure from carbonated beverages, daily aspartame intake
can average 3-11 mg/kg (0.3-1.1 mg methanol/kg), with the 99th
percentile ingesting up to 34 mg/kg (3.4 mg/kg methanol) (Stegink,
1981; Medinsky & Dorman, 1994). If aspartame were used to replace all
sucrose in the diet, its average daily ingestion would be
7.5-8.5 mg/kg which would be the equivalent to 0.75-0.85 mg
methanol/kg (Stegink et al., 1981; Davoli et al., 1986).
The average intake of methanol from natural sources varies, but
limited data suggest an average intake of considerably less than 10 mg
methanol/day (US EPA, 1977; Monte, 1984).
Estimated methanol body burdens for selected situations were
reported by Medinsky & Dormam (1994). The "background" body burden of
methanol was estimated to be 0.5 mg/kg. Fruit juices containing
12-640 mg methanol/litre would have a variable effect on body burden,
while personal garage exposure (200 mg/m3; 15 min) and self-service
refuelling (50 mg/m3; 4 min) would increase the body burdens by an
estimated 0.6 mg/kg and 0.04 mg/kg, respectively.
Methanol, either 100% or in gasoline blends (85% methanol and 15%
gasoline), has the potential to become a major automotive fuel
particularly in the USA in the next century (Kavet & Nauss, 1990;
Medinsky & Dorman, 1994). Emissions of methanol can arise from its
release as uncombusted fuel in the exhaust or from its evaporation
during refuelling and after the engine has stopped. Formaldehyde
emissions can result from the incomplete combustion of methanol fuels
(Medinsky & Dorman, 1994).
The US EPA has modelled methanol exposure levels that might occur
under specific conditions of use (Kavet & Nauss, 1990). For example,
if 100% of all automobiles were powered by methanol-based fuels,
models predict concentrations of methanol in expressways, street
canyons, railroad tunnels or parking garages ranging from a low of
1 mg/m3 to a high of 60 mg/m3. Methanol concentrations in a personal
garage during engine idle or hot soak conditions are predicted to
range from 2.9 to 50 mg/m3, while those predicted during refuelling
of vehicles ranged from 30 to 50 mg/m3. For comparison, the American
Conference of Governmental Industrial Hygienists (ACGIH) Threshold
Limit Value (TLV) for exposure to methanol over an 8-h workday is
260 mg/m3 (200 ppm) for working populations.
Some methanol exposure concentrations have been calculated for
various scenarios (traffic conditions, wind patterns, meteorological
conditions) from emission data from a few cars using methanol
dispersion models. The highest methanol concentration projected to
occur in a personal garage is 490 mg/m3 (375 ppm) during the cold
start. In public garages, assuming 100% of the vehicles were fuelled
with methanol, concentrations were projected to be as high as
200 mg/m3 (150 ppm), while in either scenarios the concentrations
would be expected to be lower than 65 mg/m3 (50 ppm). In the majority
of cases, exposure to the general public would be brief but repeated
in time (Gold & Moulif, 1988).
Most available evidence indicates that exposure to methanol
vapour from use as a motor fuel is not associated with adverse effects
(Gold & Moulif, 1988). The uncertainties in this conclusion are based
on the lack of information at reasonable projected exposure levels and
of studies examining end-points of concern in sensitive species. Lack
of complete data (dose-response, exposure) reveals that numerous
uncertainties exist in the safety/risk assessments. Small effects and
trends in behavioural and neurophysiological responses and subjective
ratings have been reported but need to be further substantiated.
6. KINETICS AND METABOLISM IN LABORATORY ANIMALS AND HUMANS
6.1 Absorption
The primary routes of methanol exposure are inhalation and
ingestion, with dermal exposure currently of much less importance in
terms of total daily intake for both the general and occupational
populations. Regardless of the exposure route, methanol distributes
readily and uniformly to all organs and tissues in direct relation to
their water content (Yant & Schrenk, 1937; Haggard & Greenberg, 1939).
Thus all exposure routes are presumed to be toxicologically equivalent
(Tephly & McMartin, 1984). No differences exist between the
capabilities for absorption of methanol among various animal species,
and blood levels are entirely predictable based on the concept that
methanol distributes uniformly to body water content.
6.1.1 Inhalation
Inhalation of methanol is the most common route of entry in an
occupational setting. Experiences in occupational health and volunteer
studies show that methanol is rapidly absorbed after inhalation
(Angerer & Lehnert, 1977; Baumann & Angerer, 1979; Ferry et al., 1980;
Sedivec et al., 1981; Heinrich & Angerer, 1982; Liesivouri &
Savolainen, 1987; Kawai et al., 1992; d'Alessandro et al., 1994).
The body burden is estimated from methanol concentration,
ventilation rate, duration of exposure and lung retention. Around
60-85% of inhaled methanol is absorbed in the lung of humans (Leaf &
Zatman, 1952; Sedivec et al., 1981). Blood methanol concentration,
frequently employed to characterize the body burden of methanol is, on
average, equal to 83% of its aqueous concentration. Urine contains
methanol concentrations 20-30% higher than blood (Yant & Schrenk,
1937; Leaf & Zatman, 1952).
Following uptake and distribution, methanol clears from the body.
In humans, clearance proceeds after either inhalation or oral exposure
with a half-life of 1 day or more for high doses (greater than 1 g/kg)
and about 3 h for low doses (less than 0.1 g/kg) with first-order
kinetics in humans, monkeys and rats (Leaf & Zatman, 1952; Teply &
McMartin, 1984).Methanol is either excreted unchanged in the urine and
breath or it enters a metabolic pathway whose ultimate product is
carbon dioxide. The time course for the disappearance of methanol from
the circulation is dependent upon the combined action of both direct
excretion and metabolism. The elimination of methanol from the blood
appears to be very slow in all species, especially when compared to
ethanol (Tephly & McMartin, 1984).
Relationships between methanol inhalation exposure,
concentrations, duration of exposure and urinary methanol
concentrations have been characterized in exposures of volunteers and
in occupational settings. Ferry et al. (1980), Sedivec et al. (1981),
and Heinrich & Angerer (1982) reported that urinary methanol
concentrations strictly depend on the duration and intensity of the
methanol exposure, suggesting that measurement of urinary methanol
concentrations would be a reliable parameter for evaluating the degree
of methanol exposure.
Sedivec et al. (1981) exposed four volunteers to methanol at
concentrations of 102, 205 and 300 mg/m3 for 8 h/day. Urine was
monitored for methanol during exposure and for 18 h afterwards. The
concentrations in urine were proportional to the air concentrations.
When exposure ceased, urinary methanol levels decreased exponentially
with a half-life of about 1.5-2 h; a mean urinary level of 0.73 mg/litre
(range 0.32-2.61 mg/litre) in 31 unexposed subjects was also
reported. Heinrich & Angerer (1982) determined methanol in blood and
urine and formic acid in urine from 20 subjects occupationally exposed
to methanol. The air concentration was on average 145 mg/m3 (111 ppm)
but varied from 49 to 303 mg/m3. An 8-h exposure resulted in methanol
levels in blood and urine of 8.9 ± 14.7 mg/litre and 21.8 ±
20 mg/litre, respectively. Formic acid concentrations were 29.9 ±
28.6 mg/litre. The corresponding normal values were < 0.6, 1.1 ± 0.9
and 12.7 ± 11.7 mg/litre.
Volunteers exposed for 6 h to 260 mg/m3 (200 ppm) methanol, the
current permissible US OSHA 8-h time-weighted average limit, were
found to have a blood methanol concentration increase from a mean of
1.8 µg/ml to 7.0 µg/ml (3.5-4 fold increase) compared to their
pre-exposure levels. Formate did not accumulate in the blood above its
background level (8.11 µg/ml) following the 6-h exposure (Lee et al.,
1992).
Franzblau et al. (1993) demonstrated the absence of formic acid
accumulation in the urine of five volunteers following 5 days of
exposure to an atmosphere containing 260 mg/m3 (200 ppm) of methanol
in a test chamber. These results indicated that there was no day-to-
day accumulation of formic acid in urine in conjunction with 5
consecutive days of near-maximal permissible airborne methanol
exposure and that measurement of formic acid in urine specimens
collected 16 h following cessation of exposure did not appear to
reflect inhalation methanol exposure on the preceding day.
Twenty-six volunteers exposed at rest to 260 mg/m3 (200 ppm) of
methanol vapour for 4 h did not show significant differences in serum
or urinary formate concentration. At the TLV of 260 mg/m3 (200 ppm)
methanol exposure did not contribute substantially to endogenous
formate formation (d'Alessandro et al., 1994).
Inhalation of from 650 to 1450 mg/m3 (500 to 1100 ppm) methanol
for periods of 3-4 h in humans yielded urine concentrations of about
10-30 mg/litre at the end of the exposure period (Leaf & Zatman,
1952). Based on their findings, it was suggested that an 8-h exposure
to 3990 mg/m3 (3000 ppm) methanol would be necessary before a gradual
accumulation of methanol would occur in the body.
6.1.2 Oral
Methanol is rapidly absorbed from the gastrointestinal tract with
peak absorption occurring in 30-60 min depending on the presence or
absence of food in the stomach (Becker, 1983).
Ingestion of methanol has been the principal route of exposure in
the many reported cases of acute poisoning (Buller & Wood, 1904; Wood
& Buller, 1904; Bennett et al., 1953; Erlanson et al., 1965; Kane et
al., 1968; Gonda et al., 1978; Naraqi et al., 1979; Swartz et al.,
1981; Jacobsen et al., 1982; Becker, 1983; Litovitz et al., 1988).
During methanol poisoning in humans, concentrations of methanol
and formic acid in blood and urine are quite variable. Concentrations
of both compounds are highly dependent upon dose, time following
exposure and concomitant ingestion of ethanol (Lund, 1948a, Gonda et
al., 1978, Jacobsen et al., 1982a). Excretion of methanol in urine is
initially high and decreases with time following exposure. Maximum
excretion of formic acid in urine may occur as late as the second or
third day following ingestion (Lund, 1948a).
Blood methanol concentrations during experimentally induced
ethanol intoxication in alcoholics during a 10-15 day period of
chronic alcohol intake showed that blood methanol levels increased
progressively from 2-27 mg/litre from the first to the 11th day of
drinking, when blood ethanol concentrations ranged between 1500 and
4500 mg/litre. Blood methanol levels decreased at the rate of 2.9 ±
0.4 mg/litre per h only after blood ethanol levels decreased to 700 to
200 mg/litre. Blood methanol disappearance lagged behind the linear
disappearance of ethanol by approximately 6-8 h and complete clearance
of methanol required several days. Methanol probably accumulates in
the blood as a result of the competitive inhibition of alcohol
dehydrogenase by ethanol and the presence of endogenously formed
methanol (Majchrowicz & Mendelson, 1971).
Oral doses of 71-84 mg methanol/kg in humans resulted in blood
levels of 47-76 mg/litre blood 2-3 h later. The urinary concentrations
of methanol rapidly reached a peak capacity in 1 h and declined
exponentially, reaching control values in 13-16 h. The urine/blood
concentration ratio was found to be relatively constant at 0.30 (Leaf
& Zatman, 1952). Leaf & Zatman (1952) monitored methanol disappearance
from the circulation of three volunteers orally administered 3, 5 and
7 ml (2.4, 4.0 and 5.6 g) (highest dose, 0.08 g/kg). Blood and urine
methanol disappearance obeyed first-order kinetics with a half-time of
about 3 h.
Aspartame (see section 5.1.3) is a widely used artificial
sweetening agent which is hydrolysed in the intestinal mucosa to 10%
methanol by weight. Beverages totally sweetened with aspartame
typically contain 0.5-0.6 mg aspartame/ml or approximately 195 mg/
350 ml soft-drink; dry mixes and puddings use about 100 mg/serving and
pre-sweetened cereal products about 60 mg/25 ml (cup). The methanol
body burden following ingestion of any of these products could vary
from 6-20 mg (Stegink et al., 1981,1983). Clearance of methanol from
human circulation after body burdens as high as 80 mg/kg follows
first-order kinetics with a half-time of about 2.5-3 h (the rate
constant for total clearance kt is 0.23-0.28/h (Stegink et al., 1981;
Kavet & Nauss, 1990).
After intake of small quantities of methanol (10-20 ml), human
subjects showed no methanol in blood after 48 h, and the concentration
of formic acid in the urine was normal (6.5-12.8 mg%) within 24 h
(Lund, 1948a). Following intake of large amounts of methanol (50 ml),
methanol was found in the blood (250-1200 mg/litre) after 48 h. Formic
acid was found in the blood (26-78 mg/litre) as well as an increased
excretion of formic acid in the urine (540-2050 mg/litre), and up to
20 500 mg/litre within 24 h. Maximum excretion of formic acid was
found to occur not later than the second or third day after intake of
methanol (Lund, 1948a).
6.1.3 Dermal
It has been known for some time that pure methanol has an
anomalously high diffusion rate through epidermis because of the
damage it produces on the stratum corneum (the thin sheath of
keratinized cells that comprise the outermost layer of the epidermis).
The permeability of epidermis for pure methanol is 10.4 mg/cm2 per h
(Scheuplein & Blank, 1971).
Skin absorption rate studies of methanol ranging from 0.031-0.241
mg/cm2 per min conducted in human volunteers showed that an average
of 0.192 mg methanol/cm2 per min is absorbed through direct contact
of the skin to methanol. Compared with absorption via the respiratory
tract, exposure of one hand to liquid methanol for only 2 min would
result in a body burden of as much as 170 mg methanol, similar to that
resulting from exposure to an approximate air concentration of
50 mg/m3 (40 ppm) methanol for 8 h (Dutkiewicz et al., 1980). It was
also reported that in the context of a 20-min immersion of one hand in
methanol, the cumulative urinary excretion of methanol over 8 h was
2 mg. However, it should be noted that the assessment of Dutkiewicz et
al. (1980) would imply that a 10-min exposure of one hand to liquid
methanol roughly corresponds to an 8-h inhalation exposure at
260 mg/m3 (200 ppm). Such an inhalation exposure was found to be
accompanied with a post-shift urinary methanol concentration of about
40 mg/litre (Sedivec et al., 1981; Kawai et al., 1991b) or 6.5
mg/litre (Franzblau et al., 1993).
The rate of absorption into the skin has been found to be higher
with M-85 (85% methanol-15% gasoline) than with pure methanol. The
gasoline was suggested to act by drying out the skin allowing the
methanol to be more readily absorbed (Machiele, 1990). In 11 children
treated for percutaneous methanol intoxication, methanol blood levels
ranged from 0.57 to 11.3 g/litre (mean 4.61 g/litre) (Giminez et al.,
1968). Methanol was identified in the urine and in peritoneal fluid
(no quantitative estimation) in an 8-month-old boy poisoned by
percutaneous absorption of methanol (Kahn & Blum, 1979).
Downie et al. (1992) reported a case of percutaneous industrial
methanol toxicity involving two workers who spent 2-3 h cleaning out a
cargo tank with methanol while wearing positive pressure breathing
apparatus. One of the workers, who suffered from a previous sunburn,
wore no protective clothing during cleaning. He experienced methanol
toxicity from percutaneous exposure and required hospitalization and
methanol poisoning treatment.
6.2 Distribution
Methanol distributes readily and uniformly to organs and tissues
in direct relation to their water content (Yant & Schrenk, 1937;
Haggard & Greenberg, 1939). The apparent volume of distribution of
methanol is 0.6-0.7 litres/kg, similar to that of ethanol. In methanol
inhalation studies conducted in dogs, Yang & Schrenk (1937) reported
that the highest concentrations of methanol were found in the blood,
vitreous and aqueous humour, bile and urine, and the lowest in bone
marrow and fatty tissue. In other animal studies, high concentrations
of methanol have been reported in the kidney, liver and gastro-
intestinal tract with smaller concentrations in brain, muscle and
adipose tissue (Bartlett, 1950).
Postmortem analysis of methanol concentrations in body fluids and
tissues reported in fatal human cases of methanol poisoning has
revealed high concentrations of methanol in cerebrospinal fluid (CSF),
vitreous humour and bile (Bennet et al., 1953; Wu Chen, 1985).
Methanol concentrations in these fluids were higher than blood
concentrations. In one study the ratio of methanol in blood to
vitreous humour was 0.82, which was similar to the ratio of ethanol in
blood to vitreous humour of 0.89 (Coe & Sherman, 1970). In tissues the
highest concentrations were found in brain, kidney, lung and spleen,
with lower concentrations in skeletal muscle, pancreas, liver and
heart (Wu Chen et al., 1985).Methanol-induced alterations in
uteroplacental blood-flow were studied in CD-1 mice and Sprague-Dawley
rats employing microdialysis as a tool for investigating the flux of
toxicants across the maternal-conceptual unit. Microdialysis probes
were inserted into the uteri of gestational day 20 rats and methanol
was administered as either an intravenous bolus (100 or 500 mg/kg) or
infusion (100 or 1000 mg/kg/hour).
In separate studies, methanol (100 or 500 mg/kg) and 3H2O
(20 µCi/kg) were administered intravenously on gestational days
20 and 14 to rats and on gestational day 18 to mice. The methanol
concentration-time data were consistent with saturable maternal
elimination and apparent first-order transfer between maternal and
conceptual compartments. At distribution equilibrium, conceptual
methanol concentrations exceeded those in the dam by approximately
25%. The initial rate of conceptual permeation of methanol was
proportional to the reciprocal of maternal blood methanol
concentration (r2 = 0.910).
The data indicated that high circulating maternal methanol
concentrations decrease the rate of presentation of methanol and
3H2O to the conceptus, and, depending on the severity of the
decrease, fetal hypoxia could also result (Ward & Pollack, 1996b).
6.3 Metabolic transformation
After uptake and distribution, most of the methanol is
metabolized in the liver to carbon dioxide (96.9%), while a small
fraction is excreted directly to the urine (0.6%) and through the
lung. In all mammalian species studied, methanol is metabolized in the
liver by sequential oxidative steps to form formaldehyde, formic acid
and CO2 (Fig. 1). However, there are profound differences in the rate
of formate oxidation in different species which determine the
sensitivity to methanol (Rietbrock, 1969; Palese & Tephly, 1975;
McMartin et al., 1977; Eells et al., 1981a, 1983).
Two enzymes are important in the oxidation of methanol to
formaldehyde, alcohol dehydrogenase and catalase. In non-human
primates and humans, alcohol dehydrogenase mediates this reaction
(Makar et al., 1968; Röe, 1982). In rats and other non-primate species
this reaction is mediated by catalase. Definitive evidence of these
differences has been provided by studies of methanol oxidation
in vivo using alternative substrates (ethanol, 1-butanol) and
selective inhibitors of catalase (3-amino-1,2,4-triazole) and alcohol
dehydrogenase (4-amino-pyrazole). The hepatic microsomal mixed-
function oxidase system (P450IIE1) has also been implicated in the
conversion of methanol to formaldehyde, but there is no definitive
information on its role in vivo (Rietbrock et al., 1966; Teschke et
al., 1975). Despite the difference in enzyme mediation, the conversion
from methanol to formate occurs at similar rates in non-human primates
and in rats (Tephly et al., 1964; Makar et al., 1968; Noker et al.,
1980; Eells et al., 1981a, 1983). The metabolism of methanol can be
significantly inhibited by co-exposure to ethanol, which acts as a
competing substrate for alcohol dehydrogenase (Jones, 1987).
Formaldehyde is oxidized to formate by several enzyme systems
including a specific formaldehyde dehydrogenase. In the reaction
catalysed by this enzyme, formaldehyde combines with reduced
glutathione to form S-formyl glutathione, which is hydrolysed in the
presence of thiolase to formate and reduced glutathione (Strittmatter
& Ball, 1955; Uotila & Koivusalo, 1974). The second step of this
reaction is irreversible (Strittmatter & Ball, 1955). Formaldehyde
dehydrogenase activity has been shown to be present in numerous
species and tissues including human liver and brain (Strittmatter &
Ball, 1955; Kinoshita & Masurat, 1958; Goodman & Tephly, 1971).
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR METHANOL
Members
Dr D. Anderson, British Industry Biological Research Association
(BIBRA) Toxicology International, Carshalton, Surrey, United
Kingdom
Dr S.A. Assimon, Contaminants Standards Monitoring and Projects
Branch, US Food and Drug Administration, Washington DC, USA
Dr H.B.S. Conacher, Bureau of Chemical Safety, Ottawa, Ontario,
Canada
Professor J. Eells, Department of Pharmacology and Toxicology,
Medical College of Wisconsin Milwaukee, USA (Chairman)
Mr J. Fawell, National Centre for Environmental Toxicology,
Marlow, Essex, United Kingdom
Dr L. Fishbein, Fairfax, Virginia, USA (Joint Rapporteur)
Dr K. McMartin, Department of Pharmacology and Therapeutics,
Louisiana State University Medical Center, Shreveport,
Louisiana, USA
Mr H. Malcolm, Institute of Terrestrial Ecology, Monks Wood,
Huntingdon, United Kingdom (Joint Rapporteur)
Dr H.B. Matthews, National Institute of Environmental Health
Sciences, Research Triangle Park, North Carolina, USA
Professor M. Piscator, Karolinska Institute, Stockholm, Sweden
(Vice-Chairman)
Dr G. Rosner, Merzhausen, Germany
Representatives of other Organizations
Professor K.R. Butterworth, BIBRA Toxicology International,
Carshalton, Surrey, United Kingdom (representing the
International Union of Toxicology)
Mr M.G. Penman, ICI Chemicals & Polymers Limited,
Middlesbrough, Cleveland, United Kingdom (representing the
European Centre for Ecotoxicology and Toxicology of
Chemicals)
Secretariat
Dr E. Smith, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland (Secretary)
Mr J.D. Wilbourn, Unit of Carcinogen Identification and
Evaluation, International Agency for Research on Cancer
(IARC), Lyon, France
ENVIRONMENTAL HEALTH CRITERIA FOR METHANOL
A WHO Task Group on Environmental Health Criteria for Methanol
met at the British Industrial Biological Research Association (BIBRA)
Toxicology International, Carshalton, Surrey, United Kingdom from 28
to 31 October 1996. Dr D. Anderson opened the meeting and welcomed
the participants on behalf of the host institute. Dr E. Smith, IPCS,
welcomed the participants on behalf of the Director, IPCS, and the
three IPCS cooperating organizations (UNEP/ILO/WHO). The Task Group
reviewed and revised the draft criteria monograph and made an
evaluation of the risks for human health and the environment from
exposure to methanol.
Dr L. Fishbein, Fairfax, Virginia, USA prepared the first draft
of this monograph. The second draft, incorporating comments received
following the circulation of the first draft to the IPCS Contact
Points for Environmental Health Criteria monographs, was also prepared
by Dr Fishbein.
Dr E.M. Smith and Dr P.G. Jenkins, both of the IPCS Central Unit,
were responsible for the overall scientific content and technical
editing, respectively.
The efforts of all who helped in the preparation and finalization
of the monograph are gratefully acknowledged.
ABBREVIATIONS
ATP adenosine triphosphate
BCF bioconcentration factor
BOD biochemical oxygen demand
COD chemical oxygen demand
CNS central nervous system
FID flame ionization detection
GC gas chromatography
MLD minimum lethal dose
MS mass spectrometry
MTBE methyl tertiary butyl ether
NAD nicotinamide adenine dinucleotide
NCAM neural cell adhesion molecule
NOAEL no-observed-adverse-effect level
THF tetrahydrofolate
TLV threshold limit value
1. SUMMARY
1.1 Identity, physical and chemical properties, analytical methods
Methanol is a clear, colourless, volatile flammable liquid with a
mild alcoholic odour when pure. It is miscible with water and many
organic solvents and forms many binary azeotropic mixtures.
Analytical methods, principally gas chromatography (GC) with
flame ionization detection (FID), are available for the determination
of methanol in various environmental media (air, water, soil and
sediments) and foods, as well as the determination of methanol and its
principal metabolite, formate, in body fluids and tissues. In addition
to GC-FID, enzymatic procedures with colorimetric end-points are
utilized for the determination of formate in blood, urine and tissues.
Determination of methanol in the workplace usually involves
collection and concentration on silica gel, followed by aqueous
extraction and GC-FID or GC-mass spectrometry analysis of the extract.
1.2 Sources of human exposure
Methanol occurs naturally in humans, animals and plants. It is a
natural constituent in blood, urine, saliva and expired air. A mean
urinary methanol level of 0.73 mg/litre (range 0.3-2.61 mg/litre) in
unexposed individuals and a range of 0.06 to 0.32 µg/litre in expired
air have been reported.
The two most important sources of background body burdens for
methanol and formate are diet and metabolic processes. Methanol is
available in the diet principally from fresh fruits and vegetables,
fruit juices (average 140 mg/litre, range 12 to 640 mg/litre),
fermented beverages (up to 1.5 g/litre) and diet foods (principally
soft drinks). The artificial sweetener aspartame is widely used and,
on hydrolysis, 10% (by weight) of the molecule is converted to free
methanol, which is available for absorption.
About 20 million tonnes of methanol were produced worldwide in
1991, principally by catalytic conversion of pressurized synthesis gas
(hydrogen, carbon dioxide and carbon monoxide). Worldwide capacity was
projected to rise to 30 million tonnes by 1995.
Methanol is used in the industrial production of many important
organic compounds, principally methyl tertiary butyl ether (MTBE),
formaldehyde, acetic acid, glycol methyl ethers, methylamine, methyl
halides and methyl methacrylate.
Methanol is a constituent of a large number of commercially
available solvents and consumer products including paints, shellacs,
varnishes, paint thinners, cleansing solutions, antifreeze solutions,
automotive windshield washer fluids and deicers, duplicating fluids,
denaturant for ethanol, and in hobby and craft adhesives. Potentially
large uses of methanol are in its direct use as a fuel, in gasoline
blends or as a gasoline extender. It should be noted that the highest
morbidity and mortality has been associated with deliberate or
accidental oral ingestion of methanol-containing mixtures.
Methanol has been identified in exhausts from both gasoline and
diesel engines and in tobacco smoke.
1.3 Environmental levels and human exposure
Emissions of methanol primarily occur from the miscellaneous
industrial and domestic solvent use, methanol production, end-product
manufacturing and bulk storage and handling losses.
Exposures to methanol can occur in occupational settings through
inhalation or dermal contact. Many national occupational health
exposure limits suggest that workers are protected from any adverse
effects if exposures do not exceed a time-weighted average of
260 mg/m3 (200 ppm) methanol for any 8-h day and for a 40-h working
week.
Current general population exposures through air are typically
10 000 times lower than occupational limits. The general population is
exposed to methanol in air at concentrations ranging from less than
0.001 mg/m3 (0.8 ppb) in rural air to nearly 0.04 mg/m3 (30 ppb) in
urban air.
Data on the occurrence of methanol in finished drinking-water is
limited, but methanol is frequently found in industrial effluents.
If the projected use of methanol as an alternate fuel or in
admixture with fuels increases significantly, it can be expected that
there will be widespread exposure to methanol via inhalation of
vapours from methanol-fuelled vehicles and/or siphoning or
percutaneous absorption of methanol fuels or blends.
1.4 Environmental distribution and transformation
Methanol is readily degraded in the environment by photo
oxidation and biodegradation processes. Half-lives of 7-18 days have
been reported for the atmospheric reaction of methanol with hydroxyl
radicals.
Many genera and strains of microorganisms are capable of using
methanol as a growth substrate. Methanol is readily degradable under
both aerobic and anaerobic conditions in a wide variety of
environmental media including fresh and salt water, sediments and
soils, ground water, aquifer material and industrial wastewater; 70%
of methanol in sewage systems is generally degraded within 5 days.
Methanol is a normal growth substrate for many soil
microorganisms, which are capable of completely degrading methanol to
carbon dioxide and water.
Methanol has a fairly low absorptive capacity on soils.
Bioconcentration in most organisms is low.
Methanol is of low toxicity to aquatic and terrestrial organisms,
and effects due to environmental exposure to methanol are unlikely to
be observed except in the case of a spill.
1.5 Absorption, distribution, biotransformation and elimination
Methanol is readily absorbed by inhalation, ingestion and dermal
exposure, and it is rapidly distributed to tissues according to the
distribution of body water. A small amount of methanol is excreted
unchanged by the lungs and kidneys.
Following ingestion, peak serum levels occur within 30-90 min,
and methanol is distributed throughout the body with a volume of
distribution of approximately 0.6 litre/kg.
Methanol is metabolized primarily in the liver by sequential
oxidative steps to formaldehyde, formic acid and carbon dioxide. The
initial step involves oxidation to formaldehyde by hepatic alcohol
dehydrogenase, which is a saturable rate-limiting process. The
relative affinity of alcohol dehydrogenase for ethanol and methanol is
approximately 20:1. In step 2, formaldehyde is oxidized by
formaldehyde dehydrogenase to formic acid/or formate depending on the
pH. In step 3, formic acid is detoxified to carbon dioxide by folate-
dependent reactions.
Elimination of methanol from the blood via the urine and exhaled
air and by metabolism appears to be slow in all species, especially
when compared to ethanol. Clearance proceeds with reported half-times
of 24 h or more with doses greater than 1 g/kg and half-times of
2.5-3 h for doses less than 0.1 g/kg. It is the rate of metabolic
detoxification, or removal of formate that is vastly different between
rodents and primates and is the basis for the dramatic differences in
methanol toxicity observed between rodents and primates.
1.6 Effects on laboratory mammals and in vitro test systems
1.6.1 Systemic toxicity
The acute and short-term toxicity of methanol varies greatly
between different species, toxicity being highest in species with a
relatively poor ability to metabolize formate. In such cases of poor
metabolism of formate, fatal methanol poisoning occurs as a result of
metabolic acidosis and neuronal toxicity, whereas, in animals that
readily metabolize formate, consequences of CNS depression (coma,
respiratory failure, etc.) are usually the cause of death. Sensitive
primate species (humans and monkeys) develop increased blood formate
concentrations following methanol exposure, while resistant rodents,
rabbits and dogs do not. Humans and non-human primates are uniquely
sensitive to the toxic effects of methanol. Overall methanol has a low
acute toxicity to non-primate animals. The LD50 values and minimal
lethal doses after oral exposure range from 7000 to 13 000 mg/kg in
the rat, mouse, rabbit and dog and from 2000 to 7000 mg/kg for the
monkey.
Rats exposed to levels of methanol up to 6500 mg/m3 (5000 ppm)
for 6 h/day, 5 days/week for 4 weeks, exhibited no exposure-related
effects except for increased discharges around the nose and eyes.
These were considered reflective of upper respiratory irritation.
Rats exposed to methanol vapour levels up to 13 000 mg/m3
(10 000 ppm) for 6 h/day, 5 days/week for 6 weeks, failed to
demonstrate pulmonary toxicity.
In the rabbit, methanol is a moderately irritant to the eye. It
was not skin-sensitizing in a modified maximization test.
Toxic effects found in methanol-exposed primates include
metabolic acidosis and ocular toxicity, effects that are not normally
found in folate-sufficient rodents. The differences in toxicity are
due to differences in the rate of metabolism of the methanol
metabolite formate. For instance, the clearance of formate from the
blood of exposed primates is at least 50% slower than for rodents.
Monkeys receiving methanol doses higher than 3000 mg/kg by gavage
demonstrated ataxia, weakness and lethargy within a few hours of
exposure. These signs tended to disappear within 24 h and were
followed by transient coma in some of the animals.
In monkeys exposed to methanol for 6 h/day for 5 days a week, 20
repeated exposures to 6500 mg/m3 (5000 ppm) methanol failed to elicit
ocular effects.
1.6.2 Genotoxicity and carcinogenicity
Methanol has given negative results for gene mutation in bacteria
and yeast assays, but it did induce chromosomal malsegregation in
Aspergillus. It did not induce sister chromatic exchanges in Chinese
hamster cells in vitro but caused significant increases in mutation
frequencies in L5178Y mouse lymphoma cells.
Methanol inhalation did not induce chromosomal damage in mice.
There is some evidence that oral or intraperitoneal administration
increased the incidence of chromosomal damage in mice.
There is no evidence from animal studies to suggest that methanol
is a carcinogen, although the lack of an appropriate animal model is
recognized.
1.6.3 Reproductive toxicity, embryotoxicity and teratogenicity
Conflicting results have been reported on the effects of
inhalation of methanol for up to six weeks on gonadotropin and
testosterone concentrations.
The inhalation of methanol by pregnant rodents throughout the
period of embryogenesis induces a wide range of concentration-
dependent teratogenic and embryolethal effects. Treatment-related
malformations, predominantly extra or rudimentary cervical ribs and
urinary or cardiovascular defects, were found in fetuses of rats
exposed 7 h/day for 7-15 days of gestation to 26 000 mg/m3
(20 000 ppm) methanol. Slight maternal toxicity was found at this
exposure level, and no adverse effects to the mother or offspring were
found in animals exposed to 6500 mg/m3 (5000 ppm), which was
interpreted as the no-observed-adverse-effect level (NOAEL) for this
test system.
Increased incidences of exencephaly and cleft palate were found
in the offspring of CD-1 mice exposed 7 h/day, on days 6-15 of
gestation, to methanol levels of 6500 mg/m3 (5000 ppm) or more. There
was increased embryo/fetal death at 9825 mg/m3 (7500 ppm) or more and
an increasing incidence of full-litter resorptions. Reduced fetal
weight was observed at 13 000 and 19 500 mg/m3 (10 000 or 15 000
ppm). The NOAEL for developmental toxicity was 1300 mg/m3 (1000 ppm)
methanol. There was no evidence of maternal toxicity at methanol
exposure levels below 9000 mg/m3 (7000 ppm).
When litters of pregnant CD-1 mice were given 4 g methanol/kg by
gavage, the incidences of adverse effects on resorption, external
defects including cleft palate, and fetal weight were similar to those
found in the 13 000 mg/m3 (10 000 ppm) inhalation exposure group,
presumably due to the greater rate of respiration of the mouse. The
mouse is more sensitive than the rat to developmental toxicity
resulting from inhaled methanol.
Transient neurological signs and reduced body weights were found
in CD-1 dams exposed to 19 500 mg/m3 (15 000 ppm) for 6 h/day
throughout organogenesis (gestational days 6-15). Fetal malformations
found at 13 000 and 19 500 mg/m3 (10 000 and 15 000 ppm) included
neural and ocular defects, cleft palate, hydronephrosis and limb
anomalies.
1.7 Effects on humans
Humans (and non-human primates) are uniquely sensitive to
methanol poisoning and the toxic effects in these species is
characterized by formic acidaemia, metabolic acidosis, ocular
toxicity, nervous system depression, blindness, coma and death. Nearly
all of the available information on methanol toxicity in humans
relates to the consequences of acute rather than chronic exposures. A
vast majority of poisonings involving methanol have occurred from
drinking adulterated beverages and from methanol-containing products.
Although ingestion dominates as the most frequent route of poisoning,
inhalation of high concentrations of methanol vapour and percutaneous
absorption of methanolic liquids are as effective as the oral route in
producing acute toxic effects. The most noted health consequence of
longer-term exposure to lower levels of methanol is a broad range of
ocular effects.
The toxic properties of methanol are based on factors that govern
both the conversion of methanol to formic acid and the subsequent
metabolism of formate to carbon dioxide in the folate pathway. The
toxicity is manifest if formate generation continues at a rate that
exceeds its rate of metabolism.
The lethal dose of methanol for humans is not known for certain.
The minimum lethal dose of methanol in the absence of medical
treatment is between 0.3 and 1 g/kg. The minimum dose causing
permanent visual defects is unknown.
The severity of the metabolic acidosis is variable and may not
correlate well with the amount of methanol ingested. The wide
interindividual variability of the toxic dose is a prominent feature
in acute methanol poisoning.
Two important determinants of human susceptibility to methanol
toxicity appear to be (1) concurrent ingestion of ethanol, which slows
the entrance of methanol into the metabolic pathway, and (2) hepatic
folate status, which governs the rate of formate detoxification.
The symptoms and signs of methanol poisoning, which may not
appear until after an asymptomatic period of about 12 to 24 h, include
visual disturbances, nausea, abdominal and muscle pain, dizziness,
weakness and disturbances of consciousness ranging from coma to clonic
seizures. Visual disturbances generally develop between 12 and 48 h
after methanol ingestion and range from mild photophobia and misty or
blurred vision to markedly reduced visual acuity and complete
blindness. In extreme cases death results. The principal clinical
feature is severe metabolic acidosis of the anion-gap type. The
acidosis is largely attributed to the formic acid produced when
methanol is metabolized.
The normal blood concentration of methanol from endogenous
sources is less than 0.5 mg/litre (0.02 mmol/litre), but dietary
sources may increase blood methanol levels. Generally, CNS effects
appear above blood methanol levels of 200 mg/litre (6 mmol/litre);
ocular symptoms appear above 500 mg/litre (16 mmol/litre), and
fatalities have occurred in untreated patients with initial methanol
levels in the range of 1500-2000 mg/litre (47-62 mmol/litre).
Acute inhalation of methanol vapour concentrations below
260 mg/m3 or ingestion of up to 20 mg methanol/kg by healthy or
moderately folate-deficient humans should not result in formate
accumulation above endogenous levels.
Visual disturbances of several types (blurring, constriction of
the visible field, changes in colour perception, and temporary or
permanent blindness) have been reported in workers who experienced
methanol air levels of about 1500 mg/m3 (1200 ppm) or more.
A widely used occupational exposure limit for methanol is
260 mg/m3 (200 ppm), which is designed to protect workers from any of
the effects of methanol-induced formic acid metabolic acidosis and
ocular and nervous system toxicity.
No other adverse effects of methanol have been reported in
humans except minor skin and eye irritation at exposures well above
260 mg/m3 (200 ppm).
1.8 Effects on organisms in the environment
LC50 values in aquatic organisms range from 1300 to
15 900 mg/litre for invertebrates (48-h and 96-h exposures), and
13 000 to 29 000 mg/litre for fish (96-h exposure).
Methanol is of low toxicity to aquatic organisms, and effects due
to environmental exposure to methanol are unlikely to be observed,
except in the case of a spill.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1 Identity
Chemical formula: CH3OH
Chemical structure: H
'
H - C - OH
'
H
Relative molecular mass: 32.04
CAS chemical name: methanol
CAS registry number: 67-56-1
RTECS number: PC 1400000
Synonyms: methyl alcohol, carbinol, wood
alcohol, wood spirits, wood
naphtha, Columbian spirits,
Manhattan spirits, colonial spirit,
hydroxymethane, methylol,
methylhydroxide,
monohydroxymethane, pyroxylic
spirit
Impurities in commercial methanol include acetone, acetaldehyde,
acetic acid and water.
2.2 Physical and chemical properties
2.2.1 Physical properties
Methanol is a colourless, volatile, flammable liquid with a mild
alcoholic odour when pure. However, the crude product may have a
repulsive pungent odour. Methanol is miscible with water, alcohols,
esters, ketones and most other solvents and forms many azeotropic
mixtures. It is only slightly soluble in fats and oils (Clayton &
Clayton, 1982; Windholz, 1983; Elvers et al., 1990).
Important physical constants and properties of methanol are
summarized in Table 1.
Table 1. Some physical properties of methanola
Appearance clear colourless liquid
Odour slight alcoholic when pure;
crude material pungent
Boiling point 64.7°C
Flash point 15.6°C (open cup)
12.2°C (closed cup)
Freezing point -97.68°C
Specific gravity 0.7915 (20/4°C)
0.7866 (25°C)
Vapour pressure
at 30°C 160 mmHg
at 20°C 92 mmHg
Henry's Law Constant (25°C) 1.35 x 10-4atm.m3/mole
Log P (octanol/water) -0.82; -0.77; -0.68
Partition constant -0.66; -0.64
Ignition temperature 470°C
Explosive limits in air lower 5.5
(% by volume) upper 44
Refractive index n20 1.3284
a Data from: Clayton & Clayton, 1982; Elvers et al., 1990;
Grayson, 1981; Howard, 1990; Windholz, 1983.
In the USA, sales grade methanol must normally meet the
following specifications:
methanol content (weight %) minimum 99.85
acetone and aldehydes (ppm) maximum 30
acid (as acetic acid) (ppm) maximum 30
water content (ppm) maximum 1.500
specific gravity (d2020) 0.7928
permanganate time, minimum 30
odour characteristic
distillation range at 101 kPa 1°C, must include
64.6°C
colour, platinum-cobalt scale, maximum 5
appearance clear-colourless
residual on evaporation, g/100 ml 0.001
carbonizable impurities, colour 30
platinum-cobalt scale, maximum 5
Grade AA differs in specifying an acetone maximum (20 ppm), a
minimum for ethanol (10 ppm), and in having a more stringent water
content specification (1.000 ppm, maximum) (Grayson, 1981).
2.2.2 Chemical properties
Methanol undergoes reactions that are typical of alcohols as a
chemical class. The reactions of particular industrial importance
include the following: dehydrogenation and oxidative dehydrogenation
over silver or molybdenum-iron oxide to form formaldehyde; the
acid-catalysed reaction with isobutylene to form methyl tertiary butyl
ether (MTBE); carbonylation to acetic acid catalysed by cobalt or
rhodium; esterification with organic acids and acid derivatives;
etherification; addition to unsaturated bonds and replacement of the
hydroxyl group (Grayson, 1981; Elvers et al., 1990).
2.3 Conversion factors
1 ppm = 1.31 mg/m3 (25°C, 1013hPa) 1 mmol/litre = 32 mg/litre
1 mg/m3 = 0.763 ppm (25°C, 1013hPa) 1 mg/litre =31.2 µmol/litre
(Adapted from Clayton & Clayton, 1982)
2.4 Analytical methods
Prior to the advent of sensitive gas chromatographic techniques,
the analysis of methanol in environmental, consumer and biological
samples was performed by procedures involving isolation of the
volatile alcohol and titrimetry. This was followed later by more
sensitive spectrophotometric methods based on the oxidation of
methanol to formaldehyde with potassium permanganate then reaction
with Schiff's reagent or rosaniline solution to produce an easily
recognizable and stable colour (Gettler, 1920; Boos, 1948; Skaug,
1956; Hindberg & Wieth, 1963; NIOSH, 1976).
The earliest procedures for the determination of methanol in
blood and urine were based on the initial distillation to isolate the
volatile alcohol (Gettler, 1920). Feldstein & Klendshog (1954)
determined methanol in biological fluids by initial microdiffusion
followed by oxidation to formaldehyde and subsequent reaction with
chromotropic acid (1,8-dihydroxy naphthalene-3,6-disulfonic acid). The
recovery ranged from 80 to 85% for less than 0.10 mg methanol. In the
procedure of Harger (1935), methanol was determined by oxidation with
bichromate to carbon dioxide and water followed by titration with a
mixture of ferrous sulfate and methyl orange. Jaselkis & Warriner
(1966) determined methanol in aqueous solution by titrimetry employing
xenon trioxide oxidation. Methanol was determined at a level of
0.03 mg with a relative standard deviation of 4%.
2.4.1 Environmental samples
The determination of methanol by primarily GC-FID procedures has
been frequently reported in ambient air, workplace air, fuels, fuel
emissions, sewage and aqueous solutions, soils, coal-gasification
condensate water and tobacco smoke.
The measurement of methanol in ambient and workplace air, usually
involves a preconcentration step in which the sample is passed through
a solid absorbent containing silica gel, Tenax GC, Porapak or
activated charcoal (NIOSH, 1976,1977,1984; CEC, 1988). It can also be
accomplished by on-column cryogenic trapping or can be analysed
directly. Direct reading infrared instruments with gas cuvettes can be
used for continuous monitoring of methanol in air (Lundberg, 1985).
2.4.1.1 Methanol in air
The use of absorption tubes to trap methanol from ambient and
workplace air with subsequent liquid or thermal desorption prior to
gas chromatographic analysis has been reported frequently. The US
National Institute of Occupational Safety and Health (NIOSH,
1977,1984) recommended the use of a glass tube (7 cm × 4 mm internal
diameter) containing two sections of 20-40 mesh silica gel separated
by a 2-mm portion of urethane foam (front=100 mg, back=50 mg). Water
is used to extract the methanol, which is separated on a 2 m × 2 mm
internal diameter glass column containing 60-80 mesh Tenax GC or the
equivalent using flame ionization detection (FID). The working range
is 25 to 900 mg/m3 (19 to 690 ppm) methanol for a 5-litre air sample.
The limit of detection has been reported to be 1.05 mg/m3 in a
3-litre air sample (NIOSH, 1976). At high concentrations of methanol
or at high relative humidity, a large silica gel tube is required
(700 mg silica gel front section). The injection, detector and column
temperatures are 200°C, 250-300°C and 80°C respectively. Positive
identification by mass spectrometry may be necessary in some cases,
and alternative gas chromatographic columns, e.g., SP-1000, SP-2100 or
FFAP, are also conformation aides.
Although GC-FID provides greater sensitivity than GC-MS, the
latter is generally considered more reliable for the measurement of
methanol in samples containing other alcohols or low molecular weight
oxygenates.Analysis of methanol in workplace air has been carried out
by head-space GC-FID using a column containing 15% Carbowax 1500 on
diatomaceous earth, 70-100 mesh operated at 100°C. The detection limit
was below 5 ml/m3 ( Heinrich & Angerer, 1982). Methanol in workplace
air was initially collected in silica gel tubes and the methanol
concentrations analysed by GC-FID equipped with a 50 m silica
capillary column containing Carbowax 20M. Additionally, methanol
vapour concentrations in the workplace have been analysed by a Miron-B
analyser with detection at a wavelength of 9.70 µm.
Methanol and other low molecular weight oxygenates have been
determined in ambient air by cryogradient sampling and two-dimensional
gas chromatography (Jonsson & Berg, 1983). Samples were initially
separated on a packed column (1,2,3-tris (2-cyanoethoxy)propane on
Chromosorb W-AW), then refocused on-line in a fused-silica capillary
cold trap, followed by on-line splitless reinjection onto a 50 m ×
0.3 mm internal diameter fused silica capillary column. The detection
limit for a typical oxygenate (3-methylbutanol) was 0.1 µg/m3 using a
3-litre sample. The detection limit for methanol was slightly higher.
Spectrophotometric methods have also been employed for the
determination of methanol in air. Aqueous potassium permanganate
acidified with phosphoric acid was used to absorb methanol from air
with the simultaneous oxidation to formaldehyde. After the addition of
p-aminoazobenzene and sulfur dioxide, the resulting pink dye was
determined spectrophotometrically at 505 nm. The limit of detection
was 5 µg methanol/ml air (Verma & Gupta, 1984).
Methanol from air was absorbed by acidified potassium
permanganate producing formaldehyde which on reaction with
4-nitroaniline produced a yellow dye determined spectroscopically at
395 nm (Upadhyay & Gupta, 1984).
Infrared spectrometry and infrared lasers have also been employed
for the determination of methanol in air (Diaz-Rueda et al., 1977;
Sweger & Travis, 1979). Methanol together with acetone, toluene and
ethyl acetate were recovered from 10 litres of air at a flow rate of
11 ml/min by passage through a tube containing 150 mg of activated
charcoal. The carbon disulfide extracts of the organic compounds were
determined by infrared at 1300 cm-1 using caesium bromide windows.
The minimum concentration of methanol detected quantitatively was
0.77 mg/m3 (0.60 ppm) and the minimum concentration required for
identification was 0.24 mg/m3 (0.18 ppm) (Diaz-Rueda et al., 1977).
Infrared lasers have been used to detect trace organic gases
including methanol. An air sample at 8 Tor was introduced to a
20-litre capacity sample cell, and laser radiation was detected
synchronously by a mercury-cadmium Te detector. The laser line
employed was P (34), the electric field was 1.40 kV/cm and the
measurement time was 2 min. The detection limit for methanol was
0.105 mg/m3 (0.08 ppm) (Sweger & Travis, 1979).
Methanol in the workplace can be measured by portable direct
reading instruments, real-time continuous monitoring systems and
passive dosimeters (NIOSH, 1976,1977,1984; Liesivouri & Savolainen,
1987; Kawai et al., 1990).
Kawai et al. (1990) described a personal diffusive badge type
that could absorb methanol vapour in linear relation to the exposure
duration up to 10 h and to exposure concentrations up to 1050 mg/m3
(800 ppm) the maximum duration and concentration tested respectively.
Additionally it was shown that the response to short-term peak
exposure was rapid enough and that no spontaneous desorption would
occur.
2.4.1.2 Methanol in fuels
Agarawal (1988) determined methanol quantitatively in commercial
gasoline via an initial extraction with ethylene glycol then by GC
utilizing a GB-1 fused silica capillary column (OV-1 equivalent, 60 m
× 0.32 mm internal diameter) and FID. The recovery of 4% methanol in
gasoline by this procedure was 95.4 ± 2.34% (SD).
In the procedure of Tackett (1987), gasoline samples were
injected directly on a Carbowax 20M column operated at 50°C for 3.0
min and then programmed to rise to 150°C at a rate of 10°C per min.
The calibration curve is linear up to 10% (v/v) methanol and the
detection limit was 0.2% employing a thermal conductivity detector.
Low molecular weight alcohols and MTBE were determined in
gasoline by GC-FID utilizing dual columns: 4.6 m × 3.2 mm o.d. column
packed with 30% m/m ethylene glycol succinate on Chromosorb P (85-100
mesh) and a 2.7 m × 3.2 mm o.d. stainless steel column packed with
Porapak P (80-100 mesh) operated at 150°C (Luke & Ray, 1984).
Gas chromatographic analyses of methanol, ethanol and tert-
butanol in gasoline have been reported by Pauls & McCoy (1981). The GC
column was 150 cm × 3 mm in o.d. stainless steel packed with Porapak R
(80-100 mesh) operated at 175°C and the injector and FID detector
temperatures were maintained at 250°C.
A direct liquid chromatographic method for the determination of
C1-C3 alcohols and water in gasoline-alcohol blends was described by
Zinbo (1984). The separation was performed on either one or two
microparticulate size-exclusion columns of ultrastyragel with toluene
as the mobile phase. The quantification of alcohols and water in the
effluent was achieved by a differential refractometer at 30°C. The
lower limits of detection for C1-C3 alcohols was 0.005 vol %. Methanol
in gasoline-alcohol blends has been determined by nuclear magnetic
resonance (Renzoni et al., 1985). The method takes advantage of a
window in the proton nuclear magnetic resonance spectrum of gasoline
that extends from a chemical shift of 2.8 to 6.8 ppm. Methanol was
quantified in gasoline by integration of the methyl singlet at
3.4 ppm. The method gave linear calibration curves in the range of
0-25% (v/v) methanol with a detection limit of less than 0.1%.
2.4.1.3 Methanol in fuel emissions
Methanol has been detected in motor vehicle emissions at levels
of 0.9 mg/m3 (0.69 ppm) and in ambient air by GC-FID utilizing a
360 cm × 0.27 cm internal diameter stainless steel column packed with
Porapak Q (50-80 mesh) operated at 150°C (Bellar & Sigsby, 1970).
Seizinger & Dimitriades (1972) determined methanol in simple
hydrocarbon fuel emissions utilizing GC with time-of-flight mass
spectrometry. The analytical procedure involved concentration of the
exhaust oxygenates drawn through a Chromosorb bed followed by GC-FID
initially on a 30 in by 1/4 in o.d. column packed with 10% 1,2,3-tris
(2-cyanoethoxy) propane (TCEP) programmed from -20°C to 110°C at
4°C/min. The second-stage column was a 45 m × 0.05 cm internal
diameter by 0.03 o.d Carbowax 20M support coated on tubular (SCOT)
column programmed from 60°C to 210°C at 10°C/min. The column effluent
was split for parallel detection with FID and mass spectrometry.
Methanol was found at levels of 0.1-0.8 mg/m3 (0.1-0.6 ppm) in
the exhaust of simple hydrocarbon fuels.
Methods for the quantification of evaporative emissions (running
losses, hot soak, diurnal and refuelling) from methanol-fuelled motor
vehicles (methanol/gasoline fuel mixtures of 100, 85, 50, 15 and 0%
methanol) have been described (Snow et al., 1989; Federal Register,
1989; Gabele & Knapp, 1993).
Methanol emissions from methanol-fuelled cars were determined by
GC employing a Quadrex 007 methyl silicone 50 m × 0.53 mm internal
diameter column with 5.0 µm film thickness. The separation was
affected isothermally at 75°C (limit of detection 0.25 µg/ml)
(Williams et al., 1990).
2.4.1.4 Methanol in sewage and aqueous solutions
Fox (1973) determined methanol at levels of 0.5-100 mg/litre
(0.5-100 ppm) in sewage or other aqueous solutions by GC-FID employing
a 0.5 m × 3.175 mm o.d. stainless steel column packed with Tenax GC
60/80 mesh and operated at 70°C isothermal.
C1-C4 alcohols in aqueous solution were determined
quantitatively by GC-FID using a 1 m × 0.32 cm stainless steel column
packed with 5% w/w Carbowax 20M on Chromosorb 101 (80-100 mesh) with a
column temperature of 65°C for methanol and ethanol and 100°C for n-
propanol and n-butanol (Sims, 1976).
Methanol and ethanol at the mg/litre level in aqueous solution
were determined by Komers & Sir (1976) utilizing a combination of
stripping and GC-FID technique. The alcohols were analysed as their
corresponding volatile nitrite on a 170 cm × 0.4 cm internal diameter
glass column containing Chromosorb 102 (80-120 mesh) operated at
104°C. Approximately 1 µg of the individual alcohol could be
determined in sample volumes of about 5 ml.
Mohr & King (1985) determined methanol in coal-gasification
condensate water by GC. Condensate water was injected directly on a 45
× 0.32 cm Porapak R column programmed from 80-200°C at 20°C/min.
A standard method for the analysis of methanol in raw, waste and
potable waters has been published by the UK Standing Committee of
Analysts (1982). The method is based on direct injection GC-FID using
a 2 m stainless steel column with 15% carbowax 1540 m chromosorb
W80-100 DMCS. The limit of detection is 0.11 mg/litre.
2.4.1.5 Methanol in soils
The biodegradation of methanol in gasolines by various soils was
determined by Novak et al. (1985). Methanol extracted in water (25%
v/v) was measured by direct injection GC-FID using a 2.1 m × 3 mm
stainless steel column packed with 0.2% Carbowax 1500 0n 80/100 mesh
Carbopak C at 120°C isothermal.
2.4.2 Foods, beverages and consumer products
Lund et al. (1981) determined methanol in orange and grapefruit
juice, fresh and canned, by GC-FID using a 1.5 × 3 mm column packed
with 50/80 mesh Porapak Q at 100°C with injector port and detector
block at 200°C.
Greizerstein (1981) utilized GC-FID and GC-MS for the analysis of
alcohols, aldehydes and esters in commercial beverages (beers, wines,
distilled spirits). Separations were carried out using a 3 m × 2 mm
internal diameter glass column packed with 30% Carbowax 20 M at 150°C.
A more satisfactory separation of methanol from the other congeners
was achieved using a 180-cm Porapak P column. Methanol was found at
levels of 6-27 mg/litre beer; 96-321 mg/litre in wines and
10-220 mg/litre in distilled spirits. Methanol in distilled liquors
and cordials has been determined by GC-FID (AOAC, 1990).
Rastogi (1993) analysed methanol content of 26 model and hobby
glues and found methanol in 12 of them by head-space GC-FID employing
capillary columns of different polarity. The polar GC column was a
Supelcowax 10, 60 m × 0.32 mm internal diameter; and the non-polar
column was a CP-Sil-5 CB, 50 m × 0.32 mm. The detection limit for
methanol was 20 mg/litre.
Methanol in wine vinegars was determined by GC-MS (Blanch et al.,
1992). Methanol with many other minor volatile components was
fractionated using a simultaneous distillation extraction technique
before GC analysis on a 4 m × 0.85 mm internal diameter micropacked
column coated with a mixture of Carbowax and bis-(2-ethylhexyl)-
sebecate (92:8), 4% on desilanized Volaspher A-2. The column
temperature was 60°C and the injector and FID detector were at 180°C.
2.4.3 Biological materials
A variety of primarily gas chromatographic methods have been
utilized for the determination of methanol in biological samples from
normal, poisoned and occupationally exposed individuals. Methanol
exposure has been measured in exhaled breath, blood and urine samples.
2.4.3.1 Methanol in exhaled air
Prior to analysis, expired air samples are normally collected in
sampling bags or glass containers or after preconcentration on Tenax
or other solid sorbents in adsorbent tubes and thermally desorbed, or
utilizing cryotraps (Franzblau et al., 1992a).
Free methanol has been detected and measured by GC in the expired
air of normal healthy humans with separations made on 1.52 m × 0.3 cm
columns filled with Anakrom ABS, 70-80 mesh coated with 2% N,N,-N,-N-
tetramethyl azeleamide and 8% behenyl alcohol at 86°C. The
concentration of methanol in nine subjects ranged from
0.06-0.32 µg/litre (Eriksen & Kulkarni, 1963). Methanol was only
infrequently detected in samples of human expired air and saliva by
Larsson (1965) employing GC-FID and a 1.75 mx 3.5 mm internal diameter
glass column containing polyethylene glycol (M=1500) 20% on Chromosorb
W.
Methanol in expired air and in head-space analysis of plasma was
determined as the nitrite ester utilizing GC-MS (Jones et al., 1983).
Condensed expired air samples were analysed on Porapak Q and the assay
of methanol nitrite ester was accomplished on a 2 m × 2 mm internal
diameter silanized glass column containing Tenax GC (30-60 mesh) at
60°C.
Krotosynski et al. (1977) analysed expired air from normal
healthy subjects using for sample preconcentration a 18 cm × 6 mm o.d.
stainless steel column containing Tenax GC. Sample analysis was
performed using GC-FID and a 91 m × 6 mm stainless steel column coated
with Emulphoron-870. Apart from methanol, 102 organic compounds were
detected.
Alveolar air of workers exposed to methanol was first collected
in gas sampling tubes and then analysed by GC-FID using a Porapak Q
(80-100 mesh) column at 150°C (Baumann & Angerer, 1979).
The detection of methanol and other endogenous compounds in
expired air by GC-FID with on-column concentration of sample and
separation on a 1.5 m × 3 mm o.d. stainless steel column packed with
Porapak Q, 80-100 mesh maintained at 35°C was described by Phillips &
Greenberg (1987).
The expired air of volunteer subjects exposed for periods of
about 90 min to atmospheres artificially contaminated with low levels
of methanol (ca. 130 mg/m3 (100 ppm)) was monitored during and
after the exposure using an atmospheric pressure ionization mass
spectrometer (API/MS) fitted with a direct breath analysis system
(Benoit et al., 1985).
A transportable Fourier Transform Infrared (FTIR) spectrometer
was utilized for the analysis of methanol vapour in alveolar and
ambient air in humans exposed to methanol vapour. The infrared
spectrum region used for methanol quantification was in the 950-1100
cm region. For the analysis of methanol in alveolar air with FTIR the
limit of detection for methanol was 0.4 mg/m3 (0.32 ppm), and for
methanol in ambient air the detection limit was 0.13 mg/m3 (0.1 ppm)
(Franzblau et al., 1992a).
2.4.3.2 Methanol in blood
A number of methods have been used to extract methanol from blood
prior to analysis including purge-and-trap, head-space analysis and
solvent extraction.
Baker et al. (1969) reported the simultaneous determination of
lower alcohols, acetone and acetaldehyde in blood by GC-FID utilizing
a 183 cm × 5 mm internal diameter column containing Porapak Q operated
at 100°C. The method did not require precipitation of protein prior to
analysis.
Methanol in whole blood and serum was analysed by GC-FID
employing 1.2 m and 1.8 m × 3 mm internal diameter glass columns
packed with 20% Hallcomid or 10% Carbowax on 60-80 mesh Diatopor TW
operated at 70°C (Mather & Assimos, 1965).
Blood serum was deproteinized and acetone and aliphatic alcohols
including methanol were determined by GC-FID using a pre-column of 3%
OV-1 on Gas Chrom Q and an analytical 30-m capillary column packed
with SPB-1 and operated at 35°C. Methanol and other alcohols were
separated in less than 3 min (Smith, 1984).
Methanol in deproteinized blood samples from occupationally
exposed workers was quantified by GC-FID employing a 1.8 m × 4 mm
internal diameter glass column packed with 60-80 mesh Carbopak B/5%
Carbowax 20M at 60°C. The detection limit for methanol was about
0.4 µg/ml (Lee et al., 1992).
Methanol in blood of occupationally exposed workers was
determined by head-space GC-FID utilizing a column containing 15%
Carbowax 1599 on diatomaceous earth, 70-80 mesh and operated at 70°C.
The detection limit was 0.6 mg/litre (Heinrich & Angerer, 1982).
The simultaneous determination of methanol, ethanol, acetone,
isopropanol and ethylene glycol in plasma by GC-FID was accomplished
using a 180 cm × 4 mm internal diameter glass column packed with
Porapak Q, 50-80 mesh. The column temperature was programmed from
199-210°C at 2°C/min, and the injection port and detector temperatures
were 210°C and 240°C respectively. The detection limit for methanol
was 0.1 nmol/ml. The procedure was recommended for methanol and
ethylene glycol intoxication cases (Cheung & Lin, 1987).
Methanol in blood from occupationally exposed workers was
determined directly without further pretreatment by GC-FID using a 4 m
× 3 mm glass column packed with 10% SBS 100 on Shimalite TPA, 60-80
mesh. The detector and oven were heated at 180°C and 60°C,
respectively (Kawai et al., 1991a).
Head-space GC-FID on methanol in blood from workers exposed at
sub-occupational exposure limits was reported by Kawai et al. (1992).
A 30 m × 0.53 mm capillary column coated with 1.0 um DB-Wax was used
with the injection port and detector heated at 200°C and the oven
temperature kept at 40°C for 1 min after the injection and then
elevated at a rate of 5°C/min to 110°C for 15 min. The detection limit
for methanol in blood was 100 µg/litre.
Leaf & Zatman (1952) utilized a colorimetric procedure for the
determination of methanol in air as well as in the blood and urine of
occupationally exposed workers in a methanol synthesis plant. The
procedure involved acid permanganate oxidation of methanol to
formaldehyde, which was then determined with a modified Schiff's
reagent. Concentrations of methanol up to 150 mg/litre were determined
to within 3%.
Determination of methanol in patients with acute methanol
poisoning was accomplished with a colorimetric procedure following
permanganate oxidation to formaldehyde and the subsequent reaction
with chromotropic acid (1,8-dihydroxy naphthalene 3,6-disulfonic
acid). Quantitative recovery of 100% was found for methanol following
the analysis of 3 ml of plasma, which required 45 min (Hindberg &
Wieth, 1963).
Accumulation of methanol in blood was detected in alcoholic
subjects during a 10-15 day period of chronic alcohol intake using
GC-FID and a 1.8 m column packed with Porapak Q, 80-100 mesh, or
Chromosorb 101 operated at 140°C (Majchrowicz & Mendelson, 1971). The
identity of methanol was also confirmed chemically using the
specificity of the colour reaction between permanganate and
formaldehyde.
Head-space GC was used to determine the concentrations of
methanol and ethanol in blood samples from 519 individuals suspected
of drinking and driving in Sweden. Methanol was determined in whole
blood without prior dilution with an internal standard. Carbopack C
(0.2% Carbowax 1500) was used as the stationary phase and the oven
temperature was 80°C (Jones & Lowinger, 1988).
Methanol in whole blood of poisoned patients was determined
without pretreatment by GC-FID using a 1800 mm × 4 mm internal
diameter glass column packed with 80-100 mesh Carbopack C/0.2% CW 1500
operated at 80°C; the detector temperature was 120°C (Jacobsen et al.,
1982a).
Serum methanol concentrations in men after oral administration of
the sweetening agent aspartame were determined by GC-MS utilizing a
fused silica capillary column 26 m × 0.22 mm internal diameter of
CPWAX 57 CB operated at 50°C isothermally (Davoli et al., 1986).
Methanol and formate in blood and urine of rats administered
methanol intravenously was determined by HPLC employing a REZEX-ROA-
organic acid column (300 mm × 7.8 mm internal diameter) and a
similarly packed pre-column (50 mm × 4.6 mm internal diameter). The
mobile phase was 0.043 N sulfuric acid with 10% acetonitrile at a flow
rate of 1 ml/min (Horton et al., 1992).
Methanol in serum has also been determined by high-field (500
MHZ) proton nuclear magnetic resonance at the 3.39 singlet peak. For
serum containing 20-500 mg of added methanol/litre, peak area was a
linear function of concentration (r=0.998). This NMR technique
permitted the determination of methanol and acetone in blood serum at
a level of less than 1mM (Bock, 1982).
Pollack & Kawagoe (1991) determined methanol in deproteinized
whole blood of rats by capillary GC-FID with direct column injection
utilizing a 15 m × 0.54 mm internal diameter fused silica capillary
column coated with Carbowax and operated at 35°C. The limit of
detection was 2 µg/ml.
2.4.3.3 Methanol in urine
Sedivec et al. (1981) determined methanol in urine in five
volunteers exposed to methanol vapour for 8 h. Head-space GC-FID was
used with a 120 cm × 3 mm column packed with Chromosorb 102, 60-80
mesh at 120°C. The detection limit of methanol was 0.1 mg/litre. The
methanol content in urine of 20 subjects occupationally exposed to
methanol was determined by head-space GC-FID utilizing a column
containing Porapak QS, 80-100 mesh and operated at 130°C. The
detection limit was 0.6 mg/litre (Heinrich & Angerer, 1982).
Methanol in the urine of exposed workers was determined by
head-space GC-FID using a 4.1 m × 3.2 mm glass column containing 10%
SBS-100 on Shimalite TPA, 60-80 mesh. The oven and injection port
temperatures were 60°C and 180°C respectively. The limit of detection
for methanol in urine was 0.1 mg/litre (Kawai et al., 1991b, 1992).
Urinary methanol as a measure of occupational exposure was
determined by GC-FID utilizing a 2 m glass column packed with Porapak
Q, 80-100 mesh. The detection limit for methanol was 0.32 mg/litre
(Liesivouri & Savolainen, 1987).
Urine concentrations of methanol in volunteers who had ingested
small amounts of methanol was determined by head-space GC-FID using
Tenax GC as the column packing (Ferry et al., 1980).
2.4.3.4 Methanol in miscellaneous biological tissues
Methanol and other alcohols have been determined in tissue
homogenates either per se or as their nitrite esters by GC-FID
employing a 1.8 m × 6 mm o.d. glass column packed with Chromosorb 101
operated at 145°C. The sensitivity was 8 µg per g of tissue (Gessner,
1970).
2.4.3.5 Methanol metabolites in biological fluids
The principal metabolite of methanol in humans and monkeys is
formate and it has been shown that accumulation of blood formate at
higher levels of methanol exposure coincides with the development of
metabolic acidosis and visual system toxicities (Clay et al., 1975;
McMartin et al., 1975; Baumbach et al., 1977; Tephly, 1991). Formate
is an endogenous product of single carbon metabolism and is normally
found in the urine of healthy individuals.
Formate has been analysed in blood and urine samples primarily by
enzymatic methods with a colorimetric or fluorimetric end-point or by
derivatization followed by analysis by GC-FID. Formate in plasma has
also been determined by isotachophoresis (Sejersted et al., 1983).
Ferry et al. (1980) measured formic acid as an ethyl ester formed
by the treatment of urine with 30% sulfuric acid in ethanol. The
samples were analysed by head-space GC-FID on a column packed with 10%
silar 10C on Chrom Q.
The analysis of formic acid in blood was performed via an initial
transformation of formic acid by concentrated sulfuric acid into water
and carbon monoxide, the latter being reduced to methane on a
catalytic column and analysed directly by GC-FID (Angerer & Lehnert,
1977; Baumann & Angerer, 1979; Heinrich & Angerer, 1982).
Urinary formic acid was determined after the methylation of the
acid and its conversion to N,N-dimethylformamide with GC-FID equipped
with a 50-m silica capillary column containing Carbowax 20M liquid
phase. The detection limit was 2.3 mg/litre (Liesivouri & Savolainen,
1987).
Franzblau et al. (1992b) found that urinary formic acid in
specimens collected 16 h following cessation of methanol exposure and
analysed by head-space GC-FID may not be an appropriate approach to
assess methanol exposure biologically.
Enzymatic methods for the determination of formate are based
primarily on the enzyme-catalysed conversion of formate to carbon
dioxide in the presence of nicotinamide adenine dinucleotide (NAD),
generating NADH as the other reaction product. NADH formation can be
subsequently measured directly or reacted in a coupled reaction to
generate a fluorescent or coloured complex.
A specific assay for formic acid in body fluids based on the
reaction of formate with bacterial formate dehydrogenase coupled to a
diaphorase-catalysed reduction of the non-fluorescent dye resazurin to
the fluorescent substance resorufin was reported by Makar et al.
(1975) and Makar & Tephly (1982). This permitted the accurate
determination of about 6 mg formate/litre blood at excitation
wavelength of 565 nm and an emission wavelength of 590 nm (Makar et
al., 1975; Makar & Tephly, 1982).
A serum formate enzymic assay based on modifications of the
formate dehydrogenase (FDH)-diaphorase procedure using NAD-diaphorase-
iodonitrotetrazolium violet to develop a red-coloured complex, which
is measured at 500 nm, was described by Grady & Osterloh (1986). The
calibration curve was linear over the formate range of 0 to
400 mg/litre.
Formate in plasma was determined by Lee et al. (1992) employing
an enzymatic procedure (Grady & Osterloh, 1986; Buttery & Chamberlin,
1988) and measured spectrophotometrically at 510 nm. The detection
limit was about 3 µg/ml.
Lee et al. (1992) determined that formate associated with acute
methanol toxicity in humans does not accumulate in blood when
atmospheric methanol exposure concentrations are below the
occupational threshold limit value of 260 mg/m3 (200 ppm) for 6 h in
exposed healthy volunteers.
d'Alessandro et al. (1994) found that serum and urine formate
determinations were not sensitive biological markers of methanol
exposure at the threshold limit value (TLV) in human volunteers.
Formate in serum was analysed by the enzymatic-colorimetric procedure
of Grady & Osterloh (1986). The sensitivity of the method was
0.5 mg/litre of formate in serum.
Buttery & Chamberlin (1988) developed an enzymatic method for the
determination of abnormal levels of formate in plasma requiring no
deproteinization and utilizing a stable colour reagent consisting of
phenazine methosulfate, p-iodonitrotetrazolium and NAD to produce a
stable red formazan colour. The precision at 1.0 and 5.0 mmol/litre
formate was 2.9% and 1.7%, respectively, within-day and 5.5% and 2.3%,
respectively, between days.
Urinary formic acid was determined using formate dehydrogenase
(FDH) in the presence of NAD. The detection limit was 0.5 mg/litre.
The normal formic acid excretion in urine is between 2.0 and
30 mg/litre (Triebig & Schaller, 1980).
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Methanol occurs naturally in humans, animals and plants (Axelrod
& Daly, 1965; CEC, 1988). It is a natural constituent of blood, urine
and saliva (Leaf & Zatman, 1952) and expired air (Erikssen & Kulkarni,
1963; Larsson, 1965; Krotosynski et al., 1979; Jones et al., 1990),
and has also been found in mother's milk (Pellizzari et al., 1982).
Humans have a background body burden of 0.5 mg/kg body weight (Kavet
& Nauss, 1990).
Levels of methanol in expired air are reported to range from 0.06
to 0.49 µg/litre (46-377 ppb) (Eriksen & Kulkarni, 1963). Methanol has
been detected in the expired air of normal, healthy non-smoking
subjects at a mean level of 0.5 ng/litre (Krotosynski et al., 1979).
It is believed that dietary sources are only partial contributors
to the total body pool of methanol (Stegink et al., 1981). It has been
suggested that methanol is formed by the activities of the intestinal
microflora or by other enzymatic processes (Axelrod & Daly, 1965). The
methanol-forming enzyme was shown to be protein carboxylmethylase, an
enzyme that methylates the carboxyl groups of proteins (Kim, 1973;
Morin & Liss, 1973).
Natural emission sources of methanol include volcanic gasses,
vegetation, microbes and insects (Owens et al., 1969; Holzer et al.,
1977; Graedel et al., 1986). Isidorov et al. (1985) identified
methanol emissions of evergreen cyprus in the forests of Northern
Europe and Asia. Methanol was identified as one of the volatile
components emitted by alfalfa (Owens et al., 1969) and it is formed
during biological decomposition of biological wastes, sewage and
sludges (US EPA, 1975; Howard, 1990; Nielsen et al., 1993).
3.2 Anthropogenic sources
The major anthropogenic sources of methanol include its
production, storage and use, principally its use as a solvent, as a
chemical intermediate, in the production of glycol ethers, and in the
manufacture of charcoal, and exhaust from vehicle engines (US EPA,
1976a,b, 1980a,b; CEC, 1988).
3.2.1 Production levels and processes
3.2.1.1 Production processes
The earliest important source of methanol ("wood alcohol") was
the dry distillation of wood at about 350°C, which was employed from
around 1830 to 1930. In countries where wood is plentiful and wood
products form an important industry, methanol is still obtained by
this procedure (ILO, 1983).
In 1880, about 1.5 million litres of wood alcohol were produced
in the USA while in 1910 the amount had increased to over 3 million
litres (Tyson & Schoenberg, 1914). However methanol produced from wood
contained more contaminants, primarily acetone, acetic acid and allyl
alcohol, than the chemical-grade methanol currently available
(Grayson, 1981; Elvers et al., 1990). Methanol was also produced as
one of the products of the non-catalytic oxidation of hydrocarbons (a
procedure discontinued in the USA in 1973), and as a by-product of
Fischer-Tropsch synthesis, which is no longer industrially important
(Grayson, 1981).
Modern industrial scale methanol production is based exclusively
on the catalytic conversion of pressurized synthesis gas (hydrogen,
carbon monoxide and carbon dioxide) in the presence of metallic
heterogenous catalysts. All carbonaceous materials such as coal, coke,
natural gas, petroleum and fractions obtained from petroleum (asphalt,
gasoline, gaseous compounds) can be employed as starting materials for
synthesis gas production (Grayson, 1981; Elvers et al., 1990).
The required synthesis pressure is dependant upon the activity of
the particular metallic catalyst employed, with copper-containing zinc
oxide-alumina catalysts being the most effective in industrial
methanol plants (Elvers et al., 1990). By convention the processes are
classified according to the pressure used: low-pressure processes,
50-100 atmospheres; medium-pressure processes, 100-250 atmospheres;
and high-pressure processes, 250-350 atmospheres. Low-pressure
technology is the most widely employed globally and accounted for 55%
of the USA methanol capacity in 1980 (Grayson, 1981).
Almost all the methanol produced in the USA is made from natural
gas. This is steam reformed to produce synthesis gas, which is
converted to methanol by low-pressure processes. A small amount of
methanol is obtained as a by-product from the oxidation of butane to
produce acetic acid and from the destructive distillation of wood to
produce charcoal (Grayson, 1981; Elvers et al., 1990).
The composition of methanol obtained directly from synthesis
without any purification or with only partial purification varies
according to the synthesis (e.g., pressure, catalyst, feedstock). The
principal impurities include 5-20% (by volume) water, higher alcohols
(principally ethanol), methyl formate and higher esters, and smaller
amounts of ethers and aldehydes (Grayson, 1981; Elvers et al., 1990).
Methanol is purified by distillation, the complexity required
depending on the desired methanol purity and the purity of the crude
methanol (Grayson, 1981; Elvers et al., 1990).
Natural gas, petroleum residues and naphtha accounted for 90% of
worldwide methanol capacity in 1980, miscellaneous off-gas sources
constituting the remaining 10%. Natural gas alone accounted for 70%,
petroleum residues 15%, and naphtha 5% (Grayson, 1981). Natural gas
feedstock accounted for 75% in the USA and 70% of global capacity in
1980. Methanol produced from residual oil accounted for approximately
15% of USA and worldwide capacity in 1980, while naphtha and coal
feedstocks accounted for approximately 5% and 2%, respectively, of
worldwide methanol capacity in 1980 (Grayson, 1981). About 90% of the
global methanol capacity is currently based on natural gas (SRI,
1992).
The production of methanol from coal, being independent of oil
and natural gas supplies, is noted to be an attractive alternative
feed stock in some quarters (Grayson, 1981; CEC, 1988). Newer
approaches to the production of methanol that have been suggested
include the catalytic conversion from carbon dioxide and hydrogen
avoiding conventional steam reforming (Rotman, 1994a) and the direct
catalytic conversion of methane to methanol (Rotman, 1994b).
3.2.1.2 Production figures
As shown in Table 2, worldwide annual capacity for methanol
production has increased over the past decades from approximately 15 ×
106 tonnes in 1979 (Grayson, 1981) to 21 × 106 tonnes in 1989
(Elvers et al., 1990) and more than 22.1 × 106 tonnes in the
beginning of 1991 (SRI, 1992). Worldwide demand was projected to rise
further to about 25.8 × 106 tonnes in 1994 (Anon., 1991; Nielsen et
al., 1993) and 30.1 × 106 tonnes in 1995 (SRI, 1992). The data
available do not allow capacity and production figures to be compared;
however, it is assumed that approximately 80% of production capacity
is utilized (Fiedler et al., 1990).
The USA and Canada are the largest methanol-producing countries.
About 85% of Canada's production is exported to the USA, Japan and
Europe (Heath, 1991). In Western Europe, Germany, the Netherlands and
the United Kingdom are the major methanol-producing countries,
accounting for 7%, 3% and over 2% of the world capacity, respectively
(SRI, 1992). The production of methanol in Germany in 1991 and 1992
amounted to 715 000 and 770 000 tonnes respectively.
The annual capacity in Eastern Europe was estimated to be 5.8 ×
106 tonnes in 1987. The production in the former USSR was 3.28 × 106
tonnes and 3.21 × 106 tonnes in 1987 and 1988, respectively (Rippen,
1990).
Table 2. Methanol production or production capacity (× 106 tonnes per year) from 1978 to 1995
Year World-wide USA Canada Western Japan Capacity/ Reference
Europe production
1978 12 3.4 3 1 capacity Grayson (1981)
production
1979 15 4.05 3.45 1.35 capacity Grayson (1981)
1980 2.5 production CEC (1988)
1981 8 production CEC (1988)
1983 15.9 5.52 (33%) 1.75 (11%) 2.53 1.27 (8%) capacity SRI (1992)
production CEC (1988)
1988 1.91 production Anderson (1993)
1989 21 capacity Elvers et al. (1990)
19 production
1990 22.3 capacity Anon. (1991);
Nielsen et al. (1993)
1991 22.1 4.42 (20%) 2.21 (10%) 2.65 (12%)a 0.22 (1%) capacity SRI (1992)
1991 2.22 0.077 production Anderson (1993)
1992 2.15 0.034 production Anderson (1993)
1992 3.66 2.15 production Reisch (1994)
1993 4.78 production Reisch (1994)
1995 30.1 capacity SRI (1992)
a Only Germany, the Netherlands and the United Kingdom.
The figures in Table 2 indicate a major shift in methanol
production from the developed countries to the developing areas. In
fact, the methanol industry underwent large structural changes during
the 1980s as a result of the discovery of large natural gas fields in
remote regions having little demand for natural gas themselves. Since
methanol production is a very suitable alternative for marketing
natural gases, a number of methanol production plants for export were
built or proposed to be built in Asia (Bahrein, Oman, Qatar, Saudi
Arabia, Indonesia, Malaysia), South America (Chile, Mexico,
Venezuela), the Caribbean (Trinidad) and in New Zealand and Norway
(Fiedler et al., 1990; SRI, 1992). The largest single train plant
based on this concept came on stream in southern Chile in 1988 with an
annual output of 750 000 tonnes (Fiedler et al., 1990).
Future trends in methanol production and demand are being driven
to a large extent by increasing demand for methyl tertiary butyl ether
(MTBE), which is used in gasoline blending as an octane enhancer and
to reduce carbon monoxide emissions (Anon., 1991; Morris, 1993;
Nielsen et al., 1993).
3.2.2 Uses
During the 1890s, the market for methanol (then better known as
wood alcohol) increased as a commercial product and as a solvent for
use in the workplace. It was included in many consumer products such
as witch hazel, Jamaica ginger, vanilla extract and perfumes (Wood &
Buller, 1904). The most notorious use of wood alcohol was and
continues to be as an adulterant in alcoholic beverages, which has led
to large-scale episodes of poisonings since 1900 (Bennett et al.,
1953; Kane et al., 1968).
Historically, in terms of commercial usage, about half of all
methanol produced has been used to produce formaldehyde. Other earlier
large-volume chemicals based on methanol include acetic acid, dimethyl
terephthalate, glycol methyl ethers, methyl halides, methylamines,
methyl acrylate and various solvent uses (Grayson, 1981; CEC, 1988;
Elvers et al., 1990; Nielsen et al., 1993).
3.2.2.1 Use as feedstock for chemical syntheses
Approximately 70% of the methanol produced worldwide is used as
feedstock for chemical syntheses. As shown in Table 3, formaldehyde,
methyl tertiary butyl ether (MTBE), acetic acid, methyl methacrylate,
and dimethyl terephthalate are, in order of importance, the main
chemicals produced from methanol. Methyl halides produced from
methanol include methyl chloride, methylene chloride and chloroform.
Nearly all the formaldehyde manufactured worldwide is produced by
oxidation of methanol with atmospheric oxygen. The annual formaldehyde
production was projected to increase at a rate of 3%, but because
other bulk products have higher growth rates, its relative importance
with respect to methanol use has decreased (Elvers et al., 1990;
Fiedler et al., 1990).
Table 3. Use pattern for methanol (as a percentage of production) according to region and year
Global Global USA USA Japan Western Europe Brazil
1979 1988 1973 1985 n.g. 1985 n.g.
Use for synthesis of:
formaldehyde 52 40 39 30 47 50 60
MTBE 4 20 8 - 5 -
acetic acid 6 9 3.4 12 10 5 -
dimethyl terephthalate 4 6.1 4 1 4 16
methyl methacrylate 4 3.7 4 6 3 2
methyl halides 8a 6.1 9 3 6 -
methyl amines 3.3 4 2 4 9
glycol methyl ethers 1.1
Direct use
solvent 10 6 6 2
fuel 6 - 5 -
Miscellaneous 14 16.9 13 25 12 11
Referenceb [1] [2] [3] [4] [4] [4] [4]
a together with methyl amines production
b Reference: [1] Kennedy & Shanks (1981); [2] Elvers et al. (1990); [3] US EPA (1980a); [4] Rippen (1990)
n.g. = year not given
MTBE has become an important octane-enhancing blending component
in gasoline, particularly in the USA where the Clean Air Act
Amendments of 1990 have prompted further steps toward reducing
emissions from motor vehicles by changing the formulations of
gasoline. This is achieved by using so-called oxygenated fuel, i.e.
fuel containing at least 2% oxygen by weight in the form of
oxygenates, but less benzene and other aromatic compounds than
conventional fuel (Health Effects Institute, 1996). MTBE is produced
by reacting methanol with isobutene in acid ion exchangers. In 1987,
MTBE (production of 1.6 × 106 tonnes) ranked 32nd among the top 50
chemicals produced in the USA (Scholz et al., 1990). In 1993, 11 ×
106 tonnes were produced, ranking MTBE ninth of the top 50 chemicals
(Reisch, 1994).
Acetic acid is produced by carbonylation of methanol with carbon
monoxide. Annual growth rates of 6% have been estimated (Fiedler et
al., 1990).
Methanol is present in a broad variety of commercial and consumer
products including shellacs, paints, varnishes, mixed solvents in
duplicating machines (95% concentration or greater), antifreeze and
gasoline deicers (generally containing 35-95% methanol), windshield
washer fluid (contains 35-90% methanol), cleansing solutions
(containing around 5% methanol), model and hobby glues and adhesives,
and Sterno ("canned heat") containing 4% methanol (Posner, 1975; US
EPA, 1980a; CEC, 1988; ATSDR, 1993).
Methanol is also used in the denitrification of wastewater,
sewage treatment application (carbon source for bacteria to aid in the
anaerobic conversion of nitrates to nitrogen and carbon dioxide), as a
substrate for fermentation production of animal feed protein (single
cell protein), as a hydrate inhibitor in natural gas, and in the
methanolysis of polyethylene terephthalate (PET) from recycled plastic
wastes (Posner, 1975; US EPA, 1980a; Kennedy & Shanks, 1981; ATSDR,
1993).
3.2.2.2 Use as fuel
Methanol is a potential substitute for petroleum. It can be
directly used in fuel as a replacement for gasoline in gasoline and
diesel blends. Methanol is in favour over conventional fuels because
of its lower ozone-forming potential, lower emissions of some
pollutants, particularly benzene and polycyclic aromatic hydrocarbons
and sulfur compounds, and low evaporative emissions. On the other
hand, the possibility of higher formaldehyde emissions, its higher
acute toxicity and, at present, lower cost-efficiency favour
conventional fuels (CONCAWE, 1995).
For use in gasoline engines, pure methanol (so-called M100 fuel)
or mixtures of 3, 15 and 85% methanol with conventional petroleum
products (M3, M15, M85) are most common. In diesel engines methanol
cannot be used as an exclusive fuel because of its low cetane number
that would impose proper ignition. Therefore, methanol is injected
into the cylinder after ignition of the conventional diesel fuel
(Fiedler et al., 1990).
3.2.2.3 Other uses
Methanol is used in refrigeration systems, e.g., in ethylene
plants, and as an antifreeze in heating and cooling circuits. However,
its use as an engine antifreeze has been replaced by glycol-based
products. Methanol is added to natural gas at the pumping stations of
pipelines to prevent formation of gas hydrates at low temperature and
can be recycled after removal of water. Methanol is also used as an
absorption agent in gas scrubbers to remove, for example, carbon
dioxide and hydrogen sulfide. According to Table 3, large amounts of
methanol are used as a solvent. Pure methanol is not usually used
alone as a solvent, but is included in solvent mixtures (Fiedler et
al., 1990).
3.2.2.4 Losses into the environment
Given the high production volume, widespread use and physical and
chemical properties of methanol, there is a very high potential for
large amounts of methanol to be released to the environment,
principally to air (US EPA, 1976a,b, 1980a,b, 1994; Nielsen et al.,
1993). Emissions of methanol primarily occur from miscellaneous
solvent usage, methanol production, end-product manufacturing, and
bulk storage and handling losses. The largest source of emissions of
methanol is the miscellaneous solvent use category.
US EPA (1980b) estimated emission factors for the release of
methanol and volatile organic compounds (VOC) from the low-pressure
synthesis of methanol from natural gas in a model plant with a
capacity of 450 000 tonnes/year. The process and capacity were typical
of those built in the late 1970s. The overall emission factors were
estimated to be: uncontrolled emissions, 1.56 kg methanol/tonne
produced; controlled emissions, 0.14 kg methanol/tonne produced
(Nielsen et al., 1993).
It was estimated that about 1% of the methanol used in the
production of formaldehyde would be released to the environment during
the production process by which formaldehyde is produced by either a
metallic silver-catalyst process or a metal oxide-catalyst process (US
EPA, 1976a; 1980b). In the oxidation-dehydrogenation process with
metallic silver catalyst, 0.89 kg methanol/tonne of 39% (by weight)
formaldehyde solution was released principally from the product
absorber vents, and 1.24 kg methanol/tonne from the fractionator
vents. The production of formaldehyde using the catalytic oxidation,
metal oxide catalyst process resulted in the release of 1.93 kg
methanol/tonne of 37% formaldehyde solution with emissions from the
absorber vent (US EPA, 1980b).
US EPA (1994) reported that methanol was the most released
chemical to the environment (air, water and land) based on the 1992
Toxic Release Inventory which utilized 81 016 individual chemical
reports from a total of 23 630 facilities (approximately 65% of
facilities reporting). The air, water and land releases of methanol
totalled 1.09 × 105 tonnes, consisting of 1.53 × 104 tonnes of
fugitive or non-point air emissions, 72 956 tonnes of stack or point
air emissions, 7444 tonnes of surface water discharges and 15 095
tonnes released to land. Additionally, 1.283 × 104 tonnes were
transferred via underground injection.
Methanol had the largest off-site transfers (51 672 tonnes) to
publicly owned treatment works (POTWs) in 1992. During the same
period, methanol ranked third largest of the Toxic Release Inventory
Chemicals with off-site transfers for treatment. The total transfers
to treatment were 18 098 tonnes, consisting of 4 tonnes for
solidification, 10 295 tonnes for incineration/thermal treatment, 1971
tonnes of incineration/insignificant fuel value; 5311 tonnes for
wastewater treatment and 147 tonnes to waste broker-waste treatment. A
total of 493 980 tonnes of methanol was treated, consisting of 260 875
tonnes treated on-site and 197 400 tonnes off-site. A total of 1510
tonnes of methanol was released to land, primarily to on-site
landfills (US EPA, 1994).
The total amount of methanol release in Canada in 1993 was
306 222 tonnes distributed as follows: air, 15 326; water, 14 248;
underground, 819 and land, 205 (Ministry of Supply & Services Canada,
1993).
Tail pipe emissions as well as evaporative emissions are
monitored by a number of agencies. Emissions and air quality modelling
results have been reported from methanol/gasoline blends in prototype
flexible/variable fuelled vehicles (US EPA, 1991; Auto/Oil Air Quality
Research Program, 1992, 1994). Motor vehicle emissions are affected in
various ways by the use of methanol fuels in production flexible/
variable fuel vehicles. Higher molecular weight hydrocarbons are
reduced and carbon monoxide is reduced under some circumstances, while
increases in methanol and formaldehyde can occur (US EPA, 1991).
Methanol has been found in significant amounts in the exhaust
from gasoline-powered vehicles as well as in diesel exhausts. Methanol
was measured at levels of 100-226 mg/kg in the exhaust emissions from
non-catalyst vehicles fuelled with isobutane/methanol/gasoline
(2/15/83; M-15). Methanol emissions from a light-duty diesel vehicle
fuelled with 95% methanol were one order of magnitude higher
(3.4 g/kg) (Jonsson et al., 1985).
Chang & Rudy (1990) reported methanol emission factors for
vehicles fuelled by M-85 (85% methanol + 15% gasoline) and M-100 (100%
methanol) in the USA. For M-85-fuelled vehicles, factors were 0.156-
0.7 g methanol/mile driven in exhaust emissions and 0.055-0.25 g
methanol/mile driven in evaporative emissions. For M-100 fuelled
vehicles, they were 0.5 g methanol/mile driven in exhaust emissions
and 0.072-0.134 g methanol/mile driven in evaporative emissions.
Methanol was found at levels of 130-800 µg/m3 (0.1 to 0.6 ppm)
in the exhaust from nine hydrocarbon test fuels, e.g., iso-octane,
iso-octene, benzene, 2-methyl-2-butene, toluene, o-xylene,
benzene/ n-pentane, toluene/ n-pentane and iso-octane/toluene/
iso-octene (Seizinger & Dimitriades, 1972).
Methanol, formaldehyde and hydrocarbon emissions from methanol-
fuelled cars were reported by Williams et al. (1990). The variable
methanol-fuelled vehicles using fuel mixtures of 100, 85, 50, 15 and
0% methanol and a dedicated methanol vehicle all gave similar emission
patterns. The organic composition of the exhaust was 85-90% methanol,
5-7% formaldehyde and 3-9% hydrocarbons.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
Methanol is released into the environment from both natural and
man-made sources, the latter being the most significant. Methanol
is released predominantly from its production and use as a solvent
in industrial processes (in extraction, washing, drying and
recrystallization operations), and to a lesser degree from a variety
of industrial processes and domestic uses (US EPA, 1980a,b; Graedel et
al., 1986; CEC, 1988; Howard, 1990; Nielsen et al., 1993).
Methanol volatilization half-lives of 5.3 and 2.6 days have been
estimated for a model river (1 m deep) and an environmental pond,
respectively (Howard, 1990).
Methanol is expected to exist almost entirely in the vapour phase
in the ambient atmosphere, based on its vapour pressure (Eisenreich
et al., 1981; Graedel et al., 1986). Because of methanol's water
solubility, rain would be expected to physically remove some methanol
from the air (US EPA, 1980a,b; Snider & Dawson, 1985).
Methanol has been found in the atmosphere (Graedel et al.,
1986). It can be the product of atmospheric alkane chemistry with
concentrations as high as 131 µg/m3 (100 ppb) being found. Methanol
is expected to become an important additional trace gas in the
atmosphere due to its projected increased use as an alternative fuel
to gasoline or in a gasoline blend (CEC, 1988; Chang & Rudy, 1990).
The miscibility of methanol in water and its low octanol/water
partition coefficient suggest high mobility in soil. Lkke (1984)
studied the adsorption of methanol onto three soil types at 6°C. The
soils tested comprised two sandy soils (organic matter contents of
0.09 and 0.1%), and a clay soil (organic matter content of 0.22%).
Methanol solutions with concentrations of 0.1, 1.0, 9 and 90 mg/litre
were used in 1-h exposure studies. Adsorption coefficients for all
soil methanol concentrations and soil types ranged from 0.13 to 0.61,
indicating methanol has a low adsorptive capacity on these soils.
However Nielsen et al. (1993) suggested that the soils used in the
Lkke (1984) study had low organic matter contents compared to typical
agricultural surface soil which can have organic matter contents of 1
to 2%, and up to 5% in some soils. A soil containing a typical amount
of organic matter might therefore be expected to retain methanol and
prevent it from reaching the subsoil.
Additionally, the relatively high vapour pressure and low
adsorptive capacity suggests significant evaporation from dry
surfaces.
4.2 Transformation
4.2.1 Biodegradation
Methanol is readily biodegradable in soil and sediments, both
under aerobic and anaerobic conditions. A large number of strains/
genera of microorganisms have been identified as capable of using
methanol as a growth substrate (Hanson, 1980; Braun & Stolp,
1985; Nielsen et al., 1993). These include Pseudomonas sp.,
Methylobacterium organophilium; Hyphomicrobium sp., Desulfovibrio;
Streptomyces sp., Rhodopseudomonas acidophilia; Paracoccus
denitrificans; Microcyclus aquaticus; Thiobacillus novellus;
Micrococcus denitrificans; Achromobacter 1L (isolated from activated
sewage sludge) and Mycobacterium 50 (isolated from activated sewage
sludge). Most microorganisms possess the enzyme alcohol dehydrogenase
which is necessary for methanol oxidation. The methanogen,
Methanosarcine barkeri can grow on and produce methane from methanol
(Hippe et al., 1979).
The following genera of methanol-oxidizing yeasts have been
reported: Pichia; Saccharomyces; Hansenula; Rhodotorula; Kloechera;
Candida; Torulopsis (Stensel et al., 1973; Hanson, 1980; Nielsen et
al., 1993). Okpokwasili & Amanchukwu (1988) isolated Candida sp.
from Niger Delta sediment which utilized methanol as a growth
substrate.
4.2.1.1 Water and sewage sludge
In a closed bottle test, according to OECD guideline 301D,
methanol was found to be readily biodegradable with 99% COD removal
after the test period of 30 days (Hüls AG, 1978). In another closed
bottle test using unadapted inoculum from domestic sewage the
degradation of methanol at concentrations of 3, 7 or 10 mg/litre in
both freshwater (settled domestic wastewater) and synthetic seawater
incubated for a maximum of 20 days under aerobic conditions was
studied by Price et al. (1974). Methanol was readily degraded in both
inocula at all concentrations with average disappearance of methanol
as follows: a) after 5 days, 76% bio-oxidation in fresh water and 69%
in salt water; b) after 10 days, 88% bio-oxidation in fresh water and
84% in salt water; c) after 15 days, 91% bio-oxidation in fresh water
and 85% in salt water and d) after 20 days, 95% bio-oxidation in fresh
water and 97% in salt water.
Matsui et al. (1988) studied the biodegradability of methanol in
a batch reactor using activated sludge from an industrial wastewater
treatment plant which was acclimatized to the wastewater originating
from a petrochemical complex in Japan. Methanol at an initial
concentration of 100 mg/litre and an acclimation period of 1 day was
found to be highly biodegradable with 91% COD removal and 92% TOC
removal achieved.
Incubation of 0.05 mg methanol/litre for 5 days in activated
sludge from a municipal sewage plant resulted in the degradation of
37% of the methanol (Freitag et al., 1985). Hatfield (1957) found that
at a feed rate of 333 or 500 mg/litre, methanol was virtually
completely oxidized (with a major portion of the BOD and COD removed)
by acclimated microorganisms within 6 h in a settled domestic sewage
inoculum.
The microbial metabolism of methanol in a model activated sludge
system monitored by Swain & Somerville (1978) revealed that methanol
was not broken down when added transiently (0.23% by volume) to the
system operating with a retention time of 11 h. However adaptation of
the sludge in such a system to 0.1% by volume occurred over a period
of several days. After 2 days acclimation, about 50% of the methanol
was utilized, and after 6 days acclimation more than 80% of the
methanol had been metabolized. There were no apparent toxic effects
caused by the addition of methanol (0.1% by volume) to the sludge
prior to and after adaptation to methanol.
The anaerobic treatment of wastes containing methanol and higher
alcohols (approximately 50:50 mix) was studied by Lettinga et al.
(1981). In batch and continuous experiments using an inoculum
consisting of sugar beet waste and active anaerobic sludge, the
breakdown of methanol began within a few days while the breakdown of
higher alcohols occurred immediately depending on the initial load of
waste applied.
Denitrification is facilitated by heterotrophic and autotrophic
bacteria. Heterotrophic bacteria require a carbon source for their
growth and cell metabolism which can be supplied by methanol (Stensel
et al., 1973; Nyberg et al., 1992; Jansen et al., 1993; Upton, 1993).
Bacteria such as the organisms of the genera Pseudomonas,
Micrococcus, Achromobacter, Spirillum, and Bacillus reduce
nitrate, nitrogen oxide and nitrous oxide under anaerobic conditions.
The addition of methanol to promote denitrification has been suggested
in situations where nitrate accumulates, and methanol has been
added as an economic exogenous organic carbon source to increase
denitrification (Stensel et al., 1973; Nyberg et al., 1992; Jansen et
al., 1993; Upton, 1993).
At a wastewater treatment plant in Malmo, Sweden, complete
denitrification was obtained after approximately one month at 10°C
after methanol was added for denitrification. Microscopic examination
revealed a growing population of budding and/or appendaged bacteria,
presumably Hyphomicrobrium spp. reaching a stable maximum at the
time when optimal nitrate removal occurred (Nyberg et al., 1992)
Upton (1993) described a pilot study in the United Kingdom
indicating that denitrification in deep-bed sand filters is a feasible
technology utilizing methanol addition. Nitrogen removals greater than
70% were possible at winter sewage temperatures.
Several other laboratory studies using a variety of methodologies
have demonstrated the rapid biodegradation of methanol by sewage
organisms. These show degradation of between 66 and 95%, and usually
greater than 80%, within five days (Kempa, 1976; Hüls AG, 1978; Matsui
et al., 1988).
4.2.1.2 Soils and sediments
Methanol is biodegradable in soils and sediments, both under
aerobic and anaerobic conditions. Methanol is a normal growth
substrate for many soil microorganisms, which are capable of
completely mineralizing methanol to carbon monoxide and water (CEC,
1988; Howard, 1990; Howard et al., 1991; Nielsen et al., 1993).
Methanol at concentrations of up to 1000 mg/litre was degraded to
non-measurable amounts within a year or less in subsurface soil and
ground water sites in Pennsylvania, New York and Virginia (USA)
believed to be previously uncontaminated. Complete degradation of
100 g methanol/litre occurred in less than 30 days in one aerobic soil
sample from a Pennsylvania site (Novak et al., 1985).
Scheunert et al. (1987) monitored the formation of 14CO2 from
labelled methanol in aerobic and anaerobic suspended soil and found
methanol to be readily degradable after 5 days incubation at 35°C.
Rates and patterns of biodegradation of methanol in surface and
subsurface soils from eight sites in New York, Pennsylvania and
Virginia in static microcosms under anaerobic conditions were
evaluated by Hickman & Novak (1989) and Hickman et al. (1989). The
rates of methanol degradation varied considerably between sites, but
the soils could be characterized into two general types, namely fast
soils, in which degradation rates were high and rates were increased
by addition of nitrate and sulfate, and slow soils, in which
biodegradation rates were low and decreased by the addition of nitrate
or sulfate and inhibition of sulfate increased degradation rates.
Biodegradation rates in subsurface soils were generally within the
range of 0.5-1.1 mg/litre per day and indicated that no acclimation
period was required. Biodegradation rates were calculated and used to
estimate a half-life of between 58 and 263 days for methanol in these
soils (Hickman et al., 1989).
Compared to other substrates studied, e.g., acetate,
trimethylamine and methylamine, methanol (at concentrations less than
3 µM) was degraded relatively slowly mainly to carbon dioxide,
principally via sulfite-reducing organisms, and could not be
considered a significant in situ precursor in surface sediments of
an intertidal zone in Maine, USA (King et al., 1983).
Methanol was found to be an important substrate for methanogenic
bacteria in anaerobic sediments (highly reduced and containing methane
and hydrogen sulfide), collected from a salt marsh located in
San Francisco Bay, California. The sediments were homogenized
anaerobically with San Francisco Bay water and 310-340 µmol methanol/
flask, resulting in 83-91% conversion to methane, carbon dioxide and
water after 3 days (Oremland et al., 1982).
A sulfate-reducing bacterium of the genus Desulfovibrio, which
is capable of degrading methanol after growth on pyruvate, malate or
fumarate, completely converted anaerobic samples of 14C-methanol to
carbon dioxide. However the 14C-label was not used as a carbon source
by the bacterium and was not assimilated into cellular material (Braun
& Stolp, 1985).
4.2.2 Abiotic degradation
4.2.2.1 Water
In a 5-day experiment, 14C-labelled methanol applied to
soil-water suspensions under both aerobic and anaerobic conditions
yielded 53.4 and 46.3% 14CO2, respectively (Scheunert et al., 1987).
Half-lives of 5.1 years and 46.6 days for the photooxidation of
methanol in water have been reported based on the measured rate data
for the reaction with hydroxyl radicals in aqueous solutions (Howard
et al., 1991). A bimolecular reaction rate constant of 8.5 × 10-13
cm3/molecule per second for the reaction of methanol and hydroxyl
radicals in water has been reported by Lemaire et al. (1982).
The rate constant for the reaction of methanol with hydroxyl
radicals in aqueous solution is approximately 1 × 109 litre/mol per
second (Gurten et al., 1984). If the hydroxyl radical concentration of
sunlit natural water is assumed to be 1 × 10-17 mol/litre (Mill et
al., 1980), the half-life of methanol would be approximately 2.2 years
(Howard, 1990).
Sediment and clay suspensions did not photo-catalyse the
degradation of methanol in aqueous solution during ultraviolet
irradiation at 300 nm. However, the addition of semi-conductor powders
such as titanium dioxide led to large increases in the yield of
formaldehyde upon irradiation, in contrast to the small amounts of
formaldehyde formed from the irradiation of 10% aqueous methanol
(Oliver et al., 1979).
Hustert et al. (1981) reported that methanol in aqueous solution
was stable when exposed to sunlight. Alcohols are generally resistant
to environmental aqueous hydrolysis (Lyman et al., 1982; Howard,
1990).
4.2.2.2 Air
Methanol reacts in the atmosphere with oxidizing species (Barnes
et al., 1982; Lemaire et al., 1982; Whitbeck, 1983; Graedel et al.,
1986; Montgomery, 1991; Nielsen et al., 1993; US EPA, 1994).
The atmospheric lifetime of methanol has been estimated to be 20
days based on the reaction of compounds with the hydroxyl radical,
and assuming a hydroxyl free radical concentration of 5 × 105
radicals/cm3 (Graedel et al., 1986). Methanol half-lives of 8.4 days
(US EPA, 1979), 8.0 days (Lemaire et al., 1982) and 7.3 days (Barnes
et al., 1982) have also been reported based on reactions at 300°K and
equations reported in Lyman et al. (1984) and Resenblatt (1990).
Gusten et al. (1984) reported that at 300 °K and atmospheric pressure,
an average hydroxyl concentration of 1 × 106 molecules/cm3 and a
reaction rate constant of 0.95 × 10-12 cm3 /mol per sec, the half-
life of methanol was 8.4 days.
Reaction of methanol with nitrogen dioxide in a smog chamber
yielded methyl nitrite and nitric acid and the surface reaction of
methanol and nitrogen dioxide was enhanced under ultraviolet light
(Akimoto & Takagi, 1986). The reaction of methanol with nitrogen
dioxide may be the major source of methyl nitrite found in polluted
atmospheres (Takagi et al., 1986; Howard, 1990). Only 4.1% of the
methanol applied to silica gel was degraded when irradiated for 17 h
at wavelengths greater than 290 nm (Freitag et al., 1985).
4.2.3 Bioconcentration
Bioconcentration factors (BCFs) of methanol experimentally
measured in aquatic organisms using a log kow value for methanol of
-0.77 and correlation equations reported in Lyman et al. (1990) ranged
from 0.01-0.51 (Nielsen et al., 1993). Based on the octanol/water
partition coefficient of -0.77, the BCF value for methanol was
estimated to be 0.2 (Howard, 1990).
Freitag et al. (1985) reported a BCF of < 10 (wet weight basis)
for the golden ide (Leuciscus idus melanotus) after 3 days exposure
to 0.05 mg methanol/litre.
Gluth et al. (1985) proposed a BCF of about 1 for the carp
Cyprinus carpio exposed to 14C-methanol for up to 72 h. The amount
of radioactivity was measured in the liver, kidneys, intestine,
muscle, blood and gills of carp exposed to methanol at 5 mg/litre. The
initial uptake of methanol into the different tissue types was the
same after 24 h and levels remained constant for over 72 h in the
liver, kidneys, gills and intestines, but dropped slightly in the
blood and muscle.
Geyer et al. (1984) calculated a BCF of 28 400 (dry weight.
basis) for the green alga Chlorella fusca exposed to 0.05 mg/litre
14C-labelled methanol for 24 h at a temperature of 20-25°C with 16 h
illumination and with agitation. Nielsen et al. (1993) suggested that
this high bioconcentration factor is anomalous compared to those for
other aquatic organisms. It may be due to the fact that methanol is
metabolized by the algae, and the 14C-label, which is measured to
calculate the BCF value, is incorporated into the algae in metabolic
forms other than methanol.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
Methanol was detected at mean ambient concentrations of 10 and
3 µg/m3 (7.9 and 2.6 ppb) at Tucson, Arizona, USA, and two remote
Arizona locations, respectively, during monitoring in 1982 of air
pollutants in the USA (Snider & Dawson, 1985). It was also detected in
rural air in Alabama (Holzer et al., 1977). Methanol was detected at
concentrations of 0.65-1.8 µg/m3 (0.5-1.2 ppb) (average 0.77 ppb
methanol plus ethanol) in Arctic air from Point Barrow, Alaska, in
September 1967 (Cavanaugh et al., 1969).
Urban air levels of methanol in the range of 10.5-131 µg/m3
(8-100 ppb) have been reported (Graedel et al., 1986). Jonsson et al.
(1985) reported significant amounts of methanol (0.59-94 µg/m3;
0.45-72 ppb) at dense traffic sites in Stockholm, Sweden. Average
ambient methanol concentrations of 5-30 µg/m3 (3.83-26.7 ppb) were
detected at five sites in and around Stockholm.
In 1994, methanol was listed as one of the 189 hazardous air
pollutants (HAPs) under the Clean Air Act Amendment of 1990, Title III
in the USA (Kelly et al., 1994). In a US EPA (1993) summary, median
methanol levels of 6-60 µg/m3 were found in 52 samples from three
locations (Boston, Houston, and Lima, Ohio) in the USA.
5.1.2 Water
Data on the occurrence of methanol in water, particularly
finished drinking-water, is limited. Methanol was identified in water
at 24 locations in the USA during the period 1974-1976 (US EPA,
1976b). The frequency of occurrence was as follows: finished drinking-
water, 12; effluents from chemical plants, 6; effluents from sewage
treatment, 4; effluents from paper production, 1; and effluents from
latex production, 1.
Methanol was detected in the USA at a mean level of 22 µg/litre
in rainwater collected during a thunderstorm in Arizona in 1982
(Snider & Dawson, 1985).
Methanol at levels of 17-80 mg/litre (17-80 ppm) was detected in
wastewater effluents from a speciality chemicals manufacturing
facility in Massachusetts, USA, but none was detected in associated
river water or sediments (Jungclaus et al., 1978). A concentration of
42.4 mg/litre were found in a leachate from the Love Canal in Niagara
Falls, New York (Venkataraman et al., 1984). Methanol at a level of
1050 mg/litre was detected in condensate waters discharged from a coal
gasification plant at North Dakota, USA (Mohr & King, 1985).
5.1.3 Food
Dietary methanol can arise in large part from fresh fruits and
vegetables where it occurs as the free alcohol, methyl esters of fatty
acids or methoxy group on polysaccharides such as pectin (Kirchner &
Miller, 1957; Casey et al., 1963; Self et al., 1963; Lund et al.,
1981; Stegink et al., 1981; Monte, 1984).
The methanol content of fresh and canned fruit juices
(principally orange and grapefruit juices) varies considerably and may
range from 1-43 mg/litre (Kirchner & Miller, 1957), 10-80 mg/litre
(Lund et al., 1981; Monte, 1984) and 12-640 mg/litre with an average
of 140 mg/litre (Francot & Geoffroy, 1956; Monte, 1984). Methanol
evolved during the cooking of high pectin foods (Casey et al., 1963)
has been accounted for in the volatile fraction during boiling and is
quickly lost to the atmosphere (Self et al., 1963). However entrapment
of the volatiles during canning is possible and probably accounts for
the elevated methanol levels of certain fruits and vegetables during
this process (Lund et al., 1981).
Fermented distilled beverages can contain high levels of
methanol, with some neutral spirits having as much as 1.5 g/litre
(Francot & Geoffroy, 1956). Methanol was found at levels of
6-27 mg/litre in beer, 96-321 mg/litre in wines and 10-220 mg/litre
in distilled spirits (Greizerstein, 1981). The methanol content in
representative beverage alcohol varied between 40 and 55 mg/litre
bourbon. This value is comparable with those reported by the
distillers. The concentration of methanol in 50% grain alcohol was
found to be approximately 1 mg/litre (Majchrowicz & Mendelson,
1971).The presence of methanol in distilled spirits is directly linked
to the pectin content of the raw materials. During the process of
making fruit spirits, pectic substances contained in different parts
of the fruit undergo degradation by pectin methylases, which can lead
to the formation of significant quantities of methanol (Bindler et
al., 1988). Concentrations of methanol permitted in brandies in the
USA, Canada and Italy range from 6-7 g/litre ethanol (Bindler et al.,
1988).
Methanol has been identified in the volatile fraction of sherry
wine vinegars (Blanch et al., 1992), lemon, orange and lime extracts,
distilled liquors and cordials (AOAC, 1980, 1990).
Methanol has been identified as a volatile component of dried
legumes with reported levels of 1.5-7.9 mg/kg in beans, 3.6 mg/kg in
split peas and 4.4 mg/kg in lentils (Lovegren et al., 1979). Methanol
has also been reported (no levels stated) in roasted filberts (Kinlin
et al., 1972) and baked potatoes (Coleman et al., 1981). It has been
detected in low-boiling volatile fractions of cooked foods, including
Brussels sprouts, carrots, celery, corn, onion, parsnip, peas and
potatoes (Self et al., 1963).
Humans can also ingest varying amounts of methanol in foods and
or drugs isolated or recrystallized from methanol, e.g., methanol is
used as an extraction solvent for spice oleoresins and hops (Lewis,
1989). Additionally, certain foods and drugs, consumed or administered
as their methyl ester, can release methanol during their metabolism
and excretion (Stegink et al., 1981; Davoli et al., 1986). For
example, 10% of the sweetening agent aspartame (L-aspartyl-L-
phenylalanine methyl ester) hydrolyzes in the gastrointestinal tract
to become free methanol. Carbonated beverages contain about 555 mg
aspartame/litre (Medinsky & Dorman, 1994), equivalent to approximately
56 mg methanol per litre.
The amount of methanol present in an average serving of beverage
sweetened by aspartame alone is considerably less than in the same
volume of many fruit and vegetable juices. For instance, tomato juice
will result in 6 times the amount of methanol exposure than
consumption of an equivalent volume of aspartame sweetened beverage
(Wucherpfennig et al., 1983).
Exposure to several industrial compounds such as methanol,
formaldehyde and acetone may contribute to increasing amounts of
formate in the body (Boeniger, 1987). Formate is present in blood at
background or endogenous levels that range from 0.07 to 0.4 mM.
Although it is essential for survival, an excess of formate, which
often occurs after intake of large doses of methanol, can cause severe
toxicity and even death (Medinsky & Dorman, 1994).
Ingestion of formate can arise from such foods as honey, fruit
syrups and roasted coffee as well as from its use as a food
preservative. Formate is also produced as a by-product of several
metabolic pathways including histidine and tryptophan degradation
(Stegink et al., 1983).
The possible utility of formic acid as a biomarker for
occupational exposure to methanol has been investigated (Angerer &
Lehnert, 1977; Baumann & Angerer, 1979; Ferry et al., 1980; Heinrich &
Angerer, 1982; Liesivouri & Savolainen, 1987; Franzblau et al., 1992b;
Lee et al., 1992; d'Alessandro et al., 1994).
5.1.4 Tobacco smoke
Methanol at levels of 180 µg/cigarette has been detected in the
vapour phase in mainstream smoke (Norman, 1977; Guerin et al., 1987).
It has been reported to represent about 2% by weight of the mainstream
smoke organic phase and particulate matter (Dube & Green, 1982).
5.2 Occupational exposure
US NIOSH (1976) estimated that 175 000 workers in the USA are
potentially exposed to methanol. As stated in Clayton & Clayton
(1982), the US Department of Labor reported that 72 occupations
involve exposure to methanol. Estimates derived from the NIOSH
1972/1974 National Occupational Hazard Survey and 1982-1983 National
Occupational Exposure Survey indicate that approximately 1-2 million
workers in the USA are potentially exposed to methanol (Howard, 1990).
In a 1978-1982 survey of solvent products associated with USA
industrial workplace exposure, methanol was identified in 9.8% of 275
solvent samples collected. The products represented solvent classes
such as thinners, degreasers, paints, inks and adhesives (Howard,
1990).Workplace concentrations in the range of 29-108 mg/m3 were
found during production of "fused collars" (Greenberg et al., 1938).
No signs or symptoms of methanol intoxication were observed.
In the vicinity of "spirit" duplicator machines operated with
methanol-based duplicator fluids, methanol concentrations of between
475 to 4000 mg/m3 were found in the breathing zone. Teacher aides and
clerical workers exposed to these concentrations experienced typical
symptoms of methanol intoxication (Kingsley & Hirsch, 1955; NIOSH,
1981; Frederick et al., 1984).
In a Japanese factory producing canned fuel containing mainly
methanol, air levels of methanol were high (Kawai et al., 1991b). A
mean geometric concentration of 600 mg/m3 (459 ppm) with a geometric
standard derivation of 4.1 was found in the breathing zone of 22
production workers (8-h sampling). This resulted in high blood and
urine levels of methanol (see section 8.1.3 for further details).
In a chemical plant, 30-min workplace concentrations ranged from
about 49 to 303 mg/m3 during the course of a shift, with a geometric
mean of 129 mg/m3. After an 8-h exposure, average methanol blood and
urine levels of 8.9 ± 14.7 and 21.8 ± 20.0 mg/litre and a mean formic
acid urine level of 29.9 ± 28.6 mg/litre were found (Heinrich &
Angerer, 1982).
Increases in blood and urine methanol and formate levels can be
measured in humans exposed to methanol vapours in the workplace at
concentrations below the ACGIH threshold limit value (TLV) of 260
mg/m3 (200 ppm). The recommended limit of 260 mg/m3 for methanol was
first proposed by Cook (1945), based on previous studies of Sayers et
al. (1942) who observed no symptoms in dogs exposed daily (7
days/week) for 379 days at concentrations between 590 and 655 mg/m3
(450 and 500 ppm). Printing office and chemical workers exposed to
approximately 130 mg/m3 (100 ppm) during the workshift exhibited a
1.5- to 2.5-fold increase in blood and urinary formate and a 15- to
20-fold increase in blood and urinary methanol at the end of the
workday, whereas unexposed workers did not exhibit an increase in
their blood and urinary methanol or formate levels (Baumann & Angerer,
1979; Heinrich & Angerer, 1982).
5.3 General population
Humans are routinely exposed to methanol from both the diet and
natural metabolic processes. Sedivec et al. (1981) reported a mean
blood methanol level of 0.73 mg/litre in 31 unexposed subjects (range:
0.32-2.61 mg/litre). Eriksen & Kulkarni (1963) measured a mean level
of 0.25 µg/litre in the expired air of 9 "normal" people (range:
0.06-0.45 µg/litre).
Methanol is available from the ingestion of dietary fruits and
vegetables, from the consumption of fruit juices and fermentation
beverages, and from the use of the synthetic sweetener aspartame,
which on hydrolysis yields 10% of its weight as free methanol, which
is available for absorption. Estimates of intakes of methanol from
these sources vary considerably. Consuming a 354 ml carbonated
beverage is approximately equivalent to a methanol intake of 20 mg.
Excluding exposure from carbonated beverages, daily aspartame intake
can average 3-11 mg/kg (0.3-1.1 mg methanol/kg), with the 99th
percentile ingesting up to 34 mg/kg (3.4 mg/kg methanol) (Stegink,
1981; Medinsky & Dorman, 1994). If aspartame were used to replace all
sucrose in the diet, its average daily ingestion would be
7.5-8.5 mg/kg which would be the equivalent to 0.75-0.85 mg
methanol/kg (Stegink et al., 1981; Davoli et al., 1986).
The average intake of methanol from natural sources varies, but
limited data suggest an average intake of considerably less than 10 mg
methanol/day (US EPA, 1977; Monte, 1984).
Estimated methanol body burdens for selected situations were
reported by Medinsky & Dormam (1994). The "background" body burden of
methanol was estimated to be 0.5 mg/kg. Fruit juices containing
12-640 mg methanol/litre would have a variable effect on body burden,
while personal garage exposure (200 mg/m3; 15 min) and self-service
refuelling (50 mg/m3; 4 min) would increase the body burdens by an
estimated 0.6 mg/kg and 0.04 mg/kg, respectively.
Methanol, either 100% or in gasoline blends (85% methanol and 15%
gasoline), has the potential to become a major automotive fuel
particularly in the USA in the next century (Kavet & Nauss, 1990;
Medinsky & Dorman, 1994). Emissions of methanol can arise from its
release as uncombusted fuel in the exhaust or from its evaporation
during refuelling and after the engine has stopped. Formaldehyde
emissions can result from the incomplete combustion of methanol fuels
(Medinsky & Dorman, 1994).
The US EPA has modelled methanol exposure levels that might occur
under specific conditions of use (Kavet & Nauss, 1990). For example,
if 100% of all automobiles were powered by methanol-based fuels,
models predict concentrations of methanol in expressways, street
canyons, railroad tunnels or parking garages ranging from a low of
1 mg/m3 to a high of 60 mg/m3. Methanol concentrations in a personal
garage during engine idle or hot soak conditions are predicted to
range from 2.9 to 50 mg/m3, while those predicted during refuelling
of vehicles ranged from 30 to 50 mg/m3. For comparison, the American
Conference of Governmental Industrial Hygienists (ACGIH) Threshold
Limit Value (TLV) for exposure to methanol over an 8-h workday is
260 mg/m3 (200 ppm) for working populations.
Some methanol exposure concentrations have been calculated for
various scenarios (traffic conditions, wind patterns, meteorological
conditions) from emission data from a few cars using methanol
dispersion models. The highest methanol concentration projected to
occur in a personal garage is 490 mg/m3 (375 ppm) during the cold
start. In public garages, assuming 100% of the vehicles were fuelled
with methanol, concentrations were projected to be as high as
200 mg/m3 (150 ppm), while in either scenarios the concentrations
would be expected to be lower than 65 mg/m3 (50 ppm). In the majority
of cases, exposure to the general public would be brief but repeated
in time (Gold & Moulif, 1988).
Most available evidence indicates that exposure to methanol
vapour from use as a motor fuel is not associated with adverse effects
(Gold & Moulif, 1988). The uncertainties in this conclusion are based
on the lack of information at reasonable projected exposure levels and
of studies examining end-points of concern in sensitive species. Lack
of complete data (dose-response, exposure) reveals that numerous
uncertainties exist in the safety/risk assessments. Small effects and
trends in behavioural and neurophysiological responses and subjective
ratings have been reported but need to be further substantiated.
6. KINETICS AND METABOLISM IN LABORATORY ANIMALS AND HUMANS
6.1 Absorption
The primary routes of methanol exposure are inhalation and
ingestion, with dermal exposure currently of much less importance in
terms of total daily intake for both the general and occupational
populations. Regardless of the exposure route, methanol distributes
readily and uniformly to all organs and tissues in direct relation to
their water content (Yant & Schrenk, 1937; Haggard & Greenberg, 1939).
Thus all exposure routes are presumed to be toxicologically equivalent
(Tephly & McMartin, 1984). No differences exist between the
capabilities for absorption of methanol among various animal species,
and blood levels are entirely predictable based on the concept that
methanol distributes uniformly to body water content.
6.1.1 Inhalation
Inhalation of methanol is the most common route of entry in an
occupational setting. Experiences in occupational health and volunteer
studies show that methanol is rapidly absorbed after inhalation
(Angerer & Lehnert, 1977; Baumann & Angerer, 1979; Ferry et al., 1980;
Sedivec et al., 1981; Heinrich & Angerer, 1982; Liesivouri &
Savolainen, 1987; Kawai et al., 1992; d'Alessandro et al., 1994).
The body burden is estimated from methanol concentration,
ventilation rate, duration of exposure and lung retention. Around
60-85% of inhaled methanol is absorbed in the lung of humans (Leaf &
Zatman, 1952; Sedivec et al., 1981). Blood methanol concentration,
frequently employed to characterize the body burden of methanol is, on
average, equal to 83% of its aqueous concentration. Urine contains
methanol concentrations 20-30% higher than blood (Yant & Schrenk,
1937; Leaf & Zatman, 1952).
Following uptake and distribution, methanol clears from the body.
In humans, clearance proceeds after either inhalation or oral exposure
with a half-life of 1 day or more for high doses (greater than 1 g/kg)
and about 3 h for low doses (less than 0.1 g/kg) with first-order
kinetics in humans, monkeys and rats (Leaf & Zatman, 1952; Teply &
McMartin, 1984).Methanol is either excreted unchanged in the urine and
breath or it enters a metabolic pathway whose ultimate product is
carbon dioxide. The time course for the disappearance of methanol from
the circulation is dependent upon the combined action of both direct
excretion and metabolism. The elimination of methanol from the blood
appears to be very slow in all species, especially when compared to
ethanol (Tephly & McMartin, 1984).
Relationships between methanol inhalation exposure,
concentrations, duration of exposure and urinary methanol
concentrations have been characterized in exposures of volunteers and
in occupational settings. Ferry et al. (1980), Sedivec et al. (1981),
and Heinrich & Angerer (1982) reported that urinary methanol
concentrations strictly depend on the duration and intensity of the
methanol exposure, suggesting that measurement of urinary methanol
concentrations would be a reliable parameter for evaluating the degree
of methanol exposure.
Sedivec et al. (1981) exposed four volunteers to methanol at
concentrations of 102, 205 and 300 mg/m3 for 8 h/day. Urine was
monitored for methanol during exposure and for 18 h afterwards. The
concentrations in urine were proportional to the air concentrations.
When exposure ceased, urinary methanol levels decreased exponentially
with a half-life of about 1.5-2 h; a mean urinary level of 0.73 mg/litre
(range 0.32-2.61 mg/litre) in 31 unexposed subjects was also
reported. Heinrich & Angerer (1982) determined methanol in blood and
urine and formic acid in urine from 20 subjects occupationally exposed
to methanol. The air concentration was on average 145 mg/m3 (111 ppm)
but varied from 49 to 303 mg/m3. An 8-h exposure resulted in methanol
levels in blood and urine of 8.9 ± 14.7 mg/litre and 21.8 ±
20 mg/litre, respectively. Formic acid concentrations were 29.9 ±
28.6 mg/litre. The corresponding normal values were < 0.6, 1.1 ± 0.9
and 12.7 ± 11.7 mg/litre.
Volunteers exposed for 6 h to 260 mg/m3 (200 ppm) methanol, the
current permissible US OSHA 8-h time-weighted average limit, were
found to have a blood methanol concentration increase from a mean of
1.8 µg/ml to 7.0 µg/ml (3.5-4 fold increase) compared to their
pre-exposure levels. Formate did not accumulate in the blood above its
background level (8.11 µg/ml) following the 6-h exposure (Lee et al.,
1992).
Franzblau et al. (1993) demonstrated the absence of formic acid
accumulation in the urine of five volunteers following 5 days of
exposure to an atmosphere containing 260 mg/m3 (200 ppm) of methanol
in a test chamber. These results indicated that there was no day-to-
day accumulation of formic acid in urine in conjunction with 5
consecutive days of near-maximal permissible airborne methanol
exposure and that measurement of formic acid in urine specimens
collected 16 h following cessation of exposure did not appear to
reflect inhalation methanol exposure on the preceding day.
Twenty-six volunteers exposed at rest to 260 mg/m3 (200 ppm) of
methanol vapour for 4 h did not show significant differences in serum
or urinary formate concentration. At the TLV of 260 mg/m3 (200 ppm)
methanol exposure did not contribute substantially to endogenous
formate formation (d'Alessandro et al., 1994).
Inhalation of from 650 to 1450 mg/m3 (500 to 1100 ppm) methanol
for periods of 3-4 h in humans yielded urine concentrations of about
10-30 mg/litre at the end of the exposure period (Leaf & Zatman,
1952). Based on their findings, it was suggested that an 8-h exposure
to 3990 mg/m3 (3000 ppm) methanol would be necessary before a gradual
accumulation of methanol would occur in the body.
6.1.2 Oral
Methanol is rapidly absorbed from the gastrointestinal tract with
peak absorption occurring in 30-60 min depending on the presence or
absence of food in the stomach (Becker, 1983).
Ingestion of methanol has been the principal route of exposure in
the many reported cases of acute poisoning (Buller & Wood, 1904; Wood
& Buller, 1904; Bennett et al., 1953; Erlanson et al., 1965; Kane et
al., 1968; Gonda et al., 1978; Naraqi et al., 1979; Swartz et al.,
1981; Jacobsen et al., 1982; Becker, 1983; Litovitz et al., 1988).
During methanol poisoning in humans, concentrations of methanol
and formic acid in blood and urine are quite variable. Concentrations
of both compounds are highly dependent upon dose, time following
exposure and concomitant ingestion of ethanol (Lund, 1948a, Gonda et
al., 1978, Jacobsen et al., 1982a). Excretion of methanol in urine is
initially high and decreases with time following exposure. Maximum
excretion of formic acid in urine may occur as late as the second or
third day following ingestion (Lund, 1948a).
Blood methanol concentrations during experimentally induced
ethanol intoxication in alcoholics during a 10-15 day period of
chronic alcohol intake showed that blood methanol levels increased
progressively from 2-27 mg/litre from the first to the 11th day of
drinking, when blood ethanol concentrations ranged between 1500 and
4500 mg/litre. Blood methanol levels decreased at the rate of 2.9 ±
0.4 mg/litre per h only after blood ethanol levels decreased to 700 to
200 mg/litre. Blood methanol disappearance lagged behind the linear
disappearance of ethanol by approximately 6-8 h and complete clearance
of methanol required several days. Methanol probably accumulates in
the blood as a result of the competitive inhibition of alcohol
dehydrogenase by ethanol and the presence of endogenously formed
methanol (Majchrowicz & Mendelson, 1971).
Oral doses of 71-84 mg methanol/kg in humans resulted in blood
levels of 47-76 mg/litre blood 2-3 h later. The urinary concentrations
of methanol rapidly reached a peak capacity in 1 h and declined
exponentially, reaching control values in 13-16 h. The urine/blood
concentration ratio was found to be relatively constant at 0.30 (Leaf
& Zatman, 1952). Leaf & Zatman (1952) monitored methanol disappearance
from the circulation of three volunteers orally administered 3, 5 and
7 ml (2.4, 4.0 and 5.6 g) (highest dose, 0.08 g/kg). Blood and urine
methanol disappearance obeyed first-order kinetics with a half-time of
about 3 h.
Aspartame (see section 5.1.3) is a widely used artificial
sweetening agent which is hydrolysed in the intestinal mucosa to 10%
methanol by weight. Beverages totally sweetened with aspartame
typically contain 0.5-0.6 mg aspartame/ml or approximately 195 mg/
350 ml soft-drink; dry mixes and puddings use about 100 mg/serving and
pre-sweetened cereal products about 60 mg/25 ml (cup). The methanol
body burden following ingestion of any of these products could vary
from 6-20 mg (Stegink et al., 1981,1983). Clearance of methanol from
human circulation after body burdens as high as 80 mg/kg follows
first-order kinetics with a half-time of about 2.5-3 h (the rate
constant for total clearance kt is 0.23-0.28/h (Stegink et al., 1981;
Kavet & Nauss, 1990).
After intake of small quantities of methanol (10-20 ml), human
subjects showed no methanol in blood after 48 h, and the concentration
of formic acid in the urine was normal (6.5-12.8 mg%) within 24 h
(Lund, 1948a). Following intake of large amounts of methanol (50 ml),
methanol was found in the blood (250-1200 mg/litre) after 48 h. Formic
acid was found in the blood (26-78 mg/litre) as well as an increased
excretion of formic acid in the urine (540-2050 mg/litre), and up to
20 500 mg/litre within 24 h. Maximum excretion of formic acid was
found to occur not later than the second or third day after intake of
methanol (Lund, 1948a).
6.1.3 Dermal
It has been known for some time that pure methanol has an
anomalously high diffusion rate through epidermis because of the
damage it produces on the stratum corneum (the thin sheath of
keratinized cells that comprise the outermost layer of the epidermis).
The permeability of epidermis for pure methanol is 10.4 mg/cm2 per h
(Scheuplein & Blank, 1971).
Skin absorption rate studies of methanol ranging from 0.031-0.241
mg/cm2 per min conducted in human volunteers showed that an average
of 0.192 mg methanol/cm2 per min is absorbed through direct contact
of the skin to methanol. Compared with absorption via the respiratory
tract, exposure of one hand to liquid methanol for only 2 min would
result in a body burden of as much as 170 mg methanol, similar to that
resulting from exposure to an approximate air concentration of
50 mg/m3 (40 ppm) methanol for 8 h (Dutkiewicz et al., 1980). It was
also reported that in the context of a 20-min immersion of one hand in
methanol, the cumulative urinary excretion of methanol over 8 h was
2 mg. However, it should be noted that the assessment of Dutkiewicz et
al. (1980) would imply that a 10-min exposure of one hand to liquid
methanol roughly corresponds to an 8-h inhalation exposure at
260 mg/m3 (200 ppm). Such an inhalation exposure was found to be
accompanied with a post-shift urinary methanol concentration of about
40 mg/litre (Sedivec et al., 1981; Kawai et al., 1991b) or 6.5
mg/litre (Franzblau et al., 1993).
The rate of absorption into the skin has been found to be higher
with M-85 (85% methanol-15% gasoline) than with pure methanol. The
gasoline was suggested to act by drying out the skin allowing the
methanol to be more readily absorbed (Machiele, 1990). In 11 children
treated for percutaneous methanol intoxication, methanol blood levels
ranged from 0.57 to 11.3 g/litre (mean 4.61 g/litre) (Giminez et al.,
1968). Methanol was identified in the urine and in peritoneal fluid
(no quantitative estimation) in an 8-month-old boy poisoned by
percutaneous absorption of methanol (Kahn & Blum, 1979).
Downie et al. (1992) reported a case of percutaneous industrial
methanol toxicity involving two workers who spent 2-3 h cleaning out a
cargo tank with methanol while wearing positive pressure breathing
apparatus. One of the workers, who suffered from a previous sunburn,
wore no protective clothing during cleaning. He experienced methanol
toxicity from percutaneous exposure and required hospitalization and
methanol poisoning treatment.
6.2 Distribution
Methanol distributes readily and uniformly to organs and tissues
in direct relation to their water content (Yant & Schrenk, 1937;
Haggard & Greenberg, 1939). The apparent volume of distribution of
methanol is 0.6-0.7 litres/kg, similar to that of ethanol. In methanol
inhalation studies conducted in dogs, Yang & Schrenk (1937) reported
that the highest concentrations of methanol were found in the blood,
vitreous and aqueous humour, bile and urine, and the lowest in bone
marrow and fatty tissue. In other animal studies, high concentrations
of methanol have been reported in the kidney, liver and gastro-
intestinal tract with smaller concentrations in brain, muscle and
adipose tissue (Bartlett, 1950).
Postmortem analysis of methanol concentrations in body fluids and
tissues reported in fatal human cases of methanol poisoning has
revealed high concentrations of methanol in cerebrospinal fluid (CSF),
vitreous humour and bile (Bennet et al., 1953; Wu Chen, 1985).
Methanol concentrations in these fluids were higher than blood
concentrations. In one study the ratio of methanol in blood to
vitreous humour was 0.82, which was similar to the ratio of ethanol in
blood to vitreous humour of 0.89 (Coe & Sherman, 1970). In tissues the
highest concentrations were found in brain, kidney, lung and spleen,
with lower concentrations in skeletal muscle, pancreas, liver and
heart (Wu Chen et al., 1985).Methanol-induced alterations in
uteroplacental blood-flow were studied in CD-1 mice and Sprague-Dawley
rats employing microdialysis as a tool for investigating the flux of
toxicants across the maternal-conceptual unit. Microdialysis probes
were inserted into the uteri of gestational day 20 rats and methanol
was administered as either an intravenous bolus (100 or 500 mg/kg) or
infusion (100 or 1000 mg/kg/hour).
In separate studies, methanol (100 or 500 mg/kg) and 3H2O
(20 µCi/kg) were administered intravenously on gestational days
20 and 14 to rats and on gestational day 18 to mice. The methanol
concentration-time data were consistent with saturable maternal
elimination and apparent first-order transfer between maternal and
conceptual compartments. At distribution equilibrium, conceptual
methanol concentrations exceeded those in the dam by approximately
25%. The initial rate of conceptual permeation of methanol was
proportional to the reciprocal of maternal blood methanol
concentration (r2 = 0.910).
The data indicated that high circulating maternal methanol
concentrations decrease the rate of presentation of methanol and
3H2O to the conceptus, and, depending on the severity of the
decrease, fetal hypoxia could also result (Ward & Pollack, 1996b).
6.3 Metabolic transformation
After uptake and distribution, most of the methanol is
metabolized in the liver to carbon dioxide (96.9%), while a small
fraction is excreted directly to the urine (0.6%) and through the
lung. In all mammalian species studied, methanol is metabolized in the
liver by sequential oxidative steps to form formaldehyde, formic acid
and CO2 (Fig. 1). However, there are profound differences in the rate
of formate oxidation in different species which determine the
sensitivity to methanol (Rietbrock, 1969; Palese & Tephly, 1975;
McMartin et al., 1977; Eells et al., 1981a, 1983).
Two enzymes are important in the oxidation of methanol to
formaldehyde, alcohol dehydrogenase and catalase. In non-human
primates and humans, alcohol dehydrogenase mediates this reaction
(Makar et al., 1968; Röe, 1982). In rats and other non-primate species
this reaction is mediated by catalase. Definitive evidence of these
differences has been provided by studies of methanol oxidation
in vivo using alternative substrates (ethanol, 1-butanol) and
selective inhibitors of catalase (3-amino-1,2,4-triazole) and alcohol
dehydrogenase (4-amino-pyrazole). The hepatic microsomal mixed-
function oxidase system (P450IIE1) has also been implicated in the
conversion of methanol to formaldehyde, but there is no definitive
information on its role in vivo (Rietbrock et al., 1966; Teschke et
al., 1975). Despite the difference in enzyme mediation, the conversion
from methanol to formate occurs at similar rates in non-human primates
and in rats (Tephly et al., 1964; Makar et al., 1968; Noker et al.,
1980; Eells et al., 1981a, 1983). The metabolism of methanol can be
significantly inhibited by co-exposure to ethanol, which acts as a
competing substrate for alcohol dehydrogenase (Jones, 1987).
Formaldehyde is oxidized to formate by several enzyme systems
including a specific formaldehyde dehydrogenase. In the reaction
catalysed by this enzyme, formaldehyde combines with reduced
glutathione to form S-formyl glutathione, which is hydrolysed in the
presence of thiolase to formate and reduced glutathione (Strittmatter
& Ball, 1955; Uotila & Koivusalo, 1974). The second step of this
reaction is irreversible (Strittmatter & Ball, 1955). Formaldehyde
dehydrogenase activity has been shown to be present in numerous
species and tissues including human liver and brain (Strittmatter &
Ball, 1955; Kinoshita & Masurat, 1958; Goodman & Tephly, 1971).
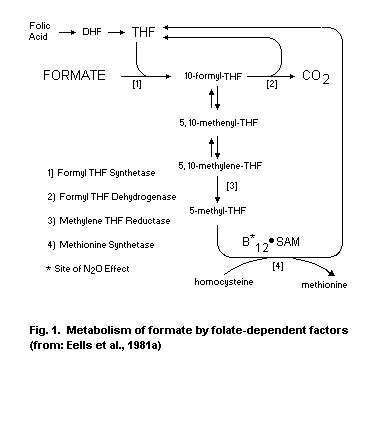 The elimination of formaldehyde in many species including
primates is extremely rapid with a half-life of approximately 1 min
(Rietbrock, 1965; McMartin et al., 1979). Malorny et al. (1965) found
that equimolar infusions of formaldehyde, formic acid and sodium
formate in dogs produced equivalent peak concentrations of formic
acid, indicating that formaldehyde was rapidly metabolized to formic
acid. In a human case of formaldehyde poisoning, toxic concentrations
of formate (7-8 mm) were detected within 30 min of ingestion,
confirming rapid metabolism of formaldehyde to formate in humans
(Eells et al., 1981b). Formaldehyde has not been detected in body
fluids or tissues following toxic methanol exposures (Makar & Tephly,
1977, McMartin et al., 1977, McMartin et al., 1980a). Formate is
oxidized to CO2 in vivo in mammalian species primarily by a
tetrahydrofolate-dependent pathway (Fig. 2). Formate enters this
pathway by combining with tetrahydrofolate (H4folate) to form
10-formyl-H4folate in a reaction catalysed by formyl-tetrahydrofolate
synthetase. 10-Formyl-H4folate may then be further oxidized to CO2
and H4folate by formyl-H4folate dehydrogenase (Kutzbach & Stokstad,
1968) (Fig. 1). Rietbrock et al. (1966) found an inverse correlation
between plasma concentrations of folate in different animal species
and the half-life of exogenously administered formate, suggesting that
folates are involved in formate metabolism. Formate metabolism in rats
and monkeys has been shown to be mediated by the folate-dependent
pathway (Makar et al., 1968; Palese & Tephly, 1975). Inhibition of
catalase with aminotriazole had no effect on formate oxidation,
whereas folate-deficiency markedly reduced formate oxidation in both
species. Tetrahydrofolate is derived from folic acid in the diet and
is the major determinant of the rate of formate metabolism (McMartin
et al., 1975).
The folate-mediated oxidation of formate proceeds about twice as
slowly in non-human primates and humans as in rats. This explains the
susceptibility of primates to the accumulation of formate, which is
seen to occur at doses of methanol greater than 0.5 g/kg (Tephly &
McMartin, 1984) (Fig. 2).There is substantial clinical and
experimental evidence that formic acid is the toxic metabolite
responsible for the metabolic and visual toxicity characteristic of
methanol poisoning. Specifically, formic acid is the toxic metabolite
responsible for the metabolic acidosis observed in methanol poisoning
in humans, in non-human primates and in folate-depleted rodents
(McMartin et al., 1975, 1977, 1980; Eells et al., 1983; Jacobsen &
McMartin, 1986; Eells, 1991; Murray et al., 1991; Lee et al., 1994).
Formic acid is believed to be the toxic metabolite responsible for the
ocular toxicity in methanol-poisoned humans (Sharpe et al., 1982), and
is also responsible for the ocular toxicity produced in non-human
primates and folate-depleted rodents (Martin-Amat et al., 1977, 1978;
Eells et al., 1983; Eells, 1991; Lee et al., 1994a,b).
The elimination of formaldehyde in many species including
primates is extremely rapid with a half-life of approximately 1 min
(Rietbrock, 1965; McMartin et al., 1979). Malorny et al. (1965) found
that equimolar infusions of formaldehyde, formic acid and sodium
formate in dogs produced equivalent peak concentrations of formic
acid, indicating that formaldehyde was rapidly metabolized to formic
acid. In a human case of formaldehyde poisoning, toxic concentrations
of formate (7-8 mm) were detected within 30 min of ingestion,
confirming rapid metabolism of formaldehyde to formate in humans
(Eells et al., 1981b). Formaldehyde has not been detected in body
fluids or tissues following toxic methanol exposures (Makar & Tephly,
1977, McMartin et al., 1977, McMartin et al., 1980a). Formate is
oxidized to CO2 in vivo in mammalian species primarily by a
tetrahydrofolate-dependent pathway (Fig. 2). Formate enters this
pathway by combining with tetrahydrofolate (H4folate) to form
10-formyl-H4folate in a reaction catalysed by formyl-tetrahydrofolate
synthetase. 10-Formyl-H4folate may then be further oxidized to CO2
and H4folate by formyl-H4folate dehydrogenase (Kutzbach & Stokstad,
1968) (Fig. 1). Rietbrock et al. (1966) found an inverse correlation
between plasma concentrations of folate in different animal species
and the half-life of exogenously administered formate, suggesting that
folates are involved in formate metabolism. Formate metabolism in rats
and monkeys has been shown to be mediated by the folate-dependent
pathway (Makar et al., 1968; Palese & Tephly, 1975). Inhibition of
catalase with aminotriazole had no effect on formate oxidation,
whereas folate-deficiency markedly reduced formate oxidation in both
species. Tetrahydrofolate is derived from folic acid in the diet and
is the major determinant of the rate of formate metabolism (McMartin
et al., 1975).
The folate-mediated oxidation of formate proceeds about twice as
slowly in non-human primates and humans as in rats. This explains the
susceptibility of primates to the accumulation of formate, which is
seen to occur at doses of methanol greater than 0.5 g/kg (Tephly &
McMartin, 1984) (Fig. 2).There is substantial clinical and
experimental evidence that formic acid is the toxic metabolite
responsible for the metabolic and visual toxicity characteristic of
methanol poisoning. Specifically, formic acid is the toxic metabolite
responsible for the metabolic acidosis observed in methanol poisoning
in humans, in non-human primates and in folate-depleted rodents
(McMartin et al., 1975, 1977, 1980; Eells et al., 1983; Jacobsen &
McMartin, 1986; Eells, 1991; Murray et al., 1991; Lee et al., 1994).
Formic acid is believed to be the toxic metabolite responsible for the
ocular toxicity in methanol-poisoned humans (Sharpe et al., 1982), and
is also responsible for the ocular toxicity produced in non-human
primates and folate-depleted rodents (Martin-Amat et al., 1977, 1978;
Eells et al., 1983; Eells, 1991; Lee et al., 1994a,b).
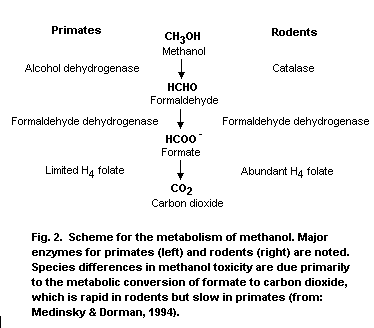 A comparative metabolism study between rodents and non-human
primates showed that formic acid concentration in blood of rats and
monkeys was similar at doses of 25, 125 and 600 mg methanol/kg, but
became substantially higher in monkeys at 3000 mg/kg. Monkeys and
rodents showed different excretion patterns for methanol. As the dose
increased, monkeys tended to excrete an increasing percentage of
methanol in urine, whereas in rats, the percentage of methanol
excreted in expired air increased. Additionally, rats excreted much
higher levels of carbon dioxide in expired air (as a percentage of
dose) than monkeys (Katoh, 1989).
In a study of formate metabolism in young swine (Makar et al.,
1990), it was found that the pig, compared to other species (mouse,
rat, monkey and humans), has extremely low levels of hepatic folates.
Furthermore, the rate of formate elimination in the pig was much lower
in the pig than in the rat. It was suggested that the pig might be
sensitive to the methanol toxicity syndrome (metabolic acidosis and
blindness).
Ward & Pallack (1996a) studied the in vitro biotransformation
of methanol in Sprague-Dawley rat and CD-1 mouse fetal livers to
assess the capability of the near-term rodent fetus to metabolize
methanol. Adult near-term rodent livers metabolized methanol to
formate (at gestational day 20) with a maximum of about 85% that in
livers from non-pregnant rodents (p < 0.05). This was consistent with
in vivo experiments (Ward & Pollack, 1996a).
Fetal rat and mouse liver was capable of metabolizing methanol
in vitro, but only at a rate of < 5% of the respective adult liver.
The difference was in fact even greater, considering the difference in
organ weight between the conceptus and the dam (about 10-fold).
Fetal mouse liver homogenates converted methanol to formaldehyde
at a significantly higher (about 40%) rate than fetal rat liver
homogenates. These data suggest that the near-term rodent fetus does
not possess a significant ability to biotransform methanol to
formaldehyde and ultimately formate in situ.
6.4 Elimination and excretion
The primary route of methanol elimination from the body is via
oxidation to formaldehyde and then to formic acid, which may be
excreted in the urine or further oxidized to carbon dioxide.
In humans, methanol is primarily eliminated by oxidation and only
2% of a 50 mg/kg dose of methanol is excreted unchanged by the lungs
and kidney (Leaf & Zatman, 1952). The small excretion of unchanged
methanol was also observed in methanol-poisoned subjects in whom the
renal and pulmonary excretory clearances of methanol were 1 and
6 ml/min, respectively (Jacobsen et al., 1982a, 1983b).
The elimination of formaldehyde in many species, including
primates, is extremely rapid with a half-life of approximately 1 min
(McMartin et al., 1979). Toxic concentrations of formate (7-8 mM) were
detected within 30 min of ingestion in a human case of formaldehyde
poisoning, confirming the rapid metabolism of formaldehyde to formate
in humans (Eells et al., 1981b).
Following uptake and distribution methanol is either excreted
unchanged (direct excretion) in urine or exhaled breath, or it enters
a metabolic pathway in the liver, whose ultimate product is carbon
dioxide. The time course of the disappearance of methanol from the
circulation is dependent upon the combined action of both direct
excretion and metabolism. The clearance from the circulation of humans
following low-level exposures to methanol administered orally
(<0.1 g/kg) (Leaf & Zatman, 1952) or by inhalation (102-300 mg/m3)
(Sedivec et al., 1981) indicated that methanol disappearance obeyed
first-order kinetics with a half-time of about 2.5-3 h in both studies
as determined by blood and urinary methanol concentrations. In general
estimated methanol dose correlated with resulting blood and urine
methanol levels after both ingestion and inhalation, and methanol
concentrations in urine were approximately 30% higher than in blood
(Leaf & Zatman, 1952).
Elimination half-lives of methanol ranging from 110-213 min were
found in human volunteers following consumption of 1000-1500 ml red
wine (95% w/w ethanol, 100 mg/litre methanol) the previous evening
(Jones, 1987). After concomitant ingestion of a very low dose of
methanol (< 2 mg/kg) and ethanol (ethanol: methanol = 10), by human
subjects, a 10 fold increase in blood methanol was observed due to the
combined ingestion of the alcohols (Jones, 1987). Jacobsen et al.
(1982a) reported that during haemolysis in 2 patients being treated
for methanol poisoning, the elimination half-lives were 219 and
197 min respectively.
At higher doses of methanol, the elimination appears to become
saturated, resulting in nonlinear elimination kinetics. In an
untreated methanol-poisoned subject, methanol elimination was
clearly zero order with a rate of 85 mg/litre per h, about half the
elimination rate of ethanol (Jacobsen et al., 1988). The rates of
elimination in two other cases appeared to be 30-50 mg/litre per h
(Kane et al., 1968).
The kidney apparently exerts no active control over the urinary
concentration of methanol. The methanol content that enters the
bladder reflects the aqueous concentration of methanol in the blood
(Yant & Schrenk, 1937; Leaf & Zatman, 1952; Sedivec et al., 1981). The
rate at which methanol clears into the urine is directly proportional
to its blood level which satisfies the condition for first-order
kinetics (Kavet & Nauss, 1990).
In the lung, a small fraction of blood-borne methanol is exhaled.
The amount of methanol that crosses the blood-air barrier is
proportional to its blood concentration (first-order kinetics) and is
governed by its blood-air partition ratio (Kavet & Nauss, 1990). In
contrast to direct renal and pulmonary excretion, the metabolic
conversion of methanol to carbon dioxide is not a linear function of
concentration (Tephly et al., 1964; Makar et al., 1968).
Elimination of methanol from the blood appears to be slow in all
species especially when compared to ethanol (Tephly & McMartin, 1984;
Tephly, 1991).
One to 7 g of methanol/litre of blood (1000-7000 mg/litre) was
found in the blood of rats following oral administration of 4 g
methanol/kg body weight, and 70% of the methanol lost was eliminated
in expired air (Haggard & Greenberg, 1939).
Following administration of a 10% methanol solution (1 g/kg)
of 14C-methanol by gavage to the rat, 89% of the administered
radioactivity was recovered after 48 h; 65% as CO2 in expired air, 3%
as methanol in urine; 3% as formic acid in urine and 4% fixed in
tissues. An oxidation rate of 25 mg/kg/h was found during the first
28 h following methanol administration (Bartlett, 1950a).
Methanol was oxidized at a constant rate of 24 mg/kg per h during
the first 28 h following intraperitoneal administration of a 10%
14C-methanol solution (1 g/kg) to male albino rats. By the end of
36 h, 77% of the methanol had been converted to 14CO2 and 24% of the
dose was excreted unchanged. About equal quantities of methanol were
eliminated by the pulmonary and renal plus faecal routes (Tephly et
al., 1964).
Comparative studies in rats and monkeys have shown that 75-80% of
a 1 g/kg dose of 14C-methanol was recovered as 14CO2; 10-18% was
excreted unchanged in expired air and 6-11% eliminated in the urine as
methanol or formate within a 24-h period (Eells et al., 1981, 1983).
Excretion of similar amounts of unchanged methanol eliminated by
pulmonary (10-15%) and renal (3-19%) routes in rats and guinea-pigs
have also been reported (Bartlett, 1950; Tephly et al., 1964).
After oral administration to dogs of a single dose of methanol
(1.97 g/kg), about 10% was excreted unchanged in the urine, over a
period of about 100 h. The methanol concentration in the organs was
nearly half as high as that found in the urine. About 20% of the
administered dose was excreted as formic acid in the urine, which
ceased after 100 h. Formic acid concentrations in tissues were about
one-half to one-quarter that found in serum (Lund, 1948b).
Oral administration of 2.38 g methanol/kg to male rabbits
resulted in 10% of methanol being excreted unchanged in the urine and
essentially no increase in formic acid in the urine. Formic acid is
oxidized almost completely in the rabbit (Lund, 1948c).
Damian & Raabe (1996) investigated the dose-dependent elimination
of formate in male CD rats employing a perfused liver system to
separate the kinetic contributions of hepatic metabolism and renal
excretion in the total elimination of formate. Formate was eliminated
from the perfused rat liver following Michaelis-Menten kinetics.
The in vitro and in vivo dose-dependent studies of formate
elimination, in conjunction with the proposed toxicokinetic model (a
central, well-mixed compartment and a urine compartment, endogenous
production of formate), indicated two main pathways of formate
elimination in the rat: (a) hepatic metabolism via Michaelis-Menten
kinetics which predominates at low levels, and (b) extremely rapid and
extensive urinary excretion that predominates at high dose levels.
Urinary excretion consists primarily of glomerular filtration with
saturable tubular reabsorption.
6.5 Modelling of pharmacokinetic and toxicokinetic data
Pharmacokinetic and toxicokinetic models have been developed in
order to gain better insight into the interspecies variation in the
uptake, metabolic fate and excretion of methanol and its metabolites,
both compartmentally and physiologically based (Horton et al., 1992;
Pollack et al., 1993; Dorman et al., 1994). As has been noted, the
elimination of formaldehyde in many species, including primates, is
extremely rapid (McMartin et al., 1979).
A pharmacokinetic model of inhaled methanol in humans and
comparison to methanol disposition in mice and rats was described by
Perkins et al. (1995). Michaelis-Menten elimination parameters (Vmax=
115 mg/litre per h; km = 460 mg/litre) were selected for input into
a semi-physiological pharmacokinetic model. Literature values for
blood or urine methanol concentrations in humans and non-human
primates after methanol inhalation were employed as input to an
inhalation disposition model that evaluated the absorption of methanol
expressed as the fraction of inhaled methanol concentration that was
absorbed. Incorporation of the kinetic parameters and absorption into
a pharmacokinetic model of human exposure to methanol, compared to a
similar analysis in rodents, indicated that, following an 8-h exposure
to 6550 mg/m3 (5000 ppm) of methanol vapour, blood methanol
concentrations in the mouse would be 13-18 fold higher than in humans
exposed to the same methanol vapour concentration. Blood methanol
concentrations in the rat under similar conditions would be 5-fold
higher than in humans. The prediction of higher concentrations in rats
was due to the greater respiration rates and consequent greater
absorption of methanol by rats.
To address the problems associated with the appropriate design of
chronic methanol studies, methanol pharmacokinetics were characterized
in male Fischer-344 rats and rhesus monkeys exposed to atmospheric
methanol concentrations ranging from 65 to 2600 mg/m3 (50-2000 ppm)
for 6 h (Horton et al., 1992). A physiologically based pharmacokinetic
(PBPK) model was then developed to simulate the in vivo time course
data. The models were used to predict the atmospheric methanol
concentration range over which the laboratory species exhibit
quantitative similarities with humans. Below 1500 mg/m3 (1200 ppm)
the model predicted all three species would exhibit similar end-of-
exposure blood methanol concentrations which would be proportional to
atmospheric concentrations. At higher concentrations the increase of
methanol in the blood of rats and monkeys was predicted to become
non-linear, whereas for humans blood methanol levels were predicted to
increase in a linear fashion (Horton et al., 1992).
Female Sprague-Dawley rats at gestational days 7, 14 and 21 and
CD-1 mice at gestational days 9 and 18 were exposed to methanol
intravenously and orally (100-2500 mg/kg) or by inhalation exposure to
1310 to 26 200 mg/m3 (1000-20 000 ppm) for 8 h and the concentrations
of methanol were measured in blood, urine and amniotic fluid (Pollack
& Brouwer, 1996). Methanol disposition was virtually unaffected by
pregnancy and the fetal methanol concentrations were approximately
similar to those in the mother. Mice accumulated methanol at a rate 2
to 3 times faster than rats, despite the two-fold higher rate of
elimination observed in the mouse.
A pharmacokinetic model described the disposition of methanol in
rats and mice with the disposition profile being partitioned into
saturable and linear metabolic elimination pathways. The saturable
pathway was evident at lower doses (100 and 500 mg methanol/kg) and
displayed classical carrier-mediated Michaelis-Menten kinetics with a
rate-limiting step. The linear pathway, which consisted of passive
elimination via pulmonary and urinary clearance of methanol in
approximately equal amounts, appeared at the highest dose (2500 mg/kg
iv) and displayed the first-order kinetics of elimination that are
characteristic of passive-diffusion mechanisms (Pollack & Brouwer,
1996).
In further studies of the comparative toxicokinetics of methanol
in pregnant and non-pregnant Sprague-Dawley rats and CD-1 mice (Ward &
Pollack, 1996a), methanol disposition in the pregnant rodent was found
to be qualitatively similar to that in non-pregnant animals. Rats
received a single dose (100 or 2500 mg/kg) of methanol either orally
(by gavage) or intravenously; mice received a single oral or
intravenous dose of 2500 mg/kg.
The maximal rate of methanol elimination (Vmax) in vivo
decreased at term in both species. Vmax in near-term rats and mice
was only 65-80% of that in non-pregnant animals. The kinetic
parameters that appeared to be most sensitive to the gestational stage
were the rate constants associated with intercompartmental transfer
(k12 and k21), although there was no obvious relationship between the
estimate of these parameters and gestational stage. The data generated
in both the in vivo and in vitro studies demonstrated that
alterations in methanol disposition associated with gestational stage
should be accounted for in the development of a toxicokinetic model
for methanol in pregnant mammals.
The examination of the toxicokinetics of intravenously
administered methanol to female Sprague-Dawley rats as a single bolus
dose of 50 or 100 mg/kg, or 2500 mg/kg administered over 2 min,
resulted in a markedly non-linear elimination of methanol from the
systemic circulation suggesting a significant capacity-limited rate of
elimination. The data from the 2500 mg/kg group was described by a
kinetic model incorporating parallel first-order and saturable
elimination processes; a portion of this apparent linear elimination
pathway was due to renal excretion of the unchanged alcohol (Pollack
et al., 1993). The blood methanol concentration-time profile was
consistent with the presence of parallel linear pathways for methanol
elimination.
The toxicokinetics of methanol in female CD-1 mice and Sprague-
Dawley rats was examined by Ward et al. (1995). Non-linear disposition
of methanol was reported in both female CD-1 mice administered a
single dose of 2.5 g methanol/kg either by gavage or intravenously (as
a 1-min infusion) and Sprague-Dawley rats receiving a single oral dose
of 2.5 g/kg. Data obtained after intravenous administration were
well-described by a one-compartment model with Michaelis-Menton
elimination. Blood methanol concentration-time data after oral
administration could be described by a one-compartment (mice) or a
two-compartment (rats) model with Michaelis-Menton elimination
from the central compartment and biphasic absorption from the
gastrointestinal tract. Kinetic parameters (Vmax for elimination),
apparent volume of the central compartment (Vc), first-order rate
constants for intercompartmental transfer (k12 and k21), and first-
order absorption rate constants for fast (kAF) and slow (Kas)
absorption processes were compared between species. Mice showed a
higher maximal elimination rate than rats (when normalized for body
weight) (Vmax = 117 + 3 mg/kg per h versus 60.7 + 1.4 mg/kg per h for
rats). Additionally, the contribution of the fast absorption process
to overall methanol absorption was larger in the mouse than in the
rat. The study demonstrated that the disposition of methanol is
similar in rats and mice, although mice eliminated methanol nearly
twice as rapidly as rats.
The pharmacokinetics of 14C-methanol and 14C-formate were
studied in normal and folate-deficient (FD) female cynomolgus monkeys
anaesthetized and exposed by lung-only inhalation to 13, 60, 260 and
1200 mg/m3 (10, 45, 200 and 900 ppm) 14C-methanol for 2 h to
determine the concentration of methanol-derived formate to the total
formate pool. The blood concentration of 14C-methanol-derived formate
from all exposures was 10-1000 times lower than the endogenous blood
formate concentration (0.1-0.2 mmol/litre) reported for monkeys and
orders of magnitude lower than levels that produce acute toxicity
(8-10 mmol/litre). This suggested that low-level exposure to methanol
would not result in elevated blood formate concentrations in humans
under short-term exposure conditions (Dorman et al., 1994) (Medkinsky
& Dorman, 1985). This was confirmed in a subsequent short-duration
inhalation study in which anaesthetized female cynomolgus monkeys were
exposed for 2 h to methanol vapour (tagged with radiolabelled carbon)
at concentrations of 13, 59, 262 and 1179 mg/m3 (10, 45, 200 and
900 ppm), and monkeys fed on a diet deficient in folic acid were
exposed to 1179 mg/m3 (900 ppm) for the same duration (Medinsky et
al., 1997). The blood levels of methanol increased in a dose-dependent
manner. Blood formate levels increased by only a small extent in both
groups of monkeys.
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1 Single exposure
7.1.1 Non-primates
The lethal oral dose of methanol for most experimental animals is
relatively high compared to the lethal dose for humans and non-human
primates. In all non-primate species that have been studied, methanol
has been shown to be the least toxic of the aliphatic alcohols
(Koivusalo, 1970). The LD50 values or minimum lethal dose for a
single oral dose of methanol have been reported to be 9 g/kg for dogs
(Gilger & Potts, 1955), 7 g/kg for rabbits (Hunt, 1902; Gilger &
Potts, 1955), 7.4-13 g/kg for rats (Gilger & Potts, 1955; Rowe &
McCollister, 1982) and 7.3-10 g/kg for mice (Gilger & Potts, 1955;
Smith & Taylor, 1982) (Table 4). These doses are 6-10 times the lethal
human dose of methanol (Tephly & McMartin, 1984; Jacobsen & McMartin,
1986; HEI, 1987).
Table 4. Single-dose oral toxicity values for methanol in animals
Species LD50 (g/kg) Reference
Rat 6.2 Kimura et al. (1971)
9.1 Welch & Slocum (1943)
9.5 MLDa Gilger & Potts (1955)
12.9 Deichmann (1948)
13.0 Smyth et al. (1941)
Mouse 0.420 Smyth et al. (1941)
7.3-10.0 Smith & Taylor (1982)
Rabbit 7.0 MLD Gilger & Potts (1955)
Dog 8.0 Gilger & Potts (1955)
Monkey 2-3 MLD Gilger & Potts (1955)
7.0 MLD Cooper & Felig (1961)
a Minimum lethal dose
Other reported oral LD50 values for methanol in Sprague-Dawley
rats varied in 14-day-old, young adult and older rats ( 7.4, 13.0 and
8.8 ml/kg respectively), suggesting that young adult rats were least
susceptible to methanol toxicity (Kimura et al., 1971).
Youssef et al. (1992) reported that the order of oral LD50 in
adult female albino rats increased as follows: 95/5%-ethanol/methanol,
pure methanol, pure ethanol, and 65/35% methanol/ethanol. Clinical
features of intoxication in treated rats generally progressed from
signs of inebriation to gait disturbances, dose-proportional decreases
in response to painful stimuli, respiratory depression and coma,
ending in death due to cardio-respiratory failure. In almost all
instances, overnight coma was followed by death of the animal. Gross
and histopathological examinations of the gastric mucosa revealed
diffuse congestion with dilation of gastric blood vessels, but with
absence of gross haemorrhage and ulceration.
Rats exposed to 1.0, 2.0 and 3.0 g methanol/kg by gavage
exhibited an altered response in an operant conditioning paradigm
designed to assess motor deficits produced by neurotoxicants. Methanol
decreased the rate of response in a dose-related fashion that
suggested impaired coordination and/or reduced endurance (Youssef et
al., 1993).
Methanol administered by gavage or intraperitoneally induced
hypothermia in Fischer and Long-Evans rats, e.g., brain temperature
decreased 1.5°C within 35 min and colonic temperature was
significantly lower (Mohler & Gordon, 1990). This occurred at dose
levels of 2-3 g/kg, which is about 20% of the reported LD50 value
of 10 g/kg in rats (Gilger & Potts, 1955).
Among 40 strains of mice, 72 h oral LD50 values ranged from 7.3
to 10.0 g/kg with a mean of 8.68 g/kg methanol for mice fed a standard
laboratory chow diet (Smith & Taylor, 1982). Methanol-dosed C57BL/GCs
(acatalasemic) mice exhibited slightly lower LD50 than Csa (normal
catalase) mice, irrespective of their folate state (7.1-8.0 versus
8.6-9.0 g/kg). Oral methanol 72-h LD50 values ranged from 6.4 to
7.3 kg for mice with folic acid deficiency (FAD) diets, depending upon
the concentration of methionine in the diet (0.2-1.8%).
Female minipigs (Minipig YU, Charles River) treated with a single
oral dose of methanol at 1, 2.5 and 5.0 g/kg body weight by gavage
showed dose-dependent signs of acute methanol intoxication, including
mild CNS depression, tremors, ataxia and recumbency, which developed
within 0.5-2.0 h and resolved by 52 h. Methanol- and formate-dosed
minipigs did not develop optic nerve lesions, toxicologically
significant formate accumulation or metabolic acidosis (Dorman et al.,
1993).
The effects of single exposures of methanol by inhalation are
summarized in Table 5. The following signs of intoxication were noted:
increased rate of respiration, a state of nervous depression followed
by excitation, irritation of the mucous membranes, loss of weight,
ataxia, partial paralysis, prostration, deep narcosis, convulsions and
death occurring from respiratory failure (Loewy & von der Heide, 1914;
Tyson & Schoenberg, 1914; Eisenberg, 1917; Weese, 1928; Scott et al.,
1933; Mashbitz et al., 1936).
Under acute inhalation conditions, folate-deficient Long-Evans
male rats exposed to 4000 mg/m3 (3000 ppm) methanol for 20 h/day did
not survive more than 4 days. Rhesus monkeys exposed to 4000 mg/m3
(3000 ppm) methanol for 21 h/day survived the 20-day exposure period
and rhesus monkeys exposed to 13 000 mg/m3 (10 000 ppm) methanol for
21 h/day survived for more than 4 days (Lee et al., 1994).
The LD50 for single intraperitoneal injections of methanol was
10.5-11.0 g/kg in Swiss albino male mice. The animals initially
entered into deep narcosis within a few minutes and death usually
occurred within 24 h following recovery from deep narcosis (Gilger et
al., 1952). The LD50 values (mmole/kg) for single intraperitoneal
administration were as follows: male Wistar rats, 237; male strain H
mice, 336; male Syrian hamster, 267 (Tichy et al., 1985). These values
were calculated to correspond to 1489, 1493 and 1499 mmole/m2 body
surface, respectively. Tichy et al. (1985) also determined LD50
values for intravenous administration of methanol. The values reported
in rats and mice were 66.5 and 147 mmole/kg, corresponding to 418 and
653 mmole/m2 body surface, respectively.
Studies of rats have indicated that there are changes in levels
of dopamine, norepinephrine, serotonin and 5-hydroxyindole acetic acid
in various brain regions after a single intraperitoneal injection of
3 g methanol/kg (Jegnathan & Namasivayam, 1989). Studies on the
steady-state level of rat brain showed that there was severe depletion
of dopamine level in the striatum but a significant increase in the
level of dopamine, serotonin and 5-hydroxyindole acetic acid in the
hypothalamus. At the same time, norepinephrine and epinephrine levels
were reduced in the hypothalamus as well as in the striatum. These
effects do not seem to be induced by metabolic acidosis. The changes
in monoamine levels are very well correlated with the blood and brain
level of methanol as shown by maintaining a higher methanol level
either by simultaneous administration of ethanol or by blocking
methanol metabolism by pretreatment with 4-methyl pyrazole and
3-amino-1,2,4-triazole. It is thus postulated that monoamine changes
induced by methanol appear to be the direct effect of methanol
per se on the monoaminergic neuronal membranes.
Table 5. Effects from single inhalation exposure to methanol
Animal Concentration Duration of Signs of Outcome Reference
ppm exposure (h) Intoxication
Mouse 72 600 54 narcosis died Weese (1928)
72 600 28 narcosis died
54 000 54 narcosis died
48 000 24 narcosis survived
10 000 230 ataxia survived
152 800 94 min narcosis Mashbitz et al. (1936)
101 600 91 min narcosis
91 700 95 min narcosis
76 400 89 min narcosis overall
61 100 134 min narcosis mortality
45 800 153 min narcosis 45%
30 600 190 min narcosis
Rat 60 000 2.5 narcosis
convulsions Loewy & Von Der Heide
22 500 8 narcosis (1914)
13 000 24 prostration
8800 8 lethargy
4800 8 none
Dog 3000 8 none
32 000 8 prostration survived
incoordination
13 700 4 none
2000 24 none
7.1.2 Non-human primates
The lethal oral dose of methanol in monkeys (Table 4) has been
shown by several investigators to be of the same order of magnitude as
the lethal dose for humans. Gilger & Potts (1955) reported a minimum
lethal dose (MLD) for methanol of 3 g/kg for the rhesus monkey
(Macaca mulatta). Clinically the signs of toxicity were similar to
those noted in humans. There was a slight initial CNS depression for
1-2 h, followed by a latent period of about 12 h, a progressive
weakness, coma and death usually in about 20-30 h. All the monkeys (4)
given a lethal dose became severely acidotic within 24 h. Two of the
animals showed signs typical of methanol amblyopia observed in humans
including dilated, unresponsive pupils and changes of the retina. One
monkey exhibited evidence of optic disc hyperaemia and retinal oedema.
Cooper & Felig (1961) reported a MLD dose of 7 g methanol/kg
administered orally to rhesus monkeys and observed inebriation,
narcosis, coma and death within 24 h (usually without a latent
period). Sixteen animals survived 6 g methanol/kg or less. Acidosis
(an increased urinary excretion of organic acids) was reported in most
cases.
Studies by McMartin et al. (1975) and Clay et al. (1975) were in
agreement with earlier studies in monkeys by Gilger & Potts (1955).
Rhesus monkeys and pigtail monkeys (Macaca nemestrina) administered
3 g methanol/kg orally, showed an initial slight CNS depression
followed by a latent period of 12-16 h, during which time the animals
showed no obvious signs of toxicity. This was followed by progressive
deterioration characterized by anorexia, vomiting, weakness,
hyperpnoea and tachypnoea followed by coma with shallow and infrequent
respiration and finally death due to respiratory failure 20-30 h after
oral administration of methanol. The gradual development of metabolic
acidosis coincided with the accumulation of formic acid in the blood
and the decrease of bicarbonate in the plasma (McMartin et al., 1975).
An attenuated but prolonged syndrome was produced in monkeys by
the administration of an initial methanol dose of 2 g/kg body weight.
and subsequent doses (0.5-1.0 g/kg at 12-24 h intervals), producing
profound ocular toxicity approximately 40-60 h after the initial
dosage (Baumbach et al., 1977; Hayreh et al., 1977; Martin-Amat et
al., 1977).
Various species exposed to methanol by inhalation have exhibited
haemorrhage, oedema, congestion and pneumonia in the lungs (Eisenberg,
1917; Weese, 1928; Tyson & Schoenberg, 1914). Albuminous and fatty
degeneration and fatty infiltration of the liver and kidneys have also
been noted (Eisenberg, 1917; Weese, 1928). Fatty degeneration of
cardiac muscle has been observed in rabbits exposed repeated over 2 to
6 months to methanol via inhalation (Eisenberg, 1917). This subchronic
exposure to methanol in rabbits was also associated with notable
central nervous system effects such as optic nerve damage, lesion and
atrophy of the cerebrum, cerebellum, medulla and pons, along with
decreases in neurocytes, Nissl's granules and in severe cases,
parenchyma cells. Repeated inhalation of methanol resulted in
hyperaemia of choroid, oedema of ocular tissue including the retina
and optic disks, and degeneration of ganglion cells and nerve fibres
in a number of species such as the dog, rabbit and monkey (Tyson &
Schoenberg, 1914). Acute exposure to methanol via inhalation, as well
as oral and dermal exposure, was associated with degeneration and
necrosis of parenchymal tissue and neurons, accompanied by capillary
congestion and oedema, and degeneration of the retina and optic nerve
in rats, rabbits and monkeys (Scott et al, 1933).
An approximate intraperitoneal methanol LD50 of 3-4 g/kg for
pigtail monkeys (Macaca nemestrina) was reported by Clay et al.
(1975). Doses of 2 and 3 g/kg produced metabolic acidosis in the
animals, while monkeys given 4 g/kg became severely acidotic and
exhibited signs of toxicity that were remarkably similar to those
reported in human poisoning (Kane et al., 1968). These animals
displayed a sharp decrease in blood pH (7.03) at 7.5-21 h after
methanol administration. Bicarbonate was the single blood electrolyte
observed to change during the course of methanol acidosis. There was a
latent period of 15-18 h prior to the onset of overt signs of
toxicity, followed by a sequence of signs beginning with behavioural
distress, coma within 24-30 h and death. This time-course parallels
that reported for humans suffering from methanol poisoning (Röe,
1955).
7.2 Short-term exposure
7.2.1 Inhalation exposure
Male and female Sprague-Dawley rats exposed to 650, 2600 and
6500 mg/m3 (500, 2000 and 5000 ppm) methanol for 6 h/day, 5 days/week
for 4 weeks, exhibited no exposure-related effects except for
increased discharges around the nose and eyes which were considered
reflective of upper respiratory tract irritation. No consistent
treatment-related effects were found for organ weight or body weights
or in histopathological or ophthalmoscopical examinations. No ocular
effects were noted in rats from 20 repeated exposures to 6500 mg/m3
(5000 ppm) (Andrews et al., 1987).
Male Sprague-Dawley rats exposed to methanol vapour at
concentrations of 260, 2600 and 13 000 mg/m3 (200, 2000 and
10 000 ppm) for 6 h/day, 5 days/week for 6 weeks, did not develop
pulmonary toxicity. No significant changes were found at the lung
surface and in lung tissue (White et al., 1983).
Rats exposed to 16.8 methanol (0.022 mg methanol/litre of air)
4 h/day for 6 months and simultaneously administered 0.7 mg
methanol/kg daily by gavage exhibited changes in blood morphology,
oxidation-reduction processes and liver function (Pavlenko, 1972).
A preliminary study reported that F-344 rats fed control and
folate-deficient diets and exposed to methanol at a concentration of
1050 mg/m3 (800 ppm) for 20 h/day; 7 days/week for 13 weeks showed
spontaneous degeneration of retina and optic nerve in both diet
groups, while Long-Evans rats did not develop such ocular lesions. The
authors suggest that F-344 rats are unsuitable for ocular toxicity
studies (Lee et al., 1990).
Mice exposed to 63 000 mg/m3 (48 000 ppm) methanol for
3.5-4 h/day up to a cumulative total of 24 h were in a state of
narcosis but survived, whereas mice became comatose when exposed to
71 000 mg/m3 (54 000 ppm) for 54 h (Pavlenko, 1972).
Rabbits exposed by inhalation to 61 mg/m3 (46.6 ppm) methanol
for 6 months (duration of exposure/day not reported) exhibited
ultrastructural changes in the photoreceptor cells of the retina and
Müller fibres (Vendilo et al., 1971).
Two male dogs exposed to methanol vapour in air at 13 000 mg/m3
(10 000 ppm) for about 3 min in each of 8-h periods/day for 100
consecutive days, exhibited no symptoms, unusual behaviour or visual
toxicity. Methanol levels in blood measured at weekly intervals showed
median values of 65 and 140 mg/litre blood (Sayers et al., 1944).
In contrast to many studies of methanol toxicity that reported no
effect of low doses, two Russian studies (Chao, 1959; Ubaidullaev,
1966) reported evidence of neurobehavioural toxicity at low doses as
shown by altered chronaximetry (chronaximetry is the ratio of the
minimum time necessary for a stimulus of twice the absolute threshold
intensity to evoke a response measured as muscle contractions in
response to an electric current applied to an animal's hind leg).
Normally, the flexor chronaxia is shorter than the extensor chronaxia,
and their ratio is stated to be relatively stable.
Chao (1959) reported that the average chronaxia ratio for rats
exposed in the high-dose group (49.77 mg/m3) for 12 h/day, 5
days/week for 3 months, differed significantly from that in the
control group of animals at week 8 of exposure. The average chronaxia
ratio returned to normal during the recovery period and the effects in
the low-dose group (1.77 mg/m3) were insignificant. Histopathological
changes found in the high-dose group, but not in the low-dose group,
included poorly defined changes in the mucous membranes of the trachea
and bronchi, hyperplasia of the submucosa of the trachea, slight
lymphoid infiltration, swelling and hypertrophy of the muscle layer of
arteries, slight degenerative changes to the liver and changes in the
neurons of the cerebral cortex (Chao, 1959).Ubaidullaev (1966)
reported that male rats exposed continuously for 90 days to a
concentration of 5.3 mg/m3 (4 ppm) of methanol vapour, exhibited
changes in chronaxia ratio between antagonistic muscles, in whole
blood cholinesterase activity, in urinary excretion of coproporphyrin
and in albumin-globulin ratio of the serum. Male rats exposed to
0.57 mg/m3 (0.4 ppm) of methanol vapour continuously for 90 days
showed no changes.
It should be noted, however, that an analysis of these studies
by Kavet & Nauss (1990) indicated that, due to flaws in the study
designs, these studies do not provide adequate evidence of an
association between neurobehavioural effects and low-level exposure to
methanol. Both studies were limited by the use of small numbers of
animals per dose group, as well as insufficient reporting of
experimental methods, study results and statistical analysis. Kavet &
Nauss (1990) also stated that the biological significance of changes
in the chronaxia ratio is uncertain.
Male and female cynomolgus monkeys (Macaca fascicularis), three
per sex per dose, that were exposed to 650, 2600 and 6500 mg/m3 (500,
2000 and 5000 ppm) methanol for 6 h/day, 5 days/week for 4 weeks
showed no upper respiratory tract irritation. Neither gross,
microscopic nor ophthalmoscopic examinations disclosed any ocular
effects in the monkeys exposed to 6500 mg/m3 (5000 ppm) (Andrews et
al., 1987).
7.3 Long-term studies
In two 12-month chronic inhalation studies, Fischer-344 rats
(20 female and 20 male animals per group) and B6C3F1 mice (30/30
female/male) were exposed to 13, 130 and 1300 mg/m3 (10, 100 and
1000 ppm) of methanol to examine toxic effects unrelated to
carcinogenesis. A concentration of 130 mg/m3 (100 ppm) was found
to be the NOEL in both species. At the highest exposure, a slightly
reduced weight gain in male and female rats and a small but not
significant increase in the relative liver and spleen weight in female
rats were observed. In mice, the body weight was significantly higher
in the highest exposure groups in both males (after 6 months) and in
females (after 9 months). In addition, the incidence and degree of
fatty degeneration of hepatocytes was significantly enhanced in the
highest exposure groups of mice. However, this could have been due to
the higher incidence of fatty degeneration in mice of great body weight.
Clinical laboratory results did not show any changes attributable to
methanol (NEDO, 1987; Katoh, 1989).
Monkeys (Macaca fascicularis) (eight females per group) were
exposed to 13, 130 or 1300 mg/m3 for periods of 22 h/day for up to 29
months. Body weight, haematological and pathological examinations did
not reveal any dose-dependent effects except for hyperplasia of
reactive astroglias in the nervous system. However, this effect was
not correlated to dose or exposure time and was found to be reversible
in a recovery test (NEDO, 1982).
7.4 Skin and eye irritation; sensitization
In a modified Magnusson-Kligman maximization test with 10 female
guinea-pigs no sensitization was found after intracutaneous or
percutaneous induction and challenge with 50% methanol solution in
distilled water or with Freud's adjuvant. No skin irritation effects
were observed. In a parallel test, a 25% formaldehyde solution was
applied in order to test for possible sensitizing effects resulting
from the metabolic transformation of methanol to formaldehyde. Again
negative test results were seen (BASF, 1979).
New Zealand White albino rabbits treated by application of 100 µl
methanol into the lower conjunctival sac according to OECD test
guidelines and Draize scoring criteria exhibited the following mean
scores of conjunctivitis, chemosis, iritis and corneal opacity after
1, 4, 24, 48 and 72 h (Jacobs, 1990).
Time after application (h): 1 4 24 48 72
Mean score of conjunctivitis: 0.89 2.00 1.67 2.28 2.22
Mean score of chemosis: 2.00 2.00 0.67 1.00 0.50
Mean score of irititis: 0.33 1.00 1.00 0.50 0.33
Mean score of corneal opacity: 0.00 0.00 0.50 0.50 0.67
This demonstrates that methanol causes significant conjunctivitis
under the conditions of this test. Initial oedema (chemosis) seen up
to 4 h had decreased significantly by 72 h. Other ocular lesions were
much less significant.
7.5 Reproductive toxicity, embryotoxicity and teratogenicity
7.5.1 Reproductive toxicity (effects on fertility)
When male Sprague-Dawley rats were exposed for 8 h/day, 5
days/week to airborne methanol concentrations of 260, 2600 or
13 999 mg/m3 (200, 2000 or 10 000 ppm) for 1, 2, 4 or 6 weeks,
significantly decreased levels of circulating free testosterone were
found among rats exposed to 260 mg/m3 for 2 and 6 weeks and to
2600 mg/m3 for 6 weeks. However, the 13 000 mg/m3 group showed
no change. Significant changes in luteinizing hormone (LH) were found
after 6 weeks in animals exposed to 13 000 mg/m3, but no changes
in follicle-stimulating hormone (FSH) were observed at the various
exposure levels (Cameron et al., 1984). Sprague-Dawley rats exposed to
260 mg/m3 for 6 h for either 1 day or 1 week showed significant
depression (59%) in serum testosterone immediately after the first
exposure, but not after 1 week of daily 6-h exposures (Cameron et
al., 1985).
In a subsequent study groups of 10 male Long-Evans hooded rats,
60 days of age and acclimatized (or not) to handling, were exposed to
0, 260, 6500 or 13 000 mg/m3 (0, 200, 5000 or 10 000 ppm) methanol
for 6 h and killed either immediately on removal from the chambers or
18 h later. Similar groups of rats, acclimatized to handling or not,
were exposed to 6500 mg/m3 during 1, 3 or 6 h and killed immediately.
Serum testosterone levels were not significantly increased at 6 or
24 h in acclimatized rats, but levels were increased in non-
acclimatized rats exposed to 6500 mg/m3 and killed after 24 h. The
serum luteinizing hormone (LH) level was increased in acclimatized
rats exposed to 13 500 mg/m3 and killed at 6 and 24 h but the LH
level was reduced in non-acclimatized rats exposed to 6500 or
13 000 mg/m3 at 6 h but not 24 h. This experiment did not confirm the
earlier report that exposure to 260 mg/m3 for 6 h reduced serum
testosterone levels. In the second experiment serum LH and
testosterone levels did not differ at any time point between controls
and rats exposed to 6500 mg/m3 (Cooper et al.,1992). Methanol
inhalation at 260 mg/m3 for 8 h/day for up to 6 weeks did not reduce
serum testosterone levels in normal Sprague-Dawley rats (Lee et al.,
1991). In Long-Evans rats fed either control or folate-reduced diets
and exposed to 1040 mg/m3 for 20 h/day for 13 weeks, no adverse
effect on testicular morphology was observed with 10-month-old rats
fed either diet. A greater incidence of testicular degeneration was
however noted with 18-month-old rats given the folate-reduced diet,
suggesting that methanol potentially accelerates the age-related
degeneration of the testes (Lee et al., 1991).
7.5.2 Developmental toxicity
The inhalation of methanol by pregnant rodents throughout the
period of embryogenesis to high atmospheric concentrations (6500 to
26 000 mg/m3; 5000 to 20 000 ppm) impaired neural tube closure and
induced a wide range of concentration-dependent teratogenic and
embryolethal effects (Nelson et al., 1985; Rogers et al., 1993; Bolon
et al., 1993, 1994). In these studies, significant increase in the
incidence of exencephaly were observed following maternal methanol
exposures of > 6500 mg/m3 (> 5000 ppm) in mice, while similar
effects were observed in rats following exposures of > 13 000 mg/m3
(> 10 000 ppm), indicating that mice are more sensitive than rats to
the embryotoxic effects of methanol.
Pregnant Sprague-Dawley rats were given by inhalation for 7 h/day
either 6500 or 13 000 mg/m3 (5000, or 10 000 ppm) methanol on days
1-19 of gestation, or 26 000 mg/m3 (20 000 ppm) methanol on days 7-15
of gestation. The blood levels of methanol in the 26 000 mg/m3 group
ranged from 8.34 to 9.26 mg/ml after 1 day of exposure and from 4.84
to 6.00 mg/ml after 10 days of exposure. Methanol induced a dose-
related decrease in fetal weights and an increase in malformations.
The highest methanol concentration (26 000 mg/m3) produced slight
maternal toxicity (slightly unsteady gait) after the initial days
of exposure, and a high incidence of congenital malformations
(p < 0.001), predominantly extra or rudimentary cervical ribs and
urinary or cardiovascular defects. Similar malformations were found in
the groups exposed to 13 000 mg/m3 but the incidence was not
significantly different from that of the controls. No increase in
malformations was found in the group exposed to 6500 mg/m3
(5000 ppm), which was suggested to be a no-observed-effect level for
this test system (Nelson et al., 1985).
Pregnant CD-1 mice were treated by inhalation to 1300, 2600,
6500, 10 000, 13 000 or 19 500 mg/m3 (1000, 2000, 5000, 7500, 10 000
or 15 000 ppm) of methanol for 7 h/day on days 6-15 of pregnancy.
Significant increases were observed in the incidence of exencephaly
and cleft palate at 6500 mg/m3 or more. Increased embryo/fetal death
was found at exposures of 10 000 mg/m3 or more, including an
increasing incidence of full-litter resorptions. Reduced fetal weight
was found at 13 000 mg/m3 or more. A dose-related increase in
cervical vertebrae was significant at 2600 mg/m3 or more. The NOAEL
for the developmental toxicity was suggested to be 1300 mg/m3 (1000
ppm) methanol in this test system. There was no evidence of maternal
toxicity at methanol exposures below 10 000 mg/m3 (Rogers et al.,
1993).
A spectrum of cephalic neural tube defects was found in near-term
(gestation day 17) CD-1 mouse fetuses following maternal inhalation of
methanol at high concentration (19 500 mg/m3; 15 000 ppm) for 6 h/day
during neurulation (gestation days 7-9). Dysraphism, chiefly
exencephaly, occurred in 15% of the fetuses, usually in association
with reduction or absence of multiple bones in the craniofacial
skeleton and ocular anomalies (prematurely open eyelids, cataracts,
retinal folds). Exposure to a high concentration of methanol
(19 500 mg/m3) injured the multiple stem populations in the
neuralating mouse embryo. Significant neural pathology may remain in
older conceptuses even in the absence of gross lesions (Bolon et al.,
1994).
Transient neurological signs and reduced body weights were found
in up to 20% of CD-1 dams exposed to 19 500 mg/m3 (15 000 ppm)
methanol 6 h/day throughout organogenesis (gestational days 6-15).
Near-term fetuses revealed embryotoxicity (increased resorptions,
reduced fetal weights and/or fetal malformations) at 13 000 and
19 500 mg/m3 (10 000 and 15 000 ppm) methanol while 3-day exposures
at 6500 mg/m3 (5000 ppm) for 6 h/day yielded no observable adverse
effects (Bolan et al., 1993). In the studies of Bolon et al. (1993,
1994), terata included neural and ocular defects, cleft palate,
hydronephrosis, deformed tails and limb (paw and digit) anomalies.
Neural tube defects and ocular lesions occurred after methanol
inhalation by pregnant CD-1 mice between gestational days 7 and 9,
while limb anomalies were induced only during gestational days 9-11;
cleft palate and hydronephrosis were observed after exposure during
either period. The spectrum of teratogenic effects depended upon both
the stage of embryonic development and the number of methanol
exposures.
Long-Evans rats administered single oral doses of 1.3, 2.6 or
5.2 ml methanol/kg by gavage on day 10 of gestation, exhibited dose-
related anomalies, e.g., undescended testes and eye defects
(exophthalmia and anophthalmia) in the offspring. At the methanol dose
of 5.2 ml/kg, the maternal weight loss was > 10%, which was the only
clinical toxic manifestation/histopathological change noted for the
dams. A significant decrease in fetal body weight (11-21%) was
associated with oral ingestion of methanol in the dams. Methanol given
acutely can produce anomalies in the offspring where there are no
apparent maternal toxic responses (Youssef et al., 1991).
Methanol was shown to impair uterine decidualization during early
pregnancy in Holtzman rats administered 1.6, 2.4 or 3.2 g methanol/kg
per day by gavage during days 1-8. Reductions in pregnant uterine and
implantation site weights seen on day 9 were the result of methanol
impedance of normal uterine decidualization as demonstrated by effects
on decidual cell response technique. Methanol (3.2 g/kg per day)
produced a non-specific maternal toxicity (reduction in body weight)
by day 9, but no effect on days 11 or 20 on embryo and fetal survival
or development were found (Cummings, 1993).
When pregnant CD-1 mice were gavaged orally with 4 g methanol/kg,
the incidences of fetal resorption, external defects (including cleft
palate) and reduced fetal weight were similar to those observed in the
13 000 mg/m3 (10 000 ppm) inhalation exposure group. Cleft palate
(43.5% per litter) and exencephaly (29% per litter) were the
predominant external defects seen following methanol exposure by oral
gavage. Methanol blood level in the gavage study was 4 mg/ml, which
was reportedly similar to the blood level at the 13 000 mg/m3
inhalation exposure group (see above) (Rogers et al., 1993).
No effects on reproductive performance were reported in a two-
generation reproductive study in F-344 rats administered 13, 130 or
1300 mg/m3 (10, 100 or 1000 ppm) methanol by inhalation for
18-20 h/day. A statistically significant decrease in brain weight was
found at the 1300 mg/m3 level in 3-, 6- and 8-week-old pups of the
F1 generation. In the F2 generation reduced brain thymus and
hypophysis weight was observed. (NEDO, 1987; Katoh, 1989).Teratology
studies with Sprague-Dawley rats exposed to 260, 1300 or 6500 mg/m3
(200, 1000 or 5000 ppm) methanol by inhalation for 22 h/day during
gestational days 7-17 revealed significant weight decreases in brain,
thyroid and thymus of the offspring resulting from maternal exposure
to 6500 mg/m3. However, no abnormal changes were detected
histopathologically. Evidence of maternal toxicity was found at this
level of exposure and toxic effects to fetuses were reported,
including death. No effects were found at 1300 kg/m3 (NEDO, 1987;
Katoh, 1989).
A pilot developmental toxicity study was conducted by Ryan et al.
(1994) to assess the utility of the folic-acid-deficient rat model, a
model that would be sensitive to methanol and potentially reflective
of the human risk/response. Methanol was administered in drinking-
water on days 6-15 of gestation at concentrations of 0.5, 1.0 and 2.0%
to three groups of 7 to 9 sperm-positive Long-Evans rats. The average
blood levels were given as 0.21, 0.26 and 0.67 mg/ml, respectively. A
dose-dependant increase in the incidence of maternal and developmental
effects was observed. For both end-points the NOEL was assumed to be
less than 0.5% methanol in drinking-water, corresponding to a blood
level of 0.21 mg/ml.
Weiss et al. (1996) studied developmental neurotoxicity of
pregnant Long-Evans rats and their newborn offspring exposed to
5900 mg/m3 (4500 ppm) of methanol by inhalation for 6 h daily,
beginning on gestation day 6, with both dams and pups then being
exposed through postnatal day 21. Although findings suggested
significant functional consequences in rats resulting from this
exposure, these consequences were considered subtle in character.
Exposure to 5900 mg methanol/m3 did not affect the suckling time
and conditioned olfactory aversion test of newborn rats. Methanol-
exposed newborn pups were less active on postnatal day 18 and more
active on postnatal day 25 than control newborn pups (motor activity
test). The study found only isolated positive results that were small
and variable. The two adult assays, the fixed-ratio wheel-running
test and the stochastic discrimination test, yielded evidence of a
significant methanol effect.
No evidence of brain damage emerged on the basis of neuro-
pathology, although differences in neural cell adhesion molecules
(NCAMs) arising from methanol exposure were observed in neonatal
cerebella (Weiss et al., 1996). Methanol treatment caused a decrease
in expression in both NCAM 140 and NCAM 180.
Further elaboration of the effects of perinatal exposure on NCAM
in Long-Evans rats exposed to 5900 mg/m3 (4500 ppm) methanol vapour
for 6 h daily (beginning on gestation day 6 with dams and pups then
exposed until postnatal day 21) were described by Stern et al. (1996).
Blood methanol concentrations from samples obtained immediately
following a 6-h exposure reached approximately 500-800 µg/ml in the
dams during gestation, and lactation average concentrations for pups
attained levels about twice those of the dams. Light-microscopic
analysis showed no significant abnormalities in the brains of the
methanol-treated animals. However, assays of NCAM in the brains of
pups sacrificed on postnatal day 4 showed staining for both the 140
and the 180 kDa isoforms to be less intense in the cerebellum of
exposed animals. NCAM differences were not apparent in animals
sacrificed after their final exposure. NCAM 140 is the primary isoform
expressed during the stages of neuronal migration and NCAM 180 is
expressed during synaptogenesis where it is critical to neuronal
plasticity, learning and memory. NCAMs are developmentally regulated
glycoproteins that serve critical roles in the formation and
maintenance of the nervous system (Stern et al., 1996).
7.5.3 Behavioural effects
Neonatal behavioural toxicity was reported in studies involving
two groups of primigravid Long-Evans rats given drinking solutions of
2% methanol either on gestational days 15-17 or 17-19, with the
average daily intake on these days amounting to 2.5 g methanol/kg.
Lack of maternal toxicity was indicated by measurements of weight
gain, gestational duration or daily fluid intake. Litter size, birth
weight and infant mortality did not differ between the two treatment
groups and the control. Pups from methanol-treated rats required
longer periods than controls to begin suckling on postnatal day 1. On
postnatal day 10, they required more time to locate nesting material
from their home cages, suggesting that prenatal methanol exposure
induced behavioural abnormalities early in life, unaccompanied by
overt toxicity (Infurna & Weiss, 1986).
Following inhalation exposure of Long-Evans rats to 19 500 mg/m3
(15 000 ppm) methanol for 7 h/day on gestational days 7-19, maternal
blood levels decreased significantly from 3.8 mg/litre on the first
day of exposure to 3.1 mg/litre on the 12th day of exposure. Methanol
transiently reduced maternal body weight by 4-7% on gestational days
8-10 and offspring body weight by 5% on post-natal days 1-3. Motor
activity, olfactory learning, behavioural thermoregulation, T-maze
learning, acoustic startle response, pubertal landmarks and passive
avoidance tests performed at the end of the exposure period failed to
reveal significant effects. Prenatal exposure to high levels of
inhaled methanol appeared to have little effect beyond post-natal day
3 in this series of tests (Stanton et al., 1995).
7.5.4 In vitro studies
Methanol is developmentally toxic to both mouse (CD-1) and rat
(Sprague-Dawley) embryos during organogenesis in whole embryo culture
(WEC), a technique which removes the confounding maternal influences
(Andrews et al., 1993). Comparable developmental stages of CD-1 mouse
and Sprague-Dawley rat embryos were exposed to methanol (0-16 mg/ml
for rat and 0-8 mg/ml for mouse embryos) for 24 h. Rat embryos were
cultured for an additional 24 h without methanol in the medium, having
a total culture time of 48 h. Concentration-dependent decreases in
somite number, head length and developmental score occurred in both
species, with significant effects in the rat at > 8 mg/ml and in
the mouse at > 4 mg/ml (Andrews et al., 1993).
In studies of 8-day mouse embryos cultured in methanol,
concentrations greater than 2 mg methanol/ml caused a significant
decrease in developmental score and crown-rump length; the 8 mg/ml
group also suffered 80% embryolethality (Andrews et al., 1993). Mouse
embryos were affected at methanol concentrations that were not
dysmorphogenic or embryotoxic in the rat following teratogenic
in vivo exposures (Rogers et al, 1993), suggesting that the higher
sensitivity of the mouse was due, at least in part, to the greater
intrinsic embryonal sensitivity of this species to methanol (Andrews
et al., 1993).
Depending on the concentration and duration of methanol exposure
(0-20 mg/ml for 6 h, 12 h, or 1 or 4 days) on embryonic CD-1 mouse
palate in serum-free organ culture, the medial epithelium either
degenerated completely or remained intact in unfused palates (either
condition would interfere with fusion) (Abbott et al., 1994). Cellular
proliferation appeared to be a specific and sensitive target for
methanol as craniofacial tissues responded to methanol with reduction
in DNA content at an exposure that did not effect total protein.
However both DNA and protein levels decreased with increasing exposure
to methanol. Methanol selectively altered the morphological fate of
the medial palatal epithelium cells and the specific effect on cell
survival was exposure dependent (Abbott et al., 1994).
7.6 Mutagenicity and related end-points
7.6.1 In vitro studies
The structure of methanol (by analogy with ethanol) does not
suggest that it would be genotoxic.
Methanol gave negative results when tested in Salmonella
typhimurium plate incorporation assays with or without metabolic
activation using strains TA98, TA100, TA1535, TA1537 and TA1538
(Simmon et al., 1977). It was also negative in the presence or absence
of metabolic activation in strains TA1535, TA100, TA1538, TA98 and
TA1537 (De Flora et al., 1984) and in a DNA repair test in E. coli
using strains WP 2, WP 67 and CM 871 in the presence or absence of
metabolic activation (De Flora et al., 1984).
Methanol (6.0% v/v) induced 3.02% chromosomal malsegregation in
Aspergillus nidulans diploid strain P1 (Crebelli et al., 1989). The
result was statistically significant at two concentrations and a dose-
response relationship was evident.
Methanol was negative for gene mutation at the ade 6 locus
in the yeast Schizosaccharomyces pombe with or without the
postmitochondrial fraction from mouse liver (Abbondandolo et al.,
1980). It was also negative in a mutagenicity test for n+1 aneuploidy
arising from meiotic disfunction of linkage group I in the fungus
Neurospora crassa (Griffiths, 1981).
Methanol did not induce sister chromatid exchanges (SCEs) in
Chinese hamster cells in vitro during treatment for 8 days to a
final concentration of 0.1% (v/v) (Obe & Ristow, 1977). Only in the
presence of S-9 mix and methanol (7.9 mg/ml) was there a significant
increase in the mutation frequency in L5178Y mouse lymphoma cells
(McGregor et al., 1985), possibly because this assay detects
chromosome damage as well as gene mutation.Methanol was negative in
two in vitro tests for cell transformation: the Syrian hamster
embryo cell (SHE) clonal system (Pienta et al., 1977) and the Rausher
leukaemia virus-infected rat embryo cell (RLV/RE system) (Heidelberger
et al., 1983).
Addition of methanol (or ethanol) to unleaded gasoline as a fuel
extender did not appear to significantly alter the genetic toxicity of
particulate exhaust particles when tested in S. typhimurium strains
TA100, TA98, TA98 NR, and TA98 DNPR with S-9 activation. In all the
alcohol-blended fuel tests, the mass of particle-associated organics
emitted from the exhaust was lower than that observed during the
control tests using gasoline alone (Clark et al., 1983).
7.6.2 In vivo studies
No increased frequencies of micronuclei in blood cells, of
SCEs, chromosome aberrations or micronuclei in lung cells, or of
synaptonemal complex damage in spermatocytes were found in mice
exposed by inhalation to 1050 or 5200 mg/m3 (800 or 4000 ppm)
methanol for 5 days (Campbell et al., 1991).
Urine from mice orally administered five daily doses of methanol
(5 g/kg total) showed no mutagenic activity, and no increase in the
incidence of abnormal sperm was reported (Chang et al., 1983). Oral
administration of 1 g methanol/kg to mice increased the incidence of
chromosomal aberrations, particularly aneuploidy and SCEs, as well as
the incidence of micronuclei in polychromatic erythrocytes (Pereira et
al., 1982).
The oral administration of 14C-labelled methanol to rats
resulted in covalent binding to haemoglobin, with binding exhibiting a
linear dose relationship between 10 and 100 µmol/kg (Pereira et al.,
1982).
B6C3F1 mice treated with five daily oral doses of 1 g
methanol/kg exhibited abnormal (banana type) sperm morphology. The
biological significance of these changes is unknown (Ward et al.,
1984). It should be noted that the above results, namely altered sperm
(Ward et al., 1984) and haemoglobin binding (Pereira et al., 1982) are
end-points not generally used for genotoxic evaluation and their
assessment in terms of mutagenicity is unclear.
There is some evidence that bone marrow cytogenetic analysis
indicated a dose-related response for structural aberrations,
especially centric fusions in mice treated with three daily
intraperitoneal methanol doses of between 75-300 mg/kg total dose
(Chang et al., 1983).
In vitro and in vivo mutagenicity studies on methanol, i.e.,
the Ames test, somatic mutation assay in CH-V79 cells, chromosome
aberrations, SCEs and the micronucleus test in mice conducted by NEDO
(1987; Katoh, 1989), were all reported to be negative.
7.7 Carcinogenicity
There have been no studies reported in the peer-reviewed
literature on the potential carcinogenicity of methanol per se in
laboratory animals.
The New Energy Development Organization (NEDO) in Japan reported
carcinogenicity studies in which B6C3F1 mice and Fischer-344 rats of
both sexes were exposed by inhalation to 13, 130 or 1300 mg/m3 (10,
100 and 1000 ppm) methanol for 20 h/day for 18 and 24 months,
respectively (NEDO, 1987; Katoh, 1989). No evidence of carcinogenicity
was found in either species. High-dosed animals had a higher, but not
statistically significant, incidence of papillary adenomas than
controls , and histopathological examination suggested that these
changes were between non-neoplastic and neoplastic changes.
Additionally, seven cases of adrenal pheochromocytoma were found in
high-dose animals compared to one case in controls. This observation
was not statistically significant according to the Fisher exact test
(Katoh, 1989).
It is unlikely that methanol is carcinogenic to mouse skin. In an
experiment using four strains of female mice (Balb/c, Sencar, CD-1 and
Swiss) to study N-nitrosomethylurea carcinogenesis, methanol was
used as a solvent control. Four groups of 20 mice of each strain
received 25 µl methanol twice weekly for 50 weeks followed by
observation for lifespan. Only one skin tumour was observed among the
80 control animals (Lijinsky et al., 1991).
7.8 Special studies
7.8.1 Effects on hepatocytes
When Garcia & Van Zandt (1969) administered repeated doses of 3
to 6 g/kg by gavage to rhesus monkeys (Macaca mulata) for 3-20
weeks, average serum levels of methanol of 4750 mg/litre were attained
within a few hours. Animals were killed at the end of treatment and
livers examined histologically. Hepatocytes showed nucleolar
segregation (zoning of nucleus), hyperplasia of endoplasmic reticulum
and swelling of mitochondria. These changes were also found in one
monkey sacrificed 12 weeks after the end of treatment.
7.8.2 Toxic interactions
Inhaled methanol potentiated the hepatotoxicity produced by
carbon tetrachloride in adult male F-344 rats. Rats were exposed to
methanol (0 or 13 000 mg/m3) 10000 ppm for 6 h, then treated 24 h
later with oral CCl4 (0.075 ml/kg). CCl4 alone produced a low level
of hepatotoxicity within 3 days. Methanol plus CCl4 resulted in
marked increases in serum aspartate aminotransferase and alanine
aminotransferase that lasted for 7 days. Methanol also exacerbated the
histological evidence of CCl4-induced centrilobular degeneration and
necrosis (Simmons et al., 1995).
Methanol exposure by inhalation induced cytochrome P4502E1
(CYP2E1), which appeared to be the principal toxicokinetic mechanism
underlying methanol potentiation of carbon tetrachloride
hepatotoxicity (Allis et al., 1996).
When dichloromethane (DCM) is metabolized carbon monoxide is
formed, leading to increased carboxyhaemoglobin (COHb) levels in
blood. Pankow & Jagielki (1993) found that in rats pretreated with
methanol, methanol doses of 790-6330 mg/kg (24.7-198 mmol/kg)
stimulated increased metabolism of DCM, as seen by further increases
in COHb levels. When methanol was administered simultaneously with
DCM, a decrease in COHb formation was seen at methanol doses of 4736
to 7900 mg/kg (148-247 mmol/kg) but not at 3162 mg/kg (98.8 mmol/kg).
Thus methanol can interact with DCM metabolism both by induction and
by competitive inhibition, the latter only at very high doses.
Poon et al. (1994) reported no significant interactive effects in
young Sprague-Dawley rats exposed to vapours of methanol/toluene
(400/110 mg/m3; 400/1100 mg/m3; 4000/110 mg/m3 and 4000/1100 mg/m3)
for 6 h/day, 5 days/week for 4 weeks. Exposure to methanol (400 to
4000 mg/m3) and to toluene (110 mg/m3 to 1100 mg/m3) or to a mixture
of both produced mild biochemical effects and histological changes in
the thyroid (moderate reduction in follicle size in the thyroids) and
nasal passages.
The biochemical, haematological and histological effects on
Sprague-Dawley rats after exposure to methanol (3000 mg/m3;
2500 ppm), gasoline (3200 ppm) and methanol/gasoline (2500/3200 ppm)
vapour 6 h/day for 4 weeks were examined by Poon et al. (1995).
Gasoline was largely responsible for the adverse effects, the most
significant of which included depression in weight gain in the males,
increased liver weight and hepatic microsomal enzyme activities in
both sexes, and suppression of uterine eosinophilia. No apparent
interactive effects between methanol and gasoline were observed.
7.8.3 Studies with exhaust emissions from methanol-fuelled engines
There are few data related to the effects of emissions from
methanol-fuelled engines. Since most such fuels will contain a
proportion of gasoline and other additives and the emissions will be
complex, the interpretation of these data in relation to methanol
toxicity is complicated.
Maejima et al. (1992, 1993 and 1994) studied the effects of
emissions from M-85 methanol-fuelled engines (methanol with 15%
gasoline), without a catalyst, on Fischer-344 rats for periods up to
12 weeks. The exhaust contained significant amounts of carbon monoxide
(89.9 ppm), oxides of nitrogen (22.9 ppm), formaldehyde (2.3 ppm) and
methanol (8.1 ppm). The effects observed were considered to be
primarily related to formaldehyde. No increase in plasma methanol or
formic acid was detected.
7.9 Mechanism of ocular toxicity
Formic acid, the toxic metabolite of methanol, has been
hypothesized to produce retinal and optic nerve toxicity by disrupting
mitochondrial energy production (Fig. 1) (Martin-Amat et al., 1977;
Sharpe et al., 1982). It has been shown in vitro to inhibit the
activity of cytochrome oxidase, a vital component of the mitochondrial
electron transport chain involved in ATP synthesis (Nicholls, 1975).
Inhibition occurs subsequent to the binding of formic acid to the
ferric haem iron of cytochrome oxidase, and the apparent inhibition
constant is between 5 and 30 mM (Nicholls, 1975). Concentrations of
formate present in the blood and tissues of methanol-intoxicated
humans, non-human primates and rodent models of methanol-intoxication
are within this range (Martin-Amat et al., 1977; Sejersted et al.,
1983; Eells, 1991).
Studies conducted in methanol-sensitive rodent models have
revealed abnormalities in retinal and optic nerve function and
morphology, consistent with the hypothesis that formate acts as a
mitochondrial toxin (Fig. 2). In these animal models, formate
oxidation is selectively inhibited by dietary (Lee et al., 1994) or
chemical (Eells et al., 1981) depletion of folate coenzymes, thus
allowing formate to accumulate to toxic concentrations following
methanol administration. Methanol-intoxicated rats developed formic
acidaemia, metabolic acidosis and visual toxicity analogous to the
human methanol poisoning syndrome (Eells, 1991; Murray et al., 1991;
Lee et al., 1994a,b).
Sixty hours after the administration of the first dose of
methanol, blood formate values ranged from 8-20 mM with blood hydrogen
carbonate values in the range of 5-12 mEq/litre and blood pH values of
6.83-7.08. Similar blood formate concentrations, hydrogen carbonate
levels and pH values were reported in methanol-intoxicated monkeys
(Martin-Amat et al., 1977) and in severe cases of human methanol
poisoning (McMartin et al., 1980a; Sejersted et al., 1983; Jacobsen et
al., 1988).
Visual dysfunction was measured as reduction in the flash evoked
cortical potential (FEP) and electroretinogram (ERG). The FEP is a
measure of the functional integrity of the primary visual pathway from
the retina to the visual cortex and the ERG is a global measure of
retinal function in response to illumination (Creel et al., 1970;
Dowling, 1987). The FEP was progressively diminished in methanol-
intoxicated rats, indicative of a disruption of neuronal conduction
along the primary visual pathway from the retina to the visual cortex
(Eells, 1991). ERG analysis in methanol-intoxicated rats revealed a
significant early deficit in b-wave amplitude, followed by a
temporally delayed lesser reduction in a-wave amplitude (Murray et
al., 1991). The b-wave of the ERG is generated by depolarization of
the Muller glial cells and reflects synaptic activity at the level of
the bipolar cells (Dowling, 1987). The b-wave of the ERG is
extremely sensitive to conditions that interfere with retinal energy
metabolism and is reduced or abolished following brief ischaemia or
the administration of metabolic poisons (Bresnick, 1989; Dowling,
1987). Both FEP and ERG alterations occurred at the same time as
accumulation of blood formate, indicative of a causal relationship
between formate-induced metabolic and visual disturbances. Similar ERG
reductions have been reported in methanol-intoxicated primates
(Ingemansson, 1983) and in human methanol intoxication (Ruedemann,
1962; Murray et al., 1991).
In addition to neurofunctional changes, bioenergetic and
morphological alterations indicative of formate-induced disruption of
retinal energy metabolism have been documented in methanol-intoxicated
rats (Murray et al., 1991; Eells et al., 1996; Garner et al.,
1995a,b). Morphological studies, coupled with cytochrome oxidase
histochemistry, revealed generalized retinal oedema, photoreceptor and
RPE vacuolation, mitochondrial swelling and a reduction in cytochrome
oxidase activity in photoreceptor mitochondria from methanol-
intoxicated rats (Murray et al., 1991; Eells et al., 1995, 1996). The
most striking structural alterations observed in the retinas of
methanol-intoxicated rats were vacuolation and mitochondrial swelling
in inner segments of the photoreceptor cells (Murray et al., 1991).
Photoreceptor mitochondria from methanol-intoxicated rats were swollen
and expanded to disrupted cristae and showed no evidence of cytochrome
oxidase reaction product. In contrast, photoreceptor mitochondria from
control animals showed normal morphology with well-defined cristae and
were moderately reactive for cytochrome oxidase reaction product.
These findings are consistent with disruption of ionic homoeostasis in
the photoreceptors, secondary to inhibition of mitochondrial function.
Biochemical measurements also showed a significant reduction in
retinal and brain cytochrome oxidase activity and ATP concentrations
in methanol-intoxicated rats relative to control animals (Eells et
al., 1995). Surprisingly, no differences from control values were
observed in hepatic, renal or cardiac cytochrome oxidase activity or
ATP concentrations in methanol-intoxicated rats. The reduction in
retinal function, inhibition of retinal, optic nerve and brain
cytochrome oxidase activity, depletion of retinal and brain ATP
concentrations, and mitochondrial disruption produced in methanol-
intoxicated rats are consistent with the hypothesis that formate acts
as a mitochondrial toxin with selectivity for the retina and brain.
Studies by Eells et al. (1996) compared the effects on retinal
function and structure of rapidly increasing formate concentrations
typical of acute methanol intoxication with low-level plateau formate
concentrations more likely to be generated by subacute or chronic
methanol exposure. Methanol-intoxicated rats that accumulated formate
concentrations of 8-15 mM developed metabolic acidosis, retinal
dysfunction, and retinal histopathological changes. Retinal
dysfunction was measured as reductions in the a- and b-waves of
the electroretinogram that occurred at the same time as blood formate
accumulation. Histopathological studies revealed vacuolation in the
retinal pigment epithelium and photoreceptor inner segments. Rats
exposed to formate concentrations ranging from 4 to 6 mM for 48 h
showed evidence of retinal dysfunction in the absence of metabolic
acidosis and retinal histopathology. These data indicated that
formate-induced retinal dysfunction in methanol-intoxicated rats can
be produced by steadily increasing concentrations of formate and,
importantly, can also be produced by prolonged exposure to lower
concentrations of formate.
Martinasevic et al. (1996) studied components of folate-dependent
formate oxidation, e.g., folate and 10-CHO-H4-folate dehydrogenase
(10-FDH), in human and rat retinae. Total folate levels in human and
rat retinal tissues were much lower than the levels in liver. However,
folate levels in human retina were only 14% of those determined in rat
retina. Comparable amounts of this 10-FDH were present in both
cellular compartments in each species. However, the amount of 10-FDH
in the human retina was approximately three times the amount found in
the rat retina. Immunohistochemical staining for 10-FDH showed that
this enzyme was preferentially localized in Müller cells. Since Müller
cells appear to represent the target for formate-induced ocular
toxicity, the authors suggested that formate oxidation reactions might
serve two roles, first a protective role and then a role in methanol-
induced toxicity in Müller cells.
Garner & Lee (1994) employing oscillatory potential analysis
showed that retinal ischaemia was not involved in methanol-induced
visual system toxicity.
The role of retinal metabolism in methanol-induced retinal
toxicity in folate-sufficient (FS) rats and folate-deficient (FR)
rats, some of which were also pretreated with disulfiram (DSF), was
examined by Garner et al. (1995). Folate-deficient rats treated with
methanol displayed elevated blood and vitreous humour formate levels
along with abnormal electroretinograms (ERG), whereas methanol-exposed
folate-deficient rats pretreated with DSF did not. Formaldehyde was
not detected in blood or vitreous humour, either with or without DSF
treatment, suggesting that formate is the toxic metabolite in
methanol-induced retinal toxicity. Additionally, intravenous infusion
of formate to levels seen in methanol toxicity did not alter ERG
levels, suggesting intraretinal metabolism of methanol to formate may
be necessary for retinal toxicity.
Studies measuring ATP synthesis in mitochondria isolated from
bovine retina and bovine heart have provided additional evidence for a
tissue-selective action of formate (Eells et al., in press). In these
studies, mitochondrial ATP synthesis was measured in the presence of
different metabolic substrates. Formate selectively inhibited ATP
synthesis in mitochondria isolated from bovine retina in the presence
of metabolic substrates supplying electrons at the level of complex I,
complex II and complex IV in the mitochondrial respiratory chain. The
inhibitory effect of formate on retinal mitochondrial ATP synthesis
was concentration-dependent, significant reductions in ATP synthesis
being produced at 10 mM formate and Ki values for inhibition ranging
from 30 to 50 mM formate. Comparative studies conducted in
mitochondria isolated from bovine heart showed little or no inhibition
of ATP synthesis at formate concentrations up to 50 mM. These findings
provide direct evidence that formate acts as retinal mitochondrial
toxin and suggest that one component of the retinotoxic actions of
formate may be due to tissue-specific differences in mitochondrial
transport mechanisms or in mitochondrial metabolism.
The apparent selective vulnerability of the retina and optic
nerve to the toxic actions of formate in methanol poisoning has been
the subject of considerable speculation (Röe, 1955; Sharpe et al.,
1982; Jacobsen & McMartin, 1986). Although methanol intoxication is
known to disrupt brain function and severe intoxication results in
coma and death, the most common permanent consequence of methanol
intoxication is blindness (Röe, 1955). Several factors may contribute
to the unique vulnerability of the retina and optic nerve to the
cytotoxic actions of formate. One component of this selectivity is
related to the differences in the distribution of formate in the eye
and the brain. Formate concentrations measured in the vitreous humour
and retinas of methanol-intoxicated rats (Eells, 1991; Eells et al.,
1996) were equivalent to or greater than corresponding blood formate
concentrations. In contrast, the concentrations of formate in the
brain were significantly lower than blood formate concentrations.
These data suggest that the toxic actions of methanol on the visual
system may be due to the selective accumulation of formate in the
vitreous humour and the retina as compared with other regions of the
central nervous system. Secondly, the retina has a very limited
metabolic capacity to oxidize and thus detoxify formate (Eells et al.,
1996). Thirdly, cytochrome oxidase activity and ATP concentrations
have been shown to be selectively reduced in the retina, optic nerve
and brain in methanol-intoxicated rats, suggesting that there may be
tissue- and cell-specific differences in mitochondrial populations and
in the actions of formate on mitochondrial function (Eells et al.,
1995). Finally, in vitro studies in isolated retinal and cardiac
mitochondria have shown that formate selectively inhibits retinal
mitochondrial ATP synthesis (Eells et al., in press). These findings
support the hypothesis that formate acts as a selective mitochondrial
toxin in the retina and establish a link between the effects of
formate in vitro and the retinal toxicity associated with formate
accumulation in methanol intoxication.
8. EFFECTS ON HUMANS
Acute oral and inhalation exposures, and to a lesser extent
percutaneous absorption of high concentrations of methanol, have
resulted in CNS depression, blindness, coma and death. The most noted
effects resulting from longer-term exposure to lower levels of
methanol have been a broad range of ocular effects.
8.1 General population and occupational exposure
The human health effects after exposure to methanol are
qualitatively the same for the general population and for those
exposed in the workplace, and will be considered together. Acute
methanol intoxication in the general population is an uncommon
occurrence, but often results in serious morbidity and mortality.
Litovitz et al. (1988) reviewed the acute methanol exposure cases
reported in the USA. In 1987, 1601 methanol poisonings were reported
to the American Association of Poison Control Centers (AAPCC). Half of
these individuals required hospitalization and the death rate was
0.375%. It was estimated that the actual annual incidence of methanol
poisonings in the USA in 1987 was about 6400 cases. Subsequent surveys
of methanol exposure cases have been conducted by the AAPCC, and these
have shown similar annual frequencies to that in 1987. These data
result from poisoning cases that are not usually reported elsewhere,
since case reports of methanol poisoning are rarely published in
today's literature. Poisoning frequency surveys are not available from
the rest of the world, but reports in the biomedical literature and in
the press would suggest a worldwide distribution of methanol poisoning
cases at least as great as in the USA.
8.1.1 Acute toxicity
Methanol (wood alcohol) has been recognized as a human toxic
agent since the end of the 19th century. Since the early part of the
20th century, many hundreds of cases of methanol intoxication have
been reported as single cases and as groups in many countries. Many of
the human cases were due to the ingestion of denatured alcohol.
The preponderance of methanol poisonings have resulted from the
consumption of adulterated alcoholic beverages, e.g., "moonshine", or
"bootleg whiskey", wood alcohol and spirits mixed with whiskey. Buller
& Wood (1904) and Wood & Buller (1904) reported 235 cases of blindness
or death primarily connected with drinking adulterated beverages or
wood alcohol products, but these also included 10 deaths involving
inhalation or absorption of methanol through the skin.
Bennett et al. (1953) described a case that occurred in Atlanta,
Georgia, USA, in 1951, when within a 5-day period, 323 people consumed
bootlegged whiskey contaminated with 35-40% methanol and 41 of them
died. Kane et al. (1968) reported the poisoning of 18 individuals, of
whom 8 died, when a diluted paint thinner containing approximately 37%
(by volume) methanol was used as an alcoholic beverage in Lexington,
Kentucky, USA.
An epidemic in the State Prison of Southern Michigan in 1979 in
which methanol diluent used in photocopying machines was used as
"home-made" spirits (containing approximately 3% methanol) resulted in
46 definite cases of methanol intoxication and 3 deaths (Swartz et
al., 1981). Methanol poisoning among 23 servicemen in an Army hospital
in Korea who had ingested bootleg sake contaminated with methanol was
reported by Keeney & Mellinkoff (1951). Tonning et al. (1956) reported
acute methanol poisoning in 49 naval personnel who consumed drinks
made from duplicating fluid containing a high concentration of
methanol.
An outbreak of acute methanol intoxication involving 28 young men
in Papua New Guinea in 1977, each of whom consumed an equivalent of
60-600 ml pure methanol, resulted in all becoming hospitalized within
8-36 h due to acute metabolic acidosis, severe visual impairment and
acute pancreatitis. Four died within 72 h after hospitalization. Of 24
who recovered, 16 showed no residual complications, 6 had bilateral
visual impairment and 2 had difficulty in speech as well as visual
impairment (Dethlefs & Naraqi, 1978; Naraqi et al., 1979).
Before 1978, many alcoholics in Sweden were reported to
supplement their intake of alcohol with readily available cleansing
solutions containing up to 80% methanol. Since 1978, the methanol
content of such solutions has been limited to 5%. However, consumption
of these solutions by alcoholics is still widely seen, exposures of
1-2 weeks being associated with blood methanol concentrations ranging
from 1000 to 2000 mg/litre (31-62 mmol/litre) (Heath, 1983).
Although ingestion of methanol historically has been shown to be
the most frequent route of poisoning, percutaneously absorption of
methanol liquids or inhalation of its vapour is as effective as the
oral route in producing methanol acute toxic syndrome in adult and
pediatric poisonings (Buller & Wood, 1904; Wood & Buller, 1904;
Giminez et al., 1968; Kahn & Blum, 1979; Dutkiewicz et al., 1980;
Becker, 1983). Giminez et al. (1968) reported 48 children intoxicated
with percutaneously applied alcohol. Thirty of these patients had
severe respiratory depression, 14 were comatose, 11 had seizures, 7
had anuria or severe oliguria and there were 12 deaths.
About 100 cases of amblyopia (impairment of vision) and death
from inhalation of wood alcohol were reported up to 1912, the majority
occurring from occupational exposure to the fumes (Tyson & Schoenberg,
1914). Toxicity has also been associated with inhalation of methanol
vapour in excess of 400 mg/m3 (300 ppm) (Becker, 1983; Frederick et
al., 1984).
Hazardous inhalation exposures of methanol can occur in the
context of intentional inhalation of volatile preparations such as
carburettor cleaners. Frenia & Schauben (1993) reported seven cases
involving four patients who had inhaled a carburettor cleaner
containing toluene (43.8%), methanol (22.3%), methylene chloride
(20.5%) and propane (12.5%). Measured blood methanol levels ranged
from 504 to 1286 mg/litre. Blood formic acid levels were 120, 193 and
480 µg/ml, respectively, in three patients. Ophthalmic examinations
revealed hyperaemic discs and decreased visual acuity in one patient.
Acute methanol toxicity in humans evolves in a fairly defined
pattern. A toxic exposure results in a transient mild depression of
the CNS, similar to that of ethanol, but to a much lesser degree. The
initial depressant period is followed by an asymptotic latent period,
which occurs most commonly about 8-24 h after ingestion of the alcohol
but may last from several hours to 2 or more days. During the latent
period the patients describe no overt symptoms or signs.
The latent period is followed by a syndrome that consists of an
uncompensated metabolic acidosis with superimposed toxicity to the
visual system. Physical symptoms typically may include headache,
dizziness, nausea and vomiting, followed in more severe cases by
abdominal and muscular pain and difficult periodic breathing (Kussmaul
breathing), which may progress to coma and death, usually from
respiratory distress. Death may occur if patients are not treated for
metabolic acidosis, and blindness may result even if treatment for
metabolic acidosis is performed (Bennett et al., 1953; Röe, 1955; Kane
et al., 1968; Tephly & McMartin, 1984; Tephly, 1991).
The neurotoxic effects of methanol on the visual system can
involve transient abnormalities such as peripapillary oedema, optic
disc hyperaemia, diminished pupillary reactions to light, and central
scotomata. Permanent ocular abnormalities include optic disc pallor,
attenuation of arterioles, sheathing of arterioles, diminished
pupillary reactions to light, diminished visual acuity, central
scotomata, and other nerve fibre bundle defects (Bennett et al., 1953;
Dethlefs & Naraqi, 1978; Kavet & Nauss, 1990). Pallor of the optic
disc is an end-stage sign of irreversible effects of the visual system
and may appear 1 to 2 months after an acute methanol dosage (or
possibly following chronic occupational exposure to methanol vapour)
(Buller & Wood, 1904; Wood & Buller, 1904; Bennett et al., 1953).
Within the general population, the range of the dose levels that
is hazardous to humans and the variable susceptibility to acute
effects are well recognized (Buller & Wood, 1904; Wood & Buller, 1904;
Bennett et al., 1953). As little as 15 ml of 40% methanol resulted in
the death of one individual while others survived following the
consumption of 500 ml of the same solution in the Atlanta, Georgia,
epidemic of 1951. There were large individual differences in the
duration of the latency period. Symptoms of methanol poisoning
appeared within a few hours or were delayed for up to 72 h. The
severity of the disease was not related to the length of the latent
period or the amount of methanol consumed (Bennett et al., 1953). (It
should be noted that in earlier reported poisoning epidemics, large
errors in dose estimates may have been made).
In another example of the range of dose levels of methanol that
are toxic, 120 ml (4 fluid ounces) of Columbian spirits, or 95 g of
methanol (Columbian spirits is basically pure methanol), was lethal in
40% of the poisoning cases. For a 70-kg person, this dose is
equivalent to about 1.4 g methanol/kg body weight (Buller & Wood,
1904). This figure is consistent with currently accepted values for
lethality, and 0.3 to 1 g/kg is considered the range of a minimum
lethal dose for untreated cases of methanol poisoning (Röe, 1955;
Erlanson et al., 1965; Gonda et al., 1978).
It has been suggested that the variability in the reaction to
methanol may have been due to the concomitant ingestion of ethanol
with methanol, which resulted in some patients having a longer latent
period prior to the onset of poisoning (Röe, 1950, 1955). Another
explanation for the variability in susceptibility to methanol
poisoning is the different levels of folate in the diet. Folate-
deficient individuals have a lesser capacity to metabolize formate, so
are more susceptible to accumulation of formate to toxic levels (see
section 8.1.7 for sensitive sub-populations).
In some clinical cases, the blood methanol level is low in the
last phase of the poisoning. In three such cases, blood methanol
concentrations were 0.275, 0.277 and 0.194 g/litre, respectively
(Erlanson et al., 1965). On the assumption that the body in diffusion
equilibrium with the blood represents about 70% of the body weight,
Röe (1982) calculated that 0.19-0.14 g/kg of methanol was present in
the body. However, low blood methanol levels do not indicate a lower
susceptibility to toxicity, i.e., blood methanol levels do not
correlate with patient prognosis (Jacobsen & McMartin, 1986). Patients
that are examined late after methanol ingestion are likely to have low
blood methanol levels, yet high accumulation of formate. Such patients
often have poor prognosis.
Acute methanol poisoning patients with blood levels of methanol
above 500 mg/litre are generally regarded as requiring haemodialysis
(Becker, 1983). The dose of methanol required to achieve this blood
concentration is very small (0.4 ml/kg body weight). This corresponds
to the ingestion of 4 ml (less than a teaspoon of 100% methanol by a
10-kg (1-year old) child and 28 ml (less than 1 fluid ounce) by a
70-kg adult (Litovitz et al., 1988).
A case was reported of a 46-year-old man who, after consuming a
beverage containing methanol, exhibited one of the highest reported
serum methanol levels (4930 mg/litre), well above those at which
ethanol treatment and haemodialysis are recommended (200 mg/litre
and 500 mg/litre, respectively). The lowest serum pH was 7.0
with a hydrogen carbonate level of 8.8 and an anion gap of 42.8.
Additionally, his visual acuity decreased to a complete loss of
vision. The patient was aggressively treated with haemodialysis and
ethanol infusion, regained his vision with a visual acuity of 20/30
bilaterally and suffered no neurological sequelae (Pambies et al.,
1993b).
An additional number of cases are particularly informative
regarding treatment of methanol intoxication and sequelae of
poisoning. A case of methanol intoxication was reported involving a
53-year-old man. Along with blindness and metabolic acidosis, this
resulted in cerebral oedema and subarachnoid haemorrhage followed by a
comatose state and subsequent death (del Carpio-O'Donavan & Glay,
1992).
A 31-year-old male alcoholic who consumed ethanol containing
methanol experienced severe signs and symptoms of poisoning. He
underwent minimal medical treatment consisting of sodium hydrogen
carbonate and peritoneal dialysis and exhibited necrosis and
haemorrhage of the (bilateral) putamen and necrosis of bilateral
subcortical white matter and post-contrast gyral enhancement at the
otherwise normal-looking areas of the cerebral cortex by the 22nd day,
as revealed by computed tomography (Hsieh et al., 1992).
A 31-year-old man entered hospital with a 370 mg/litre serum
methanol level after exhibiting the signs and symptoms of methanol
poisoning (nausea, vomiting, diffuse abdominal pain and blurred tunnel
vision) for 7 days. Following a complete regimen of treatment
consisting of hydrogen carbonate, ethanol and folate combined with a
6-h haemodialysis, which corrected the acidosis and eliminated
methanol (methanol decreased to 100 mg/litre by the second day),
permanent blindness still resulted (Vogt et al., 1993).
A case study of acute methanol poisoning in a 27-year-old man
with a previous pattern of drinking was reported by King (1992).
Following a comprehensive treatment regimen consisting of
administration of alkali, fluids and ethanol, intubation and
haemodialysis, this patient exhibited significant neurological and
physical impairment, including trauma to the vocal cords and
hypophonic voice and urinary incontinence (of central origin), along
with cognitive defects. However upon discharge his vision was normal
with no atrophy of the optic nerve.
A case of a severe methanol poisoning in a 33-year-old man with a
history of alcoholism was reported by Burgess (1992). The individual
required 21 h of dialysis to bring the serum methanol levels down to a
non-toxic level. A haemodialysis treatment usually lasts approximately
4 h but this may not be sufficient in severe poisoning. Prolonged
haemodialysis treatment should be considered in cases of severe
poisoning and also possibly for patients with compromised renal
function.
Extensive white and grey matter brain damage was seen in an
alcoholic 37-year-old man who consumed 1900 ml of windshield washer
fluid containing methanol. Both CT scan and MR imaging revealed
diffuse white matter oedema and damage throughout frontal and parietal
lobes. Bilateral changes in the basal ganglia and necrosis and
haemorrhage of putamen were also noted (Glazer & Dross, 1993).
Autopsies from victims of lethal methanol poisonings revealed
gross pathology in the visceral organs, the brain, lung, liver,
kidney and the CNS, all of which involved a variety of oedematous,
haemorrhagic and degenerative changes (Keeney & Mellinkoff, 1951;
Bennett et al., 1953; Tonning et al., 1956; Kaplan, 1962; Erlanson et
al., 1965; McLean et al., 1980; Wu Chen, 1985; Suit & Estes, 1990).
A fatal case involving a 41-year-old man who had ingested a large
quantity of methanol disclosed a broad distribution of methanol in
postmortem tissues and fluids. The highest content of methanol was
found in the kidney (5.13 g/kg) followed by the liver (4.18 g/kg),
vitreous humour (3.9 g/litre), heart (3.45 g/kg), urine
(3.43 g/litre), pericardial fluid (3.29 g/litre), blood (2.84 g/litre)
and stomach contents (2.21 g/litre) (Pla et al., 1991).
Methanol toxicity can cause brain oedema, necrosis, brain atrophy
and cerebral haemorrhage. Putaminal necrosis and haemorrhage result
from the direct toxic effects of the methanol metabolites (e.g.,
formate) and metabolic acidosis in the basal ganglia. The typical
appearance of bilateral putaminal necrosis has been described as
characteristic of methanol toxicity (Gonda et al., 1978).
Optic neuropathy and putaminal necrosis are the two main
complications of methanol poisoning generally occurring in combination
after severe poisoning of either suicidal or accidental origin (Sharpe
et al., 1982).
A case study of a woman who drank a substantial amount of
methylated spirits, which resulted in optic neuropathy and putaminal
necrosis, has been reported (Pelletier et al., 1992). The woman
exhibited tremor and rigidity, hypokinesia, altered speech and loss of
superficial and proprioceptive sensation of the lower extremities with
hyperpathia. Signs of moderate bilateral sensory neuropathy and
extrapyramidal damage persisted for 2 months as did total blindness
due to optic atrophy. Repeat CT and MRI examinations revealed the
damage to be a core lesion of the putamen with residual bilateral
putaminal hypodensity suggestive of an ischaemic and necrotic process
possibly including disruption of the blood-brain barrier.
Postmortem analysis of methanol concentrations in body fluids and
tissues reported in fatal human cases of methanol poisoning has
revealed higher concentrations of methanol in cerebrospinal fluid
(CSF), vitreous humour and bile than in blood (Bennett et al., 1953;
Wu Chen et al., 1985). In tissues, the highest concentrations were
found in brain, kidney, lung and spleen, and there were lower
concentrations in skeletal muscle, pancreas, liver and heart (Wu Chen
et al., 1985).
Postmortem signs of damage to the basal ganglia in the brain,
specifically the putamen, have been reported in several cases
(Erlanson et al., 1965; Aquilonius et al., 1978; Suit & Estes, 1990).
A number of human studies have shown that survivors of severe methanol
poisoning may suffer residual disorders as a permanent complication
(Erlanson et al., 1965; Guggenheim et al., 1971; Aquilonius et al.,
1978; McLean et al., 1980; Ley & Gali, 1983). Ley & Gali (1983)
described a case of Parkinsonian syndrome after methanol intoxication.
Co-ingestion of methanol with other solvents, e.g., methyl ethyl
ketone (MEK) (found in multiple ink cleaning products) has resulted in
a hyperosmolar coma without anion gap metabolic acidosis in one
reported case of poisoning. MEK was believed to have inhibited
methanol metabolism contributing to the low serum formate (1.3
mmol/litre) and normal anion gap despite a blood methanol level of
67 mmol/litre (Price et al., 1994).
8.1.2 Clinical features of acute poisonings
The time course of clinical effects due to acute methanol
poisoning is heavily dose-dependent. Blood methanol concentrations of
> 500 mg/litre are associated with severe acute clinical signs of
toxicity, although formate concentrations may give a better indication
of potential toxicity (National Poisons Information Service, 1993).
Thirty minutes to 2 h after ingestion of methanol, clinical
effects resemble those of mild ethanol inebriation, and drowsiness,
confusion and irritability are often noted. After a latent period,
which can range from a few hours to 30 h (but may appear as early as
1 h or as late as 72 h), the patient shows mild CNS depression
followed by abdominal pain, nausea, vomiting, hypernoea, gradually
failing vision, progressive encephalopathy, severe metabolic acidosis
and hypokalaemia; coma and death may ensue. Patients may complain of
blurred or "snowfield" vision with whiteness, spots or mistiness
within the visual field. Survivors may have permanent blindness or
various neurological sequelae. Mortality and morbidity may be more
related to the time between ingestion and therapy rather than to the
initial methanol levels, thus emphasizing the need for rapid treatment
(Mahieu et al., 1989; National Poisons Information Service, 1993;
Pambies et al., 1993a).
Metabolic acidosis associated with high anion and osmolal gaps is
considered an important laboratory indicator of methanol poisoning
(Kruse, 1992). The difference between measured and calculated
osmolality or osmolal gap permits a rough estimation of alcohol
concentrations (Pappas et al., 1985) so that specific therapy is often
initiated before results of quantitative methanol determinations are
available.
The determination of osmolal and anion gaps are readily available
techniques in the initial handling of poisoning with unknown agents
and of patients with a metabolic acidosis of unknown origin. A
combined increase in both anion and osmolal gaps has been shown to be
a sensitive marker of either ethylene glycol or methanol poisoning
(Jacobsen & McMartin, 1986). Reported earlier reference values for
osmolal gap and anion gap are -1 (+ 6) mosm/kg H2O and 16 (+ 2)
mmol/litre, respectively (Jacobsen et al., 1982b). However, Aabakken
et al. (1994) determined osmolal and anion gaps in populations that
were consecutively admitted to a hospital emergency department and
suggested that the present reference values for anion and osmolal gaps
may be too narrow. They further suggested that the values for the
osmolal gap should be 5 + 15 mosm/kg H2O (-10 to + 20 mosm/kg H2O)
and for the anion gap should be 13 + 9 mmol/litre (4-20 mmol/litre).
In their previous reports of methanol poisonings, all patients
exceeded these ranges (Jacobsen et al., 1982).
Demedts et al. (1994) hypothesized that excessive serum
osmolality gaps that are not predictive of methanol levels as
frequently seen in acute poisonings may be attributed to methodology
used to measure methanol (analysing samples using head-space GC were
compared to results found with gas-chromatography using split-mode
injections). Although the determination of increased anion gap is
suggestive of methanol poisoning, definitive evidence would be
increased blood or serum formate concentrations.
Characteristic clinical and laboratory findings in methanol
poisoning are summarized as follows:
* Physical findings
a) Kussmaul respiration (difficult, periodic breathing)
b) faint odour of methanol on breath
c) visual disturbances
d) nausea, vomiting, abdominal pain
e) altered sensation
* Laboratory findings
a) elevated anion gap
b) metabolic acidosis
c) elevated osmol gap
d) positive serum methanol and/or serum formate assay
In treating methanol poisoning a 3-step procedure is common:
1) administration of hydrogen carbonate to combat metabolic acidosis;
2) administration of ethanol to compete as a substrate for alcohol
dehydrogenase, and 3) haemodialysis to remove methanol from the blood
(Erlanson et al., 1965; Gonda et al., 1978; McCoy et al., 1979; Lins
et al., 1980; Jacobsen et al., 1982a,b; Pappas & Silverman, 1982;
Becker, 1983; Jacobsen & McMartin, 1986; Kruse, 1992; Pambies et al.,
1993a,b). Current recommendations are that ethanol treatment be
conducted for patients with blood methanol levels of 200 mg/litre or
more, while haemodialysis be used above 500 mg/litre (Jacobsen &
McMartin, 1986).
The rationale for the administration of ethanol (Röe, 1950;
Keyvan-Larijarni & Tannenbaum, 1974; McCoy et al., 1979; Becker, 1983)
is that alcohol dehydrogenase, the enzyme responsible for converting
methanol to formaldehyde and formic acid, is also involved in the
metabolism of ethanol to acetaldehyde and acetate. The conversion of
methanol to its toxic by-products is slowed in the presence of ethanol
due to competition for the enzyme.
4-Methyl pyrazole (4-MP) is a more specific inhibitor of alcohol
dehydrogenase, less toxic than pyrazole and has been shown to
dramatically inhibit production of formic acid from methanol in
experimental animals (Blomstrand et al., 1979; McMartin et al.,
1980b). Monkeys given usually lethal doses of methanol survived when
treated with 4-MP following methanol administration (McMartin et al.,
1980b). In humans the slower elimination rate and lesser degree of
toxicity of 4-MP suggested that it might be preferable to ethanol in
the treatment of methanol poisoning (Jacobsen et al., 1990). 4-MP is
currently undergoing clinical trials for treatment of methanol
poisoning.
Haemodialysis effectively removes methanol and formate from the
circulation (Erlanson et al., 1965; Gonda et al., 1978; McCoy et al.,
1979). If haemodialysis is not available, peritoneal dialysis has been
used with some success in treating acute methanol intoxication
(Keyvan-Larijarnc & Tannenberg, 1974). Discussion of the treatment of
methanol poisoning can be found in the IPCS Poisons Information
Monograph (PIM) No. 335 (IPCS, 1991).
8.1.3 Repeated or chronic exposure
In comparison to acute toxicity, reports of effects from repeated
or chronic methanol exposures have been only infrequently reported.
Information based on a limited number of case reports and even fewer
epidemiological studies (generally containing unknown levels and/or
durations of methanol exposure) suggests that extended exposure to
methanol may cause effects qualitatively similar to those observed
from relatively high levels of acute exposure, including in some cases
CNS and visual disorders (Buller & Wood, 1904; Wood & Buller, 1904;
Greenberg et al., 1938; Bennett et al., 1953; Kingsley & Hirsch, 1955;
Frederick et al., 1984).
Greenberg et al. (1938) studied 19 workers employed in the
production of "fused collars", where solutions of acetone-methanol
(3:1) were used to impregnate collars which were then steam-pressed.
Methanol concentrations in the work room were 29-33 mg methanol/m3
and 96-108 mg acetone/m3. The shortest period of employment in this
occupation was 9 months and the longest was 2 years. No CNS symptoms
or visual anomalies were observed.
Frederick et al. (1984) reported on teacher aides who worked at
or near spirit duplicators that used a 99% methanol duplicator fluid.
The exposures ranged from 1 h/day for 1 day/week to 8 h/day for 5
days/week and had occurred for 3 years. Since the introduction of the
equipment, the aides began to experience headaches, dizziness and eye
irritation, blurred vision and nausea/upset stomach while working near
the machines. Fifteen-minute breathing zone samples near 21 operating
machines contained between 475 and 4000 mg/m3 of methanol vapour.
Fifteen of these samples exceeded the NIOSH recommended 15-min
standard of 1050 mg/m3 (800 ppm). The aides were also exposed while
collating and stapling papers impregnated with the fluid up to 3 h
earlier and these exposures ranged from 235-1140 mg/m3 . The results
suggested that chronic effects may occur when methanol concentrations
exceed the threshold limit value (TLV) of 260 mg/m3 (200 ppm). The
effects reported in the study of Frederick et al. (1984) were similar
in nature but appeared less severe than those reported from acute
poisoning by methanol (Buller & Wood, 1904; Wood & Buller, 1904;
Bennett et al., 1953).
Kingsley & Hirsch (1955) reported frequent and persistent
headaches, but no visual effects or other permanent sequelae, in
clerical workers located close to spirit duplicating equipment that
used methanol-based duplicating fluid. Methanol concentrations were
reported to be as high as 490 mg/m3 in the air surrounding the
duplicating equipment after 60 min of operation and approximately
130 mg/m3 about 3 m away from the device. The methanol concentration
around the duplicating equipment always exceeded 260 mg/m3. No
information was provided concerning the number of employees exposed or
affected, nor on the actual duration of methanol exposure.
NIOSH (1981) reported that 45% of "spirit" duplicating machine
operators at the University of Washington experienced some symptoms
(blurred vision, headache, nausea, dizziness and eye irritation),
consistent with the toxic effects of methanol. Airborne methanol
concentrations of 1330 mg/m3 were measured in the vicinity of the
duplicators when windows and doors were open. No information on the
actual length of duration of methanol exposure among the employees
engaged in the duplicating machine operations were provided.
A number of other studies have measured methanol and formate in
the blood and urine of workers exposed during an 8-h day to between
100 and 200 mg/m3 of methanol vapour (Baumann & Angerer, 1979;
Heinrich & Angerer, 1982. Although these studies were predicated on
issues of occupational health related to methanol exposure, no health
effects were provided nor did the investigators imply that the workers
studied had suffered health effects.
Kawai et al. (1991b), utilizing methanol in urine as a biological
indicator of occupational exposure, compared subjective complaints and
major clinical findings among 33 methanol-exposed workers over several
8-h workshifts. Urine levels of methanol in controls were on average
1.9 ± 0.8 mg/litre (n = 91), and in 14 exposed workers pre-shift
concentrations were significantly elevated compared to controls. At
the end of the shift the urine concentrations were generally above
100 mg/litre in 8 men with a mean exposure level of 1690 mg/m3 and
30-100 mg/litre in 6 men with a mean exposure level of 550 mg/m3. The
highest exposures (breathing zone, 8-h/samples) were 4000-7000 mg/m3
and corresponding urine levels 300-500 mg/litre. The leading
subjective complaints included: dimmed vision and nasal irritation
during work, and headache, dimmed vision, forgetfulness and increased
sensitivity of the skin in the extremities when off-work. The authors
attributed the dimmed vision to the fog created by methanol vapours
and high humidity in air. No visual problems were noted when windows
were kept open and fresh air was allowed to flow in. It was also noted
that there were no complaints of photophobia (and thus perhaps no
major corneal involvement). Fundus photography revealed that the optic
discs were normal and thus the symptom of dimmed vision was not
recognized as a sign of impending retinal involvement. In three
workers with methanol exposures of 1250-2130 mg/m3, 1385-2075 mg/m3
and 155-4685 mg/m3 (953-1626 ppm, 1058-1585 ppm and 119-3577 ppm) the
reaction of pupils to light was slow in two subjects, and a third
subject had slight mydriatic pupils. The duration of service of the
workers ranged from 0.3 to 7.8 years. The exposures were high and the
methods for measurement of visual toxicity were relatively crude, but
the data did not indicate that occupational exposure to such
concentrations caused permanent damage.
The effects of methanol vapour (249 mg/m3; SD + 7 mg/m3) for
75 min on neurobehavioural measures were studied in 12 healthy young
men. The exposure produced significant increases (approximately 3
fold) in blood and urine methanol levels but no changes in plasma
formate level. Although most of the neurobehavioural end-points were
unaffected by exposure to methanol, statistically significant effects
and trends were found for a cluster of variables, including the
latency of the p-200 component of event-related potentials,
performance on the Sternberg memory task and subjective measures of
fatigue and concentration. However, the effects were small and did not
exceed the normal range (Cook et al., 1991).
8.1.4 Reproductive and developmental effects
No studies have been reported in the peer-reviewed literature on
the reproductive and developmental effects of methanol in humans.
8.1.5 Chromosomal and mutagenic effects
No studies have been reported in the peer-reviewed literature on
chromosomal or mutagenic effects of methanol in humans.
8.1.6 Carcinogenic effects
No studies have been reported in the literature on the
carcinogenicity of methanol in humans.
8.1.7 Sensitive sub-populations
Folate-deficient individuals might be at greater risk from
inhalation of low concentrations of methanol, compared to normal
individuals. Human populations that are potentially at high risk of
folate deficiency include pregnant women, the elderly, individuals
with poor-quality diets, alcoholics and individuals on certain
medications or with certain diseases (API, 1993).
It has been suggested that the metabolic acidosis due to methanol
might be exacerbated in individuals with diabetes since it is well
known that these patients suffer from diabetic ketoacidosis (Posner,
1975). However, there are no clinical or experimental data on any
interaction between methanol acidosis and diabetic ketoacidosis.
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1 Aquatic organisms
9.1.1 Microorganisms
The toxicity of methanol to each of three bacterial groups, i.e.,
aerobic heterotrophic, Nitrosomonas and methanogens (key agents in the
natural recycling of organic material in the environment and in
wastewater treatment systems), was described by Blum & Speece (1991).
The following IC50 values (mg/litre) (the concentration that
inhibited the culture by 50%) compared to the uninhibited controls
were reported: Nitrosomonas (after 24-h exposure), 880 mg/litre;
methanogens (after 48-h exposure), 22 000 mg/litre; and aerobic
heterotrophs (after 15-h exposure), 20 000 mg/litre. Methanol was
found to be completely inhibitory to ammonia oxidation by
Nitrosomonas bacteria at a concentration of 5 × 10-3 M (about
160 mg/litre) (Hooper & Terry, 1973).
A 15-min EC50 of 14 700 mg/litre for the luminescent marine
bacterium Photobacterium phosphoreum and a 4-h LC50 value of 1.0%
by volume (7690 mg/litre) have been reported (Schiewe et al., 1985).
Calleja et al. (1994) found the EC50 for the marine bacterium
Photobacterium phosphoreum in the Microtox(R) test to be
29 348 mg/litre. Rajini et al. (1989) reported a 10-min LC50 of 6%
(44 860 mg/litre) for the ciliate protozoan Paramecium caudatum.
Toxicity threshold values for methanol in the cell multiplication
inhibition test of 6600 mg/litre for the bacterium Pseudomonas
putida and > 10 000 mg/litre for the protozoa Entosiphonsulcatum
were reported by Bringmann & Kühn (1980).
An experimental EC50 value (the concentration that reduced the
maximum observed biodegradation rate by 50%) for methanol of
2.8 mol/litre (89.7 g/litre) was obtained in a system employing an
enriched mixed microbial culture derived from domestic waste water in
the USA (Vaishnav & Lopas, 1985).
9.1.2 Algae
Stratton (1987) determined the following EC50 values:
Anabaena cylindrica: 2.57% (20 300 mg/litre)
Anabaena inaequalis: 2.68% (21 179 mg/litre)
Anaebaena sp.: 3.12% (24 650 mg/litre)
Anaebaena variabilis: 3.13% (24 730 mg/litre)
Nostoc sp.: 5.48% (43 290 mg/litre)
For the green alga Chlorella pyrenoidosa an EC50 value of
28 440 mg/litre was found (Stratton & Smith, 1988). Bringman & Kühn
(1978), employing a cell multiplication test, reported a toxicity
thresholds of 8000 mg/litre for the green alga Scenedesmus
quadricauda and 530 mg/litre for the cyanobacterium (blue green
alga) Microcystis aeruginosa.
9.1.3 Aquatic invertebrates
The toxicity of methanol, as reported for a broad spectrum of
aquatic invertebrates, is summarized in Table 6. EC50 values for the
water flea (Daphnia magna) range from 13 240 to 24 500 mg/litre.
Helmstetter et al. (1996) exposed the mussel, Mytilus edulis, to
methanol concentrations of 1, 2, 3, 5 and 10% (v/v) for 96 h. All the
mussels in both the 5 and 10% exposure groups died within 13.5 h.
Sublethal narcotic effects such as slow movement and sporadic filter
feeding were reported in mussels exposed to 2 and 3%. Mussels exposed
to 1% methanol exhibited no adverse effects during the 96-h exposure
period.
9.1.4 Fish
The acute toxicity to fish is listed in Table 7. LC50 values
reported for freshwater fish species range from 10 880 to
29 700 mg/litre.
The physiological changes in the carp (Cyprinus carpio)
affected by a sub-lethal methanol concentration of 1 ml/litre
(790 mg/litre) included a significant increase in blood cortisol
levels after 6 h of exposure, but not after 24 or 72 h, significant
decreases in blood protein and cholesterol levels after 72 h of
exposure, and reduced concentration of glycogen in the liver after
72 h. Methanol did not produce significant changes in blood glucose
levels after any duration of exposure (Gluth & Hanke, 1985).
The effect of methanol on the fertilization of chum salmon
(Oncorhynchus keta) ova was examined at methanol exposure levels of
0.001% to 10% by volume (7.9 to 79 000 mg/litre) (Craig et al., 1977).
Both gametes (sperm and unfertilized ova) and fertilized eggs were
exposed to methanol for brief periods. Exposures up to and including
1% methanol did not significantly affect fertilization, survival to
hatching, hatching time, alevin size at hatch or physical deformities
among alevins, although a methanol concentration of 10% was lethal in
most cases (Craig et al., 1977).
Cuéllar et al. (1995) determined the effect of methanol on the
embryonic development of the medaka fish (Oryzias latipes). The eggs
were exposed to methanol in both Petri dishes and vials. No effects on
embryonic development were reported at a methanol concentration of
0.5%.
Table 6. Acute toxicity of methanol to aquatic invertebrates
Organism Size/age Stat/ Temp Hardness pH Parameterc Concentration Reference
flowa (°C) (mg/litre)b (mg/litre)d
Water flea <24 h stat 20 (1) 7.8-8.2 48-h EC50e >10 000 n Kuhn et al. (1989)
(Daphnia magna)
<24 h stat 20 (1) 7.8-8.2 48-h EC0e >10 000 n Kuhn et al. (1989)
<24 h stat 20 (2) 7.8-8.2 24-h EC50 >10 000 n Bringmann & Kuhn
(1982)
<24 h stat 20 (2) 7.8-8.2 24-h EC100 >10 000 n Bringmann & Kuhn
(1982)
<24 h stat 20 154.5 7.0-8.2 24-h EC50 24 500 n Bringmann & Kuhn
(1982)
48-h LC50 13 240 n Vaishnav &
Korthals (1990)
24-h EC50 21 402 n Calleja et al.
(1994)
Water flea <24 h stat 22 23±2 18-h-LC50 19 500 n Bowman et al.
(Daphnia pulex) (1981)
Water flea <24 h stat 20±2 250 7.8±0.2 24-h EC50 23 500 n Rossini & Ronco
(Daphnia obtusa) <24 h stat 20±2 250 7.8±0.2 48-h EC50 22 200 n (1996)
Brown shrimp adult stat 15 seawater 48-h LC50 1975 n Portmann & Wilson
(Crangon crangon) (1971)
adult stat+ 15 seawater 96-h LC50 1340 n Portmann & Wilson
(1971)
stat 24.5 seawater 24-h LC50 10 000 n Price et al. (1974)
Brine shrimp 24 h stat 25 seawater 24-h LC50 1578.84 n Barahona-Gomariz
(Artemia salina) et al. (1994)
48 h stat 25 seawater 24-h LC50 1101.46 n Barahona-Gomariz
et al. (1994)
Table 6. Continued
Organism Size/age Stat/ Temp Hardness pH Parameterc Concentration Reference
flowa (°C) (mg/litre)b (mg/litre)d
Brine shrimp 72 h stat 25 seawater 24-h LC50 900.73 n Barahona-Gomariz
(Artemia salina) et al. (1994)
seawater 24-h LC50 43 574 n Calleja et al.
(1994)
Glass shrimp juvenile stat 23±2 18 h-LC50 21 900 n Bowman et al.
(Palaemonetes (1981)
kadiakensis)
Streptocephalus 24-h LC50 32 681 n Calleja et al.
proboscideus (1994)
Mussel 5-7 cm flow 15±0.5 seawater 96-h LC50 15 900 m Helmstetter et al.
(Mytilus edulis) (1996)
Cockle adult stat 15 seawater 48-h LC50 7900 n Portmann & Wilson
(Cardium edule) (1971)
adult stat+ 15 seawater 96-h LC50 2610- Portmann & Wilson
7900 n (1971)
Harpacticoid, adult stat 21±1 7s 7.9 96-h LC50 12 000 n Bengtsson et al.
copepod (1984)
(Nitocra spinipes)
Scud juvenile stat 23±2 18-h LC50 19 350 n Bowman et al.
(Hyalella azteca) (1981)
Table 6. Continued
Organism Size/age Stat/ Temp Hardness pH Parameterc Concentration Reference
flowa (°C) (mg/litre)b (mg/litre)d
Rotifer 24-h LC50 35 884 Calleja et al.
(Brachionus (1994)
calyciflorus)
a stat = static conditions (water unchanged for duration of test); stat+ = semi-static conditions (test solutions
renewed every 24 h); flow = flow through conditions (concentration of toxicant continuously maintained);
s = salinity, expressed as %
b hardness expressed as mg CaCO3 litre, unless stated otherwise; (1)- total hardness = 2.4 mmol/litre;
(2)- total hardness = 2.5 mmol/litre
c All EC50 values refer to immobilization
d n = nominal concentration; m = measured concentration
e same as 24 h EC50 and EC0 values
Table 7. Acute toxicity of methanol to fish
Organism Size/age Stat/ Temp Hardness pH Parameter Concentration Reference
flow (°C) (mg/litre)b (mg/litre)c
Rainbow trout (juv) 0.813 g flow 12.7±1 46.4 7.0-8.0 24-h EC50d 13 200 m Poirier et al.
(Oncorhynchus (1986)
mykiss)
(juv) 0.813 g flow 12.7±1 46.4 7.0-8.0 96-h EC50e 13 000 m Poirier et al.
(1986)
(juv) 0.813 g flow 12.7±1 46.4 7.0-8.0 24-h LC50 20 300 m Poirier et al.
(1986)
(juv) 0.813 g flow 12.7±1 46.4 7.0-8.0 96-h LC50d 20 100 m Poirier et al.
(1986)
0.8 g stat 12 44 7.4 96-h LC50 19 000 n Mayer &
Ellersieck (1986)
(fingerlings) flow 12 96-h LC50d 20 100 m US EPA (1983)
1-6 g
Fathead minnow (28-32 d) flow 23.3±1.7 46.4 7.0-8.0 24-h EC50d 29 700 m Poirier et al.
(Pimephales 0.126 g (1986)
promelas)
(28-32 d) flow 23.3±1.7 46.4 7.0-8.0 96-h EC50e 28 900 m Poirier et al.
0.126 g (1986)
(28-32 d) flow 23.3±1.7 46.4 7.0-8.0 24-h LC50d 29 700 m Poirier et al.
0.126 g (1986)
(28-32 d) flow 23.3±1.7 46.4 7.0-8.0 96-h LC50 29 400 m Poirier et al.
0.126 g (1986)
(30 d) 0.12 g flow 24-26 45.5 7.5 96-h LC50 28 100 m Veith et al.
(1983)
Table 7. Continued
Organism Size/age Stat/ Temp Hardness pH Parameter Concentration Reference
flow (°C) (mg/litre)b (mg/litre)c
Bluegill sunfish (juv) 3.07 g flow 19.8±2.3 46.4 7.0-8.0 24-h EC50e 16 100 m Poirier et al.
(Leponimis (1986)
macrochirus)
(juv) 3.07 g flow 19.8±2.3 46.4 7.0-8.0 24-h EC50e 16 100 m Poirier et al.
(1986)
(juv) 3.07 g flow 19.8±2.3 46.4 7.0-8.0 48-h EC50e 16 000 m Poirier et al.
(1986)
(juv) 3.07 g flow 19.8±2.3 46.4 7.0-8.0 96-h EC50e 12 700 m Poirier et al.
(1986)
(juv) 3.07 g flow 19.8±2.3 46.4 7.0-8.0 24-h LC50d 19 100 m Poirier et al.
(1986)
(juv) 3.07 g flow 19.8±2.3 46.4 7.0-8.0 96-h LC50 15 400 m Poirier et al.
(1986)
1.5 g flow 25 24-h LC50d 19 230 m US EPA (1983)
1.5 g flow 25 96-h LC50 15 500 m US EPA (1983)
Guppy 2-3 months stat+ 21-23 25 7-day LC50 10 860 m Konemann (1981)g
(Poecilia Hermens &
reticulata) Leeuwangh (1982)
Golden orfe juv stat 19-21 (1) 7.0-8.0 48-h LC50 >10 000f m Juhnke &
(Leuciscus idus Lüdemann
melanotus) (1978)
juv stat 19-21 (1) 7.0-8.0 48-h LC0 7900f m Juhnke &
Lüdemann
(1978)
juv stat 19-21 (1) 7.0-8.0 48-h LC100 >10 000f m Juhnke &
Lüdemann
(1978)
Table 7. Continued
Organism Size/age Stat/ Temp Hardness pH Parameter Concentration Reference
flow (°C) (mg/litre)b (mg/litre)c
Bleak 8 cm stat 10 7s 7.9 96-h LC50 28 000 n Bengtsson
(Alburnus et al. (1984)
alburnus)
Armed bullhead adult stat+ 15 seawater 96-h LC50d 7900- Portmann &
(Agonus 26 070 n Wilson (1971)
cataphractus)
a stat = static conditions (water unchanged for duration of test)
stat+ = semi-static conditions (test solutions renewed every 24 hours)
flow = flow through conditions (concentration of toxicant continuously maintained)
s = salinity, expressed as %
b hardness expressed as mg/CaCO3/litre, unless otherwise stated; (1)- total hardness = 2.7 mmol/litre;
c n = nominal concentration
m = measured concentration
d same as 48-h LC50 or EC50 values
e effects on equilibrium, behaviour and coloration
f two laboratories following the same test protocol, same result from each laboratory
g consulted for experimental method only
9.2 Terrestrial organisms
9.2.1 Plants
Hemming et al. (1995) determined the effect of methanol on the
respiration of pepper (Capsicum annuum), tomato (Lycopersicon
esulentum) and petunia (Petunia hybrida). Whole plants were
exposed to either methanol vapour or methanol solution. The general
response to methanol was the same for the three species, with a
respiratory rate increase of up to 50% at the lower methanol
concentrations tested. The response was the same for exposure to
methanol vapour or solution. Exposure of a single leaf resulted in a
systemic response throughout the whole plant within a few hours. The
response lasted for several weeks. Decreased metabolic rates and
waterlogged appearance were reported in plants following a brief
exposure of a leaf to methanol concentrations > 30%. Root tissue
was reported to be more sensitive; a decrease in metabolic rate was
reported following brief exposures to > 10% methanol.
10. EVALUATION OF EFFECTS ON HUMAN HEALTH AND THE ENVIRONMENT
10.1 Evaluation of human health risks
10.1.1 Exposure
Methanol occurs naturally in humans, animals and plants. Humans
are routinely exposed to low levels of methanol from both the diet
(fruits, vegetables, fruit juices and foods containing the synthetic
sweetener aspartame) and metabolic processes. Human exposure to large
acutely toxic amounts of methanol via the oral route has principally
been noted in a relatively small number of individuals, generally
resulting through accidental or intentional consumption of methanol in
illicit or contaminated alcoholic beverages.
Methanol is produced in large amounts in many countries and is
extensively used as an industrial solvent, a chemical intermediate
(principally in the production of methyl tertiary butyl ether (MTBE),
formaldehyde, acetic acid and glycol ethers), as a denaturant of
ethanol and in a variety of consumer products.
The most important route of occupational exposure to methanol is
inhalation. Sources of occupational exposure include the dissipative
emissions of methanol primarily occurring from miscellaneous solvent
usage, methanol production, end-product manufacturing and bulk storage
and handling.
An increased number of people could be potentially exposed to
environmental methanol as a result of the projected expanded use of
methanol in methanol-blended gasolines. Exposures would principally
arise from exhaust, evaporative emissions and normal heating of the
engine. Simulation models based on 100% of all vehicles powered by
methanol-based fuels predict concentrations of methanol in urban
streets, expressways, railroad tunnels or parking garages ranging from
a low of 1 mg/m3 (0.77 ppm) to a high of 60 mg/m3 (46 ppm).
Predicted concentrations during refuelling of vehicles range from 30
to 50 mg/m3 (23-38.5 ppm). For comparison and reference purposes, a
current occupational exposure limit for methanol in many countries is
260 mg/m3 (200 ppm) for an 8-h working day.
There are limited data on human dermal exposure to methanol but
the potential expanded use of methanol in automotive fuels would
increase the potential for dermal exposure in a large number of
people.
10.1.2 Human health effects
Methanol is rapidly absorbed by inhalation, ingestion and dermal
exposure and is rapidly distributed to tissues according to the
distribution of body water. The dose and blood concentrations of
methanol and its metabolite formate are among the major determinants
of the resultant toxicity in humans.
The acute and short-term toxicity of methanol varies greatly
between different species, toxicity being highest in species with a
relatively poor ability to metabolize formate. Methanol has been
studied most intensively in acute high-dose oral exposures in
laboratory animals and as case reports of ingestion in humans. In
general, humans and primates respond to such exposures with transient
central nervous system (CNS) depression (intoxication), followed by an
asymptomatic latent period culminating in metabolic acidosis and
severe ocular toxicity (blindness).
Non-primate animals such as rodents do not ordinarily exhibit
metabolic acidosis or blindness on exposure to methanol although they
exhibit the general narcotic effects noted in non-human primates and
humans. The clearance of formate from the blood of exposed primates is
at least 50% slower than in rodents. Formate, an endogenous biological
substrate, is detoxified by a multi-step pathway to CO2 via a
tetrahydrofolate (THF)-dependent pathway. Species such as rodents with
high hepatic THF levels are less sensitive to the toxic effects of
methanol than species with low hepatic THF levels such as humans and
non-human primates. The faster rate of formate removal means that
rodents do not accumulate formate above endogenous levels and hence
are not susceptible to methanol-induced metabolic acidosis or ocular
toxicity.
The primary enzymatic pathway that catalyses methanol metabolism
in humans and non-human primates is alcohol dehydrogenase, while in
the rat it is the catalase-peroxidase system. Available data suggest
that methanol elimination from the systemic circulation is capacity-
limited in both rats and in humans.
Studies in humans and non-human primates exposed to
concentrations of methanol ranging from 13 to 2601 mg/m3 (10 to
2001 ppm) and the widely used occupational exposure limit of
260 mg/mg3 (200 ppm) suggest that exposure to methanol vapour during
the normal use of methanol fuel does not pose an unacceptable risk to
healthy adults. General population exposures to methanol through air
(although infrequently measured) are over 1000 times lower than
occupational limits.
Along with methanol, formate is present in blood at low
endogenous concentrations, being found naturally in some foods and
also produced as a by-product of several metabolic pathways, including
histidine and tryptophan degradation. Background levels of formate in
humans have been shown to range from 3 to 19 mg/litre (0.07-0.4 mM).
Human susceptibility to the acute effects of methanol
intoxication are extremely variable. On the basis of available human
case reports, the minimum lethal dose in the absence of medical
treatment is in the range of 0.3 to 1 g/kg. The major determinants of
human susceptibility to methanol toxicity appear to be the concurrent
ingestion of ethanol, which slows the entrance of methanol into the
metabolic pathway, and the hepatic status of THF, which governs the
rate of formate detoxification.
Some human populations are at increased risk of folate
deficiency. These include pregnant women, the elderly, individuals
with poor-quality diets, alcoholics, and individuals on certain
medications or with certain diseases.
Much fewer data are available on the health effects in humans or
laboratory animals associated with chronic or repeated exposure to
methanol. In the absence of details of exposure (e.g., duration,
concentrations), the effects of prolonged exposure are considered
qualitatively very similar to those reported for acute cases, ranging
from nausea and dizziness to blurred vision and temporary or permanent
blindness. Chronic exposure to methanol vapour concentrations of
480-4000 mg/m3 (365-3080 ppm) has resulted in headache, dizziness,
nausea and blurred vision.
There are no reports of carcinogenic, genotoxic, reproductive or
developmental effects in humans due to methanol exposures.
10.1.3 Approaches to assessment of risk
The assessment of risk from chronic exposure requires dose-
response information in the form of quantitative data from animal
studies using appropriate test species and, where available, relevant
human epidemiological and clinical data. In the case of methanol, the
assessment of the risks of exposure is confounded by the fact that
both methanol and its toxic metabolite, formate, are endogenous
metabolic intermediates in all species including humans. Therefore, it
must be assumed that there are levels of methanol exposure that do not
represent significant risk. Determining the hazards associated with
methanol exposure is additionally complicated by the fact that there
are no adequate or comprehensive data from animal tests for chronic
toxicity. Because of species differences in methanol metabolism, data
available from normal rats appear to be inappropriate for use in
characterizing the adverse effects of methanol in humans.
Investigation of folate-deficient rodent models may provide valuable
mechanistic, pharmacokinetic and toxicological effect information on
methanol, particularly with respect to acute exposures. However, the
nature of this animal model is such that it may have inherent
weaknesses for the toxicological assessment of long-term exposure
because of the adverse effects of folate deficiency itself and the
background nutritional status of these rats in chronic studies.
Similarities in the metabolism of methanol within primates suggest the
use of non-human primates may be more appropriate for determining the
nature of the hazards of methanol for humans, but adequate findings
for chronic exposure are also lacking. Human methanol exposure data
are extensive but primarily focus on acute exposure and clinical
effects associated with poisoning. Although this information from
humans does highlight the wide individual variability in the toxic
response to methanol in humans, it contains limited comprehensive
information on sub-chronic to chronic methanol exposure.
Taken together, the above considerations suggest a conventional
safety or risk assessment would not appear feasible, and would most
likely be incomplete at present. An alternative approach might be one
based on consideration of blood levels of the most toxic metabolite,
formate. Since formate occurs naturally in humans, it would seem
reasonable to assume that normal background levels should not pose any
risk to health and consequently that levels of human exposure that do
not result in levels of blood formate above background levels could be
considered to pose insignificant risk. In this respect, based on
information from limited studies in humans, it might be concluded that
occupational exposure to current exposure limits (around 260 mg/m3)
or single oral exposure to approximately 20 mg/kg body weight would
fall into this category.
10.2 Evaluation of effects on the environment
Methanol may be released into the environment in significant
amounts during its production, storage, transportation and use.
Methanol is readily degraded in the environment by photo-
oxidation. Half-lives of 7-18 days have been reported for the
atmospheric reaction of methanol with hydroxyl radicals.
Methanol is readily biodegradable under both aerobic and
anaerobic conditions in a wide variety of environmental media. Many
genera and strains of microorganisms are capable of using methanol as
a growth substrate. Generally 80% of methanol in sewage systems is
biodegraded within 5 days.
Methanol is a normal growth substrate for many soil micro-
organisms, which are capable of completely degrading methanol to
carbon dioxide and water.
Methanol is of low toxicity to aquatic and terrestrial organisms
and it is not bioaccumulated. Effects due to environmental exposure to
methanol are unlikely to be observed, unless it is released to the
environment in large quantities, such as a spill.
In summary, unless released in high concentrations, methanol
would not be expected to persist or bioaccumulate in the environment.
Low levels of release would not be expected to result in adverse
environmental effects.
11. RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH AND THE
ENVIRONMENT
11.1 Protection of human health
a) Methanol and methanol mixtures should be clearly labelled
with a warning of the acute toxicity of methanol. Labels
should use the description "methanol".
b) Storage, process and drying plants should be designed to
protect against fire and explosion risks and exposure of
personnel to methanol.
c) Workplaces where methanol is present should be provided with
adequate ventilation to minimize inhalation exposure. Where
necessary, personnel handling methanol should be provided
with suitable protective clothing to prevent skin
contamination.
d) Clinicians should be aware of the latent period and signs
and symptoms following exposure to methanol, particularly by
ingestion. Consideration associated with the existence of
sensitive subgroups should be recognized, including those at
increased risk of folate deficiency.
e) To avoid misuse, methanol used as fuel should be denatured
and should contain a colour additive.
11.2 Protection of the environment
Although methanol is rapidly degraded in the environment and is
of low acute toxicity to aquatic organisms, care should be taken to
prevent spills of large quantities of methanol. Particular care should
be taken to prevent spilled methanol reaching surface water.
12. FURTHER RESEARCH
a) Further research is needed to characterize the mechanism and
pathogenesis of methanol-induced visual toxicity.
b) There is a need for definitive studies concerning the dose-
response relationship for subtle CNS function using
neurotoxic, neurobehavioural and ocular end-points across
species at both single and repeated low-level exposures.
c) Investigation of the metabolism of methanol and formate in
target organs, including the brain, retina, optic nerve and
testes, under various exposure conditions is needed.
d) The pharmacokinetics of methanol and formate during
pregnancy should be investigated in appropriate animal
models to determine whether long-term exposure to methanol
alters maternal or fetal disposition of methanol and
formate.
e) Additional studies are required to resolve whether methanol,
formate or a combination of the two is responsible for
methanol-induced developmental toxicity.
f) Exposure models should be developed and validated to
estimate exposure concentrations and routes of exposure in
specific exposure scenarios. Ambient and personal monitoring
to determine the distribution of exposures should be
conducted.
g) Dose-effect and time-course relationships for both acute and
chronic effects of methanol or formate generated from
methanol, in humans or appropriate models, have not been
established and are essential for adequate risk assessment.
h) There is a need for studies into the nutritional, metabolic,
genetic and age-related factors that may contribute to
variation in susceptibility to methanol intoxication.
i) The genotoxic effects of methanol should be further
investigated to determine whether it is clastogenic.
j) A rapid, practical and inexpensive assay for formate in
blood and body tissues is needed for early diagnosis of
methanol poisoning.
k) Improved therapeutic measures, including the development of
4-methylpyrazole and new agents for reversing formate-
induced visual neurotoxicity, are needed.
13. PREVIOUS EVALUATION BY INTERNATIONAL BODIES
Methanol was evaluated in 1970 as an extraction solvent by the
Joint FAO/WHO Expert Committee on Food Additives.
The Committee recommended that when used as an extraction
solvent, residues should be reduced to a minimum by observing good
manufacturing practice. It was considered that the limited uses of
methanol as an extraction solvent for spice and hop oils meant that
residues from these sources were insignificant in the diet (FAO/WHO,
1971; WHO, 1971).
REFERENCES
Aabakken L, Johansen KS, Rydningen EB, Bredesen JE, Ovrebos S, &
Jacobsen D (1994) Osmolal and anion gaps in patients admitted to an
emergency medical department. Hum Exp Toxicol, 13: 131-134.
Abbondandolo A, Bonatti S, Corsi C, Corti G, Fiorio R, Leporini C,
Mazzacccaro A, & Nieri R (1980) The use of organic solvents in
mutagenicity testing. Mutat Res, 79: 141-150.
Abbott BD, Logsdon TR, & Wilke TS (1994) Effects of methanol on
embryonic mouse palate in serum-free organ culture. Teratology, 49:
122-134.
Abbott BD, Ebron-McCoy M, & Andrews JE (1995) Cell death in rat and
mouse embryos exposed to methanol in whole embryo culture. Toxicology,
97: 159-171.
Agarwal VK (1988) Determination of low relative molecular mass
alcohols in gasoline by gas chromatography. Analyst, 113: 907-909.
Akimoto H & Takagi H (1986) Formation of methyl nitrite in the surface
reaction of nitrogen dioxide and methanol: 2. Photoenhancement.
Environ Sci Technol, 20: 393-397.
Allis JW, Simmons JE, Robinson BL, McDonald A, & House DE (1992)
Induction of rat hepatic cytochrome P-450II E1 by methanol: Its role
in the enhancement of carbon tetrachloride hepatotoxicity.
Toxicologist, 12: 85.
Allis JW, Brown BL, Simmons JE, Hatch GE, McDonald A, & House DE
(1996) Methanol potentiation of carbon tetrachloride hepato toxicity:
the central role of cytochrome P450. Toxicology, 112: 131-170.
Anderson EV (1993) Health studies indicate MTBE is safe gas additive.
Chem Eng News, 71(38): 9-18.
Andrews LS, Clary JJ, Terrill JB, & Bolte HF (1987) Subchronic
inhalation toxicity of methanol. J Toxicol Environ Health, 20:
117-124.
Andrews JE, Ebron-McCoy M, Logsdon TR, Mole LM, Kavlock RJ, & Rogers
JM (1993) Developmental toxicity of methanol in whole embryo culture:
A comparative study with mouse and rat embryos. Toxicology, 81:
205-215.
Andrews JE, Ebron-McCoy M, Kavlock RJ, & Rogers JM (1995)
Developmental toxicity of formate and formic acid in whole embryo
culture: A comparative study with mouse and rat embryos. Teratology,
51: 243-251.
Angerer J & Lehnert G (1977) Occupational exposure to methanol. Acta
Pharmacol, 41: 551-556.
Anon (1991) Methanol: tight markets ahead. Chem Ind, 17: 598.
AOAC (1980) In: Horwitz W ed. Official methods of analysis of the
Association of Official Analytical Chemists, 13th ed. Washington, DC,
Association of Official Analytical Chemists, pp 9.107, 9.086-9.093.
AOAC (1990) In: Helrich K ed. Official methods of analysis of the
Association of Official Analytical Chemists, 15th ed. Washington, DC,
Association of Official Analytical Chemists, pp 702-705.
API (1993) Study of the relationship between folate studies and
methanol toxicity. Washington, DC, American Petroleum Industry
(Publication No. 4554).
Aquilonius S, Askmark H, Enoksson P, Lundberg PO, & Mostrom U (1978)
Computerized tomography in severe methanol intoxication. Br Med J,
ii: 929-930.
ASTM (1993) Standard test methods for the determination of MTBE, ETBE,
TAME, DIPE, tert-amyl alcohol and C1 to C4 alcohols in gasoline by
gas chromatography (D4815). Philadelphia, Pennsylvania, American
Society for Testing and Materials.
ATSDR (Agency for Toxic Substances Disease Registry) (1993) Methanol
toxicity. Am Fam Phys, 47: 163-174.
Auto/Oil Air Quality Improvement Research Program (1992) Emissions and
air quality modeling results from methanol/gasoline blends and
prototype flexible/variable vehicles. Atlanta, Georgia, US Gasoline
Producers, Coordinating Research Council (Technical Bulletin No.7).
Auto/Oil Air Quality Improvement Research Program (1994) Emissions
from methanol fuels and reformulated gasoline in 1993 production
flexible/variable fuel and gasoline vehicles (Technical Bulletin No.
13).
Axelrod J & Daly J (1965) Pituitary gland : enzymic formation of
methanol from S-adenosylmethionine. Science, 158: 892-993.
Baker RN, Alenty Al, & Zack JF Jr (1969) Simultaneous determination
of lower alcohols, acetone and acetaldeyhde in blood by gas
chromatography. J Chromatogr Sci, 7: 312-314.
Barahona-Gomariz MV, Sanz-Barrera F, & Sánchez-Fortún S (1994) Acute
toxicity of organic solvents on Artemia salina. Bull Environ Contam
Toxicol, 52: 766-771.
Barnes I, Bastian V, Becker KH, Fink EH, & Zabel F (1982) Reactivity
studies of organic substances towards hydroxyl radicals under
atmospheric conditions. Atmos Environ, 16: 545-550.
Bartlett GR (1950) Inhibition of methanol oxidation by ethanol in the
rat. Am J Physiol, 163: 619-621.
BASF (1979) [Report on the comparative testing on sensitizing effects
in guinea pigs, modified maximization test.] Ludwigshafen, Germany,
BASF AG, 11 pp (Unpublished report) (in German).
BASF (1980a) [Determination of the acute inhalation toxicity LD50 of
methanol (Merck min. 99.8%) as vapour at 4 hours exposure to Sprague-
Dawley rats.] Ludwigshafen, Germany, BASF AG, 14 pp (Unpublished
report) (in German).
BASF (1980b) [Determination of the acute inhalation toxicity LD50 of
methanol (Merck min. 99.8%) as vapour at 6 hours exposure to Sprague-
Dawley rats.] Ludwigshafen, Germany, BASF AG, 22 pp (Unpublished
report) (in German).
Baumann K & Angerer J (1979) Occupational chronic exposure to organic
solvents: VI. Formic acid concentration in blood and urine as an
indicator of methanol exposure. Int Arch Occup Environ Health, 42:
241-249.
Baumbach Gl, Cancilla PA, Martin-Amat G, Tephly TR, McMartin KE, Makar
Ab, Hayreh M, & Haryeh SS (1977) Methyl alcohol poisoning: IV.
Alterations of the morphological findings of the retina and optic
nerve. Arch Ophthalmol, 95: 1859-1865.
Becker CE (1983) Methanol poisoning. J Emerg Med, 1: 51-58.
Bellar TA & Sigsby JE Jr (1970) Direct gas chromatographic analysis of
low molecular weight substituted organic compounds in emissions.
Environ Sci Technol, 4: 150-156.
Bengtsson BE, Renberg L, & Tarkpea M (1984) Molecular structure and
aquatic toxicity-an example with C1-C13 aliphatic alcohols.
Chemosphere, 13(5/6): 613-622.
Bennett IL, Cary FH, Mitchell GL, & Cooper MN (1953) Acute methyl
alcohol poisoning: a review based on experiences in an outbreak of 323
cases. Medicine, 32: 431-463.
Benoit FM, Davidson WR, Lovett AM, Nacson S, & Ngo A (1985) Breath
analysis by API/MS-human exposure to volatile organic solvents. Int
Arch Occup Environ Health, 55: 113-120.
Bindler F, Voges E, & Laugel P (1988) The problem of methanol
concentration admissible in distilled fruit spirits. Food Addit
Contam, 5: 343-351.
Blanch GP, Tabera J, Sanz J, Herraiz M, & Reglero G (1992) Volatile
composition of vinegars. Simultaneous distillation-extraction and gas
chromatographic-mass spectrometric analysis. J Agric Chem, 40:
1046-1049.
Blomstrand R, Ostling-Wintzell H, Lof A, McMartin K, Tolf BR, &
Hedstrom KG (1979) Pyrazoles as inhibitors of alcohol oxidation and as
important tools in alcohol research: an approach to therapy against
methanol poisoning. Proc Natl Acad Sci (USA), 76: 3499-3503.
Blum DJW & Speece RE (1991) A data base of chemical toxicity to
environmental bacteria and its use in interspecies comparisons and
correlations. Res J Water Pollut Control Fed, 63: 198-207.
Bock JL (1982) Analysis of serum by high-field proton nuclear magnetic
resonance. Clin Chem, 28: 1873-1877.
Boeniger MF (1987) Formate in urine as a biological indicator of
formaldehyde exposure: a review. Am Ind Hyg Assoc J, 48: 900-908.
Bolon B, Dorman DC, Janszen D, Morgan KT, & Welsch F (1993) Phase
specific developmental toxicity in mice following maternal methanol
inhalation. Fundam Appl Toxicol, 21: 508-516.
Bolon B, Welsch F, & Morgan KT (1994) Methanol induced neural tube
defects in mice: pathogenesis during neuralation. Teratology, 49:
497-517.
Boos RN (1948) Quantitative colorimetric microdetermination of
methanol with chromotropic acid. Anal Chem, 20: 964-965.
Bowman MC, Oller WL, Cairns T, Gosnell AB, & Oliver KH (1981) Stressed
bioassay systems for rapid screening of pesticide residues. Part I:
Evaluation of bioassay systems. Arch Environ Contam Toxicol, 10:
9-24.
Braun M & Stolp H (1985) Degradation of methanol by a sulfate reducing
bacterium. Arch Microbiol, 142: 77-80.
Bresnick GH (1989) Excitotoxins: A possible new mechanism for the
pathogenesis of ischemic retinal damage. Arch Ophthalmol, 107:
339-341.
Bringmann G & Kühn R (1980) Comparison of the toxicity thresholds of
water pollutants to bacteria, algae and protozoa in the cell
multiplication inhibition test. Water Res, 14: 231-241.
Buller F & Wood CA (1904) Poisoning by wood alcohol. J Am Med Assoc,
43: 1058-1062.
Burbacher TM (1993) Neurotoxic effects of gasoline and gasoline
constituents. Environ Health Perspect, 101(Suppl 6): 133-141.
Burgess E (1992) Prolonged hemodialysis in methanol intoxication.
Pharmacotherapy, 12: 238-239.
Buttery JE & Chamberlain BR (1988) A simple enzymatic method for the
measurement of abnormal levels of formate in plasma. J Anal Toxicol,
12: 292-294.
Calleja MC, Persoone G, & Geladi P (1994) Comparative acute toxicity
of the first 50 multicentre evaluation of in vitro cytotoxicity
chemicals to aquatic non-vertebrates. Arch Environ Contam Toxicol,
26: 69-78.
Cameron AM, Nilsen OG, Haug E, & Eik-Nes KB (1984) Circulating
concentrations of testosterone, luteinizing hormone and follicle
stimulating hormone in male rats after inhalation of methanol. Arch
Toxicol, 7(Suppl): 441-443.
Cameron AM, Zahlsen K, Haug E, Nilsen OG, & Eik-Nes KB (1985)
Circulating steroids in male rats following inhalation of n-alcohols.
Arch Toxicol, 8(Suppl): 422-424.
Campbell JA, Howard DR, Backer LC, & Allen JW (1991) Evidence that
methanol inhalation does not induce chromosome damage in mice. Mutat
Res, 260: 257-264.
Casey JC, Self R, & Swain T (1963) Origin of methanol and dimethyl
sulfide from cooked foods. Nature (Lond), 200: 885.
Cavanaugh LA, Schadt CF, & Robinson E (1969) Atmospheric hydrocarbon
and carbon monoxide measurements at Point Barrow, Alaska. Environ Sci
Technol, 3: 251-257.
CEC (Commission of the European Communities) (1988) Solvents in common
use: Health risks to workers. Cambridge, Royal Society of Chemistry,
pp 1-7, 157-186 (Publication EUR/11553).
Chang TY & Rudy SJ (1990) Ozone-forming potential of organic emissions
from alternative-fueled vehicles. Atmos Environ, 24A: 2421-2430.
Chang LW, McMillan L, Wynne BR, Pereira MA, Colley RA, Ward JB, &
Legator MS (1983) The evaluation of six monitors from the exposure to
formaldehyde in laboratory animals. Environ Mutagen, 5: 381-387.
Chao CT (1959) [Data for determining the standard maximum permissible
concentration of methanol vapours in the atmospheric air.] Gig I
Sanit, 24: 7-12 (in Russian).
Cheung ST & Lin WN (1987) Simultaneous determination of methanol,
ethanol, isopropanol and ethylene glycol in plasma by gas
chromatography. J Chromatogr, 414: 248-250.
Clark CB, Dutcher JS, McClellan RG, Naman TM, & Seizinger DE (1983)
Influence of ethanol and methanol gasoline blends on the mutagenicity
of particulate exhaust extracts. Arch Environ Contam Toxicol, 12:
311-317.
Clay KL, Murphy RC, & Watkins WD (1975) Experimental methanol toxicity
in the primate: analysis of metabolic acidosis. Toxicol Appl
Pharmacol, 34: 49-61.
Clayton GD & Clayton FE ed. (1982) Patty's industrial hygiene and
toxicology - Volume 2C: Toxicology with cumulative index for Volume 2,
3rd ed. New York, Chichester, Brisbane, Toronto, John Wiley & Sons, pp
4527-4551.
Coe JI & Sherman RE (1970) Comparative study of postmortem vitreous
..... and blood alcohol. J Forensic Sci, 25: 185-190.
Coleman EC, Ho CT, & Chang SS (1981) Isolation and identification of
volatile compounds from baked potatoes. J Agric Food Chem, 29:
42-48.
CONCAWE (1995) Alternative fuels in the automotive market. Brussels,
CONCAWE, 67 pp (Report No. 2/95, prepared for the CONCAWE Automotive
Emissions Management Group by its Technical Coordinator, RC
Hutcheson).
Cook WA (1945) Maximum allowable concentrations of industrial
atmospheric contaminants. Ind Med, 14: 936-946.
Cook MR, Bergman FJ, Cohen HD, Gerkovich MM, Graham O, Harris RX, &
Siemann LG (1991) Effects of methanol vapor on human neurobehavioural
measures. Cambridge, Massachusetts, Health Effects Institute (Research
Report No. 42).
Cooper JR & Felig P (1961) The biochemistry of methanol poisoning: II.
Metabolic acidosis in the monkey. Toxicol Appl Pharmacol, 3:
202-209.
Cooper RL, Rehnberg GL, Goldman JM, Linder RE, Mole ML, Edwards TL,
Hein JF, & McElroy WK (1990) Effect of methanol on hormonal control of
testes in male rats. Toxicologist, 10: 211.
Cooper Rl, Mole ML, Rehnberg GL, Goldman JM, McElroy WK, Hein J, &
Stoker TE (1992) Effects of inhaled methanol on pituitary and
testicular hormones in chamber acclimated and non-acclimated rats.
Toxicology, 71: 69-81.
Costantini MG (1993) Health effects of oxygenated fuels. Environ
Health Perspect, 101(Suppl 6): 151-160.
Craig PC, Withler FC, & Morley RB (1977) Effects of methanol on the
fertilization of chum salmon (Oncorhynchus keta) ova. Environ
Pollut, 14: 85-91.
Crebelli R, Conti G, Conti L, & Carere A (1989) A comparative study on
ethanol and acetaldehyde as inducers of chromosome malsegregation in
Aspergillus nidulans. Mutat Res, 215: 187-195.
Creel DJ, Dustman RD, & Beck EC (1970) Differences in visually evoked
responses in albino versus hooded rats. Exp Neurol, 29: 298-309.
Cuéllar M, González M, & Muñoz MJ (1995) [Methanol toxicity on the
embryonic development of Oryzias latipes.] Rev Toxicol, 12:
109-113 (in Spanish).
Cummings AM (1993) Evaluation of the effects of methanol during early
pregnancy in the rat. Toxicology, 79: 205-214.
D'Alessandro A, Osterloh JD, Chuwers P, Quinlan PJ, Kelly TJ, & Becker
CE (1994) Formate in serum and urine after controlled methanol
exposure at the threshold limit value. Environ Health Perspect, 102:
178-181.
Damian P & Rabbe OG (1996) Toxicokinetic modelling of dose-dependent
formate elimination in rats: in vivo-in vitro correlations using
perfused rat liver. Toxicol Appl Pharmacol, 139: 22-32.
Davoli E, Cappellini L, Airoldi L, & Fanelli R (1986) Serum methanol
concentrations in rats and in men after a single dose of aspartame.
Food Chem Toxicol, 24: 187-189.
De Flora S, Zanacchi P, Camoirano A, Bennicelli C, & Badolati GS
(1984) Genotoxic activity and potency of 135 compounds in the Ames
reversion test and in a bacterial DNA-repair test. Mutat Res, 133:
161-198.
Deichmann WB (1948) Methanol. J Ind Hyg Toxicol, 30: 373-376.
Del Carpio-O'Donovan L & Glay J (1992) Subarachnoid hemorrhage
resulting from methanol intoxication: Demonstrated by computed
tomography. Can Assoc Radiol J, 43: 263-299.
Demedts P, Theunis L, Wauters A, Franck F, Daelemans R, & Neels H
(1994) Excess serum osmolality gap after ingestion of methanol: A
methodology-associated phenomenon? Clin Chem, 40: 1587-1590.
Dethlefs R & Naraqi S (1978) Ocular manifestations and complications
of acute methyl alcohol intoxication. Med J Aust, 2: 483-485.
Diaz-Rueda J, Sloane HJ, & Obremski RJ (1977) An infrared solution
method for the analysis of trapped atmospheric contaminants desorbed
from charcoal tubes. Appl Spectrom, 31: 298-307.
Donnelly MI & Dagley S (1980) Production of methanol from amino acids
by Pseudomonas putida. J Bacteriol, 142: 916-924.
Dorman DC, Dye JA, Nassise MP, Ekuta J, Bolon B, & Medinsky MA (1993)
Acute methanol toxicity in minipigs. Fundam Appl Toxicol, 20:
341-347.
Dorman DC, Moss OR, Farris GM, Janszen D, Bond JA, & Medinsky MA
(1994) Pharmacokinetics of inhaled 14C-methanol and methanol-derived
14C-formate in normal and folate-deficient cynomologus monkeys.
Toxicol Appl Pharmacol, 128: 229-238.
Dorman DC, Bolon B, Struve MF, La Perle KMD, Wong BA, Elswick BE, &
Welsch F (1995) Role of formate in methanol-induced exencephaly.
Teratology, 52: 30-40.
Dowling JE (1987) The electroretinogram and glial responses. In:
Dowling JE ed. The retina: An approachable part of the brain.
Cambridge, Massachusetts, Belknapp Press, pp 164-186.
Downie A, Khattab TM, Malik IA, & Samara IN (1992) A case of
percutaneous industrial methanol toxicity. Occup Med, 42: 47-49.
Dube MF & Green CR (1982) Methods of collection of smoke for
analytical purposes. Recent Adv Tob Sci, 8: 42-102.
Dutkiewicz B, Konczalik J, & Karwacki W (1980) Skin absorption and per
os administration of methanol in men. Int Arch Occup Environ Health,
47: 81-88.
Ebron-McCoy MT, Andrews JE, Kavlock RJ, & Rogers LM (1994) The
developmental toxicity of formate and formic acid in mouse and rat
embryos in whole culture (WEC). Teratology, 49: 393.
Eells JT (1991) Methanol- induced visual toxicity in the rat. J
Pharmacol Exp Ther, 257: 56-63.
Eells JT, Makar AB, Noker PE, & Tephly TR (1981a) Methanol poisoning
and formate oxidation in nitrous oxide-treated rats. J Pharmacol Exp
Ther, 217: 57-61.
Eells JT, McMartin KE, Black K, Virayotha V, Tisdell RH, & Tephly TR
(1981b) Formaldehyde poisoning: a rapid metabolism to formic acid. J
Am Med Assoc, 246: 1237-1238.
Eells JT, Black KA, Makar AB, Tedford CE, & Tephly TR (1982) The
regulation of one-carbon oxidation in the rat by nitrous oxide and
methionine. Arch Biochem Biophys, 219: 316-326.
Eells JT, Black KA, Tedford CE, & Tephly TR (1983) Methanol toxicity
in the monkey: effects of nitrous oxide and methionine. J Pharmacol
Exp Ther, 227: 349-353.
Eells JT, Salzman MM, & Trusk TC (1995) Inhibition of retinal
mitochondrial function in methanol intoxication. Toxicologist, 15:
21-23.
Eells JT, Salzman MM, Lewandowski MF, & Murray TG (1996) Formate
induced alterations in retinal function in methanol-intoxicated rats.
Toxicol Appl Pharmacol, 140: 58-69.
Eells JT, Salzman MM, Lewandowski MF, & Murray TG (in press)
Developmental and characterization of a non-primate animal model of
methanol induced neurotoxicity. In: Bengston DA & Henschel DS ed.
Environmental toxicology and risk assessment: Biomarkers and risk
assessment - Volume 5. Philadelphia, Pennsylvania, American Society
for Testing and Materials, (Publication ASTM STP No. 1306).
Egle JL & Gochberg BJ (1979) Retention of inhaled isoprene and
methanol in the dog. Am Ind Hyg Assoc J, 36: 369-373.
Eisenberg AA (1917) Visceral changes in wood alcohol poisoning by
inhalation. Am J Public Health, 7: 765-771.
Eisenreich SJ, Looney BB, & Thornton JD (1981) Airborne organic
contaminants in the Great Lakes ecosystem. Environ Sci Technol, 15:
30-38.
Elvers B, Hawkins S, & Schulz G ed. (1990) Ullmann's encyclopedia of
industrial chemistry, 5th ed. Weinheim, Germany, VCH-Verlag, vol 16A,
pp 465-486.
Eriksen SP & Kulkarni AB (1963) Methanol in normal human breath.
Science, 141: 639-640.
Erlanson P, Fritz H, Hagstam KE, Liljenberg B, Tryding N, & Voigt G
(1965) Severe methanol intoxication. Acta Med Scand, 177: 393-408.
Ewell WS, Gorsuch JW, Kringle RO, Robillard KA, & Spiegel RC (1986)
Simultaneous evolution of the acute effects of chemicals on seven
aquatic species. Environ Toxicol Chem, 5: 831-840.
FAO/WHO (1971) Toxicological evaluation of some extraction solvents
and certain other substances. Report of the Joint FAO/WHO Expert
Committee on Food Additives, 24 June-2 July 1970. Rome, Food and
Agriculture Organization, pp 105-109.
Federal Register (1989) Standards for emissions from methanol-fueled
motor vehicles and motor vehicle engines - Final rule. Fed Reg,
54(68): 40CFR Part 86.
Feldstein M & Klendshoj RE (1954) Determination of methanol in
biological fluids by microdiffusion analysis. Anal Chem, 26:
932-933.
Ferry DG, Temple WA, & McQueen EG (1980) Methanol monitoring. Int Arch
Occup Environ Health, 47: 155-163.
Fiedler E, Grossmann G, Kersebohm B, Weiss G, & Witte C (1990)
Methanol. In: Elvers B, Hawkins S, & Schutz G ed. Ullmann's
encyclopedia of industrial chemistry, 5th ed. Weinheim, VCH
Verlagsgesellschaft, vol 16A, pp 465-486.
Filley CM & Kelly JP (1993) Alcohol-and drug-related neurotoxicity.
Current Opin Neurol Neurosurg, 6: 443-447.
Fox ME (1973) Rapid gas chromatographic method for determination of
residual methanol in sewage. Environ Sci Technol, 7: 838-840.
Francot P & Geoffroy P (1956) Le méthanol dans les jus de fruits, les
boissons fermentées des alcools et spiriteux. Rev Ferment Ind Aliment,
11: 279-287.
Franzblau A, Levine SP, Burgess LA, Qu QS, Schreck RM, & D'Arcy JB
(1992a) The use of a transportable Fourier Transform infrared (FTIR)
spectrometer for the direct measurement of solvents in breath and
ambient air: I. Methanol. Am Ind Hyg Assoc J, 53: 221-227.
Franzblau A, Levine SP, D'Arcy JB, & Qu QS (1992b) Use of urinary
formic acid as a biologic exposure index of methanol exposure. Appl
Occup Environ Hyg, 7: 467-471.
Franzblau A, Lee EW, Schreck RM, D'Arcy JB, Santrock J, & Levine SP
(1993) Absence of formic acid accumulation in urine following five
days of methanol exposure. Appl Occup Environ Hyg, 8: 883-888.
Frederick LJ, Schulte PA, & Apol A (1984) Investigation and control of
occupational hazards associated with the use of spirit duplicators. Am
Ind Hyg Assoc J, 45: 51-55.
Freitag D, Ballhorn L, Geyer H, & Korte F (1985) Environmental hazard
profile of organic chemicals. Chemosphere, 14: 1589-1616.
Frenia Ml & Schauben JL (1993) Methanol inhalation toxicity. Ann Emerg
Med, 22: 1919-1923.
Gabele PA & Knapp KT (1993) A characterization of emissions from an
early model flexible-fuel vehicle. J Air Waste Manage Assoc, 43:
851-858.
Garcia JH & Van Zandt JP (1969) Proceedings of 27th Annual Meeting of
the Electron Microscope Society of America. Baton Rouge, Louisiana,
Claitor Publishers, vol 27, pp 360-361.
Garner CD & Lee EW (1994) Evaluation of methanol-induced
retinotoxicity using oscillatory potential analysis. Toxicology,
93: 113-124.
Garner CD, Lee EW, & Louis-Ferdinand RJ (1995a) Muller cell
involvement in methanol-induced retinal toxicity. Toxicol Appl
Pharmacol, 130: 101-107.
Garner CD, Lee EW, Terzo TS, & Louis-Ferdinand RJ (1995b) The role of
retinal metabolism in methanol-induced retinal toxicity. J Toxicol
Environ Health, 44: 43-56.
Gessner PK (1970) Method for the assay of ethanol and other aliphatic
alcohols applicable to tissue homogenates and possessing a sensitivity
of 1 µg/ml. Anal Biochem, 38: 499-505.
Gettler AO (1920) Critical study of methods for the detection of
methyl alcohol. J Biol Chem, 42: 311-328.
Geyer H, Politzki G, & Freitag D (1984) Prediction of ecological
behavior of chemicals: relationship between n-octanol/water partition
and bioaccumulation of organic chemicals by alga Chlorella.
Chemosphere, 13: 269-284.
Gilger AP & Potts AM (1955) Studies on the visual toxicity of
methanol: V. The role of acidosis in experimental methanol poisoning.
Am J Ophthamol, 39: 63-86.
Gilger AP, Potts AM, & Johnson JV (1952) Studies on the visual
toxicity of methanol: II. The effect of parenterally administered
substances on the systemic toxicity of methyl alcohol. Am J Ophthamol,
35(Part 2): 113-126.
Giminez ER, Vallejo NE, Roy E, Lis M, Izurieta EM, Rossi S, & Capuccio
M (1968) Percutaneous alcohol intoxication. Clin Toxicol, 1: 39-48.
Glazer M & Dross P (1993) Necrosis of the putamen caused by methanol
intoxication: MR findings. Am J Roentgenol, 160: 1105-1106.
Gluth G & Hanke W (1985) A comparison of physiological changes in
carp, Cyprinus carpio, induced by several pollutants at sublethal
concentrations: 1. The dependency on exposure times. Ecotoxicol
Environ Saf, 9: 179-188.
Gluth G, Freitag D, Hanke W, & Korte F (1985) Accumulation of
pollutants in fish. Comp Biochem Physiol, 81C: 273-277.
Gold MD & Moulif CE (1988) Effects of emission standards on methanol
vehicle-related ozone, formaldehyde and methanol exposure. Presented
at 81st Meeting of Air Pollution Control Association, Dallas, TX, June
19-24. Pittsburgh, Pennsylvania, Air Pollution Control Association.
Gonda A, Gault H, Churchill D, & Hollomby D (1978) Hemodialysis for
methanol intoxication. Am J Med, 64: 749-757.
Goodman JI & Tephly TR (1971) A comparison of rat and human liver
formaldehyde dehydrogenase. Biochim Biophys Acta, 252: 489.
Grady S & Osterloh J (1986) Improved enzymic assay for serum formate
with colorimetric endpoint. J Anal Toxicol, 10: 1-5.
Graedel TE, Hawkins DT, & Claxton LD ed. (1986) Atmospheric chemical
compounds: Sources, occurrence and bioassay. New York, London,
Academic Press, pp 512-514, 557.
Grayson M ed. (1981) Kirk-Othmer encyclopedia of chemical technology,
3rd ed. New York, John Wiley & Son, vol 15, pp 398-405.
Greenberg L, Mayers MR, Goldwater LJ, & Burke WJ (1938) Health hazards
in the manufacture of "fused collars": II. Exposure to acetone-
methanol. J Ind Hyg Toxicol, 20: 148-154.
Greizerstein HB (1981) Congener contents of alcoholic beverages. J
Stud Alcohol, 42: 1030-1037.
Griffiths AJF (1981) Neurospora and environmentally induced
aneuploidy. In: Stich HF & San RHC ed. Short-term tests for chemical
carcinogens. Berlin, Heidelberg, New York, Springer-Verlag,
pp 187-199.
Guerin MR, Higgins CE, & Greist WH (1987) The analysis of the
particulate and vapour phases of tobacco smoke. In: O'Neill IK,
Brunnemann KD, Dodet B, & Hoffmann D ed. Environmental carcinogens:
Methods of analysis and exposure measurement, Volume 9. Lyon,
International Agency for Research on Cancer, pp 115-139 (IARC
Scientific Publications No. 81).
Guggenheim MA, Couch JR, & Weinberg W (1971) Motor dysfunction as a
permanent complication of methanol ingestion. Arch Neurol, 24:
550-554.
Güsten H, Klasinc L, & Maric D (1984) Prediction of the abiotic
degradability of organic compounds in the troposphere. J Atmos Chem,
2: 83-93.
Haggard HW & Greenberg LA (1939) Studies on the absorption,
distribution and elimination of alcohol: IV. The elimination of
methylalcohol. J Pharmacol Exp Ther, 66: 479-496.
Hanson RS (1980) Ecology and diversity of methylotropic organisms. Adv
Appl Microbiol, 26: 3-29.
Harger RN (1935) A simple micromethod for the determination of alcohol
in biological material. J Lab Clin Med, 20: 746-751.
Hatfield R (1957) Biological oxidation of some organic compounds. Ind
Eng Chem, 49: 192-197.
Hayreh MS, Hayreh SS, Baumbach GL, Cancilla P, Martin-Amat G, Tephly
TR, McMartin KE, & Makar AB (1977) Methyl alcohol poisoning: III.
Ocular toxicity. Arch Ophthamol, 95: 1851-1858.
Heath A (1983) Methanol poisoning. Lancet, 1: 1339-1340.
Heath M (1991) Alternative transportation fuels: Natural gas, propane,
methanol and ethanol compared with gasoline and diesel. Calgary,
Canadian Energy Research Institute, pp 113-115.
HEI (1987) Automotive methanol vapors and human health: An evaluation
of existing scientific information and issues for future research.
Cambridge, Massachusetts, Health Effects Institute, 70 pp (Special
Report).
HEI (1996) The potential health effects of oxygenates added to
gasoline: A review of the current literature. Cambridge,
Massachusetts, Health Effects Institute, 165 pp.
Heidelberger C, Freeman AE, Pienta RJ, Sivak A, Bertram DS, Casto BC,
& Dunkel VC (1983) Cell transformation by chemical agents: A review
and analysis of the literature. Mutat Res, 114: 283-385.
Heinrich R & Angerer J (1982) Occupational chronic exposure to organic
solvents. Int Arch Occup Environ Health, 50: 341-349.
Helmstetter A, Gamerdinger AP, & Pruell RJ (1996) Acute toxicity of
methanol to Mytilus edulis. Bull Environ Contam Toxicol, 57(4):
675-681.
Hemming DJB, Criddle RS, & Hansen LD (1995) Effects of methanol on
plant respiration. J Plant Physiol, 146(3): 193-198.
Hermens J & Leeuwangh P (1982) Joint toxicity of mixtures of 8 and 24
chemicals to the guppy (Poecilia reticulata) Ecotoxicol Environ Saf,
6: 302-310.
Hertelendy ZI, Mendenhall CL, Rouster SD, Marshall L, & Weesner R
(1993) Biochemical and clinical effects of aspartame in patients with
chronic, stable alcoholic liver disease. Am J Gastroenterol, 88:
737-743.
Hickman GT & Novak JT (1989) Relationship between subsurface
biodegradation rates and microbial density. Environ Sci Technol, 23:
525-532.
Hickman GT, Novak JT, Morris MS, & Rebhun M (1989) Effects of site
variations on subsurface biodegradation potential. J Water Pollut
Control Fed, 61: 1564-1575.
Hindberg J & Wieth JO (1963) Quantitative determination of methanol in
biological fluids. J Lab Clin Med, 61: 355-361.
Hippe H, Caspari D, Fiebig K, & Gottschalk G (1979) Utilization of
trimethylamine and other N-methyl compounds for growth and methane
formation by methanosarcina barkeri. Proc Natl Acad Sci (USA), 76:
494-498.
Holzer G, Shanfield H, Zlatkis A, Bertsch W, Juarez P, Mayfield H, &
Liebich HM (1977) Collection and analysis of trace emissions from
natural sources. J Chromatogr, 142: 755-764.
Hooper AB & Terry KR (1973) Specific inhibitors of ammonia oxidation
in Nitrosomonas. J Bacteriol, 115: 480-485.
Horton VI, Higuchi MA, & Rickert DE ( 1992) Physiologically based
pharmacokinetic model for methanol in rats, monkeys and humans.
Toxicol Appl Pharmacol, 117: 26-36.
Howard PH ed. (1990) Handbook of environmental fate and exposure data
for organic compounds - Volume II: Solvents. Chelsea, Michigan, Lewis
Publishers, pp 310-317.
Howard PH, Boethling RS, Jarvis WF, Meylan WM, & Michalenko EM (1991)
Handbook of environmental degradation rates. Chelsea, Michigan, Lewis
Publishers, pp 93-94.
Hsieh FY, Leu TM, & Chia LG (1992) Bilateral putaminal necrosis caused
by methanol poisoning: A case report. Chin Med J (Taipei), 49:
283-288.
Hüls AG (1978) [Determination of the biodegradability of methanol in
the closed bootle tert (OECD Guideline 30D).] Marl, Hüls AG, 11 pp (in
German).
Hunt R (1902) The toxicity of methyl alcohol. John Hopkins Hosp Bull,
13: 213.
Hustert K, Mansour M, Parlar H, & Korte F (1981) [The EPA test - A
method for the determination of photochemical degradation of organic
compounds in aquatic systems.] Chemosphere, 10: 995-998 (in German).
ILO (1983) In: Parmeggiani L ed. Encyclopedia of occupational health
and safety, 3rd revis ed. Geneva, International Labour Office, vol 2,
pp 1356-1358.
ILO (1991) Occupational exposure limits for airborne toxic substances,
3rd ed. Geneva, International Labour Office, pp 256-257.
Infurna R & Weiss B (1986) Neonatal behavioural toxicity in rats
following prenatal exposures to methanol. Teratology, 33: 259-265.
Ingemansson SO (1983) Studies on the effect of 4-methylpyrazole on
retinal activity in the methanol poisoned monkey by recording the
electroretinogram. Acta Opthalmol, 158(Suppl): 5-24.
IPCS (1995) Poison Information Monograph No. 335: Methanol. Geneva,
World Health Organization, International Programme on Chemical Safety.
Isidorov VA, Zenkevich IG, & Ioffe BV (1985) Volatile organic
compounds in the atmosphere of forests. Atmos Environ, 19: 1-8.
Jacobs GA (1990) OECD eye irritation tests on three alcohols: Acute
toxicity data. J Am Coll Toxicol, 1: 56-57.
Jacobsen D & McMartin KE (1986) Methanol and ethylene glycol
poisonings: Mechanisms of toxicity, clinical course, diagnosis and
treatment. Med Toxicol, 1: 309-314.
Jacobsen D, Jansen H, Wiek-Larsen E, Bredesen DE, & Halvorsen S
(1982a) Studies on methanol poisoning. Acta Med Scand, 12: 5-10.
Jacobsen D, Bredesen JE, Eide I, & Ostborg J (1982b) Anion and osmolal
gaps in the diagnosis of methanol and ethylene glycol poisoning. Acta
Med Scand, 212: 17-20.
Jacobsen D, Ovrebo S, & Sejersted OM (1983a) Toxicokinetics of formate
during hemodialysis. Acta Med Scand, 214: 409-412.
Jacobsen D, Ovrebo S, Arnesen E, & Paus PN (1983b) Pulmonary excretion
of methanol in man. Scand J Clin Invest, 43: 377-379.
Jacobsen D, Webb P, Collins TD, & McMartin KE (1988) Methanol and
formate kinetics in late diagnosed methanol intoxication. Med Toxicol,
3: 418-423.
Jacobsen D, Sebastian CS, Barron SK, Carriere EW, & McMartin KE (1990)
Effects of 4-methylpyrazole, methanol/ethylene glycol antidote in
healthy humans. J Emerg Med, 8: 455-461.
Jansen JLC, Nyberg U, Aspegren H, & Andersson B (1993) Handling of
anaerobic digester supernatant combined with full nitrogen removal.
Water Sci Technol, 27: 391-403.
Jaselkis B & Warriner JP (1966) Titrimetric determination of primary
and secondary alcohols by xenon trioxide oxidation. Anal Chem, 38:
563-564.
Jeganathan PS & Namasivayam A (1989) Methanol induced monoamine
changes in hypothalamus and striatum of albino rats. Alcohol, 6:
451-454.
Jones AW (1986) Abnormally high concentrations of methanol in breath:
A useful biochemical marker of recent heavy drinking. Clin Chem, 32:
1241-1242.
Jones AW (1987) Elimination half-life of methanol during hangover.
Pharmacol Toxicol, 60: 217-220.
Jones AW & Lowinger H (1988) Relationship between the concentration of
ethanol and methanol in blood samples from Swedish drinking drivers.
Forensic Sci Int, 37: 277-285.
Jones AW, Mardh G, & Anggard E (1983) Determination and endogenous
ethanol in blood and breath by gas chromatography-mass spectrometry.
Pharmacol Biochem Behav, 18(Suppl 1): 267-272.
Jones AW, Skagerberg S, Yonekurat T, & Sato A (1990) Metabolic
interaction between endogenous ethanol studied in human volunteers by
analysis of breath. Pharmacol Toxicol, 66: 62-65.
Jonsson A & Berg S (1983) Determination of low-molecular-weight
oxygenated hydrocarbons in ambient air by cryogradient sampling and
two-dimensional gas chromatography. J Chromatogr, 279: 307-322.
Jonsson A, Persson KA, & Grigoriadis V (1985) Measurements of some low
molecular-weight oxygenated, aromatic and chlorinated hydrocarbons in
ambient air and in vehicle emissions. Environ Int, 11: 383-392.
Juhnke VI & Lüdemann D (1978) [Results of testing of 200 chemical
compounds for acute fish toxicity with the Golden Orfe test.] Z Wasser
Abwasser Forsch, 11(5): 161-164 (in German).
Jungclaus GA, Lopez-Avila V, & Hites RA (1978) Organic compounds in an
industrial wastewater: A case study of their environmental impact.
Environ Sci Technol, 12: 88-96.
Kahn A & Blum D (1979) Methyl alcohol poisoning in an 8-month old boy:
An unusual rank route of intoxication. J Pediatr, 94: 841-843.
Kane Rl, Talbert W, Harlan J, Sizemore G, & Cataland S (1968) A
methanol poisoning outbreak in Kentucky. Arch Environ Health, 17:
119-129.
Kaplan K (1962) Methyl alcohol poisoning. Am J Med Sci, 244:
170-174.
Katoh M (1989) New Energy Development Organization data. Presented at
the Methanol Vapors and Health Effects Workshop: What we know and what
we need to know - Summary Report. Washington, DC, ILSI Risk Science
Institute/US Environmental Protection Agency/Health Effects
Institute/American Petroleum Institute, p A-7.
Kavet R & Nauss KM (1990) The toxicity of inhaled methanol vapors. CRC
Crit Rev Toxicol, 21: 21-50.
Kawai T, Yasugi T, Uchida Y, & Ikeda M (1990) Personal diffusive
sampler for methanol, a hydrophilic solvent. Bull Environ Contam
Toxicol, 44: 514-520.
Kawai T, Yasugi T, Mizunuma K, Horiguchi S, Hirase Y, Uchida Y, &
Ikeda M (1991a) Simple method for the determination of methanol in
blood and its application in occupational health. Bull Environ Contam
Toxicol, 47: 797-803.
Kawai T, Yasugi T, Mizunuma K, Horiguchi S, Hirase Y, Uchida Y, &
Ikeda M (1991b) Methanol in urine as a biological indicator of
occupational exposure to methanol vapor. Int Arch Occup Environ
Health, 63: 311-318.
Kawai T, Yasugi T, Mizunuma K, Horiguchi S, Iguchi H, Uchida Y, Iwami
O, & Ikeda m (1992) Comparative evaluation of urinalysis and blood
analysis as means of detecting exposure to organic solvents at low
concentrations. Int Arch Occup Environ Health, 64: 223-234.
Kawai T, Mizunuma K, Yasugi T, Horiguchi S, Moon CS, Zhang ZW,
Miyashita K, Takeda S, & Ikeda M (1995) Effects of methanol on styrene
metabolism among workers occupationally exposed at low concentrations.
Arch Environ Contam Toxicol, 28: 543-546.
Keeney AH & Mellinkoff SM (1951) Methyl alcohol poisoning. Ann Intern
Med, 34: 331-338.
Keller AE (1993) Acute toxicity of several pesticides, organic
compounds, and a wastewater effluent to the fresh water mussel,
Anodonta imbecilis, Ceriodaphnia dubia and Pimephales promelas.
Bull Environ Contam Toxicol, 51: 696-702.
Kelly TJ, Mukund R, Spicer CW, & Pollack AJ (1994) Concentrations and
transformations of hazardous air pollutants. Environ Sci Technol,
28: 378A-387A.
Kennedy TF & Shanks D (1981) Methanol: manufacture and uses. In:
Wickson EJ ed. Monohydric alcohols. Washington, DC, American Chemical
Society, pp 19-27 (ACS Symposium Series No. 159).
Kempa ES (1976) [Oxygen needs during the degradation of waste
compounds.] Österr Abwasser Rundsch, 2: 20-25 (in German).
Keyvan-Larijarni H & Tannenberg AM (1974) Methanol intoxication:
Comparison of peritoneal dialysis and hemodialysis treatment. Arch
Intern Med, 124: 293-296.
Kim S (1973) Purification and properties of protein methylase. Arch
Biochem Biophys, 157: 476-484.
Kimura ET, Ebert DM, & Dodge PW (1971) Acute toxicity and limits of
solvent residue for sixteen organic solvents. Toxicol Appl Pharmacol,
19: 699-704.
King L (1992) Acute methanol poisoning: A case study. Heart Lung,
21: 260-264.
King GM, Klug MJ, & Lovley DR (1983) Metabolism of acetate, methanol
and methylated amines in intertidal sediments of Lowes Cove, Maine.
Appl Environ Microbiol, 45: 1848-1853.
Kingsley WH & Hirsch FG (1955) Toxicological considerations in direct
process spirit duplicating machines. Compens Med, 6: 7-8.
Kinlin TE, Muralidhara R, Pittet AO, Sanderson A, & Walradt JP (1972)
Volatile components of roasted filberts. J Agric Food Chem, 20:
1021-1028.
Kinoshita JH & Masurat T (1958) Effect of glutathione in formaldehyde
oxidation in the retina. Am J Opthalmol, 46: 42.
Kirchner JG & Miller JM (1957) Volatile water-soluble and oil
constituents of Valencia orange juice. J Agric Food Chem, 5:
283-291.
Kohl WL ed. (1990) Methanol as an alternative fuel choice: An
assessment. Washington, DC, The Johns Hopkins University School of
Foreign Service, pp 439.
Koivusalo M (1970) Methanol. In: Tremolieres J ed. International
encyclopaedia of pharmacology and therapeutics. Oxford, New York,
Pergamon Press, vol 2, section 20, pp 465-505.
Komers R & Sir Z (1976) Gas chromatographic determination at the parts
per million level of methanol and ethanol in aqueous solution.
J Chromatogr, 119: 251-254.
Konemann H (1981) Quantitative structure-activity relationships in
fish toxicity studies. Toxicology, 19: 209-221.
Krotoszynski B, Gabriel G, O'Neill HJ, & Claudio MPA (1977)
Characterization of human expired air: A promising investigative and
diagnostic technique. J Chromatogr Sci, 15: 239-244.
Krotoszynski BK, Bruneau GM, & O'Neill HJ (1979) Measurement of
chemical inhalation exposure in urban populations in the presence of
endogenous effluents. J Anal Toxicol, 3: 225-234.
Kruse JA (1992) Methanol poisoning. Intensive Care Med, 18: 391-397.
Kuhn R, Pattard M, Pernak KD, & Winter A (1989) Results of the harmful
effects of selected water pollutants (anilines, phenols, aliphatic
compounds) to Daphnia magna. Water Res, 23: 495-499.
Kutzbach C & Stokstad ELR (1968) Partial purification of a 10-formyl
tetrahydrofolate: NADP oxidoreductase from mammalian liver. Biochem
Biophys Res Commun, 30: 111.
Larsson BT (1965) Gas chromatography of organic volatiles in human
breath and saliva. Acta Med Scand, 19: 159-164.
Leaf G & Zatman LJ (1952) A study of the conditions under which
methanol may exert a toxic hazard in industry. Br J Ind Med, 9:
19-31.
Lee EW, Reader JA, Garner CD, Brady AN, & Li LC (1990) Evaluation of
methanol (M) toxicity on the visual system: spontaneous degeneration
of the retina and optic nerve in the F-344 rat. Toxicologist, 10:
155.
Lee EW, Brady AN, Brabec MJ, & Fabelt JN (1991) Effects of methanol
vapors on testosterone production and testis morphology in rats.
Toxicol Ind Health, 7: 261-275.
Lee EW, Terzo TS, D'Arcy JB, Gross KB, & Schreck RM (1992) Lack of
blood formate accumulation in humans following exposure to methanol
vapors at the current permissible exposure limit of 200 ppm. Am Ind
Hyg Assoc J, 53: 99-104.
Lee EW, Garner CD, & Terzo TS (1994a) Animal model for the study of
methanol toxicity: Comparison of folate-reduced rat responses with
published monkey data. J Toxicol Environ Health, 41: 71-82.
Lee EW, Garner CD, & Terzo TS (1994b) A rat model manifesting
methanol-induced visual dysfunction suitable for both acute and long-
term exposure studies. Toxicol Appl Pharmacol, 128: 199-206.
Lemaire J, Campbell I, Hulpke H, Guth JA, Merz W, Philp J, & von
Waldow C (1982) An assessment of test methods for photodegradation of
chemicals in the environment. Chemosphere, 11: 119-164.
Lettinga G, De Zeeuw W, & Ouberg E (1981) Anaerobic treatment of
wastes containing methanol and higher alcohols. Water Res, 15:
171-182.
Lewis RJ Sr (1989) Food additives handbook. New York, Van Nostrand
Reinhold Co., pp 291-292.
Ley CO & Gali FG (1983) Parkinsonian syndrome after methanol
intoxication. Eur Neurol, 22: 405-409.
Liesivuori J & Savolainen H (1987) Urinary formic acid as an indicator
of occupational exposure to formic acid and methanol. Am Ind Hyg Assoc
J, 48: 32-34.
Liesivuori J & Savolainen H (1991) Methanol and formic acid toxicity:
Biochemical mechanisms. Pharmacol Toxicol, 69: 157-163.
Lijinsky W, Thomas BJ, & Kovatch RM (1991) Differences in skin
carcinogenesis by methyl nitroso urea between mice of several strains.
Cancer Lett, 61: 1-5.
Linden E, Bengtsson BE, Svanberg O, & Sundstrom G (1979) The acute
toxicity of 78 chemicals and pesticide formulations against two
brackish water organisms - The bleak (Alburnus alburnus) and the
harpacticoid nitocraspinipes. Chemosphere, 11/12: 843-851.
Lins RL, Zachee P, Christiaens M, Van De Vijver F, De Waele L,
Sandra P, & De Broe ME (1980) Prognosis and treatment of methanol
intoxication. In: Holmstedt B, Lauwerys R, Mercier M, & Roberfroid M
ed. Mechanisms of toxicity and hazard evaluation. Amsterdam,
Elsevier/North-Holland Biomedical Press, pp 415-421.
Litovitz LT, Schmitz BF, Matyunas N, & Martin TG (1988) 1987 Annual
report of the American Association of Poison Control Centers National
Data Collection System. Am J Emerg Med, 6: 479-515.
Loewy A & von der Heide R (1914) [The uptake of methyl alcohol by
inhalation.] Biochem Ztg, 65: 230-252 (in German).
Lkke H (1984) Leaching of ethylene glycol and ethanol in subsoils.
Water Air Soil Pollut, 22: 373-387.
Lovegren NV, Fisher GS, Legendre MG, & Schuller WH (1979) Volatile
constituents of dried legumes. J Agric Food Chem, 27: 851-853.
Luke LA & Ray JE (1984) Gas-chromatographic method for the
determination of low relative molecular mass alcohols and methyl tert-
butyl ether in gasoline. Analyst, 109: 989-992.
Lund A (1948a) Excretion of methanol and formic acid in man after
methanol consumption. Acta Pharmacol, 4: 205-212.
Lund A (1948b) Metabolism of methanol and formic acid in dogs. Acta
Pharmacol, 4: 108-121.
Lund A (1948c) Metabolism of methanol and formic acid in rabbits. Acta
Pharmacol, 4: 99-107.
Lund ED, Kirkland CL, & Shaw PE (1981) Methanol, ethanol and
acetaldehyde contents of citrus products. J Agric Food Chem, 29:
361-366.
Lundberg P (1985) Scientific basis for Swedish occupational standards
VI. Arb Och Halsa, 32: 115-121.
Lyman WJ, Reehl WF, & Rosenblatt DH ed. (1990) Handbook of chemical
property estimation methods. Washington, DC, American Chemical
Society.
McCord CP (1931) Toxicity of methyl alcohol (methanol) following skin
absorption and inhalation. Ind Eng Chem, 23: 931-936.
McCoy H, Cipolle RJ, Ehlers SM, Sawchuk RJ, & Zaske DE (1979) Severe
methanol poisoning: Application of a pharmacokinetic model for ethanol
therapy and hemodialysis. Am J Med, 67: 804-807.
McDonald A, Sey YM, House DE, & Simmons JE (1992) Methanol
potentiation of carbon tetrachloride hepatotoxicity is dependent on
the time of carbon tetrachloride administration. Toxicologist, 12:
239.
McGregor DB, Martin R, Riach CG, & Caspary WJ (1985) Optimization of a
metabolic activation system for use in the mouse lymphoma L5178Y
tk+tk_ mutation system. Environ Mutagen, 7(Suppl 3): 10.
Machiele PA (1990) A health and safety assessment of methanol as an
alternative fuel. In: Kohl WL ed. Methanol as an alternative choice.
Washington, DC, The Johns Hopkins Foreign Policy Institute, pp
217-239.
McLean DR, Jacobs H, & Mielke BW (1980) Methanol poisoning: A clinical
and pathological study. Ann Neurol, 8: 162-167.
McMartin KE, Makar AB, Martin-Amat G, Palese M, & Tephly TR (1975)
Methanol poisoning I. The role of formic acid in the development of
metabolic acidosis in the monkey and the reversal by 4-methylpyrazole.
Biochem Med, 13: 319-333.
McMartin KE, Martin-Amat G, Makar AB, & Tephly TR (1977) Methanol
poisoning: V. The role of formate metabolism in the monkey. J
Pharmacol Exp Ther, 201: 564-572.
McMartin KE, Martin-Amat G, Noker PE, & Tephly TR (1979) Lack of a
role for formaldehyde in methanol poisoning in the monkey. Biochem
Pharmacol, 28: 645-649.
McMartin KE, Ambre JJ, & Tephly TR (1980a) Methanol poisoning in human
subjects: Role of formic acid accumulation in the metabolic acidosis.
Am J Med, 68: 414-418.
McMartin KE, Hedstrom KG, Tolf BR, Ostung-Wintzell H, & Blomstrand R
(1980b) Studies on the metabolic interactions between 4-methylpyrazole
and methanol using the monkey as an animal model. Arch Biochem
Biophys, 199: 606-614.
Maeda Y, Fujio Y, Suetaka T, & Munemori M (1988) Selective gas
chromatographic determination of trace amounts of alcohols in ambient
air. Analyst, 113: 189-191.
Maejima K, Suzuki T, Niwa H, Maekawa A, Nagase S, & Ishinishi N (1992)
Toxicity to rats of methanol-fueled engine exhaust continuously for 28
days. J Toxicol Environ Health, 37: 293-312.
Maejima K, Suzuki T, Numata H, Maekawa A, Nagase S, & Ishinishi N
(1993) Recovery from changes in the blood and nasal cavity and/or
lungs of rats caused by exposure to methanol-fueled engine exhaust.
J Toxicol Environ Health, 39: 323-340.
Maejima K, Suzuki T, Numata H, Maekawa A, Nagase S, & Ishinishi N
(1994) Subchronic (12-week) inhalation toxicity study of methanol-
fueled engine exhaust in rats. J Toxicol Environ Health, 41:
315-327.
Mahieu P, Hassoun A, & Lauwerys R (1989) Predictors of methanol
intoxication with unfavourable outcome. Hum Toxicol, 8: 135-137.
Majchrowicz E & Mendelson JH (1971) Blood methanol concentrations
during experimentally induced ethanol intoxication in alcoholics.
J Pharmacol Exp Ther, 179: 293-300.
Makar AB & Mannering GJ (1968) Role of the intracellular distribution
of hepatic catalase in the peroxidative oxidation of methanol. Mol
Pharmacol, 4: 484-491.
Makar AB & Tephly TR (1976) Methanol poisoning in the folate deficient
rat. Nature (Lond), 261: 715-716.
Makar AB & Tephly TR (1977) Methanol poisoning: VI. Role of folic acid
in the production of methanol poisoning in the rat. J Toxicol Environ
Health, 2: 1201-1209.
Makar AB & Tephly TR (1982) Improved estimation of formate in body
fluids and tissues. Clin Chem, 28: 385.
Makar AB, Tephly TR, & Mannering GJ (1968) Methanol metabolism in the
monkey. Mol Pharmacol, 4: 471-483.
Makar AB, McMartin KE, Palese M, & Tephly TR (1975) Formate assay in
body fluids: application to methanol poisoning. Biochem Med, 13:
117-126.
Makar AB, Tephly TR, Sahin G, & Osweiler G (1990) Formate metabolism
in young swine. Toxicol Appl Pharmacol, 105: 315-320.
Malorny G, Rietbrock N, & Schneider M (1965) [The oxidation of
formaldehyde to formic acid in blood: a contribution to the metabolism
of formaldehyde.] Naunyn Schmiedebergs Arch Pathol Exp Pharmacol,
250: 419-436 (in German).
Martin-Amat G, Tephly TR, McMartin KE, Makar AB, Hayreh MS, Hayreh SS,
Baumbach G, & Cancilla P (1977) Methyl alcohol poisoning: II.
Development of a model for ocular toxicity in methyl alcohol poisoning
using the rhesus monkey. Arch Ophthamol, 95: 1847-1850.
Martin-Amat G, McMartin KE, Hayreh SS, & Tephly TR (1978) Methanol
poisoning: Ocular toxicity produced by formate. Toxicol Appl
Pharmacol, 45: 201-208.
Martinasevic MK, Green MD, Baron J, & Tephly TR (1996) Folate and 10
formyltetrahydrofolate dehydrogenase in human and rat retina: relation
to methanol toxicity. Toxicol Appl Pharmacol, 141: 373-381.
Mashbitz LM, Sklianskaya RM, & Urieva FI (1936) The relative toxicity
of acetone, methyl alcohol and their mixtures: II. Their action on
white mice. J Ind Hyg Toxicol, 18: 117-122.
Mather A & Assimos A (1965) Evaluation of gas-liquid chromatography in
assays for blood volatiles. Clin Chem, 11: 1023-1035.
Matsui S, Okawa Y, & Ota R (1988) Experience of 16 years' operation
and maintenance of the Fukashiba industrial wastewater treatment plant
of the Kashima petrochemical complex: II. Biodegradability of 37
organic substances and 28 process waste waters. Water Sci Technol,
20: 201-210.
Mayer FL & Ellersieck MR (1986) Manual of acute toxicity:
Interpretation and database for 410 chemicals and 66 species of
freshwater animals. Washington, DC, US Department of the Interior,
Fish and Wildlife Service (Resource Publication No. 160).
Medinsky MA & Dorman DC (1994) Assessing risks of low-level methanol
exposure. CIIT Act, 14(7): 1-7.
Medinsky MA, Dorman DC, Bond JA, Moss OR, Janszen DB, & Everitt JI
(1997) Pharmacokinetics of methanol and formate in female cynomolgus
monkeys exposed to methanol vapours. Cambridge, Massachusetts, Health
Effects Institute (Research Report No. 77).
Menne FR (1938) Acute methyl alcohol poisoning. Arch Pathol, 26:
77-92.
Mill T, Hendry DG, & Richardson H (1980) Free-radical oxidants in
natural waters. Science, 207: 886-887.
Ministry of Supply and Services Canada (1993) Summary report 1993-A:
National pollutant release inventory-Methanol air release. Ottawa,
Ontario, Environment Canada, 11 pp.
Mohler FS & Gordon CJ (1990) Themoregulatory effects of methanol in
Fischer and Long-Evans rats. Neurotoxicol Teratol, 12: 41-45.
Mohr DH & King CJ (1985) Identification of polar organic compounds in
coal-gasification condensate water by gas-chromatography-mass
spectrometry analysis of high-performance liquid chromatography.
Environ Sci Technol, 19: 929-935.
Monte WC (1984) Aspartame: methanol and the public health. J Appl
Nutr, 36: 42-54.
Montgomery JH (1991) Groundwater chemicals desk reference. Chelsea,
Michigan, Lewis Publishers, pp 491-495.
Morin AM & Liss M (1973) Evidence for a methylated protein
intermediate in pituitary methanol formation. Biochem Biophys Res
Commun, 52: 373-378.
Morris GPL (1993) Renewed interest emerges for methanol, MTBE
projects. Chem Week, 153: 7.
Murray TG, Burton TC, Rajani C, Lewandowski MF, Burke JM, & Eells JT
(1991) Methanol poisoning: A rodent model with structural and
functional evidence for retinal involvement. Arch Ophthalmol, 109:
1012-1016.
Naraqi S, Dethlefs RF, Slobodnuik RA, & Sairere JS (1979) An outbreak
of acute methyl alcohol intoxication. Aust NZ J Med, 9: 65-68.
National Poisons Information Service (1993) Antifreeze poisoning:
Clinical effects and management. London, National Poisons Information
Service, pp 1-4.
NEDO (1982) Toxicological research of methanol as a fuel for power
station: Summary report on tests with monkeys, rats and mice. Tokyo,
Japan, New Energy Development Organization.
NEDO (1987) Toxicological research of methanol as a fuel for power
station: Summary report on tests with monkeys, rats and mice. Tokyo,
Japan, New Energy Development Organization, pp 1-296.
Nelson BK, Brightwell WS, MacKenzie DR, Khan A, Burg JR, Weigel WW, &
Goad PT (1985) Teratological assessment of methanol and ethanol at
high inhalation levels in rats. Fundam Appl Toxicol, 5: 727-736.
Nicholls P (1975) Formate as an inhibitor of cytochrome c oxidase.
Biochem Biophys Res Commun, 67: 610-616.
Nielsen IR, Malcolm HM, & Dobson SD (1993) Environmental hazard
assessment: Methanol. Garston, Watford, UK Department of the
Environment, Building Research Establishment, 45 pp.
NIOSH (1976) Criteria for a recommended standard .... Occupational
exposure to methyl alcohol. Cincinnati, Ohio, National Institute for
Occupational Safety and Health (HEW (NIOSH) Publication No. 76-148).
NIOSH (1977) NIOSH manual of analytical methods, 2nd ed. Cincinnati,
Ohio, National Institute for Occupational Safety and Health (DHEW
(NIOSH) Publication No. 77-157A).
NIOSH (1981) Health hazard evaluation report No. HETA-81-177, 178-988,
University of Washington, Seattle. Cincinnati, Ohio, National
Institute for Occupational Safety and Health.
NIOSH (1984) Method No. 2000: Methanol. In: Eller PM ed. NIOSH manual
of analytical methods. Cincinnati, Ohio, National Institute for
Occupational Safety and Health, vol 2, pp 2000/1-2000/4.
Noker PE, Eells JT, & Tephly TR (1980) Methanol toxicity: Treatment
with folic acid and 5-formyl tetrahydrofolic acid alcoholism. Clin Exp
Res, 4: 378-383.
Norman V (1977) An overview of the vapor phase, semivolatile, and
non-volatile components of cigarette smoke. Recent Adv Tob Sci, 3:
25-58.
Novak JT, Goldsmith CD, Benoit RE, & O'Brien JH (1985) Biodegradation
of methanol and tertiary butyl alcohol in subsurface systems. Water
Sci Technol, 17: 71-85.
Nyberg U, Aspegren H, Andersson B, Jansen JC, & Villadsen IS (1992)
Full-scale application of nitrogen removal with methanol as carbon
source. Water Sci Technol, 26: 1077-1086.
Obe G & Ristow H (1977) Acetaldehyde, but not ethanol induces sister
chromatid exchanges in Chinese hamster cells in vitro. Mutat Res,
56: 211-213.
Okpokwasili GC & Amanchukwu SC (1988) Petroleum hydrocarbon
degradation by candida species. Environ Int, 14: 243-247.
Oliver B, Cosgrove EG, & Carey JH (1979) Effect of suspended sediments
on the photolysis of organics in water. Environ Sci Technol, 13:
1075-1077.
Ophaswongse S & Maibach HI (1994) Alcohol dermatitis: Allergic contact
dermatitis and alcohol urtiaria syndrome. Contact Dermatitis, 30:
1-6.
Oremland RS, Marsh LM, & Polcin S (1982) Methane production and
simultaneous sulphite reduction in anoxic, salt marsh sediments.
Nature (Lond), 296: 143-145.
Owens LD, Gilbert RG, Griebel GE, & Menzies JD (1969) Identification
of plant volatiles that stimulates microbial respiration and growth in
soil. Phytopathology, 59: 1468-1472.
Palese M & Tephly TE (1975) Metabolism of formate in the rat.
J Toxicol Environ Health, 1: 13-24.
Pamies RJ, Sugar D, Rives LA, & Herold AH (1993a) Methanol
intoxication. Postgrad Med, 93: 183-194.
Pamies RJ, Sugar D, Rives L, & Herold AH (1993b) Methanol
intoxication: A case report. J Fla Med Assoc, 80: 464-467.
Pankow S & Jagielki S (1993) Effect of methanol or modifications of
the hepatic glutathione concentration on the metabolism of
dichloromethane to carbon monoxide in rats. Hum Exp Toxicol, 12:
227-231.
Pappas SC & Silverman M (1982) Treatment of methanol poisoning with
ethanol and hemodialysis. Can Med Assoc J, 126: 1391-1394.
Pappas AA, Gadsden RH, & Taylor EH (1985) Serum osmolality in acute
intoxication: A prospective clinical study. Am J Clin Pathol, 84:
74-79.
Pauls RE & McCoy RW (1981) Gas and liquid chromatographic analysis of
methanol, ethanol, t-butanol and methyl-t-butyl ether in gasoline.
J Chromatogr Sci, 19: 558-561.
Pavlenko SM (1972) [Certain common traits in the action of industrial
non-electrolyte poisons entering the body simultaneously with the
water and air.] Gig I Sanit, 37: 40-45 (in Russian).
Pearson A (1952) Acute methyl alcohol poisoning. Med J Aust, 2: 437.
Pelletier J, Habib MH, Khalil R, Salamon G, Bartoli D, & Jean P (1992)
Putaminal necrosis after methanol intoxication. J Neurol Neurosurg
Psych, 55: 234-235.
Pellizzari ED, Hartwell TD, Harris BSH III, Waddell RD, Whitaker DA, &
Erikson MD (1982) Purgable organic compounds in mother's milk. Bull
Environ Contam Toxicol, 28: 322-328.
Pereira MA, Chang LW, McMillan L, Ward JB, & Legator MS (1982) Battery
of short-term tests in laboratory animals to corroborate the detection
of human population exposures to genotoxic chemicals. Environ Mutagen,
4: 317.
Perkins RA, Ward KW, & Pollack GM (1995) A pharmacokinetic model of
inhaled methanol in humans and comparison to methanol disposition in
mice and rats. Environ Health Perspect, 103: 716-733.
Phillips M & Greenberg J (1987) Detection of endogenous ethanol and
other compounds in the breath by gas chromatography with on-column
concentration of sample. Anal Biochem, 163: 165-169.
Pienta RJ, Poiley JA, & Lebherz WB III (1977) Morphological
transformation of early passage Golden Syrian hamster embryo cells
derived from cryopreserved cultures as a reliable in vitro bioassay
for identifying carcinogens. Int J Cancer, 19: 642-655.
Pla A, Hernandez AF, Gil F, Garcia-Alonso M, & Villanueva E (1991) A
fatal case of oral ingestion of methanol: Distribution in postmortem
tissues and fluids including pericardial fluid and vitreous humor.
Forensic Sci Int, 49: 193-196.
Poirier SH, Knuth Ml, Anderson-Buchou CD, Brooke CT, Lima AR, & Shubat
PJ (1986) Comparative toxicity of methanol and N,N-dimethylformamide
to freshwater fish and invertebrates. Bull Environ Contam Toxicol,
37: 615-621.
Pollack GM & Brouwer KLR (1996) Maternal-fetal pharmacokinetics of
methanol. Cambridge, Massachusetts, Health Effects Institute, 55 pp
(Research Report No. 74).
Pollack GM & Kawagoe JL (1991) Determination of methanol in whole
blood by capillary gas chromatography with direct on-column injection.
J Chromatogr, 570: 406-411.
Pollack GM, Brouwer KLR, & Kawagoe J (1993) Toxicokinetics of
intravenous methanol in the female rat. Fundam Appl Toxicol, 21:
105-110.
Poon R, Chu IH, Bjarnason S, Potvin M, Vincent R, Miller RB, & Valli
VE (1994) Inhalation toxicity study of methanol, toluene and
methanol/toluene mixtures in rats. Toxicol Ind Health, 10: 231-245.
Poon R, Chu IH, Bjarnason S, Vincent R, Potvin M, Miller RB, & Valli
VE (1995) Short-term inhalation toxicity of methanol, gasoline and
methanol/gasoline in the rat. Toxicol Ind Health, 11: 343-361.
Portmann JE & Wilson RW (1971) The toxicity of 140 substances to the
brown shrimp and other marine animals. London, Ministry of
Agriculture, Fisheries and Food (Shellfish Information Leaflet No.
22).
Posner HS (1975) Biohazards of methanol in proposed new uses.
J Toxicol Environ Health, 1: 153-171.
Price KS, Waggy GT, & Conway RA (1974) Brine shrimp bioassay and
seawater BOD of petrochemicals. J Water Pollut Control Fed, 46:
63-77.
Price EA, d'Alessandro A, Kearney T, Olson KR, & Blanc PD (1994)
Osmolar gap with minimal acidosis in combined methanol and methyl
ethyl ketone ingestion. Clin Toxicol, 32: 79-84.
Rajini PS, Krishnakumari MK, & Majumder SK (1989) Cytotoxicity of
certain organic solvents and organophosphorus insecticides to the
ciliated protozoan Paramecium caudatum. Microbios, 59: 157-163.
Rana SVS & Kumar S (1993a) Liver function in rats treated individually
and with a combination of xylene, toluene and methanol. Toxicol Ind
Health, 9: 479-484.
Rana SVS & Kumar S (1993b) Effect of xylene, toluene and methyl
alcohol on liver collagenesis in rats. Indian J Exp Biol, 31:
782-784.
Randall TL & Knopp PV (1980) Detoxification of specific organic
substances by wet oxidation. J Water Pollut Control Fed, 52:
2117-2130.
Rastogi SC (1993) Organic solvents in model and hobby glues. Bull
Environ Contam Toxicol, 51: 501-507.
Reese E & Kimbrough RD (1994) Acute toxicity of gasoline and some
additives. Environ Geochem Health, 101(Suppl): 113-115.
Reisch MS (1994) Top 50 chemicals production rose modestly last year.
Chem Eng News, 72: 12-18.
Renzoni GE, Shankland EG, Gaines JA, & Callis JB (1985) Determination
of alcohols in gasoline/alcohol blends by nuclear magnetic resonance
spectrometry. Anal Chem, 57: 2864-2867.
Rietbrock N (1969) [Kinetics and pathways of methanol metabolism.]
Naunyn-Schmiedebergs Arch Pharmacol Exp Pathol, 263: 189-201 (in
German).
Rietbrock N, Stieren B, & Malorny G (1966) [Influence of folic acid on
methanol metabolism.]. Klin Wochenschr, 44: 1318.
Rippen G (1980) [Handbook of environmental chemicals.] Landsberg,
Ecomed Verlagsgesellschaft, 16 pp (in German).
Röe O (1948) Methanol poisoning: The ganglion cells of the retina in
cases of methanol poisoning in human beings and experimental animals.
Acta Med Scand, 26: 169-182.
Röe O (1950) The roles of alkaline salts and ethyl alcohol in the
treatment of methanol poisoning. Q J Stud Alcohol, 11: 107-112.
Röe O (1955) The metabolism and toxicity of methanol. Pharmacol Rev,
7: 399-412.
Röe O (1982) Species differences in methanol poisoning. CRC Crit Rev
Toxicol, 10: 275-286.
Roeggla G, Wagner A, Frossard M, & Roeggla H (1993) Marked variability
in methanol toxicity. Am Fam Physician, 48: 731.
Rogers JM, Mole ML, Chernoff N, Barbee BD, Turner CI, Logsdon TR, &
Kavlock RJ (1993) The developmental toxicity of inhaled methanol in
the CD-1 mouse, with quantitative dose-response modeling for
estimation of benchmark doses. Teratology, 47: 175-188.
Rossini GDB & Ronco AE (1996) Acute toxicity bioassay using Daphnia
obtusa as a test organism. Environ Toxicol Water Qual, 11(3):
255-258.
Rotman D (1994a) Lurgi unveils route to methanol from carbon dioxide
at ACS meeting. Chem Week, 154: 14.
Rotman D (1994b) New approaches rekindle methanol conversion. Chem
Week, 154: 35.
Rowe VK & McCollister SB (1982) Alcohols. In: Clayton GD & Clayton FE
ed. Parry's industrial hygiene and toxicology, 3rd ed. New York, John
Wiley and Sons, vol 2C, pp 4528-4541.
Ruedemann AD (1962) The electroretinogram in chronic methyl alcohol
poisoning in human beings. Am J Ophthalmol, 54: 34-53.
Ryan BM, Hatoum NS, Mallett ES, & Yermakoff JK (1994) A pilot toxicity
study of methanol in folate-deficient long-Evans rats. Teratology,
49: 399.
Sakanashi TM, Rogers JM, & Keen CL (1994) Influence of folic acid
intake on the developmental toxicity of methanol in the CD-1 mouse.
Teratology, 49: 368.
Sayers RR, Yant WP, & Schrenk HH (1942) Methanol poisoning exposure of
dogs to 450-500 ppm methanol vapour in air. Washington, DC, US Bureau
of Mines (Investigation Report No. 3619).
Sayers RR, Yant WP, Schrenk HH, Chornyak J, Pearce SJ, Patty FA, &
Linn JG (1944) Methanol poisoning: II. Exposure of dogs for brief
periods eight times daily to high concentrations of high methanol
vapor in air. J Ind Hyg Toxicol, 26: 255-259.
Scheunert I, Vockel D, Schnitzer J, & Korte F (1987) Bromineralization
rates of 14C-labelled organic chemicals in aerobic and anaerobic
suspended soil. Chemosphere, 16: 1031-1041.
Scheuplein RJ & Blank IH (1971) Permeability of the skin. Physiol Rev,
51: 702-747.
Schiewe MH, Hawk EG, Actor DI, & Krahn MM (1985) Use of bacterial
bioluminescence assay to access toxicity of contaminated marine
sediments. Can J Fish Aquat Sci, 42: 1244-1247.
Scholz B, Butzert H, Neumeister J, & Nierlich F (1990) Methyl tert-
butyl ether. In: Elvers B, Hawkins S, Schutz G ed. Ullmann's
encyclopedia of industrial chemistry, 5th ed. Weinheim, VCH
Verlagsgesellschaft, vol 16a, pp 543-550.
Scott E, Helz MK, & McCord CP (1933) The histopathology of methyl
alcohol poisoning. Am J Clin Pathol, 3: 311-319.
Sedivec V, Mraz M, & Flek J (1981) Biological monitoring of persons
exposed to methanol vapors. Int Arch Occup Environ Health, 48:
257-271.
Seizinger DE & Dimitriades B (1972) Oxygenates in exhaust from simple
hydrocarbon fuels. J Air Pollut Control Assoc, 22: 47-51.
Sejersted OM, Jacobsen D, Ovrebo S, & Jansen H (1983) Formate
concentration in plasma from patients poisoned with methanol. Acta Med
Scand, 213: 105-110.
Self R, Casey JC, & Swain T (1963) The low boiling volatiles of cooked
foods. Chem Ind, 1963: 863-864.
Sharpe JA, Hostovsky M, Bilbao JM, & Rewcastle NB (1982) Methanol
optic neuropathy: A histopathological study. Neurology, 32:
1093-1100.
Simmon VF, Kauhanen K, & Tardiff RG (1977) Mutagenic activity of
chemicals identified in drinking water. In: Scott D, Bridges BA, &
Sobels FH ed. Progress in genetic toxicology. Amsterdam,
Elsevier/North Holland Press, vol 2, pp 249-268.
Simmons JE & McDonald A (1994) Effect of Kupffer cell inhibition on
carbon tetrachloride hepatotoxicity in methanol pretreated rats.
Toxicologist, 14: 374.
Simmons JE, McDonald A, Seely JC & Sey YM (1995) ..... of carbon
tetrachloride hepatotoxicity by inhaled methanol: Time course of
recovery. J Toxicol Environ Health, 46: 203-216.
Sims EW (1976) Determination of trace C1-C4 alcohols in aqueous
solutions by gas chromatography. J Chromatogr Sci, 14: 65-67.
Skaug OE (1956) A rapid and extremely sensitive test for methanol in
blood and biological material. Scand J Clin Lab Invest, 8: 338-339.
Smallwood AW (1978) Analysis of formic acid in air samples. Am Ind Hyg
Assoc J, 39: 151-153.
Smith NB (1984) Determination of volatile alcohols and acetone in
serum by non-polar capillary gas chromatography after direct sample
injection. Clin Chem, 30: 1672-1674.
Smith EN & Taylor RT (1982) Acute toxicity of methanol in the folate-
deficient acatalasemic mouse. Toxicology, 25: 271-287.
Smyth HF, Seaton J, & Fischer L (1941) The single dose toxicity of
some glycols and derivatives. J Ind Hyg Toxicol, 23: 259-268.
Snider JR & Dawson GA (1985) Tropospheric light alcohols, carbonyls
and acetonitrile: Concentrations in the Southwestern United States and
Henry's law data. J Geophys Res, 90: 3797-3805.
Snow R, Baker L, Crews W, Davis CO, Duncan J, Perry N, Siudak P,
Stumpf K, Ray W, & Braddock J (1989) Characterization of emissions
from a methanol fueled motor vehicle. J Air Pollut Control Assoc,
39: 48-53.
SRI (1992) Chemical economics handbook: Marketing research report on
methanol. Menlo Park, California, SRI International.
Stanton ME, Crofton KM, Gray LE, Gordon CM, Bushnell RJ, Mole ML, &
Peale DB (1991) Assessment of offspring development and behavior
following gestational exposure to inhaled methanol in the rat.
Toxicologist, 11: 118.
Stanton ME, Crofton KM, Gray LE, Gordon CJ, Boyes WK, Mole ML, Peele
DB, & Bushnell PS (1995) Assessment of offspring development and
behaviour following gestational exposure to inhaled methanol in the
rat. Fundam Appl Toxicol, 28: 100-110.
Stegink LD, Brummel MC, McMartin KE, Martin-Amat G, Filer LJ Jr, Baker
GL, & Tephly TR (1981) Blood methanol concentrations in normal adult
subjects administered abuse doses of aspartame. J Toxicol Environ
Health, 7: 281-290.
Stegink LD, Brummel MC, Filer LJ Jr, & Baker GL (1983) Blood methanol
concentrations in one-year-old infants administered graded doses of
aspartame. J Nutr, 113: 1600-1606.
Stensel HD, Loehr RC, & Lawrence AW (1973) Biological kinetics of
suspended-growth denitrification. J Water Pollut Control Fed, 45:
249-261.
Stern S, Reuhl K, Soderholm S, Cox C, Sharma A, Balys M, Gelein R, Yin
C, & Weiss B (1996) Perinatal methanol exposure in the rat: I. Blood
methanol concentration and neural cell adhesion molecules. Fundam Appl
Toxicol, 34: 36-46.
Stratton GW (1985) The influence of solvent type on solvent-pesticide
interactions in bioassays. Arch Environ Contam Toxicol, 14: 651-658.
Stratton GW (1987) Toxic effects of organic solvents on the growth of
blue-green algae. Bull Environ Contam Toxicol, 38: 1012-1019.
Stratton GW & Smith TM (1988) Interaction of organic solvents with the
green alga Chlorella pyrenoidosa. Bull Environ Contam Toxicol, 40:
736-742.
Strittmatter P & Ball EG (1955) Formaldehyde dehydrogenase, a
glutathione-dependent enzyme system. J Biol Chem, 213: 445.
Suit PF & Estes M (1990) Methanol intoxication: clinical features and
differential diagnosis. Clevel Clin J Med, 57: 464-471.
Swain HM & Somerville HJ (1978) Microbial metabolism of methanol in a
model activated sludge system. J Appl Bacteriol, 45: 147-151.
Swartz RD, Millman RP, Billi JE, Bondar NP, Migdal SD, Simonian SK,
Monforte JR, McDonald FD, Harness JK, & Cole KL (1981) Epidemic
methanol poisoning: clinical and biochemical analysis of a recent
episode. Medicine, 60: 373-382.
Sweger DM & Travis JC (1979) An application of infrared lasers to the
selective detection of trace organic gases. Appl Spectrom, 33:
46-51.
Tackett Sl (1987) Determination of methanol in gasoline by gas
chromatography. Analyst, 112: 339-340.
Takagi H, Hatakeyama S, Akimoto H, & Koda S (1986) Formation of methyl
nitrite in the surface reaction of nitrogen dioxide and methanol:
1. Dark reaction. Environ Sci Technol, 20: 387-393.
Tephly TR (1991) Mini review-the toxicity of methanol. Life Sci, 48:
1031-1041.
Tephly TR & McMartin KE (1984) Methanol metabolism and toxicity. In:
Stegink LD & Filer LJ Jr ed. Aspartame: Physiology and biochemistry.
New York, Basel, Marcel Dekker, pp 111-140.
Tephly TR, Parks RE, & Mannering GJ (1964) Methanol metabolism in the
rat. J Pharmacol Exp Ther, 143: 292-300.
Teschke R, Masamura Y, & Lieber CS (1975) Hepatic microsomal alcohol-
oxidising system: affinity for methanol, ethanol, propanol and
butanol. J Biol Che,. 250: 7397.
Tichy M, Trcka V, Roth Z, & Krivucova M (1985) QSAR analysis and data
extrapolation among mammals in a series of aliphatic alcohols. Environ
Health Perspect, 61: 321-328.
Tonning DJ, Brooks DW, & Harlow CM (1956) Acute methyl alcohol
poisonings in 49 naval ratings. Can Med Assoc J, 74: 20-27.
Triebig G & Schaller KH (1980) A simple and reliable enzymatic assay
for the determination of formic acid in urine. Clin Chim Acta, 108:
355-360.
Tyson HH & Schoenberg MJ (1914) Experimental researches in methyl
alcohol inhalation. J Am Med Assoc, 63: 915-921.
Ubaidullaev R (1966) [Effect of small concentrations of methanol
vapours on the body of men and animals.] Gig I Sanit, 31: 9-12 (in
Russian).
UK Standing Committee of Analysts (1982) Formaldehyde, methanol and
related compounds in raw, waste and potable waters: Methods for the
examination of waters and associated materials, London, UK Standing
Committee of Analysts, Her Majesty Stationery Office.
Uotila L & Koivusalo M (1974) Formaldehyde dehydrogenase from human
liver: Purification, properties and evidence for the formation of
glutathione thiol esters by the enzyme. J Biol Chem, 249: 7653.
Upadhyay S & Gupta VK (1984) Reagent system for the spectrophotometric
determination of methanol in environmental and biological samples.
Analyst, 109: 1427-1429.
Upton J (1993) Denitrification of sewage effluents in deep bed sand
filters. Water Sci Technol, 27: 381-390.
US EPA (1975) Identification of organic compounds in effluents from
industrial sources. Washington, DC, US Environmental Protection
Agency, Office of Toxic Substances (EPA 560-3-75-002).
US EPA (1976a) Assessment of methyl alcohol as a potential air
pollution problem - Volume II. Research Triangle Park, North Carolina,
US Environmental Protection Agency (NTIS Publication No. PB-258354).
US EPA (1976b) Frequency of organic compounds identified in water.
Washington, DC, US Environmental Protection Agency (EPA 600/4-76-062).
US EPA (1977) Multimedia environmental goals for environmental
assessment - Volume II: MEG, charts and background information.
Washington, DC, US Environmental Protection Agency, pp E28-E29 (EPA
600/7-77-126b).
US EPA (1979) Atmospheric reaction products of organic compounds.
Washington, DC, US Environmental Protection Agency, 80 pp (Report No.
PB 301-384, prepared by SRI International, Menlo Park, California).
US EPA (1980a) Chemical hazard information profiles (CHIPS).
Washington, DC, US Environmental Protection Agency (EPA 560/11-80-
011).
US EPA (1980b) Organic chemical manufacturing - Volume 9: Selected
processes. Research Triangle Park, North Carolina, US Environmental
Protection Agency (EPA 450/3-80-028d).
US EPA (1983) Toxicity and metabolism studies with EPA priority
pollutants and related chemicals in freshwater organisms. Washington,
DC, US Environmental Protection Agency.
US EPA (1988) Toxic air pollutant emission factors: A compilation for
selected air toxic compounds and sources. Research Triangle Park,
North Carolina, US Environmental Protection Agency (EPA 450/2-88-006).
US EPA (1991) Urban formaldehyde and methanol concentrations for
alternative methanol vehicle scenarios. Research Triangle Park, North
Carolina, US Environmental Protection Agency (Report No. CRC-APRAC-
ME-2).
US EPA (1993) Ambient concentration summaries for Clean Air Act, Title
III: Hazardous air pollutants. Washington, DC, US Environmental
Protection Agency, p B-17 (EPA 600/R-94-090).
US EPA (1994) 1992 Toxic release inventory: Public data. Washington,
DC, US Environmental Protection Agency (EPA 745/R-94-001).
US NIOSH (1976) Recommended standard for occupational exposure to
methyl alcohol. Cincinnati, Ohio, National Institute for Occupational
Safety and Health, 136 pp.
Vaishnav DD & Korthals ET (1990) Comparative toxicities of selected
industrial chemicals to microorganisms and other aquatic organisms.
Arch Environ Contam Toxicol, 19: 624-628.
Vaishnav DD & Lopas DM (1985) Relationship between lipophilicity and
biodegradation inhibition of selected industrial chemicals. Dev Ind
Microbiol, 26: 557-565.
Veith GD, Call DJ, & Brooke LT (1983) Structure-toxicity relationships
for the fat-head minnow, Pimephales promelas: narcotic industrial
chemicals. Can J Fish Aquat Sci, 40: 743-748.
Vendilo MV, Egorov YL, & Feldman NG (1971) [The effects of methanol
and of some higher alcohols on the retina of the eyes (an electron-
microscope investigation).] Gig Tr Prof Zabol, 15: 17-21 (in
Russian).
Venkataraman ES, Ahlert RC, & Corbo P (1984) Biological treatment of
landfill leachates. CRC Crit Rev Environ Control, 14: 333-376.
Verma P & Gupta VK (1984) A sensitive spectrophotometric method for
the determination of methyl alcohol in air and water. Talanta, 31:
394-396.
Vogt MJ, Heffner JE, & Sahn SA (1993) Vomiting, abdominal pain and
visual disturbances in a 31-year old man. Chest, 103: 262-263.
Ward KW & Pollack GM (1996a) Comparative toxicokinetics of methanol in
pregnant and nonpregnant rodents. Drug Metab Dispos, 24: 1062-1070.
Ward KW & Pollack GM (1996b) Use of intrauterine microdialysis to
investigate methanol-induced alterations in uteroplacental blood flow.
Toxicol Appl Pharmacol, 140: 203-210.
Ward JB, Hokanson JA, Smith ER, Chang LW, Pereira MA, Whorton EB, &
Legator MS (1984) Sperm count, morphology and fluorescent body
frequency in autopsy workers exposed to formaldehyde. Mutat Res,
130: 417-424.
Ward KW, Perkins RA, Kawagoe JL, & Pollack GM (1995) Comparative
toxicokinetics of methanol in the female mouse and rat. Fundam Appl
Toxicol, 26: 258-264.
Weese H (1928) Vergleichende untersuchungen uber die wirksamkeit und
giftigkeit der dampfe niederer aliphatischer alkohole. Arch Exptl
Pathol Pharmacol, 135: 118-130 [in German].
Weiss B, Stern S, Soderholm SC, Cox C, Sharma A, Inglis GB, Preston R,
Balys M, Reuhl KR, & Gelein R (1996) Developmental neurotoxicity of
methanol exposure by inhalation in rats. Cambridge, Massachusetts,
Health Effects Institute (Research Report No. 73).
Welch H & Slocum GG (1943) Toxicity of methanol. J Lab Chem Med, 28:
1440-1445.
Wenzyl JE, Mills SD, & McCall JT (1968) Methanol poisoning in an
infant. Am J Dis Child, 116: 445-447.
Whitbeck M (1983) Photo-oxidation of methanol. Atmos Environ, 17:
121-126.
White LR, Martinsen ABL, & Nilsen OG (1983) Biochemical and
cytological studies of rat lung after inhalation of methanol vapour.
Toxicol Lett, 17: 1-5.
WHO (1971) Evaluation of food additives. Fourteenth report of the
Joint FAO/WHO Expert Committee on Food Additives. Geneva, World Health
Organization, p 20 (WHO Technical Report Series No. 462).
Williams RL, Lipari F, & Potter RA (1990) Formaldehyde, methanol and
hydrocarbon emissions from methanol-fueled cars. J Air Waste Manage
Assoc, 40: 747-756.
Windholz M ed. (1983) The Merck index: An encyclopedia of chemicals,
drugs and biologicals, 10th ed. Rahway, New Jersey, Merck & Co.,Inc.,
p 853.
Wood CA & Buller F (1904) Poisoning by wood alcohol. J Am Med Assoc,
43: 973-977.
Wu Chen NB, Donoghue ER, & Schaffer MI (1985) Methanol intoxication:
Distribution in postmortem tissues and fluids including vitreous
humor. J Forensic Sci, 30: 213-216.
Wucherpfennig K, Dietrich H, & Bechtel J (1983) Alcohol actual, total
and potential methyl alcohol of fruit juices. Flussiges Obst, 8:
348-354.
Yant WP & Schrenk HH (1937) Distribution of methanol in dogs after
inhalation and administration by stomach tube and subcutaneously.
J Ind Hyg Toxicol, 19: 337-345.
Yasugi T, Kawai T, Mizunuma K, Horiguchi S, Iwami O, Iguchi H, & Ikeda
M (1992) Formic acid excretion in comparison with methanol excretion
in urine of workers occupationally exposed to methanol. Int Arch Occup
Environ Health, 64: 329-337.
Youssef AF, Baggs RB, Weiss B, & Miller RK (1991) Methanol
teratogenicity in pregnant Long-Evans rats. Teratology, 43: 467.
Youssef AF, Madkour K, Cox C, & Weiss B (1992) Comparative lethality
of methanol, ethanol and mixtures in female rats. J Appl Toxicol,
12: 193-197.
Youssef AF, Weiss B, & Cox C (1993) Neurobehavioural toxicity of
methanol reflected by operant running. Neurotoxicol Teratol, 15:
223-227.
Zinbo M (1984) Determination of one-carbon to three-carbon alcohols
and water in gasoline/alcohol blends by liquid chromatography. Anal
Chem, 56: 244-247.
RESUME
1. Identité, propriétés physiques et chimiques et méthodes d'analyse
Le méthanol se présente sous la forme d'un liquide incolore et
limpide qui dégage une légère odeur alcoolique à l'état pur. Volatil
et inflammable, il est miscible à l'eau et à de nombreux solvants
organiques et forme un grand nombre d'azéotropes binaires.
Il existe un certain nombre de méthodes, principalement la
chromatographie en phase gazeuse avec détection par ionisation de
flamme, pour la recherche et le dosage du méthanol dans divers
échantillons prélevés dans l'environnement (air, eau, sol, et
sédiments) ou dans les produits alimentaires. Ces méthodes sont
également utilisées pour la recherche et le dosage du méthanol et de
son principal métabolite, le formiate, dans les liquides et les tissus
biologiques. Outre la chromatographie en phase gazeuse ave détection
par ionisation de flamme, il existe des méthodes enzymatiques
colorimétriques pour le dosage du formiate dans le sang, les urines et
les tissus.
Pour les analyses sur le lieu de travail, on commence
généralement par recueillir et concentrer l'échantillon sur gel de
silice, après quoi on procède à une extraction par l'eau, puis au
dosage proprement dit par chromatographie en phase gazeuse avec
détection par ionisation de flamme ou spectrométrie de masse.
2. Sources d'exposition humaine
Le méthanol est présent à l'état naturel chez l'Homme, les
animaux et les plantes. C'est un constituant du sang, de l'urine, de
la salive et de l'air expiré. On a mesuré des concentrations moyennes
de méthanol égales à 0,73 mg/litre dans les urines (valeurs
extrêmes:0,3-2,61 mg/litre) chez des sujets non exposés et des valeurs
allant de 0,06 à 0,32 µg/litre ont été observées dans l'air expiré.
Le méthanol et le formiate naturellement présents dans
l'organisme proviennent essentiellement de deux sources:
l'alimentation et le métabolisme. Le méthanol d'origine alimentaire
est principalement apporté par les fruits et les légumes frais ainsi
que par les jus de fruits (teneur moyenne: 140 mg/litre avec des
valeurs extrêmes de 12-640 mg/litre), les boissons fermentées
(jusqu'à 1,5 g/litre), et autres composants du régime alimentaire
(principalement les boissons non alcoolisées). L'aspartame est un
édulcorant très utilisé dont l'hydrolyse donne du méthanol absorbable
dans la proportion de 10% en poids.
En 1991, la production mondiale de méthanol a atteint environ 20
millions de tonnes, principalement par conversion catalytique de gaz
de synthèse sous pression (hydrogène, dioxyde et monoxyde de carbone).
La capacité mondiale de production devrait atteindre 30 millions de
tonnes en 1995.
Le méthanol est utilisé dans l'industrie pour la production
de nombreux produits chimiques importants, principalement le
méthyltertiobutyléther, le formaldéhyde, l'acide acétique, les éthers
méthyliques du glycol, la méthylamine, les halogénures de méthyle et
le méthacrylate de méthyle.
Le méthanol entre dans la composition de nombreux solvants du
commerce et de divers produits comme les peintures, les laques, les
vernis, les diluants pour peintures, les détachants, les antigels,
les liquides pour pare-brise, les dégivrants, les produits pour la
photocopie, les solutions destinées à la dénaturation de l'éthanol
ainsi que différent types de colles. Le méthanol pourrait également
être utilisé directement comme combustible, ou bien encore être ajouté
à l'essence à titre de combustible auxiliaire ou de diluant. Il est à
noter que les cas les plus fréquents d'intoxication, mortelle ou non,
par le méthanol, sont dus à l'ingestion volontaire ou accidentelle de
produits qui en contiennent.
On a trouvé du méthanol dans les gaz d'échappement des moteurs à
essence et des moteurs diesel ainsi que dans la fumée de tabac.
3. Concentrations dans l'environnement et exposition humaine
Les émissions de méthanol proviennent essentiellement des divers
usages qui en sont faits en tant que solvant industriel ou domestique,
des unités de production du composé lui-même ou de ses dérivés, enfin
des pertes lors du stockage ou de la manipulation.
Il peut y avoir exposition au méthanol sur le lieu de travail par
inhalation ou contact cutané. A en juger d'après les limites
d'exposition fixées par de nombreux pays, il semblerait que les
travailleurs ne courent pas de danger tant que l'exposition exprimée
en moyenne pondérée par rapport au temps ne dépasse pas 260 mg/m3
(200 ppm) par journée de 8 h et semaine de 40 h.
Actuellement la population est exposée à des concentrations qui
sont 10 000 fois inférieures aux limites d'exposition professionnelle.
En ce qui concerne l'exposition au méthanol contenu dans l'air, les
concentrations vont de 0,001 mg/m3 (0,8 parties par milliard) en
milieu rural, à près de 0,04 mg/m3 (30 parties par milliard) en
milieu urbain.
On ne possède guère de données sur la teneur en méthanol de l'eau
de boisson après traitement, mais ce composé est en tout cas souvent
présent dans les effluents industriels.
Si les prévisions d'utilisation du méthanol comme combustible de
substitution ou d'appoint augmentent de façon sensible, il faut
s'attendre à ce que l'exposition à ce composé se généralise par
suite de l'inhalation des vapeurs émises par les véhicules qui
l'utiliseront, ou encore de son siphonage ou de son absorption
percutanée lors de la manipulation de combustibles qui en
contiendront.
4. Distribution et transformation dans l'environnement
Le méthanol se décompose rapidement dans l'environnement par
photooxydation et sous l'action de processus de biodégration. Dans le
cas de la réaction atmosphérique du méthanol avec les radicaux
hydroxyle, on a mesuré une demi-vie de 7 à 18 jours.
De nombreux genres et souches de microorganismes sont capables
d'utiliser le méthanol comme substrat. Le composé est facilement
dégradé en aérobiose ou en anaérobiose dans des milieux très divers,
notamment les eaux douces ou salées, les sols et les sédiments,
les eaux souterraines, les nappes phréatiques et les effluents
industriels. En général, 70% du méthanol présent dans les eaux d'égout
est décomposé en l'espace de 5 jours.
Le méthanol sert normalement de substrat à de nombreux
microorganismes terricoles, qui sont capables de le dégrader
complètement en dioxyde carbone et en eau.
Le méthanol est médiocrement absorbé par les sols. Sa
bioaccumulation est faible dans la plupart des organismes.
Le méthanol est peu toxique pour les organismes aquatiques et
terrestres et il est peu probable que l'on observe des effets
résultant d'une exposition environnementale à ce composé, sauf en cas
de déversements dans la nature.
5. Absorption, distribution, biotransformation et élimination
Après inhalation, ingestion ou contact cutané, le méthanol est
facilement résorbé et se diffuse rapidement dans les tissus en
fonction de la répartition de l'eau dans l'organisme. Une faible
proportion est excrétée telle quelle par les poumons et les reins.
Après ingestion, les concentrations sériques maximales sont
atteintes en 30 à 90 minutes et le méthanol se répartit dans
l'organisme avec un volume de distribution d'environ 0,6 litre/kg.
Le méthanol est métabolisé principalement au niveau du foie
selon un processus oxydatif qui le transforme successivement en
formaldéhyde, acide formique et dioxyde de carbone. La première étape,
celle de l'oxydation en formaldéhyde, s'effectue sous l'action de
l'alcool-déshydrogénase hépatique; il s'agit d'une étape limitante qui
correspond à un processus saturable. L'affinité relative de l'alcool-
déshydrogénase pour le méthanol et pour l'éthanol est d'environ
20:1. Lors de la seconde étape, le formaldéhyde est oxydé par la
formaldéhyde - déshydrogénase en acide formique ou en formiate, selon
la valeur du pH. La troisième étape consiste dans la détoxication de
l'acide formique en dioxyde de carbone par des réactions dépendant de
l'acide folique.
L'élimination du méthanol présent dans le sang par la
voie urinaire ou dans l'air expiré, soit tel quel, soit après
métabolisation, se révèle être un processus lent chez toutes les
espèces, en particulier par comparaison avec l'éthanol. Ainsi, la
clairance du méthanol s'effectue avec une demi-vie de 24 h ou
davantage pour des doses inférieures à 0,1 g/kg. C'est au niveau de la
détoxication métabolique, c'est-à-dire de l'élimination du formiate,
que des différences très importantes existent entre les rongeurs et
les primates et ce sont elles qui expliquent la différence
spectaculaire de toxicité que l'on constate entre les premiers et les
seconds.
6. Effets sur les mammifères de laboratoire et les systèmes d'épreuve
in vitro
6.1 Toxicité générale
La toxicité aiguë et la toxicité à court terme du méthanol
varient beaucoup selon les diverses espèces et alles sont maximales
chez celles qui métabolisent relativement mal le formiate. En pareil
cas, le méthanol provoque une intoxication mortelle par acidose
métabolique et toxicité neuronale. En revanche, chez les animaux qui
métabolisent bien le formiate, la mort survient habituellement par
suite de la dépression du système nerveux central (coma, insuffisance
respiratoire etc.). Chez les primates sensibles (comme l'Homme et les
singes), il y a augmentation du taux sanguin de formiate après
exposition au méthanol, alors que chez les rongeurs résistants, les
lapins et les chiens, cette augmentation du taux de formiate ne se
produit pas. L'Homme et les primates non humains présentent une
sensibilité unique aux effets toxiques du méthanol. Globalement, le
méthanol est peu toxique pour les animaux autres que les primates. La
valeur de la DL50 et de la dose létale minimale pour une exposition
par la voie orale, varie de 7000 à 13 000 mg/kg chez le rat, la
souris, le lapin et le chien et de 2000 à 7000 mg/kg chez le singe.
Chez des rats exposés à du méthanol 6 h par jour, 5 jours par
semaine pendant 4 semaines, à des concentrations allant jusqu'à
6500 mg/m3 (5000 ppm), on n'a observé aucun effet imputable à
l'exposition, sauf une augmentation des écoulements au niveau du nez
et des yeux. On estime qu'il s'agissait là de la conséquence d'une
irritation des voies respiratoires supérieures.
Des rats exposés à des vapeurs de méthanol à des concentrations
pouvant atteindre 13 000 mg/m3 (10 000 ppm), 6 h par jour, 5 jours
par semaine pendant 6 semaines, n'ont pas présenté de signes de
toxicité pulmonaire.
Chez le lapin, le méthanol irrite modérément la muqueuse
oculaire. Lors d'une épreuve qui était une variante du test de
maximalisation, il n'a pas provoqué de sensibilisation cutanée.
Parmi les effets toxiques du méthanol observés chez les primates,
on peut citer l'acidose métabolique et la toxicité oculaire qui ne se
produisent en principe pas chez les rongeurs dont le taux de folate
est suffisant. Ces différences de toxicité s'expliquent par des
différences dans la vitesse de métabolisation du formiate, qui est un
métabolite du méthanol. Par exemple, la clairance du formiate sanguin
est au moins 50% plus lente chez les primates que chez les rongeurs.
Des singes qui recevaient du méthanol par gavage à des doses
dépassant 3000 mg/kg ont présenté une ataxie, de la faiblesse et une
léthargie dans les quelques heures suivant l'administration du
composé. Ces signes avaient tendance à disparaître en l'espace de 24 h
et ils étaient suivis d'un coma passager chez certains des animaux.
Chez des singes exposés à du méthanol à 20 reprises 6 h par jour
et 5 jours par semaines, à la dose de 6500 mg/m3 (5000 ppm), on n'a
pas constaté d'effets oculaires.
6.2 Génotoxicité et cancérogénicité
Les tests de mutation génétique effectués avec du méthanol sur
des bactéries et des levures ont donné des résultats négatifs, mais le
composé à provoqué une ségrégation chromosomique défectueuse chez
Aspergillus. Il n'a pas provoqué d'échanges de chromatides soeurs
dans des cellules de hamster chinois in vitro, mais il a augmenté
de façon sensible la fréquence des mutations dans des cellules
lymphomateuses de souris L5178Y.
L'inhalation de méthanol n'a pas provoqué de lésions chromo-
somiques chez la souris. Par contre, on est fondé à penser, dans une
certaine mesure, que l'administration intrapéritonéale ou buccale de
méthanol augmente l'incidence des lésions chromosomiques chez la
souris.
Rien n'indique, au vu de l'expérimentation animale, que le
méthanol soit cancérogène, mais il faut admettre qu'il n'existe pas de
modèle animal approprié pour ce genre d'étude.
6.3 Toxicité pour la fonction reproductrice, embryotoxicité et
tératogénicité
Des études concernant les effets sur les taux de gonadotrophine
et de testostérone d'une exposition au méthanol, par la voie
respiratoire, pendant des périodes allant jusqu'à 6 semaines, ont
donné des résultats contradictoires.
En faisant inhaler du méthanol à des rongeurs gravides pendant
toute la période de l'embryogénèse, on obtient toute une série
d'effets tératogènes et embryocides qui dépendent de la concentration.
Ainsi, on a observé des malformations attribuables au traitement et
consistant principalement dans la présence de côtes cervicales
surnuméraires ou rudimentaires, ou encore de malformations urinaires
ou cardiovasculaires, chez des foetus de rats exposés 7 h par jour du
7iéme au 15 ième jour de la gestation à une concentration de
26 000 mg/m3, soit l'équivalent de 20 000 ppm, de méthanol. A cette
concentration, le méthanol était légèrement toxique pour les mères. En
revanche, à la concentration de 6500 mg/m3 (5000 ppm), aucun effet
indésirable n'a été noté chez les mères ou chez leur progéniture et on
a considéré que cette valeur constituait la concentration sans effet
nocif observable (NOAEL) pour ce système d'épreuve.
Dans la progéniture de souris CD-1 exposées à du méthanol à des
concentrations supérieures ou égales à 6500 mg/m3 (5000 ppm), 7 h par
jour du 6 ième au 15 ième jour de la gestation, on a observé une
incidence accrue d'exencéphalies et de fissures de la voûte palatine.
Aux concentrations supérieures ou égales à 9825 mg/m3 (7500 ppm), les
résorptions affectant la totalité de la portée étaient également plus
fréquentes. Aux concentrations de 13 000 et 19 500 mg/m3 (10 000
ou 15 000 ppm), on a observé une réduction du poids foetal. La
concentration sans effet observable (NOAEL) sur le développement a été
évaluée à 1300 mg/m3 (1000 ppm). Aux concentrations inférieures à
9000 mg/m3 (7000 ppm), rien n'a été relevé qui puisse indiquer une
toxicité du méthanol pour les mères.
En donnant à la progéniture de ces souris CD-1 une dose de 4 g/kg
de méthanol par gavage, on a constaté que l'incidence des effets
nocifs (résorptions, fissures palatines et réduction du poids foetal)
était analogue à celle constatée dans le groupe de rats auquels on
avait fait inhaler le composé à la concentration de 13 000 mg/m3
(10 000 ppm), probablement en raison de la fréquence respiratoire plus
élevée chez la souris. La souris est plus sensible que le rat aux
effets toxiques que le méthanol inhalé exerce sur le développement.
Des signes neurologiques passagers et une réduction du poids
corporel ont été enregistrés chez des souris CD-1 gravides, exposées
6 h par jour à une concentration de 19 500 mg/m3, soit l'équivalent
de 15 000 ppm tout au long de l'organogénèse (du sixième au quinzième
jour). Parmi les malformations foetales observées aux doses de 19 500
et 13 000 mg/m3, soit 15 000 et 10 000 ppm, on peut citer des
anomalies neurales et oculaires, des fissures palatines, des
hydronéphroses et des malformations des membres.
7. Effets sur l'Homme
L'Homme (et les primates non humains) présentent une sensibilité
unique au méthanol et les effets toxiques relevés chez ces espèces
sont caractérisés par une acidémie formique, une acidose métabolique,
une toxicité oculaire, une dépression du système nerveux, la cécité,
le coma et la mort. Presque toutes les données que l'on possède sur la
toxicité du méthanol pour l'Homme, ont trait aux conséquences des
intoxications aiguës plutôt qu'à celles des intoxications chroniques.
La très grande majorité des intoxications par le méthanol résultent de
la consommation de boissons frelatées et de produits contenant du
méthanol. C'est par ingestion que se produisent la plupart de ces
intoxications, mais l'inhalation de vapeurs de méthanol sous forte
concentration et l'absorption percutanée de solutions méthanoliques
conduisent aux mêmes effets toxiques que l'ingestion. Les effets
toxiques les plus fréquemment notés à la suite d'une exposition de
longue durée, sont des effets oculaires très variés.
Les effets toxiques du méthanol sont liés aux facteurs qui
régissent la conversion du méthanol en acide formique et la
transformation ultérieure de ce dernier en dioxyde de carbone par la
voie des folates. Ces effets se manifestent lorsque la vitesse de
formation du formiate est supérieure à sa vitesse de métabolisation.
On ne sait pas avec certitude quelle est la dose mortelle pour
l'Homme. En l'absence d'intervention médicale, la dose létale minimum
se situe entre 0,3 et 1 g/kg. On ignore quelle est dose minimale à
partir de laquelle se produisent des lésions oculaires permanentes.
L'acidose métabolique est de gravité variable et alle n'est pas
forcément en bonne corrélation avec la quantité de méthanol ingérée.
Les intoxications méthanoliques se caractérisent par de grandes
variations individuelles dans la dose toxique.
Il semble que deux facteurs importants déterminent la sensibilité
humaine aux effets toxiques du méthanol: 1) l'ingestion simultanée
d'éthanol, qui retarde l'entrée du méthanol dans sa voie de
dégradation métabolique; 2) le bilan des folates hépatiques, dont
dépend la vitesse de détoxication du formiate.
Les symptômes de l'intoxication méthanolique, qui peuvent ne se
manifester qu'au bout de 12 à 24 h, consistent en troubles visuels,
nausées, douleurs abdominales et musculaires, étourdissements,
faiblesse et troubles de la conscience allant du coma au crises
cloniques. Les troubles visuels apparaissent généralement dans les
12 à 18 h suivant l'ingestion de méthanol et vont d'une légère
photophobie avec une vision floue ou voilée à une réduction importante
de l'acuité visuelle, voire à la cécité totale. Dans les cas extrêmes,
l'intoxication peut avoir une issue fatale. Sur le plan clinique, la
principale manifestation est une acidose métabolique grave par
augmentation du trou anionique. L'acidose est largement attribuée à
l'acide formique résulant de la métabolisation du méthanol.
La concentration sanguine normale du méthanol d'origine endogène
est inférieure à 0,5 mg/litre (0,02 mmol/litre), mais l'alimentation
peut accroître le taux sanguin de méthanol. En général, les effets
neurologiques centraux apparaissent lorsque la concentration sanguine
du méthanol dépasse 200 mg/litre (6 mmol/litre); les symptômes
oculaires se manifestent à partir de 500 mg/litre (16 mmol/litre) et
la mort est survenue chez des patients non traités dont les taux
sanguins initiaux de méthanol se situaient entre 1500 et
2000 mg/litre, soit 47 à 62 mmol/litre.
L'inhalation occasionnelle de vapeurs de méthanol à une
concentration inférieure à 260 mg/m3 ou l'ingestion du liquide en
quantités ne dépassant pas 20 mg/kg, ne devraient pas conduire à une
accumulation de formiate supérieure au taux endogène, s'agissant de
sujets en bonne santé ou présentant un déficit modéré en folate. Des
troubles visuels de divers types (vision floue, rétrécissement du
champ visuel, modification de la perception des couleurs et cécité
temporaire ou permanente) ont été signalés chez des travailleurs
exposés à des concentrations de méthanol dans l'air inférieures ou
égales à environ 1500 mg/m3 (1200 ppm).
On utilise largement la valeur de 260 mg/m3 (200 ppm) comme
limite d'exposition professionnelle au méthanol. Cette valeur a été
calculée pour protéger les travailleurs contre l'acidose formique
induite par le méthanol et contre les effets toxiques de ce composé
sur l'oeil et le système nerveux.
On n'a pas signalé chez l'Homme d'autres effets nocifs qu'une
légère irritation cutanée et oculaire aux concentrations très
supérieures à 260 mg/m3 (200 ppm).
8. Effets sur les êtres vivants dans leur milieu naturel
Pour les organismes aquatiques, la valeur de la CL50 varie de
1300 à 15 900 mg/litre dans le cas des invertébrés (exposition de 48 h
et de 96 h), et de 13 000 à 29 000 mg/litre dans le cas des poissons
(exposition de 96 h).
Le méthanol est peu toxique pour les organismes aquatiques et il
n'est guère probable que l'on observe des effets imputables à une
exposition environnementale, sauf en cas de déversement de méthanol
dans la nature.
RESUMEN
1. Identidad propiedades físicas y químicas y métodosanalíticos
El metanol es un líquido transparente, incoloro, volátil e
inflamable con un ligero olor alcohólico en estado puro. Se puede
mezclar con el agua y con muchos disolventes orgánicos y forma
numerosas mezclas azeotrópicas binarias.
Hay métodos analíticos, principalmente la cromatografía de
gases (CG) con detección por ionización de llama (DIL), para la
determinación del metanol en diversos medios (aire, agua, suelo y
sedimentos) y productos alimenticios, así como para la determinación
del metanol y de su principal metabolito, el formiato, en los líquidos
y tejidos corporales. Además de la CG-DIL, en la determinación del
formiato en la sangre, la orina y los tejidos se utilizan
procedimientos enzimáticos con resultados finales colorimétricos.
Para la determinación del metanol en el lugar de trabajo se suele
comenzar con la recolección y concentración en silicagel, seguida de
extracción acuosa y CG-DIL o análisis de CG-espectrometría de masa del
extracto.
2. Fuentes de exposición humana
El metanol está presente de forma natural en el ser humano, los
animales y las plantas. Es un elemento constitutivo natural en la
sangre, orina, la saliva y el aire expirado. Se ha descrito una
concentración media de metanol en orina de 0,73 mg/litro (intervalo de
0,3-2,61 µg/litro) en individuos no expuestos y una gama de 0,06 a
0,32 µg/litro en el aire expirado.
Las dos fuentes más importantes de acumulación básica de
metanol y formiato en el organismo son la alimentación y los
procesos metabólicos. El metanol está disponible en la alimentación
principalmente a partir de las frutas y hortalizas frescas, los zumos
de fruta (promedio de 140 mg/litro, margen de variación de 12 a
640 mg/litro), las bebidas fermentadas (hasta 1,5 g/litro) y los
alimentos de dieta (sobre todo bebidas no alcohólicas). El aspartame
es un edulcorante artificial muy utilizado, y al hidrolizarse el 10%
(por peso) de la molécula se convierte en metanol libre, que queda
disponible para la absorción.
En 1991 se produjeron en todo el mundo alrededor de 20 millones
de toneladas de metanol, fundamentalmente por conversión catalítica de
gas de síntesis a presión (hidrógeno, anhídrido carbónico y monóxido
de carbono). Según las proyecciones, la capacidad mundial se elevaría
a 30 millones de toneladas en 1995.
El metanol se utiliza en la producción industrial de numerosos
compuestos orgánicos importantes, sobre todo metil terbutil éter
(MTBE), formaldehído, ácido acético, éteres de metilglicol,
metilamina, haluros de metilo y metacrilato de metilo.
El metanol es un elemento constitutivo de un gran número de
disolventes y productos de consumo disponibles en el comercio, como
pinturas, gomas laca, barnices, diluyentes de pinturas, soluciones
limpiadoras, soluciones anticongelantes, líquidos limpiaparabrisas y
anticongelantes para automóviles, líquidos de multicopista,
desnaturalizante para el etanol y pegamento para actividades de
pasatiempo y artesanía. Una aplicación potencialmente en gran escala
del metanol está en su uso directo como combustible, mezclado con
gasolina o para aumentar su volumen. Hay que señalar que la mayor
morbilidad y mortalidad se ha relacionado con la ingestión oral
deliberada o accidental de mezclas con contenidos de metanol.
Se ha detectado metanol en los gases de escape de motores tanto
de gasolina como diésel y en el humo del tabaco.
3. Niveles ambientales y exposición humana
Las emisiones de metanol se derivan principalmente de los
diversos usos industriales y domésticos como disolvente, su
producción, la manufactura final y las pérdidas durante el
almacenamiento a granel y la manipulación.
Pueden darse exposiciones al metanol en los lugares de trabajo
mediante inhalación o contacto cutáneo. Muchos de los límites
nacionales de exposición para la higiene del trabajo parecen indicar
que los trabajadores están protegidos de cualquier efecto adverso si
la exposición no supera un promedio ponderado por el tiempo de
260 mg/m3 (200 ppm) de metanol en cualquier jornada de trabajo
de 8 horas y en una semana laboral de 40 horas.
La exposición general actual de la población por medio del aire
es normalmente 10 000 veces inferior a los límites ocupacionales.
La población general está expuesta al metanol en el aire a
concentraciones que oscilan entre menos de 0,001 mg/m3 (0,8 ppm)
en el aire del medio rural y cerca de 0,04 mg/m3 (30 ppm) en el
aire urbano.
Los datos sobre la presencia del metanol en el agua potable de
uso inmediato son limitados, pero con frecuencia se encuentra metanol
en efluentes industriales.
En el caso de que el uso previsto del metanol como
combustible alternativo o mezclado con otros combustibles aumente
considerablemente, cabe prever que habrá una exposición generalizada
al metanol por medio de la inhalación de vapores procedentes de los
vehículos que funcionen con él y del bombeo o la absorción percutánea
de combustibles o mezclas de metanol.
4. Distribución y transformación en el medio ambiente
El metanol se degrada fácilmente en el medio ambiente mediante
procesos de fotooxidación y biodegradación. Se han descrito semividas
de 7-18 días para la reacción atmosférica del metanol con radicales
oxhidrilo.
Hay muchos géneros y cepas de microorganismos capaces de utilizar
el metanol como sustrato de crecimiento. El metanol es fácilmente
degradable en condiciones tanto aerobias como anaerobias en una amplia
variedad de medios naturales, entre ellos agua dulce y salada,
sedimentos y suelos, agua freática, material de acuíferos y aguas
residuales industriales; el 70% del metanol de los alcantarillados se
suele degradar en un plazo de 5 días.
El metanol es un sustrato de crecimiento normal de muchos
microorganismos del suelo, que son capaces de degradarlo completamente
a anhídrido carbónico y agua.
El metanol tiene una capacidad de absorción bastante baja en los
suelos. La bioconcentración en la mayoría de los organismos es escasa.
El metanol es poco tóxico para los organismos acuáticos y
terrestres y no es probable que se observen efectos debidos a su
exposición en el medio ambiente, excepto en el caso de un derrame.
5. Absorción, distribución, biotransformación yeliminación
El metanol se absorbe fácilmente por inhalación, ingestión y
exposición cutánea y se distribuye rápidamente en los tejidos
siguiendo la distribución del agua corporal. Por los pulmones y los
riñones se excreta una pequeña cantidad de metanol sin cambios.
Tras la digestión se alcanzan niveles máximos en suero en un
plazo de 30-90 minutos, y se reparte por todo el organismo con un
volumen de distribución aproximado de 0,6 litros/kg.
El metanol se metaboliza principalmente en el hígado siguiendo
una fase oxidativa secuencial a formaldehído, ácido fórmico y
anhídrido carbónico. El paso inicial consiste en la oxidación a
formaldehído por acción de la alcohol deshidrogenasa hepática, que
es un proceso saturable limitante de la velocidad. La afinidad
relativa de la alcohol deshidrogenasa por el etanol y el metanol es
aproximadamente de 20:1. En el segundo paso, el formaldehído se oxida
por acción de la formaldehído deshidrogenasa a ácido fórmico o
formiato, en función del pH. En el tercer paso, el ácido fórmico se
destoxifica a anhídrido carbónico mediante reacciones dependientes del
folato.
La eliminación del metanol de la sangre a través de la orina y el
aire exhalado y por el metabolismo parece ser lenta en todas las
especies, especialmente si se compara con el etanol. En el proceso se
han descrito períodos de semieliminación de 24 horas o más con dosis
superiores a 1 g/kg y de 2,5-3 horas para dosis inferiores a 0,1 g/kg.
El ritmo de desintoxicación metabólica o eliminación del formiato sí
es muy distinto entre los roedores y los primates, constituyendo la
base de las enormes diferencias de toxicidad del metanol observadas
entre ambos grupos.
6. Efectos en mamíferos de laboratorio y en sistemas deensayo
in vitro
6.1 Toxicidad sistémica
La toxicidad aguda y a corto plazo del metanol varía mucho entre
las distintas especies, siendo máxima en las especies con una
capacidad relativamente escasa para metabolizar el formiato. En tales
casos de metabolismo deficiente del formiato, se produce una
intoxicación letal por metanol como consecuencia de la acidosis
metabólica y la toxicidad neuronal, mientras que en los animales que
metabolizan fácilmente el formiato la muerte suele deberse a las
consecuencias de la depresión del sistema nervioso central (coma,
insuficiencia respiratoria, etc.). En las especies de primates
sensibles (el ser humano y los monos) aumenta la concentración del
formiato en sangre tras la exposición al metanol, pero no en los
roedores, los conejos y los perros resistentes. Los primates humanos y
no humanos tienen una sensibilidad única a los efectos tóxicos del
metanol. En conjunto, el metanol tiene una toxicidad aguda baja para
los animales no primates. Los valores de la DL50 y las dosis letales
mínimas tras la exposición oral oscilan entre 7000 y 13 000 mg/kg en
ratas, ratones, conejos y perros y entre 2000 y 7000 mg/kg en el mono.
Las ratas expuestas a concentraciones de metanol de hasta
6500 mg/m3 (5000 ppm) 6 horas al día y 5 días a la semana durante
un período de 4 semanas no mostraron ningún efecto relacionado con la
exposición, salvo el aumento de la exudación alrededor de la nariz y
de los ojos. Se consideró que esto era un reflejo de la irritación de
las vías respiratorias superiores.
Las ratas expuestas a concentraciones de vapor de metanol de
hasta 13 000 mg/m3 (10 000 ppm) 6 horas al día y 5 días a la semana
durante un período de 6 semanas, no mostraron ninguna toxicidad
pulmonar.
En el conejo el metanol es moderadamente irritante de los ojos.
En una prueba de maximización modificada no se produjo sensibilización
cutánea.
Entre los efectos tóxicos observados en primates expuestos al
metanol cabe mencionar la acidosis metabólica y la toxicidad ocular,
pero estos efectos no aparecen normalmente en roedores con una
concentración suficiente de folato. Las diferencias de toxicidad se
deben a variaciones en la tasa del metabolismo del formiato,
metabolito del metanol. Por ejemplo, la eliminación del formiato de la
sangre de los primates expuestos es como mínimo un 50% más lenta que
en los roedores.
En monos que recibieron dosis de metanol superiores a 3000 mg/kg
con sonda se observó ataxia, debilidad y letargo a las pocas horas de
exposición. Estos signos mostraron una tendencia a desaparecer en un
plazo de 24 horas y los siguió un coma transitorio en algunos de los
animales.
En monos expuestos a metanol durante 6 horas al día y 5 días a la
semana, con 20 exposiciones repetidas a 6500 mg/m3 (5000 ppm) de
metanol no aparecieron efectos oculares.
6.2 Genotoxicidad y carcinogenicidad
El metanol ha dado resultados negativos en cuanto a la mutación
genética en ensayos con bacterias y levaduras, pero indujo anomalías
en la segregación cromosómica en Aspergillus. No indujo intercambios
de cromátidas hermanas en células de hámster chino in vitro, pero
provocó un aumento considerable de la frecuencia de las mutaciones en
células de linfoma de ratón L5178Y.
La inhalación de metanol no indujo daños en los cromosomas de
ratones. Hay algunas pruebas de que la administración oral e
intraperitoneal ha aumentado la incidencia de daños en los cromosomas
de ratones.
No hay ninguna prueba en estudios con animales que indique que el
metanol es carcinógeno, aunque se reconoce que se carece de un modelo
animal apropiado.
6.3 Toxicidad reproductiva, embriotoxicidad yteratogenicidad
Se han descrito resultados contradictorios en relación con los
efectos de la inhalación de metanol durante un período de hasta 6
semanas sobre las concentraciones de gonadotropina y testosterona.
La inhalación de metanol por roedores gestantes durante todo el
período de la embriogénesis induce una amplia variedad de efectos
teratogénicos y embrioletales dependientes de la concentración. En
fetos de ratas expuestas 7 horas al día durante 7-15 días de gestación
a 26 000 mg/m3 (20 000 ppm) de metanol se encontraron malformaciones
relacionadas con el tratamiento, predominantemente costillas
cervicales adicionales o rudimentarias y defectos del aparato urinario
o cardiovascular. Con este nivel de exposición se observó una ligera
toxicidad materna, pero no se detectó ningún efecto adverso para la
madre o la descendencia en animales expuestos a 6500 mg/m3 (5000
ppm), lo cual se interpretó como la concentración sin efectos adversos
observados (NOAEL) para este sistema de prueba.
Se registró una mayor incidencia de exencefalia y paladar hendido
en la descendencia de ratones CD-1 expuestos 7 horas al día durante
los días 6-15 de gestación a concentraciones de metanol de 6500 mg/m3
(5000 ppm) o más. La mortalidad embriofetal aumentó a concentraciones
de 9825 mg/m3 (7500 ppm) o más y fue mayor la incidencia de
resorciones de toda la descendencia. A concentraciones de 13 000 y
19 500 mg/m3 (10 000 y 15 000 ppm) se observó un peso fetal reducido.
La NOAEL para la toxicidad del desarrollo fue de 1300 mg/m3 (1000
ppm) de metanol. No se encontraron pruebas de toxicidad materna en la
exposición a concentraciones de metanol inferiores a 9000 mg/m3
(7000 ppm).
Cuando se administraron mediante sonda 4 g/kg de metanol a la
descendencia de ratones CD-1 gestantes, la incidencia de los efectos
adversos en la resorción, los defectos externos como el paladar
hendido y el peso del feto fue análoga a la observada en el grupo
expuesto por inhalación a 13 000 mg/m3 (10 000 ppm), posiblemente
debido al mayor ritmo de respiración del ratón. Éste es más sensible
que la rata a la toxicidad en el desarrollo provocada por el metanol
inhalado.
En hembras CD-1 expuestas a 19 500 mg/m3 (15 000 ppm) durante 6
horas diarias a lo largo de la organogénesis (días de gestación 6-15)
aparecieron signos neurológicos transitorios y una reducción del peso
corporal. Entre las malformaciones fetales registradas con 13 000 y
19 500 mg/m3 (10 000 y 15 000 ppm) cabe mencionar defectos neurales y
oculares, paladar hendido, hidronefrosis y anomalías de las
extremidades.
7. Efectos en el ser humano
El ser humano y los primates no humanos tienen una sensibilidad
única a la intoxicación por metanol, caracterizándose los efectos
tóxicos en estas especies por acidemia fórmica, acidosis metabólica,
toxicidad ocular, depresión del sistema nervioso, ceguera, coma y la
muerte. Casi toda la información disponible sobre la toxicidad del
metanol en el ser humano se refiere a las consecuencias de
exposiciones agudas más que crónicas. La inmensa mayoría de las
intoxicaciones por metanol se han debido al consumo de bebidas
adulteradas y de productos con metanol. Aunque la ingestión es con
diferencia la vía más frecuente de intoxicación, la inhalación de
concentraciones elevadas de vapor de metanol o la absorción percutánea
de líquidos metanólicos son tan eficaces como la vía oral para la
producción de efectos tóxicos agudos. La consecuencia más conocida
para la salud de una exposición a plazo más largo a niveles inferiores
de metanol es una amplia gama de efectos oculares.
Las propiedades tóxicas del metanol se basan en factores que
rigen tanto su conversión en ácido fórmico como el posterior
metabolismo del formiato a anhídrido carbónico en la ruta del folato.
La toxicidad es manifiesta si la generación de formiato continúa a un
ritmo superior al del metabolismo.
No se conoce con seguridad la dosis letal del metanol para el ser
humano. La dosis letal mínima del metanol en ausencia de tratamiento
médico está comprendida entre 0,3 y 1 g/kg. No se conoce la dosis
mínima que provoca defectos visuales permanentes.
La gravedad de la acidosis metabólica es variable y puede no
tener correlación con la cantidad de metanol ingerido. Una
característica destacada de la intoxicación aguda por metanol es la
enorme variabilidad interindividual de la dosis tóxica.
Parece que dos factores determinantes importantes de la
susceptibilidad humana a la toxicidad por metanol son: 1) ingestión
junto con etanol, que reduce el ritmo de entrada de metanol en la ruta
metabólica, y 2) la situación del folato hepático, que rige la tasa de
desintoxicación del formiato.
Los síntomas y signos de intoxicación por metanol, que pueden
aparecer solo transcurrido un período asintomático aproximado de 12 a
24 horas, son perturbaciones visuales, náuseas, dolor abdominal y
muscular, mareo, debilidad y perturbaciones de la conciencia que van
desde el coma hasta las convulsiones clónicas. Las alteraciones
visuales aparecen en general entre las 12 y las 48 horas después de la
ingestión del metanol y van desde la ligera fotofobia y la visión
brumosa o borrosa hasta una reducción acentuada de la agudeza visual y
la ceguera completa. En casos extremos se produce la muerte. La
principal característica clínica es una acidosis metabólica grave del
tipo de deficiencia de aniones. La acidosis se atribuye en gran medida
al ácido fórmico producido al metabolizarse el metanol.
La concentración normal en sangre de metanol procedente de
fuentes endógenas es de menos de 0,5 mg/litro (0,02 mmol/litro), pero
las fuentes alimenticias pueden elevarla. En general aparecen efectos
en el sistema nervioso central cuando la concentración de metanol en
sangre supera los 200 mg/litro (6 mmol/litro); se detectan síntomas
oculares por encima de 500 mg/litros (16 mmol/litro) y se han
registrado casos de letalidad en pacientes no tratados con
concentraciones iniciales de metanol del orden de 1500-2000 mg/litro
(47-62 mmol/litro).
La inhalación aguda de concentraciones de vapor de metanol por
debajo de 260 mg/m3 o la ingestión de cantidades de hasta 20 mg/kg de
metanol por parte de personas sanas o con una deficiencia moderada de
folato no debe dar lugar a la acumulación de formiato por encima de
las concentraciones endógenas.
Se ha informado de alteraciones visuales de varios tipos (visión
borrosa, reducción del campo visivo, cambios en la percepción de los
colores y ceguera temporal o permanente) en trabajadores expuestos a
concentraciones de metanol en el aire de alrededor de 1500 mg/m3
(1200 ppm) o más.
Un límite muy utilizado de exposición en el trabajo para el
metanol es el de 260 mg/m3 (200 ppm), concebido para proteger a los
trabajadores de cualquiera de los efectos de la acidosis metabólica
por ácido fórmico inducida por el metanol y de la toxicidad ocular y
del sistema nervioso.
No se ha notificado ningún otro efecto adverso del metanol en
el ser humano, salvo una ligera irritación cutánea y ocular con
exposiciones muy superiores a los 26 mg/m3 (200 ppm).
8. Efectos en los organismos del medio ambiente
Los valores de la CL50 en organismos acuáticos oscilan entre
1300 y 15 900 mg/litro para los invertebrados (48 y 96 horas de
exposición) y entre 13 000 y 29 000 mg/litro para los peces (96 horas
de exposición).
El metanol es poco tóxico para los organismos acuáticos siendo
poco probable la observación de efectos debidos a exposición ambiental
al metanol, excepto en el caso de un derrame.
A comparative metabolism study between rodents and non-human
primates showed that formic acid concentration in blood of rats and
monkeys was similar at doses of 25, 125 and 600 mg methanol/kg, but
became substantially higher in monkeys at 3000 mg/kg. Monkeys and
rodents showed different excretion patterns for methanol. As the dose
increased, monkeys tended to excrete an increasing percentage of
methanol in urine, whereas in rats, the percentage of methanol
excreted in expired air increased. Additionally, rats excreted much
higher levels of carbon dioxide in expired air (as a percentage of
dose) than monkeys (Katoh, 1989).
In a study of formate metabolism in young swine (Makar et al.,
1990), it was found that the pig, compared to other species (mouse,
rat, monkey and humans), has extremely low levels of hepatic folates.
Furthermore, the rate of formate elimination in the pig was much lower
in the pig than in the rat. It was suggested that the pig might be
sensitive to the methanol toxicity syndrome (metabolic acidosis and
blindness).
Ward & Pallack (1996a) studied the in vitro biotransformation
of methanol in Sprague-Dawley rat and CD-1 mouse fetal livers to
assess the capability of the near-term rodent fetus to metabolize
methanol. Adult near-term rodent livers metabolized methanol to
formate (at gestational day 20) with a maximum of about 85% that in
livers from non-pregnant rodents (p < 0.05). This was consistent with
in vivo experiments (Ward & Pollack, 1996a).
Fetal rat and mouse liver was capable of metabolizing methanol
in vitro, but only at a rate of < 5% of the respective adult liver.
The difference was in fact even greater, considering the difference in
organ weight between the conceptus and the dam (about 10-fold).
Fetal mouse liver homogenates converted methanol to formaldehyde
at a significantly higher (about 40%) rate than fetal rat liver
homogenates. These data suggest that the near-term rodent fetus does
not possess a significant ability to biotransform methanol to
formaldehyde and ultimately formate in situ.
6.4 Elimination and excretion
The primary route of methanol elimination from the body is via
oxidation to formaldehyde and then to formic acid, which may be
excreted in the urine or further oxidized to carbon dioxide.
In humans, methanol is primarily eliminated by oxidation and only
2% of a 50 mg/kg dose of methanol is excreted unchanged by the lungs
and kidney (Leaf & Zatman, 1952). The small excretion of unchanged
methanol was also observed in methanol-poisoned subjects in whom the
renal and pulmonary excretory clearances of methanol were 1 and
6 ml/min, respectively (Jacobsen et al., 1982a, 1983b).
The elimination of formaldehyde in many species, including
primates, is extremely rapid with a half-life of approximately 1 min
(McMartin et al., 1979). Toxic concentrations of formate (7-8 mM) were
detected within 30 min of ingestion in a human case of formaldehyde
poisoning, confirming the rapid metabolism of formaldehyde to formate
in humans (Eells et al., 1981b).
Following uptake and distribution methanol is either excreted
unchanged (direct excretion) in urine or exhaled breath, or it enters
a metabolic pathway in the liver, whose ultimate product is carbon
dioxide. The time course of the disappearance of methanol from the
circulation is dependent upon the combined action of both direct
excretion and metabolism. The clearance from the circulation of humans
following low-level exposures to methanol administered orally
(<0.1 g/kg) (Leaf & Zatman, 1952) or by inhalation (102-300 mg/m3)
(Sedivec et al., 1981) indicated that methanol disappearance obeyed
first-order kinetics with a half-time of about 2.5-3 h in both studies
as determined by blood and urinary methanol concentrations. In general
estimated methanol dose correlated with resulting blood and urine
methanol levels after both ingestion and inhalation, and methanol
concentrations in urine were approximately 30% higher than in blood
(Leaf & Zatman, 1952).
Elimination half-lives of methanol ranging from 110-213 min were
found in human volunteers following consumption of 1000-1500 ml red
wine (95% w/w ethanol, 100 mg/litre methanol) the previous evening
(Jones, 1987). After concomitant ingestion of a very low dose of
methanol (< 2 mg/kg) and ethanol (ethanol: methanol = 10), by human
subjects, a 10 fold increase in blood methanol was observed due to the
combined ingestion of the alcohols (Jones, 1987). Jacobsen et al.
(1982a) reported that during haemolysis in 2 patients being treated
for methanol poisoning, the elimination half-lives were 219 and
197 min respectively.
At higher doses of methanol, the elimination appears to become
saturated, resulting in nonlinear elimination kinetics. In an
untreated methanol-poisoned subject, methanol elimination was
clearly zero order with a rate of 85 mg/litre per h, about half the
elimination rate of ethanol (Jacobsen et al., 1988). The rates of
elimination in two other cases appeared to be 30-50 mg/litre per h
(Kane et al., 1968).
The kidney apparently exerts no active control over the urinary
concentration of methanol. The methanol content that enters the
bladder reflects the aqueous concentration of methanol in the blood
(Yant & Schrenk, 1937; Leaf & Zatman, 1952; Sedivec et al., 1981). The
rate at which methanol clears into the urine is directly proportional
to its blood level which satisfies the condition for first-order
kinetics (Kavet & Nauss, 1990).
In the lung, a small fraction of blood-borne methanol is exhaled.
The amount of methanol that crosses the blood-air barrier is
proportional to its blood concentration (first-order kinetics) and is
governed by its blood-air partition ratio (Kavet & Nauss, 1990). In
contrast to direct renal and pulmonary excretion, the metabolic
conversion of methanol to carbon dioxide is not a linear function of
concentration (Tephly et al., 1964; Makar et al., 1968).
Elimination of methanol from the blood appears to be slow in all
species especially when compared to ethanol (Tephly & McMartin, 1984;
Tephly, 1991).
One to 7 g of methanol/litre of blood (1000-7000 mg/litre) was
found in the blood of rats following oral administration of 4 g
methanol/kg body weight, and 70% of the methanol lost was eliminated
in expired air (Haggard & Greenberg, 1939).
Following administration of a 10% methanol solution (1 g/kg)
of 14C-methanol by gavage to the rat, 89% of the administered
radioactivity was recovered after 48 h; 65% as CO2 in expired air, 3%
as methanol in urine; 3% as formic acid in urine and 4% fixed in
tissues. An oxidation rate of 25 mg/kg/h was found during the first
28 h following methanol administration (Bartlett, 1950a).
Methanol was oxidized at a constant rate of 24 mg/kg per h during
the first 28 h following intraperitoneal administration of a 10%
14C-methanol solution (1 g/kg) to male albino rats. By the end of
36 h, 77% of the methanol had been converted to 14CO2 and 24% of the
dose was excreted unchanged. About equal quantities of methanol were
eliminated by the pulmonary and renal plus faecal routes (Tephly et
al., 1964).
Comparative studies in rats and monkeys have shown that 75-80% of
a 1 g/kg dose of 14C-methanol was recovered as 14CO2; 10-18% was
excreted unchanged in expired air and 6-11% eliminated in the urine as
methanol or formate within a 24-h period (Eells et al., 1981, 1983).
Excretion of similar amounts of unchanged methanol eliminated by
pulmonary (10-15%) and renal (3-19%) routes in rats and guinea-pigs
have also been reported (Bartlett, 1950; Tephly et al., 1964).
After oral administration to dogs of a single dose of methanol
(1.97 g/kg), about 10% was excreted unchanged in the urine, over a
period of about 100 h. The methanol concentration in the organs was
nearly half as high as that found in the urine. About 20% of the
administered dose was excreted as formic acid in the urine, which
ceased after 100 h. Formic acid concentrations in tissues were about
one-half to one-quarter that found in serum (Lund, 1948b).
Oral administration of 2.38 g methanol/kg to male rabbits
resulted in 10% of methanol being excreted unchanged in the urine and
essentially no increase in formic acid in the urine. Formic acid is
oxidized almost completely in the rabbit (Lund, 1948c).
Damian & Raabe (1996) investigated the dose-dependent elimination
of formate in male CD rats employing a perfused liver system to
separate the kinetic contributions of hepatic metabolism and renal
excretion in the total elimination of formate. Formate was eliminated
from the perfused rat liver following Michaelis-Menten kinetics.
The in vitro and in vivo dose-dependent studies of formate
elimination, in conjunction with the proposed toxicokinetic model (a
central, well-mixed compartment and a urine compartment, endogenous
production of formate), indicated two main pathways of formate
elimination in the rat: (a) hepatic metabolism via Michaelis-Menten
kinetics which predominates at low levels, and (b) extremely rapid and
extensive urinary excretion that predominates at high dose levels.
Urinary excretion consists primarily of glomerular filtration with
saturable tubular reabsorption.
6.5 Modelling of pharmacokinetic and toxicokinetic data
Pharmacokinetic and toxicokinetic models have been developed in
order to gain better insight into the interspecies variation in the
uptake, metabolic fate and excretion of methanol and its metabolites,
both compartmentally and physiologically based (Horton et al., 1992;
Pollack et al., 1993; Dorman et al., 1994). As has been noted, the
elimination of formaldehyde in many species, including primates, is
extremely rapid (McMartin et al., 1979).
A pharmacokinetic model of inhaled methanol in humans and
comparison to methanol disposition in mice and rats was described by
Perkins et al. (1995). Michaelis-Menten elimination parameters (Vmax=
115 mg/litre per h; km = 460 mg/litre) were selected for input into
a semi-physiological pharmacokinetic model. Literature values for
blood or urine methanol concentrations in humans and non-human
primates after methanol inhalation were employed as input to an
inhalation disposition model that evaluated the absorption of methanol
expressed as the fraction of inhaled methanol concentration that was
absorbed. Incorporation of the kinetic parameters and absorption into
a pharmacokinetic model of human exposure to methanol, compared to a
similar analysis in rodents, indicated that, following an 8-h exposure
to 6550 mg/m3 (5000 ppm) of methanol vapour, blood methanol
concentrations in the mouse would be 13-18 fold higher than in humans
exposed to the same methanol vapour concentration. Blood methanol
concentrations in the rat under similar conditions would be 5-fold
higher than in humans. The prediction of higher concentrations in rats
was due to the greater respiration rates and consequent greater
absorption of methanol by rats.
To address the problems associated with the appropriate design of
chronic methanol studies, methanol pharmacokinetics were characterized
in male Fischer-344 rats and rhesus monkeys exposed to atmospheric
methanol concentrations ranging from 65 to 2600 mg/m3 (50-2000 ppm)
for 6 h (Horton et al., 1992). A physiologically based pharmacokinetic
(PBPK) model was then developed to simulate the in vivo time course
data. The models were used to predict the atmospheric methanol
concentration range over which the laboratory species exhibit
quantitative similarities with humans. Below 1500 mg/m3 (1200 ppm)
the model predicted all three species would exhibit similar end-of-
exposure blood methanol concentrations which would be proportional to
atmospheric concentrations. At higher concentrations the increase of
methanol in the blood of rats and monkeys was predicted to become
non-linear, whereas for humans blood methanol levels were predicted to
increase in a linear fashion (Horton et al., 1992).
Female Sprague-Dawley rats at gestational days 7, 14 and 21 and
CD-1 mice at gestational days 9 and 18 were exposed to methanol
intravenously and orally (100-2500 mg/kg) or by inhalation exposure to
1310 to 26 200 mg/m3 (1000-20 000 ppm) for 8 h and the concentrations
of methanol were measured in blood, urine and amniotic fluid (Pollack
& Brouwer, 1996). Methanol disposition was virtually unaffected by
pregnancy and the fetal methanol concentrations were approximately
similar to those in the mother. Mice accumulated methanol at a rate 2
to 3 times faster than rats, despite the two-fold higher rate of
elimination observed in the mouse.
A pharmacokinetic model described the disposition of methanol in
rats and mice with the disposition profile being partitioned into
saturable and linear metabolic elimination pathways. The saturable
pathway was evident at lower doses (100 and 500 mg methanol/kg) and
displayed classical carrier-mediated Michaelis-Menten kinetics with a
rate-limiting step. The linear pathway, which consisted of passive
elimination via pulmonary and urinary clearance of methanol in
approximately equal amounts, appeared at the highest dose (2500 mg/kg
iv) and displayed the first-order kinetics of elimination that are
characteristic of passive-diffusion mechanisms (Pollack & Brouwer,
1996).
In further studies of the comparative toxicokinetics of methanol
in pregnant and non-pregnant Sprague-Dawley rats and CD-1 mice (Ward &
Pollack, 1996a), methanol disposition in the pregnant rodent was found
to be qualitatively similar to that in non-pregnant animals. Rats
received a single dose (100 or 2500 mg/kg) of methanol either orally
(by gavage) or intravenously; mice received a single oral or
intravenous dose of 2500 mg/kg.
The maximal rate of methanol elimination (Vmax) in vivo
decreased at term in both species. Vmax in near-term rats and mice
was only 65-80% of that in non-pregnant animals. The kinetic
parameters that appeared to be most sensitive to the gestational stage
were the rate constants associated with intercompartmental transfer
(k12 and k21), although there was no obvious relationship between the
estimate of these parameters and gestational stage. The data generated
in both the in vivo and in vitro studies demonstrated that
alterations in methanol disposition associated with gestational stage
should be accounted for in the development of a toxicokinetic model
for methanol in pregnant mammals.
The examination of the toxicokinetics of intravenously
administered methanol to female Sprague-Dawley rats as a single bolus
dose of 50 or 100 mg/kg, or 2500 mg/kg administered over 2 min,
resulted in a markedly non-linear elimination of methanol from the
systemic circulation suggesting a significant capacity-limited rate of
elimination. The data from the 2500 mg/kg group was described by a
kinetic model incorporating parallel first-order and saturable
elimination processes; a portion of this apparent linear elimination
pathway was due to renal excretion of the unchanged alcohol (Pollack
et al., 1993). The blood methanol concentration-time profile was
consistent with the presence of parallel linear pathways for methanol
elimination.
The toxicokinetics of methanol in female CD-1 mice and Sprague-
Dawley rats was examined by Ward et al. (1995). Non-linear disposition
of methanol was reported in both female CD-1 mice administered a
single dose of 2.5 g methanol/kg either by gavage or intravenously (as
a 1-min infusion) and Sprague-Dawley rats receiving a single oral dose
of 2.5 g/kg. Data obtained after intravenous administration were
well-described by a one-compartment model with Michaelis-Menton
elimination. Blood methanol concentration-time data after oral
administration could be described by a one-compartment (mice) or a
two-compartment (rats) model with Michaelis-Menton elimination
from the central compartment and biphasic absorption from the
gastrointestinal tract. Kinetic parameters (Vmax for elimination),
apparent volume of the central compartment (Vc), first-order rate
constants for intercompartmental transfer (k12 and k21), and first-
order absorption rate constants for fast (kAF) and slow (Kas)
absorption processes were compared between species. Mice showed a
higher maximal elimination rate than rats (when normalized for body
weight) (Vmax = 117 + 3 mg/kg per h versus 60.7 + 1.4 mg/kg per h for
rats). Additionally, the contribution of the fast absorption process
to overall methanol absorption was larger in the mouse than in the
rat. The study demonstrated that the disposition of methanol is
similar in rats and mice, although mice eliminated methanol nearly
twice as rapidly as rats.
The pharmacokinetics of 14C-methanol and 14C-formate were
studied in normal and folate-deficient (FD) female cynomolgus monkeys
anaesthetized and exposed by lung-only inhalation to 13, 60, 260 and
1200 mg/m3 (10, 45, 200 and 900 ppm) 14C-methanol for 2 h to
determine the concentration of methanol-derived formate to the total
formate pool. The blood concentration of 14C-methanol-derived formate
from all exposures was 10-1000 times lower than the endogenous blood
formate concentration (0.1-0.2 mmol/litre) reported for monkeys and
orders of magnitude lower than levels that produce acute toxicity
(8-10 mmol/litre). This suggested that low-level exposure to methanol
would not result in elevated blood formate concentrations in humans
under short-term exposure conditions (Dorman et al., 1994) (Medkinsky
& Dorman, 1985). This was confirmed in a subsequent short-duration
inhalation study in which anaesthetized female cynomolgus monkeys were
exposed for 2 h to methanol vapour (tagged with radiolabelled carbon)
at concentrations of 13, 59, 262 and 1179 mg/m3 (10, 45, 200 and
900 ppm), and monkeys fed on a diet deficient in folic acid were
exposed to 1179 mg/m3 (900 ppm) for the same duration (Medinsky et
al., 1997). The blood levels of methanol increased in a dose-dependent
manner. Blood formate levels increased by only a small extent in both
groups of monkeys.
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1 Single exposure
7.1.1 Non-primates
The lethal oral dose of methanol for most experimental animals is
relatively high compared to the lethal dose for humans and non-human
primates. In all non-primate species that have been studied, methanol
has been shown to be the least toxic of the aliphatic alcohols
(Koivusalo, 1970). The LD50 values or minimum lethal dose for a
single oral dose of methanol have been reported to be 9 g/kg for dogs
(Gilger & Potts, 1955), 7 g/kg for rabbits (Hunt, 1902; Gilger &
Potts, 1955), 7.4-13 g/kg for rats (Gilger & Potts, 1955; Rowe &
McCollister, 1982) and 7.3-10 g/kg for mice (Gilger & Potts, 1955;
Smith & Taylor, 1982) (Table 4). These doses are 6-10 times the lethal
human dose of methanol (Tephly & McMartin, 1984; Jacobsen & McMartin,
1986; HEI, 1987).
Table 4. Single-dose oral toxicity values for methanol in animals
Species LD50 (g/kg) Reference
Rat 6.2 Kimura et al. (1971)
9.1 Welch & Slocum (1943)
9.5 MLDa Gilger & Potts (1955)
12.9 Deichmann (1948)
13.0 Smyth et al. (1941)
Mouse 0.420 Smyth et al. (1941)
7.3-10.0 Smith & Taylor (1982)
Rabbit 7.0 MLD Gilger & Potts (1955)
Dog 8.0 Gilger & Potts (1955)
Monkey 2-3 MLD Gilger & Potts (1955)
7.0 MLD Cooper & Felig (1961)
a Minimum lethal dose
Other reported oral LD50 values for methanol in Sprague-Dawley
rats varied in 14-day-old, young adult and older rats ( 7.4, 13.0 and
8.8 ml/kg respectively), suggesting that young adult rats were least
susceptible to methanol toxicity (Kimura et al., 1971).
Youssef et al. (1992) reported that the order of oral LD50 in
adult female albino rats increased as follows: 95/5%-ethanol/methanol,
pure methanol, pure ethanol, and 65/35% methanol/ethanol. Clinical
features of intoxication in treated rats generally progressed from
signs of inebriation to gait disturbances, dose-proportional decreases
in response to painful stimuli, respiratory depression and coma,
ending in death due to cardio-respiratory failure. In almost all
instances, overnight coma was followed by death of the animal. Gross
and histopathological examinations of the gastric mucosa revealed
diffuse congestion with dilation of gastric blood vessels, but with
absence of gross haemorrhage and ulceration.
Rats exposed to 1.0, 2.0 and 3.0 g methanol/kg by gavage
exhibited an altered response in an operant conditioning paradigm
designed to assess motor deficits produced by neurotoxicants. Methanol
decreased the rate of response in a dose-related fashion that
suggested impaired coordination and/or reduced endurance (Youssef et
al., 1993).
Methanol administered by gavage or intraperitoneally induced
hypothermia in Fischer and Long-Evans rats, e.g., brain temperature
decreased 1.5°C within 35 min and colonic temperature was
significantly lower (Mohler & Gordon, 1990). This occurred at dose
levels of 2-3 g/kg, which is about 20% of the reported LD50 value
of 10 g/kg in rats (Gilger & Potts, 1955).
Among 40 strains of mice, 72 h oral LD50 values ranged from 7.3
to 10.0 g/kg with a mean of 8.68 g/kg methanol for mice fed a standard
laboratory chow diet (Smith & Taylor, 1982). Methanol-dosed C57BL/GCs
(acatalasemic) mice exhibited slightly lower LD50 than Csa (normal
catalase) mice, irrespective of their folate state (7.1-8.0 versus
8.6-9.0 g/kg). Oral methanol 72-h LD50 values ranged from 6.4 to
7.3 kg for mice with folic acid deficiency (FAD) diets, depending upon
the concentration of methionine in the diet (0.2-1.8%).
Female minipigs (Minipig YU, Charles River) treated with a single
oral dose of methanol at 1, 2.5 and 5.0 g/kg body weight by gavage
showed dose-dependent signs of acute methanol intoxication, including
mild CNS depression, tremors, ataxia and recumbency, which developed
within 0.5-2.0 h and resolved by 52 h. Methanol- and formate-dosed
minipigs did not develop optic nerve lesions, toxicologically
significant formate accumulation or metabolic acidosis (Dorman et al.,
1993).
The effects of single exposures of methanol by inhalation are
summarized in Table 5. The following signs of intoxication were noted:
increased rate of respiration, a state of nervous depression followed
by excitation, irritation of the mucous membranes, loss of weight,
ataxia, partial paralysis, prostration, deep narcosis, convulsions and
death occurring from respiratory failure (Loewy & von der Heide, 1914;
Tyson & Schoenberg, 1914; Eisenberg, 1917; Weese, 1928; Scott et al.,
1933; Mashbitz et al., 1936).
Under acute inhalation conditions, folate-deficient Long-Evans
male rats exposed to 4000 mg/m3 (3000 ppm) methanol for 20 h/day did
not survive more than 4 days. Rhesus monkeys exposed to 4000 mg/m3
(3000 ppm) methanol for 21 h/day survived the 20-day exposure period
and rhesus monkeys exposed to 13 000 mg/m3 (10 000 ppm) methanol for
21 h/day survived for more than 4 days (Lee et al., 1994).
The LD50 for single intraperitoneal injections of methanol was
10.5-11.0 g/kg in Swiss albino male mice. The animals initially
entered into deep narcosis within a few minutes and death usually
occurred within 24 h following recovery from deep narcosis (Gilger et
al., 1952). The LD50 values (mmole/kg) for single intraperitoneal
administration were as follows: male Wistar rats, 237; male strain H
mice, 336; male Syrian hamster, 267 (Tichy et al., 1985). These values
were calculated to correspond to 1489, 1493 and 1499 mmole/m2 body
surface, respectively. Tichy et al. (1985) also determined LD50
values for intravenous administration of methanol. The values reported
in rats and mice were 66.5 and 147 mmole/kg, corresponding to 418 and
653 mmole/m2 body surface, respectively.
Studies of rats have indicated that there are changes in levels
of dopamine, norepinephrine, serotonin and 5-hydroxyindole acetic acid
in various brain regions after a single intraperitoneal injection of
3 g methanol/kg (Jegnathan & Namasivayam, 1989). Studies on the
steady-state level of rat brain showed that there was severe depletion
of dopamine level in the striatum but a significant increase in the
level of dopamine, serotonin and 5-hydroxyindole acetic acid in the
hypothalamus. At the same time, norepinephrine and epinephrine levels
were reduced in the hypothalamus as well as in the striatum. These
effects do not seem to be induced by metabolic acidosis. The changes
in monoamine levels are very well correlated with the blood and brain
level of methanol as shown by maintaining a higher methanol level
either by simultaneous administration of ethanol or by blocking
methanol metabolism by pretreatment with 4-methyl pyrazole and
3-amino-1,2,4-triazole. It is thus postulated that monoamine changes
induced by methanol appear to be the direct effect of methanol
per se on the monoaminergic neuronal membranes.
Table 5. Effects from single inhalation exposure to methanol
Animal Concentration Duration of Signs of Outcome Reference
ppm exposure (h) Intoxication
Mouse 72 600 54 narcosis died Weese (1928)
72 600 28 narcosis died
54 000 54 narcosis died
48 000 24 narcosis survived
10 000 230 ataxia survived
152 800 94 min narcosis Mashbitz et al. (1936)
101 600 91 min narcosis
91 700 95 min narcosis
76 400 89 min narcosis overall
61 100 134 min narcosis mortality
45 800 153 min narcosis 45%
30 600 190 min narcosis
Rat 60 000 2.5 narcosis
convulsions Loewy & Von Der Heide
22 500 8 narcosis (1914)
13 000 24 prostration
8800 8 lethargy
4800 8 none
Dog 3000 8 none
32 000 8 prostration survived
incoordination
13 700 4 none
2000 24 none
7.1.2 Non-human primates
The lethal oral dose of methanol in monkeys (Table 4) has been
shown by several investigators to be of the same order of magnitude as
the lethal dose for humans. Gilger & Potts (1955) reported a minimum
lethal dose (MLD) for methanol of 3 g/kg for the rhesus monkey
(Macaca mulatta). Clinically the signs of toxicity were similar to
those noted in humans. There was a slight initial CNS depression for
1-2 h, followed by a latent period of about 12 h, a progressive
weakness, coma and death usually in about 20-30 h. All the monkeys (4)
given a lethal dose became severely acidotic within 24 h. Two of the
animals showed signs typical of methanol amblyopia observed in humans
including dilated, unresponsive pupils and changes of the retina. One
monkey exhibited evidence of optic disc hyperaemia and retinal oedema.
Cooper & Felig (1961) reported a MLD dose of 7 g methanol/kg
administered orally to rhesus monkeys and observed inebriation,
narcosis, coma and death within 24 h (usually without a latent
period). Sixteen animals survived 6 g methanol/kg or less. Acidosis
(an increased urinary excretion of organic acids) was reported in most
cases.
Studies by McMartin et al. (1975) and Clay et al. (1975) were in
agreement with earlier studies in monkeys by Gilger & Potts (1955).
Rhesus monkeys and pigtail monkeys (Macaca nemestrina) administered
3 g methanol/kg orally, showed an initial slight CNS depression
followed by a latent period of 12-16 h, during which time the animals
showed no obvious signs of toxicity. This was followed by progressive
deterioration characterized by anorexia, vomiting, weakness,
hyperpnoea and tachypnoea followed by coma with shallow and infrequent
respiration and finally death due to respiratory failure 20-30 h after
oral administration of methanol. The gradual development of metabolic
acidosis coincided with the accumulation of formic acid in the blood
and the decrease of bicarbonate in the plasma (McMartin et al., 1975).
An attenuated but prolonged syndrome was produced in monkeys by
the administration of an initial methanol dose of 2 g/kg body weight.
and subsequent doses (0.5-1.0 g/kg at 12-24 h intervals), producing
profound ocular toxicity approximately 40-60 h after the initial
dosage (Baumbach et al., 1977; Hayreh et al., 1977; Martin-Amat et
al., 1977).
Various species exposed to methanol by inhalation have exhibited
haemorrhage, oedema, congestion and pneumonia in the lungs (Eisenberg,
1917; Weese, 1928; Tyson & Schoenberg, 1914). Albuminous and fatty
degeneration and fatty infiltration of the liver and kidneys have also
been noted (Eisenberg, 1917; Weese, 1928). Fatty degeneration of
cardiac muscle has been observed in rabbits exposed repeated over 2 to
6 months to methanol via inhalation (Eisenberg, 1917). This subchronic
exposure to methanol in rabbits was also associated with notable
central nervous system effects such as optic nerve damage, lesion and
atrophy of the cerebrum, cerebellum, medulla and pons, along with
decreases in neurocytes, Nissl's granules and in severe cases,
parenchyma cells. Repeated inhalation of methanol resulted in
hyperaemia of choroid, oedema of ocular tissue including the retina
and optic disks, and degeneration of ganglion cells and nerve fibres
in a number of species such as the dog, rabbit and monkey (Tyson &
Schoenberg, 1914). Acute exposure to methanol via inhalation, as well
as oral and dermal exposure, was associated with degeneration and
necrosis of parenchymal tissue and neurons, accompanied by capillary
congestion and oedema, and degeneration of the retina and optic nerve
in rats, rabbits and monkeys (Scott et al, 1933).
An approximate intraperitoneal methanol LD50 of 3-4 g/kg for
pigtail monkeys (Macaca nemestrina) was reported by Clay et al.
(1975). Doses of 2 and 3 g/kg produced metabolic acidosis in the
animals, while monkeys given 4 g/kg became severely acidotic and
exhibited signs of toxicity that were remarkably similar to those
reported in human poisoning (Kane et al., 1968). These animals
displayed a sharp decrease in blood pH (7.03) at 7.5-21 h after
methanol administration. Bicarbonate was the single blood electrolyte
observed to change during the course of methanol acidosis. There was a
latent period of 15-18 h prior to the onset of overt signs of
toxicity, followed by a sequence of signs beginning with behavioural
distress, coma within 24-30 h and death. This time-course parallels
that reported for humans suffering from methanol poisoning (Röe,
1955).
7.2 Short-term exposure
7.2.1 Inhalation exposure
Male and female Sprague-Dawley rats exposed to 650, 2600 and
6500 mg/m3 (500, 2000 and 5000 ppm) methanol for 6 h/day, 5 days/week
for 4 weeks, exhibited no exposure-related effects except for
increased discharges around the nose and eyes which were considered
reflective of upper respiratory tract irritation. No consistent
treatment-related effects were found for organ weight or body weights
or in histopathological or ophthalmoscopical examinations. No ocular
effects were noted in rats from 20 repeated exposures to 6500 mg/m3
(5000 ppm) (Andrews et al., 1987).
Male Sprague-Dawley rats exposed to methanol vapour at
concentrations of 260, 2600 and 13 000 mg/m3 (200, 2000 and
10 000 ppm) for 6 h/day, 5 days/week for 6 weeks, did not develop
pulmonary toxicity. No significant changes were found at the lung
surface and in lung tissue (White et al., 1983).
Rats exposed to 16.8 methanol (0.022 mg methanol/litre of air)
4 h/day for 6 months and simultaneously administered 0.7 mg
methanol/kg daily by gavage exhibited changes in blood morphology,
oxidation-reduction processes and liver function (Pavlenko, 1972).
A preliminary study reported that F-344 rats fed control and
folate-deficient diets and exposed to methanol at a concentration of
1050 mg/m3 (800 ppm) for 20 h/day; 7 days/week for 13 weeks showed
spontaneous degeneration of retina and optic nerve in both diet
groups, while Long-Evans rats did not develop such ocular lesions. The
authors suggest that F-344 rats are unsuitable for ocular toxicity
studies (Lee et al., 1990).
Mice exposed to 63 000 mg/m3 (48 000 ppm) methanol for
3.5-4 h/day up to a cumulative total of 24 h were in a state of
narcosis but survived, whereas mice became comatose when exposed to
71 000 mg/m3 (54 000 ppm) for 54 h (Pavlenko, 1972).
Rabbits exposed by inhalation to 61 mg/m3 (46.6 ppm) methanol
for 6 months (duration of exposure/day not reported) exhibited
ultrastructural changes in the photoreceptor cells of the retina and
Müller fibres (Vendilo et al., 1971).
Two male dogs exposed to methanol vapour in air at 13 000 mg/m3
(10 000 ppm) for about 3 min in each of 8-h periods/day for 100
consecutive days, exhibited no symptoms, unusual behaviour or visual
toxicity. Methanol levels in blood measured at weekly intervals showed
median values of 65 and 140 mg/litre blood (Sayers et al., 1944).
In contrast to many studies of methanol toxicity that reported no
effect of low doses, two Russian studies (Chao, 1959; Ubaidullaev,
1966) reported evidence of neurobehavioural toxicity at low doses as
shown by altered chronaximetry (chronaximetry is the ratio of the
minimum time necessary for a stimulus of twice the absolute threshold
intensity to evoke a response measured as muscle contractions in
response to an electric current applied to an animal's hind leg).
Normally, the flexor chronaxia is shorter than the extensor chronaxia,
and their ratio is stated to be relatively stable.
Chao (1959) reported that the average chronaxia ratio for rats
exposed in the high-dose group (49.77 mg/m3) for 12 h/day, 5
days/week for 3 months, differed significantly from that in the
control group of animals at week 8 of exposure. The average chronaxia
ratio returned to normal during the recovery period and the effects in
the low-dose group (1.77 mg/m3) were insignificant. Histopathological
changes found in the high-dose group, but not in the low-dose group,
included poorly defined changes in the mucous membranes of the trachea
and bronchi, hyperplasia of the submucosa of the trachea, slight
lymphoid infiltration, swelling and hypertrophy of the muscle layer of
arteries, slight degenerative changes to the liver and changes in the
neurons of the cerebral cortex (Chao, 1959).Ubaidullaev (1966)
reported that male rats exposed continuously for 90 days to a
concentration of 5.3 mg/m3 (4 ppm) of methanol vapour, exhibited
changes in chronaxia ratio between antagonistic muscles, in whole
blood cholinesterase activity, in urinary excretion of coproporphyrin
and in albumin-globulin ratio of the serum. Male rats exposed to
0.57 mg/m3 (0.4 ppm) of methanol vapour continuously for 90 days
showed no changes.
It should be noted, however, that an analysis of these studies
by Kavet & Nauss (1990) indicated that, due to flaws in the study
designs, these studies do not provide adequate evidence of an
association between neurobehavioural effects and low-level exposure to
methanol. Both studies were limited by the use of small numbers of
animals per dose group, as well as insufficient reporting of
experimental methods, study results and statistical analysis. Kavet &
Nauss (1990) also stated that the biological significance of changes
in the chronaxia ratio is uncertain.
Male and female cynomolgus monkeys (Macaca fascicularis), three
per sex per dose, that were exposed to 650, 2600 and 6500 mg/m3 (500,
2000 and 5000 ppm) methanol for 6 h/day, 5 days/week for 4 weeks
showed no upper respiratory tract irritation. Neither gross,
microscopic nor ophthalmoscopic examinations disclosed any ocular
effects in the monkeys exposed to 6500 mg/m3 (5000 ppm) (Andrews et
al., 1987).
7.3 Long-term studies
In two 12-month chronic inhalation studies, Fischer-344 rats
(20 female and 20 male animals per group) and B6C3F1 mice (30/30
female/male) were exposed to 13, 130 and 1300 mg/m3 (10, 100 and
1000 ppm) of methanol to examine toxic effects unrelated to
carcinogenesis. A concentration of 130 mg/m3 (100 ppm) was found
to be the NOEL in both species. At the highest exposure, a slightly
reduced weight gain in male and female rats and a small but not
significant increase in the relative liver and spleen weight in female
rats were observed. In mice, the body weight was significantly higher
in the highest exposure groups in both males (after 6 months) and in
females (after 9 months). In addition, the incidence and degree of
fatty degeneration of hepatocytes was significantly enhanced in the
highest exposure groups of mice. However, this could have been due to
the higher incidence of fatty degeneration in mice of great body weight.
Clinical laboratory results did not show any changes attributable to
methanol (NEDO, 1987; Katoh, 1989).
Monkeys (Macaca fascicularis) (eight females per group) were
exposed to 13, 130 or 1300 mg/m3 for periods of 22 h/day for up to 29
months. Body weight, haematological and pathological examinations did
not reveal any dose-dependent effects except for hyperplasia of
reactive astroglias in the nervous system. However, this effect was
not correlated to dose or exposure time and was found to be reversible
in a recovery test (NEDO, 1982).
7.4 Skin and eye irritation; sensitization
In a modified Magnusson-Kligman maximization test with 10 female
guinea-pigs no sensitization was found after intracutaneous or
percutaneous induction and challenge with 50% methanol solution in
distilled water or with Freud's adjuvant. No skin irritation effects
were observed. In a parallel test, a 25% formaldehyde solution was
applied in order to test for possible sensitizing effects resulting
from the metabolic transformation of methanol to formaldehyde. Again
negative test results were seen (BASF, 1979).
New Zealand White albino rabbits treated by application of 100 µl
methanol into the lower conjunctival sac according to OECD test
guidelines and Draize scoring criteria exhibited the following mean
scores of conjunctivitis, chemosis, iritis and corneal opacity after
1, 4, 24, 48 and 72 h (Jacobs, 1990).
Time after application (h): 1 4 24 48 72
Mean score of conjunctivitis: 0.89 2.00 1.67 2.28 2.22
Mean score of chemosis: 2.00 2.00 0.67 1.00 0.50
Mean score of irititis: 0.33 1.00 1.00 0.50 0.33
Mean score of corneal opacity: 0.00 0.00 0.50 0.50 0.67
This demonstrates that methanol causes significant conjunctivitis
under the conditions of this test. Initial oedema (chemosis) seen up
to 4 h had decreased significantly by 72 h. Other ocular lesions were
much less significant.
7.5 Reproductive toxicity, embryotoxicity and teratogenicity
7.5.1 Reproductive toxicity (effects on fertility)
When male Sprague-Dawley rats were exposed for 8 h/day, 5
days/week to airborne methanol concentrations of 260, 2600 or
13 999 mg/m3 (200, 2000 or 10 000 ppm) for 1, 2, 4 or 6 weeks,
significantly decreased levels of circulating free testosterone were
found among rats exposed to 260 mg/m3 for 2 and 6 weeks and to
2600 mg/m3 for 6 weeks. However, the 13 000 mg/m3 group showed
no change. Significant changes in luteinizing hormone (LH) were found
after 6 weeks in animals exposed to 13 000 mg/m3, but no changes
in follicle-stimulating hormone (FSH) were observed at the various
exposure levels (Cameron et al., 1984). Sprague-Dawley rats exposed to
260 mg/m3 for 6 h for either 1 day or 1 week showed significant
depression (59%) in serum testosterone immediately after the first
exposure, but not after 1 week of daily 6-h exposures (Cameron et
al., 1985).
In a subsequent study groups of 10 male Long-Evans hooded rats,
60 days of age and acclimatized (or not) to handling, were exposed to
0, 260, 6500 or 13 000 mg/m3 (0, 200, 5000 or 10 000 ppm) methanol
for 6 h and killed either immediately on removal from the chambers or
18 h later. Similar groups of rats, acclimatized to handling or not,
were exposed to 6500 mg/m3 during 1, 3 or 6 h and killed immediately.
Serum testosterone levels were not significantly increased at 6 or
24 h in acclimatized rats, but levels were increased in non-
acclimatized rats exposed to 6500 mg/m3 and killed after 24 h. The
serum luteinizing hormone (LH) level was increased in acclimatized
rats exposed to 13 500 mg/m3 and killed at 6 and 24 h but the LH
level was reduced in non-acclimatized rats exposed to 6500 or
13 000 mg/m3 at 6 h but not 24 h. This experiment did not confirm the
earlier report that exposure to 260 mg/m3 for 6 h reduced serum
testosterone levels. In the second experiment serum LH and
testosterone levels did not differ at any time point between controls
and rats exposed to 6500 mg/m3 (Cooper et al.,1992). Methanol
inhalation at 260 mg/m3 for 8 h/day for up to 6 weeks did not reduce
serum testosterone levels in normal Sprague-Dawley rats (Lee et al.,
1991). In Long-Evans rats fed either control or folate-reduced diets
and exposed to 1040 mg/m3 for 20 h/day for 13 weeks, no adverse
effect on testicular morphology was observed with 10-month-old rats
fed either diet. A greater incidence of testicular degeneration was
however noted with 18-month-old rats given the folate-reduced diet,
suggesting that methanol potentially accelerates the age-related
degeneration of the testes (Lee et al., 1991).
7.5.2 Developmental toxicity
The inhalation of methanol by pregnant rodents throughout the
period of embryogenesis to high atmospheric concentrations (6500 to
26 000 mg/m3; 5000 to 20 000 ppm) impaired neural tube closure and
induced a wide range of concentration-dependent teratogenic and
embryolethal effects (Nelson et al., 1985; Rogers et al., 1993; Bolon
et al., 1993, 1994). In these studies, significant increase in the
incidence of exencephaly were observed following maternal methanol
exposures of > 6500 mg/m3 (> 5000 ppm) in mice, while similar
effects were observed in rats following exposures of > 13 000 mg/m3
(> 10 000 ppm), indicating that mice are more sensitive than rats to
the embryotoxic effects of methanol.
Pregnant Sprague-Dawley rats were given by inhalation for 7 h/day
either 6500 or 13 000 mg/m3 (5000, or 10 000 ppm) methanol on days
1-19 of gestation, or 26 000 mg/m3 (20 000 ppm) methanol on days 7-15
of gestation. The blood levels of methanol in the 26 000 mg/m3 group
ranged from 8.34 to 9.26 mg/ml after 1 day of exposure and from 4.84
to 6.00 mg/ml after 10 days of exposure. Methanol induced a dose-
related decrease in fetal weights and an increase in malformations.
The highest methanol concentration (26 000 mg/m3) produced slight
maternal toxicity (slightly unsteady gait) after the initial days
of exposure, and a high incidence of congenital malformations
(p < 0.001), predominantly extra or rudimentary cervical ribs and
urinary or cardiovascular defects. Similar malformations were found in
the groups exposed to 13 000 mg/m3 but the incidence was not
significantly different from that of the controls. No increase in
malformations was found in the group exposed to 6500 mg/m3
(5000 ppm), which was suggested to be a no-observed-effect level for
this test system (Nelson et al., 1985).
Pregnant CD-1 mice were treated by inhalation to 1300, 2600,
6500, 10 000, 13 000 or 19 500 mg/m3 (1000, 2000, 5000, 7500, 10 000
or 15 000 ppm) of methanol for 7 h/day on days 6-15 of pregnancy.
Significant increases were observed in the incidence of exencephaly
and cleft palate at 6500 mg/m3 or more. Increased embryo/fetal death
was found at exposures of 10 000 mg/m3 or more, including an
increasing incidence of full-litter resorptions. Reduced fetal weight
was found at 13 000 mg/m3 or more. A dose-related increase in
cervical vertebrae was significant at 2600 mg/m3 or more. The NOAEL
for the developmental toxicity was suggested to be 1300 mg/m3 (1000
ppm) methanol in this test system. There was no evidence of maternal
toxicity at methanol exposures below 10 000 mg/m3 (Rogers et al.,
1993).
A spectrum of cephalic neural tube defects was found in near-term
(gestation day 17) CD-1 mouse fetuses following maternal inhalation of
methanol at high concentration (19 500 mg/m3; 15 000 ppm) for 6 h/day
during neurulation (gestation days 7-9). Dysraphism, chiefly
exencephaly, occurred in 15% of the fetuses, usually in association
with reduction or absence of multiple bones in the craniofacial
skeleton and ocular anomalies (prematurely open eyelids, cataracts,
retinal folds). Exposure to a high concentration of methanol
(19 500 mg/m3) injured the multiple stem populations in the
neuralating mouse embryo. Significant neural pathology may remain in
older conceptuses even in the absence of gross lesions (Bolon et al.,
1994).
Transient neurological signs and reduced body weights were found
in up to 20% of CD-1 dams exposed to 19 500 mg/m3 (15 000 ppm)
methanol 6 h/day throughout organogenesis (gestational days 6-15).
Near-term fetuses revealed embryotoxicity (increased resorptions,
reduced fetal weights and/or fetal malformations) at 13 000 and
19 500 mg/m3 (10 000 and 15 000 ppm) methanol while 3-day exposures
at 6500 mg/m3 (5000 ppm) for 6 h/day yielded no observable adverse
effects (Bolan et al., 1993). In the studies of Bolon et al. (1993,
1994), terata included neural and ocular defects, cleft palate,
hydronephrosis, deformed tails and limb (paw and digit) anomalies.
Neural tube defects and ocular lesions occurred after methanol
inhalation by pregnant CD-1 mice between gestational days 7 and 9,
while limb anomalies were induced only during gestational days 9-11;
cleft palate and hydronephrosis were observed after exposure during
either period. The spectrum of teratogenic effects depended upon both
the stage of embryonic development and the number of methanol
exposures.
Long-Evans rats administered single oral doses of 1.3, 2.6 or
5.2 ml methanol/kg by gavage on day 10 of gestation, exhibited dose-
related anomalies, e.g., undescended testes and eye defects
(exophthalmia and anophthalmia) in the offspring. At the methanol dose
of 5.2 ml/kg, the maternal weight loss was > 10%, which was the only
clinical toxic manifestation/histopathological change noted for the
dams. A significant decrease in fetal body weight (11-21%) was
associated with oral ingestion of methanol in the dams. Methanol given
acutely can produce anomalies in the offspring where there are no
apparent maternal toxic responses (Youssef et al., 1991).
Methanol was shown to impair uterine decidualization during early
pregnancy in Holtzman rats administered 1.6, 2.4 or 3.2 g methanol/kg
per day by gavage during days 1-8. Reductions in pregnant uterine and
implantation site weights seen on day 9 were the result of methanol
impedance of normal uterine decidualization as demonstrated by effects
on decidual cell response technique. Methanol (3.2 g/kg per day)
produced a non-specific maternal toxicity (reduction in body weight)
by day 9, but no effect on days 11 or 20 on embryo and fetal survival
or development were found (Cummings, 1993).
When pregnant CD-1 mice were gavaged orally with 4 g methanol/kg,
the incidences of fetal resorption, external defects (including cleft
palate) and reduced fetal weight were similar to those observed in the
13 000 mg/m3 (10 000 ppm) inhalation exposure group. Cleft palate
(43.5% per litter) and exencephaly (29% per litter) were the
predominant external defects seen following methanol exposure by oral
gavage. Methanol blood level in the gavage study was 4 mg/ml, which
was reportedly similar to the blood level at the 13 000 mg/m3
inhalation exposure group (see above) (Rogers et al., 1993).
No effects on reproductive performance were reported in a two-
generation reproductive study in F-344 rats administered 13, 130 or
1300 mg/m3 (10, 100 or 1000 ppm) methanol by inhalation for
18-20 h/day. A statistically significant decrease in brain weight was
found at the 1300 mg/m3 level in 3-, 6- and 8-week-old pups of the
F1 generation. In the F2 generation reduced brain thymus and
hypophysis weight was observed. (NEDO, 1987; Katoh, 1989).Teratology
studies with Sprague-Dawley rats exposed to 260, 1300 or 6500 mg/m3
(200, 1000 or 5000 ppm) methanol by inhalation for 22 h/day during
gestational days 7-17 revealed significant weight decreases in brain,
thyroid and thymus of the offspring resulting from maternal exposure
to 6500 mg/m3. However, no abnormal changes were detected
histopathologically. Evidence of maternal toxicity was found at this
level of exposure and toxic effects to fetuses were reported,
including death. No effects were found at 1300 kg/m3 (NEDO, 1987;
Katoh, 1989).
A pilot developmental toxicity study was conducted by Ryan et al.
(1994) to assess the utility of the folic-acid-deficient rat model, a
model that would be sensitive to methanol and potentially reflective
of the human risk/response. Methanol was administered in drinking-
water on days 6-15 of gestation at concentrations of 0.5, 1.0 and 2.0%
to three groups of 7 to 9 sperm-positive Long-Evans rats. The average
blood levels were given as 0.21, 0.26 and 0.67 mg/ml, respectively. A
dose-dependant increase in the incidence of maternal and developmental
effects was observed. For both end-points the NOEL was assumed to be
less than 0.5% methanol in drinking-water, corresponding to a blood
level of 0.21 mg/ml.
Weiss et al. (1996) studied developmental neurotoxicity of
pregnant Long-Evans rats and their newborn offspring exposed to
5900 mg/m3 (4500 ppm) of methanol by inhalation for 6 h daily,
beginning on gestation day 6, with both dams and pups then being
exposed through postnatal day 21. Although findings suggested
significant functional consequences in rats resulting from this
exposure, these consequences were considered subtle in character.
Exposure to 5900 mg methanol/m3 did not affect the suckling time
and conditioned olfactory aversion test of newborn rats. Methanol-
exposed newborn pups were less active on postnatal day 18 and more
active on postnatal day 25 than control newborn pups (motor activity
test). The study found only isolated positive results that were small
and variable. The two adult assays, the fixed-ratio wheel-running
test and the stochastic discrimination test, yielded evidence of a
significant methanol effect.
No evidence of brain damage emerged on the basis of neuro-
pathology, although differences in neural cell adhesion molecules
(NCAMs) arising from methanol exposure were observed in neonatal
cerebella (Weiss et al., 1996). Methanol treatment caused a decrease
in expression in both NCAM 140 and NCAM 180.
Further elaboration of the effects of perinatal exposure on NCAM
in Long-Evans rats exposed to 5900 mg/m3 (4500 ppm) methanol vapour
for 6 h daily (beginning on gestation day 6 with dams and pups then
exposed until postnatal day 21) were described by Stern et al. (1996).
Blood methanol concentrations from samples obtained immediately
following a 6-h exposure reached approximately 500-800 µg/ml in the
dams during gestation, and lactation average concentrations for pups
attained levels about twice those of the dams. Light-microscopic
analysis showed no significant abnormalities in the brains of the
methanol-treated animals. However, assays of NCAM in the brains of
pups sacrificed on postnatal day 4 showed staining for both the 140
and the 180 kDa isoforms to be less intense in the cerebellum of
exposed animals. NCAM differences were not apparent in animals
sacrificed after their final exposure. NCAM 140 is the primary isoform
expressed during the stages of neuronal migration and NCAM 180 is
expressed during synaptogenesis where it is critical to neuronal
plasticity, learning and memory. NCAMs are developmentally regulated
glycoproteins that serve critical roles in the formation and
maintenance of the nervous system (Stern et al., 1996).
7.5.3 Behavioural effects
Neonatal behavioural toxicity was reported in studies involving
two groups of primigravid Long-Evans rats given drinking solutions of
2% methanol either on gestational days 15-17 or 17-19, with the
average daily intake on these days amounting to 2.5 g methanol/kg.
Lack of maternal toxicity was indicated by measurements of weight
gain, gestational duration or daily fluid intake. Litter size, birth
weight and infant mortality did not differ between the two treatment
groups and the control. Pups from methanol-treated rats required
longer periods than controls to begin suckling on postnatal day 1. On
postnatal day 10, they required more time to locate nesting material
from their home cages, suggesting that prenatal methanol exposure
induced behavioural abnormalities early in life, unaccompanied by
overt toxicity (Infurna & Weiss, 1986).
Following inhalation exposure of Long-Evans rats to 19 500 mg/m3
(15 000 ppm) methanol for 7 h/day on gestational days 7-19, maternal
blood levels decreased significantly from 3.8 mg/litre on the first
day of exposure to 3.1 mg/litre on the 12th day of exposure. Methanol
transiently reduced maternal body weight by 4-7% on gestational days
8-10 and offspring body weight by 5% on post-natal days 1-3. Motor
activity, olfactory learning, behavioural thermoregulation, T-maze
learning, acoustic startle response, pubertal landmarks and passive
avoidance tests performed at the end of the exposure period failed to
reveal significant effects. Prenatal exposure to high levels of
inhaled methanol appeared to have little effect beyond post-natal day
3 in this series of tests (Stanton et al., 1995).
7.5.4 In vitro studies
Methanol is developmentally toxic to both mouse (CD-1) and rat
(Sprague-Dawley) embryos during organogenesis in whole embryo culture
(WEC), a technique which removes the confounding maternal influences
(Andrews et al., 1993). Comparable developmental stages of CD-1 mouse
and Sprague-Dawley rat embryos were exposed to methanol (0-16 mg/ml
for rat and 0-8 mg/ml for mouse embryos) for 24 h. Rat embryos were
cultured for an additional 24 h without methanol in the medium, having
a total culture time of 48 h. Concentration-dependent decreases in
somite number, head length and developmental score occurred in both
species, with significant effects in the rat at > 8 mg/ml and in
the mouse at > 4 mg/ml (Andrews et al., 1993).
In studies of 8-day mouse embryos cultured in methanol,
concentrations greater than 2 mg methanol/ml caused a significant
decrease in developmental score and crown-rump length; the 8 mg/ml
group also suffered 80% embryolethality (Andrews et al., 1993). Mouse
embryos were affected at methanol concentrations that were not
dysmorphogenic or embryotoxic in the rat following teratogenic
in vivo exposures (Rogers et al, 1993), suggesting that the higher
sensitivity of the mouse was due, at least in part, to the greater
intrinsic embryonal sensitivity of this species to methanol (Andrews
et al., 1993).
Depending on the concentration and duration of methanol exposure
(0-20 mg/ml for 6 h, 12 h, or 1 or 4 days) on embryonic CD-1 mouse
palate in serum-free organ culture, the medial epithelium either
degenerated completely or remained intact in unfused palates (either
condition would interfere with fusion) (Abbott et al., 1994). Cellular
proliferation appeared to be a specific and sensitive target for
methanol as craniofacial tissues responded to methanol with reduction
in DNA content at an exposure that did not effect total protein.
However both DNA and protein levels decreased with increasing exposure
to methanol. Methanol selectively altered the morphological fate of
the medial palatal epithelium cells and the specific effect on cell
survival was exposure dependent (Abbott et al., 1994).
7.6 Mutagenicity and related end-points
7.6.1 In vitro studies
The structure of methanol (by analogy with ethanol) does not
suggest that it would be genotoxic.
Methanol gave negative results when tested in Salmonella
typhimurium plate incorporation assays with or without metabolic
activation using strains TA98, TA100, TA1535, TA1537 and TA1538
(Simmon et al., 1977). It was also negative in the presence or absence
of metabolic activation in strains TA1535, TA100, TA1538, TA98 and
TA1537 (De Flora et al., 1984) and in a DNA repair test in E. coli
using strains WP 2, WP 67 and CM 871 in the presence or absence of
metabolic activation (De Flora et al., 1984).
Methanol (6.0% v/v) induced 3.02% chromosomal malsegregation in
Aspergillus nidulans diploid strain P1 (Crebelli et al., 1989). The
result was statistically significant at two concentrations and a dose-
response relationship was evident.
Methanol was negative for gene mutation at the ade 6 locus
in the yeast Schizosaccharomyces pombe with or without the
postmitochondrial fraction from mouse liver (Abbondandolo et al.,
1980). It was also negative in a mutagenicity test for n+1 aneuploidy
arising from meiotic disfunction of linkage group I in the fungus
Neurospora crassa (Griffiths, 1981).
Methanol did not induce sister chromatid exchanges (SCEs) in
Chinese hamster cells in vitro during treatment for 8 days to a
final concentration of 0.1% (v/v) (Obe & Ristow, 1977). Only in the
presence of S-9 mix and methanol (7.9 mg/ml) was there a significant
increase in the mutation frequency in L5178Y mouse lymphoma cells
(McGregor et al., 1985), possibly because this assay detects
chromosome damage as well as gene mutation.Methanol was negative in
two in vitro tests for cell transformation: the Syrian hamster
embryo cell (SHE) clonal system (Pienta et al., 1977) and the Rausher
leukaemia virus-infected rat embryo cell (RLV/RE system) (Heidelberger
et al., 1983).
Addition of methanol (or ethanol) to unleaded gasoline as a fuel
extender did not appear to significantly alter the genetic toxicity of
particulate exhaust particles when tested in S. typhimurium strains
TA100, TA98, TA98 NR, and TA98 DNPR with S-9 activation. In all the
alcohol-blended fuel tests, the mass of particle-associated organics
emitted from the exhaust was lower than that observed during the
control tests using gasoline alone (Clark et al., 1983).
7.6.2 In vivo studies
No increased frequencies of micronuclei in blood cells, of
SCEs, chromosome aberrations or micronuclei in lung cells, or of
synaptonemal complex damage in spermatocytes were found in mice
exposed by inhalation to 1050 or 5200 mg/m3 (800 or 4000 ppm)
methanol for 5 days (Campbell et al., 1991).
Urine from mice orally administered five daily doses of methanol
(5 g/kg total) showed no mutagenic activity, and no increase in the
incidence of abnormal sperm was reported (Chang et al., 1983). Oral
administration of 1 g methanol/kg to mice increased the incidence of
chromosomal aberrations, particularly aneuploidy and SCEs, as well as
the incidence of micronuclei in polychromatic erythrocytes (Pereira et
al., 1982).
The oral administration of 14C-labelled methanol to rats
resulted in covalent binding to haemoglobin, with binding exhibiting a
linear dose relationship between 10 and 100 µmol/kg (Pereira et al.,
1982).
B6C3F1 mice treated with five daily oral doses of 1 g
methanol/kg exhibited abnormal (banana type) sperm morphology. The
biological significance of these changes is unknown (Ward et al.,
1984). It should be noted that the above results, namely altered sperm
(Ward et al., 1984) and haemoglobin binding (Pereira et al., 1982) are
end-points not generally used for genotoxic evaluation and their
assessment in terms of mutagenicity is unclear.
There is some evidence that bone marrow cytogenetic analysis
indicated a dose-related response for structural aberrations,
especially centric fusions in mice treated with three daily
intraperitoneal methanol doses of between 75-300 mg/kg total dose
(Chang et al., 1983).
In vitro and in vivo mutagenicity studies on methanol, i.e.,
the Ames test, somatic mutation assay in CH-V79 cells, chromosome
aberrations, SCEs and the micronucleus test in mice conducted by NEDO
(1987; Katoh, 1989), were all reported to be negative.
7.7 Carcinogenicity
There have been no studies reported in the peer-reviewed
literature on the potential carcinogenicity of methanol per se in
laboratory animals.
The New Energy Development Organization (NEDO) in Japan reported
carcinogenicity studies in which B6C3F1 mice and Fischer-344 rats of
both sexes were exposed by inhalation to 13, 130 or 1300 mg/m3 (10,
100 and 1000 ppm) methanol for 20 h/day for 18 and 24 months,
respectively (NEDO, 1987; Katoh, 1989). No evidence of carcinogenicity
was found in either species. High-dosed animals had a higher, but not
statistically significant, incidence of papillary adenomas than
controls , and histopathological examination suggested that these
changes were between non-neoplastic and neoplastic changes.
Additionally, seven cases of adrenal pheochromocytoma were found in
high-dose animals compared to one case in controls. This observation
was not statistically significant according to the Fisher exact test
(Katoh, 1989).
It is unlikely that methanol is carcinogenic to mouse skin. In an
experiment using four strains of female mice (Balb/c, Sencar, CD-1 and
Swiss) to study N-nitrosomethylurea carcinogenesis, methanol was
used as a solvent control. Four groups of 20 mice of each strain
received 25 µl methanol twice weekly for 50 weeks followed by
observation for lifespan. Only one skin tumour was observed among the
80 control animals (Lijinsky et al., 1991).
7.8 Special studies
7.8.1 Effects on hepatocytes
When Garcia & Van Zandt (1969) administered repeated doses of 3
to 6 g/kg by gavage to rhesus monkeys (Macaca mulata) for 3-20
weeks, average serum levels of methanol of 4750 mg/litre were attained
within a few hours. Animals were killed at the end of treatment and
livers examined histologically. Hepatocytes showed nucleolar
segregation (zoning of nucleus), hyperplasia of endoplasmic reticulum
and swelling of mitochondria. These changes were also found in one
monkey sacrificed 12 weeks after the end of treatment.
7.8.2 Toxic interactions
Inhaled methanol potentiated the hepatotoxicity produced by
carbon tetrachloride in adult male F-344 rats. Rats were exposed to
methanol (0 or 13 000 mg/m3) 10000 ppm for 6 h, then treated 24 h
later with oral CCl4 (0.075 ml/kg). CCl4 alone produced a low level
of hepatotoxicity within 3 days. Methanol plus CCl4 resulted in
marked increases in serum aspartate aminotransferase and alanine
aminotransferase that lasted for 7 days. Methanol also exacerbated the
histological evidence of CCl4-induced centrilobular degeneration and
necrosis (Simmons et al., 1995).
Methanol exposure by inhalation induced cytochrome P4502E1
(CYP2E1), which appeared to be the principal toxicokinetic mechanism
underlying methanol potentiation of carbon tetrachloride
hepatotoxicity (Allis et al., 1996).
When dichloromethane (DCM) is metabolized carbon monoxide is
formed, leading to increased carboxyhaemoglobin (COHb) levels in
blood. Pankow & Jagielki (1993) found that in rats pretreated with
methanol, methanol doses of 790-6330 mg/kg (24.7-198 mmol/kg)
stimulated increased metabolism of DCM, as seen by further increases
in COHb levels. When methanol was administered simultaneously with
DCM, a decrease in COHb formation was seen at methanol doses of 4736
to 7900 mg/kg (148-247 mmol/kg) but not at 3162 mg/kg (98.8 mmol/kg).
Thus methanol can interact with DCM metabolism both by induction and
by competitive inhibition, the latter only at very high doses.
Poon et al. (1994) reported no significant interactive effects in
young Sprague-Dawley rats exposed to vapours of methanol/toluene
(400/110 mg/m3; 400/1100 mg/m3; 4000/110 mg/m3 and 4000/1100 mg/m3)
for 6 h/day, 5 days/week for 4 weeks. Exposure to methanol (400 to
4000 mg/m3) and to toluene (110 mg/m3 to 1100 mg/m3) or to a mixture
of both produced mild biochemical effects and histological changes in
the thyroid (moderate reduction in follicle size in the thyroids) and
nasal passages.
The biochemical, haematological and histological effects on
Sprague-Dawley rats after exposure to methanol (3000 mg/m3;
2500 ppm), gasoline (3200 ppm) and methanol/gasoline (2500/3200 ppm)
vapour 6 h/day for 4 weeks were examined by Poon et al. (1995).
Gasoline was largely responsible for the adverse effects, the most
significant of which included depression in weight gain in the males,
increased liver weight and hepatic microsomal enzyme activities in
both sexes, and suppression of uterine eosinophilia. No apparent
interactive effects between methanol and gasoline were observed.
7.8.3 Studies with exhaust emissions from methanol-fuelled engines
There are few data related to the effects of emissions from
methanol-fuelled engines. Since most such fuels will contain a
proportion of gasoline and other additives and the emissions will be
complex, the interpretation of these data in relation to methanol
toxicity is complicated.
Maejima et al. (1992, 1993 and 1994) studied the effects of
emissions from M-85 methanol-fuelled engines (methanol with 15%
gasoline), without a catalyst, on Fischer-344 rats for periods up to
12 weeks. The exhaust contained significant amounts of carbon monoxide
(89.9 ppm), oxides of nitrogen (22.9 ppm), formaldehyde (2.3 ppm) and
methanol (8.1 ppm). The effects observed were considered to be
primarily related to formaldehyde. No increase in plasma methanol or
formic acid was detected.
7.9 Mechanism of ocular toxicity
Formic acid, the toxic metabolite of methanol, has been
hypothesized to produce retinal and optic nerve toxicity by disrupting
mitochondrial energy production (Fig. 1) (Martin-Amat et al., 1977;
Sharpe et al., 1982). It has been shown in vitro to inhibit the
activity of cytochrome oxidase, a vital component of the mitochondrial
electron transport chain involved in ATP synthesis (Nicholls, 1975).
Inhibition occurs subsequent to the binding of formic acid to the
ferric haem iron of cytochrome oxidase, and the apparent inhibition
constant is between 5 and 30 mM (Nicholls, 1975). Concentrations of
formate present in the blood and tissues of methanol-intoxicated
humans, non-human primates and rodent models of methanol-intoxication
are within this range (Martin-Amat et al., 1977; Sejersted et al.,
1983; Eells, 1991).
Studies conducted in methanol-sensitive rodent models have
revealed abnormalities in retinal and optic nerve function and
morphology, consistent with the hypothesis that formate acts as a
mitochondrial toxin (Fig. 2). In these animal models, formate
oxidation is selectively inhibited by dietary (Lee et al., 1994) or
chemical (Eells et al., 1981) depletion of folate coenzymes, thus
allowing formate to accumulate to toxic concentrations following
methanol administration. Methanol-intoxicated rats developed formic
acidaemia, metabolic acidosis and visual toxicity analogous to the
human methanol poisoning syndrome (Eells, 1991; Murray et al., 1991;
Lee et al., 1994a,b).
Sixty hours after the administration of the first dose of
methanol, blood formate values ranged from 8-20 mM with blood hydrogen
carbonate values in the range of 5-12 mEq/litre and blood pH values of
6.83-7.08. Similar blood formate concentrations, hydrogen carbonate
levels and pH values were reported in methanol-intoxicated monkeys
(Martin-Amat et al., 1977) and in severe cases of human methanol
poisoning (McMartin et al., 1980a; Sejersted et al., 1983; Jacobsen et
al., 1988).
Visual dysfunction was measured as reduction in the flash evoked
cortical potential (FEP) and electroretinogram (ERG). The FEP is a
measure of the functional integrity of the primary visual pathway from
the retina to the visual cortex and the ERG is a global measure of
retinal function in response to illumination (Creel et al., 1970;
Dowling, 1987). The FEP was progressively diminished in methanol-
intoxicated rats, indicative of a disruption of neuronal conduction
along the primary visual pathway from the retina to the visual cortex
(Eells, 1991). ERG analysis in methanol-intoxicated rats revealed a
significant early deficit in b-wave amplitude, followed by a
temporally delayed lesser reduction in a-wave amplitude (Murray et
al., 1991). The b-wave of the ERG is generated by depolarization of
the Muller glial cells and reflects synaptic activity at the level of
the bipolar cells (Dowling, 1987). The b-wave of the ERG is
extremely sensitive to conditions that interfere with retinal energy
metabolism and is reduced or abolished following brief ischaemia or
the administration of metabolic poisons (Bresnick, 1989; Dowling,
1987). Both FEP and ERG alterations occurred at the same time as
accumulation of blood formate, indicative of a causal relationship
between formate-induced metabolic and visual disturbances. Similar ERG
reductions have been reported in methanol-intoxicated primates
(Ingemansson, 1983) and in human methanol intoxication (Ruedemann,
1962; Murray et al., 1991).
In addition to neurofunctional changes, bioenergetic and
morphological alterations indicative of formate-induced disruption of
retinal energy metabolism have been documented in methanol-intoxicated
rats (Murray et al., 1991; Eells et al., 1996; Garner et al.,
1995a,b). Morphological studies, coupled with cytochrome oxidase
histochemistry, revealed generalized retinal oedema, photoreceptor and
RPE vacuolation, mitochondrial swelling and a reduction in cytochrome
oxidase activity in photoreceptor mitochondria from methanol-
intoxicated rats (Murray et al., 1991; Eells et al., 1995, 1996). The
most striking structural alterations observed in the retinas of
methanol-intoxicated rats were vacuolation and mitochondrial swelling
in inner segments of the photoreceptor cells (Murray et al., 1991).
Photoreceptor mitochondria from methanol-intoxicated rats were swollen
and expanded to disrupted cristae and showed no evidence of cytochrome
oxidase reaction product. In contrast, photoreceptor mitochondria from
control animals showed normal morphology with well-defined cristae and
were moderately reactive for cytochrome oxidase reaction product.
These findings are consistent with disruption of ionic homoeostasis in
the photoreceptors, secondary to inhibition of mitochondrial function.
Biochemical measurements also showed a significant reduction in
retinal and brain cytochrome oxidase activity and ATP concentrations
in methanol-intoxicated rats relative to control animals (Eells et
al., 1995). Surprisingly, no differences from control values were
observed in hepatic, renal or cardiac cytochrome oxidase activity or
ATP concentrations in methanol-intoxicated rats. The reduction in
retinal function, inhibition of retinal, optic nerve and brain
cytochrome oxidase activity, depletion of retinal and brain ATP
concentrations, and mitochondrial disruption produced in methanol-
intoxicated rats are consistent with the hypothesis that formate acts
as a mitochondrial toxin with selectivity for the retina and brain.
Studies by Eells et al. (1996) compared the effects on retinal
function and structure of rapidly increasing formate concentrations
typical of acute methanol intoxication with low-level plateau formate
concentrations more likely to be generated by subacute or chronic
methanol exposure. Methanol-intoxicated rats that accumulated formate
concentrations of 8-15 mM developed metabolic acidosis, retinal
dysfunction, and retinal histopathological changes. Retinal
dysfunction was measured as reductions in the a- and b-waves of
the electroretinogram that occurred at the same time as blood formate
accumulation. Histopathological studies revealed vacuolation in the
retinal pigment epithelium and photoreceptor inner segments. Rats
exposed to formate concentrations ranging from 4 to 6 mM for 48 h
showed evidence of retinal dysfunction in the absence of metabolic
acidosis and retinal histopathology. These data indicated that
formate-induced retinal dysfunction in methanol-intoxicated rats can
be produced by steadily increasing concentrations of formate and,
importantly, can also be produced by prolonged exposure to lower
concentrations of formate.
Martinasevic et al. (1996) studied components of folate-dependent
formate oxidation, e.g., folate and 10-CHO-H4-folate dehydrogenase
(10-FDH), in human and rat retinae. Total folate levels in human and
rat retinal tissues were much lower than the levels in liver. However,
folate levels in human retina were only 14% of those determined in rat
retina. Comparable amounts of this 10-FDH were present in both
cellular compartments in each species. However, the amount of 10-FDH
in the human retina was approximately three times the amount found in
the rat retina. Immunohistochemical staining for 10-FDH showed that
this enzyme was preferentially localized in Müller cells. Since Müller
cells appear to represent the target for formate-induced ocular
toxicity, the authors suggested that formate oxidation reactions might
serve two roles, first a protective role and then a role in methanol-
induced toxicity in Müller cells.
Garner & Lee (1994) employing oscillatory potential analysis
showed that retinal ischaemia was not involved in methanol-induced
visual system toxicity.
The role of retinal metabolism in methanol-induced retinal
toxicity in folate-sufficient (FS) rats and folate-deficient (FR)
rats, some of which were also pretreated with disulfiram (DSF), was
examined by Garner et al. (1995). Folate-deficient rats treated with
methanol displayed elevated blood and vitreous humour formate levels
along with abnormal electroretinograms (ERG), whereas methanol-exposed
folate-deficient rats pretreated with DSF did not. Formaldehyde was
not detected in blood or vitreous humour, either with or without DSF
treatment, suggesting that formate is the toxic metabolite in
methanol-induced retinal toxicity. Additionally, intravenous infusion
of formate to levels seen in methanol toxicity did not alter ERG
levels, suggesting intraretinal metabolism of methanol to formate may
be necessary for retinal toxicity.
Studies measuring ATP synthesis in mitochondria isolated from
bovine retina and bovine heart have provided additional evidence for a
tissue-selective action of formate (Eells et al., in press). In these
studies, mitochondrial ATP synthesis was measured in the presence of
different metabolic substrates. Formate selectively inhibited ATP
synthesis in mitochondria isolated from bovine retina in the presence
of metabolic substrates supplying electrons at the level of complex I,
complex II and complex IV in the mitochondrial respiratory chain. The
inhibitory effect of formate on retinal mitochondrial ATP synthesis
was concentration-dependent, significant reductions in ATP synthesis
being produced at 10 mM formate and Ki values for inhibition ranging
from 30 to 50 mM formate. Comparative studies conducted in
mitochondria isolated from bovine heart showed little or no inhibition
of ATP synthesis at formate concentrations up to 50 mM. These findings
provide direct evidence that formate acts as retinal mitochondrial
toxin and suggest that one component of the retinotoxic actions of
formate may be due to tissue-specific differences in mitochondrial
transport mechanisms or in mitochondrial metabolism.
The apparent selective vulnerability of the retina and optic
nerve to the toxic actions of formate in methanol poisoning has been
the subject of considerable speculation (Röe, 1955; Sharpe et al.,
1982; Jacobsen & McMartin, 1986). Although methanol intoxication is
known to disrupt brain function and severe intoxication results in
coma and death, the most common permanent consequence of methanol
intoxication is blindness (Röe, 1955). Several factors may contribute
to the unique vulnerability of the retina and optic nerve to the
cytotoxic actions of formate. One component of this selectivity is
related to the differences in the distribution of formate in the eye
and the brain. Formate concentrations measured in the vitreous humour
and retinas of methanol-intoxicated rats (Eells, 1991; Eells et al.,
1996) were equivalent to or greater than corresponding blood formate
concentrations. In contrast, the concentrations of formate in the
brain were significantly lower than blood formate concentrations.
These data suggest that the toxic actions of methanol on the visual
system may be due to the selective accumulation of formate in the
vitreous humour and the retina as compared with other regions of the
central nervous system. Secondly, the retina has a very limited
metabolic capacity to oxidize and thus detoxify formate (Eells et al.,
1996). Thirdly, cytochrome oxidase activity and ATP concentrations
have been shown to be selectively reduced in the retina, optic nerve
and brain in methanol-intoxicated rats, suggesting that there may be
tissue- and cell-specific differences in mitochondrial populations and
in the actions of formate on mitochondrial function (Eells et al.,
1995). Finally, in vitro studies in isolated retinal and cardiac
mitochondria have shown that formate selectively inhibits retinal
mitochondrial ATP synthesis (Eells et al., in press). These findings
support the hypothesis that formate acts as a selective mitochondrial
toxin in the retina and establish a link between the effects of
formate in vitro and the retinal toxicity associated with formate
accumulation in methanol intoxication.
8. EFFECTS ON HUMANS
Acute oral and inhalation exposures, and to a lesser extent
percutaneous absorption of high concentrations of methanol, have
resulted in CNS depression, blindness, coma and death. The most noted
effects resulting from longer-term exposure to lower levels of
methanol have been a broad range of ocular effects.
8.1 General population and occupational exposure
The human health effects after exposure to methanol are
qualitatively the same for the general population and for those
exposed in the workplace, and will be considered together. Acute
methanol intoxication in the general population is an uncommon
occurrence, but often results in serious morbidity and mortality.
Litovitz et al. (1988) reviewed the acute methanol exposure cases
reported in the USA. In 1987, 1601 methanol poisonings were reported
to the American Association of Poison Control Centers (AAPCC). Half of
these individuals required hospitalization and the death rate was
0.375%. It was estimated that the actual annual incidence of methanol
poisonings in the USA in 1987 was about 6400 cases. Subsequent surveys
of methanol exposure cases have been conducted by the AAPCC, and these
have shown similar annual frequencies to that in 1987. These data
result from poisoning cases that are not usually reported elsewhere,
since case reports of methanol poisoning are rarely published in
today's literature. Poisoning frequency surveys are not available from
the rest of the world, but reports in the biomedical literature and in
the press would suggest a worldwide distribution of methanol poisoning
cases at least as great as in the USA.
8.1.1 Acute toxicity
Methanol (wood alcohol) has been recognized as a human toxic
agent since the end of the 19th century. Since the early part of the
20th century, many hundreds of cases of methanol intoxication have
been reported as single cases and as groups in many countries. Many of
the human cases were due to the ingestion of denatured alcohol.
The preponderance of methanol poisonings have resulted from the
consumption of adulterated alcoholic beverages, e.g., "moonshine", or
"bootleg whiskey", wood alcohol and spirits mixed with whiskey. Buller
& Wood (1904) and Wood & Buller (1904) reported 235 cases of blindness
or death primarily connected with drinking adulterated beverages or
wood alcohol products, but these also included 10 deaths involving
inhalation or absorption of methanol through the skin.
Bennett et al. (1953) described a case that occurred in Atlanta,
Georgia, USA, in 1951, when within a 5-day period, 323 people consumed
bootlegged whiskey contaminated with 35-40% methanol and 41 of them
died. Kane et al. (1968) reported the poisoning of 18 individuals, of
whom 8 died, when a diluted paint thinner containing approximately 37%
(by volume) methanol was used as an alcoholic beverage in Lexington,
Kentucky, USA.
An epidemic in the State Prison of Southern Michigan in 1979 in
which methanol diluent used in photocopying machines was used as
"home-made" spirits (containing approximately 3% methanol) resulted in
46 definite cases of methanol intoxication and 3 deaths (Swartz et
al., 1981). Methanol poisoning among 23 servicemen in an Army hospital
in Korea who had ingested bootleg sake contaminated with methanol was
reported by Keeney & Mellinkoff (1951). Tonning et al. (1956) reported
acute methanol poisoning in 49 naval personnel who consumed drinks
made from duplicating fluid containing a high concentration of
methanol.
An outbreak of acute methanol intoxication involving 28 young men
in Papua New Guinea in 1977, each of whom consumed an equivalent of
60-600 ml pure methanol, resulted in all becoming hospitalized within
8-36 h due to acute metabolic acidosis, severe visual impairment and
acute pancreatitis. Four died within 72 h after hospitalization. Of 24
who recovered, 16 showed no residual complications, 6 had bilateral
visual impairment and 2 had difficulty in speech as well as visual
impairment (Dethlefs & Naraqi, 1978; Naraqi et al., 1979).
Before 1978, many alcoholics in Sweden were reported to
supplement their intake of alcohol with readily available cleansing
solutions containing up to 80% methanol. Since 1978, the methanol
content of such solutions has been limited to 5%. However, consumption
of these solutions by alcoholics is still widely seen, exposures of
1-2 weeks being associated with blood methanol concentrations ranging
from 1000 to 2000 mg/litre (31-62 mmol/litre) (Heath, 1983).
Although ingestion of methanol historically has been shown to be
the most frequent route of poisoning, percutaneously absorption of
methanol liquids or inhalation of its vapour is as effective as the
oral route in producing methanol acute toxic syndrome in adult and
pediatric poisonings (Buller & Wood, 1904; Wood & Buller, 1904;
Giminez et al., 1968; Kahn & Blum, 1979; Dutkiewicz et al., 1980;
Becker, 1983). Giminez et al. (1968) reported 48 children intoxicated
with percutaneously applied alcohol. Thirty of these patients had
severe respiratory depression, 14 were comatose, 11 had seizures, 7
had anuria or severe oliguria and there were 12 deaths.
About 100 cases of amblyopia (impairment of vision) and death
from inhalation of wood alcohol were reported up to 1912, the majority
occurring from occupational exposure to the fumes (Tyson & Schoenberg,
1914). Toxicity has also been associated with inhalation of methanol
vapour in excess of 400 mg/m3 (300 ppm) (Becker, 1983; Frederick et
al., 1984).
Hazardous inhalation exposures of methanol can occur in the
context of intentional inhalation of volatile preparations such as
carburettor cleaners. Frenia & Schauben (1993) reported seven cases
involving four patients who had inhaled a carburettor cleaner
containing toluene (43.8%), methanol (22.3%), methylene chloride
(20.5%) and propane (12.5%). Measured blood methanol levels ranged
from 504 to 1286 mg/litre. Blood formic acid levels were 120, 193 and
480 µg/ml, respectively, in three patients. Ophthalmic examinations
revealed hyperaemic discs and decreased visual acuity in one patient.
Acute methanol toxicity in humans evolves in a fairly defined
pattern. A toxic exposure results in a transient mild depression of
the CNS, similar to that of ethanol, but to a much lesser degree. The
initial depressant period is followed by an asymptotic latent period,
which occurs most commonly about 8-24 h after ingestion of the alcohol
but may last from several hours to 2 or more days. During the latent
period the patients describe no overt symptoms or signs.
The latent period is followed by a syndrome that consists of an
uncompensated metabolic acidosis with superimposed toxicity to the
visual system. Physical symptoms typically may include headache,
dizziness, nausea and vomiting, followed in more severe cases by
abdominal and muscular pain and difficult periodic breathing (Kussmaul
breathing), which may progress to coma and death, usually from
respiratory distress. Death may occur if patients are not treated for
metabolic acidosis, and blindness may result even if treatment for
metabolic acidosis is performed (Bennett et al., 1953; Röe, 1955; Kane
et al., 1968; Tephly & McMartin, 1984; Tephly, 1991).
The neurotoxic effects of methanol on the visual system can
involve transient abnormalities such as peripapillary oedema, optic
disc hyperaemia, diminished pupillary reactions to light, and central
scotomata. Permanent ocular abnormalities include optic disc pallor,
attenuation of arterioles, sheathing of arterioles, diminished
pupillary reactions to light, diminished visual acuity, central
scotomata, and other nerve fibre bundle defects (Bennett et al., 1953;
Dethlefs & Naraqi, 1978; Kavet & Nauss, 1990). Pallor of the optic
disc is an end-stage sign of irreversible effects of the visual system
and may appear 1 to 2 months after an acute methanol dosage (or
possibly following chronic occupational exposure to methanol vapour)
(Buller & Wood, 1904; Wood & Buller, 1904; Bennett et al., 1953).
Within the general population, the range of the dose levels that
is hazardous to humans and the variable susceptibility to acute
effects are well recognized (Buller & Wood, 1904; Wood & Buller, 1904;
Bennett et al., 1953). As little as 15 ml of 40% methanol resulted in
the death of one individual while others survived following the
consumption of 500 ml of the same solution in the Atlanta, Georgia,
epidemic of 1951. There were large individual differences in the
duration of the latency period. Symptoms of methanol poisoning
appeared within a few hours or were delayed for up to 72 h. The
severity of the disease was not related to the length of the latent
period or the amount of methanol consumed (Bennett et al., 1953). (It
should be noted that in earlier reported poisoning epidemics, large
errors in dose estimates may have been made).
In another example of the range of dose levels of methanol that
are toxic, 120 ml (4 fluid ounces) of Columbian spirits, or 95 g of
methanol (Columbian spirits is basically pure methanol), was lethal in
40% of the poisoning cases. For a 70-kg person, this dose is
equivalent to about 1.4 g methanol/kg body weight (Buller & Wood,
1904). This figure is consistent with currently accepted values for
lethality, and 0.3 to 1 g/kg is considered the range of a minimum
lethal dose for untreated cases of methanol poisoning (Röe, 1955;
Erlanson et al., 1965; Gonda et al., 1978).
It has been suggested that the variability in the reaction to
methanol may have been due to the concomitant ingestion of ethanol
with methanol, which resulted in some patients having a longer latent
period prior to the onset of poisoning (Röe, 1950, 1955). Another
explanation for the variability in susceptibility to methanol
poisoning is the different levels of folate in the diet. Folate-
deficient individuals have a lesser capacity to metabolize formate, so
are more susceptible to accumulation of formate to toxic levels (see
section 8.1.7 for sensitive sub-populations).
In some clinical cases, the blood methanol level is low in the
last phase of the poisoning. In three such cases, blood methanol
concentrations were 0.275, 0.277 and 0.194 g/litre, respectively
(Erlanson et al., 1965). On the assumption that the body in diffusion
equilibrium with the blood represents about 70% of the body weight,
Röe (1982) calculated that 0.19-0.14 g/kg of methanol was present in
the body. However, low blood methanol levels do not indicate a lower
susceptibility to toxicity, i.e., blood methanol levels do not
correlate with patient prognosis (Jacobsen & McMartin, 1986). Patients
that are examined late after methanol ingestion are likely to have low
blood methanol levels, yet high accumulation of formate. Such patients
often have poor prognosis.
Acute methanol poisoning patients with blood levels of methanol
above 500 mg/litre are generally regarded as requiring haemodialysis
(Becker, 1983). The dose of methanol required to achieve this blood
concentration is very small (0.4 ml/kg body weight). This corresponds
to the ingestion of 4 ml (less than a teaspoon of 100% methanol by a
10-kg (1-year old) child and 28 ml (less than 1 fluid ounce) by a
70-kg adult (Litovitz et al., 1988).
A case was reported of a 46-year-old man who, after consuming a
beverage containing methanol, exhibited one of the highest reported
serum methanol levels (4930 mg/litre), well above those at which
ethanol treatment and haemodialysis are recommended (200 mg/litre
and 500 mg/litre, respectively). The lowest serum pH was 7.0
with a hydrogen carbonate level of 8.8 and an anion gap of 42.8.
Additionally, his visual acuity decreased to a complete loss of
vision. The patient was aggressively treated with haemodialysis and
ethanol infusion, regained his vision with a visual acuity of 20/30
bilaterally and suffered no neurological sequelae (Pambies et al.,
1993b).
An additional number of cases are particularly informative
regarding treatment of methanol intoxication and sequelae of
poisoning. A case of methanol intoxication was reported involving a
53-year-old man. Along with blindness and metabolic acidosis, this
resulted in cerebral oedema and subarachnoid haemorrhage followed by a
comatose state and subsequent death (del Carpio-O'Donavan & Glay,
1992).
A 31-year-old male alcoholic who consumed ethanol containing
methanol experienced severe signs and symptoms of poisoning. He
underwent minimal medical treatment consisting of sodium hydrogen
carbonate and peritoneal dialysis and exhibited necrosis and
haemorrhage of the (bilateral) putamen and necrosis of bilateral
subcortical white matter and post-contrast gyral enhancement at the
otherwise normal-looking areas of the cerebral cortex by the 22nd day,
as revealed by computed tomography (Hsieh et al., 1992).
A 31-year-old man entered hospital with a 370 mg/litre serum
methanol level after exhibiting the signs and symptoms of methanol
poisoning (nausea, vomiting, diffuse abdominal pain and blurred tunnel
vision) for 7 days. Following a complete regimen of treatment
consisting of hydrogen carbonate, ethanol and folate combined with a
6-h haemodialysis, which corrected the acidosis and eliminated
methanol (methanol decreased to 100 mg/litre by the second day),
permanent blindness still resulted (Vogt et al., 1993).
A case study of acute methanol poisoning in a 27-year-old man
with a previous pattern of drinking was reported by King (1992).
Following a comprehensive treatment regimen consisting of
administration of alkali, fluids and ethanol, intubation and
haemodialysis, this patient exhibited significant neurological and
physical impairment, including trauma to the vocal cords and
hypophonic voice and urinary incontinence (of central origin), along
with cognitive defects. However upon discharge his vision was normal
with no atrophy of the optic nerve.
A case of a severe methanol poisoning in a 33-year-old man with a
history of alcoholism was reported by Burgess (1992). The individual
required 21 h of dialysis to bring the serum methanol levels down to a
non-toxic level. A haemodialysis treatment usually lasts approximately
4 h but this may not be sufficient in severe poisoning. Prolonged
haemodialysis treatment should be considered in cases of severe
poisoning and also possibly for patients with compromised renal
function.
Extensive white and grey matter brain damage was seen in an
alcoholic 37-year-old man who consumed 1900 ml of windshield washer
fluid containing methanol. Both CT scan and MR imaging revealed
diffuse white matter oedema and damage throughout frontal and parietal
lobes. Bilateral changes in the basal ganglia and necrosis and
haemorrhage of putamen were also noted (Glazer & Dross, 1993).
Autopsies from victims of lethal methanol poisonings revealed
gross pathology in the visceral organs, the brain, lung, liver,
kidney and the CNS, all of which involved a variety of oedematous,
haemorrhagic and degenerative changes (Keeney & Mellinkoff, 1951;
Bennett et al., 1953; Tonning et al., 1956; Kaplan, 1962; Erlanson et
al., 1965; McLean et al., 1980; Wu Chen, 1985; Suit & Estes, 1990).
A fatal case involving a 41-year-old man who had ingested a large
quantity of methanol disclosed a broad distribution of methanol in
postmortem tissues and fluids. The highest content of methanol was
found in the kidney (5.13 g/kg) followed by the liver (4.18 g/kg),
vitreous humour (3.9 g/litre), heart (3.45 g/kg), urine
(3.43 g/litre), pericardial fluid (3.29 g/litre), blood (2.84 g/litre)
and stomach contents (2.21 g/litre) (Pla et al., 1991).
Methanol toxicity can cause brain oedema, necrosis, brain atrophy
and cerebral haemorrhage. Putaminal necrosis and haemorrhage result
from the direct toxic effects of the methanol metabolites (e.g.,
formate) and metabolic acidosis in the basal ganglia. The typical
appearance of bilateral putaminal necrosis has been described as
characteristic of methanol toxicity (Gonda et al., 1978).
Optic neuropathy and putaminal necrosis are the two main
complications of methanol poisoning generally occurring in combination
after severe poisoning of either suicidal or accidental origin (Sharpe
et al., 1982).
A case study of a woman who drank a substantial amount of
methylated spirits, which resulted in optic neuropathy and putaminal
necrosis, has been reported (Pelletier et al., 1992). The woman
exhibited tremor and rigidity, hypokinesia, altered speech and loss of
superficial and proprioceptive sensation of the lower extremities with
hyperpathia. Signs of moderate bilateral sensory neuropathy and
extrapyramidal damage persisted for 2 months as did total blindness
due to optic atrophy. Repeat CT and MRI examinations revealed the
damage to be a core lesion of the putamen with residual bilateral
putaminal hypodensity suggestive of an ischaemic and necrotic process
possibly including disruption of the blood-brain barrier.
Postmortem analysis of methanol concentrations in body fluids and
tissues reported in fatal human cases of methanol poisoning has
revealed higher concentrations of methanol in cerebrospinal fluid
(CSF), vitreous humour and bile than in blood (Bennett et al., 1953;
Wu Chen et al., 1985). In tissues, the highest concentrations were
found in brain, kidney, lung and spleen, and there were lower
concentrations in skeletal muscle, pancreas, liver and heart (Wu Chen
et al., 1985).
Postmortem signs of damage to the basal ganglia in the brain,
specifically the putamen, have been reported in several cases
(Erlanson et al., 1965; Aquilonius et al., 1978; Suit & Estes, 1990).
A number of human studies have shown that survivors of severe methanol
poisoning may suffer residual disorders as a permanent complication
(Erlanson et al., 1965; Guggenheim et al., 1971; Aquilonius et al.,
1978; McLean et al., 1980; Ley & Gali, 1983). Ley & Gali (1983)
described a case of Parkinsonian syndrome after methanol intoxication.
Co-ingestion of methanol with other solvents, e.g., methyl ethyl
ketone (MEK) (found in multiple ink cleaning products) has resulted in
a hyperosmolar coma without anion gap metabolic acidosis in one
reported case of poisoning. MEK was believed to have inhibited
methanol metabolism contributing to the low serum formate (1.3
mmol/litre) and normal anion gap despite a blood methanol level of
67 mmol/litre (Price et al., 1994).
8.1.2 Clinical features of acute poisonings
The time course of clinical effects due to acute methanol
poisoning is heavily dose-dependent. Blood methanol concentrations of
> 500 mg/litre are associated with severe acute clinical signs of
toxicity, although formate concentrations may give a better indication
of potential toxicity (National Poisons Information Service, 1993).
Thirty minutes to 2 h after ingestion of methanol, clinical
effects resemble those of mild ethanol inebriation, and drowsiness,
confusion and irritability are often noted. After a latent period,
which can range from a few hours to 30 h (but may appear as early as
1 h or as late as 72 h), the patient shows mild CNS depression
followed by abdominal pain, nausea, vomiting, hypernoea, gradually
failing vision, progressive encephalopathy, severe metabolic acidosis
and hypokalaemia; coma and death may ensue. Patients may complain of
blurred or "snowfield" vision with whiteness, spots or mistiness
within the visual field. Survivors may have permanent blindness or
various neurological sequelae. Mortality and morbidity may be more
related to the time between ingestion and therapy rather than to the
initial methanol levels, thus emphasizing the need for rapid treatment
(Mahieu et al., 1989; National Poisons Information Service, 1993;
Pambies et al., 1993a).
Metabolic acidosis associated with high anion and osmolal gaps is
considered an important laboratory indicator of methanol poisoning
(Kruse, 1992). The difference between measured and calculated
osmolality or osmolal gap permits a rough estimation of alcohol
concentrations (Pappas et al., 1985) so that specific therapy is often
initiated before results of quantitative methanol determinations are
available.
The determination of osmolal and anion gaps are readily available
techniques in the initial handling of poisoning with unknown agents
and of patients with a metabolic acidosis of unknown origin. A
combined increase in both anion and osmolal gaps has been shown to be
a sensitive marker of either ethylene glycol or methanol poisoning
(Jacobsen & McMartin, 1986). Reported earlier reference values for
osmolal gap and anion gap are -1 (+ 6) mosm/kg H2O and 16 (+ 2)
mmol/litre, respectively (Jacobsen et al., 1982b). However, Aabakken
et al. (1994) determined osmolal and anion gaps in populations that
were consecutively admitted to a hospital emergency department and
suggested that the present reference values for anion and osmolal gaps
may be too narrow. They further suggested that the values for the
osmolal gap should be 5 + 15 mosm/kg H2O (-10 to + 20 mosm/kg H2O)
and for the anion gap should be 13 + 9 mmol/litre (4-20 mmol/litre).
In their previous reports of methanol poisonings, all patients
exceeded these ranges (Jacobsen et al., 1982).
Demedts et al. (1994) hypothesized that excessive serum
osmolality gaps that are not predictive of methanol levels as
frequently seen in acute poisonings may be attributed to methodology
used to measure methanol (analysing samples using head-space GC were
compared to results found with gas-chromatography using split-mode
injections). Although the determination of increased anion gap is
suggestive of methanol poisoning, definitive evidence would be
increased blood or serum formate concentrations.
Characteristic clinical and laboratory findings in methanol
poisoning are summarized as follows:
* Physical findings
a) Kussmaul respiration (difficult, periodic breathing)
b) faint odour of methanol on breath
c) visual disturbances
d) nausea, vomiting, abdominal pain
e) altered sensation
* Laboratory findings
a) elevated anion gap
b) metabolic acidosis
c) elevated osmol gap
d) positive serum methanol and/or serum formate assay
In treating methanol poisoning a 3-step procedure is common:
1) administration of hydrogen carbonate to combat metabolic acidosis;
2) administration of ethanol to compete as a substrate for alcohol
dehydrogenase, and 3) haemodialysis to remove methanol from the blood
(Erlanson et al., 1965; Gonda et al., 1978; McCoy et al., 1979; Lins
et al., 1980; Jacobsen et al., 1982a,b; Pappas & Silverman, 1982;
Becker, 1983; Jacobsen & McMartin, 1986; Kruse, 1992; Pambies et al.,
1993a,b). Current recommendations are that ethanol treatment be
conducted for patients with blood methanol levels of 200 mg/litre or
more, while haemodialysis be used above 500 mg/litre (Jacobsen &
McMartin, 1986).
The rationale for the administration of ethanol (Röe, 1950;
Keyvan-Larijarni & Tannenbaum, 1974; McCoy et al., 1979; Becker, 1983)
is that alcohol dehydrogenase, the enzyme responsible for converting
methanol to formaldehyde and formic acid, is also involved in the
metabolism of ethanol to acetaldehyde and acetate. The conversion of
methanol to its toxic by-products is slowed in the presence of ethanol
due to competition for the enzyme.
4-Methyl pyrazole (4-MP) is a more specific inhibitor of alcohol
dehydrogenase, less toxic than pyrazole and has been shown to
dramatically inhibit production of formic acid from methanol in
experimental animals (Blomstrand et al., 1979; McMartin et al.,
1980b). Monkeys given usually lethal doses of methanol survived when
treated with 4-MP following methanol administration (McMartin et al.,
1980b). In humans the slower elimination rate and lesser degree of
toxicity of 4-MP suggested that it might be preferable to ethanol in
the treatment of methanol poisoning (Jacobsen et al., 1990). 4-MP is
currently undergoing clinical trials for treatment of methanol
poisoning.
Haemodialysis effectively removes methanol and formate from the
circulation (Erlanson et al., 1965; Gonda et al., 1978; McCoy et al.,
1979). If haemodialysis is not available, peritoneal dialysis has been
used with some success in treating acute methanol intoxication
(Keyvan-Larijarnc & Tannenberg, 1974). Discussion of the treatment of
methanol poisoning can be found in the IPCS Poisons Information
Monograph (PIM) No. 335 (IPCS, 1991).
8.1.3 Repeated or chronic exposure
In comparison to acute toxicity, reports of effects from repeated
or chronic methanol exposures have been only infrequently reported.
Information based on a limited number of case reports and even fewer
epidemiological studies (generally containing unknown levels and/or
durations of methanol exposure) suggests that extended exposure to
methanol may cause effects qualitatively similar to those observed
from relatively high levels of acute exposure, including in some cases
CNS and visual disorders (Buller & Wood, 1904; Wood & Buller, 1904;
Greenberg et al., 1938; Bennett et al., 1953; Kingsley & Hirsch, 1955;
Frederick et al., 1984).
Greenberg et al. (1938) studied 19 workers employed in the
production of "fused collars", where solutions of acetone-methanol
(3:1) were used to impregnate collars which were then steam-pressed.
Methanol concentrations in the work room were 29-33 mg methanol/m3
and 96-108 mg acetone/m3. The shortest period of employment in this
occupation was 9 months and the longest was 2 years. No CNS symptoms
or visual anomalies were observed.
Frederick et al. (1984) reported on teacher aides who worked at
or near spirit duplicators that used a 99% methanol duplicator fluid.
The exposures ranged from 1 h/day for 1 day/week to 8 h/day for 5
days/week and had occurred for 3 years. Since the introduction of the
equipment, the aides began to experience headaches, dizziness and eye
irritation, blurred vision and nausea/upset stomach while working near
the machines. Fifteen-minute breathing zone samples near 21 operating
machines contained between 475 and 4000 mg/m3 of methanol vapour.
Fifteen of these samples exceeded the NIOSH recommended 15-min
standard of 1050 mg/m3 (800 ppm). The aides were also exposed while
collating and stapling papers impregnated with the fluid up to 3 h
earlier and these exposures ranged from 235-1140 mg/m3 . The results
suggested that chronic effects may occur when methanol concentrations
exceed the threshold limit value (TLV) of 260 mg/m3 (200 ppm). The
effects reported in the study of Frederick et al. (1984) were similar
in nature but appeared less severe than those reported from acute
poisoning by methanol (Buller & Wood, 1904; Wood & Buller, 1904;
Bennett et al., 1953).
Kingsley & Hirsch (1955) reported frequent and persistent
headaches, but no visual effects or other permanent sequelae, in
clerical workers located close to spirit duplicating equipment that
used methanol-based duplicating fluid. Methanol concentrations were
reported to be as high as 490 mg/m3 in the air surrounding the
duplicating equipment after 60 min of operation and approximately
130 mg/m3 about 3 m away from the device. The methanol concentration
around the duplicating equipment always exceeded 260 mg/m3. No
information was provided concerning the number of employees exposed or
affected, nor on the actual duration of methanol exposure.
NIOSH (1981) reported that 45% of "spirit" duplicating machine
operators at the University of Washington experienced some symptoms
(blurred vision, headache, nausea, dizziness and eye irritation),
consistent with the toxic effects of methanol. Airborne methanol
concentrations of 1330 mg/m3 were measured in the vicinity of the
duplicators when windows and doors were open. No information on the
actual length of duration of methanol exposure among the employees
engaged in the duplicating machine operations were provided.
A number of other studies have measured methanol and formate in
the blood and urine of workers exposed during an 8-h day to between
100 and 200 mg/m3 of methanol vapour (Baumann & Angerer, 1979;
Heinrich & Angerer, 1982. Although these studies were predicated on
issues of occupational health related to methanol exposure, no health
effects were provided nor did the investigators imply that the workers
studied had suffered health effects.
Kawai et al. (1991b), utilizing methanol in urine as a biological
indicator of occupational exposure, compared subjective complaints and
major clinical findings among 33 methanol-exposed workers over several
8-h workshifts. Urine levels of methanol in controls were on average
1.9 ± 0.8 mg/litre (n = 91), and in 14 exposed workers pre-shift
concentrations were significantly elevated compared to controls. At
the end of the shift the urine concentrations were generally above
100 mg/litre in 8 men with a mean exposure level of 1690 mg/m3 and
30-100 mg/litre in 6 men with a mean exposure level of 550 mg/m3. The
highest exposures (breathing zone, 8-h/samples) were 4000-7000 mg/m3
and corresponding urine levels 300-500 mg/litre. The leading
subjective complaints included: dimmed vision and nasal irritation
during work, and headache, dimmed vision, forgetfulness and increased
sensitivity of the skin in the extremities when off-work. The authors
attributed the dimmed vision to the fog created by methanol vapours
and high humidity in air. No visual problems were noted when windows
were kept open and fresh air was allowed to flow in. It was also noted
that there were no complaints of photophobia (and thus perhaps no
major corneal involvement). Fundus photography revealed that the optic
discs were normal and thus the symptom of dimmed vision was not
recognized as a sign of impending retinal involvement. In three
workers with methanol exposures of 1250-2130 mg/m3, 1385-2075 mg/m3
and 155-4685 mg/m3 (953-1626 ppm, 1058-1585 ppm and 119-3577 ppm) the
reaction of pupils to light was slow in two subjects, and a third
subject had slight mydriatic pupils. The duration of service of the
workers ranged from 0.3 to 7.8 years. The exposures were high and the
methods for measurement of visual toxicity were relatively crude, but
the data did not indicate that occupational exposure to such
concentrations caused permanent damage.
The effects of methanol vapour (249 mg/m3; SD + 7 mg/m3) for
75 min on neurobehavioural measures were studied in 12 healthy young
men. The exposure produced significant increases (approximately 3
fold) in blood and urine methanol levels but no changes in plasma
formate level. Although most of the neurobehavioural end-points were
unaffected by exposure to methanol, statistically significant effects
and trends were found for a cluster of variables, including the
latency of the p-200 component of event-related potentials,
performance on the Sternberg memory task and subjective measures of
fatigue and concentration. However, the effects were small and did not
exceed the normal range (Cook et al., 1991).
8.1.4 Reproductive and developmental effects
No studies have been reported in the peer-reviewed literature on
the reproductive and developmental effects of methanol in humans.
8.1.5 Chromosomal and mutagenic effects
No studies have been reported in the peer-reviewed literature on
chromosomal or mutagenic effects of methanol in humans.
8.1.6 Carcinogenic effects
No studies have been reported in the literature on the
carcinogenicity of methanol in humans.
8.1.7 Sensitive sub-populations
Folate-deficient individuals might be at greater risk from
inhalation of low concentrations of methanol, compared to normal
individuals. Human populations that are potentially at high risk of
folate deficiency include pregnant women, the elderly, individuals
with poor-quality diets, alcoholics and individuals on certain
medications or with certain diseases (API, 1993).
It has been suggested that the metabolic acidosis due to methanol
might be exacerbated in individuals with diabetes since it is well
known that these patients suffer from diabetic ketoacidosis (Posner,
1975). However, there are no clinical or experimental data on any
interaction between methanol acidosis and diabetic ketoacidosis.
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1 Aquatic organisms
9.1.1 Microorganisms
The toxicity of methanol to each of three bacterial groups, i.e.,
aerobic heterotrophic, Nitrosomonas and methanogens (key agents in the
natural recycling of organic material in the environment and in
wastewater treatment systems), was described by Blum & Speece (1991).
The following IC50 values (mg/litre) (the concentration that
inhibited the culture by 50%) compared to the uninhibited controls
were reported: Nitrosomonas (after 24-h exposure), 880 mg/litre;
methanogens (after 48-h exposure), 22 000 mg/litre; and aerobic
heterotrophs (after 15-h exposure), 20 000 mg/litre. Methanol was
found to be completely inhibitory to ammonia oxidation by
Nitrosomonas bacteria at a concentration of 5 × 10-3 M (about
160 mg/litre) (Hooper & Terry, 1973).
A 15-min EC50 of 14 700 mg/litre for the luminescent marine
bacterium Photobacterium phosphoreum and a 4-h LC50 value of 1.0%
by volume (7690 mg/litre) have been reported (Schiewe et al., 1985).
Calleja et al. (1994) found the EC50 for the marine bacterium
Photobacterium phosphoreum in the Microtox(R) test to be
29 348 mg/litre. Rajini et al. (1989) reported a 10-min LC50 of 6%
(44 860 mg/litre) for the ciliate protozoan Paramecium caudatum.
Toxicity threshold values for methanol in the cell multiplication
inhibition test of 6600 mg/litre for the bacterium Pseudomonas
putida and > 10 000 mg/litre for the protozoa Entosiphonsulcatum
were reported by Bringmann & Kühn (1980).
An experimental EC50 value (the concentration that reduced the
maximum observed biodegradation rate by 50%) for methanol of
2.8 mol/litre (89.7 g/litre) was obtained in a system employing an
enriched mixed microbial culture derived from domestic waste water in
the USA (Vaishnav & Lopas, 1985).
9.1.2 Algae
Stratton (1987) determined the following EC50 values:
Anabaena cylindrica: 2.57% (20 300 mg/litre)
Anabaena inaequalis: 2.68% (21 179 mg/litre)
Anaebaena sp.: 3.12% (24 650 mg/litre)
Anaebaena variabilis: 3.13% (24 730 mg/litre)
Nostoc sp.: 5.48% (43 290 mg/litre)
For the green alga Chlorella pyrenoidosa an EC50 value of
28 440 mg/litre was found (Stratton & Smith, 1988). Bringman & Kühn
(1978), employing a cell multiplication test, reported a toxicity
thresholds of 8000 mg/litre for the green alga Scenedesmus
quadricauda and 530 mg/litre for the cyanobacterium (blue green
alga) Microcystis aeruginosa.
9.1.3 Aquatic invertebrates
The toxicity of methanol, as reported for a broad spectrum of
aquatic invertebrates, is summarized in Table 6. EC50 values for the
water flea (Daphnia magna) range from 13 240 to 24 500 mg/litre.
Helmstetter et al. (1996) exposed the mussel, Mytilus edulis, to
methanol concentrations of 1, 2, 3, 5 and 10% (v/v) for 96 h. All the
mussels in both the 5 and 10% exposure groups died within 13.5 h.
Sublethal narcotic effects such as slow movement and sporadic filter
feeding were reported in mussels exposed to 2 and 3%. Mussels exposed
to 1% methanol exhibited no adverse effects during the 96-h exposure
period.
9.1.4 Fish
The acute toxicity to fish is listed in Table 7. LC50 values
reported for freshwater fish species range from 10 880 to
29 700 mg/litre.
The physiological changes in the carp (Cyprinus carpio)
affected by a sub-lethal methanol concentration of 1 ml/litre
(790 mg/litre) included a significant increase in blood cortisol
levels after 6 h of exposure, but not after 24 or 72 h, significant
decreases in blood protein and cholesterol levels after 72 h of
exposure, and reduced concentration of glycogen in the liver after
72 h. Methanol did not produce significant changes in blood glucose
levels after any duration of exposure (Gluth & Hanke, 1985).
The effect of methanol on the fertilization of chum salmon
(Oncorhynchus keta) ova was examined at methanol exposure levels of
0.001% to 10% by volume (7.9 to 79 000 mg/litre) (Craig et al., 1977).
Both gametes (sperm and unfertilized ova) and fertilized eggs were
exposed to methanol for brief periods. Exposures up to and including
1% methanol did not significantly affect fertilization, survival to
hatching, hatching time, alevin size at hatch or physical deformities
among alevins, although a methanol concentration of 10% was lethal in
most cases (Craig et al., 1977).
Cuéllar et al. (1995) determined the effect of methanol on the
embryonic development of the medaka fish (Oryzias latipes). The eggs
were exposed to methanol in both Petri dishes and vials. No effects on
embryonic development were reported at a methanol concentration of
0.5%.
Table 6. Acute toxicity of methanol to aquatic invertebrates
Organism Size/age Stat/ Temp Hardness pH Parameterc Concentration Reference
flowa (°C) (mg/litre)b (mg/litre)d
Water flea <24 h stat 20 (1) 7.8-8.2 48-h EC50e >10 000 n Kuhn et al. (1989)
(Daphnia magna)
<24 h stat 20 (1) 7.8-8.2 48-h EC0e >10 000 n Kuhn et al. (1989)
<24 h stat 20 (2) 7.8-8.2 24-h EC50 >10 000 n Bringmann & Kuhn
(1982)
<24 h stat 20 (2) 7.8-8.2 24-h EC100 >10 000 n Bringmann & Kuhn
(1982)
<24 h stat 20 154.5 7.0-8.2 24-h EC50 24 500 n Bringmann & Kuhn
(1982)
48-h LC50 13 240 n Vaishnav &
Korthals (1990)
24-h EC50 21 402 n Calleja et al.
(1994)
Water flea <24 h stat 22 23±2 18-h-LC50 19 500 n Bowman et al.
(Daphnia pulex) (1981)
Water flea <24 h stat 20±2 250 7.8±0.2 24-h EC50 23 500 n Rossini & Ronco
(Daphnia obtusa) <24 h stat 20±2 250 7.8±0.2 48-h EC50 22 200 n (1996)
Brown shrimp adult stat 15 seawater 48-h LC50 1975 n Portmann & Wilson
(Crangon crangon) (1971)
adult stat+ 15 seawater 96-h LC50 1340 n Portmann & Wilson
(1971)
stat 24.5 seawater 24-h LC50 10 000 n Price et al. (1974)
Brine shrimp 24 h stat 25 seawater 24-h LC50 1578.84 n Barahona-Gomariz
(Artemia salina) et al. (1994)
48 h stat 25 seawater 24-h LC50 1101.46 n Barahona-Gomariz
et al. (1994)
Table 6. Continued
Organism Size/age Stat/ Temp Hardness pH Parameterc Concentration Reference
flowa (°C) (mg/litre)b (mg/litre)d
Brine shrimp 72 h stat 25 seawater 24-h LC50 900.73 n Barahona-Gomariz
(Artemia salina) et al. (1994)
seawater 24-h LC50 43 574 n Calleja et al.
(1994)
Glass shrimp juvenile stat 23±2 18 h-LC50 21 900 n Bowman et al.
(Palaemonetes (1981)
kadiakensis)
Streptocephalus 24-h LC50 32 681 n Calleja et al.
proboscideus (1994)
Mussel 5-7 cm flow 15±0.5 seawater 96-h LC50 15 900 m Helmstetter et al.
(Mytilus edulis) (1996)
Cockle adult stat 15 seawater 48-h LC50 7900 n Portmann & Wilson
(Cardium edule) (1971)
adult stat+ 15 seawater 96-h LC50 2610- Portmann & Wilson
7900 n (1971)
Harpacticoid, adult stat 21±1 7s 7.9 96-h LC50 12 000 n Bengtsson et al.
copepod (1984)
(Nitocra spinipes)
Scud juvenile stat 23±2 18-h LC50 19 350 n Bowman et al.
(Hyalella azteca) (1981)
Table 6. Continued
Organism Size/age Stat/ Temp Hardness pH Parameterc Concentration Reference
flowa (°C) (mg/litre)b (mg/litre)d
Rotifer 24-h LC50 35 884 Calleja et al.
(Brachionus (1994)
calyciflorus)
a stat = static conditions (water unchanged for duration of test); stat+ = semi-static conditions (test solutions
renewed every 24 h); flow = flow through conditions (concentration of toxicant continuously maintained);
s = salinity, expressed as %
b hardness expressed as mg CaCO3 litre, unless stated otherwise; (1)- total hardness = 2.4 mmol/litre;
(2)- total hardness = 2.5 mmol/litre
c All EC50 values refer to immobilization
d n = nominal concentration; m = measured concentration
e same as 24 h EC50 and EC0 values
Table 7. Acute toxicity of methanol to fish
Organism Size/age Stat/ Temp Hardness pH Parameter Concentration Reference
flow (°C) (mg/litre)b (mg/litre)c
Rainbow trout (juv) 0.813 g flow 12.7±1 46.4 7.0-8.0 24-h EC50d 13 200 m Poirier et al.
(Oncorhynchus (1986)
mykiss)
(juv) 0.813 g flow 12.7±1 46.4 7.0-8.0 96-h EC50e 13 000 m Poirier et al.
(1986)
(juv) 0.813 g flow 12.7±1 46.4 7.0-8.0 24-h LC50 20 300 m Poirier et al.
(1986)
(juv) 0.813 g flow 12.7±1 46.4 7.0-8.0 96-h LC50d 20 100 m Poirier et al.
(1986)
0.8 g stat 12 44 7.4 96-h LC50 19 000 n Mayer &
Ellersieck (1986)
(fingerlings) flow 12 96-h LC50d 20 100 m US EPA (1983)
1-6 g
Fathead minnow (28-32 d) flow 23.3±1.7 46.4 7.0-8.0 24-h EC50d 29 700 m Poirier et al.
(Pimephales 0.126 g (1986)
promelas)
(28-32 d) flow 23.3±1.7 46.4 7.0-8.0 96-h EC50e 28 900 m Poirier et al.
0.126 g (1986)
(28-32 d) flow 23.3±1.7 46.4 7.0-8.0 24-h LC50d 29 700 m Poirier et al.
0.126 g (1986)
(28-32 d) flow 23.3±1.7 46.4 7.0-8.0 96-h LC50 29 400 m Poirier et al.
0.126 g (1986)
(30 d) 0.12 g flow 24-26 45.5 7.5 96-h LC50 28 100 m Veith et al.
(1983)
Table 7. Continued
Organism Size/age Stat/ Temp Hardness pH Parameter Concentration Reference
flow (°C) (mg/litre)b (mg/litre)c
Bluegill sunfish (juv) 3.07 g flow 19.8±2.3 46.4 7.0-8.0 24-h EC50e 16 100 m Poirier et al.
(Leponimis (1986)
macrochirus)
(juv) 3.07 g flow 19.8±2.3 46.4 7.0-8.0 24-h EC50e 16 100 m Poirier et al.
(1986)
(juv) 3.07 g flow 19.8±2.3 46.4 7.0-8.0 48-h EC50e 16 000 m Poirier et al.
(1986)
(juv) 3.07 g flow 19.8±2.3 46.4 7.0-8.0 96-h EC50e 12 700 m Poirier et al.
(1986)
(juv) 3.07 g flow 19.8±2.3 46.4 7.0-8.0 24-h LC50d 19 100 m Poirier et al.
(1986)
(juv) 3.07 g flow 19.8±2.3 46.4 7.0-8.0 96-h LC50 15 400 m Poirier et al.
(1986)
1.5 g flow 25 24-h LC50d 19 230 m US EPA (1983)
1.5 g flow 25 96-h LC50 15 500 m US EPA (1983)
Guppy 2-3 months stat+ 21-23 25 7-day LC50 10 860 m Konemann (1981)g
(Poecilia Hermens &
reticulata) Leeuwangh (1982)
Golden orfe juv stat 19-21 (1) 7.0-8.0 48-h LC50 >10 000f m Juhnke &
(Leuciscus idus Lüdemann
melanotus) (1978)
juv stat 19-21 (1) 7.0-8.0 48-h LC0 7900f m Juhnke &
Lüdemann
(1978)
juv stat 19-21 (1) 7.0-8.0 48-h LC100 >10 000f m Juhnke &
Lüdemann
(1978)
Table 7. Continued
Organism Size/age Stat/ Temp Hardness pH Parameter Concentration Reference
flow (°C) (mg/litre)b (mg/litre)c
Bleak 8 cm stat 10 7s 7.9 96-h LC50 28 000 n Bengtsson
(Alburnus et al. (1984)
alburnus)
Armed bullhead adult stat+ 15 seawater 96-h LC50d 7900- Portmann &
(Agonus 26 070 n Wilson (1971)
cataphractus)
a stat = static conditions (water unchanged for duration of test)
stat+ = semi-static conditions (test solutions renewed every 24 hours)
flow = flow through conditions (concentration of toxicant continuously maintained)
s = salinity, expressed as %
b hardness expressed as mg/CaCO3/litre, unless otherwise stated; (1)- total hardness = 2.7 mmol/litre;
c n = nominal concentration
m = measured concentration
d same as 48-h LC50 or EC50 values
e effects on equilibrium, behaviour and coloration
f two laboratories following the same test protocol, same result from each laboratory
g consulted for experimental method only
9.2 Terrestrial organisms
9.2.1 Plants
Hemming et al. (1995) determined the effect of methanol on the
respiration of pepper (Capsicum annuum), tomato (Lycopersicon
esulentum) and petunia (Petunia hybrida). Whole plants were
exposed to either methanol vapour or methanol solution. The general
response to methanol was the same for the three species, with a
respiratory rate increase of up to 50% at the lower methanol
concentrations tested. The response was the same for exposure to
methanol vapour or solution. Exposure of a single leaf resulted in a
systemic response throughout the whole plant within a few hours. The
response lasted for several weeks. Decreased metabolic rates and
waterlogged appearance were reported in plants following a brief
exposure of a leaf to methanol concentrations > 30%. Root tissue
was reported to be more sensitive; a decrease in metabolic rate was
reported following brief exposures to > 10% methanol.
10. EVALUATION OF EFFECTS ON HUMAN HEALTH AND THE ENVIRONMENT
10.1 Evaluation of human health risks
10.1.1 Exposure
Methanol occurs naturally in humans, animals and plants. Humans
are routinely exposed to low levels of methanol from both the diet
(fruits, vegetables, fruit juices and foods containing the synthetic
sweetener aspartame) and metabolic processes. Human exposure to large
acutely toxic amounts of methanol via the oral route has principally
been noted in a relatively small number of individuals, generally
resulting through accidental or intentional consumption of methanol in
illicit or contaminated alcoholic beverages.
Methanol is produced in large amounts in many countries and is
extensively used as an industrial solvent, a chemical intermediate
(principally in the production of methyl tertiary butyl ether (MTBE),
formaldehyde, acetic acid and glycol ethers), as a denaturant of
ethanol and in a variety of consumer products.
The most important route of occupational exposure to methanol is
inhalation. Sources of occupational exposure include the dissipative
emissions of methanol primarily occurring from miscellaneous solvent
usage, methanol production, end-product manufacturing and bulk storage
and handling.
An increased number of people could be potentially exposed to
environmental methanol as a result of the projected expanded use of
methanol in methanol-blended gasolines. Exposures would principally
arise from exhaust, evaporative emissions and normal heating of the
engine. Simulation models based on 100% of all vehicles powered by
methanol-based fuels predict concentrations of methanol in urban
streets, expressways, railroad tunnels or parking garages ranging from
a low of 1 mg/m3 (0.77 ppm) to a high of 60 mg/m3 (46 ppm).
Predicted concentrations during refuelling of vehicles range from 30
to 50 mg/m3 (23-38.5 ppm). For comparison and reference purposes, a
current occupational exposure limit for methanol in many countries is
260 mg/m3 (200 ppm) for an 8-h working day.
There are limited data on human dermal exposure to methanol but
the potential expanded use of methanol in automotive fuels would
increase the potential for dermal exposure in a large number of
people.
10.1.2 Human health effects
Methanol is rapidly absorbed by inhalation, ingestion and dermal
exposure and is rapidly distributed to tissues according to the
distribution of body water. The dose and blood concentrations of
methanol and its metabolite formate are among the major determinants
of the resultant toxicity in humans.
The acute and short-term toxicity of methanol varies greatly
between different species, toxicity being highest in species with a
relatively poor ability to metabolize formate. Methanol has been
studied most intensively in acute high-dose oral exposures in
laboratory animals and as case reports of ingestion in humans. In
general, humans and primates respond to such exposures with transient
central nervous system (CNS) depression (intoxication), followed by an
asymptomatic latent period culminating in metabolic acidosis and
severe ocular toxicity (blindness).
Non-primate animals such as rodents do not ordinarily exhibit
metabolic acidosis or blindness on exposure to methanol although they
exhibit the general narcotic effects noted in non-human primates and
humans. The clearance of formate from the blood of exposed primates is
at least 50% slower than in rodents. Formate, an endogenous biological
substrate, is detoxified by a multi-step pathway to CO2 via a
tetrahydrofolate (THF)-dependent pathway. Species such as rodents with
high hepatic THF levels are less sensitive to the toxic effects of
methanol than species with low hepatic THF levels such as humans and
non-human primates. The faster rate of formate removal means that
rodents do not accumulate formate above endogenous levels and hence
are not susceptible to methanol-induced metabolic acidosis or ocular
toxicity.
The primary enzymatic pathway that catalyses methanol metabolism
in humans and non-human primates is alcohol dehydrogenase, while in
the rat it is the catalase-peroxidase system. Available data suggest
that methanol elimination from the systemic circulation is capacity-
limited in both rats and in humans.
Studies in humans and non-human primates exposed to
concentrations of methanol ranging from 13 to 2601 mg/m3 (10 to
2001 ppm) and the widely used occupational exposure limit of
260 mg/mg3 (200 ppm) suggest that exposure to methanol vapour during
the normal use of methanol fuel does not pose an unacceptable risk to
healthy adults. General population exposures to methanol through air
(although infrequently measured) are over 1000 times lower than
occupational limits.
Along with methanol, formate is present in blood at low
endogenous concentrations, being found naturally in some foods and
also produced as a by-product of several metabolic pathways, including
histidine and tryptophan degradation. Background levels of formate in
humans have been shown to range from 3 to 19 mg/litre (0.07-0.4 mM).
Human susceptibility to the acute effects of methanol
intoxication are extremely variable. On the basis of available human
case reports, the minimum lethal dose in the absence of medical
treatment is in the range of 0.3 to 1 g/kg. The major determinants of
human susceptibility to methanol toxicity appear to be the concurrent
ingestion of ethanol, which slows the entrance of methanol into the
metabolic pathway, and the hepatic status of THF, which governs the
rate of formate detoxification.
Some human populations are at increased risk of folate
deficiency. These include pregnant women, the elderly, individuals
with poor-quality diets, alcoholics, and individuals on certain
medications or with certain diseases.
Much fewer data are available on the health effects in humans or
laboratory animals associated with chronic or repeated exposure to
methanol. In the absence of details of exposure (e.g., duration,
concentrations), the effects of prolonged exposure are considered
qualitatively very similar to those reported for acute cases, ranging
from nausea and dizziness to blurred vision and temporary or permanent
blindness. Chronic exposure to methanol vapour concentrations of
480-4000 mg/m3 (365-3080 ppm) has resulted in headache, dizziness,
nausea and blurred vision.
There are no reports of carcinogenic, genotoxic, reproductive or
developmental effects in humans due to methanol exposures.
10.1.3 Approaches to assessment of risk
The assessment of risk from chronic exposure requires dose-
response information in the form of quantitative data from animal
studies using appropriate test species and, where available, relevant
human epidemiological and clinical data. In the case of methanol, the
assessment of the risks of exposure is confounded by the fact that
both methanol and its toxic metabolite, formate, are endogenous
metabolic intermediates in all species including humans. Therefore, it
must be assumed that there are levels of methanol exposure that do not
represent significant risk. Determining the hazards associated with
methanol exposure is additionally complicated by the fact that there
are no adequate or comprehensive data from animal tests for chronic
toxicity. Because of species differences in methanol metabolism, data
available from normal rats appear to be inappropriate for use in
characterizing the adverse effects of methanol in humans.
Investigation of folate-deficient rodent models may provide valuable
mechanistic, pharmacokinetic and toxicological effect information on
methanol, particularly with respect to acute exposures. However, the
nature of this animal model is such that it may have inherent
weaknesses for the toxicological assessment of long-term exposure
because of the adverse effects of folate deficiency itself and the
background nutritional status of these rats in chronic studies.
Similarities in the metabolism of methanol within primates suggest the
use of non-human primates may be more appropriate for determining the
nature of the hazards of methanol for humans, but adequate findings
for chronic exposure are also lacking. Human methanol exposure data
are extensive but primarily focus on acute exposure and clinical
effects associated with poisoning. Although this information from
humans does highlight the wide individual variability in the toxic
response to methanol in humans, it contains limited comprehensive
information on sub-chronic to chronic methanol exposure.
Taken together, the above considerations suggest a conventional
safety or risk assessment would not appear feasible, and would most
likely be incomplete at present. An alternative approach might be one
based on consideration of blood levels of the most toxic metabolite,
formate. Since formate occurs naturally in humans, it would seem
reasonable to assume that normal background levels should not pose any
risk to health and consequently that levels of human exposure that do
not result in levels of blood formate above background levels could be
considered to pose insignificant risk. In this respect, based on
information from limited studies in humans, it might be concluded that
occupational exposure to current exposure limits (around 260 mg/m3)
or single oral exposure to approximately 20 mg/kg body weight would
fall into this category.
10.2 Evaluation of effects on the environment
Methanol may be released into the environment in significant
amounts during its production, storage, transportation and use.
Methanol is readily degraded in the environment by photo-
oxidation. Half-lives of 7-18 days have been reported for the
atmospheric reaction of methanol with hydroxyl radicals.
Methanol is readily biodegradable under both aerobic and
anaerobic conditions in a wide variety of environmental media. Many
genera and strains of microorganisms are capable of using methanol as
a growth substrate. Generally 80% of methanol in sewage systems is
biodegraded within 5 days.
Methanol is a normal growth substrate for many soil micro-
organisms, which are capable of completely degrading methanol to
carbon dioxide and water.
Methanol is of low toxicity to aquatic and terrestrial organisms
and it is not bioaccumulated. Effects due to environmental exposure to
methanol are unlikely to be observed, unless it is released to the
environment in large quantities, such as a spill.
In summary, unless released in high concentrations, methanol
would not be expected to persist or bioaccumulate in the environment.
Low levels of release would not be expected to result in adverse
environmental effects.
11. RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH AND THE
ENVIRONMENT
11.1 Protection of human health
a) Methanol and methanol mixtures should be clearly labelled
with a warning of the acute toxicity of methanol. Labels
should use the description "methanol".
b) Storage, process and drying plants should be designed to
protect against fire and explosion risks and exposure of
personnel to methanol.
c) Workplaces where methanol is present should be provided with
adequate ventilation to minimize inhalation exposure. Where
necessary, personnel handling methanol should be provided
with suitable protective clothing to prevent skin
contamination.
d) Clinicians should be aware of the latent period and signs
and symptoms following exposure to methanol, particularly by
ingestion. Consideration associated with the existence of
sensitive subgroups should be recognized, including those at
increased risk of folate deficiency.
e) To avoid misuse, methanol used as fuel should be denatured
and should contain a colour additive.
11.2 Protection of the environment
Although methanol is rapidly degraded in the environment and is
of low acute toxicity to aquatic organisms, care should be taken to
prevent spills of large quantities of methanol. Particular care should
be taken to prevent spilled methanol reaching surface water.
12. FURTHER RESEARCH
a) Further research is needed to characterize the mechanism and
pathogenesis of methanol-induced visual toxicity.
b) There is a need for definitive studies concerning the dose-
response relationship for subtle CNS function using
neurotoxic, neurobehavioural and ocular end-points across
species at both single and repeated low-level exposures.
c) Investigation of the metabolism of methanol and formate in
target organs, including the brain, retina, optic nerve and
testes, under various exposure conditions is needed.
d) The pharmacokinetics of methanol and formate during
pregnancy should be investigated in appropriate animal
models to determine whether long-term exposure to methanol
alters maternal or fetal disposition of methanol and
formate.
e) Additional studies are required to resolve whether methanol,
formate or a combination of the two is responsible for
methanol-induced developmental toxicity.
f) Exposure models should be developed and validated to
estimate exposure concentrations and routes of exposure in
specific exposure scenarios. Ambient and personal monitoring
to determine the distribution of exposures should be
conducted.
g) Dose-effect and time-course relationships for both acute and
chronic effects of methanol or formate generated from
methanol, in humans or appropriate models, have not been
established and are essential for adequate risk assessment.
h) There is a need for studies into the nutritional, metabolic,
genetic and age-related factors that may contribute to
variation in susceptibility to methanol intoxication.
i) The genotoxic effects of methanol should be further
investigated to determine whether it is clastogenic.
j) A rapid, practical and inexpensive assay for formate in
blood and body tissues is needed for early diagnosis of
methanol poisoning.
k) Improved therapeutic measures, including the development of
4-methylpyrazole and new agents for reversing formate-
induced visual neurotoxicity, are needed.
13. PREVIOUS EVALUATION BY INTERNATIONAL BODIES
Methanol was evaluated in 1970 as an extraction solvent by the
Joint FAO/WHO Expert Committee on Food Additives.
The Committee recommended that when used as an extraction
solvent, residues should be reduced to a minimum by observing good
manufacturing practice. It was considered that the limited uses of
methanol as an extraction solvent for spice and hop oils meant that
residues from these sources were insignificant in the diet (FAO/WHO,
1971; WHO, 1971).
REFERENCES
Aabakken L, Johansen KS, Rydningen EB, Bredesen JE, Ovrebos S, &
Jacobsen D (1994) Osmolal and anion gaps in patients admitted to an
emergency medical department. Hum Exp Toxicol, 13: 131-134.
Abbondandolo A, Bonatti S, Corsi C, Corti G, Fiorio R, Leporini C,
Mazzacccaro A, & Nieri R (1980) The use of organic solvents in
mutagenicity testing. Mutat Res, 79: 141-150.
Abbott BD, Logsdon TR, & Wilke TS (1994) Effects of methanol on
embryonic mouse palate in serum-free organ culture. Teratology, 49:
122-134.
Abbott BD, Ebron-McCoy M, & Andrews JE (1995) Cell death in rat and
mouse embryos exposed to methanol in whole embryo culture. Toxicology,
97: 159-171.
Agarwal VK (1988) Determination of low relative molecular mass
alcohols in gasoline by gas chromatography. Analyst, 113: 907-909.
Akimoto H & Takagi H (1986) Formation of methyl nitrite in the surface
reaction of nitrogen dioxide and methanol: 2. Photoenhancement.
Environ Sci Technol, 20: 393-397.
Allis JW, Simmons JE, Robinson BL, McDonald A, & House DE (1992)
Induction of rat hepatic cytochrome P-450II E1 by methanol: Its role
in the enhancement of carbon tetrachloride hepatotoxicity.
Toxicologist, 12: 85.
Allis JW, Brown BL, Simmons JE, Hatch GE, McDonald A, & House DE
(1996) Methanol potentiation of carbon tetrachloride hepato toxicity:
the central role of cytochrome P450. Toxicology, 112: 131-170.
Anderson EV (1993) Health studies indicate MTBE is safe gas additive.
Chem Eng News, 71(38): 9-18.
Andrews LS, Clary JJ, Terrill JB, & Bolte HF (1987) Subchronic
inhalation toxicity of methanol. J Toxicol Environ Health, 20:
117-124.
Andrews JE, Ebron-McCoy M, Logsdon TR, Mole LM, Kavlock RJ, & Rogers
JM (1993) Developmental toxicity of methanol in whole embryo culture:
A comparative study with mouse and rat embryos. Toxicology, 81:
205-215.
Andrews JE, Ebron-McCoy M, Kavlock RJ, & Rogers JM (1995)
Developmental toxicity of formate and formic acid in whole embryo
culture: A comparative study with mouse and rat embryos. Teratology,
51: 243-251.
Angerer J & Lehnert G (1977) Occupational exposure to methanol. Acta
Pharmacol, 41: 551-556.
Anon (1991) Methanol: tight markets ahead. Chem Ind, 17: 598.
AOAC (1980) In: Horwitz W ed. Official methods of analysis of the
Association of Official Analytical Chemists, 13th ed. Washington, DC,
Association of Official Analytical Chemists, pp 9.107, 9.086-9.093.
AOAC (1990) In: Helrich K ed. Official methods of analysis of the
Association of Official Analytical Chemists, 15th ed. Washington, DC,
Association of Official Analytical Chemists, pp 702-705.
API (1993) Study of the relationship between folate studies and
methanol toxicity. Washington, DC, American Petroleum Industry
(Publication No. 4554).
Aquilonius S, Askmark H, Enoksson P, Lundberg PO, & Mostrom U (1978)
Computerized tomography in severe methanol intoxication. Br Med J,
ii: 929-930.
ASTM (1993) Standard test methods for the determination of MTBE, ETBE,
TAME, DIPE, tert-amyl alcohol and C1 to C4 alcohols in gasoline by
gas chromatography (D4815). Philadelphia, Pennsylvania, American
Society for Testing and Materials.
ATSDR (Agency for Toxic Substances Disease Registry) (1993) Methanol
toxicity. Am Fam Phys, 47: 163-174.
Auto/Oil Air Quality Improvement Research Program (1992) Emissions and
air quality modeling results from methanol/gasoline blends and
prototype flexible/variable vehicles. Atlanta, Georgia, US Gasoline
Producers, Coordinating Research Council (Technical Bulletin No.7).
Auto/Oil Air Quality Improvement Research Program (1994) Emissions
from methanol fuels and reformulated gasoline in 1993 production
flexible/variable fuel and gasoline vehicles (Technical Bulletin No.
13).
Axelrod J & Daly J (1965) Pituitary gland : enzymic formation of
methanol from S-adenosylmethionine. Science, 158: 892-993.
Baker RN, Alenty Al, & Zack JF Jr (1969) Simultaneous determination
of lower alcohols, acetone and acetaldeyhde in blood by gas
chromatography. J Chromatogr Sci, 7: 312-314.
Barahona-Gomariz MV, Sanz-Barrera F, & Sánchez-Fortún S (1994) Acute
toxicity of organic solvents on Artemia salina. Bull Environ Contam
Toxicol, 52: 766-771.
Barnes I, Bastian V, Becker KH, Fink EH, & Zabel F (1982) Reactivity
studies of organic substances towards hydroxyl radicals under
atmospheric conditions. Atmos Environ, 16: 545-550.
Bartlett GR (1950) Inhibition of methanol oxidation by ethanol in the
rat. Am J Physiol, 163: 619-621.
BASF (1979) [Report on the comparative testing on sensitizing effects
in guinea pigs, modified maximization test.] Ludwigshafen, Germany,
BASF AG, 11 pp (Unpublished report) (in German).
BASF (1980a) [Determination of the acute inhalation toxicity LD50 of
methanol (Merck min. 99.8%) as vapour at 4 hours exposure to Sprague-
Dawley rats.] Ludwigshafen, Germany, BASF AG, 14 pp (Unpublished
report) (in German).
BASF (1980b) [Determination of the acute inhalation toxicity LD50 of
methanol (Merck min. 99.8%) as vapour at 6 hours exposure to Sprague-
Dawley rats.] Ludwigshafen, Germany, BASF AG, 22 pp (Unpublished
report) (in German).
Baumann K & Angerer J (1979) Occupational chronic exposure to organic
solvents: VI. Formic acid concentration in blood and urine as an
indicator of methanol exposure. Int Arch Occup Environ Health, 42:
241-249.
Baumbach Gl, Cancilla PA, Martin-Amat G, Tephly TR, McMartin KE, Makar
Ab, Hayreh M, & Haryeh SS (1977) Methyl alcohol poisoning: IV.
Alterations of the morphological findings of the retina and optic
nerve. Arch Ophthalmol, 95: 1859-1865.
Becker CE (1983) Methanol poisoning. J Emerg Med, 1: 51-58.
Bellar TA & Sigsby JE Jr (1970) Direct gas chromatographic analysis of
low molecular weight substituted organic compounds in emissions.
Environ Sci Technol, 4: 150-156.
Bengtsson BE, Renberg L, & Tarkpea M (1984) Molecular structure and
aquatic toxicity-an example with C1-C13 aliphatic alcohols.
Chemosphere, 13(5/6): 613-622.
Bennett IL, Cary FH, Mitchell GL, & Cooper MN (1953) Acute methyl
alcohol poisoning: a review based on experiences in an outbreak of 323
cases. Medicine, 32: 431-463.
Benoit FM, Davidson WR, Lovett AM, Nacson S, & Ngo A (1985) Breath
analysis by API/MS-human exposure to volatile organic solvents. Int
Arch Occup Environ Health, 55: 113-120.
Bindler F, Voges E, & Laugel P (1988) The problem of methanol
concentration admissible in distilled fruit spirits. Food Addit
Contam, 5: 343-351.
Blanch GP, Tabera J, Sanz J, Herraiz M, & Reglero G (1992) Volatile
composition of vinegars. Simultaneous distillation-extraction and gas
chromatographic-mass spectrometric analysis. J Agric Chem, 40:
1046-1049.
Blomstrand R, Ostling-Wintzell H, Lof A, McMartin K, Tolf BR, &
Hedstrom KG (1979) Pyrazoles as inhibitors of alcohol oxidation and as
important tools in alcohol research: an approach to therapy against
methanol poisoning. Proc Natl Acad Sci (USA), 76: 3499-3503.
Blum DJW & Speece RE (1991) A data base of chemical toxicity to
environmental bacteria and its use in interspecies comparisons and
correlations. Res J Water Pollut Control Fed, 63: 198-207.
Bock JL (1982) Analysis of serum by high-field proton nuclear magnetic
resonance. Clin Chem, 28: 1873-1877.
Boeniger MF (1987) Formate in urine as a biological indicator of
formaldehyde exposure: a review. Am Ind Hyg Assoc J, 48: 900-908.
Bolon B, Dorman DC, Janszen D, Morgan KT, & Welsch F (1993) Phase
specific developmental toxicity in mice following maternal methanol
inhalation. Fundam Appl Toxicol, 21: 508-516.
Bolon B, Welsch F, & Morgan KT (1994) Methanol induced neural tube
defects in mice: pathogenesis during neuralation. Teratology, 49:
497-517.
Boos RN (1948) Quantitative colorimetric microdetermination of
methanol with chromotropic acid. Anal Chem, 20: 964-965.
Bowman MC, Oller WL, Cairns T, Gosnell AB, & Oliver KH (1981) Stressed
bioassay systems for rapid screening of pesticide residues. Part I:
Evaluation of bioassay systems. Arch Environ Contam Toxicol, 10:
9-24.
Braun M & Stolp H (1985) Degradation of methanol by a sulfate reducing
bacterium. Arch Microbiol, 142: 77-80.
Bresnick GH (1989) Excitotoxins: A possible new mechanism for the
pathogenesis of ischemic retinal damage. Arch Ophthalmol, 107:
339-341.
Bringmann G & Kühn R (1980) Comparison of the toxicity thresholds of
water pollutants to bacteria, algae and protozoa in the cell
multiplication inhibition test. Water Res, 14: 231-241.
Buller F & Wood CA (1904) Poisoning by wood alcohol. J Am Med Assoc,
43: 1058-1062.
Burbacher TM (1993) Neurotoxic effects of gasoline and gasoline
constituents. Environ Health Perspect, 101(Suppl 6): 133-141.
Burgess E (1992) Prolonged hemodialysis in methanol intoxication.
Pharmacotherapy, 12: 238-239.
Buttery JE & Chamberlain BR (1988) A simple enzymatic method for the
measurement of abnormal levels of formate in plasma. J Anal Toxicol,
12: 292-294.
Calleja MC, Persoone G, & Geladi P (1994) Comparative acute toxicity
of the first 50 multicentre evaluation of in vitro cytotoxicity
chemicals to aquatic non-vertebrates. Arch Environ Contam Toxicol,
26: 69-78.
Cameron AM, Nilsen OG, Haug E, & Eik-Nes KB (1984) Circulating
concentrations of testosterone, luteinizing hormone and follicle
stimulating hormone in male rats after inhalation of methanol. Arch
Toxicol, 7(Suppl): 441-443.
Cameron AM, Zahlsen K, Haug E, Nilsen OG, & Eik-Nes KB (1985)
Circulating steroids in male rats following inhalation of n-alcohols.
Arch Toxicol, 8(Suppl): 422-424.
Campbell JA, Howard DR, Backer LC, & Allen JW (1991) Evidence that
methanol inhalation does not induce chromosome damage in mice. Mutat
Res, 260: 257-264.
Casey JC, Self R, & Swain T (1963) Origin of methanol and dimethyl
sulfide from cooked foods. Nature (Lond), 200: 885.
Cavanaugh LA, Schadt CF, & Robinson E (1969) Atmospheric hydrocarbon
and carbon monoxide measurements at Point Barrow, Alaska. Environ Sci
Technol, 3: 251-257.
CEC (Commission of the European Communities) (1988) Solvents in common
use: Health risks to workers. Cambridge, Royal Society of Chemistry,
pp 1-7, 157-186 (Publication EUR/11553).
Chang TY & Rudy SJ (1990) Ozone-forming potential of organic emissions
from alternative-fueled vehicles. Atmos Environ, 24A: 2421-2430.
Chang LW, McMillan L, Wynne BR, Pereira MA, Colley RA, Ward JB, &
Legator MS (1983) The evaluation of six monitors from the exposure to
formaldehyde in laboratory animals. Environ Mutagen, 5: 381-387.
Chao CT (1959) [Data for determining the standard maximum permissible
concentration of methanol vapours in the atmospheric air.] Gig I
Sanit, 24: 7-12 (in Russian).
Cheung ST & Lin WN (1987) Simultaneous determination of methanol,
ethanol, isopropanol and ethylene glycol in plasma by gas
chromatography. J Chromatogr, 414: 248-250.
Clark CB, Dutcher JS, McClellan RG, Naman TM, & Seizinger DE (1983)
Influence of ethanol and methanol gasoline blends on the mutagenicity
of particulate exhaust extracts. Arch Environ Contam Toxicol, 12:
311-317.
Clay KL, Murphy RC, & Watkins WD (1975) Experimental methanol toxicity
in the primate: analysis of metabolic acidosis. Toxicol Appl
Pharmacol, 34: 49-61.
Clayton GD & Clayton FE ed. (1982) Patty's industrial hygiene and
toxicology - Volume 2C: Toxicology with cumulative index for Volume 2,
3rd ed. New York, Chichester, Brisbane, Toronto, John Wiley & Sons, pp
4527-4551.
Coe JI & Sherman RE (1970) Comparative study of postmortem vitreous
..... and blood alcohol. J Forensic Sci, 25: 185-190.
Coleman EC, Ho CT, & Chang SS (1981) Isolation and identification of
volatile compounds from baked potatoes. J Agric Food Chem, 29:
42-48.
CONCAWE (1995) Alternative fuels in the automotive market. Brussels,
CONCAWE, 67 pp (Report No. 2/95, prepared for the CONCAWE Automotive
Emissions Management Group by its Technical Coordinator, RC
Hutcheson).
Cook WA (1945) Maximum allowable concentrations of industrial
atmospheric contaminants. Ind Med, 14: 936-946.
Cook MR, Bergman FJ, Cohen HD, Gerkovich MM, Graham O, Harris RX, &
Siemann LG (1991) Effects of methanol vapor on human neurobehavioural
measures. Cambridge, Massachusetts, Health Effects Institute (Research
Report No. 42).
Cooper JR & Felig P (1961) The biochemistry of methanol poisoning: II.
Metabolic acidosis in the monkey. Toxicol Appl Pharmacol, 3:
202-209.
Cooper RL, Rehnberg GL, Goldman JM, Linder RE, Mole ML, Edwards TL,
Hein JF, & McElroy WK (1990) Effect of methanol on hormonal control of
testes in male rats. Toxicologist, 10: 211.
Cooper Rl, Mole ML, Rehnberg GL, Goldman JM, McElroy WK, Hein J, &
Stoker TE (1992) Effects of inhaled methanol on pituitary and
testicular hormones in chamber acclimated and non-acclimated rats.
Toxicology, 71: 69-81.
Costantini MG (1993) Health effects of oxygenated fuels. Environ
Health Perspect, 101(Suppl 6): 151-160.
Craig PC, Withler FC, & Morley RB (1977) Effects of methanol on the
fertilization of chum salmon (Oncorhynchus keta) ova. Environ
Pollut, 14: 85-91.
Crebelli R, Conti G, Conti L, & Carere A (1989) A comparative study on
ethanol and acetaldehyde as inducers of chromosome malsegregation in
Aspergillus nidulans. Mutat Res, 215: 187-195.
Creel DJ, Dustman RD, & Beck EC (1970) Differences in visually evoked
responses in albino versus hooded rats. Exp Neurol, 29: 298-309.
Cuéllar M, González M, & Muñoz MJ (1995) [Methanol toxicity on the
embryonic development of Oryzias latipes.] Rev Toxicol, 12:
109-113 (in Spanish).
Cummings AM (1993) Evaluation of the effects of methanol during early
pregnancy in the rat. Toxicology, 79: 205-214.
D'Alessandro A, Osterloh JD, Chuwers P, Quinlan PJ, Kelly TJ, & Becker
CE (1994) Formate in serum and urine after controlled methanol
exposure at the threshold limit value. Environ Health Perspect, 102:
178-181.
Damian P & Rabbe OG (1996) Toxicokinetic modelling of dose-dependent
formate elimination in rats: in vivo-in vitro correlations using
perfused rat liver. Toxicol Appl Pharmacol, 139: 22-32.
Davoli E, Cappellini L, Airoldi L, & Fanelli R (1986) Serum methanol
concentrations in rats and in men after a single dose of aspartame.
Food Chem Toxicol, 24: 187-189.
De Flora S, Zanacchi P, Camoirano A, Bennicelli C, & Badolati GS
(1984) Genotoxic activity and potency of 135 compounds in the Ames
reversion test and in a bacterial DNA-repair test. Mutat Res, 133:
161-198.
Deichmann WB (1948) Methanol. J Ind Hyg Toxicol, 30: 373-376.
Del Carpio-O'Donovan L & Glay J (1992) Subarachnoid hemorrhage
resulting from methanol intoxication: Demonstrated by computed
tomography. Can Assoc Radiol J, 43: 263-299.
Demedts P, Theunis L, Wauters A, Franck F, Daelemans R, & Neels H
(1994) Excess serum osmolality gap after ingestion of methanol: A
methodology-associated phenomenon? Clin Chem, 40: 1587-1590.
Dethlefs R & Naraqi S (1978) Ocular manifestations and complications
of acute methyl alcohol intoxication. Med J Aust, 2: 483-485.
Diaz-Rueda J, Sloane HJ, & Obremski RJ (1977) An infrared solution
method for the analysis of trapped atmospheric contaminants desorbed
from charcoal tubes. Appl Spectrom, 31: 298-307.
Donnelly MI & Dagley S (1980) Production of methanol from amino acids
by Pseudomonas putida. J Bacteriol, 142: 916-924.
Dorman DC, Dye JA, Nassise MP, Ekuta J, Bolon B, & Medinsky MA (1993)
Acute methanol toxicity in minipigs. Fundam Appl Toxicol, 20:
341-347.
Dorman DC, Moss OR, Farris GM, Janszen D, Bond JA, & Medinsky MA
(1994) Pharmacokinetics of inhaled 14C-methanol and methanol-derived
14C-formate in normal and folate-deficient cynomologus monkeys.
Toxicol Appl Pharmacol, 128: 229-238.
Dorman DC, Bolon B, Struve MF, La Perle KMD, Wong BA, Elswick BE, &
Welsch F (1995) Role of formate in methanol-induced exencephaly.
Teratology, 52: 30-40.
Dowling JE (1987) The electroretinogram and glial responses. In:
Dowling JE ed. The retina: An approachable part of the brain.
Cambridge, Massachusetts, Belknapp Press, pp 164-186.
Downie A, Khattab TM, Malik IA, & Samara IN (1992) A case of
percutaneous industrial methanol toxicity. Occup Med, 42: 47-49.
Dube MF & Green CR (1982) Methods of collection of smoke for
analytical purposes. Recent Adv Tob Sci, 8: 42-102.
Dutkiewicz B, Konczalik J, & Karwacki W (1980) Skin absorption and per
os administration of methanol in men. Int Arch Occup Environ Health,
47: 81-88.
Ebron-McCoy MT, Andrews JE, Kavlock RJ, & Rogers LM (1994) The
developmental toxicity of formate and formic acid in mouse and rat
embryos in whole culture (WEC). Teratology, 49: 393.
Eells JT (1991) Methanol- induced visual toxicity in the rat. J
Pharmacol Exp Ther, 257: 56-63.
Eells JT, Makar AB, Noker PE, & Tephly TR (1981a) Methanol poisoning
and formate oxidation in nitrous oxide-treated rats. J Pharmacol Exp
Ther, 217: 57-61.
Eells JT, McMartin KE, Black K, Virayotha V, Tisdell RH, & Tephly TR
(1981b) Formaldehyde poisoning: a rapid metabolism to formic acid. J
Am Med Assoc, 246: 1237-1238.
Eells JT, Black KA, Makar AB, Tedford CE, & Tephly TR (1982) The
regulation of one-carbon oxidation in the rat by nitrous oxide and
methionine. Arch Biochem Biophys, 219: 316-326.
Eells JT, Black KA, Tedford CE, & Tephly TR (1983) Methanol toxicity
in the monkey: effects of nitrous oxide and methionine. J Pharmacol
Exp Ther, 227: 349-353.
Eells JT, Salzman MM, & Trusk TC (1995) Inhibition of retinal
mitochondrial function in methanol intoxication. Toxicologist, 15:
21-23.
Eells JT, Salzman MM, Lewandowski MF, & Murray TG (1996) Formate
induced alterations in retinal function in methanol-intoxicated rats.
Toxicol Appl Pharmacol, 140: 58-69.
Eells JT, Salzman MM, Lewandowski MF, & Murray TG (in press)
Developmental and characterization of a non-primate animal model of
methanol induced neurotoxicity. In: Bengston DA & Henschel DS ed.
Environmental toxicology and risk assessment: Biomarkers and risk
assessment - Volume 5. Philadelphia, Pennsylvania, American Society
for Testing and Materials, (Publication ASTM STP No. 1306).
Egle JL & Gochberg BJ (1979) Retention of inhaled isoprene and
methanol in the dog. Am Ind Hyg Assoc J, 36: 369-373.
Eisenberg AA (1917) Visceral changes in wood alcohol poisoning by
inhalation. Am J Public Health, 7: 765-771.
Eisenreich SJ, Looney BB, & Thornton JD (1981) Airborne organic
contaminants in the Great Lakes ecosystem. Environ Sci Technol, 15:
30-38.
Elvers B, Hawkins S, & Schulz G ed. (1990) Ullmann's encyclopedia of
industrial chemistry, 5th ed. Weinheim, Germany, VCH-Verlag, vol 16A,
pp 465-486.
Eriksen SP & Kulkarni AB (1963) Methanol in normal human breath.
Science, 141: 639-640.
Erlanson P, Fritz H, Hagstam KE, Liljenberg B, Tryding N, & Voigt G
(1965) Severe methanol intoxication. Acta Med Scand, 177: 393-408.
Ewell WS, Gorsuch JW, Kringle RO, Robillard KA, & Spiegel RC (1986)
Simultaneous evolution of the acute effects of chemicals on seven
aquatic species. Environ Toxicol Chem, 5: 831-840.
FAO/WHO (1971) Toxicological evaluation of some extraction solvents
and certain other substances. Report of the Joint FAO/WHO Expert
Committee on Food Additives, 24 June-2 July 1970. Rome, Food and
Agriculture Organization, pp 105-109.
Federal Register (1989) Standards for emissions from methanol-fueled
motor vehicles and motor vehicle engines - Final rule. Fed Reg,
54(68): 40CFR Part 86.
Feldstein M & Klendshoj RE (1954) Determination of methanol in
biological fluids by microdiffusion analysis. Anal Chem, 26:
932-933.
Ferry DG, Temple WA, & McQueen EG (1980) Methanol monitoring. Int Arch
Occup Environ Health, 47: 155-163.
Fiedler E, Grossmann G, Kersebohm B, Weiss G, & Witte C (1990)
Methanol. In: Elvers B, Hawkins S, & Schutz G ed. Ullmann's
encyclopedia of industrial chemistry, 5th ed. Weinheim, VCH
Verlagsgesellschaft, vol 16A, pp 465-486.
Filley CM & Kelly JP (1993) Alcohol-and drug-related neurotoxicity.
Current Opin Neurol Neurosurg, 6: 443-447.
Fox ME (1973) Rapid gas chromatographic method for determination of
residual methanol in sewage. Environ Sci Technol, 7: 838-840.
Francot P & Geoffroy P (1956) Le méthanol dans les jus de fruits, les
boissons fermentées des alcools et spiriteux. Rev Ferment Ind Aliment,
11: 279-287.
Franzblau A, Levine SP, Burgess LA, Qu QS, Schreck RM, & D'Arcy JB
(1992a) The use of a transportable Fourier Transform infrared (FTIR)
spectrometer for the direct measurement of solvents in breath and
ambient air: I. Methanol. Am Ind Hyg Assoc J, 53: 221-227.
Franzblau A, Levine SP, D'Arcy JB, & Qu QS (1992b) Use of urinary
formic acid as a biologic exposure index of methanol exposure. Appl
Occup Environ Hyg, 7: 467-471.
Franzblau A, Lee EW, Schreck RM, D'Arcy JB, Santrock J, & Levine SP
(1993) Absence of formic acid accumulation in urine following five
days of methanol exposure. Appl Occup Environ Hyg, 8: 883-888.
Frederick LJ, Schulte PA, & Apol A (1984) Investigation and control of
occupational hazards associated with the use of spirit duplicators. Am
Ind Hyg Assoc J, 45: 51-55.
Freitag D, Ballhorn L, Geyer H, & Korte F (1985) Environmental hazard
profile of organic chemicals. Chemosphere, 14: 1589-1616.
Frenia Ml & Schauben JL (1993) Methanol inhalation toxicity. Ann Emerg
Med, 22: 1919-1923.
Gabele PA & Knapp KT (1993) A characterization of emissions from an
early model flexible-fuel vehicle. J Air Waste Manage Assoc, 43:
851-858.
Garcia JH & Van Zandt JP (1969) Proceedings of 27th Annual Meeting of
the Electron Microscope Society of America. Baton Rouge, Louisiana,
Claitor Publishers, vol 27, pp 360-361.
Garner CD & Lee EW (1994) Evaluation of methanol-induced
retinotoxicity using oscillatory potential analysis. Toxicology,
93: 113-124.
Garner CD, Lee EW, & Louis-Ferdinand RJ (1995a) Muller cell
involvement in methanol-induced retinal toxicity. Toxicol Appl
Pharmacol, 130: 101-107.
Garner CD, Lee EW, Terzo TS, & Louis-Ferdinand RJ (1995b) The role of
retinal metabolism in methanol-induced retinal toxicity. J Toxicol
Environ Health, 44: 43-56.
Gessner PK (1970) Method for the assay of ethanol and other aliphatic
alcohols applicable to tissue homogenates and possessing a sensitivity
of 1 µg/ml. Anal Biochem, 38: 499-505.
Gettler AO (1920) Critical study of methods for the detection of
methyl alcohol. J Biol Chem, 42: 311-328.
Geyer H, Politzki G, & Freitag D (1984) Prediction of ecological
behavior of chemicals: relationship between n-octanol/water partition
and bioaccumulation of organic chemicals by alga Chlorella.
Chemosphere, 13: 269-284.
Gilger AP & Potts AM (1955) Studies on the visual toxicity of
methanol: V. The role of acidosis in experimental methanol poisoning.
Am J Ophthamol, 39: 63-86.
Gilger AP, Potts AM, & Johnson JV (1952) Studies on the visual
toxicity of methanol: II. The effect of parenterally administered
substances on the systemic toxicity of methyl alcohol. Am J Ophthamol,
35(Part 2): 113-126.
Giminez ER, Vallejo NE, Roy E, Lis M, Izurieta EM, Rossi S, & Capuccio
M (1968) Percutaneous alcohol intoxication. Clin Toxicol, 1: 39-48.
Glazer M & Dross P (1993) Necrosis of the putamen caused by methanol
intoxication: MR findings. Am J Roentgenol, 160: 1105-1106.
Gluth G & Hanke W (1985) A comparison of physiological changes in
carp, Cyprinus carpio, induced by several pollutants at sublethal
concentrations: 1. The dependency on exposure times. Ecotoxicol
Environ Saf, 9: 179-188.
Gluth G, Freitag D, Hanke W, & Korte F (1985) Accumulation of
pollutants in fish. Comp Biochem Physiol, 81C: 273-277.
Gold MD & Moulif CE (1988) Effects of emission standards on methanol
vehicle-related ozone, formaldehyde and methanol exposure. Presented
at 81st Meeting of Air Pollution Control Association, Dallas, TX, June
19-24. Pittsburgh, Pennsylvania, Air Pollution Control Association.
Gonda A, Gault H, Churchill D, & Hollomby D (1978) Hemodialysis for
methanol intoxication. Am J Med, 64: 749-757.
Goodman JI & Tephly TR (1971) A comparison of rat and human liver
formaldehyde dehydrogenase. Biochim Biophys Acta, 252: 489.
Grady S & Osterloh J (1986) Improved enzymic assay for serum formate
with colorimetric endpoint. J Anal Toxicol, 10: 1-5.
Graedel TE, Hawkins DT, & Claxton LD ed. (1986) Atmospheric chemical
compounds: Sources, occurrence and bioassay. New York, London,
Academic Press, pp 512-514, 557.
Grayson M ed. (1981) Kirk-Othmer encyclopedia of chemical technology,
3rd ed. New York, John Wiley & Son, vol 15, pp 398-405.
Greenberg L, Mayers MR, Goldwater LJ, & Burke WJ (1938) Health hazards
in the manufacture of "fused collars": II. Exposure to acetone-
methanol. J Ind Hyg Toxicol, 20: 148-154.
Greizerstein HB (1981) Congener contents of alcoholic beverages. J
Stud Alcohol, 42: 1030-1037.
Griffiths AJF (1981) Neurospora and environmentally induced
aneuploidy. In: Stich HF & San RHC ed. Short-term tests for chemical
carcinogens. Berlin, Heidelberg, New York, Springer-Verlag,
pp 187-199.
Guerin MR, Higgins CE, & Greist WH (1987) The analysis of the
particulate and vapour phases of tobacco smoke. In: O'Neill IK,
Brunnemann KD, Dodet B, & Hoffmann D ed. Environmental carcinogens:
Methods of analysis and exposure measurement, Volume 9. Lyon,
International Agency for Research on Cancer, pp 115-139 (IARC
Scientific Publications No. 81).
Guggenheim MA, Couch JR, & Weinberg W (1971) Motor dysfunction as a
permanent complication of methanol ingestion. Arch Neurol, 24:
550-554.
Güsten H, Klasinc L, & Maric D (1984) Prediction of the abiotic
degradability of organic compounds in the troposphere. J Atmos Chem,
2: 83-93.
Haggard HW & Greenberg LA (1939) Studies on the absorption,
distribution and elimination of alcohol: IV. The elimination of
methylalcohol. J Pharmacol Exp Ther, 66: 479-496.
Hanson RS (1980) Ecology and diversity of methylotropic organisms. Adv
Appl Microbiol, 26: 3-29.
Harger RN (1935) A simple micromethod for the determination of alcohol
in biological material. J Lab Clin Med, 20: 746-751.
Hatfield R (1957) Biological oxidation of some organic compounds. Ind
Eng Chem, 49: 192-197.
Hayreh MS, Hayreh SS, Baumbach GL, Cancilla P, Martin-Amat G, Tephly
TR, McMartin KE, & Makar AB (1977) Methyl alcohol poisoning: III.
Ocular toxicity. Arch Ophthamol, 95: 1851-1858.
Heath A (1983) Methanol poisoning. Lancet, 1: 1339-1340.
Heath M (1991) Alternative transportation fuels: Natural gas, propane,
methanol and ethanol compared with gasoline and diesel. Calgary,
Canadian Energy Research Institute, pp 113-115.
HEI (1987) Automotive methanol vapors and human health: An evaluation
of existing scientific information and issues for future research.
Cambridge, Massachusetts, Health Effects Institute, 70 pp (Special
Report).
HEI (1996) The potential health effects of oxygenates added to
gasoline: A review of the current literature. Cambridge,
Massachusetts, Health Effects Institute, 165 pp.
Heidelberger C, Freeman AE, Pienta RJ, Sivak A, Bertram DS, Casto BC,
& Dunkel VC (1983) Cell transformation by chemical agents: A review
and analysis of the literature. Mutat Res, 114: 283-385.
Heinrich R & Angerer J (1982) Occupational chronic exposure to organic
solvents. Int Arch Occup Environ Health, 50: 341-349.
Helmstetter A, Gamerdinger AP, & Pruell RJ (1996) Acute toxicity of
methanol to Mytilus edulis. Bull Environ Contam Toxicol, 57(4):
675-681.
Hemming DJB, Criddle RS, & Hansen LD (1995) Effects of methanol on
plant respiration. J Plant Physiol, 146(3): 193-198.
Hermens J & Leeuwangh P (1982) Joint toxicity of mixtures of 8 and 24
chemicals to the guppy (Poecilia reticulata) Ecotoxicol Environ Saf,
6: 302-310.
Hertelendy ZI, Mendenhall CL, Rouster SD, Marshall L, & Weesner R
(1993) Biochemical and clinical effects of aspartame in patients with
chronic, stable alcoholic liver disease. Am J Gastroenterol, 88:
737-743.
Hickman GT & Novak JT (1989) Relationship between subsurface
biodegradation rates and microbial density. Environ Sci Technol, 23:
525-532.
Hickman GT, Novak JT, Morris MS, & Rebhun M (1989) Effects of site
variations on subsurface biodegradation potential. J Water Pollut
Control Fed, 61: 1564-1575.
Hindberg J & Wieth JO (1963) Quantitative determination of methanol in
biological fluids. J Lab Clin Med, 61: 355-361.
Hippe H, Caspari D, Fiebig K, & Gottschalk G (1979) Utilization of
trimethylamine and other N-methyl compounds for growth and methane
formation by methanosarcina barkeri. Proc Natl Acad Sci (USA), 76:
494-498.
Holzer G, Shanfield H, Zlatkis A, Bertsch W, Juarez P, Mayfield H, &
Liebich HM (1977) Collection and analysis of trace emissions from
natural sources. J Chromatogr, 142: 755-764.
Hooper AB & Terry KR (1973) Specific inhibitors of ammonia oxidation
in Nitrosomonas. J Bacteriol, 115: 480-485.
Horton VI, Higuchi MA, & Rickert DE ( 1992) Physiologically based
pharmacokinetic model for methanol in rats, monkeys and humans.
Toxicol Appl Pharmacol, 117: 26-36.
Howard PH ed. (1990) Handbook of environmental fate and exposure data
for organic compounds - Volume II: Solvents. Chelsea, Michigan, Lewis
Publishers, pp 310-317.
Howard PH, Boethling RS, Jarvis WF, Meylan WM, & Michalenko EM (1991)
Handbook of environmental degradation rates. Chelsea, Michigan, Lewis
Publishers, pp 93-94.
Hsieh FY, Leu TM, & Chia LG (1992) Bilateral putaminal necrosis caused
by methanol poisoning: A case report. Chin Med J (Taipei), 49:
283-288.
Hüls AG (1978) [Determination of the biodegradability of methanol in
the closed bootle tert (OECD Guideline 30D).] Marl, Hüls AG, 11 pp (in
German).
Hunt R (1902) The toxicity of methyl alcohol. John Hopkins Hosp Bull,
13: 213.
Hustert K, Mansour M, Parlar H, & Korte F (1981) [The EPA test - A
method for the determination of photochemical degradation of organic
compounds in aquatic systems.] Chemosphere, 10: 995-998 (in German).
ILO (1983) In: Parmeggiani L ed. Encyclopedia of occupational health
and safety, 3rd revis ed. Geneva, International Labour Office, vol 2,
pp 1356-1358.
ILO (1991) Occupational exposure limits for airborne toxic substances,
3rd ed. Geneva, International Labour Office, pp 256-257.
Infurna R & Weiss B (1986) Neonatal behavioural toxicity in rats
following prenatal exposures to methanol. Teratology, 33: 259-265.
Ingemansson SO (1983) Studies on the effect of 4-methylpyrazole on
retinal activity in the methanol poisoned monkey by recording the
electroretinogram. Acta Opthalmol, 158(Suppl): 5-24.
IPCS (1995) Poison Information Monograph No. 335: Methanol. Geneva,
World Health Organization, International Programme on Chemical Safety.
Isidorov VA, Zenkevich IG, & Ioffe BV (1985) Volatile organic
compounds in the atmosphere of forests. Atmos Environ, 19: 1-8.
Jacobs GA (1990) OECD eye irritation tests on three alcohols: Acute
toxicity data. J Am Coll Toxicol, 1: 56-57.
Jacobsen D & McMartin KE (1986) Methanol and ethylene glycol
poisonings: Mechanisms of toxicity, clinical course, diagnosis and
treatment. Med Toxicol, 1: 309-314.
Jacobsen D, Jansen H, Wiek-Larsen E, Bredesen DE, & Halvorsen S
(1982a) Studies on methanol poisoning. Acta Med Scand, 12: 5-10.
Jacobsen D, Bredesen JE, Eide I, & Ostborg J (1982b) Anion and osmolal
gaps in the diagnosis of methanol and ethylene glycol poisoning. Acta
Med Scand, 212: 17-20.
Jacobsen D, Ovrebo S, & Sejersted OM (1983a) Toxicokinetics of formate
during hemodialysis. Acta Med Scand, 214: 409-412.
Jacobsen D, Ovrebo S, Arnesen E, & Paus PN (1983b) Pulmonary excretion
of methanol in man. Scand J Clin Invest, 43: 377-379.
Jacobsen D, Webb P, Collins TD, & McMartin KE (1988) Methanol and
formate kinetics in late diagnosed methanol intoxication. Med Toxicol,
3: 418-423.
Jacobsen D, Sebastian CS, Barron SK, Carriere EW, & McMartin KE (1990)
Effects of 4-methylpyrazole, methanol/ethylene glycol antidote in
healthy humans. J Emerg Med, 8: 455-461.
Jansen JLC, Nyberg U, Aspegren H, & Andersson B (1993) Handling of
anaerobic digester supernatant combined with full nitrogen removal.
Water Sci Technol, 27: 391-403.
Jaselkis B & Warriner JP (1966) Titrimetric determination of primary
and secondary alcohols by xenon trioxide oxidation. Anal Chem, 38:
563-564.
Jeganathan PS & Namasivayam A (1989) Methanol induced monoamine
changes in hypothalamus and striatum of albino rats. Alcohol, 6:
451-454.
Jones AW (1986) Abnormally high concentrations of methanol in breath:
A useful biochemical marker of recent heavy drinking. Clin Chem, 32:
1241-1242.
Jones AW (1987) Elimination half-life of methanol during hangover.
Pharmacol Toxicol, 60: 217-220.
Jones AW & Lowinger H (1988) Relationship between the concentration of
ethanol and methanol in blood samples from Swedish drinking drivers.
Forensic Sci Int, 37: 277-285.
Jones AW, Mardh G, & Anggard E (1983) Determination and endogenous
ethanol in blood and breath by gas chromatography-mass spectrometry.
Pharmacol Biochem Behav, 18(Suppl 1): 267-272.
Jones AW, Skagerberg S, Yonekurat T, & Sato A (1990) Metabolic
interaction between endogenous ethanol studied in human volunteers by
analysis of breath. Pharmacol Toxicol, 66: 62-65.
Jonsson A & Berg S (1983) Determination of low-molecular-weight
oxygenated hydrocarbons in ambient air by cryogradient sampling and
two-dimensional gas chromatography. J Chromatogr, 279: 307-322.
Jonsson A, Persson KA, & Grigoriadis V (1985) Measurements of some low
molecular-weight oxygenated, aromatic and chlorinated hydrocarbons in
ambient air and in vehicle emissions. Environ Int, 11: 383-392.
Juhnke VI & Lüdemann D (1978) [Results of testing of 200 chemical
compounds for acute fish toxicity with the Golden Orfe test.] Z Wasser
Abwasser Forsch, 11(5): 161-164 (in German).
Jungclaus GA, Lopez-Avila V, & Hites RA (1978) Organic compounds in an
industrial wastewater: A case study of their environmental impact.
Environ Sci Technol, 12: 88-96.
Kahn A & Blum D (1979) Methyl alcohol poisoning in an 8-month old boy:
An unusual rank route of intoxication. J Pediatr, 94: 841-843.
Kane Rl, Talbert W, Harlan J, Sizemore G, & Cataland S (1968) A
methanol poisoning outbreak in Kentucky. Arch Environ Health, 17:
119-129.
Kaplan K (1962) Methyl alcohol poisoning. Am J Med Sci, 244:
170-174.
Katoh M (1989) New Energy Development Organization data. Presented at
the Methanol Vapors and Health Effects Workshop: What we know and what
we need to know - Summary Report. Washington, DC, ILSI Risk Science
Institute/US Environmental Protection Agency/Health Effects
Institute/American Petroleum Institute, p A-7.
Kavet R & Nauss KM (1990) The toxicity of inhaled methanol vapors. CRC
Crit Rev Toxicol, 21: 21-50.
Kawai T, Yasugi T, Uchida Y, & Ikeda M (1990) Personal diffusive
sampler for methanol, a hydrophilic solvent. Bull Environ Contam
Toxicol, 44: 514-520.
Kawai T, Yasugi T, Mizunuma K, Horiguchi S, Hirase Y, Uchida Y, &
Ikeda M (1991a) Simple method for the determination of methanol in
blood and its application in occupational health. Bull Environ Contam
Toxicol, 47: 797-803.
Kawai T, Yasugi T, Mizunuma K, Horiguchi S, Hirase Y, Uchida Y, &
Ikeda M (1991b) Methanol in urine as a biological indicator of
occupational exposure to methanol vapor. Int Arch Occup Environ
Health, 63: 311-318.
Kawai T, Yasugi T, Mizunuma K, Horiguchi S, Iguchi H, Uchida Y, Iwami
O, & Ikeda m (1992) Comparative evaluation of urinalysis and blood
analysis as means of detecting exposure to organic solvents at low
concentrations. Int Arch Occup Environ Health, 64: 223-234.
Kawai T, Mizunuma K, Yasugi T, Horiguchi S, Moon CS, Zhang ZW,
Miyashita K, Takeda S, & Ikeda M (1995) Effects of methanol on styrene
metabolism among workers occupationally exposed at low concentrations.
Arch Environ Contam Toxicol, 28: 543-546.
Keeney AH & Mellinkoff SM (1951) Methyl alcohol poisoning. Ann Intern
Med, 34: 331-338.
Keller AE (1993) Acute toxicity of several pesticides, organic
compounds, and a wastewater effluent to the fresh water mussel,
Anodonta imbecilis, Ceriodaphnia dubia and Pimephales promelas.
Bull Environ Contam Toxicol, 51: 696-702.
Kelly TJ, Mukund R, Spicer CW, & Pollack AJ (1994) Concentrations and
transformations of hazardous air pollutants. Environ Sci Technol,
28: 378A-387A.
Kennedy TF & Shanks D (1981) Methanol: manufacture and uses. In:
Wickson EJ ed. Monohydric alcohols. Washington, DC, American Chemical
Society, pp 19-27 (ACS Symposium Series No. 159).
Kempa ES (1976) [Oxygen needs during the degradation of waste
compounds.] Österr Abwasser Rundsch, 2: 20-25 (in German).
Keyvan-Larijarni H & Tannenberg AM (1974) Methanol intoxication:
Comparison of peritoneal dialysis and hemodialysis treatment. Arch
Intern Med, 124: 293-296.
Kim S (1973) Purification and properties of protein methylase. Arch
Biochem Biophys, 157: 476-484.
Kimura ET, Ebert DM, & Dodge PW (1971) Acute toxicity and limits of
solvent residue for sixteen organic solvents. Toxicol Appl Pharmacol,
19: 699-704.
King L (1992) Acute methanol poisoning: A case study. Heart Lung,
21: 260-264.
King GM, Klug MJ, & Lovley DR (1983) Metabolism of acetate, methanol
and methylated amines in intertidal sediments of Lowes Cove, Maine.
Appl Environ Microbiol, 45: 1848-1853.
Kingsley WH & Hirsch FG (1955) Toxicological considerations in direct
process spirit duplicating machines. Compens Med, 6: 7-8.
Kinlin TE, Muralidhara R, Pittet AO, Sanderson A, & Walradt JP (1972)
Volatile components of roasted filberts. J Agric Food Chem, 20:
1021-1028.
Kinoshita JH & Masurat T (1958) Effect of glutathione in formaldehyde
oxidation in the retina. Am J Opthalmol, 46: 42.
Kirchner JG & Miller JM (1957) Volatile water-soluble and oil
constituents of Valencia orange juice. J Agric Food Chem, 5:
283-291.
Kohl WL ed. (1990) Methanol as an alternative fuel choice: An
assessment. Washington, DC, The Johns Hopkins University School of
Foreign Service, pp 439.
Koivusalo M (1970) Methanol. In: Tremolieres J ed. International
encyclopaedia of pharmacology and therapeutics. Oxford, New York,
Pergamon Press, vol 2, section 20, pp 465-505.
Komers R & Sir Z (1976) Gas chromatographic determination at the parts
per million level of methanol and ethanol in aqueous solution.
J Chromatogr, 119: 251-254.
Konemann H (1981) Quantitative structure-activity relationships in
fish toxicity studies. Toxicology, 19: 209-221.
Krotoszynski B, Gabriel G, O'Neill HJ, & Claudio MPA (1977)
Characterization of human expired air: A promising investigative and
diagnostic technique. J Chromatogr Sci, 15: 239-244.
Krotoszynski BK, Bruneau GM, & O'Neill HJ (1979) Measurement of
chemical inhalation exposure in urban populations in the presence of
endogenous effluents. J Anal Toxicol, 3: 225-234.
Kruse JA (1992) Methanol poisoning. Intensive Care Med, 18: 391-397.
Kuhn R, Pattard M, Pernak KD, & Winter A (1989) Results of the harmful
effects of selected water pollutants (anilines, phenols, aliphatic
compounds) to Daphnia magna. Water Res, 23: 495-499.
Kutzbach C & Stokstad ELR (1968) Partial purification of a 10-formyl
tetrahydrofolate: NADP oxidoreductase from mammalian liver. Biochem
Biophys Res Commun, 30: 111.
Larsson BT (1965) Gas chromatography of organic volatiles in human
breath and saliva. Acta Med Scand, 19: 159-164.
Leaf G & Zatman LJ (1952) A study of the conditions under which
methanol may exert a toxic hazard in industry. Br J Ind Med, 9:
19-31.
Lee EW, Reader JA, Garner CD, Brady AN, & Li LC (1990) Evaluation of
methanol (M) toxicity on the visual system: spontaneous degeneration
of the retina and optic nerve in the F-344 rat. Toxicologist, 10:
155.
Lee EW, Brady AN, Brabec MJ, & Fabelt JN (1991) Effects of methanol
vapors on testosterone production and testis morphology in rats.
Toxicol Ind Health, 7: 261-275.
Lee EW, Terzo TS, D'Arcy JB, Gross KB, & Schreck RM (1992) Lack of
blood formate accumulation in humans following exposure to methanol
vapors at the current permissible exposure limit of 200 ppm. Am Ind
Hyg Assoc J, 53: 99-104.
Lee EW, Garner CD, & Terzo TS (1994a) Animal model for the study of
methanol toxicity: Comparison of folate-reduced rat responses with
published monkey data. J Toxicol Environ Health, 41: 71-82.
Lee EW, Garner CD, & Terzo TS (1994b) A rat model manifesting
methanol-induced visual dysfunction suitable for both acute and long-
term exposure studies. Toxicol Appl Pharmacol, 128: 199-206.
Lemaire J, Campbell I, Hulpke H, Guth JA, Merz W, Philp J, & von
Waldow C (1982) An assessment of test methods for photodegradation of
chemicals in the environment. Chemosphere, 11: 119-164.
Lettinga G, De Zeeuw W, & Ouberg E (1981) Anaerobic treatment of
wastes containing methanol and higher alcohols. Water Res, 15:
171-182.
Lewis RJ Sr (1989) Food additives handbook. New York, Van Nostrand
Reinhold Co., pp 291-292.
Ley CO & Gali FG (1983) Parkinsonian syndrome after methanol
intoxication. Eur Neurol, 22: 405-409.
Liesivuori J & Savolainen H (1987) Urinary formic acid as an indicator
of occupational exposure to formic acid and methanol. Am Ind Hyg Assoc
J, 48: 32-34.
Liesivuori J & Savolainen H (1991) Methanol and formic acid toxicity:
Biochemical mechanisms. Pharmacol Toxicol, 69: 157-163.
Lijinsky W, Thomas BJ, & Kovatch RM (1991) Differences in skin
carcinogenesis by methyl nitroso urea between mice of several strains.
Cancer Lett, 61: 1-5.
Linden E, Bengtsson BE, Svanberg O, & Sundstrom G (1979) The acute
toxicity of 78 chemicals and pesticide formulations against two
brackish water organisms - The bleak (Alburnus alburnus) and the
harpacticoid nitocraspinipes. Chemosphere, 11/12: 843-851.
Lins RL, Zachee P, Christiaens M, Van De Vijver F, De Waele L,
Sandra P, & De Broe ME (1980) Prognosis and treatment of methanol
intoxication. In: Holmstedt B, Lauwerys R, Mercier M, & Roberfroid M
ed. Mechanisms of toxicity and hazard evaluation. Amsterdam,
Elsevier/North-Holland Biomedical Press, pp 415-421.
Litovitz LT, Schmitz BF, Matyunas N, & Martin TG (1988) 1987 Annual
report of the American Association of Poison Control Centers National
Data Collection System. Am J Emerg Med, 6: 479-515.
Loewy A & von der Heide R (1914) [The uptake of methyl alcohol by
inhalation.] Biochem Ztg, 65: 230-252 (in German).
Lkke H (1984) Leaching of ethylene glycol and ethanol in subsoils.
Water Air Soil Pollut, 22: 373-387.
Lovegren NV, Fisher GS, Legendre MG, & Schuller WH (1979) Volatile
constituents of dried legumes. J Agric Food Chem, 27: 851-853.
Luke LA & Ray JE (1984) Gas-chromatographic method for the
determination of low relative molecular mass alcohols and methyl tert-
butyl ether in gasoline. Analyst, 109: 989-992.
Lund A (1948a) Excretion of methanol and formic acid in man after
methanol consumption. Acta Pharmacol, 4: 205-212.
Lund A (1948b) Metabolism of methanol and formic acid in dogs. Acta
Pharmacol, 4: 108-121.
Lund A (1948c) Metabolism of methanol and formic acid in rabbits. Acta
Pharmacol, 4: 99-107.
Lund ED, Kirkland CL, & Shaw PE (1981) Methanol, ethanol and
acetaldehyde contents of citrus products. J Agric Food Chem, 29:
361-366.
Lundberg P (1985) Scientific basis for Swedish occupational standards
VI. Arb Och Halsa, 32: 115-121.
Lyman WJ, Reehl WF, & Rosenblatt DH ed. (1990) Handbook of chemical
property estimation methods. Washington, DC, American Chemical
Society.
McCord CP (1931) Toxicity of methyl alcohol (methanol) following skin
absorption and inhalation. Ind Eng Chem, 23: 931-936.
McCoy H, Cipolle RJ, Ehlers SM, Sawchuk RJ, & Zaske DE (1979) Severe
methanol poisoning: Application of a pharmacokinetic model for ethanol
therapy and hemodialysis. Am J Med, 67: 804-807.
McDonald A, Sey YM, House DE, & Simmons JE (1992) Methanol
potentiation of carbon tetrachloride hepatotoxicity is dependent on
the time of carbon tetrachloride administration. Toxicologist, 12:
239.
McGregor DB, Martin R, Riach CG, & Caspary WJ (1985) Optimization of a
metabolic activation system for use in the mouse lymphoma L5178Y
tk+tk_ mutation system. Environ Mutagen, 7(Suppl 3): 10.
Machiele PA (1990) A health and safety assessment of methanol as an
alternative fuel. In: Kohl WL ed. Methanol as an alternative choice.
Washington, DC, The Johns Hopkins Foreign Policy Institute, pp
217-239.
McLean DR, Jacobs H, & Mielke BW (1980) Methanol poisoning: A clinical
and pathological study. Ann Neurol, 8: 162-167.
McMartin KE, Makar AB, Martin-Amat G, Palese M, & Tephly TR (1975)
Methanol poisoning I. The role of formic acid in the development of
metabolic acidosis in the monkey and the reversal by 4-methylpyrazole.
Biochem Med, 13: 319-333.
McMartin KE, Martin-Amat G, Makar AB, & Tephly TR (1977) Methanol
poisoning: V. The role of formate metabolism in the monkey. J
Pharmacol Exp Ther, 201: 564-572.
McMartin KE, Martin-Amat G, Noker PE, & Tephly TR (1979) Lack of a
role for formaldehyde in methanol poisoning in the monkey. Biochem
Pharmacol, 28: 645-649.
McMartin KE, Ambre JJ, & Tephly TR (1980a) Methanol poisoning in human
subjects: Role of formic acid accumulation in the metabolic acidosis.
Am J Med, 68: 414-418.
McMartin KE, Hedstrom KG, Tolf BR, Ostung-Wintzell H, & Blomstrand R
(1980b) Studies on the metabolic interactions between 4-methylpyrazole
and methanol using the monkey as an animal model. Arch Biochem
Biophys, 199: 606-614.
Maeda Y, Fujio Y, Suetaka T, & Munemori M (1988) Selective gas
chromatographic determination of trace amounts of alcohols in ambient
air. Analyst, 113: 189-191.
Maejima K, Suzuki T, Niwa H, Maekawa A, Nagase S, & Ishinishi N (1992)
Toxicity to rats of methanol-fueled engine exhaust continuously for 28
days. J Toxicol Environ Health, 37: 293-312.
Maejima K, Suzuki T, Numata H, Maekawa A, Nagase S, & Ishinishi N
(1993) Recovery from changes in the blood and nasal cavity and/or
lungs of rats caused by exposure to methanol-fueled engine exhaust.
J Toxicol Environ Health, 39: 323-340.
Maejima K, Suzuki T, Numata H, Maekawa A, Nagase S, & Ishinishi N
(1994) Subchronic (12-week) inhalation toxicity study of methanol-
fueled engine exhaust in rats. J Toxicol Environ Health, 41:
315-327.
Mahieu P, Hassoun A, & Lauwerys R (1989) Predictors of methanol
intoxication with unfavourable outcome. Hum Toxicol, 8: 135-137.
Majchrowicz E & Mendelson JH (1971) Blood methanol concentrations
during experimentally induced ethanol intoxication in alcoholics.
J Pharmacol Exp Ther, 179: 293-300.
Makar AB & Mannering GJ (1968) Role of the intracellular distribution
of hepatic catalase in the peroxidative oxidation of methanol. Mol
Pharmacol, 4: 484-491.
Makar AB & Tephly TR (1976) Methanol poisoning in the folate deficient
rat. Nature (Lond), 261: 715-716.
Makar AB & Tephly TR (1977) Methanol poisoning: VI. Role of folic acid
in the production of methanol poisoning in the rat. J Toxicol Environ
Health, 2: 1201-1209.
Makar AB & Tephly TR (1982) Improved estimation of formate in body
fluids and tissues. Clin Chem, 28: 385.
Makar AB, Tephly TR, & Mannering GJ (1968) Methanol metabolism in the
monkey. Mol Pharmacol, 4: 471-483.
Makar AB, McMartin KE, Palese M, & Tephly TR (1975) Formate assay in
body fluids: application to methanol poisoning. Biochem Med, 13:
117-126.
Makar AB, Tephly TR, Sahin G, & Osweiler G (1990) Formate metabolism
in young swine. Toxicol Appl Pharmacol, 105: 315-320.
Malorny G, Rietbrock N, & Schneider M (1965) [The oxidation of
formaldehyde to formic acid in blood: a contribution to the metabolism
of formaldehyde.] Naunyn Schmiedebergs Arch Pathol Exp Pharmacol,
250: 419-436 (in German).
Martin-Amat G, Tephly TR, McMartin KE, Makar AB, Hayreh MS, Hayreh SS,
Baumbach G, & Cancilla P (1977) Methyl alcohol poisoning: II.
Development of a model for ocular toxicity in methyl alcohol poisoning
using the rhesus monkey. Arch Ophthamol, 95: 1847-1850.
Martin-Amat G, McMartin KE, Hayreh SS, & Tephly TR (1978) Methanol
poisoning: Ocular toxicity produced by formate. Toxicol Appl
Pharmacol, 45: 201-208.
Martinasevic MK, Green MD, Baron J, & Tephly TR (1996) Folate and 10
formyltetrahydrofolate dehydrogenase in human and rat retina: relation
to methanol toxicity. Toxicol Appl Pharmacol, 141: 373-381.
Mashbitz LM, Sklianskaya RM, & Urieva FI (1936) The relative toxicity
of acetone, methyl alcohol and their mixtures: II. Their action on
white mice. J Ind Hyg Toxicol, 18: 117-122.
Mather A & Assimos A (1965) Evaluation of gas-liquid chromatography in
assays for blood volatiles. Clin Chem, 11: 1023-1035.
Matsui S, Okawa Y, & Ota R (1988) Experience of 16 years' operation
and maintenance of the Fukashiba industrial wastewater treatment plant
of the Kashima petrochemical complex: II. Biodegradability of 37
organic substances and 28 process waste waters. Water Sci Technol,
20: 201-210.
Mayer FL & Ellersieck MR (1986) Manual of acute toxicity:
Interpretation and database for 410 chemicals and 66 species of
freshwater animals. Washington, DC, US Department of the Interior,
Fish and Wildlife Service (Resource Publication No. 160).
Medinsky MA & Dorman DC (1994) Assessing risks of low-level methanol
exposure. CIIT Act, 14(7): 1-7.
Medinsky MA, Dorman DC, Bond JA, Moss OR, Janszen DB, & Everitt JI
(1997) Pharmacokinetics of methanol and formate in female cynomolgus
monkeys exposed to methanol vapours. Cambridge, Massachusetts, Health
Effects Institute (Research Report No. 77).
Menne FR (1938) Acute methyl alcohol poisoning. Arch Pathol, 26:
77-92.
Mill T, Hendry DG, & Richardson H (1980) Free-radical oxidants in
natural waters. Science, 207: 886-887.
Ministry of Supply and Services Canada (1993) Summary report 1993-A:
National pollutant release inventory-Methanol air release. Ottawa,
Ontario, Environment Canada, 11 pp.
Mohler FS & Gordon CJ (1990) Themoregulatory effects of methanol in
Fischer and Long-Evans rats. Neurotoxicol Teratol, 12: 41-45.
Mohr DH & King CJ (1985) Identification of polar organic compounds in
coal-gasification condensate water by gas-chromatography-mass
spectrometry analysis of high-performance liquid chromatography.
Environ Sci Technol, 19: 929-935.
Monte WC (1984) Aspartame: methanol and the public health. J Appl
Nutr, 36: 42-54.
Montgomery JH (1991) Groundwater chemicals desk reference. Chelsea,
Michigan, Lewis Publishers, pp 491-495.
Morin AM & Liss M (1973) Evidence for a methylated protein
intermediate in pituitary methanol formation. Biochem Biophys Res
Commun, 52: 373-378.
Morris GPL (1993) Renewed interest emerges for methanol, MTBE
projects. Chem Week, 153: 7.
Murray TG, Burton TC, Rajani C, Lewandowski MF, Burke JM, & Eells JT
(1991) Methanol poisoning: A rodent model with structural and
functional evidence for retinal involvement. Arch Ophthalmol, 109:
1012-1016.
Naraqi S, Dethlefs RF, Slobodnuik RA, & Sairere JS (1979) An outbreak
of acute methyl alcohol intoxication. Aust NZ J Med, 9: 65-68.
National Poisons Information Service (1993) Antifreeze poisoning:
Clinical effects and management. London, National Poisons Information
Service, pp 1-4.
NEDO (1982) Toxicological research of methanol as a fuel for power
station: Summary report on tests with monkeys, rats and mice. Tokyo,
Japan, New Energy Development Organization.
NEDO (1987) Toxicological research of methanol as a fuel for power
station: Summary report on tests with monkeys, rats and mice. Tokyo,
Japan, New Energy Development Organization, pp 1-296.
Nelson BK, Brightwell WS, MacKenzie DR, Khan A, Burg JR, Weigel WW, &
Goad PT (1985) Teratological assessment of methanol and ethanol at
high inhalation levels in rats. Fundam Appl Toxicol, 5: 727-736.
Nicholls P (1975) Formate as an inhibitor of cytochrome c oxidase.
Biochem Biophys Res Commun, 67: 610-616.
Nielsen IR, Malcolm HM, & Dobson SD (1993) Environmental hazard
assessment: Methanol. Garston, Watford, UK Department of the
Environment, Building Research Establishment, 45 pp.
NIOSH (1976) Criteria for a recommended standard .... Occupational
exposure to methyl alcohol. Cincinnati, Ohio, National Institute for
Occupational Safety and Health (HEW (NIOSH) Publication No. 76-148).
NIOSH (1977) NIOSH manual of analytical methods, 2nd ed. Cincinnati,
Ohio, National Institute for Occupational Safety and Health (DHEW
(NIOSH) Publication No. 77-157A).
NIOSH (1981) Health hazard evaluation report No. HETA-81-177, 178-988,
University of Washington, Seattle. Cincinnati, Ohio, National
Institute for Occupational Safety and Health.
NIOSH (1984) Method No. 2000: Methanol. In: Eller PM ed. NIOSH manual
of analytical methods. Cincinnati, Ohio, National Institute for
Occupational Safety and Health, vol 2, pp 2000/1-2000/4.
Noker PE, Eells JT, & Tephly TR (1980) Methanol toxicity: Treatment
with folic acid and 5-formyl tetrahydrofolic acid alcoholism. Clin Exp
Res, 4: 378-383.
Norman V (1977) An overview of the vapor phase, semivolatile, and
non-volatile components of cigarette smoke. Recent Adv Tob Sci, 3:
25-58.
Novak JT, Goldsmith CD, Benoit RE, & O'Brien JH (1985) Biodegradation
of methanol and tertiary butyl alcohol in subsurface systems. Water
Sci Technol, 17: 71-85.
Nyberg U, Aspegren H, Andersson B, Jansen JC, & Villadsen IS (1992)
Full-scale application of nitrogen removal with methanol as carbon
source. Water Sci Technol, 26: 1077-1086.
Obe G & Ristow H (1977) Acetaldehyde, but not ethanol induces sister
chromatid exchanges in Chinese hamster cells in vitro. Mutat Res,
56: 211-213.
Okpokwasili GC & Amanchukwu SC (1988) Petroleum hydrocarbon
degradation by candida species. Environ Int, 14: 243-247.
Oliver B, Cosgrove EG, & Carey JH (1979) Effect of suspended sediments
on the photolysis of organics in water. Environ Sci Technol, 13:
1075-1077.
Ophaswongse S & Maibach HI (1994) Alcohol dermatitis: Allergic contact
dermatitis and alcohol urtiaria syndrome. Contact Dermatitis, 30:
1-6.
Oremland RS, Marsh LM, & Polcin S (1982) Methane production and
simultaneous sulphite reduction in anoxic, salt marsh sediments.
Nature (Lond), 296: 143-145.
Owens LD, Gilbert RG, Griebel GE, & Menzies JD (1969) Identification
of plant volatiles that stimulates microbial respiration and growth in
soil. Phytopathology, 59: 1468-1472.
Palese M & Tephly TE (1975) Metabolism of formate in the rat.
J Toxicol Environ Health, 1: 13-24.
Pamies RJ, Sugar D, Rives LA, & Herold AH (1993a) Methanol
intoxication. Postgrad Med, 93: 183-194.
Pamies RJ, Sugar D, Rives L, & Herold AH (1993b) Methanol
intoxication: A case report. J Fla Med Assoc, 80: 464-467.
Pankow S & Jagielki S (1993) Effect of methanol or modifications of
the hepatic glutathione concentration on the metabolism of
dichloromethane to carbon monoxide in rats. Hum Exp Toxicol, 12:
227-231.
Pappas SC & Silverman M (1982) Treatment of methanol poisoning with
ethanol and hemodialysis. Can Med Assoc J, 126: 1391-1394.
Pappas AA, Gadsden RH, & Taylor EH (1985) Serum osmolality in acute
intoxication: A prospective clinical study. Am J Clin Pathol, 84:
74-79.
Pauls RE & McCoy RW (1981) Gas and liquid chromatographic analysis of
methanol, ethanol, t-butanol and methyl-t-butyl ether in gasoline.
J Chromatogr Sci, 19: 558-561.
Pavlenko SM (1972) [Certain common traits in the action of industrial
non-electrolyte poisons entering the body simultaneously with the
water and air.] Gig I Sanit, 37: 40-45 (in Russian).
Pearson A (1952) Acute methyl alcohol poisoning. Med J Aust, 2: 437.
Pelletier J, Habib MH, Khalil R, Salamon G, Bartoli D, & Jean P (1992)
Putaminal necrosis after methanol intoxication. J Neurol Neurosurg
Psych, 55: 234-235.
Pellizzari ED, Hartwell TD, Harris BSH III, Waddell RD, Whitaker DA, &
Erikson MD (1982) Purgable organic compounds in mother's milk. Bull
Environ Contam Toxicol, 28: 322-328.
Pereira MA, Chang LW, McMillan L, Ward JB, & Legator MS (1982) Battery
of short-term tests in laboratory animals to corroborate the detection
of human population exposures to genotoxic chemicals. Environ Mutagen,
4: 317.
Perkins RA, Ward KW, & Pollack GM (1995) A pharmacokinetic model of
inhaled methanol in humans and comparison to methanol disposition in
mice and rats. Environ Health Perspect, 103: 716-733.
Phillips M & Greenberg J (1987) Detection of endogenous ethanol and
other compounds in the breath by gas chromatography with on-column
concentration of sample. Anal Biochem, 163: 165-169.
Pienta RJ, Poiley JA, & Lebherz WB III (1977) Morphological
transformation of early passage Golden Syrian hamster embryo cells
derived from cryopreserved cultures as a reliable in vitro bioassay
for identifying carcinogens. Int J Cancer, 19: 642-655.
Pla A, Hernandez AF, Gil F, Garcia-Alonso M, & Villanueva E (1991) A
fatal case of oral ingestion of methanol: Distribution in postmortem
tissues and fluids including pericardial fluid and vitreous humor.
Forensic Sci Int, 49: 193-196.
Poirier SH, Knuth Ml, Anderson-Buchou CD, Brooke CT, Lima AR, & Shubat
PJ (1986) Comparative toxicity of methanol and N,N-dimethylformamide
to freshwater fish and invertebrates. Bull Environ Contam Toxicol,
37: 615-621.
Pollack GM & Brouwer KLR (1996) Maternal-fetal pharmacokinetics of
methanol. Cambridge, Massachusetts, Health Effects Institute, 55 pp
(Research Report No. 74).
Pollack GM & Kawagoe JL (1991) Determination of methanol in whole
blood by capillary gas chromatography with direct on-column injection.
J Chromatogr, 570: 406-411.
Pollack GM, Brouwer KLR, & Kawagoe J (1993) Toxicokinetics of
intravenous methanol in the female rat. Fundam Appl Toxicol, 21:
105-110.
Poon R, Chu IH, Bjarnason S, Potvin M, Vincent R, Miller RB, & Valli
VE (1994) Inhalation toxicity study of methanol, toluene and
methanol/toluene mixtures in rats. Toxicol Ind Health, 10: 231-245.
Poon R, Chu IH, Bjarnason S, Vincent R, Potvin M, Miller RB, & Valli
VE (1995) Short-term inhalation toxicity of methanol, gasoline and
methanol/gasoline in the rat. Toxicol Ind Health, 11: 343-361.
Portmann JE & Wilson RW (1971) The toxicity of 140 substances to the
brown shrimp and other marine animals. London, Ministry of
Agriculture, Fisheries and Food (Shellfish Information Leaflet No.
22).
Posner HS (1975) Biohazards of methanol in proposed new uses.
J Toxicol Environ Health, 1: 153-171.
Price KS, Waggy GT, & Conway RA (1974) Brine shrimp bioassay and
seawater BOD of petrochemicals. J Water Pollut Control Fed, 46:
63-77.
Price EA, d'Alessandro A, Kearney T, Olson KR, & Blanc PD (1994)
Osmolar gap with minimal acidosis in combined methanol and methyl
ethyl ketone ingestion. Clin Toxicol, 32: 79-84.
Rajini PS, Krishnakumari MK, & Majumder SK (1989) Cytotoxicity of
certain organic solvents and organophosphorus insecticides to the
ciliated protozoan Paramecium caudatum. Microbios, 59: 157-163.
Rana SVS & Kumar S (1993a) Liver function in rats treated individually
and with a combination of xylene, toluene and methanol. Toxicol Ind
Health, 9: 479-484.
Rana SVS & Kumar S (1993b) Effect of xylene, toluene and methyl
alcohol on liver collagenesis in rats. Indian J Exp Biol, 31:
782-784.
Randall TL & Knopp PV (1980) Detoxification of specific organic
substances by wet oxidation. J Water Pollut Control Fed, 52:
2117-2130.
Rastogi SC (1993) Organic solvents in model and hobby glues. Bull
Environ Contam Toxicol, 51: 501-507.
Reese E & Kimbrough RD (1994) Acute toxicity of gasoline and some
additives. Environ Geochem Health, 101(Suppl): 113-115.
Reisch MS (1994) Top 50 chemicals production rose modestly last year.
Chem Eng News, 72: 12-18.
Renzoni GE, Shankland EG, Gaines JA, & Callis JB (1985) Determination
of alcohols in gasoline/alcohol blends by nuclear magnetic resonance
spectrometry. Anal Chem, 57: 2864-2867.
Rietbrock N (1969) [Kinetics and pathways of methanol metabolism.]
Naunyn-Schmiedebergs Arch Pharmacol Exp Pathol, 263: 189-201 (in
German).
Rietbrock N, Stieren B, & Malorny G (1966) [Influence of folic acid on
methanol metabolism.]. Klin Wochenschr, 44: 1318.
Rippen G (1980) [Handbook of environmental chemicals.] Landsberg,
Ecomed Verlagsgesellschaft, 16 pp (in German).
Röe O (1948) Methanol poisoning: The ganglion cells of the retina in
cases of methanol poisoning in human beings and experimental animals.
Acta Med Scand, 26: 169-182.
Röe O (1950) The roles of alkaline salts and ethyl alcohol in the
treatment of methanol poisoning. Q J Stud Alcohol, 11: 107-112.
Röe O (1955) The metabolism and toxicity of methanol. Pharmacol Rev,
7: 399-412.
Röe O (1982) Species differences in methanol poisoning. CRC Crit Rev
Toxicol, 10: 275-286.
Roeggla G, Wagner A, Frossard M, & Roeggla H (1993) Marked variability
in methanol toxicity. Am Fam Physician, 48: 731.
Rogers JM, Mole ML, Chernoff N, Barbee BD, Turner CI, Logsdon TR, &
Kavlock RJ (1993) The developmental toxicity of inhaled methanol in
the CD-1 mouse, with quantitative dose-response modeling for
estimation of benchmark doses. Teratology, 47: 175-188.
Rossini GDB & Ronco AE (1996) Acute toxicity bioassay using Daphnia
obtusa as a test organism. Environ Toxicol Water Qual, 11(3):
255-258.
Rotman D (1994a) Lurgi unveils route to methanol from carbon dioxide
at ACS meeting. Chem Week, 154: 14.
Rotman D (1994b) New approaches rekindle methanol conversion. Chem
Week, 154: 35.
Rowe VK & McCollister SB (1982) Alcohols. In: Clayton GD & Clayton FE
ed. Parry's industrial hygiene and toxicology, 3rd ed. New York, John
Wiley and Sons, vol 2C, pp 4528-4541.
Ruedemann AD (1962) The electroretinogram in chronic methyl alcohol
poisoning in human beings. Am J Ophthalmol, 54: 34-53.
Ryan BM, Hatoum NS, Mallett ES, & Yermakoff JK (1994) A pilot toxicity
study of methanol in folate-deficient long-Evans rats. Teratology,
49: 399.
Sakanashi TM, Rogers JM, & Keen CL (1994) Influence of folic acid
intake on the developmental toxicity of methanol in the CD-1 mouse.
Teratology, 49: 368.
Sayers RR, Yant WP, & Schrenk HH (1942) Methanol poisoning exposure of
dogs to 450-500 ppm methanol vapour in air. Washington, DC, US Bureau
of Mines (Investigation Report No. 3619).
Sayers RR, Yant WP, Schrenk HH, Chornyak J, Pearce SJ, Patty FA, &
Linn JG (1944) Methanol poisoning: II. Exposure of dogs for brief
periods eight times daily to high concentrations of high methanol
vapor in air. J Ind Hyg Toxicol, 26: 255-259.
Scheunert I, Vockel D, Schnitzer J, & Korte F (1987) Bromineralization
rates of 14C-labelled organic chemicals in aerobic and anaerobic
suspended soil. Chemosphere, 16: 1031-1041.
Scheuplein RJ & Blank IH (1971) Permeability of the skin. Physiol Rev,
51: 702-747.
Schiewe MH, Hawk EG, Actor DI, & Krahn MM (1985) Use of bacterial
bioluminescence assay to access toxicity of contaminated marine
sediments. Can J Fish Aquat Sci, 42: 1244-1247.
Scholz B, Butzert H, Neumeister J, & Nierlich F (1990) Methyl tert-
butyl ether. In: Elvers B, Hawkins S, Schutz G ed. Ullmann's
encyclopedia of industrial chemistry, 5th ed. Weinheim, VCH
Verlagsgesellschaft, vol 16a, pp 543-550.
Scott E, Helz MK, & McCord CP (1933) The histopathology of methyl
alcohol poisoning. Am J Clin Pathol, 3: 311-319.
Sedivec V, Mraz M, & Flek J (1981) Biological monitoring of persons
exposed to methanol vapors. Int Arch Occup Environ Health, 48:
257-271.
Seizinger DE & Dimitriades B (1972) Oxygenates in exhaust from simple
hydrocarbon fuels. J Air Pollut Control Assoc, 22: 47-51.
Sejersted OM, Jacobsen D, Ovrebo S, & Jansen H (1983) Formate
concentration in plasma from patients poisoned with methanol. Acta Med
Scand, 213: 105-110.
Self R, Casey JC, & Swain T (1963) The low boiling volatiles of cooked
foods. Chem Ind, 1963: 863-864.
Sharpe JA, Hostovsky M, Bilbao JM, & Rewcastle NB (1982) Methanol
optic neuropathy: A histopathological study. Neurology, 32:
1093-1100.
Simmon VF, Kauhanen K, & Tardiff RG (1977) Mutagenic activity of
chemicals identified in drinking water. In: Scott D, Bridges BA, &
Sobels FH ed. Progress in genetic toxicology. Amsterdam,
Elsevier/North Holland Press, vol 2, pp 249-268.
Simmons JE & McDonald A (1994) Effect of Kupffer cell inhibition on
carbon tetrachloride hepatotoxicity in methanol pretreated rats.
Toxicologist, 14: 374.
Simmons JE, McDonald A, Seely JC & Sey YM (1995) ..... of carbon
tetrachloride hepatotoxicity by inhaled methanol: Time course of
recovery. J Toxicol Environ Health, 46: 203-216.
Sims EW (1976) Determination of trace C1-C4 alcohols in aqueous
solutions by gas chromatography. J Chromatogr Sci, 14: 65-67.
Skaug OE (1956) A rapid and extremely sensitive test for methanol in
blood and biological material. Scand J Clin Lab Invest, 8: 338-339.
Smallwood AW (1978) Analysis of formic acid in air samples. Am Ind Hyg
Assoc J, 39: 151-153.
Smith NB (1984) Determination of volatile alcohols and acetone in
serum by non-polar capillary gas chromatography after direct sample
injection. Clin Chem, 30: 1672-1674.
Smith EN & Taylor RT (1982) Acute toxicity of methanol in the folate-
deficient acatalasemic mouse. Toxicology, 25: 271-287.
Smyth HF, Seaton J, & Fischer L (1941) The single dose toxicity of
some glycols and derivatives. J Ind Hyg Toxicol, 23: 259-268.
Snider JR & Dawson GA (1985) Tropospheric light alcohols, carbonyls
and acetonitrile: Concentrations in the Southwestern United States and
Henry's law data. J Geophys Res, 90: 3797-3805.
Snow R, Baker L, Crews W, Davis CO, Duncan J, Perry N, Siudak P,
Stumpf K, Ray W, & Braddock J (1989) Characterization of emissions
from a methanol fueled motor vehicle. J Air Pollut Control Assoc,
39: 48-53.
SRI (1992) Chemical economics handbook: Marketing research report on
methanol. Menlo Park, California, SRI International.
Stanton ME, Crofton KM, Gray LE, Gordon CM, Bushnell RJ, Mole ML, &
Peale DB (1991) Assessment of offspring development and behavior
following gestational exposure to inhaled methanol in the rat.
Toxicologist, 11: 118.
Stanton ME, Crofton KM, Gray LE, Gordon CJ, Boyes WK, Mole ML, Peele
DB, & Bushnell PS (1995) Assessment of offspring development and
behaviour following gestational exposure to inhaled methanol in the
rat. Fundam Appl Toxicol, 28: 100-110.
Stegink LD, Brummel MC, McMartin KE, Martin-Amat G, Filer LJ Jr, Baker
GL, & Tephly TR (1981) Blood methanol concentrations in normal adult
subjects administered abuse doses of aspartame. J Toxicol Environ
Health, 7: 281-290.
Stegink LD, Brummel MC, Filer LJ Jr, & Baker GL (1983) Blood methanol
concentrations in one-year-old infants administered graded doses of
aspartame. J Nutr, 113: 1600-1606.
Stensel HD, Loehr RC, & Lawrence AW (1973) Biological kinetics of
suspended-growth denitrification. J Water Pollut Control Fed, 45:
249-261.
Stern S, Reuhl K, Soderholm S, Cox C, Sharma A, Balys M, Gelein R, Yin
C, & Weiss B (1996) Perinatal methanol exposure in the rat: I. Blood
methanol concentration and neural cell adhesion molecules. Fundam Appl
Toxicol, 34: 36-46.
Stratton GW (1985) The influence of solvent type on solvent-pesticide
interactions in bioassays. Arch Environ Contam Toxicol, 14: 651-658.
Stratton GW (1987) Toxic effects of organic solvents on the growth of
blue-green algae. Bull Environ Contam Toxicol, 38: 1012-1019.
Stratton GW & Smith TM (1988) Interaction of organic solvents with the
green alga Chlorella pyrenoidosa. Bull Environ Contam Toxicol, 40:
736-742.
Strittmatter P & Ball EG (1955) Formaldehyde dehydrogenase, a
glutathione-dependent enzyme system. J Biol Chem, 213: 445.
Suit PF & Estes M (1990) Methanol intoxication: clinical features and
differential diagnosis. Clevel Clin J Med, 57: 464-471.
Swain HM & Somerville HJ (1978) Microbial metabolism of methanol in a
model activated sludge system. J Appl Bacteriol, 45: 147-151.
Swartz RD, Millman RP, Billi JE, Bondar NP, Migdal SD, Simonian SK,
Monforte JR, McDonald FD, Harness JK, & Cole KL (1981) Epidemic
methanol poisoning: clinical and biochemical analysis of a recent
episode. Medicine, 60: 373-382.
Sweger DM & Travis JC (1979) An application of infrared lasers to the
selective detection of trace organic gases. Appl Spectrom, 33:
46-51.
Tackett Sl (1987) Determination of methanol in gasoline by gas
chromatography. Analyst, 112: 339-340.
Takagi H, Hatakeyama S, Akimoto H, & Koda S (1986) Formation of methyl
nitrite in the surface reaction of nitrogen dioxide and methanol:
1. Dark reaction. Environ Sci Technol, 20: 387-393.
Tephly TR (1991) Mini review-the toxicity of methanol. Life Sci, 48:
1031-1041.
Tephly TR & McMartin KE (1984) Methanol metabolism and toxicity. In:
Stegink LD & Filer LJ Jr ed. Aspartame: Physiology and biochemistry.
New York, Basel, Marcel Dekker, pp 111-140.
Tephly TR, Parks RE, & Mannering GJ (1964) Methanol metabolism in the
rat. J Pharmacol Exp Ther, 143: 292-300.
Teschke R, Masamura Y, & Lieber CS (1975) Hepatic microsomal alcohol-
oxidising system: affinity for methanol, ethanol, propanol and
butanol. J Biol Che,. 250: 7397.
Tichy M, Trcka V, Roth Z, & Krivucova M (1985) QSAR analysis and data
extrapolation among mammals in a series of aliphatic alcohols. Environ
Health Perspect, 61: 321-328.
Tonning DJ, Brooks DW, & Harlow CM (1956) Acute methyl alcohol
poisonings in 49 naval ratings. Can Med Assoc J, 74: 20-27.
Triebig G & Schaller KH (1980) A simple and reliable enzymatic assay
for the determination of formic acid in urine. Clin Chim Acta, 108:
355-360.
Tyson HH & Schoenberg MJ (1914) Experimental researches in methyl
alcohol inhalation. J Am Med Assoc, 63: 915-921.
Ubaidullaev R (1966) [Effect of small concentrations of methanol
vapours on the body of men and animals.] Gig I Sanit, 31: 9-12 (in
Russian).
UK Standing Committee of Analysts (1982) Formaldehyde, methanol and
related compounds in raw, waste and potable waters: Methods for the
examination of waters and associated materials, London, UK Standing
Committee of Analysts, Her Majesty Stationery Office.
Uotila L & Koivusalo M (1974) Formaldehyde dehydrogenase from human
liver: Purification, properties and evidence for the formation of
glutathione thiol esters by the enzyme. J Biol Chem, 249: 7653.
Upadhyay S & Gupta VK (1984) Reagent system for the spectrophotometric
determination of methanol in environmental and biological samples.
Analyst, 109: 1427-1429.
Upton J (1993) Denitrification of sewage effluents in deep bed sand
filters. Water Sci Technol, 27: 381-390.
US EPA (1975) Identification of organic compounds in effluents from
industrial sources. Washington, DC, US Environmental Protection
Agency, Office of Toxic Substances (EPA 560-3-75-002).
US EPA (1976a) Assessment of methyl alcohol as a potential air
pollution problem - Volume II. Research Triangle Park, North Carolina,
US Environmental Protection Agency (NTIS Publication No. PB-258354).
US EPA (1976b) Frequency of organic compounds identified in water.
Washington, DC, US Environmental Protection Agency (EPA 600/4-76-062).
US EPA (1977) Multimedia environmental goals for environmental
assessment - Volume II: MEG, charts and background information.
Washington, DC, US Environmental Protection Agency, pp E28-E29 (EPA
600/7-77-126b).
US EPA (1979) Atmospheric reaction products of organic compounds.
Washington, DC, US Environmental Protection Agency, 80 pp (Report No.
PB 301-384, prepared by SRI International, Menlo Park, California).
US EPA (1980a) Chemical hazard information profiles (CHIPS).
Washington, DC, US Environmental Protection Agency (EPA 560/11-80-
011).
US EPA (1980b) Organic chemical manufacturing - Volume 9: Selected
processes. Research Triangle Park, North Carolina, US Environmental
Protection Agency (EPA 450/3-80-028d).
US EPA (1983) Toxicity and metabolism studies with EPA priority
pollutants and related chemicals in freshwater organisms. Washington,
DC, US Environmental Protection Agency.
US EPA (1988) Toxic air pollutant emission factors: A compilation for
selected air toxic compounds and sources. Research Triangle Park,
North Carolina, US Environmental Protection Agency (EPA 450/2-88-006).
US EPA (1991) Urban formaldehyde and methanol concentrations for
alternative methanol vehicle scenarios. Research Triangle Park, North
Carolina, US Environmental Protection Agency (Report No. CRC-APRAC-
ME-2).
US EPA (1993) Ambient concentration summaries for Clean Air Act, Title
III: Hazardous air pollutants. Washington, DC, US Environmental
Protection Agency, p B-17 (EPA 600/R-94-090).
US EPA (1994) 1992 Toxic release inventory: Public data. Washington,
DC, US Environmental Protection Agency (EPA 745/R-94-001).
US NIOSH (1976) Recommended standard for occupational exposure to
methyl alcohol. Cincinnati, Ohio, National Institute for Occupational
Safety and Health, 136 pp.
Vaishnav DD & Korthals ET (1990) Comparative toxicities of selected
industrial chemicals to microorganisms and other aquatic organisms.
Arch Environ Contam Toxicol, 19: 624-628.
Vaishnav DD & Lopas DM (1985) Relationship between lipophilicity and
biodegradation inhibition of selected industrial chemicals. Dev Ind
Microbiol, 26: 557-565.
Veith GD, Call DJ, & Brooke LT (1983) Structure-toxicity relationships
for the fat-head minnow, Pimephales promelas: narcotic industrial
chemicals. Can J Fish Aquat Sci, 40: 743-748.
Vendilo MV, Egorov YL, & Feldman NG (1971) [The effects of methanol
and of some higher alcohols on the retina of the eyes (an electron-
microscope investigation).] Gig Tr Prof Zabol, 15: 17-21 (in
Russian).
Venkataraman ES, Ahlert RC, & Corbo P (1984) Biological treatment of
landfill leachates. CRC Crit Rev Environ Control, 14: 333-376.
Verma P & Gupta VK (1984) A sensitive spectrophotometric method for
the determination of methyl alcohol in air and water. Talanta, 31:
394-396.
Vogt MJ, Heffner JE, & Sahn SA (1993) Vomiting, abdominal pain and
visual disturbances in a 31-year old man. Chest, 103: 262-263.
Ward KW & Pollack GM (1996a) Comparative toxicokinetics of methanol in
pregnant and nonpregnant rodents. Drug Metab Dispos, 24: 1062-1070.
Ward KW & Pollack GM (1996b) Use of intrauterine microdialysis to
investigate methanol-induced alterations in uteroplacental blood flow.
Toxicol Appl Pharmacol, 140: 203-210.
Ward JB, Hokanson JA, Smith ER, Chang LW, Pereira MA, Whorton EB, &
Legator MS (1984) Sperm count, morphology and fluorescent body
frequency in autopsy workers exposed to formaldehyde. Mutat Res,
130: 417-424.
Ward KW, Perkins RA, Kawagoe JL, & Pollack GM (1995) Comparative
toxicokinetics of methanol in the female mouse and rat. Fundam Appl
Toxicol, 26: 258-264.
Weese H (1928) Vergleichende untersuchungen uber die wirksamkeit und
giftigkeit der dampfe niederer aliphatischer alkohole. Arch Exptl
Pathol Pharmacol, 135: 118-130 [in German].
Weiss B, Stern S, Soderholm SC, Cox C, Sharma A, Inglis GB, Preston R,
Balys M, Reuhl KR, & Gelein R (1996) Developmental neurotoxicity of
methanol exposure by inhalation in rats. Cambridge, Massachusetts,
Health Effects Institute (Research Report No. 73).
Welch H & Slocum GG (1943) Toxicity of methanol. J Lab Chem Med, 28:
1440-1445.
Wenzyl JE, Mills SD, & McCall JT (1968) Methanol poisoning in an
infant. Am J Dis Child, 116: 445-447.
Whitbeck M (1983) Photo-oxidation of methanol. Atmos Environ, 17:
121-126.
White LR, Martinsen ABL, & Nilsen OG (1983) Biochemical and
cytological studies of rat lung after inhalation of methanol vapour.
Toxicol Lett, 17: 1-5.
WHO (1971) Evaluation of food additives. Fourteenth report of the
Joint FAO/WHO Expert Committee on Food Additives. Geneva, World Health
Organization, p 20 (WHO Technical Report Series No. 462).
Williams RL, Lipari F, & Potter RA (1990) Formaldehyde, methanol and
hydrocarbon emissions from methanol-fueled cars. J Air Waste Manage
Assoc, 40: 747-756.
Windholz M ed. (1983) The Merck index: An encyclopedia of chemicals,
drugs and biologicals, 10th ed. Rahway, New Jersey, Merck & Co.,Inc.,
p 853.
Wood CA & Buller F (1904) Poisoning by wood alcohol. J Am Med Assoc,
43: 973-977.
Wu Chen NB, Donoghue ER, & Schaffer MI (1985) Methanol intoxication:
Distribution in postmortem tissues and fluids including vitreous
humor. J Forensic Sci, 30: 213-216.
Wucherpfennig K, Dietrich H, & Bechtel J (1983) Alcohol actual, total
and potential methyl alcohol of fruit juices. Flussiges Obst, 8:
348-354.
Yant WP & Schrenk HH (1937) Distribution of methanol in dogs after
inhalation and administration by stomach tube and subcutaneously.
J Ind Hyg Toxicol, 19: 337-345.
Yasugi T, Kawai T, Mizunuma K, Horiguchi S, Iwami O, Iguchi H, & Ikeda
M (1992) Formic acid excretion in comparison with methanol excretion
in urine of workers occupationally exposed to methanol. Int Arch Occup
Environ Health, 64: 329-337.
Youssef AF, Baggs RB, Weiss B, & Miller RK (1991) Methanol
teratogenicity in pregnant Long-Evans rats. Teratology, 43: 467.
Youssef AF, Madkour K, Cox C, & Weiss B (1992) Comparative lethality
of methanol, ethanol and mixtures in female rats. J Appl Toxicol,
12: 193-197.
Youssef AF, Weiss B, & Cox C (1993) Neurobehavioural toxicity of
methanol reflected by operant running. Neurotoxicol Teratol, 15:
223-227.
Zinbo M (1984) Determination of one-carbon to three-carbon alcohols
and water in gasoline/alcohol blends by liquid chromatography. Anal
Chem, 56: 244-247.
RESUME
1. Identité, propriétés physiques et chimiques et méthodes d'analyse
Le méthanol se présente sous la forme d'un liquide incolore et
limpide qui dégage une légère odeur alcoolique à l'état pur. Volatil
et inflammable, il est miscible à l'eau et à de nombreux solvants
organiques et forme un grand nombre d'azéotropes binaires.
Il existe un certain nombre de méthodes, principalement la
chromatographie en phase gazeuse avec détection par ionisation de
flamme, pour la recherche et le dosage du méthanol dans divers
échantillons prélevés dans l'environnement (air, eau, sol, et
sédiments) ou dans les produits alimentaires. Ces méthodes sont
également utilisées pour la recherche et le dosage du méthanol et de
son principal métabolite, le formiate, dans les liquides et les tissus
biologiques. Outre la chromatographie en phase gazeuse ave détection
par ionisation de flamme, il existe des méthodes enzymatiques
colorimétriques pour le dosage du formiate dans le sang, les urines et
les tissus.
Pour les analyses sur le lieu de travail, on commence
généralement par recueillir et concentrer l'échantillon sur gel de
silice, après quoi on procède à une extraction par l'eau, puis au
dosage proprement dit par chromatographie en phase gazeuse avec
détection par ionisation de flamme ou spectrométrie de masse.
2. Sources d'exposition humaine
Le méthanol est présent à l'état naturel chez l'Homme, les
animaux et les plantes. C'est un constituant du sang, de l'urine, de
la salive et de l'air expiré. On a mesuré des concentrations moyennes
de méthanol égales à 0,73 mg/litre dans les urines (valeurs
extrêmes:0,3-2,61 mg/litre) chez des sujets non exposés et des valeurs
allant de 0,06 à 0,32 µg/litre ont été observées dans l'air expiré.
Le méthanol et le formiate naturellement présents dans
l'organisme proviennent essentiellement de deux sources:
l'alimentation et le métabolisme. Le méthanol d'origine alimentaire
est principalement apporté par les fruits et les légumes frais ainsi
que par les jus de fruits (teneur moyenne: 140 mg/litre avec des
valeurs extrêmes de 12-640 mg/litre), les boissons fermentées
(jusqu'à 1,5 g/litre), et autres composants du régime alimentaire
(principalement les boissons non alcoolisées). L'aspartame est un
édulcorant très utilisé dont l'hydrolyse donne du méthanol absorbable
dans la proportion de 10% en poids.
En 1991, la production mondiale de méthanol a atteint environ 20
millions de tonnes, principalement par conversion catalytique de gaz
de synthèse sous pression (hydrogène, dioxyde et monoxyde de carbone).
La capacité mondiale de production devrait atteindre 30 millions de
tonnes en 1995.
Le méthanol est utilisé dans l'industrie pour la production
de nombreux produits chimiques importants, principalement le
méthyltertiobutyléther, le formaldéhyde, l'acide acétique, les éthers
méthyliques du glycol, la méthylamine, les halogénures de méthyle et
le méthacrylate de méthyle.
Le méthanol entre dans la composition de nombreux solvants du
commerce et de divers produits comme les peintures, les laques, les
vernis, les diluants pour peintures, les détachants, les antigels,
les liquides pour pare-brise, les dégivrants, les produits pour la
photocopie, les solutions destinées à la dénaturation de l'éthanol
ainsi que différent types de colles. Le méthanol pourrait également
être utilisé directement comme combustible, ou bien encore être ajouté
à l'essence à titre de combustible auxiliaire ou de diluant. Il est à
noter que les cas les plus fréquents d'intoxication, mortelle ou non,
par le méthanol, sont dus à l'ingestion volontaire ou accidentelle de
produits qui en contiennent.
On a trouvé du méthanol dans les gaz d'échappement des moteurs à
essence et des moteurs diesel ainsi que dans la fumée de tabac.
3. Concentrations dans l'environnement et exposition humaine
Les émissions de méthanol proviennent essentiellement des divers
usages qui en sont faits en tant que solvant industriel ou domestique,
des unités de production du composé lui-même ou de ses dérivés, enfin
des pertes lors du stockage ou de la manipulation.
Il peut y avoir exposition au méthanol sur le lieu de travail par
inhalation ou contact cutané. A en juger d'après les limites
d'exposition fixées par de nombreux pays, il semblerait que les
travailleurs ne courent pas de danger tant que l'exposition exprimée
en moyenne pondérée par rapport au temps ne dépasse pas 260 mg/m3
(200 ppm) par journée de 8 h et semaine de 40 h.
Actuellement la population est exposée à des concentrations qui
sont 10 000 fois inférieures aux limites d'exposition professionnelle.
En ce qui concerne l'exposition au méthanol contenu dans l'air, les
concentrations vont de 0,001 mg/m3 (0,8 parties par milliard) en
milieu rural, à près de 0,04 mg/m3 (30 parties par milliard) en
milieu urbain.
On ne possède guère de données sur la teneur en méthanol de l'eau
de boisson après traitement, mais ce composé est en tout cas souvent
présent dans les effluents industriels.
Si les prévisions d'utilisation du méthanol comme combustible de
substitution ou d'appoint augmentent de façon sensible, il faut
s'attendre à ce que l'exposition à ce composé se généralise par
suite de l'inhalation des vapeurs émises par les véhicules qui
l'utiliseront, ou encore de son siphonage ou de son absorption
percutanée lors de la manipulation de combustibles qui en
contiendront.
4. Distribution et transformation dans l'environnement
Le méthanol se décompose rapidement dans l'environnement par
photooxydation et sous l'action de processus de biodégration. Dans le
cas de la réaction atmosphérique du méthanol avec les radicaux
hydroxyle, on a mesuré une demi-vie de 7 à 18 jours.
De nombreux genres et souches de microorganismes sont capables
d'utiliser le méthanol comme substrat. Le composé est facilement
dégradé en aérobiose ou en anaérobiose dans des milieux très divers,
notamment les eaux douces ou salées, les sols et les sédiments,
les eaux souterraines, les nappes phréatiques et les effluents
industriels. En général, 70% du méthanol présent dans les eaux d'égout
est décomposé en l'espace de 5 jours.
Le méthanol sert normalement de substrat à de nombreux
microorganismes terricoles, qui sont capables de le dégrader
complètement en dioxyde carbone et en eau.
Le méthanol est médiocrement absorbé par les sols. Sa
bioaccumulation est faible dans la plupart des organismes.
Le méthanol est peu toxique pour les organismes aquatiques et
terrestres et il est peu probable que l'on observe des effets
résultant d'une exposition environnementale à ce composé, sauf en cas
de déversements dans la nature.
5. Absorption, distribution, biotransformation et élimination
Après inhalation, ingestion ou contact cutané, le méthanol est
facilement résorbé et se diffuse rapidement dans les tissus en
fonction de la répartition de l'eau dans l'organisme. Une faible
proportion est excrétée telle quelle par les poumons et les reins.
Après ingestion, les concentrations sériques maximales sont
atteintes en 30 à 90 minutes et le méthanol se répartit dans
l'organisme avec un volume de distribution d'environ 0,6 litre/kg.
Le méthanol est métabolisé principalement au niveau du foie
selon un processus oxydatif qui le transforme successivement en
formaldéhyde, acide formique et dioxyde de carbone. La première étape,
celle de l'oxydation en formaldéhyde, s'effectue sous l'action de
l'alcool-déshydrogénase hépatique; il s'agit d'une étape limitante qui
correspond à un processus saturable. L'affinité relative de l'alcool-
déshydrogénase pour le méthanol et pour l'éthanol est d'environ
20:1. Lors de la seconde étape, le formaldéhyde est oxydé par la
formaldéhyde - déshydrogénase en acide formique ou en formiate, selon
la valeur du pH. La troisième étape consiste dans la détoxication de
l'acide formique en dioxyde de carbone par des réactions dépendant de
l'acide folique.
L'élimination du méthanol présent dans le sang par la
voie urinaire ou dans l'air expiré, soit tel quel, soit après
métabolisation, se révèle être un processus lent chez toutes les
espèces, en particulier par comparaison avec l'éthanol. Ainsi, la
clairance du méthanol s'effectue avec une demi-vie de 24 h ou
davantage pour des doses inférieures à 0,1 g/kg. C'est au niveau de la
détoxication métabolique, c'est-à-dire de l'élimination du formiate,
que des différences très importantes existent entre les rongeurs et
les primates et ce sont elles qui expliquent la différence
spectaculaire de toxicité que l'on constate entre les premiers et les
seconds.
6. Effets sur les mammifères de laboratoire et les systèmes d'épreuve
in vitro
6.1 Toxicité générale
La toxicité aiguë et la toxicité à court terme du méthanol
varient beaucoup selon les diverses espèces et alles sont maximales
chez celles qui métabolisent relativement mal le formiate. En pareil
cas, le méthanol provoque une intoxication mortelle par acidose
métabolique et toxicité neuronale. En revanche, chez les animaux qui
métabolisent bien le formiate, la mort survient habituellement par
suite de la dépression du système nerveux central (coma, insuffisance
respiratoire etc.). Chez les primates sensibles (comme l'Homme et les
singes), il y a augmentation du taux sanguin de formiate après
exposition au méthanol, alors que chez les rongeurs résistants, les
lapins et les chiens, cette augmentation du taux de formiate ne se
produit pas. L'Homme et les primates non humains présentent une
sensibilité unique aux effets toxiques du méthanol. Globalement, le
méthanol est peu toxique pour les animaux autres que les primates. La
valeur de la DL50 et de la dose létale minimale pour une exposition
par la voie orale, varie de 7000 à 13 000 mg/kg chez le rat, la
souris, le lapin et le chien et de 2000 à 7000 mg/kg chez le singe.
Chez des rats exposés à du méthanol 6 h par jour, 5 jours par
semaine pendant 4 semaines, à des concentrations allant jusqu'à
6500 mg/m3 (5000 ppm), on n'a observé aucun effet imputable à
l'exposition, sauf une augmentation des écoulements au niveau du nez
et des yeux. On estime qu'il s'agissait là de la conséquence d'une
irritation des voies respiratoires supérieures.
Des rats exposés à des vapeurs de méthanol à des concentrations
pouvant atteindre 13 000 mg/m3 (10 000 ppm), 6 h par jour, 5 jours
par semaine pendant 6 semaines, n'ont pas présenté de signes de
toxicité pulmonaire.
Chez le lapin, le méthanol irrite modérément la muqueuse
oculaire. Lors d'une épreuve qui était une variante du test de
maximalisation, il n'a pas provoqué de sensibilisation cutanée.
Parmi les effets toxiques du méthanol observés chez les primates,
on peut citer l'acidose métabolique et la toxicité oculaire qui ne se
produisent en principe pas chez les rongeurs dont le taux de folate
est suffisant. Ces différences de toxicité s'expliquent par des
différences dans la vitesse de métabolisation du formiate, qui est un
métabolite du méthanol. Par exemple, la clairance du formiate sanguin
est au moins 50% plus lente chez les primates que chez les rongeurs.
Des singes qui recevaient du méthanol par gavage à des doses
dépassant 3000 mg/kg ont présenté une ataxie, de la faiblesse et une
léthargie dans les quelques heures suivant l'administration du
composé. Ces signes avaient tendance à disparaître en l'espace de 24 h
et ils étaient suivis d'un coma passager chez certains des animaux.
Chez des singes exposés à du méthanol à 20 reprises 6 h par jour
et 5 jours par semaines, à la dose de 6500 mg/m3 (5000 ppm), on n'a
pas constaté d'effets oculaires.
6.2 Génotoxicité et cancérogénicité
Les tests de mutation génétique effectués avec du méthanol sur
des bactéries et des levures ont donné des résultats négatifs, mais le
composé à provoqué une ségrégation chromosomique défectueuse chez
Aspergillus. Il n'a pas provoqué d'échanges de chromatides soeurs
dans des cellules de hamster chinois in vitro, mais il a augmenté
de façon sensible la fréquence des mutations dans des cellules
lymphomateuses de souris L5178Y.
L'inhalation de méthanol n'a pas provoqué de lésions chromo-
somiques chez la souris. Par contre, on est fondé à penser, dans une
certaine mesure, que l'administration intrapéritonéale ou buccale de
méthanol augmente l'incidence des lésions chromosomiques chez la
souris.
Rien n'indique, au vu de l'expérimentation animale, que le
méthanol soit cancérogène, mais il faut admettre qu'il n'existe pas de
modèle animal approprié pour ce genre d'étude.
6.3 Toxicité pour la fonction reproductrice, embryotoxicité et
tératogénicité
Des études concernant les effets sur les taux de gonadotrophine
et de testostérone d'une exposition au méthanol, par la voie
respiratoire, pendant des périodes allant jusqu'à 6 semaines, ont
donné des résultats contradictoires.
En faisant inhaler du méthanol à des rongeurs gravides pendant
toute la période de l'embryogénèse, on obtient toute une série
d'effets tératogènes et embryocides qui dépendent de la concentration.
Ainsi, on a observé des malformations attribuables au traitement et
consistant principalement dans la présence de côtes cervicales
surnuméraires ou rudimentaires, ou encore de malformations urinaires
ou cardiovasculaires, chez des foetus de rats exposés 7 h par jour du
7iéme au 15 ième jour de la gestation à une concentration de
26 000 mg/m3, soit l'équivalent de 20 000 ppm, de méthanol. A cette
concentration, le méthanol était légèrement toxique pour les mères. En
revanche, à la concentration de 6500 mg/m3 (5000 ppm), aucun effet
indésirable n'a été noté chez les mères ou chez leur progéniture et on
a considéré que cette valeur constituait la concentration sans effet
nocif observable (NOAEL) pour ce système d'épreuve.
Dans la progéniture de souris CD-1 exposées à du méthanol à des
concentrations supérieures ou égales à 6500 mg/m3 (5000 ppm), 7 h par
jour du 6 ième au 15 ième jour de la gestation, on a observé une
incidence accrue d'exencéphalies et de fissures de la voûte palatine.
Aux concentrations supérieures ou égales à 9825 mg/m3 (7500 ppm), les
résorptions affectant la totalité de la portée étaient également plus
fréquentes. Aux concentrations de 13 000 et 19 500 mg/m3 (10 000
ou 15 000 ppm), on a observé une réduction du poids foetal. La
concentration sans effet observable (NOAEL) sur le développement a été
évaluée à 1300 mg/m3 (1000 ppm). Aux concentrations inférieures à
9000 mg/m3 (7000 ppm), rien n'a été relevé qui puisse indiquer une
toxicité du méthanol pour les mères.
En donnant à la progéniture de ces souris CD-1 une dose de 4 g/kg
de méthanol par gavage, on a constaté que l'incidence des effets
nocifs (résorptions, fissures palatines et réduction du poids foetal)
était analogue à celle constatée dans le groupe de rats auquels on
avait fait inhaler le composé à la concentration de 13 000 mg/m3
(10 000 ppm), probablement en raison de la fréquence respiratoire plus
élevée chez la souris. La souris est plus sensible que le rat aux
effets toxiques que le méthanol inhalé exerce sur le développement.
Des signes neurologiques passagers et une réduction du poids
corporel ont été enregistrés chez des souris CD-1 gravides, exposées
6 h par jour à une concentration de 19 500 mg/m3, soit l'équivalent
de 15 000 ppm tout au long de l'organogénèse (du sixième au quinzième
jour). Parmi les malformations foetales observées aux doses de 19 500
et 13 000 mg/m3, soit 15 000 et 10 000 ppm, on peut citer des
anomalies neurales et oculaires, des fissures palatines, des
hydronéphroses et des malformations des membres.
7. Effets sur l'Homme
L'Homme (et les primates non humains) présentent une sensibilité
unique au méthanol et les effets toxiques relevés chez ces espèces
sont caractérisés par une acidémie formique, une acidose métabolique,
une toxicité oculaire, une dépression du système nerveux, la cécité,
le coma et la mort. Presque toutes les données que l'on possède sur la
toxicité du méthanol pour l'Homme, ont trait aux conséquences des
intoxications aiguës plutôt qu'à celles des intoxications chroniques.
La très grande majorité des intoxications par le méthanol résultent de
la consommation de boissons frelatées et de produits contenant du
méthanol. C'est par ingestion que se produisent la plupart de ces
intoxications, mais l'inhalation de vapeurs de méthanol sous forte
concentration et l'absorption percutanée de solutions méthanoliques
conduisent aux mêmes effets toxiques que l'ingestion. Les effets
toxiques les plus fréquemment notés à la suite d'une exposition de
longue durée, sont des effets oculaires très variés.
Les effets toxiques du méthanol sont liés aux facteurs qui
régissent la conversion du méthanol en acide formique et la
transformation ultérieure de ce dernier en dioxyde de carbone par la
voie des folates. Ces effets se manifestent lorsque la vitesse de
formation du formiate est supérieure à sa vitesse de métabolisation.
On ne sait pas avec certitude quelle est la dose mortelle pour
l'Homme. En l'absence d'intervention médicale, la dose létale minimum
se situe entre 0,3 et 1 g/kg. On ignore quelle est dose minimale à
partir de laquelle se produisent des lésions oculaires permanentes.
L'acidose métabolique est de gravité variable et alle n'est pas
forcément en bonne corrélation avec la quantité de méthanol ingérée.
Les intoxications méthanoliques se caractérisent par de grandes
variations individuelles dans la dose toxique.
Il semble que deux facteurs importants déterminent la sensibilité
humaine aux effets toxiques du méthanol: 1) l'ingestion simultanée
d'éthanol, qui retarde l'entrée du méthanol dans sa voie de
dégradation métabolique; 2) le bilan des folates hépatiques, dont
dépend la vitesse de détoxication du formiate.
Les symptômes de l'intoxication méthanolique, qui peuvent ne se
manifester qu'au bout de 12 à 24 h, consistent en troubles visuels,
nausées, douleurs abdominales et musculaires, étourdissements,
faiblesse et troubles de la conscience allant du coma au crises
cloniques. Les troubles visuels apparaissent généralement dans les
12 à 18 h suivant l'ingestion de méthanol et vont d'une légère
photophobie avec une vision floue ou voilée à une réduction importante
de l'acuité visuelle, voire à la cécité totale. Dans les cas extrêmes,
l'intoxication peut avoir une issue fatale. Sur le plan clinique, la
principale manifestation est une acidose métabolique grave par
augmentation du trou anionique. L'acidose est largement attribuée à
l'acide formique résulant de la métabolisation du méthanol.
La concentration sanguine normale du méthanol d'origine endogène
est inférieure à 0,5 mg/litre (0,02 mmol/litre), mais l'alimentation
peut accroître le taux sanguin de méthanol. En général, les effets
neurologiques centraux apparaissent lorsque la concentration sanguine
du méthanol dépasse 200 mg/litre (6 mmol/litre); les symptômes
oculaires se manifestent à partir de 500 mg/litre (16 mmol/litre) et
la mort est survenue chez des patients non traités dont les taux
sanguins initiaux de méthanol se situaient entre 1500 et
2000 mg/litre, soit 47 à 62 mmol/litre.
L'inhalation occasionnelle de vapeurs de méthanol à une
concentration inférieure à 260 mg/m3 ou l'ingestion du liquide en
quantités ne dépassant pas 20 mg/kg, ne devraient pas conduire à une
accumulation de formiate supérieure au taux endogène, s'agissant de
sujets en bonne santé ou présentant un déficit modéré en folate. Des
troubles visuels de divers types (vision floue, rétrécissement du
champ visuel, modification de la perception des couleurs et cécité
temporaire ou permanente) ont été signalés chez des travailleurs
exposés à des concentrations de méthanol dans l'air inférieures ou
égales à environ 1500 mg/m3 (1200 ppm).
On utilise largement la valeur de 260 mg/m3 (200 ppm) comme
limite d'exposition professionnelle au méthanol. Cette valeur a été
calculée pour protéger les travailleurs contre l'acidose formique
induite par le méthanol et contre les effets toxiques de ce composé
sur l'oeil et le système nerveux.
On n'a pas signalé chez l'Homme d'autres effets nocifs qu'une
légère irritation cutanée et oculaire aux concentrations très
supérieures à 260 mg/m3 (200 ppm).
8. Effets sur les êtres vivants dans leur milieu naturel
Pour les organismes aquatiques, la valeur de la CL50 varie de
1300 à 15 900 mg/litre dans le cas des invertébrés (exposition de 48 h
et de 96 h), et de 13 000 à 29 000 mg/litre dans le cas des poissons
(exposition de 96 h).
Le méthanol est peu toxique pour les organismes aquatiques et il
n'est guère probable que l'on observe des effets imputables à une
exposition environnementale, sauf en cas de déversement de méthanol
dans la nature.
RESUMEN
1. Identidad propiedades físicas y químicas y métodosanalíticos
El metanol es un líquido transparente, incoloro, volátil e
inflamable con un ligero olor alcohólico en estado puro. Se puede
mezclar con el agua y con muchos disolventes orgánicos y forma
numerosas mezclas azeotrópicas binarias.
Hay métodos analíticos, principalmente la cromatografía de
gases (CG) con detección por ionización de llama (DIL), para la
determinación del metanol en diversos medios (aire, agua, suelo y
sedimentos) y productos alimenticios, así como para la determinación
del metanol y de su principal metabolito, el formiato, en los líquidos
y tejidos corporales. Además de la CG-DIL, en la determinación del
formiato en la sangre, la orina y los tejidos se utilizan
procedimientos enzimáticos con resultados finales colorimétricos.
Para la determinación del metanol en el lugar de trabajo se suele
comenzar con la recolección y concentración en silicagel, seguida de
extracción acuosa y CG-DIL o análisis de CG-espectrometría de masa del
extracto.
2. Fuentes de exposición humana
El metanol está presente de forma natural en el ser humano, los
animales y las plantas. Es un elemento constitutivo natural en la
sangre, orina, la saliva y el aire expirado. Se ha descrito una
concentración media de metanol en orina de 0,73 mg/litro (intervalo de
0,3-2,61 µg/litro) en individuos no expuestos y una gama de 0,06 a
0,32 µg/litro en el aire expirado.
Las dos fuentes más importantes de acumulación básica de
metanol y formiato en el organismo son la alimentación y los
procesos metabólicos. El metanol está disponible en la alimentación
principalmente a partir de las frutas y hortalizas frescas, los zumos
de fruta (promedio de 140 mg/litro, margen de variación de 12 a
640 mg/litro), las bebidas fermentadas (hasta 1,5 g/litro) y los
alimentos de dieta (sobre todo bebidas no alcohólicas). El aspartame
es un edulcorante artificial muy utilizado, y al hidrolizarse el 10%
(por peso) de la molécula se convierte en metanol libre, que queda
disponible para la absorción.
En 1991 se produjeron en todo el mundo alrededor de 20 millones
de toneladas de metanol, fundamentalmente por conversión catalítica de
gas de síntesis a presión (hidrógeno, anhídrido carbónico y monóxido
de carbono). Según las proyecciones, la capacidad mundial se elevaría
a 30 millones de toneladas en 1995.
El metanol se utiliza en la producción industrial de numerosos
compuestos orgánicos importantes, sobre todo metil terbutil éter
(MTBE), formaldehído, ácido acético, éteres de metilglicol,
metilamina, haluros de metilo y metacrilato de metilo.
El metanol es un elemento constitutivo de un gran número de
disolventes y productos de consumo disponibles en el comercio, como
pinturas, gomas laca, barnices, diluyentes de pinturas, soluciones
limpiadoras, soluciones anticongelantes, líquidos limpiaparabrisas y
anticongelantes para automóviles, líquidos de multicopista,
desnaturalizante para el etanol y pegamento para actividades de
pasatiempo y artesanía. Una aplicación potencialmente en gran escala
del metanol está en su uso directo como combustible, mezclado con
gasolina o para aumentar su volumen. Hay que señalar que la mayor
morbilidad y mortalidad se ha relacionado con la ingestión oral
deliberada o accidental de mezclas con contenidos de metanol.
Se ha detectado metanol en los gases de escape de motores tanto
de gasolina como diésel y en el humo del tabaco.
3. Niveles ambientales y exposición humana
Las emisiones de metanol se derivan principalmente de los
diversos usos industriales y domésticos como disolvente, su
producción, la manufactura final y las pérdidas durante el
almacenamiento a granel y la manipulación.
Pueden darse exposiciones al metanol en los lugares de trabajo
mediante inhalación o contacto cutáneo. Muchos de los límites
nacionales de exposición para la higiene del trabajo parecen indicar
que los trabajadores están protegidos de cualquier efecto adverso si
la exposición no supera un promedio ponderado por el tiempo de
260 mg/m3 (200 ppm) de metanol en cualquier jornada de trabajo
de 8 horas y en una semana laboral de 40 horas.
La exposición general actual de la población por medio del aire
es normalmente 10 000 veces inferior a los límites ocupacionales.
La población general está expuesta al metanol en el aire a
concentraciones que oscilan entre menos de 0,001 mg/m3 (0,8 ppm)
en el aire del medio rural y cerca de 0,04 mg/m3 (30 ppm) en el
aire urbano.
Los datos sobre la presencia del metanol en el agua potable de
uso inmediato son limitados, pero con frecuencia se encuentra metanol
en efluentes industriales.
En el caso de que el uso previsto del metanol como
combustible alternativo o mezclado con otros combustibles aumente
considerablemente, cabe prever que habrá una exposición generalizada
al metanol por medio de la inhalación de vapores procedentes de los
vehículos que funcionen con él y del bombeo o la absorción percutánea
de combustibles o mezclas de metanol.
4. Distribución y transformación en el medio ambiente
El metanol se degrada fácilmente en el medio ambiente mediante
procesos de fotooxidación y biodegradación. Se han descrito semividas
de 7-18 días para la reacción atmosférica del metanol con radicales
oxhidrilo.
Hay muchos géneros y cepas de microorganismos capaces de utilizar
el metanol como sustrato de crecimiento. El metanol es fácilmente
degradable en condiciones tanto aerobias como anaerobias en una amplia
variedad de medios naturales, entre ellos agua dulce y salada,
sedimentos y suelos, agua freática, material de acuíferos y aguas
residuales industriales; el 70% del metanol de los alcantarillados se
suele degradar en un plazo de 5 días.
El metanol es un sustrato de crecimiento normal de muchos
microorganismos del suelo, que son capaces de degradarlo completamente
a anhídrido carbónico y agua.
El metanol tiene una capacidad de absorción bastante baja en los
suelos. La bioconcentración en la mayoría de los organismos es escasa.
El metanol es poco tóxico para los organismos acuáticos y
terrestres y no es probable que se observen efectos debidos a su
exposición en el medio ambiente, excepto en el caso de un derrame.
5. Absorción, distribución, biotransformación yeliminación
El metanol se absorbe fácilmente por inhalación, ingestión y
exposición cutánea y se distribuye rápidamente en los tejidos
siguiendo la distribución del agua corporal. Por los pulmones y los
riñones se excreta una pequeña cantidad de metanol sin cambios.
Tras la digestión se alcanzan niveles máximos en suero en un
plazo de 30-90 minutos, y se reparte por todo el organismo con un
volumen de distribución aproximado de 0,6 litros/kg.
El metanol se metaboliza principalmente en el hígado siguiendo
una fase oxidativa secuencial a formaldehído, ácido fórmico y
anhídrido carbónico. El paso inicial consiste en la oxidación a
formaldehído por acción de la alcohol deshidrogenasa hepática, que
es un proceso saturable limitante de la velocidad. La afinidad
relativa de la alcohol deshidrogenasa por el etanol y el metanol es
aproximadamente de 20:1. En el segundo paso, el formaldehído se oxida
por acción de la formaldehído deshidrogenasa a ácido fórmico o
formiato, en función del pH. En el tercer paso, el ácido fórmico se
destoxifica a anhídrido carbónico mediante reacciones dependientes del
folato.
La eliminación del metanol de la sangre a través de la orina y el
aire exhalado y por el metabolismo parece ser lenta en todas las
especies, especialmente si se compara con el etanol. En el proceso se
han descrito períodos de semieliminación de 24 horas o más con dosis
superiores a 1 g/kg y de 2,5-3 horas para dosis inferiores a 0,1 g/kg.
El ritmo de desintoxicación metabólica o eliminación del formiato sí
es muy distinto entre los roedores y los primates, constituyendo la
base de las enormes diferencias de toxicidad del metanol observadas
entre ambos grupos.
6. Efectos en mamíferos de laboratorio y en sistemas deensayo
in vitro
6.1 Toxicidad sistémica
La toxicidad aguda y a corto plazo del metanol varía mucho entre
las distintas especies, siendo máxima en las especies con una
capacidad relativamente escasa para metabolizar el formiato. En tales
casos de metabolismo deficiente del formiato, se produce una
intoxicación letal por metanol como consecuencia de la acidosis
metabólica y la toxicidad neuronal, mientras que en los animales que
metabolizan fácilmente el formiato la muerte suele deberse a las
consecuencias de la depresión del sistema nervioso central (coma,
insuficiencia respiratoria, etc.). En las especies de primates
sensibles (el ser humano y los monos) aumenta la concentración del
formiato en sangre tras la exposición al metanol, pero no en los
roedores, los conejos y los perros resistentes. Los primates humanos y
no humanos tienen una sensibilidad única a los efectos tóxicos del
metanol. En conjunto, el metanol tiene una toxicidad aguda baja para
los animales no primates. Los valores de la DL50 y las dosis letales
mínimas tras la exposición oral oscilan entre 7000 y 13 000 mg/kg en
ratas, ratones, conejos y perros y entre 2000 y 7000 mg/kg en el mono.
Las ratas expuestas a concentraciones de metanol de hasta
6500 mg/m3 (5000 ppm) 6 horas al día y 5 días a la semana durante
un período de 4 semanas no mostraron ningún efecto relacionado con la
exposición, salvo el aumento de la exudación alrededor de la nariz y
de los ojos. Se consideró que esto era un reflejo de la irritación de
las vías respiratorias superiores.
Las ratas expuestas a concentraciones de vapor de metanol de
hasta 13 000 mg/m3 (10 000 ppm) 6 horas al día y 5 días a la semana
durante un período de 6 semanas, no mostraron ninguna toxicidad
pulmonar.
En el conejo el metanol es moderadamente irritante de los ojos.
En una prueba de maximización modificada no se produjo sensibilización
cutánea.
Entre los efectos tóxicos observados en primates expuestos al
metanol cabe mencionar la acidosis metabólica y la toxicidad ocular,
pero estos efectos no aparecen normalmente en roedores con una
concentración suficiente de folato. Las diferencias de toxicidad se
deben a variaciones en la tasa del metabolismo del formiato,
metabolito del metanol. Por ejemplo, la eliminación del formiato de la
sangre de los primates expuestos es como mínimo un 50% más lenta que
en los roedores.
En monos que recibieron dosis de metanol superiores a 3000 mg/kg
con sonda se observó ataxia, debilidad y letargo a las pocas horas de
exposición. Estos signos mostraron una tendencia a desaparecer en un
plazo de 24 horas y los siguió un coma transitorio en algunos de los
animales.
En monos expuestos a metanol durante 6 horas al día y 5 días a la
semana, con 20 exposiciones repetidas a 6500 mg/m3 (5000 ppm) de
metanol no aparecieron efectos oculares.
6.2 Genotoxicidad y carcinogenicidad
El metanol ha dado resultados negativos en cuanto a la mutación
genética en ensayos con bacterias y levaduras, pero indujo anomalías
en la segregación cromosómica en Aspergillus. No indujo intercambios
de cromátidas hermanas en células de hámster chino in vitro, pero
provocó un aumento considerable de la frecuencia de las mutaciones en
células de linfoma de ratón L5178Y.
La inhalación de metanol no indujo daños en los cromosomas de
ratones. Hay algunas pruebas de que la administración oral e
intraperitoneal ha aumentado la incidencia de daños en los cromosomas
de ratones.
No hay ninguna prueba en estudios con animales que indique que el
metanol es carcinógeno, aunque se reconoce que se carece de un modelo
animal apropiado.
6.3 Toxicidad reproductiva, embriotoxicidad yteratogenicidad
Se han descrito resultados contradictorios en relación con los
efectos de la inhalación de metanol durante un período de hasta 6
semanas sobre las concentraciones de gonadotropina y testosterona.
La inhalación de metanol por roedores gestantes durante todo el
período de la embriogénesis induce una amplia variedad de efectos
teratogénicos y embrioletales dependientes de la concentración. En
fetos de ratas expuestas 7 horas al día durante 7-15 días de gestación
a 26 000 mg/m3 (20 000 ppm) de metanol se encontraron malformaciones
relacionadas con el tratamiento, predominantemente costillas
cervicales adicionales o rudimentarias y defectos del aparato urinario
o cardiovascular. Con este nivel de exposición se observó una ligera
toxicidad materna, pero no se detectó ningún efecto adverso para la
madre o la descendencia en animales expuestos a 6500 mg/m3 (5000
ppm), lo cual se interpretó como la concentración sin efectos adversos
observados (NOAEL) para este sistema de prueba.
Se registró una mayor incidencia de exencefalia y paladar hendido
en la descendencia de ratones CD-1 expuestos 7 horas al día durante
los días 6-15 de gestación a concentraciones de metanol de 6500 mg/m3
(5000 ppm) o más. La mortalidad embriofetal aumentó a concentraciones
de 9825 mg/m3 (7500 ppm) o más y fue mayor la incidencia de
resorciones de toda la descendencia. A concentraciones de 13 000 y
19 500 mg/m3 (10 000 y 15 000 ppm) se observó un peso fetal reducido.
La NOAEL para la toxicidad del desarrollo fue de 1300 mg/m3 (1000
ppm) de metanol. No se encontraron pruebas de toxicidad materna en la
exposición a concentraciones de metanol inferiores a 9000 mg/m3
(7000 ppm).
Cuando se administraron mediante sonda 4 g/kg de metanol a la
descendencia de ratones CD-1 gestantes, la incidencia de los efectos
adversos en la resorción, los defectos externos como el paladar
hendido y el peso del feto fue análoga a la observada en el grupo
expuesto por inhalación a 13 000 mg/m3 (10 000 ppm), posiblemente
debido al mayor ritmo de respiración del ratón. Éste es más sensible
que la rata a la toxicidad en el desarrollo provocada por el metanol
inhalado.
En hembras CD-1 expuestas a 19 500 mg/m3 (15 000 ppm) durante 6
horas diarias a lo largo de la organogénesis (días de gestación 6-15)
aparecieron signos neurológicos transitorios y una reducción del peso
corporal. Entre las malformaciones fetales registradas con 13 000 y
19 500 mg/m3 (10 000 y 15 000 ppm) cabe mencionar defectos neurales y
oculares, paladar hendido, hidronefrosis y anomalías de las
extremidades.
7. Efectos en el ser humano
El ser humano y los primates no humanos tienen una sensibilidad
única a la intoxicación por metanol, caracterizándose los efectos
tóxicos en estas especies por acidemia fórmica, acidosis metabólica,
toxicidad ocular, depresión del sistema nervioso, ceguera, coma y la
muerte. Casi toda la información disponible sobre la toxicidad del
metanol en el ser humano se refiere a las consecuencias de
exposiciones agudas más que crónicas. La inmensa mayoría de las
intoxicaciones por metanol se han debido al consumo de bebidas
adulteradas y de productos con metanol. Aunque la ingestión es con
diferencia la vía más frecuente de intoxicación, la inhalación de
concentraciones elevadas de vapor de metanol o la absorción percutánea
de líquidos metanólicos son tan eficaces como la vía oral para la
producción de efectos tóxicos agudos. La consecuencia más conocida
para la salud de una exposición a plazo más largo a niveles inferiores
de metanol es una amplia gama de efectos oculares.
Las propiedades tóxicas del metanol se basan en factores que
rigen tanto su conversión en ácido fórmico como el posterior
metabolismo del formiato a anhídrido carbónico en la ruta del folato.
La toxicidad es manifiesta si la generación de formiato continúa a un
ritmo superior al del metabolismo.
No se conoce con seguridad la dosis letal del metanol para el ser
humano. La dosis letal mínima del metanol en ausencia de tratamiento
médico está comprendida entre 0,3 y 1 g/kg. No se conoce la dosis
mínima que provoca defectos visuales permanentes.
La gravedad de la acidosis metabólica es variable y puede no
tener correlación con la cantidad de metanol ingerido. Una
característica destacada de la intoxicación aguda por metanol es la
enorme variabilidad interindividual de la dosis tóxica.
Parece que dos factores determinantes importantes de la
susceptibilidad humana a la toxicidad por metanol son: 1) ingestión
junto con etanol, que reduce el ritmo de entrada de metanol en la ruta
metabólica, y 2) la situación del folato hepático, que rige la tasa de
desintoxicación del formiato.
Los síntomas y signos de intoxicación por metanol, que pueden
aparecer solo transcurrido un período asintomático aproximado de 12 a
24 horas, son perturbaciones visuales, náuseas, dolor abdominal y
muscular, mareo, debilidad y perturbaciones de la conciencia que van
desde el coma hasta las convulsiones clónicas. Las alteraciones
visuales aparecen en general entre las 12 y las 48 horas después de la
ingestión del metanol y van desde la ligera fotofobia y la visión
brumosa o borrosa hasta una reducción acentuada de la agudeza visual y
la ceguera completa. En casos extremos se produce la muerte. La
principal característica clínica es una acidosis metabólica grave del
tipo de deficiencia de aniones. La acidosis se atribuye en gran medida
al ácido fórmico producido al metabolizarse el metanol.
La concentración normal en sangre de metanol procedente de
fuentes endógenas es de menos de 0,5 mg/litro (0,02 mmol/litro), pero
las fuentes alimenticias pueden elevarla. En general aparecen efectos
en el sistema nervioso central cuando la concentración de metanol en
sangre supera los 200 mg/litro (6 mmol/litro); se detectan síntomas
oculares por encima de 500 mg/litros (16 mmol/litro) y se han
registrado casos de letalidad en pacientes no tratados con
concentraciones iniciales de metanol del orden de 1500-2000 mg/litro
(47-62 mmol/litro).
La inhalación aguda de concentraciones de vapor de metanol por
debajo de 260 mg/m3 o la ingestión de cantidades de hasta 20 mg/kg de
metanol por parte de personas sanas o con una deficiencia moderada de
folato no debe dar lugar a la acumulación de formiato por encima de
las concentraciones endógenas.
Se ha informado de alteraciones visuales de varios tipos (visión
borrosa, reducción del campo visivo, cambios en la percepción de los
colores y ceguera temporal o permanente) en trabajadores expuestos a
concentraciones de metanol en el aire de alrededor de 1500 mg/m3
(1200 ppm) o más.
Un límite muy utilizado de exposición en el trabajo para el
metanol es el de 260 mg/m3 (200 ppm), concebido para proteger a los
trabajadores de cualquiera de los efectos de la acidosis metabólica
por ácido fórmico inducida por el metanol y de la toxicidad ocular y
del sistema nervioso.
No se ha notificado ningún otro efecto adverso del metanol en
el ser humano, salvo una ligera irritación cutánea y ocular con
exposiciones muy superiores a los 26 mg/m3 (200 ppm).
8. Efectos en los organismos del medio ambiente
Los valores de la CL50 en organismos acuáticos oscilan entre
1300 y 15 900 mg/litro para los invertebrados (48 y 96 horas de
exposición) y entre 13 000 y 29 000 mg/litro para los peces (96 horas
de exposición).
El metanol es poco tóxico para los organismos acuáticos siendo
poco probable la observación de efectos debidos a exposición ambiental
al metanol, excepto en el caso de un derrame.