
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organization, or the World Health Organization.
Concise International Chemical Assessment Document 58
First draft prepared by Mr Peter Watts, Toxicology Advice & Consulting Ltd, Sutton, Surrey, United Kingdom; Mr G. Long, Health Canada, Ottawa, Canada; and Ms M.E. Meek, Health Canada, Ottawa, Canada
Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organization, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals.
World Health Organization
Geneva, 2004
The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organization (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals.
The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and Agriculture Organization of the United Nations, WHO, the United Nations Industrial Development Organization, the United Nations Institute for Training and Research, and the Organisation for Economic Co-operation and Development (Participating Organizations), following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase coordination in the field of chemical safety. The purpose of the IOMC is to promote coordination of the policies and activities pursued by the Participating Organizations, jointly or separately, to achieve the sound management of chemicals in relation to human health and the environment.
WHO Library Cataloguing-in-Publication Data
Chloroform.
(Concise international chemical assessment document ; 58)
1.Chloroform - adverse effects 2.Risk assessment 3.Environmental exposure
I.International Programme on Chemical Safety II.Series
ISBN 92 4 153058 8 (LC/NLM Classification: QV 81)
ISSN 1020-6167
©World Health Organization 2004
All rights reserved. Publications of the World Health Organization can be obtained from Marketing and Dissemination, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel: +41 22 791 2476; fax: +41 22 791 4857; email: bookorders@who.int). Requests for permission to reproduce or translate WHO publications — whether for sale or for noncommercial distribution — should be addressed to Publications, at the above address (fax: +41 22 791 4806; email: permissions@who.int).
The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement.
The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters.
The World Health Organization does not warrant that the information contained in this publication is complete and correct and shall not be liable for any damages incurred as a result of its use.
Risk assessment activities of the International Programme on Chemical Safety, including the production of Concise International Chemical Assessment Documents, are supported financially by the Department of Health and Department for Environment, Food & Rural Affairs, UK, Environmental Protection Agency, Food and Drug Administration, and National Institute of Environmental Health Sciences, USA, European Commission, German Federal Ministry of Environment, Nature Conservation and Nuclear Safety, Health Canada, Japanese Ministry of Health, Labour and Welfare, and the Swiss Agency for Environment, Forests and Landscape.
Technically and linguistically edited by Marla Sheffer, Ottawa, Canada, and printed by Wissenchaftliche Verlagsgesellschaft mbH, Stuttgart, Germany
Concise International Chemical Assessment Documents (CICADs) are the latest in a family of publications from the International Programme on Chemical Safety (IPCS) — a cooperative programme of the World Health Organization (WHO), the International Labour Organization (ILO), and the United Nations Environment Programme (UNEP). CICADs join the Environmental Health Criteria documents (EHCs) as authoritative documents on the risk assessment of chemicals.
International Chemical Safety Cards on the relevant chemical(s) are attached at the end of the CICAD, to provide the reader with concise information on the protection of human health and on emergency action. They are produced in a separate peer-reviewed procedure at IPCS. They may be complemented by information from IPCS Poison Information Monographs (PIM), similarly produced separately from the CICAD process.
CICADs are concise documents that provide summaries of the relevant scientific information concerning the potential effects of chemicals upon human health and/or the environment. They are usually based on selected national or regional evaluation documents or on existing EHCs. Before acceptance for publication as CICADs by IPCS, these documents undergo extensive peer review by internationally selected experts to ensure their completeness, accuracy in the way in which the original data are represented, and the validity of the conclusions drawn.
The primary objective of CICADs is characterization of hazard and dose-response from exposure to a chemical. CICADs are not a summary of all available data on a particular chemical; rather, they include only that information considered critical for characterization of the risk posed by the chemical. The critical studies are, however, presented in sufficient detail to support the conclusions drawn. For additional information, the reader should consult the identified source documents upon which the CICAD has been based.
Risks to human health and the environment will vary considerably depending upon the type and extent of exposure. Responsible authorities are strongly encouraged to characterize risk on the basis of locally measured or predicted exposure scenarios. To assist the reader, examples of exposure estimation and risk characterization are provided in CICADs, whenever possible. These examples cannot be considered as representing all possible exposure situations, but are provided as guidance only. The reader is referred to EHC 170.1
While every effort is made to ensure that CICADs represent the current status of knowledge, new information is being developed constantly. Unless otherwise stated, CICADs are based on a search of the scientific literature to the date shown in the executive summary. In the event that a reader becomes aware of new information that would change the conclusions drawn in a CICAD, the reader is requested to contact IPCS to inform it of the new information.
Procedures
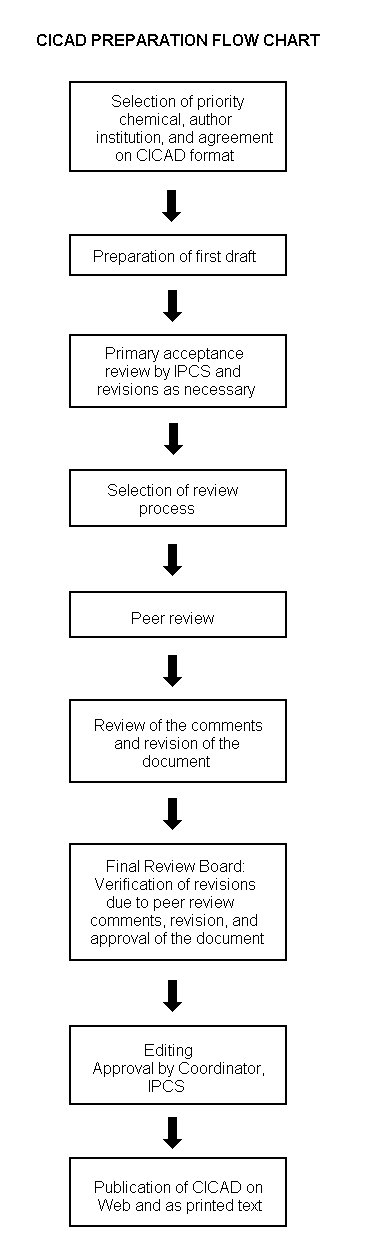
The flow chart on page 2 shows the procedures followed to produce a CICAD. These procedures are designed to take advantage of the expertise that exists around the world — expertise that is required to produce the high-quality evaluations of toxicological, exposure, and other data that are necessary for assessing risks to human health and/or the environment. The IPCS Risk Assessment Steering Group advises the Coordinator, IPCS, on the selection of chemicals for an IPCS risk assessment based on the following criteria:
Thus, it is typical of a priority chemical that
|
Advice from Risk Assessment Steering Group Criteria of priority:
Thus, it is typical of a priority chemical that
Special emphasis is placed on avoiding duplication of effort by WHO and other international organizations. A prerequisite of the production of a CICAD is the availability of a recent high-quality national/regional risk assessment document = source document. The source document and the CICAD may be produced in parallel. If the source document does not contain an environmental section, this may be produced de novo, provided it is not controversial. If no source document is available, IPCS may produce a de novo risk assessment document if the cost is justified. Depending on the complexity and extent of controversy of the issues involved, the steering group may advise on different levels of peer review:
|
The Steering Group will also advise IPCS on the appropriate form of the document (i.e., a standard CICAD or a de novo CICAD) and which institution bears the responsibility of the document production, as well as on the type and extent of the international peer review.
The first draft is usually based on an existing national, regional, or international review. When no appropriate source document is available, a CICAD may be produced de novo. Authors of the first draft are usually, but not necessarily, from the institution that developed the original review. A standard outline has been developed to encourage consistency in form. The first draft undergoes primary review by IPCS to ensure that it meets the specified criteria for CICADs.
The second stage involves international peer review by scientists known for their particular expertise and by scientists selected from an international roster compiled by IPCS through recommendations from IPCS national Contact Points and from IPCS Participating Institutions. Adequate time is allowed for the selected experts to undertake a thorough review. Authors are required to take reviewers’ comments into account and revise their draft, if necessary. The resulting second draft is submitted to a Final Review Board together with the reviewers’ comments. At any stage in the international review process, a consultative group may be necessary to address specific areas of the science. When a CICAD is prepared de novo, a consultative group is normally convened.
The CICAD Final Review Board has several important functions:
Board members serve in their personal capacity, not as representatives of any organization, government, or industry. They are selected because of their expertise in human and environmental toxicology or because of their experience in the regulation of chemicals. Boards are chosen according to the range of expertise required for a meeting and the need for balanced geographic representation.
Board members, authors, reviewers, consultants, and advisers who participate in the preparation of a CICAD are required to declare any real or potential conflict of interest in relation to the subjects under discussion at any stage of the process. Representatives of nongovernmental organizations may be invited to observe the proceedings of the Final Review Board. Observers may participate in Board discussions only at the invitation of the Chairperson, and they may not participate in the final decision-making process.
This CICAD on chloroform was drafted by Toxicology Advice & Consulting Ltd based on documentation prepared by Environment Canada and Health Canada as part of the Priority Substances Program under the Canadian Environmental Protection Act (CEPA). The objective of assessments of priority substances under CEPA is to assess potential effects of indirect exposure in the general environment on human health as well as environmental effects. Data identified as of October 1999 were considered in the source document (Environment Canada & Health Canada, 2001). A comprehensive literature search of several on-line databases and other sources was conducted in February 2003 to identify any key references published subsequent to those incorporated in the source document. Information on the nature of the peer review and the availability of the source document is presented in Appendix 1. Information on the peer review of this CICAD is presented in Appendix 2. This CICAD was approved as an international assessment at a meeting of the Final Review Board, held in Varna, Bulgaria, on 8-11 September 2003. Participants at the Final Review Board meeting are listed in Appendix 3. The International Chemical Safety Card (ICSC 0027) for chloroform, produced by the International Programme on Chemical Safety (IPCS, 2000a), has also been reproduced in this document.
Chloroform (CAS No.
The total global flux of chloroform through the environment is approximately 660 000 tonnes per year, and about 90% of emissions are natural in origin. In the late 1990s, some 520 000 tonnes were manufactured annually, mainly in the USA, the European Union, and Japan. A major use is in the production of chlorodifluoromethane (HCFC-22), which is used (in decreasing quantities) as a refrigerant and (increasingly) as a fluoropolymer feedstock. Chloroform may be released into the environment from HCFC-22 plants. The other main chloroform releases to the environment occur as a result of using chlorine-based chemicals for bleaching and disinfection purposes at pulp and paper mills and water treatment plants.
Chloroform volatilizes readily from soil and surface water and undergoes degradation in air to produce phosgene, dichloromethane, formyl chloride, carbon monoxide, carbon dioxide, and hydrogen chloride. Its half-life in air ranges from 55 to 620 days. Biodegradation in water and soil is slow. Chloroform does not bioaccumulate to any significant extent in aquatic organisms. Chloroform is detected in outdoor air, usually at concentrations below 1 µg/m3. Indoor air concentrations can be approximately 10-fold higher, but may rise to about 1000 µg/m3 temporarily during hot-water showering in a poorly ventilated shower compartment. In drinking-water, mean chloroform concentrations of about 10-90 µg/litre have been reported in Canada. Mean total intake from food, drinking-water, and air was approximately 0.6-10 µg/kg body weight per day.
Chloroform is absorbed, metabolized, and eliminated rapidly by mammals following oral, inhalation, and dermal exposure. Oxidative metabolism, mainly CYP2E1 dependent, generates carbon dioxide as well as the toxic metabolites phosgene and hydrochloric acid. Metabolism of chloroform is much faster in mice than in humans.
Neat chloroform was irritating to human and rabbit eyes and to the skin of rabbits. Inhalation of chloroform causes anaesthesia in humans. Nasal lesions have also been observed in rats and mice exposed by inhalation or via the oral route. Laboratory animal studies identify the liver and kidneys as the key target organs of chloroform’s toxic potential, and limited data suggest that the liver and kidneys are the likely target organs in humans also. Informative epidemiological studies on chloroform were not identified. In laboratory animal bioassays, chloroform induced liver and kidney tumours. In rats, the only convincing evidence of carcinogenicity was an increase in kidney tumours in males given chloroform in a corn oil vehicle or in drinking-water. Kidney tumours were also seen in male mice exposed by inhalation or by ingestion in a toothpaste vehicle. In addition, male and female mice developed liver tumours when chloroform was delivered by gavage in a corn oil vehicle. Extensive investigation of chloroform’s genotoxicity potential generally failed to identify any activity, although some studies suggest that it may be weakly genotoxic in rats. A weight-of-evidence approach suggests that chloroform does not have significant genotoxic potential. There is convincing experimental evidence that the liver and kidney tumours seen in mice are a secondary consequence of sustained cytotoxicity (presumably due to metabolites such as phosgene and hydrogen chloride) and persistent associated reparative cell proliferation. Experimental support for a similar mechanism underlying the development of kidney tumours in male rats is more limited, but the data that are available are consistent with the proposed mechanism. Reproductive and developmental studies in a range of laboratory animal species suggest that chloroform is not a specific developmental toxin and is fetotoxic only at doses that cause maternal toxicity.
On repeated inhalation exposure, the lowest reported effect level in a laboratory animal study was 9.8 mg/m3, which caused cellular proliferation in nasal passage tissues of rats and mice. For repeated oral exposure, lowest reported effect levels were similar (10-17 mg/kg body weight per day) in various species for different end-points. A physiologically based pharmacokinetic (PBPK) model and the results from a 7.5-year dog study in which mild liver toxicity (fatty cysts suggestive of disruption of hepatic metabolism of fat) was seen were used to predict the rate of chloroform metabolism in the human liver (3.8 mg/litre per hour) that would produce a tissue dose rate of toxic metabolites associated with a 5% increase in risk. This tissue dose rate would result from lifetime drinking of water containing chloroform at 37 mg/litre or lifetime exposure to 9.8 mg chloroform/m3 air. Respective lower 95% confidence limits were 12 mg/litre and 3.4 mg/m3. A tolerable daily oral intake of 0.015 mg/kg body weight per day and a tolerable concentration of 0.14 mg/m3 air are derived from these figures.
In addition, the PBPK model and the results from a study in which chloroform induced kidney tumours in male rats were used to derive analogous human rates of metabolism leading to a 5% increase in the incidence of tumours and tumour precursor lesions. These were estimated to be 3.9 and 1.7 mg/litre per hour, respectively. For the former, the 95% lower confidence limits for continuous exposure via drinking-water and via air were 2363 mg/litre and 74 mg/m3, respectively. For the latter, the metabolic rate was equivalent to continuous exposure at 1477 mg/litre water and 33.3 mg/m3 air (95% lower confidence limits were not given).
In a sample risk characterization, the margins between estimated exposure of the general population in Canada and tumorigenic and benchmark doses for cancer and non-cancer effects, respectively, for chloroform were greater than 2 orders of magnitude.
The lowest concentration reported to cause cellular proliferation in the nasal cavities of rats and mice (9.8 mg/m3) is 4298 and 1225 times higher, respectively, than the midpoint (2.28 µg/m3) and 95th percentile (8.0 µg/m3) estimates for chloroform in indoor air in Canada.
No toxicity data were identified for birds or wild mammals, but laboratory animal data indicate that atmospheric emissions of chloroform do not pose any significant risks to terrestrial wildlife. No directly relevant data were available for estimating potentially harmful concentrations in soil. For aquatic organisms, concentrations in surface waters are rarely above estimated toxicity thresholds, even for sensitive species. There is some uncertainty regarding exposure levels — and hence possible risks to aquatic organisms — near industrial leachate sources such as pulp and paper mills, water treatment plants, and landfill sites.
Chloroform (CAS No.
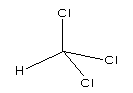
Fig. 1: Chemical structure of chloroform
At room temperature, chloroform is a clear, colourless, volatile liquid with a pleasant etheric odour. The ranges of values reported for selected physical/chemical properties are presented in Table 1. Additional properties are given in the International Chemical Safety Card (ICSC 0027) reproduced in this document.
Table 1: Physical and chemical properties of chloroform.
|
Property |
Valuea |
|
Boiling point (°C) at 101.3 kPa |
61.3 |
|
Vapour pressure (kPa) at 20 °C |
21.3 |
|
Water solubility (g/litre) at 25 °C |
7.2-9.3 |
|
Density (g/cm3) at 25 °C |
1.48 |
|
Henry’s law constant (Pa·m3/mol) at 20 °C |
304 |
|
Log Kow |
1.97 |
|
Log Koc |
1.44-2.79 |
a Data listed in source document (Environment Canada & Health Canada, 2001).
The conversion factors2 for chloroform in air at 20 °C and 101.3 kPa are as follows:
1 ppm = 4.96 mg/m3
1 mg/m3 = 0.202 ppm
In this CICAD, we have followed the convention of the source document, which is to use conversion factors at 25 °C instead of 20 °C:
1 ppm = 4.9 mg/m3
1 mg/m3 = 0.204 ppm
The general method of quantifying chloroform in water samples involves preservation of samples with sodium thiosulfate, without prior pH adjustment, and analysis by gas chromatography (GC) with electron capture detection (ECD), halogen-specific detection, or mass-selective detection. There are two recommended International Organization for Standardization methods (ISO, 1997). The first involves liquid-liquid extraction GC with ECD or other suitable detector. Pentane, hexane, petroleum ether, heptane, or xylene (for wastewater) are used as extraction solvents, and the quantification limits are 0.05-0.3 µg/litre. The second method involves static headspace GC with ECD or other suitable detector and has a quantification limit of 0.3 µg/litre. The recommended US Environmental Protection Agency (EPA) methods involve purge and trap GC with electrolytic conductivity or microcoulometric detectors (EPA Method 502) or purge and trap GC-mass spectrometry (MS) (EPA Method 524). Quantification limits are 0.02-0.2 µg/litre.
A number of analytical methods may be used to determine chloroform concentrations in air. The most common procedures use GC techniques with ECD, flame ionization detection, photoionization detection, or MS. Chloroform can be measured directly in a procedure in which air is aspirated or injected directly into the measuring instrument without pretreatment. Although these methods are simple, they can be used only when chloroform is present in the air at relatively high levels (e.g., urban source areas). In a second major method (adsorption-liquid desorption), air samples are passed through an activated adsorbing agent (e.g., charcoal or Porapak-N). The adsorbed chloroform is then desorbed with an appropriate solvent (e.g., carbon disulfide or methanol) and subsequently passed through the GC for measurement. In the adsorption-thermal desorption technique, air samples are also passed through an activated absorbing agent (e.g., Tenax-GC, Porapak-Q, Porapak-N, or carbon molecular sieve). The adsorbed chloroform is then thermally desorbed and driven into the GC column for determination. The fourth major technique (cold trap-heating) involves injection of air samples into a cold trap (liquid nitrogen or liquid oxygen is used for cooling). The trap is then heated while transferring its chloroform content into the column of a GC for measurement. Details on currently used methods may be obtained from the US Occupational Safety and Health Administration (OSHA), United Kingdom Health and Safety Executive, American Society for Testing and Materials (ASTM), US National Institute for Occupational Safety and Health (NIOSH), and US EPA.3
The sensitivity of analytical methods has improved over time; lowest detection limits reported in the source document are 0.1 µg/m3 in air (T. Dann, personal communication, 1998), 0.001 µg/litre in water (Comba et al., 1993), 0.05 µg/kg in dry food (Page & Lacroix, 1993), and 0.02 µg/kg in beverages (McNeal et al., 1995).
Based on estimated half-time and measured concentrations in different parts of the world, the total release of chloroform to the air was estimated to be 470 000 tonnes per year (Khalil & Rasmussen, 1999). A review paper published in 2003 reported that the chloroform flux through the environment is apparently constant at some 660 000 tonnes per year and that about 90% of emissions are natural in origin. This global flux consisted of 360 000 ± 90 000, 220 000 ± 100 000, <20 000, and 66 000 ± 23 000 tonnes per year from offshore seawater, soil processes, other natural sources (including volcanic activity and geological), and anthropogenic activities, respectively (McCulloch, 2003).
The natural production of chloroform by marine macroalgae has been reported (Nightingale et al., 1995; Scarratt & Moore, 1999). The release of chloroform (12 µg/m2 per day) from an organic-rich spruce forest soil, under aerobic conditions in the laboratory, suggested biogenic formation (Haselmann et al., 2000). Chloroform was found at 20-30 µg/m3 in soil air down to a depth of 160 cm, compared with a reported value of about 0.1 µg/m3 in the atmosphere. Soil spiking studies with radiolabelled chloride (Na37Cl) demonstrated natural formation in soil. Fungi were believed to play an important role in this natural production (Hoekstra et al., 1998).
One group has suggested that natural and anthropogenic sources make approximately equal contributions to atmospheric chloroform. This group measured chloroform flux at seven peatland locations and two evergreen forested bog sites in Ireland in 1998 and estimated annual global fluxes of 4700 (range 100-151 900) tonnes per year from peatland ecosystems and 24 100 (range not given) tonnes per year from total wetlands (Dimmer et al., 2001). As mentioned above, McCulloch (2003) reported an approximate global chloroform flux of 660 000 tonnes per year and estimated that about 90% of these emissions were natural in origin.
Chloroform can be released to the environment from direct processes (production, storage, transit, or use) or as a result of its formation from other substances, in processes such as paper bleaching with chlorine and water chlorination. Pulp and paper mills, municipal wastewater treatment plants, chemical manufacturing plants, and waste incinerators represent anthropogenic sources of chloroform (IPCS, 1994a). Various organic compounds present in natural waters, particularly humic and fulvic acids derived from soils and the decomposition of plant material, may contribute to the formation of chloroform (via the "haloform reaction") in areas where the drinking-water has been chlorinated (Environment Canada & Health Canada, 2001). As mentioned above, McCulloch (2003) reported that anthropogenic sources contribute about 66 000 ± 23 000 tonnes per year.
An industrial survey carried out in Canada revealed reported releases, by 23 pulp and paper mills, of 288 tonnes of chloroform into the atmosphere, 15.6 tonnes into water bodies, 0.019 tonnes into wastewater treatment plants, and 0.127 tonnes into landfills in 1996 (Environment Canada, 1997a). Chloroform generation and concentrations in effluents of these mills are reduced significantly when chlorine dioxide is substituted for elemental chlorine in the bleaching process (Solomon et al., 1994; M. Henteleff, personal communication to Environment Canada, 1999).
In 1996, total on-site environmental releases of chloroform reported to the Canadian National Pollutant Release Inventory were 208 tonnes. Almost all was released by the pulp, paper, and allied products industry; more than 96% was released to the atmosphere, with the remainder being released to water (NPRI, 1999).
Although not quantified, Canadian municipal wastewater treatment plant disinfection systems that use chlorine can be significant sources of chloroform. Chloroform is produced by the reaction between chlorine and organic precursor molecules such as fulvic and humic acids (Environment Canada, 1999a; Environment Canada & Health Canada, 2001).
Chloroform can also be released from industrial plants. A Canadian survey revealed that three facilities belonging to members of the Canadian Chemical Producers’ Association released a total of 145 kg chloroform in 1996, of which 88% was released to air (Environment Canada, 1997a). The Canadian Chemical Producers’ Association estimated that its member companies released 540 kg to the environment in 1992 (CCPA, 1992). In 1993, chloroform releases as a result of its use in HCFC-22 production were estimated to range from 31 to 1040 kg (Environment Canada & Health Canada, 2001).
Chloroform is manufactured mainly in the USA, the European Union, and Japan, the total global capacity in the late 1990s being about 520 000 tonnes per year (McCulloch, 2003). In 1995, chloroform was produced in 19 countries. The volume of production of chloroform in the USA was 229 000 tonnes in 1991 and 216 000 tonnes in 1993 (IARC, 1999). Chloroform is no longer produced in Canada (Environment Canada & Health Canada, 2001). The total production in the European Union has been estimated at 316 000 tonnes (ECSA, 1997).
Chloroform’s main use is in HCFC-22 production, and this accounts for 90-95% of its use in the European Union (Zok et al., 1998). Although use of HCFC-22 in refrigerant applications is decreasing, increasing use of HCFC-22 as the feedstock for fluoropolymers such as polytetrafluoroethylene means that demand for chloroform has remained relatively constant. Earlier use of chloroform as an anaesthetic has been largely discontinued in Canada, but it still has limited use in some dental procedures and in certain pharmaceuticals. The Montreal Protocol, as amended, means that HCFC-22 will be phased out between 2010 and 2020, effectively eliminating much of the present market for chloroform (Environment Canada & Health Canada, 2001). Worldwide, chloroform is also used in pesticide formulations, as a solvent for fats, oils, rubber, alkaloids, waxes, gutta-percha, and resins, as a cleansing agent, in fire extinguishers, and in the rubber industry (ESCA, 1997; Budavari, 2001).
Chloroform emitted to air reacts primarily with photochemically generated hydroxyl radicals in the troposphere (Kindler et al., 1995). Reaction products include phosgene, dichloromethane, formyl chloride, carbon monoxide, carbon dioxide, and hydrogen chloride (Gürtler & Kleinermanns, 1994). Experimentally derived rate constants for this reaction at 25 °C range from 1.0 × 10-13 to 2.95 × 10-13 cm3/molecule per second. Its rate of decomposition depends on a number of factors, including temperature, hydroxyl radical concentration, and the number of hours of sunshine. Estimated half-lives vary between about 55 and 620 days (Derwent & Eggleton, 1978; Singh et al., 1981; Klöpffer et al., 1988; Khalil & Rasmussen, 1999). Wet deposition is considered minor, as most will return to the air by volatilization (Diamond et al., 1994). Mass destruction rates have been estimated to be 250 000-570 000 and 120 000-260 000 tonnes per year in the northern and southern hemispheres, respectively (McCulloch, 2003).
In surface water, the principal removal process is volatilization. Modelling studies have generated estimated half-lives of 1.5 days and 9-10 days in a river and a lake, respectively (US EPA, 1984). Other models have indicated shorter half-lives in shallow, well-mixed systems with high wind velocities (Kaczmar, 1979; Lyman et al., 1982). Most studies have indicated little biodegradation after up to 25 weeks in aquatic systems under aerobic conditions (Bouwer et al., 1981; Wilson et al., 1981, 1983; Bouwer & McCarty, 1984). In groundwater, restricted volatilization and slow biodegradation (under anaerobic conditions) or no biodegradation (under most aerobic conditions) means that chloroform may be quite persistent (Environment Canada & Health Canada, 2001). The half-life by hydrolysis has been reported to be greater than 1000 years (McCulloch, 2003).
Limited studies suggest that chemical degradation in sediments is not rapid, except under anaerobic methanogenic conditions. The major degradation products under anaerobic conditions are carbon dioxide, methane, and hydrogen chloride, with smaller amounts of dichloromethane. Under anaerobic conditions, chloroform had half-lives of 12 days at 10 °C and 2.6 days at 20 °C (Van Beelen & Van Keulen, 1990). In another study carried out under anaerobic conditions, chloroform was degraded in muddy sediments with a half-life of 2-37 days, whereas no degradation could be demonstrated in sandy sediments (Van Beelen & Van Vlaardingen, 1993).
The principal fate of chloroform at the soil surface is temperature-dependent volatilization, due to its volatile nature and low soil adsorption. A microcosm study involving daily application of wastewater containing chloroform to soil found that 75% of applied chloroform volatilized to the air, while the remainder leached through the soil (Piwoni et al., 1986). Chloroform adsorption is correlated with soil clay content (Dural & Peng, 1995). Limited studies suggest that chemical degradation in soil is not rapid, except under anaerobic methanogenic conditions. The major degradation products under anaerobic conditions are carbon dioxide, methane, and hydrogen chloride, with smaller amounts of dichloromethane (Van Beelen & Van Keulen, 1990; Van Beelen & Van Vlaardingen, 1993).
The octanol/water partition coefficient (log Kow = 1.97) indicates that chloroform is unlikely to bioaccumulate to any significant extent in aquatic biota (Anderson & Lusty, 1980; Zok et al., 1998). Reported bioconcentration factors include 690 in green algae (Mailhot, 1987) and 1.4-10 in various fish (bluegill Lepomis macrochirus, rainbow trout Oncorhynchus mykiss, largemouth bass Micropterus salmoides, and channel catfish Ictalurus punctatus) (Veith et al., 1978; Anderson & Lusty, 1980; Barrows et al., 1980). Depuration is rapid, with a half-life of less than 1 day in all of the above fish species (Anderson & Lusty, 1980; Barrows et al., 1980).
Chloroform in soil or surface water volatilizes readily; at equilibrium, greater than 99% is expected to partition to the atmosphere (Zok et al., 1998; McCulloch, 2003). Due to chloroform’s water solubility, some wet deposition of atmospheric chloroform may occur, but subsequent revolatilization is likely to be extensive (Diamond et al., 1994). Chloroform is not expected to partition significantly to soils or sediments, because its affinity for organic carbon and lipids is low. Modelling has predicted that the percentage of chloroform in water transferred to bottom sediments would range from <0.06% in lakes to 8% in ponds (Anderson et al., 1985). Compartmental partitioning has been reported to be 99.1%, 0.9%, 0.01%, and 0.01% in air, water, soil, and sediment, respectively (Zok et al., 1998).
Over land, there is substantial variability in chloroform concentration. Mean levels in urban and/or industrial areas ranged up to 3.5 µg/m3, with most concentrations in the range 0.5-1.5 µg/m3 (McCulloch, 2003). Median values in air from Madeira, the Portugese coast, and the Black Forest in a 1991 report were 0.2-0.6 µg/m3 (range 0.07-8.7 µg/m3), and measurements reported in a 1975 paper quantified chloroform at 0.12-0.6 µg/m3 in rural air in the United Kingdom (McCulloch, 2003).
Chloroform was detected (i.e., above the detection limit of 0.1 µg/m3) in approximately 69% of 8807 24-h samples collected from 47 sites in seven Canadian provinces between 1989 and 1996. During this period, annual median and mean concentrations ranged from <0.1 to 0.18 µg/m3 and from 0.12 to 0.23 µg/m3, respectively. Concentrations were lowest at rural sites, higher at urban sites, and highest immediately adjacent to major roadways. Comparison of 3344 samples taken during 1989-1992 with 5463 samples taken between 1993 and 1996 indicated that chloroform concentrations were slightly lower in the more recent period. The highest 24-h average concentration detected before 1996 was 6.0 µg/m3, compared with 0.75 µg/m3 during 1996 (T. Dann, personal communication, 1998).
Atmospheric chloroform levels of 0.1-10 µg/m3 and 1.4-110 µg/m3 have been reported for urban and source-dominated areas in the USA (ATSDR, 1996).
Based on a 9-year series of measurements at the surface of the polar, middle, and tropical latitudes of both hemispheres, an average surface concentration of 0.09 µg/m3 was reported, although annual averages at some continental locations rose to 0.2 µg/m3. No significant trends were seen during the period of measurement (Khalil & Rasmussen, 1999).
Chloroform levels in the air over the southern Atlantic Ocean were reported to be 0.05-0.1 µg/m3. Equivalent figures for the northern hemisphere were 0.1-0.25 µg/m3, with lower levels of 0.04-0.07 µg/m3 above the trade wind system. These figures were based on lower troposphere samples taken aboard a ship cruising from Capetown to Bremerhaven during 1985, together with samples taken in the Azores in 1982, in Madeira in 1984, and in Bermuda in 1985 (Class & Ballschmiter, 1986).
Chloroform was detected (detection limit 3.5 µg/m3) in 11% of 24-h samples taken in 754 residences in nine Canadian provinces during 1991. The maximum concentration found was 68.6 µg/m3 (Concord Environmental Corporation, 1992; Health Canada, 1999). Chloroform was detected (detection limit 2.3 µg/m3) in 8 of 44 households in Ontario (Greater Toronto Area), Nova Scotia, and Alberta during 1996 and in 34 of 50 households (detection limit 0.22 µg/m3) in the same areas during 1997. The maximum concentration detected was 14.1 µg/m3, and the estimated mean concentration for the total of 94 samples (assuming chloroform was present at half of the appropriate detection limit for each "non-detect" sample) was 1.5 µg/m3. Single personal breathing zone samples taken from all 94 households had concentrations ranging from undetected (<0.22 µg/m3) to 94.5 µg/m3, with an overall estimated mean of 2.6 µg/m3 (Otson & Meek, 1996; Conor Pacific Environmental, 1998; Health Canada, 1999). Indoor air sampling found chloroform in 89 of 146 households in Windsor, Ontario, during 1991-1992 (detection limit unknown). In the indoor air of non-smoking households, the highest mean concentration was 5.6 µg/m3; the concentration was higher (16 µg/m3) where environmental tobacco smoke was present (OMEE, 1994). However, no differences in concentrations were seen in the air of 61 non-smoking households and 32 smoking households in New Jersey, USA, in 1962 (means 0.60 and 0.85 µg/m3, respectively; medians 0.28 and 0.23 µg/m3, respectively) (Heavner et al., 1996), and tobacco was not identified by IPCS (1994a) as an important source of environmental chloroform exposure. Mean concentrations of 0.17-43.9 µg/m3 (maximum 210 µg/m3) have been reported for indoor air in the USA (Samfield, 1992). Mean concentrations in 248 homes in Los Angeles, California, USA, in 1987 ranged from 0.9 to 1.5 µg/m3 (maximum 13 µg/m3) (Wallace, 1997).
Indoor air concentrations of chloroform may rise for short periods of time due to volatilization from hot water. In particular, chloroform concentrations in the shower compartment while taking a shower may exceed 1000 µg/m3, due to volatilization of more than 50% of dissolved chloroform (Tancrède et al., 1992; Giardino & Andelman, 1996; Health Canada, 1999).
Chloroform concentrations ranging from 0.002 to 0.015 µg/litre have been reported for the open ocean (Class & Ballschmiter, 1986; IPCS, 1994a; Zok et al., 1998). A review reported chloroform concentrations in North Sea coastal water and estuarine waters of several European countries (including France, Germany, Sweden, and the United Kingdom) in the 1980s to 1990s to range from 0.004 to 11.5 µg/litre. Typical background levels in rivers in non-industrialized areas were generally below 0.5 µg/litre, while levels of up to 10 µg/litre were detected in rivers in industrialized areas or in the vicinity of emission points from municipal wastewater treatment plants (Zok et al., 1998). Chloroform concentrations ranging from <0.01 to 70 µg/litre have been reported for European estuaries, with locally high levels related to point source discharges (McCulloch, 2003).
Analysis of 59 samples of surface water and well water in Alberta, Canada, during 1990-1995 revealed only two samples (containing 2 and 7 µg/litre) above the 1 µg/litre detection limit (Alberta Environment, 1996). Chloroform was detected (detection limit 1 µg/litre) in only a few of 321 samples of surface water from Alberta in 1990-1996, with the highest concentration reported as 2 µg/litre (Environment Canada, 1996). In water from Lake Superior, chloroform levels varied from <0.001 to 4.2 µg/litre (median 0.064 µg/litre) in 192 samples taken during 1991 (Comba et al., 1993), and a maximum level of 0.19 µg/litre was found in 293 samples taken from the Niagara River during 1990-1993 (Environment Canada, 1996). Concentrations in 107 samples of Quebec surface waters collected from 1990 to 1993 ranged from non-detectable (<0.2 µg/litre) to 44 µg/litre (MENVIQ, 1996). Across four Canadian provinces, the median value (of 984 measurements) was <0.2 µg/litre, and the 95th and 99th percentiles were <1 and 2.94 µg/litre, respectively (Environment Canada & Health Canada, 2001).
High concentrations have occasionally been reported in Canadian surface waters, particularly near pulp and paper mills using chlorine bleaching (e.g., chloroform concentrations below a mill in 1986 ranged from 80 to 200 µg/litre) (OMOE, 1990). Similarly, concentrations of up to 394 µg/litre were reported in rivers sampled in the 1970s in highly industrial US cities (IPCS, 1994a).
Rainwater concentrations of 11-17 ng/litre and up to 97 ng/litre were reported in two studies carried out in the Black Forest area in the 1990s (McCulloch, 2003).
Chloroform is the principal by-product of water disinfection processes such as chlorine-chloramine, chlorine-chlorine, and ozone-chlorine treatment. Levels vary widely depending upon concentrations of organic materials in the raw water and are also influenced by treatment method, temperature, and pH; chloroform concentrations increase in summer months, as the water moves along the distribution system, and in domestic hot water tanks (Environment Canada & Health Canada, 2001). The majority of data collected in Canada originate from measurements collected within water treatment plants and distribution systems; little information on levels at the consumer tap is available. Concentrations measured (detection limits 0.1-1.0 µg/litre) in drinking-water in several areas in Canada during the 1990s are presented in Table 2. The data were used to derive a 95th-percentile value of 166 µg/litre. Using only data from the two areas of highest concentrations (to establish a "reasonable worst case"), the 95th-percentile value was 220 µg/litre (Health Canada, 1999).
Table 2: Concentrations of chloroform in drinking-water in Canada during the 1990s.
|
Province/territory |
Period |
No. of samples |
Frequency of detection (%) |
Mean concentration (µg/litre) |
Maximum concentration (µg/litre) |
|
Newfoundland |
1995-1996 |
51 |
100 |
9.6 |
29.8 |
|
New Brunswick |
1994-1996 |
410 |
100 |
9.4 |
77.4 |
|
Quebec |
1991-1995 |
165 |
95 |
51.9 |
440 |
|
Ontario |
1991-1997 |
3332 |
98 |
35.0 |
390 |
|
Manitoba |
1990-1995 |
832 |
94 |
89.4 |
1125 |
|
Alberta |
1990-1997 |
1765 |
92 |
60.6 |
1224 |
|
Northwest Territories |
1990-1992 |
52 |
75 |
27.5 |
258 |
|
All data for 1990s |
6607 |
96 |
47.3 |
1224 |
More limited data are available from smaller national studies. In 1993, median, mean, and maximum concentrations of 13.4, 27.6, and 336 µg/litre, respectively, were recorded in 214 samples collected from 53 water treatment facilities in nine Canadian provinces; chloroform was detected (>0.2 µg/litre) in all samples. Arithmetic means among the provinces varied from 6.5 to 62.1 µg/litre and were about twice as high in summer as in winter (Williams et al., 1995; Health Canada, 1999).
Chloroform was measured at 24 µg/litre in warm water entering a shower when the cold water contained about 6 µg/litre in the winter or 12 µg/litre in the summer (Benoit et al., 1997).
In a Canadian study carried out in 1992, chloroform (detection limits ranged from 0.5 to 3.0 µg/litre) was not found in any of 61 bottles of mineral water and was detected (at 3.7 µg/litre) in only 1 of 86 spring water samples. Chloroform was detected in 10 of 35 further samples of bottled water, including carbonated, demineralized, deionized, treated, and distilled waters (Page et al., 1993).
Although only limited data were identified, chloroform does not appear to be sorbed in sediments or soils to any great extent, and so it is unlikely that it will accumulate in these media to any significant extent (Environment Canada, 1999a; Environment Canada & Health Canada, 2001).
The sources of chloroform in food are not clearly understood, although migration of chloroform from packaging solvents, glues, and inks has been documented, and transfer from surfaces cleaned with chlorinated water to lipid-containing foods contacting these surfaces is a possibility. The use of chlorinated water by bottling plants (e.g., soft drink manufacturers) may explain the presence of chloroform in some beverages. Chloroform introduced to foods as a consequence of the use of chlorinated drinking-water during food preparation probably escapes by volatilization during cooking, reducing the concentrations in the table-ready foods (Environment Canada & Health Canada, 2001).
Chloroform was detected (at up to 14.8 µg/kg) in 11 of 13 beverages purchased in Ottawa, Ontario, Canada, but not in dry foods (detection limit 0.05 µg/kg). Subsequent sampling of 47 foods and beverages found chloroform in 41 samples at concentrations ranging from 0.23 to 129 µg/kg. The highest three concentrations (50-129 µg/kg) were found in butter (Page & Lacroix, 1993).
Analysis of composite food groups prepared from groceries bought from four retailers in Windsor, Ontario, Canada, found chloroform in 5 of 33 composites (cheese/butter, canned meats, vine vegetables, soft drinks, and dehydrated soups). The maximum concentration found was 67 µg/litre (Enviro-Test Laboratories, 1992). A similar study of 35 composite groups found chloroform (detection limits were 1 µg/litre in beverages and 5 µg/kg in foods) only in soft drinks and alcoholic drinks (Enviro-Test Laboratories, 1993).
Limited data are also available from the USA. Chloroform was found in 94 of 231 table-ready foods obtained from the US Food and Drug Administration’s (FDA) market basket collection, with the highest concentration (312 µg/kg) found in cheddar cheese (Daft, 1988). In an analysis of grain-based products, chloroform concentrations ranged from 0.5 µg/kg in lasagna noodles to 3400 µg/kg in wheat (Heikes & Hopper, 1986). Chloroform was found in 10 of 18 table-ready food samples; the highest concentration was 670 µg/kg in butter (Heikes, 1987). Chloroform was present at 30-255 µg/kg in 36 butter samples collected in Washington, DC (Miller & Uhler, 1988). Analysis of 234 foods revealed chloroform (detection limit 5 µg/kg) in 44 samples, including margarine (7.3 µg/kg), butter (38.9 µg/kg), and cream cheese (110 µg/kg) (Heikes et al., 1995).
Chloroform was detected at 0.1-65 µg/litre in 40 of 42 breast milk samples from nursing mothers in five US hospitals (Erickson et al., 1980).
Deterministic estimates of average and upper-bounding estimates for daily intake have been developed in Canada based on concentrations determined in Canadian air (national surveys), food in Canada and the USA, and drinking-water (provincial and territorial data) (Environment Canada & Health Canada, 2001). These are presented in Tables 3 and 4.
Table 3: Deterministic estimates of average daily intakes for the general population.a
|
Exposure medium |
Average daily intake (µg/kg body weight per day) for age groups in the general population |
||||||
|
0-6 months |
7 months - 4 years |
5-11 years |
12-19 years |
20-59 years |
60+ years |
||
|
Outdoor air |
0.002-0.034 |
0.004-0.072 |
0.003-0.056 |
0.002-0.032 |
0.001-0.027 |
0.001-0.024 |
|
|
Indoor air |
0.559-0.744 |
1.197-1.596 |
0.933-1.244 |
0.531-0.708 |
0.456-0.608 |
0.396-0.528 |
|
|
Food |
- (included in water data) |
0.150-1.145 |
0.105-0.899 |
0.060-0.612 |
0.043-0.478 |
0.028-0.349 |
|
|
Drinking-water |
1.003-9.536 |
0.424-4.037 |
0.334-3.172 |
0.190-1.806 |
0.199-1.891 |
0.209-1.987 |
|
|
Subtotal |
1.56-10.31 |
1.78-6.85 |
1.38-5.37 |
0.78-3.16 |
0.70-3.00 |
0.63-2.89 |
|
|
Showeringb |
- |
- |
- |
0.43-4.06 |
0.36-3.40 |
0.35-3.35 |
|
a Further details on the basis for estimated figures are given in Environment Canada & Health Canada (2001).
b Inhalation and dermal intake from daily showering.
Table 4: Deterministic upper-bounding estimates of daily intake for the general population.a
|
Exposure medium |
Upper-bounding estimates of intake (µg/kg body weight per day) for age groups in the general population |
|||||
|
0-6 months |
7 months - |
5-11 years |
12-19 years |
20-59 years |
60+ years |
|
|
Outdoor air |
0.21 |
0.45 |
0.35 |
0.20 |
0.17 |
0.15 |
|
Indoor air |
16.81 |
36.02 |
28.08 |
15.97 |
13.72 |
11.92 |
|
Food |
- (included in water data) |
2.87 |
2.36 |
1.58 |
1.25 |
0.89 |
|
Drinking-water |
130.6 |
55.28 |
43.43 |
24.73 |
25.90 |
27.20 |
|
Subtotal |
147.6 |
94.62 |
74.22 |
42.48 |
41.04 |
40.16 |
|
Showeringb |
- |
- |
- |
55.64 |
46.61 |
45.90 |
a Further details on the basis for estimated figures are given in Environment Canada & Health Canada (2001).
b Inhalation and dermal intake from daily showering.
Deterministic estimates were generated using the above monitoring data and reference values for body weight, inhalation volume, and consumption of food and water. Average intake from food, drinking-water, and air varied from 0.6 to 10 µg/kg body weight per day. Upper-bounding estimates were calculated using maximum reported concentrations in water, food, and air and ranged from 40 to 95 µg/kg body weight per day (or up to 148 µg/kg body weight per day for infants fed exclusively on powdered infant formula prepared with tap water containing the maximum reported chloroform concentration). Daily showering increased estimated exposure by about 50-100% for some subgroups. Further details are given in the source document (Environment Canada & Health Canada, 2001).
In addition, probabilistic estimates of daily chloroform intake from air and drinking-water in Canada were developed for two scenarios (average population exposure and reasonable worst case), but data were considered insufficient to develop probabilistic exposure estimates from food consumption or showering. Simulations of 10 000 trials were run 5 times each using Monte Carlo random and Latin Hypercube methods. The two sampling methods gave similar estimates, and relative standard deviations (for n = 5 simulations of 10 000 trials each) of the upper-percentile estimates of intake did not exceed 5%, indicating a high degree of reproducibility. The average population scenario was based on the distribution of chloroform in 8807 outdoor air samples collected during the 1990s, the estimated geometric mean and standard deviation of an assumed lognormal distribution of chloroform in the indoor air of 754 Canadian homes, and analysis of chloroform in 6607 drinking-water samples in Canadian provinces and territories. The 95th percentiles of the distribution of intakes from inhalation and ingestion of drinking-water for five age groups of the general population (i.e., 0.5 years to 60+ years of age) ranged from 4.9 to 12.9 µg/kg body weight per day (Health Canada, 1999). The limitations of the data on the daily intake rate of total tap water by infants (EHD, 1998) prevented the development of probabilistic estimates for this subgroup.
The reasonable worst-case scenario was based on 800 outdoor air samples collected during the 1990s from four sites adjacent to major Canadian roadways, the estimated geometric mean and standard deviation of an assumed lognormal distribution of chloroform in the indoor air of 754 Canadian homes, and the distribution of chloroform in 2597 drinking-water samples from the two provinces with the highest reported concentrations. The 95th percentiles of the distribution of intakes from inhalation and ingestion of drinking-water for the same five age groups of the general population ranged from 7.0 to 19.1 µg/kg body weight per day (Health Canada, 1999). The limitations of the data on the daily intake rate of total tap water by infants (EHD, 1998) prevented the development of probabilistic estimates for this subgroup.
Chloroform was found (detection limit 0.1 µg/litre) in 54% of 979 samples of human blood collected in the USA, but concentrations were not quantified (Ashley et al., 1994). Concentrations in the urine of healthy male students in New Jersey, USA, ranged from 36.5 to 48.7 µg/litre (Youssefi et al., 1978).
The HSDB (2003) chloroform record includes a brief mention of mean time-weighted average (TWA) exposures of 13, 2, and 1 mg/m3 for production operators, drummers/bottle fillers, and maintenance/utility personnel at the Shell Chemical Company, Rocky Mountain Arsenal (a pesticide plant), levels of 10-1000 mg/m3 in a Polish pharmaceutical plant, an 8-h TWA of 77.4 mg/m3 (range 13-227 mg/m3) in a police forensic laboratory, and (during 1968-1972) levels of 34-830 mg/m3 (mean 230 mg/m3, 79 samples) in a film manufacturing plant using a solvent containing 22% chloroform (Santodonato et al., 1985).
Chloroform is well absorbed, metabolized, and eliminated rapidly by mammals after oral, inhalation, or dermal exposure (IPCS, 2000b). In humans given a single oral dose of 0.5 g chloroform, about 50-52% of the dose was absorbed, and virtually all of the absorbed dose was metabolized to carbon dioxide. Blood levels peaked after 1.5 h and declined in line with a two-compartment model with half-lives of 13 and 90 min, respectively (Fry et al., 1972). Following a single inhalation exposure to approximately 5 mg [38Cl]chloroform, volunteers absorbed about 80% (Morgan et al., 1970). The relative contributions of dermal and pulmonary uptake have been studied in individuals taking showers, using post-exposure exhaled air concentrations to estimate uptake. These were 6-21 µg/m3 for normal showering and 2.4-10 µg/m3 if exposure during showering was restricted to the inhalation route ("inhalation-only" showers). This difference was statistically significant and indicated that the contributions of dermal and inhalation exposures were approximately equivalent (Jo et al., 1990).
Species differences can be seen. When rats, mice, and monkeys were given radiolabelled chloroform at 60 mg/kg body weight by the oral route, about 90% was absorbed and exhaled in all three species in the 48 h following dosing. However, while mice excreted about 85% of the dose as exhaled carbon dioxide and 5% as unchanged chloroform, monkeys exhaled only 18% as carbon dioxide and 79% as chloroform. The rat was intermediate, with 67% exhaled as carbon dioxide and 20% as chloroform. Excretion in the urine/faeces combined accounted for only about 2-3% of the dose in mice and monkeys and about 8% in rats (D.M. Brown et al., 1974). Metabolism of chloroform is much faster in mice than in humans. For example, the mean peak rate of metabolism at an inhalation exposure of 49 mg/m3 has been predicted to be approximately 78 times lower in humans than in mice (Delic et al., 2000).
Corley et al. (1990) measured radioactivity in the exhaled air, urine, faeces, carcass and skin, and cage wash in the 48 h following a 6-h inhalation exposure of rats and mice at various chloroform concentrations (49, 440, and 1790 mg/m3 for mice; 460, 1740, and 5100 mg/m3 for rats). At the low concentration, metabolism was extensive in both species. In mice, exhaled carbon dioxide, exhaled chloroform, urine, and faeces accounted for 7.22, 0.03, 0.95, and 0.05 mg equivalents/kg body weight, respectively; in rats, these figures were 31.84, 0.76, 3.34, and 0.04, respectively. However, partial saturation of metabolism was indicated at about 1800 mg/m3; in mice, the equivalent figures were 217.85, 23.03, 21.24, and 3.84 mg equivalents/kg body weight, respectively, while in rats, the equivalent figures were 54.85, 16.15, 6.53, and 0.81 mg equivalents/kg body weight, respectively (Corley et al., 1990).
Following a 10-min inhalation exposure of mice to [14C]chloroform (dose 280 mg/kg body weight), whole-body autoradiography carried out immediately after exposure or 2 h later showed high concentrations in the fat, blood, lungs, liver, kidneys, spinal cord and nerves, meninges, and cerebellar cortex. Non-volatile radioactivity was bound in the bronchi, nasal mucosa, liver, kidneys, salivary glands, and duodenal contents. High levels of volatile or extractable radioactivity were found in testes, preputial gland, and epididymis (Bergman, 1984). Transplacental transfer has been demonstrated in rats, mice, and guinea-pigs (Nicloux, 1906; Withey & Karpinski, 1985; Danielsson et al., 1986).
Both oxidative and reductive pathways of chloroform metabolism have been identified, although data in vivo are limited. Carbon dioxide is the major metabolite of chloroform generated by the oxidative pathway in vivo. The oxidative pathway also generates reactive metabolites, including phosgene (Pohl et al., 1977; Pohl & Krishna, 1978) (determined in vitro, with phenobarbital induction), while the reductive pathway generates the dichloromethylcarbene free radical (Wolf et al., 1977; Tomasi et al., 1985; Testai & Vittozzi, 1986) (determined in vitro and in vivo, both with and without phenobarbital induction). Oxidative and reductive metabolism both proceed through a cytochrome P450 (CYP)-dependent enzymatic activation step. The balance between oxidative and reductive pathways depends on species, tissue, dose, and oxygen tension. In intact mammals, oxidative tension probably precludes any significant metabolism by the reductive pathway (Testai & Vittozzi, 1986; Ammann et al., 1998). Phosgene is produced by oxidative dechlorination of chloroform to trichloromethanol, which spontaneously dehydrochlorinates (Mansuy et al., 1977; Pohl et al., 1977). Dehydrochlorination of trichloromethanol produces one molecule of hydrochloric acid, and hydrolysis of phosgene produces another two molecules, so that three molecules of hydrochloric acid are produced in the conversion of chloroform to carbon dioxide.
The electrophilic metabolite phosgene binds covalently to nucleophilic components of tissue proteins (Pohl et al., 1980). It also interacts with other cellular nucleophiles (Uehleke & Werner, 1975) and binds to some extent to the polar heads of phospholipids (Vittozzi et al., 1991). Alternatively, phosgene reacts with water to release carbon dioxide and hydrochloric acid (Fry et al., 1972; B.R. Brown et al., 1974; D.M. Brown et al., 1974). The interaction of phosgene with glutathione results in the formation of S-chlorocarbonyl glutathione, which can either interact with an additional glutathione to form diglutathionyl dithiocarbonate (Pohl et al., 1981) or form glutathione disulfide and carbon monoxide (Ahmed et al., 1977; Anders et al., 1978). Incubation of mouse renal microsomes with glutathione increases production of these metabolites from chloroform and decreases irreversible binding to proteins and further metabolism to carbon dioxide (Smith & Hook, 1984). Reduced glutathione is capable of scavenging essentially all chloroform metabolites produced in incubations with mouse liver microsomes when chloroform concentrations are not too high (Vittozzi et al., 1991). The relative importance of the minor pathways of phosgene metabolism depends upon the availability of glutathione, other thiols, and other nucleophilic compounds, such as histidine and cysteine (see Figure 2).
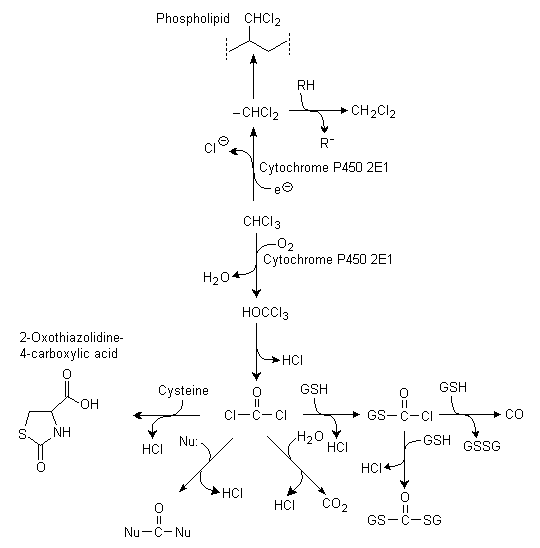
Fig. 2: Metabolism of chloroform
(GSH = glutathione; GSSG = bis(gamma-glutamyl-L-cysteinylglycine) disulfide; Nu = tissue nucleophiles; R = alkyl group)
Oxidative metabolism, with CYP2E1 (an ethanol-inducible mono-oxygenase isoenzyme system present in the liver of mammals, including humans) playing a key role, is probably the only significant in vivo pathway at low exposures, and available data indicate that oxidative metabolism has a major role in toxicity. The dominant role of CYP2E1 in metabolizing chloroform to toxic metabolites has been demonstrated in studies involving treatment of animals with enzyme inducers or inhibitors, as well as studies in mice lacking CYP2E1 (Brady et al., 1989; Guengerich et al., 1991; Nakajima et al., 1995a,b; Constan et al., 1999; see also section 8.8). Immunoinhibition studies with anti-CYP2E1 monoclonal protein have shown that CYP2E1 is responsible for 81% of the metabolism assayed at a low chloroform (0.5 mmol/litre) concentration in liver microsomes from acetone-induced rats (Brady et al., 1989). Toxicity to rat and mouse hepatocytes incubated in vitro with up to 5 mmol chloroform/litre was prevented by the addition of a CYP2E1 inhibitor or by reduced oxygen tension, underscoring the importance of oxidative metabolism in toxicity (Ammann et al., 1998). Regional distribution of liver lesions in rats and mice correlates well with the hepatic distribution of CYP2E1 and glutathione (Smith et al., 1979; Ingelman-Sundberg et al., 1988; Tsutsumi et al., 1989; Johansson et al., 1990; Dicker et al., 1991; Nakajima et al., 1995a,b).
CYP2B1 may also have a role in chloroform metabolism, although this is likely to be only minor at low tissue chloroform concentrations (studies reviewed in Environment Canada & Health Canada, 2001). However, at high tissue concentrations (e.g., resulting from an oral dose of 0.5 ml/kg body weight), chloroform hepatotoxicity was dramatically potentiated in Wistar rats treated with phenobarbital (a CYP2B1 inducer) but not in rats treated with n-hexane (a CYP2E1 inducer), compared with uninduced controls (Nakajima et al., 1995b).
A study in which rats were exposed to [14C]chloroform showed that metabolism was most active in the liver, followed by the nose and kidney. Metabolic activity was correlated with accumulation of metabolites (Löfberg & Tjälve, 1986).
The first extensive PBPK model for chloroform described liver and kidney individually as metabolic sites for chloroform. The maximum velocity of metabolism in the kidney was scaled to that in the liver, and terms were introduced to account for loss and resynthesis of metabolizing enzyme (Corley et al., 1990). This model was modified to include a description of liver cytotoxicity (Reitz et al., 1990). Later, Gearhart et al. (1993) modified the tissue:blood partition coefficients according to temperature and fitted gas uptake without the need to describe enzyme loss and resynthesis. Others subsequently incorporated absorption from the stomach as well as the gastrointestinal tract and also accounted for gastric emptying time (Dix et al., 1994; Dix & Borghoff, 1995). In 1996, kidney and liver model compartments were subdivided into regions of high and low metabolic activity (Lilly, 1996). The combination of this approach with the two-compartment absorption model of Dix & Borghoff (1995) resulted in a recent PBPK model in the "hybrid" species5 (ILSI, 1997).
Health Canada developed a model for the dog, using physiological and anatomical parameters from Brown et al. (1997), while metabolic parameters were based on the average of rat and human parameters. The fractional subvolumes for the liver were assumed to be the same as those reported for the rat by ILSI (1997) (Environment Canada & Health Canada, 2001).
Health Canada also developed a human model. Physiological parameters were derived from Brown et al. (1997), with the exception of the ventilation rate and cardiac output, which were related to an assumed breathing rate of 23 m3 air/day. ILSI (1997) was used as the source of the partition coefficients and rate constants. Liver tissue subvolumes were assumed to be the same as in the rat, while kidney was subdivided into a 70:30 cortex:non-cortex ratio. Human metabolic parameters had been determined in vitro in eight human liver samples, as reported by Corley et al. (1990). Kidney rate constants were based on the relationship of activity observed in the microsomal fraction of kidneys to the activity observed in the microsomal fraction of the liver, based on the in vitro results reported by Corley et al. (1990), but supported by data on metabolism of two known substrates of CYP2E1 by microsomal fractions of the kidney and liver from 18 humans, reported by Amet et al. (1997). As it is based on metabolized dose, the model accounts for differences in metabolism between humans and (in this case, hybrid) laboratory animals (Environment Canada & Health Canada, 2001).
Results from the human model were compared with data on total metabolized parent and exhaled chloroform reported by Fry et al. (1972), where chloroform was administered, in olive oil or gelatin capsules, to male and female volunteers. Exhaled chloroform was measured for up to 8 h following dosing, and the total percentage of the dose exhaled unchanged was calculated by extrapolation to infinite time. Human model simulations conducted using a single-compartment description of oral uptake were closer to the observations of Fry et al. (1972) than those estimated using a multi-compartment description. Therefore, while a multi-compartment description was necessary in the rat model, a single-compartment description of oral uptake was used in estimating human-equivalent concentrations (Environment Canada & Health Canada, 2001). The model was also modified to permit assessment of exposure to chloroform from all likely sources, including air, water, and food. The exposure scenario (see section 11.1.3) was modelled within a 24-h day and included inhalation, ingestion, and dermal exposure from one 10-min shower, a brief washing-up period before retiring at night, discrete periods of food and water consumption, and inhalation of chloroform at various concentrations (ICF Kaiser, 1999; Environment Canada & Health Canada, 2001).
The physiological and metabolic parameter values for rats, dogs, and humans used to exercise the PBPK model are reproduced in Table 5. In the liver, the Vmax for the metabolism of chloroform is twice as high in humans as in rats, while there is little difference in Vmax in the kidney or in the affinity (Km) in either liver or kidney (see Table 5). Further details are available in the source document (Environment Canada & Health Canada, 2001).
Table 5: PBPK model physiological and metabolic parameter values in rats, dogs, and humans.
|
Rat (ILSI, 1997) |
Rat (ILSI, 1997, modified) |
Dog |
Human |
|
|
Weights |
||||
|
Body (kg) |
0.40 |
0.40 |
15.0 |
70.0 |
|
% of body weight |
||||
|
Fat |
0.063 |
0.124 |
0.145 |
0.2142 |
|
Kidney |
0.0071 |
0.0073 |
0.0055 |
0.0044 |
|
Liver |
0.0253 |
0.0366 |
0.0329 |
0.0257 |
|
Rapidly perfused |
0.0439 |
0.0621 |
0.0836 |
0.0709 |
|
Slowly perfused |
0.77 |
0.594 |
0.548 |
0.4368 |
|
Fractional tissue subvolumes |
||||
|
Liver periportal |
0.58 |
0.58 |
0.58 |
0.58 |
|
Liver centrilobular |
0.42 |
0.42 |
0.42 |
0.42 |
|
Kidney cortical |
0.76 |
0.76 |
0.73 |
0.70 |
|
Kidney non-cortical |
024 |
0.24 |
0.27 |
0.30 |
|
Flows (litre/h) |
||||
|
Alveolar ventilation (litre/h for 1-kg animal) |
15.0 |
24.2 |
28.5 |
24.0 |
|
Cardiac output (litre/h for 1-kg animal) |
15.0 |
14.4 |
30.9 |
16.5 |
|
% of cardiac output |
||||
|
Fat |
0.05 |
0.07 |
0.07 |
0.052 |
|
Kidney |
0.25 |
0.141 |
0.173 |
0.175 |
|
Liver |
0.25 |
0.183 |
0.297 |
0.227 |
|
Slowly perfused |
0.19 |
0.336 |
0.277 |
0.249 |
|
Partition coefficients |
||||
|
Blood/air |
20.8 |
20.8 |
20.8 |
7.43 |
|
Fat/air |
203 |
203 |
203 |
280 |
|
Kidney/air |
11 |
11 |
11 |
11 |
|
Liver/air |
21.1 |
21.1 |
17.0 |
17.0 |
|
Rapidly perfused/air |
21.1 |
21.1 |
21.0 |
17.0 |
|
Slowly perfused/air |
13.9 |
13.9 |
13.9 |
12.0 |
|
Metabolic constants |
||||
|
VmaxC for liver (mg/h for 1-kg animal) |
6.44 |
6.44 |
11.025 |
15.7 |
|
Km for liver (mg/litre) |
0.543 |
0.543 |
0.496 |
0.448 |
|
VmaxC for kidney (mg/h for 1-kg animal) |
0.094 |
0.067 |
0.078 |
0.089 |
|
Km for kidney (mg/litre) |
0.543 |
0.543 |
0.496 |
0.448 |
|
Absorption rate constants for water (/h) |
||||
|
kSL (from stomach) |
2.5 |
2.5 |
NA |
5.0 |
|
kIL (from upper gastrointestinal tract) |
0.5 |
0.5 |
NA |
0.0 |
|
kSI (from stomach to upper gastrointestinal tract) |
3.5 |
3.5 |
NA |
0.0 |
|
Absorption rate constants for oil gavage (/h) |
||||
|
kSL |
1.5 |
1.5 |
1.5 |
NA |
|
kIL |
0.5 |
0.5 |
0.5 |
NA |
|
kSI |
1.8 |
1.8 |
1.8 |
NA |
Chloroform has a moderate acute oral toxicity in rats, with LD50s ranging from 0.45 to 2.0 g/kg body weight (Kimura et al., 1971; Chu et al., 1980). In mice, a wide range (36-1366 mg/kg body weight) of acute oral LD50 values has been reported (IPCS, 1994a). Acute oral administration produced narcosis and anaesthesia in rodents (IPCS, 1994a). An increase in renal cell proliferation was seen in male Osborne-Mendel and F344 rats given gavage doses of 10 and 90 mg/kg body weight, respectively (Templin et al., 1996a). In male F344 rats, a no-observed-adverse-effect level (NOAEL) and a lowest-observed-adverse-effect level (LOAEL) for serum enzyme changes indicative of liver damage following acute gavage exposure have been established as 30 and 60 mg/kg body weight, respectively (Keegan et al., 1998). Administration of 0, 67, 135, or 338 mg/kg body weight by gavage in olive oil to male Wistar rats increased, in a dose-dependent manner, the number of necrotic hepatocytes in the centrilobular region and elevated plasma alanine aminotransferase (ALAT) levels significantly (Nakajima et al., 1995b). Liver and kidney changes were seen in rats administered chloroform at 250 mg/kg body weight by gavage (Torkelson et al., 1976). Cell proliferation occurred in the liver and kidneys of male B6C3F1 mice given 150 mg/kg body weight by gavage; severe necrosis was also seen in the kidneys (Gemma et al., 1996). Hepatic necrosis was observed in male mice 48 h following a single gavage administration of 240 mg/kg body weight (Reitz et al., 1982). Minimal centrilobular enlargement was observed in male mice 4 days following intragastric administration of 66 mg/kg body weight (Moore et al., 1982). Lesions and epithelial cell proliferation were seen in the nasal passage of F344 and Osborne-Mendel rats following gavage administration of 90 mg/kg body weight in corn oil (Templin et al., 1996a).
An inhalation LC50 value (for 6-h exposure) of 9.2 g/m3 has been reported in rats (Bonnet et al., 1980). No deaths occurred when F344 rats (10 per sex per concentration) were exposed for 6 h at up to 5 g/m3, but 17/20 died at 10 g/m3 (Kasai et al., 2002). Depression of the central nervous system is a dominant symptom of acute inhalation. Rats exposed at 2.1 g/m3 for 4 h showed significant subnarcotic effects (Frantík et al., 1998).
In female OF1 mice, an inhalation LC50 value (for 6-h exposure) of 6.2 g/m3 was reported (Gradiski et al., 1978). Groups of 10 female BDF1 mice survived a 6-h exposure at up to 2.5 g/m3, but died (showing centrilobular liver necrosis) at 40 g/m3. Male mice are much more susceptible to acute chloroform inhalation toxicity, 1 of 10 and 8 of 10 dying after a single 6-h exposure at 59 and 120 mg/m3, respectively. The cause of death in the males was necrosis of the proximal tubules of the kidneys (Kasai et al., 2002).
Kidney tubule degeneration was seen in rabbits following a 24-h covered application of 1 g/kg body weight; no gross changes were seen in the liver (Torkelson et al., 1976).
Lesions and cell proliferation in the olfactory epithelium and changes in the nasal passages were seen in female F344 rats given 34 mg/kg body weight per day for 4-5 days in corn oil by gavage; after 3 weeks of administration, these effects were observed at 100 but not at 34 mg/kg body weight per day (Larson et al., 1995a; Dorman et al., 1997).
Tissue changes in the kidneys (mineralization, hyperplasia, and cytomegaly) and liver (inflammation) were seen in mice given 37 mg/kg body weight per day by gavage for 14 days (Condie et al., 1983).
F344 rats and BDF1 mice (10 per sex per species per concentration) were exposed at 0, 2.5, 5, 10, 20, or 40 g/m3, 6 h/day, 5 days/week, for 2 weeks. Rats survived at up to 5 g/m3, but all died within 2 days at 10 g/m3 and above. The female mice survived exposure at 2.5 g/m3, but deaths occurred (from day 4 onwards) at 5 g/m3. Only two male mice survived, one at 2.5 g/m3 and one at 5 g/m3. Dead rats showed lung congestion and inflammation, believed to arise as a result of cardiovascular toxicity. Deaths in mice were ascribed to necrosis of the kidney proximal tubules (males) and centrilobular necrosis of the liver (females). Surviving rats showed vacuolic changes in the proximal kidney tubules and the central area of the liver, as well as desquamation, atrophy, and disarrangement of the olfactory epithelium and oedema of the lamina propria of the nasal cavity. Surviving male mice had necrosis in the kidney proximal tubules, slight swelling and vacuolic change in the liver, and atrophy and respiratory metaplasia in the olfactory epithelium. The surviving female mice showed necrosis and vacuolic changes in the liver and necrosis and disarrangement of the olfactory and respiratory epithelia, but no kidney changes (Kasai et al., 2002).
Cell proliferation was observed in the nose (ethmoid region) of male F344 rats following inhalation exposure at 9.8 mg/m3 for 6 h/day for 4 days. At 49 mg/m3, only minimal to mild lesions were seen (Templin et al., 1996b). Following exposure to 50 mg/m3 for 6 h/day for 7 consecutive days, male F344 rats had lesions in nasal turbinates, including increased cell proliferation in central, proximal, and distal regions of the first endoturbinate, and histological changes in the central turbinate bone (Larson et al., 1994a; Mery et al., 1994).
Cell proliferation was seen in the nasal turbinates of female B6C3F1 mice exposed at 10 mg/m3, but not at 1.5 mg/m3, for 6 h/day, 7 days/week, for 3 weeks (Larson et al., 1996). Increased cell proliferation was detected in the first endoturbinate of the nasal passage in female B6C3F1 mice exposed to 49 mg chloroform/m3, 6 h/day, for 7 consecutive days (Mery et al., 1994). No microscopic damage was seen in the nasal passages of female B6C3F1 mice exposed to up to 1500 mg/m3 for 6 h/day for 7 consecutive days. Cell proliferation was not measured (Larson et al., 1994a).
Studies designed specifically to investigate cytotoxicity and regenerative cell proliferation in target organs are mentioned briefly (without experimental details) in section 8.8.
Studies designed specifically to investigate cytotoxicity and regenerative cell proliferation in target organs are mentioned briefly (without experimental details) in section 8.8.
When B6C3F1 mice were given 60 mg/kg body weight per day and above (the other doses were 130 and 270 mg/kg body weight per day) for 90 days by gavage in corn oil, both sexes showed increased liver weights and vacuolation and lipid accumulation in the liver. When chloroform was given in an Emulphor vehicle, the only effect at the lowest dose level (60 mg/kg body weight per day) was increased liver weight in females. No chloroform-related histopathological changes were observed in the kidneys (Bull et al., 1986).
In a study in which groups of 7-12 male and female CD1 mice were given 0, 50, 125, or 250 mg/kg body weight per day by stomach tube for 90 days, effects noted at all doses were increased liver weight and increased hepatic microsomal activity (in females) and (in both sexes) microscopic tissue changes in the liver (hepatocyte degeneration and focal lymphocyte collection) and kidneys (intertubular collection of inflammatory cells) (Munson et al., 1982).
When female B6C3F1 mice were exposed to chloroform in drinking-water for 90 days, fatty changes in the liver were observed at 263 mg/kg body weight per day (US EPA, 1980).
In male Osborne-Mendel rats, liver cholesterol was significantly increased when chloroform was given at 81 mg/kg body weight per day for 90 days in drinking-water (US EPA, 1980).
Effects on the liver were reported when chloroform was administered at 15 or 30 mg/kg body weight per day in a toothpaste base in gelatin capsules to male and female beagle dogs for 6 days/week for 7.5 years. Sacrifice followed a subsequent period of 19-23 weeks without treatment. The protocol included vehicle controls, untreated controls, and dogs receiving alternative (non-chloroform) toothpaste. Each group contained 8 animals of each sex, with the exception of the vehicle control group, which included 16 of each sex. At the high dose, there were significant increases in serum ALAT levels at 6 weeks of treatment. At the low dose, significant increases in ALAT levels were observed at 34 weeks and later. "Fatty cysts" of the liver were observed in both dose groups at the end of the study. In males, the incidences of fatty cysts of moderate or marked severity were 1/15, 6/7, and 6/7 in the vehicle control, low-dose, and high-dose groups, respectively. Equivalent incidence figures in the females were 0/12, 3/8, and 7/8 for the vehicle control, low-dose, and high-dose groups, respectively (see Table 6). There were no treatment-related increases in tumours (Heywood et al., 1979). The fatty cysts might reflect chronic low-grade disruption of hepatocyte function. Hepatocytes play a vital role in synthesis and transport of lipoprotein, triglyceride, and fatty acid metabolism. These fatty cysts (granulomas) are commonly seen in old dogs and probably form after the rupture or fusion of fat-laden cells. The subsequent macrophage response results in foamy aggregates in sinusoids, portal triad stroma, and hepatic venules, possibly accompanied by multinucleate giant cells and ceroid pigment accumulation resulting from lipid breakdown (D. Malarkey, personal communication to Health Canada, 2003). [LOAEL = 15 mg/kg body weight per day]
Table 6: Fatty cyst incidence in dog study.a
|
Group |
No. of dogs examined histologically |
No. of dogs with fatty cysts |
|
|
Occasional/minimal |
Moderate/marked |
||
|
Males |
|||
|
30 mg/kg body weight per day |
7 |
1 |
6 |
|
15 mg/kg body weight per day |
7 |
0 |
6 |
|
Vehicle control |
15 |
7 |
1 |
|
Untreated |
7 |
2 |
0 |
|
Alternative toothpaste without chloroform |
8 |
2 |
0 |
|
Females |
|||
|
30 mg/kg body weight per day |
8 |
0 |
7 |
|
15 mg/kg body weight per day |
8 |
2 |
3 |
|
Vehicle control |
12 |
3 |
0 |
|
Untreated |
5 |
1 |
0 |
|
Alternative toothpaste without chloroform |
7 |
0 |
0 |
a From Heywood et al. (1979).
Exposure of rats (10-12 per sex per exposure level; strain unspecified) at 120, 240, or 420 mg/m3, 7 h/day, 5 days/week, for 6 months resulted in increased relative kidney weight, cloudy swelling in the renal tubular epithelium, and focal liver necrosis in the males at all dose levels (Torkelson et al., 1976).
Nasal effects were seen in a study in which female B6C3F1 mice were exposed at 1.5, 9.8, 49, 147, or 441 mg/m3, 6 h/day, for up to 13 weeks. After 3 weeks, there was slightly increased proliferation in the nasal turbinate lamina propria at 9.8 mg/m3 and above and mild to minimal nasal lesions at 49 mg/m3 and above. After 13 weeks, nasal effects were minimal at 49 and 147 mg/m3, but cell proliferation persisted at 441 mg/m3 (Larson et al., 1996). [LOAEL = 49 mg/m3]
When BDF1 mice (10 per sex per exposure level) were exposed by inhalation at 0, 59, 123, 245, 490, or 980 mg/m3 for 6 h/day, 5 days/week, for 13 weeks, the females all survived, but reduced growth and deaths occurred in males in all chloroform groups. Increased liver and kidney weights occurred in the mice at 980 mg/m3. Male mice showed effects in the kidney (necrosis of the proximal tubules) and nasal cavity (bone thickening and degeneration of the olfactory epithelium) at all exposure levels (59 mg/m3 and above). Female mice showed nasal cavity toxicity (thickening of bone, eosinophilic changes in the olfactory and respiratory epithelia) at all exposure levels. The liver was normal in both sexes at up to 245 mg/m3, but cell atypia was seen in the females at 490 mg/m3, and swelling or necrosis occurred at 980 mg/m3 (Kasai et al., 2002). [LOAEL = 59 mg/m3]
Daily exposure of male F344 rats for 6 h/day for 13 weeks resulted in mild histological changes in nasal passages at 9.8 mg/m3 and increased cell proliferation with enhanced bone growth at 49 mg/m3. At 147 mg/m3 and above, the cortical proximal tubules of the kidneys showed increased epithelial cell proliferation. Hepatic lesions (including cell proliferation) were seen at 1470 mg/m3 (Templin et al., 1996b). [LOAEL = 9.8 mg/m3]
When F344 rats (10 per sex per exposure level) were exposed by inhalation at 0, 123, 245, 490, 980, or 1960 mg/m3 for 6 h/day, 5 days/week, for 13 weeks, growth was reduced and liver and kidney weights were increased at 245 mg/m3 and above. The nasal cavity tissues were the most sensitive target, showing mineralization and atrophy of the respiratory epithelium at all exposure concentrations and necrosis at 980 mg/m3 and above. The liver and kidneys were microscopically normal at up to 245 mg/m3, but there were vacuolic changes in the kidneys and liver cell collapse/hepatocyte loss at 490 mg/m3 and above (Kasai et al., 2002). [LOAEL = 123 mg/m3]
Chloroform induced liver tumours in mice (both sexes) when given by gavage in a corn oil vehicle and possibly in males when given (orally) in a toothpaste vehicle, but was not carcinogenic to the mouse liver when given in the drinking-water or by inhalation. Chloroform induced kidney tumours in male mice exposed by inhalation or (in one of four strains) by ingestion in a toothpaste vehicle, but not when given in corn oil. Female mice were not susceptible to chloroform-induced kidney cancer.
Chloroform induced kidney tumours in one strain of male rats treated via a corn oil vehicle and in drinking-water, but not in another strain treated via the drinking-water. Chloroform was not carcinogenic in inhalation studies or where given (orally) in a toothpaste base. Female rats were not susceptible to chloroform-induced kidney cancer. Chloroform did not induce liver tumours when given in corn oil or by inhalation, although two drinking-water studies found limited evidence of liver carcinogenicity, one in females only and a second in which only males were treated.
There were no clear descriptions of non-neoplastic lesions in the tumour target organs in the reports of these studies; consequently, a phase of research work has since been undertaken to investigate early changes in the target organs at the doses that were active in the chronic studies. In addition, re-evaluation of the kidney tissue slides has been undertaken for the Jorgenson et al. (1985) study. There has also been a very limited re-evaluation of the kidney tissues from the male rats of the NCI (1976) study.
Chloroform induced liver tumours in male and female B6C3F1 mice following gavage at 138 mg/kg body weight per day or more in a corn oil vehicle for 78 weeks (NCI, 1976), but not when given (to females only) at up to 1800 mg/litre in the drinking-water for 104 weeks (up to 263 mg/kg body weight per day) (Jorgenson et al., 1985). It did not significantly increase the incidence of liver tumours when given by gavage (in a toothpaste) at up to 60 mg/kg body weight per day, 6 days/week, for 80 weeks to ICI mice (both sexes) or male C57BL, CBA, or CF1 mice (Roe et al., 1979). Chloroform induced a statistically borderline significant increase in hepatic tumours (adenomas and carcinomas combined) in female but not in male BDF1 mice exposed by inhalation at up to 441 mg/m3, 6 h/day, 5 days/week, for 104 weeks (Yamamoto et al., 2002).
Chloroform did not induce liver tumours when given by gavage to male or female Osborne-Mendel rats at up to 200 mg/kg body weight per day for 111 weeks (NCI, 1976), in the drinking-water at up to 160 mg/kg body weight per day to male Osborne-Mendel rats (Jorgenson et al., 1985), orally in a toothpaste at up to 165 mg/kg body weight per day for 80 weeks to male or female Sprague-Dawley rats (Palmer et al., 1979), or by inhalation at up to 441 mg/m3 in male or female F344 rats exposed for 6 h/day, 5 days/week, for 104 weeks (Yamamoto et al., 2002). In a study in which Wistar rats were treated (at >100 mg/kg body weight per day for up to 185 weeks) via the drinking-water, males did not develop liver tumours, but a statistically significant increase in foci of cellular atypia (possibly representing pre-neoplastic lesions) was seen in the females. However, the relevance of this finding is difficult to determine because the control group was small (22) and the exposed females survived longer (185 weeks) than the controls (145 weeks), so there was no basis for comparing the incidence of late-developing tumours (Tumasonis et al., 1987). According to a recent abstract, administration of chloroform at 800 mg/litre in the drinking-water for 100 weeks did not induce liver cancer in a group of 78 male F344 rats. However, at the top dose (1600 mg/litre water; probably about 160 mg/kg body weight per day), the incidence of rats with hepatocellular neoplasia (adenoma or carcinoma) was increased (17% versus 5.1%; P < 0.05). The significance of this result cannot be clearly determined from the limited presentation of the data (DeAngelo et al., 2003).
No evidence of carcinogenic potential was seen in a study in which male and female wild-type and rasH2-Tg transgenic CB6F1 mice were given up to 140 (males) or 240 (females) mg/kg body weight, 5 days/week for 26 weeks, by gavage in corn oil. A range of organs was examined, including the liver. Swelling and vacuolation of hepatocytes were seen in the transgenic and non-transgenic mice, and the incidence of hepatocellular foci was increased in the female mice (transgenic and wild type) at the top dose (Sehata et al., 2002). In another study in which hemizygous transgenic (Tg.AC) mice were given the same doses of chloroform by gavage in corn oil, 5 days/week for 13 weeks, V-Ha-ras transgene expression was not induced in the liver, although there was evidence of liver tissue damage and cell proliferation (Delker et al., 1999). (These transgenic mouse model studies were primarily conducted for testing their suitability/validity as a bioassay for showing tumorigenicity of a positive "non-genotoxic-cytotoxic rodent liver and kidney carcinogen" using chloroform as a prototype model carcinogen of this category of chemical carcinogens.)
Chloroform induced kidney tubular cell carcinoma in male BDF1 mice treated by inhalation at 147 or 441 mg/m3, 6 h/day, 5 days/week, for 104 weeks. No clear response was seen at 25 mg/m3, the lowest exposure concentration (Yamamoto et al., 2002). Chloroform induced a mixture of malignant hypernephromas and benign cortical adenomas in the kidneys of male ICI mice given the compound at 60 mg/kg body weight per day for 80 weeks in a toothpaste vehicle (Roe et al., 1979). It did not induce kidney tumours in B6C3F1 mice (either sex) treated at up to 477 mg/kg body weight per day for 78 weeks by corn oil gavage (NCI, 1976), in female B6C3F1 mice ingesting up to 263 mg/kg body weight per day for 104 weeks via the drinking-water (Jorgenson et al., 1985), in female BDF1 mice exposed by inhalation at up to 441 mg/m3 for 104 weeks (Yamamoto et al., 2002), or in female ICI or male C57BL, CBA, or CF/1 mice given chloroform at 60 mg/kg body weight per day for 80 weeks in toothpaste (Roe et al., 1979).
Chloroform induced kidney epithelial tumours in male Osborne-Mendel rats treated at 180 mg/kg body weight per day for 111 weeks by gavage in corn oil (NCI, 1976) and kidney tubular cell adenomas and adenocarcinomas in male rats of the same strain given drinking-water containing chloroform at 1800 mg/litre (providing 160 mg/kg body weight per day) for 104 weeks (Jorgenson et al., 1985; see Table 7). It did not cause kidney cancer in male F344 rats when given at up to 1600 mg/litre in the drinking-water (probably about 160 mg/kg body weight per day) for 100 weeks (DeAngelo et al., 2003), in female Osborne-Mendel rats when given at up to 200 mg/kg body weight per day for 111 weeks in corn oil (NCI, 1976), in male or female F344 rats exposed by inhalation at up to 441 mg/m3, 6 h/day, 5 days/week, for 104 weeks (Yamamoto et al., 2002), in male or female Wistar rats treated at up to 2900 mg/litre drinking-water (average doses about 180-240 mg/kg body weight per day) for up to 185 weeks (Tumasonis et al., 1987), or in male or female Sprague-Dawley rats given chloroform at up to 165 mg/kg body weight per day for 80 weeks in toothpaste (Palmer et al., 1979).
Table 7: Histopathology in kidneys of male Osborne-Mendel rats in drinking-water bioassay of Jorgenson et al. (1985).a
|
Group |
Exposure time (months) |
Number of rats examined for cytotoxicity |
Percentage of rats with lesionsb |
Percentage of rats with renal adenomas and carcinomas |
|
Untreated control |
24 |
24 |
0 |
1.3 |
|
18 |
19 |
0 |
||
|
12 |
20 |
0 |
||
|
6 |
20 |
0 |
||
|
Water-matched control |
24 |
0 |
|
2.0 |
|
18 |
18 |
0 |
||
|
12 |
19 |
0 |
||
|
6 |
19 |
0 |
||
|
200 mg/litre |
24 |
0 |
1.3 |
|
|
18 |
16 |
0 |
||
|
400 mg/litre |
24 |
40 |
0 |
2.7 |
|
18 |
19 |
0 |
||
|
900 mg/litre |
24 |
48 |
50 |
6.3 |
|
18 |
10 |
58 |
||
|
12 |
20 |
33 |
||
|
6 |
20 |
25 |
||
|
1800 mg/litre |
24 |
46 |
100 |
14.0 |
|
18 |
17 |
100 |
||
|
12 |
18 |
100 |
||
|
6 |
20 |
95 |
a From Hard et al. (2000).
b Lesions indicative of tubule injury included nuclear crowding, cytoplasmic vacuolation, and faint basophilia in the mid to deep cortex.
In a study identifed only as an abstract, transgenic p53+/- (sensitive to mutagenic carcinogens) and p53+/+ wild-type mice were given chloroform at up to 140 or 240 mg/kg body weight per day (males and females, respectively) by gavage in corn oil for up to 26 weeks. The males showed renal tubular regeneration and proliferation, but there were no increases in kidney tumour incidence (Gollapudi et al., 1999). No evidence of carcinogenic potential was seen in another study in which male and female wild-type and rasH2-Tg transgenic CB6F1 mice were given chloroform at up to 140 (males) or 240 (females) mg/kg body weight, 5 days/week for 26 weeks, by gavage in corn oil. A range of organs was examined, including the kidneys (Sehata et al., 2002). In another study in which hemizygous transgenic (Tg.AC) mice were given the same doses of chloroform by gavage in corn oil, 5 days/week for 13 weeks, V-Ha-ras transgene expression was not induced in the kidneys, although there was evidence of kidney tissue damage and cell proliferation in the males (Delker et al., 1999).
The bioassay in which evidence of carcinogenicity was seen at the lowest concentration or dose, and which involved exposure similar to that of humans, was that of Jorgenson et al. (1985). Male Osborne-Mendel rats were given chloroform in the drinking-water at 0, 200, 400, 900, or 1800 mg/litre for 2 years. Group sizes were 330, 330, 150, 50, and 50 respectively, and the TWA daily doses were 0, 19, 38, 81, and 160 mg/kg body weight per day, respectively. A matched control group consisted of 50 rats given drinking-water (without chloroform) in an amount equal to that consumed by the 1800 mg/litre group. According to Environment Canada & Health Canada (2001), clinical chemistry indicated renal impairment in the controls, mild impairment in the 200 and 400 mg/litre groups, but no impairment in the 900 and 1800 mg/litre groups. This is consistent with severe chronic nephropathy in the controls as a result of ad libitum diet consumption (calorie overload) and a protective effect of reduced food consumption in higher dose groups as a result of reduced drinking-water consumption. Consistent with these results was the reduced mortality in the matched control group; mortality was inversely related to exposure concentration. Data on organ weights were not provided. The only clear dose-related adverse effect was an increase in the incidence of renal tubular cell adenomas and adenocarcinomas, with the combined incidence being significantly increased (P < 0.01) at the top dose (combined incidence 4/301, 4/313, 4/148, 3/48, and 7/50; P < 0.001 for trend) for 0, 19, 38, 81, and 160 mg/kg body weight per day, respectively. The incidence in the matched control group was 1/50 (Jorgenson et al., 1985).
Microscopic re-evaluation of kidney tissues from this study identified damage in the proximal tubular epithelial cells at 6, 12, 18, and 24 months in all rats but one (at 6 months) of the 1800 mg/litre group and about half of the rats in the 900 mg/litre group at 18 and 24 months, but not in the other groups. The changes included slightly increased basophilia, cytoplasmic vacuolation, karyomegaly, anisokaryosis, nuclear crowding, and mild tubular hyperplasia. Cytotoxic tubular lesions, occasional foci of atypical tubular hyperplasia, and incipient renal tubular tumours were all located in the mid to deep cortex (Hard & Wolf, 1999; Hard et al., 2000) (see Table 7).
Similar changes were present in males of the same strain in the NCI (1976) gavage study, although systematic re-evaluation of the tissues was not possible. Renal tumours seen in the NCI study were larger (at least twice the diameter) than those seen in the drinking-water study of Jorgenson et al. (1985) (Hard et al., 2000).
In a chronic study involving exposure of F344 rats to 0, 49, 147, or 441 mg/m3 and of BDF1 mice to 0, 25, 147, or 441 mg/m3, 6 h/day, 5 days/week, for 104 weeks, ossification of the nasal turbinates (rats) and nasal septum (mice) and atrophy and respiratory metaplasia of the olfactory epithelium were reported at all exposure concentrations (Yamamoto, 1996; Yamamoto et al., 2002). In spite of the overt toxicity and increased cell proliferation in the nose, no nasal tumours were noted in this or any of the other chronic studies.
No evidence of carcinogenic potential was seen in a study in which male and female wild-type and rasH2-Tg transgenic CB6F1 mice were given up to 140 (males) or 240 (females) mg/kg body weight, 5 days/week for 26 weeks, by gavage in corn oil. A range of organs was examined, including the nasal cavity (Sehata et al., 2002).
When male and female Osborne-Mendel rats were given average chloroform doses of 0, 100, or 200 mg/kg body weight, 5 days/week for 78 weeks, by gavage (and sacrificed at 111 weeks), the incidence of thyroid tumours was increased by treatment in the females (1/19, 8/49, and 10/46 in these groups, respectively). The increases were not statistically significant (NCI, 1976).
A few studies exist that indicate that chloroform lacks tumour-initiating activity. For example, chloroform showed no initiating activity in the liver when given as a single oral dose (180 or 360 mg/kg body weight) to male Sprague-Dawley rats and B6C3F1 mice. Phenobarbital was used as the promoter (Pereira et al., 1982; Herren-Freund & Pereira, 1986).
Chloroform at 0.6-1.8 g/litre in the drinking-water for 51-52 weeks did not promote liver or lung cancer in mice previously initiated with diethylnitrosamine or ethylnitrosourea (Pereira et al., 1985; Klaunig et al., 1986). Chloroform did not promote liver tumours when given at 1.8 g/litre in the drinking-water for 48 weeks to B6C3F1 mice and male Sprague-Dawley rats, following treatment with diethylnitrosamine or ethylnitrosourea as an initiator (and partial hepatectomy in the case of rats) (Herren-Freund & Pereira, 1986).
A study in rats suggested that chloroform in an oil vehicle might promote the development of hepatic tumours. Chloroform was given at 25-400 mg/kg body weight twice weekly for 11 weeks by gavage in olive oil to female Sprague-Dawley rats previously initiated with dimethylnitrosamine. There was a dose-related increase of ATPase-negative, gamma-glutamyl transpeptidase (GGTase)-positive, and glycogen-storing foci of cells within the liver (Deml & Oesterle, 1985, 1987). In a previous investigation of promoting activity, administration of chloroform at 180 mg/kg body weight (in tricaprylin) twice a week for 53 days as a promoter produced a small, but statistically significant, increase in the numbers of GGTase-positive foci (Pereira et al., 1982).
Chloroform administered in drinking-water at 900 or 1800 mg/litre to F344 rats for 39 weeks significantly decreased gastrointestinal tumours that were initiated with dimethylhydrazine (Daniel et al., 1989). At 1800 mg/litre in the drinking-water for 30 days, chloroform inhibited the propensity for three gastrointestinal tract carcinogens — benzo(a)pyrene, 1,2-dimethylhydrazine, and methylnitrosourea — to induce nuclear anomalies in the proximal colon of B6C3F1 mice (Daniel et al., 1991). Others demonstrated that chloroform inhibits the development of diethylnitrosamine-initiated, phenobarbital-promoted GGTase- and placental form glutathione-S-transferase-positive foci in the liver of male F344 rats (Reddy et al., 1992). Chloroform has also been reported to inhibit ethylnitrosourea-initiated liver tumour growth in young mice (Pereira et al., 1985).
The lack of initiating activity in these initiation/ promotion assays supports the weight-of-evidence conclusion that chloroform is non-genotoxic (see section 8.5).
Chloroform has been extensively studied for genotoxic potential in a range of short-term screening assays. A more detailed, tabulated presentation of available data is given in the source document (Environment Canada & Health Canada, 2001).
Chloroform gave no evidence of mutagenic activity in the vast majority of a large number of assays in Salmonella typhimurium and Escherichia coli bacteria, although two papers report weak activity in, respectively, four Salmonella strains (Varma et al., 1988) and a single Salmonella strain (Pegram et al., 1997) at toxic/ lethal concentrations. Chloroform evidently did not cause chromosome aberrations in human lymphocytes in culture. Sister chromatid exchange (SCE) assays have given mixed results, but tests for unscheduled DNA synthesis (UDS) have consistently given no evidence of activity in a range of human and laboratory animal cells. In vivo, three of four bone marrow micronuclei studies in mice were clearly negative (Gocke et al., 1981; Salamone et al., 1981; Tsuchimoto & Matter, 1981), and the fourth (Agustin & Lim-Sylianco, 1978) was equivocal. Chloroform induced micronuclei formation in the kidney (Robbiano et al., 1998) and liver (Sasaki et al., 1998) of rats and chromosome damage (aberrations) in the bone marrow of rats (Fujie et al., 1990); a hamster bone marrow chromosome aberration study also gave evidence of a weak effect (Hoechst, 1987). Weak DNA binding has been reported in the rat liver and kidney (Pereira et al., 1982) and the mouse kidney, lung, liver, and stomach following intraperitoneal injection (Colacci et al., 1991), and there have been mixed results for sperm abnormalities in mice (Topham, 1980, 1981; Land et al., 1981) and a positive SCE result in mouse bone marrow (Morimoto & Koizumi, 1983). For other end-points (e.g., UDS in rat and mouse hepatocytes, DNA adducts, methylation, strand breaks and repair in mouse liver, DNA damage in rat liver and kidney), in vivo results have been negative (Petzold & Swenberg, 1978; Diaz-Gomez & Castro, 1980; Mirsalis et al., 1982; Reitz et al., 1982; Larson et al., 1994d; Potter et al., 1996; Butterworth et al., 1998; Pereira et al., 1998).
In conclusion, most studies did not identify genotoxic potential for chloroform. Results from a few, non-standard studies indicate the possibility of a weak positive response in rats. Overall, however, the weight of evidence indicates that chloroform does not have significant genotoxic potential.
Reproductive and developmental assays involving oral exposure in mice, rats, and rabbits were identified. There was no clear evidence of teratogenic potential, and effects on the fetus were observed only at dose levels that were maternally toxic.
In a continuous-breeding protocol with CD-1 mice, there were no effects on fertility or reproduction in the F1 generation. These mice had been exposed in utero and during lactation (as a result of maternal treatment) and then by gavage at 41 mg/kg body weight per day (in corn oil) through young adulthood; at this dose, there was hepatocellular degeneration in females (EHRT, 1988). [NOAEL for fertility = 41 mg/kg body weight per day; LOAEL for toxicity = 41 mg/kg body weight per day]
Reduced food intake, slower growth, and fatty liver changes were seen in Sprague-Dawley rats given chloroform at 50 mg/kg body weight per day on days 6-15 of gestation by gavage in corn oil. At 126 mg/kg body weight per day, reduced fetal body weight was seen, but there were no teratogenic effects (Thompson et al., 1974). [NOAEL for developmental toxicity = 126 mg/kg body weight per day; maternal LOAEL = 50 mg/kg body weight per day]
In a similar protocol, chloroform caused maternal toxicity (slower growth and increased liver weight) when given at 100 mg/kg body weight per day and mild fetal toxicity (lower body weight), but no teratogenicity, at 400 mg/kg body weight per day (Ruddick et al., 1983). [NOAEL for developmental toxicity = 400 mg/kg body weight per day; maternal LOAEL = 100 mg/kg body weight per day]
Administration of chloroform at up to 50 mg/kg body weight per day by stomach tube (in corn oil) on days 6-18 of gestation resulted in reduced maternal weight gain at the top dose, but there were no dose-related effects on reproduction or fetal development (Thompson et al., 1974). [NOAEL for developmental toxicity = 50 mg/kg body weight per day; maternal lowest-observed-effect level (LOEL) = 50 mg/kg body weight per day]
Results across the few inhalation bioassays identified were fairly consistent. In Sprague-Dawley rats exposed to 0, 150, 450, or 1430 mg/m3, there was a significant decrease in maternal body weight at the lowest concentration (Schwetz et al., 1974).6 [Maternal LOEL = 150 mg/m3] When Wistar rats were exposed to identical atmospheric concentrations, there was decreased food consumption and maternal body weight gain at 150 mg/m3 (Hoechst, 1988). [Maternal LOAEL = 150 mg/m3] At 150 mg/m3 in both studies, fetal crown-rump length was significantly reduced, although not in a dose-related manner in the former study. Although adverse skeletal and visceral effects were reported by Schwetz et al. (1974), they were not dose related; no teratogenic effects were observed by Hoechst (1988). When the study was repeated at lower concentrations, reproduction and development were unaffected at 50 mg/m3, but maternal weight gain was reduced (Hoechst, 1990, 1993). [NOAEL for developmental toxicity = 50 mg/m3; LOAEL for maternal toxicity = 50 mg/m3]
Neat chloroform caused severe irritation when instilled into the rabbit eye (Duprat et al., 1976; Torkelson et al., 1976). Moderate necrosis and scab formation were seen following 24-h application of chloroform on a cotton pad to the skin of rabbits (Torkelson et al., 1976).
Tissues containing CYP2E1 are targets for chloroform toxicity, because CYP2E1 metabolizes chloroform to toxic metabolites, including hydrogen chloride and phosgene. The latter is strongly electrophilic and can covalently bind to cell proteins and to the polar heads and fatty acyl chains of phospholipids, leading to the loss of cell function and cell death (IPCS, 1994a, 2000b; Ammann et al., 1998). Continued exposure at a sufficiently high tissue concentration leads to a cycle of persistent cytotoxicity and sustained regenerative cell proliferation. There is convincing evidence that these events are critical features of the mechanistic route to the liver and kidney tumours induced by chloroform in rodents. This view is supported by the weight-of-evidence conclusion that chloroform and its metabolites lack any significant direct genotoxic potential (IPCS, 1994a, 2000b; Templin et al., 1998; Constan et al., 2002).
Effects observed most consistently at lowest concentrations or doses following repeated exposures to chloroform in rats and mice are cytotoxicity and regenerative proliferation. Target organs are the liver (centrilobular region) and kidney (cortical region). Chloroform has caused liver and kidney tumours in mice and kidney tumours in rats. In addition, chloroform has induced nasal lesions in rats and mice exposed by both inhalation and ingestion; chloroform is highly volatile, and humans will be exposed to chloroform in air. Available data from exposed workers indicate that the liver is a target in humans.
The toxicity of chloroform is attributable to metabolites (particularly phosgene) resulting from oxidative metabolism, which is probably the only significant in vivo pathway at low exposures (Testai & Vittozzi, 1986; Ade et al., 1994; Ammann et al., 1998; Environment Canada & Health Canada, 2001). The key role of CYP2E1 was demonstrated by studies in which there was no cytotoxicity or cell proliferation in the liver or kidney of CYP2E1 null Sv/129 or CYP2E1 null B6C3F1 mice at an exposure concentration that caused severe hepatic and renal lesions in the wild type of either strain (Constan et al., 1999). The organs in which chloroform-induced cytotoxicity and proliferative lesions are observed (liver, kidney, and nasal passages) correlate well with the distribution of CYP2E1 both across and within species (Löfberg & Tjälve, 1986). In Wistar rats (naive or treated with a CYP2E1 inducer), the observed centrilobular location of chloroform-induced hepatocellular damage correlated with a similar distribution of CYP2E1. Following treatment with phenobarbital (a CYP2B1 inducer), hepatic damage was spread over the centrilobular, mid-zonal, and periportal regions, consistent with a uniform distribution of CYP2B1 enzyme expression (Nakajima et al., 1995b).
The evidence that oxidative metabolites cause cytotoxicity in the mouse liver and kidney includes observation of a direct correlation between binding of metabolites to the polar heads of phospholipid molecules (a feature characteristic of oxidative metabolites) and protein binding in the liver and kidney of the DBA/2J mice (Ade et al., 1994).
The extent of chloroform-induced hepatic necrosis correlates with the extent of covalent binding to liver proteins in male and female rats and in male mice (Ilett et al., 1973; B.R. Brown et al., 1974). This covalent binding is more prevalent within the areas of necrosis (Ilett et al., 1973; Tyson et al., 1983). The results of in vitro studies are consistent, in that irreversible binding to macromolecules in rat and human liver microsomes requires prior metabolism (Cresteil et al., 1979).
In mice, covalent binding of chloroform to renal proteins and microsomes is correlated with the degree of renal tubular necrosis (Ilett et al., 1973; Smith & Hook, 1983, 1984). Strain- and sex-related differences in sensitivity of mice to nephrotoxicity are also correlated with the ability of the kidney to metabolize chloroform (Taylor et al., 1974; Clemens et al., 1979; Pohl et al., 1984; Smith et al., 1984; Mohla et al., 1988; Henderson et al., 1989; Hong et al., 1989).
The hypothesized mode of tumour induction is the same for each tumour type, but there is some variation in the amounts of supporting evidence.
Liver tumours were observed in B6C3F1 mice following administration of bolus doses by gavage in corn oil (NCI, 1976), but not following administration of the same daily doses in drinking-water (Jorgenson et al., 1985). Bolus (gavage) dosing results in a higher tissue dose rate than does continuous administration and so is likely to produce greater tissue damage. Doses at which tumours have been observed following gavage administration in corn oil in the cancer bioassay were associated with sustained toxic and proliferative responses in the liver of the same strain exposed similarly in shorter-term studies (Larson et al., 1994b,c; Pereira, 1994; Melnick et al., 1998). Sustained increases in proliferative response were not observed in the liver following short-term ingestion in drinking-water (Larson et al., 1994c), at concentrations that also did not induce increases in hepatic tumour incidence in the long-term bioassay (Jorgenson et al., 1985).
Thus, there is convincing evidence of a relationship between metabolism to reactive intermediates, cytotoxicity, regenerative proliferation, and tumour development in the B6C3F1 mouse liver.
Chloroform also induced renal tumours in BDF1 mice following inhalation (Yamamoto et al., 2002) and in ICI mice exposed by gavage in toothpaste (Roe et al., 1979), although at lower rates than liver tumours. The response is sex specific, occurring only in males. Evidence of concordance between metabolism to reactive intermediates, cytotoxicity, regenerative proliferation, and tumour development in the mouse kidney is substantial, although data on sustained enhanced proliferative response in the tumour-susceptible strains are limited. In BDF1 mice, there was an increase in the labelling index as well as sustained cytoplasmic basophilia as an indication of regeneration (Templin et al., 1996c; Kasai et al., 2002; Yamamoto et al., 2002) in the kidneys of males but not females at concentrations that induced renal tumours in males of this strain in the long-term inhalation bioassay (Yamamoto et al., 2002).
The evidence for the hypothesized mode of induction of tumours in the male rat kidney is less than that for the mouse liver and kidney, primarily because of the limited data on intermediate end-points in the only strain (Osborne-Mendel) in which kidney tumour increases were seen. These increases were reported following exposure via gavage in corn oil and via drinking-water (NCI, 1976; Jorgenson et al., 1985). Also, information on the relationship between the metabolism of chloroform and induction of renal lesions in rats is very limited. In male F344 rats, there were sustained increases in proliferative response in shorter-term studies following administration of doses similar to those that induced tumours in Osborne-Mendel rats, following administration by gavage in corn oil but not following ingestion in drinking-water (Larson et al., 1995a). An abstract reports that male F344 rats did not develop kidney tumours in a chronic drinking-water study (DeAngelo et al., 2003). Sustained increases in DNA labelling index (a quantitative measure of cell proliferation) were observed in the proximal tubules of F344 rats exposed at 147 mg/m3 and above daily and at 441 mg/m3 and above on 5 days/week (Templin et al., 1996b). However, increases in kidney tumour incidence were not observed when rats of this strain were exposed to up to 441 mg/m3, 6 h/day, 5 days/week, in the only inhalation cancer bioassay (Yamamoto et al., 2002). Based on data from short-term studies conducted primarily in F344 rats (a strain in which kidney tumours were not observed), a mode of action for carcinogenicity in the kidneys of Osborne-Mendel rats based on cytotoxicity and tubular cell regeneration is, therefore, plausible. For Osborne-Mendel rats, the results of reanalyses of the original renal tissues (Hard & Wolf, 1999; Hard et al., 2000), from both the drinking-water bioassay (Jorgenson et al., 1985) and the gavage study (NCI, 1976), have been critical. They provide strong support for the contention that the mode of induction of these tumours is consistent with the hypothesis that sustained proximal tubular cell damage is a requisite precursor lesion for chloroform-induced tumours.
In all cases where examined, therefore, sustained cytotoxicity and cellular proliferation were observed in the liver and kidney of the same strain of mice and rats exposed in a similar manner in short-term studies to concentrations or doses that induced tumours in these organs in cancer bioassays. This consistent pattern of response across species and organs is consistent with the hypothesis that, where chloroform causes tumours, toxicity and reparative hyperplasia are obligatory precursor steps. This hypothesis is generally supported by the weight of evidence indicating that chloroform has given little sign of significant genotoxic potential.
It should be noted that sustained proliferation does not inevitably lead to tumours. For example, kidney tumours were not induced in chronic bioassays in B6C3F1 mice (NCI, 1976; Jorgenson et al., 1985) and F344 rats (Yamamoto et al., 2002), although sustained increases in kidney damage and resulting proliferation were seen in these same strains/species when exposed to similar concentrations in the same manner, in shorter-term studies (Larson et al., 1994b, 1995a,b; Templin et al., 1996b). However, tumours would not necessarily be expected whenever there is an increase in cell replication. The multiple susceptibility factors that produce tumours following cytotoxicity will depend on tissue-specific factors and will likely vary between species and strains. For example, in spite of the overt toxicity and sustained increased cell proliferation in the epithelial tissue of the nose in both rats and mice, no tumours have been noted in this tissue in any chronic studies, including the inhalation bioassay in which nasal tissues were carefully evaluated (Yamamoto et al., 2002).
The hypothesized mode of carcinogenesis for chloroform is in keeping with the growing body of evidence supporting the biological plausibility that prolonged regenerative cell proliferation can be a causal mechanism in chemical carcinogenesis. Enhanced cell proliferation can lead to an increased frequency of spontaneous genetic damage either through errors that result from the infidelity of DNA replication or through the increased conversion of endogenous DNA changes into heritable genetic changes (Cohen & Ellwein, 1990, 1991, 1996; Ames et al., 1993; Cohen, 1995). Additionally, during periods of cell replication, heritable non-mutagenic modifications of the genome may occur that may lead to changes in gene expression, contributing to carcinogenesis (US EPA, 1996). This view that cell proliferation is a risk factor for carcinogenesis is not universally accepted, because strict correspondence between increased cell turnover and carcinogenic response is not always demonstrable (Melnick, 1992; Farber, 1996). However, as indicated above, in view of the complex interplay of factors involved in the carcinogenesis process, it is not surprising that acute measures of cell proliferation do not always indicate a one-to-one correlation. Among the factors to be considered are the balance between cell proliferation, differentiation, and death, proliferation in the target cell compartments compared with that of non-target cells, and the consequences of overt tissue toxicity.
In summary, chloroform has induced liver tumours in mice and kidney tumours in mice and rats. Sex and strain specificity in tumour response, concordance of cytotoxicity, regenerative proliferation and tumours, and the weight of evidence of non-genotoxicity are consistent with the hypothesis that marked, persistent cytotoxicity concomitant with a period of sustained cell proliferation likely represent a secondary mechanism for the induction of tumours following exposure to chloroform. This is consistent with a non-linear dose-response relationship for induction of tumours. This cytotoxicity is primarily related to rates of oxidation of chloroform to reactive intermediates, principally phosgene and hydrochloric acid. The weight of evidence for this mode of action is strongest for liver and kidney tumours in mice and more limited for kidney tumours in rats.
In general, chloroform elicits the same symptoms of toxicity in humans as in laboratory animals. Chloroform was used in the past to induce (exposure at 24-73 g/m3 air) and maintain (12-48 g/m3 air) medical anaesthesia. However, this practice was discontinued because it caused deaths due to respiratory and cardiac arrhythmias and failure (IPCS, 1994a). Following chloroform-induced anaesthesia, some patients suffered nausea, vomiting, prostration, jaundice, and coma due to hepatic dysfunction. At autopsy, liver necrosis and degeneration have been observed (Goodman & Gilman, 1970). There have been infrequent case reports of renal tubular necrosis and renal dysfunction resulting from the use of chloroform as an anaesthetic (Kluwe, 1981). It has been reported that a 1-h exposure at 2.5 g/m3 can cause effects, and these can be severe at 10 g/m3; exposure at less than 0.25 g/m3 might cause discomfort (Verschueren, 1983). When ingested, chloroform caused symptoms similar to those seen following inhalation. Serious illness has followed ingestion of 7.5 g. The mean lethal oral dose for an adult is estimated to be about 45 g (Winslow & Gerstner, 1978). Accidental splashing into the eyes has caused irritation, and the concentrated vapour has reportedly induced stinging sensation of the eyes (Winslow & Gertsner, 1978; IPCS, 1994a).
Toxic liver jaundice was reported in workers exposed at 80-160 mg/m3 for less than 4 months (Phoon et al., 1983), although the short exposure period makes the reliability of this conclusion uncertain. In an earlier report, these investigators associated a toxic jaundice outbreak with exposures of "more than 1950 mg/m3" for "less than 6 months" (Phoon et al., 1975). In another study, hepatitis was observed at a higher than expected frequency in workers exposed at 10-1000 mg/m3 for 1-4 years (Bomski et al., 1967).
Numerous reports have attempted to evaluate the possible relationship between chlorinated drinking-water and cancer incidence. Chloroform is one of the many by-products produced when chlorine reacts with organic material in water. Although some studies have found increased risks of bladder cancer associated with long-term ingestion of chlorinated drinking-water and cumulative exposure to trihalomethanes, results were inconsistent between men and women and between smokers and non-smokers (IPCS, 2000b). Moreover, relevant studies contain little information on specific exposure, and it is not possible to attribute any excess risk specifically to chloroform (ILSI, 1997; IARC, 1999; IPCS, 2000b). Specific risks may be due to other disinfection by-products, mixtures of by-products, other water contaminants, or other factors for which chlorinated drinking-water or trihalomethanes may serve as a surrogate (IPCS, 2000b).
The toxicity of chloroform has been studied in aquatic bacteria, algae, invertebrates, fish, and amphibians.
Microorganisms can be quite sensitive to chloroform. Anaerobic digestion of sewage sludge was inhibited at 0.1 mg chloroform/litre (Jackson & Brown, 1970). Others observed inhibition of unacclimated cultures at 0.5 mg/litre; with acclimation, concentrations up to 15 mg/litre were tolerated. This study examined methane production from acetate-enriched methanogenic cultures exposed to slug doses of chloroform at initial concentrations of 0.5, 1, 2, or 2.5 mg/litre. Seeded cultures were established in oxygen-free serum bottles operated in batch or semi-continuous (50, 25, or 12.5 days solids retention time [SRT]) mode. Methane production was inhibited at all chloroform concentrations. At 0.5 mg/litre, recovery had occurred after 3 days at all SRTs. At 1 mg/litre, recovery was slower (2.5, 4, 11, and 25 days under batch and semi-continuous 50, 25, and 12.5 days SRT, respectively). Chloroform partitioned (3.1:1) between the liquid and gas phases (68% of the initial chloroform remained in the liquid phase after equilibrium) and was also reduced gradually due to liquid washout and stripping by methane production. In a separate experiment in which chloroform was gradually administered as a daily feed, methane production was unaffected at 10 mg/litre, while there was initial inhibition followed by adaptation at 15 mg/litre. At 20 mg/litre, no recovery was observed within 80 days (Yang & Speece, 1986).
Several studies on freshwater and marine algae are available. During a 6-day exposure to chloroform, an initial reduction in cell multiplication of Microcystis aeruginosa was reported at 185 mg/litre (Bringmann & Kühn, 1977, 1978). A 48-h EC10 for biomass of 225 mg/litre (EC50 560 mg/litre) was observed for the green alga Scenedesmus subspicatus (Kühn & Pattard, 1990). Reported EC50s for end-points such as cell count, biomass, and carbon dioxide uptake (photosynthesis) include 382 mg/litre for Chlamydomonas angulosa (Hutchinson et al., 1980) and >1000 mg/litre for Selenastrum capricornutum (Cowgill et al., 1989). A very brief report of screening studies noted that the 7-day EC50 for inhibition of cell division exceeded 32 mg/litre, the highest concentration tested, for four marine phytoplankton: Skeletonema costatum (a filamentous diatom), Thalassiosira pseudonana (a unicellular diatom), Glenodinium halli (a dinoflagellate), and Isochrysis galbana (a microflagellate) (Erickson & Freeman, 1978). In a static system using S. costatum, the 5-day no-observed-effect concentration (NOEC) and EC50 for cell count were 216 and 477 mg/litre, respectively (Cowgill et al., 1989). A more recent study in which a gas-tight system was used to prevent chloroform volatilization reported a 72-h EC10 of 3.61 mg/litre (and a 72-h EC50 of 13.3 mg/litre) for growth inhibition of the freshwater green alga Chlamydomonas reinhardtii. This assay also used bipartite culture flasks to ensure adequate carbon dioxide supply (Brack & Rottler, 1994).
In the only identified test on vascular plants, no effects were seen when two duckweed species, Lemna gibba and four clones of Lemna minor, were exposed within the concentration range 28-1000 mg/litre (Cowgill et al., 1991).
Among aquatic invertebrates, the rotifer Brachionus calcyciflorus was apparently particularly sensitive, with a 1-h LC50 of 2 mg/litre, but the short exposure duration reduces confidence in this result (Snell et al., 1991). For Daphnia magna, 48-h LC50s ranged from 28.9 mg/litre (US EPA, 1978) to 353 mg/litre (Cowgill & Milazzo, 1991), with most results at the lower end of the range. In 24-h exposures in a closed vessel, EC0 and EC50 values for D. magna were determined to be 48 and 79 mg/litre (nominal concentrations), respectively (Kühn et al., 1989). Using growth of D. magna as an end-point, a 16-day EC50 of 59.8 mg/litre and a 16-day NOEC of 15 mg/litre were reported (Hermens et al., 1985). In a 21-day reproduction test in D. magna, in which organisms were exposed at nominal concentrations of 1.6-200 mg/litre in a closed vessel, the NOEC was 13 mg/litre (nominal). The medium was renewed twice weekly, and the lowest analysed concentration at the NOEC was 6.3 mg/litre (Kühn et al., 1989). In the marine pink shrimp Penaeus duorarum, 72-h LC50 and NOEC values of 81.5 and 32 mg/litre, respectively, have been reported (Bentley et al., 1979). A 24-h EC50 of 30 mg/litre for immobilization was recorded in 30-h post-hatch larvae of a marine shrimp, Artemia salina (Foster & Tullis, 1984). An acute toxicity study on oyster (Crassostrea virginica) larvae reported a 48-h EC50 of 1 mg/litre, with effects on survival at concentrations as low as 50 µg/litre (Stewart et al., 1979). However, deficiencies in the analytical aspects make the validity of the study questionable (Zok et al., 1998).
Several flow-through studies have been carried out to determine the effects of chloroform on fish. In rainbow trout Oncorhynchus mykiss, 4-day post-hatching LC50s ranged from 1.24 mg/litre (200 mg calcium carbonate/litre) to 2.03 mg/litre (50 mg calcium carbonate/litre) (Birge et al., 1979). In bluegills Lepomis macrochirus, the 96-h LC50 in a flow-through study was 18.2 mg/litre (Anderson & Lusty, 1980). In a flow-through test with measured concentrations, a 96-h LC50 of 28 mg/litre was determined in dab (Limanda limanda), a marine fish species (Pearson & McConnell, 1975). No toxicity was seen in Japanese medaka fish (Oryzias latipes) exposed, in flow-through studies, to chloroform at about 21 mg/litre for 96 h or at up to 0.151 mg/litre for 9 months, but gall-bladder lesions and bile duct abnormalities (including proliferation and hyperplasia) were observed at 1.463 mg/litre in the chronic study. Chloroform did not induce liver carcinogenicity (Toussaint et al., 2001).
Amphibians appear to be quite sensitive to chloroform. In studies of toxicity to early life stages of seven species of amphibians, involving 7-9 days of total exposure, the 4-day post-hatching LC50s ranged from 0.27 mg/litre in spring peepers Hyla crucifer to >68 mg/litre in African clawed frogs Xenopus laevis. The 4-day post-hatching LC1 and LC10 values for spring peepers were 0.0019 and 0.0177 mg/litre, respectively. The LC50 and LC10 values for the second most sensitive amphibian (the leopard frog Rana pipiens) were 4.16 and 0.383 mg/litre, respectively (Birge et al., 1980; Black et al., 1982).
Very little information on the toxicity of chloroform to terrestrial microorganisms was identified. Single application of chloroform at 1000 mg/kg to a silty loam soil caused microbial respiration to increase for several days (e.g., day 4: CO2 effluxtreatment/CO2 effluxcontrol = 1.39) before returning to control levels 6 days after treatment. The same treatment applied to sandy soils caused an initial depression in microbial respiration followed by a stimulation period (e.g., day 4: CO2 effluxtreatment/CO2 effluxcontrol = 1.77) and a return to control levels 6 days after treatment (Walton et al., 1989). Fumigation treatments with chloroform (concentration not stated) apparently did not eliminate microbial populations (Alphei & Scheu, 1993).
Chloroform toxicity data are similarly limited for terrestrial invertebrates. Two studies involved contact tests with the earthworm Eisenia fetida, in which chloroform was added to filter paper (Neuhauser et al., 1985, 1986; Callahan et al., 1994). A third study demonstrated that fumigation treatments with chloroform (concentration not stated) eliminated protozoans from the soil but did not eliminate nematode (roundworm) populations (Alphei & Scheu, 1993). None of these tests is considered to be directly relevant for estimating potentially harmful concentrations in soil.
No information on the toxicity of chloroform to birds or wild mammals was identified. Data on the effects of chloroform on laboratory animals are presented in section 8.
Chloroform toxicity data in humans are extremely limited but suggest that chloroform elicits the same symptoms of toxicity in humans as in laboratory animals. Liver and kidney toxicity have been reported. Available epidemiological data do not allow conclusions to be drawn with respect to the potential carcinogenicity of chloroform in humans.
Because of the limited nature of the available human data, hazard characterization and dose-response analysis for chloroform are based primarily on studies in laboratory animals.
Acute oral toxicity in rats is moderate. Single gavage administration to rats and mice resulted in central nervous system depression and nasal lesions in rats and liver damage in both species. Acute inhalation toxicity is low in rats and female mice, but male mice are more sensitive.
Short-term oral exposure caused nasal lesions in rats and tissue changes in the liver and kidneys of mice. Exposure by inhalation for up to 7 days resulted in a proliferative response in the nasal tissues of rats and mice.
When chloroform was administered to mice by gavage or in the drinking-water for 90 days, liver effects (increases in weight and microsomal enzyme activity, lipid accumulation and vacuolation) were observed. Liver effects were seen in dogs treated orally for 7.5 years, including increased enzyme activity and fatty cyst development. Rats exposed by inhalation for 4-6 months showed liver damage (necrosis) and increased kidney weight. Cell proliferation was seen in the nasal tissues of rats and mice inhaling chloroform for 13 weeks.
In chronic toxicity/carcinogenicity studies, chloroform induced liver tumours in mice (both sexes) when given by gavage in a corn oil vehicle. It was not carcinogenic to the mouse liver when given in the drinking-water, by gavage in a toothpaste base, or by inhalation. Chloroform induced kidney tumours in male mice exposed by inhalation or (in one of four strains) by ingestion in a toothpaste vehicle, but not when given in corn oil. In rats, chloroform did not induce liver tumours when given in corn oil or by inhalation, and there was no convincing evidence of liver carcinogenicity in a drinking-water study. Chloroform induced kidney tumours in male rats treated via a corn oil vehicle and in drinking-water, but not in inhalation studies or when given (orally) in a toothpaste base. Female rats and mice were not susceptible to chloroform-induced kidney cancer. Chronic inhalation caused ossification, necrosis, hyperplasia, and metaplasia in the nasal tissues of rats and mice, but not nasal tumours.
Only limited information was available on potential toxicity to the reproductive and developmental processes. Fertility and reproduction were normal in mice given chloroform by stomach tube. When maternally toxic doses were given to pregnant rats and rabbits, the only reported effect in the offspring was reduced fetal weight in the rats. On repeated inhalation exposure of pregnant rats, adverse effects on the fetus were seen inconsistently and only at maternally toxic exposure concentrations.
Chloroform has been extensively investigated in a range of short-term screening tests for genotoxicity. End-points studied include mutation in bacteria, yeast, and Drosophila, mutation, SCE, UDS, and cell transformation in human and laboratory animal cells in culture, chromosome damage in various tissues in rats, mice, and hamsters, DNA binding, and sperm abnormalities. Although a few in vivo studies suggested the possibility of a weak ability to damage the chromosomes of rats, results in the large majority of genotoxicity studies were negative. The weight of evidence suggests that chloroform does not have direct genotoxic potential.
Available data indicate that the target organ in populations exposed occupationally to high concentrations of chloroform is similar to that in laboratory animals (i.e., the liver), but the levels at which effects occur (i.e., dysfunction and necrosis) in humans are not well documented and are inadequate as a basis to meaningfully characterize exposure-response. Laboratory animal data have been used to develop tolerable exposures for neoplastic and non-neoplastic end-points separately. Details are given in Appendix 5.
Although chloroform has produced tumours in mice and in male rats in several bioassays, the currently available data provide convincing support for the view that the mechanism of tumour induction does not depend on direct DNA damage and that chloroform can be considered to be a non-genotoxic carcinogen. The most appropriate basis for deriving tolerable human exposures to chloroform, therefore, is the application of a suitable safety factor to a NOAEL or LOAEL from an appropriate laboratory animal study.
As the liver is an established target organ in humans, the study in which fatty cysts developed in the liver of dogs given chloroform orally at 15 mg/kg body weight per day, 6 days/week, for 7.5 years was selected as the critical study. The PBPK model developed for dogs and the dose-response data for fatty cyst induction were used to predict that, for humans, a 5% increase in risk would be associated with a mean rate of metabolism per unit centrilobular region of the liver (VMRATEL 7) of 3.8 mg/litre per hour (95% lower confidence limit = 1.3 mg/litre per hour, chi-square = 0.00, degrees of freedom = 1, P-value = 1.00). It was estimated that the liver tissue dose rate of toxic metabolites would be achieved if humans were exposed, on a lifetime basis, to chloroform at 37 mg/litre in drinking-water or at 9.8 mg/m3 in air. Respective lower 95% confidence limits for these values were 12 mg/litre and 3.4 mg/m3.
To derive a tolerable daily intake (TDI) for humans, it is common practice to apply an uncertainty factor of 100 to a NOAEL from a suitable laboratory animal study. This factor is composed of two factors of 10 each, one for extrapolation between species and a second to account for any interindividual variation in sensitivity within the human population. The first factor of 10 itself consists of two subfactors, 4 and 2.5, to account for possible toxicokinetic and toxicodynamic differences between the laboratory species and humans (Health Canada, 1994; IPCS, 1994b). Since in this case the use of the PBPK model allows for the use of figures based on metabolized tissue dose, the subfactor (4) for possible toxicokinetic differences in humans and laboratory animals is accounted for. Therefore, it is appropriate to apply an uncertainty factor of about 25 (10 for intraspecies differences in toxicokinetics and toxicodynamics and 2.5 for differences in interspecies toxicodynamics) to the figure generated by the PBPK model in order to derive a TDI. The TDI for oral exposure, based on the increase in hepatic cysts, thus would be:
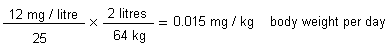
where:
The tolerable concentration (TC) for inhalation exposure would be:
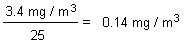
where
Although chloroform is considered to be a non-genotoxic carcinogen (and so the cancer risk would be zero at exposures lower than those that induce the critical non-neoplastic lesions), it can be useful to carry out a risk assessment based on tumour data and compare the results with the tolerable exposure figures derived from the above assessment based on non-tumour data. Using the exposure-response assessment for the combined incidence of rat renal adenomas and adenocarcinomas in Jorgenson et al. (1985) and the mean rate of metabolism in the kidney (VMRATEK 8), it was shown that VMRATEK in humans associated with a 5% increase in tumour risk (TC05), estimated on the basis of the PBPK model, is 3.9 mg/litre per hour (95% lower confidence limit = 2.5 mg/litre per hour, chi-square = 0.04, degrees of freedom = 1, P-value = 0.84). The tissue dose rate of toxic metabolites associated with this VMRATEK would result from lifetime drinking of water containing chloroform at 3247 mg/litre or continuous exposure at 147 mg chloroform/m3 in air. Respective lower 95% confidence limits for these values are 2363 mg/litre and 74 mg/m3. These figures are higher than the 12 mg/litre and 3.4 mg/m3 values derived using the dog study data, indicating that the latter are protective even against a possible human carcinogenic risk from chloroform exposure.
On similar lines, a benchmark dose (BMD) was developed for histological lesions in the rat kidney in the reanalysis of a subset of the slides from the Jorgenson et al. (1985) bioassay. The mean rate of metabolism (VMRATEK) in humans associated with a 5% increase in histological lesions characteristic of cytotoxicity was 1.7 mg/litre per hour (95% lower confidence limit = 1.4 mg/litre per hour, chi-square = 3.9, degrees of freedom = 2, P-value = 0.14). The tissue dose rate of toxic metabolites associated with this VMRATEK would result from lifetime drinking of water containing chloroform at 1477 mg/litre or from continuous exposure to chloroform at 33.3 mg/m3 in air (95% lower confidence limits were not given). Again, these figures are higher than the 12 mg/litre and 3.4 mg/m3 values derived using the dog study data, indicating that the latter are protective against the possible kidney damage that likely precedes tumour development.
The source document describes the development of a 24-h exposure scenario for Canadians that included inhalation from indoor and outdoor air and ingestion from food and tap water, together with inhalation, ingestion, and dermal exposure from a 10-min shower. The scenario was based on midpoint and 95th percentiles of concentrations in outdoor air, indoor air, air in the shower compartment, air in the bathroom after showering, tap water, and food. The midpoint values for outdoor air (background), outdoor air (commuting), indoor air, air in the shower compartment, air in the bathroom after showering, tap water, and food were 0.14 µg/m3, 0.27 µg/m3, 2.28 µg/m3, 833 µg/m3, 5 µg/m3, 47.3 µg/litre, and 0.0035 µg/g, respectively, and the corresponding 95th-percentile values were 0.31 µg/m3, 0.66 µg/m3, 8.0 µg/m3, 1950 µg/m3, 18 µg/m3, 166 µg/litre, and 0.0298 µg/g, respectively. The greatest single contributor to chloroform exposure within the 24-h period results from inhalation/dermal exposure during showering (Environment Canada & Health Canada, 2001).
The tissue doses that would result from the human exposure scenarios described above were modelled using the developed PBPK model. Daily exposure (based on 95th percentiles of concentrations in environmental media) was predicted to give rise to an estimated kidney tissue dose that was 1794 (lower 95% confidence limit 570) times lower than that associated with the tumorigenic concentration (TC05) for cancer. For non-cancer effects, the comparable margin between predicted human liver tissue concentration arising from environmental exposure and that associated with exposure at the BMD05 was 591 (lower 95% confidence limit 165). As discussed above, the tumorigenic and benchmark doses for cancer and non-cancer, respectively, are based on metabolized dose and thus account for any toxicokinetic differences between humans and (in this case, hybrid) laboratory animals. Consequently, the appropriate uncertainty factor for derivation of a tolerable intake is approximately 25. The margins between estimated exposure and tumorigenic and benchmark doses for cancer and non-cancer, respectively, for chloroform are considerably greater than 25; therefore, it was concluded that exposure of the general population is considerably lower than the level to which it is believed persons may be exposed daily over a lifetime without deleterious effect (Environment Canada & Health Canada, 2001).
Additionally, the lowest concentrations reported to induce cellular proliferation in the nasal cavities of rats and mice in short-term studies (i.e., 9.8 mg/m3) are clearly higher than the midpoint and 95th-percentile estimates of concentrations of chloroform in indoor air in Canada. These values were the same as those selected to run the human models for the kidney and liver. The midpoint and 95th-percentile estimates are 4298 and 1225 times less than the lowest value reported to induce a proliferative response in rats and mice (midpoint for indoor air = 2.28 µg/m3, 95th percentile = 8.0 µg/m3). Comparisons with midpoint and 95th-percentile estimates of concentrations during showering were considered unwarranted, since such exposures are intermittent and last for very limited periods of time during the day.
The WHO has carried out a risk assessment for swimmers who are exposed to chloroform while using indoor pools disinfected with chlorine. Preliminary data for indoor pools in several European countries indicated mean chloroform concentrations of 11-198 µg/litre (maximum 980 µg/litre). Mean concentrations in the air above the water ranged from 30 to 214 µg/m3 (maximum 1630 µg/m3). The uptake via ingestion, inhalation, and dermal routes during a swim was estimated for swimmers (children, adult recreational swimmers, and competitors) under conditions of low, moderate, and worst-case exposure. For children under moderate-exposure conditions, the total chloroform intake was estimated to be 0.035 mg/kg body weight, with 81, 14, and 5% contributed by the dermal, ingestion, and inhalation routes, respectively. Under worst-case conditions, the total uptake rose to an estimated 0.2 mg/kg body weight. For adults under moderate-exposure conditions, the estimated total uptake was 0.009 mg/kg body weight, with dermal, ingestion, and inhalation routes being responsible for 93, 1, and 6% of the total, respectively. The worst-case exposure scenario entailed an estimated total uptake of 0.047 mg/kg body weight. For competitive swimmers, estimated uptakes were similar (on a body weight basis) to those of children. It was concluded that pool users could exceed the TDI when concentrations of chloroform in the water and air are relatively high (WHO, 2000).
For many, the principal source of chloroform exposure appears to be showering. It is uncertain whether concentrations in the water at the showerhead are similar to those in the incoming cold tap water or whether concentrations measured in the water treatment plants and distribution systems are representative of the concentrations at the consumers’ taps. For indoor air, limited sampling and analysis sensitivity limits are issues. Uncertainty over ingestion from foods results from the assumption that the limited data available for a specific food item are representative of the concentrations generally found in that food item. Nevertheless, there is a high degree of certainty that chloroform is not highly concentrated in foods, since chloroform is only moderately lipophilic and does not significantly biomagnify in food-chains. Confidence in the quantitative estimates of daily ingested chloroform intakes for infants is low, due to uncertainty over the extent of breastfeeding compared with formula feeding and feeding of table-ready foods.
With respect to the toxicity of chloroform, human data are limited, but the liver is a key target organ in laboratory animals and likely to be so in humans. The interaction with other hepatotoxic agents (chemical, dietary, infectious) is probably the main cause of high interindividual variation in the sensitivity of liver to hepatotoxic chemicals, such as chloroform. Chloroform shares the putative mechanism of hepatotoxicity with many common chemicals, particularly other chlorinated hydrocarbons, which might accumulate in groundwater basins in the vicinity of chemical plants. The degree of confidence that critical effects in animal species are well characterized in the available database is high. Indeed, in numerous investigations in experimental animals by various routes of exposure, effects on the kidney, liver, and nose have been consistently observed at lowest doses. The nature of the effects has been similar and generally consistent with a mode of action that involves repeated cellular toxicity induced by oxidative metabolites and subsequent sustained regenerative proliferation. The degree of confidence in the database that supports an obligatory role of sustained cytotoxicity and persistent reparative cell proliferation in the carcinogenicity of chloroform is also high, although there are some uncertainties. The weight of evidence in this regard is strongest for hepatic and renal tumours in mice and rather more limited for renal tumours in rats.
The overall weight of evidence for the genotoxicity of chloroform is negative. A few positive results have been obtained in rats in non-standard assays.
In use of the PBPK models, the key limitations relate to the paucity of data available to serve as the basis of characterization of human metabolism, especially in the kidney. Data on possible interindividual differences within the human population are very limited.
Nearly all chloroform is released to air, but there are also some direct releases to surface water. Chloroform is also present in groundwater, particularly in the vicinity of landfills. Therefore, assessment end-points for the environmental assessment of chloroform relate to populations of terrestrial animals living near industrial sources, freshwater pelagic organisms, and groundwater-dwelling organisms (Environment Canada & Health Canada, 2001). The results of a marine risk assessment (Zok et al., 1998) are also presented here.
For each end-point, a conservative estimated exposure value (EEV9) is selected and an estimated no-effects value (ENEV10) is determined by dividing a critical toxicity value (CTV) by an application factor. A conservative quotient (EEV/ENEV) was calculated for each of the assessment end-points in order to determine whether there is potential ecological risk in the source country (Canada). If these quotients are less than 1, it can be concluded that the substance poses no significant risk to the environment, and the risk assessment is completed. If, however, the quotient is greater than 1 for a particular assessment end-point, then the risk assessment for that end-point proceeds to an analysis based on more realistic assumptions, and the probability and magnitude of effects are considered. This latter approach involves a more thorough consideration of sources of variability and uncertainty in the risk analysis. EEVs, CTVs, ENEVs, and risk quotients are summarized in Table 8.
Table 8: Summary of risk quotients for chloroform.
|
Environmental compartment |
Estimated exposure value (EEV) |
Critical toxicity value (CTV) |
Application factor |
Estimated |
Risk quotient (EEV/ENEV) |
|
Terrestrial wildlife |
110 µg/m3 |
9.8 × 103 µg/m3 |
10 |
9.8 x 102 µg/m3 |
0.11 |
|
Freshwater pelagic biota |
44 µg/litre |
65.7 µg/litre |
10 |
6.57 µg/litre |
6.7 |
|
Groundwater biota |
13.8 µg/litre |
500 µg/litre |
10 |
50 µg/litre |
0.28 |
Since chloroform does not bioaccumulate, biota are exposed via the atmosphere; given that the highest concentrations occur in air in cities, urban wildlife has the greatest potential for exposure to chloroform. Small mammals such as deer mice are likely to have the highest exposure because of their rapid respiration rate and high metabolism. Although no data have been identified for wild animals, data on effects are available for surrogates such as laboratory mammals.
For terrestrial wildlife exposed to chloroform via inhalation, the conservative EEV is 110 µg/m3, which is the highest atmospheric concentration of chloroform reported in the USA (ATSDR, 1996). This value is very conservative, being much higher than atmospheric concentrations reported for Canada. Chloroform in the atmosphere can be transported over long distances, but concentrations in Canada from this source would be much less than the EEV because of environmental transformation and dispersion.
The CTV is 9.8 × 103 µg/m3, the lowest concentration of chloroform reported to cause adverse effects in inhalation toxicity tests with laboratory animals (Templin et al., 1996b). Dividing this CTV by an application factor of 10 (to account for the extrapolation from laboratory to field conditions and interspecies and intraspecies variations in sensitivity) results in an ENEV of 9.8 × 102 µg/m3.
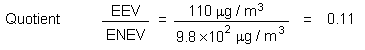
Because the conservative quotient is less than 1, it is unlikely that chloroform emissions will cause adverse effects on terrestrial wildlife in the sample country (Canada).
1) Freshwater and marine biota
The highest levels observed in Canadian surface waters have in the past been near pulp and paper mills using chlorine bleaching. The maximum concentrations in the Fraser River below the Northwood Pulp and Timber outfall in 1989 and below the Canadian Pacific Forest Products Kraft Mill in Thunder Bay in 1986 were 83 µg/litre and 200 µg/litre, respectively. Chloroform concentrations in Canadian surface water samples collected after 1989 have been much lower. The maximum reported concentration of chloroform in 984 water samples collected from British Columbia, Alberta, Ontario, and Quebec from 1990 to 1996 was 44 µg/litre, and this value is used as the EEV.
Based on the available effects data, the most sensitive freshwater pelagic biota are early life stages of spring peepers. The 4-day post-hatching LC50 for the spring peeper was 0.27 mg/litre, or 270 µg/litre (Birge et al., 1980; Black et al., 1982). Environment Canada (1997b) recommends estimating an EC25 or LC25 for the CTV and dividing by a factor of 10 to account for uncertainties arising from laboratory to field extrapolations and interspecies and intraspecies variations in sensitivity. Using an EC25 or LC25 ensures that the toxicity estimates are not model dependent, as is often the case with levels of effect below 5% (e.g., LC1) (Moore & Caux, 1997). The 4-day post-hatch LC25 for spring peepers was 65.7 µg/litre (95% confidence interval = 36.6-106 µg/litre). Dividing this value by 10 produces an ENEV of 6.57 µg/litre.
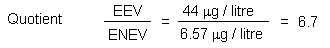
As this conservative quotient is greater than unity, it is necessary to examine the exposure and effects data more closely in order to determine the likelihood of chloroform causing harm to populations of freshwater pelagic organisms. From the 984 water samples collected from British Columbia, Alberta, Ontario, and Quebec from 1990 to 1996, the 99th- and 95th-percentile chloroform concentration values were 2.94 µg/litre and <1 µg/litre, respectively. The median value was <0.2 µg/litre. Only five of the samples contained chloroform concentrations above the ENEV value of 6.57 µg/litre: three samples (44, 31.6, and 13 µg/litre) were from Quebec, one sample (18 µg/litre) was from British Columbia, and one sample (7 µg/litre) was from Alberta. Chloroform concentrations in Canadian surface water are therefore only rarely above the ENEV.
In the toxicity study with spring peepers, the LC50, LC25, LC10, and LC1 were 270 µg/litre, 65.7 µg/litre, 17.7 µg/litre, and 1.9 µg/litre, respectively. The LC10 can be used as a good representation of threshold mortality, given that acute toxicity test protocols allow 10% mortality in control treatments. Only 2 of the 984 water samples contained concentrations substantially above the LC10 value, and a third sample contained chloroform at a concentration almost identical to the LC10 value. Other amphibians tested along with spring peepers were less sensitive. The LC10 for the second most sensitive amphibian (the leopard frog Rana pipiens) was 383 µg/litre. Other types of aquatic organisms (microorganisms, invertebrates, and fish) were less sensitive still.
Based on the available information, concentrations of chloroform in Canadian surface waters are rarely above estimated toxicity thresholds for sensitive aquatic organisms. Chloroform therefore does not appear to pose significant risks to pelagic biota in Canada.
Although there were some differences in the selection of critical studies, a similar conclusion was reached in a European risk assessment based on measured chloroform concentrations in river, coastal, and estuarine waters of several countries bordering the North Sea. A detailed review of the available toxicity data identified the lowest reliable acute toxicity value as the EC50 of 13.3 mg/litre for algae (Brack & Rottler, 1994). An assessment factor of 1000 was applied to this value to generate an acute predicted no-effect concentration (PNEC11) of 13 µg/litre. It was noted that a PNEC of 1 µg/litre would be derived by applying an assessment factor of 1000 to the less reliable 48-h EC50 of 1 mg/litre reported in oyster larvae (where effects on mortality may have been seen at concentrations as low as 50 µg/litre) (Stewart et al., 1979). For repeated exposure, the lowest reliable toxicity value was a NOEC of 3.6 mg/litre in algae (Brack & Rottler, 1994). Application of an assessment factor of 50 (according to the 2003 European Union technical guidance document [European Commission, 2003]) generated a PNEC of 72 µg/litre. This was compared with the typical predicted environmental concentrations (PECs12) (in this case derived from actual measurements) of 0.2 and 0.5 µg/litre for coastal/estuarine waters and river water, respectively. Additionally, the PNEC was compared with the worst-case PECs (again based on actual measurements) of 11.5 and 10 µg/litre for coastal/estuarine waters and river water, respectively. In all cases, the PEC/PNEC ratio was less than unity (0.0028-0.007 for typical conditions, 0.14-0.16 for worst case). With regard to seawater, the assessment noted that the sea would provide a diluting effect; thus, the PEC would be lower. Overall, it was concluded that the present concentrations of chloroform should not represent a risk to the aquatic environment. It was noted that if the PNEC was derived as 1 µg/litre from the oyster larvae study, then the PEC/PNEC ratio would still be less than unity for typical waters, but would exceed unity in worst-case situations (Zok et al., 1998).
The overall conclusion is that current concentrations of chloroform in water are not likely to cause significant toxicity to freshwater or marine organisms.
2) Groundwater-dwelling biota
No toxicity data were identified for groundwater-dwelling biota. The only available toxicity data that could reasonably be extrapolated to effects on groundwater-dwelling biota are from studies on microbial populations used in wastewater treatment. Under anaerobic conditions, Yang & Speece (1986) observed inhibition of unacclimated cultures at 500 µg/litre. A conservative ENEV of 50 µg/litre was derived by dividing this CTV by an application factor of 10. The reliability of this ENEV is uncertain, as no data were available to estimate effect levels for groundwater-dwelling invertebrates and because of the need to extrapolate from wastewater microbial populations to groundwater-dwelling populations. There are very few data available on the concentration of chloroform in groundwater not associated with the specialized conditions at a landfill site. In what may be regarded as typical of the groundwater conditions independent of the contamination found at landfill sites, 31 groundwater samples collected in British Columbia, Canada, in 1987 and 1989 were all below the 1 µg/litre detection limit (B.C. MOE, 1996). Furthermore, V. Carmichael (personal communication to Environment Canada, 1996) reported a maximum concentration of 13.8 µg chloroform/litre in 16 samples of British Columbia groundwater collected in 1992 and 1993. Using 13.8 µg/litre as the EEV, a conservative quotient would be calculated as follows:
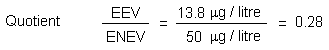
Therefore, it appears that chloroform poses little risk to groundwater-dwelling biota in Canada at locations that are not in the immediate vicinity of contaminated landfills.
Not surprisingly, the situation at some landfills in Canada is very different from the conditions existing in groundwater in general. These areas have been recognized as contaminated sites and are typically managed or have undergone remediation. They are atypical of the overall conditions that prevail and are therefore not suitable for use in assessing the impact of chloroform or other substances on the environment in general. For example, the maximum chloroform concentration first observed in groundwater at a landfill site in the Ottawa, Ontario, area in 1981 was 53 200 µg/litre (Jackson et al., 1985). This site has since undergone extensive remediation, and, in 1988, the highest concentration of chloroform in groundwater from the same sampling site was 97.1 µg/litre, while the concentration of chloroform at a sampling site approximately 50 m away was 5.8 µg/litre (C. Moralejo, personal communication to Environment Canada, 1999). The highest concentrations reported at two contaminated sites, 950 µg/litre in leachates from a chemical company landfill near Sarnia, Ontario (King & Sherbin, 1986), and 916 µg/litre in the groundwater at Ville Mercier, Quebec (Pakdel et al., 1992), were the primary figures used to determine the applicability for site remediation. Deriving quotients for these sites would not provide any further help in defining the risk that chloroform poses to the Canadian environment.
There are a number of potential sources of uncertainty in this environmental risk assessment. Although direct releases of chloroform from its use by industry are fairly well characterized, the quantity of chloroform released to the Canadian environment from wastewater treatment plants that chlorinate for disinfection is not known. Chloroform releases are highly variable, depending on the flow rate handled and on the chemical conditions at the plants. Chloroform can be produced in the environment through reactions of chlorine with organic chemicals, and the quantity released from these sources is unknown.
High surface water concentrations of chloroform were reported in the vicinity of pulp and paper mills in the 1980s. Since that time, new regulations have discouraged the use of elemental chlorine by these facilities, and it is believed that the release of chlorinated substances has dropped very significantly. For example, the total discharge of dioxins and furans from pulp and paper mills has fallen by approximately 99%. Concentrations of chloroform in water in the vicinity of pulp and paper mills have also likely decreased considerably, but few monitoring data are available. According to Environment Canada’s Environmental Effects Monitoring database, the chloroform concentration was below the 1 µg/litre detection limit in each of 85 surface water samples taken near four mills in British Columbia (Environment Canada, 1999b).
Chloroform has been reported at quite high concentrations in leachate from landfills. Concentrations as high as 950 µg/litre in leachates from a chemical company landfill near Sarnia, Ontario (King & Sherbin, 1986), have been reported, and contamination has led to reports of concentrations of 916 µg/litre in the groundwater at Ville Mercier, Quebec (Pakdel et al., 1992), and up to 25 µg/litre in contaminated groundwaters from southern Ontario (Barker, 1988). Remediation work undertaken at some landfills has considerably lowered the threat of pollution of groundwater and surface water. Uncertainty also arises from the need to extrapolate effects data from wastewater microbial populations to groundwater-dwelling populations. However, it was shown that wastewater microbial organisms acclimated readily to chloroform and subsequently were able to tolerate concentrations of chloroform up to 15 mg/litre.
Existing studies on the toxicity of chloroform to terrestrial invertebrates are few and not directly relevant for estimating potentially harmful concentrations in the soil. No information was found on the toxicity of chloroform to birds or wild mammals, but there are data on laboratory animals.
In 1998, a WHO Task Group evaluated the risks posed by compounds used for disinfection of drinking-water and their by-products. For chloroform, the Task Group concluded that the weight of evidence indicated that chloroform can induce cancer in animals only after chronic exposure to cytotoxic doses and that exposures to low concentrations of chloroform in drinking-water do not pose carcinogenic risks to humans. The NOAEL for cytolethality and regenerative hyperplasia (in the liver) in mice was 10 mg/kg body weight per day after administration of chloroform in corn oil for 3 weeks (from Larson et al., 1994c). Based on the mode of action evidence for chloroform carcinogenicity, a TDI of 10 µg/kg body weight was derived, using the NOAEL for cytotoxicity in mice and applying an uncertainty factor of 1000 (10 each for inter- and intraspecies variation and 10 for the short duration of the study) (IPCS, 2000b).
Previously, a WHO Task Group on drinking-water quality guidelines had derived a TDI of 13 µg/kg body weight, by applying an uncertainty factor of 1000 to the LOAEL of 15 mg/kg body weight per day seen in dogs given chloroform for 7.5 years. The figure was adjusted to account for 6 days/week dosing. The uncertainty factor comprised three factors of 10 to account for interspecies differences, interindividual variation, and the fact that a LOAEL was used rather than a NOAEL. Allocation of 50% of this TDI to drinking-water resulted in a guideline value for chloroform of 200 µg/litre drinking-water, assuming a person weighs 60 kg and ingests 2 litres of water per day. WHO noted that the linearized multistage model for renal tumours in rats would predict that an excess cancer risk of 1 in 100 000 would be associated with a drinking-water concentration similar to that developed on the basis of non-cancer effects (WHO, 1998).13
In a report on air quality guidelines, WHO tabulated a TDI for chloroform of 15 µg/kg body weight per day for non-carcinogenic end-points, based on the 1979 dog study. For carcinogenicity, a unit risk of 4.2 × 10-7 per µg/m3 was presented, based on the rat kidney tumour data (WHO, 1999).
The International Agency for Research on Cancer (IARC, 1999) has determined that chloroform is possibly carcinogenic to humans (Group 2B). This conclusion was reached on the basis of inadequate evidence in humans and sufficient evidence in experimental animals for the carcinogenicity of chloroform.
In a review of food additives, the Joint FAO/WHO Expert Committee on Food Additives (JECFA, 1979) concluded that chloroform should not be used as an extraction solvent.
Ade P, Guastadisegni C, Testai E, Vittozzi L (1994) Multiple activation of chloroform in kidney microsomes from male and female DBA/2J mice. Journal of Biochemical Toxicology, 9(6):289-295.
Agustin JS, Lim-Sylianco CY (1978) Mutagenic and clastogenic effects of chloroform. Bulletin: Philippine Biochemical Society, 1:17-23.
Ahmed AE, Kubic VL, Anders MW (1977) Metabolism of haloforms to carbon monoxide: I. In vitro studies. Drug Metabolism and Disposition, 5(2):198-204.
Alberta Environment (1996) Alberta Environmental Protection database. Edmonton, Alberta.
Alphei J, Scheu S (1993) Effects of biocidal treatments on biological and nutritional properties of a mull-structured woodland soil. Geoderma, 56(1-4):435-448.
Ames BN, Shigenaga MK, Gold LS (1993) DNA lesions, inducible DNA repair, and cell division: three factors in mutagenesis and carcinogenesis. Environmental Health Perspectives, 101(Suppl. 5):35-44.
Amet Y, Berthou F, Fournier G, Dreano Y, Bardou L, Cledes J, Menez J-F (1997) Cytochrome P450 4A and 2E1 expression in human kidney microsomes. Biochemical Pharmacology, 53:765-771.
Ammann P, Laethem CL, Kedderis GL (1998) Chloroform-induced cytolethality in freshly isolated male B6C3F1 mouse and F-344 rat hepatocytes. Toxicology and Applied Pharmacology, 149(2):217-225.
Anders MW, Stevens JL, Sprague RW, Shaath Z, Ahmed AE (1978) Metabolism of haloforms to carbon monoxide: II. In vitro studies. Drug Metabolism and Disposition, 6(5):556-560.
Anderson DR, Lusty EW (1980) Acute toxicity and bioaccumulation of chloroform to four species of freshwater fish. Richland, WA, Batelle Pacific Northwest Laboratory (Report 701).
Anderson L, Bayliss D, Chen CW, Coleman JP, Davidson IWF, Gray DA, Lee SD, Rosenthal S, Sakai C, Wilbur SB (1985) Health assessment document for chloroform. Research Triangle Park, NC, US Environmental Protection Agency (EPA/600/8-84/004 F).
Ashley DL, Bonin MA, Cardinali FL, McGraw JM, Wooten JV (1994) Blood concentrations of volatile organic compounds in a nonoccupationally exposed U.S. population and in groups with suspected exposure. Clinical Chemistry, 40(7):1401-1404.
ATSDR (1996) Toxicological profile for chloroform (update). Draft for public comment. Atlanta, GA, Agency for Toxic Substances and Disease Registry.
Barker JF (1988) Organic contaminants in groundwaters affected by landfill leachates. Waterloo, Ontario, University of Waterloo, Department of Earth Sciences, Institute for Groundwater Research, pp. 200-206.
Barrows ME, Petrocelli SR, Macek KJ, Carroll JJ (1980) Bioconcentration and elimination of selected water pollutants by bluegill sunfish (Lepomis macrochirus). In: Haque R, ed. Dynamics, exposure, and hazard assessment of toxic chemicals. Ann Arbor, MI, Ann Arbor Scientific Publications, pp. 379-392.
B.C. MOE (1996) SEAM database system. Victoria, British Columbia, British Columbia Ministry of Environment and Parks, Environmental Protection Service.
Benoit FM, Nicolidakis H, Cardinall C, Alleyne C, Mori B (1997) Characterization of chloroform levels in the breathing zone of showers by GC/MS. Presented at the 45th American Society for Mass Spectrometry Conference on Mass Spectrometry and Allied Topics, Palm Springs, CA.
Bentley RE, Heitmuller T, Sleight BH, Parrish PR (1979) Acute toxicity of chloroform to bluegill (Lepomis macrochirus), rainbow trout (Salmo gairdneri) and pink shrimp (Penaeus duorarum). Washington, DC, US Environmental Protection Agency, Criteria Branch (WA-6-99-1414-B) [cited in US EPA, 2003].
Bergman K (1984) Application and results of whole-body autoradiography in distribution studies of organic solvents. Critical Reviews in Toxicology, 12:59-119.
Birge WJ, Black JA, Bruser DM (1979) Toxicity of organic chemicals to embryo-larval stages of fish. Washington, DC, US Environmental Protection Agency (EPA-560/11-79-007).
Birge WJ, Black JA, Kuehne RA (1980) Effects of organic compounds on amphibian reproduction. Lexington, KY, University of Kentucky, Water Resources Research Institute (Report No. 121).
Black JA, Birge WJ, McDonnell WE, Westerman AG, Ramey BA, Bruser DM (1982) The aquatic toxicity of organic compounds to embryo-larval stages of fish and amphibians. Lexington, KY, University of Kentucky, Water Resources Research Institute (Report No. 133).
Bomski HA, Sobdewska A, Strakowski A (1967) [Toxic damage of the liver by chloroform in chemical industry workers.] Internationale Archiv für Gewerbepathologie und Gewerbehygiene, 24:127-134 (in German).
Bonnet P, Francin J-M, Gradiski D, Raoult G, Zissu D (1980) Determination of the median lethal concentration of the main chlorinated aliphatic hydrocarbons in the rat. Archives des Maladies Professionnelles de Médecine du Travail et de Sécurité Sociale, 41(6-7):317-321.
Bouwer EJ, McCarty PL (1984) Modeling of trace organics biotransformation in the subsurface. Ground Water, 22:433-440.
Bouwer EJ, Rittmann BE, McCarty PL (1981) Anaerobic degradation of halogenated 1- and 2-carbon organic compounds. Environmental Science and Technology, 15(5):596-599.
Brack W, Rottler H (1994) Toxicity testing of highly volatile chemicals with green algae — a new assay. Environmental Science and Pollution Research, 1:223-228.
Brady JF, Li D, Ishizaki H, Lee M, Ning SM, Xiao F, Yang CS (1989) Induction of cytochromes P450IIE1 and P450IIB1 by secondary ketones and the role of P450IIE1 in chloroform metabolism. Toxicology and Applied Pharmacology, 100:342-349.
Bringmann G, Kühn R (1977) Grenzwerte der schadwirkung wassergefahrdender stoffe gegen bakterien (Pseudomonas putida) und grunalgen (Scenedesmus quadricauda) im zellvermehrungshemmtest. Zeitschrift für Wasser und Abwasser Forschung, 10:87-98.
Bringmann G, Kühn F (1978) Limiting values for the noxious effects of water pollutant material to blue algae (Microcystis aeruginosa) and green algae (Scenedesmus quadricauda) in the cell multiplication inhibition test. Vom Wasser, 50:45-60.
Brown BR, Sipes IG, Sagalyn MA (1974) Mechanisms of acute hepatic toxicity: Chloroform, halothane and glutathione. Anesthesiology, 41(6):554-561.
Brown DM, Langley PF, Smith D, Taylor DC (1974) Metabolism of chloroform. I. The metabolism of 14C-chloroform by different species. Xenobiotica, 4(3):151-163.
Brown RP, Delp MD, Lindstedt SL, Rhomber LR, Beliles RP (1997) Physiological parameter values for physiologically based pharmacokinetic models. Toxicology and Industrial Health, 13:407-484.
Budavari S, ed. (2001) The Merck index, 13th ed. Whitehouse Station, NJ, Merck & Co., p. 2162.
Bull RJ, Brown JM, Meierhenry EA, Jorgenson TA, Robinson M, Stober JA (1986) Enhancement of the hepatotoxicity of chloroform in B6C3F1 mice by corn oil: implications for chloroform carcinogenesis. Environmental Health Perspectives, 69:49-58.
Butterworth BE, Templin MV, Constan AA, Sprankle CS, Wong BA, Pluta LJ, Everitt JI, Recio L (1998) Long-term mutagenicity studies with chloroform and dimethylnitrosamine in female lacI transgenic B6C3F1 mice. Environmental and Molecular Mutagenesis, 31(3):248-256.
Callahan CA, Shirazi MA, Neuhauser EF (1994) Comparative toxicity of chemicals to earthworms. Environmental Toxicology and Chemistry, 13(2):291-298.
CCPA (1992) Reducing emissions. 1992 emissions inventory and five year projections. Ottawa, Ontario, Canadian Chemical Producers’ Association.
Chu I, Secours V, Marino I, Villeneuve DC (1980) The acute toxicity of four trihalomethanes in male and female rats. Toxicology and Applied Pharmacology, 52:351-353.
Class T, Ballschmiter K (1986) Chemistry of organic traces in air. VI: Distribution of chlorinated C1-C4 hydrocarbons in air over the northern and southern Atlantic oceans. Chemosphere, 15:413-427.
Clemens TL, Hill RN, Bullock LP, Johnson WD, Sultatos LG, Vessell ES (1979) Chloroform toxicity in the mouse: Role of genetic factors and steroids. Toxicology and Applied Pharmacology, 48:117-130.
Cohen SM (1995) Role of cell proliferation in regenerative and neoplastic disease. Toxicology Letters, 82/83:15-21.
Cohen SM, Ellwein LB (1990) Cell proliferation in carcinogenesis. Science, 249:1007-1011.
Cohen SM, Ellwein LB (1991) Genetic errors, cell proliferation, and carcinogenesis. Cancer Research, 51:6493-6505.
Cohen SM, Ellwein LB (1996) Correspondence re: E. Farber, Cell proliferation as a major risk factor for cancer: a concept of doubtful validity. Cancer Res., 55: 3759-3762, 1995 [letter to editor]. Cancer Research, 56:4269-4270.
Colacci A, Bartoli S, Bonora B, Guidotti L, Lattanzi G, Mazzullo M, Niero A, Perocco P, Silingardi P, Grilli S (1991) Chloroform bioactivation leading to nucleic acids binding. Tumori, 77:285-290.
Comba ME, Palabricia VS, Kaiser KLE (1993) Tracking bleached kraft mill effluent with volatile hydrocarbons: the Jackfish Bay example. Burlington, Ontario, National Water Research Institute, Lakes Research Branch, Nearshore-Offshore Interactions Project (NWRI Contribution).
Concord Environmental Corporation (1992) Results of a national pilot survey of airborne volatile organic compounds in Canadian residences. Vols. 1 and 2. Downsview, Ontario, Concord Environmental Corporation (CE J2431).
Condie LW, Smallwood CL, Laurie RD (1983) Comparative renal and hepatotoxicity of halomethanes: bromodichloromethane, bromoform, chloroform, dibromochloromethane and methylene chloride. Drug and Chemical Toxicology, 6(96):563-578.
Conor Pacific Environmental (1998) A report on multimedia exposures to selected PSL2 substances. Prepared by Conor Pacific Environmental (formerly Bovar Environmental) and Maxxam Analytics Inc. for Health Canada, Ottawa, Ontario (Project No. 741-6705; Contract #DSS File No. 025SS.H4078-6-C574).
Constan AA, Sprankle CS, Peters JM, Kedderis GL, Everitt JI, Wong BA, Gonzalez FL, Butterworth BE (1999) Metabolism of chloroform by cytochrome P450 2E1 is required for induction of toxicity in the liver, kidney, and nose of male mice. Toxicology and Applied Pharmacology, 160:120-126.
Constan AA, Wong BA, Everitt JI, Butterworth BE (2002) Chloroform inhalation exposure conditions necessary to initiate liver toxicity in female B6C3F1 mice. Toxicological Sciences, 66:201-208.
Corley RA, Mendrala AL, Smith FA, Staats DA, Gargas ML, Conolly RB, Andersen ME, Reitz RH (1990) Development of a physiologically based pharmacokinetic model for chloroform. Toxicology and Applied Pharmacology, 103:512-527.
Cowgill UM, Milazzo DP (1991) The sensitivity of Ceriodaphnia dubia and Daphnia magna to seven chemicals utilizing the three-brood test. Archives of Environmental Contamination and Toxicology, 20(2):211-217.
Cowgill UM, Milazzo DP, Landenberger BD (1989) Toxicity of nine benchmark chemicals to Skeletonema costatum, a marine diatom. Environmental Toxicology and Chemistry, 8(5):451-456.
Cowgill UM, Milazzo DP, Landenberger BD (1991) The sensitivity of Lemna gibba G-3 and four clones of Lemna minor to eight common chemicals using a 7-day test. Research Journal of the Water Pollution Control Federation, 63(7):991-998.
Cresteil T, Beaune P, Leroux JP, Lange M, Mansuy D (1979) Biotransformation of chloroform by rat and human liver microsomes: in vitro effect on some enzyme activities and mechanisms of irreversible binding to macromolecules. Chemico-Biological Interactions, 24:153-165.
Daft JL (1988) Rapid determination of fumigant and industrial chemical residues in food. Journal of the Association of Official Analytical Chemists, 71(4):748-760.
Daniel FB, DeAngelo AB, Stober JA, Pereira MA, Olson GR (1989) Chloroform inhibition of 1,2-dimethylhydrazine-induced gastrointestinal tract tumours in the Fischer 344 rat. Fundamental and Applied Toxicology, 13:40-45.
Daniel FB, Reddy TV, Stober JA, Olson GR (1991) Site specific modulation of carcinogen-induced gastrointestinal tract nuclear anomalies in B6C3F1 mice by chloroform. Anticancer Research, 11:665-670.
Danielsson BRG, Ghantous H, Dencker L (1986) Distribution of chloroform and methyl chloroform and their metabolites in pregnant mice. Biological Research in Pregnancy, 7:77-83.
DeAngelo AB, George MH, Kilburn S, Geter DR (2003) The failure of chloroform administered in the drinking water to induce renal cell cancer in the F344/N rat. Toxicologist, S-1:84 (Abstract 405).
Delic JI, Lilly PD, MacDonald AJ, Loizou GD (2000) The utility of PBPK in the safety assessment of chloroform and carbon tetrachloride. Regulatory Toxicology and Pharmacology, 32(2):144-155.
Delker DA, Yano BL, Gollapudi BB (1999) V-Ha-ras gene expression in liver and kidney of transgenic Tg.AC mice following chemically induced tissue injury. Toxicological Sciences, 50:90-97.
Deml E, Oesterle D (1985) Dose-dependent promoting activity of chloroform in rat liver foci bioassay. Cancer Letters, 29:59-63.
Deml E, Oesterle D (1987) Dose-response of promotion by polychlorinated biphenyls and chloroform in rat liver foci bioassay. Archives of Toxicology, 60:209-211.
Derwent RG, Eggleton AEJ (1978) Halocarbon lifetimes and concentration distributions calculated using a two-dimensional tropospheric model. Atmospheric Environment, 12(6-7):1261-1269.
Diamond ML, Poulton DJ, Mackay D, Stride FA (1994) Development of a mass balance model of the fate of 17 chemicals in the Bay of Quinte. Journal of Great Lakes Research, 20(4):643-666.
Diaz-Gomez MI, Castro JA (1980) Covalent binding of chloroform metabolites to nuclear proteins: No evidence for binding to nucleic acids. Cancer Letters, 9:213-218.
Dicker E, McHugh T, Cederbaum AI (1991) Increased catalytic activity of cytochrome P-450IIE1 in pericentral hepatocytes compared to periportal hepatocytes isolated from pyrazole-treated rats. Biochimica Biophysica Acta, 1073:316-323.
Dimmer CH, Simmonds PG, Nickless G, Bassford MR (2001) Biogenic fluxes of halomethanes from Irish peatland ecosystems. Atmospheric Environment, 35:321-330.
Dix K, Borghoff SJ (1995) Comparison of gavage vehicle-dependent chloroform (CHCl3) tissue dosimetry in mice and rats. Toxicologist, 15:268 (abstract).
Dix K, Murphy JE, Borghoff SJ (1994) Vehicle dependence of chloroform (CHCl3) tissue dosimetry following oral gavage in male F344 rats. Toxicologist, 14:275 (abstract).
Dorman DC, Miller KL, D’Antonio A, James RA, Morgan KL (1997) Chloroform-induced olfactory mucosal degeneration and osseous ethmoid hyperplasia are not associated with olfactory deficits in Fischer 344 rats. Toxicology, 122(1-2):39-50.
Duprat P, Delsaut L, Gradiski D (1976) Pouvoir irritant des principaux chlorés aliphatiques sur le peau et les muqueuses oculaires du lapin. European Journal of Toxicology and Environmental Health, 9:171-177 [cited in IPCS, 1994a].
Dural NH, Peng D (1995) Competitive adsorption of chlorinated aliphatic hydrocarbons from aqueous mixtures onto soil. Cleveland, OH, Cleveland State University, Department of Chemical Engineering, pp. 528-537.
ECSA (1997) Personal communication from the European Chlorinated Solvent Association [cited in Zok et al., 1998].
EHD (1998) Exposure factors for assessing total daily intake of priority substances by the general population of Canada. Ottawa, Ontario, Health Canada, Environmental Health Directorate, Priority Substances Section (unpublished report).
EHRT (1988) Chloroform reproduction and fertility assessment in CD-1 mice when administered by gavage. Prepared by Environmental Health Research and Testing Inc., Lexington, KY, for National Toxicology Program, National Institutes of Health, Research Triangle Park, NC (Study No. NTP-86-FACB-052; NTIS Publication No. PB89-148639).
Environment Canada (1996) NAQUADAT summary report. Surface water data. Ottawa, Ontario, Environment Canada.
Environment Canada (1997a) Results of the CEPA Section 16 notice respecting the second Priority Substances List and di(2-ethylhexyl) phthalate. Hull, Quebec, Environment Canada, Commercial Chemicals Evaluation Branch, Use Patterns Section.
Environment Canada (1997b) Environmental assessments of Priority Substances under the Canadian Environmental Protection Act. Guidance manual version 1.0 — March 1997. Hull, Quebec, Environment Canada, Commercial Chemicals Evaluation Branch, Chemicals Evaluation Division (EPS 2/CC/3E).
Environment Canada (1999a) Canadian Environmental Protection Act — Priority Substances List supporting document for the environmental assessment of chloroform. Hull, Quebec, Environment Canada, Commercial Chemicals Evaluation Branch.
Environment Canada (1999b) EEM (Environmental Effects Monitoring) database. Hull, Quebec, Environment Canada.
Environment Canada, Health Canada (2000) Publication after assessment of a substance — chloroform — specified on the Priority Substances List (Subsection 77(1) of the Canadian Environmental Protection Act, 1999). Canada Gazette, Part I, 3 June 2000, pp. 1709-1711.
Environment Canada, Health Canada (2001) Canadian Environmental Protection Act, 1999. Priority Substances List assessment report. Chloroform. Ottawa, Ontario, Minister of Public Works and Government Services Canada.
Enviro-Test Laboratories (1992) Windsor area background study: analysis of food products for target organic and inorganic parameters. Prepared for the National Contaminated Sites Remediation Program, Health Protection Branch, Health and Welfare Canada, Ottawa, Ontario, 8 May 1992 (Reference No. 92-E1052.REP).
Enviro-Test Laboratories (1993) Ville-Mercier background study: analysis of food products for target organic and inorganic parameters. Prepared for the National Contaminated Sites Remediation Program, Health Protection Branch, Health and Welfare Canada, Ottawa, Ontario, 14 June 1993 (Reference No. E3-02-147.REP).
Erickson SJ, Freeman AE (1978) Toxicity screening of fifteen chlorinated and brominated compounds using four species of marine phytoplankton. In: Jolley RL, Gorchev H, Hamilton DH, eds. Water chlorination: Environmental impact and health effects. Ann Arbor, MI, Ann Arbor Scientific Publications, pp. 307-310.
Erickson MD, Harris BSH, Pellizzari ED, Tomer KB, Wadell RD, Whitaker DA (1980) Acquisition and chemical analysis of mother’s milk for selected toxic substances. Research Triangle Park, NC, Research Triangle Institute, December (Report No. 560/13-80-029).
European Commission (2003) Technical Guidance Document (Edition 2). Technical Guidance Document in support of Commission Directive 93/67/EEC on risk assessment for new notified substances, Commission Regulation (EC) No. 1488/94 on risk assessment for existing substances and Directive 98/8/EC of the European Parliament and of the Council concerning the placing of biocidal products on the market. Ispra, European Commission, Joint Research Centre. Available at http://ecb.jrc.it/php-bin/reframer.php?B=/TGD.
Farber E (1996) Correspondence re: E. Farber, Cell proliferation as a major risk factor for cancer: a concept of doubtful validity. Cancer Res., 55: 3759-3762, 1995 [reply to letter to editor]. Cancer Research, 56:4272-4274.
Foster GD, Tullis RE (1984) A QSAR between partition coefficients and the acute toxicity of naphthalene derivatives in Artemia. Aquatic Toxicology, 5(3):245-254.
Frantík E, Vodičková L, Hornychová M, Nosek M (1998) Relative subnarcotic potency of solvents predicted by partition coefficients. Central European Journal of Occupational and Environmental Medicine, 4:25-35.
Fry BJ, Taylor T, Hathway DE (1972) Pulmonary elimination of chloroform and its metabolite in man. Archives of International Pharmacodynamics and Therapeutics, 196:98-111.
Fujie K, Aoki T, Wada M (1990) Acute and subacute cytogenetic effects of the trihalomethanes on rat bone marrow cells in vivo. Mutation Research, 242:111-119.
Gearhart JM, Seckel C, Vinegar A (1993) In vivo metabolism of chloroform in B6C3F1 mice determined by the method of gas uptake: The effects of body temperature on tissue partition coefficients and metabolism. Toxicology and Applied Pharmacology, 119:258-266.
Gemma S, Facioli S, Chieco P, Sbraccia M, Testai E, Vittozzi L (1996) In vivo CHCl3 bioactivation, toxicokinetics, toxicity, and induced compensatory cell proliferation in B6C3F1 mice. Toxicology and Applied Pharmacology, 141:394-402.
Giardino NJ, Andelman JB (1996) Characterization of the emissions of trichloroethylene, chloroform, and 1,2-dibromo-3-chloropropane in a full-size experimental shower. Journal of Exposure Analysis and Environmental Epidemiology, 6(4):413-423.
Gocke E, King M-T, Eckhardt K, Wild D (1981) Mutagenicity of cosmetics ingredients licensed by the European Communities. Mutation Research, 90:91-109.
Gollapudi BB, Yano BL, Day SJ, Linscombe VA, Jackson KM, Bus JS (1999) Response of the transgenic p53+/- mouse 26-week carcinogenicity assay to chloroform. Toxicologist, 48(1-S):369 (Abstract 1740).
Goodman LS, Gilman AG (1970) The pharmacological basis of therapeutics. New York, NY, McMillan Co.
Gradiski D, Bonnet P, Raoult G, Magadur JL (1978) Toxicité aiguë comparée par inhalation des principaux solvents aliphatiques chlorés. Archives des Maladies Professionnelles de Médecine du Travail et de Sécurité Sociale, 39:249-257.
Guengerich FP, Kim DH, Iwasaki M (1991) Role of human cytochrome P-450 IIE1 in the oxidation of many low molecular weight cancer suspects. Chemical Research in Toxicology, 4:168-179.
Gürtler KR, Kleinermanns K (1994) Photooxidation of exhaust pollutants: ii. Photooxidation of chloromethanes, degradation efficiencies, quantum yields and products. Chemosphere, 28(7):1289-1298.
Hard GC, Wolf DC (1999) Re-evaluation of the chloroform 2-year drinking water bioassay in Osborne-Mendel rats indicates that sustained renal tubule injury is associated with renal tumor development. Toxicologist, 48(1-S):30 (Abstract 140).
Hard GC, Boorman GA, Wolf DC (2000) Re-evaluation of the 2-year chloroform drinking water carcinogenicity bioassay in Osborne-Mendel rats supports chronic renal tubule injury as the mode of action underlying the renal tumor response. Toxicological Sciences, 53(2):237-244.
Haselmann KF, Laturnus F, Svensmark B, Gron C (2000) Formation of chloroform in spruce forest soil — results from laboratory incubation studies. Chemosphere, 41(11):1769-1774.
Health Canada (1994) Canadian Environmental Protection Act. Human health risk assessment for Priority Substances. Ottawa, Ontario, Minister of Supply and Services Canada, 36 pp. (Catalogue No. En40-215/41E; ISBN 0-662-22126-5).
Health Canada (1999) Canadian Environmental Protection Act. Priority Substances List. Supporting documentation for chloroform: exposure assessment. Ottawa, Ontario, Health Canada, Environmental Health Directorate, Priority Substances Section, November.
Heavner DL, Morgan WT, Ogden MW (1996) Determination of volatile organic compounds and respirable suspended particulate matter in New Jersey and Pennsylvania homes and workplaces. Environment International, 22(2):159-183.
Heikes DL (1987) Purge and trap method for determination of volatile halocarbons and carbon disulfide in table-ready foods. Journal of the Association of Official Analytical Chemists, 70(2):215-226.
Heikes DL, Hopper ML (1986) Purge and trap method for determination of fumigants in whole grains, milled grain products, and intermediate grain-based foods. Journal of the Association of Official Analytical Chemists, 69(6):990-998.
Heikes DL, Jensen SR, Fleming-Jones ME (1995) Purge and trap extraction with GC-MS determination of volatile organic compounds in table-ready foods. Journal of Agricultural and Food Chemistry, 43:2869-2875.
Henderson CJ, Scott AR, Yang CS, Wolf RC (1989) Testosterone-mediated regulation of mouse renal cytochrome P-450 isoenzymes. Biochemical Journal, 278:499-503.
Hermens J, Broekhuyzen E, Canton H, Wegman R (1985) Quantitative structure activity relationships and mixture toxicity studies of alcohols and chlorohydrocarbons: Effects on growth of Daphnia magna. Aquatic Toxicology, 6(3):209-217.
Herren-Freund SL, Pereira MA (1986) Carcinogenicity of by-products of disinfection in mouse and in rat liver. Environmental Health Perspectives, 69:59-66.
Heywood R, Sortwell RJ, Noel PRB, Street AE, Prentice DE, Roe FJC, Wadsworth PF, Worden AN, Van Abbé NJ (1979) Safety evaluation of toothpaste containing chloroform. III. Long-term study in beagle dogs. Journal of Environmental Pathology and Toxicology, 2:835-851.
Hoechst (1987) Chloroform. Detection of gene mutations in somatic cells in culture. HGPRT-test with V79 cells. Frankfurt, Pharma Research Toxicology and Pathology, Hoechst Aktiengesellschaft Laboratory (Project ID 86.1085; Hoechst Report No. 87.0692; TSCA Document No. FYI-OTS-0988-0635; Microfiche No. OTS0000635).
Hoechst (1988) Inhalation embryotoxicity study of chloroform in Wistar rats. Frankfurt, Pharma Research Toxicology and Pathology, Hoechst Celanese Group. Submitted to Dow Chemical Company. Toxic Substances Control Act submission. "Initial submission: Inhalation embryotoxicity study with chloroform in Wistar rats with cover letter dated 072492" (Document ID 88-920005781; Microfiche No. OTS0544564).
Hoechst (1990) Chloroform: Supplementary inhalation embryotoxicity study in Wistar rats. Frankfurt, Pharma Development Toxicology, Hoechst Celanese Group. Submitted to Dow Chemical Company. Toxic Substances Control Act submission. "Initial submission: Chloroform: Supplementary inhalation embryotoxicity study in Wistar rats (final report) with attachments and cover letter dated 122491" (Document ID 88-920000566; Microfiche No. OTS0535017).
Hoechst (1993) Supplementary inhalation embryotoxicity study of chloroform in Wistar rats. Frankfurt, Hoechst Celanese Group. Toxic Substances Control Act submission. "Support: Supplementary inhalation embryotoxicity study of chloroform in Wistar rats — study amendments, with cover letter dated 062593" (Document ID 89-930000184; Microfiche No. OTS0535017-1).
Hoekstra EJ, de Leer EWB, Brinkmann UAT (1998) Natural formation of chloroform and brominated trihalomethanes in soil. Environmental Science and Technology, 32:3724-3729.
Hong JY, Pan J, Ning SM, Yang CS (1989) Molecular basis for the sex-related difference in renal N-nitrosodimethylamine demethylase in C3H/HeJ mice. Cancer Research, 49:2973-2979.
HSDB (2003) Hazardous substances data bank. Bethesda, MD, National Library of Medicine. Available at http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?HSDB.
Hutchinson TC, Hellebust JA, Tam D, Mackay D, Mascarenhas RA, Shiu WY (1980) The correlation of the toxicity to algae of hydrocarbons and halogenated hydrocarbons with their physical-chemical properties. Environmental Science and Research, 16: 577-586.
IARC (1999) Chloroform (Group 2B). In: International Agency for Research on Cancer (IARC) — Summaries & evaluations. Lyon, International Agency for Research on Cancer, pp. 131-182 (IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 73). Available at http://www.inchem.org/documents/iarc/vol73/73-05.html.
ICF Kaiser (1999) Development of a PBPK model for chloroform for human health risk assessment. Ruston, LA, The K.S. Crump Group, Inc., ICF Kaiser (contract report for Health Canada).
Ilett KE, Reid WD, Sipes IG, Krishna G (1973) Chloroform toxicity in mice: correlation of renal and hepatic necrosis with covalent binding of metabolites to tissue macromolecules. Experimental and Molecular Pathology, 19:215-229.
ILSI (1997) An evaluation of EPA’s proposed guidelines for carcinogen risk assessment using chloroform and dichloroacetate as case studies: Report of an expert panel. Washington, DC, International Life Sciences Institute, Health and Environmental Institute (ISBN 1-57881-002-70).
Ingelman-Sundberg M, Johansson I, Penttilä KE, Glaumann H, Lindros KO (1988) Centrilobular expression of ethanol-inducible cytochrome P-450 (IIE1) in rat liver. Biochemical and Biophysical Research Communications, 157(1):55-60.
IPCS (1994a) Chloroform. Geneva, World Health Organization, International Programme on Chemical Safety, 174 pp. (Environmental Health Criteria 163).
IPCS (1994b) Assessing human health risks of chemicals: derivation of guidance values for health-based exposure limits. Geneva, World Health Organization, International Programme on Chemical Safety (Environmental Health Criteria 170).
IPCS (2000a) Chloroform. Geneva, World Health Organization, International Programme on Chemical Safety (International Chemical Safety Card 0027).
IPCS (2000b) Disinfectants and disinfectant by-products. Geneva, World Health Organization, International Programme on Chemical Safety (Environmental Health Criteria 216).
ISO (1997) Water quality — Determination of highly volatile halogenated hydrocarbons — Gas-chromatographic methods. Geneva, International Organization for Standardization (ISO 10301:1997). Available at http://www.iso.ch/iso/en/stdsdevelopment/tc/tclist/TechnicalCommitteeStandardsListPage.TechnicalCommitteeStandardsList?COMMID=3671.
Jackson RE, Patterson RJ, Graham BW, Bahr J, Belanger D, Lockwood J, Priddle M (1985) Contaminant hydrogeology of toxic chemicals at a disposal site, Gloucester, Ontario. 1. Chemical concepts and site assessment. Ottawa, Ontario, Environment Canada, Inland Waters Directorate, National Hydrology Research Institute (NHRI Paper No. 23).
Jackson S, Brown VM (1970) Effects of toxic wastes on treatment processes and watercourses. Water Pollution Control, 69:292-313.
JECFA (1979) Chloroform. In: Summary of evaluations performed by the Joint FAO/WHO Expert Committee on Food Additives. Rome, Food and Agriculture Organization of the United Nations, and Geneva, World Health Organization, Joint FAO/WHO Expert Committee on Food Additives (JECFA Technical Report Series 648-23/19 and Food Additive Series 14-23/24). Available at http://www.inchem.org/documents/jecfa/jeceval/jec_329.htm.
Jo WK, Weisel CP, Lioy PJ (1990) Routes of chloroform exposure and body burden from showering with chlorinated tap water. Risk Analysis, 10:575-580.
Johansson I, Lindros KO, Eriksson H, Ingelman-Sundberg M (1990) Transcriptional control of CYP2E1 in the perivenous liver region and during starvation. Biochemical and Biophysical Research Communications, 173(1):331-338.
Jorgenson TA, Meierhenry EF, Rushbrook CJ, Bull RJ, Robinson M (1985) Carcinogenicity of chloroform in drinking water to male Osborne-Mendel rats and female B6C3F1 mice. Fundamental and Applied Toxicology, 5:760-769.
Kaczmar SW (1979) The occurrence and behavior of halomethanes in the aquatic environment [MSc thesis]. East Lansing, MI, Michigan State University.
Kasai T, Nishizawa T, Arito H, Nagano K, Yamamoto S, Matsushima T, Kawamoto T (2002) Acute and subchronic inhalation toxicity of chloroform in rats and mice. Journal of Occupational Health, 44:193-202.
Keegan TE, Simmons JE, Pegram RA (1998) NOAEL and LOAEL determinations of acute hepatotoxicity for chloroform and bromodichloromethane delivered in an aqueous vehicle to F344 rats. Journal of Toxicology and Environmental Health, 55(1):65-75.
Khalil MAK, Rasmussen RA (1999) Atmospheric chloroform. Atmospheric Environment, 33(7):1151-1158.
Kimura ET, Ebert DM, Dodge PW (1971) Acute toxicity and limits of solvent residue for sixteen organic solvents. Toxicology and Applied Pharmacology, 19:699-704.
Kindler TP, Chameides WL, Wine PH, Cunnold DM, Alyea FN, Franklin JA (1995) The fate of atmospheric phosgene and the stratospheric chlorine loadings of its parent compounds: CCl4, C2Cl4, C2HCl3, CH3CCl3, and CHCl3. Journal of Geophysical Research, 100(D1):1235-1251.
King L, Sherbin G (1986) Point sources of toxic organics to the upper St. Clair River. Water Pollution Research Journal of Canada, 21(3):433-446.
Klaunig JE, Ruch RJ, Pereira MA (1986) Carcinogenicity of chlorinated methane and ethane compounds administered in drinking water to mice. Environmental Health Perspectives, 69:89-95.
Klöpffer W, Haag F, Kohl E-G, Frank R (1988) Testing of the abiotic degradation of chemicals in the atmosphere: The smog chamber approach. Ecotoxicology and Environmental Safety, 15(3):298-319.
Kluwe WM (1981) The nephrotoxicity of low molecular weight halogenated alkane solvents, pesticides, and chemical intermediates. In: Hook JB, ed. Toxicology of the kidney. New York, NY, Raven Press, pp. 179-226.
Kühn R, Pattard M (1990) Results of the harmful effects of water pollutants to green algae (Scenedesmus subspicatus) in the cell multiplication inhibition test. Water Research, 24(1):31-38.
Kühn R, Pattard M, Pernak K-D, Winter A (1989) Results of the harmful effects of water pollutants to Daphnia magna in the 21 day reproduction test. Water Research, 23(4):501-510.
Land PC, Owen EL, Linde HW (1981) Morphologic changes in mouse spermatozoa after exposure to inhalational anaesthetics during early spermatogenesis. Anesthesiology, 54:53-56.
Larson JL, Wolf DC, Morgan KT, Mery S, Butterworth BE (1994a) The toxicity of 1-week exposures to inhaled chloroform in female B6C3F1 mice and male F-344 rats. Fundamental and Applied Toxicology, 22:431-446.
Larson JL, Wolf DC, Butterworth BE (1994b) Induced cytolethality and regenerative cell proliferation in the livers and kidneys of male B6C3F1 mice given chloroform by gavage. Fundamental and Applied Toxicology, 23(4):537-543.
Larson JL, Wolf DC, Butterworth BE (1994c) Induced cytotoxicity and cell proliferation in the hepatocarcinogenicity of chloroform in female B6C3F1 mice: comparison of administration by gavage in corn oil vs ad libitum in drinking water. Fundamental and Applied Toxicology, 22:90-102.
Larson JL, Sprankle CS, Butterworth BE (1994d) Lack of chloroform-induced DNA repair in vitro and in vivo in hepatocytes of female B6C3F1 mice. Environmental and Molecular Mutagenesis, 23:132-136.
Larson JL, Wolf DC, Butterworth BE (1995a) Induced regenerative cell proliferation in livers and kidneys of male F-344 rats given chloroform in corn oil by gavage or ad libitum in drinking water. Toxicology, 95:73-86.
Larson JL, Wolf DC, Mery S, Morgan KT, Butterworth BE (1995b) Toxicity and cell proliferation in the liver, kidneys and nasal passages of female F-344 rats, induced by chloroform administered by gavage. Food and Chemical Toxicology, 33(6):443-456.
Larson JL, Templin MV, Wolf DC, Jamison KC, Leininger JR, Mery S, Morgan KT, Wong BA, Conolly RB, Butterworth BE (1996) A 90-day chloroform inhalation study in female and male B6C3F1 mice: implications for cancer risk assessment. Fundamental and Applied Toxicology, 30(1):118-137.
Lilly PD (1996) A physiologically-based toxicity model of orally-administered bromodichloromethane [PhD thesis]. Chapel Hill, NC, University of North Carolina.
Löfberg B, Tjälve H (1986) Tracing tissues with chloroform-metabolizing capacity in rats. Toxicology, 39:13-35.
Lyman WJ, Reehl WF, Rosenblatt DH (1982) Handbook of chemical property estimation methods. Environmental behaviour of organic compounds. New York, NY, McGraw-Hill Book Co., pp. 9-31.
Mailhot H (1987) Prediction of algal bioaccumulation and uptake of nine organic compounds by ten physicochemical properties. Environmental Science and Technology, 21:1009-1013.
Mansuy D, Beaune P, Cresteil T, Lange M, Leroux JP (1977) Evidence for phosgene formation during liver microsomal oxidation of chloroform. Biochemical and Biophysical Research Communications, 79:513-517.
McCulloch A (2003) Chloroform in the environment: occurrence, sources, sinks and effects. Chemosphere, 50:1291-1308.
McNeal TP, Hollifield HC, Diachenko GW (1995) Survey of trihalomethanes and other volatile chemical contaminants in processed foods by purge-and-trap capillary gas chromatography with mass selective detection. Journal of the Association of Official Analytical Chemists International, 78(2):391-397.
Meek ME, Beauchamp R, Long G, Moir D, Turner L, Walker M (2002) Chloroform: exposure estimation, hazard characterization, and exposure-response analysis. Journal of Toxicology and Environmental Health, Part B, 5:283-334.
Melnick RL (1992) Does chemically-induced hepatocyte proliferation predict liver carcinogenesis? FASEB Journal, 6:2698-2706.
Melnick RL, Kohn MC, Dunnick JK, Leininger JR (1998) Regenerative hyperplasia is not required for liver tumor induction in female B6C3F1 mice exposed to trihalomethanes. Toxicology and Applied Pharmacology, 148(1):137-147.
MENVIQ (1996) Water quality database. Quebec City, Quebec, Ministère de l’environnement du Québec.
Mery S, Larson JL, Butterworth BE, Wolf DC, Harden R, Morgan KT (1994) Nasal toxicity of chloroform in male F-344 rats and female B6C3F1 mice following a 1-week inhalation exposure. Toxicology and Applied Pharmacology, 125(2):214-227.
Miller LJ, Uhler AD (1988) Volatile halocarbons in butter: elevated tetrachloroethylene levels in samples obtained in close proximity to dry-cleaning establishments. Bulletin of Environmental Contamination and Toxicology, 41:469-474.
Mirsalis JC, Tyson CK, Butterworth BE (1982) Detection of genotoxic carcinogens in the in vivo-in vitro hepatocyte DNA repair assay. Environmental Mutagenesis, 4:553-562.
Mohla S, Ahir S, Ampy FR (1988) Tissue specific regulation of renal N-nitrosodimethylamine-demethylase activity by testosterone in BALB/c mice. Biochemical Pharmacology, 37(13):2697-2702.
Moore DRJ, Caux PY (1997) Estimating low toxic effects. Environmental Toxicology and Chemistry, 16(4):794-801.
Moore DH, Chasseaud LF, Majeed SK, Prentice DE, Roe FJC, Van Abbé NJ (1982) The effect of dose and vehicle on early tissue damage and regenerative activity after chloroform administration to mice. Food and Chemical Toxicology, 20:951-954.
Morgan A, Black A, Belcher DR (1970) The excretion in breath of some aliphatic halogenated hydrocarbons following administration by inhalation. Annals of Occupational Hygiene, 13:219-233.
Morimoto K, Koizumi A (1983) Trihalomethanes induce sister chromatid exchanges in human lymphocytes in vitro and mouse bone marrow cells in vivo. Environmental Research, 32:72-79.
Munson AE, Sain LE, Sanders VM, Kauffmann BM, White KL, Page DG, Barnes DW, Borzelleca JF (1982) Toxicology of organic drinking water contaminants: trichloromethane, bromodichloromethane, dibromomethane and tribromomethane. Environmental Health Perspectives, 46:117-126.
Nakajima T, Elovaara E, Park SS, Gelboin H, Vainio H (1995a) Immunochemical detection of cytochrome P450 isozymes induced in rat liver by n-hexane, 2-hexanone and acetonyl acetone. Archives of Toxicology, 55:542-547.
Nakajima T, Elovaara E, Okino T, Gelboin H, Klockars M, Riihimäki V, Aoyama T, Vainio H (1995b) Different contributions of cytochrome P450 2E1 and P450 2B1/2 to chloroform hepatotoxicity in rat. Toxicology and Applied Pharmacology, 133:215-222.
NCI (1976) Report on carcinogenesis bioassay of chloroform. Bethesda, MD, US Department of Health, Education and Welfare, National Institutes of Health, National Cancer Institute (NTIS Publication No. PB-264 018).
Neuhauser EF, Loehr RC, Malecki MR, Milligan DL, Durkin PR (1985) The toxicity of selected organic chemicals to the earthworm Eisenia fetida. Journal of Environmental Quality, 14:383-388.
Neuhauser EF, Loehr RC, Malecki MR (1986) Contact and artificial soil tests using earthworms to evaluate the impact of wastes in soil. In: Petros JK Jr, Lacy WJ, Conway RA, eds. Hazardous and industrial solid waste: fourth symposium. Philadelphia, PA, American Society for Testing and Materials, pp. 192-203 (ASTM Special Technical Publication 886).
Nicloux MM (1906) Passage du chloroforme de la mère au foetus. Comptes Rendus des Séances de la Societé de Biologie et de ses Filiales, 60:373-375.
Nightingale PD, Malin G, Liss PS (1995) Production of chloroform and other low-molecular-weight halocarbons by some species of macroalgae. Limnology and Oceanography, 40:680-689.
NPRI (1999) National pollutant release inventory. Ottawa, Ontario, Environment Canada.
OMEE (1994) Windsor air quality study: Ambient air monitoring activities. Toronto, Ontario, Ontario Ministry of Environment and Energy.
OMOE (1990) Personal communication from N. Bazinet. Toronto, Ontario, Ontario Ministry of the Environment [cited in Canadian Council of Resource and Environment Ministers (1992) Canadian water quality guidelines: update (March 1992). Appendix X. Ottawa, Ontario, Environment Canada, Task Force on Water Quality Guidelines].
Otson R, Meek B (1996) Multimedia exposure for PSL2. Bureau of Chemical Hazards proposal. Ottawa, Ontario, Health Canada (Project No. 331312).
Page BD, Lacroix G (1993) Application of solid-phase microextraction to the headspace gas chromatographic analysis of halogenated volatiles in selected foods. Journal of Chromatography, 648:199-211.
Page BD, Conacher HBS, Salminen J, Nixon GR, Riedel G, Mori B, Gagnon J, Brouseau R (1993) Survey of bottled drinking water sold in Canada. Part 2. Selected volatile organic compounds. Journal of the Association of Official Analytical Chemists International, 76(1):26-31.
Pakdel HG, Couture C, Roy A, Masson J, Locat J, Gelinas P, Lesage S (1992) Developing methods for the analysis of toxic chemicals in soil and groundwater: The case of Ville Mercier, Quebec, Canada. In: Lesage S, Jackson RE, eds. Groundwater contamination and analysis at hazardous waste sites. New York, NY, Marcel Dekker.
Palmer AK, Street AE, Roe FJC, Worden AN, Van Abbé NJ (1979) Safety evaluation of toothpaste containing chloroform. II. Long term studies in rats. Journal of Environmental Pathology and Toxicology, 2:821-833.
Pearson CR, McConnell G (1975) Chlorinated C1 and C2 hydrocarbons in the marine environment. Proceedings of the Royal Society of London Series B, 189:305-332.
Pegram RA, Andersen ME, Warren SH, Ross TM, Claxton LD (1997) Glutathione S-transferase-mediated mutagenicity of trihalomethanes in Salmonella typhimurium: contrasting results with bromodichloromethane and chloroform. Toxicology and Applied Pharmacology, 144:183-188 [cited in ILSI, 1997].
Pereira MA (1994) Route of administration determines whether chloroform enhances or inhibits cell proliferation in the liver of B6C3F1 mice. Fundamental and Applied Toxicology, 23:87-92.
Pereira MA, Lin LC, Lippitt JM, Herren SL (1982) Trihalomethanes as initiators and promoters of carcinogenesis. Environmental Health Perspectives, 46:151-156.
Pereira MA, Knutsen GL, Herren-Freund SL (1985) Effect of subsequent treatment of chloroform or phenobarbital on the incidence of liver and lung tumours, initiated by ethylnitrosourea in 15 day old mice. Carcinogenesis, 6:203-207.
Pereira MA, Kramer PM, Ge R, Tao L (1998) Effect of chloroform and haloacetic acids on DNA methylation in liver and tumors of female B6C3F1 mice. Environmental and Molecular Mutagenesis, 31(Suppl. 29):53 (Abstract P103).
Petzold GL, Swenberg JA (1978) Detection of DNA damage induced in vivo following exposure of rats to carcinogens. Cancer Research, 38:1589-1594.
Phoon WH, Liang OK, Kee CP (1975) An epidemiological study of an outbreak of jaundice in a factory. Annals of the Academy of Medicine Singapore, 4:396-399.
Phoon WH, Goh KT, Lee LT, Tan KT, Kwok SF (1983) Toxic jaundice from occupational exposure to chloroform. Medical Journal of Malaysia, 38:31-34.
Piwoni MD, Wilson JT, Walters DM, Wilson BH, Enfield CG (1986) Behaviour of organic pollutants during rapid-infiltration of wastewater into soil: I. Processes, definition, and characterization using a microcosm. Hazardous Waste Materials, 3(1):43-55.
Pohl LR, Krishna G (1978) Deuterium isotope effect in bioactivation and hepatotoxicity of chloroform. Life Sciences, 23:1067-1072.
Pohl LR, Bhooshan B, Whittaker NF, Krishna G (1977) Phosgene: A metabolite of chloroform. Biochemical and Biophysical Research Communications, 79:684-691.
Pohl LR, Martin JL, George JW (1980) Mechanism of metabolic activation of chloroform by rat liver microsomes. Biochemical Pharmacology, 29:3271-3276.
Pohl LR, Branchflower RV, Highet RJ, Martin JL, Nunn DS, Monks TJ, George JW, Hinson JA (1981) The formation of diglutathionyl dithiocarbonate as a metabolite of chloroform, bromotrichloromethane, and carbon tetrachloride. Drug Metabolism and Disposition, 9:334-339.
Pohl LR, George JW, Satoh H (1984) Strain and sex differences in chloroform induced nephrotoxicity. Different rates of metabolism of chloroform to phosgene by the mouse kidney. Drug Metabolism and Disposition, 12(3):304-308.
Potter CL, Chang LW, DeAngelo AB, Daniel FB (1996) Effects of four trihalomethanes on DNA strand breaks, renal hyaline droplet formation and serum testosterone in male F-344 rats. Cancer Letters, 106:235-242.
Reddy TV, Daniel FB, Lin EL, Stober JA, Olson GR (1992) Chloroform inhibits the development of diethylnitrosamine-initiated, phenobarbital-promoted gamma-glutamyltranspeptidase and placental form glutathione S-transferase positive foci in rat liver. Carcinogenesis, 13:1325-1330.
Reitz RH, Fox TR, Quast JF (1982) Mechanistic considerations for carcinogenic risk estimations: Chloroform. Environmental Health Perspectives, 45:163-168.
Reitz RH, Mendrala AL, Corley RA, Quast JF, Gargas ML, Andersen ME, Staats DA, Conolly RB (1990) Estimating the risk of liver cancer associated with human exposures to chloroform using physiologically based pharmacokinetic modeling. Toxicology and Applied Pharmacology, 105:443-459.
Robbiano L, Mereto E, Morando AM, Pastore P, Brambilla G (1998) Increased frequency of micronucleated kidney cells in rats exposed to halogenated anaesthetics. Mutation Research, 413(1):1-6.
Roe FJC, Palmer AK, Worden AN, Van Abbé NJ (1979) Safety evaluation of toothpaste containing chloroform. I. Long-term studies in mice. Journal of Environmental Pathology and Toxicology, 2:799-819.
Ruddick JA, Villeneuve DC, Chu I, Valli VE (1983) A teratological assessment of four trihalomethanes in the rat. Journal of Environmental Science and Health, B18(3):333-349.
Salamone MF, Heddle JA, Katz M (1981) Mutagenic activity of 41 compounds in the in vivo micronucleus assay. In: De Serres FJ, Ashby J, eds. Evaluation of short-term tests for carcinogens: Report of the international collaborative study. Amsterdam, Elsevier/North-Holland, pp. 686-697 (Progress in Mutation Research, Vol. 1).
Samfield MM (1992) Indoor air quality data base for organic compounds. Research Triangle Park, NC, US Environmental Protection Agency, Office of Research and Development, Air and Energy Engineering Research Laboratory, 73 pp. (EPA/600/13).
Santodonato J, Bosch S, Meylan W, Becker J, Neal M (1985) Monographs on human exposure to chemicals in the workplace: Chloroform. Syracuse, NY, Syracuse Research Corporation, Center for Chemical Hazard Assessment, 40 pp. (Report No. SRC-TR-84-650) [cited in HSDB, 2003].
Sasaki T, Suzuki M, Noda K, Noguchi T, Ishida R, Oda H, Araki A, Matsushima T (1998) Mutagenicity study of carbon tetrachloride and chloroform with microbial mutagenicity test and rat liver micronucleus test. Journal of Toxicological Sciences, 23(Suppl. II):305 (Abstract P-018).
Scarratt MG, Moore RM (1999) Production of chlorinated hydrocarbons and methyl iodide by the red microalga Porphyridium purpureum. Limnology and Oceanography, 44(3):703-707.
Schwetz BA, Leong BKJ, Gehring PJ (1974) Embryo- and fetotoxicity of inhaled chloroform in rats. Toxicology and Applied Pharmacology, 28:442-451.
Sehata S, Maejima T, Watanabe M, Ogata S, Makino T, Tanaka K, Manabe S, Takaoka M (2002) Twenty-six week carcinogenicity study of chloroform in CB6F1 rasH2-transgenic mice. Toxicologic Pathology, 30:328-338.
Singh HB, Salas LJ, Smith AJ, Shigeishi H (1981) Measurements of some potentially hazardous organic chemicals in urban environments. Atmospheric Environment, 15:601-612.
Smith JH, Hook JB (1983) Mechanism of chloroform nephrotoxicity. II. In vitro evidence for renal metabolism of chloroform in mice. Toxicology and Applied Pharmacology, 70:480-485.
Smith JH, Hook JB (1984) Mechanism of chloroform nephrotoxicity: III. Renal and hepatic microsomal metabolism of chloroform in mice. Toxicology and Applied Pharmacology, 73:511-524.
Smith JH, Maita K, Sleight SD, Hook JB (1984) Effect of sex hormone status on chloroform nephrotoxicity and renal mixed function oxidase in mice. Toxicology, 30:305-316.
Smith JH, Hewitt WR, Hook JB (1985) Role of intrarenal biotransformation in chloroform-induced nephrotoxicity in rats. Toxicology and Applied Pharmacology, 79:166-174.
Smith MT, Loveridge N, Wills ED, Chayen J (1979) The distribution of glutathione in the rat liver lobule. Biochemistry Journal, 182:103-108.
Snell TW, Moffat BD, Janssen C, Persoone G (1991) Acute toxicity tests using rotifers. III. Effects of temperature, strain, and exposure time on the sensitivity of Brachionus plicatilis. Environmental Toxicology and Water Quality, 6(1):63-75.
Solomon K, Bergman H, Huggett R, Mackay D, McKague B (1994) A review and assessment of the ecological risks associated with the use of chlorine dioxide for the bleaching of pulp. Erin, Ontario, Alliance for Environmental Technology.
Stewart ME, Blogoslawski WJ, Hsu RY, Helz GR (1979) By-products of oxidative biocides: toxicity to oyster larvae. Marine Pollution Bulletin, 10:166-169 [cited in Zok et al., 1998].
Tancrède M, Yanagisawa Y, Wilson R (1992) Volatilization of volatile organic compounds from showers — 1. Analytical method and quantitative assessment. Atmospheric Environment, 26A:1103-1111.
Taylor DC, Brown DM, Kebble R, Langley PF (1974) Metabolism of chloroform: II. A sex difference in the metabolism of [14C]chloroform in mice. Xenobiotica, 4(3):165-174.
Templin MV, Jamison KC, Wolf DC, Morgan KY, Butterworth BE (1996a) Comparison of chloroform-induced toxicity in the kidneys, liver, and nasal passages of male Osborne-Mendel and F-344 rats. Cancer Letters, 104:71-78.
Templin MV, Larson JL, Butterworth BE, Jamison KC, Leininger JR, Mery S, Morgan KT, Wong BA, Wolf DC (1996b) A 90-day chloroform inhalation study in F-344 rats: profile of toxicity and relevance to cancer studies. Fundamental and Applied Toxicology, 32:109-125.
Templin MV, Jamison KC, Sprankle CS, Wolf DC, Wong BA, Butterworth BE (1996c) Chloroform-induced cytotoxicity and regenerative cell proliferation in the kidneys and liver of BDF1 mice. Cancer Letters, 108:225-231.
Templin MV, Constan AA, Wolf DC, Wong BA, Butterworth BE (1998) Patterns of chloroform-induced regenerative cell proliferation in BDF1 mice correlate with organ specificity and dose-response of tumor formation. Carcinogenesis, 19(1):187-193.
Testai E, Vittozzi L (1986) Biochemical alterations elicited in rat liver microsomes by oxidation and reduction products of chloroform metabolism. Chemico-Biological Interactions, 59:157-171.
Thompson DJ, Warner SD, Robinson VB (1974) Teratology studies on orally administered chloroform in the rat and rabbit. Toxicology and Applied Pharmacology, 29:348-357.
Tomasi A, Albano E, Biasi F, Slater TF, Vannini V, Dianzani MU (1985) Activation of chloroform and related trihalomethanes to free radical intermediates in isolated hepatocytes and in the rat in vivo as detected by the ESR-spin-trapping technique. Chemico-Biological Interactions, 55:303-316.
Topham JC (1980) Do induced sperm-head abnormalities in mice specifically identify mammalian mutagens rather than carcinogens? Mutation Research, 74:379-387.
Topham JC (1981) Evaluation of some chemicals by the sperm morphology assay. In: De Serres FJ, Ashby J, eds. Evaluation of short-term tests for carcinogens: Report of the international collaborative study. Amsterdam, Elsevier/North-Holland, pp. 718-720 (Progress in Mutation Research, Vol. 1).
Torkelson TR, Oyen F, Rowe VK (1976) The toxicity of chloroform as determined by single and repeated exposure of laboratory animals. American Industrial Hygiene Association Journal, 37:697-705.
Toussaint MW, Rosencrance AB, Brennan LM, Wolfe MJ, Hoffmann FJ, Gardner HS (2001) Chronic toxicity of chloroform to Japanese medaka fish. Environmental Health Perspectives, 109(1):35-40.
Tsuchimoto T, Matter BE (1981) Activity of coded compounds in the micronucleus test. In: De Serres FJ, Ashby J, eds. Evaluation of short-term tests for carcinogens: Report of the international collaborative study. Amsterdam, Elsevier/North-Holland, pp. 705-711 (Progress in Mutation Research, Vol. 1).
Tsutsumi M, Lasker JM, Shimizu M, Rosman AS, Lieber CS (1989) The intralobular distribution of ethanol-inducible P450IIE1 in rat and human liver. Hepatology, 10(4):437-446 [cited in ILSI, 1997].
Tumasonis CF, McMartin DN, Bush B (1987) Toxicity of chloroform and bromodichloromethane when administered over a lifetime in rats. Journal of Environmental Pathology, Toxicology and Oncology, 7(4):55-64.
Tyson CA, Hawk-Prather K, Story DL, Gould DH (1983) Correlations of in vitro and in vivo hepatotoxicity for five haloalkanes. Toxicology and Applied Pharmacology, 70:289-302.
Uehleke H, Werner T (1975) A comparative study on the irreversible binding of labelled halothane trichlorofluoromethane, chloroform, and carbon tetrachloride to hepatic protein and lipids in vitro and in vivo. Archives of Toxicology, 34(4):289-308.
US EPA (1978) Ambient water quality criteria: Chloroform. Washington, DC, US Environmental Protection Agency, Environmental Criteria and Assessment Office (PB-292 427).
US EPA (1980) Effects of chloroform in the drinking water of rats and mice: Ninety-day subacute toxicity study. Conducted by SRI International, Menlo Park, CA, for Office of Research and Development, US Environmental Protection Agency (Report No. EPA-600/1-80-030; NTIS Publication No. PB80-219108).
US EPA (1984) Health assessment document for chloroform. Washington, DC, US Environmental Protection Agency (Report No. EPA-6008-84-004A).
US EPA (1996) Proposed guidelines for carcinogen risk assessment; notice. US Environmental Protection Agency. Federal Register, 61:17960-18011.
US EPA (2003) Ecotox database. US Environmental Protection Agency. Available at http://www.epa.gov/ecotox/.
Van Beelen P, Van Keulen F (1990) The kinetics of the degradation of chloroform and benzene in anaerobic sediment from the river Rhine. Hydrobiological Bulletin, 24(1):13-22.
Van Beelen P, Van Vlaardingen PLA (1993) The mineralization of chloroform in river sediments. Netherlands Journal of Aquatic Ecology, 27(1):51-58.
Varma MM, Ampy FR, Verma K, Talbot WW (1988) In vitro mutagenicity of water contaminants in complex mixtures. Journal of Applied Toxicology, 8(4):243-248.
Veith GD, Macek KJ, Petrocelli SR, Carroll J (1978) An evaluation of using partition coefficients and water solubility to estimate bioconcentration factors for organic chemicals in fish. In: Eaton JG, Parrish PR, Hendricks AC, eds. Aquatic toxicology. Proceedings of the 3rd Annual Symposium on Aquatic Toxicology. Philadelphia, PA, American Society for Testing and Materials, pp. 116-129 (ASTM Special Technical Publication 707).
Verschueren U (1983) Handbook of environmental data on organic chemicals, 2nd ed. New York, NY, Van Nostrand Reinhold Company, pp. 367-369.
Vittozzi L, Testai E, De Biasi A (1991) Multiple bioactivation of chloroform: A comparison between man and experimental animals. Advances in Experimental Medicine and Biology, 283: 665-667.
Wallace LA (1997) Human exposure and body burden for chloroform and other trihalomethanes. Critical Reviews in Environmental Science and Technology, 27(2):113-194.
Walton BT, Anderson TA, Hendricks MS, Talmage SS (1989) Physicochemical properties as predictors of organic chemical effects on soil microbial respiration. Environmental Toxicology and Chemistry, 8(1):53-64.
WHO (1998) Guidelines for drinking-water quality, 2nd ed. Addendum to Volume 2. Geneva, World Health Organization, pp. 255-275.
WHO (1999) Air quality guidelines. Geneva, World Health Organization.
WHO (2000) Chemical hazards. In: Guidelines for safe recreational-water environments. Vol. 2. Swimming pools, spas and similar recreational-water environments. Final draft for consultation, August. Geneva, World Health Organization. Available at http://www.who.int/water_sanitation_health/bathing/bathing2/en/.
Williams DT, Lebel GL, Benoit FM (1995) A national survey of chlorinated disinfection by-products in Canadian drinking water. Ottawa, Ontario, Health Canada, Environmental Health Directorate (Report 95-EHD-197; Catalogue H46-2/95-197E; ISBN 0-662-24295-5).
Wilson JT, Enfield CG, Dunlap WJ, Cosby RL, Foster DA, Baskin LB (1981) Transport and fate of selected organic pollutants in a sandy soil. Journal of Environmental Quality, 10(4):501-507.
Wilson JT, McNabb JF, Wilson BH, Noonan MJ (1983) Biotransformation of selected organic pollutants in ground water. In: Proceedings of the 39th General Meeting of the Society of Industrial Microbiology, St. Paul, MN, 14-20 August 1982. Arlington, VA, Society of Industrial Microbiology, pp. 225-233.
Winslow SG, Gerstner HB (1978) Health aspects of chloroform. A review. Drug and Chemical Toxicology, 1:259-275.
Withey JR, Karpinski K (1985) The fetal distribution of some aliphatic chlorinated hydrocarbons in the rat after vapour phase exposure. Biological Research in Pregnancy, 6:79-88.
Wolf CR, Mansuy D, Nastainczyk W, Deutschmann G, Ullrich V (1977) The reduction of polyhalogenated methanes by liver microsomal cytochrome P-450. Molecular Pharmacology, 13:698-705.
Yamamoto S (1996) Carcinogenesis study of chloroform (inhalation). Kanagawa, Japan Bioassay Research Center, Division of Experimental Toxicology (unpublished study).
Yamamoto S, Kasai T, Matsumoto M, Nishizawa T, Arito H, Nagano K, Matsushima T (2002) Carcinogenicity and chronic toxicity in rats and mice exposed to chloroform by inhalation. Journal of Occupational Health, 44:283-293.
Yang J, Speece RE (1986) The effects of chloroform toxicity on methane fermentation. Water Research, 20(10):1273-1279.
Youssefi M, Faust SD, Zenchelsky ST (1978) Rapid determination of light and halogenated hydrocarbons in urine. Clinical Chemistry, 24(7):1109-1111.
Zok S, Boutonnet J-C, de Rooij C, Garny V, Lecloux A, Papp R, Thompson RS, van Wijk D (1998) Euro Chlor risk assessment for the marine environment OSPARCOM region: North Sea — Chloroform. Environmental Monitoring and Assessment, 52:401-424.
Environment Canada, Health Canada (2001) Canadian Environmental Protection Act, 1999. Priority Substances List assessment report. Chloroform. Ottawa, Ontario, Minister of Public Works and Government Services Canada
Paper copies of the Canadian Environmental Protection Act, 1999 Priority Substances List assessment report on chloroform are available upon request from:
Inquiry Centre
Environment Canada
351 St. Joseph Blvd.
Gatineau, Quebec
Canada K1A 0H3
1-800-668-6767
An electronic pdf version may be obtained by request from PSL.LSIP@ec.gc.ca.
Unpublished supporting documentation (Environment Canada, 1999a; Health Canada, 1999), which presents additional information, is available upon request from:
Commercial Chemicals Evaluation Branch
Environment Canada
14th Floor, Place Vincent Massey
351 St. Joseph Blvd.
Gatineau, Quebec
Canada K1A 0H3
or
Environmental Health Centre
Room 104
Health Canada
Tunney’s Pasture
Ottawa, Ontario
Canada K1A 0L2
The environmental sections of this report were produced by D. Moore and L. Pirie of the Cadmus Group, Inc. on behalf of Environment Canada and were revised by D. Caldbick and K. Taylor, Environment Canada. They were reviewed by the following members of the Environmental Resource Group, established by Environment Canada to support the environmental assessment:
P. Doyle, Environment Canada
W. Hayes, Dow Chemical
K. Kaiser, Environment Canada
Environmental sections of the assessment report and the environmental supporting documentation (Environment Canada, 1999a) were also reviewed by internal reviewers at Environment Canada — namely, P. Cureton and D. Dubé — as well as by external reviewers:
D. Averill, Water Technology International Corporation
W.J. Birge, University of Kentucky
N. Bunce, University of Guelph
D.B. Carlisle, Brez-Carlisle Inc.
L. Gammie, AQUALTA
D. Gessford, Dow Chemical Company
M.D. Kercher, University of Kentucky
D.J. Price, University of Kentucky
The health-related sections of this assessment report are based in part on the deliberations of two expert groups, in which staff of Health Canada participated. These were a Task Group on chloroform of the International Programme on Chemical Safety (IPCS, 1994a) and an Expert Panel convened by the International Life Sciences Institute (ILSI) to develop case-studies for chloroform and dichloroacetic acid in the context of the revised cancer guidelines released in 1996 by the US Environmental Protection Agency (EPA) (ILSI, 1997).
The IPCS monograph on the health effects of chloroform was published in 1994. The first draft was prepared by Dr J. de Fouw of the National Institute of Public Health and Environmental Protection, Bilthoven, Netherlands, and subsequently circulated to IPCS focal points for comment. The revised draft was subsequently finalized at a meeting of the following Task Group members, held in Geneva on 15-19 November 1993:
M.W. Anders, University of Rochester, Rochester, NY, USA
D. Anderson, British Industrial Biological Research Association, Surrey, United Kingdom
R.J. Bull, Washington State University, Pullman, WA, USA
C.D. Carrington, Food and Drug Administration (FDA), Washington, DC, USA
M. Crookes, Building Research Establishment, Watford, United Kingdom
J. de Fouw, National Institute of Public Health and Environmental Protection, Bilthoven, Netherlands (Rapporteur)
E. Elovaara, Institute of Occupational Health, Helsinki, Finland
M.E. Meek, Health Canada, Ottawa, Canada (Chair)
R. Pegram, US EPA, Research Triangle Park, NC, USA
S.A. Soliman, King Saud University-Al-Qasseem, Bureidah, Saudi Arabia
L. Vittozzi, Istituto Superiore di Sanita, Rome, Italy
P.P. Yao, Chinese Academy of Preventive Medicine, Beijing, China
The ILSI Expert Panel, which first met in September 1996, was composed of the following members:
M. Andersen, ICF Kaiser International (Chair)
G. Boorman, National Institute of Environmental Health Sciences
D. Brusick, Covance Laboratories, Inc.
S. Cohen, University of Nebraska Medical Center
Y. Dragan, McArdle Laboratory for Cancer Research
C. Frederick, Rohm & Haas Company
J. Goodman, Michigan State University
G. Hard, American Health Foundation
M.E. Meek, Health Canada
E. O’Flaherty, University of Cincinnati
The final draft of the Expert Panel report on chloroform was reviewed externally by:
C. Klaassen, University of Kansas Medical Center
R. Melnick, National Institute of Environmental Health Sciences
L. Rhomberg, Harvard Center for Risk Analysis
The outcome of these assessments has been updated and considered in the context of the approach to assessment of "toxic" under Paragraph 64(c) of the Canadian Environmental Protection Act, 1999. In addition, the physiologically based pharmacokinetic (PBPK) model for animals included in ILSI (1997) was refined and a human component developed by the K.S. Crump Group (ICF Kaiser, 1999).
The contents of the health-related sections of this assessment report and the supporting documentation (Health Canada, 1999) were prepared by the following staff of Health Canada:
R. Beauchamp
G. Long
M.E. Meek
D. Moir
L. Turner
M. Walker
The section related to genotoxicity was reviewed by D. Blakey, Environmental and Occupational Toxicology Division. The PBPK model incorporated herein was reviewed externally by M. Gargas, ChemRisk, McLaren Hart Inc.
The health-related sections on toxicity in the assessment report were reviewed externally by:
M. Andersen, Colorado State University
B. Butterworth, Chemical Industry Institute of Toxicology
J. Wiltse, Office of Water, US EPA
D. Wolf, National Health and Environmental Effects Research Laboratory, US EPA
The health-related sections of the assessment report were reviewed and approved by the Health Protection Branch Risk Management meeting of Health Canada. The entire assessment report was reviewed and approved by the Environment Canada/Health Canada CEPA Management Committee.
A draft of the assessment report was made available for a 60-day public comment period (3 June to 2 August 2000) (Environment Canada & Health Canada, 2000). Following consideration of comments received, the assessment report was revised as appropriate. A summary of the comments and responses can be obtained by request from PSL.LSIP@ec.gc.ca.
The text of the assessment report was structured to address environmental effects initially (relevant to determination of "toxic" under Paragraphs 64(a) and (b)), followed by effects on human health (relevant to determination of "toxic" under Paragraph 64(c)).
In February 2003, a comprehensive literature search was conducted by Toxicology Advice & Consulting Ltd in order to identify critical data published since publication of the source document. Databases searched included ChemIDplus (the ChemIDplus system searches and/or identifies literature from a wide range of on-line databases and databanks, including ATSDR, CANCERLIT, CCRIS, DART/ETIC, GENE-TOX, HSDB, IRIS, MEDLINE, TOXLINE Core, TOXLINE Special, and TSCA); INCHEM (the INCHEM database consolidates information from a number of intergovernmental organizations, including the Joint FAO/WHO Expert Committee on Food Additives, the Joint Meeting on Pesticide Residues, the International Agency on Research in Cancer, Chemical Inventory System, Environmental Health Criteria monographs, and Screening Information Data Sets); Registry of Toxic Effects of Chemical Substances; and EPA Toxicological Profiles.
A substantial amount of information has been published on chloroform in the last 3 years. However, judging from information presented in the above sources (usually only a title or abstract), few new papers appear to be critical in regard to the preparation of this CICAD. Critical papers were purchased, assessed, and included in the CICAD, where appropriate, by Toxicology Advice & Consulting Ltd.
The draft CICAD on chloroform was sent for review to institutions and organizations identified by IPCS after contact with IPCS national Contact Points and Participating Institutions, as well as to identified experts. Comments were received from:
M. Baril, Institut de Recherche en Santé et en Sécurité du Travail, Montreal, Canada
R. Benson, Drinking Water Program, US Environmental Protection Agency, Denver, CO, USA
R. Chhabra, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA
C. Cowles, Health and Safety Executive, Bootle, Merseyside, United Kingdom
E. Elovaara, Institute of Occupational Health, Helsinki, Finland
E. Frantik, National Institute of Public Health, Prague, Czech Republic
R. Gatehouse, Environment Australia, Canberra, Australia
P. Howe, Centre for Ecology and Hydrology, Monks Wood, United Kingdom
E. Ohanian, Health and Ecological Criteria Division, US Environmental Protection Agency, Washington, DC, USA
N. Sugawara, Japan Chemical Industry Association, Tokyo, Japan
G. Ungvary, National Centre for Public Health, Budapest, Hungary
K. Victorin, Karolinska Institute, Stockholm, Sweden
J. Weder, National Industrial Chemicals Notification and Assessment Scheme, Sydney, Australia
D. Wolf, Environmental Carcinogenesis Division, US Environmental Protection Agency, Research Triangle Park, NC, USA
K. Ziegler-Skylakakis, European Union, Luxembourg
Varna, Bulgaria
8-11 September 2003
Members
Dr I. Benchev, Sofia, Bulgaria
Dr R. Chhabra, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA
Dr C. De Rosa, Agency for Toxic Substances and Disease Registry, Centers for Disease Control and Prevention, Atlanta, GA, USA
Dr S. Dobson, Centre for Ecology and Hydrology, Monks Wood, Abbots Ripton, Huntingdon, Cambridgeshire, United Kingdom
Dr G. Dura, National Institute of Environment, József Fodor Public Health Centre, Budapest, Hungary
Dr L. Fishbein, Fairfax, VA, USA
Dr H. Gibb, National Center for Environmental Assessment, US Environmental Protection Agency, Washington, DC, USA
Dr R.F. Hertel, Federal Institute for Risk Assessment, Berlin, Germany
Mr P. Howe, Centre for Ecology and Hydrology, Monks Wood, Abbots Ripton, Huntingdon, Cambridgeshire, United Kingdom
Dr S. Ishimitsu, Division of Safety Information on Drug, Food and Chemicals, National Institute of Hygienic Sciences, Tokyo, Japan
Dr D. Kanungo, Central Insecticides Board, Directorate of Plant Protection, Quarantine & Storage, Ministry of Agriculture, Haryana, India
Dr J. Kielhorn, Fraunhofer Institute for Toxicology and Experimental Medicine, Hanover, Germany
Ms B. Meek, Environmental Health Directorate, Health Canada, Ottawa, Ontario, Canada
Dr T. Morita, Division of Safety Information on Drug, Food and Chemicals, National Institute of Hygienic Sciences, Tokyo, Japan
Mr F.K. Muchiri, Directorate of Occupational Health and Safety Services, Nairobi, Kenya
Dr L. Olsen, Biological Monitoring & Health Assessment Branch, Division of Applied Research & Technology, National Institute for Occupational Safety and Health, Cincinnati, OH, USA
Dr N. Rizov, National Center of Hygiene, Medical Ecology and Nutrition, Sofia, Bulgaria
Dr P. Schulte, Education and Information Division, National Institute for Occupational Safety and Health, Cincinnati, OH, USA
Dr J. Sekizawa, Faculty of Integrated Arts and Sciences, Tokushima University, Tokushima, Japan
Dr F. Petrova Simeonova, Sofia, Bulgaria
Dr S. Soliman, Faculty of Agriculture, Alexandria University, El Shatby, Alexandria, Egypt
Dr J. Stauber, CSIRO Energy Technology, Centre for Advanced Analytical Chemistry, Bangor, NSW, Australia
Mr P. Watts, Toxicology Advice & Consulting Ltd, Surrey, United Kingdom
Ms D. Willcocks, National Industrial Chemicals Notification and Assessment Scheme, Sydney, NSW, Australia
Dr K. Ziegler-Skylakakis, European Commission, Luxembourg
Observers
Dr S. Jacobi, Degussa AG, Fine Chemicals, Hanau-Wolfgang, Germany
Mr M. Southern, Shell International Petroleum Company Ltd, London, United Kingdom
Dr W. ten Berge, DSM, Heerlen, The Netherlands
Secretariat
Dr A. Aitio, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
Mr T. Ehara, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
|
ALAT |
alanine aminotransferase |
|
ASTM |
American Society for Testing and Materials |
|
ATPase |
adenosine triphosphatase |
|
AVCL2 |
average concentration of chloroform in the non-metabolizing centrilobular region of the liver |
|
BMC |
benchmark concentration |
|
BMD |
benchmark dose |
|
CAS |
Chemical Abstracts Service |
|
CEPA |
Canadian Environmental Protection Act |
|
CICAD |
Concise International Chemical Assessment Document |
|
CTV |
critical toxicity value |
|
CYP |
cytochrome P450 |
|
DNA |
deoxyribonucleic acid |
|
EC50 |
median effective concentration |
|
ECD |
electron capture detection |
|
EEV |
estimated exposure value |
|
EHC |
Environmental Health Criteria |
|
ENEV |
estimated no-effects value |
|
EPA |
Environmental Protection Agency (USA) |
|
FDA |
Food and Drug Administration (USA) |
|
FAO |
Food and Agriculture Organization of the United Nations |
|
GC |
gas chromatography |
|
GGTase |
gamma-glutamyl transpeptidase |
|
HCFC |
hydrochlorofluorocarbon |
|
ILO |
International Labour Organization |
|
ILSI |
International Life Sciences Institute |
|
IPCS |
International Programme on Chemical Safety |
|
Km |
substrate concentration at which the velocity of metabolic reaction is 50% of Vmax |
|
Koc |
organic carbon/water partition coefficient |
|
Kow |
octanol/water partition coefficient |
|
LC50 |
median lethal concentration |
|
LD50 |
median lethal dose |
|
LOAEL |
lowest-observed-adverse-effect level |
|
LOEL |
lowest-observed-effect level |
|
MDHS |
Methods for the Determination of Hazardous Substances (Health and Safety Executive, United Kingdom) |
|
MS |
mass spectrometry |
|
NIOSH |
National Institute for Occupational Safety and Health (USA) |
|
NOAEL |
no-observed-adverse-effect level |
|
NOEC |
no-observed-effect concentration |
|
OSHA |
Occupational Safety and Health Administration (USA) |
|
PBPK |
physiologically based pharmacokinetic |
|
PEC |
predicted environmental concentration |
|
PIM |
Poison Information Monograph |
|
PNEC |
predicted no-effect concentration |
|
SCE |
sister chromatid exchange |
|
SI |
Système international d’unités (International System of Units) |
|
SRT |
solids retention time |
|
TC |
tolerable concentration |
|
TC05 |
tumorigenic concentration05, concentration causing a 5% increase in tumour incidence over background |
|
TDI |
tolerable daily intake |
|
TWA |
time-weighted average |
|
UDS |
unscheduled DNA synthesis |
|
UNEP |
United Nations Environment Programme |
|
USA |
United States of America |
|
Vmax |
maximum velocity of metabolic reaction |
|
VRAMCOR |
maximum rate of metabolism in kidney cortex |
|
VMRATEL |
mean rate of metabolism in the liver |
|
VMRATEK |
mean rate of metabolism in the kidney |
|
WHO |
World Health Organization |
Available data indicate that the target organ in populations exposed occupationally to high concentrations of chloroform is similar to that in laboratory animals (i.e., the liver), but the levels at which effects occur (i.e., dysfunction and necrosis) in humans are not well documented and are inadequate as a basis to meaningfully characterize exposure-response. Laboratory animal data have been used to develop tolerable exposures for chloroform.
Although chloroform is carcinogenic in rodents under certain circumstances, the available data strongly support the view that chloroform induces tumours in laboratory animals as a secondary result of sustained tissue damage and persistent replicative cell proliferation, rather than as a result of direct effects on the genetic material. For such chemicals, it is probable that there is no cancer risk at exposures that do not cause the critical neoplastic damage, and tolerable exposures can be derived by application of a safety factor to a NOAEL (or LOAEL) from an appropriate study; this approach was adopted for chloroform. For comparison, tolerable exposures were also derived separately for neoplastic end-points.
Cancer
Chloroform has induced liver and kidney tumours in laboratory animals exposed by inhalation or by the oral route. There is considerable information on the potential mode of tumour induction. Available data are consistent with carcinogenicity of chloroform being a secondary consequence of sustained cytotoxicity, induced by oxidative metabolites, and associated persistent reparative cell proliferation. Hence, where chloroform caused tumours, oxidative metabolism in the target organs, sustained cytotoxicity, and persistent reparative hyperplasia are considered obligatory precursor steps.
The critical carcinogenesis bioassay is that of Jorgenson et al. (1985), in which renal tumours were observed in male Osborne-Mendel rats in an adequate and relevant study in which the route and pattern of exposure were similar to those of humans (i.e., continuously in drinking-water). In the other bioassays, liver tumours were induced in mice (males and females) only by administration of bolus doses in corn oil (NCI, 1976) or possibly (females only) following inhalation at high concentrations (Yamamoto et al., 2002). Kidney tumours have been reported in male mice following ingestion in a toothpaste vehicle (Roe et al., 1979) or inhalation (Yamamoto et al., 2002), but at concentrations in the latter study that caused severe kidney necrosis.
Unfortunately, no data on precursor lesions such as cytotoxicity or regenerative hyperplasia were collected by Jorgenson et al. (1985). A recent re-examination of a proportion of the slides from several dose groups confirmed histopathological changes consistent with the hypothesis that sustained tubular cytotoxicity and regenerative hyperplasia led to renal tubular tumour induction (Hard & Wolf, 1999; Hard et al., 2000), but data amenable for quantification of exposure-response in this investigation were limited (incidences of histological changes indicative of tubular injury in slides from the animals sacrificed at 1.5-2 years were 0, 0, 0, 50, and 100% for the 0, 200, 400, 900, and 1800 mg/litre dose groups, respectively).
In the absence of good relevant data from the cancer bioassay, the possibility that shorter-term investigations of the proliferative response might be an adequate basis for dose-response estimation was examined. A number of these investigations have studied responses in the liver and kidney of various strains of mice and rats exposed to doses and concentrations of chloroform similar to those administered in the cancer bioassays in which tumours have been observed. Unfortunately, for renal tumours in rats, most of these investigations have been conducted in the F344 rather than the Osborne-Mendel strain in which increases in renal tumours have been observed. Limited available data indicate that the proliferative response in the F344 rat is not an appropriate surrogate for characterization of exposure-response for an intermediate end-point for renal tumours in the Osborne-Mendel rat. For example, there is no indication of sex-specific variation in the proliferative response in the kidney of F344 rats (Larson et al., 1995a,b), although the increase in renal tumours in Osborne-Mendel rats is sex specific (i.e., restricted to males). In addition, in metabolic studies in F344 rats, intrarenal activation by cytochrome P450 was not implicated as a determinant of nephrotoxicity (Smith et al., 1985). Available data are also inadequate as a basis of characterization of the relative sensitivity of the two strains to cytotoxicity. In the single study in which proliferative response was examined in Osborne-Mendel rats (Templin et al., 1996a), it was concluded that they were about as susceptible as F344 rats to chloroform-induced renal injury, based on comparison 2 days following a single gavage administration. However, a statistically significant increase in labelling index was observed at a much lower dose in the Osborne-Mendel rat (10 mg/kg body weight) than in the F344 rat (90 mg/kg body weight). This latter observation may have been a function of the low value in controls for the Osborne-Mendel rats, attesting to the fact that these data are inadequate in themselves to characterize variations in sensitivity of the two strains. Rather, the results of this study contribute inasmuch as they are not inconsistent with a mode of action of induction of tumours involving tubular cell regeneration in Osborne-Mendel rats.
Since quantitative data on the incidence of precursor lesions for cancer in the strain of interest are inadequate to meaningfully characterize exposure-response, a tumorigenic concentration has been developed for this purpose, based on the incidence of tubular cell adenomas and adenocarcinomas in the bioassay of Jorgenson et al. (1985). In view of the weight of evidence for the role of oxidative metabolites in induction of requisite damage and resulting tumours, dose-response for cancer for chloroform is optimally expressed in terms of amounts or rates of formation of reactive metabolites in the target tissue. These rates have been estimated from pharmacokinetic models that include specific parameters related to metabolic rates, enzyme affinities, and enzyme tissue distribution.
Characterization of exposure-response for cancer associated with exposure to chloroform in the context of rates of formation of reactive metabolites in the target tissue is considered appropriate in view of the sufficiency of the evidence to support the following assumptions inherent in the PBPK modelling:
The "hybrid" animal model (ILSI, 1997; ICF Kaiser, 1999; Environment Canada & Health Canada, 2001) investigated four dose metrics in relation to the labelling indices (assumed to be representative of response for cytotoxicity, the intermediate end-point in induction of cancer) in the liver and kidney of exposed F344 rats. As would be expected based on the hypothesized mode of action, the fit for two of these — namely, the total amount of phosgene produced and the maximum concentration of chloroform reached in each experimental dosing interval — with proliferative response was poor. Of the other two — the mean and maximum rates of phosgene production during each experimental dosing interval — the fit with the labelling indices was best for maximum rate (ILSI, 1997).
Both maximum rate of metabolism per unit kidney cortex volume (VRAMCOR15) and mean rate of metabolism per unit kidney cortex volume during each dose interval (VMRATEK) were considered (Environment Canada & Health Canada, 2001). Although similar, the fit of the data on tumour incidence for VRAMCOR (P = 0.97) was slightly better than that for VMRATEK (P = 0.84). However, human equivalent concentrations for the former could be developed only for the 95% lower confidence limit of the tumorigenic concentration01 (TC01), since the maximum rate of human metabolism in the kidney is less than that in the rat. The maximum rate of metabolism that can be achieved in the human kidney, based on metabolic parameters included in the model (approximately 8.1 mg/litre per hour), was between the animal dose metrics associated with the benchmark concentration01 (BMC01) and the lower 95% confidence limit of the BMC05.
An exposure-response assessment was carried out for the combined incidence of renal adenomas and adenocarcinomas in the Jorgenson et al. (1985) study versus the mean rate of metabolism in the kidney (VMRATEK), fit to the following model:
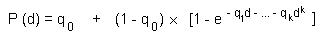
where d is dose, k is the number of dose groups, P(d) is the probability of the animal developing the effect at dose d, and qi > 0, i = 1,…, k is a parameter to be estimated. The model was fit to the incidence data, and the benchmark doses were calculated as the concentration D that satisfies:
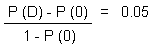
The results showed that VMRATEK in humans associated with a 5% increase in tumour risk (TC05) estimated on the basis of the PBPK model is 3.9 mg/litre per hour (95% lower confidence limit = 2.5 mg/litre per hour, chi-square = 0.04, degrees of freedom = 1, P-value = 0.84). This dose rate would result from lifetime drinking of water containing chloroform at 3247 mg/litre or continuous exposure to 147 mg chloroform/m3 in air. Respective lower 95% confidence limits for these values are 2363 mg/litre and 74 mg/m3.
For comparison (although data on dose-response were less robust than those for the cancer bioassay), a benchmark dose was developed for histological lesions in the kidney in the reanalysis of a subset of the slides from the Jorgenson et al. (1985) bioassay. The mean rate of metabolism (VMRATEK) in humans associated with a 5% increase in histological lesions characteristic of cytotoxicity was 1.7 mg/litre per hour (95% lower confidence limit = 1.4 mg/litre per hour, chi-square = 3.9, degrees of freedom = 2, P-value = 0.14). This dose rate would result from lifetime drinking of water containing 1477 mg chloroform/litre or continuous exposure to chloroform at 33.3 mg/m3 in air (95% lower confidence limits were not given).
Non-cancer effects
Repeated-dose toxicity was analysed for bolus dosing by gavage, continuous administration in drinking-water, and inhalation. Exposures were expressed as concentrations in the administered medium (for continuous administration by drinking-water and inhalation) and in mg/kg body weight, based on assumed volumes for inhalation and ingestion of drinking-water and body weights (Health Canada, 1994), with the exception of those studies in which effects were observed at site of contact (i.e., nasal lesions following inhalation).
Following exposure to chloroform by inhalation, effects at the site of contact are limiting, with proliferation in the nasal passages being reported at concentrations as low as 9.8 mg/m3 in both rats and mice for 6 or 7 h/day, for 4-7 days (Larson et al., 1996; Templin et al., 1996b). At 25 mg/m3, ossification of the nasal septum was observed in BDF1 mice exposed for 6 h/day on 5 days/week for 2 years (Yamamoto et al., 2000). At 49 mg/m3, cell proliferation and histopathological lesions were reported in the nasal passages of rats exposed for 6 h/day for 1-3 days and mice exposed for 6 h/day for 4-7 days (Mery et al., 1994; Templin et al., 1996b); ossification of the nasal turbinates was reported in rats exposed to this concentration for 6 h/day on 5 days/week for 2 years (Yamamoto et al., 2002). In one study (Larson et al., 1994b), moderate hepatic changes were observed in mice exposed at 49 mg/m3 for 6 h/day for 7 days. At concentrations of 123-147 mg/m3, effects on the kidney and liver in rats and mice, including increases in organ weights, histopathological lesions, and increases in proliferation, were observed following exposure for periods ranging from 4 days to 6 months (Torkelson et al., 1976; Larson et al., 1996; Templin et al., 1996c, 1998).
Following administration of chloroform in drinking-water, renal effects were reported at the lowest doses in rats and mice, with hepatic effects observed at higher doses. Regenerative proliferation was observed following 3 weeks’ exposure to 17 and 40 mg/kg body weight per day in rats and mice, respectively (200 mg/litre in drinking-water) (Larson et al., 1994c, 1995a). Histological alterations in the liver of F344 rats were reported at 58 mg/kg body weight per day after 4 days’ exposure (Larson et al., 1995a).
In protocols with bolus administration, the weight of the liver was affected in rats at the lowest dose following gavage in corn oil for 4 days (10 mg/kg body weight per day), while at higher doses (34 mg/kg body weight per day), there were histological changes in the liver (Larson et al., 1995a,b). At 15 mg/kg body weight per day, fatty cysts in the liver were observed in dogs exposed to chloroform in toothpaste base in gelatin capsules 6 days/week for 7.5 years. These changes are believed to possibly reflect mild toxicity to the hepatocytes (see main text for further discussion) (Heywood et al., 1979). At 34 mg/kg body weight per day, effects upon kidney and liver were reported in mice (Larson et al., 1994b); proliferation and lesions in the olfactory epithelium were observed at this dose in rats.
In summary, short-term exposure by inhalation resulted in cellular proliferation in nasal passages in rats and mice at concentrations as low as 9.8 mg/m3, with ossification being observed at slightly higher concentrations following long-term exposure. In short-term studies, moderate hepatic changes were observed in mice at 49 mg/m3; following both short- and long-term exposure at 123-147 mg/m3, there were multiple adverse effects in the kidney and liver in both rats and mice in several studies. Following ingestion in drinking-water, regenerative proliferation was observed following short-term exposure of mice to doses as low as 17 mg/kg body weight per day. Following bolus dosing, increases in proliferation in the liver of rats have been observed following short-term exposure at 10 mg/kg body weight per day and fatty cysts in the liver of dogs at 15 mg/kg body weight per day.
For oral exposure, therefore, lowest reported effect levels in various species for different end-points are similar and occur following bolus dosing. One of the lowest dose levels at which effects on liver and kidney have been observed is that in dogs reported by Heywood et al. (1979). As a result, a PBPK model in dogs was developed, since characterization of exposure-response for ingestion on the basis of this study is likely to be protective, although it should be considered in the context of an example, in view of the fact that effects on the liver of rodents have also been observed at similar dose levels.
Two dose metrics were investigated in exposure-response: the mean rate of metabolism per unit centrilobular region of the liver (VMRATEL) and the average concentration of chloroform in the non-metabolizing centrilobular region of the liver (AVCL2). The two dose metrics were selected in order to evaluate the possibility of the fatty cyst formation in the dogs being the result of either the solvent effects of chloroform or effects of a reactive metabolite.
The incidence of fatty cysts in this study (see Table 6 in section 8.3.1) versus VMRATEL and AVCL2 was fitted to the model in the manner described for the assessment of exposure-response for cancer described above. The fit of the data on the incidence of fatty cysts was better for VMRATEL (P = 1) than for AVCL2 (P = 0.45), supporting the assumption that a metabolite rather than chloroform itself was responsible for the observed effects. The mean rate of metabolism per unit centrilobular region of the liver (VMRATEL) in humans associated with a 5% increase in fatty cysts estimated on the basis of the PBPK model is 3.8 mg/litre per hour (95% lower confidence limit = 1.3 mg/litre per hour, chi-square = 0.00, degrees of freedom = 1, P-value = 1.00). This dose rate would result from lifetime drinking of water containing 37 mg chloroform/litre or from continuous exposure to chloroform at 9.8 mg/m3 in air. Respective lower 95% confidence limits for these values were 12 mg/litre and 3.4 mg/m3.
The tumorigenic and benchmark doses for cancer and non-cancer, respectively, are based on metabolized dose and thus account for toxicokinetic differences between humans and laboratory animals. An appropriate uncertainty factor for derivation of a tolerable intake for both cancer and non-cancer effects would therefore be about 25 (10 for intraspecies differences in toxicokinetics and toxicodynamics and 2.5 for differences in interspecies toxicodynamics) (Health Canada, 1994).
The tolerable daily intake (TDI) for oral exposure, based on the increase in hepatic cysts, thus would be:
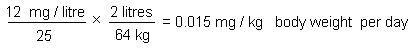
where:
The tolerable concentration (TC) for inhalation exposure would be:
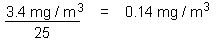
where:
Ce CICAD sur le chloroforme a été rédigé par Toxicology Advice & Consulting Ltd sur la base d’une documentation préparée par Environnement Canada et Santé Canada dans le cadre du Programme d’évaluation des produits chimiques prioritaires prévu par la Loi canadienne sur la protection de l’environnement (LCPE). Les évaluations sanitaires des substances prioritaires effectuées en application de cette loi concernent les effets que pourraient avoir ces produits sur la santé humaine en cas d’exposition indirecte dans l’environnement général ainsi que leurs effets sur l’environnement lui-même. Le document initial (Environnement Canada & Santé Canada, 2001) prend en compte les données publiées jusqu’en octobre 1999. Un dépouillement exhaustif de la littérature portant sur plusieurs bases de données en ligne et sur diverses autres sources a été effectué en février 2003 à la recherche de références importantes publiées postérieurement à celles qui sont prises en considération dans le document initial. L’appendice 1 donne des informations sur la nature de l’examen par des pairs et sur les sources documentaires. Des renseignements sur l’examen par des pairs du présent CICAD sont donnés à l’appendice 2. Ce CICAD a été adopté en tant qu’évaluation internationale lors de la réunion du Comité d’évaluation finale qui s’est tenue à Varna (Bulgarie) du 8 au 11 septembre 2003. La liste des participants à cette réunion figure à l’appendice 3. La fiche internationale sur la sécurité chimique (ICSC 0027) du chloroforme, établie par le Programme international sur la sécurité chimique (IPCS, 2000a), est également reproduite dans le présent document.
Le chloroforme (No CAS
On estime que dans l’ensemble du monde, environ 660 000 tonnes de chloroforme passent chaque année dans l’environnement et qu’à peu près 90 % des émissions sont d’origine naturelle. A la fin des années 1990, on produisait annuellement quelque 520 000 tonnes de cette substance, principalement aux Etats-Unis, dans l’Union européenne et au Japon. L’une de ses applications principales est la préparation de chlorodifluorométhane (HCFC-22) que l’on utilise comme réfrigérant (de moins en moins) et comme matière première pour la fabrication de polymères fluorés (de plus en plus). Les usines qui produisent le HCFC-22 sont susceptibles de laisser échapper du chloroforme dans l’atmosphère. Du chloroforme peut également être libéré par d’autres sources, principalement lors de l’utilisation de produits chlorés comme agents de blanchiment ou comme désinfectants dans les usines de pâte à papier et les stations de traitement de l’eau.
Le chloroforme présent sur le sol ou dans les eaux de surface s’évapore facilement et se décompose dans l’air pour donner du phosgène, du dichorométhane, du chlorure de formyle, du monoxyde et du dioxyde de carbone, et du chlorure d’hydrogène. Dans l’air, sa demi-vie va de 55 à 620 jours. Dans l’eau et le sol, il subit une lente biodégradation. Il n’y a pas de bioaccumulation notable du chloroforme dans les organismes aquatiques. On peut déceler sa présence dans l’air extérieur, généralement à une concentration inférieure à 1 μg/m3. Dans l’air intérieur, la concentration peut atteindre des valeurs environ dix fois plus fortes, et même s’élever temporairement jusqu’à 1000 μg/m3 lors d’une douche chaude si la cabine de douche est mal ventilée. Dans de l’eau de boisson, une concentration moyenne de chloroforme d’environ 10 à 90 μg/litre a été observée au Canada. La dose journalière absorbée à partir de l’eau de boisson, des aliments et de l’air inspiré a été estimée à environ 0,6-10 μg/kg de poids corporel.
Chez les mammifères, le chloroforme est rapidement absorbé, métabolisé et éliminé après ingestion, inhalation ou exposition cutanée. Le métabolisme oxydatif de cette substance, qui dépend principalement de la CYP2E1, conduit à la formation de dioxyde de carbone et de métabolites toxiques comme le phosgène et l’acide chlorhydrique. Le chloroforme est plus rapidement métabolisé par l’organisme murin que par l’organisme humain.
Non dilué, le chloroforme se révèle irritant pour la muqueuse oculaire chez l’Homme et le lapin ainsi que pour l’épiderme du lapin. Lorsqu’il est inhalé, le chloroforme a un effet anesthésiant chez l’Homme. Des lésions nasales ont également été observées chez des rats et des souris exposés par la voie respiratoire ou orale. L’expérimentation animale montre que le rein et le foie sont les principaux organes cibles de l’action toxique du chloroforme et selon des données en nombre limité, ce serait également le cas chez l’Homme. On n’a pas trouvé trace d’études épidémiologiques informatives concernant l’action toxique du chloroforme. Chez le rat, la seule preuve de cancérogénicité convaincante consiste dans une augmentation du nombre de tumeurs rénales chez des mâles qui avaient reçu du chloroforme mêlé à de l’huile de maïs ou à leur eau de boisson. On a également observé des tumeurs rénales chez des souris mâles auxquelles on avait administré du chloroforme, soit par inhalation, soit par ingestion dans de la pâte dentifrice. En outre, des tumeurs hépatiques apparaissent également chez des souris mâles et femelles lorsqu’on leur fait ingérer par gavage du chloroforme mêlé à de l’huile de maïs. Malgré de nombreuses études visant à mettre en évidence une activité génotoxique éventuelle du chloroforme, aucun signe de génotoxicité n’a été relevé, encore que selon certains travaux, le composé soit faiblement génotoxique pour le rat. Selon les éléments de preuve dont on dispose, le chloroforme ne présente pas de génotoxicité notable. On possède par contre des preuves expérimentales convaincantes que les tumeurs observées au niveau du foie et du rein chez la souris sont consécutives à des effets cytotoxiques prolongés (vraisemblablement dus à des métabolites comme le phosgène et le chlorure d’hydrogène) et à une prolifération cellulaire réparatrice persistante. Les arguments qui pourraient militer en faveur d’un mécanisme analogue dans le cas des tumeurs rénales observées chez le rat mâle sont plus limités, mais les données disponibles sont compatibles avec un tel mécanisme. Les études relatives à l’action du chloroforme sur la reproduction et le développement de diverses espèces d’animaux de laboratoire, montrent que ce composé n’a pas d’action toxique spécifique sur le développement et qu’il n’est foetotoxique qu’à des doses également toxiques pour la mère.
A la suite d’expositions répétées au chloroforme par la voie respiratoire, on a constaté que la dose la plus faible capable de produire un effet sur des animaux de laboratoire était de 9,8 mg/m3. A cette dose, on a observé une prolifération cellulaire dans les tissus des fosses nasales de rats et de souris. Dans le cas d’expositions répétées par voie orale, les doses les plus faibles produisant un effet se sont révélées du même ordre (10 à 17 mg/kg de poids corporel par jour) chez diverses espèces animales et pour des points d’aboutissement variés. Un modèle pharmacocinétique à base physiologique et les résultats d’une étude de 7,5 ans sur des chiens qui a révélé une légère hépatotoxicité (infiltrations graisseuses indicatrices d’une perturbation du métabolisme lipidique hépatique), ont été utilisés pour tenter de déterminer quelle vitesse de métabolisation du chloroforme par le foie humain (3,8 mg/litre à l’heure) produirait un rythme d’apparition de métabolites tissulaires toxiques correspondant à un accroissement de 5 % du risque. Pour que des métabolites tissulaires se forment à un tel rythme, il faudrait consommer pendant toute une vie de l’eau de boisson contenant 37 mg de chloroforme par litre ou être exposé pendant la même durée à de l’air où la concentration du composé serait de 9,8 mg/m3. Les limites inférieures de confiance à 95 % se situent respectivement à 12 mg/litre et à 3,4 mg/m3. Ces données permettent de fixer à 0,015 mg/kg de poids corporel par jour la dose ingérée tolérable et à 0,14 mg/m3 la concentration tolérable dans l’air.
Par ailleurs, on a également utilisé le modèle pharmacocinétique à base physiologique et les résultats d’une étude sur des rats dans laquelle le composé avait provoqué l’apparition de tumeurs rénales pour déterminer la vitesse de métabolisation du chloroforme qui, chez l’Homme, conduirait à une augmentation de 5 % de l’incidence des tumeurs et des lésions prétumorales. Les valeurs de ce paramètre ont été trouvées respectivement égales à 3,9 et 1,7 mg/litre à l’heure. Dans le premier cas, les limites inférieures de confiance à 95 % pour une exposition continue par l’intermédiaire de l’eau de boisson et de l’air étaient respectivement égales à 2363 mg/litre et à 74 mg/m3. Dans le second cas, la vitesse de métabolisation correspondait à la consommation continue d’eau contenant une dose de 1477 mg/litre et à l’inhalation d’air contenant 33,3 mg de chloroforme par m3 (les limites inférieures de confiance à 95 % n’ont pas été données).
Pour une caractérisation représentative du risque, on a estimé que la marge entre l’exposition estimative de la population générale canadienne et les doses de référence pour l’apparition de lésions cancéreuses et non cancéreuses est de plus de deux ordres de grandeur.
Au Canada, la concentration de chloroforme dans l’air intérieur la plus faible qui produise une prolifération cellulaire dans les fosses nasales de rats et de souris (9,8 mg/m3) est respectivement 4298 et 1225 fois plus forte que la dose estimative médiane (2,28 μg/m3) et que la dose estimative correspondant au 95ème centile (8,0 μg/m3).
On n’a pas trouvé de données sur la toxicité du chloroforme pour les oiseaux et les mammifères sauvages, mais l’expérimentation animale indique que les émissions atmosphériques de chloroforme ne représentent pas de risque important pour la faune terrestre. On ne possède pas non plus de données directement utilisables pour déterminer les concentrations dans le sol susceptibles d’être dangereuses. En ce qui concerne les organismes aquatiques, la concentration dans les eaux de surface est rarement supérieure au seuil estimatif de toxicité, même dans le cas d’espèces sensibles. Il y a une certaine indétermination au sujet des niveaux d’exposition - et par voie de conséquence, au sujet du risque pour les organismes aquatiques - à proximité de zones où des déchets industriels peuvent subir un lessivage, par exemple aux alentours d’usines de pâte à papier, de stations de traitement de l’eau et de décharges par enfouissement.
Este CICAD sobre el cloroformo, preparado por Toxicology Advice & Consulting Ltd, se basó en la documentación elaborada por los Ministerios de Medio Ambiente y de Sanidad del Canadá como parte del Programa de Sustancias Prioritarias en el marco de la Ley Canadiense de Protección del Medio Ambiente (CEPA). Las evaluaciones de sustancias prioritarias previstas en la CEPA tienen por objeto valorar los efectos potenciales para la salud humana de la exposición indirecta en el medio ambiente general, así como los efectos ecológicos. En el documento original se examinaron los datos identificados hasta octubre de 1999 (Environment Canada & Health Canada, 2001). Se realizó una búsqueda bibliográfica amplia en diversas bases de datos en línea en febrero de 2003 para localizar cualquier referencia importante publicada después de las incorporadas al documento original. La información relativa al carácter del examen colegiado del documento original y su disponibilidad figura en el apéndice 1. La información sobre el examen colegiado de este CICAD aparece en el apéndice 2. Este CICAD se aprobó como evaluación internacional en una reunión de la Junta de Evaluación Final celebrada en Varna (Bulgaria) del 8 al 11 de septiembre de 2003. La lista de participantes en esta reunión figura en el apéndice 3. También se reproduce en este documento la Ficha internacional de seguridad química (ICSC 0027) para el cloroformo, preparada por el Programa Internacional de Seguridad de las Sustancias Químicas (IPCS, 2000a).
El cloroformo (CAS Nº
La totalidad del flujo mundial de cloroformo a través del medio ambiente es de unas 660 000 toneladas al año y alrededor del 90% de las emisiones son de origen natural. A finales de los años noventa se fabricaban anualmente unas 520 000 toneladas, principalmente en los Estados Unidos, la Unión Europea y el Japón. Se utiliza sobre todo en la producción de clorodifluorometano (HCFC-22), que se usa (en cantidades cada vez menores) como refrigerante y (de manera creciente) como materia prima para la obtención de fluoropolímeros. Se pueden producir emisiones de cloroformo al medio ambiente a partir de instalaciones de clorodifluorometano. Las otras emisiones importantes de cloroformo al medio ambiente se deben a la aplicación de productos químicos clorados con fines de blanqueado y desinfección en las fábricas de pasta y de papel y en las instalaciones de tratamiento de aguas.
El cloroformo se volatiliza fácilmente a partir del suelo y del agua superficial y sufre en el aire una degradación que da lugar a fosgeno, diclorometano, cloruro de formilo, monóxido de carbono, dióxido de carbono y cloruro de hidrógeno. Sus semividas en el aire varían entre 55 y 620 días. La biodegradación en el agua y el suelo es lenta. El cloroformo no se bioacumula de manera significativa en los organismos acuáticos. Se detecta en el aire exterior, normalmente en concentraciones inferiores a 1 µg/m3. Las concentraciones en el aire de espacios cerrados pueden ser unas 10 veces más altas, pero pueden elevarse temporalmente hasta alrededor de 1000 µg/m3 durante la ducha con agua caliente en un baño poco ventilado. En el Canadá se han notificado concentraciones medias de cloroformo en el agua de bebida de unos 10-90 µg/l. La ingesta total media a partir de los alimentos, el agua de bebida y el aire fue de alrededor de 0,6-10 µg/kg de peso corporal al día.
Los mamíferos absorben, metabolizan y eliminan el cloroformo con rapidez tras la exposición oral, por inhalación y cutánea. El metabolismo oxidativo, dependiente principalmente de la CYP2E1, genera dióxido de carbono, así como los metabolitos tóxicos fosgeno y ácido clorhídrico. El metabolismo del cloroformo es mucho más rápido en los ratones que en las personas.
El cloroformo puro provocó irritación en los ojos de personas y conejos y en la piel de conejos. La inhalación de cloroformo produce anestesia en las personas. También se han detectado lesiones nasales en ratas y ratones expuestos por inhalación o por vía oral. En estudios con animales de experimentación se ha observado que los principales órganos destinatarios de la acción tóxica del cloroformo son el hígado y los riñones y hay datos limitados que parecen indicar que en las personas también son éstos los órganos más afectados. No se han encontrado estudios epidemiológicos informativos sobre el cloroformo. En biovaloraciones con animales de laboratorio, el cloroformo indujo tumores en el hígado y los riñones. En ratas, la única prueba convincente de su carcinogenicidad fue un aumento del número de tumores de riñón en los machos tras la administración de cloroformo en un excipiente de aceite de maíz o en el agua de bebida. Se observaron asimismo tumores de riñón en ratones machos expuestos por inhalación o por ingestión utilizando un dentífrico como excipiente. Además, cuando se administró a ratones machos y hembras mediante sonda cloroformo en un excipiente de aceite de maíz aparecieron tumores de hígado. En una investigación amplia de la posible genotoxicidad del cloroformo no se logró determinar ninguna actividad, aunque algunos estudios parecen indicar que podría ser ligeramente genotóxico en ratas. Un método con valor demostrativo parece indicar que el cloroformo no tiene un potencial genotóxico significativo. Hay pruebas experimentales convincentes de que los tumores de hígado y riñón observados en ratones son una consecuencia secundaria de una citotoxicidad sostenida (posiblemente debida a metabolitos como el fosgeno y el cloruro de hidrógeno) y la proliferación asociada persistente de células reparadoras. El respaldo experimental de un mecanismo similar subyacente en la aparición de tumores de riñón en ratas machos es más limitado, pero los datos disponibles son compatibles con el mecanismo propuesto. Los estudios reproductivos y del desarrollo realizados en una serie de especies de animales de laboratorio parecen indicar que el cloroformo no es una toxina especifica del desarrollo y que sólo es fetotóxico en dosis que provocan toxicidad materna.
En una exposición repetida por inhalación, la concentración más baja con efectos notificada en un estudio con animales de laboratorio fue de 9,8 mg/m3, provocando proliferación celular en los tejidos del conducto nasal de ratas y ratones. En exposiciones repetidas por vía oral, las concentraciones más bajas con efectos notificadas fueron semejantes (10-17 mg/kg de peso corporal al día) en diversas especies para distintos efectos finales. Se utilizaron un modelo farmacocinético con base fisiológica y los resultados de un estudio de 7,5 años en perros en el que se observó una toxicidad hepática leve (quistes grasos indicativos de una perturbación del metabolismo hepático de las grasas) para pronosticar la tasa de metabolización del cloroformo en el hígado humano (3,8 mg/l por hora) que produciría una tasa de dosis en los tejidos de metabolitos tóxicos asociados con un aumento del 5% del riesgo. Dicha tasa procedería de la bebida de agua con 37 mg/l de cloroformo durante toda la vida o la exposición durante toda ella a 9,8 mg de cloroformo/m3 de aire. Los límites inferiores respectivos del intervalo de confianza del 95% fueron de 12 mg/l y 3,4 mg/m3. A partir de esas cifras se obtiene una ingesta diaria por vía oral tolerable de 0,015 mg/kg de peso corporal al día y una concentración tolerable de 0,14 mg/m3 de aire.
Además, se utilizaron el modelo farmacocinético con base fisiológica y los resultados de un estudio en el que el cloroformo indujo la aparición de tumores de riñón en ratas machos para derivar tasas análogas de metabolización en las personas, con un aumento del 5% en la incidencia de tumores y lesiones precursoras de tumores. Éstas se estimaron en 3,9 y 1,7 mg/l por hora, respectivamente. En el primer caso, los límites inferiores del intervalo de confianza del 95% para la exposición continua en el agua de bebida y en el aire fueron 2363 mg/l y 74 mg/m3, respectivamente. En el segundo, la tasa metabólica fue equivalente a la exposición continua a 1477 mg/l de agua y 33,3 mg/m3 de aire (no se facilitaron límites inferiores del intervalo de confianza del 95%).
En una caracterización del riesgo de muestra para el cloroformo, los márgenes entre la exposición estimada de la población general en el Canadá y las dosis tumorígena y de referencia para el cáncer y los efectos distintos de esta enfermedad, respectivamente, fueron de más de dos órdenes de magnitud.
La concentración más baja notificada que provoca proliferación celular en la cavidad nasal de ratas y ratones (9,8 mg/m3) es 4298 y 1225 veces más elevada, respectivamente, que la estimación del punto medio (2,28 µg/m3) y el percentil 95 (8,0 µg/m3) para el cloroformo presente en el aire de espacios cerrados en el Canadá.
No se localizó información relativa a la toxicidad en aves o mamíferos silvestres, pero los datos relativos a los animales de laboratorio indican que las emisiones atmosféricas de cloroformo no plantean ningún riesgo significativo para la flora y fauna terrestres. No se disponía de ningún dato directamente pertinente para la estimación de concentraciones potencialmente peligrosas en el suelo. En los organismos acuáticos, las concentraciones en el agua superficial raramente son superiores a los umbrales de toxicidad estimados, incluso para las especies sensibles. Hay cierta incertidumbre con respecto a los niveles de exposición - y por consiguiente los posibles riesgos para los organismos acuáticos - cerca de lugares de filtración industrial, como las fábricas de pasta y de papel, las instalaciones de tratamiento de aguas y los vertederos.
ENDNOTES
See Also:
Toxicological Abbreviations
Chloroform (EHC 163, 1994)
Chloroform (HSG 87, 1994)
Chloroform (ICSC)
Chloroform (WHO Food Additives Series 14)
CHLOROFORM (JECFA Evaluation)
Chloroform (PIM 121)
Chloroform (IARC Summary & Evaluation, Supplement7, 1987)
Chloroform (IARC Summary & Evaluation, Volume 1, 1972)
Chloroform (IARC Summary & Evaluation, Volume 20, 1979)
Chloroform (IARC Summary & Evaluation, Volume 73, 1999)