
UNITED NATIONS ENVIRONMENT PROGRAMME
INTERNATIONAL LABOUR ORGANISATION
WORLD HEALTH ORGANIZATION
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 193
Phosgene
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Environmental Health Criteria 193
First draft prepared at the National Institute of Health Sciences,
Tokyo, Japan, and the Institute of Terrestrial Ecology, Monk's Wood,
United Kingdom
Published under the joint sponsorship of the United Nations
Environment Programme, the International Labour Organisation, and the
World Health Organization, and produced within the framework of the
Inter-Organization Programme for the Sound Management of Chemicals.
World Health Organization
Geneva, 1997
The International Programme on Chemical Safety (IPCS) is a joint
venture of the United Nations Environment Programme, the International
Labour Organisation, and the World Health Organization. The main
objective of the IPCS is to carry out and disseminate evaluations of
the effects of chemicals on human health and the quality of the
environment. Supporting activities include the development of
epidemiological, experimental laboratory, and risk-assessment methods
that could produce internationally comparable results, and the
development of manpower in the field of toxicology. Other activities
carried out by the IPCS include the development of know-how for coping
with chemical accidents, coordination of laboratory testing and
epidemiological studies, and promotion of research on the mechanisms
of the biological action of chemicals.
WHO Library Cataloguing in Publication Data
Phosgene
(Environmental health criteria ;193)
1.Phosgene - toxicity 2.Phosgene - adverse effects
3. Environmental exposure I.Series
ISBN 92 4 157193 4 (NLM Classification: QV664)
ISSN 0250-863X
The World Health Organization welcomes requests for permission to
reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made to
the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1997
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved. The designations
employed and the presentation of the material in this publication do
not imply the expression of any opinion whatsoever on the part of the
Secretariat of the World Health Organization concerning the legal
status of any country, territory, city or area or of its authorities,
or concerning the delimitation of its frontiers or boundaries. The
mention of specific companies or of certain manufacturers' products
does not imply that they are endorsed or recommended by the World
Health Organization in preference to others of a similar nature that
are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR PHOSGENE
PREAMBLE
ABBREVIATIONS
1. SUMMARY
1.1 Identity, physical and chemical properties, and analytical
methods
1.2 Uses and sources of human and environmental exposure
1.3 Environmental transport, distribution and transformation
1.4 Environmental levels and human exposure
1.5 Kinetics and metabolism
1.6 Effects on experimental animals and in vitro test systems
1.6.1 Single and short-term exposures
1.6.2 Non-pulmonary effects
1.7 Effects on humans
1.8 Effects on organisms in the environment
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL
METHODS
2.1 Identity
2.2 Physical and chemical properties
2.3 Analytical methods
2.4 Conversion factors
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
3.2 Anthropogenic sources
3.2.1 Production levels and processes
3.2.2 Environmental processes
3.2.3 Uses
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
4.2 Abiotic degradation
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
5.1.2 Water
5.1.3 Soil
5.1.4 Food and feed
5.2 General population exposure
5.3 Occupational exposure
5.3.1 Manufacture and use
5.3.2 Non-manufacturing occupations
6. KINETICS AND METABOLISM
6.1 Absorption
6.2 Distribution
7. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
7.1 Single and short-term inhalation exposures
7.2 Skin and eye irritation; sensitization
7.3 Long-term exposure
7.4 Reproductive and developmental toxicity
7.5 Mutagenicity and related end-points
7.6 Carcinogenicity
7.7 Immunotoxicity
7.8 Mechanism of toxicity.
8. EFFECTS ON HUMANS
8.1 General population and occupational exposure
8.2 Case reports - individual accidents
8.3 Epidemiological studies
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of human health risks
10.1.1 Exposure
10.1.2 Health effects
10.1.2.1 Evaluation of animal data
10.1.2.2 Evaluation of human data
10.1.3 Guidance value
10.2 Evaluation of effects on the environment
11. CONCLUSIONS AND RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH
11.1 Conclusions
11.2 Recommendations for protection of human health
12. FURTHER RESEARCH
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
REFERENCES
RESUME ET CONCLUSIONS
RESUMEN Y CONCLUSIONES
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the criteria
monographs as accurately as possible without unduly delaying their
publication. In the interest of all users of the Environmental Health
Criteria monographs, readers are requested to communicate any errors
that may have occurred to the Director of the International Programme
on Chemical Safety, World Health Organization, Geneva, Switzerland, in
order that they may be included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Case postale
356, 1219 Châtelaine, Geneva, Switzerland (telephone no. + 41 22 -
9799111, fax no. + 41 22 - 7973460, E-mail irptc@unep.ch).
* * *
This publication was made possible by grant number 5 U01 ES02617-
15 from the National Institute of Environmental Health Sciences,
National Institutes of Health, USA, and by financial support from the
European Commission.
* * *
The Federal Ministry for the Environment, Nature Conservation and
Nuclear Safety, Germany, provided financial support for this
publication.
Environmental Health Criteria
PREAMBLE
Objectives
In 1973 the WHO Environmental Health Criteria Programme was
initiated with the following objectives:
(i) to assess information on the relationship between exposure to
environmental pollutants and human health, and to provide
guidelines for setting exposure limits;
(ii) to identify new or potential pollutants;
(iii) to identify gaps in knowledge concerning the health effects of
pollutants;
(iv) to promote the harmonization of toxicological and
epidemiological methods in order to have internationally
comparable results.
The first Environmental Health Criteria (EHC) monograph, on
mercury, was published in 1976 and since that time an ever-increasing
number of assessments of chemicals and of physical effects have been
produced. In addition, many EHC monographs have been devoted to
evaluating toxicological methodology, e.g., for genetic, neurotoxic,
teratogenic and nephrotoxic effects. Other publications have been
concerned with epidemiological guidelines, evaluation of short-term
tests for carcinogens, biomarkers, effects on the elderly and so
forth.
Since its inauguration the EHC Programme has widened its scope,
and the importance of environmental effects, in addition to health
effects, has been increasingly emphasized in the total evaluation of
chemicals.
The original impetus for the Programme came from World Health
Assembly resolutions and the recommendations of the 1972 UN Conference
on the Human Environment. Subsequently the work became an integral
part of the International Programme on Chemical Safety (IPCS), a
cooperative programme of UNEP, ILO and WHO. In this manner, with the
strong support of the new partners, the importance of occupational
health and environmental effects was fully recognized. The EHC
monographs have become widely established, used and recognized
throughout the world.
The recommendations of the 1992 UN Conference on Environment and
Development and the subsequent establishment of the Intergovernmental
Forum on Chemical Safety with the priorities for action in the six
programme areas of Chapter 19, Agenda 21, all lend further weight to
the need for EHC assessments of the risks of chemicals.
Scope
The criteria monographs are intended to provide critical reviews
on the effect on human health and the environment of chemicals and of
combinations of chemicals and physical and biological agents. As
such, they include and review studies that are of direct relevance for
the evaluation. However, they do not describe every study carried
out. Worldwide data are used and are quoted from original studies,
not from abstracts or reviews. Both published and unpublished reports
are considered and it is incumbent on the authors to assess all the
articles cited in the references. Preference is always given to
published data. Unpublished data are only used when relevant
published data are absent or when they are pivotal to the risk
assessment. A detailed policy statement is available that describes
the procedures used for unpublished proprietary data so that this
information can be used in the evaluation without compromising its
confidential nature (WHO (1990) Revised Guidelines for the Preparation
of Environmental Health Criteria Monographs. PCS/90.69, Geneva, World
Health Organization).
In the evaluation of human health risks, sound human data,
whenever available, are preferred to animal data. Animal and
in vitro studies provide support and are used mainly to supply
evidence missing from human studies. It is mandatory that research on
human subjects is conducted in full accord with ethical principles,
including the provisions of the Helsinki Declaration.
The EHC monographs are intended to assist national and
international authorities in making risk assessments and subsequent
risk management decisions. They represent a thorough evaluation of
risks and are not, in any sense, recommendations for regulation or
standard setting. These latter are the exclusive purview of national
and regional governments.
Content
The layout of EHC monographs for chemicals is outlined below.
* Summary - a review of the salient facts and the risk evaluation
of the chemical
* Identity - physical and chemical properties, analytical methods
* Sources of exposure
* Environmental transport, distribution and transformation
* Environmental levels and human exposure
* Kinetics and metabolism in laboratory animals and humans
* Effects on laboratory mammals and in vitro test systems
* Effects on humans
* Effects on other organisms in the laboratory and field
* Evaluation of human health risks and effects on the environment
* Conclusions and recommendations for protection of human health
and the environment
* Further research
* Previous evaluations by international bodies, e.g., IARC, JECFA,
JMPR
Selection of chemicals
Since the inception of the EHC Programme, the IPCS has organized
meetings of scientists to establish lists of priority chemicals for
subsequent evaluation. Such meetings have been held in: Ispra, Italy,
1980; Oxford, United Kingdom, 1984; Berlin, Germany, 1987; and North
Carolina, USA, 1995. The selection of chemicals has been based on the
following criteria: the existence of scientific evidence that the
substance presents a hazard to human health and/or the environment;
the possible use, persistence, accumulation or degradation of the
substance shows that there may be significant human or environmental
exposure; the size and nature of populations at risk (both human and
other species) and risks for environment; international concern, i.e.
the substance is of major interest to several countries; adequate data
on the hazards are available.
If an EHC monograph is proposed for a chemical not on the
priority list, the IPCS Secretariat consults with the Cooperating
Organizations and all the Participating Institutions before embarking
on the preparation of the monograph.
Procedures
The order of procedures that result in the publication of an EHC
monograph is shown in the flow chart. A designated staff member of
IPCS, responsible for the scientific quality of the document, serves
as Responsible Officer (RO). The IPCS Editor is responsible for
layout and language. The first draft, prepared by consultants or,
more usually, staff from an IPCS Participating Institution, is based
initially on data provided from the International Register of
Potentially Toxic Chemicals, and reference data bases such as Medline
and Toxline.
The draft document, when received by the RO, may require an
initial review by a small panel of experts to determine its scientific
quality and objectivity. Once the RO finds the document acceptable as
a first draft, it is distributed, in its unedited form, to well over
150 EHC contact points throughout the world who are asked to comment
on its completeness and accuracy and, where necessary, provide
additional material. The contact points, usually designated by
governments, may be Participating Institutions, IPCS Focal Points, or
individual scientists known for their particular expertise. Generally
some four months are allowed before the comments are considered by the
RO and author(s). A second draft incorporating comments received and
approved by the Director, IPCS, is then distributed to Task Group
members, who carry out the peer review, at least six weeks before
their meeting.
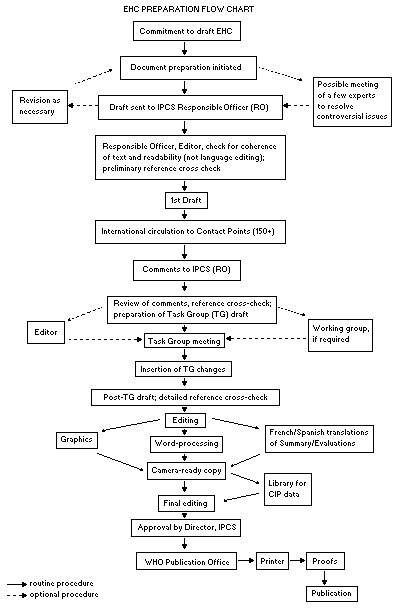 The Task Group members serve as individual scientists, not as
representatives of any organization, government or industry. Their
function is to evaluate the accuracy, significance and relevance of
the information in the document and to assess the health and
environmental risks from exposure to the chemical. A summary and
recommendations for further research and improved safety aspects are
also required. The composition of the Task Group is dictated by the
range of expertise required for the subject of the meeting and by the
need for a balanced geographical distribution.
The three cooperating organizations of the IPCS recognize the
important role played by nongovernmental organizations.
Representatives from relevant national and international associations
may be invited to join the Task Group as observers. While observers
may provide a valuable contribution to the process, they can only
speak at the invitation of the Chairperson. Observers do not
participate in the final evaluation of the chemical; this is the sole
responsibility of the Task Group members. When the Task Group
considers it to be appropriate, it may meet in camera.
All individuals who as authors, consultants or advisers
participate in the preparation of the EHC monograph must, in addition
to serving in their personal capacity as scientists, inform the RO if
at any time a conflict of interest, whether actual or potential, could
be perceived in their work. They are required to sign a conflict of
interest statement. Such a procedure ensures the transparency and
probity of the process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, it then goes for language editing, reference checking, and
preparation of camera-ready copy. After approval by the Director,
IPCS, the monograph is submitted to the WHO Office of Publications for
printing. At this time a copy of the final draft is sent to the
Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern for
health or environmental effects of the agent because of greater
exposure; an appreciable time period has elapsed since the last
evaluation.
All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR PHOSGENE
Members
Dr D. Anderson, British Industry Biological Research Institute (BIBRA)
Toxicology International, Carshalton, Surrey, United Kingdom
Dr R. Chhabra, Environmental Toxicology Program, Toxicology Branch,
National Institute of Environmental Health Sciences, Research
Triangle Park, North Carolina, USA
Dr H. Ellisa, Epidemiology Department, Rohm & Haas, Bristol,
Pennsylvania, USA
Dr B. Gilbert, FarManguinhos, FIOCRUZ, Institute of Technology and
Pharmacology, Ministry of Health, Manguinhos, Rio de Janeiro, Brazil
( Chairman)
Professor M. Jakubowski, Occupational and Environmental Hygiene
Division, Nofer Institute of Occupational Medicine, Lodz, Poland
Dr S.K. Kashyap, National Institute of Occupational Health, Meghani
Nagar, Ahmedabad, India ( Vice-chairman)
Dr R. Liteplo, Environmental Health Directorate, Health Protection
Branch, Environmental Health Centre, Tunney's Pasture, Ottawa,
Ontario, Canada
Dr E. E. McConnell, Laurdane Estates, Raleigh, North Carolina, USA
( Co-rapporteur)
Dr H. Naito, Ibaraki Prefecture University of Health Sciences,
Amimachi, Inashikigun, Ibaraki, Japan
__________
a Invited, but unable to attend.
Dr W. Popp, Universitatsklinikum Essen, Institute for Health and
Occupational Medicine, Essen, Germany
Dr R. Sram, Laboratory of Genetic Ecotoxicology, Institute of
Experimental Medicine, Videnska, Prague, Czech Republic
Dr Shou-Zheng Xue, Toxicology Programme, Shanghai Medical University,
Shanghai, People's Republic of China
Secretariat
Dr G. C. Becking, Team Leader, IPCS/IRRU, World Health Organization,
Research Triangle Park, North Carolina, USA
Ms R. Gomes, Health Canada, Environmental Health Directorate, Tunney's
Pasture, Ottawa, Ontario, Canada ( Co-rapporteur)
ENVIRONMENTAL HEALTH CRITERIA FOR PHOSGENE
A WHO Task Group on Environmental Health Criteria for Phosgene
and Selected Chloroalkyl Ethers met at the British Industrial
Biological Research Association (BIBRA) Toxicology International,
Carshalton, Surrey, United Kingdom, from 18 to 23 March 1996. Dr D.
Anderson opened the meeting and welcomed the participants on behalf of
the host institute. Dr G.C. Becking, IPCS, welcomed the participants
on behalf of Dr M. Mercier, Director of the IPCS, and the three
cooperating organizations (UNEP/ILO/WHO). The Task Group reviewed and
revised the draft criteria monograph and made an evaluation of the
risks for human health and the environment from exposure to phosgene.
Financial support for this Task Group was provided by the United
Kingdom Department of Health as part of its contribution to the IPCS.
Dr E.E. McConnell, Raleigh, North Carolina, USA, prepared the
first draft of this monograph. The draft reviewed by the Task Group,
which contained the comments received following circulation of the
draft monograph to the IPCS Contact Points for Environmental Health
Criteria monographs, was prepared by the Secretariat.
Dr G.C. Becking (IPCS, Central Unit, Inter-regional Research
Unit) and Dr P.G. Jenkins (IPCS, Central Unit, Geneva) were
responsible for the overall scientific content and technical editing,
respectively.
The efforts of all who helped in the preparation of the document
are gratefully acknowledged.
ABBREVIATIONS
CI confidence interval
L(CT)50 median lethal concentration-time product
LFP lavage fluid protein
MDI methylene-diphenyl diisocyanate
Nk natural killer
OES occupational exposure standard
PMN polymorphonuclear leukocyte
ppb parts per billion
ppm parts per million
ppt parts per trillion
PVC polyvinyl chloride
SMR standardized mortality ratio
TDI toluene diisocyanate
TLV threshold limit value
TWA time-weighted average
1. SUMMARY
1.1 Identity, physical and chemical properties, and
analytical methods
Phosgene is a highly reactive colourless gas at room temperature
and ambient pressure, and has a suffocating odour similar to mouldy
hay. The odour may be detected between 1.6 and 6 mg/m3.
Analytical methods are available for the detection of phosgene in
air and for use in industrial hygiene programmes that measure total
dose (e.g., paper tape monitors).
1.2 Uses and sources of human and environmental exposure
More than 99% of the phosgene produced is used on-site in closed
systems. It is produced by reacting equimolar amounts of anhydrous
chlorine and carbon monoxide in the presence of a carbon catalyst.
World production has been estimated to be greater than 3 million
tonnes. Environmental phosgene levels arise from industrial
emissions and thermal degradation of some chlorinated solvents and
chlorinated polymers. However, a significant source of environmental
phosgene is the photochemical oxidation of chloroethylenes such a tri-
and tetraethylene.
1.3 Environmental transport, distribution and transformation
Because of its high reactivity, inter-compartmental transport of
phosgene is expected to be limited. Removal of phosgene from ambient
air occurs by heterogeneous decomposition (surface catalysis) and slow
gas-phase hydrolysis. Long-range transport takes place and diffusion
from the troposphere to the stratosphere is believed to lead to more
rapid photolytic degradation of phosgene.
1.4 Environmental levels and human exposure
Human exposure in both the general population and occupational
setting is primarily by inhalation.
The average level of phosgene in ambient air may range from
approximately 80 to 130 ng/m3 although few data are available. In
view of the varied industrial hygiene practices worldwide it is
impossible to give an exposure figure for workers manufacturing or
using phosgene or for fire-fighters. At present the Threshold Limit
Values (time-weighted average) in 15 countries range from 0.4 and
0.5 mg/m3.
Levels of phosgene in water, soil and food have not been
reported.
1.5 Kinetics and metabolism
There are very few data on the absorption, metabolism,
distribution and fate of phosgene. The primary route of exposure is by
inhalation, the gas penetrates into the tissues of the respiratory
tract, and so only minimal amounts of phosgene are distributed in the
body. The very short half-life (0.026 seconds) in aqueous solutions
precludes a significant retention of phosgene in the body. No
information on the metabolism of phosgene has been reported. The
hydrolyic products of phosgene, i.e. hydrochloric acid and carbon
dioxide, are disposed of by the body through normal physiological
processes.
Phosgene exerts its toxicity through acylation of proteins, as
well as through the production of hydrochloric acid. The amino,
hydroxyl and sulfhydryl groups in the proteins appear to be the target
for acylation leading to marked inhibition of several enzymes related
to energy metabolism and a breakdown of the blood:air barrier.
1.6 Effects on experimental animals and in vitro test systems
1.6.1 Single and short-term exposures
In all species studied, the lung is the major target organ. The
L(CT)50 varies from 900 mg/m3-min (225 ppm-min) in the mouse to
1920 mg/m3-min (480 ppm-min) in the guinea-pig. An L(CT)50 of
1000 mg/m3-min (250 ppm-min) was reported in the monkey. In all
species the characteristic pathological feature is the delayed
clinical manifestation of pulmonary oedema, which is dose-dependent.
Pathological changes in the terminal bronchioles and alveoli at low
concentrations are typical of a pulmonary irritant, whereas at higher
exposures pulmonary oedema occurs, leading to interference with gas
exchange and death.
No long-term exposure studies of phosgene have been reported.
One study in rats showed that a single phosgene exposure of
2 mg/m3 for 4 h can result in decreased pulmonary immunocompetence as
measured by the natural killer activity of pulmonary cells. No
effects were seen at an exposure level of 0.4 mg/m3 for 4 h.
Two other studies of the effects of single exposures of phosgene
on pulmonary immunocompetence in rats and mice have been reported. In
rats infected with influenza virus after a 4-h exposure to 4 mg
phosgene/m3, there was a 10-fold increase in viral titre 1 day
post-infection, which remained significantly elevated for 4 days.
Furthermore, in rats exposed to phosgene levels between 0.2 and
4 mg/m3 for 4 h, a marked decrease in prostaglandin E2 and
leukotrienes was noted at exposure levels of 0.4 mg/m3 or more, with
a decrease in the number of alveolar macrophages and an increase in
the number of neutrophils observed at 0.4 mg/m3. In a host-
resistance assay, where mice were exposed to levels of phosgene
between 0.04 and 0.4 mg/m3 for 4 h, an increase in mortality from
Streptococcus zooepidemicus infection or an increased number of
B16/BL6 melanoma lung tumours was noted at levels of 0.1 mg/m3 or
more. Pulmonary bacterial clearance was reduced in rats exposed to
0.4 mg/m3 (0.1 ppm) phosgene for 6 h or to 0.4 mg/m3 (0.1 ppm) for
6 h/day, 5 days/week for 4 to 12 weeks. This effect was reversible
following termination of exposure.
1.6.2 Non-pulmonary effects
Phosgene exposure can result in eye and skin irritation. Studies
concerning the sensitization potential of phosgene have not been found
in the literature.
No data are available on the reproductive and developmental
effects of phosgene.
No adequate data are available for the assessment of the
mutagenicity or carcinogenicity of phosgene.
1.7 Effects on humans
The target organ in humans, as in experimental animals, is the
lung. After exposure to phosgene levels between 120 and
1200 mg/m3-min, three distinct clinicopathological phases have been
reported. The initial phase consists of pain in the eyes and throat
and tightness in the chest, often with shortness of breath, wheezing
and coughing; hypotension, bradycardia and rarely sinus arrhythmias
can occur. The second or latent phase, which is often asymptomatic,
can last as long as 24 h depending upon the level and duration of
exposure. In the third phase, pulmonary oedema may develop, leading
to death in some cases.
Populations exposed to phosgene after industrial accidents have
reported a wide variety of symptoms, including headache, nausea,
cough, dyspnoea, fatigue, pharyngeal pain, chest tightness and pain,
intense pain in the eye and severe lacrimation. In one study
pulmonary oedema occurred after a latent phase of 48 h.
The effects of long-term exposure to phosgene have been studied
in three groups of workers at two facilities, i.e., a phosgene
production plant and an uranium processing facility. In both
facilities, only limited air sampling or personal monitoring was
carried out, and worker exposures were only estimates.
An examination of the medical records of all 326 workers in the
phosgene production facility who were potentially exposed to phosgene
(up to 0.5 mg/m3, with some excursions above this value; average
0.01 mg/m3) did not yield any chronic lung problems or increased
mortality from respiratory disease compared to a group of 6228
controls. However, the lack of detail in the report on both exposure
and effects makes it difficult to draw firm conclusions from this
study.
Two groups of workers were studied at the uranium processing
plant: a cross-section of 699 workers from the over 18 000 employed
during the period, with potential exposure to phosgene levels below
0.4 mg/m3 (and 4 or 5 daily short-term excursions to > 4 mg/m3);
and a group of 106 workers known to have been involved in accidents
and exposed to levels above 200 mg/m3-min. In the group exposed
chronically to low levels of phosgene, an examination of death
certificates did not indicate an increased mortality from all causes
or from respiratory disease or lung cancer. In the group involved in
chemical accidents no increase in deaths from all causes was reported.
There were no lung cancer deaths but there was a slight increase in
the number of deaths from respiratory diseases. In view of the lack
of exposure data and the methodological characteristics of this study
the conclusions regarding the chronic effects of phosgene that can be
drawn are limited.
1.8 Effects on other organisms in the laboratory and field
No information concerning the effects of phosgene on organisms in
the environment has been reported.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL METHODS
2.1 Identity
Molecular formula: COC12
Chemical structure: CI
\
C = O
/
CI
Common Synonyms: carbonic acid dichloride, carbonyl chloride,
chloroformyl chloride, carbon oxychloride
IUPAC and CAS names: carbonic dichloride
CAS Registry number: CAS 75-44-5
RTECS Registry number: SY5600000
UN Transport number: 1076
2.2 Physical and chemical properties
Phosgene is a colourless nonflammable gas at room temperature and
ambient pressure. It has a suffocating odour and a smell reminiscent
of mouldy hay (Budavari, 1996). The recognition of the odour of
phosgene occurs at levels > 6 mg/m3 (1.5 ppm), although some
trained workers are capable of perceiving the odour at a level of
0.4 mg/m3 (0.1 ppm). The physical and chemical properties are
summarized in Table 1.
Table 1. Physical and chemical properties of phosgenea
Colour colourless
Relative molecular mass 98.92
Physical state gas
Melting point -127.8°C
Boiling point 7.56°C
Vapour pressure (20°C) 161.6 kPa
Relative vapour density (air = 1)3.42
Relative density, 20°C (water = 1)1.4
Solubility in water slight, reacts with water
Solubility in organic solvents reacts with ethanol, very
soluble in benzene,
toluene, acetic acid,
and most liquid
hydrocarbons
a From: Schneider & Diller (1989); Verschueren (1983);
Budavari (1996)
2.3 Analytical methods
A number of techniques may be used to determine phosgene
concentrations in air. These include passive dosimetry (Moore &
Matherne, 1981; Mathern et al., 1981), manual colorimetry (NIOSH,
1976), automated colorimetry (US EPA, 1986; Dangwal, 1994), gas
chromatography (Singh, 1976; Tuggle et al., 1979), infrared
spectroscopy (Esposito, 1977) and ultraviolet spectrophotometry
(Crummett & McLean, 1965). In addition, paper tape monitors capable
of detecting 5 µg/m3 have been described (Hardy, 1982). A summary of
these methods is presented in Table 2.
Table 2. Sampling and analysis of phosgene in aira
Sampling Analytical methodb Limit of detection Sample Comments Reference
methodb (range) size
Passive Direct reading (8-400 mg/m3-min) - Concentration- Moore &
dosimetry colorimetric reaction time relationship Matherne
with NBP in breathing zone (1981)
Air through Measure colour at (0.2 - 100 mg/m3) 1 litre/min Too slow NIOSH (1976)
impinger 475 nm for 25 min response for
containing DEP continuous
solution of NBP monitoring
and BA
Air bubbled into Automated colorimetry 0.004 mg/m3 1 litre/min Response time of US EPA (1986)
flowing stream of at reagent flow rate of (0-4 mg/m3) for 20 min 20 min too long
NBP-BA-DEP 0.2 ml/min for continuous
monitoring
Air through Derivative determined 0.04 mg/m3 1 litre air Extremely Wu & Gaind
impinger by reverse-phase HPLC sample sensitive but (1993)
containing needs highly
tryptamine trained staff and
equipment
Table 2 (contd).
Sampling Analytical methodb Limit of detection Sample Comments Reference
methodb (range) size
Direct sample Gas-chromatography 0.5 ml Extremely Singh (1976)
injection, - electron capture < 0.08 mg/m3 injection sensitive, needs Tuggle et al.
continuous - aluminium columns < 0.08 - 20 mg/m3 highly trained (1979)
sampling didecyl phthalate on staff and
chromosorb p equipment
Air is drawn Infrared spectroscopy, 0.1 mg/m3 2 to 5 Can be used as a Esposito et al.
directly into comparison of (0.1 to 1200 litre/min continuous (1977)
spectrophotometer absorbance at 11.8 µm mg/m3) monitor for
and reference ambient levels of
wavelength of 11.2 µmn phosgene
using 20 m variable
path length cell
a Proper medical treatment for those exposed to phosgene will depend on the concentration and length of exposure. Therefore,
any procedure used for monitoring ambient and workplace levels must give information on both parameters, preferably on a
continuous basis, since a latent period of 2-24h may occur between exposure and any warning symptoms.
b NBP = 4,4 -nitrobenzyl pyridine
DEP = diethyt phthalate
BA = N-benzylalanine
HPLC = high performance
2.4 Conversion factors
1 ppm = 4.05 mg/m3
1 mg/m3 = 0.25 ppm
at 25°C and 101.3 kPa
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Phosgene is not known to occur naturally.
3.2 Anthropogenic sources
3.2.1 Production levels and processes
Phosgene is produced by reacting equimolar amounts of anhydrous
chlorine and carbon monoxide in the presence of a carbon catalyst
(Schneider & Diller, 1989). The great majority is used directly in a
closed system.
It is difficult to give accurate production figures because more
than 99% of phosgene production is for on-site use (Schneider &
Diller, 1989). However, phosgene is manufactured in most
industrialized countries. Approximately 37% of the world's production
is in the USA (about 1 million tons); in 1989 European phosgene
production was approximately 1.2 million tonnes (Schneider & Diller,
1989).
3.2.2 Environmental processes
Phosgene in ambient air may arise from three sources:
a) direct emissions during its manufacture, handling, use and
disposal;
b) thermal decomposition, in the presence of air, of chlorinated
hydrocarbons, e.g., solvents such as chloroform, methylene chloride
(Snyder et al., 1992) and 1,2-dichloropropane (IPCS, 1993) and
polymers such as polyvinyl chloride (PVC);
c) photooxidation of chlorinated hydrocarbons, particularly
chloroform and the chloroethylenes (Rinzema, 1971; Singh, 1976; Gay et
al., 1976; Birgesson, 1987).
The thermal degradation of chlorinated hydrocarbons can occur as
a result of combustion of these materials during waste disposal, in
fires, and during welding in situations where PVC plastics are
degraded (Rinzema, 1971; Birgesson, 1982). Firefighters and welders
are at particular risk from these sources of phosgene.
A significant contribution to ambient air levels of phosgene is
the photooxidation of chloroethylenes, particularly tri- and tetra-
chloroethylene (Singh, 1976; Gay et al. 1976). It has been estimated
that such reactions may result in the worldwide formation of 350 000
tonnes of phosgene per year (Singh, 1976).
3.2.3 Uses
Initially important as an agent of chemical warfare, phosgene is
now widely used as a chemical intermediate, most often at the point of
production. The major use is in the production of aromatic
diisocyanates such as methylene diphenyl diisocyanate (MDI) and
toluene diisocyanate (TDI), which are used to produce polyurethane
foams and other polymers. Worldwide about 80% of phosgene is used for
TDI and MDI production. Other major uses of phosgene include the
production of polycarbonate, aliphatic diisocyanates, monoisocyanates
and chloroformic esters and urethanes (Schneider & Diller, 1989;
Borak, 1991). Phosgene is also used in the manufacture of some
agrochemicals, in the pharmaceutical industry, and metallurgy (US NLM,
1995).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
Phosgene can enter the atmosphere in the form of industrial
emissions or from the degradation of chlorohydrocarbons (section
3.2.2). Detectable levels have been found in ambient air (section
5.1.1). However phosgene is unlikely to be detectable in soil and
vegetation owing to heterogeneous decomposition (section 4.2).
In water, phosgene is rapidly degraded to hydrochloric acid and
carbon dioxide (Butler & Snelson., 1979), the half-life in aqueous
solution being 0.026 seconds (Manogue and Pigford, 1969).
In the atmosphere, even at high humidity levels, phosgene is only
slowly decomposed (Noweir et al., 1973; US EPA, 1986). The half-life
by homogeneous gas-phase hydrolysis of 4 g /m3 phosgene (1 ppb) in
nitrogen (at sea-level pressure, 25 C and with water vapour at a
pressure of 10 Torr) has been calculated to be 113 years (range 20 to
630 years) (Butler & Snelson, 1979). Reaction rates with activated
oxygen and hydroxyl radicals are also slow (Singh, 1976). Phosgene
is, therefore, likely to be persistent in the atmosphere and subject
to long-range transport. Diffusion to the stratosphere leads to more
rapid degradation by photolysis (Singh, et al., 1977).
4.2 Abiotic degradation
Reaction with molecules having an active hydrogen atom (e.g.,
water, primary and secondary alcohols, thiols and amines) does occur,
forming hydrochloric acid, carbon dioxide, and carbonic acid
derivatives (Butler & Snelson, 1979; Schneider & Diller, 1989), but
phosgene reacts very slowly in the gas phase with photochemically
produced hydroxyl radicals (Singh, 1976).Removal of phosgene from
ambient air occurs by two major pathways, i.e. heterogeneous
decomposition (Noweir et al., 1973) and liquid-phase hydrolysis (Singh
et al., 1977). At normal ambient temperatures the gas-phase
hydrolysis is the major pathway for phosgene degradation (Singh et
al., 1977). However, even contact with soil particles and vegetation
at ambient temperatures enhances the rate of phosgene degradation
(Noweir et al., 1973).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
It has been suggested that the primary source of atmospheric
levels of phosgene is from the thermal degradation and photo-
degradation of chlorinated solvents such as tri- and
tetrachloroethylene and PVC (Singh, 1976). Direct emissions from the
production and use of the chemical play a minor role and would most
likely affect the air levels only near the factory. Phosgene levels
in ambient air at four locations in California, USA, were reported by
Singh et al. (1977). Multiple samples (10 to 257) were taken on a
24-h basis from a single location in each area. In one rural area an
average level of 87 ng/m3 (21.7 ppt) was reported. The average
levels found in three urban areas were 117 ng/m3 (29.3 ppt),
121 ng/m3 (30.3 ppt) and 129 ng/m3 (31.8 ppt), with a peak level
in one sample of 244 ng/m3 (61.0 ppt). In three other cities in the
USA the average level of phosgene in ambient air was reported to be less
than 80 ng/m3 (20 ppt) (Singh et al., 1981).
5.1.2 Water
There are no data on levels of phosgene in water, since
hydrolysis precludes significant accumulation in this medium.
5.1.3 Soil
Data on levels of phosgene in soil are not available, since rapid
breakdown on contact with solid surfaces and moisture prevents a
significant accumulation in this medium (Noweir et al., 1973).
5.1.4 Food and feed
Although no data are available, the lack of stability in the
presence of liquid-phase water, solid surfaces, and alcohols and or
amines in foods makes contamination of food by phosgene unlikely (see
sections 4.1 and 4.2).
5.2 General population exposure
The general population is exposed to only very low levels (in the
ng/m3 range) of phosgene and this is almost entirely via contaminated
urban air. The origin of this phosgene is from decomposition of other
chlorinated compounds or, in isolated circumstances, from the
emissions of an industrial enterprise making or using phosgene without
carrying out appropriate industrial hygiene practices. Based on the
range of average concentrations (< 80-129 ng/m3) given in section
5.1.1, the estimated total daily intake of phosgene may range from
< 1.6 to 2.6 g, assuming a daily respiratory intake of 20 m3 air.
Much higher levels of phosgene exposure are possible during home use
of chemicals such as methylene chloride under conditions where the
temperature is sufficiently high to lead to degradation of this
chemical (Snyder et al., 1992).
5.3 Occupational exposure
5.3.1 Manufacture and use
Occupational exposure limits (8- or 10-h TLV) in some 15
countries range between 0.4 and 0.5 mg/m3 (ILO, 1991). Based upon
animal data, the United Kingdom has proposed an OES (8-h TWA) of
0.08 mg/m3 (0.02 ppm) and an STEL of 0.24 mg/m3 (0.06 ppm) (HSE,
1995). It is difficult to report actual values in individual
factories worldwide since levels will vary greatly depending upon the
level of industrial hygiene practiced in any particular factory.
However, even early monitoring reports indicated that exposures were
generally below the recommended TLV. In a few cases exposure levels
above 0.4 mg/m3 have been reported (Levina et al., (1966). In a
factory manufacturing phosgene, personal samplers detected levels up
to 0.08 mg/m3 (average 0.012 mg/m3), whereas fixed position samplers
(total of 56) showed levels between non-detectable and 0.52 mg/m3 in
51 samples, with excursions in a few samples to about 71 mg/m3
(NIOSH, 1976). More recent monitoring data are lacking.
5.3.2 Non-manufacturing occupations
Firefighters and workers engaged in welding and building trades
are at risk from the phosgene formed by the thermal degradation of
chlorinated hydrocarbons and PVC. The pyrolysis of Freon present in
commercial refrigeration units (Birgesson, 1982), tri- and
tetrachloroethylene (Rinzema, 1971; Andersson et al., 1975), PVC
(Brown & Birky, 1980) and methylene chloride (Snyder et al., 1992)
have all been shown to result in toxic levels of phosgene. However,
actual levels in the area of work or the breathing zone were not
quantified in these studies.
6. KINETICS AND METABOLISM
Because of the physico-chemical properties of phosgene, the
kinetics and metabolism of phosgene in animals should be similar if
not identical to those found in humans. However, because of the
highly toxic nature of phosgene, human experimental data appropriate
for use in this monograph does not exist.
6.1 Disposition of phosgene
There are very few data on absorption, metabolism, distribution
and fate of phosgene. The primary route of exposure is by inhalation.
The gas penetrates into the tissues of the respiratory tract, and so
only minimal amounts of phosgene are distributed in the body. The
very short half-life (0.026 seconds) in aqueous solutions precludes a
significant retention of phosgene in the body. No information on the
metabolism of phosgene has been reported. The hydrolytic products of
phosgene, i.e. hydrochloric acid and carbon dioxide, are disposed of
by the body through normal physiological processes (Manogue & Pigford,
1960; Thienes & Haley, 1972).
6.2 Reaction with body components
Apart from the formation of hydrochloric acid on contact, Cessi
et al. (1966) showed marked acylation of in vitro poly-L-lysine and
human albumin by phosgene. The reaction of phosgene with cysteine
yields 2-oxothiazolidine-4-carboxylic acid (Mansuy et al., 1977). The
same product is formed when chloroform (Pohl et al., 1977) or carbon
tetrachloride (Shah et al., 1979) is incubated with hepatic
microsomes.
7. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
7.1 Single and short-term inhalation exposures
For some end-points (e.g., death, lung damage), the effects of
phosgene exposure are dependent upon both the concentration and
duration of exposure; considered as a product of CT=K as stated in
Haber's law (the product of the concentration and time of exposure
required to produce a specific physiological effect is a constant).
Early workers validated this relationship using death as the
physiological end-point following acute and short-term exposures. The
validity of this concept for phosgene was tested in cats (Flury, 1921;
Flury & Zernick, 1931). Twenty cats were exposed to phosgene at
levels between 5 and 500 mg/m3 for periods of time between 0.5 and
120 min (CT values varied from 37.5 to 562 ppm-min). A plot of K
(death or survival) against T (abscissa) and C (ordinate) resulted in
an hyperbolic plot with marked deviations at both high and low
concentrations (Long & Hatch (1961). Similar results were obtained in
rats when the physiological effect was impaired gas exchange (Rinehart
& Hatch, 1964).
Based on reviews of available information (Coman et al., 1947;
Atherley, 1985) it was concluded that, for phosgene, effects
associated with the product of CT were reasonable constant only
within the middle range of concentrations (i.e., between 4 and
800 mg/m3), and for exposure times that negated the effect of an
animal holding its breath. Under these conditions and for these
effects, it was considered appropriate to express the actual dose of
phosgene, assuming equivalent respiratory volume, as CT. The CT
relationship probably does not apply to potential effects resulting
from long-term exposure to low levels of phosgene.
Single and short-term exposures are described together because of
the similar biological responses observed. Most of the numerous
animals studied are summarized elsewhere (US EPA, 1986) and
representative examples are shown (Table 3, Table 4).
In inhalation studies the lung is the primary target organ in all
species, and the characteristic pathological feature is the delayed
clinical manifestation of pulmonary oedema, which is dose dependent.
Other mucous membranes such as the eye can also be affected.
Underhill (1919, 1920) exposed dogs for 30 min to phosgene
concentrations between 176 and 480 mg/m3. At the lower
concentrations he reported that phosgene exposures resulted in
pathological lesions in the terminal bronchioles and alveoli, typical
for a pulmonary irritant. However, at the higher exposure levels
phosgene resulted in oedema leading to interference with gas exchange,
cyanosis and eventually death.
Table 3. Effects of phosgene after single exposure by inhalation
Speciesa Exposure Effects Reference
C x T C T
(mg/m3-min) (mg/m3) (min)
Rat 100 0.4 250 Widening of pulmonary intestices Diller et al. (1985)
Rat 144 144 0.4-380 Reduced pulmonary bacterial Yang et al. (1995)
clearance
Cat 150 10 15 Slight illness Flury (1921)
Mice, rats, 192 0.8 240 Increase in levels of lavage fluid Hatch et al.
hamsters protein (1986)
Rat 200 0.24 500 Increase in protein levels in Diller et al. (1985)
20 10 pulmonary lavage fluid
Rat 200 20 10 Initiation of pulmonary oedema Diller et al. (1985)
Rats (male) 240 1 240 Increase in levels of lavage fluid Currie et al.
protein. Increase in the percentage (1987a)
of polymorpho-nuclear leukocytes
(PMN)
Table 3 (contd).
Speciesa Exposure Effects Reference
C x T C T
(mg/m3-min) (mg/m3) (min)
Cat 440 44 10 L(CT)minimum Flury & Zernick
(1931)
Cat 450 10 45 L(CT)mimimum Flury & Zernick
(1931)
Guinea-pigs, 480 2.0 240 Increase in levels of lavage fluid Hatch et al.
rabbits protein (1986)
Rat 480 2 240 Increase of wet and dry lung weight Currie et al.
(1987a)
Rat 480 2 240 Decrease in pulmonary natural killer Burleson & Keys
activity (NK) (1989)
Cat 660 44 15 L(CT)100 Flury (1921)
Cat 720 12 60 L(CT)100 Flury (1921)
Mouse 900 60 15 L(CT)50 Cameron & Foss
(1941)
Table 3 (contd).
Speciesa Exposure Effects Reference
C x T C T
(mg/m3-min) (mg/m3) (min)
Monkey 1000 1000 1 L(CT)50 Diller & Zante
(1982)
Rat 1200 16 75 L(CT)50 Rinehart & Hatch
(1964)
Monkey 1320 440 3 L(CT)100 Winternitz et al.
(1920)
Dog 1800 180 10 L(CT)50 Diller & Zante
(1982)
Guinea-Pig 1920 128 15 L(CT)50 Underhill (1920)
Rat 4000 400 10 Ultrastructural changes in the Pawlowski &
bronchoalveolar region of lungs; Frosolono (1977)
cellular disruption and necrosis
Guinea-pig 6160 308 20 L(CT)100 Ong (1972)
Dog 8808 2936 3 L(CT)99 Coman et al.
(1947)
Table 3 (contd).
Speciesa Exposure Effects Reference
C x T C T
(mg/m3-min) (mg/m3) (min)
Sheep 13 300 1330 10 L(CT)50 Keeler et al.
(1990)
Rabbit 21 140 604 35 L(CT)99 Coman et al.
(1947)
a In most cases the sex of the animals was not specified
Necropsy of dogs that died shortly after single exposure to
phosgene showed frothy material around the mouth, engorgement of the
visceral vasculature (shock-like syndrome), and heavy wet congested
lungs (Winternitz et al., 1920). Microscopically, the lungs were
characterized by congestion and severe oedema. Proteinaceous fluid,
strands of fibrin, and leukocytes filled the alveoli. In dogs that
died 4 or more days after dosing, pulmonary infection (inflammation)
was the primary cause of death. Oedema, congestion and emphysema of
lesser severity were still present but there was also evidence of an
attempt to repair tissue. In dogs that died or were killed later (11
to 129 days), necropsy revealed varying degrees of lung collapse and
emphysema suggesting obliterative bronchiolitis. The epithelium of
the larger airways (trachea and bronchi) did not show evidence of
damage. The authors stated that the pathology of phosgene exposure
was similar in the goat, dog, monkey, rabbit, guinea-pig, rat and
mouse (Winternitz et al., 1920).
Concurrent studies in a different laboratory confirmed the above
results (Meek & Eyster, 1920). These authors documented a well-marked
succession of events after exposure of dogs for 30 min to phosgene
levels of 320-400 mg/m3. In the initial stage of exposure there is
direct damage to the epithelial cells lining the pulmonary airways,
with the distal ones showing the most damage. The cells are killed
(necrosis) and sloughed. This is immediately followed by effusion of
fluid into the affected airways (oedema). There is some damage to
erythrocytes in pulmonary capillaries, which aggregate, thus causing
occlusion. Gaseous exchange is interfered with, and death is the
result of hypoxia/anoxia. Other authors (Cameron & Courtice, 1946)
have also shown that pulmonary oedema is the primary cause of death in
several species after acute phosgene poisoning (440 mg/m3). The area
of lung affected appears to depend on the level of exposure. Gross et
al. (1965) postulated that at lower levels the alveolar bronchioles
and alveoli are the primary target tissues. At higher levels more
proximal respiratory tissues are at risk of developing lesions. The
relationship between the primary target site in the lung and dose of
phosgene was confirmed in rats (Diller et al., 1985). Changes within
the blood-air barrier (pulmonary oedema) were noted at phosgene levels
of 20 mg/m3 (5 ppm) or more and at durations of exposure of 10 min or
longer (50 ppm-min). The lowest dose of phosgene producing an
increase in protein levels in pulmonary lavage fluid was also
200 mg/m3 min (50 ppm-min), and that for the production of widening
within the pulmonary interstices was 100 mg/m3 min (25 ppm-min).
However, there was no apparent threshold of phosgene concentration for
these two parameters. Concentrations of phosgene studied were 0.4 to
20 mg/m3 (0.1 to 5 ppm). Changes noted at low concentrations (0.4 to
10 mg/m3; 0.1 to 2.5 ppm) were primarily located at the transition
from terminal bronchioles to the alveolar ducts, while at higher
concentrations (20 mg/m3, 5 ppm) damage to the alveolar pneumocytes
(type 1) was reported (Diller et al., 1985).
The earliest ultrastructural change observed in the
bronchoalveolar region of lungs of rats exposed to 400 mg/m3
(100 ppm) for 10 min was characterized by vesiculation of bronchiolar
epithelium immediately after exposure (Pawlowski & Frosolono, 1977).
This was followed, 30 min after the exposure, by extracellular
accumulation of serous fluid in interstitial spaces and alveoli. The
final events were cellular disruption and necrosis.
The extent of the long-term effects after acute exposure appears
to depend on the severity of the initial pathology (Coman et al.,
1947; Diller, 1985b).
The relative sensitivity of female mice and male hamsters,
rabbits, guinea-pigs and rats to 4-h phosgene exposures at
concentrations of 0.4, 0.8, 2 and 4 mg/m3 (0.1, 0.2, 0.5 and 1 ppm)
was studied by Hatch et al. (1986). As an indicator of phosgene-
induced pulmonary oedema, levels of lavage fluid protein (LFP) were
measured 18-20 h after exposure. Groups of seven or eight animals
were examined at each exposure level. Phosgene-induced changes in LFP
levels in mice, hamsters and rats occurred at phosgene levels of
0.8 mg/m3 (about 190 mg/m3 min) and above, whereas the minimal
effective dose in guinea-pigs and rabbits was 2 mg/m3 (about
480 mg/m3-min).
Rats were exposed 4 h/day, 5 days/week for 17 exposures at a
level of 0.5 or 1 mg/m3 (0.125 or 0.25 ppm) and killed on days 3, 7,
10, 13 or 17 or on days 2 or 20 after exposure. After day 7 the lung
wet weights were increased by 20-25% (p < 0.05 in the high-level
group). In the low-level group the lung wet weights increased with
exposure time but were significantly increased only at day 17. These
changes were paralleled by an increased activity of the pulmonary
glucose-6-phosphate dehydrogenase. The non-protein sulfhydryl content
in the lungs was elevated throughout exposure in both groups. All
deviations were reversible after exposure ceased. At day 17 in the
high-level group a moderate multifocal accumulation of mononuclear
cells in the walls of the terminal bronchioli and their adjacent
alveoli was observed. A minimal amount of type II alveolar cell
hyperplasia was also found in this region. Macrophages with
vacuolated cytoplasm were seen in the lumens of some alveolar ducts
and alveoli. The lesions in the lungs of the low-dose animals were
described as being minimal (Franch & Hatch, 1986). A no-observed-
effect-level could not be demonstrated in this study.
The effects of low-level acute exposure to phosgene in male rats
was also studied by Currie et al. (1987a), who exposed groups of adult
animals (250-300 g) for 4 h to 0.5 to 4 mg/m3 (0.125 to 1 ppm).
Dose-related changes in body weight, wet and dry lung weights, LFP,
total cell counts, and cell differentials were measured at the
conclusion of the exposure and 3 days after exposure, at least 10
animals being sacrificed each time. A dose-response relationship for
the measured parameters was noted. Both wet and dry lung weights
increased after exposure to 120 and 240 ppm-min, and an increase in
LFP was noted at > 60 ppm-min. The most sensitive cellular
indicator of phosgene pulmonary damage was the increase in the
percentage of polymorphonuclear leukocytes (PMN); there was a
significant increase at 60 ppm-min. Both PMN and LFP can be used as
sensitive indicators of pulmonary damage by phosgene after acute
exposure. All parameters returned to control levels 3 days after
exposure, indicating that the pulmonary damage within the dose-range
studied was reversible.
In Table 3, effects of inhalation exposure to phosgene have been
summarized according to the degree of exposure, expressed as
mg/m3-min. Pulmonary bacterial clearance appears to be the most
sensitive end-point for acute phosgene toxicity in rats (about
100 mg/m3-min). The lowest dose of phosgene that produced an
increase in protein levels in pulmonary lavage fluid and changes in
the blood-air barrier (pulmonary oedema) was 100 to 200 mg/m3-min.
This effect can be considered as the early critical effect of acute
exposure to phosgene. Data presented in Table 4 suggest that
evaluation of exposure according to Haber's law can be applied as the
basis for constructing dose-effect relationships only in the case of
acute relatively high exposures.
Studies of pulmonary physiology in animals mirror the
pathological observations (Gibbon et al., 1948; Boyd & Perry, 1960;
Long & Hatch, 1961; Rhinehart & Hatch, 1964). Progressive loss of
capacity for gas exchange is the initial and critical event. The
respiration rate is increased but there is increased resistance and
poor ventilation.
7.2 Skin and eye irritation; sensitization
Very little information on this subject is available. Skin
irritation is possible if concentrations are high enough, but the
hazard is minimal compared to the severe lung damage that can be
produced by much longer levels (Diller, 1985a; Borak, 1991). No
studies on sensitization have been reported. Eye irritation and
corneal oedema have been reported in dogs exposed to lethal
concentrations of phosgene (Winternitz et al., 1920).
Table 4. Toxicity of phosgene after repeated exposure by inhalation
Speciesa Exposure Effects Reference
Rat 0.4 mg/m3; 6 h/day, 5 days/week, Pulmonary bacterial clearance inhibited Selegrade et al.
4-12 weeks (1989)
Rat 0.5-1 mg/m3 4 h/day 5 days/week, 17 Lung wet weight increase (20-25%); GPDb Franch & Hatch (1986)
exposures activity increase; alveolar cell
hyperplasic; macrophages with vacuolated
cytoplasm
Guinea-pig 0.8 mg/m3 for 300 min daily, 5 days Pulmonary oedema in 70% of animals Cameron et al. (1942)
Cat 0.8 mg/m3 for 300 min daily, 5 days Pulmonary oedema in 70% of animals Cameron et al. (1942)
Mouse 4 mg/m3 for 300 min daily, 5 days L(CT)90 Cameron & Foss
(1941)
Rat 4 mg/m3 for 300 min daily, 5 days Pulmonary oedema in 80% of animals Cameron & Foss
(1941)
Rabbit 4 mg/m3 for 300 min daily, 5 days L(CT)20 Cameron & Foss
(1941)
Dog 96-160 mg/m3 for 30 min Up to 20-fold increase in airway Rossing (1964)
1-3 times per week, resistance
12 weeks
a Sex not specified b GPD= glucose-6-phosphate dehydrogene
7.3 Long-term exposure
Long-term exposure studies of phosgene have not been conducted.
However, there have been a limited number of studies on the effects of
phosgene following repeated exposures over periods of time (dosing 1
to 3 times per week for up to 12 weeks) (Clay & Rossing, 1964;
Rossing, 1964). Adult mongrel dogs (sex not identified) were exposed
to phosgene at concentrations between 96 and 160 mg/m3 for 30 min, 1
to 3 times per week. Rossing (1964) exposed 14 animals 3 times weekly
until increased airway resistance was noted, and then the frequency of
exposure was decreased to 1 or 2 times weekly for 12 weeks. Seven
animals died during the first 3 weeks of exposure and only three
animals survived the full 12 weeks; two of which were maintained for
12 weeks with further exposure. The lungs of all animals were
examined within 48 h after-exposure. After the initial inflammatory
reaction, the ensuing lesion consisted of chronic bronchiolitis and
emphysema that persisted for the duration of the exposure period.
After cessation of exposure, elastance dropped rapidly to normal, but
airway resistance was still elevated 11 weeks after exposure.In a
similar experiment, Clay & Rossing (1964) studied the development of
pulmonary emphysema in adult mongrel dogs after exposure to phosgene
at concentrations between 96 and 160 mg/m3 for 30 min at a rate of 1
to 3 exposures per week. Group size varied between four and seven
animals. The number of exposures varied between 1 and 25, and the
dogs were killed either immediately or up to 2 weeks after exposure.
However, in view of the low number of animals used, the experimental
design and the lack of reported dose-response information, these data
are difficult to use in assessing quantitatively the long-term risk to
humans from phosgene exposure.
7.4 Reproductive and developmental toxicity
No data were found on reproductive or developmental effects of
phosgene in experimental animals.
7.5 Mutagenicity and related end-points
No studies on the mutagenicity of phosgene have been reported.
7.6 Carcinogenicity
No adequate studies are available for the assessment of the
carcinogenicity of phosgene. In a review of the potential
carcinogenicity of 266 substances found in various workplaces, data
from one study involved 20 guinea-pigs and 20 rats that were exposed
by inhalation to phosgene for 24 and 18 months, respectively. No
pulmonary neoplasms were observed (Schepers, 1971), but information on
dosing regimen, sex or strain of animals was lacking.
7.7 Immunotoxicity
In one study, the immunotoxic effects in Fischer-344 male rats of
a single 4-h exposure to 0, 0.4, 2.0 or 4 mg phosgene/m3 were
reported (Burleson & Keys, 1989). As a measure of pulmonary
immunocompetence the authors measured the natural killer (NK)
activity of pulmonary cells on the day after phosgene exposures. At
exposures of 2 and 4 mg/m3 there was a significant decrease in
pulmonary NK activity, which was considered by the authors to be an
indication of decreased immunocompetence. At 4 mg/m3 this decrease
was still significant 4 days after exposure. A significant decrease
was also noted at 2 mg/m3 one day after exposure, but no effect was
seen at 0.4 mg/m3. Phosgene did not affect NK activity in blood
lymphocytes, but NK activity in the splenic cells was decreased 1 day
after exposure to 4 mg/m3. Effects on NK activity in lymphocytes and
splenic cells at other phosgene doses and days post-treatment were not
reported. Decreased immunocompetence in rats resulting from a 4-h
exposure to phosgene was reported by Ehrlich & Burleson (1991). After
male Fischer-344 rats (8-10 weeks old) were exposed to phosgene at
4 mg/m3 for 4 h, the animals were infected with a rat-adapted
influenza virus and the virus titre measured at 2 h and 1, 2, 3, 4, 5
and 7 days post-infection. The virus titre was measured in three
replicate experiments using three rats per group. Following an
initial decrease in the viral titre at 2 h after infection, the virus
titre increased by a factor of 10 on the day after infection and
remained significantly higher than in controls up to 4 days after
infection. The virus was cleared below detectable levels after 5 days
post-infection.
Bacterial clearance was assessed in male Fisher-344 rats exposed
to 0, 0.4 or 0.8 mg/m3 (0.1 or 0.2 ppm) phosgene for 6 h (Yang et
al., 1995). Immediately after exposure to phosgene the animals were
infected with Streptococcus zooepidemicus, and the number of
bacteria in the pulmonary lavage fluid was assessed up to 72 h later.
Pulmonary bacterial clearance was significantly reduced (p <0.05)
following exposure to 0.4 mg/m3 (0.1 ppm) phosgene (LOEL = 0.1 ppm).
Based on an analysis of other immunological parameters (e.g.,
pulmonary NK activity and pulmonary macrophage function), the authors
indicated that bacterial clearance appeared to be the most sensitive
end-point for acute phosgene toxicity in rats.
CD-1 mice were exposed to phosgene concentrations of 0.04 to
0.4 mg/m3 (0.01 to 0.1 ppm) for 4 h and infected with S. zoo-
epidimicus or inoculated with B16/BL6-melanoma tumour cells. At and
above 0.1 mg/m3 (0.025 ppm), mortality due to the infection with
S. zooepidimicus and the number of B16/BL6 melanoma tumours in the
lungs were both elevated. An 8-h exposure to 0.04 mg/m3 increased
the mortality due to S. zooepidimicus but not the number of B16/BL6
melanoma tumours (Selgrade et al., 1989).
F-344 rats were exposed to phosgene concentrations of 0.2 to
4 mg/m3 (0.05 to 1 ppm) and their lungs lavaged 0, 4, 20 or 44 h
later. At and above 0.4 mg/m3 the concentrations of prostaglandin E2
as well as of leukotrienes B4, C4, D4 and E4 were decreased by 29 to
69%. The eicosanoid concentrations after exposure to 0.4 and 1 mg/m3
returned to normal 44 h later. After exposure to 0.4 or 4 mg/m3, but
not to 0.2 mg/m3, the number of alveolar macrophages was decreased,
whereas the number of neutrophils was increased at 44 h after exposure
to 0.4 mg/m3, but not to 4 mg/m3 (Madden et al., 1991).
Pulmonary effects have also been observed in male Fisher-344 rats
exposed for a longer term to phosgene (Selgrade et al., 1995). Groups
of animals were exposed to 0, 0.4 or 0.8 mg/m3 (0, 0.1 or 0.2 ppm)
for 6 h/day, 5 days/week for 4 or 12 weeks, or to 2 mg/m3 (0.5 ppm)
for 6 h/day, 2 days/week for 4 or 12 weeks. When assessed immediately
after 4 or 12 weeks of exposure, pulmonary bacterial clearance was
inhibited following exposure to 0.4 mg/m3 (0.1 ppm) phosgene (LOEL =
0.1 ppm). This effect appeared reversible, since pulmonary bacterial
clearance was unchanged when assessed 4 weeks after a 12-week exposure
period. These subchronic effects on pulmonary bacterial clearance were
similar to those observed previously, following acute exposure to
phosgene (Yang et al., 1995). In animals administered the same dose of
phosgene, pulmonary bacterial clearance was more severely affected in
animals exposed to the higher concentration of this substance (i.e.,
2 mg/m3 (0.5 ppm), 6 h/day, 2 days/week, compared to 0.8 mg/m3
(0.2 ppm) 6 h/day, 5 days/week). A significant reduction in pulmonary
NK activity was observed following exposure to 2 mg/m3 (0.5 ppm).
7.8 Mechanism of toxicity
Although the exact mechanism of phosgene toxicity remains
unknown, it seems likely that the original hypothesis of Winternitz et
al. (1920), suggesting that hydrochloric acid was the causal agent for
the pulmonary effects noted, is incorrect. Current data indicate that
the effects from phosgene exposure result from the acylation of tissue
components, although the production of HCl may play a minor role,
particularly at high levels of exposure (Diller, 1985a). Nash &
Pattle (1971) studied the chemical reactivity of phosgene when bubbled
through aqueous solutions at various pH values, some containing
amines, phenoxide ions or sulfite. From these data it was concluded
that molecular phosgene could penetrate all layers of the blood-air
barrier, causing the observed pathology by reacting with chemical
groups in the cells. It was shown mathematically that insufficient
HCl to produce the observed effects could be generated under
physiological conditions and phosgene exposures as high as 100 mg/m3
(25 ppm).
Early evidence that acylation of amino, hydroxyl and sulfhydryl
groups was the major mechanism was reported by Potts et al. (1949).
Rats and mice exposed to 0.5 mg ketene/litre for 1.5 min showed
clinical signs and pathological lesions identical to those cause by
phosgene. Ketene is a known acrylating agent that does not break down
to a strong acid.
The effects on pulmonary ultrastructure and enzyme activities in
adult rats from exposure to phosgene for 10 min at 400 mg/m3
(100 ppm) were studied by Pawlowski & Frosolono (1977) and Frosolono &
Pawlowski (1977). Homogenates of the combined lungs from six to eight
rats were assayed for several enzyme activities in duplicate
immediately after exposure and at 30 and 60 min post-exposure. A
decrease in the enzymatic activity of 10-80% was noted at all time
periods for p-nitrophenyl phosphatase, cytochrome C oxidase, ATPase
and lactic dehydrogenase (Frosolono & Pawlowski 1977). Using the same
protocol Pawlowski & Frosolono (1977) examined the cascade of
ultrastructural changes in the terminal bronchiolar epithelium after
exposure. An immediate vesiculation of cells was followed by septal
extracellular oedema and, finally intracellular oedema, cell
disruption and necrosis. The authors suggested that the biochemical
changes preceded major ultrastructural changes in the alveolar region.
Currie et al. (1985) studied the effects on energy metabolism of
exposure to 4 mg/m3 (1 ppm) for 4 h (CT = 960 mg/m3-min) in rats.
An attempt was made to correlate the onset of pulmonary oedema with
alterations in energy metabolism. At the exposures studied, there was
a significant reduction in the respiratory control index, which
coincided with the highest level of percentage water in the lung. In
addition, a decrease in ATP concentration was noted. It was concluded
that reductions in ATP levels and Na-K-ATPase activity play a major
role in damage to the lung after phosgene exposure and prior to the
onset of oedema.
Studies by Currie et al. (1987b) have confirmed these findings at
lower doses. Rats were exposed for 4 h to 48, 120, 240, 480 or
960 mg/m3-min (12, 30, 60, 120 or 240 ppm-min). Decreased ATP
levels were noted prior to the onset of oedema at doses as low as
48 mg/m3-min (12 ppm-min) after exposure for 4 h. Further studies by
Frosolono & Currie (1985), using the same exposure regimen as Currie
et al. (1985) (i.e., 960 mg/m3-min), indicated that phosgene may
alter the level of pulmonary surfactant thus altering the homeostatic
mechanism for fluid balance in the lung.
Jaskot et al. (1991) studied the effect of inhaled phosgene on
lung anti-oxidant systems in Fischer-344 male rats. Levels of 0, 0.4,
1, 2 and 4 mg/m3 were administered for 4 h and a satellite group
received 1 mg/m3 for 8 h. Changes in glutathione (GSH) and
anti-oxidant associated enzymes (GSH peroxidase, GSH reductase,
glucose-6-phosphate dehydrogenase and superoxide dismutase) were
measured 0, 1, 2, 3 and 7 days post-exposure in groups of 12 animals
per dose. At all dose levels significant increases were noted for one
or more components of the anti-oxidant system studied. Peaking at 2
to 3 days post-exposure, the changes noted were similar to those
observed after exposure to the pulmonary irritants ozone and nitrogen
dioxide.
The role of arachidonic acid metabolites in the pathogenesis of
phosgene-induced lung injury (oedema and vascular permeability) was
studied in rabbits (Guo et al., 1990). Animals (four to six per
group) were exposed to 6000 mg/m3-min (1500 ppm-min) of phosgene and
killed 30 min after-exposure. The effects were compared to those of a
control group of eight animals. Lungs were perfused for 90 min and
cyclooxygenase- and lipoxygenase-generated metabolites of arachidonic
acid were measured. Phosgene exposure did not enhance the
cyclooxygenase metabolism of arachidonic acid but did result in a
10-fold increase in lipoxygenase metabolites (leukotriene). A marked
decrease in the gain in lung weight after phosgene exposure was
reported when the perfused lung was pretreated with leukotriene
receptor blockers. The results suggest that lipoxygenase metabolites
of arachidonic acid contribute to the phosgene-induced pulmonary
damage, but the mechanism by which phosgene stimulates the metabolism
of arachidonic acid is still unknown.
Evidence that the non-cardiogenic pulmonary oedema and mortality
resulting from phosgene inhalation was the result of an influx of
neutrophils into the lung was provided by Ghio et al. (1991). After
exposure of rats to 2 mg phosgene/m3 (0.5 ppm) for 60 min,
significant increases in the percentage of neutrophils and
concentrations of protein and thiobarbitunic acid reactive products in
bronchoalveolar lavage fluid were noted. These increases were
significantly less after treatment prior to exposure with:
cyclophosphamide to deplete the leukocytes; inhibition of the
production of the chemotaxis leukotriene B4, which directs the influx
of neutrophils into the lung; or treatment with colchicine, which
decreases leukocyte migration. These treatments in mice exposed to
8 mg phosgene/m3 (2.0 ppm) for 90 min resulted in decreased
mortality. Colchicine reduced neutrophil influx, lung injury and
mortality in mice even when administered 30 min after exposure.
Preliminary evidence that F-actin in lung cells may be a target
of phosgene was reported by Werrlein et al. (1994). In cultured ovine
pulmonary artery endothelial cells and rat airway epithelial cells
exposed to phosgene there was a dose-dependent decrease in the F-actin
content and organization. Exposure of sheep to 0.15 L(CT)50
(96 mg/m3 for 20 min) led to a decrease in the F-actin concentration
of endothelial cells. Exposure of sheep to 0.83 L(CT)50 (548 mg/m3
for 20 min) disrupted basal lamina and produced paracellular leakage
paths in the cultured cells. If they occur in vivo, these effects
of phosgene on F-actin may contribute to the decreased barrier
function and increased permeability of vascular tissues.
8. EFFECTS ON HUMANS
8.1 General population and occupational exposure
The information on the effects of high-level, short-term phosgene
exposure in humans is derived from wartime experiences as well as from
industrial accidents. General population and occupational exposures
to high levels of phosgene will be discussed together, since the
immediate effects and outcome of such exposures are identical in both
populations. As with animal experiments (see section 7.1), the acute
effects in humans are reported as a result of a combination of
exposure level and time of exposure (mg/m3-min). The exposure levels
at which perception of the odour is possible compared to those that
cause varying degrees of toxicity and death in humans are presented in
Table 5. In "trained subject" the lowest level at which the odour
(like mouldy hay) is perceived is 1.6-2 mg/m3 (NIOSH, 1976), but at
such levels workers may not detect the odour due to olfactory fatigue
(Proctor & Hughes, 1991). Under normal conditions the odour is
recognized only at levels greater than 6 mg/m3 (1.5 ppm). Signs of
irritation of mucous membranes are observed at > 12 mg/m3 (3 ppm),
early lung damage at > 120 mg/m3-min (>30 ppm-min), and death
(L(CT)50) at approximately 2000 mg/m3-min (approximately
500 ppm-min) (Diller & Zante, 1982; Diller, 1985a).
As shown in Table 5, concentrations of phosgene vapour
> 2 mg/m3 (3 ppm) will result in irritation of the eyes and nose.
Such concentrations in contact with moist skin will also lead to
irritation and erythema (Borak, 1991), but there is no evidence that
they would result in serious skin injury. At 12 mg/m3 (3 ppm) the
only effect reported on the human eye was inflammation (conjunctival
hyperaemia) (Grant & Schumann, 1993). Liquid phosgene splashed in the
eye, however, caused complete corneal opacification, conjunctival
adhesions and perforation in one victim (Grant & Schumann, 1993).
Although skin contact can result in severe burns, no reports of such
cases are available.
Table 5. Correlation of phosgene dose and effects in humansa
Effects Dose levelb
Perception of odour 1.6 mg/m3 0.4 ppm
Recognition of odour 6 mg/m3 1.5 ppm
Irritation of eyes, nose, and throat 12 mg/m3 3 ppm
Beginning lung damage >120 mg/m3-minc > 30 ppm-min
Pulmonary oedema >600 mg/m3-minc > 150 ppm-min
L(CT)1 approx 1200 mg/m3-minc approx 300 ppm-min
L(CT)50 approx 2000 mg/m3-minc approx 500 ppm-min
L(CT)100 approx 5200 mg/m3-minc approx 1300 ppm-min
a From: Diller (1985a)
b A conversion factor of 4 was used to calculate mg/m3 from the ppm value used by author.
c These values should be considered in relation to Haber's Law (see section 7.1).
With respect to pulmonary damage, three distinct
clinicopathological phases (initial reflex syndrome, clinical latent
phase and clinical oedema phase) have been reported in humans acutely
exposed to phosgene levels of 120 mg/m3-min to 1200 mg/m3-min
(30-300 ppm-min) (Diller & Zante, 1982; Diller, 1985a). During and
immediately after exposure the individual experiences pain in the eyes
and throat (an irritating or burning sensation) and tightness in the
chest, which may be accompanied by shortness of breath and coughing.
This is followed by a latent phase, which is often asymptomatic.
Depending on total dose this period may last from 1 to 24 h. The
oedema phase is manifested when enough lung is affected to become
clinically apparent, i.e. shortness of breath, productive cough,
and/or expectoration of large amounts of frothy and possibly bloody
sputum. If enough of the lung is involved the person may become
cyanotic and enter into shock. If the exposure is in the lethal
range, the early phases may be truncated, and the latent phase may be
very short or non-existent. It has been reported that radiographs
taken immediately after exposure can be used to predict the severity
of ensuing pulmonary oedema (Ardran, 1964). The author stated that an
increase in lung volume following expiration is highly predictive for
the future development of oedema.
8.2 Case reports - individual accidents
The most definitive data on both the short- and long-term effects
of acute phosgene exposure in humans is found in case reports of
industrial accidents. Such accidents may also pose a potential hazard
to adjacent communities. In Hamburg, Germany, an industrial storage
tank released 11 tonnes of phosgene into the atmosphere in 1928
(Hegler, 1928). The atmospheric conditions were conducive to the slow
spread of the gas outside the plant. No exposure levels were
reported. During a period of 5 days, over 300 people became ill, of
whom 10 died. The initial symptoms were severe irritation of the eyes
and throat, coughing, tightness of the chest, nausea and vomiting.
Autopsies of some of the victims revealed typical pulmonary lesions
and nonspecific lesions in other organs that were attributable to
local hypoxia (Wohwill, 1928). The author also felt that some
degenerative lesions in the brain and spinal cord were due directly to
phosgene, but this has not been confirmed in subsequent studies. The
only reference to the long-term effects in this population was that
there was no apparent damage to health 2 months after the accident.
In November 1966, phosgene was accidentally released from a
factory in Japan; 382 people were poisoned and 12 were hospitalized
(Sakakibara et al., 1967). Signs and symptoms observed in the 12
patients on admission were headache (9), nausea (9), cough (8),
dyspnoea (7), fatigue (7), pharyngeal pain (5), chest tightness (5),
chest pain (5), and fever (3). Lacrimation and redness of the eyes
were only observed in one patient. Seven patients showed evidence of
pulmonary oedema as revealed by chest X-rays 48 h after the exposure.
These findings indicate that pulmonary oedema may develop even 48 h
after exposure without initial symptoms of eye or nose irritation.
Other industrial accidents that have been reported usually
involved only a few people and no details of exposure levels were
given. The clinicopathological syndrome was similar in all of these
reports (Everett & Overholt, 1968; Stavrakis, 1971; Regan, 1985). The
predominant finding during and immediately following exposure was
irritation of mucous membranes. Victims described intense pain in the
eyes with profuse lacrimation. At the same time there was a burning
sensation in the throat and tightness of the chest. Coughing ensued,
often of a very severe nature. Victims sometimes did not show any
other symptoms. However, more common was the subsequent development
of pulmonary oedema, which, if sufficiently severe, resulted in death
due to interference with gas exchange. In none of the case reports of
direct phosgene exposure was any data on the actual levels of phosgene
given.
Several case studies of phosgene poisoning have been reported
where the patient was not working directly with phosgene. Such
reports include those of Seidelin (1961) on carbon tetrachloride used
to extinguish a fire, those of Glass et al. (1971) and Sjogren et al.
(1991) on the decomposition of trichlorethylene during welding and
those of Gerritsen & Buschmann (1960) and Snyder et al. (1992)
reporting possible phosgene poisoning due to the thermal degradation
of methylene chloride used to remove paint. All of these chemicals
are known to be degraded thermally to phosgene. Furthermore, the
progression of effects (clinical latent phase) after exposure, the
development of dyspnoea, chest discomfort and pulmonary oedema in all
subjects was typical of phosgene poisoning reported in case studies of
direct phosgene exposure.
There are relatively few reports on the long-term sequelae of an
acute exposure. In a review of this subject, Diller (1985a,b) found
that the vast majority of survivors of acute exposure have a good
prognosis. However, some of those exposed to high levels of phosgene
showed chronic symptoms such as shortness of breath and reduced
physical capacity, which persisted, in some cases, for the rest of
their lives. However, the severity and duration of such effects was
also related to subsequent smoking habits. Pre-existing pulmonary
disease such as emphysema was exacerbated by phosgene exposure. While
most published reports indicate that the respiratory tract is the
primary target organ for phosgene poisoning, a few reports have
indicated effects on other organs, especially the heart and brain
(Diller, 1985a,b). Neurasthenia is the most common of these
conditions associated with acute phosgene exposure. Others are an
epilepsy-like syndrome, loss of speech, peripheral Raynaud-like
syndrome and a type of paralysis characterized by disfunction of the
peroneal nerve. A causal relationship between phosgene exposure and
such effects has yet to be confirmed. It has been suggested by Diller
(1985b) that such changes are more likely a result of anoxia from the
pulmonary oedema rather than the direct action of phosgene.
8.3 Epidemiological studies
Phosgene, isopropyl alcohol, aniline and caustic soda are raw
materials used in a Russian plant for the manufacture of the
herbicide, isopropylphenyl carbamate. Levina & Kurando (1967) found
phosgene at levels of about 0.5 mg/m3 in 30% of all air samples. In
89 workers studied no mention was made of pulmonary problems.
However, methaemoglobinaemia and anaemia were detected and attributed
to exposures to aniline and the herbicide itself.
At a phosgene factory in the USA the medical records of all
workers exposed (326) and 6228 non-exposed workers were compared
(NIOSH, 1976). Limited air sampling conducted during a 2-month period
using the NBP method of analysis with 20-min sampling time (passive
dosimetry Table 2), indicated an average exposure to phosgene of
0.01 mg/m3 (ND to 0.08 mg/m3). Out of 56 fixed-position samplers
(2-h or 20-min collection) 51 showed phosgene levels of up to
0.52 mg/m3 and five samples showed levels greater than 0.55 mg/m3
(off the scale). Deaths attributable to respiratory disease and
pulmonary function (defined as "lung problems") were compared. No
chronic lung problems associated with working at these phosgene levels
were reported, nor was there any increased mortality from respiratory
disease in the exposed workers.
Polednak (1980) described a study of a cross-section of workers
exposed to phosgene at a uranium processing plant between 1943 and
1947. The study contained one group of 699 white male workers exposed
routinely to low (but undetermined) levels of phosgene with daily
episodes (4 or 5 daily) of exposures to levels above 4 mg/m3. A
second group of 106 white males involved in accidents resulting in
acute exposures to phosgene at levels estimated by the authors to be
greater than 200 mg/m3-min. This estimate was based on initial
symptoms reported and clinical data obtained immediately after
exposure. All workers reported detecting the odour of phosgene, 82
reported chest pain and dyspnoea, 25 showed x-ray and clinical
evidence of pneumonitis and 1 worker died 24 h after the exposure from
pulmonary oedema (based on clinical symptoms). The control group
contained 9352 non-exposed workers at the same facility. Those
workers employed for 2 days or more in departments where phosgene
exposure was possible were considered to be in the exposed group.
Mortality data was determined by examination of the Social Security
Administration records and coding the cause of death using the Eighth
Revision of the International Classification of Diseases. As of 1974
there was no evidence of excess mortality from diseases of the
respiratory system in the group of 699 male workers (about 30 years
after exposure). In fact standardized mortality ratios (SMR) for
death from all causes were essentially the same in both the exposed
and control groups. The Task Group noted that possible simultaneous
exposure to ionizing radiation was not taken into account.
In 1974, 30 deaths had occurred in the 106 workers exposed to
high levels of phosgene (SMR = 113). No deaths from lung cancer were
noted but three deaths (1.37 expected) were due to respiratory
diseases. One worker in this group died from pulmonary oedema 24 h
after exposure.
A follow-up of the workers from the cohort studied by Polednak
(1980) was made by Polednak & Hollis (1985). Similar trends to those
reported earlier were noted 35 years after exposure. Of the 694
workers chronically exposed, there were 14 deaths from diseases of the
respiratory tract (13.1 expected) (SMR = 107; 59-180 95% CI). In 1974,
the SMR for lung cancer within the 699 chronically exposed workers was
127 (95%, CI 66-220) and in 1979 it was 122 (72-193, 95% CI). The
slightly elevated SMR values were not significantly higher than
controls. The SMR for death from all causes was 97 (85-111 95% CI) in
the exposed workers and 101 (98-104 95% CI) in controls. In the high-
exposure group there were 41 deaths from all causes compared to 33.9
expected (SMR = 121; 86-165 95% CI). Five deaths were coded to
diseases of the respiratory tract (SMR = 266; 86-622 95% CI). In two
of these cases, bronchitis due to phosgene poisoning had been reported
in 1945 during clinical examination after exposure.
There have been no epidemiological or case reports linking the
development of reproductive or teratogenic effects in humans to acute
and/or chronic phosgene exposures.
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
No information concerning the effects of phosgene in the
laboratory and field has been reported.
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of human health risks
10.1.1 Exposure
Exposure to phosgene is primarily by inhalation. There are three
exposure situations: chronic exposure of the general population to
extremely low levels; chronic exposure in the workplace to very low
levels; and accidental acute exposure to high levels. It is likely
that the principle source of exposure to phosgene for the majority of
the general population is through the photo-degradation and thermal
degradation of chlorinated hydrocarbons, especially solvents and
polymers (e.g., tri- and tetrachloroethylene and PVC). On the basis
of limited data, average levels of phosgene in ambient air can be
expected to vary between 80 and 130 ng/m3.
Available data are inadequate to determine quantitatively the
exposure to phosgene in the workplace. Those working simultaneously
with flames (or thermal energy sources) and organochlorine solvents or
PVC can be exposed to phosgene levels well above present threshold
limit values (time-weighted average) of 0.4 mg/m3.
Accidental release of phosgene during its manufacture, use or
transport can lead to high levels of exposure for workers and for the
general population in the vicinity of the accident.
10.1.2 Health effects
Phosgene is a highly reactive chemical, hydrolysing to
hydrochloric acid, and is capable of acrylating nucleophilic groups,
such as amino, hydroxyl and sulfhydryl groups, in tissues.
In all species studied, including humans, the major target organ
is the lung. High concentrations can also cause skin and eye
irritation. For health effects after acute exposure, Haber's Law,
which states that the toxicological effect is due to the product of
exposure (C) and time (T), holds between levels of 4 and 800 mg/m3
(1 and 200 ppm) using lung disease and death as toxicological end-
points. This law does not, however, prevail for chronic exposure.
The cascade of events after acute inhalation exposure in humans and
experimental animals are similar. It occurs in a dose-related manner
and results in pulmonary oedema and death in humans, which is
dose-dependent at levels exceeding 120 mg/m3-min. Three distinct
clinicopathological phases can be recognized: pain in the eyes and
throat and tightness of the chest, often with shortness of breath,
wheezing and coughing; a latent phase that is often asymptomatic and
can last up to 24 h depending upon the concentration and duration of
exposure; and the final phase of pulmonary oedema.
10.1.2.1 Evaluation of animal data
The L(CT)50 and L(CT)100 values for single exposure vary widely
among animal species (Table 3). In all species the characteristic
pathological feature is the delayed clinical manifestation of
pulmonary oedema, which is dose-dependent. The extent of the
long-term chronic effects of acute exposure appears to depend on the
severity of the initial pathology.Single exposure of rats for 4 h to
between 0.5 and 4 mg/m3 resulted in a dose-related increase in lavage
fluid protein (LFP) concentration and an increased percentage of
polymorphonuclear leukocytes (PMN) in the alveoli. Changes in the LFP
and PMN were the most sensitive parameters occurring at 240 mg/m3-
min. These changes were reversible within 3 days after exposure.
There have been no long-term exposure studies in animals, and studies
in dogs exposed 1-3 times/week for 12 weeks are of limited value for
risk assessment in view of inadequate study design and lack of dose-
response. Available data in experimental animals are inadequate for
the assessment of the potential reproductive, developmental,
neurotoxic and carcinogenic effects resulting from phosgene exposures.
Single 4-h exposures to 0.1 mg phosgene/m3 in mice have resulted
in a demonstrable decrease in pulmonary host resistance to bacteria.
Rats exposed to 2 or 4 mg/m3 for 4 h had decreased pulmonary cell
natural killer (NK) activity, whereas no effect was seen at a level of
0.4 mg phosgene/m3 for 4 h. Increased infectivity by influenza virus
was reported in rats exposed for 4 h to 4 mg/m3. Virus titres were
not detectable 4 days after infection. Mortality was increased after
exposure to S. zooepidemicus and inoculation of B16/BL6 pulmonary
melanoma tumours in mice exposed to phosgene levels at or above
0.1 mg/m3. No effect was reported at a level of 0.04 mg/m3.
Pulmonary bacterial clearance was reduced in rats exposed to
0.4 mg/m3 (0.1 ppm) phosgene for 6 h and to 0.4 mg/m3 (0.1 ppm) for
6 h/day, 5 days/week for 4 to 12 weeks. This effect was reversible
following termination of exposure.
10.1.2.2 Evaluation of human data
As a result of industrial accidents and occupational monitoring
(levels and health status), it has been reported that some humans can
only recognize the odour of phosgene at levels of about 6 mg/m3,
making this an unacceptable parameter for early warning. After
short-term exposure, throat and eye irritation occurs at a level of
12 mg/m3 and eye irritation is noted at 16 mg/m3. The risk of
morbidity and mortality after acute exposure, is determined by the
dose (CT), not solely by concentration. It has been calculated that
doses below 100 mg/m3-min result in no effect, whereas pulmonary
oedema results from doses above 600 mg/m3-min. It should be
recognized, however, that death has been recorded at doses above
400 mg/m3-min, although with proper medical intervention death may be
prevented. Exposures for several hours at or below the odour
threshold (6 mg/m3) may result in severe tissue damage and death.
A review of the health status of workers who have recovered from
acute phosgene exposures has shown no adverse effects. However, full
recovery may take several months.
Available data on human health effects associated with chronic
exposure to phosgene are extremely limited. Epidemiological studies
of phosgene production workers and uranium workers reported no adverse
effects on human health. However, these investigations are of limited
value owing to the small numbers of exposed workers, lack of reliable
quantitative information on exposure to phosgene, concomitant exposure
to other substances, limited number of end-points examined and limited
reporting of relevant information. Available data are therefore
considered inadequate to assess the risk associated with long-term
exposure to low levels of phosgene.
10.1.3 Guidance value
Available data is considered inadequate to derive a meaningful
health-based guidance value for exposure of the general population to
phosgene. Information from epidemiological studies of occupationally
exposed workers is insufficient to characterize quantitatively
exposure-response relationships associated with potentially adverse
health effects resulting from exposure to this substance. Appropriate
studies in laboratory animals are lacking, and the available
toxicological investigations do not provide relevant data upon which
development of a credible guidance value for the long-term exposure of
humans to phosgene can be based.
Recent toxicological studies of rats subchronically exposed by
inhalation to low levels of phosgene indicate that early pulmonary
effects may occur at present TLV values. Thus, consideration by
appropriate authorities should be given to re-evaluating current
occupational exposure guidelines for this substance.
10.2 Evaluation of effects on the environment
The levels of phosgene now found in the environment would not be
expected to result in significant effects to aquatic or terrestrial
biota. However, no data were found to substantiate or refute this
hypothesis.
Damage to plants and aquatic organisms, owing to the rapid
release of hydrochloric acid, could occur in areas where accidental
release of phosgene has occurred.
11. CONCLUSIONS AND RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH
11.1 Conclusions
a) Phosgene is an extremely reactive chemical with the potential to
cause adverse effects in humans, the primary target organ being
the respiratory system.
b) Acute severe phosgene exposure primarily causes respiratory
disease (pulmonary oedema) and may result in death. Survivors
may recover completely provided they receive proper medical
support.
c) Present levels of exposure to phosgene in the general population
are extremely low and do not pose a health risk in the short
term. However, humans working with chlorinated solvents such as
trichloroethane, tetrachloroethylene and methylene chloride or
who are exposed to chlorinated hydrocarbon polymers (e.g., PVC)
in contact with flames or other thermal energy sources can be
exposed to levels of phosgene known to cause adverse effects in
humans. This could apply to firemen, welders, painter or people
working at home with the above-mentioned materials.
d) Accidental industrial releases can cause health problems in
workers and in the nearby community.
e) Workers have been shown not be at risk in closed-system
industrial facilities that manufacture or use phosgene and employ
good industrial practice.
f) No human or animal data are available on the effects of chronic
low-level exposures to phosgene.
g) No data are available concerning adverse effects on organisms in
the environment. However, accidental release would be expected
to give rise to adverse effects.
11.2 Recommendations for protection of human health
a) Present occupational exposure limits for phosgene should be
re-assessed.
b) Data should be obtained on the release of phosgene by the
incineration of chlorine-containing organic materials.
c) International and national regulations regarding transport of
phosgene should be followed in order to avoid accidental
releases.
d) Analytical methods capable of monitoring whole shift individual
exposure (e.g., paper tape monitors) should be used routinely in
the workplace.
12. FURTHER RESEARCH
a) The mechanism(s) of phosgene toxicity need to be clarified in
order to improve risk assessment and therapy.
b) Data gaps in the areas of reproduction/development toxicity,
mutagenicity and carcinogenicity following chronic low level
exposure should be addressed. More data should be gained about
the long-term effects of acute exposure.
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
No international body has evaluated the human health or
environmental risks from exposure to phosgene. Similarly, no health-
based guidance values have been developed by such groups.
Regulatory standards for phosgene established by national bodies
in some countries have been summarized by the ILO (ILO, 1991).
REFERENCES
Andersson HG, Dahlberg JA, & Wettstrom R (1975) Phosgene formation
during welding in air contaminated with perchloroethylene. Ann Occup
Hyg, 18: 129-132.
Ardran GM (1964) Phosgene poisoning [letter]. Br Med J, 1(5379):
375.
Atherley G (1985) A critical review of time-weighted average as an
index of exposure and dose, and of its key elements. Am Ind Hyg Assoc
J, 46: 481-487.
Birgesson D (1982) An unknown working environment hazard for
refrigerating equipment fitters: freon. Arbetsmiljo, 6: 48-50.
Borak J (1991) Phosgene toxicity: review and update. CEM Rep, 5:
19-22.
Boyd EM & Perry WF (1960) Respiratory tract fluid and inhalation of
phosgene. J Pharm Pharmacol, 12: 726-732.
Brown JE & Birky MM (1980) Phosgene in the thermal decomposition
products of poly(vinyl chloride): generation, detection and
measurement. J Anal Toxicol, 4: 166-174.
Budavari S ed. (1996) The Merck index: an encyclopedia of chemicals,
drugs, and biologicals, 12th ed. Rahway, New Jersey, Merck & Co., Inc.
Burleson GR & Keyes LL (1989) Natural killer activity in Fischer-344
rat lungs as a method to assess pulmonary immunocompetence:
Immunosuppression by phosgene inhalation. Immunopharmacol
Immunotoxicol, 11(2-3): 421-443.
Butler R & Snelson A (1979) Kinetics of the homogeneous gas phase
hydrolysis of CC13COC1, CC12HCOC1, CH2C1COC1, and COCL2. J Air
Pollut Control Assoc, 29: 833-837.
Cameron GR & Courtice FC (1946) The production and removal of oedema
fluid in the lung after exposure to carbonyl chloride (phosgene). J
Physiol, 105: 175-185.
Cameron GR & Foss GL (1941) Effect of low concentrations of phosgene
for 5 hours on 5 consecutive days in groups of different animals.
Washington, DC, British Embassy Defense Staff (Porton Report No. 2316,
Serial No. 63).
Cameron GR, Courtice FC, & Foss GL (1942) Effect of exposing different
animals to low concentrations of phosgene (1:5,000,000=0.9 mg/m3) for
5 hours on five consecutive days. Second report. In: First report on
phosgene poisoning: chapter VIII. Washington, DC, British Embassy
Defense Staff (Porton Report No. 2349).
Cessi C, Colombini C, & Mameli L (1966) The reaction of liver proteins
with a metabolite of carbon tetrachloride. Biochem J, 101: 46c-47c.
Clay JR & Rossing RG (1964) Histopathology of exposure to phosgene: An
attempt to produce pulmonary emphysema experimentally. Arch Pathol,
78: 544-551.
Coman DR, Bruner HD, Horn RCJr, Friedman MD, Boche RD, McCarthy MD,
Gibbon MH, & Schultz J (1947) Studies on experimental phosgene
poisoning. I. The pathologic anatomy of phosgene poisoning, with
special reference to the early and late phases. Am J Pathol, 23:
1037-1074.
Crummett WB & McLean JD (1965) Ultraviolet spectrophotometry
determination of trace quantities of phosgene in gases. Anal Chem,
37: 424-425.
Currie WD, Pratt PC, & Frosolono MF (1985) Response of pulmonary
energy metabolism to phosgene. Toxicol Ind Health, 1(2): 17-27.
Currie WD, Hatch GE, & Frosolono MF (1987a) Pulmonary alterations in
rats due to acute phosgene inhalation. Fundam Appl Toxicol, 8:
107-114.
Currie WD, Hatch GE, & Frosolono MF (1987b) Changes in lung ATP
concentration in the rat after low-level phosgene exposure. J Biochem
Toxicol, 2: 105-114.
Dangwal SK (1994) A spectrophotometric method for determination of
phosgene in air. Ind Health, 32: 41-47.
Diller WF (1985a) Pathogenesis of phosgene poisoning. Toxicol Ind
Health, 1: 7-15.
Diller WF (1985b) Late sequelae after phosgene poisoning: a
literature review. Toxicol Ind Health, 1: 129-136.
Diller WF & Zante R (1982) Dose-effect relation in the effects of
phosgene on man and animals (literature study). Zent.bl Arb.med
Arb.schutz Prophyl, 32: 360-368.
Diller WF, Bruch J, & Dehnen W (1985) Pulmonary changes in the rat
following low phosgene exposure. Arch Toxicol, 57: 184-190.
Ehrlich JP & Burleson GR (1991) Enhanced and prolonged pulmonary
influenza virus infection following phosgene inhalation. J Toxicol
Environ Health, 34: 259-273.
Esposito GG, Lillian D, Podolak GE, & Tuggle RM (1977) Determination
of phosgene in air by gas chromatography and infrared
spectrophotometry. Anal Chem, 49: 1774-1778.
Everett ED & Overholt EL (1968) Phosgene poisoning. J Am Med Assoc,
205: 243-245.
Flury F (1921) [Combat gas poisonings: I. Irritant gases.] Z Gesamte
Exp Med, 13: 1-15 (in German).
Flury F & Zernick F (1931) [Noxious gases, fumes, vapors, and
varieties of smoke and dust.] Berlin, Verlag von Julius Springer,
pp 99-102, 222-228 (in German)
Franch S & Hatch G (1986) Pulmonary biochemical effects of inhaled
phosgene in rats. J Toxicol Environ Health, 19: 413-423.
Frosolono MF & Curie WD (1985) Response of the pulmonary surfactant
system to phosgene. Toxicol Ind Health, 1: 29-35.
Frosolono MF & Pawlowski R (1977) Effect of phosgene on rat lungs
after single high-level exposure: I. Biochemical alterations. Arch
Environ Health, 32: 271-277.
Gay BW Jr, Hanst PL, Bufalini JJ, & Noonan RC (1976) Atmospheric
oxidation of chlorinated ethylenes. Environ Sci Technol, 10: 58-67.
Gerritsen WB & Buschmann CH (1960) Phosgene poisoning caused by the
use of chemical paint removers containing methylene chloride in
ill-ventilated rooms heated with kerosene stoves. Br J Ind Med, 17:
187-189.
Ghio AJ, Kennedy TP, Hatch GE, & Tepper JS (1991) Reduction of
neutrophil influx diminishes lung injury and mortality following
phosgene inhalation. J Appl Physiol, 71(2): 657-665.
Gibbon MH, Bruner HD, Boche RD, & Lockwood JS (1948) Studies on
experimental phosgene poisoning: II. Pulmonary artery pressure in
phosgene-poisoned cats. J Thorac Surg, 17: 264-273.
Glass WI, Harris EA, & Whitlock RML (1971) Phosgene poisoning: case
report. NZ Med J, 74: 386-389.
Grant WM & Schumann JS (1993) Toxicology of the eye, 4th ed.
Springfield, Illinois, Charles C. Thomas, 733 pp.
Guo Y-L, Kennedy TP, Michael JR, Sciuto AM, Ghio AJ, Adkinson NF Jr, &
Gurtner GH (1990) Mechanism of phosgene-induced lung toxicity: Role of
arachiodonate mediators. J Appl Physiol, 69(5): 1615-1622.
Hardy EE (1982) Phosgene. In: Kirk-Othmer encyclopedia of chemical
technology, 3rd ed. New York, John Wiley and Sons, vol 17, pp 416-425.
Hatch GE, Slade R, Stead AG, & Graham JA (1986) Species comparison of
acute inhalation toxicity of ozone and phosgene. J Toxicol Environ
Health, 19: 43-53.
Hegler C (1928) [On the mass poisoning by phosgene in Hamburg: I.
Clinical observations.] Dtsch Med Wochenschr, 54: 1551-1553 (in
German)
HSE (1995) Phosgene. In: Criteria document summaries: Synopses of the
data used in setting occupational exposure limits. London, Health and
Safety Executive, pp 24-25 (EH 64 1995 Supplement).
ILO (1991) Occupational exposure limits for airborne toxic substances,
3rd ed. Geneva, International Labour Office (Occupational Safety and
Health Series, No. 37).
IPCS (1993) 1,2-Dichloropropane. In: Environmental health criteria
No. 146: 1,2-Dichloropropane. Geneva, World Health Organization,
International Programme on Chemical Safety, pp 119-162.
Jaskot RH, Grose EC, Richards JH, & Doerfler DL (1991) Effects of
inhaled phosgene on rat lung antioxidant systems. Fundam Appl Toxicol,
17: 666-674.
Keeler JR, Hurt HH, Nold JB, & Lennox WJ (1990) Estimation of the
LCt50 of phosgene in sheep. Drug Chem Toxicol, 13(2-3): 229-239.
Levina MM & Kurando TB (1967) [Problems of labour hygiene and of state
of health of workers at the isopropylpenylcarbamate production.] Gig i
Sanit, 32: 25-28 (in Russian).
Levina MM, Kurando TB, Belyakov AA, Smirnova VG, & Odlyzhko SL (1966)
[Industrial hygiene problems and worker health in the monuron
industry.] Gig Tr Prof Zabol, 10: 54-56 (in Russian).
Long JE & Hatch TF (1961) A method for assessing the physiological
impairment produced by low-level exposure to pulmonary irritants. Am
Ind Hyg Assoc J, 22: 6-13.
Madden MC, Friedmann M, Keyes LL, Koren HS, & Burleson GR (1991)
Effects of phosgene exposure on lung, arachidonic acid metabolism.
Inhal Toxicol, 3: 73-90.
Manogue WH & Pigford RL (1960) The kinetics of the absorption of
phosgene into water and aqueous solutions. Am Inst Chem Eng J, 6:
494-500.
Mansuy D, Beaune P, Cresteil T, Lange M, & Leroux J-P (1977) Evidence
for phosgene formation during liver microsomal oxidation of
chloroform. Biochem Biophys Res Commun, 79(2): 513-517.
Matherne RN, Lubs PL, & Kerfoot EJ (1981) The development of a passive
dosimeter for immediate assessment of phosgene exposures. Am Ind Hyg
Assoc J, 42: 681-684.
Meek WJ & Eyster JAE (1920) Experiments on the pathological physiology
of acute phosgene poisoning. Am J Physiol, 51: 303-320.
Moore G & Matherne RN (1981) Field experiences with a phosgene
dosimeter system. Ann Am Conf Gov Ind Hyg, 1: 253-254.
Nash T & Pattle RE (1971) The absorption of phosgene by aqueous
solutions and its relation to toxicity. Ann Occup Hyg, 14: 227-233.
NIOSH (1976) Criteria for a recommended standard .... Occupational
exposure to phosgene. Cincinnati, Ohio, US Department of Health,
Education and Welfare, National Institute for Occupational Safety and
Health, 129 pp (HEW Publication No. NIOSH 76-137).
Noweir MH, Pfitzer EA, & Hatch TF (1973) Decomposition of phosgene in
air. Am Ind Hyg Assoc J, 34: 110-119.
Ong SG (1972) Treatment of phosgene poisoning with antiserum:
anaphylactic shock by phosgene. Arch Toxikol, 29: 267-278.
Pawlowski R & Frosolono MF (1977) Effect of phosgene on rat lungs
after single high-level exposure. II. Ultrastructural alterations.
Arch Environ Health, 32(6): 278-283.
Pohl LR, Bhooshan B, Whittaker NF, & Krishna G (1977) Phosgene: a
metabolite of chloroform. Biochem Biophys Res Commun, 79(3):
684-691.
Polednak AP (1980) Mortality among men occupationally exposed to
phosgene in 1943-1945. Environ Res, 22: 357-367.
Polednak AP & Hollis DH (1985) Mortality and causes of death among
workers exposed to phosgene in 1943-1945. Toxicol Ind Health, 1:
137-151.
Potts AM, Simon FP, & Gerard RW (1949) The mechanism of action of
phosgene and diphosgene. Arch Biochem, 24: 329-337.
Proctor NJ & Hughes JP (1991) In: Hathaway G, Proctor N, & Hughes J
ed. Chemical hazards of the workplace. New York, Von Nostrand Reinhold
Company, pp 475-477.
Regan RA (1985) Review of clinical experience in handling phosgene
exposure cases. Toxicol Ind Health, 1: 69-71.
Rinehart WE & Hatch T (1964) Concentration-time product (CT) as an
expression of dose in sublethal exposures to phosgene. Am Ind Hyg
Assoc J, 25: 545-553.
Rinzema LC (1971) Behavior of chlorohydrocarbon solvents in the
welding environment. Int Arch Arbeitsmed, 28: 151-174.
Rossing RG (1964) Physiologic effects of chronic exposure to phosgene
in dogs. Am J Phsiol, 207: 265-272.
Sakakibara H, Shiiki Y, Okamoto N, & Wataki A (1967) [Mass poisonings
with phosgene.] Shindan Chiryo, 55: 1433-1437 (in Japanese).
Schepers GWH (1971) Lung tumors of primates and rodents: Part II. Ind
Med, 40: 23-31.
Schneider W & Diller W (1989) Phosgene. In: Encyclopedia of industrial
chemistry, 5th ed. Weinheim, Germany, VCH Verlag, vol A19, pp 411-420.
Seidelin R (1961) The inhalation of phosgene in a fire extinguisher
accident. Thorax, 16: 91-93.
Selgrade MK, Starnes DM, Illing JW, Daniels MJ, & Graham JA (1989)
Effects of phosgene exposure on bacterial, viral, and neoplastic lung
disease susceptibility in mice. Inhal Toxicol, 1: 234-259.
Shah H, Hartman SP, & Weinhouse S (1979) Formation of carbonyl
chloride in carbon tetrachloride metabolism by rat liver in vitro.
Cancer Res, 39: 3942-3947.
Singh HB (1976) Phosgene in the ambient air. Nature (Lond), 264:
428-429.
Singh HG, Salas L, Shigeishi H, & Crawford A (1977) Urban-nonurban
relationships of halocarbons, SF6, N2O, and other atmospheric trace
constituents. Atmos Environ, 11: 819-828.
Singh HB, Salas LJ, Smith AJ, & Shigeishi H (1981) Measurement of some
potentially hazardous organic chemicals in urban environments. Atmos
Environ, 15: 601-612.
Sjogren B, Plato N, Alexandersson R, Eklund A, & Falkenberg C (1991)
Pulmonary reactions caused by welding-induced decomposed
trichloroethylene. Chest, 99: 237-238.
Snyder RW, Mishel HS, & Christensen GC (1992) Pulmonary toxicity
following exposure to methylene chloride and its combustion product,
phosgene. Chest, 101(3): 860-861.
Stavrakis P (1971) The use of hexamethylenetetramine (HMT) in
treatment of acute phosgene poisoning. Ind Med, 40: 30-31.
Thienes CH & Haley TJ (1972) The gas penetrates into the tissues of
respiratory tract and to some extent is absorbed by the lung. In:
Clinical toxicology, 5th ed. Philadelphia, Pennsylvania, Lea &
Febiger, pp 192-193.
Tuggle RM, Esposito GG, Guinivan TL, Hess TL, Lillian D, Podolak GE,
Sexton KG, & Smith NV (1979) Field evaluation of selected monitoring
methods for phosgene in air. Am Ind Hyg Assoc J, 40: 387-394.
Underhill FP (1919) The physiology and experimental treatment of
poisoning with the lethal war gases. Arch Intern Med, 23: 753-770.
Underhill FP (1920) The lethal war gases: physiology and experimental
treatment. New Haven, Connecticut, Yale University Press, pp 3-10,
40-41, 85-87, 105, 119-120, 133-137.
US EPA (1986) Health assessment document for phosgene. Washington, DC,
US Environmental Protection Agency (EPA/600/8-86/022A).
US NLM (1995) Hazard substances data base: computerized database.
Bethesda, Maryland, US National Libray of Medicine.
Verschueren K (1983) Rabbit, inhalation, mortality. In: Handbook of
environmental data on organic chemicals, 2nd ed. New York, Van
Nostrand Reinhold Co., p 534.
Werrlein RJ, Madren-Whalley JS, & Kirby SD (1994) Phosgene effects on
F-actin organization and concentration in cells cultured from sheep
and rat lung. Cell Biol Toxicol, 10: 45-58.
Winternitz MC, Lambert RA, & Jackson L (1920) The pathology of
phosgene poisoning. In: Collected studies on the pathology of war gas
poisoning. New Haven, Connecticut, Yale University Press, pp 35-66.
Wohlwill F (1928) Pathological findings of phosgene poisoning. Dtsch
Med Wochenschr, 54: 1553-1557.
Wu WS & Gaind VS (1993) Determination of phosgene (carbonyl chloride)
in air by high performance liquid chromatography with a dual selective
detection system. Analyst, 188: 1285-1287.
Yang YG, Gilmour MI, Lange R, Burleson GR, & Selgrade MK (1995)
Effects of acute exposure to phosgene on pulmonary host defenses and
resistance to infection. Inhal Toxicol, 7: 393-404.
1. RESUME ET CONCLUSIONS
1.1 Identité, propriétés physiques et chimiques et méthodes d'analyse
Le phosgène est un gaz incolore, extrêmement réactif à la
température et à la pression ambiantes, dont l'odeur suffocante
rappelle celle du foin moisi. Cette odeur est décelable à des
concentrations comprises entre 1,6 et 6 mg/m3.
Il existe des méthodes d'analyse pour la mise en évidence du
phosgène dans l'air ou qui peuvent être utilisées dans le cadre des
programmes d'hygiène et de sécurité du travail pour la mesure de la
dose totale (par ex. ruban de papier indicateur).
1.2 Usages et sources d'exposition humaine et environnementale
Le phosgène est utilisé à plus de 99% sur le lieu de production
dans des systèmes clos. On le prépare en faisant réagir, en présence
d'un catalyseur carboné, du chlore anhydre sur du monoxyde de carbone
en proportions équimoléculaires. La production mondiale est estimée à
plus de 3 millions de tonnes.
La présence de phosgène dans l'environnement est due aux
émissions d'origine industrielle et à la décomposition thermique des
solvants et des polymères chlorés. Cependant, il peut également
provenir en proportion importante de l'oxydation photochimique des
chloréthylènes, comme le trichloréthylène et le tétrachloréthylène.
1.3 Transport, distribution et transformation dans l'environnement
Le phosgène étant très réactif, il n'est vraisemblablement que
peu transporté d'un compartiment à l'autre.
Le phosgène s'élimine de l'air ambiant par une décomposition en
phase hétérogène (catalyse de surface) et une lente hydrolyse en phase
gazeuse. Il peut être transporté sur de longues distances et sa
diffusion de la troposphère vers la stratosphère accélère probablement
le processus de décomposition par photolyse.
1.4 Concentrations dans l'environnment et exposition humaine
L'exposition humaine, qu'il s'agisse de la population générale ou
de certaines catégories professionnelles, se produit par inhalation.
La concentration moyenne du phosgène dans l'air ambiant peut
varier d'environ 80 à 130 ng/m3, encore que les données soient peu
nombreuses à ce sujet. En raison de la diversité des pratiques en
matière d'hygiène du travail dans le monde, il est impossible de
donner un chiffre pour caractériser l'exposition des travailleurs qui
produisent ou utilisent du phosgène ou, plus particulièrement,
l'exposition des pompiers. A l'heure actuelle, les valeurs-seuils (en
moyenne pondérée par rapport au temps) relevées dans 15 pays s'étagent
de 0,4 à 0,5 mg/m3.
La littérature ne donne pas d'indications sur la teneur de l'eau,
du sol et des aliments en phosgène.
1.5 Cinétique et métabolisme
On ne possède que très peu de données sur l'absorption, le
métabolisme, la distribution et la destinée du phosgène. La principale
voie d'exposition est la voie respiratoire, le gaz pénétrant dans les
tissus de l'arbre pulmonaire et ne se retrouvant par conséquent qu'en
quantités minimes dans le reste de l'organisme. Sa demi-vie très brève
(0,026 secondes) en solution aqueuse exclut toute rétention importante
dans l'organisme. On ne dispose d'aucune donnée sur le métabolisme du
phosgène. Ses produits d'hydrolyse, à savoir l'acide chlorhydrique et
le dioxyde de carbone, sont éliminés de l'organisme par les processus
physiologiques normaux.
La toxicité du phosgène est due au fait qu'il provoque
l'acylation des protéines et qu'il donne naissance à de l'acide
chlorhydrique. Cette acylation se produit au niveau des groupements
amino, hydroxyles et sulfhydriles des protéines et entraîne une
inhibition marquée des enzymes qui interviennent dans le métabolisme
énergétique ainsi qu'une rupture de la barrière air/sang.
1.6 Effets sur les animaux d'expérience et les systèmes d'épreuve
in vitro
1.6.1 Exposition à court et à long terme
Chez toutes les espèces étudiées, c'est le poumon qui est
l'organe cible principal. La valeur du L(CT)50 varie de
900 mg/m3-min (225 ppm-min ) chez la souris à 1920 mg/m3-min
(480 ppm-min) chez le cobaye. Une valeur de 1000 mg/ m3
(250 ppm-min) a été obtenue chez le singe. Chez toutes les espèces,
on constate la même pathologie caractéristique, à savoir un oedème
pulmonaire dont les manifestations cliniques sont retardées et qui est
lié à la dose. Les anomalies anato-mopathologiques observées à faible
concentration au niveau des bron-chioles terminales et des alvéoles,
sont caractéristiques d'un irritant pulmonaire. En revanche, à forte
concentration, l'oedème pulmonaire qui se développe perturbe les
échanges gazeux et finit par entraîner la mort.
On n'a pas connaissance d'études consacrées à une exposition de
longue durée au phosgène.
En ce qui concerne les durées d'exposition relativement courtes,
on dispose d'une étude au cours de laquelle des rats on été exposés en
une seule fois à du phosgène pendant 4 h à la concentration de
2 mg/m3. On a observé une réduction de l'immunocompétence pulmonaire
mesurée par l'activité des cellules NK. Aucun effet n'a été constaté
lors d'une exposition de 4 h à 0,4 mg/m3.
Deux autres études ont été publiées au sujet des effets d'une
exposition unique à du phosgène sur l'immunocompétence pulmo-naire.
Au cours de ces études, effectuées sur des rats et des souris, on a
constaté que des rats infectés par le virus grippal présentaient,
après 4 h d'exposition à 4 mg de phosgène par m3, un titre viral 10
fois plus élevé 1 jour après l'infection, ce titre conservant une
valeur élevée au cours des 4 jours suivants. En outre, chez les rats
qui avaient été soumis pendant 4 h à une concentration de gaz comprise
entre 0,2 et 4 mg/m3, on pouvait constater une diminution marquée du
taux de prostaglandine E2 et de leucotriènes dès que la dose
atteignait 0,4 mg/m3, avec réduction du nombre de macrophages
alvéolaires et augmentation du nombre de neutrophiles à la
concentration de 0,4 mg/m3. Pour étudier la résistance de l'hôte, on
a soumis des souris à des concentrations de phosgène comprises entre
0,04 et 0,4 mg/m3 sur une durée de 4 h. On a constaté, selon les
cas, une augmentation de la mortalité consécutive à une infection par
Streptococcus zooepidemicus ou un accroissement des mélanomes
pulmonaires B16/BL6 à partir de 0,1 mg/m3. Chez les rats exposés à
du phosgène à raison de 0,4 mg/m3 (0,1 ppm) pendant 6 h ou à la même
dose 6 h par jour, 5 j par semaine pendant 4 à 12 semaines , on a
observé une réduction de la clairance bactérienne pulmonaire. L'effet
était réversible après cessation de l'exposition.
1.6.2 Effets non pulmonaires
Le phosgène peut provoquer une irritation oculaire et cutanée. On
n'a pas trouvé trace, dans la littérature, d'études portant sur le
pouvoir sensibilisateur du phosgène.
On ne dispose d'aucune donnée relative aux effets du phosgène sur
la reproduction et le développement.
Il n'existe pas de données suffisantes pour permettre une
évaluation du pouvoir mutagène ou cancérogène du phosgène.
1.7 Effets sur l'homme
Chez l'homme, comme chez les animaux de laboratoire, l'organe
cible est le poumon. Après exposition à des concentrations de phosgène
comprises entre 120 et 1200 mg/m3-min , on a observé trois phases
clinico-pathologiques distinctes. La phase initiale consiste en
douleurs au niveau des yeux et de la gorge accompagnées d'une
sensation de constriction thoracique, souvent avec dyspnée
concomitante, respiration sifflante et toux; il peut également y avoir
une hypo-tension, de la bradycardie et plus rarement, une arrythmie
sinusale. La seconde phase ou phase de latence, souvent
asymptomatique, peut se prolonger pendant 24 h , selon l'intensité et
la durée de l'exposition. Au cours de la troisième phase, un oedème
pulmonaire parfois mortel peut se développer. Dans des populations
exposées au phosgène à la suite d'accidents industriels, on a fait
état de symptômes très divers comme des céphalées, des nausées, de la
toux, de la dyspnée, une fatigue générale, des maux de gorge, une
sensation douloureuse de constriction thora-cique, des douleurs
oculaires intenses et une forte lacrimation. Lors d'une étude, on a
observé la survenue d'un oedème pulmonaire après une phase de latence
de 48 h.
Les effets d'une exposition au phosgène ont été étudiés chez des
groupes de travailleurs de deux usines, à savoir une unité de
production de phosgène et une unité de traitement de l'uranium. Dans
les deux cas, on n'a effectué que des prélèvements d'air et une
surveillance individuelle limités et l'exposition n'est donc connue
que de manière estimative.
L'examen du dossier médical de la totalité des 326 travailleurs
de l'unité de production de phosgène qui pouvaient avoir été exposés à
ce gaz (jusqu'à 0,5 mg/m3 avec quelques dépassements de cette valeur;
moyenne 0,01 mg/m3) n'a pas révélé de problèmes pulmo-naires
chroniques ni de surmortalité par comparaison avec un groupe témoin de
6226 personnes. Toutefois, l'absence de détails, dans le compte rendu,
au sujet de l'exposition et des effets observés ne permet guère de
tirer des conclusions définitives de cette étude.
Dans le cas des travailleurs de l'usine de traitement de
l'uranium, on a constitué deux groupes: l'un, de 699 personnes prises
parmi les 1800 employés de cette période, a fait l'objet d'une étude
transversale. Ces travailleurs avaient été vraisemblablement soumis à
une concen-tration de phosgène inférieure à 0,4 mg/m3 (avec 4 ou 5
dépassements brefs par jour jusqu'à plus de 4 mg/m3). L'autre groupe
était constitué de 106 personnes qui avaient été impliquées dans des
accidents et soumises à une exposition de plus de 200 mg/m3-min.
Dans le groupe exposé de manière chronique à de faibles concentrations
de phosgène, l'examen des certificats de décès n'a pas fait ressortir
de surmortalité due à une cause quelconque, à une affection
respiratoire ou à un cancer du poumon. Il n'y a d'ailleurs eu aucune
mortalité par cancer du poumon, mais par contre une légère
surmortalité d'origine respiratoire. On ne peut tirer de ces études
que des conclusions limitées sur les effets chroniques du phosgène et
cela, tant du fait de l'absence de données sur l'exposition que pour
des raisons d'ordre méthodologique.
1.8 Effets sur les autres êtres vivants au laboratoire et dans leur
milieu naturel
On ne possède aucun renseignement concernant les effets du
phosgène sur les diverses formes vivantes présentes dans
l'environnement.
1. RESUMEN Y CONCLUSIONES
1.1 Identidad, propiedades físicas y químicas y métodos analíticos
El fosgeno es un gas incoloro y altamente reactivo a temperatura
y presión ambientes. Posee un olor sofocante parecido al del heno
enmohecido, que puede percibirse a concentraciones comprendidas entre
1,6 y 6 mg/m3.
Existen métodos analíticos que permiten detectar el fosgeno en el
aire y se utilizan en programas de higiene industrial que miden la
dosis total (p.ej., sensores de cinta de papel).
1.2 Usos y fuentes de exposición humana y ambiental
Más del 99% del fosgeno producido se emplea in situ en sistemas
cerrados. Se produce haciendo reaccionar cantidades equimolares de
cloro anhidro y monóxido de carbono en presencia de un catalizador de
carbono. Se ha estimado que la producción mundial supera los 3
millones de toneladas.
El fosgeno ambiental procede de emisiones industriales y de la
degradación térmica de algunos disolventes clorados y polímeros
clorados. No obstante, una fuente importante de fosgeno ambiental es
la oxidación fotoquímica de cloroetilenos tales como el tri- y el
tetraetileno.
1.3 Transporte, distribución y transformación en el medio ambiente
Debido a su alta reactividad, el transporte intercompartimental
del fosgeno es en principio limitado.
La eliminación del fosgeno del aire ambiente se produce por
descomposición heterogénea (catálisis superficial) y por hidrólisis
lenta en fase gaseosa. El fosgeno es transportado a larga distancia, y
se cree que su difusión de la troposfera a la estratosfera acelera su
degradación fotolítica.
1.4 Niveles medioambientales y exposición humana
La exposición humana tanto en la población general como en el
entorno ocupacional se produce fundamentalmente por inhalación.
Se estima que la concentración promedio de fosgeno en el aire
ambiente está comprendida aproximadamente entre 80 y 130 ng/m3,
aunque hay pocos datos disponibles. Dada la diversidad de las
prácticas de higiene industrial seguidas en todo el mundo, es
imposible facilitar una cifra para los trabajadores que fabrican o
usan fosgeno o para los bomberos. Actualmente los valores umbral de
exposición (promedio ponderado en función del tiempo) en 15 países
están comprendidos entre 0,4 y 0,5 mg/m3.
No se ha informado sobre las concentraciones de fosgeno en el
agua, el suelo y los alimentos.
1.5 Cinética y metabolismo
Hay muy pocos datos sobre la absorción, el metabolismo, la
distribución y el destino del fosgeno. La principal vía de exposición
es la inhalación; el gas penetra en los tejidos del tracto
respiratorio, y sólo una mínima parte llega a distribuirse en el
organismo. Su muy breve vida media (0,026 segundos) en soluciones
acuosas evita que sea retenido significativamente por el organismo. No
se ha publicado información alguna sobre el metabolismo del fosgeno.
Sus productos hidrolíticos, p.ej. el ácido clorhídrico y el dióxido de
carbono, son eliminados por el organismo mediante los procesos
fisiológicos normales.
El fosgeno debe su toxicidad a la acilación de las proteínas, así
como a la generación de ácido clorhídrico. La acilación afecta a los
grupos amino, hidroxilo y sulfhidrilo de las proteínas, lo que da
lugar a una notable inhibición de varias enzimas relacionadas con el
metabolismo energético y a la descomposición de la barrera
sangre:aire.
1.6 Efectos en animales de laboratorio y en sistemas de
prueba in vitro
1.6.1 Exposiciones únicas y de corta duración
En todas las especies estudiadas el pulmón es el principal órgano
blanco. La (CT)L50 varía entre 900 mg/m3-min (225 ppm-min) en el
ratón y 1920 mg/m3-min (480 ppm-min) en el cobayo. Se ha informado de
una (CT)L50 de 1000 mg/m3-min (250 ppm-min) en el mono. En todas las
especies la manifestación patológica característica es la aparición
retardada sintomática de edema pulmonar, que depende de la dosis. Los
cambios anatomopatológicos observados en los bronquiolos terminales y
en los alveolos a bajas concentraciones son típicos de los irritantes
pulmonares, mientras que a exposiciones altas se produce edema, lo que
interfiere en el intercambio gaseoso y conduce a la muerte.
No se ha publicado ningún estudio sobre la exposición a largo
plazo al fosgeno.
Un estudio efectuado en ratas reveló que una sola exposición a
una concentración de 2 mg/m3 de fosgeno durante 4 horas puede
provocar una disminución de la inmunocompetencia pulmonar, a juzgar
por la actividad citotóxica natural de las células pulmonares. No se
observó ningún efecto a niveles de exposición de 0,4 mg/m3 mantenidos
durante 4 horas.
Se han publicado otros dos estudios sobre los efectos de
exposiciones únicas de fosgeno en la inmunocompetencia pulmonar de la
rata y del ratón. En ratas infectadas por el virus de la gripe tras 4
horas de exposición a 4 mg/m3 se observó que la concentración del
virus se había multiplicado por diez un día después de la infección,
manteniéndose significativamente elevados los niveles durante 4 días.
Además, en ratas expuestas a concentraciones de fosgeno comprendidas
entre 0,2 y 4 mg/m3 durante 4 horas se detectó una disminución
considerable de la prostaglandina E2 y de los leucotrienos a partir de
0,4 mg/m3, y una disminución del número de macrófagos alveolares y un
aumento del número de neutrófilos con 0,4 mg/m3. En un ensayo de
resistencia en que se expuso a ratones a niveles de fosgeno
comprendidos entre 0,04 y 0,4 mg/m3 durante 4 horas se observó un
aumento de la mortalidad por Streptococcus zooepidemicus o un
aumento del número de tumores pulmonares melanomatosos B16/BL6 a
niveles de 0,1 mg/m3 o superiores. La eliminación bacteriana pulmonar
se redujo en ratas expuestas a 0,4 mg/m3 (0,1 ppm) de fosgeno durante
6 horas o a 0,4 mg/m3 (0,1 ppm) durante 6 horas/día y 5 días/semana
por espacio de 4 a 12 semanas. Este efecto se reveló reversible tras
la terminación de la exposición.
1.6.2 Efectos no pulmonares
La exposición al fosgeno puede causar irritación de los ojos y de
la piel. No se ha hallado en las publicaciones ningún estudio
referente al potencial de sensibilización del fosgeno.
No se dispone de datos sobre los efectos del fosgeno en la
reproducción y el desarrollo.
No se dispone tampoco de datos adecuados para evaluar la
mutagenicidad o carcinogenicidad del fosgeno.
1.7 Efectos en el hombre
El órgano blanco en el hombre, como en los animales de
laboratorio, es el pulmón. Se han descrito tres fases
clinicopatológicas características tras la exposición a niveles de
fosgeno comprendidos entre 120 y 1200 mg/m3-min. La fase inicial
consiste en la aparición de dolor en los ojos y la garganta y de una
sensación de opresión torácica, a menudo con disnea, sibilancias y
tos; puede haber asimismo hipotensión, bradicardia y, rara vez,
arritmias sinusales. La que sigue a continuación es la fase latente,
porque es a menudo asintomática; puede durar hasta 24 horas según el
nivel y duración de la exposición. En la tercera fase puede aparecer
edema pulmonar, de consecuencias eventualmente mortales.
Las poblaciones expuestas al fosgeno tras accidentes industriales
han sufrido una amplia variedad de síntomas, incluidos cefaleas,
náuseas, tos, disnea, fatiga, dolor faríngeo, opresión y dolor
torácicos, dolor ocular intenso y lagrimeo grave. En un estudio se
observó edema pulmonar tras una fase latente de 48 horas.
Se han estudiado los efectos de la exposición al fosgeno a largo
plazo en tres grupos de trabajadores de dos instalaciones: una planta
de producción de fosgeno y un centro de procesamiento de uranio. En
los dos casos el muestreo del aire y la vigilancia del personal se
hicieron sólo de forma limitada, y únicamente se efectuaron
estimaciones de la exposición de los trabajadores.
El estudio de los registros médicos de los 326 trabajadores de la
planta de producción de fosgeno potencialmente expuestos a este
producto (concentraciones entre indetectables y de 0,5 mg/m3, valor
éste superado sólo esporádicamente; promedio: 0,01 mg/m3) no reveló
ni problemas pulmonares crónicos ni una mayor mortalidad por
enfermedades respiratorias en comparación con un grupo de 6228
controles. No obstante, la falta de datos de que adolece el informe en
lo que respecta a la exposición y a los efectos hace difícil extraer
conclusiones firmes del estudio.
Se estudió a dos grupos de trabajadores de la planta de
procesamiento de uranio: una muestra transversal de 699 trabajadores
de los más de 18 000 empleados durante el periodo estudiado,
potencialmente expuestos a niveles de fosgeno inferiores a 0,4 mg/m3
(con 4 ó 5 exposiciones fugaces a niveles > 4 mg/m3 cada día), y un
grupo de 106 trabajadores que se habían visto implicados en accidentes
y expuestos a niveles > 200 mg/m3-min. En el grupo expuesto
crónicamente a bajas concentraciones de fosgeno el análisis de los
certificados de defunción no mostró una mayor mortalidad por todo tipo
de causas o por enfermedad respiratoria o cáncer pulmonar. En el grupo
afectado por accidentes químicos no se registró tampoco ningún aumento
de las defunciones por todo tipo de causas; no hubo muertes por cáncer
pulmonar, pero sí un ligero incremento del número de defunciones por
enfermedades respiratorias. Habida cuenta tanto de la escasez de datos
sobre la exposición como de la metodología del estudio, sus
conclusiones tienen un valor limitado.
1.8 Efectos en otros organismos en el laboratorio y sobre el terreno
No se han publicado datos sobre los efectos del fosgeno en
organismos en el medio ambiente.
The Task Group members serve as individual scientists, not as
representatives of any organization, government or industry. Their
function is to evaluate the accuracy, significance and relevance of
the information in the document and to assess the health and
environmental risks from exposure to the chemical. A summary and
recommendations for further research and improved safety aspects are
also required. The composition of the Task Group is dictated by the
range of expertise required for the subject of the meeting and by the
need for a balanced geographical distribution.
The three cooperating organizations of the IPCS recognize the
important role played by nongovernmental organizations.
Representatives from relevant national and international associations
may be invited to join the Task Group as observers. While observers
may provide a valuable contribution to the process, they can only
speak at the invitation of the Chairperson. Observers do not
participate in the final evaluation of the chemical; this is the sole
responsibility of the Task Group members. When the Task Group
considers it to be appropriate, it may meet in camera.
All individuals who as authors, consultants or advisers
participate in the preparation of the EHC monograph must, in addition
to serving in their personal capacity as scientists, inform the RO if
at any time a conflict of interest, whether actual or potential, could
be perceived in their work. They are required to sign a conflict of
interest statement. Such a procedure ensures the transparency and
probity of the process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, it then goes for language editing, reference checking, and
preparation of camera-ready copy. After approval by the Director,
IPCS, the monograph is submitted to the WHO Office of Publications for
printing. At this time a copy of the final draft is sent to the
Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern for
health or environmental effects of the agent because of greater
exposure; an appreciable time period has elapsed since the last
evaluation.
All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR PHOSGENE
Members
Dr D. Anderson, British Industry Biological Research Institute (BIBRA)
Toxicology International, Carshalton, Surrey, United Kingdom
Dr R. Chhabra, Environmental Toxicology Program, Toxicology Branch,
National Institute of Environmental Health Sciences, Research
Triangle Park, North Carolina, USA
Dr H. Ellisa, Epidemiology Department, Rohm & Haas, Bristol,
Pennsylvania, USA
Dr B. Gilbert, FarManguinhos, FIOCRUZ, Institute of Technology and
Pharmacology, Ministry of Health, Manguinhos, Rio de Janeiro, Brazil
( Chairman)
Professor M. Jakubowski, Occupational and Environmental Hygiene
Division, Nofer Institute of Occupational Medicine, Lodz, Poland
Dr S.K. Kashyap, National Institute of Occupational Health, Meghani
Nagar, Ahmedabad, India ( Vice-chairman)
Dr R. Liteplo, Environmental Health Directorate, Health Protection
Branch, Environmental Health Centre, Tunney's Pasture, Ottawa,
Ontario, Canada
Dr E. E. McConnell, Laurdane Estates, Raleigh, North Carolina, USA
( Co-rapporteur)
Dr H. Naito, Ibaraki Prefecture University of Health Sciences,
Amimachi, Inashikigun, Ibaraki, Japan
__________
a Invited, but unable to attend.
Dr W. Popp, Universitatsklinikum Essen, Institute for Health and
Occupational Medicine, Essen, Germany
Dr R. Sram, Laboratory of Genetic Ecotoxicology, Institute of
Experimental Medicine, Videnska, Prague, Czech Republic
Dr Shou-Zheng Xue, Toxicology Programme, Shanghai Medical University,
Shanghai, People's Republic of China
Secretariat
Dr G. C. Becking, Team Leader, IPCS/IRRU, World Health Organization,
Research Triangle Park, North Carolina, USA
Ms R. Gomes, Health Canada, Environmental Health Directorate, Tunney's
Pasture, Ottawa, Ontario, Canada ( Co-rapporteur)
ENVIRONMENTAL HEALTH CRITERIA FOR PHOSGENE
A WHO Task Group on Environmental Health Criteria for Phosgene
and Selected Chloroalkyl Ethers met at the British Industrial
Biological Research Association (BIBRA) Toxicology International,
Carshalton, Surrey, United Kingdom, from 18 to 23 March 1996. Dr D.
Anderson opened the meeting and welcomed the participants on behalf of
the host institute. Dr G.C. Becking, IPCS, welcomed the participants
on behalf of Dr M. Mercier, Director of the IPCS, and the three
cooperating organizations (UNEP/ILO/WHO). The Task Group reviewed and
revised the draft criteria monograph and made an evaluation of the
risks for human health and the environment from exposure to phosgene.
Financial support for this Task Group was provided by the United
Kingdom Department of Health as part of its contribution to the IPCS.
Dr E.E. McConnell, Raleigh, North Carolina, USA, prepared the
first draft of this monograph. The draft reviewed by the Task Group,
which contained the comments received following circulation of the
draft monograph to the IPCS Contact Points for Environmental Health
Criteria monographs, was prepared by the Secretariat.
Dr G.C. Becking (IPCS, Central Unit, Inter-regional Research
Unit) and Dr P.G. Jenkins (IPCS, Central Unit, Geneva) were
responsible for the overall scientific content and technical editing,
respectively.
The efforts of all who helped in the preparation of the document
are gratefully acknowledged.
ABBREVIATIONS
CI confidence interval
L(CT)50 median lethal concentration-time product
LFP lavage fluid protein
MDI methylene-diphenyl diisocyanate
Nk natural killer
OES occupational exposure standard
PMN polymorphonuclear leukocyte
ppb parts per billion
ppm parts per million
ppt parts per trillion
PVC polyvinyl chloride
SMR standardized mortality ratio
TDI toluene diisocyanate
TLV threshold limit value
TWA time-weighted average
1. SUMMARY
1.1 Identity, physical and chemical properties, and
analytical methods
Phosgene is a highly reactive colourless gas at room temperature
and ambient pressure, and has a suffocating odour similar to mouldy
hay. The odour may be detected between 1.6 and 6 mg/m3.
Analytical methods are available for the detection of phosgene in
air and for use in industrial hygiene programmes that measure total
dose (e.g., paper tape monitors).
1.2 Uses and sources of human and environmental exposure
More than 99% of the phosgene produced is used on-site in closed
systems. It is produced by reacting equimolar amounts of anhydrous
chlorine and carbon monoxide in the presence of a carbon catalyst.
World production has been estimated to be greater than 3 million
tonnes. Environmental phosgene levels arise from industrial
emissions and thermal degradation of some chlorinated solvents and
chlorinated polymers. However, a significant source of environmental
phosgene is the photochemical oxidation of chloroethylenes such a tri-
and tetraethylene.
1.3 Environmental transport, distribution and transformation
Because of its high reactivity, inter-compartmental transport of
phosgene is expected to be limited. Removal of phosgene from ambient
air occurs by heterogeneous decomposition (surface catalysis) and slow
gas-phase hydrolysis. Long-range transport takes place and diffusion
from the troposphere to the stratosphere is believed to lead to more
rapid photolytic degradation of phosgene.
1.4 Environmental levels and human exposure
Human exposure in both the general population and occupational
setting is primarily by inhalation.
The average level of phosgene in ambient air may range from
approximately 80 to 130 ng/m3 although few data are available. In
view of the varied industrial hygiene practices worldwide it is
impossible to give an exposure figure for workers manufacturing or
using phosgene or for fire-fighters. At present the Threshold Limit
Values (time-weighted average) in 15 countries range from 0.4 and
0.5 mg/m3.
Levels of phosgene in water, soil and food have not been
reported.
1.5 Kinetics and metabolism
There are very few data on the absorption, metabolism,
distribution and fate of phosgene. The primary route of exposure is by
inhalation, the gas penetrates into the tissues of the respiratory
tract, and so only minimal amounts of phosgene are distributed in the
body. The very short half-life (0.026 seconds) in aqueous solutions
precludes a significant retention of phosgene in the body. No
information on the metabolism of phosgene has been reported. The
hydrolyic products of phosgene, i.e. hydrochloric acid and carbon
dioxide, are disposed of by the body through normal physiological
processes.
Phosgene exerts its toxicity through acylation of proteins, as
well as through the production of hydrochloric acid. The amino,
hydroxyl and sulfhydryl groups in the proteins appear to be the target
for acylation leading to marked inhibition of several enzymes related
to energy metabolism and a breakdown of the blood:air barrier.
1.6 Effects on experimental animals and in vitro test systems
1.6.1 Single and short-term exposures
In all species studied, the lung is the major target organ. The
L(CT)50 varies from 900 mg/m3-min (225 ppm-min) in the mouse to
1920 mg/m3-min (480 ppm-min) in the guinea-pig. An L(CT)50 of
1000 mg/m3-min (250 ppm-min) was reported in the monkey. In all
species the characteristic pathological feature is the delayed
clinical manifestation of pulmonary oedema, which is dose-dependent.
Pathological changes in the terminal bronchioles and alveoli at low
concentrations are typical of a pulmonary irritant, whereas at higher
exposures pulmonary oedema occurs, leading to interference with gas
exchange and death.
No long-term exposure studies of phosgene have been reported.
One study in rats showed that a single phosgene exposure of
2 mg/m3 for 4 h can result in decreased pulmonary immunocompetence as
measured by the natural killer activity of pulmonary cells. No
effects were seen at an exposure level of 0.4 mg/m3 for 4 h.
Two other studies of the effects of single exposures of phosgene
on pulmonary immunocompetence in rats and mice have been reported. In
rats infected with influenza virus after a 4-h exposure to 4 mg
phosgene/m3, there was a 10-fold increase in viral titre 1 day
post-infection, which remained significantly elevated for 4 days.
Furthermore, in rats exposed to phosgene levels between 0.2 and
4 mg/m3 for 4 h, a marked decrease in prostaglandin E2 and
leukotrienes was noted at exposure levels of 0.4 mg/m3 or more, with
a decrease in the number of alveolar macrophages and an increase in
the number of neutrophils observed at 0.4 mg/m3. In a host-
resistance assay, where mice were exposed to levels of phosgene
between 0.04 and 0.4 mg/m3 for 4 h, an increase in mortality from
Streptococcus zooepidemicus infection or an increased number of
B16/BL6 melanoma lung tumours was noted at levels of 0.1 mg/m3 or
more. Pulmonary bacterial clearance was reduced in rats exposed to
0.4 mg/m3 (0.1 ppm) phosgene for 6 h or to 0.4 mg/m3 (0.1 ppm) for
6 h/day, 5 days/week for 4 to 12 weeks. This effect was reversible
following termination of exposure.
1.6.2 Non-pulmonary effects
Phosgene exposure can result in eye and skin irritation. Studies
concerning the sensitization potential of phosgene have not been found
in the literature.
No data are available on the reproductive and developmental
effects of phosgene.
No adequate data are available for the assessment of the
mutagenicity or carcinogenicity of phosgene.
1.7 Effects on humans
The target organ in humans, as in experimental animals, is the
lung. After exposure to phosgene levels between 120 and
1200 mg/m3-min, three distinct clinicopathological phases have been
reported. The initial phase consists of pain in the eyes and throat
and tightness in the chest, often with shortness of breath, wheezing
and coughing; hypotension, bradycardia and rarely sinus arrhythmias
can occur. The second or latent phase, which is often asymptomatic,
can last as long as 24 h depending upon the level and duration of
exposure. In the third phase, pulmonary oedema may develop, leading
to death in some cases.
Populations exposed to phosgene after industrial accidents have
reported a wide variety of symptoms, including headache, nausea,
cough, dyspnoea, fatigue, pharyngeal pain, chest tightness and pain,
intense pain in the eye and severe lacrimation. In one study
pulmonary oedema occurred after a latent phase of 48 h.
The effects of long-term exposure to phosgene have been studied
in three groups of workers at two facilities, i.e., a phosgene
production plant and an uranium processing facility. In both
facilities, only limited air sampling or personal monitoring was
carried out, and worker exposures were only estimates.
An examination of the medical records of all 326 workers in the
phosgene production facility who were potentially exposed to phosgene
(up to 0.5 mg/m3, with some excursions above this value; average
0.01 mg/m3) did not yield any chronic lung problems or increased
mortality from respiratory disease compared to a group of 6228
controls. However, the lack of detail in the report on both exposure
and effects makes it difficult to draw firm conclusions from this
study.
Two groups of workers were studied at the uranium processing
plant: a cross-section of 699 workers from the over 18 000 employed
during the period, with potential exposure to phosgene levels below
0.4 mg/m3 (and 4 or 5 daily short-term excursions to > 4 mg/m3);
and a group of 106 workers known to have been involved in accidents
and exposed to levels above 200 mg/m3-min. In the group exposed
chronically to low levels of phosgene, an examination of death
certificates did not indicate an increased mortality from all causes
or from respiratory disease or lung cancer. In the group involved in
chemical accidents no increase in deaths from all causes was reported.
There were no lung cancer deaths but there was a slight increase in
the number of deaths from respiratory diseases. In view of the lack
of exposure data and the methodological characteristics of this study
the conclusions regarding the chronic effects of phosgene that can be
drawn are limited.
1.8 Effects on other organisms in the laboratory and field
No information concerning the effects of phosgene on organisms in
the environment has been reported.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL METHODS
2.1 Identity
Molecular formula: COC12
Chemical structure: CI
\
C = O
/
CI
Common Synonyms: carbonic acid dichloride, carbonyl chloride,
chloroformyl chloride, carbon oxychloride
IUPAC and CAS names: carbonic dichloride
CAS Registry number: CAS 75-44-5
RTECS Registry number: SY5600000
UN Transport number: 1076
2.2 Physical and chemical properties
Phosgene is a colourless nonflammable gas at room temperature and
ambient pressure. It has a suffocating odour and a smell reminiscent
of mouldy hay (Budavari, 1996). The recognition of the odour of
phosgene occurs at levels > 6 mg/m3 (1.5 ppm), although some
trained workers are capable of perceiving the odour at a level of
0.4 mg/m3 (0.1 ppm). The physical and chemical properties are
summarized in Table 1.
Table 1. Physical and chemical properties of phosgenea
Colour colourless
Relative molecular mass 98.92
Physical state gas
Melting point -127.8°C
Boiling point 7.56°C
Vapour pressure (20°C) 161.6 kPa
Relative vapour density (air = 1)3.42
Relative density, 20°C (water = 1)1.4
Solubility in water slight, reacts with water
Solubility in organic solvents reacts with ethanol, very
soluble in benzene,
toluene, acetic acid,
and most liquid
hydrocarbons
a From: Schneider & Diller (1989); Verschueren (1983);
Budavari (1996)
2.3 Analytical methods
A number of techniques may be used to determine phosgene
concentrations in air. These include passive dosimetry (Moore &
Matherne, 1981; Mathern et al., 1981), manual colorimetry (NIOSH,
1976), automated colorimetry (US EPA, 1986; Dangwal, 1994), gas
chromatography (Singh, 1976; Tuggle et al., 1979), infrared
spectroscopy (Esposito, 1977) and ultraviolet spectrophotometry
(Crummett & McLean, 1965). In addition, paper tape monitors capable
of detecting 5 µg/m3 have been described (Hardy, 1982). A summary of
these methods is presented in Table 2.
Table 2. Sampling and analysis of phosgene in aira
Sampling Analytical methodb Limit of detection Sample Comments Reference
methodb (range) size
Passive Direct reading (8-400 mg/m3-min) - Concentration- Moore &
dosimetry colorimetric reaction time relationship Matherne
with NBP in breathing zone (1981)
Air through Measure colour at (0.2 - 100 mg/m3) 1 litre/min Too slow NIOSH (1976)
impinger 475 nm for 25 min response for
containing DEP continuous
solution of NBP monitoring
and BA
Air bubbled into Automated colorimetry 0.004 mg/m3 1 litre/min Response time of US EPA (1986)
flowing stream of at reagent flow rate of (0-4 mg/m3) for 20 min 20 min too long
NBP-BA-DEP 0.2 ml/min for continuous
monitoring
Air through Derivative determined 0.04 mg/m3 1 litre air Extremely Wu & Gaind
impinger by reverse-phase HPLC sample sensitive but (1993)
containing needs highly
tryptamine trained staff and
equipment
Table 2 (contd).
Sampling Analytical methodb Limit of detection Sample Comments Reference
methodb (range) size
Direct sample Gas-chromatography 0.5 ml Extremely Singh (1976)
injection, - electron capture < 0.08 mg/m3 injection sensitive, needs Tuggle et al.
continuous - aluminium columns < 0.08 - 20 mg/m3 highly trained (1979)
sampling didecyl phthalate on staff and
chromosorb p equipment
Air is drawn Infrared spectroscopy, 0.1 mg/m3 2 to 5 Can be used as a Esposito et al.
directly into comparison of (0.1 to 1200 litre/min continuous (1977)
spectrophotometer absorbance at 11.8 µm mg/m3) monitor for
and reference ambient levels of
wavelength of 11.2 µmn phosgene
using 20 m variable
path length cell
a Proper medical treatment for those exposed to phosgene will depend on the concentration and length of exposure. Therefore,
any procedure used for monitoring ambient and workplace levels must give information on both parameters, preferably on a
continuous basis, since a latent period of 2-24h may occur between exposure and any warning symptoms.
b NBP = 4,4 -nitrobenzyl pyridine
DEP = diethyt phthalate
BA = N-benzylalanine
HPLC = high performance
2.4 Conversion factors
1 ppm = 4.05 mg/m3
1 mg/m3 = 0.25 ppm
at 25°C and 101.3 kPa
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Phosgene is not known to occur naturally.
3.2 Anthropogenic sources
3.2.1 Production levels and processes
Phosgene is produced by reacting equimolar amounts of anhydrous
chlorine and carbon monoxide in the presence of a carbon catalyst
(Schneider & Diller, 1989). The great majority is used directly in a
closed system.
It is difficult to give accurate production figures because more
than 99% of phosgene production is for on-site use (Schneider &
Diller, 1989). However, phosgene is manufactured in most
industrialized countries. Approximately 37% of the world's production
is in the USA (about 1 million tons); in 1989 European phosgene
production was approximately 1.2 million tonnes (Schneider & Diller,
1989).
3.2.2 Environmental processes
Phosgene in ambient air may arise from three sources:
a) direct emissions during its manufacture, handling, use and
disposal;
b) thermal decomposition, in the presence of air, of chlorinated
hydrocarbons, e.g., solvents such as chloroform, methylene chloride
(Snyder et al., 1992) and 1,2-dichloropropane (IPCS, 1993) and
polymers such as polyvinyl chloride (PVC);
c) photooxidation of chlorinated hydrocarbons, particularly
chloroform and the chloroethylenes (Rinzema, 1971; Singh, 1976; Gay et
al., 1976; Birgesson, 1987).
The thermal degradation of chlorinated hydrocarbons can occur as
a result of combustion of these materials during waste disposal, in
fires, and during welding in situations where PVC plastics are
degraded (Rinzema, 1971; Birgesson, 1982). Firefighters and welders
are at particular risk from these sources of phosgene.
A significant contribution to ambient air levels of phosgene is
the photooxidation of chloroethylenes, particularly tri- and tetra-
chloroethylene (Singh, 1976; Gay et al. 1976). It has been estimated
that such reactions may result in the worldwide formation of 350 000
tonnes of phosgene per year (Singh, 1976).
3.2.3 Uses
Initially important as an agent of chemical warfare, phosgene is
now widely used as a chemical intermediate, most often at the point of
production. The major use is in the production of aromatic
diisocyanates such as methylene diphenyl diisocyanate (MDI) and
toluene diisocyanate (TDI), which are used to produce polyurethane
foams and other polymers. Worldwide about 80% of phosgene is used for
TDI and MDI production. Other major uses of phosgene include the
production of polycarbonate, aliphatic diisocyanates, monoisocyanates
and chloroformic esters and urethanes (Schneider & Diller, 1989;
Borak, 1991). Phosgene is also used in the manufacture of some
agrochemicals, in the pharmaceutical industry, and metallurgy (US NLM,
1995).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
Phosgene can enter the atmosphere in the form of industrial
emissions or from the degradation of chlorohydrocarbons (section
3.2.2). Detectable levels have been found in ambient air (section
5.1.1). However phosgene is unlikely to be detectable in soil and
vegetation owing to heterogeneous decomposition (section 4.2).
In water, phosgene is rapidly degraded to hydrochloric acid and
carbon dioxide (Butler & Snelson., 1979), the half-life in aqueous
solution being 0.026 seconds (Manogue and Pigford, 1969).
In the atmosphere, even at high humidity levels, phosgene is only
slowly decomposed (Noweir et al., 1973; US EPA, 1986). The half-life
by homogeneous gas-phase hydrolysis of 4 g /m3 phosgene (1 ppb) in
nitrogen (at sea-level pressure, 25 C and with water vapour at a
pressure of 10 Torr) has been calculated to be 113 years (range 20 to
630 years) (Butler & Snelson, 1979). Reaction rates with activated
oxygen and hydroxyl radicals are also slow (Singh, 1976). Phosgene
is, therefore, likely to be persistent in the atmosphere and subject
to long-range transport. Diffusion to the stratosphere leads to more
rapid degradation by photolysis (Singh, et al., 1977).
4.2 Abiotic degradation
Reaction with molecules having an active hydrogen atom (e.g.,
water, primary and secondary alcohols, thiols and amines) does occur,
forming hydrochloric acid, carbon dioxide, and carbonic acid
derivatives (Butler & Snelson, 1979; Schneider & Diller, 1989), but
phosgene reacts very slowly in the gas phase with photochemically
produced hydroxyl radicals (Singh, 1976).Removal of phosgene from
ambient air occurs by two major pathways, i.e. heterogeneous
decomposition (Noweir et al., 1973) and liquid-phase hydrolysis (Singh
et al., 1977). At normal ambient temperatures the gas-phase
hydrolysis is the major pathway for phosgene degradation (Singh et
al., 1977). However, even contact with soil particles and vegetation
at ambient temperatures enhances the rate of phosgene degradation
(Noweir et al., 1973).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
It has been suggested that the primary source of atmospheric
levels of phosgene is from the thermal degradation and photo-
degradation of chlorinated solvents such as tri- and
tetrachloroethylene and PVC (Singh, 1976). Direct emissions from the
production and use of the chemical play a minor role and would most
likely affect the air levels only near the factory. Phosgene levels
in ambient air at four locations in California, USA, were reported by
Singh et al. (1977). Multiple samples (10 to 257) were taken on a
24-h basis from a single location in each area. In one rural area an
average level of 87 ng/m3 (21.7 ppt) was reported. The average
levels found in three urban areas were 117 ng/m3 (29.3 ppt),
121 ng/m3 (30.3 ppt) and 129 ng/m3 (31.8 ppt), with a peak level
in one sample of 244 ng/m3 (61.0 ppt). In three other cities in the
USA the average level of phosgene in ambient air was reported to be less
than 80 ng/m3 (20 ppt) (Singh et al., 1981).
5.1.2 Water
There are no data on levels of phosgene in water, since
hydrolysis precludes significant accumulation in this medium.
5.1.3 Soil
Data on levels of phosgene in soil are not available, since rapid
breakdown on contact with solid surfaces and moisture prevents a
significant accumulation in this medium (Noweir et al., 1973).
5.1.4 Food and feed
Although no data are available, the lack of stability in the
presence of liquid-phase water, solid surfaces, and alcohols and or
amines in foods makes contamination of food by phosgene unlikely (see
sections 4.1 and 4.2).
5.2 General population exposure
The general population is exposed to only very low levels (in the
ng/m3 range) of phosgene and this is almost entirely via contaminated
urban air. The origin of this phosgene is from decomposition of other
chlorinated compounds or, in isolated circumstances, from the
emissions of an industrial enterprise making or using phosgene without
carrying out appropriate industrial hygiene practices. Based on the
range of average concentrations (< 80-129 ng/m3) given in section
5.1.1, the estimated total daily intake of phosgene may range from
< 1.6 to 2.6 g, assuming a daily respiratory intake of 20 m3 air.
Much higher levels of phosgene exposure are possible during home use
of chemicals such as methylene chloride under conditions where the
temperature is sufficiently high to lead to degradation of this
chemical (Snyder et al., 1992).
5.3 Occupational exposure
5.3.1 Manufacture and use
Occupational exposure limits (8- or 10-h TLV) in some 15
countries range between 0.4 and 0.5 mg/m3 (ILO, 1991). Based upon
animal data, the United Kingdom has proposed an OES (8-h TWA) of
0.08 mg/m3 (0.02 ppm) and an STEL of 0.24 mg/m3 (0.06 ppm) (HSE,
1995). It is difficult to report actual values in individual
factories worldwide since levels will vary greatly depending upon the
level of industrial hygiene practiced in any particular factory.
However, even early monitoring reports indicated that exposures were
generally below the recommended TLV. In a few cases exposure levels
above 0.4 mg/m3 have been reported (Levina et al., (1966). In a
factory manufacturing phosgene, personal samplers detected levels up
to 0.08 mg/m3 (average 0.012 mg/m3), whereas fixed position samplers
(total of 56) showed levels between non-detectable and 0.52 mg/m3 in
51 samples, with excursions in a few samples to about 71 mg/m3
(NIOSH, 1976). More recent monitoring data are lacking.
5.3.2 Non-manufacturing occupations
Firefighters and workers engaged in welding and building trades
are at risk from the phosgene formed by the thermal degradation of
chlorinated hydrocarbons and PVC. The pyrolysis of Freon present in
commercial refrigeration units (Birgesson, 1982), tri- and
tetrachloroethylene (Rinzema, 1971; Andersson et al., 1975), PVC
(Brown & Birky, 1980) and methylene chloride (Snyder et al., 1992)
have all been shown to result in toxic levels of phosgene. However,
actual levels in the area of work or the breathing zone were not
quantified in these studies.
6. KINETICS AND METABOLISM
Because of the physico-chemical properties of phosgene, the
kinetics and metabolism of phosgene in animals should be similar if
not identical to those found in humans. However, because of the
highly toxic nature of phosgene, human experimental data appropriate
for use in this monograph does not exist.
6.1 Disposition of phosgene
There are very few data on absorption, metabolism, distribution
and fate of phosgene. The primary route of exposure is by inhalation.
The gas penetrates into the tissues of the respiratory tract, and so
only minimal amounts of phosgene are distributed in the body. The
very short half-life (0.026 seconds) in aqueous solutions precludes a
significant retention of phosgene in the body. No information on the
metabolism of phosgene has been reported. The hydrolytic products of
phosgene, i.e. hydrochloric acid and carbon dioxide, are disposed of
by the body through normal physiological processes (Manogue & Pigford,
1960; Thienes & Haley, 1972).
6.2 Reaction with body components
Apart from the formation of hydrochloric acid on contact, Cessi
et al. (1966) showed marked acylation of in vitro poly-L-lysine and
human albumin by phosgene. The reaction of phosgene with cysteine
yields 2-oxothiazolidine-4-carboxylic acid (Mansuy et al., 1977). The
same product is formed when chloroform (Pohl et al., 1977) or carbon
tetrachloride (Shah et al., 1979) is incubated with hepatic
microsomes.
7. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
7.1 Single and short-term inhalation exposures
For some end-points (e.g., death, lung damage), the effects of
phosgene exposure are dependent upon both the concentration and
duration of exposure; considered as a product of CT=K as stated in
Haber's law (the product of the concentration and time of exposure
required to produce a specific physiological effect is a constant).
Early workers validated this relationship using death as the
physiological end-point following acute and short-term exposures. The
validity of this concept for phosgene was tested in cats (Flury, 1921;
Flury & Zernick, 1931). Twenty cats were exposed to phosgene at
levels between 5 and 500 mg/m3 for periods of time between 0.5 and
120 min (CT values varied from 37.5 to 562 ppm-min). A plot of K
(death or survival) against T (abscissa) and C (ordinate) resulted in
an hyperbolic plot with marked deviations at both high and low
concentrations (Long & Hatch (1961). Similar results were obtained in
rats when the physiological effect was impaired gas exchange (Rinehart
& Hatch, 1964).
Based on reviews of available information (Coman et al., 1947;
Atherley, 1985) it was concluded that, for phosgene, effects
associated with the product of CT were reasonable constant only
within the middle range of concentrations (i.e., between 4 and
800 mg/m3), and for exposure times that negated the effect of an
animal holding its breath. Under these conditions and for these
effects, it was considered appropriate to express the actual dose of
phosgene, assuming equivalent respiratory volume, as CT. The CT
relationship probably does not apply to potential effects resulting
from long-term exposure to low levels of phosgene.
Single and short-term exposures are described together because of
the similar biological responses observed. Most of the numerous
animals studied are summarized elsewhere (US EPA, 1986) and
representative examples are shown (Table 3, Table 4).
In inhalation studies the lung is the primary target organ in all
species, and the characteristic pathological feature is the delayed
clinical manifestation of pulmonary oedema, which is dose dependent.
Other mucous membranes such as the eye can also be affected.
Underhill (1919, 1920) exposed dogs for 30 min to phosgene
concentrations between 176 and 480 mg/m3. At the lower
concentrations he reported that phosgene exposures resulted in
pathological lesions in the terminal bronchioles and alveoli, typical
for a pulmonary irritant. However, at the higher exposure levels
phosgene resulted in oedema leading to interference with gas exchange,
cyanosis and eventually death.
Table 3. Effects of phosgene after single exposure by inhalation
Speciesa Exposure Effects Reference
C x T C T
(mg/m3-min) (mg/m3) (min)
Rat 100 0.4 250 Widening of pulmonary intestices Diller et al. (1985)
Rat 144 144 0.4-380 Reduced pulmonary bacterial Yang et al. (1995)
clearance
Cat 150 10 15 Slight illness Flury (1921)
Mice, rats, 192 0.8 240 Increase in levels of lavage fluid Hatch et al.
hamsters protein (1986)
Rat 200 0.24 500 Increase in protein levels in Diller et al. (1985)
20 10 pulmonary lavage fluid
Rat 200 20 10 Initiation of pulmonary oedema Diller et al. (1985)
Rats (male) 240 1 240 Increase in levels of lavage fluid Currie et al.
protein. Increase in the percentage (1987a)
of polymorpho-nuclear leukocytes
(PMN)
Table 3 (contd).
Speciesa Exposure Effects Reference
C x T C T
(mg/m3-min) (mg/m3) (min)
Cat 440 44 10 L(CT)minimum Flury & Zernick
(1931)
Cat 450 10 45 L(CT)mimimum Flury & Zernick
(1931)
Guinea-pigs, 480 2.0 240 Increase in levels of lavage fluid Hatch et al.
rabbits protein (1986)
Rat 480 2 240 Increase of wet and dry lung weight Currie et al.
(1987a)
Rat 480 2 240 Decrease in pulmonary natural killer Burleson & Keys
activity (NK) (1989)
Cat 660 44 15 L(CT)100 Flury (1921)
Cat 720 12 60 L(CT)100 Flury (1921)
Mouse 900 60 15 L(CT)50 Cameron & Foss
(1941)
Table 3 (contd).
Speciesa Exposure Effects Reference
C x T C T
(mg/m3-min) (mg/m3) (min)
Monkey 1000 1000 1 L(CT)50 Diller & Zante
(1982)
Rat 1200 16 75 L(CT)50 Rinehart & Hatch
(1964)
Monkey 1320 440 3 L(CT)100 Winternitz et al.
(1920)
Dog 1800 180 10 L(CT)50 Diller & Zante
(1982)
Guinea-Pig 1920 128 15 L(CT)50 Underhill (1920)
Rat 4000 400 10 Ultrastructural changes in the Pawlowski &
bronchoalveolar region of lungs; Frosolono (1977)
cellular disruption and necrosis
Guinea-pig 6160 308 20 L(CT)100 Ong (1972)
Dog 8808 2936 3 L(CT)99 Coman et al.
(1947)
Table 3 (contd).
Speciesa Exposure Effects Reference
C x T C T
(mg/m3-min) (mg/m3) (min)
Sheep 13 300 1330 10 L(CT)50 Keeler et al.
(1990)
Rabbit 21 140 604 35 L(CT)99 Coman et al.
(1947)
a In most cases the sex of the animals was not specified
Necropsy of dogs that died shortly after single exposure to
phosgene showed frothy material around the mouth, engorgement of the
visceral vasculature (shock-like syndrome), and heavy wet congested
lungs (Winternitz et al., 1920). Microscopically, the lungs were
characterized by congestion and severe oedema. Proteinaceous fluid,
strands of fibrin, and leukocytes filled the alveoli. In dogs that
died 4 or more days after dosing, pulmonary infection (inflammation)
was the primary cause of death. Oedema, congestion and emphysema of
lesser severity were still present but there was also evidence of an
attempt to repair tissue. In dogs that died or were killed later (11
to 129 days), necropsy revealed varying degrees of lung collapse and
emphysema suggesting obliterative bronchiolitis. The epithelium of
the larger airways (trachea and bronchi) did not show evidence of
damage. The authors stated that the pathology of phosgene exposure
was similar in the goat, dog, monkey, rabbit, guinea-pig, rat and
mouse (Winternitz et al., 1920).
Concurrent studies in a different laboratory confirmed the above
results (Meek & Eyster, 1920). These authors documented a well-marked
succession of events after exposure of dogs for 30 min to phosgene
levels of 320-400 mg/m3. In the initial stage of exposure there is
direct damage to the epithelial cells lining the pulmonary airways,
with the distal ones showing the most damage. The cells are killed
(necrosis) and sloughed. This is immediately followed by effusion of
fluid into the affected airways (oedema). There is some damage to
erythrocytes in pulmonary capillaries, which aggregate, thus causing
occlusion. Gaseous exchange is interfered with, and death is the
result of hypoxia/anoxia. Other authors (Cameron & Courtice, 1946)
have also shown that pulmonary oedema is the primary cause of death in
several species after acute phosgene poisoning (440 mg/m3). The area
of lung affected appears to depend on the level of exposure. Gross et
al. (1965) postulated that at lower levels the alveolar bronchioles
and alveoli are the primary target tissues. At higher levels more
proximal respiratory tissues are at risk of developing lesions. The
relationship between the primary target site in the lung and dose of
phosgene was confirmed in rats (Diller et al., 1985). Changes within
the blood-air barrier (pulmonary oedema) were noted at phosgene levels
of 20 mg/m3 (5 ppm) or more and at durations of exposure of 10 min or
longer (50 ppm-min). The lowest dose of phosgene producing an
increase in protein levels in pulmonary lavage fluid was also
200 mg/m3 min (50 ppm-min), and that for the production of widening
within the pulmonary interstices was 100 mg/m3 min (25 ppm-min).
However, there was no apparent threshold of phosgene concentration for
these two parameters. Concentrations of phosgene studied were 0.4 to
20 mg/m3 (0.1 to 5 ppm). Changes noted at low concentrations (0.4 to
10 mg/m3; 0.1 to 2.5 ppm) were primarily located at the transition
from terminal bronchioles to the alveolar ducts, while at higher
concentrations (20 mg/m3, 5 ppm) damage to the alveolar pneumocytes
(type 1) was reported (Diller et al., 1985).
The earliest ultrastructural change observed in the
bronchoalveolar region of lungs of rats exposed to 400 mg/m3
(100 ppm) for 10 min was characterized by vesiculation of bronchiolar
epithelium immediately after exposure (Pawlowski & Frosolono, 1977).
This was followed, 30 min after the exposure, by extracellular
accumulation of serous fluid in interstitial spaces and alveoli. The
final events were cellular disruption and necrosis.
The extent of the long-term effects after acute exposure appears
to depend on the severity of the initial pathology (Coman et al.,
1947; Diller, 1985b).
The relative sensitivity of female mice and male hamsters,
rabbits, guinea-pigs and rats to 4-h phosgene exposures at
concentrations of 0.4, 0.8, 2 and 4 mg/m3 (0.1, 0.2, 0.5 and 1 ppm)
was studied by Hatch et al. (1986). As an indicator of phosgene-
induced pulmonary oedema, levels of lavage fluid protein (LFP) were
measured 18-20 h after exposure. Groups of seven or eight animals
were examined at each exposure level. Phosgene-induced changes in LFP
levels in mice, hamsters and rats occurred at phosgene levels of
0.8 mg/m3 (about 190 mg/m3 min) and above, whereas the minimal
effective dose in guinea-pigs and rabbits was 2 mg/m3 (about
480 mg/m3-min).
Rats were exposed 4 h/day, 5 days/week for 17 exposures at a
level of 0.5 or 1 mg/m3 (0.125 or 0.25 ppm) and killed on days 3, 7,
10, 13 or 17 or on days 2 or 20 after exposure. After day 7 the lung
wet weights were increased by 20-25% (p < 0.05 in the high-level
group). In the low-level group the lung wet weights increased with
exposure time but were significantly increased only at day 17. These
changes were paralleled by an increased activity of the pulmonary
glucose-6-phosphate dehydrogenase. The non-protein sulfhydryl content
in the lungs was elevated throughout exposure in both groups. All
deviations were reversible after exposure ceased. At day 17 in the
high-level group a moderate multifocal accumulation of mononuclear
cells in the walls of the terminal bronchioli and their adjacent
alveoli was observed. A minimal amount of type II alveolar cell
hyperplasia was also found in this region. Macrophages with
vacuolated cytoplasm were seen in the lumens of some alveolar ducts
and alveoli. The lesions in the lungs of the low-dose animals were
described as being minimal (Franch & Hatch, 1986). A no-observed-
effect-level could not be demonstrated in this study.
The effects of low-level acute exposure to phosgene in male rats
was also studied by Currie et al. (1987a), who exposed groups of adult
animals (250-300 g) for 4 h to 0.5 to 4 mg/m3 (0.125 to 1 ppm).
Dose-related changes in body weight, wet and dry lung weights, LFP,
total cell counts, and cell differentials were measured at the
conclusion of the exposure and 3 days after exposure, at least 10
animals being sacrificed each time. A dose-response relationship for
the measured parameters was noted. Both wet and dry lung weights
increased after exposure to 120 and 240 ppm-min, and an increase in
LFP was noted at > 60 ppm-min. The most sensitive cellular
indicator of phosgene pulmonary damage was the increase in the
percentage of polymorphonuclear leukocytes (PMN); there was a
significant increase at 60 ppm-min. Both PMN and LFP can be used as
sensitive indicators of pulmonary damage by phosgene after acute
exposure. All parameters returned to control levels 3 days after
exposure, indicating that the pulmonary damage within the dose-range
studied was reversible.
In Table 3, effects of inhalation exposure to phosgene have been
summarized according to the degree of exposure, expressed as
mg/m3-min. Pulmonary bacterial clearance appears to be the most
sensitive end-point for acute phosgene toxicity in rats (about
100 mg/m3-min). The lowest dose of phosgene that produced an
increase in protein levels in pulmonary lavage fluid and changes in
the blood-air barrier (pulmonary oedema) was 100 to 200 mg/m3-min.
This effect can be considered as the early critical effect of acute
exposure to phosgene. Data presented in Table 4 suggest that
evaluation of exposure according to Haber's law can be applied as the
basis for constructing dose-effect relationships only in the case of
acute relatively high exposures.
Studies of pulmonary physiology in animals mirror the
pathological observations (Gibbon et al., 1948; Boyd & Perry, 1960;
Long & Hatch, 1961; Rhinehart & Hatch, 1964). Progressive loss of
capacity for gas exchange is the initial and critical event. The
respiration rate is increased but there is increased resistance and
poor ventilation.
7.2 Skin and eye irritation; sensitization
Very little information on this subject is available. Skin
irritation is possible if concentrations are high enough, but the
hazard is minimal compared to the severe lung damage that can be
produced by much longer levels (Diller, 1985a; Borak, 1991). No
studies on sensitization have been reported. Eye irritation and
corneal oedema have been reported in dogs exposed to lethal
concentrations of phosgene (Winternitz et al., 1920).
Table 4. Toxicity of phosgene after repeated exposure by inhalation
Speciesa Exposure Effects Reference
Rat 0.4 mg/m3; 6 h/day, 5 days/week, Pulmonary bacterial clearance inhibited Selegrade et al.
4-12 weeks (1989)
Rat 0.5-1 mg/m3 4 h/day 5 days/week, 17 Lung wet weight increase (20-25%); GPDb Franch & Hatch (1986)
exposures activity increase; alveolar cell
hyperplasic; macrophages with vacuolated
cytoplasm
Guinea-pig 0.8 mg/m3 for 300 min daily, 5 days Pulmonary oedema in 70% of animals Cameron et al. (1942)
Cat 0.8 mg/m3 for 300 min daily, 5 days Pulmonary oedema in 70% of animals Cameron et al. (1942)
Mouse 4 mg/m3 for 300 min daily, 5 days L(CT)90 Cameron & Foss
(1941)
Rat 4 mg/m3 for 300 min daily, 5 days Pulmonary oedema in 80% of animals Cameron & Foss
(1941)
Rabbit 4 mg/m3 for 300 min daily, 5 days L(CT)20 Cameron & Foss
(1941)
Dog 96-160 mg/m3 for 30 min Up to 20-fold increase in airway Rossing (1964)
1-3 times per week, resistance
12 weeks
a Sex not specified b GPD= glucose-6-phosphate dehydrogene
7.3 Long-term exposure
Long-term exposure studies of phosgene have not been conducted.
However, there have been a limited number of studies on the effects of
phosgene following repeated exposures over periods of time (dosing 1
to 3 times per week for up to 12 weeks) (Clay & Rossing, 1964;
Rossing, 1964). Adult mongrel dogs (sex not identified) were exposed
to phosgene at concentrations between 96 and 160 mg/m3 for 30 min, 1
to 3 times per week. Rossing (1964) exposed 14 animals 3 times weekly
until increased airway resistance was noted, and then the frequency of
exposure was decreased to 1 or 2 times weekly for 12 weeks. Seven
animals died during the first 3 weeks of exposure and only three
animals survived the full 12 weeks; two of which were maintained for
12 weeks with further exposure. The lungs of all animals were
examined within 48 h after-exposure. After the initial inflammatory
reaction, the ensuing lesion consisted of chronic bronchiolitis and
emphysema that persisted for the duration of the exposure period.
After cessation of exposure, elastance dropped rapidly to normal, but
airway resistance was still elevated 11 weeks after exposure.In a
similar experiment, Clay & Rossing (1964) studied the development of
pulmonary emphysema in adult mongrel dogs after exposure to phosgene
at concentrations between 96 and 160 mg/m3 for 30 min at a rate of 1
to 3 exposures per week. Group size varied between four and seven
animals. The number of exposures varied between 1 and 25, and the
dogs were killed either immediately or up to 2 weeks after exposure.
However, in view of the low number of animals used, the experimental
design and the lack of reported dose-response information, these data
are difficult to use in assessing quantitatively the long-term risk to
humans from phosgene exposure.
7.4 Reproductive and developmental toxicity
No data were found on reproductive or developmental effects of
phosgene in experimental animals.
7.5 Mutagenicity and related end-points
No studies on the mutagenicity of phosgene have been reported.
7.6 Carcinogenicity
No adequate studies are available for the assessment of the
carcinogenicity of phosgene. In a review of the potential
carcinogenicity of 266 substances found in various workplaces, data
from one study involved 20 guinea-pigs and 20 rats that were exposed
by inhalation to phosgene for 24 and 18 months, respectively. No
pulmonary neoplasms were observed (Schepers, 1971), but information on
dosing regimen, sex or strain of animals was lacking.
7.7 Immunotoxicity
In one study, the immunotoxic effects in Fischer-344 male rats of
a single 4-h exposure to 0, 0.4, 2.0 or 4 mg phosgene/m3 were
reported (Burleson & Keys, 1989). As a measure of pulmonary
immunocompetence the authors measured the natural killer (NK)
activity of pulmonary cells on the day after phosgene exposures. At
exposures of 2 and 4 mg/m3 there was a significant decrease in
pulmonary NK activity, which was considered by the authors to be an
indication of decreased immunocompetence. At 4 mg/m3 this decrease
was still significant 4 days after exposure. A significant decrease
was also noted at 2 mg/m3 one day after exposure, but no effect was
seen at 0.4 mg/m3. Phosgene did not affect NK activity in blood
lymphocytes, but NK activity in the splenic cells was decreased 1 day
after exposure to 4 mg/m3. Effects on NK activity in lymphocytes and
splenic cells at other phosgene doses and days post-treatment were not
reported. Decreased immunocompetence in rats resulting from a 4-h
exposure to phosgene was reported by Ehrlich & Burleson (1991). After
male Fischer-344 rats (8-10 weeks old) were exposed to phosgene at
4 mg/m3 for 4 h, the animals were infected with a rat-adapted
influenza virus and the virus titre measured at 2 h and 1, 2, 3, 4, 5
and 7 days post-infection. The virus titre was measured in three
replicate experiments using three rats per group. Following an
initial decrease in the viral titre at 2 h after infection, the virus
titre increased by a factor of 10 on the day after infection and
remained significantly higher than in controls up to 4 days after
infection. The virus was cleared below detectable levels after 5 days
post-infection.
Bacterial clearance was assessed in male Fisher-344 rats exposed
to 0, 0.4 or 0.8 mg/m3 (0.1 or 0.2 ppm) phosgene for 6 h (Yang et
al., 1995). Immediately after exposure to phosgene the animals were
infected with Streptococcus zooepidemicus, and the number of
bacteria in the pulmonary lavage fluid was assessed up to 72 h later.
Pulmonary bacterial clearance was significantly reduced (p <0.05)
following exposure to 0.4 mg/m3 (0.1 ppm) phosgene (LOEL = 0.1 ppm).
Based on an analysis of other immunological parameters (e.g.,
pulmonary NK activity and pulmonary macrophage function), the authors
indicated that bacterial clearance appeared to be the most sensitive
end-point for acute phosgene toxicity in rats.
CD-1 mice were exposed to phosgene concentrations of 0.04 to
0.4 mg/m3 (0.01 to 0.1 ppm) for 4 h and infected with S. zoo-
epidimicus or inoculated with B16/BL6-melanoma tumour cells. At and
above 0.1 mg/m3 (0.025 ppm), mortality due to the infection with
S. zooepidimicus and the number of B16/BL6 melanoma tumours in the
lungs were both elevated. An 8-h exposure to 0.04 mg/m3 increased
the mortality due to S. zooepidimicus but not the number of B16/BL6
melanoma tumours (Selgrade et al., 1989).
F-344 rats were exposed to phosgene concentrations of 0.2 to
4 mg/m3 (0.05 to 1 ppm) and their lungs lavaged 0, 4, 20 or 44 h
later. At and above 0.4 mg/m3 the concentrations of prostaglandin E2
as well as of leukotrienes B4, C4, D4 and E4 were decreased by 29 to
69%. The eicosanoid concentrations after exposure to 0.4 and 1 mg/m3
returned to normal 44 h later. After exposure to 0.4 or 4 mg/m3, but
not to 0.2 mg/m3, the number of alveolar macrophages was decreased,
whereas the number of neutrophils was increased at 44 h after exposure
to 0.4 mg/m3, but not to 4 mg/m3 (Madden et al., 1991).
Pulmonary effects have also been observed in male Fisher-344 rats
exposed for a longer term to phosgene (Selgrade et al., 1995). Groups
of animals were exposed to 0, 0.4 or 0.8 mg/m3 (0, 0.1 or 0.2 ppm)
for 6 h/day, 5 days/week for 4 or 12 weeks, or to 2 mg/m3 (0.5 ppm)
for 6 h/day, 2 days/week for 4 or 12 weeks. When assessed immediately
after 4 or 12 weeks of exposure, pulmonary bacterial clearance was
inhibited following exposure to 0.4 mg/m3 (0.1 ppm) phosgene (LOEL =
0.1 ppm). This effect appeared reversible, since pulmonary bacterial
clearance was unchanged when assessed 4 weeks after a 12-week exposure
period. These subchronic effects on pulmonary bacterial clearance were
similar to those observed previously, following acute exposure to
phosgene (Yang et al., 1995). In animals administered the same dose of
phosgene, pulmonary bacterial clearance was more severely affected in
animals exposed to the higher concentration of this substance (i.e.,
2 mg/m3 (0.5 ppm), 6 h/day, 2 days/week, compared to 0.8 mg/m3
(0.2 ppm) 6 h/day, 5 days/week). A significant reduction in pulmonary
NK activity was observed following exposure to 2 mg/m3 (0.5 ppm).
7.8 Mechanism of toxicity
Although the exact mechanism of phosgene toxicity remains
unknown, it seems likely that the original hypothesis of Winternitz et
al. (1920), suggesting that hydrochloric acid was the causal agent for
the pulmonary effects noted, is incorrect. Current data indicate that
the effects from phosgene exposure result from the acylation of tissue
components, although the production of HCl may play a minor role,
particularly at high levels of exposure (Diller, 1985a). Nash &
Pattle (1971) studied the chemical reactivity of phosgene when bubbled
through aqueous solutions at various pH values, some containing
amines, phenoxide ions or sulfite. From these data it was concluded
that molecular phosgene could penetrate all layers of the blood-air
barrier, causing the observed pathology by reacting with chemical
groups in the cells. It was shown mathematically that insufficient
HCl to produce the observed effects could be generated under
physiological conditions and phosgene exposures as high as 100 mg/m3
(25 ppm).
Early evidence that acylation of amino, hydroxyl and sulfhydryl
groups was the major mechanism was reported by Potts et al. (1949).
Rats and mice exposed to 0.5 mg ketene/litre for 1.5 min showed
clinical signs and pathological lesions identical to those cause by
phosgene. Ketene is a known acrylating agent that does not break down
to a strong acid.
The effects on pulmonary ultrastructure and enzyme activities in
adult rats from exposure to phosgene for 10 min at 400 mg/m3
(100 ppm) were studied by Pawlowski & Frosolono (1977) and Frosolono &
Pawlowski (1977). Homogenates of the combined lungs from six to eight
rats were assayed for several enzyme activities in duplicate
immediately after exposure and at 30 and 60 min post-exposure. A
decrease in the enzymatic activity of 10-80% was noted at all time
periods for p-nitrophenyl phosphatase, cytochrome C oxidase, ATPase
and lactic dehydrogenase (Frosolono & Pawlowski 1977). Using the same
protocol Pawlowski & Frosolono (1977) examined the cascade of
ultrastructural changes in the terminal bronchiolar epithelium after
exposure. An immediate vesiculation of cells was followed by septal
extracellular oedema and, finally intracellular oedema, cell
disruption and necrosis. The authors suggested that the biochemical
changes preceded major ultrastructural changes in the alveolar region.
Currie et al. (1985) studied the effects on energy metabolism of
exposure to 4 mg/m3 (1 ppm) for 4 h (CT = 960 mg/m3-min) in rats.
An attempt was made to correlate the onset of pulmonary oedema with
alterations in energy metabolism. At the exposures studied, there was
a significant reduction in the respiratory control index, which
coincided with the highest level of percentage water in the lung. In
addition, a decrease in ATP concentration was noted. It was concluded
that reductions in ATP levels and Na-K-ATPase activity play a major
role in damage to the lung after phosgene exposure and prior to the
onset of oedema.
Studies by Currie et al. (1987b) have confirmed these findings at
lower doses. Rats were exposed for 4 h to 48, 120, 240, 480 or
960 mg/m3-min (12, 30, 60, 120 or 240 ppm-min). Decreased ATP
levels were noted prior to the onset of oedema at doses as low as
48 mg/m3-min (12 ppm-min) after exposure for 4 h. Further studies by
Frosolono & Currie (1985), using the same exposure regimen as Currie
et al. (1985) (i.e., 960 mg/m3-min), indicated that phosgene may
alter the level of pulmonary surfactant thus altering the homeostatic
mechanism for fluid balance in the lung.
Jaskot et al. (1991) studied the effect of inhaled phosgene on
lung anti-oxidant systems in Fischer-344 male rats. Levels of 0, 0.4,
1, 2 and 4 mg/m3 were administered for 4 h and a satellite group
received 1 mg/m3 for 8 h. Changes in glutathione (GSH) and
anti-oxidant associated enzymes (GSH peroxidase, GSH reductase,
glucose-6-phosphate dehydrogenase and superoxide dismutase) were
measured 0, 1, 2, 3 and 7 days post-exposure in groups of 12 animals
per dose. At all dose levels significant increases were noted for one
or more components of the anti-oxidant system studied. Peaking at 2
to 3 days post-exposure, the changes noted were similar to those
observed after exposure to the pulmonary irritants ozone and nitrogen
dioxide.
The role of arachidonic acid metabolites in the pathogenesis of
phosgene-induced lung injury (oedema and vascular permeability) was
studied in rabbits (Guo et al., 1990). Animals (four to six per
group) were exposed to 6000 mg/m3-min (1500 ppm-min) of phosgene and
killed 30 min after-exposure. The effects were compared to those of a
control group of eight animals. Lungs were perfused for 90 min and
cyclooxygenase- and lipoxygenase-generated metabolites of arachidonic
acid were measured. Phosgene exposure did not enhance the
cyclooxygenase metabolism of arachidonic acid but did result in a
10-fold increase in lipoxygenase metabolites (leukotriene). A marked
decrease in the gain in lung weight after phosgene exposure was
reported when the perfused lung was pretreated with leukotriene
receptor blockers. The results suggest that lipoxygenase metabolites
of arachidonic acid contribute to the phosgene-induced pulmonary
damage, but the mechanism by which phosgene stimulates the metabolism
of arachidonic acid is still unknown.
Evidence that the non-cardiogenic pulmonary oedema and mortality
resulting from phosgene inhalation was the result of an influx of
neutrophils into the lung was provided by Ghio et al. (1991). After
exposure of rats to 2 mg phosgene/m3 (0.5 ppm) for 60 min,
significant increases in the percentage of neutrophils and
concentrations of protein and thiobarbitunic acid reactive products in
bronchoalveolar lavage fluid were noted. These increases were
significantly less after treatment prior to exposure with:
cyclophosphamide to deplete the leukocytes; inhibition of the
production of the chemotaxis leukotriene B4, which directs the influx
of neutrophils into the lung; or treatment with colchicine, which
decreases leukocyte migration. These treatments in mice exposed to
8 mg phosgene/m3 (2.0 ppm) for 90 min resulted in decreased
mortality. Colchicine reduced neutrophil influx, lung injury and
mortality in mice even when administered 30 min after exposure.
Preliminary evidence that F-actin in lung cells may be a target
of phosgene was reported by Werrlein et al. (1994). In cultured ovine
pulmonary artery endothelial cells and rat airway epithelial cells
exposed to phosgene there was a dose-dependent decrease in the F-actin
content and organization. Exposure of sheep to 0.15 L(CT)50
(96 mg/m3 for 20 min) led to a decrease in the F-actin concentration
of endothelial cells. Exposure of sheep to 0.83 L(CT)50 (548 mg/m3
for 20 min) disrupted basal lamina and produced paracellular leakage
paths in the cultured cells. If they occur in vivo, these effects
of phosgene on F-actin may contribute to the decreased barrier
function and increased permeability of vascular tissues.
8. EFFECTS ON HUMANS
8.1 General population and occupational exposure
The information on the effects of high-level, short-term phosgene
exposure in humans is derived from wartime experiences as well as from
industrial accidents. General population and occupational exposures
to high levels of phosgene will be discussed together, since the
immediate effects and outcome of such exposures are identical in both
populations. As with animal experiments (see section 7.1), the acute
effects in humans are reported as a result of a combination of
exposure level and time of exposure (mg/m3-min). The exposure levels
at which perception of the odour is possible compared to those that
cause varying degrees of toxicity and death in humans are presented in
Table 5. In "trained subject" the lowest level at which the odour
(like mouldy hay) is perceived is 1.6-2 mg/m3 (NIOSH, 1976), but at
such levels workers may not detect the odour due to olfactory fatigue
(Proctor & Hughes, 1991). Under normal conditions the odour is
recognized only at levels greater than 6 mg/m3 (1.5 ppm). Signs of
irritation of mucous membranes are observed at > 12 mg/m3 (3 ppm),
early lung damage at > 120 mg/m3-min (>30 ppm-min), and death
(L(CT)50) at approximately 2000 mg/m3-min (approximately
500 ppm-min) (Diller & Zante, 1982; Diller, 1985a).
As shown in Table 5, concentrations of phosgene vapour
> 2 mg/m3 (3 ppm) will result in irritation of the eyes and nose.
Such concentrations in contact with moist skin will also lead to
irritation and erythema (Borak, 1991), but there is no evidence that
they would result in serious skin injury. At 12 mg/m3 (3 ppm) the
only effect reported on the human eye was inflammation (conjunctival
hyperaemia) (Grant & Schumann, 1993). Liquid phosgene splashed in the
eye, however, caused complete corneal opacification, conjunctival
adhesions and perforation in one victim (Grant & Schumann, 1993).
Although skin contact can result in severe burns, no reports of such
cases are available.
Table 5. Correlation of phosgene dose and effects in humansa
Effects Dose levelb
Perception of odour 1.6 mg/m3 0.4 ppm
Recognition of odour 6 mg/m3 1.5 ppm
Irritation of eyes, nose, and throat 12 mg/m3 3 ppm
Beginning lung damage >120 mg/m3-minc > 30 ppm-min
Pulmonary oedema >600 mg/m3-minc > 150 ppm-min
L(CT)1 approx 1200 mg/m3-minc approx 300 ppm-min
L(CT)50 approx 2000 mg/m3-minc approx 500 ppm-min
L(CT)100 approx 5200 mg/m3-minc approx 1300 ppm-min
a From: Diller (1985a)
b A conversion factor of 4 was used to calculate mg/m3 from the ppm value used by author.
c These values should be considered in relation to Haber's Law (see section 7.1).
With respect to pulmonary damage, three distinct
clinicopathological phases (initial reflex syndrome, clinical latent
phase and clinical oedema phase) have been reported in humans acutely
exposed to phosgene levels of 120 mg/m3-min to 1200 mg/m3-min
(30-300 ppm-min) (Diller & Zante, 1982; Diller, 1985a). During and
immediately after exposure the individual experiences pain in the eyes
and throat (an irritating or burning sensation) and tightness in the
chest, which may be accompanied by shortness of breath and coughing.
This is followed by a latent phase, which is often asymptomatic.
Depending on total dose this period may last from 1 to 24 h. The
oedema phase is manifested when enough lung is affected to become
clinically apparent, i.e. shortness of breath, productive cough,
and/or expectoration of large amounts of frothy and possibly bloody
sputum. If enough of the lung is involved the person may become
cyanotic and enter into shock. If the exposure is in the lethal
range, the early phases may be truncated, and the latent phase may be
very short or non-existent. It has been reported that radiographs
taken immediately after exposure can be used to predict the severity
of ensuing pulmonary oedema (Ardran, 1964). The author stated that an
increase in lung volume following expiration is highly predictive for
the future development of oedema.
8.2 Case reports - individual accidents
The most definitive data on both the short- and long-term effects
of acute phosgene exposure in humans is found in case reports of
industrial accidents. Such accidents may also pose a potential hazard
to adjacent communities. In Hamburg, Germany, an industrial storage
tank released 11 tonnes of phosgene into the atmosphere in 1928
(Hegler, 1928). The atmospheric conditions were conducive to the slow
spread of the gas outside the plant. No exposure levels were
reported. During a period of 5 days, over 300 people became ill, of
whom 10 died. The initial symptoms were severe irritation of the eyes
and throat, coughing, tightness of the chest, nausea and vomiting.
Autopsies of some of the victims revealed typical pulmonary lesions
and nonspecific lesions in other organs that were attributable to
local hypoxia (Wohwill, 1928). The author also felt that some
degenerative lesions in the brain and spinal cord were due directly to
phosgene, but this has not been confirmed in subsequent studies. The
only reference to the long-term effects in this population was that
there was no apparent damage to health 2 months after the accident.
In November 1966, phosgene was accidentally released from a
factory in Japan; 382 people were poisoned and 12 were hospitalized
(Sakakibara et al., 1967). Signs and symptoms observed in the 12
patients on admission were headache (9), nausea (9), cough (8),
dyspnoea (7), fatigue (7), pharyngeal pain (5), chest tightness (5),
chest pain (5), and fever (3). Lacrimation and redness of the eyes
were only observed in one patient. Seven patients showed evidence of
pulmonary oedema as revealed by chest X-rays 48 h after the exposure.
These findings indicate that pulmonary oedema may develop even 48 h
after exposure without initial symptoms of eye or nose irritation.
Other industrial accidents that have been reported usually
involved only a few people and no details of exposure levels were
given. The clinicopathological syndrome was similar in all of these
reports (Everett & Overholt, 1968; Stavrakis, 1971; Regan, 1985). The
predominant finding during and immediately following exposure was
irritation of mucous membranes. Victims described intense pain in the
eyes with profuse lacrimation. At the same time there was a burning
sensation in the throat and tightness of the chest. Coughing ensued,
often of a very severe nature. Victims sometimes did not show any
other symptoms. However, more common was the subsequent development
of pulmonary oedema, which, if sufficiently severe, resulted in death
due to interference with gas exchange. In none of the case reports of
direct phosgene exposure was any data on the actual levels of phosgene
given.
Several case studies of phosgene poisoning have been reported
where the patient was not working directly with phosgene. Such
reports include those of Seidelin (1961) on carbon tetrachloride used
to extinguish a fire, those of Glass et al. (1971) and Sjogren et al.
(1991) on the decomposition of trichlorethylene during welding and
those of Gerritsen & Buschmann (1960) and Snyder et al. (1992)
reporting possible phosgene poisoning due to the thermal degradation
of methylene chloride used to remove paint. All of these chemicals
are known to be degraded thermally to phosgene. Furthermore, the
progression of effects (clinical latent phase) after exposure, the
development of dyspnoea, chest discomfort and pulmonary oedema in all
subjects was typical of phosgene poisoning reported in case studies of
direct phosgene exposure.
There are relatively few reports on the long-term sequelae of an
acute exposure. In a review of this subject, Diller (1985a,b) found
that the vast majority of survivors of acute exposure have a good
prognosis. However, some of those exposed to high levels of phosgene
showed chronic symptoms such as shortness of breath and reduced
physical capacity, which persisted, in some cases, for the rest of
their lives. However, the severity and duration of such effects was
also related to subsequent smoking habits. Pre-existing pulmonary
disease such as emphysema was exacerbated by phosgene exposure. While
most published reports indicate that the respiratory tract is the
primary target organ for phosgene poisoning, a few reports have
indicated effects on other organs, especially the heart and brain
(Diller, 1985a,b). Neurasthenia is the most common of these
conditions associated with acute phosgene exposure. Others are an
epilepsy-like syndrome, loss of speech, peripheral Raynaud-like
syndrome and a type of paralysis characterized by disfunction of the
peroneal nerve. A causal relationship between phosgene exposure and
such effects has yet to be confirmed. It has been suggested by Diller
(1985b) that such changes are more likely a result of anoxia from the
pulmonary oedema rather than the direct action of phosgene.
8.3 Epidemiological studies
Phosgene, isopropyl alcohol, aniline and caustic soda are raw
materials used in a Russian plant for the manufacture of the
herbicide, isopropylphenyl carbamate. Levina & Kurando (1967) found
phosgene at levels of about 0.5 mg/m3 in 30% of all air samples. In
89 workers studied no mention was made of pulmonary problems.
However, methaemoglobinaemia and anaemia were detected and attributed
to exposures to aniline and the herbicide itself.
At a phosgene factory in the USA the medical records of all
workers exposed (326) and 6228 non-exposed workers were compared
(NIOSH, 1976). Limited air sampling conducted during a 2-month period
using the NBP method of analysis with 20-min sampling time (passive
dosimetry Table 2), indicated an average exposure to phosgene of
0.01 mg/m3 (ND to 0.08 mg/m3). Out of 56 fixed-position samplers
(2-h or 20-min collection) 51 showed phosgene levels of up to
0.52 mg/m3 and five samples showed levels greater than 0.55 mg/m3
(off the scale). Deaths attributable to respiratory disease and
pulmonary function (defined as "lung problems") were compared. No
chronic lung problems associated with working at these phosgene levels
were reported, nor was there any increased mortality from respiratory
disease in the exposed workers.
Polednak (1980) described a study of a cross-section of workers
exposed to phosgene at a uranium processing plant between 1943 and
1947. The study contained one group of 699 white male workers exposed
routinely to low (but undetermined) levels of phosgene with daily
episodes (4 or 5 daily) of exposures to levels above 4 mg/m3. A
second group of 106 white males involved in accidents resulting in
acute exposures to phosgene at levels estimated by the authors to be
greater than 200 mg/m3-min. This estimate was based on initial
symptoms reported and clinical data obtained immediately after
exposure. All workers reported detecting the odour of phosgene, 82
reported chest pain and dyspnoea, 25 showed x-ray and clinical
evidence of pneumonitis and 1 worker died 24 h after the exposure from
pulmonary oedema (based on clinical symptoms). The control group
contained 9352 non-exposed workers at the same facility. Those
workers employed for 2 days or more in departments where phosgene
exposure was possible were considered to be in the exposed group.
Mortality data was determined by examination of the Social Security
Administration records and coding the cause of death using the Eighth
Revision of the International Classification of Diseases. As of 1974
there was no evidence of excess mortality from diseases of the
respiratory system in the group of 699 male workers (about 30 years
after exposure). In fact standardized mortality ratios (SMR) for
death from all causes were essentially the same in both the exposed
and control groups. The Task Group noted that possible simultaneous
exposure to ionizing radiation was not taken into account.
In 1974, 30 deaths had occurred in the 106 workers exposed to
high levels of phosgene (SMR = 113). No deaths from lung cancer were
noted but three deaths (1.37 expected) were due to respiratory
diseases. One worker in this group died from pulmonary oedema 24 h
after exposure.
A follow-up of the workers from the cohort studied by Polednak
(1980) was made by Polednak & Hollis (1985). Similar trends to those
reported earlier were noted 35 years after exposure. Of the 694
workers chronically exposed, there were 14 deaths from diseases of the
respiratory tract (13.1 expected) (SMR = 107; 59-180 95% CI). In 1974,
the SMR for lung cancer within the 699 chronically exposed workers was
127 (95%, CI 66-220) and in 1979 it was 122 (72-193, 95% CI). The
slightly elevated SMR values were not significantly higher than
controls. The SMR for death from all causes was 97 (85-111 95% CI) in
the exposed workers and 101 (98-104 95% CI) in controls. In the high-
exposure group there were 41 deaths from all causes compared to 33.9
expected (SMR = 121; 86-165 95% CI). Five deaths were coded to
diseases of the respiratory tract (SMR = 266; 86-622 95% CI). In two
of these cases, bronchitis due to phosgene poisoning had been reported
in 1945 during clinical examination after exposure.
There have been no epidemiological or case reports linking the
development of reproductive or teratogenic effects in humans to acute
and/or chronic phosgene exposures.
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
No information concerning the effects of phosgene in the
laboratory and field has been reported.
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of human health risks
10.1.1 Exposure
Exposure to phosgene is primarily by inhalation. There are three
exposure situations: chronic exposure of the general population to
extremely low levels; chronic exposure in the workplace to very low
levels; and accidental acute exposure to high levels. It is likely
that the principle source of exposure to phosgene for the majority of
the general population is through the photo-degradation and thermal
degradation of chlorinated hydrocarbons, especially solvents and
polymers (e.g., tri- and tetrachloroethylene and PVC). On the basis
of limited data, average levels of phosgene in ambient air can be
expected to vary between 80 and 130 ng/m3.
Available data are inadequate to determine quantitatively the
exposure to phosgene in the workplace. Those working simultaneously
with flames (or thermal energy sources) and organochlorine solvents or
PVC can be exposed to phosgene levels well above present threshold
limit values (time-weighted average) of 0.4 mg/m3.
Accidental release of phosgene during its manufacture, use or
transport can lead to high levels of exposure for workers and for the
general population in the vicinity of the accident.
10.1.2 Health effects
Phosgene is a highly reactive chemical, hydrolysing to
hydrochloric acid, and is capable of acrylating nucleophilic groups,
such as amino, hydroxyl and sulfhydryl groups, in tissues.
In all species studied, including humans, the major target organ
is the lung. High concentrations can also cause skin and eye
irritation. For health effects after acute exposure, Haber's Law,
which states that the toxicological effect is due to the product of
exposure (C) and time (T), holds between levels of 4 and 800 mg/m3
(1 and 200 ppm) using lung disease and death as toxicological end-
points. This law does not, however, prevail for chronic exposure.
The cascade of events after acute inhalation exposure in humans and
experimental animals are similar. It occurs in a dose-related manner
and results in pulmonary oedema and death in humans, which is
dose-dependent at levels exceeding 120 mg/m3-min. Three distinct
clinicopathological phases can be recognized: pain in the eyes and
throat and tightness of the chest, often with shortness of breath,
wheezing and coughing; a latent phase that is often asymptomatic and
can last up to 24 h depending upon the concentration and duration of
exposure; and the final phase of pulmonary oedema.
10.1.2.1 Evaluation of animal data
The L(CT)50 and L(CT)100 values for single exposure vary widely
among animal species (Table 3). In all species the characteristic
pathological feature is the delayed clinical manifestation of
pulmonary oedema, which is dose-dependent. The extent of the
long-term chronic effects of acute exposure appears to depend on the
severity of the initial pathology.Single exposure of rats for 4 h to
between 0.5 and 4 mg/m3 resulted in a dose-related increase in lavage
fluid protein (LFP) concentration and an increased percentage of
polymorphonuclear leukocytes (PMN) in the alveoli. Changes in the LFP
and PMN were the most sensitive parameters occurring at 240 mg/m3-
min. These changes were reversible within 3 days after exposure.
There have been no long-term exposure studies in animals, and studies
in dogs exposed 1-3 times/week for 12 weeks are of limited value for
risk assessment in view of inadequate study design and lack of dose-
response. Available data in experimental animals are inadequate for
the assessment of the potential reproductive, developmental,
neurotoxic and carcinogenic effects resulting from phosgene exposures.
Single 4-h exposures to 0.1 mg phosgene/m3 in mice have resulted
in a demonstrable decrease in pulmonary host resistance to bacteria.
Rats exposed to 2 or 4 mg/m3 for 4 h had decreased pulmonary cell
natural killer (NK) activity, whereas no effect was seen at a level of
0.4 mg phosgene/m3 for 4 h. Increased infectivity by influenza virus
was reported in rats exposed for 4 h to 4 mg/m3. Virus titres were
not detectable 4 days after infection. Mortality was increased after
exposure to S. zooepidemicus and inoculation of B16/BL6 pulmonary
melanoma tumours in mice exposed to phosgene levels at or above
0.1 mg/m3. No effect was reported at a level of 0.04 mg/m3.
Pulmonary bacterial clearance was reduced in rats exposed to
0.4 mg/m3 (0.1 ppm) phosgene for 6 h and to 0.4 mg/m3 (0.1 ppm) for
6 h/day, 5 days/week for 4 to 12 weeks. This effect was reversible
following termination of exposure.
10.1.2.2 Evaluation of human data
As a result of industrial accidents and occupational monitoring
(levels and health status), it has been reported that some humans can
only recognize the odour of phosgene at levels of about 6 mg/m3,
making this an unacceptable parameter for early warning. After
short-term exposure, throat and eye irritation occurs at a level of
12 mg/m3 and eye irritation is noted at 16 mg/m3. The risk of
morbidity and mortality after acute exposure, is determined by the
dose (CT), not solely by concentration. It has been calculated that
doses below 100 mg/m3-min result in no effect, whereas pulmonary
oedema results from doses above 600 mg/m3-min. It should be
recognized, however, that death has been recorded at doses above
400 mg/m3-min, although with proper medical intervention death may be
prevented. Exposures for several hours at or below the odour
threshold (6 mg/m3) may result in severe tissue damage and death.
A review of the health status of workers who have recovered from
acute phosgene exposures has shown no adverse effects. However, full
recovery may take several months.
Available data on human health effects associated with chronic
exposure to phosgene are extremely limited. Epidemiological studies
of phosgene production workers and uranium workers reported no adverse
effects on human health. However, these investigations are of limited
value owing to the small numbers of exposed workers, lack of reliable
quantitative information on exposure to phosgene, concomitant exposure
to other substances, limited number of end-points examined and limited
reporting of relevant information. Available data are therefore
considered inadequate to assess the risk associated with long-term
exposure to low levels of phosgene.
10.1.3 Guidance value
Available data is considered inadequate to derive a meaningful
health-based guidance value for exposure of the general population to
phosgene. Information from epidemiological studies of occupationally
exposed workers is insufficient to characterize quantitatively
exposure-response relationships associated with potentially adverse
health effects resulting from exposure to this substance. Appropriate
studies in laboratory animals are lacking, and the available
toxicological investigations do not provide relevant data upon which
development of a credible guidance value for the long-term exposure of
humans to phosgene can be based.
Recent toxicological studies of rats subchronically exposed by
inhalation to low levels of phosgene indicate that early pulmonary
effects may occur at present TLV values. Thus, consideration by
appropriate authorities should be given to re-evaluating current
occupational exposure guidelines for this substance.
10.2 Evaluation of effects on the environment
The levels of phosgene now found in the environment would not be
expected to result in significant effects to aquatic or terrestrial
biota. However, no data were found to substantiate or refute this
hypothesis.
Damage to plants and aquatic organisms, owing to the rapid
release of hydrochloric acid, could occur in areas where accidental
release of phosgene has occurred.
11. CONCLUSIONS AND RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH
11.1 Conclusions
a) Phosgene is an extremely reactive chemical with the potential to
cause adverse effects in humans, the primary target organ being
the respiratory system.
b) Acute severe phosgene exposure primarily causes respiratory
disease (pulmonary oedema) and may result in death. Survivors
may recover completely provided they receive proper medical
support.
c) Present levels of exposure to phosgene in the general population
are extremely low and do not pose a health risk in the short
term. However, humans working with chlorinated solvents such as
trichloroethane, tetrachloroethylene and methylene chloride or
who are exposed to chlorinated hydrocarbon polymers (e.g., PVC)
in contact with flames or other thermal energy sources can be
exposed to levels of phosgene known to cause adverse effects in
humans. This could apply to firemen, welders, painter or people
working at home with the above-mentioned materials.
d) Accidental industrial releases can cause health problems in
workers and in the nearby community.
e) Workers have been shown not be at risk in closed-system
industrial facilities that manufacture or use phosgene and employ
good industrial practice.
f) No human or animal data are available on the effects of chronic
low-level exposures to phosgene.
g) No data are available concerning adverse effects on organisms in
the environment. However, accidental release would be expected
to give rise to adverse effects.
11.2 Recommendations for protection of human health
a) Present occupational exposure limits for phosgene should be
re-assessed.
b) Data should be obtained on the release of phosgene by the
incineration of chlorine-containing organic materials.
c) International and national regulations regarding transport of
phosgene should be followed in order to avoid accidental
releases.
d) Analytical methods capable of monitoring whole shift individual
exposure (e.g., paper tape monitors) should be used routinely in
the workplace.
12. FURTHER RESEARCH
a) The mechanism(s) of phosgene toxicity need to be clarified in
order to improve risk assessment and therapy.
b) Data gaps in the areas of reproduction/development toxicity,
mutagenicity and carcinogenicity following chronic low level
exposure should be addressed. More data should be gained about
the long-term effects of acute exposure.
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
No international body has evaluated the human health or
environmental risks from exposure to phosgene. Similarly, no health-
based guidance values have been developed by such groups.
Regulatory standards for phosgene established by national bodies
in some countries have been summarized by the ILO (ILO, 1991).
REFERENCES
Andersson HG, Dahlberg JA, & Wettstrom R (1975) Phosgene formation
during welding in air contaminated with perchloroethylene. Ann Occup
Hyg, 18: 129-132.
Ardran GM (1964) Phosgene poisoning [letter]. Br Med J, 1(5379):
375.
Atherley G (1985) A critical review of time-weighted average as an
index of exposure and dose, and of its key elements. Am Ind Hyg Assoc
J, 46: 481-487.
Birgesson D (1982) An unknown working environment hazard for
refrigerating equipment fitters: freon. Arbetsmiljo, 6: 48-50.
Borak J (1991) Phosgene toxicity: review and update. CEM Rep, 5:
19-22.
Boyd EM & Perry WF (1960) Respiratory tract fluid and inhalation of
phosgene. J Pharm Pharmacol, 12: 726-732.
Brown JE & Birky MM (1980) Phosgene in the thermal decomposition
products of poly(vinyl chloride): generation, detection and
measurement. J Anal Toxicol, 4: 166-174.
Budavari S ed. (1996) The Merck index: an encyclopedia of chemicals,
drugs, and biologicals, 12th ed. Rahway, New Jersey, Merck & Co., Inc.
Burleson GR & Keyes LL (1989) Natural killer activity in Fischer-344
rat lungs as a method to assess pulmonary immunocompetence:
Immunosuppression by phosgene inhalation. Immunopharmacol
Immunotoxicol, 11(2-3): 421-443.
Butler R & Snelson A (1979) Kinetics of the homogeneous gas phase
hydrolysis of CC13COC1, CC12HCOC1, CH2C1COC1, and COCL2. J Air
Pollut Control Assoc, 29: 833-837.
Cameron GR & Courtice FC (1946) The production and removal of oedema
fluid in the lung after exposure to carbonyl chloride (phosgene). J
Physiol, 105: 175-185.
Cameron GR & Foss GL (1941) Effect of low concentrations of phosgene
for 5 hours on 5 consecutive days in groups of different animals.
Washington, DC, British Embassy Defense Staff (Porton Report No. 2316,
Serial No. 63).
Cameron GR, Courtice FC, & Foss GL (1942) Effect of exposing different
animals to low concentrations of phosgene (1:5,000,000=0.9 mg/m3) for
5 hours on five consecutive days. Second report. In: First report on
phosgene poisoning: chapter VIII. Washington, DC, British Embassy
Defense Staff (Porton Report No. 2349).
Cessi C, Colombini C, & Mameli L (1966) The reaction of liver proteins
with a metabolite of carbon tetrachloride. Biochem J, 101: 46c-47c.
Clay JR & Rossing RG (1964) Histopathology of exposure to phosgene: An
attempt to produce pulmonary emphysema experimentally. Arch Pathol,
78: 544-551.
Coman DR, Bruner HD, Horn RCJr, Friedman MD, Boche RD, McCarthy MD,
Gibbon MH, & Schultz J (1947) Studies on experimental phosgene
poisoning. I. The pathologic anatomy of phosgene poisoning, with
special reference to the early and late phases. Am J Pathol, 23:
1037-1074.
Crummett WB & McLean JD (1965) Ultraviolet spectrophotometry
determination of trace quantities of phosgene in gases. Anal Chem,
37: 424-425.
Currie WD, Pratt PC, & Frosolono MF (1985) Response of pulmonary
energy metabolism to phosgene. Toxicol Ind Health, 1(2): 17-27.
Currie WD, Hatch GE, & Frosolono MF (1987a) Pulmonary alterations in
rats due to acute phosgene inhalation. Fundam Appl Toxicol, 8:
107-114.
Currie WD, Hatch GE, & Frosolono MF (1987b) Changes in lung ATP
concentration in the rat after low-level phosgene exposure. J Biochem
Toxicol, 2: 105-114.
Dangwal SK (1994) A spectrophotometric method for determination of
phosgene in air. Ind Health, 32: 41-47.
Diller WF (1985a) Pathogenesis of phosgene poisoning. Toxicol Ind
Health, 1: 7-15.
Diller WF (1985b) Late sequelae after phosgene poisoning: a
literature review. Toxicol Ind Health, 1: 129-136.
Diller WF & Zante R (1982) Dose-effect relation in the effects of
phosgene on man and animals (literature study). Zent.bl Arb.med
Arb.schutz Prophyl, 32: 360-368.
Diller WF, Bruch J, & Dehnen W (1985) Pulmonary changes in the rat
following low phosgene exposure. Arch Toxicol, 57: 184-190.
Ehrlich JP & Burleson GR (1991) Enhanced and prolonged pulmonary
influenza virus infection following phosgene inhalation. J Toxicol
Environ Health, 34: 259-273.
Esposito GG, Lillian D, Podolak GE, & Tuggle RM (1977) Determination
of phosgene in air by gas chromatography and infrared
spectrophotometry. Anal Chem, 49: 1774-1778.
Everett ED & Overholt EL (1968) Phosgene poisoning. J Am Med Assoc,
205: 243-245.
Flury F (1921) [Combat gas poisonings: I. Irritant gases.] Z Gesamte
Exp Med, 13: 1-15 (in German).
Flury F & Zernick F (1931) [Noxious gases, fumes, vapors, and
varieties of smoke and dust.] Berlin, Verlag von Julius Springer,
pp 99-102, 222-228 (in German)
Franch S & Hatch G (1986) Pulmonary biochemical effects of inhaled
phosgene in rats. J Toxicol Environ Health, 19: 413-423.
Frosolono MF & Curie WD (1985) Response of the pulmonary surfactant
system to phosgene. Toxicol Ind Health, 1: 29-35.
Frosolono MF & Pawlowski R (1977) Effect of phosgene on rat lungs
after single high-level exposure: I. Biochemical alterations. Arch
Environ Health, 32: 271-277.
Gay BW Jr, Hanst PL, Bufalini JJ, & Noonan RC (1976) Atmospheric
oxidation of chlorinated ethylenes. Environ Sci Technol, 10: 58-67.
Gerritsen WB & Buschmann CH (1960) Phosgene poisoning caused by the
use of chemical paint removers containing methylene chloride in
ill-ventilated rooms heated with kerosene stoves. Br J Ind Med, 17:
187-189.
Ghio AJ, Kennedy TP, Hatch GE, & Tepper JS (1991) Reduction of
neutrophil influx diminishes lung injury and mortality following
phosgene inhalation. J Appl Physiol, 71(2): 657-665.
Gibbon MH, Bruner HD, Boche RD, & Lockwood JS (1948) Studies on
experimental phosgene poisoning: II. Pulmonary artery pressure in
phosgene-poisoned cats. J Thorac Surg, 17: 264-273.
Glass WI, Harris EA, & Whitlock RML (1971) Phosgene poisoning: case
report. NZ Med J, 74: 386-389.
Grant WM & Schumann JS (1993) Toxicology of the eye, 4th ed.
Springfield, Illinois, Charles C. Thomas, 733 pp.
Guo Y-L, Kennedy TP, Michael JR, Sciuto AM, Ghio AJ, Adkinson NF Jr, &
Gurtner GH (1990) Mechanism of phosgene-induced lung toxicity: Role of
arachiodonate mediators. J Appl Physiol, 69(5): 1615-1622.
Hardy EE (1982) Phosgene. In: Kirk-Othmer encyclopedia of chemical
technology, 3rd ed. New York, John Wiley and Sons, vol 17, pp 416-425.
Hatch GE, Slade R, Stead AG, & Graham JA (1986) Species comparison of
acute inhalation toxicity of ozone and phosgene. J Toxicol Environ
Health, 19: 43-53.
Hegler C (1928) [On the mass poisoning by phosgene in Hamburg: I.
Clinical observations.] Dtsch Med Wochenschr, 54: 1551-1553 (in
German)
HSE (1995) Phosgene. In: Criteria document summaries: Synopses of the
data used in setting occupational exposure limits. London, Health and
Safety Executive, pp 24-25 (EH 64 1995 Supplement).
ILO (1991) Occupational exposure limits for airborne toxic substances,
3rd ed. Geneva, International Labour Office (Occupational Safety and
Health Series, No. 37).
IPCS (1993) 1,2-Dichloropropane. In: Environmental health criteria
No. 146: 1,2-Dichloropropane. Geneva, World Health Organization,
International Programme on Chemical Safety, pp 119-162.
Jaskot RH, Grose EC, Richards JH, & Doerfler DL (1991) Effects of
inhaled phosgene on rat lung antioxidant systems. Fundam Appl Toxicol,
17: 666-674.
Keeler JR, Hurt HH, Nold JB, & Lennox WJ (1990) Estimation of the
LCt50 of phosgene in sheep. Drug Chem Toxicol, 13(2-3): 229-239.
Levina MM & Kurando TB (1967) [Problems of labour hygiene and of state
of health of workers at the isopropylpenylcarbamate production.] Gig i
Sanit, 32: 25-28 (in Russian).
Levina MM, Kurando TB, Belyakov AA, Smirnova VG, & Odlyzhko SL (1966)
[Industrial hygiene problems and worker health in the monuron
industry.] Gig Tr Prof Zabol, 10: 54-56 (in Russian).
Long JE & Hatch TF (1961) A method for assessing the physiological
impairment produced by low-level exposure to pulmonary irritants. Am
Ind Hyg Assoc J, 22: 6-13.
Madden MC, Friedmann M, Keyes LL, Koren HS, & Burleson GR (1991)
Effects of phosgene exposure on lung, arachidonic acid metabolism.
Inhal Toxicol, 3: 73-90.
Manogue WH & Pigford RL (1960) The kinetics of the absorption of
phosgene into water and aqueous solutions. Am Inst Chem Eng J, 6:
494-500.
Mansuy D, Beaune P, Cresteil T, Lange M, & Leroux J-P (1977) Evidence
for phosgene formation during liver microsomal oxidation of
chloroform. Biochem Biophys Res Commun, 79(2): 513-517.
Matherne RN, Lubs PL, & Kerfoot EJ (1981) The development of a passive
dosimeter for immediate assessment of phosgene exposures. Am Ind Hyg
Assoc J, 42: 681-684.
Meek WJ & Eyster JAE (1920) Experiments on the pathological physiology
of acute phosgene poisoning. Am J Physiol, 51: 303-320.
Moore G & Matherne RN (1981) Field experiences with a phosgene
dosimeter system. Ann Am Conf Gov Ind Hyg, 1: 253-254.
Nash T & Pattle RE (1971) The absorption of phosgene by aqueous
solutions and its relation to toxicity. Ann Occup Hyg, 14: 227-233.
NIOSH (1976) Criteria for a recommended standard .... Occupational
exposure to phosgene. Cincinnati, Ohio, US Department of Health,
Education and Welfare, National Institute for Occupational Safety and
Health, 129 pp (HEW Publication No. NIOSH 76-137).
Noweir MH, Pfitzer EA, & Hatch TF (1973) Decomposition of phosgene in
air. Am Ind Hyg Assoc J, 34: 110-119.
Ong SG (1972) Treatment of phosgene poisoning with antiserum:
anaphylactic shock by phosgene. Arch Toxikol, 29: 267-278.
Pawlowski R & Frosolono MF (1977) Effect of phosgene on rat lungs
after single high-level exposure. II. Ultrastructural alterations.
Arch Environ Health, 32(6): 278-283.
Pohl LR, Bhooshan B, Whittaker NF, & Krishna G (1977) Phosgene: a
metabolite of chloroform. Biochem Biophys Res Commun, 79(3):
684-691.
Polednak AP (1980) Mortality among men occupationally exposed to
phosgene in 1943-1945. Environ Res, 22: 357-367.
Polednak AP & Hollis DH (1985) Mortality and causes of death among
workers exposed to phosgene in 1943-1945. Toxicol Ind Health, 1:
137-151.
Potts AM, Simon FP, & Gerard RW (1949) The mechanism of action of
phosgene and diphosgene. Arch Biochem, 24: 329-337.
Proctor NJ & Hughes JP (1991) In: Hathaway G, Proctor N, & Hughes J
ed. Chemical hazards of the workplace. New York, Von Nostrand Reinhold
Company, pp 475-477.
Regan RA (1985) Review of clinical experience in handling phosgene
exposure cases. Toxicol Ind Health, 1: 69-71.
Rinehart WE & Hatch T (1964) Concentration-time product (CT) as an
expression of dose in sublethal exposures to phosgene. Am Ind Hyg
Assoc J, 25: 545-553.
Rinzema LC (1971) Behavior of chlorohydrocarbon solvents in the
welding environment. Int Arch Arbeitsmed, 28: 151-174.
Rossing RG (1964) Physiologic effects of chronic exposure to phosgene
in dogs. Am J Phsiol, 207: 265-272.
Sakakibara H, Shiiki Y, Okamoto N, & Wataki A (1967) [Mass poisonings
with phosgene.] Shindan Chiryo, 55: 1433-1437 (in Japanese).
Schepers GWH (1971) Lung tumors of primates and rodents: Part II. Ind
Med, 40: 23-31.
Schneider W & Diller W (1989) Phosgene. In: Encyclopedia of industrial
chemistry, 5th ed. Weinheim, Germany, VCH Verlag, vol A19, pp 411-420.
Seidelin R (1961) The inhalation of phosgene in a fire extinguisher
accident. Thorax, 16: 91-93.
Selgrade MK, Starnes DM, Illing JW, Daniels MJ, & Graham JA (1989)
Effects of phosgene exposure on bacterial, viral, and neoplastic lung
disease susceptibility in mice. Inhal Toxicol, 1: 234-259.
Shah H, Hartman SP, & Weinhouse S (1979) Formation of carbonyl
chloride in carbon tetrachloride metabolism by rat liver in vitro.
Cancer Res, 39: 3942-3947.
Singh HB (1976) Phosgene in the ambient air. Nature (Lond), 264:
428-429.
Singh HG, Salas L, Shigeishi H, & Crawford A (1977) Urban-nonurban
relationships of halocarbons, SF6, N2O, and other atmospheric trace
constituents. Atmos Environ, 11: 819-828.
Singh HB, Salas LJ, Smith AJ, & Shigeishi H (1981) Measurement of some
potentially hazardous organic chemicals in urban environments. Atmos
Environ, 15: 601-612.
Sjogren B, Plato N, Alexandersson R, Eklund A, & Falkenberg C (1991)
Pulmonary reactions caused by welding-induced decomposed
trichloroethylene. Chest, 99: 237-238.
Snyder RW, Mishel HS, & Christensen GC (1992) Pulmonary toxicity
following exposure to methylene chloride and its combustion product,
phosgene. Chest, 101(3): 860-861.
Stavrakis P (1971) The use of hexamethylenetetramine (HMT) in
treatment of acute phosgene poisoning. Ind Med, 40: 30-31.
Thienes CH & Haley TJ (1972) The gas penetrates into the tissues of
respiratory tract and to some extent is absorbed by the lung. In:
Clinical toxicology, 5th ed. Philadelphia, Pennsylvania, Lea &
Febiger, pp 192-193.
Tuggle RM, Esposito GG, Guinivan TL, Hess TL, Lillian D, Podolak GE,
Sexton KG, & Smith NV (1979) Field evaluation of selected monitoring
methods for phosgene in air. Am Ind Hyg Assoc J, 40: 387-394.
Underhill FP (1919) The physiology and experimental treatment of
poisoning with the lethal war gases. Arch Intern Med, 23: 753-770.
Underhill FP (1920) The lethal war gases: physiology and experimental
treatment. New Haven, Connecticut, Yale University Press, pp 3-10,
40-41, 85-87, 105, 119-120, 133-137.
US EPA (1986) Health assessment document for phosgene. Washington, DC,
US Environmental Protection Agency (EPA/600/8-86/022A).
US NLM (1995) Hazard substances data base: computerized database.
Bethesda, Maryland, US National Libray of Medicine.
Verschueren K (1983) Rabbit, inhalation, mortality. In: Handbook of
environmental data on organic chemicals, 2nd ed. New York, Van
Nostrand Reinhold Co., p 534.
Werrlein RJ, Madren-Whalley JS, & Kirby SD (1994) Phosgene effects on
F-actin organization and concentration in cells cultured from sheep
and rat lung. Cell Biol Toxicol, 10: 45-58.
Winternitz MC, Lambert RA, & Jackson L (1920) The pathology of
phosgene poisoning. In: Collected studies on the pathology of war gas
poisoning. New Haven, Connecticut, Yale University Press, pp 35-66.
Wohlwill F (1928) Pathological findings of phosgene poisoning. Dtsch
Med Wochenschr, 54: 1553-1557.
Wu WS & Gaind VS (1993) Determination of phosgene (carbonyl chloride)
in air by high performance liquid chromatography with a dual selective
detection system. Analyst, 188: 1285-1287.
Yang YG, Gilmour MI, Lange R, Burleson GR, & Selgrade MK (1995)
Effects of acute exposure to phosgene on pulmonary host defenses and
resistance to infection. Inhal Toxicol, 7: 393-404.
1. RESUME ET CONCLUSIONS
1.1 Identité, propriétés physiques et chimiques et méthodes d'analyse
Le phosgène est un gaz incolore, extrêmement réactif à la
température et à la pression ambiantes, dont l'odeur suffocante
rappelle celle du foin moisi. Cette odeur est décelable à des
concentrations comprises entre 1,6 et 6 mg/m3.
Il existe des méthodes d'analyse pour la mise en évidence du
phosgène dans l'air ou qui peuvent être utilisées dans le cadre des
programmes d'hygiène et de sécurité du travail pour la mesure de la
dose totale (par ex. ruban de papier indicateur).
1.2 Usages et sources d'exposition humaine et environnementale
Le phosgène est utilisé à plus de 99% sur le lieu de production
dans des systèmes clos. On le prépare en faisant réagir, en présence
d'un catalyseur carboné, du chlore anhydre sur du monoxyde de carbone
en proportions équimoléculaires. La production mondiale est estimée à
plus de 3 millions de tonnes.
La présence de phosgène dans l'environnement est due aux
émissions d'origine industrielle et à la décomposition thermique des
solvants et des polymères chlorés. Cependant, il peut également
provenir en proportion importante de l'oxydation photochimique des
chloréthylènes, comme le trichloréthylène et le tétrachloréthylène.
1.3 Transport, distribution et transformation dans l'environnement
Le phosgène étant très réactif, il n'est vraisemblablement que
peu transporté d'un compartiment à l'autre.
Le phosgène s'élimine de l'air ambiant par une décomposition en
phase hétérogène (catalyse de surface) et une lente hydrolyse en phase
gazeuse. Il peut être transporté sur de longues distances et sa
diffusion de la troposphère vers la stratosphère accélère probablement
le processus de décomposition par photolyse.
1.4 Concentrations dans l'environnment et exposition humaine
L'exposition humaine, qu'il s'agisse de la population générale ou
de certaines catégories professionnelles, se produit par inhalation.
La concentration moyenne du phosgène dans l'air ambiant peut
varier d'environ 80 à 130 ng/m3, encore que les données soient peu
nombreuses à ce sujet. En raison de la diversité des pratiques en
matière d'hygiène du travail dans le monde, il est impossible de
donner un chiffre pour caractériser l'exposition des travailleurs qui
produisent ou utilisent du phosgène ou, plus particulièrement,
l'exposition des pompiers. A l'heure actuelle, les valeurs-seuils (en
moyenne pondérée par rapport au temps) relevées dans 15 pays s'étagent
de 0,4 à 0,5 mg/m3.
La littérature ne donne pas d'indications sur la teneur de l'eau,
du sol et des aliments en phosgène.
1.5 Cinétique et métabolisme
On ne possède que très peu de données sur l'absorption, le
métabolisme, la distribution et la destinée du phosgène. La principale
voie d'exposition est la voie respiratoire, le gaz pénétrant dans les
tissus de l'arbre pulmonaire et ne se retrouvant par conséquent qu'en
quantités minimes dans le reste de l'organisme. Sa demi-vie très brève
(0,026 secondes) en solution aqueuse exclut toute rétention importante
dans l'organisme. On ne dispose d'aucune donnée sur le métabolisme du
phosgène. Ses produits d'hydrolyse, à savoir l'acide chlorhydrique et
le dioxyde de carbone, sont éliminés de l'organisme par les processus
physiologiques normaux.
La toxicité du phosgène est due au fait qu'il provoque
l'acylation des protéines et qu'il donne naissance à de l'acide
chlorhydrique. Cette acylation se produit au niveau des groupements
amino, hydroxyles et sulfhydriles des protéines et entraîne une
inhibition marquée des enzymes qui interviennent dans le métabolisme
énergétique ainsi qu'une rupture de la barrière air/sang.
1.6 Effets sur les animaux d'expérience et les systèmes d'épreuve
in vitro
1.6.1 Exposition à court et à long terme
Chez toutes les espèces étudiées, c'est le poumon qui est
l'organe cible principal. La valeur du L(CT)50 varie de
900 mg/m3-min (225 ppm-min ) chez la souris à 1920 mg/m3-min
(480 ppm-min) chez le cobaye. Une valeur de 1000 mg/ m3
(250 ppm-min) a été obtenue chez le singe. Chez toutes les espèces,
on constate la même pathologie caractéristique, à savoir un oedème
pulmonaire dont les manifestations cliniques sont retardées et qui est
lié à la dose. Les anomalies anato-mopathologiques observées à faible
concentration au niveau des bron-chioles terminales et des alvéoles,
sont caractéristiques d'un irritant pulmonaire. En revanche, à forte
concentration, l'oedème pulmonaire qui se développe perturbe les
échanges gazeux et finit par entraîner la mort.
On n'a pas connaissance d'études consacrées à une exposition de
longue durée au phosgène.
En ce qui concerne les durées d'exposition relativement courtes,
on dispose d'une étude au cours de laquelle des rats on été exposés en
une seule fois à du phosgène pendant 4 h à la concentration de
2 mg/m3. On a observé une réduction de l'immunocompétence pulmonaire
mesurée par l'activité des cellules NK. Aucun effet n'a été constaté
lors d'une exposition de 4 h à 0,4 mg/m3.
Deux autres études ont été publiées au sujet des effets d'une
exposition unique à du phosgène sur l'immunocompétence pulmo-naire.
Au cours de ces études, effectuées sur des rats et des souris, on a
constaté que des rats infectés par le virus grippal présentaient,
après 4 h d'exposition à 4 mg de phosgène par m3, un titre viral 10
fois plus élevé 1 jour après l'infection, ce titre conservant une
valeur élevée au cours des 4 jours suivants. En outre, chez les rats
qui avaient été soumis pendant 4 h à une concentration de gaz comprise
entre 0,2 et 4 mg/m3, on pouvait constater une diminution marquée du
taux de prostaglandine E2 et de leucotriènes dès que la dose
atteignait 0,4 mg/m3, avec réduction du nombre de macrophages
alvéolaires et augmentation du nombre de neutrophiles à la
concentration de 0,4 mg/m3. Pour étudier la résistance de l'hôte, on
a soumis des souris à des concentrations de phosgène comprises entre
0,04 et 0,4 mg/m3 sur une durée de 4 h. On a constaté, selon les
cas, une augmentation de la mortalité consécutive à une infection par
Streptococcus zooepidemicus ou un accroissement des mélanomes
pulmonaires B16/BL6 à partir de 0,1 mg/m3. Chez les rats exposés à
du phosgène à raison de 0,4 mg/m3 (0,1 ppm) pendant 6 h ou à la même
dose 6 h par jour, 5 j par semaine pendant 4 à 12 semaines , on a
observé une réduction de la clairance bactérienne pulmonaire. L'effet
était réversible après cessation de l'exposition.
1.6.2 Effets non pulmonaires
Le phosgène peut provoquer une irritation oculaire et cutanée. On
n'a pas trouvé trace, dans la littérature, d'études portant sur le
pouvoir sensibilisateur du phosgène.
On ne dispose d'aucune donnée relative aux effets du phosgène sur
la reproduction et le développement.
Il n'existe pas de données suffisantes pour permettre une
évaluation du pouvoir mutagène ou cancérogène du phosgène.
1.7 Effets sur l'homme
Chez l'homme, comme chez les animaux de laboratoire, l'organe
cible est le poumon. Après exposition à des concentrations de phosgène
comprises entre 120 et 1200 mg/m3-min , on a observé trois phases
clinico-pathologiques distinctes. La phase initiale consiste en
douleurs au niveau des yeux et de la gorge accompagnées d'une
sensation de constriction thoracique, souvent avec dyspnée
concomitante, respiration sifflante et toux; il peut également y avoir
une hypo-tension, de la bradycardie et plus rarement, une arrythmie
sinusale. La seconde phase ou phase de latence, souvent
asymptomatique, peut se prolonger pendant 24 h , selon l'intensité et
la durée de l'exposition. Au cours de la troisième phase, un oedème
pulmonaire parfois mortel peut se développer. Dans des populations
exposées au phosgène à la suite d'accidents industriels, on a fait
état de symptômes très divers comme des céphalées, des nausées, de la
toux, de la dyspnée, une fatigue générale, des maux de gorge, une
sensation douloureuse de constriction thora-cique, des douleurs
oculaires intenses et une forte lacrimation. Lors d'une étude, on a
observé la survenue d'un oedème pulmonaire après une phase de latence
de 48 h.
Les effets d'une exposition au phosgène ont été étudiés chez des
groupes de travailleurs de deux usines, à savoir une unité de
production de phosgène et une unité de traitement de l'uranium. Dans
les deux cas, on n'a effectué que des prélèvements d'air et une
surveillance individuelle limités et l'exposition n'est donc connue
que de manière estimative.
L'examen du dossier médical de la totalité des 326 travailleurs
de l'unité de production de phosgène qui pouvaient avoir été exposés à
ce gaz (jusqu'à 0,5 mg/m3 avec quelques dépassements de cette valeur;
moyenne 0,01 mg/m3) n'a pas révélé de problèmes pulmo-naires
chroniques ni de surmortalité par comparaison avec un groupe témoin de
6226 personnes. Toutefois, l'absence de détails, dans le compte rendu,
au sujet de l'exposition et des effets observés ne permet guère de
tirer des conclusions définitives de cette étude.
Dans le cas des travailleurs de l'usine de traitement de
l'uranium, on a constitué deux groupes: l'un, de 699 personnes prises
parmi les 1800 employés de cette période, a fait l'objet d'une étude
transversale. Ces travailleurs avaient été vraisemblablement soumis à
une concen-tration de phosgène inférieure à 0,4 mg/m3 (avec 4 ou 5
dépassements brefs par jour jusqu'à plus de 4 mg/m3). L'autre groupe
était constitué de 106 personnes qui avaient été impliquées dans des
accidents et soumises à une exposition de plus de 200 mg/m3-min.
Dans le groupe exposé de manière chronique à de faibles concentrations
de phosgène, l'examen des certificats de décès n'a pas fait ressortir
de surmortalité due à une cause quelconque, à une affection
respiratoire ou à un cancer du poumon. Il n'y a d'ailleurs eu aucune
mortalité par cancer du poumon, mais par contre une légère
surmortalité d'origine respiratoire. On ne peut tirer de ces études
que des conclusions limitées sur les effets chroniques du phosgène et
cela, tant du fait de l'absence de données sur l'exposition que pour
des raisons d'ordre méthodologique.
1.8 Effets sur les autres êtres vivants au laboratoire et dans leur
milieu naturel
On ne possède aucun renseignement concernant les effets du
phosgène sur les diverses formes vivantes présentes dans
l'environnement.
1. RESUMEN Y CONCLUSIONES
1.1 Identidad, propiedades físicas y químicas y métodos analíticos
El fosgeno es un gas incoloro y altamente reactivo a temperatura
y presión ambientes. Posee un olor sofocante parecido al del heno
enmohecido, que puede percibirse a concentraciones comprendidas entre
1,6 y 6 mg/m3.
Existen métodos analíticos que permiten detectar el fosgeno en el
aire y se utilizan en programas de higiene industrial que miden la
dosis total (p.ej., sensores de cinta de papel).
1.2 Usos y fuentes de exposición humana y ambiental
Más del 99% del fosgeno producido se emplea in situ en sistemas
cerrados. Se produce haciendo reaccionar cantidades equimolares de
cloro anhidro y monóxido de carbono en presencia de un catalizador de
carbono. Se ha estimado que la producción mundial supera los 3
millones de toneladas.
El fosgeno ambiental procede de emisiones industriales y de la
degradación térmica de algunos disolventes clorados y polímeros
clorados. No obstante, una fuente importante de fosgeno ambiental es
la oxidación fotoquímica de cloroetilenos tales como el tri- y el
tetraetileno.
1.3 Transporte, distribución y transformación en el medio ambiente
Debido a su alta reactividad, el transporte intercompartimental
del fosgeno es en principio limitado.
La eliminación del fosgeno del aire ambiente se produce por
descomposición heterogénea (catálisis superficial) y por hidrólisis
lenta en fase gaseosa. El fosgeno es transportado a larga distancia, y
se cree que su difusión de la troposfera a la estratosfera acelera su
degradación fotolítica.
1.4 Niveles medioambientales y exposición humana
La exposición humana tanto en la población general como en el
entorno ocupacional se produce fundamentalmente por inhalación.
Se estima que la concentración promedio de fosgeno en el aire
ambiente está comprendida aproximadamente entre 80 y 130 ng/m3,
aunque hay pocos datos disponibles. Dada la diversidad de las
prácticas de higiene industrial seguidas en todo el mundo, es
imposible facilitar una cifra para los trabajadores que fabrican o
usan fosgeno o para los bomberos. Actualmente los valores umbral de
exposición (promedio ponderado en función del tiempo) en 15 países
están comprendidos entre 0,4 y 0,5 mg/m3.
No se ha informado sobre las concentraciones de fosgeno en el
agua, el suelo y los alimentos.
1.5 Cinética y metabolismo
Hay muy pocos datos sobre la absorción, el metabolismo, la
distribución y el destino del fosgeno. La principal vía de exposición
es la inhalación; el gas penetra en los tejidos del tracto
respiratorio, y sólo una mínima parte llega a distribuirse en el
organismo. Su muy breve vida media (0,026 segundos) en soluciones
acuosas evita que sea retenido significativamente por el organismo. No
se ha publicado información alguna sobre el metabolismo del fosgeno.
Sus productos hidrolíticos, p.ej. el ácido clorhídrico y el dióxido de
carbono, son eliminados por el organismo mediante los procesos
fisiológicos normales.
El fosgeno debe su toxicidad a la acilación de las proteínas, así
como a la generación de ácido clorhídrico. La acilación afecta a los
grupos amino, hidroxilo y sulfhidrilo de las proteínas, lo que da
lugar a una notable inhibición de varias enzimas relacionadas con el
metabolismo energético y a la descomposición de la barrera
sangre:aire.
1.6 Efectos en animales de laboratorio y en sistemas de
prueba in vitro
1.6.1 Exposiciones únicas y de corta duración
En todas las especies estudiadas el pulmón es el principal órgano
blanco. La (CT)L50 varía entre 900 mg/m3-min (225 ppm-min) en el
ratón y 1920 mg/m3-min (480 ppm-min) en el cobayo. Se ha informado de
una (CT)L50 de 1000 mg/m3-min (250 ppm-min) en el mono. En todas las
especies la manifestación patológica característica es la aparición
retardada sintomática de edema pulmonar, que depende de la dosis. Los
cambios anatomopatológicos observados en los bronquiolos terminales y
en los alveolos a bajas concentraciones son típicos de los irritantes
pulmonares, mientras que a exposiciones altas se produce edema, lo que
interfiere en el intercambio gaseoso y conduce a la muerte.
No se ha publicado ningún estudio sobre la exposición a largo
plazo al fosgeno.
Un estudio efectuado en ratas reveló que una sola exposición a
una concentración de 2 mg/m3 de fosgeno durante 4 horas puede
provocar una disminución de la inmunocompetencia pulmonar, a juzgar
por la actividad citotóxica natural de las células pulmonares. No se
observó ningún efecto a niveles de exposición de 0,4 mg/m3 mantenidos
durante 4 horas.
Se han publicado otros dos estudios sobre los efectos de
exposiciones únicas de fosgeno en la inmunocompetencia pulmonar de la
rata y del ratón. En ratas infectadas por el virus de la gripe tras 4
horas de exposición a 4 mg/m3 se observó que la concentración del
virus se había multiplicado por diez un día después de la infección,
manteniéndose significativamente elevados los niveles durante 4 días.
Además, en ratas expuestas a concentraciones de fosgeno comprendidas
entre 0,2 y 4 mg/m3 durante 4 horas se detectó una disminución
considerable de la prostaglandina E2 y de los leucotrienos a partir de
0,4 mg/m3, y una disminución del número de macrófagos alveolares y un
aumento del número de neutrófilos con 0,4 mg/m3. En un ensayo de
resistencia en que se expuso a ratones a niveles de fosgeno
comprendidos entre 0,04 y 0,4 mg/m3 durante 4 horas se observó un
aumento de la mortalidad por Streptococcus zooepidemicus o un
aumento del número de tumores pulmonares melanomatosos B16/BL6 a
niveles de 0,1 mg/m3 o superiores. La eliminación bacteriana pulmonar
se redujo en ratas expuestas a 0,4 mg/m3 (0,1 ppm) de fosgeno durante
6 horas o a 0,4 mg/m3 (0,1 ppm) durante 6 horas/día y 5 días/semana
por espacio de 4 a 12 semanas. Este efecto se reveló reversible tras
la terminación de la exposición.
1.6.2 Efectos no pulmonares
La exposición al fosgeno puede causar irritación de los ojos y de
la piel. No se ha hallado en las publicaciones ningún estudio
referente al potencial de sensibilización del fosgeno.
No se dispone de datos sobre los efectos del fosgeno en la
reproducción y el desarrollo.
No se dispone tampoco de datos adecuados para evaluar la
mutagenicidad o carcinogenicidad del fosgeno.
1.7 Efectos en el hombre
El órgano blanco en el hombre, como en los animales de
laboratorio, es el pulmón. Se han descrito tres fases
clinicopatológicas características tras la exposición a niveles de
fosgeno comprendidos entre 120 y 1200 mg/m3-min. La fase inicial
consiste en la aparición de dolor en los ojos y la garganta y de una
sensación de opresión torácica, a menudo con disnea, sibilancias y
tos; puede haber asimismo hipotensión, bradicardia y, rara vez,
arritmias sinusales. La que sigue a continuación es la fase latente,
porque es a menudo asintomática; puede durar hasta 24 horas según el
nivel y duración de la exposición. En la tercera fase puede aparecer
edema pulmonar, de consecuencias eventualmente mortales.
Las poblaciones expuestas al fosgeno tras accidentes industriales
han sufrido una amplia variedad de síntomas, incluidos cefaleas,
náuseas, tos, disnea, fatiga, dolor faríngeo, opresión y dolor
torácicos, dolor ocular intenso y lagrimeo grave. En un estudio se
observó edema pulmonar tras una fase latente de 48 horas.
Se han estudiado los efectos de la exposición al fosgeno a largo
plazo en tres grupos de trabajadores de dos instalaciones: una planta
de producción de fosgeno y un centro de procesamiento de uranio. En
los dos casos el muestreo del aire y la vigilancia del personal se
hicieron sólo de forma limitada, y únicamente se efectuaron
estimaciones de la exposición de los trabajadores.
El estudio de los registros médicos de los 326 trabajadores de la
planta de producción de fosgeno potencialmente expuestos a este
producto (concentraciones entre indetectables y de 0,5 mg/m3, valor
éste superado sólo esporádicamente; promedio: 0,01 mg/m3) no reveló
ni problemas pulmonares crónicos ni una mayor mortalidad por
enfermedades respiratorias en comparación con un grupo de 6228
controles. No obstante, la falta de datos de que adolece el informe en
lo que respecta a la exposición y a los efectos hace difícil extraer
conclusiones firmes del estudio.
Se estudió a dos grupos de trabajadores de la planta de
procesamiento de uranio: una muestra transversal de 699 trabajadores
de los más de 18 000 empleados durante el periodo estudiado,
potencialmente expuestos a niveles de fosgeno inferiores a 0,4 mg/m3
(con 4 ó 5 exposiciones fugaces a niveles > 4 mg/m3 cada día), y un
grupo de 106 trabajadores que se habían visto implicados en accidentes
y expuestos a niveles > 200 mg/m3-min. En el grupo expuesto
crónicamente a bajas concentraciones de fosgeno el análisis de los
certificados de defunción no mostró una mayor mortalidad por todo tipo
de causas o por enfermedad respiratoria o cáncer pulmonar. En el grupo
afectado por accidentes químicos no se registró tampoco ningún aumento
de las defunciones por todo tipo de causas; no hubo muertes por cáncer
pulmonar, pero sí un ligero incremento del número de defunciones por
enfermedades respiratorias. Habida cuenta tanto de la escasez de datos
sobre la exposición como de la metodología del estudio, sus
conclusiones tienen un valor limitado.
1.8 Efectos en otros organismos en el laboratorio y sobre el terreno
No se han publicado datos sobre los efectos del fosgeno en
organismos en el medio ambiente.